/

























































































































































































Автор: Крусь Г.Н. Волокитина З.В. Шалыгина А.М.
Теги: продукты животноводства и охоты пищевое производство пищевая промышленность
ISBN: 5-10-003440-8
Год: 2000




Текст
УЧЕБНИК
Г.Н.КРУСЬ, А.М.ШАПЫГИНА,
З.В.В0Л0КИТИНА
МЕТОДЫ
ИССЛЕДОВАНИЯ
МОЛОКА
И МОЛОЧНЫХ
ПРОДУКТОВ
УЧЕБНИКИ И УЧЕБНЫЕ ПОСОБИЯ ДЛЯ СТУДЕНТОВ
ВЫСШИХ УЧЕБНЫХ ЗАВЕДЕНИЙ
Г. Н. КРУСЬ, А. М. ШАЛЫГИНА,
3. В. ВОЛОКИТИНА
МЕТОДЫ
ИССЛЕДОВАНИЯ
МОЛОКА
И МОЛОЧНЫХ
ПРОДУКТОВ
Под общей редакцией д-ра техн, наук,
проф. А. М. Шалыгиной
Рекомендовано Министерством образова-
ния Российской Федерации в качестве учеб-
ника для студентов высших учебных заведе-
ний, обучающихся по специальности «Техно-
логия молока и молочных продуктов»
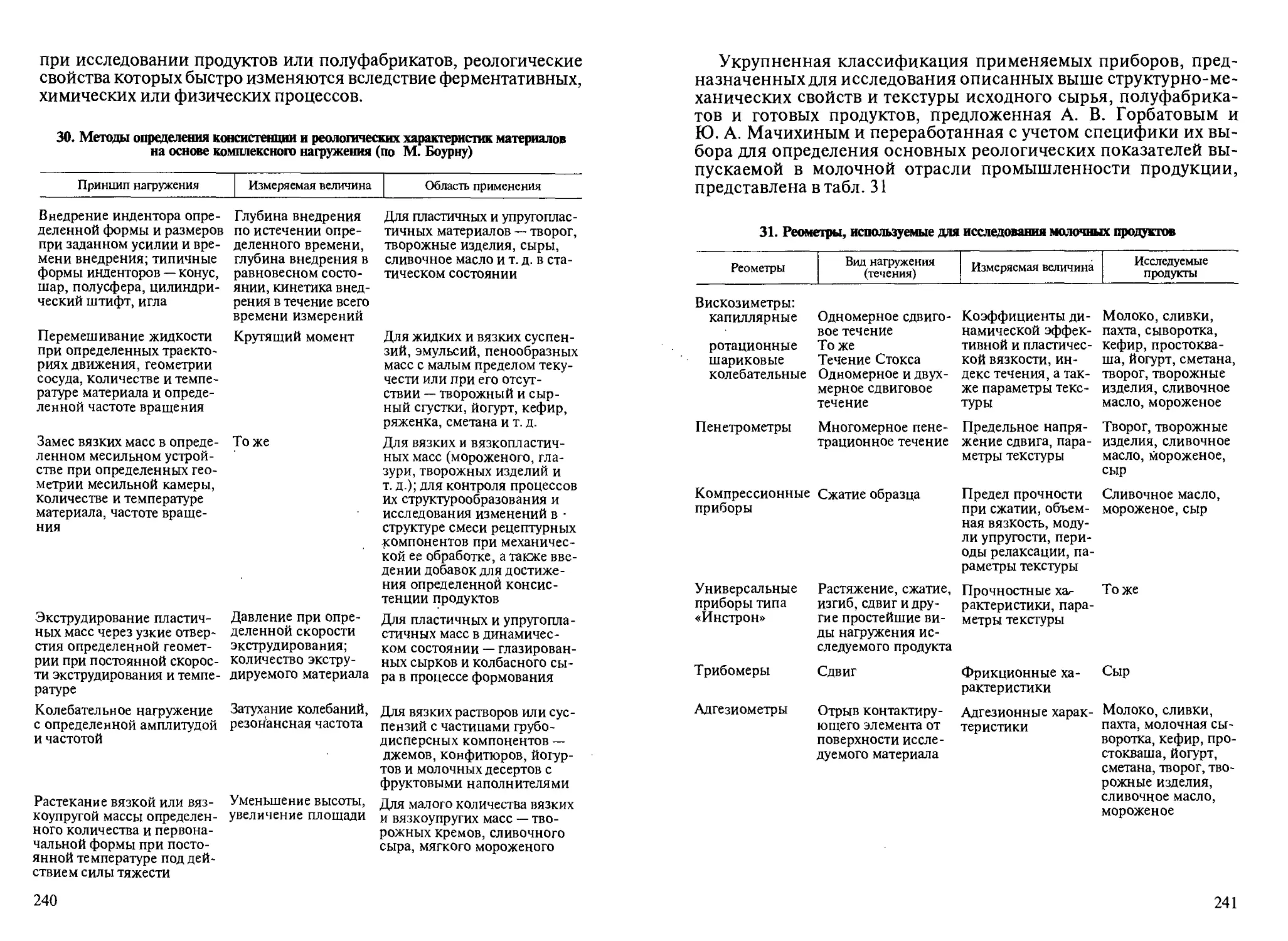
©
МОСКВА «КОЛОС» 2000
УДК 637.04/07(075.8)
ББК36.95я73
К 84
Редактор Е. Н. Соколова
Рецензенты: проф. Н. К. Ростроса (МГАВМиБ), зав. кафедрой
технологии молока и молочных продуктов проф. К. К. Полянский
(ВГТА), ведущей науч, сотрудник Н. А. Гроностайская (ВНИМИ)
Крусь Г. Н., Шалыгина А. М., Волокитина 3. В.
К84 Методы исследования молока и молочных продуктов/Под
общ. редакцией А. М. Шалыгиной. — М.: Колос, 2000. — 368 с.:
ил. (Учебники и учеб, пособия для студентов вузов)
ISBN 5-10-003440-8.
Рассмотрены спектрофотометрические, электрохимические, хромато-
графические, реологические и другие методы исследований молочных про-
дуктов. Описаны современные методы определения состава и свойств моло-
ка и молочных продуктов, содержания в них пестицидов, тяжелых металлов
и других веществ.
Учебник предназначен для студентов вузов, обучающихся по специаль-
ности «Технология молока и молочных продуктов», может быть полезен для
аспирантов, специалистов научно-исследовательских институтов и лабора-
торий предприятий, вырабатывающих молочные продукты.
УДК 637.04/07(075.8)
ББК 36.95я73
ISBN 5-10-003440-8
© Издательство «Колос», 2000
ВВЕДЕНИЕ
Исследование любого пищевого продукта — сложная аналити-
ческая задача. Из-за индивидуальности состава и многокомпонент-
ное™ продуктов необходимо приспосабливать стандартные мето-
ды к особенностям состава и физико-химической структуры про-
дукта — т. е. в каждом конкретном случае требуется проведение в
той или иной мере аналитической исследовательской работы. При
этом необходимо учитывать физическое состояние исследуемого
вещества и сопутствующих определяемому веществу компонентов.
Своеобразие состава и форм нахождения определяемых компо-
нентов в пищевых продуктах осложняет подготовку проб: необхо-
димо предварительно выделить (изолировать) компонент.
Только комплекс анализов (физико-химических, органолепти-
ческих, микробиологических и др.) дает возможность контролиро-
вать качество сырья и технологические процессы производства, а
также готовую продукцию.
Современные физико-химические (инструментальные) методы
анализа пищевых продуктов, сырья и вспомогательных материалов
характеризуются большими диапазонами обнаружения, селектив-
ностью и экспрессностью; они незаменимы при определении ульт-
рамалых количеств вещества (Ю-10 %). Кроме того, автоматизация
производства — создание поточных технологических линий, заво-
дов-автоматов, автоматизированных систем управления технологи-
ческими процессами — невозможна без применения физико-хими-
ческих методов анализа. Инструментальные методы способствуют
получению наиболее полной информации о ходе технологического
процесса и определению параметров его контроля. В то же время
они позволяют автоматизировать контроль технологического про-
цесса.
Инструментальные методы анализа широко применяют при
изучении химического состава сырья, полуфабрикатов и готовой
продукции, их физико-химических, биологических и технологи-
ческих возможностей с целью создания оптимальных технологи-
ческих процессов для переработки сырья с максимальной пользой и
наилучшими производственными показателями для получения го-
товой продукции высокого качества. Кроме того, в связи с загрязне-
нием атмосферы токсическими веществами, способными накапли-
ваться в растениях и животных организмах, необходимо определять
концентрацию таких веществ в продуктах.
3
В настоящее время в пищевой промышленности, в том числе в
молочной, широко применяют самые современные физические и
физико-химические методы анализа: электрохимические, спект-
ральные, хроматографические, реологические и др. За последние
годы получил распространение потенциометрический метод ана-
лиза в связи с использованием различных ионселективных электро-
дов, т. е. электродов с относительно высокой селективностью к оп-
ределенному иону в растворе. Изменяя состав стекла электрода, по-
лучают ионселективные электроды (более 30) с функциями щелоч-
ных металлов, галогенидов, сульфидов и др.
Все большее значение приобретают вольтамперометрические
методы анализа, и в частности полярография, основанные на ис-
пользовании процессов поляризации, возникающих на микроэлек-
троде. Создание разнообразной отечественной и зарубежной элект-
ронной аппаратуры обеспечило широкое распространение многих
видов полярографии (классической, осциллографической, диффе-
ренциальной, переменно-токовой, импульсной и др.) в практике
заводских и научно-исследовательских лабораторий.
Полярографический анализ применяют для определения таких
биологически активных соединений, как белки, аминокислоты, уг-
леводы, витамины, атакже микроэлементов и следов тяжелых метал-
лов. Можно одновременно определить несколько элементов, напри-
мер медь, свинец, олово и цинк, при их совместном присутствии.
Спектральные методы анализа — одни из самых распространен-
ных и широко применяемых методов. Они позволяют получить
наиболее полную информацию о важнейших свойствах вещества и
устанавливать содержание веществ в объектах в диапазоне от 30—
40 % до 10-3 %. К этим методам относят атомно-эмиссионный,
спектрофотометрический, атомно-абсорбционный, нефелометри-
ческий, турбидиметрический, люминесцентный, атакже рефракто-
метрический и поляриметрический. В настоящее время разработа-
но большое число фотометрических методов анализа, основанных
на способности растворов поглощать электромагнитное излучение
в видимой ультрафиолетовой (близкой и дальней) и инфракрасной
областях спектра. Их используют для определения большинства хи-
мических элементов (железа, меди, кобальта, никеля, хрома) и мно-
гих органических соединений (сахаров, белков, аминокислот и
т. д.). Фотометрические методы применяют также для определения
степени окисленности жиров, содержания пектиновых веществ,
фенольных соединений, витаминов.
С помощью ИК-спектрометрии определяют содержание белка,
жира и лактозы в молоке, пестицидов, витаминов, пищевых краси-
телей, а также контролируют технологические процессы при пере-
работке сырья. Этот метод позволяет получать достаточно полную
информацию о строении и составе веществ.
Для исследования продуктов используют флуоресцентное излу-
чение, при этом источником возбуждающего электромагнитного
4
излучения служит УФ-излучение или видимый свет с длинами волн
от 250 до 500 нм. Люминесценцию наиболее широко применяют
для идентификации и количественного определения витаминов,
белков, жиров, углеводов и других веществ.
Спектроскопия ядерного магнитного резонанса (ЯМР) и элект-
ронного парамагнитного резонанса (ЭПР) основана на резонанс-
ном взаимодействии магнитных моментов ядер и электронов, нахо-
дящихся в сильном постоянном магнитном поле с перпендикуляр-
ным полем радиочастотного или микроволнового диапазона. Глав-
ное применение метода ЯМР — это определение отдельных
компонентов (влаги, жира) без их предварительного разделения, а
также структурные исследования веществ. В последние годы появи-
лись методы импульсного ЯМР, что привело к значительному рас-
ширению возможности применения этого метода и повышению
точности определения.
С помощью методов хроматографии изучают состав и качество
молочных продуктов. Анализ основан на разделении смеси веществ
сорбционными способами в динамических условиях. Это один из
наиболее универсальных и эффективных методов разделения и ана-
лиза сложных органических и неорганических соединений.
Среди хроматографических методов широкое распространение
получила газовая хроматография. С ее помощью можно разделить,
идентифицировать и количественно определить большое число ве-
ществ: смеси углеводородов, летучие жирные кислоты, спирты,
эфиры, альдегиды, кетоны, витамины, аминокислоты, углеводы и
другие сложные соединения. Газовая хроматография позволяет
идентифицировать ароматические компоненты пищевых продук-
тов. В последнее время получили распространение газохроматогра-
фические методы определения свободных жирных кислот, остаточ-
ных количеств пестицидов в молоке.
Ионообменную хроматографию используют как метод разделе-
ния и выделения веществ, предшествующий их количественному
определению. Этот метод применяют для определения белков, ами-
нокислот, хлорида натрия в молочных продуктах и т. д.
Наряду с рассмотренными применяют и такие методы, как масс-
спектрометрию, электрофоретические и другие новейшие методы
исследования.
Использование реологических методов исследования позволяет
получать готовые продукты постоянного, заранее заданного каче-
ства; научно обосновать понятия существенных аспектов качества;
совершенствовать технологические процессы; применять высоко-
производительное непрерывнодействующее автоматически управ-
ляемое оборудование; «конструировать» те или иные виды пище-
вых продуктов и т. д.
Современные физические и физико-химические методы иссле-
дования способствуют повышению надежности технохимического
контроля на предприятиях и улучшению качества готовых продуктов.
5
Учебник «Методы исследования молока и молочных продук-
тов» написан в соответствии с программой для высших учебных
заведений по специальности «Технология молока и молочных
продуктов».
Необходимость подобного издания объясняется тем, что указан-
ная дисциплина введена впервые в учебный план подготовки инже-
неров-технологов по специальности «Технология молока и молоч-
ных продуктов» и является основой для последующего изучения
технохимического контроля производства молочных продуктов,
стандартизации, метрологии, сертификации и автоматизации тех-
нологических процессов.
Данный учебник разработан впервые. В нем рассмотрены совре-
менные методы исследования состава, свойств сырья и молочных
продуктов с учетом последних достижений отечественной и зару-
бежной науки и техники.
Авторы выражают глубокую благодарность рецензентам: проф.
Н. К. Ростросе, проф. К. К. Полянскому и канд. техн, наук Н. А. Гро-
ностайской, сотрудникам ВНИМИ за помощь при подготовке кни-
ги: д-ру техн, наук Л. П. Брусиловскому, X. Г. Хабибову, сотрудни-
кам МГУПБ: канд. хим. наук А. В. Стефанову, канд. техн, наук
С. В. Карпычеву, канд. физ.-мат. наук Ю. П. Куркину, канд. техн,
наук Г. А. Лимантову, канд. хим. наук С. А. Янковскому, канд. техн,
наук С. К. Апраксиной, а также инженеру-технологу В. П. Семено-
вой.
Г л а в a 1
ОТБОР ПРОБ И ПОДГОТОВКА
ИХ К ИССЛЕДОВАНИЮ
Пищевые продукты исследуют качественными и количествен-
ными методами измерения. При идентификации веществ выбор
методов измерения зависит от свойств вещества, его количества и
цели исследования. Общая схема измерений включает следующие
стадии: постановка задачи, выбор метода, выбор аппаратуры, отбор
пробы, подготовка пробы, проведение измерений, обработка ре-
зультатов, оформление результатов.
Масштаб применяемого метода определяется имеющимся в рас-
поряжении исследователя количеством вещества.
Масштаб метода Масса пробы, г
Макро 0,5—100
Полумикро 0,01—0,5
Микро 10~3—10-2
Ультрамикро 10“3
Чувствительность метода определяется пределами обнаружения,
т. е. минимальным количеством вещества, которое может быть об-
наружено с достаточно высокой степенью достоверности.
Получение достоверных и точных результатов при анализе мо-
лочных продуктов во многом зависит от правильной подготовки
материала к анализу. Перед анализом проводят отбор проб. Под
пробой понимают определенное количество нештучной продук-
ции, отобранное для анализа. Отбор проб молока и молочных про-
дуктов, подготовку их к анализу проводят в соответствии с
ГОСТ 26809—86. Для микробиологических анализов пробы отбира-
ют по ГОСТ 9225-84.
При подготовке образца необходимо сохранить нативные свой-
ства продукта, не допустить потерь (например, влаги), разрушения
или изменения соединений, входящих в состав продукта, а также
попадания посторонних компонентов. Материал проб должен быть
однородным. Для этого тщательно перемешивают среднюю пробу
жидких и пастообразных продуктов или измельчают и перемешива-
ют в дальнейшем твердые и сухие продукты. Чем тоньше измельче-
ние, тем выше однородность и точнее результаты анализа.
Среднюю пробу образца готовят непосредственно перед иссле-
дованием. Все операции проводят быстро во избежание потерь вла-
7
ги за счет испарения. Если продукт не относится к скоропортящим-
ся, измельченный материал можно сохранять в течение некоторого
времени в стеклянной или другой посуде, предохраняющей его от
потерь влаги.
Условие получения правильных средних величин — повтор-
ность исследования продукта одного наименования. За обязатель-
ный минимум принимают трехкратность исследований.
Стандартом предусмотрено взятие точечной и объединенной
пробы.
Точечная проба — проба, взятая единовременно из определенной
части нештучной продукции (из цистерны, фляги, от монолита мас-
ла в ящике или брикета масла и т. п.).
Объединенная проба — проба, составленная из серии точечных
проб, помещенных в одну емкость.
Точечные пробы жидких, вязких и сгущенных продуктов отби-
рают кружкой или черпаком вместимостью 0,1; 0,25; 0,5 дм3 с жест-
кой ручкой длиной от 50 до 100 см, металлической или пластмассо-
вой трубкой внутренним диаметром 9 ± 1,0 мм по всей длине и с от-
верстиями по концам.
При составлении объединенной пробы молокаи молочных про-
дуктов число точечных проб от каждой единицы тары с продукцией,
включенной в выборку, должно быть одинаковым.
Перед отбором проб молоко и жидкие молочные продукты пере-
мешивают от 1 мин (механизированный способ перемешивания во
флягах) до 15—20 мин (в железнодорожных цистернах) или путем
пятикратного перевертывания потребительской тары (бутылки и
пакеты). При отстое жира в молоке и сливках в потребительской
таре их нагревают до температуры 32 ± 2 °C на водяной бане темпе-
ратурой 38 ± 2 °C. Затем продукт из бутылок и пакетов сливают в
посуду, составляя объединенную пробу.
Точечные пробы полутвердых, твердых и сыпучих продуктов от-
бирают шпателями, ножами или специальными щупами.
Точечные пробы творога, творожных изделий, домашнего сыра
и сыров для плавления в транспортной таре отбирают щупом, опус-
кая его до дна тары; в потребительской таре — освобождают про-
дукцию от тары и тщательно перемешивают. В творожных полуфаб-
рикатах начинку отделяют от теста.
Перед тем как отбирать пробы со сгущенными молочными кон-
сервами невскрытые металлические банки массой нетто 1000 г и бо-
лее, а также фляги и бочки с продуктом переворачивают вверх дном
и оставляют в таком положении на одни сутки. До отбора проб сгу-
щенные молочные консервы перемешивают для равномерного рас-
пределения возможного осадка лактозы по всей массе продукта. В
бочках и флягах сгущенные молочные консервы перемешивают ме-
шалкой, а в потребительской таре — шпателем в течение 1—2 мин
после вскрытия тары. Если на дне банки со сгущенными молочны-
ми консервами с сахаром обнаружен осадок, банку погружают в
8
воду температурой 55 ± 5 °C и снова перемешивают до получения
однородной массы, не допуская повышения температуры продукта
выше 28 + 2 °C, затем охлаждают его до 20 + 2 °C.
Точечные пробы сухих молочных продуктов в транспортной таре
отбирают щупом из разных мест каждой единицы транспортной
тары с продукцией. От сгущенных и сухих молочных консервов в
потребительской таре точечные пробы отбирают пробником, щу-
пом или ложкой после вскрытия тары, помещают в посуду и состав-
ляют пробу для анализа.
Точечные пробы сливочного и топленого масла, пластических
сливок в транспортной таре отбирают щупом (если температура
масла ниже 10 °C, щуп нагревают в воде температурой 38 ± 2 °C); в
потребительской таре — ножом от каждого брикета.
Объединенную пробу масла помещают в водяную баню темпера-
турой 30 ± 2 °C. При постоянном перемешивании пробу нагревают
до размягчения массы и выделяют пробу, предназначенную для
анализа.
Точечные пробы сыра отбирают с двух противоположных сто-
рон каждой головки сыра, включенной в выборку, щупом, вводя
его на глубину 3/4 длины. Для оценки органолептических показа-
телей отбор точечной пробы проводят с одной стороны головки
сыра.
В табл. 1 приведены объем или масса молочных продуктов, необ-
ходимых для анализа.
1. Объем (масса) проб молочных продуктов
Продукт Объем (масса) объединенной пробы, дм3 (г) Объем (масса) пробы для анализа, дм3 (г)
Молоко, жидкие молочные про- дукты для детского питания, жидкие ЗЦМ 1 0,5
Сливки 0,5 0,1
Жидкие кисломолочные продукты — 0,1
Сметана 500 100
Творог, творожные изделия и по- луфабрикаты, домашний сыр, сыр для плавления, мороженое 500 100 (продукция без на- полнителей) 150 (продукция с на- полнителями)
Сгущенные молочные продукты 1000 300
Сухие молочные продукты 1200 200
Сливочное масло и пластические сливки — 50
Сыры — 50
Молочный сахар, пищевой и тех- нический казеин 1200 300
9
Пробы молока и молочных продуктов, предназначенные для оп-
ределения физико-химических показателей, подготавливают сле-
дующим образом.
Пробы молока, жидких заменителей цельного молока, сли-
вок, сметаны, кисломолочных напитков, мороженого перемеши-
вают путем перевертывания посуды с пробами не менее трех раз
или переливания продукта в другую посуду и обратно не менее
двух раз.
Пробы молока и молочных продуктов доводят до температуры
20 ± 2 °C.
Пробы кисломолочных напитков и сметаны, имеющие густую
консистенцию, а также пробы продуктов с отстоявшимся слоем
сливок, нагревают на водяной бане до температуры 32 ± 2 °C, после
чего охлаждают до 20 ± 2 °C.
Пробы творога, творожной массы, творожных изделий, полу-
фабрикатов и плавленых сладких сыров с наполнителями растира-
ют в ступке до получения однородной консистенции, предвари-
тельно удалив с помощью пинцета, шпателя или ложки из проб
продукции с наполнителями цукаты, изюм, орехи.
Пробы сгущенных и сухих молочных продуктов растирают в
ступке и тщательно перемешивают.
Пробы молочного сахара и казеина, предназначенные для ана-
лиза, измельчают в ступке или на лабораторной мельнице. Поро-
шок просеивают через сито с отверстиями диаметром от 0,40 до
0,50 мм без остатка.
Перед исследованием большинство продуктов (кроме некото-
рых напитков) минерализуют — освобождают от органических
соединений сухим или мокр*ым озолением. Выбор способа озоле-
ния зависит от ряда условий. Так, сухое озоление в отличие от
мокрого не требует реактивов, позволяет использовать относи-
тельно большое количество образца (5—10 г, но не более), так как
иначе наблюдаются значительные потери элементов, что важно
при низком содержании определяемого элемента или низкой
чувствительности метода, не требует постоянного наблюдения
сотрудника. Однако возможны потери некоторых элементов,
особенно в образцах, содержащих хлориды. Мокрое озоление,
как правило, дает меньше потерь элементов, но требует чистых
реактивов, большего внимания оператора и ограничено массой
образца от 2 до 5 г.
Выбор метода озоления зависит также от вида продукта, напри-
мер, продукты с высоким содержанием жира или сахара рекоменду-
ется сжигать сухим методом, а продукты, содержащие хлориды, —
мокрым методом.
В большинстве случаев сухое озоление пищевых продуктов про-
водят при температуре 450—550 °C в течение 4—16 ч. При более
низких температурах озоление затягивается, а в условиях повышен-
ных температур возможно улетучивание некоторых элементов, на-
10
пример, железа. При озолении продуктов, содержащих заметные
количества хлоридов, наблюдаются потери Fe, Sb, Pb, Al и Си при
обычных режимах сухого озоления за счет образования относитель-
но летучих хлоридов металлов. В этих случаях озоление проводят
таким образом, чтобы перевести элементы в менее летучие нитраты
или сульфаты. Чаще всего перед озолением к образцу добавляют
нитрат магния или другие соли азотной кислоты либо смачивают
образец разбавленной (1:1) азотной кислотой или разбавленной
серной кислотой. Добавление нитратов, кроме уменьшения потерь,
ускоряет озоление. В зависимости от вида элемента нитраты добав-
ляют только перед озолением, но иногда после обугливания или
после получения бурой золы. Более подробно способы сухого озо-
ления будут указываться при рассмотрении методов определения
каждого элемента.
Для проведения мокрого озоления существует около десяти ва-
риантов, но для большинства видов элементов и продуктов, в том
числе относительно богатых жирами, рекомендуется использовать
смесь трех концентрированных кислот — азотной, хлорной и сер-
ной обычно в соотношении 3:2: 1. Для низкожирных продуктов
рекомендуется смесь азотной и хлорной кислот НС1О4 в соотноше-
нии 3 : 2. Смесь азотной и серной кислот, хотя и полно минерализу-
ет продукт, но сжигание протекает очень долго.
В этом случае нет необходимости предусматривать особые меры
предосторожности, связанные с использованием хлорной кислоты.
Отдельно серную или азотную кислоту не используют из-за боль-
шой длительности минерализации. В некоторых случаях вместо
хлорной кислоты применяют пероксид водорода, добавляя иногда
перманганат калия.
Стадия озоления (минерализации) —• основной источник как
потерь контролируемых металлов, так и загрязнения ими пробы.
Особенно это относится к сухому озолению в открытых кварцевых
чашках, в общих лабораторных помещениях, с использованием об-
щих вытяжных шкафов, муфельных печей и другого оборудования
и приспособлений.
Существует довольно много разработок по реализации авто-
клавной подготовки проб, когда минерализация происходит в гер-
метичном объеме, под давлением, иногда в среде окислителя. По
времени минерализация в автоклавах обычно длится не более
1,5 ч, сведены к минимуму возможные потери из-за улетучивания
части пробы и загрязнения в результате попадания металлов в про-
бу извне. Однако такие разработки в основном применимы к ми-
неральным объектам (пробы грунтов, донного ила и т. д.) или к
биологическим объектам менее сложного состава, чем молочные
продукты (пробы растительных продуктов, сухие препараты кро-
ви, сухие ткани органов).
Минерализацию проб пищевых продуктов, в том числе и молоч-
ных, проводят в соответствии с ГОСТ 26929—88.
11
Подготовленные пробы исследуют по различным показателям в
зависимости от поставленных задач. Для этого используют инстру-
ментальные и аналитические методы.
Инструментальные методы:
спектральные методы исследования
турбидиметрия и нефелометрия
рефрактометрия
поляриметрия
ультразвуковой метод исследования
электрохимические методы исследования
хроматографические методы исследования
электрофоретические методы исследования
эбулиоскопия и криоскопия
реологические методы исследования
Аналитические методы:
методы определения состава молока и молочных продуктов: сухого вещества,
влаги, жира, белка, углеводов, хлоридов, спирта (этанола);
методы определения показателей, характеризующих свойства молока и молоч-
ных продуктов: кислотность, плотность, термоустойчивость молока, чистота моло-
ка, микробное обсеменение молока, натуральность молока и наличие фальсифици-
рующих веществ, определение маститного молока, эффективность гомогенизации
и пастеризации, индекс растворимости сухих молочных продуктов, влагоудержива-
ющая способность молочных сгустков.
Г лава 2
СПЕКТРАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
Спектральные методы исследования основаны на использова-
нии явлений поглощения (или испускания) электромагнитного из-
лучения атомами или молекулами определенного вещества.
Поглощение или испускание квантов анализируемой системой
можно рассматривать как процесс возникновения характеристи-
ческих сигналов, несущих информацию о качественном и количе-
ственном составе исследуемого вещества. Частота (длина волны)
излучения определяется составом вещества. Интенсивность анали-
тического сигнала пропорциональна количеству частиц, вызвав-
ших его появление, т. е. массе определяемого вещества или компо-
нента смеси.
Спектральные методы дают широкие возможности для наблю-
дения и исследования соответствующих аналитических сигналов в
различных областях электромагнитного спектра (табл. 2) — рент-
геновское излучение, ультрафиолетовое (УФ) излучение, види-
мый свет; инфракрасное (ИК), а также микро- и радиоволновое
излучение.
Механизмы взаимодействия излучения с исследуемым веще-
ством в каждой области различны. Наиболее важными являются
атомные и молекулярные переходы, вызываемые излучением раз-
ных областей электромагнитного спектра.
Область электромагнитного спектра Атомные и молекулярные переходы
Рентгеновская Внутренние электроны
Ультрафиолетовая: дальняя ближняя Средние электроны Валентные электроны
Видимая То же
Инфракрасная: ближняя средняя дальняя Молекулярные колебания То же Молекулярные вращения и низкочас- тотные колебания
Микроволновая Молекулярные вращения; электронный парамагнитный резонанс
Радиоволновая Ядерный магнитный резонанс
13
14
Излучение квантов в рентгеновском диапазоне обусловлено элек-
тронными переходами во внутренних электронных слоях атома, ис-
пускание квантов УФ- и видимого излучения или взаимодействие
вещества с ними — следствие перехода средних и внешних валент-
ных электронов (сфера оптических методов анализа), поглощение
ИК- и микроволновых квантов связано с переходом между колеба-
тельными и вращательными уровнями молекул, а излучение в мик-
роволновом и радиоволновом диапазоне обусловлено переходами с
изменением ориентации спинов электронов или ядер атомов.
Для решения практических задач наибольшее значение имеют
спектральные методы, в основе которых используют ультрафиолето-
вый, видимый, инфракрасный и радиоволновой диапазоны излучения.
Под воздействием этих излучений происходят электронные пе-
реходы в молекулах вещества или свободных атомах исследуемого
химического элемента (аналитический сигнал — поглощение или
испускание), а также изменения ориентации спинов атомов (ана-
литический сигнал — ядерный магнитный резонанс) или электро-
нов (аналитический сигнал — электронный парамагнитный резо-
нанс). Аналитические сигналы измеряют различными методами.
В табл. 3 приведена классификация спектральных методов. По
источнику и типу аналитического сигнала спектральные методы
разделяют на молекулярно-абсорбционную спектрометрию (МАС)
3. Классификация спектральных методов
Спектроскопия Источник аналитического сигнала Аналитический сигнал Метод
Молекулярная Молекула Поглощение (аб- сорбция) Испускание (люминесценция) Молекулярно-абсорбцион- ная спектрометрия (МАС) Молекулярно-люминес- центная спектрометрия (МЛС), или флуориметрия
Атомная Атом Поглощение [абсорбция) Испускание (эмиссия) Атомно-абсорбционная спектрометрия (ААС) Атомно-эмиссионная спектрометрия (АЭС)
Магнитного резонанса Ядро атомов (магнитный момент ядра) Электрон (маг- нитный момент электрона) Ядерный магнит- ный резонанс — ЯМР-спектр Электронный пара- магнитный резо- нанс — ЭПР-спектр Спектрометрия ядерного магнитного резонанса (ЯМР) Спектрометрия электрон- ного парамагнитного резонанса (ЭПР)
Масс-спектро- скопия* Ион Масс-спектр Масс-спектрометрия
* Масс-спектроскопия отнесена к спектральным методам условно из-за фор-
мального сходства спектров, при этом исследуемое вещество не подвергается дей-
ствию электромагнитного излучения.
15
и молекулярно-люминесцентную (МЛС), или флуориметрию; на
атомно-абсорбционную (ААС) и атомно-эмиссионную (АЭС), а
также спектрометрию ядерного магнитного резонанса (ЯМР) и
электронного парамагнитного резонанса (ЭПР).
Все спектральные методы исследования будут рассмотрены в
последующих главах учебника в соответствии с предложенной
классификацией.
МОЛЕКУЛЯРНАЯ СПЕКТРОСКОПИЯ
При изучении взаимодействия исследуемого вещества и элект-
ромагнитного излучения необходимо опираться на знание строе-
ния атомов и молекул, поскольку электронное строение молекулы
какого-либо соединения сильно отличается от строения атома.
При обычных условиях атомы находятся в основном энергети-
ческом состоянии. При облучении потоком энергии внешние элек-
троны атомов поглощают фотоны и возбуждаются. Возбужденный
электрон стремится вернуться в основное состояние; при возвраще-
нии в основное состояние происходит излучение. Спектр излуче-
ния атома является линейчатым (дискретным).
На рис. 1 представлена диаграмма энергетических уровней вне-
шних электронов молекулы. На диаграмме сплошными вертикаль-
/ \ш\ IV
Вращательные энерге-
тические уровни
Колебательные энерге-
тические уровни
Рис. 1. Диаграмма энергетических уровней молекулы:
/—поглощение; //—колебательнаядезактивация; /// — колебатель-
ная релаксация; IV— флуоресценция
16
ними линиями показаны поглощение или испускание излучения;
штриховыми линиями — безызлучательные переходы. При нор-
мальных условиях в отсутствие возбуждения такая молекула нахо-
дится в основном состоянии, обозначенном на схеме 5q. Энергия
электромагнитного излучения, поглощенная молекулой при воз-
буждении, вызывает переход на более высокий уровень 5t. Если пе-
реход заканчивается на каком-либо колебательном подуровне воз-
бужденного уровня 5Ь то молекула очень быстро переходит на уро-
вень 51; при этом избыток энергии выделяется в виде теплоты. На
диаграмме этот процесс обозначен цифрой II.
Молекула в возбужденном состоянии 5t может расходовать
энергию возбуждения по одному из следующих механизмов: пере-
дать ее другой химической частице при столкновении (колебатель-
ная релаксация III) или испустить ее в виде излучения — люминес-
ценции (флуоресценция IV).
Переходы в молекулах можно изучать, наблюдая избиратель-
ное поглощение или излучение в определенных областях элект-
ромагнитного спектра. Так, переходы между электронными
уровнями (7) наблюдаются в УФ- и видимой областях; переходы
между колебательными подуровнями в пределах одного элект-
ронного уровня находятся в ближней и средней ИК-областях.
Избирательное поглощение в дальней ИК- и микроволновой об-
ластях связано с низкоэнергетическими переходами, вызванны-
ми вращением молекул.
Различают следующие виды колебаний молекул — валентные
(периодическое движение атомов вдоль оси связи) и деформацион-
ные, при которых происходит искривление или изгиб связи и т. п.
На примере метильной группы показаны различные виды колеба-
ний молекулы (рис. 2).
Поглощение ИК-излучения при
длинах волн от 0,7 до 4 мкм отвечает ва-
лентным колебаниям между атомами
водорода и более тяжелыми атомами и
может быть использовано при иденти-
фикации функциональных групп, со-
держащих водород. Колебания атомов
в молекуле по двойным и тройным свя-
зям относятся к длинам волн от 4,0 до
Рис. 2. Колебания молекулы на примере метильной
группы:
С, А — валентные колебания соответственно симметрич-
ные и асимметричные; Н, М— деформационные колеба-
ния плоские соответственно ножничные и маятниковые;
В, К—деформационные колебания неплоские соответ-
ственно веерные и крутильные
17
6,5 мкм, а при более длинных волнах помимо валентных происхо-
дят и деформационные колебания. Поглощение при длинах волн
свыше 25 мкм соответствует колебаниям связей с участием тяжелых
металлов и групп атомов.
Следует отметить, что микроволновую область спектра можно
рассматривать как продолжение инфракрасной области спектра,
поскольку поглощение здесь связано прежде всего с вращательны-
ми переходами в молекулах, обладающих постоянным дипольным
моментом. К поглощению способны также молекулы, обладаю-
щие магнитным моментом, например О2, NO, NO2 и свободные
радикалы. В практике обычно используют спектральную область
между 8 и 40 ГГц.
МОЛЕКУЛЯРНО-АБСОРБЦИОННАЯ СПЕКТРОМЕТРИЯ
В молекулярно-абсорбционной спектрометрии исследуют ана-
литические сигналы в области от 200 до 750 нм (УФ-излучение и
видимый свет), вызванные электронными переходами внешних ва-
лентных электронов, а также поглощение излучения в ИК- и мик-
роволновой области, связанное с изменением вращения и колеба-
ния молекул.
Наиболее широкое распространение получил метод, основан-
ный на изучении поглощения в видимой области спектра в интер-
вале длин волн от 400 до 750 нм — фотометрия; а также метод, ос-
нованный на поглощении излучения в различных частях инфра-
красной области электромагнитного спектра — И К-спектромет-
рия, чаще всего используют поглощение излучения в средней
(длина волны 2,5—25 мкм) и ближней (длина волны 0,8—2,5 мкм)
ИК-области.
ФОТОМЕТРИЯ
Фотометрический метод количественного анализа основан на
способности определяемого вещества, компонента смеси или их
окрашенных форм поглощать электромагнитное излучение оп-
тического диапазона. Способность к поглощению зависит от
цветности исследуемого вещества. Цветность определяется элек-
тронным строением молекулы, обычно ее связывают с наличием
в молекуле так называемых хромофорных групп, обусловливаю-
щих поглощение электромагнитного излучения веществом в ви-
димой и УФ-областях спектра. В табл. 4 приведены органичес-
кие соединения, содержащие типичные хромофоры, их макси-
мумы поглощения и значения молярных коэффициентов погло-
щения.
18
4. Фотометрические характеристики органических соединений, содержащих
типичные хромофоры
Соединение | Хромофор [ Растворитель 1 НМ 1 1g е
Октен-3 С=С Гексан 185 3,9
230 о,з
Ацетилен с=с (Пары) 173 3,8
Ацетон с=о Гексан 188 2,9
279 1,2
Диазоэтилацетат N=N Этанол 252 3,9
371 1,1
Бутадиен С=С-С=С Гексан 217 4,3
Кротоновый альдегид с=с-с=о Этанол 217 4,2
321 1,3
Диметилглиоксим N=C-C=N Этанол 226 4,2
Октатриенол с=с-с=с-с=с Этанол 265 4,7
Декатетраенол (-С=С-)4 Этанол 300 4,8
Витамин А (-С=С-)5 Этанол 328 3,7
Кроме того, если молекула органического соединения благодаря
наличию комплексообразующих групп способна образовывать
комплексы с ионами металлов, это вызывает появление или изме-
нение окраски комплекса по сравнению с исходным соединением.
Свойства таких комплексообразующих реагентов широко исполь-
зуют на практике, в частности при определении загрязнения пище-
вых продуктов тяжелыми металлами.
Для каждого поглощающего вещества имеется определенное
распределение интенсивности поглощения по длинам волн. Кон-
центрацию поглощающего вещества проводят при длине волны, со-
ответствующей максимальному поглощению. Таким образом, по-
глощение является количественной характеристикой определяемо-
го вещества в виде аналитического сигнала.
При прохождении потока лучей через частично поглощающую
среду интенсивность прошедшего потока I согласно закону Буге-
ра — Ламберта — Бера
7=70-l(Wc,
где /0 — интенсивность падающего потока; е>. — молярный коэффициент поглоще-
ния при данной длине волны; I— толщина поглощающего слоя; С— концентрация
поглощающего вещества.
В логарифмической форме при оптической плотности раствора D
1g 7 = 1g 70 - ех/С; D = 1g (70/7) = ^1С.
Физический смысл е становится ясным, если принять /= 1 см и
С— 1 моль/л, тогда D = е. Следовательно, молярный коэффициент
поглощения равен оптической плотности одномолярного раствора
при толщине светопоглощающего слоя 1 см.
19
На практике зависимость D от концентрации определяемого ве-
щества при постоянной /ив конкретных условиях определения
изображают в виде градуировочного графика.
В соответствии с законом Бугера — Ламберта — Бера градуиро-
вочный график в координатах оптическая плотность — концентра-
ция будет линейным и прямая пройдет через начало координат.
Градуировочный график обычно строят по трем точкам, т. е. по
трем стандартным растворам, что повышает точность построения
графика и надежность определений.
При отклонениях от закона Бугера — Ламберта — Бера, т. е. при
нарушении линейной зависимости D от С, число точек на градуиро-
вочном графике должно быть увеличено. Применение градуиро-
вочных графиков — наиболее распространенный и точный метод
фотометрических измерений. Основные ограничения метода обус-
ловлены трудностями приготовления стандартных (эталонных) ра-
створов и влиянием так называемых третьих компонентов, т. е. ком-
понентов, которые содержатся в пробе и влияют на результат.
Общая схема выполнения фотометрического определения едина
и включает следующие стадии:
подготовку пробы и переведение определяемого вещества или
компонента в раствор, в реакционноспособную, в зависимости от
химизма аналитической реакции, форму;
получение окрашенной аналитической формы определяемого
вещества в результате проведения цветной реакции при оптималь-
ных условиях, обеспечивающих ее избирательность и чувствитель-
ность;
измерение светопоглощающей способности аналитической
формы, т. е. регистрация аналитического сигнала при определен-
ных условиях, отвечающих его локализации и наибольшей интен-
сивности.
Промышленностью выпускаются различные приборы молеку-
лярно-абсорбционной спектрометрии — колориметры, фотомет-
ры, фото электроколориметры, спектрофотометры и т. д., в которых
установлены различные комбинации источников света, монохро-
матизаторов и рецепторов. Приборы можно классифицировать сле-
дующим образом:
по способу монохроматизации лучистого потока — спектрофо-
тометры, т. е. приборы с призменным или решеточным монохро-
матором, позволяющие достигать высокой степени монохромати-
зации рабочего излучения; фотоэлектроколориметры, т. е. прибо-
ры, в которых монохроматизация достигается с помощью свето-
фильтров;
по способу измерения — однолучевые с прямой схемой измере-
ния (прямопоказывающие) и двухлучевые с компенсационной
схемой;
по способу регистрации измерений — регистрирующие и нере-
гистрирующие.
20
6 1
Рис. 3. Схема двухлучевого спектрометра:
1 — источник излучения (штифт Нерста или ртутная лампа для инфракрасного излучения, лампа
накаливания с вольфрамовой нитью для видимого света; водородная газоразрядная лампа для
УФ-излучения); 2— монохроматор (дифракционная решетка или призма) с щелью для регули-
рования интенсивности излучения; J — кювета с исследуемой пробой; 4 — кювета с контрольной
пробой; 5— фотоэлементы сравнения; 6 —блок регистрации (графической, оптичес-
кой и цифровой)
В настоящее время для получения спектров поглощения приме-
няют, как правило, двухлучевые спектрометры (рис. 3). Пучки моно-
хроматического излучения проходят параллельно через кюветы 3 и 4,
наполненные соответственно исследуемой и контрольной пробами.
Оба луча попадают на фотоэлементы 5, где сравниваются интенсив-
ности излучений, прошедших через контрольную пробу 10 и исследу-
емую пробу I. Результат сравнения регистрируется блоком 6.
Схема однолучевого фотометра приведена на рис. 4. Кюветы по-
очередно устанавливают на пути светового пучка. Кювету 4 запол-
няют контрольной пробой и устанавливают на пути светового пуч-
ка. Затем устанавливают стрелку гальванометра 7 на нулевое деле-
ние (нуль поглощения или 100 % пропускания) и помещают на пути
светового потока кювету с исследуемой пробой 4'. Проводят изме-
5 6
Рис. 4. Схема однолучевого фотометра:
7— источник света; 2 —линза; 3 — фильтр; 4 — кювета с конт-
рольной пробой; 4' — кювета с исследуемой пробой; 5— фото-
элемент; 6 — усилитель; 7— гальванометр
21
рения и снимают отсчет по шкале, отградуированной в единицах
оптической плотности и в процентах пропускания.
Замена светофильтров монохроматором значительно увеличи-
вает возможности фотометров. В таких приборах можно устанавли-
вать любую длину волны, что позволяет значительно повысить чув-
ствительность и точность измерения, снизить влияние примесей.
Источниками света служат в ультрафиолетовой — водородная лам-
па, в видимой области — лампа накаливания.
Применение автоматизированного, управляемого микропро-
цессором фотометра в еще большей степени расширяет возможнос-
ти спектрофотометрии: позволяет проводить измерения большого
количества образцов при различных длинах волн через различные
интервалы времени.
ИК-СПЕКТРОМЕТРИЯ
Инфракрасная спектроскопия — это метод анализа химических
соединений, при котором поглощается энергия в пределах инфра-
красного излучения (тепловое излучение). ИК-спектроскопию
применяют для определения практически любой функциональной
группы, идентификации соединений и т. п.
Различные молекулы, содержащие одну и ту же атомную груп-
пировку, дают в ИК-спектре полосы поглощения в области одной
и той же характеристической частоты. Характеристические часто-
ты дают возможность по спектру установить конкретные группы
атомов в молекуле и тем самым определить качественный состав
вещества и строение молекулы. Например, полосы в области
3000—3600 см-* могут быть приписаны только О—Н- или N—Н-
связям, полосы в области 2800—3000 см-1 — связи С—Н, появле-
ние полосы в области 3300—3500 см-1 характерно для NH2-rpyn-
пЫ, связь С=С характеризуется частотой 1650 см-1, связь ОС —
частотой 2100 см- *.
К настоящему времени изучены и систематизированы инфра-
красные спектры более чем 20 000 соединений, что существенно об-
легчает практическое проведение анализа. Для получения первых
ориентировочных данных часто пользуются так называемой картой
Колтупа, на которой указаны спектральные области многих харак-
теристических частот. Для окончательных выводов обычно требует-
ся более тщательный анализ спектра. Иногда задача качественного
анализа может быть решена простым сопоставлением спектра изве-
стного соединения и анализируемого вещества.
Количественный анализ по инфракрасным спектрам основан на
применении закона Бугера — Ламберта — Бера. Чаще всего здесь
используют метод градуировочного графика. В ИК-спектроскопии,
в отличие от других спектрометрических методов, при построении
графика на оси ординат откладывают не степень поглощения, а сте-
22
пень пропускания в процентах. Следо-
вательно, минимум на графике соответ-
ствует максимуму поглощения. Калиб-
ровочный график строят по методу ба-
зовой линии. Его сущность легко по-
нять из рис. 5, на котором приведен
участок ИК-спектра с двумя полосами
поглощения (волновые числа vA и vB).
Базовая линия проведена в основании
полосы поглощения «от горба к горбу»
(она изображена пунктиром). Коэффи-
циент пропускания определяют как от-
ношение ТА = ДДцл) или Тв = 7в/4)(в), Рис-5-Метод базовой линии
где /о(А), А(В) — интенсивность света,
прошедшего через контрольную пробу,
а А, А — интенсивность света, прошедшего через исследуемый об-
разец при волновых числах vA, vB.
По полученным данным вычерчивают градуировочный график и
проводят определения.
Использование спектрометрического ИК-метода — одно из
перспективных направлений в контроле показателей состава моло-
ка и молочных продуктов (жир, белок, влага, лактоза и др.). При ис-
пользовании этого метода практически отсутствует подготовка про-
бы продукта.
Инфракрасные анализаторы для контроля состава молока рабо-
тают в диапазоне волн 2,5—12 мкм. Метод основан на свойстве ком-
понентов молока (жира, белка, лактозы и воды) избирательно по-
глощать ИК-излучение на определенных длинах волн. Так, макси-
мумы поглощения жира наблюдаются при длинах волн 3,5 и
5,73 мкм, белка — 6,46, лактозы — 9,6, воды — 4,42 мкм.
В общем случае ИК-анализатор представляет собой однолучевой
или двухлучевой инфракрасный спектрофотометр, состоящий из
трех основных блоков: подготовки пробы, спектрофотометричес-
ких измерений, преобразования сигналов и вычислений. С помо-
щью таких анализаторов контролируют состав заготовляемого,
обезжиренного и пастеризованного молока, а также при соответ-
ствующей подготовке пробы — сливок, кисломолочных продуктов,
сгущенных продуктов и т. д.
В настоящее время разработаны ИК-анализаторы для контроля
состава молочных продуктов, основанные на работе в ближней ин-
фракрасной области спектра 0,8—2,5 мкм (БИК-анализаторы). В
БИК-анализаторах ИК-излучение поступает на образец, помещен-
ный в кювету. Излучение, диффузно отражаясь от образца, собира-
ется на специальном приемнике, где преобразуется в электричес-
кий сигнал, и усиливается в электронном блоке. С помощью встро-
енных микропроцессорных устройств осуществляются управление
и обработка данных, а по специальной программе результаты изме-
рения переводятся в цифровую форму.
23
Так, в России объединением «ЛОМО» при участии НИКТИпри-
бор, НПО «Агроприбор» и ВНИМИ создан БИК-анализатор «Ана-
ликон» для контроля массовой доли жира, белка и влажности в су-
хих молочных продуктах (сухое цельное и обезжиренное молоко и
сухой казеин).
Образец продукта помещают в кювету, на него воздействует
ИК-излучение на нескольких (до 10) фиксированных длинах
волн. Излучение диффузно отражается от образца и эталона и по-
ступает в специальный приемник, где преобразуется в электри-
ческий сигнал, который усиливается в электронном блоке, обра-
батывается встроенными микропроцессорными устройствами и
передается на ЭВМ.
Наряду с многокомпонентными БИК-анализаторами созданы
промышленные и лабораторные однокомпонентные приборы, на-
пример, для контроля влажности сухих молочных продуктов. Мето-
дика измерения влажности состоит в сравнении отражательной
способности ИК-излучения контролируемым продуктом на длинах
волн 1,95 или 1,45 мкм (поглощаемой молекулами воды) и опорной
1,75 мкм (наименее поглощаемой молекулами воды). Соотношение
излучений на указанных длинах волн пропорционально массовой
доле влаги в продуктах.
И К-анализаторы для молока и молочных продуктов выпускают-
ся рядом зарубежных фирм.
Фирма Foss-Electric (Дания) выпускала инфракрасные анализа-
торы «Милко-Скан» (модели 130 и 300) для измерения состава мо-
лока и молочных продуктов.
В последние годы фирмой предложены инфракрасные анализа-
торы «Милко-Скан» (модели S50 и FT 120), предназначенные для
использования на крупных и средних молочных заводах.
Прибор «Милко-Скан S50» предназначен для измерения массо-
вых долей жира, белка, СОМО, сухих веществ в молоке и продуктах.
Это компактный спектрофотометрический ИК-анализатор со
встроенным принтером и автоматическим нагревателем проб.
«Милко-Скан S52» измеряет два компонента, например жир и
белок или другие, избираемые потребителем, в сыром, пастеризо-
ванном или обезжиренном молоке и сливках. «Милко-Скан S53»
определяет 3 компонента в молоке и сливках. «Милко-Скан S54A»
и «Милко-Скан S54B» контролируют 4 компонента в молоке,
сливках и молочных продуктах (предусмотрено 10 калибровок, из-
бираемых потребителем, применительно к выбранным для конт-
роля молочным продуктам). Производительность анализатора 50
проб в час.
Прибор «Милко-Скан FT 120» — усовершенствованная модель
ИК-анализатора (Фурье-ИК-анализатор), имеет более высокие
метрологические и технические характеристики по сравнению с
другими моделями. Прибор обеспечивает измерение состава моло-
ка и молочных продуктов. Производительность до 120 проб в час.
24
Инфракрасный экспресс-анализатор Aegus mi 200 французс-
кой фирмы Anadis instruments предназначен для измерения соста-
ва молока и молочных продуктов. Принцип действия прибора ос-
нован на использовании спектроскопического метода в ближней
инфракрасной области спектра. Прибор представляет собой мо-
ноблочный инфракрасный спектрометр, состоящий из блока от-
бора и подготовки пробы продукта и оптико-механического элек-
тронного блока. Для отбора и подготовки пробы служат насос вы-
сокого давления (гомогенизатор) и термостат для подогрева про-
бы. Предусмотрена автоматическая мойка измерительного тракта
прибора после каждого анализа. Управление работой спектромет-
ра и отображение результатов измерений выполняется персональ-
ной ЭВМ, совместимой с цветным монитором. Контролируемые
показатели: массовые доли жира, белка, лактозы, СОМО, сухих
веществ, фруктозы, глюкозы, сахарозы, мочевины, карбоксиль-
ных кислот.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА
В МОЛОЧНОБЕЛКОВЫХ КОНЦЕНТРАТАХ МЕТОДОМ ЛОУРИ
(ПО Н. И. АРБАТСКОЙ, А. Н. КОНВОЙ, Н. А. ЛАПШИНСКОЙ)
Метод основан на образовании окрашенных продуктов при вза-
имодействии реактива Фолина с щелочными растворами белков.
Интенсивность окрашивания в основном зависит от аминокислот-
ного состава белка (от содержания в исследуемых белках тирозина и
триптофана) и измеряется на спектрофотометре или спектрофото-
колориметре.
Для определения содержания белка в растворах методом Лоури
не требуется предварительной минерализации исследуемого мате-
риала. Метод Лоури имеет свои особенности применительно к су-
хим молочнобелковым концентратам (казеинату натрия, казециту,
сухому гидролизату казеина с низкой степенью гидролиза) и сухой
сыворотке (после выделения казеина). Поскольку он обладает вы-
сокой чувствительностью и анализ ведется в сильно разбавленных
растворах, большое значение имеет тщательность перевода сухих
молочнобелковых концентратов в раствор. С этой целью навеску
продукта растворяют, добавляя i М раствор гидроксида натрия в ко-
личестве 0,1— 0,2 см3 на 100 мкг белка и нагревая на водяной бане до
температуры 50 °C.
Если концентрация белка превышает 0,04 мг/см3 и содержание
белка методом Лоури определяют при температуре 20±2 °C, то на-
рушается линейная зависимость между оптической плотностью и
содержанием белка в растворе. Линейную зависимость можно со-
хранить при более высокой концентрации белка в растворе (до
0,11 мг/см3), дополнительно нагревая пробу до 50 °C, после созда-
ния щелочной среды и добавления реактива Фолина.
25
Оптимальные концентрации исследуемых продуктов следую-
щие: для казеината натрия, казецита, сухого гидролизата казеина с
низкой степенью гидролиза — 0,1 мг/см3; для сухой казеиновой сы-
воротки — 0,4 мг/см3.
Выполнение работы. К 1 см3 исследуемого белка прибавляют
5 см3 реактива № 3, через 10 мин вносят 0,5 см3 реактива Фолина
(реактив № 4), быстро и тщательно перемешивают и через 30 мин
фотометрируют на спектрофотоколориметре при длине волны
650 нм. Содержание белка в растворе рассчитывают по градуиро-
вочному графику: вначале определяют содержание белка (в мкг)
в пробе продукта, затем делают пересчет на 100 г исследуемого
продукта.
Для построения градуировочного графика готовят серию ра-
створов белка известной концентрации (от 20 до 200 мкг/см3). С
каждым из этих растворов проводят реакцию Лоури, включая до-
полнительное подогревание. Строят градуировочный график за-
висимости оптической плотности от содержания белка в пробе (в
мкг). Поскольку разные белки содержат неодинаковое число аро-
матических аминокислот, взаимодействующих с реактивом Фоли-
на, нужно строить градуировочный график для каждого продукта,
определив предварительно содержание белка в нем методом
Кьельдаля.
Приготовление реактивов. Реактив 1 (2%-ныйраствор карбоната
натрия в 0,1 Мрастворе гидроксида натрия). 20 г карбоната натрия
растворяют в 1000 см3 0,1 М раствора гидроксида натрия.
Реактив 2 (5%>-ный раствор сульфата меди в 1%>-ном растворе
цитрата натрия). 5 г сульфата меди растворяют в 1%-ном растворе
цитрата натрия.
Реактив 3. Один объем реактива 2 смешивают с 50 объемами ре-
актива 1. Этот раствор приготавливают непосредственно перед ис-
пользованием.
Реактив 4 (реактив Фолина). 100 г вольфрамата натрия
(Na2WO4 • 2НэО) и 25 г молибдата натрия (Na2MoO4 • 2Н2О) раство-
ряют в 700 см3 дистиллированной воды. К смеси добавляют 50 см3
85%-ного раствора фосфорной кислоты и 100 см3 концентрирован-
ной соляной кислоты. К колбе присоединяют обратный холодиль-
ник и осторожно кипятят смесь в течение 10 ч. После кипячения в
колбу добавляют 150 г сульфата лития (Li2SO4), 50 см3 дистиллиро-
ванной воды и несколько капель брома. Смесь кипятят под тягой
без обратного холодильника в течение 15 мин для удаления избытка
брома. После охлаждения объем раствора доводят до 1 л дистилли-
рованной водой, фильтруют и хранят в темной склянке с притертой
пробкой. Для определения концентрации кислоты в полученном
растворе Фолина берут 1 см3 этого раствора, разводят в 10 раз дис-
тиллированной водой и титруют 0,1 М раствором гидроксида на-
трия по фенолфталеину.
26
Затем реактив Фолина разводят дистиллированной водой с та-
ким расчетом, чтобы получить 1 М раствор кислоты (т. е. приблизи-
тельно в два раза). Реактив может храниться в темной склянке дли-
тельное время.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ
НЕБЕЛКОВЫХ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ
В ПРОДУКТЕ МЕТОДОМ ЛОУРИ
Отвешивают 1 г исследуемого белка, растирают в фарфоровой
ступке с 5 см3 дистиллированной воды и переносят в химический
стакан вместимостью 100 см3. Вносят 5 см315%-ной трихлоруксус-
ной кислоты и ставят на водяную баню с температурой 45 °C на 1 ч.
Затем доводят дистиллированной водой до 50 см3, перемешивают и
фильтруют.
Исследуемый раствор в количестве 0,8 см3 смешивают с 4 см3
реактива №3 и оставляют на 10 мин в покое. Затем добавляют
0,4 см3 реактива № 4, перемешивают и через 20—40 мин измеряют
оптическую плотность при длине волны — 590—600 нм на фото-
электроколориметре КФК-2. Для определения используют кюве-
ты длиной 10 мм. Массовую долю небелковых азотсодержащих со-
единений определяют по градуировочному графику с учетом раз-
ведения пробы.
Для построения калибровочного графика берут казеин с извест-
ной массовой долей общего азота. Растворы готовят так, чтобы мас-
совая доля азотсодержащих соединений в них составляла соответ-
ственно 0,025; 0,05; 0,1; 0,15 и 0,2 %. Дальнейшую обработку прово-
дят так же, как и исследуемых образцов. Полученные величины оп-
тической плотности откладывают по оси ординат, а массовую долю
азотсодержащих соединений по оси абсцисс.
Для определения используют реактивы 1—4, приготовленные,
как указано в предыдущей работе.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА В МОЛОКЕ
ПО СВЯЗЫВАНИЮ КРАСИТЕЛЯ
Метод основан на способности белков молока при pH ниже изо-
электрической точки связывать кислый краситель, образуя с ним
нерастворимый осадок, после удаления которого измеряют опти-
ческую плотность исходного раствора красителя относительно по-
лученного раствора (фильтрата). Оптическая плотность фильтрата
уменьшается с увеличением массовой доли белка.
Оптическую плотность фильтрата определяют на фотоколори-
метре; длина кюветы 10 мм при длине волны 590 нм. Методика мо-
жет быть реализована на универсальных фотоколориметрах КФК-
27
2М, КФК-ЗМ, малогабаритном фотоколориметре КФК-5М, а так-
же приборах «Про-Милк МК-2» фирмы Foss-Electric (Дания) и
«Про-о-Мат-Ш» фирмы Funke-Gerber (Германия) и др.
Выполнение работы. В стеклянную пробирку пипеткой вносят
1 см3 молока, приливают 20 см3 раствора красителя и, закрыв про-
бирку резиновой пробкой, перемешивают ее содержимое, перево-
рачивая пробирку от 2 до 10 раз, не встряхивая.
Пробирку помещают в центрифугу и центрифугируют при час-
тоте вращения 2 5 с~1 (1500 об/мин) в течение 10 мин или при часто-
те вращения 16с1 (1000 об/мин) — в течение 20 мин. После окон-
чания центрифугирования отбирают пипеткой 1 см3 надосадочной
жидкости, помещают в мерную колбу вместимостью 50 см3, долива-
ют колбу до метки водой и перемешивают содержимое. Аналогич-
ным способом разбавляют рабочий раствор красителя в 50 раз.
На фотоэлектроколориметре или спектрофотометре измеряют
оптическую плотность разбавленного раствора красителя по отно-
шению к разбавленному содержимому мерной колбы. После каждых
24 наблюдений кювету промывают буферным раствором 1 (реактив 1).
Массовую долю белка Б (%) вычисляют по формуле
Б= 7,787) — 1,34,
где D — измеренная оптическая плотность; 7,78; 1,34 — эмпирические коэффици-
енты.
За окончательный результат измерения принимают среднее
арифметическое значение результатов вычислений двух параллель-
ных наблюдений, округляя результатдо второго десятичного знака.
Приготовление реактивов. Реактив 1 (буферный раствор). Взве-
шивают 31,70 г лимонной кислоты и 8,40 г (12-водного) или 3,4 г
(безводного) ортофосфата натрия. Результат взвешивания записы-
вают с округлением до второго десятичного знака. Реактивы поме-
щаютв колбу вместимостью 500 см3 и добавляют в нее 400 см3 воды.
Колбу нагревают до температуры не выше 70 °C. Затем содержимое
колбы перемешивают до полного растворения веществ и охлаждают
до температуры 20+2 °C.
Реактив 2 (раствор красителя). Навеску 4,60 г красителя, взве-
шенного с отсчетом до 0,01 г, помещают в колбу вместимостью
500 см3 и добавляют в нее 200 см3 воды. Колбу нагревают до темпе-
ратуры не выше 70 °C и перемешивают ее содержимое до растворе-
ния красителя. Раствор отфильтровываютчерез бумажный фильтр в
мерную колбу вместимостью 2000 см3. Фильтр промывают водой до
удаления следов красителя.
В колбу с реактивом 2 переносят реактив 1. Содержимое колбы
охлаждают до температуры 20+2 °C, после чего доливают водой до
метки, закрывают резиновой пробкой и перемешивают содержи-
мое, переворачивая колбу не менее шести раз.
28
Раствор должен иметь pH 2,3±0,1. Если pH раствора не соответ-
ствует данному значению, то добавляют соответственно концент-
рированную серную кислоту или гидроксид натрия. Раствор, раз-
бавленный в 50 раз, должен иметь оптическую плотность
(0,82±0,02) при длине волны 590 нм в кювете длиной 10 мм. Если
оптическая плотность раствора не соответствует данному значе-
нию, то добавляют реактив 1 (буферный раствор) или реактив 2 (ра-
створ красителя). Полученный раствор следует использовать толь-
ко после 12 ч выдержки. Его нужно хранить в холодильнике в буты-
ли из темного стекла не более 4 мес, еженедельно проверяя и под-
держивая необходимые pH и оптическую плотность.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ
В КАЗЕИНЕ И КАЗЕИНАТЕ НАТРИЯ ПО МЕТОДИКЕ
МЕЖДУНАРОДНОЙ ОРГАНИЗАЦИИ СТАНДАРТИЗАЦИИ (ИСО)
Сущность метода состоит в осаждении казеина уксусной кисло-
той в присутствии ацетата натрия (pH 4,6), последующем фильтро-
вании, добавлении к бесказеиновому фильтрату фенола и концент-
рированной серной кислоты и фотометрировании при длине волны
490 нм образованного комплекса лактозы с фенолом. Метод пре-
дусматривает, что наличие растворимых углеводов в указанных
продуктах составляет менее 2 %.
Для определения содержания лактозы в казеине предусматрива-
ется использовать при его растворении 0,1 г гидрокарбоната натрия
для кислотного казеина и 0,1 г пентатрифосфата натрия
(NasPsOio) — для сычужного. Кроме того, взятый для анализа казе-
ин (средняя проба в количестве 50 г) просеивают через проволочное
сито диаметром 200 мм с размером отверстий 500 мкм. Можно сред-
нюю пробу казеина измельчать на мельнице (нельзя применять мо-
лотковую дробилку). Пробы, взятые для анализа, растворяют в го-
рячей воде.
Выполнение работы. Определение начинают с отвешивания 1 г
средней пробы и помещают ее в коническую стеклянную колбу вме-
стимостью 100 см3. Затем в колбу вносят 0,1 г гидрокарбоната на-
трия для кислотного казеина или 0,1 г пентатрифосфата натрия для
сычужного.
Одновременно готовят холостую пробу, внося в коническую
колбу все те же реактивы, что и для исследуемого образца, кроме
анализируемого продукта.
В колбу добавляют 25 м3 дистиллированной воды температурой
60—65 °C и ставят колбу на водяную баню при температуре 60—
70 °C, перемешивая содержимое встряхиванием. Когда навеска
полностью растворится (обычно через 1Q—15 мин), раствор охлаж-
дают и последовательно вносят в него пипетками 15 см3 дистилли-
рованной воды, 8 см3 0,1 М раствора соляной (или серной) кислоты
29
(реактив 1) и 1 см310%-ного раствора уксусной кислоты (реактив 2).
После внесения каждого раствора колбочку закрывают пробкой и
смесь перемешивают встряхиванием. Полученный раствор остав-
ляют на 5 мин, затем вводят в него пипеткой 1 см31 М раствора аце-
тата натрия и вновь перемешивают встряхиванием.
После осаждения казеина смесь фильтруют через сухой бумаж-
ный фильтр (первые несколько миллилитров фильтрата отбрасыва-
ют). Далее в пробирку вместимостью 40 см3 с притертой пробкой
пипеткой отмеривают 2 см3 фильтрата и микропипеткой — 0,2 см3
80%-ного раствора фенола. Содержимое перемешивают встряхива-
нием. Затем, используя специальный дозатор, в течение 1 с в ту же
пробирку отмеривают 5 см3 концентрированной серной кислоты,
следя за тем, чтобы струя ее попадала не на стенки, а в жидкость, что
способствует лучшему перемешиванию. Смесь в пробирке немед-
ленно перемешивают, оставляют в покое на 15 мин, затем охлажда-
ют в течение 5 мин на водяной бане при 20 °C.
Оптическую плотность смеси измеряют при длине волны 490 нм,
используя для сравнения холостую пробу, и по градуировочному гра-
фику определяют концентрацию безводной лактозы (мкг/см3). Мас-
совую долю лактозы Л (%) в продукте рассчитывают по формуле
Л= Q • 0,005/т,
где С{ — концентрация безводной лактозы в растворе для анализа, определенная по
градуировочному графику, мкг/см3; т — масса пробы, взятая для анализа, г.
Если значение оптической плотности выше, чем верхний предел
градуировочного графика, то в анализе используют 2 см3 разбавлен-
ного раствора фильтрата вместо самого фильтрата. При этом фор-
мула расчета должна быть соответственно изменена.
Чтобы получить данные для построения градуировочного гра-
фика, готовят раствор моногидрата лактозы, для чего навеску лак-
тозы в количестве 2,105 г (соответствует 2 г безводной лактозы) по-
мещают в мерную колбочку вместимостью 100 см3 и растворяют в
дистиллированной воде, доводя ею объем раствора до метки.
Полученный раствор перемешивают и сохраняют в холодильнике
(раствор А). Пипеткой отмеривают 10 см3 раствора и вносят в мер-
ную колбочку вместимостью 100 см3, после чего наполняют ее дис-
тиллированной водой до метки (раствор Б); 1 см3 раствора Б содер-
жит 2 мг безводной лактозы. Затем готовят стандартные растворы
лактозы, отмеривая пипеткой 1,2 и 3 см3 раствора Б в три мерные
колбочки вместимостью 100 см3 и наполняют их дистиллированной
водой до метки. Концентрация безводной лактозы в полученных
стандартных растворах составляет 20, 40 и 60 мкг/см3 соответствен-
но. После этого в четыре пробирки вместимостью 40 см3 с притерты-
ми пробками отмеривают пипетками по 2 см3 каждого из трех стандарт-
ных растворов и по 2 см3 воды, проводят анализ подобно описанному.
30
Полученные величины оптических плотностей откладывают на
оси ординат, а соответствующие им концентрации безводной лак-
тозы — 20, 40 и 60 мкг/см* — на оси абсцисс и строят градуировоч-
ный график.
Приготовление реактивов. Реактив 1 (0,1 М раствор соляной или
серной кислоты) (см. приложения 5,4).
Реактив 2 (10%-ный раствор уксусной кислоты). 100 г уксусной
кислоты помещают в мерную колбу вместимостью 1000 см3 и дово-
дят водой до метки.
Реактив 3(1 Мраствор ацетата натрия). 82 г CHjCOONa ра-
створяют дистиллированной водой в мерной колбе вместимостью
1000 см3.
Реактив 4 (80%-ный раствор фенола). Емкость с кристалличес-
ким фенолом подогревают на водяной бане при температуре 45—
50 °C до расплавления кристаллов (температура плавления фенола
42 °C). Для приготовления 100 г 80%-ного фенола отвешивают 80 г
жидкого фенола и добавляют 20 см3 дистиллированной воды.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ
МЕТОДОМ ЛОРЕНСА
Принцип метода состоит в извлечении белков из исследуемого
продукта (молоко, кисломолочные напитки) путем растворения
гидроксидом натрия, последующем фильтровании, добавлении к
фильтрату фенола и концентрированной серной кислоты и фото-
метрировании образованного комплекса лактозы с фенолом на
спектрофотометре или фотоэлектроколориметре при длине волны
490 нм и черном светофильтре.
Выполнение работы. 1 г продукта смешивают с 25 см3 дистилли-
рованной воды. Добавляют 2 см3 1 М раствора гидроксида натрия
(реактив 1) и помещают в мерную колбу на 100 см3, перемешивают
до растворения белков и доводят объем дистиллированной водой до
метки. Раствор фильтруют, 1 см3 фильтрата смешивают с 1 см3 5%-
ного раствора фенола и 5 см3 концентрированной серной кислоты,
определяют оптическую плотность, используя для сравнения конт-
рольную пробу.
В качестве контрольной пробы используют смесь 1 см3 воды,
1 см3 5%-ного раствора фенола и 5 см3 концентрированной серной
кислоты.
Массовую долю лактозы в продукте рассчитывают по градуиро-
вочному графику в пробе продукта, затем пересчитывают на 100 г
продукта по формуле
Л= С/100,
где Л— массовая доля лактозы в исследуемом продукте, %; С — концентрация лак-
тозы в растворе для анализа по градуировочному графику, мкг/см3.
31
Чтобы получить данные для построения графика, из 2%-ного ра-
створа лактозы (реактив А) готовят 0,2%-ный раствор (реактив Б).
Для этого в мерную колбу вместимостью 200 см3 вносят 20 см3 реак-
тива А и доводят объем дистиллированной водой до метки, а из по-
лученного раствора (реактив Б) готовят следующие разведения
(табл. 5):
5. Концентрация лактозы и объем разведения
Объем, см1 Концентрация лактозы в разведении, мкт/см5
Реактив Б 1 Дистиллированная вода
1 99 20
2 98 40
3 97 60
4 96 80
5 95 100
10 90 200
15 85 300
20 80 400
25 75 500
30 70 600
35 65 700
40 60 800
Для фотометрирования берут 1 см3 каждого разведения, смеши-
вают с 1 см3 5%-ного раствора фенола и с 5 см3 концентрированной
серной кислоты. Определяют оптическую плотность и строят гра-
дуировочный график.
Приготовление реактива. 1 Мраствора гидроксида натрия, 70 см3
насыщенного раствора гидроксида натрия вносят в мерную колбу
вместимостью 1000 см3 и доводят объем до метки свежепрокипя-
ченной и охлажденной водой.
Насыщенный раствор гидроксида натрия готовят в соответствии
с приложением 3.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ УГЛЕВОДОВ
В ЖИДКОЙ КИСЛОМОЛОЧНОЙ СМЕСИ «МАЛЮТКА»
Метод основан на реакции углеводов с 10 М серной кислотой, из-
мерении оптической плотности окрашенного раствора на фотоэлек-
троколориметре при длине волны 450 нм с синим светофильтром.
Выполнение работы. В стеклянную пробирку пипеткой отмери-
ваются см3 дистиллированной воды и 7,5 см310 М раствора серной
кислоты (реактив 1). Хорошо перемешивают, выдерживают на ки-
пящей водяной бане в течение 20 мин, охлаждают до температуры
20 °C. Полученный раствор используют в качестве холостой пробы.
В стакане вместимостью 50см3 взвешивают Юг продукта. На-
веску растирают стеклянной палочкой с небольшим количеством
32
дистиллированной воды температурой 40—50 °C и переливают в
мерную колбу вместимостью 100 см3. Стакан ополаскивают водой,
которую затем сливают в колбу. Для осаждения белков в колбу при-
ливают по 0,5 см3 растворов ацетата цинка (реактив 2) и гексациа-
ноферрата (II) калия (реактив 3). После внесения каждого раствора
смесь перемешивают. Содержимое колбы доливают до метки во-
дой, перемешивают и спустя 5 мин фильтруют через сухой складча-
тый фильтр в сухую колбу.
В стеклянную пробирку пипеткой отмеривают 0,5 см3 фильтра-
та, 7,5 см3 10 М серной кислоты (реактив 1). Хорошо перемешива-
ют, выдерживают на кипящей водяной бане в течение 20 мин, ох-
лаждают до температуры 20 °C и фотометрируют, используя кювету
длиной 10 мм.
На фотоэлектроколориметре с синим фильтром X = 450±10 нм.
Измеряют плотность исследуемого раствора относительно холос-
той пробы. Отсчитывают показания D.
Массовую долю углеводов X(%) вычисляют по формуле
^=0Д94
где D — оптическая плотность; 0,094 и 0,8 — коэффициенты.
Расхождения между параллельными определениями массовой
доли углеводов не должны превышать 0,5 %.
Приготовление реактивов. Реактив 1 (10Мраствор серной кисло-
ты). 569 см3 концентрированной серной кислоты плотностью
1,832 г/см3 смешивают с дистиллированной водой и доводят объем
до 1000 см3.
Реактив 2 (раствор ацетата цинка). 30 г ацетата цинка взвеши-
вают и растворяют в мерной колбе вместимостью 100 см3.
Реактив 3 (раствор гексацианоферрата (II) калия). 15 г К4 [Fe (CN)g]
взвешивают, растворяют в мерной колбе вместимостью 100 см3.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЖЕЛЕЗА В МОЛОКЕ
И МОЛОЧНЫХ ПРОДУКТАХ ПО МЕТОДИКЕ
МЕЖДУНАРОДНОЙ ОРГАНИЗАЦИИ СТАНДАРТИЗАЦИИ (ИСО)
Метод основан на предварительной минерализации пробы ис-
следуемого продукта (или плазмы для сливок и сливочного масла)
смесью азотной и серной кислот, последующем восстановлении
ионов железа (III) батофенантролином с образованием окрашен-
ных комплексов железа (II) и их экстракции изоамиловым спиртом.
Оптическую плотность полученного раствора определяют на спект-
рометре при длине волны 533 нм, используя кюветы длиной 10 мм.
Массовую долю железа определяют по градуировочному графику.
33
Для минерализации анализируемых проб используют колбы вме-
стимостью примерно 70 см3 с притертыми стеклянными пробками.
Выполнение работы. Вначале минерализуют пробу. В колбу для
минерализации помещают анализируемую пробу и три стеклянные
(кварцевые) бусины. Под мощной вытяжкой устанавливают колбу в
наклонном положении и нагревают ее на горелке. Высоту пламени
регулируют для ограничения пенообразования в колбе. Допускает-
ся образование пены в горловине колбы, но без ее выплескивания.
Поддерживают слабое кипение смеси.
Одновременно проводят холостую пробу. Для этого в колбу для
минерализации вместо анализируемой пробы берут 10 см3 воды и те
же реактивы, что и для анализируемой пробы. В холостой пробе не
должно содержаться более 0,5 мгжелеза, в противном случае все ре-
активы необходимо проверить.
Когда раствор приобретает коричневую окраску, осторожно до-
бавляют 3—5 капель азотной кислоты (реактив 4) и энергично на-
гревают. Продолжают нагревание, добавляя по 5—20 капель азот-
ной кислоты, пока смесь станет бесцветной. Охлаждают до комнат-
ной температуры.
Осторожно добавляют 2 см3 воды и 1 см3 раствора пероксида во-
дорода (реактив 7). Размешивают и снова нагревают до появления
белых паров. При пожелтении раствора его охлаждают до комнат-
ной температуры. Добавляют еще 0,5 см3 раствора пероксида водо-
рода, а затем нагревают до образования белых паров. После того
как начнут образовываться белые пары, смесь нагревают еще в тече-
ние 45 мин. Затем охлаждают до комнатной температуры и осто-
рожно доливают воду до полного объема, равного примерно 20 см3.
Добавляют одну или две капли раствора перманганата калия (ре-
актив 12) до слабо-алого окрашивания смеси, затем 3 см3 раствора
хлорида гидроксиламмония (реактив 9) и тщательно перемешива-
ют. Вносят 20 см3 раствора ацетата натрия (реактив 8) и примерно
15 см3 воды, тщательно перемешивают и оставляют в покое. После
того как температура смеси будет доведена до комнатной, смесь до-
ливают водой до отметки 50 см3.
К содержимому колбы для минерализации пипеткой добавляют
4 см3 раствора батофенантролина (реактив 11) и закрывают колбу
пробкой. Энергично встряхивают колбу в течение 3 мин, следя за
тем, чтобы пробка оставалась на месте.
Охлаждают проточной водой в течение 10 мин и несколько раз
после охлаждения осторожно наклоняют колбу. Выдерживают кол-
бу на водяной бане в течение 1 ч при 25± 1 °C.
Переносят пипеткой верхний слой изоамилового спирта в кюве-
ту длиной 10 мм. Измеряют оптическую плотность изоамилового
спирта анализируемого раствора и контрольной пробы при длине
волны 533 нм, используя в качестве эталона воду. Из полученных
данных находятразность между значением, полученным для анали-
зируемого раствора, и значением, полученным для контрольной
34
пробы. Для всех определений, включая контрольную пробу, прово-
дят по два испытания.
Построение градуировочного графика. Пипеткой переносят в
шесть колб для минерализации поО; 1; 2; 3; 5 и 10 см3 стандартно-
го раствора железа (реактив 14). Доливают водой до 10 см3. В
каждую колбу добавляют по 3 см3 азотной кислоты (реактив 4) и
по 1,8 см3 раствора сульфата калия. Проводят минерализацию и
окрашивание.
Пипеткой переносят в кювету длиной 10 мм из каждой колбы
слой (верхний) изоамилового спирта. Измеряют оптическую плот-
ность изоамилового спирта при длине волны 533 нм, используя в
качестве эталона воду. Вычитают значение, полученное для воды,
из значений, полученных для других растворов.
Строят график зависимости оптической плотности от количе-
ства железа, содержащегося в рабочих стандартных растворах.
Содержание железа тре (мг/кг) в анализируемом продукте (кро-
ме сливочного масла) рассчитывают по формуле
где и, — масса железа, определяемая по градуировочному графику, мкг; т0 — масса
пробы продукта для анализа, г.
Содержание железа в сливочном масле:
т -т т>
гс гт т
где /и3 — масса плазмы, полученной из пробы масла, г; /и2 — масса пробы масла, г;
mi — масса железа в плазме, определяемая по градуировочному графику, мкг; /ио —
масса плазмы, взятая для анализа, г.
Содержание железа в продукте рассчитывают с точностью до
0,001 для молока, кефира, пахты, молочной сыворотки, сливок,
сливочного масла, молочного жира и с точностью до 0,01 — для мо-
лока сгущенного с сахаром и без сахара, молока сухого, сухой пахты,
мороженого, сыра натурального и плавленого, казеина, казеинатов
и молочного белка.
Приготовление реактивов. Все используемые реактивы должны
быть чистыми для анализа и кроме стандартных растворов железа
не содержать железа.
Реактив 1 (этанол). Концентрация раствора этанола должна со-
ставлять примерно 96 об. %. При необходимости проводят перегон-
ку в не содержащей железо перегонной установке.
Реактив 2 (диэтиловый эфир). При необходимости проводят пе-
регонку в не содержащей железо перегонной установке.
Реактив 3 (петролейный эфир). Пределы кипения 40—60 °C. При
необходимости проводят перегонку в не содержащей железо пере-
гонной установке.
35
Реактив 4 (азотная кислота, концентрированная, плотностью
1,42 г/см3). Проводят перегонку в не содержащей железо перегон-
ной установке. Первые 50 см3 дистиллята сливают. Не допускается
хранение азотной кислоты в банке из темного стекла.
Реактив 5 (серная кислота концентрированная, плотностью
1,84 г/см3).
Реактив 6(раствор сульфата калия в серной кислоте). Растворяют
25 г сульфата калия (K2SO4) в серной кислоте (реактив 5) и долива-
ют до 100 см3 той же серной кислотой. Раствор фильтруют через
стеклянный фильтр (диаметр пор 40—100 мкм).
Если имеющийся сульфат калия содержит железо, его очищают следующим об-
разом. Растворяют 40 г сульфата калия в 500 см3 воды и добавляют 3 см3 раствора
хлорида гидроксиламмония (реактив 9). Экстрагируют раствор 10 см3 раствора ба-
тофенантролина (реактив 11). Верхний слой сливают. Повторяют эти две операции,
пока верхний слой станет бесцветным. Выпаривают воду в чистой печи.
Реактив 7 (раствор пероксида водорода плотностью 1,099—
1,103 г/см3). Раствор хранят в холодильнике.
Реактив 8 (насыщенный раствор ацетата натрия). Растворяют
232,5 г безводного ацетата натрия (СН3 COONa) в 500 см3 воды.
Если имеющийся ацетат натрия содержит железо, его очищают следующим об-
разом. Растворяют 232,5 г ацетата натрия в 500 см3 воды. Фильтруют через фильтро-
вальную бумагу. Добавляют 3 см3 раствора хлорида гидроксиламмония (реактив 9).
Экстрагируют раствор 10 см3 раствора батофенантролина (реактив 11). Верхний
слой сливают. Повторяют эти две операции, пока верхний слой станет прозрачным.
Реактив 9 (раствор хлорида гидроксиламмония). Растворяют 20 г
хлорида гидроксиламмония (HONH3C1) в воде и доливают до
100 см3. Фильтруют через фильтровальную бумагу. Экстрагируют
раствор 5 см3 раствора батофенантролина (реактив 11), выдержива-
ют до нужного разделения слоев. Верхний слой сливают. Повторя-
ют эти две операции, пока верхний слой станет бесцветным.
Обычно достаточно 5-кратного экстрагирования.
При приготовлении более чем за 24 ч до использования рекомендуется повто-
рить экстрагирование батофенантролином.
Вместо раствора хлорида гидроксиламмония в качестве восстановителя можно
использовать раствор аскорбиновой кислоты свежей перегонки. Раствор аскорби-
новой кислоты получают путем растворения 10 г аскорбиновой кислоты в 100 см3
воды. Раствор экстрагируют раствором батофенантролина так же, как было описано
выше для раствора хлорида гидроксиламмония. Раствор должен храниться в холо-
дильнике. При приготовлении реактивов 6, 8 вместо 3 см3 раствора хлорида гидро-
ксиламмония можно использовать 3 см3 такого раствора аскорбиновой кислоты.
Реактив 10 (изоамиловый спирт). При необходимости перегоня-
ют на не содержащей железо перегонной установке.
Реактив 11 (раствор батофенантролина). Растворяют 83,1 мг ба-
тофенантролина (4,7 дифенил — 1,10 фенантролина (C24H)6N2) в
100 см3 изоамилового спирта (реактив 10).
36
Реактив 12 (раствор перманганата калия). Растворяют 100 мг
перманганата калия (КМпО4) в 50 см3 воды.
Реактив 13 (стандартныйраствор железа, соответствующий со-
держанию ЮООмгжелезав 1000 см3). Растворяют 7,022 г гексагидра-
та двойной соли сульфата аммония и сульфата железа
[(NH4)2Fe(SO4) • 6Н2О] в250 см3 воды. Добавляют 8 см3 серной кис-
лоты (реактив 5) и охлаждают до комнатной температуры. Долива-
ют водой до 1000 см3. В 1 см3 такого стандартного раствора содер-
жится 1 мг железа.
Допускается использовать вместо гексагидрата двойной соли сульфата аммония
и сульфата железа [2] препараты, содержащие точно 1000 мг железа. Такие препара-
ты в настоящее время выпускаются промышленностью.
Реактив 14 (стандартный раствор железа, содержащий 1мг же-
леза на 1000 см3). В день использования пипеткой переносят 1 см3
стандартного раствора железа (реактив 13) в 250 см3 воды. Добавля-
ют 1 см3 серной кислоты (реактив 5) и доливают до 1000 см3 водой. В
1 см3 такого стандартного раствора содержится 1 мкг железа.
Подготовка проб. При отборе и подготовке проб загрязнение же-
лезом не допускается.
Молоко, обезжиренное молоко и сыворотка. Пробу нагревают до
20±2 °C и тщательно размешивают. Пробу молока, в которой жир
распределен неравномерно, медленно нагревают до 40 °C, осторож-
но перемешивают, переворачивая пробирку, и быстро охлаждают
до 20±2 °C.
Пахта. При необходимости удаляют зерна жира. Температуру
пробы повышают до 20±2 °C и осторожно размешивают непосред-
ственно перед взвешиванием.
Кефир жирный и обезжиренный. Пробу нагревают до 20±2 °C и
тщательно размешивают. При отделении сыворотки энергично раз-
мешивают непосредственно перед взвешиванием пробы для анализа.
В колбу для минерализации отвешивают 10 г молока, обезжи-
ренного молока, сыворотки, пахты или кефира. Добавляют 3 см3
азотной кислоты (реактив 4) и 1,8 см3 раствора сульфата калия (ре-
актив 6). Проводят анализ.
Сливки. Пробу нагревают до 20+2 °C. Тщательно перемешивают,
не допуская при этом взбалтывания или вспенивания. Очень густые
сливки или сливки, в которых жир распределен неравномерно, для
облегчения размешивания медленно нагревают до 40 °C. Быстро ох-
лаждают пробу до 20±2 °C. Тщательно размешивают до полной од-
нородности массы. Емкость закрывают.
Отвешивают 10 г пробы с точностью до 10 мг и переносят в колбу
для минерализации. Добавляют 8 см3 азотной кислоты (реактив 4).
Колбу нагревают на водяной бане при 80—90 °C в течение 1 ч. Каж-
дые 3 мин энергично встряхивают колбу для смывания жира азот-
ной кислотой. Охлаждают до 40 °C и с помощью пипетки снимают
как можно больше жира.
37
Добавляют 15 см3 петролейного эфира (реактив 3), осторожно
размешивают и удаляют растворитель пипеткой. Процедуру повто-
ряют дважды со свежими 15-миллилитровыми порциями петролей-
ного эфира. Остаточный петролейный эфир удаляют путем нагре-
вания на водяной бане при 65 °C. Охлаждают до комнатной темпе-
ратуры. Добавляют 1,8 см3 раствора сульфата калия (реактив 6).
Проводят анализ.
Сгущенное молоко. Осторожно встряхивают емкость, часто пере-
ворачивая ее. Открывают емкость и медленно переливают молоко в
другую емкость из стекла с герметичной крышкой, стараясь собрать
в пробу весь жир или ингредиенты, приставшие к стенкам первой
емкости. Содержимое энергично размешивают и закрывают ем-
кость.
Закрытую емкость нагревают на водяной бане до 40—60 °C. Каж-
дые 15 мин емкость вынимают и энергично встряхивают. Через 2 ч
емкость снимают и охлаждают до 20±2 °C. Снимают крышку, тща-
тельно размешивают пробу ложкой или шпателем.
Сгущенное молоко с сахаром. Открывают емкость и тщательно
размешивают молоко ложкой или шпателем, перемешивая содер-
жимое круговыми движениями сверху вниз так, чтобы верхние и
нижние слои перемещались и перемешивались. Необходимо сле-
дить за тем, чтобы в пробу попадало все молоко, приставшее к стен-
кам и крышкам емкости.
Количественно переносят пробу во вторую емкость из стекла с
герметичной крышкой и закрывают контейнер. Нагревают закры-
тую емкость на водяной бане до 30—40 °C, охлаждают до 20±2 °C.
Тщательно размешивают пробу до получения однородной массы.
Закрывают емкость.
Отвешивают 2,5 г пробы сгущенного молока или сгущенного мо-
лока с сахаром с точностью до 1 мг в колбу для минерализации. До-
бавляют 4 см3 воды, 3 см3 азотной кислоты (реактив 4) и 1,8 см3 ра-
створа сульфата калия (реактив 6). Проводят анализ.
Сухие цельное и обезжиренное молоко, молочная сыворотка и пах-
та. Пробу переносят в емкость с герметичной крышкой вместимос-
тью вдвое больше объема пробы. Емкость сразу же закрывают. Тща-
тельно размешивают пробу, многократно встряхивая и переворачи-
вая емкость.
Отвешивают 1 г пробы с точностью 1 мг в колбу для минерализа-
ции. Добавляют 4 см3 воды и хорошо размешивают. Затем добавля-
ют 3 см3 азотной кислоты (реактив 4) и 1,8 см3 раствора сульфата
калия (реактив 6). Проводят анализ.
Масло. Из-за неравномерного распределения в масле железа его
определяют в плазме. Содержание железам жире мало по сравне-
нию с его содержанием в плазме и может не учитываться. Поэтому
сначала экстрагируют жир, а затем исследуют плазму масла.
Отвешивают 100 г пробы с точностью до 100 мг в сухую предва-
рительно оттарированную центрифужную пробирку. Помещают
38
пробирку на водяную баню при 45± 1 °C. Как только масло начи-
нает плавиться, проводят центрифугирование. С помощью пи-
петки отбирают как можно больше прозрачного слоя жира. Экст-
рагируют и снимают пипеткой верхний слой, добавляя 10 см3
петролейного эфира (реактив 3). Эти две операции повторяют
дважды. Остаточный петролейный эфир удаляют, нагревая пробу
на водяной бане при 65± 1 °C. Протирают насухо пробирку снару-
жи чистой бумажной салфеткой и охлаждают до 20±2 °C. Тща-
тельно перемешивают содержимое непосредственно перед взве-
шиванием пробы для анализа. Взвешивают пробирку с содержи-
мым с точностью до 100 мг.
Отвешивают 2 г плазмы с точностью до 1 мг в колбу для минера-
лизации. Добавляют 3 см3 азотной кислоты (реактив 4) и 1,8 см3 ра-
створа сульфата калия (реактив 6). Проводят анализ.
Молочный жир. Нагревают пробу до 40 °C, выдерживают при этой
температуре в течение 5 мин и осторожно размешивают. Охлажда-
ют до 20±2 °C. Отвешивают 20 г жидкой пробы с точностью до 10 мг
в колбу для минерализации. Добавляют 4 см3 воды и 8 см3 азотной
кислоты (реактив 4).
Колбу нагревают на водяной бане при 80—90 °C в течение 1 ч.
Осторожно встряхивают через каждые 3 мин, чтобы смыть жир
азотной кислотой. Охлаждают до 40 °C и с помощью пипетки удаля-
ют как можно больше жирового слоя.
Добавляют петролейный эфир (реактив 3), объем которого дол-
жен быть больше объема исследуемого раствора, осторожно раз-
мешивают и удаляют растворитель пипеткой. Дважды повторяют
процедуру со свежими 15-миллилитровыми порциями петролей-
ного эфира. Остаточный петролейный эфир удаляют нагреванием
на водяной бане при 65 °C. Охлаждают до комнатной температуры.
Добавляют 1,8 см3 раствора сульфата калия (реактив 6). Проводят
анализ.
Натуральный и плавленый сыр. С сыра удаляют оболочку, а также
оплывшие, засохшие или заплесневелые поверхностные края так,
чтобы получить пробу в том виде, в каком сыр обычно употребляют
в пищу. Пробу измельчают. Всю массу быстро перемешивают и сно-
ва быстро измельчают. Если пробу измельчить невозможно, ее тща-
тельно размешивают. Сразу же после этого предварительно обрабо-
танную пробу или ее часть переносят в емкость с герметичной
крышкой. После измельчения как можно скорее проводят анализ
пробы.
Отвешивают 1 г пробы с точностью до 1 мг в колбу для минерали-
зации. Добавляют 4 см3 воды, 3 см3 азотной кислоты (реактив 4) и
1,8 см3 раствора сульфата калия (реактив 6). Проводят анализ.
Казеин, казеинаты и молочный белок. Отобранную пробу предва-
рительно измельчают. Если пробу можно просеять через сито, то ее
используют без измельчения.
39
50 г пробы переносят в том виде, в каком она получена, в емкость
с герметичной крышкой, вместимостью вдвое больше объема по-
рошка. Емкость сразу закрывают и тщательно размешивают пробу,
многократно встряхивая и переворачивая емкость.
Отвешивают в колбу для минерализации 35 г пробы казеина, ка-
зеината или молочного белка (при этом казеин и казеинаты взвеши-
вают с точностью до 0,1 мг, а молочный белок — до 0,75 мг). Добав-
ляют 4 см3 воды, 3 см3 азотной кислоты (реактив 4) и 1,8 см3 раство-
ра сульфата калия (реактив 6). Проводят анализ.
Мороженое. Пробы помещают в емкости с герметичной крыш-
кой. Пробы, отбираемые из больших упаковок, сохраняют в емкос-
тях для пробы. Расплавление пробы выполняют в закрытой емкос-
ти, выдерживая ее на водяной бане при 45± 1 °C до перехода пробы в
жидкое состояние. Пробу размешивают встряхиванием. Охлаждают
пробу до 20±2 °C, продолжая размешивание до полного охлаждения
пробы.
Отвешивают 2,5 г пробы мороженого с точностью до 1 мг в колбу
для минерализации. Добавляют 3 см3 азотной кислоты (реактив 4) и
1,8 см3 раствора сульфата калия (реактив 6). Проводят анализ.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НИТРАТОВ И НИТРИТОВ
Фотометрический метод определения содержания нитратов и
нитритов применим для всех видов молочных продуктов.
Сущность метода определения нитритов заключается в экстра-
гировании их водой, очистке экстракта и фотометрическом измере-
нии интенсивности окраски азосоединения, образующегося при
взаимодействии нитритов с ароматическими аминами.
Нижний предел определения нитритов — 0,02 мг NO2 в 1 см3 ко-
лориметрируемого раствора. Нижний предел определения нитри-
тов в анализируемой пробе — 0,1 мг NO2 в 1 кг жидких продуктов и
0,5 мг NO2 в 1 кг твердых продуктов.
Сущность метода определения нитратов заключается в экстраги-
ровании их водой, очистке экстракта, восстановлении нитратов до
нитритов на кадмиевой колонке с последующим фотометрическим
измерением интенсивности окраски азосоединения, образующего-
ся при взаимодействии нитритов с ароматическими аминами. Ниж-
ний предел определения нитратов — 0,03 мг NO3 в 1 см3 колоримет-
рируемого раствора. Нижний предел определения нитратов в ана-
лизируемой пробе — 0,5 мг NO3 в 1 кг жидких продуктов и 1,5 мг
N О3 в 1 кг твердых продуктов.
Спектрофотометр любой марки или фотоколориметр для изме-
рения оптической плотности в диапазоне видимой области спектра
с кюветами длиной 10—40 мм.
Выполнение работы. Пробы молока, сливок, жидких кисломо-
лочных продуктов, пахты, молочной сыворотки и жидких замени -
40
телей цельного молока в количестве 100 см3 помещают в градуиро-
ванную коническую колбу вместимостью 250 см3, добавляют 50 см3
теплой (50—55 °C) дистиллированной воды и перемешивают.
Пробы твердого и плавленого сыров, сметаны, творога, творож-
ных изделий, сгущенных молочных продуктов, сливочного масла и
мороженого в количестве 20 г (творог и сыр предварительно из-
мельчают с помощью устройства для измельчения проб) помещают
в градуированную коническую колбу вместимостью 200 см3, добав-
ляют 100 см3 теплой (50—55 °C) дистиллированной воды и переме-
шивают до однородного состояния.
Пробы молока, сухих и молочных смесей для детского питания в
количестве Юг помещают в градуированную коническую колбу
вместимостью 200 см3, добавляют 100 см3 теплой (50—55 °C) дис-
тиллированной воды и перемешивают до однородного состояния.
Суспензию или эмульсию молочных продуктов в воде выдержи-
вают на водяной бане в течение 5 мин, при периодическом переме-
шивании добавляют 12 см3 раствора сульфата цинка (реактив 3) или
12 см3 ацетата цинка (реактив 2), 12 см3 раствора гексацианоферра-
та калия (реактив 1), 40 см3 аммиачного буфера (реактив 8). Выдер-
живают на водяной бане 15—20 мин, периодически встряхивая кол-
бу. Охлаждают до комнатной температуры, доводят объем дистил-
лированной водой до 200 см3 (до 250 см3 при анализе молока и жид-
ких молочных продуктов), перемешивают и фильтруют через
бумажный фильтр (фильтрат 1). Фильтрат 1 служит для определе-
ния нитратов и нитритов.
В мерную колбу на 200—250 см3 вносят 100 см3 дистиллирован-
ной воды, добавляют реактивы для осаждения в количествах, соот-
ветствующих виду продукта, выдерживают на водяной бане 15—
20 мин, охлаждают до комнатной температуры, доводят объем дис-
тиллированной водой до 200—250 см3 и фильтруют (фильтрат 2).
Для определения содержания нитритов в мерную колбу вмести-
мостью 50 см3 вносят 20 см3 фильтрата 1 и проводят цветную реак-
цию как при построении градуировочного графика. Контролем
служит раствор, в котором вместо фильтрата 1 используют воду.
Если фильтраты используемых продуктов окрашены, то в ка-
честве контрольного раствора применяют смесь без добавления
дигидрохлорида N-1 (нафтил) — этилендиамина (или 1-нафти-
ламина).
По полученному значению оптической плотности с помощью
градуировочного графика находят концентрацию нитрит-иона.
Для определения содержания нитратов в стакан или колбу вмес-
тимостью 50 см3 вносят от 5 до 20 см3 фильтрата 1 в зависимости от
предполагаемого содержания нитратов в анализируемой продук-
ции. При ожидаемой концентрации нитратов от 0 до 300 мг/кг
объем фильтрата используемого для восстановления на колонке,
составляет 20 см3, при концентрации нитратов 300—800 мг/кг —
10 см3, свыше 800 мг/кг — 5 см3. К указанному раствору добавляют
41
5 см3 аммиачного буфера (реактив 8), перемешивают, пропускают
через кадмиевую колонку со скоростью 5 см3/мин. Собирают элюат
вмерную колбу вместимостью 100 см3. Емкости из-под фильтрата и
буфера дважды промывают порциями воды по 15 см3 и также про-
пускают через слой кадмия. Затем одну из емкостей заполняют во-
дой и, если ожидаемая концентрация нитратов в образце составляет
от 0 до 50 мг/кг, продолжают элюировать до накопления около
80 см3 элюата, затем колбу удаляют. К полученному элюату добав-
ляют реагенты для проведения цветной реакции, как при построе-
нии градуировочного графика измеряют оптическую плотность по-
лученного раствора по сравнению с оптической плотностью конт-
рольной пробы продукта, полученной аналогично опытному образ-
цу, заменив фильтрат 1 на 20 см3 фильтрата 2.
Если ожидаемая концентрация нитратов в образце более 50 мг/кг,
элюацию продолжают до объема 100 см3. Для проведения цветной
реакции используют 20 см3 этого элюата (если предполагаемая кон-
центрация нитратов не превышает 1000 мг/кг) или 5—10 см3 элюата
(при концентрации нитратов в образце свыше 1000 мг/кг).
Обработка результатов. Содержание нитритов X (мг/кг) в твер-
дых продуктах (в пересчете на нитрит-ион), вычисляют по формуле
Г-Ж. г
л ' Л/й4 Л1’
где X— концентрация нитритов, мг/кг; Q — концентрация нитритов по градуиро-
вочному графику, мкг/см3; 1/ — общий объем экстракта, см3; Й2 — объем колори-
метрируемого раствора, см3; К\ — коэффициент, учитывающий влияние матрицы
молочного продукта при определении нитритов (табл. 6); М— масса навески образ-
ца для анализа, г; — объем фильтрата 1 для цветной реакции, см3.
Содержание нитритов X(мг/дм3) в жидких продуктах (в пересче-
те на нитрит-ион) вычисляют по формуле
у_СуУ2 г
“ ЙЗГ4 Л1’
где X— концентрация нитритов, мг/дм3; Из — объем образца продукта, взятый для
анализа, см3.
Содержание нитратов в твердых продуктах Y(мг/кг) (в пересчете
на нитрат-ион) вычисляют по формуле
V — 1 Гу']
где Ki — коэффициент, учитывающий влияние матрицы молочного продукта при
определении нитратов (табл. 6); С2 — концентрация нитритов по градуировочному
графику, мкг/см3; И5 — объем элюата с колонки, см3; V6 — объем колориметрируе-
мого раствора, см3; И7 — объем фильтрата, взятый для восстановления на колонку,
см3; Й8 — объем элюата для цветной реакции, см3.
42
6. Коэффициенты, учитывающие влияние матрицы продукта
при определении нитратов и нитритов
Продукт К\ (для нитритов) Кг (для нитратов)
Молоко пастеризованное 1,09 1,06
Сливки 1,05 1,07
Ке< тир жирный 0,95 1,05
Кет тир фруктовый 1,09 1,18
Ке< тир с витамином С 1,13 1,14
Простокваша 1,00 1,00
Ряженка 0,87 0,99
Сметана 1,00 1,09
Творог жирный 1,00 0,93
Творожные изделия 0,83 0,93
Паста ацидофильная 1,00 1,00
Сыры 0,96 1,05
Мороженое 1,00 0,99
Сухое молоко 1,08 1,05
Сгущенное молоко 0,99 1,05
Содержание нитратов в жидких продуктах (в пересчете на нит-
рат-ион) вычисляют по формуле
у _ 1 о <
Л)’
где Y — концентрация нитратов, мг/дм3; Iq —объем образца продукта, взятый для
анализа, см3.
Приготовление реактивов. Растворы для осаждения
белков. Реактив 1 (раствор гексацианоферрата (II) калия. 106г
гексацианоферрата (II) калия растворяют в воде и доводят объем ра-
створа до 1000 см3 водой.
Реактив 2 (раствор ацетата цинка). 220 г ацетата цинка раство-
ряют в смеси воды и 30 см3 ледяной уксусной кислоты и доводят
объем раствора до 1000 см3.
Реактив 3 (раствор сульфата цинка). 535 г сульфата цинка ра-
створяют в воде и доводят объем раствора до 1000 см3 водой.
Реактив 4 (раствор тетрабората натрия). 50 г тетрабората на-
трия растворяют в 1000 см3 теплой дистиллированной воды темпе-
ратурой 50 ± 2 °C и охлаждают до температуры 20 ± 2 °C. Объем до-
водят до метки дистиллированной водой.
Реактив 5 (раствор соляной кислоты концентрацией 2, Омоль/дм3).
160 см3 соляной кислоты (плотностью 1,19 г/см3) растворяют в мер-
ной колбе вместимостью 1 дм3 в дистиллированной воде и доводят
объем до метки.
Реактив 6 (раствор соляной кислоты концентрацией 0,1 моль/дм3).
50 см3 раствора соляной кислоты (реактив 5) растворяют в дистил-
лированной воде в мерной колбе вместимостью 1 дм3 и доводят
объем до метки.
43
Реактив 7 (раствор гидроксида натрия). 30 г гидроксида натрия
растворяют в воде и доводят объем раствора водой до метки 100 см3.
Реактив 8 (буферный раствор pH 9,6—9,7). В мерную колбу вмес-
тимостью 1000 см3 вносят около 600 см3 воды, 50 см3 концентриро-
ванной соляной кислоты, 10 г трилона Б, встряхивают и добавляют
135 см3 водного аммиака плотностью 0,88 г/см3. Перемешивают,
проверяют потенциометрически pH и доводят его при необходимо-
сти до 9,6—9,7. В раствор добавляют дистиллированную воду до
метки.
Реактив 9(раствор сульфата кадмия). 100 г сульфата или хлори-
да кадмия растворяют в воде и доводят объем раствора до 1000 см3
водой.
Реактив 10 (раствор сульфата меди). 20 г сульфата меди раство-
ряют в воде и доводят объем раствора до 1000 см3 водой.
Реактив 11 (раствор трилона Б). 33,5 г динатриевой соли этилен-
диаминтетрауксусной кислоты растворяют в воде и доводят раствор
до 1000 см3 водой.
Реактив 12 (раствор для обработки кадмиевой колонки). Смеши-
вают 50 см3 раствора трилона Б. 20 см3 раствора соляной кислоты
(реактив 6) и доводят раствор до 1000 см3.
Стандартные растворы нитрата калия. Реактив 13
(основнойраствор, содержащий нитрата калия 1 мг/см3). 1,63 г нит-
рата калия, перекристаллизованного из воды и высушенного до по-
стоянной массы при 105—110 °C, вносят в мерную колбу вместимо-
стью 1000 см3, растворяют в воде и доводят объем до метки водой.
Раствор хранят в холодильнике не более 6 мес.
Реактив 14 (рабочий раствор нитрата калия, содержащий 10 мкг/
см3). Пипеткой вносят 10 см3 основного стандартного раствора в
мерную колбу на 1000 см3, добавляют 20 см3 буферного раствора
(реактив 8) и доводят объем до метки водой. Раствор используют в
день проведения анализа.
Стандартные растворы нитрита натрия. Реактив
15(основнойраствор, содержащий нитрита натрия 0,2мг/см3). 0,15 г
нитрита натрия, перекристаллизованного из воды и высушенного
до постоянной массы при 105—110 °C, вносят в мерную колбу вмес-
тимостью 500 см3, доводят объем раствора до метки водой.
Раствор хранят в холодильнике не более недели.
Реактив 16 (рабочий раствор, содержащий нитрита натрия 2 мкг/
см3). Пипеткой вносят 10 см3 основного раствора в мерную колбу на
1000 см3, добавляют 20 см3 буферного раствора (реактив 8) и дово-
дят объем до метки водой. Раствор используют в день проведения
анализа.
Растворы для проведения цветной реакции. Ре-
актив 17(раствор сульфаниламида). В мерную колбу на 100 см3 по-
мещаются г сульфаниламида, добавляют 75 см3 дистиллированной
воды и 5 см3 концентрированной соляной кислоты. Смесь выдер-
живают на кипящей водяной бане до полного растворения сульфа-
44
ниламида, затем раствор охлаждают и доводят до метки водой. Если
необходимо, фильтруют.
Реактив 18 (раствор N-( 1-нафтил)-этилендиамина дигидрохлори-
да (НЭДА). 0,1 г НЭДА растворяют в мерной колбе вместимостью
50 см3 в дистиллированной воде и доводят объем раствора до метки.
Растворы 17 и 18 хранят в холодильнике в склянке из темного
стекла с притертой пробкой не более 1 мес.
Реактив 19 (раствор сульфаниловой кислоты). 1,5 г сульфанило-
вой кислоты растворяют в 200 см3 горячей воды (дистиллирован-
ной) и после охлаждения добавляют 50 см3 ледяной уксусной
кислоты.
Реактив 20 (раствор 1-нафтиламина). 1,2 г 1-нафтиламина ра-
створяют в 50 см3 ледяной уксусной кислоты и доводят объем ра-
створа дистиллированной водой до 200 см3.
Растворы 19 и 20 хранят в холодильнике в склянке из темного
стекла с притертой пробкой не более 1 мес.
Реактив 21 (раствор соляной кислоты). 450 см3 соляной кислоты
(плотностью 1,19 г/см3) растворяют в мерной колбе вместимостью
1 дм3 в дистиллированной воде и доводят объем до метки.
Приготовление пористого кадмия. Гранулы цинка
(250—300 шт.) распределяют по дну кристаллизатора (лучше ис-
пользовать цинковые пластинки или палочки) и заливают 1000 см3
раствора сульфата кадмия (реактив 9). Через 24 ч остаток цинка от-
деляют от кадмия, промывают кадмий водой декантацией 2—3 раза.
Образовавшийся пористый кадмий с помощью реактива 6 пере-
носят в гомогенизатор и измельчают до получения частиц размером
0,3—0,8 мм. Измельченный кадмий переносят в коническую колбу
вместимостью 250 см3 раствором соляной кислоты (реактив 6), про-
мывают и оставляют под этим раствором на 12 ч. Затем декантацией
промывают кадмий дистиллированной водой до исчезновения хло-
ридов (до отрицательной реакции с 1%-ным раствором нитрата се-
ребра), добавляют 150—200 см3 раствора сульфата меди (реактив
10), вращательными движениями колбы перемешивают содержи-
мое в течение 1 мин. Раствор сульфата меди декантируют и промы-
вают водой несколько раз, отделяя при этом мелкую фракцию кад-
мия и следя за тем, чтобы он все время был покрыт водой.
Подготовка кадмиевой колонки. Собирают систему согласно
рис. 6. На дно стеклянной колонки помещают тонкий слой стек-
лянной ваты, колонку заполняют водой и вносят суспензию кадмия
фарфоровой ложечкой на высоту 15—20 см. При заполнении ко-
лонки дают воде периодически стекать, чтобы уровень воды не
опускался ниже поверхности слоя кадмия. Слой кадмия все время
периодически перемешивают стеклянной палочкой, чтобы в ко-
лонке не осталось пузырьков газа.
Колонку, заполненную свежеприготовленным омедненным по-
ристым кадмием, обрабатывают следующим образом: пропускают
через слой кадмия смесь, состоящую из 750 см3 воды, 225 см3 рабо-
45
Рис. 6. Кадмиевая колонка:
/—резервуар; 2— носик; 3— стеклянная трубка; 4— кран
(или зажим); 5 — стеклянная вата; 6— слой кадмия; 7 — стек-
лянная колонка
чего раствора нитрата калия (реактив 14),
20 см3 буферного раствора (реактив 8) и 20
см3 раствора трилона Б (реактив 11) со ско-
ростью 6 см3/мин, после чего промывают
кадмиевую колонку (100 см3 воды) до пол-
ного удаления нитритов. Кадмий в колонке
хранят под слоем воды.
Восстановительную способность кад-
миевой колонки проверяют перед началом
работы и в конце анализа каждый день.
В резервуар колонки пипеткой вносят
20 см3 рабочего стандартного раствора нит-
рата калия (реактив 14) и 5 см3 аммиачного
буферного раствора. Устанавливают ско-
рость элюирования 3—5 см3/мин и собира-
ют элюат в мерную колбу вместимостью
100 см3. Когда резервуар опустеет, стенки
его дважды смывают водой порциями по 15 см3 и пропускают воду
через слой кадмия.
После того как соберут около 100 см3 элюата, удаляют колбу из-
под колонки, доводят объем до метки водой и перемешивают (стан-
дартный элюат).
Проводят контрольное определение, используя вместо стандар-
тного раствора нитрата калия 20 см3 воды (контрольный элюат).
В 2 мерные колбы вместимостью 50 см3 вносят пипеткой по
10 см3 стандартного и контрольного элюата и далее проводят опре-
деление нитритов. Если содержание нитрита калия, определенное
по градуировочному графику, меньше 0,27 мкг/см3 (90 %), колонку
следует регенерировать.
Регенерацию колонки проводят ежедневно. Если контроль ко-
лонки показывает ее недостаточную эффективность, колонку про-
мывают последовательно 100 см3 раствора для обработки колонки
(реактив 12), 25 см3 0,1 Моль раствора для соляной кислоты (реак-
тив 6), 50 см3 воды со скоростью 6 см3/мин. Если эффективность
колонки и после этого окажется неудовлетворительной, то кадмий
переносят из колонки в химический стакан, заливают на ночь во-
дой, затем раствором соляной кислоты концентрацией 2,0 моль/
дм3 (реактив 5) на 1 ч. Промывают 100 см3 раствора для обработки
колонки (реактив 12), затем дистиллированной водой, 50 см3 бу-
ферного раствора (реактив 8), разбавленного в 10 раз, и вновь про-
веряют восстановительную способность колонки.
46
Построение градуировочного графика для определения концентра-
ции нитритов. В 6 мерных колб вместимостью 100 см3 пипеткой вно-
сят 0 (контрольный раствор) 1,0; 5,0; 10,0; 20,0; 40,0 см3 стандартно-
го рабочего раствора нитрита натрия (реактив 16), что соответствует
0; 0,02; 0,1; 0,2; 0,4; 0,8 мкг нитрита натрия в 1 см3 раствора.
Для получения окраски используют НЭДА и сульфаниламид или
1 -нафтиламин и сульфаниловую кислоту.
При проведении реакции с НЭДА и сульфаниламидом в каждую
колбу добавляют по 10 см3 раствора сульфаниламида (реактив 17) и
по 2 см3 раствора соляной кислоты (реактив 21), перемешивают и
оставляют в темноте на 5 мин. Затем добавляют по 2 см3 раствора
НЭДА (реактив 18), доводят объем до метки дистиллированной во-
дой, перемешивают и оставляют в темноте на 10 мин.
Оптическую плотность растворов измеряют на спектрофотомет-
ре при длине волны 538 нм или фотоэлектроколориметре с зеленым
светофильтром в кюветах длиной оптического пути 10—40 мм. Кон-
тролем служит раствор, не содержащий нитритов. Измерение сле-
дует провести в течение получаса после окончания выдержки.
В случае использования сульфаниловой кислоты и 1-нафтила-
мина в колбы добавляют по 2 см3 раствора сульфаниловой кислоты
(реактив 19), 6 см3 раствора соляной кислоты (реактив 21), переме-
шивают и оставляют в темноте на 5 мин при комнатной температу-
ре. Затем добавляют по 2 см3 раствора 1-нафтиламина (реактив 20),
перемешивают, доводят дистиллированной водой до метки и вы-
держивают в темноте 1 ч. Оптическую плотность растворов измеря-
ют на спектрофотометре при длине волны 522 нм или на фотоэлект-
роколориметре с зеленым фильтром, в кюветах с длиной оптичес-
кого пути 10—40 мм. Контролем служит раствор, не содержащий
нитритов. Измерение следует провести в течение получаса после
окончания выдержки.
По полученным средним данным из двух параллельных опреде-
лений строят градуировочный график. По оси абсцисс откладывают
содержание нитритов в мкгна 1 см3 измеряемого раствора (0,02; 0,1;
0,2; 0,4; 0,8 мкг/см3), по оси ординат — соответствующие значения
оптической плотности.
МОЛЕКУЛЯРНО-ЛЮМИНЕСЦЕНТНАЯ СПЕКТРОМЕТРИЯ
ФЛУОРИМЕТРИЯ
Метод анализа, основанный на измерении интенсивности флуо-
ресценции, называется флуориметрией. Флуоресценция (люми-
несценция — испускание света) обусловлена поглощением веще-
ством света определенной длины волны. Поглощение ультрафиоле-
тового света определенными молекулами с легковозбуждаемыми
электронами приводит к флуоресценции в видимой спектральной
47
Рис. 7. Диаграмма энергетических уровней молекулы
при флуоресценции
области. Этот процесс начинается с перехода электронов с основно-
го энергетического уровня 50 на более высокие энергетические
уровни (5] и 52). Флуоресцентное излучение имеет большую длину
волны вследствие меньшей энергии фотонов, чем возбуждающее
излучение. Схема процессов, происходящих при флуоресценции,
приведена на диаграмме энергетических уровней (рис. 7). При нор-
мальных температурах осуществляются абсорбционные переходы с
низших колебательных уровней основного состояния ЛЬ на различ-
ные уровни возбужденных состояний 5] и 52. Эмиссионные перехо-
ды — это излучательные переходы с нулевого уровня состояния 5;
на любой колебательный уровень основного состояния ЛЬ- Перехо-
ды с уровня S2 (или более высоких уровней) на нулевой уровень Л)
происходит без излучения.
Дезактивация без излучения, т. е. потеря энергии возбужденны-
ми молекулами без излучения света, происходит в результате меж-
молекулярных и внутримолекулярных взаимодействий.
Флуоресценция свойственна относительно небольшому числу
соединений (прежде всего ароматическим соединениям и порфи-
ринам). Ряд соединений можно перевести во флуоресцирующие,
введя в молекулу флуоресцирующую группу, т. е. флуорофор (лю-
минофор). Реактивы для получения флуоресцирующих производ-
ных различных классов соединений приведены ниже.
Реактив
9-Изотиоцианатоакридин
Антрилдиазометан
7-Хлор-4-нитробензофуразан
Дансил хлорид
Флуорескамин
Класс соединений
Амины
Карбоновые кислоты
Амины, аминокислоты, нитрозоамины
Амины, аминокислоты, барбитураты,
карбаматы, производные мочевины,
оксистероиды
Аминокислоты, алкалоиды
48
Рис. 8. Экспериментально снятая зависимость ин-
тенсивности флуоресценции от концентрации флуо-
ресцирующего вещества
Основным преимуществом флуори-
метрии по сравнению с другими абсор-
бционными методами является высокая
селективность, так как флуоресценцией
обладает значительно меньшее число
веществ.
Интенсивность флуоресцентного излучения (/ф) пропорцио-
нальна концентрации флуоресцирующего вещества С
1ф = КС.
Однако в области относительно высоких концентраций наблю-
дается явление концентрационного тушения, происходящее при
увеличении доли безызлучательных переходов. Линейная зависи-
мость между /ф и Св этом случае нарушается (рис. 8).
Концентрационное тушение обусловливает верхний предел диа-
пазона определяемых концентраций приблизительно 10~4 М. Пре-
дел обнаружения метода составляет Ю~8 %.
Принципиальная схема типового флуориметра показана на
рис. 9. Излучение /0 источника 7 (кварцевая лампа), выделенное
первичным светофильтром 2, попадает на кювету с пробой 3. Воз-
никающее излучение /ф через вторичный светофильтр -/попадает на
фотоэлемент или фотоумножитель 5, где преобразуется в электри-
ческий сигнал, который усиливается электронным усилителем 6 и
измеряется миллиамперметром 7. При использовании линейного
участка градуировочного графика воспроизводимость флуоримет-
рических определений составляет приблизительно 5 %.
Метод применяют для чувствительного определения очень ма-
лых количеств элементов при анализе органических веществ, при
Рис. 9. Принципиальная схема флуо-
риметра:
1 — источник УФ-излучения; 2 — первич-
ный светофильтр; 3 — кювета с пробой; 4 —
вторичный светофильтр; 5 — фотоэлемент;
6— электронный усилитель; 7— миллиам-
перметр
определении малых количеств витаминов, гормонов, антибиоти-
ков, канцерогенных соединений и др.
Методом флуориметрии можно определить содержание жира в
молоке; количество микроорганизмов в сырье, продукте, смывах,
при контроле качества мойки оборудования; контролировать моло-
ко на мастит по содержанию соматических клеток и т. п.
Флуориметрический метод измерения массовой доли жира ос-
нован на способности жировых шариков при обработке их специ-
альными реагентами излучать свет под воздействием возбуждаю-
щих факторов (светового потока). Известна методика обработки
жировых шариков молока путем смешения водного раствора реа-
гента фосфина 3R-гидрохлорида и щелочи с пробой молока.
В основу работы жиромера, основанного на методе флуоресцен-
ции, положено соотношение
/ф = 2,3/0ФСжАп/,
где /ф, /0 — интенсивность светового потока флуоресценции и подаваемого от ис-
точника света; Ф и К„ — коэффициенты; Сж — массовая доля жира в продукте; / —
толщина кюветы.
Это соотношение характеризует зависимость изменения интен-
сивности флуоресценции 1$ от интенсивности подаваемого от ис-
точника света светового потока /о, массовой доли жира Сж, толщи-
ны слоя пробы в кювете /, коэффициента поглощения К и коэффи-
циента Ф (соотношение между интенсивностью излучения при
флуоресценции и поглощением излучения раствором).
Для возбуждения флуоресценции в исследуемой пробе исполь-
зуют спектральную область 320—440 нм, а измерение интенсивнос-
ти флуоресценции необходимо проводить в спектральной области
520—580 нм.
Исследование с помощью прибора ФЖМ-8, основанного на ме-
тоде флуориметрии, показало его перспективность для измерения
массовой доли жира не только в молоке, но и в сливках, сметане и
кисломолочных продуктах. Флуориметрический метод по сравне-
нию с турбидиметрическим позволяет исключить гомогенизацию
проб молока и расширить область применения прибора. Однако
этот метод контроля массовой доли жира пока не получил примене-
ния в промышленности из-за отсутствия аппаратурной базы.
Флуориметрию используют для определения микроорганизмов
и соматических клеток.
Методика определения микроорганизмов состоит в специаль-
ной подготовке пробы, в процессе которой бактерии, содержащие-
ся в молоке, отделяются от других частиц молока и окрашиваются
красителем в ярко-оранжевый цвет, в результате чего бактериаль-
ная суспензия приобретает способность флуоресцировать. Интен-
сивность флуоресценции пропорциональна числу микробов и кон-
тролируется электронным способом.
50
На основе данного метода фирмой Foss-Electric (Дания) разра-
ботан анализатор «Бактоскан». Время анализа одной пробы до
7 мин. Пределы измерения от 50 тыс. до 10 млн бактерий в 1 см3.
Прибор имеет достаточно сложную конструкцию и рассчитан на
использование в региональных центрах или на крупных предпри-
ятиях.
Флуориметрический метод контроля микроорганизмов доста-
точно универсален, имеет несложное аппаратурное оформление. В
связи с этим возможны его развитие, аттестация методики измере-
ния и создание отечественного экспресс-анализатора.
Флуориметрический оптико-электронный метод подсчета числа
соматических клеток основан на свойстве клеток флуоресцировать
при их окрашивании специальным красителем. Пробу молока сме-
шивают с красителем, смесь наносят тонким слоем (в виде пленки)
на вращающийся диск. Световой поток падает на диск, вызывая
флуоресценцию соматических клеток. Интенсивность флуорес-
ценции контролируют фотометрическим способом. На основе дан-
ного метода фирмой Foss-Electric (Дания) создан ряд приборов
(«Фоссоматик 90», «Фоссоматик 180/215» и др.), различающихся
производительностью.
Аналогичные приборы Anadis SCC выпускаются фирмой Anadis
Instruments (Франция). Принцип действия прибора основан на тех-
нике флуориметрической поточной цитометрии. Проба молока
микронасосом подается в блок подготовки пробы, где смешивается
со специальным раствором. В результате взаимодействия сомати-
ческих клеток с действующим веществом раствора последние про-
крашиваются. Далее смесь инжектируется через микроотверстие в
измерительную поточную кювету, где под действием источника ла-
зерного излучения прокрашенные соматические клетки начинают
флуоресцировать. Интенсивность излучения пропорциональна
числу соматических клеток. Излучение контролируется с помощью
детектора, сигнал которого обрабатывается микропроцессорным
электронным блоком. Контролируемая среда — сырое заготовляе-
мое молоко. Диапазон измерения от 0 до 5 млн клеток в 1 мл. Объем
пробы 4 мл.
Известен также флуоресцентный метод определения микробных
и соматических клеток, основанный на измерении интенсивности
флуоресценции АТФ, содержащейся в клетках.
Флуориметрический метод определения количества микроорга-
низмов основан на измерении содержания внутриклеточной АТФ в
микробных клетках, с помощью иммобилизированной люцифера-
зы. Метод получил название биолюминесцентного.
Общая схема определения количества микроорганизмов состоит
в следующем. АТФ, содержащаяся в микробных клетках, экстраги-
руется специальным раствором. В свободную АТФ добавляют спе-
циальный реагент (люциферин + люцифераза), и АТФ люминесци-
рует. Интенсивность продуцируемого излучения коррелирует с ко-
51
личеством АТФ, т. е. содержанием микроорганизмов в среде, и кон-
тролируется анализатором-люминомером. АТФ содержат только
живые клетки, мертвые клетки быстро теряют АТФ вследствие ав-
толизиса, поэтому с помощью данной методики определяют коли-
чество живых клеток. Реакция, катализируемая люциферазой, про-
исходит по следующей схеме (фирма Lumas (Голландия):
Mg++
Люциферин + Люцифераза + АТФ -> (Люциферин—Люцифе-
раза—АМФ) + Пирофосфат
I О2
Люциферин — Люцифераза — АМФ -> Оксилюциферин + Лю-
цифераза + СО2 + АМФ + Свет.
Биолюминесцентный метод анализа микроорганизмов создан
Н. Н. Угаровой с сотрудниками в Московском государственном
университете. Ими исследовано применение метода в медицине и
других отраслях. Проводятся работы по его использованию для кон-
троля бактериальной обсемененности молока. Во ВНИМИ при
участии Н. Н. Угаровой начаты работы по использованию биолю-
минесцентного метода для контроля содержания микроорганизмов
в молоке и молочных продуктах. Измерения с помощью биолюми-
несцентного метода достаточно просты, время анализа составляет
несколько минут.
При контроле на мастит по содержанию соматических клеток в
молоке отработана методика селективной экстракции немикроб-
ной АТФ, содержащейся в соматических клетках, с помощью спе-
циального реагента. Последний растворяет клеточную мембрану
соматических клеток, в то же время не воздействует на микробные
клетки. Экстрагированная АТФ соматических клеток отфильтро-
вывается через специальный фильтр, микробы остаются на фильт-
ре, и концентрацию АТФ соматических клеток измеряют соответ-
ствующим прибором, например М 2500.
Голландской фирмой Lumas создано несколько модификаций
биолюминесцентных анализаторов Biocounter (модели М 1500,
М 1800, М 2800).
Прибор модели Ml500 размещается в переносном чемодане, где
расположены также реагенты для подготовки пробы и пипетки для
отбора проб. Производительность прибора до 100 проб в час. Его
можно использовать для контроля микробиологической чистоты
смывов и оценки микробной массы.
Приборы моделей Ml800 (полуавтоматический) и М2800 (авто-
матический) многофункциональны, приборы пригодны для тести-
рования широкого диапазона материалов от молочного и пищевого
сырья до готовых продуктов. Они рассчитаны на работу в лаборато-
риях предприятий и исследовательских центрах. Приборы позволя-
ют контролировать бактериальную обсемененность молока, смыв-
52
ных вод при мойке оборудования, стерильность молочных продук-
тов, в том числе для детского питания, фруктовых соков и др. При-
бор М2800 имеет встроенный принтер и выход на ПЭВМ. Объем
пробы в кювете до 500 мкл. Производительность приборов:
М1800 — до 100 проб в час, М2800 — до 25 проб в минуту.
Биолюминесцентные анализаторы Biocounter (модели Ml500,
М1800, М2800) зарегистрированы в Государственном реестре
средств измерений и допущены для применения в России.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВИТАМИНА В, (ТИАМИНА)
Сущность метода заключается в освобождении связанных форм
тиамина путем кислотного и ферментативного гидролиза, хрома-
тографической очистке полученного гидролизата от соединений,
мешающих флуориметрическому определению, количественном
переводе в щелочной среде тиамина в тиохром, экстракции тиохро-
ма и измерении интенсивности флуоресценции тиохрома по срав-
нению со стандартным раствором с помощью лабораторного элект-
ронного флуориметра (ЭФ — ЗМА или других марок), обеспечива-
ющего возбуждение при длине волны 360—390 нм и флуоресцен-
цию — 400—450 нм.
Выполнение работы. Гидролиз. Тиамин определяют в двух парал-
лельных навесках продукта.
Навеску продукта (массу навески определяют с таким расчетом,
чтобы концентрация тиамина в гидролизате составляла 0,1 —
0,4 мкг/см3) помещают в колбу вместимостью 250 см3 и приливают
150 см3 раствора соляной кислоты концентрацией 0,1 моль/дм3 (ре-
актив 6).
Гидролиз проводят на кипящей водяной бане в течение 40 мин,
закрыв горловину колбы воронкой диаметром 35 м. Содержимое
колбы следует периодически перемешивать, особенно первые
5 мин. По окончании гидролиза колбу охлаждают до комнатной
температуры и насыщенным водным раствором ацетата натрия до-
водят pH гидролизата до 4,5±0,1 (потенциометрически). После это-
го в гидролизат вносят навеску амилоризина массой 0,1 г и помеща-
ют в термостат на 14—16 ч (на ночь) при температуре 37± 1 °C.
На следующий день гидролизат охлаждают, доводят объем в ци-
линдре до 250 см3 дистиллированной водой и фильтруют. В фильт-
рате определяют содержание тиамина. Если необходимо прервать
анализ на 1 —2 дня, то перед доведением объема до 250 см3 гидроли-
зат кипятят 5 мин, охлаждают, доводят объем до 250 см3 и фильтру-
ют. До проведения анализа фильтрат хранят в холодильнике в плот-
но закрытой колбе.
Очистка фильтрата от примесей. Очистку гидролизаторов от ме-
шающих определению примесей проводят на адсорбционных ко-
лонках.
53
Для перевода катионита в Н-форму перед пропусканием фильт-
рата через колонку с адсорбентом пропускают 20 см3 раствора ук-
сусной кислоты концентрацией 0,03 г/см3 (реактив 7), нагретой до
60—70 °C. Затем в две адсорбционные колонки вносят по 20 см3
стандартного раствора тиамина концентрацией 0,2 мкг/см3. После
того как весь фильтрат пройдет через колонку (скорость пропуска-
ния 15 капель в мин), адсорбент промывают 2—3 раза дистиллиро-
ванной водой порциями по 10 см3. С адсорбента тиамин элюируют
горячим раствором хлорида калия (реактив 8). Для этого в колбу от-
меривают 20 см3 элюирующего раствора, подогревают его до 60—
80 °C и пропускают через колонку порциями по 5—6 см3 (элюирую-
щий раствор необходимо подогревать несколько раз). Элюат соби-
рают в мерные цилиндры и, если объем будет несколько меньше,
его доводят водой до 20 см3. Катионит после регенерации может
быть использован повторно.
Регенерацию катионита проводят доведенным до кипения
спиртовым раствором аммиака в объеме 30 см3 (реактив 5). После
чего через колонку пропускают дистиллированную воду и перед
повторным употреблением — раствор уксусной кислоты концент-
рацией 0,03 г/дм3 (реактив 7). Такая обработка катионита бывает
недостаточна после пропускания фильтратов, содержащих много
естественных пигментов. Для удаления из катионита органичес-
ких примесей в колонку приливают раствор гидроксида натрия
концентрацией 0,1 г/см3 (реактив 9), оставляя его для контакта с
катионитом на 20—25 мин. После этого раствор удаляют из колон-
ки, к катиониту приливают новую порцию раствора щелочи. Затем
катионит промывают водой до нейтральной реакции. Перед ад-
сорбцией тиамина катионит переводят в Н-форму, как указано
выше.
Окисление тиамина в тиохром. В три конические колбы или
широкие пробирки с притертыми пробками отмеривают по 5 см3
элюата. Затем в две из них добавляют по 1,2 см3 окислительной
смеси (реактив 3), а в третью — 1,2 см3 раствора гидроксида на-
трия (реактив 9) концентрацией 0,3 г/дм3 (контроль). Все колбы
энергично встряхивают, прибавляют по 10 см3 изобутилового
спирта и снова энергично встряхивают в течение 1 мин для из-
влечения тиохрома. Водный и спиртовой слои разделяют отстаи-
ванием в темном месте или центрифугируют. Затем спиртовой
слой (верхний) сливают в кюветы для измерения интенсивности
флуоресценции.
Аналогичным образом поступают с полученным после пропус-
кания через колонку элюатом рабочего стандартного раствора тиа-
мина.
Интенсивность флуоресценции тиохрома измеряют, используя
светофильтры, обеспечивающие длины волн возбуждения 360—
390 нм и флуоресценции — 400—450 нм. Измерение начинают с
проб стандартного раствора тиамина.
54
Содержание тиамина Л^мг на 100 г продукта) вычисляют по фор-
муле
(Л-Л,)Л/Г
где А — среднее из показаний флуориметра для испытуемой пробы; At — показание
флуориметра для контроля к испытуемой пробе; М — масса тиамина во взятом для
окисления тиамина в тиохром объеме стандартного раствора, мкг; И— общий объем
гидролизата, см3; В — показание флуориметра для стандартного раствора тиамина;
В[ — показание флуориметра для контроля к стандартному раствору тиамина; И, —
объем испытуемого раствора, взятого для окисления тиамина в тиохром, см3; Л/| —
масса пробы продукта, взятой для анализа, г; 10 — пересчет из мкг/г в мг/100 г про-
дукта.
Вычисления проводят с точностью до третьего десятичного зна-
ка с последующим округлением до второго десятичного знака. За
окончательный результат испытания принимают среднее арифме-
тическое результатов двух параллельных определений.
Приготовление реактивов. Реактив I (основной стандартный ра-
створ тиамина). Навеску тиамина, высушенного в вакууме при 60—
70 °Свтечение2—3 чили в эксикаторе над серной кислотой, массой
0,1 г помещают в мерную колбу вместимостью 1000 см3, доводят
объем дистиллированной водой до метки, тщательно перемешива-
ют, переносят в склянку из темного стекла с притертой пробкой и
добавляют 0,5 см3 толуола. Концентрация основного стандартного
раствора тиамина составит 100 мкг/см3.
Раствор хранят в холодильнике не более 2 мес.
Реактив 2 (стандартный рабочий раствор тиамина). 1 см3 основ-
ного стандартного раствора тиамина, температура которого доведе-
на до комнатной, переносят в мерную колбу вместимостью 500 см3,
доводят объем дистиллированной водой до метки, тщательно пере-
мешивают. Концентрация рабочего стандартного раствора тиамина
составит 0,2 мкг/см3.
Раствор готовят в день проведения анализа.
Реактив 3 {окислительная смесь). К 2 см31 %-ного раствора гекса-
цианоферрита калия прибавляют 10 см3 раствора гидроксида на-
трия концентрацией 0,3 г/см3 и тщательно перемешивают. Смесь
пригодна к употреблению в течение 2—3 ч.
Реактив 4 (бутиловый и изобутиловый спирты). Перед проведе-
нием анализа измеряют флуоресценцию используемых спиртов в
сравнении с дистиллированной водой. В случае наличия флуорес-
ценции спирт очищают. Для этого к 1 дм3 спирта прибавляют 15—
20 г активного угля, встряхивают в течение 30 мин, оставляют на
сутки, затем декантируют, фильтруют и перегоняют на глицерино-
вой или песчаной бане при температуре 117 °C (бутанол) и 108 °C
(изобутанол).
После очистки проверяют отсутствие флуоресценции спирта
(контроль — дистиллированная вода).
55
Реактив 5(спиртовойраствор аммиака). 12 см3 раствора водного
аммиака концентрацией 0,25 г/см3 доводят до 100 см3 70%-ным
этиловым спиртом.
Реактив 6 (соляная кислота концентрацией 0,1 моль/дм3). 10, 125 г
концентрированной соляной кислоты (массовая доля НС1 36 %,
плотность 1,18 г/см3) разбавляют дистиллированной водой, доводя
объем раствора до 1000 см3.
Реактив 7 (уксусная кислота концентрацией 0,03 г/см3). 30 г ледя-
ной уксусной кислоты разбавляют дистиллированной водой, дово-
дя объем полученного раствора до 1000 см3.
Реактив 8(хлорид калия концентрацией 0,25г/см3). 250 г хлорида
калия помещают в мерную колбу на 1000 см3 и добавляют соляную
кислоту концентрацией 0,1 моль/дм3 до метки.
Реактив 9 (гидроксид натрия). Раствор гидроксида натрия раз-
личной концентрации приготавливают в соответствии с приложе-
нием 3.
Подготовка адсорбционной колонки. В нижнюю часть адсорбци-
онной колонки помещают комочек стеклянной ваты и над ним —
адсорбент. Набухший катионит переносят в колонку, которую
предварительно на 1/3 ее объема заполняют водой, чтобы исклю-
чить попадание пузырьков воздуха в пространство между частица-
ми катионита. Высота столбика катионита должна быть 10—12 см.
Необходимо помнить, что над слоем адсорбента все время должна
находиться жидкость.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВИТАМИНА В2 (РИБОФЛАВИНА)
Сущность метода заключается в освобождении связанных форм
рибофлавина путем кислотного и ферментативного гидролиза, эк-
стракционной очистке полученного гидролизата от соединений,
мешающих флуориметрическому определению, и измерении ин-
тенсивности флуоресценции рибофлавина (метод прямой флуори-
метрии) до и после восстановления рибофлавина гидросульфитом
натрия или люмифлавина (люмифлавиновый метод) по сравнению
со стандартным раствором. В последнем случае использовано свой-
ство рибофлавина в щелочной среде переходить в люмифлавин, ин-
тенсивность флуоресценции которого измеряют после извлечения
его хлороформом.
Для количественного определения рибофлавина служит лабора-
торный электронный флуориметр (ЭФ/ЗМА или других марок).
Выполнение работы. Гидролиз. Рибофлавин определяют в двух па-
раллельных навесках.
Навеску продукта (концентрация рибофлавина в гидролизате
0,04—0,20 мкг/см3) помещают в колбу вместимостью 250 см3 и
приливают 150 см3 раствора соляной кислоты концентрацией
0,1 моль/дм3. Гидролиз осуществляют на кипящей водяной бане в
56
течение 40 мин, закрыв горловину колбы воронкой диаметром
35 мм. Содержимое колбы следует периодически перемешивать,
особенно в первые 5 мин.
По окончании гидролиза колбу охлаждают до комнатной темпе-
ратуры и насыщенным водным раствором ацетата натрия доводят
pH гидролизата до 4,5±0,1 (потенциометрически).
После этого к гидролизату добавляют навеску амилоризина мас-
сой 0,10 г и помещают в термостат на 14—16 ч (на ночь) при темпе-
ратуре 37± 1 °C.
На следующий день гидролизат охлаждают, доводят его объем до
250 см3 дистиллированной водой и фильтруют. В фильтрате опреде-
ляют содержание рибофлавина. Если необходимо прервать анализ
на 1—2 дня, то перед доведением объема до 250 см3 гидролизат ки-
пятят 5 мин, охлаждают, доводят объем до 250 см3 и фильтруют.
Одновременно аналогичным образом готовят контрольную про-
бу на содержание рибофлавина в амилоризине, используя то же ко-
личество ферментного препарата и реактивов, но без исследуемой
пробы.
До проведения анализа фильтрат хранят в холодильнике в плот-
но закрытой колбе.
Дальнейший анализ проводят либо по методу прямой флуори-
метрии, либо люмифлавиновым методом.
Измерение интенсивности флуоресценции. 1. Метод прямой флуо-
риметрии. В 4 флуориметрические пробирки наливают по 10 см3
фильтрата испытуемого образца. В 2 пробирки с фильтратом добав-
ляют по 1 см3 дистиллированной воды, в 2 другие по 1 см3 рабочего
стандартного раствора рибофлавина (реактив 2). В 5-ю пробирку
наливают 10 см3 фильтрата контрольной пробы и 1 см3 дистиллиро-
ванной воды.
Во все пробирки добавляют по 1 см3 ледяной уксусной кислоты и
перемешивают содержимое. Вносят по 0,5 см3 раствора перманга-
ната калия концентрацией 0,03 г/см3 (реактив 3), смешивают и ос-
тавляют на 2 мин. Добавляют в каждую пробирку по 0,5 см3 раство-
ра пероксида водорода концентрацией 0,03 г/см3 и энергично
встряхивают для удаления избытка кислорода.
Интенсивность флуоресценции рибофлавина измеряют, ис-
пользуя светофильтры, дающие длины волн возбуждения в области
350—480 нм, флуоресценции — 475—650 нм.
После этого добавляют в каждую пробирку небольшими порциями
(по 15—25 мг) порошкообразный гидросульфит натрия, быстро пере-
мешивают и снова измеряют интенсивность. Эту операцию повторя-
ют до установления наименьшей интенсивности флуоресценции.
2. Люмифлавиновый метод. К 100 см3 фильтрата добавляют 2 см3
раствора серной кислоты концентрацией 0,3 г/см3 (реактив 5) и с
помощью пипетки или бюретки по каплям раствор перманганата
калия концентрацией 0,03 г/см3 (реактив 3), постоянно перемеши-
вая до получения малинового окрашивания.
57
Избыток перманганата калия удаляют, добавляя по каплям ра-
створ пероксида водорода.
Объем израсходованных реактивов (см3) перманганата калия,
раствора серной кислоты и пероксида водорода приплюсовывают к
первоначально взятому на окисление объему фильтрата, чтобы оп-
ределить конечный объем раствора. Полученный раствор перено-
сят в делительную воронку, добавляют 30—50 см3 хлороформа и
встряхивают в течение 1 мин. После разделения слоев хлороформ-
ный слой (нижний) отбрасывают, а водную фазу используют для
дальнейшего определения.
Фотолиз проводят в 5 конических колбах с притертыми пробка-
ми. В 4 колбы наливают по 20 см3 очищенного гидролизата, в 2 из
которых добавляют по 2 см3 рабочего стандартного раствора рибоф-
лавина (реактив 2). В 5-ю колбу наливают 20 см3 контрольной про-
бы. Во все 5 колб добавляют по 4 см3 раствора гидроксида натрия
концентрацией 7 моль/дм3 (реактив 9), закрывают колбы пробка-
ми, перемешивают содержимое и облучают их светом двух светиль-
ников с лампами накаливания по 100 Вт каждая с расстояния 30 см в
течение 40 мин. Температура окружающего воздуха должна быть не
более 25 °C. Для поддержания указанной температуры воздуха ис-
пользуют настольный вентилятор.
Немедленно по окончании облучения растворы во всех колбах
подкисляют 4 см3 ледяной уксусной кислоты, добавляют к ним по
20 см3 хлороформа, закрывают притертыми пробками и встряхивают
в течение 2 мин, избегая образования эмульсии. Затем все колбы ос-
тавляют на 10—15 мин для расслоения водной и хлороформной фаз.
После этого пипеткой отбирают по 10—12 см3 хлороформного ра-
створа (нижний слой), который фильтруют через бумажный фильтр с
безводным сульфатом натрия в флуориметрические пробирки.
Интенсивность флуоресценции люмифлавина измеряют при све-
тофильтрах, дающих длины волн возбуждения в области 350—480 нм,
флуоресценции — 475—650 нм. Контролем служит хлороформ.
Содержание рибофлавина 2Дмг на 100 г продукта) вычисляют по
формулам
по методу прямой флуориметрии
г, [(й-4)-(е-С1)И
[(в-д)-(л-4)]и2мг1о’
по люмифлавиновому методу
(А-С)М^
(В-А)Му2Уу10’
где А — среднее из показаний флуориметра для испытуемой пробы без добавления
стандартного раствора рибофлавина; А{ — то же после восстановления рибофлави-
на гидросульфитом натрия; С — показание флуориметра для контрольного опыта
на реактивы; С{ — то же после добавления гидросульфита натрия; V — общий объем
58
гидролизата, см3; £ —среднее из показаний флуориметра для испытуемой пробы с
добавлением стандартного раствора рибофлавина; — то же после восстановления
рибофлавина гидросульфитом натрия; — объем гидролизата после окисления,
см3; И2 — объем гидролизата, взятый для окисления, см3; — объем гидролизата,
взятый для облучения, см3; М— масса добавленного рибофлавина, мкг; М\ — масса
пробы продукта, взятой для анализа, г; 10 — пересчет из мкг/r в мг на 100 г продукта.
Вычисления проводят с точностью до третьего десятичного зна-
ка с последующим округлением до второго десятичного знака.
За окончательный результат испытания принимают среднее
арифметическое результатов двух параллельных определений.
Приготовление реактивов. Реактив 1 (основной стандартный ра-
створ рибофлавина). Навеску рибофлавина массой 0,020 г помещают
в мерную колбу вместимостью 1000 см3, добавляют 750 см3 дистилли-
рованной воды и 1 см3 ледяной уксусной кислоты и слегка нагревают
для лучшего растворения. После полного растворения рибофлавина
раствор охлаждают до комнатной температуры и доводят объем дис-
тиллированной водой до метки. Приготовленный раствор переносят
в склянку из темного стекла с притертой пробкой. Концентрация ос-
новного стандартного раствора рибофлавина составит 20 мкг/см3.
Раствор хранят в холодильнике не более 1 мес.
Реактив 2 (рабочий стандартный раствор рибофлавина). 5 см3 ос-
новного стандартного раствора рибофлавина, температура которо-
го доведена до комнатной, переносят в мерную колбу вместимостью
100 см , доводят объем дистиллированной водой до метки и тща-
тельно перемешивают. Концентрация рабочего стандартного ра-
створа рибофлавина составит 1 мкг/см3. Раствор готовят в день про-
ведения анализа.
Реактив 3 (перманганат калия концентрацией 0,03 г/см3). 30 г
перманганата калия растворяют в дистиллированной воде, доводя
объем полученного раствора водой до 1000 см3. Раствор хранят не
более 7 дней.
Реактив 4 (пероксид водорода концентрацией 0,03г/см3). 30 г пе-
роксида водорода растворяют в дистиллированной воде, доводя
объем полученного раствора водой до 1000 см3. Раствор хранят не
более 7 дней.
Реактив 5(серная кислота). Растворы серной кислоты различной
концентрации приготавливают в соответствии с приложением 4.
Остальные реактивы готовят так же, как описано в методике по
определению тиамина.
АТОМНАЯ СПЕКТРОСКОПИЯ
В атомной спектроскопии вещества исследуют, переводя их в со-
стояние атомного пара — атомно-абсорбционная спектроскопия
(ААС) или газообразное состояние — атомно-эмиссионная спект-
роскопия (АЭС).
59
В атомно-абсорбционной спектроскопии для возбуждения ато-
мов используют тепловую энергию. Распыляя образец в пламени,
соединения переводят в атомный пар (атомизация). Большинство
атомов, возбуждаясь, переходит на более высокий энергетический
уровень. При обратном переходе происходит выделение энергии. В
процессе облучения атомов исследуемого элемента, находящихся в
состоянии пара, линейчатым излучением того же самого элемента в
возбужденном состоянии происходит резонансное поглощение.
Этот процесс сопровождается уменьшением интенсивности линей-
чатого излучения. Измеряемое поглощение является мерой кон-
центрации свободных атомов образца.
В атомно-эмиссионной спектроскопии возбуждения происхо-
дят при помощи электрических зарядов. При этом создаются высо-
кие температуры, благодаря которым большинство атомов перехо-
дит в возбужденное состояние. Поглощение энергии этими атома-
ми невозможно, поэтому происходит эмиссия (испускание) фото-
нов возбужденных атомов.
Определение элементов в большинстве случаев — металлов в
атомной спектроскопии проводят чувствительным селективным
методом при длине волны, характерной для каждого элемента
(табл. 7).
7. Пределы обнаружения элементов методами ААС, АЭС
Элемент Длина волны, нм Предел обнаружения, млн1
ААС (В) АЭС (О)
Ag 328,07 0,001 0,008
Al 396,15 — 0,05
309,28 0,1 (О) —
As 193,70 0,03 10
Au 267,60 2
242,80 0,02 (О) —
В 518,00 — 0,05
249,68 2,5 (О) —
Ba 553,55 0,02 (О) 0,002
Be 234,86 0,002 (О) 1
Bi 306,77 — 20
223,06 0,05 —
Ca 422,67 0,002 0,0002
Cd 326,11 — 0,8
228,80 0,001 —
Co 345,35 0,03
240,72 0,002 —
Cr 425,43 — 0,004
357,87 0,002 —
Cs 455,3 0,6
852,11 0,05 —
Cu 324,5 0,004 0,01
Fe 371,99 — 0,03
248,33 0,004 —
Hg 253,65 0,5 10
К 766,49 0,003 0,00005
60
Продолжение
Элемент Длина волны, нм Предел обнаружения, млн-1
ААС (В) АЭС (О)
Li 670,78 0,001 0,00002
Mg 285,21 0,003 0,07
Мп 403,31 — 0,008
279,48 0,0008 0,002
Мо 390,30 — 0,2
313,26 0,03 (О) —
Na 589,00 0,0008 0,0005
Ni 352,45 — 0,02
232,00 0,005 —
Pb 405,78 — 0,1
283,31 0,01 —
Pf 363,47 — 0,05
247,64 0,01 —
Pt 265,94 0,05 4
Sb 252,85 — 0,6
217,58 0,03 —
Sc 196,03 0,1 100
Si 251,61 0,1(0) 3
Sn 284,00 0,1
235,48 0,05 —
Sr 470,73 0,005 0,0005
Zn 213,86 0,001 10
Примечание. Горючий газ — ацетилен, газ-окислитель, В —воздух, О —оксид
азота (I).
Пределы обнаружения элементов методом атомной спектроско-
пии достигают 10-12—Ю-14 г.
Метод атомной спектроскопии находит широкое применение в
химии, биохимии, экологии и др., а также в анализе различных видов
сырья и пищевых продуктов. Метод позволяет определить около 70
различных элементов; используется для одновременного определе-
ния большого числа элементов (многоэлементный анализ); для се-
рийного анализа, благодаря высокой чувствительности и быстроте.
АТОМНО-АБСОРБЦИОННАЯ СПЕКТРОМЕТРИЯ
Атомно-абсорбционная спектрометрия основана на измерении
поглощения электромагнитного излучения атомным «паром» анали-
зируемого вещества. Измеряют фотометрически разность интенсив-
ности излучения до и после прохождения через анализируемый обра-
зец. Ослабление излучения с интенсивностью 70до интенсивности I
для выходящего светового потока происходит по экспоненциальной
зависимости, которая идентична закону Бугера — Ламберта — Бера:
/ = 7о-10-^с,
где К~ коэффициент поглощения; / — толщина поглощающего слоя плазмы; С—
концентрация поглощающих атомов.
61
После логарифмирования этого выражения получают зависи-
мость:
А = 1g 1ОЦ= к1С,
где А — абсорбция поглощающего слоя плазмы; к — атомный коэффициент аб-
сорбции.
При постоянной толщине поглощающего слоя градуировочный
график, построенный в координатах Л—С, представляет собой пря-
мую, проходящую через начало координат.
Прибором, позволяющим осуществить метод ААС, является
атомно-абсорбционный спектрометр, имеющий следующие основ-
ные составные части:
источник света определенной длины волны, характерной для ис-
следуемого металла;
«абсорбционную ячейку», в которой происходит атомизация
пробы;
монохроматор для выделения узкой части спектра определенной
длины волны;
фотоумножитель, который детектирует, усиливает и измеряет
интенсивность результирующего светового потока;
регистрирующее и записывающее результирующий сигнал уст-
ройство.
' Источник света испускает поток луча, спектр которого характе-
рен для определяемого элемента. Этот поток фокусируется через
абсорбционную ячейку и монохроматор, где выделяется характер-
ная для исследуемого элемента область спектра. Затем поток на-
правляется в фотоумножитель и преобразуется в электрический
сигнал. Величина последнего зависит от интенсивности поступаю-
щего в фотоумножитель светового потока и регистрируется специ-
альным устройством.
Сравнивая результаты измерений в исследуемой пробе с резуль-
татами измерений в стандартных растворах, определяют содержа-
ние элемента в пробе.
В качестве источников излучения при определении содержания
рассматриваемых металлов, как правило, используют лампы с по-
лым катодом, являющиеся источниками линейных спектров. Катод
такой лампы имеет форму полого цилиндра или стакана. Объем
лампы заполнен инертным газом (неоном или иногда аргоном). Су-
ществуют многоэлементные лампы с полым катодом, например для
определения содержания меди и цинка или меди, цинка, свинца и
кадмия. Они бывают очень удобны. Их преимуществом является
снижение затрат времени на прогрев ламп. Однако такие лампы,
как правило, продуцируют излучение более низкой энергии, чем
одноэлементные лампы, что в результате приводит к ухудшению
чувствительности; могут возникать спектральные помехи, при-
62
водящие к изгибам на калибровочной кривой; продолжитель-
ность эксплуатации таких ламп существенно меньше, чем одно-
элементных.
Для атомизации пробы используют атомизаторы двух типов:
пламенные и непламенные.
При реализации пламенного варианта ААС анализируемая про-
ба распыляется в пламени горелки. Капли раствора быстро разлага-
ются до молекулярного уровня. Молекулы диссоциируют, образуя
атомы отдельных элементов. Лишь около 10 % исследуемого ра-
створа действительно попадает в пламя, что является одной из при-
чин, ограничивающих чувствительность метода. Другой важной
причиной является короткое время существования атомов элемен-
тов в пламени.
Температура пламени горючих газовых смесей, наиболее часто
используемых в ААС, приведены в табл. 8.
8. Температура пламени газов
Горючая смесь Температура пламени, ”С
Горючий газ (топливо) Газ-окислитель
Водород Воздух 1725
Пропан Воздух 1825
Ацетилен Воздух 2050
Водород Кислород 2800
Пропан Кислород 2900
Ацетилен Кислород 3200
Ацетилен Оксид азота (I) 3400
Газовые смеси могут быть стехиометрическими, обедненными (с
содержанием горючего в количестве менее стехиометрического) и
обогащенными (с содержанием горючего в количестве больше сте-
хиометрического). Обедненную смесь называют окислительной,
обогащенную — восстановительной.
Для определения таких металлов, как медь, цинк, железо, сви-
нец и кадмий, в основном применяют пламя ацетилен — воздух,
реже пропан—воздух.
Пламя ацетилен — оксид азота (I) благодаря его высокой темпе-
ратуре рекомендуется для определения элементов с высокой энер-
гией диссоциации (вольфрам, ванадий, молибден). Пламя про-
пан — воздух и водород — воздух главным образом рекомендуют
для анализа щелочных элементов. Использование горючих смесей
для определения различных химических элементов периодической
системы приведено на рис. 10.
Для количественного анализа необходимо, чтобы параметры го-
рения были постоянными. Для газовых смесей с соответствующими
температурами пламени сконструировано несколько типов газовых
горелок. При использовании горелок с предварительным смешени-
63
Ацетилен-
Ацетилен-оксид азота (Г)
- Ацетилен-оксид азота (I)
- Водород-воздух
- Пропан-воздух
- Ацетилен воздух
Рис. 10. Горючие смеси, используемые для определения различных химичес-
ких элементов периодической системы методом ААС
ем перед сгоранием происходит смешение анализируемого веще-
ства с горючим газом и окислителем, при этом расходуется неболь-
шая часть пробы (10—15 %), а остальная часть отделяется в смеси-
тельной камере и выводится. При использовании горелок полного
сгорания (рис. 11) расходуется вся проба. Чтобы более равномерно
распределить исследуемое вещество, применяют ультразвуковое
распыление.
При реализации непламенного варианта ААС чаще всего ис-
пользуют высокотемпературные печи. Это в основном графитовые
трубчатые печи, так называемые графитные кюветы, в которых ра-
зогрев происходит за счет пропускания электрического тока (элект-
ротермическая атомизация). В кювету вводят анализируемую пробу
обычно объемом 1—100 мкл, затем нагревают кювету электричес-
ким током. Температура кюветы повышается ступенчато, так что на
первой стадии достигается сравнительно низкая температура, обес-
печивающая испарение растворителя. На второй стадии температу-
ра кюветы повышается так, чтобы произошло сухое озоление про-
бы. И, наконец, кювета быстро разогревается до температуры
3000 °C, при которой происходит атомизация. На этой стадии и
производится измерение абсорбционного сигнала. На первых двух
64
стадиях через кювету пропускают
очищенный инертный газ (аргон
или азот). Но на стадии атомизации
поток инертного газа обычно оста-
навливают, с тем чтобы атомы оста-
вались в абсорбционной ячейке по
возможности дольше.
Основными достоинствами гра-
фитовых атомизаторов являются
низкие пределы обнаружения эле-
ментов и возможность анализиро-
вать очень малые объемы проб. В
среднем пределы обнаружения раз-
личных металлов при работе с графи-
товыми атомизаторами на 2—3 по-
рядка ниже, чем в пламенной ААС.
Определение элементов в атом-
но-абсорбционном методе заключа-
ется в измерении относительной ин-
тенсивности двух световых потоков.
Один из них проходит через анали-
зируемое вещество, другой является
Рис. 11. Схема горелки полного
сгорания
контрольным.
Одновременное измерение интенсивности двух световых пото-
ков, один из которых проходит через пламя с анализируемым веще-
ством, а другой нет, проводят с двухлучевыми атомно-абсорбцион-
ными спектрофотометрами. Принципиальная схема такого прибо-
ра с пламенной атомизацией анализируемого вещества представле-
на на рис. 12.
Рис. 12. Принципиальная схема двухлучевого атомно-абсорбциоииого
спектрофотометра:
7 — лампа с полым катодом; 2 — модулятор; 3~ зеркало; 4— щелевая горелка; 5— пламя; 6—
тонкая пластинка, обеспечивающая наложение двух лучей; 7— входная щель монохроматора;
8— дифракционная решетка; 9 —выходная щель; 10 — фотоумножитель; 77 — усилитель; 72 —
измерительный блок
65
Одна часть монохроматического излучения элемента от лампы с
полым катодом проходит через пламя 5и фокусируется на входной
щели 7 монохроматора. Другая часть светового потока минует пла-
мя. Затем оба световых потока совмещаются с помощью тонкой
пластинки 6. Выделенное монохроматическое излучение попадает
на фотоумножитель или фотоэлемент 10. Сила тока возрастает в
блоке 11 и регистрируется измерительным прибором 12. Раствор
поступает в пламя через горелку (атомизатор) 4. Важнейшей про-
блемой в атомной адсорбции является отделение резонансного из-
лучения элемента в пламени при данной длине волны от аналити-
ческого сигнала. Для этого падающее на поглощающий слой и кон-
трольное (не проходящее через пламя) излучение модулируют или с
помощью вращающегося диска 2 с отверстиями, или путем питания
лампы с полым катодом переменным или импульсным током. Уси-
литель 11 имеет максимальный коэффициент усиления для той же
частоты, с которой модулируется излучение полого катода. Лампы с
полым катодом обычно одноэлементны, и, чтобы определить дру-
гой элемент, нужно сменить лампу, что увеличивает время анализа.
Многоэлементные лампы, которые используют в атомно-абсорб-
ционных многоканальных спектрофотометрах, позволяют одно-
временно определять несколько элементов
В большинстве приборов раствор исследуемого вещества смеши-
вают в распылительной камере со смесью горючих газов. Затем полу-
ченную смесь подают непрерывным потоком в горелку (скорость по-
дачи 1 —2 мл/мин). В пламени горелки испаряется растворитель и об-
разуются свободные атомы образца. В качестве источника излучения
используют лампу с полым катодом, который изготовлен из того ве-
щества, которое необходимо определить. Спектр излучения источ-
ника состоит из большого числа спектральных линий, причем харак-
терная линия для данного элемента может быть не самой интенсив-
ной. Поэтому большое значение имеет правильность выбора соот-
ветствующей длины волны измерения. Чувствительность метода
зависит как от длины волны измерения, так и от концентрации ра-
створа. Применение одноэлементных ламп обеспечивает необходи-
мую чувствительность и селективность, так как атомы исследуемого
элемента поглощают излучение с длиной волны, соответствующей
излучению лампы. Как правило, применяют лампы, дающие интен-
сивное излучение необходимой длины волны.
Вариантом атомно-абсорбционной спектрометрии является ме-
тод «холодного пара», практически повсеместно используемый для
определения содержания ртути в различных объектах, в том числе в
молоке и молочных продуктах. Для реализации метода необходимо
провести разложение пробы (например в смеси KMnO4 + H2SO4)
для получения ртути в ионной форме. Затем, используя восстанови-
тель (обычно SnCl2), переводят ртуть в состояние элемента с высо-
кой летучестью при комнатной температуре. Пропуская азот или
воздух через раствор с восстановленной ртутью, направляют газо-
66
вый поток с парами ртути в абсорбционную ячейку, расположен-
ную на оптической оси атомно-абсорбционного спектрометра, и
измеряют поглощение излучения от ртутной лампы с полым като-
дом. Метод высокоселективный и чувствительный.
АТОМНО-ЭМИССИОННАЯ СПЕКТРОМЕТРИЯ
В атомно-эмиссионной спектрометрии исследуют атомно-
эмиссионные спектры, полученные в результате возбуждения ато-
мов в газообразном состоянии.
Для перевода атомов в газообразное состояние и их возбуждения
используют плазму, в качестве среды для получения плазмы приме-
няют аргон. Существует два способа получения плазмы. В одном из
них возбуждение происходит под действием электрических разря-
дов между электродами — плазма постоянного тока, а в другом —
энергия высокочастотного переменного тока передается газу с по-
мощью магнитной индукции — индуктивно-связанная плазма.
При этом создаются высокие температуры, благодаря которым
большинство атомов переходит в возбужденное состояние. Погло-
щение энергии такими атомами невозможно, поэтому при переходе
из возбужденного состояния в основное происходит эмиссия (ис-
пускание) фотонов, интенсивность которой пропорциональна чис-
лу возбужденных атомов. Количественное определение элемента
производят так же, как в атомно-абсорбционной спектрометрии.
Приборы, позволяющие осуществить метод АЭС, имеют те же ос-
новные части, что и атомно-абсорбционный спектрометр. Сравни-
тельная блок-схема АЭС и ААС-спектрометров приведена на рис. 13.
Рис. 13. Блок-схемы атомно-эмиссиониого и атомио-абсорбциоиного спектрометров:
1 — Лампа с полым катодом; 2 — анализируемые образцы д ля АЭС и ААС методов; 3 — монохро-
матор с дифракционной решеткой или фильтром; 4— фотоумножитель; 5 — усилитель; 6— ре-
гистрирующий прибор
67
Излучение анализируемого образца 2 поступает на монохроматор 3
для выделения части спектра определенной длины, а затем на фотоум-
ножитель 4, усилитель 5 и регистрирующий прибор 6. Время анализа
на современных полностью автоматизированных спектрометрах от
введения пробы до распечатки результатов составляет около 10 с.
Следует отметить, что для количественного определения щелоч-
ных и щелочноземельных элементов рекомендуется метод АЭС.
СУЩНОСТЬ МЕТОДА И ПОДГОТОВКА ПРОБ
К ОПРЕДЕЛЕНИЮ СОДЕРЖАНИЯ НАТРИЯ, КАЛИЯ, КАЛЬЦИЯ,
МАГНИЯ, ЖЕЛЕЗА, МАРГАНЦА, МЕДИ, ЦИНКА, СВИНЦА,
КАДМИЯ, КОБАЛЬТА, НИКЕЛЯ, ХРОМА
Метод включает подготовку проб способами сухой, мокрой ми-
нерализации и кислотной экстракции и непосредственное проведе-
ние количественного определения на спектрофотометре.
Раствор минерализата испытуемой пробы распыляют в воздуш-
но-ацетиленовом пламени. Металлы, находящиеся в растворе ми-
нерализата, попадая в пламя, переходят в атомное состояние и по-
глощают излучения. Адсорбция электромагнитного излучения ато-
мами анализируемого элемента пропорциональна концентрации
металла в испытуемой пробе.
Способ сухой минерализации. Способ основан на минерализации
органических веществ путем сжигания пробы сырья или продукта в
электропечи при контролируемом температурном режиме и пред-
назначен для всех видов сырья и продуктов, кроме продуктов с со-
держанием жира 60 % и более.
Подготовка к минерализации. Фарфоровые чашки или тигли пос-
ле обычной мойки дополнительно обрабатывают раствором уксус-
ной кислоты на кипящей водяной бане в течение 1 ч или промывают
горячим раствором азотной кислоты, затем промывают водопровод-
ной водой и ополаскивают дистиллированной водой. Кварцевые
чаши и тигли моют раствором азотной кислоты, затем промывают
водопроводной и ополаскивают дистиллированной водой.
В чашку или тигель в зависимости от определяемого элемента
переносят навеску продукта из подготовленной к испытаниям про-
бы, взвешенную с точностью 0,01 г при массе навески до 10 г и с точ-
ностью 0,1 г при массе навески 10 г и более (табл. 9).
9. Масса (г) или объем (см3) навески пробы
Сырье, продукция ~ | Макроэлементы | Микроэлементы
Молоко и молочные продукты:
жидкие 3—5 50—100
сухие 1 5—10
творог, сыры 1 5—20
Растительные масла и продукты их переработ- 20 20
ки, животные жиры, сливочное масло
68
Минерализация проб. Навеску средней пробы продукта замачива-
ют в 96 %-ном этиловом спирте из расчета 5 см3 спирта на 1 г сухого
вещества. Чашки (тигли) с навеской накрывают часовыми стеклами
или чашками Петри, выдерживают при комнатной температуре в
течение 12—48 ч.
Чашки переносят на электроплитку, осторожно высушивают
и, постепенно увеличивая нагрев, выдерживают до начала обуг-
ливания.
Перед обугливанием продукты с высоким содержанием сахаров
обрабатывают 20%-ным раствором серной кислоты из расчета 5 см3
кислоты на 1 г сухого вещества.
Чашки с высушенными пробами помещают в холодную электро-
печь. Пробы минерализуют постепенно, повышая температуру
электропечи на 50 °C через каждые 30 мин и доведя ее до 450 °C. При
этих условиях минерализацию продолжают до получения серой
золы. Чашу с золой вынимают из электропечи, охлаждают до ком-
натной температуры, смачивают серую золу водой и раствором
азотной кислоты (1:1) в количестве 0,5—1 см3. Затем кислоту выпа-
ривают на электроплитке со слабым нагревом до получения сухой
золы и снова помещают чашу с пробой в электропечь, постепенно
повышая температуру до 300 °C и выдерживая при этой температуре
30 мин. Минерализацию считают законченной, когда зола станет
белого или слегка окрашенного цвета, без обугленных частиц. При
наличии обугленных частиц повторяют обработку золы раствором
азотной кислоты или водой, снова доозоляют.
Полученную золу растворяют при нагревании в азотной кислоте
(1:1) из расчета 1—5 см3 кислоты на навеску в зависимости от золь-
ности продукта. Раствор выпаривают досуха. Сухой остаток раство-
ряют в 1%-ной азотной или соляной кислоте до объема 15—20 см3.
При неполном растворении золы раствор с осадком упаривают до-
суха, растворяют в минимальном объеме (5—10 см3) соляной кисло-
ты (1:1) и снова упаривают до получения влажных солей, которые
растворяют в 1 %-ной соляной кислоте до объема 15—20 см3.
Если растворы золы содержат нерастворимый осадок, объем ра-
створителя можно довести до 25—30 см3, пробу подогреть до ра-
створения. Если и в этом случае полного растворения не произой-
дет, раствор фильтруют через промытый растворителем фильтр и
осадок отбрасывают.
Способ мокрой минерализации. Способ основан на полном раз-
рушении органических веществ пробы продукта при нагревании
с серной и азотной концентрированными кислотами с добавле-
нием пероксида водорода и предназначен для исследования всех
видов сырья и продуктов, кроме сливочного масла и животных
жиров.
Подготовка к минерализации. Стеклянную посуду после обычной
мойки дополнительно промывают раствором азотной кислоты,
ополаскивают водопроводной, а затем дистиллированной водой.
69
Навеску жидких и пюреобразных продуктов взвешивают в стака-
не, берут на обеззоленный фильтр, заворачивают в него и стеклян-
ной палочкой помещают на дно колбы Кьельдаля или плоскодон-
ной колбы. Навеску сухих продуктов помещают в колбу Кьельдаля,
добавляют 15 см3 воды, перемешивают.
Минерализация проб. В колбу с пробой продукта, подготовленной
к минерализации, вносят азотную кислоту из расчета 10 см3 на каж-
дый 5 г продукта и выдерживают не менее 15 мин. Затем в колбу
вносят 2—3 стеклянных шарика для равномерности кипения, зак-
рывают грушевидной пробкой и начинают нагревать на электро-
плитке, постепенно увеличивая нагрев и упаривая содержимое кол-
бы до объема 3—5 см3.
Колбу охлаждают, вносят 10 см3 азотной кислоты, содержимое
упаривают до объема 5 см3, после чего охлаждают. Эту процедуру
повторяют 2—4 раза.
В колбу вносят 10 см3 азотной кислоты, 2 см3 пероксида водо-
рода из расчета на каждые 5 г продукта. Содержимое колбы упари-
вают до объема 5 см3, не допуская образования коричневой окрас-
ки жидкости. При появлении коричневой окраски нагревание
прекращают.
Колбу охлаждают до комнатной температуры, добавляют 5 см3
азотной кислоты и 2 см3 пероксида водорода и снова нагревают до
появления белых паров серного ангидрида. Если при этом раствор
не обесцветился, эту процедуру повторяют. Минерализацию счи-
тают законченной, если раствор после охлаждения остается бес-
цветным.
Для удаления остатков кислот в охлажденную колбу добавляют
10 см3 воды и кипятят в течение 10 мин с момента выделения белых
паров, затем охлаждают. Добавление воды и нагревание повторяют
еще 2 раза. Если при этом образуется осадок, в колбу вносят 20 см3
дистиллированной воды, 2 см3 серной кислоты, 5 см3 соляной кис-
лоты. Полученную смесь кипятят до растворения осадка, постоян-
но дополняя испаряющуюся воду. Минерализат после охлаждения
используют для анализа полностью или количественно переносят в
мерную колбу вместимостью 50 см3, доводят до метки водой и пере-
мешивают.
Способ кислотной экстракции (неполной минерализации). Способ
основан на экстракции элементов из пробы продукта кипячением с
разбавленной соляной или азотной кислотой и предназначен для
сливочного и растительного масла и сыра.
Экстракция проб продукта. В термостойкую колбу с навеской
продукта вносят цилиндром 40 см3 раствора соляной кислоты (1:1)
и такой же объем раствора азотной кислоты (1:2). Туда же добавля-
ют несколько стеклянных шариков, вставляют холодильник, поме-
щают на электроплитку, покрытую асбестом, и кипятят в течение
1,5 ч с момента закипания. Затем содержимое колбы медленно ох-
лаждают до комнатной температуры, не вынимая холодильника.
70
Колбу с экстракционной смесью сливочного масла с кислотой
ставят на холодную водяную баню для затвердевания жира. Затвер-
девший жир прокалывают стеклянной палочкой, фильтруют через
фильтр, смоченный кислотой, затем промывают фильтр 5—7 см3
дистиллированной водой. Оставшийся в колбе жир расплавляют на
водяной бане, добавляют 10 см3 раствора кислоты, встряхивают, ох-
лаждают. После охлаждения жир прокалывают и промывную жид-
кость сливают в тот же сосуд через тот же фильтр, затем фильтр про-
мывают 5—7 см3 дистиллированной воды.
Экстракционную смесь растительного масла с кислотой перено-
сят в делительную воронку. Колбу ополаскивают 10 см3 кислоты,
которую сливают в ту же воронку. После разделения слоев нижний
водный слой сливают через смоченный кислотой фильтр в кварце-
вую или фарфоровую чашку, затем фильтр промывают 5—7 см3 дис-
тиллированной воды.
Экстракционную смесь сыра с кислотой фильтруют через смо-
ченный кислотой фильтр в кварцевую или фарфоровую чашку.
Колбу ополаскивают 10 см3 кислоты, которую фильтруют через
тот же фильтр, затем фильтр промывают 5—7 см3 дистиллирован-
ной воды.
Экстракционную смесь осторожно выпаривают и обугливают на
электроплитке. Затем чашу помещают в электропечь и озоляют
смесь до исчезновения обугленных частиц.
Контрольный опыт. При проведении минерализации проб лю-
бым способом обязательно проведение контрольного опыта для
учета уровня загрязнений, которые могут возникнуть на любой ста-
дии подготовки проб. В каждой подготавливаемой серии проб кон-
трольный тигель (чашка, стакан и пр.) обязательно проводят через
все стадии подготовки вместе с тиглями, содержащими навески
продуктов, при одинаковом времени нахождения на электроплит-
ке, в муфеле, при использовании тех же реактивов и в том же коли-
честве, той же мерной посуды и т. д. Полученный в контрольном
опыте раствор анализируют в той же серии измерений, что и раство-
ры проб. Если пробы пищевых продуктов обычно анализируют в
двух повторностях, то контрольный опыт также целесообразно про-
водить в двух повторностях.
Приготовление проб к испытанию. Полученный раствор золы
можно анализировать сразу или после дополнительной подготовки,
которая необходима в следующих случаях:
концентрация элемента в растворе значительно превышает вер-
хний предел интервала рабочих концентраций. Исходный раствор
разбавляют бланковым раствором до необходимого уровня концен-
трации (бланковым называют раствор, который применяют для
приготовления анализируемых растворов проб);
концентрация элемента в исходном растворе ниже предела его
обнаружения. В таком случае концентрирование выполняют мето-
дом экстракции. Его применяют также при определении микроэле-
71
ментов на приборах, не имеющих автоматической коррекции несе-
лективного поглощения.
Экстрагировать вещества (железо, цинк, медь, марганец, кадмий,
свинец, кобальт, никель и хром) при регулировании pH можно с по-
мощью разнообразных реактивов, например диэтилдитиокарбамата
натрия (или аммония) и п-бутил ацетата (или метилизобутил кетона).
Приготовление стандартных растворов и построение градуировоч-
ного графика. Содержание элементов в испытуемых растворах опре-
деляют методом градуировочного графика.
Основные стандартные растворы (эталоны) готовят из чистых
металлов (более 99,9 %) или чистых (х.ч., ос.ч.) устойчивых соеди-
нений элементов. Для получения устойчивых при хранении раство-
ров используют растворители.
Концентрация металла в основном стандартном растворе со-
ставляет 1 г/дм3 (1000 мкг/см3). Срок хранения растворов не должен
превышать 2—3 года, при условии что растворы хранили в герме-
тичной посуде из стекла, полиэтилена высокой плотности, поли-
стирола, кварца. Разбавленные растворы концентрацией около
100 мкг/см3 обычно хранят до 1 мес, концентрацией 10 мкг/см3 — в
течение 2—3 сут. При укупоривании химической посуды необходи-
мо стараться не применять резиновые пробки, которые могут быть
источником загрязнения ряда микроэлементов. Кроме того, нельзя
хранить растворы в местах, куда попадает прямой солнечный свет.
Необходимые реактивы и схема приготовления основных стан-
дартных растворов приведены в табл. 10. Пользование таблицей про-
демонстрируем на примере подготовки основного стандартного ра-
створа для определения содержания натрия. Как показано в графе 2,
сначала необходимо подготовить хлорид натрия путем прокаливания
его при 500 °C. Для приготовления основного стандартного раствора
необходимы дистиллированная вода и хлороводородная кислота
(графа 3), количество последней определяют с учетом ее концентра-
ции в основном стандартном растворе 0,2 н. (графа 4). Для приготов-
ления основного стандартного раствора навеску хлорида натрия в ко-
личестве 2,542 г растворяют в дистиллированной воде, добавляют
рассчитанное количество соляной кислоты, объем полученного ра-
створа доводят дистиллированной водой до 1000 см3. Рабочие стан-
дартные растворы готовят путем разбавления основных стандартных
растворов до концентрации, соответствующей рабочему диапазону
концентраций и близкой к концентрации элемента в анализируемом
растворе. Для разбавления используют раствор, применяемый для
приготовления анализируемых растворов проб.
Обычно для построения градуировочного графика в области ра-
бочих концентраций достаточно 3—5 рабочих стандартных раство-
ров, при работе в линейной области — 2 раствора. При большой и
особенно знакопеременной кривизне графика необходимо увели-
чить число рабочих стандартных растворов или ограничить область
рабочих концентраций более узким диапазоном (табл. 10).
72
10. Рекомендуемые реактивы и порядок приготовления основных стандартных
растворов, концентрация металла — 1000 мкг/см3
Элемент Исходный реактив; навеска, г Схема приготовления основного стандартного раствора объемом 1000 см3 Концентрация кислоты в конец ном растворе
Натрий NaCl, прокаленный при 500 °C; 2,542 г Н2О + конц. НС1 0,02 н. НС1
Калий КС1, прокаленный при 500 °C; 1,907 г Н2О + конц. НС1 0,02 н. НС1
Кальций СаСО3, высушенный при 110 °C; 2,497 г НС1 (2 н.), нагрев + Н2О —
Магний Металл, 1,000 г MgO, прокаленный при 500 °C; 1,658 г 100 см3 разбавленной (1:100) НС1, осторожно, по каплям (!) + Н2О + 25 см3 НО (25 %-ная) + Н2О —
Железо Металл, 1,000 г 20 см3 НС1 (5 н.) + 5 см3 <оНц. HNO3 + Н2О 0,1 н. НС1
Цинк или Металл, 1,000 г ZnO, 1,245 г НС1(1:1) + Н2О HNO3 (40%-ная) + Н2О 1 н. НС1 0,1 н. HNO3
Медь Металл, 1,000 г 50 см3 HNO3 (5 н.) (упарить досуха на водяной бане) + 5 см3 конц. НС1 (упарить досуха) + разбавленная НС1 + Н2О 0,1 н. НС1
Марганец Металл, 1,000 г 10 см3 конц. HNO3 (упарить досуха на водяной бане) + 5 см3 конц. НС1 (упарить досуха) + несколько капель конц. НС1 + Н2О 0,02 н. НС1
Свинец Металл, 1,000 г Pb(NOj)2, высушен- ный при 110 °C; 1,599 г Минимальный объем HNO3 (6 н.) + Н2О + конц. НС1 Разбавленная HNO3 (1:100) 1 н. на 1 % hno3
Кадмий Металл, 1,000 г CdO, 1,142г Разбавленная HNO3 (упарить досуха и выдержать при 80— 90 °C на водяной бане) + 5 см3 НС1 (1 н.) (упарить досуха) + - разбавленная НС1 + Н2О 20 см3 HQ (5 н.)+Н2О+конц. HQ 1 н. НС1 1 н. на
Кобальт Металл, 1,000 г Минимальный объем HNO3 'упарить досуха), растворить в зазбавленной НС1 + Н2О 1 н. НС1
Никель Металл, 1,000 г Минимальный объем HNO3 (упарить досуха на водяной бане) + 5 см3 НС1 (упарить досуха) + Н2О + НС1 o,i н. на
Хром Металл, 1,000 г НС1 + Н2О Разбавленная 1 н. на
К2Сг2О2, высушенный при 150 °C; 2,830 г НС1 + Н2О 0,02 н. на
73
МЕТОДИКА ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ НАТРИЯ,
КАЛИЯ, КАЛЬЦИЯ, МАГНИЯ, ЖЕЛЕЗА, МАРГАНЦА, МЕДИ, ЦИНКА,
СВИНЦА, КАДМИЯ, КОБАЛЬТА, НИКЕЛЯ, ХРОМА
Подготовка спектрофотометра к работе и условия измерения. На-
стройку прибора проводят в соответствии с технической инструк-
цией завода фирмы-изготовителя. Рекомендуемые условия измере-
ний, а также значения предела определения приведены в табл. 11.
11. Условия атомно-абсорбционных измерений в воздупшо-ацетиленовом пламени
Элемент Длина ВОЛНЫ, нм Щель, нм Пламя Высота горелки, мм Оптималь- ный диапа- зон рабочих концентра- ций, мкг/см3 Предел опреде- ления, мкг/см3
Натрий 330,2 3,0 Окислительное 7-8 10-100 1
589,6 0,4 » 7-8 2-30 0,1
589,0 0,4 » 7-8 0,5-5 0,005
Калий 404,5 0,4 Стехиометрическое 7—8 50-1000 8
769,9 1,5 » 7-8 5-50 0,5
766,5 1,5 » 7-8 0,5-10 0,005
Магний 285,2 з,о Окислительное 10-15 0,1-10 0,001
Кальций 422,7 3,0 » 15-20 5-30 0,01
Железо 248,3 0,2 Стехиометрическое 7—8 1-10 0,01
Цинк 213,9 1,5 Окислительное 6-7 1-10 0,002
Медь 324,8 1,5 Стехиометрическое 7-8 0,005-5 0,003
Марганец 279,5 0,7 » 7-8 0,1-2 0,003
Свинец 283,3 2,0 » 7-8 0,1-2 0,02
217,0 2,0 » 7-8 0.1-2 0,01
Кадмий 228,8 1,0 Окислительное 6-7 0,02-1 0,001
Кобальт 240,7 0,2 Стехиометрическое 6-7 0,05-2 6,01
Никель 232,1 0,2 » 6-7 0,1-5 0,01
Хром 357,9 1,3 Восстановительное 6-7 0,1-5 0,005
359,4 1,3 » 6-7 0,05-5 0,005
Резонансную линию выбирают для каждого прибора после пред-
варительных сравнительных испытаний.
Настройку монохроматора проводят по максимуму излучения
источника при минимальной щели.
Вид воздушно-ацетиленового пламени устанавливают визуально.
Восстановительное пламя характеризуется слабым белесым свечени-
ем в нижней части факела, оно соответствует соотношению (4:1) по-
токов воздух — ацетилен и требует вдвое меньшего расхода ацетиле-
на при том же воздушном потоке. Стехиометрическим пламенем
считают промежуточный вариант. При определении микроэлемен-
тов, особенно кобальта, никеля и хрома условия измерения могут
быть дополнительно улучшены регулированием соотношения газов
и высоты горелки до получения максимального сигнала поглощения.
Особенности определения отдельных элементов. Натрий и калий.
В отличие от атомно-абсорбционного измерения эмиссия натрия
при длине волны дублетной линии 589 нм может давать завышен-
ные результаты из-за излучения кальция, содержащегося в исследу-
74
емой пробе. Поэтому при определении содержания натрия в подго-
товку проб включают осаждение кальция оксалатом аммония.
При концентрациях калия менее 100 мкг/см3 необходимо учи-
тывать соотношение калия и натрия в пробах и смешанных раство-
рах сравнения. Это соотношение устанавливают как на основе ожи-
даемых результатов, так и путем предварительных приближенных
определений. При концентрациях калия более 100 мкг/см3 влияни-
ем натрия можно пренебречь.
Кальций. Бланковый раствор, растворы проб и контрольные ра-
створы сравнения должны содержать 0,5 % стронция (в расчете на
металл) в виде хлорида.
Железо и цинк. Растворы проб пищевых продуктов во многих слу-
чаях требуют разбавления, которое уменьшает межэлементные и
матричные влияния. Поэтому масса навески продукта должна быть
определена с таким расчетом, чтобы не превышать границ диапазо-
на рабочих концентраций, указанных в табл. 11.
Проведение измерений и обработка результатов. Измерения про-
водят в соответствии с технической инструкцией, прилагаемой к
прибору, с учетом следующих особенностей. Поскольку система
распылителя в процессе работы может засоряться, чувствитель-
ность измерения может нарушаться. В этом случае приходится про-
водить повторные градуировки. Обычно достаточно выполнить две
градуировки — в начале и в конце измерений. В других случаях це-
лесообразно проводить измерения по следующей схеме: первая гра-
дуировка — первая серия замеров абсорбции 5—10 анализируемых
растворов; вторая градуировка — вторая серия замеров абсорбции
тех же растворов в обратном порядке; третья градуировка — третья
серия замеров. По результатам, полученным в двух сериях, находят
среднее значение.
По концентрации элемента в растворе находят его содержание в
продукте (мг/кг):
У _CVK-CkVk
Х~ М
где С — концентрация химического элемента в растворе пробы, мкг/см3; Ск — кон-
центрация химического элемента в контрольном опыте, мкг/см3; к —коэффици-
ент разбавления или концентрирования исходного раствора пробы (отношение
объема анализируемого раствора к объему раствора, взятому для разбавления или
концентрирования); И— объем исходного раствора пробы, см3; Ик — объем раство-
ра в контрольном опыте, см3; М — навеска пробы, г.
СПЕКТРОСКОПИЯ МАГНИТНОГО РЕЗОНАНСА.
МАСС-СПЕКТРОСКОПИЯ
Применение радио- и микроволновой областей электромагнит-
ного спектра в аналитической химии и физико-химических иссле-
дованиях основывается на явлениях ядерного магнитного и элект-
ронного парамагнитного резонансов.
75
Спектроскопия ядерного магнитного резонанса (ЯМР) изучает
магнитный резонанс, возникающий в результате взаимодействия
магнитного момента ядра с внешним магнитным полем. С помо-
щью метода ЯМР можно исследовать ядра с собственным моментом
количества движения (спин ядра) и связанным с ним магнитным
моментом ядра.
Согласно квантовой механике собственный момент количества
движения (спин) ядра принимает строго определенные значения.
Так как спин ядра является вектором, то он характеризуется вели-
чиной и направлением. Во внешнем магнитном поле для спина ядра
возможны две ориентации: вдоль и против направления силовых
линий внешнего магнитного поля. Каждому значению спина соот-
ветствует определенное значение энергии. Переориентация спина
ядра с изменением направления сопровождается поглощением
энергии Д Е. Такие переходы вызываются воздействием на ядро ра-
диочастотной области электромагнитного спектра. При этом ана-
лизируемая система поглощает энергию при строго фиксирован-
ных значениях частоты v, т. е. наблюдается явление резонанса. Та-
кое поглощение энергии измеряют экспериментально, ДЕ прямо
пропорционально напряженности магнитного поля в месте распо-
ложения ядра и определяется как ^.E-h-v, где h — постоянная
Планка.
Спектроскопия электронного паромагнитного резонанса (ЭПР)
изучает магнитный резонанс, возникающий в результате взаимо-
действия магнитного момента электрона с внешним высокочастот-
ным (микроволновым) магнитным полем. Метод ЭПР служит для
исследования внутримолекулярного окружения неспаренных элек-
тронов.
Теория магнитного резонанса применима не только к ядрам, но
и к электронам, поскольку последние также имеют спин и магнит-
ный момент. В отсутствие внешнего магнитного поля спины элект-
ронов беспорядочно ориентированы, а энергия электронов одина-
кова. В постоянном магнитном поле магнитные моменты электро-
нов ориентированы соответственно направлению внешнего маг-
нитного поля (рис. 14). Электроны с ориентацией спинов вдоль
поля находятся на высоком энергетическом уровне, электроны с
ориентацией против поля — на низком, более стабильном, уровне.
Если на электроны, находящиеся в однородном магнитном поле
воздействовать высокочастотным магнитным полем, направление
которого перпендикулярно направлению однородного магнитного
поля, то при определенных соотношениях между напряженностью
постоянного поля и частотой переменного поля наблюдается резо-
нансное поглощение энергии переменного поля. Оно регистриру-
ется на спектрометре в виде спектра электронного парамагнитного
резонанса — ЭПР-спектра. При количественной оценке спектра в
качестве основного аналитического параметра используют кон-
станту спин-спинового взаимодействия.
76
Рис. 14. Ориентация спинов электронов в отсутствии внешнего магнитного
поля (л) и под действием внешнего магнитного поля (6)
Масс-спектроскопия занимает особое положение среди спект-
роскопических методов. В строгом смысле слова этот метод не явля-
ется спектроскопическим, так как вещество при анализе не подвер-
гается воздействию электромагнитного излучения. Этот метод по-
лучил свое название из-за формального сходства и графического
изображения масс-спектров со спектрами спектроскопических ме-
тодов. Масс-спектроскопия основана на изучении тока от фрагмен-
тов ионов, полученных из нейтральных молекул вещества путем
воздействия на них пучка электронов.
СПЕКТРОМЕТРИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
Вещество, исследуемое методом ЯМР, помещают одновремен-
но в два магнитных поля — одно постоянное, а другое радиочас-
тотное. Измерение осуществляют на ЯМР-спектрометре (рис. 15),
основными составляющими элементами которого являются: элек-
тромагнит (в простых приборах используют постоянный магнит);
генератор радиочастотного излучения; датчик, в который поме-
щают пробирку с образцом; электронный усилитель и интегратор;
самописец.
Пробирку с анализируемым образцом 1 помещают в создаваемое
генератором радиочастотного излучения 7и магнитом 3 магнитное
поле напряженностью Яо- Вследствие поглощения энергии анали-
зируемым веществом на контуре падает напряжение, на датчик 4
поступает аналитический сигнал, усиливается усилителем сигнала
6 и подается на самописец 5 (ЯМР-спектр).
77
Рис. 15. Принципиальная схема ЯМР-спектрометра:
1 — пробирка с образцом; 2 — катушка генератора радиочастотного поля; 3 — магнит;
4 — датчик-катушка приемника радиочастотного поля; 5 — самописец для регистра-
ции ЯМР-спектр; б—детектор и усилитель сигнала; 7— генератор радиочастотного
излучения
Поле Но модулируется так, что оно меняется с определенной часто-
той и таким образом осуществляется горизонтальная развертка ЯМР-
спектра. Для записи ЯМР-спектра применяют специальные регистра-
ционные бланки, на которых показаны условия съемки спектра.
Применение метода ЯМР в анализе различных веществ связано с
использованием аналитических параметров ЯМР (схема 1).
Схема 1. Аналитические параметры и области их применения
78
В ЯМР-методе используют следующие аналитические парамет-
ры; химический сдвиг, константа спин-спинового взаимодействия,
интенсивность сигнала, время релаксации.
Химический сдвиг. Значение напряженности магнитного поля в
месте расположения ядра обычно отличается от значения напря-
женности внешнего магнитного поля Но и называется эффектив-
ной напряженностью Нэф. Для ядер с разным окружением эффек-
тивная напряженность магнитного поля различна. Это объясняет-
ся тем, что значение Нэф зависит от окружения ядра, т. е. от кон-
фигурации электронного облака окружающих ядро групп и
спинов соседних ядер. Каждой функциональной группе соответ-
ствует характерный ей диапазон значений химических сдвигов,
расположенных в более или менее узкой области спектра. Значе-
ния химических сдвигов приводятся в каталогах и атласах ЯМР-
спектров.
Изменение эффективной напряженности магнитного поля в мес-
те расположения ядра характеризуется константой экранирования о:
Яэф = Я0(1-о),
где #Эф “ эффективная напряженность магнитного поля в месте расположения
ядра п.
Поскольку измерить величину Яэф практически невозможно, в
ЯМР-спектрометрии используют разность резонансных сигналов
стандартного и анализируемого веществ. Сдвиг резонансной линии
анализируемого вещества относительно стандартного вещества на-
зывается химическим сдвигом. Значение этой величины строго со-
ответствует строению молекулы. Химический сдвиг зависит от на-
пряженности внешнего (приложенного) магнитного поля Но. В
связи с этим в зависимости от типа прибора изменяются напряжен-
ность и соответственно химический сдвиг одного и того же веще-
ства. Для того чтобы получить независимые от типа прибора резуль-
таты для химических сдвигов, ввели величину
о = (Яг-Яя)/Я0,
где а — химический сдвиг резонансной линии ядра п относительно резонансной ли-
нии ядра г.
Резонансные линии сдвигаются в области поля с меньшей или
большей напряженностью в зависимости от строения вещества. Это
объясняется различной степенью экранирования ядер в одной и той
же молекуле. Таким образом, конфигурация электронной оболочки
влияет на получаемые результаты.
Константа спин-спинового взаимодействия. Взаимодействие по-
лей ядер друг с другом через одну или несколько связей называется
спин-спиновым взаимодействием. Этот вид взаимодействия про-
79
является в расщеплении резонансного сигнала в линейчатый спектр,
причем число линий, относительные интенсивность и расстояние
между линиями можно рассчитать. Расстояние между линиями на-
зывается константой спин-спинового взаимодействия. Так как взаи-
модействие между ядрами происходит внутри молекулы, константа
взаимодействия не зависит от внешнего магнитного поля.
Интенсивность сигнала. Площадь подлинней ЯМР-спектра пря-
мо пропорциональна числу резонирующих ядер. Поэтому соотно-
шение интенсивностей сигналов в ЯМР-спектре исследуемого ве-
щества пропорционально соотношению числа ядер в чистом веще-
стве. При количественном анализе вещества методом ЯМР-спект-
рометрии нет необходимости в чистых образцах анализируемых
веществ. Это связано с тем, что прибор градуируют не по анализи-
руемому веществу, а по площади сигнала стандарта. В этом случае
на ЯМР-спектре определяют участки, соответствующие площади
сигнала одного ядра. Поскольку эта площадь известна, то по сум-
марной площади определяют общее количество ядер в молекуле.
Все рассмотренные аналитические параметры — химический
сдвиг, константа спин-спинового взаимодействия, интенсивность
сигнала используют в ЯМР-спектрометрии высокого разрешения и
применяют для определения состава и структуры химических со-
единений.
Время релаксации. Под воздействием радиочастотного поля на
ядра атомов наблюдают переходы атомов с нижнего энергетическо-
го уровня на верхний. Однако возможен переход атомов с верхнего
уровня на нижний, что сопровождается выделением энергии. Раз-
личают два типа обменных процессов: спин-решеточная релакса-
ция, когда энергия передается во внешнее окружение (на «решет-
ку»), и спин-спиновая релаксация, когда энергия передается через
систему связей другим электронам.
Методы ЯМР, в которых в качестве аналитических параметров
используют интенсивность сигналов ЯМР и время ядерной магнит-
ной релаксации, называют релаксационными. Они основаны на
том, что в присутствии парамагнитных веществ сокращается время
ядерной релаксации. Время релаксации ядер в присутствии пара-
магнитных веществ выражается уравнениями
1\ = КХС, Т2 = К2С,
где 7) и 7^ — время спин-решеточной и спин-спиновой релаксаций данных ядер;
Кх, К2 — коэффициенты релаксационной эффективности парамагнитных частиц по
отношению к данным ядрам спин-решеточной и спин-спиновой релаксации; С —
концентрация парамагнитных частиц в растворе.
По физическому смыслу К\ и К2 соответствуют скорости релак-
сации ядер при концентрации анализируемых парамагнитных час-
тиц, находящихся в 1 М растворе исследуемого вещества.
Время релаксации можно измерить несколькими методами. На-
дежным является импульсный вариант метода ЯМР — метод спи-
80
нового эха. На исследуемый образец в магнитном поле через опре-
деленные промежутки времени подают кратковременные радиоча-
стотные импульсы в области резонансного поглощения, и в прием-
ной катушке появляется сигнал спинового эха, максимальная амп-
литуда которого связана с временем релаксации определенным со-
отношением. С помощью установки спин-эха можно определять
время релаксации от 10~5 до 102 с. Для проведения обычных анали-
тических определений измеряют амплитуду сигнала резонансного
поглощения. Часто используют метод градировочного графика.
Релаксационными методами ЯМР были исследованы некоторые
пищевые продукты и вода на содержание отдельных компонентов:
влаги, жира, металлов и др. При этом использовали эксперимен-
тальные зависимости вида
Tx=f(Cy,T2~f(CyJt=f(C),
где 7), Т2 — время соответственно спин-решеточной и спин-спиновой релаксации
атомов исследуемой фазы; С — концентрация исследуемого компонента; /(—ин-
тенсивность сигнала ЯМР исследуемой фазы образца.
Линейную зависимость времени 7) от концентрации компонен-
та использовали для определения содержания парамагнитных при-
месей в воде (рис. 16). Для анализа использовали серию импульсов
180° — т — 90°. Время анализа 30 с.
Обычно зависимость времени релаксации Г1; Т2 от содержания
какого-либо компонента для нежидких сред нелинейна. Однако это
не помешало использовать их для количественного анализа пище-
вых объектов. По зависимости 7\=f (С) определили влажность
концентрата кофе (рис. 17), содержание сухих веществ и жира в
Рис. 16. Зависимость времени спии-
решеточной релаксации Tj от концен-
трации парамагнитных ионов:
I- Си(Ц); 2- Fe(III)
Рис. 17. Зависимость времени спии-
решеточиой релаксации от содер-
жания кофе
81
Рис. 18. Зависимость интенсивно-
сти сигнала ЯМР /tOT массовой
доли жира в семенах подсолнеч-
ника .Ж
сыре, воды в сухом молоке и твороге.
При определении воды в сухом мо-
локе время определения составляло
70 с, в твороге — 10 с.
Линейную зависимость интен-
сивности сигнала ЯМР It какого-
либо компонента в определенный
момент времени / от его содержания
использовали для определения со-
держания влаги, жиров и клетчатки
в сельскохозяйственных продуктах,
воды и масла —в семенах маслич-
ных культур (рис. 18).
При определении соотношения
твердой и жидкой фаз в жирах —
важного параметра при производ-
стве многих пищевых продуктов —
измеряют сигнал It дважды: в мо-
мент = 10—12 мкс и /2 = 70 мкс, а для расчета массовой доли жид-
кой фазы Ржф (%) используют выражение 7’жф = А:(/?2 -100)//^.
Полное время анализа, например, на приборе Minispec Р-20 фирмы
Bruker, зависит от требуемой точности: 2 с — 0,5 % и 6 с — 0,3 %.
Эту методику применяют для исследования твердой и жидкой фаз в
сливочном масле, маргаринах, молочном жире.
Рассмотренные релаксационные методы значительно произво-
дительнее по сравнению с базовыми методами анализа и во многих
случаях отличаются меньшей погрешностью определения, вместе с
тем они требуют использования специально подготовленных об-
разцов сравнения и иногда взвешивания пробы.
Данный метод использован сотрудниками ВНИИМСа для оцен-
ки состояния и свойств воды и жира в молочном жире, сливках, сли-
вочном масле, сычужных и плавленых сырах.
В качестве примера приведены результаты исследований по
изучению изменения состояния воды в нежирном сыре и в плав-
леных сырах с различными солями-плавителями; цитратами и
триполифосфатом натрия. Исследования проведены с помо-
щью импульсного ЯМР-релаксометра. Измеряли время спин-
спиновой релаксации Т2 и интенсивность сигнала It, которую
измеряли в относительных единицах. За единицу приняли ин-
тенсивность сигнала ЯМР натурального сыра при времени ре-
лаксации 10-1 с.
Данный метод основан на том, что релаксационные характерис-
тики ЯМР атомов воды зависят от содержания и формы связи воды
с составными частями молока. Исследования проводили по мето-
дике, описанной Б. А. Сурковым. Результаты измерений приведены
в табл. 12.
82
12. Время спин-спиновой релаксации и интенсивность сигнала ЯМР в зависимости
от вида сыра
Сыр Время спин-спиновой релаксации Тг, с
10-5 1 io-3 1 10-2 | ю-'
Интенсивность сигнала огн. ед.
Натуральный нежирный (зрелость 90 сут) Плавленый: 2,2 — 2,8 1,0
с цитратами 2,1 2,8 3,4 2,0
с триполифосфатом 2,4 5,9 17,2 2,1
натрия
Для исследованных образцов диапазон значений Т2 имеет поря-
док 10~5—10-1 с. Известно, что показатели времени релаксации в
интервале 10-3—10-1 характерны для молекулярной подвижности
воды диффузионно-усредненных структур.
Для натурального нежирного сыра обнаружены два значения
времени релаксации 10-2 и 10-1 с, а для плавленых сыров обнару-
жена величина 10"3 с, что позволяет судить о плавленых сырах как
гетерогенных системах. Компонента Тт, соответствующая времени
Ю-3 с, относится к сильно заторможенной воде, связанной с моле-
кулами белка.
На основании полученных данных построена модель распреде-
ления воды в структуре натуральных и плавленых сыров, заключаю-
щаяся в определении размеров микрообластей воды, соответствую-
щих различному времени релаксации Г2. Результаты расчета приве-
дены в табл. 13, 14.
13. Размеры микрообластей воды в образцах сыра, мкм
Сыр Я, Нг Из
Натуральный нежирный Плавленый: 0,55 — 2,1 4,9
с цитратами 0,56 0,9 2,1 6,5
с триполифосфатами 0,57 0,5 1,4 4,8
натрия
14. Содержание различных форм влаги в натуральных и плавленых сырах, %
Сыр Прочно связанной с белком (Bt) Дополнительно иммобилизован- ной в процессе плавления (В2) Удерживаемой элементами пространствен- ной сетки (2?3) Свободной (В4)
Натуральный 8,0±0,8 Плавленый: с цитратами 10,5±1,1 с триполифосфатом 8,6±0,7 0 7,7±0,8 11,5±1,3 82±1,0 78±0,5 75,6±0,5 10,0+3,0 3,7±0,4 4,3±0,5
натрия
83
Известно, что релаксация в гетерогенных системах обусловлена
не только размерами микрообластей, но также соотношением
объемной и поверхностной воды в самих микрообластях*.
Используя закономерности, приведенные в книге Лиллфорда,
определили содержание различных форм влаги в сырах.
Форма Bi характеризует воду, прочно связанную с молекулами
белка, поскольку геометрические размеры микрообластей Н\ со-
впадают с размерами мицелл казеина, формирующих белковый
каркас. Доля такой воды составляет около 9 % всей влаги. При
плавлении ее количество увеличивается до 10,5 и 8,6 % в зависи-
мости от используемой соли-плавителя, что свидетельствует о
повышении в процессе плавления гидратации молекул белков.
Это обусловлено диспергированием исходных элементов струк-
туры до субъединиц, в результате чего увеличиваются поверх-
ность белка и количество гидрофильных групп, контактирующих
с водой.
Форма Вг характерна только для плавленых сыров и составляет
для сыров с цитратами и триполифосфатом натрия соответственно
7,7 и 11,5 %. Эти результаты указывают на дополнительную иммо-
билизацию воды в процессе плавления, сопровождающую разруше-
ние пространственной сетки белкового каркаса.
Во всех исследованных образцах значительная часть влаги (75—
82 %) приходится на форму В^. Эта вода удерживается элементами
пространственной белковой структуры (макрозернами в сычужных
сырах, цилиндрическими ассоциатами, формирующими простран-
ственную сетку параказеина, или в виде пленок между флокулиро-
ванными частицами эмульсии — в плавленых). Ее доля максималь-
на в исходном натуральном сыре (82 %) и снижается в плавленых
сырах, полученных при использовании цитратов (78 %) и триполи-
фосфата натрия (75 %).
Наиболее крупные микрополости, соответствующие свободной
воде (В4), можно рассматривать как дефекты пространственной
структуры. Их образование может быть вызвано синерезисом и
схлопыванием белковой пространственной сетки. В натуральном
сыре содержание свободной воды — наибольшее (7—13 %), в плав-
леных сырах ее доля уменьшается в зависимости от качества исход-
ного сырья и природы солей-плавителей в 2,3—2,7 раза и составляет
3,7 и 4,3 % соответственно для плавленых сыров с солями-плавите-
лями цитратами и фосфатами.
Полученные данные позволяют сделать вывод об изменении со-
стояния влаги и переходе ее из одной формы связи в другую при
плавлении натурального сыра. В процессе плавления происходит
дополнительное связывание воды (7,7— 11,5 %) и увеличивается ко-
*Лиллфорд П., Кларк А., Джонс Д. Распределение воды в гетерогенных пищевых
продуктах и модельных системах/Вода в полимерах. — М., 1984.
84
личество прочно связанной воды (10,5 и 8,6 % против 8 %). При
этом содержание свободной воды уменьшается примерно в 2—3
раза, так как при плавлении возрастает гидратация молекул белков
натурального сыра.
СПЕКТРОМЕТРИЯ ЭЛЕКТРОННОГО
ПАРАМАГНИТНОГО РЕЗОНАНСА
Метод ЭПР основан на измерении поглощения веществом энер-
гии внешнего магнитного поля.
В основе ЭПР, как и ЯМР, лежит принцип магнитного резонан-
са. Однако ЭПР-сигналы в отличие от ЯМР-сигналов при одном и
том же значении напряженности постоянного магнитного поля
расположены в области более высоких частот (микроволновая об-
ласть). Это объясняется большим значением магнитного момента
электрона, чем у протона, что связано с наличием в молекуле одно-
го неспаренного электрона. Частота, при которой происходит резо-
нанс, пропорциональна напряженности внешнего магнитного
поля. ЭПР-спектр получают, изменяя как напряженность однород-
ного магнитного поля, так и его частоту. Однако на практике более
удобно сканирование по напряженности. Иногда вместо напря-
женности и частоты используют фактор пропорциональности - g-
фактор. Значение g-фактора для свободного электрона составляет
2,002319, для неспаренных электронов в молекулах —от 1 до 6.
На рис. 19 приведена принципиальная схема ЭПР-спектромет-
ра. В качестве источника излучения сверхвысокочастотного поля
при ЭПР-спектрометрии служит специальная радиолампа — клис-
трон 1. В современных приборах используют следующие частоты
СВЧ-поля: v = 9,5; 24 и 35 ГГц, при длине волны соответственно
А = 3,2; 1,25 и 0,85 см. Образец помещают в тонкие стеклянные ам-
пулы или плоские кюветы 2и размещают между полюсами электро-
магнита, где он поглощает энергию высокочастотного поля; изме-
Источник энергии Образец Детектирование и регистрация
Рис. 19. Принципиальная схема ЭПР-спектрометра:
1 — специальная радиолампа клистрон для излучения сверхчастотного поля; 2 — про-
бирка или кювета с образцом в магнитном поле; 3—детектор-полупроводниковый
диод; 4— усилитель; 5— самописец
85
ренная разность энергии детектируется полупроводниковым дио-
дом 3, усиливается на усилителе 4 и регистрируется с помощью са-
мописца 5. Площадь под ЭПР-сигналом пропорциональна числу
неспаренных электронов, содержащихся в образце.
Метод ЭПР применяют для анализа всех соединений, содержа-
щих неспаренные электроны, независимо от их агрегатного состоя-
ния. Область применения определяется конструкцией кюветы. Для
проведения кинетических исследований применяют специальные
проточные системы. Кроме того, сконструированы кюветы, позво-
ляющие получить радикалы некоторых диамагнитных соединений
(непосредственно в образце) с помощью фотолиза, радиолиза и
окислительно-восстановительных реакций. ЭПР является одним
из самых чувствительных методов, предел чувствительности состав-
ляет 10-11 моль/л.
МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометрия основана на получении ионов из нейт-
ральных молекул путем воздействия на них пучком электронов,
обладающих энергией, достаточной для ионизации. При этом
главным образом образуются положительные ионы, которые мо-
гут распадатьс'я на отдельные фрагменты. Регистрируемая зависи-
мость ионных токов от массы отдельных фрагментов называется
масс-спектром.
Молекула, возбужденная в результате взаимодействия с элект-
роном (с энергией более 103 кДж/моль), распадается с образова-
нием положительного молекулярного иона и электрона (иониза-
ция).
В большинстве случаев молекулярный ион обладает значительной
внутренней энергией и быстро распадается далее (10-8—Ю~10с) с
образованием заряженных и незаряженных фрагментов (фрагмен-
тация).
Осколочные ионы, в свою очередь, могут распадаться с обра-
зованием новых фрагментов. В некоторых случаях фрагментация
сопровождается перегруппировками. Процесс фрагментации
молекулярных ионов происходит до тех пор, пока не образуются
ионы, внутренней энергии которых недостаточно для их даль-
нейшего превращения. Образующийся молекулярный ион и его
фрагменты детектируются по их массовым числам, представляю-
щим отношение т/е, т. е. массы к заряду. Так как в большинстве
случаев образуются однозарядные ионы, то отношение т/е соот-
ветствует массе иона. Масс-спектрометры работают при высоком
вакууме, что сводит к минимуму нежелательные межмолекуляр-
ные реакции и, кроме того, благоприятствует внутримолекуляр-
ной фрагментации.
Масс-спектр представляет собой спектр линий положительно
заряженных ионов (ширина пиков на половине высоты в процес-
86
/—источникэлектронов; 2— ускоритель (пластины и щель); 3 — переменное магнитное поле;
4 — ввод образца; 5 — ионный источник для ионизации электронным ударом; 6 — патрубок для
подключения масс-спектрометра к вакуумному насосу; 7— ионный коллектор; 8 — щель; 9—
усилитель; 10 — масс-спектр; 11 — интенсивность ионного тока; а, Ь, с — пучки ионов с разными
та/е> тЬ/е, тс/е СООТВетСТВеННО
се записи остается постоянной). Этим масс-спектрометрия зна-
чительно отличается от других спектрометрических методов, для
которых характерно перекрывание полос. Несмотря на то что ре-
альной связи между масс-спектрометрией и оптической спектро-
метрией не существует, оба метода называют спектрометричес-
кими из-за формального сходства графических изображений
спектров.
Минимальным для анализа является количество вещества менее
20 мкг, предел чувствительности составляет 10~9—10-12 г.
Схема масс-спектрометра приведена на рис. 20. Исследуемый
образец через ввод 4 подают в ионный источник 5, где происходит
его ионизация под действием источника электронов 7. Образован-
ные ионы через ускоритель (щель 2) попадают в магнитное поле 3,
где разделяются на пучки ионов а, Ь, с с разными т/е (mje, ть/е, тс/ё).
Причем пучок ионов b фокусируется на щель <?и попадает в ионный
коллектор 7. Сигнал усиливается через усилитель 9 и фиксируется в
виде масс-спектра 10, где по оси абсцисс записывают отношение
т/е, а по оси ординат — интенсивность ионного тока 11.
Поскольку в масс-спектрометре используют поток газообразных
ионов, дающих четкие траектории вследствие комбинаций элект-
ростатического и магнитного полей, то очень важно, чтобы все узлы
прибора — от источника ионов до детектора — были вакуумирова-
ны. Остаточное давление не должно обычно превышать 10-3 Па
(около 10~5 мм рт. ст.). Для этого масс-спектрометр с помощью
патрубка (^подключают к вакуумному насосу.
Метод масс-спектрометрии применяют в научно-исследова-
тельской практике для идентификации соединений и установления
строения неизвестных веществ, точного определения молекуляр-
87
ной массы, определения элементного состава, анализа следовых ко-
личеств биологически активных соединений, определения амино-
кислотной последовательности пептидов, анализа многокомпо-
нентных смесей и т. п.
Контрольные вопросы и задания. 1. Сформулируйте сущность спектральных ме-
тодов исследования и дайте их классификацию. 2. Что представляет собой фотомет-
рический метод определения? Какие приборы используют для реализации этого
метода? 3. На чем основано построение градуировочного графика? 4. Что представ-
ляет собой ИК-спектрометрического метода? Какие приборы используют для реа-
лизации метода? 5. На чем основан флуориметрический метод определения? Какие
приборы используют в аналитических работах? 6. Как можно использовать флуори-
метрический метод для анализа молока и молочных продуктов? 7. Что представляет
собой ААС? Как можно использовать метод для анализа пищевых продуктов? 8. Что
представляет собой АЭС? Какие приборы применяют для реализации этого метода?
9. Какова сущность спектроскопии ЯМР- и ЭПР-резонансов? 10. Каковы возмож-
ности использования спектроскопии ЯМР и ЭПР-резонансов? 11. Каковы сущ-
ность и возможности метода масс-спектрометрии?
Глава 3
ТУРБИДИМЕТРИЯ И НЕФЕЛОМЕТРИЯ
Турбидиметрический и нефелометрический оптические методы
исследования дисперсных систем растворов связаны с рассеянием
света частицами дисперсной фазы, которое зависит от длины волны
излучения, размера и формы рассеивающих частиц и иногда от их
расположения в пространстве.
Почти все измерения связаны с видимым излучением. Пробы
освещают потоком излучения интенсивностью 10 (рис. 21), а затем
измеряют интенсивность прошедшего излучения /или определяют
интенсивность излучения, рассеянного под определенным углом
(например, 90°, 1Р 90). С ростом числа частиц в суспензии отноше-
ние 1/10 уменьшается, а отношение I„ %/Iq увеличивается до уме-
ренных концентраций. Для очень разбавленных суспензий измере-
ние под углом гораздо чувствительнее, чем измерения, когда источ-
ник и детектор находятся на одной линии, поскольку при этом
можно наблюдать слабый рассеянный свет на темном фоне. Метод,
в котором используют линейное измерение, называют турбидимет-
рией, а метод с измерением под углом 90° (или каким-либо дру-
гим) — нефелометрией.
Турбидиметрия основана на измерении интенсивности светово-
го потока, прошедшего через дисперсную систему I. Если принять
рассеянный свет за поглощенный, то можно получить соотноше-
ние, аналогичное соотношению по закону Бугера—Ламберта—Бера
для поглощения света растворами,
D = \gI0/I=tl = klC,
где D — оптическая плотность раствора; t — коэффициент мутности; 7 — толщина
слоя; к — эмпирическая константа; С — концентрация.
Так как поглощения света в данном случае практически не про-
исходит, используют понятие оптической плотности D, которая мо-
жет быть измерена на фотоэлектроколориметре. Коэффициент
мутности в данном уравнении аналогичен коэффициенту в законе
Бугера — Ламберта — Бера.
Нефелометрия основана на измерении интенсивности света,
рассеянного дисперсной системой /р. Способность частиц к рассея-
нию или отражению света определяется размером частиц и длиной
89
Рис. 21. Схема рассеяния света:
/0 — интенсивность падающего излучения; / — ин-
тенсивность прошедшего излучения; ^45’
^90 ’^р135 —интенсивности излучения, рассеян-
ного под разными углами
волны падающего света. Интенсивность светового потока, рассеи-
ваемого дисперсными частицами, определяется уравнением Рэлея:
7р = 7о[Д#И2/А47г2)(1 + cos 6)],
где F — функция от показателей преломления; N— общее число частиц; V— объем
частиц; X — длина волны падающего света; R — расстояние от детектора; 0 —угол
рассеяния.
Закономерность нарушается, если размеры частиц приближают-
ся к длине волны падающего света.
Если необходимо определить только размер частиц и их концен-
трацию, то достаточно измерить интенсивность рассеянного света
под одним углом. В этом случае уравнение Рэлея можно предста-
вить в виде
/Р = IOkCV.
В нефелометрическом методе градуировочный график может
быть построен в координатах /р — С. Более высокая чувствитель-
ность метода по сравнению с чувствительностью турбидиметричес-
кого метода объясняется прямым измерением аналитического сиг-
нала, что позволяет определять не только концентрацию и размер
частиц, но и их форму, характер взаимодействия и другие свойства.
В соответствии с уравнением Рэлея, коэффициент мутности в
турбидиметрическом анализе, можно выразить как:
t=kfNV2/k\
где к! — эмпирическая константа,
ИЛИ
D = k'NV2l/rf.
Таким образом, если взять отношение оптических плотностей
для двух дисперсных систем с одинаковым размером частиц, оно
будет равно отношению концентраций, а при одной и той же кон-
центрации отношение оптических плотностей пропорционально
размерам частиц. Размер частиц в турбидиметрическом анализе не
90
имеет такого значения, как в нефелометрическом. Однако, если
дисперсная система содержит частицы более 0,1 А, нарушается за-
кон Рэлея, что приводит к отклонению градуировочного графика от
линейности. Воспроизводимость результатов при определении ве-
ществ турбидиметрическим методом составляет 5 %.
Метод, используемый для измерения изменения мутности во
времени, получил название турбидиметрического кинетического.
Он позволяет расширить возможности обычного метода. При кине-
тическом методе для определения компонента измеряют скорость
реакции (dx/dr), которая в начальный момент протекания ее опи-
сывается уравнением
dx/dT = ^([5]ox)[C]o,
где К —константа скорости реакции; [Cj0, [5] о — начальные концентрации ана-
лизируемого вещества и реагента; х — концентрация промежуточного вещества или
продукта реакции, по которому определяют скорость реакции.
Обычно измеряют скорость реакции в начальный момент, когда
концентрация х образующегося продукта мала, что нивелирует про-
текание обратной реакции; когда минимальны побочные реакции,
а концентрация анализируемого компонента Со и реагента Во за-
метно не меняются. Следовательно, в этих условиях реакция проте-
кает как реакция псевдонулевого порядка. При х«С0 уравнение
принимает вид
бх/бт = Л5]0[С]0.
Начальную скорость реакции обычно определяют, применяя
метод фиксированной концентрации (метод варьирования време-
ни). Он заключается в измерении времени (т или Дт), необходимого
для достижения определенного изменения состава (Ах). Интегри-
руя уравнение, получим:
1/Дт = ЛС]0[5]0/Дх.
Значение 1/Дт пропорционально концентрации анализируемого
компонента [С]о при постоянных к, [5]0 и Дх ([5]0 <<[Qo, До-
предельно мало). Преобразуя это уравнение, получим:
Дх = ^T[C]q[5]q Дт.
Измеряя поглощение раствора, связанное пропорциональной
зависимостью с концентрацией, определяют содержание анализи-
руемого компонента.
Для проведения измерений в турбидиметрии и нефелометрии
используют следующие приборы.
91
Конструкции приборов для нефелометрических и флуориметри-
ческих измерений идентичны, поэтому любой флуориметр можно
использовать в качестве нефелометра. Поскольку длина волны при
рассеянии не меняется, необходимость во втором монохроматоре
или светофильтре отпадает, но если они имеются в приборе, то их
следует настроить на длину волны падающего света. Многие серий-
ные флуориметры снабжены специальными приспособлениями
для нефелометрических измерений.
Для турбидиметрических измерений можно использовать любой
фотометр или спектрофотометр. Если растворитель и рассеиваю-
щие частицы бесцветны, максимальная чувствительность достига-
ется при использовании излучения голубой или ближней ультрафи-
олетовой области. Для окрашенных систем оптимальную длину
волны лучше всего подобрать экспериментально.
Турбидиметрический метод анализа применяют в определении
состава сырья и оценке технологических процессов. Этим методом
можно измерить содержание жира в молоке, оценить степень дис-
персности жира, исследовать процесс свертывания молока, опреде-
лить готовность сгустка и сливок к сбиванию, установить концент-
рацию микроорганизмов при производстве бактериальных концен-
тратов и т. п.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЖИРА В МОЛОКЕ
ТУРБИДИМЕТРИЧЕСКИМ МЕТОДОМ
Турбидиметрический метод определения массовой доли жира
основан на использовании явления рассеивания светового потока
жировыми шариками молока. В основе математического описания
метода лежит зависимость оптической плотности молока D от мас-
совой доли жира Сж.
D = Асж,
где К— коэффициент.
В свою очередь коэффициент А" зависит от ряда факторов:
где / — толщина кюветы; / — температура пробы; — функции, характери-
зующие влияние на эффект рассеяния света размера жировых шариков и мицелл
казеина.
Для получения однозначной зависимости между оптической
плотностью и массовой долей жира в пробе молока необходимо ее
подготовить — стабилизировать размер жировых шариков (гомоге-
низировать пробу), исключить влияние размера белковых мицелл
путем добавления специального щелочного раствора, обеспечить
постоянство температуры пробы.
92
Массовую долю жира определяют, измеряя интенсивность свето-
вого потока, прошедшего через слой гомогенизированной смеси мо-
лока и растворителя белков. Световой поток принимается фотопри-
емником, электрический сигнал которого усиливается и преобразу-
ется, результаты измерений (сж, %) отражаются на цифровом табло
измерительного прибора. Световой поток имеет длину волны 600 нм.
Данная методика реализована в анализаторах для измерения
массовой доли жира «Милко-Тестер» фирмы Foss Electric (Дания) и
в отечественном приборе — цифровом жиромере ЦЖМ-1, создан-
ном НПО «Агроприбор» при участии ВНИМИ.
Выполнение работы. Подготовленная для испытания, нагретая до
40+2 °C проба молока поступает в смеситель, где смешивается с ра-
створителем. Затем смесь гомогенизируют, подают в фотометри-
ческую кювету и фотометрируют. Отсчет массовой доли жира ведут
по шкале прибора.
Отсчет показаний проводят по шкале или цифровому индикато-
ру с дискретностью отсчета не более 0,01 % массовой доли жира.
Массовую долю жира измеряют дважды в одной и той же пробе
молока. Если показания различаются не более чем на 0,05 %, то за
окончательный результат принимают среднее арифметическое двух
измерений.
Приготовление растворителя. Навески — 45 г трилона Б и 7,6 г
гидроксида натрия взвешивают с точностью до 0,1 г.
Навески (или 45 г трилона Б и 7,6 г гидроксида натрия из потре-
бительской тары) полностью переносят в колбу, растворяют в 3 дм3
дистиллированной воды, прокипяченной в течение 15 мин и ох-
лажденной до температуры 20±2 °C. Раствор в колбе тщательно пе-
ремешивают до полного растворения реактивов и выливают в бу-
тыль вместимостью 10 дм.
Объем раствора в бутыли доводят до 10 дм3 охлажденной до тем-
пературы 20±2 °C и прокипяченной в течение 15 мин дистиллиро-
ванной водой. Универсальной индикаторной бумагой проверяют
pH раствора, которая должна быть в пределах 9,5—10,0. ЕслирН ра-
створа не входит в указанные пределы, то во время приготовления
раствора допущена ошибка и следует приготовить новый раствор.
Раствор используют не ранее чем через 24 ч после приготовле-
ния. Раствор хранят в темном месте в плотно закрытой бутыли не
более 4 недель при температуре не выше 25 °C.
ОПРЕДЕЛЕНИЕ СТЕПЕНИ ДИСПЕРСНОСТИ ЖИРА
В ГОМОГЕНИЗИРОВАННОМ МОЛОКЕ И СЛИВКАХ
(ПО В. ВАЙТКУСУИЭ. КАЗЛАУСКАЙТЕ)
Метод предназначен для характеристики изменений степени
дисперсности жировой фазы в молоке с массовой долей жира от 2
до 6 % и сливках, предварительно нормализованных по жиру до
2—6 %, в технологическом процессе изготовления которых при-
93
меняются диспергирующие устройства. Сущность метода заклю-
чается в измерении оптической плотности (мутности) образца
при длинах волн — 400 и 1000 нм. Оптическую плотность изме-
ряют на предварительно подготовленном образце. Величина от-
ношения оптических плотностей при двух длинах волн (Аоо/
Diooo) характеризует изменение степени дисперсности жировой
фазы образца.
Оптическую плотность (мутность) исследуемого образца изме-
ряют на стандартном спектрофотометре СФ-4 или СФ-4А, или СФ-
26, обеспечивающем измерение оптических плотностей в диапазо-
не волн 300—1100 нм с расстоянием от образца до фоторегистриру-
ющего устройства 11,5 см. Для измерения используют прямоуголь-
ную стандартную кювету длиной 10 мм.
Выполнение работы. Если исследуют гомогенизированные
сливки, то их предварительно разбавляют обезжиренным молоком
или дистиллированной водой до массовой доли жира 2—6 %.
Отбирают 0,5 см3 пробы, разводят в соотношении Е^ООдистил-
лированной водой в мерных колбах вместимостью 250 см3 с внесе-
нием 0,5 см31 М раствора гидроксида натрия. Приготавливают две
параллельные пробы. После тщательного перемешивания выдер-
живают в течение 10 мин и отбирают для измерения 2 см3 получен-
ной смеси, вносят в прямоугольную кювету длиной 10 мм и поме-
щают в рабочий отсек измерительного прибора (СФ-4, СФ-4А,
СФ-26). Оптическую плотность образца измеряют при длинах
волн 400 и 1000 нм. Повторность измерений каждой пробы не ме-
нее двух. Отклонение между параллельными определениями оп-
тической плотности не должно превышать 0,01 по показаниям
прибора.
Определяют показатель степени дисперсности жировой фазы
образца по соотношению оптических плотностей Дюо/Дооо-
Средний диаметр жировых шариков молока рассчитывают по
формуле
dcp = 2,82 — 2,581g D^qq/ Dl000,
где dcp — средний диаметр жировых шариков, мкм; Аоо и Аооо — оптические плот-
ности образца при длинах волн 400 и 1000 нм.
Об эффективности диспергирующего воздействия судят по соот-
ношению оптических плотностей (Доо/Аооо);
Аоо/Лооо 6—4
dcp, мкм
Глубина дисперги-
рующего воздей-
ствия (по степени
дисперсности жи-
ровой фазы)
Не более 1,2
Отличная
4-2,1. 2-1
1,2-2,0 2,1—3,2 и более
Хорошая Слабая
94
Приготовление реактива. 4 г гидроксида натрия взвешивают и
вносят в мерную колбу вместимостью 100 см3, наливают 90 см3 дис-
тиллированной воды, перемешивают до полного растворения ве-
щества и заполняют дистиллированной водой до метки.
ИССЛЕДОВАНИЕ ПРОЦЕССА СВЕРТЫВАНИЯ МОЛОКА
И ОПРЕДЕЛЕНИЕ ГОТОВНОСТИ СГУСТКА
(ПО К. БУТКУСУ И Р. БУТКУСУ)
Метод основан на измерении светопропускания молока во вре-
мя его свертывания. Исследование проводят с помощью прибора
ОСС-76 (рис. 22).
Прибор состоит из измерителя и датчика. Датчик служит для оп-
ределения изменения прозрачности молока во время его свертыва-
ния. Измеритель содержит отдельные узлы и блоки: блок питания,
выдающий необходимое напряжение для нормального функциони-
рования измерителя; модулятор (триггер) — для управления свето-
вым источником (лампочкой, предназначенной для просвечивания
молока); полосовой усилитель, принимающий от датчика сигнал,
усиливающий его до необходимого уровня и преобразующий в по-
стоянное напряжение; детектор, уменьшающий воздействие слу-
чайных помех; эммитерный повторитель, передающий сигнал на
выходные клеммы и систему сигнализации.
Перед началом работы датчик подключается к измерителю. Из-
меритель включается в сеть. К измерителю присоединяется само-
пишущий прибор.
Для термостатирования проб используют ультратермостат. Его
заполняют дистиллированной водой, задают рабочую температуру
Рис. 22. Функциональная схема прибора ОСС-76
95
34 °C, включают в сеть. Вода в ультратермостате подогревается, пе-
ремешивается, и при достижении заданной температуры подогрев
автоматически отключается.
Выполнение работы. Для проведения сравнительной оценки ис-
следуемого молока по его свертываемости сычужным ферментом
при помощи прибора ОСС-76 следует сначала получить конт-
рольную кривую свертывания. Для этой цели 1,5 дм3 молока темпе-
ратурой 34 °C вносят в стакан и помещают в термостат. Во время
опыта температуру молока поддерживают постоянной. В молоко
опускают датчик на 20—30 мин. После этого в молоко вносят 5,6 см3
1%-ного водного раствора сычужного фермента активностью
100000 у. е. Включают систему сигнализации, молоко перемешива-
ется в течение 1 мин (при этом датчик оставляют в исследуемом мо-
локе). Включают самопишущий прибор.
При образовании сгустка, пригодного для разрезки, на приборе
загорается индикаторная лампочка. Самопишущий прибор вычер-
чивает кривую процесса сычужного свертывания (рис. 23).
Как видно из рис. 23, процесс сычужного свертывания подчиня-
ется определенной закономерности и имеет следующие характер-
ные фазы:
первая — происходит подготовка молока к свертыванию, ин-
тенсивность светопропускания практически не изменяется (отре-
зок ОА);
вторая — интенсивная коагуляция. Интенсивность светопро-
пускания быстро уменьшается (отрезок АВ);
третья — уплотнение и образование плотного сгустка. Уменьша-
ется интенсивность светопропускания, но медленнее, чем в преды-
дущей фазе (отрезок ВС).
Рис. 23. Кривая сычужного свертывания
молока
96
Продолжительность отдельных фаз процесса свертывания ус-
танавливают следующим образом. Проводятся касательные линии
к полученной кривой. Касательная линия фазы образования плот-
ного сгустка должна проходить через точку С, указывающую мо-
мент готовности сгустка к разрезанию при производстве сыра. Из
точек пересечения касательных строят линии, перпендикулярные
кривой. Полученные на кривой точки (А, В, С) проецируют на го-
ризонтальную ось времени и таким образом устанавливают про-
должительность отдельных стадий образования молочного сгустка
tqa, 'Cab итвс-
Кинетическими характеристиками фаз интенсивной коагуля-
ции и уплотнения сгустка являются углы наклона Zgai и tgCL^, кото-
рые выражают скорость характерных периодов процесса сверты-
вания.
Контрольная кривая сычужного свертывания молока имеет сле-
дующие параметры: продолжительность подготовки молока к свер-
тыванию (тОа) — 11,20 мин, стадии интенсивной коагуляции
(тдв) — 7,90 мин, уплотнения сгустка (твс) — 10,90 мин; угол накло-
на стадии интенсивной коагуляции (<gai) — 2,621, угол уплотнения
сгустка (^а2) — 0,487. Соотношения продолжительностей соответ-
ственно каждой из упомянутых стадий (тОА, тАВ и твс) с общим вре-
менем свертывания (Ет) 30 мин составляют 37,33; 26,33 и 36,33 %.
Для получения кривых сычужного свертывания исследуемого
молока, а также изучения отдельных факторов, влияющих на про-
цесс образования сгустка, порядок работы с прибором ОСС-76 и
проведение опыта остаются такими же, как и при получении конт-
рольной кривой свертывания.
Методика является способом получения объективных данных в
процессе свертывания молока и изучения свойств образующегося
сгустка.
Приготовление реактива. 1 г эталона сычужного фермента (ак-
тивностью 100000у.е.) растворяют в 100см3 дистиллированной
воды, подогретой до 30 °C. Раствор годен для исследований в тече-
ние 24 ч.
ОПРЕДЕЛЕНИЕ ГОТОВНОСТИ СЛИВОК К СБИВАНИЮ
(ПО Р. БУТКУСУ, Д. КАЧЕРАУСКИСУ И А. БУТКЕНЕ)
Метод основан на изменении оптической плотности сливок во
время их физической подготовки к сбиванию.
Светопропускание измеряют с помощью прибора ОСС-76, схе-
ма которого описана в предыдущей работе. Для термостатирования
проб используют ультратермостат.
Перед началом работы датчик прибора ОСС-76 соединяют с из-
мерительным блоком. Прибор включают в сеть, переключатель
«Чувствительность» устанавливают в положение «0», ставят свето-
97
Рис. 24. Кривая, характеризующая процесс
отвердевания молочного жира
фильтр оптической плотности 3 и стрелку измерительного прибора
потенциометром «Установка 0» устанавливают на «О». К прибору
ОСС-76 подсоединяют самопишущий прибор.
Алюминиевый сосуд со сливками объемом 1 дм3 опускают в воду
температурой 1—3 °C. Сливки непрерывно перемешивают для дос-
тижения температуры эксперимента. Затем сосуд помещают в ульт-
ратермостат, вводят датчик прибора ОСС-76, стрелку измери-
тельного прибора потенциометром «Установка 0» устанавливают на
«О», включают самопишущий прибор.
Определяемая прибором ОСС-76 динамика физического созре-
вания сливок в изотермических условиях в зоне температур массо-
вой кристаллизации молочного жира представлена на рис. 24.
По характеру кривой относительного светопропускания при
заданной постоянной температуре можно получить информацию
об отвердевании глицеридов в жировых шариках во время обра-
ботки сливок по заданному одноступенчатому режиму. На кри-
вой отмечаются три характерные фазы, фиксируемые прибором
ОСС-76:
первая (ОА) — кратковременное условное постоянство светоп-
ропускания сливок. Обусловлено, вероятно, пониженной тепло-
проводностью молочного жира и возникновением центров крис-
таллизации в жировых шариках;
вторая (АВ) — интенсивное (на 35 %) снижение светопропуска-
ния сливок — означает интенсивное отвердевание глицеридов;
третья (ВС) — значительное замедление изменения светопро-
пускания сливок, означающее приближение относительного фазо-
вого равновесия системы.
Продолжительность отдельных фаз процесса отвердевания гли-
церидов сливок (тОА, tab, твс) определяется следующим образом.
Чертятся касательные линии к кривой, затем из точек пересечения
касательных проводят линии, перпендикулярные кривой. Точки,
98
полученные на кривой А, В и С, проецируют на горизонтальную
ось, и таким образом получают отрезки, соответствующие про-
должительности отдельных фаз рассматриваемого процесса (тод,
тдв, твс)-
Для характеристики фазы интенсивного отвердевания (отрезок
АВ) можно рассчитать tga, который в определенной степени связан
со скоростью протекания процесса в данный период времени.
Контрольные вопросы. 1. Что представляет собой турбидиметрический метод
определения? 2. Как определяют массовую долю жира в молоке турбидиметричес-
ким методом? 3. На чем основан нефелометрический метод определения?
Глава 4
РЕФРАКТОМЕТРИЯ И ПОЛЯРИМЕТРИЯ
Рефрактометрический и поляриметрический оптические мето-
ды широко используют в практике анализа пищевых продуктов.
При прохождении через поверхность раздела двух сред световой
луч отклоняется от первоначального направления, т. е. преломляет-
ся. Величина угла отклонения зависит от концентрации и темпера-
туры среды. Угол падения и преломления связан соотношением,
которое называется показателем преломления. Рефрактометрия ос-
нована на измерении показателя преломления.
Некоторые вещества обладают оптической активностью, т. е.
они способны вращать плоскость поляризованного луча. Метод по-
ляриметрии основан на определении угла вращения поляризован-
ного луча.
РЕФРАКТОМЕТРИЯ
Если монохроматический луч А проходит через поверхность раз-
дела двух сред, то одна часть света А' отражается от поверхности раз-
дела, а другая часть В проходит через вторую среду, изменяя при
этом направление (рис. 25). Эту часть монохроматического света
называют преломленным светом. Преломление луча света описы-
вается законом Снелля:
/?1 • sin а = л2 • sin Р,
где а — угол падения; |3 — угол преломления; П[, л2 — показатель преломления 1-й и
2-й сред.
Метод рефрактометрии основан на определении показателя
преломления (рефракции). Показатель преломления зависит от
температуры и концентрации раствора, а также от длины волны
проходящего света.
Каждое вещество в смеси сохраняет преломляющую способ-
ность, и показатель преломления смеси соответствует сумме пока-
зателей преломления всех входящих в смесь компонентов.
При прохождении луча света из одной среды в другую, например
из воздуха в стекло, он направлен по прямой, когда падает перпенди-
100
кулярно на поверхность раздела двух сред. Если луч света падает под
некоторым углом, он преломляется и отношение синуса угла паде-
ния (угол между направлением падающего луча и перпендикуляром к
поверхности раздела сред) к синусу угла преломления (угол между
направлением преломленного луча к перпендикуляру поверхности
раздела) является постоянной величиной и выражается как показа-
тель преломления, в данном случае стекла по отношению к воздуху.
Если луч света А направлен под углом а из среды с меньшим по-
казателем преломления в среду с большим показателем преломле-
ния, то, изменив направление, он приближается к перпендикуляру
РР1 и угол преломления р будет меньше угла падения а. Если луч В
переходит из среды более плотной в среду менее плотную, то, пре-
ломляясь, он удаляется от перпендикуляра и занимает положение
луча А. Если при переходе из менее плотной среды в более плотную
падающий луч С образует с перпендикуляром угол а', приближаю-
щийся к 90°, то соответствующий ему луч преломления D будет да-
вать с перпендикуляром угол Р', лежащий в меньшей угловой облас-
ти. Так как угол падения не может быть больше 90°, то соответству-
ющий ему преломленный луч Л является пограничным лучом рас-
пространения света в этой среде.
Луч света Е, падающий под углом, больше предельного, не пре-
ломляется, а полностью отражается; он претерпевает «полное внут-
реннее отражение» от границы раздела, приобретая направление
ОЕ\. С правой стороны от луча D будет темнота, а с левой — свет.
При переходе к полному отражению резко возрастает яркость све-
та и это дает возможность установить направление предельного угла.
Устройство рефрактометров основано на явлении полного внут-
реннего отражения на границе раздела двух сред, из которых одна
является более плотной. Рефрактометры имеют две призмы — изме-
рительную и осветительную. Измерительная призма изготовлена из
оптического стекла с высоким известным показателем преломле-
Рис. 25. Схема преломления лучей света
101
ния. Одна из ее граней служит границей раздела, где происходит
преломление и полное внутреннее отражение. Рефрактометры раз-
личаются величиной показателя преломления измерительных
призм, поэтому величина измеряемых показателей преломления в
них находится только в определенных пределах.
Применяемые рефрактометры показывают не угол полного
внутреннего отражения, а непосредственно показатель преломле-
ния — процент сухого вещества (по сахарозе) или по условной шка-
ле — число рефракции. В зависимости от исследуемых веществ и ус-
ловий, для которых составлены таблицы, измерения выполняют
при температурах 17,5; 20 и 40 °C. Постоянную температуру поддер-
живают с помощью ультратермостата.
При рефрактометрировании в качестве источника света использу-
ют натриевое пламя, естественный дневной свет или электролампы
(75—100 В). При естественном освещении и свете электролампы
вследствие рассеивания лучей света граница светотени получается ра-
дужная, расплывчатая. Это явление устраняют с помощью компенса-
тора дисперсии (призмы Амичи), устанавливаемого перед объективом
зрительной трубы. Тогда при температуре 20 °Сполучают показатель
преломления п^, соответствующий линии D натриевого пламени.
Для определения составных частей молока и других продуктов
используют различные рефрактометры ИРФ-454, ИРФ-464 и др.
Стандартным рефрактометром является прибор Аббе (рис. 26).
Рефрактометр ИРФ-454, предназначенный для измерения показа-
теля преломления и средней дисперсии неагрессивных жидкостей и
твердых тел, служит для экспресс-анализа концентрации лактозы в
молоке и молочных продуктах, а
также сахарозы в пищевых про-
дуктах, массовой доли сухих ве-
ществ в сгущенном молоке с саха-
ром. Все измерения проводят в бе-
лом свете. Показатель преломле-
ния прозрачных сред определяют
в проходящем свете, а полупроз-
рачных — в отраженном. Сред-
нюю дисперсность вещества оп-
Рис. 26. Рефрактометр типа Аббе (универ-
сальный):
I — зеркало; 2—камера измерительной при-
змы; 3 — термометр; 4 — камера осветительной
призмы; 5, 10— винт для перемещения ком-
пенсатора и лупы; 6 — компенсатор; 7—зри-
тельная труба; окуляр; 9 — лупа; 11 — шка-
ла; 12 — алидада; 13 — винт для установления
рефрактометра наклонно
102
ределяют, пересчитывая показания шкалы по показателю преломле-
ния вещества с помощью таблиц, прилагаемых к инструкции прибора.
Кроме того, используют рефрактометры для экспресс-анализа
концентрации определенных составных частей продукта. Так, реф-
рактометр ИРФ-464 предназначен для измерения показателя пре-
ломления рассеивающих жидких сред. Его можно использовать для
определения массовой доли белка в молоке по разности показаний
для молока и сыворотки на шкале «Белок», а также других нежиро-
вых компонентов молока и жидких молочных продуктов. Анализу
подлежит молоко коровье (сырое сборное или от отдельных живот-
ных, пастеризованное, обезжиренное) кислотностью не выше 28°.
Условия эксплуатации рефрактометра: температура 10—35 °C,
влажность — 80 % при температуре 25 С. Диапазон измерения по-
казателя преломления от 1,325 до 1,360.
Принцип действия рефрактометра ИРФ-464 основан на явлении
полного внутреннего отражения при прохождении лучом света гра-
ницы раздела двух сред с разными показателями преломления. Все
измерения проводят в белом свете (дневном или электрическом).
Показатель преломления и значения по шкале «Белок» определяют
в проходящем свете.
Рефрактометр ИРФ-464 (рис. 27,а) состоит из рефрактометри-
ческого блока 4, трубы 5 и стойки 7.
Для нанесения исследуемой жидкости на измерительную при-
зму, оправа осветительной призмы с помощью рукоятки 3 откиды-
Рис. 27. Рефрактометр ИРФ-464:
а — внешний вид: 1 — окуляр; 2 — кольцо; 3 — рукоятка; 4 — рефрактометрический блок; 5—
труба; 6— винт; 7— стойка; 8— термометр в оправе; 6 — оптическая схема: 1 — осветительная
призма; 2 — измерительная призма; 3 — поворотное зеркало; 4—6 — призма прямого зрения; 7,
8— линзы объектива; 9 — шкала; 10—13 — линзы окуляра
103
вается в крайнее левое положение. В корпусе трубы установлены
окуляр, шкала, объектив, призма прямого зрения, диафрагма и по-
воротное зеркало. Окуляр 1 может быть установлен на резкость в
пределах ±5 диоптрий.
Устранение окрашенности границы раздела светотени достига-
ется поворотом кольца 2 по часовой или против часовой стрелки.
Вращая отверткой винт 6, поворачивают зеркало и устанавливают
начало отсчета (по контрольной призме или дистиллированной
воде). Для контроля температуры окружающего воздуха на стойке
закреплен термометр в оправе 8с пределом измерения от 10 до 35 °C
и ценой деления 0,5 °C.
Влияние температуры на показатель преломления исключается
самим принципом рефрактометрического метода: разность показа-
телей преломления двух жидкостей (водных растворов компонен-
тов молока), определяемых при одинаковых условиях, практически
постоянна при изменении температуры от 10 до 35 °C. Вследствие
этого рефрактометрический блок не термостатирован.
Оптическая схема рефрактометра ИРФ-464 (рис. 27, б) состоит
из рефрактометрического блока, представляющего собой освети-
тельную 1 и измерительную 2 призмы; поворотного зеркала 3;
призм прямого зрения 4—6; линз 7, <?объектива; шкалы 9; линз 10—
13 окуляра. Призма прямого зрения предназначена для устранения
окрашенности границы раздела светотени и имеет возможность по-
ворота вокруг своей оси на угол ±45°. Для установления начала отсче-
та зеркало можно повернуть относительно оси вращения на угол ± Г.
Несколько капель исследуемой жидкости помещают между граня-
ми осветительной призмы 1 с матовой гранью и измерительной
призмы 2с полированной гранью.
Так как показатель преломления жидкостей, измеряемых на
рефрактометре, меньше показателя преломления призмы 2, то лучи
всех направлений, преломившись на границе измеряемой жидко-
сти и стекла, переходят в призму 2.
При рассмотрении пучка лучей, выходящих из призмы 2 в зри-
тельную трубу, нижняя часть ее поля зрения будет освещена, а верх-
няя останется темной. В этом положении граница раздела светоте-
ни определится лучом, выходящим из призмы под предельным уг-
лом. Граница светотени, наблюдаемая в окуляр, соответствует пре-
дельному углу преломления измеряемой жидкости.
Установив границу раздела света и тени и устранив ее окрашен-
ность, выполняют отсчет.
ОПРЕДЕЛЕНИЕ МАССОВЫХ ДОЛЕЙ БЕЛКА, ЛАКТОЗЫ И СОМО
В МОЛОКЕ С ИСПОЛЬЗОВАНИЕМ РЕФРАКТОМЕТРА ИРФ-464
Метод основан на измерении показателей преломления молока
и безбелковой молочной сыворотки, полученной из того же образца
молока, разность между которыми прямо пропорциональна массо-
вой доле белка в молоке.
104
Массовую долю белка определяют по разности показателей пре-
ломления света в молоке и безбелковой сыворотке по шкале «Бе-
лок», массовую долю СОМО — по разности показателей преломле-
ния в молоке и дистиллированной воде, массовую долю лактозы —
по разности показателей преломления безбелковой молочной сы-
воротки и дистиллированной воды.
Выполнение работы. Перед началом работы рефрактометр уста-
навливают на лабораторном столе перед окном или матовой элект-
ролампой и проверяют начало отсчета по прилагаемой конт-
рольной призме или дистиллированной воде.
Для проверки по контрольной призме откидывают осветитель-
ную призму, тщательно протирают поверхности измерительной и
контрольной призм. На поверхность контрольной призмы наносят
каплю иммерсионной жидкости и накладывают контрольную при-
зму на измерительную. Иммерсионная жидкость должна равномер-
но распределяться по всей поверхности, но не выступать по краям.
Наблюдая в окуляр, прибор наводят на резкость, устанавливая гра-
ницу светотени. Поворотом кольца устраняют окрашенность гра-
ницы светотени. Отсчет данных ведут по шкале показателей пре-
ломления.
Измерения повторяют пять раз. После этого необходимо снова
наложить и протереть контрольную призму и снять три отсчета по-
казателя преломления. Если результаты отсчетов окажутся в преде-
лах первых пяти измерений, то притирка считается правильной.
Подсчитывают среднеарифметическое этих трех отсчетов.
Если начало отсчета проверяют по дистиллированной воде, то
одну или две капли воды наносят на чистую поверхность измери-
тельной призмы, опускают осветительную призму, снимают пяти-
кратный отсчет по шкале показателя преломления и подсчитывают
среднеарифметическое значение.
Для подготовки пробы наливают в 3 флакона по 5 см3 молока,
добавляют по 6 капель 4%-ного раствора хлорида кальция. Флако-
ны закрывают пробками и содержимое их перемешивают путем пе-
реворачивания флаконов.
Флаконы ставят на водяную баню, заполняя баню водой так,
чтобы ее уровень достигал половины высоты флаконов. Баню зак-
рывают, помещают на электроплитку, доводят воду в бане до кипе-
ния и кипятят не менее 10 мин. Не открывая бани, сливают горячую
воду через отверстия в крышке, наливают в баню холодную воду и
выдерживают в ней не менее 2 мин.
Открывают баню, извлекают флаконы и разрушают белковый
сгусток, энергично встряхивая флаконы. Затем их помещают в цен-
трифугу и центрифугируют не менее 10 мин. Образовавшуюся про-
зрачную сыворотку отбирают пипеткой и наносят на измеритель-
ную призму рефрактометра 1—2 капли. Закрывают измерительную
призму осветительной.
105
Наблюдая в окуляр рефрактометра, специальным корректором
убирают окрашенность границы света и тени. Для улучшения рез-
кости границы измерение проводят через 1 мин после нанесения
безбелковой сыворотки на призму, так как за это время из пробы
удаляется воздух и лучше смачивается поверхность осветительной
призмы.
По шкале «Белок» проводят не менее трех наблюдений. Удаляют
сыворотку с призмы рефрактометра, промывают ее водой и вытира-
ют фильтровальной бумагой.
Помещают на измерительную призму 2 капли исследуемого мо-
лока и проводят по шкале «Белок» не менее пяти наблюдений, так
как резкость границы света и тени у молока хуже, чем у сыворотки.
Вычисляют средние арифметические значения результатов наблю-
дений для сыворотки и молока.
Массовая доля белка в молоке (%)
5м=^-^2,
где Х[ — среднее арифметическое показаний по шкале «Белок» для молока, %; X2 —
среднее арифметическое значение показаний по шкале «Белок» для безбелковой
сыворотки, %.
Предел допустимой погрешности результата измерений состав-
ляет ±0,1 % массовой доли белка.
За окончательный результат измерения принимают среднее
арифметическое значение результатов двух параллельных вычисле-
ний массовой доли белка, округляя результат до второго десятично-
го знака.
Для определения массовой доли лактозы и СО МО в молоке на
измерительную призму рефрактометра наносят 1—2 капли дистил-
лированной воды и снимают показания по шкале «Белок».
Массовая доля лактозы (%)
л=х2~хг,
где Х} — показания по шкале «Белок» для дистиллированной воды, %.
Массовая доля СОМО (%)
СОМО = Х1~Х3.
По шкале «Белок» также можно определить массовую долю казе-
ина и сывороточных белков.
Массовая доля казеина в молоке (%)
K^iXi-X^y 1,1012,
где Хк с — показания по шкале «Белок» для бесказеиновой сыворотки, %.
106
Для получения бесказеиновой сыворотки во флакон вносят
5 см-3 молока, добавляют 10 капель 10%-ного раствора уксусной
кислоты.
Массовую долю сывороточных белков СБ (%) в молоке рассчи-
тывают по формуле
СБ=БМ — К.
Массовую долю азотистых веществ (белок и небелковые азотные
соединения) рассчитывают по формуле
А = БМ-1,0855.
Приготовление реактива (4%-ного раствора хлорида кальция). На-
веску 40,0 г хлорида кальция двухводного помещают в колбу вмес-
тимостью 1000 см3, приливают к ней 500 см3 воды и перемешивают
до полного растворения соли. Содержимое колбы нагревают до
температуры 20±2 °C и доводят водой до метки.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЖИРА
В МОЛОЧНЫХ ПРОДУКТАХ С ИСПОЛЬЗОВАНИЕМ
РЕФРАКТОМЕТРА АББЕ (ПО И. Н. ВЛОДАВЦУ}
Метод основан на определении показателя преломления раство-
ра молочного жира в растворителе с известным, более высоким по-
казателем преломления. Чем больше разница между показателем
преломления молочного жира и растворителя, тем точнее результа-
ты. Показатель преломления жидкого молочного жира находится в
пределах 1,4600—1,4640. В качестве растворителя применяют мало-
летучий и нерастворимый в воде а-монобромнафталин с показате-
лем преломления гГр = 1,6580.
По показателю преломления раствора жира в а-монобромнаф-
талине рассчитывают массовую долю жира Ж (%) по формуле
Ж = 100.4£”^ъ
т П2~П[
где d — плотность молочного жира, г/см3; V— объем взятого а-монобромнафтали-
на, см3; т — навеска масла, г; п — показатель преломления а-монобромнафталина;
Л1 — показатель преломления молочного жира; Л2 — показатель преломления ра-
створа жира в а-монобромнафталине.
На основании этой формулы составлена табл. 15 для определения
массовой доли жира в масле. Рекомендуется периодически проверять
показатель преломления применяемого а-монобромнафталина.
107
15. Зависимость массовой доли жира от показателя преломления раствора жира
в а-монобромнафталине
«2 | Ж, % II "2 | Ж,% \ | «2 | Ж,% | 1 * | Ж, %
1,5285 90,5 1,5340 80,1 1,5400 70,3 1,5455 62,7
1,5290 89,5 1,5345 79,2 1,5405 69,6 1,5460 62,0
1,5295 88,5 1,5350 78,3 1,5410 68,9 1,5465 61,4
1,5300 78,5 1,5355 77,5 1,5415 68,1 1,5470 60,8
1,5305 86,6 1,5360 76,7 1,5420 67,4 1,5475 60,4
1,5310 85,6 1,5870 75,0 1,5425 66,7 1,5480 59,5
1,5315 84,6 1,5375 74,2 1,5430 66,0 1,5485 58,9
1,5320 83,7 1,5380 73,4 1,5435 65,2 1,5490 58,3
1,5325 82,8 1,5385 72,6 1,5440 64,7 1,5495 57,7
1,5330 81,9 1,5390 71,9 1,5445 64,0 1,5500 57,1
1,5335 81,0 1,5395 71,1 1,5450 63,3 1,5505 56,5
С помощью рефрактометра Аббе (универсальный) можно делать
отсчет показателя преломления с точностью до 0,0001 и определять
высокие показатели преломления в пределах 1,3—1,7.
Выполнение работы. Масло сливочное шоколадное. Навеску масла
(10 г) помещают в алюминиевый стаканчик и нагревают на плитке
до прекращения выделения паров воды и охлаждают. После взве-
шивания и определения содержания воды в стаканчик добавляют
точно 5 см3 а-монобромнафталина и смесь тщательно перемешива-
ют стеклянной палочкой. 2—3 капли прозрачного раствора перено-
сят на призму рефрактометра и определяют показатель преломле-
ния. Массовую долю жира находят по табл. 15. Этот способ дает ре-
зультаты с точностью до ±0,5 % для сливочного масла и ±1,0 % для
шоколадного.
Кофе и какао со сгущенным молоком. 25 г сгущенного молока и
100 см3 горячей воды тщательно смешивают, охлаждают, прибавля-
ют 25 см3 раствора сульфата меди и через 5—10 мин после переме-
шивания фильтруют с отсасыванием через воронку Бюхнера через
один слой фильтровальной бумаги. Отсасывание продолжают до
получения на фильтре плотной пленки с трещинами. Осадок пере-
носят в фарфоровую ступку и высушивают в сушильном шкафу
при 100 °C, растирая его пестиком до получения сухого порошка
(около 1,5 ч).
Вносят в ступку 5 см3 а-монобромнафталина, растирают с ним
осадок 5—10 мин, переносят в воронку из тонкого бумажного филь-
тра и, прикасаясь к призме рефрактометра кончиком фильтра, на-
носят 2—3 капли раствора, определяют рефракцию. Массовую
долю жира вычисляют по формуле
X=J8,6-i^8^i
’ «2 -1,4620’
где — найденный показатель преломления раствора жира в а-монобромнафта-
лине.
108
Продолжительность определения 3 ч, точность 0,5 %.
Сухое масло. 10 г сухого масла переносят в фарфоровую ступку,
прибавляют 5 см3 а-монобромнафталина, 2—3 мин растирают в
ступке, фильтруют через бумажный фильтр и рефрактометрируют.
Массовую долю жира определяют по таблице или по формуле
Ж = 4б,5-1’65^п.
’ п2 -1,4620
Сухое молоко или сухие сливки. 5 г продукта растирают в фарфоро-
вой ступке с 5 см3 а-монобромнафталина и поступают далее, как
при определении в сухом масле. Массовую долю жира вычисляют
по формуле
\zz _ по 1,6580 ~ «2
Ж «2-1,4620’
Точность определения 1 %.
Приготовление реактива (раствора сульфата меди). 3,9283 г пере-
кристаллизованного сульфата меди CuSO4 • 5Н2О дважды растворя-
ют в мерной колбе на 1000 см3 в воде и доводят до метки водой. По-
лучают раствор сульфата меди, в I см3 которого содержится 1 мг
меди.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ В МОЛОКЕ
С ИСПОЛЬЗОВАНИЕМ РЕФРАКТОМЕТРА РЛ-2
Рефрактометр РЛ-2 (рис. 28) имеет две шкалы: показатель пре-
ломления в пределах 1,33—1,54 и содержание сухих веществ по са-
харозе до 95 %. Рефрактометр на «0» устанавливают при 20 °C.
Выполнение работы. Рефрактометр подготавливают к работе. Ка-
меры призм присоединяют штуцерами к ультратермостату и про-
пускают воду температурой 20 °C в течение 10—15 мин. Для провер-
ки нулевой точки прибора на открытую нижнюю призму наносят
1—2 капли дистиллированной воды; закрывают верхнюю призму.
Луч света направляют зеркалом 13 на осветительную призму, уста-
навливают окуляр рукояткой 10 на резкость видимости по шкале 9
и визирной линии (представляющей собой три штриха). Окуляр пе-
ремещают до совмещения визирной линии с границей светотени.
При правильной установке прибора граница светотени при 20 °C
должна совпадать с нулевым делением шкалы сухих веществ и с де-
лением = 1,333 шкалы показателей преломления. Если совпаде-
ния не происходит, то отвинчивают гайку на корпусе и, вращая
ключом винт внутри корпуса, совмещают границу светотени с нуле-
вым делением шкалы сухих веществ, соответствующим показателю
преломления rip = 1,333. Правильность установки прибора прове-
ряют 2—3 раза.
109
Рис. 28. Рефрактометр РЛ-2:
1 — основание; 2— колонка; 3 — корпус; 4 — диспер-
сионный лимб для устранения спектральной окраски
светотени; 5 —камера измерительной призмы; 6—
шарнир, соединяющий призмы; 7—осветительная
призма; 8— отверстие для регулирования шкалы реф-
рактометра; 9— шкала для отсчета; 10— рукоятка;
11 — окуляр; 12—термометр; 13—отражательное зер-
кало
Призмы насухо вытирают мягкой
тканью. Таблица для перевода коэф-
фициента преломления сыворотки на
массовую долю лактозы в молоке рас-
считана на температуру 17,5 °C, по-
этому в течение 10—15 мин пропуска-
ют воду такой температуры. Затем на
плоскость измерительной призмы на-
носят 2—3 капли сыворотки и опуска-
ют верхнюю призму. Наблюдая в оку-
ляр, вращением рукоятки 70 устанав-
ливают поле зрения на фокус (ясную видимость), устраняют рас-
плывчатость и радужность окраски границы светотени. Передвиже-
нием окуляра добиваются полного совпадения граничной линии с
визирным указателем и отсчитывают показатель преломления сы-
воротки по левой шкале рефрактометра.
Молочную сыворотку готовят, осаждая белки хлоридом кальция.
Измерение проводят 3—5 раз, берут среднее. По табл. 16 находят
массовую долю молочного сахара в молоке.
16. Зависимость массовой доли молочного сахара от коэффициента преломления
молочной сыворотки
Коэффициент преломления при 17,5 "С Массовая доля молочного сахара, % Коэффициент преломления при 17,5 'С Массовая доля молочного сахара, %
1,3406 3,77 1,3420 4,49
1,3407 3,82 1,3421 4,54
1,3408 3,87 1,3422 4,59
1,3409 3,93 1,3423 4,64
1,3410 3,98 1,3424 4,69
1,3411 4,03 1,3425 4,74
1,3412 4,08 1,3426 4,79
1,3413 4,13 1,3427 4,84
1,3414 4,18 1,3428 4,89
1,3415 4,23 1,3429 4,95
1,3416 4,28 2,3430 5,00
1,3417 4,33 1,3431 5,05
1,3418 4,38 1,3432 5,10
1,3419 4,44 1,3433 5,15
НО
Сыворотку на призму следует наносить быстро и тотчас прикры-
вать ее другой призмой во избежание испарения влаги. По оконча-
нии работы призмы очищают мягкой тканью, смоченной в дистил-
лированной воде, и вытирают насухо. Правильные значения массо-
вой доли молочного сахара в молоке рефрактометрическим мето-
дом получают при исследовании свежего молока кислотностью
16—20 °Т. При исследовании молока повышенной кислотности по-
лучают завышенные данные.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ
В СГУЩЕННОМ МОЛОКЕ С САХАРОМ
С ИСПОЛЬЗОВАНИЕМ РЕФРАКТОМЕТРА РЛ-2
Прибор подготавливают к работе так же, как указано в предыду-
щей работе.
Пробу сгущенного молока при установлении готовности варки
отбирают непосредственно из вакуум-аппарата. После быстрого
перемешивания каплю сгущенного молока наносят на призму реф-
рактометра РЛ-2. Показания рефрактометра отсчитывают при тем-
пературе 20 °C по правой шкале, соответствующей проценту сухого
остатка для цельного сгущенного молока. Для определения процен-
тного содержания влаги полученную цифру вычитают из 100.
Если массовую долю воды в продукте измеряют после его охлаж-
дения, то перед рефрактометрированием кристаллическую лактозу
растворяют. Около 30 г хорошо перемешанного сгущенного молока
помещают в широкую короткую пробирку с резиновой пробкой и
вставленным в нее термометром. Пробирку закрывают (термометр
должен быть погружен в сгущенное молоко) и опускают в воду при
температуре 75 °C до нижнего уровня пробки. Когда температура
сгущенного молока поднимется до 70 °C, его выдерживают при
этой температуре в течение 30 мин.
При нагревании пробирку периодически перевертывают, чтобы
перемешать сгущенное молоко. Пробки привязывают ниткой к
пробирке. По истечении 30 мин пробирки со сгущенным молоком
вынимают из горячей воды и помещают в воду температурой 18—
19 °C, где оставляют в покое 3—5 мин до тех пор, пока сгущенное
молоко не достигнет температуры 20 °C. Далее поступают как при
определении готовности варки. Процентное содержание влаги рас-
считывают, вычитая полученную цифру из 100.
Для определения массовой доли влаги в обезжиренном сгущен-
ном молоке с сахаром к полученной величине прибавляют 2,5 %.
ПОЛЯРИМЕТРИЯ
Атомы молекул некоторых веществ способны поляризоваться,
т. е. приобретать дипольный момент в электрическом поле. Поля-
ризация атомов обусловлена смещением в молекуле атомов разного
111
типа, что связано с несимметричным распределением в молекуле
электронной плотности — асимметрические атомы. Вещества, со-
держащие такие атомы, обладают оптической активностью. Они
способны вызвать вращение плоскости поляризации проходящего
через исследуемое вещество света. Метод исследования веществ,
основанный на измерении величины угла вращения плоскости по-
ляризации света при прохождении его через оптически активные
вещества, называется поляриметрией. Величина такого вращения в
растворах зависит от их концентрации, поэтому поляриметрию ши-
роко применяют для измерения концентрации оптически активных
веществ, например сахаров.
Асимметрический углеродный атом в сахарах делает их оптичес-
ки активными, способными вращать плоскость поляризации. Это
свойство является функцией концентрации водных растворов саха-
ра, поэтому, измеряя угол вращения а, можно определить содержа-
ние сахаров.
Характерным показателем каждого оптически активного веще-
ства является его удельное вращение [а]^ — угол вращения плос-
кости поляризации при 20 °C для линии D натриевого пламени
раствором, содержащим 100 г вещества в 100 см3, когда луч в этом
растворе проходит путь, равный 100 мм. Для сахарозы [а]^° равен
+66,5°; моногидрата лактозы +52,5°; безводной лактозы +55,3°.
Концентрация вещества (г в 100 см3 раствора) рассчитывают по
формуле
„ а 100
где а —угол вращения, град; [а)д° —удельное вращение анализируемого вещества
при температуре 20 °C; /—длина поляризационной трубки, дм.
На рис. 29 показана принципиальная схема полутеневого поля-
риметра и указано расположение плоскостей поляризации в полу-
теневых поляризаторах. Луч света поляризуется в полутеневом по-
ляризаторе 3, 4, проходит через трубку с исследуемым раствором 5и
попадает в анализатор с отсчетным устройством 6. Наблюдения
проводят с помощью зрительной трубы 7 и окуляра отсчетного уст-
ройства 8.
Плоскости поляризации двух половин полутеневых поляризато-
ров (рис. 30) Р[ и Рг составляют между собой малый угол 2а. Если
плоскость поляризации анализатора АА перпендикулярна биссект-
рисе 2а, то обе половины /и //поля зрения имеют одинаковую по-
лутеневую освещенность. При малейшем повороте анализатора от-
носительная освещенность полей зрения /и //резко меняется. Из-
мерение сводится к визуальному уравниванию яркостей двух поло-
вин поля зрения прибора и последующему считыванию показаний
по шкале вращений, снабженной нониусом.
112
Рис. 29. Принципиальная схема полуте-
невого поляриметра:
1 — источник света; 2— конденсор; 3,4— по-
лутеневой поляризатор; 5 — трубка с исследу-
емым веществом; 6 — анализатор с отсчетным
устройством; 7—зрительная труба; 8— оку-
ляр отсчетного устройства
Рис. 30. Полутеневые поляризаторы:
а — обе половины поля зрения имеют одина-
ковую полутеневую освещенность; 6, в —
неодинаковая освещенность полей зрения
При работе с поляриметром пользуются однородным желтым
светом натриевого пламени.
Сахариметр (рис. 31, а) отличается от поляриметра тем, что ана-
лизатор в нем установлен неподвижно. Для получения одинаковой
освещенности обеих половин поля зрения применен клиновой
кварцевый компенсатор, действие которого основано на измене-
нии толщины слоя кварца, компенсирующем вращение плоскости
поляризации исследуемых растворов. При работе с сахариметром
пользуются обычным белым светом.
Сахариметр имеет линейную международную сахарную шкалу,
градуированную в градусах °S. Отсчет 100° по шкале получают, когда
в кювете длиной 200 мм поляризуют раствор, содержащий 26 г чис-
той сахарозы при температуре 20 °C. Таким образом, Г междуна-
родной сахарной шкалы соответствует раствору, содержащему в
100 см3 0,26 г сахарозы. Навеска сахарозы массой 26 г и лактозы мас-
сой 33 г называется нормальной.
Г международной шкалы сахариметра соответствует 0,34615°
круговой шкалы поляриметра, а Г шкалы поляриметра соответ-
ствует 2,8889° международной шкалы сахариметра.
Оптическая схема и конструкция сахариметра показаны на
рис. 31,6. Световой поток, идущий от источника света через свето-
фильтр 2 или диафрагму 3 и конденсатор 4, 5, проходит через при-
зму-поляризатор 6, которая преобразует его в поляризованный по-
ток света. Затем поток света проходит через полутеневую пластину
7, разделяющую его на две половины линией раздела. При этом пла-
стина рассчитана и установлена так, что плоскости поляризации
обеих половин светового потока составляют равные углы с плоско-
стью поляризации анализатора 12. Поэтому анализатор пропускает
113
Рис. 31. Универсальный сахариметр СУ-4:
а — внешний вид: 1 — лупа; 2 — измерительная головка; 3 — механизм установки ко-
нуса; 4 — клин; 5 — кюветное отделение; 6— траверса; 7— оправа поляризатора; 8—
поворотная обойма; 9—осветительный узел; 10 — регулировочный винт; Л —винт
заземления; 12— вилка разъема; 13— предохранитель; 14— крышка; 15 — кнопка;
16— ручка резистора; 17— стойка; 18— основание; 19 — рукоятка клинового ком-
пенсатора; 20— зрительная труба; б— оптическая схема: 1 — источник света; 2 — све-
тофильтр; 3—диафрагма; 4, 5— конденсатор; 6 — поляризатор; 7—полутеневая
пластина; 8, 9—защитные стекла; 10— подвижный кварцевый клин; //—непод-
вижный контрклин; /2—анализатор; 13— объектив; 14— окуляр; /5—отражатель-
ная призма; 16— светофильтр; /7—шкала; 18— нониус; /9—лупы
обе половины потока и в поле зрения зрительной трубы, состоящей
из объектива 13и окуляра 14, установленной после анализатора, на-
блюдаются две одинаковой яркости половины поля, разделенные
тонкой линией и называемые полями сравнения.
При установке кюветы с раствором между поляризатором и ана-
лизатором нарушается равенство яркостей полей сравнения, так
как исследуемый раствор поворачивает плоскость поляризации на
угол, пропорциональный концентрации раствора.
Для уравнивания яркостей полей сравнения в сахариметре при-
менен клиновой компенсатор, состоящий из подвижного кварце-
вого клина левого вращения 70и неподвижного контрклина право-
го вращения 11. Перемещением подвижного клина относительно
контрклина устанавливают такую суммарную толщину клиньев по
оптической оси, при которой компенсируется угол поворота плос-
кости поляризации раствора. При этом уравниваются яркости по-
лей сравнения. Одновременно с подвижным клином перемещается
шкала 17.
По нулевому делению нониуса 18 фиксируют значение шкалы,
соответствующее состоянию одинаковой яркости полей сравнения.
Шкалу и нониус наблюдают через лупу 19. Они освещаются элект-
ролампой через отражательную призму 75и светофильтр 16.
Перед началом работы закрывают крышку кюветного отделения
без установки в нем кюветы; уравнивают яркость полей сравнения
вращением рукоятки клинового компенсатора; совмещают нулевое
деление нониуса с нулевым делением шкалы, перемещая нониус
юстировочным ключом. Если нулевой отсчет отличается от нуля не
более чем на одно деление нониуса, нуль считается установленным
правильно.
Поляриметрическую кювету перед наполнением ополаскивают
два раза испытуемым раствором. С одной стороны ее закрывают от-
шлифованным стеклом, навинчивают головку и наполняют приго-
товленным прозрачным раствором температурой 20 °C, приливая
его по стенке кюветы доверху. Образуется выпуклая поверхность
раствора. Надвигают сбоку на трубку стеклышко, удаляя лишнюю
жидкость и следя, чтобы под стеклышком не оставалось пузырьков
воздуха.
Кювету помещают с раствором в кюветное отделение и устанав-
ливают ее, вращая вокруг оси, в такое положение, чтобы линия раз-
дела полей сравнения делила поле зрения на две равные части.
Для проведения измерений необходимо уравнять яркость полей
сравнения, вращая рукоятку клинового компенсатора; считать по-
казания по шкале и нониусу.
Уравнивание яркостей полей сравнения и отсчет по шкале и но-
ниусу повторяют не менее шести раз, после чего определяют сред-
нее арифметическое. Затем рассчитывают содержание сахара (г) в
100 см3 исследуемого раствора, а в зависимости от взятой навески —
содержание сахара в продукте.
115
По окончании работы раствор из кюветы выливают, промывают
ее чистой водопроводной, затем дистиллированной водой, высу-
шивают кювету, проталкивая деревянной палочкой небольшие ко-
мочки фильтровальной бумаги.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ В МОЛОКЕ
(ПО Г. КАЧЕРАУСКЕНЕ)
Определение основано на поляриметрическом определении
массовой доли лактозы в натуральном молоке с применением уни-
версальных сахариметров СУ-3 или СУ-4. Растворы для поляримет-
рических исследований должны быть прозрачными, поэтому метод
включает осаждение белков и жира ацетатом цинка и гексациано-
ферратом (II) калия и постоянную поправку на объем осадка (пер-
вый способ) и без предварительного определения поправки (второй
способ).
Выполнение работы по первому способу. Взвешивают 32,9 г моло-
ка в стакане вместимостью 100 см3. Молоко из стакана количе-
ственно переносят в мерную колбу вместимостью 100 см3. Стакан
смывают несколько раз дистиллированной водой, которую сливают
в мерную колбу. Для осаждения белковых веществ в колбу прилива-
ют по 5 см3 растворов ацетата цинка (реактив 1) и гексацианоферра-
та (II) калия (реактив 2). После прибавления каждого раствора
смесь перемешивают, содержимое колбы доливают до метки водой,
перемешивают и спустя 5—10 мин фильтруют через сухой складча-
тый фильтр в сухую колбу.
Фильтрат поляризуют в поляриметрической кювете длиной
400 мм. Кювету заполняют фильтратом дважды, и каждый раздела-
ют по 3—5 отсчетов по шкале сахариметра. Находят среднее ариф-
метическое результатов показаний сахариметра.
Массовую долю лактозы Л (%) вычисляют по формуле
vug. Р— среднее показание шкалы сахариметра; К—поправка на объем осадка
(К= 0,9787).
Расхождение между параллельными определениями массовой
доли лактозы не должно превышать 0,2 %.
Выполнение работы по второму способу. Две порции по 65,8 г мо-
лока взвешивают в стаканах вместимостью 100 см3. Одну порцию
молока из стакана количественно переносят в мерную колбу вмес-
тимостью 100 см3, другую — в колбу вместимостью 200 см3. Стака-
ны смывают несколько раз дистиллированной водой, которую сли-
вают в мерные колбы. Для осаждения белковых веществ в колбы
приливают по 5 см3 растворов ацетата цинка (реактив 1) и гексаци-
116
аноферрата (II) калия (реактив 2). После прибавления каждого ра-
створа смесь перемешивают, содержимое колб доливают до метки
водой, перемешивают и спустя 5—10 мин фильтруют через сухой
складчатый фильтр в сухие колбы.
Фильтраты поляризуют в поляриметрической кювете длиной
400 мм. Кювету заполняют фильтратами дважды и каждый раз дела-
ют по 3—5 отсчетов по шкале сахариметра, находят среднее ариф-
метическое результатов показаний сахариметра.
Массовую долю лактозы вычисляют по формуле
•М.
где Р = 47’2 — Р\; Р[ — показание шкалы сахариметра в °S при разбавлении молока в
колбе вместимостью 100 см3; Р2 — показание сахариметра в °S при разбавлении мо-
лока в колбе вместимостью 200 см3.
Расхождение между параллельными определениями массовой
доли лактозы не должно превышать 0,2 %.
Приготовление реактивов. Реактив 1 (ацетат цинка). 300 г ацета-
та цинка взвешивают и растворяют в мерной колбе вместимостью
1000 см3.
Реактив 2 (гексацианоферрат (II) калия K4[Fe(CN)e]). 150 г гекса-
цианоферрата (II) калия навешивают и растворяют в мерной колбе
вместимостью 1000 см3.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ
В МОЛОЧНОЙ СЫВОРОТКЕ (ПО Г. КАЧЕРАУСКЕНЕ)
Содержание лактозы определяют после осветления сыворотки
растворами ацетата цинка и гексацианоферрата (II) калия на уни-
версальных сахариметрах СУ-3 или СУ-4.
33 г молочной сыворотки взвешивают с погрешностью ±0,01 г в
стакане вместимостью 100 см3. Сыворотку из стакана переносят в
мерную колбу вместимостью 100 см, смывают стакан несколько
раз водой. С помощью пипеток прибавляют в колбу по 1—3 см3 ра-
створов ацетата цинка (реактив 1) и гексацианоферрата (II) калия
(реактив 2). После прибавления каждого раствора содержимое кол-
бы осторожно перемешивают во избежание образования пузырь-
ков. Доливают колбу до метки водой, тщательно перемешивают и
спустя 5—10 мин фильтруют через сухой складчатый фильтр в су-
хую колбу.
Заполняют кювету длиной 400 мм и измеряют угол вращения ра-
створа на сахариметре. Кювету заполняют раствором дважды и каж-
дый раз делают по 5 отсчетов по шкале сахариметра. Находят сред-
нее арифметическое показаний шкалы сахариметра.
117
Массовую долю лактозы в сыворотке вычисляют по формуле
Используемые реактивы приготавливают так же, как и в преды-
дущей работе.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ
В МОЛОЧНОМ САХАРЕ (ПО Г. КАЧЕРАУСКЕНЕ)
Содержание лактозы определяют после осветления раствора мо-
лочного сахара ацетатом цинка и гексацианоферратом (II) калия на
универсальных сахариметрах СУ-3 или СУ-4.
В химическом стакане вместимостью около 100 см3 взвешивают
16,5 г молочного сахара с точностью до 0,01 г. В стакан прибавляют
около 80 см3 дистиллированной воды, содержимое перемешивают
стеклянной палочкой, доводят до кипения при непрерывном пере-
мешивании и кипятят 1—2 мин. Раствор охлаждают и количествен-
но переносят в мерную колбу вместимостью 200 см3, ополаскивая
стакан дистиллированной водой объемом 10 см3 3—4 раза. С помо-
щью пипеток прибавляют в колбу по 3 см3 раствора ацетата цинка
(реактив 1) и гексацианоферрата (II) калия (реактив 2). После при-
бавления каждого раствора содержимое колбы осторожно переме-
шивают во избежание образования пузырьков, доливают до метки
водой, вновь перемешивают и спустя 5—10 мин фильтруют через
сухой складчатый фильтр в сухую колбу. Фильтрат поляризуют в
поляриметрической трубке длиной 400 мм на универсальном саха-
риметре. Отсчет показаний выполняют не менее трех раз, и из полу-
ченных результатов берут среднее арифметическое значение.
Массовую долю лактозы в продукте вычисляют по формуле
Л=2Р— К,
где А-—для молочного сахара кристаллизата 1,2; сырца 0,5; пищевого — 0,3; рафи-
нированного и фармакопейного — 0,1.
Расхождения между параллельными определениями содержа-
ния лактозы не должны превышать 0,5 %.
Приготовление реактивов. См. предыдущую работу.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ САХАРОЗЫ
В ТВОРОЖНЫХ ИЗДЕЛИЯХ И МОРОЖЕНОМ
Метод основан на разрушении лактозы оксидом кальция и поля-
риметрическом определении сахарозы на универсальном сахари-
метре типа СУ-3 или СУ-4.
118
В стакан вместимостью 100 см3 отвешивают 26 г творожных из-
делий или 30 г мороженого. Навеску растирают стеклянной палоч-
кой с небольшим количеством дистиллированной воды температу-
рой 45±2 °C, количественно переносят в мерную колбу вместимос-
тью 200 см3 для творожных изделий и 100 см3 для мороженого, смы-
вая стакан несколько раз водой. Воду для смыва нужно брать в
количестве, равном приблизительно половине объема колбы.
Колбу с навеской охлаждают до 20±2 ° и прибавляют по 3 см3 для
творожных изделий и по 5 см3 для мороженого растворов ацетата
цинка (реактив 1) и гексацианоферрата (II) калия (реактив 2).
После добавления каждого раствора содержимое колбы осто-
рожно перемешивают во избежание образования пузырьков, дово-
дят водой до метки, снова перемешивают и спустя 10 мин фильтру-
ют через сухой складчатый бумажный фильтр в сухую колбу. 50 см3
фильтрата переносят пипеткой в мерную колбу вместимостью
100 см3, добавляют в колбу 0,3 г оксида кальция и выдерживают
колбу на кипящей водяной бане в течение 4—5 мин, перемешивая.
После этого колбу с раствором быстро охлаждают до температуры
20±2 °C и прибавляют 1—2 см3 концентрированной уксусной кис-
лоты. (Допускается неполное растворение оксида кальция.) Колбу
доливают до метки дистиллированной водой, тщательно переме-
шивают и фильтруют через сухой складчатый бумажный фильтр.
Для мороженого на молочной основе отбирают пипеткой 25 см3
фильтрата и помещают в мерную колбу вместимостью 100 см3. За-
тем добавляют 25 см3 раствора буфера (реактив 4) и 3 см3 раствора
гидроксида натрия (реактив 5) и содержимое перемешивают. Колбу
с раствором ставят на водяную баню или помещают в термостат,
выдерживают 5 мин при температуре 85±2 °C, а затем быстро ох-
лаждают до температуры 20±2 °C. Добавляют 3 см3 концентриро-
ванной уксусной кислоты, колбу доливают до метки водой и содер-
жимое тщательно перемешивают.
Для фруктового мороженого отбирают пипеткой 25 см3 фильт-
рата в мерную колбу вместимостью 100 см3. Затем доливают колбу
до метки водой и содержимое тщательно перемешивают.
Фильтрат поляризуют без светофильтра в поляриметрической
кювете длиной 400 или 200 мм. Кювету заполняют раствором дваж-
ды и каждый раз делают по 3—5 отсчетов по шкале сахариметра.
Среднее арифметическое результатов показаний шкалы сахаримет-
ра находят из 6—10 отсчетов.
Массовую долю сахарозы в творожных изделиях и фруктовом
мороженом вычисляют по формуле
S = 2PK,
где К—для творожных изделий 0,979; для мороженого: молочного — 0,988, сливоч-
ного—0,962, пломбира — 0,919, для молочного с фруктовыми наполнителями —
0,984, фруктового — 0,985.
119
Массовую долю сахарозы 5] (%) в мороженом на молочной осно-
ве (при использовании для разрушения лактозы буферного раство-
ра) вычисляют по формуле
где К\ — коэффициент пересчета, г/100 см3 (для сахариметра с длиной кюветы
400 мм К\ = 13; для сахариметра с длиной кюветы 200 мм Кх = 26); 7,5 — эмпиричес-
кий коэффициент пересчета.
За окончательный результат определения принимают среднее
арифметическое двух параллельных определений.
Расхождение между двумя параллельными определениями не
должно превышать 0,5 %.
Приготовление реактивов. Реактивы 1,2. См. предыдущие работы.
Реактив 3(раствортетрабората натрия). Навеску 19,07 гтетра-
бората натрия помещают в колбу вместимостью 1000 см3, прилива-
ют к ней 500 см3 воды, перемешивают до полного растворения соли
и доводят содержимое колбы водой до метки.
Реактив 4 (буферный раствор). 240 см3 раствора тетрабората на-
трия (реактив 3) и 88 см3 раствора гидроксида натрия (реактив 5)
вносят в колбу вместимостью 1000 см3, добавляют 200 см3 воды и
тщательно перемешивают.
Активная кислотность буферного раствора должна быть
13±О,О5 ед. pH. Если pH раствора не соответствует данному значе-
нию, то его доводят, добавляя тетраборат натрия или гидроксид на-
трия, и устанавливают по потенциометрическому анализатору.
Реактив 5 (1Мраствор гидроксида натрия). 70 см3 насыщенного
раствора гидроксида натрия помещают в мерную колбу на 1000 см3
и добавляют до метки свежепрокипяченную и охлажденную воду.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ САХАРОЗЫ
В ПЛАВЛЕНЫХ СЫРАХ (ПО Г. КАЧЕРАУСКЕНЕ И Ю. МАРГЯЛИТЕ)
Метод основан на осаждении белковых веществ плавленых сы-
ров растворами ацетата цинка и гексацианоферрата (II) калия. Про-
зрачный фильтрат обрабатывают оксидом кальция для разрушения
лактозы и других редуцирующих углеводов, а количество сахарозы в
фильтрате определяют поляриметрическим методом на универ-
сальном сахариметре типа СУ.
Выполнение работы. Для приготовления прозрачного фильтрата
из плавленых сыров с сахаром в стакан вместимостью 100 см3 взве-
шивают 26 г продукта. Навеску растирают стеклянной палочкой с
небольшим количеством дистиллированной воды температурой
60—70 °C и полностью переносят в мерную колбу вместимостью
200 см3, смывая стакан несколько раз водой.
120
Колбу с навеской охлаждают до 20±2 °C и прибавляют 5 см3 аце-
тата цинка (реактив 1), а через 2—3 мин — 5 см3 раствора гексациа-
ноферрата (II) калия (реактив 2). Следует соблюдать последова-
тельность добавления упомянутых растворов.
После добавления каждого раствора содержимое колбы осто-
рожно перемешивают круговым движением во избежание образо-
вания пузырьков водой до метки, снова перемешивают и спустя
10 мин фильтруют через сухой складчатый бумажный фильтр в су-
хую колбу. 50 см3 фильтрата приливают пипеткой в мерную колбу
вместимостью 100 см3, добавляют в колбу 0,8 г оксида кальция и
выдерживают колбу в кипящей водяной бане 4—5 мин, перемеши-
вая содержимое. После этого колбу с раствором быстро охлаждают
до температуры 20±2 °C и приливают 2 см3 концентрированной ук-
сусной кислоты (допускается неполное растворение оксида каль-
ция). Колбу доливают до метки дистиллированной водой, тщатель-
но перемешивают и фильтруют через сухой складчатый бумажный
фильтр.
Кювету сахариметра длиной 400 мм заполняют фильтратом
дважды и каждый раз делают по 3—5 отсчетов. При измерении рам-
ку сахариметра со светофильтратами поворачивают так, чтобы луч
света проходил через отверстие без светофильтра. Вычисляют сред-
нее арифметическое.
Массовую долю сахарозы вычисляют аналогично расчету пока-
зателя для творожных изделий и фруктового мороженого при
К = 0,975. За окончательный результат определения принимают
среднее арифметическое двух параллельных определений. Расхож-
дение между двумя параллельными определениями не должно пре-
вышать 0,5 %.
Приготовление реактивов. Реактив 1 (раствор ацетата цинка).
Взвешивают 60 г ацетата цинка, растворяют в дистиллированной
воде при слабом подогревании в химическом стакане вместимостью
100 см3 и переливаютв мерную колбу вместимостью 200 см3. Стакан
промывают водой. В колбу пипеткой приливают 9,0 см3 концентри-
рованной уксусной кислоты и доливают до метки дистиллирован-
ной водой.
Реактив 2 (раствор гексацианоферрата (II) калия. 30 г гексациа-
ноферрата (II) калия, взвешенного с погрешностью не более
±0,01 г, растворяют в дистиллированной воде при слабом подогре-
вании в химическом стакане вместимостью 100 см3 и переливают в
мерную колбу вместимостью 200 см3. Стакан промывают водой. Ра-
створ в колбе доливают до метки дистиллированной водой.
Контрольные вопросы и задания. 1. На чем основан рефрактометрический метод
анализа? Дайте характеристику используемым приборам. 2. На чем основан поля-
риметрический метод анализа? Дайте характеристику используемым приборам. 3.
Какие составные части продукта можно определить рефрактометрическим и поля-
риметрическим методами?
Глава 5
УЛЬТРАЗВУКОВОЙ МЕТОД ИССЛЕДОВАНИЯ
Ультразвук (УЗ) представляет собой упругие волны с частотой при-
близительно от 15—20кГцдо 109 Гц. Область частот ультразвука от 109
до 1012—1013 Гц принято называть гиперзвуком. Область частот ульт-
развука удобно подразделять на три диапазона: ультразвук низких час-
тот (1,5-IO4—105Гц), ультразвук средних частот (105— 107Гц) и об-
ласть высоких частот ультразвука (107—109 Гц). По физической при-
роде ультразвук не отличается от обычного звука, поэтому частотная
граница между УЗ и звуковыми волнами условна. Однако благодаря
более высоким частотам и, следовательно, малым длинам волн имеют-
ся особенности распространения ультразвука. Ввиду малой длины
волны ультразвука характер его распространения определяется в пер-
вую очередь молекулярной структурой среды, поэтому, измеряя ско-
рость v и коэффициент затухания а, можно судить о молекулярных
свойствах вещества. По данным v и а во многих случаях можно осуще-
ствить контроль за протеканием того или иного процесса (контроль
концентрации смесей газов, состава различных жидкостей и т. п.).
УЗ-ПРИБОРЫ ДЛЯ ОПРЕДЕЛЕНИЯ СОСТАВА И СВОЙСТВ МОЛОКА
В настоящее время разработаны УЗ-приборы для анализа соста-
ва молока. УЗ-метод контроля состава молока основан на измере-
нии изменения скоростей ультразвуковых колебаний в зависимос-
ти от массовых долей жира и сухого обезжиренного молочного ос-
татка — СОМО в молоке, определенных при температурах /] и t2 и
связанных между собой следующими соотношениями:
vM(*i) — vB(^i) °1Сж +
vM(^) = vB(y + а2Сж + 62С0,
где vM> vB — скорость ультразвука в молоке и воде; С*, Со — массовая доля в молоке и
сливках жира и СОМО соответственно; /2 — температуры пробы; аь а2, Ь\, Ь2 —
коэффициенты.
Таким образом, измеряя скорость распространения ультразвука
в одной и той же пробе молока при температурах и /2 и решая сис-
122
тему уравнений относительно Сж и Со получают данные о массовой
доле жира и СОМО. Скорость распространения ультразвука в моло-
ке рекомендуется измерять при = 41 °C и - 65 °C.
Как показали исследования, проведенные во ВНИМИ, измере-
ние скорости ультразвуковых колебаний следует проводить с по-
грешностью не более ±0,005 %, атермостатирование пробы молока
при температурах и t2 с погрешностью ±0,02 °C.
На основе ультразвукового метода в последние годы созданы
также методики измерения не только массовых долей жира и
СОМО, но и белка и плотности молока.
При измерении показателей практически не требуется специ-
альной подготовки пробы (кроме нагрева в приборе), на результаты
измерений не влияют размеры жировых шариков и мицелл белка, а
после анализа проба пригодна для дальнейшего использования.
С участием ВНИМИ на основе ультразвукового метода были со-
зданы несколько поколений ультразвуковых анализаторов: прибор
PAN-3 (СКВ Академии наук Эстонии), анализатор ФМУ-1 (СКВ
средств аналитической техники, Украина) и анализаторы нового
поколения «Лактан 1—4» и «Клевер-1М» (НПП «Сибагроприбор» и
НПП «Инфраспек», Россия).
Анализаторы на основе УЗ-метода компактны и просты в эксп-
луатации.
Анализаторы состава «Клевер-1М» и «Лактан» обеспечивают из-
мерение в молоке и сливках массовой доли жира, СОМО и плотнос-
ти. Принцип действия приборов основан на зависимости скорости
распространения ультразвуковых колебаний в молоке, от его соста-
ва. Объектом исследования может быть помимо цельного восста-
новленное молоко, консервированная проба молока, обезжирен-
ное молоко и сливки разной жирности. Диапазон измерений массо-
вой доли жира составляет от 0 до 20 % (для сливок жирностью выше
15 % перед анализом готовится разведение), СОМО — от 6 до 12 %,
плотности — от 1000 до 1040 кг/м3. В качестве консерванта для проб
молока можно использовать дихромат калия. Не рекомендуется ис-
следовать на УЗ-анализаторах нормализованное молоко, если в
него введены соли (хлорида натрия, кальция и др.).
Анализатор «Клевер-1М», внешний вид которого показан на
рис. 32, размещен в пластмассовом корпусе, состоящем из двух
разъемных частей: верхней 3 и нижней 9. В верхней части располо-
жены все основные детали анализатора, в нижней части находится
источник питания. Нижняя часть также служит подставкой, обес-
печивающей устойчивость анализатора в рабочем положении. Вер-
хняя и нижняя части соедйцены в этом положении направляющи-
ми, которые также служат контактами для электрической связи
анализатора с источником питания.
На передней панели 2 расположены: цифровой индикатор 5, дер-
жатель бдля фиксации пробозаборника 7, кнопка для выбора режима
работы анализатора 4, разъем для подключения аккумулятора 1.
123
Рис. 32. Ультразвуковой анализатор
молока «Клевер-1М»:
1 — разъем для подключения аккумулятора;
2 — передняя панель анализатора; 3 верх-
няя часть анализатора; 4 — кнопка для вы-
бора режима работы анализатора; 5 — циф-
ровой индикатор; 6— держатель; 7—про-
бозаборник; 8— патрубок; 9— нижняя
часть анализатора
В транспортном положе-
нии верхняя и нижняя части
корпуса соединяются при по-
мощи зацепов. В этом положе-
нии анализатор принимает
вид миниатюрного чемодан-
чика и становится защищен-
ным от различного рода вне-
шних воздействий.
Анализатор «Клевер-1М»
работает следующим образом.
В режиме измерения пробу молока заливают в кювету, где ее после-
довательно нагревают до двух заданных температур, при каждой из
которых определяют скорость ультразвука. На основе полученных
данных микроЭВМ автоматически вычисляет массовые доли
жира, СОМО, плотности и температуру молока. Полученные зна-
чения отображаются на цифровом индикаторе прибора.
Прибор прост в обслуживании и портативен. Температура пробы
молока может быть от 10 до 30 °C. Наряду с электропитанием прибо-
ра от однофазной сети переменного тока напряжением 220 В и часто-
той 50 Гц имеется возможность электропитания от автомобильного
аккумулятора напряжением 12—13,2 В. В этом случае прибор может
быть использован для измерений в полевых условиях.
В анализаторе «Лактан 1 —4» после установки стаканчика с пробой
молока в положение «Измерение» включают кнопку «Пуск», и на
узел управления поршневым микронасосом поступает сигнал. Про-
исходит забор пробы молока в измерительную ячейку, в которой в
течение 1,5 мин она нагревается и термостатируется при температуре
41 °C. Затем в течение 5 с при этой температуре измеряют частоту ге-
нератора, которая пропорциональна скорости распространения уль-
тразвука. После этого проба нагревается до температуры 65 °C, вновь
измеряется частота генератора. По значениям найденных частот
микроЭВМ вычисляет массовые доли жира и СОМО, значения ко-
торых отображаются на индикаторах (каждые 5 с). После определе-
ния массовых долей молоко сливается в стаканчик с пробой.
124
В результате модернизации ультразвуковых анализаторов «Лак-
тан 1—4», «Сибагроприбором» разработаны несколько моделей
ультразвукового прибора «Лактан-Супер». С помощью нового при-
бора «Лактан-Супер» можно измерять массовую долю жира, белка,
СОМО, а также плотность молока и маложирных сливок.
Проба молока микронасосом подается в измерительную ячейку
анализатора, где нагревается до заданных температур. Замеряются
параметры ультразвуковых колебаний, прошедших через пробу.
На основе этих данных с помощью встроенной микроЭВМ вычис-
ляют показатели состава молока (массовые доли жира, белка и
СОМО и плотность), которые отображаются на цифровом дисплее
прибора.
Созданы 4 модели анализатора «Лактан-Супер». Анализаторы
моделей 150 и 200 производительностью до 30 проб в час рассчита-
ны для применения на молочных заводах малой и средней мощнос-
ти, в фермерских хозяйствах. Приборы моделей 500 и 550 произво-
дительностью до 200 проб в час можно использовать на крупных
молочных предприятиях, в селекционных центрах, региональных
лабораториях.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЖИРА И СОМО
И ПЛОТНОСТИ МОЛОКА (СЛИВОК)
НА УЗ-АНАЛИЗАТОРЕ «КЛЕВЕР-1 М»
В основу работы анализатора положен принцип измерения ско-
рости распространения ультразвука, которая является функцией
массовой доли жира, СОМО, плотности и температуры молока
(сливок).
Анализатор состоит из системы приема молока (сливок), блока
нагрева и термостатирования, устройства измерения и вычисления
на базе микроЭВМ. В режиме измерения проба последовательно
подогревается до двух заданных температур.
Измеряется скорость ультразвука при каждой температуре. На
основе полученных данных микроЭВМ вычисляет значения массо-
вых долей жира, СОМО и плотности в анализируемой пробе. Ин-
формация о вычисленных значениях параметров последовательно
отображается на цифровом индикаторе анализатора.
Перед началом работы прибор приводят в рабочее положение.
Для этого открывают анализатор, отсоединяют верхнюю разъемную
часть с блоком измерения 5от нижней 9, последнюю переворачива-
ют направляющими вверх и устанавливают горизонтально на рабо-
чем месте. Вставляют пазы верхней части в направляющие нижней
части и, сохраняя вертикальное положение верхней части, до отказа
вдвигают ее по направляющим, направляя усилие на металлические
зацепы на корпусе со стороны передней панели. Фиксируют держа-
тель бпробозаборника 7в удобном для работы положении.
125
Вставляют сетевую вилку источника питания в розетку 220 В
(50 Гц) и с помощью кнопочного выключателя, расположенного в
нижней половине на источнике, включают анализатор. При этом на
индикаторе 5анализатора высвечивается его заводской номер, ана-
лизатор переходит в режим предварительного прогрева.
Проба молока при заливке ее в анализатор должна иметь темпе-
ратуру от 10 до 30 °C для получения требуемой точности измерения.
При наличии отстоявшегося слоя жира пробу молока перед ана-
лизом нагревают в водяной бане до 40—45 °C, перемешивают и ох-
лаждают до температуры 20±5 °C.
Пробы парного молока; молока, вспененного вследствие его ин-
тенсивного перемешивания; обезжиренного молока и сливок после
сепарирования, содержащих много пузырьков газа, необходимо пе-
ред измерением дегазировать, так как в противном случае могут
быть получены ошибочные результаты или сбои анализатора (выс-
вечивание символа «-с-» на индикаторе). Для освобождения пробы
от газа ее нагревают до температуры 45—50 °C и выдерживают при
этой температуре 5 мин. Затем перемешивают и охлаждают молоко
до температуры 25+5 °C, после чего проводят измерения.
При анализе сливок, массовая доля жира в которых более 15 %,
сначала готовят разведение сливок обезжиренным молоком в 5 раз
(1 часть сливок+ 4 части обезжиренного молока). Для этого 20 г
анализируемых сливок смешивают с 80 г обезжиренного молока.
Проводят измерение массовой доли жира в полученном разведении
и обезжиренном молоке на приборе «Клевер-1М».
Массовую долю жира Жс (%) в анализируемых сливках рассчи-
тывают по формуле
Жс = 5>Wp - 4^ГО,
где Жр, Жо — массовая доля жира соответственно в разведении и обезжиренном мо-
локе, %; 5 — разведение пробы в 5 раз; 4 — четыре части обезжиренного молока в
разведении.
В режиме готовности прибора (на индикаторе высвечивается
символ «Г») кнопкой 4устанавливают требуемый номер калибров-
ки и заливают пробу молока (сливок) в пробозаборник.
По истечении 10—15 мин анализатор подает однократный зву-
ковой сигнал, а на индикаторе высветится символ «Г» (режим го-
товности).
В режиме готовности для проверки работоспособности анали-
затора залить в пробозаборник дистиллированную воду темпера-
турой 25+2 °C. Объем заливаемой воды должен быть таким, что-
бы уровень воды в пробозаборнике по окончании заливки нахо-
дился чуть ниже (на 5—10 мм) верхней кромки. Анализатор авто-
матически определяет наличие пробы (воды) в измерительной
ячейке и переходит в режим измерения. Об этом свидетельствует
гашение символа «Г» (готовность) и загорание символа «—» (на-
126
грев пробы в процессе измерения) на индикаторе. Через 2,5—
3,0 мин (в зависимости от температуры воды) анализатор закан-
чивает измерение и подает короткий однократный звуковой сиг-
нал и выводит на индикатор измеренные значения жира и СОМО
в процентах, плотности (приведенной к 20 °C) в градусах арео-
метра. Абсолютные значения по дистиллированной воде должны
быть не более 0,06 % для жира, не более 0,15 % для СОМО и 0,0
для плотности. Если результат незначительно отличается от тре-
буемого, то следует дать анализатору прогреться еще в течение
нескольких минут и повторить измерение. После фиксации ре-
зультатов воду сливают из пробозаборника, при этом через 2—3 с
после слива анализатор автоматически переходит в режим готов-
ности.
В режиме готовности в пробозаборник заливают анализируемую
пробу. Уровень пробы в пробозаборнике по окончании заливки
должен находиться на 5—10 мм ниже верхней кромки. При этом
25 см3 пробы подается в измерительную кювету, анализатор автома-
тически определяет наличие пробы и переходит в режим измере-
ния. На индикаторе появляется знак «—».
Через 2,5—3 мин от начала измерений вычисленные параметры
последовательно отображаются на цифровом индикаторе. Фикси-
руют результаты измерений. Затем пробу из пробозаборника выли-
вают в кювету. После индикации «Г» (готовность) можно произво-
дить новые, измерения.
Для исключения погрешности рекомендуется промывать изме-
рительную камеру молоком «новой» пробы. Для этого в режиме «Г»
заливают молоко в пробозаборник и после появления индикации
«—» выливают пробу в кювету. После этого производят измерения
обычным порядком.
При перерыве между измерениями более 30 мин необходимо
дважды промыть измерительную камеру. Для этого заливают чис-
тую воду в пробозаборник и после установления уровня выливают в
кювету. Затем во время индикации «Г» заливают дистиллирован-
ную воду и оставляют анализатор с водой включенным до следую-
щего измерения.
По окончании работы выключают источник питания и промы-
вают анализатор следующим образом. Опускают пробозаборник в
емкость с моющим раствором («Лотос», «Астра», «Био», жидкое
мыло и т. п.), приготовленным заранее с концентрацией 1:20 и по-
догретым до температуры 50—55 °C. При этом необходимо просле-
дить, чтобы воздух из пробозаборника был вытеснен в молокопро-
вод. В качестве емкости для моющего раствора можно использовать
стакан или банку вместимостью не менее 200 см3.
Подсоединяют шприц, входящий в комплект прибора, в сжатом
положении к патрубку 8 и, прокачивая 5—7 раз шприцем моющий
раствор через измерительную камеру анализатора, промывают при-
бор. Затем таким же образом промывают анализатор дистиллиро-
127
ванной или чистой прокипяченной водой (температура 50—55 °C),
меняя воду на чистую не менее 3 раз.
По окончании мойки пробозаборник устанавливают в рабочее
положение, заливают дистиллированной или отстоянной кипяче-
ной водой комнатной температуры и оставляют в таком положении
до следующего рабочего дня.
Если не предполагается проводить анализы в течение продолжи-
тельного времени (несколько дней), то необходимо перевести ана-
лизатор в транспортное положение. Сетевую вилку источника пи-
тания вынимают из розетки.
Контрольные вопросы. 1. На чем основан УЗ-метод анализа? 2. Какие параметры
измеряют УЗ-методом? 3. Как работает ультразвуковой прибор «Клевер- 1М»?
Глава 6
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Электрохимические методы исследования основаны на исполь-
зовании процессов, происходящих в электролитической ячейке
(гальваническом элементе, цепи) — системе из электродов и элект-
ролитов, контактирующих между собой. При отсутствии электри-
ческого тока (1=0) в замкнутой гальванической цепи на межфаз-
ной границе устанавливается равновесие и потенциал достигает
равновесного значения. Если через ячейку проходит электрический
ток (7> 0), на межфазной границе равновесие не достигается и элек-
троны переходят из электрода в раствор.
В состав электролитической ячейки входят два или три электро-
да, один из которых — индикаторный или рабочий, второй — элек-
трод сравнения и третий — вспомогательный. Электрод, действую-
щий как датчик и реагирующий на фактор возбуждения, но не изме-
няющий состава раствора за время измерения, является индикатор-
ным. Если под действием тока, протекающего через ячейку,
значительно изменяется состав раствора, то электрод — рабочий.
Электрод сравнения служит для создания измерительной цепи и
поддержания постоянного значения потенциала индикаторного
(рабочего) электрода. Используемый в трехэлектродной ячейке
вспомогательный электрод (противоэлектрод) вместе с рабочим
электродом включен в электрическую цепь.
Электрические параметры (сила тока, напряжение, сопротивле-
ние) могут служить аналитическими сигналами, если они измерены
с достаточной точностью. Электрохимические методы анализа ис-
пользуют либо для прямых измерений, основанных на зависимости
«аналитический сигнал — состав», либо для индикации конечной
точки титрования в титриметрии. Электрохимические методы ана-
лиза позволяют определять концентрацию вещества в широком ин-
тервале (1 — 10-9 моль/л) с достаточной точностью и воспроизводи-
мостью.
Электрохимические методы анализа объединяют методы без
протекания электродной реакции, в которых строение двойного
электрического слоя в расчет не принимается (кондуктометрия), и
методы, основанные на электродных реакциях в отсутствие тока
(потенциометрия) или под током (вольтамперометрия, и др.).
129
КОНДУКТОМЕТРИЯ
Кондуктометрический метод основан на определении электри-
ческой проводимости веществ в различных растворах. По величине
электропроводности можно судить о концентрации растворенного
вещества.
Электропроводность раствора электролита — результат диссоци-
ации растворенного вещества и миграции ионов под действием
внешнего источника напряжения. В поле электрического тока дви-
жущиеся в растворе ионы испытывают тормозящее действие со сто-
роны молекул растворителя и окружающих противоположно заря-
женных ионов. Результатом такого тормозящего действия является
сопротивление раствора прохождению электрического тока. Элект-
ропроводность раствора определяется в основном числом, скорос-
тью (подвижностью) мигрирующих ионов, количеством переноси-
мых ими зарядов и зависит от температуры и природы растворителя.
Различают удельную (См/м) и эквивалентную (См-м2/экв) элек-
тропроводности раствора:
у = aCF(z+w+ + Z-W-);
X = (1000/Qy,
где а —степень диссоциации электролита; С — концентрация электролита; F—
число Фарадея; д+, u_, z+, Z- — скорость движения (м/с) и заряд катионов и анионов
соответственно при напряженности электрического поля (В/м).
Электропроводность раствора зависит от его концентрации. По
мере возрастания концентрации растворенного электролита увели-
чивается количество ионов-переносчиков заряда, т. е. растет удель-
ная электропроводность. Однако после достижения определенного
максимального значения удельная электропроводность начинает
уменьшаться, поскольку для сильных электролитов усиливаются
релаксационный и электрофоретический эффекты, а для слабых
электролитов уменьшается степень их диссоциации. Электропро-
водность бесконечно разбавленного раствора определяется под-
вижностью ионов в отсутствие тормозящих эффектов а«, и
С ростом концентрации эквивалентная электропроводность ра-
створа уменьшается.
Большее распространение в аналитической практике получил
метод кондуктометрического титрования, основанный на химичес-
кой реакции, в результате которой заметно изменяется электропро-
водность раствора.
На рис. 33 показаны кривые кондуктометрического титрования
кислот различной силы диссоциации и их смесей раствором силь-
130
Рис. 33. Кривые кондуктометри-
ческого титрования кислот рас-
твором NaOH:
1 — сильнодиссоциируюшая кислота;
2— слабодиссоциирующая кислота;
3— смесь сильно- и слабодиссоции-
рующих кислот
нодиссоциирующего основания.
По излому на кривой можно опре-
делить точку эквивалентности. При
проведении кондуктометрического
титрования для получения резкого
излома на кривых титрования необ-
ходимо учитывать эффект разбавле-
ния. Его можно свести к минимуму,
титруя большой объем разбавлен-
ного раствора концентрированным
раствором. Правильно подбирая
титрант и растворитель, получают
кривую титрования с резким изло-
мом и минимальную погрешность
при определении конечной точки
титрования.
Кондуктометрическое титрова-
ние обладает рядом достоинств: воз-
можно дифференцированное тит-
рование смесей некоторых кислот или оснований, титрование мут-
ных и окрашенных растворов. Нижний предел определяемых кон-
центраций 1О-4 моль/л, погрешность определений 2 %.
Электропроводность раствора измеряют в соответствующей
электролитической ячейке, представляющей собой стеклянный со-
суд с вмонтированными электродами. Конструкция ячейки для
кондуктометрических измерений должна соответствовать интерва-
лу измеряемых сопротивлений, и константа ячейки при этих изме-
рениях должна оставаться постоянной. Константа ячейки А (см-1)
определяется площадью электродов S (см2), расстоянием между
ними L (см) и зависит от формы сосуда и объема раствора А = L/S.
Непосредственно измерить константу ячейки невозможно, ее опре-
деляют, используя стандартные растворы КС1, для которых извест-
ны значения удельной электропроводности при различных темпе-
ратурах. Измерив сопротивление R ячейки, заполненной раствором
КС1, и воспользовавшись табличным значением у из соотношения
A =yR, вычисляют константу ячейки А.
Как правило, электроды, изготовленные из листовой платины,
жестко закреплены, так что расстояние между ними не изменяется.
Расстояние между электродами и их поверхность выбирают в зави-
симости от сопротивления раствора: чем выше измеряемое сопро-
тивление, тем больше должна быть площадь электродов и меньше
расстояние между ними. С учетом этого выбирают ячейку (рис. 34).
Кондуктометрические измерения можно проводить при посто-
янном или переменном токе с использованием мостовых или ком-
пенсационных измерительных схем. Измерения при постоянном
токе на практике проводят редко, поскольку точно зафиксировать
электропроводность в этих условиях нельзя из-за поляризации
131
Рис. 34. Электролитические ячейки для
измерения электропроводности раство-
ров, плохо проводящих (а) и хорошо про-
водящих (б) электрический ток
электродов. Чаще измеряют
электропроводность (сопро-
тивление) растворов с помо-
щью установок и приборов,
принципиальная схема кото-
рых включает мост Уитстона с
источником переменного тока
частотой 500— 5000 Гц.
Поскольку в схему моста
входит источник переменного
тока, балансировка моста ос-
ложнена влиянием индуктив-
ностей и емкостей всей цепи.
Выбирая оптимальные значе-
ния частоты и плотности тока,
концентрации электролита и
конструкции ячейки, можно
избежать ошибки в определе-
нии электропроводности.
Во ВНИМИ (Л. П. Брусиловский, В. П. Шидловская) проведе-
ны исследования по выявлению маститного молока кондуктомет-
рическим методом. Известно, что в маститном молоке увеличивает-
ся содержание ионов хлора и натрия, вследствие чего повышается
электропроводность молока. Результаты исследований показали,
что в маститном молоке содержание ионов хлора увеличивается в
2,2 раза, а ионов натрия в 2,8 раза по сравнению с их содержанием в
молоке здоровых коров. Соответственно удельная электропровод-
ность повышается в среднем на 37 %. Поскольку при заболевании
коров маститом резко увеличивается число соматических клеток, то
исследовали взаимозависимость между электропроводностью мо-
лока и содержанием в нем соматических клеток. По результатам ис-
следований разработана методика контроля определения мастит-
ного молока кондуктометрическим методом с пересчетом удельной
электропроводности на содержание соматических клеток. Кроме
того, во ВНИМИ разработана методика контроля нейтрализующих
веществ (соды, аммиака) в молоке кондуктометрическим методом.
ПОТЕНЦИОМЕТРИЯ
Потенциометрическим методом определяют содержание ве-
ществ в растворе, а также различные физико-химические парамет-
ры, например окислительно-восстановительный потенциал и др.
Главное достоинство потенциометрического метода — быстрота
и простота проведения измерений. Время установления равновес-
ного потенциала индикаторного электрода мало, что удобно для
изучения кинетики реакций и автоматического контроля техноло-
132
гических процессов. Используя микроэлектроды, можно прово-
дить измерения в пробах объемом до десятых долей миллилитра.
Потенциометрическим методом исследуют мутные и окрашен-
ные растворы, вязкие пасты, при этом исключаются операции
фильтрации и перегонки. Анализируемый раствор можно исполь-
зовать в дальнейших работах. Погрешность определения при пря-
мом потенциометрическом измерении составляет 2—10%, при
проведении потенциометрического титрования — 0,5—1 %.
Потенциометрический метод основан на измерении потенциа-
лов электродов, погруженных в исследуемый раствор. Эксперимен-
тально измеряют разность потенциалов (эдс).
В потенциометрии обычно применяют гальванический элемент,
включающий два электрода: индикаторный (потенциал которого
зависит от активности (концентрации) определенных ионов в ра-
створе) и сравнения (потенциал которого не зависит от концентра-
ции определяемых ионов).
Потенциометрия широко применяется для непосредственного
определения активности ионов, находящихся в растворе (иономет-
рия), а также для индикации точки эквивалентности при титрова-
нии по изменению потенциала индикаторного электрода в ходе
титрования (потенциометрическое титрование).
Метод ионометрии заключается в измерении активности раз-
личных ионов (рХ) с помощью ионоселективных электродов.
ИНДИКАТОРНЫЕ ЭЛЕКТРОДЫ
В потенциометрии используют два основных класса индикатор-
ных электродов: электронно- и ионообменные (ионоселективные).
Электроннообменные электроды, на межфазных границах кото-
рых протекают реакции с участием электронов, изготавливают из
инертных металлов (платины, золота). Потенциал, возникающий
на платиновом электроде, зависит от отношения концентраций
окисленной и восстановленной форм одного или нескольких ве-
ществ в растворе.
Ионообменные (ионоселективные) электроды, на межфазных гра-
ницах которых протекают ионообменные реакции, содержат чув-
ствительный элемент — мембрану, разделяющую внутренние ра-
створ и электрод и одновременно служащую средством электроли-
тического контакта с внешним (исследуемым) раствором. Мембра-
на обладает ионообменными свойствами. Независимо от типа
мембраны механизм действия ионоселективных электродов подчи-
нен одним и тем же общим закономерностям, различие заключает-
ся в деталях механизма переноса ионов через границу раздела двух
фаз и внутри мембраны. Если чувствительная мембрана помещена
между двумя растворами, то через нее могут перемещаться ионы
только определенного типа в направлении к раствору с меньшей ак-
133
Рис. 35. Ионоселективные электроды:
а — стеклянный; б—с твердой кристаллической мембраной; в — с жидкой мем-
браной; 1 — стеклянная мембрана; 2 — твердая кристаллическая мембрана; 3 —
внутренний электрод (токоотвод); 4— пористый диск; 5— жидкая мембрана
(ионообменник); 6— полупроницаемая инертная мембрана; 7— водная фаза
тивностью (концентрацией) подвижного иона. На поверхности
мембраны возникает потенциал, при котором прекращается даль-
нейшее перемещение ионов, и в результате устанавливается дина-
мическое равновесие.
В зависимости от типа мембраны электроды бывают стеклян-
ные, с твердой, жидкой, пленочной и другими мембранами
(рис. 35).
Стеклянный электрод (см. рис. 35, а) представляет собой сосуд
из изолирующего стекла с напаенным полым шариком-мембра-
ной 7из специального электродного стекла. В качестве внутренне-
го стандартного раствора в стеклянном электроде используют
0,1 М раствор соляной кислоты обычно с добавкой хлорида натрия
или калия. Внутренним электродом (токоотводом) служит хлорсе-
ребряный электрод, представляющий собой серебряную проволо-
ку, покрытую хлоридом серебра. В качестве электродного исполь-
зуют алюмосиликатное стекло, содержащее ионы натрия или
кальция и часто небольшие количества ионов лантанидов. Оно
способно реагировать с ионами водорода, которые могут входить в
кислородные полости решетки стекла. Между ионами водорода во
внутреннем и внешнем растворах легко устанавливается равнове-
134
сие, в результате чего возникает потенциал на границе раздела мем-
брана-раствор.
Стеклянные электроды применяют, как правило, для измерения
активности водородных ионов (pH). Выпускают также стеклянные
электроды для измерения активности ионов щелочных металлов
(натрия, калия).
Устройство ионоселективного электрода с твердой мембраной
показано на рис. 35, б. Твердая селективная мембрана /расположе-
на в нижней части корпуса электрода. Мембраны изготавливают из
обладающих ионной проводимостью кристаллических материалов
в виде пластинок или дисков. Обычно в процессе переноса заряда
участвует только один из ионов кристаллической решетки, что
обеспечивает высокую избирательность электрода. Перенос заря-
дов происходит за счет дефектов кристаллической решетки в соот-
ветствии с механизмом, при котором вакансии занимаются свобод-
ными соседними ионами. Внутренний раствор представляет собой
0,1 М раствор хлорида калия и 0,1 М раствор соли измеряемого
иона. Внутренним электродом служит хлорсеребряный электрод 3.
Наиболее распространенным из электродов с твердой мембраной
является фторидселективный электрод с мембраной из монокрис-
талла фторида лантана.
Электроды с твердыми мембранами — наиболее доступные и удоб-
ные детекторы обнаружения катионов и анионов в растворе. С помо-
щью электродов с твердой мембраной определяют активность ионов
железа, меди, хлора, кальция, магния, нитратов, нитритов и др.
Устройство электрода с жидкой мембраной показано на рис. 35, в.
Жидкая мембрана 5 представляет собой раствор электродноактив-
ного вещества в органическом растворителе, не смешивающимся с
водой. Раствор электродноактивного вещества и водная фаза /отде-
лены друг от друга полупроницаемой инертной мембраной 6. Диск
4 из пористого гидрофобного материала разделяет внутренний и
внешний (исследуемый) растворы. По своей боковой (цилиндри-
ческой) поверхности диск контактирует с органическим раствори-
телем. Под действием капиллярных сил растворитель заполняет
поры диска, осуществляя электрический контакт между водными
растворами: внутренним и исследуемым. За счет этого устанавлива-
ется равновесие между общими ионами в мембране и растворах.
Жидкие мембраны имеют ряд преимуществ перед твердыми.
Они обладают высокой скоростью обмена, малым временем уста-
новления равновесия; их использование позволяет в широких пре-
делах менять электродноактивные вещества и их концентрацию. В
зависимости от типа электродноактивного вещества различают ка-
тионные, анионные и нейтральные (хелатные) жидкие мембраны.
Конструкция электродов с пленочной мембраной аналогична кон-
струкции электродов с твердой мембраной, только вместо твердой
мембраны в корпус электрода вклеена пленочная (пластифициро-
ванная) мембрана, в которой электродноактивное соединение вве-
135
дено в полимерную матрицу, в качестве последней чаще всего ис-
пользуют поливинилхлорид. Внутри электрода находится раствор
сравнения. В качестве внутреннего электрода (токоотвода) исполь-
зуют хлорсеребряный электрод. Внутренний раствор представляет
собой 0,1 М раствор хлорида калия и 0,1 М раствор соли измеряемо-
го иона (для нитрат-селективного электрода, например, нитрат ка-
лия). Перед работой электроды с пленочной мембраной вымачива-
ют в течение суток в растворе того соединения, которое предполага-
ется определять в исследуемой пробе.
ЭЛЕКТРОДЫ СРАВНЕНИЯ
При измерении ЭДС обратимых гальванических элементов необ-
ходим полуэлемент, потенциал которого был бы известен, постоя-
нен и не зависел бы от состава исследуемого раствора. Электрод,
удовлетворяющий этим требованиям, называют электродом срав-
нения. Электрод сравнения должен быть прост в изготовлении и
сохранять практически постоянный и воспроизводимый потенциал
при прохождении небольших токов. Постоянство потенциала элек-
трода сравнения достигается поддержанием в контактирующем
внутреннем растворе постоянной концентрации веществ, на кото-
рые реагирует электрод. Наиболее распространен хлорсеребряный
электрод сравнения (Ag, AgCl|KCl), который изготавливают путем
нанесения хлорида серебра на серебряную проволоку. Электрод по-
гружают в раствор хлорида калия, который связан солевым мости-
ком с исследуемым раствором. Так как в концентрированных ра-
створах хлорид серебра растворяется с образованием хлорсеребря-
ных комплексов, растворы хлорида калия перед погружением в них
электродов обычно насыщают хлоридом серебра. При работе с
хлорсеребряным электродом необходимо следить за тем, чтобы
внутренний сосуд был заполнен насыщенным раствором К.С1.
Хлорсеребряные электроды выпускает отечественная промыш-
ленность (ЭВЛ- 1МЗ, ЭВЛ-1М1). Кроме хлорсеребряного электрода
в качестве электродов сравнения применяют каломельный и тала-
мидный электроды.
ХАРАКТЕРИСТИКА ИЗМЕРИТЕЛЬНЫХ УСТРОЙСТВ
Устройства, применяемые для измерения потенциала — потен-
циометры (pH-метр, иономер), состоят из двух блоков: измеритель-
ного (высокоомный преобразователь) и датчика (электродная сис-
тема), и могут отличаться друг от друга конструкцией как измери-
тельного блока, так и датчика.
Электродная система может быть выполнена конструктивно в
виде двух отдельных электродов (индикаторного и электрода срав-
136
нения) или в виде комбинированного электрода, где оба электрода
объединены в одном корпусе. Комбинированные электроды назы-
вают комбинированными ионоселективными датчиками (КИД).
ВНИМИ разработаны pH-метры и иономеры для исследования
молока и молочных продуктов нового поколения: рН-метр-милли-
вольтметр рН-150М; иономер лабораторный И-160, рН-метры-ионо-
меры «Экотест-120» и анализатор качества сред КС МК «Луч» и др.
рН-метр-милливолътметр pH-150 М (рис. 36) предназначен для
использования в лабораториях предприятий. В приборе применена
электродная система со стеклянным измерительным электродом и
хлорсеребряным электродом сравнения, выполненная в виде ком-
бинированного электрода. Комбинированный электрод / погружа-
ют в стаканчик с исследуемой пробой. Визуальный отсчет значений
pH проводят по цифровому индикатору 9 высокоомного преобразо-
вателя 12 с дискретностью pH 0,01. Питание прибора автономное от
встроенных батарей или аккумуляторов. Прибор может также рабо-
тать от сети напряжением 220 В и частотой 50 Гц через блок сетевого
питания, входящий в комплект прибора. Блок сетевого питания
подключается к прибору через разъем 10. Электродная система под-
ключается через гнездо 11.
При работе на рН-150М необходимо учитывать следующие об-
щие правила.
Перед погружением в буферный или контролируемый растворы
электроды промывают дистиллированной водой, остатки воды с
электрода удаляют фильтровальной бумагой.
При измерении pH показания считают после установления зна-
чения. Время установления (обычно не более 3 мин) зависит от бу-
ферной емкости раствора. В некоторых растворах время установле-
ния показаний может достигать 10 мин. Перед работой прибор на-
страивают по буферным растворам. Для приготовления буферных
растворов применяют дистиллированную воду, прокипяченную в
течение 30—40 мин для удаления растворенного диоксида углерода.
Значения pH стандартных буферных растворов приведены в
приложении 1.
Буферные растворы, полученные из фиксаналов, хранят при
температуре 20+1 °C не более 2 мес.
Рекомендуется следующий порядок настройки pH-метра по
двум буферным растворам с температурой, близкой к температуре
анализируемой среды. Кнопкой 6 задают единицы измерения pH,
вращая резистор Зна боковой стенке, устанавливают его в среднее
положение. Погружают электроды в первый буферный раствор (pH
раствора 6,88 при 20 °C), вращением оси резистора ^устанавливают
на индикаторе 6,88. Затем промывают электрод дистиллированной
водой, просушивают фильтровальной бумагой и погружают во вто-
рой буферный раствор (pH раствора 9,22 при 20 °C) вращением оси
резистора 4 на боковой стенке преобразователя устанавливают на
индикаторе значение pH буферного раствора.
137
Рис. 36. рН-метр-милливольтметр pH-150 М:
1 — комбинированный электрод; 2—стакан с исследуемой пробой; 3 — резистор для установле-
ния значения pH; -/—резистор для регулирования крутизны системы; 5— кнопка включения
питания; 6— кнопка переключения режимов измерения; 7— резистор установки температуры
раствора при ручной термокомпенсации; <?— резистор настройки по буферному раствору; 9—
цифровой индикатор; 10 — разъем подключения сетевого питания; 11 — гнездо для подключе-
ния электродной системы; 12— высокоомный преобразователь
Для проведения измерений электроды промывают дистиллиро-
ванной водой и погружают в измеряемый раствор. Отсчет показа-
ний проводят по индикатору 9.
Чтобы определить pH нескольких продуктов с близкими зна-
чениями pH, после каждого измерения электроды ополаскивают
водой и погружают в другую пробу. В тех случаях, когда опреде-
ляют pH продуктов с разными значениями pH, например молока
и кисломолочных продуктов, электроды тщательно промывают
дистиллированной водой и затем проводят исследование другой
среды.
После окончания цикла измерений электроды промывают теп-
лой водой, вытирают и погружают в дистиллированную или питье-
вую слегка подкисленную воду.
При измерении pH жирных продуктов на поверхности стеклян-
ного электрода остается жир. Поэтому стеклянный электрод обра-
батывают органическими растворителями в следующей последова-
тельности: этиловый спирт — диэтиловый спирт (или четыреххло-
ристый углерод) — этиловый спирт. Затем электрод тщательно про-
мывают дистиллированной водой.
По окончании работы с прибором электроды для измерения pH
должны оставаться погруженными в воду или в 0,1 н. раствор соля-
ной кислоты.
Для сопоставления pH и титруемой кислотности молока и мо-
лочных продуктов ВНИМИ определены усредненные соотноше-
ния (см. приложение 2).
Иономер лабораторный И-160 предназначен для измерения ак-
тивности одновалентных и двухвалентных анионов и катионов
(рХ), в том числе ионов водорода, окислительно-восстановитель-
ных потенциалов и температуры в водных растворах проб пищевых
продуктов и различных технологических растворов.
Работа прибора основана на преобразовании эдс электродной
системы и других источников эдс в пропорциональное по величине
напряжение, преобразуемое в дальнейшем в цифровой код и анало-
говый сигнал. Математические преобразования и другие функции
выполняются микропроцессором — Основным компонентом элек-
тронной схемы преобразователя. Конструктивно преобразователь
(рис. 37, а) включает трансформатор, измерительную плату с эле-
ментами измерительной схемы и плату индикации с установлен-
ным на ней цифровым дисплеем 1. Последний расположен на пере-
дней панели прибора, на которой размещены также органы выбора
режима измерения 2 и управления 3. На задней панели (рис. 37, б)
находятся разъемы для подключения измерительного 1 и вспомога-
тельного 3 электродов, персонального компьютера 4 и других ис-
полнительных устройств. Электродная система состоит из электро-
да сравнения (хлоридсеребряный) и измерительного ионселектив-
ного электрода.
139
Рис. 37. Иономер лабораторный И-160:
а — внешний вид: 7 —цифровой дисплей; 2— органы выбора режима измерения; 3 —
органы управления; (J —задняя панель: 1 — разъем для подключения измерительного
электрода; 2 — регулятор контрастности дисплея; 3 — гнездо для подключения вспомога-
тельного электрода; 4 — разъем для подключения персонального компьютера; 5 — разъем
для подключения исполнительных устройств; 6— разъем для подключения термокомпен-
сатора; 7 — шнур для подключения к сети; S— зажим заземления; 9 — выключатель сете-
вого питания
рН-Метр-иономер-«Экотест-120» (рис. 38, а) состоит из высо-
коомного преобразователя с микропроцессорным устройством 2,
датчика температуры 1 и комбинированного ионоселективного
электрода-датчика 3, предназначен для измерения активности
ионов водорода (pH), активности (рХ), концентрации различных
ионов и температуры. Прибор рассчитан на измерение 27 ионов.
Прибор создан НПП «Эконике», испытан ВНИМИ и рекомендо-
ван для контроля качества молока. Для использования в молочной
промышленности прибор комплектуется ионоселективными дат-
чиками для измерения pH, pNa, pNH4, рС1, рСа и датчиками для
измерения температуры.
Анализатор качества сред КС МК «Луч » (рис. 38, б) создан меж-
дународной компанией «Луч» и ВНИМИ. В состав прибора входят:
высокоомный преобразователь с микропроцессорным устройством
2, датчик температуры 1 и комбинированный ионоселективный
электрод 3. В комплект прибора входит набор комбинированных
ионоселективных электродов — датчиков (КИД) для измерения
pH, pNa, pNH4, рС1 и рСа, размещаемые в кейсе. Конструкция ком-
бинированных датчиков КИД унифицирована и отличается типом
чувствительного элемента. Диапазоны измерения прибора pH
(рХ) — от 1 до 14, температуры — от 0 до 100 °C. Общая продолжи-
Рис. 38. рН-метры-иоиомеры:
а — «Экотест-120»; б—анализатор качества сред КС МК «Луч»; 7 —датчик температуры; 2—
высокоомный преобразователь с микропроцессорным устройством; 3 — комбинированный
ионоселективный электрод-датчик (КИД)
141
тельность измерения 6 показателей молока — не более 20 мин. Ана-
лизатор КС МК «Луч» испытан ВНИМИ и Всероссийским НИИ
метрологии им. Д. И. Менделеева (Санкт-Петербург) и рекомендо-
ван для включения в Государственный реестр.
Сотрудниками ВНИМИ (Л. П. Брусиловский и др.) разработаны
методики ионометрического контроля активности ионов водорода,
натрия, аммония, хлора с помощью анализатора КС МК «Луч», а
также проведены исследования метода ионометрии для контроля
маститного молока (табл. 17) по концентрации ионов хлора (с пере-
счетом на соматические клетки), фальсификации молока гидро-
ксидом аммония и содой (гидрокарбонатом натрия) по концентра-
ции ионов аммония и натрия.
17. Пределы измерения рО, концентрации ионов хлора
и соответствующее им число соматических клеток в молоке
рС1 Са, мг/100 см3 Число соматических клеток в 1 см3 молока, тыс.
1,65-1,49 80-115 Не более 500
1,48-1,40 118-140 Более 500 до 1000
1,39-1,33 145-165 Более 1000 до 1500
1,33-1,00 Более 165 Более 1500
Фальсификацию молока гидроксидом аммония NH4OH опреде-
ляют по формуле
Очн4он = (Ci — С2)-1,94,
где CNH40H — концентрация гидроксида аммония в исследуемом молоке, мг/
100 см3; С, — концентрация ионов NH4 + в фальсифицированном (исследуемом)
молоке, мг/100см3; Сг — концентрация ионов NH4+ в нефальсифицированном
молоке, мг/100 см3.
Ниже приведены значения концентрации гидроксида аммония в
молоке и качественная характеристика степени фальсификации.
NH4OH, мг/100 см3
Более 2 до 5
Более 5 до 9
Более 9 до 18
Степень фальсификации молока
Слабая
Средняя
Сильная
Фальсификацию молока гидрокарбонатом натрия (NaHCO3)
определяют по формуле
СиаНСОз =(C(-Ci).3,65,
где Сцансоз — концентрация гидрокарбоната натрия в исследуемом молоке, мг/
100 см3; С{ —концентрация ионов Na+ в исследуемом молоке, мг/100см3; С2 —
концентрация ионов Na+ в нефальсифицированном молоке, мг/100 см3.
142
Ниже приведены значения концентрации гидрокарбоната на-
трия и характеристика степени фальсификации молока.
NaHCO3, мг/100 см3
Более 18 до 44
Более 44 до 88
Более 88 до 165
Степень фальсификации молока
Слабая
Средняя
Сильная
В С.-Петербургской Государственной академии холода и пище-
вых технологий К. К. Горбатовой и другими разработан метод конт-
роля термоустойчивости молока по содержанию ионов кальция,
количество которых в молоке измеряют ионометрическим методом
с использованием ионоселективного электрода. Существующий
метод определения термоустойчивости молока по тепловой пробе
длительный и трудоемкий. Разработанный метод дает возможность
проведения экспресс-анализа термоустойчивости. Разработана
шкала оценки термоустойчивости молока по содержанию ионов
кальция.
Качество молока
Хорошее (высокая термоустой-
чивость)
Удовлетворительное (средняя
термоустойчивость)
Плохое (нетермоустойчивое)
Концентрация ионов кальция, мг%
Менее 9,5
9,5-10,5
Более 10,5
ВНИМИ рекомендовано использовать ионометрический метод
для определения содержания хлорида натрия в сырах с помощью
ионоселективного электрода рС1.
ОПРЕДЕЛЕНИЕ ВОДОРОДНОГО ПОКАЗАТЕЛЯ (PH) МОЛОКА
И МОЛОЧНЫХ ПРОДУКТОВ
Метод основан на определении концентрации свободных
ионов водорода с помощью потенциометрических анализаторов
(рН-метров).
Сначала прибор проверяют по буферному раствору: включают за
30 мин до начала проверки, настраивают по буферному раствору pH
6,88 и 4,00 при температуре (20+1) °C. Перед проверкой прибора по
буферному раствору электроды необходимо тщательно промыть
дистиллированной водой, а остатки воды с электродов удалить
фильтровальной бумагой.
В стеклянный стакан вместимостью 50—100 см3 наливают
40±5 см3 буферного раствора температурой 20± 1 °C, после чего по-
гружают в него электроды и через 10—15 с снимают показания при-
бора. Если показания прибора отличаются от стандартного значе-
ния pH образцового буферного раствора более чем на 0,05, то при-
143
бор настраивают с помощью регулятора. По стандартному буфер-
ному раствору прибор необходимо проверять ежедневно.
В стакан вместимостью 50—100 см3 наливают 40+5 см3 молока
температурой 20+2 °C и погружают электроды прибора. Электроды
не должны касаться стенок и дна стакана. Через 10—15 с снимают
показания по шкале прибора. Для быстрого установления показа-
ний прибора измерения проводят при круговом перемещении ста-
канчика с молоком. Показания по прибору отсчитывают через 3—
5 с после установления стрелки. После каждого измерения электро-
ды датчика промывают дистиллированной водой. При массовых из-
мерениях pH молока остатки предыдущей пробы удаляют с
электродов следующей пробой, а электроды промывают через 3—5
измерений. В промежутках между измерениями электроды датчика
погружают в стакан с дистиллированной водой.
За окончательный результат принимают среднее арифметичес-
кое результатов двух параллельных измерений pH, расхождение
между которыми не должно превышать 0,03.
При измерении pH молочных продуктов используют определен-
ные объемы (или навески) продукта. Объем пробы молока и жидких
кисломолочных продуктов около 40 см3. Пробу заливают в стакан-
чик на 50 см3 и вносят электроды. Для измерения pH творога навес-
ку 60 г в пергаментной бумаге растирают до однородной консистен-
ции и затем вносят электроды в пробу. Для измерения pH сыра про-
бу измельчают на металлической терке, 20 г растертого образца сме-
шивают с 20 см3 дистиллированной воды в фарфоровой ступке.
Смесь переносят в химический стакан и в полученную пробу вносят
электроды.
ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ ИОНОВ КАЛЬЦИЯ
ДЛЯ ХАРАКТЕРИСТИКИ ТЕРМОУСТОЙЧИВОСТИ МОЛОКА
(ПО К. К. ГОРБАТОВОЙ И П. И. ГУНЬКОВОЙ)
Метод основан на измерении ионизированного кальция ионо-
метрическим методом с использованием электродной системы, со-
стоящей из ионоселективного рСа и хлоридсеребряного электрода
сравнения, на приборах типа pH-121, pH-340, рН-150М, «Экотест-
110» и др. Термоустойчивость молока определяют по таблице по из-
вестному содержанию ионизированного кальция в молоке, мг%.
Перед исследованием молока электродную систему градуируют
по двум стандартным растворам хлорида кальция 4 и 40 мг% с до-
бавлением в них инертных солей до ионной силы 0,07 (КС1 и NaCl в
соотношении 2:1). Концентрацию растворов хлорида кальция про-
веряют трилонометрическим титрованием с мурексидом. По разно-
сти эдс (Е4о — Е4) в растворах 40 мг% и 4 мг% определяют крутизну
электродной системы — она должна составлять 26—30 мВ. Если
крутизна меньше 26 мВ, то кальцийселективный электрод следует
144
заменить. Температура исследуемого молока должна быть приведе-
на к температуре градуировочных растворов с точностью ±0,5 °C.
После градуировки электроды погружают в исследуемое молоко
и снимают показания со шкалы прибора. Продолжительность уста-
новления показаний — 2 мин.
Концентрацию ионов кальция в молоке Смол (мг%) определяют
по формуле
= (^мол — ^)/(^40 ~ ± 0,6,
где £мол — эдс электродной системы в молоке, мВ; £4 — эдс электродной системы в
растворе концентрацией 4 мг%, мВ; £40 — эдс электродной системы в растворе кон-
центрацией 40 мг%, мВ; 0,6 — десятичный логарифм 4.
Например, при 1g Смол = 0,978 величина Смол = 9,5 мг%.
Относительная погрешность определения концентрации ион-
ного кальция в молоке составляет ±7 %. После измерений электро-
ды ополаскивают дистиллированной водой и погружают в раствор
хлорида кальция (4 мг%).
Для характеристики термоустойчивости молока пользуются ра-
нее приведенными данными.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Титрометрия — это количественный аналитический метод, зак-
лючающийся в определении концентрации определенного веще-
ства путем постепенного добавления к анализируемому раствору
реактива известной концентрации до полного завершения реакции
с определяемым веществом. Для контроля за ходом титрования и
установлением конечной точки титрования можно использовать
ионоселективные электроды, если последние чувствительны к оп-
ределяемому иону или иону титранта.
Титрант — это применяемый для титрования раствор реактива,
вступающего в реакцию с определяемым ионом или соединением.
Титрант добавляют к титруемому раствору постепенно порциями
определенного объема из точно градуированной бюретки. Концен-
трацию определяемого вещества устанавливают по объему титран-
та, необходимому для завершения реакции.
Результаты измерений методом потенциометрического титрова-
ния более точны, чем при использовании прямой потенциометрии,
так как в этом случае вблизи точки эквивалентности небольшому
изменению концентрации соответствует большое изменение по-
тенциала индикаторного электрода. В ходе титрования измеряют и
записывают эдс ячейки после добавления каждой порции титранта.
В начале титрант добавляют небольшими порциями, при прибли-
жении к конечной точке (резкое изменение потенциала при добав-
лении небольшой порции реактива) порции уменьшают. Для опре-
145
Рис. 39. Кривые потенциометрического титро-
вания:
а — зависимость потенциала электрода от объема тит-
ранта; б — дифференциальная кривая потенциометри-
ческого титрования
деления конечной точки потенцио-
метрического титрования (точки эк-
вивалентности) можно использовать
различные способы. Наиболее про-
стой способ — построение кривой
титрования: графика зависимости по-
тенциала электрода от объема титран-
та (рис. 39, а).
Другой способ состоит в расчете из-
менения потенциала (ДЕ) на единицу
изменения объема титранта, то есть
ДЕ/Д/ (рис. 39, б). Кривая титрова-
ния, построенная с использованием
этого параметра, зависящего от объема титранта, имеет четко выра-
женный максимум в конечной точке.
Ионоселективные электроды удобны для фиксирования конеч-
ной точки титрования, поскольку их показания не зависят от степе-
ни окрашенности и прозрачности раствора.
Схема установки для потенциометрического титрования приве-
дена на рис. 40. На магнитной мешалке 1 установлена ячейка с ана-
лизируемым раствором 2. В анализируемый раствор помещены ин-
дикаторный электрод 3 и хлоридсеребряный электрод сравнения 4.
Электродная система подключена к потенциометру. Титрование
осуществляется из бюретки 5, соединенной с блоком ручного (БРТ)
или автоматического (БА Т) титрования. Блок автоматического тит-
рования включает систему бюреток с электромагнитными клапана-
ми для контроля потока титранта или шприц. Плунжер последнего
приводится в рабочее состояние электродвигателем, соединенным
с микрометром. Во избежание перетитрования в обоих случаях ско-
рость титрования должна быть небольшой. Можно применять при-
способление, уменьшающее скорость добавления титранта по мере
приближения к точке эквивалентности. Хорошо зарекомендовали
себя в работе автоматические титраторы отечественного производ-
ства типа БАТ-15.
Метод потенциометрического титрования применяют для контро-
ля титруемой кислотности и массовой доли белка в молоке. Измерение
титруемой кислотности основано на нейтрализации кислот, содержа-
146
К потенцио-
Рис. 40. Схема установки для по-
тенциометрического титрования:
1 — магнитная мешалка; 2 — ячейка
для анализируемого раствора; 3— ин-
дикаторный электрод; 4— насыщен-
ный хлорсеребряный электрод срав-
нения; 5 — бюретка
Рис. 41. Титриметрический анализатор
с ручной бюреткой АТП-2:
1 — pH-метр или иономер; 2— электромагнитная
мешалка с установленной на ней ячейкой для
анализируемого раствора; 3 — блок ручного тит-
рования
щихся в продукте, раствором гидроксида натрия до pH 8,9, измерение
массовой доли белка — на формольном титровании до pH 9.
Разработаны титриметрические анализаторы двух модификаций:
АТП-1 — с полуавтоматической и АТП-2 — с ручной бюреткой.
В состав титриметрического полуавтоматического анализатора
АТП-1 входят pH-метр рХ-150, блок автоматического титрования
БАТ-15,2, магнитная мешалка, электромагнитный клапан и бюрет-
ка стеклянная модифицированная ОС-10.
Стакан с пробой продукта устанавливают на магнитную мешал-
ку, опускают в него электроды pH-метра, включают блок автомати-
ческого титрования, на задатчике которого установлено значение
конечной точки титрования. По достижении смесью pH, соответ-
ствующего конечной точке титрования, автоматически отключает-
ся клапан и процесс титрования завершается. По объему раствора
гидроксида натрия, пошедшего на титрование, определяют титруе-
мую кислотность.
В состав титриметрического анализатора с ручной бюреткой
АТП-2 (рис. 41) входят pH-метр или иономер, электромагнитная
147
мешалка и бюретка ОС-10. Титрование проводят вручную, визуаль-
но контролируя pH по цифровому индикатору измерительного
прибора. По достижении конечной точки титрования подавать гид-
роксид натрия прекращают.
ОПРЕДЕЛЕНИЕ ТИТРУЕМОЙ КИСЛОТНОСТИ МОЛОКА
И МОЛОЧНЫХ ПРОДУКТОВ С ИСПОЛЬЗОВАНИЕМ
ПОТЕНЦИОМЕТРИЧЕСКОГО АНАЛИЗАТОРА
Метод основан на нейтрализации кислот, содержащихся в про-
дукте, раствором гидроксида натрия до заданного значения pH 8,9.
Для выполнения работы используют блок автоматического титро-
вания и индикации точки эквивалентности при помощи потенцио-
метрического анализатора с диапазоном измерения pH 4—10 и це-
ной деления шкалы 0,05 ед. pH. Блок автоматического титрования
должен быть аппаратурно совместим с потенциометрическим тит-
ратором и иметь дозатор раствора (бюретку) вместимостью не ме-
нее 5 см3 с ценой деления не более 0,05 см3.
Подготовка прибора. Блок автоматического титрования подклю-
чают к анализатору в соответствии с инструкцией, прилагаемой к
блоку (см. рис. 41). Затем блок и анализатор подключают к сети и
прогревают их в течение 10 мин.
Дозатор блока автоматического титрования заполняют раство-
ром гидроксида натрия.
Блок автоматического титрования настраивают на точку эквива-
лентности и устанавливают на блоке значение pH 4,0, начиная с ко-
торого гидроксид натрия подают по каплям. Устанавливают время
выдержки после окончания титрования, равное 30 с.
Подготовка проб. Молоко, молоко с наполнителями (шоколад-
ное, кофейное), сливки, простокваша, ацидофильное молоко, ке-
фир, кумыс и другие кисломолочные продукты анализируют без
специальной подготовки. В стакан вместимостью 50 см3 отмерива-
ют 20 см3 дистиллированной воды и 10 см3 анализируемого продук-
та. Смесь тщательно перемешивают.
При анализе сливок и кисломолочных продуктов переносят ос-
татки продукта из пипетки в стакан путем промывания пипетки по-
лученной смесью 3—4 раза.
Для мороженого и сметаны в стакане взвешивают 5 г продукта.
Тщательно перемешивают продукт стеклянной палочкой, посте-
пенно добавляют к нему 30 см3 воды и перемешивают, а затем про-
водят измерения.
Для творога и творожных изделий в фарфоровую ступку вносят
5 г продукта. Тщательно перемешивают и растирают продукт пести-
ком. Затем количественно переносят продукт в стакан вместимос-
тью 100 см3, смывая его небольшими порциями воды, нагретой до
35—40 °C. Общий объем воды равен 50 см3. Затем смесь перемеши-
вают и проводят измерения.
148
Выполнение работы. В стакан с пробой продукта помещают стер-
жень магнитной мешалки и устанавливают стакан на магнитную
мешалку. Включают двигатель мешалки и погружают электроды
потенциометрического анализатора и сливную трубку дозатора
блока автоматического титрования в стакан с продуктом. Включают
кнопку «Пуск» блока автоматического титрования, а спустя 2—
3 с — кнопку «Выдержка». Раствор гидроксида натрия при этом на-
чинает поступать из дозатора блока в стакан с продуктом, нейтрали-
зуя последний. По достижении точки эквивалентности pH 8,9 и
окончании выдержки (30 с) процесс нейтрализации автоматически
прекращается, а на панели блока автоматического титрования за-
жигается сигнал «Конец». После этого отключают все кнопки. Оп-
ределяют объем раствора гидроксида натрия, затраченного на нейт-
рализацию.
Кислотность в градусах Тернера находят умножением объема
(см3) раствора гидроксида натрия, затраченного на нейтрализацию
определенного объема продукта, на следующие коэффициенты:
10 — для молока, молока с наполнителями, сливок, простоква-
ши, ацидофильного молока, кефира, кумыса и других кисломолоч-
ных продуктов;
20 — для мороженого, сметаны, творога и творожных изделий.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА В МОЛОКЕ
МЕТОДОМ ФОРМОЛЬНОГО ТИТРОВАНИЯ
С ИСПОЛЬЗОВАНИЕМ ТИТРИМЕТРИЧЕСКОГО АНАЛИЗАТОРА
Метод формольного титрования основан на нейтрализации кар-
боксильных групп моноаминодикарбоновых кислот белков раство-
ром гидроксида натрия. Объем гидроксида натрия, затраченный на
нейтрализацию, пропорционален массовой доле белка в молоке.
Выполнение работы. Блок автоматического титрования подклю-
чают к анализатору. Затем блок и потенциометрический анализатор
подключают к сети и прогревают их в течение 10 мин.
Дозатор блока автоматического титрования заполняют раство-
ром гидроксида натрия (реактив 1). Потенциометрический анали-
затор настраивают на диапазон измерения pH, включающий pH 9.
Блок автоматического титрования настраивают на точку эквива-
лентности (pH 9). Начиная с pH 4, раствор подают по каплям. После
достижения точки эквивалентности титруемый раствор выдержи-
вают 30 с.
В стакан помещают 20 см3 молока и опускают стержень магнит-
ной мешалки. Стакан устанавливают на магнитную мешалку, вклю-
чают двигатель мешалки и погружают электроды потенциометри-
ческого анализатора в молоко. Включают кнопку «Пуск» блока ав-
томатического титрования, а спустя 2—3 с — кнопку «Выдержка».
Раствор гидроксида натрия при этом начинает поступать из дозато-
ра блока в стакан с молоком, нейтрализуя последнее. По достиже-
149
нии точки эквивалентности (pH 9) и истечении выдержки (30 с)
процесс нейтрализации автоматически прекращается, а на панели
блока автоматического титрования загорается сигнал «Конец».
После этого отключают кнопки «Пуск» и «Выдержка», определяют
объем раствора щелочи, затраченный на нейтрализацию молока (до
внесения формальдегида), и вносят в стакан 5 см3 формальдегида
(реактив 2).
Выдерживают 2—2,5 мин, вновь включают кнопки «Пуск» и
«Выдержка». По окончании процесса определяют общее количе-
ство раствора, затраченное на нейтрализацию.
Параллельно проводят контрольный опыт по нейтрализации
смеси 20 см3 воды и 5 см3 раствора формальдегида.
Массовая доля белка (%)
5=(К2- Hi - Го) -0,96 + К,
где V2 — общий объем раствора, израсходованный на нейтрализацию, см3; —
объем раствора, израсходованный на нейтрализацию до внесения формальдегида,
см3; Го — объем раствора, израсходованный на контрольный опыт, см3; 0,96 — эм-
пирический коэффициент, %/см3, К— поправка к результату измерения массовой
доли белка, %.
За окончательный результат измерения принимают среднее
арифметическое двух параллельных вычислений, округляя резуль-
тат до второго десятичного знака.
Для определения поправки (А) к результатам измерения массо-
вой доли белка методом формольного титрования одновременно
измеряют массовую долю белка в одном и том же образце молока
методом формольного титрования и по Кьельдалю. Измерения вы-
полняют на средней пробе молока, полученной путем смешивания
равных по массе образцов молока, полученных из разных хозяйств.
Измерения как по Кьельдалю, так и методом формольного тит-
рования проводят в 6 повторностях.
Поправку К (%) вычисляют по формуле
А = 5к-5ф,
где Бк — среднее арифметическое значение массовой доли белка, полученное по
методу Кьельдаля, %; Ьф — среднее арифметическое значение массовой доли белка,
полученное формольным титрованием, %.
Приготовление реактивов. Реактив 1 (0,1 Мраствора гидроксида
натрия, в 1000 слР которого содержится 0,1 Мгидроксида натрия).
7 см3 насыщенного гидроксида натрия переносят в мерную колбу
на 1000 см3 и доводят объем раствора дистиллированной водой до
метки.
Реактив 2 (30%-ный раствор формальдегида). 8,1 г предваритель-
но нейтрализованного 37%-ного формальдегида смешивают с
91,9 см3 дистиллированной воды.
150
ВОЛЬТАМПЕРОМЕТРИЯ
Вольтамперометрия — высокочувствительный метод анализа
органических и неорганических веществ. Современные приборы
для вольтамперометрии позволяют снизить границу измеряемых
концентраций и добиться высокой селективности по определяемо-
му веществу. Этот метод в настоящее время довольно успешно кон-
курирует с широко распространенными методами атомно-абсорб-
ционной спектрометрии.
Вольтамперометрический метод анализа основан на использо-
вании явления поляризации микроэлектрода, получении и интер-
претации вольтамперных (поляризационных) кривых, отражаю-
щих зависимость силы тока от приложенного к рабочему электроду
напряжения. В вольтамперометрии используют два электрода: ра-
бочий поляризуемый электрод с малой поверхностью и неполяри-
зуемый электрод сравнения. Если в качестве рабочего выбран элек-
трод с постоянно обновляющейся поверхностью (например, ртут-
ный капающий электрод), то метод анализа называют полярогра-
фическим.
При прохождении постоянного тока через электролитическую
ячейку процесс характеризуется соотношением
Е = Еа — Ек + IR,
где Е — приложенное извне напряжение; Ег — потенциал анода; £к — потенциал
катода; I— сила тока в цепи; R — сопротивление электролитической ячейки.
При вольтамперометрических измерениях анализируемый ра-
створ содержит индифферентный электролит большой концентра-
ции, поэтому R ~ 1 кОм и, так как сила тока в вольтамперометрии не
превышает 10~5 А, падением напряжения на ячейке IR можно пре-
небречь.
Поскольку в вольтамперометрии один из электродов не поляри-
зуется и для него потенциал остается постоянным, подаваемое на
ячейку напряжение проявляется в изменении потенциала только
рабочего электрода. Если потенциал рабочего электрода измерять
относительно потенциала электрода сравнения, условно приняв
последний за нуль, то Е = Еа для рабочего микроанода и Е = — Ек для
рабочего микрокатода. Таким образом, регистрируемая вольтам-
перная кривая (полярограмма) отражает электрохимический про-
цесс, происходящий только на одном электроде. Если в растворе
присутствуют вещества, способные электрохимически восстанав-
ливаться или окисляться, то при наложении на ячейку линейно из-
меняющегося напряжения (скорость не превышает 200 мВ/мин)
кривая I =j{E) имеет форму волны (в отсутствие электрохимичес-
кой реакции эта зависимость линейна).
151
Рис. 42. Измерение высоты волны (а)
и пика (б) на полярограмме
На рис. 42 показана полярографическая кривая. При низких
значениях потенциала (А), величина которого недостаточна для
того, чтобы на рабочем микроэлектроде происходила электрохими-
ческая реакция, через ячейку проходит очень незначительный оста-
точный ток, обусловленный, прежде всего, током заряжения двой-
ного электрического слоя и присутствием в растворе электрохими-
чески более активных, чем анализируемое вещество, примесей.
При увеличении потенциала электрохимически активное вещество
(называемое деполяризатором) вступает в электрохимическую ре-
акцию на электроде и сила тока в результате этого резко возрастает
(В). Это так называемый фарадеевский ток. С ростом потенциала
сила тока возрастает до некоторого предельного значения, остава-
ясь затем постоянной (Q. Предельная сила тока обусловлена тем,
что в данной области потенциалов практически весь деполяризатор
из приэлектродного слоя исчерпан в результате электрохимической
реакции, а обедненный слой обогащается за счет диффузии деполя-
ризатора из объема раствора. Скорость диффузии в этих условиях
контролирует скорость электрохимического процесса в целом. Та-
кой ток называют предельным диффузионным 7д. Для того чтобы
исключить электростатическое перемещение деполяризатора (миг-
рацию) в поле электродов и понизить сопротивление в ячейке, из-
мерения проводят в присутствии большого избытка сильного элек-
тролита, называемого фоном. Являясь электрохимически индиф-
ферентным, вещество фонового раствора может вступать в хими-
ческие реакции (часто это реакции комплексообразования) с
определенным веществом. Иногда фоновый электролит одновре-
менно играет роль буферного раствора. Например, при полярогра-
фическом определении ионов меди, кадмия, цинка, кобальта, ни-
келя в качестве фона используют аммиачный буферный раствор.
Величина 5i/2 служит качественной характеристикой полярог-
рафически активного вещества и может быть определена графичес-
ки, как показано на рис. 42, а. Количественной характеристикой
152
анализируемого соединения в полярографии является величина
предельного диффузионного тока или высота волны (пика), кото-
рая является линейной функцией концентрации. Высоту полярог-
рафической кривой или пика измеряют, как показано на рис. 42, б.
Определить концентрацию деполяризатора можно различными
методами: стандартов, градуировочного графика и добавок. При
этом во всех случаях используют стандартные растворы, состав ко-
торых должен быть максимально приближен к составу анализируе-
мого раствора, условия полярографирования стандартных и анали-
зируемого растворов должны быть одинаковыми.
В методе стандартов полярографируют раствор неизвестной
концентрации и стандартный раствор. Для одних и тех же условий
анализа
Сх СстНх/ Нст,
где Сх и Сст — концентрации анализируемого и стандартного растворов; //хи Нст —
высота волны на полярограммах этих растворов.
По методу градуировочного графика регистрируют полярограм-
мы анализируемого раствора и серии стандартных растворов, стро-
ят градуировочный график в координатах Н—С, по которому для
найденного значения Ях определяют Сх.
Метод добавок может быть использован только в интервале кон-
центраций, для которых строго соблюдается линейная зависимость
Н—С. Пробу анализируемого раствора полярографируют объемом
Ух, концентрация которого Сх. На полярограмме измеряют высоту
волны Ях. Затем к анализируемому раствору добавляют определен-
ный объем VCT стандартного раствора концентрацией Сст (предпоч-
тительно, чтобы Vx >> VCT и Сх < Сст). Измеряют на полярограмме
высоту волны Я.
Полярограммы могут быть искажены за счет полярографических
максимумов — резкого возрастания тока выше предельного значе-
ния его с последующим спадом. Причины возникновения максиму-
мов различны и могут быть связаны с неравномерной поляризацией
ртутной капли и тангенциальным движением ее поверхности, что
приводит к дополнительному перемешиванию раствора. Такого
рода максимумы можно устранить введением в полярографируе-
мый раствор ПАВ: красителей (метилового красного, фуксина и
др.), высокомолекулярных соединений (агар-агара, желатина).
Кроме того, на полярограммах возникают кислородные максимумы
за счет восстановления на ртутном электроде кислорода, растворен-
ного в анализируемом растворе. Интерпретация полярограммы, ос-
ложненной волнами восстановления кислорода, затруднительна,
поэтому перед полярографированием из анализируемого раствора
кислород удаляют, продувая N2, Аг в щелочных средах можно ис-
пользовать кристаллический Na2SO3. При анализе водных раство-
153
Рис. 43. Принципиальная схема
вольтамперометрической уста-
новки:
1 — источник постоянного тока; 2 —
потенциометр; 3— гальванометр; 4—
вольтметр; 5 — электролизер
ров достаточна 5— 10-минутная деаэ-
рация, для органических раствори-
телей в течение 25—30 мин.
Анализируемый раствор полярог-
рафируют после анализа фонового
электролита и удаления из него кис-
лорода: на полярограмме должны от-
сутствовать какие-либо волны. Заре-
гистрированную в ходе анализа по-
лярограмму расшифровывают: на
ней определяют потенциал полувол-
ны Д/2 или потенциал пика Еп и вы-
соту волны или пика Н.
Принципиальная схема вольтам-
перометрической установки изобра-
жена на рис. 43. От источника тока 1
на электролитическую ячейку-элек-
тролизер 5подается ток, напряжение
которого плавно меняется с помо-
щью потенциометра 2 Силу тока, проходящего через исследуемый
раствор, измеряют гальванометром 3.
Электролитическая ячейка (электролизер), используемая в
вольтамперометрии, представляет собой сосуд вместимостью 1 —
50 см3 с погруженными в него рабочим электродом и электродом
сравнения. Электродную систему для вольтамперометрических из-
мерений выбирают таким образом, чтобы плотность тока на этих
электродах существенно различалась: на рабочем электроде плот-
ность тока должна быть велика, на электроде сравнения — ничтож-
но мала. В этом случае поляризоваться будет только рабочий элект-
род и, естественно, только на нем возможны электрохимические
процессы восстановления или окисления ионов из раствора.
Рабочий электрод, как правило, имеет очень малую поверхность
по сравнению с поверхностью электрода сравнения — это микро-
электрод, который может быть изготовлен из твердого материала
(платина, серебро, графит специальной обработки и др.) или в виде
ртутной капли, вытекающей из капилляра.
Конструктивно твердые электроды удобнее и безопаснее, чем ртут-
ные, но область их использования ограничена. Так, платиновый элек-
трод пригоден для работы при более положительных значениях потен-
циала, чем ртутный, но граница отрицательных значений потенциала
определяется значительно меньшим значением потенциала выделе-
ния водорода из водных растворов. Твердые электроды представляют
собой проволочки или стержни, запаянные в стеклянные трубки. Ра-
бочая поверхность такого электрода не превышает 0,2 см . Твердые
электроды во время работы приводятся во вращение мотором. Каж-
дый раз перед началом работы такой электрод следует промывать ра-
створом азотной кислоты (1:1), а затем многократно водой.
154
Ртутный капающий электрод в качестве рабочего микроэлектро-
да широко используют при полярографическом анализе веществ,
восстанавливающихся в области потенциалов от +0,2 до —1,9 В от-
носительно насыщенного каломельного электрода. Из стеклянного
резервуара по гибкому шлангу ртуть поступает в стеклянный капил-
ляр, из которого со скоростью, регулируемой высотой ртутного
столба и диаметром капилляра (1—10 капель в 1 с), подается в ана-
лизируемый раствор. Постоянно обновляющаяся поверхность
ртутной капли, а также высокое перенапряжение выделения водо-
рода, позволяющее электрохимически восстанавливать на таком
электроде ионы металлов, расположенных в ряду напряжений левее
водорода, делают ртутный капающий электрод одним из наиболее
применяемых. Однако работа с таким электродом требует тщатель-
ного соблюдения всех мер, предусмотренных правилами техники
безопасности при работе со ртутью.
Электроды сравнения бывают двух видов: внутренний и вне-
шний. Внутренним электродом сравнения может служить слой рту-
ти с большой поверхностью, который находится непосредственно
на дне электролитической ячейки. В качестве внешнего электрода
сравнения может служить хлорсеребряный, каломельный и др. Вне-
шний электрод сравнения соединяется с анализируемым раствором
электролитическим ключом. Потенциал внутреннего электрода
сравнения подвержен влиянию состава анализируемого раствора,
тогда как для внешнего он остается неизменным.
Электролитическая ячейка, блок питания и блок-регистратор
вольтамперной кривой — основные узлы полярографа. В полярог-
рафах различных типов плавно изменяющееся с определенной ско-
ростью (до нескольких сотых вольта в 1 с) напряжение подается на
ячейку от механического делителя напряжения. Возникающий в
ячейке ток после соответствующих преобразований регистрирует
специальное устройство. В полярографах современных моделей
имеется записывающее устройство — в ходе анализа полярограмма
записывается пером на диаграммной ленте, которая перемещается
вертикально синхронно с подаваемым напряжением. Отклонение
пера по горизонтали пропорционально току ячейки.
Если скорость изменения напряжения, подаваемого на ячейку,
велика (до нескольких десятков вольт в 1 с), визуальные и самопи-
шущие регистраторы в силу их инерционности нельзя использо-
вать, вместо них индикатором служат электронно-лучевые трубки.
Полярографические приборы, в которых скорость изменения на-
пряжения велика и полярографическая кривая регистрируется на
экране осциллографа, называют осциллографическими полярогра-
фами. На полярографическую ячейку накладывают постоянное на-
пряжение от потенциометра полярографа и переменное напряже-
ние от генератора, изменяющееся линейно во времени.
Аналитические возможности вольтамперометрического метода
анализа очень широки. Метод используют для определения неор-
ганических и органических соединений различного состава.
155
Интервал определяемых концентраций 10-2—10~6 М, нижний
предел в инверсионной вольтамперометрии достигает 10-9 М, при
определении малых концентраций погрешность не превышает 3 %.
Ограничения метода полярографического анализа связаны с ис-
пользованием ртутного электрода, поэтому в аналитической прак-
тике используют приборы с твердым электродом.
Специализированных полярографов для анализа пищевых про-
дуктов и питьевой воды нет, так как эти продукты в вольтамперо-
метрии анализируются по общим законам, и специфическим явля-
ется только стадия подготовки проб к анализу, которая, в свою оче-
редь, зависит от конкретного пищевого продукта и объекта опреде-
ления.
Так, анализатор вольтамперометрический АВА-1 предназначен
для измерения массовой концентрации ионов металлов, в частно-
сти меди, свинца, олова и ионов других металлов в водных средах и
может быть использован для аналитического контроля объектов ок-
ружающей среды, санитарного контроля и контроля сырья, пище-
вых продуктов и технологических процессов. Анализатор обеспечи-
вает измерение концентрации ионов металлов в водных растворах в
диапазоне от 5 • 10~9 до 1-Ю-5 моль/л (от 1 до 1000 мкг/л).
Анализатор типа АВА-1 состоит из измерительного блока и дат-
чика, последний представляет собой электролитическую ячейку —
электролизер (рис. 44). Он состоит из корпуса и крышки с непод-
вижными электродами и вращающимся индикаторным электро-
дом. В качестве индикаторного электрода в электролизере исполь-
зуют твердые электроды из графита, стеклоуглерода или углеси-
талла, при этом для формирования ртутно-графитового электро-
да в фоновый раствор вводят ионы ртути; хлорсеребряный
электрод сравнения отделен от исследуемого раствора катионо-
обменной мембраной; в качестве вспомогательного служит элект-
род ЭВЛ-1М3.1.
Для повышения селективности, снижения предела определения
и улучшения воспроизводимости результатов измерения в анализа-
торе АВА-1 реализованы метод инверсионной вольтамперометрии
и дифференциально-импульсный метод регистрации сигнала.
Инверсионный метод основан на предварительном электрохи-
мическом накоплении ионов определяемых металлов на поверхно-
сти вращающегося индйкаторного электрода при отрицательном
потенциале с последующим растворением их в течение короткого
промежутка времени током положительной полярности (т. е. осу-
ществляется инверсия процесса на электроде).
Дифференциально-импульсный метод регистрации сигнала
заключается в следующем. Изменяя потенциал электрода по линей-
ному закону, одновременно подают одиночные импульсы, изменя-
ющиеся во времени линейно, по форме прямоугольника, напряже-
нием 30 мВ и длительностью 0,04 с. Измерение проводят, когда ем-
костной ток сильно снижается, при этом регистрируют зависимость
156
Рис. 44. Электролизер — электролити-
ческая ячейка АЭ-4 вольтамперометри-
ческого анализатора АВА-1:
1— корпус; 2 — крышка; 3 — втулка для
электрода сравнения; -/—электрод сравне-
ния; 5 — вращающийся индикаторный элек-
трод; б— штуцер; 7— пробка; 8— вспомога-
тельный электрод; 9 — гайка; 10 — мембра-
на; 11 — отверстие для ввода добавки; 12—
отверстие для подачи инертного газа
производной тока по потенци-
алу (d//d£) от Е. Таким образом
достигают чувствительности
10-8—5-10-8 моль/дм3.
Общий измерительный
цикл анализатора АВА-1 состо-
ит из четырех стадий.
Первая стадия — электро-
химическая регенерация по-
верхности рабочего (индика-
торного) электрода, потенциал
регулируется в пределах от 0 до
+650 мВ, длительность в пре-
делах от 20 до 30 с.
Вторая стадия — электроли-
тическое накопление опреде-
ляемых металлов на поверхно-
сти индикаторного электрода в
режиме вращения электрода и поддержания отрицательного потен-
циала в пределах от 0 до 1500 мВ. Длительность стадии накопления
составляет от 30 до 2500 с.
Третья стадия — «успокоение» раствора перед съемкой вольтам-
перной кривой. Длительность стадии постоянна и составляет обыч-
но 20 с. Потенциал электрода поддерживается близким потенциалу
стадии накопления.
Четвертая стадия — измерительная. Скорость развертки задает-
ся с помощью переключателя.
По завершении получения кривой система снова переходит в
стадию регенерации, начинается следующий цикл смены стадий.
Вольтамперограмма (изменяющееся во времени напряжение) от
измерительного блока поступает на регистратор аналоговой инфор-
мации (самописец, компьютер и т. п.).
Измерение выполняют следующим образом. В электролизер
подготовленного к работе прибора помещают 20 см3 исследуемого
157
раствора. Нажимают кнопку «ячейка/эквивалент», при этом под-
ключается ячейка, а затем нажимают кнопку «пуск» и снимают
вольтамперные кривые.
При использовании метода добавок вводят добавку стандартно-
го раствора металла для увеличения его концентрации в исходном
растворе примерно в два раза. Объем добавки не должен быть боль-
шим, чтобы не исказить результаты за счет разбавления анализиру-
емого раствора (обычно Ист не более 1 см3). Снимают вольтамперо-
метрические кривые для раствора с добавкой. Вычисляют среднее
из трех значений высот (мм) пиков отдельно для исследуемого ра-
створа Я] и для раствора с добавкой Н2.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МЕДИ, СВИНЦА, КАДМИЯ, ЦИНКА
В ПРОДУКТАХ С ПОМОЩЬЮ ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО
АНАЛИЗАТОРА АВА-1
Метод основан на предварительной минерализации пробы про-
дукта, получении раствора минерализата с добавлением фонового
электролита, проведении анализа в электролизере, получении
вольтамперных кривых и измерении высоты пиков. Для количе-
ственного определения металла используют метод добавок. Анализ
проводят на вольтамперометрическом анализаторе АВА-1.
Определению содержания цинка методом инверсионной вольтам-
перометрии с использованием твердого электрода обычно мешает
присутствие в водном растворе ионов меди. В данной методике влия-
ние меди полностью устраняется введением 20-кратного по отноше-
нию к меди избытка ионов галлия. Поэтому в анализируемой пробе
необходимо предварительно определить содержание меди.
Пробы пищевых продуктов (табл. 18) подготавливают и минера-
лизуют в соответствии с ГОСТ 26929—88.
18. Масса навески при вольтамперометрическом определении содержания металлов
в молочных продуктах, г
Компонент Рекомендуемая Допустимая минимальная
Кадмий 12,5 12,5
Свинец 2,5 2,5
Медь 2,5 0,25
Цинк 2,5 0,025
Если содержание меди, цинка, свинца, кадмия определяют од-
новременно в одной пробе после сухой минерализации, то навеску
продукта выбирают по элементу с меньшим ПДК (предельно допус-
тимая концентрация металла в продукте, мг/кг).
Пробы продукта можно минерализовать несколькими способа-
ми: сухой минерализацией, мокрой минерализацией и кислотной
экстракцией (неполной минерализацией).
158
Способ сухой минерализации основан на полном разложении
органических веществ путем сжигания пробы сырья или продукта в
электропечи при контролируемом температурном режиме и пред-
назначен для всех видов сырья и продуктов, кроме продуктов с мас-
совой долей жира 60 % и более.
Способ мокрой минерализации основан на полном разрушении
органических веществ пробы продукта при нагревании с серной и
азотной концентрированными кислотами с добавлением соляной
кислоты или пероксида водорода или при нагревании только с пе-
роксидом водорода и предназначен для всех видов сырья и продук-
тов, кроме сливочного масла и животных жиров.
Способ кислотной экстракции (неполной минерализации) ос-
нован на экстракции токсичных элементов из пробы продукта ки-
пячением с разбавленной соляной или азотной кислотами и пред-
назначен для растительного и сливочного масел, маргарина, пище-
вых жиров и сыров.
При вольтамперометрическом определении содержания меди,
свинца, кадмия и цинка рекомендуется способ сухой минерализа-
ции проб продукта.
Сухую минерализацию проб молока и молочных продуктов про-
водят следующим образом.
Навеску продукта помещают в чашку или тигель. Необходимый
объем жидкого продукта отмеривают пипеткой.
К молоку, кисломолочным продуктам и молочным консервам
добавляют раствор азотной кислоты (1:1) из расчета 1 см3 на 50 г
продукта; перемешивают, помещают на электроплитку и обуглива-
ют до прекращения выделения дыма (для интенсификации обугли-
вания одновременно допускается проводить обогрев чашки с про-
дуктом инфракрасной лампой), затем чашку помещают в электро-
печь при температуре около 250 °C.
При минерализации сыров содержимое чашки смачивают азот-
ной кислотой так, чтобы вся поверхность пробы была покрыта кис-
лотой и нагревают на водяной бане для прекращения выделения па-
ров. Обработку азотной кислотой повторяют еще 2 раза. Затем обра-
ботанную пробу обугливают на электроплитке таким же образом,
как и для других молочных продуктов.
Минерализацию проб в электропечи проводят постепенно, по-
вышая температуру на 50 °C через каждые 30 мин и доводя ее до
450 °C, продолжают минерализацию при этих условиях до получе-
ния серой золы.
Чашку с золой вынимают из электропечи, охлаждают до комнат-
ной температуры и серую золу смачивают 0,5—1,0 см3 раствора
азотной кислоты. Затем кислоту досуха выпаривают на электро-
плитке со слабым нагревом и снова помещают чашку с пробой в
электропечь при температуре 250 °C, постепенно доводят темпера-
туру до 450 °C и выдерживают 1 ч. Минерализацию считают закон-
ченной, когда зола станет белого и слегка окрашенного цвета без
159
обугленных частиц. При наличии обугленных частиц повторяют
обработку золы раствором азотной кислоты или воды.
Выполнение работы. Определение содержания меди, свинца, кад-
мия. Золу, полученную методом сухой минерализации, растворяют
в азотной кислоте (реактив 2) на водяной бане, выпаривают раствор
до влажных солей, охлаждают до комнатной температуры, неболь-
шими порциями добавляют воду и растворяют пробу, количествен-
но перенося ее в мерную колбу на 25 см3, в которую добавлено 5 см3
1 М KNO3 и 0,2 см3 HNO3 (реактив 2) для создания pH пробы < 2.
Прибор подготавливают к работе и проводят пробный опыт. Рабо-
чий объем ячейки заполняют 25 см3 фонового раствора (реактив 5),
добавляют 2 капли раствора ртути (реактив 4), в том случае если
ионы ртути не были введены в фоновый раствор при приготовле-
нии. Проводят 3—4 измерения при следующих режимах:
Потенциалы накопления, успокоения
и начала развертки
Потенциалы регенерации и оконча-
ния развертки
Скорость развертки потенциала, мВ/с
Частота вращения рабочего электро-
да, об/мин
Время регенерации электрода, с
Время успокоения раствора, с
Время накопления, с (в зависимости
от концентрации определяемых эле-
ментов в растворе)
От-1,0 до—1,1 В
+0,5 В
100-200
1900-3100
30-60
10-20
10-200 (300)
По завершении 3—4 циклов измерений в фоновом растворе, в
стадии регенерации отключают ячейку.
После проведения пробного опыта из ячейки удаляют фоновый
раствор и заполняют ее 25 см3 анализируемого раствора, добавляют
2 капли раствора ртути (реактив 4), если он не был добавлен при
приготовлении. Проводят 3—4 цикла измерений на этом растворе
при тех же режимах, что и для фонового раствора.
Вводят добавку стандартного раствора исследуемого металла
так, чтобы концентрация его в исходном растворе увеличилась при-
мерно в 2 раза. Объем добавки не должен быть большим, чтобы не
исказить результат за счет разбавления анализируемого раствора
(обычно не более 1 см3). Проводят 3—4 измерения на растворе с до-
бавкой, ячейку отключают в стадии регенерации.
Измеряют высоту пиков (мм) и рассчитывают среднее арифме-
тическое значение высоты пика. Содержание металла в анализируе-
мом продукте (мг/кг)
_(Я1-Яф)СдИд 1000
х (Я2-Я1) т ’
где Яф, Hi, Нг — среднее арифметическое значение высоты пика на полярограмме
для исследуемого металла соответственно в фоновом и анализируемом растворах, а
160
также анализируемом растворе с добавкой стандартного раствора, мм; Сд — кон-
центрация стандартного раствора добавки, мг/см3; — объем стандартного раство-
ра добавки, см3; т — навеска продукта, г; 1000 — перевод г в кг.
Определение содержания цинка. Золу, полученную методом сухой
минерализации, растворяют в азотной кислоте (реактив 2) на водя-
ной бане, выпаривают раствор до влажных солей, охлаждают до
комнатной температуры, небольшими порциями добавляют фоно-
вый раствор — ацетатный буфер pH 4,6 (реактив 7) и количественно
переносят в мерную колбу на 25 см3.
Прибор подготавливают к работе и проводят пробный опыт. Рабо-
чий объем ячейки заполняют 10—25 см3 раствора фона (реактив 7) и
добавляют в него 1—2 капли раствора ртути концентрацией 10 г/л
(реактив 3) и 0,2—1,0 см3 раствора 0,001 М галлия (реактив 8). Из-
мерения проводят, используя следующие режимы работы вольтам-
перометрического анализатора (все значения потенциалов приво-
дятся относительно насыщенного хлорсеребряного электрода срав-
нения):
Потенциалы накопления, успокоения
и начала развертки, В
Потенциалы регенерации, окончания
развертки,В
Скорость развертки потенциала, мВ/с
Частота вращения рабочего электро-
да, об/мин
Время регенерации электрода, с
Время успокоения раствора, с
Время накопления, с
От —1,45 до —1,25
От +0,30 до +0,50
100-300
1900-3100
30-60
10-20
10—300 (в зависимости от кон
центрации цинка в растворе)
Высоту пика при растворении цинка регистрируют при потен-
циале -0,95 В. По завершении 3—4 циклов измерений в фоновом
растворе, в стадии регенерации отключают ячейку.
После проведения пробного опыта из ячейки удаляют фоновый
раствор, вносят 25 см3 анализируемого раствора и добавляют ра-
створ галлия (реактив 8) в количестве, обеспечивающем 20-крат-
ный избыток ионов галлия, относительно концентрации ионов
меди в ячейке и 1—2 капли раствора ртути (реактив 3). Проводят 3—
4 цикла измерений на анализируемом растворе при тех же режимах,
что и для фонового раствора.
Вводят добавку стандартного раствора цинка, который выбира-
ют так, чтобы концентрация его в анализируемом растворе увели-
чилась примерно в 2 раза. Объем добавки не должен быть большим,
чтобы не исказить результат за счет разбавления исходного раствора
(обычно не более 1 см3).
Проводят 3—4 цикла измерений на растворе с добавкой. Ячей-
ку отключают в стадии регенерации. Измеряют высоту пиков в
мм и рассчитывают среднее арифметическое значение из трех из-
мерений.
161
Содержание цинка (мг/кг) в анализируемом продукте
(Я1-Яф)СдИд их-1000
(Я2-Я1)Кх+(Я2-Яф)Ид т ’
где Яф, Яь Я2 —среднее арифметическое значение высоты пика цинка соответ-
ственно в фоновом, анализируемом растворах и анализируемом растворе с добавкой
стандартного раствора, мм; Сд — массовая доля цинка в стандартном растворе до-
бавки, мг/см3; Ид — объем раствора добавки, см3; Vx (Vx = Ио) — объем анализируе-
мого раствора в ячейке, см3; т — навеска продукта, г; ГЬ — объем раствора минера-
лизата, см3; 1 000 — пересчет г на кг.
Следует отметить, что если в ячейку для анализа берут не весь
объем приготовленного раствора минерализата (И0 = 25см3), а
только его часть (см3), тогда формула примет следующий вид;
q______(Я[ -Яф)СдИд__Ир 1000
"(^-ВД+^-Яф)^ т
Приготовление реактивов. Реактив 1 (бидистиллят). Дистилли-
рованную воду дополнительно перегоняют со щелочным раствором
перманганата калия, периодически помещая в перегонную колбу
2—3 см3 раствора перманганата калия до появления розового окра-
шивания.
Раствор перманганата калия готовят, растворяя 10 г сухой соли в
100 см3 раствора гидроксида натрия концентрацией 1 моль/л (40 г
гидроксида натрия на 1000 см3 раствора).
Реактив 2 (азотная кислота). Азотную кислоту плотностью
1042 кг/м3 (1,42 г/см3) разводят бидистиллятом в соотношении 2:3.
При использовании азотной кислоты другой плотностью изменяют
объем добавляемого бидистиллята.
Реактив 3 [(раствор концентрацией 0,05моль/л нитрата ртути
(II)]. В мерную колбу на 500 см3 вносят 10 см3 HNO3 (реактив 2),
8,35 г нитрата ртути (II), после растворения соли раствор разбавля-
ют бидистиллятом до метки. Полученный раствор имеет концент-
рацию ртути, равную 10 г/л.
Следует учесть, что если используют влажный препарат нитрата
ртути (II), то точную концентрацию раствора 10 г/л устанавливают
титрометрически по хлориду натрия.
Реактив 4 [(раствор концентрацией 1г/л нитрата ртути (II)].
Путем разбавления реактива 3 бидистиллятом в 10 раз (в соотноше-
нии 1:9) приготавливают 10—25 см3 раствора нитрата ртути (II)
(концентрация ртути 1 г/л).
Этот раствор вводят в фоновый раствор (или растворы солей ме-
таллов) при их приготовлении из расчета 1 см3 реактива 4 на 100 см3
162
фонового раствора (раствора солей металлов) либо непосредствен-
но в электролизер перед проведением измерений по 2 капли на
25 см3 анализируемого раствора.
Реактив 5 (фоновый раствор для определения содержания меди,
свинца, кадмия). Вначале приготавливают 1 М раствор нитрата ка-
лия. В мерной колбе на 250 см3 растворяют 25,3 г KNO3, доводят би-
дистиллятом до метки. Полученный раствор имеет концентрацию
нитрата калия 1 моль/л (реактив 5а). Затем приготавливают фоно-
вый раствор. В мерную колбу на 100 см3 вносят пипеткой 25 см31 М
раствора KNO3 (реактив 5а), 0,34 см3 (6—7 капель) HNO3 (реактив
2), 1 см3 раствора нитрата ртути (II) (реактив 4) и доводят до метки
бидистиллятом. Полученный фоновый раствор имеет состав, отме-
чающий 0,2 моль/л KNO3, 0,02 моль/л HNO3,0,05 ммоль/л Hg.
Реактив 6 (стандартные растворы). Основные стандартные ра-
створы из металлических меди, свинца, кадмия, цинка. 1000 г ме-
талла растворяют при нагревании в водяной бане в минимальном
количестве азотной кислоты (реактив 2), количественно переносят
в мерную колбу на 1000 см3, в которую ранее помещено 5 см3 HNO3
(реактив 2), доводят до метки бидистиллятом. Концентрация ме-
талла в основном растворе равна 1 мг/см3.
Рабочие стандартные растворы готовят путем разбавления ос-
новного стандартного раствора. Так, для десятикратного разбавле-
ния основного стандартного раствора в мерную колбу на 100 см3
вносят 10 см3 основного раствора, затем добавляют 0,34 см3 (6—7
капель) HNO3 (реактив 2) и 20 см31 М раствора KNO3, а затем дово-
дят объем раствора до метки бидистиллятом.
Стандартные растворы из готовых стандартных образцов (ГСО).
1. Растворы из ГСО меди, свинца, кадмия готовят путем после-
довательного разбавления составляющими фонового раствора (би-
дистиллятом, растворами нитрата калия и азотной кислоты).
Основной стандартный раствор меди, свинца, кадмия получают
разведением ГСО в 20 раз. При этом действительную массовую кон-
центрацию С (мкг/л) компонента в основном стандартном растворе
рассчитывают по формуле
^_СЛГ106
Ир ’
где С' — молярная концентрация компонента в исходном растворе ГСО (согласно
паспорту на ГСО), моль/л; А — молярная масса иона, г/моль; и' — отобранный
объем ГСО, см3; Йр — объем основного стандартного раствора (мерной колбы), см3;
106 — перевод г в мкг.
Каждый используемый ГСО переносят из ампулы в чистую су-
хую колбу с притертой пробкой, после чего его разбавляют рассчи-
танными объемами бидистиллята, растворов нитрата калия и азот-
ной кислоты.
Для приготовления 20-кратного разведения отобранный объем
ГСО (K'=5cm3) переносят пипеткой в мерную колбу на 100 см3,
163
после чего добавляют 0,34 см3 HNO3 (реактив 2) и 20 см3 1 М ра-
створа KNO3, а затем доводят объем раствора до метки бидистил-
лятом.
Например, для приготовления основного стандартного раствора
отобрана порция 5,0 см3 стандартного образца 2—4 с аттестованным
содержанием свинца 0,0504 ммоль/л. Содержание свинца в полу-
ченном растворе: 5,04 10~5 • 207,2 • 5 • 10° • 10-2 = 522 мкг/л.
Стандартные растворы меньшей концентрации получают мето-
дом последовательного разбавления в 10 раз основного раствора,
аналогично описанному выше.
2. Стандартные растворы цинка из ГСО готовят путем последо-
вательного разбавления бидистиллятом и азотной кислотой.
Основной стандартный раствор получают разбавлением ГСО в
10 раз. Отобранный объем ГСО (10 см3) переносят пипеткой в
мерную колбу на 100 см3, после чего добавляют 0,34 см3 (6—7 ка-
пель) HNO3 (реактив 2) и доводят объем раствора до метки биди-
стиллятом.
Например, для приготовления основного стандартного раствора
отобрана порция 10 см3 стандартного образца с аттестованным со-
держанием цинка 0,0099 ммоль/л. Содержание цинка в полученном
растворе: 9,9 • 10-6 • 65,4 • 10 • 106 • 10~2 = 64,7 мкг/л.
Стандартные растворы меньшей концентрации цинка получают
методом последовательного разбавления в 10 раз основного раство-
ра, аналогично описанному выше.
Реактив 7 (фоновый раствор для определения цинка). В качестве
фонового раствора используют ацетатный буфер с pH 4,6, который
получают, смешивая 102 см3 0,2 М уксусной кислоты и 98 см3 0,2 М
раствора ацетата калия (19,6 г ацетата калия на 1 л воды).
Уксусную кислоту очищают в герметично закрытом эксикаторе.
В его нижнюю часть наливают концентрированную уксусную кис-
лоту, подлежащую очистке, на фарфоровый вкладыш ставят квар-
цевую чашку с бидистиллированной водой объемом примерно 200
см3. Через 2—5 сут чашку вынимают, определяют плотность полу-
чившегося раствора уксусной кислоты ареометром. Очищенную та-
ким образом кислоту хранят в посуде из полиэтилена высокого дав-
ления и используют для приготовления буферного раствора. Вместо
кварцевой чашки можно применить соответствующую посуду из
полиэтилена высокого давления или фторопласта-4.
Реактив 8(раствор галлия используют при определении цинка). Ре-
актив. полученный из металлического галлия. Для приготовления
основного раствора 1,000 г металлического галлия растворяют при
нагревании на водяной бане в минимальном количестве азотной
кислоты (реактив 2) и выпаривают до влажных солей. Остаток ра-
створяют при нагревании в небольшом количестве воды и количе-
ственно переносят в мерную колбу вместимостью 1000 см3, содер-
жащую 5 см3 HNO3 (реактив 2), доводят до метки бидистиллятом.
Концентрация галлия в полученном растворе равна 0,001 г/см3.
164
(Концентрация раствора при массе, равной 69,72 г, составляет
1 моль/л галлия, а при массе, равной 0,6972 г, концентрация раство-
ра — 0,01 моль/л.)
Для получения раствора концентрацией 0,01 моль/л отмеривают
697,2 см3 основного раствора, переносят в мерную колбу на 1000 см3
и доводят объем дистиллированной водой до метки.
Растворы концентрацией 0,001 моль/л готовят разбавлением ис-
ходного раствора следующим образом. В мерную колбу вместимос-
тью 500 см3 переносят пипеткой 50 см3 исходного раствора галлия,
2 см3 HNO3 (2:3), доводят до метки бидистиллятом.
Реактив, полученный из нитрата галлия. В мерную колбу на
500 см3 вносят навеску 0,1999 г нитрата галлия 8-водного, добавля-
ют 1,5 см3 HNO3 (реактив 2) и после растворения соли раствор раз-
бавляют бидистиллятом до метки. Полученный раствор имеет кон-
центрацию ионов галлия 0,01 моль/л. При использовании безвод-
ного нитрата галлия масса навески составляет 1,279 г.
Хранение растворов. Стандартные растворы концентрацией 1 г/л
хранят в стеклянных колбах с притертой пробкой в течение 3 лет.
Стандартные растворы концентрацией более 10 мкг/л для верх-
него участка диапазона измерений содержания ионов металлов до-
пустимо готовить заранее. Стандартные растворы концентрацией
меньше 10 мкг/л готовят в день измерений.
Фоновый (стандартный) раствор хранят в колбе с притертой
пробкой не более 1 мес. Раствор азотной кислоты (2:3) хранят в кол-
бе с притертой пробкой не более 3 лет. 1 М раствор KNO3 хранят в
колбе с притертой пробкой не более 1 мес. Раствор ртути концент-
рацией 10 г/л хранят в колбе с притертой пробкой не более 3 лет в
темном месте. Раствор ртути концентрацией 1 г/л готовят неболь-
шими порциями по 10—25 см3 по мере надобности.
Контрольные, фоновые растворы и растворы небольшой кон-
центрации допускается хранить в посуде из кварца, фторопласта-4
или полиэтилена высокого давления.
Щелочной раствор перманганата калия хранят в течение 1 мес в
склянке из темного стекла.
Контрольные вопросы и задания. 1. В чем заключается сущность потенциометри-
ческого метода исследования? 2. Назовите ионоселективные электроды и дайте им
характеристику. 3. Как проводят потенциометрическое титрование? 4. В чем заклю-
чается сущность вольтамперометрического метода исследования? Как устроен
электролизер?
Глава 7
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ
Принцип хроматографического анализа был впервые предложен
М. А. Цветом в 1903—1905 гг. для разделения компонентов расти-
тельных пигментов, близких по своему строению. Поскольку при
этом методе получаются раздельные полосы окрашенных веществ,
он назвал этот метод хроматографией, что значит «цветопись».
Применение этого термина не оправдано в отношении анализа бес-
цветных веществ, однако он прочно укоренился.
В современных методах исследования хроматографией называют
процесс разделения веществ, основанный на разделении компонен-
тов смеси между неподвижной (стационарной) и подвижной (мо-
бильной) фазами. В зависимости от строения разделяемые вещества
в различной степени удерживаются той или иной фазой и вследствие
этого могут быть отделены друг от друга. Хроматографический метод
используют для разделения и очистки смесей веществ.
Под действием диффузии молекулы разделяемых веществ пере-
секают поверхность раздела фаз и в зависимости от свойств удержи-
ваются той или иной фазой. При продвижении компонентов смеси
в разделяющей среде такой процесс перехода между фазами осуще-
ствляется многократно, причем каждый раз достигается небольшой
эффект разделения (или обогащения).
Хроматографические методы классифицируют по принципу
разделения (табл. 19) в зависимости от агрегатного состояния под-
вижной фазы (элюента) (жидкостная и газовая) и по форме непод-
вижного слоя (бумажная, тонкослойная, колоночная).
19. Классификация методов хроматографии по принципу разделения
Принцип разделения Подвижная фаза (ПФ) Неподвижная фаза (НФ) Форма непод- вижного слоя Хроматография
Распределение Жидкость Жидкость на бумаге Полосы или лис- Распредели-
ты хроматогра- фической бумаги тельная бу- мажная
» Жидкость на тонком слое сорбента Стеклянные пластинки или пленки Распредели- тельная тонко- слойная
» Жидкость на твер- дом носителе Колонка Жидкостно- жидкостная (ЖЖХ)
166
Продолжение
Принцип разделения Подвижная фаза (ПФ) Неподвижная фаза (НФ) Форма непод- вижного слоя Хроматография
» Жидкость на геле или пористом носи- теле Колонка Гель-хромато- графия (экс- клюзионная)
Газ(пар) Жидкость на твер- дом носителе » Газожидкост- ная (ГЖХ)
Адсорбция Жидкость Жидкость на твер- дом носителе: по- лярном или непо- лярном адсорбенте Жидкостно- адсорбцион- ная (ЖАХ): нормально- фазовая (НФХ) или обращен- но-фазовая (ОФХ)
» Жидкость на тонком слое сорбента Стеклянные пластинки или пленки Адсорбцион- ная тонко- слойная
Газ(пар) Сухой сорбент Колонка Газоадсорбци- онная (ГАХ)
Осаждение Жидкость Жидкость на бумаге Полосы или лис- ты хроматогра- фической бумаги Осадочная бу- мажная
» Жидкость на твер- дом носителе Колонка Осадочная
Ионообмен » Ионит (катионит или анионит) » Ионообменная
Лигандообмен » Лиганд Аффинная (лигандооб- менная)
В основе газовой хроматографии лежат процессы распределения
и адсорбции. Свойства подвижной фазы (газа-носителя) имеют
второстепенное значение для процесса разделения. В жидкостной
хроматографии процесс разделения в значительной степени опре-
деляется составом подвижной фазы. В качестве подвижной фазы
используется множество веществ, поэтому для каждого специаль-
ного случая можно подобрать подходящую систему разделения. Га-
зовую хроматографию применяют главным образом для аналити-
ческих целей, в то время как жидкостную хроматографию чаще ис-
пользуют для препаративных целей.
Все более широкое применение в анализе различных малолетучих
и термически нестабильных соединений (нуклеотидов, углеводов,
пестицидов, кислот, наркотических и лекарственных препаратов и
других соединений) находит метод высокоэффективной жидкостной
хроматографии (ВЭЖХ). Это — метод разделения веществ на мелко-
зернистых сорбентах с размером частиц менее 15 мкм при повышен-
ном давлении. ВЭЖХ основывается на распределительном, адсорб-
ционном, ионообменном, аффинном принципах разделения.
167
Собственно хроматографические методы являются средством
для разделения многокомпонентных смесей, но они не позволяют
установить химическую природу веществ. Для этой цели использу-
ют детекторы — приборы для распознавания компонентов анали-
зируемой смеси, выходящих из хроматографической колонки. Сис-
тема детектирования состоит из 3 элементов — детектора, усилите-
ля и регистратора. Детектор преобразует изменение состава элюен-
та в электрический сигнал, который после усиления фиксируется
регистратором.
Существующие способы детектирования и сами детекторы
можно разделить на дифференциальные и интегральные. Диффе-
ренциальные детекторы передают мгновенное значение некото-
рой характеристики, а интегральные — суммируют количество ве-
щества за определенный промежуток времени. Дифференциаль-
ные детекторы подразделяют на концентрационные и потоковые:
первые регистрируют концентрацию вещества на выходе из хро-
матографической колонки, а вторые — произведение концентра-
ции на скорость движения, то есть массовый поток вещества. Пре-
имуществом потоковых дифференциальных детекторов является
независимость выхода сигнала от расхода элюента и слабая зави-
симость от температуры.
В хроматографии используют следующие детекторы: спектрофото-
метрические, поляриметрические, рефрактометрические, электрохи-
мические, флуориметрические, масс-спектрометрические и ядерного
магнитного резонанса.
Для анализа многих классов веществ наиболее всего подходят
два типа детекторов: спектрофотометрические (регистрируют по-
глощение в УФ- и видимой области спектра) и рефрактометричес-
кие (измеряют разность показателей преломления стандартной и
исследуемой проб).
РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ
Распределительная хроматография была предложена в 1941 г.
Мартином и Синджем, далее развита Мартином и его сотрудника-
ми в «хроматографию на бумаге», которая широко применяется в
биологических, биохимических и медицинских исследованиях.
Хроматография распределения характеризуется тем, что обе
фазы жидкие и представляют собой несмешивающиеся или частич-
но смешивающиеся жидкости. В качестве неподвижной фазы слу-
жит вода хроматографической бумаги (бумажная хроматография —
БХ) или сорбента, нанесенного на подложку в виде пластинки или
пленки и покрытого жидкой фазой (тонкослойная хроматогра-
фия — ТСХ) или упакованного в колонку (колоночная хроматогра-
фия). В качестве подвижной фазы при разделении, например ами-
нокислот, применяют органические растворители, частично сме-
168
шивающиеся с водой: фенол, креозолы, смесь бутилового спирта с
уксусной кислотой и пр. Этот метод был разработан для разделения
аминокислот, но в дальнейшем оказалось, что его можно приме-
нять для разделения и определения веществ различных классов.
БУМАЖНАЯ ХРОМАТОГРАФИЯ
Бумажная хроматография подразделяется на одномерную, двух-
мерную и круговую.
Одномерная хроматография заключается в том, что на полоску
хроматографической бумаги шириной 1,5—5 см и длиной 20—70 см
наносят исследуемый раствор (0,005—0,007 см3) на расстоянии не-
скольких сантиметров от верхнего края бумаги, который погружают
в растворитель (хроматограмма нисходящая), или — исследуемый
раствор наносят на расстоянии нескольких сантиметров от нижнего
края бумаги, который погружают в ванночку с растворителем, а вер-
хний конец закрепляют (хроматограмма восходящая).
При получении хроматограммы (рис. 45) лист бумаги подвеши-
вают в камере, погрузив верхний край в кювету с элюентом. Под
действием силы тяжести и капиллярных сил растворитель продви-
гается сверху вниз и по достижении края листа стекает на дно каме-
ры. Чтобы поток был равномерным, нижний край листа нарезают
зубцами.
Восходящую хроматограмму (рис. 46) вследствие слабой меха-
нической прочности бумаги подвешивают на специальных держа-
телях. Растворитель поднимается до верхнего края листа бумаги за
счет капиллярных сил. При этом все компоненты анализируемой
смеси остаются в пределах листа. Это особенно важно в том случае,
когда разделяют вещества с неизвестной подвижностью. Тогда по
полученной хроматограмме можно определить коэффициент рас-
пределения R(f) всех компонентов смеси. К недостаткам метода от-
носится низкое разрешение малоподвижных компонентов. Про-
блему в этом случае решают, применяя метод нисходящей хрома-
тографии в режиме протока, когда процесс продолжается сколь
Рнс. 45. Камера для нисходящей хрома-
тографии:
а — общий вид камеры; б — расположение
листа бумаги; /—кювета для элюента; 2~
стеклянная палочка для фиксации листа бу-
маги в лодочке; 3 — антисифонная палочка;
4 — подставка; 5 — лист бумаги; 6 — камера;
7 — зубцы на бумаге
169
Рис. 46. Выполнение восходящей хрома-
тографии:
а, б— способы фиксации бумаги в держателе;
в, г — способы размещения бумаги в камере
угодно долго. В результате веще-
ства даже с очень низкими R(f)
имеют достаточный для хороше-
го разделения пробег. Для стан-
дартизации результатов одновре-
менно хроматографируют конт-
рольную смесь или окрашенные
вещества с известными R(f).
Как только растворитель до-
стигает намеченного рубежа,
лист извлекают из камеры, от-
мечают положение фронта растворителя и высушивают. Поскольку
большинство веществ лишено окраски, их положение на хроматог-
рамме выявляют подходящим способом. Например, вещества, не-
сущие радиоактивную метку, выявляют при помощи счетчиков им-
пульсов; вещества, имеющие собственную флуоресценцию, выяв-
ляют при облучении УФ-светом (365 нм). Остальные вещества
обычно обнаруживают по окрашенным продуктам, которые образу-
ются под действием специфических реагентов. Так, аминокислоты
и полипептиды образуют с нингидрином хромофор, имеющий фи-
олетовую окраску.
Положение пятна, получаемого в результате распределения ис-
пытуемого вещества, выражают коэффициентом распределения.
Он представляет собой отношение скорости движения (см) испыту-
емого вещества к скорости движения (см) фронта растворителя на
полосе бумаги за определенный промежуток времени.
Например, испытуемое вещество (аминокислота) продвинулось
от места нанесения на 20 см (место расположения пятна), за этот же
промежуток времени фронт растворителя прошел 30 см, следова-
тельно: R(f) = 20/30 = 0,66.
Применяя для разделения вещества на бумаге один подвижный
растворитель, можно получить одномерную хроматограмму рас-
пределения. На такой хроматограмме вещества распределяются ли-
нейно, в виде цепочки из цветных пятен. Одномерная хроматограм-
ма чрезвычайно удобна для идентификации одной или нескольких
аминокислот, для проверки чистоты вещества или для исследова-
ния состава несложных смесей. Для сложной смеси (например гид-
ролизат белков) в результате распределения не удается получить
170
раздельные пятна для каждой аминокислоты, так как не найден ра-
створитель, в котором все аминокислоты имели бы достаточно раз-
личающиеся коэффициенты распределения. В связи с этим пятна,
соответствующие нескольким аминокислотам, на одномерной хро-
матограмме могут перекрываться.
Этого недостатка нет в двухмерной хроматографии, полученной
путем последовательного пропускания через хроматографическую
бумагу, содержащую разделяемые вещества двух различных раство-
рителей в двух взаимно перпендикулярных направлениях.
Для выполнения двухмерной хроматограммы берут лист хрома-
тографической бумаги размером 50 х 50 см или более, в один из уг-
лов его на расстоянии 5 см от каждого края наносят каплю испытуе-
мой смеси (около 20 см3). Затем верхний край бумаги помещают в
первый подвижный растворитель.
Полученную линейную хроматограмму просушивают и, не про-
являя, помещают в ванночку со вторым растворителем тем концом
бумаги, на котором расположена линейная непроявленная хрома-
тограмма. При продвижении второго растворителя разделяются ве-
щества с одним и тем же коэффициентом распределения на одно-
мерной хроматограмме. После того как фронт растворителя про-
двинется почти через весь лист, не достигнув края примерно на
5 см, хроматограмму вынимают, просушивают и проявляют.
При выполнении двухмерной хроматограммы необходимо подо-
брать такие два растворителя, которые давали бы наиболее сильно
различающиеся коэффициенты распределения. Для бумажной хро-
матографии пригодны также круглые фильтры, на которых раство-
ритель продвигается от середины к краям.
От середины круглого фильтра вырезают узкую полосу («язык»),
загибают ее вниз и наносят на середину фильтра смесь веществ.
Подготовленный таким образом фильтр кладут на края плоской
чашки, наполненной растворителем, при этом «язык» погружают в
жидкость (рис. 47). Растворитель засасывается вверх, и на круглом
фильтре образуются концентрические круги отдельных веществ.
Для повышения скорости продвижения растворителя круглый
фильтр диаметром около 30 мм кладут в эксикатор. Крышку закры-
вают пробкой с капиллярной трубкой —
пипеткой. Трубка заканчивается на не-
сколько миллиметров выше бумаги и
дает примерно 10—12 капель в минуту
Рис. 47. Прибор для круговой хроматографии:
1 — фильтр; 2— стеклянные чашки
Растворитель
171
при использовании бумаги средней мягкости. На середину круглого
фильтра наносят исследуемый раствор. Смесь жидкости подается
каплями с постоянной скоростью на бумагу.
Если хотят попутно сравнить вещества, то дают испытуемый ра-
створ не прямо на середину, а на маленький концентрический круг
вблизи середины. Отдельные смеси веществ находятся после завер-
шения распределения на отдельных круглых секторах.
Круговой метод обладает рядом преимуществ по сравнению с
другими методами бумажной хроматографии:
хроматографирование происходит достаточно быстро. Круговая
хроматограмма диаметром 20 см готова приблизительно за 2 ч, диа-
метром 32 см — за 4—5 ч;
достигается высокий эффект разделения. Резкость разделения
значительно выше, чем при восходящем или нисходящем методах;
определение различных веществ существенно облегчается.
Можно работать с большим числом проявителей, используя отдель-
ные секторы, вырезанные из готовых хроматограмм;
можно работать с относительно большими объемами исследуе-
мого раствора, так как вещества разделяются в процессе хроматог-
рафирования на большие круги;
вещества, распределяемые на круглых хроматограммах, можно
определять количественно.
Круговой метод БХ значительно обогащает и расширяет возмож-
ности применения хроматографического анализа на бумаге. Он мо-
жет использоваться в комбинации с колоночной хроматографией.
Для количественного определения веществ применяют фото-
метрирование на денситометре или спектрофотометре СФ-4, име-
ющем специальную приставку (адаптер); измерение площади пя-
тен; элюирование.
ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
При тонкослойной хроматографии компоненты исследуемого
раствора перемещаются в тонком слое сорбента, нанесенного на
стеклянную пластинку и покрытого жидкой фазой. Схема прибора
для подготовки пластин к тонкослойной хроматографии приведена
на рис. 48.
Он состоит из металлического стола или металлической доски с
ровной поверхностью и бортиками, не позволяющими сдвигаться
стеклянным пластинкам, которые кладут на стол или доску. Высота
бортика не должна превышать толщину стеклянных пластин. Пять
стеклянных пластин (точно 20 х 20 см) толщиной не менее 4 мм
должны плотно входить между бортиками стола. Их укладывают
последовательно на поверхность стола или доски без промежутков.
Перед нанесением сорбента стеклянные пластины тщательно
моют, обезжиривают и сушат.
172
Рнс. 48. Прибор для тонкослойной хро-
матографии:
а — поперечный разрез станка при наложе-
нии тонкого слоя; б— положение стеклян-
ных пластинок и станка; 1 — стеклянная пла-
стина; 2 — бортик; 3 — станок; 4 — направля-
ющая; 5 — ручка
з
б
1 2
пластинка. В полости бруска раз-
Для нанесения тонкого и
равномерного по толщине слоя
сорбента используют специ-
альный станок из латунного
хромированного квадратного
бруска, полого внутри. С пра-
вой стороны к торцу бруска
привинчивается направляющая
мещается притертая по наружной поверхности трубка, заглушенная
по краям. Заглушки трубки, выступающие на 5—8 мм за обрез брус-
ка, имеют бортики, которые фиксируют трубчатый вкладыш ци-
линдрической полости бруска. Вкладыш легко проворачивается на
180° рукояткой, укрепленной сбоку выступа вкладыша. Цилиндр
вкладыша имеет окно, соответствующее окну в бруске прибора.
Нижняя поверхность бруска, которая при нанесении слоя сорбента
соприкасается со стеклом, тщательно отполирована.
Образцы и растворители необходимо подготовить. Жирные кис-
лоты глицеридов без предварительного омыления переводят в ме-
тиловые эфиры метанолом в диэтиловом эфире в присутствии гид-
роксида калия.
Для разделения нейтральных липидов и их гидролизатов реко-
мендуют различные растворители и смеси (петролейный эфир (или
гексан) + бензол; гексан + тетралин; петролейный эфир + диэтило-
вый эфир и др.).
В качестве сорбентов при тонкослойной хроматографии исполь-
зуют силикагель в смеси с гипсом и оксидом алюминия, оксид алю-
миния, кизельгур (для разделения триглицеридов, кетокислот,
жирных кислот применяют кизельгур, который пропитывают высо-
кокипящими фракциями нефти).
Сорбент (25 г) насыпают в сухую фарфоровую ступку, подлива-
ют 35 см3 воды и растирают до получения однородной массы. До-
бавляют еще 15 см3 воды, непрерывно помешивая кашицу до смета-
нообразного состояния. Сорбент наносят на приготовленные и уло-
женные на столе стеклянные пластины специальным станком, в ко-
торый сорбент наливают из ступки через верхнее окно. После
равномерного распределения сорбента во внутреннем цилиндре его
сразу поворачивают за ручку на 180 °C. Окно цилиндра совпадает с
173
нижним окном бруска станка, который берут за оба конца и равно-
мерно, не прижимая, проводят по всем пластинам.
Слой сорбента сушат на воздухе под вентилятором в течение
10 мин, затем в сушильном шкафу при 10 °C в течение 30 мин. На
слой сорбента на расстоянии 15 мм от края пластины карандашом
или иглой наносят точки старта. В 10 см от старта отмечают линию
фронта. Исследуемое вещество наносят (за один прием 5 мм3) пи-
петкой вместимостью 10 мм3 с делениями 1 мм3. Лучше в одну точку
наносить образец несколько раз.
Затем на расстоянии 1 см от края наносят эталонную смесь из
красителей (масляный желтый, судан красный). Для последующего
разделения пластины устанавливают в гнезда стеклянных, гермети-
чески закрывающихся камер. На дне камер имеются кюветы с ра-
створителями, в которые погружают пластину на глубину 5 мм. Раз-
деление проводят при 18—20 °C в темном помещении. Хроматог-
раммы проявляют, разбрызгивая из тонкой форсунки специальные
реактивы. Затем поступают, как при проведении распределитель-
ной хроматографии на бумаге.
ЖИДКОСТНО-ЖИДКОСТНАЯ
ХРОМАТОГРАФИЯ
В жидкостно-жидкостной хроматографии (ЖЖХ) неподвижную
фазу помещают в колонку, затем вносят в нее анализируемую смесь
и элюируют подходящим растворителем. При продвижении по ко-
лонке компоненты смеси удерживаются сорбентом в соответствии с
их физико-химическими свойствами и, следовательно, мигрируют
с разной скоростью. Из колонки разделяемые вещества смеси выхо-
дят в определенной последовательности и могут быть собраны в
виде отдельных фракций.
При использовании колонки конечный результат зависит не
только от того, насколько принцип разделения (распределение, ад-
сорбция, ионообмен) соответствуют свойствам анализируемых ве-
ществ, но и от других факторов: свойств системы сорбент—элюент,
условий элюирования (скорость потока, температура, вязкость
элюента), конструкции и размеров колонки, нагрузки колонки (ко-
личество пробы), качества ее упаковки, размеров частиц сорбента и
исследуемых веществ, качества подготовки пробы и др.
В ЖЖХ низкого давления компоненты смеси разделяют на хро-
матографической колонке при нормальном (гидростатическом)
или несколько повышенном давлении.
Если в качестве неподвижной фазы используют мягкие сорбен-
ты (сефадексы, биогели, агарозы, полистирольные гели), то хрома-
тографию проводят при атмосферном давлении. Этот вид хроматог-
рафии применяют при переработке больших объемов жидкости при
условии, что не ставится задача достижения высокого разрежения,
свойственного ВЭЖХ.
174
ВЭЖХ принципиально мало отличается от классической коло-
ночной хроматографии. Однако вследствие небольших размеров
частиц сорбента и их однородности разделяющая способность
ВЭЖХ существенно выше. Из-за высокого рабочего давления (до
40 МПа) приборы для ВЭЖХ отличаются от приборов для класси-
ческой колоночной хроматографии высокими качеством и стоимо-
стью.
К растворителям для ВЭЖХ также предъявляют следующие тре-
бования: высокое качество очистки; отсутствие взвешенных час-
тиц; обеспечение максимальной селективности разделения; мини-
мальная вязкость; отсутствие комплексообразующих ионов, спо-
собных вызвать коррозию металлических частей насоса.
ГЕЛЬ-ХРОМАТОГРАФИЯ
Гель-хроматография (молекулярно-ситовая хроматография) —
метод, основанный на различной способности молекул разного раз-
мера проникать в поры нейтрального геля, который служит непод-
вижной фазой.
Схематично принцип работы молекулярных сит показан на
рис. 49. В качестве неподвижной фазы служит пористый гель с по-
рами определенного диаметра. Крупные молекулы не диффундиру-
в
000 0000 0 0 0 0000 ООО 0 0 0 0 000 0 000 0 00 оооо ООО 0 0 0 000 00 00 000 0 000 000 0 00 0 000 оооо ооо 0 0 0 000 оооо 000 оооо 000 000 0 0 0 0 ООО
ооо
ООО о
ООО
0000
000
000 0
,0 00
•.•0.0.0V
О 0 0 0
0 0 0
0 0 0
Рис. 49. Схема разделения веществ методом гель-хроматографии
175
ют в поры гранул и элюируются быстрее, молекулы небольшого
размера удерживаются в порах гранул и элюируются позднее.
Общий объем колонки V складывается из объема растворителя
между гранулами геля V$, объема растворителя в гранулах геля
(объема пор) Vn и объема матрицы VM: V=V0+ Vn+ VM.
Молекулы, не проникающие в гранулы геля, элюируются в объе-
ме Vo. Молекулы, свободно проникающие в гранулы, элюируются в
объеме Vo + Vn. Молекулы, для которых доступна лишь часть пор
гранул, элюируются в объеме Vq< Vx< Vo+ Vp. Величина VK не пол-
ностью отражает свойства того или иного вещества, так как в значи-
тельной степени зависит от общего объема V. По аналогии с распре-
делительной хроматографией поведение вещества на колонке мож-
но охарактеризовать коэффициентом распределения Кр, соответ-
ствующим доле неподвижной жидкой фазы, в которой находится
данное вещество: Кр = (Vx - Ио)/ Vn.
На практике этим параметром пользоваться сложно, поскольку
нелегко определить точное значение Vn. Легче рассчитать величину
V— Vq. Эта разность превышает Ип на объем матрицы VM и вместе с
тем пропорциональна общему объему колонки V. Таким образом
можно ввести константу Кх, также соответствующую доле непод-
вижной фазы, вкоторой находится данное вещество: Кх - (Vx — Vq)/
(V- Ко).
Хотя Кк неидентична коэффициенту распределения, с ее помо-
щью можно описать результаты эксперимента независимо от раз-
меров колонки и, таким образом, сопоставить результаты многих
опытов. Результаты экспериментов можно нормировать при помо-
щи величин Их/Ри V-JVq. Однако в отличие от Кх эти величины за-
висят от свойств геля.
Хроматографические свойства геля определяются природой
матрицы и прежде всего пористостью ее структуры, в то время как
химическое строение каркаса имеет второстепенное значение. В за-
висимости от способности набухать в воде или в органических ра-
створителях материалы для гель-хроматографии подразделяются на
гидрофильные и органофильные (гидрофобные).
Декстрановые гели, или сефадексы, представляют собой полиса-
харидные цепи, сшитые эпихлоргидрином. Благодаря наличию
множества гидроксильных групп такая матрица хорошо удерживает
воду. Степень набухания геля уменьшается по мере возрастания
степени сшитости. По мере уменьшения размеров пор в зону элюи-
рования попадают вещества все с меньшей молекулярной массой.
Чем меньше степень сшитости, тем мягче гель и, следовательно, тем
меньше рабочее давление в колонках, а значит, и скорость подачи
элюента.
Полиакриламидные гели получают сополимеризацией акрила-
мида и сшивающего агента N, N' -метилен-бис-акриламида. В за-
висимости от условий полимеризации (соотношения мономеров,
концентрации) получают сферические гранулы полимера с различ-
ие
ними диаметрами пор. При высокой концентрации сшивающего
агента образуется высокопористый гель с высокой механической
Прочностью (макропористый гель).
Агарозные гели (сефароза и биогель) получают из линейного
полисахарида — агарозы, построенного из остатков молекул D-га-
лактозы и 3,6-ангидро-Ь-галактозы. Гель с очень большими пора-
ми образуется самопроизвольно при охлаждении горячего раство-
ра агарозы. При этом происходит продольная агрегация цепей ага-
розы, вследствие чего образуется гель с очень большими порами.
Так как структура геля формируется главным образом за счет водо-
родных связей, область применения геля ограничена: при повы-
шенной температуре или воздействии реагентов, разрушающих
водородные связи (например, мочевины), структура геля наруша-
ется. Другим недостатком этих гелей является чувствительность к
микрооргани змам.
Сравнительно новым является ультрагель, основой которого
служит каркас из полиакриламида, а промежуточное пространство
заполнено гелем агарозы. Благодаря небольшим размерам гранул и
их высокой механической прочности эти материалы можно ис-
пользовать при более высокой скорости подачи элемента, чем при-
нято для сефадексов и биогелей.
Сефакрил является сополимером декстрана. Гранулы геля обла-
дают высокой механической прочностью, химически устойчивы
(стабильны в области pH 3—11, выдерживают стерилизацию в авто-
клаве при pH 7 и температуре до 120 °C); вследствие жесткой струк-
туры устойчивы к действию органических растворителей. При пе-
реходе от воды к полярным органическим растворителям объем
геля изменяется незначительно. Перевод сефакрила из водной сре-
ды в органический растворитель и обратно показан на схеме 2. Этот
процесс осуществляется одинаково для всех хроматографических
материалов.
Гидрогель — сильносшитый полимер, хорошо удерживающий
воду, может быть использован при давлении до 25 МПа. В кислой
или щелочной средах на гидрогеле возможна адсорбция заряжен-
ных веществ.
Схема 2. Перевод гелей из водного растворителя в органический и обратно
177
Производные декстрана — сефадекс LH-20 и сефадекс LH-60 -т
сефадексы G-25 и G-50, у которых гидроксильные группы этерифи-
цированы (образуют простые эфиры) гидроксипропильной груп-
пой. Вследствие этого гели обладают как гидрофильными, так и ли-
пофильными свойствами. Из смеси полярных и неполярных ра-
створителей (хлороформ—метанол, бутанол—вода) гель избира-
тельно впитывает полярные компоненты, т. е. возникает
положение, характерное для распределительной хроматографии.
Поэтому характер взаимодействия разделяемых веществ с сорбен-
том в значительной степени зависит от состава элюента.
Полистирольные гели (биобедс) являются сополимерами стирола
и дивинилбензола; диаметр пор геля в значительной степени зависит
от природы растворителя. Полистирольные гели сильно набухают в
неполярных растворителях (толуол, четыреххлористый углерод), ко-
торые по этой причине часто используют в качестве элюентов. По-
скольку гель легко деформируется, удовлетворительных результатов
можно достичь лишь при небольших скоростях потока.
Стирагель — сильносшитый макропористый полистирольный
гель, вследствие высокой механической прочности его применяют
при работе в режиме высокоэффективной жидкостной хроматогра-
фии. В качестве растворителей рекомендуется использовать тетра-
гидрофуран, диметилформамид, дихлорметан и ароматические уг-
леводороды.
Пористые стекла, стойкие ко всем типам растворителей, приме-
няют в жидкостной хроматографии при высоком давлении. Адсорб-
ционные свойства, характерные для обыкновенных кремнеземов, у
пористых стекол незначительны и могут быть сведены к минимуму
при помощи дополнительной обработки.
Гель-хроматографию используют для фракционирования и вы-
деления белков, жиров, углеводов, нуклеиновых кислот, полиме-
ров, субклеточных частиц, вирусов, для обессоливания образцов,
отделения низкомолекулярных радиоактивных веществ, для опре-
деления мольных масс биополимеров.
ГАЗОВАЯ ХРОМАТОГРАФИЯ
Газовая хроматография (ГХ) — метод разделения летучих ве-
ществ: газов (при нормальной температуре) или паров (при повы-
шенной температуре). В качестве неподвижной фазы в ГХ исполь-
зуют твердые материалы (насадочные или набивные колонки),
твердые материалы, покрытые слоем жидкости, или же капилляры
с нанесенным на внутреннюю поверхность слоем жидкости (капил-
лярные колонки). В качестве подвижной фазы применяют газ-но-
ситель, переносящий разделяемые вещества через колонку. Анали-
зируемая смесь разделяется за счет различного времени удержива-
ния веществ в неподвижной фазе.
178
Между жидкостной и газовой хроматографией не существует
принципиальной разницы. ГХ отличается лишь свойствами под-
вижной фазы: высокой скоростью диффузии газа-носителя и его
свойством сжиматься. Упрощенное уравнение Ван-Деемтера для
описания высоты теоретической тарелки сохраняет свое значение и
в газовой хроматографии:
Н = А +B/n+ Cv,
где Я—высота, эквивалентная теоретической тарелке, Л —параметр, учитываю-
щий турбулентную диффузию; В— параметр, учитывающий продольную диффу-
зию; v — линейная скорость газа-носителя в колонке; С— параметр, учитывающий
массопередачу.
Параметр А учитывает турбулентную диффузию, возникающую
за счет многообразия путей, по которым молекулы разделяемых ве-
ществ проходят между частицами сорбента. При расчете капилляр-
ных колонок, где удерживание происходит только в пленке жидко-
сти на стенке колонки, этим слагаемым пренебрегают. Положение
минимума на графике Ван-Деемтера, а следовательно, максималь-
ная разделяющая способность колонки в ГХ зависят от следующих
параметров: количества неподвижной фазы (качество упаковки,
толщина слоя), размера частиц или диапазона размеров частиц ма-
териала-носителя, размеров колонки, вязкости газа-носителя
(а следовательно, и температуры колонки).
В зависимости от состояния фаз различают газотвердофазную
или газоадсорбционную хроматографию (ГАХ) и газожидкост-
ную (распределительную) хроматографию (ГЖХ). Тип непод-
вижной фазы, механизм разделения и возможности обоих мето-
дов сопоставлены в табл. 20. При количественной оценке резуль-
татов разделения методом ГХ большое значение имеет форма
пика. Симметричность пика зависит от растворимости анализи-
руемых веществ в жидкой (неподвижной) фазе. В свою очередь
растворимость определяется зависимостью давления паров ра-
створенного вещества от его концентрации в жидкой фазе. При
постоянной температуре эта зависимость имеет вид изотермы.
Если давление пара вещества, растворенного в жидкой фазе, рас-
тет с повышением температуры линейно, будет линейная изотер-
ма. В этом случае на графике элюирования получают симметрич-
ный пик. Пик обычно симметричен, если разделяемые вещества и
жидкости, применяемые в качестве неподвижной фазы, принад-
лежат к одному классу (пример идеальной смешиваемости). От-
клонения от случая идеальной смешиваемости приводят к искрив-
лению изотермы и образованию несимметричных пиков с размы-
той восходящей («фронт») или нисходящей («хвост») ветвью.
Этот эффект становится особенно заметным при увеличении на-
грузки на колонку.
179
20. Варианты газовой хроматографии
Метод Неподвижная фаза Принцип разделения Область применения метода
Газожидкостная
(распределитель-
ная) хроматогра-
фия (ГЖХ)
Жидкая фаза, нане-
сенная на инертный
материал-носитель
Газоадсорбционная Адсорбент
хроматография
Распределение _ Анализ газов и паров
жидкостей; методи-
ческие возможности
выше, чем для ГАХ
Адсорбция Анализ газов и паров
жидкостей
Время удерживания зависит от вероятности попадания молекул
вещества в подвижную фазу. При этом компоненты с высоким
давлением паров и соответственно низкой растворимостью в не-
подвижной фазе удерживаются слабее. Напротив, вещества с низ-
жим давлением пара и высокой растворимостью элюируются по-
зднее.
Если в процессе разделения температуру повышают, то говорят о
газовой хроматографии при программируемой температуре.
В конструктивном отношении приборы для ГХ проще по срав-
нению с приборами для ВЭЖХ, так как вместо дорогостоящего на-
соса высокого давления в ГХ применяют обычный баллон с газом-
носителем. Схема прибора для ГХ приведена на рис. 50.
В ГХ применяют три типа колонок: насадочные, изготовленные
из стеклянных или металлических трубок (нержавеющая сталь),
U-образной формы.
Рис. 50. Схема газового хроматографа:
1 — баллон с газом-носителем; 2 — игольчатый вентиль баллона; 3 — регулятор потока газа-но-
сителя (дроссель); 4—дозирующее устройство (блок ввода пробы); 5 — колонка; 6 — термостат;
7—ротаметр; 8 — детектор (пламенно-ионизационный); 9, 10 — система для подачи газов в де-
тектор (водород и воздух в случае ПИД); 11— электронный блок обработки сигнала (интегра-
тор); 12— самописец
180
В отличие от жидкостной и в газовой хроматографии широкие
колонки заполнять сложнее, чем узкие. Поэтому необходимо обя-
зательно оптимизировать условия эксперимента (т. е. добиваться
максимальной производительности процесса) за счет выбора мате-
риала-носителя, жидкой неподвижной фазы, условий эксплуата-
ции колонки. Необходимо, чтобы неподвижная фаза прочно удер-
живалась на материале-носителе при повышенной температуре.
Колонки, теряющие неподвижную фазу (явление «истощения» ко-
лонки), непригодны для препаративной ГХ, поскольку целевое ве-
щество будет содержать следы жидкой фазы. Разделение стремятся
проводить при возможно более низкой температуре (ниже точки
кипения жидкой фазы).
Для препаративной ГХ более всего подходят пламенно-иониза-
ционный детектор (ПИД) и детектор по теплопроводности (ДТП),
причем ПИД предпочтительнее. Поскольку детектирование в ПИД
основано на сжигании элюата, в ячейку направляют, только часть
потока. Когда детектор начинает показывать наличие в элюате це-
левого вещества, основной поток направляют в охлаждаемый сбор-
ник на конденсацию. В случае ДТП деление потока также обяза-
тельно, так как сигнал детектора нелинеен при высокой концентра-
ции вещества.
Небольшие количества вещества вводят вручную шприцем. Для
ввода больших количеств веществ используют автоматизированные
системы ввода пробы.
Газовую хроматографию применяют для получения чистых ве-
ществ, выделения веществ (жирных и органических кислот, карбо-
нильных и хлорорганических соединений), концентрация которых
в смеси мала, выделения метаболитов в биохимии.
ОПРЕДЕЛЕНИЕ ФРАКЦИЙ МОЛОЧНЫХ БЕЛКОВ МЕТОДОМ
ГЕЛЬ-ХРОМАТОГРАФИИ (ПО К. БУТКУСУ И В. БУТКЕНЕ)
Метод основан на разделении белков, элюируемых через гель
декстрана (сефадекс).
Сухой сефадекс суспензируют в дистиллированной воде или в
соответствующем буфере в течение 48—72 ч. Мельчайшие частицы
геля декантируют. Колонку укрепляют в строго вертикальном по-
ложении. В основание колонки вставляют перфорированный диск
из плексигласа пористостью № 1, на него насыпают песок слоем
0,5 см и на песок кладут диск из фильтровальной бумаги. Над ко-
лонкой укрепляют стеклянную воронку, помещая в нее стеклянную
палочку таким образом, чтобы ее кончик частично закрывал выход
из воронки, и заполняют ее разведенным раствором сефадекса.
Стеклянная палочка должна настолько прикрывать выход из во-
ронки, чтобы суспензия сефадекса могла просачиваться в колонку
тонкой струйкой. После того как слой сефадекса осядет в колонке
181
на высоту 2—5 см, открывают кран колонки для медленного истече-
ния жидкости, колонку продолжают наполнять сефадексом. Крите-
рием правильного наполнения является наличие совершенно гори-
зонтальной поверхности сефадекса.
После того как все частицы осели, на гель помещают диск филь-
Йювальной бумаги. Колонку промывают буферным раствором,
ри работе с колонками следует помнить, что нельзя допускать
подсыхания верхнего слоя геля.
При аккуратной работе колонка с сефадексом может быть ис-
пользована многократно. Иногда со временем уменьшается ско-
рость фильтрования жидкости через колонку, что может быть выз-
вано значительным измельчением части геля и накоплением заг-
рязнений. Следует заново наполнить колонку, обмыв (путем декан-
тации) бывший в употреблении препарат сефадекса от мельчайших
частиц и загрязнений.
После пропуска через колонку одного белка ее промывают бу-
ферным раствором (2—3 объема колонки), после чего она может
быть использована для другого опыта.
При помощи перестального насоса в колонку вводят 20 см3 ис-
следуемого (обезжиренного) молока. Элюация проводится ацетат-
ным буфером (pH 6,25) по 60 см3/ч. Компоненты белка в элюате по-
стоянно определяются при длине волны 280 м. Результаты регист-
рируются самописцем.
Отдельные компоненты на графике отделяют перпендикуляра-
ми и измеряют площади под пиками. По относительной массе оп-
ределяют количество отдельных компонентов в процентах.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ СВОБОДНЫХ ЛЕТУЧИХ
ЖИРНЫХ КИСЛОТ МЕТОДОМ ГАЗОЖИДКОСТНОЙ ХРОМАТОГРАФИИ
(ПО С. УРБЕНЕ И В. ЗАВЕЦКЕНЕ)
Метод основан на выделении свободных летучих жирных кислот
(СЛЖК) при дистилляции водным паром, разделении их на хрома-
тографе.
Для анализа берут 75 см3 молока, 50 см3 кислых молочных про-
дуктов, 75 г масла, 25 г созревшего сыра, до 50 г сыра из-под пресса.
Нужное количество исследуемого продукта переносят в термо-
стойкую колбу, наливают до 150 см3 дистиллированной воды, до-
бавляют 0,5—1 см3 концентрированной ^ЗОди 1 см3 стандартного
раствора энантовой кислоты (реактив 1). Полученный раствор пе-
регоняют с водяным паром. Процесс кипения регулируют так, что-
бы уровень жидкости в колбе сохранялся постоянным. Следует на-
брать 250 см3 дистиллята. Время дистилляции — около 55 мин.
Дистиллят нейтрализуют 0,1 н. раствором гидроксида натрия для
образования солей летучих кислот. Общее количество СЛЖК можно
выразить объемом (см3) 0,1 н. гидроксида натрия, который необхо-
дим для нейтрализации кислот, выделенных из 100 г продукта.
182
Дистиллят испаряют в водяной бане при температуре 60 °C. Ос-
тавшееся минимальное количество дистиллята из колбы объемом
250 см3 переносят в бутылочку от пенициллина и подвергают пол-
ному испарению.
Для хроматографического разделения соли необходимо перевес-
ти в кислотное состояние. Для этого к соли добавляют по 2 капли
ортофосфорной кислоты и ацетона (растворитель).
Анализ проводят на газовом хроматографе «Хром-5» с пламен-
но-ионизационным детектором, на колонке из нержавеющей ста-
ли. В качестве твердого носителя используют хроматон NAW-
HMDS 0,200—0,250 мм. Жидкая фаза — 10 % диэтиленгликольади-
пинат-Lac — 22—446 с добавлением 2 % ортофосфорной кислоты
(d= 1,87). 1—10 см3 готового раствора свободных жирных кислот
(реактив 2) вводят в хроматограф с помощью микрошприца.
Условия хроматографирования: температура колонки 100—
150 °C, инжектора 170 °C; давление азота 0,1 МПа; скорость водоро-
да 30 см3/мин, воздуха 500 см3/мин.
Режим программирования температуры — I: 100—124 °C
(4°С/мин);П: 124— 150°С(6°С/мин);Ш: 150 °C const 4 мин; охлаж-
дение — 2 мин.
Для установления абсолютного количества вещества в пробе
удобно пользоваться методом внутреннего стандарта. Метод не тре-
бует точной дозировки и основан на сравнении площадей пиков ве-
щества — метки (в нашем случае — энантовая кислота) и определя-
емого компонента. В приготовленный стандартный раствор добав-
ляют точно отмеренное количество раствора энантовой кислоты.
По хроматограммам стандартного раствора с энантовой кисло-
той определяют градуировочную константу Л" для каждой кислоты
по формуле
где — площадь пика энанта, мм2; 5Х — площадь пика кислоты, мм2; Иэ — концен-
трация энанта, мг%; Их — концентрация кислоты, мг%.
В анализируемую смесь необходимо добавить такое же количе-
ство стандартного раствора. Зная градуировочные константы для
каждой кислоты, можно подсчитать массу любой кислоты.
Приготовление реактивов. Реактив 1 (стандартныйраствор энан-
товой кислоты). Взвешивают около 100 см3 энантовой кислоты, ра-
створяют в ацетоне и разбавляют в мерной колбе до 100 см3 дистил-
лированной водой.
Реактив 2 (стандартныйраствор жирных кислот)’. Состав и кон-
центрация этого раствора должны быть близкими составу СЛЖК
исследуемого продукта. Взвешивают нужное количество кислот,
добавляют небольшое количество ацетона для полного растворения
кислот и доливают дистиллированной водой до 100 см3.
183
ОПРЕДЕЛЕНИЕ ОТДЕЛЬНЫХ ОРГАНИЧЕСКИХ КИСЛОТ
МЕТОДОМ ГАЗОЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Метод основан на переводе свободных органических кислот
(типа яблочной, молочной, щавелевой, винной, лимонной) и их со-
лей в летучие этиловые или метиловые эфиры с последующим опре-
делением их содержания методом газожидкостной хроматографии.
Выполнение работы. Обезжиренные молочные продукты в коли-
честве 2—10 см3 вносят в грушевидную колбу со шлифом, добавля-
ют адипиновую кислоту (стандарт) в количестве 2—5 мг, взвешен-
ную на аналитических весах с точностью до 0,0001 г на полиэтиле-
новой пластине, и упаривают на роторном испарителе при 60 °C до-
суха (при затруднении в колбу добавляют 1 см3 бензола или
ацетона).
Молоко, кисломолочные напитки в количестве 5—10 см3 вносят
в центрифужную пробирку, добавляют адипиновую кислоту (стан-
дарт) в количестве 2—5 мг, взвешенную на аналитических весах с
точностью до 0,0001 г на полиэтиленовой пластине, проводят трех-
кратную экстракцию по 10 см3 гексана с последующим центрифу-
гированием в течение 10 мин при 4000 об/мин и отделением гекса-
нового слоя. Нижнюю часть из делительной воронки переносят в
грушевидную колбу и упаривают досуха на роторном испарителе
при 60 °C.
Сухие кисломолочные продукты, сыр, творог в количестве 0,5—
1,0 г с точностью до 0,0001 г вносят в центрифужную пробирку, до-
бавляют адипиновую кислоту (стандарт) в количестве 2—5 мг, взве-
шенную на аналитических весах с точностью до 0,0001 г на поли-
этиленовой пластине, проводят трехкратную экстракцию при
20 см3 гексана с последующим центрифугированием в течение
10 мин при 4000 об/мин и отделением гексанового слоя. К осадку
приливают 50 см3 80%-ного водного раствора этанола, выдержива-
ют в течение 10—12 ч, отделяют экстракт, переносят в круглодон-
ную колбу и упаривают на роторном испарителе при 60 °C досуха.
К сухому остатку, полученному вышеописанными методами,
приливают 5,0 см3 этерифицирующей смеси, выдерживают при
комнатной температуре с обратным холодильником не менее 15 ч,
приливают 1 см3 четыреххлористого углерода или хлороформа,
встряхивают в течение 30 с, добавляют 10 см3 дистиллированной
воды, встряхивают в течение 30 с и выдерживают до полного рас-
слоения.
Подготовка хроматографической колонки. Навеску инертного но-
сителя в количестве 50 г взвешивают на технических весах с точнос-
тью до 0,01 г в круглодонной колбе, приливают 200 см3 метанола
(этанола), в котором растворено 0,5 см3 о-фосфорной кислоты,
тщательно перемешивают, выдерживают в течение 2—3 ч и спирт
отгоняют на роторном испарителе при температуре 80 °C до су-
хого состояния сорбента. К высушенному сорбенту с нанесенной
184
о-фосфорной кислотой приливают 200 см3 хлороформа, в котором
предварительно растворяют 7,5 гдиэтиленгликольсукцината, и вы-
сушивают на роторном испарителе до исчезновения запаха хлоро-
форма. Приготовленным сорбентом заполняют 2 стеклянные наса-
дочные колонки длиной 2—3 м и внутренним диаметром 3 мм. Ко-
лонки подсоединяют к хроматографу и проводят кондиционирова-
ние при постоянном давлении небольшим потоком газа-носителя
(гелий, азот или водород) 2 ч при 80 °C, 2 ч при 120 °C, 4 ч при
180 °C, 8 ч при 200 °C.
Разделение метиловых (этиловых) эфиров органических кислот
проводят в следующем режиме: температура колонки 100—200 °C со
скоростью 6—10°С/мин, испарителя 220 °C, пламенно-ионизиру-
ющего детектора 210 °C; расход газа-носителя 30—40 см3/мин.
2—3 мкл хлороформного раствора метиловых (этиловых) эфи-
ров органических кислот вводят в испаритель хроматографа и элю-
ируют из колонки газом-носителем. Идентификацию индивиду-
альных метиловых (этиловых) эфиров проводят по времени удер-
живания метиловых (этиловых) эфиров органических кислот-мет-
чиков и методом добавки.
Массовая доля отдельной органической кислоты (%) в про-
дукте
Я1Сст-100А'
с ЛсС„р ’
где К — поправочный коэффициент для данной кислоты; Сст — масса навески
стандарта, мг; Спр — масса навески продукта, мг; Ас — площадь пика стандарта, в
относительных единицах; Л] — площадь пика данной кислоты, в относительных
единицах.
Приготовление реактива. Этерифицирующую смесь —7%-ный
раствор хлорида водорода в этаноле или метаноле готовят следую-
щим образом: 4 объемные части спирта смешивают с 1 объемной
частью концентрированной соляной кислоты. Раствор используют
свежеприготовленным.
АДСОРБЦИОННАЯ ХРОМАТОГРАФИЯ
Адсорбционная хроматография — процесс разделения веществ,
основанный на различной способности компонентов исследуемой
смеси поглощаться поверхностью данного адсорбента. В зависимо-
сти от агрегатного состояния подвижной фазы метод носит назва-
ние жидкостно-адсорбционной или газоадсорбционной хромато-
графии (см. табл. 19).
Пористые материалы обладают свойством более или менее проч-
но сорбировать различные вещества. Если колонку, заполненную
пористым материалом, промывают растворителем, несущим ана-
185
лизируемую смесь, то компоненты смеси с различной прочностью
удерживаются на поверхности адсорбента (например, обесцвечива-
ние растворов с помощью активированного угля или его использо-
вание в качестве фильтрующего материала в противогазе).
Адсорбция — это результат проявления дисперсионных элект-
ростатических сил.
Адсорбенты бывают полярные и неполярные. В жидкостно-ад-
сорбционной хроматографии (ЖАХ) разделение на полярном ад-
сорбенте называется хроматографией с нормальными фазами
(НФХ), разделение на неполярном адсорбенте — хроматографией с
обращенными фазами (ОФХ). В газо-адсорбционной хроматогра-
фии (ГАХ) или с помощью полярного растворителя полярные ве-
щества могут быть вытеснены с поверхности адсорбента (десорби-
рованы) при повышении температуры.
Между полярной поверхностью адсорбента и полярными (или
способными поляризоваться) группами или участками молекул
возможно специфическое взаимодействие (диполь-дипольное или
образование водородной связи). Достаточно часто высокоактивные
адсорбенты обладают каталитическими свойствами и способны
трансформировать вещества в процессе разделения (хемосорбция).
Например, третичные спирты могут подвергаться дегидратации
или изомеризации. Многие вещества, обычно не чувствительные к
действию света, в адсорбированном состоянии разлагаются на све-
ту. Поэтому многие адсорбенты рекомендуется использовать в час-
тично инактивированном состоянии.
Для НФХ в качестве сорбента используют силикагель и оксид
алюминия (для хроматографии при нормальном давлении размер
частиц 40—60 мкм, при высоком — 3, 5, 7, 10 мкм), для фракцио-
нирования неполярных и малополярных веществ, контроля ка-
чества (главным образом методом ВЭЖХ); для ОФХ — модифи-
цированный силикагель (для фракционирования полярных ве-
ществ), производные агарозы (для фракционирования белков по
гидрофобным свойствам; разделение проводится в водной среде,
что важно при работе с белками). Адсорбентом заполняют ко-
лонку (колоночная хроматография) или наносят на пластину
(ТСХ).
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НИЗКО- И ВЫСОКОМОЛЕКУЛЯРНЫХ
ЖИРНЫХ КИСЛОТ В СЫРАХ МЕТОДОМ ЖИДКОСТНО-
АДСОРБЦИОННОЙ ХРОМАТОГРАФИИ
(ПО ГАРПЕРУ)
Метод основан на выделении низко- и высокомолекулярных
жирных кислот из сыра, разделении их на колонке с адсорбентом с
последующим извлечением их растворителем.
В сыре определяют летучие жирные кислоты (уксусную, пропи-
оновую, масляную), группу высокомолекулярных жирных кислот и
186
группу нелетучих кислот (янтарную, молочную), исходя из того, что
для выделения из смеси отдельных кислот или их групп требуется
точный объем растворителя (предельный). Чтобы обеспечить пол-
ное выделение, предельный объем растворителя для одной кислоты
должен отличаться от объема другой не менее чем на 50 см3. Обраба-
тывая колонку с силикагелем после адсорбции на нем исследуемых
кислот, можно различным количеством растворителя разделить их
на группы (табл. 21).
21. Объем растворителя, необходимый для выявления жирных кислот
Кислота
Объем растворителя, см3
«-Олеиновая 50
«-Пальмитиновая 50
«-Лауриновая 50
«-Капроновая 80
«-Каприловая 85
«-Каприновая 90
«-Валерьяновая 110
«-Масляная 155
Пропионовая 230
Уксусная 285
Молочная 440
Муравьиная 410
Янтарная 440
Непосредственно перед анализом готовят растворитель —
смесь хлороформа и л-бутилового спирта. Хлороформ предвари-
тельно 2—3 раза промывают равными объемами воды при 25 °C,
фильтруют через сухой фильтр. л-Бутиловый спирт перегоняют,
добавляя гидроксид калия, используя фракции, полученные при
115—117 ’С.
Приготавливают 0,5; 1; 5; 20 и 40%-ные растворы л-бутилового
спирта в хлороформе и переливают их в посуду из темного стекла с
притертой пробкой.
К 5 г сыра в ступке добавляют 20%-ный раствор H2SO4 до pH
1,7—2,0. Водой доводят объем жидкости до 3 см3 и растирают тща-
тельно с Юг сухого силикагеля. Полученную смесь растирают в
ступке с таким количеством хлороформа, чтобы масса приобрела
текучесть, и без потерь переносят в колонку. Ступку обмывают хло-
роформом (всего хлороформа идет 40—50 см3). Верхний слой в ко-
лонке закрывают фильтровальным кружком и добавляют около
25 см3 хлороформа.
Когда жидкость в колонке поднимется на 2—4 см3 выше верх-
него слоя, подливают последовательно растворитель: 25 см3 0,5%-
ного раствора; 50 см31%-ного; 75 см3 5%-ного; 100 см3 20%-ногои
100 см3 40%-ного. Кран у колонки открывают и под него подстав-
187
ляют химические стаканы или колбочки, в которые последова-
тельно собирают по 5 см3 фильтрата, отмечая пробы текущими
номерами. Затем пробы титруют 0,1 н. раствором КОН в абсо-
лютном этиловом спирте с индикатором 0,02%-ным раствором
крезолового красного в абсолютном этиловом спирте. По полу-
ченным данным титрования строят график, откладывая по оси аб-
сцисс номера проб, по оси ординат объем щелочи, пошедшей на
титрование.
Разделение кислот обнаруживают прерыванием кислотности
при титровании (в пробах по 5 см3). Для идентификации кислот и
определения точности количественного метода к сыру последова-
тельно добавляют 0,1 н. растворы натриевых солей масляной, про-
пионовой, уксусной кислот, а также их смесь. Количественное об-
наружение добавленных кислот должно быть около 98 %, за исклю-
чением уксусной (96 %). Высшие жирные кислоты в сырах находят-
ся обычно в пробах при порядковом титровании их в пределах
33—75 номеров; масляная — от 33—35 до 53—55, пропионовая — от
53—55 до 63—65 и уксусная — от 63—65 до 73—75.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВИТАМИНА А
МЕТОДОМ ЖИДКОСТНО-АДСОРБЦИОННОЙ ХРОМАТОГРАФИИ
Метод основан на щелочном гидролизе жиров, гидролизе вита-
мина А до свободной формы, экстракции диэтиловым эфиром
неомыляемого остатка, выпаривании эфира, растворении остатка
в растворителе, используемом в качестве основного компонента
подвижной фазы (элюента) и определении витамина А методом
ВЭЖХ.
Так как жирорастворимые витамины легко разрушаются под
действием света, кислорода воздуха и других факторов, во время
анализа необходимо соблюдать специальные меры предосторожно-
сти, защищающие витамины от воздействия этих факторов: опреде-
ление проводят, предохраняя от света, в присутствии антиоксидан-
та, все операции от взятия навески продукта до получения результа-
та выполняют в течение рабочего дня.
Выполнение работы. Навеску молока или молочных продуктов
(20 г), взятую с точностью до 0,01 г, помещают в круглодонную
колбу, соединенную шлифом с обратным холодильником, добав-
ляют 40—60 см3 этилового спирта и 0,2 г аскорбиновой кислоты.
Затем в колбу вносят 5—7 см3 50%-ного раствора гидроксида ка-
лия. Для продуктов (масло сливочное и др.) с высоким содержани-
ем жира (более 40 %) на навеску образца 3—5 г берут 4—8 см3 ще-
лочи.
После добавления щелочи смесь кипятят 30 мин на водяной
бане с обратным водяным холодильником, охлаждают, перелива-
ют в делительную воронку и добавляют воду до окончательной
188
концентрации спирта около 30—35 %. Признак полного омыле-
ния — прозрачная смесь после добавления воды. При образовании
мути необходимо повторить омыление с новой навеской, увеличив
объем щелочи для омыления. Затем смесь экстрагируют эфиром
4 раза: сначала объемом, равным объему добавленной воды, а затем
объемами на 20 % меньшими. Колбу после омыления ополаскивают
эфиром.
Объединенный эфирный экстракт отмывают от щелочи во-
дой, контролируя pH по фенолфталеину. К экстракту добавляют
8—10 г безводного сульфата натрия, выдерживают 20—30 мин в
темном месте и медленно фильтруют через слой безводного суль-
фата натрия в круглодонную колбу, промывая сульфат натрия и
фильтр эфиром. Затем эфир полностью отгоняют под вакуумом
на роторном испарителе при температуре не выше 25—30 °C. Су-
хой неомыляемый остаток немедленно растворяют в объеме ра-
створителя так, чтобы концентрация витамина А была в пределах
3—5 мкг/см3.
В колонку хроматографа вводят последовательно равные
объемы (0,01—0,1 см3) испытуемого раствора, полученного выше
описанным методом, и стандартного. Пик витамина на хрома-
тограмме испытуемого раствора идентифицируют с пиком вита-
мина на хроматограмме стандартного раствора. Содержание ви-
тамина определяют по результатам двух хроматографических
анализов.
Содержание витамина А в продукте (мг/100 г) рассчитывают по
формуле
у_ЯисСстИг100
Н„ И2 а 1000 ’
где Нис — средняя высота (или площадь) пика витамина А на хроматограмме пробы
испытуемого раствора, мм (или мм2); Н„ — средняя высота (или площадь) пика ви-
тамина А на хроматограмме стандартного раствора, мм или мм2; С„ — масса вита-
мина А в стандартном растворе, введенном в колонку, мкг; К] — общий объем испы-
туемого раствора, см3; И2 — объем испытуемого раствора, вводимый в колонку хро-
матографа, см3; а — навеска образца продукта, г; 100 — пересчет на 100 г продукта;
1000 — пересчет на мг.
При использовании в качестве стандарта ретинола ацетата в фор-
муле будут те же обозначения, кроме Яст = Яис + ст — Яис, где Яис + ст —
средняя высота (или площадь) пика витамина А в образце с вне-
сенным ретинола ацетатом; Сст — масса витамина А в стандартном
растворе с внесенным ретинола ацетатом (мкг), введенного в ко-
лонку.
Результаты определений рассчитывают до третьей значащей
цифры.
За результат измерений принимают среднее арифметическое
значение результатов двух параллельных определений.
189
Приготовление реактива (стандартного раствора). Взвешивают
0,02 г витамина А или 0,025 г ретинола ацетата с точностью до
0,0001 г, растворяют в этиловом спирте и доводят объем в мерной
колбе до 100 см3 (основной стандартный раствор). 2 см3 раствора
переносят в мерную колбу на 100 см3 и доводят объем этиловым
спиртом (рабочий стандартный раствор). Содержание витамина А в
рабочем стандартном растворе в пересчете на ретинол приблизи-
тельно 4—4,5 мкг/см3 (13—15 МЕ/см3).
При использовании в качестве стандартного раствора ретинола
ацетата в масле навеску препарата рассчитывают, исходя из требуе-
мой концентрации рабочего раствора.
Стандартный раствор хранят при 4 °C в течение 10 дней. В день
использования определяют концентрацию в рабочем стандартном
растворе, измеряя оптическую плотность раствора на спектрофо-
тометре при длине волны 325 нм в кювете толщиной 1 см и прове-
ряя чистоту стандартного раствора хроматографически (на хрома-
тограмме должен присутствовать один пик витамина, помимо пи-
ков, не удерживаемых на колонке примесей, содержащихся в ра-
створителе). Содержание витамина А (мкг/см3) рассчитывают по
формуле
Y _ 2)40000
Аа~Ё~ ’
где D — оптическая плотность испытуемого раствора при длине волны 325 нм про-
тив этилового спирта; 10 000 — содержание витамина А в 1 см3 1%-ного раствора,
мкг; Е — 1830 для витамина А при использовании кюветы толщиной 1 см.
При использовании в качестве стандарта ретинола ацетата со-
держание в растворе пересчитывают на витамин А (ретинол) по
формуле
Y . Л-10000-0,87
Е
где Е— 1550 для ретинола ацетата при использовании кюветы толщиной 1 см;
0,87 — коэффициент пересчета на витамин А.
При использовании в качестве стандарта ретинола ацетата к на-
веске образца продукта добавляют точный объем рабочего стандарт-
ного раствора ретинола ацетата (навеска образца должна быть равна
навеске образца без добавления рабочего стандартного раствора).
Затем навеску образца с добавленным рабочим стандартным ра-
створом подвергают щелочному гидролизу и проводят экстракцию
так же, как описано выше для навески испытуемого образца. Сухой
неомыленный остаток растворяют в том же объеме растворителя. В
этом случае навеску образца, объем рабочего стандартного раство-
ра, добавляемого к навеске, объем растворителя рассчитывают, ис-
190
ходя из того, что приблизительная концентрация витамина в испы-
туемом растворе должна быть около 3 мкг/см3, в растворе исследуе-
мого образца с добавленным рабочим стандартным раствором око-
ло 5 мкг/см3.
ОСАДОЧНАЯ ХРОМАТОГРАФИЯ
В осадочной хроматографии основным фактором, определяю-
щим разделение и выделение веществ из раствора смеси компонен-
тов, является процесс образования осадков, различающихся раство-
римостью. Осадочную хроматографию проводят как на бумаге, так и
в колонке. В верхней части колонки выделяются осадки веществ, ме-
нее растворимых, в нижней части — более растворимых. Смесь, за-
полняемая колонку, состоит из носителя — высокодисперсного ве-
щества (сорбент) и осадителя — вещества, образующего с исследуе-
мым ионом характерно окрашенное труднорастворимое соединение.
При пропускании исследуемого раствора через хроматографи-
ческую колонку с сорбентом происходит обмен ионов, входящих в
состав сорбента, на ионы хроматографируемого раствора. Последо-
вательная сорбируемость (сорбционный ряд) на хроматографирую-
щем оксиде алюминия представлена для некоторых видов в следую-
щем порядке: Sn2+ >Fe3+ >Pg2+ >Cu2+ >Zn2+ >Ni2+.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МЕДИ, ОЛОВА,
СВИНЦА, ЦИНКА МЕТОДОМ ОСАДОЧНОЙ ХРОМАТОГРАФИИ
(ПО К. М. ОЛЬШАНОВОЙ, Н. М. МОРОЗОВОЙ И В. Н. КОПЫЛОВОЙ)
Метод основан на озолении молочного продукта сухим или мок-
рым сжиганием, определении металла на хроматографической ко-
лонке.
Медь. Безводный оксид алюминия (для хроматографии) тщатель-
но смешивают с порошкообразной рубеановодородной кислотой в
соотношении 100:1. Смесь помещают в укрепленную в штативе ко-
лонку (трубка длиной 10 см и диаметром 3—5 мм с небольшим отвер-
стием внизу, прикрытым тампоном ваты), тщательно уплотняя стек-
лянной палочкой. Заготовляют несколько колонок. Затем в колонку
вносят пипеткой 0,2 см3 (4 капли) исследуемого раствора.
В верхней части колонки образуется черная или серая (в зависи-
мости от концентрации меди) зона, что указывает на наличие ионов
меди. Тогда исследуемый раствор разбавляют: к 1 см3 раствора при-
бавляют 1 см3 воды, разбавленный вдвое раствор хроматографиру-
ют на другой такой же колонке. При положительной реакции на
ионы меди его снова разбавляют, добавляя к 1 см3 разбавленного в
2 раза раствора 1 см3 воды (разбавление в 4 раза по отношению к
первоначальному объему раствора).
191
Последовательные разбавления производят до тех пор, пока при
внесении раствора в колонку не будет образовываться темная зона
ионов меди. Содержание меди в продукте (мг/кг) рассчитывают по
формуле
v л л 0,42л-7
Хм - 0,42л + 100~,
где п — разбавление раствора; 0,42 — предельное количество меди в 1 кг продукта,
которое обнаруживается хроматографическим методом (установлено эксперимен-
тально), мг; 0’4qq 7 — поправка на сорбцию и другие факторы.
Присутствие других ионов не мешает исследованию.
Олово. На фильтровальную бумагу марки «синяя лента» наносят
1—2 капли 5%-ного водного раствора ксантогената калия и пипет-
кой 0,01—0,04 см3 исследуемого раствора. После этого добавляют
1—2 капли раствора ксантогената. При наличии ионов олова после
впитывания раствора на бумаге остается желтое пятно. Тогда иссле-
дуемый раствор разбавляют дистиллированной водой, подкислен-
ной соляной кислотой в 2, 4 или более раз, как указано при опреде-
лении ионов меди, до тех пор, пока ионы олова не будут обнаружи-
ваться хроматографически на фильтровальной бумаге в виде жел-
тых пятен. Количество олова в продукте (мг/кг) вычисляют по
формуле
у -14„ . 14ц-4
где 14 — предельное количество олова в 1 кг продукта, которое можно обнаружить
хроматографически, мг; — поправка на сорбцию и другие факторы.
Присутствие ионов меди мешает определению. В этом случае
медь отделяют от олова, добавляя к 1 см3 исследуемого раствора
1 см3 концентрированного раствора NaOH. Осадок Си(ОН)2 от-
фильтровывают, промывают водой. Фильтрат, содержащий олово в
виде Na2SnO2, сильно подкисляют соляной кислотой, переводя
олово в форму катиона Sn2+. Объем фильтрата доводят выпаривани-
ем до 2 см3, исследуют на присутствие ионов олова и рассчитывают
по формуле.
Свинец. На фильтровальную бумагу марки «синяя лента» нано-
сят 0,04 см3 насыщенного водного раствора родизоната натрия и
0,04 см3 исследуемого раствора. После впитывания раствора в центр
пятна вносят 1 —2 капли раствора родизоната натрия. После впиты-
вания его в присутствии ионов свинца на бумаге появляется розово-
фиолетовое кольцо. Чтобы лучше наблюдать зону, можно в центр
проявленного пятна внести 1—2 капли 2 н. раствора уксусной кис-
лоты, розово-фиолетовое кольцо переместится к периферии хрома-
тограммы.
192
При появлении ионов свинца раствор разбавляют, как указано
при определении ионов меди и олова, до тех пор, пока не будет по-
являться окрашивание с родизонатом. Содержание свинца в про-
дукте (мг/кг) рассчитывают по формуле
Хс=0,Зи+^,
где 0,3 — предельное количество свинца в 1 кг продукта, которое может быть обна-
ружено хроматографически, мг; — поправка на сорбцию и другие факторы.
Реакция специфична, присутствие других ионов не мешает.
Цинк. К 1 см5 исследуемого раствора приливают 1 см3 0,0018 н.
раствора соли кобальта, 0,2 см3 полученного раствора вносят в ко-
лонку с оксидом алюминия, как при определении меди. После впи-
тывания адсорбентом раствора хроматограмму проявляют, прили-
вая 4—6 капель раствора тетрароданомеркуроата аммония и 1 —
2 капли 2 н. раствора азотной кислоты. В присутствии ионов цинка
в верхней части колонки образуется голубая зона.
Ионы меди и железа маскируют зону ионов цинка, так как при
пропускании проявителя вверху колонки образуется желто-зеленая
зона ионов меди и красно-бурая ионов железа, между ними в виде
серо-фиолетовой зоны расположены ионы цинка. В этом случае
хроматограмму промывают 5—7 каплями 2 н. раствора азотной кис-
лоты. После этого, если обнаруживаются ионы цинка, раствор пос-
ледовательно разбавляют в 2 раза: к 1 см3 исследуемого раствора
приливают 0,5 см3 0,0018 н. раствора соли кобальта и 0,5 см3 воды,
раствор хроматографируют до тех пор, пока цинк больше не будет
обнаруживаться. Количество цинка в продукте (мг/кг) рассчитыва-
ют по формуле
X -2и ।
где п — разбавление; 2 — предельное количество цинка в 1 кг продукта, которое
можно обнаружить хроматографически, мг; — соответствующая поправка на
процессы сорбции и другие факторы.
ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ
Ионообменная хроматография основана на различной способ-
ности разделяемых ионов в растворе к ионному обмену с ионитом
(неподвижная фаза), который представляет собой нерастворимую
полимерную матрицу, несущую химически связанные ионогенные
группы. Противоионы удерживаются на матрице за счет сил элект-
ростатического взаимодействия и могут обмениваться на ионы раз-
деляемой смеси, присутствующие в подвижной фазе.
193
По химическому строению матрицы ионообменники подразделя-
ются на следующие группы: синтетические смолы на основе поли-
стирола или полиакриламида, сефадексы на основе сшитого декстра-
на, сефарозы — на основе агарозы, целлюлозные ионообменники на
основе микрокристаллической или сшитой микросферической цел-
люлозы, неорганические иониты на основе поверхностно-модифи-
цированных силикагелей.
Тип ионообменника определяется природой функциональных
ионогенных групп.
Ионит является слабокислотным при наличии карбоксильных
или оксифенильных групп; сильнокислотным — при наличии суль-
фогрупп (—SO3H); слабоосновным при наличии аминогрупп раз-
личной степени замещения; сильноосновным при наличии групп
четвертичных аммониевых оснований.
На сильные и слабые иониты разделяют по аналогии с делением
на слабые и сильные кислоты и основания, это деление отчасти от-
ражает степень диссоциации ионогенных групп. Свойство не следу-
ет путать с емкостью, которая для ионитов определяется абсолют-
ным числом функциональных групп и их доступностью. Сильный
ионит имеет полную емкость в большом диапазоне значений pH ра-
створа, слабый ионит достигает полной емкости лишь в определен-
ной области pH. Так, карбоксиметилцеллюлоза (КМЦ) при pH < 3
уже не несет диссоциированных карбоксильных групп и полностью
лишена ионообменных свойств. Полной емкости КМЦ достигает
при pH > 6.
Ионообменную хроматографию проводят в водной среде. В ка-
честве примера ионообменного процесса можно привести опера-
цию умягчения воды, при которой ионы кальция и магния обмени-
ваются на ионы натрия. Помимо обычного обмена ионов на иони-
тах можно проводить разделение заряженных частиц, прежде всего
биополимеров (белки, нуклеиновые кислоты), некоторые из кото-
рых обладают амфотерными свойствами. Именно в этой области
главным образом применяют ионообменную хроматографию. Так
как суммарный заряд таких макромолекул зависит от pH среды,
хроматографию проводят в буферных растворах. В большинстве
случаев pH среды в процессе разделения поддерживают постоян-
ным. Разделение различных типов ионов возможно при условии,
что компоненты смеси по-разному взаимодействуют с заряженны-
ми группами ионита.
Если несколько повысить ионную силу элюента путем добавле-
ния нейтральных солей (например, NaCl), то ионы элюента будут
конкурировать с компонентами смеси. Вследствие этого один или
несколько компонентов частично десорбируются и начнут медлен-
но мигрировать по колонке в подвижной фазе. При повышении
концентрации соли скорость миграции возрастает, одновременно
десорбируются более прочно удерживаемые вещества. При некото-
ром предельном значении ионной силы элюента все компоненты
194
образца полностью десорбируются и мигрируют по колонке со ско-
ростью движения подвижной фазы. Следовательно, важно найти
оптимальное значение ионной силы, при котором достигается наи-
большее разделение целевых компонентов смеси. В большинстве
случаев при постоянной ионной силе буфера не удается элюировать
все компоненты смеси, присутствующие в образце. Поэтому обще-
принятым приемом в ионообменной хроматографии является элю-
ирование в градиенте ионной силы.
Иониты на основе синтетических смол производят в виде гранул
диаметром от 5 мкм до 2 мм. Гранулы меньшего диаметра предназ-
начены для проведения анализов при повышенном давлении (на-
пример, анализ аминокислот, углеводов). Гранулы большего диа-
метра используют в технологических процессах, для которых необ-
ходима высокая скорость элюирования.
Сефадексы (Sephadex) готовят путем введения функциональных
групп в сефадексы G = 25 или G = 50.
Иониты из сефарозы готовят на основе агарозы, сшитой при по-
мощи молекул 2,3-дибромпропанола. За счет макропористой
структуры гранулы ионообменной агарозы обладают достаточной
механической прочностью.
Ионообменную хроматографию проводят в соответствии со
схемой 3.
В области анализа при помощи синтетических ионитов решают
следующие задачи: определение общего содержания солей в раство-
ре (путем ионообмена и последующего определения концентрации
ионов водорода);
удаление посторонних ионов при неорганическом анализе (на-
пример, Fe3+, Al3+, Ni2+, F- идр.);
анализ гидролизатов белков, олигонуклеотидов, полисахаридов;
определение хлорида натрия в соленых молочных продуктах,
уровня загрязнения окружающей среды.
В препаративном масштабе при помощи синтетических ионитов
решают другие задачи:
деионизация воды (деминерализованную воду часто ошибочно
называют дистиллированной);
удаление ионогенных примесей, присутствующих в органических
растворителях; очистка биополимеров, например отделение детерген-
тов и амфолитов, присутствующих в растворах белковых препаратов;
замена противоионов в буферных растворах;
получение препаратов радиоактивных изотопов;
использование в качестве катализаторов многих технологичес-
ких процессов — этерификации, омыления, дегидратации, гидра-
тации, полимеризации, инверсии сахаров, перегруппировки, алки-
лирования, циангидринного синтеза, образования ацетатов, нитро-
вания, эпоксидирования.
Иониты на основе сефадекса и целлюлозы применяют для пре-
паративного выделения белков, пептидов, олигонуклеотидов; вы-
195
Схема 3. Выполнение операций в ионообменной хроматографии
деления полисахаридов и липидов, несущих заряженные группы;
иммобилизации ферментов.
Иониты на основе агарозы используют в тех же целях, что и се-
фадексы; кроме того, на них можно фракционировать вещества с
молекулярной массой до 106 г/моль.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ОТДЕЛЬНЫХ АМИНОКИСЛОТ
МЕТОДОМ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ
Основано на выделении белков, их гидролизе до аминокислот и
количественном определении на аминокислотном анализаторе.
В пробе молока или молочных продуктов определяют массовую
долю белков, жиров и влаги. При исследовании жидких продуктов
196
учитывают содержащуюся в них воду в процессе приготовления
гидролизующей смеси таким образом, чтобы концентрация соля-
ной кислоты была 6 М.
Если в исследуемом продукте содержится более 5 % жиров, то
его обезжиривают: продукт экстрагируют 10-кратным (к массе на-
вески) количеством смеси этанол — хлороформ (соотношение 1 :2)
2 раза.
После обезжиривания остаток подсушивают на воздухе и опре-
деляют содержание белков.
Рассчитывают массу навески образца продукта для гидролиза,
исходя из соотношения масс белка и соляной кислоты 1: 1000 и при
условии содержания белка в пробе не менее 5 мг.
Навеску продукта или обезжиренного остатка, подготовленного
к гидролизу, взятую с точностью 0,0002 г, помещают в стеклянную
ампулу с оттянутым концом, заливают расчетным количеством со-
ляной кислоты (при исследовании жидких продуктов берут расчет-
ное количество концентрированной соляной кислоты и доводят до
концентрации 6М). Ампулу запаивают, устанавливают в строго
вертикальном положении в металлический патрон или фарфоро-
вый стакан с парафином и помещают в сушильный шкаф с заранее
отрегулированной температурой ПО ±2 °C. Нагревание проводят
непрерывно в течение 24 ч. Затем ампулу охлаждают до комнатной
температуры.
Ампулу вскрывают и удаляют соляную кислоту. Если в гидроли-
зате образовался видимый осадок, его отцентрифугируют или от-
фильтровывают с последующим доведением фильтрата в мерной
колбе на 25 см3 до точного объема.
Соляную кислоту удаляют одним из следующих способов:
помещают ампулу в вакуумный эксикатор над гранулированным
гидроксидом натрия (NaOH) на 12—18 ч;
гидролизат количественно переносят в грушевидную колбу,
ополаскивая ампулу дистиллированной водой, и выпаривают на ро-
торном испарителе при температуре не выше 60 °C.
Остаток переносят количественно в мерную колбу с помощью
цитратного буфера pH 2,2 или 0,02 М раствора соляной кислоты и
доводят до метки. Полученный раствор гидролизата анализируют
на аминокислотном анализаторе, при необходимости разбавляют
или фильтруют.
В случае, если анализ не проводят немедленно, осадок освобож-
дают от следов соляной кислоты, добавляя к нему дистиллирован-
ную воду или повторно выпаривая на роторном испарителе или в
вакуумном эксикаторе. Операцию повторяют до полного исчезно-
вения запаха соляной кислоты. Образец хранят в холодильнике при
температуре не выше 5 °C, перед анализом его разбавляют цитрат-
ным буфером до необходимой концентрации. Обнаруженный оса-
док отфильтровывают через плотный фильтр.
197
Массу аминокислоты в 100 г образца, подготовленного к гидро-
лизу (мг), рассчитывают по формуле
к_яю-б-рюо
Х VM
где К— объем пробы, вводимой в аминокислотный анализатор, см3; А — массовая
доля аминокислоты в объеме пробы, вводимой в аминокислотный анализатор, мг;
Р— общее разведение; М— масса навески продукта, взятой на гидролиз, г; 10~б —
коэффициент перевода нанограммов в миллиграммы; 100 — коэффициент перево-
да в процентную концентрацию.
Массу аминокислоты в 100 г белка (мг) рассчитывают по формуле
„*100
1 Б ’
где Б— содержание белка в подготовленном к гидролизу образцу продукта, %;
100 — коэффициент перевода в процентную концентрацию.
Массу аминокислоты в 100 г продукта (мг) рассчитывают по
формуле
где В— содержание белка в продукте, %; 100 — коэффициент перевода в процент-
ную концентрацию.
Результаты определений рассчитывают до второго десятичного
знака и округляют до первого знака после запятой. За окончатель-
ный результат исследования принимают среднее арифметическое
результатов двух параллельных определений.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ХЛОРИДА НАТРИЯ
В СЫРАХ, БРЫНЗЕ, СОЛЕНЫХ ТВОРОЖНЫХ ИЗДЕЛИЯХ,
СЛИВОЧНОМ МАСЛЕ МЕТОДОМ ИОНООБМЕННОЙ
ХРОМАТОГРАФИИ С КАТИОНИТОМ
Метод основан на выделении хлорида натрия из молочных про-
дуктов и определении его в катионообменной колонке.
15 г катионита КУ-2 (в пересчете на безводный катионит), взве-
шенного с погрешностью не более 0,1г, помещают на 5 ч в химичес-
кий стакан с дистиллированной водой. Набухший катионит пере-
носят в стеклянную трубку длиной 700—800 мм с внутренним диа-
метром 12— 15 мм или в бюретку вместимостью 50 см3, на дно кото-
рых кладут стеклянную вату или другой пористый материал.
Через колонку пропускают, регулируя с помощью крана,
100 см3 раствора соляной кислоты (70 г/дм3) со скоростью 2—3 кап-
198
ли в секунду. Затем катионит промывают с той же скоростью дис-
тиллированной водой до нейтральной реакции по метиловому
оранжевому.
Каждую последующую порцию жидкости необходимо прили-
вать, как только уровень ее в колонке достигнет верхнего края кати-
онита.
Необходимо следить, чтобы мениск жидкости никогда не опус-
кался ниже верхнего края катионита.
Ионообменную колонку регенерируют пропусканием через нее
50 см3 раствора соляной кислоты (50 г/дм3) со скоростью 2—3 капли
в секунду, с последующим промыванием дистиллированной водой
с той же скоростью до нейтральной реакции по метиловому оран-
жевому.
Между двумя процессами регенерации допускается исследовать
20 проб сыра, брынзы, творожных изделий или 100 проб сливочного
масла. В случае меньшего числа определений колонку следует реге-
нерировать ежедневно.
Пригодность катионита для проведения анализа проверяют пе-
риодически, пропуская через катионообменную колонку 5 см3 ра-
створа хлорида натрия, с последующим промыванием катионита
дистиллированной водой в количестве 50 см3. Фильтрат вместе с
промывными водами титруют раствором гидроксида натрия. Объем
гидроксида натрия, пошедший на титрование, может отличаться не
более чем на 0,2 см3 от взятых 5 см3 раствора хлорида натрия.
Взвешивают 2 г сыра, брынзы или творожных изделий с погреш-
ностью не более 0,01 г в фарфоровом тигле, предварительно высу-
шенном. Тигель с продуктом высушивают в сушильном шкафу при
постепенном повышении температуры до 120—140 °C. Высушен-
ную массу осторожно обугливают при постоянном повышении тем-
пературы. По окончании выделения дыма нагревание усиливают и
обугливание продолжают до получения остатка темно-серого цвета.
Обугленную массу осторожно измельчают стеклянной палочкой и
обрабатывают 4—5 порциями дистиллированной воды температу-
рой 80—90 °C.
Жидкую часть осторожно переводят по стеклянной палочке на
бумажный фильтр и фильтруют в коническую колбу. Остаток в тиг-
ле и на фильтре промывают дистиллированной водой температурой
70—80 °C до прекращения реакции последних порций фильтрата с
нитратом серебра. Для этого небольшую порцию фильтрата в про-
бирке подкисляют 1—2 каплями азотной кислоты и прибавляют 1 —
2 капли раствора нитрата серебра.
Вытяжку переносят в подготовленную катионообменную колон-
ку и пропускают со скоростью 3—4 капли в секунду. После этого
колонку промывают 50 см3 дистиллированной воды. Фильтрат вме-
сте с промывными водами титруют раствором гидроксида натрия в
присутствии 2—3 капель метилового оранжевого до получения со-
ломенно-желтого окрашивания.
199
Для выделения хлорида натрия взвешивают 5 г сливочного масла
с погрешностью не более 0,01 г в стакане вместимостью 100 см3. За-
тем пипеткой приливают в стакан 50 см3 дистиллированной воды.
Содержимое стакана нагревают до расплавления сливочного масла,
тщательно перемешивают и оставляют в покое до поднятия жира
наверх и его застывания. При необходимости охлаждения, стакан
после поднятия наверх слоя жира помещают в холодную дистилли-
рованную воду.
Стеклянной палочкой делают в слое сливочного масла отвер-
стие, через которое пипеткой отбирают 10 см3 вытяжки и переносят
в колонку, фильтруют со скоростью 3—4 капли в секунду. С той же
скоростью колонку промывают 50 см3 дистиллированной воды.
Фильтрат вместе с промывными водами титруют раствором гидро-
ксида натрия в присутствии 2—3 капель метилового оранжевого до
получения соломенно-желтого цвета.
Массовую долю хлорида натрия (%) вычисляют по формуле
в сыре, брынзе и соленых творожных изделиях:
= V- 0,292;
в масле:
Х = V- 0,585,
где V— объем раствора гидроксида натрия, израсходованный на титрование, см3;
0,292 и 0,585 — титр раствора гидроксида натрия, пересчитанный на хлорид натрия,
умноженный на 100 и деленный на величину массы навески продукта.
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,2 % для
сыра, брынзы, творожных изделий и 0,1 % — для сливочного масла.
АФФИННАЯ ХРОМАТОГРАФИЯ
Аффинная хроматография — метод разделения биологически
активных веществ, основанный на их специфическом взаимодей-
ствии с лигандами, ковалентно связанными с нерастворимым носи-
телем (матрицей). В качестве лигандов используют соединения,
взаимодействие которых с разделяемыми соединениями основано
на биологической функции последних.
Схематично механизм разделения в аффинной хроматографии
представлен на рис. 51. Лиганд L фиксирован на матрице, целевое
вещество Sсвязывается лигандом и извлекается из раствора. На ста-
дии элюирования комплекс разрушается и целевое вещество вновь
переходит в раствор.
Взаимодействие (вещество — лиганд) должно быть специфичес-
ким и обратимым. Обратимость процесса характеризуется констан-
той диссоциации.
200
Рис. 51. Механизм разделения веществ в аффинной
хроматографии:
а — иммобилизация лиганда (ковалентно); б — связывание
целевого вещества (нековалентно) и удаление сопутствующих
примесей; в — десорбция целевого вещества
Лиганд должен иметь реакционноспособные функциональные
группы, при помощи которых осуществляется его связь с матрицей,
при этом должна сохраняться биоспецифическая активность ли-
ганда. Если лиганд имеет несколько таких групп, его иммобилиза-
ция должна происходить с участием той из них, которая не входит в
участок, взаимодействующий с целевым веществом. Активные цен-
тры многих биологически активных веществ (например, фермен-
тов) часто локализованы в середине глобулы и недоступны для не-
больших молекул лигандов, непосредственно связанных с матри-
цей. Поэтому между матрицей и лигандом обычно встраивают до-
полнительный блок — «спейсер». В качестве матрицы служат гели
агарозы или полиакриламида.
Для взаимодействия выпускают материалы с различными реак-
ционноспособными группами. Тип геля выбирают по виду функци-
ональных групп лиганда. Известны матрицы, на которых лиганды
могут быть иммобилизованы только с помощью конденсирующих
агентов*.
* В качестве конденсирующих агентов используют водорастворимые производные
карбодиимида, например, М-этил-Ы'-(3'-диметиламинопропил)-карбодиимидгид-
рохлорид и др.
201
Аффинную хроматографию проводят в водных буферных ра-
створах, подбирая оптимальные условия для каждого конкретного
случая. Можно создать такие условия (значение pH раствора и кон-
центрация соли), при которых взаимодействие целевого вещества с
лигандом будет наиболее сильным.
Среди других методов выделения веществ аффинная хромато-
графия занимает особое место, поскольку процесс идет крайне спе-
цифически с использованием биологической активности целевого
вещества. Эта особенность позволяет концентрировать целевые ве-
щества из больших объемов раствора.
Из гелей на основе агарозы часто применяют сефарозу. Благода-
ря крупным порам внутренняя поверхность гранул доступна как для
молекул лигандов, так и для молекул целевых веществ. Матрица
агарозы имеет незначительную неспецифическую сорбцию. Части-
цы сефарозы мало сжимаемы, вследствие чего обеспечиваются хо-
рошие гидродинамические свойства колонки.
Аффи-гель — агарозный и полиакриламидный гель, модифи-
цированный разнообразными функциональными группами. По
сравнению с агарозными гелями материалы на основе полиакри-
ламида имеют следующие преимущества: крайне незначительную
неспецифическую сорбцию; биологическую инертность (устой-
чивы к действию ферментов); повышенную химическую и термо-
устойчивость.
Последовательность выполнения операций приведена на схеме 4.
Если целевое вещество слишком прочно связано с лигандом,
рекомендуется прервать процесс элюирования в тот момент, ког-
да элюент полностью вытеснен буферным раствором. Спустя не-
которое время (от 20 мин до 2 ч) целевое вещество будет десорби-
ровано и может быть элюировано с колонки. Заряженные веще-
ства элюируют в градиенте ионной силы. При очень высоком
сродстве в состав буфера вводят анионы, разрушающие водород-
ные связи (СЮ4 <CF3COO“<SCN“<CC13COCF).
Поскольку элюирование проводят в солевых буферах, фракции
вначале обессоливают, например методом гель-хроматографии, а
затем высушивают лиофильно.
После завершения эксперимента сорбент промывают буферным
раствором 0,1 М трис-НС1, pH 8,5 (10 объемов колонки), содержа-
щим 0,5 моль/л NaCl, а затем стартовым буфером.
Готовый гель хранят на холоде в присутствии консервантов.
Аффинную хроматографию применяют для выделения следую-
щих классов веществ: аналогов субстратов ингибиторов, кофакто-
ров (при этом роль лигандов играют ферменты); антигенов вирусов
клеток (лиганды — антитела); полисахаридов, гликопротеинов кле-
ток (лиганды — лектины); гистонов, полимераз (лиганды — нукле-
иновые кислоты); рецепторов, белков-переносчиков (гормоны, ви-
тамины); белков, специфически взаимодействующих с мембраной
клетки, лектинов (лиганды — клетки).
202
Контрольные вопросы. 1. В чем заключается сущность хроматографических Ме-
тодов? 2. Что представляет собой адсорбционная хроматография? 3. Каковы особен-
ности гель-хроматографии? 4. На чем основано разделение веществ в ионообмен-
ной хроматографии? 5. В чем состоят особенности газовой хроматографии?
Заполнение колонки 2—10 см влажного геля.
2
Колонку промывают трехкратным объемом буфера до постоянного
значения pH (процесс уравновешивания).
з
Если проба растворена не в элюенте, необходимо заменить буфера
методом гель-фильтрации. Если анализируемые вещества прочно
связаны с лигандом, объем пробы не столь важен. Вещества, слабо-
удерживаемые лигандом, следует наносить в небольшом объеме
(примерно 5 % от объема колонки).
«л
Элюирование можно проводить в градиенте ионнои силы.
Схема 4. Выполнение операций в аффинной хроматографии
203
Глава 8
ЭЛЕКТРОФОРЕТИЧЕСКИЕ МЕТОДЫ
Электрофорез — метод разделения веществ, основанный на яв-
лении миграции заряженных микрочастиц в жидкой среде под дей-
ствием внешнего электрического поля.
Существует три различных электрофоретических метода (схема 5).
Под собственно электрофорезом обычно понимают зональный
электрофорез (ЗЭ), два других называют методами изоэлектрофо-
кусирования (ИЭФ) и изотахофореза (ИТФ).
Схема 5. Электрофоретические методы
204
Электрофорез применяют главным образом для разделения ве-
ществ, молекулы которых различаются по электрофоретической под-
вижности, т. е. отношению скорости электрофореза (скорости пере-
мещения заряженных частиц вещества) к напряженности электричес-
кого поля, которое зависит от свойств заряженных частиц окружаю-
щей их среды. Путем изменения внешних условий (например, pH
среды, температуры, силы тока, состава и концентрации буферного
раствора или носителя) создают подходящие условия для разделения.
Вследствие того что при разделении на молекулы действуют только
электростатические силы, электрофорез считают «мягким» методом и
поэтому часто применяют для работы с лабильными веществами.
Электрофорез можно проводить в растворе, но из-за неизбежно-
го выделения теплоты и возникающей в связи с этим тепловой кон-
векции процесс, как правило, проводят на носителе. Вследствие не-
которых сопутствующих явлений (адсорбция, несоизмеримость
размеров высокомолекулярных соединений и пор носителя) введе-
ние носителя ограничивает область применения метода. Однако
свойства носителя иногда используют для повышения эффективно-
сти разделения: например, при электрофорезе в градиенте полиак-
риламидного геля фракционирование осуществляется не столько за
счет различной электрофоретической подвижности веществ,
сколько за счет различия в их молекулярных массах.
ЗОНАЛЬНЫЙ ЭЛЕКТРОФОРЕЗ
Зональный электрофорез (ЗЭ) — это метод разделения заряжен-
ных частиц в электрическом поле, основанный на том, что частицы
с разными соотношениями заряд/масса мигрируют с различными
скоростями.
В зависимости от знака заряда молекулы вещества мигрируют в
электрическом поле по направлению к аноду или катоду (рис. 52).
Рис. 52. Камера для зонального
электрофореза:
а — вид сбоку; б~ вид сверху; 1—
электроды (анод и катод); 2— бу-
ферный раствор в кювете; 3 — слой
геля на пластинке (в центре пластин-
ки показаны стартовые зоны анали-
зируемой смеси); 4 — мостики из
фильтровальной бумаги; 5—охлаж-
даемая проточной водой металли-
ческая пластина для отвода избыточ-
ной теплоты
205
Результаты этого процесса регистрируются на электрофореграмме
(по аналогии с хроматографией).
Ранее использовали один и тот же буфер в слое носителя и в
электродных камерах, т. е. разделение вели в непрерывной буфер-
ной системе. В настоящее время этот прием еще применяют при
электрофорезе на бумаге и пластинках. При электрофорезе в пре-
рывистой буферной системе (различные буферы в слое носителя и в
электродных камерах) быстро мигрирующие вещества образуют бо-
лее узкие зоны. Электрофорез в прерывистой буферной системе ис-
пользуют главным образом в гель-электрофорезе. ЗЭ обычно про-
водят на бумаге, пластинках и в гелях в водных буферных растворах.
При электрофорезе в электродных камерах происходит электро-
лиз раствора и вследствие этого изменяется состав буфера. Поэтому
электроды располагают так, чтобы они не касались носителя, а кон-
такт между ними осуществлялся при помощи полосок фильтро-
вальной бумаги (см. рис. 52). Электродная камера разделена на два
отсека, которые соединяются дополнительным мостиком из фильт-
ровальной бумаги. Подбирая соответствующий объем электродных
камер или перекачивая буфер насосом от анода к катоду, поддержи-
вают постоянными концентрацию и значение pH буфера в двухка-
мерной системе. Рекомендуется также проводить деполяризацию
электродов после каждого электрофоретического разделения.
Материалы-носители подразделяются на две группы:
первая — бумага, целлюлоза, ацетилированная целлюлоза, ага-
роза и материалы для ТСХ (например, силикагель);
вторая — крахмал и полиакриламид.
Эффективность разделения зависит не только от суммарного за-
ряда молекул анализируемых веществ, но и от размеров молекул.
Определяющим параметром является соотношение заряд — масса.
Носители первой группы относительно инертны и слабо влияют
на эффективность разделения. Материалы второй группы обладают
пористой структурой, что существенно влияет на качество разделе-
ния. Поскольку размеры пор соизмеримы с размером макромоле-
кул, то можно разделять вещества с одинаковыми суммарными за-
рядами, но с разными молекулярными массами (например при
ионообменной хроматографии).
Наибольший интерес вызывает разделение амфотерных веществ
(например, аминокислот, белков). Амфотерные вещества в кислой
среде присоединяют протон и ведут себя как катионы. В щелочной
среде они депротонируются и приобретают свойства анионов. В
изоэлектрической точке (pl) они становятся цвиттерионами с нуле-
вым суммарным зарядом, т. е. нейтральными молекулами, в кото-
рых противоположные заряды пространственно разделены (pl —
характеристическая константа для определенного вида молекул).
Суммарный заряд таких веществ зависит от pH среды в широком
диапазоне. При электрофорезе они могут менять направление и
скорость миграции.
206
Чем выше концентрация буферного электролита, тем меньше
электрофоретическая подвижность разделяемых веществ. Это
объясняется тем, что электрический ток переносится всеми присут-
ствующими в растворе ионами. Чем выше концентрация буферного
электролита, тем большая доля электрического тока переносится
его ионами, тем медленнее мигрируют молекулы образца. К тому же
следует учесть, что ионы разделяемых веществ окружены противо-
ионами буферного электролита, при увеличении концентрации бу-
фера ионы образца экранируются слоем противоионов и их элект-
рофоретическая подвижность падает.
Если два материала-носителя, в данном случае целлюлоза и вода,
с различными диэлектрическими свойствами вступают в контакт, то
они приобретают полярные заряды. В электрическом поле молекулы
воды медленно мигрируют к катоду, а вместе с ними увлекаются все
растворенные вещества. С некоторыми допущениями считают, что
все отрицательно заряженные ионы также смещаются к катоду, т. е.
движутся в «неправильном» направлении. При определении абсо-
лютной электрофоретической подвижности вещества вместе с образ-
цом вносят нейтральные соединения, например мочевину или глю-
козу. По завершении электрофореза зону нейтрального соединения
принимают за «истинную» стартовую линию. Явление переноса ней-
тральных веществ в электрическом поле называют электроосмосом.
В зависимости от типа электрофореза используют источник пи-
тания с регулируемым напряжением до 500 В, но чаще — до 1200 В.
Для достижения оптимального разделения необходимо обеспе-
чить равномерное распределение теплоты в носителе (особенно в
слое геля при гель-электрофорезе). Это обеспечивается либо эф-
фективным отводом теплоты с помощью внешнего теплоносителя,
либо применением источника, стабилизируемого по току или на-
пряжению. В процессе электрофореза меняется сопротивление но-
сителей за счет испарения воды и сила тока увеличивается. Это вле-
чет за собой еще более интенсивное испарение воды и в случае элек-
трофореза на бумаге приводит к ее пересыханию и обугливанию.
При электрофорезе на бумаге или пластинках при напряжении до
300 В перегрева не наблюдается. При высоком напряжении или
электрофорезе в геле рекомендуется поддерживать постоянной
силу тока и тем самым стабилизировать выделение теплоты.
ЭЛЕКТРОФОРЕЗ НА БУМАГЕ И В ТОНКОМ СЛОЕ
Электрофорез на бумаге позволяет экстрагировать вещества из
соответствующих зон или пятен и использовать для дальнейшей ра-
боты; обнаруживать вещества, используемые в бумажной хромато-
графии; проводить фракционирование в двух направлениях.
Для электрофореза на бумаге используют специальные сорта бу-
маги, характеризующиеся следующими свойствами: достаточной
207
механической прочностью; удовлетворительным удерживанием
достаточного количества электролита и образца.
При электрофорезе зоны расширяются за счет диффузии, осо-
бенно характерной для низкомолекулярных соединений. Однако
диффузию можно уменьшить за счет повышения напряжения элек-
трического поля, предусмотрев возможность отвода избыточной
теплоты. При высоковольтном электрофорезе эту роль выполняют
жидкие теплообменники, которые одновременно служат изолиру-
ющим материалом.
Камера для высоковольтного электрофореза на бумаге по Мих-
лю является практичной и удобной. В такой камере проводят элект-
рофорез при напряжении до 3 кВ и силе тока до 200 мА. В качестве
теплоносителя и изолирующей среды в камеру помещают гептан,
толуол, хлорбензол. Так как эти растворители токсичны и горючи,
необходимо соблюдать меры противопожарной безопасности —
размещать стеклянные камеры только в вытяжном шкафу в поддоне
достаточной вместимости.
Наряду с камерами погружного типа применяют камеры для
электрофореза в тонком слое с охлаждаемыми пластинами, в кото-
рых лист бумаги помещают между двумя изолирующими пленками.
Электрофорез в тонком слое проводят на стеклянных пластинках,
покрытых слоем носителя. По сравнению с полосками бумаги плас-
тины более удобны в обращении.
Для проведения электрофореза в тонком слое используют при-
бор с охлаждаемыми пластинами, аналогичный прибору, изобра-
женному на рис. 52. Пластины с носителем помещают на изолиро-
ванный блок (обычно алюминиевый), охлаждаемый проточной во-
дой. Чтобы исключить высушивание слоя носителя в процессе
электрофореза, его покрывают стеклянной пластиной. Мостики,
соединяющие носитель с электродными камерами, одновременно
служат прокладками между слоем носителя и верхней стеклянной
пластиной. Наиболее часто используют готовые пластины размера-
ми 10x20 и 20x20 см, покрытые силикагелем, кизельгуром, цел-
люлозой (аналогично пластинам для тонкослойной хроматогра-
фии). В некоторых случаях применяют носители с примесью люми-
нофора. При проведении электрофореза в тонком слое предпочти-
тельней использовать летучие буферы (смеси из муравьиной и
уксусной кислот, аммиака, пиридина, триэтиламина в разных соот-
ношениях и сочетаниях), что позволяет избежать побочных реак-
ций при последующем детектировании и выделять вещества в мик-
роколичествах путем экстракции из пятна или зоны.
Исследуемые образцы многократно (увеличивается нагрузка на
стартовую зону) наносят капилляром на сухую пластину в виде пятна
диаметром не более 12 мм. Между нанесениями образца раствори-
тель выпаривают при помощи фена, избегая перегрева. Пластины
равномерно опрыскивают рабочим буфером из пульверизатора до
почти прозрачного состояния (влажная пластина должна иметь вид
208
матового стекла). Затем устанавливают электродные мостики. Прибор
накрывают крышкой, включают охлаждение и проводят электрофорез
при напряжении 900—1000 В в течение 15 мин. Пластины быстро вы-
сушивают и обрабатывают соответствующим проявителем.
Электрофорез на бумаге и в тонком слое применяют для иссле-
дования фракций, полученных при колоночной хроматографии,
ферментативных гидролизатов белков, метаболитов, а также для
разделения аминов, аминокислот, пептидов и белков, нуклеотидов,
фенолов, нафтолов, фенолкарбоновых кислот, красителей, неорга-
нических соединений.
ГЕЛЬ-ЭЛЕКТРОФОРЕЗ
Вместо целлюлозы и силикагеля можно использовать мягкие
гели. Ниже приведены основные рабочие стадии проведения элект-
рофореза в слое геля: приготовление гелей и подготовка образца
—> электрофоретическое разделение —> детектирование —> анализ ре-
зультатов и оформление их в рабочем журнале.
Из множества гелей на практике применяют только два — гели
агарозы и полиакриламида. С помощью агарозы можно анализиро-
вать сложные биополимеры, например ферментные комплексы,
липопротеиды, ДНК и РНК. Несмотря на небольшую концентра-
цию агарозы (не более 0,2 %), необходимую для получения крупно-
пористого геля, гели агарозы характеризуются достаточно высокой
механической прочностью.
В зависимости от способа приготовления геля и типа буферной
системы различают несколько вариантов метода:
электрофорез в геле полиакриламида (ПААГ);
диск-электрофорез (диск-ПААГ) в прерывистой буферной сис-
теме;
электрофорез в геле полиакриламида в присутствии додецил-
сульфата натрия (Д СН - ПААГ);
электрофорез в градиенте пористого полиакриламидного геля.
Электрофорез в геле полиакриламида используют для эффектив-
ного разделения и идентификации биополимеров (полипептидов,
белков, нуклеиновых кислот). Гель формируют в виде блоков или
столбиков, которые располагают в электрофоретической камере вер-
тикально (рис. 53). В последнее время в основном применяют элек-
трофорез в блоке, так как можно сопоставлять несколько образцов.
При диск-электрофорезе в ПААГ гель также формируют в виде
блока (пластинки) или столбиков, однако составляют из двух слоев
различной пористости — крупнопористого (концентрирующего) и
мелкопористого (разделяющего) геля. Его формируют в две стадии.
Вначале деаэрируют и полимеризуют разделяющей гель, затем насла-
ивают разбавленный раствор мономеров (слой высотой 1 см) с боль-
шим значением pH и еще раз полимеризуют. Буфер в электрод-
209
Рнс. 53. Камера для электрофореза в верти-
кальном блоке геля:
1— камера; 2—электроды; 3 — перистальтичес-
кий насос; 4 — змеевик для подачи теплоносителя
(хладагента); 5 — слой геля между двумя стеклян-
ными пластинками; <5—изолирующая про-
кладка; 7— верхняя электродная камера
ных камерах по значению pH примерно соответствует рабочему
гелю, но содержит более слабую по сравнению с соляной кислоту
(например, глицин).
После нанесения пробы на электроды подают напряжение. При
этом все анионы (С1~) начинают двигаться к аноду. Йоны глицина
мигрируют в слой концентрирующего геля, и раствор в этой облас-
ти приобретает pH, близкий по значению к изоэлектрической точ-
ке, т. е. ионы глицина практически теряют заряд. Зона, где находит-
ся образец, становится почти нейтральной, электропроводность
резко падает, возникает высокий градиент напряженности поля,
что, в свою очередь, вызывает ускорение миграции молекул белка.
Скорость миграции ионов хлора намного превышает скорость
миграции белков. Поэтому увеличивается градиент напряженности
поля, еще больше возрастает скорость миграции молекул белка. Че-
рез некоторое время молекулы белка попадают в область высокой
концентрации ионов хлора, т. е. в область с небольшим градиентом
напряженности поля, и их подвижность резко падает. В итоге белки
концентрируются на границе между двумя зонами — иона хлора и
иона глицина. Поскольку в рабочем (разделяющем) геле среда бо-
лее кислая, равновесие глицин — глицин Н+ сильно сдвинуто впра-
во, что компенсирует нехватку ионов слабой кислоты. Дальнейшее
разделение идет в обычных условиях.
При электрофорезе в ДСН-ПААГ критерием степени очистки бел-
ков является наличие одной зоны. При этом белки разделяются строго
по молекулярной массе. Предварительно белки в образце растворяют в
детергенте ДСП. При взаимодействии ДСП с полипептидными цепя-
ми образуется комплекс ДСН-белок, характеризующийся постоян-
ным зарядом на единицу молекулярной массы. Этот метод позволяет с
достаточной точностью и небольшими затратами определять молеку-
лярную массу белков. Для облегчения задачи выпускают разнообраз-
ные наборы белков-маркеров с известными молекулярными массами.
Стандартную смесь вносят по обе стороны от точки нанесения иссле-
дуемого белка. После проведения электрофоретического разделения
по электрофоретической подвижности комплекса путем экстраполя-
ции определяют молекулярную массу исследуемого белка.
210
Электрофорез в градиенте пористого ПААГ используют для раз-
деления нативных и денатурированных биополимеров. Единствен-
ным условием его применения является одноименный заряд разде-
ляемых веществ. Градиент пористости геля формируют путем пода-
чи в рабочую камеру раствора мономеров, концентрация которых
плавно уменьшается. При этом пористость геля возрастает в на-
правлении снизу вверх (в случае белков — от анода к катоду).
В градиенте ПААГ разделяют вещества по молекулярным массам в
области 50—5000 г/кмоль. Электрофоретическая подвижность разде-
ляемых веществ при этом имеет второстепенное значение. Особенно
узкие зоны формируются при высоком напряжении, так как в этом
случае заряженные вещества под действием высокого поля буквально
вдавливаются в мелкопористую сетку геля. Этим самым исключается
возможность расширения зон вследствие броуновского движения.
Для приготовления геля кроме штатива для фиксации электро-
форетической камеры необходимы пластинки или трубочки. Для
получения градиентных гелей нужны также перистальтический на-
сос и градиентный смеситель. Получать гели самостоятельно нео-
бязательно, можно пользоваться готовыми пластинками.
Электрофорез проводят в камере с охлаждаемой горизонтальной
пластиной или вертикальной камере. Последняя состоит из следую-
щих деталей: нижней электродной камеры (выравнивание темпера-
туры в камере проводится за счет принудительной циркуляции жид-
кости), теплообменника из инертного материала, верхней камеры (в
ней обычно фиксируют верхнюю часть пластинки с рабочим гелем).
Чаще всего используют источник питания, обеспечивающий на-
пряжение до 500 В и силу тока до 400 мА.
Гель окрашивают красителем. Поскольку молекулы красителя
заряжены, гель можно обесцвечивать электрофоретически при на-
пряжении 50 В. В местах, не содержащих исследуемое вещество,
гель обесцвечивается. Количественную оценку проводят спектро-
фотометрически при помощи сканирующего денситометра.
После усадки гелей в водном этаноле или ацетоне их высушива-
ют между двумя листами целлофана в вакууме при слабом нагреве.
Гель-электрофорез применяют для разделения всех классов за-
ряженных веществ, например белков, ферментных комплексов, ви-
русов, олигонуклеотидов и нуклеиновых кислот; определения мо-
лекулярных масс биополимеров; анализа белков на микроуровне
(антигенов при количественном иммуноэлектрофорезе).
ЭЛЕКТРОФОРЕЗ В СВОБОДНОМ ПОТОКЕ
При электрофорезе в свободном потоке электролит (буфер) пере-
мещается в вертикальном направлении (перпендикулярно направле-
нию электрического поля). Заряженные частицы под действием
электрического поля мигрируют в горизонтальном направлении и
211
Рис. 54. Электрофорез в свободном по-
токе:
/ — градиент электрического поля; 2— бу-
ферный раствор; 5 — разделяемая смесь;
4 — направление потока электролита; 5—
электрод; 6— коллектор для сбора фракций
одновременно увлекаются по-
током буфера. В итоге разделен-
ные вещества распределяются в
потоке в соответствии с их элек-
трофоретической подвижнос-
тью и элюируются из прибора в
различных фракциях.
В основе метода лежат те же принципы, что и для непрерывного
зонального электрофореза, однако здесь поток электролита пере-
двигается перпендикулярно электрическому полю (рис. 54). Зоны
стабилизируются при помощи потока достаточно разбавленного
электролита.
Электрофорез в свободном потоке проводят в приборе Ханни-
га. Смесь разделяемых веществ и рабочий буфер непрерывно по-
дают в верхнюю часть камеры между двумя охлаждаемыми стек-
лянными пластинами, расстояние между которыми 0,6 мм. Ком-
поненты смеси мигрируют под действием электрического поля в
соответствии с их суммарным зарядом в направлении электро-
дов. Одновременно они передвигаются под действием потока
электролита в направлении сверху вниз. В нижней части поток
разделяется на 48 отдельных потоков. В стандартных условиях
компоненты смеси элюируются в одних и тех же зонах, поэтому
можно работать в непрерывном режиме, накапливая вещества в
сборниках коллектора.
Электрофорез в свободном потоке применяют для препаратив-
ного разделения заряженных частиц, в том числе коллоидных, суб-
клеточных частиц и клеток.
ИЗОЭЛЕКТРИЧЕСКОЕ ФОКУСИРОВАНИЕ
С помощью изоэлектрического фокусирования (ИЭФ) по изо-
электрическим точкам (ИЭТ) разделяют амфотерные вещества, в
частности белки. Сущность метода заключается в том, что молеку-
лы белков мигрируют под действием электрического поля в среде с
линейным и стабильным градиентом pH до достижения области
pH, соответствующей их ИЭТ.
212
Изоэлектрическое фокусирование отличается от зонального
электрофореза тем, что разделение осуществляется не в буфере с по-
стоянным значением pH, а в среде с линейным градиентом pH. Зна-
чение pH минимально вблизи анода, максимально — вблизи катода.
В области высоких значений pH аминокислоты и белки заряже-
ны отрицательно и мигрируют в электрическом поле в направлении
к аноду, т. е. в область низких значений pH. В этой области молеку-
лы белка перезаряжаются за счет протонирования, вследствие чего
направление движения изменяется на противоположное. В конеч-
ном счете молекулы попадают в область, где их суммарный заряд
равен нулю, вследствие чего они утрачивают электрофоретическую
подвижность. Молекулы, находившиеся в начале процесса в кислой
области (низкие значения pH), мигрируют по направлению к като-
ду. При повышении pH среды они также попадают в зону, где утра-
чивают заряд и электрофоретическую подвижность. Когда же моле-
кулы белков покидают область ИЭТ вследствие диффузии, они
вновь приобретают заряд и начинают передвигаться под действием
электрического поля до достижения ими прежнего положения. Чем
выше напряженность поля, тем меньше влияние диффузии, тем уже
зоны разделяемых веществ.
Главное условие эффективного разделения белков — наличие
стабильного градиента pH среды. В связи с тем что белки обладают
амфотерными свойствами, необходимо, чтобы амфолиты-носите-
ли — вещества, с помощью которых формируется градиент pH, об-
ладали высокой буферной емкостью. Амфолиты-носители пред-
ставляют собой многокомпонентную смесь изомеров и гомологов
алифатических поли аминополикарбоновых кислот, сульфокислот
и фосфоновых кислот, изоэлектрические точки которых располага-
ются в широкой области значений pH.
При отсутствии электрического поля раствор амфолитов-носи-
телей имеет значение pH, равное 7. В электрическом поле амфоли-
ты распределяются в соответствии со значениями их изоэлектри-
ческих точек. Например, вещества с минимальными значениями р!
(высокий отрицательный заряд) располагаются в кислой области
вблизи анодного раствора. При попадании в зону электролита
(вследствие диффузии) молекулы амфолитов заряжаются положи-
тельно и под действием поля вновь возвращаются в исходную об-
ласть, где таким образом формируется pH, близкий их изоэлектри-
ческой точке. По аналогичному механизму в прилегающем слое
концентрируются амфолиты-носители с более высокими pl, фор-
мируя тем самым область с более высоким значением pH. Этот про-
цесс распространяется вдоль рабочей камеры вплоть до катодного
электролита. Каждый раз, когда молекулы амфолитов-носителей
диффундируют в соседнюю зону, они вновь под действием поля
возвращаются в исходное положение. В результате в системе фор-
мируется плавный градиент pH, который остается устойчивым до
тех пор, пока находится в электрическом поле.
213
При отсутствии информации о значениях изоэлектрических то-
чек белков разделение проводят в широком диапазоне значений
pH — от 2 до 11, не ставя задачу достижения высокой степени разре-
шения. Таким образом определяют pl отдельных компонентов сме-
си. Затем в зависимости от распределения изоэлектрических точек
выбирают минимальный диапазон pH для последующего опыта.
При благоприятных обстоятельствах удается разделять вещества с
разницей в значениях pl около 0,01.
ИЭФ применяют для аналитического разделения (мкг) пепти-
дов, белков, нуклеотидов, органических кислот, ионов металлов и
препаративного разделения белков (г); накопления следовых коли-
честв веществ (мкг) из больших объемов пробы; определения элект-
рофоретической подвижности.
Необходимым условием проведения ИЭФ является наличие вы-
сокого напряжения при низкой ионной силе раствора. Однако
именно в этих условиях усиливается электроосмос. Отрицательное
воздействие на эффективность разделения веществ оказывают так-
же примеси солей, занесенные вместе с реактивами (гели для ИЭФ
следует готовить из особо чистых реактивов).
Для проведения ИЭФ более всего подходит полиакриламидный
гель с низкими электроосмотическими свойствами.
Продолжительность эксперимента зависит от напряженности
поля и характера изменения рН-градиента.
При быстром изменении градиента белок с pH, близким к изо-
электрической точке, еще сохраняет заряд и достаточно высокую
подвижность. При медленном изменении градиента белки имеют
небольшой суммарный заряд даже в области pH, далекой от изо-
электрической точки. Следовательно, скорость их миграции в ко-
нечную зону невысока. По этой причине для проведения экспери-
мента при медленном изменении pH требуется вдвое больше време-
ни (3 ч) по сравнению с разделением с быстрым изменением гради-
ента (1,5 ч).
Препаративное изоэлектрическое фокусирование проводят в
вертикальных колонках (в градиенте плотности сахарозы, глицери-
на, этиленгликоля) или в слое инертного материала. В качестве та-
ких материалов используют гранулированные гели (pH градиент
формируют с помощью амфолитов).
Процесс выделения биологически активных веществ, в частно-
сти белков, представленный на схеме 6, начинается, как правило, с
проведения экстракции. Затем целевое вещество осаждают (на-
пример, сульфатом аммония) в области изоэлектрической точки
при высокой ионной силе (при этом низкомолекулярные примеси
остаются в растворе) и адсорбируют на ионообменниках. После-
дние используют в статическом режиме (Batch-методика), т. е.
ионит в активированной форме суспендируют в растворе, содержа-
щем целевое вещество. После достижения равновесия вещество от-
деляют фильтрованием, тщательно промывают, а затем десорбиру-
214
Схема 6. Выделение целевого вещества из сырья биологического происхождения
ют вместе с сопутствующими белками буфером с высокой ионной
силой. На этой стадии помимо частичной очистки, в том числе и от
солей, достигается также и концентрирование целевого вещества.
Далее раствор белка обессоливают.
Многие белки адсорбируются (при высокой ионной силе) на
гидрофобных гелях (например, октил- или фенил-сефарозе), отку-
да они могут быть десорбированы дистиллированной водой. Этот
метод также позволяет достигнуть значительного обогащения сме-
си целевым веществом. Большую часть сопутствующих примесей
обычно отделяют методом гель-хроматографии. Условия разделе-
ния удается оптимизировать, если известна молекулярная масса це-
левого вещества. После гель-хроматографии получают разбавлен-
ный раствор целевого вещества в буфере с низкой ионной силой.
215
Следующей стадией является очистка методом ионообменной
хроматографии. Условия фракционирования удается оптимизиро-
вать, если известна изоэлектрическая точка целевого вещества. При
нанесении препарата на ионит компоненты смеси удерживаются
матрицей, затем их элюируют избирательно в градиенте ионной
силы или значений pH. Буферный раствор заменяют для последую-
щей обработки или операций контроля методами мембранной
фильтрации, диализа или гель-хроматографии. Способы последую-
щей обработки или контроля зависят от молекулярной массы целе-
вого вещества. При молекулярной массе до 3000 используют методы
ТСХ или БХ, электрофоретические методы; при молекулярной
массе более 3000 — электрофорез в ПААГ или изоэлектрическое
фокусирование. В процессе выделения целевые вещества следует
растворять преимущественно в летучих буферных растворах. Об-
разцы хранят в замороженном состоянии (при —30 °C) до последую-
щей обработки или высушивают лиофильно (если предстоит дли-
тельное хранение). Наряду с перечисленными методами применя-
ют и аффинную хроматографию, главным образом на завершающей
стадии очистки. В последнее время все более широкое распростра-
нение находит метод ВЭЖХ высокого давления, в основном для
очистки низкомолекулярных веществ.
ОПРЕДЕЛЕНИЕ ФРАКЦИЙ БЕЛКОВ МОЛОКА
С ПРИМЕНЕНИЕМ ЭЛЕКТРОФОРЕЗА НА БУМАГЕ
(ПО Е. А. ЖДАНОВОЙ И И. Н. ВЛОДАВЦУ)
Основано на движении заряженных частиц белков в буферном
растворе на фильтровальной бумаге под действием внешнего элек-
тростатического поля.
Выполнение работы. Исследование раствора казеина. В химичес-
кий стакан вместимостью 1-50 см3 вносят 50 см3 обезжиренного мо-
лока, нагревают до 40 °C и осаждают казеин 1 н. раствором уксусной
кислоты, приливая ее осторожно при помешивании до образования
хлопьевидного (зернистого) осадка и просветления над ним сыво-
ротки. Осадку дают отстояться, сыворотку осторожно сливают. К
осадку добавляют 50 см3 воды, дают отстояться, промывную воду с
осадка сливают, стараясь не затрагивать осадка. Промывку прово-
дят 5 раз. Осадок растворяют в 25 см3 вероналевого буфера (реактив 1)
и под вакуумом фильтруют через бактериальный фильтр Зейца.
Концентрация казеина в буферном растворе составляет около 5 %.
На полоски фильтровальной хроматографической бумаги разме-
ром 40 х 400 мм точно в центре или несколько ближе к краю, обра-
щенному к катоду, простым карандашом проводят стартовую ли-
нию и еще две линии на расстоянии 70 мм от краев. На одном из
концов полоски бумаги карандашом записывают условия опыта.
Полоску бумаги пропускают через кювету с буферным раствором.
216
Края полоски на расстоянии 50—60 мм должны остаться сухими.
Излишки жидкости удаляют, положив полоску между листами
фильтровальной бумаги.
Конец чистого предметного стекла шириной 2,5 см погружают в
исследуемый раствор, дают стечь излишкам раствора и приклады-
вают край стекла под некоторым углом к стартовой линии, оставляя
в таком положении на несколько секунд, чтобы раствор мог перей-
ти на полоску. Затем заряжают электрофорекамеру. Вынув штатив,
помещают полоски бумаги таким образом, чтобы на полочках, оги-
бая перемычку, они располагались одна на другой. Концы полосок
совмещают.
Штатив с полосками бумаги помещают в электрофорекамеру,
концы полосок должны быть погружены в раствор буфера. Камеру
закрывают крышкой, запирают и включают электропитание. Регу-
лятор напряжения устанавливают на 200 В, включают миллиампер-
метр. Напряжение тока должно составлять 250—320 В, продолжи-
тельность электрофореза — 16—18 ч, сила тока 0,5—0,7 мА на каж-
дую полоску. По окончании электрофоретического разделения
электропитание отключают, снимают крышку с камеры и высуши-
вают бумажные полоски в сушильном шкафу 10—20 мин при темпе-
ратуре 105 °C, подвешивая их за концы зажимами или на специаль-
ном штативе.
После высушивания полоски бумаги окрашивают раствором
бромфенолового синего (реактив 2), погружая их в эмалированную
ванночку с красителем. Через 20 мин раствор красителя сливают.
Полоски бумаги промывают несколько раз 5%-ным раствором ук-
сусной кислоты, пока промывная жидкость и участки бумаги, не
содержащие белка, не станут совершенно бесцветными. Вынув по-
лоски из ванночки, их высушивают в сушильном шкафу при темпе-
ратуре 105 °C. Для отчетливой видимости окрашенных пятен белко-
вых фракций электрофореграмму держат несколько секунд над рас-
крытой банкой с концентрированным раствором аммиака.
Для относительной количественной оценки содержания отдель-
ных фракций простым карандашом разбивают электрофореграмму
линиями, параллельными стартовой черте, на полоски по 0,5 см
шириной. Проставляют на полосках порядковые номера и разреза-
ют электрофореграмму по линиям. Полоски помещают в сухие про-
бирки, указывая на них номер полоски. В пробирки приливают по
3 см3 0,01 н. раствора гидроксида натрия. Через каждые 15 мин жид-
кость в пробирках перемешивают, закрыв их пробками и несколько
раз перевертывая. Раствор окрашивается в синий цвет.
Через 1 ч приступают к фотометрированию растворов на прибо-
ре типа ФЭК-М в кюветах 30 мм с зеленым светофильтром при дли-
не волны 620 нм. Данные показаний об относительном содержании
красителя в различных полосках бумаги, пропорциональные числу
белковых фракций, изображают графически столбчатой диаграм-
мой, в которой слева направо по порядку номеров располагают
217
столбики, соответствующие отдельным полоскам бумаги. Высота
этих столбиков в масштабе соответствует найденному фотометри-
чески содержанию красителя в каждой полоске.
Для большей наглядности вершины столбиков соединяют не-
прерывной линией. Для количественной оценки содержания от-
дельных фракций суммируют высоты всех столбиков, относящихся
к одной и той же фракции. При этом высоту столбиков на границе
двух различных фракций делят для подсчета пополам.
При разбивке электрофореграммы на полоски обязательно
включают участки, не содержащие белка. Результаты колориметри-
рования показывают, что полоски бумаги без белка слегка окраше-
ны. Высота столбиков, соответствующих таким полоскам, усредня-
ется. Это значение принимают за нулевое (отсутствие белка) и вы-
соту остальных столбиков отсчитывают от этого значения.
Исследование сывороточных белков. Обезжиренное молоко
(0,5 дм3) нагревают до 40 °C и казеин осаждают 1 н. раствором ук-
сусной кислоты. Отстоявшуюся сыворотку фильтруют через бумаж-
ный складчатый фильтр и нейтрализуют 1 н. раствором аммиака до
pH 6,5 (по индикаторной бумажке). Добавляют на 100 см3 сыворот-
ки 70 г сухого кристаллического сульфата аммония и тщательно пе-
ремешивают до его полного растворения. Осажденные белки от-
фильтровывают через складчатый бумажный фильтр.
Оставшийся на фильтре белковый слой снимают осторожно
шпателем и переносят в целлофановый мешочек, который погру-
жают в стакан с дистиллированной водой и помещают в холодиль-
ник при температуре 8—10 °C. Диализ против дистиллированной
воды проводят 24 ч при частой ее смене и 24 ч против вероналового
буфера (ионная сила 0,1; pH 8,5), не содержащего мочевины (реак-
тив 1), или только против дистиллированной воды. В мешочке об-
разуется раствор сывороточных белков. Его фильтруют через бакте-
риальный фильтр Зейца и подвергают электрофорезу на бумаге, как
раствор казеина.
На основании колориметрических исследований отдельных
фракций белков в казеине и сыворотке молока устанавливают коли-
чественное соотношение между ними в молоке или сыворотке.
Если показания оптической плотности при колориметрировании
фореграммы казеина молока составляют для а-казеина 0,50,0-казе-
ина 0,83, у-казеина 0,08, то относительное соотношение их находят
по формуле
где а — показание по фотоколориметру оптической плотности каждой фракции;
б — сумма показаний всех фракций.
Приготовление реактивов. Реактив 1 (вероналовый буфер; ионная
сила 0,1; pH 8,6). 0,01 моля (20,6 г) мединала (диэтилбарбитурово-
218
кислого натрия), 0,01 моля (1,84 г) веронала (диэтибарбитуровой
кислоты) и 400 г мочевины растворяют в 1 дм3 воды.
Реактив 2 (раствор бромфенолового синего), а) 0,2 г бромфеноло-
вого синего и 1 г хлорной ртути растворяют в 100 см3 2%-ного ра-
створа уксусной кислоты; б) 2 г хлорида аммония, 10 г каломели и
1 г бромфенолового синего растворяют в 1 дм3 воды. Профильтро-
ванный раствор применяют через сутки.
ОПРЕДЕЛЕНИЕ ФРАКЦИЙ БЕЛКОВ МОЛОКА
С ПРИМЕНЕНИЕМ ДИСК-ЭЛЕКТРОФОРЕЗА
(ПО К. БУТКУСУ И А. АНТАВИЧЮСУ)
Метод основан на движении заряженных частиц‘белков в ком-
бинированной системе буферных растворов с применением раз-
личной пористости акриламидного геля под действием внешнего
электростатического поля.
Выполнение работы. Образцы белков готовят следующим обра-
зом. Казеин осаждают из молока 1 н. уксусной кислотой при pH 4,6.
Осадок промывают несколько раз подкисленной до pH 4,6 дистил-
лированной водой и лиофилизируют. Лиофилизированный осадок
48 ч диализируют против дистиллированной воды и 48 ч — против
реактива 5. После диализа готовят 0,5%-ный белковый раствор в бу-
фере (реактив 5) с 7 М мочевиной.
Сывороточные белки из сыворотки осаждают сульфатом аммо-
ния до насыщенного раствора и на ночь ставят в холодильник. Ра-
створ фильтруют через бумажный фильтр. Осадок диализируют 48 ч
против дистиллированной воды и лиофилизируют. Перед электро-
форезом образец еще раз диализируют 48 ч против дистиллирован-
ной воды и 48 ч — против буфера (реактив 5). После диализа гото-
вят 0,5%-ный белковый раствор в 25%-ном растворе сахарозы на
буфере (реактив 5). Диализ проводят в холодильнике при 2—4 °C.
При той же температуре хранят и приготовленные образцы.
Диск-электрофорез приготовленных образцов белков проводят
в стеклянных трубках внутренним диаметром 5 мм и длиной 75 мм.
В качестве электродной камеры используют конический полиэти-
леновый стакан вместимостью 0,5 дм3. Нижней электродной каме-
рой служит стеклянный цилиндр, а электродами — платиновая
проволока. В качестве источника постоянного тока применяют
универсальный источник питания УИП-1. Электрофорез проводят
при силе тока 2,5 мА на трубку в течение 2 ч. Для одинакового и рав-
номерного освещения при полимеризации гелей используют ящик
с люминесцентными лампами.
В верхний слой крупнопористого геля можно вносить не более
0,15 мг белка. Это количество можно варьировать от 0,05 до 0,15 мг
в зависимости от фракционного состава исследуемых белков. Для
нерастворимых в воде белков сыра это количество можно увели-
219
чить, а для казеина молока — уменьшить. Во время проведения
опыта или серии опытов масса вносимого в гели белка должна быть
одинаковой, так как от этого зависит точность опыта. При внесении
в гель больше 0,2 мг белка количественная оценка без разрезания
геля невозможна.
После электрофореза колонку геля извлекают из трубки и 30 мин
фиксируют в растворе 7%-ной уксусной кислоты. Не связанная с
белком краска отмывается 7%-ной уксусной кислотой.
Окрашенный гель помещают в специальную кювету и заливают
7%-ным раствором уксусной кислоты. При фотометрировании кю-
вету с гелем фиксируют, чтобы луч проходил только через центр ко-
лонки геля. Можно применять кювету и другой конструкции.
Для количественной оценки используют быстродействующий
фотометр. Масштаб записи —1:5 при скорости передвижения бу-
маги 60 мм/мин и частоте вращения ведомого вала 3 об/мин.
Отдельные фракции на графике отделяют перпендикулярами и
устанавливают площади под пиком весовым методом. По относи-
тельной массе определяют количества белка отдельных фракций в
процентах.
Относительную электрофоретическую подвижность ОЭП от-
дельных фракций определяют по мигрирующей вместе с рибофла-
вином самой подвижной фракции, принимая длину пути ее мигра-
ции за единицу, по следующей формуле:
оэп=£
где Р — длина пути мигрирования фракции с рибофлавином, см; Д— длина пути
мигрирования отдельной фракции, см.
Приготовление реактивов. Колонка геля состоит из трех слоев:
нижнего мелкопористого (концентрация акриламида 10 %), сред-
него крупнопористого (концентрация акриламида 5 %) и верхнего
также крупнопористого геля, содержащего белок (табл. 22).
22. Состав мелкопористого и крупнопористого гелей
Номер реактива 10%-ный мелко- пористый гель, см5 Номер реактива 5%-ный крупно- пористый гель, см5
1 1,5 5 2,0
2 4,3 6 6,4
3 1,2 Дистиллированная 5,6
вода
4 5,0 7 2,0
Примечание. Реактивы 1—4 — для нижнего слоя геля; реактивы 5—7 — для сред-
него и верхнего.
220
Реактив 1 (pH 8,5). 48 см3 1 н. соляной кислоты, 36,6 г трис
(2-амино-2-гидроксиметилпропандиол-1,3), 0,23 см3 ТЕМЕД
(NXNXN\NX — тетраметилэтилендиамин), до 100 см3 дистиллиро-
ванной воды.
Реактив 2. 28 г акриламида, 0,735 г метиленбисакриламида, 42 г
мочевины, до 100 см3 дистиллированной воды. Для сывороточных
белков в реактив 2 мочевину не вводят.
Реактив 3. 0,05 г персульфата аммония, до 100 см3 дистиллиро-
ванной воды.
Реактив 4.42 г мочевины, до 100 см3 дистиллированной воды.
Реактив 5 (pH 6,7). 48 см3 1 н. соляной кислоты, 5,98 г трис,
0,46 см3 ТЕМЕД, до 100 см3 дистиллированной воды.
Реактив 6. 10 г акриламида, 25 г метиленбисакриламида, до 100
см3 дистиллированной воды.
Реактив 7. 4 мг рибофлавина, до 100 см3 дистиллированной
воды.
Реактив 8. Электродный буферный раствор, величина pH кото-
рого 8,3 — 6 г трис, 28,8 г глицина, до 1000 см3 дистиллированной
воды, перед электрофорезом разводят в пропорции 1:10 дистилли-
рованной водой.
Контрольные вопросы. 1. В чем заключается сущность электрофореза? 2. Каковы
особенности зонального электрофореза? 3. Как проводят гель-электрофорез? 4. Что
представляет собой электрофорез в свободном потоке?
Г лава 9
ЭБУЛИОСКОПИЯ И КРИОСКОПИЯ
Методы эбулиоскопии и криоскопии основаны на измерении
повышения точки кипения и соответственно понижения точки за-
мерзания раствора анализируемого вещества по сравнению с чис-
тым растворителем. Оба явления объясняются понижением давле-
ния пара раствора относительно чистого растворителя.
Повышение температуры кипения и понижение температуры
замерзания растворов зависят от концентрации (так называемые
коллигативные эффекты). Для очень разбавленных растворов спра-
ведливо следующее уравнение:
Мп=КС- 103/ДТ,
где М„ — молекулярная масса; К— эбулиоскопическая (или криоскопическая) по-
стоянная, К= R Т2/(103Нр); С — концентрация раствора; Д Т— соответственно по-
вышение температуры кипения или понижение температуры замерзания; R — уни-
версальная газовая постоянная; Т— абсолютная температура кипения или замерза-
ния растворителя; Нр — удельная теплота испарения растворителя.
Эбулиоскопическая Кэ и криоскопическая Кк постоянные рав-
ны соответственно повышению точки кипения и понижению точ-
ки замерзания раствора, содержащего 1 моль вещества, т. е.
6,02 1023 недиссоциированных частиц в 1 кг растворителя. Точки
кипения и замерзания наиболее часто применяемых растворите-
лей, а также молярные постоянные приведены в табл. 23.
Криоскопию чаще, чем эбулиоскопию, применяют для опреде-
ления молекулярной массы, вследствие более высоких значений
криоскопических постоянных по сравнению с эбулиоскопически-
ми, а также более простых конструкций приборов для криоскопии.
23. Криоскопические и эбулиоскопические постоянные некоторых растворителей
Растворитель Температура плавления, °C Температура кипения, С
"С/(мо ль кг)
Вода 0 100 1,86 0,52
Ледяная уксусная кислота 17 118 3,90 3,07
Диоксан 12 102 4,80 3,45
Хлороформ — 61 4,98 3,80
Бензол 5 80 5,49 2,64
Фенол 41 181 7,30 3,60
Камфора 179 204 40 6
222
Концентрацию растворов макромолекул, например растворов
биологически активных веществ, характеризуют осмоляльнос-
тью или осмолярностью, т. е. отношением количества растворен-
ных частиц к 1 кг воды (моль/кг) или к 1 л раствора (моль/л) со-
ответственно. Несмотря на незначительное различие между обе-
ими величинами, чаще используют осмоляльность, так как в
этом случае учитывается уменьшение объема раствора за счет
макромолекул.
Большинство криоскопических приборов работает следующим
образом: образец (около 2 см3) помещают в охладительную смесь и
охлаждают до определенной заранее температуры (на 5—7 °C ниже
точки замерзания). Затем вызывают мгновенную кристаллизацию
при помощи быстрого и эффективного перемешивания. На графи-
ке зависимости температуры от времени моменту кристаллизации
растворителя соответствует плато, а моменту кристаллизации ра-
створа — точка перегиба. Температуру, соответствующую точке пе-
региба, используют при расчетах.
ОПРЕДЕЛЕНИЕ ТОЧКИ ЗАМЕРЗАНИЯ МОЛОКА
Метод основан на измерении понижения точки замерзания мо-
лока по сравнению с точкой замерзания бидистиллированной воды.
Его используют для определения натуральности молока.
Выполнение работы. Точка замерзания молока должна опреде-
ляться не ранее чем через 3 ч после дойки при кислотности молока
не выше 19—20 °Т. В консервированных пробах молока точку за-
мерзания не определяют. Для анализа используют ручной криоскоп
с метастатическим термометром типа ТЛ-1 (рис. 55), который встав-
ляют в пробирку и при помощи манжета фиксируют на расстоянии
13—15 мм от конца термометра до дна пробирки. Нулевую точку
термометра настраивают переливанием ртути из запасного резерву-
ара в основной, погружая нижний основной резервуар в пробирку с
бидистиллированной водой температурой 0 °C и руководствуясь
инструкцией, приложенной к метастатическому термометру. Ме-
ниск столбика ртути при 0 °C должен находиться в средней части
шкалы, в пределах делений от 2 до 4. Нулевая точка термометра оп-
ределяется ежедневно в начале и по окончании работы по точке за-
мерзания бидистиллированной свежевскипяченной и охлажденной
воды. Термометр рекомендуется держать в вертикальном положе-
нии, погруженным в пробирку с дистиллированной водой темпера-
турой 0—10 °C, а перед работой необходимо выдерживать не менее
1 ч в таящем льду. Мыть термометр следует в пробирке с дистилли-
рованной водой температурой от 0 до 2 °C. Во время перенесения
термометра в следующую пробу столбик ртути не должен подни-
маться выше делений, а на мешалке и термометре не должно быть
кристалликов льда.
223
Рис. 55. Ручной криоскоп с метастати-
ческим термометром:
1 — сосуд (изолированный) или широко-
горлый термос; 2— пробирка для пробы;
3 — пробирка для приготовления кристал-
ликов льда; 4 — металлическая проволока с
петлей; 5 — пробка для пробирки с пробой;
6 — термометр метастический типа ТЛ-1;
7 — лупа; 8—термометр стеклянный техни-
ческий; 9 — мешалка для пробы; 10— ме-
шалка для охлаждающей смеси; 11 — крыш-
ка охлаждающего сосуда; 12— манжет
Для градуировки термомет-
ров применяют растворы хло-
рида натрия с теоретическими
точками замерзания - 0,422 и
-0,621 °C (реактив) и в соот-
ветствии с методикой проведе-
ния анализа устанавливают их
точки замерзания, которые
применяют для расчета уточ-
ненного значения точки за-
мерзания молока. Термометр
градуируют один раз в пять-
шесть месяцев для данной ну-
левой точки термометра.
Подготовленную пробу молока, градуировочные растворы
хлорида натрия (реактив) или бидистиллированную воду нали-
вают в пробирку до метки и охлаждают в сосуде первичного ох-
лаждения до 1—1,5 °C. Пробирку с пробой и вставленным (точ-
но вертикально!) метастатическим термометром помещают в
охлаждающий сосуд с постоянно поддерживаемой во время ис-
пытания температурой — 4 °C. В течение всего времени опреде-
ления следует помешивать пробу перемещением мешалки
вверх — вниз со скоростью — одно перемещение в секунду. Го-
ризонтальная петля мешалки не должна подниматься выше про-
бы. При падении столбика ртути термометра на 1 — 1,1 °C ниже
предполагаемой точки замерзания в пробирку с пробой через от-
верстие вводят кристаллики льда, после чего прекращают поме-
шивать на 4—5 с. Когда столбик ртути начнет подниматься, про-
должают помешивание пробы в течение 25 с, а затем — на 60 с
прекращают.
Спустя 90 с после введения кристалликов льда, когда столбик
ртути обычно останавливается, пробу три раза помешивают, затем
224
слегка постукивают по термометру около точки остановки столбика
ртути, после чего с помощью лупы отсчитывают показания на шка-
ле. При этом глаз наблюдателя должен находиться на уровне гори-
зонтальной касательной к мениску столбика ртути так, чтобы
штрих шкалы в точке отсчета был виден прямолинейно. После пер-
вого отсчета все операции (помешивание, постукивание и отсчет)
повторяют еще два раза через 20 с каждую. Показания на метастати-
ческом термометре отсчитывают при помощи лупы с точностью
0,001 °C. Разность в показаниях второго и третьего отсчетов не дол-
жна превышать 0,003 °C.
За результат показания термометра принимают среднее арифме-
тическое результатов второго и третьего отсчетов.
Разность между показаниями на метастатическом термометре
точек замерзания бидистиллированной воды и градуировочных ра-
створов (или молока) составляет точку замерзания пробы.
В том случае, если проба преждевременно замерзает, не достиг-
нув необходимой температуры переохлаждения, измерение прекра-
щают. При проведении серии определений точки замерзания моло-
ка температура в рабочем помещении не должна изменяться более
чем на 1 °C.
За результат анализа принимают среднее арифметическое ре-
зультатов двух параллельных определений, допускаемые расхож-
дения между которыми не должны превышать 0,005 °C. Если
расхождения превышают установленные, то анализ следует пов-
торить.
Уточненное значение точки замерзания молока (°C)
/=0!6^22(/м_?1) + 0)422)
где — установленная точка замерзания молока, °C; й — установленная точка за-
мерзания раствора хлорида натрия с теоретической точкой замерзания -0,422 °C,
°C; /2 — установленная точка замерзания раствора хлорида натрия с теоретической
точкой замерзания —0,621 °C, °C.
Массовая доля добавленной в молоко воды (%)
x=4z£.ioo,
h
где t — уточненное значение точки замерзания исследуемого молока, °C; /3 — значе-
ние точки замерзания натурального молока или точка замерзания сравнительной
пробы, °C.
В среднем точка замерзания молока повышается от добавления в
него 1 % воды на 0,005 °C (табл. 24).
225
24. Таблица пересчета точки замерзания молока на массовую долю воды, добавленной
в молоко
Массовая деля добавленной воды, % Точка замерзания натурального молока или сравнительной пробы, ‘С
-0,550 -0,540 -0,530 -0,520 -0,510
1 -0,545 -0,534 -0,524 -0,515 -0,505
2 -0,539 -0,529 -0,519 -0,510 -0,500
3 -0,534 -0,524 -0,514 -0,504 -0,495
4 -0,528 -0,518 -0,508 -0,499 -0,490
5 -0,523 -0,513 -0,503 -0,494 -0,485
6 -0,517 -0,508 -0,498 -0,489 -0,479
7 -0,512 -0,502 -0,493 -0,484 -0,474
8 —0,506 -0,497 -0,488 -0,478 -0,469
9 -0,501 -0,491 -0,482 -0;473 -0,464
10 -0,495 -0,486 -0,477 -0,468 -0,459
15 -0,468 -0,459 -0,451 -0,442 -0,434
20 -0,440 -0,432 -0,424 -0,416 -0,408
25 -0,413 -0,405 -0,398 -0,390 -0,383
Приготовление реактива. Хлорид натрия перед приготовлением
градуировочных растворов высушивают при температуре 300 °C в
течение 1 ч или при 130 °C в течение 24 ч и охлаждают до комнатной
температуры в эксикаторе.
Растворяют 0,6892 г хлорида натрия в 100 г бидистиллированной
воды или 0,6861 г хлорида натрия вносят в мерную колбу вместимос-
тью 100 см3 и объем доводят до метки бидистиллированной водой тем-
пературой 20 °C. Теоретическая точка замерзания раствора -0,422 °C.
Растворяют 1,0206 г хлорида натрия в 100 г бидистиллированной
воды или 1,0152 г хлорида натрия вносят в мерную колбу вместимос-
тью 100 см3. Объем доводят до метки бидистиллированной водой тем-
пературой 20 °C. Теоретическая точка замерзания раствора —0,621 °C.
Градуировочные растворы хранят в герметически закрывающихся
полиэтиленовых бутылках, заполненных доверху, при температуре 5—
8 °C не более 2 мес. Перед употреблением градуировочный раствор пе-
ремешивают осторожным перевертыванием и вращением бутылки.
Порцию градуировочного раствора вливают прямо в пробирку.
Контрольные вопросы. 1. Какова сущность метода эбулиоскопии? 2. На чем ос-
нован метод криоскопии? 3. Для чего используется криоскопия?
Глава 10
РЕОЛОГИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЙ*
Реология как наука о течении и деформации реальных тел изуча-
ет их поведение при механическом нагружении. Под действием
внешней нагрузки в любом продукте возникают деформации и на-
пряжения, которые зависят от состава и строения выбранных
объектов исследования, являясь мерой сил внутреннего взаимодей-
ствия между элементами их структуры.
По виду механических нагружений и вызываемых ими деформа-
ций можно классифицировать основные реологические свойства:
сдвиговые свойства, проявляющиеся при воздействии касательных
напряжений; компрессионные — при воздействии нормальных на-
пряжений и поверхностные — при сдвиге или отрыве продукта от
твердой поверхности.
Свойства можно описать множеством структурно-механичес-
ких характеристик: сдвиговые — коэффициентами динамической
(ньютоновской), эффективной и пластической вязкости, предель-
ным напряжением сдвига, индексом течения и др.; компрессион-
ные — модулями упругости и пластичности, различными прочно-
стными характеристиками и параметрами текстуры; поверхност-
ные — адгезионными и когезионными характеристиками и коэф-
фициентами внешнего трения.
Структурно-механические характеристики (СМХ) используют
для оценки консистенции продукта как одного из основных показа-
телей его качества. Оценка консистенции продукта осуществляется
либо путем измерения СМХ на специальных приборах (реометрах),
либо путем сенсорной (органолептической) оценки, т. е. субъек-
тивной оценки сопротивляемости и деформации продукта.
Сенсорная оценка консистенции, которую можно характеризо-
вать как эмпирическую характеристику деформационного поведе-
ния продукта, была известна до широкого применения реологичес-
кого анализа и используется до настоящего времени. Однако ре-
зультаты сенсорной оценки зависят от квалификации дегустатора,
тщательности проведения контроля с условием выполнения опре-
* Глава написана канд. техн, наук, ст. науч, сотрудником А. Ф. Зиминым.
227
деленных правил, гарантирующих точность и воспроизводимость
результатов, и при отсутствии специально подобранных и обучен-
ных экспертов, часто носят субъективный характер.
Оценку консистенции продукта инструментальными методами
(измеряя его СМХ) проводят следующим образом.
1. В зависимости от видов и интенсивности механического воз-
действия (нагружения во времени) определяют различные СМХ, из
которых выбирают наиболее чувствительную к изменению структу-
ры продукта при его деформации. Выбранная СМ является реоло-
гическим показателем консистенции (измеряемой величиной) для
данного продукта.
2. Предварительно проводят определение «эталонного» значе-
ния СМХ для каждого вида продукта по существующим методикам
оценки качества продукта. При этом в качестве «эталонного» при-
нимают значение СМХ продукта высшего качества.
3. Сравнивают величину выбранного реологического показателя
для исследуемого образца продукта с «эталонным» для него значе-
нием СМХ и по их разности судят о консистенции продукта.
В зависимости от поставленной задачи полученные результаты
могут быть использованы для определения качества готового про-
дукта, регулирования параметров технологического процесса про-
изводства, служить исходными данными при конструировании тех-
нологического оборудования и т. п.
В молочной промышленности вырабатывают широкий ассорти-
мент продуктов, которые по своей структуре представляют собой
различные реологические тела — от упруго-пластичных твердых тел
до истинно-вязких (ньютоновских) жидкостей.
СЫРЬЕ И МОЛОЧНЫЕ ПРОДУКТЫ
КАК РЕОЛОГИЧЕСКИЕ ТЕЛА
Молочные продукты, включая сырье и полуфабрикаты, в зави-
симости от состава, дисперсного строения и структуры обладают
различными реологическими свойствами и текстурными отличи-
тельными признаками (табл. 25).
Так как не всегда при определенном виде деформации тела одно-
временно проявляются все его реологические свойства, то для пол-
ной количественной оценки реологических свойств тела необходи-
мо применять различные методы нагружения (рис. 56). Инструмен-
тальное определение реологических констант требует правильного
выбора методов измерений и приборов (реометров).
Под действием внешней нагрузки в теле возникают деформации
и напряжения, являющиеся мерой сил внутреннего взаимодей-
ствия между элементами тела, взаимосвязь между которыми описы-
вается посредством реологических констант (коэффициентов урав-
нений деформации и течения) — различных структурно-механи-
ческих характеристик.
228
о
X
§
св
X
т
S
сх
Е
в
2
X
*
£
а>
3
X
X
X
Е
X
е
s
§
§
о
§
&
а>
g
§
Е
S
зХ
X
§
о
£*
X
м
X
зХ
*
о
о|
ё
а
о
Си
О
св
S
а>
о
X
X
S
о
3
о 3
£ S
В «
Я &
°-5
и
3 ®
X а>
&“
га
X
X
X
а>
3
х
X
св в
S
X
XJ
X
св
й
X
ё
X
X
О
О
X
5
X -
S.8
S о
3 х
схй
2 о
S
о
св
О
о
СП
О
5
ё
гЗ X
С1* 4>
2 3
на аз
сп О
5
>5.0
2- 2?’Х
•9* 2 св
Г) ч
- й
g О Я §
<5'° 5 s
а>
X
О
о
а
о
о
2
X
X
М
X
О
о х
« X
о V
5 £
О й
<5 с
о
X
±г* л
С д О
о V Си
св
X
о
i
С
к
св
229
l^=tga.
2в
Рис. 56. Виды нагружений и деформаций выделенного элементарного куба продукта:
1 — сложное трехмерное; 2— простейшие виды: 2а —одноосное растяжение; 26— одноосное
сжатие; 2в — чистый сдвиг; 2г — простой сдвиг; 2д — изотропное сжатие (гидростатическое дав-
ление)
Свойства упругих изотропных тел (сливочное масло при отрица-
тельных температурах, карамель, печенье, ядра орехов). Для них ха-
рактерны следующие реологические константы: модуль сдвига — G
(Па); модуль объемной упругости — Еу (Па); модуль Юнга, или мо-
дуль продольной упругости, — Е (Па); коэффициент Пуассона — ц.
Взаимосвязь перечисленных констант определяется следующи-
ми соотношениями:
при одноосном растяжении — сжатии
(l/E)[ox-p(oy + oz)j;
Еу=(1/Е)[оу-ц(ог + оД];
Ег=(1/Е)[ог-ц(ох + оу)];
при простом сдвиге
Уху = ®xy/G= 2 (1 + p)0^/£;
Ууг= —2(1+ p)0yj/£;
Угх = 0jx/G’ = 2(1+ \1)В^/Е,
230
где: е,- — относительная деформация — отношение абсолютной линейной деформа-
ции к начальному размеру тела (продукта), соответственно по осям х, у и z; <3j — нор-
мальное напряжение — напряжение, действующее по нормали к поверхности про-
дукта в тех же координатах; у,у — относительная деформация (отношение абсолют-
ной сдвиговой деформации к начальному расстоянию между слоями), рассматрива-
емая в соответствующей плоскости для выбранных объемных координат;
0,у — касательное напряжение, действующее по касательной к поверхности продук-
та в соответствующей плоскости.
Из теории упругости известно, что по двум константам можно
вычислить остальные:
r_ Е ЗЕЕ, _3£у(1-2ц).
l(i7^-9£v-£- 2(1+ц) ’
Е — Е _ EG 2(1+й) .
v 3(1-2ц) 9G-3£ 3(1+2ц)’
_E-1G _l-£/(3£v)_ ЗД,-2С
Ц 2G 2 2(3£v+G)’
Свойства жидких и жидкообразных продуктов. К жидкостям отно-
сятся такие вещества, у которых при постоянном напряжении сдви-
га наблюдается течение, т. е. деформация с постоянной или пере-
менной скоростью. Свойства жидкостей проявляются и у пластич-
ных тел после повышения предела текучести.
Например, при простом сдвиге ньютоновской жидкости (таких,
как молоко, сливки с массовой долей жира менее 20 %, пахта, мо-
лочная сыворотка) с напряжением 0 возникает деформация с опре-
деленной скоростью у . Отношение напряжения сдвига к скорости
деформации 0 /у является реологической константой жидкости и
называется ньютоновской (динамической) вязкостью:
П=0/у.
Реологическое уравнение состояния ньютоновской жидкости
имеет вид
0=т]у.
Для неньютоновских жидкостей (кисломолочные продукты: ке-
фир, творог, творожные изделия, сметана и др.) вязкость — функ-
ция скорости сдвига, поэтому ее называют кажущейся или эффек-
тивной вязкостью т)эф (Па с).
Инструментальное определение эффективной вязкости предус-
матривает измерение пар значений (0;у) в обширной области для
изображения кривой течения и функции вязкости.
231
Рис. 57. Кривые течения (а) и функции вязкости (б):
1 — ньютоновской жидкости; 2 — дилатентной жидкости; 3 — псевдопластичной
жидкости; 4 — нелинейного пластичного тела; 5 — линейного пластичного тела
Для характеристики жидкостей используют кривые течения — ре-
ограммы, которые представляют собой зависимость напряжения
сдвига от скорости сдвиговой деформации вусловиях простого сдвига.
Реограммы ньютоновских жидкостей представляют собой пря-
мую линию 1 (рис. 57), проходящую через начало координат. Все
кривые течения, которые отклоняются от прямой линии, соответ-
ствуют неньютоновским жидкостям. При этом кривая 2 показывает
дилатентное течение, характерное в основном концентрированным
дисперсным системам, при котором с увеличением скорости де-
формации наступает «затруднение сдвига»; кривая 3 показывает
псевдопластичное течение, что характерно для «сдвигового размяг-
чения» вследствие разрушения структуры с увеличением скорости
деформации; кривая ^показывает нелинейное пластичное течение,
характерное для большинства пластичных тел (при высокой кон-
центрации дисперсной фазы вследствие образования простран-
ственной структуры возникает предел текучести 0О). Линейный уча-
сток кривой 5характерен для бингамовских тел и соответствует иде-
альному пластичному течению.
Для ньютоновских и неньютоновских жидкостей применяют
математические модели, которые достаточно точно описывают их
поведение в интересующей области напряжений, при этом модели
неньютоновских жидкостей содержат по крайней мере две реологи-
ческие характеристики материала. Наиболее широкое распростра-
нение получила обобщенная модель, описываемая уравнением Гер-
шеля—Балкли, по которому можно представить девять кривых те-
чения (табл. 26):
232
26. Значения постоянных в уравнении Гершеля—Балкли
№ п/п Предельное напряжение сдвига Индекс течения Вязкость Название тела
1 0 оо ОО Упругое
2 >0 0 >0 Пластичное
3 >0 1 >0 Пластично-вязкое
4 0 <1 >0 Псевдопластичное
5 0 >1 >0 Дилатентное
6 >0 <1 >0 Нелинейное пластичное
7 >0 >1 >0 Нелинейное дилатентное
8 0 1 >0 Истанно-вязкое
9 0 0 0 Идеальная жидкость
Q—Qq+Byyn, (1)
где: 0 — напряжение сдвига между слоями продукта, Па; 0о — предельное напряже-
ние сдвига, Па; Bi — коэффициент консистентное™, пропорциональный вязкости,
Па с"; п — индекс течения.
Свойства твердообразных продуктов. Твердые тела в зависимости
от упругости бывают гуковскими и негуковскими.
Гуковское тело — это идеально упругое тело, состояние которого
описывается уравнением Гука
9 = Gy.
После снятия нагрузки, отдавая накопленную энергию, Гуковс-
кое тело без запаздывания возвращается в исходное состояние.
Поведение негуковских тел не соответствует поведению идеаль-
но упругого тела.
Для негуковского твердого тела с нелинейной упругостью пола-
гают
0/у = G* const.
При этом модуль сдвига является функцией деформации, что ха-
рактерно, например, для пористых пенообразных пищевых продук-
тов, в частности, натуральных сыров, мороженого.
По аналогии с неньютоновскими жидкостями вводят понятие
эффективного модуля упругости (7Эф- При напряжении, не превы-
шающем предел текучести или прочности твердого тела, соотноше-
ние между напряжением сдвига 0 и деформацией у можно описать с
помощью эмпирической формулы
0 = 6эфГ-
233
Реологическое поведение простейшего вязкоупругого тела мож-
но описать с помощью закона Кельвина
При снятии напряжения (6 = 0) и интегрирования в пределах от
Утах до у и по времени от 0 до т получают экспоненциальную функ-
цию для релаксации деформации
7 = 7тахехр(—Ст/т]),
или
7 = 7тахехр(-т/7),
где T=t\/G— период релаксации напряжений, с.
Если среду нагрузить постоянным напряжением 01 при т > 0, то
у=®4-[1-ехр(-т/Т)],
т. е. деформация во времени постепенно увеличивается, стремясь к
значению 01/Спри т -> <».
Входящая в эти уравнения величина периода релаксации имеет
большое значение при исследовании физико-механических свойств,
особенно при малых напряжениях и времени действия напряжения
того же порядка, как период релаксации. В зависимости от периода
релаксации деформации тела можно разделить на два рода. Когда
продолжительность действия силы меньше периода релаксации,
энергия, подводимая к системе, вызывает упругие деформации по
всему объему. Когда продолжительность действия силы больше пе-
риода релаксации, процесс протекает с накоплением энергии и вле-
чет за собой остаточные деформации.
Таким образом, течение реальных твердых тел наблюдается пос-
ле превышения критического напряжения — предела текучести.
При дальнейшем нагружении достигается предел прочности, при
превышении которого твердое тело разрушается. Это явление на-
блюдается в таких процессах, как дробление или резание, и поэтому
имеет технологическое значение. Если разрушение происходит без
существенного изменения формы — говорят о хрупком разруше-
нии. Если разрушению предшествует значительное изменение фор-
мы — говорят о вязком разрушении.
Простейшим случаем пластичного твердого тела является тело
Бингама. При нагрузке ниже предела текучести (0 < 0О) оно ведет
себя как гуковское тело (0 = бу), выше (0 > 0О) — как жидкость
234
(0=TlmY +®о) (см. уравнение 1 и табл. 26). Такие деформационные
свойства наблюдаются у сливочного масла при t> 14 °C, плавленых
сыров и сыров типа чедер.
Некоторые физико-механические свойства материалов. Твер-
дость — это комплексное свойство негуковских тел оказывать со-
противление проникновению другого, более твердого тела вслед-
ствие необратимых (упругой и вязкой) деформаций.
Твердость нельзя выразить как физическую величину с одно-
значной размерностью. Она представляет собой некоторый техни-
ческий параметр, который выражается в относительных величинах
в зависимости от метода измерений.
Твердость определяется следующими методами за счет внедре-
ния шара (твердость по Бриннелю), конуса (твердость по Роквел-
лу), пирамиды (твердость по Викерсу).
Коэффициент твердости рассчитывают по величине силы и гео-
метрическим параметрам остаточной деформации (площади шаро-
вого сегмента и т. п., глубине внедрения).
Мягкость — свойство, противоположное твердости.
Хрупкость — свойство твердых тел достигать разрушения без
пластичной деформации. У негуковых тел хрупкое разрушение на-
ступает только при высоких скоростях деформации или низких
температурах, когда теряют действие вязкостные свойства.
Когезия — сопротивление тела разрушению, связанному с пре-
одолением сил взаимодействия между атомами и молекулами на
поверхности раздела.
Адгезия — свойство, которое основывается на взаимодействии
двух различных тел на границе раздела фаз и вызывает сцепление
тел. При разделении тел необходимо преодолеть силы сцепления.
Прочность соединения двух тел из различных материалов зависит
от площади и состояния поверхности контакта между ними.
Липкость — свойство пограничного слоя вязких или пластичных
материалов оказывать сопротивление разделению находящихся в
контакте поверхностей. Оно основывается на адгезии материалов
на поверхности раздела и когезии самого материала.
Внешнее трение — сопротивление относительному перемеще-
нию двух находящихся в соприкосновении поверхностей твердых
тел. Для начала скольжения необходимо приложить нагрузку, пре-
вышающую силы трения покоя.
Консистенция — степень плотности (твердости) продукта. В за-
висимости от консистенции продукты по-разному деформируются
при избранных видах нагрузки и скорости. Результаты измерений
обычно дают в относительных единицах, характерных для приме-
няемых наиболее часто приборов (консистометров), имеющих уп-
рощенную конструкцию.
Воспроизводимость показателей консистенции гарантирована
только в том случае, если все условия измерений постоянны, осо-
бенно форма образца и его размеры, вид нагружения и его скорость.
235
Обобщение результатов на другие условия измерения невозможно,
данные с различных типов приборов несопоставимы.
На основе реометрического анализа при использовании более
сложных приборов деформационные свойства материала, связан-
ные с консистенцией, можно достаточно полно описать реологи-
ческими характеристиками или уравнениями состояния.
Текстура, по определению проф. М. Боурна, — физико-струк-
турные свойства вещества, в частности продукта, воспринимаемые
органами слуха, зрения и осязания и вызывающие у человека опре-
деленные ощущения при потреблении (откусывании, разжевыва-
нии, проглатывании). Комплекс ощущений при потреблении
пищи, который называется органолептическим, приводит потреби-
теля к предпочтению одних или отказу от других пищевых продук-
тов. Для создания высококачественных пищевых продуктов необ-
ходимо целенаправленно влиять на их органолептические свойства.
Консистенция и вязкость относятся к текстуре и представляют
собой два из множества возможных ее отличительных признаков
(схема 7).
Схема 7. Классификация сенсорной оценки качества и текстуры пищевых продуктов
по М. Боурну
236
При анализе текстуры определяют кинестетические признаки
продукта, связанные с мышечными ощущениями.
Инструментальные измерительные методы для определения
отдельных кинестетических признаков можно разделить на три
группы:
методы точного измерения реологических величин — коэффи-
циента вязкости, предела текучести, модуля упругости, прочности
на растяжение и др.;
эмпирические методы, при которых продукты подвергают вос-
производимой деформации или нагрузке при помощи измеритель-
ных приборов, не позволяющих точно определить реологические
свойства. Результаты измерений представляют собой параметры
консистенции. Они хорошо коррелируют с признаками текстуры,
полученными при органолептической оценке;
иммитационные методы, при которых пищевые продукты в спе-
циальных измерительных приборах подвергают испытаниям, ими-
тирующим реальные нагрузки при приеме пищи, например с помо-
щью циклических нагрузок имитируется процесс разжевывания
пробы. Цель такого анализа текстуры — измерение параметров, ко-
торые соответствуют признакам текстуры продукта, полученным
сенсорными методами.
МЕТОДЫ ИЗМЕРЕНИЙ И ИЗМЕРИТЕЛЬНЫЕ ПРИБОРЫ
Для экспериментального определения реологических параметров
продуктов или текстурных показателей консистенции существует
множество методов, которые различаются по области применения
(лабораторные и производственные), виду измеряемой величины (на-
пример, реологические характеристики продукта и показатели его
консистенции), принципам нагружения, степени автоматизации и др.
Для практического выбора метода измерения учитывают необ-
ходимое количество проб, точность и продолжительность измере-
ний и другие факторы, которые зависят от конкретных конструк-
тивных решений измерительного прибора.
Большое число реологических методов измерений предназначе-
но для лабораторных исследований. Кроме лабораторных методов
измерений для фундаментальных научных исследований реологи-
ческих характеристик материалов (в том числе и специальных) с
высокой точностью, для многократно повторяющихся исследова-
ний предпочтение отдается тем методам и приборам, которые по-
зволяют провести измерения и обработку их результатов быстро и с
минимальной зависимостью от субъективных факторов. Промыш-
ленностью ряда зарубежных стран выпускаются такие приборы, из-
мерения на которых полностью автоматизированы с помощью ком-
пьютера, одновременно математически обрабатывающего исход-
ные данные измерений в соответствии с выбранными моделями и
уравнениями деформации и течения исследуемых продуктов, а так-
237
же комплексные реологические лаборатории под названием «авто-
матизированное рабочее место реолога».
Эффективное и качественное управление процессами производ-
ства пищевых продуктов часто требует контроля реологических по-
казателей в производственных условиях. Использование лабора-
торных методов при этом сопряжено с большими затратами време-
ни и труда и возможно только для очень длительных процессов.
Методы измерений в производственном процессе требуют ис-
пользования большей частью несложных принципов одномерного
нагружения продукта (простой сдвиг, одноосное растяжение —
сжатие и т. д.), охватывающих измерение конкретных показателей
консистенции или характерных величин, которые связаны с выб-
ранными реологическими свойствами.
Одномерное стационарное сдвиговое течение может быть реали-
зовано при капиллярном, плоскопараллельном, цилиндрическом и
торсионном течении. Измерение одномерного сдвига лежит в осно-
ве принципа действия стандартных реометров (табл. 27).
27. Реометры одномерного сдвигового течения
Реометр Вид течения Область применения, требования к количеству исследуемого материала
Капиллярный виско- Капиллярное
зиметр:
постоянного давления переменного давления высокого давления Вискозиметр с каналом в Между параллель- Для ньютоновских жидкостей при малых градиентах сдвига; малое ко- личество материала Для неньютоновских жидкостей в технологических процессах; большое количество материала Для высоковязких и пластичных сред, а также при высоких градиентах сдвига Для неньютоновских жидкостей в
виде щели широкой или ными плоскостя- технологических процессах; большое
кольцевой ми количество материала
Ротационный вискози- метр:
с соосными цилин- Цилиндрическое Для ньютоновских и неньютонов-
драми Куэтта ских жидкостей в качестве лабора-
с параллельными Торсионное торных приборов; малое количество материала
плоскостями:
типа конус—плоскость Торсионное Для ньютоновских и неньютонов-
типа сфера—сфера между конусом и пластиной Торсионное ских жидкостей при постоянном градиенте сдвига в измерительном зазоре; очень малое количество ма- териала Для ньютоновских и неньютонов-
между шаром и сферической оболочкой ских жидкостей в качестве лабора- торных приборов; малое количество материала
238
Известны реометры, принцип измерения которых основан на
течении Стокса вокруг падающих шариков (табл. 28). Расчет скоро-
сти сдвига для падающих шариков в узкой трубе чрезвычайно сло-
жен, поэтому константы прибора определяют посредством калиб-
ровки с помощью жидкости с известной вязкостью.
Для количественного определения вязкоупругих характеристик
используют реометры, основанные на одномерном осциллирую-
щем сдвиговом течении (табл. 29).
28. Реометры течения Стокса
Реометр . Вид течения Область применения
Вискозиметр с падающим шариком и широкой труб- кой (диаметр шарика во много раз меньше диамет- ра трубки) Вискозиметр с падающим шариком и узкой трубкой По закону Стокса Для прозрачных ньютоновских вокруг шарика жидкостей с использованием различных шариков; для ненью- тоновских жидкостей примене- ние ограничено По модифицирован- Для прозрачных и полупрозрач- ному закону Стокса ных ньютоновских жидкостей; около шарика в для лабораторных приборов кольцевом зазоре
Вискозиметр с толкаемым шариком и узкой трубкой То же Для ньютоновских и неньюто- новских жидкостей; для лабора- торных приборов
29. Реометры одномерного осциллирующего сдвигового течения
Тип колебаний
Рабочие органы реометра
Область применения
Вынужденные враща- тельные с малой ампли- тудой и переменной частотой Плоскость—ПЛОС- КОСТЬ, конус—плос- кость, коаксиальные цилиндры Для количественного определе- ния динамических модулей и постоянных времени вязкоупру- гих жидкостей
Свободные вращатель- ные с начальными усло- виями То же Для определения реологических характеристик вязкоупругих жид- костей
Вынужденные за счет эксцентриситета оси вращения Плоскость—плос- кость, эксцентрич- ные цилиндры Для количественного определе- ния динамических модулей и постоянных времени вязкоупру- гих жидкостей
Вынужденные из-за относительного откло- нения оси вращения Полусфера—полу- сферическая оболоч- ка, конус — плос- кость, цилиндр—ци- линдр Тоже
Для исследования сложных неньютоновских жидкостей приме-
няют методы (табл. 30), которые дают быстрые, воспроизводимые
результаты измерений. Такие методы приобретают особое значение
239
при исследовании продуктов или полуфабрикатов, реологические
свойства которых быстро изменяются вследствие ферментативных,
химических или физических процессов.
30. Методы определения консистенции и реологических характеристик материалов
на основе комплексного нагружения (по М. Боурну)
Принцип нагружения
Измеряемая величина
Область применения
Внедрение индентора опре-
деленной формы и размеров
при заданном усилии и вре-
мени внедрения; типичные
формы инденторов — конус,
шар, полусфера, цилиндри-
ческий штифт, игла
Перемешивание жидкости
при определенных траекто-
риях движения, геометрии
сосуда, количестве и темпе-
ратуре материала и опреде-
ленной частоте вращения
Глубина внедрения
по истечении опре-
деленного времени,
глубина внедрения в
равновесном состо-
янии, кинетика внед-
рения в течение всего
времени измерений
Крутящий момент
Для пластичных и упругоплас-
тичных материалов — творог,
творожные изделия, сыры,
сливочное масло и т. д. в ста-
тическом состоянии
Замес вязких масс в опреде- То же
ленном месильном устрой-
стве при определенных гео-
метрии месильной камеры,
количестве и температуре
материала, частоте враще-
ния
Экструдирование пластич-
ных масс через узкие отвер-
стия определенной геомет-
рии при постоянной скорос-
ти экструдирования и темпе-
ратуре
Давление при опре-
деленной скорости
экструдирования;
количество экстру-
дируемого материала
Колебательное нагружение
с определенной амплитудой
и частотой
Затухание колебаний,
резонансная частота
Растекание вязкой или вяз-
коупругой массы определен-
ного количества и первона-
чальной формы при посто-
янной температуре под дей-
ствием силы тяжести
Уменьшение высоты,
увеличение площади
Для жидких и вязких суспен-
зий, эмульсий, пенообразных
масс с малым пределом теку-
чести или при его отсут-
ствии — творожный и сыр-
ный сгустки, йогурт, кефир,
ряженка, сметана и т. д.
Для вязких и вязкопластич-
ных масс (мороженого, гла-
зури, творожных изделий и
т. д.); для контроля процессов
их структурообразования и
исследования изменений в •
структуре смеси рецептурных
компонентов при механичес-
кой ее обработке, а также вве-
дении добавок для достиже-
ния определенной консис-
тенции продуктов
Для пластичных и упругопла-
стичных масс в динамичес-
ком состоянии — глазирован-
ных сырков и колбасного сы-
ра в процессе формования
Для вязких растворов или сус-
пензий с частицами грубо-
дисперсных компонентов —
джемов, конфитюров, йогур-
тов и молочных десертов с
фруктовыми наполнителями
Для малого количества вязких
и вязкоупругих масс — тво-
рожных кремов, сливочного
сыра, мягкого мороженого
240
Укрупненная классификация применяемых приборов, пред-
назначенных для исследования описанных выше структурно-ме-
ханических свойств и текстуры исходного сырья, полуфабрика-
тов и готовых продуктов, предложенная А. В. Горбатовым и
Ю. А. Мачихиным и переработанная с учетом специфики их вы-
бора для определения основных реологических показателей вы-
пускаемой в молочной отрасли промышленности продукции,
представлена в табл.31
31. Реометры, используемые для исследования молочных продуктов
Реометры Вид нагружения (течения) Измеряемая величина Исследуемые продукты
Вискозиметры: капиллярные ротационные шариковые колебательные Одномерное сдвиго- вое течение Тоже Течение Стокса Одномерное и двух- мерное сдвиговое течение Коэффициенты ди- Молоко, сливки, намической эффек- пахта, сыворотка, тивной и пластичес- кефир, простоква- кой вязкости, ин- ша, йогурт, сметана, деке течения, а так- творог, творожные же параметры текс- изделия, сливочное туры масло, мороженое
Пенетрометры Многомерное пене- трационное течение Предельное напря- Творог, творожные жение сдвига, пара- изделия, сливочное метры текстуры масло, мороженое, сыр
Компрессионные приборы Сжатие образца Предел прочности Сливочное масло, при сжатии, объем- мороженое, сыр ная вязкость, моду- ли упругости, пери- оды релаксации, па- раметры текстуры
Универсальные приборы типа «Инстрон» Растяжение, сжатие, изгиб, сдвиг и дру- гие простейшие ви- ды нагружения ис- следуемого продукта Прочностные ха^ То же рактеристики, пара- метры текстуры
Трибомеры Сдвиг Фрикционные ха- Сыр рактеристики
Адгезиометры Отрыв контактиру- ющего элемента от поверхности иссле- дуемого материала Адгезионные харак- Молоко, сливки, теристики пахта, молочная сы- воротка, кефир, про- стокваша, йогурт,
сметана, творог, тво-
рожные изделия,
сливочное масло,
мороженое
241
ОПРЕДЕЛЕНИЕ СТРУКТУРНО-МЕХАНИЧЕСКИХ
ХАРАКТЕРИСТИК ЖИДКИХ (УСЛОВНО-НЬЮТОНОВСКИХ)
И ЖИДКООБРАЗНЫХ МОЛОЧНЫХ ПРОДУКТОВ
МЕТОДАМИ КАПИЛЛЯРНОЙ ВИСКОЗИМЕТРИИ
К условно-ньютоновским жидкостям относят пастеризованное
или стерилизованное молоко жирностью не более 3,6 %, обезжи-
ренное молоко, пахту, творожную и подсырную сыворотки и све-
жие напитки на их основе.
Их течение с достаточной точностью описывается уравнением для
истинно-вязких жидкостей, подчиняющихся закону И. Ньютона
0=ПУ,
где т) — ньютоновская (динамическая) вязкость, Па • с; у — структурно-механи-
ческая характеристика.
К жидкообразным продуктам относят сливки жирностью до
40 %, кисломолочные напитки, не обогащенные белком и без до-
бавления стабилизаторов консистенции, сырный и творожный сгу-
стки. Они практически не имеют предельного напряжения сдвига,
т. е. обладают текучестью при любых значениях 0 > 0 и аномаль-
ной — неньютоновской вязкостью. Их течение описывается урав-
нением Оствальда — де Валле для псевдопластичных жидкостей
е=в;(т/7,)",
где Д' — коэффициент эффективной вязкости (Па с) при единичном значении
скорости сдвига (у L = 1с-1) — структурно-механическая характеристика; п — индекс
течения (структурно-механическая характеристика), показывающий меру отклоне-
ния течения от описываемого законом Ньютона.
Вязкостные характеристики вышеперечисленных продуктов из-
меряют преимущественно капиллярными вискозиметрами.
В качестве примера капиллярных вискозиметров рассмотрим
устройство и принцип измерения вязкости вискозиметрами Ост-
вальда и Убеллоде (рис. 58, а, б). Эти вискозиметры представля-
ют собой U-образные, установленные вертикально трубки, в
одно колено которых впаян капилляр. У вискозиметра Оствальда
определенное количество жидкости из левого резервуара от мет-
ки А до В перетекает в правый за счет гидростатического давле-
ния. В вискозиметре Убеллоде для истечения жидкости необхо-
димо в одном колене создать давление или вакуум, это предопре-
деляет его использование для исследования жидкообразных про-
дуктов.
Особенно удобен в применении для жидкообразных продуктов
и, в частности, слабоструктурированных (кефир, сырный и творож-
242
Рис. 58. Принципиальные схемы вискозиметров для определения вязкости ньютонов-
ских жидкостей и жидкообразных систем:
а — Оствальда; б — Убеллоде: 1 — шарик для измерения объема протекающей через капилляр
жидкости; 2 — капилляр; 3 — шарик для сбора жидкости; в — с двумя параллельно работающими
капиллярами: 1 — широкая трубка; 2 — капилляр; 3 — водяная рубашка; г — вискозиметр Энгле-
ра: 1 — латунная ванна; 2 —крючки для контроля уровня исследуемой жидкости; 3 — капилляр-
ный насадок; 4— стержень игольчатого клапана; 5—крышка ванны; б—термометр; 7—водяная
баня; 8— электронагреватель; 9— мешалка; 10— перепускная трубка; И — сливной кран; 72-
стойка; 13 — основание прибора; 14— электропривод мешалки; 15— кулачок-толкатель; 76-
трансформатор; 77— выпрямитель переменного тока; 18 — переключатель терморегулятора;
19— мерный приемник жидкости; 20—термометр
ный сгустки) вискозиметр типа ВК-4, работающий в горизонталь-
ном положении, что позволяет исключить поправки на гидроста-
тическое давление столба жидкости, а его конструкция — умень-
шить кратность механического воздействия. В этих вискозиметрах
для обеспечения течения жидкости через капилляр создается раз-
режение.
Для под держания заданной температуры исследуемой жидкости
вискозиметры Оствальда и Убеллоде помещают в жидкостной ульт-
ратермостат, вискозиметры типа ВК имеют собственную водяную
баню. Кроме названных имеются различные модификации капил-
лярных вискозиметров.
Методика измерения с помощью капиллярных вискозиметров
принципиально одинакова для любых конструкций приборов. Они
могут работать как абсолютные, когда величину вязкости и кон-
243
станту прибора определяют в случае течения за счет гидростатичес-
кого напора по формулам
где к= — постоянная прибора; h — удельный гидростатический напор, м;
D—диаметр капилляра, м; V— объем протекающей исследуемой жидкости, м3; / —
длина капилляра, м; р — плотность исследуемой жидкости, кг/м3; т — продолжи-
тельность истечения объема Киселедуемой жидкости,
или в случае принудительного течения за счет создаваемой разности
давлений
т1=Т^7 т=^рт’
где к=лР4(128Г7)~1 — постоянная прибора; р — разность гидростатического давле-
ния на входе и выходе капилляра.
Иногда на практике капиллярные вискозиметры используют как
относительные приборы.
В случае течения продукта за счет гидростатического напора его
вязкость
где Пз — коэффициент динамической вязкости дистиллированной воды при темпера-
туре исследуемого продукта, Па • с; рв — плотность дистиллированной воды при той
же температуре, кг/м3; тв — время истечения объема дистиллированной воды при
указанных выше параметрах и равенстве объемов воды и исследуемого продукта, с.
В случае принудительного течения за счет создаваемой разности
давления р
П=Пв~-
|’В
Для жидкообразных продуктов вискозиметрические данные, ап-
проксимируемые уравнением Оствальда—де Валле, предпочти-
тельно обрабатывать на графиках в консистентных переменных:
0С — консистентная переменная — «напряжение сдвига»;
32И 32ИС
л55т = л5г='^" ~ консистентная переменная — «градиент ско-
рости».
244
Результаты измерений заносят в соответствующий для каждого
типа приборов журнал наблюдений:
для вискозиметра Оствальда
Продукт № опыта Температу- ра t, "С Плотность продукта р, кг/м3 Время ис- течения т, с Константа прибора к, м’/с2 Вязкость Т], Па с
для вискозиметра Убеллоде
Продукт № опыта Температура Избыточное давление или вакуум Р, Па Время истечения т, с Безразмер- ная кон- станта при- бора, к Вязкость я, Па с
для горизонтальных вискозиметров (типа ВК-4)
Продукт № опыта Темпера- тура t, ‘С Диаметр капилля- ра Д м Величина вакуума р, Па Объем протека- ния V, м3 Время истече- ния т, с Скорость сдвига у, С'1 Напря- жение сдвига 0„ Па
По полученным данным на масштабированную логарифмичес-
кую бумагу с осями X=Igy и Y= lg0c наносят все эксперименталь-
ные точки и графически аппроксимируют их в первом приближе-
нии прямой линией таким образом, чтобы число точек сверху и
снизу было одинаково, не считая тех, через которые проходит ли-
ния. Чтобы точнее определить положение прямой, пользуются ме-
тодом наименьших квадратов.
Уравнение прямой в логарифмических шкалах
у = ах + Ь.
Для определения коэффициентов а и b берут две неэксперимен-
тальные точки, принадлежащие этой прямой и максимально уда-
ленные друг от друга, и составляют систему уравнений
У\-ах\+Ь
у2=ах2+Ь.
Решив эту систему, находят
Х1-Х2
245
и
b =Ji — ах\,
или
Ь=у2-ах2.
Полученные значения коэффициентов подставляют в уравнение
прямой и записывают его в виде
lg0c=tflgy+lg£
и после потенцирования находят
в котором а = п — индексу течения (первая реологическая кон-
станта), а
Из последнего выражения определяют значение второй реоло-
гической константы (Па • с) — коэффициента эффективной
вязкости при единичной скорости сдвига.
Помимо вышеперечисленных, теоретически обоснованных вис-
козиметров с длинными капиллярами существуют вискозиметры с
короткими капиллярами-насадками, не имеющие достаточного те-
оретического обоснования. После измерения на них времени исте-
чения жидкости для расчета вязкости используют различные эмпи-
Еические формулы, которые не всегда дают достоверный результат,
[аиболее сопоставимые результаты вискозиметрии для данного
типа приборов получают на вискозиметрах Энглера (рис. 58, г).
Вискозиметры Энглера применяют для определения коэффици-
ентов кинематической и динамической вязкости молочных продук-
тов, условно относящихся к ньютоновским жидкостям, и сопоста-
вительного текстурного анализа жидкообразных молочных продук-
тов в широком диапазоне изменения их температур.
Вязкость v3 испытуемой жидкости, измеряемая прибором, ха-
рактеризуется градусами Энглера (°Е)
V3 Ък/^д.в,
где тж — определяемое время истечения через насадок прибора 200 мл испытуемой
жидкости, с; тд в — время истечения такого же объема дистиллированной воды
при 20 °C.
246
Время истечения 200 см3 дистиллированной воды при 20 °C со-
ставляет 50—52 с, что соответствует величине кинематической вяз-
кости v = 10~6, м2/с.
Таким образом, градус Энглера представляет собой относитель-
ную величину вязкости, которую по соответствующим эксперимен-
тальным формулам можно привести к истинным значениям вязкости.
При исследованиях данные и результаты опытов заносят в жур-
нал наблюдений
Продукт № опыта Темпера- тура t, °C Время исте- чения тх, с Расчетное значение ВЯЗКОСТИ V . °Е Кинемати- ческая ВЯЗКОСТЬ V, м2/с Плот- ность продукта р, кг/м3 Ньютонов- ская (дина- мическая) ВЯЗКОСТЬ Т|, Па с
По средним значениям при каждой из выбранных температур
необходимо вычислить вязкость v3(°E) по предыдущей формуле,
приняв время истечения дистиллированной воды тд в равным 51с,
что обеспечит погрешность измерений на различных вискозимет-
рах Энглера не более 2 %.
Пересчитывают v3 в кинематическую вязкость, пользуясь фор-
мулами
Убеллоде
v=fo,O732v3-^^-\lO“4,
I °Е I
или Фогеля
v=0,01v3-7,6^1-^-10-4.
Определяют плотность исследуемого продукта при заданной
температуре. Рассчитывают динамическую вязкость по формуле
T]=Vp.
ОПРЕДЕЛЕНИЕ СТРУКТУРНО-МЕХАНИЧЕСКИХ ХАРАКТЕРИСТИК
ЖИДКООБРАЗНЫХ И ТВЕРДООБРАЗНЫХ МОЛОЧНЫХ ПРОДУКТОВ
МЕТОДАМИ РОТАЦИОННОЙ ВИСКОЗИМЕТРИИ
К жидкообразным продуктам, которые целесообразно исследо-
вать на ротационных вискозиметрах, относят сливки жирностью от
20 до 40 %, йогурт, кефир, простоквашу, ряженку, ацидофилин,
жидкие смеси для мороженого, кисломолочные напитки как обога-
щенные белком, так и со стабилизаторами консистенции.
247
Рнс. 59. Ротационные вискозиметры:
а — РВ-8 — системы Воларовича: 1 — корпус со станиной; 2 — асбестовый сосуд; 3 — вставной
сосуд для термостатирующей жидкости; -/—термопара; 5— отражательное кольцо; 6 — электро-
нагревательный элемент; 7—шкив; 8— тормозное приспособление; 9 — шкала отсчета; 70-
стрелка; И — барабан; 12— подшипник; 13— крышка прибора; 14— обойма для стакана; 75-
ротор; 16— стакан; 77— мешалка; 18— установочный винт; 6 —вискозиметр «Реотест»: 7 — ста-
нина; 2 — термостатная емкость; 3 — ротор; 4— стакан; 5 — соединение измерительного вала с
валом привода; б— термометр; 7— измерительный вал; 8— потенциометр; 9 — пружинный ди-
намометр (торсион); 10— вал привода; 77 —ступенчатый регулятор динамометра; 72 —частото-
мер; 13— шкала логометра; 14— стопорные рукоятки фиксаторов; 15 — шкала ступени частоты
вращения; 16 — рукоятка переключения ступени
К твердообразным молочным продуктам относят творог, тво-
рожные изделия, сливки жирностью более 40 %, мороженое, пу-
динги и прочие молочные десерты. Они обладают ярко выражен-
ным предельным напряжением сдвига Оо, и их течение описывается
уравнением Шведова — Бингама для пластично-вязкого тела
О = Оо + ПплУ,
где Пга — пластическая вязкость, Па • с.
248
В настоящее время конструкций ротационных вискозиметров
насчитывается несколько сотен, которые можно разделить на две
основные группы. К первой группе относятся приборы, имеющие
постоянный момент вращения ротора при переменной частоте вра-
щения. Вторую группу составляют приборы, имеющие постоянную
частоту вращения ротора при переменном вращающем моменте.
В России и странах СНГ наибольшее распространение получили
вискозиметры системы проф. Воларовича РВ-8, относящиеся к
первой группе, и вискозиметры «Реотест», относящиеся ко второй
(рис. 59).
Исследование процесса течения грубодисперсных систем, к ко-
торым относят творог и творожные изделия, проводят на вискози-
метрах РВ-8.
Методика измерения структурно-механических характеристик
для ротационных вискозиметров системы Воларовича сводится к
подготовке прибора, измерениям и разборке. Подготовка включает
установку вискозиметра по уровню, определение силы трения в
подшипниках, заполнение рабочего объема исследуемым материа-
лом и его термостатирование. Во время измерений меняют массу
грузов М(кг) и для каждого груза определяют частоту вращения ро-
тора ^(максимальная частота вращения может достигать 2—-2,5 об/с).
Каждый опыт состоит из 30—40 замеров, проводимых несколько
раз с постепенным увеличением и уменьшением массы грузов. Од-
новременно строят реограммы N Затем прибор разбирают
для мытья и определения высоты продукта Л(м) по оставленным им
следам на роторе. После этого определяют константы опыта по
формулам
ШКО ' _ ту- ту /у
\ Лн Лв -Ли ~£\в J
/ (2nRlh+n2R2 14);
k\ =2nRHh I
2 2л ’
где g — ускорение свободного падения, м/с2; Лшк — радиус шкива, м; Ян — внутрен-
ний радиус стакана, м; Ав — радиус ротора, м; h — глубина погружения ротора в про-
дукт, м.
Вычисляют эффективную вязкость для каждой опытной точки
по уравнению:
т)Эф = kM/N.
249
Строят график зависимости эффективной вязкости от ок-
ружной скорости со = 2hRbNb логарифмических шкалах. Через
полученные экспериментальные точки проводят прямую ли-
нию так, чтобы она делила относительные отклонения расстоя-
ний между точками и прямой на равные части. Составляют
уравнение этой прямой, определяют темп разрушения структу-
ры т, значение коэффициента эффективной вязкости по фор-
муле
Th4>=^w/wi) т=В**т>
где В — эффективная вязкость при фиксированном значении окружной скорости,
Па - с; и»! — фиксированное единичное значение окружной скорости боковой по-
верхности ротора коаксиально-цилиндрического вискозиметра (w, = 1 м/с); %—
безразмерная окружная скорость (ее числовое значение).
После определения по полученным экспериментальным дан-
ным индекса течения п = 1 - т рассчитывают инвариантную для
любых коаксиально-цилиндрических ротационных вискозиметров
реологическую константу — Bq по формуле
у1лЯв[1-(Ав/Ян)2/л]
2^1
п-1
учитывающей закон изменения градиента скорости.
Затем вычисляют реологические константы — предельное на-
пряжение сдвига 0о и пластическую вязкость г)пл, ориентируясь на
график зависимости N(M). При этом точки для вычислений берут
непосредственно с кривой. 0О и 1)^ определяют в следующей после-
довательности. Предельное напряжение сдвига0о (Па) соответству-
ет отрезку, отсекаемому кривой N(M), по оси абсцисс (Мо). Его оп-
ределяют по формуле
0о =
Напряжение сдвига на поверхности ротора
0 = к$М.
Пластическую вязкость в зависимости оттого, распространяет-
ся ли сдвиг на всю толщину кольца продукта (в зазоре между рото-
ром и стаканом), рассчитывают по одной из приведенных ниже
формул.
Первый случай — сдвиг распространяется на всю толщину коль-
250
ца, когда масса нагрузки Мо больше массы нагрузки М\ = £10о. В
этом случае пластическую вязкость определяют по формуле
Ппл = (кМ—k2Q0)/N.
Все расчеты пластической вязкости, когда сдвиг распространя-
ется на всю толщину кольца исследуемого продукта, рекомендуется
сводить в таблицу:
Константа опыта к = *2 =
N М кМ kfla кМ-кг^о Ппл = (*3/-*20о)/Л'
Второй случай (наиболее часто встречающийся при течении в за-
зоре творога и творожных изделий) — сдвиг не распространяется на
всю толщину кольца, когда масса нагрузки Мо меньше массы на-
грузки Мр
Ппл = (6о/Л0[1/4л)][М/Мо) - 1 - 1п(М/М0)]
или
Ппл = (0о/Л9Д^Ж);
F(M/M0) = (1/4л)[(М/М0) - 1 - 1п(М/М0)]-
Значение F(M/M0) определяют по табл. 32.
32. Определение числовых значений функций 1\М/Мй) — ДО/9о) в зависимости
от соотношений М/Мй = 9/0#
М/Мо ЦМ/М0) м/м0 RM/Mv) м/м0 ЦМ/Мп)
1,1 0,000373 3,5 0,099255 5,9 0,246880
1,2 0,001407 3,6 0,104971 6,0 0,255312
1,3 0,002995 3,7 0,110749 6,1 0,261954 ,
1,4 0,005056 3,8 0,116585 6,2 0,268618
1,5 0,007523 3,9 0,122475 6,3 0,275303
1,6 0,010345 4,0 0,128418 6,4 0,282008
1,7 0,013478 4,1 0,134411 6,5 0,288732
1,8 0,016888 4,2 0,140452 6,6 0,295475
1,9 0,020544 4,3 0,146537 6,7 0,302246
2,0 0,024419 4,4 0,152666 6,8 0,309014
2,1 0,028494 4,5 0,158835 6,9 0,315812
2,2 0,032750 4,6 0,165044 7,0 0,322624
2,3 0,037171 4,7 0,171291 7,1 0,329453
2,4 0,041742 4,8 0,177573 7,2 0,336299
2,5 0,046452 4,9 0,183890 7,3 0,343158
2,6 0,051288 5,0 0,190241 7,4 0,350034
2,7 0,056246 5,1 0,196623 7,5 0,356924
251
Продолжение
м/м0 НМ/М0) м/м0 ЦМ/М0) м/м0 дм/мо)
2,8 0,061307 5,2 0,203036 7,6 0,363828
2,9 0,066472 5,3 0,209478 7,7 0,370746
3,0 0,071733 5,4 0,215948 7,8 0,377677
3,1 0,077081 5,5 0,222446 7,9 0,384621
3,2 0,082512 5,6 0,228970 8,0 0,391578
з,з 0,088022 5,7 0,235519 8,1 0,398548
3,4 0,093604 5,8 0,242094 8,2 0,405528
8,3 0,412493 8,9 0,454716 9,5 0,497272
8,4 0,419527 9,0 0,461785 9,6 0,504397
8,5 0,426543 9,1 0,468863 9,7 0,511530
8,6 0,433571 9,2 0,475925 9,8 0,518672
8,7 0,440609 9,3 0,483051 9,9 0,525822
8,8 0,447657 9,4 0,490156 10,0 0,532981
Для облегчения расчетов по предпоследней формуле следует вы-
бирать отношения М/Мо. Расчет удобно сводить в таблицу.
N М м/м0 G0/N Ппл = (Оо/ЛОДМ/Мо)
Для оценки пластичности продукта с текстурной точки зрения
представляет интерес степень разрушения структуры Я (%), кото-
рую определяют по формуле Хайтона
Я=^Ц^--100,
eg
где ej — предельное напряжение сдвига практически не разрушенной структуры
продукта, Па; eg — предельное напряжение сдвига разрушенной структуры, Па.
Хотя по сравнению с вискозиметрами РВ-8 у вискозиметров
типа «Реотест» и более узкая область применения, они за счет доста-
точно большой универсальности, резкого снижения трудоемкости
исследований и эргономичности получили самое широкое распро-
странение в научной практике. Поэтому на их конструкции и реа-
лизуемой методики реометрии остановимся несколько подробнее.
Ротационный вискозиметр «Реотест» (рис. 59, б) состоит из четы-
рех основных узлов: станины с синхронным электроприводом, блока
многоступенчатого редуктора с переключателем скоростей и изме-
рительного блока, которые объединены в общем корпусе, и измери-
тельной головки, состоящей из четырех сменных цилиндрических
роторов, двух сменных стаканов со съемным дном и водяной бани.
В полностью укомплектованный стенд для проведения иссле-
дований помимо прибора входят ультратермостат и самопишущий
логометр.
252
Предварительно выбирают соответствующую измерительную
головку для определения сдвиговых характеристик того или иного
продукта, затем фиксируют при помощи цангового зажима выбран-
ный измерительный ротор на выходном валу измерительного бло-
ка. Исследуемый продукт помещают в соответствующий стакан с
герметично закрытым съемным дном. Стакан герметично фиксиру-
ют в водяной бане и коаксиально ротору по направляющей при по-
мощи винтового зажима фиксируют на измерительном блоке. Во-
дяную баню подключают к ультратермостату и термостатируют
продукт до установления требуемой температуры.
Соответствующую измерительную головку выбирают, руковод-
ствуясь табл. 33.
33. Пределы измеряемых реологических характеристик, соответствующих индексам
измерительных головок
Индекс измери- тельных головок Соотноше- ния радиу- сов рото- ров и ста- канов RyRH Область напряже- ний 0 Напряже- ние сдви- га 0. Па Скорость де^юрма- ции у, с-' Эффектив- ная (дина- мическая) ВЯЗКОСТЬ Т], Па с Исследуемый продукт
51 0,98 I 11 11-110 55-550 1,5-1310 1,5-1310 0,01-75 0,05-375 Мороженое, сливки жирнос- тью 20—40 %
S1 0,94 I II 12-120 60-600 0,5-437 0,5-437 0,03-240 0,15-1200 Сгущеное моло- ко с сахаром, высокожирные сливки (жир- ность более 40 %)
Sj 0,81 I II 16-160 80-800 1/6-146 1/6-146 0,12-1000 0,6-5000 Творожные кре- мы, кисломо- лочные десерты, сметана
н 0,81 I II 60-600 300-3000 1/6-146 1/6-146 0,4-3600 2-18000 Сливочное мас- ло, творожные
пасты
Константы выбраной измерительной годовки определяют в за-
висимости от жесткости торсиона I—II (положение рукоятки на ли-
цевой панели измерительного блока) из табл. 34.
34. Константы измерительных головок
Тип измерительной головки Объем загружаемого продукта, V • 10й, м3 Константы роторов, Z, Па/ед. шкалы
при жесткости торсиона — I при жесткости торсиона —• II
51 25 1,19 5,92
52 30 1,23 6,16
5з 50 1,69 8,45
Н 17 5,89 29,29
253
Через специальную розетку, которая находится на оборотной
стороне станины прибора, в случае необходимости подключают са-
мопишущий логометр. Селекторный переключатель на лицевой па-
нели станины вискозиметра в этом случае устанавливают в положе-
ние 5, а при визуальном считывании показаний со стрелочного ло-
гометра измерительного блока — в положение А.
Прибор подключают к сети напряжением 220 В частотой 50 Гц.
Задают требуемую частоту вращения ротора, начиная с минималь-
ной, с помощью переключателя скоростей за счет установки рычага
в положение, соответствующее данным табл. 35.
35. Определение частоты вращения ротора вискозиметра «Реотест» по ступеням
Ступень Частота вращения, N, с~' Ступень Частота вращения, N, с-1
1а 0,00926 1в 0,00463
2а 0,01660 2в 0,00833
За 0,02778 Зв 0,01389
4а 0,05000 4в 0,02500
5а 0,08330 5в 0,04167
6а 0,15000 6в 0,07500
7а 0,25000 7в 0,12500
8а 0,45000 8в 0,22500
9а 0,75000 9в 0,37500
10а 1,35000 10в 0,67500
Па 2,25000 11в 1,12500
12а 4,0500 12в 2,02500
Для переключения от ступени 1 до ступени 12 требуется 2,5 обо-
рота рукоятки. Промежуточные ступени соответствуют меньшему
повороту и индицируются на табло блока многоступенчатого ре-
дуктора. Другую частоту вращения ротора можно установить в про-
цессе измерений.
Отклонение показаний измерителя частоты, расположенного на
измерительном блоке, от величины 50 Гц требует внесения поправ-
ки при расчете частоты вращения ротора по формуле
^д = #//50,
где 7V„ — действительная частота вращения ротора; N— частота вращения ротора
(по табл. 35);/— индицируемая частота переменного тока, Гц.
Измеряемое значение напряжения сдвига при различных скоро-
стях вращения ротора выбранной измерительной головки в соот-
ветствии с жесткостью торсиона вычисляют по формуле
0 = 2а,
где а — показания стрелочного логометра; Z — константа ротора.
254
Для построения реограммы — кривой течения Na(9) обычно бы-
вает достаточно 12 экспериментальных точек в диапазоне от мини-
мальной до максимальной частоты вращения ротора.
Все данные измерений заносятся в журнал наблюдений.
№ п/п Напряжение сдвига в, Па Действительная частота вращения ротора N , с_| Окружная скорость вращения ротора, <о = мс~* Эффективная вязкость, Пэф, Па • с Температу- ра продукта, /,'С
По окончании опыта измерительную головку отсоединяют от
измерительного блока, разбирают, удаляют исследованный про-
дукт, моют водой, сушат и окончательно обезжиривают при помо-
щи спирта.
Эффективную вязкость при каждой частоте вращения ротора
рассчитывают по формуле
Пэф = ЩМц,
где к — константа измерительной головки;
,, 1-(Ц,/Лн)2
К 4л
где соотношение Ав/Лн Для соответствующей измерительной головки берут из
табл. 33.
Первоначально строят график зависимости Na (0) и, используя
эту реограмму, проводят графоаналитическую обработку опытных
данных в соответствии с методикой, приведенной для вискозимет-
ра РВ-8, исключая случай, когда сдвиг распространяется на всю
толщину кольца продукта между ротором и стаканом вискозиметра.
Это связано с тем, что соотношение радиусов измерительных голо-
вок S2, и Н для пластично-вязких продуктов изначально мало
(см. табл. 33).
Для случая, когда сдвиг распространяется не на всю толщину
кольца используют для расчетов табл. 32, заменяя в столбцах:
в 1-м — Мна 0; во 2-м — M/M0Ha 0/0о; в 3-м — F(M/Mq) на F(0/0O).
В чисто технологических целях для оценки качества продукта,
влияния внесения ПАВ и протекания технологического процесса
можно использовать консистентную переменную — скорость сдви-
га, рассчитанную из условий течения ньютоновских жидкостей, из
табл. 36.
В результате полученные данные намного проще аппроксими-
ровать уравнениями состояния, входящими как частные случаи в
уравнение Гершеля—Балкли, и по выбранным наиболее чувстви-
тельным к структурным изменениям в продуктах реологическим
константам проводить анализ происходящих в продукте качествен-
ных изменений.
255
ОПРЕДЕЛЕНИЕ ПРЕДЕЛЬНОГО НАПРЯЖЕНИЯ
СДВИГА ПЛАСТИЧНО-ВЯЗКИХ МОЛОЧНЫХ ПРОДУКТОВ
И ТВЕРДОСТИ СЫРА ПРИ ПОМОЩИ ПЛАСТОМЕТРОВ
И ПЕНЕТРОМЕТРОВ
Наряду с ротационными, капиллярными и шариковыми виско-
зиметрами существует огромное количество различных приборов
для определения реологических свойств при относительном сдвиге
слоев исследуемого продукта.
Так, для изменения предельного напряжения сдвига 0О использу-
ют приборы, принцип действия которых основан на погружении ин-
дентора в исследуемую среду. Для исследования молочных продук-
тов получили широкое применение пластометры и пенетрометры с
инденторами в виде конусов с различными углами при вершине.
Измерение предельного напряжения сдвига 0О (Па) основано на
определении глубины погружения индентора определенной массы
в продукт за время не менее 180 с (формула Агранат — Воларовича)
0о = кт/№,
где fc — константа конуса, зависящая от угла 2а при его вершине, Н/кг; т — суммар-
ная масса конуса, дополнительных грузов и подвижных частей прибора, оказываю-
щих прямое силовое воздействие на продукт, кг; h — глубина погружения конуса, м.
При определении предельного напряжения сдвига предполага-
ется, что продукт течет вдоль образующей поверхности конуса. В
момент погружения индентора в продукт напряжение сдвига беско-
нечно велико (площадь касания равна нулю), скорость индентора
равна нулю, а ускорение соответствует ускорению силы тяжести.
Поскольку сила, действующая на конус (сила тяжести), за все время
погружения остается постоянной, а площадь соприкосновения ко-
нуса с продуктом увеличивается, то напряжение сдвига в системе
конус — продукт уменьшается.
При остановке конуса погружение будет предельным и внешняя
сила уравновешивается пластичной прочностью структуры. В этом
случае предельное напряжение сдвига рассчитывают как отноше-
ние силы, действующей вдоль «смоченной» боковой поверхности
конуса, к площади «смоченной» поверхности.
Принципиальную теорию конических пластометров впервые
разработал акад. П. А. Ребиндер. Согласно теоретическим уравне-
ниям были получены следующие константы в зависимости от угла
при вершине конуса:
2а 30° 40° 45° 60° 89°20" 90°
к. 9,4 5,2 4,1 2,1 0,82 0,67
Однако при измерении предельного напряжения сдвига одного
и того же продукта конусами с различными углами при вершине час-
то не получаются одинаковые величины.
256
257
Рис. 60. Полуавтоматический пенетро-
метр ПП-5Ц с цифровой индикацией
показаний
На основе сопоставления
экспериментальных данных с
данными ротационной виско-
зиметрии для получения инва-
риантных характеристик плас-
тично-вязких молочных про-
дуктов был выбран индентор с
углом при вершине конуса 2а,
равным 60°.
Для измерения предельного
напряжения сдвига наиболь-
шее распространение получили полуавтоматические пенетромет-
ры. Одним из таких современных приборов является пенетрометр
ПП-5Ц с цифровой индикацией показаний (рис. 60). Он выполнен
в фигурном корпусе, в нижней половине которого расположены
блок управления и электронный измерительный блок, а в верх-
ней — электропривод перемещения индентора и оптико-электрон-
но-механическая измерительная система. На нижней половине
корпуса имеется площадка для установки емкости с исследуемым
продуктом. При опускании конуса фиксация момента его касания
поверхности продукта осуществляется автоматически. С этого мо-
мента на двойном цифровом табло высвечиваются текущие значе-
ния глубины погружения индентора с точностью до 0,01 мм. По ис-
течении 5 с в левой части табло регистрируется величина пенетра-
ционного внедрения индентора в продукт, а через 180 с в правой ча-
сти табло — полная глубина погружения.
В исходное состояние индентор возвращается нажатием кнопки
управления реверсивным двигателем и обнуления показаний на
табло.
Перед проведением измерений прибор с помощью регулировоч-
ных винтов устанавливают строго вертикально по уровню. Продукт
плотно, без пустот закладывают в цилиндрическую емкость и шпа-
телем, вровень с верхним краем емкости, осторожно выравнивают
поверхность продукта, снимая излишки. Затем продукт термоста-
тируют до требуемой температуры и фиксируют емкость с ним на
площадке пенетрометра. Нажимают кнопку «Пуск». И по истече-
нии 180 с записывают зафиксированные глубины внедрения ин-
дентора. Показания правой части табло из мм переводят в метры и
рассчитывают предельное напряжение сдвига по приведенной
выше формуле. Максимальная точность измерений достигается в
258
диапазоне 30—45 мм глубины погружения индентора, которая регу-
лируется массой дополнительных грузов, размещающихся на осно-
вании индентора. Максимально измеряемая глубина погружения
индентора в продукт 50 мм.
Величину пенетрации (глубину йп (мм) внедрения индентора оп-
ределенной формы и массы за определенное время — 5 с) применя-
ют для экспресс-анализа быстро протекающих процессов. Для от-
носительного сопоставления влияния различных технологических
параметров удобно пользоваться величиной 1 / /^.
Сыры, как натуральные, так и плавленые, относятся к упруго-
пластичным, упруго-вязким или упруго-пластично-вязким систе-
мам, сдвиговое течение в которых без нарушения сплошности невоз-
можно, и при внедрении конуса имеют место пластические деформа-
ции смятия или разрушения. В этом случае, при помощи пенетро-
метра измеряют предельное давление 0П, которое рассчитывают
путем деления силы Р, действующей на продукт, на площадь сечения
конуса, проходящей по поверхности продукта
п" г т2 “ г 12 ’
%[/Hgaj nptgaj
где a — половинный угол при вершине конуса (2а/2).
Этот показатель характеризует микротвердость продукта.
Для определения макротвердости целесообразно заменить ко-
нусный индентор на полусферический, тогда ее рассчитывают по
формуле
О' - mg /,ч
°П-ЯЛ(2А-Л)’ (1)
где R — радиус шаровой сферической поверхности, м.
ОПРЕДЕЛЕНИЕ КОМПРЕССИОННЫХ ХАРАКТЕРИСТИК СЫРОВ
Деформационному поведению сыров наиболее адекватны про-
стые математические модели стандартных линейных тел (рис. 61),
первая из которых в случае осевого сжатия для пастообразных
плавленых сыров описывается общим реологическим урав-
нением
о+т£^=£я(е+то^),
(2)
а вторая, для остальных плавленых и натуральных сыров, уравнени-
ем Олдройда
259
a+TeJ-=£e+Te£lB7’ (3)
где о — нормальное напряжение, Па; хе — компрессионная характеристика — пе-
риод релаксации напряжения при постоянной деформации, с; do/dx — скорость
увеличения напряжения, Па/с; Ец — компрессионная характеристика — релакса-
ционный модуль упругости, определяемый соотношением между упругими и вяз-
ко-пластичными характеристиками продукта, Па; е — относительная линейная де-
формация; та — компрессионная характеристика — период релаксации деформа-
ции при постоянном напряжении, с; de/dx — скорость деформации, с-1; Е, Ei~
компрессионные характеристики — условно-мгновенный и равновесный модули
упругости, Па.
Интегрируя уравнение (2) при граничных условиях de /<1т=ёф =
=const, е=ё*т и ot = o, получают закономерность изменения напря-
жения во времени при постоянной скорости деформации упруго-
вязкого тела.
Рнс. 61. Основные механические модели стандартных линейных тел:
а — упруго-вязкого тела с релаксацией деформаций и напряжений; б — упруго-пластично-вяз-
кого тела с релаксацией деформаций; элементы моделей; 1 — пружина (упругое тело Гука); 2 —
цилиндр и поршень с отверстиями (вязкое тело Ньютона); 3 — пара трения скольжения (плас-
тичное тело Сен-Венана); в — релаксационные характеристики; 1 — упруго-вязкого тела; 2 — уп-
руго-пластично-вязкоготела
260
о(т)=Л[1-еф (-т/t6)]+£re.t,
(4)
где
Л=£лЁ.(та-те} (5)
При т << те разложением экспоненты в ряд находят
о(т)=(Л/те + £/?Ё.)т, (6)
прит >> ТЕ
o(t)=^+£re,t. (7)
Графическое отображение уравнения (6) представляет собой
прямую, проходящую через начало координат, а уравнения (7) так-
же представляет собой прямую, отсекающую на оси ординат отре-
зок А (рис. 61, в, зависимость 1).
Методика расчета постоянных достаточно проста. Так, графо-
аналитически из уравнения (7) определяют А как отрезок, отсекае-
мый от оси ординат, a ER — по тангенсу угла наклона этой прямой.
Из уравнения (6) для участка прямой о (т), выходящей из начала ко-
ординат, вычисляют
из уравнения (5)
те=Л/[о(т)/т--Е'кЁ.],
та=те + Л/(£ЛЁ.),
(8)
(9)
т. е. находят все реологические константы — компрессионные ха-
рактеристики.
Интегрируя уравнение (3), при тех же граничных условиях, как и
уравнение (2), получают закон изменения напряжения во времени
при постоянной скорости деформации упруго-пластичного тела
о (т)=+ Дё* .
(Ю)
Графическая интерпретация этого уравнения показана на рис. 61, в
(зависимость 2), а его графоаналитическая обработка не вызывает
затруднений, так как зависимость о (т) при т->0 отсекает отрезок на
оси ординат, равный Е^, а дальнейшие вычисления постоянных
элементарны.
Для определения компрессионных характеристик сыров ис-
пользуют дефометры различных конструкций.
261
Рис. 62. Прибор для определения компрессионных характеристик сыров:
1 — корпус; 2 — выдвижной столик; 3 — тензобалка, 4 — тензодатчики; 5— верхняя и нижняя
пластины; 6 — штанга-гайка; 7— винт; 8— электродвигатель с редуктором; 9 — индикатор;
10— поперечина-поводок
Принципиальная схема полуавтоматического дефометра пока-
зана на рис. 62. Он имеет корпус, состоящий из верхней и нижней
частей. В верхней части установлен столик с тензометрической бал-
кой, которая расположена в фиксированном положении на при-
змах. На тензометрической балке в средней ее части жестко закреп-
лена бобышка для установки и крепления нижней круглой пласти-
ны. Подвижная штанга-гайка получает возвратно-поступательное
равномерное движение от вращающегося винта, жестко соединен-
ного с электродвигателем через редуктор и ступенчатый вариатор,
позволяющий создавать различные скорости движения штанги-
гайки. В ее нижней части закреплены пластина поводка, служащего
опорой для ножки микрометрического индикатора, и верхняя рабо-
чая круглая пластина. На корпусе прибора смонтированы включа-
тель и переключатель реверса электродвигателя.
262
Тензометрическая балка воспринимает усилие сжатия при ма-
лых собственных деформациях. На нее наклеены тензодатчики и
тензокомпенсаторы, которые подключены к различным плечам мо-
стовой измерительной схемы. Изгиб тензометрической балки вы-
зывает деформацию растяжения тензодатчиков, вследствие чего из-
меняется их электрическое сопротивление и нарушается баланс мо-
ста. Электрический сигнал моста усиливается тензоусилителем и
записывается шлейфовым осциллографом на самопроявляющейся
фотоленте.
До проведения измерений предварительно градуируют прибор с
помощью стандартных грузов. Для этого с помощью электроприво-
да верхнюю площадку поднимают в максимально верхнее положе-
ние. Балансируют тензометрический мост, устанавливают показа-
ния микрометрического индикатора на нуль. Включают протяжку
фотобумаги на осциллографе и начинают нагружать нижнюю пло-
щадку грузами от минимальной массы до 1 кг, записывают показа-
ния микрометрического индикатора, тем самым определяя стрелку
прогиба Дй тензобалки в зависимости от массы грузов, и строят гра-
дуировочный график Дй = /(/и).
Градуировочный график усилия Р= mg прогиба тензобалки на
диаграмме фотобумаги получают автоматически.
Образцы сыра подготавливают цилиндрической высечкой тако-
го же диаметра, как и диаметр круглых пластин, между которыми
помещают исследуемый продукт. Начальная высота Яо (м) образца
для упрощения расчетов должна быть равной 10,0 мм.
Образец помещают на нижнюю площадку и опускают верх-
нюю до соприкосновения с образцом. Без нагрузки устанавлива-
ют «зайчик» осциллографа при помощи реохорда на нуль. Для
визуализации процесса деформации ножку микрометрического
индикатора устанавливают на пластину поводка и выставляют
его показания на нуль. Во время опыта при включенном приво-
де механизма подъема — опускания верхней пластины записыва-
ют на фотобумаге осциллографа диаграмму усилие — время.
Опыты проводят при различной скорости w (м/с) сжатия образ-
цов.
Относительную деформацию сжатия е. вычисляют по формуле
где А(т) = нт — абсолютная деформация образца за время т (с) при скорости сжатия и.
Скорость деформации определяют по формуле
е. =еф /т.
263
Напряжение сжатия с учетом градуировочной диаграммы рас-
считывают по формуле
где F= nlP/4 — площадь основания образца диаметром D (м), м2.
По полученным данным строят графики зависимости а (т) и рас-
считывают константы компрессионных характеристик, учитывая
выбранную математическую модель и используя графоаналитичес-
кие методы применительно к формулам (5)—(10).
Контрольные вопросы и задания. 1. Для чего необходимо знание структурно-ме-
ханических характеристик молока и молочных продуктов? 2. Что представляют со-
бой основные реологические свойства и структурно-механические характеристики
молочных продуктов? 3. Дайте классификацию сенсорной оценки качества и тек-
стуры молочных продуктов. 4. Дайте классификацию реометров и определите об-
ласть их применения. 5. Опишите схемы ротационных вискозиметров и обоснуйте
необходимость получения инвариантных величин сдвиговых структурно-механи-
ческих характеристик при использовании различных конструкций приборов. 6. На-
зовите механические модели пастообразных продуктов, плавленых и натуральных
сыров.
Глава 11
АНАЛИТИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ СОСТАВА МОЛОКА
И МОЛОЧНЫХ ПРОДУКТОВ
ИЗМЕРЕНИЕ ВЛАЖНОСТИ
И СОДЕРЖАНИЯ СУХОГО ВЕЩЕСТВА
Для измерения влажности продуктов используют в основном
термогравиметрический метод (ТГ-метод). Термогравиметричес-
кий метод контроля влажности продуктов основан на взвешивании
продукта до и после высушивания (выпаривания) и вычислении
массовой доли влаги В (%) в соответствии с соотношением
5=^LZ^..1OO,
т\
где mi, тг — масса пробы продукта до и после высушивания, г.
Массовую долю сухого вещества С (%) в продукте определяют по
формуле
С= 100-5.
Температуру высушивания (выпаривания) продукта выбирают с
таким расчетом, чтобы в процессе сушки не изменялись составные
части продукта.
Для высушивания (выпаривания) используют различного типа
устройства, в которых продукт сушат нагретым от электронагрева-
теля воздухом в сушильных шкафах, непосредственно электронаг-
ревателем (контактная сушка), с помощью инфракрасного излуче-
ния (ИК-излучения) или СВЧ-энергии. Взвешивание и вычисле-
ние выполняют вручную или автоматически. Использование ИК-
излучения и СВЧ-энергии наиболее предпочтительно для создания
автоматических термогравиметрических (ТГ) влагомеров, обеспе-
чивающих контроль процесса взвешивания и автоматизацию рас-
чета массовой доли влаги.
Высушивание проб продукта с использованием сушильных
электрических шкафов используют, как правило, когда возникают
разногласия при оценке качества продукта. Для равномерного и
быстрого прогревания и высушивания продукта применяют раз-
личные материалы: промытый речной песок, двойной слой марли,
обезвоженное топленое масло, парафин. В лабораторной практике
широко применяют контактную сушку (выпаривание) на электро-
265
нагревателях (аппарат сушильный АПС-1, устройство ПИВИ-1.1
выпариватель влаги ВВМ-1 и т. п.).
ВНИМИ проведены испытания устройств, разработаны методи-
ки-проведения анализов и даны соответствующие рекомендации по
режимам (температура и время высушивания) при определении
влажности (табл. 37).
Аппарат Сушильный АПС-1 обеспечивает сушку проб продукта
выпариванием влаги из тонкослойного образца, находящегося
между нагретыми до рабочей температуры плитами устройства. В
аппарат входят блок высушивания, состоящий из верхней и нижней
нагревательных плит, блок контроля и управления. С помощью ап-
парата АПС-1 определяют влажность творога, сыров, сухих молоч-
ных продуктов, теста и других пищевых продуктов.
При определении массовой доли влаги навеску продукта (5 г)
равномерно распределяют в бумажном пакете, помещают пакет
между плитами устройства и высушивают в течение определенного
времени.
37. Режим сушки молочных продуктов в ТГ-влагомерах
ТГ-влагомер Продукт Температура суш- ки, “С Продолжи- тельность сушки, мин
Аппарат сушильный АПС-1 Сухое цельное молоко 140-142 2
(контактная сушка) Сухое обезжиренное молоко 140-142 3
Влагомер ЭВЛАС-1 Сухое цельное молоко 120 10
инфракрасной сушки Сухое обезжиренное молоко 100 6
Сухой казеин 102 7
Устройство ПИВИ-1.1, Творог 150-152 5
АПС-1 (контактная сушка) Сыр 160-162 6
Влагомер ЭВЛА С-1 обеспечивает измерение влажности сухих сы-
пучих молочных продуктов, а также других материалов раститель-
ного и минерального происхождения.
Пробу продукта известной массой высушивают в течение задан-
ного времени с помощью инфракрасной лампы и взвешивают оста-
ток пробы после удаления влаги. Изменение массы вычисляется ав-
томатически. Взвешивание продукта до сушки, в процессе сушки и
после нее контролируется встроенными в прибор весами. Результа-
ты измерения влажности и массы продукта, продолжительности
сушки и ее температуры отображаются на цифровом индикаторе.
ВНИМИ проведены испытания анализатора при контроле сухого
цельного и обезжиренного молока, сухого казеина и молочной сы-
воротки и даны рекомендации по методике проведения измерений.
Влажность сливочного масла контролируют с помощью комп-
лекта устройств, состоящего из выпаривателя влаги ВВМ-1 и мас-
лопробных весов СМП-84М.
266
J
Рис. 63. Инфракрасный (галогенный) влагомер (HR-73):
1 — подвижной стол; 2 — чашка для исследуемого продукта; 3 — исполнительный блок; 4 —
микропроцессорный блок; 5 — распечатка
Выпариватель влаги ВВМ-1 состоит из нагревателя и электронного
блока. Нагреватель имеет два гнезда для установки стаканов с навеска-
ми масла, датчик измерения температуры и контрольный термометр.
Электронный блок обеспечивает стабилизацию заданной температу-
ры процесса выпаривания. Он имеет индикатор нагрева, кнопки пуска
таймера и индикаторы начала и конца выпаривания. Устройство
ВВМ-1 позволяет выпаривать влагу одновременно из двух проб.
Маслопробные весы СМП-84М представляют собой одночашеч-
ные весы с неравноплечим коромыслом и гирями-рейтерами. Они
имеют стойку с отсчетной шкалой, футляр, коромысло, процент-
ную шкалу, арретир, серьгу и навеску.
Для определения влаги в сливочном масле берут навеску 5 или
10 г, взвешивают в специальном алюминиевом стакане. Стакан по-
мещают в гнездо выпаривателя, термостатируют и выпаривают вла-
гу. Далее стакан повторно взвешивают на весах, шкала которых гра-
дуирована в процентах влаги.
В настоящее время разработаны влагомеры, использующие для
сушки ИК-излучение.
Так, исполнительный блок 3 инфракрасный галогенного* влаго-
мера HR-73 (рис. 63) снабжен подвижным столом 1, на котором ус-
*В приборе для высушивания продукта использована галоген-вольфрамовая
лампа накаливания, в которую для увеличения срока службы лампы вводят пары
галогена, например йода.
267
тановлена чашка 2 для исследуемого продукта. После размещения
продукта на чашке 2 стол 1 перемещается в галогенную сушилку бло-
ка 3. Работа прибора осуществляется в автоматическом режиме. Про-
ба продукта взвешивается на встроенных в блок 3 весах и высушива-
ется. Температура и продолжительность сушки задаются с помощью
микропроцессорного блока 4, который также обрабатывает результа-
ты взвешивания продукта до и после сушки и выдает их на печать 5.
ОПРЕДЕЛЕНИЕ МАССОВЫХ ДОЛЕЙ ВЛАГИ
И СУХОГО ВЕЩЕСТВА В ПИТЬЕВОМ МОЛОКЕ, МОРОЖЕНОМ,
СЫРАХ, ТВОРОГЕ И ТВОРОЖНЫХ ИЗДЕЛИЯХ
Метод основан на высушивании навески продукта при температуре
102 ± 2 °C до постоянной массы в сушильном электрическом шкафу
типа СЭШ или другом аналогичного типа. По полученным данным
вычисляют массовую долю влаги и сухого вещества. Метод использу-
ют при возникновении разногласий в оценке качества продукта.
Выполнение работы. Стеклянную бюксу с 20—30 г хорошо промы-
того и прокаленного песка и стеклянной палочкой, не выступающей
за края бюксы, помещают в сушильный шкаф и выдерживают при
102 ± 2 °C в течение 30—40 мин. После этого бюксу вынимают из су-
шильного шкафа, закрывают крышкой, охлаждают в эксикаторе
40 мин и взвешивают с погрешностью не более 0,001 г. В эту же бюксу
пипеткой вносят 10 см3 молока или 5—10 г мороженого, или 3—5 г
сыра, творога, творожных изделий, взвешенных с погрешностью не
более 0,001 г, закрывают крышкой и немедленно взвешивают.
Затем содержимое тщательно перемешивают стеклянной палоч-
кой и открытую бюксу нагревают на водяной бане при частом пере-
мешивании содержимого до получения рассыпающейся массы. За-
тем открытую бюксу и крышку помещают в сушильный шкаф с тем-
пературой 102 ± 2 °C. По истечении 2 ч бюксу вынимают из сушиль-
ного шкафа, закрывают крышкой, охлаждают в эксикаторе 40 мин и
взвешивают.
Последующие взвешивания выполняют после высушивания в
течение 1 ч до тех пор, пока разность между двумя последователь-
ными взвешиваниями будет равна или менее 0,001г. Если при од-
ном из взвешиваний после высушивания будет установлено увели-
чение массы, для расчетов принимают результаты предыдущего
взвешивания.
Массовая доля сухого вещества (%)
(mi-ffio)lOO
т-т0 ’
где mi — масса бюксы с песком, стеклянной палочкой и навеской исследуемого
продукта после высушивания, г; то — масса бюксы с песком и стеклянной палоч-
кой, г; т — масса бюксы с песком, стеклянной палочкой и навеской исследуемого
продукта до высушивания, г.
268
Расхождение между параллельными определениями не должно
быть более 0,1 % для молока и 0,2 % — для мороженого, сыра, тво-
рога и творожных изделий. За окончательный результат для каждо-
го исследуемого продукта принимают среднее арифметическое двух
параллельных определений.
Массовая доля влаги в продуктах (%)
В= 100- С,
где С — массовая доля сухого вещества, %.
Массовая доля сухого обезжиренного вещества (%)
Со = С-Ж,
где С — массовая доля сухого вещества, %; Ж— массовая доля жира, %.
Подготовка песка. Песок просеивают через сито с отверстиями
диаметром 1 — 1,5 мм и отмачивают питьевой водой. Затем при-
ливают соляную кислоту (1 : 1) столько, чтобы песок был полнос-
тью покрыт ею, помешивают толстой стеклянной палочкой, дают
отстояться в течение 10 ч. Слив соляную кислоту, промывают пе-
сок питьевой водой до нейтральной реакции (определяют по лак-
мусовой бумажке), затем дистиллированной водой, высушивают
и прокаливают. Хранят песок в банке, плотно закрытой проб-
кой.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ СУХОГО ВЕЩЕСТВА
В ПИТЬЕВОМ МОЛОКЕ И КИСЛОМОЛОЧНЫХ НАПИТКАХ
УСКОРЕННЫМ МЕТОДОМ
Метод основан на высушивании пробы продукта в сушильном
шкафу до постоянной массы при температуре 105 °C. Для равномер-
ного и быстрого прогрева высушиваемой пробы на дно бюксы по-
мещают два слоя марли.
В металлическую бюксу на дно укладывают два кружка марли, вы-
сушивают с открытой крышкой при 105 °C в течение 20—30 мин и,
закрыв крышкой, охлаждают в эксикаторе 20—30 мин, затем взвеши-
вают. В подготовленную бюксу пипеткой вносят 3 см3 исследуемого
продукта, равномерно распределяя его по всей поверхности марли и,
закрыв крышкой, взвешивают. Затем открытую бюксу и крышку по-
мещают в сушильный шкаф при 105 °C на 60 мин, после чего бюксу
закрывают, охлаждают и взвешивают. Высушивание и взвешивание
продолжают через 20—30 мин до получения разницы между массами
двух последовательных взвешиваний не более 0,001 г. Сухой остаток
на поверхности марлевого кружка должен иметь равномерный свет-
269
ло-желтый цвет. Массовую долю влаги, сухого вещества и сухого
обезжиренного вещества определяют так же, как и в предыдущем
методе. Расхождение между параллельными определениями долж-
но быть не более 0,2 %. За окончательный результат принимают
среднее арифметическое двух параллельных определений.
ОПРЕДЕЛЕНИЕ МАССОВЫХ ДОЛЕЙ ВЛАГИ
И СУХОГО ВЕЩЕСТВА В МОРОЖЕНОМ
УСКОРЕННЫМ МЕТОДОМ
Метод основан на предварительном разведении пробы мороженого
дистиллированной водой, последующем выпаривании влаги при тем-
пературе 180 ± 2 °C на нагревательном приборе и досушивании в су-
шильном шкафу до постоянной массы при температуре 110 ± 2 °C.
Металлическую бюксу высушивают с открытой крышкой при
110 ± 2 °C в течение 20—30 мин и, закрыв крышкой, охлаждают в эк-
сикаторе 20—30 мин, затем взвешивают. В подготовленную бюксу
отвешивают 1 г мороженого с погрешностью не более 0,01 г и при-
бавляют пипеткой 1 см3 дистиллированной воды. Легким покачива-
нием бюксы ее содержимое перемешивают до получения однород-
ной массы и равномерного распределения по дну. Затем бюксу с на-
веской ставят на нагревательный прибор, накрытый железной плас-
тинкой, температура поверхности которой 180 ± 2 °C. Содержимое
бюксы выпаривают до легкого пожелтения остатка, получающегося в
виде пористой массы, при интенсивном кипении, после чего бюксу
помещают в сушильный шкаф с температурой ПО+ 2°C. Через
10 мин бюксу вынимают из сушильного шкафа, закрывают крыш-
кой, охлаждают в эксикаторе и взвешивают. Высушивание и взвеши-
вание продолжают до получения разницы в массе между двумя пос-
ледовательными взвешиваниями не более 0,01 г.
Массовые доли влаги и сухого вещества вычисляют так же, как и
при исследовании пастеризованного молока. Расхождение между
параллельными определениями должно быть не более 0,5 %. За
окончательный результат принимают среднее арифметическое двух
параллельных определений.
ОПРЕДЕЛЕНИЕ МАССОВЫХ ДОЛЕЙ ВЛАГИ
И СУХОГО ВЕЩЕСТВА В СЫРАХ, ТВОРОГЕ
И ТВОРОЖНЫХ ИЗДЕЛИЯХ
УСКОРЕННЫМИ МЕТОДАМИ
Первый метод. Основан на выпаривании влаги из тонкослойного
образца продукта, находящегося между нагретыми до рабочей тем-
пературы плитами нагревательного устройства. В качестве нагрева-
тельного устройства применяют прибор Чижовой, сушильный ап-
парат АПС-1 и другие, аналогичные им.
270
Для определения массовой доли влаги в продукте пакеты (одно-
или двухслойные) из газетной бумаги размером 150x150мм
складывают по диагонали, загибают углы и края примерно на
15 мм.
При определении массовой доли влаги в сыре, твороге и творож-
ных изделиях пакет вкладывают в листок пергамента, несколько
большего размера, чем пакет, не загибая краев. Готовые пакеты вы-
сушивают в приборе в течение 3 мин при той же температуре, при
которой будет высушен исследуемый продукт, после чего их охлаж-
дают и хранят в эксикаторе.
Подготовленный пакет взвешивают с погрешностью не более
0,01 г, отвешивают в него 5 г исследуемого продукта с погрешнос-
тью не более 0,01 г, распределяя равномерно по всей внутренней
поверхности пакета. Пакет с навеской закрывают, помещают в при-
бор между плитами, нагретыми до требуемой температуры, и вы-
держивают необходимое время (табл. 38).
38. Температура и продолжительность обработки пробы продукта
Продукт Температура нагревания нижней плиты прибора, "С Продолжительность выдержки, мин
Творог, творожные изделия и паста 150-152 5
Сыр после прессования 160-162 6
Сыр зрелый 150-155 7
Сыр плавленый 160-162 8
Одновременно можно высушивать два пакета. При высушива-
нии продуктов с относительно высокой влажностью, таких, как
творог и творожные изделия, в начале сушки во избежание разрыва
пакета верхнюю плиту прибора приподнимают и поддерживают в
таком положении до прекращения обильного выделения паров, ко-
торое обычно длится 30—50 с. Затем плиту опускают и продолжают
высушивание в течение времени, установленного для данного про-
дукта. Пакеты с высушенными пробами охлаждают в эксикаторе 3—
5 мин и взвешивают.
Массовая доля влаги в продукте (%)
^=(туп).1ОО,
где т — масса пакета с навеской до высушивания, г; — масса пакета с навеской
после высушивания, г; 5 — масса навески продукта, г.
Расхождение между параллельными определениями не должно
быть более 0,5 %. За окончательный результат принимают среднее
арифметическое двух параллельных определений.
Массовая доля сухого вещества в продукте (%)
С= 100-в.
271
Второй метод. Основан на высушивании влаги на нагреватель-
ном приборе из образца продукта, находящегося в среде обезвожен-
ного топленого масла или парафина.
В сухой алюминиевый стакан (без крышки) кладут кружок пер-
гамента, закрывающий дно стакана и на 0,5 см нижнюю часть его
стенок. Взвешивают в стакане от 5 до 8 г обезвоженного топленого
масла или парафина и 5 г испытуемого продукта погрешностью не
более 0,01 г.
С помощью специального металлического держателя или щип-
цов алюминиевый стакан осторожно, особенно вначале, нагревают,
поддерживая спокойное и равномерное кипение, не допуская вспе-
нивания и разбрызгивания. Нагревание ведут до прекращения от-
потевания холодного зеркала или часового стекла, поддерживаемо-
го над стаканом. Признаком окончания испарения воды служит
прекращение вспенивания и треска и появление легкого побуре-
ния. После высушивания стакан охлаждают на чистом, гладком ме-
таллическом листе и взвешивают.
Массовую долю влаги (%) вычисляют по формуле
В=(^2/П1) Юо
m-niQ
где т — масса алюминиевого стакана с обезвоженным топленым маслом или пара-
фином, пергаментом и навеской продукта до нагревания, г; гщ — масса алюминие-
вого стакана с обезвоженным топленым маслом или парафином, пергаментом и на-
веской продукта после удаления влаги, г; т0 — масса алюминиевого стакана с обез-
воженным топленым маслом или парафином, пергаментом, г.
Расхождение между параллельными определениями не должно
быть более 0,5 %. За окончательный результат принимают среднее
арифметическое двух параллельных определений.
Массовую долю сухого вещества в продукте вычисляют так же,
как и по первому методу.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ
В СЛИВОЧНОМ МАСЛЕ
Первый метод. Основан на высушивании пробы масла в сушиль-
ном шкафу при температуре 102 ± 2 °C. Его применяют при возник-
новении разногласия в оценке качества.
Пробу масла нагревают до температуры не выше 30 °C, обеспечи-
вающей гомогенное состояние при перемешивании механической
мешалкой или вручную. Затем охлаждают до комнатной температу-
ры (около 20 °C) при постоянном перемешивании. 12—30 г песка
помещают в чашку. Чашку с песком и стеклянной палочкой сушат в
сушильном шкафу при температуре 102 ± 2 °C в течение 1 ч, затем
охлаждают в эксикаторе до комнатной температуры (около 20 °C) и
взвешивают с погрешностью не более 0,001 г.
272
В чашку отвешивают от 5 до 10 г масла с погрешностью не более
0,001 г, тщательно перемешивают с песком. Ставят чашку в сушиль-
ный шкаф и сушат при температуре 102 ± 2 °C не менее 2 ч. Затем
содержимое чашки охлаждают в эксикаторе до комнатной темпера-
туры (около 20 °C) и взвешивают с погрешностью не более 0,001 г.
Последующие взвешивания проводят после высушивания в те-
чение 1 ч до тех пор, пока разность между массами двух последую-
щих взвешиваний будет не более 0,001 г.
Если после повторного высушивания масса увеличится, для рас-
чета берут результаты предыдущего взвешивания.
Массовую долю влаги (%) в масле вычисляют по формуле
m-ntQ
где т — масса чашки с песком и стеклянной палочкой и маслом до высушивания, г;
mt — масса чашки с песком и стеклянной палочкой и маслом после высушивания, г;
то — масса чашки с песком и стеклянной палочкой, г.
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,1%.
Песок просеивают через сито с диаметром отверстий 1,5 мм, за-
тем через сито с диаметром 1,0 мм. Берут ту часть, которая оста-
лась на втором сито, промывают питьевой водой, после чего кипя-
тят в течение 30 мин с 25%-ным раствором соляной кислоты, про-
мывают питьевой, затем дистиллированной водой до отрицатель-
ной реакции на хлориды, после чего просушивают на воздухе и
прокаливают в муфельной печи или на электрической плитке
500+ 25 °C.
Второй метод. Основан на выпаривании влаги из пробы масла на
нагревательном приборе (плитке, выпаривателе влаги ВВМ-1) с
последующим определением массовой доли влаги в масле на масло-
пробных весах СМП-84. Метод широко используется в лаборатор-
ной практике.
Масло без наполнителей. В сухой алюминиевый стакан отвешива-
ют 5 или 10 г исследуемого топленого или сливочного масла с по-
грешностью не более 0,01 г.
С помощью специального металлического держателя или щип-
цов алюминиевый стакан осторожно, особенно вначале, нагревают,
поддерживая спокойное и равномерное кипение, не допуская вспе-
нивания и разбрызгивания. Нагревание проводят до прекращения
отпотевания холодного зеркала или часового стекла, поддерживае-
мого над стаканом.
Признаком окончания испарения воды служит прекращение
вспенивания и треска и появление легкого побурения. После высу-
шивания стакан охлаждают на чистом, гладком металлическом лис-
те и взвешивают.
273
Массовую долю влаги (%) вычисляют по формуле
5=К^).юо,
«о
где т — масса алюминиевого стакана с навеской продукта до нагревания, г; гщ —
масса алюминиевого стакана с навеской продукта после удаления влаги, г; т0 — на^
веска продукта, г.
Расхождение между параллельными определениями не должно
быть более 0,1 % для топленого масла, 0,2 % — для сливочного мас-
ла. За окончательный результат принимают среднее арифметичес-
кое двух параллельных определений.
Масло с наполнителями. Алюминиевый стакан с тремя бумажны-
ми роликами на дне (фильтровальную бумагу разрезают на полосы
шириной 7—8 мм и длиной 620 мм и свертывают каждую полосу в
отдельности с помощью палочки в виде ролика, который не должен
быть очень тугим) помещают в сушильный шкаф с температурой
102 ± 2 °C. Через 1 ч стакан вынимают из сушильного шкафа, ох-
лаждают в эксикаторе, взвешивают с погрешностью не более 0,01 г
и отвешивают в нем с той же погрешностью 10 г масла.
С помощью металлического держателя или щипцов алюминие-
вый стакан с маслом осторожно, особенно вначале, нагревают, под-
держивая спокойное и равномерное кипение, не допуская вспени-
вания и разбрызгивания. Нагревание проводят до прекращения от-
потевания холодного зеркала или часового стекла, поддерживаемо-
го над стаканом.
Признаком удаления влаги служит прекращение образования
пузырьков на роликах. После высушивания стакан охлаждают на
чистом гладком металлическом листе и взвешивают.
Массовую долю влаги (%) в масле вычисляют по формуле
^J^O.100,
т — масса алюминиевого стакана с бумажными роликами и навеской продукта до
нагревания, г; т1 — масса алюминиевого стакана с роликами и навеской продукта
после удаления влаги, г; 10 — масса навески продукта, г.
Расхождение между параллельными определениями не должно
быть более 0,2 %. За окончательный результат принимают среднее
арифметическое двух параллельных определений.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ СОМО
В МАСЛЕ БЕЗ НАПОЛНИТЕЛЕЙ
Метод основан на удалении влаги из образца масла двумя мето-
дами — выпариванием и высушиванием в сушильном шкафу (в ар-
274
битражных случаях) и последующей экстракцией из сухого веще-
ства масла жира бензином, этилом или петролейным эфиром. По-
дученный остаток взвешивают и рассчитывают массовую долю
СОМО в масле.
Первый метод. Массовую долю СОМО в масле определяют после
установления в нем массовой доли влаги путем выпаривания на на-
гревательном приборе.
В алюминиевый стакан вкладывают стеклянную палочку и
взвешивают. В стакан отвешивают навеску исследуемого масла —
10 г сливочного или 20 г топленого, взвешенную с погрешностью
не более 0,01 г. Массовую долю влаги определяют так же, как в
масле без наполнителей. Остаток в алюминиевом стакане после
определения массовой доли влаги слабо нагревают до расплавле-
ния жира, приливают 50 см3 бензина или этилового эфира, смесь
тщательно перемешивают палочкой и оставляют в покое на 3—
5 мин для осаждения осадка. Плавающие на поверхности бензи-
на частицы, не осаждающиеся на дно, указывают на неполное
выпаривание влаги. В этом случае определение должно быть пов-
торено.
После отстаивания осадка бензино-жировой раствор осторожно
сливают, не взмучивая осадка и оставляя в стакане 1 —2 см3 бензи-
но-жирового раствора. Обработку осадка бензином повторяют три
раза. Остаток в стакане нагревают на слабом пламени спиртовки
или на электроплитке до полного удаления бензина. Полное удале-
ние бензина устанавливают по рассыпчатости остатка при переме-
шивании его стеклянной палочкой. Стакан с содержимым охлажда-
ют до комнатной температуры и взвешивают.
Массовая доля СОМО в масле без наполнителей (%)
где т\ — масса стакана после удаления бензино-жирового раствора, г; т0 — масса
пустого стакана со стеклянной палочкой, г; т — масса стакана со стеклянной па-
лочкой и навеской масла, г.
Массовая доля СОМО в соленом масле (%)
С01=С0-Сс,
где Со — массовая доля СОМО в соленом масле (вместе с солью), %; Сс — массовая
доля соли в соленом масле, %.
Второй метод. Пробу нагревают до температуры не выше 30 °C,
обеспечивающей гомогенное состояние при перемешивании меха-
нической мешалкой или вручную. Затем охлаждают до комнатной
температуры (около 20 °C) при постоянном перемешивании. Чашку
и тигель высушивают при температуре 102 ± 2 °C в течение 1 ч, за-
275
тем охлаждают в эксикаторе до комнатной температуры (около
20 °C) и взвешивают с погрешностью не более 0,0001 г. В охлажден-
ную чашку отвешивают около 10 г пробы с погрешностью не более
0,0001 г. Осторожно нагревают чашку, растапливают масло, про-
должают нагревание до тех пор, пока масло не перестанет пениться,
не допуская при этом перегрева.
Чашку с содержимым охлаждают в эксикаторе до комнатной тем-
пературы (около 20 °C). Добавляют от20 до 25 см3 петролейного эфи-
ра и растворяют жир легким перемешиванием, переливают раствор и
осадок в тигель и фильтруют при помощи вакуумного насоса.
Обработку петролейным эфиром и переливание раствора и осад-
ка в тигель повторяют пять раз. Чашку и тигель сушат в течение 2 ч
при температуре 102 ± 2 °C. Последующие взвешивания проводят
после высушивания в течение 30 мин до тех пор, пока разность меж-
ду двумя последовательными взвешиваниями будет не более 0,001 г.
Сушку, охлаждение и взвешивание повторяют через 30 мин до
получения постоянной массы (изменение массы не должно превы-
шать 0,001 г).
Массовую долю СОМО (%) вычисляют по формуле
= (т2-т1)+(т4-т3)
0 т ’
где т2 — масса тигля с осадком, г; mj — масса пустого тигля, г; т4 — масса чашки с
осадком, г; т3 — масса пустой чашки, г; т — масса навески масла, г.
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,1 %.
ИЗМЕРЕНИЕ ЖИРНОСТИ
В настоящее время для определения жира в молоке и молочных
продуктах наиболее широко применяют два метода: кислотный и
гравиметрический.
Для проведения исследования кислотным методом используют
серную кислоту и изоамиловый спирт. Под действием серной кис-
лоты казеинаткальцийфосфатный комплекс молока переходит в
растворимое соединение казеина с серной кислотой. При добавле-
нии изоамилового спирта понижается поверхностное натяжение
жировых шариков, с поверхности жировых шариков удаляется обо-
лочка. Реакции ускоряются нагреванием и центрифугированием.
Анализ выполняют в специальном приборе — жиромере. После
центрифугирования жир выделяется в виде сплошного слоя и объем
его измеряют в градуированной части жиромера. Применяют жиро-
меры трех типов: для определения жира в молоке и молочных про-
276
дуктах, в сливках и молочных продуктах с высоким содержанием
жира, в обезжиренном молоке и пахте.
Жиромер для молока и молочных продуктов показывает содер-
жание жира в процентах массы при навеске 11 г продукта (10,77 см3
молока), жиромер для сливок и молочных продуктов с высоким со-
держанием жира — при навеске продукта 5 г, а жиромер для обез-
жиренного молока и пахты — при навеске продукта 22 г.
Гравиметрический метод определения жира основан на его из-
влечении (экстракции) органическими растворителями и последу-
ющем определении количества жира в экстракте.
Для экстракции жира применяют растворители с низкой темпе-
ратурой кипения, удаление которых из жира не представляет зат-
руднений. Чаще всего используют петролейный, этиловый, диэти-
ловый, серный и другие эфиры, а также хлороформ, дихлорэтан.
Петролейный эфир имеет преимущества перед другими раствори-
телями, так как меньше извлекает веществ, сопутствующих жирам.
Вода, содержащаяся в продукте, препятствует диффузии жира из
материала в растворитель. Поэтому перед экстракцией жира иногда
прибегают к обезвоживанию материала.
Известно несколько модификаций гравиметрического метода: г
по Розе—Готлибу, по Шмидт-Бондзинскому — Вацлаву, по Можо-
нье, экспресс-метод.
КИСЛОТНЫЙ МЕТОД
Проведение работы. Молоко коровье (сырое, пастеризованное
различных видов, кроме нежирного, стерилизованное, для детского
питания).
В два молочных жиро мера (типов 1 —6 ил и 1 —7), стараясь не сма-
чивать стенок горловины, наливают дозатором по 10 см3 серной
кислоты (плотностью от 1810 до 1820 кг/м3) и осторожно, чтобы
жидкости не смешивались, добавляют пипеткой до 10,77 см3 моло-
ка, приложив кончик пипетки к горловине жиромера под углом.
Уровень молока в пипетке устанавливают по нижнему уровню ме-
ниска.
Молоко из пипетки должно вытекать медленно. После опорож-
нения пипетку отнимают от горловины жиромера не ранее чем че-
рез 3 с. Выдувание молока из пипетки не допускается. Дозатором
добавляют в жиромеры по 1 см3 изоамилового спирта. Уровень сме-
си в жиромере должен быть на 1—2 мм ниже основания горловины
жиромера, для чего можно добавлять несколько капель дистилли-
рованной воды.
Для повышения точности измерений, особенно для молока низ-
кой плотности, рекомендуется сначала взвешивать 11 г молока с
точностью до 0,005 г, затем приливать серную кислоту и изоамило-
вый спирт. Жиромеры закрывают сухими пробками, вводя их в гор-
277
ловину жиромеров немного глубже чем наполовину. Жиромеры
встряхивают до полного растворения белковых веществ, перевора-
чивая не менее 5 раз так, чтобы жидкости в них полностью переме-
шались.
Для обеспечения проведения измерений поверхность пробок
для укупорки жиромеров натирают мелом.
Жиромеры устанавливают пробкой вниз на 5 мин в водяную
баню при температуре 65 ± 2 °C. Вынутые из бани жиромеры встав-
ляют симметрично, один против другого, в стаканы центрифуги
градуированной частью к центру. При нечетном числе жиромеров в
центрифугу помещают жиромер, наполненный водой вместо моло-
ка, серной кислотой и изоамиловым спиртом в том же соотноше-
нии, что и для анализа.
Центрифугирование длится 5 мин. По окончании жиромеры по-
очередно вынимают из центрифуги и движением резиновой пробки
регулируют столбик жира так, чтобы он находился в градуирован-
ной части жиромера.
Снова погружают жиромеры пробками вниз на 5 мин в водяную
баню при температуре 65 ± 2 °C, при этом уровень воды в бане дол-
жен быть несколько выше уровня жира в жиромере. По истечении
5 мин жиромеры вынимают из водяной бани и быстро отсчитывают
содержание жира. При этом жиромер держат вертикально, граница
жира должна находиться на уровне глаз. Движением пробки уста-
навливают нижнюю границу столбика жира на нулевом или целом
делении шкалы жиромера. От него отсчитывают число делений до
нижнего уровня мениска столбика жира с точностью до наимень-
шего деления шкалы жиромера. Граница раздела жира и кислоты
должна быть резкой, а столбик жира прозрачным. При наличии
«кольца» (пробки) буроватого или темно-желтого цвета, различных
примесей в столбике жира или размытой нижней границы измере-
ние проводят повторно.
При анализе гомогенизированного или восстановленного моло-
ка массовую долю жира в нем определяют в соответствии с выше-
описанными требованиями, но трехкратно центрифугируют и на-
гревают между каждым центрифугированием в водяной бане при
температуре 65 ± 2 °C в течение 5 мин.
При использовании центрифуги с подогревом жиромеров до-
пускается один раз центрифугировать в течение 15 мин с последую-
щей выдержкой в водяной бане при температуре 65 ± 2 °C в течение
5 мин.
Кефир, простокваша, ряженка, ацидофилин, сметана, творог,
творожные изделия, кисломолочные продукты для детского питания,
сливки, мороженое. Содержание жира в этих продуктах определяют,
как и для молока, в соответствии с требованиями, указанными в
табл. 39, а также и следующими дополнительными условиями:
последовательность операций при заполнении жиромерй —
отвешивание продукта в жиромер с отсчетом до 0,005 г, добавле-
278
ние воды (при необходимости), серной кислоты и изоамилового
спирта;
серную кислоту в жиромер с водой добавляют осторожно, слегка
наклонив жиромер;
при определении содержания жира в сливках, сметане, твороге,
творожных изделиях и мороженом жиромеры с исследуемой сме-
сью подогревают перед центрифугированием в водяной бане при
частом встряхивании до полного растворения белка;
при определении содержания жира в сливках, сметане и молоч-
ном мороженом уровень смеси в жиромере устанавливают на 4—
5 мм ниже основания горловины жиромера, при определении со-
держания жира в сливочном мороженом и пломбире — на 6—10 мм.
Сыры (натуральные и плавленые). Условия проведения измерений
соответствуют требованиям табл. 39.
39. Условия для проведения исследований различных продуктов
Продукт Тип жиромера Объем, масса образца для анализа Объем добавлен- ной во- ды, см! Плотность серной кис- лоты, кг/м3 Объем серной кислоты^ см3 Число центри- фугиро- ваний
Молоко всех ви- дов, кроме нежир- ного, негомогени- зированное 1-6; 1-7 10,77 см3; 11,00 г — От 1810 до 1820 10 1
Молоко всех ви- дов, кроме нежир- ного, гомогенизи- рованное 1-6; 1—7 10,77 см3; 11,00 г — Ог18Ю до 1820 10 3
Кисломолочные продукты из него- могенизирован- ного молока 1-6; 1-7 11,00 г — От 1810 до 1820 10 1
Кисломолочные продукты из гомо- генизированного молока, в том чис- ле для детского питания 1-6; 1-7 11,00 г От 1810 до 1820 10 3
Сливки негомоге- низированные и сметана из него- могенизирован- ных сливок с мас- совой долей жира не более 40 %; творог, творож- ные изделия без сахара 1-40 5,00 г 5 От 1810 до 1820 10 1
Сливки негомоге- низированные с массовой долей жира более 40 % 1-40 2,50 г 7,5 От 1810 до 1820 10 1
279
Продолжение
Продукт Тип жиромера Объем, масса образца для анализа Объем добавлен- ной во- ды, см3 Плотность серной кис- лоты, кг/м3 Объем серной кислоты, см3 Число центри- фугиро- ваний
Сливки гомогени- зированные и сме- тана из гомогени- зированных сли- вок 1-40 5,00 г 5 От 1810 до 1820 10 3
Творожные изде- лия с сахаром 1-40 5,00 г 5 От 1800 до 1810 10 1
Мороженое мо- лочное и люби- тельских видов с массовой долей жира не более 5 % из гомогенизиро- ванной смеси 1-6; 1-7 5,00 г От 1500 до 1550 16 4
Мороженое сли- вочное и люби- тельских видов с массовой долей жира от 5 до 10%, из гомогенизиро- ванной смеси 1-6; 1-7; 1-40 5,00 г От 1500 до 1550 16 4
Мороженое сли- вочное и люби- тельских видов с массовой долей жира от 5 до 10%, из негомогенизи- рованной смеси 1-6; 1-7; 1-40 5,00 г От 1500 до 1550 16 1
Мороженое плом- бир и любитель- ских видов с мас- совой долей жира более 10 % 1-6; 1-7; 1-40 4,00 г 5,00 г — От 1500 до 1550 16 4
Сыры натураль- ные и плавленые 1-6; 1-7 1,50 г — От 1500 до 1550 19 1
Масло сливочное с наполнителями 1-40 2,50 г — От 1500 до 1550 16 1
Масло сливочное без наполнителей (производствен- ный метод), кро- ме соленого масла — — — — — —
Молоко нежирное 2 и пахта -0,5; 2-1,0 10,77 см3 х 2 — От 1810 до 1820 20 3
Сыворотка (после сепарирования) 2-0,5 10,77 см3 х 2 — От 1780 до 1800 20 3
280
В два жиромера отвешивают по 1,5 г сыра с отсчетом до 0,005 г,
затем приливают дозатором по 10 см3 серной кислоты, доливают
по 9 ± 1 см3 так, чтобы уровень жидкости был от 4 до 6 мм ниже
основания горловины жиромера. Дозатором добавляют в жироме-
ры по 1 см3 изоамилового спирта. Жиромеры закрывают пробка-
ми, помещают в водяную баню при температуре 65 ± 2 °C и выдер-
живают там при частом встряхивании до полного растворения бел-
ка в течение 50—70 мин. Если за это время белок растворился не-
полностью, температуру водяной бани при повторном опре-
делении можно довести до 73 ± 3 °C. Показания жиромера при
этом снимают после пятиминутной выдержки жиромеров в водя-
ной бане при температруре 65 ± 2 °C. Далее измерения проводят,
как и для молока.
Масло с наполнителями. Условия проведения измерений соответ-
ствуют требованиям табл. 39.
В два жиромера отвешивают по 2,5 г масла с отсчетом до 0,005 г,
приливают дозатором по 10 см3 серной кислоты, доливают по
6 ± 1 см3 серной кислоты так, чтобы уровень жидкости был от 4 до
6 мм ниже основания горловины жиро мера. Дозатором добавляют в
жиромеры по 1 см3 изоамилового спирта. Закрывают жиромеры
пробками и помещают их в водяную баню при температуре
65 ± 2 °C. Жиромеры выдерживают в водяной бане при частом
встряхивании до полного растворения белка. Далее измерения про-
водят, как и для молока.
Молоко нежирное, пахта. Условия проведения измерений соот-
ветствуют требованиям табл. 39.
В два жиромера, горловины которых со стороны градуирован-
ной части закрыты пробками, осторожно, стараясь не смочить
горловину, отмеривают серную кислоту. Затем отмеривают ис-
следуемый продукт в каждый жиромер при помощи пипетки вме-
стимостью 10,77 см3 (по два раза), осторожно сливая его по стен-
ке жиромеров. Дозатором добавляют в жиромеры по 2 см3 изо-
амилового спирта, закрывают их большими пробками и встряхи-
вают до полного растворения белковых веществ, время от
времени переворачивая. Затем жиромеры устанавливают боль-
шой пробкой вниз на 5 мин в водяную баню температурой
65 ± 2 °C. После водяной бани жиромеры центрифугируют три
раза по 5 мин или два раза по 10 мин. В центрифуге их устанавли-
вают градуированной частью к центру. Между центрифугирова-
ниями жиромеры термостатируют по 5 мин в водяной бане при
температуре 65 ± 2 °C.
После первого центрифугирования для облегчения регулирова-
ния уровня жира в жиромере маленькую пробку слегка приоткры-
вают, не вынимая ее полностью. С помощью большой пробки ус-
танавливают верхний уровень жидкости в градуированной части
жиромера. Затем меньшее отверстие плотно закрывают. Обычно
281
после первого центрифугирования заметного отделения жира не
наблюдают.
После второго центрифугирования и выдерживания в водяной
бани проверяют положение уровня жидкости. После третьего цен-
трифугирования вынимают из жиромеров маленькие пробки, жи-
ромеры помещают на 5 мин в водяную баню при температуре
65 ± 2 °C и следят, чтобы уровень жидкости не поднимался выше
делений шкалы.
Вынув жиромер из бани и регулируя большой пробкой, устанав-
ливают нижнюю границу жира на нулевом или ближайшем целом
делении шкалы и быстро отсчитывают содержание жира.
Молочная сыворотка. Для очистки сыворотки от белковых частиц
пробу нагревают до 35 ± 5 °C и фильтруют через ватный фильтр или
не менее чем через три слоя марли.
Массовую долю жира в молочной сыворотке измеряют так же,
как и в нежирном молоке. За результат измерений принимают сред-
нее арифметическое значение результатов двух параллельных на-
блюдений.
Показания жиромера при измерениях в молоке, в том числе не-
жирном; кисломолочных продуктах, в том числе сметане, твороге,
сливках (с массовой долей жира не более 40 %), сливочном мороже-
ном, пломбире, пахте и молочной сыворотке, соответствуют массо-
вой доле жира в этих продуктах в процентах.
Массовую долю жира Ж (%) в молочном мороженом и сыре вы-
числяют по формуле
в сливках с массовой долей жира более 40 % и в сливочном масле с
наполнителями — по формуле
где Р— показания жиромера, %; т — масса навески, г; 11 и 5 — массы навесок про-
дуктов, которые используют для градуировки жиромеров (11— для жиромеров ти-
пов 1-6,1-7; 5 — для жиромеров типа 1-40), г.
Массовую долю жира в сырье в пересчете на сухое вещество
Жс (%) вычисляют по формуле
ж _Ж100
с 100-5’
где В — массовая доля влаги в сырье, %; 100 — коэффициент пересчета массовой
доли жира на 100 г продукта.
282
Массовую долю жира в сливочном масле без наполнителей вы-
числяют по формулам
Жх = 100-(5+ Со);
Ж2 = 100-(5+ Со+ Q),
где — массовая доля жира в масле без наполнителей всех видов, кроме солено-
го, %; В — массовая доля влаги в масле, %; Жг — массовая доля жира в соленом мас-
ле, %; Со — массовая доля обезжиренного сухого вещества в масле, %; С{ — массо-
вая доля соли в масле, %; 100 — коэффициент пересчета массовой доли жира на
100 г продукта.
Приготовление реактивов. Реактив 1 {серная кислота). Концент-
рированную серную кислоту разбавляют водой до требуемой плот-
ности. Определяют плотность имеющейся H2SO4 ареометром и по
приложению находят процентное содержание в ней H2SO4. Объем
воды Ив (см3), необходимый для разведения, рассчитывают по
формуле
у -у.( di c> fl
КВ V2-C2
где Й! —объем кислоТы, подлежащей разведению, см3; dj—плотность кислоты,
подлежащей разведению, г/см3; С[ — концентрация H2SO4 в кислоте, подлежащей
разведению, %; — плотность кислоты после разведения, г/см3; Сг — концентра-
ция H2SO4 в кислоте после разведения, %.
Если требуется получить известное количество разведенной кис-
лоты (У2), то объем воды для разведения находят по формуле
у =у,(
а объем концентрированной кислоты, подлежащей разведению,
определяют по формуле
И1=И2-ИВ.
Серную кислоту разводят в тонкостенной термоустойчивой кол-
бе, приливая кислоту в воду, а не воду в кислоту. Вносят в колбу все
количество воды, необходимое для разведения кислоты. При смеши-
вании серной кислоты с водой происходит сильное разогревание,
поэтому при приливании кислоты колбу помещают в таз с водой.
Держа колбу левой рукой, не вынимая колбу из воды, правой ру-
кой осторожно небольшими порциями приливают серную кислоту,
помешивая содержимое колбы. После приливания всей серной
кислоты раствор охлаждают до 20 °C и проверяют плотность арео-
метром.
283
Если температура серной кислоты при измерении плотности от-
личается от 20 °C, то к отсчету ареометра вносят поправку: на каж-
дый градус температуры ниже 20 °C из найденной величины плот-
ности серной кислоты вычитают, а выше 20 °C — прибавляют сле-
дующие величины:
Плотность 1,30—1,67 1,67—1,70 1,70—1,80 1,80—1,84
Поправка 0,0010 • 0,0011 0,0012 0,0014
Реактив 2(изоамиловый спирт). Плотность изоамилового спирта
должна быть 0,8108—0,8115 г/см3, температура кипения 128—
132 °C, в пределах указанных температур должно перегоняться 95 %
его. Плотность определяют пикнометром или ареометром.
ГРАВИМЕТРИЧЕСКИЙ МЕТОД
МЕТОД РОЗЕ — ГОТЛИБА
Сущность метода заключается в экстрагировании жира из амми-
ачно-спиртового раствора молока диэтиловым и петролейным
эфирами, выпариванием растворителей и взвешиванием остатка.
• Жир из молока извлекают диэтиловым и петролейным эфирами
после растворения белковых веществ концентрированным водным
раствором аммиака в присутствии этилового спирта. Аммиак пере-
водит казеинаткальцийфосфатный комплекс в растворимые аммо-
нийные соли и, понижая вязкость смеси, способствует быстрому
растворению жира эфирами. Этиловый спирт, способствуя удержа-
нию в водном растворе растворимых в спирте экстрактивных ве-
ществ, облегчает переход жира из водной фазы в эфирную и препят-
ствует образованию желеобразной смеси при интенсивном взбал-
тывании.
Диэтиловый и петролейный эфиры растворяют жир. Петролей-
ный эфир, не смешиваясь с водой, способствует удалению следов
воды из этилово-эфирного жирового раствора, что предотвращает
переход в него нежировых составных частей молока, остающихся в
водной фазе. После отделения эфиро-жирового слоя, отгонки ра-
створителей и высушивания остатка взвешивают чистый жир.
Для экстрагирования жира применяют прибор — экстрактор
(рис. 64), который представляет собой пробирку из прочного стекла
с фторопластовой пробкой. Для слива эфирного раствора жира слу-
жит сифон, вставленный в фторопластовую пробку. Трубка сифона
выполнена из нержавеющей стали и заканчивается изгибом вверх
(крюком) для предохранения попадания внутрь нее воды.
Для экстрагирования применяют также специальные сосуды и
колбы с притертыми или корковыми пробками. Перед употребле-
нием корковые пробки обрабатывают сначала диэтиловым, а затем
петролейным эфиром, после чего помещают на 20 мин в воду при
284
Рис. 64. Экстрактор:
1 — трубка из нержавеющей стали диаметром Змм; 2 — фторо-
пластовая пробка
температуре 60 °C и выдерживают в ней до ох-
лаждения для насыщения водой. Используют
не высушивая.
Выполнение работы. Пробу молока, сливок
или кисломолочных напитков доводят до
20 ± 2 °C, тщательно перемешивают до одно-
родной консистенции, чтобы диспергировать
жир. При этом пробу сильно не встряхивают,
чтобы не вызвать ее вспенивания или сбива-
ния молочного жира.
Если диспергирование жира в молоке,
сливках и кисломолочных напитках вызыва-
ет затруднение, пробу при осторожном перемешивании медленно
нагревают до 40 ± 2 °C, тщательно перемешивают и быстро охлаж-
дают до 20 ± 2 °C.
Колбу для перегонки вместе с материалом, облегчающим кипе-
ние (стеклянные шарики или кусочки карборунда), высушивают в
сушильном шкафу при (102 + 2)°С или в вакуум-сушильном шкафу
при 70—75 °C и давлении 6,66 Па в течение 0,5—1 ч, охлаждают в
эксикаторе до 20 ± 2 °C и взвешивают.
При проведении первого экстрагирования масса навески про-
дукта с массовой долей жира до 5 % — от 10 до 11г. Массу навески
продукта тр (г) с массовой долей жира более 5 % вычисляют по
формуле
т
_ 50
Р Ж{
где — массовая доля жира, установленная кислотным методом, %.
Навеску продукта помещают в экстрактор, сосуд или колбу для
экстрагирования. В сосуд с творогом добавляют от 6 до 8 см3 дис-
тиллированной воды, в сосуд с сухим молоком 9—19 см3 дистилли-
рованной воды. Затем экстрактор с навеской творога или сухого мо-
лока ставят на водяную баню с температурой воды 38 ± 2 °C и пере-
мешивают до получения однородного раствора.
Затем вводят 1,5 см3 25%-ного раствора аммиака и тщательно пе-
ремешивают, приливают 10 см3 этилового спирта и аккуратно сме-
шивают, не закрывая. Добавляют 25 см3 диэтилового эфира, зак-
285
рывают экстрактор и интенсивно встряхивают, переворачивая его
в течение 1 мин. При необходимости охлаждают сосуд в проточ-
ной воде.
Осторожно вынимают пробку и добавляют 25 см3 петролейного
эфира, предварительно сполоснув пробку и горлышко экстрактора
так, чтобы растворитель попал внутрь. Закрывают экстрактор,
встряхивают, переворачивая его в течение 30 с, и центрифугируют в
течение 15 ± 1 мин или оставляют в покое до тех пор, пока верхний
слой жидкости не станет прозрачным и четко не отделится от вод-
ного слоя. Вынимают пробку, ополаскивают ее и горлышко экст-
рактора смешанным растворителем, сливая его внутрь экстрактора,
и аккуратно переливают по возможности большую часть эфирного
слоя декантацией в колбу для перегонки.
Для облегчения декантации в экстрактор можно добавить дис-
тиллированную воду с целью поднятия поверхности раздела между
двумя слоями. Ополаскивают внутреннюю и внешнюю части гор-
лышка экстрактора смешанным растворителем, сливая его в колбу
для перегонки. Добавляют 5 см3 этилового спирта в экстрактор,
ополаскивая внутреннюю поверхность и перемешивая содержимое.
Второе и третье экстрагирование проводят, как первое, исполь-
зуя 15 см3 диэтилового и 15 см3 петролейного эфиров.
При анализе обезжиренного молока третье экстрагирование не
проводят.
Для определения массы экстрагированного жира из колбы для
перегонки тщательно удаляют выпариванием и перегонкой как
можно больше растворителей и этилового спирта. После исчезно-
вения запаха растворителей нагревают колбу в горизонтальном по-
ложении в сушильном шкафу при 102 ± 2 °C или в вакуумном шка-
фу при 70—75 °C и давлении 6,66 Па в течение 1 ч. Колбу помещают
в эксикатор для охлаждения до 20 ± 2 °C и взвешивают. В последую-
щем колбу взвешивают после высушивания при тех же режимах в
течение 30—60 мин до тех пор, пока разница между массами после-
довательных взвешиваний не будет более 0,0003 г. Если при одном
из взвешиваний после высушивания будет увеличение массы, для
расчетов принимают результаты предыдущего взвешивания.
Чтобы установить, что все экстрагированное вещество является
жиром, в колбу для перегонки добавляют 15—25 см3 петролейного
эфира, осторожно нагревают и взбалтывают до полного растворе-
ния экстрагированного вещества.
При неполном растворении экстрагированного вещества из кол-
бы для перегонки полностью экстрагируют жир путем нескольких
промывок петролейным эфиром и три раза ополаскивают вне-
шнюю сторону горлышка колбы растворителем. Далее эфир высу-
шивают в сушильном шкафу, колбу охлаждают и взвешивают.
Разность между массой колбы с жиром и окончательной массой
после экстрагирования жира петролейным эфиром представляет
собой массу экстрагированного жира.
286
Одновременно с определением жира в молоке, сливках и молоч-
ных продуктах без сахарозы проводят контрольный опыт с 10 см3
дистиллированной воды, используя те же реактивы.
Массовую долю жира в пробе Ж2 (%) вычисляют по формуле
(т1-то)-(/ПЗ-/П4) |QQ
т
где mi — масса колбы с жиром, высушенная до постоянной массы, г; тг — масса
колбы без жира, высушенная до постоянной массы, г; т3 — масса колбы в конт-
рольном опыте после анализа, г; т< — масса колбы в контрольном опыте до анали-
за, г; т — масса навески, г.
За окончательный результат испытания принимают среднее
арифметическое результатов двух параллельных определений.
Сходимость результатов анализа для молока, кисломолочных
напитков, творога должна быть не более 0,02 %; сливок — не более
0,3 %; сухого молока с массовой долей жира (м. д. ж.) от 10 до
15 % — не более 0,05 %, с м. д. ж. — от 15 до 25 % — не более 0,15 %,
с м. д. ж. — от 25 до 30 % — не более 0,2 %.
Приготовление реактивов. Все реактивы должны быть чистыми
для анализа (ч. д. а.), не должны оставлять осадка больше, чем при
контрольном опыте. Если масса осадка при контрольном опыте
превышает 0,0005 г, реактивы очищают перегонкой с 1 г молочного
жира на 1 см3 растворителя.
Для проверки наличия пероксидов в диэтиловом эфире в колбу с
притертой пробкой (предварительно ополоснутую диэтиловым
эфиром) вносят 1 см3 свежеприготовленного 10%-ного йодита ка-
лия на 10 см3 диэтилового эфира. Все это встряхивают и выдержива-
ют в течение 1 мин. Слой эфира при этом не должен желтеть.
Растворитель приготавливают непосредственно перед использо-
ванием путем смешения равных объемов диэтилового и петролей-
ного эфиров. Кроме того, применяют спирт этиловый ректифико-
ванный высшей очистки.
МЕТОД ШМИДТ-БОНДЗИНСКОГО — РАЦЛАВА
Метод предназначен для определения массовой доли жира в на-
туральных и плавленых сырах и используется при возникновении
разногласий в оценке качества продукта. Метод заключается в обра-
ботке сыра соляной кислотой, добавлении спирта и последующей
экстракции жира из кислотно-спиртовой смеси диэтиловым и пет-
ролейным эфирами, выпаривании растворителей и взвешивании
остатка.
Выполнение работы. Колбу высушивают в сушильном шкафу
(или вакуум-сушильном) в течение 30—60 мин, предварительно по-
местив в нее небольшое количество стеклянных шариков, кусочков
287
фарфора или кусочков карборунда. Затем колбу охлаждают в экси-
каторе и взвешивают с точностью 0,0001 г.
Отобранную пробу сыра измельчают в фарфоровой ступке и
тщательно перемешивают.
Около 2 г измельченной пробы сыра помещают в бюксу или на ча-
совое стекло, взвешивают с отсчетом до 0,0001 г и переносят в сухую
плоскодонную колбу или экстракционную колбу. Пробу сыра для
испытания допускается взвешивать на целлюлозной пленке, кото-
рую затем складывают и вместе с пробой сыра помещают в колбу.
В колбу с испытуемой пробой наливают 9 ± 1 см3 соляной кисло-
ты и выдерживают ее в кипящей водяной бане при постоянном
встряхивании до тех пор, пока сыр полностью не растворится. Кол-
бу выдерживают в кипящей водяной бане еще в течение 20 мин, а
затем охлаждают до температуры 20 ± 2 °C в холодной водопровод-
ной воде.
Если обработку сыра соляной кислотой проводят в экстракцион-
ной колбе, то в нее наливают 10 см3 этилового спирта и осторожно
тщательно перемешивают. Если это выполняют в плоскодонной
колбе, то обработанную соляной кислотой пробу переносят в экст-
ракционную колбу, ополаскивая первоначальную емкость последо-
вательно 10 см3 этилового спирта, 25 см3 диэтилового эфира и 25
см3 петролейного эфира, собирая смывную жидкость в экстракци-
онную колбу.
После внесения 25 см3 диэтилового эфира экстракционную колбу
закрывают притертой пробкой, сильно встряхивают при постоянном
переворачивании в течение 1 мин. Затем осторожно вынимают проб-
ку и добавляют в колбу 25 см3 петролейного эфира, используя первые
5—10 см3 для ополаскивания пробки и внутренней стороны горлови-
ны колбы. Затем закрывают колбу притертой пробкой и встряхивают
при постоянном переворачивании в течение 30 с.
Оставляют колбу в покое до тех пор, пока верхний слой жидко-
сти не будет чистым и четко отделенным от нижнего слоя. (Если
четкого разделения слоев не достигается, то жидкость центрифуги-
руют, используя экстракционную колбу.) Вынимают пробку, опо-
ласкивают ее и внутреннюю поверхность горловины колбы 5—
10 см3 смешанного растворителя так, чтобы он стекал в колбу. Пос-
ле этого верхний слой осторожно переносят путем декантации или
при помощи сифонной трубки в плоскодонную колбу. Если верх-
ний слой переносят путем декантации, то для улучшения разделе-
ния слоев можно внести небольшое количество воды.
Внешнюю и внутреннюю поверхность горловины колбы или
кончик и нижнюю часть сифонной трубки ополаскивают 5—10 см3
смешанного растворителя, при этом растворитель с внешней сторо-
ны горловины экстракционной колбы должен стекать в плоскодон-
ную колбу, а с внутренней стороны — в экстракционную колбу.
Проводят вторую экстракцию, добавляя при этом по 15 см3 ди-
этилового и петролейного эфиров. Третью экстракцию выполняют
288
так же, как и вторую, только без ополаскивания колбы. Осторожно
выпаривают или постепенно отгоняют из плоскодонной колбы
максимальное количество растворителей по мере удаления диэти-
лового и петролейного эфиров, повышая температуру водяной бани
от 30 до 60 °C.
После исчезновения запахов растворителей колбу нагревают,
поместив ее на 1 ч в сушильный шкаф (или вакуум-сушильный).
Затем охлаждают в эксикаторе до температуры 20 ± 2 °C и взвеши-
вают с отсчетом до 0,0001 г. В последующем колбу взвешивают
после высушивания в течение 30—60 мин до тех пор, пока разница
между массами последовательных взвешиваний не будет более
0,001 г. В случае увеличения массы колбы с содержимым после по-
вторного высушивания для расчета берут результат предыдущего
взвешивания.
Для проверки полноты растворения экстрагированной фракции
в колбу добавляют 20 ± 5 см3 петролейного эфира, при этом колбу
постепенно нагревают до температуры не выше 60 °C при постоян-
ном перемешивании содержимого колбы круговыми движениями
до полного растворения жира.
Если экстрагированная фракция не растворяется в петролей-
ном эфире полностью, то содержание нерастворимого осадка оп-
ределяют после удаления жира теплым петролейным эфиром. Об-
работку эфиром повторяют не менее трех раз. Перед каждой де-
кантацией дают осесть на дно нерастворимому остатку. После
полного удаления жира колбу с нерастворимым остатком подогре-
вают в водяной бане, постепенно повышая ее температуру от 30 до
60 °C с целью наиболее полного удаления петролейного эфира.
После исчезновения запаха петролейного эфира колбу с нераство-
рившимся остатком сушат в сушильном шкафу (или вакуум-су-
шильном) в течение 1 ч, охлаждают до температуры 20 ± 2 °C и
взвешивают с точностью до 0,0001 г. Одновременно с определени-
ем массовой доли жира проводят контрольный опыт с 10 см3 дис-
тиллированной воды.
Если масса сухого остатка в колбе после высушивания превыша-
ет 0,0005 г, то реактивы следует проверить на чистоту или заменить.
Массовая доля жира (%) в натуральном или плавленом сыре
ЖЬ-^2)-(^з-^).1оо
4 "% ’
где mt — масса колбы с жиром последнего взвешивания, г; m2 — масса пустой колбы
или с сухим нерастворимым остатком, г; т3 — масса колбы после последнего взве-
шивания в контрольном опыте, г; — масса пустой колбы или с сухим нераствори-
мым остатком в контрольном опыте, г; — масса испытуемой пробы, г.
За окончательный результат испытания принимают среднее
арифметическое значение результатов двух параллельных опреде-
лений, расхождение между которыми не должно превышать 0,2 %.
289
Требования к реактивам те же, что и в методе Розе—Готлиба.
Используют также кислоту соляную (ч. д. а. плотностью 1,125 г/см3)
и спирт этиловый ректификованный технический.
МЕТОД МОЖОНЬЕ
Данным методом определяют содержание жира в сухих детских
продуктах типа «Солнышко».
Метод основан на экстракции жира из продукта смесью петро-
лейного и этилового эфиров после обработки продукта аммиаком и
этиловым спиртом. 1,0 г продукта переносят в плоскодонную колбу
или в колбу Можонье, в которую добавляют пипеткой 5,0 см3 дис-
тиллированной воды, 1,5 см3 водного аммиака и тщательно переме-
шивают, встряхивая. Затем в эту же колбу вносят 10,0 см3 ректифи-
кованного спирта, закрывают пробкой и еще раз встряхивают. В
колбу добавляют цилиндром 25 см3 этилового эфира и взбалтывают
20—30 с. Затем добавляют 25 см3 петролейного эфира и снова взбал-
тывают 20—30 с. Далее содержимое колбы выливают в делительную
воронку или разделяют слои центрифугированием.
При разделении слоев центрифугированием экстракционную
колбу помещают в стакан центрифуги и вращают с частотой враще-
ния 5 с-1 (300 об/мин). После разделения слоев в течение 5 ± 1 мин
нижний водно-спиртовой слой сливают в ту же колбу, а верхний
слой — эфирный, сливают в ранее взвешенные чашки и выпарива-
ют эфир из чашки на песочной бане.
Повторяют экстракцию, добавляя по очереди 5,0 см3 спирта,
25 см3 этилового эфира, 25 см3 петролейного эфира с 1 каплей фе-
нолфталеина и тщательно взбалтывают после добавления каждого
реактива. Смесь снова сливают в делительную воронку, отстаивают
в течение 5 ± 1 мин, после разделения слоев эфир сливают в ту же
чашку, что использовалась при первой экстракции.
Если необходимо поднять разделительную линию между эфир-
ным раствором и остальным содержимым, добавляют 1 см3дистил-
лированной воды. Выпаривают эфир из чашки на песочной бане до
исчезновения запаха эфира. Осадок высушивают в вакуум-сушиль-
ном шкафу при температуре 135 ± 2 °C в течение 5—6 мин. Чашки
охлаждают в эксикаторе в течение 5—6 мин и быстро взвешивают.
Массовая доля жира (%)
^=(«2-^1).1ОО
т
где т\ — масса пустой чашки, г; т2 — масса чашки с экстрагированным жиром, г;
т — масса пробы, г.
За окончательный результат принимают среднее арифметичес-
кое двух параллельных определений, сходимость которых не долж-
на превышать 0,05 %.
290
Проверка чистоты эфиров. Отмеривают 50 см3 этилового эфира и
50 см3 петролейного эфира в отдельные чашки для жира. Эфир вы-
паривают, чашки охлаждают и взвешивают. Осадок не должен пре-
вышать 0,0005 г. Непосредственно перед использованием готовят
смешанный растворитель из равных объемов диэтилового и петро-
лейного эфиров.
Подготовка чашек для жира. Чашки для жира нагревают в вакуум-
сушильном шкафу при температуре 100 ± 5 °C в течение 10 ± 2 мин
и охлаждают в эксикаторе в течение 5—10 мин.
ЭКСПРЕСС-МЕТОД
Метод предназначен для определения жира в сухом цельном мо-
локе и основан на экстракции жира из сухого цельного молока ме-
тилхлоридом, выпаривании и высушивании растворителя и взве-
шивании остатка.
Пробу сухого молока массой 10 г переносят в коническую кол-
бу вместимостью 250 см3, прибавляют 50 см3 метилхлорида и
встряхивают на качалке с частотой 107 колебаний в минуту в те-
чение 11 мин. Экстракт фильтруют через бумажный фильтр, от-
бирают 25 см3 фильтрата и помещают в чашку. Удаляют раство-
ритель упариванием, остаток высушивают при 105 °C, выдержи-
вают 30 мин, затем помещают на 30 мин в эксикатор, а затем
взвешивают. Массовую долю жира (%) в продукте рассчитывают
по формуле
Х=^^-$-100,
т И2
где —масса чашки с жиром после высушивания, г; — масса чашки без жира, г;
т — навеска продукта, г; — объем добавленного растворителя, см3; й2 — объем
фильтрата, взятого для высушивания, см3.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА
Ранее были рассмотрены следующие методы определения белка:
фотометрический, рефрактометрический, формольного титрова-
ния с использованием титрометрического анализатора, в данной
главе будут рассмотрены метод Кьельдаля и расчетный метод.
Универсальным методом определения белка и других азотистых
соединений в пищевых продуктах является метод Кьельдаля, сущ-
ность которого заключается в переводе путем минерализации азота
аминокислот в аммиак с последующим его количественным опре-
делением и пересчетом на белок.
Метод Кьельдаля используют в качестве эталонного при разра-
ботке других методов определения белка, в том числе и инструмен-
291
тальных. Метод используют в качестве арбитражного при возник-
новении разногласий в оценке качества продукта.
Массовую долю общего белка Б (%) вычисляют до третьего деся-
тичного знака по формуле
^_W(M-Ko)-6,38
т
где 1,4 — количество азота, эквивалентное 1 см3 раствора соляной кислоты концен-
трацией 0,1 моль/дм3, мг/см3; N— коэффициент пересчета на фактическую кон-
центрацию раствора соляной кислоты; — объем раствора соляной кислоты, из-
расходованный на титрование дистиллята в основном анализе, см3; Vo — объем ра-
створа соляной кислоты, израсходованный на титрование дистиллята в конт-
рольном анализе, см3; 6,38 — коэффициент пересчета массовой доли общего азота
на массовую долю общего белка; т — масса молока, взятая на анализ, г.
МЕТОД КЬЕЛЬДАЛЯ
Органические вещества молока при нагревании с концентриро-
ванной серной кислотой окисляются до воды, диоксида углерода.
При этом азот аминокислот образует аммиак, который с серной
кислотой дает сульфат аммония. Для более быстрого и полного
сжигания прибавляют катализаторы — оксид ртути, селен, сульфат
меди и др. По объему аммиака, определяемого титрованием кисло-
той, устанавливают количество общего азота умножением после-
днего на принятый коэффициент 6,38, находят содержание общего
белка в молоке.
В колбу Кьельдаля помещают последовательно несколько стек-
лянных бусинок или кусочков фарфора, около 10 г сульфата калия,
0,04 г сульфата меди. В бюксу с крышкой отмеривают 5 см3 молока,
крышку закрывают и взвешивают. Молоко из бюксы переливают в
колбу. Пустую бюксу вновь взвешивают и по разнице между массой
бюксы с молоком и массой пустой бюксы устанавливают массу взя-
того молока. В колбу добавляют 20 см3 серной кислоты, вливая ос-
торожно по стенкам колбы, смывая с них капли молока. Колбу зак-
рывают грушеобразной стеклянной пробкой и осторожно круговы-
ми движениями перемешивают содержимое колбы.
Колбу ставят на нагревательный прибор в наклонном положе-
нии под углом 45° и осторожно нагревают до тех пор, пока не пре-
кратится пенообразование и содержимое колбы не станет жидким.
Затем сжигание продолжают при более интенсивном нагревании.
Степень нагревания считают достаточной, когда кипящая кислота
конденсируется в середине горловины колбы Кьельдаля.
Время от времени содержимое колбы перемешивают, смывая
обуглившиеся частицы со стенок колбы. Нагревание продолжают
до тех пор, пока жидкость не станет совершенно прозрачной и
практически бесцветной (при применении в качестве катализатора
окиси ртути) или слегка голубоватой (при применении в качестве
292
катализатора сульфата меди). После осветления раствора нагрева-
ние продолжают в течение 1,5 ч, после чего колбе дают остыть до
комнатной температуры. Добавляют 150 см3 дистиллированной
воды и несколько кусочков свежепрокаленной пемзы, перемеши-
вают и снова охлаждают.
В коническую колбу отмеривают 50 см3 раствора борной кисло-
ты (реактив 1)*, добавляют 4 капли индикатора и перемешивают.
Коническую колбу соединяют с холодильником с помощью ал-
лонжа и резиновой пробки так, чтобы конец аллонжа был погружен
в раствор борной кислоты в конической колбе. Колбу Кьельдаля со-
единяют с холодильником при помощи каплеуловителя, проходя-
щего через одну пробку с делительной воронкой. Градуированным
цилиндром отмеривают 80 см3 раствора гидроксида натрия (реак-
тив 3) (при применении в качестве катализатора красного оксида
ртути используют раствор гидроксида натрия, содержащий сульфид
натрия) и через делительную (или капельную) воронку вносят его в
колбу Кьельдаля. Сразу же после выливания раствора закрывают
кран делительной воронки для избежания потери образующегося
аммиака.
Содержимое колбы Кьельдаля осторожно смешивают круговы-
ми движениями и нагревают до кипения. При этом необходимо из-
бегать пенообразо вания.
Продолжают перегонку до тех пор, пока жидкость не начнет
булькать. При этом регулируют степень нагрева так, чтобы время
дистилляции было не менее 20 мин. Убедиться в полноте перегонки
аммиака можно путем дополнительной перегонки в новую порцию
борной кислоты (20 см3) в течение 5 мин. Окраска раствора борной
кислоты должна оставаться без изменения. При перегонке не до-
пускают нагревания раствора борной кислоты в конической колбе.
Слишком сильное охлаждение (ниже 10 °C) также нежелательно,
так как оно может вызвать переброс жидкости из конической колбы
в колбу Кьельдаля.
Перед окончанием перегонки опускают коническую колбу так,
чтобы конец аллонжа был над поверхностью раствора борной кис-
лоты, и продолжают перегонку в течение 1—2 мин.
После прекращения нагревания отсоединяют аллонж. Ополаски-
вают внешнюю и внутреннюю поверхности аллонжа небольшим ко-
личеством дистиллированной воды, сливая ее в коническую колбу.
Дистиллят титруют раствором соляной кислоты (реактив 2) до
перехода зеленого цвета в серый. При избытке титранта раствор
приобретает фиолетовый цвет. Параллельно проводят конт-
рольный опыт так же, как и основной, применяя 5 см3 дистиллиро-
ванной воды вместо молока. Количество повторностей конт-
рольного опыта должно быть не менее 5. Контрольный анализ
’Приготовление реактивов см. «Ускоренный метод».
293
проводится в каждой серии определений количества белка и при
каждой замене реактивов.
За окончательный результат испытания при анализе по Кьельда-
лю принимают среднее арифметическое результатов двух парал-
лельных определений, допускаемое расхождение между которыми
не должно превышать 0,03 %.
УСКОРЕННЫЙ МЕТОД
Ускорение анализа по сравнению с традиционным методом
Кьельдаля достигается за счет более интенсивной минерализации
пробы. С этой целью пробы минерализуют с использованием пе-
роксида водорода в кварцевых пробирках, помещенных в имеющий
изоляцию алюминиевый блок с шестью гнездами. После минерали-
зации пробы кварцевую пробирку непосредственно присоединяют
к аппарату для отгонки аммиака.
Выполнение работы. В кварцевую пробирку помещают последо-
вательно несколько стеклянных бусинок или кусочков фарфора,
около 10 г сульфата калия, 0,04 г сульфата меди. В бюксу отмерива-
ют 5 см3 молока, закрывают крышкой и взвешивают. Молоко из
бюксы переливают в кварцевую пробирку. Пустую бюксу вновь
взвешивают и по разнице между массой бюксы с молоком и массой
пустой бюксы устанавливают массу взятого молока. В пробирку до-
ливают 20 см3 серной кислоты, вливая осторожно по стенке про-
бирки, смывая с нее капли молока.
Осторожно круговыми движениями перемешивают содержимое
пробирки. Затем вносят в пробирку 20 см3 пероксида водорода, не
допуская вспенивания.
Пробирку ставят в гнездо алюминиевого блока (рис. 65), поме-
щенного на электроплитку. Регулятор нагрева плитки устанавлива-
ют в среднее положение. После прекращения вспенивания содер-
жимого пробирки регулятор нагрева плитки переводят в положе-
ние, соответствующее максимуму. Нагревание продолжают до тех
пор, пока жидкость не станет прозрачной и бесцветной или слегка
Рис. 65. Алюминиевый блок для сжига-
ния:
7 — блок; 2—теплоизоляция (асбест или
асбоцемент); 3 — гнездо для пробирки
294
Рис. 66. Прибор для отгонки аммиака:
/ — электрическая плитка; 2— коническая колба вместимостью 2000 см3; 3 — делительная во-
ронка; /—каплеуловитель; 5—кварцевая пробирка; 6— холодильник; 7—колба коническая
вместимостью 250 см3
голубоватой. Затем пробирку охлаждают и присоединяют к пере-
гонному аппарату (рис. 66).
В коническую колбу вместимостью 250 см3 отмеривают мер-
ным цилиндром 20 см3 раствора борной кислоты (реактив 1), до-
бавляют 3—4 капли раствора двойного индикатора (реактив 4).
Отмеривают мерным цилиндром 60 см3 раствора гидроксида на-
трия (реактив 3) и осторожно, не допуская выбросов, переливают
его через делительную воронку в пробирку. Кран воронки сразу
закрывают. Открывают зажим на линии подачи пара из коничес-
кой колбы вместимостью 2000 см3 и направляют пар в пробирку.
Перегонку ведут до достижения объема конденсата от 50 до 70 см3.
Конденсат титруют раствором соляной кислоты (реактив 2) до пе-
рехода зеленого цвета в серый. При избытке титранта раствор при-
обретает фиолетовый цвет.
Параллельно проводят контрольный анализ, используя вместо
молока 5 см3 дистиллированной воды. Количество повторностей
контрольного анализа должно быть не менее 5. Контрольный ана-
лиз проводят периодически при замене хотя бы одного из реакти-
вов, кроме соляной кислоты.
При ускоренном анализе допускаемое расхождение между двумя
параллельными определениями не должно превышать 0,1% массовой
доли белка. За окончательный результат испытания принимают сред-
нее арифметическое результатов двух параллельных определений.
295
Приготовление реактивов. Реактив 1 (раствор борной кислоты).
40 г борной кислоты растворяютв мерной колбе на 1000 см3 дистил-
лированной водой.
Реактив 2 (0,1 Мраствор соляной кислоты). В 1000 см3 раствора
должно содержаться 3,647 г НС1. Расчет проводят, руководствуясь
приложением 5.
Реактив 3 (раствор гидроксида натрия). 500 г гидроксида натрия
растворяют в 1000 см3, а при ускоренном анализе — в 500 см3 дис-
тиллированной воды.
Реактив 4 (двойной индикатор). 2 г метилового красного и 1 г ме-
тиленового голубого растворяют в 1000 см3 96%-ного этилового
спирта.
РАСЧЕТНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ БЕЛКА В МОЛОКЕ
При выводе расчетной формулы для определения белка в молоке
(Бы) исходили из постоянства соотношения между составными час-
тями молока белок: лактоза: зола = 9: 13:2
£м = 0,075Д, + 0,0984/ + 0,085,
где Жм —массовая доля жира в молоке, %; ^—плотность молока, выраженная в
градусах ареометра.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ УГЛЕВОДОВ
Для определения содержания углеводов применяют методы йо-
дометрический, Бертрана, феррицианидный.
Методы используют для определения углеводов в молочных про-
дуктах, в рецептуру которых входит сахар: йодометрический ме-
тод — в творожных изделиях, кремах, кисломолочных продуктах,
мороженом и шоколадном масле; метод Бертрана — в фруктовом
масле, плодово-ягодном и ароматическом мороженом и мороже-
ном на молочной основе, изготовляемом с использованием инверт-
ного сиропа; ускоренный феррицианидный метод — в кисломолоч-
ных продуктах с плодово-ягодными наполнителями.
ЙОДОМЕТРИЧЕСКИЙ МЕТОД
Метод основан на окислении редуцирующих сахаров (лактоза,
глюкоза), содержащих альдегидную группу, йодом в щелочной сре-
де. Массовую долю сахарозы определяют по разности между коли-
чеством взятого и неизрасходованного йода, определяемого титро-
ванием тиосульфатом натрия.
Метод является арбитражным.
296
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЛАКТОЗЫ
Исследования начинают с приготовления фильтрата. 25 г молока
с точностью до 0,01 г отвешивают в мерную колбу вместимостью
500 см3, прибавляют до половины колбы дистиллированную воду и
10 см3 реактива 1,4 см3 М раствора гидроксида натрия. Жидкость пе-
ремешивают после добавления воды и каждого реактива. Доводят до
метки водой (при температуре 20 °C), перемешивают, оставляют на
30 мин. Отстоявшуюся жидкость фильтруют в сухую колбу через
складчатый бумажный фильтр, удаляя первые 10—20 см3 фильтрата.
12,5 г сухого молока растворяют в химическом стакане в неболь-
шом количестве горячей воды, тщательно растирая комочки. Пере-
носят без потерь в мерную колбу вместимостью 100 см3, доводят до
метки водой, ополаскивая ею стакан. 25 см3 раствора вносят в мер-
ную колбу вместимостью 500 см3, добавляют 200 см3 воды, 10 см3 ра-
створа Фелинга (реактив 1) и 4 см31 н. раствора гидроксида натрия.
50 см3 фильтрата, что соответствует 2,5 г молока или 0,312 г сухо-
го молока, переносят пипеткой в коническую колбу на 250—300 см3
с притертой или резиновой пробкой. Приливают пипеткой или из
бюретки 25 см3 0,1 раствора йода (реактив 2) и медленно при непре-
рывном перемешивании приливают из бюретки 37,5 см3 0,1 н. ра-
створа гидроксида натрия. Закрыв колбу пробкой, оставляют ее в
темном месте на 20 мин при температуре 20 °C. Затем прибавляют
8 см3 0,5 н. раствора соляной кислоты и титруют выделившийся йод
0,1 н. раствора тиосульфата натрия (реактив 3) сначала без прибав-
ления индикатора до получения светло-желтого раствора, затем
прибавляют 1 см31%-ного раствора крахмала и продолжают титро-
вание каплями до момента, когда исчезнет синяя окраска.
Для контрольного опыта в другую такую же колбу отмеривают
пипеткой 25 см3 0,1 н. раствора йода, 25 см3 воды и добавляют при
непрерывном перемешивании 37,5 см3 0,1 н. раствора гидроксида
натрия и, закрыв колбу пробкой, оставляют в темном месте на
20 мин при температуре 20 °C и дальше определение проводят как в
первой колбе.
Массовую долю лактозы в молоке Л (%) рассчитывают по формуле
д= 0,01801(И-ИЮ0 0.97_ 0,699 _уу
где — объем 0,1 н. раствора тиосульфата натрия, пошедшее на титрование йода в
контрольном опыте, см3; И— объем 0,1 н. раствора тиосульфата натрия, пошедшее
на титрование йода при определении в фильтрате, см3; т — масса молока в 50 см3
фильтрата, равная 2,5 г; 0,97 — поправка, установленная эмпирически; 0,01801 —
масса лактозы, моногидрата, соответствующая 1 см3 0,1 н. раствора йода, г.
Массовую долю лактозы в сухом молоке рассчитывают по
формуле
Л= 5,578 (^ - V).
297
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ САХАРОЗЫ
Выполнение работы. 5 г творожных изделий, кисломолочных
продуктов, мороженого с сахаром и кремов (10 г молочных продук-
тов, содержащих менее 10 % сахара) взвешивают в стакане вмести-
мостью 100 см3. В стакан с продуктом прибавляют 25 см3 воды; со-
держимое стакана тщательно растирают оплавленной стеклян-
ной палочкой и количественно переносят в мерную колбу вмести-
мостью 250 см3. Содержимое стакана смывают несколько раз водой
температурой 20 ± 2 °C, количество которой не превышает полови-
ны объема колбы.
Затем в колбу прибавляют 5 см3 раствора Фелинга (реактив 1) и
2 см3 1 н. раствора гидроксида натрия, содержимое колбы хорошо
перемешивают и оставляют в покое на 5 мин. Если жидкость в колбе
над осадком окажется мутной, то в колбу приливают дополнитель-
но еще несколько капель раствора Фелинга (реактив 1).
После появления над осадком прозрачного слоя жидкости, ука-
зывающего на полноту осаждения, колбу доливают водой до метки
и содержимое колбы тщательно перемешивают. Колбу оставляют в
покое на 20—30 миндлятого, чтобы дать возможность жидкости от-
стояться, после чего прозрачную жидкость, находящуюся над осад-
ком, фильтруют через сухой складчатый бумажный фильтр в сухую
колбу. Первые 25—30 см3 фильтрата отбрасывают.
Для приготовления фильтрата из шоколадного масла Юг его
взвешивают в стакане вместимостью 100 см3. В стакан с маслом вно-
сят пипеткой 50 см3 воды, нагретой до температуры 45 ± 5 °C. После
того как масло расплавилось, содержимое стакана тщательно пере-
мешивают и переносят в сухую делительную воронку. После отста-
ивания жира большую часть водяного слоя сливают из воронки в
сухую колбу.
40 см3 водной вытяжки (соответствует 8 г испытуемого масла)
пипеткой переносят в мерную колбу вместимостью 250 см3 и допол-
няют колбу водой приблизительно до половины объема. В колбу с
раствором водной вытяжки из масла приливают 5 см3 7,3 н. раство-
ра соляной кислоты (реактив 4) и 2 см31 н. раствора гидроксида на-
трия. Содержимое колбы перемешивают, доливают водой до метки
и после вторичного тщательного перемешивания оставляют на 20—
30 мин для отстаивания. Прозрачный слой жидкости, находящийся
над осадком, фильтруют через сухой складчатый бумажный фильтр
в сухую колбу.
Для определения редуцирующей способности фильтрата до ин-
версии 25 см3 фильтрата вносят пипеткой в коническую колбу с
притертой пробкой вместимостью 250 см3. Затем пипеткой прили-
вают в колбу 25 см3 0,1 н. раствора йода (реактив 2) и из бюретки при
непрерывном помешивании — 37,5 см3 0,1 н. раствора гидроксида
натрия. Затем колбу закрывают притертой пробкой и оставляют в
покое в темном месте.
298
Через 20 мин в колбу приливают 8 см3 0,5 н. раствора соляной кис-
лоты и титруют выделившийся йод 0,1 н. раствора тиосульфата на-
трия (реактив 3). После перехода цвета титруемого раствора из бурого
в желтоватый в колбу прибавляют 1 см31 % -ного раствора крахмала и
титрование продолжают до исчезновения синей окраски.
После титрования записывают объем тиосульфата натрия, из-
расходованного на титрование выделившегося йода.
Для определения редуцирующей способности фильтрата после
инверсии другие 25 см3 фильтрата приливают пипеткой в коничес-
кую колбу вместимостью 250 см3 с притертой пробкой. Колбу зак-
рывают пробкой с пропущенным через нее термометром так, чтобы
ртутный резервуар находился в жидкости, и нагревают в водяной
бане до температуры 65 ± 3 °C. Приоткрыв пробку, приливают в
колбу 2,5 см3 7,3 н. раствора соляной кислоты (реактив 4). Для ин-
версии жидкость перемешивают и держат в водяной бане при тем-
пературе 68 ± 2 °C. Через 10 мин после приливания соляной кисло-
ты колбу вынимают из водяной бани и, не вынимая термометра,
быстро охлаждают до температуры 20 ± 2 °C.
После прибавления одной капли раствора метилового оранжевого
в колбу при непрерывном помешивании приливают по каплям 1 н.
раствора гидроксида натрия до наступления слабокислой реакции
(переход окраски раствора от розовой до желтой). Термометр выни-
мают из колбы после промывания его первыми каплями раствора
гидроксида натрия. Пипеткой приливают в колбу 25 см3 0,1 н. ра-
створа йода (реактив 2), а из бюретки при непрерывном помешива-
нии — 37,5 м3 0,1 н. раствора гидроксида натрия. Затем колбу закры-
вают притертой пробкой и оставляют в покое в темном месте. Через
20 мин в колбу приливают 8 см3 0,5 н. раствора соляной кислоты и
титруют выделившийся йод 0,1 н. раствором тиосульфата натрия (ре-
актив 3). После перехода цвета титруемого раствора из бурого в жел-
товатый в колбу прибавляют 1 см31 %-ного раствора крахмала и тит-
рование продолжают до исчезновения синей окраски.
После окончания титрования записывают объем тиосульфата
натрия, пошедший на титрование выделившегося йода. Конец тит-
рования устанавливают по резкому переходу синей окраски в блед-
но-розовую, обусловленную наличием метилового оранжевого.
Массовую долю сахарозы в продукте S(%) вычисляютпо формуле
(И-К2)Л/сы . _ -100-0,99
<$_ К 1 z/ Na2S2O3/S
~ т ’
где V\ — объем раствора тиосульфата натрия, израсходованный на титрование до ин-
версии, см3; Vi — объем раствора тиосульфата натрия, израсходованный на титрование
после инверсии,см3; McNa2S2p2/s —массовая концентрация тиосульфата натрия,
г/см3; 0,99 — коэффициент, найденный эмпирическим путем; т — навеска продукта,
соответствующая 25 см3 фильтрата, взятого для титрования, г (т — 0,5 г при первона-
чальной навеске 5 г и разведении до 250 см3; т — 1,0 г при первоначальной навеске 10 г
и разведении до 250см3; т — 0,8г при первоначальной навеске Юг и разведении до
50 см3, из которых взято 40 см3 и разведено водой до 250 см3).
299
Массовую концентрацию раствора тиосульфата натрия устанав-
ливают следующим образом: в коническую колбу вместимостью
500—750 см3 с притертой пробкой вносят 1—2 г йодита калия, ра-
створяют его в 2—3 см3 воды, прибавляют 5 см3 соляной кислоты,
разбавленной 1: 5, 20 см3 0,1 н. раствора дихромата калия. Закрыв
колбу пробкой, содержимое тщательно перемешивают, дают ра-
створу постоять 5 мин, после этого титруют раствором тиосульфата
(массовую концентрацию которого устанавливают), приливая его
из бюретки постепенно, все время перемешивая жидкость. Когда
коричневый цвет раствора перейдет в желтовато-зеленый, в колбу
добавляют 1 см31 %-ного раствора крахмала и для более четкого оп-
ределения окончания титрования 250—300 см3 воды. Титрование
продолжают, приливая тиосульфат натрия по каплям до резкого пе-
рехода раствора от синего до светло-зеленого, обусловленного
ионами трехвалентного хрома.
Массовую концентрацию раствора тиосульфата натрия вычис-
ляют по формуле
„ 0,0171-20
MCNa2S2O3/S_ V
где 0,0171 — массовая концентрация сахарозы, соответствующая 1 см3 0,1 н. раство-
ра тиосульфата натрия, г/м3; 20 — объем 0,1 н. раствора дихромата калия, см3; V—
объем раствора тиосульфата натрия, израсходованный на титрование 20 см3 0,1 н.
раствора дихромата калия, см3.
За окончательный результат определения принимают среднее
арифметическое результатов двух параллельных определений, вы-
численных до десятых долей процента. Расхождение между двумя
параллельными определениями не должно превышать 0,5 %.
Для проверки точности приготовленных растворов проводят
контрольный анализ. Контрольные образцы молочных продуктов
приготовляют из сахара-рафинада, молока или творога.
Например, в два стаканчика отвешивают 1,3 г сахарозы и 3,7 г творога, добавля-
ют небольшое количество дистиллированной воды. Содержимое стакана тщательно
растирают оплавленной стеклянной палочкой и количественно переносят эти ком-
поненты в мерную колбу вместимостью 250 см3, заполняя ее примерно на 1/2 объе-
ма. Далее анализ проводят так, как описано выше. Взятые навески соответствуют
творожному изделию, содержащему 26 % сахарозы. Если отклонение установлен-
ных результатов сахарозы по сравнению с фактическим ее содержанием не превы-
шает ± 0,3 %, такие растворы можно применять для контроля молочных продуктов.
Если отклонение результатов сахарозы в контрольном образце превышает ± 0,3 %,
то необходимо менять растворы или внести соответствующую поправку в результа-
ты, полученные описанным выше йодометрическим методом.
Приготовление реактивов. Реактив 1 (раствор Фелинга). 69,26 г
перекристаллизованного сульфата меди, не содержащего железа,
взвешивают и растворяют в мерной колбе вместимостью 1000 см3.
Реактив 2 (0,1 н. раствор йода). 20—25 г йодита калия, взве-
шенного в стакане вместимостью 100 см3, растворяют в возмож-
300
но малом количестве воды. В стакан с раствором йодита калия
прибавляют 12,7 г металлического йода. Жидкость перемешивают
до полного растворения йода и количественно переносят в мер-
ную колбу вместимостью 1000 см3, затем объем доводят водой до
метки.
Реактив 3(0,1 н. раствор тиосульфата натрия). 24,8 г тиосульфа-
та натрия переносят в мерную колбу вместимостью 1000 см3, ра-
створяют, прибавляют 0,2 г безводного карбоната натрия и объем
раствора доводят водой до метки. Для приготовления раствора тио-
сульфата используют бидистиллированную или дистиллированную
свежепрокипяченную воду. Охлаждают воду в колбе с закрытой
пробкой, через которую проходит хлоркальциевая трубка, напол-
ненная кусочками натронной извести.
Реактив 4 (7,3н. раствор соляной кислоты). К 120 см3 соляной
кислоты плотностью 1,19 г/см3 добавляют 80 см3 воды.
МЕТОД БЕРТРАНА
Метод основан на способности редуцирующих сахаров — лакто-
зы, глюкозы, фруктозы восстанавливать в щелочной среде двухва-
лентную медь (CuSO4 — жидкость Фелинга) в одновалентную
(красный осадок). Осадок оксида меди (I) окисляют железоаммо-
нийными квасцами до CuSO4. При этом трехвалентное железо вос-
станавливается до двухвалентного, количество которого определя-
ют титрованием перманганатом калия.
Выполнение работы. Определение содержания лактозы начина-
ют с приготовления фильтратов. Для этого 25 г молока переносят
количественно в мерную колбу вместимостью 500 см3.
12,5 г сухого молока в небольшом стаканчике растворяют в воде
температурой 50—60 °C, тщательно растирая комочки, и переносят
в мерную колбу на 100 см3, ополаскивая стаканчик и сливая воду в
ту же колбу. Охладив содержимое колбы до 20 °C, доводят до метки
водой, перемешивают. 25 см3 восстановленного молока переносят в
мерную колбу вместимостью 500 см3. 2,5 г кристаллизата или 1,25 г
сахара-сырца и рафинированного растворяют в стакане с водой при
нагревании. Раствор переносят в мерную колбу вместимостью
500 см3, ополаскивая стакан водой, сливая ее в ту же колбу, долива-
ют воды до половины колбы.
В колбу с одним из продуктов прибавляют 300 см3 воды, 10 см3
раствора Фелинга 1 (реактив 1), 4 см3 1 н. раствора гидроксида
натрия, 5 см3 5%-ного раствора фторида натрия. Жидкость пере-
мешивают после добавления каждого реактива, доводят ее темпе-
ратуру до 20 °C, доливают водой до метки и, снова перемешав, ос-
тавляют на 30 мин. Отстоявшуюся жидкость фильтруют через су-
хой складчатый фильтр в колбу, отбрасывая первые порции
(20-25 см3).
301
100 см3 прозрачного фильтрата переносят пипеткой в коничес-
кую колбу вместимостью 250—300 см , приливают по 25 см3 раство-
ров Фелинга 1 и 2 (реактивы 1и 2) и кипятят 6 мин с момента заки-
пания, поместив колбу на нагревательный прибор на проволочной
сетке, покрытой асбестовым картоном с круглым отверстием, соот-
ветствующим диаметру дна колбы. Выпадает красный осадок заки-
си меди.
Жидкость отсасывают водоструйным насосом через трубку с ас-
бестовым фильтром Аллина или стеклянным фильтром со впаян-
ной пластинкой из пористого стекла и фильтруют ее в колбу Бунзе-
на через воронку Бюхнера, сливая на фильтр по стеклянной палоч-
ке. При этом надо стараться не переносить осадок на фильтр и не
взбалтывать раствор в колбе. Осадок в колбе и на фильтре не должен
оставаться без жидкости, так как при соприкосновении с воздухом
закись меди может окислиться.
Осадок оксида меди (I) в конической колбе промывают 3—4
раза (порциями по 20—30 см3) водой, декантируя жидкость че-
рез тот же фильтр. Оставив на фильтре немного жидкости над
осадком, отключают насос. Колбу с фильтратом и промывными
водами удаляют и под фильтр подставляют колбу Бунзена, не
включая насоса. Осадок оксида меди (I), оставшийся в колбе,
растворяют в 40—50 см3 раствора железоаммонийных квасцов
(реактив 3) и небольшими порциями переливают на фильтр и
слегка помешивают стеклянной палочкой для растворения по-
павшего на фильтр осадка оксида меди (I). Когда большая часть
меди растворится, начинают фильтрацию при слабом отсасыва-
нии (весь осадок оксида меди (I) должен быть растворен). Для
полного растворения осадка можно прибавить еще немного ра-
створа железоаммонийных квасцов. После стекания последних
капель раствора колбу и фильтр 3—4 раза промывают неболь-
шими порциями холодной воды, которую собирают в колбу с
фильтратом. Содержимое колбы Бунзена зеленоватого цвета
титруют раствором перманганата калия (реактив 4) до слабо-ро-
зовой окраски.
Одновременно в тех же условиях ставят контрольный опыт со
всеми реактивами, но вместо фильтрата берут воду. Объем КМпО4,
пошедший при титровании в контрольном опыте, вычитают из
объема израсходованного на титрование в рабочей пробе.
Массу меди (г), восстановленной 100 см3 фильтрата, рассчиты-
вают по формуле
Cu=VMc
КМПО4/С11
где V— объем раствора перманганата калия, затраченный на титрование, см3;
^cKMnO4/Cu — массовая концентрация перманганата калия, г/см3.
302
Массу лактозы в растворе, зная массу меди, можно найти по
справочным данным. Массовую долю Л (%) рассчитывают по
формуле
#=•^•100,
т ’
где т\ — масса молочного сахара, г; т — масса продукта, соответствующая 100 см3
фильтрата (5 г молока; 0,625 г сухого молока);
в кристаллизате
Л=В_-
J 0,5’
в сахаре-сырце
Л=^-
J 0,25
Для контроля реактивов рекомендуется применять раствор хи-
мически чистой лактозы (навески 2,5 и 1,25 г). Определение про-
водят, как описано выше, и массовую долю лактозы находят по
формуле
#=^100,
где тл — навеска химически чистой лактозы, г; тс — навеска исследуемого молочно-
го сахара, г; g— масса лактозы для химически чистой и определяемой лактозы, мг.
Определение содержания сахарозы в мороженом плодово-ягод-
ном, ароматном и на молочной основе начинают с приготовления
фильтратов по методике приготовления фильтрата из творожных
изделий, мороженого с сахаром и кремов, в фруктовом масле — по
методике приготовления фильтрата из шоколадного масла (йодо-
метрический метод).
Для определения редуцирующей способности фильтрата до ин-
версии в коническую колбу вместимостью 250 см3 пипеткой прили-
вают 50 см3 фильтрата, а также последовательно по 25 см3 растворов
Фелинга 1 и 2 (реактивы 1 и 2). После перемешивания колбу с ра-
створом помещают на асбестовую сетку с вырезанным в ней круг-
лым отверстием диаметром 40—50 мм и нагревают. Раствор кипятят
ровно 6 мин с момента его закипания. При этом выпадает красный
осадок оксида меди (I).
Не переводя осадок на фильтр, жидкость из конической колбы
декантируют при слабом отсасывании на воронку типа ВФ или в
колбу с трубкой с асбестовым фильтром. Для предохранения оксида
меди (I) от окисления осадок на дне колбы все время должен быть
покрыт жидкостью, для чего колбу с осадком под держивают во вре-
мя фильтрования в наклонном положении. По окончании фильт-
303
рования жидкости Фелинга осадок оксида меди (I) промывают не-
сколько раз водой, отфильтровывая каждую порцию воды после
кратковременного отстаивания через тот же фильтр.
К промытому осадку оксида меди (I) в колбу приливают не-
большими порциями при постоянном помешивании 30 см3 ра-
створа железоаммонийных квасцов (реактив 3) до полного раство-
рения осадка. Содержимое конической колбы количественно пе-
реносят на тот же фильтр и фильтруют в другую чистую колбу. Ос-
таток на фильтре промывают 15 см3 раствора железоаммонийных
квасцов (реактив 3). Коническую колбу из-под осадка оксида меди
(I) и фильтр дополнительно промывают три-четыре раза неболь-
шими порциями воды, нагретой до температуры 80 ± 5 °C. По
окончании промывания фильтрат с промывными водами титру-
ют раствором перманганата калия (реактив 4) до слабо-розового
окрашивания, отмечая количество раствора, пошедшего на титро-
вание.
Для определения редуцирующей способности фильтрата после
инверсии 20 см3 фильтрата пипеткой приливают в чистую кони-
ческую колбу вместимостью 250 см3. Закрывают колбу с фильтра-
том пробкой с пропущенным через нее термометром так, чтобы
ртутный резервуар находился в жидкости, и нагревают в водяной
бане до температуры 65 ± 5 °C. Приоткрыв пробку, приливают в
колбу 2,5 см3 7,3 н. раствора соляной кислоты для инверсии, жид-
кость перемешивают и держат в водяной бане при температуре
68 ± 2 °C.
Через 10 мин после приливания соляной кислоты колбу выни-
мают из водяной бани и вместе с термометром быстро охлаждают до
температуры 20 ± 2 °C. При непрерывном помешивании вносят в
колбу одну каплю метилового оранжевого и приливают по каплям
1 н. раствора гидроксида натрия до наступления слабокислой реак-
ции. Термометр вынимают из колбы после промывания его первы-
ми каплями раствора гидроксида натрия.
По окончании нейтрализации фильтрата проводят те же опера-
ции с раствором, что и до инверсии сахарозы.
Массу меди Си (мг), восстановленной 20 см3 фильтрата после
инверсии, вычисляют по формуле
Cu=(r2-iWc,,., _ /г ,
( 2 2,5д КМПО4/С11’
где Vi — объем раствора перманганата калия, израсходованный на титрование
20 см3 фильтрата после инверсии, см3; И1/2,5 — объем раствора перманганата ка-
лия, израсходованный на титрование 20 см3 фильтрата до инверсии, см3;
^скмпО4/Си — массовая концентрация раствора перманганата калия, г/см3.
Массу сахарозы (мг) в 20 см3 фильтрата находят по количеству
меди в приложении 6 (табл. 1).
304
Массовую долю сахарозы в продукте 5) (%) вычисляют по
формуле
51=^-100,
где т\ — масса сахарозы, содержащаяся в 20 см3 фильтрата (см. приложение 6), мг;
т — масса продукта, соответствующая 20 см3 фильтрата, мг.
Для определения массовой доли общего сахара в ароматическом
и плодово-ягодном мороженом находят массу меди, восстановлен-
ной 20 см3 фильтрата до инверсии, по формуле
Си(доинверсии)=^МСкмп04/Си.
По приложению 6 (табл. 2) находят соответствующую массу ин-
вертного сахара wii (мг).
Массовую долю общего сахара в пересчете на инвертный сахар
S2 (%) вычисляют по формуле
(^•1,053+т.) о
z т ’
где ms — масса сахарозы, содержащаяся в 20 см3 фильтрата, г; mt — масса инвертно-
го сахара, содержащаяся в 20 см3 фильтрата, г; т — масса продукта, соответствую-
щая 20 см3, г; 1,053 — коэффициент для перевода сахарозы в инвертный сахар.
Приготовление реактивов. Реактив 1 (раствор Фелинга 1). 69,26 г
перекристаллизованного сульфата меди, не содержащей железа,
взвешивают и растворяют в мерной колбе вместимостью 1000 см3.
Реактив 2 (раствор Фелинга 2). 365 г соли винной кислоты тарт-
рата калия-натрия растворяют при слабом нагревании в 600 см3
воды и фильтруют в мерную колбу вместимостью 1000 см3, куда за-
тем приливают приготовленный отдельно раствор гидроксида на-
трия (103 г гидроксида натрия растворяют в 200 см3 воды). Объем
раствора доводят водой до 1000 см3.
Реактив 3 (железоаммонийные квасцы). К 250 см3 насыщенного
на холоде раствора железоаммонийных квасцов приливают 25 см3
серной кислоты. Раствор перемешивают, охлаждают, переносят в
мерную колбу вместимостью 1000 см3, разбавляют водой до гор-
лышка колбы и после охлаждения до температуры 20 ± 2 °C доводят
водой до метки. В момент определения раствор должен иметь тем-
пературу 20 ± 2 °C.
Реактив 4 (раствор перманганата калия). 1 см3 раствора должен
соответствовать приблизительно 10 мг меди. 4,98 г перманганата
калия взвешивают и количественно переносят в мерную колбу вме-
стимостью 1000 см3, растворяют и объем доводят водой 1000 см3.
305
Раствор кипятят в течение 5 мин, охлаждают и фильтруют через ас-
бестовый фильтр, предварительно Промытый тем же раствором,
хранят в бутылке из темного стекла.
Массовую концентрацию раствора перманганата калия устанав-
ливают следующим образом: 0,25 г оксалата натрия (аммония) взве-
шивают, количественно переносят в коническую колбу и растворя-
ют в 100 см3 воды. После прибавления в колбу 2 см3 серной кислоты
раствор нагревают до температуры 80 ± 2 °C и титруют раствором
перманганата калия до появления розового окрашивания.
Массовую концентрацию перманганата калия ^cKMnO4/Cu (г/см3)
вычисляют по формуле
м _тКЛООО
KMnO4/Cu V ’
где т — масса навески оксалата натрия (аммония), г; К— коэффициент пересчета
оксалатов на медь для оксалата натрия К = 0,9488, для оксалата аммония К= 0,8951;
V— объем перманганата калия, израсходованный на титрование, см3.
УСКОРЕННЫЙ ФЕРРИЦИАНИДНЫЙ МЕТОД
Метод основан на способности редуцирующих сахаров окис-
ляться и восстанавливать в щелочной среде гексацианоферрат (III)
калия в гексацианоферрат (II) калия К3 [Fe (CN)6],
Выполнение работы. Анализ начинают с приготовления фильтра-
та из кисломолочных продуктов с плодово-ягодными наполнителя-
ми, применяя навеску 10 г.
Затем проводят инверсию. 50 см3 фильтрата переносят в мерную
колбу вместимостью 100 см3, прибавляют 5 см3 соляной кислоты,
затем нагревают раствор в водяной бане до температуры 68—70 °C и
выдерживают при той же температуре в течение 8 мин. Температуру
контролируют термометром, опущенным в исследуемый раствор.
После инверсии охлажденный раствор осторожно нейтрализуют по
метиловому оранжевому до желто-оранжевого окрашивания, при-
ливая по капле 20%-ный раствор гидроксида натрия, а к концу ней-
трализации — 1%-ный раствор гидроксида натрия. Нейтрализован-
ный раствор сахаров доливают до метки дистиллированной водой.
Для определения редуцирующей способности фильтрата после
инверсии предварительно определяют общий сахар. В коничес-
кую колбу вместимостью 100 см3 вносят 20 см3 1%-ного раствора
гексацианоферрата (III) калия и 5 см3 2,5 н. раствора гидроксида
натрия, прибавляют 1 каплю раствора хлоргидрата метиленового
голубого и нагревают до кипения. Кипящую смесь осторожно тит-
руют полученным раствором сахаров, медленно прибавляя его по
капле. При этом гексацианоферрат (III) калия переводится в гек-
сацианоферрат (II) калия. Первая избыточная капля раствора са-
306
хара приводит к исчезновению синей окраски. Раствор приобретает
слабо-желтую окраску гексацианоферрата (II) калия. Титрование в
этот момент прекращают. Появление фиолетовой окраски после ос-
тывания раствора во внимание не принимают. Результат получается
наиболее точным, если на титрование расходуется 5—6 см3 раствора
сахара.
Для окончательного определения общего сахара к смеси гексаци-
аноферрата (III) калия и гидроксида натрия приливают из бюретки
на 0,2—0,3 см3 меньше полученного раствора сахара, чем было израс-
ходовано при ориентировочном определении. Нагревают смесь-до
кипения, кипятят 55—60 с, затем прибавляют 1 каплю раствора хлор-
гидрата метиленового голубого и дотитровывают раствором сахара из
бюретки до исчезновения синей окраски.
Массовую долю общего сахара Х{%) вычисляют по формуле
(20,12+0,035И)^Г-250-100-100
Л~ mV-50-iQQQ
где 20,12 и 0,035 — эмпирически установленные коэффициенты; V— объем израс-
ходованного при повторном титровании раствора сахара, см3; К— поправка для
1 %-ного раствора гексацианоферрата (III) калия; 250 — объем, до которого разведе-
на навеска, см3; 100 —объем фильтрата после инверсии, см3; 100 — коэффициент
пересчета на 100 г продукта; т — масса навески продукта, г; 50 — объем фильтрата
для инверсии, см3; 1000 — коэффициент пересчета из мг в г.
За результат анализа принимают среднее арифметическое ре-
зультатов двух параллельных определений. Допускаемое расхожде-
ние между параллельными определениями не должно превышать
0,5 %. Вычисление проводят до первого десятичного знака.
Приготовление реактива. Для приготовления 1 %-ного раствора
гексацианоферрата (III) калия в коническую колбу вместимостью
200 или 250 см3 вносят 50 см3 приготовленного раствора гексациа-
ноферрата (III) калия, прибавляют 20 см310%-ного раствора суль-
фата цинка и 20 см3 10%-ного раствора йодита калия (не содержа-
щего йода), взбалтывают содержимое колбы и немедленно титруют
выделившийся йод 0,1 н. раствором сульфита натрия в присутствии
крахмала в качестве индикатора. Поправку к 1 %-ному раствору гек-
сацианоферрата (III) калия вычисляют до четвертого десятичного
знака по формуле
„ К О,3291
А—ОЗ-’
где V— объем 0,1 н. раствора сульфита натрия, пошедший на титрование выделив-
шегося йода, см3; 0,3291 — массовая концентрация гексацианоферрата (III) калия,
соответствующая 1 см3 0,1 н. раствора сульфита натрия, г/см3; 0,5 — масса гексаци-
аноферрата (III) калия в 50 см3 1 %-ного раствора, г.
Раствор гексацианоферрата (III) калия хранят в темной посуде.
307
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ
ХЛОРИДА НАТРИЯ
Содержание хлорида натрия в сырах, брынзе и соленых творож-
ных изделиях определяют с нитратом серебра без озоления или с
озолением пробы продукта. Метод является арбитражным.
Выполнение работы. С сычужного сыра срезают поверхностный
слой толщиной до 10 мм, у бескоркового сыра — до 2 мм. Рассоль-
ный сыр при необходимости помещают на сетчатую подставку или
фильтровальную бумагу, покрывают крышкой и выдерживают в за-
висимости от вида сыра 2—4 ч при температуре 20 ± 5 °C. Пробу
протирают через терку, помещают в фарфоровую ступку и тщатель-
но перемешивают.
Пробы соленых творожных изделий растирают в ступке до полу-
чения однородной консистенции.
Без озоления. 5 г продукта, взвешенного с погрешностью не более
0,01 г, помещают в стакан вместимостью 100 см3 с носиком, прили-
вают 50 см3 дистиллированной воды, нагретой до 90 °C. Продукт хо-
рошо растирают стеклянной палочкой и содержимое стакана коли-
чественно переносят в мерную колбу вместимостью 100 см3, смывая
дистиллированной водой, нагретой до 70—80 °C.
Мерную колбу с содержимым охлаждают до 20 °C, доливают дис-
тиллированной водой до метки, хорошо перемешивают и фильтру-
ют через сухой фильтр в чистую сухую колбу. Если фильтрат полу-
чается мутный, его переливают обратно в мерную колбу и фильтро-
вание повторяют.
В коническую колбу пипеткой приливают 50 см3 фильтрата,
прибавляют 5—8 капель раствора хромата калия и фильтрат титру-
ют раствором нитрата серебра (реактив 1) при постоянном взбалты-
вании до появления слабого кирпично-красного окрашивания, не
исчезающего при взбалтывании и измельчении палочкой крупных
частиц осадка. Массовая доля хлорида натрия в соленых творожных
изделиях (%)
_июо
и-50 ’
где V— объем раствора нитрата серебра, 1 см3 которого соответствует точно 0,01 г
хлорида натрия, израсходованный на титрование 50 см3 фильтрата, см3; т — масса
навески продукта, г.
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,2 %.
Для определения хлорида натрия в сливочном масле пробу на-
гревают до температуры не выше 30 °C, обеспечивающей гомоген-
ное состояние при смешивании механической мешалкой или вруч-
ную. Затем охлаждают до температуры 20 ± 5 °C при постоянном
308
перемешивании. Взвешивают около 5 г приготовленной пробы с
погрешностью не более 0,001 г в коническую колбу. Осторожно до-
бавляют к пробе 100 см3 кипящей дистиллированной воды. Дают
постоять от 5 до 10 мин, перемешивают круговыми движениями.
После охлаждения до температуры 50—55 °C вносят 2 см3 ра-
створа хромата калия (50 г/см3) и перемешивают содержимое не-
сколько раз. Если масло кислосливочное (pH менее 6,5), то перед
титрованием добавляют на кончике шпателя карбонат кальция и
перемешивают круговыми движениями. Титруют раствором нит-
рата серебра (реактив 1) при непрерывном помешивании до тех
пор, пока не появится оранжево-коричневое окрашивание, не ис-
чезающее в течение 30 с. Параллельно проводят контрольный
опыт, используя 5 см3 дистиллированной воды, вместо 5 г сливоч-
ного масла.
С озолением. На часовом стекле или в бюксе взвешивают от 1,8 до
2,2 г измельченных сыра, брынзы или соленых творожных изделий
с погрешностью не более 0,001 г и переносят в коническую колбу.
Пипеткой добавляют 25 см3 раствора нитрата серебра (реактив 1).
затем при помощи градуированного цилиндра приливают 25 см3
азотной кислоты и тщательно перемешивают.
Смесь нагревают в вытяжном шкафу до кипения, добавляют
10 см3 раствора перманганата калия и поддерживают реагирующую
смесь в слабокипящем состоянии.
Если реагирующая смесь изменяет окраску от темно-коричне-
вого до светло-желтой или бесцветной, то добавляют еще 5—10 см3
раствора перманганата калия. Наличие избытка последнего (ко-
ричневая окраска смеси) показывает, что органическое вещество
полностью разложилось. Удаляют избыток перманганата калия,
добавляя щавелевую кислоту или глюкозу до исчезновения корич-
невой окраски. Затем приливают 100 см3 дистиллированной воды
и 2 см3 раствора железоаммонийных квасцов и тщательно переме-
шивают.
Избыточный объем нитрата серебра (реактив 1) титруют раство-
ром роданита калия или аммония до появления красно-коричнево-
го окрашивания, не исчезающего в течение 30 с. Параллельно про-
водят контрольный опыт при использовании 2 см3 дистиллирован-
ной воды вместо 2 г сыра, брынзы или соленых творожных изделий.
Массовая доля хлорида натрия в сливочном масле, сыре, брынзе
или соленых творожных изделиях (%)
_5,85C(Pj-K0)
2 т ’
где 5,85 — коэффициент для выражения результатов в виде процентного содержа-
ния хлорида натрия; С — малярная концентрация титрованного раствора роданита
калия или роданита аммония, моль/дм3' Ио — объем раствора роданита калия, ис-
пользованный в контрольной пробе, см3' Ki — объем раствора роданита калия, ис-
пользованный при анализе продукта, см3; т — масса навески продукта, г.
309
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,07 %.
Приготовление реактива. Для приготовления раствора нитрата
серебра — 2,906 г нитрата серебра, взвешенного с погрешностью не
более 0,001 г, переносят небольшими порциями в мерную колбу
вместимостью 100 см3, заполняют колбу дистиллированной водой
приблизительно до 2/3 объема, вращением колбы перемешивают ее
содержимое до полного растворения реактива, доливают колбу дис-
тиллированной водой до метки и вновь тщательно перемешивают.
Титр раствора уточняют по хлориду натрия. 1 см3 раствора должен
соответствовать 0,01 г хлорида натрия.
ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ
ЭТИЛОВОГО СПИРТА
Метод определения спирта в кефире и кумысе основан на изме-
рении при помощи пикнометра относительной массы рг2о) водно-
спиртовой смеси, полученной из продукта путем отгонки.
Относительной массой водно-спиртовой смеси ^20) называется
отношение массы этилового спирта, определенной при температу-
ре 20 °C, к массе воды в том же объеме и при той же температуре.
Вначале подготавливают пикнометр, который тщательно про-
мывают последовательно слабым спиртовым раствором щелочи,
водой, хромовой смесью и вторично водой, после чего высушива-
ют при температуре 100—105 °C, охлаждают в эксикаторе и взве-
шивают.
В колбу для перегонки отвешивают 100 г продукта с погрешнос-
тью не более 0,1 г, добавляют по каплям раствор гидроксида натрия
(калия) до нейтральной или слабощелочной реакции (по лакмусо-
вой бумажке), помещают туда несколько стеклянных капилляров и
закрывают колбу пробкой. Затем соединяют колбу с обратным хо-
лодильником и медленно проводят перегонку при умеренном на-
гревании. В качестве приемника применяют мерную колбу вмести-
мостью 100 см3. Отгонку прекращают после заполнения колбы при-
близительно на 2/3 объема.
При получении не очень чистого раствора его количественно пе-
реносят в чистую колбу для перегонки, в которой объем раствора
доводят водой приблизительно до 100 см3, и вторично перегоняют.
По окончании перегонки мерную колбу с водно-спиртовой смесью
дополняют водой до метки и тщательно перемешивают. В пикно-
метр, предварительно взвешенный и подготовленный, приливают
(пипеткой или трубкой с оттянутым капилляром) из мерной колбы
водно-спиртовую смесь до уровня немного выше метки и проводят
определение. Так же, как и раствор, в него приливают воду.
310
Пикнометр с водой подвешивают на тонкой нити к стеклянной па-
лочке, положенной на кольцо штатива, и опускают в стакан с водой,
которая должна быть приблизительно на одном уровне с водой пикно-
метра. Для поддержания постоянной температуры (30,0 ± 0,2 °C) ста-
кан помещают в термостат.
Через 40 мин с помощью фильтровальной бумаги или трубки с
оттянутым капилляром мениск пикнометра устанавливают точно
на метке, после чего пикнометр закрывают пробкой, вынимают из
стакана, тщательно вытирают снаружи фильтровальной бумагой и
взвешивают.
Водное число пикнометра Р (масса воды в объеме данного пик-
нометра при 20 °C) вычисляют по формуле
Р = Ш2 — «11,
где mi — масса пустого пикнометра с пробкой, г; mi — масса пикнометра с водой и
пробкой, г.
Относительную массу раствора этилового спирта вычисляют
по формуле
где т3'— масса пикнометра с водно-спиртовой смесью, г.
Расхождение между параллельными определениями относи-
тельной массы раствора отгона должно быть не более 0,0002. Мас-
совую долю этилового спирта в продукте находят по относительной
массе (см. приложение 7).
Контрольные вопросы. 1. Что определяют термогравиметрическим методом? 2.
На чем основано определение массовой доли жира? 3. Как определяют массовую
долю белка по Кьельдалю? 4. Какими способами определяют массовую долю угле-
водов?
Глава 12
АНАЛИТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
СВОЙСТВ МОЛОКА И МОЛОЧНЫХ ПРОДУКТОВ
•
ОПРЕДЕЛЕНИЕ КИСЛОТНОСТИ
ТИТРУЕМАЯ КИСЛОТНОСТЬ
Метод основан на нейтрализации кислот, содержащихся в про-
дукте, раствором гидроксида натрия в присутствии индикатора фе-
нолфталеина. Титруемую кислотность выражают в градусах Терне-
ра (Т) или для масла в градусах Кеттстофера (°К).
Один градус Тернера (°Т) соответствует объему (см3) водного ра-
створа гидроксида натрия концентрацией 0,1 моль/дм3, необходи-
мый для нейтрализации 100 г (100 см3) исследуемого продукта.
Один градус Кеттстофера (°К) соответствует объему (см3) водно-
го раствора гидроксида натрия концентрацией 0,1 моль/дм3, необ-
ходимый для нейтрализации 5 г сливочного масла или его жировой
фазы, умноженный на 2.
При определении титруемой кислотности молока, сливок и кис-
ломолочных напитков в колбу вместимостью 100 или 250 см3 отме-
ривают 20 см3 дистиллированной воды (40 см3 для молока с напол-
нителями), 10 см3 анализируемого продукта и 3 капли фенолфтале-
ина. При анализе сливок и кисломолочных напитков переносят ос-
татки продукта из пипетки в колбу путем промывания пипетки
полученной смесью 3—4 раза. Смесь тщательно перемешивают и
титруют раствором гидроксида натрия до появления слабо-розово-
го окрашивания, соответствующего контрольному эталону окрас-
ки, не исчезающего в течение 1 мин. Для молока с наполнителями с
целью более точного установления конца титрования рядом с тит-
руемой пробой ставят контрольную колбу.
В неокрашенном мороженом и сметане кислотность определяют
следующим образом: в колбу вместимостью 100 или 250 см3 отвеши-
вают 5 г продукта, добавляют 30 см3 воды и 3 капли фенолфталеина.
Смесь тщательно перемешивают и титруют, как пробу молока.
Кислотность окрашенного мороженого определяют так: отве-
шивают в колбу вместимостью 250 см3 5 г мороженого, добавляют
80 см3 дистиллированной воды и 3 капли фенолфталеина. Смесь
тщательно перемешивают и титруют, как пробу молока. Для опре-
деления конца титрования окрашенного мороженого колбу с титру-
емой смесью ставят на белый лист бумаги и рядом помещают колбу
со смесью: 5 г образца мороженого и 80 см3 воды.
При определении кислотности творога или творожных изделий
в фарфоровую ступку вносят 5 г продукта, тщательно перемешива-
312
ют и растирают пестиком. Затем прибавляют небольшими порция-
ми 50 см3 воды, нагретой до температуры 35—40 °C и 3 капли фенол-
фталеина. Смесь перемешивают и титруют, как пробу молока.
Для определения кислотности сливочного масла в колбе вместимо-
стью 50 и 100 см3 отвешивают 5 г сливочного масла, нагревают колбу в
водяной бане или сушильном шкафу при температуре 50 ± 5 °C до
расплавления масла, вносят 20 см3 нейтрализованной смеси спирта с
эфиром, 3 капли фенолфталеина и титруют, как пробу молока.
Для определения кислотности жировой фазы сливочного масла в
колбе вместимостью 50 или 100 см3 взвешивают 5 г жира, подготов-
ленного следующим образом. В сухой чистый стакан вместимостью
250 см3 отвешивают около 150 г исследуемого масла. Стакан поме-
щают в водяную баню или сушильный шкаф при температуре
50 ± 5 °C и выдерживают до полного расплавления и разделения
масла на жир и плазму. После чего вынимают из водяной бани (су-
шильного шкафа) и осторожно сливают верхний слой жира, фильт-
руя его через бумажный фильтр в колбу вместимостью 250 см3. Ана-
лиз проводят, как указано выше.
Для определения кислотности плазмы сливочного масла в
плоскодонную колбу вместимостью 100 см3 приливают 20 см3 дис-
тиллированной воды и 10 см3 плазмы, подготовленной следую-
щим образом.
Оставшуюся в стакане после слива жира плазму переносят в жиро-
мер, который плотно закрывают пробкой, помещают в центрифугу и
центрифугируют 5 мин с частотой вращения 1000 об/мин. Затем жи-
ромер помещают в стакан с холодной водой градуированной частью
вверх и выдерживают до застывания молочного жира, отделившегося
от плазмы в процессе центрифугирования. Свободную от жира плаз-
му осторожно выливают в сухой чистый стакан вместимостью 100 см3
и тщательно перемешивают стеклянной палочкой.
Смесью плазмы и дистиллированной воды 3—4 раза промывают
пипетку, затем прибавляют 3 капли фенолфталеина и титруют, как
пробу молока.
Кислотность в градусах Тернера (°Т) находят умножением объе-
ма (см3) раствора гидроксида натрия, затраченного на нейтрализа-
цию кислот, содержащихся в определенном объеме продукта, на
следующие коэффициенты:
10 — для молока, молока с наполнителями, сливок, простоква-
ши, ацидофильного молока, кефира, кумыса, других кисломолоч-
ных продуктов, а также плазмы сливочного масла;
20 — для мороженого, сметаны, творога и творожных изделий.
Кислотность сливочного масла и его жировой фазы в градусах
Кеттстофера (°К) находят умножением на два объема раствора гид-
роксида натрия, затраченного на нейтрализацию кислот, содержа-
щихся в 5 г продукта.
Допускаемая погрешность результата анализа при принятой до-
верительной вероятности Р = 0,95 составляет:
313
± 1,9 °Т — для молока, молока с наполнителями, сливок, про-
стокваши, ацидофильного молока, кефира, кумыса, других кисло-
молочных продуктов и мороженого;
± 2,3 °Т — для сметаны;
± 3,6 °Т — для творога и творожных изделий;
± 0,1 °К — для масла сливочного и его жировой фазы;
± 0,5 °Т — для плазмы сливочного масла.
Расхождение между двумя параллельными определениями не
должно превышать:
2,6 °Т — для молока, молока с наполнителями, сливок, просто-
кваши, ацидофильного молока, кефира, кумыса, других кисломо-
лочных продуктов и мороженого;
3,2 °Т — для сметаны;
5,0 °Т — для творога и творожных изделий;
0,1 °К — для сливочного масла и его жировой фазы;
0,6 Т — для плазмы сливочного масла.
За окончательный результат анализа принимают среднее ариф-
метическое значение результатов двух параллельных определений,
округляя результат до второго десятичного знака. При большем рас-
хождении испытание повторяют с четырьмя параллельными опре-
делениями. При этом расхождение между средним арифметичес-
ким значением результатов четырех определений и любым значе-
нием из четырех результатов определения не должно превышать:
1,8 Т — для молока, молока с наполнителями, сливок, просто-
кваши, ацидофильного молока, кефира, кумыса, других кисломо-
лочных продуктов и мороженого;
2,3 °Т — для сметаны;
3,6 Т — для творога и творожных изделий;
0,1 °К — для сливочного масла и его жировой фазы;
0,5 °Т — для плазмы сливочного масла.
При большем расхождении приготовляют заново все реактивы,
проводят государственную проверку используемых приборов и по-
вторяют испытание с четырьмя параллельными определениями. В
этом случае при наличии расхождения больше вышеуказанных зна-
чений выполнение данной работы поручают оператору более высо-
кой квалификации.
ПРЕДЕЛЬНАЯ КИСЛОТНОСТЬ
Анализ основан на нейтрализации кислот, содержащихся в про-
дукте, избыточным количеством гидроксида натрия в присутствии
индикатора фенолфталеина. При этом избыток щелочи и интен-
сивность окраски в полученной смеси обратно пропорциональны
кислотности молока.
Выполнение работы. Для определения предельной кислотнос-
ти готовят рабочие растворы: в мерную колбу вместимостью
1000 см3 отмеривают необходимый объем 0,1 М раствора гидрок-
314
сида натрия, добавляют 10 см3 фенолфталеина и дистиллирован-
ную воду до метки.
Объем раствора гидрок- 80 85 90 95 100 105 ПО
сида натрия, см3
Кислотность, Т 16 17 18 19 20 21 22
В несколько пробирок вносят по 10 см3 приготовленного раство-
ра гидроксида натрия и 5 см3 молока. Содержимое пробирки пере-
мешивают, перевертывая. Если содержимое пробирки обесцвечи-
вается, то кислотность данной пробы молока будет выше соответ-
ствующего данному раствору градуса.
Приготовление контрольных эталонов окраски.
а) для молока и сливок — в колбу вместимостью 100 или 250 см3 от-
меривают 10 см3 молока или сливок, 20 см3 дистиллированной воды и
1 см3 раствора сульфата кобальта. Смесь тщательно перемешивают.
Срок хранения эталона не более 8 ч при комнатной температуре;
б) для смеси этилового спирта и диэтилового эфира — к 10 см3
спирта добавляют 10 см3 диэтилового эфира и 1 см3 раствора суль-
фата кобальта. Смесь тщательно перемешивают;
в) для сливочного масла, его жировой фазы — к 5 г расплавлен-
ного масла добавляют 20 см3 нейтрализованной смеси спирта и
эфира и 1 см3 раствора сульфата кобальта. Смесь перемешивают;
г) для плазмы сливочного масла — к 10 см3 плазмы добавляют
20 см3 воды. Полученной смесью 3—4 раза промывают пипетку и
добавляют 1 см3 раствора сульфата кобальта. Смесь перемешивают.
Смесь этилового спирта и диэтилового эфира готовят непосред-
ственно перед измерением кислотности сливочного масла или его
жировой фазы следующим образом. В колбу вместимостью 50 см3
приливают по 10 см3 спирта и эфира, 3 капли фенолфталеина и ней-
трализуют смесь раствором щелочи до появления слабо-розового
окрашивания, не исчезающего в течение 1 мини соответствующего
контрольному эталону окраски.
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ
АРЕОМЕТРИЧЕСКИЙ МЕТОД
Плотность заготовляемого коровьего молока, пастеризованного
(цельного, белкового, витаминизированного, обезжиренного) и сте-
рилизованного определяют при 20 ± 5 °C. Плотность пастеризован-
ного коровьего молока с повышенным содержанием жира, напитков
с наполнителями, сливок, пахты, сыворотки, молокаи других живот-
ных и кисломолочных продуктов определяют при 20 ± 2 °C.
Измерения в заготовляемом молоке проводят не ранее чем через
2 ч после дойки; кисломолочные продукты анализируют в подго-
товленной смеси до сквашивания.
315
Перед определением плотности пробы с отстоявшимся слоем сли-
вок ее нагревают до 35 + 5 °C, перемешивают и охлаждают до 20 ± 2 °C.
Ареометры типа AM и АМТ и необходимую стеклянную аппара-
туру тщательно моют моющими растворами, ополаскивают дистил-
лированной или кипяченой питьевой водой, а остатки влаги удаля-
ют льняной тканью или полотенцем. Вымытые приборы и аппара-
туру сушат на воздухе до полного высыхания. При массовых анали-
зах допускается ополаскивать цилиндр после каждого определения
молоком, отобранным для очередного определения плотности ис-
следуемой пробы молока.
Ареометры, термометры и мешалки, подготовленные к измере-
ниям, хранят в цилиндрах, накрытых покровным стеклом или по-
лиэтиленовым чехлом.
Пробу объемом 250 или 500 см3 тщательно перемешивают и ос-
торожно, во избежание образования пены, переливают по стенке в
сухой цилиндр вместимостью 250 см3, который следует держать в
слегка наклонном положении. Если на поверхности пробы в ци-
линдре образовалась пена, ее снимают мешалкой.
Цилиндр с исследуемой пробой устанавливают на ровной гори-
зонтальной поверхности и измеряют температуру пробы Ц. Подго-
товленный к измерениям ареометр берут за верхнюю часть стержня,
свободную от шкалы, и медленно опускают в исследуемую пробу.
Когда до предполагаемой отметки ареометрической шкалы оста-
нется 3—4 мм, его оставляют в свободно плавающем состоянии,
следя за тем, чтобы ареометр не касался стенок цилиндра.
Цилиндр с пробой располагают удобно по отношению к источ-
нику света для отсчета показаний по шкале плотности и шкале тер-
мометра.
Первый отсчет показаний плотности pj проводят визуально со
шкалы ареометра через 3 мин после установления его в непод-
вижном положении. Затем ареометр осторожно приподнимают
на высоту до уровня балласта и снова опускают, оставляя его в
свободно плавающем состоянии. После установления его в не-
подвижном состоянии, проводят второй отсчет показаний плот-
ности р2- При отсчете показаний плотности глаз должен нахо-
диться на уровне мениска. Отсчет показаний проводят по верхне-
му краю мениска.
Отсчет показаний по ареометрам типов AM и АМТ для молока
проводят с точностью 0,5 кг/м3. В ареометрах общего назначения
типов АОН-1 и АОН-2 — с точностью 1 кг/м3.
Затем измеряют температуру t2 пробы.
Расхождение между повторными определениями плотности
(последовательно одно определение за другим в одной и той же про-
бе) не должно превышать 0,5 кг/м3 для ареометров типов AM и АМТ
и 1,0 кг/м3 для ареометров типов АОН-1 и АОН-2.
При проведении массовых измерений плотности молока допус-
кается прикасаться нижним концом ареометра, извлекаемого из
316
молока, к внутренней поверхности цилиндра; после стекания с аре-
ометра молока прибор погружают в другой цилиндр с новой пробой
молока, не допуская засыхания молока на поверхности ареометра.
Измерения повторяют.
Если возникают разногласия в оценке качества при определении
плотности молока, применяют арифметический метод, заключаю-
щийся в том, что пробу нагревают до 40 ± 2 °C, выдерживают при
этой температуре в течение 5 ± 1 мин, затем охлаждают ее до
20 ± 2 °C и измеряют плотность молока.
За среднее значение температуры t исследуемой пробы прини-
мают среднее арифметическое результатов двух показаний t\ и 6.
При этом руководствуются следующим правилом: если дробная
часть среднего арифметического значения температуры равна или
меньше 0,25, то ее не учитывают; если она равна или больше 0,75, то
ее округляют до единицы; если она больше 0,25, но меньше 0,75, ее
округляют до 0,5.
За среднее значение показаний ареометра при температуре t
(Pep) исследуемой пробы молока принимают среднее арифметичес-
кое результатов двух показаний pi и р2, руководствуясь теми же пра-
вилами округления.
Если проба во время определения плотности имела температуру
выше или ниже 20 °C, то результаты определения плотности при
температуре /должны быть приведены к 20 °C в соответствии с при-
ложениями 8 и 9.
По таблицам в левой крайней графе находят строку со значением
Pep, а в последующих графах таблиц — температуру I. На пересече-
нии соответствующей строки и графы находят значение плотности
молока при 20 °C, которое принимается за окончательный результат.
Если заготовляемое или обезжиренное коровье молоко имеет
температуру от 10 до 15 °C, то для определения его фактической
плотности к полученному значению плотности пробы этого молока
Рср добавляют поправку, найденную по приложениям 8, 9.
Результат определения плотности молока с учетом погрешности
метода должен быть представлен в виде
Рт ± ЛРа,
где рт —значение плотности, приведенной к 20 °C, кг/м3, найденное по приложе-
ниям 8, 9; Дра — погрешность определения плотности молока ареометрическим ме-
тодом, не более + 0,5 кг/м3.
Допускаемое расхождение между результатами определения
плотности молока р^0 одним типом ареометров в различных усло-
виях (в разное время, в разных местах и разными операторами) не
должно превышать 0,8 кг/м3.
317
ПИКНОМЕТРИЧЕСКИЙ МЕТОД
Метод предназначен для проведения научных и эксперимен-
тальных исследований по определению плотности молока и сгу-
щенных молочных консервов.
При подготовке к анализу пикнометры (не менее двух) моют мо-
ющими растворами и тщательно ополаскивают дистиллированной
водой. После этого их высушивают в электропечи при 110 ± 10 °C не
менее 30 мин, вынимают и выдерживают при комнатной темпера-
туре не менее 30 мин и взвешивают.
Затем пикнометры снова помещают в электропечь и выдержи-
вают при той же температуре не менее 15 мин, вынимают из элек-
тропечи, выдерживают в помещении не менее 30 мин и взвеши-
вают. Разность между результатами двух взвешиваний каждого
пикнометра не должна превышать 3 • 10~7 кг для весов 2-го клас-
са точности и 5 • 10-6 кг для весов 4-го класса точности. Если ука-
занное условие не соблюдается, то высушивание следует пов-
торить.
За значение массы гирь, уравновешивающих каждый пустой
пикнометр, принимают среднее арифметическое результатов двух
взвешиваний тх.
Пикнометры заполняют при помощи шприца свежекипяченой и
охлажденной до комнатной температуры дистиллированной водой
немного выше отметки на их шейке и закрывают пробками, поме-
щают в стакан с водой, так чтобы вода покрывала заполненную
часть пикнометров, и опускают стакан в термостат. Пикнометры
выдерживают в термостате при 20,00 ± 0,05 °C в течение 30 мин.
Температуру воды контролируют термометром с ценой деления
0,01 °C, опущенным в термостат. Пикнометры вынимают из термо-
стата, доводят при помощи шприца и фильтровальной бумаги уро-
вень воды до отметки на их шейке (по верхнему краю мениска).
Внутреннюю поверхность шейки пикнометров выше отметки тща-
тельно промокают фильтровальной бумагой, не касаясь уровня
воды в пикнометрах, вытирают снаружи досуха полотенцем и остав-
ляют в витрине весов не менее 20 мин. После этого пикнометры
взвешивают. Опыт повторяют не менее 3 раз для каждого пикномет-
ра. За значение массы гирь, уравновешивающих каждый пикнометр
с водой, принимают среднее арифметическое результатов всех взве-
шиваний m2- Значение массы гирь, уравновешивающих пустой
пикнометр и пикнометр с водой, определяют повторно для каждого
пикнометра после 20 определений плотности молока и сгущенных
молочных консервов. Воду выливают из пикнометров и высушива-
ют их.
Пробу молока и сгущенных молочных консервов нагревают до
40 ± 2 °C, выдерживают в течение 5 ± 1 мин и охлаждают до
20 ± 2 °C. Подготовленные к измерениям пикнометры (не менее
двух) заполняют при помощи шприца предварительно тщательно
318
перемешанной исследуемой пробой заготовляемого коровьего или
обезжиренного молока немного выше отметки на их шейке и зак-
рывают пробками. Пикнометры термостатируют и уровень иссле-
дуемой пробы до отметки на их шейке доводят с помощью пипетки.
За значение массы гирь, уравновешивающих каждый пикнометр с
молоком, принимают среднее арифметическое результатов двух
взвешиваний ту
При помощи воронки заполняют пикнометры исследуемой про-
бой сгущенных молочных консервов, уровень до отметки доводят
пипеткой.
После проведения измерений из пикнометров выливают про-
дукт, промывают, высушивают, закрывают пробкой и хранят до
проведения новых исследований.
Плотность молока и сгущенных молочных консервов р] (кг/м3)
при 20 °C и нормальном давлении 0,1 МПа вычисляют по формуле
Р1 = 4--,(Рв -рвоз)+Рвоз,
где От], ту m-i — массы гирь, уравновешивающих, соответственно, пустой пикно-
метр, пикнометр с водой и пикнометр с молоком, кг; рв — плотность воды
(рв - 998,20 кг/м3); рвоз — плотность воздуха (рвоз — 1,2 кг/м3).
Аналогично определяют плотность р2 той же пробы молока с по-
мощью второго пикнометра.
Допускаемое расхождение между двумя результатами определе-
ния плотности не должно превышать по абсолютному значению
0,3 кг/м3 — для молока и 10 кг/м3 — для сгущенных молочных кон-
сервов. Если расхождения превышают допустимые, то проводят по-
вторные контрольные измерения плотности.
За плотность рср молока при 20 °C принимают среднее арифме-
тическое результатов двух полученных значений плотности молока
Pi и р2 при 20 °C.
При округлении вычисленных значений плотности рср при 20°С
руководствуются правилом: если шестая отбрасываемая цифра,
считая слева направо, равна или больше 5, то последняя сохраняе-
мая цифра увеличивается на единицу; если же шестая отбрасывае-
мая цифра, считая слева направо, меньше 5, то последняя сохраняе-
мая цифра не меняется.
Результат определения плотности молока и сгущенных молоч-
ных консервов при 20 °C записывают в виде
Рср ± Арп,
где рср — среднее арифметическое значение плотности при 20 °C, полученное пик-
нометрическим методом, кг/м3; Дрп — погрешность определения плотности пикно-
метрическим методом: ±0,2кг/м3 — для молока, ± 10кг/м3—для сгущенных мо-
лочных консервов.
319
Допускаемые расхождения между результатами определения
плотности молока пикнометрическим и ареометрическим метода-
ми не должны превышать 1,0 кг/м3 при 20 °C.
ОПРЕДЕЛЕНИЕ ТЕРМОУСТОЙЧИВОСТИ МОЛОКА
Под термоустойчивостью молока понимают свойство продукта
выдерживать воздействие высоких температур без видимой коагу-
ляции белков.
Термоустойчивость молока определяется термоустойчивос-
тью казеина, которая связана с солевым равновесием в молоке, а
именно с соотношением растворимых форм следующих солей:
катионов кальция и магния, с одной стороны, и анионов фос-
форной (фосфатов) и лимонной (цитратов) кислот, с другой сто-
роны.
Повышение концентрации ионов кальция и магния приводит к
снижению термоустойчивости молока, что обусловлено уменьше-
нием отрицательного заряда мицелл казеина и, как следствие, на-
рушением их гидратной оболочки. В результате мицеллы казеина
легко агрегатируют. При воздействии высоких температур крупные
агломераты могут коагулировать.
С увеличением числа ионов кальция и магния в молоке агрега-
ция мицелл казеина при нагревании усиливается, а возможность
свертывания молока возрастает. И наоборот, снижение в молоке
концентрации катионитов способствует переходу части кальция,
связанного с мицеллой, в растворимое состояние, происходит де-
загрегация мицелл, что способствует повышению термоустойчиво-
сти молока.
Известные методы определения термоустойчивости молока ос-
нованы либо на непосредственном тепловом воздействии на про-
дукт (тепловая проба), либо на определении содержания ионов
кальция в молоке (проба по К. К. Горбатовой и П. И. Гуньковой),
либо на внесении веществ, вызывающих частичную денатурацию
казеина (алкогольная проба), либо на частичном смещении солево-
го равновесия в молоке в неблагоприятную для устойчивости казеи-
на сторону (кальциевая и фосфатная пробы).
Кроме того, сотрудниками ВНИМИ создан специальный изме-
ритель термоустойчивости «Термол-1», предназначенный для сор-
тировки молока, направляемого на стерилизацию, и в производстве
детских молочных продуктов.
Прибор «Термол-1» создан предприятием ИСТ (Санкт-Петер-
бург) и ВНИМИ.
Принцип его работы основан на том, что пробу молока в ячейке
ступенчато нагревают до заданной температуры, выдерживают в те-
чение определенного времени, после чего подвергают анализу гид-
320
ромеханическим методом с целью выявления коагуляции белка. В
качестве гидрозапирающего элемента используют насадку с дрос-
сельным отверстием, которая свободно пропускает несвернувшееся
молоко, но легко забивается хлопьями коагулированного белка.
Диапазон сканирования температур — от 80 до 140 °C с дискретнос-
тью 10 °C. Объем пробы — не более 25 см3 для всего диапазона тем-
ператур. Продолжительность выдержки пробы при каждой темпе-
ратуре — 0,5, 1 и 2 мин. Общее время контроля молока на термоус-
тойчивость составляет 23 мин.
Прибор выдает информацию (значение температуры в °C) в
цифровой форме на табло и звуковой сигнал в момент термокоагу-
ляции. Конструктивно он состоит из единого блока, включающего
узел нагрева и устройство управления. Может применяться как в
лабораториях, так и в производственных помещениях.
ВНИМИ разработана и утверждена в установленном порядке
методика определения термоустойчивости молока с помощью при-
бора «Термол-1». В соответствии с методикой предложены крите-
рии оценки термоустойчивости молока с использованием этого
прибора (табл. 40).
40. Показания прибора для молока разных групп
Группа молока Температура, "С Выдержка, мин
I 140 и выше 2,0
II От 120 до 130 2,0
III От 100 до НО 1,0
IV От 80 до 90 1,0
V Ниже 80 0,5
Так, молоко I и II групп пригодно для стерилизации соответ-
ственно при температурах 140 °C и выше и 120—130 °C.
ТЕПЛОВАЯ ПРОБА
(ПО Т. ВЛАДЫКИНОЙ И В. ВАЙТКУСУ)
Тепловая проба с использованием глицериновой бани заключа-
ется в установлении продолжительности теплового воздействия на
молоко или молочный продукт от момента погружения испытуемой
пробы в глицерин с высокой температурой до начала видимой коа-
гуляции молочных белков. В качестве глицериновой бани применя-
ют ультратермостат, заполненный глицерином.
Метод используют для определения термоустойчивости моло-
ка и молочных продуктов с массовой долей сухих веществ не бо-
лее 30 %, в том числе жира не более 20 %. Метод предназначен
для исследовательских и арбитражных промышленных испы-
таний.
321
Выполнение работы. Ультратермостат заполняют глицерином на
45—50 мм ниже крышки. Подставку ультратермостата закрепляют
ниже уровня глицерина на 110 мм. Глицерин разогревают до
70— 100 °C с помощью электрокипятильника. Затем включают ульт-
ратермостат в электросеть и доводят температуру глицерина до тре-
буемой (в зависимости от вида испытуемого продукта), например
для испытания проб молока сырого (заготовляемого) и подвергну-
того тепловой обработке (пастеризованного, стерилизованного при
130 ± 0,2 °C), для испытания проб молока сгущенного стерилизо-
ванного без сахара и стерилизованного концентрированного (при
120 ±0,2 °C). Электролампу укрепляют над рабочим отверстием
ультратермостата.
Пробирки устанавливают в пробиркодержателе, наполовину за-
полняют молоком (молочным продуктом), закрывают резиновыми
пробками и зажимают верхней планкой пробиркодержателя. Про-
биркодержатель должен иметь деревянную ручку.
Из одного образца молока или молочного продукта подготавли-
вают две параллельные пробы, которые размещают в разных про-
биркодержателях. Первый пробиркодержатель с пробами устанав-
ливают вертикально на подставку ультратермостата с глицерином,
нагретым до требуемой температуры, и включают секундомер. Спу-
стя 10— 15 мин рядом с первым пробиркодержателем ставят второй
и включают другой секундомер.
За состоянием проб наблюдают визуально в защитных очках до
начала видимой коагуляции молочных белков (появления хлопьев).
Для этого первый пробиркодержатель извлекают из глицерина че-
рез каждые 3 мин в течение первых 30 мин от начала испытания, за-
тем через 2 мин до конца испытания. При наблюдении пробирко-
держатель быстро наклоняют на 20—40° к горизонтальной плоско-
сти так, чтобы проба перетекала из одного конца пробирки в дру-
гой. Продолжительность одного наблюдения не должна превышать
5 с. Для каждой пробы устанавливают свое время начала видимой
коагуляции молочных белков.
За состоянием проб второго пробиркодержателя наблюдают
через каждые 10 мин, и только в конце испытания за 1 —3 мин до
начала видимой коагуляции проб первого пробиркодержателя —
через каждую минуту. Для каждой пробы второго пробиркодер-
жателя также устанавливают свое время начала видимой коагу-
ляции.
Показатель термостойкости молока или молочного продукта по
тепловой пробе — время (мин) от момента погружения пробирко-
держателя с пробами в глицерин с высокой температурой до начала
видимой коагуляции молочных белков (табл. 41).
За окончательный результат принимают среднее арифметичес-
кое результатов двух параллельных определений, допускаемое рас-
хождение между которыми не должно превышать 3 мин. Относи-
тельная предельная погрешность определения показателя термо-
стойкости ± 5 %.
322
41. Термостойкое!» молока и молочных продуктов, мин
Группа Качество Молоко сырое и подвергнутое тепловой обработке при 130 °C Молоко сгущенное стери- лизованное без сахара и стери- лизованное концентри- рованное при 120 *С
I Хорошее 60 Более 40-60 Более 60
II Удовлетворительное 30-40 20—60
III Плохое 40 Менее Менее 20
Примечание. В числителе приведены данные для летнего времени, в знаменате-
ле — для осенне-весеннего и зимнего.
АЛКОГОЛЬНАЯ ПРОБА
Основана на воздействии этилового спирта на белки молока и
сливок, которые полностью или частично денатурируют при сме-
шивании равных объемов молока или сливок со спиртом.
Термоустойчивость молока и сливок по алкогольной пробе оп-
ределяют при помощи водного раствора этилового спирта с объем-
ной долей этилового спирта 68, 70, 72, 75 и 80 %. Чем большую кон-
центрацию спирта выдерживает молоко не свертываясь, тем оно
термоустойчивее.
Метод используют для определения термоустойчивости сырья, а
также подвергнутых тепловой обработке молока и сливок с массо-
вой долей жира не более 40 %.
Выполнение работы. Пробу сливок перед проведением алкогольной
пробы подогревают в стакане на водяной бане до температуры в преде-
лах 43 ± 2 °C, перемешивают и охлаждают до температуры 20 ± 2 °C. В
чистую сухую чашку Петри наливают 2 см3 исследуемого молока или
сливок, приливают 2 см3 этилового спирта требуемой объемной доли,
круговыми движениями смесь тщательно перемешивают. Спустя
2 ± 0,1 мин, наблюдают за изменением консистенции анализируемых
молока или сливок. Если на две чашки Петри при стекании анализи-
руемых смесей молока или сливок со спиртом не появились хлопья,
считается, что они выдержали алкогольную пробу.
В зависимости от того, какой раствор этилового спирта не выз-
вал осаждения хлопьев в испытуемых молоке и сливках, их подраз-
деляют на группы, указанные ниже:
Группа Объемные доли этилового спирта, %
I 80
II 75
III 72
IV 70
V 68
323
Приготовление реактива. Водный раствор этилового спирта гото-
вят в соответствии с табл. 42.
42. Объемы этилового спирта и воды при температуре 20 °C
для получения 1 дм3 водно-спиртового распора (с учетом сжатия раствора
в процессе приготовления)
Объемная доля этило- вого спирта в получен- ном раство- ре, % Объемная доля спирта в исходном растворе, см3
98 97 96 95 94
спирт | вода спирт вода спирт вода спирт вода спирт вода
68 694 336 701 328 708 319 716 310 723 302
69 704 326 711 317 719 308 726 299 734 290
70 714 315 722 306 729 297 737 288 745 279
71 724 304 732 295 740 287 747 277 755 268
72 735 294 742 285 750 275 758 266 766 257
73 745 283 753 274 760 265 768 255 777 245
74 755 273 763 263 771 253 779 244 787 234
75 765 261 773 252 781 242 789 233 798 223
76 776 251 784 241 792 231 800 221 809 212
77 786 240 794 230 802 220 811 210 819 200
78 796 230 804 219 812 209 821 199 830 189
79 806 218 814 208 823 198 832 187 840 177
80 816 207 825 197 833 187 842 176 851 166
81 827 196 835 186 844 176 853 165 862 154
82 837 186 845 175 854 164 863 154 872 143
После приготовления водного раствора спирта необходимо про-
верить его плотность или объемную долю спирта ареометрами при
20,0 ±0,1 °C.
Плотность, кг/м3
890,4
885,5
880,5
872,8
859,3
Объемная доля спирта, %
68
70
72
75
80
Результаты измерения плотности или объемной доли водно-
спиртового раствора не должны отличаться от заданной величины
более чем 0,25%-ной объемной доли спирта.
КАЛЬЦИЕВАЯ ПРОБА
В пробирку отмеривают пипеткой 10см3молокаи0,5см31%-ного
раствора хлорида кальция, тщательно перемешивают содержимое и
помещают пробирку в кипящую баню на 5 мин. После этого вынима-
ют, охлаждают и наблюдают, образовались ли в пробирке хлопья бел-
ка. Видимая коагуляция белка свидетельствует о том, что молоко не-
термоустойчивое, оно не выдержит стерилизации и свернется.
324
Для приготовления реактива (1%-ного раствора хлорида каль-
ция) 4 г безводного СаС12 растворяют в 96 см3 воды или 7,9 г
СаС1 • 6Н2О растворяют в 92 см3 воды.
ФОСФАТНАЯ ПРОБА
В пробирку отмеривают пипеткой 10 см3 молока и 1 см3 дигидро-
фосфата калия КН2РО4 (68,1 г на 1 дм3 воды) и, перемешав содер-
жимое пробирки, погружают ее в кипящую баню на 5 мин. После
охлаждения наблюдают за изменением консистенции молока. Коа-
гуляция белка от едва заметных до явно отличимых хлопьев указы-
вает на пониженную стабильность молока к нагреванию.
ОПРЕДЕЛЕНИЕ ЧИСТОТЫ МОЛОКА
Основано на отделении механической примеси из дозированной
пробы молока путем процеживания через фильтр и визуального
сравнения наличия механической примеси на фильтре с образцом
сравнения.
Фильтр вставляют в прибор гладкой поверхностью кверху. Из
объединенной пробы отбирают 250 см3 хорошо перемешанного мо-
лока, которое подогревают до температуры 35 + 5 °C и выливают в
сосуд прибора.
По окончании фильтрования фильтр вынимают и помещают на
лист пергаментной или другой непромокаемой бумаги.
В зависимости от количества механической примеси на фильтре
молоко подразделяют на три группы чистоты путем сравнивания
фильтра с образцом.
Группа чистоты
Первая
Вторая
Третья
Характеристика
На фильтре отсутствуют частицы меха-
нической примеси. Допускается для
сырого молока наличие на фильтре
не более двух частиц механической
примеси
На фильтре имеются отдельные части-
цы механической примеси (до 13 час-
тиц)
На фильтре заметный осадок частиц
механической примеси (волоски, час-
тицы корма, песка)
Примечание. Цвет фильтра должен соответствовать цвету молока в соответ-
ствии с требованиями НТД. При изменении цвета фильтра молоко, независимо от
количества имеющейся на фильтре механической примеси, относят к третьей
группе чистоты.
325
ОПРЕДЕЛЕНИЕ МИКРОБНОЙ ОБСЕМЕНЕННОСТИ
МОЛОКА
МЕТОД С МЕТИЛЕНОВЫМ ГОЛУБЫМ
Метод основан на восстановлении метиленового голубого окис-
лительно-восстановительными ферментами, выделяемыми в моло-
ко микроорганизмами. По продолжительности обесцвечивания ме-
тиленового голубого оценивают бактериальную обсемененность
сырого молока.
В пробирки наливают по 1 см3 рабочего раствора метиленового
голубого и по 20 см3 исследуемого молока, закрывают резиновыми
пробками и смешивают путем медленного трехкратного перевора-
чивания пробирок. Пробирки помещают в редуктазник с водой
температурой 37 ± 1 °C.
Вода в редуктазнике или водяной бане после погружения проби-
рок с молоком должна доходить до уровня жидкости в пробирке или
быть немного выше. Температуру воды поддерживают в течение
всего времени определения 37 ± 1 °C. Для предотвращения влияния
на свет редуктазник плотно закрывают крышкой. Момент погруже-
ния пробирок в редуктазник считают началом анализа. Наблюдение
за изменением окраски ведут через 40 мин, 2,5 и 3,5 ч с начала про-
ведения анализа. Окончанием анализа считают момент обесцвечи-
вания окраски молока. При этом остающийся небольшой кольце-
образный окрашенный слой вверху (шириной не более 1 см) или
небольшую окрашенную часть внизу пробирки (шириной не более
1 см) в расчет не принимают. Появление окрашивания молока в
этих пробирках при встряхивании не учитывают.
В зависимости от продолжительности обесцвечивания молоко
относят к одному из четырех классов, указанных в табл. 43.
43. Характеристика молока по классам
Класс молока Продолжительность обесцвечивания,ч Ориентировочное количество бактерий в 1 см’ молока, КОЕ
Высший Более 3,5 До 300 тыс.
I 3,5 От 300 тыс. до 500 тыс.
II 2,5 От 500 тыс. до 4 млн
III 40 мин От 4 млн до 20 млн
МЕТОД С РЕЗАЗУРИНОМ
Метод основан на восстановлении резазурина окислительно-
восстановительными ферментами, выделяемыми в молоко микро-
организмами. По продолжительности изменения окраски резазу-
рина оценивают бактериальную обсемененность сырого молока.
326
' Пробу с резазурином следует проводить не ранее чем через 2 ч
после доения.
В пробирки наливают по 1 см3 рабочего раствора резазурина и по
10 см3 исследуемого молока, закрывают резиновыми пробками и
смешивают, медленно трехкратно перевертывая пробирки. Про-
бирки помещают в редуктазник с температурой воды 37 ± 1 °C. При
отсутствии редуктазника можно использовать водяную баню, поме-
щенную в термостат с требуемой температурой. Уровень воды в ре-
дуктазнике или водяной бане после погружения пробирок с моло-
ком должен соответствовать уровню жидкости в пробирке или быть
немного выше, его поддерживают в течение всего времени опреде-
ления.
Пробирки с молоком и резазурином на протяжении анализа за-
щищают от прямых солнечных лучей (редуктазник должен быть
плотно закрыт крышкой). Время погружения пробирок в редуктаз-
ник считают началом анализа.
Появление окрашивания молока и в этих пробирках при встря-
хивании не учитывают.
По истечении 1 ч пробирки вынимают из редуктазника. Про-
бирки с молоком, имеющие серо-сиреневую окраску до сиреневой
со слабым серым оттенком, оставляют в редуктазнике еще на
30 мин. В зависимости от продолжительности обесцвечивания
или изменения цвета молоко относят к одному из четырех классов,
указанных в табл. 44.
44. Характеристика молока по классам
. Класс молока Продолжительность обесцвечивания или изменения цвета, ч Окраска молока Ориентировочное количество бактерий, в 1 см5 молока, КОЕ
Высший 1,5 Серо-сиреневая до сирене- вой со слабым серым от- тенком До 300 тыс.
I 1 Серо-сиреневая до сирене- вой со слабым серым от- тенком От 300 тыс. до 500 тыс.
II 1 Сиреневая с розовым от- тенком или ярко-розовая От 500 тыс. до 4 млн
III 1 Бледно-розовая или белая От 4 млн до 20 млн
Молоко, имеющее через 1,5 ч окраску, соответствующую I классу, относят к
высшему классу.
327
ОПРЕДЕЛЕНИЕ НАТУРАЛЬНОСТИ МОЛОКА
И НАЛИЧИЯ ФАЛЬСИФИЦИРУЮЩИХ ВЕЩЕСТВ
ОПРЕДЕЛЕНИЕ ПРИМЕСИ ВОССТАНОВЛЕННОГО МОЛОКА
В ЦЕЛЬНОМ
Основано на выделении из восстановленного молока комплекса
белка с лактозой, образующегося в процессе сушки молока. Казеин
осаждается кислотой, осадок промывают от несвязанной лактозы,
обезжиривают и получают окрашенное соединение при растворе-
нии выделенного комплекса в гидроксиде натрия (калия).
Подготовка проб. Для проведения исследований приготавливают
несколько образцов молока, содержащих различное количество
восстановленного молока. Для приготовления восстановленного
молока взвешивают 8 г сухого обезжиренного молока и растворяют
его в 92 см3 подогретой до 40 °C дистиллированной воды. Затем го-
товят 3 опытных образца, которые представляют собой смесь цель-
ного и восстановленного молока в соотношении 80:20, 90: 10 и
95 : 5. Исследуют полученные смеси. Контрольной пробой служит
натуральное молоко.
Выполнение работы. 25 см3 подготовленного образца молока
вносят в центрифужную пробирку, добавляют 25 см3 дистиллиро-
ванной воды и осаждают казеин. Для этого в пробирку вносят 4 см3
10%-ного раствора уксусной кислоты (реактив 1), перемешивают и
центрифугируют при 3000—4000 об/мин в течение 10 мин, центри-
фугат удаляют.
Полученный осадок промывают водой для полного удаления не-
связанной лактозы. Для этого к осадку прибавляют 25 см3 дистил-
лированной воды, тщательно растирают содержимое пробирки и
центрифугируют в течение 10 мин, центрифугат удаляют.
Промытый осадок обезжиривают последовательно ацетоном и
диэтиловым эфиром. Для этого к осадку добавляют 25 см3 ацетона,
растирают и центрифугируют в течение 5 мин, центрифугат удаля-
ют. Затем к осадку добавляют 25 см3 диэтилового эфира, тщательно
растирают осадок, смесь центрифугируют 3 мин, центрифугат уда-
ляют.
Обезжиренный осадок растворяют в 3%-ном растворе гидро-
ксида натрия или калия (реактив 2). Последний берут из расчета
4 см3 на 1 г осадка. Отбирают в пробирку 1 см3 полученного ра-
створа, нагревают на водяной бане при 85 °C в течение 5 мин и на-
блюдают изменение окраски раствора. Желто-оранжевая окраска
раствора свидетельствует о наличии в исследуемой пробе примеси
восстановленного молока, зеленовато-желтая — о натуральности
молока.
Приготовление реактивов. Реактив 1 (10%-ный раствор уксусной
кислоты). 10 см3 ледяной уксусной кислоты доводят до 100 см3 дис-
тиллированной водой.
328
Реактив 2 (3%-ныйраствор гидроксида натрия или калия). 3 г гид-
роксида натрия или калия растворяют в 97 см3 дистиллированной
воды, свободной от диомида углерода.
ОПРЕДЕЛЕНИЕ ПРИМЕСИ СОДЫ
(КАРБОНАТА ИЛИ БИКАРБОНАТА НАТРИЯ)
КАЧЕСТВЕННЫЙ МЕТОД
Метод основан на изменении окраски раствора индикатора
бромтимолового синего при добавлении его в молоко, содержащее
соду, карбонат или бикарбонат натрия.
Выполнение работы. В сухую или ополоснутую дистиллирован-
ной водой пробирку, помещенную в штатив, наливают 5 см3 испы-
туемого молока и осторожно по стенке добавляют 7—8 капель ра-
створа бромтимолового синего (реактив 1). Через 10 мин наблюда-
ют за изменением окраски кольцевого слоя, не допуская встряхива-
ния пробирки.
Одновременно ставят контрольную пробу с молоком, не содер-
жащим соды. Желтая окраска кольцевого слоя указывает на отсут-
ствие соды в молоке. Появление зеленой окраски различных оттен-
ков (от светло-зеленого до темно-зеленого) свидетельствует о при-
сутствии соды в молоке.
Приготовление реактива. Навеску бромтимолового синего мас-
сой 0,1 г переносят в мерную колбу вместимостью 250 см3 и долива-
ют до метки этиловым спиртом.
КОЛИЧЕСТВЕННЫЙ МЕТОД
Метод основан на озолении молока и определении щелочности
золы путем титрования.
Навеску молока массой 10 г помещают в предварительно промы-
тый соляной кислотой, водой и прокаленный до постоянной массы
тигель. Тигель с молоком помещают на водяную баню и выпарива-
ют. Затем проводят обугливание на электроплите и озоление навес-
ки до золы белого цвета в муфельной печи при температуре
500 ±50 °C. Не допускается обработка золы кислотами или перокси-
дом водорода.
После окончания озоления в тигель приливают 10 см3 раствора
соляной кислоты концентрацией 0,1 моль/дм3 и переносят раствор
количественно в коническую колбу вместимостью 150 см3. Тигель
ополаскивают дважды прокипяченной водой (25 см3), сливая ее в ту
же колбу. Содержимое колбы нагревают на электроплитке до слабо-
го кипения и кипятят в течение 1 мин.
Раствор охлаждают до температуры 20 ± 2 °C, добавляют 2—3
капли раствора фенолфталеина на 3—5 капель нейтрализованного
329
насыщенного раствора хлорида кальция. Затем титруют раствором
гидроксида натрия концентрацией 0,1 моль/дм3 до получения розо-
вого окрашивания, устойчивого в течение 1 мин.
Массовую долю соды X (%) в пересчете на карбонат натрия вы-
числяют по формуле
(И-К2АГ)-0,0106100 0 025
т ’ ’
где Ц — объем добавленного раствора соляной кислоты концентрацией 0,1 моль/
дм3, см3; И2 — объем раствора гидроксида натрия концентрацией 0,1 моль/дм3, см3;
К— коэффициент поправки к концентрации на раствор гидроксида натрия;
0,0106 — коэффициент пересчета на массовую долю карбоната натрия; т — масса
навески молока, г; 0,025 — массовая доля соды в натуральном коровьем молоке, %.
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений, допускае-
мые расхождения между которыми не должны превышать 0,01 %.
Суммарная погрешность измерений массовой доли соды в молоке
Д = ± 0,008 %.
ОПРЕДЕЛЕНИЕ НАЛИЧИЯ АММИАКА
Метод основан на изменении цвета выделенной молочной сыво-
ротки при ее взаимодействии с реактивом Несслера.
Минимальное значение определяемой массовой доли аммиака
составляет 0,006—0,009 %.
В стакан отмеривают цилиндром 20 ± 2 см3 молока и нагревают в
течение 2—3 мин на водяной бане при температуре 40—45 °C. В по-
догретое молоко вносят 1 см3 водного раствора с объемной долей
уксусной кислоты 10 %. Смесь оставляют в покое на 10 мин для
осаждения казеина.
Пипеткой (с ваткой на нижнем конце для предотвращения попа-
дания казеина) отбирают 2 см3 отстоявшейся сыворотки и перено-
сят в пробирку. В ту же пробирку прибором для отмеривания жид-
костей или пипеткой с резиновой грушей добавляют 1 см3 реактива
Несслера и содержимое сразу же перемешивают, наблюдая при
этом в течение не более 1 мин изменение окраски смеси.
Появление лимонно-желтой окраски смеси указывает на при-
сутствие аммиака в количестве, характерном для молока. Появле-
ние оранжевой окраски различной интенсивности указывает на на-
личие аммиака в молоке выше его естественного содержания.
ОПРЕДЕЛЕНИЕ НАЛИЧИЯ ПЕРОКСИДА ВОДОРОДА
Метод основан на взаимодействии пероксида водорода с йодом
калия, выделении йода, дающего с крахмалом синее окрашивание.
Чувствительность метода составляет 0,001 % пероксида водорода.
330
Выполнение работы. В пробирку помещают 1 см3 исследуемого
молока, прибавляют, не перемешивая, две капли раствора серной
кислоты (реактив 1) и 0,2 см3 крахмального раствора йодида калия
(реактив 2). Через 10 мин наблюдают за изменением цвета раствора
в пробирке, помещенной в штатив, не допуская ее встряхивания.
Появление в пробирке отдельных пятен синего цвета свидетель-
ствует о присутствии пероксида водорода в молоке.
Приготовление реактивов. Реактив 1 (раствор серной кислоты).
Цилиндром отмеривают одну объемную часть серной кислоты и
смешивают ее в стакане с тремя объемными частями воды.
Реактив 2 (крахмальный раствор йодида калия). Навеску крах-
мала массой 3 г растворяют в стакане в 20 см3 воды и приливают в
Колбу к 80 см3 кипящей воды. После охлаждения до температуры
18—25 °C к крахмальному раствору добавляют навеску йодида ка-
лия массой 3 г, растворенную в 5—10 см3 дистиллированной
воды. Раствор хранят в холодильнике не более 5 сут. Перед про-
ведением анализа раствор проверяют с использованием кипяче-
ного молока.
ОПРЕДЕЛЕНИЕ ПРИМЕСИ МАСТИТНОГО МОЛОКА
Молоко от животных, больных маститом, определяют кос-
венным методом на основании изменений состава и свойств
молока.
Так, молоко коров, больных маститом, имеет пониженную кис-
лотность (до 6—10 °Т), на этом основана проба молока на мастит с
индикатором бромтимоловым синим. К 0,5 см3 молока в фарфоро-
вой чашке прибавляют 5 капель 0,2%-го спиртового раствора (в
60%-ном этаноле) бромтимолового синего. Молоко от здоровых
животных дает желто-зеленую окраску, от больных — изменяется
от сине-зеленой до темно-синей.
В 1 см3 нормального молока содержится менее 500 тыс. сомати-
ческих клеток (лейкоцитов и клеток тканей вымени), при мастите
их число возрастает в 20—100 раз. Контроль маститного молока
обычно проводят по числу соматических клеток, которое определя-
ют косвенным методом, а именно, вискозиметрическим. Метод ос-
нован на том, что при добавлении к молоку поверхностно-актив-
ных веществ, например препарата «Мастоприм», последний взаи-
модействует с соматическими клетками, при этом вязкость смеси
повышается. Чем больше соматических клеток в молоке, тем боль-
ше вязкость.
Увеличение вязкости определяют визуально по консистенции
сгустка смеси молока с препаратом «Мастоприм» (визуальный ме-
тод), а также на вискозиметре: по времени истечения смеси через
капиллярное отверстие (условная вязкость).
331
ОПРЕДЕЛЕНИЕ ЧИСЛА СОМАТИЧЕСКИХ КЛЕТОК
ВИЗУАЛЬНЫМ МЕТОДОМ
В лунку пластинки ПМК-1 вносят 1 см3 тщательно перемешан-
ного молока и добавляют 1 см3 водного раствора препарата «Мас-
топрим». Молоко с препаратом интенсивно перемешивают дере-
вянной, пластмассовой или стеклянной палочкой в течение 10 с.
Полученную смесь из лунки при непрерывным интенсивном пере-
мешивании поднимают палочкой вверх на 50—70 мм, после чего в
течение не более 60 с оценивают результаты анализа.
Число соматических клеток в исследуемом молоке устанавлива-
ют по консистенции молока в соответствии с требованиями
табл. 45.
45. Характеристика молока по числу соматических клеток
Характеристика консистенции молока Число соматических клеток в 1 см3 молока Сорт молока
Однородная жидкость или слабый сгусток, который слегка тянется за палочкой в виде нити До 500 тыс. Высший
Выраженный сгусток, при перемешивании которого хорошо видна выемка на дне лун- ки пластинки. Сгусток не выбрасывается из лунки От 500 тыс. до 1 млн I, п
Плотный сгусток, который выбрасывается палочкой из лунки пластинки Свыше 1 млн Несортовое
Приготовление реактива. Для получения водного раствора препа-
рата «Мастоприм» 2,5 г препарата вносят в мерную колбу или ци-
линдр вместимостью 100 см3 и доливают до метки дистиллирован-
ной водой (или питьевой свежекипяченой водой), нагретой до тем-
пературы 30—35 °C. Раствор перед применением взбалтывают до
равномерного распределения осадка.
Срок годности раствора — 1 сут при температуре хранения 10—30 °C.
ОПРЕДЕЛЕНИЕ ЧИСЛА СОМАТИЧЕСКИХ КЛЕТОК
НА ИНДИКАТОРЕ ИСКМ-1
Индикатор соматических клеток молока ИСКМ-1 состоит из
двух блоков: исполнительного и измерительного (рис. 67).
Исполнительный блок представляет собой капиллярное устрой-
ство, включающее: воронку, капилляр с нормированным отверсти-
ем, перекрываемый механическим клапаном с помощью ручки, и
стакан для сбора вытекающей из воронки смеси.
Измерительный блок конструктивно представляет собой метал-
лический корпус с расположенными на его лицевой и задней пане-
332
4
5
Рис. 67. Индикатор соматических клеток молока ИСКМ-1:
1 — стакан; 2 — механический клапан; 3 — капилляр; 4 — воронка; 5 — кнопка пуска; 6 — ручка
для закрытия клапана; 7 — кнопка включения прибора в сеть; 8 — цифровой индикатор; 9 — све-
тодиоды для фиксации переключения показаний; 10 — кнопка переключений показаний
лях элементами индикации и управления. На лицевой панели на-
ходятся кнопка включения прибора в сеть, кнопка переключения
показаний (время/клетки), светодиоды для фиксации переключе-
ния показаний и цифровой индикатор.
Пробу молока смешивают с «Мастопримом» вручную в специ-
альной пробирке, затем смесь сливают в воронку с капилляром и
ручным клапаном. Время истечения определяют с помощью элект-
ронного блока индикации. Датчик фиксации времени истечения
пробы работает на И К-излучении.
Выполнение работы. В специальную пробирку вносят 5 см3 3,5%-
ного водного раствора препарата «Мастоприм» и 10 см3 исследуе-
мого молока, тщательно профильтрованного через четыре слоя
марли, и закрывают пробирку пробкой. Смесь молока перемешива-
ют вручную с «Мастопримом», десятикратно переворачивая про-
бирку. Затем смесь сливают в воронку, нажимают кнопку пуска.
Определяют время истечения смеси через капилляр с помощью
электронного блока индикации.
Время истечения фиксируется на цифровом индикаторе. Время
истечения пересчитывают на число соматических клеток, нажав
кнопку для переключения показаний 10 «время/клетки». Число со-
матических клеток указано в тысячах на 1 см3 молока. Кроме того, к
прибору прилагается таблица для пересчета времени истечения
пробы на число соматических клеток (табл. 46).
333
46. Таблица для пересчета времени истечения смеси (молоко + «Мастоприм»)
иа число соматических клеток
Показание вис- козиметра, с Число клеток в 1 см’ смеси, тыс. Показание вис- козиметра, с Число клеток в 1 см3 смеси, тыс. Показание вис- козиметра, с Число клеток в 1 см5 смеси, тыс.
12,0 90 15,8 223 19,6 346
12,2 97 16,0 230 19,8 351
12,4 104 16,2 237 20,0 357
12,6 111 16,4 244 20,2 363
12,8 118 16,6 251 20,4 368
13,0 125 16,8 258 20,6 374
13,2 132 17,0 265 20,8 380
13,4 139 17,2 272 21,0 383
13,6 146 17,4 279 21,2 391
13,8 153 17,6 286 21,4 396
14,0 160 17,8 293 21,6 403
14,2 167 18,0 300 21,8 408
14,4 174 18,2 306 22,0 414
14,6 181 18,4 311 22,2 420
14,8 188 18,6 317 22,4 425
15,0 195 18,8 323 22,6 431
15,2 202 19,0 329 22,8 437
15,4 209 19,2 334 23,0 443
15,6 216 19,4 340 23,2 448
23,4 454 29,2 675 35,0 917
23,6 460 29,4 683 35,2 925
23,8 465 29,6 692 35,4 933
24,0 471 29,8 700 35,6 942
24,2 477 30,0 708 35,8 950
24,4 482 30,2 717 36,0 958
24,6 488 30,4 725 36,2 967
24,8 494 30,6 733 36,4 975
25,0 500 30,8 742 36,6 983
25,2 508 31,0 750 36,8 992
25,4 517 31,2 758 37,0 1000
25,6 525 31,4 767 37,2 1006
25,8 533 31,6 775 37,4 1011
26,0 542 31,8 783 37,6 1017
26,2 550 32,0 792 37,8 1022
26,4 558 32,2 800 38,0 1028
26,6 567 32,4 808 38,2 1033
26,8 575 32,6 817 38,4 1039
27,0 583 32,8 825 38,6 1044
27,2 592 33,0 833 38,8 1050
27,4 600 33,2 842 39,0 1056
27,6 608 33,4 850 39,2 1061
27,8 617 33,6 858 39,4 1067
28,0 625 33,8 867 39,6 1072
28,2 633 34,0 875 39,8 1078
28,4 642 34,2 883 40,0 1083
28,6 650 34,4 892 40,2 1089
28,8 658 34,6 900 40,4 1094
29,0 667 34,8 909 40,6 1100
40,8 1105 46,6 1263 52,4 1383
41,0 1111 46,8 1267 52,6 1388
41,2 1117 47,0 1271 52,8 1392
41,4 1122 47,2 1275 53,0 1396
334
Продолжение
Показание вис- козиметра, с Число клеток в 1 сМ3 смеси, тыс. Показание вис- козиметра, с Число клеток в 1 см3 смеси, тыс. Показание вис- козиметра, с Число клеток в 1 см3 смеси, тыс.
41,6 1128 47,4 1279 53,2 1400
41,8 1133 47,6 1283 53,4 1404
42,0 1139 47,8 1288 53,6 1409
42,2 1144 48,0 1292 53,8 1413
42,4 1150 48,2 1296 54,0 1417
42,6 1155 48,4 1300 54,2 1421
42,8 1161 48,6 1304 54,4 1425
43,0 1167 48,8 1308 54,6 1429
43,2 1172 49,0 1313 54,8 1434
43,4 1178 49,2 1317 55,0 1438
43,6 1183 49,4 1321 55,2 1442
43,8 1189 49,6 1325 55,4 1446
44,0 1194 49,8 1329 55,6 1450
44,2 1200 50,0 1333 55,8 1454
44,4 1205 50,2 1338 56,0 1459
44,6 1211 50,4 1342 56,2 1463
44,8 1216 50,6 1346 56,4 1467
45,0 1222 50,8 1350 56,6 1471
45,2 1228 51,0 1354 56,8 1475
45,4 1233 51,2 1358 57,0 1479
45,6 1239 51,4 1363 57,2 1484
45,8 1244 51,6 1367 57,4 1488
46,0 1250 51,8 1371 57,6 1492
46,2 1254 52,0 1375 57,8 1496
46,4 1258 52,2 1379 58,0 1500
За окончательный результат анализа принимают среднее ариф-
метическое результатов двух параллельных определений.
Во время исследования температура помещения, в котором про-
водят исследования, должна быть 10—30 °C.
После проведения анализа смеси для каждой исследуемой пробы
молока воронку и капилляр 2—3 раза промыть дистиллированной во-
дой и 4—5 раз продуть с помощью резиновой груши. После очистки
сосуда прибор считается подготовленным для дальнейших анализов.
Приготовление реактива. 3,5 г препарата «Мастоприм» вносят в мер-
ную колбу или мерный цилиндр вместимостью 100 см3 и доливают до
метки дистиллированной водой, подогретой до 30—35 °C. Раствор пе-
ред применением взбалтывают до равномерного распределейия осадка.
Срок годности раствора — 1 сут при температуре хранения 10—30 °C.
ОПРЕДЕЛЕНИЕ ЭФФЕКТИВНОСТИ ГОМОГЕНИЗАЦИИ
МЕТОД ОТСТАИВАНИЯ ЖИРА
Сущность метода состоит в определении процентного отноше-
ния нестабильной жировой фазы к общему количеству жировой
фазы в эмульсии после выдерживания ее определенное время при
установленных условиях.
335
Жировые молочные эмульсии стандартизируют по жиру до 2,5—
3,0 % обезжиренным молоком или дистиллированной водой.
Подготовленную пробу жировой молочной эмульсии наливают в
цилиндры вместимостью 250 см3, закрывают фольгой и выдержива-
ют 24 или 48 ч (в зависимости от стабильности исследуемой пробы)
при температуре 6—8 °C. Эмульсии более высокой стабильности,
например гомогенизированное молоко, выдерживают 48 ч. После
истечения времени выдерживания образцов содержимое цилиндра
разделяют на два слоя: верхний и нижний. Для этого применяют
пипетку вместимостью 2 см3 с надетой резиновой трубкой. Пипетку
погружают в цилиндр, зажимая пальцами трубку, чтобы часть верх-
него слоя эмульсии не попадала в пипетку, и отбирают сифонным
способом нижний слой эмульсии объемом 150 см3, т. е. 60 % общего
объема. Стабильность жировой молочной эмульсии (отстаивание
жира) С (%) вычисляют по формуле
•100,
где Ж3 — массовая доля жира в эмульсии, %; Жл — массовая доля жира в нижнем
(менее жирном) слое эмульсии, %; Ц, — объем нижнего (менее жирного) слоя
эмульсии, см3; Йо6щ — общий объем пробы, размещенной в цилиндре, см3.
При необходимости массовую долю жира в верхнем слое жиро-
вой молочной отстоявшейся эмульсии определяют расчетным пу-
тем по формуле
угу
в ’
где Жв — массовая доля жира в верхнем слое жировой молочной отстоявшейся
эмульсии, %; Ив — объем верхнего (более жирного) слоя эмульсии, см3.
Жировая молочная эмульсия обладает высокой стабильностью,
если процент отстаивания жира в ней после 48-часового выдержи-
вания не превышает 10.
МЕТОД ЦЕНТРИФУГИРОВАНИЯ ЖИРА
Сущность метода состоит в определении объема выделившейся в
жиромере недостаточно диспергированной жировой фазы при ус-
тановленном режиме центрифугирования.
Выполнение работы. Молоко до и после гомогенизации с массо-
вой долей жира, превышающей 3 %, нормализуют обезжиренным
молоком или дистиллированной водой по жиру до 2,5 %.
Сливки до и после гомогенизации нормализуют обезжиренным
молоком или дистиллированной водой по жиру до 2,5 %.
336
Стерилизованное концентрированное молоко, сгущенное цель-
ное молоко с сахаром, сухое цельное молоко, смеси для мороженого
нормализуют дистиллированной водой по жиру до 2,5 %.
Подготовленные образцы молока и молочных продуктов разводят
рабочим раствором (реактив 1) в соотношении 1 : 3. После тщатель-
ного перемешивания отбирают 20 см3 полученной смеси и вносят в
молочный жиромер. Оставшуюся часть жиромера заполняют дис-
тиллированной водой в количестве 1,0—1,5 см3, т. е. наливают
столько, чтобы выделившийся слой недостаточно диспергированной
жировой фазы находился в проградуированной части жиромера. Жи-
ромеры перевертывают, ставят в водяную баню, выдерживают 10 мин
при температуре 65 °C, после чего центрифугируют в течение 5 мин.
Сразу после остановки центрифуги (без применения тормоза)
жиромеры вынимают и, не переворачивая, отсчитывают по шкале
объем недостаточно диспергированной жировой фазы. На каждую
пробу исследуемого образца делают по два параллельных определе-
ния. Находят среднее арифметическое показание жиромеров. Рас-
хождение между параллельными определениями не должно превы-
шать одного малого деления, т. е. 0,1 % по жиромеру.
Рассчитывают эффективность диспергирования жира Э (%) по
следующей формуле:
э=Г1-А1.юо,
где Nt — объем выделившейся в жиромере недостаточно диспергированной жиро-
вой фазы для гомогенизированного молока, деления шкалы молочного жиромера;
No — объем выделившейся в жиромере жировой фазы для негомогенизированного
молока, деления шкалы молочного жиромера.
В случае отсутствия возможности определения объема выделив-
шейся в жиромере жировой фазы в негомогенизированном молоке
показатель No определяют из следующего соотношения:
No = 6ЖМ,
где Жм — массовая доля жира в исследуемой нормализованной пробе образца.
Установленный показатель эффективности диспергирования
жира в смесях для мороженого с массовой долей жира до 10 % умно-
жают на коэффициент 2, а в смесях для мороженого с массовой до-
лей жира, превышающей 10 %, умножают на коэффициент 1,5.
Зависимость качества питьевого пастеризованного молока по
степени дисперсности жировой фазы от показателя эффективности
диспергирования жира приведена ниже.
Эффективность дисперги- Не менее 60 50—60 40—45 Менее 40
рования жира в молоке, %
Качество питьевого пасте- Отличное Хорошее Удовлетво- Плохое
ризованного молока рительное
337
Эффективность диспергирования жира в продуктах с разной
массовой долей жира, с учетом результатов производственной про-
верки, будет следующей (%): пастеризованного коровьего моло-
ка — 50; сливок с массовой долей жира 8 и 10 % — 45, 20 % — 40,
35 % — 30; сливок для производства сметаны с массовой долей жира
20 % - 40,25 и 30 % - 35.
Разработанный метод является простым, нетрудоемким, не тре-
бует дополнительного оборудования (приборов) и дефицитных ре-
активов. Экспресс-метод можно использовать на всех молочных за-
водах. Время проведения анализа — 15 мин. Безвозвратные потери
нормализованных молока и молочных продуктов составляют не бо-
лее 15 см3 на полный анализ одного образца.
Приготовление реактива. Вскрывают одну упаковку сухого пре-
парата для определения эффективности диспергирования жира.
Содержимое без потерь высыпают в мерную колбу вместимостью
1000 см3, добавляют дистиллированную воду до метки, закрывают
притертой пробкой и хорошо перемешивают. Полученный раствор
(рабочий раствор) применяют для разведения молока и молочных
продуктов при оценке их качества по степени дисперсности жиро-
вой фазы.
ОПРЕДЕЛЕНИЕ ЭФФЕКТИВНОСТИ ПАСТЕРИЗАЦИИ
В молочной промышленности для определения эффективности
пастеризации молока, сливок, пахты, сыворотки, а также сырья, ис-
пользованного при выработке творога, сметаны, сливочного масла,
кисломолочных напитков и других молочных продуктов, применя-
ют пробы на пероксидазу и фосфатазу.
ПРОБА НА ПЕРОКСИДАЗУ
С ХЛОРИДОМ ПАРАФЕНИЛЕНДИАМИНА
Метод основан на разложении пероксида водорода ферментом
пероксидазой, содержащейся в молоке и молочных продуктах. Ос-
вобождающийся при разложении пероксида водорода активный
кислород окисляет парафенилендиамин, образуя соединение сине-
го цвета.
Выполнение работы. В пробирку отмеривают или отвешивают
анализируемый продукт: пастеризованное молоко, кисломолочные
напитки и напитки с наполнителями — 5 см3, сливки, сметану, тво-
рог, творожные изделия и пасты, сливочное масло — 2—3 см3 (г).
Затем добавляют 2—3 см3 дистиллированной воды.
Кисломолочные напитки с плодово-ягодными наполнителями
фильтруют через бумажный фильтр.
338
Для получения 2—3 см3 плазмы масла 50 г сливочного масла рас-
плавляют при температуре не выше 50 °C, затем охлаждают и зас-
тывший слой жира удаляют.
После добавления воды анализируемые продукты тщательно ра-
стирают стеклянной палочкой, затем приливают 2,5 см3 буферной
смеси (реактив 1), тщательно перемешивают стеклянной палочкой
и помещают в водяную баню с температурой воды 35 ± 2 °C, где вы-
держивают 3—5 мин, чтобы содержимое пробирки довести до этой
температуры. Затем добавляют 6 капель 0,5%-ного раствора перок-
сида водорода (реактив 2) и 3 капли раствора хлорида парафенилен-
диамина (реактив 3), перемешивают вращательными движениями
содержимое пробирки после добавления каждого реактива. Снова
помещают пробирку в водяную баню и наблюдают изменение ок-
раски жидкости.
При отсутствии фермента пероксидазы в молоке и молочных
продуктах цвет содержимого пробирки не меняется. Следователь-
но, молоко и молочные продукты подвергались пастеризации при
температуре не ниже 80 °C.
При наличии пероксидазы в молоке, сливках, сливочном масле
содержимое пробирок окрашивается в темно-синий цвет. При нали-
чии пероксидазы в кисломолочных продуктах и кислосливочном
масле содержимое пробирок приобретает серо-фиолетовую окраску,
постепенно переходящую в темно-синюю. Следовательно, молоко и
молочные продукты или не были пастеризованы, или подвергались
пастеризации при температуре ниже 80 °C, или были смешаны с не-
пастеризованными продуктами. Чувствительность метода позволяет
обнаружить добавление не менее 5 % непастеризованных молочных
продуктов к пастеризованным.
Приготовление реактива. Реактив 1 (буферная смесь). 97 г дифос-
фата натрия и 0,65 г лимонной кислоты, взвешенных с погрешнос-
тью не более 0,0002 г, растворяют дистиллированной водой в мер-
ной колбе вместимостью 500 см3, доливают до метки и перемешива-
ют. Буферную смесь следует сохранять в склянке из темного стекла
с плотно закрытой пробкой.
Реактив 2 (0,5%-ный раствор пероксида водорода). Имеющийся
концентрированный раствор в зависимости от содержания в нем
пероксида водорода разводят водой, предварительно прокипячен-
ной и охлажденной. Раствор быстро разлагается, поэтому его гото-
вят в небольшом количестве.
Растворы следует хранить в склянке из темного стекла в прохлад-
ном месте.
Для определения содержания пероксида водорода в концентри-
рованном растворе 3—4 г концентрированного пероксида водоро-
да, взвешенного с погрешностью не более 0,01 г, переносят без по-
терь в мерную колбу вместимостью 250 см3, доводят водой до метки
и перемешивают. 10 см3 приготовленного раствора переносят в ко-
ническую колбу вместимостью 250 см3, добавляют 50 см3 воды,
339
10 см3 разбавленной серной кислоты (1: 4) и содержимое титруют
0,1 н. раствором перманганата калия до розового окрашивания, не
исчезающего в течение 1 мин. Параллельно проводят контрольный
опыт в тех же условиях, с тем же количеством реактивов и воды (без
пероксида водорода).
Массовую долю пероксида водорода Х(%) вычисляют по формуле
(И-И]) 0,0017 25-100
Х---------т ’
где V— объем 0,1 н. раствора перманганата калия, израсходованного на титрование
раствора пероксида водорода, см3; И| — объем 0,1 н. раствора перманганата калия,
израсходованного на контрольное титрование, см3; 0,0017 — масса пероксида водо-
рода, соответствующая 1 см3 0,1 н. раствора перманганата калия, г; т — навеска пе-
роксида водорода, г.
Проверка пригодности реактива 3 (2%-ного раствора соляно-
кислого парафенилендиамина). Для проверки раствора, хранивше-
гося более 1—2 дней в пробирке кипятят 5 см3 молока, охлажда-
ют, приливают 2,5 см3 буферной смеси, перемешивают и поме-
щают на 3—5 мин в водяную баню с температурой воды 35 ± 2 °C.
Затем прибавляют 6 капель 0,5%-ного раствора пероксида водо-
рода и 3 капли раствора хлорида парафенилендиамина, переме-
шивают и снова помещают в водяную баню. Появление темно-
синей или серовато-синей окраски указывает на непригодность
раствора.
ПРОБА НА ПЕРОКСИДАЗУ
С ЙОДИСТОКАЛИЕВЫМ КРАХМАЛОМ
Метод основан на разложении пероксида водорода ферментом
пероксидазой. Освобождающийся при разложении пероксида во-
дорода активный кислород окисляет йодид калия, освобождая йод,
образующий с крахмалом соединение синего цвета.
Выполнение работы. Отмеривание или взвешивание анализи-
руемых продуктов и воды, подготовку плазмы масла проводят,
как указано в пробе на пероксидазу, с хлоридом парафениленди-
амина.
В пробирку с указанным количеством продукта и воды прилива-
ют 5 капель раствора йодистокалиевого крахмала (реактив 1) и 5 ка-
пель 0,5%-ного раствора пероксида водорода (реактив 2), враща-
тельными движениями перемешивают содержимое пробирки после
добавления каждого реактива. Затем определяют наличие перокси-
дазы по изменению окраски.
Если применяют отдельно раствор крахмала и йодида калия, то
поступают следующим образом: в каждую пробирку с продуктами
приливают 0,5 см3 1%-ного раствора крахмала, 2 капли 10%-ного
340
раствора йодида калия и 5 капель 0,5%-ного раствора пероксида во-
дорода, перемешивают содержимое пробирок после добавления
каждого реактива, затем определяют наличие пероксидазы по изме-
нению окраски.
При отсутствии пероксидазы в молоке и молочных продуктах цвет
содержимого пробирки не изменится. Следовательно, молоко и мо-
лочные продукты пастеризовали при температуре не ниже 80 °C.
При наличии пероксидазы в молоке, сливках, сливочном масле
содержимое пробирок приобретает темно-синюю окраску. При
наличии пероксидазы в кисломолочных продуктах и кислосли-
вочном масле содержимое пробирок не более чем через 2 мин при-
обретает серовато-синюю окраску, постепенно переходящую в
темно-синюю. Следовательно, продукты или не пастеризовали,
или пастеризовали при температуре ниже 80 °C, или смешивали с
непастеризованными молочными продуктами. Появление окрас-
ки в пробирках более чем через 2 мин после добавления йодисто-
калиевого крахмала и пероксида водорода не указывает на отсут-
ствие пастеризации, так как может вызываться разложением реак-
тивов.
Чувствительность метода позволяет обнаружить добавление
не менее 5 % непастеризованных молочных продуктов к пастери-
зованным, а для напитков с плодово-ягодными наполнителя-
ми — 0,5 %.
Приготовление реактивов. Реактив 1 (раствор йодистокалиевого
крахмала). 3 г крахмала взвешивают с погрешностью не более 0,01 г
и смешивают с 5—10 см3 дистиллированной холодной воды до по-
лучения однородной массы. Отдельно в колбе доводят до кипения
100 см3 дистиллированной воды и при непрерывном помешивании
приливают воду к разведенному крахмалу, не допуская образования
комков. Полученный раствор доводят до кипения. После охлажде-
ния к раствору крахмала прибавляют 3 г йодида калия, перемеши-
вая до растворения кристаллов соли.
Раствор йодистокалиевого крахмала является нестойким реак-
тивом, поэтому приготовлять его следует в небольшом количестве и
сохранять в темном прохладном месте не более 2 дней.
Для определения пригодности раствора йодистокалиевого крах-
мала, хранившегося более 2 дней, перед употреблением его необхо-
димо проверить. Для этого в пробирке нужно вскипятить 5 см3 моло-
ка, охладить, прилить 5 капель раствора йодистокалиевого крахмала
и 5 капель 0,5%-ного раствора пероксида водорода и перемешать.
Появление темно-синей или серовато-синей окраски указывает на
непригодность раствора.
Допускается вместо йодистокалиевого крахмала применять от-
дельно приготовленные 1%-ный раствор крахмала и 10%-ный ра-
створ йодида калия.
Реактив 2 приготавливают, как указано в пробе на пероксидазу,
с солянокислым парафенилендиамином.
341
ПРОБА НА ФОСФАТАЗУ ПО РЕАКЦИИ
С 4-АМИНОАНТИПИРИНОМ (АРБИТРАЖНЫЙ МЕТОД)
Основана на гидролизе динатриевой соли фенилфосфорной
кислоты ферментом фосфатазой, содержащейся в молоке и молоч-
ных продуктах. Выделившийся при гидролизе свободный фенол в
присутствии окислителя дает розовое окрашивание с 4-аминоанти-
пирином.
Выполнение работы. Для проведения анализа пастеризованного
молока, сливок, кисломолочных напитков, простокваши дополни-
тельной подготовки не требуется. Творог и сметану необходимо
развести дистиллированной водой. Для этого 1 г творога и сметаны
помещают в пробирку и тщательно перемешивают с 2 см3 дистил-
лированной воды.
Кисломолочные напитки с плодово-ягодными наполнителями
необходимо профильтровать через бумажный фильтр с активным
углем (2 г активного угля на 25 см3 продукта). Сыворотку и фильтра-
ты кисломолочных напитков с плодово-ягодными наполнителями
следует нейтрализовать 1 н. раствором гидроксида натрия до pH 6,
контролируя по универсальной индикаторной бумажке.
К 3 см3 молока, сливок, кефира, простокваши, сыворотки или
предварительно подготовленных для анализа продуктов добавляют
2 см3 рабочего раствора субстрата (реактив 1). Затем перемешивают
содержимое пробирки и ставят в водяную баню, нагретую до 40—
45 °C на 30 мин. В пробирку, вынутую из водяной бани, добавляют
5 см3 осадителя системы цинк—медь (реактив 2), тщательно пере-
мешивают содержимое пробирки и снова ставят в водяную баню с
температурой 40—45 °C на 10 мин. Вынув пробирку из бани, произ-
водят визуальное сравнение содержимого пробирки испытуемого
продукта с контрольным опытом.
Контрольным опытом для всех продуктов является аналогичная
реакция с кипяченым молоком. Если контрольный опыт с кипяче-
ным молоком дает слабо-розовое окрашивание, динатрийфенил-
фосфат подлежит дополнительной очистке.
При отсутствии фермента фосфатазы в молоке и молочных про-
дуктах окраска содержимого пробирки (раствора, отделившегося от
осажденного белка) аналогична содержимому пробирок конт-
рольного опыта. Следовательно, молоко и молочные продукты пас-
теризовали при температуре не ниже 63 °C.
При наличии фосфатазы в молоке и молочных продуктах содер-
жимое пробирок (растворы) окрашено от розового до темно-крас-
ного цвета. Следовательно, молоко и молочные продукты не под-
вергались пастеризации или подвергались пастеризации при тем-
пературе ниже 63 °C или были смешаны с непастеризованными мо-
лочными продуктами.
При оценке реакции учитывают только цвет, но не прозрачность
раствора. Чувствительность метода позволяет обнаружить добавле-
342
ние непастеризованных молочных продуктов к пастеризованным:
0,3 % в молоке, сливках, кисломолочных напитках, 0,5 % в твороге
и сметане и 1 % в напитках с плодово-ягодными наполнителями и
сыворотке.
Творог и сметану на пастеризацию исходного сырья — молока и
сливок определяют по фосфатазе не позднее 7 сут с момента выра-
ботки творога, сметаны — не позднее 5 сут. Продукты из молочной
сыворотки проверяют путем контроля исходного сырья (молока,
сливок, творога, сыворотки).
Приготовление реактивов. Реактив 1 (рабочий раствор субстра-
та). Готовят непосредственно перед определением реакции сме-
шиванием раствора Аи Б (1 : 9). Рабочий раствор пригоден для ра-
боты в течение 8 ч при хранении его в склянке из темного стекла.
1. Приготовление раствора А — 1,25 г динатриевой соли фенил-
фосфорной кислоты взвешивают с погрешностью не более 0,0002 г,
растворяют в 100 см3 основного буферного раствора (pH 10 ± 0,2),
который приготовляют следующим образом. 40 г хлорида аммония,
взвешенного с погрешностью не более 0,01 г, растворяют в 100—
200 см3 дистиллированной воды, добавляют 348 см3 25%-ного вод-
ного аммиака и доводят до 1000 см3 дистиллированной водой. При
необходимости динатриевую соль фенилфосфорной кислоты очи-
щают от свободного фенола промыванием этиловым эфиром до
полного удаления фенола. Сушить реактив при комнатной темпе-
ратуре под тягой.
2. Приготовление раствора Б — 0,8 г 4-аминоантипирина взве-
шивают с погрешностью не более 0,0002 г, растворяют в 900 см3 ди-
стиллированной воды.
Растворы А и Б должны быть бесцветными и храниться в склян-
ках из темного стекла в холодильнике. Срок хранения не более
1 мес. Пожелтевшие растворы для работы непригодны.
Реактив 2 (осадитель системы цинк—медь). 30 г сульфата цинка и
6 г сульфата меди, взвешенных с погрешностью не более 0,01 г, ра-
створяют в 1000 см3 дистиллированной воды.
ПРОБА НА ФОСФАТАЗУ ПО РЕАКЦИИ
С ФЕНОЛФТАЛЕИНФОСФАТОМ НАТРИЯ
Метод основан на гидролизе фенолфталеинфосфата натрия фер-
ментом фосфатазой. Освобождающийся при гидролизе фенолфта-
леин в щелочной среде дает розовое окрашивание.
Выполнение работы. В пробирку отмеривают 2 см3 анализируе-
мого продукта, 2 см3 дистиллированной воды (в пастеризованное
молоко воду не добавляют) и раствор фенолфталеинфосфата натрия
(реактив 1). 1 см3 — в пастеризованное молоко и сливки и 2 см3 — в
кисломолочные напитки.
343
После добавления дистиллированной воды и реактива содер-
жимое пробирки закрывают пробкой и взбалтывают. Затем про-
бирку помещают в водяную баню с температурой воды от 40 до
45 °C и определяют окраску содержимого пробирки через 10 мин и
через 1 ч.
При отсутствии фермента фосфатазы в молоке и молочных про-
дуктах окраска содержимого пробирки не изменяется. Следова-
тельно, молоко и молочные продукты подвергались пастеризации
при температуре не ниже 63 °C. При наличии фосфатазы в молоке и
молочных продуктах содержимое пробирки приобретает окраску от
светло-розовой до ярко-розовой. Следовательно, молоко и молоч-
ные продукты не подвергались пастеризации или подвергались пас-
теризации при температуре ниже 63 °C, или были смешаны с непас-
теризованными продуктами.
Чувствительность метода позволяет обнаружить добавление не
менее 2 % непастеризованных молочных продуктов к пастеризо-
ванным.
Приготовление реактива. Для приготовления 0,1 %-ного раствора
фенолфталеинфосфата натрия 0,1 г порошкообразного фенолфта-
леинфосфата натрия, взвешенного с погрешностью не более
0,0002 г, растворяют в мерной колбе вместимостью 100 см3 с не-
большим количеством аммиачной буферной смеси, затем доливают
буферную смесь до метки и перемешивают.
Аммиачную буферную смесь готовят смешиванием 80 см3 1 М
раствора аммиака с 20 см31 М раствора хлорида аммония (pH 9,8).
МЕТОД ОПРЕДЕЛЕНИЯ КИСЛОЙ ФОСФАТАЗЫ
Кислая фосфатаза инактивируется при температуре пастериза-
ции молока и сливок 85 °C и выдержке не менее 30 мин, 90 °C и вы-
держке не менее 5 мин и кипячении; при температуре термической
обработки сливок 103 °C и выдержке 15—20 с.
Метод основан на свойстве кислой фосфатазы катализировать
при pH 4,00 ± 0,05 гидролиз динатриевой соли фенилфосфорной
кислоты с образованием фенола и фосфата натрия. Фенол с 4-ами-
ноантипирином при добавлении осадителя системы цинк—медь в
условиях щелочной реакции образует окрашенное соединение, из-
меняющее интенсивность окраски от слабо-розовой до темно-виш-
невой (в зависимости от концентрации фенола). По разнице в ин-
тенсивности окраски опытных и контрольных проб определяют ак-
тивность кислой фосфатазы (соблюдены ли режимы термической
обработки).
Выполнение работы. Проведение анализа начинают с приготов-
ления опытной пробы. Исследуемое молоко или сливки вносят по
1 см3 в 3 пробирки. В одну из пробирок с молоком или сливками до-
бавляют 10 см3 раствора динатриевой соли фенилфосфорной кис-
344
лоты и 4-аминоантипирина (реактив 1), перемешивают, помещают
в водяную баню или ультратермостат температурой 36 ± 1 °C. Че-
рез одинаковые промежутки времени, например 1 мин, вышеука-
занную операцию проводят последовательно с остальными 2 про-
бирками. Продолжительность выдерживания каждой из 3 проби-
рок в водяной бане или ультратермостате, контролируемая по се-
кундомеру, составляет 90 мин. Затем вносят из бюретки
вместимостью 25 см3 или отмеривают пипеткой по 1 см3 осадителя
системы цинк—медь (реактив 2), 0,6 см3 раствора с концентраци-
ей аммиака 3 моль/дм3 (реактив 3) через те же промежутки време-
ни, как и при смешивании с раствором соли фенил фосфорной
кислоты и 4-аминоантипирина, перемешивают после добавления
каждого реактива, фильтруют и выдерживают 60 мин при темпера-
туре 20 ± 5 °C.
Контрольную пробу готовят следующим способом: в 3 пробирки
вносят по 10 см3 раствора динатриевой соли фенилфосфорной кис-
лоты и 4-аминоантипирина (реактив 1), выдерживают, подобно
опытным пробам, при температуре 36 ± 1 °C в течение 90 мин. Про-
бирки вынимают из водяной бани или ультратермостата, добавляют
по 1 см3 осадителя системы цинк—медь (реактив 2), 1 см3 исследуе-
мых молока или сливок и 0,6 см3 раствора с концентрацией аммиа-
ка 3 моль/дм3 (реактив 3) (перемешивание выполняют после каж-
дой добавки), фильтруют и выдерживают 60 мин при температуре
20 ± 5 °C.
Во избежание попадания в содержимое пробирок постороннего
фенола пробирки типа П2 закрывают фольгой, перемешивают,
дважды переворачивая их.
При полной инактивации кислой фосфатазы в молоке или
сливках окраска опытных проб не отличается от окраски конт-
рольных проб. Следовательно, молоко и сливки подвергались пас-
теризации при температуре 85 °C с выдержкой не менее 30 мин,
90 °C с выдержкой не менее 5 мин и кипячению; сливки подверга-
лись термической обработке при температуре 103 °C с выдержкой
15-20 с.
В зависимости от активности кислой фосфатазы в молоке и
сливках окраска опытных проб изменяет цвет от слабо-розового (но
более яркого, чем окраска контрольных проб) до темно-вишневого.
Следовательно, наличие активности кислой фосфатазы свидетель-
ствует о несоблюдении режимов термической обработки.
Приготовление реактивов. Реактив 1 (раствор динатриевой или
фенилфосфорной кислоты и 4-аминоантипирина). В мерную колбу
вместимостью 100 см3 вносят 0,300 ± 0,001 г динатриевой соли фе-
нилфосфорной кислоты и 0,021 ±0,001 г 4-аминоантипирина,
80 см3 дистиллированной воды, содержимое растворяют, добавля-
ют 10 см3 буферного раствора, доводят дистиллированной водой до
метки и перемешивают. Раствор готовят непосредственно перед
проведением анализа, хранят в темном месте не более 2 ч.
345
Буферный раствор готовят следующим способом: в мерную кол-
бу вместимостью 1000 см3 вносят 72 см3 ледяной уксусной кислоты,
доводят до метки дистиллированной водой и перемешивают. Затем
подщелачивают концентрированным раствором гидроксида на-
трия до pH 3,78 ± 0,05.
Реактив 2 (осадитель системы цинк—медь). В мерную колбу вме-
стимостью 1000 см3 вносят 150,00 ±0,05 г сульфата цинка и
30,00 ± 0,05 г сульфата меди, растворяют в дистиллированной воде,
доводят объем до метки дистиллированной водой и перемешивают.
Реактив 3 (раствор аммиака концентрацией Змоль/дм3). В мер-
ной колбе вместимостью 1000 см3 разбавляют 333 см3 аммиака с
массовой долей 25 % до метки дистиллированной водой.
Раствор аммиака хранят в плотно закупоренной посуде. Перед
взятием аммиака содержимое бутылки перемешивают.
ОПРЕДЕЛЕНИЕ ДИСПЕРСНОСТИ ВЛАГИ
СЛИВОЧНОГО МАСЛА
Метод основан на изменении цвета индикатора бромфенол-
синего при соприкосновении с капельками влаги сливочного
масла.
Выполнение работы. Образец сливочного масла подогреть до
температуры 5 °C. Специальным проволочным ножом из монолита
масла вырезают брус размером 6 х 6 см и толщиной 2—3 см. На све-
жий срез пинцетом плотно прикладывают индикаторную бумажку
и выдерживают 15—30 с. Индикаторную бумагу пинцетом снимают
и опускают в обезвоженный расплавленный парафин для фиксации
образовавшихся пятен.
По количеству сине-фиолетовых точек и пятен, характеру их
распределения при сравнении с эталоном судят о распределении
влаги в сливочном масле, определяют класс дисперсности:
класс I — масло с хорошо диспергированной влагой, точки от-
сутствуют. Масло стойкое, подлежит длительному хранению;
класс II — масло с удовлетворительно диспергированной вла-
гой — на индикаторной бумаге видно несколько (3—5) равномерно
распределенных точек диаметром 0,3—1,0 мм. Масло средней стой-
кости;
класс III — масло с плохо диспергированной влагой — на инди-
каторной бумаге видно много неравномерно распределенных точек
различной величины диаметром более 1,0 мм. Масло нестойкое;
класс IV — плохое распределение влаги — на индикаторной бу-
мажке видно очень много точек и пятен диаметром более 3 мм. Мас-
ло нестойкое.
Приготовление реактивов. Реактив 1 (раствор бромфенолсинего).
Отвешивают 0,25 г бромфенолсинего и растворяют в 100 см3 95%-
346
кого спирта-ректификата. Затем добавляют 1 см3 3%-ного раствора
соляной кислоты. Приготовленный раствор должен иметь pH 3.
Кислотность раствора определяют с помощью универсальной ин-
дикаторной бумаги.
Индикаторные бумажные полоски. Беззольную фильтровальную
бумагу режут на полоски размером 5 х 3 см. Нарезанные полоски
погружают в раствор бромфенолсинего, выдерживают 1—1,5 мин,
затем просушивают в темноте при температуре 35—40 °C (в сушиль-
ном шкафу или термостате).
Хранить индикаторные бумажные полоски следует в закупорен-
ной бутылке из темного стекла или в темном помещении при ком-
натной температуре и относительной влажности не выше 70 %.
ОПРЕДЕЛЕНИЕ ИНДЕКСА РАСТВОРИМОСТИ
СУХИХ МОЛОЧНЫХ ПРОДУКТОВ
Основано на измерении объема нерастворившегося осадка в вос-
становленной пробе сухого молочного продукта. Выделение нера-
створившегося осадка проводят в пробирке вместимостью 10 см3 на
центрифуге с частотой вращения 1000 об/мин.
В мензурку вместимостью 100 см3 взвешивают пробу исследуе-
мого продукта с отсчетом результата до 0,01 (г):
12,5 — сухого цельного молока 25%-ной жирности сухих кисло-
молочных продуктов, сухого заменителя цельного молока (ЗЦМ)
для телят, молока регенерированного для молодняка сельскохозяй-
ственных животных;
12,0 — сухого цельного молока 20%-ной жирности;
10,5 — сухого молока 15%-ной жирности, в том числе «Смоленс-
кое»;
9,0 — сухого обезжиренного молока;
16,0 — сухих сливок;
37,0 — сухой смеси для сливочного, сливочно-белкового, сли-
вочно-кофейного мороженого;
32,0 — сухой смеси для молочного мороженого;
48,0 — сухой смеси для мороженого «Пломбир Домашний».
Обычно массу пробы для восстановления сухих молочных про-
дуктов указывают в нормативной документации на конкретный вид
продукта.
Пробу продукта растворяют порциями воды температурой
40 ± 2 °C, тщательно растирая комочки стеклянной палочкой, дово-
дят объем водой до 100 см3 и выдерживают в течение 15—20 мин при
температуре 18—25 °C. Проводят параллельно два измерения.
Восстановленный продукт перемешивают, заполняют им цент-
рифужные пробирки до метки «10 см3» и закрывают пробками.
Пробирки обертывают фильтровальной бумагой и помещают в пат-
роны центрифуги, располагая пробками к центру симметрично
347
одна против другой. Пробирки центрифугируют в течение 5 мин.
По окончании центрифугирования, при отсутствии четкой грани-
цы, насадочную жидкость сливают, оставляя над осадком ее слой
высотой около 5 мм. Затем доливают в пробирки воду температурой
18—25 °C до метки «10 см3», перемешивают содержимое пробирок
палочкой, закрывают пробками и центрифугируют в течение 5 мин.
Поочередно вынимают их из центрифуги и отсчитывают объем
осадка до ближайшего наименьшего деления пробирки, держа ее
пробкой вниз в вертикальном положении так, чтобы верхний уро-
вень находился на уровне глаз. При неровном размещении осадка
отсчет проводят по средней линии между верхним и нижним поло-
жениями. Индекс растворимости выражают в кубических санти-
метрах сырого осадка по шкале пробирки.
За окончательный результат измерения принимают среднее
арифметическое значение результатов двух параллельных измере-
ний, округленное до первого десятичного знака.
ОПРЕДЕЛЕНИЕ ВЛАГОУДЕРЖИВАЮЩЕЙ СПОСОБНОСТИ
СГУСТКОВ МОЛОЧНОКИСЛЫХ КУЛЬТУР И ЗАКВАСОК
МЕТОДОМ ЦЕНТРИФУГИРОВАНИЯ ПО МЕТОДИКЕ ВНИМИ
Влагоудерживающую способность сгустков молочнокислых
культур и заквасок устанавливают на центрифуге при факторе раз-
деления 1000.
На лабораторных центрифугах разных марок указанный фактор
достигается различной частотой вращения п, поскольку он зависит
от радиуса внутреннего ротора (расстояния от оси вращения цент-
рифуги до поверхности центрифугируемого образца R).
Следует установить радиус ротора имеющейся центрифуги и
выбрать необходимую частоту вращения:
R, м п, об/мин R, м п, об/мин
0,05 4200 0,18-0,19 2200
0,10 3000 0,20-0,21 2100
0,11 2800 0,22-0,23 2000
0,12 2700 0,24-0,25 1900
0,13 2600 0,26-0,28 1800
0,14 2500 0,29-0,30 1700
0,15-0,16 2400
0,17 2300
10 см3 разрушенного сгустка вносят в центрифужную пластмас-
совую пробирку вместимостью 15 см3 и центрифугируют при уста-
новленной частоте вращения в течение 5 мин. После остановки
центрифуги в образце измеряют объем выделившейся сыворотки
путем декантации ее в градуированную стеклянную центрифужную
348
пробирку на 10 см3 или мензурку. По количеству выделившейся сы-
воротки судят о способности сгустков к влагоотдаче. Результаты
выражают в количестве миллиметров сыворотки, полученной из
10 см3 сгустков (см3/10 см3).
Сгустки культур и заквасок с влагоотдачей от 3,5 до 5,5 см3 сыво-
ротки рекомендуются для приготовления творога. Сгустки с влаго-
удерживающей способностью до 2,5 см3 сыворотки пригодны для
производства кисломолочных напитков и сметаны.
Контрольные вопросы. 1. На чем основаны методы определения термоустойчи-
вости молока? 2. Каким образом выявляют маститное молоко? 3. Как определяют
эффективность гомогенизации? 4. Как можно определить, подвергалось молоко па-
стеризации или нет?
ПРИЛОЖЕНИЯ
Приложение 1
Значение pH буферных распоров
Темпера- тура, *С 0,05 М раст- вор тетра- оксилата калия Насыщенный при 20 *С раствор гид- ротартрата калия 0,05 М раствор гцдрофгалата калия 0,025 М раствор дигидрофосфата калия и 0,025 М раствор гидро- фосфата натрия 0,01 М раствор тетрабората натрия
0 1,666 4,003 6,984 9,464
5 1,668 —- 3,999 6,951 9,395
10 1,670 — 3,998 6,923 9,332
15 1,672 — 3,999 6,900 9,276
20 1,675 — 4,002 6,881 9,225
25 1,679 3,557 4,008 6,865 9,180
30 1,683 3,552 4,015 6,853 9,139
35 1,688 3,549 4,024 6,844 9,102
40 1,694 3,547 4,035 6,838 9,068
45 1,700 3,547 4,047 6,834 9,038
50 1,707 3,549 4,060 6,833 9,011
55 1,715 3,554 4,075 6,834 8,985
60 1,723 3,560 4,091 6,836 8,962
70 1,743 3,580 4,126 6,845 8,921
80 1,766 3,609 4,164 6,859 8,885
90 1,792 3,650 4,205 6,877 8,850
95 1,806 3,674 4,227 6,886 8,833
Приложение 2
Усредненные соотношения между pH и титруемой кислотностью, пределы изменения
pH молока и молочных продуктов
Таблица 1
Титруе- мая кис- лот- ность, °Т Молоко
заготовляемое пастеризованное топленое
среднее пределы pH значение pH среднее пределы pH значение pH среднее пределы pH значение pH
16 6,75-6,72 6,73 6,7-6,66 6,68 6,64-6,59 6,61 17 6,71-6,67 6,69 6,65-6,71 6,63 6,58-6,54 6,56 18 6,67-6,61 6,64 6,6-6,55 6,57 6,53-6,48 6,5 19 6,60-6,55 6,58 6,54-6,49 6,51 6,47-6,42 6,44 20 6,54-6,49 6,52 6,48-6,43 6,45 6,41-6,37 6,39 21 6,48-6,44 6,46 6,42-6,38 6,4 6,36-6,32 6,34 22 6,43-6,39 6,41 6,37-6,32 6,32 6,31-6,26 6,28 23 6,38-6,34 6,36 6,3-6,26 6,28 6,25-6,21 6,23 24 6,33-6,29 6,31 6,25-6,21 6,23 6,2-6,16 6,18 25 6,28-6,24 6,26 6,2-6,16 6,18 6,15—6,11 6,18 26 6,23-6,19 6,21 6 15—6,11 6,13 6,16-6,06 6,08 27 6,16-6,14 6,16 6,1—6,06 6,08 6,05-6,01 6,03
350
Таблица 2
Титруемая кислотность, т Сливки пастеризованные
10%-ной жирности 20%-ной жирности
пределы pH среднее значение pH пределы pH среднее значение pH
16 6,67-6,61 6,63 6,62-6,58 6,6
17 6,6-6,55 6,57 6,57-6,52 6,54
18 6,54-6,49 6,51 6,51-6,46 6,48
19 6,48-6,42 6,45 6,45-6,4 6,42
20 6,41-6,35 6,39 6,39-6,34 6,36
21 6,43-6,3 6,33 6,33-6,28 6,3
22 6,29-6,24 6,27 6,27-6,22 6,24
23 6,24-6,18 6,21 6,21-6,16 6,18
24 6,17-6,12 6,15 6,15-6,10 6,12
25 6,11-6,06 6,09 6,09-6,04 6,06
26 6,05-6,0 6,03 6,03-5,98 6,0
Таблица 3
Титруемая кислотность, “Т Кефир и творожный сгусток Ацидофилин Простокваша обыкновенная Ряженка
50 5,38 5,37 5,3 5,04
55 5,25 5,23 5,15 4,90
60 5,14 5,10 5,00 4,77
65 5,04 4,96 4,86 4,65
70 4,94 4,82 4,73 4,55
75 4,85 4,69 4,60 4,45
80 4,76 4,57 4,47 4,37
85 4,68 4,46 4,37 4,30
90 4,60 4,36 4,28 4,23
95 4,54 4,38 4,21 4,18
100 4,48 4,20 4,14 4,13
105 4,42 4,14 4,08 4,09
ПО 4,36 4,08 4,02 4,05
115 4,31 4,02 3,98 —
120 4,26 3,97 3,94 —
125 — 3,92 3,91 —
130 — 3,88 3,88 —
135 — 3,84 — —
140 — 3,82 — —
145 — 3,8 — —
150 — 3,78 — —
351
Таблица 4
Титруемая кислотность, °Т Творог
жирный (18 %) | диетический (11 %)
150 — 4,54
155 — 4,48
160 — 4,42
165 — 4,38
170 — 4,33
175 — 4,27
180 4,62 4,22
185 4,54 4,17
190 4,48 4,12
195 4,43 4,08
200 4,38 4,04
205 4,33 —
210 4,28 —
215 4,24 —
220 4,20 —
225 4,16 —
23Q 4,13 —
235 4,10 —
240 4,08 —
245 4,06 —
250 4,04 —
255 4,02 —
260 4,00 —
Таблица 5
Продукт Способ производства pH
в конце сквашивания при выпуске в реализацию
Кефир Резервуарный Термостатный 4,65-4,50 4,85-4,75 4,65-4,25 4,65-4,25
Ацидофилин Резервуарный Термостатный 4,70-4,55 4,80-4,70 4,70-3,95 4,70-3,95
Ряженка Резервуарный Термостатный 4,40-4,30 4,45-4,35 4,35-4,05 4,35-4,05
Простокваша обыкно- венная Термостатный 4,55-4,65 4,50-4,05
Сгусток для производ- ства творога Кислотно-сычужный 5,05-5,15 —
Сгусток для производ- ства сыра домашнего Кислотно-сычужный 4,70-4,90 —
Закваска для сметаны, тво- — рога, простокваши обыкно- венной 4,55-4,65 —
Закваска для кефира — 4,50-4,55 —
352
Приложение 3
Приготовление растворов гидроксида натрия
Раствор различной концентрации приготавливают из насыщенного раствора
гидроксида натрия. Для получения насыщенного раствора NaOH растворяют в рав-
ном по объему количестве воды, например, 500 г NaOH растворяют в 500 см3 воды,
предварительно прокипяченной для удаления СО2. Так как раствор при этом силь-
но разогревается, растворение выполняют в фарфоровой чашке, добавляя NaOH к
воде постепенно с непрерывным перемешиванием. Раствор отвечает приблизитель-
но 16 н. раствору NaOH. Переливают раствор в емкость с резиновой пробкой, дают
отстояться осадку Na2CO3 (одну-две недели). Для разбавления используют свеже-
прокипяченную, охлажденную воду, защищенную от действия СО2 воздуха проб-
кой с хлоркальциевой трубкой. Титр приготовленного раствора гидроксида натрия
устанавливают по 0,1 М НС1 или по 0,05 М H2SO4.
Для приготовления 0,1 М раствора гидроксида натрия 7 см3 насыщенного ра-
створа гидроксида натрия переносят в мерную колбу на 1000 см3 и доводят объем
водой до метки.
1, 2 и 0,05 М раствор приготавливают, пользуясь расчетом, как при приготовле-
нии 0,1 М раствора.
0,01 и 0,02 М раствор NaOH готовят из 0,1 М разведением в 10 и 5 раз в мерной
колбе.
Приложение 4
При определении объема исходной серной кислоты, необходимой для разведе-
ния, пользуются приведенной ниже зависимостью концентрации серной кислоты
от ее плотности. „
Приготовление растворов серной кислоты
Плотность, г/см3 Концен- трация H2SO,, % Плотность, г/см5 Концен- трация H2SO„ % Плотность, г/см3 Концен- трация H2SO„ % Плотность, г/см5 Концен- трация H2SO4, %
0,999 0,09 1,266 35,71 1,535 63,43 1,799 87,60
1,000 1,57 1,276 36,87 1,545 64,26 1,804 88,30
1,019 3,03 1,286 38,03 1,555 65,08 1,809 89,05
1’029 4,49 1,296 39,19 1,565 65,90 1,814 90,05
1,039 5,96 1,306 40,35 1,575 66,71 1,815 90,20
1,048 7,37 1,316 41,50 1,585 67,59 1,816 90,40
1,058 8,77 1,326 42,66 1,595 68,51 1,817 90,60
1,068 10,19 1,336 43,74 1,605 69,43 1,818 90,80
1,077 11,60 1,346 44,82 1,615 70,32 1,819 91,00
1,087 12,99 1,356 45,88 1,625 71,16 1,820 91,25
1,097 14,35 1,366 46,94 1,635 71,99 1,822 91,70
1,107 15,71 1,376 48,00 1,645 72,82 1,825 92,30
1,117 17,01 1,386 49,06 1,655 73,64 1,827 92,75
1’127 18,31 1,396 50,11 1,665 74,51 1,829 93,43
1Д37 19,61 1,406 51,15 1,675 75,42 1,831 94,25
1,147 20,91 1,416 52,15 1,685 76,30 1,832 94,60
1,157 22,19 1,426 53,15 1,695 77,17 1,833 95,00
1,167 22,47 1,436 54,07 1,705 78,04 1,834 95,60
1,177 24,76 1,446 55,03 1,715 78,92 1,835 97,00
1,187 26,04 1,456 55,97 1,725 79,80 1,838 99,70
1,197 27,32 1,466 56,90 1,735 80,68 1,839 99,95
1,207 28,58 1,476 57,83 1,745 81 56 — —
1,217 29,84 1,486 58,74 1,755 82,44 — —
1,226 31,11 1,496 59,70 1,764 83,32 — —
1,236 32,28 1,505 60,65 1,774 84,50 — —
1,246 33,43 1,515 61,59 1,784 85,70 — —
1,256 34,57 1,525 62,53 1,794 86,90 — —
353
Растворы готовят из концентрированной химически чистой H2SO4 — молеку-
лярная масса 98,08.
Для приготовления 1 М раствора на 1000 см3 требуется 98,08 г H2SO4. Ареомет-
ром определяют плотность исходной кислоты при температуре 20 “С и вычисляют
объемы (см3) этой кислоты и воды, необходимые для приготовления заданного koj
личества 1 М кислоты. Например, при плотности 1,833 концентрация H2SO4 со-
ставляет 95%. Для приготовления 1000 см3 раствора следует взять 100-98,08/
95 = 103,2 г или 56,2 см3 (103,2/1,833) H2SO4 и 943,8 см3 дистиллированной воды.
0,1 и0,01 М растворы приготавливают разведением водой 1 М раствора (0,1 М —
10 см3 раствора 1 М кислоты разводят водой в мерной колбе на 100 см3; 0,01 М —
10 см3 1 М раствора разводят водой в мерной колбе на 1000 см3).
Для получения разбавленных растворов H2SO4 (более низкой плотности, чем
исходная кислота) необходимо приготовить раствор кислоты плотностью 1,815 из
имеющейся кислоты большей плотности. Определяют плотность имеющейся кис-
лоты и, используя правило смешения («правило креста»), устанавливают массовые
части кислоты и воды, необходимые для приготовления раствора заданной плотно-
сти. Например, в серной кислоте плотностью 1,833 концентрация H2SO4 составляет
95 %, а в серной кислоте заданной плотности 1,815 — 90,2 %. Используя правило
смешения,
90,2-0 = 90,2
95 - 90,2 = 4,8
находим, что для получения 90,2%-ной серной кислоты плотностью 1,815 смешива-
ют 90,2 весовые части 95%-ной кислоты с 4,8 весовыми частями (4,8 см3) воды.
Приложение 5
Для приготовления растворов соляной кислоты пользуются зависимостью кон-
центрации НО от плотности.
Приготовление растворов реактивов соляной кислоты (НС1)
Плотность, г/см’ Концентрация HCI Плотность, г/см3 Концентрация HCI
% I г/дм3 % I г/дм3
1,0032 1 10,03 1,1083 22 243,8
1,0082 2 20,16 1,1187 24 268,5
1,0181 4 40,72 1,1290 26 293,5
1,0279 6 61,67 1,1392 28 319,0
1,0376 8 83,01 1,1493 30 344,8
1,0474 10 104,7 1,1593 32 371,0
1,0574 12 126,9 1,1691 34 397,5
1,0675 14 149,5 1,1789 36 424,4
1,0776 16 172,4 1,1885 38 451,6
1,0878 18 195,8 1,1980 40 479,2
1,0980 20 219,6 — — —
Для приготовления 1000 см3 раствора НС1 заданной молярности требуется взять
исходную НС1 в количестве
v_ ХМ-1000
354
где И— объем исходной НО, необходимый для приготовления раствора НО, см3;
л —молярность приготовляемого раствора НО; М— молекулярная масса НО
(36,47), г; 1000 — коэффициент для перевода объема в см3; С — концентрация НО в
исходной кислоте (находят по плотности исходной НО), г/дм3.
Отмеривают рассчитанный объем кислоты и количественно переносят в мер-
ную колбу на 1000 см3, доводят объем до метки водой.
Например, требуется приготовить 1000 см3 0,1 М раствора НО. Плотность ис-
ходной НС1, определенная по ареометру при температуре 20 °C, составляет 1,1885.
По плотности находят содержание НС1 в исходной кислоте, равное 451 г/дм3 .
Объем исходной НС1, который необходимо взять для приготовления 0,1 М раствора
НС1, составляет
к=0Д26;47Л000 = 8 1смЗ
451
В мерную колбу на 1000 см3 вносят 8,1 см3 исходной НС1 и доводят до метки водой.
Приложение 6
Пересчет массы восстановленной меди на массу сахарозы и инвертного сахара, мг
Т аблица 1
Медь | Сахароза 11 Медь | Сахароза 11 Медь | Сахароза | | Медь | Сахароза
80,8 39,9 113,7 56,4 146,5 73,0 179,4 90,0
81,7 40,4 114,5 56,9 147,4 73,5 180,3 90,4
82,6 40,9 115,4 57,3 148,3 74,0 181,2 90,7
833 41,3 116,3 57,8 149,2 74,5 182,0 91,4
84,4 41,7 117,7 58,2 150,1 74,9 182,9 91,8
85,2 42,2 118,1 58,7 15L0 75,4 183,8 92,2
86; 1 42,6 119,0 59,2 151,8 75,9 184,7 92,7
87,0 43,0 119,9 59,6 152,7 76,4 185,6 93,2
87,9 43,5 120,8 60,0 153,6 76,9 186,5 93,7
88,8 44,0 121,6 60,5 154,5 77,3 187,4 94,1
89,7 44,4 122,5 60,9 155,4 77,8 188,3 94,6
90,6 44,8 123,4 61,4 156,3 78,3 189,1 95,1
91,5 45,2 124,3 61,8 157,2 78,7 190,0 95,6
92,3 45,5 125,2 62,3 158,1 79,1 190,9 96,0
93,2 46,1 126,1 62,7 159,0 79,7 191,8 96,5
94,1 46,5 127,0 63,2 159,8 80,1 • 192,7 97,0
95,0 47,0 127,9 63,6 160,7 80,5 193,6 97,5
95,9 47,4 128,8 64,1 161,6 80,9 194,5 97,9
96,8 47,9 129,6 64,5 162,5 81,4 195,4 98,4
97,7 48,3 130,5 65,0 163,4 81,9 196,2 98,9
98,6 48,8 131,4 65,5 164,3 82,3 197,1 99,4
99,4 49,2 132,3 65,8 165,2 82,7 198,0 99,8
100,3 49,7 133,2 66,3 166,1 83,2 198,9 100,3
101,2 50,2 134,1 66,8 166,9 83,7 199,8 100,8
102,1 50,5 135,0 67,3 167,8 84,1 200,7 101,3
103,0 51,0 135,9 67,6 168,7 84,5 201,6 101,7
103,9 51,5 136,8 68,1 169,7 85,0 202,5 102,2
104,8 52,0 137,6 68,6 170,5 85,5 203,4 102,7
105,7 52,4 138,5 69,1 171,4 85,9 204,2 103,2
106,6 52,9 139,4 69,5 172,3 86,3 205,1 103,6
107,4 53,3 140,3 69,9 173,2 86,8 206,0 104,1
108,3 53,7 141,2 70,4 174,0 87,3 206,9 104,6
109,2 54,1 142,1 70,9 174,9 87,7 207,8 105,1
110,1 54,6 143,0 71,3 175,8 88,3 208,7 105,5
ш;о 55,1 143,9 71,7 176,7 88,6 209,6 106,0
111,9 55,6 144,7 72,2 177,6 89,1 210,5 106,5
112,8 56,0 145,6 72,7 178,5 89,5 211,3 107,0
355
Продолжение
Медь | Сахароза 11 Медь | Сахароза | | Медь | Сахароза 11 Медь | Сахароза
212,2 107,4 243,3 124,3 274,4 141,2 305,5 158,6
213,1 107,9 244,2 124,7 275,3 141,6 306,4 159,1
214,0 108,5 245,1 125,2 276,2 142,1 307,2 159,6
214,9 109,0 246,0 125,7 277,1 142,6 308,1 160,2
215,8 109,4 246,9 126,2 277,9 143,1 309,0 160,6
216,7 109,9 247,8 126,6 278,8 143,6 309,9 161,1
217,6 110,4 248,6 127,1 279,7 144,1 310,8 161,6
218,4 110,9 249,5 127,7 280,6 144,7 311,7 162,1
219,3 111,3 250,4 128,1 281,5 145,2 312,6 162,6
220,2 111,8 251,3 128,6 282,4 145,6 313,5 163,1
221,1 112,3 252,2 129,1 283,3 146,2 314,4 163,7
222,0 112,8 253,1 129,6 284,2 146,7 315,2 164,2
222,9 113,2 254,0 130,0 285,0 147,1 316,1 164,6
223,8 113,7 254,9 130,5 285,9 147,6 317,0 165,2
224,7 114,2 255,7 131,0 286,8 148,1 317,9 165,7
225,6 114,7 256,6 131,5 287,7 148,7 318,8 166,2
226,4 115,1 257,5 131,9 288,6 149,1 319,7 166,6
227,3 115,6 258,4 132,4 289,5 149,6 320,6 167,2
228,2 116,1 259,3 133,0 290,4 150,1 321,6 167,7
229,1 116,6 260,2 133,5 291,3 150,7 322,3 168,1
230,0 117,0 261,1 134,0 292,2 151,1 323,2 168,6
230,9 117,5 262,0 134,4 293,0 151,6 324,1 169,1
231,8 118,0 262,8 134,8 293,9 152,1 325,0 169,7
232,7 118,5 263,7 135,4 294,8 152,7 325,9 170,1
233,5 118,9 264,6 135,8 295,7 153,1 326,8 170,6
234,4 119,4 265,5 136,3 296,6 153,7 327,7 171,2
235,3 119,9 266,4 136,8 297,5 154,2 328,6 171,7
236,2 120,4 267,3 137,3 298,4 154,7 329,4 172,1
237,1 120,8 268,2 137,7 299,3 155,2 330,4 172,7
238,0 121,4 269,1 138,2 300,1 155,7 331,2 173,2
238,9 121,9 270,0 138,8 301,0 156,2 332,1 173,7
239,8 122,4 270,8 139,3 301,9 156,8 333,0 174,3
240,6 122,8 271,7 139,7 302,8 157,2 333,9 174,8
241,5 123,3 272,6 140,2 303,7 157,7 334,8 175,3
242,4 123,8 273,5 140,7 304,6 158,2 335,7 175,8
Таблица 2
Медь | Инвертный сахар 11 Медь | Инвертный сахар
43,5 22,5 54,2 28,1
44,4 23,0 55,1 28,6
45,3 23,5 55,9 29,0
46,2 23,9 56,8 29,5
47,1 24,4 57,7 30,0
48,0 24,9 58,6 30,4
48,8 25,3 59,5 30,9
49,7 25,8 60,4 31,4
50,6 26,2 61,3 31,8
51,5 26,7 62,2 32,3
52,4 27,2 63,0 32,7
53,3 27,6 63,9 33,1
356
Приложение 7
Таблица для определения массовой доли спирта по относительной массе (d}§) водно-
спиртовых смесей v '
Относитель- ная масса водно-спирто- вой смеси , 20 «20 Массовая доля спирта в про- дукте, % Относитель- ная масса водно-спирто- вой смеси г 20 “20 Массовая доля спирта в про- дукте, % Относитель- ная масса водно-спирто- вой смеси г 20 «20 Массовая доля спирта в про- дукте, %
1,000 0,00
0,9999 0,5 0,9969 1,66 0,9939 3,33
8 10 8 71 8 38
7 16 7 77 7 44
6 21 6 82 6 50
5 26 5 88 5 56
4 32 4 93 4 61
3 37 3 98 3 67
2 42 2 2,04 2 73
1 48 1 09 1 78
0 53 0 15 0 84
9989 59 9959 20 9929 90
8 64 8 26 8 96
7 69 7 32 7 4,02
6 74 6 37 6 08
5 80 5 43 5 14
4 85 4 48 4 20
3 90 3 54 3 26
2 96 2 59 2 31
1 1,01 1 65 1 37
0 06 0 70 0 43
9979 12 9949 76 9919 49
8 17 8 82 8 55
7 23 7 87 7 61
6 28 6 93 6 67
5 34 5 98 5 73
4 39 4 3,04 4 79
3 44 3 10 3 85
2 50 2 16 2 91
1 55 1 21 1 97
0 60 0 27 0 5,03
Приложение 8
Таблица приведения плотности коровьего молока к 20 °C
Плотность молока рф, кт/м1 Плотность, приведенная к 20 ’С, кт/м3, при температуре молока t, °C
15,0 15,5 16,0 16,5 17,0 17,5 18,0
1025,0 1023,4 1023,6 1023,7 1023,9 1024,0 1024,2 1024,4
1025,5 1023,9 1024,1 1024,2 1024,4 1024,5 1024,7 1024,9
1026,0 1024,4 1024,6 1024,7 1024,9 1025,0 1025,2 1025,4
1026,5 1024,9 1025,1 1025,2 1025,4 1025,5 1025,7 1025,9
1027,0 1025,4 1025,6 1025,7 1025,9 1026,0 1026,2 1026,4
1027,5 1025,9 1026,1 1026,2 1026,4 1026,5 1026,7 1026,9
357
Продолжение
Плотность молока Рср, кг/м3 Плотность, приведенная к 20 ’С, кт/м5, при температуре молока t, "С
15,0 15,5 16,0 16,5 17,0 17,5 18,0
1028,0 1026,4 1026,6 1026,7 1026,9 1027,0 1027,2 1027,4
1028,5 1026,9 1027,1 1027,2 1027,4 1027,5 1027,7 1027,9
1029,0 1027,4 1027,6 1027,7 1027,9 1028,0 1028,2 1028,4
1029,5 1027,9 1028,1 1028,2 1028,4 1028,5 1028,7 1028,9
1030,0 1028,4 1028,6 1028,7 1028,9 1029,0 1029,2 1029,4
1030,5 1028,9 1029,1 1029,2 1029,4 1029,5 1029,7 1029,9
1031,0 1029,4 1029,6 1029,7 1029,9 1030,0 1030,2 1030,4
1031,5 1029,9 1030,1 1030,2 1030,4 1030,5 1030,7 1030,9
1032,0 1030,4 1030,6 1030,7 1030,9 1031,0 1031,2 1031,4
1032,5 1030,9 1031,1 1031,2 1031,4 1031,5 1031,7 1031,9
1033,0 1031,4 1031,6 1031,7 1031,9 1032,0 1032,2 1032,4
1033,5 1031,9 1032,1 1032,2 1032,4 1032,5 1032,7 1032,9
1034,0 1032,4 1032,6 1032,7 1032,9 1033,0 1033,2 1033,4
1034,5 1032,9 1033,1 1033,2 1033,4 1033,5 1033,7 1033,9
1035,0 1033,4 1033,6 1033,7 1033,9 1034,0 1034,2 1034,4
1035,5 1033,9 1034,1 1034,2 1034,4 1034,5 1034,7 1034,9
1036,0 1034,4 1034,6 1034,7 1034,9 1035,0 1035,2 1035,4
Продолжение
Плотность молока Рср, кг/м3 Плотность, приведенная к 20 °C, кг/м3, при температуре молока t, °C
18,5 19,0 19,5 20,0 20,5 21,0 21,5
1025,0 1024,5 1024,7 1024,8 1025,0 1025,2 1025,3 1025,5
1025,5 1025,0 1025,2 1025,3 1025,5 1025,7 1025,8 1026,0
1026,0 1025,5 1025,7 1025,8 1026,0 1026,2 1026,3 1026,5
1026,5 1026,0 1026,2 1026,3 1026,5 1026,7 1026,8 1027,0
1027,0 1026,5 1026,7 1026,8 1027,0 1027,3 1027,3 1027,5
1027,5 1027,0 1027,2 1027,3 1027,5 1027,7 1027,8 1028,0
1028,0 1027,5 1027,7 1027,8 1028,0 1028,2 1028,3 1028,5
1028,5 1028,0 1028,2 1028,3 1028,5 1028,7 1028,8 1029,0
1029,0 1028,5 1028,7 1028,8 1029,0 1029,2 1029,3 1029,5
1029,5 1029,0 1029,2 1029,3 1029,5 1029,7 1029,8 1030,0
1030,0 1029,5 1029,7 1029,8 1030,0 1030,2 1030,3 1030,5
1030,5 1030,0 1030,2 1030,3 1030,5 1030,7 1030,8 1031,0
1031,0 1030,5 1030,7 1030,8 1031,0 1031,2 1031,3 1031,5
1031,5 1031,0 1031,2 1031,3 1031,5 1031,7 1031,8 1032,0
1032,0 1031,5 1031,7 1031,8 1032,0 1032,2 1032,3 1032,5
1032,5 1032,0 1032,2 1032,3 1032,5 1032,7 1032,8 1033,0
1033,0 1032,5 1032,7 1032,8 1033,0 1033,2 1033,3 1033,5
1033,5 1033,0 1033,2 1033,3 1033,5 1033,7 1033,8 1034,0
1034,0 1033,5 1033,7 1033,8 1034,0 1034,2 1034,3 1034,5
1034,5 1034,0 1034,2 1034,3 1034,5 1034,7 1034,8 1035,0
1035,0 1034,5 1034,7 1034,8 1035,0 1035,2 1035,3 1035,5
1035,5 1035,0 1035,2 1035,3 1035,5 1035,7 1035,8 1036,0
1036,0 1035,5 1035,7 1035,8 1036,0 1036,2 1036,3 1036,5
Продолжение
Плотность молока Рср5 кг/м3 Плотность, приведенная к 20 °C, кг/м3, при температуре молока t, °C
22,0 22,5 23,0 23,5 24,0 24,5 25,0
1025,0 1025,6 1025,8 1026,0 1026,1 1026,3 1026,4 1026,6
1025,5 1026,1 1026,3 1026,5 1026,6 1026,8 1026,9 1027,1
1026,0 1026,6 1026,8 1027,0 1027,1 1027,3 1027,4 1027,6
1026,5 1027,1 1027,3 1027,5 1027,6 1027,8 1027,9 1028,1
1027,0 1027,6 1027,8 1028,0 1028,1 1028,3 1028,4 1028,6
1027,5 1028,1 1028,3 1028,5 1028,6 1028,8 1028,9 1029,1
1028,0 1028,6 1028,8 1029,0 1029,1 1029,3 1029,4 1029,6
1028,5 1029,1 1029,3 1029,5 1029,6 1029,8 1029,9 1030,1
1029,0 1029,6 1029,8 1030,0 1030,1 1030,3 1030,4 1030,6
1029,5 1030,1 1030,3 1030,5 1030,6 1030,8 1030,9 1031,1
1030,0 1030,6 1030,8 1031,0 1031,1 1031,3 1031,4 1031,6
1030,5 1031,1 1031,3 1031,5 1031,6 1031,8 1031,9 1032,1
1031,0 1031,6 1031,8 1032,0 1032,1 1032,3 1032,4 1032,6
1031,5 1032,1 1032,3 1032,5 1032,6 1032,8 1032,9 1033,1
1032,0 1032,6 1032,8 1033,0 1033,1 1033,3 1033,4 1033,6
1032,5 1033,1 1033,3 1033,5 1033,6 1033,8 1033,9 1034,1
1033,0 1033,6 1033,8 1034,0 1034,1 1034,3 1034,4 1034,6
1033,5 1034,1 1034,3 1034,5 1034,6 1034,8 1034,9 1035,1
1034,0 1034,6 1034,8 1035,0 1035,1 1035,3 1035,4 1035,6
1034,5 1035,1 1035,3 1035,5 1035,6 1035,8 1035,9 1036,1
1035,0 1035,6 1035,8 1036,0 1036,1 1036,3 1036,4 1036,6
1035,5 1036,1 1036,3 1036,5 1036,6 1036,8 1036,9 1037,1
1036,0 1036,6 1036,8 1037,0 1037,1 1037,3 1037,4 1037,6
Приложение 9
Таблица приведения плотности обезжиренного молока к 20 °C
Плотность молока Рср’ кг/м3 Плотность, приведенная к 20 °C, кт/м1, при температуре молока t, 'С
15,0 15,5 16,0 16,5 17,0 17,5 18,0
1028,0 1026,7 1026,8 1027,0 1027,1 1027,2 ' 1027,4 1027,5
1028,5 1027,2 1027,3 1027,5 1027,6 1027,7 1027,9 1028,0
1029,0 1027,7 1027,8 1028,0 1028,1 1028,2 1028,4 1028,5
1029,5 1028,2 1028,3 1028,5 1028,6 1028,7 1028,9 1029,0
1030,0 1028,7 1028,8 1029,0 1029,1 1029,2 1029,4 1029,5
1030,5 1029,2 1029,3 1029,5 1029,6 1029,7 1029,9 1030,0
1031,0 1029,7 1029,8 1030,0 1030,1 1030,2 1030,4 1030,5
1031,5 1030,2 1030,3 1030,5 1030,6 1030,7 1030,9 1031,0
1032,0 1030,7 1030,8 1031,0 1031,1 1031,2 1031,4 1031,5
1032,5 1031,2 1031,3 1031,5 1031,6 1031,7 1031,9 1032,0
1033,0 1031,7 1031,8 1032,0 1032,1 1032,2 1032,4 1032,5
1033,5 1032,2 1032,3 1032,5 1032,6 1032,7 1032,9 1033,0
1034,0 1032,7 1032,8 1033,0 1033,1 1033,2 1033,4 1033,5
1034,5 1033,2 1033,3 1033,5 1033,6 1033,7 1033,9 1034,0
1035,0 1033,7 1033,8 1034,0 1034,1 1034,2 1034,4 1034,5
1035,5 1034,2 1034,3 1034,5 1034,6 1034,7 1034,9 1035,0
1036,0 1034,7 1034,8 1035,0 1035,1 1035,2 1035,4 1035,5
1036,5 1035,2 1035,3 1035,5 1035,6 1035,7 1035,9 1036,0
1037,0 1035,7 1035,8 1036,0 1036,1 1036,2 1036,4 1036,5
1037,5 1036,2 1036,3 1036,5 1036,6 1036,7 1036,9 1037,0
1038,0 1036,7 1036,8 1037,0 1037,1 1037,2 1037,4 1037,5
358
359
Продолжение
Плотность молока Рср, кг/м3 Плотность, приведенная к 20 ’С, кг/м3, при температуре молока t, С
18,5 19,0 19,5 20,0 20,5 21,0 21,5
1028,0 1027,6 1027,7 1027,9 1028,0 01028,1 1028,3 1028,4
1028'5 1028,1 1028,2 1028,4 1028,5 1028,6 1028,8 1028,9
1029'0 1028,6 1028,7 1028,9 1029,0 1029,1 1029,3 1029,4
1029,5 1029 Л 1029,2 1029,4 1029,5 1029,6 1029,8 1029,9
юзо'о 1029,6 1029,7 1029,9 1030,0 1030,1 1030,3 1030,4
юзоЗ 1030,1 1030,2 1030,4 1030,5 1030,6 1030,8 1030,9
1031^0 1030,6 1030,7 1030,9 1031,0 1031,1 1031,3 1031,4
1031',5 1031Д 1031,2 1031,4 1031,5 1031,6 1031,8 1031,9
1032,0 1031,6 1031,7 1031,9 1032,0 1032,1 1032,3 1032,4
1032'5 1032,1 1032,2 1032,4 1032,5 1032,6 1032,8 1032,9
1033,0 юз2;б 1032,7 1032,9 1033,0 1033,1 1033,3 1033,4
1033,5 1034,0 1033Д 1033,6 1033,2 1033,7 1033,4 1033,9 1033,5 1034,0 1033,6 1034,1 1033,8 1034,3 1033,9 1034,4
1034'5 1034,1 1034,2 1034,4 1034,5 1034,6 1034,8 1034,9
1035'0 юз4;б 1034,7 1034,9 1035,0 1035,1 1035,3 1035,4
1035,5 1035,1 1035,2 1035,4 1035,5 1035,6 1035,8 1035,9
1036^0 1935,6 1035,7 1035,9 10,36,0 1036,1 1036,3 1936,4
1036,5 1036,1 1036,2 1036,4 1036,5 1036,6 1036,8 1036,9
1037,0 1036,6 1036,7 1036,9 1037,0 1037,1 1037,3 1037,4
10373 1037,1 1037,2 1037,4 1037,5 1037,6 1037,8 1037,9
1038',0 1037,6 1037,7 1037,9 1038,0 1038,1 1038,3 1038,4
Продолжение
Плотность молока Рср* кг/м3 Плотность, приведенная к 20 °C, кг/м3, при температуре молока t, С
22,0 22,5 23,0 23,5 24,0 24,5 25,0
1028,0 1028,5 1028,7 1028,8 1028,9 1029,0 1029,2 1029,3
1028,5 1029,0 1029,2 1029,3 1029,4 1029,5 1029,7 1029,8
1029,0 1029,5 1029,7 1029,8 1029,9 1030,0 1030,2 1030,3
1029,5 1030,0 1030,2 1030,3 1030,4 1030,5 1030,7 1030,8
1030,0 1030,5 1030,7 1030,8 1030,9 1031,0 1031,2 1031,3
1030,5 1031,0 1031,2 1031,3 1031,4 1031,5 1031,7 1031,8
1031,0 1031,5 1031,7 1031,8 1031,9 1032,0 1032,2 1032,3
1031,5 1032,0 1032,2 1032,3 1032,4 1032,5 1032,7 1032,8
1032,0 1032,5 1032,7 1032,8 1032,9 1033,0 1033,2 1033,3
1032,5 1033,0 1033,2 1033,3 1033,4 1033,5 1033,7 1033,8
1033,0 1033,5 1033,7 1033,8 1033,9 1034,0 1034,2 1034,3
1033,5 1034,0 1034,2 1034,3 1034,4 1034,5 1034,7 1034,8
1034,0 1034,5 1034,7 1034,8 1034,9 1035,0 1035,2 1035,3
1034,5 1035,0 1035,2 1035,3 1035,4 1035,5 1035,7 1035,8
1035,0 1035,5 1035,7 1035,8 1035,9 1036,0 1036,2 1036,3
1035,5 1036,0 1036,2 1036,3 1036,4 1036,5 1036,7 1036,8
1036,0 1036,5 1036,7 1036,8 1036,9 1037,0 1037,2 1037,3
1036,5 1037,0 1037,2 1037,3 1037,4 1037,5 1037,7 1037,8
1037,0 1037,5 1037,7 1037,8 1037,9 1038,0 1038,2 1038,3
1037,5 1038,0 1038,2 1038,3 1038,4 1038,5 1038,7 1038,8
1038,0 1038,5 1038,7 1038,8 1038,9 1039,0 1039,2 1039,3
Приложение 10
Таблица поправок для определения фактической плотности коровьего молока
в диапазоне температур 10—15 °C
Темпертура молока t при измерении плотности, ’С Значение поправки, кг/м3, при температуре заготовляемого молока, °C
10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5 14,0 14,5
15,0 1,6 1,4 1,3 1,1 1,0 0,8 0,6 0,5 0,3 0 2 15,5 1,8 1,6 1,4 1,3 1,1 1,0 0,8 0,6 0,5 0 3. 16,0 1,9 1,8 1,6 1,4 1,3 1,1 1,0 0,8 0,6 0,5 16,5 2,1 1,9 1,8 1,6 1,4 1,3 1,1 1,0 0,8 0,6 17,0 2,2 2,1 1,9 1,8 1,6 1,4 1,3 1,1 1,0 0,8 17,5 2,4 2,2 2,1 1,9 1,8 1,6 1,4 1,3 1,1 1,0 18,0 2,6 2,4 2,2 2,1 1,9 1,8 1,6 1,4 1,3 1,1 18,5 2,7 2,6 2,4 2,2 2,1 1,9 1,8 1,6 1,4 1,3 19,0 2,9 2,7 2,6 2,4 2,2 2,1 1,9 1,8 1,6 1,4 19,5 3,0 2,9 2,7 2,6 2,4 2,2 2,1 1,9 1,8 1,6 20,0 3,2 3,0 2,9 2,7 2,6 2,4 2,2 2,1 1,9 1,8 20,5 3,4 3,2 3,0 2,9 2,7 2,6 2,4 2,2 2,1 1,9 21,0 3,5 3,4 3,2 3,0 2,9 2,7 2,6 2,4 2,2 2,1 21,5 3,7 3,5 3,4 3,2 3,0 2,9 2,7 2,6 2,4 2,2 22,0 3,8 3,7 3,5 3,4 3,2 3,0 2,9 2,7 2,6 2,4 22,5 4,0 3,8 3,7 3,5 3,4 3,2 3,0 2,9 2,7 2,6 23,0 4,2 4,0 3,8 3,7 3,5 3,4 3,2 3,0 2,9 2,7 23,5 4,3 4,2 4,0 3,8 3,7 3,5 3,4 3,2 3,0 2,9 24,0 4,5 4,3 4,2 4,0 3,8 3,7 3,5 3,4 3,2 3,0 24,5 4,6 4,5 4,3 4,2 4,0 3,8 3,7 3,5 3,4 3,2 25,0 4,8 4,6 4,5 4,3 4,2 4,0 3,8 3,7 3,5 3,4
Приложение 11
Таблица поправок для определения фактической плотности обезжиренного молока
в диапазоне температур 10—15 °C
Температура молока t при измерении плотности,"С Значение поправки, кг/м3, при температуре молока, "С
10,0 10,5 п,о 11,5 12,0 12,5 13,0 | 13,5 14,0 14,5
15,0 1,3 1,2 1,1 1,0 0,8 0,7 о,6 0,4 о,з 0,1
15,5 1,4 1,3 1,2 1,1 1,0 0,8 0,7 0,6 0,4 0,3
16,0 1,6 1,4 1,3 1,2 1,1 1,0 0,8 0,7 0,6 0,4
16,5 1,7 1,6 1,4 1,3 1,2 1,1 1,0 0,8 0,7 0,6
17,0 1,8 1,7 1,6 1,4 1,3 1,2 1,1 1,0 0,8 0,7
17,5 2,0 1,8 1,7 1,6 1,4 1,3 1,2 1,1 1,0 0,8
18,0 2,1 2,0 1,8 1,7 1,6 1,4 1,3 1,2 1,1 1,0
18,5 2,2 2,1 2,0 1,8 1,7 1,6 1,4 1,3 1,2 1,1
19,0 2,3 2,2 2,1 2,0 1,8 1,7 1,6 1,4 1,3 1,2
19,5 2,5 2,3 2,2 2,1 2,0 1,8 1,7 1,6 1,4 1,3
20,0 2,6 2,5 2,3 2,2 2,1 2,0 1,8 1,7 1,6 1,4
20,5 2,7 2,6 2,5 2,3 2,2 2,1 2,0 1,8 1,7 1,6
21,0 2,9 2,7 2,6 2,5 2,3 2,2 2,1 2,0 1,8 1,7
21,5 3,0 2,9 2,7 2,6 2,5 2,3 2,2 2,1 2,0 1,8
22,0 3,1 3,0 2,9 2,7 2,6 2,5 2,3 2,2 2,1 2,0
22,5 3,3 3,1 з,о 2,9 2,7 2,6 2,5 2,3 2,2 2,1
23,0 3,4 3,3 3,1 3,0 2,9 2,7 2,6 2,5 2,3 2,2
23,5 3,5 3,4 3,3 3,1 3,0 2,9 2,7 2,6 2,5 2,3
24,0 3,6 3,5 3,4 з,з 3,1 3,0 2,9 2,7 2,6 2,5
24,5 3,8 3,6 3,5 3,4 3,3 3,1 3,0 2,9 2,7 2,6
25,0 3,9 3,8 3,6 3,5 3,4 3,3 3,1 3,0 2,9 2,7
360
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Брусиловский Л. П. Инструментальные методы и экспресс-анализаторы для кон-
троля состава и качества молока и молочных продуктов. — М.: Молочная промыш-
ленность, 1997. — 48 с.
Геккелер К., Экштайн X. Аналитические и препаративные лабораторные мето-
ды/ Пер. с нем. — М.: Химия, 1994. — 416 с.
Горбатов А. В. Реология мясных и молочных продуктов. — М.: Пищевая про-
мышленность, 1979.— 383 с.
Инихов Г. С., Врио Н. П. Методы анализа молока и молочных продуктов. — М.:
Пищевая промышленность, 1971. — 423 с.
Мачихин Ю. А., Мачихин С. А. Инженерная реология пищевых материалов. —
М.: Легкая и пищевая промышленность, 1981. — 216 с.
Методы анализа состояния и защита окружающей среды в мясной и молочной
промышленности /Н. В. Макаров, И. С. Анцыпович, А. В. Степанов и др. — М.: Аг-
ропромиздат, 1989. — 151 с.
Методы исследования молока и молочных продуктов. Литовский филиал
ВНИИМСа и республиканского правления НТО пищевой промышленности. — М.:
АгроНИИТЭИММП, 1988.- 173 с.
Практикум по физико-химическим методам анализа /Под ред. О. М. Петрухи-
на. — М.: Химия, 1987. — 248 с.
Продукты из обезжиренного молока, пахты и молочной сыворотки /А. Г. Храм-
цов, Э. Ф. Кравченко, К. С. Петровский и др.; Под ред. А. Г. Храмцова и П. Г. Нес-
теренко. — М.: Легкая и пищевая промышленность, 1982. — 296 с.
Рогов И. А., Горбатов А. В., Свинцов В. Я. Дисперсные системы мясных и молоч-
ных продуктов. — М.: Агропромиздат, 1990. — 320 с.
Структурно-механические характеристики пищевых продуктов/Под ред.
А. В. Горбатова. — М.: Легкая и пищевая промышленность, 1982. — 296 с.
Юинг Г Инструментальные методы химического анализа /Пер. с англ. — М.:
Мир, 1989. — 608 с.
ОГЛАВЛЕНИЕ
Введение...................................................................3
Глава 1. ОТБОР ПРОБ И ПОДГОТОВКА К ИССЛЕДОВАНИЮ............................7
Глава 2. СПЕКТРАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ.................................13
Молекулярная спектроскопия................................................16
Молекулярно-абсорбционная спектрометрия...................................18
Фотометрия................................................................18
ИК-спектрометрия..........................................................22
Определение массовой доли белка в молочнобелковых концентратах ме-
тодом Лоури (по Н. И. Арбатской, А. Н. Конвой, Н. А. Лапшинской).......25
Определение массовой доли небелковых азотсодержащих соединений
в продукте методом Лоури...............................................27
Определение массовой доли белка в молоке по связыванию красителя.27
Определение массовой доли лактозы в казеине и казеинате натрия по ме-
тодике Международной организации стандартизации (ИСО)..................29
Определение массовой доли лактозы методом Лоренса......................31
Определение массовой доли углеводов в жидкой кисломолочной смеси
«Малютка»..............................................................32
Определение содержания железа в молоке и молочных продуктах по ме-
тодике Международной организации стандартизации (ИСО)..................33
Определение содержания нитратов и нитритов.............................40
Молекулярно-люминесцентная спектрометрия..................................47
Флуориметрия..............................................................47
Определение содержания витамина В! (тиамина)...........................53
Определение содержания витамина В2 (рибофлавина).......................56
Атомная спектроскопия.....................................................59
Атомно-абсорбционная спектрометрия........................................61
Атомно-эмиссионная спектрометрия..........................................67
Сущность метода и подготовка проб к определению содержания натрия,
калия, кальция, магния, железа, марганца, меди, цинка, свинца, кадмия,
кобальта, никеля, хрома................................................68
Методика определения массовой доли натрия, калия, кальция, магния,
железа, марганца, меди, цинка, свинца, кадмия, кобальта, никеля, хрома.74
Спектроскопия магнитного резонанса. Масс-спектроскопия....................75
Спектрометрия ядерного магнитного резонанса...............................77
Спектрометрия электронного парамагнитного резонанса.......................85
Масс-спектрометрия........................................................86
Глава 3. ТУРБИДИМЕТРИЯ И НЕФЕЛОМЕТРИЯ.....................................89
Определение массовой доли жира в молоке турбидиметрическим методом..92
Определение степени дисперсности жира в гомогенизированном молоке
и сливках (по В. Вайткусуи Э. Казлаускайте) ..............................93
363
Исследование процесса свертывания молока и определение готовности сгуст-
ка (по К. Буткусу и Р. Буткусу)...................................95
Определение готовности сливок к сбиванию (по Р. Буткусу, Д. Качераус-
кису и А. Буткене)................................................97
Глава 4. РЕФРАКТОМЕТРИЯ И ПОЛЯРИМЕТРИЯ............................100
Рефрактометрия....................................................100
Определение массовых долей белка, лактозы и СОМО в молоке с использо-
ванием рефрактометра ИРФ-464..................................104
Определение массовой доли жира в молочных продуктах с использованием
рефрактометра Аббе (по И. Н. Влодавцу)........................107
Определение массовой доли лактозы в молоке с использованием рефрак-
тометра РЛ-2................................................. 109
Определение массовой доли влаги в сгущенном молоке с сахаром с использо-
ванием рефрактометра РЛ-2.....................................111
Поляриметрия.......................................................111
Определение массовой доли лактозы в молоке (по Г. Качераускене).116
Определение массовой доли лактозы в молочной сыворотке (по Г. Качераус-
кене) ........................................................117
Определение массовой доли лактозы в молочном сахаре (по Г. Качераускене) ... 118
Определение массовой доли сахарозы в творожных изделиях и мороженом ... 118
Определение массовой доли сахарозы в плавленых сырах (по Г. Качераус-
кене и Ю. Маргялите)..........................................120
Глава 5. УЛЬТРАЗВУКОВОЙ МЕТОД ИССЛЕДОВАНИЯ.......................122
УЗ-приборы для определения состава и свойств молока..............122
Определение массовой доли жира и СОМО и плотности молока (сливок) на
УЗ-анализаторе «Клевер-1М».......................................125
Глава 6. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ................................129
Кондуктометрия...................................................130
Потенциометрия...................................................132
Индикаторные электроды...........................................133
Электроды сравнения..............................................136
Характеристика измерительных устройств...........................136
Определение водородного показателя (pH) молока и молочных продуктов .. 143
Определение концентрации ионов кальция для характеристики термо-
устойчивости молока (по К. К. Горбатовой и П. И. Гуньковой)...144
Потенциометрическое титрование...................................145
Определение титруемой кислотности молока и молочных продуктов с ис-
пользованием потенциометрического анализатора.................148
Определение массовой доли белка в молоке методом формольного титрова-
ния с использованием титриметрического анализатора............149
Вольтамперометрия................................................151
Определение содержания меди, свинца, кадмия, цинка в продуктах с по-
мощью вольтамперометрического анализатора АВА-1...............158
Глава 7. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ..................166
Распределительная хроматография..................................168
Бумажная хроматография...........................................169
Тонкослойная хроматография.......................................172
Жидкостно-жидкостная хроматография...............................174
Гель-хроматография...............................................175
Газовая хроматография............................................178
364
Определение фракций молочных белков методом гель-хроматографии
(по К. Буткусу и В. Буткене)..................................181
Определение содержания свободных летучих жирных кислот методом
газожидкостной хроматографии (по С. Урбенен В. Завецкене).....182
Определение содержания отдельных органических кислот методом газо-
жидкостной хроматографии......................................184
Адсорбционная хроматография......................................185
Определение содержания низко- и высокомолекулярных жирных кислот
в сырах методом жидкостно-адсорбционной хроматографии (по Гарперу).... 186
Определение содержания витамина А методом жидкостно-адсорбцион-
ной хроматографии..................................................188
Осадочная хроматография...............................................191
Определение содержания меди, олова, свинца, цинка методом осадочной
хроматографии (по К. М. Олыиановой, Н. М. Морозовой и В. Н. Копыловой) ... 191
Ионообменная хроматография............................................193
Определение содержания отдельных аминокислот методом ионообменной
хроматографии.................................................196
Определение содержания хлорида натрия в сырах, брынзе, соленых тво-
рожных изделиях, сливочном масле методом ионообменной хромато-
графии с катионитом...........................................198
Аффинная хроматография...........................................200
Г л а в а 8. ЭЛЕКТРОФОРЕТИЧЕСКИЕ МЕТОДЫ...............................204
Зональный электрофорез ..........................................205
Электрофорез на бумаге и в тонком слое...........................207
Гель-электрофорез................................................209
Электрофорез в свободном потоке..................................211
Изоэлектрическое фокусирование...................................212
Определение фракций белков молока с применением электрофореза на бу-
маге (по Е. А. Ждановой и И. Н. Влодавцу).....................216
Определение фракций белков молока с применением диск-электрофо-
реза (по К. Буткусу и А. Антавичюсу)..........................219
Глава9. ЭБУЛИОСКОПИЯ И КРИОСКОПИЯ................................222
Определение точки замерзания молока..............................223
Г л а в а 10. РЕОЛОГИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЙ..................227
Сырье и молочные продукты как реологические тела.................228
Методы измерений н измерительные приборы.........................237
Определение структурно-механических характеристик жидких (условно-
ньютоновских) и жидкообразных молочных продуктов методами капил-
лярной вискозиметрии..........................................242
Определение структурно-механических характеристик жидкообразных и твер-
дообразных молочных продуктов методами ротационной вискозиметрии.... 247
Определение предельного напряжения сдвига пластично-вязких молочных
продуктов и твердости сыра при помощи пластометров и пенетрометров.257
Определение компрессионных характеристик сыров................259
Гл а ва 11. АНАЛИТИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ СОСТАВА
МОЛОКА И МОЛОЧНЫХ ПРОДУКТОВ......................................265
Измерение влажности и содержания сухого вещества.................265
Определение массовых долей влаги и сухого вещества в питьевом молоке,
мороженом, сырах, твороге и творожных изделиях................268
365
Определение массовой доли сухого вещества в питьевом молоке и кисломо-
лочных напитках ускоренным методом.............................269
Определение массовых долей влаги и сухого вещества в мороженом ускорен-
ным методом................................................. 270
Определение массовых долей влаги и сухого вещества в сырах, твороге
и творожных изделиях ускоренными методами.....................270
Определение массовой доли влаги в сливочном масле.............272
Определение массовой доли СОМО в масле без наполнителей.......274
Измерение жирности...............................................276
Кислотный метод..................................................277
Гравиметрический метод...........................................284
Метод Розе—Готлиба............................................284
Метод Шмидт-Бондзинского—Вацлава..............................287
Метод Можонье.................................................290
Экспресс-метод................................................291
Определение массовой доли белка..................................291
Метод Кьельдаля...............................................292
Ускоренный метод..............................................294
Расчетный метод определения белка в молоке....................296
Определение массовой доли углеводов..............................296
Йодометрический метод............................................296
Определение массов&й доли лактозы.............................297
Определение массовой доли сахарозы............................298
Метод Бертрана...................................................301
Ускоренный феррицианидный метод..................................306
Определение массовой доли хлорида натрия......................308
Определение массовой доли этилового спирта....................310
Глава 12. АНАЛИТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ СВОЙСТВ
МОЛОКА И МОЛОЧНЫХ ПРОДУКТОВ......................................312
Определение кислотности..............................................
Титруемая кислотность................................................
Предельная кислотность..............................................
Определение плотности................................................
Ареометрический метод................................................
Пикнометрический метод...............................................
Определение термоустойчивости молока.................................
Тепловая проба (по Т. Владыкиной и В. Вайткусу)......................
Алкогольная проба....................................................
Кальциевая проба.....................................................
Фосфатная проба......................................................
Определение чистоты молока...........................................
Определение микробной обсеменениости молока..........................
Метод с метиленовым голубым.......................................
Метод с резазурином...............................................
Определение натуральности молока и наличия фальсифицирующих веществ....
Определение примеси восстановленного молока в цельном................
Определение примеси соды (карбоната или бикарбоната натрия)..........
Качественный метод................................................
312
312
314
315
315
318
320
321
323
324
325
325
326
326
326
328
328
329
329
Количественный метод.............................................329
Определение наличия аммиака.........................................330
Определение наличия пероксида водорода..............................330
Определение примеси маститного молока...............................331
Определение числа соматических клеток визуальным методом.........332
Определение числа соматических клеток на индикаторе ЙСКМ-1.......332
Определение эффективности гомогенизации.............................335
Метод отстаивания жира..............................................335
Метод центрифугирования жира........................................336
366
Определение эффективности пастеризации..............................338
Проба на пероксидазу с хлоридом парафенилендиамина.................338
Проба на пероксидазу с йодистокалиевым крахмалом....................340
Проба на фосфатазу по реакции с 4-аминоантипирином (арбитражный метод).... 342
Проба на фосфатазу по реакции с фенолфталеинфосфатом натрия........343
Метод определения кислой фосфатазы.................................344
Определение дисперсности влаги сливочного масла....................346
Определение индекса растворимости сухих молочных продуктов.........347
Определение влагоудерживающей способности сгустков молочнокислых культур
и заквасок методом центрифугирования по методике ВНИМИ.............348
Приложения.........................................................350
Список рекомендуемой литературы....................................362
Учебное издание
КРУСЬ ГАЛИНА НИКОЛАЕВНА
ШАЛЫГИНА АЛЕКСАНДРА МИХАЙЛОВНА
ВОЛОКИТИНА ЗИНАИДА ВЛАДИМИРОВНА
МЕТОДЫ ИССЛЕДОВАНИЯ МОЛОКА
И МОЛОЧНЫХ ПРОДУКТОВ
Учебник для вузов
Художественный редактор В. А. Чуракова
Технические редакторы М. Н. Кулешова, Н. Н. Зиновьева
Корректор Л. Г. Новожилова
Лицензия № 010159 от 06.03.97 г.
Сдано в набор 14.01.2000. Подписано в печать 13.06.2000. Формат 60 х 88 */16-
Бумага офсетная № 1. Гарнитура Таймс. Печать офсетная.
Усл. п.л. 22,54. Усл. кр.-отт. 22,54.
Усл. изд. л. 24,47. Изд. № 038. Тираж 3000 экз.
Заказ 1659 «С» № 052.
Федеральное государственное ордена Трудового Красного Знамени
унитарное предприятие «Издательство «Колос»
107807, ГСП-6, Москва, Б-78, Садовая-Спасская ул., 18.
Типография ОАО «Внешторгиздат». 127576, Москва, Илимская, 7.
ISBN 5-10-003440-8
9 785100 034407