/

































































































































































































































































































Автор: Рогов И.А. Антипова Л.В. Глотова И.А.
Теги: продукты животноводства и охоты пищевое производство обработка продуктов исследования мяса
ISBN: 5-10-003612-5
Год: 2001




Текст
УЧЕБНИК
Л.В.АНТИПОВА, И.А.ГЛОТОВА, И.А.РОГОВ
МЕТОДЫ ИССЛЕДОВАНИЯ МЯСА И МЯСНЫХ ПРОДУКТОВ
УЧЕБНИКИ И УЧЕБНЫЕ ПОСОБИЯ ДЛЯ СТУДЕНТОВ ВЫСШИХ УЧЕБНЫХ ЗАВЕДЕНИЙ
Л. В. АНТИПОВА, И. А. ГЛОТОВА, И. А. РОГОВ
МЕТОДЫ ИССЛЕДОВАНИЯ
МЯСА И МЯСНЫХ ПРОДУКТОВ
Допущено Министерством образования Российской Федерации в качестве учебника для студентов высших учебных заведений, обучающихя по специальности «Технология мяса и мясных продуктов» и направлению подготовки дипломированных специалистов «Технология сырья и продуктов животного происхождения»
©
МОСКВА «КОЛОС» 2001
УДК 637.07(076.5)
ББК 36-1:36.92я73
А72
Редактор Куркина Н. В.
Рецензенты: кафедра технологии мяса и мясных продуктов Северо-Кавказского ГТУ (канд. техн, наук С. И. Постников, канд. техн, наук Ю. И. Куликов), кафедра аналитической химии Воронежского государственного университета (д-р техн, наук, проф. В. Ф. Селеменев)
Антипова Л. В., Глотова И. А., Рогов И. А.
А72 Методы исследования мяса и мясных продуктов. — М.: Колос, 2001. — 376 с.: ил. (Учебники и учеб, пособия для студентов высш. учеб, заведений).
ISBN 5-10-003612-5.
Приведены теоретические основы и различные физические, химические, физико-химические, биохимические и микробиологические методы анализа состава, функционально-технологических и структурномеханических свойств, показателей качества и безвредности мясного сырья и продуктов.
Для студентов вузов, обучающихся по специальности «Технология мяса и мясных продуктов» и направлению «Технология сырья и продуктов животного происхождения».
УДК 637.07(076.5)
ББК 36—1:36.92я73
ISBN 5-10-003612-5
© Издательство «Колос», 2001
ОГЛАВЛЕНИЕ
Глава 1. Анализ химических компонентов мяса и мясных продуктов..................9
1.1. Белки, пептиды и аминокислоты..........................................9
1.2. Липиды .............................................................. 34
1.3. Углеводы............................................................. 43
1.4. Фосфорорганические соединения.........................................47
1.5. Вода................................................................. 49
Лабораторная работа № 1. Определение суммарных белков в тканях животных ускоренным фотометрическим методом на основе минерализации проб......... 55
Лабораторная работа №2. Определение белков в тканях животных фотометрическими методами без минерализации проб................................. 60
I. Определение массовой доли белка методом Лоури...................... 63
2. Определение массовой доли белка биуретовым методом................. 64
3. Определение массовой доли белка методами, основанными на связывании красителей............................................................ 65
4. Определение массовой доли белка методами УФ-спектрофотометрии...... 67
Лабораторная работа № 3. Анализ фракционного состава белков на основе их растворимости.............................................. 69
Лабораторная работа № 4. Количественное определение актомиозина......... 71
Лабораторная работа №5. Количественное определение коллагена............ 73
1. Определение коллагена в мясе по методу В. Г. Воловинской........... 76
2. Определение коллагена на основе предварительного гидролиза......... 77
Лабораторная работа № 6. Определение гемоглобина крови.................. 80
1. Выделение кристаллов гемоглобина................................... 82
2. Определение гемоглобина по качественной реакции на гемовую группу (проба Тейхмана)...................................................... 83
3. Количественное определение растворенного гемоглобина в плазме крови. 83
Лабораторная работа № 7. Определение форм гемоглобина (пигментов) на основе спектральных характеристик.................................... 84
1. Определение оксигемоглобина ....................................... 86
2. Определение гемоглобина............................................ 86
3. Определение метгемоглобина......................................... 86
Лабораторная работа № 8. Количественное определение гемоглобина и органического железа в крови................................................ 87
Лабораторная работа № 9. Количественное определение пигментов мяса...... 89
1. Определение общего содержания пигментов.............................91
2. Определение форм миоглобина (пигментов) на основе спектральных характеристик мяса.................................................... 92
Лабораторная работа № 10. Количественное определение аминного азота .... 92
1. Медный способ ..................................................... 95
3
2. Нингидриновый способ............................................... 96
3. Способ формольного титрования...................................... 97
Лабораторная работа AL1 II. Количественное определение триптофана ....... 98
Лабораторная работа № 12. Количественное определение пролина ............101
Лабораторная работа АЗ 13. Определение белков и белковых веществ меюдом гель-фильтрапии..........................................................104
Лабораторная работа № 14. Анализ белков методами ионообменной хроматографии ................................................................109
1. Разделение белков сыворотки крови на ДЭАЭ-целлюлозе................111
2. Разделение белков сыворотки крови на КМ-иеллюлозе.................112
Лабораторная работа № 15. Анализ аминокислот на автоматическом аминокислотном анализаторе....................................................113
Лабораторная работа №° 16. Анализ аминокислот методом бумажной хроматографии ................................................................118
Лабораторная работа № 17. Определение аминокислот методом тонкослойной ионообменной хроматографии на пластинах..................................123
Лабораторная работа № 18. Определение экстрактивных веществ..............128
1. Определение карнозина в безбелковом экстракте по диазореакции......129
2. Определение дипептидов хроматографией на бумаге....................131
3. Определение дипептидов с помощью тонкослойной хроматографии на пластинках «Силуфол»..................................................133
Лабораторная работа № 19. Количественное определение суммарных липидов в животных тканях........................................................134
1. Гравиметрический метод в аппарате Сокслета.........................135
2. Гравиметрический метод в пленочном испарителе .....................136
3. Колориметрический метод............................................137
Лабораторная работа №20. Определение фракционного состава жиров .........138
1. Фракционирование пищевых топленых жиров кристаллизацией............140
2. Определение температуры плавления фракций .........................140
Лабораторная работа №° 21. Определение фосфолипидов......................141
1. Качественная реакция на лецитин....................................142
2. Хроматографическое разделение фосфолипидов..........................142
Лабораторная работа №° 22. Определение холестерина.......................144
1. Получение кристаллов холестерина...................................147
2. Цветные реакции на холестерин......................................148
3. Количественное определение холестерола.............................150
Лабораторная работа №23. Определение жирнокислотного состава животных жиров...........................................................152
Лабораторная работа №° 24. Количественное определение гликогена в животных тканях........................................................157
Лабораторная работа №°25. Количественное определение молочной кислоты..................................................................161
1. Метод определения содержания молочной кислоты по цветной реакции с вератролом .......................................................163
2. Метод определения содержания молочной кислоты по цветной реакции с ««уш-оксидифенилом................................................166
Лабораторная работа № 26. Количественное определение пектина в мясных продуктах................................................................169
1. Определение пектина термогравиметрическим методом по пектату кальция.............................................................170
2. Определение пектина фотометрическим методом по реакции с карбазолом..........................................................172
4
Лабораторная работа ЛЬ27. Количественное определение целлюлозы ......... 173
1. Определение массовой доли целлюлозы в модификации А. И. Ермакова .. 174
2. Определение массовой доли целлюлозы в модификации И. С. Лурье .....175
Лабораторная работа ЛЬ 28. Определение фосфорорганических соединений и их производных..............................................................177
1. Определение массовой доли АТФ.......................................178
2. Определение массовой доли креатинфосфата ...........................180
Лабораторная работа ЛЬ 29. Определение массовой доли влаги в мясе и мясных продуктах................................................................181
1. Определение массовой доли влаги высушиванием при температуре (103 ±2) °C ..........................................................183
2. Определение массовой доли влаги высушиванием при температуре (150 • 2) С ..........................................................183
3. Определение массовой доли влаги высушиванием в аппарате САЛ.........184
4. Определение массовой доли влаги высушиванием в устройстве Я10-ФВУ..184
Лабораторная работа Ns 30. Определение активности воды...................185
Контрольные вопросы и задания......................................188
Глава 2. Физические, физико-химические и структурно-механические свойства мяса и мясных продуктов.............................................................190
2.1. Физические свойства...................................................191
2.2. Теплофизические свойства .............................................196
2.3. Функционально-технологические свойства ...............................199
2.4. Структурно-механические свойства......................................202
Лабораторная работа № 1. Определение цветности мяса и мясных продуктов..204
Лабораторная работа Ns 2. Определение цветности твердых животных жиров..209
Лабораторная работа Ns 3. Определение акустических свойств мяса и мясных продуктов................................................................211
Лабораторная работа Ns 4. Исследование теплофизических характеристик мяса и мясных продуктов ......................................................215
Лабораторная работа Ns 5. Определение микроструктурных показателей мяса.223
Лабораторная работа Ns 6. Определение влагосвязывающей способности (ВСС) мяса...............................................................230
1. Метод прессования ..................................................231
2. Метод центрифугирования.............................................232
Лабораторная работа Ns 7. Определение основных функционально-технологических свойств мясных фаршей ........................................233
1. Определение влагоудерживающей способности...........................234
2. Определение жироудерживающей способности............................234
3. Определение эмульгирующей способности и стабильности эмульсии.......236
4. Определение влаго- и жироудерживающей способностей и устойчивости
фаршевой эмульсии в одной навеске (метод Р. М. Салаватулиной и др.) .236
Лабораторная работа Ns 8. Определение гелеобразующей способности животных и растительных белков....................................................238
1. Получение гелей.....................................................240
2. Определение физических показателей студней..........................241
Лабораторная работа Ns 9. Определение структурно-механических свойств мяса и мясных продуктов ......................................................245
1. Определение усилий среза............................................252
2. Определение сдвиговых свойств на коническом пластомере .............252
3. Определение предельного напряжения сдвига и эффективной вязкости на ротационном вискозиметре РВ-8М.....................................254
5
4. Определение предельного напряжения сдвига и эффективной вязкости на ротационном вискозиметре типа «Реотест».............................256
5. Определение вязкости на вискозиметре Энглера........................259
6. Определение адгезионных свойств модельных фаршей ...................261
Контрольные вопросы н задания........................................263
Глава 3. Биохимические свойства животных тканей................................265
3.1. Механизм автолиза и автолитические превращения мышечной ткани.........266
3.2. Биохимические свойства крови..........................................271
3.3. Биохимическая активность животных тканей .............................274
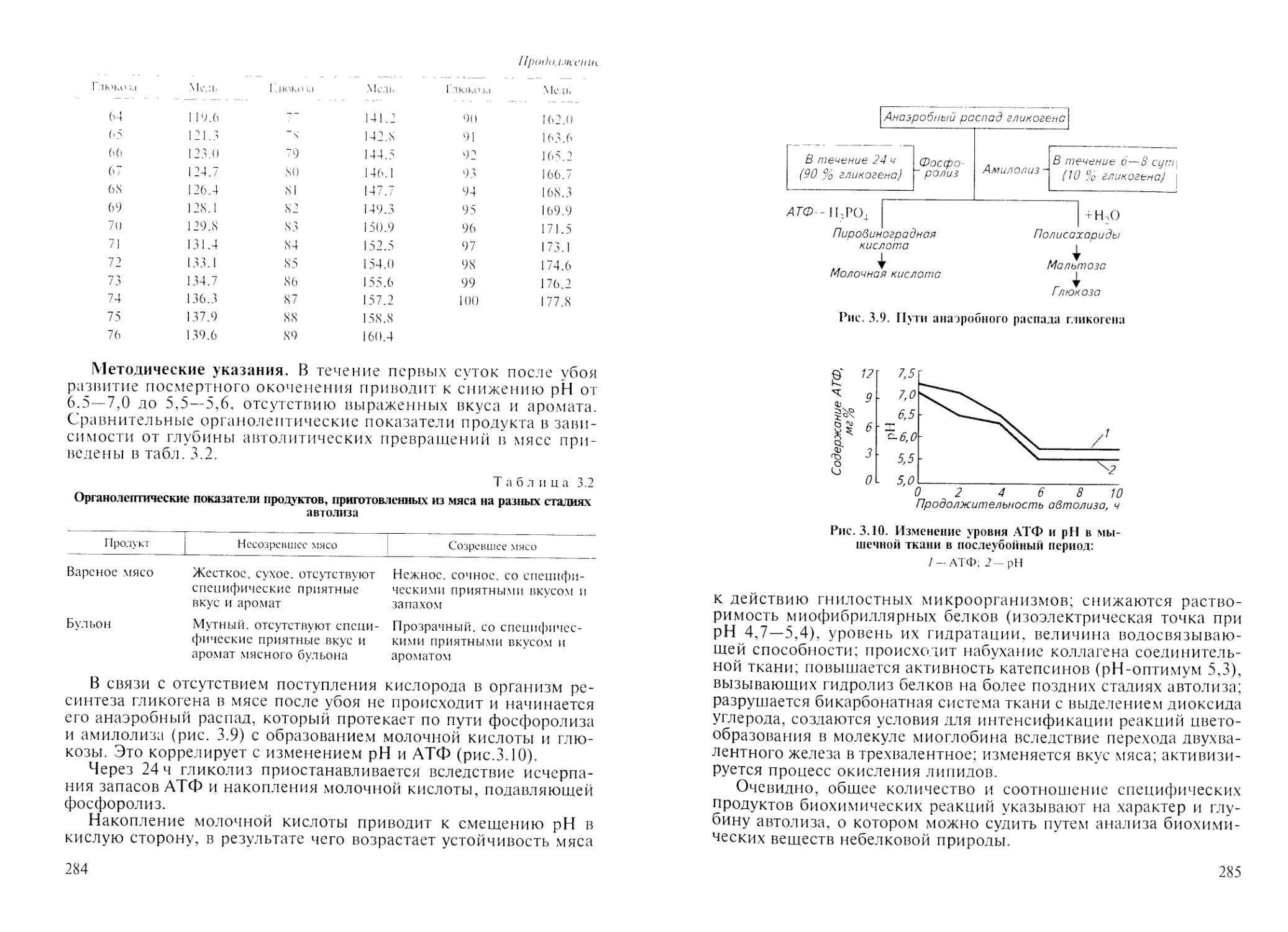
Лабораторная работа Ns 1. Оценка глубины и характера автолитических превращений мышечной ткани методами биохимического анализа небелковых веществ..................................................................282
1. Определение pH .....................................................286
2. Количественное определение гликогена и молочной кислоты в мышечной ткани................................................................. 287
3. Определение АТФ.....................................................287
4. Количественное определение глюкозы в вытяжке мышечной ткани по методу Бертрана.....................................................287
Лабораторная работа Ns 2. Определение активности катепсинов мышечной ткани....................................................................288
Лабораторная работа №3. Определение компонентов системы свертывания крови....................................................................293
1. Действие кальция на систему свертывания крови и ее фракций..........294
2. Определение тромбина................................................294
Лабораторная работа Ns 4. Определение молокосвертываюшей активности пепсина..................................................................296
Лабораторная работа № 5. Определение инсулина поджелудочной железы.......297
1. Качественные реакции на инсулин.....................................298
2. Количественное определение инсулина.................................299
Лабораторная работа Ns 6. Определение гормонов шитовидной железы и надпочечников ..............................................................302
1. Определение тиреоидина по качественной реакции с иодидом калия.305
2. Определение адреналина надпочечников................................305
Контрольные вопросы.................................................306
Глава 4. Пищевая ценность и качество мяса и мясных продуктов ..................308
4.1. Пищевая ценность......................................................310
4.2. Качество..............................................................325
Лабораторная работа Ns 1. Органолептическая оценка мяса и мясных продуктов................................................................348
Лабораторная работа Ns 2. Определение показателей биологической ценности расчетным методом........................................................354
Лабораторная работа Ns3. Определение переваримости мяса и мясных продуктов................................................................358
Лабораторная работа Ns 4. Определение качественных показателей животных жиров....................................................................360
1. Органолептический анализ............................................363
2. Определение массовой доли влаги.....................................364
3. Определение кислотного числа........................................365
4. Определение степени окислительной порчи жира........................366
Лабораторная работа Ns 5. Определение биологической ценности животных жиров....................................................................374
1. Анализ ненасыщенных жирных кислот спектрофотометрическим методом .............................................................377
6
2. Определение содержания витаминных примесей в пищевых животных жирах..................................................................382
Лабораторная работа Ар 6. Определение свежести мяса и мясных продуктов .385
I. Органолептический анализ ..........................................388
2. Физико-химический анализ...........................................389
3. Определение свежести мяса по микроструктурным показателям .........395
Лабораторная работа 7. Определение степени кулинарной готовности мяса и мясных продуктов .....................................................399
Контрольные вопросы и задания.......................................402
Глава 5. Контаминанты мяса и мясных продуктов ................................404
5.1. Общая характеристика контаминантов мяса и мясных продуктов...........404
5.2. Методы контроля безопасности мяса и мясных продуктов ................415
Лабораторная работа Ns 1. Определение микробных контаминантов в мясе......425
1. Приготовление и бактериологический анализ препаратов.................438
2. Приготовление посевов и определение физиолого-биохимических и культуральных признаков микроорганизмов....................................439
3. Ускоренное обнаружение бактерий на основе люминесцентно-серологических методов.........................................................450
Лабораторная работа Ns 2. Определение микробных контаминантов в колбасных изделиях и продуктах из мяса........................................454
1. Определение общего количества микроорганизмов в 1 г продукта.......458
2. Определение бактерий группы кишечной палочки в 1 г продукта........459
3. Определение бактерий из рода Salmonella............................461
4. Определение протея.................................................462
5. Определение коагулазоположительных стафилококков...................463
6. Определение сульфитредунируюших клостридий.........................464
Лабораторная работа Ns 3. Определение антибиотиков......................466
Лабораторная работа № 4. Определение гормонов...........................468
Лабораторная работа Ns 5. Определение пестицидов........................471
1. Определение хлорорганических пестицидов методом ТСХ................472
2. Определение ФОП энзимохроматографическим методом...................475
Лабораторная работа Ns 6. Определение фенолов в копченых мясных продуктах.478
1. Определение границ проникновения фенолов...........................480
2. Количественное определение фенолов в колбасных изделиях............480
Лабораторная работа № 7. Определение бенз(а)пирена в копченых мясных продуктах...............................................................483
1. Качественное определение бенз(а)пирена.............................485
2. Количественное определение бенз(а)пирена...........................485
Лабораторная работа Ns 8. Определение нитратов и нитритов...............486
1. Определение нитрат- и нитрит-ионов ионометрическим методом.........488
2. Определение нитритов и нитратов фотометрическим методом............490
Лабораторная работа Ns 9. Определение токсичных элементов...............498
1. Реакции качественного обнаружения токсичных элементов..............506
2. Качественное и количественное определение мышьяка по Зангер—Блеку....509
3. Количественное определение свинца нефелометрическим методом .......510
4. Определение массовой доли свинца, кадмия, меди, цинка методом атомноабсорбционной спектроскопии...........................................512
5. Количественное определение токсичных элементов полярографическим методом ...............................................................519
6. Количественное определение токсичных элементов колориметрическими методами..........................................................527
7
Лабораторная работа № 10. Экспресс-определение радионуклидов методами радиометрии.............................................................541
1. Экспресс-определение объемной и удельной активности у-излучаюши.х нуклидов с помошыо радиометра СРП-68-01 ........................... 543
2. Экспресс-определение объемной и удельной активности [З-излучаюши.х нуклидов методом прямого измерения толстых проб.....................544
Лабораторная работа № 11. Определение удельной суммарной р-радиоактив-ности мяса, костей и мясных продуктов по удельной активности зольных остатков................................................................545
Лабораторная работа 12. Определение радионуклидов радиохимическими методами................................................................551
1. Определение стронция-90 фосфатным методом .........................555
2. Определение цезия-137 с предварительным концентрированием в виде ферроцианида никеля-цезия.............................................558
3. Определение цезия-137 сурьмяно-иодидным методом....................562
Контрольные вопросы.................................................565
Рекомендуемая литература .....................................................566
Предметный указатель..........................................................568
Г л а в a 1 АНАЛИЗ ХИМИЧЕСКИХ КОМПОНЕНТОВ МЯСА И МЯСНЫХ ПРОДУКТОВ
Пищевые продукты представляют собой сложный комплекс химических веществ, в состав которых входят белки, липиды, углеводы, витамины, минеральные соли и вода. Каждая группа веществ выполняет свои определенные функции в жизнедеятельности организма. В процессе приготовления пищи входящие в нее ингредиенты подвергаются биохимическим и физико-химическим превращениям, создавая структуру, вкус, цвет и запах пищевых продуктов.
1.1. БЕЛКИ, ПЕПТИДЫ И АМИНОКИСЛОТЫ
Поступающие с пищей белки в организме человека выполняют важнейшие функции, многие из которых незаменимы. Известно, что белковое голодание в течение нескольких дней приводит к серьезным заболеваниям, а длительное отсутствие в пище белков вызывает смерть человека. Белки содержатся *во всех продуктах питания, но массовая доля их весьма различна. Например, в мясе — 18—22 мае. %, рыбе — 17—20, яйце — 20—36, молоке — 3,5, ржаном хлебе — 7,8 мае. %.
Важность информации о количественном содержании белков связана с определением потенциальных возможностей продуктов питания в покрытии физиологических потребностей организма человека, норма которых составляет около 100 г белка в сутки.
Белки сами по себе не являются незаменимыми компонентами пищи человека. Для нормального питания и поддержания здоровья необходимы содержащиеся в них незаменимые аминокислоты, обязательность наличия которых в пищевых рационах связана с тем, что они не синтезируются животными организмами. В связи с этим весьма важно их качественное и количественное соотношение. Белки, содержащие все незаменимые аминокислоты, называют полноценными. Если в белке нет хотя бы одной незаменимой аминокислоты, то он считается неполноценным. Постоянная нехватка полноценного белка в пище ведет к возникновению анемии, отечности тканей, развитию дегенеративных изменений почек, печени и поджелудочной железы,
9
нарушению умственных способностей, вызывает тяжелые необратимые нарушения физиологических функций.
Большинство белков мяса относится к полноценным, что делает их обязательным компонентом пищи.
Количественное содержание и физико-химические свойства белков определяют поведение пищевых систем под воздействием воды, электролитов, pH среды, окислителей и восстановителей, нагревания и т. д., что имеет весьма важное значение в формировании заданных функционально-технологических и органолептических свойств сырья, полуфабрикатов и готовых мясных продуктов, включая формирование коагуляционно-денатурационной структуры фаршевых изделий, сваривание и гидротермический распад коллагена при доведении продуктов до кулинарной готовности.
Уникальные биологические функции и технологическое значение белков в производстве мясных продуктов тесно связаны с особенностями их химического строения и пространственной структуры, отличающихся разнообразием, динамичностью и наличием внутримолекулярных взаимодействий, способностью изменяться под воздействием внешних факторов в водно-солевых растворах и водных растворах полярных растворителей, восстанавливать исходное состояние, вступать в различные реакции, включая биока-талитические процессы.
В состав мяса и мясопродуктов входят простые и сложные белки, в том числе водо-, соле- и щелочерастворимые, обеспечивающие, например, такие важные функции, как удержание воды, набухаемость и растворимость, а также сложные белки-пигменты, отвечающие за цвет продукта. Белки различаются не только химическим и пространственным строением, но и размерами частиц, а также формой молекул. Последняя включает две группы — фибриллярные и глобулярные, отличающиеся физико-химическими свойствами, прежде всего растворимостью в воде, водно-солевых растворах и водных растворах полярных растворителей, а также способностью к денатурации, гидролизу и другим превращениям.
Белки мяса и мясопродуктов принято разделять по морфологическому признаку клеток мышечных тканей животных (рис. 1.1). Саркоплазматические, миофибриллярные белки и белки стромы обеспечивают функциональность пищевой системы в получении мясопродуктов, а группа ядерных белков самостоятельного технологического значения не имеет.
Фракция суммарных белков саркоплазмы составляет 20—25 % количества всех мышечных белков. Установлено, что белки саркоплазмы способны образовывать гель, особенно в присутствии АТФ. При высоких концентрациях Са2+ гель разжижается. Это связано с присутствием в саркоплазме фрагментов саркоплазматического ретикулума. Очищенные от примесей белки саркоплазмы способность желировать утрачивают.
10
Белки клетки мышечной ткани животных
Рис. 1.1. Белки клетки мышечной ткани животных
Миоген представляет собой комплекс миогенов А, В и С, различающихся кристаллической формой. Обычно под миогеном подразумевается вся миогеновая фракция. Миоген составляет около 20 % всех белков мышечного волокна. Он растворяется в воде, образуя гомогенные растворы небольшой вязкости с массовой долей 20—30 %. Температура денатурации свободного от солей миогена 55—60 °C, изоэлектрическая точка (ИЭТ) лежит в интервале pH 6,0—6,5. С течением времени часть миогена переходит в нерастворимое состояние.
Миоальбумины составляют около 1—2% белков мышечного волокна. Растворимы в воде и нерастворимы в кислой среде, так как имеют изоэлектрическую точку около pH 3,0—3,5; температура денатурации 45—47 °C.
Глобулин X составляет около 20 % общего количества белковых веществ мышечного волокна. Растворим в солевых растворах даже очень низкой концентрации, температура денатурации при pH 6,5 около 50 °C, при pH 7,0 — около 80 °C, изоэлектрическая точка лежит в интервале pH 5,0—5,2.
Миоглобин — хромопротеид, составляющий в среднем 0,6—1,0 % общего количества белков. Он состоит из белковой части — глобина и простетической группы — гема. На одну молекулу миоглобина приходится одна группа гема. В миоглобине не обнаружено цистина. Миоглобин хорошо растворим в воде. Температура денатурации около 60 °C. Денатурация миоглобина сопровождается отщеплением простетической группы. Миоглобин способен присоединять оксид азота, сероводород и кислород за счет дополнительных связей. В последнем случае образуется оксимиоглобин светло-красного цвета, который переходит с течением времени в метмиоглобин коричневого цвета. При этом железо отдает один
И
(коричневый) Fe3*
Рис. 1.2. Взаимопревращения пигментов мяса
электрон. При действии восстановителей метмиоглобин снова образует миоглобин. Последний окрашен в пурпурно-красный цвет и обусловливает естественную окраску мышечной ткани, интенсивность которой зависит от его содержания и соотношения форм пигментов белка. Изменение цветности мяса и мясопродуктов (рис. 1.2) происходит под влиянием микрофлоры, теплового воздействия, посола, света и других факторов. Количество пигментов, глубина их превращений и образование форм соответствующей окраски играют значительную роль в получении продуктов высокого качества. При переходе миоглобина в метмиоглобин пурпурно-красная окраска мяса меняется на коричневую. Она наиболее заметна, когда в мет-форму переходит более 50 % миоглобина. Это свойство широко используется для определения сроков хранения мяса путем выявления соотношения различных спектральных форм миоглобина, а также при регулировании цветности мясопродуктов.
Миозин — фибриллярный белок с асимметрией молекулы 10:1, составляет около 40 % белков клетки мышечной ткани. Гетеро-генен. Обычно под миозином подразумевается вся миозиновая фракция. Миозин — полноценный, хорошо переваримый белок. Совершенно чистый миозин растворим в воде и образует вязкий раствор с массовой долей до 4 % белка. Растворы солей щелочных металлов небольшой молярной концентрации (0,25— 0,04 моль/дм3) осаждают миозин из его растворов; в солевых растворах повышенной молярной концентрации (до 0,6 моль/дм3) он растворяется. Температура денатурации миозина около 45— 50 °C (у птицы около 51 °C); изоэлектрическая точка определяется при pH 5,4. Биологические функции миозина связаны с координированным движением живых организмов и автолитическими превращениями мышечных тканей после убоя животных.
Актин составляет 12—15% всех мышечных белков и является основным компонентом тонких нитей. Этот белок существует в двух формах — глобулярной (Г-форма) и фибриллярной (Ф-фор-ма). В растворах с низкой ионной силой актин существует в виде мономера с относительной молекулярной массой около 47 000.
12
при повышении ИОННОЙ UK1J1D1 pav 1 ztv- 1|7H JNU^ V! r<
уровня Г-актин полимеризуется в Ф-актин, очень похожий на нить. Г-актин представляет собой одну полипептидную цепочку, сложенную в глобулу. Быстрая полимеризация актина происходит также при добавлении ионов Mg2*. При этом образуется двунитчатая спираль, каждая составляющая в которой напоминает нить бус, закрученных одна вокруг другой. Актин относится к полноценным и легкоусвояемым белкам.
Актомиозин — это сложный комплекс, который формируется при добавлении раствора актина к раствору миозина и сопровождается увеличением вязкости раствора. Поскольку цепь Ф-актина содержит много молекул Г-актина, каждая нить Ф-ак-тина может связывать большое число молекул миозина. Рост прекращается при добавлении АТФ или в присутствии ионов Mg2+. Содержание актомиозина указывает на глубину автолитических превращений в процессе трупного окоченения и позволяет опосредованно судить о функциональности мясного сырья в процессе технологической обработки.
Тропомиозин — белок палочковидной формы с относительной молекулярной массой около 70 000, постоянно присутствующий в структуре тонких (актиновых) филаментов. Биологическая роль тропомиозина сводится к регулированию взаимодействия актина и миозина в процессе мышечного сокращения. Массовая доля тропомиозина составляет 10—12 % всех белков миофибрилл или 2,5 % белков мышц. Растворим в воде, но из мышечной ткани водой не извлекается. Изоэлектрическая точка определяется при pH 5,1.
Тропонин представляет собой сферическую молекулу с относительной молекулярной массой 76 000, включающей три субъединицы, аминокислотный состав которых полноценен.
Весьма важной группой сложных белков являются нуклеопротеиды, играющие первостепенную роль в жизнедеятельности организма, в частности в явлениях наследственности. Простети-ческой группой нуклеопротеидов служат нуклеиновые кислоты. Они нерастворимы в воде, но растворяются в щелочах. В их состав входит простой белок, как правило протамин или гистон. При полном гидролизе нуклеопротеидов образуются ос-аминокислоты, рибоза и дезоксирибоза, фосфорная кислота и азотистые основания (пуриновые и пиримидиновые). Массовая доля нуклеопротеидов в мышечной ткани составляет 0,207—0,245 %, где они входят в состав рибосом и саркоплазматического ретикулума. В основном это рибонуклеопротеиды, функции которых связаны с синтезом белков. Нуклеопротеидами богаты ткани мозга, где они представлены нейроглобулином (дезоксирибонуклеопротеидом) и нейро-стромином (рибонуклеопротеидом). Нуклеопротеиды являются полноценными белками, однако, как отмечалось выше, самостоятельного технологического значения не имеют и входят в состав мышечных клеток.
13
В состав перечисленных белков входят все аминокислоты, включая важнейшую из них — триптофан, что послужило основой для оценки количественного содержания полноценных белков в сырье и продуктах.
Из белков стромы важная роль отводится коллагену, эластину и ретикулину, по наличию которых судят о прочностных свойствах соединительных тканей. Это протеиноиды, являющиеся фибриллярными белками упроченной структуры, нерастворимые в обычных растворителях.
Уникальными свойствами обладает коллаген. Фибриллы коллагеновых нитей состоят из субъединиц, называемых тропоколлагеном, в котором /^-группы всех аминокислот находятся на внешней стороне молекулы и мало участвуют в стабилизации структуры. Характерным признаком коллагена является высокое содержание пролина и нестандартной аминокислоты — гидроксипролина (рис. 1.3, а), сумма которых составляет около 21 %. На определении 4-гидроксипролина (оксипролина) основаны многие методы количественного анализа коллагена. Нестандартная аминокислота — гидроксилизин (рис. 1.3, б) также может служить средством идентификации коллагенов.
Коллаген способен сильно набухать в водных растворах, при этом масса его увеличивается в 1,5—2 раза. По этому свойству он уступает лишь миозину мышечной ткани. Высокая гидратация коллагена связана с содержанием в его структуре значительных количеств диамино- и аминодикарбоновых кислот. При смещении pH в кислую или щелочную сторону от НЭТ на-бухаемость коллагена резко увеличивается, при этом масса белка в состоянии полного набухания может достигнуть от 400 до 1000 % к массе сухого белка. Способность коллагена к набуханию имеет большое значение для мясного, желатинового и кожевенного производств.
Вторым важным белком стромы мышечных волокон и соединительных тканей является эластин. Это, по существу, многокомпонентная система, представленная сложными белками —
он
I з
НС,---СНэ
ls 3 1
н2с; ^сн-соон
H а
ОН
H,N-СН,-СН-СН,-СН,-CH-COON 6'| 4'3’2 1
ОН
Рис. 1.3. Структурные формулы нестандартных аминокислот, входящих в состав коллагена:
а — гидроксипролин; б— гидроксилизин
14
гликопротеинами. Подобно коллагену эластин ooiai иищии^» .. аланином. Тропоэластин отличается от тропоколлагена большим содержанием лизина, но малым — пролина. Суммарное содержание глицина, аланина, валина и пролина в эластине составляет почти 70 %. Из-за малого содержания кислых и основных аминокислот молекула эластина практически неполярна. В водной среде цепи эластина принимают форму глобул. Гидрофобные цепи аминокислот, образующие соответствующие связи, спрятаны внутри молекулы, окруженной водой. В результате свободная энергия системы минимальна. Точная структура эластина, к сожалению, пока не идентифицирована. В то же время установлено, что он очень устойчив: не растворяется в холодной и горячей воде, солевых растворах, разбавленных растворах кислот и щелочей. Даже концентрированная серная кислота оказывает на него слабое воздействие. Он не образует желатин, практически не расщепляется пищеварительными ферментами.
Ретикулин также входит в состав стромы мышечных волокон и соединительных тканей. Подобно коллагену и эластину он является гликопротеином, неполярен, очень устойчив, плохо усваивается организмом.
Таким образом, группа соединительнотканных белков имеет общие свойства и структурные признаки. Именно они используются в исследовательской практике для оценки пищевой ценности сырья и продуктов путем их количественного анализа, например, при определении сорта мяса, а также пищевой ценности по соотношению триптофана и оксипролина.
В соответствии с современной теорией питания роль соединительнотканных белков пересмотрена. Установлено положительное влияние ингредиентов соединительной ткани на процесс пищеварения. Показано, что коллаген и эластин обладают свойствами пищевого волокна, проявляют радиопротекторные свойства, активно стимулируют секреторную и двигательную функции желудка и кишечника, оказывают благоприятное действие на состояние и функцию полезной кишечной микрофлоры. На основе коллагена и других белков соединительной ткани создаются биологически ценные пищевые продукты и добавки с лечебно-профилактическим эффектом. Для рационального использования сырьевых ресурсов и создания биологически полноценных продуктов, обеспечивающих здоровье человека, весьма важно дозированное введение соединительных тканей в рецептурные композиции, что возможно лишь на основе количественного анализа соответствующих ингредиентов.
Гистидинсодержащие дипептиды являются специфической составной частью скелетной мускулатуры. Из мышечной ткани животных и человека они были выделены в начале века русским биохимиком В. С. Гулевичем. Установлено, что эти вещества выполняют ряд важных функций в процессе обмена веществ и энергии
15
при жизни, участвуя в процессах окислительного фосфорилирования, происходящих в мышцах при образовании макроэргических фосфатных соединений (АТФ и креатинфосфата). Входя в состав мышечной ткани, гистидинсодержащие дипептиды стимулируют секреторную функцию пищеварительных желез. К ним относятся: карнозин — дипептид, состоящий из остатков р-аланина и гистидина, и ансерин — гомолог карнозина, р-аланил-1-метилгистидин (метилированный карнозин).
р==^---СНэ-СН-СООН
N. NH NH I co-(ch2)2-nh2
Карнозин
р: —: СН2-СН-СООН
N^^N-СНз NH
CO-(CH2)2-NH2
Ансерин
В скелетных мышцах убойных животных содержание карнозина и ансерина колеблется в пределах 0,014—1,000 %.
Кровь — жидкая соединительная ткань животного организма, которая циркулирует в артериях, венах и капиллярах. Представляет собой непрозрачную жидкость красного цвета со слабощелочными свойствами (pH 7,3—7,5), специфического запаха и солоноватого вкуса. На долю крови приходится в среднем у крупного рогатого скота 7,5 % живой массы, у свиней — 4,5, у овец — 7 и у птицы — 8 %. Кровь вместе с лимфой и тканевой жидкостью, окружающей клетки, является внутренней средой организма, которая объединяет органы с тканями и выполняет ряд весьма важных прижизненных функций (доставка молекулярного кислорода и питательных веществ к клеткам организма, освобождение тканей от углекислоты и продуктов распада). Дыхательная, питательная, выделительная, защитная, регуляторная функции крови обеспечиваются определенными ее компонентами, а в целом кровь представляет собой сложную многокомпонентную систему. После убоя животных кровь частично остается в капиллярах и потому является неотъемлемой составляющей мяса. В настоящее время кровь приобретает огромное значение и как самостоятельное сырье для производства антианемических продуктов, фракции крови используются для структурирования пищевых систем, придания окраски продуктам, получения эмульсий, обогащения продуктов органическим железом, которое в 4—6 раз быстрее усваивается организмом по сравнению с другими источниками. В связи с вышеизложенным анализ веществ и компоентов крови необходим для расширения спектра максимального и рационального использования этого важного сырья.
16
Кровь состоит из жидкой части — плазмы и взвешенных в ней форменных элементов. К форменным элементам относятся: эритроциты (красные кровяные тельца), лейкоциты (белые кровяные тельца), тромбоциты (кровяные пластинки, бляшки). В крови разных видов животных массовая доля форменных элементов неодинакова: в среднем у крупного рогатого скота 33 %, у мелкого — 28, у свиней — 43,6 % к массе крови.
Кровь, лишенная форменных элементов (например, центрифугированием с соблюдением мер предосторожности против свертывания), представляет собой плазму. На долю растворимых веществ плазмы крови приходится 9—10 % ее массы, из них около 7 % составляют белки, остальная часть состоит из липидных компонентов, азотистых и безазотистых экстрактивных и минеральных веществ.
Плазма крови, из которой выделен белок фибриноген (предшественник фибрина), называется сывороткой.
Белки крови как пищевое сырье эффективнее, чем другие белки, могут восстанавливать белки плазмы и гемоглобин в организме. В связи с этим среди пищевых белков одно из первых мест принадлежит именно им. Массовая доля белков в цельной крови зависит от вида, возраста, упитанности, условий предубойного содержания животных и в среднем составляет: у крупного рогатого скота 17,41 %, баранов — 16,59, свиней — 2,25 %. При этом в среднем 6,8—7,3 % белков находится в плазме, 30,3—32,7 % — в форменных элементах. Основная масса белков крови представлена альбуминами, глобулинами, фибриногеном и гемоглобином.
По аминокислотному составу наиболее полноценным является фибриноген, в структуре которого содержится 3,5 % триптофана, 7,0 % фенилаланина, 2,6 % метионина.
Фибриноген — основной компонент системы свертывания крови. Он нерастворим в воде, но хорошо растворим в разбавленных растворах нейтральных солей и щелочах, осаждается сульфатом магния и хлоридом натрия ранее, чем наступает полное насыщение. Таким образом, фибриноген близок по своим свойствам к глобулинам. Фибриноген быстро усваивается.
Сывороточные альбумин и глобулин также содержат полный набор незаменимых аминокислот, хотя и в меньшем количестве. Особенно мало содержание триптофана в сывороточном альбумине. Гемоглобин нельзя отнести к полноценным белкам, так как в нем отсутствует изолейцин. Однако содержание других незаменимых аминокислот в нем довольно высокое. Поэтому в сочетании с другими белками крови £го можно рассматривать как важнейший источник жизненно необходимых аминокислот.
17
Рис. 1.4. Схема гема
таким ооразом, пищевая и биологическая ценность крови тесно связана с наличием природного пигмента — гемоглобина. Это сложный белок, состоящий из окрашенной простети-ческой группы гема (рис. 1.4) и бесцветной белковой части — глобина. Молекула гемоглобина по своей форме приближается к сфере диаметром около 5,5 нм, относительная молекулярная масса 64 500, ИЭТ при pH 5,5. Гемоглобин в кислой и щелочной средах диссоциирует на гем и глобин. Так, если добавить раствор гемоглобина к смеси ацетон — НС1, то глобин осаждается, а гем остается в растворе.
Особенно полезна информация о количестве гемоглобина как
источника органического железа, усвояемость которого прямо пропорциональна содержанию гемовых пигментов (табл. 1.1).
Таблица 1.1
Формы железа и его усвояемость для различных видов мяса
Мясо и мясопродукты Массовая доля железа, % Коэффициент усвояемости железа, %
гемового негемового
Говядина 20,2 79,8 18
Свинина 15,3 84,7 16
Баранина 18,3 81,7 17
Птица 18,3 81,7 17
Печень 28,0 72,0 14
Кровь 69,3 30,7 31
Концентрация гемоглобина в крови зависит от вида животного и количества эритроцитов. Гемоглобин разных животных разнится аминокислотной последовательностью. Гемоглобин выполняет функцию дыхательного белка — переносчика кислорода. При воздействии на гемоглобин окислителей железо гема переходит в трехвалентную форму и образует подобно миоглобину метгемоглобин. Изменение валентности связано с изменением цвета продукта от ярко-алого до коричневого, что имеет прямое технологическое значение. Некоторые фракции белков плазмы крови обладают желирующими свойствами. Сывороточный альбумин спосо
18
бен желировать плазму. с>тот факт открываем huddiv использования крови для пищевых целей, особенно при получении структурированных продуктов, и ставит перед технологами задачу отработки и внедрения методов исследования компонентов крови.
Использование крови и ее фракций для пищевых целей, в том числе для создания нетрадиционных лечебно-профилактических и специальных продуктов, актуально для решения задач рационального использования ресурсов, организации безотходных технологий на мясокомбинатах, улучшения экологических условий производства.
В аналитической практике известно достаточно много методов определения белков. Наиболее распространены метод Кьельдаля и его известные модификации, основанные на минерализации проб и количественном определении азота.
Получившие в последнее время распространение ускоренные фотометрические методы (рис. 1.5) имеют существенные преимущества по сравнению с классическим методом Кьельдаля. Основные из них — простота и быстрота выполнения — позволяют использовать эти методы для массовых анализов и проведения оперативного контроля качества сырья и готовой продукции по содержанию белка.
Эти методы весьма перспективны, особенно для однородных систем, и основаны на непосредственном фотометрирова-нии пробы.
Метод Лоури основан на реакции взаимодействия фенольного реактива Фолина—Чокалтеу с щелочными растворами белков, приводящей к образованию продуктов реакции синего цвета. Интенсивность окраски зависит от содержания в исследуемом белке тирозина и триптофана. Оптическую плотность окрашенных растворов измеряют при длине волны 750 нм. Метод рекомендуется для определения массовой доли белка в сильно разбавленных растворах, при наблюдении за ходом ферментативных процессов, для определения белков сыворотки крови, яичного альбумина, белков молока, фракционированных растительных белков, белков митохондриальных фракций и мембран.
Рис. 1.5. Экспресс-методы определения белков в мясе и мясных продуктах
19
,____х.с. w|jdJuiidHnn сине-фиолетовой
окраски при воздействии на белки сульфата меди в присутствии щелочи. Природа белка почти не влияет на формирование окрашенного биуретового комплекса, который возникает за счет присоединения ионов меди к пептидным связям белка. Оптическую плотность окрашенных растворов измеряют при длине волны около 540 нм. Преимущества метода — возможность применения стандартного белка, например сывороточного альбумина, для построения калибровочного графика, воспроизводимость и точность.
Методы определения массовой доли белка по связыванию красителей основаны на способности белков при pH ниже изоэлектрической точки (pH 2—4) присоединять кислые красители с образованием нерастворимых комплексов, после удаления которых измеряют оптическую плотность исходного раствора красителя относительно полученного раствора (фильтрата):
Белок + Краситель —> Белок-краситель (осадок) +
+ Краситель (раствор несвязавшегося красителя).
Оптическая плотность фильтрата уменьшается с увеличением массовой доли белка. Методы рекомендуются для количественного определения белка в образцах сырого мяса, а также в продуктах кулинарной степени готовности после различных видов технологической обработки (варки, запекания, копчения и т. д.).
Методы УФ-спектроскопии основаны на способности белковых растворов поглощать свет при длине волны около 280 нм благодаря присутствию в структуре белков аминокислот триптофана, тирозина и фенилаланина. Результаты определения прямо пропорциональны содержанию этих аминокислот в белках. Преимущества метода — малая трудоемкость, несложная подготовка проб, быстрота и простота анализа; предел чувствительности 10— 20 мкг/см3. Методы рекомендуются для количественной оценки содержания белков в элюатах после разделения белковых смесей методом электрофореза или хроматографии, определения белков в мясных, яичных и бобовых продуктах.
К высокоточным методам относятся хроматографические.
Хроматография — метод разделения смесей газов, жидкостей, растворенных веществ путем сорбции в динамических условиях. В простейшем варианте разделение происходит при прохождении потока анализируемой смеси через колонку, содержащую сорбент. Вследствие различной сорбируемости компонентов смеси достигается их разделение по высоте сорбента при повторяющихся циклах сорбция — десорбция.
Известно несколько подходов к классификации методов хроматографии с использованием различных признаков:
20
по средам, в которых проводится разделение (жидкостная, газожидкостная, газовая);
по механизму разделения (молекулярная или адсорбционная, ионообменная, осадочная, комплексообразовательная, окислительно-восстановительная);
по форме проведения процесса (колоночная, капиллярная, хроматография на бумаге и в тонком слое);
по способу проведения процесса (фронтальная, вытеснительная, проявительная).
В табл. 1.2 хроматографические методы систематизированы по подвижным и неподвижным фазам, форме проведения процесса и принципу разделения.
Таблица 1.2
Классификация методов хроматографии по принципу разделения
Методы хроматографии Подвижная фаза (ПФ) Неподвижная фаза (НФ) Форма неподвижного слоя Принцип разделения
Жидкостная:
твердо-жидкостная Жидкость Твердая Колонка Адсорбционный
жидкостно-жидкостная » Жидкость » Распределительный
ионообменная » Твердая » Ионный обмен
Газовая:
газоадсорбционная Газ » » Адсорбционный
газожидкостная » Жидкость » Распределительный
Гель-хроматография » » » По размерам молекул
Хроматография в тонком слое » Твердая Тонкий слой Адсорбционный
Хроматография на бумаге » Жидкость Хроматографическая бумага Распределительный
Аффинная » Лиганд Колонка Адсорбционный
По области применения хроматографические методы делятся:
на аналитические (установление качественного и количественного составов смесей);
неаналитические (исследование физико-химических характеристик сорбатов — давления пара, теплоты растворения, коэффициентов активности);
21
v1 чемие веществ в чистом и особо чис-
том виде);
промышленные (получение индивидуальных веществ, например лекарственных препаратов, в больших количествах).
Каждый хроматографический метод по мере развития сопровождался возникновением новых вариантов. Например, адсорбционная и распределительная хроматография осуществляется на колонках (колонки могут иметь различную форму и конструкцию), фильтровальной бумаге и в тонком слое сорбента, нанесенном на стеклянную пластинку. В зависимости от этих факторов различают следующие варианты: колоночная, хроматография на бумаге, хроматография в тонком слое.
Жидкостная хроматография (ЖХ) — метод разделения и анализа сложных смесей, в котором подвижной фазой является жидкость. Метод ЖХ характеризуется более широким кругом анализируемых объектов, чем газовая хроматография, поскольку большинство веществ не обладает достаточной летучестью, многие неустойчивы при высоких температурах (особенно высокомолекулярные соединения) и разлагаются при переходе в газообразное состояние. В методе ЖХ разделение проводят, как правило, при комнатной температуре. Особенности различных вариантов ЖХ обусловлены физико-химическими свойствами жидкой подвижной фазы.
Газовая хроматография (ГХ) — метод разделения и определения летучих соединений. Компоненты анализируемой смеси распределяются между неподвижной фазой (твердое вещество или жидкость) и газом-носителем (подвижная фаза).
Твердая неподвижная фаза применяется в газоадсорбционной хроматографии (ГАХ). Разделение обусловлено адсорбционными свойствами наполнителя колонки по отношению к разделяемым соединениям, преимущественно газам. Распространенные адсорбенты — силикагель, активированный уголь, молекулярные сита и цеолиты.
Жидкая неподвижная фаза применяется в газожидкостной хроматографии (ГЖХ). Она распределяется в виде тонкой пленки на поверхности инертного твердого носителя. Большой выбор жидких фаз, позволяющий работать в широком температурном диапазоне (20—400 °C), делает ГЖХ наиболее селективным хроматографическим методом разделения не только жидких, но и некоторых твердых соединений. Низкие пределы обнаружения, экс-прессность, точность и простота операций обеспечили методу ГЖХ широкое применение для анализа сложных объектов.
Гель-хроматография — метод разделения веществ, основанный на различии размеров их молекул. Метод известен под названиями «гель-проникающая», «эксклюзионная» и «молекулярно-ситовая хроматография». Последнее название наиболее полно отражает сущность метода, однако более распространен термин «гель-проникающая хроматография».
22
В качестве неподвижной фазы применяют гели, имеющие определенный размер пор; подвижной фазы — водные иди органические элюенты. Наиболее простое объяснение механизма разделения в гель-хроматографии состоит в том, что молекулы анализируемых веществ распределены между неподвижным растворителем в порах сорбента и элюентом, протекающим через слой неподвижной фазы. Молекулы с размером, позволяющим проникать в поры сорбента при движении вдоль колонки, задерживаются в порах. Молекулы, имеющие размеры большие, чем поры, не проникают в сорбент и вымываются из колонки со скоростью движения элюента. Молекулы, проникающие в поры всех размеров, движутся наиболее медленно. Снижение скорости движения компонентов вдоль колонки прямо связано с количеством пор, в которые способны диффундировать распределяемые частицы.
Таким образом, методом гель-хроматографии можно разделять смеси веществ в зависимости от размеров их молекул. Выход компонентов из колонки происходит в порядке снижения их молекулярных масс. Вытекающие из колонки растворы анализируют различными методами, например спектрофотометрическим, фотометрическим, титриметрическим.
Наиболее широкое распространение среди носителей для гель-хроматографии белков получили сорбенты, приготовленные на основе декстрана (сефадексы, сефакрилы, молселекты), полиакриламида (биогели Р, акрилексы), агарозы (сефарозы, биогели А) и др. Набухая в воде, они образуют гели.
Номер в маркировке сефадекса означает его пористость. Выбор определенной марки сефадекса определяется молекулярной массой исследуемого белка (чем больше соответствие размеров молекул и величины пор, тем выше селективность), а степень зернения зависит от поставленной задачи. Для обессоливания растворов белков и их концентрирования обычно используют сефадексы G-25 и G-50 (грубый или средний). При разделении же смеси белков пользуются сефадексами тонкого или сверхтонкого зернения. Чем мельче частицы геля, тем эффективнее происходит разделение, но тем меньше скорость протекания раствора через колонку.
Гели готовят путем насыщения водой или элюентом в течение 5—20 ч соответствующего гелеобразователя (сефадекс, полиакриламид и др.). Продолжительность и температура насыщения указаны на упаковке препарата.
Ионообменная хроматография основана на динамическом стехиометрическом обмене ионов между анализируемым раствором и ионообменником (ионитом). Этот метод широко применяется при анализе, разделении и очистке органических и неорганических соединений, ионизирующих в водных и неводных растворителях. Объем удерживания зависит от изменения свободной энергии реакции ионного обмена. Соотношение концентраций
23
ооменивающихся ионов в растворе и в фазе сорбента определяется ионообменным равновесием.
При фракционировании белков методом ионообменной хроматографии большое внимание уделяют выбору ионообменника (природе материала и емкости ионита) и буферного раствора (величине pH и ионной силы, природе буфера и буферной емкости), при котором осуществляется сорбция веществ.
При работе с белками в качестве сорбентов используют иониты, обладающие высокой степенью гидрофильности.
Белки могут быть разделены как на катионитах, так и на анионитах. Выбор типа ионита определяется изоэлектрическими точками хроматографируемого материала и устойчивостью белка в определенной зоне значений pH.
Эффективная сорбция белков происходит при значениях pH, отстоящих от р! не менее чем на единицу. В области pH < pl — 1 белки можно исследовать на катионитах, а в области pH > pl + 1 — на анионитах. Изменение pH в направлении к ИЭТ способствует десорбции белков. При работе с белками используют буферные растворы с низкой ионной силой, но высокой буферной емкостью. Для этого пользуются буферными растворами, рК которых отстоит от величины pH, используемой в эксперименте, не более чем на 0,3—0,5 единицы pH. Хроматографию на анионитах ведут в таких системах, где диссоциируемым компонентом является катион (буферы: трис, пиридин, имидазол и др.), а для катионитов — анион (ацетатный, фосфатный, бикарбонатный буфер и др.).
При ионообменной хроматографии смесь белков сорбируется в верхней части колонки и затем вытесняется веществами, уменьшающими их сорбцию на ионите. Понижение сорбции осуществляют повышением ионной силы раствора и (или) изменением его pH. Изменение pH и ионной силы элюирующего буферного раствора можно проводить путем создания ступенчатого или градиентного элюирования.
Разделение аминокислот методом ионообменной хроматографии проводят обычно на синтетических смолах — катионитах, представляющих собой сополимеры стирола с дивинилбензо-лом. Роль ионогенных группировок в них играют сульфогруппы (—SO3H). Такие смолы являются сильными электролитами и диссоциируют при любых значениях pH на БО3-ионы, прочно связанные со смолой, и противоионы Н+, переходящие в раствор. Во избежание закисления буферных растворов, окружающих смолу, ионы Н+ в ней замещают на ионы Na+, т. е. используют смолу в форме SO3Na. Для того чтобы осуществить сорбцию аминокислот на смоле путем обмена с ионами Na, их переводят в форму Na-катионитов. С этой целью аминокислоты помещают обычно в Na-цитратный буферный раствор при pH 2,2.
24
При этом значении pH все аминокислоты несут положительный заряд и находятся в виде катионов
NHt
R\
соон
Степень прочности сорбции и десорбция на смоле разных аминокислот, определяемая главным образом величинами зарядов молекул, различна. Наиболее прочно на смоле сорбируются диаминокислоты и наименее прочно — дикарбоновые аминокислоты. Разделение аминокислот при ионообменной хроматографии происходит при десорбции их со смолы элюирующими буферными растворами, отличающимися от исходного буферного раствора большими величинами pH и (или) ионной силы. Обычно для элюирования кислых и нейтральных аминокислот используют Na-цитратные буферные растворы с pH 3,25 и 4,25. Для десорбции со смолы наиболее прочно связанных (основных) аминокислот используют более щелочной Na-цитратный буферный раствор со значением pH 5,28 и более высокой ионной силой (0,35 моль/дм3).
Разделение аминокислот на колонках в настоящее время проводят автоматически в специальных приборах, называемых анализаторами аминокислот. Погрешность определения на этих приборах для многих аминокислот достигает 0,1 %. Для работы на этих анализаторах требуется 0,3—1 мг белка.
Метод широко используется при определении состава свободных аминокислот и связанных в структуре белков и пептидов.
Хроматография в тонком слое (ХТС) — один из простейших методов хроматографического анализа, разделение компонентов при котором происходит при перемещении подвижной фазы через нанесенный на подложку (пластинку) тонкий слой сорбента. Продвижение элюента (подвижная фаза) по сорбенту (неподвижная фаза) обеспечивается капиллярными силами.
Хроматография в тонком слое основана на принципах распределительной и адсорбционной хроматографии.
В качестве носителей при тонкослойной хроматографии чаще всего используют измельченный силикагель или порошок целлюлозы, а также оксид алюминия, кизельгур, крахмал, сефадекс и др. В последнее время широкое распространение получил метод разделения аминокислот на пластинках, покрытых ионообменной смолой. В качестве растворителей используют системы с органическими растворителями различной степени полярности. Тонкослойная хроматография имеет ряд преимуществ по сравнению с хроматографией на бумаге: она характеризуется быстротой разделения (30—60 мин в зависимости от размеров пластинки), большей чувствительностью метода (при-
25
мерно в 1U раз) и устойчивостью слоя сорбента по отношению к агрессивным проявителям и нагреванию.
Хроматографию на пластинках проводят в закрытых стеклянных камерах, предварительно насыщенных парами растворителя, восходящим способом, при комнатной температуре или при нагревании, одномерным или двухмерным способом. Последний (второй растворитель движется по пластинке в направлении, перпендикулярном к первому) используют для повышения эффективности метода. Часто тонкослойную хроматографию сочетают с электрофорезом (метод пептидных карт). В этих случаях пробу наносят в виде небольшого пятнышка (<У~2—Змм) в один из углов пластинки на расстоянии 20 мм от ее краев. При одномерном хроматографировании пробы наносят на пластинку в виде полос.
Многие коммерческие сорбенты для тонкослойной хроматографии содержат флуоресцентные красители, что позволяет идентифицировать разделяемые соединения, поглощающие электромагнитные волны в ультрафиолетовой области спектра.
В настоящее время разделение аминокислот (гидролизатов белков) методом тонкослойной хроматографии осуществляют на пластинках, покрытых тонким слоем ионообменной смолы полистирольной природы с сульфокислотными группировками (типа Дауэкс 50x8) или ионообменной целлюлозой. Такие пластинки выпускаются промышленностью, например «Фикси-он 50 х 8» (Венгрия), или могут быть приготовлены непосредственно в лаборатории.
Сочетание процессов ионообменной и тонкослойной хроматографии при разделении на пластинках «Фиксион 50 х 8» обусловливает высокую разрешающую способность этого метода.
Пластинки могут быть использованы для разделения аминокислот, олигопептидов, аминов и др.
Метод хроматографического разделения на бумаге основан на различной растворимости разделяемых веществ в двух мало смешивающихся жидкостях, одна из которых удерживается бумагой, а другая подвижна. Неподвижной фазой служат полосы фильтровальной бумаги, которые, будучи помещенными во влажную камеру, удерживают до 20—22 % влаги, оставаясь внешне сухими. В качестве подвижной фазы обычно используют органический растворитель, насыщенный влагой. Чем больше растворимость аминокислот или пептидов в неподвижной фазе, тем медленнее они движутся при продвижении органического растворителя по бумаге и наоборот. Скорость движения будет определяться коэффициентом распределения, характерным для вещества в данной системе растворителей:
где сНф, <?пф — соответственно концентрация в неподвижной и подвижной фазе. 26
Следовательно, относительное расположение анализируемых веществ на хроматограмме для данной системы растворителей постоянно и характеризуется величиной коэффициента скорости движения.
у п расти
’ (1-2)
° фр
где 5р1СТВ — расстояние. пройденное растворенным соединением; 5фр — расстояние, пройденное фронтом растворителя.
Тем не менее следует учитывать, что воспроизводимость значения Rf зависит от качества бумаги, постоянства температуры, степени чистоты растворителей, однотипности процедур и аппаратуры и т. д.
При разделении аминокислот и пептидов обычно пользуются трехкомпонентными системами: к насыщенному влагой органическому растворителю добавляют кислоты, основания, некоторые спирты, кетоны и др. Это приводит, во-первых, к повышению растворимости воды в подвижной фазе (увеличению гидрофильности системы), во-вторых, к изменению диссоциации кислых и основных групп разделяемых соединений. Вследствие этого кислоты замедляют движение оснований, а основания — кислот.
На практике для разделения аминокислот и пептидов основного характера используют системы, содержащие фенол и крезол, для нейтральных — смеси с бутиловым спиртом и уксусной кислотой или с амиловым спиртом, а для кислых аминокислот и пептидов — системы, содержащие соединения основного характера (обычно пиридин). Если соединение плохо растворимо в подвижной фазе и остается на стартовой линии, следует увеличить гидрофильность системы, например, добавив муравьиную кислоту, метанол или формамид. Если же вещество хорошо растворимо в подвижной фазе и движется вместе с фронтом растворителя, следует использовать органический растворитель с более выраженными гидрофобными свойствами, например изоамиловый, бензиловый спирты и др.
Для лучшего разделения соединений с близкими значениями Rf> а также для увеличения величин часто проводят хроматографирование в нескольких (обычно двух) системах растворителей, пропуская второй растворитель в том же направлении, что и первый. Это приводит к уплотнению пятен разделяемых соединений в направлении, перпендикулярном к пропускаемому растворителю, и способствует их лучшему разделению. Более полного разделения аминокислот и пептидов можно добиться при двухмерной хроматографии (второй растворитель пропускают в направлении, перпендикулярном к первому) или при сочетании хро-
27
монографии (одно направление) и электрофореза (второе взаимно перпендикулярное направление). Последний метод носит название метода «пептидных карт» или «отпечатка пальцев».
При повторном разделении в системах растворителей положение пятна на хроматограмме устанавливают путем определения значений коэффициента относительной скорости движения
у
г> _ рас гв
(1.3)
kJCI
где 5 в — расстояние, пройденное растворенным соединением; 5СТ —расстояние. пройденное стандартным веществом (смесью).
В качестве стандартного можно использовать одно из веществ разделяемой смеси. Получаемые значения Аст являются более воспроизводимыми, чем величина R?.
Разделение веществ методом бумажной хроматографии проводят в специальных камерах. Для получения одномерной хроматограммы на полоску хроматографической бумаги шириной 1,5—5 см и длиной 20—70 см наносят исследуемый раствор (0,005—0,007 см3) на расстоянии нескольких сантиметров от верхнего края бумаги, который погружают в растворитель (нисходящая хроматограмма), или исследуемый раствор наносят на расстоянии нескольких сантиметров от нижнего края бумаги, который погружают в ванночку с растворителем, а верхний конец закрепляют (восходящая хроматограмма).
При получении нисходящей хроматограммы (рис. 1.6) лист бумаги подвешивают в камере, погрузив верхний край в кювету с растворителем. Под действием силы тяжести и капиллярных сил растворитель продвигается сверху вниз и по достижении края листа стекает на дно камеры. Чтобы поток был равномерным, нижний край листа нарезают зубцами.
Восходящую хроматограмму (рис. 1.7) вследствие слабой механической прочности бумаги подвешивают на специальных держателях. Растворитель поднимается до верхнего края листа бумаги за счет капиллярных сил. При этом все компоненты анализируемой смеси остаются в пределах листа. Это особенно важно в том случае, когда разделяют вещества с неизвестной подвижностью. Тогда по полученной хроматограмме можно определить
Рис. 1.6. Камера для получения нисходящей хроматографии:
я —общий вид камеры; б— расположение листа бумаги; 7 — кювета для растворителя; 2— стеклянная палочка для фиксации листа бумаги; 3— антисифонная палочка; 4— подставка; 5 — лист бумаги; 6 — камера; 7 — зуб-
а б цы на бумаге
28
коэффициент распределения Rf всех компонентов смеси.
Как только растворитель достигает намеченного рубежа, лист извлекают из камеры, отмечают положение фронта растворителя и высушивают. Поскольку большинство веществ лишено окраски, их положение на хроматограмме выявляют подходящим способом. Например, вещества, несущие радиоактивную метку, выявляют при помощи счетчиков импульсов; вещества, имеющие собственную флуоресценцию, выявляют при облучении УФ-светом (365 нм). Остальные вещества обычно обнаруживают
Рис. 1.7. Камера для выполнения восходящей хроматографии:
а. б — способы фиксации бумаги в держателе; в, г—способы размещения бумаги в камере
по окрашенным продуктам, которые образуются под действием специфических реагентов. Так, аминокислоты и полипеп-
тиды образуют с нингидрином хромофор, имеющий фиолетовую окраску.
Аффинная хроматография основана на установлении обратимых молекулярных взаимодействий, присущих биологически активным веществам. Этой способностью обладают иммунные,
ферментные и гормональные системы; белки, которые могут переносить различные малые молекулы (витамины, жирные кислоты и др.) после связывания этих молекул за счет сродства, и нуклеиновые кислоты, способные соединяться между собой или с
некоторыми белками.
В аффинной хроматографии используют нерастворимый носитель, на котором иммобилизуется соединение, называемое лигандом. Он особым образом связывает подлежащий очистке продукт, находящийся в подвижной, обычно жидкой фазе. Лиганд удерживается за счет ковалентных связей, иногда пользуются ионным обменом, адсорбцией, микроинкапсулированием и др.
Аффинная хроматография — разновидность адсорбционной, при которой связывание происходит в соответствии со специфическими свойствами двух молекул. Она основана на разных взаимодействиях: ионных, водородных, гидрофобных и других в зависимости от конформации и размера молекул.
В любом случае методы хроматографии основываются на всех возможных различиях молекул. Поскольку эти методы требуют соответствующего материально-технического оснащения, при вы-
29
к^пкрс! hoi о варианта или способа следует обращать внимание на доступность оборудования, стоимость и дефицитность реактивов.
Для практики анализа белковых веществ хроматографическими методами рекомендуется использовать данные табл. 1.3, которые дают информацию о предпочтительности того или иного метода при определении белков, пептидов, аминокислот.
Таблица 1.3
Краткое руководство к выбору методов хроматографии
Хроматографический метод | Белки Пептиды Аминокислоты
1. Распределительная на бумаге — 4- +
2. Адсорбционная в тонком слое — 4~ 4~ 4—f-
3. Проникающая +++ 4- —
4. Газожидкостная — 4—h
5. Ионообменная + 4-4- +++
6. Аффинная 4-4-4- + —
Примечание: «—» — не применяются; «+» — применяются, «++» — достаточно часто применяются; «+++» — часто применяются, наиболее предпочтительны.
С развитием современной теории питания, необходимостью разработки биологически полноценных белковых пищевых продуктов на основе комбинирования и имитации знания и практический навык в анализе белковых веществ имеют огромное значение для решения актуальных задач отрасли. В настоящее время существует множество подходов и методов хроматографии, однако лишь некоторые из них доступны и нашли достаточно широкое распространение.
Для создания продуктов с заданным составом аминокислот, расчета показателей биологической ценности необходима информация о полном составе аминокислот, т. е. их сумме в составе белков, пептидов и свободных аминокислот.
Путем анализа свободных аминокислот можно прогнозировать свойства готовых изделий, так как многие из них являются сильными вкусообразователями. Экстрактивные азотистые вещества, присутствующие в мясе, по химической природе большей частью пептиды, также участвуют во вкусообразовании.
Таким образом, возникает необходимость общего и дробного анализов белковых веществ.
Наибольшее распространение получила колоночная хроматография для проведения проникающей или ионообменной хроматографии, а также распределительная хроматография на бумаге и адсорбционная в тонком слое.
30
Простейшее устройство для хроматографического разделения на колонках изображено на рис. 1.8.
Колонки — полые стеклянные трубки, размер которых зависит от цели работы и способа разделения. В основание колонки припаивают пористую стеклянную пластинку или перфорированный диск. Необходимо следить за тем, чтобы «мертвое» пространство под диском, между ним и основанием колонки, а также внутренний диаметр шлангов, по которым выходящий из колонки элюат поступает в коллектор фракций, было минимальным. В противном случае произойдет смешивание уже разделенных веществ. Коммерческие колонки снабжены специальными приспособлениями — концевыми адапторами переменной длины, которые позволяют регулировать длину колонки и сводят к минимуму перемешивание элюируемых фракций.
Заполнив колонку до нужной высоты, закрывают нижний кран (зажим) и дают суспензии осесть, не допуская «высыхания» наполнителя в колонке (для этого над верхним слоем сорбента всегда должен находиться слой растворителя). Следят также за тем, чтобы верхний слой наполнителя имел гладкую горизонтальную поверхность. Для уменьшения взмучивания верхнего слоя при внесении в колонку образца над ним иногда помешают кружок фильтровальной бумаги или поролона. Колонку закрывают пробкой с отводом или со стеклянной трубкой и присоединяют к ре-
Рис. 1.8. Принципиальная схема устройства для колоночной хроматографии:
1 — сосуд с раствором высокой концентрации; 2— сосуд-смеситель; 3— магнитная мешалка; 4— перистальтический насос; 5— колонка; 6— УФ-детектор; 7— коллектор фракций
31
зервуару, содержащему элюирующий раствор. Для поддержания постоянного давления и сохранения постоянной скорости тока жидкости через колонку используют склянку Мариотта или насосы различных конструкций.
Образец можно вносить в колонку несколькими способами. Самый простой из них заключается в следующем: осторожно удаляют жидкость с поверхности наполнителя, оставляя слой в 1—2 мм; при помощи пипетки осторожно вносят образец и, открыв нижний кран, дают ему впитаться; остатки образца над сорбентом смывают небольшой порцией элюента; после того как он впитается поверхностью наполнителя, добавляют новые порции элюирующего раствора, создавая слой в 5—10 см. Другой способ внесения образца, при котором избыток жидкости не удаляют из колонки, заключается в увеличении плотности раствора образца путем добавления сахарозы до концентрации 0,5 моль/дм3. Раствор образца при этом спускают под слой растворителя, в результате чего он быстро впитывается в верхний слой наполнителя.
После нанесения образца колонку соединяют с верхним резервуаром, путем изменения рабочего давления устанавливают необходимую скорость истечения элюирующего раствора и с помощью коллектора начинают сбор фракций. Собирать фракции элюата необходимо с момента нанесения образца на колонку. Фракции можно собирать в пробирку по объему (с помощью сифонов), по определенному количеству капель или через определенные промежутки времени.
Элюирование с колонок, как правило, проводят растворами, изменяя pH, ионную силу (концентрацию) или оба показателя одновременно. При этом градиент pH и ионной силы может быть ступенчатым или непрерывным (плавным). При создании ступенчатого градиента пользуются серией буферных растворов, пропускаемых через колонку последовательно один за другим. При этом виде элюирования каждый из буферных растворов пропускают через колонку до тех пор, пока концентрация белка в вытекающем из колонки элюате, пройдя через максимум, не снизится почти до исходных фоновых значений.
При непрерывном градиенте элюирования изменение ионной силы и (или) pH элюирующего раствора происходит постепенно, по линейной или нелинейной зависимости от объема протекающей жидкости. Линейное изменение ионной силы или pH элюирующего раствора происходит тогда, когда эти параметры изменяются пропорционально объему протекающей жидкости. Получить линейный градиент можно с помощью прибора, состоящего из двух соединенных между собой одинаковых сосудов, установленных на одном уровне (рис. 1.9, а). В сосуде 1 находится буферный раствор с определенным значением ионной силы (или pH), которое должно быть достигнуто к концу опыта, в сосуде 2, из которого раствор поступает непосредственно в колонку, вначале находится исходный буферный раствор в том же объеме. Часто приме-
32
в
Рис. 1.9. Приборы для создания градиента концентрации: а — линейного; б — «выпуклого»; в — «вогнутого»
няют «выпуклый» или «вогнутый» градиенты, при которых ионная сила раствора увеличивается или уменьшается соответственно экспоненциальной зависимости. Форму этих градиентов легко получить с помощью простых устройств, изображенных на рис. 1.9, б, в.
33
Рис. 1.10. Установка для колоночной хроматографии с автоматическим сбором фракций:
1 — микроиасос; 2—пульт управления; 3колонка; 4— вращающийся коллектор — сборник фракций
Стекающий из колонки растворитель собирают небольшими порциями через определенные промежутки времени с помощью специальных автоматических коллекторов — сборников фракций (рис. 1.10). Содержимое каждой пробирки подвергают анализу, определяя концентрацию компонентов смеси (чаще всего определяют оптичес-
кую плотность раствора). По полученным данным строят график зависимости концентрации компонента (или оптической плотности) от объема прошедшего через колонку раствора.
1.2. ЛИПИДЫ
К липидам относятся природные органические соединения, нерастворимые в воде и растворимые в органических растворителях (хлороформе, эфире, бензоле и др.). В организме животного липиды выполняют важнейшие биологические функции: входят в состав клеточных мембран и других биологически активных структур, служат энергетическим материалом, выполняют защитную роль, а некоторые из них — функции светочувствительных пигментов, гормонов и т. п.
Название одной из групп липидов, а именно жиров (от греч. «липос» — жир), взято для обозначения класса в целом. Липиды — сборная группа химических соединений, не имеющая единой химической характеристики. В целом их можно рассматривать как класс органических соединений, большинство из которых принадлежит к сложным эфирам многоатомных или специфически построенных спиртов и высших жирных кислот.
Существуют различные классификации липидов. В зависимости от состава, строения и роли в организме сложилась следующая классификация липидов.
34
Простые липиды. Представлены двухкомпонентными веществами — сложными эфирами высших жирных кислот с глицерином, высшими или полициклическими спиртами. К ним относятся: жиры (триглицериды) — сложные эфиры высших жирных кислот и трехатомного спирта — глицерина; воски — сложные эфиры высших жирных кислот и высших спиртов и стериды — сложные эфиры высших жирных кислот и полициклических спиртов — стеролов.
Сложные липиды. Состоят из многокомпонентных молекул, компоненты которых соединены химическими связями различного типа. К ним относятся: фосфолипиды, состоящие из остатков высших жирных кислот, глицерина или других многоатомных спиртов, фосфорной кислоты и азотистых оснований различной природы; гликолипиды, в состав которых наряду с многоатомным спиртом и высшей жирной кислотой входят также углеводы.
Неомыляемая фракция липидов. В нее входят свободные высшие жирные кислоты, высшие спирты и полициклические спирты (стеролы), производные стеролов — стероиды, жирорастворимые витамины, высшие гомологи предельных углеводородов и другие соединения.
Из простых липидов наибольшее практическое значение имеют нейтральные жиры, широко встречающиеся в биологических объектах. В некоторых органах и тканях животных их массовая доля достигает 90 %. Животные жиры более разнообразны по набору высших жирных кислот по сравнению с растительными. В их составе чаще встречаются высшие жирные кислоты с числом углеродных атомов от 20 до 24.
Наиболее часто и в большей пропорции в животных жирах (табл. 1.4) встречаются олеиновая (в жирах ее более 30 %) и пальмитиновая кислоты (от 15 до 50 %).
Таблица 1.4
Массовая доля основных жирных кислот в животных жирах, %
Кислота Говяжий Бараний Свиной Куриный
Пальмитиновая 27,0-29,0 25,0-27,0 25,0-35,0 24,0-37,0
Стеариновая 24,0-29,0 25,0-31,0 12,0-16,0 4,0-7,0
Миристиновая 2,0-2,5 2,0-4,0 1,0 0,1
Олеиновая 43,0-44,0 36,0-43,0 41,0-51,0 37,0-43,0
Линолевая 2,0-5,0 3,0-4,0 3,0-11,0 18,0-23,0
Линоленовая 0,3-9,7 0,4-0,9 0,3-0,6 —
Арахидоновая 0,09-0,2 0,27-0,28 До 2,0 0,3
Животные липиды имеют различную температуру плавления (табл. 1.5), которая зависит от степени непредельности входящих в их состав жирных кислот.
35
Г а б л и ц а 1.5
Температура плавления и иодное число некоторых животных жиров
Жир Температура плавления. С ' Полное число
Бараний Говяжий Свиной Гусиный Конский Коровье масло 44-55 31—46 40—50 33-47 28—40 46—66 26-34 - 30—43 71-86 28-30 25-27
Животные жиры представляют собой смесь однокислотных (или простых) и разнокислотных (или смешанных) триглицеридов, представленных в разных соотношениях. В них также присутствует небольшая доля ди- и моноглицеридов, а также свободных жирных кислот.
Триглицериды образуют оптические и геометрические изомеры, так как во многих случаях имеют асимметричный углеродный атом в остатке глицерина и одну или несколько двойных связей в радикалах кислотных остатков. Характерно, что непредельные высшие жирные кислоты в триглицеридах находятся, как правило, в цис-конфигурации, что сказывается на форме молекулы (рис. 1.11).
Кроме нейтральных триглицеридов в состав животных жиров входят липоиды, качественный состав которых представлен фосфатидами, стеридами и стероидами.
Фосфатиды или глицерофосфолипиды — сложные эфиры глицерина, высших жирных кислот, фосфорной кислоты и азотистого основания.
Остаток линолевой кислоты
Остаток стеариновой кислоты
Остаток арахидоновой кислоты
Остаток глицерина
• Атом углерода
Атом кислорода
•—• Простая связь
•Z» Двойная связь
Рис. 1.11. Структура и форма молекулы триглицерида
36
В зависимости от характера азотистого основания среди фосфатидов различают фосфатидилхолин (лецитины), фосфатидил-коламин (кефалины), фосфатидилсерин и фосфатидилтреонин:
СНэ-О-СО-(СНД14-СН;
I
С Н - о - С О - (С Н 2) I (, - с Н;
ch2-o-p-o-ch2-ch2-n+-4:H;
\г \н3
Лецитин (фосфатидилхолин)
С Н 2 -О -С С) - (С Н 2) и.-С Н;
СН-О-СО-(СНЛ14-СН;
I
CH2-O-P-O-CH2-CH2-NH3
Кефалин (фосфатидилколамин)
СНэ-О-СО-(СНР16-СН,
I
СН-О-СО-(СН2)7-СН=СН-(СН2)7-СН3
СН2—О—Р—О—СН2—СН—СООН
° 0 NH;
Фосфатидилсерин
СН2-0-С0-(СН2)22-СН3
СН-О-СО-(СН2)7-СН=СН-(СН2)7-СН3
CH,-O-P-O-CH-CH-COOH
' </ V I
и 0 СН3 NH3
Фосф атидилтре онин
Лецитины наиболее распространены в природе. Некоторые фосфатиды, открытые сравнительно недавно, не содержат азотистого основания, место которого в молекуле в этом случае занимают глицерин и его производные:
ch2-o-co-r сн2он
СН—О—СО—R СНОН
I /
СНэ-О-Р-О-СН2 // \ о он
Фосфатидилглицерин
\ /0Н
CH2-O-CO-R СН2—О—Р—О—сн2
СН—О—СО—R' СНОН СН—О—СО—R
СН2-О-Р-О-СН2 CH2-O-CO-R
0^ Х0Н
Дифосфатидилглицерин (кардиолипин)
37
Обладая асимметричным строением (2-й углеродный атом остатка глицерина всегда асимметричен), фосфатиды оптически активны и образуют соответствующие стереоизомеры. Вместе с тем им свойственна изомерия за счет перестановки остатков высших жирных кислот из а- в [3-положение или наоборот.
Фосфатиды — важная группа липидов, которые обязательно следует включать в рацион питания. Они способствуют лучшему усвоению жиров, препятствуют ожирению печени, необходимы для профилактики атеросклероза. Потребность человека в фосфолипидах составляет 5 г в сутки. Из продуктов животного происхождения ими богаты печень, мозги, желтки яиц, сливки; из растительных — нерафинированное подсолнечное масло и бобовые.
В «сыром» жире также содержатся стероиды, которые широко распространены в природе, многочисленны (до 20 тыс. соединений) и выполняют разнообразные функции в организме. В основе их строения лежит циклическая группировка атомов, состоящая из восстановленного фенантрена (полностью восстановленный фенантрен называют пергидрофенантреном) и циклопентана. Эта циклическая группировка называется циклопентанопергидрофенантреном или стераном. Общий углеродный каркас стероидов имеет следующий вид:
где X— ОН или OR.
Стероиды делят на две группы: высокомолекулярные циклические спирты — стеролы и их сложные эфиры — стериды.
С химической точки зрения стеролы — полициклические, одноатомные, ненасыщенные спирты гидроароматического ряда. Стериды — сложные эфиры специфически построенных циклических спиртов (стеролов) и высших жирных кислот, относятся к группе простых липидов и входят в их омыляемую фракцию.
Массовая доля стеролов в жирах, как правило, относительно невелика и составляет менее 0,5 % к массе жира (табл. 1.6), иногда достигая 1 %.
38
Г а б л и ц а 1.6
Массовая доля стеролов в животных жирах, %
Жир Общие j Свободные Эте р । к|) 11 ц 11 ро ван н ые
Свиной 0.07-0.12 0,07-0.12 Следы
Говяжий 0.07 0.07 —
Бараний 0.03 0.03 —
Коровьего молока 0.07 0.07 —
Печени трески 0,52 0.27 0.25
Основным стеролом жиров животных и человека является холестерин (3{3-оксихолестен или Д^-холестен-Зр-ол), тривиальное название происходит от греч. chole — желчь и stereos — твердый.
Структурная формула холестерина
Холестерин присутствует во всех животных липидах, в крови и яичном желтке и отсутствует (содержится в незначительном количестве) в липидах растений. Холестерин является структурным компонентом клетки, участвует в обмене желчных кислот, а также гормонов. В организме человека в печени и других тканях синтезируется 70—80 % холестерина от его общей массы (250 г на 65 кг массы тела) и около 20 % поступает с пищей. Массовая доля холестерина в некоторых животных продуктах приведена ниже.
Продукт
Массовая доля холестерина, %
Масло сливочное
Яйца
Сыр
Мясо
Рыба
0,17—0,21 0,57
0,28-1.61 0,06-0,10 0,03-0,06
Холестерин представляет важную медико-биологическую проблему в развитии и предотвращении патологических процессов в
39
организме. Средняя концентрация холестерина в плазме крови составляет от 1.9 до 2,1 г/дм1. Повышенный уровень холестерина в крови (более 2.6 г/дм-') приводит к гиперхолестеринемии, к развитию атеросклероза, поражающего внутренние стенки сосудов. Наиболее опасные его формы — ишемическая болезнь сердца и нарушение мозгового кровообращения. Нарушение липидного обмена происходит при диабете и связано также с гиперхолестеринемией. Многочисленными исследованиями установлена четкая корреляция между ожирением, связанным, как правило, с перееданием, и повышенным синтезом холестерина в организме. Еще одна неприятность, связанная с уровнем холестерина, — желчно-каменная болезнь. Пересыщение желчи холестерином ведет к образованию камней (преимущественно холестериновых) в желчном пузыре и желчных протоках. Исследования последних лет показали, что в этом случае не столько важен общий уровень холестерина в желчи, сколько изменение ее фазового состава.
Однако и значительное падение содержания холестерина в плазме крови также может привести к заболеваниям, но уже другого характера. Для взрослого человека допустимой низшей границей, принятой за норму, считается 1,5 г/дм3. С дальнейшим уменьшением концентрации холестерина возрастает риск таких заболеваний, как повышение активности щитовидной железы, поражение коры надпочечников, истощение и т. д.
На уровень холестерина в организме человека заметное влияние оказывает состав пищевых жиров. Если в рационе много растительных масел, то уровень холестерина уменьшается; употребление животных жиров в большом количестве ведет к повышению концентрации холестерина в плазме крови. Чтобы его нейтрализовать, необходимо включать в диету 2 г ненасыщенных жиров на 1 г насыщенных. Нельзя злоупотреблять салом, сливочным маслом, сливками и тому подобными продуктами. Насыщенные жиры, входящие в их состав, не должны превышать 10 % рациона.
В целом массовая доля липоидов, входящих в состав животных жиров, включая фосфатиды, стерины и стероиды, невелика и составляет десятые и сотые доли процента.
Окраска животных жиров зависит от наличия каротиноидов — пигментов, окрашивающих жиры в желтый цвет и одновременно служащих провитаминами. Массовая доля каротинов в жирах зависит от условий кормления животных, достигая максимума к осени при пастбищном откорме. В жирах присутствуют такие жирорастворимые витамины, как A, D, Е и К, однако массовая доля двух последних незначительна. Суммарная доля витаминных примесей служит показателем пищевой ценности жиров.
Соотношение триглицеридов, липоидов и свободных жирных кислот в составе мясных продуктов зависит от сырьевого источника (табл. 1.7).
40
Г а б л и ц а 1.7
Массовая доля липидов в мясе различных животных, г на 100 г съедобной части
Мясо Триглицериды (алкилгли-иериды) । Фосфо-| липиды Холестерин Полиненасышейные жирные
линоле- : вая КИСЛОТЫ
линоленовая арахидоновая
Говядина 13,10 0,80 0,07 0,35 0.12 0,017
Баранина 15,30 0,88 0,07 0.33 0,14 0,016
Свинина 32,00 0,84 0,07 3,28 0,22 0,14
Роль липидов в технологии мясопродуктов многофункцио-
нальна. Они могут использоваться как самостоятельный продукт питания (шпик), как пищевые животные жиры, как добавка в вареные колбасы в виде шпика и белково-жировых эмульсий; могут входить в состав самостоятельных пастообразных продуктов повышенной пищевой ценности на основе эмульсий, а также использоваться в качестве смесей для внутрикишечного зондового питания, источник липидов в которых тонко эмульгирован.
В связи с необходимостью сбалансированного питания исследование жира сводится не только к определению его массового содержания, но и к анализу жирно-кислотного состава, пищевой, биологической ценности и других показателей.
Методы количественного определения суммарных липидов в сырье и пищевых продуктах разнообразны и отличаются способами анализа, приемами экстракции, применяемыми экстрагентами, подготовкой образцов к анализу, продолжительностью и условиями экстрагирования и т. д.
По способам анализа методы делятся на две группы: методы определения массовой доли жира непосредственно в объекте и методы, связанные с предварительным извлечением липидов или жира.
К первой группе относятся методы ядерного магнитного резонанса, инфракрасной спектроскопии, турбидиметрии, ультразвуковые и др.
Во вторую группу входят методы, в которых липиды или жир сначала переводят в органическую фазу с последующим их количественным определением гравиметрическим или другим способом.
Полное извлечение липидов из клеток или тканей представляет собой довольно трудную задачу, поскольку они являются весьма гетерогенной группой соединений, находящихся в клетках как в свободном, так и в связанном состоянии. Причем в последнем случае липиды образуют более или менее прочные комплексы с гидрофильными компонентами клетки (белками, углеводами и др.). В образовании этих комплексов участвуют ван-дер-
41
ваальсовы силы, гидрофобные взаимодействия, водородные и ковалентные связи. Преобладание того или иного типа связей обусловливает различную экстрагируемость связанных липидов из биологического материала.
В зависимости от степени экстрагируемости из клеток липиды условно разделяют на свободные, связанные и прочносвязанные. Свободные липиды легко переходят в слабополярные растворители (петролейный и диэтиловый эфир, хлороформ, бензол и др.), способные разрушать комплексы, образованные с помощью гидрофобных взаимодействий и ван-дер-ваальсовых сил. Для извлечения связанных липидов, удерживаемых в комплексах водородными связями (например, в липопротеидах мембран), применяют более полярный растворитель (метанол или этанол) в смеси со слабополярными. Прочносвязанные липиды, удерживаемые ковалентными связями, выделяют после гидролиза комплекса слабыми растворами кислот или щелочей в органическом растворителе.
По приемам экстракции и полноте извлечения липидов или жира эти способы можно разделить на три подгруппы:
методы экстракции «сырого» жира из измельченного обезвоженного материала неполярным или слабополярным растворителем;
методы экстракции «сырого» жира из измельченного невысу-шенного материала полярным растворителем или его смесью со слабополярным или полярным растворителем;
методы экстракции «сырого» жира из бесклеточного или полностью разрушенного (гидролизом, гидродинамической кавитацией) клеточного материала полярным растворителем или его смесью со слабополярным растворителем.
Отличительной особенностью методов 1-й подгруппы является извлечение главным образом только свободных липидов, которые сорбированы и механически удерживаются гелевой частью клетки. Применяя эти методы, как правило, используют специальные приборы и аппараты, в том числе аппарат Сокслета.
Главная особенность методов 2-й подгруппы — обеспечение оптимальных условий для максимального извлечения липидов из клеточного материала, не подвергнутого разрушению химическим или физическим способом. Однако отсутствие специальных экстракторов приводит, как правило, к потере экстракта при отделении липидов от нелипидной части и других операциях.
Методы 3-й подгруппы можно условно отнести к приемам, обеспечивающим практически полное выделение липидов, в том числе и прочносвязанных. Условность связана с тем, что химическое разрушение клеточного материала при гидролизе изменяет фракционный состав выделяемых липидов, а физическое — не 42
затрагивает липиды, связанные с белками ионной связью, в том числе через ионы двухвалентньпх металлов. К методам 3-й подгруппы относятся методы концентрирования жира в бутиромерах с помощью поверхностно-активных веществ. В последнее время при оценке полного содержания липидов, а также количества прочносвязанных липидов в различных пищевых продуктах и биологических объектах используют метод динамической кавитации.
1.3. УГЛЕВОДЫ
К классу углеводов относят органические соединения, содер-. о лу
жашие альдегидную R—или кетонную R—группу и н сн2он
несколько спиртовых гидроксилов. Их элементарному составу соответствует общая формула СотН7/гО;;, однако название класса и эмпирическая формула не отражают химический характер и особенности строения этих соединений, обусловливающие многообразие свойств углеводов. Это предопределяет характер участия углеводов в процессах жизнедеятельности и построении тканей животных и растений. Основными физиологическими функциями углеводов являются структурная, энергетическая и метаболическая.
Как правило, в животных организмах большинство углеводов играют роль энергетических субстратов, при окислении которых выделяется энергия, необходимая для протекания химических реакций, и используются как резервные. Наряду с этим промежуточные продукты окисления углеводов используются для синтеза многих других органических соединений.
Углеводы делят на две группы: простые и сложные. Простые углеводы не подвергаются гидролизу, сложные гидролизуются с образованием простых углеводов.
Среди сложных углеводов выделяют группы олигосахаридов и полисахаридов. Олигосахариды — сахароподобные сложные углеводы, характеризуются сравнительно невысокой молекулярной массой, хорошей растворимостью в воде, легкой кристаллизацией и, как правило, сладким вкусом.
Полисахариды — высокомолекулярные сложные углеводы с молекулярной массой порядка сотен тысяч — отличаются друг от друга химической природой повторяющихся моносахаридных единиц, степенью разветвления и длиной цепи. Полисахариды не содержат свободных редуцирующих групп, поэтому не обладают восстанавливающей способностью. Полный гидролиз полисахаридов в присутствии кислот или специфических ферментов приводит к образованию моносахаридов, обладающих редуцирующими свойствами.
43
Различают гомополисахарилы, содержащие остатки монополисахарида одного вида, и гетерополисахариды, состоящие из остатков моносахаридов двух или более видов, регулярно или нерегулярно чередующихся в молекуле.
Полисахариды либо нерастворимы в воде, либо образуют растворы, по свойствам напоминающие коллоидные, что обусловлено высокой молекулярной массой растворенных частиц. Полисахариды не образуют явно оформленных кристаллов, лишь некоторые из них обладают псевдокристаллическим строением. Сладкий вкус для них не характерен.
К числу наиболее важных природных гомополисахаридов принадлежат крахмал, гликоген, клетчатка, декстран и хитин.
Гликоген, содержащийся в мясе и мясопродуктах, служит резервным питательным веществом, вследствие чего за ним сохранилось название «животный» крахмал. Массовая доля гликогена в печени животных достигает 20 %, в мышцах — 4 %. Содержание углеводов зависит от степени упитанности животного. В мышцах плохо откормленных, истощенных, голодных и больных животных гликогена в 2—3 раза меньше, чем в мышцах животных нормального физиологического состояния.
Гликоген имеет много общих свойств с крахмалом. Например, гликоген дает цветную реакцию с иодом. При взаимодействии крахмала и гликогена с иодом образуются комплексные адсорбционные соединения, окрашенные в реакции с крахмалом в синий, а с гликогеном — в красно-бурый цвет. Различие в цвете комплексов обусловлено некоторыми особенностями химической структуры крахмала и гликогена.
Гликоген сравнительно хорошо растворяется в горячей воде с образованием сильно опалесцирующих растворов. Как и крахмал, гликоген высаливается из коллоидного раствора при 33 °C сульфатом аммония или сульфатом натрия, подобно белкам осаждается двойным объемом спирта и эфиром в виде белого хлопьевидного осадка.
Промежуточными продуктами гидролиза гликогена являются декстрины и мальтоза, конечным — D-глюкоза. Гликоген оптически активен, причем удельное вращение его растворов близко к таковому для крахмала.
Структурная формула гликогена (рис. 1.12) идеально удовлетворяет его прижизненным функциям как глюкозное депо, служащее источником энергии. При интенсивной работе, когда доступ кислорода к тканям затруднен, быстрое окисление с выделением энергии протекает по анаэробному пути (гликогенолиз). При этом дефицит энергии восполняется за счет отщепления и окисления фосфорного эфира глюкозы сразу с нескольких ветвей молекулы гликогена.
При жизни организма образовавшийся в результате гликогенолиза лактат с помощью регуляторных механизмов снова вовлекается в метаболизм по схеме (рис. 1.13).
44
Рис. 1.12. Структурная формула гликогена
Гликоген»* Глюкоза
2 АТоЦ 2 Лактат Кровь 6 АТФ 2 Лактат
Гликолиз
Гликогенолиз
Рис. 1.13. Схема взаимодействия скелетных мышц и печени в процессе интенсивной мышечной работы
В период восстановления лактат, поступивший из мышц в кровь, превращается в печени в глюкозу крови. На образование одной молекулы глюкозы из двух молекул лактата расходуется шесть молекул АТФ. Глюкоза поступает с кровью обратно в мышцы и откладывается в запас в виде гликогена. В послеубой-ный период лактат накапливается в мышцах и выступает фоном для развития автолитических превращений мышечной ткани, формирования предшественников букета вкуса и аромата, присущего созревшему мясу. В связи с небольшой массовой долей гликогена в мясе и мясопродуктах основные нормы потребления углеводов в питании человека восполняются растениями. Однако в технологии переработки и хранения мяса он играет весьма существенную роль по ряду причин:
1. Полисахарид гликоген непосредственно влияет на формирование функционально-технологических характеристик мясного сырья (ВУС, ЖУС, ВСС, липкость, эмульгирующая способность, структурно-механические свойства), так как его автолитические превращения в послеубойный период являются пусковым механизмом для развития автолиза мяса, сопровождающегося конформационными и гидролитическими изменениями мышечных и соединительнотканных белков, что оказывает решающее влияние
45
па консистенцию мяса и мясных продуктов, приобретение нежности и сочности, накопление химических предшественников вкуса и аромата кулинарно обработанного созревшего мяса (свободных аминокислот, нуклеотидов, низкомолекулярных пептидов, органических кислот и других соединений).
2. Продукты распада гликогена — моносахариды (глюкоза, декстроза и др.) наряду с поваренной солью и нитритом натрия являются важными ингредиентами посолочных смесей при производстве колбасных и деликатесных цельномышечных изделий для достижения эффекта стабилизации окраски продуктов.
В связи с этим анализ гликогена, определение глубины его распада и образующихся продуктов необходимо проводить в технологических целях.
С развитием технологии значительно возрос ассортимент мясных продуктов, включая традиционные и комбинированные. Для образования мясных структурированных систем, придания им специфических свойств и вкуса, повышения выхода все шире используются различные полисахариды и другие углеводы, не характерные для мяса.
Особого внимания заслуживает применение в рецептурных композициях мясопродуктов физиологически полезных балластных веществ. Это главным образом структурные (целлюлоза, гемицеллюлоза, пектин и т. д.) и неструктурные (альгинаты, камеди и т. д.) вещества. Полисахариды растений в соответствии с современной научно обоснованной теорией адекватного питания должны строго дозироваться.
Целлюлоза — прочное, волокнистое, водонерастворимое соединение, фибриллы которого образуют каркас растительных клеток. Этот внеклеточный структурный полисахарид — самый распространенный в природе биополимер. Линейная неразвет-вленная цепь целлюлозы состоит из 10 000 и более остатков £>-глюкозы, связанных друг с другом (1-4)-гликозидными связями в p-конфигурации. Такой вид соединения мономеров не гидролизуется а-амилазой и другими ферментами желудочно-кишечного тракта.
Полисахариды гемицеллюлоз относятся к гетерополимерам разной степени ветвления. В их состав входят ксиланы, ксилоглюканы, арабинаны, различные галактаны и маннаны. Углеводный состав гидролизатов гемицеллюлоз пищевого и непищевого сырья в основном идентичен. Он включает гексозы (глюкозу, галактозу, маннозу, глюкуроновую и галактуроновую кислоты) и пентозы (ксилозу, арабинозу и рамнозу).
Пектин также относится к группе полисахаридов. Его макромолекулы представляют собой цепь, построенную из остатков Д-галактуроновой кислоты, соединенных по месту 1,4-углеродных атомов a-связью. В состав молекул входят свободные и метилированные карбоксилы, частично нейтрализованные кальцием и маг-46
нием. Пектиновые вещества содержатся в срединной пластинке клеточной стенки и выполняют роль связующего компонента Водные растворы этих веществ обладают желирующими и геле образующими свойствами. В цементирующий матрикс растительных клеток входит также полимер лигнин, обладающий хорошими сорбционными свойствами.
Свойствами балластных веществ обладают и негидролизуюшп-еся в кишечнике гликозоаминогликаны (мукополисахариды), содержащиеся в межклеточном веществе животных тканей. Наибольшее количество этих структурных полисахаридов содержится в соединительной ткани, легких и крови.
К балластным веществам животного происхождения относят нерастворимый полисахарид хитин. Из него состоят панцири омаров, крабов, а также многих насекомых. В некоторых странах уже освоено производство этого биополимера.
Ряд ученых считают физиологически полезными веществами и отдельные продукты реакции Майяра. Образующиеся в процессе тепловой обработки белков и редуцирующих сахаров меланоидины резистентны к действию пищеварительных ферментов.
Получило развитие производство синтетических биополимеров, которые при употреблении в пищу могут выполнять функции балластных веществ. К ним относятся многие фармацевтические препараты, а также различные виды модифицированной целлюлозы, применяемой в качестве пищевой добавки.
Анализ этих веществ необходим при расчете сырья рецептурных композиций, для придания продуктам лечебно-профилактических свойств, обеспечения физиологических норм питания и поддержания здоровья человека.
1.4. ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Фосфорорганические соединения при жизни животных играют роль запасников энергии. После убоя животных многие фракции этих веществ участвуют в образовании специфического вкуса мясных продуктов. Особая роль отводится креатинфосфату (КрФ), аденозинтрифосфорной кислоте (АТФ) и их производным.
Креатин (метилгуанидинуксусная кислота) является обязательной составной частью поперечнополосатой мускулатуры. Содержание креатина в скелетных мышцах достигает 400— 500 мг%, в сердечной мышце креатина в 2—3 раза меньше. Креатин присутствует также в ткани мозга (около 100 мг%) и в значительно меньших количествах в паренхиматозных органах (10—50 мг%).
47
о мышечной ткани креатин содержится как в свободном виде, гак и в виде фосфорилированного производного (креатинфосфа-га. фосфокреатина) — макроэргического соединения, представляющего собой источник легкоутилизируемой энергии:
NH2
C=NH
N—CH}
I
CH2
COOH
Креатин
NH~P=O
C=NH
OH
N —CH;
CH2
COOH
Креатинфосфат
Креатинфосфат участвует в обратимом переносе фосфорилированного остатка с креатинфосфата на АДФ, реакция катализируется креатинкиназой (АТФ: креатинфосфотрансфераза, КФ 2.7.3.2):
Креатинкиназа
Креатинфосфат + АДФ Креатин + АТФ
Это единственная ферментативная реакция, протекающая с участием креатинфосфата.
Соотношение количеств свободного креатина и креатинфосфата в мышечной ткани зависит от физиологического состояния мышцы. При сокращении происходит распад АТФ, но за счет креатинкиназной реакции при наличии креатинфосфата происходит ее регенерация, что в итоге приводит к увеличению свободного креатина и уменьшению креатинфосфата. В покое синтез АТФ превалирует над распадом и приводит к синтезу и накоплению в ткани креатинфосфата.
Креатинфосфат, являясь источником энергии для мышечных сокращений, представляет собой важнейший компонент мышечной ткани. По сравнению с АТФ креатинфосфат характеризуется более высоким потенциалом переноса высокоэнергетических фосфатных групп. Если имеющееся в мышце количество АТФ может поддерживать ее сократительную активность на протяжении долей секунды, то в целом резерва энергии АТФ 48
И крсатш [фосфат я хватает на Н)--Д2с мышечной рабты Далпнсп шее понижение шергстичсскш о шнснниаиа мьпишл пршюжы >-стимулированию еликони за и i лhkoiенолиза, иикла карбоноььо КИСЛОТ и ОКИСЛИ 1СЛЫ101 о фосфорилирования. В среднем КОН центрация креатинфосфата в скелетных мышцах позвоночных >. 4—5 раз превышав! концентрацию АТФ.
Уровень креатинфосфата зависит от упитанности животною в степени тренированности мыши. Определение крсатинфосфага п мышцах имеет большое значение лля выявления уровня физичсс кого развития и физиологическою состояния животною. Масео вая доля крсатинфосфата в мышцах больных живошых. как пре» вило, значительно снижается.
Аденозинтрифосфорная кислот (А ГФ)
являясь универсальным источником энергии для биохимичес ких реакций, занимает центральное место в обмене веществ. Гак, организм человека для гликолитических реакций расходует около 0,5 кг АТФ в сутки. При дефосфорилировании 1 моль АТФ освобождает 33,6—42,0 кДж энергии.
Массовая доля АТФ в мышцах составляет в среднем 30,0— 45,0 мг%. Уровень АТФ является также показателем физического и физиологического состояния животного, снижаясь при мышечном утомлении и дегенеративных процессах. В послеубойный период эти вещества вносят существенный вклад в формирование вкуса мяса при созревании.
1.5. ВОДА
Вода — важнейший компонент всех пищевых продуктов. Воду нельзя рассматривать просто как инертный компонент или универсальный растворитель для пищевых веществ. Она является не только преобладающим компонентом большинства пищевых продуктов, но и оказывает предопределяющее влияние на многие их качественные характеристики, особенно на сроки хранения.
Массовая доля влаги в мясе и мясных продуктах колеблется в широких пределах (табл. 1.8).
49
I a b j i! н ;t !
Массовая доля влаги в некоторых пищевых продуктах
Массовая толя влаги. Ч | Продукт
40—70 Копчености. свежие сосиски, ветчина, окорок, ливерная
колбаса и немецкие колбаски
50—60 Жирное мясо
70—S0 Свежие бобовые (фасоль, бобы, горох) и мясо (говядина,
баранина, конина, птина). Рыбные продукты (рыба, моллюски. ракообразные)
Интенсивные исследования структуры пока еще не привели к созданию удовлетворительной модели, которая объясняла бы все свойства воды, водных растворов и содержащих влагу твердых тел, в частности пищевых продуктов. Вода в пищевых продуктах может находиться в свободной и связанной форме.
Согласно классификации П. А. Ребиндера, в основу которой положена энергия связи, связь влаги с продуктом может быть:
химической;
физико-химической (адсорбционной, осмотической, структурной);
механической (влага в микро- и макрокапиллярах, средний радиус которых соответственно 10~7 м и менее и более 10-7 м, а также влага смачивания, находящаяся на поверхности).
Влага смачивания и влага макропор имеет весьма непрочную связь с продуктом и может быть удалена механическими способами (отжатием на прессах или под действием центробежной силы в центрифугах). Такая влага называется свободной. Свободную влагу также можно удалить путем высушивания или вымораживания.
Свободная влага, являясь растворителем органических и неорганических соединений, участвует во всех биохимических процессах, протекающих при хранении и переработке мясного сырья.
По величине энергии связи различают следующие формы связи влаги; химически связанная и физико-химически связанная (адсорбционно-связанная, осмотически-связанная, капиллярно-связанная).
Химически связанная влага наиболее прочная и представляет собой воду гидратов, связанную в виде гидроксильных ионов, и конструкционную воду кристаллогидратов, связанную значительно слабее. Химическое связывание влаги в строго определенных молекулярных соотношениях происходит при химической реакции (гидратации). При этом вода входит в состав образованного вещества. При кристаллизации из раствора вода входит в структуру кристалла целыми молекулами. Для нарушения
50
этой связи сушка недостаточно эффективна, необходимо применить прокаливание или химическое воздействие.
Связь влаги в продуктах часто осуществляется межмолекулярными силами (ван-дер-ваальсовы силы). По данным Б. В. Дерягина и И. И. Абрикосовой, ван-дер-ваальсовы силы взаимодействия имеют место на расстоянии до 0,1 —1,2 мкм. т. е. значительно большем самих молекул. Энергия ван-дер-ваальсового взаимодействия колеблется в пределах 0,42—4,20 кДж/моль.
Особым видом межмолекулярного взаимодействия является водородная связь. Она проявляется между ковалентно связанным атомом водорода и электроотрицательными атомами (кислород, фтор, азот), которые принадлежат к той же или другой молекуле. Энергия водородной связи равна 21,0— 29,4 кДж/моль, что лишь на один порядок меньше энергии химического взаимодействия.
Формы физико-химической связи разнообразны. Адсорбционно-связанная влага обусловлена взаимодействием молекул адсорбента и молекул воды и удерживается у поверхности раздела коллоидных частиц с окружающей средой. Обладая большой поверхностью, коллоидные структуры имеют высокую адсорбционную способность. Большую часть адсорбционно-связанной влаги в животных тканях мясопродуктов составляет влага, которая образует сольватную оболочку молекул белковых веществ и гидрофильных коллоидов. Часть адсорбционной влаги входит в состав сольватных оболочек гидрофобных коллоидов. Адсорбция вл-ши сопровождается выделением теплоты, которая называется теплотой гидратации.
Осмотически связанная влага является свободной в том смысле, что ей соответствует весьма малая энергия связи. Влага присоединяется без выделения теплоты и сжатия системы. Осмотически связанная влага диффундирует внутри тела в виде жидкости через стенки клеток благодаря разности концентрации внутри и вне клеток.
К капиллярно-связанной относится влага микрокапилляров. Эта часть воды находится в капиллярах (порах), средний радиус которых 10-7 м и менее. Капиллярная влага перемещается в теле как в виде жидкости, так и в виде пара. Различают два состояния капиллярной влаги: стыковое, когда влага разобщена в виде манжеток (защемленная вода), и канатное состояние, когда клинья жидкости соединены между собой, образуя непрерывную жидкую пленку, обволакивающую дисперсные частицы тела.
Связанная влага по своим свойствам значительно отличается от свободной: она не замерзает при низких температурах (вплоть до минус 40 °C); не растворяет электролиты, имеет плотность, вдвое превышающую плотность свободной воды, не всегда удаляется из продукта при высушивании (химически связанная) и т. д. Связанная влага в отличие от свободной недоступна микроорганизмам.
51
। iv ммм_\ .i.i>t in).uiiiiciui>i развитии микрофлоры в нишевых про лукгах свободную волу полностью удаляют или переводят в свя ванную, добавляя влагосвязывающие компоненты (соли, функии-опальные добавки. полисахариды и г. л.).
Для характеристики состояния влаги в продукте все шире применяют показатель активности волы ьш являющийся интегральной характерноiикой. Активность волы влияет на жизнедеятельность микроор!анпзмов, на биохимические, физико-химические реакции и процессы, протекающие в продукте. От величины активности волы зависят сроки хранения мяса и мясопродуктов, стабильность мясных консервов, формирование цвета и запаха, а также потери в процессе термообработки и хранения. Из общего количества волы, содержащейся в пищевом продукте, бактерии, плесени, дрожжи могут использовать для своей жизнедеятельности лишь определенную «активную» часть. Термин «активность воды», введенный Скоттом в 1953 г. в отношении пищевых продуктов, позволил установить взаимосвязь между состоянием слабосвязанной влаги продукта и возможностью развития в нем микроорганизмов.
Активность воды определяется как отношение парциального давления водяного пара над поверхностью продукта к давлению насыщенного водяного пара при той же температуре:
-Р/Р, = РОВ/100, (1.4)
где р — парциальное давление; д0 — давление насыщенного водяного пара: РОВ — равновесная относительная влажность.
Активность воды — это характеристика самого продукта, обусловленная химическим составом и его гигроскопическими свойствами, РОВ — характеристика окружающей среды, находящейся в гигротермическом равновесии с продуктом. Активность воды служит качественной характеристикой связи влаги в продукте. Так, энергия связи влаги с материалом равна
Е= -RT\n(p/p^ = -ДТ1пфо, (1.5)
где R— газовая постоянная; Т — температура. К.
Чем прочнее связана влага с материалом, тем меньше величина р, и наоборот, для свободной воды р достигает значения р0 и становится равным 1, а энергия связи Е равна 0.
Зависимость активности воды от влагосодержания продукта при постоянной температуре носит название изо-
термы (рис. 1.14). При удалении влаги из продукта (десорбции) и получении влаги продуктом (адсорбции) изотермы совпадают только в начальных точках, т. е. имеет место сорбционный гистерезис.
52
При изменении содержания воды происходят глубокие изменения в специфических свойствах пишевой системы, что отражается на ряде показателей.
Для каждого вида микроорганизмов существуют максимальное, минимальное и оптимальное значения активности воды. Удаление аы от оптимального значения приводит к торможению жизненных процессов, присущих микроорганизмам. При достижении определенной максимальной или минимальной величины активности воды прекращается жизнедеятельность микроорганизмов, что не говорит о гибели клетки. Минимальные значения активности воды для различных микроорганизмов приведены ниже.
Рис. 1.14. Пример зависимости равновесной влажности продукта от относительной влажности воздуха:
/ — десорбция: 2— адсорбция
Микроорганизмы
Грамотри нательные палочки
Большинство кокков и лактобацилл
Большинство дрожжей
Большинство плесеней
Большинство галофильных бактерий
Ксерофильные плесени
Осмофильные дрожжи
Активность воды а,,
1,00—0,95 0.95-0,91 0.91-0.88
0.88-0,80
0.80-0.75
0.75-0,65
0,65-0,60
За 1,0 принимается активность дистиллированной воды. Активность воды для свежего мяса равна 0,99. Активность воды имеет большое практическое значение. В табл. 1.9 приведены ее числовые значения для некоторых мясопродуктов.
Таблица 1.9
Значения активности воды для некоторых мясопродуктов
Продукт Массовая доля влаги, % Активность воды
Мясо Колбасы: 70-74 0,96-0,99
вареные 62-72 0.96-0,98
полукопченые 40-55 0.94-0,97
варено-копченые 40-43 0,90-0.93
сырокопченые 24-30 0,78-0,85
53
/Активность воды можно изменять, подоирая сырье и рецептуры с учетом используемого количества поваренной соли и жира. Создание оптимальных условий обезвоживания колбас в процессе созревания является еще одной возможностью регулирования активности воды. В созревших колбасах рост нежелательных микроорганизмов сдерживается сочетанием низкой активности воды, анаэробной среды, низкого значения pH. наличием хлорида и нитрата натрия и молочнокислой микрофлоры.
При использовании пищевых добавок степень их воздействия на активность воды уменьшается в следующем порядке: поваренная соль, полифосфат, цитрат, аскорбиновая кислота, глю-козо-5-лактон, ацетат, тартрат, глицерин, лактоза, молочный белок, жир. При этом микромолекулы с большей степенью диссоциации приводят к большему снижению активности воды, чем макромолекулярные вещества.
Для определения активности воды в мясопродуктах применяют различные методы. В гравиметрических методах выходной величиной является изменение массы пробы или вспомогательного гигроскопического материала за счет сорбции им влаги. В гигрометрических методах определяют изменение физических или электрофизических параметров гигроскопического материала (геометрические размеры, электропроводность, диэлектрическую проницаемость). Все эти методы являются косвенными. К прямым относится манометрический метод непосредственного измерения давления водяного пара с помощью манометров (жидкостных, емкостных и др.). Этот метод является основным при проведении исследовательских работ и принят за эталонный.
У нас в стране и за рубежом разработан ряд приборов, предназначенных для определения активности воды. В МГУПБ разработана вакуумная установка, с помощью которой определяют активность воды (рис. 1.15, а). Методика работы на установке следующая: в колбу помещают измельченный исследуемый продукт массой (40—50)10~3 кг. В другую колбу наливают (30—50)10 6 м3 дистиллированной воды. После закрепления колб на установке при открытых кранах для удаления воздуха и растворенных газов из влажного продукта и дистиллированной воды производят вакуумирование (120—180 с). Затем краны закрывают и установку тер-мостатируют. После термостатирования перемещение жидкости в камере прекращается и устанавливается равновесие давлений паров над дистиллированной водой и продуктом, разница между которыми (Ар) определяется U-образным манометром. Активность воды рассчитывают по формуле
аа = Ри/Ро = (Ро~ &Р)/Р^ (1 -6)
где — равновесное давление паров над продуктом; р0— равновесное давление паров над дистиллированной водой.
54
Рис. 1.15. Устройства для определения активности воды в пищевых продуктах:
я — конструкции И. А. Рогова и У. Ч. Чоманова; I — U-образный манометр; 2—колба с дистиллированной водой; 5 —термопары; 4—вакуумные краны; 5 —ловушка; 6 — колба с исследуемым продуктом; 7 — воздушный термостат; 8 — вакуум-насос; о—конструкции И. А. Рогова, В, Д. Косого. В. П. Кулагина: 1 — U-образный манометр; 2 — трубка с рабочей жидкостью; 3— емкость с исследуемым продуктом; 4~ термопара; 5 — потенциометр для замера и фиксации температуры; 6~ соленоидные клапаны; 7— вакуум-насос; 8 — потенциометр; 9 — мостовая схема; Ш- емкостный датчик; // — эбонитовая втулка
С помощью установки в течение 7—9 мин определяют величину активности воды в мясных продуктах. Для измерения равновесного давления паров воды над продуктом применяют жидкостный манометр с кремнийорганической жидкостью ПФМС-2/5 л. Недостатками данной установки являются недостаточная надежность и точность замера, продолжительное время достижения равновесного давления, невозможность использовать ее при повышенных температурах и непосредственно в цехах.
В настоящее время эти недостатки устранены благодаря тому, что все показания измерения активности воды снимаются при помощи емкостного датчика, вмонтированного в одно из колен U-образного манометра (рис. 1.15, б).
Имеются и другие конструкции приборов и их модификации. Перспективны в определении активности воды хроматографические и ЯМР-методы.
Лабораторная работа № 1
ОПРЕДЕЛЕНИЕ СУММАРНЫХ БЕЛКОВ В ТКАНЯХ ЖИВОТНЫХ УСКОРЕННЫМ ФОТОМЕТРИЧЕСКИМ МЕТОДОМ НА ОСНОВЕ МИНЕРАЛИЗАЦИИ ПРОБ
Цель работы. Освоить ускоренный фотометрический метод определения общего белка в мясе и мясопродуктах на основе проведения предварительной минерализации проб.
55
Задачи. Провести предварите.!иную минерализацию проб и рассчитать массовую долю общею белка в анализируемых образцах. Определить массовую .толю суммарных белков в образцах органов и дкатieii убойных животных.
Объекты исследования. Образцы мышечной, соединительно!! юани, внутренних органов или кровь убойных животных.
Материалы, реактивы и оборудование. Серная кислота плотностью 1840 кг/м3; раствор соляной кислоты молярной концентрацией 1 моль/дм3; пероксид водорода; сульфат аммония; хлорат кальция; гидроксид натрия; фенол; нитропруссид натрия; тиосульфат натрия; гипохлорит или дихлоризоцианурат натрия; иолид калия; карбонат натрия; установка Кьельдаля; фотоэлек-гроколориметр.
Приготовление реактивов. Р е а к т ив 1. 10 г фенола и 0,05 г нитропруссида натрия растворяют в дистиллированной воде в мерной колбе вместимостью 1000 см3 и объем раствора доводят дистиллированной водой до метки.
Реактив 2.5 г гидроксида натрия растворяют в дистиллированной воде в мерной колбе вместимостью 1000 см3. После охлаждения добавляют исходный раствор гипохлорита натрия из расчета достижения его массовой концентрации 0,42 г/дм3 или 0,2 г дихлоризоцианурата натрия. Объем раствора доводят дистиллированной водой до метки.
Приготовленные растворы хранят в темной посуде в холодильнике не более 2 мес.
Исходный раствор гипохлорита натрия. В стакане вместимостью 500 см3 перемешивают 150 г хлорной извести с 250 см3 дистиллированной воды. В другом стакане растворяют 105 г карбоната натрия в 250 см3 дистиллированной воды. Оба раствора сливают в одну емкость при постоянном перемешивании. Полученную суспензию оставляют на 1— 2 сут для отстаивания, затем надосадочную жидкость сливают и отфильтровывают. Массовая доля активного хлора в полученном реактиве находится в пределах 6—10 %. Его можно хранить в склянке из темного стекла до 1 года. Для точного определения массовой доли активного хлора 1 см3 прозрачного фильтрата разбавляют в конической колбе вместимостью 100 см3 до объема 40—50 см3, прибавляют 2 г иодида калия и 10 см3 раствора соляной кислоты молярной концентрацией 1 моль/дм-5. Образовавшийся иод оттитровывают раствором тиосульфата натрия молярной концентрацией 0,1 моль/дм3, приготовленным из фиксанала, до исчезновения вишневой окраски (1 см3 раствора тиосульфата натрия молярной концентрацией 0,1 моль/дм3 соответствует 0,00355 г хлора).
Определение гипохлорита натрия в исходном растворе. Перед приготовлением реактива 2 необходимо определить массу гипохлорита натрия в исходном растворе, учитывая неустойчивость его при хранении. По объему из-56
расходованного на титрование тиосульфата натрия определяют объем раствора гипохлорита натрия, необходимый для приготовления реактива 2.
Пример расчета. Объем раствора тиосульфата натрия молярной концентрацией ОД моль/дм5. израсходованный на титрование 1 см5 исходного раствора гипохлорита натрия, составляет 12.09 см5. Эквивалентная масса гипохлорита натрия равна половине относительной молекулярной массы гипохлорита натрия 74,4:2 = 37.2 г. Масса гипохлорита натрия в исходном растворе составляет 1,209 • 37,2 = 44.97 г.
Необходимый объем (см5) исходного раствора гипохлорита натрия
%= (1000 • 0.42)/44,97 = 9.4.
Методические указания. Контроль и оценка качества мяса и мясных изделий по массовой доле белка как при проведении научно-исследовательских работ, так и в условиях производственных лабораторий требуют наличия ускоренных, нетрудоемких и достаточно точных методов анализа.
Классическим методом определения массовой доли белков в мясе и мясопродуктах является метод Кьельдаля, предложенный для определения общего азота в различных материалах в 1883 г. Почти за целое столетие его применения появилось много модификаций, во многих из которых сохранились все основные стадии оригинального метода Кьельдаля — минерализация, отделение аммиака дистилляцией и титрование.
Минерализацию проводят нагреванием навески с концентрированной серной кислотой в присутствии катализатора (ртутно-кадмиевая соль, сульфатная смесь или пероксид водорода). Выделившийся аммиак вступает в реакцию с избытком концентрированной серной кислоты с образованием сульфата аммония:
2NH3 + H2SO4 -э (NH4)2SO4.
Для выделения аммиака сульфат аммония разлагают концентрированным гидроксидом натрия:
(NH4)2SO4 + 2NaOH 2NH3 + 2Н2О + Na2SO4.
Выделившийся аммиак поглощается титрованными растворами серной кислоты:
2NH3 + H2SO4 (NH4)2SO4.
Метод проводят на специальной установке, представленной на схеме (рис. 1.16).
Избыток серной кислоты оттитровывают гидроксидом натрия и по количеству связанной кислоты вычисляют количество поглощенного аммиака или соответствующее ему количество азота.
57
Рис. 1.16. Установка для отгонки аммиака:
I — парообразователь; 2 — каплеуловитель; воронка; 4- отгонная колба; 5— холодильник; 6 —приемная колба
Используемые на практике методы количественного определения белков основаны на анализе составных частей макромолекул или исследовании некоторых физических свойств их растворов, которые находятся в прямой зависимости от концентрации.
В связи с этим определенный интерес представляют фотометрические методы, которые отличаются высокой чувствительностью и точностью, требуют значительно меньших затрат времени, чем классический метод Кьельдаля. Для фотометрического анализа применяют различные типы фотоэлектроколо
риметров и спектрофотометров, которые удобны в работе и выпускаются отечественной промышленностью.
В группе методов определения суммарных белков в животных тканях на основе минерализации проб основное время занимает минерализация пробы, продолжительность которой благодаря подбору эффективных катализаторов составляет 2—2,5 ч. Последующее проведение цветной реакции и фотометрирование не превышают 30—40 мин, при этом одновременно можно анализировать серию проб.
Часто массовую долю белка в тканях и продуктах определяют по массовой доле азота, которая является характерным показателем элементарного состава белков. Массовая доля азота для многих белков близка к 16 %, поэтому массовое содержание белковых веществ вычисляют, умножая полученную массу азота на коэффициент 6,25, который получают путем деления: 100: 16 = 6,25. Для определения массового содержания белков соединительной ткани пользуются коэффициентом 5,62, принимая во внимание, что массовая доля азота в коллагене 17,8 %. При использовании метода небелковый азот продуктов не учитывается.
Подгототовка проб к анализу. Исследуемые образцы тщательно измельчают (ножом или на мясорубке). В колбу Кьельдаля вместимостью 50 см3 вносят 0,2 см3 сыворотки крови или взвешивают на аналитических весах 0,15—0,20 г ткани (мышц, сухожилий, почек, печени и др.). При помощи кусочка стекла навеску опускают на дно колбы. Добавляют 1—2 см3 концентрированной серной кислоты, 1 г смеси сульфата меди и сульфата калия в качестве катализатора. Содержимое колбы нагревают в вытяжном шкафу. Когда смесь приобретает коричневую окраску, колбу снимают с
58
огня, охлаждают при комнатной температуре, добавляю! 2—3 см' раствора пероксида водорода с массовой долей 30 % и продолжаю! нагревать до получения бесцветного минерализата. Последний используют для количественного определения белка.
Порядок проведения анализа. Минерализат охлаждают, количественно переносят в мерную колбу вместимостью 250 см3. объем доводят до метки дистиллированной водой, содержимое перемешивают.
В мерную колбу вместимостью 100 см3 вносят 5 см3 полученного раствора минерализата, повторно доводят объем до метки дистиллированной водой. Для проведения цветной реакции 1 см3 вторично разбавленного минерализата вносят в пробирку, добавляют последовательно 5 см3 реактива 1 и 5 см3 реактива 2, содержимое пробирки перемешивают. Одновременно готовят контрольный раствор, используя при этом контрольный минерализат (проба с использованием дистиллированной воды). Через 30 мин определяют оптическую плотность растворов на фотоэлектроколориметре с красным светофильтром. Измерение проводят в сравнении с контрольным раствором.
Построение калибровочного графика. Для построения графика используют стандартный раствор сульфата аммония, для приготовления которого берут 0,236 г сульфата аммония, предварительно высушенного до постоянной массы при температуре 60 °C, вносят в мерную колбу вместимостью 500 см3, растворяют в дистиллированной воде и доводят объем до метки. Этот раствор является стандартным и содержит 0,1 мг азота в 1 см3.
В мерные колбы вместимостью по 100 см3 вносят следующие объемы стандартного раствора (см3): 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 4,5; 5,0. После доведения объемов растворов в колбах дистиллированной водой до метки получают серию рабочих растворов концентрацией 1,0; 1,5; 2,0; 2,5; 3,0; 3,5; 4,0; 4,5; 5,0 мкг азота в 1 см3.
Для проведения цветной реакции в пробирки помещают по 1 см3 рабочего раствора, добавляют 5 см3 реактива 1 и 5 см3 реактива 2, перемешивают и через 30 мин измеряют величину оптической плотности на спектрофотометре при длине волны 625 нм или на фотоэлектроколориметре с красным светофильтром с толщиной поглощающего свет слоя 1 см по отношению к контрольному опыту. Опыт повторяют три раза. Для каждого определения готовят новый стандартный раствор.
По полученным средним данным из трех стандартных растворов строят калибровочный график (рис. 1.17), откладывая на оси абсцисс концентрацию азота (с, мкг/см3), а на оси ординат — соответствующую ей оптическую плотность (Д) при длине волны 750 нм. Калибровочный график должен проходить через начало координат.
По полученной величине оптической плотности с помощью калибровочного графика определяют концентрацию азота.
59
Массовая доля белка (%)
Рис.1.17. Вид калибровочного графика для определения белка фотометрическим методом
Х= [с -250- 100/(ш -5 - 1 106)](100 -6.25), (1.7)
где с — концентрация азота, найденная по калибровочному графику, мкг/см3; 250 — объем минерализата после первого разведения, см3; 100 —объем минсрали-зата после вторичного разведения, см3; т~ масса навески, г; 5 — объем разбавленного минерализата для вторичного разведения, см3; 1 — объем раствора, взятый для проведения цветной реакции, см3; 106 — множитель для перевода микрограммов в граммы; 100 — коэффициент для перевода в проценты; 6,25 — коэффициент для пересчета на белок.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы, форма которой приведена ниже.
Объект исследования Оптическая плотность раствора Концентрация азота по калиб-ровочному графику, мкг/см' Массовая доля в образце, %
азота белка
На основании анализа результатов делают выводы и заключение по работе.
Лабораторная работа Ns 2
ОПРЕДЕЛЕНИЕ БЕЛКОВ В ТКАНЯХ ЖИВОТНЫХ ФОТОМЕТРИЧЕСКИМИ МЕТОДАМИ БЕЗ МИНЕРАЛИЗАЦИИ ПРОБ
Цель работы. Освоить ускоренные фотометрические методы определения общего белка в мясе убойных животных, птицы, мясном фарше, мясных изделиях, готовых к употреблению, без предварительной минерализации проб.
Задачи. Определить массовую долю суммарных белков в образцах органов и тканей убойных животных и рассчитать массовую долю общего белка в объектах исследования.
60
Объекты исследования. Образцы мышечной ткани разных видов убойных животных и птицы, мясных фаршей, мясных изделий, ш-товых к употреблению (колбаса, окорок, печеночный наш тез и г. л.)
Материалы, реактивы и оборудование. Бумажные фильтры; расг-вор тартрата калия-натрия молярной концентрацией 1 моль/дмраствор гидроксида натрия молярной концентрацией 1 моль/дм3: биуретовый реактив (0.9 г тартрата калия-натрия: 3.0 i сульфата мели и 5,0 г иодила калия в ЮООсм3 раствора гидроксила натрия молярной концентрацией 0.2 моль/дм3): дистиллированная вода: сывороточный альбумин кристаллический: раствор красителя амидочерного 10В (0,6 г амидочерного ЮВ, 21 г лимонной кислоты и 2,5 см3 пропионовой кислоты в ЮООсм3 дистиллированной воды): раствор мочевины молярной концентрацией 8 моль/дм3; раствор гидроксида натрия молярной концентрацией 2 моль/дм3; раствор карбамила, содержащий гидроксид натрия (480,48 г карбамида и 80,0 г гидроксида натрия растворяют в ЮООсм3 дистиллированной воды); раствор лимонной кислоты молярной концентрацией 0,1 моль/дм3; раствор красителя оранжевого кислого 12; мерные колбы вместимостью 100 см3; пипетки вместимостью 0,1—5,0 см3; мерные цилиндры вместимостью 100—250 см3; колбы Эрленмейера вместимостью 100 см3: центрифуги лабораторные; спектрофотометр.
Методические указания. Перед выполнением работы следует внимательно ознакомиться с основами фотометрических методов, изложенных в теоретической части настоящей главы. Рекомендуется освоить несколько фотометрических методов без предварительной минерализации проб, основанных на соответствующих цветных реакциях. При фотометрировании в УФ-области спектра следует учесть, что при длинах волн 240—300 нм светопоглоще-ние белков почти полностью определяется ароматическими аминокислотами (триптофаном, тирозином и фенилаланином), при 205 нм — пептидными связями. В табл. 1.10 показана доля поглощения отдельных хромофоров «среднего» белка в общем спектре при различных длинах волн.
Таблица 1.10
Доля поглощения отдельных хромофоров «среднего» белка в общем спектре
Хромофоры Доля поглощения, %, при длине волны
205 нм 220 нм 250 нм 280 нм
Пептидная группа 78,0 32,0 2,9 0.0
Аргинин 2.0 0.0 0,0 0.0
Цистин 0.6 1,0 11,8 2,3
Цистеин 0,1 0,1 о,о 0.0
Метионин 0,6 0,8 0.0 0,0
Гистидин 2.4 7,1 0,0 0,0
Фенилаланин 6,5 3.7 9.4 0,0
Тирозин 4.3 20,0 15,0 31.9
Триптофан 5,5 36,0 61.0 65,7
61
Рис. 1.18. Спектр поглощения альбумина бычьей плазмы крови при pH 7,5
Рис. 1.19. УФ-спектры белков мяса:
/ — свиного (Ащах = 242 нм); 2 — крупного рогатого скота (лп1;1Х = 240 нм)
Для количественного анализа наиболее часто используют поглощение при 280 нм, так как в этом случае белки имеют максимум поглощения, обусловленный содержанием в них ароматических аминокислот — триптофана и тирозина. На рис. 1.18 показан спектр поглощения типичного белка — альбумина бычьей плазмы крови. Для количественного определения белков спектрофотометрическим методом предварительно строят калибровочный график с использованием раствора чистого белка концентрацией от 0,05 до 2,00 мг/см3.
УФ-спектры белков мяса показаны на рис. 1.19.
Подготовка проб к анализу. 1. При определении массовой доли белка в образцах мышечной ткани, мясных фаршах, мясных изделиях методом Лоури, биуретовым методом или методом, основанным на связывании красителей белками, образец предварительно тщательно измельчают сначала ножом на часовом стекле или на мясорубке, а затем на гомогенизаторе.
Для приготовления щелочного экстракта 15 г гомогенизированного образца взвешивают в колбе Эрленмейера вместимостью 100 см3, прибавляют 20 см3 дистиллированной воды и 10 см3 раствора гидроксида натрия молярной концентрацией 1 моль/дм3. С помощью воды суспензию переносят в мерную колбу вместимостью 100 см3, доводят до метки дистиллированной водой и хранят 12 ч в холодильнике. После этого 75 см3 экстракта (из верхней части колбы) фильтруют через смоченный дистиллированной водой бумажный фильтр диаметром 15 см, удаляя первые 15 см3 фильтрата.
2. При определении массовой доли белка в образцах мышечной ткани убойных животных, соленых мясных фаршей методами 62
УФ-спектрофотометрии образны предварительно тшазельно измельчают сначала ножом на часовом стекле ичи на мясорубке, а затем на гомогенизаторе.
1. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА МЕТОДОМ ЛОУРИ
1.1. МЕТОД ЛОУРИ В МОДИФИКАЦИИ ДЭВЕНИ И ГЕРГЕЙ (1976 г.)
Приготовление реактивов. Реактив А. Раствор Na,СО-, массовой долей 2 % в растворе NaOH молярной концентрацией 0,1 моль/дм3.
Реактив В. Раствор CuSO4 • Н,0 массовой долей 0,5 % в растворе тартрата натрия с массовой долей 1 %.
Реактив С. Получают смешиванием 60 см3 реактива А и 1 см3 реактива В (готовят непосредственно перед определением).
Реактив Д. Непосредственно перед использованием реактив Фолина—Чокалтеу титруют по фенолфталеину раствором гидроксида натрия известной молярной концентрации и по полученным результатам разбавляют до концентрации раствора 1 моль/дм3 (приготовление реактива Фолина см. главу III, лаб. раб. № 2).
Порядок проведения анализа. К 0,2 см3 исследуемого раствора, содержащего 5—100 мкг белка, прибавляют 1 см3 реактива С, смешивают и через 10 мин быстро вносят 0,1 см3 реактива Д, встряхивают и оставляют при температуре (20 ± 5) °C на 30 мин, а затем снимают показания на спектрофотометре при Л == 750 нм.
Построение калибровочного графика. Содержание белка в растворе определяют по калибровочному графику, который строят с использованием раствора тирозина известной концентрации. Для приготовления стандартного раствора 20 мг кристаллического тирозина растворяют в 200 см3 раствора соляной кислоты молярной концентрацией 0,2 моль/дм3. Калибровочный график строят, используя растворы тирозина с рекомендуемой массовой концентрацией:
Содержание тирозина, мкг Объем раствора тирозина. см; Объем раствора соляной кислоты молярной концентрацией 0,2 моль/дм’, см; Значение экстинкции
10 0,1 0,9 0,113
20 0,2 0,8 0,208
30 0,3 0,7 0,316
40 0.4 0,6 0,401
50 0,5 0,5 0,511
60 0,6 0,4 0,614
70 0,7 0,3 0.706
80 0.8 0,2 0,817
90 0,9 0,1 0,900
63
Рис. 1.20. Калибровочный график для определения белка по метолу . Гоури в модификации Дэвешь Гергей
По результатам определения строят калибровочный трафик (рис. 1.20). откладывая на оси абсцисс концентрацию стандартных растворов (мкг/смЛ тирозина, а на оси ординат — значения оптической плотнос-
ти при 750 нм. По графику находят концентрацию тирозина, которая соответствует найденной оптической плотности.
1.2. МЕТОД ЛОУРИ В МОДИФИКАЦИИ ЛАСТЫТЬ (1978 г.)
Приготовление реактивов. Реактив А. Смесь растворов: Na2CO3 молярной концентрацией 1 моль/дм3, NaOH молярной концентрацией 1 моль/дм3 и CuSO4 с массовой долей 0,5 % (содержит 1 % тартрата калия-натрия) в соотношении 10:5:1.
Порядок проведения анализа. В мерную колбу вместимостью 100 см3 вносят 2—5 см3 щелочного экстракта белков образца, приготовленного в соответствии с прописью подготовки проб, прибавляют 15 см3 раствора тартрата калия-натрия молярной концентрацией 1 моль/дм3 и доводят объем дистиллированной водой до метки. В зависимости от массового содержания азота для фотометрического определения используют 0,2 см3 (или более) приготовленного раствора. Объем доводят до 10 см3 с помощью реактива А. По истечении 30 мин измеряют оптическую плотность раствора на спектрофотометре при Л = 750 нм. Для определения содержания азота строят калибровочный график.
2. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА БИУРЕТОВЫМ МЕТОДОМ
Приготовление реактивов. Биуретовый реактив. 0,9 г тартрата калия-натрия, 3,0 г сульфата меди и 5,0 г иодида калия растворяют в 1000 см3 раствора гидроксида натрия молярной концентрацией 0,2 моль/дм3.
Порядок проведения анализа. Щелочной экстракт белков объемом 2 см3 смешивают с 15 см3 биуретового реактива. После 64
Рис. 1.21. Калибровочный график для определения белка биуретовым мето
ДОМ
i I
30 мин инкубирования смеси при 37 СС светопоглошсние измеряют на спектрофотометре при к = 550 нм.
Контрольный опыт готовят аналогично, используя вместо образца 1 см3 дистиллированной волы.
Построение калибровочного графика. Для построения i рафика готовят ряд последовательных разведений стандартного раствора белка, в качестве которого используют кристаллический сывороточный альбумин: 0,01 г белка растворяют в 100 см3 дистиллированной воды, отбирают 1 см3 и доводят объем до 10 см? и т. д.
С пробами из каждого разведения белка проводят биуретовую
реакцию в условиях, соответствующих описанию метода, и измеряют оптическую плотность окрашенных растворов. Затем строят график пример которого приведен на рис. 1.21.
3. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА МЕТОДАМИ, ОСНОВАННЫМИ НА СВЯЗЫВАНИИ КРАСИТЕЛЕЙ
3.1. МЕТОД С ИСПОЛЬЗОВАНИЕМ АМИДОЧЕРНОГО 10В
Приготовление реактивов. Раствор красителя амидочерного 10В. 0,6 г амидочерного 10В, 21г лимонной кислоты и 2,5 см3 пропионовой кислоты растворяют в 1000 см3 дистиллированной воды.
Порядок проведения анализа. Смешивают 2 см3 щелочного экстракта, содержащего мясные белки, с 25 см3 раствора красителя амидочерного 10В.
После проведения цветной реакции в течение 1 ч раствор центрифугируют в течение 10 мин при 100 с~1. Абсорбцию прозрачного супернатанта определяют на спектрофотометре при л = 615 нм. Массовую долю белка определяют по калибровочному графику.
3.2. МЕТОД С ИСПОЛЬЗОВАНИЕМ ОРАНЖЕВОГО КИСЛОГО 12
Приготовление реактивов. Раствор красителя оранжевого кислого 12.1,300 г очищенного красителя раство; ряют в фосфатном буфере молярной концентрацией 0.05 моль/дм3 и доводят объем до 1 дм3.
65
Ф о с ф a i н ы й б у ф с р м о л я р н о if к о н и с н 1 р а -и и е if 0,05 моль/дм' (pH 1.8—1.9). 3.4 г КНфО ,. 3.4 см ; Н.РО4 (массовой долей <35 Ф). 6()см' уксусной кислочы. 1 см; пропионовой кислоты и 2 г щавелевой кислоия растворяют в ' 800 см3 Н?О и затем доводят водой до 1 дм'. Щавелевую кислоту и КН->РО4 предварительно растворяют отдельно в горя чей воде.
Порядок проведения анализа. К навеске пробы, содержащей около 4 г белка, добавляют из мерной колбы 250 см3 раствора лимонной кислоты концентрацией 0.1 моль/дм3 и гомогенизируют в течение 2—3 мин. Лимонная кислота, употребляемая для эмульгирования белков мяса, облегчает связывание кислого красителя белками.
Взвешивают 2—4 г разбавленного образца с точностью до 0,01 г в 50-миллилитровые поликарбонатные центрифужные пробирки, добавляют воду до 5 г, применяя градуированную пипетку, а затем 25 см3 раствора красителя, закрывают пробкой и энергично встряхивают. Оставляют на 30 мин или дольше для взаимодействия красителя с белком и агрегации образовавшихся комплексов. Если смесь мутная, то ее центрифугируют и фильтруют. Определяют концентрацию свободного несвязав-шегося красителя, измеряя абсорбцию при 2. = 475 нм. Анализ удобно проводить в специальной проточной кювете с толщиной светопоглощающего слоя 0,75 мм. Для расчета используют стандартный график, который строят на основе анализа мяса методом Кьельдаля того же состава, что и анализируемый образец. Это связано с тем, что содержание ионогенных групп аминокислот в разных белках различно.
Как видно из рис. 1.22, между содержанием белка, определенным по методу Кьельдаля, и концентрацией свободного красителя наблюдается линейная зависимость. Рекомен-
Содержание протеина (по Кьельдалю), мг /см
дуемый предел концентраций свободного красителя составляет 0,3—0,6 мг/см3. Следует подчеркнуть, что конечный объем, а также количество добавляемого красителя должны быть постоянными.
Рис. 1.22. Отношение содержания свободного красителя к общему содержанию белка в образце свиной колбасы, определенному по методу связывания красителя
66
4. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ БЕЛКА МЕТОДАМИ УФ-СПЕКТРОФОТОМЕТРИИ
4.1. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ БЕЛКА В ГОВЯДИНЕ
Порядок проведения анализа. Образец массой 15 г гомогенизируют с 250 см3 раствора лимонной кислоты молярной концентрацией 0,1 моль/дм3 в гомогенизаторе в течение 2—3 мин.
К гомогенизированной порции (в пробирке) прибавляют 9,5 см3 раствора мочевины молярной концентрацией 8 моль/дм3 в растворе гидроксида натрия молярной концентрацией 2 моль/дм3, встряхивают, центрифугируют при частоте вращения 83 с"1 в течение 7 мин для отделения жира и измеряют оптическую плотность при X = 243 нм.
Массовую долю белка определяют, используя калибровочный график или уравнение регрессии, для чего предварительно определяют массовую долю белка по методу Кьельдаля.
Уравнение регрессии, например, для соленой говядины имеет вид
у= 17,32л-+ 1,025, (1.8)
где у — массовая доля белка, %; х — оптическая плотность (х = D при Л = 243 нм).
4.2. МЕТОД ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ БЕЛКА В МЯСЕ ПТИЦЫ
Порядок проведения анализа. При определении массовой доли белка в курином мясе методами УФ-спектрофотометрии предварительно тщательно измельченный образец массой 1,5 г суспендируют в гомогенизаторе в течение 5 мин со 100 см3 раствора лимонной кислоты, затем с помощью пипетки вносят 0,5 см3 суспензии в центрифужную пробирку, добавляют 9,5 см3 раствора карбамида, содержащего гидроксид натрия, и после осторожного перемешивания центрифугируют в течение 3 мин при частоте вращения 116—117 с . Заполняют кварцевую кювету с толщиной светопоглощающего слоя 1 см прозрачной жидкостью и измеряют оптическую плотность на спектрофотометре при к = 241 нм.
В качестве контрольного раствора используют смесь из 0,5 см3 раствора лимонной кислоты молярной концентрацией 0,1 моль/дм3 и 9,5 см3 щелочного раствора мочевины.
Массовую долю белка определяют, используя калибровочный график или приведенное выше уравнение регрессии (1.8). Пробы с различной массовой долей белка рекомендуется готовить путем смешивания гомогенизированного куриного мяса со свиным жиром в различных соотношениях.
67
-4.3 МЕ70Д ОПРЕДЕЛЕНИЯ МАССОВОЙ ДОЛИ БЕЛКА В СВИНИНЕ И ГОВЯДИНЕ
Порядок проведения анализа. Анализ проводится ио той же методике (см. и. 4.2). но с использованием других длин волн и других калибровочных графиков. Возможно также использование уравнении регрессии:
для свинины
у =—13.7282 + 63.8762а; (1.9)
для говядины
у = 6,9999 + 34,8326х. (1.10)
Результаты определения массовой доли белка в объектах исследования с использованием метода Лоури, биуретового метода и метода связывания красителей белками рекомендуется оформить в виде таблицы, форма которой приведена ниже.
Объект исследования Оптическая плотность раствора Концентрация азота по калибровочному графику, мкг/см’ Массовая доля белка в образце, %
1
Результаты определения массовой доли белка в объектах исследования с использованием методом УФ-спектрофотометрии рекомендуется свести в таблицу;
Объект исследования (наименование образца)
Оптическая плотность раствора
Массовая доля белка, %
По результатам предварительной теоретической подготовки и проведенных определений делают выводы и составляют общее заключение по работе. На базе литературных данных и результатов собственных исследований рекомендуется дать сравнительную характеристику сырья и продуктов по содержанию белка, а также прокомментировать преимущества и недостатки предлагаемых модификаций фотометрических методов анализа белков мяса и мясопродуктов.
68
Лабораторная работа № 3
АНАЛИЗ ФРАКЦИОННОГО СОСТАВА БЕЛКОВ НА ОСНОВЕ ИХ РАСТВОРИМОСТИ
Цель работы. Определить фракционный состав белков .мяса и мясопродуктов на основе их способности к растворению.
Задачи. Провести предварительное выделение фракций методами экстракции и количественно определить белки в экстрактах.
Объекты исследования. Мышцы различных видов животных (говядина, свинина, баранина) аналогичных анатомических участков, образцы соединительнотканных образований и внутренних органов.
Материалы, реактивы и оборудование. Дистиллированная вода: солевой раствор Вебера; раствор гидроксида натрия массовой долей 10%; сывороточный альбумин; биуретовый реактив; фотоэлектроколориметр (ФЭК-56М); настольная центрифуга; часовое стекло; пипетки; пробирки; технические весы; мерные цилиндры вместимостью 10 см3.
Приготовление реактивов. Биуретовый реактив. 1,5 г CuSO4 5Н2О и 6,0 г NaKC4H4O6 • 4Н2О растворяют в 500 см3 воды, при постоянном перемешивании добавляют 300 см3 раствора гидроксида натрия массовой долей 10 %. Общий объем доводят дистиллированной водой до 1000 см3.
Методические указания. Белки мяса и мясопродуктов можно разделить на растворимые в воде (белки саркоплазмы), в солевых растворах (белки миофибрилл) и нерастворимые в водно-солевых растворах, условно называемые белками стромы.
Водорастворимые белки саркоплазмы включают миоген, миоглобин (природный пигмент), миоальбумин, глобулин X.
К солерастворимым (миофибриллярным) белкам относятся миозин, актин, тропомиозин, тропониновый комплекс.
Фракция стромы включает белки, входящие в состав сарколеммы и внутримышечной соединительной ткани, которая связывает мышечные волокна в пучки, а также белки ядер. Фракция стромы объединяет белки: коллаген, эластин, ретикулин, а также гликопротеиды — муцины и мукоиды. Последние представляют собой слизистые белки, выполняющие защитные функции и облегчающие скольжение мышечных пучков. Все эти белки можно извлечь щелочными растворами. На практике их часто называют щелочерастворимой белковой фракцией мышечной ткани.
Количественное соотношение различных фракций, их состояние определяют не только технологические свойства сырья и продуктов, но и их биологическую ценность.
Для анализа белковых веществ используют много методов, включая различные модификации метода Кьельдаля, фотометрические методы с использованием спектрофотометров и фотоэлектроколориметров.
69
Среди ускоренных фотометрических методов большое признание получила биуретовая реакция, достоинства которой уже были отмечены в лабораторной работе J\g 2 при анализе белков.
На основе информации о количественном соотношении белковых фракций можно провести реальное прогнозирование функционально-технологических свойств сырья, особенно при получении продуктов заданного качества.
Подготовка проб к анализу. Мышцы различных животных (говядина, свинина, баранина) аналогичных анатомических участков тщательно освобождают от жира и прирезей соединительной ткани. Навеску массой 3—4 г тщательно измельчают ножом на часовом стекле. К навеске измельченного сырья приливают дистиллированную воду в соотношении I : 6 (по массе) и ведут экстракцию на холоде при О °C в течение 30 мин. Затем центрифугированием при частоте вращения 83 с3 в течение 5 мин отделяют осадок. Надосадочную жидкость осторожно декантируют и используют для количественного определения белков.
Осадок количественно переносят в фарфоровую ступку, растирают с песком и приливают солевой раствор Вебера в соотношении 1 : 6 к первоначальной навеске мышечной ткани. Экстракцию проводят при 0 °C в течение 20 мин. По истечении указанного времени экстракт отделяют центрифугированием в течение 10 мин при частоте вращения 83 с-1. Надосадочную жидкость декантируют и используют для количественного определения солерастворимых белков, а осадок подвергают обработке.
Далее осадок количественно переносят в термостойкую пробирку, добавляют 10 см3 раствора гидроксида натрия массовой долей 10 % и нагревают на водяной бане температурой до 96 °C. Полученный раствор после охлаждения центрифугируют при частоте вращения 83 с-1 в течение 5 мин. Надосадочную жидкость декантируют и используют для количественного определения белков стромы.
Порядок проведения анализа. Для проведения цветной реакции к 1 см3 исследуемых растворов белков (фракции: водорастворимая, солерастворимая и щелочерастворимая) добавляют 4 см3 биуретового реактива. Смесь оставляют в покое при температуре (20 ± 5) °C в течение 30 мин. По истечении времени образования окрашенного комплекса измеряют оптическую плотность раствора при длине волны 540—560 мкм на фотоэлектроколориметре с зеленым светофильтром.
Для того чтобы определить содержание белка в любой из фракций мышечной ткани, нужно снять показания оптической плотности Р1? найти это значение на оси ординат калибровочного графика и определить соответствующую концентрацию на оси абсцисс. Построение калибровочного графика — см. лабораторную работу № 2.
70
Массовую долю воло-. соле- и щелочерас гворимых белковых фракций (сс) определяют ио формуле
А'^ 100(с V )/т. (1.11)
где с — концентрация белка, найденная по калибровочному графику. мг,/см': Г— объем пробы после экстрагирования соответствующей белковой фракции, см': /и —масса навески мышечной ткани, мг.
Подготовив образцы мышечной ткани определенного вида животных, проведя фракционирование, измерив оптическую плотность, выполняют расчеты и оформляют результаты в виде таблицы:
После заполнения таблицы рекомендуется проанализировать данные для мяса от одного и от нескольких видов животных, различных тканей и сформулировать заключение по данной работе.
Лабораторная работа № 4
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АКТОМИОЗИНА
Цель работы. Приобрести практический навык количественного анализа белков актомиозинового комплекса в образцах мышечной ткани различных видов убойных животных и птицы.
Задачи. Экстрагировать солерастворимую белковую фракцию мышечной ткани, осадить белки, гравиметрически определить и рассчитать массовую долю актомиозинового комплекса.
Объекты исследования. Образцы мышечной ткани одного или различных видов убойных животных и птицы на различных стадиях автолиза, полученные от аналогичных анатомических участков.
Материалы, реактивы и оборудование. Раствор Вебера; ацетатный буферный раствор (pH 4,8); раствор СН3СООН массовой долей 1 %; сушильный шкаф; лабораторный потенциометр — pH-метр; весы аналитические; стаканчик для титрования; беззольные фильтры; ступка охлажденная.
Методические указания. Количественное соотношение различных белковых фракций мышечной ткани характеризует степень И глубину автолитических превращений. С одной стороны, количественное содержание актомиозина свидетельствует о степени
71
i рунного окоченения, ограничивающею применение мясного сырья. с другой лас г информацию о необходимости использования дополнительных средств, расширяющих его технологические возможности.
В свежей мышечной ткани растворимость миофибриллярных белков максимальна, поскольку актин и миозин существуют в виде отдельных фракций. Мышца в результате этого расслаблена, имеет высокую гидрофильность. С развитием автолитических процессов в период посмертного окоченения актин и миозин образуют комплекс, и мышца укорачивается, теряет влагу.
Актомиозин, извлекаемый из мышцы солевыми растворами, выпадает в осадок при диализе или разведении экстрактов, имеет изоэлектрическую точку при pH 5,5 и составляет около 55 % всех белков мышцы.
Количественное соотношение различных фракций, их состояние во многом определяют как технологические свойства сырья, так и его биологическую ценность.
Миофибриллярные белки мышечной ткани на различных стадиях автолиза определяют по методу Баленовича — Штрауба. Белки актомиозинового комплекса осаждают из супернатанта солерастворимых белков, полученных при экстрагировании навески ткани раствором Вебера путем снижения молярной концентрации хлорида калия до 0,05—0,06 моль/дм3 при определенных значениях pH раствора.
Для снижения молярной концентрации солей применяется десятикратное разведение супернатанта охлажденной до 0 °C дистиллированной водой.
Подготовка проб к анализу. Навеску мышечной ткани массой 3—4 г тщательно измельчают ножом на часовом стекле. К навеске измельченного сырья добавляют дистиллированную воду в соотношении 1 : 6 (по массе) и ведут экстракцию на холоде при 0 °C в течение 30 мин. Затем центрифугированием при частоте вращения 83 с'1 в течение 5 мин отделяют осадок. Надосадочную жидкость осторожно декантируют и удаляют.
Осадок количественно переносят в фарфоровую ступку, растирают с песком и добавляют солевой раствор Вебера в соотношении 1 : 6 к первоначальной навеске мышечной ткани. Экстракцию проводят при 0 °C в течение 20 мин. По истечении указанного времени экстракт отделяют центрифугированием в течение 10 мин при частоте вращения 83 с-1. Надосадочную жидкость декантируют и используют для количественного определения актомиозинового комплекса миофибриллярных белков.
Порядок проведения анализа. 1. Для снижения концентрации солей в экстракте белков солерастворимой фракции к их экстракту объемом 2 см3 добавляют дистиллированную воду температурой 0 °C в соотношении 1 : 9.
72
2. Исследуют две зоны осаждения белков актомиозинового комплекса: при pH 5,2 и 7,0.
В первом случае к разведенному раствору добавляют из расчета на каждый I см3 экстракта солерастворимых белков (в данном случае 2 см3) по 0,5 см3 ацетатного буфера pH 4,8, контролируя и доводя pH раствора по лабораторному потенциометру — pH-метру до 5,2,
Во втором случае разведенный охлажденной дистиллированной водой раствор центрифугата осторожно по каплям нейтрализуют раствором уксусной кислоты массовой долей I %, доводя pH раствора по потенциометру — pH-метру до 7,0.
Осажденный актомиозин после некоторого уплотнения в растворе при отстаивании на холоде отделяют фильтрованием через предварительно высушенный до постоянной массы и взвешенный с точностью до 0,0001 г фильтр. Массу осадка определяют после его высушивания на фильтре при 105 °C до постоянной массы.
Массовую долю фракции актомиозина (%) рассчитывают по формуле
Х= 100[(m1-m0)H]/2w, (1.12)
где /И] — масса фильтра с сухим остатком, г; т0 — масса высушенного фильтра, г; К—объем раствора Вебера, добавленный для экстрагирования, см3; 2 — объем экстракта белка, взятый для осаждения, см3; т — масса навески образна ткани, г.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы:
Образец Сроки и условия хранения (при исследовании образцов мышечной ткани на разных стадиях автолиза) Массовое содержание актомиозиновой фракции
мг/г %
По результатам проведенных определений делают выводы и формулируют общее заключение по работе.
Лабораторная работа № 5
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ КОЛЛАГЕНА
Цель работы. Приобрести навык в определении количественного содержания коллагеновых белков в образцах различных видов тканей убойных животных и птицы.
Задачи. Экстрагировать коллаген и количественно определить его на основе минерализации, а также путем гидролиза коллагено
73
вых iio.iiiueiiiидов с последующим проведением качественной реакции на оксипролин и фотометрированием.
Объекты исследования. Образцы мышечной и соединительной ткани убойных животных и птицы.
Материалы, реактивы и оборудование. Раствор соляной кислоты молярной концентрацией 12 моль/дм3: хлорид олова (II); растворы гидроксида натрия молярной концентрацией 2,5 и 6 моль/дм3; насыщенный раствор карбоната натрия; раствор сульфата меди молярной концентрацией 0,01 моль/дм3; раствор пероксида водорода массовой долей 6 %; раствор серной кислоты молярной концентрацией 3 моль/дм3; раствор пара-л и метил аминобензальдегида в пропиловом спирте массовой долей 5 %; спиртовой раствор фенолфталеина массовой долей 1 %, растворы гидроксида натрия массовой долей 0,1 и 0,2 %; раствор уксусной кислоты массовой долей 20%; лед; универсальный индикатор; бумажные фильтры; вата; воронка Бюхнера; водяная баня; ступки фарфоровые; центрифуга лабораторная; центрифужные пробирки; стеклянные стаканы; стеклянные колбы вместимостью 100 и 200 см3; установка для минерализации проб; колба Кьельдаля со шлифом из стекла пирекс или молибденового; обратный шариковый холодильник; колбонагреватель; стеклянные ампулы; стеклянные палочки; пластинка из молочно-белого стекла; спектрофотометр или фотоэлектроколориметр; автоклав.
Методические указания. Количественное определение коллагена в животных тканях проводят либо по методу В. П. Воло-винской, основанному на экстрагировании фракции коллагеновых белков и последующем определении в экстрактах белкового азота (см. лабораторную работу № 1), либо по количественному содержанию аминокислоты оксипролина, специфической для коллагена (см. лабораторную работу № 10). Метод основан на предварительном гидролизе коллагеновых полипептидов с последующим проведением качественной цветной реакции на оксипролин и фотометрированием. Включает следующие основные операции; гидролиз ткани соляной кислотой в присутствии хлорида олова, нейтрализацию кислоты и удаление олова, окисление оксипролина, развитие цветной реакции с шз/ш-диметил-аминобензальдегидом, измерение интенсивности окраски фотометрированием.
Результаты, полученные по этому методу, приведены в табл. 1.11.
Расчет количества белка ведут на основании известного массового содержания оксипролина (23 % к сумме аминокислот) в коллагенах.
Для выделения оксипролина мышечную ткань подвергают гидролизу с применением кислоты или щелочи. Продолжительность гидролиза имеет большое значение: недостаточная приводит к неполному освобождению оксипролина, а избыточная — к частичному его разрушению.
74
Таблица 1.11
Массовая доля (%) оксипролина и соединительнотканных белков в разных видах тканей
Образен Оксипролин Соедините/! ьноткан ныс белки
Поясничная мышца свиней
0.046 0.371
Шейная часть туши крупного рога- 0.322 2.600
того скота Филей крупного рогатого скота 0.220 1,800
Сырые связки убойных животных 10.56 85.200
Добавление хлорида олова II (не менее 3/4 количества белка в навеске) необходимо для предотвращения образования гуминовых веществ.
При нейтрализации и подщелачивании гидролизата используют карбонат натрия, чтобы предотвратить образование ионов олова в щелочной среде и облегчить его удаление в виде гидрата олова.
Окисление оксипролина пероксидом водорода в щелочной среде приводит к образованию продуктов, дающих с /шрд-диметил-аминобензальдегидом цветную реакцию. Следы пероксида водорода могут понизить интенсивность окраски или придать раствору неспецифический оттенок, поэтому его избыток разрушают нагреванием на водяной бане при 80 °C. При этом рекомендуется добавлять мочевину, образующую довольно стойкое соединение с пероксидом водорода CO(NH2)2H2O2.
Интенсивность образовавшейся розовой окраски измеряют спектрофотометрически или колориметрически. Окраска устойчива к течение 60—90 мин. В пределах до 25 мкг оксипролина поглощение подчиняется закону Бугера—Ламберта—Бера. Специфичность реакции очень высока.
Из всех аминокислот, содержащихся в кислотном гидролизате белков мяса, определению мешает только тирозин. По данным Е. Вербицкого и Ф. Е. Деасраджа, окраска тирозина составляет 1,3 % окраски, развиваемой оксипролином. Поэтому используется поправка на тирозин — 0,072 единицы оптической плотности на 1 см3 кислотного гидролизата, соответствующего 40 мг мяса:
211112 0^928 40 00() = 2
20 100 100
Полная поправка с учетом окраски самого гидролизата, соответствующей 0,01 ± 0,003, составляет 0,082 единицы оптической плотности на 40 мг мяса в 1 см3 гидролизата.
75
Для проведения цветной реакции в зависимости от ожидаемого содержания соединительной ткани в продукте следуе т брать разные объемы испытуемого раствора. Если, например, на анализ взята препарированная длиннейшая спинная мышца свиньи, то объем исследуемого раствора может быть равен 1 см3 (при разведении гидролизата до 100 см3). Массовая доля соединительной ткани в указанной мышце составляет 0,2—0.4 %. Если же взят образец, содержащий больше соединительной ткани, то на цветную реакцию нужно значительно меньше испытуемого раствора. Объем раствора доводят до 1 см3 дистиллированной водой.
Подготовка проб к анализу. Ткани животных предварительно режут на маленькие кусочки и взвешивают согласно нижеприведенным рекомендациям в соответствии с принятым методом определения.
1.ОПРЕДЕЛЕНИЕ КОЛЛАГЕНА В МЯСЕ ПО МЕТОДУ В. П. ВОЛОВИНСКОЙ
Порядок проведения анализа. Измельченный образец ткани массой Ю г растирают с 50 см3 раствора хлорида натрия массовой долей 0,6 %. Полученную смесь переносят в центрифужную пробирку, ополаскивая ступку и пестик 50 см3 того же раствора. Смесь экстрагируют в течение I ч, затем центрифугируют в течение 15 мин при частоте вращения 33—50 с-1. Жидкую фракцию удаляют, а к осадку прибавляют 100 см3 раствора гидроксида натрия массовой долей 0,2 %. Через 12—24 ч после тщательного перемешивания жидкую фракцию отделяют центрифугированием. Остаток ткани обрабатывают щелочью еще 2 раза, каждый раз добавляя по 50 см3 раствора гидроксида натрия массовой долей 0,2 %.
Остаток ткани из пробирки количественно переносят в стакан, смывая раствором гидроксида натрия массовой долей 0,1 % (объем раствора гидроксида натрия 100 см3). Содержимое стакана нагревают при умеренном кипении в течение 30 мин, по возможности сохраняя объем жидкости в стакане периодическим добавлением горячей воды. Затем частицам ткани дают возможность осесть на дне стакана и в горячем виде жидкость осторожно декантируют с осадка в мерную колбу вместимостью 200 см3 через воронку с небольшим количеством ваты, предварительно проверенной на содержание азота.
Остаток в стакане вместе с ватой, через которую проводилось фильтрование, при кипении обрабатывают водой, для чего в стакан наливают 100 см3 горячей дистиллированной воды и при умеренном кипении раствор сгущают за 1,5—2,0 ч до 1/3 первоначального объема. Сгущенный раствор через 15—20 мин декантируют с осадка через вату в ту же мерную колбу вместимостью 200 см3.
76
Аналогично при кипении остатки ткани обрабатывают водой еше два раза. После последней экстракции ваду промываю! водой и промывные воды присоединяют к общему объему эксдрак га.
Собранный в мерную колбу экстракт подкисляют раствором уксусной кислоты массовой долей 20 % до pH 4,8. нагревают до 50 °C и после охлаждения доводят водой до метки. При подкисле нии выпадает небольшое количество белков, од которых освобождаются фильтрованием через бумажный фильтр. Из фильтра берут 20 см3 и после минерализации определяю! азот согласно описанию лабораторной работы № 1.
Массовую долю азота в коллагене (%) вычисляют по формуле
N = (а 200 100)/(20ш),
(1.13)
где а —массовая доля азота в 20 см3 фильтрата, %: т - масса навески ткани, г.
Коэффициент пересчета азота на коллаген равен 5,62.
2.ОПРЕДЕЛЕНИЕ КОЛЛАГЕНА НА ОСНОВЕ ПРЕДВАРИТЕЛЬНОГО ГИДРОЛИЗА
Порядок проведения анализа. Навеску мяса массой 4 г вносят в колбу Кьельдаля вместимостью примерно 100 см3 со шлифом для присоединения обратного холодильника. К навеске добавляют 7см3 воды, 10см3 соляной кислоты молярной концентрацией 12 моль/дм3 и 0,7 г хлорида олова (II).
К колбе присоединяют пришлифованный шариковый холодильник и включают воду. Нагревание ведут на воздушной или песчаной бане. Для воздушной бани удобно использовать колбо-нагреватель (электроплитку с коническим углублением). С момента закипания жидкость нагревают ровно 7 ч, после чего плитку выключают и отставляют от колбы. Еще теплую колбу отсоединяют от холодильника и содержимое переносят, смывая дистиллированной водой, в мерную колбу вместимостью 100 см3. Исходную колбу несколько раз ополаскивают водой (общий объем промывных вод должен быть не более 2/3 объема колбы).
Для ускорения процесса гидролиз проводят в автоклаве. Навеску мяса массой 4 г вносят в ампулу вместимостью 25 см3 через расширенную часть горлышка (обрезают запаянный конец пустой ампулы так, чтобы образовалась воронка). К навеске добавляют 7 см3 воды, 10 см3 концентрированной соляной кислоты и 0,7 г хлорида олова (II). Ампулы запаивают на горелке и помещают в автоклав температурой 112—114 °C на 3,5 ч, после чего их можно хранить длительное время.
Содержимое ампулы переносят в мерную колбу вместимостью 100 см3, остатки смывают дистиллированной водой (общий объем промывных вод должен быть не более 2/3 объема колбы).
77
(.'одержимое мерной колбы независимо от способа гидролиза нейтрализуют раствором гидроксида натрия молярной концентрацией 6 моль/дм3. а затем доводят насыщенным раствором карбоната натрия до pH 8.0—8.2. После образования белого осадка начинают контролировать pH. используя pH-метр со стеклянным электродом или универсальный индикатор и фенолфталеин.
При работе по последнему способу из колбы отбирают тонкой стеклянной палочкой одну каплю жидкости и помещают ее на пластинку из молочно-белого стекла. Добавляют несколько капель дистиллированной воды и одну каплю универсального индикатора. После того как по универсальному индикатору pH жидкости будет близок к 7,5. добавляют несколько кубических сантиметров раствора карбоната натрия. Обработка считается законченной, если капля раствора, смешанная с водой, окрасится в чуть заметный розовый цвет (по фенолфталеину).
Колбу с жидкостью быстро охлаждают, объем доводят до метки дистиллированной водой и оставляют в покое в течение 30 мин. Затем осадок отфильтровывают через плотный бумажный фильтр на воронке Бюхнера при слабом разрежении.
Если фильтр мутный, его вторично фильтруют через тот же самый фильтр. Фильтрат светло-желтого цвета должен быть совершенно прозрачным. Одновременно гидролизуют, а затем нейтрализуют и фильтруют несколько проб.
В пробирки отмеряют пипетками по 1 см3 растворов в указанном порядке: исследуемый фильтрат, раствор сульфата меди молярной концентрацией 0,01 моль/дм3, раствор гидроксида натрия молярной концентрацией 2,5 моль/дм?, раствор пероксида водорода массовой долей 6%. Одновременно проводят три контрольных опыта, в которых исследуемый фильтрат заменяют 1 см3 дистиллированной воды.
Растворы в пробирках перемешивают, периодически встряхивая, в течение 5 мин. Затем пробирки помещают на водяную баню при 80 °C на 5 мин, часто и энергично встряхивая их. При этом пробирки должны быть глубоко погружены в воду. По окончании нагревания пробирки охлаждают на водяной бане со льдом и добавляют в каждую при перемешивании 4 см3 раствора серной кислоты молярной концентрацией 3 моль/дм3, а затем при сильном перемешивании 2 см3 раствора идря-диметиламинобензальдегида.
Далее пробирки помещают на водяную баню при 70 °C на 16 мин, после чего охлаждают водопроводной водой (под краном) и измеряют оптическую плотность раствора на спектрофотометре при длине волны 560 нм или фотоэлектроколориметре с зеленым светофильтром. Измерения сравнивают с контрольным опытом. Предварительно измеряют оптическую плотность двух контрольных растворов (одного относительно другого). Третий контрольный раствор служит запасным, его используют только в случае расхождения в показаниях первых двух растворов.
78
Построение калибровочного графика. Рас 1 воры оксинролина концентрацией 0.5—25.0 мкг/см подвергают всем операциям согласно описанию метода, начиная с гидролиза, так как 15 процессе последнего некоторое количество оксипролина разрушается (рис. 1.23).
По калибровочному графику находят концентрацию оксипролина в объеме жидкости, находящейся в пробирке после добавления /ш/м-диметиламино-бензальдегида (10 см3).
Массовую долю оксипролина в исследуемом образце (мг
D
Рис. 1.23. Калибровочный график для определения оксипролина:
/ -- без гидролиза; 2 -- после кислотною гидроди м
рассчитывают но формуле
с 100 100
Ут
(1Л4)
где с — концентрация оксипролина в 10 см3 окрашенного раствора, определенная по калибровочному графику, мг; 100 —объем раствора, полученный после нейтрализации. см3; 100 — множитель для перевода в проценты; И—объем раствора, взятый для цветной реакции, см3; т — масса навески мяса, г.
Предварительно из величины оптической плотности, отнесенной к 1 см3 гидролизата, соответствующего 40 мг мяса, вычитают поправку, равную 0,082 единицы оптической плотности.
Количество оксипролина пересчитывают на белки соединительной ткани, умножая на коэффициент 8,07.
При содержании оксипролина до 25 мкг в 10 см3 окрашенного раствора в исследуемом образце можно работать без калибровочного графика, пользуясь следующей формулой:
Р-0,01 100 100 0,164 Ут
(1.15)
где D — оптическая плотность; 0,01 — количество оксипролина, соответствующее 0,164 оптической плотности; 100 —объем раствора после нейтрализации; 100 — множитель для перевода в проценты; 0.164 — величина оптической плотности, полученная для чистого оксипролина; И—объем раствора, взятый для проведения цветной реакции, см3; т — масса навески мяса, г.
Величина оптической плотности до 0,3—0,4 единицы пропорциональна концентрации оксипролина.
79
Для пересчета содержания соединительнотканных белков в .нас товые доли от общею содержания белков используют уравнение
где 8.07 — коэффициент пересчета оксипродина на бедки соединительной ткани: д —массовая доля оксипролина. %; 6.25 — коэффициент пересчета на белки: .У — массовая доля общего азота в мясе, %.
Оформление результатов. Полученные экспериментальные и расчетные данные оформляют в виде таблицы:
Образец или вид ткани
азота
Массовая доля, %
коллагена
По результатам экспериментальных исследований формулируют выводы и делают общее заключение по работе. При этом рекомендуется сравнить полученные данные с литературными, а также указать преимущества и недостатки используемых подходов в анализе коллагена.
Лабораторная работа № 6
ОПРЕДЕЛЕНИЕ ГЕМОГЛОБИНА КРОВИ
Цель работы. Освоить методы определения гемоглобина крови убойных животных на основе его биохимических свойств.
Задачи. Получить и изучить кристаллы гемоглобина из крови убойных животных. Провести качественные реакции на гемовую группу гемоглобина крови. Определить концентрацию растворенного гемоглобина в плазме крови.
Объекты исследования. Стабилизированная кровь и ее фракции от различных видов животных, полученные в условиях производства; цельная кровь и плазма, выделенная в лабораторных условиях или при промышленной переработке крови.
Материалы, реактивы и оборудование. Физиологический раствор (раствор хлорида натрия массовой долей 0,85 %); ацетатный буфер pH 4,6 (4,9 см3 раствора CH.COONa • ЗН2О молярной концентрацией 0,2 моль/дм3 и 5,1 см3 раствора СН3СООН молярной концентрацией 0,2 моль/дм3); раствор пероксида водорода массовой долей 0,3 %; спиртовой раствор бензидина массовой долей 0,1 %; этанол 96 об.%; ледяная уксусная кислота; кристаллический хлорид натрия; стеклянные палочки; пробирки; пипетки; капельницы; штатив для проби
80
рок: фо i оэлектроколориме i р. часы, микроскоп: предметы^ стекла: покровные стекла.
Методические указания. Работ состоит из ipex этапов. каждый из которых может быть использован в экспериментальной практике самостоятельно.
Гемоглобин разных животных различается белковой частью — глобином, который характеризуется различным сочетанием и соотношением аминокислот, поэтому форма кристаллов гемоглобина неодинаковая.
Метод определения кристаллов гемоглобина основан на предварительном водном гемолизе эритроцитов с последующим осаждением кристаллического гемоглобина раствором этилового спирта.
Гемоглобин — сложный белок, который состоит из белка глобина и простетической группы — гема. Последний, в свою очередь, является сложным органическим соединением, содержащим четыре пиррольных кольца и двухвалентное железо.
Присутствие кристаллов гемина говорит о наличии гемоглобина в исследуемом объекте (рис. 1.24).
В основу количественного определения растворенного в плазме крови гемоглобина положена его способность окислять в присутствии пероксида водорода бензидин в окрашенное соединение (л-хинондиимид). Интенсивность окраски среды прямо пропорциональна содержанию гемоглобина и определяется фотометрированием.
сн3 сн=сн? СН, сн=сн?
СН3 СН? СН? СН,
I I
СН? СН?
I I
соон соон
Рис. 1.24. Хлоргемин:
а — структурная формула; б— кристаллы (х 500)
81
иищслпнИЬ КРИСТАЛЛОВ ГЕМОГЛОБИНА
Подютовка проб. Сiабилизированную кровь живошых (10 см ') центрифугируют в зсчсние 5- X .мим при частоте вращения 25—42 с Плазму (налосадочиую жидкость) осторожно отбирают пинеткой и переносят в чистую сухую пробирку (ее можно использовать для выполнения и. 3 данной работы). Осадок эритроцитов используют для выделения кристаллов гемоглобина.
Порядок проведения анализа. В сухие чистые пробирки предварительно помещают но 1.5 см3 дистиллированной воды. а затем добавляют ио 0,5 см3 эритроцитов и смесь тщательно перемешивают стеклянной палочкой. В результате происходит гемолиз эритроцитов — нарушение целостности оболочки. К гемолизованным эритроцитам добавляют 10 капель этанола 96 об.% и охлаждают (для этого используют смесь льда и хлорида натрия) в течение 40—60 мин. После этого каплю смеси наносят на предметное стекло, накрывают покровным стеклом и микроскопируют (используя объектив с увеличением в 40 раз). Аналогичные операции проводят е кровью всех видов животных.
Оформление результатов. При нагревании гемоглобина в кислой среде небелковая группа — гем отщепляется и в присутствии солей (NaCl) переходит в геми н, в котором железо трехвалентно.
= N4 N = —N4 ,N —
'fZ W— ci / s г X
/ X z X
N N— N N —
II II I II
Результаты наблюдений фиксируют в виде зарисовок с указанием объекта и применяемого увеличения (пример представ-
лен на рис. 1.25).
После сопоставления рисунков студенты самостоятельно делают заключение по работе, акцентируя внимание на форме, размерах и количестве кристаллов.
Рис. 1.25. Кристаллы гемоглобина морской свинки (8x40)
2. ОПРЕДЕЛЕНИЕ ГЕМОГЛОБИНА ПО КАЧЕСТВЕННОЙ РЕАКЦИИ НА ГЕМОВУЮ ГРУППУ(ПРОБА ТЕЙХМАНА)
Подготовка проб. В качестве проб используют стабилизированную кровь убойных животных без постороннего запаха, цвета и коагулянтов.
Порядок проведения анализа. На предметное стекло наносят каплю крови и высушивают при температуре не выше 60 ЭС. К подсушенной крови прибавляют несколько кристаллов хлорида натрия и 1—2 капли ледяной уксусной кислоты, перемешивают, затем накрывают покровным стеклом и осторожно нагревают до начала кипения (но не кипятят).
После охлаждения гемин выделяется в виде коричневых ромбоидальных кристаллов, которые рассматривают под микроскопом при кратности увеличения не менее х500. Операции проводят с кровью всех видов животных.
Результаты наблюдений фиксируют в виде зарисовок с указанием объекта и применяемого увеличения.
После сопоставления рисунков студенты самостоятельно делают заключение по работе, отмечая особенности строения кристаллов.
3. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ РАСТВОРЕННОГО ГЕМОГЛОБИНА В ПЛАЗМЕ КРОВИ
Подготовка проб. Для выполнения всех этапов работы используют плазму после отделения форменных элементов из цельной крови в соответствии с рекомендациями (см. п. 1) либо свежую плазму, полученную путем сепарирования крови в производственных условиях.
Порядок проведения анализа. В две пробирки вносят по 4 см3 ацетатного буфера, 2 см3 раствора пероксида водорода, 2 см3 раствора бензидина. В одну из пробирок (проба) добавляют 0,04 см3 плазмы крови, а в другую (контроль) — 0,04 см3 физиологического раствора. Смеси в пробирках перемешивают и спустя 3 мин фотометрируют при 680 нм.
При наличии гемоглобина интенсивность синего окрашивания возрастает в течение 4—5 мин, поэтому оптическую плотность необходимо измерить два раза и записать максимальную величину. Через 5—6 мин после фотометрирования окрашивание приобретает лилово-бурый цвет, и величина оптической плотности при 680 нм уменьшается.
Массовую концентрацию гемоглобина в плазме крови находят с помощью калибровочного графика.
Построение калибровочного графика. Исходя из того, что в крови содержится в среднем 16,5 мг/см3 гемоглобина, для приготовления
83
основного сщнларгною раствора к 0.1см' крови добавляю! 16.4см’ дистиллированной волы. В 1 см? этого раствора со* держичся i mi гемо!лобина. Его используют, разбавляя дистиллированной водой, перед каждым построением калибровочного графика для приготовления рабочих растворов крови с содержанием гемоглобина 0.51 1,0: 2.5: 5.0: 10: 20: 30 мг/см3. По 0.04 см3 каждого рабочего раствора переносят в отдельные пробирки, куда добавляют все необходимые реактивы для количественного определения гемоглобина. Фотометрирование проводят так же, как и в случае пробы с плазмой крови и физиологическим раствором. По результатам измерений строят калибровочный график — зависимость оптической плотности рабочих растворов крови от массовой концентрации гемоглобина в этих растворах.
Для определения концентрации гемоглобина вычисляют разность между оптической плотностью опытной (Рф и контрольной (Dq) проб. По величине полученной разности находят соответствующее значение массовой концентрации гемоглобина на калибровочном графике.
Номер опыта, измерения Наименование и характеристика образца плазмы крови (вид убойных животных, возраст, физиологическое состояние, способ первичной обработки крови и т. д.) Оптическая плотность Разность оптической плотности пробы и контроля Массовая концентра ция гемоглобина, мг/см’
пробы (плазмы крови) контроля (физиологического раствора)
По окончании работы студенты самостоятельно делают выводы и формулируют заключение к работе. При этом дают сравнительную характеристику экспериментальным данным, оценивают возможности и ограничения используемых подходов и методов.
Лабораторная работа № 7
ОПРЕДЕЛЕНИЕ ФОРМ ГЕМОГЛОБИНА (ПИГМЕНТОВ) НА ОСНОВЕ СПЕКТРАЛЬНЫХ ХАРАКТЕРИСТИК
Цель работы. Приобрести практический навык определения пигментов крови убойных животных на основе спектроскопических методов анализа.
Задачи. Получить дериваты (формы) гемоглобина (оксигемоглобин, метгемоглобин, карбоксигемоглобин); снять спектры поглощения гемоглобина и его дериватов и идентифицировать формы гемоглобина путем изучения характеристических полос поглощения в соответствующих спектрах.
84
В с Г) Г В Г
| Красный Желтый Зелены)) Фиолеювын ; — н
) 1 1 II 1 II 1 II 1 1 1 1 II 1 II 1 II 1
Рис. 1.26. Спектры поглощения:
7— солнечный спектр; 2 — оксигемоглобин; 3 — гемоглобин; 7 — метгемоглобин (нейтральный); В, С, D, Е, F-- характеристические линии спектра
Методические указания. Гемоглобин и его производные (формы) обладают способностью поглощать излучение различной длины волны и давать характерные спектры поглощения. Если между источником света и спектроскопом поместить сосуд с водным раствором гемоглобина или его производных, то эти вещества будут поглощать часть лучей определенной длины волны, в результате чего в этих местах появятся черные полосы (рис. 1.26), идентификация которых позволит судить о наличии соответствующих пигментов.
Спектроскопическое исследование пигментов крови можно проводить с помощью карманного спектроскопа.
Объекты исследования. Стабилизированная кровь убойных животных различных сроков хранения, получаемая в условиях производства.
Материалы, реактивы и оборудование. Свежеприготовленный раствор K3[Fe(CN)6] массовой долей 5 %; реактив Стокса; пробирки; пипетки; спектроскоп (спектрофотометр).
Приготовление реактивов. Реактив Стокса.1 часть сульфата железа и 2 части виннокаменной кислоты растворяют в 15 частях дистиллированной воды; перед употреблением добавляют аммиак до слабощелочной реакции. Раствор должен быть прозрачным и окрашен в зеленый цвет.
85
1. ОПРЕДЕЛЕНИЕ ОКСИГЕМОГЛОБИНА
Порядок проведения анализа. При идентификации оксигемоглобина в пробирку к капле крови прибавляют 2 см3 дистиллированной воды и полученный раствор желтовато-розового цвета рассматривают в спектроскопе, помещая пробирку между источником света и спектроскопом. О наличии оксигемоглобина судят по двум тонким полосам поглощения в желто-зеленой части спектра между так называемыми фраунгоферовыми линиями D и £, т. е. в областях с длинами волн 578,1 и 541,7 нм.
2. ОПРЕДЕЛЕНИЕ ГЕМОГЛОБИНА
Порядок проведения анализа. При идентификации гемоглобина к раствору крови (см. п. 1) прибавляют 5—8 капель свежеприготовленного раствора K3[Fe(CN6)] (реактив Стокса). Двухвалентное железо, входящее в состав реактива Стокса, в этих условиях окисляется, переходит в трехвалентное, в результате чего образуется гемоглобин. Светло-синий цвет жидкости становится более темным, с фиолетовым оттенком, а в спектре появляется одна широкая полоса между линиями Z) и Е. Наиболее темная ее часть соответствует длине волны 555—558 нм.
3. ОПРЕДЕЛЕНИЕ МЕТГЕМОГЛОБИНА
Порядок проведения анализа. При идентификации метгемоглобина (МНЬ) к раствору крови (см. п. 1) прибавляют 5—7 капель свежеприготовленного раствора K3[Fe(CN)6] массовой долей 5 % и взбалтывают. Жидкость приобретает бурую окраску. При этом обнаруживаются три полосы поглощения: две тонкие в желто-зеленой части спектра между линиями D и Е и третья (наиболее характерная) в красной части спектра между линиями С и D с длиной волны 630 нм.
Оформление результатов. Данные по изучению спектров гемоглобина и его производных рекомендуется оформить в виде таблицы:
Пигменты (цвет) Форма гемоглобина Характеристика спектров поглощения
По результатам экспериментальных исследований формулируют выводы и делают заключение по работе.
86
Лабораторная работа № 8
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГЕМОГЛОБИНА И ОРГАНИЧЕСКОГО ЖЕЛЕЗА КРОВИ
Цель работы. Фотометрирование крови убойных животных и фракции форменных элементов.
Задачи. Определить и рассчитать массовые доли гемоглобина и органического железа в цельной крови и фракции форменных элементов крови убойных животных.
Объект исследования. Стабилизированная кровь различных видов животных, полученная в условиях производства.
Материалы, реактивы и оборудование. Ацетонциангидрин (3 ампулы по 0,5 см3 или 0,47 г); смесь реактивов (3 флакона по 1,2 г), в том числе в каждом флаконе: калия гексацианоферрата (II) тригидрат — 0,2 г, натрия гидрокарбонат — 1,0 г, стандартный раствор (СО) гемиглобинцианида; спектрофотометр; фотоэлектроколориметр; пипетки с делениями вместимостью 0,02 и 5см3; колба мерная вместимостью ЮООсм3; пробирки; штатив для пробирок.
Приготовление реактивов. Общее количество стандартного набора реактивов рассчитано на приготовление Здм3 трансформирующего раствора (600 анализов). Используя 1/3 набора, можно готовить 1,0 дм3 трансформирующего раствора для проведения 200 анализов.
Трансформирующий раствор (1 дм3). Содержимое одной ампулы с ацетонциангидрином и одного флакона со смесью калия гексацианоферрата (II) тригидрат с гидрокарбонатом натрия количественно переносят в мерную колбу и доводят дистиллированной водой до 1,0 дм3. Содержимое осторожно перемешивают, избегая образования сильной пены. Раствор желтого цвета, прозрачный.
Раствор стабилен при хранении в посуде из темного стекла при комнатной температуре в течение нескольких месяцев. При появлении осадка или при обесцвечивании раствор непригоден к употреблению.
Стандартный раствор (СО) гемиглобинцианида. Представляет собой стерильный прозрачный раствор оранжево-красноватого цвета. Окраска раствора зависит от содержания гемиглобинцианида. Концентрация гемиглобинцианида в растворе находится в диапазоне 0,6—1,0 г/дм3, что соответствует при разведении крови в 251 раз концентрации гемиглобинцианида в крови 150,6—251,0 г/дм3.
Методические указания. Метод основан на взаимодействии гемоглобина крови с железосинеродистым калием и окислении его в метгемоглобин (гемиглобин), образующий с анто-циангидрином окрашенный комплекс гемиглобинцианида. Ин-
87
।снсивность окраски гемиглооинпианила пропорциональна кони чес гну гемоглобина.
Относительная молекулярная масса гемоглобина точно установлена, известна также масса железа в молекуле. В связи с этим количество органического железа можно легко рассчитать, зная массовую концентрацию гемоглобина в растворе.
Порядок проведения анализа. В пробирку с 5.0 см3 трансформирующего раствора добавляют О,О2см3 свежей стабилизированной крови убойных животных (разбавление в 251 раз). Содержимое пробирки тщательно перемешивают и оставляют на 10—20 мин. Затем проводят измерение оптической плотности раствора на любом из вышеуказанных приборов при длине волны 540 нм (520— 560 нм) в кювете толщиной слоя 1 см против контрольной пробы (трансформирующего раствора).
Массовую концентрацию гемоглобина (НЬ) в крови (г%) определяют по калибровочному графику, построенному по стандартному раствору гемиглобинцианида.
Построение калибровочного графика. Ампулы с раствором СО гемиглобинцианида предварительно термостатируют при (20,0 ± ± 0,5) °C в течение 30 мин, после чего их встряхивают и готовят 5 стандартных растворов при помощи градуированной пипетки с делениями 1-го или 2-го класса точности вместимостью 5,0 см3 в соответствии с данными приведенной ниже таблицы.
Номер пробирки Объем, см; Фактор разведения Массовая концентрация гемоглобина в крови, г/дм'
СО гемиглобинцианида трансформирующего раствора
1 1 4 0,2 0,2 х СО* х 251
3 2 3 0,4 0,4 х СО* х 251
3 3 2 0,6 0,6 х СО* х 251
4 4 1 0,8 0,8 х СО* х 251
5 5 — 1,0 СО* х 251
* СО — массовая концентрация гемиглобинцианида в растворе стандартного образца, г/дм3; 251 — коэффициент разведения.
Максимальная погрешность приготовления стандартных растворов по описанной методике составляет: 1,5 % при использовании пипетки 2-го класса и 0,94 % при использовании пипетки 1 -го класса точности.
Измерение оптической плотности растворов проводят по мере возрастания их концентрации против контрольной пробы в условиях, аналогичных измерениям опытной пробы.
По полученным данным строят калибровочный график в координатах: по оси абсцисс — массовая концентрация гемоглобина, а по оси ординат — оптическая плотность раствора СО ге-
88
миглобинцианида. При смене партии реактивов калибровочный график строят заново.
Массовую концентрацию гемоглобина (г%) можно также определить по формуле
X = Е^-СК 0.001, Е
(1-17)
где Е . £—экстинкции соответственно опытной пробы и стандартного раствора; Ф—концентрация гемиглобинцианида в стандартном растворе, мг%; к—коэффициент разведения крови; Л7=251; 0,001 — коэффициент пересчета количества гемоглобина из мг% в г%.
Относительная молекулярная масса гемоглобина 64 000, в его структуре имеется один атом железа (атомная масса 56). После количественного определения гемоглобина рассчитывают концентрацию органического железа путем составления простой пропорции.
Полученные экспериментальные и расчетные данные сводят в таблицу:
Наименование образца, вид убойных животных
гемоглобина
Массовая доля, %
органического железа
По результатам экспериментальных исследований формулируют выводы и делают общее заключение по работе.
Лабораторная работа № 9
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПИГМЕНТОВ МЯСА
Цель работы. Приобрести практический навык при определении общего количества и различных форм пигментов в мясе и мясопродуктах.
Задачи. Экстрагировать пигменты и измерить светопоглоще-ние растворов с последующим расчетом общего содержания пигментов в образцах и соотношения пигментов по светопоглоще-нию проб.
Объекты исследования. Мышечная ткань разных видов животных или мясо с различным содержанием мышечной ткани, а также субпродукты I категории.
Материалы, реактивы и оборудование. Раствор ацетона объемной долей 94 % (к 470 см3 ацетона добавляют 30 см3 дистиллированной воды); раствор хлорацетона (СН3СОСН2С1) объемной до-
89
ien SO ('< (к 400 см’ апсгона добавляю!' 00 см' .шспшироваинои воды и 11) см' коннсн i рирова! и юи соляной кислоты): концентрированная соляная кислота: спектрофотометр: пипетки мерные вместимостью 1 см’: колбы плосколонные вместимостью 50см\ бумажные фильтры: чашки Петри.
Методические указания. Пигменты мяса и мясопродуктов хорошо растворимы в воде и органических растворителях и легко экстрагируются, придавая соответствующим растворам определенный цвет. Интенсивность окраски прямо пропорциональна количеству пигментов. Измерив светопоглощение. легко определить концентрацию пигментов но калибровочному графику или используя рекомендуемые в конце работы расчетные формулы.
Принцип получения спектров поглощения окрашенных веществ, в частности пигментов мяса и крови, состоит в том, что световая энергия, проходя через раствор пигмента, поглощается этим раствором. Происходит абсорбция (поглощение света), которую можно визуально обнаружить спектроскопически.
Формы миоглобина (Mb, МЬО2 и MetMb) можно идентифицировать при определении спектральных характеристик образца с помощью спектрофотометрических методов анализа.
Например, водный раствор Mb характеризуется специфическим спектром поглощения в видимой области. Максимум поглощения находится при длине волны 555 нм. Переход Mb в МЬО2, MetMb или какую-либо другую форму сопровождается изменением спектра и в целом сдвигом максимума в сторону большей или меньшей длины волны (рис. 1.27).
Использование метода отражения позволяет анализировать формы миоглобина непосредственно в образце мяса без предварительной экстракции, что обеспечивает сохранение соотно-
Рис. 1.27. Спектры поглощения производных миоглобина:
1 — оксимиоглобин; 2 — миоглобин; 3 — мет-миоглобин
шения пигментных производных.
Измерив поглощение монохроматического пучка света и получив процент отражения, рассчитывают коэффициенты поглощения (К) и рассеивания (S). Относительные количества пигментов мяса определяют из отношения К/S при соответствующих длинах волн:
X = 525 нм — максимум поглощения для трех форм пигмента: дезокси-, окси- и мет-миоглобина;
90
X — 572 нм —- максимум поглощения для лсзоксимио)до би на (Mb):
X — 474 нм — максимум поглощения для окси-(МЬОЗ и мет-миоглобина (MetMb).
1. ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ ПИГМЕНТОВ
Подготовка проб. При количественном определении общего содержания пигментов из образцов разных видов мяса одного и того же срока хранения вырезают кусочки массой 5,000 ±0,001 г.
Порядок проведения анализа. Подготовленные образцы мяса помещают в стеклянные пробирки, заливают водным раствором ацетона объемной долей 94 % и гомогенизируют в течение 2 мин. В каждую пробирку к гомогенату добавляют 0,5 см3 концентрированной соляной кислоты, пробирки встряхивают, закрывают пробкой и выдерживают в темном месте в течение 2 ч, периодически перемешивая смесь. После этого растворы отфильтровывают через бумажный фильтр в колбу вместимостью 50 см3, а осадок промывают раствором хлорацетона объемной долей 80 %, доводя объем дистиллированной водой до метки.
Светопоглощение полученного раствора определяют на спектрофотометре при длине волны 540 нм. В качестве контрольной пробы используют хлорацетон. Для определения массовой доли пигментов в исследуемом образце пользуются калибровочным графиком.
Построение калибровочного графика. Используют готовые очищенные препараты миоглобина либо свежие мышцы убойных животных. Подготовку образцов к количественному определению общего содержания пигмента ведут аналогично описанию метода, нарезая для экстрагирования кусочки мышц фиксированной массы от 1 до Юг. Количественное содержание миоглобина оценивают, исходя из известных данных о среднем содержании миоглобина (мг в 1 г) в мышцах крупного рогатого скота и свиней:
Мышцы крупного рогатого скота:
Мышцы свиней:
скелетные 3,7
сердца 2,1
скелетные:
красные 1,44
белые 0,79
сердца 0,92
Строят калибровочный график, откладывая по оси абсцисс массовую концентрацию миоглобина (мг/см3 раствора), а по оси ординат — светопоглощение раствора при длине волны 540 нм.
91
2. ОПРЕДЕЛЕНИЕ ФОРМ МИОГЛОБИНА (ПИГМЕНТОВ) НА ОСНОВЕ СПЕКТРАЛЬНЫХ ХАРАКТЕРИСТИК МЯСА
Подготовка проб. Каждый из исследуемых образцов мяса и мясопродуктов нарезают ломтиками толщиной 4—5 мм перпендикулярно к направлению мышечных волокон, а затем вырезают кусочки в соответствии с размерами кювет используемого прибора.
Порядок проведения анализа. Вырезанные кусочки помещают в соответствующие по размеру кюветы измерительного прибора (спектрофотометра СФ-10, СФ-17 или другой марки). Снимают показания спектрофотометра по поглощению монохроматического пучка света и проценту отражения. Соотношение пигментов и содержание относительных количеств пигментов мяса определяют расчетным путем.
Относительные количества метмиоглобина (^MetMb) и дезоксимиоглобина (XMb) определяют по формулам:
^ме.мь = K/S5n : K/S525; (1.18)
An, = K/sm к/^- (119)
где К—коэффициент поглощения; 5—коэффициент рассеивания при соответствующих длинах волн.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы:
Образец Исходный цвет Массовая доля (%) пигментов мяса и мясопродуктов при длине волны
суммарных (540 нм) МЬО, (474 нм) Mb (572 нм) MctMb (650 нм)
По окончании работы и оформления результатов студенты самостоятельно делают выводы и формулируют заключение, акцентировав внимание на соотношении отдельных пигментов, сопоставляя данные с цветом образцов, сроками их хранения, видом мяса.
Лабораторная работа № 10
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АМИННОГО АЗОТА
Цель работы. Освоить титриметрические и фотометрические методы количественного определения суммарных аминокислот в тканях убойных животных.
Задача. Определить суммарное количество аминного азота.
92
Объекты исследования. Ткани животных (свежие или после хранения в рекомендуемых режимах); образцы мышечной ткани убойных животных на разных стадиях автолиза; гидролизаты белков крови, мышечной, соединительной ткани, кератинсодержа-шего сырья, комбинированных белковых систем.
Методические указания. Необходимость количественного определения аминокислот возникает в ходе аминокислотного анализа белков, изучения свободных аминокислот, определения активности протеолитических ферментов и в других случаях. Предлагаемые способы определения могут быть использованы при анализе биологических материалов животного или растительного происхождения, отдельных аминокислот или их смесей, а также белковых гидролизатов.
При определении количества аминного азота используют различные способы.
Медный способ. Основан на способности аминокислот образовывать с ионами Сп2+ прочные растворимые комплексы, в которых каждый ион меди связан с двумя молекулами аминокислот;
NH, ^0
/ о-с^
R-СН ^CH-R
\ / / о=с-о nh2
Количество меди, вошедшей в состав таких комплексов, определяют затем иодометрически. При выполнении анализа к определенному объему раствора аминокислоты прибавляют при слабощелочной реакции избыток суспензии фосфата меди в боратном буфере, в результате чего после взбалтывания в раствор переходят медные соли большинства аминокислот. Избыток фосфата меди удаляют фильтрованием, к прозрачному фильтрату прибавляют уксусную кислоту и иодид калия. Образовавшийся при этом ацетат меди восстанавливают иодидом калия.
2Cu(CH3COO)2 + 4KJ -> 2CuJ + 4СН3СООК + J2.
Выделившийся иод титруют раствором гипосульфита натрия:
К + 2Na?S?0, -э 2NaJ + Na?S4O,.
Z z Z 3 Z 4 0
Таким образом, количество гипосульфита натрия, пошедшего на титрование, пропорционально исходному содержанию аминокислот в растворе. При этом 1 см3 раствора Na?S2O3 молярной концентрацией 0,01 моль/дм3 соответствует 0,28 мг аминного азота.
93
Небольшое количество аммиака и высокие концентрации мочевины не мешают определению аминного азота медным способом.
Нингидриновый способ. Нингидрин, являющийся сильным окислителем, вызывает окислительное дезаминирование аминокислоты, приводящее к образованию диоксида углерода, соответствующего альдегида, воды и конденсированного соединения («пурпура Руэмана») сине-фиолетового цвета с максимумом поглощения около 580 нм.
Аминокислота Нингидрин СН «Пурпур Руэмана»
О
Поскольку оптическая плотность при этой длине волны практически линейно зависит от числа исходных аминогрупп, на основе нингидриновой реакции разработан удобный колориметрический способ количественного определения аминного азота. При нагревании с органическими растворителями альдегиды не образуются, а радикалы аминокислот входят в состав производных «пурпура Руэмана», присоединяясь к атому азота. Таким образом, в этом случае цвет образующихся окрашенных продуктов зависит от природы аминокислоты. Пролин и оксипролин (иминокислоты) в процессе реакции с нингидрином образуют продукты ярко-оранжевого цвета с максимумом поглощения около 440 нм.
Способ формольного титрования. Основан на способности аминокислот реагировать с нейтральным раствором формальдегида с образованием метиленаминокислот. Этот метод применяется для определения карбоксильных групп аминокислот, которые титруют раствором щелочи, предварительно блокировав аминогруппы формальдегидом. В ходе реакции с формальдегидом аминогруппа теряет основные свойства, при этом образуется метиленаминокислота, которая оттитровывается щелочью:
R Н R
H-C-NH2 + СО -► H-C-N=CH2 + Н2о ;
СООН Н СООН
R R
I I
Н—С—N=CH2 + NaOH —► H-C-N=CH2 + Н2О
СООН COONa
94
Определяя количество карбоксильных грхнн пированием, можно одновременно определи|ь и количество о-аминогрупп, так как количество карбоксильных групп эквивалентно количеству а-аминогрупп. связанных формальдегидом. При организации лабораторного практикума можно использовать один или несколько методов.
Подготовка проб. При использовании в качестве объектов исследования животных тканей готовят водяную вытяжку белковых фракций. Ткань освобождают от жира и прорезей соединительной ткани. Навеску массой 3—4 г тщательно измельчают ножом на часовом стекле. К навеске измельченного сырья приливают дистиллированную воду в соотношении 1 : 6 (по массе) и ведут экстракцию на холоде при О °C в течение 30 мин. Затем центрифугированием при частоте вращения 83 c J в течение 5 мин отделяют осадок. Надосадочную жидкость осторожно декантируют и используют для количественного определения аминокислот.
При использовании в качестве объектов исследования белковых гидролизатов их фильтруют, фильтраты используют для количественного определения аминокислот.
1. МЕДНЫЙ СПОСОБ
Материалы, реактивы и оборудование. Суспензия фосфата меди: раствор тимолфталеина: 0,25 г в 100 см3 раствора этанола массовой долей 50%; раствор NaOH молярной концентрацией 1 моль/дм3; ледяная уксусная кислота; раствор KJ массовой долей 15 %; раствор Na2S2O3 молярной концентрацией 0,01 моль/дм3; раствор крахмала массовой долей 1 %.
Приготовление реактивов. Суспензия фосфата меди. Непосредственно перед использованием к одному объему раствора А добавляют два объема раствора Б, тщательно перемешивают и прибавляют два объема раствора В. Хранят не более 2—3 дней.
Раствор А: 27,3 г хлорида меди (I) растворяют в 1 дм3 воды.
Раствор Б: 64,5 г двузамещенного фосфата натрия растворяют в 500 см3 воды, добавляют 7,2 г NaOH и затем доводят объем водой до 1 дм3.
Раствор В (боратный буфер pH 8,6—8,8): 57,21 г буры (Na2B4O7 • ЮН2О) растворяют в 1,5 дм3 воды, добавляют 100 см3 раствора НО "молярной концентрацией 1 моль/дм3 и доводят объем водой до 2 дм3.
Порядок проведения анализа. В мерную колбу вместимостью 25 см3 помещают либо 2 см3 нейтральной вытяжки, либо 2 см3 гидролизата белка, либо 2 см3 раствора аминокислоты, 1—2 капли раствора тимолфталеина и по каплям добавляют раствор NaOH до светло-голубого окрашивания (pH около 9). Затем в колбу приливают 10 см3 хорошо перемешанной суспензии фосфата меди и
95
объем доводят водой до метки. Содержимое мерной колбы тщательно перемешивают и оставляют на 5—10 мин при комнатной температуре, а затем фильтруют через плотный фильтр в сухую коническую колбу вместимостью 50 см3. Если фильтрат окажется мутным, повторно проводят фильтрование, иначе результаты определения будут завышены. 10 см3 прозрачного фильтрата отбирают в сухую колбу, добавляют 0,4 см3 ледяной уксусной кислоты и 4 см3 раствора K.J массовой долей 15%. Выделившийся иод титруют раствором гипосульфита натрия до светло-желтой окраски, прибавляют 1—2 капли раствора крахмала и осторожно титруют до исчезновения сине-фиолетовой окраски. Определения проводят в двух повторностях и вычисляют среднее значение объема гипосульфита натрия (Иоп), пошедшего на титрование. Параллельно при тех же условиях обрабатывают контрольную пробу, содержащую вместо раствора аминокислоты 2 см3 воды. Контроль проводят для определения возможного наличия растворимых соединений меди в используемой суспензии. Объем раствора гипосульфита натрия, пошедшего на титрование контроля, обозначают Йк.
Содержание аминного азота (мг) в 2 см3 исследуемого раствора определяют по формуле
С=0,28(Еоп- Гк)К-2,5, (1.20)
где К— коэффициент пересчета на раствор гипосульфита натрия молярной концентрацией 0,01 моль/дм3.
Если исследуемый раствор содержит только одну аминокислоту (например, глицин), то для определения ее массовой доли величину С умножают на коэффициент М/14, где М— молекулярная масса данной аминокислоты (для глицина М = 75).
2. НИНГИДРИНОВЫЙ СПОСОБ
Материалы, реактивы и оборудование. Фосфатный буфер (pH 6,5; 0,5 моль/дм3); абсолютный ацетон; смесь абсолютных ацетона и этанола (1:1 по объему); раствор нингидрина массовой долей 0,5 % в д-бутаноле; насыщенный раствор бариевой воды.
Порядок проведения анализа. В пробирку с пришлифованной пробкой помещают 0,1см3 исследуемой пробы, 0,1см3 буфера, 1,5 см3 смеси ацетона со спиртом и 2 см3 нингидрина. Содержимое пробирки перемешивают и закрывают пробкой. Пробирки помещают на 15 мин на водяную баню при температуре кипения. Затем пробирку охлаждают под струей воды, объем окрашенной 96
жидкост доводят ацетоном но !<> см ". нсрсмспшвакм п вонори-мегрирую! при .тмине воины 5Ы) нм iipoiHi> кошрояя. содержаще-го вместо пробы 0.1 см? воны.
Содержание аминного азота иди аминокислот рассчи i ыванн по калибровочному i рафику.
Построение калибровочного графика. Используют стандартный раствор глицина с содержанием 1 мг/см?. Из него готовят ряд разведений: 1 см? раствора внося ! в мерную пробирку вместимостью 10 см3 и доводят объем дистиллированной водой до метки, затем проводят окрашивание в соответствии с описанием метода и строят график зависимости отической плотности от концентрации глицина. Чувствительность метола от 5 до 25 мкг аминного азота в пробе (0.1 см3). что соответствует приблизительно 20—120 мкг глицина.
3. СПОСОБ ФОРМОЛЬНОГО ТИТРОВАНИЯ
Материалы, реактивы и оборудование. Складчатый бумажный фильтр; сухой хлорил бария; спиртовой раствор фенолфталеина массовой долей 0.1 %; раствор соляной кислоты (0.01 моль/дм3); раствор гидроксида калия (0,05 моль/дм3); формольная смесь; колбы мерные вместимостью 100 см3; капельницы; воронка стеклянная; бюретка; часы; насыщенный раствор бариевой воды.
Приготовление реактивов. Формольная смесь. К 50 см3 раствора формальдегида объемной долей 40 % добавляют 10 капель раствора фенолфталеина массовой долей 0,1 % и по каплям раствор гидроксида калия молярной концентрацией 0,1 моль/дм3 до образования слабо-розовой окраски.
Порядок проведения анализа. К 25 см3 исследуемой пробы в колбе прибавляют 1 г хлорида бария, 10 капель раствора фенолфталеина и перемешивают. К смеси приливают насыщенный раствор бариевой воды до появления едва заметного розового окрашивания. Колбу закрывают и оставляют на 15 мин. Затем разбавляют водой до объема 100 см3 и фосфаты и карбонаты бария отфильтровывают через складчатый бумажный фильтр. Готовят две колбы вместимостью по 100 см3. В одну вносят 40 см3 фильтрата, что соответствует 10 см3 раствора аминокислот, во вторую — 40 см3 дистиллированной воды. Прибавляют по 10 капель раствора фенолфталеина и раствор соляной кислоты (осторожно) до окрашивания растворов (контроля и пробы) в едва заметный розовый цвет. Затем в обе колбы вносят по 10 см3 формольной смеси и титруют из бюретки раствором гидроксида калия до появления едва заметной розовой окраски. Цвет контроля и пробы должен быть одинаковым.
97
Co. ic ржи 11 и с амиши,! v u и) i ti Lor,см ) рассчи i biiMioi по формхле
c - ( 1- I (,)W/ I.
где (, и Г -обьсмы раствора гидроксида кадия, затраченные соотвекч вснно на шарование пробы и контроля. им’: А— коэффициент пересчета на раствор шдро-ксида калия молярной концешрацией 0.05 моль/дм': С—масса аминокислот, эквивалентная 1см5 (0.05 .моль/дм’) раствора гидроксида калия (0.7 мг): Р - объем исследуемой пробы, взятой для анализа, см’.
Оформление результатов. Полученные экспериментальные и расчетные данные рекомендуется оформить в виде таблицы:
Обье кт ис ел слова н ия
Ciюсоб О1 (ределення принцип метода
Содержание аминного азота, мг/см’
Затем следует самостоятельно их проанализировать и сформулировать выводы по данной работе. Следует отметить трудоемкость и точность методов, сравнить экспериментальные данные, полученные различными способами, между собой и с данными литературы.
Лабораторная работа №11
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ТРИПТОФАНА
Цель работы. Приобрести навык количественного определения триптофана в тканях и мясе животных и птицы.
Задачи. Освоить методы подготовки проб, провести качественную реакцию на триптофан и рассчитать его количество.
Объекты исследования. Образцы мышечной ткани или мяса животных и птицы, колбасные изделия, полуфабрикаты.
Материалы, реактивы и оборудование. Ацетон (х. ч.); раствор гидроксида натрия молярной концентрацией 3 моль/дм3; раствор пя/ш-диметиламинобензальдегида массовой долей 2,5 % в растворе серной кислоты массовой долей 10 %; раствор нитрата натрия массовой долей 2 %; соляная кислота концентрированная; раствор этанола (50 об.%); универсальный индикатор; спиртовой раствор фенолрота массовой долей 0,1 %; кристаллический триптофан (х. ч.); дистиллированная вода; раствор H2SO4 массовой концентрацией 3 моль/дм3; центрифуга лабораторная; водяная баня; спектрофотометр или фотоэлектроколориметр; стеклянные пипетки; колбы; стаканы; ампулы; стеклянные палочки.
98
Методические указания. Зршпофан г> юс га iочно оо. п>шом количестве содержится в белках животного происхождения. Триптофан инее! гетероциклическую структуру, содержит иминогруппу (— N Н —):
----1 — СН2-СН-СООН
nh2
н
В основу метода определения триптофана положена цветная реакция между продуктами его распада, образующимися при обработке триптофана концентрированной соляной кислотой и пара-диметиламинобензальдегидом в присутствии нитрата натрия.
Для выделения триптофана обезжиренную мышечную ткань подвергают щелочному гидролизу. В процессе кислотного гидролиза и в присутствии жира при щелочном гидролизе триптофан разрушается. Метод определения триптофана в мясе состоит из следующих основных операций: обезжиривания навески мяса, гидролиза обезжиренной мышечной ткани гидроксидом натрия, нейтрализации гидролизата и фильтрования, окисления триптофана и развития цветной реакции с ипра-диметиламинобензальдегидом, фиксации окраски этанолом и измерения ее интенсивности.
При количественном определении триптофана в образцах используют калибровочный график.
Подготовка проб. Образец измельченного мяса или ткани массой (4,000 ± 0,0002) г помещают в центрифужный стакан вместимостью не менее 100 см3, добавляют 4 см3 дистиллированной воды и тщательно растирают. Для обезжиривания заливают ацетоном, доводя до объема 40 см3, и перемешивают. Смесь выдерживают 30 мин при температуре (20 ± 5) °C и центрифугируют в течение 20 мин при частоте вращения 41,7 с-1. Жидкость сливают с осадка и нагревают при 80 °C на водяной бане, перемешивая осадок стеклянной палочкой.
Остаток количественно переносят в стеклянную пробирку с пришлифованной пробкой или ампулу, смывая остатки дистиллированной водой (4 см3). Затем приливают 20 см3 (3 моль/дм3) раствора гидроксида натрия. Пробирку закрывают пробкой, а ампулу запаивают, подписывают каждый образец и гидролизуют в течение суток при температуре 112—114 °C.
Для ускорения процесса гидролиза можно использовать автоклавирование при 0,1 МПа в течение 2—3 ч.
Порядок проведения анализа. Полученные гидролизаты охлаждают и количественно переносят в колбу вместимостью 100 см3, доливая дистиллированную воду до 2/3 объема. Раствор нейтрали
99
зуют сначала небольшим объемом (3 моль/'дм?) раствора H-,SO4. а затем более разбавленными ее растворами. Нейтрализацию ведут в присутствии индикаторов (универсальной индикаторной бумаги или фенол фтал е и н а).
Колбу охлаждают, объем доводят до метки, а смесь фильтруют через бумажный фильтр (гидролизат должен быть совершенно прозрачным!).
Для проведения цветной реакции в мерную посуду вместимостью 100 см3 с пришлифованной пробкой приливают пипетками растворы в последовательности: I см3 исследуемого фильтрата, 0,5 см3 раствора /znpn-диметиламинобензальдегида (при встряхивании!), 0,2 см3 раствора NaNO? массовой долей 2 % (при встряхивании!) и 28 см? концентрированной соляной кислоты (под тягой). Растворы оставляют при температуре (20 ± 5) °C на 30 мин для развития окраски, а затем доводят до метки раствором этанола массовой долей 50 %.
Одновременно проводят два основных и два контрольных определения. В последних исследуемый фильтрат заменяют 1 см3 дистиллированной воды.
Интенсивность синей окраски измеряют на спектрофотометре при длине волны 610 нм или на фотоэлектроколориметре с красным светофильтром в отношении контрольного раствора.
Концентрацию триптофана в объеме жидкости, взятой для цветной реакции (1 см3), определяют по калибровочному графику.
Построение калибровочного графика. Для построения графика готовят растворы, содержащие известное количество триптофана. Три навески химически чистого триптофана массой по 50 мг растворяют в 20 см3 раствора гидроксида натрия молярной концентрацией 3 моль/дм3. Растворы триптофана гидролизуют в условиях, аналогичных опыту, и фильтруют, доводя предварительно объем до 50 см3, 1 см3 полученного раствора содержит 1 мг триптофана. Затем готовят ряд стандартных растворов в соответствии с приведенными ниже рекомендациями, проводят цветную реакцию, измеряют оптическую плотность и строят график зависимости оптической плотности раствора от массового содержания триптофана.
Рекомендуемые объемы стандартного раствора триптофана для построения калибровочного графика:
Содержание триптофана, мкг Объем, см5 Содержание триптофана, мкг Объем, см3
стандартного раствора ВОДЫ стандартного раствора воды
100 0,1 0,9 400 0,4 0,6
200 0,2 0,8 500 0,5 0,5
300 0,3 0,7 600 0,6 0,4
100
По средним данным трех серий опытов строят калибровочный график (рис. 1.28).
Массовую долю триптофана (мг%) в исследуемом образце рассчитывают по формуле
„ с 50 100
где с — концентрация триптофана, найденная по калибровочному графику, мкг/см’; 50 — объем раствора, полученный после нейтрализации и разведения, см3: 100 — коэффициент пересчета в проценты; V— объем раствора, взятый для цветной реакции, см3; т — масса навески мяса, г.
Рис. 1.28. Калибровочный график для определения триптофана:
1 — без гидролиза; 2 — после щелочного гидролиза
Оформление результатов. Полученные экспериментальные и расчетные данные рекомендуется оформить в виде таблицы:
Объект исследования
Концентрация триптофана по калибровочному графику, мкг/см’’
Содержание триптофана в образце, мг%
При формулировании выводов по работе следует сравнить полученные данные с литературными, а также сделать сравнительный анализ результатов для различных видов мяса.
Лабораторная работа № 12
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПРОЛИНА
Цель работы. Приобрести практический навык в определении пролина (оксипролина) в образцах мышечной ткани или мяса разных видов убойных животных, а также в мясопродуктах, готовых к употреблению.
Задачи. Получить гидролизат белков в анализируемых пробах; провести качественную реакцию на оксипролин и фотометриро-вание с последующим расчетом его массовой доли.
Объекты исследования. Мышечная или соединительная ткань, мясо различных сортов, а также мясопродукты.
Материалы, реактивы и оборудование. Бумага индикаторная универсальная; кипятильные камни; бумага фильтровальная лабораторная или фильтры бумажные; фольга алюминиевая; хлорамин Т; хлорамин Б; раствор соляной кислоты молярной кон-
101
иен I paunei'1 6 моль/дмраствор гидроксила мафия молярной концентрацией К) моль/лм?: пропанол: кислота лимонная: кислота уксусная, ацетат натрия: /ш/^-лиметиламинобензальлегит: кислота хлорная объемной долей не менее 40 %: вода дистиллированная: стандартный препарат оксипролина: мясорубка или электромясорубка бытовая с отверстиями решетки диаметром от 3 до 4 .мм; весы лабораторные; баня водяная и песчаная; колба стеклянная гидролизная вместимостью до 300 см3: холодильники стеклянные водяные или воздушные; электронагревательное устройство с песчаной баней; колбы мерные вместимостью 100 и 250 см3: воронки стеклянные; пробирки стеклянные; пипетки мерные вместимостью 2, 5 и 10 см3; спектрофотометр или фотоэлектроколориметр с интерференционным фильтром (рабочая длина кювет 30—50 мм, зеленый светофильтр).
Приготовление реактивов. Реактив для окисления. 1,41 г хлорамина Т или Б растворяют в 10 см3 воды и последовательно добавляют 10 см3 пропанола и 80 см3 буферного раствора. Раствор должен быть свежеприготовленным.
Цветной реактив: Юг Аш/гя-диметиламинобензальде-гида растворяют в 35 см3 хлорной кислоты, непрерывно перемешивая. Полученный раствор смешивают с 65 см3 пропанола. Раствор следует готовить в день использования.
Буферный раствор (pH 6,0): 50 г лимонной кислоты; 12 см3 уксусной кислоты массовой долей 96 %; 120 г ацетата натрия; 34 г гидроксида натрия растворяют в мерной колбе вместимостью 1000 см3 и доводят до метки дистиллированной водой. Полученный раствор смешивают с 200 см3 воды и 300 см3 пропанола.
Методические указания. Определение оксипролина предусмотрено нормативной документацией на ряд мясных продуктов, так как этот показатель позволяет судить о пищевой ценности мяса и изделий из него.
Метод основан на выделении оксипролина при кислотном гидролизе пробы продукта, проведении цветной реакции с продуктами ее окисления и спектрофотометрическом измерении интенсивности развивающейся окраски. После определения рассчитывают массовую долю пролина. В качестве объектов исследования рекомендуется использовать ткани и мясо различных анатомических участков туш животных, колбасы разных сортов и различные полуфабрикаты.
Подготовка проб. Образец мышечной или другой ткани мяса, мясопродуктов массой не менее 50 г пропускают через мясорубку не менее двух раз до тонкого измельчения и тщательно перемешивают.
В коническую колбу вместимостью 300 см3 помешают около 4 г продукта, взвешенного с точностью не менее ± 0,0002 г. Добавляют 100 см3 (6 моль/дм3) раствора соляной кислоты и кипятильные 102
камни для равномерного кипения. Колбу соединяют е воздушным или водяным холодильником и нагревают на песчаной бане в течение 7 ч при гидролизе проб вареных и полукопченых мясных продуктов, в течение 8 ч — при гидролизе проб мяса и сырокопченых мясных продуктов.
Теплый гидролизат фильтруют в мерную колбу вместимостью 250 см3. смывая гидролизную колбу и фильтр дистиллированной водой. Объем колбы доводят водой до метки и смесь тщательно перемешивают.
Порядок проведения анализа. Гидролизаты мясных продуктов разбавляют, доводя концентрацию оксипролина до 1—4 мкг/см3. Для этого 4—10 см3 гидролизата (в зависимости от предполагаемого содержания оксипролина в испытуемом продукте) вносят в мерную колбу вместимостью 100 см3 и нейтрализуют раствором гидроксида натрия молярной концентрацией 10моль/дм? по индикаторной бумаге до pH 6,0. Объем доводят водой до метки и перемешивают. Переносят 4 см3 этого раствора в пробирку и добавляют 2 см3 реактива для окисления, перемешивают и оставляют при комнатной температуре на 20 мин. Затем добавляют 2 см3 цветного реактива, перемешивают и пробирку закрывают алюминиевой фольгой. Одновременно готовят два контрольных раствора, используя вместо фильтрата дистиллированную воду. Пробирки быстро переносят на водяную баню температурой (60 ± 0,5) °C и нагревают в течение 15 мин, затем их охлаждают в течение 3 мин водой или льдом и не позднее чем через 30 мин измеряют оптическую плотность опытного раствора на спектрофотометре при длине волны 558 нм относительно контрольного. Предварительно измеряют оптическую плотность контрольных растворов одного относительно другого.
Построение калибровочного графика. Для построения графика готовят основной раствор оксипролина. 100 мг стандартного препарата оксипролина растворяют в воде в мерной колбе вместимостью 100 см3, добавляют каплю раствора соляной кислоты молярной концентрацией 6 моль/дм3 и доводят водой до метки. В день использования берут 1 см3 основного раствора в мерную колбу вместимостью 100 см3 и доводят водой до метки, тщательно перемешивая. 1 см3 этого раствора содержит 0,01 мг оксипролина.
Затем готовят четыре стандартных раствора путем разбавления 5, 10, 20 и 30 см3 основного раствора оксипролина водой в мерных колбах вместимостью 100 см3 для получения соответственно растворов следующих концентраций: 0,5; 1; 2 и 3 мкг/см3.
Далее измеряют оптическую плотность стандартных растворов оксипролина при длине волны 558 нм. Всю операцию повторяют, используя для определения по 4 см3 четырех разбавленных стандартных растворов оксипролина.
По полученным средним данным строят калибровочный график, откладывая на оси ординат величины оптической плотности,
103
л ни осп абсцисс коицсн i paiHHo оксинро.нша. По 01 меченным точкам проводят прямую линию.
Массовую долю оксипролина (б ) вычисляю! iso формуле
с 250 100 100 mV 106
(1.23)
где с — концентрация оксипро.гина. найденная по калибровочном) графи-к\. мт/см'; 250 — общий объем гидролизата, ем': 100 —объем гидролизата после нейтрализации, см’: 100 — коэффициент пересчета в проценты; т — масса навески образца, взятая для анализа, г; И—объем гидролизата, взятый для нейтрализации. см’; 106 — коэффициенг пересчета мкг в г.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы:
Объе кт 11 седелованим
Оптическая плот- । ность раствора ,
Концентрация окси прол и на по катибровочному iрафику, мкг/см'
Массовая доля оксипролина в образце, сг
По результатам проведенных исследований делают выводы и общее заключение по работе. При этом расчетные данные рекомендуется сравнить с данными литературы, провести оценку и сравнение содержания оксипролина в различных сырых образцах и продуктах.
Лабораторная работа № 13 ОПРЕДЕЛЕНИЕ БЕЛКОВ И БЕЛКОВЫХ ВЕЩЕСТВ МЕТОДОМ ГЕЛЬ-ФИЛЬТРАЦИИ
Цель работы. Приобрести практический навык при анализе белков мяса методом гель-фильтрации.
Задачи. Подготовка проб к анализу; калибрование хроматографической колонки и разделение веществ с выделением белковых фракций.
Объекты исследования. Мясо и мясопродукты.
Материалы, реактивы и оборудование. Стеклянные пипетки; фильтровальная бумага; лед; натрий-фосфатный или калий-фосфатный буфер (растворы молярной концентрацией 0,05 моль/дм3, содержащие раствор КС1 молярной концентрацией 0,05, pH 6,5); сефадекс G-100; белки известной молекулярной массы: яичный альбумин 45 000 Да или бычий сывороточный альбумин 68 000 Да; раствор НСЮ4; раствор КОН молярной концентрацией 5 моль/дм3; этанол 80 и 96 об.%; водяная баня; хроматографическая колонка; коллектор фракций; колбы вместимос
104
тью 20U—500 см ': юмоюнизатор: стеклянные пробирки: члпкп Петри: центрифуга лабораторная: фарфоровые чашки: стеклянные палочки.
Методические указания. В анализе белков наибольшее применение нашел метод гель-фильтрации. Для успешного использования метода необходимо правильно подготовить колонку с исследуемым для разделения материалом.
Сухой сефадекс суспендируют в 200-кратном объеме воды и оставляют на 48 ч при комнатной температуре для полного набухания. Процесс набухания можно ускорить, проводя его на водяной бане при температуре кипения в течение 5 ч.
В колонку (размером 1,5x50 см) вносят относительно густую суспензию полностью набухшего геля. Для предотвращения образования пузырьков воздуха в слое геля в колонке суспензию сефадекса перед заполнением колонки можно деаэрировать с помощью водоструйного насоса в колбе Бунзена или вносить его в колонку при температуре, значительно превышающей комнатную (50—60 °C). Сефадексу в колонке дают отстояться, затем с целью уравновешивания и достижения постоянной высоты столба геля колонку промывают 3—5 объемами буферного раствора. Гидростатическое давление для сефадекса G-100 не должно превышать 50 см при высоте столба жидкости в колонке 20—50 см (давление при разделении образца и при заполнении колонки должно быть одинаковым). Для проверки равномерности заполнения через колонку можно пропустить раствор окрашенного белка, например цитохрома с, или голубого декстрана. При этом окрашенная зона должна быть компактной и двигаться по колонке параллельно ее основанию.
Сначала определяют свободный объем колонки (Ио). Для этого через колонку пропускают 1 см3 (1 мг/см3) раствора голубого декстрана в растворе сахарозы (0,5 моль/дм3). В качестве растворителя как для голубого декстрана, так и для исследуемых белков применяют тот же буферный раствор, которым уравновешена колонка. Им же элюируют белки с колонки после нанесения анализируемой смеси. Следует заметить, что поскольку положительно заряженные частицы могут частично взаимодействовать с сефадексом (за счет имеющихся карбоксильных групп), то для элюирования обычно используют растворы с ионной силой выше 0,02.
Вытекающий из колонки буферный раствор собирают в пробирки порциями по 1—3 см3. Регистрацию объема элюата, прошедшего через колонку, начинают с момента нанесения образца на колонку. Многократное использование колонки с сефадексом приводит к замедленному протеканию жидкости через нее. В этом случае гель извлекают из колонки, отмучивают его от мелких частиц, а колонку заполняют заново.
Сефадексы можно хранить в виде суспензии или порошка.
105
Суспензию сефадекса следует хранить в холодильнике вместе с антисептиками: азидом натрия массовой долей 0.02 С. мсртиола-том или хлороформом. Для перевода геля в сухое состояние сефадекс сначала промывают водой для удаления солей, а затем выдерживают несколько минут в 2-кратном объеме этанола (50 об. %): после фильтрования через воронку Бюхнера сефадекс выдерживают в 2-кратном объеме этанола (96 об.%). обработку последним повторяют несколько раз. Затем осадок высушивают в термостате при 60-80 °C.
Подготовка проб. Пробы можно готовить следующими способами:
1. Навеску мышечной ткани массой 3—5 г или какого-либо другого образца тщательно освобождают от жира, соединительной ткани и измельчают ножницами на холоде. К кашице добавляют 5 объемов раствора НС1О4 молярной концентрацией 0,5 моль/дм3. ткань гомогенизируют или тщательно растирают в ступке (примерно 30 мин). Суспензию центрифугируют, центри-фугат сливают, а остаток вновь суспендируют в 1 — 1,5 объема раствора НСЮ4. Промывные воды соединяют с основным центри-фугатом и для удаления НС1О4 добавляют раствор КОН молярной концентрацией 5 моль/дм3. Необходимый объем щелочи рассчитывают и после нейтрализации проверяют pH по универсальному индикатору. Для более полного осаждения перхлората калия цент-рифугат охлаждают (можно оставить на ночь в холодильнике). Осадок КС1О4 удаляют центрифугированием или фильтрованием и промывают очень небольшим объемом хорошо охлажденной воды, фильтрат упаривают досуха на роторном испарителе (температура 40 °C) или водяной бане (температура бани не более 50— 60 °C). Сухой остаток используют в качестве пробы.
2. Экстракт получают при дополнительной обработке ткани спиртом. Это дает возможность получить экстракт, не содержащий солей, которые ухудшают качество хроматограмм. Способ сводится к следующему. Около 5 г сырья или продукта помещают в чашку Петри, стоящую во льду, очищают от жира, соединительной ткани и кровеносных сосудов. Навеску ткани массой 2—3 г измельчают ножницами на холоде до состояния грубой кашицы. Последнюю переносят с 5 объемами этанола (96 об.%) в гомогенизатор, стоящий во льду. Содержимое гомогенизатора перемешивают стеклянной палочкой и гомогенизируют в течение 10 мин. Гомогенат сливают в центрифужный стакан. Гомогенизатор ополаскивают 5 см3 этанола (96 об.%) и промывные воды присоединяют к основному гомогенату. Последний оставляют при комнатной температуре на 20 мин, затем центрифугируют в течение 8—10 мин при частоте вращения 50 с-1. Надосадочную жидкость сливают в мерную колбу вместимостью 200 см3, а осадок трижды промывают 15 см3 этанола (80 об.%), каждый раз сливая промывные воды в мерную колбу. К спиртовому экстракту, находящемуся в колбе,
106
приливают 160- -]SOcm? хлороформа так. чюбы мениск жнл.коси! был на I.5--2см ниже края горлышка колбы. Колбу закрывали пробкой (лучше стеклянной), встряхиваю! в течение 10—15 мни. помешают в холодильник и оставляют там до тех нор. пока четко не обозначится граница между водным и сииртохлороформным слоями. Водный (верхний) слой жидкости, содержащий аминокислоты. отбирают пипеткой с загнутым конном, используя резиновую грушу, и переносят в фарфоровую чашку. В колбу добавляют дистиллированную воду (2—3 см3). а в случае необходимости хлороформ, чтобы уровень жидкости в колбе был на 1,5—2 см ниже края горлышка колбы. Содержимое колбы встряхивают и оставляют для расслаивания. Верхний слой жидкости отсасывают и присоединяют к жидкости, находящейся в фарфоровой чашке. Эту операцию повторяют еще 2 раза. Содержимое фарфоровой чашки упаривают на роторном испарителе при температуре около 40 °C или водяной бане (температура бани 50—60 °C). В дальней шем оба способа обработки ткани совпадают.
Порядок проведения анализа. Перед использованием подготовленной к работе колонки с сефадексом проводят ее калибрование, т. е. пропускают через нее смесь двух окрашенных компонентов — высокомолекулярного с коэффициентом разделения К = 0 и низкомолекулярного 0^=1. При этом преследуют две цели: определить параметры колонки (Ин. Ив) и проверить качество ее заполнения. Параметры определяют по форме и степени компактности окрашенных зон. В качестве высокомолекулярного окрашенного вещества обычно используют голубой декстран — линейный водорастворимый полиглюкан молекулярной массой около 2 000 000 Да, несущий ковалентно присоединенные хромофорные группы. Низкомолекулярным окрашенным соединением служит какая-либо соль, содержащая окрашенные катионы (кобальт, никель) или анионы (бихромат).
Калибрование проводят следующим образом. Открывают кран колонки так, чтобы жидкость вытекала медленно (5—7 капель в минуту), и дают раствору, находящемуся над поверхностью геля, почти полностью впитаться. Под колонку помещают мерную пробирку для сбора элюата. На поверхность геля наносят 0,2—0,3 см3 водного раствора голубого декстрана и окрашенной соли. Наслаивание проводят осторожно, по каплям, по всей поверхности геля. Затем открывают кран и дают раствору проникнуть в гель. Кран закрывают и на поверхность геля осторожно наслаивают 0,2 см3 элюента и опять, открывая кран, дают войти этому раствору в гель. Операцию повторяют еще два раза. Как только окрашенные зоны начнут передвигаться по столбику сефадекса, свободную часть колонки заполняют элюентом, а затем в ходе элюирования его уровень поддерживают примерно постоянным, заполняя колонку почти доверху. Поддерживая с помощью крана низкую скорость элюирования (5—10 капель в минуту), собирают элюат пор-
107
киями ио 2 см-' в пронумерованные мерные пробирки. В процессе элюирования визуально оценивают качество заполнения колонки; при однородном заполнении и сохранении горизонтальной поверхности геля окрашенные соединения (особенно голубой декстран) перемещаются по столбику сефадекса в виде компактных не-перекрываюшихся зон.
По окончании элюирования всех окрашенных веществ кран колонки закрывают и проводят колориметрирование окрашенных фракций элюата: фракции, содержащие голубой декстран, — с красным светофильтром, а фракции, содержащие бихромат, — с зеленым. Затем строят график элюирования при калибровании колонки, откладывая на оси абсцисс номера фракций (или объем в см3), а на оси ординат — значения экстинкций. На графике отмечают величины и Ив.
Для разделения белков на колонке с сефадексом подготовленную пробу осторожно, с учетом вышеприведенных рекомендаций наносят на колонку пипеткой и отмечают время начала опыта. После проникновения пробы в среду сефадекса подключают элюирующий раствор.
Вытекающий из колонки буферный раствор собирают в пробирки порциями по 1—3 см3. Объем элюата, прошедший через колонку, регистрируют, начиная с момента нанесения образца на колонку.
Содержание белка во фракциях определяют спектрофотометрически при длине волны 280 нм.
По окончании анализа колонку промывают несколькими объемами буферного раствора. Строят профиль элюирования отдельных белковых фракций (рис. 1.29). При этом на оси абсцисс откладывают номера пробирок (фракций) или объем прошедшей через колонку жидкости, а на оси ординат — значения оптической плотности фракций. Белки, принадлежащие к одной фракции (пик на профиле элюирования), объединяют. О числе белковых фракций судят по количеству пиков на графике. Количественное определение белка ведут по калибровочному графику.
Рис. 1.29. Пример графика элюирования при фракционировании белков: с J по XI — белковые фракции
108
Построение калибровочного графика. Для количественного определения белка спектрофотометрическим методом предварительно строят калибровочный график с использованием раствора чистого белка концентрацией от 0.05 до 2,00мг/см3, пример которого приведен на рис. 1.30.
Полученные результаты рекомендуется оформить в виде графика элюирования (см. рис. 1.29) и таблицы результатов, форма которой приведена ниже.
Рис. 1.30. Пример калибровочного графика для определения концентрации белка в растворе спектрофотометрическим методом
Образен
Масса навески
Количество белковых фракции
Массовое содержание белков
суммарное
каждой из фракций
После этого студенты делают самостоятельные выводы и заключение по работе, акцентируя внимание на полученных экспериментальных данных и их сравнительной оценке с данными литературы.
Лабораторная работа № 14 АНАЛИЗ БЕЛКОВ МЕТОДАМИ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ
Цель работы. Приобрести практический навык при анализе белков методами ионообменной хроматографии.
Задачи. Подготовка проб, ионообменной смолы и хроматографической колонки к анализу, разделение белков, расчет количества белка в анализируемых материалах.
Объекты исследования. Кровь и ее фракции, а также мясо различных видов животных и птицы, мясопродукты.
Методические указания. Перед использованием ионитов для хроматографического разделения белков их предварительно обрабатывают. Иониты промышленного изготовления после набухания, во-первых, фракционируют по размеру частиц (однородность частиц сорбентов по размеру — одно из важных условий успешной хроматографии) и, во-вторых, подвергают «циклиза-
109
шш - И'.-рсво.1\ и\ и ; о. шои формы в лр\т у К). Kai iioihi 1 ы персы > ДЯ1 115 Nj -форМи Г Н форМХ 11.111 наоборот. <1 аниониты - из <'1 -формы в Oil -формх или наоборот. В процессе обработки стабилизируется структура ионита, и функциональные группы становятся более доступными. Одновременно ионит освобождается от примесей.
Ионообменники можно использовать многократно. Поэтому ио окончании работы их следует регенерировать. Для этого к использованному сорбенту добавляют 30-кратный объем раствора NaOH молярной концентрацией 0,2—0,5 моль/дм'. перемешивают до получения однородной суспензии и фильтруют на воронке Бюхнера. Обработку щелочью проводят дважды. После этого ионит промывают водой до нейтральной реакции фильтрата. Регенерированный ионообменник уравновешивают соответствующим буферным раствором.
Ионообменники на основе целлюлозы можно хранить во влажном состоянии непродолжительное время. Аниониты лучше хранить в солевой форме (не в ОН -форме), а катиониты — в солевой и в Н+-фор ме. При хранении набухших ионообменников рекомендуется добавлять толуол. При добавлении толуола (несколько капель на 1 дм3) следует помнить, что толуол поглощает электромагнитные волны при 280 нм, а растворимость его в некоторых буферных растворах, например в трис-буфере, достаточно высока.
В целом же следует отметить, что ионообменная хроматография белков менее предпочтительна, чем гель-фильтрация.
В работе в качестве ионита рекомендуется использовать диэтиламиноэтилцеллюлозу (ДЭАЭ-целлюлозу) и (или) кар-боксиметилцеллюлозу (КМ-целлюлозу ). ДЭАЭ-целлюлоза благодаря своим функциональным группам — диэтиламиноэтиль-ным остаткам [—СН2 — СН2 — N = (С2Н5)2] в водном растворе обладает свойствами слабого анионита; используется для хроматографического фракционирования кислых и нейтральных белков. Фракционирование белков сыворотки крови можно проводить с использованием градиентного или ступенчатого элюирования. Сорбцию белков на колонке осуществляют при pH 5,65. При этом значении pH белки сыворотки крови могут сорбироваться на слабых анионитах.
Карбоксиметилцеллюлоза — слабый катионит, содержащий в качестве ионогенных групп карбоксиметильные остатки СН2 — СОО , связанные с гидроксильными группами целлюлозы эфирными связями. Используется для разделения основных и нейтральных белков.
Фракционирование белков сыворотки крови на КМ-целлюлозе рекомендуется проводить путем создания ступенчатого элюирования. Это обусловлено достаточно большими различиями в сродстве отдельных белковых фракций к иониту. Сорбция белков на НО
колонке ос\шееib.imcгея при pH -1.6 рсырюы.ш буферщ При гя ком значении pH белки сыворотки крови сорбируются на слабых катионитах.
Подготовка проб. Для подготовки рекомендуется использовать следующие способы. Для получения сыворотки 2—3 см? кротят помешают в сухую центрифужную пробирку и оставляют на 0.5—1 ч. Тонкой стеклянной палочкой осторожно обводят стенки пробирки для отделения сгустка, центрифугируют и сыворотку сливают в чистую пробирку. С целью удаления солей ее диализируют против исходного буферного раствора. Для этого сыворотку разводят два раза исходным буферным раствором, помещают в специальный мешок и диализируют против того же буфера в течение не менее 12 ч (можно оставлять на ночь); при использовании в качестве объекта мяса белки предварительно экстрагируют согласно описи методов, указанных в лабораторной работе № 3.
1 .РАЗДЕЛЕНИЕ БЕЛКОВ СЫВОРОТКИ КРОВИ НА ДЭАЭ-ЦЕЛЛЮЛОЗЕ
Материалы, реактивы и оборудование. Раствор НО молярной концентрацией 0,5 моль/дм3; раствор NaOH молярной концентрацией 0,5 моль/дм3; ДЭАЭ-целлюлоза; натрий-ацетатный буфер — растворы молярной концентрацией 0,02; 0,1 и 0,5 моль/дм3 (pH 5,6); раствор NaCl молярной концентрацией 0,3 моль/дм3, приготовленный на натрий-ацетатном буфере молярной концентрацией 0,02 моль/дм3; раствор NaCl молярной концентрацией 0,5 моль/дм3, приготовленный на натрий-ацетатном буфере молярной концентрацией 0,5 моль/дм3.
Порядок проведения анализа. Для подготовки сорбента берут около 10 г ДЭАЭ-целлюлозы, обрабатывают и переводят в ОН~-фор-му. После удаления мелких и крупных частиц ДЭАЭ-целлюлозу суспендируют дважды в избытке натрий-ацетатного буферного раствора (0,1 моль/дм3, pH 5,65), выдерживая каждый раз в буфере по 30 мин. Избыток жидкости удаляют декантацией. После второго суспендирования смесь вносят в колонку.
Рекомендуемый размер колонки 1,5x20 см. По окончании заполнения колонки ионит уплотняют либо поршнем, либо давлением воздуха до получения постоянной высоты столба геля. Затем колонку уравновешивают исходным элюирующим буферным раствором — натрий-ацетатным буфером молярной концентрацией 0,1 моль/дм3 (pH 5,6), медленно пропуская его через колонку (при данных размерах колонки 5—6 капель в 1 мин) до тех пор, пока pH выходящего из колонки раствора не будет равен исходному. Объем раствора, необходимого для уравновешивания, составляет около 8 общих объемов колонки. Затем на колонку наносят 50—100 мг белка в объеме 1—3 см3. Пробу вводят микропипеткой
111
пли микрошприцем. :пя чего отсоединяю! капельную воронку, но кран воронки нс закрывают, чтобы не прекратить ноток жидкости. Смесь разделяемых веществ подают в колонку' в гот момент, когда сдой растворителя над сорбентом опустится до верхней его границы. Затем сверху осторожно наливают растворитель на высоту 1—2 см над поверхностью сорбента и поддерживают этот уровень. Колонку промывают исходным буферным раствором до тех пор. пока оптическая плотность элюата не станет равной примерно 0.05, после чего переходят к фракционированию белков.
Скорость протекания белков через колонку не должна превышать 15—20 см3/ч. Элюат собирают порциями по 3 см3. Элюирование белков с колонки проводят буферными растворами с линейным градиентом NaCl, меняя молярную концентрацию последнего от 0 до 0,3 моль/дм3. Прибор для создания линейного градиента представлен на рис. 1.9, а. В смеситель помещают 250 см3 натрий-ацетатного буфера (0,02 моль/дм3, pH 5,6), а в сосуд 1—250 см3 этого же раствора, но содержащего раствор NaCl молярной концентрацией 0,3 моль/дм3. Наиболее прочно адсорбированные белки дополнительно элюируют раствором NaCl молярной концентрацией 0,5 моль/дм3 в ацетатном буфере (0,5 моль/дм3, pH 5,6).
Элюаты в пробирках анализируют на спектрофотометре при длине волны 280 нм. На основании данных, полученных при определении содержания белка в отдельных порциях элюата, строят профиль элюирования, определяют число белковых фракций и рассчитывают массовое содержание белка (см. лабораторную работу № 2).
2 . РАЗДЕЛЕНИЕ БЕЛКОВ СЫВОРОТКИ КРОВИ НА КМ-ЦЕЛЛЮЛОЗЕ
Материалы, реактивы и оборудование. Раствор НС1 молярной концентрацией 0,5 моль/дм3; раствор NaOH молярной концентрацией 0,5 моль/дм3; КМ-целлюлоза; натрий-ацетатный буфер — растворы молярной концентрацией 0,02 (pH 4,6), 0,05 (pH 5,2) и 0,08 моль/дм3 (pH 6,0); фосфатный буфер — раствор молярной концентрацией 0,1 моль/дм3 (pH 7,0); фосфатный буфер-раствор молярной концентрацией 0,1 моль/дм3, приготовленный на растворе поваренной соли молярной концентрацией 0,5 моль/дм3 (pH 8,3).
Порядок проведения анализа. Для подготовки сорбента берут около 10 г КМ-целлюлозы, обрабатывают и переводят в Н+-фор-му. Гранулярную или волокнистую КМ-целлюлозу рекомендуется отмывать после обработки кислотой или щелочью фильтрованием на воронке Бюхнера или центрифугированием, сферическую — декантацией в химическом стакане. В случае необходимости КМ-целлюлозу фракционируют по размеру частиц.
112
1 Io. ti o i овка и >а i I о. 1 не н 11 с колонки 'щымсром 1.5 i(l е\: описл ны в п. ]. .чанной работы. Использую г исминыи t <-с цниовыию буферный раствор — натрин-алic iа 1 шип отфыр полярной концентрацией 0.02 моль/дм-' (pH 4.6).
На колонку наносят 70—100 хи белка в объеме 1 — 3 см'. Колонку промывают исходным буферным рас, вором до гсх пор. пока оптическая плотность o.noaiа нс станет равной 0.02.
Фракционирование бе jkob сыворотки крови на КМН осуществляют с помощью ступенчатого элюирования (см. рис. 1.8). подавая на колонку последовательно следующие растворы: натрип-апетатный буфер («стартовый буфер») (0.02 моль/дм'. pH 4,6); натрий-ацетатный буфер (0.05 моль/дм'. pH 5.2): натрий-ацетатный буфер (0.08 моль/дм5, pH 6.0): фосфатный буфер (0,1 моль/дм5, pH 6,0) и фосфатный буфер (0.1 моль/дм3. pH 8,3), содержащий раствор NaCl молярной концентрацией 0,5 моль/дм'. Каждый новый раствор полаю) на колонку только после того, как полностью элюируется пик. вымываемый предыдущим раствором. Скорость тока приблизительно составляет 50 см'/ч. Элюат собирают порциями по 2—3 см ’.
Элюаты в пробирках анализируют спсктрофотомстрически при длине волны 280 нм. На основании данных, полученных при определении содержания белка в отдельных порциях элюата, строят профиль элюирования, определяют число белковых фракций и рассчитывают массовое содержание белка фотометрически (см. лабораторную работу № 2).
Полученные результаты используют для построения профиля элюирования белков и расчетов количества белка в пиках. После этого рекомендуется самостоятельно сформулировать выводы и заключение по работе, акцентируя внимание на полученных экспериментальных данных и сравнивая их с данными литературы.
Лабораторная работа № 15
АНАЛИЗ АМИНОКИСЛОТ НА АВТОМАТИЧЕСКОМ АМИНОКИСЛОТНОМ АНАЛИЗАТОРЕ
Цель работы. Приобрести практический навык определения состава аминокислот в белках мяса и мясопродуктов на автоматическом аминокислотном анализаторе.
Задачи. Изучить конструкцию прибора и принципы его работы; провести гидролиз анализируемых образцов; получить аминограммы после разделения смеси аминокислот с последующим расчетом их количества.
Объекты исследования. Мясо и мясопродукты.
Материалы, реактивы и оборудование. Соляная кислота молярной концентрацией 6 моль/дм3; буферные растворы pH 2,2; 3,28;
ИЗ
4.25. 5.28: смесь муравьиной, уксусной кис.101 и воды в сосн-nuLiicHHit 7:5: 15; аминоанааизатор: водяная баня, иснаршсль; стеклянные пинетки: стеклянные палочки: стаканы; фарфоровая чашка.
Приготовление реактивов. Буферный раствор для нанесения образна (pH 2,2). 11.6 г хлорида натрия и 0.8 см3 концентрированной соляной кислоты растворяют в 1 дм3 дистиллированной воды.
Буферные растворы для э л ю и р о в а н и я а м и -нокислот (pH 3,28; 4,25; 5.28). Количественное соотношение компонентов указано в таблице, приведенной ниже.
Состав буферных растворов -V2S ± pH
4,25 5.2S
Лимонная кислота одноводная, г 70.5 2S2.5 123,0
Соляная кислота (плотность 1190 г/см3), см3 61,6 167.5 32,5
Гидроксид натрия, г 40,0 160,0 70,0
Этанол, см3 200,0 — —
Детергент (барий 35). г 10,0 10,0 10,0
Вода, см3 До 5000 До 5000 До 5000
Методические указания. Состав аминокислот в белках различных мясных продуктов определяют методом ионообменной хроматографии на автоматическом аминокислотном анализаторе ААА-881 (Чехия) или на каком-либо другом. Для разделения аминокислот применяют хроматографическую колонку, заполненную катионообменной смолой, со ступенчатым элюированием тремя натрий-цитратными буферными растворами с различным значением pH (3,28; 4,25; 5,28).
Метод включает обязательный предварительный гидролиз белков кислотой или щелочью с целью получения свободных аминокислот. Их затем определяют с помощью ионообменной хроматографии на колонках, заполненных твердым носителем, химически соединенным с заряженными группами, которые обеспечивают электростатическое взаимодействие с исследуемым объектом.
Аминокислоты исследуемого раствора обратимо связываются ионообменником, а затем элюируются буферным раствором, вызывая десорбцию аминокислот. Для элюирования аминокислот используют буферные растворы, pH и ионная сила которых постепенно повышаются. При этих условиях аминокислоты выходят на колонки в следующем порядке: кислые, нейтральные и основные.
114
Рис. 1.31. Схема автоматического аминокислотного анализатора:
] — cncie.Ma подачи образца: 2. 2', 2" — сосуды с буферным и растворами; 3. 6—насосы; 4 — ионообменная колонка; 5- сосуд с раствором нингидрина; 7 — смеситель; <S’ — капилляр; 9 — спектрофотометр; 10 — самописец
Принципиальная схема аминокислотного анализатора представлена на рис. 1.31.
Выходящий из колонки элюат поступает в смеситель, содержащий нингидрин, который подается насосом. Для развития цветности в результате качественной реакции смесь проходит через специальный капилляр, погруженный на водяную баню температурой 100 °C. Окрашенный раствор пропускают через спектрофотометр с длиной волны 570 нм для измерения интенсивности окраски, которая фиксируется самописцем в виде пиков. По расположению пиков судят о наличии индивидуальных аминокислот в гидролизате, а по площади пиков — об их содержании. Пример хроматограммы аминокислот (аминограммы) приведен на рис. 1.32. Для расчетов необходимо предварительно получить стандартную аминограмму.
Рис. 1.32. Хроматограмма аминокислот
115
Подютовка проб. При oi i pc.ic.i с я и 11 суммарных ампнокпс.ин (свободных и связанных) проводя! пиролиз OC/IKOB !1 пептидов, содержащихся в мясе. Для этого образен измельченного мяса массой 150—250 мг (белка должно быть не менее 25—50 мг) помешаю! в стеклянные ампулы или термостойкие пробирки с пришлифованными пробками и добавляю! 25 см' раствора соляной кислоты молярной концентрацией 6 моль/дм3. Затем ампулы запаивают (или плотно закрывают пробирки пробками) и выдерживают термостате при 114—115 С is течение суток. При этом необходимо помнить, что при кислотном гидролизе триптофан разрушается полностью, серин и треонин — на 5 — 10%, цистин, цистеин, метионин — частично. Перед гидролизом образцы следует пометить. Приготовленные пробы устанавливают в термостат и оставляют под наблюдение и контроль лаборанта до следующего занятия.
На втором этапе но окончании гидролиза образец фильтруют через стеклянный фильтр, испаряют избыток соляной кислоты. Для этого 5 см3 гидролизата помещают в роторный испаритель и упаривают досуха при температуре 40 °C. После этого в сухой остаток приливают 1,5 см3 дистиллированной воды и снова упаривают, повторяя операцию дважды. Освобожденный от соляной кислоты остаток растворяют в 10 см3 буферного раствора (pH 2,2).
При определении только свободных аминокислот предварительно получают безбелковые экстракты тканей или продуктов.
Экстракт мышечной ткани получают в соответствии с описанием, указанным в лабораторной работе № 2. После упаривания на роторном испарителе сухой остаток растворяют в смеси НСООН-СН3СООН-Н2О в соотношении 7:5:13с таким расчетом, чтобы 1 см3 раствора соответствовал 1,5—2,0 г ткани.
При определении аминокислот в жидких материалах, например в крови убойных животных и ее фракциях, к 1—2 см3 образца добавляют равный объем раствора СН3СООН молярной концентрацией 0,04 моль/дм3, нагревают на водяной бане, доводят до кипения и кипятят 2—3 мин, охлаждают и осадок белка отделяют центрифугированием. Центрифугат сливают и упаривают досуха. Сухой остаток растворяют в 0,1—0,2 см3 смеси муравьиной, уксусной кислот и воды (7:5:13), нерастворившийся осадок отбрасывают.
Порядок проведения анализа. Раствор анализируемого материала объемом 0,5 см3 вручную или с помощью насоса вводят в колонку и запускают систему. Продолжительность собственно анализа 5—6 ч (время регламентируется конструкционными особенностями аминокислотного анализатора). Строят аминограмму.
Построение стандартной аминограммы. Перед хроматографическим разделением опытных образцов для получения стандартной хроматограммы проводят анализ стандартной смеси аминокислот. Причем смесь аминокислот готовят так, чтобы молярная концентрация каждой аминокислоты была равна 1 ммоль/дм3. Для этого 116
навеску каждый аминоквелоi ы (\п) бер\ i в кыдичес i не. численно равном се молекулярной массе. Кроме юю. бср\ г coo i ветствую-шую навеску хлорила аммония лля получения сiакларi но, о пика аммиака, который образуется в гидролизате в результате распада амидов, содержащихся в белках.
Приготовление стандартного раствора аминокислот рекомендуется проводить в соответствии с данными таблицы:
Огностельная ; Массовая доля Содержание в ! 1 см расIвор;), mi
Аминокискна молекулярная 1 a so'i а. с< - - 1
масел \ аминокпело! ы ! азом I
Лизин 146.13 19.17 0.14613 0.02801
Гистидин 155.19 27.10 0.15519 0.04206
Аргинин 174.24 32.18 0.17424 0.05604
Аспарагиновая кис- 133.12 10,53 0.13312 0.01402
лота
Треонин 119,12 1 1.75 0.11912 0.01400
Серин 105.09 13.33 0,10509 0.01401
Глутаминовая кис- 147.12 9.52 0.14712 0.01401
лота
Пролин 115.12 12,17 0.11512 0.01401
Глицин 75.05 18.67 0.07505 0.01401
Аланин 89.12 15,73 0,08912 0.01402
Цистеин 121.15 11.56 0.12115 0.01389
Валин 117,09 11.96 0.11709 0.01401
Метионин 149,20 9.39 0.14920 0,01401
Изолейцин 131,20 10.60 0.13120 0.01401
Лейцин 131,20 10.60 0.13120 0,01401
Тирозин 181.19 7,74 0,18119 0.01403
Фенилаланин 165.19 8.48 0,16519 0.01403
Хлорид аммония 53.49 26,16 0.53449 0,01401
Изучают полученную аминограмму. Измеряют площади пиков с помощью потенциометра или вычисляют путем умножения высоты пика на его ширину, взятую на половине его высоты.
Массовую долю аминокислот рассчитывают по формуле
X=(SnM-5Q- 10-?)/(5>),
(1.24)
где 5П — площадь пика соответствующей аминокислоты на полученной аминограмме, см2; М — молекулярная масса аминокислоты. Да; 50 — объем раствора, полученный после кислотного гидролиза, см3; 10”3—концентрация аминокислоты в стандартном растворе, моль/дм3; 5СТ —площадь пика стандартного раствора аминокислоты, см , т — масса навески образца, г.
117
Содержание аминокислот в мышечной ткани выражают в микромолях. миля и грам м - процент ах на влажную массу ткани или в процентах к небелковому или общему азоту, в сыворотке крови — в микромолях, миллиграмм-процентах или в процентах к небелковому азоту.
Полученные экспериментальные и расчетные данные сводят в таблицу, форма которой приведена ниже.
Наименование и характеристика образна
Содержание аминокисло! в образце
мкмоль/дм!
Сравнив данные с действующими научно обоснованными нормами питания, полезно проанализировать потенциальные возможности сырья или продуктов для покрытия суточной потребности в незаменимых аминокислотах, дать рекомендации по рациональному их использованию.
Лабораторная работа №16
АНАЛИЗ АМИНОКИСЛОТ МЕТОДОМ БУМАЖНОЙ ХРОМАТОГРАФИИ
Цель работы. Приобрести практический навык анализа аминокислот мяса и мясопродуктов методами бумажной хроматографии.
Задачи. Подготовить хроматографические камеры и пробы для хроматографирования, проявить бумагу и идентифицировать аминокислоты.
Объекты исследования. Мясо, мясопродукты, кровь и ее фракции.
Материалы, реактивы и оборудование. Раствор НСЮ4 молярной концентрацией 0,5 моль/дм3; раствор КОН молярной концентрацией 5 моль/дм3; раствор нингидрина в ацетоне массовой долей 0,2 %; стандартные растворы аминокислот молярной концентрацией 0,025 моль/дм, этанол (на 50 см3 этанола добавляют 0,02 см3 насыщенного раствора CuSO4); раствор изопропанола массовой долей 10 %; диазореактив; хроматографические камеры; бумага; пульверизатор; ножницы; фотоэлектроколориметр или спектрофотометр.
Приготовление реактивов. Бутанолуксусная смесь. Смесь нормального бутанола, уксусной кислоты и воды в соотношении 4:1:5 (верхний слой) и 8:3:1 (нижний слой) тщательно встряхивают в делительной воронке и после расслаивания используют верхний слой.
118
Д и а ’> о р е а к г и в: смешиваю! о ши обьсм рас i вора сульфаниловой кислоты (к 0.9 г кислоты прибавим-. 9 см' концентрированной НС1 и 90 см' 1LO) и пять объемов свежеприготовленного раствора NaNO. массовой долей 5 сс. объем доводят водой до 50 см3.
Методические указания. Метод основан на распределении соединения между двумя жидкими фазами. Хроматографическая бумага обладает свойством поглощать воду из атмосферы и задерживать ее между своими целлюлозными волокнами. Эту воду можно рассматривать как один из растворителей (как неподвижную фазу). Когда по бумаге под действием капиллярных сил движется неводный растворитель (подвижная фаза), молекулы вещества, нанесенного на бумагу, распределяются между двумя фазами в соответствии с их коэффициентом распределения. Чем выше растворимость вещества в подвижной фазе, тем дальше оно продвинется по бумаге вместе с растворителем, и наоборот.
Как правило, при бумажной хроматографии неподвижная фаза является водной, а в качестве подвижной используются органические растворители.
Метод хроматографического разделения на бумаге уступает колоночным методам по ряду показателей, однако имеет в ряде случаев определенное преимущество.
Определение свободных аминокислот сводится к хроматографическому анализу безбелковых фильтратов на бумаге и определению аминокислот после обработки хроматограмм нингидрином.
В присутствии нингидрина отдельные аминокислоты проявляются в виде пятен, окрашенных в голубой, фиолетовый, желтый или другой цвет (в зависимости от химической структуры аминокислоты).
Идентификацию аминокислот в смеси проводят путем сопоставления распределения известных аминокислот (стандартов).
Подготовка проб. 1. Суммарные аминокислоты получают путем предварительного гидролиза (см. лабораторную работу № 15).
2. Свободные аминокислоты извлекают из образцов раствором спирта (75—80 об.%), нагретого до температуры 60—70 °C на водяной бане. Полученную смесь тщательно растирают в течение 15 мин и фильтруют через вставленный в воронку складчатый фильтр в чашку для выпаривания, которую используют в качестве приемника.
Порядок проведения анализа. Для анализа используют хроматографическую бумагу ватман № 1, FN № 3 (можно № 2) и др. Вырезают полосы длиной около 50 см и шириной 18—50 см (размер бумаги определяется размером исследуемых проб и величиной хроматографической камеры). В 5—10 см от конца полоски (зависит от высоты лодочки) простым карандашом проводят стартовую линию и отмечают точки нанесения раствора в 3 см от краев хроматограммы и в 2,5 см одна от другой. Для лучшего разделения об-
119
1.33 Форма Фмажнои по.юсы. обеспечивающая .пч шее |чн имение аминокис.101 (размеры даны в мм):
разной с большим набором аминокислот край бумаги, на который наносят гидролизат. вырезают. как это показано на рис. 1.33.
Чашку с экстрактом помешают на водяную баню при температуре кипения и полностью выпаривают спирт. Полученный сухой остаток растворяю! в растворе изопропанола массовой долей 10 % с таким расчетом, чтобы 1 см3 раствора соответствовал 1,5—2 г ткани. Раствор наносят на .хроматограмму в количестве, эквивалентном 30—50 мг ткани. На ту же хроматограмму рядом с исследуемым раствором наносят растворы известных аминокислот — «свидетелей». Стандартные растворы глутаминовой кислоты и аланина (или любых других аминоотдельные точки (обычно 3—5 точек) в строго
в
кислот) наносят определенном количестве. По этим точкам строят стандартные калибровочные графики для количественного определения аминокислот в экстракте ткани.
Построение калибровочного графика. Для каждой аминокислоты строят свой калибровочный график. В ряд точек хроматограммы наносят стандартные растворы аминокислот в количестве 0,05; 0,1; 0,15 и 0,2 мкмоль каждой. В качестве стандартных растворов удобно пользоваться смесями аминокислот известного состава, хорошо разделяющимися в данных условиях и содержащими определенные количества аминокислот. Стандартные смеси готовят из стандартных растворов отдельных аминокислот молярной концентрацией 0,025 моль/дм3, приготовленных на растворе изопропилового спирта массовой долей 10%, смешиванием по 1 см3 (0,5 см3) раствора каждой аминокислоты. Стандартные растворы аминокислот наносят на ту же хроматограмму, что и опытные пробы. Если размеры хроматограммы не позволяют этого, полоски бумаги, используемые для хроматографии стандартных и опытных растворов, вырезают из одного и того же листа хроматографической бумаги.
После проявления хроматограммы окрашенные участки обводят простым карандашом, вырезают, измельчают ножницами и помещают в пробирки. Окрашенный продукт элюируют 5 см3 перегнанного метанола (предварительно к 500 см3 метанола добавляют 0,2 см3 насыщенного раствора нитрата меди) или 5 см3 этилового спирта (в этом случае вместо нитрата меди добавляют суль
120
фат мс/нп. Пробы до колориметрирования необходимо не риодически встряхивать и держать в темноте. Элюаты начинаю! колориметрировать, котла в раствор перейдет краска из наиболее интенсивно окрашенных пятен (обычно через 30—60 мин). Определение проводят на фотоэлектроколориметре с зеленым светофильтром (500 нм) в кювете толщиной 0.5—1.0 см. Для контроля вырезают чистый участок хроматограммы на том же уровне, что и проявленные пятна, и обрабатывают его так же. как и опытные пробы.
Калибровочный график строят, откладывая на оси абсцисс концентрацию аминокислоты, а на оси ординат — величины оптической плотности.
Исследуемый раствор наносят микропипеткой с оттянутым в капилляр и изогнутым концом вместимостью 0,1 см3, полуавтоматической пипеткой или калиброванным капилляром. Диаметр наносимых пятен не должен превышать 0,3—0,5 см (объем каждой капли около 0,003—0,005 см3). При нанесении раствора в одну и ту же точку бумаги соответствующее место каждый раз подсушивают током теплого воздуха. Если расстояние между точками нанесения превышает 2,5 см, раствор можно наносить в виде полосы.
Для идентификации аминокислот в исследуемом гидролизате наряду с опытными пробами (или предварительно, до анализа опытных проб) на хроматограмму наносят растворы аминокислот — «свидетелей». После установления местоположения пятен отдельных аминокислот — «свидетелей» в дальнейшем последние можно наносить на хроматограмму в виде смесей. Смеси должны иметь известный состав и хорошо разделяться при данных условиях хроматографирования. Этим требованиям удовлетворяют смеси аминокислот следующего состава:
1. Цистеиновая кислота, гистидин, аспарагиновая кислота, глицин, глутаминовая кислота, аланин, триптофан, метионин, фенилаланин, лейцин.
2. Цистеиновая кислота, лизин, аргинин, серин, треонин, аланин, пролин,тирозин, валин, изолейцин.
Смеси «свидетелей» помещают в крайние точки стартовой линии хроматограмм по 0,05—0,1 мкмоль каждой аминокислоты в пятно.
В качестве растворителей для хроматографии аминокислот обычно используют последовательно две системы: д-бутанол — уксусная кислота — вода в соотношении 4:1:5 (верхний слой) и д-бутанол — уксусная кислота — вода в соотношении 8:3:1.
Для лучшего разделения сложной смеси аминокислот, имеющих близкие величины Rf, каждый растворитель пропускают по хроматограмме несколько раз (обычно дважды). В этом случае фронт каждого последующего растворителя должен продвигать
121
ся несколько ъгшшс. чин нрелылушсго. Перед использованием кажло! о следующею раствори геля хрома i oi раммы вынимаю! из камеры и сушат на воздухе.
Хрома I or рам му проявляю! раствором нингидрина массовой долей 0.2 Сс. приготовленным на nepci нанном ацетоне. К раствору нингидрина прибавляют 4 см? воды и I см?ледяной уксусной кислоты. Этот раствор готовят непосредственно перед проявлением. Раствор наливают в ванночку и равномерно пропитывают им хроматограмм}'. Затем ее сушат под тягой и выдерживают в темноте при комнатной температуре около 12 ч. В этих условиях с нин-тдрином реагируют все сх-аминокислоты (все другие аминокислоты дают реакцию при более высокой температуре — около 100 °C). Проявление хроматограмм при качественной хроматографии можно ускорить, помещая последние на 10—15 мин в термостат при 60 °C. Окраска, полученная при проявлении аминокислот нингидрином, неустойчивая. Ее можно закрепить, опрыскивая проявленную хроматограмму из пульверизатора раствором ацетона, содержащим нитрат меди. К 100 см3 ацетона прибавляют 1 см3 насыщенного раствора Cu(NO3X и 0,02 см3 концентрированной азотной кислоты.
Сопоставляя положение аминокислот гидролизата с положением аминокислот — «свидетелей», определяют аминокислотный состав гидролизата.
Наряду с нингидриновым реактивом существуют и другие реактивы, дающие цветные продукты с некоторыми аминокислотами. Это свойство избирательного окрашивания часто используют в хроматографии для идентификации отдельных аминокислот. Так, для определения соединений с гуанидиновой группой (аргинин) используют реакцию Сакагуши. Хроматограмму смачивают раствором 8-оксихинолина в ацетоне с массовой долей 0,1 % и после ее подсушивания на воздухе слегка опрыскивают из пульверизатора раствором гипобромита (1см3 брома в 500 см3 раствора NaOH молярной концентрацией 0,5 моль/дм3). Наблюдается оранжевое окрашивание.
Реакция Эрлиха. Производные индола и оксииндола (триптофан, 5-окситриптофан, 5-окситриптамин, 5-оксииндолуксусная кислота) с реактивом Эрлиха дают лиловую окраску. Хроматограмму погружают в раствор, содержащий диметиламинобензальдегид массовой долей 10 % в концентрированной НО с четырьмя объемами ацетона. Окраска появляется через 20 мин при комнатной температуре.
Реакция Паули. Соединения, содержащие имидазольное кольцо (гистидин, метилгистидин, гистамин, карнозин), а также тирозин дают красно-оранжевое окрашивание после обработки диазореактивом. Хроматограмму из пульверизатора слегка опрыскивают сначала диазореактивом, а затем раствором Na2CO3 массовой долей 10 %. Окраска устойчивая.
122
I 1еречислснные реакции можно примеичи. nocic нроявленю-; хроматограммы нингидрином.
Для обнаружения материала, имеющего пептидные связи, id не лающего нингидриновую реакцию (например, циклические пентилы), применяют реакцию Райдона и Смита. С этой целью .хорошо высушенную хроматограмму смачивают в смеси спирта и эфира (1 : I). Избыток растворителя удаляют фильтровальной бумагой. Влажную хроматограмму помешают в замкнутое пространство над свежеприготовленной смесью раствора НО массовой долей 10% и раствором КМпО4 массовой долей 1 % (в соотношении 1 : 1) на 5—15 мин. Затем хроматограмму погружают в смесь раствора KJ молярной концентрацией 0.05 моль/дм3 и насыщенного раствора о-толуидина (или бензидина) в уксусной кислоте массовой долей 2 % (1 : 1). На месте пептидов и белка появляются синие пятна. Через 1—3 мин хроматограмму промывают раствором СН.СООН массовой долей 2 % и высушивают на воздухе. Окраска устойчивая. Для пептидов эта реакция более чувствительна, чем нингидриновая. Реакцию дают также пуриновые и пиримиди новые основания, нуклеозиды, нуклеотиды и др.
Количественное содержание аминокислот рассчитывают по калибровочным графикам, построенным после хроматографирования стандартных растворов аминокислот, взятых в различных количествах.
Содержание аминокислот в мышцах рассчитывают в микромолях, миллиграмм-процентах на массу ткани или процентах к небелковому или общему азоту.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы. Самостоятельно анализируют их и формулируют выводы по работе.
Наименование и характс-ристика образца _____
Качественный состав аминокислот
Количество аминокислот
Лабораторная работа №17
ОПРЕДЕЛЕНИЕ АМИНОКИСЛОТ МЕТОДОМ ТОНКОСЛОЙНОЙ ИОНООБМЕННОЙ ХРОМАТОГРАФИИ НА ПЛАСТИНАХ
Цель работы. Приобрести практический навык количественного определения аминокислот методом тонкослойной ионообменной хроматографии на пластинах.
Объекты исследования. Мясо и мясопродукты.
Материалы, реактивы и оборудование. Хроматографические пластины, уравновешивающий буферный раствор (pH 4,1); разде-
123
ляюнше оуфсрныс расiворы (pH 3.5: 5.23; 6.0); кадмин-ннщиа-риновый прояви 1ель: хрома ioi рафическая камера; пульверизатор; микропицстки: сушильный шкаф.
Приготовление реактивов. У’ р а в н о в с ш и в а ю ш и it б у -ф е р н ы й р а с т в о р м о л я р н о й к о н ц е и т р а п и е и 0.02 моль/лм?. 1,4 г кристаллической лимонной кислоты. 0.8 i NaOH. 1.2 см3 концентрированной НС1 и 70.0 см3 этанола растворяют в воде и доводят объем до 1 дм? (pH 4.1).
Разделяю щи й буфе р и ы й р а с т в о р м о л я р-ной концентра ц и е й 0.4 моль/дм3. 84,0 г кристаллической лимонной кислоты, 16,0 г NaOH и 4,0 см3 концентрированной НС1 растворяют в воде и доводят объем до 1 дм3 (pH 3,5).
Р а з д е л я ю щ и й б у ф е рн ый раствор для основных и а р о м атических аминокислот м о -л я р н о й концентра ц пей 0,3 моль/дм3. 24,6 г лимонной кислоты, 14 г NaOH и 6,5 см3 концентрированной НО растворяют в 1 дм3 Н2О (pH 5,23).
Р а з д’е л я ю щ и й б у ф е р н ы й раствор для обнаружения триптофана молярной концентрацией 1,5 моль/дм3. 7,0 г лимонной кислоты, 4,0 г NaOH. 81,9 NaCl, 100 см3 метилцеллозольва растворяют в воде и доводят объем до 1 дм3 (pH 6,0).
К а д м и й-н и н г и д р и н о в ы й проявитель. Раствор А; 1 г перекристаллизованного нингидрина растворяют в смеси ацетона (100 см3) и ледяной уксусной кислоты СН.СООН (10 см3).
Раствор В: 1 г ацетата кадмия растворяют в 100 см3 раствора сн.соон массовой долей 50 %.
Йеред проявлением смешивают 100 см3 раствора А и 20 см3 раствора В.
Методические указания. При хроматографии аминокислот в тонком слое в качестве ионообменника обычно используют сильнокислый сульфополистирольный катионит «Дауэкс 50 х 8» или близкие к нему типы смол в №+-форме. Подготовленную к работе смолу наносят тонким слоем на инертную подложку (тонкослойный вариант), а затем на поверхность слоя ионообменника на пластинке наносят анализируемую смесь аминокислот в буферном растворе с низкой ионной силой и pH не выше 3,0, при котором все протеиногенные аминокислоты заряжены положительно. При нанесении аминокислот в форме катионов они связываются с сулъфополистиролом, замещая часть ионов натрия. Прочность удержания аминокислот на ионообменике зависит от их основности: наиболее прочно связываются лизин, гистидин, аргинин, наименее прочно — аспарагиновая и глутаминовая кислоты. После нанесения смеси пропускают элюирующий буферный раствор через тонкий слой смолы. Для элюирования используют цитратный буфер с более высокой ионной силой и pH выше 3,0. В этих условиях происходят постепенная нейтрализация положительного
124
заряда и ослаолснис ионных взаимолсисгвии: кроме того. \вели ченпс концентрации попов натрия в элюир\тотем растворе гакжс приволпт к вытеснению катионов аминокислот из связей с ионо-обменником. Таким образом, в процессе ионообменной хроматографии аминокислоты сразу же разделяются на три большие группы: наиболее быстро через слои смолы движутся кислые аминокислоты. затем нейтральные и наиболее медленно — основные По мере протекания через ионообменник элюирующего раствора происходит постепенное разделение нейтральных аминокислот-, поскольку на прочность взаимодействия с катионитом оказывают влияние не только ионные, но и гидрофобные взаимодействия. Полистирольная (гидрофобная) матрица ионообменника достаточно избирательно взаимодействует с радикалами нейтральных аминокислот, поэтому все они, несмотря на близость величин заряда их молекул, разделяются в процессе хроматографии. Из всех нейтральных аминокислот наиболее прочно сорбируются на матрице те, которые содержат бензольные кольца, т. е. тирозин и фенилаланин. По окончании процесса хроматографии аминокислоты выявляют с помощью нингидринового проявителя.
Для тонкослойной ионообменной хроматографии аминокислот используют пластинки «Фиксион 50x8» (Венгрия), в которых слой катионита «Дауэкс 50x8» толщиной 0.25 мм закреплен на пластмассовой подложке. Хроматография на пластинках «Фиксион 50x8» позволяет сочетать принцип ионного обмена с преимуществами тонкослойного варианта и отличается высокой чувствительностью, быстротой разделения и простотой процедуры. Она незаменима при сравнительных анализах следовании свободных аминокислот, изучении первичной . структуры пептидов.
Пластинки проявляют в а темноте при комнатной темпе- W ратуре или в термостатах при 105 °C в течение 10 мин.
На пластинках «Фиксион 50 х 8» при однократном пропускании растворителя может быть разделено 14—16 амино-кислот, входящих в состав бел-ка (рис. 1.34, а). В случае недо- 9 статочно хорошего разделения
белковых гидролизатов, ис-
Асп
Тре+сер
Тли
Цис Гли Ала
(Про)
Вал
4
♦
Рис.1.34. Разделение стандартной смеси аминокислот на пластинках «Фиксион 50x8»: а — одномерное; б — димерное
Мет
Иле Лей
Тир Фен
Гис Лиз Арг
б
а
125
смеси аминокисло! одномерным способом прибегают к димерной хроматографии, пропуская тот же иди другой растворитель в направлении, перпендикулярном предыдущему. На практике поступают следующим образом. В две точки стартовой линии хроматограммы помешают исследуемый раствор. После одно
кратного пропускания растворителя пластинку сушат, отрезают от нее часть, соответствующую крайней стартовой точке хроматограммы, и проявляют нингидрином. В случае плохого разделения оставшуюся большую часть пластинки поворачивают на 90° и снова помещают в тот же самый или другой растворитель. Проводят повторное хроматографирование во втором направлении. После высушивания пластинку также проявляют нингидрином (рис. 1.34, б).
Для оценки количественного содержания аминокислот в исследуемом образце белка наряду с гидролизатом в отдельные точ-
ки хроматограммы наносят растворы аминокислот — «свидетелей». Стандартные растворы аминокислот наносят в несколько (4—6) точек хроматограммы из расчета 0,01—0.06 мкмоль в пятно, триптофан — до 0,1 мкмоль.
pH образца должен быть ниже 3,0 (обычно его доводят до 2,0). Часто образец наносят на пластинку в натрий-цитратном или фосфатно-цитратном буфере pH 2,2. Для этого гидролизат или экстракт белка (= 1 мг) растворяют в 1 см3 смеси, состоящей из двух частей этанола 96 об.% и 1 части фосфатно-цитратного буфера (pH 2,2).
Подготовка проб. Пробы мяса и мясопродуктов готовят в соответствии с рекомендациями по подготовке проб, описанными в лабораторной работе № 15.
Для обнаружения триптофана предварительно проводят щелочной гидролиз белка. Гидролизат разбавляют водой и подкисляют концентрированной НС1 до pH 5,0.
Порядок проведения анализа. Ионообменную смолу пластинок перед употреблением уравновешивают соответствующим буфером. Для этого пластинку накладывают обратной стороной на стекло того же размера. На верхний край пластинки с помощью
стеклянных палочек (или резинового кольца) прикрепляют 4 слоя фильтровальной бумаги, сложенной так, чтобы короткая часть (1,5—2 см) прикрывала слой смолы, а длинная была загнута на обратную сторону, т. е. к стеклу (рис. 1.35). Пластинку
Рис. 1.35. Уравновешивание пластинки «Фиксион 50 х 8» буферным раствором:
/ — пластинка; 2—стекло; 3 — фильтровальная бумага; 4 — стеклянная палочка (или резиновое кольцо)
126
вместе со стеклом пометают в камеру для восходящей хроматографии так. чтобы нижний край пластинки был погружен в буферный раствор на 1 — 1.5 см. Пластинка должна быть расположена в камере под углом к поверхности буфера.
Уравновешивание пластинок проводят в закрытой камере при комнатной температуре в течение 15—18 ч буферным раствором pH 3,28 молярной концентрацией 0,02 моль/дм3 следующего состава:
Лимонная кис.юта 1.4 г
NaOH 0.8 г
НС1 (37%-ная) 1.2 см3
Вода До 1 дм’
Все буферные растворы следует готовить на деионизированной воде.
По окончании уравновешивания пластинки вынимают из камеры, снимают фильтровальную бумагу и сушат их на воздухе. Пластинки можно хранить неограниченно долгое время.
Для разделения аминокислот исследуемый образец наносят на пластинку капилляром (следует избегать повреждения слоя смолы) на стартовую линию, расположенную в 2—3 см от нижнего края в виде пятна или полосы. В случае необходимости на пластинку наряду с опытной пробой наносят стандартные растворы аминокислот в том же буфере в количестве 0,01 — 0,03 мкмоль каждой аминокислоты на пятно. Пластинку с нанесенным образцом помещают в хроматографическую камеру, на дно которой слоем 1 см наливают буферный натрий-цитратный раствор (pH 3,28).
Разделение аминокислот проводят в закрытой камере, насыщенной парами растворителя, восходящим способом при температуре 50 °C. После окончания хроматографии (60—90 мин) растворитель должен подняться по пластинке примерно на 18 см. Пластинку вынимают из камеры, сушат на воздухе и проявляют раствором нингидрина. Для проявления пластинку осторожно опрыскивают из пульверизатора раствором нингидрина в ацетоне массовой долей 0,1 %. Для получения стойкой окраски хроматограмму можно проявлять нингидриновым реактивом, содержащим кадмий.
Идентификацию аминокислот в исследуемых образцах проводят, руководствуясь порядком расположения на хроматограмме стандартных аминокислот. Порядок расположения аминокислот при одномерной хроматографии на пластинках «Фиксион 50 х 8» в направлении от старта к фронту: аргинин, лизин, гистидин, фенилаланин, тирозин, лейцин, изолейцин, метионин, валин, пролин (пятно желтого цвета), аланин, глицин, цистеин, глутаминовая кислота, треонин + серин, аспарагиновая кислота.
127
\рОМ<1 Н Н рЯММЫ ШрИСОВЫ BJ1O Г. О1МСЧЛЯ расположение ВСС\ !•; > 4 В В.'! 3 И I! Ы X ЛМИНОКИСЛО Г. И Bl f >\ал в н о с ра В Н 11 ва К) I 1ПЛро.!П!а : ь; pa зличных образцов мяса и мясопродуктов по содержанию аминокисло г.
Для количественного определения аминокислот после проявления нингидрином полосу .хроматограммы, cootbciствуюшую каждой из аминокислот (is кислой среде растворителей лучше других разделяются пятна лизина, аргинина и валина), вырезают и сканируют на денситометре при 510 нм. Результаты сканирования оценивают либо по площадям полученных пиков, которые рассчитывают путем умножения высоты пика на его ширину на уровне половины высоты, либо по массам пиков. Для этого пики переводят на кальку. Пики на кальке вырезают и взвешивают. Для определения количества аминокислот строят калибровочный график зависимости площади пика (или его массы) от содержания аминокислоты (мкмоль) для стандартных растворов аминокислот.
Построение калибровочного графика. Для построения калибровочного графика используют стандартный раствор аминокислот, который готовят в соответствии с рекомендациями, приведенными выше (см. лабораторную работу № 15).
Результаты экспериментов и расчетов сводят в таблицу. Самостоятельно формулируют выводы и заключение по работе.
Наименование и характеристика образца
Перечень идентифицирован н ых а м и и о кислот
Сол е р жа н и е а м и н о к и ел оты
в образце, мг/г
Лабораторная работа № 18
ОПРЕДЕЛЕНИЕ ЭКСТРАКТИВНЫХ ВЕЩЕСТВ
Цель работы. Освоить различные методы количественного определения некоторых специфических азотсодержащих экстрактивных веществ мышечной ткани убойных животных (гистидин-содержаших дипептидов — карнозина и ансерина).
Задачи. Получить безбелковый экстракт пептидов из мышечной ткани и определить количество гистидинсодержащих дипептидов на основе фотометрических и хроматографических методов.
Объекты исследования. Образцы мышечной ткани различных видов сельскохозяйственных животных и птицы.
Методические указания. Карнозин и ансерин хорошо растворимы в воде, осаждаются этанолом, обладают буферным действием, в растворе дают щелочную реакцию, оптически активны. Карнозин дает цветную реакцию с диазореактивом.
Гистидинсодержащие дипептиды определяют в безбелковом мышечном экстракте по цветной реакции с нингидрином после
128
разделения их методом хроматографии на бумаге иди ионообменной хроматографии. Карнозин, кроме юго. может быть определен непосредственно в безбелковом мышечном экстракте или на бумажной хроматограмме по цветной реакции Паули с диазотированной сульфаниловой кислотой. Часто колориметрическому определению дипептидов предшествует выделение их из безбелково-го мышечного экстракта в виде ртутного осадка.
Метод характеризуется более высокой чувствительностью по сравнению с хроматографией на бумаге и может быть использован также для микроопределения дипептидов после их модификации с помощью флуоресцентной метки — 7-хлор-4-нитро-2-окси-1,3-ди-азолия (НБД-хлорида). НБД-хлорид ковалентно связывается с аминогруппами аминокислот и пептидов и образует производное желтого цвета. Реакция протекает по следующей схеме:
Все вышеперечисленные методы применяются для анализа экстрактивных веществ и могут быть использованы самостоятельно или в сочетании в зависимости от цели и технического оснащения лаборатории.
1. ОПРЕДЕЛЕНИЕ КАРНОЗИНА В БЕЗБЕЛКОВОМ ЭКСТРАКТЕ ПО ДИАЗОРЕАКЦИИ
Материалы, реактивы и оборудование. Раствор сульфаниловой кислоты; свежеприготовленный раствор NaNO2 массовой долей 5 %; раствор диазотированной сульфаниловой кислоты; карбонат натрия безводный и его раствор массовой долей 10 %; карнозин — стандартный раствор 0,5 мг/см3; пипетки; пробирки; стеклянные стаканы.
Приготовление реактивов. Раствор сульфаниловой кислоты. 0,9г сульфаниловой кислоты растворяют в 9см3 концентрированной НС1 и доводят объем дистиллированной водой до 100 см3.
Раствор диазотированной сульфаниловой кислоты. В мерную колбу вместимостью 100 см3, стоящую во льду, помещают 6 см3 раствора сульфаниловой кислоты, приливают 6 см3 свежеприготовленного раствора NaNO2 массовой до-
129
леи 5 ч. перемешиваю! и оставляют на 5 мин. Прибавляю! при перемешивании еще 24 см ’ раствора \a.\O массовой долей 5 с< и через 5 мин доводят объем водой до 100 см'. Каждый раз !огов>н свежий раствор (на льду хранится в шчение 5—6 ч).
Подготовка проб. При определении пептидов но диазореакиии предварительно готовя т безбелковые экстракты.
Экстракты мышечной ткани освобождают оз белков хлорной кислотой или этанолом. Во втором случае безбелковый мышечный экстракт не содержит солей.
При осаждении хлорной кислотой навеску мышечной ткани массой 3—5 г освобождают от жира, соединительной ткани и измельчают ножницами на холоде. К кашице добавляют 5 объемов охлажденного раствора НС1О4 молярной концентрацией 0.5 моль/дм3, гомогенизируют и тщательно растирают в ступке. Суспензию центрифугируют it течение 20 мин, надосадочную жидкость собирают, осадок вновь суспензируют в 1 — 1,5 объема хлорной кислоты. Промывные воды соединяют с центрифу-гатом. Хлорную кислоту удаляют в виде перхлората калия. Полученный центрифугат упаривают на роторном испарителе при температуре 40—45 °C.
При осаждении белков этанолом навеску мышечной ткани массой 2—3 г, измельченную ножницами на холоде, переносят в гомогенизатор с 5 объемами охлажденного этанола 96 об.%; гомогенат сливают зз центрифужный стакан, оставляют при комнатной температуре на 20 мин, затем центрифугируют в течение 10 мин при частоте вращения 3000 с -1. Надосадочную жидкость сливают в колбу с узким горлом (при обработке 2—3 г мышц объем колбы 200 см3, при получении экстракта из большой навески можно пользоваться делительной воронкой), осадок трижды промывают 15 см3 раствора этанола 80 об.%, сливая центрифугат в колбу. К спиртовому экстракту приливают хлороформ (так, чтобы верхняя граница жидкости была в узкой части колбы), встряхивают в течение 15 мин и оставляют для отстаивания. Когда четко обозначится граница между водным и спиртохлороформным слоями, водный слой отсасывают шприцем, а в колбу доливают воду (до прежнего уровня), встряхивают и оставляют для расслаивания. Водный слой отсасывают и присоединяют к первому. Эту операцию повторяют еще два раза. Объединенный водный раствор упаривают на роторном испарителе.
Порядок проведения анализа. Безбелковый экстракт после упаривания на роторном испарителе растворяют в 1—2 см3 воды. Отбирают различные объемы этого раствора, доводят водой до 2 см3, приливают 3 см3 диазореактива, тщательно перемешивают и оставляют на 5 мин. Затем добавляют 3 см3 раствора карбоната натрия массовой долей 10 %, перемешивают и через 10 мин колориметрируют при длине волны 490 нм. Следует убедиться в том, что интенсивность образующейся окраски пропорциональна количеству исследуемого раствора, взятого для определения.
130
Г одержанне карноита в обьск но, июле, н >в?.цця ia j ра ж a 101 в микромолях п ш \iii । шграмм-процсн га\ на ! г 'i канн и опрсдсля-юге помощью калибровочною i рафика.
Построение калибровочного графика. Ioiobmi оанааршыи раствор. содержащий or U.05 до 0.5 мкмоль карношна toi 10 до 100 мк1) в пробе. Производят окрашивание но описанию соогвет-ствующею меюда и снимаю! показания эксiинкиии. Cipoai i рафик функции I) - /(с). 1 де с - копией 1 рання карнозина.
2. ОПРЕДЕЛЕНИЕ ДИПЕПТИДОВ ХРОМАТОГРАФИЕЙ
НА БУМАГЕ
Материалы, реактивы и оборудование. Фенол перегнанный. водонасыщенный; кислота соляная концентрированная; ацетон: раствор нингидрина в ацетоне массовой долей 0,5 %; хроматографическая бумага: стандартные расгворы карнозина и ансерина молярной концентрацией 0.01 моль/дм3; смесь для элюирования (раствор NaHCO, молярной концентрацией 0,1 моль/дм3 смешивают с этанолом в соотношении 1:1); диазотированная сульфаниловая кислота; растворы Na2CO. массовыми долями 20 и 1 %; стеклянные пипетки; воронки;" стаканы; пробирки; хроматографические камеры.
Подготовка проб. При определении карнозина и ансерина методом хроматографии навеску мышечной ткани массой 3—5 г обрабатывают точно так же, как это описано для определения сво бодных аминокислот (см. лабораторную работу № 15).
При исследовании карнозина непосредственно в безбелковом экстракте ткани или в случае осаждения дипептидов в виде ртутных солей этот метод несколько изменяют; водный экстракт после стадии осаждения упаривают не досуха, а до объема 1—2 см3.
Порядок проведения анализа. Дипептиды и составляющие их аминокислоты хорошо разделяются методом бумажной хроматографии в водонасыщенном феноле или системе д-бутанол — пропанол — раствор аммиака массовой долей 20 % в соотношении 2 ; 2 ; 1. Хроматограмму обрабатывают раствором нингидрина или диазотированной сульфаниловой кислотой (для выявления карнозина и гистидина).
На дно хроматографической камеры помещают кристаллизатор или чашку Петри с водонасыщенным раствором фенола. Туда же ставят стаканчик со смесью концентрированной НО и воды в соотношении 3:1, камеру закрывают и оставляют для насыщения парами растворителя.
Для проведения хроматографического разделения на листе хроматографической бумаги нужного размера на расстоянии 4 см от нижнего края проводят стартовую линию, на которую наносят исследуемый раствор, содержащий дипептиды. Безбелко-
131
вый экстрак! ткани после упаривания на роторном испарите ле чате всею растворяю! в смеси НСООН — СН.СООН — НХ) в соотношении 7:5: 13 пли в растворе изопропанола массовой долей 10 С/с. чтобы 1 с.м? раствора соответствовал 1.5—2 г ткани, после чего нанося! на хроматограмму небольшими порциями (0.01—0.03 см3). подсушивая бумагу теплым воздухом. На эту же хроматограмму наносят стандартные растворы карнозина и ансерина, содержащие по 0.03—0.05 мкмоль в пятне. Бумагу сшивают в виде цилиндра (следят, чтобы края не соприкасались) и помещают в хроматографическую камеру в строго вертикальном положении. Когда фронт растворителя дойдет до верхнего края бумаги, хроматограмму вынимают и высушивают под тягой в течение 2—3 сут. Следы фенола на хроматограмме удаляют горячим ацетоном. Для этого сухую хроматограмму сворачивают трубочкой, перевязывают ниткой, помещают в цилиндр, заливают ацетоном при температуре кипения и оставляют на 10—15 мин; промывание повторяют 2—3 раза, а затем высушивают под тягой.
К 9 см3 раствора нингидрина в ацетоне массовой долей 0,5 % прибавляют 1 см^ воды. Раствор наливают в ванночку и равномерно пропитывают им хроматограмму. Затем ее сушат под тягой и помещают в термостат при 110—120 °C на 5—10 мин. При этом пятна карнозина и ансерина приобретают серо-голубую окраску.
Количественное определение дипептидов можно проводить либо путем сопоставления интенсивности и размера проявленных пятен опытной пробы с пятнами стандартных растворов (в случае их несоответствия определение повторяют, изменяя соответственно количество взятых растворов), либо путем элюирования окрашенных пятен с последующим фотометрическим определением окраски элюатов аналогично количественному определению аминокислот. В этом случае окрашенные пятна элюируют в темноте в течение 2—3 ч смесью раствора NaHCO3 молярной концентрацией 0,1 моль/дм3 и этанола в соотношении 1 : 1, а затем колориметрируют на фотоэлектроколориметре при длине волны 582 нм.
При проявлении на хроматограмме карнозина диазореактивом хроматограмму обрабатывают из пульверизатора диазотированной сульфаниловой кислотой, для чего слегка влажную хроматограмму опрыскивают раствором Na2CO3 массовой долей 20 %. Пятна карнозина приобретают интенсивную оранжевую окраску, окрашенные пятна элюируют раствором карбоната натрия массовой долей 1 %, колориметрируют при длине волны 500 нм. Пользуясь калибровочными графиками, рассчитывают содержание дипептидов в исследуемой пробе.
132
3. ОПРЕДЕЛЕНИЕ ДИПЕПТИДОВ С ПОМОЩЬЮ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ НА ПЛАСТИНКАХ «СИЛУФОЛ»
Материалы, реактивы и оборудование. Пластинки «Силу-фол»: изопропанол: концентрированный NH4OH: спиртовой раствор НБД-хлорида массовой долей 0,8 %: насыщенный раствор CH3COONa в этаноле: хроматографические камеры: раствор нингидрина массовой долей 1 %: спектрофотометр.
Подготовка проб. Подготовка колонки для бумажной хроматографии и пластинок «Силуфол» — см. лабораторные работы № 16, 17.
Порядок проведения анализа. Безбелковый экстракт ткани в количестве 5—10 мкл наносят на пластинки «Силуфол» на расстоянии 1,5—2 см от краев и 1,5 см между пятнами, а также наносят стандартные растворы дипептидов (5—10 нмоль/дм3). Проводят восходящую хроматографию в системе изопропанол — аммиак — вода (7:2: 1). Камеру предварительно насыщают парами растворителя. Пластинки сушат под тягой. Пятна дипептидов выявляют, опрыскивая пластинку раствором нингидрина. Затем пластинки сушат в струе теплого воздуха и выдерживают при 70 °C в течение 10 мин. Пятна аминокислот проявляются при комнатной температуре, пятна дипептидов — при нагревании. Окраску сканируют.
Для получения НБД-производных дипептидов к 0,23 см3 спиртового экстракта ткани добавляют 0,05 см3 смеси НБД-хлорида и насыщенного раствора CH3COONa в соотношении 4:1. Пробу инкубируют в течение 20 мин при 75 °C, охлаждают и наносят аликвоты (4—10 мкл) на пластинку «Силуфол». Хроматографию проводят в системе изопропанол — вода (7 : 2). Сканируют при длине волны 470 нм, сравнивая показания с растворами с известным содержанием пептидов.
Содержание специфических дипептидов (карнозина, ансерина) в мышечной ткани выражают в микромолях и (или) в милли-грамм-процентах на влажную массу ткани.
Полученные экспериментальные и расчетные данные оформляют в виде таблицы.
Наименование и харак-________________Содержание дипептидов в образце
теристика образца мкмоль/дм’ мг?
После обобщения экспериментальных данных самостоятельно делают выводы и заключение по работе.
133
Лабораторная работа № 19
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СУММАРНЫХ ЛИПИДОВ В ЖИВОТНЫХ ТКАНЯХ
Цель работы. Освоить методы количественно!о определения суммарных липидов в животных тканях.
Задачи. Провести экстракцию липидной фракции в аппарате Сокслета и определить массовую долю суммарных липидов гравиметрически и (или) колориметрически.
Объекты исследования. Мясо различных видов убойных животных аналогичных анатомических участков или одного животного от разных отрубов мясной туши, субпродукты I и II категорий, яичный желток, жировые ткани.
Материалы, реактивы и оборудование. Петролейный или диэтиловый эфир: смесь хлороформа, метанола и воды (2:1: 8); безводный бензол; хлороформ; концентрированная серная кислота; прокаленный песок; фильтровальная бумага; водяная баня; термометр; часовое стекло; аппарат Сокслета; сушильный шкаф; бюксы; пленочный испаритель, колбы; аналитические весы; магнитная мешалка; центрифуга лабораторная; фосфованилиновый реактив.
Приготовление реактивов. Фосфованилиновый реактив. 400 мг ванилина, перекристаллизованного из хлороформа или этанола, растворяют в 40 см3 воды и доводят объем до 200 см3 раствором Н3РО4 массовой долей 85 %. Получают раствор ванилина молярной концентрацией 0,013 моль/дм3 в растворе фосфорной кислоты молярной концентрацией 11,8 моль/дм3. Реактив можно длительно хранить в темной склянке при комнатной температуре.
Методические указания. Большинство липидов легко окисляются вследствие наличия в их структуре двойных связей, поэтому для их экстракции следует применять более чистые органические растворители, не содержащие следов окислителей. Склянки с растворителями хранят в металлических шкафах, вдали от нагревательных приборов, в темноте.
Для экстракции и хранения растворов липидов используют посуду из темного стекла. Колбы, делительные воронки и другую посуду при работе с липидами закрывают только пришлифованными стеклянными пробками.
Для экстракции липидов применяют смесь из двух-трех растворителей. Суммарные липиды извлекают чаще всего смесью хлороформа и метанола в объемных соотношениях 2 : 1, 1 : 1 и 1 : 2 или этанола и диэтилового эфира 3:1, 1:1. Наиболее распространенным способом является метод Фолча — экстракция смесью хлороформа и метанола (2: 1), позволяющий извлекать 90—95 % всех клеточных липидов. Преимуществом метода Фолча является то, что используемая смесь растворителей не проявляет селективности по отношению к тем или иным группам липидов.
134
Рис. 1.36. Аппарат Сокслета:
/ — приемная кояба: 2— ipyoKa лая 110с 1 \ иления паров рас i вори ic.ia in колбы в экс:р;1кюр. о - .хололилвник. 4 — аксчрактор: 5 -- сифоппа;! трубка. 6—б\ма/кпая гичьы с навеской исслел\емого материала
Для определения массовой доли суммарных липидов предварительно обезвоженный образец ткани экстрагируют петролейным эфиром, диэтиловым эфиром или смесью эфира и этанола. Полное извлечение липидов из тканей достигается путем многократной экстракции летучим растворителем в аппарате Сокслета (рис. 1.36). Все части аппарата плотно соединяют между собой при помощи шлифов. Пары эфира из экстракционной колбы при нагревании поднимаются по трубке экстрактора и попадают в холодильник. Здесь они охлаждаются, сгущаются и каплями стекают в экстрактор, где в бумажной гильзе находится исследуемый материал. Гильза постепенно наполняется эфиром, который экстрагирует жир из ткани. Когда уровень эфира поднимется выше верхнего колена сифонной трубки, эфир вмес-
те с растворенным в нем жиром стечет в нижнюю часть аппарата — экстракционную колбочку. Жир остается в колбочке, а пары эфира вновь поднимаются и экстрагируют новую порцию жира. Исследуемое вещество, подвергаясь многократной повторной экстракции, полностью обезжиривается. Жир перемещается в колбочку.
Массовую долю экстрагированных липидов определяют гравиметрически или колориметрически.
1. ГРАВИМЕТРИЧЕСКИЙ МЕТОД В АППАРАТЕ СОКСЛЕТА
Подготовка проб. Образец мяса тщательно измельчают ножом на часовом стекле, взвешивают навеску массой (3,0000 ± 0,0002) г и помещают в тарированный бюкс с прокаленным до постоянной массы песком.
Стеклянной палочкой перетирают мясо с песком и высушивают в сушильном шкафу при 100—105 °C до постоянной массы. Вычисляют массовую долю влаги в исследуемом образце. Сухой остаток используют для определения суммарных липидов.
Порядок проведения анализа. Сухую пробу количественно переносят в предварительно взвешенную гильзу из фильтровальной бумаги, гильзу повторно взвешивают и помещают в экстрактор аппарата Сокслета.
В экстракционную колбу аппарата наливают эфир и соединяют все части прибора. Прибор помещают на водяную баню. Холо-
135
лильник соединяют с водопроводным краном, включают волу и нагревают баню до 45—50 °C. Ориентировочная продолжительность экстра! ирования 4—6 ч. Конец экстрагирования определяют. поднося к экстрактору часовое стекло, куда стекает немного эфира. Экстрагирование считается законченным, если после испарения эфира на стекле не остается налета жира.
По окончании экстрагирования гильзу вынимают из экстрактора, высушивают до постоянной массы и вычисляют массовую долю липидов в исходной навеске по формуле
Л, = 100(/ш - т4)/(т} - /д0), (1.25)
где т. — масса гильзы с навеской до обезжиривания, г; т4 — масса гильзы с навеской после обезжиривания, г; — масса бюкса с песком и навеской до высушивания. г: /»0 — масса бюкса с песком, г.
• Массовую долю липидов (% к массе абсолютно сухого вещества) определяют по формуле
Х2 = 1ОО(л7?3 - т4)/(т3 — т5), (1-26)
где т5 — масса гильзы, г.
2. ГРАВИМЕТРИЧЕСКИЙ МЕТОД В ПЛЕНОЧНОМ ИСПАРИТЕЛЕ
Подготовка проб. Навеску массой 100 г очищенной от жира и пленок сердечной мышцы или печени измельчают при температуре 2—4 °C и гомогенизируют в органическом растворителе с частотой вращения 116,6—133,3 с-1 при комнатной температуре 1—2 мин. Гомогенат переносят в колбу с притертой пробкой и заливают тремя объемами смеси хлороформ — метанол в соотношении 2:1. Экстракцию липидов проводят при комнатной температуре при постоянном перемешивании с помощью механической или магнитной мешалки в течение 30 мин.
Суспензию центрифугируют с частотой вращения 33,3 м~’ в течение 15—20 мин, органическую фазу отделяют, а остаток переносят в прежнюю колбу и повторяют экстракцию еще 2 раза в тех же условиях. Объединенные экстракты упаривают в вакууме, остаток липидов растворяют в 5—10 см3 смеси хлороформ — метанол — вода (6:3: 0,45).
Для удаления нелипидных примесей используют сефадекс G-25, который предварительно освобождают от мелких частиц и высушивают, суспендируют в трех объемах смеси хлороформ — метанол — вода (6:3: 0,45) и оставляют набухать в течение 10—12 ч. Набухшим сефадексом заполняют колонку размером 2x15 см, на дно которой предварительно помещают промытую 136
стекловату. После заполнения на поверхность сефадекса также кладут стекловату и готовую колонку промывают одним объемом смеси. Для очистки на колонку наносят экстракт липидов (20-30 %) и элюируют их 1,5—2 объемами той же смеси. В элюате содержатся практически все липиды, нелипидные примеси остаются на колонке и могут быть смыты с нее смесью хлороформ — метанол (I : 1).
Очищенный от нелипидных примесей экстракт переносят в колбу пленочного испарителя, растворитель отгоняют, к остатку добавляют 10 см3 безводного бензола, тщательно перемешивают и отгоняют бензол. Липиды, освобожденные от следов воды, растворяют в небольшом объеме свежеперегнанного бензола, переносят в пробирку с притертой пробкой и хранят в темноте на холоде.
Порядок проведения анализа. Круглодонную колбу для пленочного испарителя доводят до постоянной массы, помещают в нее отмеренный объем липидного экстракта, содержащий 10—20 мг липидов, и растворитель упаривают в токе азота или в пленочном испарителе. Колбу с осадком липидов доводят до постоянной массы в вакуум-эксикаторе над КОН. Сухую массу липидов рассчитывают по разности постоянной массы колбы с осадком и пустой колбы. Взвешивание проводят на аналитических весах.
3. КОЛОРИМЕТРИЧЕСКИЙ МЕТОД
Подготовка проб. К одному яичному желтку добавляют 50 см3 смеси хлороформа, метанола и воды (2:1: 0,8) и экстрагируют при постоянном перемешивании с помощью магнитной мешалки в течение 30 мин. Центрифугируют 20 мин с частотой вращения 33,3 с-1 и отделяют органическую фазу, содержащую липиды. Остаток заливают новой порцией растворителя и операцию повторяют еще несколько раз. Липидные фракции объединяют.
Для освобождения от нелипидных примесей экстракт переносят в делительную воронку, добавляют 20 % воды от его объема, перемешивают и после разделения водной и органической фаз последнюю переносят в колбу пленочного испарителя и отгоняют растворитель в вакууме. Для высушивания к остатку добавляют 5—7 см3 безводного бензола, перемешивают и отгоняют бензол, процедуру повторяют несколько раз. Осадок липидов растворяют в точном объеме (5—10 см3) свежеперегнанного хлороформа, переносят в пробирку с притертой пробкой и хранят в темноте на холоде.
Порядок проведения анализа. В пробирку с пришлифованной пробкой помещают 50 нм3 липидного экстракта (для контроля в пробирку берут 50 нм3 хлороформа) и испаряют на водяной бане. Добавляют 0,2 см3 концентрированной H2SO4 и нагревают на во-
137
ляной оане при icMiicpaiypc кипения в течение 10 мин. Пробирки охлаждают, добавляют в них но 2.5 см-’ фосфованилинового реактива, перемешивают и выдерживают при 37 °C в течение 15 мин: колориметрируют против контроля при длине волны 540 нм. Содержание липидов рассчитывают по калибровочному графику.
Построение калибровочного графика. В качестве стандартного используют раствор высокоочишенного абрикосового или оливкового масла в хлороформе. Берут аликвотное количество стандартного раствора и готовят разведение в соответствии с чувствительностью метода от 10 до 120 мкг. Затем проводят качественную реакцию согласно описанию метода и колориметрируют при длине волны 540 нм против контроля. После снятия показаний оптической плотности для всех концентраций строят график D = f[c), где с — концентрация липидов.
Студенты выполняют расчеты в соответствии с экспериментальными данными. Результаты оформляют в виде таблицы.
Наименование и источник образце! Массовая доля суммарных липидов
% к массе образца % к массе абсолютно сухого вещества
Экспериментальные данные сравнивают с литературными и полученными разными методами, самостоятельно делают выводы и формулируют заключение.
Лабораторная работа № 20
ОПРЕДЕЛЕНИЕ ФРАКЦИОННОГО СОСТАВА ЖИРОВ
Цель работы. Установить состав и массовую долю фракций животных жиров на основе их способности к кристаллизации и
плавлению.
Задачи. Выделить низко-, средне- и высокотемпературные фракции пищевых топленых жиров разных видов и сортов; определить температуры плавления фракций жира и рассчитать их
массовые доли.
Объекты исследования. Пищевые животные жиры разных видов и сортов.
Материалы, реактивы и оборудование. Капилляр, открытый с двух концов' термометр; штатив с кольцом; стакан вместимостью 500 см , мешалка; вакуум-насос; колба Бунзена; термостат; весы аналитические; прибор для определения температуры плавления.
138
Методические указания. Ирл мг.неипо'ч их-Ш/Ьчении жиры способны крис галлизова 1 ься. -Ую свойство лсжт. например, в основе получения маргарина. Tpniпиперины жирных кисло! в расплавленном состоянии нс имеют nisei а. вкуса и запаха. На способности различных жировых фракции плави i вся. кристаллизоваться при определенной температуре основан .метод их разделения.
Пониженная усвояемость и высокая температура плавления ограничиваю! применение говяжьего и бараньего жиров в качестве пищевых продуктов. Значительная часть них жиров используется на технические пели. Повышения потребительских свойств говяжьего и бараньего жиров можно достигнуть за счет разделения их на фракции, содержащие различные по температуре плавления триглицериды.
Переход в капельно-жидкое состояние совершается не мгновенно, а в пределах некоторого интервала температур, в котором плавятся отдельные компоненты смеси. У насыщенных жирных кислот температура плавления возрастает с увеличением молекулярной массы. У ненасыщенных жирных кислот на температуру плавления влияют не столько двойные связи, сколько их положение в цепи и пространственное расположение отдельных частей молекулы. Определенное влияние на изменение температуры плавления оказывает полиморфизм.
Методы определения температуры плавления заключаются в постепенном нагревании твердого жира до момента расплавления, который характеризуется по прозрачности, подвижности, освет-ленности и т. п. На практике температура плавления устанавливается по температуре, при которой жир становится подвижным.
Существует два метода определения температуры плавления: по сползанию капли жира в капилляре с расширением и по поднятию жира в открытом капилляре.
При разделении жировых фракций на основе кристаллизации используют различные подходы.
Классический метод проведения процесса фракционирования животных жиров заключается в медленном охлаждении расплавленного жира, в результате чего из говяжьего и бараньего жиров получают легкоплавкую фракцию — олео-маргарин или олео-ойл, из свиного — лярд-ойл, а также твердую фракцию — олео-стеарин.
Олео-маргарин имеет цвет от светло-желтого до желтого, приятный вкус, напоминающий сливочное масло, его можно использовать в домашних условиях, для производства высококачественной маргариновой продукции, в хлебопечении. Костный жир, полученный из цевок крупного рогатого скота, фракционируют с целью выделения легкоплавкой фракции для производства смазочных масел, не замерзающих при низкой температуре.
Обнаружение и расчет массы фракций весьма полезны при оценке биологической ценности жиров.
139
Подготовка проб. Образцы пищевых гонленых жиров мае сой по 35—40 г помещают в предварительно высушенные и взвешенные пробирки и нагревают в камере ультратермостата UTU-2. защищенной от естественного света, до температуры 65—70 °C. Первую стадию кристаллизации проводят при температуре 40—45 °C в течение 24 ч для наиболее полного разделения твердой и жидкой фракций. Для обеспечения равномерной кристаллизации содержимое пробирок рекомендуется периодически плавно перемешивать.
1. ФРАКЦИОНИРОВАНИЕ ПИЩЕВЫХ ТОПЛЕНЫХ ЖИРОВ КРИСТАЛЛИЗАЦИЕЙ
Порядок проведения анализа. Оставшуюся жидкой при температуре 40—45 °C жировую фракцию отфильтровывают вакуум-насосом в колбу Бунзена, помещают в высушенную и взвешенную пробирку и термостатируют далее, снизив температуру в термостате на Ю °C.
Операции по разделению фракций жира повторяют, ступенчато проводя снижение температуры до 20—22 °C. Продолжительность каждой стадии кристаллизации 30—40 мин.
Выход каждой фракции жира определяют гравиметрически.
Массовый выход фракций жира (%) определяют по формуле
М ~~ |(ш2 — /?73)/ш1]1ОО, (1-27)
где т2 — масса пробирки с соответствующей фракцией жира, г; /и, — масса пустой пробирки, г; т} — масса исходной пробы жира, взятой на фракционирование, г; 100 — коэффициент для перевода в проценты.
2. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ ФРАКЦИЙ
Порядок проведения анализа. Пробу исследуемой фракции жира, полученной по п. 1, нагревают в пробирке на водяной бане до полного расплавления. Чистую сухую, открытую с двух концов капиллярную трубочку из тонкого стекла с внутренним диаметром 1,0—1,2 мм (длина капилляра 50—60 мм) погружают одним концом в расплавленный жир так, чтобы высота его в капилляре была равна 10 мм. Затем капилляр с жиром выдерживают на льду в течение 10 мин.
После этого капилляр прикрепляют к термометру с ценой деления шкалы 0,1 °C тонким резиновым кольцом так, чтобы столбик жира находился на одном уровне с ртутным шариком термометра. Термометр с капилляром опускают в стакан с дистиллированной водой на глубину 3—4 см. Начальная температура воды в стакане должна быть 15—18 °C. Следят, чтобы в незаполненный конец ка-
140
Рис. 1.37. У становка для определения температуры плавления в открытом с двух концов капилляре.
1 -- механическая или магнитная мешалка: 2 — термометр: л — капилляр
пилляра не попала вода. При непрерывном перемешивании магнитной мешалкой воду в стакане нагревают сначала со скоростью приблизительно 2°С/мин, а по мере приближения к ожидаемой температуре плавления —- не более чем 1 °С/мин. Общий вид установки показан на рис. 1.37.
В качестве температуры плавления фиксируют ту, при которой жир в капилляре начинает подниматься. Эксперимент повторяют не менее двух раз, за результат принимают среднее арифмети
ческое двух параллельных опытов, результаты которых должны отличаться не более чем на 0,5 °C.
Результаты экспериментов оформляют в виде таблицы.
Вил жира Число фракций Выход фракций!, % Температура, °C
плавления фракции кристаллизации
Затем самостоятельно анализируют полученные результаты, формулируют выводы и заключение по работе.
Лабораторная работа № 21
ОПРЕДЕЛЕНИЕ ФОСФОЛИПИДОВ
Цель работы. Освоение методов выделения и фракционирования фосфолипидов на примере яичного желтка.
Задачи. Провести качественную реакцию на лецитин; освоить методы фракционного осаждения и хроматографического разделения фосфолипидов; охарактеризовать фосфолипидную фракцию анализируемого материала.
Объект исследования. Яичный желток.
Материалы, реактивы и оборудование. Фильтровальная бумага; химические стаканы; пробирки; капельницы; воронки; водяная баня; пипетки; этанол; ацетон; метанол; хлороформ; петролейный эфир; «-бутанол; фосфорномолибденовая кислота; ледяная уксусная кислота; нингидрин; AI2O3; хроматографическая колонка; тонкослойная пластина.
141
Mei одические указания. В qv нь. д ломы .
ЖПТСЯ 99- -94.? ' г 1 pill .1И11ирИ Ю.н О, । .; ь ; КГ;| >|..К!; |фИ\г и фосфатиды (фосфоЛИНИЛЫ ) С общ?!!
(:•
(II " С R
I - ()
I //
( Н -()-( к
I он
I /
(НО р О
X о в
где О-С— R.J и О—С— R, — остатки радикалов жирных кислот (линолевой, линоленовой, пальмитиновой, стеариновой и т.д.); В — остаток азотистого основания или аминокислоты серина.
Методы их определения основаны на специфичности физико-химических свойств.
Работа состоит из двух этапов, каждый из которых рекомендуется изучить и освоить практически.
1 .КАЧЕСТВЕННАЯ РЕАКЦИЯ НА ЛЕЦИТИН
Подготовка проб. Для осуществления качественной реакции на лецитин яичный желток выпаривают досуха на водяной бане при температуре кипения. Сухой остаток используют для анализа.
Порядок проведения анализа. В химический стакан вносят 200—300 мг высушенного и растертого яичного желтка, прибавляют 15 см3 горячего этанола и все перемешивают. Через 10—15 мин смесь охлаждают и фильтруют в сухую пробирку. В другую сухую пробирку наливают 2—3 см3 ацетона и по каплям добавляют полученный фильтрат. Наблюдают появление мути, а затем выпадение осадка лецитина, что указывает на нерастворимость лецитина в ацетоне. Если в пробирку к 2—3 см3 фильтрата прибавлять по каплям дистиллированную воду, то образуется стойкая эмульсия, что позволяет выделить лецитин из смеси липидов.
2 . ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ ФОСФОЛИПИДОВ
Подготовка проб. Для хроматографического анализа фосфолипидов яичные желтки тщательно отделяют от белка, добавляют 400 см3 ацетона и смесь гомогенизируют в течение 10 мин. Гомогенат оставляют в холоде на ночь. Осадок отделяют центрифуги
142
рованием с частотой вращения 50 с 'в геченне 20 мин. суспендируют в 400 см' ацетона и снова отделяют иенiрифегировани-ем. Обработку ацетоном повторяют. Затем осадок наносят тонким слоем на фильтровальную бумагу и сушат до исчезновения запаха ацетона в течение 1 — 2 ч.
Высушенный порошок заливают 200 см3 смеси хлороформ — метанол (1:1). Экстрагирование фосфолипидов проводят в течение 30 мин на вибровстряхивателе. Раствор центрифугируют с частотой вращения 50 с"1 в течение 20 мин. Центрифугат отделяют и сохраняют. Обработку осадка повторяют. Если центрифугирование не дает полного просветления надосадочной жидкости, проводят фильтрование на воронке Бюхнера. Цен-трифугаты (фильтраты) объединяют и выпаривают на роторном испарителе.
После выпаривания смесь фосфолипидов растворяют в 50 см3 петролейного эфира и добавляют 20-кратный объем (1000 см3) ацетона. Перемешивают и оставляют на ночь. Затем осадок отфильтровывают на воронке Бюхнера, растворяют в 50 см3 петролейного эфира и вновь повторяют осаждение. Растворы в пет-ролейном эфире обычно бывают мутными. После повторного осаждения и фильтрования осадок промывают 200 см3 ацетона прямо на фильтре.
2.1 . РАЗДЕЛЕНИЕ ФОСФОЛИПИДОВ НА КОЛОНКЕ С А12оз
Порядок проведения анализа. Колонку диаметром 2,5 см и высотой не менее 50 см заполняют за 1—2 сут до разделения фосфолипидов. На дно помещают плотный слой стекловаты толщиной 1 см, затем наливают суспензию, состоящую из 170— 190 г А12О2 в смеси с метанолом и хлороформом (1 : 1). При заполнении следует слегка постукивать по колонке деревянной палочкой для оседания и получения равномерного слоя адсорбента.
Сверху можно положить бумажный фильтр.
Полученную смесь фосфолипидов (10—15 г) растворяют в смеси метанол — хлороформ (1 : 1) и наносят на колонку. Фосфатидилхолин с примесью лизофосфатидилхолина и сфингомиелина вымывается 500 см3 смеси метанол — хлороформ (1 : 1). Первые 150 см3 и последние 100 см3 отбрасывают. После выхода фосфа-тидилхолина колонку промывают 100 см3 той же смеси. Фосфа-тидилэтаноламин с примесью фосфатидилсерина отмывается 50 см3 смеси этанол — хлороформ — вода (5:2: 2). Первые 200 см3 отбрасывают. Оба элюата упаривают на роторном испарителе досуха и получают приблизительно 5 г лецитина (фосфатидилхолина) и 2,5 г кефалина (фосфатидилэтаноламина).
143
2.2 АНАЛИЗ ФОСФОЛИПИДОВ С ПОМОЩЬЮ ТОНКОСЛОЙНОЙ ХРОМАТОГРАФИИ
Порядок проведения анализа. Для тонкослойной хроматографии применяют пластинки «Силуфол» (150 x150 мм) и стандартные камеры. В качестве растворителя используют смесь хлороформ — метанол — вода в соотношении 65 : 25 : 4.
На две пластинки на расстоянии 1,5 см от их узкого края микропипеткой наносят хлороформные растворы фракций фос-фатидилхолина (в одно пятно) и фосфатидилэтаноламина (в другое пятно) в количестве 1—5 мкмоль. Пластинки помещают в камеру (предварительно за 2—3 ч для насыщения камеры в нее помещают растворитель). После разделения одну пластинку проявляют спиртовым раствором фосфорномолибденовой кислоты массовой долей 5 % (с фосфатидилхолином образуются синие пятна на желтом фоне), другую — раствором нингидрина на //-бутаноле массовой долей 0,3 %, содержащем уксусную кислоту массовой долей 3 %. Все фосфолипиды, содержащие NH2-rpynny (фосфатидилэтаноламин, лизофосфатидилэтанола-мин, фосфатидилсерин), проявляются в виде розовых пятен на сером фоне.
Студенты фиксируют в тетради условия и эффект, полученный от реакций, включая качественную реакцию на лецитин и разделение фосфолипидов, анализируют экспериментальные данные, формулируют выводы и составляют отчет о работе.
Лабораторная работа № 22 ОПРЕДЕЛЕНИЕ ХОЛЕСТЕРИНА
Цель работы. Приобрести практический навык определения холестерина в мясопродуктах (мозг, шпик, колбасные изделия и т. д.).
Задачи. Провести подготовку к анализу; получить и исследовать кристаллы холестерина; определить содержание холестерина в объектах исследования.
Объекты исследования. Ткань головного мозга, желчь, яичный желток, сыворотка или плазма крови убойных животных.
Методические указания. Холестерин нерастворим в воде, разбавленных кислотах и щелочах, но растворим в жирах и их растворителях: эфире, хлороформе и бензоле. Холестерин кристаллизуется из этанола в виде пластинок с перламутровым блеском, жирных на ощупь. Кристаллы содержат одну молекулу воды, имеют
144
температуру плавления it, (. и о 111 ос >п с я к числ\ наиболее термос габиаьпых органических вещешв. Иг и’рира. хлороформа и бензола холестерин кристаллизуется в виде безводных тонких игл с шелковистым блеском. Растворы холестерина в хлороформе и эфире оптически активны, - -31,59.
В то время как эфиры холестерина не абсорбируют воду, свободный холестерин может поглотить ее массу, в 500 раз превышающую собственную. Высокая массовая доля воды в мозге связана с высокой массовой долей холестерина в этом органе.
Холестерин является хорошим изолятором электрического тока. Уменьшение содержания холестерина в организме ниже физиологической нормы приводит к анестезии и ряду нервных и психических расстройств. Он является фактором воспроизводства, увеличивающим плодовитость животных, обладает способностью связывать яды и токсины, в связи с чем играет важную роль в формировании иммунитета; предупреждает гемолиз эритроцитов, вызываемый лецитином.
Вместе с тем химическая стойкость холестерина и практически полная нерастворимость в тканевых жидкостях приводят с возрастом к аккумулированию его в организме, что является фактором риска ряда заболеваний, в связи с этим требуется контроль его уровня в продуктах питания и пищевых рационах.
Для обнаружения холестерина применяется ряд качественных цветных реакций, химизм которых связан с переводом его из вторичного спирта в непредельный углеводород путем дегидратирования.
Для получения растворов холестерина в хлороформе к сухим остаткам холестерина, выделенного из образцов нервной ткани и желчи на этапе подготовки пробы, добавляют обезвоженный хлороформ из расчета получения раствора холестерина с массовой долей 0,3 %.
При действии концентрированной серной кислоты стеролы дегидратируются и окисляются с образованием смеси сложных продуктов.
Раствор холестерола в хлороформе дает с уксусным ангидридом и концентрированной серной кислотой красное окрашивание, переходящее затем в синее и зеленое. Таким образом, при наличии серной кислоты происходят дегидратация и окисление холестерола. В результате две молекулы холестерола, потерявшие две молекулы воды, соединяются между собой по третьему атому углерода. Образовавшиеся непредельные углеводороды с сопряженными двойными связями дают различные производные с серной кислотой и уксусным ан-
145
i и.ipi’.lo\i i’Oi ,iacno схеме
После обработки хлороформенного раствора холестерола равным объемом серной кислоты массовой долей 92 % он окрашивается в кроваво-красный цвет, переходящий затем в пурпурный. Отстоявшийся слой серной кислоты приобретает слабо-зеленую флуоресценцию. Чувствительность реакции 0,001 мае.%.
В результате дегидратации холестерина реакция с уксусным ангидридом и концентрированной серной кислотой приводит к образованию окрашенной в зеленый цвет сульфокислоты — хо-лестерилена.
Смесь раствора холестерина в хлороформе с уксусным ангидридом и концентрированной серной кислотой окрашивается в быстро исчезающий красный цвет, затем в синий и, наконец, в более стойкий зеленый цвет. При незначительной массовой доле холестерина в растворе сразу образуется зеленое окрашивание.
146
CJi
(CH;COM)
- CH;CO()H
сщ-соо
- СН;-СООН
Эта цветная реакция служит и для количественного определения холестерина в крови или сыворотке крови.
При проведении лабораторного практикума рекомендуется ознакомиться и освоить все рекомендуемые методы определения холестерина.
1. ПОЛУЧЕНИЕ КРИСТАЛЛОВ ХОЛЕСТЕРИНА
Материалы, реактивы и оборудование. Гипс; этанол 96%-ный; эфир; фарфоровая ступка; стеклянная пластинка размером 10 х хЮсм, сушильный шкаф; термометр; микроскоп; предметное стекло; пипетка.
Подготовка проб. При анализе тканей тщательно растирают 3—5 г ткани в фарфоровой ступке с 6—Юг гипса до гомогенной массы, которую распределяют стеклянной палочкой или скальпелем тонким слоем по стеклянной пластинке и высушивают в сушильном шкафу при 60 °C.
Порядок проведения анализа. К сухому остатку неочищенного холестерина добавляют спиртоэфирную смесь (соотношение спирта и эфира 1 : 2 по объему) из расчета получения раствора массовой долей холестерина 2 %.
На предметное стекло наносят пипеткой 1—2 капли эфирно-спиртового раствора холестерина и испаряют до выпадения кристаллов.
147
Рис. 1.38 Крист i.ibi \o.ieciepnna (8x40)
Кристаллы холестерина микроскопиру-ют, используя объектив с увеличением х 40. Операции выполняют с холестерином, взятым из нервной ткани различных видов животных. Результаты наблюдений оформляют
в виде зарисовок с указанием объекта и применяемого увеличения. Пример показан на рис. 1.38.
2. ЦВЕТНЫЕ РЕАКЦИИ НА ХОЛЕСТЕРИН
Материалы, реактивы и оборудование. Концентрированная серная кислота; раствор серной кислоты массовой долей 92 % (плотность 1,76 г/см3); формалин; уксусный ангидрид; безводный хлороформ; пробирки с притертыми пробками; штатив.
Подготовка проб. Высушенную мозговую ткань с гипсом в соответствии с методикой подготовки проб (см. п. 1) соскабливают скальпелем со стекла и измельчают в ступке. К сухому порошку в пробирке добавляют 5—6 см3 хлороформа и экстрагируют в течение 5—10 мин. Для лучшей экстракции содержимое пробирки периодически встряхивают. Полученный экстракт в хлороформе фильтруют в сухую пробирку и используют для проведения качественных реакций на холестерол.
При использовании в качестве объекта исследования желчи пробу объемом 20 см3 выпаривают досуха на водяной бане при температуре кипения. К сухому остатку добавляют 5—10 см3 эфира, тщательно перемешивая стеклянной палочкой. Эфирный экстракт фильтруют, испаряют досуха и используют для проведения качественных реакций на холестерин.
При анализе яичного желтка его выпаривают досуха на водяной бане при температуре кипения. Для получения хлороформенного экстракта к 1 г сухого остатка добавляют 5—6 см3 хлороформа и экстрагируют в течение 5—10 мин. Для лучшей экстракции содержимое пробирки периодически встряхивают. Полученный экстракт в хлороформе фильтруют в сухую пробирку и используют для проведения качественных реакций.
2.1. РЕАКЦИЯ С СЕРНОЙ КИСЛОТОЙ (РЕАКЦИЯ ЗАЛКОВСКОГО)
Порядок проведения анализа. В 2 пробирки с притертыми пробками вносят по 2 см3 полученного из разных источников раствора холестерина в хлороформе массовой долей 0,3 %, добавляют по 148
2см? раствора серной кислоты массовой .имей (11.101 нос 1 ью 1,76 г/см-) и встряхиваю!.
После разделения слоев хлороформа и серной кислоты фиксируют изменение окраски.
2.2. РЕАКЦИЯ СО СМЕСЬЮ ФОРМАЛИНА И КОНЦЕНТРИРОВАННОЙ СЕРНОЙ КИСЛОТЫ
Порядок проведения анализа. В пробирку с притертой пробкой помещают 2 см3 раствора холестерина в хлороформе массовой долей 0,3 %, добавляют 2 см3 смеси концентрированной серной кислоты с формалином (на 50 г концентрированной серной кислоты 1 г формалина) и встряхивают. После разделения слоев хлороформа и серной кислоты фиксируют изменение окраски.
2.3. РЕАКЦИЯ С УКСУСНЫМ АНГИДРИДОМ И КОНЦЕНТРИРОВАННОЙ СЕРНОЙ КИСЛОТОЙ (РЕАКЦИЯ ЛИБЕРМАНА —БУХАРДА)
Порядок проведения анализа. В пробирку с притертой пробкой помещают 2 см3 раствора холестерина в хлороформе массовой долей 0,3 %, добавляют 5—10 капель уксусного ангидрида и несколько капель концентрированной серной кислоты. Фиксируют изменение окраски смеси.
После выполнения экспериментальной части работы студенты кратко описывают в тетради условия и эффект качественных реакций, самостоятельно анализируют экспериментальные данные, формулируют выводы и заключение по работе. Наблюдения рекомендуется представить в виде таблицы:
Реакция Источник холестерина Полученный эффект Выводы
Реакция с серной кислотой (реакция Залковского)
Реакция со смесью формалина и концентрированной серной кислоты
Реакция с уксусным ангидридом и концентрированной серной кислотой (реакция Либермана — Бухарда)
149
3. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ХОЛЕСТЕРОЛА
Материалы, реактивы и оборудование. Раствор хлорного железа (0,125 г FeCl; • бН-,0 растворяют в 100 см3 ледяной уксусной кислоты); этилацетат "(СН;—СОО—С2Н5); уксусный ангидрид; хлороформ; стандартный раствор (1,8 г/дм3) холестерола (180 мг холестерола растворяют в 100 см3 этилового этанола); реактив А; штатив с пробирками; пипетки; микропипетки; цилиндры; колба; термостат (37 °C); фотоэлектроколориметр.
3.1. РЕАКЦИЯ С ХЛОРНЫМ ЖЕЛЕЗОМ
Подготовку проб см. п. 2 настоящей работы.
Порядок проведения анализа. Метод основан на взаимодействии хлорного железа со стеролами с образованием продукта пурпурного цвета в присутствии этилацетата и серной кислоты.
В пробирку с пришлифованной пробкой помещают I см3 раствора стеролов в хлороформе, приливают 2 см3 этилацетата, затем 2 см3 раствора хлорного железа, перемешивают и добавляют 3 см3 концентрированной H2SO4, смесь снова осторожно перемешивают и оставляют на 30 мин при комнатной температуре. Окрашенный раствор колориметрируют при длине волны 540 нм. Количество холестерина определяют по калибровочному графику.
Построение калибровочного графика. В качестве стандартных используют растворы эргостерола или холестерола в хлороформе с содержанием этих веществ от 10 до 100 мкг, проводят качественную реакцию в соответствии с описанием метода, колориметрируют при длине волны 540 нм и строят график D где с — концентрация холестерола (эргостерола).
3.2. МЕТОД ЛИБЕРМАНА —БУХАРДА
Подготовку проб см. п. 2 настоящей работы.
Порядок проведения анализа. Стеролы в присутствии дегидратирующего агента (серной кислоты) реагируют с уксусным ангидридом, образуя конденсированные продукты зеленого цвета.
В пробирку с пришлифованной пробкой помещают 1 см3 раствора стеролов в хлороформе, прибавляют 0,7 см3 уксусного ангидрида и 0,1 см3 концентрированной H9SO4. Содержимое пробирки перемешивают, доводят его объем до 5 см3 хлороформом и оставляют на 15—20 мин в темноте. Окрашенный раствор колориметрируют против контроля при длине волны
150
610 нм. Содержание стеронов определяю! по калибровочному графику.
Построение калибровочного графика. В качестве стандартного используют растворы эргостерола или холестерола в хлороформе с содержанием этих веществ от 50 до 250 мкг. Затем проводят цветную реакцию согласно описанию метода, колориметрируют при длине волны 610 нм и строят график D
3.3. ОПРЕДЕЛЕНИЕ ХОЛЕСТЕРОЛА В ПЛАЗМЕ (СЫВОРОТКЕ) КРОВИ
Подготовка проб. Плазму или сыворотку крови получают центрифугированием или сепарированием с частотой вращения 25—42 см1 соответственно стабилизированной или дефибриниро-ванной крови убойных животных в течение 5—8 мин.
Приготовление реактивов. Реактив А: смесь ледяной уксусной кислоты (30 см3), уксусного ангидрида (150 см3) и концентрированной серной кислоты (30 см3) в соотношении 1:5:1 необходимо готовить при постоянном охлаждении. Серную кислоту следует добавлять в последнюю очередь, очень медленно, постоянно перемешивая. Хранить реактив А следует в холодильнике в посуде из темного стекла с притертой пробкой.
Порядок проведения анализа. В сухую пробирку вносят 0,1 см3 плазмы (сыворотки) крови, быстро добавляют 2,1 см3 реактива А, немедленно перемешивают, 10 раз встряхивают и помещают в термостат при 37 °C. Через 20 мин смесь в пробирке окрашивается в зеленый цвет. Фотометрируют против воды при использовании красного светофильтра.
Молярную концентрацию холестерола в исследуемой пробе определяют по оптической плотности с помощью калибровочного графика.
Построение калибровочного графика. Из стандартного раствора холестерола (1,8 мг холестерола в 1 см3) отбирают по 0,05; 0,1; 0,15; 0,20; 0,25 см3 и добавляют реактив А до объема 2,2 см3 в каждой пробирке. При этом в пробах будет содержаться соответственно 0,09; 0,18; 0,27; 0,36; 0,45 мг/см3 холестерола. После добавления реактива А пробирки энергично встряхивают и помещают в термостат при 37 °C, через 20 мин смесь фотоколориметрируют с использованием красного светофильтра. Строят график D=flc).
Молярную концентрацию холестерола в плазме (сыворотке) крови (ммоль/дм3) определяют по формуле
М = с- 1000-0,0258, (1.28)
где с — концентрация холестерола в пробе, найденная по калибровочному графику, мг/см3; 1000 — коэффициент пересчета на объем пробы 1дм2; 0,0258 — коэффициент пересчета в систему СИ.
151
Ргл'чьпиы ж/неримсн гальныч нес. iv.iob.ihhi! фиксирую i. с? \i ОС i i) Ч i С.Ч Ь । b ) (j)(';[1 . 1 И jj\ К > i ВЫВОДЫ И juK.ikHciilk' НО paoo'lC. _)к<
периmcii’i\uiijn>iс чанные сводят в чаблицх;
I IjioteiiooiHiic и к -
СрИСПЦО ojpj'ilki
Оптическая птюгност!. пробе
I
Концентрация холестерина к. пробе по ю.тброг,очному график) . мг/см ’
Молярная КОНЦиНГра пня чолежерина в Не еле чуемом обра/це. ммоль/лм
Лабораторная работа № 23
ОПРЕДЕЛЕНИЕ ЖИРНОКИСЛОТНОГО СОСТАВА ЖИВОТНЫХ ЖИРОВ
Цель работы. Приобрести практический навык при анализе жирнокислотного состава животных жиров.
Задачи. Провести подготовку проб методом экстракции и путем переэтерификации глицеридов в метиловые эфиры жирных кислот, проанализировать жирные кислоты методом газожидкостной хроматографии и идентифицировать эфиры жирных кислот.
Объекты исследования. Образцы мяса различных видов и сортов, субпродукты 1 и II категорий различных животных, шпик и жир-сырец с различных анатомических участков, жиропродукты.
Материалы, реактивы и оборудование. Абсолютный метанол; хлороформ; раствор гидроксида калия массовой долей 10 % в абсолютном метаноле; гексан; раствор фенолфталеина в этаноле массовой долей 1 %; диэтиловый эфир; безводный сульфат натрия; раствор метилата натрия массовой долей 5 %; колба с воздушным холодильником; делительная воронка; конические колбы вместимостью 50 см3; склянка с притертой пробкой; песчаная баня; вакуум-сушильный шкаф и газовый хроматограф.
Методические указания. Важным показателем пищевой ценности жиров является их жирнокислотный состав. Такие полинена-сыщенные жирные кислоты, как линолевая (С18.2) и линоленовая (С18.3), не синтезируются в организме. Линолевая кислота является предшественником образующейся в организме арахидоновой кислоты (С20.4). Полиненасыгценные жирные кислоты относятся к незаменимым компонентам пищи. Они обладают витаминной активностью, принимают участие в регуляции многих процессов в организме и образовании клеточных мембран.
Хроматография — физико-химический метод разделения жидких или газообразных смесей, который основан на различии в распределении разделяемых веществ между двумя фазами. При анализе жиров методом хроматографии в качестве подвижных фаз используют жидкости или газы. Особую популярность получили газожидкостные хроматографы. Разделение анализируемых соеди-152
нении основано на различной рас i воримосги комионенюв газо вой смеси в жидкой неподвижной фазе, которая нанесена на твердый носитель, заполняющий колонку.
Исследуемые вещества движутся по колонке с помощью газа-носителя (азот, аргон, гелий). нс вступающего в реакцию с компонентами анализируемой пробы и не растворяющегося в жидкой фазе. Газ подается из баллонов постоянно и поддерживается на определенном уровне при помощи специального игольчатого вентиля. Скорость подачи газа измеряют, определяя время истечения заданного объема. Жидкая фаза в ГЖХ должна быть нелетучей, устойчивой к температуре, при которой производят анализ, иметь невысокую вязкость, хорошо растворять компоненты разделяемой смеси. В качестве жидкой неподвижной фазы часто используют органические соединения с высокой температурой кипения. Выбор фазы зависит от природы исследуемого вещества. При разделении соединений с равной полярностью и различными точками кипения используют неполярную жидкую фазу — сквалан, высоковакуумные смазки апиезон L и апиезон М. Для разделения веществ с различной полярностью (жирных кислот, спиртов) применяют полярные жидкости — полиэтиленгликоли, сложные эфиры углеводов и производные этилендиаминов.
Неподвижную жидкую фазу закрепляют на инертном гранулированном твердом носителе и помещают в узкую стальную или стеклянную колонку, через которую пропускают подвижную фазу. Хроматографическая колонка представляет собой спиральные трубки диаметром 0,2—1 см и длиной 1—20 см.
Твердый носитель жидкой фазы должен быть химически инертным по отношению к анализируемому веществу, обладать относительно большой поверхностью (оптимальная поверхность носителя 5—20 м2/г). Чаще всего в качестве носителей используют природные алюмосиликаты, цеолит-545, силикагель (хромо-сорб), силоцел С-22 в форме шариков или пластинок.
Схема газожидкостного хроматографа приведена на рис. 1.39.
Рис. 1.39. Схема газожидкостного хроматографа:
1 — пламенно-ионизационный детектор; 2—усилитель; 3 —самопишущий потенциометр; 4— интегратор; 5 — газовый баллон с носителем; 6 — хроматографическая колонка; 7—регулятор температуры в термостате; 8— термостат
153
Гак как образец должен поступать в козонку в iазообразном виде, то между дозатором и колонкой устанавливают подогреватель. обеспечивающий мгновенное испарение образца, температура в котором поддерживается на 30—50 °C выше, чем температура в колонке.
Ввиду того что скорость продвижения компонентов смеси но хроматографической колонке зависит от температуры, колонку помещают в термостат (жидкостный, паровой, воздушный). Чаше всего используется воздушный термостат, позволяющий работать в области высоких температур.
Образующаяся газовая смесь попадает в верхнюю часть колонки и перемещается вдоль нес газом-носителем. Температура колонки поддерживается на уровне, обеспечивающем газообразное состояние анализируемой смеси. Выходящие из колонки компоненты попадают в детектор и регистрируются в виде полос на ленте самописца.
Сущность метода состоит в том, что каждый компонент разделяемой смеси при определенной температуре в парообразном состоянии с определенной скоростью с помощью газа-носителя перемещается по колонке, заполненной порошкообразным носителем, пропитанным неподвижной фазой (нелетучей жидкостью).
Хроматограмма смеси представляет собой кривую в виде ряда пиков, число которых соответствует числу разделяемых компонентов. Чем больше площадь пика, тем больше содержится данного вещества в пробе. Последовательность выхода компонентов зависит в основном от коэффициентов их распределения между жидкой неподвижной и газообразной подвижной фазами.
Количество вещества определяют по площади (иногда по высоте) пика на диаграммной ленте самописца. Площадь пика измеряют планиметрическим способом либо умножением высоты пика h на ширину Ь, которую он имеет на половине высоты (рис. 1.40). Некоторые газожидкостные хроматографы (ГЖХ) снабжены интегратора-
Рис. 1.40. Измерение площади пика
ми, которые измеряют относительное время удержания и определяют площади пиков.
При анализе смесей жирных кислот их переводят в метиловые эфиры, которые являются более летучими, чем свободные жирные кислоты, и не обладают способностью димеризоваться. Кроме того, при хроматографировании эфиров облегчается разделение насыщенных жирных кислот и ненасыщенных с тем же числом углеродных атомов. Эфиры насыщенных жирных кислот с прямой цепью углеродных атомов выходят из колонки в порядке повышения их температуры кипения. При постоянных условиях работы время и порядок выхода кислот из колонки всегда одинаковы.
154
Этот метод основан на предварительном кипячении пробы липидов в абсолютном метаноле с метилатом назрия с последующим экстрагированием полученного продукта диэтиловым эфиром.
При анализе глицерилов получают метиловые эфиры методом переэтерификации.
Подготовка проб. Сначала проводят предвари iельную экстракцию липидов. Для этого 40 г измельченного образна помешают в колбу с герметически закрывающейся пробкой, добавляют 130 см3 метанола, перемешивают и измельчаю! на гомогенизаторе в течение 1—2 мин до получения однородной массы. Затем к гомогенату добавляют 65 см3 хлороформа и встряхивают в течение 10 мин, после чего к смеси добавляют еще 65 см3 хлороформа и вновь встряхивают, но уже в течение 5 мин. К полученной смеси приливают 65 см3 дистиллированной воды и встряхивают в течение 30 с. Содержимое колбы фильтруют через бумажный фильтр под небольшим разрежением на воронке Бюхнера. Во время фильтрации желательно подавать струю диоксида углерода или азота на гомогенат, находящийся в воронке.
Остаток вместе с фильтром и небольшим кусочком фильтровальной бумаги, которым вытирают воронку, переносят в ту же смесительную колбу и повторно экстрагируют 100 см3 хлороформа в течение 10 мин. Смесь фильтруют в общую колбу. Колбу и остаток промывают 50 см3 хлороформа и весь фильтрат собирают в мерный цилиндр вместимостью 500 см3. Слои разделяют в делительной воронке, отбирают нижний хлороформный слой, выпаривают на роторном испарителе и получают жир, подлежащий дальнейшему анализу. Для этого жирные кислоты переводят в свободное состояние и получают их метиловые эфиры по одному из описаний, предложенных ниже.
1. Пробу липидов массой 0,5 г помещают в колбу вместимостью 150 см3, добавляют 10 см3 абсолютного метанола, вводят 0,05 см3 раствора метилата натрия массовой долей 5 %, присоединяют воздушный холодильник и кипятят на песчаной бане при температуре 75—80 °C в течение 1,5 ч. Затем отгоняют избыток этанола, содержимое колбы переносят в делительную воронку, ополаскивая колбу 2 раза 5 см3 диэтилового эфира. В воронку добавляют 2 см3 воды и 3 раза экстрагируют метиловые эфиры диэтиловым эфиром, каждый раз используя по 10 см3. Эфирные вытяжки промывают водой до нейтральной реакции по фенолфталеину, сушат безводным сульфатом натрия и фильтруют. Растворитель отгоняют, эфиры сушат в вакуум-сушильном шкафу при комнатной температуре, а затем хранят в герметически закрытых склянках на холоде в темноте.
2. Пробу липидов массой 0,1 г помещают в толстостенную колбу с притертой пробкой, добавляют 5 см3 абсолютного метанола и 0,22 см3 раствора гидроксида калия в абсолютном метаноле массовой долей 10 %. Колбу закрывают пробкой, которую прочно при-
155
кручпваюг медной проволокой, помешают на водяную оаню при температуре кипения и нагревают в течение 5—7 мин. После охлаждения в колбу добавляют 10см? воды. Полученную эмульсию количественно переносят в делительную воронку и метиловые эфиры экстрагируют гексаном (трижды по 20 см3). Объединенный гексановый экстракт промывают несколько раз водой порциями по 20 см3 до нейтральной реакции но фенолфталеину.
При метилировании жирных кислот в реакционную смесь можно добавить гидроксид калия из расчета 1.1 см3 раствора гидроксида калия в абсолютном метаноле массовой долей 10% на 0,1 г жира и содержимое нагреть в аналогичных условиях в течение 1 мин.
Порядок проведения анализа. В хроматограф вводят образец. Для этого пробу метиловых эфиров набирают в инъекционный шприц, прокалывают им колпачок дозатора и вводят содержимое. Момент ввода пробы фиксируется на нулевой линии.
Самописец прибора вычерчивает хроматограмму. Если в смеси отсутствует та или иная жирная кислота, то на хроматограмме будет отсутствовать соответствующий ей пик.
Для качественной идентификации параллельно при тех же условиях (скорость движения ленты, скорость газа-носителя, температура и т. д.) вычерчивают хроматограмму известной стандартной кислоты, чаще всего пальмитиновой.
В случае проведения анализа на хроматографе с плазменноионизационным детектором метиловые эфиры жирных кислот разделяют при следующих условиях: колонку длиной 3 м и внутренним диаметром 3 мм заполняют 15%-ным полиэтиленгли-кольсукцинатом на цеолите-545 с размером зерна 60—80 меш (число отверстий сита в одном квадратном дюйме), скорость газа-носителя (азота) 30 см3/мин. Разделение осуществляют в режиме линейного программирования температуры при скорости нагревания 4 °С/мин от 80 до 202 °C и в изотермическом режиме — при 202 °C. Пробу объемом 1—2 мкл вводят в хроматограф микрошприцем на 10 мкл.
Качественный анализ компонентов смеси метиловых эфиров жирных кислот липидов проводят, используя метчики идентифицированных эфиров чистых кислот. Количество каждого компонента в данной пробе определяют следующим образом. На хроматограмме проводят базовую линию, соединяющую основания всех пиков. Затем измеряют высоту каждого пика и его ширину на половине высоты. После этого вычисляют площадь пика (чаще по площади треугольника, можно использовать и другие способы расчета площадей пиков). Сумму площадей всех пиков метиловых эфиров кислот принимают за 100%. Зная, сколько образца было введено в колонку хроматографа, можно рассчи-156
тать количесзво каждой кислоты, содержащейся в данном образце. но формуле
(1.29)
где 5(1 — площадь пика определяемого компонента; Z5tl —сумма площадей пиков всех компонентов.
Полученные экспериментальные данные фиксируют в тетради, анализируют результаты, формулируют заключение по работе. Экспериментальные данные рекомендуется оформить в виде таблиц, формы которых приведены ниже.
Образец, источник получения Жирная кислота Формула Массовая доля, % к сумме
Жирные кислоты, % к сумме Объекты исследования
1 2 3 4 5 6
Насыщенные (И)
Мононенасыщенныс (М) Полиненасыщепные (П) Соотношение Н : М : П Неидентифицированные компоненты
Лабораторная работа № 24 КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГЛИКОГЕНА В ЖИВОТНЫХ ТКАНЯХ
Цель работы. Приобрести практический навык количественного определения гликогена в животных тканях.
Задачи. Получить гликоген из анализируемых образцов тканей; провести качественную реакцию с антроновым реактивом; определить светопоглощение окрашенных растворов с последующим определением концентрации гликогена по калибровочному графику.
Объекты исследования. Мышечная ткань и печень крупного рогатого скота.
Материалы, реактивы и оборудование. Растворы гидроксида калия массовыми долями 30 и 50 %; растворы этанола массовыми
157
.юлями 9>. 9(1. (я) и 80 с'(: э<|)ир: концентрированная Н-,80, (х. я.): антрон; микоген (или глюкоза); свежеприготовленный раствор антрона; центрифуга лабораторная; сифон; стеклянные пипетки; водяная баня, стеклянные пробирки; дел; спектрофотометр или фото эл е кт р о к о л о р и м е т р.
Приготовление реактивов. Р а с г в о р а н г р о н а: 0,2 г антрона помещают в мерную колбу вместимостью 100 см3, растворяют и доводят до метки раствором серной кислоты массовой долей 95 %. После выдержки в течение 1 ч раствор готов к применению.
Методические указания. Количество гликогена в свежих мышцах указывает на упитанность животного, а динамика количественного изменения гликогена в процессе хранения и переработки свидетельствует о глубине автолитических превращений.
Количество гликогена легко определить по цветной реакции с антроном. Метод сравнительно легкий и пригоден для массовых определений.
Метод состоит из следующих основных операций: гидролиза белков щелочью, выделения гликогена из раствора этанолом, промывания гликогена и его растворения, реакции с антроном (развитие окраски) и измерения интенсивности окраски.
При нагревании образцов тканей с концентрированным раствором щелочи происходит гидролиз белков, при этом гликоген выделяется из клеток. Гликоген не растворяется в этаноле, он выпадает в осадок при добавлении довольно большого его объема. Добавление нескольких капель концентрированной серной кислоты способствует осаждению гликогена вследствие образования сульфата аммония.
Нагревание промытого осадка гликогена с антроном (высокоспецифичным реактивом на углеводы), растворенным в концентрированной серной кислоте, приводит к гидролизу гликогена до глюкозы вследствие каталитического действия серной кислоты и к развитию окраски при реакции между глюкозой и антроном по схеме
По интенсивности окраски судят о количестве глюкозы. Развившаяся окраска пропорциональна количеству взятой глюкозы в пределах от 10 до 100 мкг в пробе.
158
Окрашенный от зеленого ло сине-зеленого цвета продукт реакции имеет максимум поглощения при длине волны 620 нм. Интенсивность окраски раствора измеряют на спектрофотометре или фотоэлектроколориметре с красным светофильтром с максимумом пропускания при той же длине волны.
Подготовка проб. При использовании в качестве объекта исследования мышечной ткани в центрифужные пробирки размером 25 х 160 мм вносят 5 см3 раствора гидроксида калия массовой долей 50 % и около 2.5 г пробы, взвешенной на технических весах с точностью до ± 0,01 г. Пробирки помещают в специальный штатив, вместе с которым их ставят в водяную баню при температуре кипения. После образования однородного раствора (после того как все частички мяса растворились) пробирки охлаждают, добавляют 5 см3 дистиллированной воды, осаждают гликоген 40 см3 этанола 96 об.% и вносят несколько капель концентрированной серной кислоты.
При анализе печени крупного рогатого скота в пробирку из молибденового стекла (или стекла пирекс) размером 20 х 150 мм помещают навеску тщательно измельченного образца массой 1,0 г, добавляют 30 см3 раствора гидроксида калия массовой долей 30 % и нагревают на сильно кипящей водяной бане в течение 20 мин.
По истечении времени пробирку охлаждают в холодной воде, содержимое количественно переносят в мерную колбу и доводят объем дистиллированной водой до метки. Вместимость мерной колбы подбирают в зависимости от возможного содержания гликогена в печени. Если анализируют измельченную при охлаждении печень только что убитого животного, то выбирают колбу вместимостью 200—500 см3, для анализа печени, хранившейся длительное время, используют колбу вместимостью 25— 50 см3. Гидролиз ведут в течение 24 ч.
Порядок проведения анализа. По истечении 24 ч содержимое пробирок центрифугируют в течение 30 мин при частоте вращения 50 с-1, жидкость над осадком сливают тонким сифоном или осторожно удаляют пипеткой. Осадок последовательно промывают 15 см3 раствора этанола с объемными долями 60, 80 и 96 %, а затем эфиром.
Для отделения промывных жидкостей пробирки с содержимым центрифугируют 30 мин. После каждого центрифугирования жидкость над осадком сливают, пользуясь вышеуказанным приемом. При обработке эфиром центрифугирования не требуется, после 10 мин настаивания эфир сливают из пробирки, а его следы удаляют на водяной бане.
Каждую порцию промывной жидкости добавляют не сразу, а в два приема. Сначала вносят небольшую часть жидкости и тщательно распределяют в ней осадок, перемешивая его стеклянной палочной, а затем добавляют оставшийся объем, тщательно смывая в пробирку приставшие к палочке частицы.
159
После удаления следов эфира к остатку юбавлякн 5см? юрячси волы, раствор перемешивают вращательным движением пробирки и нейтрализуют соляной кислотой по лакмусу (сначала двумя каплями концентрированной H0SO4, а затем H2SO4 массовой долей 2 %).
Из пробирки раствор гликогена переносят в мерную колбу соответствующей вместимости (см. подготовку проб). Для проведения цветной реакции в две пробирки молибденового стекла размером 30x200 мм помещают аликвотные части раствора гликогена (например, 0,5 и 1,5 см3), доливают до 5 см3 дистиллированной водой и погружают в водяную баню со льдом. Осторожно, непрерывно помешивая, приливают тонкой струей 10 см3 свежеприготовленного раствора антрона. Затем жидкости еще раз осторожно перемешивают вращательным движением и пробирки помещают на 10 мин в сильно кипящую водяную баню. По окончании нагревания пробирки быстро охлаждают в воде со льдом и измеряют оптическую плотность по отношению к контрольному раствору. Образовавшаяся зеленая окраска при использовании реактивов высокой чистоты устойчива в течение 4—5 ч.
Контрольный опыт проводят при аналогичных условиях, помещая в пробирку 5 см3 дистиллированной воды и 10 см3 раствора антрона.
Оптическая плотность контрольного раствора, измеренная относительно серной кислоты, не должна превышать 0,08.
Построение калибровочного графика. Готовят стандартный раствор на основе тщательно очищенного и высушенного в эксикаторе гликогена, полученного из тканей только что убитого кролика, или пользуются химически чистым препаратом глюкозы. Для перевода глюкозы в гликоген результаты определения делят на 1,11.
Навеску гликогена массой 16 мг растворяют в теплой дистиллированной воде в мерной колбе вместимостью 200 см3, охлаждают до комнатной температуры, доводят объем до метки и тщательно перемешивают. 1 см3 стандартного раствора содержит 0,08 мг гликогена. Из раствора готовят серию рабочих растворов для построения калибровочного графика. Рекомендуемые объемы стандартного раствора гликогена для построения калибровочного графика приведены ниже.
Масса гликогена, мг Объем,см’
раствора гликогена | воды антрона
0.01 0,125 4,875 10,0
0,02 0,25 4,75 10,0
0,04 0,50 4.50 10,0
0,06 0,75 4,25 10,0
0,08 1,00 4,00 10,0
0,12 1,50 3,50 10,0
0,16 2,00 3,00 10,0
0,20 2,50 2,50 10,0
160
Для построения калибровочного графика необходимо провести не менее трех серий определении, используя каждый раз вновь приготовленный раствор гликогена или пчюкозы.
При помощи калибровочного графика находят концентрацию гликогена во взятом на цветную реакцию растворе, а затем пересчитывают на содержание его в мясе (мг %) по формуле
а -25 100
Ут
(1.30)
где а — содержание гликогена по калибровочному графику, мг: 25 - - вместимость мерной колбы, в которой был растворен осадок гликогена; 100— множитель перевода в проценты; И—объем исследуемого раствора, взятого на цветную реакцию, см’: т — масса навески мяса. г.
Полученные экспериментальные и расчетные данные сводят в таблицу:
Образец Используемый метод определения 01 п плеская плотность раствора Содержание гликогена ио калибровочному графику Массовая доля гликогена в образце, %
Опираясь на теоретические знания, самостоятельно делают выводы и формулируют заключение.
Лабораторная работа № 25 КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ МОЛОЧНОЙ КИСЛОТЫ
Цель работы. Приобрести практический навык определения количества молочной кислоты.
Задачи. Выделить молочную кислоту из мышечной ткани методом экстракции; провести качественные реакции; определить оптическую плотность окрашенных растворов и рассчитать массовое содержание молочной кислоты в исследуемом образце.
Объекты исследования. Мясо или мышечная ткань различных видов животных разных сроков хранения.
Методические указания. В аналитической практике применяются различные методы определения молочной кислоты, наиболее распространенными среди которых являются методы, основанные на качественных реакциях с последующим фотометрированием.
Метод определения по цветной реакции с вератролом является сравнительно быстрым и пригодным для массовых определений. Он состоит из следующих основных операций: осаждения белков, углеводов, нагревания с серной кислотой, развития цветной реакции с вератролом, измерения интенсивности окраски.
161
Белки осаждаю] раствором митафосфорнол кислота массовой долей 5/с. Промежуточные продукты распада белков мсгафос форной кислотой не осаждаются.
Для осаждения углеводов и отчасти промежуточных продуктов, распада белка применяют сульфат меди и гидрат окиси кальция, углеводы осаждаются в виде сахаратов.
При нагревании с концентрированной серной кислотой молоч ная кислота превращается в ацетальдегид:
СН; СН;
СНОН + H2SO4 —► с = о
соон н
Образовавшийся ацетальдегид при реакции с вератролом дает соединение красного цвета.
Окрашенный в красный цвет продукт реакции имеет максимум поглощения при длине волны 520 нм. Интенсивность окраски раствора измеряют на спектрофотометре или фотоэлектроколориметре со светофильтром с максимумом пропускания при вышеуказанной длине волны (зелено-желтый или зеленый).
Фильтрат должен быть прозрачным. Для того чтобы убедиться в полном удалении белков, можно поместить несколько кубических сантиметров фильтрата в пробирку и добавить к нему равный объем раствора сульфосалициловой кислоты массовой долей 20 % (в присутствии следов белка появляется муть).
Метод количественного определения молочной кислоты с л-оксидифенилом основан на измерении интенсивности окраски соединения, образующегося в процессе реакции ацетальдегида с л-оксидифенилом в присутствии серной кислоты.
Белки удаляют осаждением трихлоруксусной кислотой, а углеводы — гидроксидом кальция в присутствии сульфата меди; уксусный альдегид, образующийся из молочной кислоты при нагревании с серной кислотой, дает в присутствии меди цветную реакцию с л-оксидифенилом (окрашивается в фиолетовый цвет).
Ацетальдегид образуется при нагревании молочной кислоты с минеральными кислотами. При его взаимодействии с двумя молекулами «-оксидифенила образуется 1,1-ди(оксидифенил)этан, который в присутствии H2SO4 окисляется в продукт фиолетового цвета с максимумом поглощения при длине волны 574 нм. Состав окрашенного производного неизвестен.
162
СНа'НОШ'ООН
СЖСНО + 2НО
1,1 - - дп(окспд11феш i.i )эт;ш
Метод позволяет определить молочную кислоту в количествах от 0.03 до 0,2 мкмоль в пробе.
1. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МОЛОЧНОЙ КИСЛОТЫ ПО ЦВЕТНОЙ РЕАКЦИИ С ВЕРАТРОЛОМ
Материалы, реактивы и оборудование. Свежеприготовленный раствор метафосфорной кислоты массовой долей 5 %; насыщенный на холоде раствор медного купороса и затем разведенный водой в соотношении 1:1; растертый в порошок гидроксид кальция; раствор вератрола массовой долей 0,125 % в растворе этанола 96 об.%; концентрированная серная кислота (пригодность последней устанавливают по отсутствию желто-зеленой окраски при стоянии в течение нескольких минут смеси, состоящей из 3 см3 кислоты с 0,1 см3 спиртового раствора вератрола массовой долей 0,125 %); молочная кислота или лактаты (цинка, лития) для построения калибровочного графика; раствор сульфосалициловой кислоты массовой долей 20%; спиртовой раствор а-нафтола массовой долей 10%; колбы Эрленмейера вместимостью 50 см3; бумажные фильтры; стеклянные пробирки; штатив; микропипетки; спектрофотометр.
Подготовка проб. Мясо или мышечную ткань предварительно измельчают и помещают в стакан или бюкс с притертой крышкой. Навеску образца берут из стакана или бюкса по разнице масс с точностью до ±0,1 г и помещают в мерную колбу вместимостью 100 см3, в которую добавляют 40 см3 дистиллированной воды, закрывают пробкой и встряхивают в течение 5 мин для равномерного распределения мяса и жидкости. Затем в ту же колбу добавляют 10 см3 раствора свежеприготовленной метафосфорной кислоты массовой долей 5 %, хорошо встряхивают, доводят объем водой до метки и через 15 мин фильтруют.
163
Порядок проведения анализа. Фильтра! обьемом 10 см? помешают в колбочку Эрленмеиера вместимостью примерно 50см? и добавляю! к нему 2.5 см3 раствора медного купороса и 3 г растертого в порошок гидроксида кальция. Смесь должна приобрести интенсивный бирюзовый цвет. При зеленовато-голубой окраске, указывающей на недостаточную щелочную реакцию, добавляют еще небольшое количество гидроксида кальция. Смесь оставляют на 1 ч.
время от времени взбалтывая.
По истечении 1 ч жидкость отфильтровывают от осадка через бумажный фильтр. Фильтрат должен быть прозрачным и бесцветным. С целью проверки полноты осаждения углеводов проводят реакцию с а-нафтолом. Для этого несколько кубических сантиметров фильтрата помещают в пробирку, добавляют одну каплю раствора а-нафтола массовой долей 10% и наслаивают 1 см3 концентриро-
ванной серной кислоты, в присутствии сахара на границе появляется красно-фиолетовое кольцо. В случае положительной реакции измеряют количество фильтрата и вновь повторяют осаждение, используя уже половину объема раствора медного купороса и полови
ну гидроксида кальция по отношению к первоначальным объемам. Избыток медного купороса влияет на устойчивость окраски.
Для реакции с серной кислотой берут пипеткой точно отмеренный объем фильтрата, переносят в пробирку размером 30 х 200 мм из молибденового (или термоустойчивого) стекла. Как правило, берут 0,05—0,5 см3 фильтрата в зависимости от содержания мо-
лочной кислоты в мясе. Небольшие объемы фильтрата (0,05— 0,1 см3) отбирают микропипеткой. Если исследуют парное мясо, то для реакции можно использовать 0,5 см3 фильтрата если исследуют мясо после суточного созревания — 0,05—0,1 см3.
Если для анализа взято менее 0,5 см3 фильтрата, то его доводят дистиллированной водой до объема 0,5 см3. Пробирки помещают в
ледяную воду и по каплям при встряхивании приливают из микробюретки 3 см3 концентрированной серной кислоты. Затем пробирки помещают в водяную баню при температуре кипения на 4 мин (по секундомеру), после чего охлаждают в ледяной воде 2 мин.
Пробирки ставят в штатив и в каждую микропипеткой добавляют 0,1 см3 вератрола. По прошествии 20 мин измеряют оптическую плотность раствора, окрашенного в розовый цвет, на спектрофотометре при длине волны 520 нм по отношению к воде в кювете с толщиной слоя 1 см. Контрольный раствор готовят одновременно с опытным, заменяя фильтрат, содержащий молочную кислоту, соответствующим объемом воды. Измерение проводят также по отношению к воде.
Одновременно можно проводить анализ 5—6 проб мяса, что составит с параллельными определениями 10—12 пробирок.
Цветная реакция с вератролом достаточно устойчива. Фильтраты после осаждения белков можно хранить в холодильнике при 4 °C в течение нескольких дней.
164
При расчете из величины оптической плотности опытною раствора вычитают величину оптической плотности контрольного раствора. Затем по калибровочному графику находят концентрацию молочной кислоты в 3,6 см3 опытного раствора.
Построение калибровочного графика. Готовят стандартный раствор на основе молочной кислоты или ее солей (лактата лития или цинка), содержащий 0.25 мг молочной кислоты в 1 см3. Каждую из солей берут из расчета на молочную кислоту, например, при использовании лактата цинка (CH3CHOHCOO)?Zn навеску массой 169 мг растворяют в воле в мерной колбе вместимостью 500 см3.
Для построения графика готовят серию растворов. Рекомендуемые объемы раствора молочной кислоты для построения калибровочного графика приведены ниже.
Maccci молочной кислоты, мг Объем, ем' расгвора молочной | oj, киоють1 кислоты |
0,100 0,40 0,10 3,0
0,080 0.32 0,18 3,0
0.060 0,24 0,26 3,0
0,040 0.16 0,34 3,0
0,020 0,08 0,42 3.0
0,010 0,04 0,46 3,0
Опыты повторяют три раза. Каждый раз готовят новый стандартный раствор. Для всех растворов в пробирках каждой серии получают величины оптической плотности, по которым после вычитания показаний контрольного опыта (все спектральные измерения делают в сравнении с водой) строят калибровочный график зависимости оптической плотности от концентрации.
Содержание молочной кислоты (мг %) рассчитывают по формуле
п-12,5-100-100
V 10m
(1-31)
где а — содержание молочной кислоты в 3,6 см3 опытного раствора, мг; 12,5 — объем раствора, обработанный гидроксидом кальция, см3; 100 — объем, в кото-Ром содержится навеска мяса и лге/иа-фосфорная кислота; 100 — коэффициент перевода в проценты; V— объем фильтрата после осаждения углеводов, взятый на цветную реакцию, см3; 10 —объем фильтрата после осаждения белков л/е/иа-фосфорной кислотой, взятый на осаждение углеводов, см3; т — масса навески мяса, г.
165
2. МЕТОД ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ МОЛОЧНОЙ КИСЛОТЫ ПО ЦВЕТНОЙ РЕАКЦИИ С ЛАРА-ОКСИДИФЕНИЛОМ
Материалы, реактивы и оборудование. /7щ9<7-оксидифснил; гидроксид натрия (х. ч.); реактив /ш/л7-оксидифенила; растворы тиосульфата меди массовыми долями 4 и 20%; порошок гидроксида кальция; концентрированная серная кислота (х. ч.); водный раствор трихлоруксусной кислоты массовой долей 10 %; лактат цинка (или лития) или титрованная молочная кислота; центрифуга лабораторная; пробирки стеклянные; водяная баня; микробюретки; спектрофотометр (фотоэлектроколориметр).
Приготовление реактивов. Реактив пар a-о к с и д и ф е -н и л а: раствор лд/?«-оксидифенила массовой долей 1,5 % в растворе гидроксида натрия массовой долей 0,5 %.
Подготовка проб. При использовании качественной реакции с //я/>я-оксидифенилом в ступку вносят 10 см3 раствора трихлоруксусной кислоты массовой долей 10% и 2—4 г измельченного мяса и растирают его в течение 10 мин. Образовавшуюся суспензию переносят в мерную колбу вместимостью 50 см3, используя для этого сначала 20 см3 раствора трихлоруксусной кислоты массовой долей 10%, а затем несколько кубических сантиметров дистиллированной воды.
Колбу оставляют на 30 мин при комнатной температуре, встряхивая через каждые 10 мин, затем объем доводят дистиллированной водой до метки, колбу закрывают пробкой, хорошо перемешивают содержимое, переносят в центрифужные пробирки, центрифугируют с частотой вращения 50 с-1 в течение 10—15 мин. Центрифугат сливают в сухую колбу, отбирают 25 см3 прозрачной жидкости, переносят в мерную колбу вместимостью 100 см3 и объем доводят дистиллированной водой до метки.
Порядок проведения анализа. Для осаждения углеводов к 2 см3 разбавленного центрифугата прибавляют 1 см3 раствора сульфата меди массовой долей 20 %, дистиллированной водой доводя объем жидкости до 10 см3, приливая воду пипеткой или из бюретки, добавляют 1 г растертого в порошок гидроксида кальция, энергично встряхивают, оставляют в покое на 30 мин, время от времени встряхивая, а затем центрифугируют. Центрифугат сливают в колбу.
Для проведения цветной реакции аликвотную часть (например, 1 см3 центрифугата) переносят в пробирку из молибденового стекла (или стекла пирекс) размером примерно 25 x 200 мм, добавляют 1 каплю раствора сульфата меди массовой долей 4 % и ставят в ледяную воду (обычно проводят ряд определений). При помешивании добавляют из микробюретки 6 см3 концентрированной серной кислоты, помещают пробирку на 5 мин в водяную баню при температуре кипения, после чего охлаждают в холодной воде до 20 °C.
166
В пробирку добавляют 0.1 см? раствора /ш/лт-оксидифенила. очень тщательно и осторожно перемешивают, после чего ставят пробирку на 30 мин в водяную баню при 30 °C. изредка слегка встряхивая. По прошествии этого срока пробирку помещают в сильно кипящую водяную баню на 90 с. затем охлаждают в холодной воде и измеряют интенсивность окраски, пользуясь спектрофотометром или фотоэлектроколориметром. В первом случае измерения проводят при длине волны 560 нм. во втором — со светофильтром с минимумом поглощения при той же длине волны. Прибор должен быть включен заранее, измерение проводят по отношению к контрольному раствору. Толщина кюветы 1 см.
Контрольный опыт с реактивами проводят, начиная с момента осаждения углеводов, для чего используют вместо 2 см3 центрифугата мышечной ткани 0,3 см3 раствора трихлоруксусной кислоты и 1,7 см3 дистиллированной воды. Строят калибровочный график.
Построение калибровочного графика. Для приготовления стандартного раствора можно пользоваться титрованным раствором молочной кислоты или растворами ее солей (цинковой, литиевой или другой). При использовании лактата лития навеску массой 133,3 мг переносят в мерную колбу вместимостью 500 см3, растворяют в 5 см3 серной кислоты, разбавленной в соотношении 1 : 10, объем доводят дистиллированной водой до метки (раствор № 1). 1 см3 раствора № 1 содержит 0,250 мг молочной кислоты. 20 см3 этого раствора пипеткой переносят в мерную колбу вместимостью 250 см3, и объем доводят дистиллированной водой до метки (раствор № 2). 1 см3 раствора № 2 содержит 0,020 мг молочной кислоты.
Для построения графика готовят серию растворов. Рекомендуемые объемы раствора № 2 для построения калибровочного графика приведены ниже.
Масса молочной кислоты, мкг Объем, см:
раствора № 2 воды
2 0,10 0,90
4 0,20 0,80
6 0.30 0,70
8 0,40 0,60
10 0,50 0,50
12 0,60 0,40
0 0 1.00
0 0 1,00
0 0 1,00
167
C ирт отопленными растворами проводят цветную реакцию. Измерение оптической плотности проводят но отношению к дистиллированной! воде. Среднюю арифметическую величину оптической плотности растворов последних трех пробирок (см. табл.) вычитают из величины оптической плотности каждого основного раствора. Данные наносят на миллиметровую бумагу, соединяя точки прямой линией.
По калибровочному графику находят концентрацию молочной кислоты в объеме раствора, взятом на цветную реакцию.
Содержание молочной кислоты в мясе (мг%) определяют по формуле
v_ с 10 100 -50 100
' 1.2.25,».100 ’ <'.32)
где с -- концентрация молочной кислоты, найденная по калибровочному графику в соответствии с полученной оптической плотностью, мг/см3; 10 —объем раствора при осаждении углеводов, см3; 100 — объем разбавленного центрифугата. см3; 50 — объем смеси при осаждении белков трихлоруксусной кислотой, см3; 100 — коэффициент перевода в проценты; 1 — объем раствора после осаждения углеводов, взятый для проведения цветной реакции, см3; объем разбавленного центрифугата, взятый для осаждения углеводов, см’; 25 — объем разбавленного центрифугата, взятый для разбавления, см3; т — масса навески мяса, г; 1000 — множитель для перевода в миллиграммы.
Ошибка метода составляет примерно ± 5 %.
Если точно следовать указанной методике, то можно пользоваться упрощенной формулой
Х= —-100. (1.33)
т
Полученные экспериментальные и расчетные данные оформляют в виде таблицы:
Объект Метод определения Оптическая плотность Концентрация молочной кислоты по калибровочному графику, мг/см3 Содержание молочной кислоты, мг%
Проанализировав результаты, студенты должны сделать выводы о длительности хранения и глубине созревания мяса, сформулировать общее заключение по работе.
168
Лабораторная работа № 26
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПЕКТИНА В МЯСНЫХ ПРОДУКТАХ
Цель работы. Приобрести практический навык определения пектина в мясопродуктах (или растениях).
Задачи. Получить и количественно определить растворимый пектин методом осаждения или фотометрически на основе качественной реакции.
Объекты исследования. Комбинированные (или лечебно-профилактические) мясопродукты, содержащие пектиновые вещества, а также различные источники пектиновых веществ (свекла, морковь, тыква, кабачки и т.д.).
Методические указания. В связи с актуальностью проблемы контроль количества балластных веществ, особенно при производстве комбинированных продуктов питания, приобретает особое значение.
В основе химической структуры пектиновых веществ лежит высокомолекулярная полигалактуроновая кислота, которая состоит из остатков галактуроновой кислоты, связанных кислородным мостиком между первым и четвертым углеродными атомами.
В растениях пектиновые вещества содержатся в виде протопектина, химическая природа которого еще недостаточно изучена. Полагают, что пектин связан с арабаном клеточной стенки, с целлюлозой и с ионами металлов; нерастворимый протопектин переходит в растворимый под влиянием разбавленных кислот или фермента протопектиназы. Растворимый пектин представляет собой сложный эфир полигалактуроновой кислоты и метилового спирта следующего строения:
СООСН, СООСН; соосн,
I I I
Растворимый пектин расщепляется разбавленными щелочами или при каталитическом воздействии фермента пектинэстеразы (пектазы). В результате гидролиза отщепляются метанол и пектиновая (полигалактуроновая) кислота. Пектиновая кислота расщепляется ферментом полигалактуроназой, в результате гидролиза которой образуется галактуроновая кислота.
169
В настоящей работе предлагается воспользоваться одним in двух методов определения пектина: путем осаждения по пектату кальция: по характерной для урановых кислот реакции с карбазолом в концентрированной серной кислоте. В первом случае предварительно проводят гидролиз пектиновых веществ с последующим осаждением полигалактуроновой кислоты в виде пектата кальция. Второй метод предусматривает действие концентрированной H2SO4 на пектиновые вещества с образованием фурфурола, который вступает во взаимодействие с карбазолом и образует окрашенное соединение. Применение бората способствует быстрому развитию окраски, повышая ее стабильность (сохраняется 16 ч), снижая влияние различных окислителей. Перед реакцией пектиновых веществ с карбазолом необходимо проводить деметоксилирование их раствором гидроксида натрия, что значительно повышает воспроизводимость и чувствительность анализа.
1.ОПРЕДЕЛЕНИЕ ПЕКТИНА ТЕРМОГРАВИМЕТРИЧЕСКИМ МЕТОДОМ ПО ПЕКТАТУ КАЛЬЦИЯ
Материалы, реактивы и оборудование. Раствор цитрата аммония массовой долей 10%; раствор NaOH молярной концентрацией 0,1 моль/дм3; раствор уксусной кислоты молярной концентрацией 1 моль/дм3; раствор хлорида кальция молярной концентрацией 2 моль/дм3; раствор AgNO3 молярной концентрацией 0,1 моль/дм3; колбы Эрленмейера вместимостью 250 и 500 см3; фарфоровая ступка; мерные колбы вместимостью 500 см3; водяная баня; шюттель-аппарат; центрифуга; обратный холодильник; бумажные фильтры; бюксы; сушильный шкаф; аналитические весы.
Подготовка проб. 25 г свежего материала растирают с битым стеклом в ступке до совершенно однородной массы и переносят в колбу Эрленмейера, заливают 100 см3 воды, нагретой до 45 °C (не выше), и выдерживают (при периодическом взбалтывании) 30 мин при указанной температуре на водяной бане. Затем колбу плотно закрывают каучуковой пробкой и энергично взбалтывают в течение 15—20 мин в шюттель-аппарате. После этого содержимое колбы центрифугируют и собирают прозрачный раствор пектина, который исследуют на желеобразуюшую способность. В плотном остатке определяют количество нерастворимого пектина (протопектина). При определении протопектина плотный остаток подвергают дополнительной двукратной обработке водой; второй раз заливают 75 см3 дистиллированной воды, третий — 50—60 см3. Промывные воды используют для добавочного извлечения из них растворимого пектина. Растворимый пектин определяют пектатным методом. В остатке определяют протопектин.
170
Промытый плотный остаток количественно переносят в коническую колбу с 50 см' раствора соляной кислоты молярной концентрацией 0.3 моль/дм'. Колбу соединяют с обратным холодильником и нагревают в течение 30 мин на водяной бане при температуре кипения. Гидролизат центрифугируют. плотную часть промывают горячей водой. Центрифугат и промывные воды собирают в мерную колбу вместимостью 500 см3.
Твердый остаток вместе с фильтром количесгвенно переносят в коническую колбу и заливают 50—70 см3 цитрата аммония массовой долей 10 % и помещают на 30 мин в водяную баню при температуре кипения. Фильтруют в ту же мерную колбу (вместимостью 500 см3). осадок промывают горячей водой и охлаждают. Содержимое колбы доводят водой до метки. Полученная вытяжка содержит протопектин и используется для количественного анализа.
Порядок проведения анализа. В колбу Эрленмейера или химический стакан вместимостью 500 см3 отмеряют пипеткой Мора 25 см3 фильтрата и приливают 100 см3 раствора NaOH молярной концентрацией 0,1 моль/дм3. оставляют на 30 мин. За это время происходит омыление растворимого пектина, который переходит в натриевую соль пектиновой кислоты. Затем добавляют 50 см3 раствора уксусной кислоты молярной концентрацией 1 моль/дм3 и получают свободную пектиновую (полигалактуро-новую) кислоту.
К полученной таким образом пектиновой кислоте через 5 мин прибавляют 50 см3 раствора хлорида кальция молярной концентрацией 2 моль/дм3 и оставляют на 1 ч. За это время выпадает осадок пектата кальция. Осадок промывают горячей водой на взвешенном фильтре до тех пор, пока не исчезнет реакция на хлор с каплей раствора AgNO3. Осадок вместе с фильтром помещают в бюкс и доводят до постоянной массы в сушильном шкафу при 100 °C.
При расчете массу осадка уменьшают на 8 %, т. е. вносят поправку на содержание в нем кальция. Содержание пектиновой кислоты (%) вычисляют по формуле
лИ-92 mV\
(1.34)
где а — масса пектата кальция. г; И—объем водного гидролизата протопектина (начальный объем водной вытяжки), см3; 92 — коэффициент пересчета в проценты (с поправкой на содержание кальция в пектате); т — масса навески исследуемого растительного материала, г; Г — объем фильтрата, взятого для омыления и осаждения в нем пектата кальция, см3.
Пример. Навеска моркови массой 50 г измельчена, перенесена в мерную колбу вместимостью 250 см3 и доведена водой до метки, т. е. получен объем И Для определения взято 25 см3 фильтрата (объем Г]). Масса пектата кальция после доведения до постоянной массы равна 0,0256 г.
171
Для определения массовой .толп пектина ('< ) полученные маннам лол--; линяю 1 в формулу
2. ОПРЕДЕЛЕНИЕ ПЕКТИНА ФОТОМЕТРИЧЕСКИМ МЕТОДОМ ПО РЕАКЦИИ С КАРБАЗОЛОМ
Материалы, реактивы и оборудование. Раствор соляной кислоты молярной концентрацией 0,3 моль/дм3; раствор цитрата аммония массовой долей I %; раствор NaOH молярной концентрацией 0,05 моль/дм3; раствор НС1 молярной концентрацией 0,05 моль/дм3; концентрированная H?SO4; раствор карбазола массовой долей 2 % в абсолютном этаноле; галактуроно-вая кислота; ступка фарфоровая; колбы конические вместимостью 100—250 см3; мерные колбы вместимостью 25, 250, 500 см3; водяная баня; пробирки стеклянные; сосуды со льдом; спектрофотометр.
Подготовка проб. Навеску свежего продукта (плодов, овощей) массой 10 г тщательно растирают со стеклом в ступке до однородной массы, переносят водой в коническую колбу или центрифужные пробирки, добавляют около 100 см3 нагретой до 50 °C воды и оставляют на 30 мин на водяной бане (лучше энергично взбалтывать в течение 15—20 мин, после чего центрифугировать), сливают жидкость через фильтр в мерную колбу вместимостью 250 см3. Остаток повторно заливают водой, промывают фильтр и полученный фильтрат доводят водой до метки. Получают вытяжку водорастворимых пектиновых веществ.
Остаток из центрифужных пробирок переносят раствором НО молярной концентрацией 0,3 моль/дм3 в коническую колбу вместимостью 50 см3, закрывают пробкой с обратным холодильником и помещают на 10 мин в водяную баню при температуре кипения. После этого смесь фильтруют через складчатый фильтр в мерную колбу вместимостью 250 см3, остаток промывают несколькими порциями воды. Фильтр и осадок переносят в экстракционную колбу, заливают 50 см3 раствора цитрата аммония массовой долей 1 % и выдерживают 30 мин на водяной бане при температуре кипения. Затем все смывают водой в ту же мерную колбу, фильтрат охлаждают и доводят водой до метки. При этом получается вытяжка, которая содержит нерастворимый протопектин и пектиновую кислоту.
В дальнейшем оба раствора анализируют одинаково.
Порядок проведения анализа. Для метоксилирования отбирают по 10 см3 вытяжки в мерную колбу вместимостью 50 см3, добавля-172
ют 10 см' раствора NaOH молярной концентрацией 0,05 моль/дм? и выдерживают при комнатной температуре 30 мин. после чего добавляют Нем- раствора НС1 молярной концентрацией 0,05 моль/дм3 и доводят водой до метки.
В три пробирки вместимостью 10 см3. помешенные в сосуды со льдом, отбирают по 0,5 см3 исследуемого экстракта и осторожно по каплям приливают 3 см3 концентрированной H2SO4 (не допуская перегрева смеси). Затем пробирки нагревают в течение 6 мин на водяной бане при температуре кипения и снова охлаждают льдом. В две пробирки добавляют по 0,1 см3 раствора карбазола массовой долей 2 % и все пробирки помещают на 10 мин в водяную баню при температуре кипения. После охлаждения измеряют оптическую плотность растворов в кювете слоем 0,5—1,0 см при длине волны 535 нм.
Контролем служит проба без карбазола.
Содержание пектиновых веществ рассчитывают по калибровочному графику.
Построение калибровочного графика. Готовят стандартный раствор галактуроновой кислоты и используют для получения разведений, обеспечивающих концентрацию от 0,5 до 50 мкг/см3. Затем проводят окрашивание каждого из разбавленных растворов в условиях, описанных в методе, измеряют оптическую плотность при длине волны 532 нм в кювете слоем 0,5—1,0 см и строят график D
Полученные расчетные и экспериментальные данные рекомен- • дуется свести в таблицу:
Образец
Метод определения
Массовая доля молочной кислоты, %
Затем студенты должны самостоятельно сделать выводы и сформулировать заключение.
Лабораторная работа № 27
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЦЕЛЛЮЛОЗЫ
Цель работы. Приобрести практический навык определения целлюлозы в комбинированных мясных продуктах и растительных материалах.
Задачи. Выделить целлюлозу (клетчатку) из образцов и рассчитать массовое содержание целлюлозы.
Объекты исследования. Мясорастительные продукты или растения.
Методические указания. Массовую долю целлюлозы определяют термогравиметрическим методом в двух вариантах.
173
Первый ; них (Bupnaii 1 А. И. Ермакова) основа!! на окис.киши и paciBopciiiii! ра личных bcihcci в. сон\ 1с гвуюпшх клетчатке. обработанных азо! нои кислоюй в л анонс и водным раствором щелочи. От.шчаен'я простотой. скоростью выполнения и достаточной точностью.
Сущность inoporo (вариант И. С. Лурье) сосюит в удалении кислого- и щелочерастворимых веществ и количественном определении остатка, который условно принимается за сырую клетчатку. Последняя содержит часть лигнина, гемицеллюлоз и пентозаны. В этом методе на результат анализа влияют многие факторы, поэтому для получения воспроизводимых результатов следует строго придерживаться описания метода.
1. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЦЕЛЛЮЛОЗЫ В МОДИФИКАЦИИ А. И. ЕРМАКОВА
Материалы, реактивы и оборудование. Раствор гидроксида натрия молярной концентрацией 0,1 моль/дм3; раствор уксусной кислоты молярной концентрацией 1 моль/дм3: раствор хлорида кальция молярной концентрацией 2 моль/дм3: смесь четырех объемов этанола с одним объемом азотной кислоты (относительной плотностью 1,4); эфир серный; раствор гидроксида натрия или калия массовой долей 1,25%.
Подготовка проб. При определении клетчатки в колбы вместимостью 200—300 см3 взвешивают по 1—3 г измельченного растительного продукта. Массу навески изменяют в зависимости от содержания в исследуемом материале целлюлозы. Если в растительном продукте массовая доля клетчатки 10—20 %, то берут навеску от 1,5 до 2 г, 40—50 % — от 1 до 1,3 г, менее 10 % — от 2,5 до 3 г.
Порядок проведения анализа. К навеске добавляют по 50 см3 смеси этанола и азотной кислоты, колбы соединяют с обратным холодильником на резиновой пробке и помещают на водяную баню при температуре кипения на 1 ч. Колбы в воду не погружают, кипение должно быть равномерным. По окончании нагревания дают осесть осадку, раствор декантируют на стеклянный фильтр, на поверхность которого насыпают 1—2 см3 мелкого толченого стекла и отсасывают. Фильтр вместе со стеклом перед фильтрованием предварительно просушивают. Жидкость декантируют очень осторожно, чтобы не взмутить осадка. К осадку, оставшемуся в колбе, прибавляют еще 50 см3 смеси этанола с кислотой и вновь нагревают в течение 30 мин. После вторичной декантации через тот же стеклянный фильтр осадок в колбе промывают маленькими порциями этанола (96 об.%). Затем к промытому осадку в колбе прибавляют 50 см3 гидроксида натрия массовой долей 1,25 % и смесь нагревают на плитке. Щелочной раствор вместе с осадком переносят на стеклянный фильтр, отсасывают и промы
174
вают два раза но 10— 15 см-1 горячей дистиллированной водой и 1—2 раза этанолом (96 об.9с). Стеклянный фильтр с чистой белой клетчаткой сушат до постоянной массы при 105 °C (обычно в течение 2 ч). Для ускорения сушки осадок после промывания этанолом рекомендуется промыть еще серным эфиром.
Массовую долю целлюлозы определяют по формуле
X = т1~ -юо.
т
где т, — масса сухого фильтра со стеклом и осадком целлюлозы, г; /ъ() •-- масса сухого фильтра со стеклом, г: т — масса навески объекта исследования, г.
Фильтрование следует вести в горячем состоянии, иначе образуются очень вязкие растворы. При замедлении фильтрования перемешивают стеклянный слой, который снимает с фильтровальной пластинки труднопроницаемый слой. Необходимо предотвращать просасывание воздуха через осадок, поэтому на фильтре всегда должен быть слой жидкости. Как только вся жидкость прошла через фильтр, отсасывание тотчас же прекращают. С целью наиболее полного удаления лигнина при обработке навески исследуемого материала к азотной кислоте добавляют раствор уксусной кислоты массовой долей 80 % и кристаллическую трихлоруксусную кислоту.
2. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЦЕЛЛЮЛОЗЫ В МОДИФИКАЦИИ И. С. ЛУРЬЕ
Материалы, реактивы и оборудование. Раствор серной кислоты массовой долей 1,25 %; раствор гидроксида натрия или калия массовой долей 5 %; раствор соляной кислоты массовой долей 10 %; этанол ректификованный; эфир этиловый; стеклянные пипетки; водяная баня; фильтры бумажные; колбы Эр-ленмейера; стаканы; электрическая плита; водоструйный насос; стеклянная воронка.
Подготовка проб. При определении массовой доли сырой клетчатки навеску растительного препарата массой 2—3 г берут с точностью ± 0,01 г и помещают в стакан вместимостью 500 см3, на котором отмечен уровень 200 см3.
Порядок проведения анализа. В стакан с пробой помещают 200 см3 раствора серной кислоты массовой долей 1,25 %, ставят на плитку и содержимое доводят до кипения. Суспензию кипятят при постоянном перемешивании стеклянной палочкой в течение 30 мин. По мере выкипания жидкости уровень в стакане поддерживают приблизительно постоянным, периодически добавляя кипящую дистиллированную воду. Затем суспензии
175
дают отстояться. Далее, собрав установку (рис. 1.41). с помощью водоструйного насоса отсасывают еще горячую жидкость.
Стеклянную воронку 6 диаметром около 5 см обтягивают шелковой сеткой 7и наклеивают на нее смоченный в горячей воде кружок фильтровальной бумаги, равный диаметру воронки. Изогнутой стеклянной трубкой 4 и толстостенной резиновой трубкой 5 воронку соединяют с колбой 3 для фильтрования, которую, в свою очередь, присоединяют к водоструйному насосу 7 толстостенной резиновой трубкой 2. Как только фильтр плотно пристанет к шелковой сетке, воронку переворачивают фильтром вниз и осторожно вводят в
Рис.1.41. Установка для определения массовой доли клетчатки
стакан до соприкосновения с поверхностью горячей жидкости. Жидкость отсасывают до тех пор, пока ее высота над осадком не будет равна примерно 10 мм. После этого пинцетом снимают фильтр с воронки, переносят его на внутреннюю стенку стакана и струей горячей дистиллированной воды промывают фильтр и стенки стакана от приставших к ним частиц осадка. Удалив из стакана промытый фильтр, в стакан до метки наливают горячую дистиллированную воду, размешивают в ней осадок, дают отстояться и отсасывают воду через фильтр еще раз. Операцию отмывания горячей дистиллированной водой повторяют еще три раза, каждый раз меняя фильтр.
В стакан с промытым осадком отмеривают 50 см3 раствора гидроксида натрия или калия массовой долей 5 %, доливают кипящей дистиллированной водой до метки и кипятят в течение 30 мин. За
тем жидкость над осадком отсасывают и промывают еще два раза дистиллированной водой. После последнего отсасывания воды осадок переносят с помощью дистиллированной воды, подкисленной 2—3 каплями раствора соляной кислоты, на высушенный при температуре 100—105 °C и взвешенный фильтр. Осадок на фильтре промывают последовательно дистиллированной водой, этанолом и эфиром до тех пор, пока фильтрат не станет бесцветным. Фильтр с осадком высушивают в предварительно взвешенном бюксе в сушильном шкафу при температуре 100—105 °C до постоянной массы.
Массовую долю целлюлозы — клетчатки (%) рассчитывают по формуле
К = т{ 100/т, (1.36)
где /??] — масса остатка на фильтре, г; т — масса навески объекта исследования, г.
176
Полученные эксперимснзальные и расчетые данные рекомендуется представить в виде таблицы:
Образец
1 Ктюльлемый ж-тол
Массовая лота целлюлозы. сс
i
Затем студентам необходимо самостоятельно сформулировать выводы и сделать общие заключения по работе.
Лабораторная работа № 28 ОПРЕДЕЛЕНИЕ ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ И ИХ ПРОИЗВОДНЫХ
Цель работы. Приобрести практический навык и освоить методы определения фосфорорганических соединений в мышечной ткани.
Задачи. Экстрагировать и определить массовую долю фосфорорганических соединений в анализируемых образцах на основании фотометрических методов.
Объекты исследования. Образцы мышечной ткани разных видов животных аналогичных анатомических участков на разных стадиях автолиза.
Методические указания. После убоя животного в мышечной ткани под воздействием ферментов органические фосфаты распадаются, в связи с чем уменьшается содержание органического фосфора и увеличивается количество более простых азотистых продуктов и неорганических фосфатов.
Фосфорорганические соединения входят в группу экстрактивных веществ и в технологии мясных продуктов являются пищевыми добавками, отвечающими за вкус. Важнейшими из них являются креатин, креатинфосфат и креатинин:
ОН
NH2
C=NH
N-CH3
СН2
СООН
Креатин
NH-Р=О
C=NH °Н
N—СН3
СН2
СООН
NH---
C=NH
N—СН3
сн2
со---
Креатинфосфат
Креатинин
177
К группе фосфорорганических соединении oiiiocaioi 1акжс АТФ и ее производные: АДФ. АМФ.
Принципы .методов определения фосфорсодержащих органических соединений основаны на способности неорганического фосфата давать цветные реакции.
Модифицированный кол ори.метрически и .метод Фиске — Су-барроу применяется при определении АТФ. креатининфосфата и других соединений.
Метод основан на измерении нарастания массовой концентрации неорганического фосфора в безбелковом фильтрате после кислотного гидролиза.
Неорганический фосфор определяют колориметрически. Суть метода состоит в том, что при обработке раствора, содержащего фосфор, молибдатом аммония в кислой среде образуется фосфор-номолибденовая кислота Н7| Р(Мо,О7)6].
Н3РО4 + ]2(NH4)MoO4 дй H7[P(Mo2O7)6| + (NH4)?SO4 + Н2О.
При взаимодействии с восстановителем фосфорномолибдено-вая кислота образует смесь комплексов, содержащих молибден в различных валентностях. Смесь растворима в воде и называется молибденовой синью.
Интенсивность синей окраски пропорциональна концентрации фосфора в растворе и может быть измерена фотоколориметрически.
Модификации метода связаны с применением различных восстановителей (амидола, аскорбиновой кислоты и др.) при соответствующей кислотности среды.
Приготовление реактивов. Раствор эйконогена массовой долей 0,25 % в 100 см3 воды растворяют 15 г NaHCO3, 0,5 г Na2CO3 и 0,25 г эйконогена. Раствор фильтруют, перед использованием разбавляют в 4 раза дистиллированной водой.
Подготовка проб. Образцы мышечной ткани помещают на 15— 20 мин в охлажденных металлических чашках в морозильную камеру холодильника, регулятор которого за 20—30 мин до начала опыта устанавливают на максимальный холод.
Из замороженной ткани готовят навески массой (1,0000 ± ± 0,0002) г и быстро растирают в предварительно охлажденных фарфоровых ступках с охлажденным кварцевым песком в соотношении 1 : 1(по массе). Гомогенат используют для определения АТФ и креатинфосфата.
1. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ АТФ
Материалы, реактивы и оборудование. Фарфоровая ступка с пестиком или гомогенизатор; кварцевый песок; штатив; пробирки центрифужные и мерные вместимостью 10 см3; настольная цент-178
рифуга: водяная баня: фотоэлскгроколоримшр: раствор зрихлор-уксусной кислоты (ТХУ) массовой долей 10 X; растворы ацетата ртути массовой долей 0.5 и 20 сс\ растворы соляной кислоты молярной концентрацией 0.1 и 2,0 моль/лмД спиртовые растворы фенолфталеина массовой долей 0.5 и 1 ci: раствор молибдата аммония массовой долей 2.5 % в растворе серной кислоты молярной концентрацией 2.5 и 5 моль/см3; раствор аскорбиновой кислоты массовой долей 0,1 %.
Порядок проведения анализа. Подготовленный образец помешают в мерную пробирку, доводят объем раствором трихлоруксусной кислоты массовой долей 10 % до метки, перемешивают, выдерживают 30 мин при (20 ± 5) °C и фильтруют через бумажный фильтр. В центрифужную пробирку помещают 2 см3 фильтрата, добавляют 0,5 см3 раствора ацетата ртути массовой долей 20 %, тщательно перемешивают и через 15 мин центрифугируют в течение 10 мин при частоте вращения 50 с-1.
Надосадочную жидкость осторожно сливают, а осадок дважды промывают, добавляя по 2 см3 раствора ацетата ртути массовой долей 0,5 %. Полученный осадок растворяют в 1 см3 раствора соляной кислоты молярной концентрацией 0,1 моль/дм3, доводят дистиллированной водой до объема 3 см3 и тщательно перемешивают.
По 1 см3 полученной смеси помещают в две мерные пробирки вместимостью 10 см3. Первую пробирку используют для определения легкогидролизуемой фракции фосфора (ЛГФ), вторую — для определения неорганического фосфора (НФ). В первую пробирку добавляют I см3 раствора соляной кислоты молярной концентрацией 2 моль/дм3 и нагревают на водяной бане при температуре кипения в течение 7 мин, охлаждают, добавляют каплю фенолфталеина и нейтрализуют раствором гидроксида натрия молярной концентрацией 5 моль/дм3 до появления едва заметного розового окрашивания.
В обе пробирки вносят по 1,2 см3 раствора молибдата аммония массовой долей 2,5 % в растворе серной кислоты молярной концентрацией 2,5 моль/дм3 и по 1 см3 раствора аскорбиновой кислоты массовой долей 0,1 % (в методе Фиске—Субарроу в качестве восстановителя применяется 1-амино-2-нафтол-4-сульфоновая кислота или эйконоген), доводят дистиллированной водой до метки и перемешивают. Через 20 мин содержимое каждой пробирки колориметрируют в фотоэлектроколориметре против каждой из контрольных проб при красном светофильтре (А. = 670 нм). Полученные значения оптической плотности фиксируют. Для расчета используют значения оптической плотности, соответствующие диапазону наибольшей чувствительности шкалы прибора. Количество АТФ определяют по калибровочному графику.
Построение калибровочного графика. Готовят стандартный раствор однозамещенного фосфата калия, 1 см3 которого содер-
179
Ain 0,025 Mi фосфора (0.1 u99 г КП JJ()4 помещаю! в мерную колбу вместимостью 1 дм1 и доводят объем дистиллированной водой до метки). В каждую из зрех мерных пробирок вместимостью Юсм; помещают 0,5; 1,0 и 2,0 см3 стандартного раствора КН?РО4 и проводя! те же реакции, что и для исследуемых проб. После развития специфического окрашивания растворы колориметрируют и строят график зависимости оптической плотности от концентрации н е о р г а н и ч е с к о г о ф о с ф о р а.
Массовую долю АТФ (мг%) рассчитывают по формуле
X = 10( £лгф« Ио ПЖЕнфШ V2K), (1.37)
где 10 — коэффициент пересчета в мг%; £ЧГФ и £Нф — экстинкции проб при определении фракций соответственно лсгкогйдролизуемого и неорганического фосфора; а — масса фосфора в 10 см3 стандартного раствора, мкг; ф ~ объем исследуемого раствора, см3; ф — 10 см3; И, — объем исследуемого раствора после растворения осадка, см3; К = 3 см3; т — масса навески мышечной ткани, г; К, —объем исследуемого раствора, взятого для осаждения АТФ. см3; И2 = 2см3; И) — объем исследуемого раствора, взятый для колориметрирования, см3; К = 1 см3.’
2. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ КРЕАТИНФОСФАТА
Материалы, реактивы и оборудование. Раствор ацетата ртути массовой долей 25 % в растворе уксусной кислоты массовой долей 2 %; раствор ТХУ массовой долей 6 %; раствор NH4OH массовой долей 10%; стандартный раствор КН2РО4, содержащий 2 мг фосфора в 1 см3 раствора (перед использованием разбавляют дистиллированной водой в 100 раз); раствор эйконогена массовой долей 0,25 %. Материалы и оборудование см. в п. 1 настоящей работы.
Порядок проведения анализа. В центрифужную пробирку помещают 2 см3 раствора ацетата ртути массовой долей 25 %, приготовленного на растворе уксусной кислоты массовой долей 2 %, и охлаждают ледяной смесью или в холодильнике.
К гомогенату мышечной ткани в охлажденной ступке добавляют 20 см3 охлажденного раствора ТХУ массовой долей 6 %, содержимое ступки перемешивают и выдерживают 5—7 мин.
Раствор фильтруют в предварительно охлажденные пробирки, 2 см3 прозрачного фильтрата переносят в подготовленную центрифужную пробирку с охлажденным раствором ацетата ртути, добавляют 1—2 капли раствора фенолфталеина и содержимое пробирки нейтрализуют раствором NH4OH массовой долей 10 % до появления слабого розового окрашивания, избегая добавления избытка аммиака. Пробирку оставляют на 15 мин в ледяной смеси (или холодильнике), периодически помешивая содержимое стеклянной палочкой, которую не вынимают из пробирки.
Параллельно готовят контрольную пробу в которой вместо трихлоруксусного фильтрата используют 2 см3 раствора ТХУ массовой долей 6 %.
180
Через 15 мин содержимое опытной и кон [рольной пробирок центрифугируют при частоте вращения 50 с 1 в течение 20 мин.
Надосадочную жидкость переносят в мерные колбы вместимостью 50 см3 и используют для определения креатинфосфата. В мерную колбу с надосадочной жидкостью из контрольной пробирки вносят 2 см3 стандартного раствора КН2РО4. Затем в обе колбы (опыт и контроль) одновременно добавляют по 2 см3 раствора молибдата аммония массовой долей 2,5 %, приготовленного на^ растворе серной кислоты концентрацией 5 моль/дм3, и 0,5 см3 раствора эйконогена (1 -амино-2-нафтол-сульфоновой кислоты).
Содержимое колб доводят дистиллированной водой до метки и через 30 мин фотоколориметрируют с красным светофильтром при А,= 670 нм против дистиллированной воды.
Массовую долю креатинфосфата (мг%) определяют по формуле
JT = (Пк2б - 100 20)/(Z)on/77 • 2), (1.38)
где DK и Don — оптическая плотность соответственно контрольной и опытной проб; b — масса неорганического фосфора в 2 см3 стандартного раствора, мг; Ь = 0,04 мг; 100 — коэффициент для пересчета в мг%; 20 — объем раствора ТХУ. см3; т — масса исходной навески мышечной ткани, мг; 2 — объем фильтрата, взятый для исследования, см3.
После измерения оптической плотности окрашенных растворов фосфорорганических соединений выполняют расчеты, а результаты оформляют в виде таблицы. Самостоятельно формулируют выводы и общие замечания по работе.
Образец
Массовая доля в мышечной ткани, мг% Выводы
АТФ креатинфосфата
Лабораторная работа № 29 ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ В МЯСЕ И МЯСНЫХ ПРОДУКТАХ
Цель работы. Приобрести навык в освоении экспресс- и арбитражного методов определения массовой доли влаги в мясе и мясных продуктах
Задачи. Определить массовую долю влаги в мясе и мясных продуктах термогравиметрическим методом.
Объекты исследования. Мясо и мясные продукты различных ассортиментных групп, включая копченые, варено-копченые, вареные, фаршированные колбасы, мясные хлеба, сосиски, сардель
181
ки. продукты из свинины, оаранииы. говядины, мяса и гиды, других видов скота, зельцы. студни, паштеты, фаршевые консервы и т. д.
Материалы, реактивы и оборудование. Этанол: электрическая мясорубка с диаметром отверстий решетки от 3 до 4 мм: шкаф сушильный электрический с терморегулятором: сушильный аппарат САЛ; весы лабораторные: баня водяная: стаканчики для взвешивания (бюксы) или бюксы металлические диаметром 50 мм и высотой 25—35 мм: эксикатор; палочки стеклянные: предварительно обработанный песок речной или кварцевый; устройство Я10-ФВУ.
Методические указания. Влажность продуктов — весьма важный показатель при оценке качества мясных продуктов, который влияет на сохранность, выход, консистенцию и другие технологические характеристики. В аналитической практике применяются различные методы и их модификации, в основе которых лежит гравиметрическое определение.
Приготовление песка. Песок просеивают сначала через сито с диаметром отверстий 1,5 мм, а затем через сито с диаметром отверстий 0,3 мм, промывают водопроводной водой до тех пор, пока вода перестанет мутнеть. Затем песок заливают двойным объемом разбавленной (1:1) соляной кислоты и выдерживают в течение суток, периодически перемешивая. После обработки кислотой песок промывают водой до нейтральной реакции промывных вод на лакмус, высушивают при 150—160 °C до постоянной массы и хранят в закрытой склянке.
Подготовка проб. Пробы продуктов освобождают от оболочек и измельчают.
Пробы колбасных изделий, вареных, варено-копченых, копчено-запеченных, запеченных и жареных продуктов, фаршевых консервов, а также соленого бекона два раза измельчают на бытовой или электрической мясорубке и тщательно перемешивают.
Пробы сырокопченых колбас дважды измельчают на электрической мясорубке или нарезают острым ножом на круглые ломтики толщиной не более 1 мм, после чего их режут на полоски и рубят ножом так, чтобы размер частиц пробы не превышал 1 мм, все тщательно перемешивают.
Пробы паштетов, студней и зельцев измельчают на бытовой или электрической мясорубке один раз и тщательно перемешивают.
Подготовленную пробу помещают в стеклянную банку с притертой пробкой вместимостью 200—400 см3, заполнив ее полностью, и хранят при температуре от 3 до 5 °C в течение 24 ч.
182
1. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ ПРИ ТЕМПЕРАТУРЕ (103 ±2) С
Порядок проведения анализа. В бюкс помешают песок в количестве. примерно в 2—3 раза превышающем навеску продукта, стеклянную палочку длиной немного большей диаметра бюкса (чтобы она нс мешала закрыть бюкс крышкой) и высушивают в сушильном шкафу в открытом бюксе при температуре (ЮЗ ± 2) °C в течение 30 мин. Затем бюкс закрывают крышкой, охлаждают в эксикаторе до комнатной температуры и взвешивают. Во взвешенный бюкс с песком вносят навеску продукта массой 5 г и повторно взвешивают. К содержимому приливают 5 см3 этанола и перемешивают стеклянной палочкой.
Помещают бюкс на водяную баню (80—-90 °C) и, помешивая палочкой, нагревают до исчезновения запаха этанола. Затем пробу высушивают в течение 2 ч в сушильном шкафу при температуре (103 ± 2) °C, охлаждают в эксикаторе и взвешивают.
Высушивание продолжают до постоянной массы. Каждое повторное взвешивание проводят после высушивания в течение 1ч при температуре (103 ±2) °C. Результаты двух последовательных взвешиваний не должны отличаться более чем на 0,1 % массы навески. Взвешивание проводят на весах с погрешностью не более ± 0,001 г. Массовую долю влаги рассчитывают по разнице массы проб:
где тх, т2 — масса бюкса с песком, стеклянной палочкой и навеской соответственно до и после высушивания, г; in — масса бюкса с песком и стеклянной палочкой после высушивания, г.
2. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ ПРИ ТЕМПЕРАТУРЕ (150 ±2) °C
Порядок проведения анализа. В бюкс помещают песок в количестве, примерно в 2—3 раза превышающем навеску продукта, стеклянную палочку и высушивают в сушильном шкафу при температуре (150 ±2) °C в течение 30 мин. Затем бюкс закрывают крышкой, охлаждают в эксикаторе до комнатной температуры и взвешивают. Далее в бюкс с песком вносят навеску продукта 3 г, взвешивают повторно, тщательно перемешивают с песком стеклянной палочкой и высушивают в сушильном шкафу в открытом бюксе при температуре (150 ± 2) °C в течение I ч. Затем бюкс закрывают крышкой, охлаждают в эксикаторе до комнатной температуры и взвешивают. Взвешивание проводят на весах с погрешностью не более 0,0002 г.
183
3. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ В АППАРАТЕ САЛ
Порядок проведения анализа. Перед началом работы сушильный аппарат САЛ прогревают в течение 10—15 мин при напряжении 150—200 В. После прогрева ламп устанавливают напряжение 100—105 В. что обеспечивает температуру в зоне сушки 135-140 °C.
В бюкс помещают песок в количестве, примерно в 2—3 раза превышающем навеску продукта, стеклянную палочку длиной несколько большей диаметра бюкса (чтобы она не мешала закрыть бюкс крышкой) и высушивают в сушильном аппарате САЛ при температуре 135—140 °C в течение 10 мин. Затем бюкс закрывают крышкой, охлаждают в эксикаторе до комнатной температуры и взвешивают. Во взвешенный бюкс с песком вносят навеску продукта 2 г, повторно взвешивают и перемешивают стеклянной палочкой. Затем бюкс помешают в аппарат САЛ и высушивают при температуре 135—140 °C в течение 20 мин, охлаждают в эксикаторе и взвешивают. Взвешивание проводят на весах с погрешностью не более ± 0,0002 г.
Расчет ведут по формуле (1.39).
4. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ В УСТРОЙСТВЕ Я10-ФВУ
Порядок проведения анализа. Во взвешенный со стеклянной палочкой бюкс вносят навеску продукта массой от 1,8 до 2,2 г, добавляют от 1,8 до 2,2 см3 дистиллированной воды, тщательно перемешивают стеклянной палочкой, равномерно распределяя содержимое по дну бюкса. Открытый бюкс с пробой устанавливают в держателе 10 блока высушивания 8 устройства Я10-ФВУ (рис. 1.42) и высушивают 16—18 мин при температуре (163 ± 2) °C и скорости движения воздуха (3,6 ±0,1) м/с. Затем бюкс извлекают из блока высушивания, закрывают крышкой, помещают в одну из секций блока охлаждения 12, охлаждают в потоке воздуха комнатной температуры в течение 5—7 мин при скорости движения воздуха (5 ± 1) м/с и взвешивают. Результаты фиксируют с точностью до четвертого десятичного знака.
Массовую долю влаги (%) вычисляют по формуле
(тх — т2) 100 т} — т0
(1-40)
где Wj —масса бюкса с песком, палочкой и навеской до высушивания, г; т2 — масса бюкса с песком, палочкой и навеской после высушивания, г; /и0 — масса бюкса с песком и палочкой, г.
184
Рис. 1.42. Общий вид (я) и устройство аппарата Я10-ФВУ (б):
/ — таймер; 2—сигнальная лампа; 3, б. 7—тумблеры вентилятора и регуляторов температуры и электрической сети; 4, 5—регуляторы вентилятора и температуры; 8— блок высушивания; 9— нагреватель; I0--держатель бюкса в блоке высушивания; /7 —бюкс; 12— блок охлаждения; 13 — вентиляторы; 14— изолирующая перегородка; 75—диффузор
За окончательный результат принимают среднее арифметическое двух параллельных определений.
Расхождение между результатами параллельных определений не должно превышать 0,5 %. Окончательный результат вычисляют с точностью до 0,1 %.
Экспериментальные данные оформляют в виде таблицы, анализируют полученные результаты и формулируют заключение по работе.
Образец, источник получения
Способ и условия определения показателя
Массовая доля влаги, %
Лабораторная работа № 30
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ВОДЫ
Цель работы. Освоить метод определения активности воды (аы) в мясе и мясных продуктах.
Задачи. Закрепить представления о роли воды как важнейшего компонента биосистем мяса и мясопродуктов; экспериментально определить величину показателя аы для разных образцов мясного сырья, вторичных продуктов убоя, мясных продуктов.
185
3. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ В АППАРАТЕ САЛ
Порядок проведения анализа. Перед началом работы сушильный аппарат САЛ прогревают в течение 10—15 мин при напряжении 150—200 В. После прогрева ламп устанавливают напряжение 100—105 В. что обеспечивает температуру в зоне сушки 135—140 °С.
В бюкс помещают песок в количестве, примерно в 2—3 раза превышающем навеску продукта, стеклянную палочку длиной несколько большей диаметра бюкса (чтобы она не мешала закрыть бюкс крышкой) и высушивают в сушильном аппарате САЛ при температуре 135—140 °C в течение 10 мин. Затем бюкс закрывают крышкой, охлаждают в эксикаторе до комнатной температуры и взвешивают. Во взвешенный бюкс с песком вносят навеску продукта 2 г, повторно взвешивают и перемешивают стеклянной палочкой. Затем бюкс помешают в аппарат САЛ и высушивают при температуре 135—140 °C в течение 20 мин, охлаждают в эксикаторе и взвешивают. Взвешивание проводят на весах с погрешностью не более ± 0,0002 г.
Расчет ведут по формуле (1.39).
4. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ ВЫСУШИВАНИЕМ В УСТРОЙСТВЕ Я10-ФВУ
Порядок проведения анализа. Во взвешенный со стеклянной палочкой бюкс вносят навеску продукта массой от 1,8 до 2,2 г, добавляют от 1,8 до 2,2 см3 дистиллированной воды, тщательно перемешивают стеклянной палочкой, равномерно распределяя содержимое по дну бюкса. Открытый бюкс с пробой устанавливают в держателе 10 блока высушивания 8 устройства Я10-ФВУ (рис. 1.42) и высушивают 16—18 мин при температуре (163 ± 2) °C и скорости движения воздуха (3,6 ±0,1) м/с. Затем бюкс извлекают из блока высушивания, закрывают крышкой, помещают в одну из секций блока охлаждения 12, охлаждают в потоке воздуха комнатной температуры в течение 5—7 мин при скорости движения воздуха (5 ± 1) м/с и взвешивают. Результаты фиксируют с точностью до четвертого десятичного знака.
Массовую долю влаги (%) вычисляют по формуле
(тх — т2) 100 т} — т0
(1-40)
где /г?) — масса бюкса с песком, палочкой и навеской до высушивания, г; — масса бюкса с песком, палочкой и навеской после высушивания, г; /и0 — масса бюкса с песком и палочкой, г.
184
Рис. 1.42. Общий вид (я) и устройство аппарата Я10-ФВУ (б):
/ — таймер; 2—сигнальная лампа; 3, б. 7—тумблеры вентилятора и регуляторов температуры и электрической сети; 4, 5—регуляторы вентилятора и температуры; 8— блок высушивания; 9— нагреватель; I0--держатель бюкса в блоке высушивания; /7 —бюкс; 72—блок охлаждения; 13 — вентиляторы; 14— изолирующая перегородка; 75—диффузор
За окончательный результат принимают среднее арифметическое двух параллельных определений.
Расхождение между результатами параллельных определений не должно превышать 0,5 %. Окончательный результат вычисляют с точностью до 0,1 %.
Экспериментальные данные оформляют в виде таблицы, анализируют полученные результаты и формулируют заключение по работе.
Образец, источник получения Способ и условия определения показателя Массовая доля влаги, %
Лабораторная работа № 30
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ВОДЫ
Цель работы. Освоить метод определения активности воды (аы) в мясе и мясных продуктах.
Задачи. Закрепить представления о роли воды как важнейшего компонента биосистем мяса и мясопродуктов; экспериментально определить величину показателя аы для разных образцов мясного сырья, вторичных продуктов убоя, мясных продуктов.
185
Сравнивают полученные результаты для различных видов мясных продуктов, самостоятельно формулирую! выводы и заключение по работе.
Контрольные вопросы и задания
1. В чем состоят биологические функции белков'.’
2. Какова технологическая функциональность белков в производстве мясных продуктов?
3. Как разделяют белки мяса и мясопродуктов по морфологическому признаку клеток животных тканей?
4. Охарактеризуйте фракционный состав белков мышц.
5. Какие белки мышечной ткани относятся к водорастворимым, солерастворимым, щелочерастворимым?
6. Каковы физико-химические свойства и структурные признаки белков различных фракций? Чем обусловлены их прижизненные функции и каково их пищевое значение?
7. Какие функции выполняют миофибриллярные белки?
8. Какова роль соединительнотканных белков в рационах?
9. Как можно разделить основные белковые фракции мышечной ткани?
10. Какие гистидинсодержащие дипептиды являются специфической составной частью скелетной мускулатуры? В чем заключаются их технологическое значение и влияние на пищевую ценность мяса и мясопродуктов?
11. Каков морфологический и химический состав крови?
12. Дайте биохимическую характеристику крови.
13. Какое пищевое значение имеет кровь?
14. Дайте характеристику гемоглобина.
15. Каким способом можно выделить гемоглобин из крови?
16. Какие методы определения белков применяют в аналитической практике? Дайте их сравнительную оценку, укажите преимущества и недостатки.
17. Перечислите хроматографические методы определения белков и белковых веществ.
18. В чем сущность анализа белков методами гель-хроматографии, ионообменной хроматографии, хроматографии на бумаге, тонкослойной хроматографии?
19. Какими методами можно определить свободные аминокислоты и связанные в структуре белков и пептидов?
20. Каковы особенности подготовки проб для количественного определения аминокислот?
21. Каковы биологические функции липидов?
22. Охарактеризуйте методы практического определения суммарных липидов в животных тканях.
23. В чем состоит принцип определения суммарных липидов методом Сокслета?
24. Какова химическая природа холестерина? Ответ подтвердите структурной формулой, уравнениями реакций.
25. В чем состоит сущность определения холестерина в животных тканях?
26. Перечислите качественные реакции, характерные для холестерина.
27. Охарактеризуйте физиологические функции стеролов (на примере холестерина).
28. Почему необходимо контролировать содержание холестерина в рационах?
29. Каковы прижизненные функции и технологическое значение гликогена и продуктов его распада?
30. На чем основаны методы качественного и количественного определения гликогена, молочной кислоты?
188
31. Назовите полисахариды. перспективные пая применения в технологии мясных продуктов обшего и специального назначения в качестве функциональных и физиологически активных добавок.
32. Назовите важнейшие фосфорорганические соединения животных тканей. Какова их роль при жизни и в послсубойный период?
33. Какими методами определяют фосфорорганические соединения?
34. Перечислите и охарактеризуйте формы связи влаги в мясных продуктах.
35. Какими методами можно определить массовую долю влаги в мясе и мясных продуктах?
36. Что такое показатель активности воды9
37. Как показатель активности воды можно использовать для прогнозирования стабильности свойств мяса и мясных продуктов при хранении?
Глава 2
ФИЗИЧЕСКИЕ, ФИЗИКО-ХИМИЧЕСКИЕ И СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА МЯСА И МЯСНЫХ ПРОДУКТОВ
В технологических процессах продукты подвергаются внешним воздействиям, интенсивность которых зависит от сопротивляемости сырья, т. е. его физических характеристик. Величины сопротивляемости особенно важны при проведении процессов с использованием высококонцентрированных источников энергии (инфракрасный и высокочастотный нагревы, высокоскоростная механическая обработка, ультразвук, обработка давлением и др.).
Характеристика продукта складывается из комплекса физических свойств. Отдельные свойства, например электропроводность, не отражают поведения материала даже в простейшем процессе электроконтактного нагрева. Поэтому для эффективного решения технологических задач необходимо знание динамики изменения структурно-механических, биохимических и других свойств продукта.
Всестороннее изучение свойств сырья, полуфабрикатов и готовой продукции, т. е. одновременное исследование структурно-механических, физико-химических, электрофизических, биохимических, микробиологических, гистологических и других характеристик, необходимо при обязательной оценке пищевой ценности. Только путем сопоставления и совместного рассмотрения полученных данных можно получить ответ на вопрос о возможности применения на практике новых способов обработки животного сырья, имеющего столь сложный состав и пищевое назначение.
Комплексное исследование свойств мясопродуктов необходимо при обосновании новых физических способов обработки, позволяющих интенсифицировать, а в некоторых случаях и механизировать пассивные технологические процессы.
Особо важное значение приобретает изучение взаимосвязи и взаимовлияния, казалось бы, различных на первый взгляд характеристик. Так, нежность мяса принято характеризовать совокупностью механических свойств. В то же время изменение механических параметров мяса при его хранении зависит от тангенса угла диэлектрических потерь при частотах порядка 104— 105 Гц. Установлена взаимосвязь между структурно-механическими и электрофизическими (tgS и е) свойствами животного жира. При этом
190
наблюдается иодная аналогия характерных точек фазовых превращении при изменении температуры. Такие аналогии значительно облегчают исследование комплексных характеристик продукта, так как позволяют на основе знаний одних свойств делать прогнозы о характере изменения других, а также создавать приборы контроля и управления с обратной связью, основываясь. например, на измерении неэлектрических величин электрическими способами.
Использование комплекса физических методов позволяет по-новому решать ряд технологических проблем на более высокой ступени организации и интенсификации процессов, получить новые высококачественные продукты.
Физические свойства мясопродуктов лежат в основе разработки моделей взаимодействия энергетического поля с продуктом, создания безотходных технологий, высокопроизводительного оборудования, гибких автоматизированных производств, а также систем автоматического проектирования (САПР). Значительные различия численных значений физических величин обусловлены чрезвычайной сложностью строения и состава мяса, а также их нестабильностью вследствие биологического происхождения (порода, пол, возраст животного, степень автолиза, введение в мясопродукт различных ингредиентов при последующей обработке и т. д.). Эти различия достаточно велики. Они проявляются в ходе технологического процесса, когда продукт также претерпевает большие изменения. Так, пластические свойства мясного фарша в процессе термической обработки в результате коагуляционно-денатурационных изменений становятся упругими, в процессе посола резко увеличивается электропроводность и т. д.
Очень важно установить закономерности между численными значениями свойств и качественными показателями продукции как на конечных, так и на отдельных стадиях технологического процесса, применяя соответствующие методы исследования.
2.1. ФИЗИЧЕСКИЕ СВОЙСТВА
Мясо и мясопродукты в связи со сложностью микроструктуры имеют большую оптическую плотность. Поглощение и рассеивание излучения определяются в основном четырьмя процессами: резонансным поглощением излучения молекулами сухого вещества, а также молекулами структурной и связанной влаги; рассеиванием излучения, обусловленным флуктуациями плотности вещества, а также рассеиванием излучения на молекулах белков, полисахаридов, ионах, на взвешенных коллоидных частицах, клетках, частицах пигментов на оптических неоднородностях — капиллярах и порах.
191
Оптические характеристики могут быть спектральными и интегральными. В первом случае они характеризуют явления, происходящие при определенной длине волны излучения л. во втором—для длин волн л = 0 - Для аналитических целей используют спектральные характеристики, для инженерной практики — интегральные.
Структура пищевых продуктов в большинстве случаев такова. что отражение от них является в основном диффузным (рассеянным во все стороны). Отражательную способность продукта можно изучать при помощи коэффициента отражения рл, который показывает отношение светового потока /у, отраженного исследуемым образцом, к световому потоку F\~ упавшему на образец:
рХ=С2//ф (2.1)
Таким образом, определение коэффициента отражения сводится к определению отношения двух отсчетов по шкале измерительного прибора (гальванометра).
При исследовании диффузно отражательных образцов в качестве эталонов используют диффузно отражательные поверхности с известным спектральным коэффициентом отражения, например свеженапыленный оксид магния (полный коэффициент отражения 0,97—0,98).
Оптические свойства мяса играют весьма важную роль в оценке цветности. Объективно измерение цвета мяса служит для оценки его пригодности как сырья для переработки; качества готового продукта; правильности хода технологических процессов; дополнения или контроля правильности органолептических оценок.
В технологических исследованиях объективное измерение цвета чаще всего применяют в качестве второй по важности (после измерения активной кислотности) качественной проверки испытуемого образца. В исследованиях процессов, связанных с сохранением окраски (например, посол мяса), оно выдвигается на первое место.
Мясо имеет специфический цвет благодаря пигменту миоглобину. Все нормальные мышцы содержат миоглобин, но в разном количестве. Большая реактивность миоглобина проявляется в посмертный период, вследствие чего он может давать производные различного цвета. Схема этих превращений представлена на рис. 2.1.
Кроме миоглобина и его производных на цвет мяса влияет ряд других факторов, таких, как системы ахроматические и слабо поглощающие светлые лучи (внутритканевый жир, соединительная ткань); кислотность, изменяющаяся в период посмертного окоченения; поверхностная дегидратация и т. п.
192
Рис. 2.1. Схема взаимопревращений дериватов миоглобина
Для определения цвета продуктов в отраженном монохроматическом свете используют универсальный монохроматор УМ-2 и спектрофотометры. Измерение коэффициентов отражения при длинах волн 627, 635 и 650 нм дает возможность установить образование метмиоглобина.
Отношение оптических плотностей при определенных длинах волн ^545 /^650 и D582 /Z>652 в некоторых случаях (хранение в неправильных условиях, например смена температур) может указывать на изменения в окраске мяса. Величины D545 и Z)582 являются мерой интенсивности окраски мяса.
По отражению поверхности образца можно определить интенсивность окраски различных видов мяса, а также некоторых колбасных и других продуктов.
Неоднородность в строении мышечных волокон мяса ведет к различному поглощению звука отдельными элементами, т. е. наблюдается анизотропия затухания звука. Основными характеристиками акустического поля являются частота колебаний, скорость звука, амплитуда, волновое и удельное акустическое сопротивление среды, звуковое давление, интенсивность звука.
Удельное акустическое сопротивление является важным пара
193
метром — харакшри nei своишва срслв; но ошошснию к проходящей через нее волне:
рс~=р/и. (2.2)
где р —плотность среды, кг/м-': с —скорость ивка, м/с; /> — jhvkoboc давление. МПа; и -- колебательная скорость, м/с.
Энергия звуковых колебаний, проходящая нормально к поверхности продукта через единицу площади за 1 с. является интенсивностью звука:
/=/>2/рг (2.3)
Интенсивность звука оценивают по отношению к величине предела слышимости человеческого уха, г. е. определяют силу звука (дБ).
1 дБ = 101g (//ф), (2.4)
где /п — предел слышимости, Вт/м2 (/ = 10~12 Вт/м2).
Поглощение звука в жидкостях обусловлено вязкостью среды, а также теплопроводностью. Полный коэффициент поглощения
а = аг+ае, (2.5)
где щи щ — коэффициенты поглощения, обусловленные соответственно вязкостью и теплопроводностью среды.
Распространение звуковых волн в среде сопровождается потерями на рассеивание, которые внешне проявляются в повышении температуры среды (табл. 2.1).
Т а б л и ц а 2.1
Относительное повышение температуры продуктов при распространении звуковых волн
Продукт Относительное повышение температуры, °C Продукт Относительное повышение температуры, °C
Яичный: Жир 12,5
альбумин 1,0 Печень 4.5
коагулированный 1,0 Мозги 4,75
белок —
желток 5,5
Примечание. Относительное повышение температуры яичного белка принято за единицу. Продолжительность обработки продуктов ультразвуком 30 с; /= 750 кГц; 100 Вт.
194
Аномальные отклонения коэффициент поглощения обнаружены при ультразвуковой обработке ряда органических и биологических жидкостей. Эти отклонения вызваны объемной вязкостью, являющейся функцией изменения объема в местах сжатия и расширения жидкости. При этом характер молекулярного поглощения энергии зависит от продолжительности восстановления равновесия молекулярных процессов за один полупериод колебания. Исключение составляет костная ткань, которая в диапазоне частот 500 кГц — 2 МГц не дает отклонений от классической теории.
Показатели поглощения и глубина проникновения для некоторых животных тканей при обработке частотой 1 МГц приведены в табл. 2.2.
Таблиц а 2.2
Акустические характеристики животных тканей
Продукт а, м 1/с/, м Продукт (X. М’1 1/а, м
Вода 0,03 30,0 Печень 17 0,06
Плазма крови 0,7 1,3 Почки 22 0,05
Кровь 2.0 0,5 Жировая ткань 13 0,08
Скелетные мышцы 20-25 0,045 Костная ткань 302 0,0033
(при частоте
800 кГц)
Коэффициент поглощения зависит от частоты ультразвукового поля: линейно возрастает с увеличением частоты независимо от вида ткани, а при облучении суспензий линейно возрастает с увеличением концентрации. Кроме того, он зависит от диаметра частиц суспензии. Характерно, что наиболее резкое затухание колебаний наблюдается при размерах частиц 1 —Юмкм. Анизотропия поглощения ультразвука особенно сильно проявляется у тканей, состоящих из чередующихся слоев с различными свойствами (шкура, жировые прослойки и др.). В этом случае затухание акустической энергии зависит от направления ультразвука — вдоль или поперек слоев. Акустические характеристики различных животных тканей представлены в табл. 2.3.
Таблица 2.3
Удельное акустическое сопротивление животных тканей
Образец Температура. °C Скорость звука с 10’. м/с Плотность р. кг/м’’ Удельное акустическое сопротивление р с. Па с/м
Ткань:
мышечная (говядина) 16-20 1,575-1,578 1033-1048 1,79
жировая (свинина) 16-20 1,444 930 1,32
мозговая (свинина) 16-20 1,506 1026 1,55
Печень 16-20 1,553 1064 1,63
Кость (плотная масса) 16-20 3,37 185 6,23
195
2.2. ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА
Аналитическая теория теплопроводности представляет собой теорию распространения теплоты в различных неравномерно нагретых телах, которые рассматриваются как сплошные среды, непрерывно заполняющие пространство, без учета молекулярного строения и молекулярных свойств вещества.
В соответствии с этим тела характеризуются так называемыми макросвойствами. К ним относятся коэффициенты теплопроводности и температуропроводности, удельная теплоемкость, объемная масса, вязкость вещества, коэффициенты диффузии и т. д.
Коэффициент температуропроводности а является основным тепловым параметром при неустановившемся во времени режиме. В этом случае наряду с коэффициентом теплопроводности X на распределение температуры в теле существенное влияние оказывают удельная теплоемкость с и плотность р, связанные между собой соотношением
«=V(cpp), (2.6)
которое показывает, что коэффициент температуропроводности характеризует соотношение между двумя тепловыми свойствами тела: способностью проводить и аккумулировать теплоту.
Теплофизические свойства различных тел зависят от их химического состава, микроструктуры, пористости, влажности, предварительной термообработки, температуры и т. д.
Зависимость тепловых свойств веществ от большого количества взаимно связанных факторов делает эксперимент практически единственным источником получения данных для определения этих свойств. Одновременно с этим эксперимент является источником дополнительной информации о поведении веществ, что позволяет углубить существующие физические представления о механизмах переноса теплоты, поскольку они относятся обычно не к реальным телам, а к их идеализированным моделям. Модельные представления о веществе дают возможность построить соответствующие расчетные методы для определения некоторых тепловых свойств (рис. 2.2).
Рис. 2.2. Расчетные методы для определения тепловых свойств веществ
196
Большинство экспериментальных методов основывается на наблюдении за температурным полем в исследуемом теле при нагревании (охлаждении). Применительно к стационарным условиям используют закон Фурье
(2.7)
и дифференциальное уравнение теплопроводности для одномерного температурного поля
справедливое для тел, физические свойства которых не зависят от температуры,
где к — константа, численные значения которой определяются геометрической формой исследуемого тела; £=1.2, 3 соответственно для пластины, цилиндра и шара; г —температура; г—текущая координата; // — нормаль к изотермической поверхности.
Приведенные уравнения справедливы для твердых тел. Для жидкостей и газов они могут быть использованы, если отсутствуют другие способы переноса теплоты (конвекцией, излучением и т.д.). Эти уравнения не имеют общего решения. Получены их частные решения применительно к телам определенной геометрической формы при конкретно заданных условиях однозначности, которые и используются при постановке экспериментов.
Решив уравнения (2.7) и (2.8) для тел простой геометрической формы при граничных условиях первого рода, можно найти коэффициент теплопроводности из соотношения
где —температуры на изотермических поверхностях, соответствующих
этим диаметрам; К— коэффициент формы, который выражается в виде зависимостей соответственно для неограниченных плоского и цилиндрического слоев исследуемого вещества. Его можно определить из соотношений:
(2.Ю)
(2.И)
где 5 —толщина плоского слоя, м; 5 = г2 — /^ (или 5 = .v2-.v1); Ер — расчетная
поверхность, нормальная к направлению теплового потока, м2; ф и ф — соответственно внутренний и наружный диаметры цилиндрического слоя исследуемого вещества, м; / — длина цилиндрического слоя, м.
197
При исследовании тепловых параметров методом нестационарного теплового потока используются решения дифференциальных уравнении, которые имеют вид
dQ
Эт
а/ J э2г к + \ c)t}
Ср — — л —~—г --------- —
Р Эт Э/-2 Г дг
(2.13)
На основной стадии процесса теплопроводности изменение температуры во времени приобретает упорядоченный характер и математически описывается более простыми функциями, чем на начальной стадии, так как изменение температуры в каждой точке тела перестает зависеть от начальных условий.
Теория теплопроводности на начальной стадии процесса позволяет из эксперимента найти одновременно несколько тепловых свойств. Теория теплопроводности на основной стадии процесса позволяет построить методики исследования для отдельных тепловых свойств и для их комплекса. Решения для этой стадии имеют различный вид в зависимости от рода граничных условий.
Метод нестационарной теплопроводности позволяет в ряде случаев проводить измерения при непрерывном изменении температуры до желаемого ее значения. Это дает возможность получить сразу непрерывный ряд значений измеряемого теплового параметра в широком диапазоне температур, в то время как во всех стационарных методах такой ряд может быть получен из отдельных опытов, соответствующих различным стационарным тепловым режимам, число которых обычно ограничено. Измерения тепловых параметров различных веществ проводят при относительно небольших перепадах температур, что приближает их средние значения к истинным. Поэтому нестационарные методы предпочтительны для исследования тепловых параметров влажных материалов.
К недостаткам нестационарных методов относятся трудности получения точно регулируемого во времени изменения температуры, а также учета соответствия действительных граничных условий эксперимента принятым в теории. Учесть подобное обстоятельство очень трудно, но более важно, чем в стационарных методах.
Методы исследования тепловых свойств при установившихся и неустановившихся тепловых режимах позволяют на основании одного опыта найти какой-либо один тепловой параметр. Если необходимо иметь данные по ряду физических свойств, то такой 198
комплекс физических параметров может быть получен путем комбинации двух пли нескольких приборов. Это связано с использованием нескольких образцов из исследуемого материала и трудностями сохранения идентичности свойств при их изготовлении, с увеличением погрешностей, а также затрат времени на проведение измерений. Поэтому в настоящее время стараются одновременно получить данные нескольких тепловых свойств из одного опыта, на одной установке и одном образце. Такие методы получили название комплексные, дающие наиболее полное представление о тепловых свойствах и поведении исследуемого вещества, кроме того, они позволяют сократить время на проведение экспериментов. Они могут базироваться на теориях начальной и основной стадий процессов нестационарной теплопроводности, на их совокупности, а также на процессах теплопроводности, протекающих в условиях установившихся тепловых режимов.
2.3. ФУНКЦИОНАЛЬНО-ТЕХНОЛОГИЧЕСКИЕ СВОЙСТВА
Мясное сырье многокомпонентно, вариабельно по составу и свойствам, что значительно сказывается на качестве готовой продукции. В связи с этим особенно важное значение приобретает информация о функционально-технологических свойствах различных видов основного сырья и его компонентах, влиянии вспомогательных материалов и внешних факторов на характер их изменения.
Под функционально-технологическими свойствами (ФТС) мясного сырья понимают совокупность показателей, характеризующих уровни эмульгирующей, водосвязывающей, жиро-, водопоглощающей и гелеобразующей способностей, структурно-механические свойства (липкость, вязкость, пластичность и т. д.), сенсорные характеристики (цвет, вкус, запах), величину выхода и потерь при термообработке различных видов сырья и мясных систем. Перечисленные показатели имеют приоритетное значение при определении степени приемлемости мяса для производства пищевых продуктов.
Под функциональными свойствами изолированных белков принято понимать широкий комплекс физико-химических характеристик, определяющих их поведение при переработке и хранении, обеспечивающих желаемую структуру, технологические и потребительские свойства готовых продуктов.
Физическая структура и свойства не подвергнутого термической обработке мясного фарша близки к классическим эмульсиям.
В классическом определении под эмульсией понимают дисперсные системы с жидкой дисперсионной средой и жидкой дисперс-
199
Рис. 2.3. Типы водно-жировых’эмульсий: а — прямяя; б — обратная
ной фазой, диспергированные в коллоидном состоянии. Жир— неполярное вещество и плохо (на 0,5 %) растворимо в воде. Однако при определенных условиях (наличие эмульгаторов и стабилизаторов, высокие температуры, ультразвуковые и импульсные воздействия) в системах жир — вода могут образовываться водно-жировые эмульсии прямого (жир в воде) и обратного (вода в жире) типа (рис. 2.3).
Стойкость эмульсий во многом зависит от наличия в системе эмульгаторов — веществ, в состав которых входят полярные и неполярные группы.
В мясной эмульсии, образованной в результате интенсивного механического измельчения тканей, дисперсная система состоит из дисперсной фазы — гидратированных белковых мицелл и жировых частиц различных размеров и из дисперсионной среды — раствора белков и низкомолекулярных веществ. В мясной эмульсии белок и вода образуют матрицу, которая окружает жир, т. е. колбасный фарш — эмульсия жира в воде, при этом солерастворимые белки являются эмульгаторами и стабилизаторами эмульсии (рис. 2.4). Белки мышечных волокон по убыванию величины эмульгирующей способности (ЭС) располагаются в последовательности: актин (без NaCl), миозин, актомиозин, саркоплазматические белки, актин в растворе соли молярной концентрацией 0,3 моль/дм3.
Рис. 2.4. Схематическое изображение мясной эмульсии
200
Мясные эмульсин подобного рода относятся к коагуляционным структурам, частицы которых связаны силами межмолекулярного взаимодействия в единую пространственную сетку (каркас). Сопоставление ЭС различных высокомолекулярных веществ показывает, что во всех случаях они стабилизируют эмульсии, образуя трехмерные сетчатые структуры с близкими геометрическими свойствами. Стабилизация эмульсий, обусловленная особыми структурно-механическими свойствами адсорбционных межфазных слоев, может привести к повышению устойчивости этих дисперсных систем вплоть до полного фиксирования. Такая стабилизация носит универсальный характер и необходима при получении высокоустойчивых (особенно концентрированных) эмульсий.
При технологической обработке мясного сырья со свойствами белков связаны следующие взаимодействия: белок —белок (гелеобразование); белок —вода (набухание, водосвязывающая и жироудерживающая способности), а также поверхностно-активные свойства — образование и стабилизация пен и эмульсий.
Мясной фарш — сложная гетерогенная система, функциональные свойства которой зависят от соотношения тканей, содержания в них специфических белков, жиров, воды и морфологических компонентов.
В составе мяса мышечная ткань оказывает значительное влияние на ФТС, так как состоит из комплекса белков, имеющих структурные отличия. При получении мясопродуктов от функциональных свойств мышечных белков зависит эффективность образования мясных эмульсий. Количественное содержание белка в системе, его качественный состав, условия среды предопределяют степень стабильности получаемых мясных систем, влияют на уровень водосвязывающей, жиропоглощающей и эмульгирующей способности, структурно-механические и органолептические характеристики.
Количественное содержание наиболее важного функционального белка — миозина в мышечной ткани составляет 54—60 %. Его молекулы имеют выраженную ферментативную активность, легко взаимодействуют между собой и актином, обладают высокой водосвязывающей, гелеобразующей и эмульгирующей способностями.
На характер взаимодействия в системе белок — вода оказывают влияние такие факторы, как растворимость белковых систем, концентрация, вид, состав белка, степень нарушения нативной конформации, глубина денатурационных превращений, pH системы, наличие и концентрация солей в системе. Знание и направленное использование особенностей связывания влаги различным белок-содержащим сырьем позволяют прогнозировать и регулировать выход, уровень потерь влаги при термообработке и органолептические характеристики продукта.
201
Влагоудерживающая способность (ВУС). как и растворимость, одновременно зависит от степени взаимодействий как белков с водой, так и белка с белком, а также от конформации и степени денатурации белка. В связи с этим тепловая обработка оказывает сильное влияние на влагоудерживающую способность белков, что, в свою очередь, сказывается на массовом выходе готовых изделий.
В реальных многокомпонентных мясных системах поведение белка как основного стабилизирующего компонента рецептуры рассматривают во взаимосвязи как с другими компонентами (жир, вода, минеральные вещества, морфологические элементы), так и с изменяющимися в процессе технологической обработки сырья условиями среды.
При изготовлении вареных колбас, сосисок, сарделек, мясных хлебов для направленного регулирования ФТС мясных фаршевых систем кроме поваренной соли используют пищевые фосфаты — смеси различных солей фосфорной кислоты в количестве 0,3—0,4 % к массе фарша. Фосфаты действуют как синергисты поваренной соли, вызывая изменение величины pH среды, повышая ионную силу растворов и связывая ионы кальция в системе актомиозинового комплекса, обеспечивают интенсивное набухание мышечных белков, увеличивают уровень водосвязывающей, влагоудерживающей и эмульгирующей способностей.
Особенно эффективно использование фосфатов при переработке размороженного и тощего мяса, сырья с нарушениями нормального хода автолиза. В последние годы в связи с увеличением объемов мясного сырья с нарушениями нормального хода автолиза возникла необходимость расширения диапазона pH фосфатных препаратов, используемых в отечественной промышленности, с 6,9 до 9,0.
Экспериментально установлено, что вареные колбасы имеют в среднем приемлемое качество и удовлетворительную органолептическую оценку при устойчивости фаршевой эмульсии не ниже 85 %, влагоудерживающей способности — приблизительно 85 % общего содержания влаги в фарше, или около 90—92 % связанной влаги в сыром фарше, и жироудерживающей способности — на уровне 95 % содержания жира в фарше.
2.4. СТРУКТУРНО-МЕХАНИЧЕСКИЕ СВОЙСТВА
Структурно-механические (реологические) свойства характеризуют поведение мяса и мясопродуктов в условиях напряженного состояния, основными показателями которого при приложении силы являются напряжение, величина и скорость деформации. В зависимости от характера приложения усилий свойства делятся на сдвиговые (касательные напряжения), компрессионные
202
(нормальные напряжения растяжения -- сжатия) и поверхностные на границе раздела с другим магсриа.юм (нормальные и касательные).
В реальных условиях имеет месю сочоание всех свойств, в то же время в зависимости от направленности процесса превалирует одно из них.
Сдвиговые реологические свойства: предельное напряжение сдвига 0О (Па), вязкость эффективная (Па - с) и пластическая р (Па • с), период релаксации т (с) — наиболее полно отражают внутреннюю сущность объекта, поэтому их принято считать основными. С их помощью рассчит ывают течение продуктов в трубах, рабочих органах машин и аппаратов, определяют необходимые усилия для перемещения продукта, оценивают качество продукта, обосновывают оптимальные технологические условия процесса.
К основным компрессионным (объемным) свойствам относятся модуль упругости Е (Па), равновесный модуль ER (Па), период релаксации деформации при постоянном напряжении тф (с), относительная деформация е. Эти параметры необходимы для расчета процессов шприцевания, формования, дозирования и течения по трубопроводам пластично-вязких продуктов. Обьемные свойства можно также использовать для оценки качества пластично-вязких (фарши) и унругоэластичных (колбасные изделия) продуктов.
Особое место среди структурно-механических характеристик занимают поверхностные свойства (адгезия, коэффициент внешнего трения и др.). Они характеризуют усилие при взаимодействии между поверхностями контакта при нормальном отрыве или сдвиге. Для пищевых материалов различают три основных вида отрыва: адгезионный, когезионный и адгезионно-когезионный, или так называемый смешанный отрыв.
Поверхностные характеристики необходимы для выбора и разработки новых видов контактирующих материалов с продуктом для оборудования, тары, трубопроводов и т. д., поверхности которых должны обладать малой адгезией и минимальным сопротивлением при движении продукта. Кроме того, величины поверхностных свойств частично могут характеризовать консистенцию продукта.
Структурно-механические свойства отражают внутреннее строение (структуру) и состав вещества. Наиболее полно они характеризуют структуру, которая может быть коагуляционной и конденсационно-кристаллизационной. Для мясопродуктов наиболее распространен коагуляционный тип структуры, которая является следствием взаимодействия между частицами вещества на основе сил Ван-дер-Ваальса через дисперсионную среду. Структурам такого типа присуща тиксотропия, т. е. способность восстанавливать свои свойства после снятия напряжения или даже после
203
разрушения. Очевидно, что структурно-механические свойства коагуляционных систем значительно зависят от содержания воды, размеров частиц и прослоек, их физико-химических свойств. Для технологии особенно важна зависимость структурно-механических свойств от изменения размеров частиц, например при измельчении мяса в процессе приготовления колбасного фарша, и других факторов. С помощью приборов и оценки структурно-механических свойств мясных фаршей можно контролировать любую технологическую стадию и управлять качеством продукции.
Лабораторная работа № 1
ОПРЕДЕЛЕНИЕ ЦВЕТНОСТИ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Приобрести практический навык определения цветности мяса и мясных продуктов.
Задачи. Освоить методику определения цветности на монохроматоре путем снятия спектральных характеристик и расчета оптической плотности по коэффициенту отражения.
Объекты исследования. Образцы мышечной ткани различных анатомических участков разных видов животных и птицы неодинаковых сроков хранения, мясные продукты кулинарной готовности.
Материалы, реактивы и оборудование. Миллиметровая бумага; монохроматор УМ-2 (или спектрофотометр); ртутно-кварцевая лампа СВДШ-250.
Методические указания. Для исследования цветности мяса и мясных продуктов в отраженном свете используют монохроматор УМ-2 с лампой накаливания напряжением 12 В, мощностью 40 Вт и ахроматическим конденсором с фокусным расстоянием 90 мм. Напряжение стабилизируют феррорезонансным или электронным стабилизатором.
Монохроматор при снятии спектров отражения используют в комплекте со специальной приставкой, снабженной кольцевым селеновым фотоэлементом, эталоном сравнения и кюветами для измеряемых образцов диаметром 30 мм. Селеновый фотоэлемент воспринимает отраженный свет от эталона или измеряемого образца. Возникший поток измеряется зеркальным гальванометром.
Оптическая схема монохроматора приведена на рис. 2.5.
Свет, прошедший через входную щель, попадает на объектив коллиматора и параллельным пучком проходит через диспергирующую призму. Под углом 90° к падающему пучку света размещают выходную трубу монохроматора. При повороте призменного сто-
204
8
7
6 5 4
3
2 1
Рис. 2.5. Оптическая схема монохроматора:
/ — источник света; 2 — защитное стекло кожуха лампы; 3— конденсор; 4— защитное стекло входной щели; 5—призма сравнения; 6 —щель; 7—объектив коллиматора; 8 — диспергирующая призма; 9— объектив зрительной трубы; 10— съемная выходная щель; 11 — защитное стекло выходной щели
лика на различные углы относительно падающего пучка света в выходной щели получают свет различной длины волны, проходящий через призму при минимуме отклонения.
Патрубок со щелью можно заменить патрубком зрительной трубы со сменными окулярами. Первый патрубок применяют для выполнения измерений, второй — для градуировки прибора.
Фокусное расстояние объектива для каждой длины волны изменяется при помощи маховичка. Зависимость фокусировки от длины волны приведена в аттестате прибора.
Сменные фильтры в револьверной оправе предназначены для того, чтобы освещение указателя при работе в каждой области спектра производилось светом той же длины волны.
В качестве источника света при исследовании используют лампу накаливания; при градуировке применяют неоновую и ртутную лампы, прилагаемые к монохроматору.
Для измерений в отраженном свете к монохроматору необходимо присоединить специальное приспособление. Это может быть фотометрическая сфера или приставка с кольцевым селеновым фотоэлементом, которая более проста в изготовлении.
В качестве эталона для построения градуировочного графика и проведения анализа используют молочное стекло с известным коэффициентом отражения, откалиброванное по оксиду магния, или свежеприготовленный оксид магния.
Построение градуировочного графика. После крепления и установки на рельсы к монохроматору подсоединяют плато. Через трансформатор, находящийся на плато, подают питание лампочкам для освещения шкалы и указателя в патрубке зрительной трубы. На рельс перед входной щелью монохроматора устанавливают
205
Рис. 2.6. Градуировочный график к монохроматору
неоновую лампу мощностью 40 Вт и перед нею для проектирования источника света на щель монохроматора — ахроматический конденсор с фокусным расстоянием 90 мм.
('троят градуировочный график (рис. 2.6) на миллиметровой бумаге размером примерно 40x40 см. Графиком или таблицей. составленной по этому графику, пользуются при измерении исследуемых образцов.
Для построения градуировочного графика источник света рекомендуется установить на расстоянии 410 мм от плоскости щели, а первую плоскость конденсора -- на расстоянии 256 мм. Неоновую лампу включают в сеть переменного тока 127 В (в случае необходимости через трансформатор).
Ставят диафрагму в боковое продольное отверстие коллиматора, ограничивая высоту щели 2 мм. Наблюдают за тем, чтобы свет лампы хорошо заполнил входную щель, ширина которой должна быть 0,01—0,02 мм. Коллиматорную трубу монохроматора приводят в положение, указанное в аттестате прибора при длине волны 585,2 нм.
Для проведения градуировки в выходную трубу монохроматора вставляют патрубок зрительной трубы с одним из сменных окуляров. Указатель освещают через оранжевый светофильтр. В этой области спектра наблюдают линию неона (585,2 нм). Вращая барабан измерительного механизма и наблюдая в окуляр спектроскопа, приводят спектральную линию неона (585,2 нм) в центральное по
ложение, совместив ее с указателем в окуляре.
Если отсчет совпадает с данными, приведенными в аттестате монохроматора, то градуировочный график можно построить по аттестату. При расхождении результатов градуируют заново. Для этого после нескольких отсчетов неоновую лампу выключают и снимают, а на ее место ставят ртутно-кварцевую лампу евдш -250, подключают ее к плато и зажигают. Обращаться с включенной лампой нужно осторожно, так как давление в ней достигает 2,9 • 106 Па. Ширина входной щели должна быть приблизительно 0,01—0,02 мм, высота 2 мм. Центрируют источник света на рельсе без конденсора, затем устанавливают конденсор по центру так, чтобы объектив коллиматора был равномерно заполнен светом. За линиями ртути наблюдают через окуляр спектроскопа. Вращая барабан, приводят нужную спектральную линию в центральное положение, совмещая ее с указателем в окуляре. Для освещения указателя применяют соответствующий светофильтр. При подводе линии пользуются данными ат
206
тестата и дополнительно при необходимости справочной литературой. выбирая только сильные линии.
Подготовка проб. При определении цвета на монохроматоре УМ-2 мышечную ткань разрезают на ломтики толщиной 4—5 мм перпендикулярно направлению мышечного волокна. Из нарезанных ломтиков остро отточенным пробником вырезают образцы. Диаметр пробника должен быть равен диаметру кюветы (30 мм). Вырезанные образцы помещают в чашки Петри, закрывают и выдерживают в темноте не менее 10 мин. Небольшая выдержка образцов на воздухе необходима для превращения миоглобина в оксимиоглобин, а гемоглобина — в оксигемоглобин. В пределах от 10 мин до 4 ч пробы пригодны для измерения.
Для определения интенсивности окраски из каждой пробы мяса делают 4—5 срезов. В последующем среднеарифметическое измерение 4—5 срезов от каждый пробы является окончательным результатом определения. Для выполнения измерения образцы осторожно, не касаясь поверхности, переносят в кюветы, которые закрепляют в приставке.
При работе на спектрофотометре образцы готовят аналогично, используя пробник диаметром 48—50 мм (диаметр кювет 48 мм). Образцы помещают в металлические кюветы для измерения отражения.
Порядок проведения анализа. Для снятия спектральных кривых, характеризующих цветность исследуемого образца, измерения проводят в широкой области спектра через 2—3 нм в участках, где наблюдаются характерные изменения спектральной кривой, и через 5—10 нм в менее характерных участках. Для определения интенсивности окраски измерение проводят при одной, двух или трех длинах волн.
Ширину входной и выходной щелей можно изменять для разных длин волн, подбирая наиболее пригодные. Однако определенные затруднения в работе связаны с тем, что измерения на монохроматоре проводят в темном помещении. Постоянные щели значительно упрощают работу: приемлемая ширина выходной щели 0,1 мм и входной 0,2 мм при работе с приставкой с кольцевым селеновым фотоэлементом высотой 12 мм.
За 10 мин до начала определений включают источник света (лампу накаливания), лампочку осветителя гальванометра и осветительные лампочки на корпусе монохроматора. Перед выходной щелью устанавливают кювету с эталоном, снимают отсчет по шкале гальванометра. Затем на место кюветы с эталоном ставят кювету с испытуемым образцом и снова делают отсчет. После определения при одной длине волны микрометрическим винтом поворачивают барабан, устанавливают нужную длину волны и снова определяют отражение света эталоном и образцом.
При работе с шаровой приставкой и фотоэлектронным умножителем удобнее всего использовать ширину входной щели
207
Рис. 2.8. Кривые отражения некоторых мясных продуктов, снятые на спектрофотометре СФ-10:
1,2— копченая и вареная колбасы; 3 — яичный порошок; 4 — сыр Российский
Рис. 2.7. Кривые отражения среза мышечной ткани, снятые на спектрофотометре СФ-10:
1 — в процентах отражения; 2 — в единицах оптической плотности
0,1 мм, а выходной такую же или меньше. Последнюю подбирают после включения блока питания с гальванометром и установки эталона против отверстия шара.
В зависимости от отклонения светового указателя гальванометра ширину щели увеличивают, начиная примерно с 0,02 мм, до тех пор, пока указатель гальванометра не остановится на делении шкалы примерно 60—68. При вычислении коэффициента отражения предварительно из показаний, полученных для образца и эталона, вычитают показание, полученное для черного тела.
На основании характера спектральной кривой того или иного продукта выбирают 2—3 длины волны, при которых в дальнейшем измеряют интенсивность окраски (например, интенсивность окраски говяжьего мяса определяют при длинах волн 545, 582 и 650 нм).
Коэффициент отражения рх вычисляют путем деления числа, полученного при измерении образца, на число, полученное при измерении эталона для одной и той же длины волны и одних и тех же условий измерения. Коэффициент отражения получают по отношению к эталону. Зная отражение эталона, вводят поправку. Например, если коэффициент отражения эталона равен 0,85, то поправочный множитель будет 1,176.
Коэффициенты отражения, выраженные в процентах, переводят в оптическую плотность по формуле
П-1 100
й-lg— (2.14)
208
Результаты оптической плотности выражают при длине волны 545 нм (D545) и 582 нм (Z)-82), а также в виде отношений
А45 As2
-----14 -----
Д55О Дз50
(2.15)
Результаты оформляют в виде кривых отражения (или изменений оптической плотности), примеры которых показаны на рис. 2.7 и 2.8. Затем делают вычисления и по результатам составляют заключение по работе, сопоставляя данные с визуальной оценкой продуктов.
Лабораторная работа № 2
ОПРЕДЕЛЕНИЕ ЦВЕТНОСТИ ТВЕРДЫХ ЖИВОТНЫХ ЖИРОВ
Цель работы. Приобрести практический навык определения цветности животных жиров в отраженном свете на фотометре ФТ-2.
Задачи. Измерить отражательную способность образцов твердых животных жиров.
Объекты исследования. Твердые животные жиры разных видов.
Материалы, реактивы и оборудование. Спектрофотометр ФТ-2 или других аналогичных конструкций; лопатки из плексигласа.
Методические указания. Метод основан на фотометрическом измерении отражательной способности образцов жира. Перед началом работы фотометр включают в сеть для прогрева (примерно на 10 мин), после чего проверяют коэффициент яркости градуировочной пластины, являющейся промежуточным эталоном (если коэффициент яркости пластины был установлен не более чем за 3 дня до опыта, то пользуются имеющимися данными; если перерыв больше, то вновь делают проверку). Затем заполняют кювету исследуемым жиром и проводят определение.
В этом методе белизна образцов условно характеризуется величиной монохроматического коэффициента яркости в зеленой части спектра, т. е. в области высокой видимости (измерение со светофильтром с длиной волны 510 нм). Второе число показывает отношение отражения в двух участках спектра и как бы характеризует желтизну образца. Чем меньше отношение и выше показание при 510 нм, тем белее образец.
Пример. У образца 1 отражение света при светофильтре ^эф = 410 нм равно 47,1 %, при ^эф — 510 нм — 57,4 %, отношение отражений 1,22; у образца 2 — соответственно 72,2 %; 73,5 % и 1,02. Следовательно, образец 2 обладает большей белизной, чем образец 1.
209
Рис. 2.9. Кювета (7) и вкладыш (2) для измерения цветности жира
Подготовка проб. При определении цветности твердых животных жиров в отраженном свете на фотометре ФТ-2 образец жира должен иметь температуру около 20 °C. Его помещают в кювету (глубина кюветы 6 мм, диаметр 30 мм) из плексигласа (рис. 2.9) и лопаточкой из того же материала выравнивают поверхность. Если за 1—3 приема поверхность становится ровной, жир удаляют из кюветы и заменяют новой порцией того же образца. Многократное выравнивание поверхности в некоторых случаях может повлиять на достоверность результатов.
Если исследуют легкоплавкий жир, который при указанной температуре име
ет жидкую консистенцию, можно с некоторой степенью погрешности наполнить кювету жиром непосредственно после охлаждения в холодильнике. Все операции необходимо выполнять очень быстро.
Порядок проведения анализа. При измерении цветности свиного жира подготовленную кювету с исследуемым образцом помещают в кассету для отражающих образцов. Ручку «светофильтра» ставят против цифры, соответствующей светофильтру с эффективной длиной волны 410 нм, и проводят определение так, как это указано для отражающих образцов. После записи отсчета ручку переводят на цифру, соответствующую светофильтру с эффективной длиной волны 510 нм, и аналогично снимают показания, устанавливая прибор на нуль по показаниям градуировочной пластины для светофильтра с длиной волны 510 нм, снова снимают и записывают данные шкал трех правых ручек прибора.
Результаты, характеризующие цветность свиного жира, записывают в виде двух чисел, одно из которых является результатом измерения отражательной способности образца со светофильтром с эффективной длиной волны 510 нм, другое представляет собой отношение отражательной способности образца, измеренной со светофильтром ХЭф = 510 нм, к отражательной способности образца, определенной со светофильтром Лэф = 410 нм.
Поскольку в процессе хранения в холодильнике говяжий жир иногда приобретает зеленоватую окраску, дополнительно опреде
ляют интенсивность окраски как естественного, так и позеленев
шего жира.
При определении интенсивности окраски говяжьего жира измерение проводят со светофильтром с эффективной длиной волны 460 или 475 нм.
Кювету из плексигласа с образцом жира помещают в кассету для отражающих образцов. Ручку «светофильтра» ставят против
210
цифры, соответствующей светофильтру с эффективной длиной волны 460 нм, или против ручки, соответствующей светофильтру ХЭф = 475 нм, и измеряют, как указано выше. Полученный на шкалах трех правых ручек отсчет характеризует интенсивность окраски образца.
Различные образцы говяжьего жира характеризуются примерно следующими величинами отражения (%): интенсивно-желтый — 38, желтый — 47, светло-желтый — 54.
Для определения наличия в образце зеленоватого оттенка измерения проводят с двумя светофильтрами: с ХЭ()) = 460 нм и ХЭф = 440 нм или ХЭф = 475 нм и Хэф = 440нм. Метод определения такой же, как для свиного жира.
Полученные результаты выражают отношением показаний, полученных со светофильтрами 460/440 или со светофильтрами 475/440. В первом случае образцы жира с зеленоватым оттенком характеризуются отношением > 1, а желтые <1, во втором случае — зеленоватые образцы > 1,10, желтые < 1,10.
В табл. 2.4 приведены возможные показания при определении зеленоватого оттенка в ряде образцов говяжьего жира.
Таблица 2.4
Пример определения цветности говяжьего жира
Образец жира Р4«/Рд4(| Р4-5/Р440
Желтый 0,98 1,07
Желтый со слабым зеленоватым оттенком 1,01 1,И
Зеленый 1,05 1,15
После снятия спектральных кривых и расчетов делают выводы и формулируют заключение, сопоставляя данные с результатами визуальной оценки анализируемых жиров.
Лабораторная работа № 3 ОПРЕДЕЛЕНИЕ АКУСТИЧЕСКИХ СВОЙСТВ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Приобрести практический навык определения акустических свойств мяса и мясных продуктов.
Задачи. Подготовить пробы и определить скорость распространения и коэффициент поглощения ультразвука для мяса, мясных продуктов, вторичных продуктов убоя.
Объекты исследования. Образцы мышечной ткани различных анатомических участков разных видов убойных животных и птицы, вторичных продуктов убоя и мясопродуктов.
211
Материалы, реактивы и оборудование. Установка для определения акустических характеристик; нож.
Методические указания. Ультразвуковые и звуковые колебания представляют собой механические колебания упругой среды, распространяющиеся с определенной скоростью и обладающие известной энергией. Ультразвуками называются звуковые волны с частотами от 2 • 104 до 1013 Гп. Ультразвуки генерируются механическими и электромеханическими излучателями. Длина волны является важной характеристикой ультразвука и определяется расстоянием между двумя следующими друг за другом сгущениями или разрежениями при распространении акустических волн в среде.
Звуковые и ультразвуковые колебания нетождественны, так как с повышением частоты изменяются свойства упругих колебаний и соответственно их воздействие на вещество. Характерными свойствами ультразвуковых волн являются отражение, фокусирование и способность образовывать лучи. Ультразвуковые колебания обладают большой механической энергией, которая и определяет эффект их применения в промышленности.
Ультразвук применяется для интенсификации ряда технологических процессов в пищевой, в том числе мясной, промышленности: для эмульгирования, экстракции, диффузии и др.
Ультразвук перспективно применять для контрольно-измерительных целей. Изучение ультразвуковых свойств животных тканей также открывает перспективы применения ультразвука для комплексного (неразрушающего) контроля качества мяса и мясных продуктов на основе экспрессного получения информации об их химическом составе, структуре, механических и термодинамических свойствах. Применяемое излучение безвредно для продуктов, оборудование надежно, выполнено в портативном виде и доступно по цене.
К основным физическим величинам, измеряемым опытными образцами приборов, относятся скорость распространения и коэффициент поглощения ультразвука. Поглощение звука — явление необратимого перехода энергии звуковой волны в другие виды энергии, в частности тепло. Коэффициент поглощения звука а определяется как обратная величина того расстояния, на котором амплитуда звуковой волны падает в е раз.
Зависимость скорости распространения ультразвука в мясных продуктах от температуры, структуры, физиологического состояния и состава мышечной ткани открывает разнообразные возможности для исследования ряда показателей: массовой доли влаги и ее фазового состава, липидов, соотношения белок : жир и т. д.
Изучение эффекта ослабления ультразвука в животных тканях и сравнительная оценка коэффициента ослабления ультразвука, коррелирующего с его частотой, позволяют судить о диаметре мышечных волокон и других структурных характеристиках мяса и мясных продуктов.
212
3
Рис. 2.10. Принципиальная схема устройства для определения акустических характеристик продуктов:
/— генератор опорного сигнала; 2—юнератор сигнала низкой частоты; 3 — частотомер; 4 и б— соответственно излучающий и приемный электроакустические преобразователи; 5 — измерительная камера; 7— измеритель разности фаз (фазовый детектор)
Скорость распространения и коэффициент поглощения ультразвука в мясе и мясопродуктах измеряют с помощью устройства, принципиальная схема которого представлена на рис. 2.10.
Физическая сущность метода определения акустических свойств состоит в расчете скорости распространения ультразвука по сдвигу фаз между опорным сигналом и сигналом с приемного электроакустического преобразователя (рис. 2.11). С практической точки зрения удобно фиксировать не сдвиг фаз, а частоту, которая с большой точностью может быть определена частотомером. Подбором соответствующей частоты добиваются того, чтобы по длине измерительной ячейки /укладывалась одна волна (рис. 2.12), т. е. смещение фазы в точке В было бы равно нулю.
Рис. 2.11. Сдвиг фаз между опорным сигналом и сигналом с приемного электроакустического преобразователя:
с, — степень деформации; х, У — координаты
Рис. 2.12. Волновой процесс в измерительной ячейке:
с, — степень деформации; х — координата; / — длина ячейки; л —длина волны
213
Длина волны л связана со скоростью ультразвука v и его частотой v известным соотношением
(2.16)
Если длина измерительной ячейки прибора / = Л, то скорость ультразвука легко рассчитать, экспериментально определив его частоту и длину измерительной ячейки прибора, по формуле
V = V/.
(2.17)
Для практического определения коэффициента поглощения ультразвука в мясе и мясных продуктах используют зависимость длины волны ультразвука от коэффициента его поглощения в животных тканях, согласно которой коэффициент поглощения ос связан с величиной девиации частоты 5^ соотношением
5/п +в
(2.18)
где л—длина волны; В — постоянная, которая характеризует потери энергии, нс обусловленные поглощением ультразвука в исследуемом продукте.
Подготовка проб. Вырезают образцы мышечной ткани (вдоль и поперек волокон), субпродуктов, вторичных продуктов убоя в соответствии с размерами измерительной ячейки экспериментальной установки.
Порядок проведения анализа. Подготовленный образец помещают в измерительную ячейку прибора. Меняя частоту звуковых волн, определяют акустические характеристики исследуемых образцов мяса и мясопродуктов: рассчитывают скорость ультразвука v по формуле (2.17) и коэффициент поглощения ос по формуле (2.18).
Постоянную величину В определяют путем калибровки прибора по средам с известной скоростью ультразвука.
Результаты измерения и расчетов представляют в виде таблицы:
Наименование, краткая характеристика образца Частота ультразвука, Гц (кГц) Скорость ультразвука, м/с Коэффициент поглощения ультразвука, м-'
214
Лабораторная работа № 4 ИССЛЕДОВАНИЕ ТЕПЛОФИЗИЧЕСКИХ ХАРАКТЕРИСТИК МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Освоить методы и практически определить некоторые теплофизические свойства (теплопроводность, теплоемкость, температуропроводность) мяса и мясных продуктов.
Объекты исследования. Образцы мяса разных видов и сортов, вторичных продуктов убоя и мясных продуктов.
Материалы, реактивы и оборудование. Лабораторная установка для определения тсплофизических показателей: нож.
Методические указания. Рассмотрим систему тел, состоящую из полуограниченного цилиндра В (теплоприемника) и плоскопараллельных пластин Л/р А и Л/э (рис. 2.13). Начальная температура системы равна Го, температура'нагревателя /н постоянна. Один из спаев дифференциальной термопары помещен в теплоприемник, другой — в нагреватель. При этом начальное показание /Vo гальванометра (7, включенного в цепь термопары, соответствует разности температур tl{ -- Zo.
При соприкосновении нагревателя со свободной поверхностью системы температура в точке О системы начинает увеличиваться и показания гальванометра будут уменьшаться с течением времени т так, как это изображено на рис. 2.14. Изменение показаний N гальванометра G со временем т связано с изменением относительной температуры 6 в точке О системы соотношениями
где г— температура в точке О системы; tit — температура нагревателя.
Рис. 2.13. Схема измерительного стенда Для определения теплофизических характеристик
Рис. 2.14. График изменения разности температур нагревателя и теплоприемника в точке О
215
Если на рис. 2.13 h} = //2 = 0. т. е. система состоит юлько из одной исследуемой пластины А и теплонриемника В. то метод носит название метод двух временных точек.
Наиболее простое выражение для уравнения
О =./т. (2.21)
описывающего процесс изменения относительной температуры 0 в точке О системы, будет получено в том случае, если точка О помещена на границе сред А и В. В этом случае уравнение (2.21) примет следующий вид:
0 = Z//H = (1 + сс) [ erfc (у) — aerfc (Зу) + ... | = В (а, у). (2.22)
В уравнении (2.22)
= УГ«' <2-24>
дУ’ <2'25)
у = WX (2'26)
erfc (у) = 1 — erf (у), (2.27)
где а — коэффициент температуропроводности исследуемого образца; с — безразмерный параметр, который определяют при помощи рабочих таблиц; /. — коэффициент теплопроводности исследуемого образца; т —время: 2 Г _ 2
erf(y) = -г )е dy — интеграл Гаусса; — коэффициент теплопроводности ул о
теплонриемника; ah — коэффициент температуропроводности теплоприемни-ка; Л —толщина слоя исследуемого образца; Ь~ тепловая активность или теп-лоусвояемость,
b = x/Ja = jCX = Cja. (2.28)
Как видно из рис. 2.13, один из спаев дифференциальной термопары размещен внутри системы, а другой — в нагревателе постоянной температуры /н. При этом показание NQ гальванометра G, включенного в цепь термопары, соответствует разности тем
216
ператур /и - г(|. При соприкосновении с нагревателем температура системы увеличивается, и показания гальванометра 6'уменьшаются. Измерение теплофизических характеристик сводится к фиксированию двух промежутков времени: Дт^т, —т, и Дт2 = т2 —т,_ соответствующих двум заданным изменениям показаний ’гальванометра: Л/V] = /V] — и ДЛ'2 = /Vj — Ли (см. рис. 2.14).
Полученные значения Ат, и Дт2 позволяют найти все теплофизические характеристики исследуемого материала.
Лабораторная установка (рис. 2.15) состоит из трех основных узлов: теплоприемника с исследуемым объектом, нагревателя и измерительной схемы, включающей термопару, гальванометр и реостат.
Теплоприемниками могут служить цилиндры из оргстекла, эбонита, резины, цемента, мрамора и т. д. Боковая поверхность теплоприемника должна иметь хорошую тепловую изоляцию.
Нагреватель должен обеспечивать постоянство температуры соприкасающейся с ним поверхности системы тел на протяжении всего эксперимента. Возможно применение различных типов нагревателей: закрытого водяного, открытого водяного, электрического и радиационного.
Закрытый водяной нагреватель представляет собой цилиндр, дно которого выполнено из меди толщиной 2—3 мм (рис. 2.16). Диаметр дна нагревателя должен быть несколько больше диаметра
Рис. 2.15. Схема экспериментальной установки:
7, 5—соответственно водяной и воздушный термостаты;
2 — теп.топриемник; 4 — исследуемый образен; 5— нагреватель; 6 — дифференциальная термопара; 7—гальванометр;
8 — реостат
217
те пл о приемника. Постоянство
4
Рис. 2.16. Закрытый водяной нагреватель
температуры в нем поддерживается струей воды, подаваемой из гермостата. Температура воды в термостате устанавливается на 10—15 °C выше температуры окружающей среды с помощью контактного термометра и поддерживается с точностью ± 0,05 °C. Найденные значения теплофизических характеристик в таком узком температурном интервале можно считать независящими от температуры.
Вода из термостата по трубе 7 поступает внутрь цилиндра и отдает свое тепло медному дну 6 нагревателя, затем выдавлива
ется через отверстия 5 в перегородке 4 и возвращается по трубе 3 обратно в термостат. Спай термопары расположен в закрытом патрубке 1. Прижимное устройство 2 обеспечивает тепловой контакт между соприкасающимися плоскостями системы.
Электрическая измерительная схема состоит из дифференциальной термопары, гальванометра и реостата, включенного последовательно с гальванометром и служащего для установления начального деления NQ прибора. Один спай дифференциальной термопары расположен в точке О системы, другой — в нагревателе постоянной температуры.
Подготовка проб. Для определения теплофизических характеристик используют образцы из мяса разных видов и сортов, вторичных продуктов убоя, мясных продуктов. Для подготовки проб вырезают образцы, форма и размер которых соответствуют размерам поверхности контакта теплоприемника в лабораторной установке.
Порядок проведения анализа. В кювету теплоприемника лабораторной установки помещают исследуемый образец мяса (мясопродукта). Выводят нагреватель на рабочий режим. С помощью секундомера фиксируют два промежутка времени:
Ati=t2~~ti (2.29)
и
Дт2 = т3-Т1,
(2.29а)
соответствующие двум заданным изменениям показаний галь-218
ванометра:
Л/Vj = Л'! - N\
(2.30)
и
AN2 = N\~
Пользуясь полученными значениями Ат, и Ат9, рассчитывают теплофизические характеристики исследуемого образца.
Коэффициент температуропроводности
а —-------
4рАТ]
(2.32)
Коэффициент теплопроводности
bzh bh Г
2у/рМ1 2
(2.33)
Объемная теплопроводность
X _ 2Ь^рМ^ а
h
Теплоусвояемость, или тепловая активность,
= Ье, 4а
(2.34)
(2.35)
где £ —постоянная теплоприемника. характеризующая его тепловую активность; р и е — безразмерные параметры, которые берут из рабочих таблиц, выражающих зависимость р и е от найденных в опыте значений АТ] и Дт2.
Рабочие таблицы составляются для фиксированных значений Nx/N0, N2/Nq, N./Nq и имеют вид
Лт2
Ат,
= /1(П
(2.36)
Дч
Ат,
(2.37)
Измеряя промежутки времени Ат, и Ат2 в соответствии с рабочей таблицей, т. е. в соответствии с фиксированными выбранны
219
ми значениями Л^/Л^, M,//Vo и Лп/Л^. находят значения параметров р и е, входящих в расчетные формулы.
Рекомендации по определению тепловой активности b теплоприемника и рабочие таблицы для определения безразмерных параметров р и е приведены ниже.
Определение тепловой активности теплоприемника. Формула для вычисления коэффициента температуропроводности не содержит постоянных, характеризующих прибор. Таким образом, метод определения коэффициента температуропроводности относится к абсолютным методам. В формулу, по которой вычисляется коэффициент теплопроводности, входит величина Ь, характеризующая тепловую активность теплоприемника. Для экспериментального определения постоянной величины b необходимо иметь эталонную пластинку, для которой известны либо значение ее коэффициента теплопроводности Хо, либо значение ее объемной теплоемкости Со = (ср)0, либо значение ее тепловой активности
(2.38)
Поместив в прибор эталонную пластинку А толщиной h и проводя измерения величин ДТ[ и Дт2 в соответствии с рабочей таблицей, пример которой приведен ниже, вычисляют искомое значение постоянной по одной из следующих формул:
b = 2Хох/рДт1 е/г (2.39)
= (cl)oA • 2ел//?Дт1 b = Ь0/г. (2.40) (2.41)
Таблица 2.5
Зависимости к г - /г(К); р = ft(K) для N}/No = 0,90; N2/N0 = 0,75; = 0,50
к £ Р К £ Р К £ Р
3,46 3,00 2,22 3,53 2,66 2,13 3,60 2,37 2,05
3,47 2,95 2,21 3,54 2,61 2,12 3,61 2,33 2,04
3,48 2,90 2,19 3,55 2,56 2,11 3,62 2,30 2,025
3,49 2,85 2,18 3,56 2,52 2,10 3,63 2,27 2,012
3,50 2,80 2,17 3,57 2,48 2,08 3,64 2,24 2,00
3,51 2,75 2,15 3,58 2,44 2,07 3,65 2,21 1,99
3,52 2,71 2,14 3,59 2,40 2,06 3,66 2,18 1,98
220
Продолжение
к . X. " . . х х. L ... ... L _ К Е Р
3,67 2,17 1.97 4.12 1,40 1,615 4,96 0.909 1,20
3.68 2,12 1.96 4.13 1.39 1.61 4.98 0.902 1,195
3,69 2.09 1.95 4.14 1,38 1,60 5,00 0,895 1,185
3,70 2.08 1,94 4.15 1.37 1.59 5,02 0,888 1,18
3,71 2.03 1,93 4,16 1,36 1,59 5,04 0,881 1,17
3,72 2.00 1,92 4.17 1,35 1,58 5,06 0,874 1,16
3,73 1,97 1,91 4,18 1,34 1.575 5,08 0,867 1,16
3,74 1,95 1,90 4,19 1,33 1,57 5,10 0,860 1,15
3.75 1,92 1,89 4,20 1,32 1,56 5,12 0,854 1,14
3,76 1,90 1,88 4,22 1,30 1,55 5,14 0,84 1,135
3,77 1,88 1,87 4.24 1,29 1,54 5,16 0,842 1,13
3,78 1,86 1,86 4,26 1,27 1,53 5,18 0,836 1,12
3,79 1,84 1,85 4,28 1,26 1,52 5,20 0,83 1,115
3,80 1,82 1,84 4,30 1,25 1,51 5,22 0,824 1,01
3,81 1,80 1,83 4,32 1,23 1,49 5.24 0,818 1,10
3,82 1,79 1,825 4,34 1,2 1,48 5,26 0,812 1,09
3,83 1,77 1,82 4,36 1,20 1,47 5,28 0,806 1,09
3,84 1,75 1,815 4,38 1,19 1,46 5,30 0,800 1,08
3,85 1,74 1,81 4,40 1,18 1,45 5,32 0,792 1,07
3,86 1,72 1,80 4,42 1,17 1,44 5,34 0,784 1,06
3,87 1,71 1,79 4,44 1,16 1,43 5,36 0,776 1,05
3,88 1,70 1,78 4,46 1,15 1,42 5,38 0,766 1,039
3,89 1,68 1,775 4,48 1,14 1,42 5,40 0,760 1,032
3,90 1,67 1,77 4,50 1,13 1,41 5,42 0,756 1,027
3,91 1,66 1,765 4,52 1,12 1,40 5,44 0,752 1,021
3,92 1,65 1,76 4,54 1,10 1,39 5,46 0,748 1,015
3,93 1,63 1,75 4,56 1,09 1,38 5,50 0,740 1,005
3,94 1,62 1,74 4,58 1,08 1,37 5,52 0,736 1,00
3,95 1,60 1,735 4,60 1,07 1,36 5,54 0,732 0,995
3,96 1,59 1,73 4,62 1,06 1,35 5,56 0,728 0,99
3,97 1,58 1,725 4,64 1,05 1,34 5,58 0,724 0,98
3,98 1,57 1,72 4,68 1,03 1,32 5,60 0,720 0,97
3,99 1,55 1,71 4,70 1,02 1,31 5,62 0,715 0,97
4,00 1,54 1,70 4,72 1,00 1,30 5,64 0,710 0,96
4,01 1,53 1,69 4,74 1,00 1,29 5,66 0,705 0,96
4,02 1,52 1,68 4,76 0,99 1,28 5,68 0,700 0,95
4,03 1,50 1,675 4,78 0,98 1,27 5,70 0,695 0,942
4,04 1,49 1,67 4,80 0,97 1,26 5,72 0,691 0,94
4,05 1,48 1,66 4,82 0,962 1,25 5,74 0,687 0,93
4,06 1,47 1,65 4,84 0,954 1,245 5,76 0,683 0,93
4,07 1,45 1,64 4,86 0,946 1,24 5,78 0,679 0,92
4,08 1,44 1,635 4,88 0,938 1,23 5,80 0,675 0,91
4,09 1,43 1,63 4,90 0,93 1,22 5,82 0,671 0,91
4,10 1,42 1,625 4,92 0,923 1,22 5,84 0,667 0,90
4,11 1,41 1,62 4,94 0,916 1,21 5,86 0,663 0,89
221
Прода/жение
_ Л J Р .LE. ! Л . ! К .... [.... Е ! .1. Р
5.88 0.659 0.89 6.50 0,540 0.71 7.35 0,392 0.45
5.90 0,655 0.88 6.55 0.530 0.69 7.40 0.385 0.44
5.92 0.651 0.88 6.60 0.520 0.67 7.45 0.378 0.42
5,94 0.647 0,87 6.65 0.510 0.66 7.50 0.37 0.41
5,96 0,643 0.86 6.75 0.490 0.63 7.55 0,36 0.40
5,98 0,639 0,86 6,80 0.485 0.62 7.60 0.35 0.38
6.00 0,635 0.85 6,85 0,477 0,60 7.65 0,345 0.37
6,05 0.625 084 6,90 0,470 0.59 7.70 0,34 0.36
6.10 0,615 0,82 6,95 0,460 0,57 7.75 0,33 0.35
6.15 0,608 0.81 7,00 0,450 0,55 7,80 0,32 0,33
6.20 0,600 0,80 7,05 0,440 0,54 7,85 0,31 0.31
6,25 0,590 0,78 7,10 0,430 0,52 7,90 0,30 0,29
6,30 0,580 0,77 7,15 0,425 0,51 7.95 0,29 0,28
6,35 0,570 0,75 7.20 0,420 0,50 8,00 0,28 0.26
6,40 0,560 0,74 7,25 0,410 0,48
6,45 0,550 0,72 7,30 0,400 0,46
Отметим, что если в качестве эталонной пластинки взять тонкий слой дистиллированной воды, объемную теплоемкость которой с достаточной степенью точности можно принять равной 4,19 • 106 Дж/(м3 • град), то определение коэффициента теплопроводности также является абсолютным.
Для вычисления коэффициента теплопроводности можно пользоваться зависимостью
р f Ат
г = 4= = Л Р = fy{K). (2.42)
Результаты экспериментов оформляют в виде таблицы:
Наименование и характеристика образца Номер опыта Т..С T2, с Т„ С
Результаты расчетов оформляют также в виде таблицы рекомендуемой формы:
Наименование и характеристика образца Дт,, с Ат„ с К — Ат,/Ат, Е р а 10s. м2/с X, Вт/(м • град)
222
Сопоставляют и анализируют полученные значения тепло-физических характеристик образцов мяса, субпродуктов I и II категорий и мясопродуктов; самостоятельно формулируют выводы по работе.
Лабораторная работа № 5
ОПРЕДЕЛЕНИЕ МИКРОСТРУКТУРНЫХ ПОКАЗАТЕЛЕЙ МЯСА
Цель работы. Приобрести практический навык определения микроструктуры мяса на основе оптических методов.
Задачи. Подготовка ультратонких срезов мяса и прямое микроскопирование входящих в него тканей.
Объекты исследования. Образцы мышечной ткани разных видов убойных животных и птицы с фиксированным сроком хранения (рекомендуемые режимы хранения — температура 4 °C, относительная влажность воздуха 80 %).
Материалы, реактивы и оборудование. Нейтральный раствор формалина; спирт-ректификат объемной долей 50, 70, 96 % и абсолютный (безводный) этанол, смесь этанол — эфир (1:1); растворы целлоидина массовой долей 2, 6, 12 %, приготовленные на смеси спирта с эфиром; обезвоживающая жидкость; хлороформ; пихтовый бальзам; гематоксилин Эрлиха; водный (или спиртовой) раствор соляной кислоты массовой долей 1 %; раствор нашатырного спирта объемной долей 20—25 %; водный раствор эозина массовой долей 1 %; карбол-ксилол; смесь Ван-Гизона; судан III или IV; колбы или широкогорлые пробирки; банки с широкими горлами и притертыми пробками; деревянные кубики-колодки для наклеивания пропитанного целлоидином материала; микротом; микроскоп; фильтровальная бумага; предметное стекло; яичный белок с глицерином; препаровальная игла; покровное стекло.
Приготовление реактивов. Нейтральный раствор формалина. Для нейтрализации формалин наливают в стеклянную банку с притертой крышкой, на дно банки насыпают порошкообразный мел или магнезию (100 г на 1 дм3 формалина). Содержимое сосуда несколько раз взбалтывают. Через сутки формалин приобретает нейтральную реакцию. На практике исходный нейтральный раствор формалина принимают за 100%-ный и из него непосредственно перед фиксацией готовят необходимые рабочие растворы массовой долей 10 и 20% в соответствии с описанием метода. В качестве растворителя при этом используют водопроводную воду.
Растворы этанола (50 и 70 об.%). Чтобы получить раствор этанола с необходимой объемной долей спирта, исходный
223
спирт-ректификат (96 об.%) разбавляют дистиллированной водой до объема 100 см3. Необходимый для разведения объем исходного спирта определяют по формуле
где а — необходимая объемная доля спирта; b — исходная объемная доля спирта
Абсолютный спирт (безводный). Готовят из этанола объемной долей 96 % путем извлечения из него воды с помощью обезвоженного сульфата меди (выход абсолютного спирта составляет 70 %). Для обезвоживания кристаллический сульфат меди прокаливают в фарфоровой чашке, постоянно помешивая, до приобретения им белого цвета (можно пользоваться готовым безводным сульфатом меди). Прокаленный остывший сульфат меди засыпают в банку с широким горлом с раствором этанола (96 об.%) небольшими порциями (5 % к объему этанола), а затем подсыпают его в небольшом количестве ежедневно в течение 3—4суг до тех пор, пока сульфат меди перестанет менять цвет (останется белым). Приготовленный этанол хранят на осадке сульфата меди. Контроль проводят по признакам: порошок сульфата меди в абсолютном спирте не синеет, а при добавлении ксилола спирт остается прозрачным.
Растворы целлоидина массовой долей 4—6 и 8—12 %. Готовят из смеси абсолютного этанола и чистого (медицинского) эфира в соотношении 1:1. Используют отмытую от эмульсии кино- или рентгеновскую пленку на нитроцеллулоид-ной основе, которая почти не оставляет зольных остатков, после смывания с нее эмульсия прозрачна, имеет слегка желтоватый цвет и хорошо горит.
Для заливки материала в целлоидин в лаборатории необходимо иметь набор одинаковых банок с широкими горлами и притертыми пробками. Из них составляют рабочую батарею с растворами этанола объемными долями 50, 70, 96 и 100 %, со смесью спирта с эфиром, растворами целлоидина I (массовая доля 4—6 %) и II (массовая доля 8—12 %).
Деревянные кубики-колодки для наклеивания пропитанного целлоидином материала. Готовят, как правило, из березы или бука. Колодки подвергают предварительной обработке с целью извлечения дубильных и красящих веществ для обеспечения длительного хранения блоков с наклеенным материалом в растворе спирта объемной долей 70 %. Для этого колодки сначала в течение нескольких часов вываривают в растворе соды массовой долей 2 %, затем в течение нескольких недель или месяцев выдерживают в растворе этанола (70—96 об.%), периодически меняя его до тех пор, пока он не перестанет окрашиваться, и сушат в термостате.
224
Гематоксилин Эр л и х а. 20 см-1 раствора гематоксилина массовой долей 10 % в растворе этанола объемной долей 96 %, 80 см3 раствора этанола объемной долей 96 %, 100 см3 -глицерина, 100 см-1 дистиллированной воды. 3 см3 ледяной уксусной кислоты, 3 г алюмокалиевых квасцов помещают в банку с широким горлом вместимостью не менее 500 см3, завязывают марлей и оставляют на свету для созревания в течение недели.
Раствор эозина (массовой долей 0,25—0,5 %). Сухой эозин растворяют в воде или растворе этанола объемной долей от 40 до 70 %.
К а р б о л-к с и л о л. В теплом сосуде смешивают 1 часть кристаллической карболовой кислоты (температура плавления 42 °C) с 4—5 частями ксилола (или скипидара).
Смесь В а н-Г изона. Смесь гематоксилина Вейгарта с пикрофуксином.
Гематоксилин Вейгарта: смеси Вейгарта I и Вейгарта II.
Вейгарт I: раствор гематоксилина массовой долей 1 % в растворе этанола (96 об. %).
Вейгарт II: 4 см3 раствора FeCl^ • 6НЭО массовой долей 50 %, 1 см3 концентрированной соляной кислоты плотностью 1,15— 1,19 г/см3 и 9 см3 дистиллированной воды.
Судан III (IV). К 0,5 г сухого Судана прибавляют 100 см3 смеси, состоящей из раствора этанола (70 об.%) и ацетона, в соотношении 1:1, настаивают несколько дней при комнатной температуре, изредка взбалтывая, фильтруют и хранят в стеклянной таре.
Обезвоживающая жидкость. Может иметь один из рекомендуемых составов (см3): 1) раствор этанола (96 об.%) — 50; ацетон — 30; хлороформ — 10; эфир — 5; ледяная уксусная кислота —5; 2) этанол абсолютный — 60; ацетон — 20; хлороформ — 10; эфир — 10.
Методические указания. Метод разработан Всероссийским научно-исследовательским институтом мясной промышленности и рекомендован как ускоренный при анализе мяса, который в сочетании с органолептическими показателями позволяет в течение 40—60 мин получить полное представление о состоянии, степени свежести и созревания мяса. В настоящее время внедрен ГОСТ 50372—92 «Мясо. Метод гистологического исследования». Он позволяет раньше, чем физико-химические и биохимические методы, судить об изменениях, происходящих в тканях мяса в процессе прижизненных изменений, технологической обработки и хранения. Результаты гистологического анализа отличаются высокой достоверностью и в ряде случаев могут быть дополнены данными физико-химических, биохимических, органолептических, микробиологических или других исследований.
Процесс гистологического исследования включает в себя следующие основные этапы: фиксацию образцов; подготовку проб к
225
изготовлению срезов тканей: изготовление срезов (микротоми-рование): окраску и помещение срезов под покровное стекло: микроскопию готовых препаратов и обработку результатов исследования.
Фиксация — обработка материала с целью сохранения тканевой структуры такой, какой она была во время отбора образца, и предотвращения ее дальнейших изменений. Чаще всего для этой цели используют формалин (раствор формальдегида массовой долей 40 %).
При фиксации материал промывают проточной водой. Это необходимо для удаления формалина перед дальнейшей обработкой, что обеспечивает равномерность дальнейшего окрашивания срезов.
Микротомирование необходимо для придания анализируемому материалу однородности и скрепления тканей. Обычно для этого используют замораживание (уплотняющая среда — замерзающая вода) или целлоидин. Заключение материала в целлоидин дает возможность получать более тонкие срезы, а также исследовать рыхлые, распадающиеся ткани.
Заключение срезов под покровное стекло предполагает предварительное окрашивание с целью оптического дифференцирования структурных элементов клеток и тканей. Для этого применяют различные красители, контрастные по цвету и избирательные к различным тканевым структурам. Различают два подхода к окрашиванию — прогрессивный (срезы обрабатывают красителем до момента их достаточного окрашивания) и регрессивный (срезы сначала перекрашивают, а затем избыток красителя удаляют). В практике чаще применяют регрессивный способ окраски. Окрашенные срезы исследуют под микроскопом.
Подготовка проб. Образцы размером 30x 30x 30 мм и массой 35—40 г берут от мясной туши, полутуши или ее части. Во время отбора образцов одновременно оценивают органолептическое состояние исследуемого мяса и фиксируют данные в тетради.
Образец вырезают острым ножом перпендикулярно поверхности (в глубь мышц), чтобы одна из сторон образца была наружной поверхностью туши или ее части, а другая — поверхностью разруба или разреза. При этом избегают сдавливания мышц, обмывания и очистки этих поверхностей, а также прикосновения к ним посторонних предметов или пальцев рук. Каждый образец подлежит маркировке.
Для быстрой фиксации материала вырезанные из образцов пробы помещают в небольшую колбу или широкую пробирку, заливают четырьмя или пятью объемами нейтрального водного раствора формалина (объемной долей 10—20 %) и подогревают на пламени горелки, не доводя до кипения. При появлении пузырьков воздуха подогрев прекращают, содержимое осторожно встряхивают и снова подогревают до появления пузырьков воздуха. Так
226
повторяют трижды (в течение 1—2 мин). Критерием окончательной фиксации проб служит одинаковый серый цвет кусочков как на поверхности, так и в центре. При комнатной температуре для полной фиксации требуется 18—24 ч.
Порядок проведения анализа. Из фиксированного материала вырезают небольшие кусочки размером 10x5x4 мм. толщина проб не должна превышать 4 мм, а объем обезвоживающей жидкости должен составлять не менее 15—20 см3 на одну пробу.
Последовательно обезвоживают пробы в растворах этанола объемными долями 50, 70, 96 % (два раза) и абсолютном спирте в течение 24 ч, а затем пропитывают раствором целлоидина массовой долей 4—6 % (I) в течение 6—10 сут или 8 — 12 % (II) — 3—5 сут.
По окончании пропитывания пробы наклеивают на кубики и уплотняют в парах хлороформа или эфира. Для этого материал из густого целлоидина переносят непосредственно на деревянные кубики (целлоидин берут с избытком) и помещают в эксикатор, куда одновременно ставят одну-две открытые банки с хлороформом. По окончании уплотнения (4—6 ч) блоки переносят для хранения в раствор спирта (70 об.%). Изготавливать срезы из материала, заключенного в целлоидин, желательно через 10—24 ч, но не раньше 3—4 ч после помещения блоков в спирт.
Для ускорения процесса пробы исследуемых материалов толщиной 2 мм фиксируют и одновременно обезвоживают при 37 °C последовательно в жидкостях: I — 1 ч, II—1,5 ч; в абсолютном спирте — 3 ч (первая порция — 1,5 ч, вторая — 1,5 ч), в смеси спирта и эфира — 30 мин. Затем пробы при той же температуре погружают в растворы целлоидина массовыми долями 2, 6 и 12 % на 18 ч каждый. Далее пробы наклеивают на деревянные кубики, подсушивают на воздухе в течение 20—40 мин и погружают для уплотнения в хлороформ на 30—40 мин. Хранят блоки в растворе спирта (70 об.%)
Для изготовления тонких срезов применяют специальный прибор-микротом, который состоит из станины, микроматричного устройства, держателей пробы и ножа. Микротом закрепляют на краю стола, слева устанавливают баллон с диоксидом углерода вертикально вентилем вниз. Перед началом резки открывают вентиль баллона. Держатель ножа перемещают в направлении к себе до отказа и опускают микротомный столик при помощи рукоятки микрометрического винта. Зафиксированную и промытую в воде пробу мяса кладут на столик микротома и обильно смачивают водой из пипетки. В держателе закрепляют микротомный нож и при помощи микрометрического винта поднимают предметный столик до соприкосновения образца мяса с ножом. Нож отводят от себя, прижимая образец к столику, и начинают его замораживать, выпуская диоксид углерода небольшими порциями. Объект замораживают до появления металлического звука при постукивании
227
по нему пинцетом или скальпелем. Краем ножа с одновременной подачей предметного столика при помощи микровинта вверх выравнивают поверхность образца. Затем нож возвращают в рабочее положение. Разрезание должно производиться средней частью лезвия ножа. Установив регулятор в положение 15 или ЗОмкм. приступают к изготовлению срезов в плоскости, параллельной продольной оси мышечных волокон.
Полученные срезы снимают с ножа кисточкой или пальцем в направлении к лезвию и переносят в кристаллизатор с водопроводной водой на несколько секунд для расправления. Под неповрежденный срез быстро подводят предметное стекло, обработанное яичным белком с глицерином для наклеивания, и извлекают срез из воды, удерживая его на середине стекла препаровальной иглой. Затем срез накрывают плотной сухой фильтровальной бумагой (три-четыре слоя) и, проглаживая бумагу ребром ладони, наклеивают срез на предметное стекло. Затем срезы окрашивают, обезвоживают, осветляют и заключают в бальзам под покровное стекло.
При использовании санного микротома целлоидиновый блок фиксируют на предметном столике специальным зажимом (продольная ось пробы должна располагаться параллельно продольной оси микротома). Нож устанавливают в держателе под острым углом микротома (обычно угол 13—15°). Предварительно рекомендуется выровнять поверхность целлоидинового блока. Нож и пробу во время разрезания постоянно смачивают раствором спирта (70 об.%). Полученные срезы кисточкой расправляют на микротомном ноже, перемещают его к краю и, подхватывая снизу, переносят в раствор спирта (70 об.%).
Для быстроты и экономии реактивов при окрашивании используют 8 биологических стеклянных стаканов вместимостью 40 см3 и высотой 85—90 мм с крышками, закрепленными в штативе. Переносить предметные стекла с наклеенными срезами следует аккуратно, чтобы не перенести раствор из одного стакана в другой. В ходе окраски срезы промывают водой в больших чашках-кристаллизаторах.
Срезы, фиксированные целлоидином или замороженные, окрашивают по одной схеме. При этом важно выдерживать последовательность и время обработки (мин): гематоксилин Эрлиха — 3—5; водопроводная вода — 1—2; водный или спиртовой (объемная доля этанола 70 об.%) раствор соляной кислоты массовой долей 1 % — 5—30; подкисленная аммиаком водопроводная вода (одна капля раствора нашатырного спирта объемной долей 10— 25 % на 100см3 воды—до приобретения срезом синего оттенка) — 2; водный раствор эозина массовой долей 1 % — 1; дистиллированная вода — 1; раствор спирта объемной долей 50 % — 1, 70 % — 1, 96 % — 1; карбол-ксилол — 1; ксилол — 1.
228
Окрашенные препараты микроскопируют. При этом фиксируют: ядра окрашены в темно-синий, светло-синий или лиловый цвет, мышечные волокна и соединительнотканные прослойки принимают различные тона красного или розового цвета.
При необходимости подробного изучения микроструктуры соединительных и жировой тканей используют избирательные методы окрашивания по Ван-Гизону и Судану (III или IV). Схемы представлены ниже.
Для соединительных тканей (метод Ван-Газона)'.
гематоксилин Вейгерта (Эрлиха) водопроводная вода смесь Ван-Гизона 3—5 мин 1 мин 5—10 мин
дистиллированная вода этанол (96 об.%) карбол-ксилол ксилол 1 мин 1 мин 1 мин 1 мин
Окрашенные срезы помещают под покровное стекло в пихтовый бальзам. В поле зрения наблюдают коллагеновые волокна, окрашенные в красный, протоплазму — в желтый, ядра — в коричневый или темно-синий цвет.
Для жаровой ткани'.
этанол (70 об.%) 0,5—1,0мин
судан III или IV 5,0—20,0 мин
Окрашенные срезы промывают водопроводной водой (10— 30 мин), помешают в гематоксилин Эрлиха или Майера (2— 3 мин).
Перекрашенные в гематоксилине срезы дифференцируют в водном растворе соляной кислоты массовой долей 1 % (до розового цвета) и промывают в водопроводной воде (20—30 мин), перекладывают в дистиллированную воду (3—5 мин), извлекают на предметное стекло, наносят капли глицерина или глицерин-жела-тина и накрывают покровным стеклом.
В результате окраски жир приобретает оранжево-красный цвет, ядра — темно-синий.
Исследование препарата начинают с его просмотра при небольшом увеличении, а затем применяют большее увеличение.
Наблюдая в микроскоп, устанавливают параллельно шкалы объект- и окуляр-микрометров и совмещают их нулевые отметки. Затем определяют, сколько делений объект-микрометра точно совпадает с делениями окуляр-микрометра.
229
Цену деления окуляр-микрометра определяют по формуле
ас т
Л/ =
(2.44)
где г/— отсчи! ан ное число деления по шкале объскт-микромс1ра: (.--известное значение одного деления шкалы объект-микрометра (Юмкмк /?---соответствующее число деления шкалы окуляр-микрометра.
П р и м с р. В 32 делениях объект-микрометра полностью укладывается 16 делений окуляр-микрометра: значение одного деления шкалы объект-микрометра известно: 0.01 мм или 10 мкм. Цена деления шкалы окуляр-микрометра составит
,, 3210 _А
М = - =20 мкм.
16
Зная цену одного деления окуляр-микрометра при заданном увеличении, приступают к измерению объектов. При этом соответствующее длине измеряемого объекта число делений окуляр-микрометра необходимо умножить на 20 мкм (цена одного деления).
Составляют протокол гистологических исследований, в котором приводят зарисовки и характеристику микроструктуры проб. При этом обращают внимание на состояние мышечных волокон, клеточных ядер, соединительнотканных образований, выраженность исчерченности мышечного волокна. Полученные данные сопоставляют с данными органолептической оценки.
Лабораторная работа № 6
ОПРЕДЕЛЕНИЕ ВЛАГОСВЯЗЫВАЮЩЕЙ СПОСОБНОСТИ(ВСС) МЯСА
Цель работы. Приобрести практический навык определения способности мяса и мясного сырья связывать воду.
Задачи. Подготовить модельный мясной фарш и определить его способность связывать воду методами прессования и центрифугирования.
Объекты исследования. Образцы мышечной ткани убойны,х животных (птицы) разных видов и сортов. В качестве объектов сравнения рекомендуется использовать образцы имеющих технологическое значение жировой, соединительной тканей с различных анатомических участков туши животных, вторичного мясного сырья (субпродукты II категории, мясо механической дообвалки и т. д.).
Материалы и оборудование. Груз массой 1 кг; планиметр; полиэтиленовые пробирки; центрифуга лабораторная; фильтровальная бумага; стеклянные палочки; стеклянные (или плексигласовые) пластинки.
230
Методические указания. На практике ВСС чаше всего определяют с помощью прессования или центрифугирования.
Метод прессования основан на выделении волы испытуемым образцом при легком его прессовании, сорбции выделяющейся воды фильтровальной бумагой и определении количества отделившейся влаги по плошали пятна, оставляемого ею на фильтровальной бумаге. Достоверность результатов обеспечивается трехкратной повторностью определений.
Метод центрифугирования основан на выделении жилкой фазы под действием центробежной силы из исследуемого объекта, находящегося в фиксированном положении. Количество последней зависит от степени взаимодействия влаги с «каркасной фазой» объекта. Метод условен. Достоверность результатов может быть обеспечена при грех-четырехкратной повторности определений.
По заданию преподавателя рекомендуется составить модельные композиции фарша из различных видов сырья.
Подготовка проб. Пробы мышечной ткани животных разных видов и сортов массой по 200—250 г отбирают в колбасном цехе на участке обвалки и жидовки мяса или жилуют в соответствии с нормируемыми показателями массового содержания соединительной ткани и жира.
При жиловке говядину любой упитанности разделяют на три сорта в зависимости от массовой доли соединительной ткани и жира. К высшему сорту относят мышечную ткань без жира и соединительной ткани; к I сорту — мышечную ткань, в которой допускается наличие соединительной ткани в виде пленок не более 6 % к массе мяса; ко II сорту — мышечную ткань, содержащую до 20 % соединительной ткани и жира.
При жиловке свинину разделяют в зависимости от массового содержания жировой ткани на три сорта: нежирную, содержащую не более 10% жировой ткани; полужирную — 30—50 % жировой ткани; жирную — более 50 % жировой ткани.
Пробы субпродуктов I и II категорий массой по 50—100 г отбирают в цехе обработки субпродуктов или на соответствующих участках цеха первичной обработки скота.
Жилованную говядину, свинину, субпродукты I и II категорий тщательно измельчают на волчке или мясорубке с диаметром отверстий решетки 2—3 мм; гомогенизаторе. Замороженное мясо механической обвалки (или дообвалки) предварительно размораживают.
1. МЕТОД ПРЕССОВАНИЯ
Порядок проведения анализа. При определении ВСС этим методом навеску мясного фарша массой 0,3 г взвешивают на торзионных весах на кружке из полиэтилена диаметром 15—20 мм (диаметр кружка должен быть равен диаметру чашки весов),
231
после чего ее переносят на беззольный фильтр, помещенный! на стеклянную или плексигласовую пластинку так. чтобы навеска оказалась под кружком.
Сверху навеску накрывают такой же пластинкой, что и нижнюю. устанавливают на нее груз массой 1 кг и выдерживают в течение 10 мин. После этого фильтр с навеской освобождают от груза и нижней пластинки, а затем карандашом очерчивают контур пятна вокруг спрессованного мяса.
Внешний контур вырисовывается при высыхании фильтровальной бумаги на воздухе. Площади пятен, образованных спрессованным мясом и адсорбированной влагой, измеряют планиметром.
Размер влажного пятна (внешнего) вычисляют по разности между общей площадью пятна и площадью пятна, образованного мясом. Экспериментально установлено, что 1 см2 площади влажного пятна фильтра соответствует 8,4 мг влаги.
Массовую долю связанной влаги в образце вычисляют по формулам
л-j = (Л7 — 8,45) 100/т(), (2.45)
х2 = (./И- 8,45) 100/Л/, (2.46)
где А'] —- массовая доля связанной влаги в мясном фарше, % к массе мяса: лз — го же, % к обшей влаге; М — общая масса влаги в навеске, мг; S — площадь влажного пятна, мг; /и0 —масса навески мяса, мг.
2. МЕТОД ЦЕНТРИФУГИРОВАНИЯ
Порядок проведения анализа. При определении ВСС данным методом образцы мяса массой около 4 г помещают в полиэтиленовую пробирку с перфорированным вкладышем, укрепленным таким образом, чтобы был обеспечен необходимый зазор для стекания жидкости. Пробы центрифугируют в течение 20 мин при частоте вращения 100 с-1. После центрифугирования пробы взвешивают и к массе пробы добавляют массу веществ, содержащихся в отделенной центрифугированием жидкости. Эту массу веществ определяют высушиванием при 105 °C до постоянной массы. Для расчета количества связанной влаги необходимо иметь данные о содержании влаги в объекте.
Массовую долю связанной влаги (%) рассчитывают по формуле х= (//?, + т3 — т2) 1ОО//ио, (2.47)
где т0, /«] — масса навески соответственно до и после центрифугирования, г;
— масса сухого остатка выделившейся жидкости, г; т2 — масса сухого остатка в навеске, г.
232
Экспериментальные данные рекомендуется оформить в виде таблицы:
Образец
Состав модельного фарша
ВСС. определенная по методу
прессования центрифугирования
Сравнивая компонентный состав мясных фаршей, делают выводы и самостоятельно формулируют заключение по работе.
Лабораторная работа № 7
ОПРЕДЕЛЕНИЕ ОСНОВНЫХ ФУНКЦИОНАЛЬНО-ТЕХНОЛОГИЧЕСКИХ СВОЙСТВ МЯСНЫХ ФАРШЕЙ
Цель работы. Приобрести практический навык определения ВУС, ЖУС, ЭС и СЭ в мясных системах.
Задачи. Подготовить модельные мясные фарши и определить ВУС, ЖУС, ЭС и СЭ гравиметрическими и рефрактометрическими методами.
Объекты исследования. Мясные фарши, составленные из различного мясного и немясного сырья в произвольных пропорциях.
Материалы, реактивы и оборудование. Молочный жиромер; стеклянные палочки; бюкс; сушильный шкаф; бумажный фильтр; фарфоровая ступка; прокаленный песок; а-монобромнафталин; складчатый бумажный фильтр; рефрактометр; консервные банки; водяные бани.
Методические указания. Влагоудерживающая способность мясного фарша определяется как разность между массовой долей влаги в фарше и количеством влаги, отделившейся в процессе термической обработки, а жироудерживающая способность — как разность между массовой долей жира в фарше и количеством жира, отделившимся в процессе термической обработки.
Отношение объема эмульгированного масла к общему его объему в системе называют эмульгирующей способностью. В это определение входит и понятие стабильности эмульсии, проявляющейся за промежуток времени, начиная от окончания эмульгирования до момента измерения при фиксированных условиях проведения эксперимента.
Устойчивость фарша характеризуется количеством влаги и жира, связанных фаршевой эмульсией, и определяется отношением массы выделившегося в процессе тепловой обработки бульона и жира к массе фарша, взятого на исследование.
Возможность последовательного определения в одной навеске нескольких функциональных показателей (метод Р. М. Салавату-
233
линой и др.) позволяет снизить погрешность за счет неоднородности химическою состава и лабильности свойств сырья. При этом определение и расчет устойчивости фаршевой эмульсии, ВУС и ЖУС по массе фактически связанных компонентов фаршевой эмульсии производится в условиях, максимально приближенных к производственным. Мегодика отличается просто!ой и высокой воспроизводимостью результатов.
Подготовка проб. Осуществляется в соответствии с рекомендациям!! к лабораторной работе № 6.
1. ОПРЕДЕЛЕНИЕ БЛАГО УДЕРЖИВАЮЩЕЙ СПОСОБНОСТИ
Порядок проведения анализа. Навеску тщательно измельченного мяса массой 4—6 г равномерно наносят стеклянной палочкой на внутреннюю поверхность широкой части молочного жиромера. Его плотно закрывают пробкой и помещают узкой частью вниз на водяную баню при температуре кипения на 15 мин, после чего определяют массу выделившейся влаги по числу делений на шкале жиромера.
Влагоудерживающая способность мяса (%)
ВУС=В-ВВС, (2.48)
влаговыделяющая способность мяса (%)
ВВС^ши-1 • 100, (2.49)
где В — общая массовая доля влаги в навеске, %; а — цена деления жиромера: а = 0,01 смп — число делений на шкале жиромера; т — масса навески, г.
2. ОПРЕДЕЛЕНИЕ ЖИРОУДЕРЖИВАЮЩЕЙ СПОСОБНОСТИ
Порядок проведения анализа. Предварительно рассчитывают ВВС по п. 1, находят массу мяса, оставшегося в жиромере, с точностью ± 0,0001 г. Мясо помещают в бюкс и высушивают до постоянной массы при температуре 150 °C в течение 1,5 ч. После высушивания берут навеску массой (2,0000 ± 0,0002) г, помещают в фарфоровую ступку, куда добавляют 2,5 г (1,6 см3) мелкого прокаленного песка и 6 г (4,3 см3) а-монобромнафталина. Содержимое ступки тщательно растирают в течение 4 мин и фильтруют через складчатый бумажный фильтр.
Испытуемый раствор (3—4 капли) равномерно наносят стеклянной палочкой на нижнюю призму рефрактометра. Призмы закрывают, скрепляют винтом. Луч света направляют при по-234
моши зеркала на призму рефрактометра, устанавливая зрительную трубу так. чтобы были отчетливо видны пересекающиеся нити (алиала). Алиалу передвигают io iex пор. пока граница между освещенной и темной частями нс совпадет с точкой пересечения нитей, отсчитывают показатель преломления. Одновременно определяют показатель преломления а-мо-нобромнафталина.
Определения повторяют несколько раз. используя при расчете средние данные.
Жироудерживающая способность мяса (О)
ЖУС - 100. (2.50)
где у, — массовая доля жира в навеске после термообработки. '7; g, - ю же. до термообработки, Ы.
Массовая доля жира в навеске (%)
у --- 11 04(x(/7j - //-,)/??[| /т. (2.51)
где а — коэффициент, характеризующий такое содержание жира в растворителе, которое изменяет показатель преломления на 0.0001 %; /у и //, -- показатели преломления соответственно чистого растворителя и испытуемого раствора; — масса 4,3 см3 о.-монобромнафталина, г; ш — масса навески, i.
Коэффициент о. устанавливают опытным путем при сопоставлении результатов определения массовой доли жира методами Сокслета и рефрактометрическим.
а = £ф/(104Д< (2.52)
уф = (т • 100)/тр, (2.53)
где £ф — массовая доля жира в фильтрате. %; Ал — разность между показателями преломления чистого растворителя и испытуемого фильтрата; т — масса жира в навеске, определенная в аппарате Сокслета, г; /?/ —масса навески растворителя, г.
Коэффициент о. для некоторых продуктов приведен ниже.
Продукт Мясной порошок Сосиски: свиные русские Колбаса ливерная Коэффициент а 0,0470 0.0375 0.0369 0,0394 235
3. ОПРЕДЕЛЕНИЕ ЭМУЛЬГИРУЮЩЕЙ СПОСОБНОСТИ И СТАБИЛЬНОСТИ ЭМУЛЬСИИ
Порядок проведения анализа. Навеску измельченного мяса массой 7 г суспензируют в 100 см3 воды в гомогенизаторе (или миксере) при частоте вращения 66,6 с~' в течение 60 с. Затем добавляют 100 см3 рафинированного подсолнечного масла и смесь эмульгируют в гомогенизаторе или миксере при частоте вращения 1500 с-1 в течение 5 мин. После этого эмульсию разливают в 4 калиброванные центрифужные пробирки вместимостью по 50 см3 и центрифугируют при 500 с 1 в течение 10 мин. Далее определяют объем эмульгированного масла.
Эмульгирующая способность (%)
ЭС = уЬ100, (2.54)
где И,—объем эмульгированного масла, см3; И—общий объем масла, см3.
Стабильность эмульсии определяют путем нагревания при температуре 80 °C в течение 30 мин и охлаждения водой в течение 15 мин. Затем заполняют эмульсией 4 калиброванные центрифужные пробирки вместимостью по 50 см3 и центрифугируют при частоте вращения 500 с-1 в течение 5 мин. Далее определяют объем эмульгированного слоя.
Стабильность эмульсии (%)
СЭ = ~-100, (2.55)
где И, — объем эмульгированного масла, см3; И2 — общий объем эмульсии, см3.
4. ОПРЕДЕЛЕНИЕ БЛАГО- И ЖИРОУДЕРЖИВАЮЩЕЙ СПОСОБНОСТЕЙ И УСТОЙЧИВОСТИ ФАРШЕВОЙ ЭМУЛЬСИИ В ОДНОЙ НАВЕСКЕ (МЕТОД Р. М. САЛАВАТУЛИНОЙ и др.)
Порядок проведения анализа. Образцы фарша массой 180—200 г, помещенные в герметично закрытые консервные банки № 3, взвешивают и подвергают тепловой обработке при режимах, соответствующих производственным (варка в водяной бане при температуре 78—80 °C в течение 1 ч, охлаждение в проточной воде до температуры 12—15 °C).
Затем консервные банки вскрывают, выделившийся бульон и скопившийся жир переносят в предварительно взвешенные алюминиевые бюксы. После удаления бульона и жира фарш промокают фильтровальной бумагой и взвешивают.
236
Бюксы с бульоном помещают в сушильный шкаф и сушат до постоянной массы при 103—105 °C. Определяют массовую долю влаги, выделившейся при тепловой обработке фарша, и влагоудерживающую способность фарша.
Из бюксов с остатками бульона и жира экстрагируют жир 10— 15 см3 растворителя (смесь хлороформа с этанолом в соотношении 1 : 2). Экстрагирование жира проводят в течение 3—4 мин с трехчетырехкратной повторностью. Установив массовую долю оставшегося жира после тепловой обработки фарша, рассчитывают жироудерживающую способность.
Устойчивость фаршевой эмульсии (% к массе фарша)
т — птг.,
УЭ = . -100; (2.56)
т
т
УЭ = —-100; (2.57)
т
т = т6н-тб; (2.58)
(2.59) где т — масса навески фарша, г; —масса всего отделившегося бульона с жиром, г; /пс — масса сгустка фарша после термообработки, г; /ибн — масса герметизированной консервной банки с навеской фарша, г; — масса консервной банки, г.
Влагоудерживающая способность (% к массе фарша)
тк
ВУС = W - • 100, (2.60)
т
где W— массовая доля влаги в фарше. %; та — масса в исследуемом бульоне, г; тб2 — масса исследуемого бульона с жиром, г.
Жироудерживающая способность фарша (% к массе фарша)
Я7б1 /иж
жус = ж,,,- Тжж. (2 61)
/Ид т
где Жф — массовая доля жира в фарше, %; /иж — масса жира в исследуемом бульоне, г.
Экспериментальные данные для различных вариантов модельных фаршей оформляют в виде таблицы:
Массовая доля компонентов в составе модельного фарша, % ВУС. % • ЖУС, % эс, % сэ, %
237
По результатам определений делают выводы о технолот-ческой функциональности сырья и формулируют обшее заключение по работе.
Лабораторная работа № 8
ОПРЕДЕЛЕНИЕ ГЕЛЕОБРАЗУЮЩЕЙ СПОСОБНОСТИ ЖИВОТНЫХ И РАСТИТЕЛЬНЫХ БЕЛКОВ
Цель работы. Приобрести практический навык определения ге-леобразуюшей способности.
Задачи. Получить гели (студни) миофибриллярных белков и проанализировать их.
Объекты исследования. Фракция миофибриллярных белков мышечной ткани; продукт гидролиза коллагена — пищевой желатин; плазма крови; образцы растительных белковых препаратов различной степени очистки и технологических форм на основе сои, чечевицы или других культур; агар (или агароид); пектин.
Материалы, реактивы, оборудование. Солевой раствор Вебера; поваренная соль; марлевый фильтр; сахар; плитка электрическая; воронки лабораторные; вата; приборы Валента и Тарр—Бейкера; ватерпас; стеклянный сосуд; прокаленный песок; грибовидная насадка; медицинский шприц; поршень; этанол; резервуар; груз (100—500 г).
Методические указания. В коллоидной химии гелями называют твердообразные дисперсные системы, внутри которых распределена жидкость. В отечественной литературе гели, образованные из растворов органических высокомолекулярных соединений, называют студнями. В соответствии с этими названиями иногда термин «структурообразование» заменяют на «гелеобразование», или «студнеобразование».
Склонностью к образованию коагуляционных структур обладают асимметричные (нитевидные или лентовидные) частицы с высоким, более 100, осевым соотношением (отношение длины к ширине). Даже в небольших концентрациях они способны образовывать сплошной рыхлый пространственный каркас в виде единого агрегата благодаря неравномерному распределению центров коагуляции по концам частиц. В петлях образующегося каркаса фиксируется дисперсионная среда. Такое структурообразование называется застудневанием, а образующаяся дисперсная система — лиогелем или студнем.
Переход жидкости в лиогель сопровождается изменением структурно-механических свойств системы; возникает жесткость, обусловленная наличием в системе непрерывного каркаса, в котором цепные частицы соединены локальными связями в центрах наибольшей лиофобности.
238
При напряжении выше предела прочности структурный каркас деформируется в результате смешения частиц относительно друг друга и контактов между ними, что внешне выражается в течение всей системы.
Эмульсионная природа мясных фаршей обусловливает высокую концентрацию белков в адсорбционных стабилизирующих слоях. Как правило, концентрация белка достаточно высокая и превышает критическую концентрацию гелеобразования. Это предопределяет возможность формирования гелевых структур в межфазных слоях и обусловливает физическую и химическую стабилизацию жира и влаги в мясопродуктах из тонкоизмель-ченного фарша, обеспечивая тем самым качество продукта. Готовые продукты приобретают свойства гелей, в которые включены капельки жира. При этом гелеобразование предусматривает формирование непрерывной белковой сетки, имеющей определенную степень упорядоченности.
Среди белков животных тканей основную роль в формировании структуры мясных эмульсий и последующем термотропном гелеобразовании играет миозин.
При достаточно высокой степени измельчения и под воздействием термообработки коллаген хорошо гидролизуется с образованием глютина и желатоз, которые обладают выраженной водосвязывающей и застудневающей способностью, что позволяет частично стабилизировать свойства готовых мясных изделий при использовании коллагенсодержащего сырья в виде белковых препаратов, эмульсий и гидролизатов.
Все белки плазмы крови способны образовывать гели при нагревании. Фибриноген имеет выраженную гелеобразующую способность, переходя под воздействием внешних факторов в фибрин и образуя пространственный каркас. Введение в плазму неплазменных белков, клетчатки, пектина существенно увеличивает прочность гелей.
Белки яйца обладают высокими гелеобразующими свойствами, особенно в присутствии альбуминов сыворотки крови и других компонентов.
Растительные белковые добавки в комбинированных пищевых системах проявляют свойства, аналогичные структурообразующим мышечным белкам нежирного мяса. Знания механизмов образования, способов практического получения и анализа свойств студней необходимы в технологической практике.
Работа состоит из двух этапов: получения гелей и исследования их свойств.
Экстракцию миофибриллярной фракции белков проводят солевым раствором Вебера или раствором поваренной соли эквивалентной молярной концентрации. Масса образца мышечной ткани 100 г.
239
Подготовка проб. Готовят исходный раствор образца желатина массовыми долями 10, 15 или 25 % в пересчете на обеззоленное сухое вещество (в соответствии с полученным заданием).
Массу навески желатина (г) для приготовления 1000 г исходного стандартного раствора заданной массовой долей абсолютно сухого обеззоленного вещества вычисляют по формуле
Х=т- 1000/1100 -(PTZ+3)|. (2.62)
где Щ— массовая доля влаги. %: з — массовая доля золы в образце желатина, %.
Навеску, взвешенную с точностью +0,01 г, помещают в колбу с пришлифованной пробкой, приливают необходимый объем дистиллированной воды, плотно закрывают колбу пробкой и оставляют для набухания в течение 1—2 ч. Для полного растворения колбу с набухшим желатином помещают в термостат, повышая температуру от 40 до 75 °C, колбу с содержимым периодически встряхивают. Приготовленный раствор фильтруют через два слоя марли.
Плазму крови получают сепарированием или центрифугированием цельной стабилизированной крови в течение 5—8 мин при частоте вращения барабана 25—42 с”1. Плазму (надосадочную жидкость) отделяют декантацией.
Растворы агара готовят следующим образом: к навеске сухого агара массой 1,7 г (анфельция) или 2,5 г (фурцелларан), взвешенной с точностью ± 0,001 г, добавляют объем дистиллированной воды из расчета, чтобы общая масса раствора была 200 г, и оставляют для набухания не менее чем на 1 ч, после чего нагревают на водяной бане с обратным холодильником до полного растворения агара.
Для агара из фурцелларана готовят раствор с сахаром, добавляя в смесь после полного растворения агара 140 г сахара, и продолжают нагревать, доводя всю массу до кипения. Массу кипятят в течение 2—3 мин, затем взвешивают и продолжают нагревать до тех пор, пока масса агарно-сахарного раствора не будет доведена до 200 г.
Если раствор содержит нерастворимые примеси, его фильтруют в горячем состоянии через воронку с сухой ватой.
1.ПОЛУЧЕНИЕ ГЕЛЕЙ
Порядок проведения анализа. Для получения гелей миофибрил-лярных белков раствор миофибриллярных белков помещают в лабораторные стаканы и нагревают на водяной бане, визуально фиксируя температуру и время формирования геля.
Растворы желатина различной массовой долей (0,5—5 %) помещают в соответствующую лабораторную посуду, оставляют для гелеобразования при заданной температуре из рекомендуемого температурного интервала (0—10—20—30 °C). Через каждые 20—30 мин визуально фиксируют образование геля.
Готовят смесь из плазмы крови и натурального (морковного или тыквенного) сока с мякотью при соотношении компонентов 1 : 1 по 240
объему, оставляют для телеобразования при температуре 16—22 °C. Каждые 20—30 мин визуально фиксируют образование геля.
Приготовленный раствор агара, агароида или агаро-сахарный раствор разливают в подготовленные сухие стаканы, которые затем помещают в горизонтально установленный сосуд с плоским дном (например, кристаллизатор), заполненный водой температурой 20 °C. Затем стаканы с агаро-сахарным раствором термостати-руют при 30—60 °C. Причем уровень воды в сосуде должен быть немного выше уровня раствора в стаканах. Стаканы с раствором выдерживают в сосуде при температуре 20 °C, поддерживая ее добавлением холодной или теплой воды.
При использовании сухих растительных белковых препаратов их предварительно гидратируют в следующих условиях: соотношение белковый препарат — вода равно 1 : (2—2,5) для муки, 1 : 3 для концентрата, 1 :4 для изолята, температура воды 15—25 °C, продолжительность обработки в куттере или мешалке 1—3 мин.
Визуально фиксируют образование геля и промежуток времени, прошедший до гелеобразования. По результатам испытаний различных пищевых систем строят диаграмму, иллюстрирующую зависимость скорости гелеобразования от вида и состава дисперсионной среды.
2.ОПРЕДЕЛЕНИЕ ФИЗИЧЕСКИХ ПОКАЗАТЕЛЕЙ СТУДНЕЙ
Порядок проведения анализа. Для исследования образцов гелей берут пять стаканов диаметром 4,0—4,5 см вместимостью по 100 см3. На них наносят метки, соответствующие объему 30 см3, и
далее используют для определения прочностных характеристик на приборе Валента (рис. 2.17).
Для определения студнеобразующей способности образцов на приборе Тарр-Бейкера формование гелей проводят в стаканах конической формы объемом по 50 см3, а температуры плавления студней — в двух пробирках для каждого образца.
Рис. 2.17. Прибор Валента:
ватерпас; 2— штатив; 3 — сосуд для груза; 4— площадка для сосуда; 5— шток; 6— передвижной кронштейн; 7— насадка для испытуемого студня; 8— стакан; 9—основание; 10 — регулировочный винт
241
2.1. ОПРЕДЕЛЕНИЕ ПРОЧНОСТИ СТУДНЯ НА ПРИБОРЕ ВАЛЕНТА
Порядок проведения анализа. Стаканы с образовавшимся студнем ставят на основание прибора Валента, горизонтально установленного при помощи ватерпаса. На поверхность студня осторожно опускают грибообразную насадку. Площадь поверхности, на которую давит насадка, равна 2 см2. В сосуд, помещенный на площадку, медленно насыпают сухой промытый и прокаленный песок до тех пор, пока насадка, надавливая на студень, не прорвет его. Масса подвижной системы, состоящей из грибовидной насадки, штока, площадки и сосуда для груза, должна быть 90—100 г. Насадка должна быть изготовлена из антикоррозионного металла с полированной шаровой поверхностью. Нагрузку следует подавать равномерно, приблизительно по 10—12 г/с.
Перед опытом рекомендуется проверить равномерность подачи нагрузки. Для этого в стакан в течение 1 мин насыпают песок с принятой скоростью, затем взвешивают стакан с нагрузкой. При отклонении от рекомендуемых значений проверку повторяют, соответственно изменив скорость подачи груза (песка).
Прочность студня при измерении показателя на приборе Валента (г/см2)
CK = m/S, (2.63)
где т — масса песка, сосуда и стержня с насадкой и площадкой, г; 5—площадь поверхности насадки; 5= 2 см2.
2.2. ОПРЕДЕЛЕНИЕ СТУДНЕОБРАЗУЮЩЕЙ СПОСОБНОСТИ НА ПРИБОРЕ ТАРР—БЕЙКЕРА
Порядок проведения анализа. Основой метода является определение максимальной прочности студня на разрыв. Определение проводят на приборе Тарр—Бейкера (рис. 2.18), который состоит из стеклянного стандартного поршня 9, напорного сосуда с водой 4, буферного сосуда 8, манометра 5, заполненного четыреххлористым углеродом, подкрашенным иодом. Шкала манометра имеет диапазон от 0 до 90 см с ценой деления 1 см. Система прибора снабжена кранами 7, 2, 3, 6 и 7.
В качестве стандартного поршня используют обычный медицинский шприц. Основным рабочим органом является площадка поршня 10, размеры которой должны точно соответствовать указанным на рис. 2.18. Рабочая поверхность поршня должна быть 242
Рис. 2.18. Прибор Тарр—Бейкера:
Я — измерительная часть; Б — поршень; В— чашечка; /'—стакан
совершенно чистой. После каждого измерения поршень следует промыть этанолом и высушить. Поршень хранят в этаноле, а перед употреблением вытирают.
Перед анализом регулируют скорость подачи воды из напорного резервуара краном 3 так, чтобы при полностью открытом кране 2 столб четыреххлористого углерода поднимался примерно на 40 см за 1 мин. При дальнейшем анализе положение крана 3 не изменяют. Стандартный поршень обильно смазывают глицерином, чтобы на холостом ходу он свободно скользил по корпусу.
Стакан с желе устанавливают так, чтобы его центральная ось совпала с осью поршня, который осторожно опускают на поверхность желе. Закрывают кран 7 и открывают последовательно краны 6 и 2. В момент, когда поршень прорвет поверхность желе, кран 4 закрывают и отсчитывают высоту столба четыреххлористого углерода (см) по разности уровней в обоих коленах манометра. Затем кран 2 закрывают, а кран 7 открывают и восстанавливают положение поршня для последующего определения. Кран 7 используют для слива воды.
Студнеобразующую способность (°ТБ) находят по максимальному значению разницы уровня четыреххлористого углерода (табл. 2.6).
243
Т а блиц а 2.6
Студнеобразующая способность пищевых систем по Тарр—Бейкеру
Высота ; столба ; Градусы ст\; птсобразуюшей способност и пищевых систем при десятых долях. СМ ’
CCL см" 0 1 ) з •4 5 6 ' 7 8 0
0 — — -— 83 96 107 117 125 133 142
10 149 156 162 168 175 180 186 192 197 202
20 207 212 227 221 225 229 233 237 241 246
30 250 254 258 262 265 268 272 276 280 284
40 287 290 293 296 299 302 305 308 311 314
50 318 321 324 327 329 335 338 341 344 344
60 347 350 352 354 357 360 363 365 368 370
70 372 375 378 381 383 386 388 390 392 394
80 396 398 401 404 406 408 410 412 414 417
90 419 421 424 426 428 430 431 433 436 438
2.3.'ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ СТУДНЯ
Порядок проведения анализа. Испытуемый раствор наливают в две пробирки приблизительно до половины их высоты и закрывают резиновыми пробками. Переводят находящийся в пробирках раствор или пищевую смесь в студень.
Пробирки со студнем устанавливают в стакан с водой температурой 20 °C и с погруженным в него термометром. Стакан помещают в водяную баню той же температуры. Баню подогревают так, чтобы повышение температуры воды в стакане на 1 °C происходило за 2—3 мин. Через каждые 3—5 °C повышения температуры одну из пробирок вынимают из стакана и, наклоняя ее, наблюдают, не расплавился ли студень. Температуру, при которой содержимое пробирки перейдет в жидкое состояние, отмечают как температуру плавления студня. Расхождение между параллельными определениями не должно превышать 1 °C.
Результаты исследований физических свойств гелей оформляют в виде таблицы:
Состав пищевых систем, характеристика дисперсной фазы и дисперсионной среды Прочностные свойства Температура плавления студня, °C
прочность студня, г/см студнеобразующая способность, °ТБ
Сопоставляя полученные результаты, дают сравнительную оценку желирующих свойств различных белков и пищевых систем, формулируют выводы.
244
Лабораторная работа № 9
ОПРЕДЕЛЕНИЕ СТРУКТУРНО-МЕХАНИЧЕСКИХ СВОЙСТВ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Приобрести практический навык определения прочностных, реологических и адгезионных свойств мяса, мясных фаршей и белковых систем вторичных ресурсов.
Задачи. Измерить и рассчитать усилия среза, предельного напряжения сдвига, эффективной, условной вязкости и липкости в различных объектах исследования.
Объекты исследования. Мясо (кусковое), мясные фарши, растворы клея и желатина, кровь и ее фракции.
Оборудование. Нож; мясорубка; конический пластомер; ротационный вискозиметр; вискозиметр Энглера; лабораторная установка для определения адгезионных свойств; прибор для определения липкости.
Методические указания. Лабораторная работа состоит из нескольких этапов, каждый из которых можно использовать независимо или в совокупности.
Усилие среза характеризует прочность и жесткость системы, которые тесно связаны с качественным составом белков в мясе и стадиями автолиза мышечной ткани.
Метод определения усилия среза основан на измерении давления, необходимого для разрушения образца путем среза в камере постоянного объема. Усилие среза определяют на приборе ПМ-3, принципиальная схема которого показана на рис. 2.19.
О реологических характеристиках мясных фаршей и готовых продуктов можно судить на основе определения предельного напряжения сдвига. Указанный показатель позволяет оценить прочность структуры и консистенцию продукта.
Сдвиговые свойства проявляются при касательном смещении слоев продукта, который может представлять собой жидкую или
Рис. 2.19. Схема прибора ПМ-3 для определения усилий среза:
7 — электромотор; 2—привод; 3— рейка; 4— смещающийся хомут;
5 — рабочий орган; 6 — тензобалка; 7—тензодатчик: 8— электронный потенциометр
245
твердообразную систему, а также твердое тело, т. с. неразрушенную структуру (целые ткани мяса, кость, сыр. твердый жир и ирд. Приборы для измерения величин сдвиговых свойств нишевых систем имеют определенную специфику.
Конические пластометры получили широкое распространение в реологических исследованиях из-за простоты устройства и надежности в работе. Устройство приборов такого типа показано на рис. 2.20 (а, б). Предельное напряжение сдвига Оо (Па) определяют по глубине погружения конуса.
В настоящее время насчитывается несколько сотен конструкций ротационных вискозиметров, которые можно разделить на две основные группы. К первой группе относятся приборы, имеющие постоянный момент вращения ротора при переменной частоте вращения, ко второй — приборы, имеющие постоянную частоту вращения ротора при переменном вращающем моменте.
В России и странах СНГ наибольшее распространение получили вискозиметры РВ-8 системы проф. Воларовича, относящиеся к первой группе, и вискозиметры «Реотест», относящиеся ко второй. При исследовании структурно-механических свойств с помощью ротационных вискозиметров особо важное значение приобретают правильный выбор и учет размеров рабочих органов прибора, а также математическая модель для обобщения экспериментальных данных.
Принципиальные схемы ротационных вискозиметров представлены на рис. 2.21. Их рабочие органы могут иметь одну геометрическую форму: коаксиальные цилиндры (см. рис. 2.21, а), сферы или полусферы (см. рис. 2.21, б), два конуса (см. рис. 2.21, в), две плоскопараллельные пластины (см. рис. 2.21, г), два плоских кольца (см. рис. 2.21, д') или два конических кольца (см. рис. 2.21,/с). Рабочий зазор или рабочий орган может быть комбинированным, т. е. состоять из нескольких различных поверхностей: цилиндр — диск (см. рис. 2.21, е), цилиндр — полусфера (см. рис. 2.21, ж), ко-
Рис. 2.20. Принципиальные схемы пенетрометров:
« — конический пластомер Воларовича: 1 — кювета с исследуемым продуктом; 2 — конус со штангой и поперечиной; 3— обойма для осевого перемещения штанги с фиксатором; 4— индикатор часового типа для измерения перемещения штанги; б — пластинчатый пластомер Жуховицкого и Гуткина: 7 —подставка с кронштейном; 2—плоский нож (200x 25 мм) со штоком; 3— индикатор или линейка для регистрации перемещений штока
246
г
д
M____
Рис. 2.21. Принципиальные схемы ротационных вискозиметров
нус — диск (см. рис. 2.21, з), цилиндр — конус (см. рис. 2.21, и), цилиндр — конус — диск (см. рис. 2.21, л) и пр. Между рабочими поверхностями находится исследуемый продукт. Измеряется сила сопротивления внутри продукта при вращении одной из поверхностей.
Вискозиметры различают по конструктивным особенностям и способу привода одного из цилиндров. На рис. 2.22 показана принципиальная схема прибора «Реотест-2» — структурного ротационного вискозиметра, который используется как для определения динамической вязкости ньютоновских жидкостей, так и для
проведения глубоких реологических исследований неньютоновских
жидкостей. Им можно измерить следующие показатели текучести: структурную вязкость, пластичность (предел текучести) и тиксотропию.
Ротационный вискозиметр «Реотест-2» (см. рис. 2.22) состоит из четырех основных узлов: станины с синхронным электроприводом, блока многоступенчатого редуктора с переклю-
Рис. 2.22. Ротационный вискозиметр «Реотест-2»:
1 — станина; 2 — термостатная емкость; 3— ротор; 4—стакан; 5—соединение измерительного вала с валом привода: 6—термометр; 7—измерительный вал; 8 — потенциометр; 9 — пружинный динамометр (торсион); 10 — вал привода; // — ступенчатый регулятор динамометра; 12 — частотомер; 13— шкала ло-гометра; 14— стопорные рукоятки фиксаторов; 15 — шкала ступени частоты вращения; 16 — рукоятка переключения ступени
247
чателем скоростей и измерительного блока, которые объединены в общем корпусе, а также измерительной головки, состоящей из
четырех сменных цилиндрических роторов, двух сменных стаканов со съемным дном и водяной! бани.
В полностью укомплектованный стенд для проведения исследований помимо прибора входят ультратермостат и самопишущий логометр.
Для измерения вязкости жидкостей используют капиллярные вискозиметры. Теория капиллярной вискозиметрии основана на том допущении, что поток в приборе ламинарный, скольжение на
стенке отсутствует, скорость сдвига в точке зависит от нагружения в той же точке.
Жидкообразные продукты не имеют предельного напряжения
сдвига, их течение начинается при сколь угодно малых напряжениях сдвига. Обычно, за исключением истинно вязких жидкостей, эти продукты имеют слабую структурную сетку и обладают аномалией течения. Один и тот же продукт в зависимости от интенсивности механического воздействия, массовой доли сухих веществ или температуры часто может переходить из одной группы в другую, например жидкообразные в твердообразные. Реологические свойства жидкообразных продуктов используются при расчете ма-
шин и аппаратов, а
Рис. 2.23. Вискозиметр Энглера
также в комплексе с другими показателями при оценке процессов, связанных с переработкой животного сырья и оценкой качества продуктов.
В лабораторной практике технохими-ческого контроля качества некоторых продуктов мясной и перерабатывающей промышленности, в частности растворов желатина, клея, получил распространение капиллярный вискозиметр Энглера.
Вискозиметр Энглера (рис. 2.23) состоит из двух вставленных один в другой металлических сосудов 3 и 7. Внешний сосуд 7 является термостатирующей баней, куда наливают воду или минеральное масло. Для равномерного поддержания температуры жидкость в сосуде 7 перемешивают мешалкой 5. Внутрь резервуара 3 опускают термометр 1. На внутренней поверхности резервуара имеются три указателя уровня 6, которые находятся в одной плоскости и служат отметкой для измерения объема жидкости и контроля вертикального положения прибора. Правильным считается положение, когда все три указателя уровня 6 будут едва видны над
248
поверхностью жидкости. В нижней части сосудов находится отверстие 8 для слива испытуемой жидкости, которое закрывается стержнем 2, проходящим через крышку 4 прибора. Для приема сливаемой жидкости предусмотрена измерительная колба 9 с двумя отметками: на 100 и 200 см3.
Метод основан на определении отношения времени истечения испытуемого продукта при заданной температуре ко времени истечения того же объема воды при 20 °C.
Измерение вязкости позволяет охарактеризовать реологические свойства клеевых и желатиновых растворов, которые зависят от молекулярной массы, формы молекул продуктов деструкции коллагена и степени их гидратации. Эти факторы влияют на процесс гелеобразования при охлаждении водных растворов желатина, прочность и температуру плавления студней.
Общим для всех приборов этого типа является наличие капилляра, устройства для измерения расхода или объема жидкости и системы, обеспечивающей создание гидростатического давления. Для измерения вязкости ньютоновских и не очень вязких неньютоновских жидкостей в качестве капилляра можно использовать трубку диаметром от долей до 2—3 мм. Получаемые результаты, как правило, инвариантны, т. е. не зависят от диаметра трубки.
Наиболее простые, традиционные и вместе с тем универсальные капиллярные вискозиметры Оствальда и Уббелоде имеют капилляр и два полых шарика для жидкости. Движущая сила процесса истечения (перепад давлений) в вискозиметре Оствальда обусловлена разностью высот жидкости, в вискозиметре Уббелоде — вакуумом или давлением в одном колене трубки. При измерениях приборы обычно помещают в водяную баню.
Имеются и другие конструкции вискозиметров такого типа.
Для точного измерения весьма важно правильно подготовить пробу, учитывая особенности конструкции и эксплуатации прибора.
Особое место среди структурно-механических свойств занимает такое поверхностное свойство, как липкость (адгезия). Оно характеризует усилие взаимодействия между поверхностями конструкционного материала и продуктом при нормальном отрыве или сдвиге. При этом для большинства мясных и молочных продуктов липкость обусловливает величину усилия внешнего трения.
Липкость — это физическое явление, возникающее при соприкосновении тел. Она возникает при разделении этих тел как усилие, противодействующее разделению (отрыву). Исследование липкости как характеристического свойства сырья и продуктов в технологии мяса имеет большое значение. Например, исследование липкости колбасного фарша позволяет определить оптимальное время куттерования. На практике это свойство мяса оценивают обычно по прилипаемости фарша к поверхности руки. Таким
249
же ооразом по состоянию поверхности мяса можно оценить его водосвязывающую способность. Липкость исследуют также объективными методами, измеряя усилие, необходимое для отрыва от испытуемой поверхности соответственно подобранной пластины. Мерой липкости является величина усилия, приходящаяся на единицу поверхности контакта. Липкость связана с другими явлениями и свойствами продуктов: адгезией, когезией, вязкостью и поверхностным трением. Адгезия проявляется в виде усилия, действующего на границе двух соприкасающихся фаз, и зависит от величины притяжения, действующего между частицами обеих фаз.
Качественно адгезию можно охарактеризовать двумя способами: нарушением контакта одновременно на всех участках площади (рис. 2.24, а, г, д') или же путем последовательного отрыва отдельных участков — расслаиванием, отдиранием (рис. 2.24. б, в). Оба способа определения адгезионной прочности нашли практическое применение. При первом способе разрушающую нагрузку прилагают в направлении как перпендикулярном плоскости контакта поверхностей, так и параллельном ей и обычно относят к единице площади поверхности контакта; при втором — определяют силу, необходимую для расслаивания склейки, и относят к единице длины. Очень часто адгезию, определяемую при расслаивании, характеризуют не силой, а работой, которую необходимо затратить на разделение фаз.
Подготовка проб. Для определения усилий среза образцов на приборе ПМ-3 на специальном устройстве для вырезания образцов (рис. 2.25) легким нажимом ломтика сырого или вареного продукта на вращающийся трубчатый нож вырезают ровный цилиндрический образец диаметром 10 мм. Полученный образец извлекают с помощью выталкивателя.
Для определения сдвиговых свойств мышечной ткани на коническом пластомере вырезают образцы ткани вдоль и поперек мышечных волокон, по форме и размеру соответствующие кювете прибора, на установках другого типа — образцы, соответствующие размерам кюветы используемой установки.
Для определения сдвиговых свойств мясных продуктов на ротационных вискозиметрах и адгезионных свойств на различных лабораторных установках готовят мясной фарш, последовательно измельчая мясо различных животных на волчке (мясорубке), куттере, гомогенизаторе или другой машине тонкого измельчения. Диаметр отверстий решетки и продолжительность тонкого измельчения задает преподаватель.
При исследовании реологических свойств крови убойных животных рекомендуется дать сравнительную оценку вязкости цельной крови и ее фракциям, для чего цельную стабилизированную кровь разделяют на фракции плазмы и форменных
250
Рис. 2.24. Принципиальные схемы приборов для измерения адгезионной прочности:
« — для нормального отрыва: / — адгезионный; 2 —когезионный; 3 — смешанный; 4— схема устройства для осуществления отрыва; б — для расслаивания жестких материалов: 1— внецентро-вое растяжение; 2— изгиб для плиточного и листового материалов; 3— центральный изгиб для листового материала; 7— консольный изгиб для листового материала; « — для расслаивания гибких материалов: 1—3— от жесткой подложки под углом 90°: 4 — от жесткой подложки под углом 180°; 5— от гибкой подложки; г — для сдвигового разрушения: 1 — при растяжении одностороннего соединения; 2—то же. двустороннего; 3— при сжатии соединения цилиндра со стержнем; д — для сдвигового разрушения при кручении: /—по торну цилиндров; 2—ио кольцевой поверхности торна полых цилиндров; 3— по боковой поверхности цилиндра и стержня
Рис. 2.25. Устройство для вырезания образцов: /—трубчатый нож; 2— выталкиватель; 3— привод электродвигателя
элементов сепарированием или центрифугированием в течение 5—8 мин при частоте вращения барабана 25—42 с !.
Для исследования реологических свойств клея и желатина готовят стандартные растворы: желатина — массовой долей 10%. клея — массовой долей 15 %.
1.ОПРЕДЕЛЕНИЕ УСИЛИЙ СРЕЗА
Порядок проведения анализа. Для определения усилий среза включают прибор ПМ-3 тумблером ВК в электросеть. Рукояткой выводят стрелку прибора на нуль и совмещают отверстия в пластине рабочего органа и смещающегося хомута (см. рис. 2.18). Подготовленный образец мяса осторожно помещают в образовавшееся цилиндрическое отверстие, вставляют прижимные пластины с ножевой поверхностью на конце в направляющие для срезания излишков мяса и фиксирования образца. Нажатием кнопки «Пуск» приводят в движение привод рабочего органа, смещающаяся пластина которого производит срез образца.
Усилие, необходимое для среза образца, передается тензо-балке и через тензодатчик фиксируется в виде пика на ленте потенциометра.
Экспериментально полученные данные оформляют в виде таблицы:
Наименование и характеристика образца Усилие среза
2. ОПРЕДЕЛЕНИЕ СДВИГОВЫХ СВОЙСТВ НА КОНИЧЕСКОМ ПЛАСТОМЕРЕ
Порядок проведения анализа. Прибор устанавливают по уровню с помощью трех винтов в станине. Готовят модельный материал соответствующей консистенции. Исследуемый образец помещают в кювету прибора. Выравнивают металлической линейкой поверхность модельного фарша так, чтобы масса в сосуде находилась на уровне с его краями. Кювету с исследуемым образцом устанавливают на столик прибора и поднимают вверх до соприкосновения поверхности с острием конуса. Для значительных перемещений ослабляют стопорный винт механизма подъема столика и свободно перемещают столик; для точной регулировки положения поверхности материала относительно острия конуса пользуются микрометрической гайкой. При необходимости контролируют и устанавливают нулевое положение индикатора. Нажимают пусковую кнопку, включают секундомер и, слегка придерживая в на-
252
чальныи момент штангу, опускают конус. Но мере погружения конуса в материал через каждую минуту фиксируют глубину погружения рифленого конуса по индикатору. Следует считать, что погружение заканчивается через 3—5 мин, так как по истечении этого времени конус опускается на незначительную глубину, чем практически можно пренебречь. Длительность погружения 180— 300 с соответствует наибольшему периоду релаксации используемых для исследования материалов.
Для каждого исследуемого образца следует выполнить 5—6 измерений.
Фиксируют величину угла 2а при вершине конуса, константу конуса К (м/кг), общую массу /иобш (кг) штанги, конуса и дополнительного груза при его наличии.
При использовании дополнительного груза строят тарировоч-ную кривую пружины индикатора в виде зависимости между массой грузов и вызываемых ими перемещений штока индикатора
<2'64)
Для каждого значения величины перемещения штока индикатора /?инд фиксируют соответствующую массу груза т . Расчетное значение массы грузов определяют из выражения
то6„, = + т«т- <2-65)
Для каждого образца вычисляют значения предельного напряжения сдвига 0 (Па) при фиксированной длительности погружения:
к—й— (166)
Находят среднеарифметическое значение предельного напряжения сдвига для каждого из вариантов исследуемых образцов
Z0,
ео----т~’ (2.67)
где / — количество измерений.
Для каждого варианта исследуемого образца строят график зависимости глубины погружения конуса прибора от длительности погружения.
Экспериментальные величины и расчетные данные вносят в таблицы рекомендуемых ниже форм:
253
Угол при вершине Константе. кон\ ел X. Общая масс.: ш ьг 1 Масса дополните.п>-
конуса 2</. грал У кг ного груза ( . кг
у—
Чапменов iнi:е и характерна шка образна /1.1 и тел вноси в погружения конуса, с 1 дубина потру жения h. м 1 Ipe.ie.iBHoe напряжение сдвига 6. Па Среднеарифметическое значение предельного напряжения сдвига 0.. Па
1 30
60
120
180
240
3. ОПРЕДЕЛЕНИЕ ПРЕДЕЛЬНОГО НАПРЯЖЕНИЯ СДВИГА И ЭФФЕКТИВНОЙ ВЯЗКОСТИ НА РОТАЦИОННОМ ВИСКОЗИМЕТРЕ РВ-8М
Порядок проведения анализа. Методика измерения структурно-механических характеристик для ротационных вискозиметров системы Воларовича сводится к подготовке прибора, измерениям и разборке. Подготовка включает установку вискозиметра по уровню, определение силы трения в подшипниках, заполнение рабочего объема исследуемым материалом и его термостатирование. Для определения реологических характеристик исследуемый образец массой 15—17 г помещают в наружный цилиндр вискозиметра, находящийся в крайнем нижнем положении. В случае изучения зависимости реологических свойств исследуемых образцов от температуры включают термостат и материал обрабатывают в течение 20—30 мин при температуре, заданной преподавателем. Наружный цилиндр при помощи подъемного устройства приводят в рабочее положение и фиксируют с помощью винта. Излишки образца убирают скальпелем, шпателем или ножом. Конкретные условия определения сдвиговых свойств (масса грузов на блоке, число оборотов шкива) апробируют применительно к изучаемому объекту. Устанавливают фиксированный груз на блоке, освобождают шкив со стопора, одновременно включая секундомер. После остановки системы и прекращения вращения шкива секундомер выключают. Фиксируют массу груза т (г), число оборотов шкива п и время вращения шкива.
Во время измерений меняют массу грузов М (кг) и для каждого груза определяют частоту вращения ротора N (максимальная частота вращения может достигать 2—2,5 с-1). Каждый опыт состоит из 30—40 замеров, проводимых несколько раз с
254
постепенным увеличением и уменьшением массы грузов. Одновременно строят реограммы N = а{М).
После этого определяют константы опыта по формулам:
; (2.68)
Аф “ Лвк^А2^2/? + л2Яв2/4); (2.69)
Ад 2лЯ2Л/(Д11К£); (2.70)
Л2 = 1п(А„//?в) 2л (2.71)
где g —ускорение свободного падения, м/с2; /?шк — радиус шкива, м; /^ — внутренний радиус стакана, м; Ки — радиус ротора, м; // — глубина погружения ротора в продукт, м.
Вычисляют эффективную вязкость для каждой опытной точки по уравнению
Ъф = Ш/Л/ (2.72)
По окончании измерений приводят прибор в исходное положение в следующем порядке: освобождают блок от нагрузки; подъемное устройство приводят в нижнее положение; освобождают внешний цилиндр с термостатирующим устройством от крепления и выгружают исследуемый образец; отмечают высоту контакта рифленой поверхности внутреннего цилиндра с исследуемым материалом h (см); тщательно моют и сушат оба цилиндра.
Строят график зависимости эффективной вязкости от окружной скорости со = 2nRKN в логарифмических шкалах. Через полученные экспериментальные точки проводят прямую линию так, чтобы она делила относительные отклонения расстояний между точками и прямой на равные части. Составляют уравнение этой прямой, определяют темп разрушения структуры т. Значение коэффициента эффективной вязкости определяют по формуле
Пэф = ^(со/со.Г2 = В^т, (2.73)
где В — эффективная вязкость при фиксированном значении окружной скорости, Па с; СО] — фиксированное единичное значение окружной скорости боковой поверхности ротора коаксиально-цилиндрического вискозиметра (од = 1 м/с); со, — безразмерная окружная скорость (ее числовое значение).
После определения по полученным экспериментальным данным индекса течения п = 1 — т рассчитывают инвариантную для любых коаксиально-цилиндрических ротационных вискозимет
255
ров реологическую константу В,, по формуле
д; = в
| И„|1 - (Я„/Л„ )2 " ||”
'l 2(0, I
(2.74)
учитывающей закон изменения градиента скорости.
Затем вычисляют реологические константы — предельное напряжение сдвига 0О и пластическую вязкость г|1П, ориентируясь на график зависимости N(M). При этом точки для вычислений берут непосредственно с кривой. 0О и определяют в следующей последовательности. Предельное напряжение сдвига 0О (Па) соответствует отрезку, отсекаемому кривой Л;(Л/), по оси абсцисс (Л/о). Его определяют по формуле
0О = £ОЛ/О. (2.75)
Напряжение сдвига на поверхности ротора
е = к„М. (2.76)
Пластическую вязкость в зависимости от того, распространяется ли сдвиг на всю толщину кольца продукта (в зазоре между ротором и стаканом), рассчитывают по одной из приведенных ниже формул.
Первый случай — сдвиг распространяется на всю толщину кольца, когда масса нагрузки MQ больше массы нагрузки Мх = /Cj0o. В этом случае пластическую вязкость определяют по формуле
рпл = (Ш-^20о)Ж (2.77)
Все расчеты пластической вязкости, когда сдвиг распространяется на всю толщину кольца исследуемого продукта, рекомендуется сводить в таблицу:
Константа опыта к = к,=
N М кМ кМ -- к^
4. ОПРЕДЕЛЕНИЕ ПРЕДЕЛЬНОГО НАПРЯЖЕНИЯ СДВИГА И ЭФФЕКТИВНОЙ ВЯЗКОСТИ НА РОТАЦИОННОМ ВИСКОЗИМЕТРЕ ТИПА «РЕОТЕСТ»
Порядок проведения анализа. Предварительно выбирают соответствующую измерительную головку, руководствуясь табл. 2.7 для определения сдвиговых характеристик того или иного про
256
дукта, затем фиксируют при помощи цангового зажима выбранный измерительный ротор на выходном валу измерительного блока.
Т а б а и ц а 2."
Пределы измеряемых реологических характеристик, соответствующих индексам измерительных головок
Индекс измерительных j ГОЛОВОК | 1 Соотношения радиусов роторов и ста конто в RJ Область, напряжений 0 Напряжение сдвига 0. Па Скорость деформации ! . . тУ Оффекшвная ’ (динамическая) 1 ВЯЗКОСТЬ )], Па с
ф 0.98 1 11 — 110 1.5-1310 0.01-75
11 55—550 1.5-1310 0,05 — 375
ф 0.94 1 12-120 0.5-437 0,03-240
11 60-600 0,5-437 0,15-1200
ф 0.81 1 16- 160 1/6-146 0,12-1000
11 80-800 1/6-146 0,6-5000
и 0,81 I 60-600 1/6-146 0,4-3600
II ЗОО-ЗООО 1/6—146 2-18000
Для наполнения цилиндрического измерительного устройства в мерный бачок вносят рекомендуемый объем (массу) исследуемого образца:
Тип измерительной головки Объем (масса) образца, см3 (г)
5] 52 ф Н
25 30 50 17
Константы выбранной измерительной головки определяют в зависимости от жесткости торсиона I—II (положение рукоятки на лицевой панели измерительного блока) из табл. 2.8.
Таблица 2.8
Константы измерительных головок
Тип измерительной головки
Константы роторов Z. Па/ел. шкалы
при жесткости торсиона I при жесткости торсиона II
Ф 1,19 5,92
Ф 1,23 6,16
Ф 1,69 8,45
н 5.89 29,29
Исследуемый продукт помещают в соответствующий стакан с герметично закрытым съемным дном и коаксиально ротору по направляющей при помощи винтового зажима фиксируют на измерительном блоке.
257
При необходимости выполнения измерений при различных значениях температуры стакан герметично фиксируют в водяной бане, которую подключают к ультратермостату, и продукт термо-статируют 10—20 мин до установления требуемой температуры.
Прибор подключают к сети напряжением 220 В. частотой 50 Гн Задают требуемую частоту вращения ротора, начиная с минимальной, с помощью переключателя скоростей за счет установки рычага в положение, соответствующее данным табл. 2.9.
Г а б л 11 и а 2 9
Определение частоты вращения ротора вискозиметра < Реотест-2» по ступеням
Ступень Частота вращения .V. с ! ( Ступень Частота вращения .V. с
1а 0,00926 1в 0.00463
2а 0.01660 2 в 0.00833
За 0,02778 Зв 0.01389
4а 0,05000 4в 0.02500
5 а 0,08330 5 в 0.04167
6 а 0,15000 6 в 0.07500
7а 0,25000 7 в 0.12500
8а 0.45000 8 в 0.22500
9а 0,75000 9 в 0.37500
10а 1,35000 1 Ов 0,67500
11а 2.25000 11в 1,12500
12а 4,0500 12в 2,02500
Для переключения от ступени 1 до ступени 12 требуется 2,5 оборота рукоятки. Промежуточные ступени соответствуют меньшему повороту и индицируются на табло блока многоступенчатого редуктора. Другую частоту вращения ротора можно установить в процессе измерений.
Отклонение показаний измерителя частоты, расположенного на измерительном блоке, от величины 50 Гц требует внесения поправки при расчете частоты вращения ротора по формуле
= Ж/50,
(2.78)
где N — действительная частота вращения ротора; N — частота вращения ротора (по табл. 2.9); /— индуцируемая частота переменного тока. Гц.
Измеряемое значение напряжения сдвига при различных скоростях вращения ротора выбранной измерительной головки в соответствии с жесткостью торсиона вычисляют по формуле
6 = Za,
(2.79)
где Z— константа ротора; а — показания стрелочного логометра. 258
Для построения реограммы — кривой течения N (0) обычно бывает достаточно 12 экспериментальных точек в диапазоне от минимальной до максимальной частоты вращения ротора.
Эффективную вязкость при каждой частоте вращения ротора рассчитывают по формуле
Ч.,ф = ^0/М. (2.80)
где к - константа измерительном головки:
где соотношение тля соответствующей измерительной головки берут
из табл. 2.7.
Все данные измерений заносят в журнал наблюдений:
Номер опыта Напряжение сдвига 0. Па Действительная частота вращения ротора Ду c-i Окружная скорость вращения ротора со = 2кД АХ м • с‘1 Эффективная i -г ч _ вязкость п 1 П, . с ,'1’ 1 продукта t. С 1 1 (Л L
По окончании опыта измерительную головку отсоединяют от измерительного блока, разбирают, удаляют исследованный продукт, моют водой, сушат и окончательно обезжиривают при помощи спирта.
5. ОПРЕДЕЛЕНИЕ ВЯЗКОСТИ НА ВИСКОЗИМЕТРЕ ЭНГЛЕРА
Порядок проведения анализа. Перед началом работы прибор устанавливают с помощью регулирующих винтов так, чтобы три указателя уровня 6 (см. рис. 2.23) находились в горизонтальной плоскости. Перед каждым определением прибор и его отверстие Для стока £ следует тщательно промыть спиртом, бензином и высушить продуванием воздуха.
С помощью вискозиметра Энглера устанавливают время истечения дистиллированной воды. Для этого в тщательно промытый и высушенный сосуд 3 для испытуемой жидкости при закрытом сточном отверстии 8 немного выше указателей уровня 6 наливают Дистиллированную воду температурой 20 °C. Нагревая термоста-тирующую баню 7, во внутреннем сосуде 3 в течение 10 мин поддерживают температуру 20 °C; затем слегка поднимают стержень 2 и выпускают немного воды. Таким образом, все сточное отверстие заполняется водой. Излишки воды отсасывают из сосуда пипеткой, чтобы уровень ее находился на высоте указателя уровня 6.
259
Под сточное отверстие прибора ставят измерительную колбу 9. Установив прибор, закрывают его крышкой 4, придерживая при этом рукой стержень 2, закрывающий сточное отверстие 8.
Убедившись, что температура воды равна 20 °C, быстро, не трогая прибора, поднимают рукой запирающий стержень 2 и по секундомеру точно отмечают время заполнения колбы 9 до черты, указывающей объем 200 см3. Это время должно быть равно не менее 50 и не более 52 с. Среднее для данного прибора время, принимаемое за единицу, определяют, исходя из трех последовательных измерений, разница между которыми должна быть не более 0,5 с. Такую проверку делают перед каждым новым опытом.
В сосуд 3 немного выше указателя уровня наливают испытуемый образец, подогретый на 2—3 °C выше температуры опыта.
В термостатирующую баню 7 наливают воду на 0,2—0,3 °C выше температуры опыта и поддерживают ее, перемешивая содержимое мешалкой 5.
Подняв немного стержень 2, дают стечь излишкам образца настолько, чтобы его уровень совпадал с верхними точками указателя уровня 6 и сточная труба б'была полностью заполнена. После этого прибор закрывают крышкой 4 и под сточное отверстие 8 ставят измерительную колбу 9.
Температуру испытуемого образца проверяют при перемешивании. Для этого вращают вокруг стержня 2 крышку прибора 4 с термометром 1. Когда будет достигнута требуемая температура, выдерживают еще 5 мин, а затем быстро поднимают стержень 2, закрывающий сточное отверстие 8, одновременно включив секундомер. Последний останавливают в тот момент, когда в измерительной колбе 9 уровень жидкости достигнет отметки 200 см3 (пена в расчет не принимается). Отметка 100 см3 на измерительной колбе служит ориентировочно для контроля времени истечения.
Вязкость в градусах Энглера вычисляют по формуле
°г 6
Е20’ = - ’ (2.82)
ч
где г2 — время истечения образца, с; — время истечения воды, с.
Для перевода градусов Энглера в Па • с пользуются эмпирической формулой
20е f к ото 5,9806 Л
г) = 6,92211о„-----— р, (2.83)
k r )
где в20 — вязкость, Па-с; г|оп — вязкость при температуре опыта, °Е; р — плотность жидкости при температуре опыта, г/см3.
Полученные экспериментальные данные оформляют в виде таблицы рекомендуемой формы:
260
Наименование и характеристика образна, условия эксперимента
Номер ; опьиа
Время ис течения образца.
Сре.тнеарифмесю ческое значение истечения образна, с
Время ис- Сре.тнеарпфмсти-течения ческое значение волы, с ' истечения волы, с
Расчетные данные сводят в таблицу:
Наименование и характеристика образца
Условия эксперимент------
Вязкое п>
6. ОПРЕДЕЛЕНИЕ АДГЕЗИОННЫХ СВОЙСТВ МОДЕЛЬНЫХ ФАРШЕЙ
6.1. ОПРЕДЕЛЕНИЕ ЛИПКОСТИ МЯСНОГО ФАРША НА ЛАБОРАТОРНОЙ УСТАНОВКЕ
Основным элементом лабораторной установки (рис. 2.26) являются технические весы 4, над одной из тарелок которых устанавливают скамеечку 1 так, чтобы они не соприкасались. На скамеечку помещают испытуемый образец 2 и прикрывают его измерительной пластинкой 3, которую прикрепляют к коромыслу весов прочной ниткой. На другой тарелке весов помещают химический стакан 6. Над весами 4 устанавливают бутыль Мариотта 5 с водой.
Испытуемый образец 2 помещают на скамеечку / лабора
торной установки и накрывают На пластину в течение заданного времени устанавливают груз определенной массы. Затем груз снимают, открывают кран бутыли Мариотта, наполняя стакан водой. Кран закрывают в момент отрыва пластины от поверхности образца. Далее уравновешивают весы, определяя массу воды в стакане.
Условия эксперимента (высоту слоя продукта, величину нагрузки, продолжительность ее воздействия, массовую долю влаги и жира в модельном фарше) фиксируют в тетради.
его измерительной пластиной.
Рис. 2.26. Лабораторная установка для определения липкости (по С. Тышкевичу)
261
Адгезия (Па) определяется как удельная сила нормального отрыва пластины от продукта
_ 9.81/77
(2.84)
гае сила отрыва. Н: 5’, — юометричсская площадь пдасгины. м2; di — масса груза. Ki.
Экспериментальные и расчетные данные оформляют в виде таблицы:
Образен Массовая л влаги ОЛЯ МОЛСЛЬНО! жира о фарша, % Условия эксперимента пищевых Высота \ iMacCil Продолжптель- добавок 1 слояо°- , греза, кг ность действия уразиа, мм гру;а, мин
6.2. ОПРЕДЕЛЕНИЕ ЛИПКОСТИ МЯСНОГО ФАРША НА ПРИБОРЕ СОКОЛОВА—БОЛЬШАКОВА
Метод основан на определении величины усилия, необходимого для разделения двух поверхностей, связанных (склеенных) испытуемым фаршем.
Перед началом работы выдвигают рабочий орган / прибора (рис. 2.27) и включают прибор в электросеть. На нижнюю пластину 2 помещают испытуемый образец массой 2 г. Нажатием кнопки «Пуск» включают электродвигатель. При этом втулка 8 вместе
Рис. 2.27. Схема прибора Соколова—Большакова для определения липкости мясного фарша
262
со штоком 4. грузом 9 и верхней пластиной 3 с помощью приспособления /перемещается вниз. При достижении верхней пластиной 3 поверхности образца последний подпрессовывается в течение 30 с под действием груза 9 постоянной массы для данного исследуемого продукта.
По истечении установленного времени, регулируемого реле 5. электродвигатель автоматически включается на обратный ход. вследствие чего втулка 8 начинает двигаться вверх. Усилие, необходимое для отрыва верхней пластины от образца, фиксируется на электронном самопишущем приборе 6.
Конкретные условия определения липкости (масса груза, время пребывания образца под нагрузкой) должны быть предварительно апробированы применительно к изучаемому объекту.
Далее фиксируют условия проведения эксперимента и данные самопишущего устройства.
Контрольные вопросы и задания
1. Для решения каких технологических задач требуется изучение физических характеристик мяса и мясопродуктов'.’
2. Укажите основные физические характеристики мяса и мясопродуктов. Как они связаны с особенностями их макро- и микроструктуры?
3. Какие физические свойства мяса играют важную роль в оценке цветности?
4. Какое практическое значение имеет объективная опенка оптических свойств мяса9
5. Чем обусловлена анизотропия затухания звука в мясе и цсльномышечных мясных продуктах?
6. Каковы преимущества и перспективы применения ультразвука для анализа мяса и мясных продуктов?
7. Как практически определить акустические характеристики мяса и мясопродуктов?
8. Чем обусловлен эффект поглощения звука в животных тканях?
9. Назовите основные теплофизические свойства мяса и мясопродуктов и методы их экспериментального исследования. В чем их преимущества и недостатки?
10. Какие комплексные методы исследования тсплофизических свойств пищевых продуктов вы знаете9
11. Что понимают под функционально технологическими свойствами мясного сырья9
12. Что принято понимать под функциональными свойствами изолированных белков и белковых систем?
13. Что такое эмульсия9
14. Какие факторы влияют на функциональные свойства мясных фаршевых эмульсий?
15. Охарактеризуйте способы стабилизации функциональных свойств мясных фаршей.
16. Какие свойства относятся к структурно-механическим свойствам мяса и мясопродуктов?
17. Дайте характеристику основных сдвиговых реологических, компрессионных свойств пластично-вязких продуктов. При расчете каких технологических процессов учитывают эти параметры?
18. Какое практическое значение имеют поверхностные свойства мясопродуктов?
19. Как можно практически определить цветность мяса и мясопродуктов, пищевых животных жиров?
263
20. Какова сущность экспери.менгальных методов определения акустических свойств мяса и мясопродуктов?
21. В чем состоит метод двух временных точек определения теплофизически.х характеристик'.’
22. Какие основные этапы включает процесс гистологического анализа мяса? Каковы сто преимущества перед физико-химическими и биохимическими методами исследований'.’
23. Каковы правила отбора и подготовки проб для определения микроструктурных показателей мяса'.’
24. Какие приборы используют при получении срезов животных тканей для гистологических исследований9
25. Какие методы используют при определении функционально-технологических свойств мяса? Каковы преимущества метода последовательного определения функциональных показателей в одной навеске'.’
26. Как практически определить водосвязывающую, влагоудерживающую, жироудерживающую, эмульгирующую способности и стабильность фаршевых эмульсий?
27. Охарактеризуйте способность различных белков животных ткано/! к образованию гелей.
28. Какие приборы используют для определения физических показателей гелей?
29. Как определить структурно-механические свойства мяса, мясопродуктов и вторичных продуктов убоя?
30. Что такое вязкость жидкости?
31. Как влияет температура на вязкость жидкости9
32. Какие типы вискозиметров вы знаете?
33. Каковы особенности капиллярных вискозиметров9
34. Принцип действия конического пластомера.
35. Устройство и принцип действия ротационных вискозиметров.
36. Какие технологические факторы влияют на структурно-механические свойства мяса и мясных продуктов?
Глава 3 БИОХИМИЧЕСКИЕ СВОЙСТВА ЖИВОТНЫХ ТКАНЕЙ
Рациональное использование животного сырья основано на знании не только химического состава, но и особенностей протекания биохимических процессов в органах и тканях при жизни животного, после его смерти и при технологической обработке.
Прижизненные процессы многообразны и управляемы, протекают в строгом взаимном соответствии. Большую роль в них играют биологически активные вещества -- ферменты и гормоны. Многие ткани животных синтезируют в той или иной мере эти вещества, обеспечивая жизнедеятельность организма. После убоя животных эти ткани служат источниками для получения биологически активных веществ и удаляются в процессе разделки туш, например поджелудочная железа, гипофиз, слизистые оболочки желудков и кишечника и др.
После смерти животного, с прекращением обмена веществ, основным биохимическим процессом при переработке сырья биологического происхождения является автолиз (от греч. autos — сам и lysis — растворение), основу которого составляют ферментативные процессы, при жизни связанные с функцией координированного движения. Автолизом называется процесс распада веществ и тканей под действием ферментов самих тканей. При жизни животного он возможен только в патологических случаях, например при недостаточном снабжении какого-либо органа кровью.
В послеубойный период свойства всех тканей животного организма значительно изменяются, особенно существенны изменения мышечной ткани, в которой ферменты (протеазы, карбогидразы, эстеразы, ферменты гликолиза и др.) катализируют реакции распада. Процесс становится необратимым, протекающим только в одном направлении. Особая роль в процессе автолиза принадлежит внутриклеточным протеазам — катепсинам.
Итак, при переработке убойных животных ткани можно разделить на две группы: к первой относятся те, в которых протеолиз происходит под действием внутриклеточных протеаз типа катепсинов (мышечная ткань, печень и др.); ко второй — ткани, в которых синтезируются внеклеточные протеазы типа пепсина, трипсина, химотрипсина, карбоксипептидазы, дипептидазы, аминопептидазы (слизистая оболочка желудков, подже-
265
дудочная железа и т. д.), а также гормоны. Для первой группы тканей весьма важен период автолиза, так как именно он определяет функционально-технологические свойства сырья.
Ко второй группе относятся животные ткани, нашедшие широкое применение в медицине и перерабатывающих отраслях АПК для получения медицинских и технических препаратов.
3.1. МЕХАНИЗМ АВТОЛИЗА И АВТОЛИТИЧЕСКИЕ ПРЕВРАЩЕНИЯ МЫШЕЧНОЙ ТКАНИ
Эффективное и рациональное использование сырья предусматривает сохранение его качества на всех этапах переработки, начиная от транспортирования животных на мясокомбинат и их убоя до хранения готовой продукции. Определяющим условием для формирования биохимических свойств мяса и качества продуктов являются уровень и характер развития автолитических процессов в животных тканях. Изменения, происходящие в послеубойный период в мышечной ткани, имеют важное практическое значение, оказывают существенное влияние на пищевую ценность мяса и его потери в процессе технологической обработки.
Современные представления о механизме автолитических процессов базируются на ферментативной природе посмертных изменений мышечной ткани, установленных в 30-х годах И. А. Сморо-динцевым. Наиболее важное значение отводится двум основным ферментным системам. Одна из них, очевидно, связана с функцией движения и регулирует сокращение — расслабление, другая —-катализирует непрерывный распад главных структурных элементов мышечного волокна по типу гидролиза. Роль каждой из этих двух систем в развитии этапов автолиза различна, но их функции тесно связаны между собой.
Начиная с момента убоя в мышечной ткани под действием тканевых ферментов непрерывно происходят изменения. В основе всех изменений, приводящих к развитию посмертного окоченения, его последующему разрешению и завершению созревания мяса с приобретением им функциональных и технологических свойств, лежат ферментативные реакции.
В послеубойный период с развитием окоченения главную роль играют ферменты сокращения — расслабления, вызывающие изменения сократительного аппарата — миофибрилл, распад АТФ и других несущих в себе запас энергии веществ, а также обеспечивающие синтез АТФ за счет превращения углеводов, и прежде всего гликогена.
В сложных процессах автолитических превращений мышечной ткани важная роль принадлежит изменениям углеводной системы.
266
Накопление продуктов их автоли i ическог о раснала смцественно влияет на физико-химические и биохимические изменения белков мышечной ткани.
В комплексе биохимических превращении, происходящих в начальный период авюлиза мыши, наиболее шчшлнво проявляются превращения гликогена и изменения органических фосфорных соединений.
Автолитические превращения гликогена связаны с его фосфоролитическим распадом и дальнейшим процессом анаэробного гликолиза, который приводит к необратимому накоплению молочной кислоты и снижению pH мышечной ткани. Кроме того, мышечный гликоген в процессе автолиза подвергается интенсивному гидролитическому амилолитическому распаду, ведущему к накоплению в мышечной ткани редуцирующих углеводов. Таким образом, автолитический распад мышечного гликогена обусловлен двумя параллельно протекающими процессами: анаэробным гликолизом с образованием молочной кислоты и гидролизом с образованием редуцирующих углеводов. Общие закономерности протекающих процессов, сопровождающиеся накоплением или распадом специфических биохимических веществ, показаны на рис. 3.1 —3.3.
Методы химического и биохимического анализов тканей наиболее предпочтительны при оценке глубины и характера автолитических превращений тканей мяса.
Состояние автолиза тканей легко определить по внешним признакам визуально. Сразу после убоя мышечная ткань расслаблена, поскольку в клетках содержится АТФ, уровень которой поддерживается в результате ряда хаотично протекающих реакций. Через несколько часов ранее расслабленные мышцы теряют свою гибкость, эластичность, становятся твердыми и непрозрачными. Наступает посмертное окоченение, которое сохраняется в течение 24—28 ч. По истечении 24—28 ч все более заметными становятся признаки разрушения клеточных структур: ядра сморщиваются, появляются поперечные разрывы мышечных волокон. Накопление специфических химических веществ в процессе автолиза служит основой органолептической оценки состояния сырья.
Можно выделить ряд объективных биохимических, физико-химических и морфологических признаков оценки этапов после-убойного созревания мяса.
Так, процесс созревания мяса связан с переходом части водорастворимых бел-
600
500
у 400
300
го 8 200
юо
S о
О 12 24 36 48 60 72
Продолжительность созревания мяса,ч
Рис. 3.1. Изменение содержания глюкозы (7) и гликогена (2) при созревании мяса
267
Продолжительность созревания мяса, ч
Рис. 3.2. Изменение содержания молочной кислоты при созревании мяса при температурах:
1 - 34 °C; 2—4°С
г
8°о.х>
Продолжительность созревания мяса, ч
Рис. 3.3. Изменение содержания фосфора при созревании мяса:
/- - неорганического; 2— органического
ков мышечных волокон в коагулированное состояние, причем особенно сильный сдвиг в соотношении общего аминного и аммиачного азота водной вытяжки мяса при 4 °C приходится на 6-й час после убоя животного. Массовая доля коагулирующих белков водной вытяжки составляет 60 % общего азота вытяжки.
Отношение миозина к миогену уменьшается почти в три раза к 24-му часу и остается затем постоянным (рис. 3.4). Отношение азота растворимого белка к белковому азоту к 24-му часу уменьшается в два раза — с 0,6 до 0,3 и стабилизируется. Общее количество экстрактивных веществ держится на стабильном уровне в течение 10 сут. Массовая доля молочной кислоты составляет 0,7— 0,8 %; редуцирующих сахаров — 0,2 %; pH 5,6—5,7 (данные для однотипных животных, например коровы в возрасте 6—7 лет средней упитанности).
Наиболее важной составной частью мяса по объему, питательной и биологической ценности является поперечнополосатая мышечная ткань туш убойных животных. В этой связи важно знать не только последовательность послеубойных изменений мышечной ткани, но и иметь достоверный метод их определения.
Важно отметить, что в процессе биохимических превращений белковых и азотистых экстрактивных веществ в мясе накапливаются химические предшественники вкуса и аромата мяса.
_ « —
0 24 48 72 96 120
Продолжительность созревания мяса, ч
Рис. 3.4. Изменение отношения миозина к миогену и гликогена к молочной кислоте при созревании мяса:
1 — миозин/миоген; 2— гликоген/молочная кислота
268
Интенсивный обмен белков в животных тканях требует наличия системы, осуществляющей интенсивный распад белковых веществ. В связи с ведущим значением биохимических ферментативных процессов для направленного регулирования свойств мясного сырья и готовых продуктов большое практическое значение имеют тканевые ферментные системы мяса. Действие протеолитических ферментов в животных тканях в послеубойный период направлено на расщепление собственных компонентов и структур клеток.
Протеиназы, осуществляющие гидролиз белков, подразделяют на экзо- и эндопептидазы. Первые расщепляют концевые участки полипептидных цепей, вторые — внутренние участки цепи. Экзопептидазы содержатся как в саркоплазме и саркоплазматическом ретикулуме, так и в лизосомах, тогда как внутриклеточные эндопептидазы находятся, как правило, в лизосомах клеток.
Прижизненная роль внутриклеточных протеолитических ферментов многогранна: они участвуют в катаболизме белков и доставке пластического материала для биосинтетических реакций, образовании и распаде физиологически активных веществ, оказывают прямое воздействие на энзиматический аппарат клетки посредством активации зимогенов или протеолитической модификации самих ферментов и т. д.
В автолитических превращениях мышечной ткани наиболее важное значение имеет деятельность двух основных ферментных систем. Одна из них связана с функцией движения, другая — катализирует непрерывный распад главных структурных элементов мышечного волокна, в том числе актомиозинового комплекса. Главная роль отводится катепсинам.
Катепсины — кислые протеиназы, проявляют максимальную активность при pH 2,0—5,0, находятся в органах, тканях и локализованы в лизосомах, которые представляют собой внутриклеточные пузырьки диаметром около 5,5 мкм, ограниченные мембраной.
Катепсины являются типичными протеиназами и вызывают деструкцию высокомолекулярных белков. С деятельностью катепсинов, которые во втором периоде автолиза освобождаются из лизосом и активируются кислой реакцией среды клетки, тесно связаны изменения свойств белков, предшествующие релаксации мышц.
В настоящее время в мышечной ткани идентифицирован ряд ферментов эндопептидазного действия — катепсины Вх, D, Н, L, G и экзопептидазы — катепсины А, В2 и С.
Катепсин Вх— тиоловая эндопептидаза, активируется SH-coe-Динениями, имеет оптимум активности при pH 6,0, проявляет более высокую по сравнению с коллагеназой способность к гидролизу коллагена в кислой среде и напоминает папаинтиоловую протеиназу растительного происхождения, широко используемую для мягчения мяса.
269
Катепсин D карбоксильная эндопептидаза, проявляющая активность при pH 2.8—4.0. расщепляет низкомолекулярные пептиды. имеет сродство к пептидным связям, образованным гидрофобными боковыми радикалами, и поэтому сходен с пепсином — пищеварительным ферментом, выделяемым слизистой оболочкой желудка. Катепсин Е отличается от катепсина D субстратной специфичностью. более кислым характером и лабильностью. Катепсин //относится к эндоаминопептидаза.м. способен гидролизовать белки и пептиды с максимальной активностью при pH 6.0.
Катепсин / — тиоловая эндопептидаза, присутствует в лизосомальной фракции, гидролизует различные белки с максимальной активностью при pH 5,0.
Лизосомальные экзопептидазы были открыты позднее эндопептидаз. В настоящее время установлено, что они наиболее активны на последних стадиях расщепления белковых молекул, когда под действием эндопептидаз образуется много новых концевых групп.
Катепсин А — лизосомальная карбоксипептидаза, проявляет наибольшую активность по отношению к пептидам при pH около 5,6—6,9, не способен гидролизовать крупные молекулы белка, однако вместе с катепсином D проявляет синергизм по отношению к белкам мышц. Отличительной чертой катепсина А является способность гидролизовать синтетический субстрат карбобен-зокси-£-глютамил-/-тирозин.
Катепсин В^ — относительно неспецифическая карбоксипептидаза, активируемая сульфгидрильными соединениями; гидролизует полипептиды до свободных аминокислот с оптимумом pH 5,5—5,6, осуществляет глубокий гидролиз полипептидных фрагментов, образующихся в результате действия эндопептидаз.
Катепсин С—типичная тиоловая экзопептидаза, которая расщепляет пептиды и их производные с оптимумом pH 5,0—6,0. Являясь своеобразной аминопептидазой, принимает участие в деградации белка в комплексе с другими катепсинами или выполняет функции трансфераз.
В саркоплазме, митохондриях и рибосомах клеток выделен ряд протеиназ, проявляющих максимальную активность в нейтральной среде (pH 7,0—8,0) при наличии ионов калия. Кальцийзави-симые тканевые протеиназы получили название кальпаинов.
Лизосомальные протеиназы — катепсины и кальцийзависимые протеиназы — кальпаины участвуют в автолитической деструкции тканей двояким образом: непосредственно воздействуя на основные компоненты клеточных элементов и путем активации других протеолитических ферментов.
Главенствующую роль в деградации белков играют катепсины £, В, Н и D, при этом важное место в процессе внутриклеточного протеолиза отводится катепсину D.
Действуя на разные субстратные фрагменты, катепсины ока-270
зывают существенное влияние на структуру белковых компонентов. Это вносит определенный вклад в диссоциацию образовавшихся на этапе посмертного окоченения белковых агрегатов, ведет к появлению свободных сульфгидрильных групп и частичному восстановлению свойств мышечной ткани, утраченных в процессе окоченения.
В результате действия катепсинов на белки при правильном развитии автолитических процессов мясо приобретает нежность. сочность, выраженные вкус и аромат. Характер и глубина автолитических изменений в мясе влияют на его качество и пищевую ценность.
Изучение и целенаправленное использование биохимических свойств тканевых ферментных систем необходимы для регулирования и интенсификации технологических процессов получения свежего мяса и продуктов его переработки.
При получении продуктов с заданными свойствами управление технологическими процессами осуществляется путем воздействия на основные ферментные системы мяса внешних факторов: температуры, pH среды и т. д., для чего необходимо знать условия проявления активности катепсинов.
Таким образом, имея объективную информацию о характере и глубине изменений структурных компонентов тканей, активности катепсинов, можно создать условия максимального использования животных тканей для получения качественных мясных продуктов.
3.2. БИОХИМИЧЕСКИЕ СВОЙСТВА КРОВИ
Кровь убойных животных — один из важнейших источников высокоценного животного белка. По содержанию белка кровь практически не отличается от мяса и содержит лишь на 5—10% больше воды. Реакция среды слабощелочная, почти нейтральная. Кровь обладает способностью к пенообразованию и образованию эмульсий. Коэффициент переваримости крови составляет 94—96 %, т. е. она почти полностью усваивается организмом. Кроме того, наличие в ней биологически активных веществ делает ее более полноценным источником продуктов питания.
В производстве мяса и мясопродуктов используют цельную кровь, плазму (фракцию крови без форменных элементов) и сыворотку (плазму без фибриногена). Особенностью цельной крови и плазмы является свертывание.
При глубоких повреждениях кровь обильно вытекает из сосудов и через некоторое время свертывается в результате превращения растворимого белка плазмы фибриногена в нерастворимый фибрин. Механизм свертывания крови очень сложен и регулируется с помощью каскадной системы реакций, протекающих с участием ферментов и без них (рис. 3.5).
271
Поврежденные ткани
Кровяные пластинки
освобождают
распадаются и освобождаю т
тромбопластин
ером бон 11 та рн ый фа ктор
Белки
Протромбокиназа
Протромбин
>Тромбин + пептидные фрагменты
Фибриноген
Тромбин катализирует реакцию
— Фибрин-мономер + пептиды
Полимеризация мономера
▼
Фибрин-полимер
Рис. 3.5. Последовательность реакций, приводящих к свертыванию крови
«Пусковым механизмом» свертывания крови является тканевый фактор тромбопластин, являющийся липопротеидом, который проявляет протеолитическую активность, но охарактеризован еще недостаточно. При повреждении сосудов он освобождается и инициирует свертывание. В результате реакций тромбопластина с ионами Са2+ и некоторыми белками плазмы (проакцелерином и проконвертином) образуется фермент тромбокиназа.
Второй путь образования тромбокиназы связан с распадом тромбоцитов и выделением из них тромбопластина, который впоследствии взаимодействует с ионами Са2+ и некоторыми глобулинами плазмы.
Последняя стадия механизма свертывания — протеолиз фибриногена тромбином с образованием нерастворимого фибрина.
Итак, тромбин — важный фермент системы свертывания крови, активно гидролизующий фибриноген. Способность тромбина специфически расщеплять связь между остатками аргинина и глицина свидетельствует о сходстве тромбина с трипсином. Тромбин имеет молекулярную массу 33 700 и состоит из двух полипептид-ных цепей (А и В).
Последовательность аминокислот вокруг серина в активном центре тромбина Gly—Asp—Ser—Gly—Gly—Pro аналогична их последовательности в активном центре сериновой протеиназы поджелудочной железы. Как и панкреатические сериновые протеиназы, тромбин синтезируется в виде неактивного зимогена — протромбина, который имеет молекулярную массу 66 000. Про
272
тромбин синтезируется в печени при наличии достаточного коли-чества витамина К.
' Переход протромбина в тромбин эффективно протекает ири
участии фермента тромбокиназы в присутствии ионов Са-' и других факторов сверлывания.
В результате иротеолитического растепления связи аргинин — треонин с аминоконца протромбина высвобождается фрагмент молекулярной массой 32 000 (рис. 3.6). Последующее расщепление связи аргинин — лейцин приводит к высвобождению активного тромбина.
В результате серии протеолитических реакции протромбин превращается в активную форму — тромбин с ярко выраженной специфичностью к разрыву пептидных связей между аргинином и глицином. Такая специфичность действия тромбина обеспечивает превращение фибриногена в фибрин в процессе свертывания крови.
Таким образом, свертывание крови включает эффективно рст у-лируемую серию превращений неактивных зимогенов в активные ферменты, что в итоге приводит к образованию тромбина, а затем фибрина. За небольшим исключением, все активируемые компоненты свертывания представляют собой сериновые протеиназы. Все они ингибируются подобно трипсину диизопропилфторфосфатом и гидролизуют пептидные связи, образованные карбоксильными группами остатков аргинина и глицина.
Свертывание крови в значительной степени затрудняет ее промышленную переработку. Для исключения или замедления свертывания крови выводят или ослабляют активизацию одного из компонентов системы. Вещества, вызывающие целевое действие, | называются антикоагулянтами или стабилизаторами. Химическая । природа и механизм их действия на свертывающую систему раз-
। личны. В связи с этим антикоагулянты подразделяются на две
большие группы — физиологические и нефизиологические. Действие их, как правило, очень быстрое. Стабилизаторы вводят сразу
Отщепляемый фрагмент
7 Карбоксиглутаматы локализованы в этой области
1 Алд-27 4
~А X
Arg -323^- Участки, в которых происходит расщепление
582
Рис. 3.6. Структура протромбина:
в результате растепления двух пептидных связен (Arg-274 — Thr-275 и Arg-323 — 1 1е-324) образуется тромбин; А- и В-пепи тромбина соединены дисульфидной связью
273
после изъятия крови, ло начала образования сгустка. Физиологические стабилизаторы предотвращают прижизненное свертывание крови, инактивируя важнейшие сиспемы свертывания. К ним oi носятся гепарин, антитромбопластин. ан титромбин и др.
При пропускании крови, например, через слой ионообменной смолы 50 сс кальция заменяется налрием. Достаточно снизим, концентрацию вдвое для того, чтобы кровь не свертывалась. Существуют и другие подходы к стабилизации крови.
Кровь убойных животных широко применяется для производства мясопродуктов. Это связано с тем. что белки плазмы крови кроме высокой пищевой и биологической ценности имеют высокие функциональные свойства: высокую эмульгирующую, водо связывающую способность и обладают способностью к геле-, пено- и волокнообразованию, что обусловлено наличием в ее составе альбуминов и фибриногена.
3.3. БИОХИМИЧЕСКАЯ АКТИВНОСТЬ ЖИВОТНЫХ ТКАНЕЙ
При жизни животных некоторые органы и ткани способны накапливать и секретировать биологически активные вещества, которые выполняют важнейшие функции в поддержании жизнедеятельности. После убоя сельскохозяйственных животных из этих источников получают ферментно-эндокринное сырье, которое в дальнейшем идет на производство медицинских препаратов. Последние, в свою очередь, широко используются в теоретической науке, медицине, фармацевтической промышленности, сельском хозяйстве и при переработке пищевого сырья. По механизму действия, свойствам, физиологическому и лечебному эффекту эти вещества подразделяют на гормоны (гормональные препараты) и ферменты (ферментные препараты). Гормоны выделяют главным образом из эндокринных желез (желез внутренней секреции). К такому сырью относятся гипофиз, паращитовидные железы, щитовидная, поджелудочная, половые железы и надпочечники. Ферментные препараты получают преимущественно из слизистой оболочки желудков свиней и сычугов крупного рогатого скота, молочных ягнят и телят, слизистой оболочки тонкого отдела кишечника, а также поджелудочной и слюнной желез.
Важнейшими условиями правильной организации сбора ферментно-эндокринного сырья являются быстрое извлечение его из туши животного, соблюдение санитарно-гигиенических требований, предотвращающих загрязнение и микробное инфицирование.
Под каталитическим воздействием ферментов осуществляются процессы гидролиза, фосфоролиза, переноса различных групп (метильных радикалов, остатков фосфорной кислоты и т. д.),
274
окисления, восстановления и т.п. При посредстве ферментов реализуется влияние как внутренних, генетических, так и внешних. природных факторов на развитие организма. Незначительные различия в строении ферментов определяют видовые особенности организмов, а в нарушении биосинтеза некоторых из них лежат причины возникновения наследственных и других заболеваний.
Будучи выделены из клеток, ферменты не утрачивают способности выполнять каталитическую функцию. На этом основано их практическое применение в пищевой, легкой и фармацевтической промышленности. В связи с этим ткани животных, в которых активно синтезируются ферменты, имеют практическое значение в качестве источников для их получения. Определенный интерес представляют ткани низкой нишевой ценности, клетки которых синтезируют ферменты.
Традиционно с этой целью используют слизистые оболочки, богатые пищеварительными ферментами, которые в метаболизме животного организма выполняют функции гидролитического распада пищевых веществ: секрет слюнных желез —слюна (особое значение в функции которой принадлежит амилазе, а также лизоциму): желудочный сок, содержащий пепсин, ренин и липазу (первые два активируются соляной кислотой желудочного сока); секрет поджелудочной железы, желчь и кишечный сок, богатые трипсином, химотрипсином, липазой, амилазой, рибонуклеазой и др.
Пепсин, трипсин и химотрипсин выделяются железистыми клетками в виде неактивных предшественников — зимогенов: пепсиногена. трипсиногена и химотрипсиногена. Их активные центры блокированы фрагментами полипептидной цепи, после отщепления которых ферменты приобретают активность.
В группе протеолитических ферментных препаратов животного происхождения наибольший практический интерес представляют ферментные препараты, вырабатываемые из поджелудочной железы убойных животных, а также пепсин, вырабатываемый из слизистой оболочки сычугов крупного рогатого скота.
Поджелудочная железа содержит комплекс ферментов, около 50 % которых составляют трипсин и химотрипсин. Они выделены в кристаллическом виде и всесторонне изучены. Комплексный препарат ферментов поджелудочной железы называют панкреатином.
Отечественная промышленность вырабатывает достаточно широкий ассортимент ферментных препаратов для медицины, препаративной практики, пищевой и перерабатывающей промышленности.
Гормонами (от греч. «хормао» — побуждаю, возбуждаю, поощряю) называют группу химических соединений, вырабатываемых преимущественно железами внутренней секреции, не имеющими прямого пищевого значения и оказывающими ре-
275
гулируюшсе влияние на процессы обмена вещесзв и функционирование органов и тканей.
Они регулирую! размножение, рост и развитие организма, влияки на дифференцирование тканей, формирование функции. на поддержание гомеостаза центральной нервной системы, участвуют в адаптационных реакциях на внешние раздражители (стресс) и 'I. д.
Гормоны взаимодействуют со специфическим для данного юр-мона рецептором (чаше всего это белки), вызывая ряд биохимических изменений в клетке. В зависимости от механизма взаимодействия гормонов с рецептором все гормоны можно разделить на лве группы: гормоны, не проникающие в клетку и взаимодействующие с рецепторами на наружной стороне мембраны, и гормоны, проникающие внутрь клетки и взаимодействующие с рецепторами в цитоплазме клеток.
В клетке находятся специальные механизмы, которые прекращают действие гормонов, т. с. влияют на обратимое соединение гормон — рецептор. Разрушение гормонов происходит в печени.
Эндокринные заболевания возникают не только при гипер-или гипофункции эндокринных желез, но также при интенсивном распаде гормонов или при нарушении функции рецепторов, без которых гормон не проявляет своего действия.
Гормоны интегрируют обмен веществ, регулируя соподчинен-ность и взаимосвязь разнообразных химических реакций в различных органах и тканях и организме в целом. В свою очередь, деятельность желез внутренней секреции, продуцирующих гормоны, контролируется центральной нервной системой.
Гормоны действуют в организме следующим образом: изменяют проницаемость мембран для определенных веществ, например глюкозы и аминокислот; регулируют активность отдельных ферментов путем аллостерического воздействия; регулируют синтез ферментов, действуя на генетический аппарат клетки; влияют на образование белковой части фермента (или на распад ферментов) и образование коферментов.
По химической природе гормоны млекопитающих можно разделить на три большие группы: белки и полипептиды; стероиды; прочие гормоны, в том числе амины, образующиеся в ходе метаболизма из аминокислот.
Наиболее изученным и важным является гормон инсулин, химическая и пространственная структура которого расшифрована и показана на рис. 3.7.
Некоторые полипептидные гормоны, в том числе инсулин и глюкагон, подобно протеолитическим ферментам желудка и поджелудочной железы синтезируются в клетках эндокринных желез в виде неактивных предшественников, или прогормонов. Пример — инсулин, превращающийся в активную форму путем ферментативного отщепления части полипептидной цепи. В процессе превращения 276
Рис. 3.7. Структура проинсулина и инсулина:
пппт-,^, ™ с*Р\кг>'Ра проинсулина быка: б — третичная структура 1 <. tepa. Al—А_1 — цепь А; В1-В30 — цепь В (пунктиром показана зона связывания с рецептором)
проинсулина в инсулин С-нептид отщепляется пол воздействием специфического фермента, ускоряющего гидролиз пептидных связей по остаткам аргинина в позициях 31 и 60 (см. рис. 3.7. а).
На рис. 3.7, б на примере третичной структуры пунктиром показана зона связывания протомера с рецептором. В четвертичной структуре гексамерной молекулы каждый блок соответствует протомеру: один блок отсутствует, чтобы показать взаимодействие ионов пинка с тремя радикалами гистидина и иона кальция с тремя радикалами глутаминовой кислоты. Источники основных представителей каждой из групп гормонов представлены ниже.
Гормоны
Пептидные
Тиролиберин
Кортикотропин Вазопрессин Инсулин Глюкагон
Стероидные
Кортизол
Р-Эстрадиол
Тестостерон Прогестерон
Амины
Адреналин Тироксин
Место секреции
Гипоталамус
Передняя доля гипофиза
Задняя доля гипофиза
Поджелудочная железа То же
Кора надпочечников
Яичники
Семенники
Желтое тело
Мозговой слой надпочечников Щитовидная железа
В последнее время предпринята попытка классифицировать гормоны, исходя не только из их химической природы, но и из их происхождения и физиологической функции.
Известны биологические (испытание на животных) и химические способы определения гормонов. Реакции качественного определения специфичны для каждой группы гормонов.
В результате реакции иода с крахмалом образуется соединение синего цвета.
К качественным реакциям на гормоны мозгового слоя надпочечников (адреналин, норадреналин) относятся реакции с хлоридом железа (III), с диазореактивом и йодатом калия.
Количественные методы определения гормонов основаны на проведении специфических реакций с образованием окрашенных или флуоресцирующих продуктов, определяемых колориметрически или флуориметрически. Метод количественного определения адреналина по Фолину основан на определении интенсивности синего окрашивания, возникающего при взаимодействии адреналина с солями фосфорновольфрамовой и фосфорномолибденовой кислот. Флуориметрический метод состоит в том, что некоторые вещества при ультрафиолетовом облучении начинают флуоресцировать, т. е. сами становятся источником нового (вторичного) излучения (например, количественное определение адреналина и норадреналина в моче). Излучение регистрируют и количественно измеряют 278
Рис. 3.8. Принципиальная схема флуориметра:
1 - ис 1<>чник Уф-пз.[учения, -’—первичный светофипвтр: .?- кюветв с крооои.' -/ вчоричныи светофи.чыр: 5- фогов.1емен 1: 6 — впей । ровный xcii.inie.il>. 7 - миллиам-
1 icp не i р
при помощи специального прибора — флуориметра (рис. 3.8). Интенсивность флуоресценции
пропорциональна концентрации вещества в растворе.
Пептидные гормоны. Важнейшими являются окситоцин, вазопрессин, гастрин, глюкагон, инсулин, адренокортикотропин и тиреотропин.
Окситоцин и вазопрессин — 9-членные полипептиды аналогичной структуры:
ГЛ II----II СП
тир----цис
тир----иис
S 9
-лей ------ r.Tii(NH?)
Окситоцин
7 S 9
про-----;]рг------гли(Т4Н2)
Вазопрессин
Они выделены из задней доли гипофиза. Окситоцин вызывает сокращение мышечных волокон, способствует нормальному протеканию родов. Вазопрессин в известной мере сходен с окситоцином по функциональной активности: стимулирует сокращение гладких мышц сосудов, а также участвует в регуляции водного баланса организма и осмотического давления плазмы крови.
Гастрин — 17-членный пептид, выделяемый слизистой оболочкой желудка. Он стимулирует секрецию желудочного сока, способствует выделению секрета поджелудочной железы, усиливает тонус и сокращение мышц желудка и тонкого кишечника.
Глюкагон — 29-членный пептид, синтезируется в а-клетках островковой части поджелудочной железы, способствует деструкции углеводов в процессе гликогенолиза.
Адренокортикотропин —- 39-членный пептид, продуцируемый передней долей гипофиза, оказывает разностороннее действие: повышает активность фосфорилазы, липазы и глюкозо-6-фос-фатдегидрогеназы, усиливает синтез белков и нуклеиновых кислот и т. д. Выполняет в организме основную функцию — регуляцию биосинтеза кортикостероидов надпочечными железами.
279
TiipcoipoiiHH 6c нэк. выделяемый передней долей гипофиза. I Ipc.ic гавляс 1 собой i ликонроicon с молекулярной массой 2S состоящий из двух субьсдинпп с молекулярными массами 1 3 600 и 14 700. исключительно ooiаты.х дисульфидными мостиками (5 и б coo I вс1 ствснно). Тиреот рои и и с гимулирует доп с. явность щи говид-нои железы. В свою очередь, выделение тирсофолина регулирус 1ся но принципу обратной связи юрмонами щитовидной железы.
Стероидные гормоны. 11рслс 1 авляюг собой производные полициклических спиртов — стеролов, у которых окислена боковая пень. В корковом слое надпочечников продуцируется более 40 различных стероидов, которые носят общее название «кортикостероиды». Восемь из них являются стероидными юрмонами коры надпочечников. Наибольшее распространение и значение в организме имеют кортикосгерои. 17-оксикортикостерон (гидрокортизон) и альдостерон (в сумме они составляют 4/5 всех кортикостероидов коры надпочечников):
В мужских и женских половых железах, а также в надпочечных железах синтезируется 10 стероидов, обладающих свойствами половых гормонов. Важнейшее значение из них имеют тестостерон (мужской половой гормон) и эстрадиол (женский половой гормон).
Тестостерон
Эстрадиол
280
Эстрашол 11ринадлежит к группе эстрогенов (женских половых гормонов), которые влияют на функционирование почти всех тканей в opi анизмах иозвоночных. Основное их действие — стимуляция роста и созревания органов размножения самок и поддержание их способности к воспроизведению. Эстрадиол оказывает также влияние на обмен углеводов, белков и нуклеиновых кисло'!, активизируег ряд ферментов лимоннокисло!о никла.
Прочие гормоны. Из гормонов, нс являющихся ио своей химической природе пептидами и стероидами, но широко встречающихся у человека и животных, хорошо известны адреналин (феноламин). тироксин (карбоновая кислота с карбоксилом в боковой цепи) и простагландины (производные полиеновых 2()-\тлеродных жирных кислот), а также аналогичные им соединения.
Тироксин продуцируется щитовидной железой и являемся представителем группы полз ирон и нош
он
H-C-NH?
СООН
L - 3,3 - динод-тиронин
L - 3,5,3 -трппод-тн ронин
L-Тиронин 7.-Тироксин
(3.5,3'. 5-тет-раполтирошш)
Биосинтез тиреоидных гормонов осуществляется в щитовидной железе при посредстве особого белка — тиреоглобулина путем конденсации двух молекул дииодтирозина в молекулу тетра-иодтиронина. Связываясь со специфическими глобулинами крови, иодтиронины доставляются к клеткам организма, где проявляют свое действие.
Механизм действия иодтиронинов изучен недостаточно. Считают, что эти гормоны влияют на скорость метаболизма и потребление кислорода тканями. В итоге усиления окислительных
281
процессов повышается интенсивность расщепления углеводов, жиров и белков в процессе обмена веществ с освобождением большого количества энергии.
Простагландины — соединения с широким спектром гормонального действия, синтезируются из производных полиеновых 20-углеродных жирных кислот во многих тканях человека и животных, преимущественно в репродуктивных органах.
Простагландины выполняют разнообразные физиологические и фармакологические функции. В связи с этим они находят все более широкое применение при создании принципиально новых лекарственных средств. Это связано с тем, что механизм их действия сводится к усилению или ослаблению влияния многих других гормонов на фундаментальные стороны обмена веществ на генетическом и других уровнях. Считают, что простагландины являются модуляторами гормонорецепторных комплексов, т. е. способны изменять их активность, опосредуя действие гормонов.
Нестимулированный уровень гормонов очень низок (в пределах микромолярных и ниже концентраций). В силу этого их выделение, идентификация и количественное определение связаны с определенными трудностями.
Лабораторная работа № 1
ОЦЕНКА ГЛУБИНЫ И ХАРАКТЕРА АВТОЛИТИЧЕСКИХ ПРЕВРАЩЕНИЙ МЫШЕЧНОЙ ТКАНИ МЕТОДАМИ БИОХИМИЧЕСКОГО АНАЛИЗА НЕБЕЛКОВЫХ ВЕЩЕСТВ
Цель работы. Приобрести практический навык в оценке технологической пригодности мяса на основе анализа небелковых веществ мышечной ткани.
Задачи. Определить количественное содержание гликогена, неорганических фосфорных соединений, АТФ, глюкозы в мясе, определить органолептические показатели и оценить технологическую пригодность мяса.
Объекты исследования. Образцы мышечной ткани различных видов убойных животных и птицы разных сроков хранения массой по 50 г. Хранение образцов осуществляется при температуре 4 °C и относительной влажности воздуха 80 %.
Материалы, реактивы и оборудование. Весы технические; водяная баня; электроплитка; асбестовая сетка; масляный насос; штатив; скальпель; кварцевый песок или толченое стекло; бумажные фильтры; порошковый асбест; песочные часы на 3 мин; термометр; индикаторная бумага; фарфоровые ступки; колбы конические вместимостью 150 см3; колбы мерные вместимостью 50 и 100 см3; часовые стекла; воронки; колбы Бунзена с резиновыми
282
пробками, в которые вставлены стеклянные фильтры № 2 или 3; бюретки; пинетки объемные вместимостью 5. 10 и 20см-\ стеклянные палочки с резиновыми наконечниками; раствор медного купороса (реактив А); щелочной раствор сегнетовой соли (раствор Б); раствор сульфата окисного железа; раствор КМпО4 молярной концентрацией 0.1 моль/дм3: раствор ZnSO4 массовой долей 15%; раствор желтой кровяной соли массовой долей 10%; СаССБ; раствор HCI массовой долей 17%; насыщенный раствор Na?COs: толуол в капельнице.
Приготовление реактивов. Реактив А (раствор сульфат меди): 40 г CuSO, • 5Н2О растворяют в 1 дм3 воды и фильтрую!.
Раствор Б (щелочной раствор сегнетовой соли): 150 г NaOH растворяют в воле, добавляют 20 г Сегнетовой соли и доводят объем водой до 1 дм3.
Раствор суд ь ф а т а окисного ж е л с з а: 50 г Fe?(SO4 ). растворяют в 400—500 см3 воды, добавляют 110 см3 концентрированной H2SO4 и доводят объем водой до 1 дм3.
Р а с т в о р КМпО4 м о л я р ы о й конце н г р а и и е й 0,1 моль/дм3; готовят из фиксанала или (при его отсутствии) растворяют 5 г КМпО4 в 1дм3 горячей воды, кипятят 30 мин и после охлаждения переливают в темную склянку: через 2 дня определяют титр по щавелевой кислоте.
Т а б л и ц а 3.1 Масса глюкозы (мг), соответствующая массе меди (мг) по методу Бертрана
Глюкоза | Мель Глюкоза Медь | Глюкоза Медь
10 20.4 28 55,3 46 88,2
11 2.4 29 57,2 47 90,0
12 54,3 30 59,1 48 91.8
13 26,3 31 60,9 49 93,6
14 28,3 32 62,8 50 95,4
15 30,2 33 64,6 51 97,1
16 32,2 34 65,5 52 98,9
17 34,2 35 68,3 53 100,6
18 36,2 36 70.1 54 102,3
19 38.1 37 72,0 55 104,1
20 40,1 38 73.8 56 105,8
21 42,0 39 75,7 57 107.6
22 43.9 40 77,5 58 109.3
23 45,8 41 79,3 59 111,1
24 47.7 42 81.1 60 112,8
25 49.6 43 82.9 61 114,5
26 51,5 44 84,7 62 116.1
27 53.4 45 86.4 63 117.9
283
Продолжение
ЛЮКО Мель Г.1юкоы Медь Глюкоз.। Медь
6-4 1 19.6 -- 141.2 90 162.0
65 121.3 142.S 91 163.6
66 123.0 79 144.5 92 165.2
67 124.7 SO 146.1 93 166.7
68 126.4 Si 147.7 94 168.3
69 128.1 82 149.3 95 169.9
70 129.8 S3 150.9 96 171.5
71 131.4 84 152.5 97 173.1
72 133.1 8 5 154.0 98 174.6
73 134.7 S6 155.6 99 176.2
74 136.3 87 157.2 100 177.8
75 137.9 8 <8 158.8
76 139.6 89 160.4
Методические указания. В течение первых суток после убоя развитие посмертного окоченения приводит к снижению pH от 6.5—7,0 до 5,5—5,6, отсутствию выраженных вкуса и аромата. Сравнительные органолептические показатели продукта в зависимости от глубины автолитических превращений в мясе приведены в табл. 3.2.
Таблиц а 3.2
Органолептические показатели продуктов, приготовленных из мяса на разных стадиях автолиза
Продукт
Несозревшее мясо
Созревшее мясо
Вареное мясо Жесткое, сухое, отсутствуют Нежное, сочное, со спеиифи-специфические приятные ческими приятными вкусом и вкус и аромат запахом
Бульон Мутный, отсутствуют специ- Прозрачный, со специфичес-фические приятные вкус и кими приятными вкусом и аромат мясного бульона ароматом
В связи с отсутствием поступления кислорода в организм ре-синтеза гликогена в мясе после убоя не происходит и начинается его анаэробный распад, который протекает по пути фосфоролиза и амилолиза (рис. 3.9) с образованием молочной кислоты и глюкозы. Это коррелирует с изменением pH и АТФ (рис.3.10).
Через 24 ч гликолиз приостанавливается вследствие исчерпания запасов АТФ и накопления молочной кислоты, подавляющей фосфоролиз.
Накопление молочной кислоты приводит к смещению pH в кислую сторону, в результате чего возрастает устойчивость мяса 284
Анаэробный распад гликогена
В течение 24 ч Г (90 % гликогена) ~ рол'из
Фосфо-
В течение 6—8 сут\ Амилолиз- (Ю % гликогена)
АТФ--\\:УГ)^
Пировиноградная кислота
Полисахариды
Молочная кислота
Мальтоза
Глюкоза
Рис. 3.9. Пути анаэробного распада гликогена
Продолжительность автолиза, ч
Рис. 3.10. Изменение уровня АТФ и pH в мышечной ткани в послеубойный период:
1- АТФ; 2 — pH
к действию гнилостных микроорганизмов; снижаются растворимость миофибриллярных белков (изоэлектрическая точка при pH 4,7—5,4), уровень их гидратации, величина водосвязывающей способности; происходит набухание коллагена соединительной ткани; повышается активность катепсинов (рН-оптимум 5,3), вызывающих гидролиз белков на более поздних стадиях автолиза; разрушается бикарбонатная система ткани с выделением диоксида углерода, создаются условия для интенсификации реакций цвето-образования в молекуле миоглобина вследствие перехода двухвалентного железа в трехвалентное; изменяется вкус мяса; активизируется процесс окисления липидов.
Очевидно, общее количество и соотношение специфических продуктов биохимических реакций указывают на характер и глубину автолиза, о котором можно судить путем анализа биохимических веществ небелковой природы.
285
Количественное определение |.иокозы в мышечной ткани проводят меюдом Бер 1 рана. Метод основан на восстановлении щелочного раствора оксида меди (11) в. оксид меди (1) и учете последнего путем воздействия на него раствором схльфата железа (трсхвалентного). сильно подкисленного серной кис.дотои. Количество воссгановленно!о при этом железа определяемся пированием перманганатом калия. Процесс сводится к следующим реакциям:
2СиО 4 редуцирующий сахар •( и О:
Сиф + Fe, (S()4), 4- Н ,SO4 = 2CliSO4 4 2FcSO4 4 H2O:
lOFeSO, + 2КМпО, 4- 8FFSO. = 5Fc, (SO J, + 2MnSO, + 8H,O.
Из уравнений следует, что 1 см’ раствора перманганата калия молярной концентрацией 0.1 моль/дм-’ соответствует 6.35 мг меди. Зная количество миллилитров раствора КМпО4, пошедшего на титрование сульфата железа, находят, какому количеству миллиграммов меди оно соответствует, и определяют количество сахара в исследуемом растворе по табл. 3.1.
Выполнение работы по оценке глубины и характера автолитических превращений мышечной ткани определенного вида убойных животных или птицы осуществляется комплексно бригадами студентов по 2—3 человека.
Каждой бригаде выдается задание по изучению динамики изменения одного-двух конкретных биохимических показателей при рекомендованных сроках хранения. Бригады выбирают методы исследования, обосновывают их выбор, проводят законченный цикл исследований в соответствии с конкретными методиками.
1. ОПРЕДЕЛЕНИЕ pH
Подготовка проб. Для определения pH мяса готовят водную вытяжку в соотношении 1 :10, для чего навеску образца мяса массой (10,00 ± 0,02) г тщательно измельчают (ножницами или на мясорубке), помещают в химический стакан вместимостью 100 см1 * 3 и экстрагируют дистиллированной водой в течение 30 мин при температуре окружающей среды и периодическом помешивании стеклянной палочкой. Полученный экстракт фильтруют через складчатый бумажный фильтр и используют для определения pH.
Порядок проведения анализа. pH водного экстракта мышечной ткани определяют на потенциометре (pH-метре) любой марки. Результаты фиксируют.
286
2 .КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГЛИКОГЕНА И МОЛОЧНОЙ КИСЛОТЫ В МЫШЕЧНОЙ ТКАНИ
Подготовка проб. Проводят в соответствии с методическими указаниями лабораторных работ № 24, 25. 28 главы 1.
Порядок проведения анализа. Выполняют в соотвс ic гвии с рекомендациями к лабораторным работам № 24. 25 главы 1.
3. ОПРЕДЕЛЕНИЕ АТФ
Порядок проведения анализа. Ведут в точном соответствии с рекомендациями к лабораторной работе N_’ 28 главы 1.
4. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ГЛЮКОЗЫ В ВЫТЯЖКЕ МЫШЕЧНОЙ ТКАНИ ПО МЕТОДУ БЕРТРАНА
Подготовка проб. Для приготовления вытяжки сахаров (глюкозы) навеску предварительно измельченной мышечной ткани массой (10.00 ± 0.02) г тщательно растирают в ступке с 1—2 г кварцевого песка и количественно переносят в коническую колбу вместимостью 150 см3 с 50 см3 воды. Затем колбу ставят на 30 мин на водяную баню при температуре 70—80 °C. Из охлажденной смеси удаляют белки и коллоидные вещества, мешающие определению сахаров, путем добавления в колбу 5 см3 раствора сульфата цинка массовой долей 15 % и 5 см3 раствора желтой кровяной соли массовой долей 10 %. Смесь перемешивают, фильтруют через бумажный складчатый фильтр в мерную колбу вместимостью 100 см3 и промывными водами доводят до указанного объема. На этом этапе работу прерывают, добавив в вытяжку несколько капель толуола и закрыв колбу пробкой.
Порядок проведения анализа. В коническую колбу помещают 20 см3 вытяжки, добавляют по 20 см3 реактивов А и Б, ставят полученную смесь на электроплитку (на асбест), нагревают до кипения и кипятят 3 мин. В результате выпадает красный осадок оксида меди (Г). Жидкость над осадком должна иметь синий цвет; если она бесцветна, нужно взять меньший объем вытяжки — 10 или 15 см3, доведя объем дистиллированной водой до 20 см3.
Колбу Бунзена со стеклянным фильтром закрепляют при помощи лапок в штативе и присоединяют к масляному насосу. Покрывают фильтр слоем асбеста толщиной 1 — 1,5 мм, для чего разводят порошковый асбест в горячей воде, выливают на фильтр и отфильтровывают при разрежении.
Сливают жидкость над осадком Си2О декантацией (не взбалтывая) по стеклянной палочке на стеклянный фильтр и осторожно отсасывают (не досуха, чтобы осадок, попавший на стеклянный фильтр, не окислялся). Затем осадок в колбе и на фильтре не-
287
сколько раз промывают горячей дистиллированной водой до иол ново обесцвечивания промывных вол. стекающих с фнлыра.
Колбу отделяют от фильтра, тшшсльно се промывают вначале водопроводной водой. а затем дистиллированно!!, вставляю! пробку с фильтром. Осадок в колбе и на фильтре растворяют сульфатом железа в серной кислоте, добавляя его каждый раз небольшими порциями и отсасывая насосом. После растворения осадка колбу и фильтр промывают холодной дистиллированной водой до тех пор. пока кислая реакция не исчезнет в промывных водах. Содержимое колбы титруют раствором перманганата калия до слаборозового окрашивания.
Массовую долю глюкозы (%) рассчитывают по формуле
дИ 100
ЙтТЙООО ’
(3.1)
где //— количество глюкозы во взято!! вытяжке, мг: К—объем всей вытяжки, см'; 100 —перевод в проценты; И,—объем вытяжки, взятой для определения. смД т — масса навески, г; 1000 —перевод миллиграммов в граммы.
П р и м е р. При определении глюкозы в 20 см' экстракта на титрование было израсходовано 15 см3 раствора перманганата калия молярной концентрацией 0.1 моль/дм3. Масса меди составляет 6,36 15 = 954 мг.
По табл. 3.1 эта масса меди соответствует 50 мг глюкозы, следовательно, содержание глюкозы в исследуемом растворе 250 мг%.
Полученные экспериментальные данные представляют в виде таблицы:
Наименование и характеристика образца мышечной ткани pH Содержание. мг%
Г I молочной гликогена КИСЛ0Гы глюкозы АТФ
По совокупности показателей делают вывод и формулируют заключение.
Лабораторная работа № 2 ОПРЕДЕЛЕНИЕ АКТИВНОСТИ КАТЕПСИНОВ МЫШЕЧНОЙ ТКАНИ
Цель работы. Освоить методику определения протеолитической активности катепсинов мышечной ткани убойных животных.
Задачи. Получить экстракт катепсинов из мышечной ткани и определить протеолитическую активность катепсинов фотометрическим методом.
288
Объект исследования. Мышечная ткань одного или разных видов животных (коров, свиней, баранов), содержащая комплекс ферментов.
Материалы, реактивы и оборудование. Дистиллированная вола: бумажные фильтры; водяные бани и термостаты: раствор казеината натрия или гемоглобина массовой долей 2 У с pH 3.0; 4,0; 5,0; 6,0; раствор ТХУ массовой долей 2 %: раствор карбоната натрия молярной концентрацией 0,5 моль/дм?; рабочий раствор реактива Фолина: фотоэлектроколориметр.
Приготовление реактивов. О с н о в н о й раствор Фолина: 100 г вольфрамата натрия и 25 г молибдата натрия вносят в круглодонную колбу вместимостью 2 дм3 с пришлифованным обратным холодильником, добавляют 700 см3 дистиллированной воды, 50 см3 раствора ортофосфорной кислоты плотностью 1,869 г/см3, массовой долей 85 % и 100 см3 концентрированной соляной кислоты; смесь кипятят на слабом огне на асбестовой сетке в течение 10 ч, охлаждают, переносят в колбу Эрленмейера, стенки колбы и холодильник ополаскивают 50 см3 воды, затем туда же добавляют 150 г сульфата лития и 5 капель брома. Открытую колбу нагревают и содержимое кипятят на слабом огне в течение 15—20 мин под тягой, чтобы удалить пары брома (раствор должен иметь желтую окраску, если раствор зеленый, то обработку бромом повторяют). После охлаждения объем раствора доводят дистиллированной водой до 1 дм3 и фильтруют через трубку Аллина, заполненную стекловатой. Концентрацию кислоты проверяют титрованием разбавленного в десять раз реактива Фолина раствором гидроксида натрия молярной концентрацией 0,1 моль/дм3 по фенолфталеину. Приготовленный раствор хранят в склянке из темного стекла.
Рабочий раствор Фолина: готовят разведением основного раствора дистиллированной водой в два раза.
Раствор субстрата (казеината натрия или гемоглобина массовой долей 2 %): 2 г воздушно-сухого казеината натрия или гемоглобина растворяют в 90 см3 универсального буферного раствора молярной концентрацией 0,1 моль/дм3 с заданным значением pH. Затем раствор переносят в мерную колбу вместимостью 100 см3 и доводят объем до метки тем же буферным раствором.
Универсальный буферный раствор молярной концентрацией 0.1 моль/дм3: готовят смешиванием равных объемов растворов А, В и С и различных объемов раствора гидроксида натрия молярной концентрацией 1 моль/дм3 (40 г NaOH растворяют в 1 дм3 воды). Получают буферные растворы с pH от 1,8 до 12,0.
Раствор А (раствор уксусной кислоты молярной концентрацией 0,1 моль/дм3); 5,7 см3 ледяной уксусной кислоты растворяют в 1 дм3 дистиллированной воды.
289
Рас г в о р В (раствор ортофосфорной кислоты молярной концентрацией 0.1 моль/дм3): 6.45 см3 ортофосфорной кислоты растворяют в 1 дм3 дистиллированной воды.
Раствор С (раствор ортофосфорной кислоты молярной концентрацией 0.1 моль/дм3): 6,18 см3 ортофосфорной кислоты растворяют в 1 дм3 дистиллированной воды.
Методические указания. Для анализа рекомендуется использовать мышечную ткань на разных стадиях автолиза. При изучении влияния температуры на активность катепсинов субстраты (растворы стандартных белков) прогревают до температур соответственно 20, 40, 50, 60 °C.
При изучении влияния pH на ферментативную активность мышечного экстракта субстрат растворяют и pH доводят раствором НС! молярной концентрацией 0,3 моль/дм3 до 3,0; 4,0; 5,0; 6,0.
Для определения активности протеиназ используют метод Ан-сона в модификации Е. Каверзневой, в котором о каталитических свойствах судят по степени расщепления стандартных белков с образованием низкомолекулярных продуктов: пептидов и аминокислот, в частности, по накоплению тирозина.
За единицу протеолитической активности (ПА) принято количество фермента, которое за 1 мин при 30 °C катализирует переход в неосаждаемое трихлоруксусной кислотой (ТХУ) состояние такого количества субстрата, которое содержит 1 мкмоль тирозина (1,181 мг).
Протеолитическая активность экстракта катепсинов выражается числом протеолитических единиц в 1 см3 ферментного раствора (ед/см3).
Метод определения протеолитической активности может также успешно применяться и при анализе пищеварительных ферментов: пепсина, панкреатина, химотрипсина и брипсина.
Подготовка проб. Мышечную ткань очищают от жировой и соединительной тканей, измельчают сначала на мясорубке, а затем гомогенизируют. Навеску измельченного сырья смешивают с дистиллированной водой в соотношении 1 : 20 и экстрагируют при (20 ± 5) °C при периодическом перемешивании смеси в течение 30 мин. Затем смесь фильтруют через бумажный фильтр, фильтрат используют в качестве вытяжки протеолитических ферментов — катепсинов.
Порядок проведения анализа. Субстрат (раствор казеината натрия или гемоглобина массовой долей 2 % с заданным значением pH) объемом 2 см3 помещают в пробирку, прогревают до нужной температуры (в соответствии с конкретным заданием) и прибавляют 2 см3 исследуемого экстракта протеолитических ферментов мышечной ткани. При этом фиксируют время внесения экстракта. Смесь выдерживают в течение 15 мин, а затем добавляют 4 см3 раствора ТХУ массовой долей 2 %, встряхивают и выдерживают 20 мин (количество вносимого фермента рассчитывают так, чтобы в смеси присутствовал избыток субстрата).
290
Параллельно с опытной готовят контрольную пробу, смешивая реактивы в обратной последовательности. Для этого в контрольную пробирку помещают 2 см3 ферментной вытяжки, добавляют 4 см3 раствора ТХУ массовой долей 2 %, встряхивают и выдерживают 15 мин. Затем добавляют еще 2 см3 субстрата, встряхиваю! и выдерживают 20 мин при заданной температуре.
Реакционные среды (контроль и опыт) фильтруют через бумажный фильтр. В чистые сухие пробирки отбирают по 1 см3 прозрачного фильтрата и 5 см3 раствора NaCO, молярной концентрацией 0,5 моль/дм3 и 1 см3 рабочего раствора реактива Фолина. Смесь встряхиваю! и выдерживают в течение 20 мин. После этого окрашенный раствор фотоколориметрируют против контроля при длине волны 670 нм с красным светофильтром (№ 9) в кюветах с толщиной светопоглощающего слоя 10 мм.
Протеолитическая активность (ед/см3)
ПА = (4£>К)/(ТЭ • 7), (3.2)
где D — оптическая плотность раствора; А'—степень разведения; ТЭ —тирозиновый эквивалент, определяемый по калибровочному графику для данного реактива Фолина; Г—продолжительность гидролиза, мин; Т= 15 мин.
Построение калибровочного графика. Для расчета протеолитической активности строят калибровочный график по тирозину, по которому затем вычисляют тирозиновый эквивалент ТЭ, т. е. ту оптическую плотность, которая соответствует 1 мкмоль тирозина в 1 см3 стандартного раствора. ТЭ необходимо устанавливать для каждой новой партии реактива Фолина и каждого фотоэлектроколориметра.
Готовят раствор тирозина молярной концентрацией 10~3 моль/дм3, для чего 181,19мг чистого тирозина растворяют в растворе НС1 молярной концентрацией 0,2 моль/дм3 в мерной колбе вместимостью 1 дм3 (готовят две параллельные пробы). Исходный раствор тирозина используют для приготовления дальнейших разведений в соответствии с приведенными ниже рекомендациями, для чего в мерную колбу вместимостью 50 см3 вносят от 1 до 15 см3 исходного раствора тирозина и объем доводят до метки раствором НО молярной концентрацией 0,2 моль/дм3:
Объем исходного раствора тирозина, см3 Объем раствора НС! молярной концентрацией 0,2 моль/дм’’ Концентрация тирозина
моль/дм3 мкмоль/см3
1,0 49,0 0,2 • 10~4 0.02
2,0 48,0 0,4- 10~4 0,04
4,0 46,0 0,8 • 10~4 0,08
5,0 45,0 1,о- ю-4 0,10
7,5 42,5 1,5- 10~4 0,15
10,0 40,0 2,0 IO”4 0,20
15,0 35,0 3,0- 10~4 0.30
291
Затем в пробирки вносят ио 1 см3 раствора тирозина разных концентраций. Добавляют в них при постоянном помешивании по 5 см3 раствора карбоната натрия молярной концентрацией 0.5 моль/дм3 и 1 см’ рабочего раствора Фолина. Контрольный опыт готовят точно так же. но вместо раствора тирозина берут 1 см3 дистиллированной воды. Дают реакционной смеси постоять 20 мин.
Интенсивность окраски опытного раствора измеряют по отношению к контрольному при Л = 656—670 нм в кюветах с толщиной светопоглощающего слоя 10 мм. По средним данным, полученным из двух опытов, строят калибровочный график. На оси абсцисс откладывают количество тирозина (мкмоль/см3), на оси ординат — соответствующие значения оптической плотности D.
Экспериментальные данные сводят в таблицу:
Объект D ПА. ел/см’ Условия реакции
pH температура. ‘С
По полученным данным строят графические зависимости активности катепсинов/(Д ), где А — исследуемый фактор. Примеры показаны на рис. 3.11.
После построения графиков студенты самостоятельно описывают полученные результаты, делают выводы и формулируют заключение.
Рис. 3.11. Примеры экспериментальных зависимостей протеолитической активности (ПА) катепсинов от температуры (а) и pH среды (б)
292
Лабораторная работа № 3 ОПРЕДЕЛЕНИЕ КОМПОНЕНТОВ СИСТЕМЫ СВЕРТЫВАНИЯ КРОВИ
Цель работы. Приобрести практический навык в определении компонентов системы свертывания крови.
Задачи. Изучить действие ионов кальция на свертываемость крови и ее фракций и количественно определить содержание тромбина.
Объекты исследования. Стабилизированная кровь различных видов животных, получаемая в условиях производства; плазма и сыворотка крови, полученные в лабораторных условиях или при промышленной переработке крови убойных животных.
Материалы, реактивы и оборудование. Раствор хлорида кальция массовой долей 3 %; раствор хлорида натрия массовой долей 0,85 %; штатив с пробирками; химический стакан вместимостью 200—400 см3; стеклянные палочки; пипетки вместимостью 1, 5 и 10 см3; настольная центрифуга; термостат; холодильник; водяная баня.
Методические указания. В технологической практике весьма важно не допустить свертывания крови, так как кровь является весьма ценным сырьевым источником в получении продуктов пищевого, лечебно-профилактического и медицинского назначения. В связи с этим определение факторов свертывания, улучшение механизма их действия лежат в основе разработки новых подходов глубокой переработки крови. Одними из главных компонентов системы свертывания крови являются тромбин и кальций. Действие первого связано с гидролизом фибриногена, а другой участвует практически на всех стадиях биохимических превращений, активизируя ферментные системы.
Появление тромбина в среде свидетельствует о начале развития завершающей стадии свертывания крови. Немаловажная информация связана с установлением действия кальция на белки системы свертывания крови. Свертывание крови включает эффективно регулируемую серию превращений неактивных зимогенов в активные ферменты, которая в итоге приводит к образованию тромбина, а затем и фибрина. Протеолиз фибриногена тромбином с образованием нерастворимого фибрина — заключительная стадия механизма свертывания.
На практике часто используют различные приемы для предотвращения свертывания крови, которые сводятся к исключению одного из факторов системы свертывания.
При промышленной переработке крови широко применяют стабилизаторы, действие которых выводит Са2+ из системы свертывания. В качестве стабилизаторов используют оксалаты, цитраты, фосфаты, сульфаты и др. При действии этих реагентов образуются нерастворимые кальциевые соли.
293
Немаловажное значение имеет количество образовавшегося тромбина, так как именно он осуществляет переход фибриногена в фибрин с образованием сгустка.
Подготовка проб. Стабилизированную кровь животных хранят в холодильнике и используют для получения фракций.
Для получения плазмы в лабораторных условиях стабилизированную кровь животных (50 см3) центрифугируют в течение 5—8 мин при частоте вращения 25—42 с"1. Плазму (надосадочную жидкость) осторожно отбирают пипеткой и переносят в чистую сухую пробирку.
Для получения сыворотки нестабилизированную (или дестабилизированную добавлением раствора хлорида кальция массовой долей 3 %) кровь объемом 150—200 см3 помещают в химический стакан и ставят на 1 ч в термостат при 37 °C, а затем переносят на холод.
Для лучшего отделения сгустка крови от сыворотки обводят сгусток по краям стакана стеклянной палочкой. Через 4—5 ч стояния на холоде сыворотка в виде прозрачной жидкости отделяется от кровяного сгустка. Отстоявшуюся сыворотку осторожно отбирают пипеткой.
Кровь, плазму и сыворотку используют для анализа.
1. ДЕЙСТВИЕ КАЛЬЦИЯ НА СИСТЕМУ СВЕРТЫВАНИЯ КРОВИ И ЕЕ ФРАКЦИЙ
Порядок проведения анализа. Готовят 6 пробирок для крови каждого вида животных. В одну пробирку помещают 2—3 см3 стабилизированной крови, в другую —также 2—3 см3 стабилизированной крови и 5—6 капель раствора хлорида кальция массовой долей 3 %, в третью — 5—6 см3 плазмы, в четвертую — 5—6 см3 плазмы и 5—6 капель раствора хлорида кальция, в пятую — 5—6 см3 сыворотки крови, в шестую — 5—6 см3 сыворотки крови и 5—6 капель раствора хлорида кальция. Пробирки слегка встряхивают и ставят на водяную баню при 37 °C. Через 10—15 мин их вынимают и отмечают, где произошло свертывание.
Результаты проведенных опытов оформляют в виде таблицы:
Номер пробирки Субстрат Условия опыта Результат
Заключение студенты формулируют самостоятельно.
2. ОПРЕДЕЛЕНИЕ ТРОМБИНА
Порядок проведения анализа. В качестве источника тромбина используют сыворотку крови, из которой готовят ряд разведений. В качестве источника фибриногена используют свежую плазму крови, предварительно нагретую до температуры 50—60 °C с по
294
следующей инкубацией в течение 30 мин. За единицу активности тромбина принимают минимальный объем тромбина (см3), который вызывает свертывание.
Готовят 11 пробирок для крови животных. Во все пробирки, кроме первой, помещают по 1 см3 раствора хлорида натрия массовой долей 0.85 %. В первую и вторую пробирки добавляют по 1 см3 неразбавленной сыворотки (раствор тромбина). Содержимое второй пробирки тщательно перемешивают. 1 см3 смеси из второй пробирки переносят в третью. Содержимое третьей пробирки также перемешивают и 1 см3 смеси переносят в четвертую пробирку и так далее до десятой пробирки, из которой 1 см3 жидкости удаляют. Одиннадцатая пробирка содержит 1 см3 раствора NaCl и служит контролем.
Во все 11 пробирок добавляют по 2 см3 плазмы, предварительно инактивированной и разведенной в 10 раз раствором NaCl массовой долей 0,85 % (раствор фибрина). Штатив с пробирками помещают в холодильник на 24 ч.
По истечении времени, осторожно наклоняя пробирки, определяют, в каких пробирках произошло свертывание. Количество тромбина вычисляют в той пробирке, где образовался еле заметный сгусток.
Количество тромбина X (ед.) рассчитывают по формуле
Az=l/K (3.3)
где V— объем тромбина сыворотки крови в пробирке с минимальным образованием сгустка, см3.
Пример. Образование сгустка зафиксировано в первых восьми пробирках. В девятой и последующих пробирках свертывания нс произошло. В восьмой пробирке объем сыворотки составил 0,0078 см3. Составляем пропорцию:
объему сыворотки 0,0078 см3 соответствует 1 ед. тромбина,
объему сыворотки 1 см3 — X ед. тромбина.
Х= 1/0,0078 = 128 ед.
В норме 1 см3 сыворотки содержит 62,5—250,0 ед. тромбина.
Полученные экспериментальные данные сводят в таблицу рекомендуемой формы:
Показатель Номер пробирки
1 2 3 4 5 6 7 8 9 10
Объем тромбина в
пробирке
Количество тромбина
Осадок фибрина + + + + + + + + —
(+, -)*
* Знак «+» в таблице означает, что в пробирке выпал осадок фибрина, а знак «—» — что его нет.
295
Лабораторная работа № 4 ОПРЕДЕЛЕНИЕ МОЛОКОСВЕРТЫВАЮЩЕЙ АКТИВНОСТИ ПЕПСИНА
Цель работы. Приобрести пракг ич'ескшг навык в получении и анализе пепсина.
Задачи. Получить пепсиновый экстракт слизистой оболочки желудков свиней или сычугов крупного рогатого скота и определить молокосвертывающую активность (MCA).
Объект исследования. Свежая, высушенная пли замороженная слизистая оболочка желудков свиней или сычугов крупного рогатого скота.
Материалы, реактивы и оборудование. Соляная кислота: бумажные фильтры: натуральное обезжиренное молоко; секундомер; фарфоровая ступка: стаканы или колбы вместимостью ‘100-250 см3.
Методические указания. Пепсин — пищеварительный! фермент, гидролизующий белки и проявляющий максимум активности при pH 2,0—4.0. Субстратная специфичность фермента проявляется в сродстве к разрыву пептидных связей, образованных гидрофобными радикалами.
Пепсин находит широкое применение при лечении желудочных заболеваний. Технические препараты пепсинов широко используют в пищевой технологии. Самый большой спрос пепсиновые препараты нашли в молочной промышленности при получении творога, творожных масс и различных видов сыров благодаря свойству специфически гидролизовать казеины молока, вызывая их коагуляцию.
При гидролизе пептидов пепсин проявляет высокую специфичность, имея ярко выраженное сродство к гидролизу пептидных связей, образованных карбоксильной группой фенилаланина и аминогруппой другой ароматической аминокислоты. При гидролизе белков фермент проявляет более широкую специфичность за счет функционирования в активном центре «первичных» и «вторичных» участков связывания субстрата. Он расщепляет пептидные связи, образованные тирозином, фенилаланином, триптофаном, а также лизином, гидролизует сложноэфирные связи. Продуктами гидролиза являются в основном пептиды и незначительная доля аминокислот. Он активно свертывает казеин молока, в связи с чем широко применяется в технологии белковых продуктов, вызывая коагуляцию белков молока.
Технология получения препарата пищевого пепсина на производстве включает следующие стадии: измельчение сырья (слизистой оболочки желудков свиней или сычугов крупного рогатого скота), экстрагирование и активацию фермента (pH 2,0), отделение экстракта от твердого остатка, высаливание или осаждение 296
фермента, сушку, измельчение, обезжиривание, просеивание и стандартизацию но активности.
Определение молокосвертывающей активности препаратов лежи! I! основе их стандартизации и расчета дозировки при получении продуктов с заданными свойствами. Методика состоит в фиксировании (по секундомеру) промежутка времени от внесения препарата в субстрат (молоко) до начала коагуляции казеина при проведении ферментативной реакции в стандартных условиях.
Подготовка проб. Слизистую оболочку желудков свиней или сычугов крупного рогатого скота измельчают, настаивают со слабым раствором кислоты (раствор НС1 концентрацией 0.01 моль/дм3) в соотношении 1 : 20 в течение 30 мин. Затем смесь фильтруют через бумажный фильтр, а фильтрат используют в качестве пепсинового экстракта.
Порядок проведения анализа. 100 см3 свежего натурального обезжиренного молока подогревают до 35 °C и прибавляют 1 см3 ферментной вытяжки слизистой оболочки желудков, фиксируя время внесения по секундомеру. Через 1—2 мин стеклянной палочкой периодически проверяют начало образования сгустка белков, которое также фиксируют по секундомеру.
Молокосвертывающую активность (ед/см3) рассчитывают по формуле
МСА = 2400- 100/(тИ), (3.4)
где 2400 — продолжительность свертывания молока, приведенная к стандартному времени коагуляции в производственных условиях (40 мин), с; 100 — объем пробы молока, см3; г — время образования сгустка в молоке, с; И—объем ферментной вытяжки, см3.
Температуру молока рекомендуется измерять в диапазоне 30— 55 °C, pH 5,5-7,5.
Студенты самостоятельно производят расчеты и представляют отчет по выполнению всех операций, формулируют заключение на основе построения графиков зависимостей: МСА=/(рН) и MCA =/(/).
Лабораторная работа № 5
ОПРЕДЕЛЕНИЕ ИНСУЛИНА ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ
Цель работы. Освоить методы получения и определения гормона инсулина.
Задачи. Получить экстракт инсулина и провести качественный и количественный анализы экстракта.
Объект исследования. Свежая или замороженная поджелудочная железа крупного рогатого скота или свиней.
297
Методические указания. В клинико-биохимических исследованиях применяют методы качественного и количественного определения гормонов, отклонения в их содержании используют для диагностики заболеваний.
Среди гормонов, синтезируемых поджелудочной железой, наиболее важное значение имеют инсулин и глюкагон. Являясь веществом белковой природы, инсулин реагирует с образованием специфически окрашенных веществ, которые служат качественными реакциями и могут быть использованы для обнаружения его в экстрактах.
Количественное определение инсулина осуществляется химическим, так называемым фибриллярным, методом, сущность которого состоит в том, что при нагревании инсулина в кислом растворе при 100 °C образуется тиксотропный гель, обладающий сильным двойным лучепреломлением. Путем нагревания в кислой среде при 100 °C и последующего охлаждения до — 80 °C образуется устойчивая фибриллярная модификация инсулина в растворе. Кристаллизующийся продукт, регенерированный из фибрилл, не отличается от исходного инсулина по физическим, химическим и биологическим свойствам.
Метод определения активности инсулина in vitro основан на специфической реакции удлинения инсулиновых фибрилл за счет нативного инсулина и определении их массы.
Принцип метода сводится к получению стандартного фибриллярного инсулина, который готовят из кристаллического инсулина активностью 24—25 ИЕ в 1 мг. Заготовленный фибриллярный инсулин используют в качестве затравки при определении нативного инсулина.
Испытуемые растворы инсулина после затравки выдерживают в течение 24 ч при 48 °C. За этот срок инсулин осаждается, принимая фибриллярную форму.
Фибриллы отделяют от раствора центрифугированием, затем промывают различными растворителями на центрифуге для очистки инсулина от балластных белков и примесей. Полученный осадок сушат над оксидом фосфора (V) и взвешивают или производят спектрофотометрическое определение суспензии инсулина.
Масса инсулина за вычетом массы добавленного для затравки фибриллярного инсулина соответствует содержанию инсулина в испытуемом растворе активностью 24 ИЕ в 1 мг.
При определении спектрофотометрическим методом небольших количеств инсулина получают более точные результаты, чем при гравиметрическом определении.
1. КАЧЕСТВЕННЫЕ РЕАКЦИИ НА ИНСУЛИН
Материалы, реактивы и оборудование. Весы технические; подкисленный этанол; бумажный фильтр; концентрированная азотная кислота; раствор гидроксида натрия массовой долей
298
10 %: раствор сульфата мели массовой долей 1 %: реактив Фоля: реактив Миллона.
Подготовка проб. Свежую или замороженную поджелудочную железу тщательно измельчают и экстрагируют подкисленным этанолом на холоде в соотношении 1 : 10 в течение 10 мин. Для проведения качественных реакций на инсулин используют фильтрованный экстракт поджелудочный железы.
Порядок проведения анализа. Для проведения р е а к и и и Геллера к 1 см3 концентрированной азотной кислоты осторожно по стенке пробирки приливают равный объем раствора инсулина. Пробирку наклоняют под углом 45° так, чтобы жидкости не смешивались. На границе двух жидкостей фиксирую! образование белого аморфного осадка в виде небольшого кольца.
Для проведения биуретовой р е а к ц и и к 1 см3 раствора инсулина добавляют 0,5 см3 раствора гидроксила натрия массовой долей 10 % и 0.1 см3 раствора сульфата меди массовой долей 1 %. Фиксируют изменение окраски.
Для проведения р е а к ц и и Фоля к 0,5 см3 раствора инсулина добавляют 0,5 см3 реактива Фоля и смесь кипятят. Через 1—2 мин термической обработки фиксирую! появление бурого или черного осадка сульфида свинца.
Для проведения реакции Миллона к 1 см3 раствора инсулина добавляют 2—3 капли реактива Миллона и осторожно нагревают. Фиксируют красное окрашивание раствора белка или появление белого осадка белка, который при нагревании приобретает красную окраску.
Наблюдения за проведением качественных реакций на инсулин студенты аккуратно и точно фиксируют в тетради (описывают условия и эффект реакций), формулируют выводы. Результаты полученных исследований оформляют в виде таблицы:
Название и механизм качественной реакции Условия и реактивы, используемые в реакции Полученный эффект
2.КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ИНСУЛИНА
Материалы, реактивы и оборудование. Растворы этанола объемными долями 63, 80, 95 %; серная кислота; вибрационный аппарат; марля; центрифуга лабораторная; центрифужные стаканы; кристаллический инсулин активностью 24 ИЕ: стеклянные палочки; стеклянный капилляр; резиновая трубка; сосуд, соединенный с водоструйным насосом; растворы соляной кислоты молярной концентрацией 0,05 и 1 моль/дм3: метанол; спектрофотометр; шприц; ампула из молибденового стекла; гомогенизатор; термос; сухой лед.
299
Подготовка проб. 200 г измельченной поджелудочной железы заливают раствором этанола объемной долей 80 %, подкисленным серной кислотой до pH 2.0, в соотношении 1 : 2. смесь перемешивают и экстрагируют в течение 3 ч на вибрационном аппарате, контролируя pH через каждый час экстракции.
Полученную массу фильтруют через два слоя марли, осадок отжимают. Раствор тотчас помешают в стаканы вместимостью 200 см3 и центрифугируют в течение 30 мин при частоте вращения 42—50 с '. Если полученный центрифугат недостаточно прозрачен, то его центрифугируют вторично.
Порядок проведения анализа. После окончания центрифугирования раствор сливают, измеряют его объем и переносят 150 см3 в центрифужный стакан, в который добавляют 2,2 см3 стандартного раствора фибриллярного инсулина, содержащего 4 мг инсулина активностью 24 ИЕ. Раствор тщательно перемешивают стеклянной палочкой, которую оставляют в центрифужном стакане, и помещают на 24 ч в термостат температурой 48 °C. Затем образовавшийся осадок центрифугируют в течение 45 мин (при 16с1 — 20 мин и при 3,3 с~' — 25 мин).
Прозрачный раствор из центрифужного стакана осторожно, не захватывая осадка, отбирают стеклянным капилляром, соединенным резиновой трубкой с сосудом, в котором водоструйным насосом создан небольшой вакуум.
Полученный осадок инсулина для освобождения от балластных белков промывают с последующим центрифугированием четырьмя растворами: первый — смесью, состоящей из 99 см3 раствора этанола объемной долей 63 %, 1 см3 раствора соляной кислоты молярной концентрацией 1 моль/дм3 и 1 г хлорида аммония (3 раза по 5 см3); второй — смесью, состоящей из 99 см3 раствора этанола объемной долей 65 % и 1 см3 раствора соляной кислоты молярной концентрацией 1 моль/дм3 (1 раз 5 см3)'; третий — абсолютным ацетоном (2 раза по 5 см3); четвертый — смесью, состоящей из 100 см3 метанола и 1 см3 раствора соляной кислоты молярной концентрацией 1 моль/дм3 (2 раза по 5 см3).
Полученный осадок промывают в центрифужных пробирках вместимостью 15—20 см3. Для этого осадок из центрифужного стакана после тщательного перемешивания стеклянной палочкой с 5 см3 промывной смеси (первый раствор) количественно переносят в центрифужную пробирку. Центрифужный стакан с палочкой смывают 5 см3 промывного раствора и используют этот раствор для промывки осадка в центрифужной пробирке после центрифугирования и отбора прозрачной надосадочной жидкости. Осадок промывают всеми четырьмя растворами, внимательно следя за сохранностью осадка и полнотой его переноса из центрифужного стакана. При промывке растворами центрифугирование проводят в течение 7—8 мин при частоте вращения 25 с-1.
Для спектрофотометрических измерений полученный осадок 300
после последней промывки подкисленным метанолом центрифугируют, удаляют надосадочную жидкость и тщательно перемешивают с 5 см3 раствора соляной кислоты молярной концентрацией 0,05 моль/дм-\ содержащей 0.1 % хлорида аммония.
Оптическую плотность полученной суспензии измеряют в ультрафиолетовой области спектра. Спектрофотометрические измерения суспензии с толщиной слоя 0.2 мм (кювета с вкладышем) проводят на спектрофотометре при длине волны 275 нм по отношению к растворителю (раствор соляной кислоты молярной концентрацией 0.05 моль/дм3 с 0,1%-ным хлоридом аммония). С помощью калибровочного графика по оптической плотности определяют содержание инсулина, а затем производят расчет на 100 г поджелудочной железы.
Построение калибровочного графика. Калибровочный график строят по величинам оптической плотности суспензий, содержащих известное количество фибриллярного инсулина.
Пример исходных данных для построения калибровочного графика по средним величинам трех опытов приведен ниже.
Концентрация Оптическая плотность в опытах
инсулина, мг I II И! средняя
2 0,046 — 0.047 0,047
4 0.082 0,087 — 0,084
6 0.132 0.142 — 0,140
8 0.188 0.192 0.200 0,192
10 0.235 0,243 0,255 0,245
12 0.285 0,294 0,303 0,294
14 0.330 0,342 0,357 0,340
16 — 0,398 0,403 0.400
Для приготовления стандартного раствора инсулина 0,5 г порошкообразного инсулина активностью 24 ИЕ в 1 мг растворяют в 25 см3 раствора соляной кислоты молярной концентрацией 0,05 моль/дм3 (pH 2,0). 2 см3 полученного раствора вносят шприцем в ампулы (длина 23 см, диаметр 4 мм) из молибденового стекла так, чтобы не был смочен оттянутый конец ампул, который запаивают.
Глобулярный инсулин в кислой среде переводят в фибриллярный последовательным воздействием высокой (100 °C) и низкой (— 78...— 80 °C) температур. Для этого запаянные ампулы с инсулином помещают на 2 мин на водяную баню при температуре кипения и затем быстро переносят в смесь, состоящую из ацетона и сухого льда, имеющую температуру — 80 °C, где выдерживают 1 мин; при этом раствор полностью замораживается.
301
Операцию нагревания и замораживания повторяют 8 раз. Запаянные ампулы с фибриллярным инсулином можно хранить длительное время.
При приготовлении раствора фибриллярного инсулина для затравки после вскрытия ампулы проводят гомогенизацию, т.е. пропускают гель инсулина через иглу или капилляр длиной 20—25 см 6 раз, затем раствор нагревают в ампуле вместимостью 5 см3 при 100 °C и замораживают при минус 80 °C. Гомогенизацию с последующим нагреванием и замораживанием рекомендуется проводить дважды, после чего раствор гомогенизируют еще 2 раза.
Полученный фибриллярный инсулин после разведения раствором соляной кислоты молярной концентрацией 0,05 моль/дм3 в соотношении 1:10 используют для затравки исследуемого инсулина.
Для приготовления охлаждающей смеси в термос вместимостью 2,5 дм3 наливают 1,5 дм3 технического ацетона, а затем добавляют измельченный сухой лед (до наполнения термоса). После измерения температуры термос закрывают пробкой.
Затем строят калибровочный график по величинам оптической плотности суспензий, содержащих известное количество фибриллярного инсулина; фиксируют результаты количественного определения инсулина в экстрактах поджелудочной железы крупного рогатого скота, свиней (мг на 100 г сырой ткани) из расчета активности инсулина 24 ИЕ в 1 мг.
Далее студенты самостоятельно формулируют заключение по работе с учетом полученных данных при проведении качественных реакций на инсулин.
Лабораторная работа № 6
ОПРЕДЕЛЕНИЕ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И НАДПОЧЕЧНИКОВ
Цель работы. Приобрести практический навык в анализе тиреоидина и адреналина.
Задачи. Провести качественные реакции на тиреоидин и адреналин; определить адреналин в исследуемых образцах желез.
Объекты исследования. Свежая или высушенная ткань щитовидной железы; свежие, замороженные или высушенные надпочечники крупного рогатого скота или свиней; препараты гормонов (медицинские); тиреоидин, адреналин.
Материалы, реактивы и оборудование. Раствор NaOH массовой долей 10 %; раствор H2SO4 массовой долей 10 %; раствор крахмала массовой долей 1 %; раствор KJO3 массовой долей 2 %; раствор FeCl3 массовой долей 1 %; раствор адреналина массовой долей 1 %; раствор сульфаниловой кислоты массовой долей 1 %; раствор нитрата натрия массовой долей 5 %; раствор Na2SO3 массовой долей 10 %; раствор адреналина (1 : 1000); лимонная или щавелевая 302
кислота; раствор адреналина (содержимое ампулы растворяют в 100 см? воды); раствор уксусной или ортофосфорной кислоты массовой долей 10%; флуорископ; стандартный раствор адреналина |в мерную колбу вместимостью 25 см3 отмеривают 1 см3 раствора адреналина (1 : 1000) и доводят до метки водой. 1 см3 такого раствора содержит 0.04 мг адреналина]; свежеприготовленный раствор Na2SO, массовой долей 10 %; реактив Фолина (см. главу 3, лабораторную работу № 2); штатив с пробирками; центрифуга; бумажные фильтры; лакмус; фарфоровая ступка с пестиком; газовая горелка; пробирки; мерные пипетки; колба; капельница.
Методические указания. Гормоны шитовидной железы представляют собой иодированные производные аминокислоты тирозина. Иод поступает в организм с водой. В тканях щитовидной железы он окисляется, принимает форму молекулярного иода и присоединяется к тирозиновому кольцу, образуя моно-иодтирозин. Затем в результате полимеризации иодтирозина образуются гормоны шитовидной железы — тироксин и трииод-тиронин:
J J
J J
CH2-CH(NH2)-COOH
Тироксин
CH2-CH(NH2)-COOH
Трииодтиронин
Эти процессы происходят в молекулах тиреоглобулина, содержащегося в дольках железы в составе коллоида. Более 90 % органически связанного иода организма выделяется в виде тироксина.
Гормоны щитовидной железы являются иодсодержащими соединениями. При их разрушении образуется иодид калия, который окисляется йодатом калия в кислой среде с образованием молекулярного иода;
5KJ + KJO3 + 6НС1 -» 3J2 + 6КС1 + ЗН2О.
В результате реакции иода с крахмалом образуется соединение синего цвета.
Адреналин — гормон, синтезируемый в мозговом веществе надпочечных желез, по химической структуре является
303
амином-1 -(3.4-диоксифенил)-2-метиламиноэтанолом (произвол-н ы м п и р о к а те х и н а):
он
НО
CHOHCFH-NH-CH
При добавлении к раствору адреналина хлорида железа (III) жидкость окрашивается в изумрудно-зеленый цвет вследствие образования комплексного соединения типа фенолята железа:
ОН NH-CH?
Адреналин обладает слабощелочной реакцией, легко окисляется на воздухе с образованием аденохрома, вследствие чего раствор окрашивается в красный цвет.
При взаимодействии диазореактива с адреналином жидкость окрашивается также в красный цвет вследствие образования сложного соединения типа азокрасителя.
В кислой среде с йодатом калия адреналин образует соединение красно-фиолетового цвета.
Адреналин, окисляясь кислородом воздуха, при добавлении щелочи дает флуоресцирующие продукты.
Количественное определение адреналина основано на колориметрическом определении интенсивности синего окрашивания, возникающего при взаимодействии адреналина с реактивом Фолина. Входящие в этот реактив соли при взаимодействии с адреналином восстанавливаются с образованием более низких оксидов металлов, комплексы которых окрашиваются в синий цвет.
Подготовка проб. Щитовидные железы убойных животных измельчают и высушивают при температуре 70—80 °C.
Надпочечники крупного рогатого скота или свиней измельчают и экстрагируют адреналин дистиллированной водой, подкисленной лимонной или щавелевой кислотой из расчета на 1кг надпочечников 1,5дм3 дистиллированной воды и 9—Юг кислоты. После 2 ч экстракции массу подогревают до 90—95 °C 304
для коагуляции оелков. которые отделяю! фильтрованием или центрифугированием. Фильтрат или налосадош!мю жидкость используют для качественно!о и количественного определении адреналина.
1.ОПРЕДЕЛЕНИЕ ТИРЕОИДИНА ПО КАЧЕСТВЕННОЙ РЕАКЦИИ С ИОДИДОМ КАЛИЯ
Порядок проведения анализа. В фарфоровой ступке тщательно растирают 100—200 мг измельченной и высушенной ткани щитовидной железы, переносят в колбу для гидролиза, добавляют 5 см3 раствора NaOH и 5 см? воды. Кипятят на асбестовой сетке в течение 10—15мин. К Зсм3 охлажденного гидролизата добавляю! раствор H2SO4 до кислой реакции на лакмус. После подкисления приливают 5 капель раствора крахмала и 1 см3 раствора KJO;. Выделяющийся иол дает синее окрашивание с крахмалом.
Этапы проведения и условия развития цветной реакции студенты фиксируют в тетради, самостоятельно делая соответствующие выводы.
2. ОПРЕДЕЛЕНИЕ АДРЕНАЛИНА НАДПОЧЕЧНИКОВ
2.1. КАЧЕСТВЕННЫЕ РЕАКЦИИ
Порядок проведения анализа. Для проведения реакции с хлоридом железа (1П) в пробирку вносят 1 см3 экстракта адреналина, добавляют 1 каплю раствора FeCls массовой долей 1 %, перемешивают и наблюдают появление изумруднозеленой окраски. Затем добавляют 1 каплю раствора NaOH массовой долей 10% и фиксируют появление вишнево-красного окрашивания.
Для проведения реакции с д и а з о р е а к т и в о м к 1 см3 раствора сульфаниловой кислоты добавляют 1 см3 раствора нитрита натрия массовой долей 5 % (смесь лиазореактива), 1,5 см3 экстракта адреналина и 1 см3 раствора Na2CO, массовой долей 10 %. Смесь перемешивают и наблюдают окрашивание раствора в красный цвет.
Для проведения реакции с йодатом калия к 0,5 см3 экстракта адреналина прибавляют 1 см3 раствора KJO3 массовой долей 10%, 10 капель раствора уксусной или орто-фосфорной кислоты и смесь подогревают до температуры 60— 65 °C. Фиксируют появление интенсивного красно-фиолетового окрашивания.
Для наблюдения флуоресценции продуктов окисления адреналина к 10 каплям дистиллированной воды до
305
бавляют 6 капель раствора тдроксида натрия массовой долей 10 % и 6 капель экстракта адреналина Поместив пробирку перед включенным флуорископом, наблюдаю! зеленую флуоресценцию продуктов окисления адреналин;!.
2.2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ АДРЕНАЛИНА ПО ФОЛИНУ
Порядок проведения анализа. Готовят две мерные сухие пробирки вместимостью 10 см'. В первую вносят I см' стандартного раствора адреналина, во вторую — 1 см' исследуемою раствора, добавляют но 4 см-’ свежеприготовленно!о раствора Na2CO3 массовой долей 10 % и по 0,5 см' реактива Фолина. Содержимое обеих пробирок встряхивают. Жидкость постепенно окрашивается в синий цвет, достигающий наибольшей интенсивности через 3—5 мин.
Далее объем жидкости в пробирках доводят до метки раствором Na2CO3 массовой долей 10 %. Содержимое пробирок перемешивают “и окраску исследуемого раствора колориметрически сравнивают с окраской стандартного раствора адреналина.
Массовую концентрацию адреналина в исследуемом растворе (мг/см3) рассчитывают по формуле
С-С„Ет/ЕсГ (3.5)
где С() — массовая концентрация адреналина в стандартном растворе (0.04 мг/см3): Еоп и Ест — экстинкция соответственно исследуемого и стандартного растворов адреналина.
Наблюдения за проведением качественных реакций на адреналин, результаты количественного определения и расчета содержания адреналина в экстракте надпочечников убойных животных студенты аккуратно и точно фиксируют в тетради (описывают условия и эффект реакции) и с помощью качественных реакций формулируют выводы о результатах изучения строения гормонов.
Контрольные вопросы
1. Что такое автолиз? Перечислите и охарактеризуйте основные его этапы.
2. Какова роль ферментов в развитии автолиза?
3. Что такое катепсины9
4. Как можно выделить и определить катепсины?
5. Какие внешние факторы влияют на активность катепсинов?
6. Какова динамика изменения биохимических и функциональных свойств при созревании мяса и его последующем хранении?
7. Как практически оценить технологическую пригодность мяса, используя методы биохимического анализа?
306
8. Каков механизм свертывания крови?
9. Какие факторы являются основными в системе свертывания крови'.’
10. Какую роль играет кальций в системе свертывания крови'?
11. Какое сырье относится к ферментно-эндокринному и каково его значение?
12 Какие продуценты животных гормонов вы знаете?
13. Какие продуценты животных ферментов имеют практическое значение при переработке животных9
14. Какую химическую природу имеет инсулин'? Как можно его обнаружить'?
15. В чем сущность метода количественного анализа инсулина'?
16. Какими свойствами обладают ферменты поджелудочной железы?
17. Какие животные ферменты используют в мясной промышленности9
18. Какие свойства присуши пепсину9
19. Как определить молокосвертывающую активность пепсина'.’
20. Какие основные гормоны щитовидной железы и надпочечников вы знаете'? На чем основаны методы их качественного и количественною определений?
Глава 4
ПИЩЕВАЯ ЦЕННОСТЬ И КАЧЕСТВО МЯСА И МЯСНЫХ ПРОДУКТОВ
Мясо и мясопродукты — традиционная и одновременно уникальная составная часть пищевых рационов. Уникальность мяса состоит в высокой энергоемкости, сбалансированности аминокислотного состава белков, наличии биологически активных веществ и высокой усвояемости, что в совокупности обеспечивает нормальное физическое и умственное развитие человека.
Под понятием «качество» подразумевают широкий спектр свойств, характеризующих пищевую и биологическую ценность пищевых продуктов, а также органолептические, структурно-механические, функционально-технологические, санитарно-гигиенические и прочие характеристики продукта, степень их выраженности.
В большинстве случаев значение этих показателей зависит в первую очередь от состава сырья, биохимических изменений в процессе технологической обработки, внешних воздействий, а также от используемых аддитивов. С точки зрения качественных показателей пищевой продукт должен содержать компоненты, необходимые организму человека для нормального обмена веществ.
Современные представления о количественных и качественных потребностях человека в пищевых веществах отражены в концепциях сбалансированного и адекватного питания. Согласно первой концепции в процессе нормальной деятельности человек нуждается в определенных количествах энергии и комплексе пищевых веществ: белках, аминокислотах, углеводах, жирах, жирных кислотах, минеральных элементах, витаминах, причем многие из них являются незаменимыми, т. е. не вырабатываются в организме, но необходимы ему для жизнедеятельности. Вторая доказывает, что компоненты питания должны быть в строгом соотношении, притом именно оно определяет в итоге усвояемость пищи и регулирует питание на уровне гомеостаза.
Потребности человека в пищевых веществах с учетом суммарной энергетической ценности представлены в виде формулы сбалансированного питания (табл.4.1).
308
Г а б л и ц а 4 1
Формула сбалансированного питания взрослого человека
Пищевые вещее!ва ; Дневная потребность Пищевые вещества : Дневная потребность
Вода. I 1750-2200 витамин А (различные 1.5-2.5
В том числе: формы)
питьевая (вода. чай. 800—1000 каротиноиды •> — >
кофе и т. д.) витамин Е (различные 10-20
в супах 250-500 формы)
в продуктах питания 700 витамин К (различные 0.2-0.3
Белки, г <80-100 формы)
в том числе животные 50 липоевая кислота 0.5
Незаменимые аминокис- инозит, г 0.5-0.1
лоты. г: Углеводы, г 400-500
триптофан 1 В том числе:
лейцин 4-6 крахмал 400-450
изолейцин 3-4 моно- и дисахариды 50-100
валин 3-4 Органические кислоты (ли- 2
треонин 2-3 монная, молочная и др.), г
лизин 3—5 Балластные вещества, г 25
метионин 2-4 Жиры, г 80-100
фенилаланин 2-4 В том числе:
Полу- и заменимые амино- растительные 20-25
кислоты. г: незаменимые полине- 2-6
гистидин 1.5-2 насышенные жирные
аргинин 5—6 кислоты
цистин 2-3 холестерин 0,3-0.6
тирозин 3-4 фосфолипиды 5
аланин 3 Минеральные веще-
серин 3 ства, мг:
глутаминовая кислота 16 кальций 800-1000
аспарагиновая кислота 6 фосфор 1000-1500
пролин 5 натрий 4000-6000
гл и ко кол 3 калий 2500-5000
Витамины, мг; хлориды 5000-7000
витамин С 50-70 магний 300-500
тиамин (Bp 1,2-2.0 железо 15
рибофлавин (В2) 2,0-2.5 цинк 10-15
ниацин (РР) 15-25 марганец 5-10
пантотеновая кис- 5-10 хром 0,20-0,25
лота (В3) медь 2
витамин В() 2-3 кобал ьт 0,1-0.2
витамин Вр 0,002-0,005 молибден 0.5
биотин 0,15-0,30 селен 0,5
холин 500-1000 фториды 0,5-1.0
рутин (Р) 25 иодиды 0,1-0,2
фолацин (В9) 0,2-0,46 Энергетическая ценность:
витамин D (различные 0.0025-0,01 ккал 2850
формы) кДж 11900
309
В связи с этим для характеристики качества и пищевой ценности продукта необходима информация о его общем химическом и элементном составе, степени соответствия каждого компонента, формуле сбалансированного питания, способности перевариваться и усваиваться организмом.
4.1. ПИЩЕВАЯ ЦЕННОСТЬ
По общепринятой терминологии в понятие «пищевая ценность» входят количественное соотношение пищевых веществ в продукте и суммарная энергетическая ценность, органолептические характеристики изделия и способность веществ перевариваться и усваиваться организмом (рис. 4.1).
Энергетическая ценность дает представление о той части энергии, которая выделяется из пищевых веществ в процессе их биологического окисления в организме. Необходимая энергетическая ценность пищи для людей разного пола, возраста, массы, рода деятельности колеблется от 2850 до 20875 кДж/сут. В зависимости от вида мяса и его состава мясопродукты имеют различную энергоемкость — от 147,5 до 1662,5 кДж на 100 г продукта.
Зная уровень усвоения пищевых веществ в организме (белок — 84,5 %, жир — 94 %, углеводы — 95,65 %) и величину теплоты сгорания компонентов пищи, можно рассчитать энергетическую ценность продукта.
Таким образом, зная общий химический состав и массу продукта, а также энергетическую ценность пищевых веществ, можно рассчитать пищевую ценность мясных изделий в энергетическом выражении.
Рис. 4.1. Показатели пищевой ценности
310
Наряду с этими свойствами пищевые вещества являются источником биологически необходимых, незаменимых веществ. С этих позиций весьма важными являются показатели биологической ценности белка (рис. 4.2). Понятие биологической ценности (БЦ) характеризует качество белкового компонента продукта, обусловленное как степенью сбалансированности состава аминокислот, так и уровнем переваримости и ассимиляции белка в организме.
В среднем взрослый человек в течение суток должен получать с пищей 1 — 1,2 г белка на 1 кг массы тела. Однако он нуждается не просто в белке, а в белке определенного состава. Белки, содержащиеся в различных продуктах, неравноценны. Из 20 аминокислот 8 являются незаменимыми, в отличие от других они не синтезируются в организме. По этой причине 30 % суточного белкового рациона человека должны составлять полноценные белки, содержащие все незаменимые аминокислоты, годовая потребность человека в полноценном белке — 20 кг.
Если в рацион входит несколько взаимообогащающих неполноценных белков, они должны поступать в организм одновременно и в определенном соотношении. В организме накопления аминокислот нет, а синтез белка происходит только при наличии всех незаменимых аминокислот в заданной количественной пропорции. Главным признаком полноценных белков является то, что в состав их молекул наряду с прочими аминокислотами входят радикалы незаменимых аминокислот (валина, лейцина, изолейцина, триптофана, метионина, лизина, фенилаланина, треонина). Четыре аминокислоты (тирозин, цистеин, аргинин и гистидин) считают условно незаменимыми. Следует отметить, что дефицит незаменимых аминокислот в питании может привести к нарушению здоровья человека.
На основе многолетних медико-биологических исследований ФАО/ВОЗ (1973 г.) был предложен критерий для определения качества белка — эталон, сбалансированный по незаменимым аминокислотам (табл. 4.2) и в наибольшей степени отвечающий потребностям организма. Часто за эталон принимают белки молока или яйца.
311
I а б /I и ii a 4 2
Эталон определения качества белка, сбалансированный по незаменимым аминокислотам, г/100 г белка
Вид ос iK.i
\1OJOK л алии ФАО В() 5
Аминокислн .1
Турции-.'
Я1Н1О ’Г я лете
женское : коровье . 1 л в ;рос 1Ы\ л Лс!
— . . — _ _ - -1 — .
1 1 >0.1С И 1111 н 6.9 6 .4 6.4 4.2 2.8
Лейцин 9.4 8.9 9.9 ТО 6.6
Валин 7.4 6.6 6.9 4.8 3.5
Фенилаланин 5.8 4.6 4.9 / . О 6.3
Тирозин 4.1 5.5 5.1 7.3 6.3
Цистин 2.3 2.1 0.9 2.6 2.5
Метионин з. 3 2 э 2.4 2.6 2.5
Треонин 5.0 4.6 4.6 3.5 3.4
Триптофан 1.6 1.6 1.4 1.1 1.1
Лизин 6.9 6.3 7.8 5.1 5.8
На основании сопоставления результатов определения количества незаменимых аминокислот в исследуемом продукте с данными эталона можно расчетным путем определить индекс биологической ценности или так называемый аминокислотный скор. Применительно к мясным изделиям расчет скора ведут либо для всех незаменимых аминокислот, либо для трех наиболее дефицитных: лизина, триптофана и суммы серосодержащих (метионин + цистин).
Следует отметить, что дефицит незаменимых аминокислот зависит как от качественного состава самого сырья (например, белок крови содержит мало метионина и изолейцина), так и от степени воздействия на белок различных внешних факторов. Например, при жестких режимах термообработки и щелочного гидролиза ряд аминокислот разрушается.
Кроме определения аминокислотного скора некоторые исследователи применяют и другие методы расчета потенциальной биологической ценности белка (индекс Озера, индекс Кор-пачи, показатель Митчелла и др.), причем наиболее простым и распространенным является метод расчета величины качественного белкового показателя (КП Б), представляющего собой отношение количества триптофана к оксипролину. Метод дает возможность установить соотношение мышечных и соединительнотканных белков.
Необходимо отметить, что по аминокислотному составу и аналитическому расчету показателей биологической ценности можно иметь представление лишь о потенциальной ценности белкового компонента, так как организм человека использует не
312
все. что поступает в него с нишей, а только то. что после переваривания в пищеварительном тракте всасывается через стенки кишечника в кровь.
Вторым компонентом, количественно преобладающим в составе мяса, является жир, представленный в основном триглицеридами. Биоло! ическая роль триглицеридов состоит в том, что они являются источниками энергии и. кроме того, содержат несинтези-руемые в организме человека высоконспредельные жирные кислоты и жирорастворимые витамины, роль которых в физиологии весьма велика.
В суточном потреблении взрослого человека (80—100 г, в том числе 20—25 г растительных жиров) должно содержаться 2—6 г полиненасыщенных жирных кислот. 35 г олеиновой кислоты и 20 г насыщенных жирных кислот. Соотношение между количеством полиненасыщенных и насыщенных жирных кислот должно составлять 0,3 : 0,35.
Определение уровня биологической ценности липидов можно произвести расчетным путем, сопоставляя потребное количество каждого из незаменимых компонентов в формуле сбалансированного питания с его содержанием в продукте.
При определении биологической ценности жиров большое значение имеет наличие и количественное содержание «триады» так называемых незаменимых жирных кислот. Подобно незаменимым аминокислотам они синтезируются ограниченно или не синтезируются в животных организмах совсем.
Полиненасыщенные кислоты. Из полиненасыщенных жирных кислот к биологически активным относятся линолевая, линоленовая и арахидоновая.
Линолевая кислота (цис-9, цис-12-октадиеновая кислота)
СН3(СН2 )4 - СН = СИ - сн2 — СН = (СН2 )7СООН
имеет две изолированные двойные связи. Светло-желтая маслянистая жидкость с температурой плавления от 267,8 до 268 К. Содержится в большинстве растительных масел, составляя в глицеридах значительную часть: подсолнечном 20 %, хлопковом 45 % и т. д. В жирах животного происхождения ее содержание колеблется от 2 до 20 %.
Линоленовая кислота (цис-9, цис-\2, цмс-15-окта-триеновая кислота)
сн3 - сн2 - СН = СН - сн2 — СН = = СН - сн2 - СН = СН - (СН2 )7СООН
содержит три изолированные двойные связи в z^/c-конфигура-иии. Бесцветная жидкость после затвердевания имеет температу
313
РУ плавления от 261.7 до 262 К. легко окисляется уже ири комшн ной температуре, окисление сопровождается полимеризацией, приводящей к образованию пленок в тонком слое. Наиболее распро странена в натуральных жирах триеновой кислоты, массовое содержание ее в растительных маслах составляет от долей до 50 %.
Из восьми возможных изомеров в природных жирах, по-вили-мому, встречается только щ/с-щ/сыщс-изомер.
Арах и д о н о в а я к и с л о т а (5, 8.11, 14-эйкозатетраено-вая кислота)
СН,(СН J4-CH = СН - СН,-СН - СН - СН, -СН = = CH - СН, - CH = СН - (СН2 )?СООН
содержит четыре двойные связи в изолированном положении. Светлая жидкость при глубоком охлаждении затвердевает, температура плавления 223,5 К.
Арахидоновая кислота присутствует в липидах мозговой ткани, в жирах крупного рогатого скота и свиней. В жирах растений эта кислота не обнаружена. Предполагают, что она синтезируется в печени животного и человека из линолевой и линоленовой кислот. Легко изменяется под действием кислорода воздуха, превращаясь в окисленную форму.
Перечисленные полиненасыщенные жирные кислоты являются жизненно необходимыми веществами, обладают витаминной активностью. Смесь этих кислот получила название витамина F. Недостаток этих кислот в пище приводит к отставанию животных в росте, у них появляются дерматиты, наблюдаются выпадение волос и другие признаки витаминной недостаточности. При длительном исключении витамина F из пищи животные погибают. Человек также нуждается в витамине F, так как недостаток его в пище тоже приводит к кожным заболеваниям, выпадению волос и т. д. Считают, что полноценная по содержанию витамина F пища должна иметь в своем составе 0,1 % арахидоновой или 1 % линолевой и линоленовой кислот (табл. 4.3).
Таблица 4.3
Содержание полиненасыщенных кислот в некоторых жирах
Жир Массовая доля жирных кислот,%
линолевой линоленовой арахидоновой
Шпик свиней, находившихся
на кормовом рационе:
ПЛОХОМ хорошем 12,54 12,04 0,82 0,56 1,04 0,38
Жир: вареного окорока 13,30 0,71 0,85
говяжий 2,50 0,58 0,13
свиной 11,15 0,36 0,59
314
Витамины. Представляют собой биологически активные вещества. обеспечивающие нормальное течение биохимических и фи зиологических процессов в живом организме. В жировой ткани присутствуют жирорастворимые витамины групп A. D. Е. К. Содержание двух последних незначительно.
Вита м ин А — один из немногих каротиноидов, найденных в тканях растений и животных. При этом различают витамин Аг встречающийся в тканях всех животных и рыб, и А2, встречающийся только в тканях пресноводных рыб. Структурная формула витамина А( представлена ниже:
16 17 |()
Н;С\ /СН’Н Н СН;Н
iii'i
Н?С т 1 5 С — С=С—с=~с
I , II 7 S <) 10
Н ,С С; ^.С—-СН;
чсн2 1S
20
Н Н СН, н 111'1
О-С- С—С-СНД)Н
И 12 13 14 15
Структура витамина А2 отличается от структуры витамина А( наличием добавочной двойной связи в fj-ионовом кольце между третьим и четвертым углеродными атомами. Биологическая активность витамина А2 составляет 40 % биологической активности витамина Ар
Витамин Aj встречается в трех следующих формах: в виде спирта (ретинола), альдегида (ретиналя) и кислоты (ретиеновой).
Витамин D (кальциферол) представляет собой ряд родственных соединений, обладающих антирахитической активностью. Известно несколько видов витамина D: D2, D3, D4, D5, D6, D7, которые имеют сходное строение. Все они могут быть получены путем облучения определенных провитаминов D (стероидов) ультрафиолетовым светом.
Наиболее распространенными витаминами являются D2 (эргокальциферол) и D3 (холекальциферол). Эргокальциферол образуется при облучении ультрафиолетовым светом эргостерола, а холекальциферол — при облучении 7-дегидрохолестерина. Структурные формулы витаминов D2 и D3 представлены ниже:
315
Эти две формы витамина D различаются по точкам плавления: витамин D2 плавится при 388—390 К, a D. — при 355—358 К. Кальциферолы весьма чувствительны к действию кислорода воздуха и света, особенно при нагревании.
В и т а мин Е по химической природе представляет собой группу родственных соединений — токоферолов, молекулы которых состоят из двух компонентов: кольца, являющегося производным бензохинона, и изопреноидной боковой цепи, сходной с боковыми цепями витаминов А и К. Наибольшей биологической активностью среди токоферолов обладает о-токоферол. структурная формула которого приведена ниже:
СН;
I СН; СН; СН; СН;
С. I I I I
Н;С-С^ С С-(СНД;-СН-(СНД;-СН-(СНД;-СН
I II I - I
но-с^ .С ХСН, СН;
с с I Н? СН3 Токоферол представляет собой прозрачную маслянистую жидкость светло-желтого цвета, нерастворимую в воде, хорошо растворимую в растительных маслах, этаноле, серном и петро-лейном эфирах.
Витамин К по химической природе представляет собой группу соединений, молекулы которых состоят из двух компонентов: циклической структуры хинонного строения, имеющей метильную группу (2-метил-1,4-нафтохинона), и боковой цепи. В природе встречаются витамины К с различным числом изопреновых звеньев в боковой цепи.
Длинная боковая цепь витамина К[ является остатком высокомолекулярного алифатического спирта фитола, входящего в состав хлорофилла.
Витамин К2 (мультипренилменахинон) синтезируется различными бактериями или трансформируется из нафтохинонов в организме. Биологическая активность витамина К, примерно в 2 раза ниже биологической активности витамина
Витамин К содержится в большинстве распространенных продуктов питания, причем в растениях присутствует витамин Кр а в продуктах животного происхождения — различные формы витамина К2.
Большинство животных жиров имеют цвет, за исключением свиного и козьего. Окраска зависит от наличия каротиноидов — пигментов, окрашивающих жиры в желтый цвет и одновременно служащих провитаминами. Например, под действием фермента каротиназы кишечника каротины превращаются в витамин А. Количество каротинов в жирах зависит от условий откорма живот
316
ных. Максимальное количество каротинов в жирах отмечаете^ при пастбищном откорме к осени. Витамины группы Е кро'-’е пищевого значения выполняют роль природных актиоксидачтог Благодаря наличию большого количества непредельных химических связей в своей структуре при благоприятных условиях еищ-мин Е легко окисляется, предохраняя жир от порчи.
Суммарное содержание витаминных примесей служит показателем биологической ценности жиров.
Количественное соотношение белков и жиров в составе продукта влияет на усвояемость тех и других компонентов. При повышенном содержании жира тормозится отделение желудочного сока, замедляется переваривание белков пепсином и трипсином, изменяется обмен некоторых веществ, подавляются система свертывания крови и процесс ассимиляции витаминов. Установлено, что оптимальным соотношением жира и белка в пищевых продуктах является 1(0,8): 1,0. Определенное соотношение белка и жира в мясе играет значительную роль в формировании биологической ценности мясопродуктов и позволяет с научной точки зрения подойти к вопросам рационального использования сырья и обоснованного создания рецептур и технологий.
Химическая оценка биологической ценности белков важна и необходима, но пассивна, поскольку отражает лишь потенциальную возможность белка в удовлетворении потребностей человека и животных. Конечный же результат зависит от особенностей структуры белка и атакуемости его со стороны пищеварительных протеиназ (пепсина, химотрипсина, трипсина и др.).
Под действием пищеварительных ферментов белковые вещества расщепляются на отдельные фрагменты (аминокислоты и пептиды), которые проникают через стенку кишечника и ассимилируются организмом. Биоактивность характеризует способность продукта стимулировать процессы внутреннего обмена веществ, секреторную функцию. Таким образом, соотносительная зависимость между биологической ценностью белков и их аминокислотным составом может быть справедлива лишь при условии достаточно высоких скоростей переваривания ферментами пищеварительного тракта, усвояемости компонентов и их биоактивности. В связи с этим перечисленные показатели являются составными частями биологической оценки пищевых продуктов. Биологическая доступность белка и степень его усвоения зависят от многих факторов. В частности, они обусловлены природой белка и его структурой: например, белки соединительной ткани расщепляются хуже, чем мышечные; нативные — хуже, чем денатурированные. Изменение физической структуры мяса (степени дисперсности за счет измельчения) и биохимической структуры белка (денатурация) повышает доступность компонентов действию пищеварительных ферментов. Образование надмолекулярных белковых структур в результате взаимодействия белковых частиц друг с дру-
317
юм или с молекулами некоторых других веществ понижает их б иол оги чес кую дос ту и н ость.
Скорость переваривания белков отдельными ферментами желудочно-кишечного тракта представляет интерес при разработке рационов питания для здорового и особенно для больного человека, а также при определении пищевой ценности отдельных продуктов.
На рис.4.3—4.5 представлены типичные примеры графических зависимостей переваримости белков в различных пище вых продуктах пепсином, трипсином, а также системой пепсин — трипсин.
Определение степени расщепления и усвояемости белкового компонента мяса, как правило, производят двумя методами: в опытах in vitro и in vivo. В опытах in vitro в системах пепсин — трипсин либо с использованием реснитчатой инфузории Tetra-chymena periformis в известной степени моделируется процесс переваривания белков в желудочно-кишечном тракте. Однако получить достоверное представление о биологической ценности белкового компонента и продукта можно лишь на основе опытов,
Рис. 4.3. Переваримость белков различных пищевых продуктов пепсином (in vitro):
а — рыбные продукты: 1 — сельдь атлантическая; 2 —треска; 3— килька; б—мясные продукты: 7 —баранина; 2—говядина; 3 — свинина; в —крупяные продукты и мучные изделия: 7 — манная каша; 2— пшенная каша; 3 — хлеб ржаной; 4 — хлеб пшеничный; г — сырые и вареные продукты: 7, 3 — яйцо вареное и сырое; 2, 6—фасоль вареная и сырая; 4, 5—соя вареная и сырая; д — молочные продукты: 7 — сыр степной; 2 — казеин; 3 — белковый продукт белип
318
Длительность гидролиза, ч
Рис. 4.4. Переваримость белков различных пищевых продуктов трипсином (in vitro): а — рыбные продукты: /—сельдь атлантическая; 2— треска; 3— килька; о—мясные продукты: J — баранина; 2 — говядина; 3 — свинина; в~ крупяные продукты и мучные изделия: 1 — манная каша; 2 — пшенная каша; 3 — хлеб ржаной; 4— хлеб пшеничный; г~ сырые и вареные продукты: /. 3— яйпо вареное и сырое; 2, 6 — фасоль вареная и сырая; 4, 5—соя вареная и сырая; д — молочные продукты: 1 — сыр степной; 2 — казеин; 3 — белковый продукт белип
проводимых на животных, и наблюдений за человеком, определяя степень фактической утилизации пищевых веществ в организме в процессе обмена веществ по характеру адсорбции белка и изменению ростовесовых показателей. На практике используют: коэффициент эффективности белка (КЭБ), биологическую ценность (БЦ), истинную переваримость (ИП), коэффициент использования белка (КИБ).
ИП характеризует главным образом способность белка распадаться под действием протеолитических ферментов пищеварительного тракта и всасываться через слизистую оболочку кишечника. БЦ учитывает ту часть азота, которая фактически используется, и зависит преимущественно от сбалансированности изучаемых белков по аминокислотному составу и от одновременности введения этих метаболитов в кровеносную систему. КИБ — это отношение связанного азота к азоту, поглощенному с пищей, т. е. суммарная оценка, принимаемая в расчет при оценке биологической ценности и переваримости, а КЭБ — отношение прироста массы к потребленному белку.
319
Длительность гидролиза, ч
Рис. 4.5. Переваримость белков различных пищевых продуктов системой пепсин -трипсин (стрелка обозначает момент введения в пепсиновый перевар трипсина):
а —рыбные продукты: / — сельдь атлантическая: 2 —треска: 5—килька; о—мясные продукты: / — баранина; 2—говядина; 3—свинина мясная; «--крупяные продукты и мучные изделия: /—манная каша; 2—пшенная каша; 3 —хлеб ржаной; 4 —хлеб пшеничный; г-сырые и вареные продукты: 1. 3 — яйцо вареное и сырое; 2. 6—фасоль вареная и сырая; 4. 5—соя вареная и сырая; д - молочные продукты: / — сыр степной; 2—казеин; 3 — бочковый продукт белиц
Биологические методы базируются на принципе азотного баланса в процессе жизнедеятельности организма. Поступивший в организм азот расходуется по двум направлениям. Первый после всасывания в пищеварительном тракте поступает в кровеносную систему (переваримый азот), удерживается организмом или ката-болизируется (используется как источник энергии) и выделяется с мочой, а второй — выбрасывается с фекалиями. Наконец, постоянное новообразование белков в организме из имеющихся происходит с потерей азота, которая не зависит от пищевого азота (эндокринный азот мочи). Исходя из азотных фракций, можно установить различные соотношения, позволяющие характеризовать превращения белков в организме.
Различия в усвояемости влияют на утилизацию белков, в связи с чем при расчете безопасных уровней потребления обычных смесей пищевых белков вводят поправки на усвояемость. Поскольку оценки безопасных уровней потребления основаны на данных, полученных при использования белков молока, яиц, мяса и рыбы.
320
усвояемость других ослков выражают через усвояемость ослков вышеперечисленных продуктов.
Для определения усвояемости белка и смеряют содержание азота в пище и кале в опытах in vivo. Предполагаемую (ПУ) и целинную (ИУ) усвояемости белка — азола (7) рассчитывают в соответствии с формулами (4.1) и (4.2):
Ц\ (/ /•') 100/7:
11У j / (/ ф) • 1001/7.
(4.1)
(4.2)
где / — общее количество а».ога г. потребленной нише" / - количесию ;noia. выде ляемою с калом, на фоне тест-диетьг /1 — ко.шчсс гно азота, выделяемою с калом. на фоне бс шелковой листы.
В табл. 4.4. представлены данные усвояемости белков некоторых нишевых продуктов человеком.
3 а б л и ц а 4.4
Усвояемость белков (%) некоторых пищевых продуктов человеком
Источник белка j Истинная усвояемость 1 Усвояемость относ, дтп.юнных бел
Яйца 97 + 3 100
Молоко, сыр 95 ± 3 100
Мясо, рыба 94 ± 3 100
Кукуруза 85 ± 6 89
Рис полированный 88 ± 4 93
Пшеница цельная 86 ± 5 90
Пшеница очищенная 96 ±4 101
Мука овсяная 86 ± 7 90
Мука соевая 86 + 7 90
Просо 97 83
Горох зеленый 88 93
Масло арахисовое 95 100
Бобы 78 82
Кукуруза + бобы 78 82
Кукуруза + бобы + молоко 84 88
Биологические методы достаточно объективны, но дороги в исполнении, требуют длительного времени и большого количества материалов для анализа. Химические методы в связи с этим имеют существенные преимущества. Наиболее известны подходы, основанные на определении аминокислотного состава, когда выделяют лимитирующие аминокислоты, а затем их сравнивают со стандартным белком. При этом подсчитывают сумму лимитирующих аминокислот и выражают ее в процентах от суммы всех аминокислот либо сравнивают соотношение не
321
заменимых аминокислот с тем же показателем в «идеальном» белке. При эчом. безусловно, учитывают соотношение всех незаменимых аминокислот.
В последнее время стали очень популярны ферментативные методы, поскольку они позволяю'! искусственно создать условия, максимально приближенные к условиям живого организма. Например. переваримость белков in vitro широко используется в аналитической практике.
Для определения соответствия аминокислотного состава белков потребностям человека предложен ряд индексов, каждый из которых дает лишь ограниченную опенку. Из них особое внимание заслуживает отношение суммы незаменимых аминокислот Н (мг) к общему содержанию белкового азота О (г). Чем ниже величина Н/О. тем выше содержание неспецифичсского азота. Для белков молока и яиц отношение Н/О составляет 3,1—3,25, для мяса — 2,79—2,94, для пшеницы — 2.
Более полное представление о биологической ценности белков дает показатель так называемого аминокислотного скора. Для каждой из незаменимых аминокислот лимитирующей качество белка является та, показатель аминокислотного скора которой является наименьшим.
Показатель аминокислотного скора устанавливает предел использования азота данного вида белка для пластических целей. Избыток других содержащихся в белке аминокислот может использоваться как источник для неспецифического азота или для энергетических нужд организма.
Количество углеводов в созревшем мясе составляет около 1,0—1,5 %. Их роль в основном связана с участием в биохимических процессах созревания мяса, формирования вкуса, аромата, изменения консистенции, величины pH, нежности и т. д., т. е. углеводы опосредованно влияют на качество мясных изделий и их биологическую ценность.
В соответствии с теорией адекватного питания часть балластных веществ пищи (клетчатка, пектин и др.), которые относят к пищевым волокнам, выполняет весьма важную физиологическую функцию. Благодаря специфическим функциональным свойствам пищевые волокна активно участвуют в регуляции биохимических процессов в органах пищеварения и выведении из организма токсических веществ, поступающих с водой, пищей и воздухом. Пищевые волокна могут использоваться для профилактики ряда заболеваний, в первую очередь таких, как атеросклероз, сахарный диабет, ожирение, ишемическая болезнь сердца, заболевания толстой кишки и др. В связи с необходимостью создания специальных продуктов питания вопрос применения балластных веществ типа пшеничных отрубей, яблочного пектина, целлюлозы, метилцеллюлозы, карбоксиметилцеллюлозы приобретает важное значение. Необходимо отметить, что функ
322
ции нищевых волокон, кроме указанных веществ растительного происхождения, выполняют и соединительнотканные белки тканей животных.
Витамины, микро- и макроэлементы, а также вещества, стимулирующие секреторно-моторную деятельность пищеварительного тракта (экстрактивные вещества, ферменты), являются необходимой составной частью мяса, и поступление их с пищей — необходимое условие для нормального развития и функционирования организма.
В состав сырого мяса входит полный набор водорастворимых (В,, В2, РР, В6, пантотеновая кислота, биотин, фолиевая кислота, BjС) и жирорастворимых (A, D, Е, К, F) витаминов, регулирующих рост и физиологические процессы. Однако при тепловой обработке часть витаминов теряется, а оставшееся количество не удовлетворяет потребностям организма. Недостающая часть витаминов обычно компенсируется высоким их содержанием в других компонентах рациона питания или путем целенаправленного обогащения мясных продуктов этими компонентами (витаминизацией).
Минеральные вещества мышечной ткани (соединения калия, натрия, магния, железа, цинка) участвуют во многих обменных процессах и в образовании буферных систем, влияют на степень растворимости и набухания белков, и, следовательно, наличие их также имеет значение при определении пищевой ценности продукта, причем наибольший интерес представляет содержание натрия, кальция и железа. Количество витаминов и минеральных веществ регламентируется формулой сбалансированного питания (см. табл. 4.1).
Азотистые экстрактивные и другие органические вещества участвуют в создании специфического аромата и вкуса мяса, способствуют улучшению органолептических показателей и тем самым стимулируют секреторную функцию пищеварительного аппарата. Влияние органолептических характеристик на пищевую ценность продукта состоит в том, что, воздействуя на органы чувств человека, они возбуждают аппетит и улучшают секреторно-моторную деятельность пищеварительного тракта, что во многом определяет покупательский спрос. Реакция человека на продукт зависит от внешнего вида, цвета, вкуса, запаха, консистенции (нежности) и сочности готового изделия, при этом результаты органолептической оценки зачастую бывают решающими при определении качества продукции, особенно новых видов. Органолептические показатели меняются в зависимости от природы продукта, его химического состава, степени биохимических изменений (например, созревания мяса), условий технологической обработки (измельчение, посол, варка, копчение и т. д.), использования специй, пищевых и вкусовых добавок.
323
Рис. 4.6. Показатели пищевой ценности продуктов питания
Структурно-механические свойства (консистенция, жесткость, механическая прочность), обусловленные пространственным распределением белков, липидов и воды в продукте, формой и прочностью связей между ними, предопределяют органолептические показатели, характер и степень разрушения продукта в процессе разжевывания. В то же время этот фактор обеспечивает удельную поверхность контакта и физическую доступность частиц пищевых веществ действию ферментов, т. е. переваримость.
Полная характеристика критериев оценки пищевой ценности продуктов представлена на рис. 4.6.
324
4.2. КАЧЕСТВО
Качество сырья и мясных продуктов характеризуется сложным комплексом химических, биохимических, физико-химических. гистологических и других характеристик.
Применительно к мясоперерабатывающей промышленности конкретное технологическое содержание понятия «качество» связано с такими критериями, как органолептические свойства, пищевая ценность, гигиенические и токсикологические состояния и технологические показатели.
Номенклатура показателей (признаков, параметров) качества (ПК) включает единичные ПК, каждый из которых характеризует одно свойство объекта; групповые ПК, применяемые для характеристики нескольких свойств, и комплексные (обобщенные) ПК. отражающие качество объекта в целом. Кроме того, используется понятие «относительный показатель», определяемый соотношением аналогичных ПК сравниваемых объектов.
На рис. 4.7 приведена типовая классификация показателей качества.
Группа эргономических показателей характеризует систему продукт — потребитель — окружающая среда и включает следующие показатели: гигиенические, антропометрические, физиологические, психофизиологические и психологические.
Гигиенические показатели отражают соответствие продукта санитарным нормам (отсутствие токсичных, канцерогенных и других вредных для здоровья человека веществ).
Антропометрические показатели характеризуют объекты относительно размеров человека. Они должны обеспечивать удобство транспортирования, хранения, реализации в сфере обращения и использования продукта потребителем.
Физиологические показатели оцениваются применительно к возможностям и потребностям организма человека. При разработке композиционных продуктов особое внимание уделяется сбалансированности химического состава.
Психофизиологические показатели характеризуют восприятие продукта с помощью органов чувств: зрения, осязания, обоняния, вкуса, иногда слуха, а также силовых и других физиологических способностей человека. Эту группу показателей называют также психофизическими. При определении величины показателя учитывается пороговая возможность человека к восприятию запаха, вкуса, к тактильным (осязательным) ощущениям.
Эстетические показатели качества отражают товарный вид, включая целостность композиции, совершенство производственного исполнения, индивидуальные особенности товара (художественное оформление, форма, упаковка, товарные знаки и информационная выразительность), выделяющие его среди аналогов.
325
Рис. 4.7. Показатели качества продуктов
Патентно-правовые показатели обеспечивают патентную чистоту и защищенность объекта в стране и за рубежом. Это может касаться способа получения, состава продукта или устройства для его изготовления.
326
Показатели унификации и стандартизации характеризуют степень преемственности показателе!’ новою продукта по отношению к аналогам. Эти показатели служа! гарантией качества и отражают техническое совершено 1 во объекта, но могут играть и консервативную роль, являясь тормозом при внедрении новых разработок.
Экологические показатели характеризуют степень вредного влияния объекта па окружающую среду при хранении и использовании.
Показатели назначения характеризуют социальную ориентацию и целевую функцию товара. К ним относятся: показатель общественной целесообразности выпуска данной продукции, отражающий потребность населения в продукте и неудовлетворенный спрос; показатель социального адреса и потребительского класса, характеризующий предназначение товаров конкретным группам потребителей. Показатель соответствия продукта оптимальному ассортименту отражает место продукта в фактическом и прогнозируемом ассортименте. Показатель сопутствующих социальных эффектов ориентирует производство на выпуск товаров с измененными свойствами в соответствии с новыми запросами потребителей, например низкокалорийных, витаминизированных, обогащенных биологически ценными компонентами и т. д. Показатель морального износа служит основанием для исключения из ассортимента выпускаемых товаров некоторых изделий, на которые снижается спрос.
Показатели функционального назначения отражают сферы использования продукта и универсальность применения. К показателям соответствия выполнению основных функций относится полезность продуктов, а к выполнению вспомогательных функций — содержательность информации, которую несут товарные этикетки, например сведения о составе, полезности, способах употребления, условиях хранения и сроках годности продуктов.
Технологические показатели отражают материалоемкость, трудоемкость, энергоемкость производства продукции, а также возможность утилизации отходов, т. е. употребления их с пользой для народного хозяйства, например для пищевых, кормовых, технических или иных целей.
Экономические показатели рассчитывают с учетом затрат на разработку, изготовление, хранение и потребление продукции.
Показатели сохраняемости и транспортабельности в товароведении называют также показателями надежности. Они характеризуют свойства продуктов сохранять стандартное качество при перевозках и в течение гарантийных сроков хранения при соблюдении условий, установленных в нормативно-технической документации.
Показатели безопасности потребления отражают соответствие
327
гигиенических показа гелем государственным и международ ным норма! ивам: санитарным дравидам, стандартам отечественным и И СО.
Поэтому перед предприятиями, работающими в новых условиях рыночной экономики, сюит задача быстрого повышения эффективности произволетвенного цикла на основе ею интенсификации. при этом особую роль играет совершенствование технологических процессов, которые характеризуются большим разнообразием качественных показателей сырья и значительной сложностью происходящих в нем физико-химических и биохимических изменений, разной продолжительностью, дискретностью, слабым техническим оснащением и экологическим несовершенством.
Задачу оптимального управления технологическими процессами в* мясной промышленности можно решить с помощью сплошного контроля качества сырья, полуфабрикатов и готовой продукции на основе применения специальных методов контроля. Это достигается физическими неразрушающими методами, предусматривающими воздействие на объект контроля различных веществ, физических полей, или регистрацией этих полей, имитируемых самим контролируемым объектом. Неразрушающий контроль как часть интроскопии (т. е. внутривидения) предусматривает выполнение таких задач, как измерение физико-химических параметров, определение химического состава, структуры, прочностных характеристик, внутреннего строения и т. д. Выделяют девять видов неразрушающего контроля, в том числе магнитный, электрический, вихретоковый, радиоволновый, тепловой, оптический, радиационный, акустический и контроль проникающими веществами.
Целесообразно одновременно использовать несколько взаимно дополняющих и дублирующих методов для решения конкретных задач.
В структуре затрат на контроль качества у нас в стране в последние годы произошли существенные изменения в сторону увеличения объемов средств на оценку сырья и контроль за его изменениями в процессе технологической обработки по сравнению с затратами средств на анализ готовой продукции.
Научный подход к организации эффективного производства в мясной промышленности должен базироваться на системах, обеспечивающих гарантированное качество готовой продукции с учетом медико-биологической его оценки и санитарно-ветеринарной экспертизы сырья.
Перспективным направлением сохранения качества сырья, его повышения при технологической обработке, прогнозирования качества готовой продукции является внедрение автоматизированных систем управления качеством технологических процессов на базе ЭВМ и информационного обеспечения, ха-
328
растеризующего процесс или состояние ооьекта управления ио совокупности его параметров.
Приведенные на рис. 4.8 факторы качества применительно к мясу и мясным продуктам свидетельствую! о достаточно высоком уровне сложности проблемы измерения и опенки показате лей, что сдерживает разработку объективных методов и приборов для определения их качества и внедрения в мясную промышленность АСУ I II.
Характерной особенностью мясного сырья и мясных продуктов является то, что их качество не может быть описано какой-либо одной или несколькими характеристиками. Полное описание качества мясных продуктов требует использования десятков показателей, значимость которых может быть сравнима между собой. В настоящее время частью показателей пренебрегают, отчего существенно страдает полнота оценки.
Для оценки качества мясных продуктов предложен ряд моделей на основе ряда характеристических показателей. Наиболее распространенной является модель, предложенная А. М. Бражниковым, согласно которой иерархическая классификация свойств мясной продукции разделена на четыре группы:
критические свойства, однозначно определяющие безопасность мясных продуктов; к ним относятся санитарно-гигиенические свойства и содержание вредных веществ;
существенные свойства, которые в большей мере характеризуют ценность мясных продуктов (биологическая ценность и органолептические характеристики);
второстепенные свойства, значительно меньше влияющие на оценку качества продукта, хотя для отдельных видов продуктов
Рис. 4.8. Факторы качества пищевых продуктов
329
они могут быть достаточно важными (содержание свободной влаги. стойкость при хранении и др.):
свойства, слабо влияющие на качество, наличие которых желательно, но не обязательно (внешний вил. упаковка, этикетки и т. д.).
На основе приведенной классификации предложена следующая модель оценки качества мясных продуктов:
1=1 l=~ i = q ' i = n
к = Mh X mhikhl + Mc X mcikci + Md £ "huKn
Z=l i=l+] /-^+1 i=q=\
(4.3)
где К— критерий качества; — весомость каждого свойства, относящегося к критической группе; — значение свойств, относящихся к критической группе, выраженное в относительных единицах; Л/Л, Л/у—относительная весомость
соответственно существенной, второстепенной и «слабой» групп свойств, причем Мь > Мс > М(/ и Mh + Л/, + Л/(/ = 1; /ил/, md. т,н — относительная весомость каждого /-го свойства; для каждой группы свойств;
Е =1;
/ = |
(4.4)
/ = <7
Ё та =4
i = Z
i = n
I mdi = \-i=q+\
(4.5)
(4.6)
Величины Мит устанавливают методом экспертных оценок, весомость всех свойств, относящихся к группе критических, можно определить как
/=/
Z = 1 •
/=1
(4.7)
Из выражения (4.3) видно, что если хотя бы одно из критических свойств равно нулю, то К= 0, т. е. продукт не может быть использован на пищевые цели.
Безразмерную величину, показывающую численное значение каждого свойства, определяют по формуле
(4.8)
П - г»ЭТ
где rz — показатель z-ro свойства; Р{ ~ эталонное значение z-ro свойства.
Данная модель является моделью смешанного типа, так как сочетает в себе мультипликационную и аддитивную формы свертывания.
330
Г. Б. Чижовым (1980 1.) разрабоtana адшпивная модель. представляющая собой выражение
Q “ (4.9)
где О — обобщенная численная характеристика качества: /<--ко эффициент значимости показателей качества; </ --безразмерное числовое выражение показателей качества.
Для удобства в Додели принято, чзо относительные значения показателей качеству выражаются долями единицы и что
^""1‘ (4.10)
Выбор каждой из величин зависит от набора показателей качества объекта, цели оценки и относительной значимости каждого показателя в данной их совокупности.
Менее распространенными являются модели векторною типа, а также специализированные модели для оценки качества различных видов продукции.
Одним из наименее разработанных вопросов качества мясных продуктов является определение набора единичных характеристик, систематизация которых пока не сложилась.
На основе анализа имеющихся литературных данных можно назвать следующие наиболее важные группы свойств для всех мясных объектов: органолептические характеристики, показатели биологической ценности и приспособленность к хранению, а также характеристики безвредности. Каждый конкретный тип мясного сырья должен иметь индивидуальный набор показателей качества. Специфической, но разной по набору свойств для сырья и полуфабрикатов является группа технологических показателей, определяющих направление и технологию их переработки.
В то же время, как отмечалось выше, состояние продукта описывается сложным комплексом химических, биохимических, физико-химических, гистологических, реологических и других характеристик, для объективного описания всей их совокупности необходимо одновременное и взаимосвязанное использование целого ряда характеристик и методов их определения, основанных на самых различных принципах.
Необходимо отметить, что показатели, подлежащие измерению при оценке качества мясной продукции, можно объединить в три группы: показатели, поддающиеся прямой объективной оценке (содержание соли, фракционный состав и т. д.); показатели, поддающиеся косвенной объективной оценке, и показатели, не поддающиеся объективной приборной оценке (товарный вид, вкус и т. д.). Для измерения показателей последней группы существу
331
ют методы экспертной оценки, основанные на теории вероятностей и математической! статистике и являющиеся достаточно iочными, но трудоемкими. В то же время приборное обеспечение для объективных методов оценки первых двух труни показателей в промышленных условиях находится пока еще на недостаточном уровне.
Контроль качества продуктов питания, как правило, основан на сочетании органолептических и инструментальных (или других несенсорных) методов. В оценке качества приоритетными методами являются органолептические. По сложившимся понятиям, инструментальное исследование обеспечивает достоверность и объективность результатов. Корреляцию между органолептическими и инструментальными показателями изучают для того, чтобы обосновать применение того или иного несенсорного метода для характеристики цвета, вкуса, запаха или консистенции продукта.
Органолептическая (сенсорная) оценка, проводимая с помощью органов чувств человека, — наиболее древний и широко распространенный способ определения качества пищевых продуктов, осуществляемый при непосредственном участии дегустаторов. Органолептический метод быстро и при правильной постановке анализа объективно и надежно дает общее впечатление о качестве продуктов.
При этом необходимо использовать научно обоснованные методы отбора дегустаторов и оценки продуктов, выполнять требования, предъявляемые к помещению, освещению, и другие условия проведения дегустационного анализа.
Современный уровень исследования качества продовольственных товаров немыслим без дегустационного анализа, проводимого с использованием балловых шкал.
Органолептические свойства — это свойства (вкус, запах, консистенция, окраска, внешний вид и т. д.) объектов, оцениваемые с помощью чувств человека. Органолептический анализ пищевых и вкусовых продуктов проводится посредством дегустаций, т. е. исследований, осуществляемых с помощью органов чувств дегустатора без измерительных приборов. На рис. 4.9 приведена классификация органолептических показателей соответственно воспринимаемым органам чувств человека.
Показатели качества, определяемые с помощью зрения:
внешний вид — общее зрительное ощущение, производимое продуктом;
форма — соединение геометрических свойств (пропорций) продукта;
цвет — впечатление, вызванное световым импульсом, определенное доминирующей длиной световой волны и интенсивностью;
блеск — способность продукта отражать большую часть лучей, падающих на его поверхность, в зависимости от гладкости поверхности продукта;
332
Рис. 4.9. Классификация органолептических показателей качества продуктов
прозрачность — свойство жидких продуктов, определяемое степенью пропускания света через слой жидкости определенной толщины.
Показатели качества, определяемые с помощью глубокого осязания (нажима):
консистенция — свойство продукта, обусловленное его вязкостью и определяемое степенью деформации во время нажима;
плотность — свойство сопротивления продукта нажиму;
эластичность — способность продукта возвращать первоначальную форму после нажима, не превышающего критической величины (предела эластичности).
Показатели качества, определяемые обонянием:
запах — впечатление, возникающее при возбуждении рецепторов обоняния, определяемое качественно и количественно;
аромат — приятный естественный характерный запах исходного сырья (молока, фруктов, специй и др.);
«букет» — приятный запах, развивающийся под влиянием сложных процессов, происходящих во время созревания, брожения и ферментации (например, «букет» выдержанного вина).
Показатели качества, определяемые осязанием (в полости рта):
сочность — впечатление, возникающее под действием соков продукта во время разжевывания (например, продукт сочный, малосочный, суховатый, сухой);
однородность — впечатление, вызванное размерами частиц продукта (однородность шоколадной массы, начинок конфет);
консистенция — осязание, связанное с густотой, клейкостью продукта, силой нажима (консистенция жидкая, сиропообразная, густая, плотная); она чувствуется при распределении продукта на языке;
333
волокнистость - впечатление, вызываемое волокнами, окали-накипим и сопротивление при разжевывании продукта, которое можно ощущать качественно и количественно (например, мясо с г о н к и м и в о л о к н а м и):
крошливость--свойство [верного продукта крошиться при раскусывании и разжевывании, обусловленное слабой степенью сцепления между частицами;
нежность — условный термин, оценивается как сопротивление, которое оказывает продукт при разжевывании (например, мягкое яблоко, хрустящий огурец, нежное мясо);
терпкость — чувство, вызванное тем, что внутренняя поверхность полости рта стягивается и при этом появляется сухость во рту;
вкус — чувство, возникающее при раздражении рецепторов и определяемое как качественно (сладкий, соленый, кислый, горький), так и количественно (интенсивность вкуса);
флевор, или вкусность, — комплексное впечатление вкуса, запаха и осязания при распределении продукта в полости рта, определяемое как качественно, так и количественно.
Способность к осязанию зависит от внешних факторов и индивидуальных особенностей дегустаторов. При отрицательной температуре осязательная восприимчивость рецепторов снижается. С возрастом осязание человека обычно ослабевает, но в меньшей степени по сравнению с другими органами чувств. Фактор возраста не является определяющим. В зависимости от природных данных, образа жизни, питания, привычек, характера труда, тренированности сенсорных органов с возрастом человека может повышаться чувствительность обоняния, вкуса, осязания, значительно реже — слуха и зрения.
Ученые разных стран разработали классификацию терминов, характеризующих консистенцию. В качестве примера можно привести фрагмент классификации параметров консистенции, показанный на рис. 4.10.
— Твердый- Хрустящий, хрупкий, мучнистый
Влажный, сухой, липкий
— Твердый — — Мягкий— Грубый, нежный
Эластичный, пенистый, пластичный
Влажный, сухой, липкий, водянистый
Однородн ый. зерн исты й
Плотный, пастообразный, крошащийся
Влажный, сухой, липкий, водянистый
Сгустившийся, однородный
— Жидкий
Водянистый, мягкий
Кремообразный, маслянистый, жирный
Липкий
Рис. 4.10. Классификация параметров консистенции пищевых продуктов (по данным К. Помпеи)
334
Разработаны методики органолептической оценки механических параметров консистенции (табл. 4.5).
Г а б л и и а 4.5
Методика органолептического анализа механических параметров консистенции
Порядок оценки
Парами ip
Твердость Поместить образец между зубами и нажать с равномерным усилием, оценить силу, потребовавшуюся для этого
Сцепление Поместить образец между зубами и оценить величину деформации перед откусыванием
Эластичность Поместить образец между зубами (если продукт полужидкий, то между языком и небом) и слегка нажать; затем прекратить давление и оценить степень и быстроту возвращения первоначальной формы
Клейкость Поместить образец на язык и прижать языком к небу, оценить силу, необходимую для отделения продукта от нёба с помощью языка
Хрупкость Поместить образец между зубами и нажать с равномерным усилием, пока он не расколется и не рассыплется; оценить силу, с которой это происходит
Пережсвываемость Поместить образец между зубами и жевать с частотой одно нажатие в секунду с постоянным усилием; подсчитать число нажатий, необходимых для измельчения продукта до степени, позволяющей его проглотить
Вязкость для продуктов:
полужидких Положить образец в рот и тереть его языком по нёбу, подсчитать число движений, необходимых для того, чтобы измельчить продукт
жидких Поместить ложку с образцом перед ртом и втянуть жидкость на язык; оценить силу, необходимую для втягивания жидкости с определенной постоянной скоростью
Консистенция продукта воспринимается потребителем как сумма вкуса, запаха и ощущений.
Консистенция не только взаимосвязана с вкусовыми свойствами и запахом продукта, но также влияет на усвояемость или характеризует свежесть. Например, о безупречной свежести охлажденного мяса судят по запаху и эластичности мышечной ткани.
Для создания хорошей консистенции мясных продуктов применяют функциональные добавки: загустители, студнеобразова-тели, эмульгаторы, стабилизаторы, пенообразователи и другие вещества. Механизм их действия состоит в изменении коллоидных свойств продуктов. Среди них наибольшее распространение получили различные пектины, желатин, крахмал и его модификации, агар и агароид, целлюлоза и модифицированная целлю-
335
.юза. альгинат морских водорослей. лецитины, хитозаны, конденсированные фосфаты и полифосфаты.
При проведении дегустаций к помещениям предъявляют особые требования. Рекомендуется иметь два изолированных помещения: специально оборудованное для работы дегустаторов и подготови-гельное. предназначенное для подготовки образцов для дегустации. Помещение для работы дегустаторов должно быть защищено от шума и вибрации; хорошо вентилируемо, но без сквозняков, хорошо освещено, предпочтительно рассеянным дневным светом без проникновения прямых солнечных лучей. Освещенность рабочих мест должна быть равномерной и составлять не менее 500 лк. Освещение не должно искажать цвет оцениваемого продукта.
Помещение для дегустаций рекомендуется окрашивать в светлые, спокойные для глаз тона; оно должно быть чистым, без посторонних запахов. Температура воздуха в помещении должна быть (20 ± 2) °C, относительная влажность воздуха — (70 ± 5) %.
Рабочие места дегустаторов следует располагать так, чтобы дегустаторы не оказывали влияния друг на друга и не отвлекались при проведении оценки. Рекомендуются кабины или столы (ширина 50—60 см, длина 80—90 см, высота 75—80 см) с перегородками (высота 50 см, длина 40 см), а также удобные стулья. При отсутствии перегородок места дегустаторов предпочтительнее размещать одно за другим. На столе дегустатора должны быть: дегустационные листы; карандаш или ручка; тарелки (белые, без рисунка), стаканы или чашки; нож и вилка из нержавеющей стали; салфетка; посуда для отходов; нейтрализующие средства для восстановления вкусовой чувствительности (белый хлеб, некрепкий и негорячий чай или минеральная вода).
В подготовительном помещении должны размещаться следующие оборудование и инвентарь: шкафы для хранения посуды; столовые приборы; рабочий инвентарь и др.; рабочие столы для подготовки проб; холодильники; мойка для посуды с горячей и холодной водой; посуда и неокисляемые столовые приборы; деревянная или металлическая игла для определения запаха в толще продуктов (неразрезанных); весы с наибольшим пределом взвешивания 1000 г; приборы для измерения температуры (термометры с диапазоном измерения от 0 до 100 °C); оборудование для измельчения и термической обработки.
Органолептические показатели могут указывать на степень развития автолитических процессов, происходящих при хранении, а также на свежесть, характер и глубину развития микробиологических процессов.
Обычно гнилостная порча начинается на поверхности, а затем проникает в толщу мяса, причем скорость порчи зависит от температуры и влажности окружающей среды, состояния поверхности (корочка подсыхания, порезы) и гистологической структуры, вида бактерий, возбуждающих гнилостный распад.
336
Различные виды порчи взаимосвязаны. Ослизнение, протекающее при повышенных температурах и относительной влажности воздуха более 90%. сопровождается сплошным ростом бактерий. Плесени, развивающиеся в кислой среде, сдвигают pH в щелочную сторону и подготавливают условия для жизнедеятельности гнилостных микроорганизмов.
В результате развития гнилостной микрофлоры происходит распад белка с образованием как первичных, так и вторичных продуктов гидролиза, оказывающих существенное влияние на органолептические показатели и пищевую ценность мяса.
В ходе превращения белковых веществ в мясе накапливаются карбоновые жирные (уксусная, масляная, муравьиная) и оксикислоты, амины, альдегиды, а также неорганические соединения (Н?О, NH3, СО2, N-,. H2S) и вещества, изменяющие вкус и запах (фенол, крезол, индол, скатол, меркаптан). Биологическая ценность мяса снижается за счет распада белковых веществ. Процесс гнилостной порчи частично затрагивает и липидную фракцию.
Изменение цвета обусловлено образованием мет- и сульфомиоглобина, появлением пигментации желто-зеленого цвета и обесцвеченных участков под воздействием пероксида водорода и специфических пигментов, выделяемых некоторыми микроорганизмами. Консистенция мяса ухудшается, возрастает его рыхлость.
Испортившееся мясо может стать причиной пищевых отравлений: токсикоинфекций, возникающих в результате употребления продукта, содержащего сальмонеллы, кишечную, дизентерийную палочки и протей, и интоксикаций вследствие наличия в продуктах ядов (токсинов), выделяемых некоторыми видами микроорганизмов (стафилококков, стрептококков, палочки ботулинуса) в процессе их жизнедеятельности.
Одним из быстрых методов определения свежести мяса является разработанный во ВНИИМПе метод гистологического анализа, который в сочетании с органолептическими показателями позволяет в течение 40—60 мин получить полное представление о состоянии и степени свежести мяса.
Результаты гистологического анализа отличаются высокой достоверностью, в целом ряде случаев их можно дополнить данными физико-химических, биохимических, органолептических, микробиологических и других исследований.
Гистологический метод позволяет проводить исследования поверхностных и глубинных слоев мяса раздельно и таким образом устанавливать локализацию изменений и увязывать их с изменением определенных структур мышечной ткани мяса.
Метод гистологического анализа мяса, вошедший в ГОСТ 19496—74, позволяет определить начало снижения качества мяса в результате воздействия гнилостной микрофлоры на 3—4 дня раньше, чем в нем обнаружатся органолептические и физико-химические признаки порчи. При этом в поверхностных слоях
337
мяса в местах развития гнилостной микрофлоры четко выявляются изменения структуры ядер.
В результате гистологического исследования мяса, полученного от здоровых животных, содержащихся в хороших санитарно-гигиенических условиях, при строгом соблюдении технологии разделки, охлаждения и хранения туш (при температуре хранения 2—4 °C) и взятого через 2—3 сут после убоя животного, в поверхностных слоях мяса обнаружена хорошо выраженная подсохшая корочка — уплотненный слой соединительнотканных фасций и прилегающих к ним утонченных мышечных волокон с плохо различимой поперечной исчерченностью. В местах разруба поверхностный слой также уплотнен.
В глубоколежащих слоях мышц структура ядер, поперечная и продольная исчерченность хорошо выражены, окраска равномерная. Это свидетельствует о хорошей сохранности структур мышечной ткани и наличии условий, которые исключают возможность размножения гнилостной микрофлоры, сопровождающейся изменением микроструктуры мяса.
Изменение структуры ядер мышечных волокон свидетельствует о первоначальных признаках снижения качества мяса под воздействием ферментов развивающейся в его поверхностных слоях гнилостной микрофлоры и говорит о начавшемся процессе гнилостного разложения тканей мяса.
При хороших органолептических показателях по гистологическим показателям такое мясо относят к свежему, но не подлежащему длительному хранению и транспортированию.
Микроструктурные изменения, характеризующие порчу мяса, распространяются на большую глубину, чем размножающаяся гнилостная микрофлора, что указывает на значительно опережающее проникновение в глубь мяса ферментов последней и имеет большое диагностическое значение при установлении границ порчи мяса.
Когда процесс гнилостного разложения мяса проникает глубоко внутрь, в мышечных волокнах отмечаются множественный распад миозиновых и актиновых протофибрилл, деструкция мембран саркоплазматического ретикулума и базальной мембраны сарколеммы, слияние разрушенного содержимого рядом лежащих мышечных волокон с образованием клеточного детрита.
Таким образом, в микроструктурных изменениях при порче мяса прослеживается определенная последовательность, позволяющая объективно оценить степень его свежести:
гомогенизация структуры ядер и их сморщивание присущи свежему мясу, но не подлежащему длительному хранению;
набухание мышечных волокон и начало лизиса их внутренних структур свойственны мясу сомнительной свежести, перед его использованием следует проводить санитарную зачистку в местах порчи;
338
полный лизис внутренних структур мышечных волокон говорит о том, что мясо несвежее и подлежит утилизации.
Таким образом, изменения мышечной ткани при порче носят диффузный характер и отличаются глубоким распадом всех структур мышечных волокон. Это положено в основу микроструктурной дифференциации процессов гнилостного разложения мяса и локально развивающихся автолитических процессов, лежащих в основе созревания мяса.
Общие органолептические признаки свежести мяса, субпродуктов, мяса птицы и кроликов приведены в табл. 4.6—4.8.
В процессе переработки и хранения жировой ткани убойных животных или выделенных из нее жиров под влиянием биологических и физико-химических факторов происходят разнообразные превращения. Контакт жировой ткани мяса с кислородом воздуха, водой, микроорганизмами, металлами и т. п. вызывает физико-химические и биологические процессы, изменяющие свойства жирового сырья и тканей мяса. Интенсивность изменений зависит как от свойств сырья, так и от условий хранения. Окислительные и гидролитические процессы могут вызвать порчу жиров (рис. 4.11). В результате изменяется их химический состав, ухудшаются органолептические показатели и пищевая ценность. Процессы гидролиза и окисления часто протекают одновременно, усиливая изменения жира.
Рис. 4.11. Схема порчи жиров
339
о
Табл и ц а 4.6
Признаки свежести мяса и субпродуктов
Показатель
Характерные признаки мяса или субпродуктов
свежих
сомнительной свежести
несвежих
Внешний вид и цвет Покрыта подсохшей корочкой бледно- Местами увлажнена, слегка Сильно подсохшая, покрытая
поверхности туши розового или бледно-красного цвета; у размороженных туш красного цвета; жир мягкий, частично окрашен в ярко-красный цвет липкая, потемневшая слизью серовато-коричневого цвета или плесенью
Мышцы на разрезе Слегка влажные, не оставляют влажного пятна на фильтровальной бумаге; цвет, свойственный данному виду мяса: для говядины — от светло-красного до темно-красного, для свинины — от светло-розового до красного, для баранины — от красного до красно-вишневого, для ягнятины — розовый Влажные, оставляют влажное пятно на фильтровальной бумаге, слегка липкие, темно-красного цвета. У размороженного мяса с поверхности разреза стекает слегка мутноватый мясной сок Влажные, оставляют влажное пятно на фильтровальной бумаге, липкие, красно-коричневого цвета. У размороженного мяса с поверхности разреза стекает мутный мясной сок
Консистенция На разрезе мясо плотное, упругое; образующаяся при надавливании пальцем ямка быстро выравнивается На разрезе мясо менее плотное и менее упругое; образующаяся при надавливании пальцем ямка выравнивается медленно (в течение 1 мин); жир мягкий, у размороженного мяса слегка рыхлый На разрезе мясо дряблое; образующаяся при надавливании пальцем ямка нс выравнивается; жир мягкий, у размороженного мяса рыхлый, осалившийся
Запах Специфический, свойственный каж- Слегка кисловатый или с от- Кислый или затхлbiii, или
дому виду свежего мяса тенком затхлости сл або гн ил остн ы й
Продолжение
Показатель Характерные признаки мяса или субпродуктов
свежих сомнительной свежести несвежих
Состояние жира Говяжий жир имеет белый, желтоватый или желтый цвет; консистенция твердая, при раздавливании крошится; свиной — белый или бледно-розовый цвет; мягкий, эластичный; бара- Имеет серовато-матовый оттенок, слегка липнет к пальцам; может иметь легкий запах осаливания Имеет серовато-матовый оттенок, при раздавливании мажется. Свиной жир может быть покрыт небольшим количеством плесени. Запах прогорк-
ний — белый цвет, консистенция плотная. Жир не должен иметь запаха осаливания или прогоркания лый
Состояние сухожилий Сухожилия упругие, плотные, поверхность суставов гладкая, блестящая. У размороженного мяса сухожилия мяг- Сухожилия менее плотные, матово-белого цвета. Поверхность суставов слегка Сухожилия размягчены, сероватого цвета. Поверхность суставов покрыта слизью
кие, рыхлые, окрашенные в ярко-красный цвет покрыта слизью
Прозрачность и аромат бульона Прозрачный, ароматный Прозрачный или мутный, с запахом, не свойственным свежему бульону Мутный, с большим количеством хлопьев, с резким неприятным запахом
LU -£*• KU T а б л и ц а 4 Органолептические показатели мяса (тушек) птицы различной степени свежести
Показатель Характерные признаки мяса (тушек) птицы
свежих сомнительной свежести несвежих
Внешний вид и цвет: клюва Глянцевитый Без глянца Без глянца
слизистой оболочки Блестящая, бледно-розового Без блеска, розовато-серого цве- Без блеска, серого цвета, по-
ротовой полости цвета, незначительно увлаж- та, слегка покрыта слизью. Воз- крыта слизью и плесенью
глазного яблока нена Выпуклое, роговица блестя- можно наличие плесени Невыпуклое, роговица без Провалившееся, роговица без
поверхности тушки щая Сухая, беловато-желтого цве- блеска Местами влажная, липкая под блеска Покрыта слизью, особенно под
подкожной и внут- та с розовым оттенком, у нежирных тушек желтовато-серого цвета с красноватым оттенком, у тощих — серого цвета с синюшным оттенком Бледно-желтого или желтого крыльями, в паху и в складках кожи; беловато-желтого цвета с серым оттенком Бледно-желтого или желтого крыльями, в паху и в складках кожи; беловато-желтого цвета с серым оттенком, местами с темными или зеленоватыми пятнами Подкожная - бледно-желтого
ренней жировой цвета цвета цвета, а внутренняя — желто-
ткани серозной оболочки Влажная, блестящая, без ели- Без блеска, липкая, возможно вато-белого цвета с серым оттенком Покрыта слизью, возможно
зи и плесени наличие небольшого количества слизи и плесени наличие плесени
Продолжение
Показатель Характерные признаки мяса (тушек) птицы
свежих j сомнительной свежести i | несвежих
Мышцы на разрезе Слегка влажные, нс оставляют влажного пятна на фильтровальной бумаге, у кур и индеек — бледно-розового цвета, у уток и гусей — красного Влажные, оставляют пятно на фильтровальной бумаге, слегка липкие, более темного цвета, чем у свежих тушек Влажные, оставляют пятно на фильтровальной бумаге, липкие. более темного цвета, чем у свежих тушек
Консистенция Мышцы плотные, упругие, при надавливании пальцем образующаяся ямка быстро выравнивается Мышцы менее плотные и менее упругие, чем у свежих, при надавливании пальцем образующаяся ямка выравнивается медленно (в течение 1 мин) Мышцы дряблые, при надавливании пальцем образующаяся ямка нс выравнивается
Запах Специфический, свойственный свежему мясу птицы Затхлый в грудобрюшной полости Гнилостный на поверхности тушки и внутри мыши, наиболее выражен в грудобрюшной полости
Прозрачность и аромат бульона Прозрачный, ароматный Прозрачный или мутноватый с легким неприятным запахом Мутный, с большим количеством хлопьев и резким неприятным запахом
i 5
Органолептические показатели мяса (тушек) кроликов различной степени свежести
X
X
*
о
н с_> и X
X
ч X со
:Х X X а о X
CQ
СО
О о
X а: 2 Е
о а
L0
н О и
I £ X
:Х О СО о CL X X
'X О X
* IX
о о X
X X
а X <33
о О
с О.
О
X
*
о
б
О
О 3
л
б ю
X
о
X
X QJ о о
с
X
X б
ь
X
2
с
С
О X
2
о О
о
X
*
п 02
2 Cs е; и <33 F-о о о I
о.
I о X
о
Е-
CJ 2 с X
о
о X о <33 о.
о
X н о СО X
О
О со
о
X о
о е; о ю о
:Х О X г> О О. о о
о б р" £Х G3 X
3 X а
3
3 6 2 5 х х S '4 ^>3 3 2 ах 2
к
X X о
о X о
X о
I X о со
>х о со о
:£ X
О 2
х •&
X а о с О
344
Степень порчи жиров исследуют не только органолептическими. но и различными химическими методами. Результаты определений обычно характеризуют условными единицами — кислотным, перекисным и другими числами (ГОСТ Р 51487—99). Гидролитическая порча жиров характеризуется накоплением свободных жирных кислот. Это может быть как следствием автолиза, так и результатом действия других факторов: кислот, щелочей, оксидов металлов и других неорганических катализаторов, а также ферментов микроорганизмов.
Под влиянием тканевых липаз наблюдается гидролитический распад триглицеридов, в результате чего отмечается нежелательное для качественной характеристики жира накопление свободных жирных кислот, выражающееся в повышении кислотного числа жира. В свежей жировой ткани, только что извлеченной из туши, кислотное число невелико и не превышает 0,05—0,2.
Скорость и глубина гидролиза жира зависят от температуры (рис. 4.12).
Появление в жире при гидролитическом распаде небольшого количества высокомолекулярных жирных кислот не вызывает изменения вкуса и запаха продукта. При наличии в составе триглицеридов низкомолекулярных кислот при гидролизе могут образовываться капроновая и масляная кислоты, обладающие неприятным запахом и специфическим вкусом, резко ухудшающими органолептические свойства продукта.
В топленых жирах автолитического расщепления жира, как правило, не наблюдается. Это объясняется инактивацией содержащейся в жировой ткани липазы при достижении температуры 60 °C в процессе вытопки. Гидролитическая порча топленого жира возможна при наличии влаги, обсеменении микрофлорой, неполной денатурации белков при вытопке жира или в присутствии неорганических катализаторов.
В процессе хранения и переработки жиров возможны их окис-
лительные изменения, которые могут протекать с различной скоростью, глубиной, иметь различную направленность в зависимости от природных свойств жира и условий окисления.
Окисление жиров (автоокисление) протекает при низких температурах в присутствии газообразного кислорода.
Рис.4.12. Изменение кислотного числа почечного свиного жира-сырца в процессе хра-
нения при температуре:
7 —22 °C; 2-4,4 °C
Продолжительность гранения, ч
345
Продолжительность хранения, ч
Рис. 4.13. Накопление пероксидов при окислении топленого свиного жира при 90 °C
О начале и глубине окисления жира судят по величине перекисного числа. В свежем жире пероксидов нет. На начальных стадиях окисления в течение некоторого времени химические и органолептические показатели жира почти не изменяются. Этот период, имеющий для различных жиров разную продолжительность, называют индукционным. После окончания индукционного периода жир начинает портиться (рис. 4.13), что сопровождается увеличением перекисного числа и изменением органолептических свойств жира.
Наличие индукционного периода объясняется малым количеством частиц с повышенной кинетической энергией (возбужденных или свободных радикалов) в начале процесса.
Продолжительность индукционного периода зависит от массовой доли естественных (каротиноиды, токоферолы, лецитин, витамины А и К) или искусственных (производные фенола,
содержащиеся в коптильном дыме, некоторые природные специи или их экстракты, бутилоксианизол, бутилокситолуол) антиокислителей, природы жира и условий хранения. Механизм действия антиокислителей состоит в их более активном взаимодействии со свободными радикалами и кислородом воздуха, за счет чего радикалы выводятся из сферы реакции и цепь обрывается.
Задача адекватной оценки качества мясных продуктов на основе большого количества единичных характеристик в настоящее время в основном решена. Схема методов показана на рис. 4.14. Проблемой остается набор этих характеристик, которые разнообразны и не систематизированы.
Для создания программного обеспечения, оперативности и
точности оценок, использования показателя качества в экономических расчетах необходимо формализовать критерии качества, т. е. представить их в виде массива цифровых данных, отражающих как величину отдельных показателей, так и функциональные связи между ними. Такие задачи могут быть решены на базе квалиметрической оценки качества. В результате ряда упорядоченных операций по выбору, измерению и оценке свойств исследуемого объекта квалиметрия дает возможность получить показатель его качества в виде некоторой цифровой величины, что позволяет использовать ее в алгоритме управления технологическим процессом.
346
Группа свойств Единичные свойства Методы исследования Показатели качества
— Конспс- — — экспертные — балловая опенка
тенция — реологические - — усилие среза — степень пенетра
Органолептичес кие характерис- —экспертные — балловая оценка
тики спектрофото- метрические — экспертные разложение по осям интенсивность при доминирующей длине волны интенсивность свето- поглошения вытяжки и т.д. балловая оценка
хроматогра- концентрация
— Вкус фические —«шарп» -метод— — экспертные летучих веществ — условные единицы балловая оценка
— Внешний вид- — экспеотные — балловая опенка
Рис. 4.14. Схема методов исследования для оценки органолептических показателей качества мясных продуктов
В последнее десятилетие автоматизация процессов пищевых технологий привела к созданию устройств, позволяющих регистрировать накопление, распад и взаимодействие различных веществ и изменение их состояния при самых низких концентрациях. Эти устройства, получившие названия «сенсоры», уже достаточно широко используются на различных этапах производства мясной продукции. В зарубежной и отечественной литературе термины «органолептическая оценка» и «сенсорный анализ» часто воспринимают как синонимы. Современный уровень развития органолептики требует разделения этих понятий. Способы измерения количества химических соединений в пищевом продукте с помощью сенсоров называют сенсорной технологией оценок.
Сенсоры контролируют большее количество параметров (цвет, температуру, массу и влажность), чем органы человека, причем бесконтактным способом. При этом могут быть использованы видеосистемы, работающие как в видимой области, так и в области у-лучей. В частности, с помощью рентгеновского излучения определяют наличие в продукте загрязняющих веществ. Для поточного контроля продукции предлагается также использовать ВЧ- и звуковое излучение, ближнее ИК-излучение.
347
Преимущества сенсоров — эффективный, непрерывный, неразрушающий контроль качества, применяемый в труднодоступных местах: при высоком уровне производительности труда; не повышают себестоимость продукции.
В качестве биосенсоров предлагается использовать иммобилизованные ферменты и клетки, вызывающие превращения определяемых веществ с последующей! электрохимической регистрацией продуктов ферментативных реакций. Преимущества биосенсоров — точное определение содержания вещества, быстрота измерения, возможность многократного использования.
В настоящее время получили распространение потенциометрические электроды, которые вводят в продукт и при помощи них прослеживают кинетику образования или распределения какого-либо химического компонента, характеризующего протекание процессов, изменения качества продукта. Установлено, что распределение глюкозы в свежем мясе характеризует микробиологическую обсемененность поверхности мяса. Вводя биодатчики в мясо, измеряя токовые параметры, а по ним с помощью калибровочных кривых распределение концентрации глюкозы в мясе, можно быстро определить его качество.
Конструкционно биосенсоры представляют собой потенциометрические, пьезокристаллические, оптические, акустические или электромагнитные датчики, регистрирующие различные качественные характеристики продукта.
Существует мнение, что будущее принадлежит сенсорным методам. Они будут основными при контроле технологических процессов и оценке качества сырья и готовой продукции. Измерительные устройства, основанные на регистрации электрических сигналов биологических систем, позволяют значительно расширить существующие способы контроля и сделать сенсорную технологию оценки качества неотъемлемым элементом автоматизированных систем управления технологическими процессами.
Лабораторная работа № 1
ОРГАНОЛЕПТИЧЕСКАЯ ОЦЕНКА МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Приобрести практический навык органолептической оценки мяса и мясных продуктов.
Задачи. Подготовить образцы мяса и мясных продуктов, провести органолептическую их оценку.
348
Объекты исследования. Мясные продукты — фаршированные, вареные, полукопченые, варено-копченые, сырокопченые, ливерные и кровяные колбасы, мясные хлеба, сосиски, сардельки, зельцы. студни, холодцы, паштеты, а также продукты из свинины, говядины, баранины, мяса птицы и других животных, полуфабрикаты. кулинарные изделия, мясные бульоны.
Оборудование. Набор посуды; столовые приборы; деревянные (или металлические) иглы; термометры с диапазоном измерения 0—100 °C; мясорубка: водяная баня; электрическая плитка.
Методические указания. Для выполнения работы в лабораторных условиях необходимо максимально соблюдать рекомендации по условиям и оснащению помещения.
Органолептическая оценка проводится для установления соответствия органолептических показателей качества продуктов требованиям нормативно-технической документации, а также для определения показателей новых видов мясной продукции при постановке ее на производство.
Органолептическая оценка проводится для определения внешнего вида, цвета, вкуса, аромата, консистенции и других показателей посредством органов чувств.
Органолептическая оценка осуществляется студентами при непосредственной консультации преподавателя или дегустаторов, имеющих опыт работы по оценке качества мясной продукции.
Перед проведением органолептической оценки студенты знакомятся с требованиями нормативно-технической документации к качеству оцениваемой продукции.
Образцы продукции дегустируют в следующей очередности: в первую очередь оценивают продукты, обладающие слабо выраженным (тонким) ароматом, менее соленые и острые, затем — продукты с умеренным ароматом и соленостью, после этого — продукты с сильно выраженным ароматом, соленые и острые.
В последнюю очередь оценивают изделия в подогретом виде (сосиски, сардельки и т. д.) и термически обработанные (пельмени, котлеты и другие полуфабрикаты); порядок их представления определяется также степенью выраженности аромата и вкуса.
В работе предлагается провести органолептическую оценку мясных продуктов по девятибалловой шкале. При этом предварительно знакомятся с перечнем установленных показателей и характеристикой этих показателей для каждого балла избранной системы оценок (см. формы 1—3).
349
Д| о Оценка органолептических показателей мяса Ф о р м а 1
\ Консистенция 1
№ образца Внешний вид Запах (аромат) Вкус (нежность, i Сочность Общая оценка
жесткость) качества (балл)
Положительные показатели качества мяса
Очень приятный Очень приятный Очень вкусный Очень нежная Очень сочное Отлично (9)
и сильный
Очень хороший Приятный и силь- Вкусный Нежная Сочное Очень хоро-
ный шо (8)
Хороший Приятный, но не- Достаточно вкус- Достаточно неж- Достаточно соч- Хорошо (7)
достаточно силь- ный ная нос
ный
Недостаточно хо- Недостаточно аро- Недостаточно Недостаточно Недостаточно Выше сред-
роший матный вкусный нежная сочное него (6)
Средний (удовл.) Средний (удовл.) Средний (удовл.) Средняя (удовл.) Среднее (удовл.) Среднее (5)
Отрицательные показатели качества мяса
Немного непри- Без аромата Безвкусный Жестковатая Суховатое Ниже сред-
влекательный (приемл.) (приемл.) (приемл.) (приемл.) него (4)
(приемл.)
Неприятный Немного непри- Немного непри- Немного жесткая Немного сухое Плохо
(приемл.) ятный (приемл.), ятный (приемл.) (приемл.) (приемл.) (приемл.) (3)
посторонний
(приемл.)
Неприятный, пло- Плохой, посто- Плохой, неприят- Жесткая Сухое Плохо
хой (неприемл.) ронний ный (неприемл.) (неприемл.) (неприемл.) (неприемл.) (2)
(неприемл.)
Очень неприят- Очень неприят- Очень плохой, Очень жесткая Очень сухое Очень плохо
ный, очень пло- ный, посторон- очень неприят- (совершенно (совершенно (совершенно
хой (совершенно ний (совершен- ный (совершен- неприемл.) неприемл.) неприемл.) (1)
неприемл.) но неприемл.) но неприемл.)
Замечания:
Ф о р м а 2
Оценка органолептических показателей мясного бульона
№ образца Внешний вид Запах (аромат) Вкус Наваристост ь Общая опенка качест ва (балл)
Очень приятный Очень хороший Хороший Недостаточно хороший Средний (удовл.) Положительные показатели качества бульона Очень приятный и Очень вкусный Очень наваристый сильный Приятный и сильный Вкусный Наваристый Приятный, но недо- Достаточно вкус- Достаточно нава- статочно сильный ный ристый Недостаточно аромат- Недостаточно вкус- Недостаточно на-ный ный варистый Средний (удовл.) Средний (удовл.) Средний (удовл.) Отлично (9) Очень хорошо (8) Хорошо (7) Выше среднего (6) Среднее (5)
Отрицательные показатели качества бульона
Немного неприят- Без аромата (приемл.) Безвкусный Слабо наваристый Ниже среднего
ный (приемл.) (приемл.) (приемл.) (приемл.) (4)
Неприятный Немного неприятный, Немного неприят- Ненаваристый Плохо (приемл.) (3)
очень слабый посто- ный (приемл.) (приемл.)
ронний (приемл.)
Неприятный, пло- Плохой, посторонний Плохой, неприят- Водянистый Плохо
хой (неприемл.) (неприемл.) ный (неприемл.) (неприемл.) (неприемл.) (2)
Очень неприятный, Очень плохой, силь- Очень плохой Как вода (совер- Очень плохо (совер-
очень плохой (совер- ный посторонний (со- (совершенно шенно неприемл.) шенно неприемл.) (1)
шенно неприемл.) вершенно неприемл.) неприемл.)
Замечания:
ж о Е
*
и
2
с
5
Е
JX
X
к
sX
□
X
X
5
X
к
X
X S о I
5 а И
3 о
)Х
X
& й
§ «
х _
о 3Х
2 о г
о о X
X с.
X
S о
<о о
о
о
X
Е
С
Е X
I =х о х
X
о
I
352
Подготовка проб. Проводят согласно требованиям нормативно-технической документации на соответствующие виды продукции.
Перед подачей на дегустацию их кодируют цифрами или буквами. Проводят либо «закрытую», либо «открытую» дегустацию. В последнем случае преподаватель (или дегустатор) дает краткую информацию о представленном образце продукции.
Порядок проведения анализа. Сначала оценивают целый (неразрезанный). а затем разрезанный продукт.
При оценке целого продукта визуально путем наружного осмотра определяют внешний вид, цвет и состояние поверхности. Фиксируют запах на поверхности продукта. При необходимости определения запаха в глубине продукта берут специальную деревянную или металлическую иглу, вводят ее в толщу продукта, затем быстро извлекают и определяют запах, оставшийся на поверхности иглы.
Далее определяют консистенцию путем надавливания шпателем или пальцем.
При оценке разрезанного продукта показатели определяют в следующей последовательности:
перед проведением оценки мясные изделия освобождают от упаковки, оболочки и шпагатов (клипсов), удаляют из них кости (если они имеются) и с помощью острого ножа режут тонкими ломтиками так, чтобы обеспечить характерный для данного продукта вид и рисунок на разрезе;
цвет, вид и рисунок на разрезе, структуру и распределение ингредиентов определяют визуально на только что сделанных поперечном и (или) продольном разрезах продукции;
запах, аромат, вкус и сочность оценивают опробованием мясных продуктов, нарезанных на ломтики. При этом выделяют специфический запах, аромат и вкус; отсутствие или наличие постороннего запаха, привкуса; степень выраженности аромата пряностей и копчения;солености:
консистенцию продуктов определяют надавливанием, разрезанием, разжевыванием, размазыванием (паштеты). При определении консистенции устанавливают плотность, рыхлость, нежность, жесткость, крошливость, упругость и однородность массы (паштеты).
Запах, вкус, сочность сосисок и сарделек определяют в разогретом виде, для чего их опускают в теплую воду (50—60 °C) и доводят до кипения. Сочность сосисок и сарделек в натуральной оболочке можно также определять проколом. В местах прокола в сочной продукции должна выступить капля жидкости.
Мясные консервы оценивают в разогретом или холодном виде в зависимости от рекомендуемого способа употребления в пищу данного продукта. В первом случае после внешнего осмотра закрытую банку погружают в спокойно кипящую воду на 20—30 мин в зависимости от размера банки и вида консервов.
353
Нагретые консервы сразу же подаюг дня органотептической опенки, не допуская их остывания.
Содержимое банок помещают в чистую сухую тарелку.
При оценке качества консервов, употребляемых в холодном виде, продукт перед подачей на исследование нарезают, чтобы не изменились цвет ломтиков и их товарный вид. Минимальная толщина ломтиков должна быть такой, чтобы обеспечить их целостность.
Вскрытые банки (и крышки) после опорожнения промывают горячей водой и подвергают осмотру (при необходимости).
Продукцию оценивают по девятибалловой системе, если она предусмотрена нормативной документацией, или в виде описания — на соответствие показателей качества требованиям стандартов и технических условий.
При оформлении собственных результатов анализа обмениваться мнениями не разрешается.
В процессе органолептической опенки каждый участник вносит свои оценки и замечания в дегустационный лист рекомендуемой формы:
Дегустационный лист
Фамилия, инициалы Дата « _________________________»г.
Организация__________________________________________________________
Продукт Опенка продукта по девятибалловой системе Другие замечания
Внешний вид Цвет Запах, аромат Консистенция Вкус Сочность Общая опенка, балл
Подпись_________________________
Лабораторная работа № 2
ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЕЙ БИОЛОГИЧЕСКОЙ ЦЕННОСТИ РАСЧЕТНЫМ МЕТОДОМ
Цель работы. Освоить методы определения биологической ценности мяса и мясных продуктов расчетным путем.
Задачи. Рассчитать скор незаменимых аминокислот в мясе, мясных или комбинированных продуктах; выявить лимитирующие аминокислоты, оценить среднюю величину избытка скора незаменимых аминокислот и рассчитать коэффициент утилитарности и показатель «сопоставимой избыточности».
354
Объекты исследования. Мясо различных видов животных, птицы, мясные продукты, субпродукты, колбасные изделия и консервы.
Оборудование. Микрокалькулятор.
Методические указания. В качестве исходных для расчетов используют экспериментально полученные данные при анализе состава аминокислот мясных продуктов или данные из справочной литературы (табл. 4.9).
Т а б л и ц а 4.9
Состав незаменимых аминокислот в некоторых видах мяса и мясных продуктах, г на 100 г белка
т
11родукт Валин Изо- ! лейцин| । Лейцин Лизин Метионин Тре-онин Триптофан Фенилаланин
Говядина (мышечная ткань) 5.3 4,3 7,5 8,0 2,7 4,0 1,2 4.1
Свинина (мышечная ткань) 5.5 4,7 7.5 7.9 2,3 4,7 1.3 3,9
Конина 1 категории 5.1 4.0 7,6 8,9 2.4 4,7 1,4 4.3
Куры 1 категории 4.8 3,8 7.7 8.7 2,5 4.8 1.6 4.0
Печень говяжья 5.6 5,3 9.0 5,1 2,9 4,8 1,6 5,7
Почки говяжьи 5,5 2,9 6.8 8.3 1,5 1,8 1.3 3.8
Рубец говяжий 3,8 3.4 6,0 5,0 1,6 3,5 0,9 3,4
Селезенка говяжья 4.7 7,4 6,1 9,4 2,4 3,3 1.4 2.5
Колбаса вареная докторская 5,2 4,2 7,1 7.3 2,7 4,1 1,1 3,9
Колбаса полукопченая минская 6,9 4,9 7,2 7,2 2,7 3,5 1.0 2,9
Колбаса сырокопченая сервелат 5,5 4,5 7,6 8,4 з,о 4,2 1,5 3,9
Яйцо куриное целое 6,0 4,7 8,5 7,1 з,з 4,8 1,6 5,1
Желатин пищевой 2.0 1,3 2,7 4,3 0,1 1,4 0,1 1,7
Используя данные аминокислотного состава, студенты расчетным путем определяют показатели биологической ценности продуктов. Для сопоставления результатов расчет рекомендуется вести по нескольким объектам.
Подготовка к расчету. Студенты внимательно изучают перечень показателей биологической ценности и выписывают формулы для их определения.
Аминокислотный скор. Выписывают данные о содержании незаменимых аминокислот в исследуемых мясных продуктах и рекомендации ФАО/ВОЗ применительно к «идеальному» (стандартно-
355
му) белку. Аминокислотный скор определяют ио формуле
а
пр
Ж?
100.
(4.11)
где ЛЛ' — содержание незаменимой аминокислоты в 1 г исследуемого белка, мг; /1Л'ст — содержание той же аминокислоты в 1 г «идеального» (стандартного) белка, мг; 100 — коэффициент пересчета в проценты.
Лимитирующей биологическую ценность аминокислотой считается та, скор которой наименьший.
Коэффициент различия аминокислотного скора (КРАС, %). Показывает среднюю величину избытка аминокислотного скора незаменимых аминокислот по сравнению с наименьшим уровнем скора какой-либо незаменимой аминокислоты (избыточное количество незаменимых аминокислот, не используемых на пластические нужды):
КРАС =
ЕД РАС п
(4.12)
где АРАС — различие аминокислотного скора аминокислоты;
ДРАС = С, + С„„„, (4.13)
здесь Cz — избыток скора аминокислоты; Cnijll — минимальный из скоров незаменимых аминокислот исследуемого белка по отношению к эталону, %; п — количество незаменимых аминокислот.
Биологическую ценность (БЦ) пищевого белка (%) определяют по формуле
БЦ = 100-КРАС. (4.14)
Коэффициент утилитарности аминокислотного состава имеет практическое значение, так как возможность утилизации аминокислот организмом предопределена минимальным скором одной из них.
Коэффициент утилитарности у-й незаменимой аминокислоты (доли единицы) рассчитывают по формуле
<4.15)
где ф—скор j-й незаменимой аминокислоты по отношению к физиологически необходимой норме (эталону), %;
С = (Aj/A3J) • 100,
(4.16)
здесь А — содержание j-й незаменимой аминокислоты в продукте, г/100г белка; А — содержание j-й незаменимой аминокислоты, соответствующее физиологически необходимой норме (эталону), г/100 г белка.
356
Коэффициент утилитарности у-й незаменимой аминокислоты используют для расчета коэффициента утилитарности аминокислотного состава (С/), который является численной характеристикой, достаточно полно отражающей сбалансированность незаменимых аминокислот по отношению к эталону:
7 = 1
Меньшая возможность утилизации незаменимых аминокислот в составе белка пищевого продукта организмами наблюдается тогда, когда их скоры максимальны или наиболее близки к максимуму.
Общее количество незаменимых аминокислот в белке оцениваемого продукта, которое из-за взаимонесбалансированности по отношению к эталону не может быть утилизировано организмом, служит для оценки сбалансированности состава незаменимых аминокислот по показателю сопоставимой избыточности (г), который определяется по формуле
= (4.18) к ~ (-^у (-лтппДэу)- (4.19) 7 = 1 Результаты расчетов оформляют в виде таблицы следующей формы:
Аминокислота Содержание, мг Скор, % ' КРАС. % БЦ, % и
в стандартном белке в исследуемом образце
на 1 г на 1 г белка азота на 1 г на 1 г белка азота
Изолейцин 40 50 Лейцин 70 440 Лизин 55 340 Метионин + цис- 35 220 тин Фенилаланин + 60 380 + тирозин Треонин 40 250 Триптофан 10 60 Валин 50 310 Всего 360 2250
357
Студенты рассчитывают скор каждой из незаменимых амино кислот, при этом фиксируют, по каким аминокислотам биологическая ценность лимитирована, дают оценку средней величины избытка аминокислотного скора незаменимых аминокислот по сравнению с наименьшим уровнем скора конкретной аминокислоты; делают заключение о биологической ценности белковых продуктов.
Лабораторная работа № 3 ОПРЕДЕЛЕНИЕ ПЕРЕВАРИМОСТИ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Освоить ферментативный метод определения биологической ценности мяса и мясных продуктов in vitro.
Задачи. Определить переваримость белков мяса, мясных продуктов, других пищевых белковых систем пищеварительными ферментами; провести сравнительную оценку переваримости мяса исследуемых образцов.
Объекты исследования. Образцы мяса различных видов убойных животных и птицы аналогичных анатомических участков, субпродуктов I и 11 категорий, других вторичных продуктов убоя скота, препараты растительных белков, мясные продукты различных ассортиментных групп кулинарной готовности, комбинированные и другие белковые продукты.
Материалы, реактивы и оборудование. Раствор соляной кислоты молярной концентрацией 0,02 моль/дм3; пепсин кристаллический (активность не менее 2500 ед/мг белка); раствор гидроксида натрия молярной концентрацией 5 моль/дм , бикарбонатный буфер pH 8,2 —8,6; трипсин кристаллический (активность 8 ТЕ в 1 г препарата); тирозин; прибор для определения переваримости in vitro; спектрофотометр или фотоэлектроколориметр; охлажденный раствор вольфрамата натрия массовой долей 10 %; нож или мясорубка; часовое стекло; аналитические весы.
Методические указания. Основой метода является ферментативный гидролиз в условиях, при которых доступность атакуемых пептидных связей определяется не только свойствами белка, но и дополнительными факторами, связанными со структурой и химическим составом пищевого продукта.
Метод заключается в последовательном воздействии на белковые вещества исследуемого продукта системой протеиназ, состоящей из пепсина и трипсина, при непрерывном перемешивании и удалении из сферы реакции продуктов гидролиза диализом. Это позволяет избежать ингибирования пищеварительных ферментов низкомолекулярными пептидами и свободными аминокислотами.
358
Рис.4.15. Схема прибора для определения переваримости белков под действием пищеварительных ферментов (in vitro):
1 — меша Ич.I. 2 и 2 - - соо'1ве1ственно внешний и внутренний сосуды: 4-- ио.'1\проницаем .я мембрана
Гидролиз проводят в специальном приборе (рис. 4.15), состоящем из нескольких ячеек, каждая из которых имеет наружный и внутренний сосуды, разделенные полупроницаемой мембраной.
Подготовка проб. Образцы мясных про-
дуктов измельчают ножом на часовом стекле или мясорубке. На аналитических весах взвешивают навеску пробы продукта, исходя
из расчета содержания в ней около 150 мг белка.
Порядок проведения анализа. Пробу продукта помещают во внутренний сосуд прибора. Туда же вносят 15 см3 раствора соляной кислоты концентрацией 0,02 моль/дм3 (pH 1,2). В наружный сосуд в целях соблюдения изотонии вводят 60 см3 того же раствора. Внутренний сосуд вставляют в наружный так, чтобы нижняя поверхность его дна погружалась в раствор при условии равенства уровней жидкостей во внутреннем и наружном сосудах. Пробы инкубируют в термостате при 37 °C. После уравнивания температуры во всей системе во внутренний сосуд вносят 15 мг кристаллического пепсина. Концентрация фермента при этом равна 1 мг/см3, т. е. соответствует средней концентрации его в желудочном содержимом на высоте переваривания. Реакцию проводят при перемешивании жидкости мешалкой при частоте вращения 1 с-1. Через каждый час из сосудов отбирают пробы: из внутреннего 0,1 см3, из наружного 1 см3. После этого в сосуды вносят объем раствора соляной кислоты (0,02 моль/дм3), равный объему взятой пробы.
Для остановки протеолиза и осаждения непереваренного белка пробу из внутреннего сосуда разбавляют в 10 раз охлажденным раствором вольфрамата натрия массовой долей 10 %. После центрифугирования продукты гидролиза определяют методом Лоури (см. главу 1, лабораторную работу № 2).
Для проведения дальнейшего гидролиза продуктов пепсино
вого перевара трипсином жидкость из наружного сосуда заменяют равным объемом раствора NaHCO3 молярной концентрацией 0,02 моль/дм3 (pH 8,2). Пепсиновый перевар во внутреннем сосуде нейтрализуют при достаточно быстром перемешивании 0,4 см3 раствора NaOH (2 моль/дм3), после чего к нему прибавляют 15 см3 раствора NaHCO3 (0,02 моль/дм3). После уравнивания температуры во внутренний сосуд вносят 15 мг кристаллического трипсина.
Последующие процедуры проводят аналогично определению атакуемости белков пепсином.
359
Степень атакуемости белков в исследуемом продукте оценивают но нарастанию продуктов гидролиза в результате ферментативного переваривания. Значения концентрации тирозина, определенные но калибровочному графику (см. главу 3. лабораторную работу № 2). пересчитывают на обшиб объем жидкости наружного и внутреннего сосудов, а затем эти значения суммируют. Из концентрации тирозина, характеризующей степень гидролиза, вычитают показатели, полученные в контрольных опытах: первый опыт —раствор фермента, второй опыт —взвесь анализируемого продукта в буферном растворе.
Расчеты проводят по формуле
А'=/1 — 7?—С. (4.20)
где К — нарастание продуктов гидролиза вследствие действия протеолитического фермента, мкг/см3; А — концентрация продуктов гидролиза в переваре, мкг/с.м'; В — концентрация тех же продуктов во взвеси пищевого продукта, мкг/см’; С — концентрация тех же продуктов в растворе фермента, мкг/см3.
Накопление продуктов гидролиза, определяемое но цветной реакции Лоури, выражают в микрограммах тирозина на 1 г сухого вещества.
Результаты экспериментов и расчетов оформляют в виде таблицы:
Краткая характеристика продукта Накопление продуктов ферментативного гидролиза (мкг/см’) при длительности гидролиза, ч
пепсином трипсином
1 2 3 4 5 6
По полученным данным строят графики зависимости накопления продуктов ферментативного гидролиза от продолжительности гидролиза, самостоятельно формулируют заключение. При этом обращают внимание на влияние способов тепловой обработки на переваримость белков мясных продуктов.
Лабораторная работа № 4 ОПРЕДЕЛЕНИЕ КАЧЕСТВЕННЫХ ПОКАЗАТЕЛЕЙ ЖИВОТНЫХ ЖИРОВ
Цель работы. Освоить методы анализа качественных показателей пищевых животных жиров на основе химических, физико-химических и физических методов анализа.
360
Задачи. Ознакомиться с методами отбора проб пищевых животных жиров; провести органолептическое исследование: определить физико-химические показатели качества (массовая доля влаги, кислотное число) и сделать заключение о сорте исследуемого жира: оценить степень окислительной порчи жира с помощью различных методов, составить заключение о степени доброкачественности исследуемого жира.
Объекты исследования. Жиры и жиропродукты, полученные из сырья различными технологическими способами.
Методические указания. Качество жиров определяется по органолептическим, физико-химическим показателям, а также по так называемым стандартным числам.
Кислотное число — один из основных показателей качества жиров. В процессе производства этот показатель характеризует глубину гидролитического распада, а в процессе хранения — указывает на окислительную порчу жира наряду с другими более характерными показателями, а также является косвенным показателем соблюдения температурного режима при сборе и подготовке жира-сырца к вытопке.
Пищевые животные жиры, выпускаемые промышленностью, относятся к высшему, первому сортам и сборному. Сорт жира устанавливают в зависимости от органолептических показателей, массовой доли влаги и кислотного числа.
О степени окислительной порчи жира судят по перекисному числу, которое характеризуется количеством иода, выделяемого в кислой среде из иодида калия под действием пероксидов, содержащихся в 100 г жира.
Для определения содержания пероксидов обычно применяют различные варианты йодометрического метода, которые различаются количеством и соотношением применяемых растворителей и уксусной кислоты, способом введения иодида калия, продолжительностью взаимодействия с жиром до титрования раствором тиосульфата натрия, способом выражения перекисного числа. Метод основан на способности пероксидов в кислой среде окислять иодид калия с освобождением молекулярного иода, который затем определяют титрованием раствором тиосульфата натрия, используя в качестве индикатора крахмал. Химизм протекающих реакций приведен ниже:
KJ + СН3СООН -► HJ + СНзСООК;
СНз(СН2)6СН-СН = СН(СН2)7СООН + 2HJ -►
О-ОН
СН3(СН2)6 CH-CH = СН(СН2)7СООН + Н2О + J2;
ОН
J2 + 2Na2S2O3-*-2NaJ + Na2S4O6
361
Эксперимент желательно проводить так, чтобы после смешения всех реагентов получился гомогенный раствор и обеспечивалась одинаковая продолжительность реакции от момента введения иодида калия до титрования, так как количество иода увеличивается с течением времени.
Хотя многие гидропероксиды токсичны, в индукционном периоде количество их крайне незначительно. Такой жир пригоден в нишу. В зависимости от перекисного числа определяют степень свежести жира: жир с перекисным числом до 0,03 % иода считается свежим; от 0,03 до 0,06 % — непригоден для хранения, но его можно употреблять на пищевые цели. При значении перекисного числа от 0,06 до 0,1 % иода жир считается сомнительной свежести, а при перекисном числе более 0,1 % — испорченным и непригодным в пищу.
В результате окислительной порчи в жире накапливаются альдегиды, кетоны, низкомолекулярные жирные кислоты, оксикислоты и др. Многие из этих продуктов токсичны для человека.
Для наблюдения за окислением жиров при их получении, хранении и в процессе переработки следует правильно выбрать метод контроля. Наиболее универсальным, пригодным для всех случаев является определение содержания пероксидов. Однако положительная проба на пероксиды не всегда является доказательством того, что жир испорчен, так как пероксиды не имеют ни вкуса, ни запаха. Поэтому при исследовании более глубоких стадий окисления, приводящих к накоплению вторичных продуктов, нельзя ограничиваться определением какой-либо одной группы веществ, так как распад пероксидов идет по-разному, что зависит от жирнокислотного состава жира, температурных условий, наличия или отсутствия ингибиторов. Для количественной оценки содержания карбонильных соединений в жирах определяют тиобарбитуровое и бензидиновое числа. Оба метода достаточно чувствительны, но дают хорошие результаты только при анализе жиров с одинаковым составом жирных кислот. Для количественной оценки содержания в жирах полиоксикислот используют определение суммарного содержания продуктов окисления, нерастворимых в петро-лейном эфире.
В связи с изложенным для характеристики степени окисленно-сти жиров использовать только один какой-либо показатель недостаточно. Для этого помимо химических методов применяют современные инструментальные методы — ультрафиолетовую спектроскопию с максимумом поглощения в интервале длин волн 226—300 нм и инфракрасную спектроскопию — при частотах в области 984—988 см~‘.
При выполнении лабораторных работ или при анализе жиров в производственных лабораториях рекомендуется освоить и использовать несколько методов из приведенных ниже.
362
Органолептическим исследованием при окислительной порче обнаруживают признаки прогоркания или осваивания жира.
При прогоркании в жире наканливакнея альдегиды, кеюны. низкомолекулярные жирные кислоты, эфиры и др. Жир приобретает зеленоватый или желтый! цвет, резкий! неприятный запах и острый горький вкус.
Осаливание характеризуется образованием в жире оксикисло!. а также продуктов полимеризации и конденсации жирных кислот. Жир теряет свою естественную окраску, обесцвечивается, становится более плотным, приобретает мажущуюся консистенцию, неприятный салистый запах. Температуры плавления и застывания повышаются.
Степень свежести (доброкачественности) жиров устанавливают органолептическим исследованием, определением кислотного числа. качественной реакцией на свободные жирные кислоты, качественным и количественным определением пероксидов, альдегидов и кетонов, люминесцентным исследованием и другими методами. По степени свежести жиры делятся на свежие, свежие, не подлежащие хранению, сомнительной свежести и испорченные.
В работе предлагается изучить несколько различных способов определения качественных показателей жиров, каждый из которых может быть использован независимо или в совокупности для повышения объективности оценки.
Подготовка проб. Отбор проб проводят на глубине не менее 50 см от поверхности. От партии жира в брикетах, стаканчиках, банках и другой потребительской упаковке точечные пробы отбирают массой до 50 г после вскрытия или снятия упаковки. Точечные пробы, помещенные в чистую сухую банку, представляют собой объединенную пробу. Масса объединенной пробы должна быть не менее 600 г.
1. ОРГАНОЛЕПТИЧЕСКИЙ АНАЛИЗ
Материалы и оборудование. Шпатели металлические; предметное стекло; пробирки из бесцветного стекла с внутренним диаметром 13—17 мм и высотой 150 мм; баня водяная; термометр стеклянный технический с диапазоном измерения 0—100 °C.
Порядок проведения анализа. Запах и вкус определяют в средней пробе жира при температуре 20 °C. При определении вкуса пробы не проглатывают. Эти показатели должны быть характерными для данного вида жира, вытопленного из доброкачественного сырья. Для жиров высшего сорта посторонние запах и вкус не допускаются. Для жиров I сорта допускается приятный поджаристый запах и вкус. Сборные жиры могут обладать поджаристыми запахом и вкусом, а также запахом бульона и шквары.
363
К о н систем ц и ю определяют в обшей пробе надавливанием металлическим шпателем на жир при температуре 15—20 °C. Она должна быть независимо от сорта для говяжьего и бараньего жира плотной или твердой (для курдючного мазеобразной), для свиного и конского жира мазеобразной или плотной, для костного сборного жира жидкой, мазеобразной или плотной.
Цвет устанавливают при температуре 15—20 °C. Для этого жир наносят на предметное стекло (лучше на пластинку молочного стекла) толщиной около 5 мм. Исследование проводят в отраженном дневном рассеянном свете. Различают следующие цвета и оттенки испытуемого жира: например, желтый, светло-желтый, светло-желтый с зеленоватым оттенком и т. д.
При порче цвет жира приобретает темно-серые, желтые, коричневые, зеленоватые тона или же обесцвечивается. Характерным признаком порчи жира являются неравномерность, пестрота окраски, он становится мутным, с затхлым, кислым, прогорклым и сальным запахом, горьким вкусом, мажущейся консистенцией.
Для определения прозрачности в пробирку помещают жир с таким расчетом, чтобы, будучи расплавленным, жир заполнил не менее половины пробирки. Затем пробирки с жиром помещают на водяную баню для расплавления жира. Расплавленный жир температурой 60—70 °C рассматривают в дневном рассеянном проходящем свете. При наличии в жире пузырьков воздуха пробирке дают постоять при вышеуказанной температуре в течение 2—3 мин, после чего определяют прозрачность.
Жиры высшего и I сортов должны быть прозрачными. Для сборного жира допускается мутноватость.
2. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ВЛАГИ
Оборудование. Бюксы стеклянные или металлические; эксикатор; шкаф лабораторный сушильный; весы лабораторные.
Порядок проведения анализа. Бюкс высушивают в сушильном шкафу при температуре 102—105 °C в течение 30 мин, охлаждают в эксикаторе и взвешивают с погрешностью не более 0,0002 г. Вносят 2—3 г исследуемого жира, взвешивают и сушат в сушильном шкафу при такой же температуре до постоянной массы.
При исследовании жира, взятого сразу же после вытопки, первое взвешивание проводят после высушивания в течение 1 ч, последующие — через каждые 30 мин. Если исследуемый жир находился на хранении, первое взвешивание проводят после высушивания в течение 30 мин, последующие — через 15 мин. Постоянная масса считается достигнутой, если ее уменьшение при двух последних взвешиваниях не превышает 0,0002 г. Если после очередного взвешивания будет установлено увеличение массы, то для расчета берут наименьшую массу бюкса с жиром.
364
Массовую долю влаги (%) определяют по формуле
X = |(/л, — /щ) • 100|///z.
(4.21)
где т}. т, — массы бюксов с жиром соответственно до и после высушивания. 1. /// — масса навески исследуемого жира. г.
Разница между результатами параллельных определении не должна превышать 0,05 %.
3. ОПРЕДЕЛЕНИЕ КИСЛОТНОГО ЧИСЛА
Материалы, реактивы и оборудование. Колбы конические вместимостью 150—200 см3; весы лабораторные; водяная баня; нейтрализованная эфирно-спиртовая смесь в соотношении 2 : 1 (одну часть этанола смешивают с двумя частями этилового эфира и нейтрализуют раствором гидроксида калия или натрия молярной концентрацией 0,1 моль/дм3 до бледно-розовой окраски по фенолфталеину); спиртовой раствор фенолфталеина массовой долей 1 %; раствор гидроксида калия или натрия молярной концентрацией 0,1 моль/дм3.
Порядок проведения анализа. В конической колбе вместимостью 150—200 см3 взвешивают 3—5 г исследуемого жира с погрешностью не более 0,001 г. Жир расплавляют на водяной бане, приливают 50 см3 нейтрализованной эфирно-спиртовой смеси (ее объем не менее чем в 10 раз должен превышать навеску жира) и взбалтывают. Добавляют 3—5 капель спиртового раствора фенолфталеина массовой долей 1 %. Полученный раствор при постоянном встряхивании быстро титруют раствором гидроксида калия молярной концентрацией 0,1 моль/дм3 до появления отчетливой розовой окраски, не исчезающей в течение 1 мин. Если при титровании жидкость мутнеет, то в колбу добавляют 5—10 см3 эфирно-спиртовой смеси и взбалтывают до исчезновения мути или же колбу с содержимым слегка нагревают на водяной бане, затем охлаждают до комнатной температуры и заканчивают титрование.
Кислотное число (мг КОН) вычисляют по формуле
Х2 = (Е^-5,61)/ш,
(4.22)
где V— объем раствора гидроксида калия или натрия молярной концентрацией 0,1 моль/дм3, израсходованный на титрование, см3; К— поправка к раствору щелочи для пересчета на точный (0,1 моль/дм3) раствор; 5,61 — количество миллиграммов гидроксида калия, содержащегося в 1см3 (0,1 моль/дм3) раствора; т — масса навески жира, г.
Расхождение между результатами двух параллельных определений не должно превышать 0,1.
365
Экспериментальные данные органолептической и физико-химической опенки качества нишевых животных жиров оформляют в виде таблицы:
Пока спель
i lopM.i лля сор 1 а жира данною вила
ныа/кто
uepiioi о
Значения ноказа-ic/icii. хетаноь-ленных экспериментальным путем
1
Цвет при температуре 15- 20 С
Запах и вкус
Прозрачность в расплавленном состоянии
Консистенция при 15—20 °C Массовая доля влаги. % Кислотное число, мг КОН
4. ОПРЕДЕЛЕНИЕ СТЕПЕНИ ОКИСЛИТЕЛЬНОЙ ПОРЧИ ЖИРА
Материалы, реактивы и оборудование. Фарфоровые ступки с пестиком; стеклянные палочки; пробирки химические; водяная баня; пробки резиновые; колбы^ конические с притертыми пробками вместимостью 200—250 см3; часы песочные на 3 мин или секундомер; мерная колба вместимостью 25 см3; фильтры бумажные; воронки делительные вместимостью 100 и 150 см3; цилиндры мерные; концентрированная соляная кислота плотностью 1,19 г/см3 и ее 10%-ный раствор; раствор флороглюцина в эфире массовой долей 1 %; раствор флороглюцина в ацетоне массовой долей 1 %; концентрированная серная кислота; насыщенный раствор резорцина в бензоле; смесь изооктана (СН3)3СС4Н9 и абсолютного свободного от альдегидов этанола в соотношении 1:1; свежеприготовленный раствор бензидина Cj2H12N2 массовой долей 0,5 %; бутанол; сульфат натрия; спиртовой раствор гидроксида калия молярной концентрацией 2 моль/дм3; метиловый оранжевый; петро-лейный эфир; этанол; смесь этанола с хлороформом в соотношении 1:1; гексан; свежая кровь убойных животных; спиртовой раствор гваяковой смолы; воздушный холодильник; раствор нейтрального красного массовой долей 0,01 %; хлороформ; ледяная уксусная кислота; насыщенный свежеприготовленный раствор иодида калия; раствор крахмала массовой долей 1 %; раствор гипосульфита натрия молярной концентрацией 0,01 моль/дм , флуорископ; весы технические и аналитические; фотоэлектроколориметр; pH-метр; спектрофотометр; сушильный шкаф.
Приготовление реактивов. Раствор бензидина С12Н.2N2 массовой долей 0,5 %: растворяют навеску бензидина в смеси (I: I по объему) абсолютного этанола и ледяной уксусной кислоты.
366
4.1. РЕАКЦИЯ С НЕЙТРАЛЬНЫМ КРАСНЫМ
В фарфоровую ступку помешают 0.5—1 г исследуемого жира, заливают раствором нейтрального красного массовой долей 0.01 %, растирают пестиком в течение 1 мин. Раствор нейтрального красного сливают, а его остатки смывают водопроводной водой. Оценивают окраску жира.
Степень окислительной порчи жира определяют по табл. 4.10, полученные результаты фиксируют в тетради.
Т а б а и и а 4.10
Показатели свежести жиров по реакции с нейтральным красным
Показатели свежести Окраска жира
свиного и бараньего говяжьего
Свежий Свежий, но нс подлежащий хранению Сомнительной свежести Испорченный От желтой с зеленоватым оттенком до желтой От темно-желтой до коричневой От коричневой до розовой От розовой до красной От желтой до коричневой От коричневой до коричнево-розовой От коричнево-розовой до розовой От розовой до красной
4.2. КАЧЕСТВЕННАЯ РЕАКЦИЯ НА ПЕРОКСИДЫ (ПО ВИНТИЛЕСКУ И ПОПЕСКУ)
В пробирку помешают около 5 г жира, расплавляют его на водяной бане, добавляют 5 капель свежей крови, 5—10 капель спиртового раствора гваяковой смолы и 5 см3 дистиллированной воды. Пробирку закрывают пробкой и встряхивают. При наличии пероксиды расщепляются с освобождением атомарного кислорода, который окисляет гваяковую смолу. В результате появляется голубая окраска, интенсивность которой зависит от количества пероксидов.
4.3. ОПРЕДЕЛЕНИЕ ПЕРЕКИСНОГО ЧИСЛА
В коническую колбу с притертой пробкой вносят около 0,8 г жира, взвешенного с точностью не более 0,0002 г, расплавляют на водяной бане и по стенке колбы, смывая следы жира, приливают по 10 см3 хлороформа и ледяной уксусной кислоты. Быстро добавляют 0,5 см3 насыщенного свежеприготовленного раствора иодида калия. Закрывают колбу пробкой, смешивают содержимое колбы вращательными движениями и ставят в темное место на 3 мин.
367
Затем в колбу вливают 100 см3 дистиллированной воды, в которую заранее добавляют I см3 раствора крахмала массовой долей 1 сс. Титруют раствором гипосульфита натрия молярной концентрацией 0.01 моль/дм? до исчезновения синей окраски.
Для проверки чистоты реактивов проводят контрольное определение (без жира). Реактивы считают пригодными для анализа, если на контрольное определение пошло не более 0.07 см3 раствора гипосульфита натрия молярной концентрацией 0,01 моль/дм3.
Перекисное число (%JJ вычисляют по формуле
Х, = |(Р- Vj)K-0.00127- 100|/ш, (4.23)
где И — объем раствора тиосульфата натрия молярной концентрацией 0,01 моль/дм3, израсходованный на титрование при проведении основного опыта с навеской жира, см3; И] —объем (0.01 моль/дм3) раствора тиосульфата натрия, израсходованный на титрование при проведении контрольного опыта (без жира), см3; К— коэффициент поправки к раствору тиосульфата натрия для пересчета на точный (0,01 моль/дм3) раствор; 0.00127 — количество граммов иода, эквивалентное 1 см3 (0,01 моль/дм3) раствора тиосульфата натрия: т — масса навески исследуемого жира, г.
Перекисное число (в миллиэквивалентах активного кислорода на 1 кг жира) вычисляют по формуле
Д2 = [(К- ^)N- 1000]/™,
(4.24)
где Л'—концентрация раствора тиосульфата натрия, г/дм3; 1000 — коэффициент перевода в килограммы.
Разница между результатами параллельных определений не должна превышать 0,005.
Степень окислительной порчи жира в зависимости от перекисного числа определяют по табл. 4.11.
Таблица 4.11
Степень окислительной порчи жира в зависимости от перекисного числа
Перекисное число
Процент иода Л/кв активного кислорода на 1 кг жира Степень окислительной порчи
До 0,03 До 1,05 Свежий
От 0,03 до 0,06 От 1,05 до 2,10 Свежий, не подлежащий хранению
От 0,06 до 0,1 От 2,10 до 3,00 Сомнительной свежести
Более 0,10 Более 3,00 Испорченный
368
4 4 КАЧЕСТВЕННЫЕ РЕАКЦИИ НА АЛЬДЕГИДЕ!
Метол основан на способности альлешлов в кислой среде i?cг\ пять в реакцию конденсации с многоатомными фенолами (флороглюцином. резорцином и др.). образуя окрашенные соединения.
Реакция с ф.юроглюиином в эфире (по Крейеу). В пробирку номе щают 3—5 1 жира, расплавляют его на водяной бане, добавляю'! такие же обьемы концентрированной, соляной кислоты плотностью 1.19 г/см 1 и раствора флороглюцина в эфире массовой долей 1 %. Пробирку- закрывают резиновой пробкой и энергично встря хивают. При наличии альдегидов нижний! слой в пробирке окрашивается в красный цвет'.
Реакция е флороглюцином в ацетоне (по Видману). В пробирке расплавляют 3—5 г жира, добавляют к нему такой! же объем раствора флороглюцина в ацетоне массовой долей 1 / и 2—3 капли концентрированной серной кислоты, закрывают резиновой пробкой и встряхивают. При наличии альдегидов нижний слот! содержимого it пробирке окрашивается в красный цвет
Реакция е резорцином в бензоле (по Видману). К 3—5 г расплавлен ного it пробирке жира добавляют такие же объемы концентрированной соляной кислоты и насыщенного раствора резорцина в бензоле. Пробирку закрывают резиновой пробкой и встряхивают. При наличии альдегидов появляется красно-фиолетовое окрашивание.
4.5. ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ
Работу выполняют в темном помещении. В пробирке из бесцветного стекла расплавленный жир помещают под углом 45° в поток ультрафиолетовых лучей флуорископа. Жир доброкачественный флуоресцирует серо-желтым, сомнительной свежести — слабо-розовым или голубым, испорченный — красно-фиолетовым или фиолетовым цветом.
Шпик можно исследовать без предварительной вытопки. Свежий шпик флуоресцирует чисто-белым цветом, а соединительнотканные прослойки — ярко-фиолетовым. Шпик подозрительной свежести проявляет тусклое розово-фиолетовое или красно-фиолетовое свечение, недоброкачественный — тусклое коричнево-фиолетовое.
4.6. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ (АЛЬДЕГИДОВ)
На аналитических весах взвешивают около 1 г жира в мерную колбу вместимостью 25 см3 и доводят до метки смесью изооктана (СН3)3СС4Н9 и абсолютного, свободного от альдегидов, этанола в соотношении 1:1.
369
Если раствор жира окажется слегка мутным, его отфильтровывают через бумажный фильтр. Затем в кювете фотоэлектроколориметра с толщиной слоя 2 см определяют пропускание раствора в процентах по отношению к смеси растворителей при л = 350 нм (светофильтр № 1). Полученное значение показывает пропускание раствора жира. Затем в две пробирки отбирают пипеткой по 5 см3 в одну — раствор жира, в другую — смесь растворителей. В каждую пробирку добавляют по 0,5 см3 свежеприготовленного раствора бензидина C]2H]2N2 массовой долей 0,5 %, содержимое встряхивают, выдерживают 15 мин и определяют пропускание раствора жира в процентах по отношению к смеси растворителей, обработанной бензидином. Полученное значение включает суммарное пропускание самого жира и содержащихся в нем альдегидов.
Показания снимают несколько раз и берут среднее арифметическое значение в процентах.
Содержание альдегидов (мг% коричного альдегида) рассчитывают по формуле
Х= [(£>! -1,1 - D) • 0.039 • 5 • 100]/2/н,
(4.25)
где D, D. — оптическая плотность раствора жира соответственно до и после обработки бензидином; 1,1 — коэффициент, выражающий отношение общего объема взятых растворов (5 см3 раствора жира + 0.5 см3 раствора бензидина) к 1 см’ раствора жира; 0.039 — фактор для коричного альдегида — бензидина при Х = 350 нм: 5 — объем раствора жира, взятый для обработки бензидином, см3; 2 -толщина слоя жира, см; т — масса исследуемого жира. г.
4.7. ОПРЕДЕЛЕНИЕ СТЕПЕНИ ОКИСЛЕННОСТИ ЖИРОВ С ФЛОРОГЛЮЦИНОМ (ПО КРЕЙСУ—ЛУРИ)
Навеску жира массой 5—10 г взвешивают в химическом стаканчике на технических весах, переносят в делительную воронку вместимостью 100 см3 и по разнице определяют массу взятой пробы.
В воронку приливают 5 см3 концентрированной НО, содержимое встряхивают в течение 5 мин, добавляют 5 см3 эфирного раствора флороглюцина массовой долей 1 % и оставляют на 5 мин. Затем вновь энергично встряхивают и дают отстояться до полного расслоения двух фаз: интенсивно окрашенного кислого слоя и жировой фракции (может быть тоже слегка окрашена). Окрашенный нижний кислый слой осторожно сливают в другую делительную воронку (вместимостью 100—150 см3), добавляют 5 см3 бутанола, встряхивают, приливают 25 см3 дистиллированной воды и все тщательно перемешивают. В результате происходит расслоение на нижний водный слой, обычно не окрашенный или слабо-окрашенный, и верхний бутаноловый слой с растворенным окрашенным комплексом.
370
Водную фазу сливают, а оставшийся бутаноловый слой начинают промывать водой, предварительно добавляя каждый раз но 5 см3 бутанола. Промывают порциями воды по 25 см3 пять-восемь раз до доведения pH промывной воды до 5,0 по рН-мет-ру. Бутаноловая фракция содержит небольшие капельки воды в виде эмульсии. Для ее осушения добавляют 0,1—0,3 г сульфата натрия до получения прозрачного окрашенного слоя. Светлый слой раствора сульфата натрия сливают, а оставшийся слой бутаноловой фракции переносят в мерный цилиндр и замеряют ее объем. Спекгр поглощения снимают на спектрофотометре в кювете толщиной 1 см при длине волны 548 нм.
Удельное поглощение вычисляют по формуле
£ = DV/(m- 100), (4.26)
где D — оптическая плотность; V — объем бутаноловой фракции; т — масса навески, г.
4.8. ОПРЕДЕЛЕНИЕ СУММАРНОГО СОДЕРЖАНИЯ ПРОДУКТОВ ОКИСЛЕНИЯ, НЕРАСТВОРИМЫХ В ПЕТРОЛЕЙНОМ ЭФИРЕ
На аналитических весах в колбу взвешивают около 5 г исследуемого жира, добавляют 20 см3 спиртового раствора гидроксида калия молярной концентрацией 2 моль/дм3, присоединяют воздушный холодильник и омыляют на водяной бане в течение 1 ч. Затем холодильник отсоединяют, а колбу погружают на водяную баню и отгоняют спирт до полного исчезновения запаха.
Мыло растворяют в 30 см3 горячей дистиллированной воды, добавляют метиловый оранжевый и разлагают при встряхивании горячим раствором соляной кислоты объемной долей 10 %, который прибавляют в избытке.
После охлаждения в колбу приливают медленной струей 50 см3 петролейного эфира для извлечения нормальных жирных кислот и перемешивают круговым движением. При этом оксикислоты частично осаждаются на стенках. Колбу оставляют в покое на 12—14 ч. Из раствора выделяются нерастворимые в петролейном эфире продукты окисления жирных кислот, которые накапливаются на границе раздела эфирного раствора и водного кислого слоя.
Содержимое колбы переносят в делительную воронку и отстаивают в течение 20—40 мин до полного разделения слоев. Кислый водный слой спускают из воронки, а петролейный экстракт фильтруют через бумажный фильтр. Делительную воронку и колбу тщательно промывают петролейным эфиром до полного удаления нормальных жирных кислот, пропуская эфирный раствор через тот же фильтр. Затем так же тщательно промывают и фильтр. О полноте промывания судят по отсутствию жирного пятна на фильтровальной бумаге.
371
Нерастворимые в петролейном эфире продукты окисления остаются на стенках делительной воронки, в колбе и на фильтре. Их растворяют горячим этанолом, вливая его в делительную воронку и колбу. Затем, пропуская через тот же фильтр, собирают фильтрат в заранее взвешенную колбу. Если кислоты плохо растворяются в этаноле, то применяют смесь этанола с хлороформом в соотношении 1:1. Растворитель отгоняют, а остаток сушат при 100 °C до постоянной массы. Взвешивание производят через каждые 30 мин.
Массовую долю продуктов окисления (%) вычисляют по формуле
Х=(а- 100)//д, (4.27)
где а — масса продуктов окисления, г; ш — масса исследуемого жира, г.
4.9. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ПЕРВИЧНЫХ И ВТОРИЧНЫХ ПРОДУКТОВ ОКИСЛЕНИЯ ПО УФ-СПЕКТРАМ
Электронные спектры широко используются в химии липидов для качественного и количественного анализов.
Максимум поглощения для некоторых представителей липидов в ультрафиолетовой и видимой областях приведен в табл. 4.12.
Таблица 4.12
Максимум поглощения для некоторых соединений в ультрафиолетовой и видимой областях спектра
Вид спектра Соединения Максимум поглощения, нм
Ультрафиолетовый Первичные продукты окисления Вторичные продукты окисления Полиненасыщенные кислоты Токоферолы Госсипол 232 250-300 230-233; 268-269 256-298 237; 290; 366
Видимый Каротиноиды а-Каротин [3-Каротин 400-500 477 483
Для количественного измерения обычно пользуются законом Бугера—Ламберта—Бэра, который указывает на прямую пропорциональность между концентрацией и оптической плотностью раствора. Концентрация растворенного вещества (г/дм3 или моль/дм3) в непоглощающем свет растворителе
C=D/(Kd),
(4.28)
где D — оптическая плотность анализируемого раствора; К— коэффициент поглощения растворенного вещества при длине волны, выбранной для анализа; d— толщина слоя, см.
372
При проведении анализа раствора неизвестной концентрации следует определить коэффициент поглощения чистого вещества при выбранных для анализа длинах волн.
Иногда используют величину Е, характеризующую оптическую плотность раствора, концентрация которого выражена числом граммов в 100 см3 раствора в отличие от D. где концентрация выражена в г/дм3. Тогда оптическая плотность раствора массовой долей 1 % в слое толщиной 1 см
Е.1% = 10Е.
1см
(4.29)
Отношение величины светового потока /, прошедшего через среду, к величине падающего на нее потока /0 называют пропусканием’.
Ul.= T,
(4.30)
где 7—пропускание, доли единицы.
Между оптической плотностью и величиной пропускания существует зависимость:
D=W/T).
(4.31)
Пробу исследуемого жира массой около (1,0 ± 0,0002) г растворяют в мерной колбе вместимостью 100 см3 в гексане, свободном от ароматических веществ, и доводят до метки. Полученный раствор концентрацией около 1 г/дм3 анализируют на спектрофотометре и снимают спектр жира.
Идентификацию жира проводят следующим образом: появление полосы поглощения при длине волны 232 нм указывает на присутствие первичных продуктов окисления; появление полосы поглощения при длине волны 272 нм — на присутствие карбонильных соединений (вторичных продуктов окисления).
Результаты определения окислительной порчи жира оформляют в виде таблицы:
Показатели, характеризующие степень окислительной порчи жира
Реакция с нейтральным красным
Значение показателей, установленных экспериментальным путем, или эффект качественных реакций
Вывод
Свежий; свежий, не подлежащий хранению; сомнительной свежести; испорченный
Качественная реакция на пероксиды (по Винтилеску и Попеску)
Наличие или отсутствие пероксидов
373
Продо.ю/сение
Пок шrefill. .\jp.iKiepn jyioiiiiic степень
окислительно!! порчи жире
Значение показа- । 1 слеп, установлен- ! пых акспсримен- ( тальным пхтем. или эффект качествен- ' ны.\ реакции ।
Вывод
Перекисное число. сс J,
Свежий: свежий, не подлежащий храпению: сомнительной свежести: испорченный
Качественные реакции на альдегиды: реакция с флороглюцином в эфире (по Крейсу) реакция с флороглюцином в ацетоне (по Видману) реакция с резорцином в бензоле (по Видману)
Люминесцентный анализ
Наличие или отсутст вне альдегидов
Жир доброкачественный; сомнительной свежести; испорченный
Содержание карбонильных соединений (альдегидов), мг% коричного альдегида
Степень окисленности жиров с флороглюцином (по Крейсу—Лури) Суммарное содержание продуктов окисления, нерастворимых в петролейном эфире, %
На основании анализа органолептических, физико-химических показателей, полученных экспериментально, а также показателей окислительной порчи жира студенты самостоятельно делают выводы о доброкачественности жиров.
Лабораторная работа № 5 ОПРЕДЕЛЕНИЕ БИОЛОГИЧЕСКОЙ ЦЕННОСТИ ЖИВОТНЫХ ЖИРОВ
Цель работы. Освоить методы практического определения биологической ценности пищевых животных жиров.
Задачи. Определить содержание полиненасыщенных (эссенциальных) жирных кислот, количественно оценить содержание жирных кислот с сопряженными двойными связями (ди-, три-, тетра-
374
и пентаеновых). количественно определить жирорастворимые витамины (каротиноиды, токоферолы) и провести сравнительную оценку биологической ценности различных видов нишевых животных жиров.
Объекты исследования. Образны пищевых животных жиров различных видов (говяжий, бараний, свиной, конский, костный и сборный).
Методические указания. Для изучения различных характеристик жиров, контроля их качества в процессе технологической обработки и хранения, а также оценки биологической ценности используют ряд физических методов.
Полиненасыщенные жирные кислоты можно определять спектрофотометрическим методом в ультрафиолетовой области спектра. Метод качественного и количественного определения полиненасыщенных жирных кислот в различных жировых продуктах получил широкое распространение в связи с достаточной экспрессностью и высокой специфичностью.
Ненасыщенные жирные кислоты обладают различной способностью поглощать световую энергию в области спектра ниже 200 нм. Но эта область спектра (область вакуумного ультрафиолета) недоступна для широкого круга исследований, поэтому различия в светопоглошении кислот нельзя использовать для аналитических целей. Однако сопряженные системы из двух, трех и более двойных связей поглощают свет в ультрафиолетовой области при длине волны более 220 нм (область, легкодоступная для измерения). Изомеризация нагреванием в щелочной среде приводит к возникновению сопряженных систем.
На рис. 4.16 показаны спектры поглощения изомеризованных полиненасыщенных жирных кислот с характерными полосами поглощения для систем из двух, трех и четырех двойных связей.
Рис. 4.16. Кривые поглощения чистых метиловых эфиров полиненасыщенных кислот после щелочной изомеризации при 180 °C:
1 — линолевой; 2— линоленовой; 3 — арахидоновой
375
Анали j является эмпирическим, то 1 ребуе г особого внимания к условиям его проведения. Должны строго соблюдаться продолжительность изомери зации.дозировка и копнен грация применяемых реаген'1 ов и i. л.
Визуальный люминесненгныи анализ является экспрессным, но недостаточно обьсктивным. Применяется в основном при анализе биологически активных веществ, например витаминов. Флуоримегрическис методы довольно длительны и позволяют анализировать продукты, только разделяя их на составляющие. При выделении анализируемых витаминов в свободном виде с помощью кислот, щелочей или ферментативного гидролиза, омыления, актоклавирования и других способов можез происходить частичное разрушение анализируемого вещества, которое влияем на конечные результаты флуориметрического анализа.
Спектральные люминесцентные методы анализа пищевых продуктов обладают явными преимуществами по сравнению с визуальными люминесцентными и флуориметрическими методами. Они в достаточной степени объективны, что выгодно отличает их от визуальных люминесцентных и других органолептических методов анализа пищевых продуктов.
Отсутствие контакта с анализируемым объектом, практическая безынерционность светового луча как возбуждающего света, так и света люминесценции, относительная простота измерительного устройства, возможность автоматизации измерений, высокая чувствительность, точность и специфичность — весь этот комплекс характеристик спектральных люминесцентных методов анализа создаст возможность использования их в самых разнообразных направлениях, связанных с экспресс-контролем качества и количественного определения биологически активных веществ в пищевых продуктах.
Линолевая, линоленовая и арахидоновая кислоты люминесии-руют в виде маслянистых растворов.
Характерной особенностью линолевой кислоты является наличие на ее спектре возбуждения люминесценции наиболее интенсивного максимума при X = 325 нм. Для линоленовой и особенно арахидоновой кислот большей интенсивностью обладает максимум при ?v= 355 hm. Сравнение спектров возбуждения люминесценции полиненасыщенных жирных кислот со спектрами их поглощения позволяет установить между ними определенную связь. Все максимумы, наблюдающиеся на спектрах возбуждения люминесценции полиненасыщенных жирных кислот, сдвинуты в длинноволновую область по сравнению с максимумами поглощения, которые находятся при длинах волн меньше 210 нм.
Таким образом, при непосредственном возбуждении люминесценции маслянистых растворов полиненасыщенных жирных кислот (но не их разбавленных растворов) максимальное возбуждение, т. е. максимальное активное поглощение, наблюдается в той 376
области спектра, в которой происходи! незначи1сльное йен донн -ние молекулами этих кислот.
По характеру спектра люминесценции можно сулить о наличии или отсутствии жирных кислот в пищевых продуктах. Особенно информативным в этом отношении является спектр линоленовой кислоты, на котором кроме полосы при /. = 400 нм. присущей всем трем неокисленным полиненасышенны.м кислотам, обычно наблюдается дополнительная коротковолновая полоса, расположенная вблизи л = 390 нм.
Своеобразие спектра поляризации арахидоновой кислоты, на котором выявляется как область положительной поляризации (имеющаяся у двух других кислот), так и область отрицательной поляризации (отсутствующая на соответствующих спектрах линолевой и линоленовой кислот), может явиться дополнительным аргументом при решении вопроса об идентификации этой кислоты.
Для идентификации жирных кислот очень важно определить ход температурной зависимости интенсивности люминесценции, на котором температурной области плавления каждой кислоты соответствует резкое изменение интенсивности люминесценции.
Данная лабораторная работа предусматривает использование физических методов в анализе биологической ценности жиров по наличию ненасыщенных жирных кислот и витаминных примесей.
1. АНАЛИЗ НЕНАСЫЩЕННЫХ ЖИРНЫХ КИСЛОТ СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
1.1. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПОЛИНЕНАСЫЩЕННЫХ ЖИРНЫХ КИСЛОТ
Материалы, реактивы и оборудование. Этиленгликоль, гидроксид калия в таблетках (массовая доля основного вещества 85 %); метанол или этанол; аналитические весы; ультратермостат; изолированная баня; мешалка; стеклянные стаканы; круглодонные колбы из молибденового стекла или стекла пирекс; стеклянные пробирки; мерные колбы вместимостью ЮООсм3; спектрофотометр.
Подготовка проб. Жиры предварительно подвергают изомеризации. Для этого готовят раствор из 8 г гидроксида калия в 100 см3 этиленгликоля. Если массовая доля основного вещества в гидроксиде калия ниже 85 % (но не менее 81 %), то навеску соответственно увеличивают. Сначала этиленгликоль сушат, для чего навеску помещают в круглодонную колбу из стекла пирекс (или молибденового) вместимостью 150—200 см3 и погружают в нагретый до 100 °C ультратермостат (или баню), повышают температуру до 190 °C и выдерживают при этой температуре в течение 10 мин, а затем снижают температуру.
377
По достижении 120 °C к эгилсш ликолю в колбе осторожно ио частям прибавляют гидроксид калия, встряхивают до растворения щелочи и снова помешают в юрмостат. поднимают температуру до 190 °C и выдерживают при этой температуре 10 мин. Затем колбу вынимают, охлаждаю! и при необходимости хранят раствор в холодильнике при 4 °C не более суток.
В приготовленном растворе контролируют массовую долю КОН.
Для этого нейтрализуют 90 см? метанола по фенолфталеину. К нейтрализованному спирту прибавляют Юг раствора КОН в этиленгликоле, перемешивают и титруют раствором соляной кислоты молярной концентрацией 1 моль/дм3 до исчезновения розовой окраски. Массовую долю гидроксида калия рассчитывают по формуле
У = (УЬ- 5,61>, (4.32)
где Г'—объем раствора соляной кислоты молярной концентрацией 1 моль/дм'. см'; /? — поправка; т — масса раствора, г
Если массовая доля КОН будет выше 6,6 %, то добавляют этиленгликоль, высушенный нагреванием при 190 °C, если ниже 6,5 %, то готовят новый раствор.
Исследуемые образцы изомеризуют при (180 ± 0,5) °C в растворе гидроксида калия в этиленгликоле массовой долей 6,5—6,6 %.
Для доведения температуры до (180 ± 0,5) °C используют ультратермостат или изолированную баню, снабженную автоматическим устройством с контактным термометром и электрической или ручной мешалкой. Ультратермостат или баню наполняют минеральным маслом или силиконовой жидкостью, температура вспышки которой не ниже 300 °C.
Для проведения щелочной изомеризации в стаканчики из стекла пирекс (или молибденового стекла) размером 14 х 10 мм взвешивают с точностью до четвертого знака на аналитических весах 0,1 г исследуемого образца. В пробирки с притертыми пробками размером 20 х 150 мм взвешивают на технических весах 11 г раствора гидроксида калия в этиленгликоле, устанавливают в штатив и погружают в жидкость ультратермостата (или бани), нагретую до (180 ±0,5) °C так, чтобы глубина погружения пробирки в жидкость составляла 11,5 см.
Пробирки с содержимым нагревают в течение 20 мин, затем в них переносят содержимое стаканчиков с навесками жира. Две пробирки оставляют в качестве контрольных. Вынимают каждую пробирку из бани, энергично перемешивают в течение 5 с и снова ставят в баню. К концу первой минуты нагревания пробирки вынимают из бани и проверяют раствор. Если раствор прозрачен, то пробирки вновь ставят в баню. Если раствор не прозрачен, что указывает на неполное омыление, пробирки встряхивают 2—3 раза и снова помещают в баню. За 1 мин нагревания встря-378
хивание повторяют до тех пор. пока омыление не пройде г поднос тью и раствор не станет прозрачным.
Через 25 мин нагревания при (I SO ± 0.5) (\ считая с момента опускания образца в щелочной раствор этиленгликоля, пробирки вынимают и быстро охлаждают, помещая их в сосуд с холодной водой. Затем содержимое пробирок количественно переносят в мерные колбы вместимостью 100 см? с притертыми пробками, пользуясь метанолом или этанолом, желательно абсолютным, объем доводят до метки и используют прозрачные растворы для измерения оптической плотности.
Порядок проведения анализа. Спектрофотометрические измерения проводят на спектрофотометре в отношении контрольного раствора при следующих длинах волн: 233, 262, 268, 274, 308, 315 и 322 нм. Отсчет снимают в единицах оптической плотности или процентах пропускания. Желательно, чтобы оптическая плотность измеряемых растворов находилась в пределах 0,2—0,8. Этого добиваются либо разведением, либо используют кюветы подходящей длины, либо одновременно оба приема.
При исследовании некоторых животных жиров на спектро фотометре с использованием кюветы с толщиной светопоглощающего слоя 1 см следует применять разведения, приведенные в табл. 4.13.
Таблица 4.13
Рекомендуемые разведения при определении полиненасыщенных жирных кислот в животных жирах спектрофотометрическим методом
Длина волны, при которой производится измерение, нм Разведение (отношение исходного раствора к спирту') Концентрация.г/дм;
Свиной жир
233 1:15 0,0625
262. 268, 274 3 : 7 0,3
308, 315, 322 Без разведения (исходный раствор) Говяжий, бараний и костный жиры 1,0
233 1 : 7 0,125
262. 268. 274 1 : 1 0.5
308. 315, 322 Без разведения (исходный раствор) 1,0
Во всех случаях, когда применяют разведение, обязательно точно так же разводят и контрольный раствор.
Далее рассчитывают коэффициент удельного поглощения образца при каждой длине волны.
379
Коэффициент удельного поглощения образца рассчитывают но формуле
К= D/(cl).
(4.33)
где D — измеренная величина оптической плотности: с — концентрация образца в окончательно разведенном растворе. г/д.м;: / — толщина свстопоглошающсто слоя кюветы, см.
При расчете используют поправки. В коэффициенте удельного поглощения при Л = 233 нм учитывают постоянную поправку 0,03, зависящую от поглощения карбоксильных групп. Коэффициенты удельного поглощения при длинах волн 268 и 315 нм учитывают поправку на абсорбцию фона. Однако в тех случаях, когда какая-либо полиненасыщенная кислота присутствует в сравнительно больших количествах и после изомеризации дает оптическую плотность > 1, поправку на абсорбцию фона во избежание ошибки делать не рекомендуется, и в этих случаях у = лж и К, = к]15.
Пример. Рассчитать содержание полиненасыщенных жирных кислот в свином топленом жире.
Обозначения, принятые в расчете:
К\, К2 и Х3 — коэффициенты удельного поглощения соответственно линолевой, линоленовой и арахидоновой кислот;
/ — толщина слоя раствора в кювете — 1,0 или 2,0 см;
оптическая плотность при следующих длинах волн: Dm = 0,769; 7ф6, = 0,205; Z)268 = 0,2 5 2; Z)274 = 0,194; /ф)8 = 0,3 00; Z>315 = 0,398; Z>322 = 0,192;
с — концентрация раствора при измерении соответствующей оптической плотности, г/дм3: с233 = 0,075; с262 = с268 = с274 = 0,3 00; сЗО8 = с315 = с322 = 1,000.
Массовую долю ненасыщенных жирных кислот рассчитывают в соответствии с формулами
Х= 1,086Х,- 1.324Х2 + 0,40Х3;
Y= 1,980Х2-4,92Х3;
Z=4,69X3,
(4.34)
(4.35)
(4.36)
где X, Y, Z— массовые доли соответственно линолевой, линоленовой и арахидоновой кислот, %.
1.2. ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ С СОПРЯЖЕННЫМИ ДВОЙНЫМИ связями
Подготовка проб. Навеску жира массой 0,2 г взвешивают на аналитических весах в стеклянный стаканчик или чашечку. Жир растворяют в углеводородном растворителе (н-гексане, циклогексане или изооктане) и доводят объем до 100 см3.
Порядок проведения анализа. Метод применяется для определения количества ди-, три-, тетра- и пентаеновых кислот с со-380
пряженными двойными связями, присутствующими в исследуемых жирах.
Оптическую плотность измеряют на спектрофотометре при длинах волн 233. 262. 268. 274. 308. 315, 322 нм в отношении растворителя. Желательно, чтобы оптическая плотность находилась в пределах 0,2—0,8. Для этого подбирают кювету с подходящей толщиной оптического слоя, при необходимости растворы разбавляют. Для установления наличия максимума при характерной длине волны измерения проводят при нескольких длинах волн перед и после ожидаемого максимума поглощения. Если в характерной области спектра максимум не определен, то считают, что в этой области спектра соединение отсутствует.
По полученным величинам оптической плотности рассчитывают удельные коэффициенты поглощения для соединений с сопряженными двойными связями.
Расчет ведут для каждой длины волны по формуле
Г=ф (4.37)
где D — измеренная величина оптической плотности; с — концентрация раствора, используемого для измерения (концентрированного или разбавленного), г/дм3; / — толщина свстопоглошаюшего слоя, см.
Экспериментально полученные данные и результаты расчетов сводят в таблицу.
Показатель Образцы жира
ГОВЯЖИЙ свиной бараний
Коэффициенты удельного поглощения полиненасыщенных жирных кислот:
линолевой А/
линоленовой А/
арахидоновой А/
Коэффициенты удельного поглощения для соединений с сопряженными двойными связями:
диеновых
триеновых
тетраеновых
пентаеновых
Массовая доля полиненасыщенных жирных кислот, %:
линолевой
линоленовой
арахидоновой
В заключение дают сравнительную оценку биологической ценности пищевых животных жиров.
381
2. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВИТАМИННЫХ ПРИМЕСЕЙ В ПИЩЕВЫХ ЖИВОТНЫХ ЖИРАХ
2.1. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КАРОТИНОИДОВ ПО ВИДИМЫМ СПЕКТРАМ
Материалы, реактивы и оборудование. Водяная баня или гермостат; спектрофотометр.
Подготовка проб. Образцы топленых животных жиров пометают в пробирках на водяную баню или в термостат и нагревают до температуры, превышающей температуру плавления на 5—10 °C.
Порядок проведения анализа. Метод определения содержания каротиноидов по видимым спектрам основан на том. что при небольшом содержании хлорофиллов или их отсутствии спектр масла снимают непосредственно в видимой области в диапазоне длин волн 400—500 нм.
Расплавленную пробу исследуемого жира помещают в хроматографическую кювету толщиной светопоглощающего слоя 1 см и снимают спектр на спектрофотометре в интервале длин волн 400—500 нм, определяют оптическую плотность при длине волны наиболее интенсивного максимума.
Содержание каротиноидов (г в 100 см3 жира в пересчете на [3-каротин) определяют по формуле
у - Da
\QdKcx ’ (438)
где Z) — оптическая плотность при А.П1ач; а — разведение (для чистого жира а = 1); d — толщина светопоглощающего слоя, см; Кст — коэффициент поглощения чистого (3-каротина; Л’ст = 250.
2.2. РАЗДЕЛЕНИЕ И ИДЕНТИФИКАЦИЯ КАРОТИНОИДОВ МЕТОДОМ ТСХ
Материалы, реактивы и оборудование. Фарфоровые ступки; обезвоженный сульфат натрия; кристаллический карбонат натрия; бензин; петролейный эфир или ацетон; вакуумный фильтр; воронка Бюхнера; этанол; бихромат калия.
Подготовка проб. Для разделения каротиноидов успешно применяют метод ТСХ на оксиде магния или оксиде алюминия II степени активности толщиной 1 мм.
Навеску продукта (от 2 до 2,5 г в зависимости от содержания каротиноидов) растирают в ступке с обезвоженным сульфатом натрия до получения сухого порошка. Для предотвращения окис-ляемости витаминов рекомендуется добавить несколько кристал-382
лов карбоната натрия. Для экстракции каротиноидов обычно применяют бензин. петролсйный эфир (70—100'С) или ацетон. Пробу экстрагируют до иодного извлечения пигментов. для чего обычно достаточно пяти-шести экстракций. Экстракт рекомендуется освободить от взвешенных частиц фильтрованием под вакуумом (можно на воронке Бюхнера), промыть водой (лучше использовать подсоленную) и обработать щелочью (что дает возможность в результате омыления освободиться от хлорофилла и других омыляемых пигментов и различных сложных эфиров). Дальнейшую очистку каротиноидов можно провести методом колоночной хроматографии. Очищенный экстракт сгущают под вакуумом до небольшого объема.
Порядок проведения анализа. Полученный экстракт объемом оз 0,2 до 1 см3 наносят на стеклянные пластинки, покрытые оксидом магния или алюминия II степени активности, и подвергают разделению в одной из систем углеводородных (подвижных) растворителей, которые приведены ниже.
Определяемое вешсство Сорбент Система растворите ieii Соотношение но объему
Каротиноиды Оксид магния, оксид алюминия 11 степени активности Ацетон -- петролсйный эфир Петролсйный эфир — бензол — метанол 1 :3 или 2:4S 60 :10 : 1
Разделение обычно занимает 30—35 мин. Большинство каротиноидов окрашено и легко обнаруживается при дневном свете в виде ярко окрашенных и резко очерченных зон. Каждую зону выделяют и элюируют петролейным эфиром. Для лучшего элюирования каротиноидов рекомендуется добавить 1—2 капли этанола. Идентификацию каротиноидов проводят по максимумам поглощения в видимой области спектра в интервалах длин волн от 400 до 500 нм, величинам путем сравнения со стандартными растворами «свидетелей», в качестве которых обычно используют ос- и |3-каротиноиды, выделенные из моркови; ликопин идентифицируют по ИК-спектру.
Содержание каротиноидов определяют спектрофотометрически, используя стандартную кривую по бихромату калия.
Построение калибровочного графика. Для построения кривой берут 0,072 г дважды перекристаллизованного бихромата калия и растворяют в 100 см3 воды. 1 см3 раствора соответствует 0,00416 мг каротина. Для построения графика готовят растворы с различной массовой долей бихромата и анализируют на спектрофотометре. Затем строят график зависимости D = f(c).
383
2.3, КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ТОКОФЕРОЛОВ
Материалы, реактивы и оборудование. Гидроксил калия: пирогаллол: диэтиловый эфир: бензол; пластинки с силикагелем марок КСК или АСК: хлороформ; реактив Эммери—Энгеля.
Приготовление реактивов. Р е а к т и в Э м м е р и—Э и г е л я: I объем спиртового раствора с/.-. сх'-липирилила массовой долей 0.25% смешивают с 1 объемом спиртового раствора FeCK массовой долей 0,1 %.
Ста н д а р т н ы й а ц е т а г н ы й раствор (pH 4.62) готовят путем смешивания равных объемов раствора уксусной кислоты молярной концентрацией 0.2 моль/дм3 и раствора гидроксила натрия молярной концентрацией 0,2 моль/дм3.
Подготовка проб. 3 г жира омыляют спиртовым раствором КОН массовой долей 10 % в присутствии пирогаллола. Неомыляемую фракцию экстрагируют диэтиловым эфиром. После отгонки растворителя (в атмосфере инертного газа) полученный остаток неомыляемых веществ растворяют в 5 см3 бензола и подвергают разделению на пластинках с закрепленным слоем силикагеля.
Порядок проведения анализа. Метод количественного определения токоферолов в жировых продуктах основан на выделении неомыляемых веществ и разделении их в тонком слое силикагеля. В качестве углеводородных (подвижных) растворителей рекомендуется использовать хлороформ или смесь хлороформ — циклогексан в соотношении 2:1. Способ позволяет не только отделить токоферолы от сопутствующих веществ, но и провести четкое разделение изомеров токоферола.
Для хроматографирования используют пластинки, покрытые слоем силикагеля (марки КСК или АСК) с 15 % гипса и активированные при 110 °C в течение 2 ч. В качестве подвижного растворителя используют хлороформ. Хроматограмма развивается в темноте (обычно за 35—45 мин). Для проявления хроматограммы пластинку опрыскивают реактивом Эммери—Энгеля, состоящего из равных объемов спиртового раствора а-, ot'-ди пиридила массовой долей 0,25 % и спиртового раствора FeCl3 массовой долей 0,1 %. Токоферолы окрашиваются этим реактивом в розовый цвет. Положение пятен определяют с помощью стандартного раствора ос-токоферола и по величинам R,.
Для количественного определения токоферолов пластинку делят на две части, на каждую из которых наносят бензольный раствор неомыляемых веществ соевого масла. После развития хроматограммы и подсушивания пластинки левую половину закрывают стеклом, а правую опрыскивают реактивом Эммери— Энгеля. С левой части пластинки на высоте окрашенных пятен токоферолов соскребают слой силикагеля, из которого разделенные вещества элюируют с помощью этанола, сорбент удаляют центрифугированием.
384
Массовую долю токоферолов в пробе жира (Ф) определяют по калибровочному графику и пересчитывают на о.-токоферол по формуле
Х= (5>1)/( И//?),
(4.39)
где 5 —объем бензольного раствора неомыляемых веществ, см': Л - содержание токоферолов, найденное по калибровочному графику, мг/см5: И—объем нанесенного на пластинку бензольного раствора неомылясмы.х веществ, см5; /// — навеска жира. г.
Построение калибровочного графика. Количественные определения отдельных изомерных форм токоферолов проводят колориметрическим методом по калибровочной кривой, составленной по синтетическому ацетату а-токоферола, который омыляют и выделяют описанным выше способом. Остаток растворяют в этаноле для получения раствора, содержащего 1 г/дм3 а-токоферола, из которого затем путем соответствующих разведений готовят растворы с содержанием а-токоферола от 0,005 до 0,05 г/дм3 для проведения колориметрической реакции. К 4 см3 элюата добавляют 0,5 см3 спиртового раствора массовой долей а-, а'-дипиридила 0,25 % и 0,5 см3 спиртового раствора FeCl3 массовой долей 0,1 %. Через 5 мин измеряют оптическую плотность окрашенного раствора при длине волны 520 нм против контрольного раствора, состоящего из 4 см3 этанола, 0,5 см3 раствора а-, а'-дипиридила и 0,5 см3 раствора ЕеС13.
Результаты определений сводят в таблицу рекомендуемой формы:
Массовая доля витаминных примесей, %
Жир
ГОВЯЖИЙ свиной бараний
Каротиноиды (в пересчете на р-каротин) Токоферолы (в пересчете на а-токоферол)
После оформления таблицы самостоятельно формулируют заключение о сравнительной биологической ценности различных видов животных жиров по содержанию в них витаминных примесей.
Лабораторная работа № 6
ОПРЕДЕЛЕНИЕ СВЕЖЕСТИ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Освоить методы определения свежести мяса и мясных продуктов.
Задачи. Провести отбор проб мяса и субпродуктов; оценить мясо и субпродукты различных видов скота, птицы и кроликов
385
органолептическим спосооом; определить свежесть мяса и мяс пых продуктов на основе физико-химического и микроструктурного анализов.
Объекты исследования. Мясо в тушах, полутушах. четвертинах. а также отдельные отруба или мякотные ткани мяса различных видов.
Методические указания. В работе приведены традиционные и экспресс-методы определения свежести мяса и мясных продуктов. которые могут применяться независимо или в совокупности.
Органолептические методы предусматривают определение внешнего вида и цвета, консистенции, запаха, состояния жира и сухожилий, прозрачности и аромата бульона. При этом каждый образец анализируют отдельно.
Окраска мяса обусловлена в основном наличием пигмента мышечной ткани — миоглобина. Красная окраска поверхности свежего мяса на глубину до 4 см образуется за счет оксимиоглобина (МЬОД. Более глубокие слои мяса окрашены в пурпурнокрасный цвет.
При длительном хранении на воздухе или сильном бактериальном обсеменении потемнение тканей возможно вследствие образования метмиоглобина (MetCO2). Обесцвечивание или специфическое изменение окраски (зеленый, желтый, розовый или серый пигменты) образуются как за счет химических превращений миоглобина, так и под действием микробиальных процессов.
Консистенция мяса тесно связана с состоянием белков актина и миозина — основных компонентов миофибрилл, которые являются рабочими органами движения мышц.
Метод определения продуктов первичного распада белков в бульоне основан на осаждении белков нагреванием, образовании в фильтрате комплексов сульфата меди с продуктами первичного распада белков, выпадающих в осадок.
Физико-химическая оценка свежести мяса основана на определении нескольких показателей.
Метод количественного определения летучих жирных кислот основан на выделении их из мяса после хранения и определении их массовой доли титрованием дистиллята гидроксидом калия (натрия).
Накопление в мясе аминокислот и аммиака — наиболее характерный и постоянный признак его порчи. Для определения амино-аммиачного азота мясную вытяжку предварительно освобождают от белков и титруют в два приема: по первому смешанному индикатору (равная смесь спиртовых растворов нейтральрота и метиленового голубого массовой долей 0,1 %)
386
до pH 7.0 лля нейтрализации кислых продуктов, а затем после добавления нейтрального формалина но второму смешанному индикатору (1 часть раствора тимолблау массовой долей 0.1 % и'З части раствора фенолфталеина массовой долей 1 % в растворе спирта объемной долей 50%) до pH 9.0 для определения аммиачного и аминного азота.
В аминокислотах оба водорода аминной группы замещаются углеводородным радикалом, в результате чего щелочная функция аминокислоты теряется при сохранении кислой:
СН. - CHNH - СООН + неон СН.— СН2 — N =
= сн-соон + н2о.
При взаимодействии формалина с солями аммония выделяется эквивалентное количество свободной кислоты:
4NH.C1 + 6HCOH = (CH.bN, + 4НС1 + 6Н7О. 4 х j. 7 О 4 Z
В свежем мясе содержание амино-аммиачного азота не превышает 80 мг%, в мясе подозрительной свежести составляет от 81 до 130 мг% и в несвежем — более 130 мг%.
Наиболее распространены модификации метода определения амино-аммиачного азота по А. М. Софронову и по Г. В. Коло-ботскому.
В начальных стадиях порчи мяса соотношение между окисными и закисными соединениями резко меняется, величина потенциала платинового электрода снижается в соответствии с накоплением окисляющихся веществ. Окислительно-восстановительный потенциал — также один из объективных показателей свежести субпродуктов.
По величине окислительно-восстановительного потенциала вытяжек можно отличить мясо здоровых животных от мяса животных, убитых в патологическом состоянии.
Окислительно-восстановительный потенциал вытяжек из мяса крупного рогатого скота колеблется в следующих пределах: созревшее годное мясо — от 380 до 421 мВ, несвежее — от — 204 до — 303 мВ. Мясо, вытяжки из которого имеют окислительно-восстановительный потенциал ниже нуля по отношению к нормальному водородному электроду, находится в той или иной стадии порчи. Окислительно-восстановительный потенциал свежего говяжьего мяса здоровых животных равен 363—391 мВ, свиного — 320-350 мВ, в мясе больных животных окислительно-восстановительный потенциал снижается до 120—200 мВ.
Безусловные преимущества имеет метод определения свежести мяса по микроструктурным показателям.
387
1. ОРГАНОЛЕПТИЧЕСКИЙ АНАЛИЗ
Материалы, реактивы и оборудование. Нож; стакан; мерный цилиндр вместимостью 25 см’ и с диаметром дна 20 мм; вата; про бирки; раствор сульфата мели массовой долей 5 сс.
Подготовка проб. Oi каждой исследуемой мясной туши или сс части отбирают три пробы массой не менее 200 г: у зареза, против 4-—5-го шейных позвонков, из мыши в области лопатки, в области бедра из толстых частей мышц. От замороженных или охлажденных блоков мяса и субпродуктов или от отдельных блоков сомнительной свежести также отбирают пробы целым куском массой не менее 200 г.
Для получения однородной пробы каждый образец отделяют от кости и отдельно пропускают через мясорубку с диаметром отверстий решетки 2 мм. Полученный фарш тщательно перемешивают.
Для определения прозрачности и аромата бульона 20 г полученного фарша взвешивают на лабораторных весах с погрешнос тью не более 0,2 г и помещают в коническую колбу вместимостью 100 см3, добавляют 60 см3 дистиллированной воды, тщательно перемешивают, закрывают часовым стеклом и помещают на водяную баню при температуре кипения.
Порядок проведения анализа. Мясо осматривают при естественном освещении. При осмотре отмечают состояние и цвет поверхности мяса, цвет жира. Регистрируют наличие или отсутствие корочки подсыхания, обращают внимание на наличие сгустков крови, загрязненности, плесени и личинок мух. Для установления внешнего вида и цвета мышечной ткани в глубинных слоях рекомендуется сделать надрез мяса ножом и определить цвет и внешний вид поверхности свежего разреза. Наличие липкости устанавливают ощупыванием. Увлажненность поверхности мяса на разрезе определяют путем прикладывания к разрезу полоски фильтровальной бумаги. Если мясо свежее, то на бумаге не останется пятна, при порче мяса бумага становится влажной или липкой.
Консистенцию мяса определяют путем легкого надавливания пальцем на свежий срез. При этом фиксируют наличие и скорость восстановления поверхности. Результаты фиксируют.
При определении запаха вначале анализируют поверхностный слой исследуемых проб, а затем свежий разрез мяса. При осмотре туши или ее частей особое внимание обращают на запах слоев мышечной ткани, прилегающей к кости. Данные фиксируют.
Состояние жира оценивают в туше в момент отбора образцов. Цвет, запах и консистенцию жира устанавливают в соответствии с рекомендациями лабораторной работы № 4 настоящей главы.
Состояние сухожилий определяют в туше в момент отбора образцов. Ощупыванием сухожилий устанавливают их упругость, плотность и состояние суставных поверхностей. У свежих туш су
388
хожилия упругие, плотные, поверхность суставов гладкая слепящая. У размороженного мяса сухожилия мягкие, рыхлые, окрашены в ярко-красный цвет. В ел алии сомнительной свежести сухожилия менее плотные, имеют матово-белый цвет. Суставные поверхности слегка покрьиы слизью. В несвежем состоянии сухожилия размягчены, сероватою цвета, а суставные поверхности покрыты слизью.
Запах мясного бульона определяют в процессе нагревания до 80—85 °C в момент появления паров, выходящих из приоткрытой колбы.
Прозрачность бульона определяют визуально. Для этого берут 20 см2 3 бульона, наливают в мерный цилиндр диаметром 20 мм и вместимостью 25 см3 и рассматривают.
По результатам анализа и в соответствии с данными табл. 4.6 делают заключение о свежести мяса или субпродуктов.
Заключение о степени свежести мяса птицы и кроликов делают по результатам органолептической опенки и в соответствии с данными табл. 4.7 и 4.8.
При определении продуктов первичного распада белков приготовленный горячий бульон фильтруют через плотный слой ваты толщиной не менее 0,5 см в пробирку, помещенную в стакан с холодной водой. В пробирку наливают 2 см3 фильтрата и 3 капли (0,3 см3) раствора сульфата меди массовой долей 5 %. Пробирку встряхивают 2—3 раза и ставят в штатив. Через 5 мин отмечают результат анализа.
Результаты наблюдений сравнивают с данными табл. 4.6—4.8 и заносят в таблицу рекомендуемой формы:
Образец
Внешний Консистен- I о вид н цвет пня I ‘п</
Состояние жира
Состояние । Прозрачное™, и сухожили!! аромат бульона
На основании сравнения опытной органолептической оценки каждого образца с показателями свежего мяса студенты фиксируют отклонения (если такие имеются); сравнивая полученные результаты. самостоятельно делают выводы о качестве бульона.
2. ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ
Материалы, реактивы и оборудование. Стаканы, пробирки, воронки стеклянные; градуированные пипетки; бумага фильтровальная или фильтры бумажные; формалин; тимоловый синий массовой долей 0,1 %; фенолфталеин массовой долей 1 %; прибор для отгонки летучих веществ; микробюретки и капельницы; цилиндры мерные вместимостью до 250 см3; раствор серной кислоты массовой долей 2 %; растворы гидроксида калия или гидроксида натрия молярной концентрацией 0,1 моль/дм3; гидроксид бария; спиртовой раствор фенолфталеина массовой долей 1 %; сифон.
389
2.1. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЛЕТУЧИХ ЖИРНЫХ КИСЛОТ
Подготовка проб. Для получения однородной средней пробы образцов мяса каждый образец отдельно трижды пропускаю! через мясорубку с диаметром отверстий решетки 2 мм. Фарш тщательно перемешиваю! и из него берут навески. Допускается измельчение пробы в ступке изогнутыми ножницами до состояния фарша.
Порядок проведения анализа. Для анализа используют прибор для отгонки летучих веществ с помощью водяного пара (рис. 4.17). Навеску мясного фарша массой (25 ± 0,01) г помещаю! в круглодонную колбу 3. Туда же приливают 150 см3 раствора серной кислоты массовой долей 2 %. Содержимое колбы перемешивают и колбу закрывают пробкой 2. Под холодильник 4 подставляют коническую колбу 5 вместимостью 250 см3, на которой отмечают объем 200 см3. Дистиллированную воду в плоскодонной колбе I доводят до кипения и паром отгоняют летучие жирные кислоты до тех пор, пока в колбе 5 не соберется 200 см3 дистиллята. Во время отгона колбу 3 с навеской подогревают. Весь объем дистиллята титруют в колбе 5раствором гидроксида калия (натрия) молярной концентрацией 0,1 моль/дм3 с индикатором (фенолфталеином) до появления неисчезающей малиновой окраски.
Параллельно при тех же условиях проводят контрольный опыт для определения расхода щелочи, пошедшей на титрование дистиллята с реактивом без мяса.
Рис. 4.17. Прибор для отгонки летучих веществ из мяса с помощью водяного пара
390
Содержание летучих жирных кислот (mi КОН на 100 1 мяса) вычисляют по формуле
А - |( У - 5.61 1001 /т. (4.40)
где ) — объем расгвора нмроксида кадия (натрия) молярной концентраппер 0,1 .моль/дмУ израсходованного на титрование 200см’ дистиллят;! из мяса, см'; у _объем раствора гидроксида калия (натрия) молярной концентрацией 0.1 моль/дм'. израсходованного на титрование 200см’ дистиллята контроля, см'; /—поправка к титру раствора гидроксила калия (натрия) молярной концентрацией 0.1 моль/дм’; 5.61 — масса гидроксида калия, содержащаяся в 1 см' раствора молярной концентрацией 0.1 моль/дм’. мг; in— масса навески пробы, г.
За результат исследования принимают среднее арифметическое двух параллельных определений. Вычисления проводят с точностью не более 0,01 мг КОН.
В свежем мясе содержится летучих жирных кислот до 4 мг КОН, в мясе сомнительной свежести — от 4,1 до 9 мг КОН, в несвежем мясе — свыше 9 мг.
В свежем мясе тушек нежирной птицы содержится летучих жирных кислот до 4,5 мг КОН, в мясе сомнительной свежести — от 4,51 до 9 мг КОН, а в несвежем мясе — свыше 9 мг КОН.
Мясо кролика считают свежим, если в охлажденном мясе содержится летучих жирных кислот до 2,25 мг КОН, в замороженном —- до 4,50 мг КОН. Мясо считают сомнительной свежести, если в охлажденном мясе содержится летучих жирных кислот 2,25—9,00 мг КОН, в замороженном — 4,50—13,50 мг КОН; в несвежем — соответственно более 9,00 и 13,50 мг КОН.
2.2. МЕТОД ОПРЕДЕЛЕНИЯ АМИНО-АММИАЧНОГО АЗОТА (ПО Г. В. КОЛОБОТСКОМУ)
Подготовка проб. Для приготовления вытяжки в колбу помещают 25 г мясного фарша и 100 см3 дистиллированной воды. Смесь взбалтывают в течение 3 мин, затем отстаивают и вновь взбалтывают 2 мин. Экстракт фильтруют через 3—4 слоя марли.
Порядок проведения анализа. В мерную колбу вместимостью 100 см3 отбирают 40 см3 фильтрата экстракта, для осаждения белков последовательно добавляют раствор алюминиевых квасцов массовой долей 10 % и насыщенный раствор гидроксида бария общим объемом, примерно равным или немного большим объема мясной вытяжки.
Предварительно устанавливают объем гидроксида бария, необходимый для нейтрализации определенного объема раствора алюминиевых квасцов массовой долей 10 %. 10 см3 этого раствора титруют насыщенным раствором гидроксида бария по фенолфталеину и рассчитывают объем реактивов, необходимых для осаждения белков.
391
Объем в колбе доводят дистиллированной водой до метки и жидкости дают отстояться в течение 10 мин.
Во вторую колбу вместимостью 100 см3 (контроль) помешают те же объемы растворов алюминиевых квасцов и гидроксида бария, что и для осаждения белков, объем в колбе доводят дистиллированной водой до метки и также дают отстояться в течение 10 мин.
Исследуемую вытяжку после осаждения белков и контрольный раствор фильтруют через бумажный фильтр, после чего в фильтратах определяют содержание амино-аммиачного азота.
В коническую колбу помещают 20 см3 вытяжки и добавляют 0,3 см3 первого смешанного индикатора, состоящего из смеси равных объемов спиртовых растворов нейтральрота и метиленового голубого массовой долей 0,1 %. Затем смесь титруют раствором гидроксида натрия молярной концентрацией 0,1 моль/дм3 до нейтральной реакции, т. е. до перехода окраски фильтрата из сине-фиолетовой в зеленую. В ту же колбу приливают 10 см3 формалина, предварительно оттитрованного до нейтральной реакции по тому же индикатору, и 0,5 см3 второго смешанного индикатора [1 часть раствора тимолового синего массовой долей 0,1 % и 3 части раствора фенолфталеина массовой долей 1 % в растворе этанола (50 об. %)]. Содержимое колбы окрашивается в сине-фиолетовый цвет. Фильтрат вновь титруют раствором гидроксида натрия молярной концентрацией 0,1 моль/дм3. По мере прибавления щелочи фильтрат приобретает вначале ярко-зеленый цвет, а затем, при последующем титровании — сине-фиолетовый. Переход цвета фильтрата от ярко-зеленого до сине-фиолетового следует считать окончанием формольного титрования. Параллельно проводят контрольный опыт. В колбу помещают 20 см3 контрольного раствора и титруют так же, как и исследуемый раствор.
Содержание амино-аммиачного азота (мг на 100 г мяса) вычисляют по формуле
Х= [1,4- 100- 100- 100(У; — Г2) • 100]/(25 • 40-20) =
= 70(Г1-Г2), (4.41)
где Yv }/ —объемы раствора гидроксида натрия молярной концентрацией 0,1 моль/дм3. пошедшие на титрование исследуемого фильтрата и контрольного раствора, см3.
2.3. МЕТОД ОПРЕДЕЛЕНИЯ АМИНО-АММИАЧНОГО АЗОТА (ПО А. М. СОФРОНОВУ)
Подготовка проб. Предварительно готовят вытяжку из мясного фарша согласно рекомендациям п. 2.2 настоящей работы.
Порядок проведения анализа. В колбу помещают 10 см3 профильтрованной вытяжки, приготовленной в соотношении мя
392
со — вода I : 4. Приливают 40 см-'дистиллированной воды и три капли спиртовою раствора фенолфталеина массовой долей 1 %. Вытяжку нейтрализуют раствором гидроксида натрия молярной концентрацией 0.1 моль/дм3 до слабо-розовой окраски. Затем в колбу добавляют 10 см3 формалина, нейтрализованного по фенолфталеину, и содержимое колбы титруют раствором гидроксида натрия молярной концентрацией 0.1 моль/дм3 до слабо-розового цвета.
Содержание амино-аммиачного азота рассчитывают по формуле
%= 1,4 К
(4.42)
где И—объем раствора гидроксида натрия молярной концентрацией 0.1 моль/дм5. пошедший на второе титрование.
В доброкачественном мясе содержится до 1,26 мг амино-аммиачного азота на 10 см3 вытяжки (в мясе кроликов —от 0,98 до 1,82 мг), в мясе подозрительной свежести — от 1,27 до 1,68 мг (в мясе кроликов — от 1,90 до 2,5 мг), в несвежем мясе — более 1,68 мг (в мясе кроликов — более 2,5 мг).
2.4. ОПРЕДЕЛЕНИЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ПОТЕНЦИАЛА
Подготовка проб. Предварительно готовят вытяжку из мясного фарша согласно рекомендациям п. 2.2 настоящей работы.
Порядок проведения анализа. В стакан с исследуемой мясной вытяжкой, приготовленной в соотношении 1 : 4, подвешивают 5 сухих платиновых электродов, причем электроды не должны соприкасаться со стеклом. В другой стакан наливают жидкость сравнения — стандартный ацетатный раствор; в нее погружают платиновый электрод и вносят хингидрон. Стандартный ацетатный раствор соединяют с исследуемой жидкостью агаровым сифоном.
Техника определения окислительно-восстановительного потенциала, по существу, не отличается от потенциометрического определения показателя концентрации водородных ионов. Электроды присоединяют к потенциометру. В цепь включают дополнительное сопротивление не менее 50 000 0м. Потенциалы электродов фиксируют через 30—60 мин. Рекомендуется определить величину нескольких электродов (не менее пяти) и вычислить среднеарифметическое.
По экспериментально измеренной разности между потенциалом платинового электрода в мясной вытяжке и потенциалом хингидронного электрода в ацетатном растворе (±П) определяют величину окислительно-восстановительного потенциала (мВ):
гЯ=±П + 436.
(4.43)
393
2.5. ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ СВЕЖЕСТИ МЯСА
Подготовка проб. Для определения флуоресценции вырезают плоские кусочки мяса в соответствии с размерами кюветы флуорископа.
Для анализа водной вытяжки готовят экстракт из мяса в соотношении 1 : 4 (25 г мяса и 100 см3 воды). Экстракт освобождают от белков нагреванием и фильтруют через бумажный фильтр.
Порядок проведения анализа. Определение флуоресценции мяса проводят в темной комнате. После нагревания кварцевой лампой в течение 10 мин мясную пластинку помешают под ультрафиолетовые лучи. Угол падения лучевого потока должен быть около 45°. Исследователь, наблюдающий за свечением, должен быть обязательно в защитных очках. Свежее мясо крупного рогатого скота флуоресцирует красно-бархатным цветом, баранина — темно-коричневым, свинина — светло-коричневым, телятина — коричневым, конина — ржаво-коричневым. В испорченном мясе на общем грязно-темном фоне наблюдается свечение в виде желтых точек.
Люминесцентный анализ можно проводить и с водной вытяжкой из мяса в разведении 1 : 4. Для облучения 5 см3 экстракта помещают в кварцевые стаканчики или пробирки из бесцветного стекла. Экстракт свежего мяса флуоресцирует желтоватым цветом, несвежий экстракт в зависимости от степени разложения — от интенсивного сине-зеленого до молочно-голубого (по М.М. Данилову и А. А. Кондратенко).
Результаты химической или физико-химической оценки свежести образцов мяса фиксируют в таблице:
Показатели свежести мяса Свежее мясо Значение показателей, установленных экспериментальным путем для образцов
говядины свинины баранины
Содержание летучих жирных кислот, мг КОН
Содержание амино-аммиач-ного азота:
мг%
мг/10см3 вытяжки
Окислительно-восстановительный потенциал, мВ
Не более 4
Не более 80
Не более 1,26 3,63-3,91
Сравнивая экспериментальные данные, для каждого образца фиксируют отклонения (если таковые имеются), по результатам исследований формулируют заключение.
394
3. ОПРЕДЕЛЕНИЕ СВЕЖЕСТИ МЯСА ПО МИКРОСТРУКТУРНЫМ ПОКАЗАТЕЛЯМ
Материалы, реактивы и оборудование. Микроюм замораживающий с металлическим шлангом; набор ножей и принадлежности для их заточки; диоксид углерода в баллонах; микроскоп МБИ-3 или другой аналогичной! марки; микрометр окулярный; объект-микрометр; осветитель (ОИ-7, ОИ-9); спиртовка; ножницы; нож секционный; пинцеты анатомические; иглы препаровальные или зонды зубоврачебные; кристаллизаторы вместимостью 100 см?; цилиндры мерные вместимостью 100 см3; колбы Эрленмейера вместимостью 100 см?; стекла
предметные и покровные; стаканчики стеклянные с крышками размером 40 х 20 х 85 мм вместимостью 35 см3; штатив металлический с отверстиями овальной формы размером 40 х 20 мм; бальзам пихтовый или канадский; квасцовый гематоксилин Эрлиха; глицерин дистиллированный; кислота карболовая кристаллическая (фенол); кислота соляная; кислота уксусная ледяная; квасцы алюмокалиевые; карбоксилол (орто); масло вазелиновое медицинское; этанол ректификованный; тушь черная; тимол или камфара; карбонат кальция, бумага фильтровальная; водный раствор (х. ч.) формалина массовой долей 10%; эозин водорастворимый; белок яичный.
Подготовка проб. От исследуемой мясной туши или ее части острым ножом отрезают образец мяса в виде куба размером 30 x 30 x 30 мм. Образец вырезают в направлении, перпендикулярном поверхности туши, в глубь мышц так, чтобы одна из сторон образца была наружной поверхностью туши или ее части, а другая — поверхностью разруба или распила. Образец вырезают из мест, наиболее быстро подвергающихся порче, не нарушая товарного вида туши или ее части; из шейной части, включая зарез; у мест разруба грудной кости — из глубокой грудной мышцы на уровне четвертого-пятого ребра; из мест разруба лонного сращения; из других мест туши или ее частей,
сомнительных по свежести.
Порядок проведения анализа. Из каждого образца вырезают ку-
сочек мяса размером 30х15х х 4 мм таким образом, чтобы в него вошли поверхность разруба или распила, наружная поверхность туши и все лежащие ниже нее ткани на глубину до 30 мм. Мышечные волокна в вырезанном кусочке мяса должны быть расположены параллельно плоскости разреза (рис. 4.18).
Наружная поверхность
Поверхность разруба
Рис. 4.18. Образец мяса для приготовления гистологических препаратов
395
Вырезанные из всех образцов кусочки мяса подвергают быс трои фиксации водным раствором нейтрального формалина массовой долей П) сс. Для этого кусочки мяса с предварительно прикрепленной пергаментной этикеткой и номером образца помещают в небольшую колбу пли широкую пробирку, заливают 4—5 объемами формалина и подогревают на пламени горелки, не доводя до кипения. При появлении пузырьков воздуха подогрев прекращают, содержимое осторожно встряхивают и снова подогревают до появления пузырьков воздуха. Так повторяют три-четыре раза.
После фиксации формалин выливают, в колбочку вставляют стеклянную воронку, а кусочки мяса промывают холодной проточной водой в течение 2 мин. Промытые кусочки мяса режут на замораживающем микротоме в плоскости, параллельной продольной оси волокон, получая срезы толщиной 15—30 мкм.
С микротомного ножа с помощью кисточки срезы переносят в небольшой кристаллизатор с водопроводной водой на несколько секунд для распрямления. Под неповрежденный срез быстро подводят предметное стекло для наклеивания, обработанное яичным белком с глицерином (2:1), и извлекают срез из воды, удерживая его на середине стекла препаровальной иглой. Затем срез накрывают сухой фильтровальной бумагой (3—4 слоя) и, прижимая бумагу ребром ладони, наклеивают его на предметное стекло.
Наклеенные таким образом срезы окрашивают квасцовым гематоксилином Эрлиха в течение 3—4 мин и промывают в воде в течение 2 мин. Для удаления избытка гематоксилина срезы помещают в раствор соляной кислоты массовой долей 1 % (до появления розового цвета), затем в аммиачную воду с целью нейтрализации соляной кислоты (до посинения срезов) с последующей промывкой их в воде в течение 2 мин. Затем срезы окрашивают водным раствором эозина массовой долей 1 % в течение 1 мин с последующим ополаскиванием в воде. Далее срезы обезвоживают двумя порциями этанола, последовательно погружая их в каждую порцию на 1 мин. Осветляют срезы в карбоксилоле и отмывают в ксилоле, выдерживая в каждом растворе по 1 мин. Подготовленные таким образом срезы заключают в пихтовый или канадский бальзам под покровное стекло.
Проводят исследование гистопрепаратов. Для этого подготовленные и окрашенные срезы рассматривают под микроскопом сначала при малом увеличении объектива (х!0), затем при среднем (х40) и в отдельных случаях под иммерсией (х90). Полученные результаты сравнивают с данными, приведенными в табл. 4.14.
396
Характеристика микроструктуры мяса при оценке его свежести
397
Продолжение
I
398
При и с ч а н и я: 1. На всех этапах созревания составные элементы волокна сохраняют восприятие к окраске. 2. В несвежем мясе микроструктурные показатели созревания мяса становятся неразличимыми в местах порчи, так как нал ними доминируют микроструктурные изменения, характеризующие несвежее мясо.
Результаты микроструктурных исследований свежести образцов мяса и субпродуктов студенты сравнивают с данными табл. 4.14. фиксируют отклонения (если таковые имеются) от характеристик свежего мяса и формулируют заключение по работе.
Данные заносят в таблицу:
Показал еть
Образны
говядины > свинины ।
баранины
Наличие и состояние корочки подсыхания Состояние структуры ядер мышечных волокон
Состояние поперечной и продольной исчерченное™ мышечных волокон Способность ядер и мышечных волокон к окрашиванию
Наличие микрофлоры и границы ее распространения
Лабораторная работа № 7
ОПРЕДЕЛЕНИЕ СТЕПЕНИ КУЛИНАРНОЙ ГОТОВНОСТИ МЯСА И МЯСНЫХ ПРОДУКТОВ
Цель работы. Освоить методы определения степени кулинарной готовности мяса и мясных продуктов.
Задачи. Определить кулинарную готовность мясных продуктов арбитражным методом. Дать оценку кулинарной готовности вареных мясных продуктов или колбасных изделий.
Объекты исследования. Вареное мясо, мясные продукты, колбасные изделия.
Материалы, реактивы и оборудование. Весы лабораторные с наибольшим пределом взвешивания 200 г; потенциометр; фотоэлект-ролориметр или спектрофотометр; ультратермостат или водяная баня, обеспечивающие регулирование температуры от 30 до 99 °C; воронки; колбы мерные вместимостью 500 и 1000 см3; пипетки, колбы, пробирки; палочки стеклянные; бумага фильтровальная лабораторная; груша резиновая; кислота лимонная; цитрат натрия 5-водный; свежеприготовленный раствор динатриевой соли фенилфосфорной кислоты концентрацией 2 г/дм3; растворы трихлоруксусной кислоты концентрацией 50 и 200 г/дм3; раствор гидроксида натрия молярной концентрацией 0,5 моль/дм3; вода дистиллированная; фенол; толуол; вольфрамат натрия 2-водный; молибдат натрия; сульфат лития 1-водный; кислота ортофосфорная плотностью 1,72 г/см3; кислота соляная плотностью 1,19 г/см3; бром.
399
Приготовление реактивов. Ц и г р а т и ы й б у ф е р (pH 6.5у I <88 г цитрата натрия и 0.588 г лимонной кислоты растворяют в дистиллированной воде в мерной колбе вместимостью 1 дм3, доводят дистиллированной водой до метки и перемешивают, затем добавляют 1 см? толуола. Раствор хранят в холодильнике при (4 ± 1) 'С не более ] 2 сут.
С г а и д а р т и ы й р а с т в о р ф е и о л а: взвешиваю! 2 i фенола (с точностью до третьего десятичного знака), растворяю! в воде в мерной колбе вместимостью 1 дм3. доводят объем дистиллированной водой до метки и перемешиваю!. Отбирают пинеткой с помощью резиновой груши 5 см3 раствора в колбу вместимостью 500 см3. добавляют около 300 см3 дистиллированно!! воды, вносят 25 г кристаллической трихлоруксусной кислоты. После растворения содержимое колбы доводят до метки дистиллированной водой и перемешивают. Полученный раствор содержит 20 мкг фенола в 1 см3.
Реактив Фолин а: см. главу 3. лабораторную работу № 2.
Методические указания. Принятые режимы тепловой обработки вареных колбас и мясных продуктов предусматривают инактивацию тканевых ферментов. В случае разногласий в оценке кулинарной готовности вареных продуктов прибегают к использованию методов, позволяющих определить остаточную активность ферментов.
Арбитражный метод основан на фотометрическом определении в продукте интенсивности развивающейся окраски, зависящей от остаточной активности кислой фосфатазы, выраженной массовой долей фенола.
Подготовка проб. Образцы продуктов из свинины освобождают от жировой ткани и шкурки, пробы вареных колбас, сосисок и сарделек — от оболочки и шпика. Пробы дважды измельчают на мясорубке, тщательно перемешивают, помещают в стеклянную или пластмассовую банку с крышкой и хранят при температуре (4 ± 2) °C до окончания анализа.
Порядок проведения анализа. От каждой пробы отбирают две навески по 1 г, взвешенные с точностью до 0,001 г, переносят в две пробирки, одна из которых является опытной, а вторая — контрольной.
В пробирки вносят по 10 см3 цитратного буфера pH 6,5, тщательно перемешивают стеклянной палочкой и настаивают в течение 20 мин при комнатной температуре, периодически перемешивая.
В контрольную пробирку добавляют 5 см3 раствора трихлоруксусной кислоты концентрацией 200 г/дм3, перемешивают и добавляют 5 см3 раствора динатриевой соли фенилфосфорной кислоты концентрацией 2 г/дм3, выдерживают в течение 10 мин и фильтруют.
В опытную пробирку вносят 5 см3 раствора динатриевой соли фенилфосфорной кислоты концентрацией 2 г/дм3. Пробирку по
400
мешают в х'льтратермостат при температуре (39 ± 1) С на 1 ч. затем добавляют 5ум? раствора трихлоруксусной кислоты концентрацией 200 I /дм'. выдерживаю! 10.мин и фильтрую!.
Для проведения цветной реакции из контрольной и опытной проб отбирают по 2.5с.м? безбелкового фильтрата. В каждую пробирку добавляют 5см? раствора гидроксида натрия молярной концентрацией 0.5 моль/лм3. перемешивают, выдерживают 10 мин. добавляют 1.5 см3 реактива Фолина, разбавленного дистиллированной водой в соотношении 1 : 2. смесь вновь перемешивают.
Через 30 мин измеряют оптическую плотность растворов по отношению к раствору трихлоруксусной кислоты на фотоэлектроколориметре при к = 600 нм в кювете с расстоянием между рабочими гранями 10 мм или на спектрофотометре при той же длине волны в кювете аналогичного размера.
Содержание фенола определяют по калибровочному графику.
Построение калибровочного графика. В пробирки вносят следующие объемы стандартного раствора фенола: 0: 0.25; 0.5; 1,0: 1,5; 2,0 см3, что соответствует массе фенола: 0: 5; 10; 20; 30: 40 мкг. Объем в каждой пробирке доводят до 2,5 см3, добавляя соответствующий объем раствора трихлоруксусной кислоты концентрацией 50 г/дм3 (2,5; 2,25; 2,0; 1,5; 1,0; 0,5 см3) и перемешивая. В каждую пробирку вливают 5 см3 раствора гидроксида натрия молярной концентрацией 0,5 моль/дм3, перемешивают, выдерживают 10 мин и добавляют 1,5 см3 реактива Фолина, разбавленного дистиллированной водой в соотношении 1 : 2. Смесь вновь перемешивают.
Через 30 мин измеряют оптическую плотность растворов по отношению к раствору трихлоруксусной кислоты на фотоэлектроколориметре при л = 600 нм в кювете с расстоянием между рабочими гранями 10 мм или на спектрофотометре при той же длине волны в кювете аналогичного размера.
По полученным средним данным трех стандартных растворов на миллиметровой бумаге размером 20 х20 см строят калибровочный график,^ откладывая на оси абсцисс содержание фенола (мкг в 1 см3 окрашенного раствора), а на оси ор-
динат — значение соответствующей оптической плотности D. Калибровочный график должен проходить через начало координат. Пример калибровочного графика приведен на рис. 4.19.
Рис. 4.19. Калибровочный график для определения массовой доли фенола с помощью фотоэлектроколориметра
401
Массовую долю фенола (%) вычисляю! по формоле
(т} - ш->) 20 100 т 2.5 I06
(4.44)
где и т, - масса фенола соответственно в щштнои и кошрольнои нробир ка.\. найденная по калибровочному графику, мкг: 20 - разведение. см': тмасс.! анализируемой пробы, г: 2.5 — объем фильтрата. собранный для цветной реакции. см'; 11г - коэффициент пересчета в |раммы.
Вычисления проводят до четвертого десятичного знака.
Результаты экспериментальных работ по анализу эффективности тепловой обработки вареных мясных продуктов рекомендуется оформить в виде таблицы:
Образец
Массовая доля фенола. % фактическая по нормативной документации
Полученные данные студенты сравнивают с нормативными показателями для каждого вида продукции, самостоятельно делают выводы и формулируют заключение.
Контрольные вопросы и задания
1. Какова сущность формулы сбалансированного питания'?
2. Какова структура пищевой ценности продуктов?
3. Что понимается под определением «энергетическая ценность продуктов питания»?
4. Каковы основные критерии оценки биологической ценности пищевых продуктов?
5. Какие способы и методы определения биологической ценности вы знаете? В чем их сущность?
6. Что понимают в современной интерпретации под определением «качество продуктов питания»?
7. Охарактеризуйте систему показателей качества продуктов. Какие из них наиболее важны и почему'?
8. Приведите пример математической модели оценки качества мясных продуктов.
9. Что относится к органолептическим показателям качества и каковы подходы к их оценке?
10. По каким параметрам оценивается консистенция продуктов?
11. Какова сущность органолептической и сенсорной оценки качества пищевых продуктов?
12. В чем сущность микроструктурного метода анализа свежести мяса и мясных продуктов?
13. Как определить свежесть мяса методом органолептической оценки9
14. Какие факторы влияют на качество мяса и мясных продуктов?
15. Какие документы оформляют при органолептической оценке качества продуктов?
402
16. В чем с\щнисн> миологических xicsomoh or 1 ре,ie. 1 снiгя пенност нишевых продуктов?
17. Перечислите критерии биологическом нснносш продуктов с приведением расчетных формул.
18. По каким показателям оцениваю, качество нишевых жиров? Как можно практически их определить'?
19. Каков механизм окислительной иорчи жиров и какими мыодамп можно определить продукты окисления жира?
20. В чем сеть гидролитической порчи жиров/? Какие продукты образеются в результате этой реакции и как их можно определить'?
21. Каковы критерии оценки биологической ценност жиров'?
22. Какие методы применяют при оценке свежести мяса и мясных продуктов? Каковы преимущества и недостатки этих методов'?
23. Что такое кулинарная готовность продуктов'? Каковы принципы ее определения'?
24. Представьте общую схему анализа качества пищевых продуктов.
Глава 5 КОНТАМИНАНТЫ МЯСА И МЯСНЫХ ПРОДУКТОВ
Приобретая конкретный мясной продукт, потребитель прежде всего оценивает его товарные качества — внешний вид и свежесть, однако ему подчас совершенно неизвестно о другой важнейшей его характеристике — экологической безопасности, которая характеризуется наличием в нем веществ, способных вызвать специ фическую и неспецифическую токсичность.
Непригодность в пищу продукта, изготовленного из сырья, но лученного от здоровых животных, обусловлена, как правило, ис ключительно внешними источниками. Например, пищевые добавки, используемые в мясном производстве (нитриты, фосфаты, антиокислители, коптильные препараты), в больших дозах могут быть причиной нарушения процессов жизнедеятельности, в связи с чем возникает необходимость строго дозировать их в соответ ствии с пороговой концентрацией.
Другой группой химических веществ, способных вызвать отравления, являются пестициды, гормоны, антибиотики, радионуклиды, содержащиеся в сырье и материалах, а также соли тяжелых металлов (цинка, олова, свинца), которые могут попасть в продукт при контакте с тарой (консервы) или оборудованием. Количество этих веществ регламентируется нормативно-техническими документами.
5.1. ОБЩАЯ ХАРАКТЕРИСТИКА КОНТАМИНАНТОВ МЯСА И МЯСНЫХ ПРОДУКТОВ
Потенциально опасные токсиканты мяса делятся на две большие группы. К первой группе относятся вещества, которые попадают в организм животного с водой и кормом. Такие вещества более или менее прочно связываются в системе метаболизма с органами и тканями сельскохозяйственных животных и могут сохраняться в них достаточно длительное время. К этой группе токсикантов относятся устойчивые неорганические ионы тяжелых и переходных металлов, радионуклиды, а также сложные органические вещества: гормоны, антибиотики и пестициды, способные не только сохраняться в мясных продуктах определенное время, но и
404
вследствие химико-ферментативных и окислительных реакций превращаться в структурные аналоги, многие из которых представляют опасность для организма человека. Например, дехлорирование в структуре пестицида ДДТ вовсе не приводит к снижению токсичности. Теряя содержащийся хлор, пестицид ДДТ превращается в свои аналоги — ДДД и ДДЕ, отрицательно действующие на здоровье человека.
Вторая группа токсикантов включает те химические вещества, которые могут образовываться в мясном продукте в результате разложения тканей либо как продукты жизнедеятельности микрофлоры. Например, в условиях длительного хранения липиды могут образовывать пероксиды и эпоксиды; при нарушении режимов технологической обработки (копчение) могут накапливаться канцерогенные вещества — 3.4-бенз(а)пирен, фенол; при использовании некоторых электрофизических, микробиологических и ферментативных процессов также могут образовываться вещества с токсическим эффектом. К ним относятся нитрозамины, появляющиеся в результате разложения нитритных консервантов и азотсодержащих групп в аминокислотах белков мяса, пирены (бенз(а)пирен) и полихлорированные бифенилы — конечные и весьма стойкие продукты биохимической трансформации органических препаратов первой группы, а также афлатоксины — результат жизнедеятельности патогенных микроорганизмов при соответствующей нежелательной бактериальной контаминации. Формально в эту группу можно отнести также микроорганизмы, наличие которых оценивается по микробиологическим показателям.
Контаминация продуктов животного происхождения болезнетворными микроорганизмами наблюдается вдоль всей «пищевой» цепи; от кормов до готового пищевого продукта (рис. 5.1). В настоящее время насчитывается 18 видов бактерий, 26 видов паразитов, включая простейшие, 9 групп вирусов, 4 группы биотоксинов, 9 групп химических веществ, 3 группы биологически активных веществ, различные токсические растения, грибы, пищевые добавки и т. д., которые играют роль этиологических факторов пищевых отрав-
Корма
Разведение Транспор- Мясо- Обработ- Хранение Потре-
мясного тирование комби- ка мясных и продажа битель
скота на нот продуктов
фермах
Рис. 5.1. Схема «пищевой» пени
405
пений человека. Однако около 80% пищевых отравлений вызвано микроорганизмами, большинство из которых имеют зоонозную природу (например, сальмонеллы, иерсинии. кампилобактерии и др.). За содержанием в мясных продуктах вредных веществ, относящихся к первой группе, необходим тщательный инструментальный контроль. Содержание токсикантов второй группы можно регулировать вплоть до предупреждения их образования, обеспечивая правильные режимы технологической обработки и хранения продукции.
Важным условием получения экологически чистых продуктов является использование экологически чистого сырья. Под последним следует понимать растительное и животное сырье, произведенное в условиях, не допускающих попадания в него вредных или нежелательных компонентов из окружающей среды.
Источниками химических веществ в мясном сырье в основном являются корма и вода. Учитывая низкую [порядка (1—5) • 106 г/дм3] растворимость органических токсикантов в воде, потребляемую животными и птицей воду можно рассматривать как источник загрязнения токсичными элементами и органическими веществами с низкой степенью разложения и высоким кумулятивным эффектом. Пестициды, антибиотики, гормоны попадают в организм животного либо путем непосредственного введения лекарственных средств, либо с кормами. Остаточное содержание таких элементов и веществ в мясе и мясных продуктах зависит от полученной дозы токсиканта на стадии выращивания скота и птицы, скорости его выведения из организма, а также скорости окисления и распада самого вещества.
Металлы являются одним из главных источников загрязнения окружающей среды. В результате выбросов металлургических заводов, сжигания топлива тяжелые металлы отравляют атмосферу, воду, почву и, как следствие, попадают в организм животных и человека. Характерная черта распределения тяжелых металлов в биосфере — весьма значительные колебания концентраций. Усиливающееся загрязнение тяжелыми металлами создает в ряде мест серьезную опасность для здоровья населения.
Наиболее часто в пищевых продуктах встречается свинец, который обладает сильно выраженными токсикологическими и кумулятивными свойствами. Повышенное содержание свинца в окружающей среде связано главным образом с техногенным загрязнением воздуха, почвы и воды. Источниками загрязнения являются энергетические установки, работающие на угле, жидком топливе, двигатели внутреннего сгорания, в которых используется топливо с добавлением антидетонатора — тетраэтилсвинца.
Повышенная загрязненность свинцом отмечается в промышленных районах и городах. Выбросы металлургических заводов, химических предприятий, отходящие газы автомобильного транспорта, попадая в почву, увеличивают содержание свинца в растениях в зонах, прилегающих к автотрассам, в десятки раз. Скарм-406
ливание гравоядным травы или сена из придорожных и пригородных зон приводит к накоплению свинца в организме животных. Часть свинца может выводиться из организма с молоком, и в этом случае молоко становится опасным для употребления в пищу, а часть —• накапливаться в органах и тканях животного. При поступлении в большом количестве может возникнуть острое отравление, при незначительных дозах, но частом потреблении — хроническое (у человека при ежедневном поступлении 2 мг отравление развивается через несколько недель), в результате чего повреждается мозг, развивается рак.
Мышьяк в чистом виде ядовит только в больших количествах. Соединения мышьяка (мышьяковистый ангидрид, арсениты, арсенаты) чрезвычайно опасны и токсичны, обладают высокой степенью аккумуляции. Основную опасность представляет техногенное загрязнение окружающей среды соединениями мышьяка вокруг металлургических заводов, предприятий, перерабатывающих цветные металлы, сжигающих бурые угли. В зоне их действия создается высокая концентрация мышьяковистого ангидрида и других соединений мышьяка в воздухе, происходит их накопление в воде, почве, растениях с последующим перераспределением сначала в органы и ткани животных, потребляющих загрязненные корм, воду, а затем в молоко и мясо.
Вторым источником загрязнения продуктов животноводства мышьяком являются лечебные мышьяковистые препараты (осар-сол, новарсенол, миарсенол, атоксил, аминорсен и др.), акарициды (арсенит натрия, кальция и др.), антигельминтики (арсенат олова, марганца, калия и др.). Применение указанных веществ в животноводстве длительное время или в высоких дозах может привести к их накоплению в получаемых от животных мясе, молоке, а при противочесоточных обработках — в шерсти. Человек принимает ежедневно с пищей около 1,2—2,0 мг мышьяка, что близко к максимально допустимому количеству. При потреблении продуктов, содержащих повышенную концентрацию мышьяка, создается опасность интоксикации и других отрицательных последствий. Соединения мышьяка обладают высокой степенью материальной кумуляции, и поэтому их поступление с пищей в повышенных количествах может привести к острой или хронической интоксикации, развитию злокачественных новообразований. Известны массовые случаи рака кожи у людей, возникающие в результате использования одежды, изготовленной из шерсти, содержащей соединения мышьяка после противочесоточной обработки овец мышьяковистыми препаратами. Карциномы, индуцированные мышьяком, возникают главным образом в коже, а также в легких и печени.
Соединения кадмия довольно широко распространены в окружающей среде. Наибольшие количества их встречаются в почве (в среднем 0.1 мг/т). В более высокой концентрации кадмий со
407
держится в минеральных удобрениях (особенно в фосфорсолсржа -тих) и нексиорых футииидах (до 4.5 Ф). Значительным источником загрязнений являются арматура и пластмасса, окрашенные кадмиевыми соединениями и используемые в пищевой промышленности для машин и оборудования. Токсичность кадмия проявляется весьма сильно, в связи с чем лот металл рассматривается в числе приоритетных загрязнителей. Имеются данные об эмбрио-тропном и канцерогенном действии кадмия. Этот металл способен замешать цинк it энзиматических системах, необходимых для формирования костных тканей, что сопровождается тяжелыми заболеваниями. Кадмий обладает высоким коэффициентом биологической кумуляции (период биологической полужизни 19—40 лет), в связи с чем возникает реальная угроза неблагоприятного воздействия его на население даже при низких дозах.
Ртутные соединения относятся к наиболее опасным глобальным загрязнителям биосферы. Они содержатся в большом количестве в стоках химических заводов (главным образом предприятий, производящих гидроксид натрия, ацетальдегид), бумажных и целлюлозных производств. Их много в продуктах сжигания каменного угля, в результате сжигания которого в атмосферу ежегодно выбрасывается около 3000т ртути. Соединения ртути являются действующей основой многих пестицидов, используемых для протравливания семян растений, некоторых лекарственных препаратов (каломель, сулема, ртутные мази).
В почве ртутные соединения находятся преимущественно в виде менее токсичного сульфида ртути или могут вноситься в нее с протравленными семенами в виде очень ядовитых ртуть-органических соединений, используемых в растениеводстве как фунгициды (гранозан, агрозан, агронал, меркургексан, меркур-бензол и др.).
В природе существует цепочка передачи ртутных соединений: промышленные выбросы, смывы с полей -э водоемы -э зоопланктон, ракообразные, моллюски, рыбы, морские животные (кормовая мука из рыб, морских животных)—> домашние животные —> человек. Скармливание животным рыбы, рыбной муки, других кормов, содержащих соединения ртути, например зерна, обработанного ртутными пестицидами (гранозан, меркургексан и др.), сопровождается длительным (до 60 дней) выделением ртути с молоком, а также может вызвать ее накопление в большом количестве в органах и тканях животных, употребляемых в пищу. Органические соединения ртути — стойкие вещества, очень медленно разрушающиеся и выводящиеся из организма. Они способны накапливаться в организме человека в опасных концентрациях, имея период полураспада примерно 70 дней. Особую опасность представляют метилртуть и алкильные соединения, обладающие высокой токсичностью (с преимущественным действием на центральную нервную систему, 408
почечный эпителий. печень), эмбриотоксическим (мергворожда-емоеть) и мутагенным (эмбриональные уродства) действием.
Цинк входит в состав разнообразных биокатализаторов. Он из бирательно поглощается растениями и животными, концентрируется 15 органах размножения, участвует is биохимических процессах белкового. \тневодного и жирового обмена веществ. В то же время цинк мигрирует среди металлов, поступающих в окружающую среду с технологическими и бытовыми отходами. Суммарная масса выбросов цинка превысила производство этого металла перед второй мировой войной. Валовое содержание цинка в гумусовом горизонте почв СНГ колеблется от 20 до 80 мкг/г. Влияние высоких концентраций пинка проявляется преимущественно в синергическом действии, усиливая эффект других загрязнителей. Заболевания, связанные с загрязнением цинком, недостаточно изучены, хотя в литературе имеются данные, которые говорят о том, что цинк поражает органы дыхания, печень и почки.
Загрязнение продуктов животноводства радиоактивными веществами может происходить в результате непосредственного их воздействия на животных естественных природных источников (сухие и мокрые атмосферные осадки), ионизирующих излучений (первичные и вторичные космические излучения) или вследствие включения радиоактивных веществ в абиотические (почва, вода) или биотические (флора, фауна) компоненты биосферы. В последних случаях передача радиоактивных веществ осуществляется по цепочке: почва (вода)—> растения-э животные-ч> продукты животноводствачеловек. Ионизирующее излучение этих источников различно. В некоторых районах (главным образом за счет выхода на поверхность земли радиоактивных руд, пород) доза радиоактивного излучения может превышать среднемировой фон в 100—500 раз.
В связи с широким использованием ядерной энергии в окружающую среду поступает дополнительное количество радиоактивных веществ. Загрязнителями организма животных и продуктов животноводства могут быть искусственные источники ионизирующих излучений: ядерные и термоядерные взрывы, выбросы из реакторов с термоядерными процессами, отходы атомной промышленности, радиоактивные изотопы, используемые в сельском хозяйстве и других сферах деятельности человека.
Из большого количества радиоактивных веществ наиболее опасными для биологических объектов являются стронций-90 и цезий-137.
Стронций — щелочноземельный элемент второй группы периодической системы элементов Д. И. Менделеева. Он имеет ряд радиоактивных изотопов — от стронция-81 до стронция-97. В ра-диотоксикологическом плане наибольший интерес представляют стронций-89 и стронций-90, образующиеся при делении урана в ядерных реакторах, а также при взрывах атомных бомб как продукты ядерного деления.
409
Стронций-90 — [3-излучатель. имеющий период полураспада 28 лет и энергию [3-частиц 0.54 МэВ. Претерпевая [3-распад. он превращается в дочерний радиоактивный элемент иттрий-90. который находится вместе с ним в равновесном по радиоактивности состоянии. Период полураспада иттрия-90 составляет 64.2 ч. максимальная энергия [3-частиц — 2.18 МэВ.
Являясь аналогом кальция, стронций при поступлении в организм включается в минеральный обмен, его соединения растворимы в воде.
При выпадении на поверхность земли в виде сухих, а чаще мокрых осадков (вместе с атмосферными), в виде радиоактивных отходов в связи с широким использованием атомной энергии в мирных целях стронций-90 включается в компоненты биосферы (почву, воду, растения, животных), мигрирует но биологическим цепочкам и с продуктами растительного и животного происхождения может попасть в организм человека.
В организме стронций-90 хорошо всасывается в желудочно-кишечном тракте, значительные количества его откладываются в скелете. Это приводит к облучению не только самих костей и костного мозга, но и других тканей. Всасывание стронция-90 из желудочно-кишечного тракта колеблется от 5 до 100 % и зависит от многих факторов (рациона, физико-химических свойств соединения, возраста животных и человека и физиологического состояния организма). Значительно больше стронция всасывается из кишечника у молодых животных. Это связано с более высокой потребностью их организма в щелочноземельных элементах, необходимых для построения скелета. Добавка кальция к рациону с целью уменьшить усвоение стронция-90 эффективна только для молодых животных, а для взрослых и старых существенного значения не имеет.
Изотопы стронция имеют скелетный тип распределения. При любом пути поступления в организм они избирательно откладываются в костях. В мягких тканях стронция-90 накапливается не более 1 %. Стронций-90 концентрируется в участках костей, обладающих наибольшей зоной роста. В компактном веществе кости концентрация стронция-90 всегда больше, чем в губчатом. С возрастом животных эта разница уменьшается.
Из организма стронций-90 выделяется при пероральном поступлении в основном с калом, а при ингаляционном — с мочой. Период полувыведения стронция-90 из мягких тканей составляет 2,5—8,5 сут, а из костей — 90—154 сут. Стронций выделяется и с молоком. После перорального поступления количество его в молоке в 8—10 раз ниже, чем после внутривенного или внутрибрюшинного.
Благодаря специфике отложения стронция-90 создаются такие условия, когда облучается не весь организм, а преимущественно скелет и костный мозг.
410
Цезии — элемен! первой ipyiniw периодической системы элементов Д. И. Менделеева. Большинство его химических соединений (хлориды, нитраты. карбонаты) растворимы в возе, поэтому хорошо всасыванием в желудочно-кишечном rpaKie. разносятся кровью по организму и быс тро выводятся из него.
Из радиоактивных изотонов пезия напболыпую биологическую опасность представляет цезий-137. ядра которого при p-распаде излучают (Бчастипы и у-квашы. Период полураспада цезия-137 равен 30 юлам. Является продуктом деления ядер тяжелых элементов.
Глобальные осадки радиоизотопов, в том числе и цезий-37, выпадают в течение ряда лет после ялерного взрыва, загрязняя всю биосферу (воздух, воду, почву и растительность). Степень загрязненности почвы зависит не только от количества годовых атмосферных осадков, но и от локальных условий — тина почв, вида и густоты растительности и агротехнической обработки почвы. Цезий-137 сорбируется почвой значительно сильнее, чем стронций-90, и поэтому выносится из нее с урожаем растительности во .много раз меньше. Органические вещества в почве затрудняют корневое поглощение радиоизотопов. Из влажных почв растения извлекают значительно больше цезия-137, чем на суходольных участках.
Цезий-137 поступает в растения как через корневую систему, так и через наземные их части. Переработка и подготовка кормов к скармливанию могут значительно изменить в них концентрацию радионуклидов. С кормом, водой, почвой, воздухом цезий-137 поступает в организм животных в основном через пищеварительный тракт и дыхательные нуги, а в организм человека он поступает с продуктами питания животного и растительного происхождения, а также с водой и воздухом.
Степень всасывания цезия-137 в желудочно-кишечном тракте достигает 100 %, так как он не образует труднорастворимых соединений. Молодые животные усваивают его быстрее, чем старые. Характер метаболизма цезия-137 своеобразен, сходен с обменом калия и определяется его физико-химическими свойствами. Отмечена исключительно высокая скорость обмена радиоизотопа в звене кровь — органы — ткани. Быстрое снижение концентрации его в крови после поступления в нее объясняется тем, что, с одной стороны, происходит интенсивное включение его в органы и ткани, а с другой — выведение через органы выделения или молочную железу.
Больше всего цезия накапливается в мышцах, сердце, печени, почках и меньше — в коже, крови и жировой ткани. При длительном или хроническом поступлении цезия-137 отмечается постоянное увеличение общего содержания его в организме, а затем наступает состояние равновесия, когда ежедневное поступление его уравновешивается выведением.
411
..... v. ми its. оаршшна ii свинина ) максима п
пая кониин грация этого ранна hi ан она в Оарашшс: в мшч.шне к 2 раза, а в свинине в 3 pa sa меньше, в оленине в 10 par больше, нем 15 мясе других в,идов животных.
Использование в пишу продуктов животноводе!ва. содержащих радиоактивные вещее ша в больших дозах, может вызвать у людей нарушение функций эндокринной. кроветворной, сердечно-сосудистой. иммунной. нервной, половши дыхательной и других систем с развитием тяжелых заболеваний (лейкемии, злокачественных новообразований, дистрофии, ожирения и др.).
Авария на Чернобыльской АЭС резко обострила воздействие ионизирующей радиации на огромные контингенты людей, проживающих на обширных территориях и отличающихся по возрасту, полу, исходному состоянию здоровья и условиям жизни. Следствием радиационной ситуации после аварии на Чернобыльской АЭС является повышение заболеваемости в целом за счет потребления загрязненных радионуклидами продуктов питания, загрязнения почвы, воды и воздуха, воздействия ионизирующей радиации на организм.
Обеспечение радиационной безопасности при повышенном содержании изотопов во внешней среде неразрывно связано с нормированием и контролем концентрации радиоактивных веществ в объектах окружающей среды и организме человека. Для ограничения радиационного облучения человека установлены гигиенические нормативы содержания радионуклидов (цезия-137 и строн-ция-90) в продовольственном сырье и пищевых продуктах, которые разработаны на основе предельно допустимых суточных доз (ПДС) их поступлений в организм в составе пищевых рационов.
Выборочный анализ мясных продуктов показывает, что в 1—5 % образцов содержатся токсичные элементы и соединения в количествах, неприемлемых для безопасного потребления. В силу чрезвычайной важности и глобального характера проблемы контроля за остатками стимуляторов и химических токсикантов в пище в законодательстве развитых стран наблюдается тенденция ужесточения требований к контролю за содержанием указанных веществ в пищевом сырье, расширению перечня контролируемых показателей, снижению предельно допустимых остаточных уровней этих препаратов. Аналитический контроль за уровнем содержания опасных веществ в продуктах осуществляют аккредитованные при соответствующих органах исследовательские сертификационные лаборатории.
Контроль безопасности пищевых продуктов на территории РФ осуществляется системой сертификации в соответствии с правилами ГОСТ Р, согласованными с требованиями международных стандартов в ИСО/МЭК в области стандартизации, а также с собственными стандартными правилами, разработанными с учетом специфики объектов сертификации и обеспечивающими безопас-412
носгь .1.1>! юрытя ц жи аш ж1 мщ ( ср, ификапия мяса и мясных продуктов являснш (юяжгн.жнон Нерен проведением сертификационных испытаний проводят идентификацию продукции на соотвеic 1 вис указанному наименованию по органолептическим и физико-химическим показателям, предусмотренным нормативными документами (НД) на продукцию.
Для сертификации продукцию мясной промышленности разделяют на 6 групп: мясо, колбасные изделия, полуфабрикаты и кулинарные изделия, консервы, жиры и желатин. При этом сертификация продукции в мясной промышленности отличается рядом специфических особенностей:
зависимостью показателей безопасности продукции от безо-пас ности с е л ьс ко хо з я й ст ве и н о го сыр ья;
зависимостью качества мясного сырья от экологического благополучия региона, производящего сырье;
зависимостью качества готовых мясных изделий от качества применяемых специй, пряностей, пряно-вкусовых ароматизаторов и других вспомогательных материалов;
наличием мясной продукции как длительных сроков хранения, так и скоропортящейся.
Содержание веществ, вредных для здоровья человека, в продуктах питания строго нормируется и не должно превышать предельно допустимых уровней, установленных гигиеническими требованиями к качеству и безопасности продовольственного сырья и пищевых продуктов (СанПиН 2.3.2.560—96).
Пищевые продукты и продовольственное сырье подвергаются обязательной ветеринарно-санитарной экспертизе, проводимой государственной ветеринарной службой в соответствии с действующими ветеринарно-санитарными правилами и с оформлением ветеринарного свидетельства, выдаваемого органами государственной ветеринарной службы. Только после ветеринарносанитарной экспертизы проводится санитарно-гигиеническая оценка продовольственного сырья и пищевых продуктов животного происхождения. Новыми санитарными правилами не допускается наличие в продовольственном сырье и пищевых продуктах паразитарных организмов и патогенных микроорганизмов, вызывающих инфекционные болезни животных и человека. Действующие гигиенические нормативы по микробиологическим показателям включают контроль четырех групп микроорганизмов;
мезофильные аэробные и факультативно-анаэробные микроорганизмы и бактерии группы кишечных палочек (санитарно-показательная группа);
условно-патогенные микроорганизмы и сульфитредуцирую-щие клостридии;
патогенные микроорганизмы, в том числе сальмонеллы;
микроорганизмы порчи — в основном дрожжи и плесневые грибы.
413
ч ntn>.iurA> !1>с i пенно! о сырья и 11111 нс вых продуктов
нормируется содержание пес i инидов: гсксахлорииклогексана (с/.-, |3-, у-изомеры). .1,11 и его мегабоидов В продуктах животноводства регламентируется содержание ветеринарных лечебных препаратов. а также антибиотиков. применяемых для откорма, лечения it профилактики заболеваний скота и шины. В мясе и мясных продуктах кон гредируются допущенные к применению в животноводстве кормовые антибиотики — гризин. бацитрацин и лечебные антибиотики тетрациклиновой группы и левомицетин.
В продуктах растительного происхождения необходимо контролировать содержание следующих микотоксинов: афлатоксина В,, дезоксиниваленола. зеараленола, патулина, а в молоке и молочных продуктах — афлатоксина М(. В новых санитарных правилах содержание микотоксинов в мясе, мясных продуктах, яйцах и яй-пенродуктах не регламентируется.
В соответствии с новыми санитарными правилами в продукции отечественного животноводства не контролируется содержание гормональных препаратов. В импортных мясе и мясных продуктах содержание гормональных препаратов, антибиотиков и ветеринарных средств учитывается в экспертном порядке по сертификату страны-экспортера и фирмы-производителя с учетом рекомендаций Объединенного комитета экспертов по пищевым добавкам ФАО/ВОЗ. При необходимости (в конфликтных ситуациях) гормональные препараты в мясных и молочных продуктах определяются в арбитражном порядке. Регламентируется содержание азотсодержащих соединений, в частности нитрозаминов (суммы нитрозодиэтил- и нитрозодиметиламина), и дополнительно для копченых мясных продуктов — полициклических ароматических углеводородов (бенз(а)пирена).
Для ограничения радиационного облучения человека установлены гигиенические нормативы содержания радионуклидов (цезия-137 и стронция-90) в продовольственном сырье и пищевых продуктах.
В табл. 5.1. приведен перечень наиболее часто встречающихся токсикантов мясных продуктов, который объединяет представителей разных классов с существенно разными физико-химическими свойствами и остаточным регламентированным содержанием вещества от 0,5 мкг до 200 мг в 1 кг продукта.
Таблица 5.1
Контаминанты мясных продуктов
Контаминанты Химический класс ПДК, мг/кг
Медь Цинк Свинец Ртуть
Токсичный элемент 5,0
То же 70,0
» 0,5
» 0,03
414
Продолжение
КоН I.IMHII.IHHJ Химически)! кии ПДК. мг/кг
Олово • > 200.0
Хром 0.5
Кадмии O.OS
М ышьяк 0.1
ДИЭТПЛС ГИЛ ЬОЭС Iрол Гормон —
Эстрадиол » 0.0005
Тестостерон 0,015
Нитрозоди этил амин Нитрозами н 0,001
Нитрозоди метилам и > i » 0,001
Тетрациклин Антибиотик < 0,01*
Левомицетин » < 0,01*
Стрептомицин » < 0,01*
Гризин < 0.50*
Бацитрацин » < 0,02*
(Бензил) пенициллин » < 0,01*
ДДТ Хл о рс од е ржа щи й 0,1
пестицид
ДДД То же 0,1
ДДЕ » 0,1
Гексахлорциклогексан » 0,1
Альдрин » Не допускается
Цезий-137 Радионуклид 160-320 Бк/кг
Стронций-90 » 50—200 Бк/кг
* Допустимое содержание антибиотиков приведено в ед/г.
5.2. МЕТОДЫ КОНТРОЛЯ БЕЗОПАСНОСТИ МЯСА И МЯСНЫХ ПРОДУКТОВ
Оптические колориметрические методы контроля токсичных элементов достаточно просты и используются как для их качественного обнаружения, так и количественного определения. Они предусматривают получение растворов токсичных элементов и проведение одной или нескольких химических реакций с развитием специфического окрашивания.
Распространенными методами количественного определения токсичных элементов в пищевых продуктах являются атомно-абсорбционная спектрометрия и вольтамперометрия, связанная с использованием ртутного капающего электрода (полярография).
Атомно-абсорбционная спектрометрия предложена Уолшем в 1955 г. Метод сразу получил признание в связи с рядом многих своих достоинств.
415
Наиболее вероятным изменением энср!етичсского состояния атома при возбуждении является его переход на уровень, ближайший к основному энергетическому состоянию, т. е. резонансный переход. Если на невозбужденный атом направить излучение с частотой, равной частоте резонансного перехода, кванты света будут поглощаться атомами и интенсивность излучения будет уменьшаться. Использование этих явлении составляет физическую основу атомно-абсорбционной спектрометрии.
Принципиальная схема атомно-абсорбционного спектрофотометра приведена на рис. 5.2.
В атомно-абсорбционном методе анализа в качестве источников излучения чаще всего используют специальные газоразрядные лампы с полым катодом. Конструкция ламп такова, что в спектре испускания интенсивно проявляются спектральные линии атомов, входящих в состав материала катода, или веществ, специально помещенных в полость катода. Изменяя материал катода или состав помещаемого в полость катода вещества, можно получать спектры испускания различных атомов. Обычно каждая лампа для атомно-абсорбционного анализа дает спектр испускания атомов какого-либо одного элемента (табл. 5.2).
Для определения нескольких элементов в пробе необходимо иметь набор ламп на различные элементы, поскольку лампы, позволяющие определять сразу несколько элементов, пока не нашли широкого применения в практике атомно-абсорбционного анализа. Таким образом, несколько элементов определяют при последовательной замене ламп, используя их поочередно в качестве источников излучения.
Рис. 5.2. Принципиальная схема двухлучевого атомноабсорбционного спектрофотометра:
7 —лампа с полым катодом; 2—модулятор; 3—зеркало; 4— щелевая горелка; 5— пламя; 6—тонкая пластинка, обеспечивающая наложение двух лучей; 7— входная щель монохроматора; .У—дифракционная решетка; 9—выходная щель; 10 — фотоумножитель; // — усилитель; /2— измерительный блок
416
Г а б л и ц а 5.2
Длины волн резонансных линий и максимальная сила тока для ламп с полым катодом
О и рслс.т я с мы и ал с мс i i t Диша воины резонансной линии, нм Максимальная сила тока. мА
Са 422.7 20
Cd 228.8 10
Со 240.7 25
С г 357.9 20
Си 324.7 20
Fc 248.3 20
Mg 285.2 20
Мп 279,5 20
Ni 232,0 20
Pb 217.0 15
Zn 213,8 15
Мерой поглощения излучения служат обычно показания прибора, прокалиброванного в единицах оптической плотности или пропускания, при условии, что первое измерение соответствует 100% пропускания. Оптическая плотность прямо пропорциональна концентрации поглощающих атомов, поэтому калибровочный график обычно строят в координатах: оптическая плотность — концентрация определяемого вещества.
Методики атомно-абсорбционного определения разработаны более чем для 70 элементов (Mg, Zn, Си, Са, Pb, Fe, Ag, Ni, Hg, Cd, Bi и др.), в том числе токсичных.
Атомно-абсорбционный спектральный анализ получил широкое распространение в практике вследствие многих своих достоинств. Однако метод также имеет ряд ограничений. Атомно-абсорбционным методом не определяются элементы, резонансные линии которых лежат в далеком ультрафиолете (углерод, фосфор, галогены и др.). Необходимость растворения пробы также можно рассматривать как недостаток, поскольку эта операция удлиняет анализ. Однако работа с растворами упрощает выбор эталона и обеспечивает высокую воспроизводимость результатов. К существенным недостаткам метода относится невозможность одновременного определения нескольких элементов, хотя для этого имеются все предпосылки.
Вольтамперометрия основана на изучении поляризационных, или вольтамперных кривых (кривых зависимости силы тока от напряжения), которые получаются, если при электролизе раствора анализируемого вещества постепенно повышать напряжение и фиксировать при этом силу тока. Электролиз следует проводить с использованием легкополяризуемого электрода с неболь-
417
j.iioii поверхностью, на котором происходит элсктровосстанов-ление или электроокисление вещества.
Изменение внешней ЭДС в системе, где катодом служит ртутный капающий электрод, а анодом — практически неполяризуемый каломельный электрод, будет полностью идти на изменение потенциала катода. Если в растворе нет веществ, способных восстанавливаться под действием электрического тока, сила тока / будет пропорциональна приложенному напряжению £ (закон Ома):
1 = E/R. (5.1)
где R — сопротивление.
В присутствии веществ, способных восстанавливаться на ртутном электроде в области исследуемых напряжений, вид кривой зависимости тока от напряжения существенно изменится. По достижении потенциала восстановления ионы начнут разряжаться на ртутном катоде нередко с образованием амальгамы:
М"+ + пе~ + Hg = M(Hg), (5.2)
где M(Hg) — условное обозначение амальгамы металла, которая образуется при растворении выделившегося металла в ртути.
В результате процесса сила тока в цепи начнет возрастать, и концентрация восстанавливающихся ионов у поверхности ртутной капли уменьшится. Однако за счет диффузии из массы раствора к поверхности капли доставляются новые порции ионов. Сила тока в цепи будет зависеть от скорости диффузии, которая пропорциональна разности концентраций ионов металла в массе раствора (cfj и в приэлектродном слое (см). Сила полярографического тока I будет пропорциональна этой разности:
I = С - Ч,), (5.3)
где км — постоянная, характеризующая процесс диффузии ионов металла из раствора в приэлектродный слой.
Концентрация восстанавливающегося иона в глубине раствора постоянна, так как электролиз идет при очень небольшой силе тока (порядка 10-5 А), а концентрация в прикатодном слое близка к нулю. Поэтому разность концентраций, определяющая скорость диффузии при данной температуре, будет постоянна, что и приведет к постоянной скорости поступления ионов к катоду. Наступившее состояние равновесия будет характеризоваться постоянной силой тока, не изменяющейся при дальнейшем увеличении напряжения. Этот постоянный ток, контролируемый диффузией, 418
называют диффузионным и обозначают I Исходя из уравне ния (5.3) и допущения с,, = 0 сида диффузионного тока
Г/ = АЗ,4 <5
Сида диффузионного тока прямо пропорциональна концентрации восстанавливающегося иона в массе раствора. При сочетании уравнений (5.3) и (5.4) получим
/ = </-А;Л| (5.5)
или
_ Л/ - /
Ci ,z (5.6)
Лм
Концентрация атомов металла на поверхности амальгамы, образовавшейся в результате процесса, пропорциональна силе полярографического тока:
/
= <5-7> 'mi
где /^ — постоянная, характеризующая процесс диффузии атомов металла с поверхности в глубину ртутной капли.
Напряжение, при котором начинается электролиз, зависит прежде всего от природы восстанавливающихся ионов. Оно зависит также от концентрации этих ионов в растворе, от присутствия в растворе других электролитов, а также от того, находится ли определяемый металл в виде хлорида, нитрата, аммиачного комплекса и т. д. Имеет значение также характер анодного процесса.
В самом деле, напряжение разложения электролита представляет собой алгебраическую разность потенциалов анода и катода*:
Г = £,-4 (5.8)
Запишем уравнение Нернста для реакции (5.2):
£. „ 0,058, см
£ = Л« + дг18ту <5-9)
где см, «^ — концентрации ионов металла в водном растворе вблизи поверхности
ртутной капли и атомов металла в поверхностном слое ртутной капли.
* Падением напряжения в растворе можно пренебречь, так как обычно оно невелико.
419
Подставляя значения см и с, из \равнении (5.6) и (5.7) в уравнение Нернста (5.9). получим
0,05(5 , /
--------(Т-------------
п = . - /
С/
(5.10)
Это уравнение называют уравнением полярографическом волны.
Потенциалом полуволны называют то значение потенциала, при котором полярографический ток достигает половины предельного значения, т. с.
(5.1 Г)
Типичная зависимость силы тока от приложенного напряжения (полярограмма) показана на рис. 5.3. Потенциал полуволны является качественной характеристикой иона в растворе данного фонового электролита, и его определение составляет основу качественного полярографического анализа.
Связь диффузионного тока Idc концентрацией иона см и другими величинами выражается в виде уравнения Ильковича:
/d=6Q5nD^2/n2/2ty('cy[. (5.12)
где сила тока, мкА; п — количество электронов, участвующих в электрохимическом восстановлении определяемого иона; D— коэффициент диффузии определяемого иона, см2 • с~ '; т — масса ртути, вытекающей из капилляра за I с, мг с”1; /—период капания, с; ^ — концентрация восстанавливающегося иона, ммоль/дм3.
Среди величин, входящих в это уравнение, труднее всего поддается экспериментальному определению коэффициент диффузии D, а использование соответствующих справочных данных не всегда возможно. Поэтому коэффициент пропорциональности между концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. Действительно, при постоянных условиях полярографирования D, т и t постоянны и уравнение примет соответствующий вид
Е1/2 е
Рис. 5.3. Полярограмма:
1 — остаточный ток; 2 — диффузионный ток
4=4- (5.13)
Линейная зависимость (5.13) является основой количественного полярографического анализа.
420
Принципиальная схема полярографической установки представлена на рис. 5.4. Анализируемый раствор находится в электролизере 1. на дно которого помешен слой ртути, являющийся анодом. Часто в качестве анода используют насыщенный каломельный электрод (НКЭ). Катодом служит ртутный капающий электрод 2, соединенный с резервуаром ртути 3. Внешнее напряжение, подаваемое на электроды, можно плавно менять с помощью реохорда 5 или де
Рис. 5.4. Схема полярографической установки:
1 — электролизер; 2— ртутный капающий электрод (катод); 3 — резервуар ртути; 4- гальванометр; 5— реохорд
лителя напряжения и измерять при этом гальванометром 4 силу тока, проходящего через раствор.
Как уже отмечалось, на-
пряжение, которое подается на электроды, будет практически целиком определять потенциал катода (капающего ртутного электрода).
В вольтамперометрии с успехом применяют также твердые микроэлектроды, изготовляемые из благородных металлов или графита. Основными достоинствами твердых электродов являются возможность работы в более положительной области потенциалов (до 1,3 В), чем с ртутным электродом (ртутный капающий электрод используется в области примерно от плюс 0,3 до минус 2,0 В) и их нетоксичность (пары ртути, как известно, чрезвычайно ядовиты, и работа с ртутным электродом требует строгого соблюдения специальных правил техники безопасности).
Однако использование твердых электродов также имеет свои трудности, связанные главным образом с обновлением поверхности электродов. Стационарные твердые электроды не нашли широкого
применения в практике из-за медленного установления предельного тока, невысокой чувствительности и других недостатков.
Наиболее широко в количественном полярографическом анализе применяется метод калибровочного графика на основе уравнения (5.10). График строят по данным полярографирования нескольких стандартных растворов. На оси ординат откладывают высоту полярографической волны, пропорциональную силе диффузионного тока, а по оси абсцисс — концентрацию анализируемого вещества. В соответствии с уравнением (5.10) калибровочный график должен представлять собой прямую линию, проходящую через начало координат. Метод дает точные результаты при
421
н.кляш с i poi ои идет ичности условий i юлярш рафирования стандартных растворов и неизвесiнои пробы. К условиям полярогра-фировання относя гхсловия работы капилляра, температуру и среду (фоновый электролит). Метол калибровочного графика наиболее трудоемкий, но и наиболее точный.
При анализе некоторых хорошо изученных систем, для которых применимость уравнения (5.10) установлена вполне надежно, час то применяемся менее трудоемкий метод стандартных растворов. В этом методе в строго одинаковых условиях снимают полярограммы стандартного и анализируемого растворов и из пропорции. основанной на уравнении (5.10). рассчитывают неизвестную концентрацию с.:
(5.14)
где — концентрация стандартного раствора; /у. и /у, — высота водны при поля-poi рафировании соответственно анализируемого и стандартного растворов.
Метод применим только в условиях строгой стандартизации у с л о в и й п о л я р о гра ф и ро ва н и я.
В количественной полярографии широко распространен также
метод добавок.
Пусть при полярографировании исследуемого раствора сила диффузионного тока
/Л = Ч- (5.15)
Добавим к этому раствору известное количество стандартного раствора сст и снова определим диффузионный ток:
/Л.+ст - (СЛ. + Сст).
(5.16)
При почленном делении уравнений (5.15) и (5.16) получим
__1X _ ^х
/ ~ с + с (5.17)
1 Л' + СГ с.\' т сст
откуда
е = е
v Сст , (5.18)
1 X + СТ 7 X
По соотношению (5.14) находим концентрацию анализируемого раствора.
422
Вольтамперометрический метод достаточно универсален и применим к многочисленному кругу объектов. Основными достоинствами метола являются быстрота анализа, возможность оиретелс-ния нескольких веществ в смеси без предварительного раз.тс-ления. достаточно высокая точность и применимость к анализу небольших содержании определяемого элемента. Погрешность полярографического метода в обычных условиях составляет ± 2 сс для растворов концентрацией 1() ?- 10 4 моль/дм? и около ± 5 (с для более разбавленных. При сочетании вольтамперометрического метола с методами экстракции, хроматографии и т.п. предел обнаружения снижается еще больше.
Для определения сложных химических контаминантов используют две группы аналитических методов. К первой относятся традиционно применяемые спектральные (например, флуориметрия) или хроматографические методы: тонкослойная хроматография (ТСХ), высокоэффективная жидкостная хроматография (ВЭЖХ). жидкостная хроматография с масс-спектрометрическим детектированием (ЖХМС). газожидкостная хроматография (ГЖХ).
Вторую группу составляют современные иммунные методы: им-муноферментный метод (ELISA), радиоиммунный метод (R1A).
Среди требований, предъявляемых к методам экспресс-мо-ниторинга продовольствия, основными являются чувствительность и селективность метода, а также время и стоимость выполнения анализа.
Метод ТСХ характеризуется недостаточной чувствительностью и экспрессностью. Хроматографические методы с масс-спектро-метрическим окончанием отличаются высокой стоимостью оборудования и малопригодны для массовых анализов в силу сложности выполняемых операций и длительной подготовки проб. Реализация методов ГХМС, ЖХМС и ВЭЖХ требует высокой квалификации персонала.
Методы контроля первой и второй групп различаются по чувствительности определения в 5—10 раз, а продолжительность анализа одной пробы для методов первой группы составляет в среднем не менее 30 мин, не считая подготовки проб, осуществляемой, как правило, в десятки стадий.
Наиболее удачное сочетание чувствительности, экспрессности и стоимости анализа отличает иммуноферментный метод, позволяющий быстро проводить скрининг пищевой продукции по максимальному числу показателей. Техническая реализация данного метода состоит в том, что экстракт из анализируемого образца пропускают через аффинную колонку, надетую, например, на шприц, в результате чего полностью отделяется анализируемое вещество. Последующее его смещение путем капельного прибавления специальных ферментсодержащих конъюгатов позволяет по развитию цветной окраски и сравнению со стандартом простым спектрофотометрированием определять содержание веществ на
423
уpworiv ,i.u u.i .мм/м (tn/см ). 15 настоящее время ряд зарубежных фирм освоили серийный выпуск специальных наборов и методов определения этих веществ но типу экспресс-анализа.
В табл. 5.3 приведены перечень и характеристика базовых аналитических методов. применяемых для количественного определения сложных химических контаминантов мясных продуктов.
Г а б л л на 56
Характеристика аналитических методов количественного определения контаминантов
Мею.i ।ice.'ie -лов. шин I j 1 Определяемое венке"! во 1 । - - . . _ . _ j Предел оона ! [П-ження L ПЛ К. мг кг Время анализа (включая по. 1-югонку проб), 1
вэжх Афлатоксин В, 10 мкг/кг 0.005 14
Флуорл.мет рия Бенз(а)ппрсн 0.2 мкг/кг 0.001 ('
тех Тетрациклин 0.1 мкг/кг 0.01* 5
Флуориметр ИЯ 1 МКГ/К1 0.0 г 4
ELISA 6 и г/г 0.0 г 3
ELISA Левомицетин 1 нг/г (мкг/кг) 0.01 1
тех » 0.5 мкг/кг 0,01* 5
гжх Эстадио.т 17р 0,2 нг/г 0.0005 10
R.IA 4 нг/г 0.0005 8
тех Ди31 иле гил ьбэстрол 0.01 мг/кг — 12
гжх » 0.5 мкг/кг — 12
гжх ДДТ 0.007 мг/кг 0.1 8
тех ддт 10 мкг/к г 0.1 5
* ПДК антибиотиков приведены в сд/г.
Контроль содержания радионуклидов в мясном сырье и продуктах осуществляют на основе современных экспресс-методов радиометрии и радиохимии.
Безвредность пищевых продуктов оценивают также специальными медико-биологическими методами, в частности путем введения водной вытяжки из продукта внутривенно, внутрибрюшинно и под мозговую оболочку экспериментальным животным, в эмбрион куриного яйца с последующим инкубированием последнего и определением патологических изменений в развитии животных и характере их поведения. Исследование общего состава белка сыворотки и фагоцитарной активности лейкоцитов крови животных или человека при длительном кормлении опытным рационом также дает возможность отметить отклонения в иммунобиологической реактивности организма. В некоторых случаях токсичность изучают в опытах на культуре ткани — на клетках почек эмбриона человека по характеру цитологического действия субстрата.
Таким образом, опыты in vitro — на культуре ткани и in vivo — на экспериментальных животных (в хроническом и остром опы-424
тах). так же как и комплекс химико-микроОиологических методов. позволяют установить наличие юксических веществ в пищевых продуктах, реализовал, эффективные пути обезвреживания н обеспечить их безопасность для здоровья человека.
Актуальным является не только контроль, но и разработка рекомендаций по совершенствованию технологических процессов. обеспечивающих минимальное попадание токсических элементов с продуктами питания в организм человека. Технологическая обработка мясного сырья играет немаловажную роль, так как такие технологические параметры и процессы, как степень измельчения сырья, продолжительность посола, применение отдельных компонентов при посоле, подбор рационального вида термической обработки, могут изменить нс только конечные органолептические показатели готового продукта, но и показатели его безвредности.
В данной главе рассмотрены методы, нашедшие наибольшее распространение и доступные для учебных, производственных и аккредитованных лабораторий. В связи с этим специалистам мясной отрасли необходим практический навык в анализе различных токсичных веществ для совершенствования технологии и управления технологическими процессами.
Лабораторная работа Ns 1
ОПРЕДЕЛЕНИЕ МИКРОБНЫХ КОНТАМИНАНТОВ В МЯСЕ
Цель работы. Освоить практические методы определения микробных контаминантов в мясе на основе бактериологического анализа.
Задачи. Отбор проб мяса для проведения микробиологических анализов: изучение и реализация схемы бактериологического исследования мяса; санитарная оценка мяса.
Объекты исследования. Образцы мяса различных видов убойных животных и птицы.
Материалы, реактивы и оборудование. Стерильные ножницы: стерильные (стеклянные) стаканы или колбы: электрический гомогенизатор; фарфоровая ступка: спиртовка: петли для засева; пастеровские пипетки: предметные стекла; покровные стекла; фильтровальная бумага; карандаши по стеклу: скальпель; пинцет; пипетки мерные с делениями вместимостью I см?; лупа ручная; пробирки Уленгута: агглютиноскоп и вогнутое зеркало (от микроскопа); микроскоп; чашки Петри; термостат; штативы большие (для постановки линейной агглютинации); автоклав: аппарат Коха; ватно-марлевый фильтр: культуры сальмонеллезных и кишечных бактерий на скошенном агаре; мясо, в которое инъецирован смыв агаровой культуры сальмонеллезных бактерий; генциан
425
фиолетовый (генцианвиолст) или кристаллический фиолетовый (кристаллвиолет): фенол: этанол: иод: иодил калия: сафранин и его водный раствор с массовой долей 2 С: фуксин и его водный раствор: синь метиленовая и ее водный раствор массовой долей 0.5 %; раствор эозина (бактериологического) массовой долей 2 С: лактоза; маннит: карболовый раствор генцианвиолета или крис-таллвиолета (для окраски мазков по Граму): раствор Л юголя: масло вазелиновое.
Питательные среды. Мясо-пептонный агар (МПА): мясо-пептонный бульон (МПБ): элективные среды (Эндо. Левина, Плос-кирева. Хейфеца); среды обогащения (селенитовый Ф-бульон: среды Мюллера. Кауфмана. Киллиана: хлористомагниевая среда «М»); среда Симмонса; желатин; бульон, основной раствор Хот-тингера: агар с кровью; висмут-сульфитный агар; среда Китта— Тароцци; набор флуоресцирующих иммунных сывороток; физиологический раствор.
Приготовление реактивов для окраски мазков, растворов, смесей, питательных сред. Карболовый раствор генцианвиолета (для окраски по Граму): 1 г генцианвиолета растирают в ступке с 2 г кристаллической карболовой кислоты (фенола). Во время растирания небольшими порциями прибавляют 10 см3 этанола (96 об.%). После полного растворения краски прибавляют при постоянном помешивании 100 см3 дистиллированной воды и фильтруют через бумажный фильтр. Растворы карболового генцианвиолета нестойкие и хранению не подлежат.
Раствор Лю гол я: в 10 см3 дистиллированной воды растворяют 2 г иодида калия и добавляют 1 г кристаллического иода. Раствор выдерживают в течение 5—6 ч до полного растворения иода, после чего прибавляют 290 см3 дистиллированной воды. Хранят раствор в склянке из темного стекла.
Красящая фильтровальная бумага (для видоизмененной окраски по Граму в модификации Синева): в 100 см3 этанола (96 об.%) растворяют 1 г кристаллвиолета и 1 см3 глицерина. Краску сливают в лоток. Полоски фильтровальной бумаги длиной 30—50 см и шириной 2,0—2,5 см погружают на несколько секунд в краску так, чтобы она окрасилась с обеих сторон. Пинцетом вынимают окрашенные полоски, дают стечь краске и подвешивают их на шпагате для высушивания. Сушат полоски на воздухе при температуре 18—23 °C. Высушенные полоски хранят в закрытой банке из темного стекла.
Раствор генцианвиолета (для окраски капсул по методу Ребигера): 15—20 г генцианвиолета растворяют в 100 см3 раствора формалина массовой долей 40 %. Раствор оставляют на 8—10 ч при температуре 20 °C, а затем фильтруют, после чего он готов к употреблению.
Раствор сафранина массовой долей 2% (для окраски капсул методом Ольта). Сафранин растворяют в воде, 426
доведенной до кипения, и фильтрую! через бумажный фильтр. Раствор готовят непосредственно перед употреблением.
Водны й р а с т в о р ф у к с и н a Ц и л я: 15 ступке растирают 1 г основного кристаллическою фуксина. 5 г кристаллической карболовой кислоты (фенола) и 0.5 см-: пиперина. Во время растирания приливают небольшими порциями 10 см3 этанола (96 об.%). После того как краска полностью растворится, прибавляют при постоянном перемешивании 100 см3 дистиллированной воды. Затем раствор краски фильтруют. Фуксин Циля стоек, поэтому его хранят во флаконах из темного стекла с притертой пробкой.
Мясо-пептонный агар (МПА): к ЮООсм3 мясопептонного бульона перед стерилизацией добавляют 20 г агара и кипятят на слабом огне при постоянном помешивании до полного растворения. Мясо-пептонный агар, охлажденный до 50—55 °C, осветляют яичным белком (из расчета один белок на 1000 см3 МПА), помещают в автоклав, не завинчивая его крышку, или в аппарат Коха на 1 ч. чтобы белок свернулся. Оседая, он увлекает за собой взвешенные частицы. Горячий МПА фильтруют через ватно-марлевый фильтр, устанавливают pH 7,0— 7,4, разливают во флаконы или пробирки и 20 мин стерилизуют в автоклаве при 120 °C.
М я с о-п е п т о н н ы й бульон (М П Б) из м я с н о й воды: к 1дм3 мясной воды добавляют Юг пептона и 5г хлорида натрия, устанавливают pH на уровне 7,3—7,4, кипятят в течение 20 мин и фильтруют. Стерилизуют 20 мин при давлении 105 Па.
Мясная вода: к 1 кг мясного фарша, приготовленного из говядины высшего сорта, добавляют 2 дм3 водопроводной воды и настаивают в холодильнике в течение 24 ч; затем кипятят в течение 30 мин при постоянном помешивании и фильтруют через ватно-марлевый фильтр. К полученному фильтрату добавляют воду до первоначального уровня. Стерилизуют 20 мин при давлении Ю3 Па.
Основной раствор Хотт ингера:! 000 г мяса, освобожденного от жира и сухожилий, нарезают кусками размером 1—2 см и опускают небольшими порциями в кастрюлю с двойным объемом кипящей водопроводной воды. Кипятят в течение 15—20 мин, пока цвет мяса не станет серым; это указывает на то, что белки свернулись. Мясо вынимают шумовкой и измельчают на мясорубке. Оставшуюся жидкость фильтруют и в ней устанавливают pH 8,0 с помощью раствора гидроксида натрия массовой долей 10%. Фарш опускают в отфильтрованную жидкость и охлаждают до 40 °C в открытой кастрюле, затем добавляют очищенную от жира, соединительной ткани и дважды измельченную поджелудочную железу массовой долей 10 % к полученной взвеси (на 1000 см3 жидкости 100 г железы) или сухой панкреатин массо-
427
вон нолей Ню--1,0 1 <_ (в зависимоеth oi сю акзивнос i и г Полученную смесь хорошо размешиваю!, после чего снова нолшелачиваю! раствором гпл.роксила натрия массовой лолеи 10 с7 но pH 7.8—8.0. Подщелачивание повторяют через 30 мин. Отсутствие сдвша реакции смеси указывает на ее доброкачественность.
После доведения pH до 7.8—8.0 смесь переливают в бутыль с хорошо подобранной резиновой пробкой с таким расчетом, чтобы 1/3 часть бутыли оставалась свободной. Затем добавляют хлороформ объемной долей 1—3% (15 холодное время гола — меньше, чем в теплое), закрывают бутыль пробкой и несколько раз встряхиваю!. после чего вынимают пробку для удаления избытка хлороформа и тут же снова закрывают.
Через 1—2 ч после добавления поджелудочной железы или панкреатина проверяют реакцию, устанавливают pH 7,4—7.6 и оставляют смесь при комнатной температуре на 7—16 сут до образования аморфного осадка. Первые 3—4 сут переваривания ежедневно проверяют реакцию среды и поддерживают pH на уровне 7,4—7.6. В течение этого времени встряхивают жидкость не менее трех раз в сутки так, как указано выше. В дальнейшем встряхивать можно реже. За 1—2 сут до окончания переваривания встряхивание прекращают, чтобы гидролизат отстоялся. Конец переваривания характеризуется следующими признаками: на дне бутылки собирается аморфный осадок; жидкость над осадком просветляется и принимает соломенно-желтый цвет; гидролизат легко фильтруется; реакция на триптофан с бромной водой должна быть положительной (в пробирку наливают 3—4 см3 фильтрованного гидролизата, добавляют три-четыре капли бромной воды); при наличии триптофана жидкость принимает розово-фиолетовый цвет, в то время как контрольная пробирка с гидролизатом без бромной воды имеет желтое окрашивание. Раствор гидролизата массовой долей 5 % должен содержать 1100—1200 мг% общего азота. После гидролиза жидкость фильтруют через полотняный или бумажный фильтр, сливают в бутыль и стерилизуют в течение 30 мин при температуре 120 °C.
Приготовленную среду можно хранить в течение 5 мес. Перед употреблением жидкость фильтруют.
Бульон Хоттингера: к 100см3 основного раствора Хоттингера добавляют 500 см3 водопроводной воды, 3 г хлорида натрия и 0,12 г двузамещенного фосфата калия. Полученный раствор кипятят в течение 10 мин, фильтруют, устанавливают pH 7,2—7,4 и стерилизуют в течение 20 мин при 120 °C.
Мясо-пептонный бульон (МПБ) на основе бульона Хоттингера; основной раствор Хоттингера разводят в 5—10 раз (до соломенного цвета). К 1 дм3 разведенного бульона добавляют 10 г пептона и 5 г хлорида натрия. Затем устанавливают pH на уровне 7,3—7,4, кипятят 20 мин и фильтруют. Стерилизуют 20 мин при давлении 10" Па.
428
ф и з п о .1 о г и ч с с к п й р а с г в о р: 1S.5 г \. ч. хлорида натрия растворяют в ЮООсм' дистиллированной волы. Раствор доводят до кипения, охлаждают, фильтруют через бумажный фильтр, разливают в колбы или пробирки и стерилизуют в автоклаве в течение 20 мин при 120 СС.
Сред а Л е в и н а: к 100 см-' расплавленного мясо-пептонного агара pH 7.0—7.4 добавляют 2 см-' предвари тельно подогретого на водяной бане водного раствора метиленовой сини массовой долей 0,5 76, 1,5 см ' раствора эозина (бактериологического) массовой долей 2 76, 2 г лактозы и 0.2 г двузамещенного фосфата калия. Растворы красок готовят на дистиллированной воде и стерилизуют в течение 1 ч при 100 °C. После добавления всех компонентов в указанном порядке среду тщательно перемешивают, разливают в чашки и подсушивают. Готовая среда должна быть красно-фиолетового цвета.
Среда Левина может быть приготовлена из сухой стандартной среды по методике, указанной на этикетке.
Среда Эндо и бактоагар П л ос ки рева: готовят из сухих стандартных сред. Способ приготовления указывается на этикетке.
Среда Мюллера: вначале готовят растворы тиосульфата натрия и Люголя. Для приготовления раствора тиосульфата натрия в мерном цилиндре со 100 см3 дистиллированной воды растворяют 50 г тиосульфата натрия. Раствор переливают в бутыль и стерилизуют текучим паром в течение 30 мин.
Для приготовления раствора Люголя в 100 см3 дистиллированной воды растворяют 25 г металлического иода и 20 г иодида калия.
Для приготовления среды Мюллера в стерильные флаконы помещают по 4,5 г мела и стерилизуют их сухим паром в течение 1ч. Затем наливают в каждый флакон по 90 см3 бульона Хоттингера, содержащего 130—150 мг% аминного азота, устанавливают pH 7,2—7,4 и стерилизуют 30 мин при температуре 120 °C. После стерилизации вновь устанавливают pH 7,2—7,4, для чего проверяют в одном из флаконов и определяют необходимый для подтитровки данного объема среды объем соляной кислоты или гидроксида натрия. Перед употреблением в асептических условиях добавляют по 2 см3 раствора Люголя и по 10 см3 раствора тиосульфата натрия.
Среда Кауфмана: к 500 см3 стерильной среды Мюллера добавляют 25 см? стерильной желчи крупного рогатого скота и 5 см3 водного раствора бриллиантовой зелени массовой долей 0,1 %. Смесь хорошо взбалтывают, разливают в стерильные флаконы, но не стерилизуют.
Селенитовый Ф-б у л ь о н: готовят из двух основных растворов —А и Б. Раствор А (pH 6,9—7,1): 5 г пептона фирмы «Спофа» (Чехия) или «Рихтер» (Венгрия), 7 г безводного двузаме-
429
________ наipn>i. ? i однозамешснного фосфата натрия, 4г х.ч. лактозы растворяют в 100см' дистиллированной воды; стерилизую! 30 мин ири 11? (Раствор Б; раствор кислого селенита натрия массовой долей 10 сс готовят на стерильной воде непосредственно перед употреблением.
При изменении серии любого из входящих в среду компонентов (пептона, кислою селенита натрия, фосфаюв) предварительно проводят полтитровку. для чего экспериментально определяют точную пропорцию двузамешенного безводного фосфата натрия и однозамешснного фосфата натрия, которая с используемыми образцами пептона и селенита назрия лаез pH нс выше 6.9—7.1. что регулируется изменением соотношения фосфатов.
Для приготовления селенитовой среды к 50 см3 раствора А в стерильных условиях добавляют 2 см3 раствора Б. Среду разливают в стерильные флаконы или колбы, но нс стерилизуют.
Хлор и с т о м а г н и е в а я с р е д а М (модифицированная); готовят из трех основных растворов; А. Б и В. Раствор А; 0,42 г сухого ферментативного пептона, 0.7 г хлорида натрия, 0,15 г однозамещенного фосфата калия. 4 см' дрожжевого автолизата растворяют в 90 см3 дистиллированной волы. Раствор Б; 3,6 г кристаллического хлорида магния растворяют в 9 см3 дистиллированной воды. Раствор В: 0,09 см3 водного раствора бриллиантовой зелени массовой долей 5 %.
Для приготовления хлорисгомагниевой среды М (модифицированной) растворы А, Б и В смешивают и стерилизуют в течение 30 мин при температуре 112,5 °C и давлении 5 • 104 Па.
Концентрированную среду готовят, удваивая содержание всех компонентов, кроме дистиллированной воды.
При отсутствии дрожжевого диализата допускается применять дрожжевой экстракт.
Дрожжевой экстракт: в 2000 см3 дистиллированной воды растворяют 1000 г прессованных хлебопекарных дрожжей. Полученную суспензию стерилизуют 30 мин текучим паром, затем отстаивают в холодильнике при 4 °C в течение 5—6 сут. Жидкость над осадком декантируют, добавляют 2,5 см3 раствора кристаллви-олета массовой долей 0,01 %, разливают во флаконы или пробирки и вновь стерилизуют при 100 °C в течение 30 мин. Экстракт хранят в холодильнике при 4—6 °C в течение двух недель.
Среда Киллиана: к 100см3 стерильного питательного бульона (pH 6,8—6.9) в стерильных условиях добавляют 1см3 раствора бриллиантовой зелени массовой долей 0,1%, Раствор бриллиантовой зелени: к 0,1 г бриллиантовой зелени добавляют 100 см3 дистиллированной воды, наливают во флаконы с резиновой или корковой пробкой и помещают в термостат при 37 °C на сутки.
Среда Крум вид е—О лькеницкого (в модификации Ковальчука): 2 г глюкозы, 3,5 г сахарозы, 15 г лактозы, 0,6 г 430
соли Мора. 1 г тиосульфата натрия и 10 г мочевины растворяют в 1000 см3 дистиллированной воды. Соль Мора и тиосульфат натрия предварительно растворяют в 5—7 см? дистиллированной воды в пробирках. Затем углеводы и мочевину также растворяют в 10 см3 дистиллированной воды в колбе на водяной бане. До стерилизации pH устанавливают на уровне 7.4—7.6 и добавляю! 46 г сухой среды с сахарозой и индикатором «ВР». Раствор размешивают и кипятят до расплавления агара, разливают в пробирки по 5—6 см3. Среду, разлитую в пробирки, стерилизуют текучим паром 2 дня по 30 мин или при 112 °C в течение 20 мин. После стерилизации среду скашивают так. чтобы остался небольшой столбик (нс менее 3 см).
П е и т о н н а я в о л а: к 1000 см3 дистиллированной воды добавляют 10 I пептона и 5 г хлорида натрия, кипятят до растворения пептона, фильтруют и устанавливают pH 7.2—7.4. после чего стерилизуют 30 мин ири 120 °C.
Цитрат н а я и л а з м а крови кролик а: в пробирку вносят 2 см3 раствора цитрата натрия массовой долей 5% и 8 см3 только что полученной крови кролика. Цитратную кровь ставят на 18—20 ч в холодильник при 4—6 °C или центрифугируют с частотой вращения 50 с 1 в течение 15 мин, в результате чего над осадком эритроцитов формируется прозрачный слой жидкости желтоватого цвета — плазмы, которую разводят физиологическим раствором в соотношении 1:4.
Агар с кровью: к 100 см3 расплавленного стерильного мясо-пептонного агара, охлажденного до 45 °C, стерильно прибавляют 5—10 см3 стерильно взятой дефибринированной крови. Готовую среду разливают в стерильные чашки Петри, дают ей застыть, а затем подсушивают в термостате при 37 °C. Слой агара должен быть равномерно окрашен в красный цвет.
Среда Китт а—Т а р о ц и и: свежую печень крупного рогатого скота разрезают на куски массой по 50—60 г, заливают равным объемом воды и кипятят 30 мин при постоянном помешивании. Печеночный экстракт фильтруют через ватно-марлевый фильтр и смешивают с мясо-пептонным бульоном в соотношении 1:3. Смесь нагревают до температуры кипения, добавляют хлорид натрия из расчета 1,25 г на 1000 см3 среды и pH устанавливают на уровне 7,6—7,8, после чего кипятят в течение 15 мин и фильтруют через бумажный фильтр.
В пробирки кладут мелко нарезанные кусочки печени по 1,5— 2 г и заливают смесью печеночного экстракта с мясо-пептонным бульоном. На поверхность среды наслаивают 0,5—1 см3 вазелинового масла. Среду стерилизуют в течение 30 мин при 120 °C.
Среда Эндо: к 100 см3 расплавленного мясо-пептонного агара (МПА) добавляют 1 г лактозы, растворенной в 5 см-5 водопроводной воды, прокипяченной в течение 5 мин. МПА охлаждают до 60—70 °C и к нему добавляют смесь растворов основного
431
.. vy.iDipHid tiaipMM. для приготовления смеси 0,5 г сульфита натрия растворяют в 5 см3 стерильной воды, кипятят в течение 5 мин и добавляют 1 см3 спиртового раствора основного фуксина массовой долей 10 %. После перемешивания среду разливают в чашки Петри. Готовая среда Эндо в чашках должна быть бледно-розового цвета.
Среду Эндо готовят в день ее использования. Ее можно приготовить также из сухой готовой среды.
Среда КОДА (сухая готовая): готовят согласно инструкции. указанной на этикетке.
Среда X Б (хинозол-бромкрезол-пурпурная): для приготовления бромкрезол-пурпура к 0,8 г порошка добавляют 50 см3 ректификованного этанола. Через день раствор готов к употреблению. Раствором можно пользоваться в течение месяца со дня приготовления.
Для приготовления хинозола 0.1 г порошка растворяют в 100 см3 стерильной дистиллированной воды. При хранении свойства его не изменяются. Срок хранения не ограничен.
Краски хранят в темном месте при комнатной температуре.
Дрожжевой автолизат: в 600 см3 стерильной воды растворяют 1 г однозамещенного фосфата калия и 0,1 г сульфата магния, затем добавляют 100 г прессованных хлебопекарных дрожжей и взбалтывают до получения суспензии. В колбу с суспензией добавляют 8—10 см3 хлороформа, закрывают ватной пробкой и накрывают колпачком из парафинированной бумаги (для предотвращения испарения). Затем колбу помещают в термостат при 45—47 °C на 10 сут. Далее кипятят на водяной бане в течение 20 мин, фильтруют и стерилизуют при давлении 5 - 104Па в течение 15 мин. Готовый дрожжевой автолизат хранят в холодильнике при 4—6 °C в течение двух недель.
Среда Хейфеца: в 1 дм3 водопроводной воды растворяют Юг пептона, 5г хлорида натрия и 5г маннита, устанавливают pH на уровне 7,4—7,6, нагревают до кипения, фильтруют, добавляют 1 см? спиртового раствора розоловой кислоты массовой долей 5 % и 2,5 см3 раствора метиленовой сини массовой долей 0,1 %, стерилизуют однократно, нагревая 20 мин в аппарате Коха, или кипятят в колбе, закрытой ватной пробкой, в течение 5 мин. Среду разливают в стерильные пробирки размером 19 х 180 мм по 7—9 см3. Цвет среды в пробирках должен быть красно-фиолетовым.
Среда Хейфеца двойной концентрации: готовят аналогично, уменьшив объем водопроводной воды до 500 см3 и увеличив объем, разливаемый в пробирки, до 10 см3.
Раствор метиленового голубого массовой долей 0,1 %: 0,1г порошкообразного метиленового голубого заливают 100 см3 дистиллированной воды. Через сутки раствор готов к употреблению. При хранении свойства его не изменяются, срок хранения не ограничен.
432
Краски хранят в темном месте при комнатной темнератчре.
С р е л а К с с с л е р а (модифицированная): к I дм? дистиллированной воды добавляют Юг пептона. 50 см-' желчи, кипятят 30 мин, фильтруют через вату, добавляют 2,5 г лактозы и доводят объем дистиллированной водой до первоначального (1дм3), добавляют 2см; водного раствора генпианвиолета массовой долей 1 %. Среду разливают в пробирки с поплавками по 10 см3 и стерилизуют при давлении 5- 104 Па в течение 15 мин. Готовая среда имеет фиолетовый цвет. Допускается замена поплавков клочками стерильной ваты.
Среда С и м м о н с а: в 1000 см3 дистиллированной воды растворяют 5 г хлорида натрия, 0,2 г сульфата магния, 1,5 г фосфата натрия — аммония, 2 г двузамещенного фосфата калия, 5 i нейтрального цитрата натрия, 20 г агара. Раствор фильтруют, добавляют 40 мл раствора бромтимолового синего (1 : 500). Среду разливают в пробирки по 5—6 л в каждую, стерилизуют при температуре 120 °C в течение 20 мин, после чего скашивают. Готовая среда должна быть оливкового цвета.
Молочно-солевой агар массовой долей 6,5 %: к 1 дм3 расплавленного мясо-пептонного агара добавляют 6,5 г х. ч. хлорида натрия, устанавливают pH 7,4, стерилизуют в автоклаве 20 мин при давлении 105Па. К 100 см3 расплавленного и охлажденного до 45 °C солевого агара добавляют 10 см3 стерильного обезжиренного молока, разливают по чашкам Петри. Хранят при 4—9 °C в течение недели со дня приготовления.
Ж е л т о ч н о-с о левой агар (среда Чистович): к 150 см3 расплавленного и охлажденного до 45 °C солевого агара мссовой долей 6,5 % стерильно добавляют 50 см3 желточного раствора, взбалтывают и разливают по чашкам Петри. Хранят при 4—9 °C в течение недели со дня приготовления.
Желточный раствор: к 200 см3 стерильного физиологического раствора стерильно добавляют один яичный желток, раствор взбалтывают. Хранят при 4—9 °C в течение одной недели со дня приготовления.
Среда циклосериновая (СПС): к 1 дм3 питательной основы (бульон Хоттингера, бульон Вайнберга, сухая кито-пептоно-дрожжевая среда) добавляют последовательно 5 см3 раствора сульфата железа массовой долей 10%, 10 см3 раствора сульфита натрия (кристаллического) массовой долей 10% и 40 см3, раствора D-циклосерина массовой долей 1 %. Все перечисленные компоненты готовят отдельно на стерильной дистиллированной воде. Не следует их смешивать вместе перед добавлением к основе, так как может образоваться осадок. Среду хранят при 4—9 °C не более недели со дня приготовления.
Среда Вильсон а—Б л е р а: к 100 см3 расплавленного и охлажденного до 80 °C мясо-пептонного агара добавляют 1 г глюкозы (pH не ниже 7,2), 10 см3 раствора сульфита натрия массовой
433
долей 20 % и 1 см' раствора хлорида железа массовой долей 8 %. Смесь разливают в стерильные пробирки столбиками высотой 10 см3.
Раствор хлорида железа готовят на стерильной дистиллированной воде: раствор сульфита натрия стерилизуют в течение I ч текучим паром.
Методические указания. Вследствие высокого содержания влаги и белков мясо здоровых животных является благоприятной средой для развития микрофлоры, вызывающей гнилостную порчу. В обычных условиях убоя стерильного мяса не бывает, в нем идентифицируются все группы микроорганизмов: бактерии, микромицеты, лучистые грибки, дрожжи и фильтрующиеся вирусы.
Санитарное состояние мяса и его устойчивость к гнилостному разложению зависят от соблюдения санитарно-гигиенических требований при выращивании и заготовке скота, транспортировании, первичной переработке и производстве мясных продуктов. У истощенных и утомленных животных понижается устойчивость организма, и бактерии из кишечника и лимфоузлов проникают в кровь и ткани. В этом случае в мясе обнаруживают кишечную палочку, палочку протея, стафилококки и анаэробные палочки. При различных заболеваниях животных и птицы мышцы и внутренние органы нередко обсеменены микроорганизмами. Продукты убоя этих животных (птицы) могут вызвать у человека инфекционные заболевания или пищевые отравления.
Обсеменение мяса микроорганизмами может происходить и в процессе переработки скота: при съемке шкуры, извлечении внутренних органов, обескровливании, зачистке, шпарке, а также при использовании грязного инструмента, низком уровне личной гигиены работников.
С целью предотвращения микробной кросс-контаминации мяса в процессе переработки рекомендуется делать акцент на выявление потенциально опасного пищевого сырья и ингредиентов, которые могут содержать токсические вещества, патогенные бактерии или большое количество микроорганизмов, вызывающих порчу продуктов; обнаружение вдоль всей технологической цепи источников и конкретных точек, где может возникнуть контаминация продукта; предотвращение условий, при которых возможны выживание и рост микроорганизмов.
Учитывая скоропортящийся характер сырья и благоприятные естественные условия развития микрофлоры в мясе, контроль общей микробиологической обсемененности и определение наличия патогенных бактерий и бактериальных токсинов являются обязательным этапом исследования сырья и готовой продукции.
Бактериологический анализ проводят в следующих случаях:
при подозрении на остропротекающие инфекционные заболевания, обнаружении в мышцах единичных некротических очагов, наличии патологических изменений в мышцах туши и во внутрен-434
них органах; при поражении отдельных лимфа! ических узлов или органов, нескольких органов и удовлетворительной упитанности туши; при мыте; при беломышечной болезни и кегозах; при маститах, эндометритах, параметритах коров и овец; во всех случаях вынужденного убоя животных независимо от причин убоя и принадлежности животных; при отравлении или подозрении на отравление ядовитыми веществами химического или растительного происхождения; при подозрении на сальмонеллез; при желудочно-кишечных заболеваниях; при заболеваниях органов дыхания; при обширных ожогах, кровоизлияниях и небольших кровоизлияниях в подкожной клетчатке, во внутренних органах, на слизистых оболочках; при отеках внутренних органов и частей туши; при жировом перерождении печени; при наличии гнойных очагов в печени, почках, селезенке и легких; при желтушном окрашивании всех тканей туши, исчезающем в течение двух суток; при сомнительной свежести мяса или других продуктов и невозможности установить их доброкачественность органолептическим путем и в других случаях.
Кроме указанных случаев бактериологическое исследование мяса может проводиться также по требованию ветеринарного или медико-санитарного надзора.
Параллельно с бактериологическим анализом в лаборатории проводят биохимические, органолептические и физико-химические исследования. Это позволяет сделать более обоснованное заключение о предубойном состоянии животного и порядке реализации продуктов убоя.
Бактериологическое исследование мяса проводят по схеме, приведенной на рис. 5.5.
Придерживаясь этой схемы, можно сравнительно быстро дать заключение о наличии в мясе возбудителей основных микробных инфекций, вызываемых аэробами (сибирской язвы, рожи свиней, пастереллеза, листериоза, кокковых инфекций), а также бактерий рода сальмонелла (Salmonella) и условно-патогенных микроорганизмов, вызывающих пищевые отравления.
Лабораторная работа состоит из ряда этапов, объем выполнения и перечень которых конкретизируются с учетом имеющихся в вузе специализаций, объема и специфики рабочих программ, и включает в себя элементы, максимально приближенные к условиям и требованиям бактериологического анализа на предприятиях.
Исследование мяса и мясопродуктов на наличие микроорганизмов основано на бактериоскопии мазков-отпечатков из глубоких слоев образцов и посеве на простые и элективные (избирательные) среды и среды обогащения. В зависимости от результатов бактериоскопии и характера роста на питательных средах проводят исследования на наличие определенных микробов.
435
Рис. 5.5. Схема бактериологического исследования мяса
Рекомендуемый план исследований, проводимых студентами на занятии по бак гериоло! ическому исследованию мяса, предусматривает:
изучение характера роста бактерии на мясо-пептонном агаре. Цветным карандашом по донышку чашки студенты обводят несколько колоний, готовят из них мазки, окрашиваю! по Граму, микроскопируют:
определение обшей бактериальной загрязненности мяса по числу колоний, выросших на мясо-пептонном агаре;
изучение характера роста бактерий на среде Эндо или других элективных средах. Студенты обводят цветным карандашом несколько колоний, похожих по внешнему виду на колонии бактерий сальмонеллезной группы;
подсчет колоний на среде Эндо;
приготовление мазков из подозрительных колоний на среде Эндо, окрашивание по Граму, определение морфологии бактерий;
исследование бактерий из подозрительных колоний на подвижность:
постановка предметной агглютинации со взвесью микробных тел из подозрительных колоний;
высев микробов из подозрительных колоний на пестрый ряд, в 2—3 пробирки на скошенный агар и в 2—3 пробирки со средой для определения образования индола.
Примерный план лабораторной работы может быть расширен дополнительными заданиями (см. п. 3 настоящей работы), демонстрацией засеянных питательных сред и результатов серологических реакций или, наоборот, сужен в зависимости от целей, особенностей учебного плана, наличия курсов по углубленной подготовке и т. д.
Подготовка проб. Отбор проб для анализа ведут в соответствии с действующей нормативной документацией в производственных условиях. При анализе в учебной лаборатории в образцах мяса устанавливают патологоанатомические и органолептические характеристики. После этого проводят бактериоскопию мазков-отпечатков из глубоких слоев и посев на простые и элективные (избирательные) среды и среды обогащения, спектр которых зависит от имеющейся материальной базы и конкретного задания.
Для этого каждый образец перед посевом освобождают от видимых жировой и соединительной тканей, погружают на 2—3 мин в этанол и два раза обжигают с поверхности. Затем стерильными ножницами из глубины различных мест каждого образца вырезают кусочки размером не менее 2,0 х 1,5 х 2,5 см; лимфатические узлы разрезают пополам. Затем все вырезанные кусочки измельчают стерильными ножницами.
Для посева готовят две пробы по 15 г каждая, одна из которых состоит из кусочков мышц и лимфатических узлов, другая — из кусочков паренхиматозных органов (печень, почки и селезенка).
437
Каждую пробу о1дедьно пометают в стерильный стакан (колбу) гомогенизатора для приготовления взвеси. Для этого в стакан (колбу)добавляют по 15 см? физиологического раствора, объем которого соответствует массе каждой пробы, и обрабатывают в электрическом гомогенизаторе. Сначала исследуемый материал измельчают с небольшой частотой вращения, а затем с большей частотой вращения не более 2.5 мин (в зависимости от числа оборотов).
1 см3 приготовленной взвеси содержит 0.5 г продукта.
При органолептической и визуальной оценке следует обращать внимание на цвет, консистенцию, запах, состояние поверхности, а также наличие изменений структуры тканей, нехарактерные включения и другие показатели. Результаты органолептической и визуальной оценки фиксируют в тетради. В зависимости от результатов органолептических исследований с преподавателем (или специалистом) намечают план дальнейшего бактериологического анализа.
1.ПРИГОТОВЛЕНИЕ И БАКТЕРИОЛОГИЧЕСКИЙ АНАЛИЗ ПРЕПАРАТОВ
Порядок проведения анализа. В зависимости от характера патологических изменений и предполагаемого диагноза из проб готовят от 2 до 10 мазков-отпечатков. Препараты сушат на воздухе, фиксируют и окрашивают одновременно по Граму и раствором сафранина массовой долей 2 % (метод Ольта) или раствором Ребигера.
1.1. ОКРАСКА МАЗКОВ ПО ГРАМУ
Общепринятая модификация. На фиксированный мазок помещают полоску фильтровальной бумажки и наливают карболовый раствор генцианвиолета или кристаллвиолета. Выдерживают 1—2 мин, после чего снимают бумажку, сливают краску, мазок промывают водой и наносят на него раствор Люголя (мазок чернеет). Через 1—2 мин раствор сливают и наливают этанол на 0,5—1 мин. Затем мазок вновь промывают водой и дополнительно окрашивают водным раствором фуксина или раствором сафранина в течение 1—2 мин. Затем мазок промывают водой и высушивают фильтровальной бумагой.
Окраска мазков по Граму в модификации Синева. Предусматривает использование окрашенных полосок приготовленной фильтровальной бумаги вместо карболового раствора генцианвиолета. Для окрашивания мазков на фиксированный мазок накладывают полоску фильтровальной бумаги, пропитанной спиртовым раствором генцианвиолета, и наносят две-три капли ‘
438
воды, которые полностью впитываются бумагой, последняя плотно прилегает к стеклу. Выдерживают 2 мин. затем бумагу удаляют пинцетом и в дальнейшем мазок окрашивают по Граму по общепринятой схеме.
1.2. ОКРАСКА КАПСУЛ ПО МЕТОДУ ОЛЬТА
Мазки окрашивают водным раствором сафранина массовой долей 2 % в течение 1—3 мин (лучше при нагревании) и быстро промывают водой, высушивают фильтровальной бумагой.
Препараты микроскопируют. Наличие в мазках грамположи-тельных палочек с обрубленными концами, палочек или цепочек с капсулами, при обнаружении теней в мазках из лимфатических узлов свиней со специфической для сибирской язвы патологоанатомической картиной дает основание для предварительного заключения о бактериоскопическом выявлении микробов, характерных для возбудителя сибирской язвы.
1.3. ОКРАСКА КАПСУЛ ПО МЕТОДУ РЕБИГЕРА
Мазки окрашивают и фиксируют одновременно. Нефиксированные мазки окрашивают раствором генцианвиолета в течение 15—20 с, быстро промывают водой и высушивают фильтровальной бумагой.
При окраске по методу Ребигера сибиреязвенные бациллы окрашиваются в темно-фиолетовый цвет, а капсулы — в красно-фиолетовый.
Результаты бактериологического анализа представляют в виде таблицы рекомендуемой формы:
Образец Опенка органолептических показателей Способ окраски препарата Зарисовка микроскопического поля препарата Описание микроскопической картины
Студенты самостоятельно делают выводы о наличии признаков исследуемых возбудителей и строят дальнейший план бактериологического анализа.
2 .ПРИГОТОВЛЕНИЕ ПОСЕВОВ И ОПРЕДЕЛЕНИЕ ФИЗИОЛОГО-БИОХИМИЧЕСКИХ И КУЛЬТУРАЛЬНЫХ ПРИЗНАКОВ МИКРООРГАНИЗМОВ
Порядок проведения анализа. Готовят посевы из исследуемого материала на мясо-пептонный агар, на агар Эндо (по секторам) или на другую элективную среду, на среду накопления и в конденсационную воду скошенного агара (по Шукевичу).
439
Полученные на .лапе подготовки проб взвеси отстаивают 10 мин. Из верхней части налосадочной жидкости пипеткой Пастера или петлей вносят в чашку с мясо-пептонным агаром и элективной средой (Эндо. Левина) одну-две капли взвеси (или одну петлю) и тщательно втирают материал в поверхность предварительно подсушенных сред.
Одновременно с посевом на плотные среды проводят посев материала для накопления сальмонелл в одну из сред обогащения (селенитовый Ф-бульон. Мюллера, Кауфмана. Киллиана, хлорис-томагниевую среду «М»). Для этого 20 см3 взвеси из мыши и лимфатических узлов вносят в один флакон (колбу), а 20 см3 взвеси из паренхиматозных органов — в другой. В каждый флакон наливают по 50 см3 среды обогащения.
При отсутствии гомогенизатора допускается посев кусочка пробы размером не менее 2,0 х 1,5 х 2,5 см путем нанесения отпечатков разными сторонами пробы на поверхность питательной среды в чашках с предварительно подсушенным мясо-пептонным агаром и элективной средой (Эндо, Левина).
Посевы помещают в термостат при температуре 37 °C. Через 18 ч чашки с первичными посевами на плотных средах извлекают из термостата и просматривают визуально, при необходимости — через лупу или под малым увеличением микроскопа.
При отсутствии роста через 18 ч посевы выдерживают в термостате дополнительно еще 6 ч.
На мясо-пептонном агаре отыскивают колонии, характерные для сибирской язвы, рожи, пастереллеза, листериоза, кокковых инфекций и др., а на чашках с элективными средами (Эндо, Левина) — колонии, характерные для бактерий группы кишечных палочек.
При обнаружении в мазках микробов, подозрительных на сибирскую язву, посевы на элективные среды и среды обогащения не проводят.
2.1. ОБНАРУЖЕНИЕ АЭРОБНЫХ БАКТЕРИЙ
Бациллы сибирской язвы. Сущность метода заключается в определении характерной морфологии и характера роста на питательных средах.
Бациллы сибирской язвы неподвижны, в организме образуют капсулы, а во внешней среде при доступе кислорода и температуре 12—42 °C — споры. При микроскопии мазков из патологического материала обнаруживают крупные грамположительные палочки, соединенные в короткие цепочки; иногда цепочки имеют форму бамбуковой трости.
При окраске раствором сафранина массовой долей 2 % сибиреязвенные бациллы окрашиваются в кирпично-красный цвет, а капсулы и тени (следы распада бактерий) — в светло-желтый.
440
При окраске раствором Ребигера бациллы антракса окрашиваются в тсмно-фиоле'ювый цвет, а капсулы — в краснофиолетовый.
На МПА ч срез 16—24 ч бациллы растут в виде серо-белых, шероховатых. с бахромчатыми краями колоний, напоминающих при осмотре через лупу головку медузы или львиную гриву. Из подозрительной колонии делают посев в пробирку с МПБ.
На МПБ сибиреязвенные бациллы растут в виде крупных хлопьев, оседающих на дно пробирки к концу первых суток, с образованием осадка, напоминающего комок ваты. Бульон остается прозрачным. Из бульонной культуры готовят препарат «раздавленная», или «висячая», капля. При микроскопировании препарата под средним увеличением микроскопа обнаруживают неподвижные сибиреязвенные палочки.
Иногда возникает необходимость отличать сибиреязвенные бациллы от сапрофитных бацилл, которые морфологически очень похожи. В этом случае чистую культуру, выросшую на МПА или МПБ, исследуют: на капсулообразование in vitro, на гемолитическую активность, на тест «жемчужное ожерелье», на чувствительность к сибиреязвенному фагу или на свертываемость желтка куриного яйца.
Основанием для подтверждения диагноза сибирской язвы являются: наличие в мазках капсулообразующих палочек, а из колоний грамположительных — неподвижных бацилл; характерный рост на питательных средах; положительная реакция преципитации и положительный результат биологической пробы.
Бактерии рожи свиней, листериоза и пастереллеза. Сущность метода заключается в определении специфического роста этих микроорганизмов на мясо-пептонном агаре и их дифференциации по морфологическим, культуральным и биологическим свойствам.
На мясо-пептонном агаре они растут в виде мелких прозрачных колоний. Из этих колоний готовят мазки, окрашивают по Граму, исследуют на подвижность и проводят посев на мясо-пептонный агар и мясо-пептонный бульон.
Для проведения анализа пробы на каталазу к суточной культуре добавляют I см3 раствора пероксида водорода массовой долей 10 %. Листерии, выделяя каталазу, расщепляют пероксид водорода с образованием пузырьков газа.
При необходимости проводят дополнительную дифференциацию листерий от возбудителя рожи свиней с применением дополнительных сред. Так, бактерии листериоза в отличие от бактерий рожи свиней на желатине дают медленный рост в виде узловатой нити с неровными краями и пушистыми отростками; желатин не разжижают; ферментируют салицин; вызывают [3-гемолиз на агаре с кровью; на мясо-пептонном печеночном агаре с добавлением 0,5 % глюкозы, 3 % глицерина и 0,01 % тел-
441
.чилпл pativi n виде мелких черных колонии; вызываю! кератоконъюнктивит у морских свинок.
О наличии бактерий сулят по характеристикам, представленным в табл. 5.4.
Т а 6 л п и а 5.4
Морфологические и культуральные свойства возбудителей рожи свиней, листериоза и пастереллеза
Пока затечь Характеристика бактерии
рожи свиней I листериоза i пастереллеза
Рост на МПА Мелкие, росинча- Мелкие, росинчатые Мелкие, росинча-
тыс прозрачные колонии прозрачные колонии, через 2—3 сут — помутнение колоний тые. слегка опалесцирующие. прозрачные колонии: через 2—3 сут приобретают серовато-белый цист
Рост на МПБ Слабое помутнение, поднимающийся при встряхивании осадок Слабое помутнение с выпадением слизистого осадка, при встряхивании которого образуется косичка Равномерное помутнение с осадком
Мазки-отпе- Неспорообразую- Неспорообразующие Неспорообразую-
чатки из ма- щие, тонкие, пря- короткие палочки с за- щие мелкие, бипо-
териала мые или слегка изогнутые палочки, иногда нити кругленными концами: расположены поодиночке, попарно, в форме римской цифры V или в виде палисада лярно окрашивающиеся палочки
Мазки из Короткие, прямые Короткие, прямые. Мелкие палочки.
культуры или изогнутые палочки; при хроническом течении инфекции — короткие тонкие палочки и удлиненные цепочки и нити овоидные палочки, иногда почти кокки; располагаются кучками или поодиночке биполярность почти всегда отсутствует
Окраска по Граму Положительная Положительная Отрицательная
Подвижность Неподвижны Подвижны лишь в молодой 6—20-часовой культуре, выращенной при температуре 20-22 °C Неподвижны
Проба на каталазу Отрицательная Положительная Не ставят
442
Бактерии кокковой группы (стафилококки, стрептококки). Сущность метода заключается в определении морфологии, характера роста на питательных средах и способное!и отдельных стафилококков коагулировать нитратную плазму крови кролика тюсг воздействием фермента коагулазы.
В зависимости oi пигмента, образующеюся на питательных средах, различают золотистый, белый и лимонно-желтый стафилококки (St. aureus, album, citreus). Из различных серологических групп стрептококков (А. В. D. Н) в патолоыш животных и человека имеют значение Str. liaemolyticus. Str. viridans. Str. faecalis. Стафилококки и стрептококки — аэробы или факультативные анаэробы шаровидной формы, располагаются в виде единичных кокков, скоплений диплококков и в других сочетаниях, не имеют капсул и жгутиков, не образуют спор, хорошо растут на обычных питательных средах, окрашивание по Граму даст положительную реакцию.
Золотистый и другие виды стафилококков, а также некоторые стрептококки обладают патогенными свойствами и продуцируют токсины.
Наличие на чашках с мясо-иептонным агаром мелких прозрачных или мутноватых колоний, иногда образующих различные пигменты, вызывает подозрение на присутствие возбудителей кокковых инфекций (диплококкоза. стафилококкоза, стрептококкоза).
Дифференциальный диагноз ставят на основании микроскопического исследования (грамположительные, неподвижные, неспорообразующие, круглые клетки, располагающиеся поодиночно, цепочками, гроздями и в виде ланцетовидных диплококков), а также определения характера роста на питательных средах.
На мясо-пептонном бульоне стафилококки и диплококки дают равномерное помутнение с выпадением обильного осадка.
При росте стрептококков на бульоне с 2 % глюкозы бульон остается прозрачным, а на дно пробирки выпадает осадок.
Для установления патогенности стафилококков ставят реакцию коагулирования плазмы крови. Плазму крови кролика, приготовленную соответствующим образом, разливают в две стерильные пробирки по 0,5 см\ В одну из них вносят петлей суточную агаровую культуру испытуемого стафилококка, другая пробирка служит контрольной. Внесенную культуру тщательно перемешивают, после чего обе пробирки помешают в термостат при температуре 37 °C. Затем пробирки осторожно, избегая встряхивания, просматривают. Результаты реакции коагуляции регистрируют в течение 2—4 ч и через 24 ч. Штаммы стафилококков, продуцирующие фермент плазмокоагулазу, вызывают свертывание плазмы, вследствие чего она превращается в студнеобразную массу, не выливающуюся при перевертывании пробирки. Свертывание плазмы обозначают знаком «+» с указанием времени, в течение которого произошла реакция.
443
Бактерии рода ca.ihvone.i.ia (Salmonella). Наиболее |ру|оемкая часть бактериологического исслелования мяса и мясопродуктов -установление вила бактерии, вызывающих нишевые токсикоин-фекции. к которым относятся сальмонеллы. Бактерии различных видов рода Salmonella по морфологическим и культуральным свойствам друз- от друга не отличаются: помимо этого они имеют общие признаки с бактериями рода Е. сой (кишечная группа) и рода Shigella (дизентерийная группа).
Сущность метода выявления сальмонелл заключается в определении их характерного роста на элективных (избирательных) средах и установлении их специфических ферментативных и серологических свойств.
Сальмонеллы выявляют в четыре последовательных этапа: первичный посев, обогащение, носе]? со среды обогащения и подтверждение.
На первом этапе из взвеси исследуемого материала делают посев на плотные элективные среды. Эти среды выдерживают в термостате при 37 °C в течение 18—24 ч и определяют наличие колоний типичных или подозрительных на сальмонеллы.
На плотных элективных средах сальмонеллы растут в виде характерных колоний, на среде Эндо — в виде круглых бесцветных или слегка розоватых прозрачных (полупрозрачных) мелких колоний. На среде Левина сальмонеллы растут в виде прозрачных бледных нежно-розовых или розовато-фиолетовых колоний.
На втором этапе (этапе обогащения) для накопления сальмонелл проводят посев на жидкие селективные среды (среды Мюллера, Киллиана, Кауфмана, селенитовый Ф-бульон, хлористомаг-ниевую среду «М») и выдерживают в термостате при температуре 37 °C. Оптимальная температура для накопления сальмонелл на селенитовом Ф-бульоне 43 °C.
На третьем этапе осуществляют пересев из жидких сред на плотные селективные диагностические среды, которые после тер-мостатирования исследуют на присутствие колоний типичных или подозрительных на сальмонеллы.
На четвертом этапе проводят подтверждение наличия бактерий из рода сальмонелл определением морфологических, биохимических и серологических свойств.
При наличии роста микроорганизмов на питательных средах берут три—пять подозрительных колоний с каждой чашки. Из части колоний готовят мазок, окрашивают по Граму, микроскопируют. Одновременно исследуют на подвижность в «висячей» капле. Оставшуюся часть колонии растирают в конденсационной воде скошенного агара или в одной-двух каплях добавленного к агару стерильного бульона. Эта взвесь служит исходным материалом для биохимической и серологической типизации.
Сальмонеллы относятся к одному из 12 родов семейства бактерий Enterobacteriacae. По серологической типизации систематизи-444
ровано око.ю ЙЮО ccpoi нтш a.,ь\io11c.i.i. Они встречаются (обитаю!; в кишечнике животных и человека. а !акжс во внеш Heii срс.ш. Морфологически ли палочки имеют закругленные концы, иногда овально!! формы, длиной 2—I и шириной 0.5 мкм. Подвижны, окрашивание но Гфаму лает отрицательную реакцию, спор и капсул не образуют. Аэробы или факультативные анаэробы. Оптимальная реакция среды для роста — слабощелочная (pH 7,2—7,5). оптимальная температура роста — 37 С. хотя сальмонеллы хорошо растут и при комнатной температуре, и даже при низких плюсовых температурах (5—8 °C). По росту на простом агаре и обычных жидких питательных средах сальмонеллы почти неразличимы. На мясо-пептонном агаре гладкие формы (5-формы) этих бактерий образуют круглые, полупрозрачные, выпуклые, иногда со слегка вдавленным центром и влажные колонии с легким металлическим блеском. Шероховатые (7?-формы) растут в виде неровно округленных, шероховатых, тусклых и сухих колоний. На скошенном агаре растут пышно, образуя в конденсационной воде сильную муть, на мясо-пептонном бульоне они вызывают равномерное помутнение среды, желатин не разжижают, индол не образуют, молоко не ферментируют.
На плотных селективных средах сальмонеллы растут в виде характерных колоний: на среде Плоскирева — бесцветные мелкие колонии; на висмут-сульфитном агаре — черные или коричневые колонии с характерным металлическим блеском с прокрашиванием участка среды под колонией в черный цвет. Лишь некоторые серологические типы сальмонелл из группы С растут на висмутсульфитном агаре в виде нежных светло-зеленых или крупных серовато-зеленых колоний.
Сальмонеллы продуцируют эндотоксины — глюцидолипоидо-полипептидные комплексы, тождественные с соматическим антигеном бактерий, термостабильные. При парентеральном введении они высокотоксичны.
Вместе с большой общностью морфологических и культуральных характеристик, а также токсинообразования бактерии рода Salmonella отличаются друг от друга по биохимическим и антигенным (серологическим) свойствам. Эти различия положены в основу методов типизации (установления видов) бактерий рода Salmonella.
Условно-патогенные бактерии. Определенную роль в пищевых токсикоинфекциях могут играть некоторые бактерии, объединяемые под названием «условно-патогенные». К ним относят группы кишечной палочки и протея, которые могут быть источниками пищевых отравлений. Морфологически они представляют собой палочки с закругленными концами или овальной формы, длиной 1—4 и шириной 0,5—0,6 мкм. За исключением некоторых видов, эти бактерии подвижны, окрашивание по Граму дает отрицательную реакцию, спор и капсул не образуют, аэробы хорошо растут на обычных питательных средах.
445
Название «кишечная палочка- носи г собирательный .характер, виды отличаются друг от друга культуральными, биохимическими, серологическими и патогенными свойствами.
Биохимически кишечные палочки весьма активны. Все они расщепляют лактозу, глюкозу, маннит, мальтозу, декстрозу, галактозу и ксилозу: разжижают желатин, редуцируют нитраты в нигриты, подавляющее большинство образует индол, но не разданы г инозита и не образует сероводорода. Для выделения кишечной палочки из различных объектов и дифференциации их подгрупп в лабораторных условиях используют элективные среды Эндо. Левина, Плоскирева и Хейфеца.
При выявлении бактерий из рода кишечной палочки (рода Escherichia) определяют морфологию, характер роста на элективных средах с лактозой, неспособность образовывать цитохромо-ксидазу, утилизировать цитрат, образовывать сероводород и способность продуцировать индол.
На среде Эндо бактерии из рода кишечной палочки растут в виде красных с металлическим блеском (или без блеска), розовых с красным центром или белых колоний; на среде Левина — в виде темно-фиолетовых блестящих колоний; на среде Плоскирева — в виде кирпично-красных с глянцевой поверхностью колоний.
Для определения подвижности культуры готовят препарат «раздавленная капля» и микроскопируют. Бактерии кишечной палочки чаще подвижны.
При наличии колоний, характерных для бактерий кишечной палочки (рода Escherichia), их выделяют из других сходных микроорганизмов по биохимическим свойствам (табл. 5.5).
Таблица 5.5
Биохимическая дифференциация бактерий групп кишечной палочки и протея
Биохимические дифференцирующие свойства Род микроба
Биохимические варианты Escherichia Pr. vulgaris Pr. mi-rabilis
1 2 3 4 5 6
Образование сероводорода на трехсахарном агаре с солью Мора — — — — — — + +
Образование газа на глюкозе, на трехсахарном агаре с солью Мора 446 + + + + +
Продолжение
Род микроба
Биохимические дифференцирующие свойства Pi. mi-rabilis
j Био.мкмщ 12 сскис 3 варианты Escherichia Pr. vulgaris
1 4 5 6
Ферментация раствора лактозы массовой долей 10 % + + + + — — — —
Образование индола + + — — + + + —
Расщепление мочевины — — — — — — + +
Утилизация цитрата (на среде Симмонса) — — — — — — Различно Различно
Подвижность +/- +/- +/- - +/- +/- +/- + +
Ферментация мальтозы + + + + + + + —
Ферментация ман- + + + + + + — —
нита
Примечание. Под знаком «+» в таблице обозначен положительный результат; знаком «—» — отрицательный результат; знаком «+/—» — положительный или отрицательный результат.
Бактерии рода Proteus. Сущность метода заключается в определении морфологии, роста на питательных средах, способности гидролизовать мочевину, образовывать сероводород и отсутствии ферментации маннита.
Бактерии группы протея также имеют различную антигенную структуру.
На обычных средах бактерии рода Proteus растут в виде вуалеобразного налета (Я-форма), при микроскопировании которого находят полиморфные грамотрицательные подвижные палочки. Это указывает на присутствие вульгарного протея. Однако могут быть определены изолированные колонии средней величины, нежные полупрозрачные с розоватым центром (О-форма). При микроскопировании этих колоний палочки лишены жгутиков и неподвижны.
Для подтверждения наличия протея (Я-форма) проводят посев в конденсационную воду скошенного агара (по Шукевичу). Для обнаружения О-форм делают посев на среде Плоскирева. На этой среде протей растет в виде прозрачных колоний с характерным запахом. Общую характеристику признаков выявления см. в табл. 5.5.
447
2.2. ОБНАРУЖЕНИЕ: АНАЭРОБОВ
К анаэробам, представляющим большую опасность для людей и животных, относят палочку ботулиниум (Cl. botulinum) и нерфрингенс (Cl. pertringens).
Cl. botulinum — слабоиодвижная палочка длиной 4—8 и шириной 0,6—0,8 мкм. окрашивание по Граму дает положительную реакцию. Спора обычно располагается на конце палочки, в связи с чем споровые формы микроба имеют вид теннисной ракетки или короткой свечи с пламенем. Вегетативные формы Cl. botulinum инактивируются при 80 °C в течение 30 мин, а споры не погибают при кипячении даже в течение 4—5 ч. Палочка ботулиниум отно сится к группе сапрофитных почвенных микробов, широко распространенных в природе. Она присутствует в почве, листьях, траве, сене, овощах, фруктах, на поверхности тела и в кишечнике крупных рыб, кишечном канале человека и животных, навозе и т. д. Различают шесть серотипов этого возбудителя, которые обладают различной патогенностью по отношению к животным и человеку. Последние заболевают ботулизмом только при проникновении в их организм токсинов, накопившихся в продуктах и кормах.
При наличии анаэробных условий в продуктах и кормах животного и растительного происхождения Cl. botulinum продуцирует токсин, в среднем по активности в семь раз превышающий столбнячный. Образование токсина происходит при температуре выше 20 °C. Однако имеются сведения, что он может образовываться и при 10 °C, исключение составляет серотип Е, который может продуцировать токсин даже при температуре 3,3 °C. Содержание в продуктах поваренной соли 6 % и более тормозит образование токсина, а при содержании соли более 10 % токсин вообще не образуется. Для образования токсина оптимальной является нейтральная среда (pH 6,95—7,0). Кислая среда препятствует развитию возбудителя ботулизма, поэтому в продуктах, где происходит молочнокислое брожение (моченые яблоки, кислая капуста, соленые огурцы и томаты), токсин развивается слабо. Однако кислая среда не разрушает токсин, а щелочная способна его разрушить. Токсин не обладает и высокой термостабильностью. При температуре 80 °C и выше он разрушается в течение 30—60 мин, а при 100 °C — через 10—15 мин. Для образования ботулинического токсина требуется 5—7 сут, поэтому употребление в пищу свежих продуктов после тепловой стерилизации к отравлению не приводит.
Cl. perfringens — короткая, спорообразующая, неподвижная, грамположительная палочка, анаэроб. Существуют 6 типов Cl. perfringens, обозначаемых начальными буквами латинского алфавита. Некоторые представители этих типов могут быть патогенными. Типы В, С, D, £ являются возбудителями энтеротоксемии различных животных, а тип С служит также возбудителем некротического энтерита у людей.
448
В отличие от ботулизма нишевые заболевания, связанные с заражением продуктов Cl. perfringens. по-видимому, следует о тнести к токсикоинфекциям.
Сущность выявления анаэробов заключается в определении их способности расти в отсутствие кислорода воздуха, морфологии возбудителей, роста на питательных средах и на выявлении патогенности возбудителей путем заражения лабораторных животных.
Удовлетворительные анаэробные условия создаются в жидкой питательной среде, где в качестве восстановителя применяют мясо, печень, которые одновременно служат и источником питания.
Среда должна быть расфасована во флаконы или пробирки таким образом, чтобы площадь поверхности среды по абсолютной величине не превышала 1/10 ее объема и поверхность ее была изолирована от кислорода воздуха.
Диагноз на анаэробные инфекции ставят на основании бактериоскопии, посева материала на питательные среды и биопроб на лабораторных животных.
Для бактериоскопии готовят 2—5 мазков-отпечатков из каждой пробы и окрашивают по Граму или метиленовой синью, а при необходимости выявления спор или капсул сибиреязвенных бацилл — по методу Ребигера. При микроскопировании обращают внимание на форму, наличие спор и капсул и расположение отдельных микроорганизмов.
Для посева на питательные среды пробы обжигают и навеску массой около 10 г растирают в стерильной ступке с физиологическим раствором в соотношении 1 : 2.
По 3—5 см3 приготовленной взвеси засевают в четыре большие пробирки с мясной средой Китт—Тароцци, залитой слоем вазелинового масла толщиной 0,5 см, предварительно прогретой на водяной бане при температуре кипения в течение 20—30 мин, а затем быстро охлажденной до температуры не ниже 50 °C. Посевы перед термостатированием прогревают при температуре 80 °C в течение 20 мин (две пробирки); при исследовании на Cl. botulinum типа Е одну пробирку нагревают при температуре 60 °C в течение 15 мин (при этом сохраняются споры Cl. botulinum типа Е), а другую — при 80 °C в течение 20 мин. Остальные пробирки не нагревают.
При подозрении на Cl. botulinum для выявления типа Е две пробирки — одну непрогретую и одну прогретую до 60 °C — выдерживают при температуре 28 °C. Другие две пробирки (непрогретую и прогретую при 80 °C) инкубируют при температуре 37 °C для выявления остальных анаэробов.
Термостатирование проводят в течение 5—10 сут, за ростом наблюдают ежедневно. При обнаружении роста проводят микроскопическое исследование.
449
полученные результаты оформляю! в виде протокола, в юмором фиксируют морфологические, культуральные и другие признаки выявленных микроорганизмов. Все сведения представляю! is таблице рекомендуемой формы:
i Микроор! ннизмы
Обри sen ! - - - ------- --------- ------- . _
I аэробные анаэробные
---------------г_-----------------------------------
j
j Санн1арная оценка j мяса
По итогам экспериментальных исследований студенты самостоятельно делают вывод о пригодности мяса для использования на пищевые цели.
3 . УСКОРЕННОЕ ОБНАРУЖЕНИЕ БАКТЕРИЙ НА ОСНОВЕ ЛЮМИНЕСЦЕНТНО-СЕРОЛОГИЧЕСКИХ МЕТОДОВ
3.1. ВЫЯВЛЕНИЕ ВОЗБУДИТЕЛЕЙ РОЖИ СВИНЕЙ. ЛИСТЕРИОЗА, САЛЬМОНЕЛЛЕЗА В КУЛЬТУРЕ ПРИ ПОМОЩИ ФЛУОРЕСЦИРУЮЩИХ АНТИТЕЛ
Порядок проведения анализа. Сущность метода заключается в определении способности антител иммунных сывороток при соединении через карбамидные группы со специальными красителями (флуорохромами) вступать в специфическую связь с соответствующими антигенами. Образующийся при этом комплекс антиген — антитело флуоресцирует и легко определяется при люминесцентной микроскопии.
Метод может быть применен в качестве экспрессного и для ускоренной идентификации микробов. Экспресс-метод проводится в начале исследования наряду с бактериоскопическим и не требует выделения чистой культуры микробов из исследуемого материала. Полученный в течение 2—6 ч ответ является предварительным, сигнальным, требующим подтверждения бактериологическим методом.
Метод люминесцирующих сывороток для идентификации выделенной культуры микробов применяют для ускорения исследования, исключающего в ряде случаев определение культуральных и биохимических свойств, серологические реакции и типирование.
Используют 18—24-часовые чистые или смешанные культуры, из которых готовят по три мазка на отдельных предметных стеклах. Один из мазков используют для обработки рожистой люми-несцирующей сывороткой, два других — для обработки листериозными люминесцирующими сыворотками.
450
Перед применением флуоресцирующую сыворотку разводят физиологическим раствором с фосфатным буфером (pH 7.4) до рабочего разведения, указанного на этикетке ампулы. Сыворотки в рабочем разведении могут храниться при температуре 2—4 °C в течение 2 сут.
Мазки готовят средней густоты (несколько десятков микробов в поле зрения микроскопа), размером не более 1 см2. Место нанесения культуры очерчивают специальным карандашом с обратной стороны предметного стекла, подсушивают мазки на воздухе, маркируют и фиксируют этанолом в течение 15 мин; мазки для фиксации погружают в вертикальном положении в стеклянный сосуд с этанолом. Каждое стекло отделяют металлической проволочкой или стеклянной соломкой.
После фиксации и испарения этанола мазки ополаскивают физиологическим раствором с фосфатным буфером (pH 7,4). На слегка подсушенный мазок наносят одну-две капли соответствующей люминесцирующей сыворотки в рабочем разведении.
Мазки с сывороткой помещают во влажную камеру (чашки Петри с влажной фильтровальной бумагой на дне) и выдерживают в термостате при температуре 37 °C в течение 30 мин.
Мазки допускается ополаскивать физиологическим раствором или дистиллированной водой без фосфатного буфера.
Затем сыворотку отмывают, погружая мазки в кювету, содержащую физиологический раствор с фосфатным буфером (pH 7,4), на 20 мин. Раствор в кювете периодически помешивают и меняют через 10 мин.
Отмытые мазки ополаскивают дистиллированной водой и высушивают на воздухе. Для быстроты высушивания можно использовать вентилятор.
На поверхность мазка наносят небольшую каплю глицерина с буфером pH 8,0, накрывают покровным стеклом, излишки глицерина удаляют.
На покровное стекло наносят нефлуоресцирующее иммерсионное масло или его заменитель и проводят люминесцентную микроскопию.
О наличии возбудителей судят по интенсивности свечения. Диагностическую оценку выражают в крестах согласно представленным ниже характеристикам: «+ + ++» — яркая, сверкающая зеленая люминесценция морфологически типичных бактерий с более интенсивным свечением по их периферии (положительная); «+ ++» — отчетливо выраженная, достаточно яркая, зеленоватая люминесценция морфологически типичных бактерий с более интенсивным свечением по их периферии (положительная); «++» — недостаточно яркая желто-зеленая люминесценция, периферический ободок выявляется с трудом (сомнительная); «+» —люминесценция очень слабая, морфология бактерий различается с трудом (отрица-
451
гельная); «—» — люминесценция отсутствует. видны лишь тени бактерий (отрицательная).
Видовую принадлежность обнаруженного возбудителя устанавливают по определенной сыворотке, которая вызывает специфическое свечение исследуемой культуры интенсивностью нс менее чем на «+ + + ».
3.2. ЛЮМИНЕСЦЕНТНО-СЕРОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ВОЗБУДИТЕЛЕЙ РОЖИ СВИНЕЙ И ЛИСТЕРИОЗА
В МЯСЕ И ПАРЕНХИМАТОЗНЫХ ОРГАНАХ
Порядок проведения анализа. Материал для исследования должен быть свежим. Для люминесцентного микроскопирования из паренхиматозных органов готовят взвесь на физиологическом растворе хлорида натрия в соотношении 1 : 5. После осаждения крупных частиц взвеси из разных участков ее поверхности готовят по два-три мазка на каждый вид сыворотки.
Обработку препаратов и люминесцентное микроскопирование проводят так же, как при исследовании культур (см. п. 3.1).
Диагностическая оценка интенсивности свечения бактерий рожи свиней и листериоза состоит в том, что при обнаружении возбудителя с типичной морфологией, специфическим свечением интенсивностью не ниже чем на «+++», ставят положительный люминесцентно-серологический диагноз соответственно той сыворотке, которая вызвала свечение возбудителя.
При сомнительных или отрицательных результатах (слабое свечение на «+» или «++», свечение микробов с атипичной морфологией или отсутствие свечения возбудителя в мазках) диагноз уточняют люминесцентным микроскопированием культур, выделенных из посевов патологического материала.
3.3. ЛЮМИНЕСЦЕНТНО-СЕРОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ САЛЬМОНЕЛЛ
Порядок проведения анализа. Для исследования могут быть использованы мазки-отпечатки из органов и тканей, пробы мяса после подращивания в термостате в течение 4—6 ч, а также мазки, приготовленные из бактериальных культур (как чистых, так и смешанных).
Сухую флуоресцирующую сальмонеллезную сыворотку и альбумин, меченный родамином, перед применением растворяют каждый отдельно дистиллированной водой в объеме, указанном на этикетке ампулы.
При использовании сыворотки вместе с альбумином для приготовления рабочей смеси флуоресцирующую сальмонеллезную сы-452
воротку и альбумин, меченный родамином, разводят каждый отдельно раствором хлорида натрия молярной концентрацией 0,15 моль/дм3 (pH 7,2—7.4) на одно разведение меньше, чем их рабочие титры, указанные на этикетках препаратов. Разведенную флуоресцирующую сыворотку и альбумин, меченный родамином. смешивают в равных объемах. Рабочую смесь хранят в пробирке с плотной резиновой пробкой в темном месте при температуре 4 °C. Различают прямой и непрямой способы обнаружения сальмонелл.
Прямой способ. Исследованию подлежат молодые чистые или смешанные культуры, выращенные в жидких или лучше на плотных элективных средах после появления макроскопически видимого роста.
Из двух-трех типичных для сальмонелл колоний готовят мазки на предметных стеклах соответственно числу сывороток, которые должны быть использованы для исследования.
Препараты помещают в чашки Петри с влажной фильтровальной бумагой на дне, на каждый мазок наносят одну-две капли сыворотки в рабочем разведении или сыворотку с альбумином, меченным родамином, сыворотку распределяют по всей поверхности мазка и выдерживают при комнатной температуре в течение 30 мин или в термостате при температуре 37 °C в течение 15 мин. Затем сыворотку отмывают дважды по 10 мин раствором хлорида натрия молярной концентрацией 0,15 моль/дм3. Отмытые мазки ополаскивают дистиллированной водой и высушивают на воздухе или в термостате.
Обработку и микроскопирование мазков ведут аналогично п. 3.2.
В свежих пробах мяса и паренхиматозных органов при отсутствии патологических изменений типичные формы сальмонелл можно обнаружить в редких случаях, поэтому люминесцентное микроскопирование проводят после накопления сальмонелл. Для этого от всех органов отрезают кусочки размером 2 х 3 х 4 см и помещают их в закрытых чашках в термостат на 4—6 ч для подращивания. Затем их исследуют при отрицательном результате первичной микроскопии.
Готовят две серии мазков-отпечатков (по три мазка в каждой серии). Одну серию мазков окрашивают по Граму и исследуют по общепринятой методике. Вторую серию окрашивают люминесци-рующими сальмонеллезными сыворотками или смесью люминес-цирующей сальмонеллезной сыворотки с альбумином, меченным родамином. Окрашивание проводят аналогично окрашиванию сальмонелл в культуре.
Непрямой способ. На первом этапе после фиксации мазков этанолом на мазки из культур или мазки-отпечатки из мяса или органов наносят пастеровской пипеткой одну-две капли поливалентной сальмонеллезной сыворотки, разведенной физиологическим раствором в соотношении 1 : 32. Мазки помещают в чашки Петри
453
с влажной фильтровальной бумагой и выдерживают 30 мин при комнатной температуре или 15 мин при температуре 37 3С.
Несвязавшуюся иммунную сыворотку отмывают в течение 5 мин водопроводной водой, мазки высушивают на воздухе или в термостате при температуре 37 СС.
На втором этапе для окраски используют рабочую смесь люми-несцирующей сыворотки против у-глобмлина кролика (меченного ФИТЦ) или рабочую смесь люминесцируюшей сыворотки с альбумином, меченным родамином, которую готовят заранее.
Обработку и микроскопирование мазков проводят по 3.2.
При диагностической оценке интенсивности свечения сальмонелл учитывают, что при микроскопировании мазков из культуры или мазков-отпечатков из органов и тканей бактерии сальмонелл дают яркое изумрудно-зеленоватое свечение по периферии бактерий с хорошо выраженной центральной темной зоной. Фон препарата темный с оранжево-красным окрашиванием посторонней микрофлоры, тканевых элементов и других органических частиц (при применении альбумина, меченного родамином), содержащихся в исследуемых препаратах.
Характерное свечение контура (ободка) при визуальном осмотре оценивается в крестах: «++++» — сияющее зелено-желтое свечение; «+++>> —яркое желто-зеленое свечение; «++» —умеренное желто-зеленое или белое свечение отчетливо заметного контура; «+» — слабое белое свечение различимого контура; «—» —- клетки в виде сероватых теней, контур отсутствует или слабо заметен на отдельных участках периферии клеток (не у всех микробов).
Положительным результатом обнаружения сальмонелл считается свечение типичных для них форм, оцениваемое не ниже чем на «++» при условии, что в контрольных препаратах, окрашенных рабочим разведением гетерологической сыворотки, оно оценивается отрицательно, а также при отрицательных результатах окрашивания сальмонеллезными флуоресцирующими сыворотками гетерологических бактерий (кишечной палочки, протея).
Свечение на «+» оценивается как сомнительное, и исследование повторяют.
Результаты ускоренного анализа возбудителей студенты оформляют в виде протокола. Самостоятельно делают выводы и формулируют общее заключение по работе.
Лабораторная работа Ns 2
ОПРЕДЕЛЕНИЕ МИКРОБНЫХ КОНТАМИНАНТОВ В КОЛБАСНЫХ ИЗДЕЛИЯХ И ПРОДУКТАХ ИЗ МЯСА
Цель работы. Приобрести практический навык в анализе микробных контаминантов колбасных изделий и продуктов кулинарной готовности на основе бактериологического анализа.
454
Задачи. Отбор и подготовка проб, определение обшей микробной обсемененносги и анализ на выявление отдельных наиболее опасных возбудителей токсикоинфекпий.
Объекты исследования. Вареные, полукопченые, варено-копченые колбасные изделия, сосиски, сардельки, продукты кулинарной готовности из мяса на основе цельномышечной ткани (деликатесная продукция в ассортименте), студни, паштеты, мясные хлеба.
Материалы, реактивы и оборудование: см. лабораторную работу № 1 настоящей главы: среды Вильсона—Блера, «ХБ». Кесслера, КОДА. Хейфеца двойной концентрации, Крумвиде— Олькеницкого в модификации Ковальчука, СЦС: цитратная плазма крови кролика; молочно-солевой и желточно-солевой агар; Н-сыворотки.
Подготовка реактивов: см. лабораторную работу № 1 настоящей главы.
Методические указания. Обсеменение колбасных изделий микрофлорой происходит в основном через сырье, оборудование, инвентарь, тару и т. п. По количественному и качественному составам микрофлора сырого колбасного фарша разнообразна. Общее количество микроорганизмов в 1 г сырого фарша вареных колбас, например, составляет 0,6 • 103— 1,4- 1(Р.
В готовых колбасах или копченых изделиях не должно быть патогенной и условно-патогенной микрофлоры.
Бактериологический анализ колбасных изделий и продуктов из мяса включает определение: общего количества микроорганизмов; бактерий группы кишечной палочки; бактерий рода Salmonella; бактерий рода Proteus; коагулазоположительных стафилококков; сульфитредуцирующих рода Clostridium perfringens.
Обнаружение кишечной палочки и протея в глубоких слоях продукта указывает на нарушение технологии изготовления и прежде всего температурного режима. Наличие в колбасных изделиях кишечной палочки свидетельствует о неудовлетворительных санитарно-гигиенических условиях технологического процесса и обязывает принять незамедлительные меры по их улучшению.
При наличии кишечной палочки и протея, но при хороших органолептических показателях вареные и полукопченые колбасы направляют на переработку на низшие сорта с повторной проваркой. Сыровяленые и сырокопченые изделия в этом случае дополнительно выдерживают 10—12 сут и повторно исследуют в лаборатории на наличие микрофлоры. При отрицательном результате продукцию реализуют без ограничений, при положительном — перерабатывают на вареные виды колбас.
При обнаружении в колбасных изделиях аэробных сапрофитов (В. subtilis, В. mesentericus) или спорообразующих непатогенных анаэробов (В. putrificus, В. sporogenes и др.), но при хороших органолептических показателях продукцию выпускают без ограничений.
455
... .и ля-.mu и продуктов из мяса рекомендуется
проводить по следующему плану:
бактериосконическое исследование колбасных изделии или продуктов из мяса;
посев на среду Эндо для определения обсемененноети ее бактериями группы кишечной палочки;
посев для учета общего количества микроорганизмов в 1 г продукта;
посев в конденсационную волу скощенного агара (по Шукеви-чу) с целью выявления протея.
По истечении времени (24—48 ч) рекомендуется организовать анализ по плану:
изучение характера роста микрофлоры на питательных средах;
подсчет общей бактериальной обсемененности продукта;
приготовление мазков из подозрительных колоний с окрашиванием по Граму и микроскопированием;
определение подвижности микроорганизмов;
пересев на одну из сред накопления (Мюллера, Киллиана или Кауфмана);
изучение биохимических и антигенных свойств выросшей культуры и идентификация вида микроорганизма.
Предложенный план может быть изменен преподавателем с учетом специфики учебного процесса и материально-технической базы вуза.
Микробиологическое исследование колбасных изделий заключается в приготовлении мазков-отпечатков из поверхностных и глубинных слоев батона и посеве на питательные среды с последующим изучением полученной культуры и подсчетом количества микробных тел в 1 г продукта.
Для бактериоскопического исследования пробы берут непосредственно из-под оболочки и из середины батона. Если колбасное изделие без оболочки, то срезают верхний слой на 1—2 мм. Стерильными ножницами вырезают два кусочка колбасы и прикладывают к поверхности предметного стекла. Подсушивают, фиксируют их над пламенем горелки, окрашивают по Граму и микроскопируют. В случае порчи колбас накопление микрофлоры отмечается в мазках-отпечатках из поверхностных слоев.
Для выявления аэробов и анаэробов, а также для подсчета общего количества микробных тел в 1 г готового продукта готовят взвесь, которая служит исходным материалом для посева на питательные среды.
Определение общего количества микробов в колбасных изделиях служит дополнительным методом установления их свежести. Наличие более 1,5 млн микробов в 1 г продукта свидетельствует о его порче.
456
Подготовка проб. Учитывая, что микробы развиваются в колбасных изделиях неравномерно (погнездно). пробы для приготовления взвеси отбирают как можно с большей площади продукта.
Отбор точечных проб в условиях производства для бактериологического анализа проводят в соответствии с действующей нормативной документацией.
В зависимости от вида продукта объединенную пробу массой 50 г составляют из точечных проб следующим образом:
колбасные изделия в оболочке и продукты из свинины, баранины и говядины помещают в металлический или эмалированный тазик (тарелку), тщательно протирают ватным тампоном, смоченным этанолом, и дважды обжигают над пламенем. Затем батоны разрезают вдоль стерильным ножом или скальпелем на две половинки, не рассекая оболочку противоположной стороны батона. Пробу отбирают из нескольких участков центральной части и из-под оболочки обеих половинок батона;
из свиных, бараньих, говяжьих продуктов на костях и из бекона пробы вырезают стерильным инструментом из различных участков обожженного образца на глубине 2—3 см от поверхности, предпочтительно ближе к кости;
изделия без оболочки (мясные хлебы, паштеты, студни и другие изделия) исследуют с поверхности и в глубине продукта.
Тампоны помещают в пробирки, заполненные на 3/4 их высоты средой «ХБ>>, Хейфеца или 5 см3 среды Кесслера.
Для анализа глубинных участков продукта образцы помещают в металлический или эмалированный тазик (тарелку), смачивают этанолом и обжигают. Затем делают продольный разрез и отбирают навеску методом, указанным для колбасных изделий и продуктов в оболочке, составляя из них одну объединенную пробу для каждого образца в отдельности, которую помещают в предварительно взвешенный стерильный бюкс или чашку Петри.
Из объединенной пробы каждого образца в стерильную посуду (пергамент) берут навеску массой 20 г с точностью ±0,1 г.
Навеску помещают в стерильную колбу (стакан) гомогенизатора для приготовления испытуемой взвеси. Для этого в колбу добавляют раствор стерильной пептонной воды массовой долей 1 % в соотношении 1 : 4 и гомогенизируют в электрическом смесителе. Сначала материал измельчают на кусочки с замедленной скоростью вращения ножей, затем при частоте вращения 250—333 с-1 в течение 2.5 мин.
При отсутствии гомогенизатора допускается готовить взвесь путем растирания 20 г продукта в стерильной фарфоровой ступке с 2—3 г стерильного песка, постепенно приливая 80 см3 раствора стерильной пептонной воды массовой долей 0,1 %. При растирании проб вареных изделий мажущейся консистенции (ливерные, кровяные колбасы) стерильный песок можно не добавлять.
457
_________________________ Livpii.ibHOH iралуированной пипеткой отбирают взвесь после 15 мин выдержки при комнатной температуре.
1 см? приготовленной взвеси содержи г 0.2 i продукта.
1. ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА МИКРООРГАНИЗМОВ В 1 г ПРОДУКТА
Сущность метода заключается в способное nt мезофильных аэробов и факультативных анаэробов расти на ниппельном агаре при температуре (37.0 ± 0,5) С с образованием колоний, видимых при увеличении в 5 раз. Метод не распространяемся на сырокопченые колбасы.
Порядок проведения анализа. Для определения общего количества микроорганизмов микропилегкой берут 0.1см3 взвеси из верхнего слоя жидкости, выливают на середину стерильной чашки Петри и заливают 12—15 см3 остуженного мясо-неитонного агара (45—50 °C), равномерно распределяя его по всей поверхности. Чашку помещают в термостат и спустя 48 ч подсчитывают общее количество колоний на поверхности среды и в глубине.
Мясо-пептонный агар расплавляют на водяной бане и охлаждают до температуры 45 °C. Стерильные чашки Петри раскладывают на столе, подписывают наименование анализируемого продукта, дату посева и количество посеянного продукта.
Из каждой пробы должно быть сделано не менее двух посевов, различных по объему, взятых с таким расчетом, чтобы на чашках выросло от 30 до 300 колоний. При этом на одной чашке Петри проводят посев 0,1 г, а на другой — 0,01 г продукта.
Для посева 0,1 г продукта готовят первое разведение испытуемой взвеси: стерильной пипеткой с широким конном отбирают 5 см3 испытуемой взвеси, переносят ее в пробирку с 5 см3 стерильного физиологического раствора или пептонной воды. Конец пипетки должен быть опущен ниже поверхности раствора, не прикасаясь к стенкам пробирки, чтобы избежать смывания бактерий с наружной стороны. 1 см3 полученного раствора содержит 0,1 г испытуемого продукта.
Другой стерильной пипеткой тщательно перемешивают содержимое пробирки продуванием, отбирают 1 см3 и переносят в стерильную чашку Петри, слегка приоткрывая крышку.
Для посева 0,01 г продукта готовят следующее разведение: стерильной пипеткой тщательно перемешивают содержимое пробирки, отбирают 1 см3 и переносят в пробирку с 9 см3 стерильного физиологического раствора. 1 см3 испытуемого раствора вторичного разведения содержит 0,01 г испытуемого продукта. 1 см3 этого раствора переносят в стерильную чашку Петри так, как описано выше. При необходимости таким же образом готовят последующие разведения.
458
После внесения развечения анализируемой в шеей в чашки Петри последнюю ыливанн 12 15см' расп.тав.1еН|Ччо и охлажденного лптате шншч) ш ар?. при ф. ымбпгювашш чрась пробирки или буч быки. Lie он содержигоя. быстро смешивали с мясо-пептонным питательным агаром, осюрожно наклоняя или вращая чашку по поверхности сюда. Необходимо избсы.э. образования пузырьков воздуха, нсзалитых участков дна чашки 1 Icrpii. попадания среды на края и крышку чашки.
Для того чтобы помешан, развилию на поверхноыи aiapa сно-рообразующи.х микробов и бактерий группы ирогся в //-форме, допускается наслоение расплавленного и охлажденного до температуры 45—50 °C голодного aiapa толщиной 3--4 мм.
После застывания агара чашки Петри перевертывают и помещают в термостат температурой 37 °C на 48 ч. Затем подсчитывают общее количество колоний бактерий, выросших на чашках.
Колонии, выросшие как на поверхности, гак и в глубине агара, подсчитывают при помощи луны с пятикратным увеличением или специальным прибором с луной. Для этого чашку кладут вверх дном на черный фон и каждую колонию отмечаю! со стороны дна тушью или чернилами для стекла.
Для определения общего количества микробов в 1 г продукта подсчитанное количество колоний умножают на степень разведения анализируемого продукта.
За окончательны!! результат определения количества бактерий в 1 г анализируемого продукта принимают среднее арифметическое результатов подсчета двух чашек, разной массы продукта.
2. ОПРЕДЕЛЕНИЕ БАКТЕРИЙ ГРУППЫ КИШЕЧНОЙ ПАЛОЧКИ В 1 г ПРОДУКТА
Метод основан на способности бактерий группы кишечной палочки расщеплять глюкозу и лактозу. При этом в средах «ХБ», Хейфеца и КОДА образуются кислые продукты, меняющие цвет индикаторов, а в среде Кесслера в поплавке вследствие расщепления лактозы образуется газ.
Порядок проведения анализа. Для установления характера микрофлоры по 0,1 см3 взвеси наносят на поверхность мясо-пептонного агара и среды Эндо, равномерно распределив ее по всей площади. После суточного термостатирования изучают морфологию выросших колоний, а из подозрительных на кишечную палочку или на сальмонеллы готовят мазки, окрашивают по Граму и микроскопируют. При необходимости идентификации микробов взвесь пересевают на среду накопления и типизируют по биохимическим и серологическим свойствам.
В пробирки, содержащие по 5 см3 среды «ХБ», среды Хейфеца двойной концентрации или среды КОДА, вносят стерильной пи-
459
песков вместимостью ?—10см? с широким концом ио 5 см' испытуемой взвеси. Дня анализа можно также использовать среду Кесслера (10 см3).
Пробирки со средами «ХБ». Кесслера. Хейфеца и КОДА помещают в термостат температурой (37 ± 0.5) °C на 18 —20 ч.
Посевы смывов, отобранных тампонами с поверхности изделии без оболочки, выдерживают при температуре 43 СС (для обнаружения повторного бактериального загрязнения).
При росте бактерий группы кишечной палочки среды «ХБ» и КОДА окрашивают в желтый цвет, среда Хейфепа приобретает также желтый цвет, который может изменяться до салатно-зеленого, на среде Кесслера в поплавке образуется газ.
Для окончательного заключения о наличии в продукте бактерий группы кишечной палочки проводят высев со среды Кесслера (забродившие пробирки) или Хейфепа (с измененным цветом среды) в чашки Петри со средой Эндо или Плоскирева, или Левина. Чашки Петри помешают в термостат температурой 37 °C на 18— 20 ч, после чего посевы просматривают. На среде Эндо бактерии группы кишечной палочки образуют темно-красные колонии с металлическим блеском или розово-красные без блеска, на среде Плоскирева — кирпично-красные с глянцевой поверхностью, на среде Левина — темно-фиолетовые или фиолетово-черные блестящие колонии. Из подозреваемых колоний готовят мазки, которые окрашивают по Граму.
Специфическое изменение сред «ХБ» и КОДА не требует дальнейшего подтверждения.
При заведомо высокой обсемененности анализируемый продукт массой не более 0,25 г помещают в пустую пробирку, в которую закладывают комочек стерильной фильтровальной бумаги размером 5 х 5 см, и стерильной стеклянной палочкой или фламбированной проволокой проталкивают материал до дна (не уплотняя). В пробирку наливают среду «ХБ», КОДА или Хейфеца (нормальной концентрации), заполняя ее на 3/4 высоты пробирки. Последнюю помещают в термостат температурой 37 °C на 8—10 ч. При росте бактерий группы кишечной палочки на средах «ХБ» и КОДА среда изменяет свой цвет из фиолетово-пурпурного в желтый, на среде Хейфеца — из красно-фиолетового в желтый, который затем может меняться до салатно-зеленого.
Пробы, отобранные с поверхности изделий без оболочки тампонами, анализируют аналогично.
Обнаружение грамотрицательных не образующих спор палочек, специфически изменяющих цвет жидких дифференциальнодиагностических сред и образующих характерные колонии на элективных средах с лактозой, указывает на наличие бактерий группы кишечной палочки.
460
3. ОПРЕДЕЛЕНИЕ БАКТЕРИИ ИЗ РОДА SALMONELLA
Сущность метола заключается в определении характерного роста сальмонелл на элективных средах. При необходимости рекомендуется проводить иденз ификапию биохимических и серологических свойств.
Порядок проведения анализа. Навеску продукта массой 25 г. взятую из объединенной пробы, вносят во флакон Сокслета, содержащий 100 см3 среды обогащения (Мюллера. Кауфмана, хло-ристомагниевой среды «М»). Жидкость во флаконе должна подняться до отметки 125 см3. Флаконы тщательно встряхивают и помещают в термостат температурой 37 °C. Через 16—24 ч после тщательного перемешивания с помощью бактериологической петли (диаметр 0.4—0,5 мм) или пастеровской пипетки проводят высев из среды обогащения в чашки Петри с предварительно подсушенной средой Эндо, Плоскирева, Левина или Вильсона— Блера (по выбору).
Чашки с посевами помещают в термостат температурой 37 °C. Посевы, выращенные на средах Эндо, Плоскирева и Левина, просматривают через 16—48 ч, на висмут-сульфитном агаре — через 24—48 ч.
На среде Эндо бактерии из рода сальмонелл образуют бесцветные или с розовым оттенком колонии.
На среде БФА сальмонеллы образуют крупные, гладкие, красноватого оттенка прозрачные колонии (колонии сальмонеллы Salm, tuphi suis, как и на среде Эндо, мелкие). Бактерии группы кишечной палочки образуют колонии желто-зеленоватого цвета. Бактерии группы протея дают рост через 72 ч.
На среде Плоскирева сальмонеллы растут в виде бесцветных колоний, но они более плотные и несколько меньшего размера, чем на среде Эндо.
На среде Левина сальмонеллы растут в виде прозрачных, бледных, нежно-розовых или розовато-фиолетовых колоний. На висмут-сульфитном агаре сальмонеллы растут в виде черных или коричневых колоний с характерным металлическим блеском. При этом наблюдается прокрашивание в черный цвет участка среды под колонией. Исключение составляют некоторые серологические типы из группы С, которые на этой среде растут в виде нежных светло-зеленых или крупных серовато-зеленых колоний.
Изолированные колонии, характерные для бактерий из рода Salmonella, пересевают на среду Крумвиде—Олькеницкого в модификации Ковальчука штрихом по скошенной поверхности и уколом в столбик. Посевы помещают на 12—16 ч в термостат температурой 37 °C.
При росте бактерий из рода сальмонелл цвет скошенной поверхности среды Крумвиде—Олькеницкого в модификации Ко-
461
........................................ ... - .V Г1 I IL \С~ 1 andiriикают но наличию зрсшин и разрыве столбика агара, серо-водородобра м юшие вы мывани шмсмнение сюлбика.
Другие грамотринатс юные бактерии лаюг следующие изменения цвета среды:
бактерии 1 руины кишечнои палочки вся среда окрашивается в синий или сине-зеленый цвет с образованием газа или без него:
бактерии и; i руины протея - среда окрашивается в ярко-красный цвете може т образоват ься черный осадок:
шиге.тлы и возбудители брюшною гифа -- косяк окрашивается в розовый цвет, столбик — в синий или сине-зеленый.
Вместо среды Крумвиде —Олькениикого в модификации Ковальчука допускается посев на углеводные среды в короткий пестрый ряд, включая среды с глюкозой, лактозой, сахарозой, маннитом и мальтозой, полужидкий агар уколом (для определения подвижности) и бульон Хоттингера (для определения образования индола и сероводорода).
Для дальнейшей идентификации бактерий готовят мазки, которые окрашивают по Граму, микроскопируют и изучают серологические свойства микроорганизмов путем постановки пробной агглютинации на предметном стекле с агглютинирующей адсорбированной поливалентной сальмонеллезной й-сывороткой. При получении положительной реакции на стекле с поливалентной сывороткой проводят идентификацию с помощью монорецептор-ных агглютинирующих й-сывороток.
Установив серологическую группу, к которой относятся исследуемые бактерии, с помощью /-/-сывороток можно определить тип бактерий.
Обнаружение подвижных (кроме S. pullorum и S. gallinarum) грамотрицательных палочек, дающих характерный рост на элективных средах, не ферментирующих лактозу и сахарозу, ферментирующих глюкозу и маннит с образованием кислоты и газа (Salm, typhi suis не ферментирует маннит), дающих положительную реакцию агглютинации с монорецепторными О- и //-сальмо-неллезными сыворотками, указывает на наличие бактерий из рода Salmonella.
4. ОПРЕДЕЛЕНИЕ ПРОТЕЯ
Метод основан на определении морфологии и роста на питательных средах, способности гидролизовать мочевину и образовывать сероводород.
Порядок проведения анализа. Для определения присутствия протея вносят 0,1 см3 взвеси в конденсационную воду скошенного мясо-пептонного агара (по Шукевичу), термостатируют 18—24 ч и изучают полученную культуру.
462
для подтверждения наличия протея в //-форме 0,5 см3 анализируемой взвеси вносят в конденсационную воду свежескошенного мясо-пептонного агара, разлитого в широкие пробирки, не касаясь поверхности среды (метод Шукевича). Пробирки помещают в термостат температурой 37 °C строго вертикально. Через 18—24 ч посевы просматривают. Обращают внимание на образование ползучего вуалеобразного налета с голубым оттенком: на скошенном мясо-пептонном агаре культура поднимается из конденсационной жидкости вверх ио поверхности среды. При появлении характерного роста микробов рода Proteus окрашенные по Граму мазки микроскопируют и изучают подвижность микробов в «раздавленной» или «висячей капле».
Для обнаружения нерояшихся О-форм можно проводить посев на поверхность среды Плоскирева. О-форма протея растет на этой среде в виде прозрачных колоний, слегка подщелачивающих среду, окрашивая ее в желтый цвет. После пересева на среду Крумвиде—Олькеницкого в модификации Ковальчука при наличии бактерий из группы протея среда окрашивается в ярко-красный цвет (вследствие расщепления мочевины) и может образовываться черный осадок с возможным разрывом агарового столбика (вследствие образования сероводорода).
Обнаружение в среде полиморфных грамотрицательных палочек, образующих характерный рост на средах в Я-форме (подвижные) и О-форме (неподвижные), ферментирующих глюкозу и мочевину и не ферментирующих лактозу и маннит, указывает на наличие бактерий из рода Proteus.
5. ОПРЕДЕЛЕНИЕ КОАГУЛАЗОПОЛОЖИТЕЛЬНЫХ СТАФИЛОКОККОВ
Сущность метода заключается в определении морфологии, характера роста на питательных средах и в способности отдельных стафилококков продуцировать лецитиназу и коагулировать цитратную плазму крови кролика под воздействием фермента коагулазы.
Порядок проведения анализа. Из разведения анализируемой взвеси продукта (1 : 10) в физиологическом растворе или пептонной воде для выявления пигмента проводят посевы на молочносолевой агар, содержащий 6,5 % хлорида натрия, или желточносолевой агар, содержащий 6,5 % хлорида натрия, для выявления лецитиназной активности.
Взвесь объемом 0,2 см3 наносят на поверхность агара и равномерно растирают по всей поверхности агаровой среды.
Посевы термостатируют в течение 24 ч при температуре 37 °C и в течение 24 ч выдерживают при комнатной температуре.
На поверхности питательной среды колонии стафилококка имеют вид плоских или слегка выпуклых блестящих колоний с
463
ровным краем. При этом на молочно-солевом агаре лучше проявляется пигмент (эмалево-белый или золотистый), а на желточно-солевом агаре колонии стафилококков могут обра зовывать радужный венчик, что является одним из признаков их патогенности.
Из подозрительных колоний готовят препараты, которые окрашивают по Граму. При наличии стафилококков в препарате грамположительные мелкие кокки располагаются в виде неправильных гроздьев.
Для подтверждения признаков патогенности стафилококков ставят реакцию плазмокоагуляции. В пробирку с 0,5 см3 цитратной плазмы крови кролика, разведенной физиологическим раствором в соотношении 1 : 4, вносят петлю чистой суточной культуры стафилококка и ставят в термостат при температуре 37 °C. Результаты реакции плазмокоагуляции фиксируют через 3—4 ч (не встряхивая пробирку) и оставляют в термостате на сутки для окончательного заключения.
Для постановки реакции плазмокоагуляции можно использовать также сухую цитратную плазму крови кролика.
Реакцию считают положительной, если плазма коагулируется в сгусток.
Для определения количества стафилококков учитывают колонии стафилококков, давшие положительную реакцию плазмокоагуляции. При расчете на 1 г продукта количество подсчитанных колоний умножают на степень разведения и на количество посевного материала.
6. ОПРЕДЕЛЕНИЕ СУЛЬФИТРЕДУЦИРУЮЩИХ КЛОСТРИДИЙ
Сущность метода заключается в специфическом росте клостридий перфригненс в средах СЦС и Вильсона—Блера, на которых в результате восстановления сульфита натрия в сульфат натрия происходит взаимодействие с хлоридом железа и фиксируется почернение среды за счет сульфида железа.
Порядок проведения анализа. Анализируемую взвесь объемом 1 см3 стерильной пипеткой вносят в пробирку с 9 см3 жидкой сульфит-циклосериновой среды (среды Вильсона—Блера), затем проводят последовательные пересевы на аналогичные объемы среды, в результате чего получают возрастающие десятикратные разведения суспензии. Инкубируют в течение 8—12 ч при 46 °C. При наличии роста сульфитвосстанавливающих клостридий фиксируют почернение среды.
Почернение среды Вильсона—Блера могут вызвать многие энтеробактерии. Для подтверждения роста сульфитвосстанавливающих клостридий используют пересев в пробирки со средой Кит-та—Тароцци, предварительно прогретой в течение 25 мин в водя-464
ной бане ири температуре кипения и быстро охлажденной до 45 С' Термостатирование посевов проводят при (37 ±0,5) “'С. ежедневно в течение 5 сут проверяя в них помутнение среды, выделение газа, появление постороннего запаха, иногда разложение кусочков печени. Сразу после появления признаков роста готовят микроскопический препарат. Для этого материал берут пастеровской пипеткой со дна пробирки. При микроскопировании отмечают грамположи-тельные палочки, образующие овальные споры.
У спорообразующих грамположительных микроорганизмов выявляют каталазную активность с помощью раствора пероксида водорода концентрацией 30 г/дм3. Отсутствие пузырьков газа при добавлении к капле культуральной жидкости такого же объема раствора пероксида водорода позволяет считать, что в посевах присутствуют микроорганизмы из рода клостридий.
При отсутствии спор в микроскопическом препарате положительной пробы на каталазу и наличии в посевах смешанной микрофлоры 1—2 капли накопительной среды переносят в стерильную чашку Петри и заливают расплавленной и охлажденной до 45 °C средой Вильсона—Блера. Застывшую поверхность плотной среды заливают холодным агаром. Посевы термостатируют в течение 24—48 ч при (37 ± 0,5) °C. Появление в нижнем слое агара черных или коричневых колоний свидетельствует о присутствии в посевах сульфитвосстанавливающих клостридий.
За положительный титр клостридий (сульфит-восстановите-лей) принимают то максимальное разведение суспензий, в посеве которого произошло почернение среды. Например, если характерные изменения наблюдаются в пробирках с разведением 10“’, то считают, что в исследуемом продукте будет 10 (или 1 • 101) клеток в 1 г; если характерные изменения наблюдаются в пробирках с разведением 10-2, то считают, что в исследуемом продукте 100 (или 1 • 102) микробных клеток в 1 г.
Результаты бактериологического исследования колбасных и мясных изделий оформляют в виде протокола, форма которого представлена ниже. При этом фиксируют морфологические и культуральные признаки выявленных микроорганизмов, сопоставляют результаты исследований с требованиями соответствующей нормативной документации, дают обоснованную оценку состояния мясных продуктов.
Характеристика образца Обшее количество микроорганизмов, КОЕ/г Количество стафилококков, КОЕ/г Наличие
бактериЯ группы кишечной палочки (коли-формы) патогенных микроорганизмов (бактерий из рода Salmonella) бактерий из рода Proteus сульфит-редуцирую-ших клостридий
+/- +/- +/-
465
Лабораторная работа № 3
ОПРЕДЕЛЕНИЕ АНТИБИОТИКОВ
Цель работы. Приобрести практический навык в определении остаточных количеств антибиотиков в мясе, вторичных продуктах убоя скота и мясных продуктах.
Задачи. Установить наличие антибиотиков в продуктах убоя животных; провести их качественное определение и по результатам исследований дать оценку мяса, вторичных продуктов убоя скота, мясных продуктов кулинарной готовности.
Объекты исследования. Мышечная ткань разных видов убойных животных и птицы, внутренние органы (почки) убойных животных, мясные продукты различных ассортиментных групп.
Материалы, реактивы и оборудование. Мясо-пептонный агар с pH 7,0—7,3; спиртовка; ножницы; пинцет, пастеровские пипетки; бумажные диски, пропитанные 0,25 ЕД пенициллина (тетрациклина); тест-культура микроорганизмов в лиофильно высушенном состоянии (Sar. lutea, St. aureus, Вас. subtilis — паспортизованные штаммы).
Методические указания. Наличие остаточных количеств антибиотиков в пищевых продуктах животного происхождения может отрицательно сказываться на здоровье человека. Это обусловлено способностью антибиотиков воздействовать сенсибилизирующе и приводить к анафилактическим и аллергическим реакциям у человека, вызывать дисбактериозы пищеварительного тракта и формирование антибиотикоустойчивых штаммов патогенных микроорганизмов. Не исключена возможность токсического, тератогенного и мутагенного действия.
Наиболее сильными аллергенами считаются пенициллин, стрептомицин, олеандомицин. Высокой сенсибилизирующей способностью обладают пенициллин, стрептомицин и тилозин.
Несмотря на то что использование антибиотиков разрешено, производители мяса и молока должны гарантировать, что остаточное содержание антибиотиков в их продукции не превышает максимально допустимых уровней.
Согласно действующим санитарным нормам и правилам предельное содержание в мясе и мясных продуктах левомицетина и тетрациклина должно быть менее 0,01 ед/г, гризина — менее 0,5 ед/г, бацитрацина — менее 0,02 ед/г. Для мяса птицы и молока нормируется также содержание стрептомицина (менее 0,5 ед/г) и пенициллина (менее 0,01 ед/г). В зависимости от вида продуктов максимальное содержание антибиотиков не должно превышать (мг/кг); бензилпенициллина 0,004—0,05, спектиномицина 0,2—5, дигидрострептомицина 0,2—1, неомицина 0,5—5; гентамицина 0,1 — 1, хлор- и окситетрациклина 0,1—0,6, сефтиофура 0,2—4.
Контроль за наличием остаточных количеств антибиотиков необходим на всех стадиях производства, особенно в готовой про-466
дукиии. Установленный законодакльс гвом мнопгхлран порядок предусматривает время обязательной выдержки животных перед убоем, во время которой содержание антибиотиков в крови и тканях животных постепенно снижается до безопасного уровня.
Присутствие антибиотиков в продуктах животного происхождения затрудняет их бактериологическое исследование, нарушает технологию производства сырокопченых колбас. Для решения практических задач ветсанэкспертизы часто бывает достаточно проводить качественное исследование продуктов животноводства на наличие антибиотиков и других ингибирующих рост и размножение микроорганизмов веществ, без учета их количества и вида.
В основе метода исследования продуктов убоя животных на наличие антибиотиков лежит способность многих видов антибиотиков задерживать рост микроорганизмов. Рекомендуется использовать одну из тест-культур микроорганизмов в лиофильно высушенном состоянии: паспортизованные штаммы Sar. lutea, St. aureus. Вас. subtilis.
Подготовка проб. Перед исследованием тест-культуры выращивают на мясо-пептонном бульоне или другой жидкой питательной среде в течение суток при соответствующей температуре. Если культуры находятся на агаровой среде, хранить их нужно при температуре 4—10 °C, пересевая на свежую питательную среду через 15—30 сут. Из образцов готовят навески массой 2—3 г.
Порядок проведения анализа. На пластинчатый мясо-пептонный агар пастеровской пипеткой наносят 2—3 капли бульонной тест-культуры (или смыва с агаровой культуры) микроорганизмов и тщательно распределяют по его поверхности. Затем на поверхность агара на одинаковом расстоянии друг от друга и от краев чашки Петри помещают три анализируемых пробы мяса (или продукта) массой 2—3 г и бумажный диск, пропитанный 0,25 ЕД пенициллина (тетрациклина). Чашку ставят сначала в холодильник при температуре 4—5 °C на 3—5 ч (для диффузии антибиотиков из мяса в питательную среду), а затем в термостат при температуре 37 °C на 15—20 ч. При наличии антибиотиков в пробе вокруг кусочка мяса образуется зона задержки роста микроорганизмов. Для контроля ее сравнивают с зоной задержки роста вокруг бумажного диска, пропитанного пенициллином (тетрациклином).
Полученные в ходе исследования результаты оформляют в виде протокола, форма которого приведена ниже:
Характеристика образца
Реакция тест-культуры микроорганизмов на наличие антибиотиков
Мышечная ткань
Внутренние органы (печень, почки и т. л.)
Мясные продукты
+/-+/-+/-
467
.. ..v. npvu.Msmti iвенной локализации ангиои-
отиков в соответствующих органах, лают опенку мяса и мясопродуктов по результатам исследований.
Лабораторная работа № 4
ОПРЕДЕЛЕНИЕ ГОРМОНОВ
Цель работы. Изучить и освоить серийный анализ мясных продуктов на содержание гормональных препаратов метолом EL1SA с использованием стандартных наборов для иммунофер-ментного анализа.
Задачи. Подготовить пробы и определить остаточное содержание гормональных препаратов в мясе и мясных продуктах.
Объекты исследования. Мясо и мясные продукты из говядины, свинины и птицы.
Материалы, реактивы и оборудование. Набор для иммунофер-ментной очистки экстрактов, включающий иммуноаффинную колонку (кат. NSJ 2154, «Randox Lab. Ltd». Англия) на 10 определений с максимальной поглощающей емкостью по диэтилстильбэстролу 50 нг/колонку; концентрированный буферный раствор для промывки колонки; вспомогательный набор для определения стильбенов иммуноферментным методом (кат. NSJ 2152) на 96 определений, включающий: микроплашку на 12x8 мм разборных ячеек с сенсибилизированными к стильбенам антителами; буферный раствор для промывки плашки и разбавления проб; белковый конъюгат; субстрат для окрашивания пробы; набор стандартных растворов концентрацией стильбена от 0 до 10 нг/см3 и спектрофотометр для определения оптической плотности среды в ячейках плашек Strip Reader EL-301 производства «Bio-Тек Instruments» (США): метил-третбутиловый или серный эфир; хлороформ; раствор гидроксида натрия молярной концентрацией 1 моль/дм3; раствор фосфорной кислоты молярной концентрацией 6 моль/дм3; раствор соляной кислоты молярной концентрацией 1 моль/дм3; раствор этанола (70 об.%); бидистиллированная вода.
Приготовление реактивов. Буферный раствор для промывки колонки. К одной части концентрированного раствора моющего буфера добавляют 19 частей бидистилли-рованной воды.
Буферный раствор для хранения колонки. К одной части концентрированного раствора буфера для хранения добавляют 4 объема бидистиллированной воды.
Белковый конъюгат. Концентрированный конъюгат разбавляют в 15 000 раз (см. инструкцию к прибору).
Субстраты АиБ для окрашивания пробы (см. инструкцию к прибору). Перед применением субстраты смешивают в соотношении 1:1.
468
Методические указания. Так называемые гормональные технологии» привлекают внимание товаропроизводителей мяса из-за известного положительного влияния некоторых гормонов на быстрый прирост массы скота (на 5—25 %). Несмотря на запрет применения гормонов в России, в ряде европейских стран и на Американском континенте гормоны используются достаточно широко. Рост импорта мяса у нас в стране делает проблему гормонального контроля весьма актуальной.
Для аналитического определения остатков гормональных препаратов используют две основные группы методов: хроматографические (тонкослойная хроматография — ТСХ: высокоэффективная жидкостная хроматография — ВЭЖХ; жидкостная хроматография с масс-спектрофотометрическим детектированием — ЖХМС; газовая хроматография с масс-спектрофотометрическим окончанием — ГХМС) и современные иммунные методы (иммуноферментный метод — ELISA и радиоиммунный метод — R1A).
Среди требований, предъявляемых к методам экспресс-мони-торинга продовольствия, главное место занимают чувствительность и селективность метода, а также время и стоимость анализа. Некоторые характеристики методов представлены в табл. 5.6.
Т а б л и ц а 5.6
Максимальный уровень содержания гормонов в мясных продуктах, определяемый различными методами
Метод Оп редел яе мое нс u iec гво Предел обнаружения, мкг/кг ПДК, мг/кг Время анализа с подготовкой проб.ч
гжх Эстрадиол 17(3 0.2 0,0005 10
RIA » 4 0,0005 8
ТСХ Диэтил стильбэстрол 0.01 Не допускается 7
ГЖХ » 0,5 То же 12
EL1SA » 1 » 5
Как видно из данных табл. 5.6, метод ELISA, широко используемый в европейских странах для контроля безопасности мяса и мясопродуктов, имеет явные преимущества.
Анализ структурных особенностей сложных органических соединений показывает, что развитие метаболических процессов в живых организмах, а также развитие дальнейших процессов приводят к уменьшению концентрации, например, самого диэтил-стильбэстрола и появлению родственных химических метаболитов, отличающихся от исходного вещества наличием различных органических заместителей, т. е. через определенное время в зависимости от химической устойчивости токсиканта он либо исчезнет в продукте, либо трансформируется, а токсичность вновь об-
469
rl.веществ co структурой, подобной исходному веще С1ву. окажшея б.пнкой к (окснчности первоначально!о вещества ч го также опасно для человека.
Предлагаемый к изучению и освоению шммунофермен гныи мегог оодалае) как называемой перекрестной чувсгшпсльнос тьк>. г. с.. являясь исключи 1ельно специфичным методом применительно к какому-либо веществу. позволяв! определить всю 1 руину родственных соединении, что повышает его ценность для реальной сертификации пищевой продукции ио максимально возможному числу вредных токсикантов.
Подготовка проб. Гомогенизируют 2,5 г образца мяса или мясопродукта с 15 см3 мет и.ттрегбутилового пли серного эфира. гомогенат перемешивают и отделяют центрифугированием при частоте вращения 33,3 с”1 в течение 10 мин или отстаиванием 12 см3 эфирного слоя, который испаряют в токе сжатого воздуха. Сухой остаток растворяют в 1 см’ хлороформа, добавляют 2 см3 раствора гидроксила натрия молярной концентрацией I моль/дм3, раствор перемешивают и отделяют 1 см3 водного слоя с последующим добавлением еще 1 см3 раствора гидроксида натрия молярной концентрацией I моль/дм3 к слою хлороформа и отделением 1 см' водного экстракта. Водный экстракт нейтрализуют, добавляя к 2 см3 экстракта 200 мкдм3 раствора фосфорной кислоты молярной концентрацией 6 моль/дм) и раствор соляной кислоты молярной концентрацией I моль/дм3.
Порядок проведения анализа. Весь объем нейтрализованного раствора наносят на иммуноферментную колонку' после ее кондиционирования, которую для этого промывают 15 см3 предварительно разбавленного раствора моющего буфера (расход менее 3 см3/мин). Экстракт пропускают через колонку самотеком, промывают колонку 5 см3 разбавленного раствора моющего буфера и 5 см3 бидистиллированной воды (расход менее 5 см3/мин). Элюируют фракцию стильбенов, промывая колонку 3 см3 раствора этанола (70 об. %). Колонку окончательно промывают 10 см3 раствора этанола (70 об. %).
С помощью пипетки в лунки плашки вносят по 100 мкдм3 разбавленного буферного и 25 мкдм3 анализируемого растворов с иммуноферментной колонки (или стандартного образна). Затем плашку выдерживают в темноте при комнатной температуре (19—25 °C) в течение 1ч. С помощью пипетки добавляют по 75 мкдм3 разбавленного раствора конъюгата в каждую лунку плашки, выдерживают в течение 30 мин, промывают плашку 10—12 раз разбавленным буферным раствором и высушивают. Затем добавляют по 25 мкдм3 смеси субстратов А и Б в соотношении 1:1, выдерживают плашку в темноте при комнатной темпе; ратуре в течение 20 мин и добавляют в каждую лунку по 50 мкдм3 раствора соляной кислоты молярной концентрацией 1 моль/дм3. При этом наблюдают за изменением голубого окрашивания ра-470
Рис. 5.6. Зависимость оптической плотности комплекса диэтилсгильбэстрола с конъюгатом Randox при длинах воли 450 (У) и 495 нм (2) от концентрации стильбена в растворе этанола 70об.гс (при использовании в качестве стандарта растворов гексаэстрола концентрациями 0,05; 0.24; 0.56; 0.95 и 4.6нг/см3)
створа в лупках платки на желтое. Оптическую плотность растворов в лунках измеряют при длине волны на спектрофотометре 450 нм. Содержание гормонов определяют по калибровочному графику.
Построение калибровочного графика. Для построения графика используют стандартные растворы известной концентрации гормона, которые помещают в лунки плашки и окрашивают в аналогичных с исследуемой пробой условиях. Пример калибровочного графика определения гормонов представлен на рис. 5.6.
Экспериментальные данные оформляют в виде таблицы:
Образен
Оптическая плотность раствора в лунке
Концентрация гормонов, полученная по калибровочному графику, нг/см:
Лабораторная работа № 5
ОПРЕДЕЛЕНИЕ ПЕСТИЦИДОВ
Цель работы. Освоить методы контроля остаточных количеств пестицидов в мясе, вторичных продуктах убоя скота и мясных продуктах.
Задачи. Определить наличие хлорорганических пестицидов (ХОП) хроматографией в тонком слое (ТСХ) и фосфорорганических пестицидов (ФОП) энзимохроматографией.
Объекты исследования. Образцы мышечной ткани разных видов убойных животных и птицы: субпродукты [ и И категорий; мясные продукты различных ассортиментных групп; пишевые животные жиры.
Методические указания. Необходим строгий контроль остаточных количеств ХОП, ФОП и их метаболитов в продуктах животноводства, в частности в мясе и мясных продуктах, на соответствие санитарно-гигиеническим нормам. В соответствии с требованиями безопасный уровень содержания хлорорганических пес-
471
тицидов (ДДТ и его метаболитов. iексахлорциклогексана) в продукте должен составлять не более ОД мг/кг продукта, а предельно допустимая концентрация большинства хлорорганичес-ких, фосфорорганических и триазиновых пестицидов — 0.01 — 1 мг/кг продукта.
Метод ТСХ для определения остаточных количеств пестицидов в пищевых продуктах считается перспективным с точки зрения экспрессное™ и избирательности. Его широко используют для контроля за остаточным содержанием пестицидов в пищевом сырье и готовой продукции.
Метод основан на экстрагировании препаратов из исследуемого материала органическими растворителями, очистке экстрактов и последующем хроматографировании в тонком слое сорбентов. Подвижным растворителем служит гексан или гексан в смеси с ацетоном. Места локализации пестицидов обнаруживают после опрыскивания пластинок раствором аммиаката серебра с последующим ультрафиолетовым облучением пластинок «Силуфол», содержащих о-толуидин. Количественное определение проводят визуальным сравнением или измерением площадей пятен пробы и стандартных растворов. Этим методом можно определить ДДТ, гексахлоран, альдрин, кельтан, гептахлор, метоксихлор, эфир-сульфонат и другие препараты. Наименьшее содержание пестицида, выявляемое в мясе, органах и жире, 0,02 мг/кг.
Определение ФОП энзимохроматографическим методом (метод М. В. Письменной) основано на экстрагировании ФОП из исследуемой пробы органическим растворителем, очистке экстракта охлажденным ацетоном и определении препарата на пластинке методом хроматографии в тонком слое с ферментным проявлением без активации или после активации. Фосфорные (ДДВФ. дибром и др.) и тиофосфорные эфиры (рицид, циодрин и др.) определяют без активации, а тиофосфорнокислые эфиры (метафос, метилнитрофос, пиразофос и др.), дитиофосфаты (карбофос, фталофос, фозалон, рогор и др.) и эфиры фосфорной кислоты (хлорофос) необходимо предварительно активировать. Препараты, угнетающие холинэстеразу, проявляются в виде белых пятен на голубом фоне.
1.ОПРЕДЕЛЕНИЕ ХЛОРОРГАНИЧЕСКИХ ПЕСТИЦИДОВ МЕТОДОМ ТСХ
Материалы, реактивы и оборудование. Пластинки для хроматографии; хроматографическая камера (можно использовать эксикатор); прибор для отгонки растворителей; ртутно-кварцевая лампа ПРК-4; баня водяная; пульверизатор стеклянный для опрыскивания пластинок; прибор для встряхивания; микропипетки для нанесения стандартных растворов; пипетка или шприц для нанесе-472
ния пробы: хроматографическая колонка размером 20 x 400 мм: ступка: ножницы; колбы с притертыми пробками вместимостью 250 и 500 см3: воронка диаметром 6 см: круглодонная колба со шлифом вместимостью 250—300 см3; цилиндры мерные вместимостью 50 и 100 см3; пипетки вместимостью 1, 5 и 10 см3: чашки для выпаривания: сульфат натрия безводный; гексан; петролей-ный эфир (температура кипения 40—70сС): диэтиловый эфир; бензол; ацетон; стандартные образцы гексахлорана, гептахлора, эфирсульфоната и др.; стандартные растворы ядохимикатов; проявляющие реактивы № 1 или 2.
Приготовление растворов и реактивов. Стандартные образцы гексахлорана, гепта хлор а, эфирсульфоната и других ядохимикатов. 10 мг пестицида растворяют в гексане в мерной колбе вместимостью 100 см3 и доводят до метки этим же растворителем. Хранят в стеклянной посуде с притертыми пробками в холодильнике.
Проявляющий реактив № 1. 0,5 г нитрата серебра растворяют в 5 см3 дистиллированной воды, прибавляют 7 см3 аммиака и доводят объем раствора до 100 см3 ацетоном. В готовый раствор добавляют 0,2 см3 пероксида водорода. Раствор хранят в колбе с притертой пробкой в течение 3 сут. Расход раствора на пластинку размером 9 х 12 см — 8—10 см3.
Проявляющий реактив №2. 0,5 г нитрата серебра растворяют в 5 см3 дистиллированной воды, прибавляют 10 см3 2-феноксиэтанола и доводят объем раствора ацетоном до 200 см3, затем добавляют 6 капель раствора пероксида водорода массовой долей 30 %.
Подготовка проб. Навеску мяса массой 20 г тщательно измельчают в ступке ножницами, смешивают с безводным сульфатом натрия и помещают в колбу с притертой пробкой. Экстрагируют в течение 1,5 ч при встряхивании, дважды приливая по 50 см3 смеси гексана (или петролейного эфира) с ацетоном 1:1.
Подготовка хроматографической колонки. В нижнюю часть колонки помещают стекловату или 500 мг обезжиренной ваты, засыпают 70 см3 силикагеля АСК, уплотняют его постукиванием по колонке, промывают 50 см3 гексана или петролейного эфира.
Порядок проведения анализа. Экстракт фильтруют через воронку с бумажным фильтром, заполненным на 2/3 безводным сульфатом натрия. Растворитель отгоняют, а сухой остаток растворяют в 20 см3 гексана и переносят в хроматографическую колонку. После впитывания экстракта в сорбент пестициды элюируют 110 см3 смеси бензола с гексаном в соотношении 3 : 8 порциями по 25—30 см3. Элюат собирают в круглодонную колбу со шлифом вместимостью 250—300 см3. Через 10 мин после впитывания последней порции растворителя сорбент отжимают с помощью груши. Элюат отгоняют до объема 0,1 см3.
473
Для хроматографирования используют пластинку заводского изготовления типа «Силуфол» или готовят накануне с использованием в качестве сорбента оксида алюминия или силикагеля КГК. Пластинки «Силуфол» обрабатывают о-толуилином. Для этого их погружают в раствор о-толуидина в ацетоне массовой долей 0,1 с’с. налитого I? камеру для хроматографирования. После того как фронт растворителя поднимется до верхнего края пластинок, их вынимают и сушат на воздухе. Хранят в эксикаторе.
На хроматографической пластинке на расстоянии 1.5 см от края отмечают линию старта (на которую наносят растворы, подлежащие разделению), а на расстоянии 10 см от нее —линию фронта растворителей (до которой должен подняться растворитель в процессе хроматографирования).
На стартовую линию шприцем или пипеткой наносят экстракт в одну точку так, чтобы диаметр пятна не превышал 1 см. Остаток экстракта в колбе смывают тремя порциями (по 0,2 см3) диэтилового эфира, которые наносят в центр первого пятна (после высыхания). Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, содержащие 10, 5 и 1 мкг исследуемых препаратов. Хроматографическую камеру насыщают парами растворителя. Для этого на дно камеры наливают растворитель слоем толщиной около 0,5 см и выдерживают 30 мин. Пластинки с нанесенными растворами устанавливают в камере в вертикальном положении или под углом 80—85°. Нижний край пластинки (со стороны стартовой линии) погружают в растворитель на 0,5 см. При использовании пластинок с оксидом алюминия или силикагелем в качестве подвижного растворителя применяют гексан или смесь гексана с ацетоном (6: 1), при использовании пластинок «Силуфол» — подвижный растворитель — раствор ацетона в гексане массовой долей 1 %, а в случае, если пластинки «Силуфол» обработаны о-толуидином, — гексан с диэтиловым эфиром (49: 1). После того как растворитель поднимется до фронтальной линии, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Затем ее орошают проявляющим реактивом и облучают УФ-лучами в течение 10—15 мин (расстояние от лампы ПРК-4 — 20 см). При наличии хлорорганических пестицидов на пластинке появляются пятна серо-черного цвета. При использовании пластинок «Силуфол», обработанных о-толуиди-ном, их сразу после хроматографирования облучают УФ-лучами в течение нескольких минут. При наличии указанных пестицидов появляются пятна сине-голубого цвета.
Вид и количество пестицида определяют, сравнивая величины и площади пятна пробы и стандартных растворов. Величина R^— это отношение фронта вещества (расстояние в сантиметрах от линии старта до центра пятна) и фронта растворителя (расстояние от линии старта до линии фронта). Она служит качественной характеристикой каждого вещества и используется для его идентификации.
474
Содержание у-шроргшшчегких пестицидов в пес ю.1\?мом ма-териале (мг/кН рассчи i ываю! по формсде
иг .. с.ыгржаш.с препарат: в ciапдартном растворе, mki : 5. площадь пятна ii[V"4.. -.!\i ; /и - масса проби. в ;ятоп .тля ана.тта. г: ф - • плошать пятна стан-дар: но: > раствора. м\г
2. ОПРЕДЕЛЕНИЕ ФОП ЭНЗИМОХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ
Материалы, реактивы и оборудование. Пластинки для хроматографии на основе силикагеля КСК; прибор для отгонки растворителей; баня водяная; хроматографическая камера; ртутно-кварцевая лампа ПРИ -4; эксикаторы; пульверизатор стеклянный; ступка; ножницы; делительная воронка, воронки диаметром 5 см (фильтровальные); мерные пробирки на шлифах вместимостью 5—10 см3; колбы на шлифах вместимостью 50, 100 и 250 см3; пипетки вместимостью 1, 5 и 10 см3; мерные колбы вместимостью 25 и 100 см3; микропипетки для нанесения стандартных растворов; пипетка или шприц для нанесения пробы; ацетон; хлорид метилена; сульфат натрия безводный; гексан; бром; хлороформ; бензол; аммиак; раствор тиосульфата (серноватистокислого) натрия массовой долей 2,5 %; стандартные образцы ФОП и приготовленные из них стандартные растворы.
Приготовление растворов и реактивов. Основной стандартный раствор (раствор А). 10 мг пестицида растворяют в 100 см3 ацетона.
Рабочие стандартные растворы. Получают разбавлением основного раствора так, чтобы в 1 см3 содержалось 0,5 мкг (раствор Б) и 0,1 мкг (раствор В) пестицида.
Пластинки для хроматографирования. Для 12 стеклянных пластинок размером 9х 12 см берут 35 г силикагеля, просеянного через сито 100 меш (число отверстий на 1 кв. дюйм), и 2 г сульфата кальция, просушенного при 160 °C в течение 6 ч, тщательно растирают в фарфоровой ступке. Порциями прибавляют 90 см3 дистиллированной воды и размешивают до однородной массы. 10 г суспензии наносят на пластинку и равномерно распределяют по ее поверхности. Сушат в горизонтальном положении в течение 18—20 ч при комнатной температуре, хранят в эксикаторе.
Ферментный раствор. Получают из печени крупного рогатого скота (свежую, однократно замороженную и хранимую в холодильнике печень можно использовать в течение 6 мес). 1 г печени растирают в ступке с 9 см3 буферного раствора и фильтруют через вату. К 1 см3 фильтрата прибавляют 4 см3
475
буферного раствора. Ферментный раствор используют для опрыскивания пластинок: готовят непосредственно перед употреблением.
Б у ф е р н ы й р а с т в о р (pH 8,69). Смесь ортофосфорной (2,1 см3), уксусной (2.3 см3) и борной (2,47 г) кислот доводят дистиллированной водой до 1 дм3. Для получения буферного раствора pH 8 ,69 к 100 см3 этой смеси прибавляют 65 см3 раствора гидроксида натрия молярной концентрацией 0,2 моль/дм3.
Проявляющие реактивы (один из двух).
Реактив 1. Смесь растворов дважды диазотированного о-диани-зидина и 2-нафтилацетата. Готовят два раствора: № 1 — 10 мг дважды диазотированного о-дианизидина растворяют в 100 см3 дистиллированной воды, № 2 — 125 мг 2-нафтилацетата растворяют в 100 см3 этанола. Перед проявлением смешивают 16 см3 раствора № 1 и 4 см3 раствора № 2.
Реактив 2. 10 мг индоксилацетата (или броминдоксиоксидата) растворяют в 6 см3 этанола. К раствору прибавляют 6 см3 буферного раствора (pH 8,69), 1 см3 водного раствора феррицианида калия K3Fe(CN)6 молярной концентрацией 0,05 моль/дм3 и 1 см3 раствора ферроцианида калия [K4Fe(CN)6] • ЗН0О.
Для проявления пластинок используют свежеприготовленный раствор.
Подготовка проб. Навеску мяса массой 25 г тщательно измельчают ножницами в ступке, переносят в колбу, прибавляют 60 см3 ацетона и экстрагируют в течение 20 мин, периодически встряхивая.
Порядок проведения анализа. Экстракт фильтруют через бумажный фильтр в делительную воронку. Экстракцию повторяют с 30 см3 ацетона. К объединенному экстракту приливают 150 см3 дистиллированной воды, перемешивают и препараты экстрагируют хлоридом метилена дважды по 50 см3, встряхивая по 2—3 мин. Органическую фазу объединяют, пропуская через воронку с 10—15 г безводного сульфата натрия, отгоняют хлорид метилена (при температуре 30—35 °C) до объема 0,5—1 см3, досуха испаряют на воздухе. Сухой остаток смывают 5 см3 охлажденного при 0 °C ацетона, фильтруют через бумажный фильтр в мерную пробирку, выдерживают 10—15 мин при комнатной температуре и доводят объем в пробирке точно до 5 см3.
На стартовую линию хроматографической пластинки поочередно с интервалом около 2 см наносят 10 нм3 стандартного раствора В (0,001 мкг пестицида), 2 нм3 пробы, 10 нм3 стандартного раствора Б (0,005 мкг), Юмксм3 пробы. Пластинку помещают в хроматографическую камеру, в которую предварительно за 30 мин до исследования наливают соответствующий подвижный растворитель. В качестве последнего при определении большинства ФОП используют хлороформ, для хлоро-476
фоса — смесь ыжсана с ацетоном (1:1). для ротора и ангио — смесь бензола и ацетона (3 : 2). Когда фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и растворитель испаряют. После этого, если необходимо, проводят активацию (окисление). Для эфиров тиофосфорной и дитпофос-форной кислот окисление осуществляют парами брома, водным раствором брома или действием ультрафиолетовых лучей, для эфиров типа хлорофоса — аммиаком.
Окис л е н и е в и а р а х б р о м а. Пластинки на 1 мин помещают в эксикатор, насыщенный парами брома. После этого их выдерживают 60 мин на воздухе для удаления избытка брома и подвергают дальнейшей обработке.
Окисление водным раствором брома. Пластинки опрыскивают насыщенным водным раствором брома до слабого увлажнения слоя. После 15 мин экспозиции при комнатной температуре избыточный бром удаляют легким опрыскиванием пластинок раствором тиосульфата натрия массовой долей 2,5 %.
Активация У Ф-л у ч а м и. Проводят так же, как при определении хлорорганических соединений.
Активация аммиаком. Пластинки опрыскивают водным раствором аммиака (1 : 4) до слабого увлажнения, а затем выдерживают 15 мин при комнатной температуре.
После активации и высушивания при комнатной температуре пластинки опрыскивают свежеприготовленным ферментным раствором и выдерживают 40—60 мин при температуре 38 °C в термостате, насыщенном парами воды. В термостат предварительно ставят чашки Петри с водой. Затем их опрыскивают проявляющим раствором и снова помещают в термостат при той же температуре. Пестициды проявляются в течение 10—30 мин в виде белых пятен на голубом фоне.
Количественное определение ФОП проводят сравнением площади пятна пробы с наиболее близкой к ней по величине площади стандарта. Содержание пестицида в исследуемом материале (мг/кг) рассчитывают по формуле
aS2k] • 1000
(5-20)
где а — содержание препарата в стандарте, мкг; ф и ф — площадь пятна пробы и стандарта, мм2; ф — общий объем экстракта, см3/ И2 — объем экстракта, нанесенного на пластинку, мкл/нм3; т — масса пробы, взятой для анализа, г.
По окончании экспериментальных работ анализируют результаты, делают вывод о преимущественной локализации остаточных пестицидов в органах и тканях убойных животных, дают оценку
477
- .............. 11[,и.пм(|м iio содержа
ншо оспаточных количеси) Х()11 и ФОП. Данные заносят в таблицу рекомендуемой формы:
( \и< рл IHIK-. \и м
Харли анк । i'k.i < 'opj ! i.i \()ll ФОН
I n'P'1,1 ! IIKHOC >, I К I I! 4Ul Ki H( ip M. I III liHOC i <|>U KT 11ЧСС КОС
Говядина
Свинина
Баранина
Мясо птицы
Субпродукт! 1 Kaieiopiiii
Субпродукты 11 каты ори и
Мясоиродх к 1 ы
Лабораторная работа № 6
ОПРЕДЕЛЕНИЕ ФЕНОЛОВ В КОПЧЕНЫХ МЯСНЫХ ПРОДУКТАХ
Цель работы. Освоить методы качественного обнаружения и количественного определения суммарных фенолов в колбасных и копченых изделиях.
Задачи. Определить зоны проникновения фенолов при копчении мясных продуктов различных ассортиментных групп; установить суммарное содержание фенолов в копченых колбасных изделиях различного группового ассортимента; по результатам исследований дать санитарно-гигиеническую оценку копченым мясным изделиям.
Объекты исследования. Копченые колбасные изделия различного группового ассортимента или копчености.
Материалы, реактивы и оборудование. Раствор ацетона массовой долей 50 %; раствор тетрабората натрия массовой долей 0,5 %; раствор 4-аминоантипирина массовой долей 2 %: раствор гексацианоферрата калия (II) массовой долей 8 %; гваякол (для построения калиброчного графика); раствор персульфата аммония массовой долей 20 %; раствор карбоната натрия массовой долей 1 %; проявитель; фильтровальная бумага; дистиллированная вода; фотоэлектроколориметр ФЭК-56М или КФК-2; мерные колбы вместимостью 50 см3; мерный цилиндр вместимостью 150 см3; пипетки вместимостью 1, 5, 10 см3; колориметрические пробирки; коническая колба вместимостью 250 см3; стеклянная палочка; бумажный фильтр «синяя лента»; весы технические, вибровстряхива-тель; гидроксид натрия NaOH и его водный раствор молярной концентрацией 0,1 моль/дм3; серная кислота H2SO4 и ее раствор массовой долей 25 %; сульфат цинка ZnSO4 и его водный раствор массовой долей 0,45 %; нитрит натрия NaNO2 и его свежеприго-478
товленныи водный раствор массовой долей и.? мд гидроксид аммония NH4OH и его раствор массовой долей 10 стандартный водный раствор фенола (с = 1 мг/см').
Методические указания. Фенольные соединения обладают токсическим и даже канцерогенным действием, в связи с чем количество их в пищевых продуктах должно быть сведено до минимума. Для гарантии экологической чистоты нишевых продуктов необходимо строю контролировать содержание фенолов.
При копчении фенолы сначала интенсивно накапливаются в поверхностном слое. В дальнейшем проникновение их в колбасу значительно замедляется. Одновременно происходит диффузия фенольных компонентов дыма во внутренние слои колбасы. На последней стадии копчения и сушки количество фенольных соединений в поверхнос! ном слое уменьшается почти наполовину и заметно возрастает во всех внутренних слоях колбасного батона, фронт проникновения фенольных соединений внутрь колбасного батона тесно связан с химическим составом сырья и технологическими режимами производства копченых изделий и характеризует качество копчения. Например, фенолы хорошо растворяются в жире. В жировой ткани их в 1,5 раза больше, чем в мышечной. В процессе холодного копчения в копченостях накапливается в среднем 15 мг % (9—24 мг %) фенолов.
При изучении вопроса о скорости и характере проникновения фенолов в колбасные изделия удобно пользоваться методом отпечатков, разработанным сотрудниками кафедры аналитической химии Воронежской государственной технологической академии, который основан на развитии качественной реакции фенолов в присутствии карбоната натрия и специального проявителя.
Количественное определение фенолов в колбасных изделиях проводят одним из колориметрических методов. Суммарное определение содержания фенолов основано на измерении оптической плотности окрашенного раствора, цвет которого возникает в результате качественной реакции:
ОН + 4K;|Fe(CN)6| + 4NH4()H
0+ 3K4[Fe(CN)b[ + (NH4)4Fe(CN)6 + 4H2O
479
Другой метол суммарного определения содержания фенолов основан на получении нитрозосоелинений при взаимодействии фенола с нитритом натрия. В результате реакции нитрозосоединение образует с избытком аммиака продукт, окрашенный в желтый цвет, который затем фотоэлектроколориметрпруют.
Подготовка проб. Образцы продуктов (не менее 500 г) дважды измельчают на мясорубке.
Перед определением фенолов в копченых изделиях проводят органолептическую оценку продуктов. При этом осматривают поверхность колбасного батона, отмечают вид колбасной оболочки, групповой ассортимент и наименование колбасы (с помощью преподавателя). Путем визуальной оценки устанавливают цвет, состояние поверхности на разрезе, запах и вкус. Данные фиксируют в таблице результатов.
1.ОПРЕДЕЛЕНИЕ ГРАНИЦ ПРОНИКНОВЕНИЯ ФЕНОЛОВ
Приготовление реактивов и материалов. Ф и л ь т р о в а л ь н а я бумага для определения границ п р о н и к н о в е-ния фенолов. Фильтровальную бумагу погружают на 20— 30 с в раствор Na9CO3 массовой долей I %, после чего ее сушат на воздухе и хранят.”
Проявитель для определения границ проникновения фенолов. 1г 4-аминоантипирина растворяют в 50 см3 раствора этанола (96 об. %).
Порядок проведения анализа. Полученный со среза копченой колбасы отпечаток проникших фенолов должен быть видимым и отличаться по цвету от фона предварительно подготовленной фильтровальной бумаги. Подготовленную фильтровальную бумагу опрыскивают из пульверизатора проявителем и плотно прижимают к срезу колбасного батона. Спустя 20—30 с бумагу отделяют от поверхности среза колбасы и опрыскивают раствором персульфата аммония массовой долей 20 %. Через 2 мин на бумаге образуется отчетливый, окрашенный в розовый цвет отпечаток границ проникновения фенолов. Отпечаток зарисовывают, измеряют с точностью до 0,1 мм границы проникновения или аккуратно вырезают ножницами и вносят в таблицу результатов.
2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ФЕНОЛОВ В КОЛБАСНЫХ ИЗДЕЛИЯХ
2.1. КОЛОРИМЕТРИЧЕСКИЙ МЕТОД НА ОСНОВЕ ЦВЕТНОЙ РЕАКЦИИ С 4-АМИНОАНТИПИРИНОМ И ГЕКСАЦИАНОФЕРРАТОМ КАЛИЯ (II)
Навески измельченных колбас массой 3—5 г помещают в малый сосуд гомогенизатора, заливают раствором ацетона массовой долей 50 % в соотношении 1 :4 (по объему) и гомогенизируют в
480
течение 5 мин. а затем фильтруют. К 5 см? прозрачного раствора добавляют 20 см3 раствора тетрабората натрия массовой нолей 0,5 %. 0.5 см3 раствора 4-аминоантипирина массовой долей 2 с’с и 0.25 см3 раствора гексацианоферрата калия (II) массовой долей 8 %. Все реагенты перемешивают и по истечении 15 мин измеряют оптическую плотность (интенсивность окраски) на фотоэлектроколориметре ФЭК-56М с зеленым светофильтром при длине волны 510 нм в кювете с толщиной слоя 1 см.
Параллельно готовят контрольную пробу, в которой вместо исследуемого фильтра используют 5 см3 раствора ацетона массовой долей 50 %.
Содержание суммарных фенолов (мг/100 г) вычисляют ио формуле
Я100Л
где В— объем ацетонового экстракта, см-’; 100 — коэффициент пересчета на 100г продукта; А — содержание фенолов в 5 см3 окрашенного раствора, определенное по калибровочному графику; И—объем фильтрата, взятый для анализа, см3; т — масса навески продукта, г.
Для проведения расчетов готовят растворы гваякола в диапазоне концентраций от 1 до 100 мкг/см3 и строят график в координатах: оптическая плотность (Л) — концентрация гваякола.
2.2. КОЛОРИМЕТРИЧЕСКИЙ МЕТОД, ОСНОВАННЫЙ
НА ПОЛУЧЕНИИ НИТРОЗОСОЕДИНЕНИЙ ПРИ ВЗАИМОДЕЙСТВИИ ФЕНОЛА С НИТРИТОМ НАТРИЯ
В коническую колбу помещают 15 г копченой колбасы, добавляют 50 см3 дистиллированной воды, закрывают пришлифованной стеклянной или корковой пробкой и встряхивают на вибро-встряхивателе в течение 15 мин. Содержимое конической колбы фильтруют в мерную колбу вместимостью 50 см3 и доводят дистиллированной водой до метки. Для осаждения белков 10 см3 полученного раствора переносят в колориметрическую пробирку, добавляют пипеткой 4 см3 раствора ZnSO4 массовой долей 0,45 %, 1 см3 раствора NaOH молярной концентрацией 0,1 моль/дм3 и выдерживают 5 мин на водяной бане при температуре кипения, после чего раствор фильтруют. В колориметрическую пробирку помещают 5 см3 фильтрата, добавляют 0,25 см3 раствора H9SO^массовой долей 25 % и 2,5 см3 раствора NaNO2 массовой долей 0,5 %. Содержимое пробирки нагревают в течение 5 мин на водяной бане, охлаждают и добавляют 5 см3 раствора NH4OH массовой долей 10 %. Оптическую плотность окрашенного в желтый цвет раствора измеряют на фотоэлектроколориметре ФЭК-56М при
481
....... u пшцинои раоочсго слоя
3 см. Содержание фенола в пробе определяют по калибровочному графику, построенному по стандартным растворам фенола.
Построение калибровочного графика. В четыре мерные колбы вместимостью 50 см' отмеряют пипеткой 2.5: 5.0: 7.5 и 10 см стандартного раствора фенола (с= 1 мг/см3) и доводят дистиллированной водой до метки. В четыре колориметрические пробирки помещают по 5 см3 фенола, добавляют 1 см3 раствора NaOH молярной концентрацией 0,1 моль/дм3, 0,25 см3 раствора H,SO4 массовой долей 25 % и 2,5 см3 раствора NaNCC массовой долей 5 10 ? %. Содержимое пробирок перемешивают стеклянной палочкой, нагревают на водяной бане при температуре кипения. охлаждают и добавляют в каждую пробирку 5 см3 раствора NH4OH массовой долей 10%. Растворы тщательно перемешивают и через 15 мин измеряют оптическую плотность окрашенных в желтый цвет растворов в кюветах с толщиной рабочего слоя Зсм при длине волны ?>. = 400 нм. В качестве раствора сравнения используют раствор, содержащий все компоненты, кроме фенола. Каждое измерение выполняют три раза. По результатам измерений строят калибровочный график D =f(c).
Содержание фенолов (мг %) рассчитывают по формуле
Х = с- 5Q/m 100, (5.22)
где с — концентрация фенолов в водной вытяжке, найденная по калибровочному графику, мг/см3; 50 — объем водной вытяжки, см3; w— .масса навески продукта, г.
Полученные результаты сводят в таблицу:
Групповой ассортимент продуктов
Органолептические показатели ! cVMMapHOe
Цвет Запах Вкус Вид оболочки, со- стояние и окраска 1 % ,, Ю ’ поверхности мг/100г
Границы проникновения фенолов (отпечатки)
Полукопченые
Варено-копченые
Сырокопченые
На основании полученных результатов студенты самостоятельно формулируют выводы и делают общее заключение по работе с учетом отмеченных органолептических показателей и количественного содержания фенольной фракции в мясных продуктах.
482
Лабораторная работа № 7
ОПРЕДЕЛЕНИЕ БЕНЗ(А)ПИРЕНА В КОПЧЕНЫХ МЯСНЫХ ПРОДУКТАХ
Цель работы. Освоить методы качественного и количественного определения полициклических ароматических yiлеволоролов (бенз(а)нирена) в копченых мясных продуктах на основе флуоресцентно-спектральных методов.
Задачи. Изучить методы подготовки проб к определению полициклических ароматических угле водородов; | бен з(а)п ирена | в копченых мясных продуктах; провести качественное определение бенз(а)пирена (БП) флуоресцентно-спектральным методом с использованием эффекта Э. В. Шпольского; установить количественное содержание БП в копченых мясных продуктах флуоресцентно-спектральным метолом в одной из модификаций! (с помощью добавок или внутреннего стандарта); по результатам исследований дать санитарно-гигиеническую оценку копченым мясным продуктам на соответствие требованиям гигиенических нормативов по содержанию полициклических ароматических углеводородов.
Объекты исследования. Колбасные изделия различного группового ассортимента: вареные, варено-копченые, полукопченые, сырокопченые, а также копчености.
Материалы, реактивы и оборудование. Петролсйный эфир: смесь хлороформ — петролсйный эфир (1:2); //-октан; жидкий азот; колонка стеклянная длиной 120—140 мм; хроматографическая колонка (или пластинка); сосуд Дьюара; ртутно-кварцевая лампа ДРШ-250. ДРШ-50 (или ПРК-2); фильтр УФС-1 (или УФС-2); спектрограф ИСП-51; эталонное вещество (1,12-бензперилен), чистый бенз(а)пирен; мясорубка; этанол; этиловый эфир; дистиллированная вода; безводный Na2SO4: оксид алюминия; бензол.
Методические указания. Полициклические ароматические углеводороды (ПАУ) присутствуют в продуктах растительного происхождения, копченых колбасах и копченостях. Одним из наиболее известных представителей ПАУ является бенз(а)пирен (БП). содержание которого в копченых и полукопченых колбасах колеблется от 1 мкг до нескольких десятков микрограммов на 1 кг продукта, а в вареных — от 0,2 до 1,0 мкг/кг.
Для количественного определения канцерогенных ПАУ в пищевых продуктах широкое применение нашли люминесцентные методы исследования. Определение бенз(а)пирена и других канцерогенных ПАУ проводится по тонкой структуре спектра флуоресценции при низкой температуре.
Флуоресцентно-спектральный метод определения бензапирена включает несколько этапов: извлечение из навески продукта
4X3
фракции, содержащей ПАЗ': очистку полученной (фракции os принесен и хроматографическое разделение ПАУ: качественное определение БП и других ПАУ но спектрам люминес пеннин при le.Miiepaiypc жидкою азота: количественное онрс деление БП с помощью одной из модификаций спектрально флуоресцентно!о метола (метолом добавок или методом bhvi-ренпего стандарта).
При выполнении всех лаион выделения, очистки и фракционирования ПАУ предел чувствительности метола равен 0.1 — 0.2 мкг/кг. Погрешность опыта составляет ± 10—15 %.
Качественное определение БП проводят спектральным методом с использованием эффекта Э. В. Шпольского. При температуре минус 196 °C получают спектры люминесценции отдельных фракций ПАУ, растворенных в нормальных парафиновых углеводородах. Спектры имеют тонкую структуру и называются квази-линейчатыми.
Подготовка проб. Из копченого продукта предварительно готовят фарш путем измельчения на мясорубке. К 1 кг фарша приливают 1 дм3 этанола, добавляют 150—-250 г КОН (в зависимости от содержания жиров в продукте) и кипятят 1,5— 2 ч для омыления липидов. Затем приливают 3—5-кратный объем дистиллированной воды и экстрагируют неомыляемые вещества этиловым эфиром. Первая порция эфира должна быть в 4—5 раз больше объема обрабатываемого раствора. Последующие три-четыре порции эфира должны быть в 3 раза больше первой.
Эфирный экстракт несколько раз промывают дистиллированной водой, подкисляя первую порцию воды, потом сушат над безводным Na2SO4. Эфир отгоняют, остаток растворяют в бензоле и пропускают через колонку длиной 120—140 мм, заполненную оксидом алюминия. Адсорбированные в колонку ПАУ, отделенные от других неомыляемых веществ, элюируют бензолом до тех пор, пока не прекратится выделение фракции с синей флуоресценцией. Бензол отгоняют из элюата, а остаток фракционируют колоночной или тонкослойной хроматографией.
Выделенную смесь ПАУ, содержащую некоторые примеси, растворяют в 10—15 см3 петролейного эфира и наносят на заполненную оксидом алюминия колонку диаметром 10—14 мм и высотой 120—140 мм. Флуоресцирующие фракции ПАУ сначала элюируют петролейным эфиром, а затем с добавлением бензола. Бенз(а)пирен содержится в III, IV или V фракциях. Для более четкого отделения бенз(а)пирена можно повторить фракционирование колоночным методом или в тонком слое оксида алюминия. При использовании второго метода в качестве растворителя служит смесь хлороформ — петролейный эфир в соотношении 1 : 2.
4X4
1. КАЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ БЕНЗ(А)ПИРЕНА
Порядок проведения анализа. Для качественного определения БП используют смесь, состоящую из I см' бензольного экстракта и 2 см? //-октана. Пробирку со смесью помешают в сосуд Дьюара с жидким азотом. Возбуждают люминесценцию с помощью ртутнокварцевой лампы ДРШ-250 (ДРШ-50 или ПРК -2). пропуская УФ-излучение через фильтр УФС-1 или УФС-2. При определении только БП (если другие фракции ПАУ нс определяют) можно пользоваться также УФС-3 или УФС-4. Для записи спектра обычно используют спектрограф ИСП-51 с камерой / = 270 мм. В спектре замороженного //-октанового раствора БП имеются характерные квазилинии при длинах волн 403.0 и 408.5 нм.
2. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ БЕНЗ(А)ПИРЕНА
Порядок проведения анализа. Количественное определение БП проводят с помощью флуоресцентно-спектрального метода. Оно может быть выполнено с использованием одной из двух модификаций: с помощью добавок и установкой прибора по фону, создаваемому люминесцирующими примесями, содержащимися в исследуемом экстракте, или с помощью внутреннего стандарта.
При определении бенз(а)пирена с помощью первой из указанных модификаций исследуемый раствор сравнивают не с раствором чистого бенз(а)пирена, а с таким же исследуемым, но сильно разбавленным раствором при добавлении в него определенного количества чистого бенз(а)пирена (массовый излишек), на свечение которого посторонние вещества влияют так же. как и в исследуемом растворе.
При определении БП с помощью внутреннего стандарта в бенз(а)пиреновую фракцию вводят чистый эталон, дающий хороший квазилинейчатый спектр в аналогичных условиях, причем в спектре этого вещества не должно быть линий, перекрывающихся с аналитическими линиями бенз(а)пирена. Таким веществом (стандартом) обычно служит 1,12-бензперилен. В спектре Э. В. Шпольского «-октанового раствора 1.12-бензперилена есть четкая линия при л. = 406,3 нм, которая располагается между соответствующими линиями бенз(а)пирена, а вблизи аналитических линий бензапирена в спектре 1,12-бензперилена заметные линии отсутствуют. Стандарт вводят для сравнения и в эталонные растворы БП. В исследуемом растворе измеряют отношение интенсивности линии БП и добавленного вещества. Пользуясь ранее определенным по «растворам-свидетелям» отношением интенсивности линий для известных концентраций БП и вещества-стандарта и зная количество добавленного стандарта, находят количество БП в бенз(а)пире-новой фракции, выделенной из продукта.
485
Полученные рсзулышы сводят в ыбоину:
Групповом jL'cop'i имен : конечных ii'.icinn
Co'iep.+.jHiie оен в onnpcHi мкг кг
Вареные
Поаукопчсные
Варено-копченые
Сырокопченые
Полученные данные анализируют, формулируют выводы и дают санитарно-гигиеническую опенку копченых мясных продуктов.
Лабораторная работа № 8
ОПРЕДЕЛЕНИЕ НИТРАТОВ И НИТРИТОВ
Цель работы. Освоить ионометрический и фотометрический методы определения нитратов и нитритов в мясе, вторичных продуктах убоя скота, мясных изделиях.
Задачи. Изучить ионометрический и фотометрические методы количественного определения нитратов и нитритов: установить содержание нитрат- и нитрит-ионов в мясе, вторичных продуктах убоя скота, мясных изделиях различного группового ассортимента; по результатам исследований дать санитарно-гигиеническую оценку мяса, вторичных продуктов убоя скота, мясных продуктов кулинарной готовности.
Объекты исследования. Мясо различных видов убойных животных и птицы; субпродукты I и II категорий, колбасные изделия, продукты из свинины, говядины, баранины, мяса птицы; консервы, при изготовлении которых применяют нитрит натрия.
Методические указания. Среди перечня токсических и вредных веществ, обнаруживаемых в сырье и продуктах, большое практическое значение имеет определение нитрат- и нитрит-ионов, источниками которых служат корма животных и собственно нитрит, добавляемый для имитации цвета при производстве мясных продуктов.
Проблема производства экологически чистых продуктов питания связана с реализацией инструментальных методов контроля вредных веществ, применяемых в условиях производства и имеющих достаточную точность и экспрессность. Существующие фотоколориметрические, хроматографические, спектрофотометрические и химические методы определения нитратов и нитритов не отвечают в полной мере требованиям и условиям производственных лабораторий. Методики имеют ряд недостатков: длительность, использование токсичных и дефицитных реактивов, дорогостоящей аппаратуры, определенный уровень требований к квалификации оператора для выполнения работ и т. д. По сравнению
486
с перечисленными ионоселективный метол определения нитрат-и нитрит-ионов имеет ряд достоинств, прежде всего связанных с малой продолжительностью, точностью и простотой определения, а также компактностью приборов.
В зависимости от уровня материальной базы в аналитической практике могут быть применены те или иные методы. Принципы и основная суть их изложены ниже.
Ионометрический метод определения нитрат- и нитрит-ионов предусматривает использование ионоселективного (нитратного) электрода типа ЭМ-Л 0.-01 путем индикации и измерения ЭДС электрода на ионометре И-130 (или нитратомере). Ионометр предназначен для измерения активности ионов водорода (pH), одновалентных и двухвалентных анионов и катионов (рХ), окислительно-восстановительных потенциалов в цифровой форме и в виде сигналов постоянного тока. Содержание нитрат-ионов можно фиксировать без предварительного измерения pH. На точность измерения не влияет присутствие фосфора, белков и жиров. Не рекомендуется проводить определение в объектах, содержащих хлорид натрия в массовых концентрациях более 3,5 %.
Измерение ЭДС и определение концентрации нитратов проводят в водной вытяжке, полученной из пробы продукта после предварительной экстракции при интенсивном перемешивании смеси с последующим фильтрованием.
Для исследования растворов с небольшими концентрациями нитрат- и нитрит-ионов используют метод добавок. В 50 см^ вытяжки измеряют ЭДС, затем в нее вводят нитрат калия так, чтобы массовая концентрация нитрата увеличилась до значений, соответствующих предварительно построенному калибровочному графику. По разнице значений рассчитывают искомую величину.
Для определения содержания нитритов их окисляют персульфатом аммония до нитратов. Разность между найденным суммарным содержанием нитрат-ионов и начальной концентрацией нитрат-ионов равна концентрации нитрит-ионов.
Фотометрические методы применяют в ряде модификаций, каждая из которых имеет практическое значение в анализе мясных продуктов и основана на той или иной химической реакции с образованием специфически окрашенных растворов. Например, применяется метод, основанный на реакции нитрита с N-1-нафтилэтилендиамином дигидрохлорида и сульфаниламидом в фильтрате с удаленным белком с последующим фотометри-рованием или визуальным определением интенсивности окраски. При фотоколориметрическом определении интенсивности окраски метод соответствует международному стандарту и применяется при разногласиях в оценке.
Используют также метод, основанный на реакции нитрита с реактивом Грисса (смесь растворов сульфаниловой кислоты и а-нафтиламина в уксусной кислоте) в фильтрате с удаленным
487
белком с последующим и змерением интенсивноеги окраски на ф от о к од ори м е тре.
Подготовка проб. С колбасных изделии снимают оболочку, с фаршированных колбас и языков в шпике — поверхностный слой шпика и оболочку, с окороков, лопаток, рулетов, корейки и гр\-линки — поверхностный слой шпика. Затем пробы дважды измельчают на мясорубке с отверстиями решетки диаметром от 3 до 4 мм. Продукты, полностью состоящие из шпика с промежуточными слоями мышечной ткани (ветчина в форме, прессованный бекон и аналогичные им), измельчают полностью.
Полученный фарш тщательно перемешивают, помещают в стеклянную или пластмассовую банку вместимостью от 200 до 400 см3, заполнив ее полностью, закрывают крышкой. Пробу хранят при (4 ± 2) °C до окончания анализа. Анализ проводят не позднее чем через 24 ч после отбора проб. Пробу сырых продуктов анализируют сразу после измельчения.
1. ОПРЕДЕЛЕНИЕ НИТРАТ- И НИТРИТ-ИОНОВ ИОНОМЕТРИЧЕСКИМ МЕТОДОМ
Материалы, реактивы и оборудование. Ионометр И-130 или нитратомер; ионоселективный электрод на NCh-ионы; электрод сравнения — хлорсеребряный; весы технические и аналитические; конические колбы вместимостью 250 см3; химические стаканы вместимостью 50 см3; мерный цилиндр вместимостью 100 см3; мерная колба вместимостью 1 дм3; пипетки вместимостью 5 и 10 см3; нитрат калия, ч. д. а.; водный раствор сульфата цинк£1 массовой долей 0,45 %; водный раствор сульфата калия концентрацией (1/2 K2SO4) 1 моль/дм3; водный раствор гидроксида натрия NaOH (0,1 моль/дм3); водный раствор персульфата аммония (NH4)S2O3 массовой долей 8 %.
С целью определения рабочего диапазона концентраций нит-рат-ионов предварительно строят калибровочный график.
Построение калибровочного графика. Навеску нитрата калия массой 10,1 г растворяют в дистиллированной воде в мерной колбе вместимостью 1 дм3, доводят содержимое колбы водой до метки. Получают раствор нитрата калия молярной концентрацией 10-1 моль/дм3 (pNO3 = 1). Методом последовательного разбавления из полученного раствора готовят серию стандартных растворов нитрата калия концентрацией 10“2, 10-3, 10'4 и Ю-3 моль/дм3 (pNO3 равны соответственно 2, 3, 4 и 5).
В пять химических стаканов отбирают по 50 см3 стандартных растворов нитрата калия, в каждый стакан добавляют по 1 см3 раствора сульфата калия. Погрузив электроды в стаканы, в каждом растворе регистрируют ЭДС элемента, составленного из нит-488
ратселективищ о и хлорсерсбряногн электродов. Перед началом измерений электроды промывают несколько раз дистиллированной водой. Измерения выполняют, переходя от разбавленных растворов к концентрированным. По полученным измерениям строят калибровочный график в координатах E = /'(pNO;). откладывая по оси ординат значения Е(мВ), по оси абсцисс — соответствующие значения pNO-.
Порядок проведения анализа. Для определения нитрат-ионов в коническую колбу вместимостью 250 см; помешают навеску продукта массой 10—20 г, взятую с точностью 0,01 г. добавляют 100 см3 дистиллированной воды (подогретой до 50—60 °C) и экстрагируют в течение 30 мин при непрерывном перемешивании. Содержимое колбы охлаждают и фильтруют через бумажный фильтр в коническую колбу. В полученном мутном растворе осаждают белки. Для этого добавляют к фильтрату 2,5 см3 раствора гидроксида натрия молярной концентрацией 0,1 моль/дм3 и 10см? раствора сульфата цинка массовой долей 0,45 %, нагревают 5 мин на водяной бане при температуре кипения, охлаждают колбу и полученный раствор фильтруют через бумажный фильтр. Фильтрат и промывные воды после промывания осадка белков на фильтре собирают в мерную колбу вместимостью 100 см3 и доводят объем до метки раствором сульфата калия молярной концентрацией 1 моль/дм3. В прозрачном фильтрате измеряют ЭДС, по величине которой на калибровочном графике находят начальное содержание нитрат-ионов в растворе.
Для определения нитрит-ионов их окисляют персульфатом аммония до нитратов. К 25 см3 фильтрата добавляют 0,5 см3 раствора персульфата аммония массовой долей 8 %, энергично перемешивают и через 5 мин измеряют ЭДС, по величине которой находят концентрацию нитрат-ионов после окисления нитрит-ионов, используя калибровочный график. Разность между найденным суммарным содержанием нитрат-ионов и начальной концентрацией нитрат-ионов равна концентрации нитрит-ионов, содержащихся в исследуемом растворе.
Содержание нитрат-ионов (мг %) в мясе и мясных продуктах находят по формуле
“ъЖюо,
т
(5.23)
где 62 — молярная масса эквивалента нитрат-ионов. г/.моль: с — концентрация нитрат-ионов до окисления, найденная по калибровочному графику, моль/дм3; 100 —объем фильтрата, см3; т — навеска измельченного мяса, г.
Содержание нитрит-ионов (мг %) в мясе и мясных продуктах
489
находят но формуле
глс 4b - мо.ъчрнзя мясся якниваясн Iа нитрит-ионов. i мол,: <. - - нонпсн iра -ция нитрат-ионов после окисления., наиленная по калиоровоянело' i рафик;. моль/дм': 100 - объем фильтрата, см'.
2. ОПРЕДЕЛЕНИЕ НИТРИТОВ ФОТОМЕТРИЧЕСКИМ МЕТОДОМ
2.1. МЕТОД ОПРЕДЕЛЕНИЯ НИТРИТОВ ПО РЕАКЦИИ С N-1-НАФТИЛЭТИЛЕНДИАМИНОМ ДИГИДРОХЛОРИДА
Материалы, реактивы и оборудование. Мясорубка: весы лабораторные; водяная баня; фотоэлектроколориметр или спектрофотометр; фильтровальная бумага: колбы мерные вместимостью 100, 200 и 1000 см3; пипетки мерные: калия гексацианоферрат; ацетат свинца; ледяная уксусная кислота; тетраборат натрия Na2B4O7 • 10Н?О; нитрит натрия; растворы для осаждения белков: реактив Карреза I, реактив Карреза II. насыщенный раствор буры: растворы для проведения цветной реакции: раствор I, раствор II. раствор III, основной, рабочий и стандартные растворы нитрита натрия для построения калибровочного графика.
Приготовление реактивов. Реактив Карреза I: 106 г гексацианоферрата калия растворяют в дистиллированной воде и доводят объем раствора до 1000 см3. Реактив хранят в склянке из темного стекла не более месяца.
Реактив Карреза II: 220 г ацетата цинка и 30 см3 ледяной уксусной кислоты растворяют в дистиллированной воде и доводят объем раствора до 1000 см3. Реактив хранят не более месяца.
Насыщенны й^ раствор б у р ы: 50 г тетрабората натрия растворяют в 1000 см3 теплой дистиллированной воды и охлаждают до (20 ± 2) °C.
Растворы для проведения цветной реакции.
Раствор I. Растворяют, подогревая на водяной бане. 2 г аминобензола сульфамида NH2C6H4SO0NH, в 800 см3 воды. Охлаждают, при необходимости фильтруют и добавляют, помешивая, 100 см3 концентрированной соляной кислоты (р20 1,19 г/см3), затем доливают водой до 1000 см3.
Раствор II. Растворяют в воде 0,25 г N-1-нафтилэтилендиамина дигидрохлорида C10H7NHCH2CH2NH2 • 2НС1, доливают водой до 250 см3. Полученный раствор хранят "в холодильнике, в хорошо укупоренной бутыли из коричневого стекла не более недели.
490
Раствор 1П. Разбавляют 4-45 с.х[-; кон лен 1 рированнои соляной кислоты (р0(1 1.19 I /см') полон ло 1СЮ1) см'.
Порядок проведения анализа. Образен для анализа помешают в коническую колбу вместимостью 300 см'. Для освобождения от белков добавляю!' иоследовагечьно 5 см' насыщенно! о раензора тетрабората натрия (буры) и 100см-' волы при температуре не ниже 70 °C.
Нагревают колбу на кипящей бане в течение 15 мин. периодически встряхивая.
Дают колбе с содержимым остыть до комнатной гемперату-ры и добавляют последовательно 2 см? реактива Карреза I и 2 см3 реактива Карреза И. тщательно перемешивая после каждого добавления.
Переливают содержимое в мерную колбу вместимостью 200 см\ доливают водой до метки и перемешивают. Содержимое колбы выдерживают в течение 30 мин при комнатной температуре.
Осторожно сливают верхний слой жидкости и фильтруют его через гофрированную фильтровальную бумагу, получая прозрачный раствор.
Пипеткой переносят часть фильтрата объемом не более 25 см3 в мерную колбу вместимостью 100 см3 и доливают водой до 60 см3.
Добавляют 10 см3 раствора I для цветной реакции, затем 6 см3 раствора III для цветной реакции, перемешивают и оставляют на 5 мин в темноте при комнатной температуре.
Добавляют 2 см3 раствора II для цветной реакции, перемешивают и оставляют на 3—10 мин в темноте при комнатной температуре. Затем разбавляют водой до метки.
Измеряют показатель спектрального поглощения раствора на фотоэлектроколориметре или спектрофотометре при длине волны около 538 нм.
Построение калибровочного графика. Для приготовления основного раствора нитрита натрия взвешивают навеску реактива, содержащую точно 1 г нитрита натрия, растворяют в воде, количественно переносят в мерную колбу вместимостью 500 см3, доводят водой до метки и перемешивают.
Массу навески (г) для химически чистого реактива с массовой долей основного вещества 99 % вычисляют по формуле
100-1
99
1.0101.
Для приготовления рабочего раст вора 5 см-' основного раствора переносят в мерную колбу вместимостью 1000 см '. доводят водой до метки и перемешивают. Из полученного рабочего раствора готовят серию стандартных растворов: 5, 10 и 20 см3 рабочего раствора пипеткой вносят в три мерные колбы вместимостью 100 см3, доводят водой до метки и перемешивают.
491
Полученные стандартные растворы содержат в 1 см’ со<лж-ы сгвенно 2.5: 5.0 и 10.0 mki нисрича натрия.
Готовят чри серии стандартных растворов, начиная каждый раз с приготовления основного раствора из новой навески нитрита натрия. Рабочий и стандартные растворы нитрита натрия нестойки. поэтому их готовят неиосрелствснно перед построением калибровочного графика.
Для построения калибровочного графика в четыре мерные колбы вместимостью 100 см-' пипеткой вносят: в первую колбу .тля приготовления контрольного раствора 10 см3 волы, а в остальные — по 10 см3 стандартных растворов, содержащих 2.5: 5.0 и 10,0 мкг нитрита натрия в 1 см3 раствора.
В каждую колбу добавляют по 50 см3 воды, ио 10 см3 раствора I и по 6 см3 раствора III для проведения цветной реакции. Растворы в колбах перемешивают и выдерживают в темном месте в течение 5 мин. Добавляют 2 см3 раствора II для проведения цветной реакции, перемешивают и выдерживают 3—10 мин в темном месте при (20 ±2) °C.
Растворы в колбах доводят водой до метки и перемешивают. Измеряют интенсивность красной окраски на спектрофотометре при Х = 538 нм или фотоэлектроколориметре с зеленым светофильтром в кювете с толщиной поглощающего свет слоя 1 см в отношении контрольного раствора.
По полученным средним данным из трех стандартных растворов на миллиметровой бумаге размером 25 х 25 см строят калибровочный график. На оси абсцисс откладывают концентрацию нитрита натрия (мкг в 1 см3 окрашенного раствора); на оси ординат — соответствующую оптическую плотность. Калибровочный график должен проходить через начало координат. Пример калибровочного графика приведен на рис. 5.7.
Если полученная оптическая плотность превышает максимальную оптическую плотность на калибровочном графике, то цветную реакцию проводят с меньшим объемом фильтрата.
Содержание нитритов (мг/кг) в пересчете на нитрит-ион вычисляют по формуле
сГ)С т Е,
Рис. 5.7. Пример калибровочного графика для определения содержания нитрита натрия
(5.25)
где с — концентрация нитрита натрия, найденная по калибровочному графику, мкг/с.м'; ф — общий объем экстракта. см3; ГГ — объем колориметрируемого раствора, см3; т — масса навески образца для анализа, г; И, — объем фильтрата, взятый для проведения цветной реакции, см’.
492
2.2. МЕТОД ОПРЕДЕЛЕНИЯ НИТРАТОВ ПО РЕАКЦИИ С N-1-НАФТИЛЭТИЛЕНДИАМИНОМ ДИГИДРОХЛОРИДА
Материалы, реактивы и оборудование. См. п. 1.2 настоящей лабораторной работы, а также кадмиевая колонка: раствор сульфата кадмия концентрацией 30 г/дм-\ раствор соляной кислоты молярной концентрацией 0.1 моль/лмП буферный аммиачный раствор pH 9.6—9,7: эталонный раствор нитрата калия: цинковые стержни длиной около 15 см и диаметром 5—7 мм.
Приготовление реактивов. Р а с г в о р с у л ь ф а г а кал -м и я к о н и е и т р а ц и с й 30 г/лм3. Навеску сульфата кадмия 3CdSO4 XH?O массой 37 г растворяют в воле в мерной колбе вместимостью 1000см? и доводят объем до метки дистиллированной водой.
Раствор с о л я н о й кислот ы м о л я р н о й к о Till е н т р а ц и е й 0,1 моль/дм3. Разбавляют дистиллированной водой 8 см3 концентрированной соляной кислоты (р2()= 1,19 г/см3) до объема 1000 см3.
Буферный^ аммиачный раствор pH 9,6—9,7. Разбавляют 20 см3 концентрированной соляной кислоты (р20 = = 1,19 г/см3) в 500 см3 воды. Перемешивают и добавляют 10 г этилендиаминтетраацетата натрия [CH2N(CH2COOH)CH?COONa]zx х 2Н?О и 55 см3 концентрированного аммиака (р20 ='0,88 г/см?). Доливают водой до 1000 см3, перемешивают и проверяют pH.
Нитрат калия, эталонный раствор. Растворяют в воле 1,465 г нитрата калия KNO3 и разбавляют до 100 см3 в мерной колбе. С помощью пипетки приливают 5 см3 полученного раствора в мерную колбу вместимостью 1000 см3 и разбавляют до метки. Полученный раствор содержит 73,25 мг нитрата калия на 1 см3.
Эталонный раствор готовят в день проведения анализа.
Порядок проведения анализа. Анализ состоит из подготовки кадмиевой колонки, проверки ее восстановительной способности, из подготовки пробы с целью освобождения от белков, перевода нитрата в нитрит и колориметрического измерения.
Подготовка кадмиевой колонки. Помещают 3—5 цинковых стержней в раствор сульфата кадмия, налитого в химический стакан (для одной кадмиевой колонки достаточно 1 дм3 раствора сульфата кадмия).
Удаляют губчатый металлический осадок кадмия с цинковых стержней каждые 1—2 ч, помешивая ими в растворе или потирая их друг о друга.
Через 6—8 ч сливают раствор и дважды промывают осадок 1 дм3 воды, чтобы кадмий был постоянно покрыт слоем жидкости.
Помещают осадок кадмия с 400 см3 раствора соляной кислоты в лабораторную мешалку и перемешивают Юс. Затем возвращают содержимое мешалки в химический стакан.
493
Рис. 5.8. Кадмиевая колонка:
1 ре up J- носик. .<•- с ick.ihhh.i.i ipeoKu: 4 kpu:i ihtii ' V I vb .Ml llUtM 15.11... о V.IC'O k:i ~ . I ck. i ,11II I.. . .'.e l- ч Д.
Несколько раз перемешивают осадок ка iOiM стеклянной палочкой и оставляют его на ночь пол слоем раствора соляной кислоты, после чего перемешивают еще раз. чюбы удали i в все пузырьки газа из кадмия.
Сливают раствор и промываю! кадмий два раза, используя каждый раз по 1 дм’ воды.
На дно кадмиевой колонки помешают стекловолокно (рис. 5.8).
Смывают кадмий в колонку водой до тех пор. пока слой кадмия не достигнет высоты 17 см. При заполнении колонки дают воде сте-
кать время от времени, следя, чтобы уровень волы не опускался ниже поверхности слоя кадмия. Удаляют пузырьки газа (например, с помощью швейной иглы). Вода должна вытекать со скоростью не более 3 см3/мин.
Промывают кадмиевую колонку последовательно 25 см3 раствора соляной кислоты, 50 см3 воды и 25 см3 аммиачного буферного раствора, разбавленного в соотношении 1 : 9. Не следует давать жидкости в воронке опускаться ниже верхней части капиллярной вводной трубки кадмиевой колонки.
Проверка восстановительной способности кадмиевой колонки. С помощью пипетки приливают 20 см3 эталонного раствора нитрата калия и одновременно 5 см3 аммиачного буферного раствора в резервуар на верху кадмиевой колонки. Собирают вытекающую жидкость в мерной колбе вместимостью 100 см3.
Когда резервуар почти полностью опустеет, обмывают его стенки два раза, используя каждый раз по 15 см3 воды.
После того как последняя порция воды стечет в колонку, полностью заполняют резервуар водой.
После того как наберется около 100 см3 стекшей жидкости, удаляют колбу из-под колонки и доливают водой до метки.
С помощью пипетки приливают 10 см3 элюата в мерную колбу вместимостью 100 см3 и далее проводят цветную реакцию с последующим фотометрическим измерением интенсивности окраски.
Для этого добавляют 10 см3 раствора I для цветной реакции, затем 6 см3 раствора III, перемешивают и оставляют на 5 мин в темноте при комнатной температуре.
Добавляют 2 см3 раствора II, перемешивают и оставляют на 3— 10 мин в темноте при комнатной температуре. Затем разбавляют водой до метки.
494
Измеряют показатель спектрального поглощения раствора на фотоэлектроколориметре или спектрофотометре с оптической длиной камеры 1 см и при длине волны около 538 нм.
Если концентрация нитрита в элюате по калибровочному графику составляет менее 0.9 мкг нитрита натрия на 1 см3 (т. е. 90 сс от теоретической величины), кадмиевую колонку нельзя использовать для анализа. Кадмий необходимо перенести в химический стакан и залить его на ночь раствором соляной кислоты молярной концентрацией 0,05 моль/дм3, промыть его водой и повторить операции по подготовке колонки.
Освобождение пробы от белков. Проводят в соответствии с методикой, приведенной в п. 2.1 настоящей работы.
Пере в о д н и т р а т а в н и т р и т. С помощью пипетки приливают в резервуар на верху колонки 20 см3 освобожденного от белков фильтрата и одновременно 5 см3 аммиачного буферного раствора.
Собирают жидкость, стекающую из колонки, в мерную колбу вместимостью 100 см3.
Когда резервуар почти полностью опустеет, обмывают его стенки два раза, используя каждый раз по 15 см3 воды. После того как последняя порция воды стечет в колонку, полностью заполняют резервуар водой. После того как наберется около 100 см3 стекшей жидкости, удаляют колбу из-под колонки и доливают водой до метки.
Колориметрическое измерение. С помощью пипетки переносят произвольный объем элюата, но не более 25 см3 в мерную колбу вместимостью 100 см3 и доливают водой до 60 см3.
Добавляют 10 см3 раствора I для цветной реакции, затем 6 см3 раствора III для цветной реакции, перемешивают и оставляют на 5 мин в темноте при комнатной температуре.
Добавляют 2 см3 раствора II для цветной реакции, перемешивают и оставляют на 3—10 мин в темноте при комнатной температуре. Затем разбавляют водой до метки.
Измеряют показатель спектрального поглощения раствора на фотоэлектроколориметре или спектрофотометре с оптической длиной камеры 1 см и при длине волны около 538 нм.
Далее необходимо воспользоваться калибровочным графиком и рекомендациями раздела «Порядок проведения анализа» п. 2.1 настоящей лабораторной работы.
Содержание нитратов в твердых продуктах (мг/кг) (в пересчете на нитрат-ион) вычисляют по формуле
рЬЬИ, , «’К,к
(5.26)
где с — концентрация нитритов по калибровочному графику, мкг/см3; И,—общий объем экстракта, см3; й, — объем элюата с колонки, см3; — объем колориметрируемого раствора, см/ т — масса навески образца для анализа, г; И4 — объем фильтрата, взятый для восстановления на колонку, см3; I/ — объем элюата Для цветной реакции, см3; Д'— концентрация нитритов, мг/кг.
495
2.3. МЕТОД ОПРЕДЕЛЕНИЯ НИТРИТА НАТРИЯ ПО РЕАКЦИИ ГРИССА
Материалы, реактивы и оборудование. Мясорубка: весы набора [орные: баня водяная: колбы .мерные вмссчимостыо 100. 200 см': стакан химический: конические колбы вместимостью 100см-': .воронки стеклянные: фильтры беззольные бумажные: фотоэлек-гроколориметр или спектрофотометр: раствор гидроксила натрия молярной концентрацией 0.1 моль/дм-': раствор сульфата цинка массовой долей 0,45 Sc; водный раст вор аммиака молярной концентрацией 3,0 моль/дм ': раствор соляной кислоты молярной концентрацией 0,1 моль/дм': реактив Грисса: раствор сравнения, содержащий 1 мкг нитрита натрия в 1 см3: рабочий раствор нитрита натрия: кислота сульфаниловая безводная, ч.д. а. (или х. ч.). ос-нафтиламин, х. ч.
Приготовление реактивов. Р е а к г и в Грис с а. Готовят растворы 1 и 2 и смешивают их равные объемы. В случае появления при смешивании растворов розовой окраски добавляют цинковую пыль, взбалтывают и фильтруют. Готовят непосредственно перед употреблением.
Раствор 1. 0,5г сульфаниловой кислоты растворяют в 150 см3 раствора уксусной кислоты молярной концентрацией 2 моль/дм3.
Раствор 2. 0,2г а-нафтиламина кипятят с 20см3 воты. раствор фильтруют и прибавляют к фильтрату 180 см3 раствора уксусной кислоты молярной концентрацией 2 моль/дм3. Раствор хранят в темной склянке.
Раствор сравнения. Готовят с использованием стандартного и рабочего растворов нитрита натрия.
Для приготовления стандартного раствора нитрита натрия взвешивают навеску нитрита натрия, содержащую 1 г основного вещества. Массу навески (г) для химически чистого реактива с массовой долей основного вещества 99 % вычисляют по формуле
Навеску переносят в мерную колбу вместимостью 1000 см3 и доводят дистиллированной водой до метки. Для приготовления рабочего раствора нитрита натрия 10 см3 основного раствора переносят в мерную колбу вместимостью 500 см3 и доводят водой до метки. Рабочий раствор нитрита натрия используется для построения калибровочного графика.
Для приготовления раствора сравнения 5 см3 рабочего раствора переносят в мерную колбу вместимостью 100 см3 и доводят водой до метки. 1 см3 раствора сравнения содержит 0,001 мг (или 1 мкг) нитрита натрия.
496
Порядок проведения анализа. Взвешиваю! 20 г подготовленной к анализу пробы с точностью не более 0.01 г. помещают в химический стакан, заливают 35—40 см3 дистиллированной воды, нагретой до (55 ± 2)’С и настаивают, периодически перемешивая, в течение 10 мин. Затем вытяжку фильтруют через фильтр в мерную колбу вместимостью 200 см3. Навеску несколько раз промывают и переносят на фильтр, вновь промывают водой, затем раствор охлаждают и доводят водой до метки.
Для приготовления вытяжки сырокопченых продуктов из свинины, баранины, говядины и сырокопченых колбас навеску массой 20 г заливают 200 см3 предварительно нагретой до (55 ± 2) СС дистиллированной воды и настаивают, периодически помешивая, в течение 30 мин. Затем вытяжку фильтруют через фильтр, не перенося осадка на фильтр.
20 см3 вытяжки помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 раствора гидроксида натрия молярной концентрацией 0,1 моль/дм3 и 40 см3 раствора сульфата цинка массовой долей 0.45 % для осаждения белков. Смесь в колбе нагревают в течение 7 мин на водяной бане при температуре кипения, после чего охлаждают, доводят до метки водой, перемешивают и фильтруют через обеззоленный бумажный фильтр.
Параллельно проводят контрольный анализ на реактивы, помещая в мерную колбу вместимостью 100 см3 вместо 20 см3 вытяжки 20 см3 дистиллированной воды.
В коническую колбу вместимостью 100 см3 наливают 5 см3 про; зрачного фильтрата, полученного после осаждения белков, 1 см3 раствора аммиака, 2 см3 раствора соляной кислоты, 2 см3 дистиллированной воды и, для усиления окраски, 5 см3 раствора сравнения, содержащего 1 мкг нитрита натрия в 1 см2 Затем в колбу приливают 15 см3 реактива Грисса и через 15 мин измеряют интенсивность окраски на спектрофотометре при длине волны 538 нм или на фотоэлектроколориметре с зеленым светофильтром (№ 6) в кювете с толщиной поглощающего свет слоя 2 см в отношении раствора сравнения.
Массовую долю нитрита (%) вычисляют по формуле
.. _ М] -200-100 100-30
Х = -ДПУУЮ <5-27)
где Л/] — массовая концентрация нитрита натрия, найденная по калибровочному графику, мкг/см3: т — масса навески продукта, г; 106 — коэффициент перевода в граммы.
Построение калибровочного графика. В 6 мерных колб вместимостью 100 см3 каждая пипеткой вносят следующие объемы рабочего раствора: 0; 1,0; 2,0; 4,0; 6,0; 8,0 см3. В первую колбу рабочий раствор не вносят, используя ее как контрольную.
497
В каждую колбу добавляют 5 см1 раствора аммиака. 10 см' раствора соляной кислоты, доводят водой до метки и перемешивают. В конические колбы вместимостью 100см3 пинеткой переносят по 15 см3 приготовленных растворов. 15 см3 реактива Грисса и после 15 мин выдержки при комнатной температуре измеряют интенсивность розовой окраски на спектрофотометре при л = 538 нм или фотоэлектроколориметре с зеленым светофильтром (№ 6) в кювете с толщиной поглощающего свет слоя 2 см в отношении раствора сравнения.
Готовят три серии стандартных растворов, начиная каждый раз с приготовления основного раствора из новой навески нитрита натрия.
По полученным средним данным из трех стандартных растворов на миллиметровой бумаге размером 25 х 25 см строят калибровочный график. На оси абсцисс откладывают массовую концентрацию нитрита натрия (мкг/см3), а на оси ординат — соответствующие значения оптической плотности. Калибровочный график должен проходить через начало координат.
Полученные результаты анализа сводят в таблицу рекомендуемой формы:
Образец
Используемый метод
Содержание, мг/кг
нитритов
нитратов
Полученные результаты студенты сравнивают с предельно допустимыми значениями для данного вида продуктов, делают выводы и формулируют общее заключение по работе.
Лабораторная работа № 9
ОПРЕДЕЛЕНИЕ ТОКСИЧНЫХ ЭЛЕМЕНТОВ
Цель работы. Освоить методы подготовки проб, качественного и количественного определения токсичных элементов в мясе, вторичных продуктах убоя скота, мясных изделиях.
Задачи. Подготовить пробы к определению токсичных элементов способами сухой, мокрой минерализации и кислотной экстракции; определить массовую долю тяжелых металлов и мышьяка в мясных продуктах колориметрическими, нефелометрическим, атомно-абсорбционным и полярографическим методами.
Объекты исследования. Образцы мышечной ткани различных видов убойных животных и птицы; субпродукты I и II категорий, колбасные изделия, продукты из свинины, говядины, баранины, мяса птицы, мясные консервы и пищевые животные жиры.
498
Методические указания. Лабораторная работа но определению токсикантов является комплексной и включает в себя несколько этапов, каждый из которых может использоваться как самостоятельно, так и в комплексе. Тяжелые металлы и мышьяк в биологических объектах прочно связаны с белками. Для их определения необходимо разрушить эти комплексы (альбуминаты). Цель достигается сухой, мокрой минерализацией или кислотной экстракцией (в случае анализа жировых продуктов) исследуемого материала.
Способ сухой минерализации основан на полном разложении органических веществ путем сжигания образца сырья или продукта в электропечи при контролируемом температурном режиме. Этот способ рекомендуется использовать при подготовке проб всех видов мясного сырья и продуктов, кроме жиров, для определения содержания свинца, кадмия, меди, цинка методом атомноабсорбционной спектрометрии.
Способ мокрой (кислотной) минерализации основан на полном разрушении органических веществ пробы продукта при нагревании с серной и азотной концентрированными кислотами с добавлением хлорной кислоты или пероксида водорода и может быть использован для исследования всех видов мясного сырья и продуктов, кроме животных жиров.
Способ мокрой минерализации применяется при подготовке проб для качественных реакций обнаружения свинца и цинка, качественного и количественного определений мышьяка, определения олова в мясном сырье и продуктах, за исключением жировых, а также для определения содержания меди во всех видах продуктов, включая жиры.
Каждый из способов имеет свои преимущества и недостатки.
При мокром озолении все металлы без потерь переходят в раствор, однако для пищевых продуктов, содержащих большое количество органических веществ и имеющих незначительную зольность, приходится брать достаточно большую навеску (10— 20) г. Расход кислоты для обугливания оказывается достаточным, чтобы внести в анализируемый раствор дополнительное количество минеральных элементов, а это влияет на количественные результаты.
Поэтому для многих пищевых продуктов лучше применять осторожное сухое озоление. Хорошие результаты дает применение для предварительного обугливания инфракрасной лампы мощностью 250 или 500 Вт. Озоление проводят при температуре 450—550 °C до постоянной массы. Остаток растворяют в 1—2 см3 соляной кислоты, добавляют 3—5 см3 дважды дистиллированной воды и нагревают до растворения. Раствор переносят в колбу вместимостью 50 см3 и доводят водой до метки. При температуре озоления не выше 550 °C сохраняются следующие элементы: натрий, калий, кальций, магний, железо, цинк, медь,
499
кобальт, марганец и олово. В незначительных количествах происходят потери таких элементов, как свинец, серебро и мышьяк.
При неполном обугливании мель и железо адсорбируются углеродом и полностью не экстрагируются. Если зола имеет кислую реакцию и продукт богат фосфорными соединениями. то осадок растворяют в соляной кислоте, отфильтровывают и остаток дополнительно обугливают. Щелочная зола может взаимодействовать с керамикой тигля и образовывать нерастворимые кремнистые соединения. Поэтому для озоления пищевых продуктов лучше использовать платиновые или кварцевые тигли. При исследовании минерального состава животных жиров сухое озоление приводит к значительным потерям меди и железа.
Способ кислотной экстракции (неполной минерализации) основан на экстракции токсичных элементов из пробы продукта при кипячении с разбавленными растворами соляной или азотной! кислоты. Способ может быть использован при подготовке проб для определения содержания в животных жирах меди колориметрическим методом, а также других токсичных элементов методами атомно-абсорбционной спектроскопии и вольтамперометрии.
Рекомендации по выбору конкретных методов определения токсичных элементов в мясе и мясных продуктах и соответствующих способов подготовки проб приведены в табл. 5.7.
Таблица 5.7
Методы определения токсичных элементов в мясе и мясных продуктах
Токсичный элемент Метод определения Способ минерализации проб
Свинец Колориметрический (качественные реакции) Нефелометрический Атомно-абсорбционной спектрометрии Полярографический Мокрая Сухая » »
Мышьяк Колориметрический Сухая или мокрая
Кадмий Полярографический Сухая
Ртуть Колориметрический Деструкция закрытым или открытым способом
Медь Колориметрический (качественные реакции) Атомно-абсорбционной спектрометрии Полярографический Колориметрический (количественное определение) Мокрая Сухая » Сухая или мокрая
Цинк Колориметрический (качественные реакции) Полярографический Мокрая Сухая
500
При высоком содержании гоксичных металлов и мышьяка в мясе и продуктах убоя животных их присутствие можно обнаружить при проведении с минерализатом несложных качественных цветных или других специфических реакций.
Методы качественного обнаружения свинца в мпнерализатс основаны на его растворении в ацетате аммония с последующей постановкой цветной реакции с дитизоном или микрокристалло-скопических реакций.
Метод выявления меди основан на экстрагировании се из минерализата хлороформом в виде диэтилдитиокарбамината меди, последующего вытеснения из этого соединения в водный слой ртутью, где она и обнаруживается соответствующими цветными реакциями.
Метод обнаружения цинка основан на экстракции цинка из минерализата хлороформом, связывании ионов кадмия и меди (мешающих обнаружению цинка) тиосульфатом натрия или мочевиной, образовании окрашенного соединения цинка с дитизоном —дитизоната цинка.
Метод качественного и количественного определения мышьяка (по Зангер—Влеку) основан на восстановлении мышьяка до мышьяковистого водорода AsH3, который при взаимодействии с хлоридом HgCl7 или бромидом ртути HgBiy образует окрашенное в желтый или ж’елтовато-коричневый цвет соединение.
Нефелометрический оптический метод исследования дисперсных систем растворов связан с рассеянием света частицами дисперсной фазы, которое зависит от длины волны излучения, размера и формы рассеивающих частиц и иногда от их расположения в пространстве. Для осуществления нефелометрического метода анализа ионы определяемого элемента или определяемое вещество переводят в малорастворимое соединение, способное образовывать относительно устойчивую дисперсную систему в начальный период формирования осадка.
Количественное определение свинца нефелометрическим методом основано на получении сульфата свинца, растворении его в ацетате аммония и последующем взаимодействии с хроматом калия, сопровождающемся образованием малорастворимого в воде хромата свинца РЬСгО4.
Атомно-абсорбционная спектрометрия основана на измерении поглощения электромагнитного излучения атомным «паром» анализируемого вещества. Разность интенсивности излучения до и после прохождения через анализируемый образец измеряют фотометрически. Ослабление излучения с интенсивностью /0 до интенсивности I для выходящего светового потока происходит по экспоненциальной зависимости, которая идентична закону Бугера—Ламберта—Бера:
/= /0- (5.28)
где К~ коэффициент поглощения: / — толщина поглощающего слоя: с — концентрация поглощающих атомов.
501
После .км арнфмирования этого выражения получают зависимое ГЬ
/1 = lg IJl= к/с. (5.29)
i.ic Л — абсорбция iioiлощаюшс!о слоя плазмы; А -- атомный коэффициент абсорбции.
При постоянной голшине поглощающего слоя калибровочный график, построенный в координатах /1 — с. представляет собой прямую, проходящую через начало координат.
Определение массовой доли токсичных элементов (свинца, кадмия, меди, цинка) методом атомно-абсорбционной спектроскопии включает подготовку проб способами сухой, мокрой минерализации или кислотной экстракции и непосредственное проведение количественного определения на спектрофотометре.
Раствор минерализата испытуемой пробы распыляют в воз-душно-апетиленовом пламени. Металлы, находящиеся в растворе минерализата, попадая в пламя, переходят в атомное состояние и поглощают излучения. Адсорбция электромагнитного излучения атомами с анализируемого элемента пропорциональна концентрации металла в испытуемой пробе.
Полученный раствор золы можно анализировать сразу или после дополнительной подготовки, которая необходима в следующих случаях:
если концентрация элемента в растворе значительно превышает верхний предел интервала рабочих концентраций. Исходный раствор разбавляют бланковым раствором до необходимого уровня концентрации (бланковым называют раствор, который применяют для приготовления анализируемых растворов проб);
если концентрация элемента в исходном растворе ниже предела его обнаружения. В этом случае концентрирование выполняют методом экстракции.
При эксплуатации атомно-абсорбционных спектрофотометров в лабораториях необходимо соблюдать следующие правила техники безопасности:
1. К работе допускаются лица, знакомые с конструкцией и описанием прибора, правилами его эксплуатации, прошедшие инструктаж по технике безопасности и расписавшиеся в журнале.
2. Студенты, прошедшие инструктаж, допускаются к работе только в присутствии преподавателя или ответственного за лабораторию.
3. Все работы следует проводить только при включенной вытяжке.
502
4. Необходимо тщательно следить за исправным состоянием вентилей, регулирующих доступ газа и воздуха.
5. Во время работы горелка должна быть закрыта специальным ультрафиолетовым защитным стеклом.
6. Профилактический осмотр, чистку прибора, замену ламп и пр. можно проводить только после отключения установки от сети.
Полярографический метод определения токсичных элементов основан на предварительной сухой минерализации пробы продукта, получении раствора минерализата с добавлением фонового электролита, проведении анализа в электролизере, получении полярографических кривых и измерении высоты пиков. Для количественного определения металлов используют метод добавок.
Аналитические возможности метода вольтамперометрического титрования очень широки. Электролитическая ячейка (электролизер), используемая в вольтамперометрии, представляет собой сосуд вместимостью 1—50 см-’ с погруженным в него рабочим электродом и электродом сравнения. Электролитическим сосудом может быть обычный химический стакан или сосуд специальной конструкции, если он предназначен для работы без контакта с атмосферой. Систему электродов для вольтамперометрического титрования выбирают таким образом, чтобы плотность тока на этих электродах существенно различалась: на рабочем электроде плотность тока должна быть велика, на электроде сравнения — ничтожно мала. В этом случае поляризоваться будет только рабочий электрод и только на нем возможны электрохимические процессы восстановления или окисления ионов из раствора. Рабочий электрод, как правило, имеет очень малую поверхность по сравнению с поверхностью электрода сравнения — это микроэлектрод, который может быть изготовлен из твердого материала (платины, серебра, золота, графита специальной обработки и др.) или в виде ртутной капли, вытекающей из капилляра. Конструктивно твердые электроды более удобны и безопасны, чем ртутные, но область их использования ограничена.
Ртутный капающий электрод широко используют в качестве рабочего микроэлектрода при полярографическом анализе веществ, восстанавливающихся в области потенциалов + 0,2 — — 1,9 В относительно насыщенного каломельного электрода. Из стеклянного резервуара по гибкому шлангу ртуть поступает в стеклянный капилляр, из которого со скоростью, регулируемой высотой ртутного столба и диаметром капилляра (1 — 10 капель в 1 с), подается в анализируемый раствор. Постоянно обновляющаяся поверхность ртутной капли, а также возможность электрохимически восстанавливать на таком электроде ионы
503
металлов, расположенных в ряду напряжений левее водорода, высокая точность и воспроизводимость результатов измерении делают ртутный капающий электрод одним из наиболее широко используемых для анализа токсичных элементов в мясе и мясных продуктах.
Минимальная концентрация токсичных элементов в растворе, определяемая полярографическим методом, составляет (мкг/см3): меди 0,1; свинца 0,06; кадмия 0.02; цинка 0,2.
Однако работа с таким электродом требует тщательного соблюдения всех мер, предусмотренных правилами техники безопасности при работе с ртутью: электролизер с ртутным капающим электродом должен быть установлен в эмалированную или пластмассовую кювету, во время работы ртуть должна находиться в плотно закрытых сосудах или под слоем водного раствора. Отработанную ртуть необходимо хранить в толстостенных банках.
Практическое значение имеют и различные колориметрические методы определения токсичных элементов.
Например, колориметрический метод определения ртути основан на деструкции анализируемой пробы мяса или мясных продуктов смесью азотной и серной кислот, осаждении ртути иодидом меди и последующем колориметрическом определении в виде тетраиодомеркурата меди путем визуального сравнения со стандартной шкалой. Минимальная определяемая масса ртути составляет 0,15 мкг в колориметрируемом объеме пробы.
Фотоколориметрические методы определения меди и мышьяка в мясе и мясных продуктах основаны на минерализации пробы и последующем измерении интенсивности окраски раствора соответствующего комплексного соединения: меди — с диэтилдитиокарбаматом натрия (желтого цвета), мышьяка — с диэтилдитиокарбаматом серебра в хлороформе.
Минимальная масса токсичных элементов в колориметрируемом объекте, определяемая фотоколориметрическими методами, составляет: меди 5 мкг, мышьяка 2,5 мкг при использовании поглощающего раствора с моноэтаноламином и 5 мкг — с уротропином.
Подготовка проб. Состоит в тщательном измельчении навески исследуемого сырья или продукта массой 100—250 г и последующей минерализации.
Рекомендуемая масса навески для качественного обнаружения токсичных элементов 100 г.
Рекомендуемые массы навесок образцов в зависимости от определяемого токсичного элемента и метода его количественного определения приведены в табл. 5.8.
504
Рекомендуемые массы навесок (г) для определения токсичных элементов различными методами
>
505
1. РЕАКЦИИ КАЧЕСТВЕННОГО ОБНАРУЖЕНИЯ ТОКСИЧНЫХ ЭЛЕМЕНТОВ
1.1. МИНЕРАЛИЗАЦИЯ ПРОБ
Материалы, реактивы и оборудование. Колба Кьельдаля вместимостью 500 см': лабораторный штатив; электроплитка или газовая горелка; ступка; фарфоровая луночка; пинцет, ножницы; делительная воронка; стакан вместимостью 200 с\ф; мерная колба вместимостью 200 см3; мерный цилиндр вместимостью 100 см3; пипетка; бумажный фильтр; концентрированные серная и азотная кислоты; разбавленная азотная кислота (1:1); формалин; раствор хлорной кислоты массовой долей 42 %; раствор дифениламина в серной кислоте (0,5 г дифениламина растворяют в смеси, состоящей из 100 частей х. ч. концентрированной серной кислоты и 20 частей дистиллированной воды).
Порядок проведения анализа. Навеску исследуемого материала массой 100 г тщательно измельчают в ступке, помещают в колбу Кьельдаля и приливают по 25 см3 дистиллированной воды, концентрированных азотной и серной кислот (можно заранее приготовить смесь из этих ингредиентов и прилить сразу 75 см3). Для ускорения минерализации добавляют 25 см3 раствора хлорной кислоты массовой долей 42 %. При отсутствии хлорной кислоты минерализацию можно проводить и без нее. Колбу закрепляют в лабораторном штативе в вертикальном положении, над ней фиксируют делительную воронку с разбавленной азотной кислотой (1 : 1). После прекращения вспенивания колбу нагревают на газовой горелке или плитке (при постепенном усилении нагревания) до начала потемнения жидкости. Затем при постоянном нагревании по каплям добавляют разбавленную азотную кислоту, пока содержимое колбы не станет бесцветным и не будет изменяться по цвету при добавлении HNO3. После этого продолжают нагревание без добавления HNO3 до появления белых паров SO2. Далее колбу охлаждают и ее содержимое переносят в стакан вместимостью 200 см3, несколько раз сполоснув водой.
Наличие оксидов азота в минерализате, мешающих дальнейшему исследованию, определяют при помощи раствора дифениламина в серной кислоте, В фарфоровой луночке каплю минерализата смешивают с раствором дифениламина. В присутствии окислов азота появляется синее окрашивание, которое исчезает при добавлении формалина. К нагретому до кипения минерализату по каплям приливают формалин до прекращения выделения пузырьков газа. Окончание денитрации определяют пробой с дифениламином. Остатки формалина удаляют нагреванием жидкости в течение 5—10 мин. Остывшую жидкость разбавляют водой до 180— 190 см3 и оставляют на 18—20 ч при комнатной температуре. Свинец и барий в виде сульфатов выпадают в осадок. Его отфильтро
506
вывают на бумажном фильтре. Филыраг в мерной ко im доводят водой до 200 см? и исслсдз !<м на созержшшс юкииных • каментов (медь, цинк, мышьяк и др.).
При небольшой массе исследуемого Marepnaij. можно брать 25 или 50г. соответственно чхжныиив обьем реакшвов. используемых для минерализации
1.2. Qi 1РЕДЕГ1ЕНИЕ СВИНЦА
Материалы, реактивы и оборудование. Бумажный фильтр с осадком, полученным после фильтрования минерализата: микроскоп: спиртовка; колба: пробирки: предметные стекла; глазные пипетки: раствор ancnaia аммония (насыщенный раствор ацетата аммония разбавляю! равным обьемом воды и на 1 дм3 раствора добавляют 30 см' ледяной уксусной кислоты); раствор серной кислоты молярной концентрацией 0,81 моль/дм3; раствор дитизона в хлороформе массовой долей 0.01 %; раствор уксусной кислоты массовой долей 30 %; хлорил пезия: иолид калия; нитрит калия: раствор ацетата меди массовой долей 1 %.
Порядок проведения анализа. Бумажный фильтр с осадком, полученным после фильтрования минерализата. промывают 15—20 см3 раствора Н >SO4 молярной концентрацией 0.1 моль/дм3, а затем 10 см3 воды (жидкость не собирают). Осадок на фильтре обрабатывают кипящим раствором ацетата аммония (от 0,5 до 10 см3 в зависимости от массы осадка). Сульфат свинца растворяется и переходит в фильтрат, который собирают в пробирку.
Реакция с дитизоном. В пробирке смешивают встряхиванием 1—2 см3 фильтрата с равным объемом раствора дитизона в хлороформе массовой долей 0,01 %. При наличии свинца (pH 7—10) слой органического растворителя окрашивается в пурпурно-красный цвет.
Микрокристаллические реакции. По 0,5 см3 фильтрата распределяют на двух предметных стеклах и упаривают на пламени спиртовки.
1. К остатку добавляют 2—3 капли раствора уксусной кислоты массовой долей 30 %. С одной стороны капли вносят 2—3 кристаллика хлорида цезия, а с другой — несколько кристаллов иодида калия. При наличии свинца через несколько минут под малым увеличением микроскопа обнаруживают желто-зеленые игольчатые кристаллы, часто собранные в пучки и сфероиды.
2. К остатку прибавляют 1—2 капли раствора ацетата меди массовой долей 1 %, упаривают досуха и наносят 2—3 капли раствора уксусной кислоты массовой долей 30 %. На край капли помещают несколько кристаллов нитрита калия. При наличии свинца через несколько минут под микроскопом выявляются черные или коричневые кристаллы в виде кубов.
507
1.3. ОПРЕДЕЛЕНИЕ МЕДИ
Материалы, реактивы и оборудование. Фильтрат минерализата (после отделения сульфатов цинка и бария): лабораторный штатив; делительная воронка; колба; пробирки; раствор гидроксида аммония NH4OH массовой долей 25 %, тетрарода-номеркурат аммония (5 г хлорида ртути и 5 г роданида аммония растворяют в 6 см3 воды); спиртовой раствор 2,4-динитрофенола массовой долей I % (индикатор); раствор ферроцианида калия массовой долей 5 %; раствор хлорида кадмия массовой долей 2 %; раствор соляной кислоты молярной! концентрацией 6 моль/дм3; раствор хлорида ртути HgCI2 массовой долей I %; хлороформный раствор диэтилдитиокар'бамата свинца РЬ(ДДТК)2; хлороформ, сульфат цинка.
Приготовление реактивов. X л о р о ф о р м н ы й раствор д и э т и л д и т и о к а р б а м а т а св и н ц а РЬ(ДДТК)2. 0,5 г ацетата свинца растворяют в воде, добавляют 25 см3 раствора нитрата калия массовой долей 10% и 0,5 г растворенного в воде Ха(ДДТК). Образовавшийся белый осадок [РЬ(ДДТК)Д экстрагируют хлороформом; водный раствор, свободный от белого осадка [РЬ(ДДТК)2], отбрасывают, а хлороформный слой фильтруют, после чего к нему добавляют хлороформ, доводя объем до 250 см3.
Порядок проведения анализа. В делительную воронку помещают 10 см3 минерализата, прибавляют несколько капель спиртового раствора 2,4-динитрофенола массовой долей 1 % и по каплям раствор гидроксида аммония массовой долей 25 % до появления желтого окрашивания. Приливают 5 см3 хлороформного раствора диэтилдитиокарбамата свинца и энергично встряхивают. Хлороформный слой окрашивается от желтого до коричневого цвета (возможно, за счет естественного содержания меди в мясе и органах). Хлороформный экстракт промывают раствором соляной кислоты молярной концентрацией 6 моль/дм3, а затем дистиллированной водой. Добавляют к нему по каплям (периодически встряхивая) раствор хлорида ртути (0,5—1 см3) массовой долей 1 % до обесцвечивания. Приливают 0,5—1 см3 воды, встряхивают и отделяют водный слой. Делят его на две равные части, переносят в пробирки и проводят две реакции.
В первую пробирку добавляют 0,2 г сульфата цинка и несколько капель раствора тетрароданомеркурата аммония. При наличии меди осадок окрашивается в розово-лиловый цвет.
Во вторую пробирку вносят 10 капель раствора хлорида кадмия массовой долей 2 % и 1—2 капли раствора ферроцианида калия массовой долей 5 %. При наличии меди осадок окрашивается в лиловый цвет.
508
1.4. ОПРЕДЕЛЕНИЕ ЦИНКА
Материалы, реактивы и оборудование. Универсальная индикаторная бумага: насыщенный раствор тиомочевины или насыщенный раствор тиосульфата натрия: ацетатный буфер (смесь 4.2 г ацетата натрия и 3.2 г уксусной кислоты, доведенная до ! дм3 дистиллированной водой): раствор дитизона в хлороформе массовой долей 0.01 %; хлороформ: раствор гидроксида калия (или натрия) массовой долей 10 %.
Порядок проведения анализа. В пробирку вносят 0,5 см3 минерализата, прибавляют 0,25 см3 насыщенного раствора тиосульфата натрия (или тиомочевины) для связывания ионов кадмия, по каплям добавляют раствор гидроксида калия массовой долей 10 % до pH смеси 5,0—5,5 (pH устанавливают по универсальной индикаторной бумаге). Добавляют 1 см3 ацетатного буфера, 2—3 капли раствора дитизона в хлороформе массовой долей 0,01 % и 1 см3 хлороформа. Смесь энергично встряхивают. При наличии цинка в смеси зеленый цвет хлороформного слоя переходит в розовый или красно-фиолетовый (в зависимости от его количества).
2. КАЧЕСТВЕННОЕ И КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ МЫШЬЯКА ПО ЗАНГЕР—БЛЕКУ
Материалы, реактивы и оборудование. Прибор Зангер—Блека; 6 колб к прибору вместимостью 50 см3; раствор хлорида олова массовой долей 10% в серной кислоте молярной концентрацией 7,5 моль/дм3 или соляной кислоте молярной концентрацией 6 моль/дм3 (при растворении подогревают); раствор серной кислоты массовой долей 20 %; купированный мелкогранулированный цинк [гранулы цинка на несколько секунд (до потемнения) погружают в раствор сульфата меди массовой долей 0,05 %, а затем промывают дистиллированной водой]; бромо- или хлорортутная бумага (полоски фильтровальной бумаги смачивают спиртовым раствором хлорида или бромида ртути массовой долей 5 % и высушивают при комнатной температуре в вытяжном шкафу); вата, смоченная раствором ацетата свинца массовой долей 5 % и высушенная на воздухе; стандартный раствор мышьяка.
Приготовление стандартного раствора мышьяка. 0,1321 г перекристаллизованного мышьяковистого ангидрида растворяют в как можно меньшем объеме раствора гидроксида натрия массовой долей 10 %. Полученный раствор переносят в мерную колбу вместимостью 100 см3, нейтрализуют серной кислотой в присутствии фенолфталеина и доводят объем дистиллированной водой до 10 см3. Из нее отбирают 1 см3 в мерную колбу вместимостью 100 см3 и доводят объем водой до метки. 0,1 см3 полученного второго раствора содержит 0,001 мг мышьяка. В 6 колб
509
1’ik.5.9. Прибор Зангер -Блекл для определения мышьяка:
. р. Г'м ’ киж\\ р>. <o.U!i.i-i>ii - h.h.l пропи 1 ;н1Н;>я .'Uici.t-
ъ>'1 сыпни. jijif(j). ? pe.iki[ионная Ko.ioa
прибора Зангер —Блока последовательно вносят 0.1 (1-я колба). 0.3: 0.5: 0.7: 1 и 2.5 см3 стандартного раствора мышьяка.
Порядок проведения анализа. В коническую колбу прибора Зангер—Блека (рис. 5.9) вносят 2 см3 минерализата или 5 г измельченного продукта, 10 см3 раствора серной кислоты массовой долей 20 %, 5 см3 дистиллированной воды, 1 см3 раствора хлорида олова массовой долей 10 % (в серной или соляной кислоте) и около 2 г купированного мелкогра-нулированного цинка. Колбу закрывают хорошо пришлифованной насадкой, в которую помещают
комочек ваты, смоченной раствором ацетона массовой долей 5 % и высушенной на воздухе (для поглощения сероводорода). Выше размещают бромо- или хлорортутную бумагу. Колбу с насадкой оставляют на 1 ч, после чего реактивную бумагу снимают и осматривают. При наличии в пробе мышьяка бумага окрашивается в желтый или коричневый цвет.
Для количественного определения мышьяка в исследуемом материале анализ проводят в колбах, в которых вместо минерализата находится разный объем стандартного раствора. Из полученных окрашенных реактивных бумажек составляют стандартную шкалу, в которой интенсивность окраски возрастает в соответствии с количеством мышьяка (точно известным), выделившимся из стандартного раствора. Приготовленная стандартная шкала может храниться в темном месте в течение 5—6 мес, и ее можно использовать многократно. Полученную после исследования продуктов убоя животных окрашенную хлоро- или бромортутную бумажку сравнивают со стандартной шкалой, устанавливают количество связанного ею мышьяка, а затем проводят расчет на 100 г продукта в естественном состоянии.
3. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ СВИНЦА НЕФЕЛОМЕТРИЧЕСКИМ МЕТОДОМ
Материалы, реактивы и оборудование. Нефелометр или фотоэлектроколориметр любой марки с толщиной рабочего слоя 3 см, светофильтром Х = 490нм; центрифуга; электропечь; весы технические; прибор Чижовой; фарфоровый тигель диаметром Зсм; колориметрические пробирки; градуированные пипетки вместимостью 5 и 10 см3; водный раствор ацетата аммония массовой до-510
лей 3 %: серная кислота, разбавленная 1:1 (р= 1,62 г/см3): водный раствор хромата калия массовой долей 1 %: стандартный водный раствор нитрата свинца с концентрацией свинца в растворе 1 мг/см3 (раствор 1).
Порядок проведения анализа. Сухая минерализация проб для определения содержания свинца нефелометрическим методом имеет некоторые особенности.
Взвешивают навеску мяса массой 5,00 г, удаляют влагу на приборе Чижовой, переносят навеску в тигель, добавляют 2 см3 раствора H?SO4 и помещают в электропечь, температуру которой повышают до 500—550 °C. Навеску озоляют до образования золы белого цвета.
Тигель после сухой минерализации пробы вынимают из печи, остывшую золу обрабатывают 12 см3 раствора ацетата аммония массовой долей 3 %. Содержимое тигля переносят в пробирку с оттянутым конусом и центрифугируют. Затем отбирают 10 см3 раствора в колориметрическую пробирку, добавляют 1 см3 раствора К?СгО4 массовой долей 1 %, взбалтывают и измеряют оптическую "плотность мутных растворов. По калибровочному графику находят массу свинца в пробе тх (мг).
Содержание (мг%) свинца в мясе и мясопродуктах рассчитывают по формуле
Л'=(/д1»-100, (5.30)
где /И) — масса свинца, найденная по калибровочному графику, мг; т — масса навески мяса, г.
Построение калибровочного графика. Для построения калибровочного графика пипеткой отбирают 10 см3 стандартного раствора 1, переносят в мерную колбу вместимостью 1дм3 и доводят раствором ацетата аммония массовой долей 3 % до метки. Получают стандартный раствор 2 с концентрацией свинца 0,01 мг/см3. Из раствора 2 готовят серию рабочих растворов для определения свинца в соответствии с приведенными ниже рекомендациями.
Реактивы для приготовления Номера пробирок
серии рабочих растворов 0 1 2 3 4 5
Стандартный раствор 2, см3 0 12 3 4 5
Раствор ацетата аммония, см3 10 9 8 7 6 5
Раствор хромата калия, см-’ Во все пробирки по 1 см3
Масса свинца, мг 0 0,002 0,003 0,004 0,006 0,010
Оптическую плотность растворов измеряют при X = 490 нм в кюветах с длиной рабочего слоя 3 см. По полученным данным строят калибровочный график в координатах: оптическая плотность — масса свинца.
511
4. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ СВИНЦА, КАДМИЯ, МЕДИ, ЦИНКА МЕТОДОМ АТОМНО-АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
4.1. МИНЕРАЛИЗАЦИЯ ПРОБ
4.1.1. Способ сухой минерализации
Материалы, реактивы и оборудование. Вода дистиллированная: вода бидистиллированная или деионизированная; растворы азотной (I : I), соляной (I : I), серной (1:1) и уксусной кислот (1:19) по объему; этанол ректификованный; чаши или тигли кварцевые вместимостью 50, 100, 250 см3 или чашки (тигли) фарфоровые № 2—4; пипетки; стакан вместимостью 1000 см3; стекла часовые; весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г (для взятия навесок массой меньше Юг) и 500 г или 1 кг (для взятия навесок массой Юг и более); шкаф сушильный лабораторный; электропечь сопротивления камерная лабораторная; лампа инфракрасная мощностью 250 или 500 Вт; щипцы тигельные; электроплитка бытовая или горелка газовая; баня водяная.
Подготовка посуды. Новую или сильно загрязненную лабораторную посуду (колбы, пипетки, чаши или тигли) после обычной мойки в растворе любого моющего средства промывают водопроводной и ополаскивают дистиллированной водой.
Непосредственно перед использованием посуду дополнительно обрабатывают горячим раствором азотной кислоты (1 : 1), ополаскивают дистиллированной водой, обрабатывают горячим раствором соляной кислоты (1 : 1), ополаскивают 3—4 раза дистиллированной водой и 1—2 раза бидистиллированной или деионизированной водой, а затем сушат. Обработку горячим раствором кислоты проводят следующим образом: посуду помещают в термостойкий химический стакан вместимостью ЮООсм3, заливают раствором кислоты и нагревают до кипения. Затем выдерживают до полного охлаждения и моют, как указано выше.
Вместо обработки посуды одним из растворов кислот допускается выдерживание чаш или тиглей с раствором уксусной кислоты на водяной бане при температуре кипения в течение 1 ч.
Порядок проведения анализа. В чашу (чашку, тигель) берут навеску продукта из подготовленной к испытаниям пробы. Необходимый объем жидкого продукта отмеряют пипеткой. Значения массы навески (или объема пробы) см. в табл. 5.8.
Чашу с навеской помещают на водяную баню при температуре кипения, или в сушильный шкаф (доводя его температуру до 150 °C), или на электроплитку и удаляют влагу. Затем осторожно обугливают содержимое чаши на газовой горелке или электроплитке до прекращения выделения дыма, не допуская сильного воспламенения и выбросов. Чашу помешают в электропечь, на-512
гретую до 250 С' (продукцию, содержащую более 26 У сахарог нагревают в нечи до 150 СО.
Для инзенсификании процесса обугливания рскоменлуечся'
a) naipcBaii, чашу с навеской продукта. одновременно пеною зуя инфракрасную лампу:
б) лобавпiь в чашу с навеской эшнол и’> расчеia 5 см' на 1 1 о хого вещества, закрыть часовым стеклом. выдержан» 24 -48 ч. а к тем проводить обугливание;
в) для продуктов, содержащих 20--60 сс жира, в навеску ло бавить раствор азотной кислоты (I : 1) из расчета 1- 1.5см на каждые Юг навески, выдержан» 15 мин. а затем проводин» ooyi ли ван не.
После окончания обутливания минерализацию проб проводя: в электропечи, постепенно (на 50’С через каждые 30 мин) повышая температуру до 450 С. Минерализацию продолжают при этой температуре до получения серой золы. Допускается минсрализа ция зерна и зернопродуктов при температуре 500 °C.
Чашу с золотт вынимают из электропечи черен 10—15 : озоления, охлаждают до комнатной температуры и смачиваю: содержимое ио каплям минимальным объемом раствора азотной кислоты.
Кислоту выпаривают досуха на водяной бане с последующей выдержкой в сушильном шкафу при температуре до 140 °C , либо под инфракрасной лампой, либо на электроплитке со слабым нагревом. После охлаждения чашку с навеской снова помещают в охлажденную электропечь. Постепенно доводят температуру до 300 °C и выдерживают в течение 0,5 ч. Указанный цикл повторяют несколько раз. Минерализацию считают законченной, если зола станет белого или слегка окрашенного цвета, без обугленных частиц. При наличии обугленных частиц повторяют обработку золы раствором азотной кислоты или водой.
Параллельно в двух чашках проводят минерализацию добавляемых к навеске реактивов для контроля их чистоты.
4.1.2. Способ мокрой минерализации
Материалы, реактивы и оборудование. Весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г, 500 г или I кг для взятия навесок массой Юг и более; электроплитка бытовая или горелка газовая; штатив химический; баня водяная; колбы Кьельдаля или плоскодонные вместимостью 250 см3; стаканы вместимостью 50 см3; цилиндры вместимостью 5—50 см3; воронки; пипетки: колбы мерные вместимостью 50 и 100cmj; колбы конические; шарики стеклянные (для обеспечения равномерности кипения); фильтры обеззоленные диаметром II см; кислота азотная, х. ч., концентрированная и ее раствор (I : 5 по объему); кис-
513
.юта серная концентрированная: кислота соляная. \. ч. и си раствор концентрацией 10 г/дм5: кислота хлорная концентрированная и ее раствор концентрацией 570 г/дм3: пероксид водорода, с ул ьф а т г и др а з и н а.
Подготовка посуды. Чистую стеклянную посулу дополнительно промывают раствором азотной кислоты, ополаскивают водопроводной, а затем дистиллированной водой.
Порядок проведения анализа. Навеску подготовленной к выполнению анализа твердой или пастообразной пробы массой, указанной в табл. 5.9. берут на обеззоленный фильтр, заворачивают в него и стеклянной палочкой помешают на дно колбы Кьельдаля или плоскодонной колбы.
Навеску сухих продуктов (желатин, сухие яичные продукты) помещают в колбу Кьельдаля, добавляют 15 см3 воды, перемешивают. Желатин затем оставляют на 1 ч для набухания.
В колбу с пробой продукта, подготовленной к минерализации, вносят азотную кислоту из расчета 10 см3 на каждые 5 г продукта и выдерживают не менее 15 мин. Затем в колбу вносят 2—3 стеклянных шарика для равномерности кипения, закрывают грушевидной стеклянной пробкой и начинают нагревать на электроплитке, постепенно увеличивая нагрев и упаривая содержимое колбы до объема 3—5 см3.
Колбу охлаждают, вносят 10 см3 азотной кислоты, содержимое упаривают до объема 5 см3, после чего снова охлаждают. Процедуру повторяют 2—4 раза.
В колбу вносят по 10 см3 азотной кислоты, 5 см3 серной кис; лоты, 4 см3 хлорной кислоты (или вместо хлорной кислоты 4 см3 пероксида водорода) из расчета на каждые 5 г пробы. Изменять последовательность внесения кислот не допускается. Хлорную кислоту всегда добавляют последней. Содержимое колбы упаривают до объема около 5 см3, не допуская окрашивания жидкости в коричневый цвет. При появлении коричневой окраски нагревание прекращают.
Колбу охлаждают до комнатной температуры, добавляют 5 см3 азотной кислоты и 2 см3 хлорной кислоты (или вместо хлорной кислоты 2 см3 пероксида водорода) и снова нагревают до появления белых паров серного ангидрида. Если при этом раствор не обесцветился, процедуру повторяют. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным.
Для удаления остатков кислот в охлажденную колбу добавляют 10 см3 воды и кипятят 10 мин с момента выделения белых паров, затем охлаждают. Добавление воды и нагревание повторяют еще два раза. Если при этом образуется осадок, в колбу вносят 20 см3 воды, 2 см3 серной кислоты и 5 см3 соляной кислоты. Полученную смесь кипятят до растворения осадка, постоянно пополняя испаряющуюся воду. Минерализат после охлаждения используют 514
для анализа без разбавления или количественно переносят водой в мерную колбу вместимостью 50 см3, доводят до метки дистиллированной водой и перемешивают.
Кислотная минерализация проб пищевых жиров для определения содержания меди имеет следующие особенности. Пробу продукта в колбе Кьельдаля, подготовленную к минерализации, нагревают 7—8 ч на электроплитке до образования вязкой массы, охлаждают, добавляют 25 см3 азотной кислоты и bhobi> осторожно нагревают, избегая бурного вспенивания. После прекращения вспенивания колбу с содержимым охлаждают, добавляют еще по 25 см3 азотной и 12 см3 хлорной кислоты и нагревают до получения бесцветной жидкости. Если в процессе нагревания жидкость темнеет, к ней периодически добавляют по 5 см? азотной кислоты и продолжают процесс до завершения минерализации. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным.
Для удаления остатков кислот в охлажденную колбу добавляют 10 см3 воды и кипятят 10 мин с момента выделения белых паров, затем охлаждают. Добавление воды и нагревание повторяют еще два раза. Если при этом образуется осадок, в колбу вносят 20 см3 воды, 2 см3 серной кислоты и 5 см3 соляной кислоты. Полученную смесь кипятят до растворения осадка, постоянно пополняя испаряющуюся воду. Минерализат после охлаждения используют для анализа без разбавления или количественно переносят водой в мерную колбу вместимостью 50 см3, доводят до метки дистиллированной водой и перемешивают.
4.1.3. Способ кислотной экстракции (неполной минерализации)
Материалы, реактивы и оборудование. См. п. 4.1.2 настоящей работы, а также колбы конические вместимостью 250 см3; воздушный холодильник; делительная воронка; азотная кислота концентрированная, х. ч. и ее раствор (1 : 2 по объему).
Подготовка посуды. См. п. 4.1.2 настоящей лабораторной работы.
Порядок проведения анализа. В термостойкую колбу с навеской продукта, масса которого указана в табл. 5.9, вносят цилиндром 40 см3 раствора соляной кислоты (1 : 1 по объему).
В колбу добавляют несколько стеклянных шариков, присоединяют к ней холодильник, помещают на электроплитку, покрытую асбестом, и кипятят в течение 1,5 ч с момента закипания. Затем содержимое колбы медленно охлаждают до комнатной температуры, не вынимая холодильника.
Колбу с экстракционной смесью (жир с кислотой) помещают в холодную водяную баню. Затвердевший жир прокалывают стеклянной палочкой, водный слой фильтруют через фильтр, смочен
515
ный раствором кислоты, используемой для экстракции, в сосуд, выбор которого определяется дальнейшим ходом анализа и зависит от определяемого элемента (реакционную колбу, колбу Кьельдаля, кварцевую или фарфоровую чашу). Оставшийся в колбе жир расплавляют на водяной бане, добавляют 10 см3 раствора используемой кислоты, встряхивают и охлаждают. После охлаждения жир прокалывают и промывную жидкость сливают в тот же сосуд через тот же фильтр. Затем фильтр промывают 5—7 см3 воды.
В случае подготовки экстрактов к колориметрическому определению содержания меди экстракционную смесь фильтруют в колбу Кьельдаля. При использовании для экстракции раствора азотной кислоты содержимое колбы упаривают на электроплитке до объема 5—7 см3, колбу охлаждают, добавляют 1 см3 хлорной кислоты и кипятят до получения бесцветного или слабо-окрашенного раствора.
При использовании для экстракции раствора соляной кислоты в колбу вносят 5 см3 азотной кислоты, упаривают на электроплитке до объема 5—7 см3, колбу охлаждают, добавляют 4 см3 азотной кислоты и 1 см3 хлорной кислоты и кипятят до получения бесцветного или слабоокрашенного раствора.
При определении меди в растительных маслах и маргарине экстракты не обрабатывают.
Для удаления остатков кислот в охлажденную колбу с экстракционной смесью добавляют 10 см3 воды и кипятят 10 мин, а затем охлаждают. Добавление воды и нагревание повторяют еще два раза. Полученный раствор количественно переносят в мерную колбу. Объем раствора доводят водой до метки и перемешивают.
При использовании полярографического и атомно-абсорбционного анализа экстракционную смесь фильтруют в кварцевую или фарфоровую чашу. Жидкость осторожно выпаривают, а затем обугливают на электроплитке. Затем содержимое чаши минерализуют в электропечи, постепенно (на 50 °C через каждые 30 мин) повышая температуру до 450 °C. Минерализацию продолжают при этой температуре до получения серой золы.
Параллельно в двух колбах проводят экстракцию и подготовку экстрактов к анализу добавляемых к навеске реактивов для контроля их чистоты.
4.2. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ТОКСИЧНЫХ ЭЛЕМЕНТОВ МЕТОДОМ КАЛИБРОВОЧНОГО ГРАФИКА
Материалы, реактивы и оборудование. Атомно-абсорбирующий спектрофотометр; водяная баня; цинк или сульфат цинка; медь; свинец или нитрат свинца; кадмий; соляная и азотная кислоты; концентрированная соляная кислота, раствор (1:1 по объему) и раствор молярной концентрацией 1 моль/дм3; растворы азотной 516
кислоты молярной концентрацией 5моль/лм' и массовой долей 40%: основные и рабочие стандартные растворы пинка, меди, свинца, кадмия.
Приготовление основных и рабочих стандартных растворов. Основные стандартные растворы (эталоны) готовят из чистых металлов (более 99,9 %) или чистых (х. ч.. ос. ч.) устойчивых соединений элементов. Для получения устойчивых при хранении растворов используют растворители.
Концентрация металла в основном стандартном растворе составляет 1 г/дм3 (1000 мкг/см3). Срок хранения растворов не должен превышать 2—3 лет при условии хранения в герметичной посуде из стекла, полиэтилена высокой плотности, полистирола, кварца. Разбавленные растворы концентрацией около 100 мкг/см0 обычно хранят до 1 мес. концентрацией 10 мкг/см3 — в течение 2 — 3 сут.
Необходимые реактивы и схема приготовления основных стандартных растворов приведены в табл. 5.9.
Таблиц а 5.9
Реактивы и порядок приготовления основных стандартных растворов, концентрация металла 1000 мкг/см3
Элемент Масса навески исходного реактива, г Схема приготовления основного стандартного раствора объемом 1000 см'' Концентрация кислоты в основном стандартном растворе
Цинк Zn (1.000 г) HCI (1:1) + Н2О 1 моль/дм3 НО
ZnO (1.245 г) HNO3 (массовая доля 40 %) + Н2О 0,1 моль/дм3 HNO3
Медь Си (1,000 г) 50 см3 HNO3 молярной концентрацией 5 моль/дм3 (упарить досуха на водяной бане) + 5 см3 концентрированной НО (упарить досуха) + разбавленная НО + Н,0 1 моль/дм3 НО
Свинец РЬ (1,000 г) Pb (NO3 )2, высушенный при 1'10 °C (1.599 г) Минимальный объем HNO3 молярной концентрацией 6 моль/дм' + Н2О + концентрированная НО Разбавленная NHO3 (1 : 100) 1 моль/дм3 НО 1 % HNO,
Кадмий Cd (1.000 г) Разбавленная NHC). (упарить досуха и выдержать при 80—90 “С на водяной бане) + 5 см3 НС1 молярной концентрацией 1 моль/дм3 (упарить досуха) + разбавленная но + н2о 1 моль/дм3 НО
CdO (1,142 г) 20 см-3 НО молярной концентрацией 5 моль/дм3 + Н2О + концентрированная НО 1 моль/дм3 НО
517
Поясним пользование таблицей на примере подготовки стан лартного раствора для определения массовой доли пинка. Для приготовления основного стандартного раствора необходимы дистиллированная вода и соляная кислота, объем которой определяют с учетом ее концентрации в основном стандартном растворе 1 моль/дм3. Для приготовления основного стандартного раствора навеску цинка массой 1,000 г растворяют в рассчитанном объеме разбавленной соляной кислоты (1:1 по объему), объем полученного раствора доводят дистиллированной водой до 1000 см3.
Рабочие стандартные растворы готовят путем разбавления основных стандартных растворов до концентрации, соответствующей рабочему диапазону концентраций и близкой к концентрации элемента в анализируемом растворе. Для разбавления используют раствор, применяемый для приготовления анализируемых растворов проб.
Порядок проведения анализа. Проводят настройку прибора в соответствии с технической инструкцией фирмы-изготовителя. Настройку монохроматора проводят по максимуму излучения источника при минимальной щели.
Рекомендуемые условия измерений, а также значения предела определения приведены в табл. 5.10.
Таблиц а 5.10
Условия атомно-абсорбционной спектрометрии в воздушно-ацетиленовом пламени
Элемент Длина волны, нм Щель, нм Пламя Высота горелки, мм Оптимальный диапазон рабочих концентраций, мкг/см’ Предел определения, мкг/см;
Цинк 213,9 1,5 Окислительное 6-7 1-10 0.002
Медь 324,8 1,5 Стехиометрическое 7-8 0,005-5 0,003
Свинец 283,3 2,0 » 7-8 0,1-2 0,02
217,0 2,0 » 7-8 0,1-2 0,01
Кадмий 228,8 1,0 Окислительное 6-7 0,02-1 0,001
Измерения проводят в соответствии с технической инструкци-
ей, прилагаемой к прибору.
Поскольку система распылителя в процессе работы может засоряться, чувствительность измерения может нарушаться. В этом случае приходится проводить повторные градуировки. Обычно достаточно выполнить две градуировки — в начале и в конце измерений. В других случаях целесообразно проводить измерения по следующей схеме: первая градуировка — первая серия замеров абсорбции (5—10 анализируемых растворов); вторая градуировка — вторая серия замеров абсорбции тех же растворов в обратном порядке; третья градуировка — третья серия замеров. По результатам, полученным в двух сериях, находят среднее значение.
518
По концентрации элемента в растворе находят его содержание в продукте (мг/кг):
где г —концентрация химического элемента и растворе пробы, найденная по калибровочному графику, мкг/см': 1—объем исходного раствора пробы, см’; Л — коэффициент разбавления или концентрирования исходного раствора пробы (отношение объема анализируемого раствора к объему раствора, взятому для разбавления или концентрирования): — концентрация химического элемента в контрольном опыте, мкг/см': I/. — объем раствора в контрольном опыте, ему /и — масса навески пробы, г.
Построение калибровочного графика. Обычно для построения калибровочного графика в области рабочих концентраций достаточно 3—5 рабочих стандартных растворов, при работе в линейной области — 2 раствора. При большой и особенно знакопеременной кривизне графика необходимо увеличить число рабочих стандартных растворов или ограничить область рабочих концентраций более узким диапазоном (см. табл. 5.9).
Поочередно фотометрируют стандартные растворы не менее трех раз каждый, начиная с наименее концентрированного. После каждого стандартного раствора устанавливают нулевое поглощение прибора по дистиллированной воде. По результатам измерений строят калибровочный график в координатах: абсорбция — концентрация определяемого элемента (мкг/см3).
5. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ТОКСИЧНЫХ ЭЛЕМЕНТОВ ПОЛЯРОГРАФИЧЕСКИМ МЕТОДОМ
5.1. МИНЕРАЛИЗАЦИЯ ПРОБ
Минерализацию проб для определения массовой доли свинца, кадмия, меди, цинка проводят сухим способом (см. 4.1 настоящей лабораторной работы).
5.2. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МЕДИ, СВИНЦА И КАДМИЯ
Материалы, реактивы и оборудование. Полярограф марки ПУ-1 или других марок, обеспечивающих возможность работы в режиме переменного тока; баня водяная; pH-метр; электроплитка; пипетки мерные; фильтры обеззоленные; линейка; пробирки мерные вместимостью 10 и 15 см3; воронки делительные; набор ареометров без шара с пределом измерений от 700 до 1840 кг/м, вода дистиллированная и бидистиллированная; фоновые электро
519
литы A ii Б: раствор aiornoii кислоты (1:1 но обьему): ортофос-форная кислота молярной концентрацией 1.3 моль/лм\ хлорная кислота молярной концентрацией 0.7 моль/лм': лимонная кислота: раствор тимолового синего: растворы дитизона в хлороформе концентрацией 0.01: 0.2 и 1 г,/дм3: раствор соляной кислоты молярной концентрацией 0.2 моль/дм'; хлороформ: сульфат мели: нитрат свинца: контрольные растворы; основные, рабочие и стандартные растворы сульфата мели, нитрата свинца, кадмия: азот газообразный или другой инертный газ массовой долей кислорода не более 0,001 %: для очистки инертного газа от кислорода: гидроксид калия, ч. д. а., гранулированный и раствор концентрацией 600 г/дм-'; пирогаллол А. ч.л. а.; раствор пирогаллола концентрацией 250 г/дм3; аммиак водный особой чистоты или ч.д. а., очищенный методом изотермической перегонки; для очистки аммиака методом изотермической перегонки: эксикатор; чашка фарфоровая; гидроксид натрия или калия гранулированный.
Подготовка посуды и материалов. Лабораторную стеклянную посуду промывают хромовой смесью, водой, азотной кислотой плотностью 1,40 г/см3, несколько раз дистиллированной и дважды бидистиллированной водой и сушат. Затем посуду моют раствором дитизона концентрацией 0,01 г/дм3. Даже при незначительном изменении окраски обработку дитизоном проводят несколько раз. Сначала посуду заполняют раствором дитизона концентрацией 0,30 г/дм3 и выдерживают 30 мин, после чего промывают хлороформом и вновь повторяют обработку, используя раствор дитизона концентрацией 0,01 г/дм3. Далее посуду моют хлороформом и высушивают на воздухе в вытяжном шкафу.
Очистка инертного газа от кислорода. При наличии кислорода более 0,001 % газ пропускают через поглотительную смесь, состоящую из растворов пирогаллола и гидроксида калия в соотношении 1 : 5.
Очистка аммиака от примесей методом изотермической перегонки. На дно эксикатора помещают несколько кусочков гидроксида калия или натрия и приливают 500 см3 водного аммиака, а на фарфоровой сетке устанавливают фарфоровую чашку с 250 см3 бидистиллированной воды. Эксикатор закрывают крышкой и оставляют на 5 сут. В чашке получают очищенный раствор аммиака концентрацией 130—150 г/дм3. Концентрацию аммиака в растворе уточняют на основе измеренных ареометром показателей плотности.
Приготовление реактивов. Фоновый электролит А. Смешивают растворы ортофосфорной кислоты молярной концентрацией 1,3 моль/дм3, хлорной кислоты молярной концентрацией 0,7 моль/дм3 и бидистиллированную воду в соотношении 3 : 2 :5.
Фоновый электролит Б (раствор соляной кислоты молярной концентрацией 0,1 моль/дм3). Пипеткой переносят
520
8.2 см' соляном кислоты плотностью 1.19 г/см '1 в мерную кол бу вместимостью ЮООсм3 и доводят бидистиллированной водой до метки.
Основной раствор сульфата, меди CuSO4 5Н?О концентрацией меди 1 000 мкг/см3. Навеску х. ч. CuSOJ х х5Н-,0 1.965 г (/' = MCuso4-5H2oMzu = 3,929) растворяют в 50 см3 H?SO} (1 : 20). переносят в мерную колбу вместимостью 500 см' и доводят объем раствора до метки дистиллированной водой.
Рабочий раствор сульфата меди концентрацией меди 100 мкг/см3. 10 см3 исходного раствора переносят в мерную колбу вместимостью 100 см3 и доводят объем раствора до метки дистиллированной водой.
Стандартные растворы меди. Готовят четыре стан; дартных раствора, содержащих соответственно 1, 2, 4 и 8 мкг/см3 меди, для чего в мерные колбы вместимостью 100 см3 переносят соответственно 1,00; 2,00; 4,00 и 8,00 см3 рабочего раствора сульфата меди, доводят объем раствора до метки дистиллированной водой и тщательно перемешивают.
Основной раствор нитрата свинца концентрацией свинца 1000 мкг/см3. Навеску х. ч. Pb( NO3)? 1,599 г (F = = 1,599) растворяют в дистиллированной воде
с добавлением 1 см3 концентрированной азотной кислоты, переносят в мерную колбу вместимостью ЮООсм3 и доводят объем раствора дистиллированной водой до метки.
Рабочий раствор нитрата свинца концентрацией свинца 100 мкг/см-. 10 см3 основного раствора переносят в мерную колбу вместимостью 100 см3 и доводят объем раствора до метки дистиллированной водой.
Стандартные растворы свинца. Готовят пять стандартных растворов, содержащих соответственно 5, 10, 15, 20 и 25 мкг/см3 свинца, для чего в мерные колбы вместимостью 100 см3 переносят соответственно 5,00; 10,00; 15,00; 20,00 и 25,00 см3 рабочего раствора соли свинца, доводят объемы до метки дистиллированной водой и тщательно перемешивают.
Основной раствор кадмия. 1,000 г металлического кадмия помещают в коническую колбу вместимостью 250 см3 и растворяют при нагревании на электроплитке в 25 cmj разбавленной (1:1) азотной кислоты. Раствор выпаривают на электроплитке со слабым нагревом до объема 3 см3, приливают 15 см3 соляной кислоты плотностью 1,19 г/см3 и вновь выпаривают до того же объема. Выпаривание повторяют еще два раза, добавляя каждый раз по 5 см3 соляной кислоты. После охлаждения добавляют 50 см3 соляной кислоты плотностью 1,19 г/см3, количественно переносят раствор в мерную колбу вместимостью 1000 см3 и доводят бидистиллированной водой до метки.
Раствор хранят не более 1 года. Концентрация кадмия в основном растворе 1 мг/см3.
521
С т а н д а р т и ы е р а с т в о р ы к а д м и я. Готовят последовательным разбавлением в 10. 100 и 1000 раз основного раствора кадмия. При изменении концентрации кадмия в испытуемом растворе с использованием фонового электролита А разбавление проводят бидистиллированной водой, в остальных случаях — соответствующим фоновым электролитом.
Контрольные растворы. Готовят, используя все реактивы и растворы, аналогично приготовлению испытуемого раствора.
Порядок проведения анализа. Для приготовления испытуемого раствора при использовании фонового электролита А золу после сухой минерализации пробы растворяют в тигле при нагревании на водяной бане в 5 см3 разбавленной (1 : 2) азотной кислоты. Раствор выпаривают до влажных солей. К осадку в тигле добавляют 2 см3 разбавленной хлорной кислоты и нагревают на водяной бане в течение 5 мин. Раствор охлаждают, добавляют по 3 см3 ор-тофосфорной кислоты и воды, тщательно перемешивают и дают отстояться осадку. Раствор фильтруют в мерную пробирку через обеззоленный фильтр, предварительно промытый фоновым электролитом. Остатки золы в тигле и на фильтре смывают водой, доводя объем до 10 см3.
Измерение на полярографе проводят в режиме переменного тока ртутно-капельным электродом в электролизере вместимостью 5 см3. Выбирают режим работы в соответствии с инструкцией, прилагаемой к полярографу, при следующем напряжении относительно донной ртути:
Определяемый элемент Напряжение. В
Медь От — 0,1 до — 0.5
Свинец От — 0,4 до — 0.8
Кадмий От —0.6 до—1,0
Прямое полярографирование используют в тех случаях, когда массовая доля определяемого элемента в пробе обеспечивает получение четкого пика на полярограмме, а состав элементов в золе не создает помех.
Определение проводят следующим образом. В две конические колбы вместимостью 10 или 25 см3 помещают по 4 см3 контрольного или испытуемого раствора. В первую колбу добавляют 1 см3 соответствующего фонового электролита или бидистиллированной воды (при работе с фоновым электролитом А) и в течение 10 мин пропускают через раствор азот или любой другой инертный газ.
Раствор немедленно переносят в электролизер, предварительно промытый дистиллированной водой, фоновым электролитом и полярографируемым раствором, полярографируют и измеряют высоту пика свинца.
522
Во вторую колбу вносят добавку --стандартный раствор в та ком объеме, чтобы высока ника свинна на полярограмме пример но удвоилась по сравнению с первоначальной. Следует вносить tic более I см3 добавки, чтобы предотвратить изменение концентрации фонового электролита и зольных элементов. Затем в колбу добавляют фоновый! электролит или бидистиллированную воду (при работе с фоновым электролитом А) в объеме, необходимом для доведения его до 5 см-1. Пропускают инертный газ. полярогра-фируют в тех же условиях и измеряют высоту пика свинца.
Полярографирование с предварительным внесением определяемого элемента в испытуемый раствор используют при анализе образцов с низкой массовой долей свинца или в те.х случаях, когда на полярограмме вследствие помех из-за сложного элементного состава золы в области пика свинца наблюдается только нечеткий изгиб.
Определение проводят следующим образом. В две конические колбы вместимостью 10 или 25 см3 помещают по 4 см3 контрольного или испытуемого раствора и добавляют минимальную массу определяемого элемента для свинца и кадмия (0,2—0,5 мкг), которая обеспечила бы получение на полярограмме четкого пика. Далее поступают, как при прямом полярографировании. Необходимые объемы жидкости отбирают только пипетками.
Содержание токсичных элементов (мг/кг) или массовую концентрацию (мг/дм3) определяют по высоте пиков, измеренных на полярограммах с помощью линейки с точностью до 1 мм, и вычисляют соответственно по следующим формулам:
при прямом полярографировании
при полярографировании с предварительным внесением элементов в полярографируемый раствор
X -
------ — ----
(Н2-Нф “JP>’
(5.34)
X =
vy
(5.35)
523
i.ic m, — масса свинца, добавленного перед вторым полярографированисм. мм; //, — высота пика свинца, полученного при первом полярографировании. мм: Г, -обшиб объем испытуемого раствора, приготовленного из озоленной навески, ему /Л — высота пика свинца, полученного при втором полярографировании. мм: Г, -объем испытуемого раствора, взятого для полярографировании. см': //у — масса свинца в контрольном растворе, мкг: /// — масса навески продукта, взятая для озоления. г: И—объем продукта, взятый для озоления, см': //у — масса свинца, предварительно добавленная для получения четкого пика свинца, мкг.
Вычисления проводят до третьего десятичного знака.
В случаях, когда при полярографировании испытуемых растворов не удается получить четкий пик свинца или кадмия в связи с возникновением помех вследствие присутствия в золе мешающих элементов, проводят предварительное экстракционное выделение свинца или кадмия.
При этом золу растворяют в тигле при нагревании на водяной бане в 5 см3 разбавленной (1:2) азотной кислоты. Раствор охлаждают, добавляют 25 см3 бидистиллированной воды и 2 г лимонной кислоты. После растворения лимонной кислоты вносят 1 см3 раствора тимолового синего и доводят pH раствора примерно до 8,8, медленно вливая аммиак в пробу при охлаждении ее в ледяной бане. Цвет раствора должен измениться от красного через желтый до зеленовато-синего. Если при подщелачивании раствором аммиака образуется осадок, то количество добавляемой лимонной кислоты увеличивают.
Раствор количественно переносят в делительную воронку вместимостью 250 или 500 см3 и, смывая несколько раз тигель бидистиллированной водой, доводят объем приблизительно до 150 см3.
Свинец или кадмий несколько раз экстрагируют раствором дитизона в хлороформе порциями по 5 см3 до стабилизации цвета, каждый раз встряхивая его в делительной воронке в течение 2 мин. Сначала используют раствор дитизона концентрацией 1 г/дм3, затем —- 0,2 г/дм3.
Дитизоновые экстракты собирают в делительной воронке, промывают 50 см3 бидистиллированной воды и переносят в другую делительную воронку. Водный слой промывают 5 см3 хлороформа и смывные воды присоединяют к объединенным дитизоновым экстрактам.
В делительную воронку с дитизоновыми экстрактами .добавляют 30 см3 раствора соляной кислоты молярной концентрацией 0,2 моль/дм3, встряхивают в течение 2 мин и оставляют до разделения слоев. Дитизоновый раствор в хлороформе отбрасывают.
Оставшийся в делительной воронке раствор соляной кислоты промывают 5 см3 хлороформа. Хлороформ отбрасывают. Раствор фильтруют в тигель через обеззоленный фильтр, предварительно 524
промытый раствором соляной кислоты молярной концентрацией 0.2 моль/дм3. Делительную воронку и фильтр промывают два раза бидистиллированной водой порциями по 10 см3. Промывные воды присоединяют к раствору соляной кислоты, выпаривают на электроплитке при слабом нагреве до 1 см\ а затем досуха на водяной бане. Остаток растворяют в 10 см3 фонового электролита Б. Далее проводят прямое полярографирование или полярографи-рование с предварительным внесением определяемого элемента (свинца или кадмия) в испытуемый раствор.
5.3. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЦИНКА
Материалы, реактивы и оборудование. См. 5.2 настоящей лабораторной работы, а также цинк гранулированный или оксид цинка, растворы соляной кислоты плотностью 1,39 г/см3 и разбавленный (1:1 по объему); растворы хлорида аммония и аммиака молярной концентрацией 1 моль/дм-1; основной и стандартные растворы цинка; фоновый электролит А; контрольные растворы.
Подготовка посуды и материалов. См. 5.2 настоящей лабораторной работы.
Приготовление реактивов. Фоновый электролит А (раствор концентрацией хлорида аммония NH4C1= 1 моль/дм3 и аммиака NH3= 1 моль/дм3): 53,49 г хлорида аммония растворяют в небольшом объеме бидистиллированной воды и переносят в мерную колбу вместимостью 1000 см3. В колбу добавляют водный раствор аммиака в таком объеме, который содержит 17 г аммиака. Необходимый объем раствора аммиака рассчитывают на основе измеренных ареометром показателей плотности (от 63 до 75 см3 водного аммиака ос. ч. или от 120 до 130 см3 очищенного изотермической перегонкой аммиака ч. д. а.). Объем в колбе доводят бидистиллированной водой до метки.
Основной раствор цинка. 1.1,000 г гранулированного цинка растворяют в 7 см3 разбавленной (1:1) соляной кислоты, количественно переносят в мерную колбу вместимостью 1000 см3 и доводят объем раствора бидистиллированной водой до метки.
2. 1,242 г оксида цинка, высушенного при (104 ± 1)°С до постоянной массы, растворяют в 3,65 см3 соляной кислоты плотностью 1,39 г/см3, количественно переносят в мерную колбу вместимостью 1000 см3 и доводят объем раствора бидистиллированной водой до метки.
Основной раствор хранят не более 1 года. Концентрация цинка в основном растворе 1 мг/см3.
525
С г а и д а р т н ы е р а с т в о р ы и и н к а. Готовят в лень использования последовательным разбавлением основного раствора цинка в 10. 100 и 1000 раз фоновым электролитом А.
К о и г роль и ы е рас т в о р ы. См. 5.2 настоящей лабораторной работы.
Порядок проведения анализа. Золу после сухой минерализации растворяют в тигле при нагревании на водяной бане в 10 см3 разбавленной (1 : 1) соляной кислоты. Растворы нейтрализуют водным раствором аммиака по универсальной индикаторной бумаге и количественно переносят в мерную колбу вместимостью 50 см3, смывая тигель несколько раз бидистиллированной водой. К раствору добавляют водный раствор аммиака в таком объеме, который содержит 0,85 г аммиака. Необходимый объем этого раствора рассчитывают по его плотности, измеренной ареометром. Объем раствора в колбе доводят бидистиллированной водой до метки, перемешивают и фильтруют через обеззоленный фильтр, предварительно промытый фоновым электролитом А.
Концентрация цинка в испытуемом растворе должна быть от 0.2 до 10 мкг/см3. При более высокой концентрации раствор дополнительно разводят фоновым электролитом.
Измерения проводят на полярографе в режиме переменного тока с ртутно-капельным электродом в электролизере вместимостью 5 см3. Полярограмму записывают при напряжении от —1,0 до —1,4 В относительно донной ртути, выбирая режим работы в соответствии с инструкцией, прилагаемой к полярографу.
В две конические колбы вместимостью 10 или 25 см3 вносят по 8 см3 контрольного или испытуемого раствора и по 1 см3 раствора сульфита натрия. В первую колбу добавляют 1 см3 бидистиллированной воды. Раствор немедленно переносят в электролизер, предварительно промытый дистиллированной водой, фоновым электролитом А и полярографируемым раствором, исследуют и измеряют высоту пика цинка.
Во вторую колбу вносят добавку — стандартный раствор цинка в таком количестве, чтобы высота пика цинка на полярограмме примерно удвоилась по сравнению с первоначальной. Следует вносить не более 1 см3 добавки, чтобы предотвратить изменение концентрации фонового электролита и зольных элементов. Затем в колбу приливают бидистиллированную воду в объеме, необходимом для доведения его до 10 см3. Далее поступают так же, как с раствором без добавки. Необходимые объемы жидкости отбирают только пипетками.
Содержание цинка (мг/кг) или массовую концентрацию (мг/дм3) определяют по высоте пиков, измеренных на полярограммах с помощью линейки с точностью до 1 мм, и вычисляют соответственно по формулам (5.32) и (5.33).
526
6. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ТОКСИЧНЫХ ЭЛЕМЕНТОВ КОЛОРИМЕТРИЧЕСКИМИ МЕТОДАМИ
6.1. ОПРЕДЕЛЕНИЕ МЕДИ С ДИЭТИЛДИТИОКАРБАМАТОМ
НАТРИЯ
Материалы, реактивы и оборудование. Весы лабораторные; баня водяная; фотоэлектроколориметр; часы песочные на 1 мин; штатив химический; воронки делительные; колбы мерные вместимостью 25. 50, 100, 500 и ЮООсм3; пипетки; палочки стеклянные; стаканы, цилиндры мерные вместимостью 250 и 500 см3; эксикатор; вода дистиллированная; разбавленный раствор аммиака (2 ; 3 по объему); цитрат аммония; разбавленная соляная кислота (1:1 по объему);^ раствор диэтилдитиокарбамата натрия концентрацией 10 г/дм3 (хранят не более 7 сут в посуде из темного стекла); сульфат меди, дважды перекристаллизованный и высушенный в эксикаторе до постоянной массы; хлороформ или четыреххлористый углерод; трилон Б; этанол; раствор фенолфталеина в этаноле концентрацией 10 г/дм3; фильтры обеззоленные; серная кислота плотностью 1,84 г/см3; основной раствор меди концентрацией 1 мг/см3; смешанный раствор три-лона Б и цитрата аммония.
Приготовление реактивов. Основной раствор меди концентрацией 1 мг/см3. Сульфат меди дважды перекристаллизовывают и высушивают в эксикаторе до постоянной массы. Навеску сульфата меди массой 3,929 г растворяют дистиллированной водой в мерной колбе вместимостью ЮООсм3, добавляют 1 см3 серной кислоты плотностью 1,84 г/см3 и доводят объем водой до метки.
Смешанный раствор трилона Б и цитрата аммония. 100г цитрата аммония и 25г трилона Б, взвешенных с погрешностью не более ±0,1 г, помещают в мерную колбу вместимостью 500 см3, растворяют и доводят объем раствора до метки дистиллированной водой. Содержимое колбы перемешивают. Раствор переносят в делительную воронку вместимостью ЮООсм3, добавляют 0,5см3 раствора диэтилдитиокарбамата натрия и 50 см3 растворителя (хлороформа или четыреххлористого углерода). Воронку интенсивно встряхивают в течение 1 мин и оставляют в покое до разделения слоев. Нижний слой сливают и отбрасывают. В делительную воронку вносят 50 см3 растворителя, встряхивают в течение 1 мин и после разделения слоев нижний слой сливают и отбрасывают. Последнюю операцию повторяют до получения бесцветного нижнего слоя. Раствор хранят не более 2 мес.
Порядок проведения анализа. Золу, приготовленную сухой минерализацией (см. 4.1.1), растворяют в 5 см3 разбавленной (1 ; 1 по объему) соляной кислоты, нагревая на водяной бане при температуре кипения.
527
При ожидаемом содержании меди в растворе зоны, большем чем 40 мкг. его количественно переносят в мерную колбу вместимостью 50 см-' и доводят объем раствора дистиллированной водой io метки: ири ожидаемом содержании меди в растворе золы, меньшем 40 mki . раствор золы используют для последующего испытания целиком, без дополнительного разведения.
Раствор, полученный в результате мокрой минерализации (см. 4.1.2) или кислотной экстракции (см. 4.1.3). используют для проведения испытания без дополнительной обработки.
В каждую делительную воронку вносят 10 см' смешанного раствора цитрата аммония и трилона Б, две капли раствора фенолфталеина, раствор перемешивают, нейтрализуют, добавляя ио каплям раствор аммиака до появления окраски, охлаждают и объем доводят дистиллированной водой до 100 см \ Затем в делительные воронки вводят 2 см5 раствора диэтилдитиокарбамата натрия концентрацией 10 г/дм’ и 15 см ' растворителя (хлороформа или четыреххлористого углерода). Воронки интенсивно встряхивают в течение 1 мин и оставляют в покое до разделения слоев. Нижний слой сливают в мерную колбу вместимостью 25 см5. В делительные воронки вливают 10 см5 растворителя, встряхивают в течение 1 мин и после разделения слоев нижний слой сливают в ту же мерную колбу. В случае необходимости объем раствора в колбе доводят до метки с помощью растворителя и перемешивают. Контрольный раствор готовят аналогично, без введения раствора меди. Содержимое колб с растворами сравнения и контрольным раствором фильтруют через сухой бумажный фильтр в кюветы. Оптическую плотность растворов сравнения измеряют по отношению к контрольному раствору на фотоэлектроколориметре при X = (440 ± 5) нм или на спектрофотометре при длине волны Л =440 нм в кюветах с расстоянием между гранями соответственно 20 и 10 мм. При анализе жировых продуктов расстояние между гранями кювет должно быть соответственно 50 и 20 мм.
При определении меди в растворах с ожидаемым содержанием в них меди, большим 40 мкг, в делительную воронку вместимостью 250 см5 вносят аликвотный объем испытуемого раствора, содержащий от 10 до 40 мкг меди, и добавляют реактивы в той же последовательности, что и в раствор сравнения; при определении меди в растворах с ожидаемым содержанием в них меди, меньшим 40 мкг, содержимое колбы Кьельдаля или чашки с раствором золы количественно переносят в делительную воронку вместимостью 250 см5 с помощью дистиллированной воды.
Контрольный раствор готовят из контрольной пробы аналогично. Оптическую плотность испытуемого раствора измеряют по отношению к контрольному раствору.
По полученному значению оптической плотности с помощью калибровочного графика находят массу меди.
528
Построение калибровочного графика. Для построения калибровочного графика 1 см3 основного раствора меди помешают в мерную колбу вместимостью 100 см3. объем доводят до метки дистиллированной водой. Затем в делительные воронки вместимостью 250 см3 помещают 0.5; 1: 2; 3 и 4 см3 раствора, что соответствует 5, 10, 20, 30 и 40 мкг меди. В каждую делительную воронку помещают 10 см3 смешанного раствора цитрата аммония и три-лона Б. две капли раствора фенолфталеина, раствор перемешивают, нейтрализуют, добавляя по каплям раствор аммиака до появления окраски, охлаждают и добавляют дистиллированную воду до объема около 100 см3. Затем в делительные воронки вводят 2 см3 раствора диэтилдитиокарбамата натрия и 15 см3 растворителя (хлороформа или четыреххлористого углерода). Воронки интенсивно встряхивают в течение 1 мин и оставляют стоять до разделения слоев. Нижний слой сливают в мерную колбу вместимостью 25 см3. В делительные воронки помешают 10 см3 растворителя, встряхивают в течение 1 мин и после разделения слоев нижний слой сливают в ту же мерную колбу. В случае необходимости объем раствора в колбе доводят до метки с помощью растворителя и перемешивают. Контрольный раствор готовят аналогично, без введения раствора меди. Содержимое колб с растворами сравнения и контрольным раствором фильтруют через сухой бумажный фильтр в кюветы. Оптическую плотность растворов сравнения измеряют по отношению к контрольному раствору на фотоэлектроколориметре при длине волны Хтах = = (440 ± 5) нм или на спектрофотометре при X = 440 нм в кюветах с расстоянием между гранями соответственно 20 и 10 мм. При анализе жировых продуктов рекомендуемое расстояние между гранями кювет соответственно 50 и 20 мм.
Калибровочный график строят, откладывая на оси абсцисс массу меди (мкг), введенную в растворы сравнения, на оси ординат — соответствующие значения оптической плотности.
Содержание меди Х1 (мг/кг) или массовую концентрацию Х2 (мг/дм3) при анализе растворов с использованием объема, взятого для анализа, вычисляют по формулам
v т{ 50
¥|=ДШ; <5-
v _ т}- 50
= Ди ' Ц
где /«J — масса меди, найденная по калибровочному графику, мкг; 50 — общий объем минерализата, см3; К] — объем минерализата, взятый для анализа, см3; т — масса навески продукта, взятой для минерализации, г; V— объем продукта, взятый для минерализации, см3.
529
Содержание мели X (мг/кг) или массовую концентрацию Л', (мг/лм3) в анализе с использованием всей минерализованной пробы вычисляют по формулам
V т\
=—• Х.Х)
т
v т\
Н=у- (5.39)
где — масса меди, найденная по калибровочному графику, мкг; ///—масса на вески продукта, взятой для минерализации, г; I' объем продукта, взятый для минерализации, см3.
6.2. ОПРЕДЕЛЕНИЕ МЫШЬЯКА С ДИЭТИЛДИТИОКАРБАМАТОМ СЕРЕБРА В ХЛОРОФОРМЕ
Материалы, реактивы и оборудование. Прибор для отгонки и поглощения мышьяка; фотоэлектроколориметр или спектрофотометр; весы лабораторные с наибольшим пределом взвешивания 200 г и 1 кг; воронка Бюхнера; воронки стеклянные; эксикатор; колбы конические вместимостью 250 см3; конус взаимозаменяемый КШ-29/32; поглотительный прибор с пористой стеклянной пластинкой № 2; трубки стеклянные цилиндрические и дрот глухой; палочки стеклянные; колбы мерные вместимостью 100 и 1000 см3; пробирки мерные вместимостью 100 см3; фильтры обез-зольные с синей лентой диаметром 7 см; вата; этанол ректификованный; фенолфталеин, раствор в этаноле массовой долей 0,1 %; соляная кислота плотностью 1,19 г/см3 и ее раствор молярной концентрацией 0,3 моль/дм3; раствор серной кислоты молярной концентрацией 1 моль/дм3; кислота азотная (1 : 1); вода дистиллированная; раствор иодида калия 150 г/дм3; раствор дихлорида олова 200 г/дм3 в соляной кислоте плотностью 1,19 г/см3; цинк гранулированный; хлорид кальция двуводный гранулированный, прокаленный; сульфат натрия безводный, прокаленный; раствор гидроксида натрия молярной концентрацией 2 моль/дм3; гидроксид калия гранулированный; ангидрид мышьяковистый; натрий мышьяковистый двузамещенный, семиводный или стандарт-титр; сульфат меди пятиводный, дважды перекристаллизованный и высушенный в эксикаторе до постоянной массы; моноэтанол-амин, ч., или гексаметилентетрамин (уротропин), ч., натрия N, N-диэтилдитиокарбамат; нитрат серебра; ацетат свинца (II), раствор 150 г/дм3; хлороформ.
Приготовление реактивов. Диэтилдитиокарбамат серебра. Раствор, содержащий 1,7 г нитрата серебра в 100 см3 воды, помешивая, медленно приливают к раствору, содержащему 2,3 г диэтилдитиокарбамата натрия в 100 см3 воды. При этом тем
530
пература растворов должна быть не более ИКС. Образовавшийся лимонно-желтый осадок диэтилдитиокарбамата серебра отфильтровывают на воронке Бюхнера и тщательно промывают водой с несколькими каплями соляной кислоты молярной концентрацией 0,3 моль/дм3 до исчезновения реакции на серебро. Осадок разрыхляют стеклянной палочкой и высушивают в эксикаторе над хлоридом кальция в темноте до постоянной массы при комнатной температуре. Сухое вещество хранят в темноте не более 6 мес.
П огл ощ а ю ш и й р а ст вор. 0,2 г диэтилдитиокарбамата серебра растворяют в^ 100 см3 хлороформа, в который предварительно добавлен 1,0 см3 моноэтаноламина или 1,0 г уротропина. Раствор с уротропином используют только для продуктов с массовой долей мышьяка более 0,1 мг/кг. Для работы используют только свежеприготовленный раствор.
Все основные реактивы допускается хранить в течение года.
Построение калибровочного графика. В шесть цилиндров или поглотительных приборов с пористой стеклянной пластинкой наливают по 10 см3 поглощающего раствора. В трубки 2 с расширением (рис. 5.10) помещают слой ваты, пропитанной ацетатом свинца, затем 5—6 гранул гидроксида калия. Отверстие трубки закрывают слоем ваты; пропитанной ацетатом свинца.
В шесть реакционных колб вместимостью 250 см3 вносят соответственно 0; 0,25; 0,5; 1,0; 1,5; 2,0 см3 рабочего раствора мышьяка, т. е. соответственно 0; 2,5; 5; 10; 15; 20 мкг мышьяка.
В каждую реакционную колбу добавляют 25 см3 соляной кис; лоты плотностью 1,19 г/см3, 2,5 см3 раствора иодида калия, 1,5 см3 раствора дихлорида олова. Объем колбы доводят водой до 100 см3, приливают 1 см3 раствора сульфата меди концентрацией 10 г/дм3, все тщательно перемешивают и выдерживают 10—15 мин. Затем в каждую реакционную колбу вносят 5 г гранулированного цинка, после чего быстро надевают на колбу соединительную трубку с капилляром, конец которого погружен в цилиндр с поглощающим раствором или поглотительным прибором с пористой стеклянной пластинкой, в который налит поглощающий раствор. Образовавшийся мышьяковистый водород отгоняют в течение 1 ч. В случае помутнения поглощающего раствора его фильтруют через ватный тампон, помещенный в носик воронки.
Оптическую плотность растворов сравнения измеряют по отношению к поглощающему раствору на фотоэлектроколориметре при длине волны Хтах = (520 ± 10) нм в кюветах с расстоянием между рабочими гранями 20 мм или спектрофотометре при длине волны 520 нм в кювете с расстоянием между рабочими гранями 5 или 10 мм.
Калибровочный график строят, откладывая по оси абсцисс массы мышьяка (мкг), введенные в растворы сравнения, а по оси ординат — соответствующие значения оптической плотности.
531
Порядок проведения анализа. При приготовлении испытуемых и контрольного растворов золу, полученную сухой минерализацией (см.4.1.1). осторожно растворяют в 30—50 см3 раствора соляной кислоты концентрацией 0.3 моль/дм3 и, избегая разбрызгивания, добавляют соляную кислоту плотностью 1,19 г/см3 из расчета 4 см3 кислоты на 1 г оксида магния, добавленной в пробу перед озолением. Если зола плохо растворяется, ее подогревают с соляной кислотой на водяной бане. Полученный раствор золы используют для последующего количествен
ного определения.
Раствор, полученный в результате мокрой минерализации или кислотной экстракции, используют без дополнительной обработки.
Контрольный раствор готовят из контрольной пробы, исполь
зуя все реактивы и растворы, аналогично приготовлению испытуемых растворов. Контрольную пробу готовят, используя применяемые для минерализации реактивы, прибавляя их в тех же количествах, объемах и последовательности, что и при минерализации пробы, но без добавления испытуемой пробы.
Прибор для отгонки и поглощения мышьяка собирают в соответствии с рис. 5.10. Прибор состоит из реакционной колбы 1 вместимостью 250 см3, соединительной трубки 2 (внешний диаметр 4 мм) с расширением, шлифом и капилляром и цилиндра 3 (внутренний диаметр 11мм) с поглощающим раствором (или поглотительного прибора 4 с пористой стеклянной пластинкой 5 для поглощения раствора). Перед употреблением прибор промывают разбавленной азотной кислотой (1 : 1), а затем водой.
Для проведения измерений в реакционные колбы прибора вносят испытуемый и контрольный растворы, затем операции повторяют аналогично операциям, проводимым при построении калибровочного графика.
По полученным значениям оптической плотности графика находят массу мышьяка.
Содержание X (мг/кг) и массовую концентрацию Х} (мг/дм3) мышьяка вычисляют по формулам
Рис. 5.10. Прибор для отгонки и поглощения мышьяка
/И] — т2
(5.40)
т
532
где т}. т. — массы мышьяка соответственно в испытуемом и контрольном растворах."найденные по калибровочному графику, мкг; т — масса навески продукта. взятая для минерализации, г; Г—объем продукта, взятый для минерализации. см;.
Вычисления проводят до третьего десятичного знака.
6.3. ВИЗУАЛЬНОЕ КОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ РТУТИ
Материалы, реактивы и оборудование. Аппарат для встряхивания колб; баня водяная; бюретка; весы лабораторные общего назначения с наибольшим пределом взвешивания 200 и 1000 г; воронки; воронка делительная; колбы конические; колбы мерные вместимостью 100, 500 и ЮООсм3; палочки стеклянные; пипетки; пробки мерные; пробирки для колориметрирования из бесцветного стекла; стаканы вместимостью 50 см3; штатив химический; цилиндры мерные вместимостью 25, 50, 100, 250, 500, ЮООсм3; сосуд стеклянный цилиндрический вместимостью 5дм3 (для приготовления взвеси иодида меди); холодильник ХШ-1-300, 400, 500-29/32 ХС; бумага индикаторная универсальная (pH 1 — 10); бумага фильтровальная лабораторная; фильтры обеззоленные диаметром 5 и 9 см (синяя лента); ацетон; хлорид бария и его раствор концентрацией 200 г/дм3; вода дистиллированная; иод кристаллический, растворы 2,5 и 3,5 г/дм3 в растворе иодида калия 30 г/дм3; иодид калия, раствор 30 г/дм3; калий надсернокислый и его раствор концентрацией 10 г/дм3; кислота азотная ос.ч., кислота серная ос.ч.; сульфат меди пятиводный и его растворы концентрациями 100 и 200 г/дм3; мочевина и ее раствор концентрацией 200 г/дм3; сульфат натрия безводный, раствор 10 г/дм3 и насыщенный; сульфат натрия, раствор 1,25 моль/дм3; этанол; дихлорид ртути или стандарт-титр; дииодид ртути или стандарт-титр; взвесь иодида меди; основной и стандартный растворы ртути; составной раствор сульфата меди и сульфита натрия.
Приготовление реактивов. Основной раствор ртути. 0,135 г хлорида ртути (II) [или 0,226 г иодида ртути (II)] количественно переносят в мерную колбу вместимостью 1 дм3 и доводят до метки при постоянном перемешивании раствором иода концентрацией 2,5 г/дм3.
Аналогично готовят основной раствор ртути из стандарт-титра. Вскрывают стандарт-титр, содержащий 0,135 г хлорида ртути (II) [или 0,226 г иодида ртути (II)], количественно переносят в мерную колбу вместимостью 1 дм3 и доводят до метки раствором иода кон-
533
центранией 2.5 г/дм3. 1 см3 основного раствора содержит 100 мкг ртути. Раствор хранят в склянке с притертой пробкой в защищенном от света месте в течение 3 мес.
Стандартные растворы рту т и. Непосредственно перед определением ртути 1 см1 основного раствора ртути помещают в мерные колбы вместимостью 100 и 200 см3 и доводят объем в обеих колбах до метки раствором соляной кислоты молярной концентрацией 0,5 моль/дм3. Полученные растворы содержат соответственно 1 и 0,5 мкг ртути в 1 см3.
Составной раствор сульфата меди и сульфита натри я. Смешивают растворы сульфата меди концентрацией 100 г/дм3 и сульфита натрия молярной концентрацией 1,25 моль/дм3 в соотношении 1 : 5. Смесь перемешивают в конической колбе вместимостью 100 см3 до получения прозрачного раствора и используют немедленно. При появлении мути раствором пользоваться нельзя.
Иодид меди (взвесь). Для получения 1 дм3 взвеси 212 г иодида калия растворяют в 2 дм3 воды, смешивают с 800 см3 раствора сульфата меди концентрацией 200 г/дм3 в стеклянной банке вместимостью не менее 5 дм3 и оставляют до полного осаждения осадка (от 30 до 50 мин). С образовавшегося осадка декантируют жидкость. Осадок многократно промывают водой (по 2—Здм3) до светло-желтого цвета надосадочной жидкости. Для удаления розового оттенка осадок отбеливают. Для этого в сосуд со взвесью добавляют сначала от 10 до 20 см3 раствора сульфита натрия концентрацией 1,25 моль/дм3, а затем — насыщенный раствор сульфата натрия от 10 до 20 см3 для коагуляции осадка. Если взвесь отбелена недостаточно и плохо осаждается, следует еще раз добавить перечисленные растворы. Надосадочную жидкость сливают декантацией, а осадок переносят на двойной фильтр, сделанный из лабораторной фильтровальной бумаги и плотно уложенный в воронку диаметром 250 мм, промывают на фильтре водой до почти отрицательной реакции на сульфат-ион (проба фильтрата с раствором хлорида бария не должна давать осадка). Фильтр прокалывают стеклянной палочкой, осадок смывают водой в мерную колбу и доводят объем до 1 дм3.
Взвесь считается приготовленной правильно, если имеет белый цвет и оседает в течение 15—20 мин. Хранят взвесь в темной склянке не более 1 мес.
Порядок проведения анализа. При определении колориметрическим методом предварительно проводят деструкцию пробы закрытым или открытым способом. Для анализа используют измельченную пробу массой 40 г. Параллельно ставят контроль на реактивы, учитывая его при расчете конечного результата.
Закрытый способ. Проведение деструкции закрытым способом рекомендуется при анализе жировых продуктов с использованием установки, схема которой приведена на рис. 5.11.
534
Рис. 5.11. Устройство для деструкции:
I — 1ьник: 2 капельная аоропка. а -реакционная колба
В реакционную колбу установки помещают исследуемую пробу, добавляют реактивы, количество которых указано в табл. 5.1 I. и соединяют с обратным холодильником. Колбу выдерживают 15— 20 мин при комнатной температуре, а затем помещают на водяную баню температурой около 70 °C. постепенно доводя ее до температуры кипения. Время пребывания колбы на водяной бане зависит от вида продукта. В случае бурного течения реакции (наполнение реакционной колбы и холодильника бурыми парами оксидов азота) через боковую капельную воронку или холодильник в колбу добавляют порциями по 5—10 см3 кипящей воды, но
так, чтобы общий объем приливаемой воды не превышал 30— 50 см3. Деструкцию проводят до полного просветления придонного слоя. По окончании деструкции колбу снимают с бани и охлаждают в течение 15 мин, а затем промывают 50 см3 горячей воды через холодильник, который затем отсоединяют от колбы. Горячий деструктат фильтруют в колбу вместимостью 500 см3, в которую предварительно наливают 20 см3 раствора мочевины через увлажненный водой двойной бумажный фильтр, уложенный в воронку диаметром 100—150 мм. Реакционную колбу и фильтр несколько раз промывают кипящей водой. Общий объем фильтрата и смывных вод должен составлять около 300 см3.
Открытый способ. Деструкцию проводят в термостойкой конической колбе вместимостью 750 см3. Пробу равномерно распределяют по дну колбы, добавляют реактивы, количество которых указано в табл. 5.11.
В колбу с пробой вносят последовательно этанол, воду и азотную кислоту. Колбу закрывают воронкой диаметром 25 мм, содержимое перемешивают и выдерживают при комнатной температуре в зависимости от вида продукта от 20 до 30 мин. Серную кислоту наливают в стакан вместимостью 50 см3 и осторожно по каплям добавляют в колбу с пробой через воронку. Скорость внесения серной кислоты должна постоянно поддерживать реакцию разложения азотной кислоты, но при этом необходимо следить за тем, чтобы не происходило выделения оксидов из колбы, так как при бурном течении реакции возможны потери ртути. По окончании внесения серной кислоты колбу оставляют в вытяжном шкафу при комнатной температуре до прекращения выделения бурых паров оксидов азота, но не более чем на 30 мин. Затем колбу помещают на водяную баню. В первые 15 мин пребывания колбы на водяной
535
Г а б л и п а 5.11
Рекомендации по проведению деструкции проб при колориметрическом определении ртути
Операция и вид продукта Масса навески, г Добавление, см' Выдерживание при комнатной температуре, мин Добавление п концентри- : Выдержива-рованной ! Н|1с П!1И ком" серной кие- ! натнои тсмпс' лоты.см' ; РЭТУРС, мин Температура и время нагрева на водяной бане,'С
этанола воды концентрированной азотной кислоты
Деструкция закрытым способом
Контроль на ре- ~ — 15’° 15-° “ 15.0 |5 70 °C - 15 мин
активы1 100 °C — 90 мин
Растительные 40,0 - - 40,0 разбавленной (1 : 2) азотной кис- 20,0 по кап- - 100 °C-60 мин
масла, марга- лоты перемешивают, выдерживают Лям до ПОЛНОГ() ос.
рин, жировые продукты2 при комнатной температуре 30 мин, |,рТпР1„|(| пп„ DVl.'iCllIlZI 111/11 помещают на водяную баню 70 °C, донного слоя выдерживают до прекращения вспенивания, а затем вновь помешают на баню 100°С на 60 мин, охлаж.ниог до комнатной температуры Деструкция открытым способом
Контроль на ре- - 1,0 15,0 15,0 порциями по 20 20,0 по кап-До полного 100 °C-45 мин
активы 1 2—3 см3 лям прскрашс-
ния выделения океидов азота
Продолжение
Операция и вид продукта Масса навески, г Добавление, см’ Выдерживание при комнатной температуре, мин Добавление концентрированной серной кислоты, см1 Выдерживание при комнатной температуре, мин Температура и время нагрева на водяной бане,°C
этанола воды концентрированной азотной кислоты
Мясо и мясо птицы, печень, почки и другие внутренние органы, мясные продукты и продукты мяса птицы, яйцепро-дукты3 40,0 1,0 15,0 15,0 порциями по 2-3 см3 20 20,0 по кап- лям До полного прекращения выделения оксидов азота 100 °C — 45 мин
1 Количество реактивов в контроле должно быть таким же, как и в исследуемом продукте.
2 Навеску жиров перед деструкцией предварительно обрабатывают 10,0 см3 надсернокислого натрия и перемешивают.
3 При наличии в продукте более 30 % жира перед деструкцией в колбу вносят 10,0 см3 надсернокислого калия. Объем серной кислоты в этом случае увеличивается до 25—30 см3.
бане возможно бурное разложение азотной кислоты с большим выделением оксидов азота. В это время необходимо особенно внимательно следить за ходом реакции. При бурном течении реакции (выделение окислов азота или сильное ценообразование) в колбу добавляют кипящую воду порциями но 5—10 см3 так, чтобы обшиб объем добавленной воды не превышал 30—50 см3, или на 3—5 мин снимают ее с бани.
Деструкцию проводят до полного осветления придонного слоя жидкости в колбе, но нс менее 45 мин. Колбу снимают с бани и горячий деструктат фильтруют в колбу вместимостью 500 см3, в которую предварительно наливают 20 см3 раствора мочевины через увлажненный водой двойной бумажный фильтр, уложенный в воронку диаметром 100—150 мм. Колбу из-под де-структата и фильтр несколько раз промывают кипящей водой. Общий объем деструктата и промывных вод доводят приблизительно до 300 см3.
При проведении количественного анализа в колбу с охлажденным деструктатом добавляют 15 см3 взвеси иодида меди. Содержимое колбы перемешивают три раза с интервалом 5 мин и оставляют до полного осаждения осадка. Если образующийся осадок окрашен в ярко-розовый или кирпично-красный цвет, что свидетельствует о содержании ртути в образце более 25 мкг, добавляют еще 15 см3 иодида меди или анализ повторяют, соответственно уменьшив навеску образца и количество реактивов для деструкции.
Через 1 ч максимально возможную часть надосадочной жидкости сливают, стараясь не взмутить осадок, и отбрасывают. К осадку добавляют 5 см3 раствора сульфата натрия концентрацией 10 г/дм3, взбалтывают и переносят на увлажненный водой однослойный бумажный фильтр (синяя лента), плотно уложенный в воронку диаметром не более 35 мм. Края фильтра должны выступать из воронки не более чем на 5 мм. Колбу из-под осадка несколько раз ополаскивают раствором сульфата натрия концентрацией 10 г/дм3 и сливают на тот же фильтр, с тем чтобы весь осадок был перенесен на фильтр.
Когда вся жидкость профильтруется, осадок на фильтре промывают 10 см3 смеси ацетона с раствором сульфата натрия концентрацией 10 г/дм3 в соотношении 1:1. После того как смесь полностью пройдет через фильтр, осадок и фильтр вновь промывают раствором сульфата натрия концентрацией 10 г/дм3. Осадок проливают до исчезновения желтой окраски промывных вод и до pH не менее 5 (по универсальной индикаторной бумаге). Промывные воды отбрасывают. Остаток жидкости из узкой части воронки удаляют полоской фильтровальной бумаги, а осадок подсушивают на фильтре в течение 15 мин. Затем его обрабатывают на фильтре раствором иода концентрацией 538
3.5 г/дм3 в зависимости от цвета осадка. Для этого необходимый объем раствора иода, указанный в табл. 5.12, помещают в цилиндр или пробирку и обработку фильтра ведут небольшими порциями, нанося жидкость по краю фильтра. Полученный фильтрат доводят до выбранного объема. Фильтрат можно хранить в пробирках с притертыми пробками в темном месте в течение суток.
Т а б л и ц а 5.12
Ориентировочная схема визуального колориметрического определения ртути
Цвет осадка Примерное содержание ртути в образце, мкг Объем раствора иода концентрацией 3,5 г/дм' для растворения ртути. см’ Объем, взятый для колориметрирования, см''
Белый 0.0-0,5 6,0 6,0
Белый 0.5-5,0 10.0 3.0 и 6.0
Белый с розовым оттен- 5,0-15,0 15,0 0.5; 1,0 или 2,0
ком
Бледно-розовый 5,0—25,0 25,0 0,5; 1,0 или 2,0
Ярко-розовый Более 25,0 25.0 0.5 и 1,0
Содержание ртути (мкг/г) вычисляют по формуле
х = (w2 - mQK
(5-42)
где — массы ртути соответственно в аликвотном объеме, взятом для колориметрирования. и в контрольном опыте, определенные по градуировочной шкале, мкг; И—объем раствора иода, использованный для растворения ртути, см3; V= 3,5 г/дм3; И — аликвотный объем, см3; т — масса образца, взятая для деструкции, г.
Вычисление проводят до третьего десятичного знака.
Для визуального колориметрического определения ртути раствор или его аликвотный объем, подобранный в соответствии с табл. 5.12, помещают в пробирки и доводят объем раствором иода концентрацией 2,5 г/дм3 до 6 см3. Затем прибавляют из бюретки по 3 см3 составного раствора, закрывают пробками, перемешивают и выдерживают в темном месте (не менее 15 мин) до полного осаждения тетраиодмеркурата меди.
Колориметрическое определение ртути проводят путем визуального сравнения цвета осадка в пробирках с пробой с цветом осадка в пробирках градуировочной шкалы (табл. 5.13).
539
Т а б л 11 ц а 5.13
Рекомендуемая схема приготовления градуировочной шкалы для визуального
(колориметрического) определения ртути
Объем стандартного раствора , Объем раствора ио.та кон-ртхти. см пентрацпей 2.5 г дм', см'
_____:т
Содержание ртути, мкг
0.00 6.00 0.00
0.15 5.85 0.15
0.25 5.75 0.25
0.50 5.50 0,50
0.75 5.25 0.75
1.00 5.00 1.00
1.25 4,75 1.25
1.50 4,50 1.50
1,75 4.25 1,75
2.00 4,00 2.00
Построение градуировочной шкалы. В мерные пробирки для колориметрирования вносят точные объемы стандартного раствора ртути и раствора иода, указанные в табл. 5.13. Затем добавляют из бюретки по 3 см3 составного раствора, закрывают пробками и тщательно перемешивают. Выдерживают в защищенном от света месте (не менее 15 мин) до полного осаждения осадка тетраиод-меркурата меди. Для этого пробирки располагают под углом 25—30° так, чтобы осадок оставался на дне пробирки, а надосадочная жидкость переместилась к пробке.
Полученные результаты сводят в таблицу рекомендуемой формы (см. ниже). Анализируют, дают оценку мяса и мясных продуктов по содержанию токсичных элементов. При исследовании вторичных продуктов убоя делают вывод о преимущественной локализации токсичных элементов в органах и тканях убойных животных. При этом указывают используемый метод определения.
Содержание токсичных элементов, мг/кг
Наименование и краткая характеристика образцов РЬ As Cd Hg Си Zn
пдк фак-ти-чес-кое пдк фак-ти-чес-кое пдк фак-ти-чес-кое пдк фак-ти-чес-кое пдк фак-ти-чес-кое пдк фак-ти-чес-кое
Говядина Свинина Баранина Мясо птицы Субпродукты:
I категории
II категории Мясные продукты
540
Лабораторная работа №10 ЭКСПРЕСС-ОПРЕДЕЛЕНИЕ РАДИОНУКЛИДОВ МЕТОДАМИ РАДИОМЕТРИИ
Цель работы. Приобрести практический навык радиационного контроля мяса, вторичных продуктов убоя скота, мясных продуктов экспресс-методами радиометрии.
Задачи. Отбор, подготовка проб и экспрессное определение суммарной объемной (ОА) и удельной (УА) активности у- и Р-излучающих нуклидов.
Объекты исследования. Мясо и кость различных видов убойных животных и птицы; субпродукты I и II категорий, колбасные изделия, продукты из свинины, говядины, баранины, мяса птицы, мясные консервы, пищевые животные жиры, яйца.
Материалы, реактивы и оборудование. Радиометр СРП-68-Ol, КРК.-1, РКБ4-1еМ или другой марки; кювета эмалированная; стеклянные стаканы вместимостью 1 дм3; пакеты для радиоактивных отходов; полиэтиленовые пакеты; раствор формалина массовой долей 4—5 %.
Методические указания. Для практического освоения методов контроля радиационной загрязненности продуктов питания следует уяснить ряд традиционных понятий и определений радиометрии.
Радиоактивность — испускание ионизирующих излучений при самопроизвольном превращении радиоактивных ядер.
Активность радионуклида — отношение числа dN самопроизвольных превращений ядер данного радионуклида, происходящих за интервал времени dt, к этому интервалу времени;
А = dN/dt. (5.43)
Единица активности — беккерель (Бк) — одно ядерное превращение в 1 с.
В зависимости от реального состава радионуклидов в измеряемых пробах разработаны два варианта экспресс-метода; для определения объемной (ОА) и удельной (УА) активности у- и {3-излу-чающих нуклидов.
Экспрессное определение объемной и удельной активности у-излучающих нуклидов в пищевых продуктах проводят с помощью радиометра СРП-68-01.
Экспрессное определение объемной и удельной активности Р-излучаюших нуклидов в воде, продуктах питания, продукции животноводства проводят методом прямого измерения толстых проб.
Методика обеспечивает достоверность и единство измерений объемной (ОА) и удельной (УА) активности ^-излучающих нуклидов с максимальной энергией более 100 кэВ при использовании радиометров КРК-1, РКБ4-1еМ, КРВП-ЗАБ, ДП-100, СПР-68-01 и их аналогов для массовых экспрессных измерений количества
541
радиоактивных веществ в различных объектах с содержанием нс менее 1 • 10 ~9 Ки/кг и 1 • 10“10 Ки/дм3 при контроле воды. Предел погрешности измерения равен 50%.
Перед измерением предварительно проводят подготовку приборов в соответствии с прилагаемыми инструкциями и с учетом правил и требований техники безопасности по их эксплуатации.
Предел основной относительной погрешности (5) при измерении объемной и удельной активности в пробах с постоянным составом нуклидов для наиболее чувствительного [3-радиометра РКБ4-1еМ определяется по формуле (5.44) и составляет 40 %:
8 = 8о + -ЮО, (5.44)
(Ц]
где 8(| — погрешность методики исходного раствора и приготовления образцовой меры: t/() — удельная (объемная) активность образцов меры; ^--удельная (объемная) активность образцовой меры.
При контроле проб воды, растительности, продуктов питания (кроме мяса, молока, грибов), почвы предел основной относительной погрешности определен для следующего состава нуклидов:
137Cs —2 %;
144Се + 144Pv - 51 %;
1()6Ru + 106Rh — 12%;
95Zr + 95Nb — 31 %;
90Sr + 90Y —4%.
При контроле проб мяса, молока, грибов предел основной погрешности определен для следующего состава нуклидов:
137Cs —30%;
144Се + I44Pv- 36%;
106 Ru+ 106Rh-9%;
95Zr + 95Nb — 22 %;
90Sr + 90Y—3%.
При измерении суммарной объемной или удельной активности |3-излучающих нуклидов в пробах с неизвестным процентным составом нуклидов предел погрешности для радиометра РК.Б4-1еМ с блоком детектирования БДЖБ-02 составляет 60 %.
Подготовка проб. Объективность и точность результатов анализа зависят от правильного выполнения отбора и подготовки проб к анализу.
Образцы проб отбирают от однородной партии.
Продукция считается однородной по уровню загрязнения, если результаты измерений, проведенных в разных точках, различаются не более чем в два раза.
Для проведения лабораторных исследований из объединен-542
ной пробы беруг в необходимом количестве среднюю пробу, которая должна характеризовать радиоактивное загрязнение всего образца.
Пробы мяса (без жира) от туш или полутуш отбирают кусками по 30—50 г в области четвертого-пятого шейного позвонка, лона гки. бедра и толстых частей спинных мышц. Общая масса пробы должна составлять 0,2—0,3 кг.
Масса костей для исследования должна составлять 0.3—0.5 кг (позвоночник и второе-третье ребро).
Пробы из внутренних органов животных отбирают массой 0,1—0,2 кг частично (печень, почки, селезенка, легкие) или целиком (щитовидная железа).
Пробы мяса птицы отбирают в количестве 1/4 тушки (куры, индейки, утки, гуси) или одной целой тушки (цыплята).
Масса проб мясных полуфабрикатов, готовых мясных продуктов и колбасных изделий соответствует массе проб мяса.
Яйца отбирают на птицефабриках или птицефермах по 5 —10 шт. от одной фермы, от упакованных партий — по Зшт. от каждой тысячи яиц.
Количество проб продуктов, используемых для лабораторного анализа, определяется величиной партии и составляет при массе партии от 1 до 500 кг — 1 образец, от 500 кг до 3 т — 2 образца, от 3 до 5 т — 3 образца, от 5 до 10 т — 5 образцов, от 10 до 20 т — 6 образцов, от 20 т и более — 10 образцов.
Отобранные средние пробы взвешивают, упаковывают в чистую сухую тару, соответствующую виду продукта (целлофан, пергамент, полиэтиленовые пакеты, стеклянная или полиэтиленовая посуда), наклеивают этикетки с указанием продукта или сырья, мощности дозы его у-излучения, массы, даты и места отбора. Пробы мяса, рыбы, молока при длительном транспортировании консервируют раствором формалина массовой долей 4—5 %.
1. ЭКСПРЕСС-ОПРЕДЕЛЕНИЕ ОБЪЕМНОЙ И УДЕЛЬНОЙ АКТИВНОСТИ у-ИЗЛУЧАЮЩИХ НУКЛИДОВ С ПОМОЩЬЮ РАДИОМЕТРА СРП-68-01
Порядок проведения анализа. После подготовки к работе радиометра СРП-68-Ol в соответствии с инструкцией к прибору проводят измерения в следующей последовательности:
измеряют фон в свинцовой защите не менее 5 см (при уровне фона меньше 25 мкР/ч допускается проводить измерения без защиты). При измерении фона в свинцовой защите на дно колодца ставят банку с пробой. При использовании сосуда типа Маринелли его надевают на детектор и записывают показания N, (мкР/ч).
В пробу (по центру банки) погружают детектор (щуп) радиометра, покрытый полиэтиленовым пакетом, чтобы нижний
543
торец щупа не доходил до дна банки на 2—3 см. При использовании сосуда Маринелли последний с пробой устанавливают на детектор и записывают показания радиометра. обусловленные фоном и активностью измеряемой пробы N (мкР/ч). Объемную и удельную активность пробы рассчитывают по формуле
* -
(5.45)
где Р — коэффициент псрсечета. Для радиометра СРП-68-01 для всех продуктов при измерении в трехлитровом сосуде Маринелли значение Р составляет 2,2 • 10~3 (8,2-10), при измерении животных или туш —6,5 10~’ (2,4 • 10s) дм3 (кг) мкРч-1 • Бк”1 [или дм3 (кг) • мкР ч1 • Ки”1].
2. ЭКСПРЕСС-ОПРЕДЕЛЕНИЕ ОБЪЕМНОЙ И УДЕЛЬНОЙ АКТИВНОСТИ Р- ИЗЛУЧАЮЩИХ НУКЛИДОВ МЕТОДОМ ПРЯМОГО ИЗМЕРЕНИЯ ТОЛСТЫХ ПРОБ
Перед анализом проводят подготовку радиометров КРК-1 или РКБ4-1еМ к работе в соответствии с инструкцией, прилагаемой к прибору. Определяют скорость счета фона радиометра с пустой измерительной кюветой или в измерительной кювете с «чистой» фоновой пробой. Кювету с подготовленной пробой помещают в свинцовую защиту на вторую полку сверху измерительной этажерки (для ДП-100 и КРК-1). Включают радиометр и определяют скорость счета N' пробы несколько (л) раз, вычисляют ее среднее значение N{.p .
Объемную [Бк/дм3 (Ки/дм3)] или удельную [Бк/кг (Ки/кг)] активность пробы рассчитывают по формуле
Q = —Н;—-
(5.46)
где /’—чувствительность радиометра в измеряемой пробе.
Чувствительность Р радиометров при измерении ОА и УА проб мяса и мясных продуктов, яиц, яичного порошка, питьевой воды, водопроводной, колодезной воды (радионуклидный состав 137Cs, 134Cs 100 %) составляет для радиометров ДП-100 — 1,7- IO”5 (0,6 • 106); КРК-1 — 1,6 - 10-4 (0,6- 107); КРВП-ЗАБ-3,2 • 10“4 (1,2 • 107); РКБ4-1еМ с блоком БДЖБ-07 - 2,5 • 10~5 (0,9 • 106) дм3 (кг) • с^'Бк"'1 [или дм3 (кг) • с-1 Ки-1].
Полученные результаты рекомендуется оформить в виде таблицы.
544
-к ' ек’..урч1.. ,, Li.k -ь (. ффнф \ j
Ч \ I ' С ! I 1Ч - I I < ч f- < • [ . ! . , [ ] |
,. , ? i: я: । \.сг117_ и .. ' , - , •
/I ч - ; ’ 1 . * ' - - 1 ' '
t h ';' г ’ t ч 7 L : I . ч , ‘ ’ р -• i ц ।; , , .
,<’! х [J \;1-,Г Ч \ \|И " I
НО' 4i' I, pt \ ЛМ ч
VlC.lbflir: (1107. \| П.1Я I ЯКФЯЯ(‘КЯ| 7- 11 I. Л ЯП Ю: 1111 \ HVkHKKK.
\
v:
\;
' 4c.lbH;i!l (Ol)l,K\i I Ki Я ) ЯКГИИНОС I I, Д Il J l\ 4Ill<)iil!l\ H\ K.lll Ю к
,v: л;
В таблице фиксируют массу и характеристику образцов мж пых продуктов, отобранных для контроля удельной пли обьс* ной активности у- и р-излучаюших нуклидов, результаты нз\ь рений и расчетов. Полученные данные анализируют, делают заключение о радиационном загрязнении или радиационной безопасности исследуемых мясных продуктов, сравнивая полученны? значения с предельно допустимыми.
Лабораторная работа №11
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ СУММАРНОЙ р-РАДИОАКТИВНОСТИ МЯСА, КОСТЕЙ И МЯСНЫХ ПРОДУКТОВ ПО УДЕЛЬНОЙ АКТИВНОСТИ ЗОЛЬНЫХ ОСТАТКОВ
Цель работы. Приобрести практический навык в определении радионуклидов путем радиометрии зольных остатков проб.
Задачи. Отбор и подготовка проб, определение удельной суммарной ^-радиоактивности по удельной активности зольных остатков.
Объекты исследования. Образны мяса и вторичных продуктов убоя различных видов животных и птицы: мясные продукты в ассортименте: яйца и яйцепродукты.
Материалы, реактивы и оборудование. Радиометр; стандартные алюминиевые подложки: весы аналитические или торзионные; полиэтиленовые пакеты: нож: мясорубка: тигли; чашки фарфоровые: эксикатор с прокаленным CaCI2: ступка с пестиком: весы технические; шпатели; стеклянные палочки; асбестовые пластинки: газовые горелки или электроплитки; электропечь: сушильный шкаф на 105—ИО °C: респиратор «Лепесток»; вата; марля; инфракрасная лампа; этанол: эталонный препарат 40К (200—-300 мг KCI).
545
Методические указания. При малой радиоактивности объектов экспресс-методом обнаружить ее не удается, поэтому суммарную [3-активность определяют по зольному остатку пробы. Предвари тельно пробу концентрируют (озоляют).
Навеску полученной золы определенной массы (200 или 300 мг) помещают на стандартную алюминиевую подложку площадью 2,5 см2, определяют скорость счета Л'о (имп/мин) и рассчитывают суммарную удельную [3-радиоактивность золы, а затем через коэффициент озоления и суммарную удельную [3-радиоактивность пробы.
Суммарная [3-радиоактивность отражает удельную радиоактивность (Ки/кг, Ки/дм?) объектов ветеринарного надзора, обусловленную смесью [З-излучающих изотопов неизвестного состава. Определение ее позволяет быстро (в течение дня) получить сведения об уровне загрязнения объекта радиоактивными веществами.
Входящий в состав исследуемых объектов калий за счет содержания в нем естественного радиоактивного изотопа калия-40 (0,0119%) создает удельную радиоактивность порядка п • 10~9 Ки/кг сырой пробы. Суммарная [3-активность объектов при дефиците калия в пробе обусловливается повышенным содержанием искусственных радиоизотопов — продуктов ядерного деления, среди которых могут быть радиоизотопы высокой радиотоксичности (например, иод-131, стронций-90, цезий-137 и др.). В этом случае уровень суммарной удельной радиоактивности объекта будет выше того, который обусловлен изотопом калий-40. Поэтому, определив суммарную [3-активность пробы, необходимо сопоставить полученные данные с расчетной активностью пробы за счет калия-40 (табл. 5.14).
Таблица 5.14
Удельная [3-активность (п • 10 “ Ки/кг) некоторых кормов и продуктов, обусловленная калием-40
Продукт
Молоко коровье Говядина Свинина
Мясо кролика Рыба
Удельная [3-активность
1,0
2,5
1,9
2,9
1.9
Подготовка проб. Образец пробы должен быть типичным для объекта, а масса (объем) — достаточной, чтобы после концентрирования получить количество золы, необходимое для определения суммарной [3-радиоактивности и проведения радиохимического анализа. Сроки отбора проб определяются видом продукта, сезонностью его получения и уровнем радиоактивности (табл. 5.15).
546
Т а блиц а 5.15
Сроки и нормы отбора проб для исследования на радиоактивность
Ооъект 1 Сроки отбора проб Масса пробы, кг
Мясо Весна и осень 2-3
Кости То же 0.5
Концентрированные корма Осень (по мере поступления) 1-2
Молоко Ежеквартально 5—6
Рыба свежая По мере поступления 3,0
Вола Весна и осень Необходимый
объем
При отборе проб следует соблюдать установленные правила.
Пробы мяса берут из нежирной части туши, не снижая ее товарных качеств. Для анализа можно использовать мышцы шеи или конечностей. Однотипность отбираемых проб мяса позволяет сопоставить получаемые результаты при исследовании мяса животных различных видов, пород и возрастов.
Однотипность следует соблюдать и при отборе проб костей, так как отложения остеотропных радиоизотопов (например, стронция-90) неравномерные не только в различных костях скелета. но и в разных участках одной и той же кости. Для исследования удобно брать последние ребра, препарируя их с плевральной поверхности, что позволяет сохранить товарный вид туши.
При небольшой массе птицы на анализ берут 3—4 тушки, отделяют мясо от костей и делают среднюю пробу. Мышцы и кости исследуют отдельно.
Яйца отбирают от кур из одного птичника, содержащихся в одинаковых условиях и при одинаковом рационе. Для анализа берут 2—4 десятка яиц, объединяют в среднюю пробу. Всю пробу яиц разделяют перед анализом на съедобную часть (белок и желток) и скорлупу, которые исследуют отдельно. Загрязненное с поверхности яйцо включать в пробу не следует. Пробу яиц транспортируют в целом виде в упаковке, обеспечивающей их сохранность.
Прием и предварительную обработку доставленных в лабораторию проб проводят в специальном помещении, оборудованном вытяжными и сушильными шкафами, муфельными печами, приспособлениями для мытья тары, посуды и при необходимости — проб.
Материал перед взятием средней пробы тщательно перемешивают.
Мясо предварительно измельчают ножом или в мясорубке. Масса средней пробы должна быть такова, чтобы масса полученного зольного остатка была достаточна для выполнения двух радиохимических исследований (40—60 г). Рекомендации приве-
547
юны в табл. 5.1b. Далее проводят предварительную обработку проб, заключающуюся в их минерализации. Используемые при пом методы могут быть различны в зависимости от вила исследуемого материала, химической природы определяемых радионуклидов и схемы радиометрического анализа.
Tao.i и на 5.I6
Примерный выход золы из некоторых видов проб
Проб;, Выход золы. Ч к сыром массе
Мясо 1.0—1.5
Молоко 0,7—1.2
Кость 35.0—50.0
Вода Различный в зависимости от минерализации
Процесс минерализации проб состоит из 3 последовательных этапов — высушивания, обугливания и озоления.
Высушивание про б. Пробы мяса, отделенные от жира, сухожилий и костей, измельчают, взвешивают, подсушивают при комнатной температуре, а затем сушат на лотках в сушильном шкафу до постоянной массы.
Кости отделяют от мягких тканей, костного мозга, измельчают, взвешивают и сушат в сушильном шкафу при температуре 100—150 °C в течение 2—3 ч.
Концентрирование водных проб достигается упариванием с последующим озолением и радиометрией сухого остатка.
Обугливание проб. После установления постоянной массы пробы сухой остаток обугливают путем прокаливания на электроплитах в вытяжном шкафу. Пробы мяса и рыбы можно сжигать на металлических противнях или больших сковородах. Полученный обугленный материал уже в меньшем объеме переносят в фарфоровые чашки или тигли и продолжают обугливание.
Фарфоровые тигли и чашки, предназначенные для озоления проб, тщательно моют, высушивают, нумеруют (можно раствором хлорного железа), прокаливают в муфельных печах до постоянной массы, затем охлаждают в эксикаторах и взвешивают. В прокаленные и взвешенные тигли или чашки помещают навески пробы необходимой массы или объема.
Процесс обугливания считают законченным при прекращении вспучивания пробы и исчезновении дыма.
Озоление проб. Обугленные сухие остатки озоляют в муфельных печах при температуре 400—450 °C, а пробы костей — при 600—800 °C. В процессе озоления температуру в муфельной печи повышают постепенно во избежание возгорания материала и потери радионуклидов цезия-137, свинца-210, иода-131. Продолжительность озоления зависит от количества и вида органических 548
соединений в пробе: оптимальным временем для растительных проб следует считать 2—4 ч. для мяса, молока. костей и корнеклубнеплодов — 15—25 ч. Внешним признаком готовности золы является ее цвет — светло-серый. Для достижения такого состояния зольного остатка может быть затрачено значительное время, а это. в свою очередь, связано с потерями определяемых радионуклидов. Для ускорения процесса озоления в золу периодически по каплям вносят такое количество царской водки, чтобы капли кислоты не стекали на дно и стенки фарфоровой посуды. Для более быстрого выжигания органических частиц золу в фарфоровых чашках рекомендуется периодически перемешивать. Если после указанного времени термической обработки зола нс приобрела светло-серого цвета, ее доозоление проводят в процессе радиохимического анализа после внесения в пробу носителей.
После некоторого остывания озоленные пробы переносят из муфеля в эксикатор, охлаждают до комнатной температуры и взвешивают. Вычитая из обшей массы тигля с золой массу тигля, получают массу полученной золы пробы, которую сравнивают с данными, приведенными в табл. 5.16.
Рассчитывают коэффициент озоления пробы, который будет использован для пересчета радиоактивности золы на радиоактивность сырой пробы.
Для твердых проб коэффициент озоления рассчитывают по формуле
^03 = —’ (5.47)
т} v 7
где т2 — масса полученной золы, г; тх — масса взятой сырой навески для озоления, г.
При определении коэффициента озоления следует иметь в виду, что получаемая зола гигроскопична, поэтому сразу же после ее взвешивания отбирают навеску золы на радиохимический и радиометрический анализы. При проведении указанных исследований в более поздние дроки золу взвешивают повторно с повторным расчетом коэффициента озоления.
Готовую золу растирают до мелкого порошка обратным (узким) концом пестика в той же чашке или тигле.
Порядок проведения анализа. Суммарную [3-активность проб определяют, подвергая радиометрическому измерению непосредственно зольные остатки. Для этого на аналитических весах взвешивают 200—300 мг золы, наносят на стандартные алюминиевые подложки, смачивают этанолом, равномерно распределяют и сушат под инфракрасной лампой. Затем проводят испытание на установках ДП -100 или УМФ-1500 в течение времени, необходимого для получения результата с заданной точностью.
549
При подготовке радиометра к эксплуатации проверяют работу пересчетной схемы. Устанавливают по вольтметру рабочее напряжение счетчика. Определяют скорость счета фона. Измерение проводят в течение 30 мин в тех же условиях, в которых будет проведена радиометрия проб. Скорость счета фона (имп/мин) вычисляют по формуле
.. .N
ЛФ=— (3.48)
где Л'—общее число сосчитанных импульсов от фона: г — продолжительность счета импульсов, мин.
После радиометрии проб скорость счета фона необходимо определить повторно. При заметном изменении фона для расчетов берут среднее двух измерений.
Под счетчик на расстоянии 3—5 мм от окна счетчика размещают эталонный препарат 40К (200 или 300 мг КО), приготовленный на стандартной алюминиевой подложке площадью 2,5 см2. Определяют число зарегистрированных импульсов от эталона за время счета (/), обычно равное 20 мин, и вычисляют УОэт (имп/мин).
Строго при одних и тех же условиях (расстояние от препарата до счетчика, размер и материал подложки, рабочее напряжение счетчика и т. п.), стандартных для данной радиометрической установки, измеряют скорость счета от препаратов зольных остатков (по 200 или 300 мг) всех исследуемых проб. Время счета выбирают в зависимости от заданной величины относительной статистической ошибки счета.
Скорость счета (имп/мин) от препарата с фоном определяют по формуле
лг, N
N =— (5.49)
Скорость счета от каждого препарата без фона (имп/мин) определяют по формуле
^0=^-% (5.50)
Используя полученные данные радиометрии эталона и препарата и известные данные об эталоне 40К, рассчитывают удельную [3-радиоактивно‘сть исследуемой пробы (Ки/кг или Ки/дм3) по формуле
(7V - Л%)/фЛ03 /Ч — -------------- / к С I \
где N и Л/ — скорость счета соответственно пробы и фона, имп/мин; А/п — коэффициент перехода от активности, выраженной в импульсах в 1 мин, к активности, выраженной в кюри (коэффициент связи); Кт — коэффициент озоления, равный массе золы в граммах, полученной при озолении 1 кг продуктов или 1 дм3 молока, воды; /И] — масса навески, взятой для радиометрических исследований, г.
550
Коэффициент связи определяют ио хлориду калия, для чего на аналитических весах взвешивают точно такую же навеску высушенного хлорида калия х.ч.. что и навеску золы (200—300 мг). Измерение радиоактивности проводят в аналогичных условиях.
Полученные данные сводят в таблицу.
Характерце:ика и масса образна
Масса золы ирои. I
Коаффиш1ен i
озоления
1 Угельная суммарная । р радиоактивность
пробы, Ки/кг
Лабораторная работа № 12
ОПРЕДЕЛЕНИЕ РАДИОНУКЛИДОВ РАДИОХИМИЧЕСКИМИ МЕТОДАМИ
Цель работы. Приобрести практический навык в определении радионуклидов на основе радиохимических методов.
Задачи. Отбор и подготовка проб, определение стронция-90 фосфатным методом, определение цезия-137 сурьмяно-иодным методом.
Объекты исследования. Мясо и вторичные продукты убоя различных животных и птицы; мясные продукты в ассортименте; яйца и яйце продукты.
Методические указания. Суммарную [3-радиоактивность определяют, как правило, при анализе наиболее загрязненных радиоактивными веществами объектов. Основным методом определения радиоактивности в объектах ветеринарного надзора является радиохимический анализ, который позволяет дать полную и объективную характеристику радиоактивной загрязненности объекта отдельными радиоизотопами, выделенными из пробы. При радиохимическом анализе в пробах определяют содержание наиболее опасных в биологическом отношении радиоизотопов: стронция-90, цезия-137 и др.
При выборе методов определения того или иного радиоизотопа следует учитывать как индивидуальные особенности исследуемых проб, так и характер их радиоактивного загрязнения.
Радиоактивные изотопы обладают теми же химическими свойствами, что и стабильные, и выделяют их так же, как и стабильные изотопы тех же элементов. При содержании в пробе нескольких радиоактивных элементов сначала их разделяют на химические группы, а затем на отдельные элементы и определяют их радиоактивность. Значительные трудности при этом связаны с очень малым содержанием радионуклидов в исследуемом материале, так как свойства одних и тех же веществ, взятых в ультрамалых и больших количествах, могут отличаться. Эти особенности радио
551
химическою ana.in sa учитываются уже при отоорс ирод. Объем (масса) оюирасмою ма1ериала должен обеспечивать возможность выяснения радионуклида из пробы. Для этою c.ic.iyci ориен 1 ироваться на кол ичсс гвенный выход золы при обогащении пробы путем озоления сухою осника. В среднем на один радиохимический анализ требуется К)—-50 г золы исслс-рсмоп пробы.
Большое значение имеет соблюдение режимов термической обработки исследуемого материала. Высушивание проводят при (S0—100 °C до постоянной массы. Продолжительность высушивания после каждого контрольною высушивания нс более 1 ч. Взвешивание проводят после охлаждения проб ло комнатной температуры.
Особенно важно соблюдать температурный режим при озолении пробы. Озоление проводят в электропечи после обхллпвания высушенной пробы на электроплитке или газовой горелке. Некоторые радионуклиды возгоняются или разрушаются при высоких температурах. Так. например, сублимация цезия-137 происходит при температуре 450 °C и выше. Поэтому пробу для этого анализа о зол я юг при температуре нс выше 400 °C. Для выделения из пробы стронция-90 озоление рекомендуется проводить при температуре 900—1000 °C. так как стронций не разрушается от действия этих температур, а удаление из зольного остатка калия-40, цезия-137 и других гермолабильных радиоизотопов в данном случае даже желательно, так как способствует получению радиохимически чистого стронция-90.
Поведение ультрамалых количеств вещества при растворении, нагревании, осаждении может не совпадать с известными для данного элемента свойствами. Отмечают различия в летучести, повышенной адсорбции на поверхностях, способности образовывать коллоидные растворы. Многие из особенностей поведения элементов в ультрамалых количествах еще недостаточно изучены, поэтому удобнее проводить выделение данного изотопа в таких количествах, в которых поведение его при той или иной химической реакции будет проявляться типично и обеспечит более полное отделение его от примесей.
Поэтому в пробу золы до ее растворения вносят точно известное количество так называемого носителя.
Носителем служит элемент одноименный или сходный по химическим свойствам с радиоактивным изотопом, извлекаемым из пробы. Носители используют в форме титрованных растворов с точно известной концентрацией (мг/см3). Введенный в пробу носитель, увеличивая массу извлекаемого элемента, позволяет увлечь за собой одноименный (или сходный) радиоизотоп по всем этапам анализа, чем достигается наиболее полное его извлечение.
552
Но химическому выходу носи геля судя т о полноте выделения радиоизотопа из пробы. Этот показатель использую! при расчете радиоактивности объекта. Химический выход (,\. в.) носителя определяют как отношение массы выделенного носителя (мг) в конце анализа к массе внесенного носителя в пробу золы перед анализом.
Носители бывают изотопные, изоморфные, инертные и удерживающие.
Изотопный носитель — стабильный элемент того же изотопа (радиоактивного), который требуется выделить из пробы. Так. при выделении из пробы иттрия-90 в качестве носителя в пробу вводят стабильный иттрий в виде раствора (нитрата или хлорида).
Изоморфный носитель —- стабильный химический элемент, сход ный но химическим свойствам с выделяемым радиоизотопом. На пример, стронций-89 при добавлении хлорида бария осаждается вместе с образующимся сульфатом бария.
Инертный носитель — стабильный элемент, который не сходен по химическим свойствам с выделяемым из пробы радиоизотопом. но способен перевести его из одной фазы в другую (например. из раствора в осадок). Так. стронций-90 можеа быть выделен из раствора вместе с осадком гидроксида железа.
Удерживающий носитель - стабильный химический элемент того радиоактивного изотопа, который необходимо отделить от выделяемого радиоизотопа. Например, чтобы отделить лантан-140 при выделении иттрия-90, в пробу вносят раствор соли стабильного лантана.
При добавлении к пробе изотопного носителя создаются наиболее благоприятные условия для выделения изотопа с данным носителем. Однако слишком большое количество носителя нежелательно. При внесении изоморфного носителя можно сначала выделить радиоизотоп с этим носителем, а затем отделить радиоизотоп от носителя. В результате применения инертного носителя в ходе анализа можно полностью извлечь радиоизотоп из смеси, а в последующих реакциях быстро освободить радиоактивный изотоп от носителя. Добавление удерживающих носителей способствует выделению радиохимического изотопа.
Носители вводят в пробу в виде титрованных растворов определенных веществ. На один анализ обычно требуется 10—30 мг носителя (в пересчете на металл).
Для приготовления титрованных растворов изотопных, изоморфных и удерживающих носителей в зависимости от выбранной методики выделения радиоизотопа из пробы используют нитраты или хлориды стронция, цезия, иттрия, лантана, церия и др.
Хранить титрованные растворы рекомендуется в мерных колбах с притертыми или хорошо пригнанными резиновыми пробками. Длительное хранение их нежелательно, так как в случае испарения раствора со временем может измениться его титр.
553
Для определения строниия-90 в мясе, костях и других мясных продуктах используют фосфатный метод, который основан на предварительном выделении иттрйя-90. находящегося в пробе в равновесном по радиоактивности состоянии со стронпием-90. Его выделяют из раствора золы пробы в виде фосфата иттрия по реакции
УСЕ + ЕЕРО, = УРО, + ЗНС1.
В последующем проводят радиохимическую очистку иттрия-90 вместе со стабильным иттрием путем осаждения его в виде гидроксида и оксалата иттрия.
Цезий-137 определяют сурьмяно-иодидным методом.
Метод основан на предварительном концентрировании цезия-137 с носителем из кислотного раствора золы пробы в виде комплексного соединения ферроцианида никеля-цезия по реакции
K4[Fe(CH)6 ] + Ni(NO3 )2 + 2CsCl =
- + 2KNO3 + 2HC1.
Затем цезий осаждают из кислого раствора треххлористой сурьмой и иодидом аммония в виде сурьмяно-иодида цезия по реакции
3CsCl + 9NH4J + 2SbCl3 = Cs^--2J9- + 9NH4C1.
Ф
Осадок сурьмяно-иодида цезия отфильтровывают, высушивают, по выходу носителя определяют химический выход цезия-137, радиометрируют и рассчитывают радиоактивность пробы (Ки/кг).
Метод позволяет выделить цезий-137 в радиохимически чистом состоянии из больших объемов растворов золы пробы с высоким содержанием солей. Кроме того, из одной навески золы можно одновременно выделить цезий-137 и стронций-90, что позволяет ускорить радиохимический анализ и провести его при ограниченном количестве золы пробы, что очень существенно при анализе молока и мяса, имеющих очень малый выход золы при концентрировании пробы.
Существует также другая модификация анализа цезия-137 сурьмяно-иодидным методом. Она основана на осаждении цезия-137 вместе со стабильным цезием треххлористой сурьмой и иодидом аммония из солянокислого раствора золы пробы в виде соединения постоянного состава Cs3Sb2I9 по реакции
3CsCl + 2SbCl3 + 9NH4J = — З8-^9 + 9NH4C1.
Ф
554
1. ОПРЕДЕЛЕНИЕ СТРОНЦИЯ-90 ФОСФАТНЫМ МЕТОДОМ
Материалы, реактивы и оборудование. Радиометр: весы аналитические; муфельная йены сушильный шкаф; электроплитка или газовая горелка; химические стаканы термостойкие вместимостью 100 и 500 см3: пипетки вместимостью 1 и 5 см\ воронки; фильтры «синяя лента»: фильтровальная бумага: стеклянные палочки: алюминиевые подложки стандартные: тигли фарфоровые; титрованные растворы носителей иттрия и стронция: раствор соляной кислоты молярной концентрацией 6 моль/дм3: вола дистиллирован пая; аммиак безугольный; насыщенный раствор щавелевой кисло ты; аммиачная вода; этанол.
Приготовление реактивов. П р и г о г о в л с н и е г и г р о -ванного раствора с т а б и л ь н о г о стр о н и и я и определение титра п о с у л ь ф а т у с т р о и и и я. На аналитических весах взвешивают 4 г хлорила стронция или 6 i нитрата стронция [SrCl2 или Sr(NO.)J. Навеску помешают в мерную колбу вместимостью 100 см3, растворяют в небольшом объеме раствора соответствующей кислоты (соляной или азотной) молярной концентрацией 2 моль/дм3 и доводят дистиллированной водой до метки. Колбу закрывают пробкой и раствор тщательно перемешивают. Титр приготовленного раствора проверяют конечной реакцией при выделении данного изотопа из исследуемого материала, для чего в 3 или 5 стаканов вместимостью 100 см3 вносят по 1 см3 приготовленного раствора стронция. В каждый стакан вносят 30 см3 дистиллированной воды, 5 см3 раствора серной кислоты массовой долей 5 % и равный объем этанола. Полученную смесь тщательно перемешивают стеклянной палочкой. В результате выпадает осадок сульфата стронция, который отстаивают в течение нескольких часов (удобнее оставить на ночь), а затем отфильтровывают через бумажные фильтры «синяя лента». Полученные осадки на фильтрах высушивают в сушильном шкафу, осторожно переносят в фарфоровые тигли, прокаленные до постоянной массы, обугливают на электроплитке или газовой горелке и озоляют в муфельной печи при температуре 800 °C до постоянной массы. Во избежание поглощения воды и СО7 из воздуха тигли с прокаленными осадками охлаждают в эксикаторе, взвешивают и определяют массу каждого осадка. Расхождение по массе отдельных осадков не должно превышать 1 %. Среднее арифметическое 3—5 полученных значений масс принимают за титр раствора-носителя стронция по его соединению SrSO4 (мг/см3).
Следовательно, в 1 см3 приготовленного раствора содержится такое количество SrSO4, какое соответствует средней массе выделенного осадка. Для выражения титра раствора по металлическому стронцию нужно величину титра по SrSO4 умножить на 0,477.
555
П р и г о т о в л с н и с т н т р о в а н н ы х рас т в о р о в ini' р и я и о п р с л е л ен не т и т р а п о о к с и л у и т т р и я. Для приготовления титрованного раствора носителя необхолпмо рассчитать массу соли стабильного изотопа. Пример расчета приведен ниже.
Пример. Требуется приготовить 100 см? раствора-носителя из стабильного нитрата иттрия Y(NO;); • 6Н.0 с содержанием ме-шллического иттрия 40 мг в 1 см3 раствора." Молекулярная масса Y(NO.)3'6H2O равна 383 г. Атомная масса иттрия равна 88.92 г (округленно’89 г). Для расчета необходимого количества соли нитрата иттрия на 1 см3 раствора составляют пропорцию:
89 г- 383 г
40 мг-Аг.
40-383-103 89-103
= 172 мг,
где К)3 — перевод граммов в миллиграммы.
На 100 см3 раствора требуется 172 • 100 = 17 200 мг = 17,2 г.
Берут навески 17,2 г нитрата или 14 г хлорида иттрия, помещают их в мерную колбу вместимостью 100 см3, растворяют в небольшом объеме азотной или соляной кислоты молярной концентрацией 2 моль/дм3 и доводят объем дистиллированной водой до метки. Закрывают колбы пробками и растворы тщательно перемешивают.
Для установления титра отбирают по 1 см3 приготовленного раствора в три химических стакана вместимостью 100 см3, прибавляют по 25—30 см3 дистиллированной воды и нагревают до 60—70 °C. В каждый стакан добавляют по 10 см3 насыщенного раствора щавелевой кислоты и, приливая по каплям аммиак, доводят pH содержимого стакана до 1,5. В осадок выпадает оксалат иттрия (лантана). Некоторое время дают растворам отстояться для формирования осадков оксалатов. Проверяют полноту осаждения, прибавляя по каплям насыщенный раствор щавелевой кислоты. Если в просветленной части раствора не обнаружено помутнение, то осаждение оксалатов считают полным, если образуется помутнение, то необходимо добавить минимальный объем насыщенного раствора щавелевой кислоты. Растворы оставляют на 4—5 ч для более полного выпадения осадка оксалатов (можно оставить растворы на ночь, закончив титрование на следующий день). После отстаивания и охлаждения осадки фильтруют через бумажные фильтры «синяя лента», перенося полностью осадок на фильтр. Смывать остатки осадка со стенок стакана можно фильтратом. Фильтры с осадками высушивают в сушильном шкафу, переносят в фарфоровые тигли, предварительно прокаленные и взвешенные. Затем обугливают на электроплитке и прокаливают в электропечи
556
при температуре 900—1000 СС в течение часа (до постоянной массы). Подучают осадок оксида иттрия (Y.O.) Тигли с оксидом иттрия охлаждают в эксикаторе, взвешивают и определяю! массу каждого осадка. Среднее значение масс трех осадков принимают за титр приготовленного раствора, который указывает, какое число миллиграммов Y2O, содержится в 1 см3 раствора.
Порядок проведения анализа. Берут навеску золы массой 20—25 г в термостойкий стакан. Вносят титрованные растворы носителей: иттрия (60—70 мг по Y2O3) и стронция (200 мг по SrSO4). Приливают 150—200 см3 раствора соляной кислоты молярной концентрацией 6 моль/дм? и кипятят при помешивании 1? течение 30 мин.
Нерастворившийся осадок отфильтровывают, промывают на фильтре раствором соляной кислоты молярной концентрацией 2 моль/дм3 и отбрасывают. Фильтрат упаривают до минимального объема (до получения влажных солей) и добавляют 300—400 см3 горячей дистиллированной воды. Из горячего раствора осаждают фосфат иттрия, для чего добавляют разбавленный в соотношении 1 : 5 безугольный аммиак при энергичном помешивании до установления pH 3—4 (для проб молока и мяса) и pH 2—3 (для проб костей). При этом иттрий-90 отделяется от стронция-90. Фиксируют время отделения иттрия от стронция.
Раствор с выпавшим рыхлым осадком фосфата иттрия подогревают в течение 1—2 мин и фильтруют. Осадок фосфата иттрия на фильтре несколько раз промывают горячей водой, а фильтрат отбрасывают.
Осадок на фильтре растворяют в минимальном объеме горячего раствора соляной кислоты молярной концентрацией 2 моль/дм3. Приливают к раствору хлорида иттрия 1/2 объема насыщенного раствора щавелевой кислоты. Устанавливают pH раствора 1,5—2,0 добавлением безугольного аммиака. В осадок выпадает оксалат иттрия. Эту операцию проводят с целью дополнительной очистки иттрия от элементов второй аналитической группы (стронция, кальция и др.). Раствор с осадком оставляют на 20—30 мин для укрупнения кристаллов и формирования осадка.
Остаток оксалата иттрия отфильтровывают через обеззоленный фильтр «синяя лента» и промывают на фильтре несколько раз теплой водой. Осадок на фильтре помещают в фарфоровый тигель, подсушивают, сжигают и прокаливают в муфельной печи при температуре 600—700 °C в течение 1 ч.
Прокаленный осадок карбоната иттрия растворяют в 100 см3 горячего раствора соляной кислоты молярной концентрацией 2 моль/дм3 и кипятят в течение 10 мин для удаления СО-,. Осаждают гидроксид иттрия Y(OH)3 безугольным аммиаком при pH 7 и выше.
Осадок гидроксида иттрия отфильтровывают через обеззоленный фильтр и промывают 1—2 раза горячей аммиачной водой.
557
Фильтрат отбрасывают. Осадок на фильтре растворяют в 50см? горячего раствора соляной кислоты молярной концентрацией 2 моль/дм'. Приливают к раствору 10 см' насыщенного раствора щавелевой кислоты и безутольным аммиаком устанавливают pH на уровне 1.5—2.0. 13 результате выпадает белый кристаллический осадок оксалата иттрия. Это нереосаждение иттрия проводят с целью лучшей его очистки. Дают раствору с осадком постоять в течение 20—30 мин. затем фильтруют через обеззоленный фильтр «синяя лента». Осадок на фильтре промывают 2—3 раза торячей водой.
Фильтр с осадком помещают в фарфоровый тигель, подсушивают, сжигают и прокаливают в муфельной печи при температуре 800—1000 °C в течение 1 ч. Получают оксид иттрия (У7О3). Осадок тт тигле измельчают' стеклянной палочкой, взвешивают, определяют химический выход носителя иттрия, помещают на стандартную алюминиевую подложку, смачивают 4—5 каплями спирта, высушивают (под лампой пли в сушильном шкафу) и проводят радиометрию. Удельную радиоактивность стронция-90 ио иттрию-90 в исследуемой пробе вычисляют аналогично расчету при приготовлении титрованных растворов-носителей для радиохимического анализа.
2. ОПРЕДЕЛЕНИЕ ЦЕЗИЯ-137 С ПРЕДВАРИТЕЛЬНЫМ КОНЦЕНТРИРОВАНИЕМ В ВИДЕ ФЕРРОЦИАНИДА НИКЕЛЯ-ЦЕЗИЯ
Материалы, реактивы и оборудование. Радиометр; весы аналитические; муфельная печь; сушильный шкаф; электроплитка или газовая горелка; химические стаканы термостойкие вместимостью 100 и 500 см3, чашки фарфоровые; пипетки вместимостью 1 и 5 см3; воронки; фильтры «синяя лента», фильтровальная бумага; центрифуга; баня песчаная; тигли фарфоровые; стеклянные палочки; алюминиевые подложки стандартные; титрованные растворы носителей цезия, иттрия; соляная кислота концентрированная и растворы молярной концентрацией 2 и 6 моль/дм3; раствор ферроцианида калия KJFe(CH)6] массовой долей 10 %; раствор нитрата никеля Ni(NOp7 массовой долей 10%; вода дистиллированная; насыщенный раствор треххлористой сурьмы SbCl3 в ледяной уксусной кислоте (740 мг SbCF на 1 см3 кислоты); кислота уксусная ледяная; иодид аммония или иодид калия; этанол.
Приготовление титрованного раствора стабильного цезия и определение его титра. Для приготовления титрованного раствора-носителя необходимо рассчитать массу соли стабильного изотопа. Пример расчета приведен ниже.
558
Пример. Требуется приготовить 100 см3 раствора-носителя стабильного цезия с содержанием металлического цезия 30 мг/см3. Молекулярная масса CsNO; 194.91г (округленно 195 г). Атомная масса цезия 133 г. Для расчета соли нитрата цезия на 1 см3 приготовленного раствора указанной концентрации составляют пропорцию:
133 г - 195 г
30 мг — X г.
30 195-103
133-103
= 43,9 мг.
На 100 см3 раствора требуется 43,9- 100 = 4390 мг = 4,39 г нитрата цезия.
Навеску нитрата цезия массой 4,4 г помещают в мерную колбу вместимостью 100 см3, растворяют раствором азотной кислоты молярной концентрацией 2 моль/дм-’ и доводят дистиллированной водой до метки. Титр приготовленного раствора устанавливают экспериментально.
Навеску хлорида цезия (CsCl) 3,8 г или нитрата цезия (CsNO3) 4,4 г вносят в мерную колбу вместимостью 100 см3, растворяют в небольшом объеме соляной или азотной кислоты молярной концентрацией 2 моль/дм3 и доводят объем дистиллированной водой до метки. Закрывают колбу пробкой и раствор тщательно перемешивают.
Для определения титра раствора по висмут-иодиду цезия в три химических стакана вместимостью по 50 см3 помещают по 1 см3 приготовленного раствора-носителя. В каждый стакан добавляют по 2 см3 ледяной уксусной кислоты, нагревают до 50—70 °C и осаждают висмут-иодид цезия путем добавления (по каплям) осадителя — раствора иодида висмута (BiJ3) объемом 1 см3. Для приготовления осадителя 5 г оксида висмута (Bi2O3) и 17 г иодида калия (KJ) растворяют в 50 см3 ледяной уксусной кислоты.
Стаканы с выпавшим осадком висмут-иодида цезия (Cs3Bi2J9) выдерживают на водяной бане в течение 30—40 мин при температуре кипения, затем охлаждают, отстаивают и отфильтровывают через предварительно высушенные и взвешенные фильтры «синяя лента». Осадки на фильтрах высушивают в сушильном шкафу при температуре 140—150 °C до постоянной массы, взвешивают и определяют массу каждого осадка.
Среднее из полученных значений принимают за титр раствора по висмут-иодиду цезия (Cs3Bi2J9), который указывает, какое количество миллиграммов висмут-иодида цезия содержится в 1 см3 раствора. Для выражения титра по сурьмяно-иодиду цезия (Cs3Bi2J9) необходимо полученное значение титра по висмут-иодиду цезия (Cs3Bi2J9) в миллиграммах умножить на 0,91.
559
Порядок проведения анализа. Навеску золы пробы (15—20 г) помешают в термостойкий стакан или фарфоровую чашку. Приливают титрованные растворы носителей: цезия (100—150 мг по СьДФЦ) и иттрия (50—60 мг Y.OJ (приготовление титрованных раса воров итгрия см. в и. 1 настоящей лабораторной работы).
Проводят доозолснис пробы. Для итого смачиваю! золу концентрированной соляной кислотой из расчет;! 2—3 см? кислоты на 1 г золы, подсушивают на электроплитке или песчаной бане в вытяжном шкафу. Охлаждают содержимое стакана и снова приливают концентрированную соляную кислоту с последующим выпариванием досуха. Этим достигается наиболее полная минерализация пробы.
Проводят растворение юлы. К сухому sic татку приливаю] 150—200 см3 раствора соляной кислоты молярной концентрацией 6 моль/дм3. кипятят 30 мин и дают осадку осесть на дно. Фильтруют раствор, нс перенося осадок на фильтр
Фильтр с частичным осадком переносят в стакан с оставшимся нерастворившимся осадком и заливают 150—200 см3 раствора соляной кислоты молярной концентрацией 2 моль/дм?. Кипятят 30 мин при помешивании.
Теплый раствор фильтруют, осадок на фильтре промывают горячим раствором соляной кислоты молярной концентрацией 2 моль/дм3. Оставшийся нсрастворившийся осадок отбрасывают, а фильтраты объединяют.
Кислотный раствор золы слегка подогревают и осаждают из него цезий, добавляя при помешивании стеклянной палочкой 5 см3 водного раствора ферроцианида калия К4[ Fe(CN)6] массовой долей 10% и 5 см3 водного раствора нитрата никеля Ni(NO/)2 массовой долей 10 %. В осадок выпадает С$эМ1|Ре(СМ)6] — ферроцианид никеля-цезия. Для укрупнения кристаллов осадок оставляют в покое не менее чем на 3 ч (лучше оставить на ночь). Затем осадок фильтруют через складчатый фильтр «синяя лента» (двойной) или центрифугируют. Оставшийся на фильтре осадок промывают 20—30 см3 раствора соляной кислоты молярной концентрацией 2—3 моль/дм3. Объединенные фильтры оставляют для определения стронпия-90 любым рекомендуемым методом.
Фильтр с осадком подсушивают в сушильном шкафу, помешают в фарфоровый тигель и прокаливают в муфельной печи при температуре 400—450 °C в течение 1 ч. Прокаленный осадок переносят в химический стакан объемом 100 см3, приливают 40 см3 дистиллированной воды и упаривают до 20 см3.
Нсрастворившийся осадок отфильтровывают через фильтр «синяя лента» и промывают на фильтре 3—5 см3 дистиллированной воды.
Полученный фильтрат доводят концентрированной соляной кислотой до молярной концентрации по НС1 3 моль/дм3 (к 20 см3 фильтрата добавляют 7 см3 концентрированной соляной кислоты).
Раствор охлаждают на ледяной бане и приливают 0.2—0.3 см? насыщенного раствора треххлористой сурьмы в ледяной уксусной кислоте. Добавляют к раствору 3 г иодида аммония (или иодида калия) в сухом виде. Раствор интенсивно перемешивают стеклянной палочкой до выпадения осадка сурьмяно-иодида цезия. Дают осадку отстояться в течение 1 ч, а затем фильтруют через фильтр «синяя лента» или центрифугируют.
Осадок на фильтре промывают несколько раз небольшими порциями (по 5—7 см3) ледяной уксусной кислоты (до исчезновения желтой окраски вытекающего фильтрата), а затем еще раз 1—2 см3 этанола и высушивают на фильтре при температуре 80-90 °C.
Цезий в виде Cs,SbJ9 переносят на взвешенную стандартную подложку, смачивают 2—3 каплями этанола, равномерно распределяют на подложке, высушивают и взвешивают. Определяю! массу осадка без подложки.
Удельную радиоактивность исследуемой пробы определяют на радиометре.
Удельную радиоактивность исследуемой пробы, обусловленную содержанием цезия-137 (Ки/кг или Ки/дм3), рассчитывают по формуле
<5-52)
где Л'о — скорость счета от препарата без фона, имп/мин; KCD — коэффициент связи счетной установки: Кт — коэффициент озоления пробы; т — масса навески золы, взятой на анализ, г; х. в. — химический выход носителя; Р — поправочный коэффициент на самопоглошение р-частиц в препарате, определяется по табл. 5.17.
Таблица 5.17
Поправка на поглощение р-частиц цезия-137 в образце
Масса препарата, мг Р Масса препарата, мг Р
10 0,936 50 0.743
15 0,915 55 0.721
20 0,881 60 0,702
25 0.857 70 0.664
30 0.838 75 0.648
35 0,807 80 0.631
40 0.787 90 0.598
45 0,763 100 0.569
560
561
3. ОПРЕДЕЛЕНИЕ ЦЕЗИЯ-137 СУРЬМЯНО-ИОДИДНЫМ МЕТОДОМ
Материалы, реактивы и оборудование. Радиометр: весы аналитические; сушильный шкаф: электроплитка или газовая горелка; химические стаканы термостойкие вместимостью ЮОсм3; чашки фарфоровые; пипетки; воронки; фильтры «синяя лента»; центрифуга;^ баня ледяная: пробирки центрифужные вместимостью Юсм3; алюминиевые подложки стандартные; титрованные растворы-носители цезия, иттрия; азотная кислота концентрированная; раствор пероксида водорода массовой долей 30%; соляная кислота концентрированная и растворы молярной концентрацией 3 моль/дм3; иодид аммония NH4J кристаллический и насыщенный (свежеприготовленный) раствор (17г NH4J на Юсм3 воды); сульфит натрия Na7SO3; насыщенный раствор треххлористой сурьмы SbQ3 в ледяной уксусной кислоте (740 мг SbCl3 на 1 см3 кислоты); кислота уксусная ледяная; этанол.
Приготовление титрованного раствора стабильного цезия и определение его титра по сурьмяно-иодиду цезия. Для приготовления титрованного раствора 3,2 г хлорида цезия (CsCl) или 3,7 г нитрата цезия (CsNO3) помешают в мерную колбу вместимостью 100 см3, растворяют в растворе соляной кислоты молярной концентрацией 3 моль/дм3, доводят объем дистиллированной водой до метки и тщательно перемешивают.
Для определения титра раствора в три химических стакана вместимостью 50 см3 вливают по 4 см3 приготовленного титрованного раствора, по 3 см3 свежеприготовленного насыщенного водного раствора иодида аммония (NHJ) и охлаждают на ледяной бане. К охлажденным растворам прибавляют по 0,2 см3 насыщенного раствора хлорида сурьмы (III) в ледяной уксусной кислоте при постоянном перемешивании (с трением стеклянной палочки о стенки стаканов) до выпадения осадка, который отстаивают в течение 1—2 ч, а затем пропускают через предварительно высушенные до постоянной массы фильтры. Осадки на фильтрах промывают 3 раза ледяной уксусной кислотой порциями по 2 см3 и 1 раз этанолом, высушивают в сушильном шкафу при температуре 90 °C до постоянной массы. Охлажденные в эксикаторе фильтры с осадками взвешивают на аналитических весах и определяют чистую массу каждого осадка. Среднее значение масс осадков и будет показателем титра по сурьмяно-иодиду цезия (мг/см3).
Порядок проведения анализа. Навеску золы пробы (10—20 г) помещают в термостойкий стакан или фарфоровую чашку и вносят титрованные растворы носителей цезия (100—150 мг в расчете на Cs3Sb2J9), иттрия (50—60 мг по Y3O3). Проводят доозоление пробы. Для этого смачивают золу концентрированной азотной кислотой с добавлением нескольких капель пероксида водорода, подсушивают. Затем смачивают золу концентрированной соляной кис-562
лотой (из расчета 2—3 см3 на 1 г золы) и упаривают досуха. Эту операцию повторяют 2—3 раза, чтобы полностью удалить азотную кислоту. Анализ проводят в вытяжном шкафу.
Проводят растворение золы. К сухому остатку при нагревании приливают 25—30 см-\ раствора соляной кислоты молярной концентрацией 3 моль/дм3. Необходимо следить за тем, чтобы раствор не упарился и в связи с этим не изменилась его кислотность. Нерастворившийся осадок отфильтровывают через фильтр «синяя лента» в химический стакан вместимостью 100 см3. Стакан, в котором проводилось растворение золы, смывают 2—3 раза небольшими порциями (по 2—3 см3) раствора соляной кислоты молярной концентрацией 3 моль/дм3. Общий объем фильтрата не должен превышать 40—50 см3. Осадок на фильтре отбрасывают, а фильтрат используют для анализа.
Стакан с раствором помешают на ледяную баню. Если выпадет осадок избыточных солей, то раствор осторожно переливают в другой стакан, не перенося осадок. В охлажденный раствор вносят 2—3 см3 насыщенного (свежеприготовленного) раствора NH4J (17rNH4J на Юсм3 воды). Если при этом появится темно-коричневая окраска из-за выделения свободного иода, то добавляют сухую соль NaSO3 до образования светло-коричневой окраски раствора. Прибавляют 0,2 см3 насыщенного раствора треххлористой сурьмы в ледяной уксусной кислоте и интенсивно перемешивают стеклянной палочкой до выпадения осадка. Если осадок не выпадет, то необходимо добавить 2—3 капли раствора треххлористой сурьмы или несколько кристаллов иодида аммония и интенсивно перемешать.
Раствору с осадком сурьмяно-иодида цезия дают постоять 20—30 мин, а центрифугируют в пробирках вместимостью Юсм3 (раствор с осадком из стаканчика переносят в пробирку в несколько приемов, последней порцией раствора над осадком в пробирке смывают кристаллы со стенок стакана и снова центрифугируют). Осадок в пробирке промывают 2—3 раза порциями по 5—7 см3 ледяной уксусной кислоты и центрифугируют. Затем осадок промывают 1—2 см3 этанола и снова центрифугируют.
Осадок сурьмяно-иодида цезия можно отфильтровать через фильтр «синяя лента», предварительно высушенный при температуре 90 °C до постоянной массы и взвешенный. Осадок на фильтре промывают небольшими порциями ледяной уксусной кислоты до прекращения окрашивания вытекающего фильтрата. Осадок на фильтре высушивают в сушильном шкафу при температуре не выше 90 °C, переносят на предварительно взвешенную подложку, смачивают этанолом, равномерно распределяют по подложке, высушивают под лампой, определяют массу навески препарата и проводят радиометрию.
Если осадок отделен центрифугированием, то его следует подсушить в пробирке при температуре не выше 90 °C и перенести
563
(сначала сухой осадок, затем остаток на стенках пробирки с помощью небольшого количества этанола) на предварительно взвешенную подложку, равномерно распределить, высушить, взвесить, определить массу препарата и провести радиометрию на радиометрической установке, отградиурованной по цезию-137.
Удельную радиоактивность исследуемой пробы рассчитывают аналогично п. 3 настоящей лабораторной работы.
В отчете студенты составляют акт отбора проб продуктов для радиологического исследования, в котором указывают: дату, наименование населенного пункта, района; название учреждения, должности, фамилии людей, производивших отбор проб и присутствовавших при отборе; место (хозяйство, участок, поле, бригада, ферма) и время отбора; название продукта; место и время получения продукта (для привозных — номер и дату документа, по которому получена данная партия); общую массу продукта, из которого отобрана проба; опись взятых проб (с указанием пробы, ее номера и массы — сырой и высушенной); куда направляется и для какой цели; подписи.
Оформляют сопроводительные этикетки к пробам, отобранным для радиохимического анализа, при отправке для исследования в лабораторию, а также этикетки на хранение измельченной золы проб, подготовленных для проведения радиохимического анализа.
Данные, полученные радиохимическими методами (или одним из них), оформляют в виде протокола, в котором фиксируют массу и характеристику образцов мясопродуктов, отобранных для контроля удельной радиоактивности, массу золы проб, коэффициент озоления, результаты измерений и расчетов уровней радиоактивной загрязненности исследуемых объектов. Рекомендуемая форма протокола представлена ниже.
Масса и характеристика образца мяса или мясопродуктов Масса золы проб,г Коэффициент озоления Удельная радиоактивность (Ки/кг) пробы, обусловленная содержанием
стронция-90 цезия-137
Полученные данные анализируют, делают заключение об уровне загрязненности объектов контроля стронцием-90, цезием-137 и о радиационной безопасности исследуемых продуктов.
564
Контрольные вопросы
1. Каковы источники загрязнений токсикантами мяса и мясных продуктов?
2. Что относится к основным контаминантам мяса и мясных продуктов?
3. Какова химическая природа контаминантов мяса и мясных продуктов.’
4. Что называется предельно допустимой концентрацией?
5. Что относится к микробным контаминантам?
6. Какие этапы включает в себя бактериологический контроль мяса, мясных продуктов'.’
7. В чем суть метода ускоренного обнаружения бактерий?
8. Каким методом определяют антибиотики в мясе и мясных продуктах'.’
9. В чем суть метода определения гормонов'.’
10. Что такое пестициды и почему их относят к группе контаминантов мяса?
11. Какими методами анализа определяют пестициды в мясе'.’
12. В чем суть метода определения пестицидов методом тонкослойной хроматографии?
13. В чем преимущества анализа пестицидов энзимо-хроматографическим методом?
14. Как можно определить границы проникновения фенолов в копченых мясных продуктах?
15. В чем суть методов количественного определения фенолов? Каковы разновидности методов?
16. Как определить бенз(а)пирен в мясных продуктах?
17. Какова роль нитритов и нитратов в технологии мясных продуктов?
18. В чем суть ионометрического метода определения нитрит- и нит-рат-ионов?
19. Какими фотометрическими методами определяют нитраты и нитриты? На чем они основаны?
20. Какие элементы относятся к токсическим и почему?
21. Какие методы используются при анализе токсических элементов? В чем их сущность?
22. Каковы особенности подготовки проб при анализе токсических элементов?
23. Каковы подходы при анализе свинца?
24. Как определяют кадмий в мясе и мясных продуктах?
25. В чем суть методов качественного обнаружения и количественного определения меди?
26. В чем суть метода определения мышьяка по Зангер—Влеку? Какие существуют модификации?
27. Для чего проводят предварительную деструкцию проб при анализе ртути?
28. Что такое радионуклиды? Каковы их источники и действие на организм человека?
29. Что относится к экспресс- и массовым методам определения радионуклидов?
30. В чем сущность и разновидности радиохимических методов определения радионуклидов?
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Арутюнян Н. С., Аеишева Е. А. Лабораторный практикум по химии жиров. — М.: Пищевая промышленность, 1979. — 176 с.
Биохимия'. Практикум/Н. Е. Кучеренко. Ю. Д. Бабенюк. А. Н. Васильев и др. — Киев: Высшая школа, 1988. — 125 с.
Биохимия'. Сборник задач и упражнений/Н. Е. Кучеренко, Ю. Д. Бабенюк. А. Н. Васильев и др. — Киев: Высшая школа. 1988. — 104 с.
Гурова Н. В., Токаев Э. С.. Гуров А. Н. Методы определения эмульсионных свойств белков. — М.: АгроНИИТЭТИММП, 1994. — 32 с.
Землянухин А. А. Малый практикум по биохимии, — Воронеж: Изд-во ВЕУ, 1985. - 128 с.
Коренман Я. И. Практикум по аналитической химии. Титриметри-ческие методы анализа. — Воронеж: Изд-во ВГУ, 1986. — 244 с.
Коренман Я. И. Практикум по аналитической химии. Оптические методы анализа. — Воронеж: Изд-во ВГУ, 1989. — 228 с.
Коренман Я. И. Практикум по аналитической химии. Электрохимические методы анализа. — Воронеж: Изд-во ВГУ, 1992. — 191 с.
Коренман Я. И. Практикум по аналитической химии. Хроматографические методы анализа. — Воронеж. Изд-во ВГУ. 2000. — 336 с.
Лабораторный практикум по технологии переработки жиров/Н. С. Арутюнян, Л. И. Янова, Е. А. Аршиева и др. — М.: Агропромиздат, 1991. — 160 с.
Лурье И. С. Технохимический контроль сырья в кондитерском производстве. — М.: Агропромиздат, 2001. — 351 с.
Манихин Ю.А., Манихин С. А. Инженерная реология пищевых материалов. — М.: Легкая и пищевая промышленность, 1981. — 216 с.
Павловский П. Е., Пальмин В. В. Биохимия мяса. — М.: Пищевая промышленность, 1975. — 343 с.
Практикум по биохимии/Г. А. Соловьева и др.; под ред. С. Е. Северина, Г. А. Соловьевой. — 2-е изд., перераб. — М.: Изд-во МГУ. 1989. - 508 с.
Практикум по биохимии/Н. В. Алексахина, Н. Н. Зайцева. Н. П. Мешкова и др.; под ред. Н. П. Мешковой и С. Е. Северина. — М.: Изд-во МГУ, 1979. -429 с.
Практикум по биохимии/Т. М. Ермохина, М. В. Пахомова. И. А. Крашенинников и др. — М.: Изд-во МГУ, 1991. Ч. 1. — 190 с.
Практикум по ветеринарной радиобиологии/А. Д. Белов, А. С. Косенко, В. В. Пак и др.; под ред. А. Д. Белова. — М.: Агропромиздат, 1988. - 240 с.
Практикум по ветеринарно-санитарной экспертизе с основами технологии продуктов животноводства/Под ред. В. А. Макарова. — М.: ВО «Агропромиздат», 1987. — 271 с.
566
Рогов НА.. Горбатов А. В.. Сепиров В. Я. Дисперсные сислемы мясных и молочных продуктов. — М.: .Агропромиздат. 1990. - 320 с.
Рогов И. А.. Забашта А. /'.. Казю.шн Г. П. Общая технология мяса и мясопродуктов. — М.: Колос. 2000. — 368 с.
Родина Г. Т.. Вуке Г. А. Дегустационный анализ продуктов. Мл Колос. 1994. — 192 с.
Саааватуаина Р. №.. Алиев С. А.. Любченко В. Н. Новый метод определения основных функциональных свойств фарша//Мясная индустрия СССР. - 1983. - №'9. - С. 26- 27.
СанПиИ 2.3,560—96. Гигиенические требования к качеству и безопасност продовольственного сырья и пищевых продуктов. — М.: Изд-во стандартов. 1997. — 267 с.
Структурно-механические харак i еристики пищевых продуктов/ Под рет. Л. В. Горбатова. — М.: Легкая и пищевая промышленность. 1982. - 296 с.
Технохимическии контроль производства мяса и мясопродуктов/ Н. К. Журавская. Б. Е. Гутник. Н. Л. Журавская и др. — М.: Колос. 2001.- 476 с.
Филиппович Ю. Б.. Егорова Т.А., Севастьянова ГА. Практикум по общей биохимии. — М.: Высшая школа. 1982. -- 318 с.
предметный указатель
А
Автолиз 7. 265
Аденозинтрифосфорная кислота 49
Адреналин 303. 305
Активность воды 52, 54, 187
Актин 12
Актомиозин 13
Аминограмма 116
Аминокислоты 5, 10
— свободные 26, 30
Анализ фракционного состава
белков 69—71
— аминокислот 113
Аниониты 110
Ансерин 16
Антикоагулянты 273
Б
Безвредность 310
Белки миофибриллярные 10
— мышечные 12
— неполноценные 9
— плазмы крови 19, 271
— полноценные 9
— саркоплазмы 10
— стромы 14
В
Взаимодействие Ван-дер-Ваальсо-во 51
Вискозиметр ротационный 247, 256
— Энглера 248, 259
Витамин А 315
D 315
Е 316
К 316
Влага свободная 49
— связанная 50
----адсорбционно 51
----капиллярно 51
---- осмотически 51
Влагоудерживающая способность
202, 233, 234
Вода 49
Водосвязывающая способность
230, 232
Вольтамперометрия 415
Вязкость пластическая 203
— эффективная 203
Г
Гель-хроматография 22, 30
Гем 18
Гемицеллюлоза 46
Гемоглобин 17, 18, 80, 91
Гидроксилизин 14
Гидроксипролин 14
Гистон 13
Гликоген 6, 44, 45, 267, 285
Гликолиз 284
Глицерофосфолипиды 36
Глобулин X 11
Глюкоза 45, 46
д
Дипептиды гистидинсодержащие 15
Дифосфатидилглицерин 37
Ж
Жиры животные 35
568
3
Закон Фурье 197
Застудневание 238
И
Индекс Корпачи 312
— Озера 312
Инсулин 276, 279, 297
Интенсивность звука 194
Ионообменники 110
К
Карбамид 61
Кардиолипин 37
Карнозин 16
Каротиноиды 40
Каротины 40
Катепсины 269—271, 288
Катиониты 110
Кефалин 37
Кислота аденозинтрифосфорная
49, 267
— арахидоновая 314
— аскорбиновая 54
— галактуроновая 46
— линолевая 313
— линоленовая 313
— метилгуанидинуксусная 47
Клетчатка 44
Коллаген 6, 14, 15, 74
Контаминация 405
Коэффициент отражения 192, 208
— разделения 107
— различия аминокислотного скора 356
— распределения 29
— температуропроводности 196
— теплопроводности 198
— утилитарности 357
— эффективности белка 318
Крахмал 44
Креатин 47, 48
Креатинфосфат 48
Кровь 16, 19, 271
Л
Лецитин 37
Лиганд 29
Лиогель 238
Липиды 6. 34. 35
— простые 35
— сложные 35
- неомыляемая фракция 35
Липкость 249, 261
Липоиды 40
М
Метод Ван-Гизона 229
— Воловинской 76
— Зангер — Блека 509
— Кьельдаля 57, 58
— Либермана-Бухарда 150
— Лоури 19, 63
— в модификации Ластыть 64
----Дэвени — Гергей 63
— пептидных карт 28
— Салаватулиной 236
— Фолча 134
Миоальбумины 11
Миоген 11
Миоглобин 11,12
Миозин 12
Модуль упругости 203
Монохроматор 204, 207
Н
Нингидрин 94, 96
Нуклеопротеиды 13
О
Окраска препаратов по Граму 438
----по методу Ольта 439
------Ребигера 439
Олигосахариды 43
Определение аденозинтрифосфор-ной кислоты 178
— адреналина по Фолину 306
— активности воды 185—188
— актомиозина 71—73
— аминокислот 123—128
— аминного азота 92—98
------медным способом 93, 95
— белка по связыванию красителей 65, 66
— белков суммарных 55, 60—68
— влаги 181 — 185
— влагосвязывающей способности
----методом прессования 230
------центрифугирования 232
569
— водоудерживающей способности 236
— гелеобразующей способности белков 238
— гемоглобина 80—84, 87. 89
— гликогена 157—161
— глюкозы по методу Бертрана 287
— дипептидов 131. 133
— жирнокислотного состава жиров 152-157
— жироудерживающей способности 234
— креатинфосфата 180
— липидов 135—137
— липкости 261, 262
— молочной кислоты 161 — 168
— органического железа крови
87-89
— пектина 169—173
— пигментов мяса 89—92
— пролина 101 — 104
— прочности студня 242
— стабильности эмульсии 236
— студнеобразующей способнос-
ти 242
— температуры плавления жиров 140-144
— триптофана 98—101
— форм гемоглобина 84—86
— фосфолипидов 141 — 144
— фосфорорганических соедине-
ний 177-181
— фракционного состава жиров 138-141
— холестерина 144—152
— целлюлозы 173—177
----методом Ермакова 174
------Лурье 175
— экстрактивных веществ 128—133
П
Панкреатин 275
Пектин 46
Пенетрометр 246, 153
Плазма крови 17, 40
Полисахариды 43
Полярография 415
Предельное напряжение сдвига
203
Прибор Валента 241
— Зангер — Блека 509
— Соколова — Большакова 262
— Тарр — Бейкера 243
Проба Тейхмана 83
Протамин 13
Р
Радиоактивность 541
Разделение белков ----на ДЭАЭ-целлюлозе 11 1 ----на КМ-целлюлозе 1 12
Реактив Стокса 84
Реакция Геллера 299
— Грисса 496
— Залковского 148
— Либермана — Бухарда 149
— Миллона 299
— Паули 122
— Райдона и Смита 123
— Сакагуша 122
— Смита 123
— Фоля 299
— Эрлиха 122
Ретикулин 15
С
Свертывание крови 272
Скор аминокислотный 310, 312, 335
Смесь Ван Гизона 223, 225
Соединения фосфорорганические 47
Созревание мяса 45, 267
Сопротивление удельное акустическое 193
Сорбент 31
Спектроскопия 415
Среда Вильсон — Блера 433
— Кауфмана 429
— Китта — Тароцци 431
— Крумвиде — Олькеницкого 430
— Мюллера 429
— Симмонса 433
— Эндо 429
Стериды 38
Стероиды 38
Студень 238
Сыворотка крови 17
Т
Теплоемкость удельная 196
Триглицериды 36, 40
570
Тромбин 272. 294
Тропомиозин 13
Тропонин 13
У
Углеводы 43
Уравнение Клапейрона — Менделеева 186
— Нернста 419
— Ильковича 420
Установка для отгонки аммиака 58
— Тышкевича 261
Ф
Фибриноген 17, 293
Фиксация 226, 229
Фосфатидилглццерин 37
Фосфатидилсерин 37
Фосфатидилтреонин 37
Фосфатидилхолин 37
Фосфатиды 38
Фракционный состав белков 69
---жиров 69
X
Характеристики мяса акустические 211 ---микроструктурные 223
----теплофизические 215
Холестерин 39. 40
Хроматограмма восходящая 28
— нисходящая 28
Хроматография адсорбционная в тонком слое 30
— аффинная 21. 30
•— газовая 21. 22, 30
— газожидкостная 30
- гель-проникающая 22
— жидкостная 21, 22. 30
— ионообменная 23, 25, 30
— тонкослойная 21, 26, 30
Хромопротеид 11
ц
Целлюлоза 173
Циклизация 109
Ч
Число кислотное 361
— перекисное 361
Э
Эластин 14
Элюирование 32, 108, 113
Элюит 112
Эмульсия 199
Энергия связи 50
Учебное издание
Людмила Васильевна Антипова Ирина Анатольевна Глотова Иосиф Александрович Рогов
МЕТОДЫ ИССЛЕДОВАНИЯ МЯСА И МЯСНЫХ ПРОДУКТОВ
Учебник для вузов
Художественный редактор В. А. Чуракова
Технические редакторы Н. Н. Лопашова, Н. Н. Зиновьева Корректор Л. А. Котова
Лицензия № 010159 от 06.03.97 г.
Сдано в набор 01.03.2001. Подписано в печать 23.10.2001.
Формат 60x88*/,л. Бумага офсетная № 1. Гарнитура Ньютон.
Печать офсетная. Уел. п. л. 35,28. Уч.-изд. л. 38,70. Изд. № 045.
Тираж 3000 экз. Заказ № 457 «С» №047.
Государственное ордена Трудового Красного Знамени унитарное предприятие «Издательство «Колос», 107996, ГСП-6, Москва, Б-78, ул. Садовая-Спасская, 18.
Типография ОАО «Внешторгиздат», 127576, Москва, Илимская, 7.
Уважаемые читатели!
В издательстве «Колос» в 2001 г. вышел в свет учебник для вузов
Курочкин А. А., Ляшенко В. В.
ТЕХНОЛОГИЧЕСКОЕ ОБОРУДОВАНИЕ ДЛЯ ПЕРЕРАБОТКИ ПРОДУКЦИИ ЖИВОТНОВОДСТВА
Под редакцией В. М. Баутина
Рассмотрены технологическое оборудование для переработки продукции животноводства, а также вспомогательные машины, обеспечивающие реализацию производственного цикла изготовления молочных и мясных продуктов. Приведены технологические расчеты и справочные данные по основным видам оборудования.
Для студентов высших сельскохозяйственных учебных заведений по агроинженерным специальностям.
По всем вопросам обращаться по адресу:
107996, ГСП-6, Москва, Б-78, ул. Садовая-
Спасская, д. 18 (м. «Красные ворота»),
тел. 207-65-18,
факс 207-28-70.
Уважаемые читатели!
В издательстве «Колос» в 2001 г. вышел в свет учебник для вузов
Крусь Г. Н., Шалыгина А. М., Волокитина 3. В.
МЕТОДЫ ИССЛЕДОВАНИЯ МОЛОКА И МОЛОЧНЫХ ПРОДУКТОВ
Под редакцией А. М. Шалыгиной
Рассмотрены спектрофотометрические, электрохимические, хроматографические, реологические и другие методы исследований молочных продуктов. Описаны современные методы определения состава и свойств молока и молочных продуктов, содержания в них пестицидов, тяжелых металлов и других веществ.
Для студентов вузов, обучающихся по специальности «Технология молока и молочных продуктов», может быть полезен для аспирантов, специалистов научно-исследовательских институтов и лабораторий предпрятий, вырабатывающих молочные продукты.
По всем вопросам обращаться по адресу:
107996, ГСП-6, Москва, Б-78, ул. Садовая-
Спасская, д. 18 (м. «Красные ворота»),
тел. 207-65-18,
факс 207-28-70.
Уважаемые читатели!
В издательстве «Колос» в 2001 г. вышла в свет книга
Бредихин С. А., Бредихина О. В., Космодемьянский Ю. В., Никифоров Л. Л. ТЕХНОЛОГИЧЕСКОЕ ОБОРУДОВАНИЕ МЯСОКОМБИНАТОВ
В книге приведены общие сведения о технологическом оборудовании мясокомбинатов: структура, классификация, основные параметры, требования и организация эксплуатации оборудования. Рассмотрено основное технологическое оборудование, применяемое для переработки мясного сырья. Даны описание, устройство, принципы работы оборудования и его технические характеристики. В конце каждой главы приведены требования безопасности при эксплуатации оборудования.
Для работников мясной промышленности, может быть также использована для профессионального обучения рабочих на производстве.
По всем вопросам обращаться по адресу:
107996, ГСП-6, Москва, Б-78, ул. Садовая-
Спасская, д. 18 (м. «Красные ворота»),
тел. 207-65-18,
факс 207-28-70.
Уважаемые читатели!
В издательстве «Колос» в 2001 г. вышла в свет книга
Рогов И. А., Забашта А. Г., Казюлин Г. П.
ОБЩАЯ ТЕХНОЛОГИЯ МЯСА И МЯСОПРОДУКТОВ
Приведены характеристики состава и свойств сырья. Описаны технологические процессы переработки скота, птицы, кроликов. Даны основы холодильной обработки мяса и мясопродуктов, переработки крови, обработки эндокринно-ферментного сырья, субпродуктов, шкур, кишок, кератин содержаще го сырья.
Для специалистов мясной промышленности, может быть использована в качестве учебного пособия для вузов.
По всем вопросам обращаться по адресу:
107996, ГСП-6, Москва, Б-78, ул. Садовая-
Спасская, д. 18 (м. «Красные ворота»),
тел. 207-65-18,
факс 207-28-70.