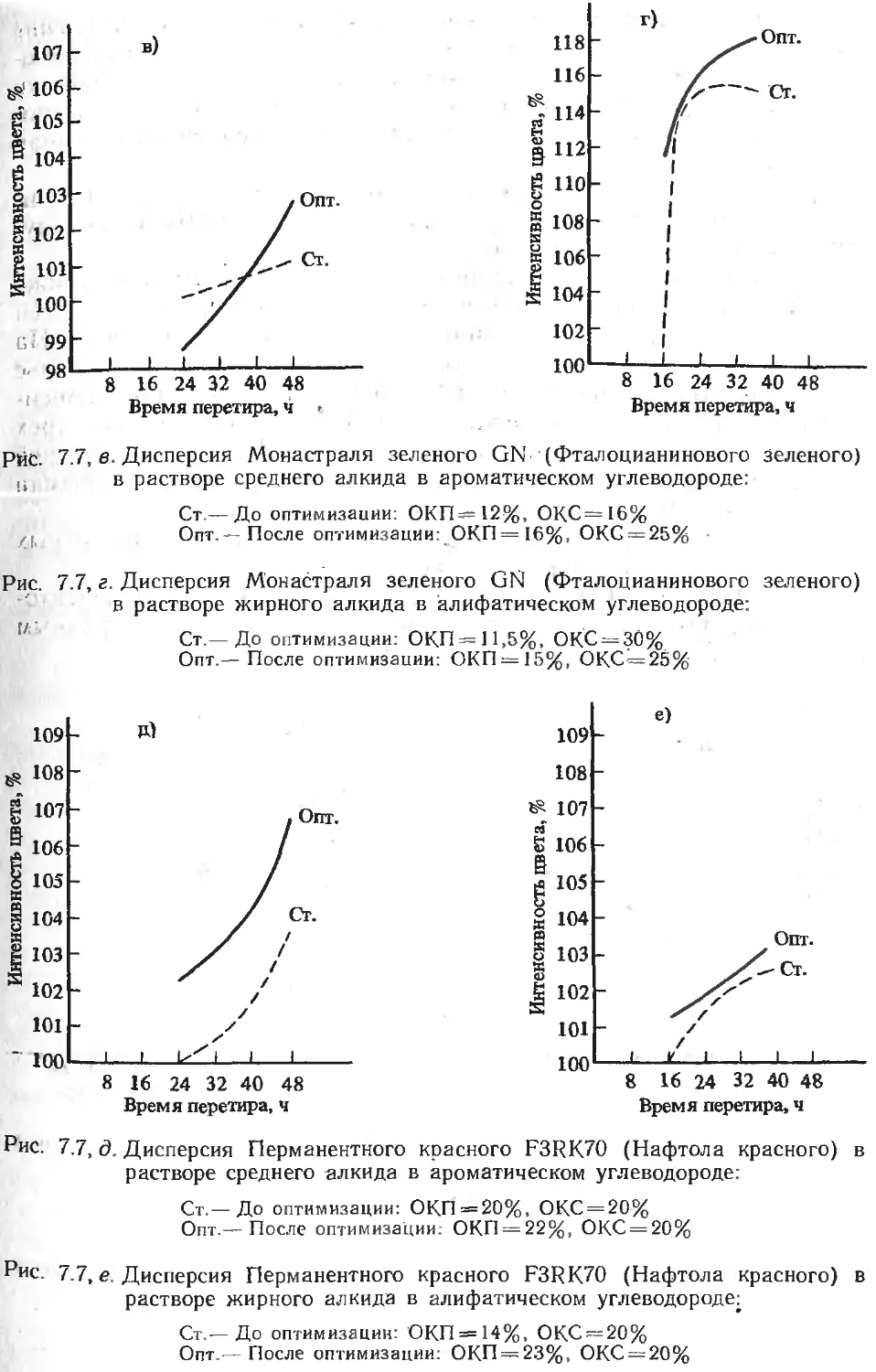
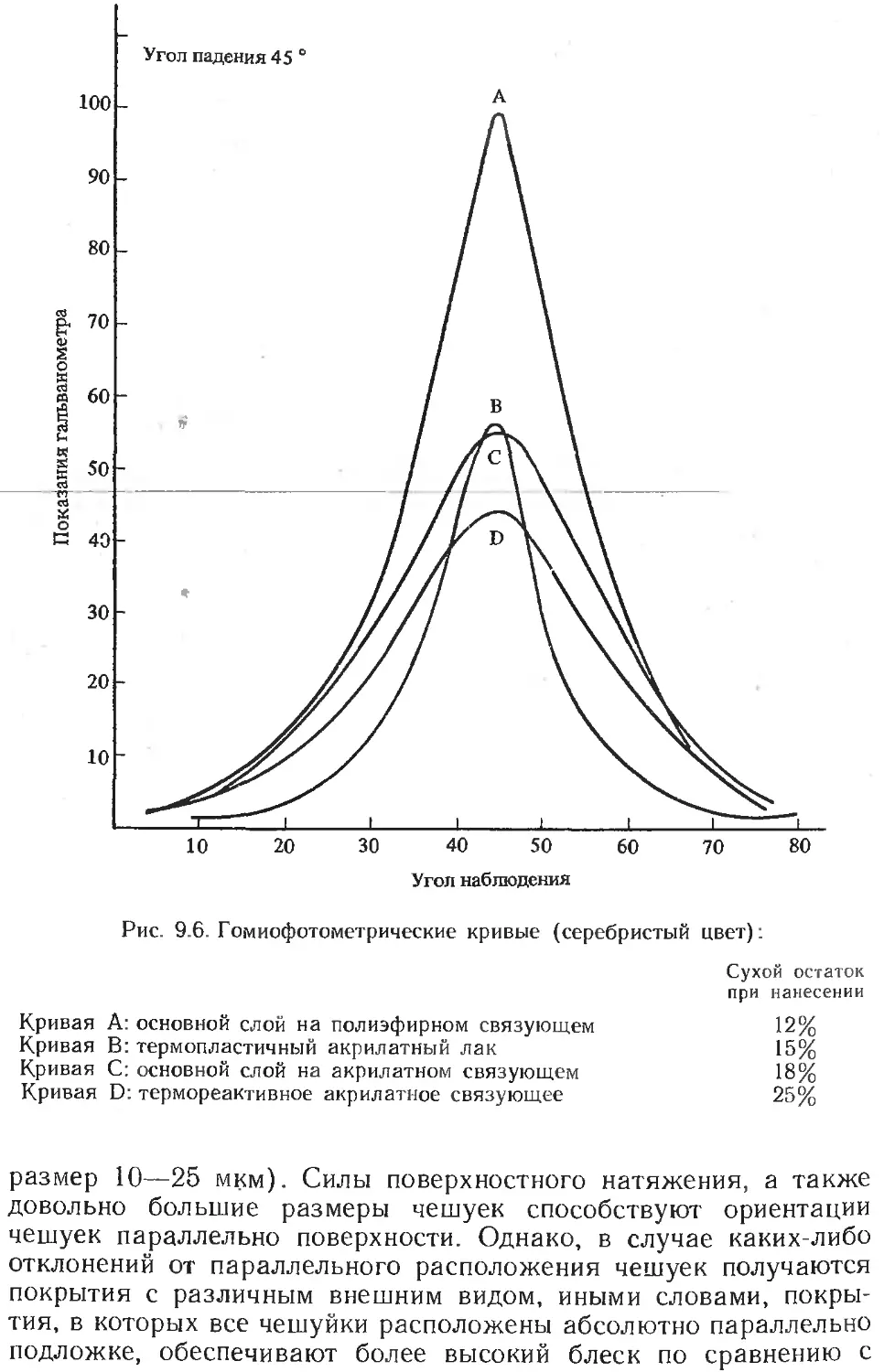
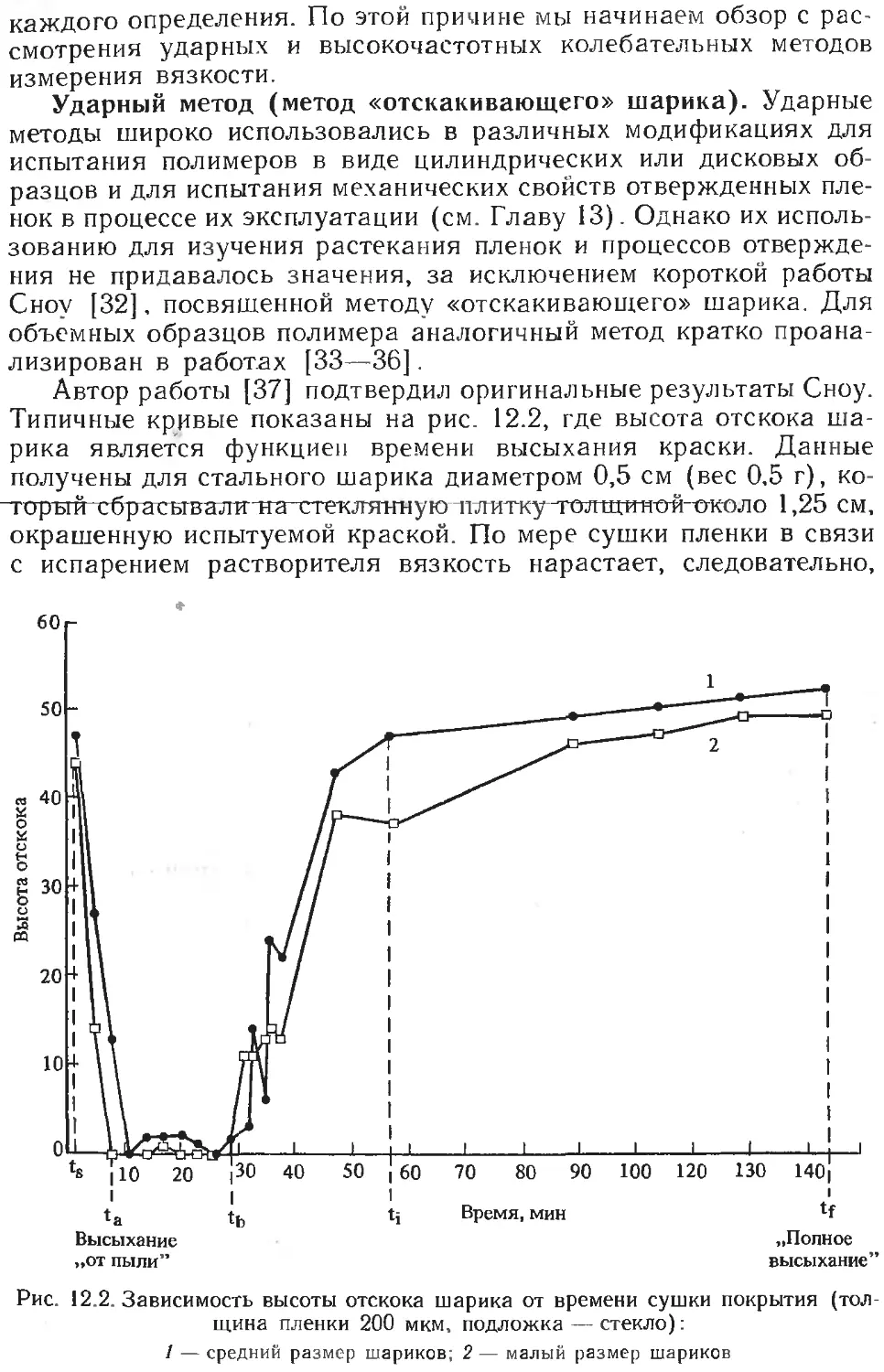
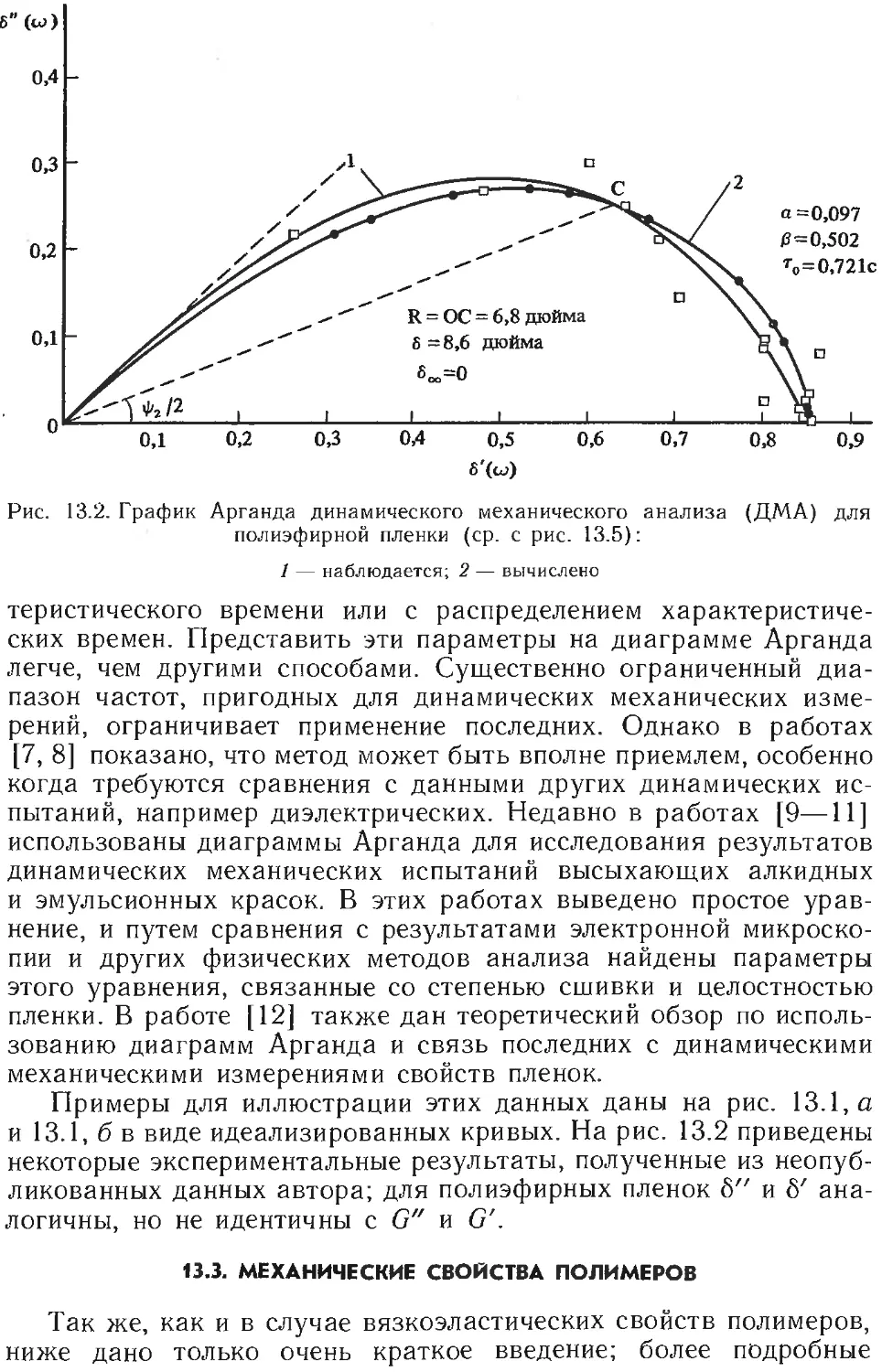
/



























































































































































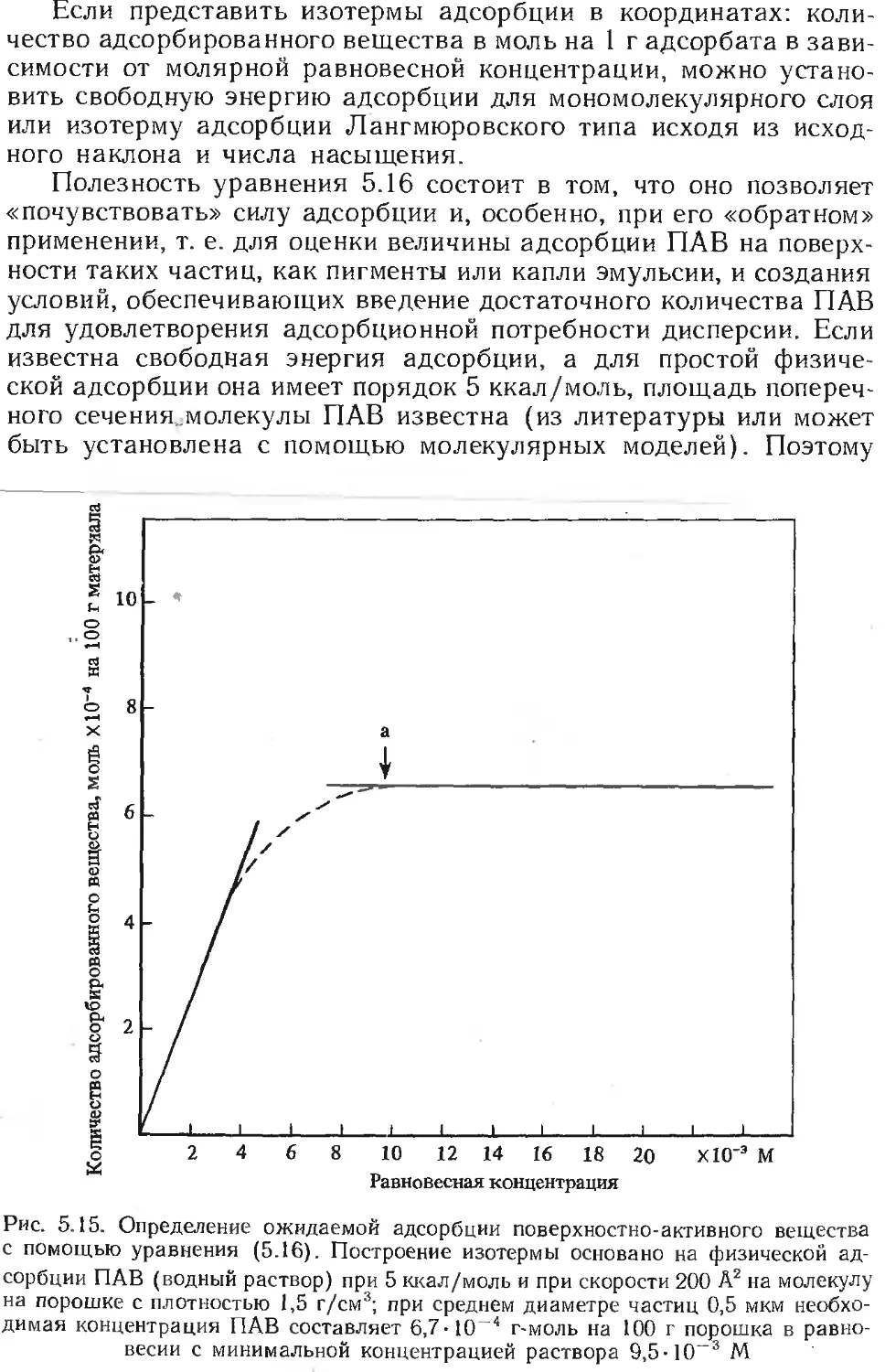






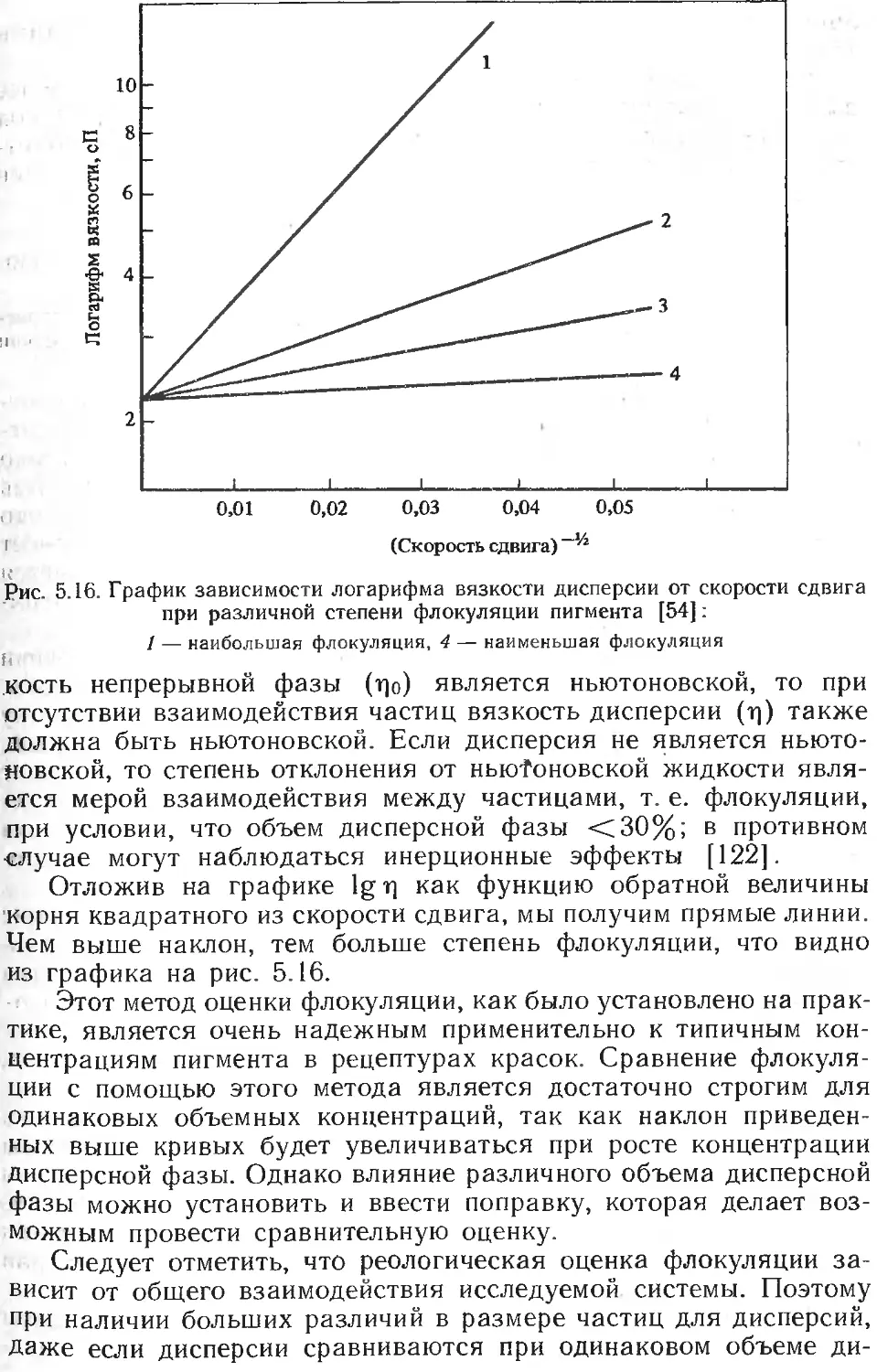










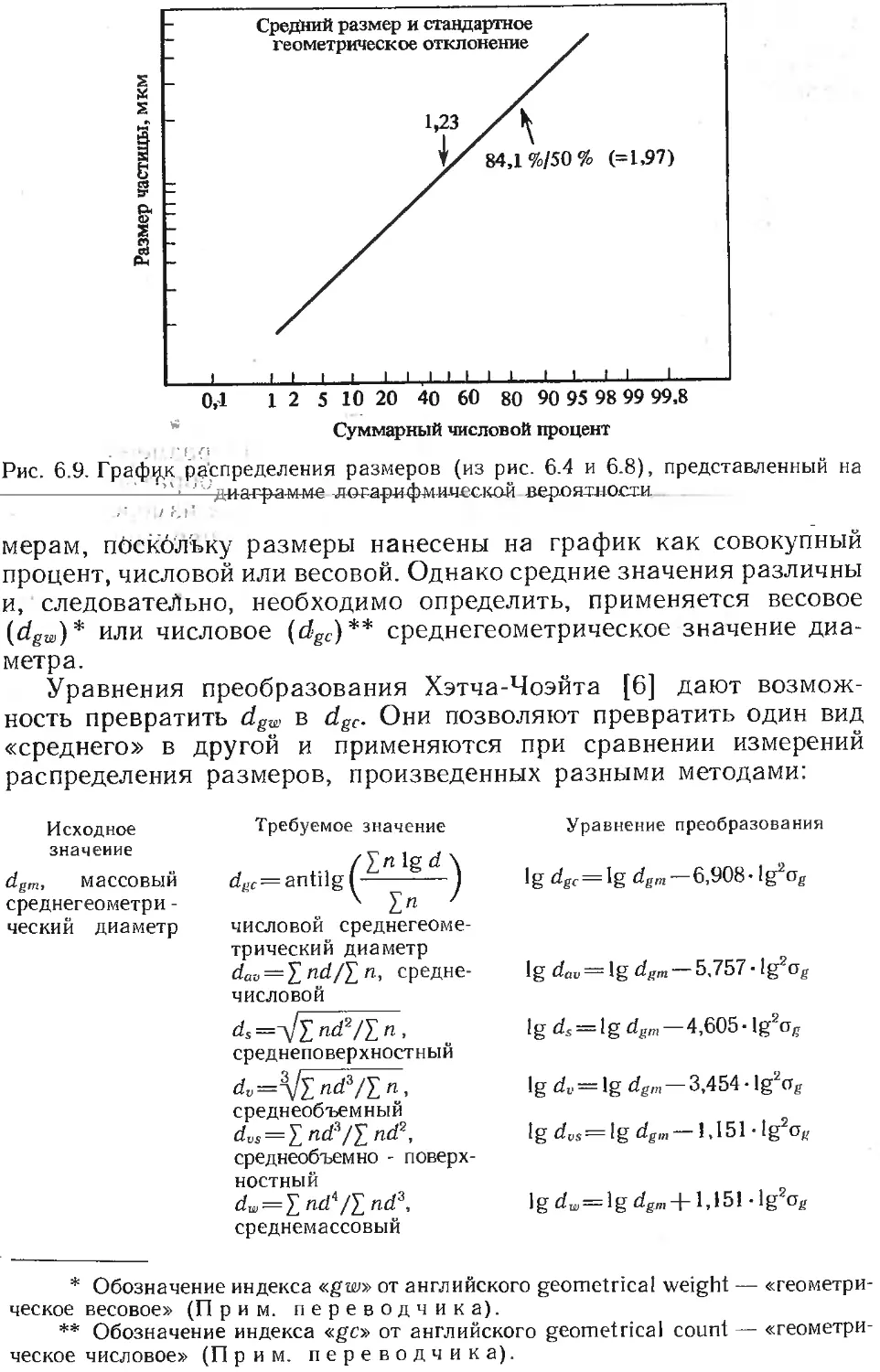






















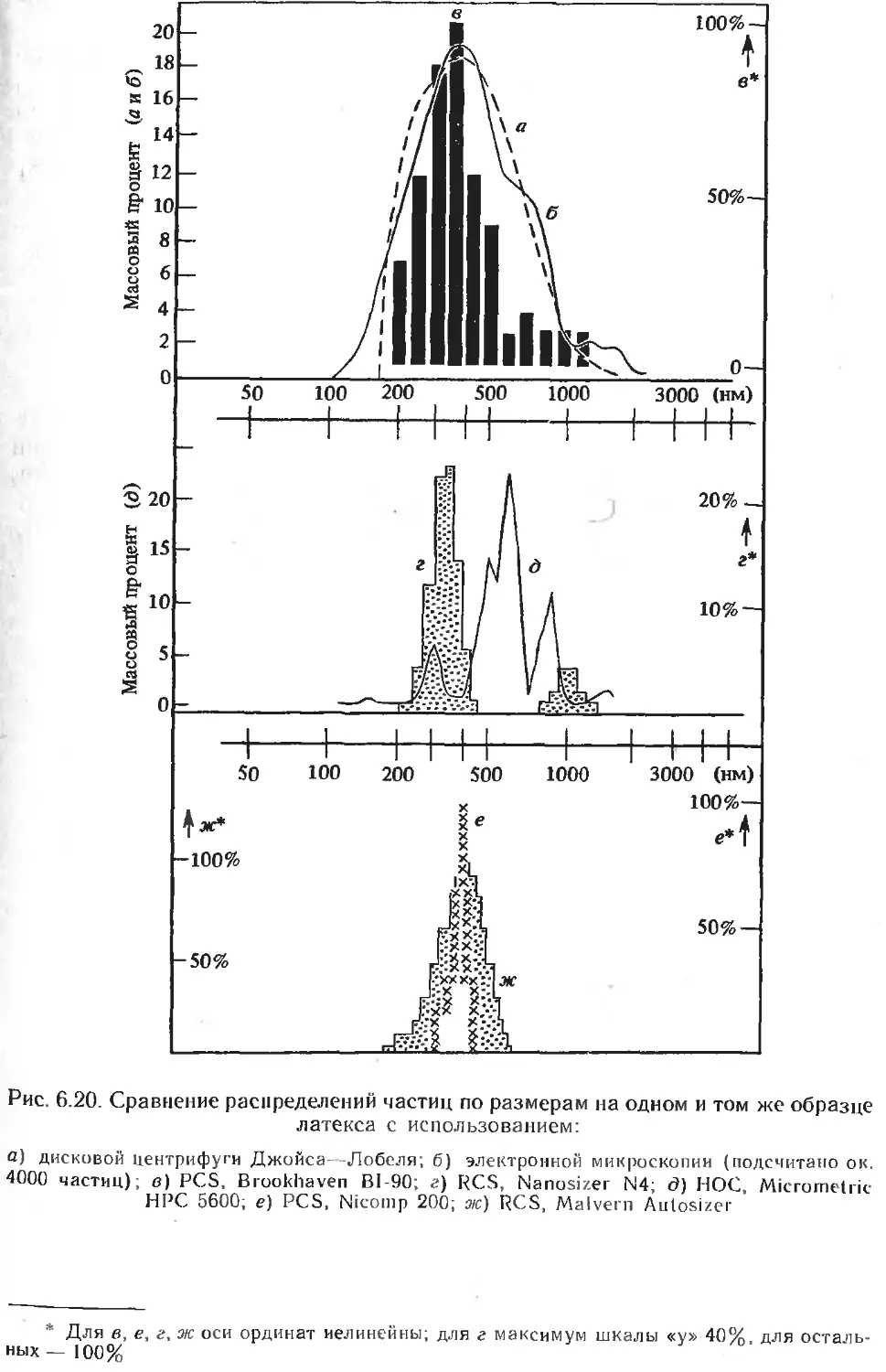





















































































































































































































































































































Текст
ЛАКОКРАСОЧНЫЕ
МАТЕРИ АЛ Ы
И ПОКРЫТИЯ
ТЕОРИЯ И ПРАКТИКА
Под редакцией Р. Ламбурна
Перевод с английского под редакцией
д-ра хим. наук, проф. л. н. машляковского
, и д-ра техн, наук А. м. ФРОСТА
САНКТ-ПЕТЕРБУРГ
«ХИМИЯ»
САНКТ-ПЕТЕРБУРГСКОЕ ОТДЕЛЕНИЕ
1991
PAINT AND
SURFACE COATINGS:
Theory and Practice
Editor:
R. LAMBOURNE
Technical Manger
INDCOLLAG (Industrial Colloid Advisory Group)
Department of Physical Chemistry
University of Bristol
ELLIS HORWOOD LIMITED
Publishers • Chichester
Halsted Press: a division of
JOHN WILEY & SONS
New York • Chichester • Brisbane • Toronto
Рецензент: д-р техн, наук, проф. А. Д. Яковлев
УДК 667.6
Лакокрасочные материалы и покрытия. Теория и практика:
Пер. с англ./Под ред. Р. Ламбурна — СПб.: Химия, 1991.— 512 с.—
Пер. изд.: Великобритания, 1987.— ISBN 5—7245—0446—4
Книга представляет собой фундаментальный труд по теории и практике
производства и применения лакокрасочных материалов, отражающий мировой
уровень развития отрасли. Большое внимание уделено рассмотрению состава и
важнейших свойств готовых лакокрасочных материалов и покрытий на их основе.
Подробно описаны современные методы исследования механических и декора-
тивных свойств покрытий, контроль внешнего вида и оценки долговечности. Осо-
бенно интересны и ценны сведения по перспективным материалам (краскам водо-
разбавляемым, с высоким сухим остатком, радиационно-отверждаемым покры-
тиям и др.; добавкам для придания декоративных, защитных, специальных
свойств), а также данные по физикохимии и реологии дисперсных систем. Широко
показано применение лакокрасочных материалов в строительстве, автомобиле-
и судостроении.
Для научных и инженерно-технических работников, занятых производством
лакокрасочных материалов и применением их в разных отраслях народного хо-
зяйства.
Табл. 22. Ил. 89. Библиогр.: 710 назв.
„ 2804090000—098
Л —050(01)-91 98~9’
ISBN 5—7245—0446—4
© 1987 R. Lambourne/Ellis Horwood Limited
© Перевод на русский язык, Л. Н. Машля-
ковский, А. М. Фрост, Е. А. Фрост,
И. А. Луковский, В. Ю. Репкин, А. И. Ярцев
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Вниманию читателей предлагается перевод книги, написанной
группой ученых и специалистов ведущих английских лакокрасоч-
ных фирм (Ай-Си-Ай, ППГ-Индастри) и Бристольского универ-
ситета. Книга издана под редакцией Р. Ламбурна, являющегося
крупным специалистом в области лакокрасочных материалов.
Книгу можно рассматривать как фундаментальный труд по
теории и практике производства и применения лакокрасочных
материалов и покрытий; она охватывает практически все вопросы,
связанные с составом лакокрасочных материалов (гл. 1), химией
и технологией пленкообразователей (гл. 2), пигментов (гл. 3),
целевых добавок (гл. 4). В ней описываются физическая химия
и важнейшие свойства пигментных дисперсий, включая вопросы
смачивания, флокуляции и стабилизации (гл. 5), а также опреде-
ления размера диспергированных частиц (гл. 6).
Отдельные главы посвящены описанию общих проблем техно-
логии и оборудования для производства лакокрасочных мате-
риалов (гл. 7).
Главы с 8 по 11 посвящены описанию свойств готовых лако-
красочных материалов и покрытий на их основе для разнообраз-
ных областей применения. При этом наиболее подробно изложен
материал, посвященный краскам и покрытиям для строительных
работ и автомобилестроения.
В гл. 12 серьезное внимание уделено вопросам реологии лако-
красочных материалов; здесь приведены весьма ценные сведения
о методах исследования реологических свойств и о взаимосвязи
реологии с процессом производства и хранения лакокрасочных
материалов. В главах 13—16 приведено описание методов иссле-
дования механических свойств покрытий, оценки их декоративных
свойств, включая многообразные методы цветовых измерений,
контроля внешнего вида покрытий и оценки их долговечности.
Особо следует отметить высокую информативность предлагаемых
методов исследования, базирующихся на высокоточном обору-
довании при соответствующем программном обеспечении с по-
мощью компьютерной техники.
Слишком большой объем книги потребовал внести при ее
переводе ряд сокращений. Так, из текста перевода исключены
разделы, посвященные свойствам и применению масел и масля-
ных лаков, фенолформальдегидных и амино-смол, эпоксидных,
полиуретановых, силиконовых и ряда других материалов. Опу-
щена также глава, посвященная описанию свойств растворителей
и разбавителей и некоторые другие разделы.
Подробные сведения по указанным вопросам можно найти
в отечественной литературе, список которой прилагается к дан-
ной книге.
Несмотря на многие достоинства книги, она не свободна от
ряда недостатков.
Прежде всего следует отметить неодинаковый методологиче-
ский подход, форму и глубину изложения материала разных глав,
так как они написаны разными авторами. Весьма глубокими по
содержанию, теоретическому и прикладному значению являются
главы 5, 6, 7. Очень полезный материал содержится в главах 4,
14, 15, где описаны целевые добавки к лакокрасочным материа-
лам, а также методы исследования оптических свойств покрытий.
Вместе с тем имеются главы (например, главы 2 и 3), где материал
был изложен с химической точки зрения весьма конспективно,
что и послужило одной из причин сокращения материала этих глав.
Хотя книга претендует на комплексное рассмотрение теории
и практики производства и применения лакокрасочных материа-
лов и покрытий, нельзя однозначно утверждать, что эта задача
авторами выполнена -в полной мере.
Имеются отдельные разделы, которые либо слабо освещены,
либо совсем не получили освещения. Так, большой объем книги
посвящен применению лакокрасочных материалов в строитель-
стве (гл. 8) и автомобилестроении (гл. 9 и 10), в то время как их
применению в других отраслях техники отведено всего 28 с. Со-
вершенно недостаточный объем отведен применению лакокрасоч-
ных материалов в судостроении и судоремонте (глава 11, всего
19 с), причем мало внимания уделено современной тенденции
развития этого направления лакокрасочной технологии. В книге
не представлены данные по исследованию таких очень важных
характеристик лакокрасочных покрытий, как проницаемость и
адгезия.
В некоторых случаях авторы применяют термины и обозначе-
ния, которые не находят применения или отсутствуют в отечествен-
ной научно-технической литературе (например, «среднегармони-
ческий диаметр» — гл. 6 и др.). Схема коррозионного процесса
(гл. 11) представлена весьма упрощенно.
Несмотря на ряд недостатков, следует признать, что книга
содержит много ценных и полезных сведений для специалистов,
работающих в области различных направлений технологии лако-
красочных материалов и покрытий. Она отражает в целом миро-
вой уровень развития отрасли, так как Великобритания относится
к числу ведущих стран с широко развитой лакокрасочной про-
мышленностью, а фирма Ай-Си-Ай занимает лидирующее поло-
жение в мире по объему производства лакокрасочной продукции.
Л. Н. Машляковский,
А. М. Фрост
ПРЕДИСЛОВИЕ К АНГЛИЙСКОМУ изданию
Уже давно я чувствовал необходимость написания книги, ко-
торая помогла бы выпускникам учебных заведений, начинаю-
щим работать в лакокрасочной промышленности, использовать
полученные теоретические знания в прикладной науке и техноло-
гии производства лакокрасочных материалов и покрытий. Хотя
по технологии издано много прекрасных книг, насколько мне из-
вестно, отсутствуют монографии, посвященные научным основам
химии и физики покрытий. Многие из стандартных технологиче-
ских руководств в настоящее время устарели (и не издаются),'
поэтому представлялась оправданной попытка написания книги,
которая, я надеюсь, восполнит существующий пробел. Тем не
менее, не без колебаний я решился на издание, охватывающее
такую сложную технологию. Эта область знаний настолько ши-
рока, что редко признанный эксперт в одном из ее аспектов будет
чувствовать себя достаточно уверенным в другом. Поэтому мне
казалось, что один автор не справится с задачами, поставленными
перед данным изданием, и я обратился к друзьям и коллегам,
работающим в промышленности, с просьбой написать отдельные
главы по проблемам, в которых они имели все необходимые зна-
ния и опыт. К счастью, мне удалось получить согласие на участие
в написании книги наиболее квалифицированных специалистов
с большим опытом работы в лакокрасочной промышленности.
Однако из-за ограниченного объема книга не могла быть
всеобъемлющей. Поэтому мне пришлось принять такую ее струк-
туру, которая бы позволила уделить больше внимания вопросам
физики красок и физической химии дисперсий по сравнению с
большинством других подобных книг. Это же послужило причи-
ной сокращения объема (а иногда и глубины) описания отдельных
технологий. Например, если главы, посвященные окраске автомо-
’билей и строительным лакокрасочным материалам, представлены
довольно подробно, глава о красках общего промышленного на-
значения изложена кратко с целью проиллюстрировать многообра-
зие предъявляемых промышленностью требований и те проблемы,
которые приходится решать в связи с этим технологам при про-
изводстве лакокрасочных материалов.
В главах, в которых рассматриваются теоретические основы
технологии, авторам было предложено критически проанализи-
ровать современное состояние науки и технологии в соответствую-
щей области. Это нашло отражение в обширном списке ссылок
на оригинальные работы, опубликованные преимущественно в
течение последних 10 лет. Мне кажется, что указанные работы
будут полезным источником информации для читателей, заинте-
ресованных в дальнейшем углублении своих знаний.
Следует отметить, что кроме авторов в издание этой книги
внесли вклад и другие участники. Я хотел бы поблагодарить док-
тора Гордона Феттиса, научного руководителя отдела красок
фирмы Ай-Си-Ай, миссис Милли Коэн (Ай-Си-Ай) и миссис Катю
Слэттерн (Бристольский университет), напечатавшим большую
часть рукописи.
Р. Ламбурн, апрель 1986 г.
СОСТАВ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
И ИХ ПРИМЕНЕНИЕ — ОБЩЕЕ ВВЕДЕНИЕ
Р. Лаллбурн
1.1. КРАТКИЙ историческим обзор
Предполагают, что первобытные люди иговляли первые
краски около 25 000 лет тому назад. Они были охотниками и жи-
телями пещер и, очевидно, под влиянием вдохновения на каменных
стенах своих пещер делали наброски животных, на которых охо-
тились, и раскрашивали их. Создавая эти образы, люди, вероятно,
думали, что их власть над добычей возрастет.
Химический анализ пещерных рисунков, обнаруженных в Аль-
тамире (Испания) и Ласкауксе (Франция), показывает, что ос-
новными пигментами, которые использовали художники времен
палеолита, были оксиды железа и марганца. Они обеспечивали
получение трех основных цветов, найденных в большинстве пещер-
ных картин, а именно: черного, красного и желтого, наряду с про-
межуточными оттенками. Возможно, йспользовались также уголь
после сжигания древесины, желтый карбонат железа и мел. Стран-
но, что в Ласкауксе, где естественная окраска камня использо-
валась как бледный фон, нет и следов применения белого пигмен-
та, который в наше время является наиболее широко используе-
мым. Однако белые пигменты встречаются в некоторых доистори-
ческих картинах в Африке.
Эти земляные пигменты измельчали в тонкий порошок пести-
ком в ступке. Считают, что в качестве ступок использовали при-
родные камни с углублениями, а кости служили пестиками, о чем
свидетельствовали находки таких предметов, окрашенных пиг-
ментами. Порошкообразные пигменты, по-видимому, смешивали
с водой, костным мозгом, животными жирами, яичным белком
или с растительными сахарами и получали краски. Их наносили
«тыканием» пальца или с помощью примитивных тампонов или
кистей из волос, меха животных или мха. Пещерные рисунки со-
хранились потому, что они располагались глубоко внутри пещер,
входы в которые впоследствии оказались плотно закрыты. Эти
краски обладают очень плохой долговечностью, а связующие
служили просто для того, чтобы приклеить пигменты к стенам
пещер.
В период, приблизительно, между 3000 и 600 гг. до н. э. егип-
тяне значительно развили искусство приготовления красок. Они
разработали более широкую цветовую гамму пигментов, которые
включали синие цвета, лазурит (смешанные кристаллы силиката
натрия и сульфида натрия) и азурит (химически аналогичный
малахиту). В этот период начали применять красную и желтую
охры (оксид железа), желтый трисульфид мышьяка, зеленый
малахит (основной карбонат меди), лампоь^ю сажу и белый пиг-
ментный гипс (сульфат кальция). Первый синтетический пигмент,
известный сегодня как Египетский голубой, был получен почти
5000 лет назад. Его приготовили путем прокаливания извести,
карбоната натрия, малахита и кремнезема при температуре свы-
ше 830 °C. Египтянам же принадлежит разработка первых кра-
сочных лаков. Их готовили путем осаждения растворимых орга-
нических красителей на неорганическую (минеральную) основу
и «фиксирования» их химическим путем с образованием нераст-
воримого соединения. Вначале для этих целей был использован
красный краситель, полученный из корней растения Марены
(красильной). В настоящее время из-за низкой светостойкости
он нигде больше не используется за исключением художествен-
ных красок («розовый крапп»). Однако и сегодня красочные лаки
по-прежнему представляют важную группу пигментов. Древние
египтяне начали использовать свинцовый сурик в защитных крас-
ках для древесины, однако более широкое применение он получил
у римлян. В качестве пленкообразователей почти исключительно
использовали природные смолы, расплавы восков, поскольку необ-
ходимые растворители были неизвестны. Льняные и другие высы-
хающие масла были известны, однако нет никаких доказательств,
подтверждающих их применение в красках.
Греки и римляне в период между 600 г. до н. э. и 400 г. н. э.
почти наверняка знали, что лакокрасочные покрытия могут вы-
полнять как защитные, так и декоративные функции при окраске
объектов. В это время начали использовать лаки на основе высы-
хающих масел. Однако только в XIII в. хорошие защитные свой-
ства высыхающих масел начали признавать в Европе. В средне-
вековье многие картины, особенно на дереве, защищали лакиро-
ванием. Лаки приготовляли растворением соответствующих смол
в горячем льняном, конопляном или ореховом маслах, которые
со временем склонны к потемнению.
К концу XVIII в. спрос на краски всех типов возрос до такой
степени, что стало экономически выгодно организовывать произ-
водство лаков и красок для продажи. В 1833 г. Дж. В. Нэйл реко-
мендовал производителям в целях безопасности при производстве
лаков и красок всегда иметь при себе помощника: «Никогда не
делай ничего в спешке или небрежно... Нервные или робкие люди
не годятся ни в производители, ни в помощники; наибольшее
число несчастных случаев происходит вследствие спешки, страха
или опьянения». Это предостережение является свидетельством
увеличения как масштабов производства, так и опасностей исполь-
зования открытых реакторов для производства лаков.
Промышленная революция оказала большое влияние на раз1
витие лакокрасочной промышленности. Возрастающее примене-
ние железа и стали в строительстве и технике обусловило потреб-
ноСть в противокоррозионных грунтовках, которые бы замедляли
или предотвращали ржавление и коррозию. В связи с этим были
разработаны свинец- и цинксодержащие краски. Интересно от-
метйть, что одна из простейших красок на. основе свинцового
сурика, диспергированного в льняном масле, является по-преж-
нему, вероятно, одной из лучших противокоррозионных грунтовок
для конструкционной стали. Свинецсодержащие краски постепен-
но вытесняются не потому, что были разработаны лучшие, а из-за
их токсичности.
Ускорение научно-технического прогресса, начиная с XVIII в.
до настоящего времени, оказало растущее влияние на производ-
ство лаков и красок. Берлинская лазурь — первый искусственный
пигмент, химизм получения которого был понят — открыта в
1704 г. Использование скипидара в качестве растворителя красок
впервые описано в 1740 г. Металлические сиккативы для ускоре-
ния высыхания растительных масел начали применять в 1840 г.
Основа химии формальдегидных смол заложена в период
между 1850 и 1890 гг., хотя они не применялись в красках вплоть
до двадцатого века. Подобно этому в 1877 г. было открыто, что
нитроцеллюлозу можно сделать безопасной для применения в
качестве пластиков или пленок путем пластификации ее камфо-
рой, однако только после первой мировой войны ее начали ис-
пользовать в значительных количествах в производстве красок.
Настоятельная необходимость их применения была вызвана мас-
совым производством автомобилей. Огромные количества нитро-
целлюлозы производили для взрывчатых веществ во время войны.
В конце войны с уменьшением потребности во взрывчатых ве-
ществах для нитроцеллюлозы необходимо было найти другое
применение; массовое производство автомобилей обеспечило не-
обходимый рынок. Война ускорила использование открытий хи-
мии и рост химической промышленности. Появились новые цвет-
ные синтетические пигменты и красители, а в 1918 г. начали
использовать новый белый пигмент, диоксид титана, который
должен был полностью заменить свинцовые белила. Диоксид
титана при первоначальном применении в красках повысил бе-
лизну и укрывистость, или кроющую способность красок, однако
он же вызывал более быстрое разрушение лакокрасочных покры-
тий вследствие его фотоактивности. Последующие исследования
позволили преодолеть эту проблему и разработать современные
пигментные формы диоксида титана, которые можно применять
в любых лакокрасочных композициях без опасения ухудшить
эксплуатационные свойства покрытий.
Последующие главы этой книги будут в основном посвящены
рассмотрению прогресса в лакокрасочной промышленности, ко-
торый наблюдался в XX в., и особенно в последние 50 лет, когда
произошли наибольшие изменения.
1.2. ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
Термины «paint» и «surface coating» часто используются как
взаимозаменяемые. Термин «surface coating» является более
общим и применим к любому материалу, который может быть
использован в виде тонкой сплошной пленки на поверхности.
Термин «paint» (краска, лакокрасочное покрытие) традиционно
использовался для описания пигментированных материалов с
целью отличить их от прозрачных пленок, которые более правиль-
но называть лаками («lacquers» или «varnishes»). В данной книге
будут рассматриваться главным образом «paints» (то есть пиг-
ментированные лакокрасочные материалы и покрытия); однако,
как мы увидим, современные процессы окрашивания могут вклю-
чать получение составных систем, в которых общее покрытие
состоит из нескольких тонких пленок, причем не все они могут
быть пигментированными. Мы будем использовать оба термина
в соответствии с контекстом, в котором обсуждаются конкретные
лакокрасочные композиции.
Лакокрасочные покрытия выполняют двоякую функцию. Они
обеспечивают удовлетворение эстетических требований, выпол-
няют защитные функции, или и те и другие одновременно. Напри-
мер, при окраске автомобилей лакокрасочное покрытие должно
улучшить внешний вид автомобиля, то есть придать ему цвет
и блеск, а если корпус изготовлен из малоуглеродистой стали,
то оно должно обеспечить и защиту от коррозии. Если же корпус
изготовлен из стеклопластика, тогда функция покрытия будет
только декоративной. Существуют очень веские экономические
причины, по которым лучше окрашивать только наружную по-
верхность изделий, например из пластмасс, вместо окраски во
всем объеме за счет использования пигментированных материа-
лов при их изготовлении. Преимущества применения лакокрасоч-
ных покрытий особенно ясны в тех случаях, когда необходимо
иметь широкий набор цветовых эффектов. Эта тема будет рас-
смотрена в главах, посвященных лакокрасочным материалам
и покрытиям целевого назначения (гл. 8—11).
При рассмотрении природы красок станет совершенно ясно,
что очень важное значение имеет взаимосвязь между покрытием
и подложкой. Различны требования к краскам для древесины и
для металла. Более того, методы нанесения и сушки красок могут
сильно различаться. При разработке рецептуры краски для кон-
кретной цели важно знать, где, в каких условиях будет эксплуа-
тироваться окрашенное изделие, каковы требования к физиче-
ским и механическим характеристикам. Исследователь должен
знать, каким методом краска будет наноситься и отверждаться.
Так, краски для изделий из литой стали должны обладать хоро-
шим сопротивлением к разрушению при ударе (то есть отслаива-
нию) , в то время как к покрытиям для жестяных банок предъяв-
ляЬтся требование высокой гибкости. Эти различные требования
будут описаны в гл. 8—11, посвященных различным областям
использования лакокрасочных материалов.
Уже давно поняли, что трудно, если вообще возможно вполне
удовлетворить разнообразным требованиям, используя только
один слой краски. Достаточно перечислить требования, предъяв-
ляемые к типичному покрытию. Обычно требуются многие, если
не вое, из следующих свойств: укрывистость, цвет, глянец, шеро-
ховатость поверхности (текстура), адгезия к подложке, необхо-
димый механические или физические свойства, химическая стой-
кость) защита от коррозии и все то, что вкладывается в понятие
долговечность. Долговечность — очень важный показатель, к
которому мы будем часто возвращаться. Число различных слоев,
из которых состоит система покрытия, будет определяться типом
подложки и условиями эксплуатации окрашенного объекта. Ти-
пичная система глянцевого покрытия может состоять из грунто-
вочного слоя, промежуточного слоя и верхнего слоя. Каждый
из этих слоев может быть получен из пигментированных компо-
зиций, причем для формирования каждого слоя может быть ис-
пользовано неоднократное нанесение соответствующего лакокра-
сочного материала. Например, может быть один слой грунтовки,
промежуточный слой, полученный двукратным нанесением, и
верхний слой также двукратного нанесения. Назначение каж-
дого из слоев и, соответственно, их состав сильно различаются.
Грунтовка должна укрыть подложку и обеспечить достижение
хорошей адгезии между подложкой и промежуточным слоем.
Она может также дополнительно повышать укрывистость, но
это не является ее основной функцией. Промежуточный слой
имеет два назначения: обеспечить укрывистость подложки и вы-
равнивание поверхности, по которой будет нанесен верхний слой.
Гладкую поверхность получают путем шлифования шлифовочной
шкуркой после высушивания каждого слоя. Затем наносят верх-
ний слой, который также вносит вклад в укрывистость и обеспе-
чивает соответствующий эстетический эффект, например, цвет
и глянец. В целом покрытия должны обеспечить защиту деревян-
ных, металлических и других подложек, на которые они нанесены.
Целесообразно рассмотреть взаимосвязь между этими слоями.
Механические и физические свойства каждого из слоев весьма
различны. О назначении грунтовки обеспечить адгезию уже упо-
миналось. Кроме этого, может появиться необходимость умень-
шить напряжения, которые возникают в покрытии при отвержде-
нии и вследствие появления хрупкости верхнего (и промежуточ-
ного) слоя в результате процесса старения, или воспринять
напряжения, возникающие при различных движениях подложки.
Известно, что мягкие сорта дерева, используемые для оконных
рам, расширяются и сжимаются в сухих (летом) и влажных (зи-
мой) условиях почти на 10% поперек волокна, но намного меньше
вдоль волокон. Цвет промежуточного слоя должен приблизитёль-
но соответствовать цвету верхнего слоя. Обычно содержание
пигментов в нем высокое, в противоположность верхнему слою.
Причина этого в том, что от верхнего слоя требуется обеспечение
максимального глянца и растяжимости. Использовать лакокра-
сочные материалы верхнего слоя в промежуточных слоях поэтому
нецелесообразно. /
Очень часто грунтовка должна обеспечить защиту от фэрро-
зии. Поэтому при использовании по стальным подложка^ она
должна содержать в своем составе химически активный антикор-
розионный пигмент. Коррозионная защита может быть достигнута
дополнительно другими методами, в частности химической обра-
боткой подложки. Поэтому обычно технология получения покры-
тий включает предварительную химическую обработку металла,
главным образом алюминиевых или железных подложек. Послед-
ние наиболее часто обрабатывают фосфатным раствором, который
обеспечивает образование кристаллического фосфатного слоя.
Поэтому последующий слой грунтовки наносится на кристалли-
ческий неорганический слой, а не прямо на чистую поверхность
металла.
Поверхности редко бывают тем, чем кажутся. За исключением
благородных металлов, на поверхности почти всех других метал-
лов, по которым наносятся покрытия, находится не собственно
металл, а оксидный слой. Поэтому чистота поверхности может
быть неизвестна. Так как состояние поверхности сильно влияет
на адгезионную прочность лакокрасочного покрытия, очень важ-
но иметь это в виду. Поскольку большинство поверхностей «за-
грязнены» и качество их не всегда одно и то ж&, необходимо, чтобы
лакокрасочные материалы и получаемые покрытия не были очень
чувствительны к загрязнениям и непостоянству состава поверх-
ностей. Такие материалы должны допускать присутствие на по-
верхности различных загрязнений в умеренных количествах; их
часто называют «устойчивыми к загрязнениям» («robust»). Одна-
ко это не означает, что промышленные технологические процессы
окраски не требуют предварительной обработки поверхностей,
например, обезжиривания и упомянутой выше химической об-
работки.
Разрушение покрытий при их эксплуатации в различных усло-
виях обусловлено в основном изменениями в химической природе
пленкообразователя с последующими изменениями в его механи-
ческих свойствах. Поэтому усиленно проводятся исследования
по созданию полимеров и смол с целью повышения долговечности
покрытий при их эксплуатации. Разработка новых более каче-
ственных пигментов может повысить срок службы покрытий, но
в большинстве случаев наиболее слабым звеном в системе покры-
тия является пленкообразователь. Одним из следствий этого яв-
ляется необходимость разработки материалов и покрытий для
конкретных областей использования. Этот подход позволяет из-
бежать компромиссов, на которые приходится идти в случае
материалов общего назначения. По экономическим или другим
причинам наилучший материал для данного конкретного приме-
нения может быть недоступен, вследствие чего неизбежен- ком-
промисс между ценой и эксплуатационными качествами. Дейст-
вительно, соотношение цена — эффективность конкретной лако-
красочной композиции обычно является доминирующим по срав-
нению с другими факторами на рынке промышленных красок.
| 1.3. КОМПОНЕНТЫ КРАСОК
В табл. 1.1 представлен состав красок и указаны назначения
основных компонентов. '
В; последующих специальных главах будет показано, что на-
личие каждого из компонентов не обязательно во всех красках.
Например, краски для глянцевых покрытий не содержат наполни-
телей, которые являются грубодисперсными неорганическими
частицами. Последние используются в красках для матовых по-
крытий, например в автомобильной промышленности.
Природа полимеров или смол для красок различного целевого
назначения сильно различается. Это обусловлено различиями
Таблица 1.1. Состав красок
Компоненты красок
Типичная функция
Раствор свя-
зующего
(гомогенная
фаза)
Полимер или смола
(связующее)
Растворитель или раз-
бавитель
Пигмент
(дисперсная
фаза)
Добавки (могут отно-
ситься как к гомогенной,
так и к дисперсной фа-
зам)
Основной пигмент (тон-
кодисперсные частицы,
органические или неор-
ганические)
Наполнитель (грубодис-
персные частицы неор-
ганической природы)
Обеспечивает создание сплошной пленки,
изолирование или же защиту покрывае-
мой’ поверхности. Варьируется по хими-
ческому составу в зависимости от области
использования покрытия
Обеспечивает возможность нанесения
краски. Отсутствует в некоторых компози-
циях, таких как порошковые краски и
100%-ные полимеризующиеся системы
Компоненты в незначительных количест-
вах, разнообразные по природе и эффек-
там, например катализаторы, сиккативы,
добавки, улучшающие розлив
Обеспечивает укрывистость, цвет и другие
оптические или визуальные эффекты. Наи-
более часто используется для эстетических
целей. В грунтовках пигмент может обес-
печивать противокоррозионные свойства
Имеет многочисленные функции, включая
повышение укрывистости .(в дополнение
к основному пигменту); для облегчения
шлифовки, например, поверхностей грун-
товки
в методах нанесения и отверждения, природе подложек и усло-
виями эксплуатации покрытий. Так, краски для архитектурных
сооружений («декоративные» или «строительные») должны при-
меняться на месте при умеренных температурах (7—30 °C в за-
висимости от климата и географического района) Они «высы-
хают» или «отверждаются» по одному из двух механизмов: за
счет окисления на воздухе или испарения разбавителя (воды),
сопровождающегося коалесценцией латексных частиц связую-
щего. Многие промышленные процессы окраски требуют приме-
нения тепла или других видов облучения (УФ-, ИК-, ускоренными
электронами) для стимулирования химических реакций, таких
как свободнорадикальная полимеризация или поликонденсация,
которые необходимы, чтобы превратить жидкие полимеры в силь-
но сшитые твердые пленки. Обычно наиболее часто в этих про-
цессах используют «термоотверждаемые» пленкообразователи,
которые часто являются смесями двух различных по химической
природе олигомеров, например, алкидных с аминосмолами. Между
процессами окислительного высыхания и термоотверждения
имеется сходство в том, что в обоих случаях используются низко-
молекулярные полимеры, которые в процессе отверждения сши-
ваются и превращаются в весьма сложные высокомолекулярные
продукты. Наряду с этим можно получить покрытия обоих ука-
занных выше типов, не прибегая к сшиванию. В случае декора-
тивных или строительных красок таковыми являются эмульсион-
ные краски, в которых связующее находится в виде частичек вы-
сокомолекулярного полимера, взвешенных в водной среде. Лаки,
используемые в автомобильной промышленности, могут быть
растворами высокомолекулярных полимеров. В обоих случаях
нет необходимости в сшивании для достижения удовлетворитель-
ных свойств пленки.
1.3.1. Полимерные
или олигомерные пленкообразователи
Органическая химия пленкообразователей подробно описана
в гл. 2. Здесь, тем не менее, будет полезно перечислить некоторые
типы полимеров и олигомеров, которые нашли применение как
пленкообразователи, и указать на общие области их применения.
Пленкообразователи или связующие можно классифицировать
в соответствии с их молекулярной массой. Так низкомолекуляр-
ные полимеры (олигомеры), неспособные формировать твердые
пленки в обычных условиях без дальнейших химических реакций,
образуют один класс. Высокомолекулярные полимеры, способные
формировать качественные пленки без дополнительных хими-
ческих превращений, образуют второй класс. Примеры таких по-
лимеров и олигомеров показаны ниже:
\ Низкомолекулярные
Маапяносмоляные связующие
Алкиды
Полиуретаны
Уретановые масла
Аминрсмолы
Фенольные смолы
Эпоксидные смолы
Ненасыщенные полиэфиры
Хлорированный каучук
Высокомолекулярные
Нитроцеллюлоза
Виниловые полимеры
Акриловые полимеры
Неводные дисперсионные полимеры
Латексы:
поливинилацетатные (ПВА);
акриловые;
стирол/бутадиеновые
I 1.3.1.1. Низкомолекулярные пленкообразователи
Мдсляносмоляные связующие. Их получают нагреванием ра
стительных масел с природными смолами, такими как канифоль,
ископаемые смолы, например, копалы и смола каури. Они могут
также включать фенольные смолы, модифицированные маслами.
В значительной степени масляносмоляные связующие были вы-
теснены алкидными и подобными им смолами, но многие из них
могут обеспечить качество покрытий, сравнимое с получаемым
с помощью современных связующих, особенно в специфических
областях применения, таких как краски промежуточного слоя
для строительных покрытий. Они труднее поддаются стандарти-
зации по сравнению с конденсационными полимерами и менее
удобны для современного промышленного производства.
Алкиды. Они представляют собой полиэфиры, полученные
реакцией триглицеридов растительных масел, полиолов (напри-
мер, глицерина) и двухосновных кислот или их ангидридов (на-
пример, фталевого ангидрида). Классифицируют алкиды по содер-
жанию растительного масла (для описания вводится понятие
«жирность») на три большие группы: тощие, средние и жирные
алкиды, что приблизительно соответствует содержанию масла
45%, 45—60%, 60%. Вариации жирности обычно определяются
типом растительного масла и областью применения материала.
Так для глянцевых декоративных покрытий с максимальным сро-
ком службы в условиях атмосферных воздействий необходимо ис-
пользовать жирное алкидное связующее на высыхающем масле,
таком как льняное или соевое (т. е. ненасыщенные триглицериды).
Высыхающее масло обеспечивает способность пленкообразова-
теля давать твердую пленку. В этом случае превращение низко-
молекулярного жидкого полимера в высокосшитую твердую плен-
ку обусловлено окислительной полимеризацией. Для жирных
алкидов характерна способность растворяться в алифатических
углеводородах. Напротив, тощие алкиды обычно получают из
насыщенных триглицеридов (таких как кокосовое масло). Они
не растворяются в алифатических, но растворимы в высококипя-
щих ароматических углеводородах. Хотя тощие алкиды и могут
образовывать лаковые пленки, но последние имеют низкие темпе-
ратуры размягчения и их необходимо дополнительно сштъ! для
достижения удовлетворительных свойств пленки. Для этого обыч-
но такие алкиды используют в смеси с аминосмолами и сшивают
за счет конденсационных процессов при термоотверждении./В та-
ких композициях дискуссионно: сшивает ли аминосмола фалкид
или алкид пластифицирует высокосшитую аминосмолу. Обычно
предпочитают первое объяснение, так как доля алкида /всегда
больше, чем аминосмолы. Обычно весовое соотношение /алкид/
аминосмола колеблется между 2:1 и 4:1. Подобные системы ис-
пользуются для окраски промышленных изделий. Так аЛкидно-
меламиноформальдегидные композиции уже много лет исполь-
зуются в автомобильной промышленности. Алкидно-мочевино-
формальдегидные. композиции нашли применение для окраски
бытовых изделий, хотя для обоих случаев разработаны новые
материалы, которые частично вытеснили их, особенно в тех слу-
чаях, когда'к покрытиям предъявляются более жесткие требо-
вания.
Полиуретаны, уралкиды и уретановые масла. По структуре
эти материалы напоминают алкиды, в которых сложноэфирные
мн с- о
связи частично или полностью замещены уретановыми________у_
* О
К полиуретанам также относятся двухупаковочные составы для
окраски промышленных изделий и ремонтной окраски, в которых
отверждение достигается в результате взаимодействия между
свободными изоцианатными группами одного' из компонентов
с гидроксильными группами другого. Преимущества уретановых
масел и уралкидов обусловлены стойкостью уретановой связи
к гидролизу. В красках для декоративных покрытий с целью
обеспечения максимального срока службы общепринято исполь-
зовать связующие из смеси жирного алкида и уралкида.
Аминосмолы. Наиболее типичными представителями амино-
смол являются продукты реакции мочевины или меламина (1,3,
5-триаминотриазина) с формальдегидом. Эти смолы получают
в присутствии спирта, который обеспечивает регулирование их
молекулярной массы и степени разветвленности в необходимых
пределах. Влияние такой модификации сказывается на раствори-
мости и реакционной способности получаемых смол. Такие поли-
меры обычно рассматривают как образующиеся из промежуточ-
ных гидроксиметильных производных меламина или мочевины;
последующие реакции конденсации этих производных приводят
к образованию сложных высокоразветвленных олигомерных про-
дуктов. Отверждение или сшивку их и превращение в твердые
пленки (обычно в сочетании с алкидами или другими олигоме-
рами) осуществляют при повышенных или комнатной темпера-
турах. В обоих случаях для,быстрого отверждения необходимо
присутствие кислотного катализатора.
\ Аминосмолы могут также ыть использованы для отвержде-
ния акриловых олигомеров. В этом случае в структуру акрилового
пленкообразователя вводят небольшое количество (обычно не-
сколько процентов) другого мономера, например, N-бутоксиметил-
акриламида. Последний обеспечивает появление в акриловом со-
полимере реакционноспособных функциональных групп, за счет
которых происходит последующая сшивка. Правильный выбор
мономеров позволяет получать внутренне пластифицированные
акриловые олигомеры и избежать использования добавок пласти-
фикаторов. Эти смолы представляют особенный интерес в тех
случаях, когда требуется получение высококачественных, с высо-
кими эксплуатационными свойствами покрытий и, в частности,
с хорошей адгезионной прочностью и гибкостью.
Фенольные смолы. Реакция формальдегида с фенолом приво-
дит к получению ряда смол, которые в сочетании с другими смо-
лами или высыхающими маслами находят применение в защит-
ных покрытиях. Производится два основных типа фенольных
смол — новолачные и резольные. Новолаки представляют собой
низкомолекулярные линейные продукты конденсации формальде-
гида и фенолов с алкильными заместителями в пара-положении.
Если алкильный заместитель содержит четыре или более углерод-
ных атомов, смола способна растворяться в маслах. Резолы яв-
ляются продуктами реакции незамещенных фенолов с формаль-
дегидом. Поскольку в этом случае реакция может протекать как
в пара- так и в тиета-положения фенольного кольца, молекулы
резолов очень разветвлены и по мере протекания, реакции могут
превращаться в жесткие стеклоподобные продукты. Фенольные
смолы обычно повышают химическую стойкость композиций,
в которых они используются. Они всегда применяются в комби-
нации с другими пленкообразующими. При этом фенольный ком-
понент может либо прореагировать с другим пленкообразовате-
лем, либо просто образовать с ним смесь. Так, фенольная смола
(например, новолак) после взаимодействия с канифолью или
с ее эфиром может быть затем смешана с полимеризованным
высыхающим маслом; полученное связующее пригодно для грун-
товок в строительстве, либо в непигментированном виде в масля-
ных лаках. Композиции на основе фенольных смол находят при-
менение там, где требуется химическая стойкость, например, для
защиты трубопроводов и резервуаров.
Эпоксидные смолы. Использование эпоксидной или оксирано-
вой группы —СН-СН2 для синтеза смол или сшивки связую-
О
щих хорошо известно. Большую группу эпоксидных смол получают
на основе реакции эпихлоргидрина и бисфенола А (дифенилол-
пропана). Эти смолы можно этерифицировать ненасыщенными
жирными кислотами и получить эпоксиэфиры. Последние являют-
ся самостоятельными пленкообразующими и напоминают высы-
хающие на воздухе алкиды. Они характеризуются лучшей хиыи-
ческой стойкостью по сравнению с алкидами, но в некоторых
случаях менее долговечны, чем жирные алкиды. Однако последнее
обстоятельство может оказаться полезным, например, при созда-
нии «самоочищающихся» покрытий. В этом случае пленки пигмен-
тируют оксидом титана в анатазной форме с необработанной
поверхностью. Разрушение при ультрафиолетовом облучении вы-
зывает эрозию поверхностных слоев пленки, что известно как
явление «меления». Такие пленки постепенно разрушаются под
влиянием атмосферных воздействий и всегда имеют обновляю-
щуюся белую поверхность.
Эпоксидные смолы также применяются в комбинации с амино-
формальдегидными или фенольными смолами. Эпокси-алкидные
смолы получаются при использовании эпоксидных смол как по-
лиолов в смеси с менее функциональными полиолами, например
глицерином. Эпоксидная группа позволяет использовать широкий
круг реакций при отверждении, поэтому возможно большое число
двухупаковочных композиций. Одним из наиболее популярных
методов сшивки является использование реакции с полиамидами.
Это тот же метод отверждения, что и в эпоксидных клеях. Сшива-
ние происходит в результате присоединения конечных аминогрупп
полиамида к эпоксидной группе. Реакция медленно протекает
при комнатной температуре. Для сшивки по эпоксидным группам
используют также полиамины или катализируемую кислотами
полимеризацию с образованием сшивок за счет простых эфирных
связей. Большинство из этих продуктов используют в промыш-
ленности.
Ненасыщенные полиэфиры. Ненасыщенные полиэфиры обла-
дают тем преимуществом перед ранее описанными связующими,
что они полностью превращаются в полимер, поскольку исполь-
зуемый растворитель является способным к полимеризации моно-
мером. Самые простые и распространенные полиэфиры получают
из смесей малеинового и фталевого ангидридов с гликолями,
например пропиленгликолем. Полученные смолы растворяют в
стироле. Обычно для инициирования радикальной сополимери-
зации винильного мономера с полиэфирмалеинатом при умерен-
ных температурах используют систему переходный металл —
органический гидропероксид, а при более высоких температурах —
разложение диацилпероксида. Ненасыщенные полиэфиры широко
применяются как в пигментированном, так и в непигментирован-
ном виде для отделки древесины. Из полиэфиров на основе изо-
фталевой и терефталевой кислот могут быть приготовлены хими-
чески стойкие покрытия для резервуаров. Другой класс химически
стойких покрытий получается на основе хлорированных поли-
эфиров. В этом случае в состав полиэфира входит хлорэндико-
вый или ХЭТ ангидрид, используемый вместо фталевого ангид-
рида.
Хлорированный каучук. Хлорированный каучук представляет
собой пленкообразующее с широким диапазоном молекулярной
массы — от 3500 до почти 20000. Его получают хлорированием
каучука в растворе; промышленный продукт содержит около
65% хлора. Он используется как основное связующее в красках
естественной сушки, к которым предъявляются требования хими-
ческой стойкости и высокой долговечности. Из-за хрупкости поли-
мера при использовании в лакокрасочных материалах хлориро-
ванный каучук необходимо пластифицировать. Хлорированные
каучуки также используют в комбинации с другими смолами,
с которыми они совмещаются, например алкидами. Лакокрасоч-
ные материалы на основе хлорированного каучука нашли при-
менение для окраски зданий, каменных кладок, плавательных
бассейнов, разметки дорог, в судостроении.
1.З.1.2. Высокомолекулярные пленкообразователи
Почти все высокомолекулярные полимеры получают радикаль-
ной полимеризацией смесей виниловых, акриловых или метакри-
ловых мономеров. Последние полимеризуют в растворе, суспен-
зии или дисперсии. Дисперсионную полимеризацию проводят в
углеводородном разбавителе (неводные дисперсии, НВД) или
в водной среде («эмульсионные полимеры»). Протекание реакций
в этих системах сильно различается, как будет показано в гл. 2.
Важным исключением из сказанного выше является нитроцел-
люлоза. Этот полимер получают прямым нитрованием целлюлозы
в присутствии серной кислоты. Доступны различные марки нитро-
целлюлозы, различающиеся между собой по степени нитрования,
которая, в свою очередь, определяет растворимость ее в различных
растворителях. Нитроцеллюлоза, используемая в красках, лаках
для древесины и т. п., должна обладать меньшей молекулярной
массой, чем у исходной целлюлозы, чтобы достичь требуемой
вязкости в обычно используемых растворителях.
В большинстве случаев нет необходимости в дополнительной
сшивке высокомолекулярных полимеров для достижения необхо-
димых свойств пленок. Однако, некоторые растворимые полимеры
средней молекулярной массы сшивают по реакционноспособным
группам, имеющимся в полимерной цепи. На физические свойства
пленок из высокомолекулярных полимеров способ их получения
или физическая структура полимера влияют в незначительной
степени. Так, автомобильные покрытия, полученные из растворов
акриловых полимеров и из неводных дисперсий, в целом невоз-
можно различить несмотря на то, что метод нанесения, условия
формирования покрытий и т. д. могут сильно различаться. В боль-
шинстве случаев выбор материала определяется стоимостью всего
процесса получения покрытия, а не только ценой материала.
Необходимость в обеспечении конкретных требований к покрытию
нужно учитывать при выборе из альтернативных составов.
Использование водных латексов(эмульсионных полимеров)
в производстве красок быстро растет. Кроме го.мополимера поли-
винилацетата (ПВА) как связующего в матовых красках и деко-
ративных красках для фасадов, в настоящее время появились
новые системы с использованием внутренней пластификации (на-
пример, включение пластифицирующего сомономера). В послед-
нее время начали использовать латексы акриловых и метакрило-
вых сополимеров. Лучшие эксплуатационные свойства позволили
использовать эти краски для наружных покрытий каменных кла-
док. Одной из причин быстрого роста производства эмульсионных
красок является необходимость уменьшения загрязнения атмо-
сферы и защиты окружающей среды.
1.3.2. Пигменты
1.3.2.1. Основные пигменты
Основные пигменты представляют собой твердые микрочасти-
цы, распределенные в связующем или пленкообразователе. Мы
будем проводить различие между ними и вспомогательными
пигментами, наполнителями, удешевляющими добавками и т. п.
Основные пигменты вносят главный вклад в одну или несколько
основных функций, например, цвет, кроющую способность, анти-
коррозионные свойства. Вспомогательные пигменты, удешевляю-
щие добавки, хотя и важны, но в общем не вносят большого вклада
в эти свойства. Их назначение — снижение стоимости, но они
могут также влиять на другие свойства, менее очевидные, чем
цвет или укрывистость. Так, они могут усиливать укрывистость,
влиять на глянец, облегчать шлифовку.
Основной применяемый пигмент — диоксид титана. Это
обусловлено главным образом требованиями моды: на рынке
декоративных красок в последние годы наибольшим спросом
пользуются белые и пастельные тона, а не насыщенные. Подобная
картина, хотя и в меньшей степени, наблюдается и в других обла-
стях потребления, например, в автомобильной промышленности.
Раньше автомобили окрашивали в черный цвет и было трудно
достать автомобили других цветов. В настоящее время купить
черный автомобиль гораздо труднее. Диоксид титана в пигмент-
ной форме является высококачественным пигментом. Наиболее
часто используют рутильную модификацию с высоким показа-
телем преломления. Этот пигмент производится с необходимыми
размерами частиц и дисперсностью. Хотя обычно поверхность
кристаллов диоксида титана считают химически инертной, однако
она фотоактивна и в пигментной форме ее защищают покрытием
с целью свести до минимума фотохимическую активность на гра-
нице раздела связующее — пигмент. Типичные покрытия поверх-
ности TiO2 содержат диоксид кремния и оксид алюминия в раз-
личных соотношениях. Помимо уменьшения фотохимической ак-
тивности они могут улучшать диспергируемость пигмента в
связующем. Были проведены обширные исследования диоксида
титана и в настоящее время в промышленности производится
ряд марок, пригодных для использования в различных типах
красок. Использование TiO2 и других белых пигментов подробно
изложено в гл. 3, а его оптические свойства обсуждаются в
гл. 14 и 15.
Цветные пигменты можно разделить на две большие группы —
неорганические и органические. Вследствие появления законов,
регламентирующих обращение с токсичными материалами и их
использование, многие неорганические пигменты, традиционно
использовавшиеся лакокрасочной промышленностью, были за-
менены на другие менее токсичные. Так, за исключением неболь-
шого числа выпускаемых в промышленности композиций, свинец-
содержащие пигменты всех типов (например, хроматы, оксиды,
карбонаты) были в основном заменены и ожидается, что свинец
во всех его формах и хроматы будут полностью исключены из
всех красок. Исключение этих пигментов вызывает появление
проблем в тех случаях, когда цветные пигменты используются
с двойной целью — придания цвета и антикоррозионых свойств
пленке.
Выбор и использование пигментов подробно рассматривается
в гл. 3. Физика цвета анализируется в гл. 15, в которой рассматри-
ваются принципы и практика подбора цветов. Ранее подгонка
цвета была исключительно искусством очень квалифицированных
колористов. В настоящее время, хотя они еще полностью не заме-
нены, наблюдается постепенный переход к инструментальному
подбору цвета. Эта тенденция особенно усилилась с появлением
сложных колориметров и компьютеров. Некоторые широко исполь-
зуемые пигменты приведены ниже:
Цвет Неорганические Органические
Черный Сажа, карбонат меди, ди- Анилиновый черный
оксид марганца
Желтый Хроматы свинца, цинка и Никелевый желтый азо-
бария; сульфид кадмия, ок- пигмент
сиды железа
Синий, фио- Ультрамарин, берлинская Фталоцианиновый голу-
летовый лазурь, кобальт синий бой, Индантр'еновый голу-
бой, Карбазоловый фио-
летовый
Зеленый Оксид хрома Фталоцианиновый зеле-
ный
Красный Красный оксид железа, селе- Толуидиновый красный,
нид кадмия, свинцовый сурик, хинакридоны
красные кроны
Белые Диоксид титана, оксид цинка,
оксид сурьмы, основной кар-
бонат свинца.
В красках для автомашин наблюдался рост потребления в
качестве пигмента чешуйчатого алюминия с целью получения
привлекательных покрытий с металлическим блеском. Размеры
чешуек и их ориентация в красочной пленке определяют оптиче-
ские эффекты, которые невозможно достичь другими путями.
Однако в таких красках существуют трудности с контролем ка-
чества, применением и составлением цветов. Тем не менее причины
этих затруднений и пути управления ими известны, а продолжаю-
щееся использование таких материалов полностью оправдывается
эстетически.
1.3.2.2. Удешевляющие добавки, наполнители
и вспомогательные пигменты
Все эти три термина применяют к широкому кругу материалов,
которые вводят в состав красок для самых разнообразных целей.
Они относительно дешевы и поэтому могут быть использованы
вместе с основными пигментами для достижения определенных
эффектов. Например, было бы технически трудно и непозволитель-
но дорого производить хорошую эмульсионную белую краску
с матовым эффектом, используя в качестве пигмента только лишь
диоксид титана. Последний не эффективен как матирующий агент,
да и вообще не предназначен для этой цели. Намного выгоднее
использовать наполнитель с грубодисперсными частицами, такой
как карбонат кальция в сочетании с TiCX, для достижения необ-
ходимой белизны и укрывистости в матовых или полу.матовых
материалах (например, матовые латексные декоративные краски
верхнего или промежуточного слоя или грунтовки). Подобные
добавки обычно не вносят вклада в цвет и в большинстве случаев
важно, чтобы они были бесцветными. Размер частиц удешевляю-
щих добавок колеблется от долей микрона до нескольких десят-
ков микрон; их показатель преломления обычно близок к показа-
телю преломления органического связующего, в который их вво-
дят, и поэтому их вклад в укрывистость за счет рассеяния света
мал. Добавки пластинчатого типа, такие как слюда мокрого
помола, могут влиять на водопроницаемость пленок и поэтому
многие из них способствуют повышению коррозионной стойкости.
Часто используются различные виды талька (например, в авто-
мобильных грунтовках) с целью улучшения способности пленки
к шлифовке перед нанесением верхнего слоя. Многие обычно
используемые удешевляющие добавки имеют природное проис-
хождение и подвергаются различной степени очистке в зависи-
мости от их целевого использования. Хотя делается все возмож-
ное для обеспечения стабильности свойств этих добавок, все же
по сравнению с основными пигментами их свойства менее постоян-
ны; имеют место вариации формы, размера частиц, дисперсности
(распределения по размерам частиц). Ниже дан перечень типич-
ных неорганических наполнителей:
Химическая природа
Сульфат бария
Карбонат кальция
Сульфат кальция
Силикаты
Т нп
Бариты, бланфикс
Мел, кальцит, осажденный мел
Гипс, ангидрит, осажденный сульфат кальция
Кремнезем (диоксид кремния), диатомит,
глина, тальк, слюда
В последние годы были попытки приготовить синтетические
полимерные наполнители для специальных целей, в частности,
для замены части TiCb в красочной пленке. Один из таких мате-
риалов «spindrift», полученный в Австралии, находится в форме
полимерных шариков (бисера) диаметром до 30 мкм, которые
содержат внутри себя воздушные пузырьки субмикронных раз-
меров и небольшое количество пигментной формы TiCb. Введение
воздушных пузырьков внутрь полимера, образующего бисер,
влияет на эффективность рассеяния света диоксидом титана по
одному из двух механизмов. Если пузырьки очень малы
(<0,1 мкм), может уменьшиться средний эффективный показа-
тель преломления полимерной матрицы и вследствие этого уси-
литься рассеяние света диоксидом титана. Если пузырьки боль-
шие (~0,8 мкм), они способны рассеивать свет сами. Другие
методы введения пустот в пигментированные пленки с целью ис-
пользования способности пузырьков рассеивать свет заключают-
ся в эмульгировании маленьких капелек летучей жидкости в вод-
ных латексных красках, которые в результате испарения после
коалесценции приводят к появлению в пленке полостей. Еще один
метод заключается в использовании некоалесцирующих латекс-
ных частиц вместе с коалесцирующими латексами, что приводит
к появлению в пленке пустот с острыми выступами. Все эти ме-
тоды преследуют цель повысить кроющую способность и, следо-
вательно, снизить стоимость без потери сплошности и других
свойств пленки. Эти методы не получили широкого распростра-
нения, главным образом по экономическим соображениям. Тем
не менее, они расширяют возможности при разработке рецептур
лакокрасочных материалов для конкретных целей.
1.3.3. Растворители
Растворители используются в лакокрасочных составах для
двух основных целей: для приготовления краски и для обеспече-
ния возможности ее нанесения на различные поверхности. Это
может казаться очевидным, однако важно представлять себе,
что растворитель не влияет на свойства лакокрасочной пленки
в процессе ее длительной эксплуатации. Хотя нельзя сказать,
что в свежесформированной пленке остатки растворителя не
влияют на твердость и другие свойства.
Термин растворитель часто используется для жидкостей, ко-
торые не растворяют полимерное связующее, и в этих случаях
более правильно использовать термин раз авитель. назначение
разбавителя то же, что и растворителя. В водных системах вода
может быть истинным растворителем для некоторых компонентов,
однако не являться растворителем для основного пленкообразо-
вателя. Примером могут служить декоративные водоэмульсион-
ные краски. Чаще всего в этих случаях принято говорить о «вод-
ной фазе» композиции, имея в виду, что хотя вода и не растворяет
пленкообразователь, однако является основным компонентом
жидкой дисперсионной среды.
В качестве растворителей используют очень широкий ряд
органических жидкостей, причем тип растворителя зависит от
природы пленкообразователя.
В последние два десятилетия большие усилия были направ-
лены на изучение термодинамики растворов. Это позволило раз-
работать намного более точные методы выбора растворителя.
Эти методы основаны на лучшем понимании природы молекуляр-
ных притяжений в жидкостях и признании аддитивности молеку-
лярных притяжений в системах из нескольких растворителей.
Редко, чтобы один и тот же растворитель можно было использо-
вать для разных случаев. Поэтому новейшие методы, основанные
на концепции параметра растворимости, позволяют осуществлять
более рациональный выбор смесей растворителей в соответствии
с возникающей необходимостью.
Растворяющая способность — не единственный критерий, по
которому выбирают растворитель. Другими важными факторами
являются скорость испарения, запах, токсичность, горючесть,
стоимость. Эти факторы могут иметь неодинаковый вес в зависи-
мости от того, как используются лакокрасочные материалы. Если
нанесение осуществляется в промышленных условиях, то пробле-
мы, связанные с запахом, токсичностью и горючестью разрешимы,
хотя и не всегда. Необходимость установки дорогого рекупера-
ционного оборудования или камер сжигания может исключить
использование некоторых растворителей и, следовательно, не^
которых типов лакокрасочных композиций. С токсичностью тесно
связаны и вопросы загрязнения окружающей среды. Принятие
во многих странах законов, защищающих человека и окружаю-
щую среду, оказало сильное влияние на развитие лакокрасочной
промышленности и повлияло как на поставщиков сырья, так и
на потребителей лакокрасочных материалов. В Северной Америке
очень резко возросло производство водных лакокрасочных ма-
териалов и, соответственно, снизилось использование органиче-
ских растворителей. Вполне вероятно, что эта тенденция будет
продолжаться.
Другими видами лакокрасочных материалов, альтернативных
органорастворимым, являются 100%-ные полимеризующиеся
системы и порошковые краски. В первом случае мономер, такой
как стирол, выполняет роль растворителя в композиции, а затем
превращается в полимер в процессе отверждения. В случае порош-
ковых красок растворители могут использоваться лишь на ран-
них стадиях в процессе производства красок, однако затем они
удаляются и возвращаются в производственный цикл, так что
у потребителя красок не возникает проблем, связанных с раство-
рителями.
1.3.4. Добавки
Наиболее простая лакокрасочная композиция, состоящая
из пигмента, диспергированного в связующем, и жидкой фазы
(растворителя или нерастворителя), на практике имеет легко
обнаруживаемые недостатки. Они проявляются в ограничениях
химического и физического характера и должны быть устранены
или, по крайней мере, сведены до минимума прежде, чем лако-
красочный материал будет поставлен потребителю.
Некоторые из главных недостатков, о которых следует упомя-
нуть,— это оседание пигмента и образование пленки на поверх-
ности жидкого лакокрасочного материала в таре; аэрация и со-
хранение пузырьков при нанесении; образование «кратеров» на
покрытии, натеков и сморщивания лакокрасочной пленки; всплы-
вание пигмента и изменение цвета покрытия. Эти недостатки
охватывают лишь небольшое число из возможных. Видимо, целе-
сообразно здесь кратко описать явления кратерообразования,
сморщивания, образования натеков, всплывания пигментов и из-
менения цвета. Более детально они рассмотрены в гл. 4, в которой
описаны некоторые наиболее распространенные типы добавок
в краски.
«Кратерообразование» — появление маленьких, круглых уг-
лублений на поверхности пленки.
«Сморщивание» — образование морщинистой поверхности
пленки, которая высыхает за счет окисления.
«Образование натеков» — образование неровного покрытия
в результате избыточного стекания краски на вертикальной по-
верхности.
«Расслаивание» — термин, используемый для описания раз-
личий в цвете, которые могут появляться в лакокрасочной пленке
из-за спонтанного разделения компонентов пигментной части
после нанесения.
«Кистевая аномалия» — изменение цвета пленки вследствие
всплывания пигмента после нанесения.
Чтобы избежать этих недостатков, необходимо понять при-
чину их появления и найти способы преодоления. В некоторых
случаях эти недостатки могут быть устранены небольшими из-
менениями в рецептуре. Например, сморщивание обусловлено
дисбалансом между скоростями реакций окислительного сшива-
ния в поверхностном слое и внутри пленки. Положительный эф-
фект достигается изменением состава сиккатива, вчастности
введением в него активного сиккатива, содержащего способствую-
щий окислению переходный металл, такой как кобальт, и «пря-
мого» сиккатива, например свинцового и циркониевого, который
увеличивает скорость сшивания, но не катализирует окислитель-
ный процесс. В других случаях простым изменением рецептуры
невозможно устранить недостатки. Поэтому были разработаны
специальные добавки. В настоящее время существует большое
разнообразие добавок для большинства красок — нротивоосади-
тельные, предохраняющие от образования поверхностной пленки
при храпении и т. п.
Проблемы изменения цвета из-за всплывания и разделения
пигментов связаны с коллоидной устойчивостью пигментных дис-
персий и могут быть обусловлены рядом причин. Сепарация пиг-
ментов, проявляемая во всплывании, происходит в результате
различий в размерах частиц составного пигмента и может быть
преодолена совместной флокуляцией этих пигментов в данной
системе. Другой метод стабилизации системы может заключать-
ся во введении небольшого количества очень тонкодисперсного
наполнителя, такого как оксид алюминия, с поверхностным за-
рядом частиц* противоположным мелким частичкам пигмента,
чтобы обеспечить совместную флокуляцию с последними.
Появление проблемы изменения цвета вследствие «кистевой
аномалии» указывает на флокуляцию, протекающую по мере
высыхания пленки. Под влиянием усилия сдвига, когда кистью
проводят по краске, пигмент редиспергируется и оттенок краски
становится бледнее. Это обусловлено увеличением вторичного
рассеяния падающего света из-за дефлокуляции белого пигмента.
Образование кратеров и натеков обусловлено другими аспек-
тами, связанными с химией поверхности и реологией. В первом
случае эффект вызывается локальным изменением поверхност-
ного натяжения пленки. В предельных случаях это может при-
вести к неполному смачиванию подложки, часто называемому
термином «сморщивание». Образование натсков, с другой сто-
роны, связано с объемными свойствами пленки, на которые мо-
жет влиять коллоидная стабильность композиции. Идеальные,
коллоидно устойчивые дисперсии склонны проявлять ньютонов-
ское поведение, т. е. их вязкость не зависит от скорости сдвига.
Это значит, что на вертикальной поверхности ньютоновская жид-
кость с соответствующей вязкостью, требуемой для нанесения
кистью (примерно 0,5 Па-с), будет обладать чрезмерной теку-
честью, если только вязкость не возрастет быстро в результате
испарения растворителя. Напротив, при составлении композиций
может возникнуть необходимость обеспечения не ньютоновского
поведения, когда при малых усилиях сдвига вязкость материала
очень высока. Таким образом, можно избежать образования на-
теков, используя любой из этих эффектов или их комбинацию.
1.4. МЕТОДЫ НАНЕСЕНИЯ
Существуют четыре основных метода нанесения лакокрасоч-
ных материалов: а) нанесение кистью, валиком, тампоном или
ракелем; б) распыление, например, пневматическое, безвоздуш-
ное, с подогревом, электростатическое; в) струйный облив, напри-
мер, окунание, налив, валиком, обратным валиком; г) электро-
осаждение.
Метод нанесения часто диктуется потребителем и каждый тип
лакокрасочного материала готовится с учетом требований, предъ-
являемых методом нанесения. Нанесение кистью или ручным ва-
ликом — основной метод нанесения декоративных строительных
красок и красок для текущего ремонта стальных конструкций и
сооружений. Он также важен при ремонтных работах в судострое-
нии, хотя при строительстве судов могут использоваться другие
методы (например безвоздушное распыление).
Нанесение распылением — наиболее распространенный метод.
Он используется при окраске автомобилей на заводах и при по-
вторных окрасках после повреждений; в деревообрабатывающей
промышленности (например, мебельной) и в быту. Различные
виды распыления делают этот метод нанесения весьма распро-
страненным. Методы струйного облива в основном применяются
для листовых материалов (например, картона) и покрытий для
рулонного металла (алюминия или стали) на заводе-изготови-
теле, где они очень ценятся из-за высоких скоростей окраски.
Электроосаждение, как метод окраски, получило широкое
распространение в последние два десятилетия. Оно стало основ-
ным методом грунтования стальных корпусов автомобилей. Про-
цесс окраски, включающий обезжиривание, фосфатирование,
электроосаждение грунтовки и последующее нанесение распыле-
нием шпатлевки и верхних слоев покрытия значительно повысил
уровень коррозионной защиты и внешний вид покрытий.
Электроосаждение возможно в случаях, когда корпус авто-
мобиля является либо анодом, либо катодом. В последние годы
принято считать, что катодное электроосаждение обеспечивает
лучшую защиту от коррозии.
Методы нанесения будут подробнее описаны в последующих
главах.
Глава 2
ОРГАНИЧЕСКИЕ ПЛЕНКООБРАЗОВАТЕЛИ
Дж. Бентли
2.1. ВВЕДЕНИЕ
В первой главе были приведены основные типы низко- и высоко-
молекулярных полимеров, используемых для получения покры-
тий. В этой главе более подробно будет описана их химия, вклю-
чая получение. Во введении кратко рассматриваются вопросы
теории образования и отверждения полимеров. Механизмы, ха-
рактерные для каждого типа пленкообразующего полимера, бо-
лее подробно-обсуждены в последующих разделах этой главы.
Будут рассмотрены также отдельные классы пленкообразующих
полимеров и факторы, определяющие выбор пленкообразователи
для конкретного применения. Свойства и применение каждого
типа пленкообразователей вновь детально рассматриваются в
последующих главах.
Для пленкообразующего использован ряд взаимозаменяемых
терминов. Термины «пленкообразователь», «связующее» озна-
чают тот очевидный факт, что этот компонент содержит в себе
и связывает воедино другие компоненты микроскопических раз-
меров и обеспечивает образование сплошной пленки покрытия.
Термины «смола» или «масляный лак» — более старые термины,
относящиеся к тому времени, когда преобладало использование
природных полимеров как пленкообразователей в виде их раство-
ров в растворителях или в маслах, и когда химия и состав этих
компонентов были недостаточно известны. В наше время, когда
понятна природа используемых веществ, наряду с широким при-
менением сложных синтетических полимеров, используемых так-
же в промышленности пластмасс и клеев, но специально приспо-
собленных для лакокрасочных производств, гораздо более пра-
вильно использовать термин «полимерный пленкообразующий
компонент». Использование взаимозаменяемых старых и новых
названий также встречается и в технологии производства пленко-
образователей, где термин «котел» относится к реактору поли-
меризации, а «мешалка» — к смесителю.
Пленкообразующие полимеры получают в присутствии рас-
творителя или же без него. Так как полимеры без растворителя
обычно представляют собой либо очень вязкие жидкости, либо
хрупкие твердые тела, при хранении и при производстве лако-
красочных материалов с ними практически всегда имеют дело
в виде растворов (или дисперсий) в значительном количестве
растворителя или разбавителя. Единственным исключением яв-
ляются жидкие олигомерные продукты для лакокрасочных мате-
риалов с высоким сухим остатком и твердые олигомеры для по-
рошковых красок.
Большинство используемых полимеров образуют истинные
растворы и растворитель является вторым компонентом. Но в не-
которых случаях, обусловленных методом синтеза полимера или
его конечным использованием, полимер находится в виде диспер-
сии очень мелких частиц в нерастворителе. Примерами могут
служить водные и неводные дисперсии, материалы, используемые
в электроосаждении, и ряд других водоразбавляемых систем.
В некоторых случаях могут использоваться смешанные системы
раствор — дисперсия; например, раствор, содержащий мицелляр-
ную полимерную дисперсию, микроэмульсию или микрогель.
Вязкость очень сильно зависит от того, растворен полимер
или же диспергирован. Обычно дисперсии менее вязкие при оди-
наковых содержаниях сухого остатка, чем растворы. Существен-
но, что если вязкость раствора увеличивается при увеличении
молекулярного веса, то вязкость эмульсий или дисперсий не за-
висит от него. Для любого полимера в растворе или дисперсии
вязкость растет при увеличении содержания сухого остатка; при
повышении температуры вязкость уменьшается.
Обычно потребителя интересует способность материала обес-
печить конечные защитные и декоративные свойства, а не его
состав. В табл. 1.1 гл. 1 были перечислены функции компонентов
краски, а в табл. 2.1 показано, какой вклад три основные компо-
нента — пленкообразователь, пигмент и растворитель — вносят
в наиболее важные свойства типичной глянцевой краски.
При всей условности данных этой таблицы ее основное на-
значение — подчеркнуть более весомый вклад системы связую-
щее — растворитель.
Обычно из полимеров различной природы можно приготовить
множество композиций с широким спектром свойств и различной
стоимостью, что позволяет удовлетворить самые разнообразные
требования потребителей. Окончательный выбор лакокрасочных
материалов для потребителя в конце концов будет определяться
с учетом следующих факторов: эксплуатационные свойства и
стоимость лакокрасочного материала; истинная стоимость, вклю-
Таблица 2.1. Вклад основных компонентов краски в ее свойства
Свойство Пленкообразователь
Пигмент
Растворитель
Способность к нанесе-
нию'
Скорость отверждения
Стоимость
Механические свойства
Долговечность
Цвет
Основной
Основной
Основной
Основной
Основной
Незначительный
Незначительный
Отсутствует
Основной
Незначительный
Основной
Основной
Основной
Значительный
Незначительный
чающая общую стоимость окраски; стоимость оборудования,
трудозатраты, энергозатраты на отверждение.
Промышленность непрерывно стремится производить новые
лакокрасочные материалы с лучшими эксплуатационными свой-
ствами. Стимулирующее влияние на поиск новых материалов
оказывают требования гарантий качества в таких разных обла-
стях применения, как декоративные ремонтные краски, автомо-
билестроение и окраска рулонного металла. Не менее важны и
такие факторы, как наличие законов, контролирующих степень
загрязнения, доступность и стоимость энергии, периодические
избытки и нехватка природного и маслосодержащего сырья.
Появляются новые требования, связанные, например, с заменой
цельнометаллических корпусов автомобилей на металлопласт-
массовые композиты, что определяет потребность в высококаче-
ственных покрытиях для пластиков.
Выбор растворителя или разбавителя, его количество, а также
вязкость готового лакокрасочного материала зависят от природы
полимера и метода нанесения. Метод нанесения обычно предъ-
являет ограничения по температуре кипения растворителя и
скорости испарения, например, для обеспечения способности
хорошо наноситься распылением или кистью. Если полимер мо-
жет быть синтезирован в присутствии небольшого количества
растворителя или вообще без него, то химик может не заботить-
ся о растворителе, необходимом для хорошего нанесения. Алкиды
или Аолиэфиры, например, легко могут быть смешаны на самой
последней стадии с высоко- или низкокипящим растворителем.
Однако, в случае акриловых полимеров, обычно необходимо ис-
пользовать растворитель или их смесь с низкой способностью к
передаче цепи и с такой температурой кипения, чтобы обеспечить
возможность кипения реакционной смеси и тем самым отвод теп-
лоты полимеризацции. Эта температура должна также обеспечить
возможность выбора доступного инициатора, способствующего
эффективной полимеризации.
Механические свойства полимеров обычно улучшаются с уве-
личением молекулярного веса вплоть до некоторого значения,
после достижения которого они стабилизируются. Напротив,
вязкость растворов полимеров непрерывно растет с увеличением
молекулярного веса. Это налагает ограничение при составлении
лакокрасочных композиций. Так, для синтеза полимера с опти-
мальными эксплуатационными свойствами и способностью к на-
несению необходимо тщательно задать и контролировать его
молекулярную массу. Для достижения хороших механических
свойств и долговечности для многих полимеров необходимы вы-
сокие молекулярные веса, поэтому требуется применение значи-
тельных количеств растворителя, чтобы обеспечить хорошую
способность лакокрасочного материала к нанесению, если поли-
мер собираются использовать в виде раствора при этом молеку-
лярном весе. Примером подобной системы может быть лак, вы-
сыхающий только вследствие испарения растворителя с образо-
ванием пленки полимера без изменения молекулярного веса или
каких-либо химических реакций. Таковы ранее использовавшиеся
лаки, например, шеллачный лак или политуры, а в настоящее
время пластифицированная нитроцеллюлоза и термопластичные
акриловые лаковые системы, применяемые как в автомобиле-
строении, так и в системах для повторной окраски.
Наиболее простой путь преодоления противоречия между
молекулярной массой и необходимой вязкостью — использование
дисперсных систем, примерами которых являются производимые
в настоящее время в больших количествах декоративные водные
эмульсии, а также дисперсии полимеров для систем с высоким
сухим остатком, неводные дисперсии и органозоли. Дисперсии
позволяют, кроме того, применять более дешевые, менее токсич-
ные разбавители, и они особенно ценны тогда, когда законом
установлены допустимые пределы содержания летучих органиче-
ских веществ в газовых выбросах предприятий. Но приготовле-
ние этих систем достаточно сложно, и они более ограничены по
составу, чем растворы полимеров.
Во многих лакокрасочных системах удается избежать огра-
ничений, налагаемых молекулярной массой полимера, за счет
использования реакций «сшивания» или «отверждения». Такие
системы состоят из одного или нескольких реакционноспособных
полимеров относительно низкой молекулярной массы, способных
к химическим превращениям после нанесения с образованием
полимера с высокой или бесконечной молекулярной массой. Сши-
вание предполагает наличие полифункциональности; в результате
каждая молекула исходных низкомолекулярных полимеров соеди-
няется с рядом других молекул, приводя к образованию бесконеч-
ной сетки в готовом покрытии.
Методы, применяемые для формирования пленки с типичными
полимерными системами, приведены ниже:
Метод Внешний агент Типичная полимерная система
Испарение раствори- теля Отсутствует или теп- лота Лаковые системы
Отверждение в окру- жающей среде Кислород Влага Алкиды, модифицированные маслами Уретаны, отверждаемые вла- гой воздуха
Отверждение в парах другого вещества Амин Смеси гидроксилсодержащих акрилатов с изоцианатами
Двухупаковочные со- Отсутствует или теп- Двухупаковочная система
ставы лота эпоксид — амин
Облучение ИК-, УФ-, ускоренные электроны Фотоотверждаемые ненасы- щенные полиэфиры
Термоотверждение Сушильная камера Смеси алкидных и азотсодер- жащих пленкообразователей. Термоотверждаемые акрило-
вые полимеры
В каждом случае, за исключением лаковых систем, полимер-
ные системы содержат в своей структуре свободные реакционно-
способные группы, соответствующие выбранному, методу отвер-
ждения. Реакции отверждения будут рассмотрены в данной главе
применительно к конкретным пленкообразователям.
Могут применяться комбинированные методы; например,
горячую сушку в печи можно использовать для ускорения испа-
рения растворителя. При этом различие между термопластичными
и термореактивными акриловыми полимерами становится менее
выраженным. Подобно этому горячая сушка ускоряет отвержде-
ние алкида, способного медленно высыхать на воздухе.
Электроосажденные пленки обычно необходимо дополнительно
отверждать при повышенной температуре; при электроосаждении
полимер переводится в нерастворимую форму и выделяется на
подложке, однако без дополнительного сшивания полученная
пленка остается мягкой и не вполне устойчивой к действию других
агентов.
Полимеры обычно классифицируют в соответствии с методом
их получения из мономеров либо путем поликонденсации, либо
цепной аддитивной полимеризации.
Поликонденсация имеет место в тех случаях, когда молекулы
исходных моломеров практически все соединяются в большие
молекулы уже на ранней стадии реакции; эти большие молекулы
сохраняют реакционную способность и продолжают соединяться
друг с другом, так что средняя молекулярная масса увеличивается
со временем, но после того, как реакция началась, выход поли-
мера уже не зависит от времени. В этой реакции обычно участвуют
два разных мономера с различными функциональными группами,
способными к взаимодействию друг с другом. Часто, хотя и не
обязательно, в каждом элементарном акте реакции выделяется
какая-то небольшая молекула, например вода. Типичными пред-
ставителями полимеров, получаемых поликонденсацией, явля-
ются алкиды и полиэфиры. В этом случае важное значение
имеет концепция функциональности. Для того, чтобы про-
межуточные продукты, образующиеся на каждой стадии реакции,
могли участвовать в последующих реакциях, каждая молекула
мономера должна содержать как минимум две реакционноспособ-
ные функциональные группы.
Цепная аддитивная полимеризация характеризуется тем, что
высокомолекулярный полимер образуется уже с момента начала
цепной реакции, а концентрация мономера постепенно уменьшает-
ся в течение всего периода полимеризации; в отличие от поли-
конденсации выход полимера увеличивается со временем. Поли-
меризация инициируется определенными активными соедине-
ниями, способными разорвать одну из связей в мономере, и может
быть радикальной, электрофильной или нуклеофильной. При про-
изводстве лакокрасочных материалов обычно используется только
радикальная цепная полимеризация, хотя могут применяться
полимеры, синтезированные ионной полимеризацией.
Примером наиболее хорошо известных пленкообразователей,
получаемых цепной аддитивной полимеризацией, являются акри-
ловые полимеры.
Ниже приведены типы полимеров, используемых в технологии
лакокрасочных материалов и
той классификации:
Тип полимера
Природные смолы
Модифицированные природные по-
лимеры
Полимеры, полученные поликонден-
сацией
Полимеры, полученные цепной ад-
дитивн ой пол и мер иза цие й
Полимерные простые эфиры
покрытии, с учетом вышеупомяну-
Подгруппа
Смолы, клеи, канифоль
Целлюлоза, крахмал, нитроцеллюлоза
Сложные полиэфиры, алкидные смолы,
формальдегидные смолы (мочевино-, мел-
амино-, феноло-), эпоксидные смолы
Акриловые полимеры и сополимеры, вини-
ловые полимеры
Полиэтиленоксиды и полиэтиленгликоли
Полимерные простые эфиры выделены в отдельную группу
отчасти для констатации того факта, что такие материалы, как
полиэтиленгликоль, можно синтезировать как поликонденсацией,
так и аддитивной полимеризацией, а также для избежания даль-
нейшей путаницы. Эпоксидные смолы получают поликонден-
сацией, однако следует иметь в виду, что эпоксидная группа
может участвовать в реакциях как ступенчатой, так и цепной
аддитивной полимеризации.
2.2. ПРИРОДНЫЕ ПОЛИМЕРЫ
Раньше связующие для покрытий получали в основном на
основе природных масел, клеев и смол, комбинируя которые по-
лучали целый ряд лаков как неотверждаемых, так и способных
к автоокислению и высыханию.
В настоящее время триглицериды масел по-прежнему доста-
точно широко применяются для синтеза модифицированных мас-
лами алкидов и в меньшей степени в других производных модифи-
цированных жирных кислот, таких как эпоксиэфиры. Применение
других природных продуктов сейчас крайне ограничено. Некото-
рые из природных смол еще применяются в масляносмоляных
связующих. Из модифицированных природных полимеров по-
прежнему широко применяются производные целлюлозы, особен-
но «нитроцеллюлоза», которую более правильно называть нитра-
том целлюлозы.
2.3. АЛКИДЫ
Алкиды были одними из первых синтетических полимеров,
использованных в технологии покрытий. Оказалось очень полез-
ным химически связать масла или жирные кислоты масел в струк-
туру сложного полна ира и тем самым существенно улучшить
механические свойства, скорость высыхания и долговечность
этих связующих по сравнению с маслами и масляносмоляными
связующими. И хотя в настоящее время для более жестких усло-
вий эксплуатации имеются более сложные полимеры с улучшен-
ными свойствами, алкиды по-прежнему занимают большую долю
в общем объеме производства пленкообразователей. Это объяс-
няется использованием хотя бы частично для их производства
возобновляемого природного сырья и, главным образом, благо-
даря возможности изготовления огромного разнообразия компо-
зиций. Жирные алкиды, иногда используемые в смеси с другими
модифицированными алкидами для увеличения твердости пленок
или изменения реологических свойств, остаются основным типом
пленкообразователей в органорастворимых лакокрасочных мате-
риалах для декоративных покрытий. Тощие алкиды по-прежнему
используются в значительных количествах в лакокрасочных ма-
териалах горячей сушки для автомобильной промышленности и
общего промышленного назначения, где их применяют в сочета-
нии с меламиноформальдегидными отверждающимися смолами.
Масла не способны непосредственно взаимодействовать с
полиэфиром и поэтому их невозможно включить в структуру ал-
кида без предварительной модификации. Можно использовать
жирные кислоты, полученные омылением масел, особенно при
применении глицерина, однако это дорогой путь. Разработан и
широко применяется так называемый моноглицеридный метод,
в котором на первой стадии осуществляется модификация масла.
В этом методе глицерин (или другой полиол) и масло в молярном
соотношении 2:1 взаимодействуют при температуре, примерно,
240 °C в присутствии основного катализатора (гидроксида натрия,
свинцового глета и др.), образуя «моноглицерид», моноэфир жир-
ной кислоты и глицерина:
СНгООС— СНгОН СНгООС—ф— СНгОН
СНООС—ф— Ь2 ^нон Катализатор 2 СНОН + 1 /1 СНООС •
CH 2ООС -—^|/— 1 СНгОН 1 СНгОН 1 CHZOH
Масло Глицерин сс-Моиоглидерид р-Мои огл и не ряд
—оос—-ф— — остаток жирной КИСЛОТЫ.
На практике реакция не доходит до завершения и в продукте
присутствует равновесная смесь веществ, включающая масло,
полиол, моно- и диглицериды. Если в реакции используют другой
полиол вместо глицерина, например пентаэритрит для жирных
алкидов, аналогично проводят стадию получения «моноглице-
рида» с таким количеством полиола, чтобы на этой стадии полу-
чить основной продукт с двумя гидроксильными группами. Дости-
жение достаточной глубины реакции проверяют по растворимости
в спирте: в нем не должно наблюдаться выделения свободного
масла. Иногда с такими полиолами, как пентаэритрит, проверка
растворимости в спирте не дает хороших результатов, в этом
случае реакцию проводят в небольшом объеме в широкогорлой
колбе с необходимым соотношением «моноглицерида» и поли-
основной кислоты для того, чтобы проверить возможность полу-
чения необходимого конечного продукта без опасности гелеобра-
зования. Некоторые масла нельзя долго выдерживать при повы-
шенной температуре на этой стадии, чтобы свести к минимуму
увеличение вязкости масла; по этой же причине следует стандар-
тизовать скорость нагрева и время реакции, что обеспечивает
воспроизводимое и предсказуемое поведение на следующей стадии.
Синтез конечного пленкообразователя осуществляют путем
прибавления дополнительного количества полиола и двухоснов-
ной кислоты на так называемой стадии «уплотнения». На этой
стадии протекает реакция полиэтерификации; образующуюся
в качестве побочного продукта воду необходимо удалять. Эта
реакция приводит к постепенному формированию структуры по-
лимера (алкида) и, следовательно, сопровождается увеличением
вязкости. Реакцию в растворителе ведут до тех пор, пока не будет
достигнута необходимая вязкость продукта при определенной
концентрации и температуре:
сн2оос^р—
П СНОН + п [ноос----соон] —*
СН2ОН
ноос---
СН2ООС
соосн
I
СН2ООС
оос-Д/-----остаток жирной кислоты.
Описанный выше моноглицеридный метод применяют для
получения жирных алкидов. В случае менее жирных алкидов,
для которых вместо глицерина необходимы другие полиолы, часто
используют более простой, так называемый «жирнокислотный
метод» с прямой загрузкой всех ингредиентов, включая жирную
кислоту (не масло). Возможны и другие методы, например, аци-
долиз, когда масло реагирует сначала с полиосновной кислотой
до взаимодействия с полиолом; этот метод не получил широкого
распространения. Если необходимо в структуру алкида включить
тунговое масло, применение моноглицеридного метода опасно
из-за возможности чрезмерного повышения вязкости; возможным
вариантом является использование смеси жирной кислоты с
маслом. Например, реакцию смеси тунгового масла со значитель-
ным количеством жирной кислоты, полиола и двухосновной кисло-
ты проводят в одну стадию. Если концентрация масла невысока,
при такой переработке происходит переэтерификация масла в
степени, достаточной для обеспечения его взаимодействия с поли-
эфиром и образования прозрачного конечного продукта. Предло-
жен также метод производства алкидов [1, 2], заключающийся
в ступенчатом добавлении ингредиентов, что обеспечивает дости-
жение более предпочтительного молекулярного веса, распределе-
ния реакционноспособных групп и улучшение некоторых свойств.
Считается, что он особенно эффективен при синтезе алкидных
смол на талловом масле.
2.3.1. Состав алкидов
Каждый из основных компонентов алкида вполне определенно
влияет на его свойства [3]. Содержание масла выражают через
«жирность», причем жирные алкиды содержат свыше 60% масла,
средние — от 40 до 60%, а тощие — менее 40%. Жирные алкиды
обычно получают используя высыхающие масла. Они растворимы
в алифатических растворителях, характеризуются низкой вяз-
костью, медленно высыхают на воздухе, образуя мягкие эластич-
ные пленки гс посредственными долговечностью и сохранением
глянца.
Уменьшение содержания масла вызывает необходимость ис-
пользования ароматических растворителей и обуславливает бо-
лее высокую вязкость растворов смол при меньшем содержании
сухого остатка. Жирные и средние алкиды, для синтеза которых
использовались высыхающие или полувысыхающие масла, спо-
собны к окислительному высыханию на воздухе при комнатной
температуре. В иных случаях для образования твердых пленок
необходима термообработка со сшивающей смолой. Средние и
тощие алкиды дают твердые менее гибкие пленки с хорошими
химической стойкостью и сохранением глянца, обычно высыхаю-
щие при повышенной температуре.
Ароматические кислоты, такие как фталевая или изофтале-
вая кислоты и малеиновый ангидрид, способствуют повыше-
нию твердости, химической стойкости и долговечности алкид-
ных покрытий. Напротив, длинноцепные двухосновные ки-
слоты, такие как азелаиновая кислота, иногда используются для
пластификации алкидов и обеспечения гибкости. Преимущест-
венное использование фталевого ангидрида в рецептурах алки-
дов обусловлено его низкой стоимостью и доступностью. Допол-
нительным преимуществом фталевого ангидрида по сравнению
с двухосновными кислотами является выделение при синтезе вдвое
меньшего количества воды. Первая стадия реакции фталевого
ангидрида, приводящая к образованию так называемого «полу-
эфира», экзотермична.
Обычно используют по крайней мере трифункциональные
полиолы для того, чтобы обеспечить разветвление и введение в
алкид гидроксильных групп для последующей реакции. В общем
они способствуют получению и сохранению хорошего цвета по-
крытий, но различаются по влиянию на химическую стойкость
и атмосферостойкость. Алкиды общего назначения получают с
использованием фталевого ангидрида, а свойства их модифици-
руют за счет варьирования полиола. Влияние различных поли-
олов можно легко объяснить изменением структуры. Так в ряду:
глицерин — триметилолэтан — триметилолпропан увеличивается
растворимость в алифатических углеводородах, уменьшается
вязкость, а эластичность пленок возрастает, что обусловлено уве-
личением длины боковой цепи в спирте. Иными словами, варьиро-
вание полиола может иметь существенный эффект, обусловленный
в значительной мере не только тем, что изменение его молекуляр-
ной массы влияет на содержание фталевого ангидрида в готовом
алкиде, и, следовательно, на твердость пленки.
Вначале канифоль использовали в алкидах как модификатор
вследствие ее низкой стоимости. Однако ее можно добавлять как
и другие моноосновные кислоты, например бензойную, с целью
уменьшения жирности, не увеличивая при этом вязкость. Кани-
фоль увеличивает скорость высыхания и твердость пленки, но
ухудшает атмосферостойкость. В настоящее время ее используют
только в алкидных грунтовках.
В тех случаях, когда необходимо обеспечить растворимость
тощих алкидов в алифатических растворителях, в качестве по-
лиола можно использовать триметилолпропан, хотя он и умень-
шает твердость пленки, а в качестве модифицирующей однооснов-
ной кислоты — трет-бутил бензойную кислоту. Эффективность
этих соединений обусловлена присутствием в их структуре али-
фатических боковых групп.
Изофталевая кислота по сравнению с ортофталевой обеспе-
чивает получение более высокомолекулярных, быстрее высыхаю-
щих смол и более твердых и долговечных пленок. Ее труднее
ввести в структуру алкида, так как она остается твердой в реак-
ционной смеси до тех пор, пока по крайней мере одна из ее
карбоксильных групп не прореагирует. Подобно ортофталевой,
изофталевая кислота возгоняется, однако из-за более низкой
растворимости изофталевой кислоты в ксилоле, используемом
для удаления реакционной воды, это может привести к закупорке
трубопроводов при синтезе. Терефталевая кислота очень мало
используется для синтеза алкидов из-за ее очень плохой раство-
римости в реакционных смесях и низкой реакционной способности.
Особо необходимо упомянуть о таком соединении как Cardura
Е 10 (Shell Chemicals), представляющем собой глицидиловый
сложный эфир разветвленной кислоты (Versatic Acid, Shell
Chemicals). Последняя является «третичной» или триалкилуксус-
ной кислотой с общим числом углеродных атомов равным ПГ
Благодаря эпоксидной группе это соединение быстро и полностью
реагирует с карбоксильными группами при температурах выше
150 °C; при включении в структуру алкида этого соединения
достигается тот же эффект, что и в случае невысыхающего масла
или, соответственно, жирной кислоты. Cardura Е 10 обеспечивает
хороший цвет, сохранение глянца и очень хорошую стойкость
к пожелтению и образованию пятен [4, 5]. Такие алкиды могут
использоваться в эмалях горячей сушки или в нитроцеллюлозных
лаках. Необходимо соблюдать осторожность при производстве
алкидов с использованием Cardura Е 10 и фталевого ангидрида,
так как реакция протекает быстро с выделением тепла (примерно,
110 кДж на эпоксидный эквивалент). Для подобных алкидов
разработаны специальные приемы составления композиций, ко-
торые также позволяют контролировать строение полимера [6, 7].
Cardura Е 10 имеет специальное применение для уменьшения
кислотного числа смол, например, когда требуется нулевое кислот-
ное число смол при использовании с фотокатализатор.ом, в крас-
ках, содержащих металлические пудры, или с некоторыми сик-
кативами.
* R1
К2-----СООСНг—сн—сн2,
R3
Cardura Е 10 (Shell Chemicals)
где Ri, Ro, Rs— алкильные группы, одна из которых метильная.
В качестве полиолов в алкидных рецептурах вместе с обыч-
ными полиолами могут применяться эпоксидные смолы или поли-
этиленгликоли. Введение эпоксидной смолы повышает водостой-
кость, химическую устойчивость, твердость и прочность, но уве-
личивает стоимость и может привести к мелению при эксплуатации
покрытия вне помещений. Полиэтиленгликоли придают водорас-
творимость или вододиспергируемость алкиду, который может
использоваться при производстве глянцевых красок (а также
как поверхностно-активное вещество) [8].
Другой путь придания водорастворимости или самоэмульги-
руемости заключается в получении алкида с высоким кислотным
числом за счет использования тримеллитового ангидрида или
диметилолпропионовой кислоты с последующей частичной ней-
трализацией щелочью, аммиаком или амином. Наиболее обще-
известный метод производства водорастворимых алкидов, содер-
жащих тримеллитовый ангидрид, заключается в том, что вначале
реагируют все компоненты за исключением этого ангидрида и
образуется полимер с кислотным числом ниже 10, а затем добав-
ляют тримеллитовый ангидрид при пониженной температуре,
примерно при 175 °C. В этом случае тримеллитовый ангидрид
присоединяется за счет одной сложноэфирной связи по гидро-
ксильным группам полиэфира, а две остающиеся карбоксильные
группы могут участвовать в солеобразовании и обеспечении за
счет этого растворимости в воде. Этот метод называют методом
«раскрытия кольца», так как реакция протекает в одну стадию
по ангидридной группе.
При производстве водорастворимых или водоэмульгируемых
алкидных смол необходимо тщательно составлять рецептуры,
чтобы избежать как повышенной чувствительности к воде конеч-
ной пленки, так и плохой стабильности композиции при хранении.
Диметилолпропионовая кислота, хотя и в меньшей степени, также
используется для получения алкидов с высоким кислотным числом.
Такие смолы более устойчивы к гидролизу. При их получении
обычно применяют жирные кислоты высыхающих масел, а ком-
позиции применяются в красках, высыхающих на воздухе или
при повышенных температурах, в сочетании с водорастворимыми
меламиноформальдегидными смолами.
Так как алкиды растворяются в ароматических или алифати-
ческих растворителях в зависимости от их жирности, они допу-
скают дополнительное использование спиртов, сложных эфиров
или кетонов, которые иногда необходимо применять в составах.
Могут быть приготовлены алкиды специально для применения
в промышленных ваннах для окунания с использованием три-
хлорэтилена.
2.3.2. Реакции и строение алкидов
Основной реакцией при синтезе алкидов, приводящей к обра-
зованию полимера, является реакция этерификации между ки-
слотой и спиртом, то есть поликонденсация. В случае получения
алкидов эту реакцию проводят обычно без катализаторов. Воз-
можны побочные реакции, включающие образование простых
эфиров полиолов, особенно на длительной стадии получения моно-
глицеридов. В связи с тем, что к образованию простых эфиров
наиболее склонен пентаэритрит, необходимо отбирать партии
пентаэритрита с низким содержанием димера.
Причиной получения более низкомолекулярных алкидов при
использовании фталевого ангидрида, а не изо- или терефталевой
кислот, является тенденция к образованию внутримолекулярных
кольцевых структур в ортофталатных сложных эфирах. Из-за
этого при расчетах эффективная функциональность фталевого
ангидрида всегда принимается меньше двух.
Имеются данные о том, что некоторые особые свойства ал-
кидов, включая способность к нанесению и высыханию, обуслов-
лены присутствием в них «микрогелей» [9]. Хотя это кажется
вполне возможным, практически трудно регулировать содержа-
ние микрогеля при синтезе алкидов, если только не вводить спе-
циально в композицию «микрогель», приготовленный отдельно
[10, 11]. Высказано предположение, что гелеобразование ал-
кидов происходит через выделение частиц микрогеля, после до-
стижения достаточного количества которых происходит их коалес-
ценция в «макрогель».
2.3.3. Контроль процесса синтеза алкидов
Реакцию получения алкидов необходимо контролировать по
вязкости и кислотному числу путем отбора проб. Измерение вяз-
кости разбавленных образцов можно проводить, используя про-
бирку Гарднера с пузырьком воздуха.
Гораздо быстрее и с большей точностью измерение вязкости
проводят, используя подогреваемую воронку и тарельчатый вис-
козиметр [12]. Интервалы между отборами проб в конце про-
цесса синтеза обычно уменьшают из-за увеличения скорости
повышения вязкости, особенно в случае тощих алкидов.
В случае хорошо изученных процессов можно осуществлять
выборочный контроль вязкости и кислотного числа; при их от-
клонении от контрольных значений делают корректировку, добав-
ляя небольшие количества кислоты или полиола. При синтезе
с растворителем и при возможности уплотнения масла, коррек-
тировку осуществляют изменяя интенсивность кипения раство-
рителя на достаточно ранних стадиях процесса. Для регулирова-
ния скорости нарастания вязкости можно также изменять темпе-
ратуру синтеза.
При достижении необходимых показателей для остановки
реакции смесь охлаждают и разбавляют. Разбавление необхо-
димо еще и потому, что алкидная смола после охлаждения ста-
новится вязкой и твердой.
Для вновь разработанных рецептур при выборе температур-
ного режима в случае жирных алкидов применяют более высокие
температуры для уплотнения масла и ускорения реакции, а в слу-
чаях менее жирных алкидов — более низкие температуры для
облегчения контроля конечной стадии. Можно рассчитать кислот-
ное число при гелеобразовании (см. следующий раздел) в про-
цессе синтеза. Эти данные позволяют изменить как состав реак-
ционной смеси, так и режим синтеза.
2.3.4. Описание алкидов
В технических условиях на алкиды указывается тип масла,
жирность, растворитель, а иногда двухосновная кислота и ее про-
цент в композиции.
Жирность определяется процентным весовым содержанием
масла в расчете на теоретический выход алкида. Для составов,
не содержащих глицерина, учитывают содержание жирных ки-
слот. В обоих случаях это основной показатель, от которого за-
висят эластичность пленок, растворимость, способность к высы-
ханию (при использовании высыхающих масел).
Как и для других смол определяют содержание сухого остат-
ка, вязкость, кислотное число и, если необходимо, гидроксильное
число в мг КОН на 1 г смолы.
Избыточное содержание гидроксильных групп, в отличие от
гидроксильного числа, определяемого экспериментально, рассчи-
тывается по молекулярной формуле и выражается как избыток
гидроксильных групп в процентах по сравнению с их количеством,
необходимым для полного реагирования с присутствующей поли-
основной кислотой. Избыточный процент гидроксильных групп
можно использовать как композиционный параметр при расчете
рецептур с требуемыми кислотным числом при гелеобразовании
или средней функциональностью.
Кислотное число при гелеобразовании можно рассчитать
теоретически и определить практически экстраполяцией зависи-
мость кислотного числа от величины обратной вязкости по мере
протекания процесса синтеза алкида.
2.3.5. Составление рецептур алкидов
Разработка рецептур алкидов включает расчетные методы,
использование предыдущего практического опыта и контрольные
синтезы, подтверждающие получение требуемого продукта. При
этом жирность и тип масла (жирной кислоты) в основном опре-
деляются областью применения, а полиол и двухосновная кисло-
та — совокупностью других требований, включая растворимость,
доступность, стоимость.
Метод средней функциональности — это один из методов
расчета рецептуры. При значениях F = 2 или более происходит
гелеобразование.
/?грсл!юс = 2-Общее число эквивалентов кислоты/Общее число молей.
При применении этого уравнения к маслосодержащим рецепту-
рам, вначале необходимо выразить количество молей масла в
молях составляющих его жирных кислот и глицерина.
Другой метод включает расчет степени завершенности реак-
ции в момент гелеобразования, наиболее удобно выражаемой
как кислотное число при гелеобразовании, которое должно быть
приблизительно на пять единиц меньше, чем требуемое кислотное
число в готовой смоле. В этом случае для регулирования кислот-
ного числа при гелеобразовании необходимо изменять содержа-
ние гидроксильных групп, выражаемое в виде избытка ОН. После
разработки теории гелеобразования были предложены различные
уравнения, например, Карозерсом, Флори, Штокмайером. Имеют-
ся специальные руководства по расчету рецептур алкидов [13, 14].
Часто используется одно из уравнений Штокмайера:
2 (Z&B)2
р =----------------------.
(Z,f^-ZM) (lifB-lgB)
где р — степень конверсии кислотных групп при гелеобразовании; /. g—функ-
циональность карбоксил- и гидроксилсодержащих соединений соответственно;
А, В — число молей присутствующих карбоксил- и гидроксилсодержащих соеди-
нений.
Эффективность различных уравнений была проверена экспери-
ментально [15]. В случае фталевого ангидрида необходимо при-
нимать функциональность меньше двух, учитывая его реальное
поведение при синтезе. Уравнения, предложенные Вейдерхорном
[16], более просты, удобны и менее сложны для расчетов в тех
случаях, где "они применимы. В перечисленных источниках чита-
тель найдет .примеры всех возможных уравнений.
При составлении рецептур тощих алкидов может возникнуть
необходимость использовать одноосновную кислоту, что опреде-
ляется требованиями по растворимости и содержанию гидроксиль-
ных групп. Применение уплотненного масла в жирных алкидах
диктуется необходимостью увеличить молекулярную массу и по-
лучить более высокую вязкость в тех случаях, когда этого невоз-
можно достичь другими методами; результатом будет повышение
экспериментального кислотного числа при гелеобразовании до
значений, превышающих расчетное.
Благодаря доступности микрокомпьютеров можно проводить
более сложные расчеты, которые ограничиваются только способ-
ностью пользователя правильно составить уравнения и написать
программы на языке машины. Поэтому вполне возможно написать
программу для компьютера, включающую параметры всех обычно
используемых в качестве сырья веществ, чтобы иметь возмож-
ность составлять рецептуры алкидов с различной жирностью и
содержанием гидроксилов, рассчитывать кислотное число при
гелеобразовании, среднюю функциональность и ряд других по-
казателей, используя упомянутые выше уравнения. Очень важно,
что с помощью компьютера, особенно если уравнения нельзя
решить по-другому, можно быстро повторять расчеты при изме-
нении факторов и изучать их влияние, прежде чем приступить
к эксперименту. В настоящее время составлены довольно слож-
ные и быстро решаемые программы [17].
23.6. Модифицированные алкиды
При замене части двухосновной кислоты в рецептуре алкида
на диизоцианат, например толуилендиизоцианат (ТДИ), можно
приготовить уралкиды; в этом случае расчет рецептуры проводят
аналогично обычным алкидам. Жирные уралкиды используют
в составах для декоративных покрытий для придания повышенной
прочности и скорости высыхания. Хотя возможна полная замена
двухосновной кислоты (с получением уретанового масла), но
обычно такие алкиды получают с уменьшенным содержанием
двухосновной кислоты, проводя синтез до низкого значения кис-
лотного числа, но высокого содержания остаточных гидроксиль-
ных групп, с последующей обработкой разбавленной смолы диизо-
цианатом. Изоцианатные группы взаимодействуют с гидроксиль-
ными с образованием уретановых связей, но, в отличие от кислот,
участвующих в образовании сложноэфирных связей, реакция
протекает экзотермически при более низких температурах и без
выделения побочного продукта. Важно, чтобы все изоцианатные
группы полностью прореагировали, учитывая высокую токсич-
ность свободных изоцианатов. Контроль в процессе синтеза мож-
но осуществлять титрованием амином с окончательной провер-
кой с помощью ИК-спектроскопии. Небольшие остаточные коли-
чества изоцианата можно удалить путем добавления низкомоле-
кулярного спирта. Для приготовления уралкидов необходимо
выбрать такой катализатор алкоголиза при получении моногли-
церида, который не способствовал бы нежелательным побочным
реакциям изоцианата, например образованию аллофаната. Таки-
ми катализаторами являются оксид кальция или растворимое
кальциевое мыло.
Другим видом модификации жирных алкидов является моди-
фикация полиамидными смолами, используемая для придания
тиксотропности готовым растворам смол. Для этих целей обычно
используют полиамидные смолы, получаемые из димеров жирных
кислот и диаминов. Физические свойства таких алкидов можно
очень тонко регулировать, а такие характеристики, как прочность
геля, скорость восстановления геля, способность к нестеканию
полученных красок сильно зависят как от состава, так и от техно-
логии получения алкида, а также от количества и типа полиамида
и степени завершенности реакции [18]. При приготовлении ал-
кидов этого типа или составлении содержащих их рецептур необ-
ходимо избегать применения или попадания в композицию поляр-
ных, например, спиртовых разбавителей. Последние разрушают
реологическую структуру, в которую водородные связи почти
наверняка вносят значительный вклад. Обычно вводят около
50% полиамида, а в процессе синтеза останавливают реакцию
на ее последней стадии в такой момент, чтобы достигнуть прозрач-
ности и оптимальных свойств геля.
Модификация силиконами жирных алкидов придает весьма
высокую способность сохранения блеска при их использовании
в ремонтных составах (красках для текущего ремонта). Исполь-
зуются те же самые силиконовые смолы с гидроксильными или
метоксильными функциональными группами, которые применяют
для модификации полиэфиров. В общем степень улучшения
свойств пропорциональна количеству этого дорогого модифика-
тора. Поэтому следует тщательно взвесить соотношение стоимости
с одной стороны, и достигаемое увеличение долговечности —
с другой.
Для модификации алкидов можно использовать как реак-
ционноспособные, так и нереакционноспособные маслораствори-
мые фенолоформальдегидные смолы. Они повышают твердость
пленки и улучшают водо- и химстойкость, но вызывают пожел-
тение. Подобные модифицированные алкиды полезно использо-
вать в грунтовках вместо эпоксидных смол, как более дешевые,
но менее химически стойкие.
Модификация алкидов виниловыми мономерами используется
для повышения скорости высыхания, увеличения твердости, улуч-
шения цвета, водо- и щелочестойкости. Однако при этом пони-
жается способность к сохранению блеска и устойчивость к рас-
творителям, причем последнее существенно затрудняет приготов-
ление составов, пригодных для быстрого нанесения повторных
слоев. Винилированные алкиды обычно используются для покры-
тий по металлам. В качестве модификаторов обычно используют
стирол или винилтолуол и реже—метилметакрилат. Модифика-
ция включает свободнорадикальную цепную полимеризацию
мономера в присутствии алкида в условиях, способствующих
прививке к ненасыщенным жирнокислотным остаткам алкида.
Обычно в рецептуру вводят по крайней мере некоторое количе-
ство дегидратированного касторового масла, поскольку наличие
остатков кислот с сопряженными двойными связями способствует
необходимой прививке; применение пероксидных инициаторов,
таких как ди-трет-бутилпероксид, также облегчает прививку
за счет отрыва атома водорода.
Возможны разные способы получения винилированных алки-
дов; наиболее распространенный из них заключается в винилиро-
вании алкида путем введения мономера и инициатора в раствор
смолы при 150—160 °C. Менее распространены способы, когда
винилируют масло до получения алкида или же винилируют моно-
глицерид перед поликонденсацией с двухосновной кислотой.
Последние способы позволяют использовать для разбавления
низкокипящие или хлорированные растворители, в которых реак-
ция винилирования была бы невозможна. В тех случаях, когда
возникают затруднения в достижении необходимой степени при-
вивки, что приводит к образованию мутных растворов смол, в
структуру алкида можно включать малеиновый ангидрид или
метакриловую кислоту и тем самым обеспечить введение допол-
нительных центров прививки.
Возможна также модификация путем предварительной кон-
денсации с меламиноформальдегидными (МЛ) смолами. Обычно
для многокомпонентных лакокрасочных составов вязкость мини-
мальна, если компоненты не прореагировали друг с другом до
стадии отверждения. Однако для ряда систем проведение предва-
рительного химического взаимодействия компонентов повышает
стабильность, совместимость или улучшает способность к отвер-
ждению даже в случае, когда оба полимера совместимы. Для
меламино-алкидных композиций, отверждаемых при невысоких
температурах, предварительная конденсация алкида и мелами-
ноформальдегида может облегчить.отверждение и в то же время
уменьшить тенденцию меламиноформальдегидного компонента
к самоконденсации при использовании каталитических систем
с высокой кислотностью.
2.4. ПОЛИЭФИРЫ
Термин полиэфир обычно применяют для полиэфирных смол
с карбоксильными или гидроксильными функциональными груп-
пами, не содержащих масел. Полиэфирные смолы получают
взаимодействием ди- или полиатомных спиртов и ди- или трехос-
новных кислот или ангидридов. Могут использоваться и одно-
основные кислоты. В этом разделе полиэфиры, содержащие лаури-
новую или стеариновую кислоты, для удобства будут называться
алкидами, а содержащие только алифатические кислоты с более
короткими цепями, включая низкомолекулярные разветвленные
синтетические жирные кислоты, и одноосновные ароматические
кислоты, такие как бензойная кислота, будут называться поли-
эфирами.
Ассортимент полиэфиров включает как низкомолекулярные,
для использования в композициях с высоким сухим остатком,
так и более высокомолекулярные, как с гидроксильными, так и
с карбоксильными функциональными группами. Ввиду отсутствия
в их составе более дешевых масел они дороже алкидов. Было
потрачено много усилий для достижения оптимальных свойств
и на поиски новых видов сырья. Последнее относится главным
образом к новым полиолам, позволяющим создавать материалы
с высокой долговечностью, необходимой при использовании, на-
пример, в верхних покрытиях автомобилей и при окрашивании
стальной спиральной ленты. Была предложена концепция «анхи-
мерного» эффекта [19], согласно которой для обеспечения устой-
чивости к гидролизу и стабильности сложного эфира у углерод-
ного атома в [3-положении к гидроксильной группе должны отсут-
ствовать атомы водорода. Этому требованию удовлетворяют
такие полиолы, как циклогександиметанол (ЦГДМ) и 2,24-три
метил-1,3-пентандиол (ТМПД), в которых либо уменьшено коли-
чество атомов водорода в [3-положении, либо они стерически за-
труднены, а также триметилолпропан (ТМП), неопентилгликоль
(НПГ) и Диол 204 (Union Carbide).
НОСН2—( )—СН2ОН цгдм
СН3 ОН СН3
НОСНг—С CH—СН—СНз ТМПД
I
СНз
СНз СНз
НОСНг—С СН2—ОСО—С—СНгОН Диол 204
(^Нз <^Нз
2.4.1. Состав
Состав полиэфиров определяется аналогично алкидам путем
расчетов средней функциональности и дополнительно значения
кислотного числа при гелеобразовании по методу Штокмейера
[13]. Кроме того, для теоретически ожидаемого гидроксилсодер-
жащего полиэфира можно рассчитать молекулярную массу по
формуле:
__ Масса реагентов
£Л4— ££а+(Масса реагентов• AV/56 100)
где М — общее число молей в рецептуре, — общее исходное число эквива-
лентов кислоты в рецептуре, AV — кислотное число.
Жесткость и эластичность полиэфиров можно регулировать
либо путем добавления к «более мягким» алифатическим двухос-
новным кислотам «более жестких» ароматических кислот, либо
введением в смесь полиолов «более жесткого» цйклогександиме-
танола (ЦГДМ) вместо «более мягких» алифатических полиолов.
При использовании изофталевой кислоты (ИФК), а не ортофтале-
вого ангидрида, могут возникнуть проблемы, связанные с возмож-
ностью кристаллизации из готовых растворов полиэфиров. Один
из путей управления этим явлением заключается во введении в
рецептуру небольшого количества терефталевой кислоты (ТФК)
с целью нарушения регулярности молекулярной цепи полиэфира.
Рецептуры органорастворимых полиэфиров пригодны для та-
ких областей использования, как покрытия рулонного металла,
автомобильные грунтовки и лаковые покрытия; покрытия по дре-
весине могут быть составлены на основе смеси «жестких» и «мяг-
ких» кислот (ИФК и адипиновой кислоты) с диолами и триолами,
такими как НПГ и ТМП. С целью удешевления или дополнитель-
ного увеличения эластичности в рецептуру могут вводиться фта-
левый ангидрид или гликоли с более длинной цепью. Возможны
добавки одноосновных кислот, таких как пеларгоновая или бен-
зойная. Такие полиэфиры обычно сшивают МЛ смолой при горя-
чей сушке или полифункциональным изоцианатом при комнатной
температуре, например в двухупаковочных композициях. Введение
триола в зависимости от состава композиции приводит к развет-
влению и появлению гидроксильных групп вдоль цепи; в тех слу-
чаях, когда требуется большая молекулярная масса полиэфира,
например при использовании для покрытий рулонного металла,
количество вводимого триола минимально.
В полиэфирах для лакокрасочных материалов с высоким сухим
остатком общее содержание полиола в композиции увеличивают,
хотя количество триола может быть уменьшено; процесс ведут
до получения низкомолекулярного продукта с более высоким
содержанием гидроксильных групп. Для придания водораствори-
мости составляют композиции, обеспечивающие получение высо-
кого кислотного числа. Процесс часто ведут в две стадии, как и
в случае синтеза водоразбавляемых алкидов, когда после завер-
шения поликонденсации проводят обработку тримеллитовым ан-
гидридом, сопровождающуюся раскрытием кольца. Для получе-
ния высококислых водорастворимых полиэфиров используют
также диметилолпропионовую кислоту.
Поскольку полиэфиры для порошковых красок должны иметь
температуру размягчения обычно более 40 °C, они чаще всего не
содержат пластифицирующих двухосновных кислот с длинными
цепями, а содержат одну или больше ароматических кислот и по
возможности простой диол. Они могут иметь высокое кислотное
число для отверждения эпоксидных смол или низкое кислотное
число и высокое содержание гидроксильных групп для отвержде-
ния меламиноформальдегидных смол.
2.4.2. Получение
Для получения полиэфиров используется та же технология,
что и для синтеза алкидов с небольшой корректировкой, учиты-
вающей используемое сырье. Разнообразные составы можно
приготовить из исходных твердых компонентов, которые необхо-
димо вначале тщательно расплавить. При использовании изофта-
левой кислоты могут возникнуть затруднения в получении прозрач-
ного продукта даже при полном завершении реакции. Они еще
более усугубляются, если присутствует менее растворимая и менее
реакционноспособная терефталевая кислота. Поскольку ряд гли-
колей, используемых для получения полиэфиров (например,
этиленгликоль, неопентилгликоль), легко теряются с реакционной
водой, при производстве полиэфиров с воспроизводимыми свой-
ствами требуются реакторы с ректификационными колоннами
[20, 21]. Была предложена технология с использованием давле-
ния, позволяющего повысить температуру синтеза и следователь-
но скорость реакции без существенной потери гликоля, особенно
при наличии в рецептуре этиленгликоля и пропиленгликоля. При
необходимости получения высокомолекулярных полиэфиров по-
лезно на заключительной стадии реакции использовать вакуум
для удаления остаточной реакционной воды и увеличения моле-
кулярной массы. Этот метод используется при производстве во-
локнообразующих полиэфиров [22]. При синтезе высокомолеку-
лярных полиэфиров очень важно принять меры, предотвращаю-
щие потерю гликоля, так как даже небольшие его потери приводят
к нарушению исходной рецептуры и ограничению молекулярной
массы.
Для увеличения скорости реакции часто используют катали-
заторы. Их надо выбирать с учетом того, что катализаторы могут
ухудшать цвет полиэфира. Было показано, что весьма практичны
оловосодержащие катализаторы [34].
2.4.3. Модификация
Для улучшения свойств полиэфиров, особенно долговечности,
используют модификацию силиконами. Возможна модификация
как органорастворимых [24], так и водоразбавляемых [25] поли-
эфиров. В первом случае при использовании для защиты рулон-
ного металла это стало общепринятой практикой. В органораство-
римых композициях модификацию силиконами проводят на второй
стадии после получения полиэфира, используя от 20% до 50%
силикона. В случае водоразбавляемых смол с высоким кислотным
числом модификацию силиконами можно осуществлять после по-
лиэтерификации до стадии обработки тримеллитовым ангидридом.
Обычно используют силиконовые смолы с метоксильными груп-
пами; в процессе модификации выделяется метанол, по количе-
ству которого при определенных условиях можно контролировать
реакцию. Степень завершенности реакции, если судить по коли-
честву замещенных метоксигрупп, составляет от 70% до 80%.
2.4.4. Ненасыщенные полиэфиры
Их получают из полиолов и двухосновных кислот, которые со-
держат определенное количество кислоты с двойной связью, рас-
творенных в активном мономере. Большинство этих полиэфиров
линейны и содержат малеиновый ангидрид как источник ненасы-
щенности. В технологии покрытий их применяют в основном в по-
крытиях по древесине и в ремонтных двухупаковочных лако-
красочных материалах, включая ремонтные шпатлевки. При ис-
пользовании для получения тонкослойных покрытий встречались
затруднения в преодолении ингибирующего влияния воздуха на
реакцию отверждения у поверхности пленки, что ограничивало
их использование. Однако расширение ассортимента активных мо-
номеров и возросшее применение радиационного отверждения по-
зволило в значительной мере преодолеть эту проблему. Для ра-
диационного отверждения используют акрилированные ненасы-
щенные полиэфиры, а не полиэфирмалеинаты. Однако для этих
целей в настоящее время с ними конкурируют акриловые и урета-
новые олигомеры.
Обычно в рецептуру ненасыщенных полиэфиров входят орто-
и изофталевые кислоты наряду с малеиновым ангидридом и соот-
ветствующие алифатические гликоли, такие как диэтиленгликоль,
который вводят как сореагент для регулирования гибкости. Коли-
чество малеинового ангидрида может составлять от 25% до
75% (мольн.) по отношению к другим кислотам. В процессе син-
теза происходит изомеризация цис-малеинатных остатков в транс-
фумаратные, и это имеет важное значение для последующего
отверждения, так как транс-фумаратная конфигурация более
активна в сополимеризации со стиролом. Степень изомеризации
может достигать 100%, но она изменяется в зависимости от
состава реакционной смеси и условий реакции.
Метод акрилирования, упомянутый выше, заключается в полу-
чении насыщенного гидроксилсодержащего полиэфира и его
этерификации на последней стадии акриловой кислотой.
Синтез ненасыщенных полиэфиров осуществляют аналогично
насыщенным [18]. Однако при использовании вместо ортофтале-
вого ангидрида менее реакционноспособных изо- и терефталевой
кислот для получения продуктов лучшего качества нужно исполь-
зовать многостадийный процесс, при котором на первой стадии
реагирует ароматическая кислота, а затем — малеиновый ангид-
рид [26, 27]. Поскольку ненасыщенные полиэфиры растворяются
в мономерах, способных к термической полимеризации, при раз-
бавлении необходимо, чтобы температура была как можно более
низкой. Для предотвращения полимеризации при разбавлении и
обеспечения стабильности при хранении, в смеситель, еще до вве-
дения в него смолы, необходимо добавлять ингибитор, такой как
п-трет-бутилкатехол. Кроме того, смолу можно охладить до
затвердевания, раздробить и затем уже растворить в мономере.
В качестве мономеров традиционно применяют стирол, а также
винилтолуол, метилметакрилат и некоторые аллиловые простые и
сложные эфиры, например, диаллилфталат. Окончательное от-
верждение осуществляется по механизму радикально-цепной поли-
меризации, инициируемой окислительно-восстановительной систе-
мой при комнатной температуре, хотя возможно и термическое
инициирование. Окислительно-восстановительная система двух-
упаковочная и состоит из органического пероксида или гидро-
пероксида в качестве одного компонента и восстановителя (амина
или соли высшей жирной кислоты), смешанного со смолой (см.
следующую главу).
Кислород ингибирует полимеризацию и поэтому поверхность
пленок ненасыщенных разбавленных стиролом полиэфиров после
отверждения липкая. Это можно практически полностью устра-
нить либо изолируя поверхность от кислорода введением воска,
либо включением в композицию химических групп, которые реа-
гируют с кислородом. Для этой цели используют простые аллиль-
ные группировки, которые вводят в систему добавлением аллиль-
ных мономеров [28], либо в цепь полиэфира путем его реакции
с аллильными соединениями, такими как аллилглицидиловый
эфир или диаллиловый эфир триметилолпропана [29]. Оксиал-
лильные группы являются единственными из известных, которые
по реакционной способности при окислительном сшивании при-
ближаются к природным высыхающим маслам. Радиационно-
отверждаемые композиции, менее чувствительны к ингибированию
воздухом, так как из-за большой концентрации радикалов ско-
рость основной реакции выше, чем скорость растворения кисло-
рода в пленке; кроме того, мономеры и олигомеры, используемые
в таких композициях, обладают повышенной функциональностью.
2.5. АКРИЛОВЫЕ ПОЛИМЕРЫ
Акриловые полимеры широко используются благодаря их пре-
восходным свойствам, таким как прозрачность, прочность, хими-
ческая устойчивость и атмосферостойкость. К ним относятся
полимеры, содержащие в структуре акриловые и метакриловые
сложные эфиры наряду с другими винильными ненасыщенными
соединениями. Они могут быть как термопластичными, так и термо-
реактивными, причем при получении последних в рецептуру вклю-
чают мономеры с дополнительными функциональными группами,
способными после образования исходного полимера к дальнейшим
реакциям с образованием сшивок. Большое значение имеет сопо-
лимеризация винильных и акриловых мономеров, так как в этом
случае имеются намного большие возможности, чем при поликон-
денсации, управлять строением полимера и придавать ему спе-
циальные свойства. В разных публикациях достаточно полно об-
суждаются вопросы получения и использования акриловых поли-
меров в покрытиях [30, 31].
В зависимости от свойств, которые мономеры придают конечно-
му полимеру или сополимеру, их. можно классифицировать на
«твердые», «мягкие» или «реакционноспособные». Твердыми моно-
мерами, например, являются метилметакрилат, стирол, винилаце-
1ат. Акрилаты более «мягкие», чем метакрилаты; к «мягким»
мономерам относятся: этилакрилат, 2-этилгексилакрилат, а также
длинноцепные метакрилаты. Реакционноспособные мономеры мо-
гут иметь гидроксильные группы, например, гидроксиэтилакрилат.
Достаточной реакционной способностью обладают акриламид и
особенно глицидилметакрилат. Реакционноспособны также кислые
мономеры; метакриловую кислоту часто вводят в неоольших коли-
чествах, так как кислотные группы могут улучшить диспергиро-
вание пигментов и катализировать отверждение сополимера.
Метилметакрилат как твердый мономер придает стойкость к
бензину, УФ-облучению, обеспечивает сохранение блеска. Поэтому
его используют в сополимерах для верхних покрытий, особенно
при окраске автомобилей. Бутилметакрилат, более мягкий моно-
мер, придающий очень хорошую влагостойкость материалам хо-
лодной сушки, но его пластифицирующий эффект ограничен.
Он придает хорошую межслойную адгезию, стойкость к раство-
рителям, превосходную устойчивость к УФ-облучению и сохране-
ние блеска. Этилакрилат обладает хорошими пластифицирующими
свойствами, но пары мономера весьма токсичны и обладают не-
приятным запахом. Его сополимеры довольно устойчивы к'УФ-об-
лучению и хорошо сохраняют блеск.
Практически акриловые полимеры для покрытий редко явля-
ются гомополимерами, а представляют собой сополимеры твер-
дых и мягких мономеров. Твердость полимера характеризуется
температурой стеклования (Тс), и для конкретного сополимера
его Тс можно рассчитать по уравнению \/TG= Wt/TGi -|- W2/TG)
и т. д., где TG[, TG4 являются температурами 7С гомополимеров
составляющих мономеров в К, a Wi, W2 — их массовые доли.
Для термоотверждаемых полимеров такая рассчитанная Гс не бу-
дет являться 7С конечной пленки, так как сшивание приведет к
дальнейшему повышению Тс, и это необходимо иметь в виду.
Хотя при сополимеризации могут быть получены полимеры раз-
личной структуры (статистические, чередующиеся, блочные или
привитые), для покрытий в подавляющем большинстве случаев
используются статистические сополимеры. Их статистический
характер определяет также то, что явления тактичности и кристал-
лизации, столь важные для объемных свойств полимеров, в этих
полимерах для покрытий практически не проявляются. А наиболее
часто встречающиеся структурные эффекты у этих полимеров
заключаются в разделении фаз и эффектах доменов, которые про-
исходят либо случайно, либо их заранее планируют.
2.5.1. Состав и синтез
При выборе рецептуры акриловых смол состав мономеров мож-
но определить исходя из требований срока службы, стоимости и
функционального назначения [30, 31]. Соотношения твердого и
мягкого мономера будут определяться требуемой величиной Тс,
которая наряду с молекулярной массой довольно четко опреде-
ляет возможность применения сополимера в покрытиях. Основ-
ная цель — достигнуть этих необходимых параметров по Тс и
М и высоких конверсий. Основными факторами, влияющими
на~М при синтезе полимера, являются природа инициатора и его
Таблица 2.2. Типичные непредельные мономеры
Мономер Строение Температура стеклования полимера Тс, °C
Бутилметакрилат О СН3 т- II । Л-С4Н9 —о—с—с=сн2 22
Этилакрилат С2Н5ООС —сн=сн2 — 22
2-Этилгексилакри- С2Н5 с4н0—сн—сн2—оос—сн=сн2 -70
лат 2-Гидроксипропил- мета кри л ат ОН сн3 СНз—СН—СН2ООС—с=сн2 76
Метакриловая кис- лота О СНз нос—с=сн2 210
Метилметакрилат о СНз СНз—О—С—с=сн2 105
Стирол сн2=сн— 100
Винилацетат О СН2=СНОССНз 30
концентрация, температура, концентрация мономера и агентов
передачи цепи в случае их применения. Обычно полимеризацию
проводят в присутствии до 50% растворителя при кипячении с
обратным холодильником. Таким образом, температура реакции
определяется природой используемого растворителя. Обычно коли-
чество инициатора колеблется между 0,1 и 4%, а температура
инициированной полимеризации — от 90 до 150 °C. Показано, что
довольно часто среднечисленная молекулярная масса обратно
пропорциональна корню квадратному из концентрации инициа-
тора при данной температуре.
Полимеризацию акриловых мономеров можно осуществлять в
массе, суспензии, эмульсии, дисперсии или в растворе. Однако
при синтезе полимеров, предназначенных для получения покры-
тий, наиболее часто используют методы полимеризации в растворе,
в эмульсии и в дисперсии.
Возможны несколько вариантов полимеризации в растворе.
Наиболее прост одностадийный вариант — нагревание мономера,
растворителя и инициатора до достижения необходимой степени
превращения. Однако, винильная полимеризация высоко экзо-
термична (50—70 кДж/моль) и эта теплота обычно отводится
холодильником при кипении смеси. При одностадийном процессе
реакция может быть очень экзотермичной, а это опасно. Поэтому
используют методы, когда мономер и инициатор, или только
инициатор вводят в реакционную систему при кипении посте-
пенно в течение 1—3 ч для того, чтобы обеспечить умеренное выде-
ление тепла и более контролируемое протекание реакции. Техни-
чески и с точки зрения безопасности наиболее предпочтителен
метод, когда в кипящий растворитель постепенно добавляют
мономер и инициатор. Он, кроме того, обеспечивает лучшую воз-
можность контролировать М и молекулярно-массовое распреде-
ление. При этом мономер и инициатор либо предварительно смеши-
вают и затем постепенно вводят в реакцию, либо, для полной
безопасности, каждый из этих компонентов дозируют из разных
емкостей. При этом должен быть обеспечен хороший контроль
скорости введения каждого компонента. После завершения поли-
меризации продукт можно дополнительно разбавить раствори-
телем, однако процесс всегда проводят в присутствии 30—40%
растворителя с целью снижения вязкости смеси, улучшения пере-
мешивания и отвода тепла. В большинстве случаев необяза-
тельно после конденсатора устанавливать сепаратор для отделе-
ния воды. Однако это нужно делать в тех случаях, когда наряду
с полимеризацией протекает реакция конденсации, например, при
синтезе некоторых сополимеров, содержащих акриламид.
Тип растворителя зависит от строения акрилового полимера.
На растворимость полиакрилатов влияет природа боковых групп.
Полимеры с короткими боковыми цепями сравнительно полярны и
растворяются в кетонах, сложных эфирах или смесях из простых
эфиров и спиртов. При увеличении длины боковой цепи могут
быть использованы ароматические растворители. При выборе рас-
творителя следует также учитывать тип вводимого сшивающего
агента, метод сшивания и другие требования, обусловленные
методом применения.
Контроль за процессом синтеза обычно проводят, измеряя
вязкость и содержание сухого остатка реакционной смеси, глав-
ным образом для того, чтобы знать степень конверсии. В этих про-
цессах уже практически невозможно после начала полимеризации
управлять молекулярной массой. Во многих случаях для заверше-
ния реакции требуется дополнительно вводить одну или несколько
порций инициатора. Молекулярную массу полимера можно опреде-
лять методом гель-хроматографии или рассчитывать из данных
измерений его приведенной вязкости в разбавленных растворах в
дихлорэтане.
2.5.2. Термопластичные акриловые полимеры
Термопластичные акриловые полимеры применяют особенно
широко для автомобильных верхних покрытий как при производ-
стве автомобилей, так и при последующих ремонтных окрасках.
В этих случаях необходимо очень жестко контролировать молеку-
лярную массу и ММР полимера, используемого для приготовле-
ния лаков и, особенно, композиций с металлами, поскольку ориен-
тация алюминиевых чешуек в красочной пленке зависит от реоло-
гии композиций. В свою очередь, ориентация чешуек сильно влияет
на достижение специфических эффектов цветового тона пленки.
При слишком больших молекулярных массах полимера получают
лакокрасочные материалы с низким сухим остатком и, кроме того,
возможны дефекты покрытий при нанесении, заключающиеся в
образовании паутинообразной сетки. В случае низкой молеку-
лярной массы ухудшаются прочность пленки, механические свой-
ства и срок службы. Было показано, что в случае полиметил-
метакрилатных лаков оптимальная молекулярная масса состав-
ляет примерно 80 тыс. [32].
Гомо- или.сополимеры ПММА для этих целей можно получить
полимеризацией в смесях углеводородов с кетонами в присутствии
пероксидных инициаторов в одну стадию при повышенном давле-
нии в случае необходимости [33]. Пленки ПММА необходимо
пластифицировать для повышения сопротивления растрескиванию
при пониженной температуре, адгезии и гибкости. Раньше в каче-
стве пластификаторов применяли бутилбензилфталат или низкомо-
лекулярные сложные полиэфиры. Но фталатные пластификаторы
могут улетучиваться, приводя к появлению хрупкости пленки.
Кроме того, при использовании внешних пластификаторов возни-
кают определенные трудности, связанные с необходимостью устра-
нения образующихся микротрещин и необходимостью полировки
покрытий. В связи с этим были проведены дополнительные
исследования. Использование ацетобутирата целлюлозы позво-
лило применить шлифовку песком наряду с горячей сушкой,
однако наиболее хорошие результаты были получены после раз-
работки двухфазных полимерных пленок [34]. Пленки с такой
структурой можно получить, например, путем смешения ММА —
бутилакрилатного сополимера с раствором гомополимера ПММА.
При этом образуются внешне прозрачные, но двухфазные пленки
с повышенной устойчивостью к образованию микротрещин.
В случае акриловых лакокрасочных материалов на основе
ММА использование мономеров, обеспечивающих внутреннюю
пластификацию, позволяет достичь оптимальных соотношений
между гибкостью и твердостью. Можно также вводить небольшие
количества кислотного мономера, например метакриловой кисло-
ты (МАК), с целью увеличения полярности и тем самым смачивае-
мости пигментов и адгезии. Для повышения адгезии вместо МАК
можно вводить мономеры с аминогруппой, например, диметилами-
нометилметакрилат, однако они могут вызвать небольшое пожел-
тение пленок.
Термопластичные акриловые полимеры в смеси с нитратцел-
люлозой и пластифицирующими алкидами можно применять для
внешних автомобильных покрытий низкотемпературной сушки
как при окраске автомобилей, так и при ремонте.
2.5.3. Термореактивные акриловые материалы
Термореактивные акриловые материалы были разработаны с
целью преодоления недостатков термопластичных полимеров.
Они характеризуются улучшенной химической и щелочестойко-
стыо, повышенным содержанием сухого остатка, растворимостью
в более дешевых растворителях, возможностью эксплуатации при
более высоких температурах, так как они менее склонны к размяг-
чению. Термореактивные материалы могут быть либо самоотверж-
дающимися, либо отверждаться в присутствии специально введен-
ных реакционноспособных полимеров или отвердителей. Большин-
ство из них относится ко второму типу.
Обычно молекулярная масса термореактивных акриловых
материалов составляет 20—30 тыс., а отвердители и сами полиме-
ры являются полифункциональными (функциональность больше
двух). Содержание мономера, обеспечивающего появление функ-
циональных групп в макромолекулярной цепи, колеблется в пре-
делах 5—25% (масс). Главное, чем руководствуются при разра-
ботке полностью отверждаемых акриловых систем.— это образо-
вание бесконечной сетки из сшивок между цепями макромо-
лекул полимера. Вследствие этого исходный низкополярный и
низковязкий полимер после отверждения превращается в пол-
ностью нерастворимый полимер с бесконечной молекулярной
массой.
Термореактивные акриловые полимеры с гидроксильными
функциональными группами не способны к самоотверждению.
Поэтому их необходимо смешивать, например, с азотсодержа-
щими смолами при создании материалов горячей сушки или с
аддуктами изоцианатов при получении двухупаковочных материа-
лов, отверждаемых при комнатной температуре, а также ремонт-
ных красок низкотемпературного отверждения.
Гидроксильные группы вводят в макромолекулярные цепи
сополимера за счет использования в исходной смеси мономеров
гидроксиалкил (мет) акрилатов, например, гидроксиэтилакрилата в
количестве до 25% масс [35]. При этом обычно в смесь моно-
меров вводят небольшой процент кислотного мономера с целью
достижения значения кислотного числа готового полимера 5—
10 и обеспечения катализа реакций отверждения азотсодержащи-
ми смолами. Регулируя соотношение компонентов, можно получить
необходимые твердость, гибкость и долговечность покрытий.
При этом необходимо обеспечить совместимость и стабиль-
ность при хранении с выбранной аминосмолой. Подобные компози-
ции с меламиноформальдегидными смолами (МЛ) используются
для нанесения последнего слоя при окраске автомобилей, причем
соотношение акрилового и меламиноформальдегидного компонен-
тов обычно равно 80—20 и 70—30. Реакции сшивания те же, что,
и у аминосмол с гидроксилсодержащими материалами. Акрило-
вомеламиноформальдегидные композиции превосходят алкидно-
меламиноформальдегидные по цвету и стойкости покрытия в усло-
виях атмосферных воздействий, но не обязательно по блеску.
При ремонтной окраске автомобилей на заводах для понижения
температуры сушки в композиции вводят кислотные катализаторы,,
такие как полуэфиры ангидридов дикарбоновых кислот или кислых
фосфатов. В этом отношении акриловомеламиноформальдегид-
ные композиции также превосходят алкидномеламиноформаль-
дегидные.
Гидроксилсодержащие акриловые полимеры получают по вы-
шеописанной технологии, обычно в ароматических растворите-
лях или в сложных эфирах, вводя постепенно в смесь мономеров
при кипении растворителя пероксидный инициатор, такой как
трет-бутилпербензоат. Для лучшего регулирования молекулярной
массы могут быть использованы агенты передачи цепи.
Акриловые полимеры с гидроксильными группами также полу-
чаются путем гидроксилирования карбоксилсодержащего поли-
мера этилен- или пропиленоксидами. Межмолекулярным сшивкам
можно придать дополнительную гибкость, удаляя гидроксильные
группы от основной цепи макромолекулы в результате реакции
с капролактоном гидроксильных групп, введенных ранее в полимер
в процессе сополимеризации с гидроксилсодержащим мономером.
Особое место среди самоотверждающихся термореактивных
композиций занимают сополимеры, полученные из акриламида с
последующей обработкой формальдегидом и затем подвергнутые
алкоксилированию [36, 37]. При этом образуются концевые ал-
коксиметильные реакционноспособные группы, обычно бутокси-
метильные. Эти группы реагируют почти так же, как и при их на-
хождении в азотсодержащих смолах; они улучшают совме-
стимость полимера с алкидными, эпоксидными смолами и нитро-
целлюлозой. Такие сополимеры можно смешивать с акриловыми
полимерами и затем отверждать. Ниже приведены типичные
реакции отверждения N-бутоксиметиламидных сополимеров:
Полимер—СО—NH—СН2ОВи-|- НО—Полимер-->
Полимер—СО—NH—СН2О—Полимер + ВиОН
Полимер—СО—NH—CH2OBu + ВиОСН2—NH—СО—Полимер >
Полимер—CONHCHqNHCO—Полимер-)- BuOCHsOBu
Возможны три способа получения таких полимеров. Вначале
проводят сополимеризацию мономеров, включая акриламид, а за-
тем последовательное взаимодействие амидных групп с формаль-
дегидом и спиртом. Другой способ заключается в том, что рас-
творители, включая спирт, мономеры и формальдегид, загружают-
ся сразу, а реакции полимеризации и оксиметилирования проте-
кают одновременно. Наконец, N-алкоксиметилакриламид можно
синтезировать еще до стадии полимеризации. В большинстве
случаев в этих композициях формальдегид используют в спирто-
вом, а не в водном растворе (например, 40% раствор формаль-
дегида в бутаноле), либо в виде параформальдегида. Реакция
амида с формальдегидом может катализироваться кислотами или
щелочами. При проведении реакции образования простого эфира
необходимо удалять реакционную воду азеотропной перегонкой.
Избыток спирта, который необходим как реагент и для сольвата-
ции акриламида, после завершения реакции должен оставаться в
качестве растворителя для обеспечения стабильности полимера.
Полимеризацию инициируют пероксидами и вводят агенты пере-
дачи цепи, так как конечный продукт из-за его способности к сши-
ванию должен быть низкомолекулярным.
Термореактивные акриламидные полимеры могут содержать
стирол для повышения стойкости к щелочам, моющим средствам
и солевому туману, акриловые сложные эфиры для повышения
гибкости и акрилонитрил для повышения стойкости к раствори-
телям и жесткости. Отсюда видно, что можно приготовить боль-
шое число композиций для различных областей применения,
как для бытовых целей, так и в промышленности для покрытия
рулонного металла. Отверждение катализируется вводимым в
смесь кислотным мономером. Для достижения очень высокой
стойкости покрытий к моющим средствам и коррозионной стой-
кости используют добавки эпоксидных смол. Применяется также
модификация силиконами для повышения атмосферостойкости.
Возможны материалы с высоким кислотным числом, которые
после нейтрализации аминами пригодны для использования в
водоразбавляемых системах [38, 39].
Получены карбоксилсодержащие термореактивные акриловые
полимеры, которые могут быть сшиты диэпоксидами. Они были
рекомендованы для покрытий верхнего слоя автомобилей и доволь-
но долго применялись для окраски промышленных изделий,
рулонного металла и барабанов [40]. В настоящее время они в
основном вытеснены гидроксилсодержащими и акриламидными
термореактивными акриловыми полимерами.
Возможна модификация готовых акриловых полимеров, па-
пример, конденсация акрилового полимера с высоким кислотным
числом с эпоксидным соединением Cardura Е 10 (Shell Chemicals)
с целью присоединения к полимеру пластифицирующих боковых
цепей [4]. Можно поступить и по-другому — вначале синтези-
ровать аддукт непредельной кислоты с Cardura Е 10 и использо-
вать его в качестве мономера при сополимеризации. Проводилась
также модификация гидроксилсодержащих акриловых полимеров
насыщенными или ненасыщенными жирными кислотами с целью
пластификации или же придания способности к высыханию на
воздухе.
2.6. ЭМУЛЬСИОННЫЕ ПОЛИМЕРЫ
Эмульсионные полимеры в настоящее время занимают первое
место по использованию в лакокрасочной промышленности среди
других полимеров, главным образом, из-за продолжающегося
роста применения водоэмульсионных красок в быту. Такие поли-
меры не содержат функциональных групп, коалесцируют при
комнатной температуре и термопластичны. Однако известны и
полимеры с функциональными группами, которые используются в
водоразбавляемых сшивающихся лакокрасочных материалах.
Можно отметить, что эмульсионные полимеры широко исполь-
зуются также в качестве адгезивов и в текстильной промышленно-
сти. Эмульсионной полимеризацией получают поливинилхлорид
и синтетические каучуки.
Эмульсии представляют собой двухфазные системы из двух
несмешивающихся жидкостей, в которых маленькие капельки
одной образуют дисперсную фазу в другой непрерывной фазе.
По терминологии, принятой в промышленности полимеров, терми-
ны эмульсионная полимеризация и эмульсионный полимер приме-
няются к процессу и к конечному продукту полимеризации моно-
меров в воде в присутствии ПАВ и водорастворимых инициаторов
с образованием стабильных дисперсий из очень маленьких частиц.
Для описания готовой дисперсии полимеров используется также
эквивалентный термин латекс. Частички полимеров имеют обычно
размер 0,1—0,5 мкм, так что в литре эмульсии может содержаться
1016 индивидуальных частиц с площадью поверхности 2000 м2.
Эмульсионная полимеризация — одна из разновидностей диспер-
сионной полимеризации. Другая разновидность дисперсионной по-
лимеризации — это неводная дисперсионная полимеризация
(НДП). Подобные полимеры также широко используются в ла-
кокрасочной промышленности. Известны и другие полимеры, по-
лучаемые дисперсионной полимеризацией, которые мало при-
меняются в технологии покрытий [41, 42]. Среди них можно
упомянуть неионностабилизированные латексы, в которых стаби-
лизирующими группами могут быть, например, цепи полиэтилен-
гликоля [43], механизм стабилизации которыми такой же, как и
в неводных дисперсиях (см. 2.7).
Типичная рецептура для эмульсионной полимеризации [44]
кроме воды содержит примерно 50 % смеси мономеров, состав-
ленной с учетом получения необходимой Тс, ПАВ и другие коллоид-
ные добавки, инициатор, буферные добавки для регулирования
pH и фунгициды. В эмульсионной полимеризации используются
такие «твердые» мономеры как винилацетат, метилметакрилат,
стирол и газообразный винилхлорид. «Мягкими» мономерами
являются бутилакрилат, 2-этилгексилакрилат, «винилверсатат»
(Shell Chemicals) [5], сложные эфиры малеиновой кислоты и
газообразные мономеры — этилен и винилиденхлорид.
Наиболее подходят для этих целей мономеры с умеренно низкой
растворимостью в воде. Мономеры с очень низкой растворимостью
трудно использовать. Могут использоваться и другие мономеры,
например, кислоты, такие как акриловая и метакриловая, и моно-
меры, способствующие повышению адгезии. Важно, чтобы пленки
коалесцировали после-испарения разбавителя. Минимальная тем-
пература пленкообразования (МТП) лакокрасочной композиции
является характеристикой, определяемой Тс полимера, а также
наличием ПАВ и негомогенностью полимерной композиции у
поверхности [45]. Полимеры с более высокими Тс, которые не
коалесцируют при комнатной температуре, могут коалесцировать
при введении в композицию временного пластификатора или
коалесцирующей добавки, такой как бензиловый спирт. Для эмуль-
сии обычно определяют МТП, а не Тс полимера, так как трудно
учесть все факторы, влияющие на МТП. Наиболее часто МТП
лежит в пределах 0—10 °C. Для лакокрасочных материалов с
большим количеством пигментов и наполнителей требуется эмуль-
сия с более низким МТП.
В качестве поверхностно-активных веществ используются
анионные, катионные и неионные ПАВ, существенным признаком
которых является то, что их молекулы обладают дифильными груп-
пами, одна из которых гидрофильна и обеспечивает водораство-
римость, а другая — гидрофобная, водонерастворимая. Строение,
растворимость, местонахождение и относительные размеры этих
двух групп по отношению ко всей структуре определяют поверх-
ностную активность данного соединения.
Роль ПАВ заключается [46], во-первых, в создании условий
для полимеризации мономера, а во-вторых,— в стабилизации об-
разующихся частиц полимера. При концентрациях ПАВ в воде
выше определенного значения молекулы ПАВ ассоциируют с обра-
зованием мицелл. Эти мицеллы способны солюбилизировать моно-
мер. В эмульсионной полимеризации часто используют смесь
анионных и неионных ПАВ. Катионные ПАВ используются редко.
В качестве защитных коллоидов используют такие водораство-
римые полимеры, как поли (мет) акриловую кислоту или ее сопо-
лимеры, так называемый поливиниловый спирт (частично гидро-
лизованный поливинилацетат) или производные целлюлозы, на-
пример гидроксиэтилцеллюлозу. Свойства этих коллоидов зависят
от молекулярной массы, степени разветвления и состава (содер-
жания карбоксильных или гидроксильных групп, обеспечивающих
водорастворимость). В процессе эмульсионной полимеризации к
ним могут прививаться растущие цепи образующегося полимера,
особенно в случае производных целлюлозы, или же может про-
исходить расщепление их макроцепей. Они участвуют в процессах,
регулирующих размер частиц, и влияют на реологию готового
лакокрасочного материала, в частности, на тиксотропность и
строение геля. В эмульсионной полимеризации обычно различают
две характерные стадии: стадию затравки и стадию подачи мате-
риала. На первой стадии смесь воды, ПАВ и защитного коллоида
нагревают до температуры 85—90 °C и добавляют 5—10% смеси
мономеров наряду с частью инициатора (обычно водораствори-
мого персульфата). На этой стадии образуются первые частички
полимера.
Реакционная смесь на стадии затравки содержит стабилизи-
рованные ПАВ капли мономера, инициатор, небольшое количе-
ство мономера в растворе и ПАВ как в растворе, так и в виде ми-
целл. Радикалы образуются в растворе при распаде инициатора и
инициируют полимеризацию небольшого количества мономера,
растворенного в воде. Затем растущая полимерная цепь прони-
кает в мицеллы и вызывает полимеризацию находящегося там
мономера. Обрыв происходит тогда, когда следующий растущий
полимерный радикал входит в мицеллу. На стадии затравки,
когда больше -не добавляется никаких реагентов, исходные моно-
мер и инициатор почти полностью превращаются в полимер,
а число частиц примерно соответствует числу мицелл, образован-
ных введенным ПАВ. Количество ПАВ, которое не связано с
маленькими частичками полимера или с капельками мономера,
невелико.
На стадии подачи материала в реакционную смесь добавляют
оставшийся мономер и раствор инициатора. Источником мономера
в реакционной среде являются капельки эмульгированного моно-
мера. Полимеризация протекает по мере того, как мономер диф-
фундирует из капелек мономера через водную фазу в уже сформи-
ровавшиеся растущие полимерные частички. В это же время в эти
частички набухшего в мономере полимера проникают радикалы,
вызывая как обрыв, так и повторное инициирование полимери-
зации. По мере увеличения размеров частичек остающийся ПАВ
из водной фазы поглощается поверхностью этих частичек и
стабилизирует дисперсию. На рис. 2.1. показаны частицы, прини-
мающие участие в процессе полимеризации. Частицы стабилизи-
руются от флокуляции и коалесценции вследствие взаимного
отталкивания поверхностных зарядов, возникающих из анион-
ного ПАВ.
На завершающей стадии полимеризации для более полной кон-
версии может быть введена еще одна порция инициатора (раз-
лагающегося при повышенной температуре или окислительно-
восстановительная система), после чего систему охлаждают и при
необходимости вводят биоцид.
Олигомерный аниои-радикал
Рис. 2.1. Структурные образования, участвующие в процессе эмульсионной поли-
меризации
Эмульсионную полимеризацию труднее контролировать, чем
полимеризацию акриловых мономеров в растворе. Важное значе-
ние для хорошего эмульгирования реагентов на первой стадии,
отвода тепла через охлаждающие стенки реактора и распределе-
ния добавленного мономера на второй стадии имеет эффективное
перемешивание. В то же время композиции могут быть чувстви-
тельными к сдвигу, поэтому следует избегать очень интенсив-
ного перемешивания. Для поддержания необходимой температуры
реакции после первоначального нагрева следует охлаждать смесь.
Определенное затруднение вызывает также наличие комочков
полимера и его налипание на стенки реактора. Кинетика эмуль-
сионной полимеризации довольно сложна, и для изучения ее меха-
низма были проведены многочисленные исследования [47]. В на-
стоящее время разработаны методы, позволяющие регулировать
структуру образующихся частичек, в частности, получать частички
с взаимопроникающими трехмерными сетками, частицы с види-
мыми раздельно ядровой и оболочковой структурами, получать
микрогелевые структуры с целью модификации механических
свойств или регулирования реологии подобно тому, как в микро-
гелях из раствора [41, 48, 49].
Хотя можно получать эмульсии, используя только твердые
мономеры за счет добавления так называемых внешних пласти-
фикаторов в лакокрасочные материалы, все применяемые в настоя-
щее время эмульсии внутренне пластифицированы путем сополи-
меризации с мягким мономером. Стиролсодержащие эмульсии,
мало используются для получения покрытий в Великобритании,
хотя они и применяются в остальных европейских странах. Эмуль-
сии общего назначения обычно получают на основе винилаце-
тата, пластифицированного акрилатом, например бутил- или
2-этилгексилакрилатом, или диалкилмалеинатом. Покрытия по-
добного состава обладают хорошей стойкостью к мелению, но
плохой устойчивостью к щелочам и гидролизу. Для улучшения
последних свойств в качестве пластифицирующего мономера
можно использовать винилверсатат (Shell Chemicals) [50]. Ком-
позиции, содержащие только акриловые мономеры, в частности
метилметакрилат и акриловые пластифицирующие мономеры,
обычно приводят к более качественным эмульсиям, за исключе-
нием меньшей устойчивости покрытий к мелению.
При использовании газообразных мономеров требуется обору-
дование, способное работать под давлением. Эти мономеры можно
применять для получения более дешевых эмульсий, например,
сополимеризацией винилхлорида и этилена с винилацетатом.
Особенно удешевляет композиции применение этилена, но он на-
столько «мягкий» мономер, что обычно используют винилхлорид
как «твердый» мономер наряду с винилацетатом. Полимеры,
содержащие винилхлорид и винилиденхлорид, представляют боль-
шой интерес для получения противокоррозионных грунтовок.
Винилхлорид — канцерогенный мономер, поэтому готовые эмуль-
сии необходимо тщательно обрабатывать паром для удаления
следов мономера из продукта.
Эмульсии характеризуют по содержанию сухого остатка, вяз-
кости, минимальной температуре пленкообразования [51] и устой-
чивости к замораживанию — оттаиванию. Также проводят оценку
внешнего вида пленок на отсутствие комочков полимера. В случае
эмульсий, содержащих винилацетат и винилхлорид, для пред-
отвращения гидролиза необходимо контролировать pH.
Для модификации реологических свойств в рецептуру могут
вводиться небольшие количества непредельных кислот. Для повы-
шения адгезии используются глицидилметакрилат и аминосодер-
жащие мономеры, а в тех случаях, когда требуются особенно
высокие показатели, например для повышения адгезии грунто-
вок к дереву и к старым глянцевым покрытиям, были рекомендо-
ваны непредельные ацетоуксусные сложные эфиры [52] и моче-
вины [53].
Термореактивные полимеры получают по аналогии с описан-
ным выше, но в их состав входит гидроксилсодержащий мономер
и они обычно не содержат защитного коллоида.
2.7. ДИСПЕРСИОННАЯ ПОЛИМЕРИЗАЦИЯ В НЕВОДНЫХ СРЕДАХ
Другим типом полимерных дисперсий, используемых в техно-
логии покрытий, являются неводные дисперсии, которые внешне
имеют много сходных свойств с эмульсионными полимерами в
водной среде. В этом случае полимер, обычно акриловый, диспер-
гирован в неводной среде, чаще всего в алифатическом угле-
водороде [54]. Неводные дисперсии акриловых полимеров в на-
стоящее время применяются, главным образом, в автомобильных
лакокрасочных материалах, хотя описан и ряд других областей
применения. Можно получить полимерные дисперсии с различной
структурой частиц, включая слоевые, гетерогенные и ячеистые
[42]. Размеры частиц в полимерных неводных дисперсиях, как и в
водных дисперсиях полимеров, составляют 0,1—0,5 мкм. Механизм
цепной аддитивной полимеризации также подобен ранее описан-
ному для акриловых полимеров. Однако, в отличие от эмульсион-
ных полимеров, в которых частицы стабилизированы за счет оттал-
кивания поверхностных зарядов, обусловленных применением ани-
онных ПАВ, единственный возможный способ стабилизации
неводных дисперсий заключается в так называемой стерической
стабилизации, которая предусматривает наличие на межфазной
границе полимерных частичек адсорбированного сольватирован-
ного полимерного слоя. Стабилизация за счет заряда имеет не-
большое значение в средах, применяемых для неводной полиме-
ризации из-за значений диэлектрических констант, которые на
один или два порядка меньше, чем у воды. При стерической
стабилизации силы отталкивания, которые препятствуют флоку-
ляции при соударении частиц, появляются в результате увеличе-
ния локальной концентрации сольватированного полимера в точке
контакта. Реакция системы в ответ на это заключается в устране-
нии этого избытка локальной концентрации путем разделения
частиц (рис. 2.2.). Стерическая стабилизация может быть также
эффективна и в средах с высокой диэлектрической постоянной,
где обычно применяется стабилизация за счет заряда, в том слу-
чае, если используются неионногенные стабилизаторы [43].
Наиболее эффективные стабилизаторы, используемые при дис-
персионной полимеризации, получены на основе блок- или при-
витых сополимеров [55]. Для получения привитых сополимеров,
которые могут применяться в неводной дисперсионной полимери-
зации, были разработаны специальные методы [56]. Подобные
привитые стабилизаторы на основе поли-12-гидроксистеариновой
кислоты легко получаются и эффективны в неводной диспер-
сионной полимеризации, протекающей как по цепному, так и по
ступенчатому механизмам. Промышленная 12-гидроксистеарино-
Рис. 2.2. Изображение стериче-
ского отталкивания, обусловлен-
ного растворимыми цепями, при-
соединенными к дисперсным
частицам
12-Гвдроксистеариновая Поли ГС
соон
кислота
Поли ГС
Метакрилат (МА)
ГМА
СООН —*• По™ ГС
Рис. 2.3. Схематические изображения образования «гребневидного» стабилизатора
из поли- 12-гидроксистеариновой кислоты
вая кислота содержит 8—15% пальмитиновой и стеариновой
кислот, которые ограничивают молекулярную массу при поли-
конденсации, что приводит к образованию полимера со средне-
численной молекулярной массой 1500—2000, содержащего одну
карбоксильную группу. Такой олигомер может быть превращен
в «макромономер» при реакции карбоксильной группы с глицидил-
метакрилатом и этот «макромономер» затем подвергнут сополи-
меризации с равным по массе количеством метилметакрилата или
аналогичного мономера с образованием полимера с молекуляр-
ной массой 10—20 тыс. Такой полимер затем служит как гребне-
образный стабилизатор, в котором имеются 5—10 растворимых
боковых цепей, отходящих от закрепленной главной цепи макро-
молекулы (рис. 2.3).
Образование привитого полимера и способ стабилизации дис-
персных частиц вследствие адсорбции нерастворимой прикрепляе-
мой части полимера схематически показаны на рисунке 2.3. Про-
изводные поли-12-гидроксистеариновой кислоты также применя-
ются как диспергирующие вещества [12]. Обзор специальных
методов получения «макромономеров», необходимых для синтеза
привитых диспергаторов определенного строения, приведен в
работе [57]. Полимеризацией в неводных дисперсиях можно полу-
чить практически все полимеризационные пленкообразователи.
Даже мономеры, которые трудно полимеризуются в растворе,
могут быть легко заполимеризованы в дисперсии (так же как и в
случае водоэмульсионной полимеризации). Основное ограничение
для применяемых мономеров заключается в том, чтобы образую-
щийся полимер был не растворим в используемой среде. Так,
если средой являются алифатические углеводороды, количество
используемого длинноцепного пластифицирующего мономера
должно быть ограничено. Поскольку полимеризация почти пол-
ностью происходит внутри частиц, состоящих из набухшего в моно-
мере полимера, скорость полимеризации намного больше по срав-
нению с полимеризацией в растворе из-за уменьшения диффузион-
но-контролируемой скорости обрыва.
Подобно эмульсионной полимеризации, неводные дисперсии
получают обычно в 2 стадии. На стадии затравки разбавитель
вместе с частью диспергатора и мономера нагревают в присут-
ствии инициатора с образованием исходной тонкой дисперсии с
низкой концентрацией. Затем в течение нескольких часов загру-
жают оставшийся мономер, диспергатор и инициатор для завер-
шения роста частиц. Для контроля молекулярной массы обычно
добавляют небольшое количество агента передачи цепи. В зависи-
мости от температуры реакции, инициаторами могут быть либо
азо-, либо пероксидные соединения.
Как и в случае эмульсионной полимеризации, на качество ди-
сперсии может отрицательно влиять способ подпитки мономером,
хотя, в отличие от эмульсионной полимеризации, мономер пол-
ностью растворим в непрерывной фазе. Обычно процесс проводят
при рециркуляции разбавителя и рекомендуют смешивать допол-
нительно вводимый мономер с возвратным охлажденным дистил-
лятом. Возможны также непрерывные методы получения. Этот
метод позволяет очень хорошо контролировать размер частиц и
при правильно выбранном диспергаторе и его распределении
между двумя стадиями можно легко получить дисперсии с части-
цами одного размера, гораздо легче, чем при эмульсионной поли-
меризации. Подобно этому, используя соответствующие способы
регулирования, можно получить частички различных размеров,
способные к более эффективной упаковке, что позволяет готовить
дисперсии с высоким сухим остатком, вплоть до 85% (объемн.)
[58, 59].
Неводные дисперсии акриловых полимеров находят примене-
ние в автомобильных термоотверждаемых лакокрасочных мате-
риалах; в этом случае в исходную смесь мономеров включают
гидроксилсодержащие мономеры. К полученной полимерной
дисперсии можно добавить более сильный растворитель для
растворения части или всего диспергированного полимера. Таким
образом можно готовить широкий ряд композиций, от таких,
в которых полимер находится в истинном растворе или в виде
набухшего в растворителе геля, до устойчивых ненабухших ча-
стичек полимера. Неводные дисперсии этого типа применяются
потому, что присутствие набухшего нерастворимого полимера
оказывает сильное влияние на скорость испарения растворите-
лей и на скорость повышения вязкости в процессе испарения.
Эти эффекты позволяют регулировать в широких пределах тол-
щину покрытий при нанесении распылением, уменьшить образо-
вание наплывов и хорошо управлять ориентацией металлических
пигментов в пленке. Действительно, недавно было установлено,
что для эффективного управления реологическими характери-
стиками при нанесении наиболее целесообразно получать органи-
ческие «микрогели» и смешивать их с растворами термореак-
тивных гидроксилсодержащих акриловых полимеров. Для этих
целей в качестве мономеров могут использоваться глицидил-
метакрилат и метакриловая кислота, реагирующие в процессе
синтеза в присутствии аминного катализатора, приводя, в ко-
нечном счете, к частичкам, которые абсолютно нерастворимы в
органических растворителях. Эти частички можно модифициро-
вать другим полимером после получения ядра микрогеля и окон-
чательно разбавить смесью сильных растворителей.
2.8. ВОДОРАЗБАВЛЯЕМЫЕ МАТЕРИАЛЫ
Хотя водоразбавляемые системы применялись и раньше, при-
нятие законов в ряде стран привело к увеличению их использо-
вания с целью уменьшения загрязнения воздушной среды раство-
рителями. Этому требованию отвечают кроме упомянутых еще три
вида прогрессивных лакокрасочных материалов — с высоким су-
хим остатком, радиационно-отверждаемые и порошковые. Для
того, чтобы заменить органические растворители на воду, необ-
ходимо изменить состав пленкообразователя в сторону увеличения
его гидрофильности за счет введения в его структуру групп, обеспе-
чивающих водорастворимость, применения ПАВ или обоих фак-
торов. В таких системах полимер может находиться либо в раство-
ре, либо в виде дисперсии. Использование воды в качестве рас-
творителя или разбавителя увеличивает время сушки и может вы-
звать необходимость в регулировании влажности распылительной
камеры. Кроме того, из-за более высокой теплоты испарения воды
при горячей сушке требуются большие затраты энергии. Если
после отверждения гидрофильные группы не разрушатся или не
дезактивируются, повышенная чувствительность готового покры-
тия к воде создает в ряде случаев проблемы. Несмотря на указан-
ные недостатки, замена органических растворителей водой повы-
шает безопасность для потребителя как с точки зрения пожаро-
опасности, так и токсичности, уменьшает или устраняет опас-
ность загрязнения окружающей среды летучими веществами при
сушке. Эти преимущества могут перевесить приведенные недо-
статки. Электроосаждаемые лакокрасочные материалы представ-
ляют собой особый класс водоразбавляемых систем.
Водоразбавляемые материалы, получаемые только с исполь-
зованием ПАВ, были рассмотрены в разделе, посвященном эмуль-
сионной полимеризации. В данном разделе рассмотрены системы,
солюбилизация или диспергирование которых происходит благо-
даря наличию гидрофильных групп в основной цепи полимера.
В этом случае, как правило, полимер получают в отсутствие воды
и только после достижения конечной молекулярной массы его
переводят в воду. Конечно, полимеры, не содержащие полярных
групп, можно эмульгировать добавлением ПАВ. Однако обычно
этого не делают, за исключением тех случаев, когда применяют
смесь пленкообразователей и один из них «солюбилизирует»
другие, выполняя роль эмульгатора для всех присутствующих
полимерных компонентов. Такой подход можно применить, напри-
мер, в том случае, когда для отверждения необходимо присутствие
нерастворимого отвердителя. Известны полимеры, полностью рас-
творимые в воде без добавления солеобразующих добавок, напри-
мер полиэтиленоксид или полиэтиленгликоль, поливинилпирроли-
дон, полиакриламид и сополимеры с большим содержанием этих
веществ. Полиэтиленгликоль благодаря наличию концевых гид-
роксильных групп широко используется в качестве реагента для
придания алкидам растворимости или диспергируемое™ в воде.
Полиэтиленоксид используется как водорастворимая часть мно-
гих ПАВ. Другие водорастворимые полимеры, включая поливини-
ловый спирт, акриловые полимеры с высоким содержанием кис-
лоты, производные целлюлозы — применяют в качестве защит-
ных коллоидов при эмульсионной полимеризации. Некоторые
продукты конденсации с участием формальдегида, в том числе
гексаметоксиметилмеламин и некоторые фенолформальдегиды,
водорастворимы и поэтому им отдается предпочтение как сшиваю-
щим агентам в водоразбавляемых системах по сравнению с дру-
гими полимерами [60].
Основной способ достижения растворимости или диспергируе-
мое™ в воде — синтез полимера с кислотными или аминогруппами
в молекулярной цепи, растворимость которых можно повысить
образованием соли при добавлении летучего амина или кислоты.
При растворении такого полимера в воде солевые группы иони-
зируются и противоионы переходят в объем раствора. Преиму-
щество применения летучих основания или кислоты заключается
в уменьшении гидрофильности готовой пленки вследствие удале-
ния солеобразующего агента. Гидрофильность можно дополни-
тельно уменьшить или устранить, если эти группы способны затем
принимать участие в реакциях сшивания. В общем необходимо
отметить, что многие хорошие водоразбавляемые композиции
представляют собой дисперсии (коллоидные или мицеллярные), а
не растворы просто потому, что таким путем можно получи™
наибольшее содержание сухого остатка и самую низкую вязкость.
Можно синтезировать полимеры с более высоким содержанием
кислотных или аминогрупп,' чем необходимо. Тогда «степень
нейтрализации» с солеобразующими аминами или кислотой бу-
дет составлять 40—70%. Свойства таких полимеров можно
очень тонко регулировать и получать оптимальные вязкость,
стабильность, устойчивость к оседанию и способность к нанесению.
Первыми водоразбавляемыми связующими были нейтрализо-
ванные щелочью малеиновые аддукты, наиболее простыми из ко-
торых являлись малеинизированные масла. Взаимодействие
малеинизированных жирных кислот с основными цепями поли-
мера по-прежнему имеет существенное значение при создании
водоразбавляемых систем. Так, на основе эпоксидных смол,
этерифицированных малеинизированными жирными кислотами,
льняного масла или их смесью с другими жирными кислотами с
последующей нейтрализацией, можно получить связующие как
для обычного нанесения распылением, так и для электроосажде-
ния. Малеинизированный полибутадиен можно использовать как
основу для создания водоразбавляемых систем. Водоразбавляе-
мые композиции на основе масел и полибутадиена способны
отверждаться вследствие окисления при повышенной температуре
без добавок, специальных отвердителей. Водоразбавляемые ал-
кидные и полиэфирные композиции, полученные специально с
высоким значением кислотного числа для придания водораство-
римости, уже упоминались ранее. Термореактивные кислые
акриловые композиции, получаемые, например, сополимериза-
цией малеинового ангидрида или акриловой кислоты с другими
ненасыщенными мономерами, также уже были описаны. В эпок-
сидные композиции можно ввести карбоксильные группы за счет
взаимодействия с ангидридами дикарбоновых кислот или с
тримеллитовым ангидридом с образованием полиэфиров, как опи-
сано для алкидов и полиэфиров [61].
Водоразбавляемые пленкообразователи могут отверждаться
как самостоятельно (алкиды, термореактивные акриловые поли-
меры), так и при добавлении растворимых или совместно эмуль-
гируемых водонерастворимых сшивающих смол. Полностью
растворимые сшивающие смолы вводят в композицию на любой
стадии. Примерами таких смол являются меламиноформаль-
дегидные, мочевинные и фенольные смолы. Недавно были разра-
ботаны [3-гидроксиалкиламиды, которые специально применяются
как сшиватели для карбоксилсодержащих водоразбавляемых ком-
позиций и характеризуются низкой экологической вредностью.
Их получают взаимодействием алкиловых сложных эфиров дикар-
боновых кислот с алканоламинами в присутствии основного ката-
лизатора. Типичным примером является взаимодействие диметил-
адипата с дигидроксипропаноламином [62]. Их реакционная
способность при отверждении сравнима с аминосмолами, однако
сшивание происходит за счет образования сложноэфирных свя-
зей с карбоксильными группами:
Me Me
I I
RCO—N—CH—OH-|-HOOC—Полимер > RCO—N—CH—OOC—Полимер-f-H2O.
В эпоксидные смолы можно ввести аминные функциональные
группы путем взаимодействия со вторичными аминами и затем с
уксусной кислотой с образованием соли, что обеспечивает повы-
шение водорастворимости [61]:
О
—СН^СНз + HNR, > —CH—СН2—NR2 сн=соон „
ОН
I
——CH—СН2—NR2 СНзСОО-.
н
Для отверждения этих композиций необходимо добавлять
такие сшивающие смолы как меламиноформальдегидные, феноль-
ные или блокированные изоцианаты. Водоразбавляемые эпоксид-
ные смолы применяются в качестве грунтовок по металлу и
покрытий консервных банок [63] В последнем случае наиболее
широко используются водоразбавляемые привитые сополимеры
эпоксидных смол с акрилатами [64].
В качестве термоотверждаемых водоразбавляемых компо-
зиций применяются специально приготовленные акриловые эмуль-
сии. Их получают путем обычной эмульсионной полимеризации.
В состав мономеров входит гидроксилсодержащий мономер,
причем синтез обычно проводят без защитного коллоида, а только
в присутствии ПАВ. Эти системы обычно называют «безколлоид-
ными» латексами.
Водорастворимые смолы можно модифицировать силиконами;
преимущество таких продуктов было показано на примере алки-
дов и полиэфиров, модификацию которых проводили путем пред-
варительного взаимодействия силиконов с полиолом. Модифика-
цию силиконами готовых акриловых латексов можно проводить
на последней стадии после завершения эмульсионной полимери-
зации [22].
При использовании водоразбавляемых материалов возникают
специфические проблемы, которые необходимо учитывать при раз-
работке рецептур. Так, продукты конденсации могут быть склонны
к гидролизу. На реакцию отверждения может отрицательно
влиять присутствие вещества, использовавшегося при синтезе
для повышения водорастворимости смол, либо за счет величи-
ны pH, либо, как в случае высыхающих на воздухе алкидов, за счет
нейтрализации аминов, замедляющих процесс окислительного
сшивания [65]. Присутствие аминов может отрицательно влиять
на сохранение цвета пленки.
Большинство водорастворимых композиций содержит опреде-
ленное количество смешиваемых с водой органических раствори-
телей [66], которые необходимо было использовать при синтезе
смолы еще до введения воды. Их присутствие не вызывает
сильного загрязнения окружающей среды, и содержание в атмо-
сфере летучих компонентов не превышает установленных норм,
вместе с тем они позволяют регулировать такие свойства компози-
ций, как стабильность, способность к высыханию и реологию.
В состав дисперсионных композиций также может входить орга-
нический растворитель для облегчения коалесценции. Присутствие
смешиваемых с водой растворителей может также способствовать
эмульгированию в дисперсных системах, как это хорошо известно
в так называемых «микроэмульсиях».
2.9. МАТЕРИАЛЫ ДЛЯ ЭЛЕКТРООСАЖДЕНИЯ
Пленкообразователи для электроосаждения представляют со-
бой особый класс водоразбавляемых пленкообразователей [67].
В этом случае полимер находится в водной среде, а при прохож-
дении тока у одного из электродов в области электрического
граничного слоя полимер теряет стабильность и осаждается на
электроде. Количество осаждающегося полимера растет во вре-
мени, что приводит к образованию изолирующего слоя, который в
конечном итоге ограничивает дальнейшее осаждение. Процесс
электроосаждения был назван электрофоретическим осаждением,
хотя в настоящее время признано, что электрофорез играет не-
большую роль в этом процессе.
При анодном осаждении отрицательно заряженный полимер
осаждается на аноде, а при катодном электроосаждении — поло-
жительно заряженный полимер осаждается на катоде. По сравне-
нию с другими, этот способ окраски позволяет получать очень
равномерное покрытие наружных поверхностей и осаждение
краски внутри частично закрытых областей. Степень использо-
вания краски высокая и процесс можно почти полностью автома-
тизировать. Указанные преимущества не достигаются при более
простых процессах окунания. Так как пленки могут быть получены
только на поверхности металлов и их толщина ограничена, мате-
риалы для электроосаждения используются в качестве грунтовок
или в некоторых случаях для получения однослойных покрытий.
Непигментированные прозрачные композиции используются для
нанесения покрытий на металлические блестящие изделия. В на-
стоящее время в промышленности применяются как анодный,
так и катодный процессы [68—70]. После появления новых лако-
красочных материалов для катодного нанесения они вытесняют
анодные композиции, особенно в автомобилестроении.
Почти все типы полимеров, которые были описаны ранее в
разделе «водоразбавляемые пленкообразователи», можно сделать
пригодными для электроосаждения. В большинстве случаев плен-
кообразователи, которые могут быть использованы для электро-
осаждения, находятся в виде устойчивых мицеллярных диспер-
сий или дисперсий микрочастиц с гидроксильными ионогенными
группами, обеспечивающими коллоидную стабилизацию. Однако,
дисперсии с неионной стерической стабилизацией могут быть
также нанесены электроосаждением [71].
Для достижения требуемого качества покрытия защи-
щаемой поверхности состав композиции должен обеспечивать
высокое электрическое сопротивление осажденной пленки. Ком-
позиция может содержать органический растворитель для облегче-
ния процесса диспергирования, повышения стабильности диспер-
сии и для улучшения розлива при нанесении и отверждении покры-
тия. Природа нейтрализующей кислоты или основания имеет
очень важное значение как для стабильности дисперсии, так и для
процесса нанесения покрытия электроосаждением. На практике
для анодных систем в качестве нейтрализующих агентов исполь-
зуются щелочи или амины, а для катодных — молочная или
уксусная кислоты.
Первые пленкообразующие для анодного электроосаждения
представляли собой малеинизированные масла и производные
масел, затем появились конденсаты винилированного и алкид-
ного типов и, наконец, сложные эфиры эпоксидных смол на
основе малеинизированных жирных кислот. Поскольку компози-
ции на основе эпоксидных смол очень хорошо зарекомендо-
вали себя в качестве грунтовок по металлу, эпоксидные свя-
зующие играют основную роль как в анодных, так и в катодных
системах, особенно при окраске автомобилей.
Были разработаны и нашли промышленное применение для
электроосаждения алкидные и акриловые композиции. Основой
алкйдных композиций для анодного процесса являются алкиды с
высокими значениями кислотного числа, в частности полученные
с применением тримеллитового ангидрида, которые могут быть
модифицированы высыхающими маслами. Они могут отверждать-
ся при нагревании за счет автоокисления или же, в случае моди-
фикации невысыхающими маслами, а также в случае безмасля-
ных алкидов, за счет отверждения совместно эмульгируемыми
с ними или растворимыми меламиноформальдегидными смолами.
Акриловые композиции предложены как для анодного, так и для
катодного процессов. В первом случае в них вводят большее
чем обычно "количество кислоты, например акриловой [50, 72],
а в последнем—сополимеризующийся аминосодержащий моно-
мер, например диметиламиноэтилметакрилат [73] или глициди-
ловый мономер, который затем подвергают взаимодействию
с амином [74].
Катодные эпоксидные композиции могут содержать первичные
или вторичные аминогруппы. Взаимодействием эпоксидных смол с
аминами или с четвертичными солями аминов получают аддукты
с концевыми вторичными, третичными или четвертичными аммо-
ниевыми группами. Хотя получить такие продукты и нелегко,
однако были разработаны соответствующие методы. Они вклю-
чают взаимодействие избытка первичных диаминов с эпоксидной
смолой [75] или же первоначальное блокирование первичных
аминогрупп кетонами и затем реакцию с эпоксидами за счет
других функциональных групп [76], как показано ниже:
О
О
2 N Н2—R—N Н2 + С Н2—С Н—R'—С Н—С Н2
ОН ОН
I I
N Н2—R—N Н—С Н а—С Н—R '—С Н—С Н 2—N Н—R—N Н 2;
R'
R—NH2 + O=q\
\r„
н+
1^0
R'
rnh2+o=c^
XR'
Вследствие того, что их пленки обладают щелочными свой-
ствами, такие композиции медленно отверждаются меламино-
выми или фенольными отвердителями, но весьма эффективно —
блокированными изоцианатами, которые стабильны при перера-
ботке, но деблокируются при температурах отверждения [77,
78]. Этими сшивающими агентами можно отверждать и акриловые
катионные композиции для однослойных покрытий в тех случаях,
когда более важны требования к цвету, чем к коррозионной
стойкости.
2.10. МАТЕРИАЛЫ С ВЫСОКИМ СУХИМ ОСТАТКОМ
Такие материалы позволяют уменьшить содержание органи-
ческих растворителей и загрязнение окружающей среды. При их
применении можно получить дополнительную экономию, так как на
испарение растворителей требуется меньше энергии. Однако в
целом такие системы не обязательно должны быть более деше-
выми, особенно если для их приготовления были использованы
более дорогие исходные компоненты, что в ряде случаев неиз-
бежно.
Для получения лакокрасочных материалов с высоким сухим
остатком существует ряд путей. Одним из видов таких материа-
лов со 100%-м сухим остатком являются порошковые краски.
Можно использовать реакционноспособные разбавители (напри-
мер ненасыщенные мономеры) вместо растворителей, как в случае
ненасыщенных полиэфиров и радиационно-отверждаемых пленко-
образователей. Можно добавлять реакционноспособный разбави-
тель вместо растворителя также и в высыхающие алкиды. Этот
разбавитель является винильным мономером, который входит в
структуру алкидной пленки при высыхании за счет радикальной
окислительной сополимеризации. Используются также полифунк-
циональные мономеры и новые соединения, например ненасыщен-
ные меламиноформальдегидные смолы, полученные специально
для разбавления алкидов.
Обычно материалами с высоким сухим остатком называют
такие лакокрасочные материалы, в которых содержание сухого
остатка выше, чем в обычно применяемых —то есть не 40—60%,
а 60—80%.
В принципе можно немного повысить сухой остаток любой
краски, используя ее при более высокой вязкости или нагревая
для уменьшения вязкости. Однако для значительного увеличения
сухого остатка при сохранении хорошей способности материала
к нанесению необходимо разрабатывать полностью новые компо-
зиции, как, например, в случае растворов низкомолекуляр-
ных полимеров, когда для достижения необходимой вязкости при
нанесении требуется меньше растворителя. Для композиций с
высоким сухим остатком потребовалась разработка новых поли-
меров. Из-за низкой молекулярной массы полимеры должны
обладать способностью к отверждению, образовывать пленки с
хорошими свойствами. Кроме того, низкая молекулярная масса
определяет необходимость принятия специальных мер для получе-
ния нужного количества реакционноспособных функциональных
групп и их распределения.
Говоря о материалах с высоким сухим остатком, мы встреча-
емся с новой терминологией, включая понятия «олигомерные»
и «телехелатные» полимеры. Первое понятие используют для опи-
сания низкомолекулярных полимеров с такой молекулярной
массой, когда только начинают проявляться специфические
свойства, характерные для полимерного состояния. Олигомер
может иметь молекулярную массу 1—5 тыс., в то время как мо-
лекулярная масса обычного термореактивного полимера равна
10—40 тыс., а термопластичного 80—100 тыс. [79]. Понятие
«телехелатный» относится к полимерным молекулам, обладаю-
щим двумя концевыми функциональными группами. Синтез их
для материалов с высоким сухим остатком описан в работе [80].
Для достижения высокого сухого остатка при низкой вязкости
необходимо хорошо знать зависимость между молекулярной
массой и вязкостью компонентов краски, а также взаимодействие
между компонентами (олигомером, сшивающим агентом, раство-
рителем, пигментом). В настоящее время для этого существует
неплохая теоретическая база [81], хотя и признается необходи-
мость дальнейших исследований. Требуется тщательно контро-
лировать молекулярную массу и молекулярно-массовое распре-
деление.
В случае обычных лакокрасочных материалов для получения
трехмерных пленок в реакциях сшивания достаточно участия
сравнительно небольшого числа функциональных групп. В красках
с высоким сухим остатком для достижения той же молекулярной
массы необходимо, чтобы прореагировало намного большее коли-
чество функциональных групп. Такое увеличение количества по-
лярных функциональных групп обычно повышает вязкость, при-
водя к частичной утрате выигрыша в снижении вязкости за счет
уменьшения молекулярной массы. Тем не менее, необходимо,
чтобы в каждой олигомерной молекуле содержалось по крайней
мере две функциональные группы.
Разработаны лакокрасочные материалы с высоким сухим ос-
татком на основе акриловых и полиэфирных олигомеров. При по-
лучении акриловых олигомеров встретились определенные труд-
ности. Дело в том, что необходимость применения высоких кон-
центраций агента передачи цепи вызывает появление остаточного
очень неприятного запаха. Высокие концентрации при синтезе при-
водят к возрастанию вклада реакций передачи цепи на полимер
и тем самым к нежелательно широкому молекулярно-массовому
распределению и ухудшению реологических свойств. Полиэфир-
ные композиции для материалов с высоким сухим остатком от-
носительно проще. Были разработаны хорошие составы с
использованием тех же компонентов, что и при получении более
высокомолекулярных полиэфиров. В случае полиэфиров проще
обеспечить их бифункциональность. Предложены и новые алкид-
ные составы ^использованием продукта Cardura Е (Shell Chemi-
cals) [7]. Упомянутые выше составы на основе акрилатов и поли-
эфиров содержат гидроксильные функциональные группы и для
их отверждения в качестве сшивающих агентов наиболее часто
применяют гексаметоксиметилмеламиновые смолы. Причины
этого рассмотрены в работе [82]. С учетом более высокого содер-
жания функциональных групп в олигомере, количество аминосо-
держащей смолы в композиции может достигать 50% и более
по сравнению с 10—40% в обычных лакокрасочных материалах.
Для получения материалов с высоким сухим остатком могут
быть использованы и другие системы, например композиции на
основе полиэфиров и аддуктов изоцианатов. Получение полиэфи-
ров с необходимым для этих целей строением описано в [83].
Аналогично и эпоксидно-полиамидные системы можно отнести к
системам с высоким сухим остатком в том случае, когда исполь-
зуются низкомолекулярные жидкие диэпоксиды. В этом случае
можно применять и моноэпоксидные соединения в качестве реак-
ционноспособных разбавителей.
2.11. РАДИАЦИОННО-ОТВЕРЖДАЕМЫЕ ПЛЕНКООБРАЗОВАТЕЛИ
Радиационное отверждение включает отверждение пучком
электронов, ультрафиолетовым (УФ) и инфракрасным (ПК) из-
лучением. Все эти методы используются в промышленности. Росту
их применения способствуют меньшее потребление энергии, луч-
шая защита окружающей среды и, особенно, применимость при
защите покрытиями деревянных, бумажных и пластмассовых
подложек. Для инициирования цепной полимеризации при отверж-
дении УФ-излучением применяют фотоинициаторы, а в случае
I4K — термические инициаторы. При отверждении электронным
пучком нет необходимости добавлять инициатор, так как электро-
ны благодаря их собственной высокой энергии генерируют ради-
калы в полимеризующемся слое. Для этих целей используют
обычно материалы с высоким сухим остатком, а типичная пленко-
образующая система состоит из смеси форполимера и реакцион-
носпособного разбавителя. Типичными форполимерами являются
ненасыщенные полиэфиры, применяемые вместе с соответствую-
щими винильными или акриловыми мономерами в качестве раз-
бавителей. Обзор механизмов отверждения под влиянием УФ-,
ИК-излучения и электронного пучка, а также соответствующих
композиций дан в работах [84, 85].
Помимо уже описанных ранее ненасыщенных полиэфиров,
для этих целей используются также эпоксидные и полиуретановые
пленкообразователи. Эпоксиакрилаты получают реакцией диано-
вых эпоксидных смол с акриловой кислотой. Полиуретановые
пленкообразователи для фотоотверждения синтезируют взаимо-
действием ди- или полифункциональных изоцианатных аддуктов
с гидроксиэтилакрилатом.
Определенное применение нашли также акрилаты простых и
сложных полиэфиров, получаемые реакцией этерификации гидрок-
силсодержащих простых или сложных полиэфиров акриловой
кислотой. При получении всех акрилированных пленкообразова-
телей необходимо тщательно контролировать содержание ингиби-
тора, чтобы предохранить от преждевременной Полимеризации.
Кроме того, может потребоваться разбавление готового продукта
мономером для достижения требуемой текучести. Основное до-
стоинство форполимеров этого типа — низкая вязкость.
К мономерам, применяемым в качестве реакционноспособных
разбавителей для регулирования вязкости, предъявляются опреде-
ленные требования по вязкости, сольватирующей способности,
летучести и фоточувствительности. Механизмы отверждения до-
статочно сложны и для каждой конкретной системы существует
оптимальное соотношение олигомер — мономер при данной дозе
облучения, необходимое для достижения требуемых характе-
ристик пленки. Акрилаты обладают наибольшей скоростью от-
верждения и обычно они полифункциональны. Из мономеров
широко применяются гександиолдиакрилат и триметилолпропан-
триакрилат. В настоящее время в промышленности производится
широкий ряд таких многофункциональных мономеров. По реак-
ционной способности ненасыщенных групп в радиационно-отверж-
даемых системах мономеры располагаются в следующий ряд:
акрилаты > метакрилаты > аллильные > винильные.
Фотоинициаторами для отверждения в УФ и видимой области
ненасыщенных систем должны быть генераторы свободных ради-
калов, например ароматические кетоны. Применяют также и
сочетания кетонов с аминами [86]. Реакции отверждения те же,
что и описанные ранее при полимеризации акриловых соеди-
нений.
Последним достижением в этой области являются эпоксидные
смолы, отверждаемые по катионному механизму, в которых приме-
няют фотоинициаторы, выделяющие под воздействием облучения
кислоты Бренстеда. Такие композиции основаны, главным обра-
зом, на циклоалифатических эпоксидных смолах и алифатических
диэпоксидах. Диановые эпоксидные смолы ухудшают способность
к отверждению подобных систем.
2.12. ПОРОШКОВЫЕ СОСТАВЫ
Порошковые композиции обладают тем преимуществом по
сравнению с обычными лакокрасочными материалами, что при на-
несении не выделяют никаких загрязняющих окружающую среду
растворителей. Кроме того, благодаря использованию при нанесе-
нии метода электростатического распыления, потери материала
невелики. Приготовление порошковых композиций характеризу-
ется рядом особенностей из-за отсутствия в них растворителей
[87]. Большинство порошков получают измельчением экструдиро-
ванных расплавов, вследствие чего термореактивные композиции
должны выдерживать процесс плавления (при экструдировании
осуществляют также пигментирование). Все используемые компо-
ненты, и особенно готовые композиции, должны быть твердыми и
стеклообразными при комнатной температуре, что требуется для
хорошего измельчения и сохранения сыпучести порошка при хра-
нении. В связи с этим, смесь не должна размягчаться или агломе-
рировать при температуре ниже 40 °C. При разработке рецептур
и в производстве необходимо обращать внимание на такие факто-
ры, как размер частиц, показатели текучести расплава, удельное
сопротивление.
Для получения порошковых красок широко используются
эпоксидные композиции на основе диановых смол, отверждаемые
аминами (особенно производными дициандиамида) или феноль-
ными отвердителями. Применяют также насыщенные полиэфиры
в смеси с эпоксидами. Для улучшения розлива и адгезии можно
проводить модификацию димеризованными жирными кислотами,
а для повышения твердости вводить эпоксидные смолы, которые
содержат разветвленные продукты, полученные эпоксидированием
новолаков. Для отверждения эпоксидных порошков используют
также ангидриды дикарбоновых кислот и кислые полиэфиры.
Покрытия на основе таких композиций не желтеют и превосходят
по эксплуатационным характеристикам эпоксидные покрытия,
отвержденные аминами.
Полиэфирные порошковые композиции могут быть на основе
гидроксилсодержащих полиэфиров и отверждаться меламинофор-
мальдегидами или блокированными изоцианатами или же на осно-
ве кислых полиэфиров, отверждаемых эпоксидными смолами или
триглицидилизоциануратом. Последний отвердитель обеспечивает
очень хорошую долговечность и стабильность цвета. Для получе-
ния блокированных изоцианатов используют капролактам, хотя
его выделение при отверждении пленки создает определенные
проблемы.
Разработаны акриловые композиции, которые однако характе-
ризуются умеренными эксплуатационными свойствами и осложне-
ниями при хранении. Применяют также термопластичные порош-
ковые композиции на основе, например, найлона 11 или най-
лона 12, поливинилхлорида, ацетата и бутирата целлюлозы,
полиэтилена. Порошки можно диспергировать в воде и затем
наносить на изделия. Примером таких систем являются так назы-
ваемые суспензионные лакокрасочные материалы или водные по-
рошковые суспензии, которые можно наносить электроосажде-
нием (покрытия, наносимые электроосаждением порошков).
Глава У
ПИГМЕНТЫ ДЛЯ КРАСОК
Дж. Ф. Ролинсон
3.1. ВВЕДЕНИЕ
Настоящая глава, посвященная пигментам для красок, пред-
назначается для людей, незнакомых с лакокрасочной промыш-
ленностью. Обсуждаемые вопросы изложены сжато, чтобы дать
возможность начинающему технологу по лакам и краскам полу-
чить основные представления о роли пигментов в красках. Более
подробные сведения можно получить из коммерческих изданий,
а также из многих книг и статей, уже опубликованных по этому
вопросу.
Вначале даны определения пигментам и перечислены требова-
ния, предъявляемые к их качеству. Приведена классификация
пигментов и объяснена терминология, используемая в лакокра-
сочной промышленности. Обращено внимание на токсичность
пигментов, их влияние на состояние окружающей среды и лако-
красочную промышленность; Затем перечислены свойства пигмен-
тов и предложены некоторые основные правила, которые можно
использовать при выборе пигментов для лакокрасочных материа-
лов.
3.2. ОПРЕДЕЛЕНИЯ
Пигменты совместно с пленкообразователем могут быть
использованы в лакокрасочных материалах для придания по-
следним:
1) эстетической привлекательности — цвета и укрывистости
или прозрачности и специальных эффектов;
2) защитных свойств — атмосферостойкости и коррозионной
стойкости;
3) вспомогательных свойств — повышения прочности пленки,
твердости, огневой защиты, придания поверхности антиконден-
сационных и противоскользящих свойств и т. д.
Пигменты можно определить как вещества, представляющие
собой твердые частицы, практически нерастворимые в используе-
мых пленкообразователе, растворителях и разбавителях, способ-
ные к диспергированию в компонентах краски и обеспечиваю-
щие достижение оптимальных свойств, предъявляемых к лако-
красочным материалам и покрытиям.
3.3. ТРЕБОВАНИЯ К КАЧЕСТВУ ПИГМЕНТОВ
3.3.1. Внешний вид
Среди пигментов можно выделить два класса — истинные
пигменты и наполнители. Первые применяются для придания
цвета, белизны или укрывистости, тогда как наполнители могут
быть добавлены в лакокрасочный материал для изменения глянца
или блеска, оказывая незначительное влияние на цвет или укры-
вистость. При более высоких концентрациях наполнители воз-
действуют на цвет и укрывистость так же, как и на другие важ-
ные физические свойства покрытия. Наполнители используют
для изменения таких важных свойств, как реологические и седи-
ментационные характеристики жидкой краски и стекание пленки
после нанесения. Они также добавляются в лакокрасочный ма-
териал из экономических соображений.
Первое и главнейшее требование к пигментам заключается в
том, что они должны придавать цвет прозрачной или непрозрачной
пленке; важным свойством является также красящая способ-
ность.
Часто требуется, чтобы субстрат был затемнен, при этом не-
обходимо использовать пигменты, придающие укрывистость.
В случаях других отделок используют цветные полупрозрачные
или прозрачные пигменты.
3.3.2. Стойкость
Важно, чтобы используемые в красках пигменты были стойки
к воздействию других компонентов краски, воды, растворителей,
химикатов, которые воздействуют на пленку. Если пленкообразо-
ватель требует нагревания для отверждения, то пигменты должны
быть термостабильными.
3.3.3. Долговечность
Пигменты должны быть долговечны в смысле сохранения
цвета, т. е. не должны ни обесцвечиваться, ни темнеть. Кроме
того, пигменты не должны ухудшать, и даже могут увеличивать
способность связующего образовывать когезионную твердую,
эластичную пленку с хорошей адгезией к подложке. Таким образом
получается прочная, долговечная, износостойкая пленка, защи-
щающая поверхность и увеличивающая срок ее службы. Стой-
кость чистой пленки к внешним воздействиям возрастает много-
кратно после введения пигментов. Лакокрасочное покрытие
должно быть достаточно толстым, чтобы обеспечить максималь-
ную защиту, и это свойство заметно улучшается при правильном
пигментировании.
Таблица 3.1. Пигменты для лакокрасочных материалов — белые, цветные и наполнители
Классификация по химическому строению
Основные цвета
Нррргяничрг.кие пигменты Карбонаты Белые, бесцветные Оксиды Белые, зеленые, желтые, крас- ные, коричневые — Сульфиды, сульфоселениды Белые, желтые, оранжевые, крас- ные — Сульфаты Бесцветные
। Ариламид — Моноазо [ . . A'ini/nunourarianuMi.ip — Силикаты Белые, бесцветные, голубые — Ферроцианиды Синие — Хроматы, молибдаты Желтые, красные — Сажа Черная — Металлические Алюминиевый, бронзовый — Синтетические смешан- ные комплексы оксидов Различные цвета (керамика) — Ацетоариламид Желтый — Нафтанилид Красный — Нафтол Красный, оранжевый
।—Азопигменты — * лраииыи, желтым, ирдпжсьыи - - и»,-глпт^<иаг'г/>1а плгти t/UCTltTv ОЧЛГ/ЛО гчггл nort Г ТиГРТЯ TTTTllUpr Tt'TTP 'rnUpntll К'г^ЯГ’МГ.ГР
1 — Диазо Гетероциклические — Диарилиды Желтые, красные, оранжевые — Пиразолом Оранжевый, красный — Изоиндолинон Желтый, оранжевый — Фталоцианин Синий, зеленый — Хинакридон Оранжевый, красный, фуксино- вый, фиолетовый —• Перилеи Красный, каштановый — - Диоксазин Фиолетовый — Тиоиндиго Каштановый — Антралиримидин Желтый — Инадантрон Синий — Флав антрон Желтый — Дибромантантрон Красный — Пирантрон Красный — Перинон Оранжевый — Полимерные гранулы Бесцветные наполнители Перламутровые (поверх- Многоцветные ностно-обработанные слюды с перламутровым эффектом)
Органические 1 Полициклические
пигменты Кубовые пигменты 1 Антрахиноновые пигменты
*“ - Прочие пигменты *
Таблица 3.2. Основные свойства пигментов различной химической природы
Свойство в краске Пигментный рутильный диоксид титана Оксиды железа Синие ка- лий-свинцо- вые хроматы Берлинская лазурь (феррофер- рицнанид) Сажа Моноазо Диазо Азоконден- сационные Металло- азотонеры Фталоциа- ниновые Поликличе- ские, вклю- чая кубовые и хинакридон
Цвет 5 4 5 4 5 5 5 5 5 5 5
Укрывистость 5 5 (кроме про- зрачных) 5 5 5 3 4—3 4—2 4—3 2 4—3
Красящая способность 5 3—2 3 5 5 5 5 4 5 5 4
Термостабильность Стойкость к растворителям: 5 5 (кроме желтых) 4 4 5 3 4 5 4 5 4
к растворению 5 5 5 5 5 2 3 5 4 5 4
к выцветанию 5 5 5 5 5 2 5 5 5 5 5
к потерям при распылении Стойкость к: 5 5 5 5 5 2 3 5 5 5 5—4
кислотам 5 5 4—3 3 5 5 5 5 2 5 5
щелочам 5 5 2 2 5 5 5 5 2 5 5
химическим агентам Светостойкость при: 5 5 4 2 5 5 5 5 2 4 2
насыщенных тонах 5 5 4 4 5 5 3 4 3 5 5—4
средних тонах 5 5 4 3 5 3 2 3 2 5 5—4
разбавленных тонах 5 5 2 2 5 2 2 2 2 4 5—4
Диспергируемость 5 5 (кроме про- зрачных) 3* 3 3 4 4 4 4 3 3
Текучесть 5 5 5 4 4 3 2 3 3 2 3
Стабильность к флокуляции 4 4 (кроме красных) 5 4 2 4 4 4 4 4—2 4—3
Цена Низкая Низкая Низкая Низкая Низ- Низ- Сред- Сред- Сред- Сред- Высокая
кая кая няя няя няя няя
2 — плохо, 3 — удовлетворительно. 4 — хорошо, 5 — отлично
* Имеются легкодиспергируемые типы.
Защита от коррозии лакокрасочными покрытиями имеет
наиболее важное значение и она сильно зависит как от пигмен-
тов — ингибиторов коррозии, так и собственно от технологии
приготовления антикоррозионных красок.
3.4. КЛАССИФИКАЦИЯ ПИГМЕНТОВ
Существующие пигменты после их-смачивания пленкообразо-
вателем можно подразделить на белые, цветные и бесцветные или
наполнители. Цветные пигменты традиционно подразделяют на
неорганические и органические, но развитие технологии привело
к тому, что различие в свойствах этих пигментов быстро сти-
рается.
Весьма тонкие процессы обработки поверхности пигментов
наряду с особенностями современных технологий их производства
обеспечивают выпуск пигментов, для каждого из которых необ-
ходим свой индивидуальный способ получения.
Однако легче начать с простой классификации:
Неорганические пигменты — сюда входят белые пигменты, на-
полнители и ряд цветных пигментов, как синтетических, так и
природных. »
Органические пигменты — в настоящее время это, в основном,
синтетические пигменты.
Общие свойства неорганических и органических пигментов
представлены ниже:
Свойства пигментов Неорганических Цвет Иногда тусклый Кроющая способность Обычно высокая Красящая способность Обычно низкая Стойкость к действию раство- Хорошая рнтелей — «стойкость к вы- мыванию» Стойкость к действию хими- Различная катов Термостойкость Большей частью хорошая Долговечность Обычно хорошая Цена Обычно невысокая Органических Обычно яркий Относительно низкая Обычно высокая Изменяется в широких пределах — от хорошей до плохой Различная Различная Различная Различная, но иногда очень высокая
Дальнейшая классификация неорганических и органических
пигментов по химической природе дана в табл. 3.1, а типичные
свойства — в табл. 3.2.
3.S. НОМЕНКЛАТУРА ПИГМЕНТОВ
Технологи полакам и краскам идентифицируют группы пигмен-
тов, употребляя традиционную терминологию для химических
классов пигментов, приведенных в табл. 3.1. Они используют
традиционные наименования, например, «берлинская лазурь»,
которые возникли в промышленности, а также используют номер
по Colour Index.
Colour Index [1] — важная международная система кодиро-
вания, поскольку она является логической и исчерпывающей и
дает ясную идентификацию определяемых типов пигментов.
Colour Index (третье издание, второе уточнение, 1982 г.)
публикуется Обществом красильщиков и колористов (Society
of Dyers and Colourists), совместно с Американской Ассоциа-
цией химиков-текстильщиков и колористов (American-Association
of Textile Chemists and Colorists). Это очень полезное справочное
издание дает общую информацию о пигментах, классифицируя
их по следующим признакам:
Наименование группы, тип цвета и номер по Colour Index (CI).
Имеющиеся материалы сгруппированы согласно тому, являются
они пигментами или растворимыми красителями, и в группы по
цветам. Номер отвечает всем материалам с одинаковым химиче-
ским строением в определяемом цветовом семействе. Например:
CI Пигмент желтый 3 — желтый пигмент, и эта запись отличает
данную группу от, скажем, CI Растворимый желтый 3, который
является красителем.
Химическое строение для каждого пигмента дано по номеру
строения по Colour Index. Так, в вышеприведенном примере CI
Пигмент желтый 3 имеет номер строения по CI 11710. Все желтые
пигменты, которые имеют одинаковое химическое строение, полу-
ченные при взаимодействии 4-хлор-2-нитроанилина с 2-хлорацето-
анилидом, даны под номером 11710.
Коммерческое или торговое наименование. Пигменты, имею-
щиеся на рынке, внутри каждой цветовой группы перечисляются
под их коммерческими или торговыми наименованиями. Обычное
наименование Пигмента желтого 3 — «Ариламид желтый 10 G».
«G» в наименовании подчеркивает зеленый оттенок желтого
цвета, а «10» обозначает, что пигмент на 10 единиц зеленее, чем
«Ариламид желтый G», который представляет собой Пигмент
желтый 1. Эти желтые пигменты также называют «Желтые ганза».
Colour Index перечисляет более 700 типов используемых цвет-
ных пигментов, что составляет более 5000 различных пигментов
всех назначений, в том числе — для типографских красок, пласт-
масс, резины, цемента и бумаги:
Пигменты по Colour Index Число перечисленных химических типов Пигменты но Colour Index Число перечисленных химических типов
Белые 33 Желтые 163
Черные 32 Оранжевые 54
Коричневые 26 Красные 244
Синие 66 Фиолетовые 49
Зеленые 36 Металлические 6
Из всех этих пигментов, за исключением наполнителей, до
80% общего объема производства и потребления приходится
на долю примерно 30-ти. Некоторые основные крупнотоннажные
пигменты приведены ниже:
Пигменты Тип
С1 Пигмент белый 6
CI Пигмент красный 101
CI Пигмент желтый 42
CI Пигмент голубой
15 : 1, 15 : 2, 15 : 3, 15 : 4
CI Пигмент красный 49 : 1
CI Пигмент красный 57 : 1
3.6.
Диоксид титана
Красный железоокисный пигмент
Желтый железоокисный пигмент
Фталоцианин медн
Моноазокраситель Литоль красный (бариевая
соль кислого азокрасителя), используется в ти-
пографских красках
Мопоазокраситель Литоль рубиновый тонер
(кальциевая соль кислого азокрасителя), исполь-
зуется в типографических красках
ГРУППЫ ПИГМЕНТОВ
Большая группа современных цветных пигментов своим появ-
лением обязана технологии красителей, которые используются
при окраске волокон и тканей. Эти красители, по их природе,
должны быть растворимы на определенной стадии процесса
крашения. Такая технология часто может быть приспособлена к
приготовлению нерастворимых цветных порошков, пригодных к
использованию в качестве пигментов для лакокрасочных мате-
риалов. Согласно их происхождению, можно рассмотреть сле-
дующие группы пигментов: а) тонеры, б) литоли *, в) красочные
лаки, г) пигменты, модифицированные канифолью, д) нафтолы и
пигменты типа Нафтола AS **, е) пигменты «BON», ж) кубовые
пигменты, з) керамические пигменты, и) пигменты, полученные
флашинг-процессом.
3.6.1. Тонеры
Тонеры — это такие материалы, которые, в отличие от раство-
римых красителей, нерастворимы в воде, большинстве смол,
растворителей и разбавителей. Этот термин используется в Аме-
рике для обозначения всех цветных пигментов, кроме красочных
лаков, но в Европе это название обычно закрепляется за особой
группой пигментов, называемых «металлическими тонерами».
Тонеры получают путем реакции определенных растворимых
кислых азокрасителей с основаниями с получением металли-
ческих солей, нерастворимых в воде; примерами могут служить
бариевые, кальциевые или марганцевые тонеры. Светостойкость
частично определяется ионом металла, который также влияет
и на цвет, и изменяется следующим образом:
Na Ba Са Sr Мп
Увеличение светостойкости->
Ораижевый^Бордо
* Регистрационное наименование фирмы BASF (ФРГ).
** Регистрационное наименование фирмы Hoechst AG (ФРГ).
Типичный тонер — Пигмент красный 48 : 2, кальциевая
соль продукта взаимодействия 2-хлор-4-толуидин-5-сульфо-
кислоты с p-гидроксинафтойной кислотой. Обычное название
этого пигмента — «Кальциевый красный 2В тонер».
Некоторые тонеры обрабатываются канифолью при приготов-
лении; присутствие канифоли при осаждении позволяет получить
меньшие частицы, обеспечивая большую прочность и прозрач-
ность (см. 3.6.4).
3.6.2. Литоли
Литоли — историческое название класса металлических тоне-
ров. Они в основном используются в типографских красках, а
само название произошло от термина «литография». Использо-
вание литолей в красках ограничивается дешевыми малодолго-
вечными системами из-за низкой термо- и светостабильности,
которая характерна для литолей. Пример — Пигмент красный
49: 1 (Литоль красный), бариевая соль продукта взаимодей-
ствия 2-нафтиламин-1 -сульфокислоты с 2-нафтолом.
У
3.6.3. Красочные лаки
Многие растворимые кислые или основные красители нельзя
превратить в простые нерастворимые тонеры, но они могут быть
осаждены или сорбированы на неорганической основе, такой как
оксид алюминия в смеси с сульфатом бария (бланфикс), которая
выступает как носитель. При этом получается пигмент.
Другие красочные лаки представляют собой комплексы алюми-
ния или кальция с гидроксиантрахиноновыми красителями.
Например, Пигмент красный 60 : 1, «Пигмент алый ЗВ лак»,
имеет сложное строение и представляет собой бариевую соль
моноазокрасителя «Протравной красный 9», наполненный гид-
роксидом алюминия и оксидом цинка; Пигмент красный ВЗ,
«Ализариновый крапплак» или «Ализариновый красный В» —
комплекс дигидроксиантрахинона с кальцием или алюминием,
с небольшими добавками сульфированного касторового масла
(турецкое красное масло) и фосфата алюминия или каль-
ция.
Чтобы проиллюстрировать широту терминологии, применяемой
в промышленности пигментов, можно упомянуть Пигмент крас-
ный 53 ; 1. Это пигмент для типографских красок, известный также
как «Красный лак С» или «Красный лак С тонер». Это бариевая
соль кислого моноазокрасителя, и в строгом смысле определе-
ний, данных выше, является тонером, но не истинным красочным
лаком.
3.6.4. Пигменты, модифицированные канифолью
Модификация канифолью представляет собой обработку реа-
гентов, используемых при производстве пигментов, канифолью
(абиетиновой кислотой) или резинатом щелочного или щелочно-
земельного металла, полученного взаимодействием кипящей ка-
нифоли, например с гидроксидом натрия. Модификация кани-
фолью наиболее широко используется для металлических тоне-
ров. Введение резината натрия в процесс при осаждении соли
металла благоприятствует получению частиц меньших размеров и
дает более яркий пигмент с повышенной стойкостью и прозрач-
ностью по сравнению с вариантом синтеза без канифоли. Метод
часто используется для улучшения диспергируемости пигментов.
Модифицированная канифоль, например тетрагидроабиетино-
вая кислота, с более высокими температурой размягчения и
устойчивостью к окислению позволяет получать более качествен-
ные пигменты с лучшей механической стойкостью и стабиль-
ностью красок при хранении.
Пигменты, модифицированные канифолью, широко исполь-
зуются в производстве типографских красок, где важное значе-
ние имеет высокая прозрачность.
3.6.5. Пигменты на основе нафтола.
Нафтола AS* и нафтанилидов
Все эти пигменты являются красными моноазопигментами.
Простые красные моноазопигменты представляют собой продукты
взаимодействия 2-нафтола с первичными ароматическими ами-
нами.
Пигменты на основе нафтола и Нафтола AS. Ряд красных пиг-
ментов этого типа получается при замене 2-нафтола на 2-гид-
рокси-3-нафтойную кислоту (кислоту BON**).
ОН
Кислота BON
соон
Некоторые их этих пигментов иногда называют BON-арилами-
дами, поскольку ариламид получается при реакции BON-кислоты
с амином (см. 3.6.6).
Пример: Пигмент красный 2 (Нафтол красный) — продукт
реакции 2,5-дихлоранилина с 2-гидрокси-З-нафтанилидом:
Торговая марка арилида 2-оксинафтойной кислоты (Пр и меч пере-
водчика).
** Сокращение от английского Beta-Oxy-Naphtoic Acid (Примеч. пере-
водчика).
Нафтанилидные пигменты. Этот термин происходит от при-
сутствия 2-гидрокси-З-нафтанилидной группировки (или ее заме-
щенных вариантов), продукта взаимодействия BON-кислоты с
первичным ароматическим амином:
2 - Гидро кси - 3-н а фта пи лид
Следовательно, вышеупомянутый Пигмент красный 2 является
также и нафтанилидным пигментом.
Все эти термины широко используются в промышленности
пигментов, лаков и красок и иллюстрируют неупорядоченность
в номенклатуре групп пигментов, что придает особое значение
Colour Index и системам нумерации.
3.6.6. BON-пигменты
Это красные или каштановые азопигменты и металлические
тонеры, в которых BON-кислота используется как фрагмент в
химической структуре пигмента. «BON» — нестрогий термин, пра-
вомерность которого подвергается сомнениям рядом экспертов.
Однако, многие технологи в области лаков и красок продолжают
его использовать.
3.6.7. Кубовые пигменты
Термин «Кубовые» пришел из промышленности красителей,
где ткани окрашивались в кубах. Кубовые красители превраща-
ются в пигментную форму в результате ряда сложных хими-
ческих и физико-химических превращений.
Многие кубовые пигменты являются производными антрахи-
нона, например Индантрон синий, Флавантрон и Антрапирими-
дин желтые, Пирантрон красный, Перинон оранжевый и Антра-
хинон красный. Дибромантантрон красный такж,е классифици-
руется как антрахиноновый пигмент. К кубовым пигментам также
относят Тиоиндиго и Перилен.
Эти сложные полициклические пигменты дороги и исполь-
зуются в красках для высококачественных покрытий и в автомо-
бильных эмалях. Их употребление оправдывается высокой термо-,
химстойкостью, стойкостью к растворителям и хорошей долго-
вечностью при насыщенных и разбавленных тонах. Однако
отдельные из этих пигментов, в частности Флавантрон желтый,
при высоких концентрациях темнеют на свету.
3.6.8. Керамические пигменты
(синтетические смешанные фазовые оксиды]
Керамические пигменты, называемые так, поскольку они были
разработаны для окрашивания керамических глазурей в фарфо-
ровой промышленности, являются смешанными фазовыми пиг-
ментами на основе оксидов металлов, в которых инородный ион
включен в данную кристаллическую решетку. Шпинель —
MgAKOi или рутил — ТЮ2 приобретают различную окраску, если
ионы кобальта, никеля, хрома, железа или меди замещают ионы
металла в основной кристаллической структуре.
Многие синие керамические пигменты имеют основную струк-
туру шпинели, представляя собой смешанные оксиды кобальта
и алюминия. Коричневые керамические пигменты имеют струк-
туру рутила с титаном, хромом и сурьмой. Хорошо известные
титанаты никеля имеют рутильную структуру с никелем и сурь-
мой в кристаллической решетке; они окрашены в желтый цвет.
Керамические пигменты получаются в результате прокали-
вания при исключительно высоких температурах. Поэтому они
имеют очень высокую термостабильность и превосходные свето-,
погодо- и химическую стойкость. Некоторые из них имеют яркие
чистые оттенки, приближаясь в этом отношении к органическим
пигментам.
В отличие от большинства неорганических цветных пигмен-
тов, они не обладают высокой кроющей способностью. Они имеют
низкую красящую способность, а некоторые — обусловливают
невысокий глянец покрытий при нормальной объемной концентра-
ции пигмента. Их применяют главным образом в покрытиях для
рулонного или листового металла и некоторых автомобильных
эмалях.
3.6.9. Пигменты, полученные флашинг-процессом
Флашинг — это процесс, когда пигмент переводится из суспен-
зии в воде во вторую, не смешивающуюся с водой жидкость,
например масло, смолу, растворитель с использованием механи-
ческого перемешивания. Эффективность переноса зависит от срод-
ства пигмента к воде, маслу, смоле, растворителю. Хороший
флашинг-процесс может обеспечить получение более тонких дис-
персий по сравнению с обычными методами диспергирования,
например на бисерных мельницах. При этом получаются пасты
с большей красящей способностью и яркостью цвета, но с худшей
укрывистостью. Это объясняется тем, что пигменты сохраняют
свой первоначальный очень малый размер частиц, который был
достигнут при их производстве, и не образуют более крупных
агрегатов, что обычно происходит при сушке.
3.7. ТОКСИЧНОСТЬ
Требования к безопасности пигментов для окружающей среды
и людей постоянно возрастают. Термин «безопасность» трудно
определить, когда речь идет о здоровье людей и охране природы,
поскольку содержание понятий «безопасность» и «вредность»
для людей, животных, растений и окружающей среды постоянно
меняется.
В последние несколько лет пристальное внимание в некото-
рых европейских странах уделяется в этой связи двум основным
группам пигментов. Это хроматы свинца и цинка и кадмиевые
пигменты. Пока только хроматы цинка показали свою очевидную
вредность для здоровья людей и были отнесены к канцерогенам.
Обращение со всеми этими пигментами в соответствии с реко-
мендациями изготовителей сводит их воздействие и риск для здо-
ровья к минимуму.
Иногда необходимо менять сорта пигментов, поскольку может
оказаться, что компоненты или побочные продукты сомнительны
в отношении безопасности.
В качестве примера можно привести отнесение к потенциаль-
ным канцерогенам хлорированных бис-фенолов, которые могут
получаться при производстве фталоцианиновых пигментов. Про-
цесс синтеза следует модифицировать таким образом, чтобы
такие побочные продукты или не получались, или же была бы
обеспечена безопасность для людей и окружающей среды при
обращении с ними.
Выражается беспокойство по поводу слива отходов в реки,
озера и моря или сжигания их в атмосфере. Любой новый процесс
может изменить тонкое равновесие в природе, и производителям
пигментов и красок все дороже обходится производство безвред-
ных материалов, удовлетворяющих требованиям потребителя.
3.8. СВОЙСТВА ПИГМЕНТОВ
Основной цвет и стойкость пигментов определяются их хими-
ческой природой, но оттенок, интенсивность, укрывистость, кра-
сящая способность и долговечность зависят от других факторов
сложным и запутанным образом. Основные из этих факторов:
химическая природа, кристаллическая структура, форма частиц,
размер частиц и его распределение, показатель преломления,
природа поверхности пигментов и покрытий на пигментных части-
цах, диспергируемость, цвет и другие свойства пленкообразо-
вателя.
3.8.1. Химическая природа
Цвет пигмента определяется избирательным поглощением
и отражением видимого света с различной длиной волны (от 0,4
до 0,7 мкм). Синие пигменты имеют такой цвет потому, что они
отражают волны в синей части спектра падающего белого света
и поглощают остальные волны; черные пигменты поглощают все
длины волн падающего света почти полностью; белые — отра-
жают все видимые длины волн.
Различные характеристики отражения и поглощения пигмен-
тов обусловлены расположением электронов в их молекулах, их
энергией и частотой колебаний.
В результате поглощения молекулой кванта света происхо-
дит возбуждение электронов и переход электрона в молекуле
с орбитали с более низкой энергией Е} на орбиталь с более высокой
энергией Е%. Возбужденный электрон впоследствии возвращается
тем же_.лудем на уровень с меньшей энергией, при этом избыток
энергии рассеивается, обычно в виде тепла.
Длина волны поглощенного света (л) определяется как раз-
ность энергий (£) между двумя упомянутыми орбиталями:
Е—Ег — Е\ — hc/'i.,
где /г — константа Планка, с — скорость света.
Конкретная молекула пигмента имеет ограниченное число
орбиталей, причем каждая орбиталь обладает собственной харак-
теристической энергией. Это значит, что упомянутая выше раз-
ность энергий Е имеет некоторые определенные значения. Следова-
тельно, молекула пигмента поглощает свет только при некоторых
длинах волн, определяемых разностью энергий Е и характеристи-
ками взятого образца. Волны другой длины отражаются и опреде-
ляют цвет пигмента. Дальнейшие объяснения природы цвета
даны в гл. 14.
В случае органических пигментов установлено, что их цвет
определяется присутствием в структуре определенных групп.
Эти группы называются хромофорами, а группы, которые усили-
вают цвет — ауксохромами:
Хромофоры . >С—С< >С=О >С—S >С—NH —N—N— —N=C< —NO
Ауксохромы- —OH — NH, — NHR —NR, —SO2H —COOH —NO?
—CH3 —Cl —Bi-
Ниже проиллюстрировано влияние хромофоров и ауксохро-
мов на цвет органических молекул;
СНз—СО—СНз бесцветный
СНз—СО—СО—СНз • - желтый
— оранжевый
но
-NO2 — глубокий желтый
-N=N-
— глубокий красный
Очень удачное объяснение природы цвета органических соеди-
нений можно найти в «Органической химии» (авторы Л. Физер и
М. Физер).
Укрывистость пигмента обусловлена преимущественно его
химической природой. Так, диоксид титана рассеивает свет, тогда
как сажа — поглощает. Оба эти явления определяют укрывистость
в лакокрасочной пленке. Цветные пигменты имеют различную
кроющую способность, что вызвано поглощением и рассеива-
нием в различных областях видимого спектра.
Неорганические пигменты обычно имеют большую кроющую
способность, чем органические того же цвета, ввиду различий
в показателях преломления в областях рассеивания видимого
спектра (см. стр. 97).
Химическая природа пигментов также предопределяет их
стабильность к действию тепла, растворителей, кислот, щелочей
и других химических веществ.
Простые моноазопигменты (красные, оранжевые и желтые),
например, несмотря на то, что имеют хорошую кислотостойкость
и хороший срок службы при насыщенных тонах (но не в разбав-
ленных тонах, где они обесцвечиваются), в то же время чувстви-
тельны к теплу и растворителям. Это ограничивает использова-
ние их только в красках, высыхающих на воздухе и содержащих
слабые растворители, и только для более насыщенных тонов.
Более сложные и дорогие пигменты — продукты азоконден-
сации (красные, оранжевые, коричневые и желтые) имеют такие
же свойства, как и азопигменты во всех отношениях, кроме того,
что они имеют исключительную термостойкость и стабильность
к растворителям, и, таким образом, могут быть использованы
в красках горячей сушки.
Берлинская лазурь стойка к кислотам, но теряет цвет в щелоч-
ных условиях, превращаясь в коричневый гидроксид железа,
а синий ультрамарин — сложный алюмосиликат натрия, содержа-
щий серу — чувствителен к кислотам, обесцвечиваясь при их
воздействии. Фталоцианиновый голубой, с другой стороны, ста-
билен в большинстве условий.
3.8.2. Кристаллическая структура
Пигменты могут быть кристаллическими или некристалличе-
скими (аморфными) веществами. Иными словами, атомы внутри
каждой молекулы пигмента расположены упорядоченно или бес-
Таблица 3.3. Примеры кристаллических форм полиморфных материалов
Пигмент Кристаллическая форма Свойства
Фталоцианиновый голубой (Пигмент голубой 15:1, 15:2, 15:3, 15:4) Хинакридон Пигмент фиолето- вый 19 Хромат свинца с сульфатом свинца Пигмент желтый 34 Диоксид титана (Пигмент белый 6) ♦ Стабилизированная альфа (Пигмент голубой 15:1 и 15:2) Бета (Пигмент голубой 15:3 и 15:4) Гамма Альфа и Дельта Бета Гамма Орторомбическая (включая 30—50% сульфата свинца и стабилизированная) Моноклинная (включая 20—45% сульфата свинца) Красновато-голубой Зеленовато-синий Не используется как пигмент Не используется как пигмент Фиолетовый Красный Бледно-желтый Лимонный 1. Показатель преломления 2,55; 2. Очень белый; 3. Некоторая тенденция к мелению при внеш- нем воздействии 1. Показатель преломления 2,76, что дает большую крою- щую способность по сравнению с анатазом; 2. Чуть более жел- тый, чем анатаз; 3. Большая ус- тойчивость к мелению при внеш- нем воздействии Не используется как пигмент
Рутил Брукит
порядочно. Некоторые кристаллические формы могут быть устой-
чивее других, а некоторые кристаллы вообще не могут исполь-
зоваться в качестве пигментов., Менее стабильные кристаллы
иногда модифицируются введением, скажем, другого иона в кри-
сталлическую решетку, превращаясь в более стабильные.
Примеры полиморфных пигментов: фталоцианиновый голубой,
хинакридоны, хроматы свинца, диоксид титана (см. табл. 3.3).
Современная технология получения пигментов дает возмож-
ность контролировать форму и размер кристаллов, так что можно
достичь требуемого цвета и полезных эксплуатационных харак-
теристик.
3.8.3. Форма частиц
Первичная форма пигментных частиц определяется химиче-
ской природой, кристаллической структурой или ее отсутствием,
и условиями, в которых пигмент получается в природе или изго-
тавливается синтетически. Пигменты в первичной форме могут
быть сферическими, узловидными — неправильными сферами,
Сферы
Узлы
Кубы
Иглы
Пластины
Рис 3 1. Формы частиц
кубическими, игольчатыми — игло- и стержнеподобными, пластин-
чатыми— листоподобными. Эти формы показаны на рис. 3.1.
Однако, пигменты обычно поставляются в виде агрегатов или
агломератов Агрегированные частицы трудно разделить, так как
они прочно скреплены или даже соединены в результате, напри-
мер, спекания при производстве или сушке пигментов. Агло-
мераты — это свободные ассоциаты первичных частиц, которые
легко разрушаются при диспергировании пигмента в компонен
тах лакокрасочного материала.
С агрегированными или агломерированными частицами легче
обращаться при приготовлении красок, чем с исходными части-
цами, поскольку последние могут быть настолько малы и поэтому
легки, что могут «пылить» при воздействии на них.
Производители пигментов предлагают «непылящие» марки
пигментов, у которых форма и размер агломератов или агрега-
тов подобраны таким образом, чтобы облегчить обращение с ними.
Состояние агломерации или агрегации является ключевым
фактором, влияющим на диспергируемость пигментов.
Форма и размер первичных частиц пигментов влияет на их
способ упаковки в красочной пленке. Так, известно, что должным
образом диспергированные игольчатые частицы упрочняют пленку
подобно стекловолокну в армированных стеклопластиках. Пиг-
менты с частицами пластинчатой формы, такие как алюминий
и слюда, имеют тенденцию к образованию частично перекрываю-
щихся пластинчатых структур (по типу черепицы на кровле), что
повышает стойкость покрытия к проникновению воды.
Форма частиц может также влиять на оттенок цвета пигмента.
Показано [2], что медьсодержащие р-фталоцианиновые пигменты
(Пигмент синий 15:3) имеют различные оттенки в зависимости
от степени отклонения от изометрии к игольчатости.
3.8.4. Размер и распределение частиц по размерам
Размер частиц обычно выражается как средний диаметр пре-
обладающих частиц, при этом форма первичной частицы пигмента
предполагается сферической (см. гл. 6).
Распределение частиц по размерам, как правило, выражается
в виде весового процента, учитывая все фракции от самых мел-
ких частиц до самых грубых. Типичные диапазоны таковы:
Для органических пигментов — от 0,01 до 1,00 мкм;
Для неорганических пигментов —от 0,10 до 5,00 мкм;
(кроме сажи, у которой диа-
пазон) — от 0,01 до 0,08 мкм;
Для диоксида титана — от 0,22 до 0,24 мкм;
при среднем размере частиц — около 0,23 мкм
Стоит отметить, что хотя у наполнителей частицы являются,
как правило, крупными, в то же время их размер может колебаться
от исключительно малого (например, у осажденного диоксида
кремния) в очень широком диапазоне, включая частицы до 50 мкм.
Очевидно, что размер и распределение частиц по размерам
являются иными способами выражения средней свободной пло-
щади поверхности пигмента и числа первичных пигментных частиц
в единице его массы. Если данный пигмент заменить другим
с сильно отличающимся распределением частиц по размерам,
то предсказания основных характеристик, основанные на концеп-
ции объемной концентрации пигмента и критической объемной
концентрации, вероятно, не будут удовлетворительными. Обще-
принятый параметр «маслоемкость 1 рода» (вес в граммах рафи-
нированного льняного масла, которого достаточно для образова-
ния пасты со 100 г пигмента) прямо зависит от распределения
частиц по размерам, хотя существенно влияют также и такие
факторы как степень агрегирования пигмента, плотность упа-
ковки и смачиваемость маслом. ,
Агрегация и агломерация увеличивают эффективный размер
частиц большинства пигментов по меньшей мере до 20 мкм. Такое
скопление может содержать до миллиона первичных частиц.
Размер частиц пигментов имеет первостепенное влияние на
основные свойства лакокрасочных материалов, в которых эти
пигменты используются. По этой причине большинство исследо-
ваний, проводимых изготовителями пигментов, направлены на то,
чтобы приблизиться к оптимальному размеру частиц.
Например, размер частиц пигментов влияет на укрывистость.
Для белых пигментов это обусловлено увеличением рассеивания
света с уменьшением размера до тех пор, пока не будет достигнут
оптимальный для рассеяния размер. Дальнейшее уменьшение
размера уменьшит рассеяние и увеличит светопроницаемость.
Для цветных пигментов также существует оптимальный диапа-
зон размеров частиц для достижения наилучшей укрывистости,
но следует учитывать как рассеяние, так и поглощение света.
Диоксид титана — очень хороший кроющий пигмент. Во-первых,
он имеет высокий показатель преломления (2,76); во-вторых, мож-
но достичь оптимального для светорассеяния размера частиц,
который составляет половину среднего значения длины волны
дневного света в воздухе — 0,25 мкм.
Понять влияние размера частиц на укрывистость можно,
рассмотрев кусок льда. В виде массивной глыбы материал прозра-
чен, но в виде порошка он становится непрозрачным и белым.
На границе раздела лед — воздух ввиду различия их показателей
преломления возникает многократное преломление. Размер частиц
также влияет на оттенок цвета. Это наиболее ярко можно про-
демонстрировать на примере сферического оксида железа, у кото-
рого частицы меньшего размера (0,09—0,12 мкм) имеют желтый
оттенок красного цвета, а большего (0,17—0,70 мкм)—усили-
вающийся синий оттенок.
В общем, более мелкие частицы пигментов (менее 0,40 мкм) —
более яркие, с более чистыми оттенками.
Другое свойство пигментов, на которое может повлиять раз-
мер частиц — это интенсивность окраски. В целом, для органи-
ческих пигментов, чем мельче частицы для одного и того же класса
пигментов, тем выше интенсивность окраски, но эта зависимость
различна для различных пигментов.
Блеск покрытия также зависит от размера частиц, высокий
блеск обычно достигается при размерах частиц пигмента, не пре-
вышающих 0,3 мкм (при правильно выбранной концентрации).
Больший размер частиц уменьшает блеск, вплоть до получения
матовой пленки.
Долговечность некоторых пигментов, главным образом низко-
молекулярных органических веществ, тоже зависит от размера
частиц. Например, ариламиды желтые (Пигменты желтые 1 и 3)
могут быть с большими или малыми частицами, обладая свой-
ствами, приведенными ниже:
Интенсивность окраски
Кроющая способность
Светостойкость
Частицы крупного
размера
ниже
выше
лучше
Более мелкие
частицы
выше
ниже
хуже
3.8.5. Показатель преломления
Показатель преломления (п}) материала имеет ключевое зна-
чение для проявления им пигментных свойств. Чем больше раз-
ница между П\ пигмента и среды, в которой он диспергирован,
тем больше укрывистость.
Рассмотрение показателей преломления некоторых типичных
наполнителей и белых пигментов показывает, что диоксид титана
имеет более высокую кроющую способность, чем оксид цинка,
тогда как тальк и карбонат кальция прозрачны в покрытиях:
Среда Л1 Наполнители Белые пигменты П>
Воздух 1,00 Карбонат кальция 1,58 Литопон 30% (суль- 1,84
Вода 1,33 Китайская глина (си- 1,56 фид цинка/сульфат бария) Оксид цинка 2,01
Пленкообра* зователи 1,40— 1,60 ликат алюминия) Тальк (силикат маг- ния) Бариты (сульфат ба- рия) 1,55 1,64 Сульфид цннка Диоксид титана: анатазный рутильный 2,37 2,55 2,76
Помимо того, что рутильный диоксид титана имеет самый
высокий показатель преломления ггь он может быть приготовлен
с оптимальным для светорассеяния размером частиц 0,25 мкм;
посредством этого достигается улучшение укрывистости.
Для цветных пигментов показатель преломления пигмента
в непоглощающей или сильно отражающей части спектра дает
представление о пигменте как об укрывающем материале. На-
пример, Пигмент желтый 1 (Ариламид желтый G) имеет мень-
шую кроющую способность, чем Пигмент желтый 34 (Хромат
свинца желтый); причем все остальные (кроме разницы показа-
телей преломления) факторы примерно одинаковы.
Изготовители пигментов стараются увеличить кроющую спо-
собность органических красных и желтых пигментов в основном
путем регулирования размеров частиц.
3.8.6. Природа поверхности пигментов
и покрытий на пигментных частицах
Природа поверхности пигментной частицы является ее наи-
более важной особенностью. Ее полярность определяет сродство
к растворам алкидов, полиэфиров, акриловых полимеров и т. д.,
а также к водным растворам и дисперсиям пленкообразователей.
Она определяет легкость деагрегирования пигментов и, следо-
вательно, влияет на диспергирование пигментов и стабильность
готовой жидкой краски.
Рассмотрение природы некоторых пигментов до модификации
их поверхности добавками проиллюстрирует коренную разницу
в полярности поверхности, связанную с химическим строением
пигментов:
Фталоцианин меди голубой
(Пигмент голубой 15:3)
Полихлорфталоцианиновый зеленый
(Пигмент зеленый 7)
Фталоцианиновый голубой имеет атомы водорода по пери-
метру плоской молекулы, тогда как зеленый — атомы хлора.
Очевидно, что последний имеет более плоскую молекулу. Однако,
более детальное изучение пигментов [3] показало, что разница
в полярности более резко выражена. Фталоцианиновый голубой
состоит из «пачек» квадратных плоских молекул, сложенных
подобно колоде игральных карт. Оси пачек наклонены к плоско-
сти молекулы под углом, зависящим от кристаллической моди-
фикации. Такое расположение молекул приводит к образованию
ацикулярной или иглоподобной структуры. В кристалле существуют
сильно отличающиеся поверхности — неполярные атомы водорода
вдоль иглы со сравнительно полярными л-связями бензольных
колец и атомы азота и меди на поверхностях на каждом из концов
Иглы. В подтверждение этого, синие р-фталоцианиновые пиг-
1менты, имеющие более длинные иглы, как показано, являются
относительно менее полярными, чем изометрические кубические
(кристаллы р-фталоцианина меди. Они также более гидрофоб-
ны [2].
В противоположность, в периленимиде (Пигмент красный 179)
сильно полярные имидокислотные группы находятся на перифе-
рии молекулы, а число атомов водорода относительно невелико
по сравнению с фталоцианиновым голубым. В самом деле, дока-
зано, что периленимид относительно более гидрофилен, чем не-
полярный фталоцианиновый голубой:
В силу ионного строения молекул неорганические пигменты
в целом значительно более полярны и гидрофильны, чем орга-
нические. Пигмент белый 6 (рутильный диоксид титана), напри-
мер, имеет компактную решетку, которую можно рассматривать
как коробку с ионами титана в углах и кислорода внутри и между
ионами титана (рис. 3.2). Некоторые из этих атомов кислорода
входят в ОН-группы, а они образуют сильные водородные связи
или даже могут участвовать в обмене с кислотными или основ-
ными гидроксилами, имеющимися в компонентах краски [4].
Поверхностная обработка пигментов. Модификация поверх-
ности пигментов осуществляется изготовителем или целенаправ-
ленно (с целью изменения свойств), или как побочный эффект,
обусловленный добавлением веществ с целью облегчения произ-
водства самих пигментов. Некоторые добавки делают и то, и дру-
гое. Свойства, которые стараются улучшить путем введения
добавок, таковы: смачиваемость и диспергируемость в выбран-
ных смолах и растворителях или разбавителях; стойкость к фло-
куляции; стабильность краски; долговечность покрытия.
Наиболее яркие примеры пигментов, обработанных с целью
улучшения долговечности — это хромат свинца и диоксид титана.
Рис. 3.2. Рутильиый диоксид титана (Пигмент
белый 6)
На желтый и красный хромат свинца наносят покрытия различ-
ной толщины, плотности и однородности, с использованием
оксида алюминия, диоксида титана и кремния, фосфатов метал-
лов с целью улучшения свето-, термо- и химостойкости.
Диоксид титана обычно обрабатывают оксидом алюминия,
диоксидом кремния и органическими соединениями — жирными
кислотами и аминами для улучшения диспергируемости во взя-
тых типах лакокрасочных материалов, укрывистости и интен-
сивности окраски, долговечности покрытия, сохранения блеска,
цвета и стойкости к мелению, а также для защиты связующего.
.В свете последнего, фотоактивная природа диоксида титана,
которая может способствовать разрушению полимерной пленки,
нивелируется нанесением покрытия на кристалл диоксида ти-
тана. Однако, этот пигмент хорошо поглощает Уф-излучение,
и это свойство обеспечивает защиту полимеров в пленках, которые
могли бы деструктировать под воздействием этого изучения.
Поэтому тип и качество покрытия, наносимого на кристаллы TiO2,
определяется необходимостью достижения компромиссов между
двумя этими явлениями.
Другим примером, где модификация строения кристалла и
поверхности — главный фактор, определяющий свойства пигмен-
тов, являются фталоцианиновые голубые пигменты. а-Форма
Пигмента голубого 15 нестабильна и в растворителях, таких как
ксилол, при температурах выше 95 °C превращается в 0-форму.
Стабилизированная a-форма широко применяется в лакокрасоч-
ной промышленности (Пигмент голубой 15:1). Стабилизация
достигается введением атома хлора в молекулу. Пигмент голу-
бой 15:2 — стабилизированная a-форма фиалоцианинового
голубого, который дополнительно модифицируется поверхност-
ными покрытиями, что дает стойкость к флокуляции и улучшен-
ный розлив красок.
Модификация канифолью с использованием абиетиновой
кислоты или резината натрия ингибирует рост кристаллов и при-
меняется главным образом при синтезе пигментов реакцией азо-
сочетания. Это благоприятствует получению меньших частиц
пигментов, что приводит к высокой интенсивности окраски и хоро-
'шей прозрачности.
Обработка аминами, также широко используемая для моди-
фикации азопигментов, таких как Пигмент желтый 3 (Акрил-
амид желтый) улучшает диспергируемость, например в алкид-
ных эмалях (растворитель — уайт-спирит) холодной сушки [5].
Однако, обработка аминами может замедлить процесс окисли-
тельного отверждения. Поэтому и в данном случае, чтобы достичь
хороших свойств пигмента, необходимо компромиссное решение.
Пигменты могут также быть обработаны замещенными произ-
водными основного пигмента. Так, фталоцианиновые пигменты,
обработанные фталоцианинметиленаминами, более стабильны
и имеют лучшую диспергируемость и стойкость к локуляции
в красках с органическими растворителями.
Детальная технология поверхностной обработки пигментов
остается неясной для технологов-лакокрасочников, в част-
ности, потому, что изготовители пигментов не желают выдавать
детальную информацию об особых сортах пигментов. Это значит,
что технолог не знает точно, что за пигмент он покупает. Каждый
пигмент должен быть проверен в каждой конкретной рецептуре
лакокрасочного материала. Следовательно, каждый пигмент и тип
краски следует рассматривать как уникальную систему, так что
разработка и производство красок становится искусством. Это
достижимо только опытным путем, особенно если используются
сочетания пигментов, как обычно и бывает. Увеличение разно-
образия специальных красок сопровождается быстрым ростом
числа сортов пигментов, используемых для специальных целей.
Искусство технолога-лакокрасочника заключается в достижении
компромисса между сложностью и простотой, который должен
обеспечить необходимое потребителю качество и коммерческий
успех изготовителю. '
* 3.8.7. Диспергируемость
Процесс диспергирования включает разделение под действием
механических сил агломератов и агрегатов пигментов на первич-
ные частицы, вытеснение поглощенного воздуха и адсорбиро-
ванной воды, смачивание и обволакивание поверхности пигмента
дисперсионной средой. В идеальном случае каждая первичная
частица, выделяемая при диспергировании, также стабилизи-
руется против флокуляции. Стабилизация пигментных частиц
в красках на органических растворителях часто происходит в силу
стерических препятствий. Молекулы сольватированного поли-
мера, адсорбированные на поверхности пигмента, действуют как
физический барьер для повторной ассоциации.
Полярность поверхности пигмента и сродство к молекуляр-
ным группам диспергируемой смолы, а также природа и концен-
трация растворителя влияют на диспергируемость и последую-
щую стабильность.
Преимущества, заложенные в пигмент при его производстве,
могут быть утеряны, если частицы пигмента должным образом
не диспергированы и не стабилизированы в краске, так, чтобы
получающаяся сухая пленка покрытия содержала только дефлоку-
лированные частицы, равномерно распределенные по всей пленке.
Некоторые пигменты легко диспергируются, если их пра-
вильно используют. Определенные азопигменты быстро диспер-
гируются в алкидных смолах (растворитель — уайт-спирит), да-
вая максимальную интенсивность. Другие, желтые ариламиды и
некоторые красные пигменты — например, сульфоселенид кад-
мия и хромомолибдаты — могут измельчиться до очень малых
размеров, что приведет к потере интенсивности или изменению
оттенка, или к тому и другому. Так называемые высокодекора-
тивные пигменты — кубовые и хинакридоновые — часто диспер-
гируются труднее. Более продолжительное диспергирование
часто дает более высокую интенсивность окраски, хотя это часто
сопровождается изменением оттенка, так как размер частиц
пигментных агломератов уменьшается.
Необходимо учитывать различные параметры, влияющие на
диспергирование. Хафнер [6] и Шафер [7] советуют тщательно
конструировать мелющее оборудование и подводить минималь-
ное количество энергии для разрушения пигментных агломера-
тов, что необходимо для достижения требуемого блеска и яркости
цвета. Отмечено, что совместное диспергирование сильно отли-
чающихся типов пигментов, таких как красный хромомолибдат
(Пигмент красный 104) с хинакридоном (Пигментом красным 122)
не дает наилучших результатов. Органический пигмент хинакри-
дон в отличие от неорганического красного молибдата требует
более продолжительного диспергирования для достижения тре-
буемых свойств. Поэтому последний может быть поврежден.
В таком случае целесообразно раздельное диспергирование.
Дальнейшее обсуждение диспергирования можно найти в гл. 7.
3.8.8. Цвет и другие свойства пленкообразователя
Полимеры, используемые в лакокрасочных материалах, под-
робно рассмотрены в гл. 2. Особенно разнообразен цвет смол
.разных типов. Пленки нитроцеллюлозы и водных латексов почти
•бесцветны, тогда как алкиды, модифицированные маслами —
желтые или коричневые. Показатели преломления также раз-
личны, хотя изменяются в узком диапазоне от 1,4 до 1,6. Поэтому
суммарное влияние большого разнообразия смол, в которых
диспергированы пигменты, на цвет и укрывистость также очень
разнообразно.
и Вдобавок, различные смолы и растворители или разбави-
тели имеют различное сродство к пигментам, что связано с раз-
личием в полярности. Эффективность разрушения агломератов,
смачивание частиц и их последующая стабилизация отличаются
от одной смолы к другой. Хафнер [6] описал диспергируемость
Пигментов фиолетовых 19 и 23 (хинакридон и диоксазин фиоле-
товые) в трех различных смолах; двух тощих алкидах, растворен-
ных в ксилоле,— один на основе ненасыщенных синтетических
жирных кислот, другой на основе дегидратированного касторо-
вого масла — и в высокореакционноспособном бутанолизиро-
ванном меламиноформальдегиде в бутаноле. Он показал, что
яркость цвета и реологические свойства в значительной степени
зависят от смолы.
Калуза |4] обсуждает работы многиЗГ^авторов, которые ясно
[продемонстрировали, что полярный диоксид титана диспергиру-
ется и стабилизируется более легко в карбоксилсодержащих
полимерах; гидроксильные группы образуют более слабые связи.
Он также указывает на важность концентрации, размера поли-
мерных молекул и их сложности и на влияние природы раство-
рителя на стабилизацию диспергированного пигмента.
3.9. ВЫБОР ПИГМЕНТОВ
При выборе пигментов для лакокрасочных материалов следует
учитывать тип материала и область конечной эксплуатации по-
крытия. Всегда принимается во внимание рыночная конъюнктура.
Таблица 3.4. Применение основных
Типы красок Диоксид титана Оксиды железа Хроматы свиица
Декоративные * водоэмульсионные естественной сушки: насыщенные тона 5 5 4*
промежуточные 5 5 4*
разбавленные 5 5 2*
Декоративные и краски общего назначения на осно- ве высыхающих на воздухе жирных алкидов в сла- бых растворителях типа уайт-спирита: насыщенные тона 5 5 4*
промежуточные 5 5 4*
разбавленные 5 5 2*
На основе высыхающих на воздухе алкидов средней 5 5 5
жирности, алкидов модифицированных винилтолуо- лом или стиролом, горячей сушки в сильных раство- рителях Ремонтные отделочные и автомобильные «лаки». Быстросохнущие нитроцеллюлозные и акриловые материалы в сильных растворителях: насыщенные тона и промежуточные 5 5 5
разбавленные 5 5 2
Автомобильные и «сшивающиеся» краски общего назначения горячей сушки типа алкид/меламино- формальдегид, термореактивные акриловые/мел- аминоформальдегид и термопластичные акриловые: насыщенные тона и промежуточные 5 5 5
разбавленные 5 5 2
Защитные краски для промышленных установок — 5 5 4
химически отверждаемые изоцианатные, эпокси- аминные Примечания. 2 — плохо; 3 — удовлетворительно; 4 — хорошо, 5- — отлично
* Не используются в Европе в декоративных красках ** Слишком дороги для
Известны лакокрасочные материалы и покрытия для строитель-
ства___архитектурные или декоративные краски (см. гл. 8),
автомобильные в том числе ремонтные краски (см. гл. 9), краски
общепромышленного назначения и т. д.
3.9.1. Тип лакокрасочного материала
Наиболее распространенная классификация лакокрасочных
материалов с точки зрения их пигментирования — по типу пленко-
образователя и механизму отверждения. Это связано с тем, что
на многие (но не все) пигменты влияет химическое строение
смолы, растворителей, разбавителей и других компонентов лако-
красочного материала, а также нагревание.
пигментов в красках
Берлин- ская лазурь Сажа Моноазо- пигменты S S ? Л- Пигменты — продукты азоконден- сации Металличе- ские азо- пигменты Фтало- цианино- вые пиг- менты Полицикли- ческие пиг- менты в т. ч. кубовые и хинакридон
2 4 2 2 2 От 4 до 2 4 5**
2 4 3 3 От 4 до 2 От 4 до 3 5 5**
2 4 2 2 4 2 5 5
5 4 2 2 4** От 4 до 2 4(Б) 5**
3 4 3 3 От 4 до 2** От 4 до 3 5 5**
2 4 2 2 4** 2 5 5
4 4 2 3 2** 4—3 5 5**
4
2
4 3 2 От 4 до 2 От 4 до 2 4(Б)
4 2 2 От 4 до 2 От 4 до 2 5
5**
5
4 4 2 2
2 4 2 2
2 4 2 2
3 От 4 до 2 4(Б) 4**
2 От 4 до 2 5 5
2 2 5 5
(Б) — «бронзовеет»
в насыщенных тонах
широкого использования.
Так, например, такой красный пигмент моноазонафтола, как
толуидиновый красный (Пигмент красный 3), пригоден для полу-
чения насыщенных цветов в алкидных красках естественной
сушки растворенных в уайт-спирите или в водно-дисперсионных
красках, в которых разбавителем является вода. Такой пигмент
может, однако, выцветать в случае промежуточных или разбав-
Таблица 3.5. Упрощенный указатель для
Система плепкообразова- тель — растворитель Механизм отверждения Глубина цвета Красный
Краски естест
Водные дисперсии сопо- лимеров винилацетата, Вода испаря- ется и пленка Насыщен- ный Нафтол красный (Пиг-
мент красный 3, 4, 6);
полиакрилатов и сополи- латекса коа- Пигмент оранжевый
меров стирола с акрила тами (называемые латек- сами и используемые в эмульсионных красках) * лесцирует Средний (Пигмент красный 48, 57) Нафтиланилиды крас- ные (Пигмент красный 2, 5, 7, 9, 112)
Алкиды жирные и сред- Испарение Разбавлен- Красный железоокисный
ние в уайт-спирите растворителя с последующей сшивкой с участием кис- лорода возду- ха ный пигмент (Пигмент крас- ный 101); Хинакридон (Пигмент фиолетовый 19; Пигмент красный 122); Дибромметантро- ны красные (Пигмент красный 168)
Алкид/меламинофор-
мальдегиды в ксилоле
или бутаноле
Испарение
растворителя
с последую-
щим пленко-
образованием
под действием
тепла
Насыщен-
ный
Термоотверждае
Хром красный (Пигмент
красный 104) с хинакри-
доном (Пигментом фио-
летовым 19) или с ме-
таллотонером (Пигмен-
том красным 122); Пе-
рилен красный
ленных тонов, он может также вымываться растворителями
(ввиду плохой устойчивости к действию растворителей) Это
особенно важно, если верхнее покрытие белое; оно может в резуль-
тате стать розовым.
Чувствительность к растворителям и плохая термостабиль-
ность этого пигмента препятствует его использованию, напри-
выбора пигментов
Желтый Зеленый Синий Коричневый Черный Белый
ценной сушки
Диазопигмент желтый (Пигмент желтый 13, 83); Ариламид жел- тый (Пигмент желтый 1, 3, 73, 74) Ариламид желтый (Пигмент желтый 1, 3, 73, 74) Желтый железо- окисный пигмент (Пигмент желтый 42); Флавантрон желтый (Пигмент желтый 112); Ан- трапиримидий желтый (Пигмент желтый 108); Изоиндолинон желтый (Пигмент желтый 109, ПО); Никель азо-жел- тый (Пигмент зе- леный 10) Фталоциа ниновый голубой (Пигмент голубой 15:1) Фталоцианино- вый зеленый (Пигмент зе леный 7, 36); Фталоциани- новый голубой (Пигмент го- лубой 15; 1) с ацетоацета риламидами жел- тыми (Пигмент желтый 3, 74) Красный железоокис- ный пигмент (Пигмент красный 101) с Пиг- ментом черным 6,7 Красный железоокис- ный пиг- мент (Пиг- мент крас- ный 101) с Пигментом черным 6, 7 Сажа (Пигмент черный 6, 7) Диоксид титана (Пиг- мент бе- лый 6); каолин Карбо- нат каль- ция
мые краски Желтый железо- окисный пигмент (Пигмент желтый 42), Хром жел- тый Фталоциани- новый зеленый Железо си- нее (Пиг- мент синий 27); Фта- лоцианино- вый голу- бой (Пиг- мент голу- бой 15:1); Индантрон синий То же
Система пленкообразова-
тель — растворитель
Механизм
отверждения
Глубина
цвета
Красный
Термопластичные акри-
ловые в ксилоле или
бутаноле
Испарение
растворителя
с последую-
щим пленко-
образованием
под действием
тепла
Насыщен- Пигмент красный 123,
ный 179, 190, 224; Тиоиндиго
(Пигмент красный 88);
Красный железоокисный
пигмент (Пигмент крас-
ный 101)
Быстро
Нитроцеллюлозные и ак-
риловые лаки в аромати-
ческих углеводородах,
высших спиртах, эфирах,
кетонах
После испаре-
ния раствори-
теля остается
сухая пленка
Промежу-
точный
Красный хром (Пигмент
красный 104) с хинакрц-
доном (Пигментом фио-
летовым 19) или с [Ди-,
азопигментом красным
(Пигментом красным
144, 214)]; Перилены:
красные (Пигмент крас-
ный 123, 179); Тиоинди-
го (Пигмент красный
88)
Полиэфирные, полиуре-
тановые, эпоксидные смо-
лы, растворенные в аро-
матических катонах, эфи-
рах, спиртах
Химическое
сшивание
Разбавлен-
ный
Химически отвер
Красный железоокисный
пигмент (Пигмент крас-
ный 101); Хинокридоиы
(Пигмент фиолетовый
19, Пигмент красный
122); Дибромантантрон
(Пигмент красный 168);
[Диазопигмент красный
(Пигмент красный 144,
214)]
Примечание. В квадратные скобки заключены пигменты, имеющие ограниченное
Продолжение
\ ЖЁЛТЫЙ Зеленый Синий Коричневый Черный Белый
[Диарилид жел- тый (Пигмент Пигмент зеле- Фталоци- Красный Сажа Диоксид
ный 7, 36 или аниновый железоокис- (Пигмент титана
желтый, 13, 83)] Фталоциани- голубой ный пиг- черный (Пиг-
новый голубой (Пигмент мент (Пиг- 6, 7) мент бе-
(Пигмент го- голубой мент крас- лый 6)
лубой 15:1) с 15:1); Ин- ный 101);
хромом жел- дантрон си- Хинокридо-
тым (Пигмен- ний (Пиг- ны (Пиг-
том желтым мент синий мент фио-
34) или с 60) летовый 19,
[Диазопигмен- Пигмент
том желтым красный
(Пигментом 122); Ди-
желтым 155)]; броман-
[Брауншвейг- тантрон
ская зелень (Пигмент
(Пигмент зе- красный
леный 15)] 168); [Диа-
сохнущие зопигмент
красный (Пигмент
Изоиндолином красный
желтый (Пигмент желтый 109, 110); Желтый железо- окисный пигмент То же 144. 214)]
(Пигмент желтый
42); [Бензимида- золов (Пигмент
желтый 120, 151, 154)]; Никель азо-желтый (Пиг- мент зеленый 10)
ждаемые краски
Желтый железо-
окисный пигмент
(Пигмент желтый
42); Флавантрон
желтый (Пигмент
желтый 112);
Антрапиримидин
желтый (Пигмент
желтый 108);
[Изоиндолинон
желтый (Пигмент
желтый 109, НО)]
применение для высококачественных красок.
мер, в алкидно-меламино ормальдегидных эмалях, где исполь-
зуемые растворители могут вызвать вымывание пигмента, а нагрев
вание, используемое для отверждения,— его возгонку. Для
получения такой эмали красного цвета лучше использовать крас-
ный диазопигмент, например Пигмент красный 144 или красный
бензимидазолоновый пигмент (Пигмент красный 208). Для дости-
жения наилучших свойств можно применить смесь двух пигмен-
тов, таких как хромомолибдат свинца с хинакридоном пурпур-
ным (смесь Пигмента красного 104 с Пигментом красным 122).
Тип смолы, таким образом, является ключевым фактором для
выбора пигмента, поскольку он определяет выбор растворителей,
разбавителей и других компонентов. Способ пленкообразова-
ния — второй важный фактор.
Общие принципы выбора пигментов представлены в табл. 3.4
и 3.5.
3.9.2. Свойства красок
Следующие соображения относятся к применению красок и
требованиям потребителя к долговечности пленки, ее стойкости,
цене краски.
Например, стойкость к воздействию растворителей и хим-
стойкость при высокой долговечности покрытий требуются в авто-
мобильной промышленности. Для таких покрытий применяют
высококачественные пигменты, и в случае органических пигмен-
тов они обычно более дорогие, т. к. обычно включают более
сложные органические молекулы для стабилизации свойств пиг-
ментов.
Типичные пигменты, используемые в автомобильных высоко-
качественных эмалях, описаны в гл. 9.
Когда требуются специальные свойства, такие как коррозион-
ная стойкость, необходимо применять пигменты — ингибиторы
коррозии. Последние обычно входят в грунтовки.
3.9.3. Укрывистость и прозрачность
Ограничив область выбора рассмотрением природы пленко-
образователя и требуемой долговечности, необходимо иметь в виду,
кроме того, следующие соображения — требуется ли, чтобы плен-
ка была равномерно окрашенной, имела специальные эффекты
или прозрачность.
Для получения хорошо укрывающей пленки требуются крою-
щие пигменты. Для достижения специальных эффектов, таких
как «металлические» или «перламутровые» оттенки и т. п., тре-
буются прозрачные или полупрозрачные пигменты совместно
с чешуйчатой алюминиевой пудрой и слюдой.
Когда требуется хорошая укрывистость, ее легче достичь
в случае черных, коричневых, синих и зеленых цветов путем при-
менения, соответственно, сажи, оксидов железа, фталоцианиновых
голубых и зеленых пигментов. В белых красках почти всегда при-
меняют рутильный диоксид титана. Наиболее трудно придать
укрывистость ярким красным, оранжевым, желтым цветам. Укры-
вающие, но тусклые желтые и красные цвета дают желтые и
красные оксиды железа. Использование хроматов свинца — наи-
более простой путь для получения хорошо укрывающих ярких
желтых и красных цветов, но в Европе, Японии и США их стре-
мятся избегать в верхних покрытиях из-за токсичности шести-
валентного хрома и свинца.
Результаты поисков ярких, укрывающих и долговечных крас-
ных и желтых пигментов отражены в большом числе пигментов,
предлагаемых изготовителями.
Существует более 240 типов красных и более 160 — желтых
пигментов, классифицированных в Colour Index, тогда как си-
них — менее 70 (см. разд. 3.5).
«Прозрачные» цветные краски применяются в тех случаях,
когда требуется, чтобы подложка была видна, но защищена.
Так называемые «высококачественные материалы для древе-
сины», которые после нанесения не скрывают структуру твердой
и мягкой древесины (или ее текстуру), также требуют применения
прозрачных пигментов.
Окрашенные прозрачные покрытия можно получить путем
применения прозрачных или малых количеств полуукрывающих
пигментов. К прозрачным пигментам относят прозрачные жел-
тые и красные оксиды железа, которые представляют собой очень
тонко измельченные частицы укрывающих пигментов. Полу-
прозрачные — это органические красные, желтые, зеленые и
синие пигменты, которые ярче и прочнее оксидов железа и упо-
требляются в меньших количествах вместе с невсплывающими
алюминиевыми пигментами, с тем, чтобы получить эффект «свер-
кания» или другие специальные эффекты, требуемые, скажем,
в автомобильной промышленности.
3.9.4. Цвет и смешивание пигментов
(
Простейшие (в смысле пигментирования) краски — это та-
кие, в которых для придания желаемого цвета используется
только один пигмент. Например, в высокоглянцевых алкидных
черных или белых эмалях для дверей или оконных рам, в качестве
единственного пигмента используют сажу или диоксид титана,
ч Более сложное пигментирование требуется, чтобы удовлетво-
рить следующим требованиям:
1) получить оттенок промежуточный между теми, которые
получаются от применения индивидуальных пигментов;
2) получить краски с меньшим глянцем для полуглянцевых
и матовых покрытий, когда для эффекта матирования можно
применять наполнители;
3) получать специальные реологические свойства жидкой,
краски.
Органические и неорганические цветные пигменты можно
(смешивать, получая «вычитающиеся» цвета:
желтый + синий = зеленый черный + белый = серый
красный + белый = розовый красный + желтый -|- белый =
= бледно-оранжевый
прозрачный красный оксид-(-алюминиевые чешуйки = «металличе-
ский» коричневый
Правильным принципом является составление рецептур кра-
ток настолько простых, насколько возможно, с использованием
|минимального числа пигментов. В идеале следует смешивать
в краске только пигменты с одинаковой стойкостью и долговеч-
ностью. Это помогает обеспечить сохранение цветового оттенка
лакокрасочной пленки в течение ее эксплуатации, хотя в общем
она может тускнеть или темнеть. Бледно-оранжевый цвет, полу-
ченный из малых количеств красного и желтого с белым будет
обесцвечиваться до бледно-желтого, если Толуидиновый красный
(Пигмент красный 3) использован с Ариламидом желтым (Пиг-
ментом желтым 74) и диоксидом титана (Пигментом белым 6).
Лучше примёнять Ариламид красный (Пигмент красный 112).
Цвета, полученные при смешивании пигментов, часто менее
чистые и яркие, чем у одного пигмента. Например, смесь желтого
пигмента с зеленым оттенком, Ариламида желтого 10G (Пигмента
желтого 3) с медьсодержащим р-фталоцианиновым голубым
(Пигментом голубым 15:3), дает приемлемый зеленый цвет, но
он будет слегка менее чистым и ярким, чем у Фталоцианинового
зеленого (Пигмента зеленого 36).
Для получения максимального эффекта чистоты и яркости
цвета лучше использовать смежные оттенки цветов цветового
спектра. Например, для получения ярко-оранжевого цвета из двух
пигментов смешивать желтый цвет с красным оттенком с крас-
ным цветом с желтым оттенком лучше, чем зеленовато-желтый
с синевато-красным. Последнее даст бледновато-оранжевый цвет.
Так, например, Пигмент желтый 1 (красный оттенок) плюс Пиг-
мент красный 168 (желтоватый) дает лучший оранжевый цвет,
чем Пигмент желтый 3 (зеленоватый) плюс Пигмент красный 112
(слегка синеватый). Однако, сохранение свойств этих пигментов
неодинаково: Пигмент красный 168 более светостоек, чем Пиг-
мент желтый 1, и оранжевый цвет со временем станет менее жел-
тым. Здесь необходим компромисс, и он заключается в том, что
Пигмент красный 112, менее светостойкий, чем Пигмент крас-
ный 168, следует использовать с Пигментом желтым 1. Это соче
тание сохранит оранжевый цвет, хотя он может тускнеть.
Другое соображение при смешивании пигментов заключа-
ется в том, что большие различия в размере частиц и природе
поверхности пигментов могут привести к флокуляции, давая не-
равномерное окрашивание. Пигмент сажа (размер частиц 0,01 —
0,08 мкм), смешанный с диоксидом титана (размер частиц
\)'25 мкм), часто дает флокулирующую смесь серого цвета.
V Использование специальных перламутровых пигментов, та-
ких как синтетические модифицированные слюды, включает
в себя принцип «аддитивного» смешения цветов, например:
красный + зеленый = «белый»
Следует соблюдать осторожность при использовании таких
пигментов в сочетании с классическими неорганическими и орга-
ническими цветными пигментами, где наблюдается «вычитатель-
ное» цветосмешение.
3.9.5. Количество пигментов в краске
Это количество определяется интенсивностью и прочностью
пигмента, требуемой укрывистостью, уровнем глянца, стабиль-
ностью и долговечностью. При этом важны два показателя:
объемная концентрация пигментов (ОКП) и весовое (объем-
ное) соотношение пигмент : связующее (П : С).
Обычно автомобильные эмали и краски общего назначения
имеют низкую ОКП, тогда как ОКП строительных красок в зави-
симости от подложки и требуемой стойкости изменяется от 10
до 90%. Низкокачественные краски с высокой ОКП приготавли-
вают с использованием смеси малых количеств такого первичного
пигмента, как анатазный диоксид титана с большим количеством
наполнителя, например карбоната кальция, с получением краски
для окрашивания, например, потолков.
Глава 4
ДОБАВКИ В КРАСКИ
. Р. А. Джеффе и В. Джонс
4.1. ВВЕДЕНИЕ
В данной главе под добавками понимаются такие вещества,
которые, будучи введенными в рецептуру красок в небольших
количествах, тем не менее оказывают значительное влияние на
их свойства. Обычно к добавкам не относят те компоненты, кото-
рые рассматривают как часть основной рецептуры. Например,
в рецептуру .лакокрасочных материалов для текстурированных
покрытий и покрытий пониженной горючести входят специаль-
ные компоненты. Хотя их количества относительно невелики,
однако для приготовления указанных материалов недостаточно
просто добавить эти компоненты. Поэтому такие вещества не
рассматриваются в этой главе.
Мы надеемся, что краткие комментарии, которые будут даны
в каждом подразделе, окажутся полезными, однако следует
помнить, что:
а) Любое перечисление никогда не может быть всеобъемлющим,
учитывая появление новых разработок.
б) Иногда добавка является очень специфической для конкрет-
ной композиции, будучи эффективной в одних типах красок и
бесполезной в других.
в) Применение добавок очень часто приводит к нежелательным
вторичным эффектам, особенно, если их вводить в избытке или
необдуманно.
г) Необходимо позаботиться о том, чтобы применение1 добавок
не привело к «спиральной рецептуре». Например, ПАВ вводят
для облегчения смачивания поверхности, однако они вызывают
вспенивание. Последнее уменьшают, добавляя силиконовый
раствор, но он способствует кратерообразованию, которое, в свою
очередь, преодолевают введением ПАВ.
д) Может встретиться какая-то композиция, которая разраба-
тывалась в течение большого промежутка времени, возможно,
несколькими исследователями, и содержащая поэтому целый ряд
добавок. В случае, если при ее применении возникнут дополни-
тельные затруднения, может оказаться гораздо выгоднее исклю-
чить эти добавки, а не искать еще другие. Мы довольно часто
исключали из известных композиций добавки и оставляли только
базовую рецептуру. С точки зрения современных требований они
не всегда были действительно необходимы. Некоторые из них
продолжали вводить по традиции, хотя первоначально их при-
меняли для преодоления определенных недостатков при изготов-
лении конкретной партии лакокрасочного материала.
1е) Добавки обычно применяют для устранения недостатков кра-
сок, но описания этих недостатков часто неточные. Поэтому вна-
чале необходимо четко убедиться в том, что это именно тот не-
достаток и та добавка, с помощью которой его можно преодолеть.
Вспенивание и биологическая защита представляют две области,
где возможно появление проблем при невнимательном подходе.
Упомянутые в тексте добавки снабжены ссылками к перечню
в конце этой главы, где можно найти названия поставщиков и их
представителей в Великобритании. Это не полный перечень
поставщиков добавок — давая его мы сознаем, что рискуем по-
терять друзей. Дополнительную информацию и подробные адреса
можно найти в таких публикациях, как Polymers Paint Colour
year Book (издаваемый Fuel and Metallurgical Journals Ltd,
Redhill, Surrey).
Многие из перечисленных продуктов имеют торговые наиме-
нования.
4.2. УСИЛИТЕЛИ ПРОТИВОКОРРОЗИОННЫХ СВОЙСТВ
Известно много веществ, которые предлагаются в качестве
усилителей .противокоррозионных свойств обычных противо-
коррозионных пигментов. Типичный пример — производные тан-
ниновой кислоты Kelate [45/34], Albarex [44/15] —это отрабо-
танный наполнитель, используемый для частичной замены истин-
ных противокоррозионных пигментов, таких, как фосфат цинка.
Alcophor 827 [27], представляющий собой цинковую соль орга-
нического азотистого соединения, также усиливает действие
основных антикоррозионных пигментов. Ferrophos [42] рекомен-
дуют для частичной замены цинковой пыли в грунтовках, обога-
щенных цинком.
4.3. ПЕНОГАСИТЕЛИ
Вододисперсионные краски стабилизируют ПАВ и коллои-
дами, которые, к сожалению, способствуют стабилизации воз-
душных пузырьков, вводимых при производстве или при нанесе-
нии красок и, вследствие этого, приводят к образованию устой-
чивых пен. В неводных красках (как и вообще в жидких) может
также наблюдаться образование воздушных пузырьков. Известны
антивспениватели как для определенного класса лакокрасочных
материалов, так и для общего употребления. Иногда антивспе-
нивающие добавки вводят в два приема — первый в начале произ-
водства, а второй непосредственно перед расфасовкой.
Обычно антивспениватели обладают высокой поверхностной
активностью и хорошей подвижностью, хотя и не растворяются
во вспенивающейся жидкости. Обычно их действие основано на
понижении поверхностного натяжения в окрестности пузырьков,
что приводит к их слиянию в более крупные и менее устойчивые
пузырьки, которые затем разрушаются. Наиболее простые из
этих добавок представляют собой растворы какого-то одного
вещества, например пихтового масла, дибутилфосфата или спир-
тов с короткой цепью (Св—Сю). С другой стороны, они могут
быть сложными композициями неопределенного состава, состоя-
щими из минеральных или силиконовых масел, нанесенных на
мелкоизмельченный диоксид кремния в присутствии ПАВ. Мно-
гие антивспениватели действуют благодаря несовместимости,
создавая вследствие этого центры, на которых может начаться
разрушение пузырьков. Медленное увлажнение или эмульгиро-
вание антивспенивателей часто является причиной того, что они
теряют свою эффективность при длительном процессе производ-
ства или при-продолжительном хранении.
Испытание добавки на эффективность может быть затруд-
нительным, так как обычно применяемые методы испытания
встряхиванием или перемешиванием редко могут полностью отра-
зить то, что происходит в реальных производственных условиях.
И поэтому часто превосходная добавка для одной композиции
оказывается бесполезной в другой. Тем не менее, ниже приведены
в качестве примера некоторые из имеющихся в продаже добавок:
Foamaster NDW На основе минерального масла [18]
Бик 031 Бик 069 На основе минерального масла (устойчив до 100 °C и выше) [8/39] Минеральные масла, спирты, мыла [8/39]
Bevaloid GS32 Perenol Е2 На основе силиконового масла [5] Не силиконовая добавка для эпоксидных си- стем [27]
Пеногаситель 1512 М Минеральное масло, диоксид кремния, силикон [28]
Пеногаситель L409 На основе силикона (для алкидных латексов горячей сушки) [20]
Пеногаситель L413 EFKA Schwegofoam На основе силикона/диоксида кремния, для вод- ных "систем [20] [22/26] [52/12]
4.4. ДОБАВКИ, ПРЕПЯТСТВУЮЩИЕ ОСАЖДЕНИЮ
При решении вопроса о предотвращении осаждения пигмен-
тов прй хранении органорастворимых лакокрасочных материалов
часто приходится идти на компромисс. Хорошие диспергаторы
и дефлокулянты повышают блеск и укрывистость, но отрица-
тельно влияют на осаждение. При регулировании конечных реоло-
гических характеристик красок также необходим компромисс
между устойчивостью к осаждению и ухудшением блеска. Такая
хорошо известная, но тем не менее полезная добавка на стадии
диспергирования, как соевый лецитин, может очень эффективно
препятствовать осаждению. Тем не менее, при превышении опта-
мального ее содержания возможно очень сильное расслоение
системы.
< Запатентовано много веществ, придающих небольшую тиксо-
тропность. До недавнего времени в рецептуры обычно включали
менее одного процента стеарата алюминия. Однако это создавало
проблемы при использовании в промышленности. Более удобны
в обращении карбонаты кальция с нанесенным сверху слоем стеа-
рата (например, Winnofil и Sturcal). Другим представителем
веществ,' используемых для предотвращения осаждения, явля-
ются модифицированные гидрированные касторовые масла. Их
обычно выпускают в виде студней в растворителе и подбирают
к конкретной краске с учетом температуры перетира, чтобы из-
бежать образования «зернистых» частиц при хранении (Thixomen,
Crayvallac, Thicotrol, MPA).
Модифицированные клеи, например Bentone 34 и Perchem 44,
также вызывают определенное структурирование, но их необ-
ходимо активировать полярным растворителем, таким как спирт.
При этом можно получить различные структуры. Считают, что
Byk Anti-terra 203 более надежен в качестве загустителя. Не-
давно появился ряд новых структурирующих веществ на основе
указанных клеев без полярного растворителя, которые легче при-
менять (Bentone SD1 и SD2, Easigel).
Следует помнить, что часто для усиления эффекта исполь-
зуют смесь добавок, препятствующих осаждению. Так тонко-
дисперсные пирогенные диоксиды кремния (например, Аэросил
и Cab-O-Sil) эффективны сами по себе или в смеси с Byk Anti-
terra 203.
Так как многие из вышеупомянутых добавок влияют на вяз-
кость при малых скоростях сдвига (удерживая вследствие этого
пигменты в виде суспензии, несмотря на гравитационные силы),
их используют также для других целей, например против обра-
зования натеков, регулирования розлива и изменения цвета вслед-
ствие всплывания пигмента.
В латексных красках осаждение не представляет проблему
ввиду специфики их рецептуры. Для водных красок других типов
важно правильно выбрать коллоидную систему и, кроме того,
разумно использовать при пигментировании бентонит, пироген-
ные диоксиды кремния или же целлюлозные или синтетические
полимерные загустители. Ниже приведены производители доба-
вок, препятствующих осаждению:
Стеарат алюминия [21];
Соевый лецитин [10/61, 9];
Winnofil [30];
Sturcal [57];
Thixomen [30];
Crayvallac [14];
Thixotrol [41];
MPA [41];
Beniones [55];
Perchem [43];
Byk Anti-terra [8/39];
Аэросил [17];
Бентонит [55];
Easigel [43];
EFKA [22/26].
4.5. ДОБАВКИ, ПРЕПЯТСТВУЮЩИЕ ОБРАЗОВАНИЮ
ПОВЕРХНОСТНОЙ пленки при хранении красок
Сиккативы в красках воздушной автоокислительной сушки
имеют важное значение для правильного регулирования соотно-
шения между процессами высыхания в поверхностном слое и во
всем объеме. К сожалению, они могут также вызывать образо-
вание поверхностной пленки при хранении лакокрасочных мате-
риалов. Раньше для борьбы с этим явлением применяли 1—2%
пихтового масла или дипентена, а для более сложных случаев —
фенольные антиоксиданты, такие как гваякол (менее 0,1%).
Затем они были в основном заменены более удобными оксимами.
В настоящее время широко применяют бутиральдоксим и осо-
бенно метилэтилкетоксим в количестве около 0,2% от веса краски.
Циклогексаноноксим является порошком и применяется благо-
даря слабому запаху. Будучи летучими, оксимы удаляются из
-нл-енки -на ранних стадиях и поэтому незначительно замедляют
процесс высыхания. Однако эта летучесть может быть и недостат-
ком, поскольку после вскрытия емкости и повторной герметиза-
ции при хранении уже может образовываться поверхностная
пленка.
Некоторые антиоксиданты при хранении могут вызывать по-
терю способности краски к высыханию, поэтому необходимо про-
верять их влияние как на способность к высыханию, так и на
образование поверхностной пленки. Ниже приведены некоторые
такие добавки:
Бутиральдоксим [53/40]; Дипентен [37, 59];
Метилэтилкетоксим [53/40, 6/11 ]; Гваякол [7,49];
Циклогексаноноксим [53/40, 4]; Exkins 1, 2 и 3 [53/40].
Пихтовое масло [37, 59];
4.6. ИНГИБИТОРЫ КОРРОЗИИ ТАРЫ
Водные краски всех типов даже при упаковке в лакирован-
ные емкости из белой жести способны вызывать коррозию, осо-
бенно на внутренних спаях, стойках и т. д.
Такие ингибиторы как нитрит натрия и бензоат натрия, часто
вводимые совместно при концентрациях около 1%, довольно
хорошо известны из опыта их применения в автомобильных анти-
фризах. Возможным недостатком применения этих солей является
их отрицательное влияние на водостойкость покрытий.
Эффективными ингибиторами коррозии данного типа явля-
ются Ser-Ad FA 179 [53/40] при концентрации меньше 0,3% и
Borchicor Antirust D [6/11]. Они обладают тем преимуществом,
что переходят в нерастворимую форму в процессе пленкообразо-
вания.
4.7. ДОБАВКИ, ПОГЛОЩАЮЩИЕ ВЛАГУ
И ПРЕДОТВРАЩАЮЩИЕ ГАЗОВЫДЕЛЕНИЕ
Для обеспечения стабильности некоторых красок при хране-
нии важно поддерживать низкое содержание влаги. Так, чтобы
избежать сшивания и газовыделения при хранении в емкостях
отверждаемых влагой полиуретановых красок, необходимо при-
менять только те пигменты, из которых почти полностью удалена
адсорбированная влага.
Добавка TI [4] представляет собой мономерный изоцианат,
который жадно реагирует с водой и поэтому используется для
обезвоживания пигментов на стадии диспергирования. Добавку
OF [4] (триэтилортоформиат) вводят в краски для того, чтобы
регулировать влияние остаточной влаги при хранении.
Другой группой материалов, в которых присутствие влаги
может вызывать проблемы при хранении, являются алюминий-
содержащие краски и грунтовки с цинковой пылью. В них воз-
можна реакция металла с влагой и выделение водорода, вызываю-
щего повышение давления в закрытой емкости. В этих случаях
применяют такие добавки, как Silosiv Al и ZN1 [25], хотя они
и не выполняют свою миссию, если были применены очень влаж-
ные растворители или связующие.
4.8. ДИСПЕРГАТОРЫ
Одной из главных задач при изготовлении красок является
оптимизация диспергирования пигментов в связующем. По мере
возможности этого добиваются без привлечения добавок, однако
некоторые связующие плохо смачивают пигменты, а некоторые
пигменты трудно смачиваются (см. гл. 5—7). Чтобы сократить
процесс диспергирования, избежать ухудшения укрывистости,
цвета и глянца может потребоваться применение диспергаторов.
Выбор диспергаторов в самом широком смысле этого слова очень
большой, причем они часто пригодны только для конкретных
связующих, пигментов и растворителей в определенной рецеп-
туре. Так, первыми в качестве диспергаторов начали применять
металлические мыла, которые обычно добавляли в краску на за-
ключительной стадии ее изготовления в качестве сиккативов.
Однако было обнаружено, что при добавлении их в пигментную
пасту значительно улучшается диспергирование. В настоящее
рремя в качестве добавок при диспергировании продолжают
применять октоаты кальция и цинка.
Некоторые из диспергаторов применимы как для водных, так
И для органорастворимых систем. Они могут быть анионными,
катионными, неионными или амфотерными. Обычно для них харак-
терно присутствие закрепляющейся группы, которая притягива-
ется к поверхности пигмента и специфична для определенных
групп пигментов, а также полимерной или, по крайней мере,
более высокомолекулярной части, которая совместима со связую-
щим и растворителями.
По их применению можно дать мало общих рекомендаций
за исключением того, что обычно смачиватель вводят на ранней
стадии, чтобы создать наилучшую возможность контакта с по-
верхностью пигмента и избежать конкуренции с другими жид-
кими компонентами композиции или, что еще хуже, необходи-
мости вытеснять их. Во-вторых, если не хотят, чтобы сами диспер-
гаторы были вытеснены с поверхности пигмента, целесообразно
добавлять другие жидкие компоненты к пигментной пасте на
более поздних стадиях с осторожностью и при перемешивании.
Из большого разнообразия таких добавок, один ряд «сверх-
диспергаторов» типа Solsperse ярко демонстрирует насколько
они могут быть эффективными и, в равной мере, специфическими.
Для этого ряда диспергаторов обычно необходимо, чтобы мостико-
-образующ-ая молекула, во-первых, была той же природы, что и
диспергируемый пигмент, а во-вторых, чтобы она сильно притя-
гивалась к стабилизирующей молекуле с полярностью, соответ-
ствующей полярности растворителя. При достижении этого на-
полнение пигментом пигментной пасты может быть выше, чем
при диспергировании в обычных связующих. Ниже приведены
некоторые из добавок, выпускаемых промышленностью:
Бик 104S (органическое соединение Для водных и орга- [8/39]
с силиконом) Диспербик 163 (катионный ПАВ) норастворимых систем Для систем с раство- [8/39]
Lactimon (анионный ПАВ) рителем Для систем с раство- [8/39]
Solsperse (полимерные ПАВ) рителем Для систем с раство- [31]
Centrol 3FDB (на основе соевого рителем Для систем с раство- [Ю/61]
лецитина) Colorol Е (модифицированный сое- рителем Для эмульсионных [15]
вый лецитин) Tamol 731 (диизобутилен-малеино- красок Для эмульсионных [50]
вый диспергатор) Dispex А40 (полиакрилат аммония) красок Для водных красок [3]
4.9. СИККАТИВЫ
Так как сиккативы являются неотъемлемой частью лакокра-
сочных материалов, их можно было бы не рассматривать как
добавки. Но для удобства они включены в этот раздел.
Реакции окисления и полимеризации при высыхании красок
идут и без катализатора, но в присутствии определенных орга-
нических соединений металлов они сильно ускоряются. Катали-
тическая активность зависит от способности катиона металла
легко окисляться из стабильного состояния с низшей валент-
костью в менее устойчивое состояние с высшей валентностью.
Поэтому все сиккативы многовалентны. На практике используют
смесь металлических сиккативов. Хотя в качестве сиккативов
в способных к автоокислению водных красках можно с опреде-
ленной эффективностью использовать неорганические соли,
например нитрат кобальта, до сих пор подавляющее их число
представляет собой металлические производные органических
кислот («мыла»), которые эмульгируются в водных системах.
В частности, раньше для получения так называемых линоле-
атов и резинатов свинца проводили взаимодействие оксида свинца
с жирными кислотами льняного масла или с канифолью. Они
были достаточно эффективны, но не стабильны при хранении
и с непостоянным содержанием металла. Наибольшей популяр-
ностью пользуются устойчивые металлические сиккативы с по-
стоянным составом, обычно получаемые на основе октановой
кислоты, из изомеров которой наиболее доступна 2-этилгекса-
новая кислота. Кислотный радикал обеспечивает растворимость,
а катион металла является наиболее важной частью сиккатива.
Из исследованных свыше 40 переходных металлов примерно около
десяти оказались достаточно активными.
Кобальт до сих пор наиболее эффективен в сиккативах. Исполь-
зование одного этого сиккатива приводит к эффективному вы-
сыханию с поверхности, оставляя текучим подпленочный слой,
что вызывает сморщивание толстых пленок. Самостоятельно его
применяют редко, например в алюминийсодержащих красках
холодной сушки (когда наносят очень тонкие пленки и когда
свинец привел бы к потускнению алюминия) и в некоторых крас-
ках горячей сушки.
Свинец — традиционно наиболее широко используемый металл
с тех пор, как было замечено, что свинцовые пигменты способ-
ствуют высыханию. Сиккатив объемного действия. По-прежнему
используется в сочетании с другими металлами, если только
требования по токсичности и возможность образования пятен
сульфидов на покрытии не исключают его применение.
Марганец — второй металл не из группы кобальта очень ак-
тивный в сиккативах. Ускоряет высыхание с поверхности и в объ-
еме. Его недостаток обусловлен цветом, так как светло-коричне-
вый двухвалентный марганец переходит преимущественно в
темно-коричневый трехвалентный марганец. Кроме того, если для
большинства других сиккативов введение их избыточного коли-
чества не приносит никакой пользы, то в случае марганцевых
Скорость высыхания заметно уменьшается при отклонении содер-
жания сиккатива от оптимального.
Железосодержащие сиккативы являются устаревшими и имеют
плохой цвет. Их применяли в дешевых эмалях темного цвета
горячей сушки. Практически не используют в красках естествен-
ной сушки.
Цинк используют в виде мыл или пигмента — оксида цинка,
вводимых в количествах, типичных для сиккативов. Он замед-
ляет начальную скорость высыхания с поверхности, но ускоряет
объемное высыхание. Использование цинка приводит к большой
твердости конечной пленки.
Кальций в качестве основного сиккатива малоэффективен,
Как дополнительный сиккатив к свинцово-кобальтовым системам
он препятствует осаждению свинца при хранении и способствует
ускорению сушки при низкой температуре. Несколько лет тому
назад применение кальциевого сиккатива помогло решить пробле-
му, связанную с помутнением высохших алкидных пленок.
Церий был предложен несколько лет тому назад взамен свинца,
Для цериевых сиккативов характерно объемное действие. Хотя
они и эффективны, но заметное пожелтение пленки ограничивает
их применение.
Ванадий. Замечания те же, что и для церия. Сиккатив вызы-
вает изменение цвета пленки и характеризуется потерей актив-
ности при хранении. Практически не используется.
Барий, так же как и кальций, образует вспомогательные сик-
кативы. В настоящее время отсутствуют законодательные ограни-
чения, связанные с токсичностью, однако допустимые количества'
бария в красках для игрушек и т. п. жестко контролируются.
Барий применяют в некоторых системах сиккативов, не содержа-
щих свинец. Использование в будущем пока неясно.
Цирконий широко рекомендуют вместо свинца в сиккативах
объемного действия, хотя еще и не выяснены причины его актив-;
ности. Для облегчения замены свинцовых сиккативов цирконие-
вые выпускают в сочетании с кобальтовыми. Они удовлетворяют
требованиям по токсичности.
Алюминий. Через каждые несколько лет появляются сообще-
ния о нетоксичных, с необычными свойствами алюминиевых!
сиккативах объемного действия для красок, не содержащих сви-
нец. Однако заменить свинец на алюминий не очень просто. Для
этих сиккативов необходимо обычно специально готовить связую-
щее. Их достоинства — хороший цвет, хорошее ускорение высы-
хания в объеме, твердость пленок. .
4.10. РЕГУЛЯТОРЫ ЭЛЕКТРИЧЕСКИХ СВОЙСТВ
Необходимость модификации электрических свойств жидких
красок может возникнуть по следующим основным причинам:
а) чтобы избежать накопления статического электричества и свя-
занного с этим появления искры и возгорания при хранении и
транспортировке (для этого в углеводородные растворители
могут вводиться антистатические добавки, повышающие прово-
димость) ; б) для уменьшения электрического сопротивления
красок с неполярными растворителями с целью обеспечения
хорошей наносимости при электростатическом распылении.
В первом случае обычно во все углеводородные растворители
добавляют 0,3% антистатической добавки, например ASA3 [54].
Для электростатическго распыления выбирают растворители
с оптимальным удельным сопротивлением. Если необходимо
увеличить проводимость, вводят небольшие количества таких
веществ, как Бик ES80 [8/39],Ransprep [48] или Lankrostat [38].
Желательно применять также простые эфиры гликолей вслед-
ствие их хорошей растворяющей способности, полярности, высо-
кой проводимости, хорошей способности к электростатическому
распылению. Подобные растворители (Exxates 600 и 700) одно-
временно характеризуются высоким удельным сопротивлением.
4.11. ИНГИБИТОРЫ КОРРОЗИИ
При нанесении водных лакокрасочных материалов, например
водоразбавляемых грунтовок непосредственно на черные ме-
таллы, пленка может быть испорчена ржавчиной, что обычно
проявляется в виде скоплений светло-коричневых пятен. Эту так
называемую «флеш-коррозию» можно предотвратить использо-
ванием ингибиторов. Обычно применяют те же добавки, что и для
защиты от коррозии тары при хранении, но при немного большей
концентрации:
Prestantil 448 [36/15]; Ингибитор коррозии 179 [1/35].
4.12. ДОБАВКИ, ПРЕДОТВРАЩАЮЩИЕ ФЛОТАЦИЮ И ФЛОКУЛЯЦИЮ
Большинство цветных красок немного изменяют свой цвет
при сушке вследствие миграции одного из пигментов к поверх-
ности. Проблем бы почти не возникало, если бы это происходило
всегда в одинаковой степени.
Термин «floating» обычно применяют для описания явления
образования в покрытии цветных полос или крапчатости, в то
время как «flooding» означает заметное изменение исходного
однородного цвета, который становится более светлым в местах
механического воздействия (например, на частично высохшую
пленку). Вследствие высокой вязкости затвердевающей пленки
исходный цвет в этих местах больше не восстанавливается.
Оба эти эффекта проявляются в случае присутствия в рецеп-
туре двух или более пигментов. Считают, что второй эффект пред-
ставляет собой более яркую разновидность первого. Уменьшить
оба эти эффекта можно путем тщательного совместного диспер-
гирования пигментов, чтобы в случае флокуляции она происхо-
дила для всех пигментов только совместно. С этой целью часто
полезно на стадии диспергирования вводить мелкодисперсные
наполнители, такие как оксид алюминия [17] и модифицирован-
ные осажденные карбонаты кальция. Имеются данные, что Дис-
пербик 160 [8/39] выравнивает поверхностные электрические
заряды органических пигментов, хотя требуемая степень вырав-
нивания и выше, чем обычно ожидаемая от введения добавки.
С другой стороны, может оказаться полезным применение веществ
типа Anti-terra [8/39], вызывающих небольшую регулируемую
флокуляцию.
В качестве регулирующих добавок в колеровочные краски
используют различные силиконсодержащие продукты, например
Dow Corning РАЗ [19/34] или несиликоновые добавки, такие
как продукт Henkel’s 963 [27].
Важное значение имеет правильный выбор добавок для кон-
кретной композиции. Авторы предложили простой метод проверки,
позволяющий оценить как эффективность, так и необходимое
количество подобных добавок. Он заключается в наблюдении
за отсутствием образования ячеек Бенарда на небольших пор-
циях краскй“после ТГх~падения на стеклянную плиту.
4.13. КОНСЕРВАНТЫ ДЛЯ ХРАНЕНИЯ КРАСКИ В ТАРЕ
*
Латексные и другие водные краски особенно подвержены
порче под воздействием микроорганизмов. Важно не допускать
загрязнений еще при их производстве, однако все равно для пред-
отвращения их порчи необходимо вводить антисептики. Сейчас
уже почти не встречаются случаи, когда приходится сталкиваться
с дурно пахнущими продуктами разложения клея и казеина, од-
нако заражение бактериями по-прежнему может вызвать газо-
образование в емкости для хранения, а ферментативное разру-
шение целлюлозных загустителей — уменьшение вязкости.
Лучше всего биоцид добавлять в первую порцию загружаемой
воды, тем самым обеспечивая его присутствие уже на началь-
ной стадии производства. Поэтому такие добавки должны хорошо
растворяться в воде.
Хотя ртуть- и оловоорганические соединения эффективны и
применяются до сих пор, наметился определенный поворот в сто-
рону не содержащих металлов органических биоцидов вследствие
возрастания требований к безвредности. Определяющее значе-
ние имеет область использования лакокрасочного материала.
При использовании в быту важное значение имеют низкое раздра-
жающее действие и низкая светочувствительность. В случае свет-
лых тонов необходимы биоциды, не вызывающие пожелтения.
Из выпускаемых продуктов хорошо зарекомендовали себя
производные бензизотиазолинона (например, типа Proxel [32]),
замещенные оксазолидины (например, Nuosept 95 [21]) и смеси
гетероциклов с веществами, выделяющими формальдегид, или
без них (например, Mergal K6N и Mergal К7 [29]).
4.14. БИОЦИДЫ
В соответствующих условиях большинство лакокрасочных
покрытий способствует развитию плесени, вызывающей появле-
ние характерных черных пятен в местах соединения стен и потолка
в ванных комнатах, а также с наружной стороны зданий. Порча
наружных поверхностей может вызываться зеленоватыми мор-
скими водорослями. Добавлением биоцидов в краски можно за-
щитить покрытия от развития плесени, но' от них нельзя ожидать
слишком многого, если поверхность постоянно обрабатывается
питательными веществами. Авторы встречались с проблемами
сохранения чистоты окрашенных стен, которые были обильно
покрыты сиропом на сахарном заводе в Барбадосе.
Обычно содержание биоцидов не превышает 5% и поэтому
часто их рассматривают как добавки.
В отличие от хорошо растворимых в воде биоцидов, приме-
няемых в эмульсионных красках в качестве консервантов для
хранения в таре, биоциды, применяемые для защиты покрытий,
должны практически не растворяться в воде. В противном слу-
чае они будут вымываться водой из пленки, что снизит эффек-
тивность защиты.
Запатентованные средства часто представляют собой смеси
биоцидов для обеспечения наилучшей защиты от широкого круга
грибков и морских водорослей. Испытания на эффективность
действия против грибков и водорослей не простые, однако произ-
водители биоцидов помогают правильно выбрать тип и количество
биоцида для конкретного лакокрасочного материала и условий
эксплуатации покрытия.
Выпускаемые биоциды:
Acticide АРА [58]; Merga 1 588 [29];
Parmetol А23 [56]; Proxel [32].
Nopcocide N40 [18];
4.15. ИНСЕКТИЦИДЫ
Представляется интересным использовать инсектициды в по-
крытиях против бытовых мух. При этом нужно решить две проб-
лемы. Во-первых, для обеспечения доступности активного компо-
нента к севшему насекомому концентрация его должна быть
высокой, а композиция обеспечивать получение матового покры-
тия. Вторая проблема, связанная с конечным эффектом, более
серьезна. Дело в том, что там, где много летающих насекомых,
люди предпочитают, чтобы они летали, а не падали мертвыми
в их пищу. Поэтому такие краски применяют в специальных слу-
чаях, например для уничтожения тараканов в корабельных трю-
мах. Нельзя забывать и об инсектицидах, применяемых в пропи-
точных композициях для защиты древесины. Для этого приме-
няют органические соединения металлов, например трибутил-
оловооксид [2], октоат цинка [21, 39] и нафтенат меди [16].
В качестве инсектицидов для красок предложены также хло-
рированные ароматические соединения, например, 6-хлорэпокси-
гидроксинафталин и 1,1-дихлор-2,2-бис-(и-хлорфенил) этан.
4.16. ДОБАВКИ, ПОВЫШАЮЩИЕ БЕЛИЗНУ ПОКРЫТИИ
Эти вещества поглощают УФ-излучение, а излучают энергию
в области видимых длин волн. Если излучение попадает в сине-
фиолетовую область, добавки повышают белизну и устраняют
склонность к пожелтению. Хотя такие добавки широко исполь-
зуют в производстве бумаги и моющих средств, в технологии
покрытий они мало применяются из-за короткого срока их дей-
ствия и стоимости. Примером таких веществ является Uvitex
ОВ [13].
Такие добавки не следует путать с применением, например
-фиолетового пигмента, который вводят для устранения желтизны.
В этом случае может появиться нежелательный серый оттенок,
что часто исключает его применение.
4.17. ДЕЗОДОРАНТЫ
Большинство красок при высыхании пахнет. Умеренный запах
латексных красок не вызывает особого беспокойства, но запах
органорастворимых материалов значительно сильнее. Он объясня-
ется, в первую очередь, выделением растворителей, а в случае
красок, высыхающих вследствие окислительных процессов, по-
является дополнительный устойчивый запах из-за химических
реакций. Добавку, которая бы полностью устранила эти запахи,
ищут до сих пор.
В общем случае необходимо избегать применения в исходной
рецептуре сильно пахнущих компонентов, например выбирая
растворители с меньшим запахом. Вместе с тем можно смягчить
или изменить запах из-за указанных двух причин, применяя
промышленные пахучие вещества. Однако по опыту авторов боль-
шинство людей предпочитает запах первоначальной краски смеси
запахов, которая получается при введении в нее дезодорантов.
В конце главы перечислены производители дезодорантов под
номерами [7, 24, 47, 62].
4.18. ДОБАВКИ, ПОГЛОЩАЮЩИЕ УФ-ИЗЛУЧЕНИЕ
Многие пигменты выцветают, а связующие деструктируют
под воздействием облучений, особенно ультрафиолетового. Много
лет тому назад было установлено, что нанесение слоя лака замед-
ляет выцветание непрочных красок, но, к сожалению, непигмен-
тированные лаковые пленки сами быстро разрушаются. Недавно
было показано, что поглотители УФ-излучения (подобные исполь-
зуемым в косметических кремах против загара) повышают срок
службы таких защитных лаковых покрытий. Еще более успеш-
ным оказалось сочетание светостабилизаторов с УФ-поглотите-
лями. При этом достигается защита на двух стадиях. УФ-погло-
титель (около 1%) превращает вредное коротковолновое излу-
чение в тепловую энергию, а светостабилизатор (стерически
затрудненный амин) захватывает образующиеся свободные
радикалы, которые являются причиной деструкции пленки. Эта
технология позволила использовать систему наружного покрытия
в автомобилестроении типа «основное покрытие плюс лаковое
покрытие» и решить проблемы подпленочного меления, расслоения
и растрескивания верхнего лакового покрытия.
Ниже даны некоторые из используемых добавок:
Tinuvin 900
Tinuvin 901
Sanduvor 3206
Sanduvor 3046
УФ-поглотитель [13]
Затрудненный амин [13]
УФ-поглотитель [51]
Затрудненный амин [51]
лава
ФИЗИЧЕСКАЯ ХИМИЯ ДИСПЕРСИИ
А. Дороукковский
5.1. ВВЕДЕНИЕ
Физическая химия дисперсий охватывает как производство
полимерных дисперсий, например латексов, так и получение пиг-
ментных суспензий. При получении латексов важное значение
имеет кинетика полимеризации, закономерности которой не при-
менимы к диспергированию пигментов. С другой стороны, при
диспергировании пигментов могут рассматриваться определен-
ные межфазные взаимодействия, неприменимые к процессам
образования латексов. Однако, с коллоидной точки зрения, после
образованггя дисперсии физическая химия как полимерных частиц,
так и пигментных дисперсий одна и та же.
В этой главе основное внимание уделено рассмотрению физи-
ческой химии дисперсий, в которых дисперсная фаза уже полу-
чена ранее, например, как в случае с пигментами, хотя анало-
гичные рассуждения применимы и для полимерных частичек, если
они составляют дисперсную фазу.
Для простоты будем рассматривать краску как коллоидную
дисперсию пигмента (дисперсная фаза) в растворе полимера
(непрерывная фаза). В эмульсионных красках как полимер, так
и пигмент находятся в дисперсной фазе. Хотя химическое строе-
ние полимера имеет важное значение для свойств красок и покры-
тий, не менее важным является и качество диспергирования
пигмента в полимере.
Можно сказать, что практически большинство проблем, свя-
занных с производством и применением красок, обусловлено
состоянием пигментной дисперсии. Диспергирование пигментов
влияет на оптические свойства, например цвет [ 1 ], на розлив [2],
долговечность [3], укрывистость [4], блеск [5], стабильность
при хранении [6].
Для получения «хорошей» дисперсии коллоидных частиц из
сухого порошка необходимо осуществить ряд процессов. Их услов-
но можно подразделить на две отдельные операции, которые
в действительности могут протекать одновременно: 1) погруже-
ние и смачивание пигмента; 2) распределение и коллоидная
стабилизация пигмента. За этими простыми названиями опера-
ций может скрываться много сложных процессов, требующих
детального рассмотрения. Пример «простого» вопроса: «Какой
растворитель — ароматический или алифатический — лучше при-
менять для диспергирования диоксида титана в растворе алкида?»
5.2. ПОГРУЖЕНИЕ И СМАЧИВАНИЕ ПИГМЕНТА
Предположим, что мы имеем отдельную твердую частицу пиг-
мента, погружающуюся в жидкость. Это можно представить
схематически в виде трехстадийного процесса:
Стадия 1
Частица на воздухе
Стадия 3
Заполнение полости
частицей
Пар
Жидкость
Стадия 2
Создание полости
в жидкости
Пар
Жидкость
Если поверхность частицы равна А, а свободная энергия на
единицу поверхности утп, то общую энергию этих трех стадий
можно представить следующим образом (рис. 5.1):
Стадия 1: Поверхностная энергия частицы равна Дутп.
Стадия 2: Энергия частицы в паровой фазе плюс работа,
затраченная на создание пустоты в жидкости,
объем и поверхность которой равны таковым
у частицы пигмента:
^Ттп+^Тжп-
Стадия 3: Работа, затраченная на заполнение полости в жид-
кости частицей равна:
^Ттж—^Ттп—^Тжп
Тогда изменение общей энергии при погружении частицы
будет равно сумме стадий 2-|—3—1:
Дб=Дутж—^Ттп- (5.1)
Из уравнения Юнга следует Ттп = Тжт+Тжп cos 6. После под-
становки в уравнение 5.1 изменение энергии будет равно:
Дб= —Дужп cos 0. (5.2)
Из уравнения Юнга косинус краевого угла смачивания (0) равен:
cos 6 =
Ттп~Ттж
Тжп
(5-3)
Тогда, при условии, что 6 <90°, уменьшение ужп приведет к умень-
шению 6 и улучшит смачивание. Следовательно, алифатический
Рис. 5.1. Графическое изображение удель-
ной свободной энергии на границе раздела
пар/жидкость/твердое тело
углеводород более предпочтителен, чем ароматические раство-
рители, так как ужп алифатического Сужг1 ароматического.
Если, однако, добавить ПАВ, оно адсорбируется на межфазной
границе с воздухом, что приводит к уменьшению ужп, а при ад-
сорбции на поверхности частицы — уменьшению утж. Оба эти
эффекта улучшают смачивание.
Но, если 6 = 0, как в случае поверхности с высокой энергией
(например, поверхности Т1О2), лучше, чтобы ужп была максималь-
ной, т. е. в этом случае лучше применять ароматический раство-
ритель, а не алифатический.
Проницаемость агломератов. Если рассматривать пустоты
между порошкообразными частицами как простые капилляры
с кажущимся радиусом г, тогда поверхностное давление, необхо
димое для проникновения жидкости в капилляр, равно:
р- -2УжпсозО (5.4)
Следовательно, проникновение жидкости произойдет само-
произвольно (гравитационные эффекты не учитываются), только
если 0<90° и если давление внутри капилляров не возрастет
настолько, что воспрепятствует вхождению жидкости.
Таким образом, для облегчения проникновения жидкости в
агломераты желательно сделать ужп максимальной и уменьшить 0.
Но, поскольку изменения ужп и 0 взаимосвязаны, это условие
трудно выполнить. Введение ПАВ уменьшает как ужП, так и 0,
особенно в водных средах. Поэтому оценить, какой из этих эффек-
тов будет' доминирующим, лучше всего опытным путем.
Представленная выше картина с использованием поверхност-
ной энергии твердого тела, находящегося в равновесии с паром
и. жидкостью, значительно упрощена. Хертжес и Витрот [7]
исследовали смачивание агломератов и показали, что только при
0 = 0 возможно добиться полного смачивания порошкообразных
агломератов.
Уравнение скорости проникновения жидкости внутрь агло
мерата было получено Вошбурном [8]:
dl rVxnC0S 6
—'— -------- > (b.c>)
dt 4 т]
где dl/dt скорость проникновения жидкости с вязкостью т) в капилляр с радиусом г
и .длиной I.
Для плотного слоя порошка принято пользоваться «эффектов
ным радиусом пор» или «фактором кривизны». Поэтому г/4 можно
заменить на фактор К, который считают постоянным для конкрет-
ной упаковки частиц. Тогда уравнение 5.5 будет иметь вид:
р KfoxncosO
л
(5.5а)
Из уравнения (5.5а) видно, что для облегчения проницаемости
порошка необходимы: максимальное произведение ужпсозб,
минимальная вязкость г] и максимальное значение К, например,
рыхло упакованные агломераты пигмента.
Уравнение Вошбурна описывает систему, в которой стенки
трубки покрыты двойной пленкой, т. е. поверхностная энергия
пленки такая же, как и энергия поверхности в объеме вещества.
Гуд [9] распространил уравнение Вошбурна для случая, когда
поверхность свободна от адсорбированного пара:
/2 = К/
' Тжп cos04-77e — П(|=(»'
ч
(5.56)
где Пе — давление растекания адсорбированной пленки, находящейся в равно-
весии с насыщенным паром, а /7р=о) давление растекания для пленки в нулевой
момент времени.
Вернемся к вопросу, в алифатическом или ароматическом
углеводороде лучше диспергировать TiOs. Ходя в случае с TiOs
трудно принять определенную удельную энергию поверхности из-за
различного строения поверхностного слоя, состоящего из сме-
шанных гидроксидов алюминия, кремния и титана, тем не менее
она высока и для простоты может быть приравнена к удельной
поверхностной энергии воды [10]. Поэтому cos0=1 как для аро-
матического, так и алифатического углеводородов, если только
одна из этих жидкостей не является аутофобной [11]. Следо
Таблица 5.1. Адгезионное напряжение (Ужпс05®) и скорость измельчения
рутильного диоксида титана на лабораторной шаровой мельнице
Среда Адгезионное напряжение (?жпС05°) Время достижения степени диспергирования, ч
3* (по Хегману) 5* (по Хегману)
5% изомеризованного каучука 21 9 0,9 2,0
5% алкида 16,3 1,0 1,7
10% алкида 14,4 1,7 2,5
10% изомеризованного каучука 12,3 2.0 4,0
* Примечание. Степень диспергирования по Хегману; 0—грубая или плохая
дисперсия, 8 — тонкая или наилучшая дисперсия.
вательно, чтобы добиться максимума ужпсо5 0, лучше исполь-
зовать ароматический углеводород.
В табл. 5.1 приведены данные, полученные Кроулом [12],
по влиянию ужп cos 0 на время перетира.
S3. ДЕАГЛОМЕРАЦИЯ [МЕХАНИЧЕСКОЕ РАЗРУШЕНИЕ АГЛОМЕРАТОВ]
Пигментная форма диоксида титана представляет собой по-
рошок, состоящий из рыхлых агломератов с диаметром от 30
до 50 мкм. Нанесение покрытия на поверхность пигмента при его
производстве оказывает большое влияние на уменьшение коге-
зионных сил порошка и за счет этого облегчает процесс механи-
ческого разъединения (или дезагрегации) [13]. Трудно определить
тот момент перетира или диспергирования, когда рыхлые агло-
мераты полностью разрушатся до более мелких частиц после
смачивания Ьсей доступной поверхности; поэтому обычно исполь-
зуют эмпирический контроль за процессом, как описано в гл. 7.
— Хотя -имеется -много публикаций, пытающихся объяснить
модификацию поверхности пигментов при их производстве, на-
пример поверхности TiO2 [14, 15], тем не менее в этой области
много секретов и не все понятно. Химический состав пигмента
и его поверхностного покрытия обычно представляют в виде со-
держания А120з, SiO2, ZnO, TiO2 и т. д., даже если покрытие со-
стоит из смешанных гидроксидов, которые очевидно более пра-
вильно представлять в виде МХ(ОН)У, где М может представлять
собой смесь Al, Si, Ti, Zn и т. д. Поверхность может быть обра-
ботана полиолами для облегчения последующего производства
красок. Основная цель обработки рутильного TiO2 — дезактиви-
ровать поверхность пигмента, которая в противном случае будет
ускорять деструкцию пленкообразователя в атмосферных усло-
виях. Кроме того, она облегчает процесс диспергирования.
Сущность перетира при производстве пигментных паст в дей-
ствительности заключается не в измельчении, а в диспергирова-
нии пигмента до размера первичных частиц, полученных на стадии
производства пигмента. Некоторые из «первичных» частиц со-
стоят из сростков кристаллов ТЮ2, образовавшихся на стадии
модификации поверхности пигмента при его получении, и остаются
неизменными после завершения стадии перетира, о чем свидетель-
ствует анализ размера частиц до и после введения пигмента
в краску (см. рис. 5.2). Среднечисловой размер частик (с!ЙГ =
==0,16 мкм) и распределение частиц по размерам (<т= 1,52) до
перетира оказались теми же самыми, что и полученные с помощью
седиментационного анализа после перетира в шаровой мельнице
(для перевода среднемассового размера частиц в среднечисловой
использовали соответствующее уравнение Хэтч-Чоэта; d,,, • =
= 0,16 мкм; с/А„„ = 0,3 мкм при о= 1,52, что близко к измеренным
значениям размера dgm=0,36 мкм, 0=1,5).
a)
Рис. 5.2. Анализ размера частиц TiO2 (логарифмические зависимости вероятных
размеров частиц):
а) но данным электронной микроскопии до диспергирования; </„с = 0,16 мкм, о = 1,52, по-
этому d(Iln = 0,3; 1=1 — при подсчете всех частиц (включая очевидные агрегаты); • — при
подсчете только единичных кристаллов (при условии что видно более половины периметра)
б) поданным рентгеновского седиментационного анализа после диспергирования; </g,n =
= 0,36, а=1,5
5.4. ДИСПЕРГИРОВАНИЕ — КОЛЛОИДНАЯ СТАБИЛИЗАЦИЯ
Для получения устойчивой коллоидной дисперсии недоста-
точно только смачивания частиц веществом непрерывной фазы.
Важно учитывать, что в пигментных дисперсиях всегда имеются
силы притяжения между частицами. Это силы Лондона, Ван-дер-
Ваальса или (поверхностные). Причиной появления этих сил явля-
ются силы притяжения, действующие между атомами, из которых
состоят частицы. Полярные вещества оказывают электростатиче-
ские воздействия на другие диполи (силы Киисома [16]), а поляр-
ные молекулы могут притягивать неполярные за счет наведенных
диполей (силы Дебая [17]). Существование притяжения между
неполярными атомами или молекулами не поддавалось объясне-
нию до тех пор, пока не было высказано предположение, что
в электронном облаке, окружающем ядро, могут наблюдаться
локальные флуктуации плотности заряда. Это приводит к возник-
новению дипольного момента, частота флуктуаций которого
совпадает с частотой флуктуаций заряда. Если рядом имеется
другой атом, то он поляризуется и взаимодействует с первым.
Лондон [18] показал возможность использования такого под-
хода для расчета взаимодействия между двумя одинаковыми
и различными атомами. Анализ Лондона основан на представ-
лении о мгновенных диполях, период колебания которых состав-
ляет от 10“15 до 10~16 сек. Если два атома удалены друг от друга
на расстояние больше определенной величины, то за промежуток
времени, пока электрическое поле от одного диполя вызовет поля-
ризацию другого атома, в первом уже произойдут изменения.
Между двумя данными диполями будет наблюдаться плохая
корреляция; в таком случае между атомами действуют «запазды-
вающие» силы Ван-дер-Ваальса.
В общем случае можно ожидать проявления сильных не-
запаздывающих сил на расстояниях меньших 10 нм и запазды-
вающих сил на больших расстояниях.
5.4.1. Силы, действующие между макроскопическими телами
Силы, действующие между двумя телами в дисперсиях, обычно
называют поверхностными силами. Они обусловлены не только
атомами на поверхности вещества, но и атомами, находящимися
в его объеме. Гамакер [19] рассчитал эти силы. Их можно пред-
ставить следующим образом:
Еа= -Л/48л ^l/d2+l/(d+<5)2-2/(d+y)2] ! (5.6)
если d»6, то VA=— б2Д/32лД4; (5.6а)
d<6 VA= —Д/48л[1/<72—7/62]; (5 66)
d<<5 VA=—Д/48лД2, (5.6в)
yz___энергия притяжения для двух пластин толщиной б на расстоянии друг
от друга равном 2d; А — константа Гамакера для вещества, из которого состоят
пластины.
Гамакер [20] также показал, что энергия притяжения (E.t)
между двумя одинаковыми сферами радиуса а, расстояние.между
поверхностями которых d, равна:
УА= -Д/6[2/(52-4) +2/S2 + ln((S2-4)/S2)], (5.7)
где 5 = 2 + d /а.
Если d^La, то
VA ~ — Aa/I2d,
и для сфер с радиусами а\, а2:
У _ —Аща?
А 6d (ai +а2)
Можно рассчитать силы притяжения, зная размер
среду, расстояние между ними, и выразить результаты
(5.7а) '
(5.76)
частиц,
в виде
энергии притяжения (VA). Однако константа Гамакера А полу-
чена для вакуума. При рассмотрении энергии притяжения между
двумя частицами в жидкости необходимо ее модифицировать
с учетом окружающей среды. Гамакер показал, что частицы веще-
ства Ai в среде вещества А-> имеют общую константу Гамакера
A—AiA-^2 — 2~у/АлА2. Если имеются два разных вещества-—
1 и 2 в среде 3, то модифицированная константа Гамакера равна:
А132 = (А |{2-Л^2) (Atf-Atf). (5.8)
• Перечень значений констант Гамакера для обычных веществ
дан в работе [21]. Грегори [22] показал, как на основании про-
стых экспериментальных измерений можно приблизительно рас-
считать константу Гамакера.
.Следует обратить внимание на то, что эти силы всегда явля-
ются силами притяжения. При необходимости получения боль-
шей точности можно вводить поправки, например уточнение
Волда с учетом адсорбционных слоев [22] или Казимира и Пол-
дера с учетом запаздывания сил притяжения. [24, 25]. Но простое
рассмотрение опубликованных значений констант Гамакера пока-
зывает, что они могут изменяться на порядок и поэтому редко
оправдана необходимость использования усложняющих поправок
с целью учета непостоянных трудно уловимых эффектов, таких
как в работах Волда [23] и Винсента [26].
Хотя между двумя одинаковыми частицами всегда действует
сила притяжения, Виссер [27] отмечает, что при определенных
условиях можно получить отрицательную константу Гамакера
в трехкомпонентной системе, когда Ли <Лзз<Л22 или Ли >Лзз>
>Л22, где Лзз представляет собой индивидуальную константу
Гамакера данной среды. Важно отметить, что и в этом случае
одинаковые частицы Ли, находящиеся в среде Л33, также как
и Ач2 в среде Л33 по-прежнему будут притягиваться, но частицы
Ли и Л22 — отталкиваться друг от друга. Примером подобного
типа отсутствия ассоциации, как полагают, могут быть частицы
политетрафторэтилена и графита в воде.
Чтобы получить «коллоидную дисперсию» необходимо создать
условия, чтобы энергия отталкивания (Vr) между частицами
была больше энергии притяжения и в результате сложения этих
двух энергий оставалась бы значительная результирующая энер-
гия отталкивания. Поскольку энергии отталкивания и притяже-
ния зависят от расстояния между частицами, важно знать рас-
пределение частиц, особенно в красках, которые с коллоидной
точки зрения рассматриваются как «концентрированные» си-
стемы. Можно показать, что в случае плотной упаковки сфери-
ческих частиц, отношение расстояний между поверхностями
частиц (S) к расстоянию между центрами частиц (с) связано
с общим объемом зависимостью:
S/c= 1 — (ОКП/0,74)|/3,
(5.9)
где ОКП—объемная концентрация пигмента, %.
Ниже приведены значения ОКП и расстояния между поверх-
ностями сферических частиц:
Для монодисперсных сферических частиц любого размера
среднее расстояние S равно одному диаметру частицы при ОКП
равном 9,25%. Поэтому среднее расстояние между частицами
для ТЮг при такой концентрации равно приблизительно 200 нм,
а для меньших частиц при той же ОКП это расстояние будет про-
порционально меньше.
Если сравнить энергии отталкивания и притяжения, помня,
что для частиц, находящихся в Броуновском движении, средняя
энергия перемещений при комнатной температуре равна 3/2 kT,
то для их стабилизации необходимо иметь энергетический барьер
(Емакс), значительно превышающий эту величину (рис. 5.3).
На кривой общей энергии может наблюдаться «вторичный
минимум», соответствующий энергии, которая характеризует
возможность существования слабой флокуляции. Она называ-
ется «слабой флокуляцией» (Кмин) или «вторичной минималь-
ной флокуляцией» в отличие от более сильной флокуляции,
которая происходит при дальнейшем сближении частиц и иногда
называется «первичной флокуляцией». Следует заметить, что
иногда в литературе [28] пользуются терминологией, отличной
Рис. 5.3. Типичная зависимость общей энергии от расстояния между частицами,
показывающая наличие «вторичного минимума» (VMll„ флокуляции)
от используемой в лакокрасочной промышленности; например,
термин «коагуляция» в литературе употребляется для описания
флокуляции, в то время как обычно этот термин в промышлен-
ности применяют тогда, когда наблюдается необратимая ассо-
циация латексных или эмульсионных частиц.
5.4.2. Стабилизация дисперсий с помощью заряда
Чтобы получить стабильную коллоидную дисперсию, необхо-
димо придать частицам энергию отталкивания, достаточную для
предотвращения флокуляции (при требуемой концентрации ча-
стиц, т. е. ОКП). Эта энергия отталкивания может быть обуслов-
лена кулоновскими силами в соответствии с теорией двойного
электрического слоя или за счет «стерической стабилизации».
Создать заряд на поверхности можно различными способами:
1) предпочтительной адсорбцией ионов, 2) диссоциацией поверх-
ностных групп, 3) изоморфным замещением, 4) адсорбцией
полиэлектролитов, 5) накоплением электронов.
Наиболее распространенным способом получения заряжен-
ной поверхности частицы является предпочтительная адсорбция
ионов, например адсорбция ионов Ag+ или J- на золях гало-
генидов серебра или ионогенных ПАВ на пигментах. Диссоциа-
|ция поверхностных групп наи олее часто используется в случае
латексов, которые приобретают заряд вследствие диссоциации
сульфатных или карбоксильных групп. Теория стабилизации
коллоидных частиц зарядом развивалась довольно долго. Ее куль-
минацией было появление теории двойного электрического слоя,
которая изложена Вервеем и Овербиком в их книге [29]. Этому
вопросу посвящено много прекрасных обзоров [30, 31] и статей,
поэтому нет необходимости здесь подробно рассматривать теорию.
В общих чертах, согласно теории двойного электрического слоя
у плоской поверхности находится прилегающий адсобрционный
|Слой ионов, называемый «слоем Штерна». Плоскость (в слое
Штерна), проходящая через центр гидратированных ионов (при
отсутствии специфической адсорбции), называется «внешней
плоскостью Гельмгольца» (или плоскостью Штерна) и непосред-
ственно связана с гидратированным радиусом адсорбированных
ионов.
Если имеет место и специфическая адсорбция, при которой
обычно происходит предварительная дегидратация иона, пло-
скость, проходящую через центр этих ионов, называют «внутрен-
ней плоскостью Гельмгольца». Необходимо различать эти две
плоскости, поскольку при специфической адсорбции ионы распола-
гаются ближе у поверхности, чем гидратированные ионы в слу-
чае ее отсутствия.
За слоем Штерна (или компактным слоем) располагается
диффузный слой, называемый «слоем Гойя» (или Гойя-Чеп-
мена)
При суммировании зарядов в слое Штерна, диффузном слое и
на поверхности общий заряд равен нулю вследствие электроней-
тральности. Потенциалы непосредственно у твердой поверхности
(Фо), у «внутренней плоскости Гельмгольца» (фД и у «внешней
плоскости Гельмгольца» (фг/) являются абстрактными понятиями,
на что указывает Ликлима [30]; они не являются зета-потенциа-
лом, который можно установить экспериментально и который
определяют как потенциал у «плоскости скольжения» в ионной
атмосфере вокруг частицы.
Однако, хотя зета-потенциал не идентичен потенциалу у пло-
скости Гельмгольца, для простых систем, таких как мицеллы,
монослои ПАВ на частицах и др., существуют веские основания
приравнять их [32].
5.4.3. Зета-потенциалы
Зета-потенциал можно рассчитать на основании эксперимен-
тально найденной электрофоретической подвижности частиц.
Уравнение, используемое для рассчета зета-потенциалов по най-
денным значениям подвижностей, включает отношение радиуса
частицы (а) к толщине двойного слоя (1 //г). Для 10-3 М водного
раствора при 25 °C, содержащего электролит 1:1, 1//г=1Х
X Ю-® см.
Для других типов электролитов и концентраций значения k
изменяются пропорционально, поскольку для водного раствора
симметричного электролита толщина двойного слоя l/fe = 3X
Х10-8/2д/с см, где z— валентность, а с — молярность.
При /га>200 подвижность (ц) связана с зета-потенциалом
(g) уравнением Смолуховского:
ц = е£/4лх),
где е — диэлектрическая постоянная среды, a i) — ее вязкость.
Если подвижность измеряется в мкм-с—‘/вольт'см-1> тогда
для водных растворов £=12.8 мкВ на единицу подвижности.
При ka <0,1 применяют уравнение Хюккеля:
eg
и=----.
блт]
Для 200 < ka<0,1 необходимо пользоваться усовершенство-
ванными вычислениями О’Брайена и Уайта [33] по сравнению
с более ранними зависимостями Вайерсема с сотр. [34], связы-
вающими подвижность и зета-потенциал.
Переход от электрофоретической подвижности к зета-потен-
циалу основан на предположении о том, что частицы приблизи-
тельно сферические. Если же частицы не сферические, то при
небольших значениях ka, величины зета-потенциалов, вычислен-
ные исходя из данных ) подвижности, вызывают большие сомне-
ния. Если частица состоит из совокупности маленьких сфер и если
ka (для малой сферы) велико, то применимо уравнение Смолу-
ховского.
Таким образом, величина зета-потенциала часто несет в себе
некоторую неопределенность и поэтому многие исследователи
предпочитают приводить экспериментально установленную элек-
трофоретическую подвижность.
5.4.4. Измерение электрофоретической подвижности
Электрофоретическую подвижность малых частиц можно
определить путем микроэлектрофореза, измеряя время, необхо-
димое для перемещения малых частиц на определенное расстоя-
ние, или же методом движущейся границы. Метод микроэлектро-
фореза обладает многими преимуществами и наиболее часто
применяется, хотя иногда предпочтительнее использовать метод
движущейся границы [35].
Другой метод, основанный на электроосаждении частиц, был
разработан Франклином [36]. Хотя данные, полученные по этому
методу, хорошо согласуются с результатами микроэлектрофо-
Рис. 5.4. Схематическое изображение простой электрофоретической ячейки, сделанной из предметных стекол и крышек, склеенных
эпоксидной смолой. Электродное отделение сделано из стеклянных трубок с уширениями на конце
реза, метод теоретически неоооснован, так как электроосажде-
ние дисперсии основано на электрокоагуляции и не зависит от
электрофореза [37].
При исследовании подвижности заряженных частиц в микро-
электрофоретической ячейке можно обнаружить, что частицы
передвигаются с сильно отличающимися скоростями, причем не-
которые из них движутся даже в противоположном направлении.
Этот эффект обусловлен явлением электроосмоса в ячейке. Истин-
ную электрофоретическую подвижность можно определить только
в «стационарных условиях», когда электроосмотический поток
компенсируется гидродинамическим потоком. Эти условия за-
висят от формы микроэлектрофоретической ячейки, которая
может быть круглой или прямоугольной. Для быстрых и точных
измерений известно множество разных микроэлектрофоретиче-
ских ячеек [38] и различных усовершенствований, например
установка братьев Рэнк [39], оснащенная лазерным освещением,
вращающейся призмой, видеокамерой и монитором. Подроб-
ности, касающиеся измерения электрофоретической подвижности,
можно найти в работах Смита [32], Джеймса [40] и Хантера [41].
При необходимости можно сделать и очень простую ячейку,
пригодную для наблюдений с помощью обычного микроскопа,
используя стеклянные предметные стекла и стеклянные крышки
двух размеров, как показано на рис. 5.4.
5.4.5. Объяснение коллоидной стабильности
При приближении двух заряженных поверхностей они начи-
нают оказывать друг на друга электростатическое воздействие,
как только их двойные слои начнут перекрываться. В случае оди-
наково заряженных поверхностей результатом такого взаимодей-
ствия будет отталкивание. При качественном рассмотрении не-
обходимо принимать во внимание ряд факторов.
Согласно теории двойного электрического слоя межчастичное
притяжение уменьшается обратно пропорционально расстоянию
между частицами и не зависит от содержания электролита в не-
прерывной фазе, в то время как кулоновское (или электростати-
ческое) отталкивание уменьшается по экспоненциальному закону
в пределах толщины \/k Дебая-Хюккеля ионной атмосферы.
Слой Штерна не участвует непосредственно во взаимодей-
ствии, но его косвенное влияние на величину i|?d очень большое.
В теории двойного слоя двойные слои рассматриваются как чисто
диффузные, но в действительности эта теория применяется к двум
диффузным участкам двух взаимодействующих двойных слоев.
Поверхностный потенциал необходимо заменить на потенциал
Штерна x]?d, который, в свою очередь, для непористых веществ за-
меняется на зета-потенциал.
Типичные толщины двойных слоев при разных концентрациях
электролита приведены ниже:
Молярная концентрация
электролита в воде при 25 °C
icr5
10~4
10“3
ю-2
Ю-1
Толщина двойного слоя \/k
в см•10 8
1000
300
100
30
10
Толщины двойных слоев рассчитываются по уравнению:
(5.10)
ЕгЁоЛТ
где F— константа Фарадея; RT = 2,5-Ю10 эрг при 298 К; егео— относительная
диэлектрическая.; проницаемость и диэлектрическая проницаемость в вакууме;
z;, с, — заряд и число всех ионов в растворе.
В схематическом виде влияние концентрации электролита на
толщину двойного слоя изображено на рис. 5.5.
Рис. 5.5. Схематическое изображение влияния концентрации электролита на тол-
щину двойного слоя l/k
10 2 моль/л
5.4.5.1. Динамика взаимодействия
Очень многое зависит от времени, в течение которого осущест-
вляется взаимодействие частиц. Если скорость сближения велика,
на перестройку двойных слоев остается очень мало или совсем
нет времени. При медленном сближении двойные слои будут все
время находиться в равновесном состоянии.
В последнем случае согласно теории двойного электрического
слоя энергия отталкивания может быть рассчитана с помощью
обратимой термодинамики как изотермическая обратимая работа,
затраченная на то, чтобы перенести частицу из бесконечности
на расстояние И.
Основная идея заключается в том, что при равновесном столк-
новении потенциал должен оставаться постоянным, так как., он
определяется адсорбцией определяющих заряд ионов,- а химиче-
ские потенциалы зафиксированы их значениями в объеме. Такое
взаимодействие принято называть взаимодействием при «постоян-
ном потенциале».
Если взаимодействие протекает настолько быстро, что не успе-
вает произойти перестройка ионной адсорбции/десорбции, такое
взаимодействие будет происходить при «постоянном заряде»;
в этом случае поверхностный потенциал фо не постоянен, а повы-
шается в процессе перекрывания.
После всего сказанного следует заметить, что величины VR
для этих двух типов взаимодействий мало отличаются, но в слу-
чае постоянного заряда обычно получаемые значения VR больше,
чем при постоянном потенциале.
Для двух малых сферических частиц радиуса а при постоян-
ном заряде энергия отталкивания может быть определена согласно
[29] по уравнению:
(5.11)
а при постоянном потенциале:
, -T(S-2) х
l/R=ео^2 /-------) р, (5.12)
где:
₽
-е-2’) (14-я)
е-t(S-2)
2&
S = R/cr, R — расстояние между центрами двух частиц.
1 Юскольку значения у и р находятся всегда в пределах от 0,6
до 1,0, их влиянием во многих случаях можно принебречь [29,
с. 152]. Поэтому уравнение (5.13) является хорошим приближе-
нием для вычисления свободной энергии электростатического
отталкивания (Овербик отмечает, что не существует точного
уравнения [30]):
[4DT ,1-
—— т J In [1 +ехр (— kH)] ~ (5.13)
[^RT I2
—— -у' exp (—kH), (5.13а)
zr J
где у' —тангенс (zFtyo/4RT); е, — относительная диэлектрическая проницаемость
среды; ко — диэлектрическая проницаемость в вакууме; R, Т, F, г — имеют обыч-
ные значения;
= f2^c'2- . (5.136)
e.reoRT
Для случая взаимодействия двух частиц с разными поверх-
ностными потенциалами (гроь Ф02) и разными радиусами сц и а?
(при условии, что fe//>10 и -фо<50 мВ) Хогг с сот. [42] дает
выражение: ,
Рис. 5.6. Зависимость общей энергии взаимодействия (1/о6,ц = РА) от расстоя-
ния между частицами при постоянных значениях размера частиц и содержания
электролита и изменении поверхностного потенциала (использованы уравнения
5.13 и 5.7):
И — 10 —3; a — IC~5 см; I: фо = 2(> мВ; 2: фо = 25 мВ; 3: фр = 30 мВ; 4: фо — 35 мВ; 5: фо —
= 40 мВ
Рис. 5.7. Зависимость общей энергии (Уо6ш= PR+ РА) от расстояния между части-
цами при постоянных значениях содержания электролита и поверхностного по-
тенциала и изменении радиуса частицы (использованы уравнения 5.13 и 5.7):
М= 10“3; тро = 35 мВ; 1: а = 2,5-10“5 см; 2: а=1,0-10-5 см; 3: а = 7,5-10-6 см; 4: а =
= 5•10 _ 6 см
Рис. 5.8. Зависимость общей энергии (Уо61ц= PR+ РА) от расстояния между части-
цами при постоянных значениях размера частиц и поверхностного потенциала и
изменении содержания электролита (использованы уравнения 5.13 и 5.7):
а=10~5 см; 1|)о = 35 мВ; I: 10-1 М; 2: 10 2 М; 3: 10~3 М; 4: IO-4 М
На рис. 5.6, 5.7, 5.8 приведены зависимости Нобщ = И? + НА
от расстояния между поверхностями частиц (Н) при постоянных
значениях размера частиц и содержания электролита, но при
изменении поверхностного потенциала; при постоянных значениях
поверхностного потенциала и содержании электролита, но при
изменении размера частиц; при постоянных значениях размера
частиц и поверхностного потенциала, но при изменении содержа-
ния электролита.
5.4.5.2. Флокуляция электролитом
Из данных рис. 5.8 для + VA видно, что добавление
электролита приведет к флокуляции дисперсии, стабилизирован-
ной зарядом. Это явление было установлено в конце XIX в., когда
Шульц обнаружил, что способность противоиона вызывать фло-
куляцию тем больше, чем выше его валентность. Это было под-
тверждено Харди и в настоящее время известно как правило
Шульца-Харди.
Используя подход Овербика [31], можно сказать, что диспер-
сия будет нестабильной, когда
Voom — Гр-Г Уд=0 (5.14а)
и дУобШ/дН = 0 для того же значения И. Таким образом,
д Уо6ш/дН = д Ук/дН + <5 VfJdH = 0.
Используя уравнения 5.13 и 5.7а, где расстояние между по-
верхностями частиц равно d = H, мы получим:
д УоСт/дН= - k rR - (1 /Н) VA=0,
и поэтому критическому состоянию соответствует kH=\. Заме-
няя Н на k в уравнении 5.14а, получим выражение для критиче-
ского состояния в виде:
feKp»T = 24nereo(4/?7’/f )2е“' (•y')'2/4z2 = 2,03-10~5 (y')2/z2A в единицах СГС,
где е, — диэлектрическая постоянная воды при 25 °C (78,5); s0 = s,74n= 1/4л
(е,= 1 в безразмерных единицах СГС, но не в единицах СИ); е = 2,718;
= 8,314-107 эрг Г"1 моль1; Г = 298 К; F= 2,892• 1014 моль”1.
Для воды при 25 °C (в случае симметричного z — z электро-
лита) можно написать уравнение 5.136 в виде:
Подставив в него /гкрИт, получим выражение для критической
концентрации (Мкрит), вызывающей флокуляцию:
мкрит= 3,7'106 2g (v )4 , моль/л. (5.146)
z А
Если поверхностный потенциал высокий (фо» 100 мВ), тогда
y' = tg (zFtyo/ART) -И. Следовательно, теория двойного электри-
ческого слоя предсказывает, что способность индифферентных
электролитов вызывать флокуляцию должна быть обратно про-
порциональной валентности в шестой степени, т. е. 1 /z6; поэтому
отношения концентраций одно-, двух- и трехвалентных ионов,
вызывающие флокуляцию, будут равны 1/16 : 1/26 : 1/36 или
729 : И : 1-
Для низких поверхностных потенциалов (ф0<25 мВ) можно
приравнять поверхностный потенциал и зета-потенциал и упро-
стить уравнение (5.146):
В этом уравнении g имеет размерность мВ, так как (4RT/F) =
= 102,8 мВ при 25 °C и для малых значений х. Следо-
вательно отношения концентраций одно-, двух- и трехвалентных
ионов, вызывающих коагуляцию, будут равны 1 /12 : 1/22 : 1/З2
или 100 : 25 : 11.
На практике наблюдалось хорошее совпадение с предсказа-
ниями теории двойного слоя. Но поскольку данные при изучении
флокуляции зависят не только от условий равновесия, но и от
кинетических факторов и специфических ионных эффектов, необ-
ходимо осторожно применять этот упрощенный подход, так как
даже ионы с одной и той же валентностью образуют ряды с раз-
личной эффективностью по способности вызывать флокуляцию.
Примером может служить лиотропный ряд (Гофмейстера), в ко-
тором концентрация ионов, вызывающих флокуляцию дисперсии,
уменьшается вследствие гидратации ионов в ряду:
Li+ >Na+ >К+ > Rb+ > Cs+.
Таким образом, присутствие электролитов в' эмульсионных
красках является очень важным фактором. Пигментные пасты,
приготовление которых основано только на сообщении зарядов
частицам, например гексаметафосфатом натрия, очень склонны
к флокуляции; иногда она происходит даже при добавлении ла-
текса, поскольку в нем может содержаться значительное коли-
чество электролита. Обычно после приготовления таких дисперсий
их дополнительно стабилизируют добавлением водорастворимых
коллоидов (полимеров), таких как натриевая соль карбокси-
метил целлюлозы, которую можно было бы назвать только «загу-
стителем», тем не менее она повышает стабильность дисперсий
по отношению к присутствующему электролиту благодаря своему
«защитному коллоидному» действию.
5.4.5.3. Стабилизация зарядом в средах
с низкой диэлектрической постоянной
Стабилизация за счет заряда очень важна в средах с высокими
Диэлектрическими постоянными, например в водных растворах.
В неводных растворах и особенно в неполярных системах с низ-
кими диэлектрическими постоянными отталкивание частиц за счет
заряда обычно имеет небольшое значение [43]. Были попытки
Рис. 5.9. Зависимость общей энергии от расстояния в неводной среде [46]
объяснить коллоидную стабильность в среде с низкой диэлектри-
ческой константой, например в п-ксилоле [44], с использованием
представлений о заряде.
Осмонд [45] отмечает, что применение в этом случае теории
двойного слоя со всеми необходимыми поправками, учитываю-
щими неполярную неводную среду, сводится к рассмотрению
простого кулоновского отталкивания между двумя заряженными
сферами в инертном диэлектрике (игнорируется существование
ионов в растворе).
Рассмотрение расчетов Ликлема [46] взаимодействия между
двумя сферическими частицами с радиусом 0,1 мкм, например
частицами ТЮг, в среде с низкой диэлектрической проницаемостью
(рис. 5.9) показывает, что при зета-потенциале частиц 45 мВ и
при 9% ОКП (т. е. среднее расстояние между ними 0,2 мкм) макси-
мальный энергетический барьер, препятствующий сближению
частиц и флокуляции, равен только 4/гТ. При 20% ОКП (среднее
расстояние между частицами 0,1 мкм) он равен всего лишь 2/гТ,
что явно недостаточно.
Таким образом, поскольку зависимость между энергией оттал-
кивания и расстоянием в неполярной среде очень «пологая»
(см. рис. 5.9), концентрированные дисперсии (т. е. расстояние
между частицами мало) трудно стабилизировать зарядом. Но при
очень низкой концентрации, когда межчастичные расстояния
велики, заряд может обеспечить коллоидную стабилизацию. Хотя
электрические взаимодействия и не играют существенную роль
в стабилизации концентрированных неводных дисперсий в средах
с низкими диэлектрическими постоянными, однако они могут
вызвать флокуляцию в таких системах, если смешать две диспер-
сии с противоположным зарядом частиц.
5.5. СТЕРИЧЕСКАЯ (ИЛИ ПОЛИМЕРНАЯ) СТАБИЛИЗАЦИЯ
Другим источником появления энергии отталкивания, необхо-
димой для стабилизации коллоидных частиц как в водных, так
и в неводных (включая неполярные) средах, является «стери-
ческая» или «энтропийная» стабилизация. В 1966 г. Овербик
отмечал, что «теория защитного (энтропийного) действия нахо-
дится по-прежнему в примитивном состоянии» [47]. С тех
пор в понимании стерической стабилизации достигнут большой
прогресс и этому вопросу посвящены подробные обзоры
[48—50].
В работе [51] дан очень хороший обзор современного уровня
представлений в этой области. Важной особенностью стериче-
ской стабилизации является то, что она не представляет собой
обычные стерические препятствия, которые рассматривают в орга-
нической химии, когда конфигурация молекулы препятствует
реакции. В этом случае энергетические изменения обусловлены
потерей энтропии. Ее иногда называют полимерной стабилиза-
цией, но поскольку неполимерные вещества также могут обеспе-
чить стабилизацию, применяют указанное выше название, а также
и ряд других, например, неионная, энтропийная стабилизация.
В простейшем виде стерическую стабилизацию можно наглядно
представить как стабилизацию, обусловленную сольватирован-
ным слоем из олигомерных или полимерных молекул, необра-
тимо присоединенных к поверхности частицы, который образует
«сольватированную» оболочку толщиной 6.
При приближении частиц друг к другу настолько, что сольва-
тированные оболочки начинают перекрываться или перераспре-
делять их сегментальную плотность в зоне перекрывания, проис-
ходит локальное увеличение концентрации полимера (см.
рис. 5.10), что приводит к возникновению осмотического давле-
ния растворителя в этой системе. Следовательно, источник энер-
гии отталкивания эквивалентен неидеальному компоненту свобод-
ной энергии разбавления.
Величину адсорбированного слоя полимера вокруг частицы
можно установить путем измерения вязкости дисперсии при раз-
личных скоростях сдвига [52]. На основании этих измерений
получают определенные представления об эффективности адсор-
бированного слоя, изучая наклон кривой зависимости логарифма
вязкости дисперсии от квадратного корня из обратной величины
скорости сдвига [53]. Чем больше наклон, тем больше степень
флокуляции. Если вязкость не зависит от сдвига, то при конкрет-
ных условиях флокуляция отсутствует.
Рис. 5.10. Схематическое изображе-
ние перекрывания при стерической
стабилизации: а) Оттевилл [76],
б) Дорожковский и Ламбурн [69]
Проверка применимости этого метода для измерения толщины
адсорбционного слоя была сделана путем синтеза ряда олиго-
эфиров с известной длиной цепи, адсорбция которых на основной
поверхности пигмента была возможна только по конечным карбок-
сильным группам. Измеренная «барьерная толщина» оказалась
очень близкой к той, которая была получена при адсорбции
олигоэфиров по концевым группам [54], что подтвердило эффек-
тивность метода.
Однако количество адсорбированного полимера — не един-
ственный фактор, определяющий стабилизацию [55]. Меньшие
адсорбционные слои могут быть более эффективными, чем боль-
шие слои [56], заметно влияет на эффективность стабилизирую-
щего слоя разветвление цепи [54]. Например, очень эффективна
поли-12-гидроксистеариновая кислота, использовавшаяся как
растворимая часть в привитом сополимере [57].
Подобно многим другим, теория стерической стабилизации
постепенно развивалась. Истоки ее возникновения имеют теперь
лишь историческое значение: зародилась ли она в концепции
«защитных коллоидов» выдвинутой Зигмонди [58] в 1901 г. при-
менительно к золям золота, или в ранних попытках рассчитать
эффект уменьшения энтропии при адсорбции молекул, подобных
рассмотренным Макором [59]. Тем не менее, стерическую стаби-
лизацию использовали ремесленники, включая производителей
красок, еще до того, как ученые смогли понять и объяснить это
явление.
По-видимому, наибольший вклад в понимание стерической
стабилизации внес Фишер [60], поскольку он предусматри-
вал важную роль растворителя в стерической стабилизации, кото-
рую не смогли в должной мере оценить даже более поздние
исследователи [61].
В настоящее время происхождение стерической стабилиза-
ции объясняют двумя факторами, которые не обязательно вно-
сят одинаковый вклад: 1) ограничением объема (т. е. конфигу-
рация адсорбированных молекул уменьшается вследствие при-
сутствия ограничительной плоскости VVi?); 2) смешением или
осмосом (т. е. взаимопроникновением или сжатием адсорбиро-
ванных слоев, приводящим к увеличению концентрации поли-
мера VM).
Считают, что вклад этих двух факторов аддитивен, т. е. Vc6lu =
VVR+Вм, хотя долю каждого из них оценить трудно, так как для
этого необходимо знать конфигурацию адсорбированного поли-
мера и, что более важно, распределение плотности сегментов
перпендикулярно поверхности частицы.
Мейер [62] и Хесселинк [63, 64] сделали попытку рассчитать
это распределение сегментов и, следовательно, относительный
вклад каждого из этих двух источников энергии отталкивания.
Однако имеются возражения, что при таком подразделении суще-
ствует опасность получения некорректных данных из-за двойного
подсчета [65].
. Было предпринято несколько неудачных попыток измерения
энергии отталкивания экспериментальным путем [66—71], но
оказалось, что теоретическое рассмотрение и сложные математи-
ческие расчеты, выполненные Майером и Хесселинком, дают более
точные значения энергий отталкивания, чем экспериментальные
данные [69]. Упрощенная оценка энергии отталкивания (с уче-
том только осмотического фактора VRM) лучше коррелирует с экс-
периментальными данными.
Наппер показал в ряде экспериментальных работ [72—74],
что в стерической стабилизации важную роль играет раствори-
тель и в любом случае при рассмотрении стерической стабилиза-
ции следует учитывать рост осмотического фактора. Следова-
тельно, энергию отталкивания при взаимодействии двух сольват-
ных оболочек стабилизированных молекул, можно записать в
простой форме:
р ) р, (5.15)
Vip
где с — концентрация полимера в сольватном барьере; р — плотность полимера;
Vi — парциальный молярный объем растворителя; — энтропийный и энталь-
пийный параметры Флори [75]; V — увеличение концентрации сегментов в виде
толщины сольватного барьера б.
В соответствии с моделью Оттвила и Волкера [76]:
Р=(б —/г/2).(За + 2б + /г/2),
где а — размер частиц; /г — расстояние между поверхностями.
Согласно модели Дорожковского и ЛамОурна |09j:
_ nx2Rt (37?,-х) (2ft,-х) 4 (R3t-r3)+xR, (х-3/?,)
2 (R't-r3)+xR, (x-3R,)
где fti=a + 6; г —а и x=8—h/2.
Можно получить практически более пригодное соотношение,
если в уравнении 5.15 заменить я]?!—kt на я]и (1 —6/Г), где 0 —
температура, определенная Флори [75] для случая, когда второй
переменный коэффициент равен нулю (т. е. имеет место тот
идеальный случай, когда молекулы не оказывают влияния друг
на друга).
Таким образом, сразу становится понятным, что при тета-
температуре (1—0/7') обращается в нуль (0 = 7’), энергия от-
талкивания исчезает, и возникает энергия притяжения, вызываю-
щая флокуляцию, как это было установлено Наппером. Более
ргого, если- -7^<UJ,--to не только -нес.происходит отталкивания, но,
наоборот, энергия отталкивания превращается в энергию притя-
жения.
Необходимо учитывать величину с2, которая соответствует
концентрации полимера в «стерическом барьере». Если эта ве-
личина мала, может возникнуть флокуляция, что в действитель-
ности и наблюдалось на ряде примеров.
Наппер [77] подразделил стерическую стабилизацию на «эн-
тропийную» и «энтальпийную» в зависимости от того, является ли
величина энтропии положительной или отрицательной. Например,
при стерической стабилизации алифатических углеводородов
длинными полигидроксистеариновыми цепями флокуляция вы-
зывается охлаждением; в случае же энтальпийной стабилизации
флокуляция возникает при нагревании (как в случае цепных
молекул полиэтиленгликоля в воде).
Исследователь должен точно знать, какого характера будет
флокуляция, энтропийного или энтальпийного и не будет ли проис-
ходить стабилизация [78]; с практической точки зрения это полез-
ное подразделение.
Другой простой способ достижения тета-точки заключается
в изменении растворяющей способности непрерывной фазы, т. е.
в добавлении в систему нерастворителя. Наппер [79] проиллю-
стрировал пример использования различной растворяющей спо-
собности для достижения начала флокуляции при постоянной
температуре и еще раз продемонстрировал особую важность
осмотических эффектов при рассмотрении стерической стаби-
лизации;
5.6. ПОЛНАЯ ФЛОКУЛЯЦИЯ И ПОЛНАЯ СТАБИЛИЗАЦИЯ
Стерическая стабилизация достигается в результате адсорб-
ции полимера на поверхности частиц. Влияние свободного поли-
мера в растворе на коллоидную ста ильность ыло исследовано
Наппером, который ввел термины «полная стабилизация» и «пол-
ная флокуляция» [80]. Рассмотрение этого явления приведено
в работе [51, гл. 17].
Концепция «полной флокуляции» была развита в работах
[81, 82]. Однако в работе [83] было показано, что чем выше кон-
центрация свободного полимера в растворе, тем в большей мере
падает флокулирующее действие добавленного полимера.
Существуют различные представления о явлении полной фло-
куляции [84—86], но основная концепция базируется на рас-
смотрении поведения неадсорбирующих поверхностей и молекул
полимера в растворе.
По мере того, как поверхности коллоидных частиц сближа-
ются до расстояния меньшего, чем размеры частиц полимера
в растворе, последние активно выталкиваются из пространства
между частицами, где остается только растворитель. С точки зре-
ния термодинамики это не является благоприятным, поскольку
приводит к осмосу растворителя в раствор полимера, вследствие
чего частицы еще больше слипаются, т. е. флокулируют.
Наппер и Фейгин [80] распространили механизм полной
флокуляции на явление полной стабилизации, рассмотрев пове-
дение в равновесных условиях двух инертных плоских пластин,
погруженных в раствор неадсорбирующегося полимера, и иссле-
довав распределение молекул полимера, находящихся в контакте
с поверхностями и в объеме раствора, с точки зрения статистики.
Они показали, что концентрация полимерных сегментов, контак-
тирующих с плоской, неадсорбирующей поверхностью, ниже, чем
в объеме раствора. Определив «предельную свободную энергию»
как функцию расстояния между пластинами, выраженного в вели-
чине диаметра молекул полимера, они показали, что по мере сбли-
жения двух пластин вначале сопротивление возрастает, поскольку
молекулы полимера оказывают это сопротивление, а при более
тесном контакте пластин они испытывают притяжение друг к другу
(см. рис. 5.11).
Авторы использовали метод Дерягина для превращения кри-
вых потенциальной энергии для случая плоских пластин в соответ-
Рис. 5.11. Схематическое изображение полной флокуляции (а) и стабилизации (б)
dh
Рис. 5.12. Переход от плоской
пластины к сферической поверх-
ности по Дерягину
ствующие кривые для взаимодействия неплоских поверхностей
(например, сфера — среда) путем введения математического
коэффициента пересчета, как это показано на рис. 5.12.
Винсет и др. исследовали флокулирующий эффект добавки
полимера в стерически стабилизированные латексы [87] и объ-
яснили свои выводы с помощью полуколичественной теории,
что схематически представлено на рис. 5.13. Они показали, что
величина энергии отталкивания адсорбированной полимерной
оболочки -(77LL) уменьшается в присутствии полимера в непрерыв-
ной фазе в соответствии с уравнением:
LL LL.O ,„„РР
G — G —п/УЛз ,
„LL.O „ _
где G — энергия выталкивания полимерной оболочки в отсутствие полимера
в растворе; Gpp — энергия взаимодействия двух полимерных глобул в растворе;
п — количество полимерных глобул, находящихся в растворе.
Эта теория предсказывает, что при добавлении полимера про-
исходит флокуляция между частицами полимера при концен-
трации Ф^5 тогда, когда распрямленные полимерные глобулы
в растворе приходят в тесный контакт друг с другом; Ф**— кон-
центрация, при которой имеет место однородная плотность сег-
ментов и которая возникает тогда, когда полимерные глобулы
сжимаются до их тета-конфигурации при дальнейшем добавле-
нии полимера.
Рис. 5.13. Механизм полной флокуляции по Винсенту с сотр. [88]
Таким образом:
М М
®ps =-------я---и ®PS =-------------5----,
/>* <S2>'/27Vp t>** <£2>0Л Wp
где д| — молекулярная масса полимера с плотностью р; N — число Авогадро,
7^2^ А — радиус спирали, Ь* и Ь** —упаковочные факторы (8 и 2,52 для плот-
ной кубической упаковки; 5,6 и 1,36 для плотной гексагональной упаковки, со-
оответственно).
При практическом использовании эта теория дает приемлемое
соответствие с экспериментом как для водных, так и для невод-
ных систем [88]. Шотьен и Флир [89] предложили механизм
полной флокуляции — стабилизации, подобный механизму Нап-
пера, основанный на теории конформации полимерных молекул.
Как Наппер [80], так и Шотьен и Флир [89] предсказали
высокие значения энергии полной стабилизации для коллоидных
частиц при определенном содержании свободного полимера,
однако эти две теории отличаются по ряду аспектов. Например,
Шотьен и Флир предсказали более высокий эффект стабилиза-
ции в случае слабой растворяющей способности растворителей
(т. е. тета-растворителей), в то время как Наппер отдает пред-
почтение хорошей растворяющей способности.
Недостаток теории полной стабилизации состоит в том, что
она распространяется на исследования растворов полимеров
с достаточно высокой концентрацией и предпочтительно с высо-
кой молекулярной массой для получения хорошей пигментной
дисперсии. Совершенно очевидно, что это не всегда соответствует
встречающимся на практике условиям.
Не следует думать, что теория полной стабилизации явля-
ется ошибочной, однако она не является, вероятно, всеобъемлю-
щей по той причине, что теория концентрированных растворов
полимеров еще недостаточно разработана. Шотьен и Флир [90],
в отличие от Наппера, пересмотрели свою оценку падения энергии
полной стабилизации, приведя ее в согласие с данными Винсента
с сотр., которые с полной очевидностью показали, что не суще-
ствует полной стабилизации, а указали только на отсутствие
Полной флокуляции при высоких концентрациях полимеров.
Концепция полной флокуляции имеет тесную связь со свой-
ствами красок. Из нее следует, например, что если даже очень
хорошая дисперсия пигмента получена в разбавленном растворе,
то последующее добавление к ней даже совместимого не адсор-
бирующегося полимера может вызвать флокуляцию. Следует
отметить, что флокуляция пигментов может происходить и при
Добавлении растворителя, т. е. при разбавлении краски перед
нанесением.
Нужно также обратить внимание на то, что различные методы
Нанесения, которые требуют соответствующего разбавления кра-
сок до необходимой вязкости, могут привести к различной степени
флокуляции пигментов, что, в свою очередь, может привести к по-
явлению различий в цвете покрытий даже несмотря на то, что
исходная дисперсия может быть полностью дефлокулирована
при низком содержании полимера в непрерывной фазе.
Полная флокуляция может иметь место в случае систем, ста
билизированных зарядом, например в латексах, при добавлении
«инертных» загустителей, которые, как показал Сперри [91],
могут оказать влияние на седиментационные процессы в краске.
5.7. АДСОРБЦИЯ
Для достижения высокой стабильности дисперсий при стери-
ческой стабилизации важно зафиксировать молекулы стабилиза-
тора на поверхности частиц.
Чем прочнее они удерживаются на поверхности частиц, тем
лучше их соответствующее действие и имеется больше возможно-
стей принимать различные конфигурации, как, например, в случае
полной адсорбции таких молекул, как полигидроксистеариновая
кислота, которая известна как очень хороший стабилизатор [57].
Если, например, адсорбированные молекулы закрепляются на
поверхности частиц и могут перемещаться около поверхности, то
при перекрывании сольватированных слоев повышение концен-
трации частично перераспределялось бы по поверхности, что
привело бы к уменьшению энергии отталкивания по сравнению
с ситуацией, когда перераспределение по поверхности невоз-
можно. Кроме того, если адсорбция стабилизирующих молекул
недостаточно прочна, то увеличение осмотического давления
может быть обусловлено не только поверхностным перераспре-
делением, но также и десорбцией стабилизатора, в результате
чего силы отталкивания между частицами становятся очень
малыми или совсем исчезают
Но если силы адсорбции молекул стабилизатора достаточно
велики, так что молекулы «пригвождены» к поверхности частицы,
эффективно образуя псевдо- или несольватпый слой, тогда при
тесном соприкосновении с другим подобным слоем не будет про-
исходить снижения энтропии полимерных цепей. Единственное
что будет достигнуто в этом случае — растяжение поверхности
частицы на некоторое малое расстояние. Потенциал притяжения
в этом случае будет возникать за счет новой поверхности и к со-
стоянию частиц будет применима композиционная константа
Гамакера, характеризующая потенциал притяжения при отсут-
ствии энергии отталкивания
При разработке стабилизаторов для дисперсий прежде всего
нужно, чтобы молекула стабилизатора обладала способностью
прочно связываться с поверхностью предпочтительно своим
концом, оставляя возможность оставшейся части молекулы быть
сольватированной в наилучшем растворителе. Примером может
служить реакционноспособный ста илизатор, описанный 1омпсо-
ном с сотр. [92]. Связывание может происходить и за счет кислот-
но-основного взаимодействия в неполярной среде, как в случае
реакции жирных, кислот, например поли-12-гидроксистеариновой
кислоты, с пигментом, содержащим покрытие из оксида алюминия.
Иногда невозможно обеспечить взаимодействие стабилиза-
тора с поверхностью частицы, в этом случае необходимые требо-
вания по прочному закреплению и максимальной сольватации
могут быть достигнуты только в результате физической адсорб-
ции при использовании специальных сополимеров, растворенных
в геми-растворителях (например блок- или графт-полимеры),
когда одна часть молекулы осаждается на поверхность частицы
(т. е. прикрепляющаяся часть несольватирована), а остающаяся
сольватированная ее часть образует стабилизирующий барьер,
как описано в [57].
5.7.1. Изотермы адсорбции
Изотермы адсорбции весьма полезны для понимания процес-
сов, происходящих в пигментных дисперсиях. Например, можно
ожидать, что если пигмент ТЮ2 диспергирован в бутилацетате,
ксилоле или уайт-спирите при использовании одного и того же
диспергатора [54], то энергия притяжения в соответствии со зна-
чениями констант Гамакера для соответствующих растворителей
должна быть расположена в следующем порядке: Гд>Й|>-Гд,
где VI — энергия притяжения в уайт-спирите, Уд — энергия
притяжения в ксилоле; Уд — энергия притяжения в бутилацетате.
Так как стабилизатор полностью адсорбирован (т. е. толщина
барьера постоянна), он должен обеспечить наилучшую стабиль-
ность в растворе бутилацетата. Однако вискозиметрические
измерения показывают, что наблюдается обратная последова-
тельность, т. е. дисперсия с наименьшей флокуляцией получается
в уайт-спирите, затем в ксилоле и наихудшая — в бутилацетате.
Изучение изотерм адсорбции сразу же позволяет установить,
что поверхностная концентрация наименьшая в случае бутил-
ацетата (см. рис. 5.14), а следовательно наиболее слабые стаби-
лизирующие барьеры образуются в растворе бутилацетата.
Изотермы адсорбции можно легко определить, помещая опре-
деленное количество пигмента в известном количестве раствора
полимера в стеклянные банки с маленькими стеклянными бусин-
ками (диаметр примерно 0,75 см) для ускорения диспергирова-
ния и помещая банку примерно на 24 ч на вращающиеся валики.
Последующее центрифугирование дает возможность отделить не-
прерывную фазу, после чего количество адсорбированного мате-
риала может быть определено, как описано в [54].
Альтернативный метод определения изотерм заключается
в том, что пигмент помещают в хроматографическую колонку и
Рис. 5.14. Изотермы адсорбции димера 1.2-гидроксистеариновой кислоты на ТЮ2:
/ — в уайт-спирите; 2 — в ксилоле; 3 — в бутилацетате [54]
пропускают через него разбавленный раствор адсорбата, как
описано Краулом [93].
Преимущество хроматографического метода состоит в том,
что он дает возможность легко определить избирательную и обра-
тимую адсорбцию. Метод хорошо работает в случае органиче-
ских пигментов, однако в случае пигментной ТЮ? скорость проте-
кания через слой пигмента очень мала, что делает этот метод на-
много менее привлекательным, чем кажется на первый взгляд.
5.7.2. Свободная энергия адсорбции и изотермы адсорбции
Предположим, что мы исследуем частицу, погруженную в
жидкость, например в воду, и представим поверхность частицы
состоящей из большого числа адсорбционных центров; обозначим
такой центр, занятый растворителем, символом «5». Если мы
введем в жидкость X молекул растворяемого вещества, то для
адсорбции на поверхности они должны вытеснять молекулы воды,
занимающие этот центр. Обозначим через SX адсорбцию раство-
ряемого вещества на центре и через 5(НгО) — адсорбцию моле-
кул воды. Мы можем представить это в виде равновесного про-
цесса:
/г, /г,
S(H2O)+X SX + H2O; К=-г-,
/г2 «2
где К — разделительная константа или константа равновесия
для растворенного вещества между поверхностью частиц и раство-
рителем, связанная со свободной энергией адсорбции (Абадс)
уравнением:
AGMc= —RT 1п к.
Используя обозначения Гласстона [94] мы можем выразить
в следующем виде:
/мольная доля растворенногоX /мольная доля раство-Х
Увёщества на поверхности / урителя в объеме /
/мольная доля раствори-Х /мольная доля растворен-Х
(теля на поверхности ) уного вещества в объеме )
Предположим, что поверхность имеет а участков на 1 г адсор-
бента, которые заняты молекулами растворителя в отсутствие
растворенного вещества, и что часть %i из них, занимается раство-
ренным веществом при его растворении в растворителе. Тогда:
„ (xt/a) (мольная доля растворителя в объеме)
К=----------------------------------------------------------
(а — Xi у/мольная доля растворен-Х
у а ) уного вещества в объеме )
Если мы ограничим наши рассмотрения низкими (начальными)
значениями адсорбции (т. е. когда Х|-Са), то для разбавленного
раствора получим:
„ (Х(/а) (1)
(1) (N) '
и следовательно:
ДСада=-/?Г1п(-^-),
где N—мольная, доля растворенного вещества с концентрацией с (моль/г)
- Мс
в жидкости с молекулярной массой М и плотностью р, так что /V = -— и,
следовательно, для водного раствора М = с/55,5.
Пусть х/т — количество адсорбированного растворенного
вещества, приходящееся на 1 г адсорбента, моль/г; оно пропор-
ционально доле участков, занятых растворенным веществом,
, х>
т, е. х/т=р-^-< где р—константа пропорциональности.
.Если поверхность полностью покрыта мономолекулярным
слрем растворенного вещества, тогда х>/а=1 и х/т = р, т. е.
равно максимальной адсорбции (или высоте плато). Так, для
водных растворов:
AG„w^-/?rin(^;p55-5).
п о х/т
При низкой концентрации раствора —характеризует ис-
ходный наклон (IS) изотермы адсорбции, следовательно:
Абадс=— RTln
4S -55,5
к Р
(5.16)
Если представить изотермы адсорбции в координатах: коли-
чество адсорбированного вещества в моль на 1 г адсорбата в зави-
симости от молярной равновесной концентрации, можно устано-
вить свободную энергию адсорбции для мономолекулярного слоя
или изотерму адсорбции Лангмюровского типа исходя из исход-
ного наклона и числа насыщения.
Полезность уравнения 5.16 состоит в том, что оно позволяет
«почувствовать» силу адсорбции и, особенно, при его «обратном»
применении, т. е для оценки величины адсорбции ПАВ на поверх-
ности таких частиц, как пигменты или капли эмульсии, и создания
условий, обеспечивающих введение достаточного количества ПАВ
для удовлетворения адсорбционной потребности дисперсии. Если
известна свободная энергия адсорбции, а для простой физиче-
ской адсорбции она имеет порядок 5 ккал/моль, площадь попереч-
ного сечения.молекулы ПАВ известна (из литературы или может
быть установлена с помощью молекулярных моделей). Поэтому
Рис. 5.15. Определение ожидаемой адсорбции поверхностно-активного вещества
с помощью уравнения (5.16). Построение изотермы основано на физической ад-
сорбции ПАВ (водный раствор) при 5 ккал/моль и при скорости 200 А2 на молекулу
на порошке с плотностью 1,5 г/см3; при среднем диаметре частиц 0,5 мкм необхо-
димая концентрация ПАВ составляет 6,7-10 4 г-моль на 100 г порошка в равно-
весии с минимальной концентрацией раствора 9 5-Ю"3 М
можно построить исходные наклонный участок и участок плато
изотермы адсорбции. Соединив наклонный участок и участок
плато плавной кривой в области близкой к насыщению (как по-
казано пунктиром на рис. 5.15), получим изотерму адсорбции
с небольшой долей погрешности и, следовательно, установим
требуемое минимальное количество ПАВ.
Представления о Абаде, приведенные выше, основаны на упро-
щенной модели, в которой поверхность адсорбции состоит из
отдельных адсорбционных участков, которые заняты либо моле-
кулами растворителя, либо растворенного вещества, например
такого, как простое поверхностно-активное вещество. Эти пред-
ставления полезны для понимания изотермы адсорбции и иллю-
стрируют важное значение начального наклонного участка, а так-
же числа насыщения (которое также используется применительно
к адсорбции полимеров). Однако, расчет Абадс для молекул поли-
меров, которые могут занимать различное число участков в зави-
симости от их конформации, не может быть сделан, если не при-
нимать во внимание конфигурацию полимерных цепей. Подобные
условия были соблюдены Коралом с сотр. [95], которые опре-
делили «изостерические» теплоты адсорбции полимеров, исходя
из изотерм адсорбции.
Так, если изотермы адсорбции измерены при различных тем-
пературах, то можно установить энтальпию адсорбции (А//),
используя уравнение Хюккеля, аналогичное уравнению Клаузиуса-
Клайперона:
ДЯ HS — HL dln(aL/as) _
~R~~~~R~~~~d (i/т) ’ (517)
где и — парциональные молярные энтальпии полимера на адсорбате и
растворе; и aL — соответствующие активности полимера на поверхности
и в растворе.
В очень разбавленных растворах а, =с, и если предположить,
что активность полимера на поверхности не изменяется, когда
количество его поддерживается постоянным, то приведенное выше
уравнение примет вид:
А// _ dine
R ~ d (1/Т)
(5.17а)
Следовательно, измерив изотермы адсорбции при разных темпе-
ратурах, взяв одинаковые количества адсорбирующегося вещества
при разных температурах и отложив на графике в логарифми-
ческой шкале соответствующую равновесную концентрацию ра-
створа как функцию обратного значения абсолютной темпера-
туры, можно получить прямую линию, наклон которой равен
ДЯ//?.
5.7.3. Адсорбция и температура
Количество полимера, адсорбируемого из раствора, часто
увеличивается с повышением температуры, что указывает на эндо-
термичность процесса. Однако известны также случаи, когда ад-
сорбция при увеличении температуры снижается [96] или не за-
висит от нее [97].
По мнению Корала и др. [95], процесс адсорбции не должен
быть эндотермическим в случае физической адсорбции простых
молекул на чистых поверхностях, так как в этом случае измене-
ние энтропии адсорбции должно быть отрицательным, а необхо-
димая энтальпия также должна быть отрицательной (вследствие
уменьшения свободной энергии адсорбции). Однако в случае
полимеров следует рассматривать систему в целом. Адсорбция
полимерной молекулы на нескольких участках требует, чтобы
несколько молекул растворителя переходило с поверхности в ра-
__створ. Поступательная энтропия молекулы полимера наряду
с вращательной и колебательной энтропией теряется при ад-
сорбции молекулы вследствие частичного ограничения ее сегмен-
тальной подвижности. Вследствие этого десорбирующиеся моле-
кулы растворителя увеличивают свою поступательную энтропию,
которая в сумме намного больше, чем энтропия молекул поли-
мера. Конечным результатом является общий выигрыш в энтро-
пии в системе при адсорбции полимерных молекул, вытесняющих
молекулы растворителя, даже если процесс является эндотерми-
ческим.
Качество растворителя, в котором растворен полимер, также
влияет на количество полимера, адсорбированного субстратом.
Обычно, чем хуже растворитель, тем большее количество поли-
мера адсорбируется [95], т. е. адсорбцию можно связать с пара-
метром растворимости [98]. Однако адсорбция полимера зави-
сит не только от взаимодействия полимера и растворителя, но
также от взаимодействия субстрат — растворитель [99]. Можно
также уменьшить адсорбцию полимера на субстрате путем добав-
ления небольших количеств нерастворителя к раствору поли-
мера, если растворитель сильно взаимодействует с субстра-
том [100].
5.7.4. Скорость адсорбции и равновесие
Трудно определить скорость адсорбции, поскольку она зави-
сит от многих факторов, особенно от концентрации [101], однако
обычно считают, что она протекает быстро [102, 103]. По вопросу
адсорбции полимеров можно обратиться к работе Липатова и Сер-
геевой [104]. Однако в большинстве практических случаев наи-
больший интерес представляет состояние равновесной адсорб-
ции в системе из смеси компонентов; в случае адсорбции поли-
мера из раствора действует простои принцип — первым адсор-
бируется тот компонент, который первым достиг поверхности,
т е. адсорбция контролируется диффузией [99, 105, 106]. Более
мелкие по размерам (молекулы первыми достигают поверхности
и адсорбируются, а затем их вытесняют более крупные молекулы,
достигающие поверхности позже, адсорбция которых более пред-
почтительна [Ю7]. Явление преимущественной адсорбции моле-
кул с более высокой молекулярной массой довольно часто при-
водит к заметному максимуму на изотермах адсорбции полимеров
[56]. При диспергировании пигментов, т. е. при приготовлении
пигментных паст, важно создать такие условия, чтобы происхо-
дила преимущественная адсорбция полимера в самом начале,
так как процесс вытеснения других компонентов с поверхности
пигментов может быть очень медленным.
Это иногда отчетливо проявляется на практике, например в
случае, когда пигмент первоначально диспергируют в алкиде,
модифицированном полиэтиленгликолем, а другие полимерные
компоненты добавляются после. При этом наблюдается быстрый
переход пигмента в водную фазу, как, например, при промывке
кисти моющим раствором от прилипшей к ней свежеприготовлен-
ной краски на масляной основе. Однако, если пигмент вначале
диспергировать в других полимерных компонентах, а затем до-
бавить алкид, модифицированный полиэтиленгликолем, то в этом
случае потребуется примерно четыре недели для того, чтобы по-
явилась способность кисти отмываться. За это время алкид,
модифицированный полиэтиленгликолем, вытесняет другие ком-
поненты с поверхности пигмента [108].
Другой пример подобного медленного перераспределения
адсорбированных молекул иногда проявляется в виде медлен-
ного изменения цвета при хранении, когда цвет создается сме-
шением различных пигментных дисперсий (колеровочных паст),
особенно если они приготовлены в разных связующих. Одним
из путей решения этой «цветовой» проблемы является пропуска-
ние готовой краски через дисперсионную мельницу (например
мельницу Сасмейера). Деформации сдвига в мельнице, а также
повышение температуры ускоряют перераспределение ранее ад-
сорбированных веществ и поэтому предотвращают изменение
цвета.
Стереорегулярность также является важным фактором при
адсорбции полимеров. Майамото с сотр. [109] установили, что
изотактический полиметилметакрилат лучше адсорбируется из
раствора в одном и том же растворителе по сравнению с синдио-
тактическим при одинаковых молекулярных массах. Синдиотакти-
ческий полиметилметакрилат, в свою очередь, более прочно адсор-
бируется, чем атактический, что было показано с помощью
метода тонкослойной хроматографии при проявлении этилацета-
том [110].
5.7.5. Специфическая адсорбция
Адсорбция полимера из раствора зависит от природы раство-
рителя и «активности» поверхности.
При обычной физической адсорбции полимеров можно интуи-
тивно предположить, что в данном гомологическом ряду преиму-
щественно адсорбируются образцы с более высокой молекуляр-
ной массой. Специфическая адсорбция может нарушить влияние
молекулярной массы. Типичным примером является преимуще-
ственная адсорбция низкомолекулярных полимеров с более вы-
сокой полярностью, таких, как фталевые полуэфиры, входящие
в состав жирных алкидов. Уолбридж и др. [111] показали, что
адсорбция этих продуктов может быть объяснена с точки зрения
кислотно-основного взаимодействия, в котором основаниями явля-
ются поверхность ТЮг и металл сиккатива. Они установили, что
общее поведение системы при флокуляции — дефлокуляции за-
висит от порядка введения сиккатива и димерной жирной кислоты
в обычную белую эмаль на основе жирного алкида, а также от
относительной силы кислоты и возможности образования необра-
тимых связей карбоксильных групп с поверхностью пигмента,
что, в свою очередь, зависит от температуры дисперсии. Поверх-
ность различных пигментов может связывать кислоты (или осно-
вания) подобно ионообменным смолам. Соломон и др. [112]
исследовали кислотные центры на поверхности минеральных
наполнителей и пришли к выводу, что они сравнимы по силе
с кислотными центрами катализаторов «крекинга». Наличие
подобных центров, которые проявляются при действии тепла,
оказывает очень сильное влияние на химические реакции в поли-'
мерных соединениях, особенно в неполярных средах.
5.8. СКОРОСТЬ ФЛОКУЛЯЦИИ
Смолуховский [ИЗ] показал, что для однородных сфериче-
ских частиц, между которыми отсутствуют силы притяжения или
отталкивания и которые слипаются друг с другом при столкно-
вении, скорость уменьшения количества частиц может быть вы-
ражена уравнением: —dN/<М=К1\Р. После интегрирования
получаем:
1/W=1/№>+№, (5.18)
где N — количество частиц; No — начальное количество частиц, т. е. N = No при
времени Zo; К — константа скорости.
Если мы определим «время полужизни» А/г как время, необхо-
димое для того, чтобы количество частиц уменьшилось в два раза
по сравнению с исходным, т. е. 2N — N0 при 6/г, то, подставляя
эти величины в уравнение (5.18), получим:
2/NB=l/NB+Ki>h
или
/1/2=l//(Wo. (5.18а)
Если константа скорости контролируется диффузией, тогда
К=Кд=46773т],
Где k — постоянная Больцмана, Т — температура, rj — вязкость среды.
В табл. 5.2 приведено время для частиц различных размеров
при различном объемном содержании пигментов, необходимое
для уменьшения количества частиц вдвое, в соответствии с кине-
тическим уравнением быстрой флокуляции Смолуховского. Урав-
нение быстрой флокуляции основано на броуновском движении
частиц в среде при комнатной температуре и типичной вязкости
краски (1 пуаз). При флокуляции типичная глянцевая краска
(TiOs, ОКП=15%) возвращается во флокулированное состоя-
ние через 0,5 с после активного возмущения, например, при на-
нёсении кистью. Даже если вязкость связующего значительно
больше, время, необходимое для повторной флокуляции после
механического воздействия, будет по-прежнему очень мало.
По тем же причинам при использовании аэросила (мелкодисперс-
ного кремнезема) в качестве структурирующего агента, его тре-
буется значительно меньшее количество по сравнению с "ПОг, и
структурообразование в результате флокуляции происходит
почти мгновенно.
Предпринимались попытки ревизии и уточнения уравнения
Смолуховского с учетом сил межмолекулярного взаимодействия
частиц и гидродинамического взаимодействия [114—118]. Хотя
эти исследования показали, что константа скорости должна быть
приблизительно в 2 раза меньше, чем по уравнению Смолухов-
ского, все же не удалось опровергнуть предположение, что в реаль-
ных системах флокуляция пигментов протекает с большой ско-
ростью.
Гедан и др. [119] измерили быструю флокуляцию частиц
полистирола. Хотя, вопреки уравнению Смолуховского, было
показано, что скорость образования дублетов, триплетов и квад-
руплетов одинакова, тем не менее до сих пор считается, что теория
Смолуховского вполне справедливо описывает скорость флоку-
Таблица 5.2. Время полужизни для частиц различных размеров при быстрой
флокуляции в среде с вязкостью 1 пуаз при 25 °C
Время полужизни ОКП, % Время полужизни ОКП, % (аэросил,
(П/2), С (TiOa, ~ 0,2 мкм) (б/s), мкс ~ 0,014 мкм)
7,6 1 2400 1
1,5 5 480 5
0,76 10 240 10
0,5 15 160 15
0,38 20 120 20
ляции дисперсий и подтверждается многими экспериментами
[114, 115].
Седиментация и флокуляция. Пигменты обычно имеют более
высокую плотность, чем растворы полимеров в красках, и под
действием силы тяжести они стремятся к оседанию в соответ-
ствии с законом Стокса. Этот закон может быть представлен
в виде уравнения:
где частицы с кажущимся диаметром d (диаметр Стокса) опускаются на рас-
стояние h в среде с вязкостью ц за время t под действием силы тяжести g при
наличии разности плотностей частиц и среды Др.
При хорошей дефлокуляции дисперсия частиц оседает в соот-
ветствии с вышеприведенным уравнением, образуя твердый, плот-
ный осадок, трудно поддающийся редиспергированию. Однако
скорость седиментации менее 10~6 см/с в большинстве случаев
перекрывается диффузией и конвекцией, и можно считать, что
седиментация при этом отсутствует. Если флокуляция имеет
место, седиментация пигмента протекает быстро и образуется
мягкий, объемистый и легко редиспергируемый при перемеши-
вании осадок.
При седиментации конечный объем осадка зависит от степени
флокуляции: чем больше флокуляция, тем больше объем осадка.
В предельном случае объем осадка может быть равен общему
объему, что не следует смешивать с состоянием дефлокуляции,
когда седиментация может отсутствовать. Объем осадка зависит
не только от флокуляции, но также от формы частиц и распреде-
ления по размерам [120]. Следует помнить, что объем осадка
используется только как относительный показатель степени фло-
куляции в подобных системах.
Другим простым способом определения степени флокуляции
пигмента является нанесение капли дисперсии на фильтроваль-
ную бумагу и измерение соотношения размеров пигментного пятна
к пятну растворителя; чем больше это отношение, тем более дефло-
кулирована дисперсия.
Смит и Пембертон [121] описали вариант метода «пятна»,
который, как они считают, дает лучшие результаты.
Обычно для определения флокуляции по седиментации или
по измерению Rf необходимо уменьшить вязкость краски путем
добавления растворителя; в противном случае седиментация
протекает слишком медленно. Но разбавление следует проводить
осторожно, поскольку в этом случае вносится новый источник
флокуляции вследствие десорбции или полной флокуляции, или
того и другого вместе.
Флокуляция может быть более просто исследована в жидком
состоянии по данным вискозиметрии [52]. Например, если вяз-
I______I _____I______I ______I I
0,01 0,02 0,03 0,04 0,05
(Скорость сдвига) —1/2
Рис. 5.16. График зависимости логарифма вязкости дисперсии от скорости сдвига
при различной степени флокуляции пигмента [54]:
1 — наибольшая флокуляция, 4 — наименьшая флокуляция
кость непрерывной фазы (т]о) является ньютоновской, то при
отсутствии взаимодействия частиц вязкость дисперсии (т]) также
должна быть ньютоновской. Если дисперсия не является ньюто-
новской, то степень отклонения от ньютоновской жидкости явля-
ется мерой взаимодействия между частицами, т. е. флокуляции,
при условии, что объем дисперсной фазы <30%; в противном
случае могут наблюдаться инерционные эффекты [122].
Отложив на графике 1g г] как функцию обратной величины
корня квадратного из скорости сдвига, мы получим прямые линии.
Чем выше наклон, тем больше степень флокуляции, что видно
из графика на рис. 5.16.
Этот метод оценки флокуляции, как было установлено на прак-
тике, является очень надежным применительно к типичным кон-
центрациям пигмента в рецептурах красок. Сравнение флокуля-
ции с помощью этого метода является достаточно строгим для
одинаковых объемных концентраций, так как наклон приведен-
ных выше кривых будет увеличиваться при росте концентрации
дисперсной фазы. Однако влияние различного объема дисперсной
фазы можно установить и ввести поправку, которая делает воз-
можным провести сравнительную оценку.
Следует отметить, что реологическая оценка флокуляции за-
висит от общего взаимодействия исследуемой системы. Поэтому
при наличии больших различий в размере частиц для дисперсий,
даже если дисперсии сравниваются при одинаковом объеме ди-
сперсной фазы, дисперсия с частицами меньшего размера и олее
слабым взаимодействием между частицами будет в большей
степени проявлять свойства неньютоновской жидкости (т. е. будет
более флокулированной). Это происходит потому, что силы
взаимодействия между частицами пропорциональны размеру
частиц в первой степени, в то время как число частиц на единицу
объема пропорционально кубу радиуса. Следовательно, общая
«сила» большого числа слабых связей между малыми частицами
больше, чем малое число сильных связей между большими ча-
стицами.
Предпринимались попытки оценить силы взаимодействия
между частицами на основе реологических измерений путем опре-
деления критического напряжения сдвига [123], однако оценка
оказалась весьма неопределенной [124].
Степень флокуляции пигмента можно также определить по
измерению цветовых характеристик [125], оседанию [111] или
при непосредственном визуальном наблюдении под микроскопом,
как показано на' рис. 5.4. Флокуляцию в сухой пленке краски
труднее определить, поскольку при получении тонких срезов
пленки краски под воздействием микротома может изменяться
состояние дисперсии пигмента [126]. Для преодоления этого
затруднения был разработан метод получения сколов при замо-
раживании с последующим кислородным отжигом [127]; однако
при концентрациях более 12% визуальное наблюдение стано-
вится более затруднительным.
Альтернативный метод определения флокуляции пигмента
был разработан Балфуром и Хердом [128], использовавшими
измерение коэффициентов отражения в ИК-Диапазоне, который
более чувствителен к частицам больших размеров. Однако ни
один из этих методов не позволяет установить наличие флоку-
ляции пигментов непосредственно как в жидком, так и в твердом
состоянии. Эрмитедж [129] описал метод рассеивания протонов,
который позволяет измерить состояние флокуляции пигментов
непосредственно, независимо от природы фазы (т. е. сухой или
жидкой краски).
В заключение отметим, что кажущийся простым вопрос, за-
данный в начале данного раздела, относительно того, в чем лучше
диспергировать пигмент, в алифатических или ароматических
углеводородных растворителях, в действительности оказывается
чрезвычайно сложным, поскольку он содержит взаимоисключаю-
щие положения, а именно:
1. Необходимо максимально повышать у cos 6 для систем,
что находится в зависимости от природы подложки и концен-
трации алкида. Для ТЮг, имеющего большую поверхностную
энергию, по всей вероятности лучше использовать арома-
тический растворитель, так как cos 6= 1 для обоих раствори-
телей.
2. Из рассмотрения энергии притяжения становится очевид-
ным, что для TiO2 более предпочтительным является использо-
вание ароматического растворителя, поскольку значение кон-
станты Гамакера будет более точным.
3. При рассмотрении природы адсорбционного слоя и его
влияния на стерическое отталкивание можно заключить, что луч-
ше использовать алифатический растворитель (уайт-спирит),
поскольку в этом случае возможна адсорбция большего коли-
чества полимера и в результате будет получена более стабильная
дисперсия при небольших различиях в растворимости сольват-
ного слоя, чем при использовании ароматического растворителя
(толуола).
В целом положение 3 более существенно, чем положения 1
и 2 вместе взятые.
Глава 6
РАЗМЕР ЧАСТИЦ И ЕГО ИЗМЕРЕНИЕ
А. Дорожковский
6.1. ВВЕДЕНИЕ
Чтобы понять поведение и свойства диспергированных частиц,
таких как пигменты в лакокрасочных материалах, необходимо
иметь сведения о размере частиц и его распределении. Лако-
красочный материал может содержать пигменты, наполнители,
капли эмульсии и частицы латекса.
На вопрос: «Чем является частица?» и, следовательно, «Ка-
ков ее размер?» не всегда легко ответить, что видно из рис. 6.1.
Ответ на этот вопрос вызывает другой вопрос: «Какое свой-
ство необходимо исследовать?» Измерение размера частиц обычно
является способом решения той или иной задачи и редко исполь-
зуется как самоцель. При любых измерениях важно подобрать
наиболее пбдходящий метод из имеющихся. Если измеряется
адсорбция ПАВ, следует измерять площадь поверхности; при
рассмотрении седиментации измерение размера частиц следует
производить объемным (весовым) методом, а не числовым.
Если частицы являются одинаковыми правильными сферамй,
то для определения их размера и точного описания распределе-
ния частиц по размерам необходимо лишь знать их диаметр.
Однако на практике такое случается исключительно редко. Форма
Рис. 6.1. Что такое частица? (12-кратное увеличение)
частиц часто неправильная; обычно они имеют различные диа-
метры, и поэтому определение их размеров, достаточное для од-
них условий, может быть недостаточным для других. Следова-
тельно, важно правильно выбрать метод определения размера
частиц. Например, переменный размер агломератов, показанный
на рис. 6.1, не связан с размером монодисперсных первичных
частиц, из которых состоят эти агломераты.
6.2. ОПРЕДЕЛЕНИЯ
Сферические частицы хорошо описываются их диаметрами,
но несферические можно измерять различными способами. Неко-
торые из них показаны на рис. 6.2 [1].
Диаметр Ферета — расстояние между двумя касательными
на противоположных сторонах частицы, перпендикулярными
направлению, в котором рассматривается частица.
Диаметр Мартина — длина линии, параллельной тому же
направлению, разделяющей профиль частицы на две равных по
площади части.
Максимальный и минимальный линейные диаметры — два
очевидных линейных измерения, которые могут быть использо-
ваны. Эти значения можно объединить, чтобы дать одно значе-
ние в виде корня квадратного из их произведения, что дает лучшее
представление о размере, чем любое из значений, взятое отдельно.
Однако, такое измерение в известной степени трудоемко, и часто
используют специальные шкалы в виде ряда кругов различного
диаметра, наносимые на фотографии (или окуляр микроскопа);
при этом частицы неправильной формы можно приравнять к кругу
эквивалентной площади (или периметра). Иными словами, диа-
метр проекции частицы — это диаметр круга, равновеликого
проекции частицы. Типичными примерами являются шкала Пат-
Рис. 6.2. Диаметр несферической частицы:
^1—диаметр Ферета; dz—диаметр Мартина; dz— макси-
мальный линейный диаметр; — минимальный линейный
диаметр; — диаметр круга эквивалентной площади
терсона-Кэйвуда, где нанесен ряд кругов в арифметической про-
грессии, и шкала Портона, где площади возрастают в геометри-
ческой прогрессии с основанием 2°15 [2].
6.2.1. Требования к счету частиц
Часто возникает вопрос, как много частиц следует подсчитать,
чтобы получить характерное распределение по размерам. Интуи-
тивно можно понять, что это должно зависеть от диапазона раз-
личий частиц по размерам. Если частицы монодисперсны, то
будет достаточно малого числа измерений. Чем выше полидисперс-
ность, тем большее потребуется число измерений. На рис. 6.3
представлен для примера результат измерения среднего размера
частиц в зависимости от числа измеренных частиц. Видно, что
после 350 измерений средний размер мало меняется вплоть до
1000 измерений.
__-Время--или усталость— обычные критерии для ограничения
числа измеряемых частиц. На практике ASTM [3] рекомендует
измерять не более 25 частиц каждой формы и, как минимум,
10 частиц каждого размера.
Предложена методика для минимизации числа измерений при
сохранении верной картины [4], получившая название «сокра-
щенное многократное профилирование». Это модификация ме-
тода «послойного исследования» [5]. При использовании этого
метода следует наблюдать как минимум 10 частиц в каждом типе
размеров, которые оказывают значительное влияние на кривую
Число подсчитанных частиц
Рис. 6.3. Зависимость среднего размера от числа частиц
Таблица 6.1. Пример измерения частиц по методу Сичела [4]
Номер про- филя Число частиц в каждом диапазоне размеров
0—1 1—2 2—3 3—4 4—5 5—6 6—7 7—8 8—9 9—10 10—11 11 — 12 Итого
1 0 5 11 34 41 24 5 3 1 0 1 0 125 2 17 4 2 1 1 0 0 16 30 553100 15 ,4 0 2 210 6 5 1 3 2 118 6 0 112 0 4 7 0 2 10 3 8 0 110 2 9 0 10 1 10 о 0 0 0
'Итого в столбце: 2 12 11 34 41 24 14 12 11 10 8 1 180 Итого на 1 профиль: 0,2 6 11 34 41 24 4,7 4 2,2 1,3 0,8 0,1 129,3 % от общего числа (частота): 0,15 4,64 8,51 26,3 31,7 18,6 3,6 3,1 1,7 1,01 0,62 0,08 100
Примечание: Общее число считаемых частиц равно 180 (эквивалентно подсчету -1250 частиц).
распределения частиц по размерам. Метод проиллюстрирован
в табл. 6.1.
В первом профиле была найдена площадь и проведены соот-
ветствующие измерения для интервалов 2—3, 3—4, 4—5, 5—
6 единиц. Второй профиль был изучен аналогично, с той разни-
цей, что считались частицы большие, чем 5—6 и меньшие, чем
2—3 единицы. Процесс повторяется до тех пор, пока критерий
Сичела не будет удовлетворять большинству типов размеров.
В итоге при счете только 180 частиц уже получается надежность
результатов, эквивалентная счету 1250 частиц при другом методе
(десять профилей дают 1250 частиц, т. е. 125-10).
Важнейшие критерии счета частиц заключаются в том, чтобы
изучаемый образец был типичным и чтобы на результаты не ока-
зывал влияния способ приготовления образцов. Следовательно,
недостаточно измерить большое число частиц с одной электрон-
ной микрофотографии; более точные результаты можно полу-
чить при измерении меньшего числа частиц, но с многих различ-
ных микрофотографий. Считаемые частицы должны быть типич-
ны для представляемой выборки, их распределение должно быть
случайным, независимо от формы или размера, и агломераты
должны быть разрушены. Частое требование для одной частицы
заключается в том, чтобы более половины ее периметра было
видно при измерении. Это, однако, исключает возможность иссле-
дования спекшихся частиц, как, например, в случае диоксида
титана (см. рис. 5.2).
6.2.2. Средний размер частиц и способы его выражения
Если построить графическую зависимость размера частиц
от числа частиц этого размера (частота), получится график рас-
пределения частиц по размерам, или гистограмма, зависящая
от способа построения. Хотя этот путь очень информативен, но
неудобен для использования, поэтому требуется простой способ
для конечного выражения диапазона размеров, т. е. среднего раз-
мера. Существует много способов выражения средних величин,
что зависит от того свойства, которое принимается во внимание.
Некоторые часто встречающиеся определения даны ниже, а их
положение на кривой показано на рис. 6.4.
Основной диаметр Наиболее часто наблюдаемый диаметр
Медианный диаметр Значение диаметра, для которого масса частиц,
имеющих большие или меньшие размеры,
составляет 50%.
Среднечисловой (dQ0) диа- Сумма всех диаметров, деленная на общее
"метр число "частиц:^" (тй) /27й
Среднегеометрический (de) Корень и-ой степени из произведения диа-
диамстр метров п измеренных частиц: dg=‘\]dtd2...dll.
Обычно определяется как lg de=£n()g d) /£п.
Среднегармонический диа- Обратная величина измеренных диаметров:
метр l/dn = X (n/d) /£п.
Рис. 6.4. Различные определения «среднего»
Среднеповерхностный (ds)
диаметр
Среднеобъемныи (dv) диа-
метр
Средний объем но-поверх-
ностный (dM) (ИЛИ сред-
ний Саутера) диаметр
Среднемассовый (dm) диа-
метр или средний Де-
Броукера
Среднепротяженный диа-
метр (dt)
ds=^£ nd2/£n.
dv—-y/^ nd'/'/ n (часто называется мас-
совым срединным диаметром).
Средний размер, определяемый величиной
поверхности, отнесенный к единице объема:
dus = X nd? nd2.
dw = £ п“/Т. nd3-
Средняя величина di =£ nd2/£ nd сравнима
co среднечисловым и среднегеометрическим
диаметрами, он представляет сумму площадей
поверхности, деленную на сумму диаметров
Примечание: Индексы «ап», «g», «s», «п», «ns», «и>» происходят соответственно
от английских слов average — средний, geometrical — геометрический, surface —
поверхностный, volume — объемный, volume-surface — объемно-поверхностный,
weight — весовой (Прим, переводчика).
6.2.3. Кривые размер — частота его наблюдения
Для большинства определений размеров частиц, полученные
кривые размер — частота следуют закону вероятности. Обычное
уравнение вероятности применимо к распределению, которое
симметрично относительно вертикальной оси, иногда называемому
Гауссовским распределением. Поскольку распределения разме-
ров часто «косые» или асимметричные, нормальный закон к ним
неприложим (см. рис. 6.5).
К счастью, в большинстве случаев асимметричные кривые
можно сделать симметричными, если размер откладывать на лога-
Рис. 6.5. Кривые распределения:
1 — симметричное (нормальное распределение); 2 — асимметричное распределение
Рис. 6.6. Типичное симметричное
распределение (гауссовское или
нормальное)
рифмической шкале (частота остается линейной). Такой вид
распределения известен как логарифмическое нормальное [6].
Уравнение кривой нормального распределения (рис. 6.6) в
применении к распределению размер — частота таково:
Г (d) = —--------exp
о \/2л
(d-dmf2l
2а2 J’
(6-1)
где F(d) —частота, с которой наблюдается диаметр; п — общее число наблю-
дений; dav - среднечисловой диаметр; о — стандартное отклонение, о =
=^/l.n(d — dav)2/'£n.
Константы dav п о полностью определяют кривую распределе-
ния для серии наблюдений. Таким образом, если размеры частиц
нанесены на сетку «арифметической вероятности», то совокупная
кривая представляет собой прямую линию, где средняя величина
(50%-ная величина) — простое среднечисловое dav- стандартное
отклонение о взято как о=(84,1% размера минус 50% раз-
Рис. 6.7. Гауссовская кривая (из рис. 6.6), построенная на основании диаграммы
арифметической вероятности
Рис. 6.8. Преобразование асимметричного
распределения (из рис. 6.4) в симметрич-
ный график с использованием логарифми-
ческой шкалы для размера частиц (мкм)
Логарифм размера, мкм
мера) = (50% размера минус 15,9% размера) (если график по-
строен с отрицательным наклоном, рис. 6.7).
Асимметричную кривую разделения, где размер частиц
нанесен на линейной шкале (рис. 6.3), можно превратить в сим-
метричную, если диаметры частиц нанести на логарифмическую
шкалу (рис. 6.8), т. е. уравнение (6.1) примет вид:
F (d) —---------exp
lgog-^л
— (Igrf —Igrfg)2
2 1g2 og
(6-2)
где de—среднее геометрическое; as определяется из равенства:
, 0g4—igrfg)2]
Z«
Параметры lgdg и 1g og называются средним логарифмиче-
ским геометрическим диаметром и логарифмическим геометри-
ческим стандартным отклонением соответственно. Они очень
важны, поскольку полностью определяют логарифмическое нор-
мальное распределение размеров, которое типично для процесса
диспергирования [7].
Простой способ построения графика логарифмического нор-
мального распределения размеров заключается в использовании
специальной, логарифмически вероятностной, масштабно-коорди-
натной сетки (рис. 6.9), где по оси ординат наносится размер
частиц, а по оси абсцисс — совокупный весовой (или числовой)
процент. Значение ds составляет 50% от величины распределе-
ния, a og — 84,1% величины, деленной на 50%-ную величину
(или 15,9% величины, деленной на 50%-ную величину, при отри-
цательном наклоне графика).
Стандартное геометрическое отклонение всегда одинаково
в логарифмическом нормальном распределении частиц по раз-
. •' I. О
Рис. 6.9. График распределения размеров (из рис. 6.4 и 6.8), представленный на
-------------— диаграмме логарифмической вероятности
мерам, поскольку размеры нанесены на график как совокупный
процент, числовой или весовой. Однако средние значения различны
и, следоватеЛьно, необходимо определить, применяется весовое
(dgw)* или числовое (dgc)** среднегеометрическое значение диа-
метра.
Уравнения преобразования Хэтча-Чоэйта [6] дают возмож-
ность превратить dgw в dgc. Они позволяют превратить один вид
«среднего» в другой и применяются при сравнении измерений
распределения размеров, произведенных разными методами:
Исходное
значение
dgrnt массовый
среднегеометри -
ческий диаметр
Требуемое значение
, ...
dfiC = antilgl------)
' Т.п '
числовой среднегеоме-
трический диаметр
== Z п, средне-
числовой
среднеповерхностный
/£п,
среднеобъемный
dvs = ^nd3/l nd\
среднеобъемно - поверх-
ностный
dw = Y nd'/Y nd3,
среднемассовый
Уравнение преобразования
1 g dgc = lg dgm — 6,908 • 1 g2oe
lg dl:c — 1g dsm — 5,757 lg2ofi
lg ds = 1 g de,„ — 4,605 lg2ofi
lg dt. = lg ds,„ — 3,454 -Ig’og
lg d»s=lg dg,„ —1,151 lg2o(,
lgdw = lgdgm+1,151 -lg2Og
* Обозначение индекса «gw» от английского geometrical weight — «геометри-
ческое весовое» (Прим, переводчика).
** Обозначение индекса «gc» от английского geometrical count — «геометри-
ческое числовое» (Прим, переводчика).
dgC, медианный dav
среднегеометри- d„
ческий. диаметр,
числовой dvs
dgm
d^y
lg d™ = lgdgC+1,151 -lg2aE
1g ds = 1 g dgC + 2,303 • Ig’og
lg dv = 1g dgC+ 3,454- lg2oE
lgrf„ = lg4r + 5,757-lg2Og
1 g dgr„ = 1 g dgC+6,908 • I g2Og
lg rf»=lg dac + 8,023• lg2Og
В преобразовании распределений размеров из числовых в мас-
совые возможны ошибки, так как наибольшие и наиболее тяже-
лые частицы часто присутствуют в статистически малых количе-
ствах. Джексон и др. [8] вычислили ошибки, которые вероятны
при таких преобразованиях, и показали, какие шаги следует
предпринять для того, чтобы эти ошибки были минимальны.
6.3. МЕТОДЫ ИЗМЕРЕНИЯ ЧАСТИЦ
Следует отметить, что трудность измерения размера частиц,
и особенно распределения частиц по размерам, обратно пропор-
циональна самому размеру; особенно сложен для измерений наи-
более часто встречающийся в лакокрасочной промышленности
субмикронный диапазон размеров частиц.
Вероятно, основным методом измерения все еще является
визуальное наблюдение в микроскоп или, для мелких частиц,
электронномикроскопическое фотографирование. При использо-
вании других применяются калибровочные стандартные мате-
риалы, например монодисперсные латексы. Появление современ-
ных зеркальных анализаторов устранило трудоемкость и утоми-
тельность таких измерений.
Методы измерения размеров частиц можно разделить на мно-
жество общих способов, а последние можно далее подразделить
на различные модификации, как показано в табл. 6.2.
Таблица 6.2. Методы измерения размеров частиц
Методы Диапазон размеров Распре’-' деление по раз- мерам Только средний размер
1. Методы прямого наблюдения а) микроскопия > 1000 нм Да б) электронная микроскопия/SEM— 5 нм — 5 мкм Да ТЕМ 2. Методы седиментации а) самопроизвольная седиментация > 100 мнм Да (или расслоение) б) ускоренная седиментация — центрифугирование/ультрацент- > 50 нм Да рифугирование — отмучивание 5—10 мкм Да — декантация 1—50 мкм Да
П родолжение
Методы Диапазон размеров Распре- деление по раз- мерам Только средний размер
3. Хроматографические методы а) гель-проникающая хроматография > 3 нм Да (ГПХ) б) разделение по гидродинамическим 90—1500 нм Да свойствам и по размерам в) фракционирование в потоке 10—1000 нм Да 4. Методы ситового анализа а) механические - просеивание . >20 мкм Да — ультрафильтрация 0,1—5 мкм Да б) электрические — воздействие электрического поля 0,5—500 мкм Да 5. Оптические методы (светорассеяние) 50—300 нм Да а) турбидиметрия (определение мут- 50—300 нм Да -—ности)- - — отклонениеугла падающего све- <500 нм Да та^ ’ Юл — приближение к максимуму и ми- > 200 нм Да* нимуму — степень поляризации <500 нм Да* — - асимметрия <200 нм Да — изменение длины волны 95—1000 нм Да* Да — жидкостная ультрамикроскопия 100 нм — 2 мкм Да б) дифракция Фраунхофера 4 -1800 мкм Да в) квантовая корреляционная спек- 10 нм—3000 нм Да* троскопия 6. Методы определения по площади по- верхности а) газовая адсорбция (теория ВЕТ) <5 мкм Да б) макромолекулярная адсорбция — адсорбция красителя 5 нм—100 мкм Да — титрование с помощью мыла 5 нм—100 нм Да в) калориметрия (метод Харкинса- Да Юра)
* Позволяет определить распределение по размерам.
6.3.1. Прямые измерения
Микроскопия и электронная микроскопия являются, вероятно,
такими методами измерения, которые позволяют установить пол-
ное распределение частиц по размерам. Монодисперсные частицы
латексов или золей золота, используемые в качестве стандартов
в ряде методов, проверены вышеуказанными методами и приняты
как эталоны в других способах измерений. Существуют хорошо
проверенные методы счета, в частности упомянутые в начале
настоящей главы, и множество путей их упрощения, например
использование современных анализаторов изображения [9, 10],
таких как Quantimet 920 [11], Magiscan 2 [12] или Zeiss Kontron.
Эти приборы могут напрямую присоединяться к оптическому
или электронному микроскопу или работать просто по фотогра-
фиям измеряемых частиц.
Зеркальные анализаторы состоят из двух частей: ячейки для
преобразования оптического изображения в электрический сиг-
нал и компьютера, который расшифровывает сигнал для получе-
ния количественной наглядной информации.
Развертка изображения может увеличить его четкость или
контрастность и дает возможность изучить различные частицы
и их параметры, такие как площадь поверхности, периметр, диа-
метр Ферета или Мартина, радиус круга эквивалентной площади
или периметра. Фактически существует только одно ограниче-
ние — программное обеспечение компьютера. Световой, луч мо-
жет быть использован для исключения или выделения*слипшихся
частиц. К сожалению, зеркальные анализаторы ' Очень дороги,
дороже некоторых типов электронных микроскопов'.’ Их точность,
однако, зависит от надлежащего приготовления.образца.
При приготовлении образца для прямого микроскопического
или электронномикроскопического измерения следует позабо-
титься о том, чтобы не привнести аппаратурные артефакты, на-
пример агрегирование частиц.
Мягкие или жидкие частицы особенно трудно подготовить
для электронномикроскопического изучения. Для получения ка-
чественных образцов, пригодных для такого исследования, требу-
ется замораживать пробу с последующим измельчением на холоду
[13]. Если это производится при высокой концентрации (напри-
мер, 15%) мягких частиц, то может произойти их коалесценция
даже при скоростях охлаждения более 1000 град/сек [14] с полу-
чением непригодных для анализа эмульсий. В таких условиях
следует разбавить эмульсию по меньшей мере до нескольких
процентов перед измельчением. Даже при успешном фракциони-
ровании при замораживании для получения правильного размера
частиц следует использовать специальную аппаратуру [15], так
как не во всех фракциях будет представлен истинный диаметр
частиц.
6.3.2. Седиментометрические методы
-I
Измерение размера частиц путем седиментации дает полный
анализ распределения по размерам, причем как при самопроиз-
вольной (под действием только сил тяжести), так и при ускорен-
ной (путем центрифугирования) седиментации. Измеряемый
диаметр — это диаметр Стокса, который представляет собой диа-
метр сферы той же плотности и скорости оседания, что и оцени-
ваемая частица.
Эти измерения всегда производятся при высоком разбавле-
нии, поэтому следует позаботиться о хорошем диспергировании
(дефлокуляции) частиц.
Простейший способ получения распределения частиц по раз-
мерам на легкодоступном дешевом оборудовании с точностью
от 2 до 5% — это использование пипетки Андреасена [16] (см.
рис. 6.10), состоящей из цилиндрического седиментационного
сосуда (объем около 550 мл) с 10-ти мл пипеткой, снабженной
двухходовым краном, которая позволяет отбирать пробы с опре-
деленной глубиной, обычно 20 см. Хорошо диспергированная
суспензия, не более чем с 1%-ным содержанием дисперсной фазы
помещается внутрь сосуда, который термостатируется на водяной
бане. Образцы дисперсии медленно отбираются через равные
промежутки времени (обычно время отбора вдвое больше интер-
вала между отборами проб), после чего определяется содержа-
ние твердой фазы на единицу объема.
Вычисления размера частиц основаны на определении началь-
ной концентрации частиц (нулевое время) и концентрации через
промежуток времени t.
Вычисление диаметра Стокса d производится по формуле:
й=Г——I7 . ю4. мкм, (6.3)
L(a-p)gd
где с — кажущаяся плотность частиц, г/см3 (эквивалентна истинной плотности
непористых частиц); р — плотность дисперсионной среды, г/см3; т; — абсолют-
ная вязкость дисперсной среды; h — расстояние (см), на которое падает частица
за время t (с); g— ускорение силы тяжести, см/с2.
Таким образом можно получить график зависимости суммар-
ного весового процента частиц от их диаметра. Данный метод
является стандартным британским тестом и полностью описан
в работе [17]. Диапазон размеров частиц обычно от 1 до 100 мкм,
Рис. 6.10. Схематическое представление
пипетки Андреасена
но если соблюдать нео ходимую осторожность, то метод можно
использовать для определения распределения по размерам тяже-
лых субмикронных частиц, например частиц пигментного ди-
оксида титана. Однако, из-за большого времени седиментации
частиц TiOz размером около 0,2 мкм, с целью ускорения измерения
пипетка Андреасена часто модифицируется таким образом, чтобы
было возможно отбирать пробы с различных глубин [18]. Анализ
ошибок, возможных при седиментационных измерениях, проведен
Сваровским и Алленом [19].
Видоизменением седиментационных измерений под действием
силы тяжести является использование ареометра [20]. В этой
методике измеряется плотность суспензии в точке ниже поверх-
ности суспензии, в центре тяжести поплавкового ареометра,
имеющего диапазон измерения плотности от 0,995 до 1;038 г/см3.
Ареометр помещается в седиментационный цилиндр; содержа-
щий хорошо диспергированную суспензию околосбО т? пигмента
в 200 мл дисперсионной среды; плотность дисперсии измеряется
через возрастающие экспоненциально промежутки: .'времени одно-
временно с измерениями в контрольном цилиндре? содержащем
дисперсионную среду, точность поддержания температуры
±0,1 °C. Процент пигмента, остающегося в суспензии через t
минут на уровне, где ареометром измерялась плотность су-
спензии, составляет [(/? — —В)] -100, где /?о и R—пока-
зания ареометра в начальный момент и в момент времени t, со-
ответственно; В — показания ареометра для дисперсионной среды.
" Диаметр Стокса определяется из уравнения (6.3), которое
может относиться к проценту пигмента, остающегося в суспен-
з'ии. Метод в основном применим к частицам диаметром от 1
до 15 мкм или даже меньшим, если их плотность выше.
п Использование ареометра предполагает, что разница плот-
ностей суспензии и дисперсионной среды пропорциональна кон-
центрации пигмента, и что плотность измеряется на уровне где-то
около центра тяжести ареометра. Положение этого уровня в из-
вестной степени неопределенное. Более того, ареометр погружа-
ется по мере протекания седиментации, так что глубина, на которой
производится измерение, увеличивается. Удаление и повторное
введение ареометра в суспензию также нарушает процесс осажде-
ния. Для преодоления этих трудностей можно использовать ма-
ленькие стеклянные тела с различной плотностью, которые погру-
жают в суспензию [21,22]. Они находятся в равновесии на уровне,
где их плотность равна плотности суспензии, которая, как пред-
полагают, пропорциональна концентрации. Поскольку длина этих
тел около I см, то точность измерения уровня достаточно высока.
Недостаток этого метода заключается в том, что стеклянные
тела обычно невидимы и их необходимо перемещать к боковой
поверхности сосуда с помощью магнита, что вносит возмущения
в дисперсию.
Рис. 6.11. Схема статического
рентгеновского аппарата:
1 — рентгеновская трубка; 2 —
образец; 3 — прибор для изме-
рения уровня; 4 — рентгенов-
ский детектор; 5 — платформа
переменной высоты
Другой подход к измерению размеров путем простой седимен-
тацйТГ на “бснОТе^закона Стокса^— использование седиментацион-
ных весов [23]. Современные методики используют микровесы,
например Cahn или Sartorius [24, 25], что дает возможность
автоматической записи и вычисления размеров с помощью про-
стых компьютерных программ. Установлено, что этот метод более
точен и дает более строгий анализ по сравнению с методикой,
используемой при работе с пипеткой Андреасена. Однако он го-
раздо дороже и лимитируется необходимостью иметь опреде-
ленную разницу плотностей частиц и дисперсионной среды, кроме
того, дисперсия должна быть дефлокулирована.
При исследовании размера частиц седиментационным мето-
дом производится аналитическое определение концентрации
нелетучих веществ в тщательно отобранной пробе пипеткой, а в
методике с использованием «седиментационных весов» — путем
взвешивания in situ. Другой удобный метод непрерывного опре-
деления концентрации — это измерение ослабления потока излу-
чения, проходящего через образец. В этом случае ослабление
должно быть пропорционально массе образца, через который про-
ходит луч. Поглощение видимого света может использоваться
для цветных пигментов, которые дают незначительное свето-
рассеяние. Однако для частиц с сильным светорассеянием, на-
пример двуокиси титана, следует использовать рентгеновское
излучение. Мэрли [26] описывает такой аппарат (показанный
схематически на рис. 6.11). Примером промышленного прибора,
работающего на этом принципе, может явиться Sedigraph 5000ЕТ.
Все вышеупомянутые методы основаны на седиментации под
действием силы тяжести и потому занимают много времени в
случае малых частиц. Для ускорения седиментации можно при-
менить центрифугование, например использовать дисковую цент-
рифугу Джойса-Лебла, а для более мелких частиц — ультра-
центрифугу [27]. Центрифуга Джойса-Лебла [28] имеет полый
дискообразный прозрачный цилиндр, который вращается с зара-
нее выбранной скоростью. Измеряемые частицы вводят в диск
с использованием специальных приспособлений, препятствую-
щих начальному оседанию частиц. Для определения размера
частиц используется модифицированное соотношение Стокса:
/ = 9т]/2ш'2с(2 Др 1п(г2/г|),
где t — время, прошедшее с момента введения образца; л — вязкость центри-
фугируемой жидкости; w — скорость вращения диска; d — эквивалентный диа-
метр частицы; Др — разность плотностей частицы и жидкости; и — исходный
радиус центрифугируемой жидкости; гг — радиус на глубине отбора проб.
В первых аппаратах такого типа для изготовления диска
использовали специальные материалы, поэтому природа центри-
фугируемой жидкости была сильно ограничена. Более современ-
ная модель («Центрифуга Джойса-Лебла дисковая 4») имеет
стойкий к растворителям диск; изготовитель обещает получение
сравнительных кривых распределения частиц в диапазоне раз-
меров от 0,01 до 60 мкм (в зависимости от плотности) при не-
прерывной записи продвижения частиц через луч света. Истинные
кривые распределения можно получить при калибровке прибора
с помощью стандартных образцов, но Требуется тщательная
интерпретация данных [29].
Проблема исследования светорассеивающих частиц, для ко-
торых ослабление света на единицу массы образца сильно зави-
сит от размера частиц, разрешена Хорнби и Тунсталлом [30],
использовавшими рентгеновские лучи, как указывалось выше.
Они применили этот принцип к конструированию дисковых цент-
рифуг, которые легко позволили им измерить мелкие частицы,
и использовали этот метод для сравнения распределения частиц
двуокиси титана, полученных при измельчении пигмента на раз-
личных видах измельчающего оборудования [26].
Отмучивание [31]. Следствием того факта, что частицы про-
ходят через жидкость, является вывод, что жидкость проходит
через частицы; это является основой отмучивания и измерения
проницаемости. Отмучивание (в противоположность седимента-
ции) происходит, когда жидкость (чаще газ) проходит через
слой порошка и, таким образом, может использоваться для опре-
деления распределения по размерам. Основой метода является
способность быстротекущей жидкости поддерживать частицы
меньше определенного размера. Здесь применяются следующие
уравнения:
\ ро / \ Ро /
для ламинарного течения для турбулентного течения
где (в единицах СГС): р и ро — плотность частиц и жидкости; d — диаметр
частиц; V — скорость жидкости; V,,, — максимальная скорость частиц; К5 и Ki —
константы, зависящие от формы частиц.
Значения последних констант таковы [32]:
Форма частицы К/
Сферическая 54,5 24,5
Неправильная (кварц) 36.0 50,0
Частицы могут быть фракционированы по размерам путем
изменения количества воздуха, проходящего через сопло, или
путем изменения размера камеры отмучивания. Таким образом
можно получить полное распределение по размерам, собирая раз-
личные фракции в отдельные приемники. Методы отмучивания
сейчас непопулярны, так как процессом обычно трудно управ-
лять [33].
Измерение проницаемости. Первый стандартный аппарат;
в котором используется способность газа проникать через слой
порошка, по предложению Кармана [34] создали Ли и Нурз [35].
Гуден и Смит. [86] модифицировали его; они использовали номо-
грамму для упрощения подсчета частиц и прибор стал выпускаться
промышленноопод названием «Субситовый анализатор Фишера».
Прибор'Д^ет; хорошие результаты в диапазоне размеров от 1
до 50 мкм, но не может использоваться ниже этих пределов, по-
скольку механизм течения в порошках субмикронных частиц
в основном молекулярный и не ламинарный, что является осно-
вой определения размеров частиц методом суб-ситового анализа.
Рижден [37] показал, как производить подсчеты для моле-
кулярного течения и для скопления частиц, которые могут дать
представление о «естественной» пористости при низких скоростях
потока и в которых составляющие частицы действуют как инди-
видуальные. Печоукас и Гэйдж [38], Карман и Малхерби [39];
а также Хутто и Дэвис [40] усовершенствовали аппарат; послед’
ние показали, что их результаты хорошо согласуются с измере-
ниями площади поверхности по теории ВЕТ.
f
6.3.3. Хроматографические методы
Гель-проникающая хроматография (ГПХ) стала широко при-
нятым методом измерения размеров молекул полимеров. Прин-1
цип измерения основан на рассмотрении молекулы полимера как
«частицы» определенного размера, а молекулярно-массовые
распределения калибруются относительно стандарта — молеку-
лярной массы полистирола, соответствующего данному объему;
Метод широко используется для определения молекулярных масс
через молекулярные объемы [41, 42]. Мы не предлагаем обсу-
ждать этот подход, так как свернутая молекулярная цепь, состав-
ляющая «частицу», не является частицей в контексте настоящей
главы. Существует много отличных серийных приборов и много
книг и статей по этому вопросу. Приборы для ГПХ дороги, и нет
необходимости в применении этого метода для определения моле-
Рис. 6.12. Схематическое представление гидродинамической хроматографии (а)
и хроматографии по исключению размера (6)
кулярно-массового распределения, так как имеются дешевые
видоизменения ГПХ для таких исследований на основе тонко-
слойной хроматографии [43, 44].
Применение ГПХ для более крупных, твердых частиц известно
как «эксклюзивная» или «гидродинамическая» хроматография.
Хотя эти два типа хроматографии отличаются по своим прин-
ципам, они могут применяться одновременно в хроматографи-
ческой колонке. Они схематически представлены на рис. 6.12.
В гидродинамической хроматографии, ввиду того, что более
крупные частицы находятся дальше от стенок капилляра, они
подвергаются воздействию более быстрого потока в центре ка-
нала по сравнению с меньшими частицами, чьи центры находятся
ближе к медленному потоку у стенки канала.
В эксклюзивной хроматографии малая частица может «найти
убежище» от градиента скорости в порах, недоступных для боль-
ших частиц. В обоих случаях большие частицы проходят через
колонку быстрее, чем меньшие; измерения производятся по вре-
мени их удержания как функции размера, как и в ГПХ-анализе.
Хотя имеется много работ по хроматографическому измерению
размеров частиц [45—49], эти методики пока не стали общими
и не нашли применения для исследования пигментов. Однако не-
давно начат выпуск прибора, основанного на этих принципах,
для анализа латексов («Flow Sizer HDC 5600»), который дает
полный анализ распределения по размерам частиц латексов от 30
до 1500 нм с разрешением до 5% от размера частицы. Колонка
его содержит катионообменную смолу [50], ограничивая таким
образом анализ для анионных латексов. Колонка стабилизи-
руется продолжительным циркулированием элюента, после чего
прибор готов к работе. Хотя принцип фракционирования для
измерения размера прост, устройство прибора сложное, требую-
щее достаточно мощного мини-компьютера для обработки сиг-
нала детектора. Фракционирование в потоке [51] —метод раз-
деления частиц по размерам, и, следовательно, метод измерения
размеров, основанный на использовании поля, воздействующего
на суспензию, текущую в узкой трубке (рис. 6.13). Приложенное
Поток -«—]
1 Поле
* (например, гравитационное)
— Градиент
-tt .. потока
Рис. 6.13. Принцип фракционирования потока в поле
поле может иметь термический градиент [52], а также быть гидро-
статическим или гравитационным полем. Киркланд и сотр. [53,
54] описали методику, использующую аппарат с переменным гра-
витационным полем, названный TDE-SFFF, который позволил
получить распределение по размерам в диапазоне <0,01--
1,00 мкм:.;дл^-.. различных пигментов: диоксид титана, сажи,
Фталоцианинового голубого, а также различных латексов.
— По- сравнению с аналогичными приборами с постоянным при-
ложенным-: пешем, TDE-SFFF дает более быстрый анализ и более
высокую чувствительность детектирования благодаря повышен-
ной разрешающей способности. Время удерживания коррелирует
с логарифмом размера частиц (чем последний больше, тем выше
время удерживания), причем плотность частиц одинакова. В слу-
чае, если образец содержит различные компоненты с разными
плотностями, точная корреляция невозможна. Тем не менее, ка-
чественные изменения в составе могут быть «отпечатками паль-
цев» образца.
6.3.4. Методы ситового анализа
Просеивание — это старый, хорошо проверенный метод фрак-'
ционирования частиц по размерам; он, однако, не так прост, как
может показаться. Хотя разработаны микросита с диаметром
отверстий менее 5 мкм [55], метод обычно используется для фрак-
ционирования частиц от 20 мкм до 125 мкм, с использованием
стандартных тканых проволочных сит.
Вес частиц, собранных между двух сит в конце просеивания,
может быть легко определен. Однако, номинальный диаметр
отверстия не может представлять собой «размер сечения» сита.
Просеивание — неидеальный процесс, и упомянутый размер сле-
дует определять независимо, используя для точных исследова-
ний известный материал.
Сита, используемые для ситового анализа, стандартизованы.
Их номинальные диаметры располагаются в ряды прогрессий.
Они могут изготавливаться из тканых проволочных сеток, как
в британском стандарте BS 410 (1969), или быть микроячеистыми
ситами, изготавливаемыми методами гальванопластики (ASTM
EI61—607). Диаметр отверстий стандартного сита не может быть
строго одинаковым и поэтому должен характеризоваться распре-
делением по диаметру отверстий сита.
Классификация на ситах зависит от многих факторов, не за-
висящих от размеров ячейки, например от нагрузки на частицу,
встряхивания, тенденции частиц к агломерации, количества
частиц с размерами, близкими к размеру ячейки и т. д. Лешков-
ский [56] предполагает, что лучше строить график зависимости
количества просеянных частиц от медианного размера отверстий
сита, а не от величины ячеек. Сито следует характеризовать его
медианной ячейкой, ее доверительным интервалом и стандарт-
ным отклонением, полученным при калибровке.
Особую тщательность следует соблюдать при очистке тонких
сит; для них (например, для сит ниже 200 мкм) следует избегать
применения щеток, так как можно легко повредить ячейки; такие
сита целесообразно очищать ультразвуком. Сверхтонкие сита
требуют применения специальной технологии мокрого просеива-
ния [57, 58].
Принципы просеивания. При просеивании порошок разделя-
ется на две части: одна проходит через ячейки, другая остается
на сите. Процесс усложняется для несферических частиц, кото-
рые проходят через отверстия в сите только тогда, когда распо-
лагаются в благоприятном направлении.
Процесс просеивания можно разбить на две стадии: 1) от-
сеивание частиц, значительно меньших размера ячеек, что про-
исходит достаточно быстро; 2) отделение частиц с размерами,
близкими к размеру ячейки, что происходит постепенно и ни-
когда не достигается полностью.
Общий подход заключается в отделении мелких частиц и в
определении «конечной точки» просеивания, когда отделение
более крупных частиц практически достигает предела. Выбор
сухого или мокрого просеивания зависит от характеристик мате-
риала. Очень мелкие частицы быстрее отделяются при мокром
просеивании, которое может также уменьшить разрушение рых-
лых материалов, но может дать результаты, отличные от резуль-
татов сухого просеивания.
Существуют два альтернативных метода определения «конеч-
ной точки», которая указывает на завершение просеивания час-
тиц, близких к размеру ячейки: 1) просеивание до тех пор, пока
скорость процесса не уменьшится до определенного веса (про-
цента) в минуту; 2) просеивание в течение определенного вре-
мени.
Первый метод лучше, но с целью упрощения процесса для
стандартных испытаний можно использовать второй метод при
условии, что выбранное время просеивания оптимально.
И сухое, и мокрое просеивание, а также устройство механи-
ческого вибратора для сит описано в британском стандарте BS
Рис. 6.14. Схематическое представление принципа Коултера:
1 — чувствительная зона; 2 — вакуум; 3— главный усилитель; 4 — пороговый фильтр;
5 — вибрационный усилитель; 6 — электроды; 7 — осциллограф; 8 — счетчик; 9 — печа-
тающее устройство; 10 — горизонтальная развертка
1796 (1976). Обзор процессов просеивания, а также теория и
практика просеивания рассмотрены в работах [59, 60].
Ариетти [61] расширил методику просеивания для анализа
распределения по размерам частиц в субмикронном диапазоне
размеров, используя для этого микропористую фильтрацию.
Результаты исследований хорошо согласуются с данными элек-
тронной микроскопии.
Электрическое сопротивление (принцип Коултера). Принцип
Коултера позволяет измерить частицы размером от нескольких
сотен микрон до 1 мкм, а при большой тщательности измерений —
даже до 0,4 мкм.
Метод первоначально использовался для подсчета клеток
крови [62—64]. Предложены модификации [65], позволившие
использовать этот метод также для определения объема клетки.
Принцип действия заключается в том, что между двумя электро-
дами, помещенными в два разделенных резервуара, которые
связаны стеклянной чувствительной зоной (отверстие), протекает
постоянный ток (см. рис. 6.14). Ртутный сифон временно выво-
дится из равновесия с помощью вакуума. При закрывании крана
суспензия засасывается через отверстие за счет возвращения
ртути в равновесие. Электрические контакты замыкаются ртутью
при прохождении через отверстие определенного точного коли-
чества суспензии. При этом изменяется сопротивление электро-
литз, что вызывает колебания напряжения, пропорциональные
объему частиц. Эти вызванные частицами колебания усиливаются
и подсчитываются в измерительном контуре. Калибровка при-
бора выполняется для определенного раствора электролита,
который используется обычно со стандартными монодисперс-
ными латексами с известным диаметром частиц. Калибровочный
коэффициент k определяется из соотношения:
dc=kt'/3,
где de — известный диаметр; — пороговый уровень для средних импульсов,
генерируемых суспензией с известным размером частиц.
При наличии откалиброванного таким образом прибора со
стандартным латексом, константа k будет сохраняться для всех
других пороговых размеров при использовании того же элек-
тролита.
Корректировка случайностей. Измерение импульсов осно-
вано на предположении, что каждый импульс вызывается одной
частицей. Однако, в чувствительную зону могут входить вместе
две частицы, давая непропорциональный сигнал. При проведении
измерений в пределах определенных скоростей подсчета влияние
случайностей может быть минимизировано со статистической
точки зрения, и оно может быть оценено экспериментально при
проведении исследований в сильно разбавленных суспензиях.
Принцип Коултера является очень распространенным, надеж-
ным и удобным методом определения размеров частиц более
0,6 мкм, что проверено более чем в тысяче работ [66, 67]. Particle
Pata Inc. [68] производит простой прибор для' таких работ,
Elzone 180, который может соединяться с дополнительным микро-
процессором и (или) легко совмещаться с различными большими
компьютерами. Минимальный размер частиц, который можно из-
мерить этим методом (примерно 2% диаметра отверстия), огра-
ничивается невозможностью создать соответствующие отверстия,
меньшие 12—15 мкм, а не электронной частью прибора.
6.3.5. Оптические методы (светорассеяние]
Хороший обзор теории светорассеяния применительно к раз-
меру частиц сделан Керкером [69], а общие методы, используе-
мые для измерения размеров частиц, обсуждены Коллинзом
и др. [70].
1 Причиной рассеяния света является его повторное излучение
благодаря колебанию диполей в поляризуемых частицах, полу-
ченных при воздействии осциллирующего электрического поля
луча света. Поляризуемость на единицу объема в любой момент
времени в каждой точке может быть выражена как сумма постоян-
ного (возрастающего с рефракцией) и переменного слагаемого,
которое дает рассеяние.
Если мы ограничимся сферическими, неадсорбирующими и
невзаимодействующими частицами, то светорассеяние в основ-
ном определяется двумя факторами: отношением размера частиц
к длине волны падающего света в среде (с//Х) и относительным
показателем преломления, m = n1/n2, где п\ и и2— показатели
преломления частиц и дисперсионной среды соответственно. На
практике разбавленная дисперсия облучается узким интенсив-
ным лучом монохроматического света, и измеряется интенсив-
ность рассеянного света под некоторым углом 0 от падающе-
го луча.
Существует три наиболее общих подхода к измерению раз-
мера частиц:
1) по мутности (пропусканию), где 0=180° и измеряется
интенсивность света;
2) интенсивность света измеряется под некоторым фиксиро-
ванным углом (обычно 90°);
3) интенсивность рассеянного света измеряется как функция
угла.
Измерения обычно выполняют при бесконечном разбавлении,
так что можно применять теории Рэлея (когда с//Х<^1), Рэлея-
Дебая [кргда («1 — и2)-с/Д«1] или Лоренца-Мая [когда
(«I — и2), которые справедливы для одного рассеиваю-
щего. центра. Для рассеивателя Рэлея интенсивность рассеян-
ного света I не зависит от угла:
_ 16n4d6 (и? — и!)2
х2Х4(и2 -риг)2
где х — расстояние между образцом и детектором.
Интенсивность рассеяния зависит от угла для рассеивателя
Рэлея-Дебая, а для рассеивателя Лоренца-Мая получается еще
более сложная зависимость от угла. Концентрированные ра-
створы дают многократное рассеяние, и теория в этом случае
неопределенна, за исключением полуэмпирических соображений.
Преимущества и недостатки методик пропускания, диссиметрии,
максимума — минимума, отношения поляризации и др. приве-
дены в упоминавшемся обзоре [70].
Недавнее расширение турбидиметрического подхода дало
возможность также определять распределение частиц по раз-
мерам [71]. Вдобавок существует метод, запатентованный ком-
панией Tioxide International [72], согласно которому спектро-
фотометрически измеряется пропущенный свет при трех различ-
ных длинах волн. Это позволяет измерять средний размер частиц
и его стандартное отклонение, что полезно при определении эффек-
тивности помола. Ноббс [73] недавно описал метод, использую-
щий тонкопленочную технологию, что является модификацией
Рис. 6.15. Схематическое представление измерения размеров по методу дифракции
фраунхофера (интенсивность на каждом элементе детектора равна сумме интен-
сивностей от частиц данного размера):
1 — большая частица; 2 - лазерный луч; 3 — малая частица; 4 — многоэлементпый коль-
цевой детектор (вид сбоку); частицы А и В одинакового размера (следовательно, их углы
дифракции равны)
обычных измерений светорассеяния, но дает возможность изме-
рить кажущийся размер частиц при высоких концентрациях.
Также интересны измерения показателя преломления дисперсии,
из которых можно получить диаметр частицы [74].
Дифракция Фраунхофера. По мере увеличения размера час-
тицы и приближения его к длине волны источника света X, коли-
чество света, рассеянного в направлении падающего луча, уве-
личивается и становится намного больше, чем количество света,
рассеянного в других направлениях. Когда размер частицы d
много больше X, теория дифракции Фраунхофера (FD) описы-
вает свойства частицы в отношении рассеяния света в направ-
лении падающего луча, что можно рассматривать как предель-
ный случай теории Лоренца-Мая. Теория FD показывает, что
интенсивность рассеяния (дифракционная картина) пропорцио-
нальна, а величина угла рассеяния обратно пропорциональна
размеру частицы (рис. 6.15). При этом используется Фурье-
преобразованная линза (линза, расположенная между части-
цами и детектором таким образом, что детектор находится в
фокальной плоскости линзы).
Типичный прибор FD показан на рис. 6.16.
Поскольку геометрия и расположение линзы в приборе удовлет-
воряют требованиям теории FD [75], дифракционная картина
движущейся частицы будет стационарной.
Детектор, состоящий из ряда световых сенсоров, анализи-
рует распределение световой энергии (источник—луч мало-
мощного лазера) на определенной площади, и микропроцессор
вычисляет распределение частиц по размерам.
Приборы этого типа, например PDPS11-C, который можно
использовать для анализа различных проб, предназначены для
точного измерения распределения частиц по размерам, начиная
с нескольких микрон. Они могут быть полезны в определении
2
Рис. 6.16. Схема'типичного прибора для измерения размеров частиц аэрозолей по
методу дифракции Фраунхофера:
1 — лазер; 2— частицы аэрозоля; 3 — многоэлементный детектор; 4— усилительный
блок; 5 — микропроцессор^??—"блок" записи и регистрации^"? —"линзы для Фурье-пре-
образования; S — распылительное сопло; 9 — линзы для уширения луча
размера частиц красочных аэрозолей, получающихся при на-
несении лакокрасочных материалов методами распыления, в опре-
делении распределения частиц в жидкостях (приборы Malvern
3600Е ED, Microtrac, HIAC).
Корреляция анализов размеров частиц, выполненных с по1
мощью приборов HIAC, Coulter Counter, Sedigraph, Quanti-
met 720 и Microtrac приведена в работе [76].
Фотон-корреляционная спектроскопия. Другой подход к ис1
пользованию светорассеяния для определения размера частиц —
фотон-корреляционная спектроскопия (PCS) * или квазиупругое
светорассеяние [75].
С появлением в начале 60-х годов лазеров, которые давали
интенсивное когерентное монохроматическое излучение, появи-
лась возможность использовать для измерения размера частиц
время-корреляционные функции. Последние — способ описания
флуктуаций некоторого свойства (в данном случае числа испу-
щенных фотонов) методами статистической механики. Такой
анализ требует когерентного монохроматического излучения и
исследует флуктуации последнего, связанные со случайным пере-
мещением светорассеивающих центров в малом объеме, что дает
информацию о коэффициенте диффузии таких центров.
При изучении интенсивности светорассеяния на временной
основе найдено, что она будет колебаться около среднего значе-
ния, если светорассеивающие частицы участвуют в случайном
* Сокращение от английского «photon correlation spectroscopy» (Прим,
переводчика).
Рис. 6.17. Блок-схема измерителя частиц BI-90:
/ — фокусирующие линзы; 2— калиброванные зазоры; 3 — трубка катодных лучей; 4 —
усилитель; 5 — фотоумножитель; 6 — ячейка; 7—лазер; 8 принтер; 9 — компьютер;
10 — коррелятор
(Броуновском) движении. Рассеянное электрическое поле —
функция положения частицы и, следовательно, постоянно изме-
няется. Интенсивность (пропорциональная площади электриче-
ского поля) также колеблется во времени. При измерении ука-
занных флуктуаций возможно определить, используя автокорре-
ляционную теорию для определения коэффициента диффузии для
частиц, как эти флуктуации затухают за более продолжительные
промежутки времени. Это, в свою очередь, может быть соотне-
сено через уравнение. Стокса-Эйнштейна с диаметром частицы,
если сделать определенные предположения относительно формы
частиц, и известна вязкость среды.
Типичная экспериментальная установка приведена на рис. 6.17.
Интенсивность рассеянного света — усредненная величина и
является функцией индивидуальных свойств, так же как давле-
ние газа — интегральный результат бомбардировки газовыми
молекулами стенок сосуда.
г Для разбавленных суспензий с частицами, меньшими X, спра-
ведливо равенство:
(/,(</)> =.K/VM2.P(G)B(C), (6.4)
'где —интенсивность рассеяния, усредненная во времени; q — вектор
волновой амплитуды флуктуации рассеяния; К — оптическая константа; М —
масса частицы; N — число частиц, вносящих вклад в светорассеяние; Р— фактор
формы частицы; 0 — угол светорассеяния; В — концентрационный фактор;
С — концентрация частиц.
Флуктуации, связанные с термическим возбуждением, могут
быть разложены на различные частоты, и при любом угле рас-
сеяния, обусловленного данной флуктуацией, вектор волновой
амплитуды может быть выражен в виде:
q = (4шг/Х) sin 0/2,
где п — показатель преломления.
Масса сферической частицы пропорциональна с/3, следова-
тельно, параметр М2 в уравнении (6.4) приведет к параметру d6
при рассеянии. Фактор формы частицы, Р(0), известен для
простых форм и для размеров, меньших X. Однако, в пределе,
при стремлении к нулю, факторы формы частиц стремятся к еди-
нице. Следовательно, угловые PCS-измерения с экстраполяцией
к нулевому углу требуются для полидисперсных образцов с круп-
ными частицами. Если частицы являются макромолекулами и
находятся в растворе, уравнение (6.4) можно использовать для
определения молекулярного веса.
Как было найдено, методика PCS очень надежна и эквива-
лентна электронной микроскопии для определения размера моно-
дисперсных частиц [77]. Однако, для полидисперсных систем
метод гораздо более проблематичен, так как информация о рас-
пределении, получается из анализа сумм показательных функций,
которые вносят вклад в измеряемую автокорреляционную функ-
цию. Существуют различные математические подходы к решению
этой задачи. Наиболее распространенный из них — это «накопи-
тельный анализ», который дает два параметра размеров: сред-
ний диаметр и фактор полидисперсности [78]. Если измерения
не экстраполированы к нулевому углу и концентрации, кажу-
щийся размер зависит от угла и концентрации.
В настоящее время PCS-измерения, за исключением моно-
дисперсных систем, относительно нечувствительны к распределе-
нию по размерам. Показано, что разделение на два пика воз-
можно, если соотношение размеров больше, чем примерно 2:1,
хотя в этом отношении постоянно делаются усовершенствования.
Часто приборы изображают гистограммы, которые дают
классификацию размеров вдоль оси абсцисс; однако, показатель
вдоль оси ординат не называют обычно «процент количества
частиц данного размера», а используют термины: «процент отно-
сительного светорассеяния» или «распределение частиц по раз-
мерам (в произвольных единицах)».
Распределение частиц по размерам зависит от величины пара-
метра прибора, который весьма чувствителен к присутствию пыли.
В некоторых приборах влияние частиц пыли уменьшено за счет
процесса вычитания этого эффекта, основанного на использо-
вании подхода с задержкой базовой линии, что эффективно исклю-
чает вклад в рассеяние частиц, размер которых больше заранее
установленного.
В основе PCS лежит предположение (как и вообще для свето-
рассеяния), что все частицы гомогенны по составу, так что не
требуется даже знать их показатель преломления (т. е. требуется
знать последний только для непрерывной фазы). Это на практике
означает, что нельзя измерить средний размер частиц смеси двух
различных латексов разных полимеров, например, смеси поли-
стирольного и акрилового латексов.
Существует много промышленных приборов для PCS с исполь-
зованием фиксированного угла (обычно 90°), например Coulter
Electronics Nanosizer [79] или более новые модели серии N4,
которые могут измерять при различных углах. Brookharen Instru-
ments Corp. (США) производит прибор В1—90 с фиксированным
углом и противопыльным фильтром и прибор «автоматический
гониометр» В1—200 SM с диапазоном углов от 15 до 160°, кото-
рый можно использовать совместно с коррелятором В1—2020.
фирма Malvern Instruments производит систему «4600» — фотон-
корреляционный анализатор частиц с переменным углом и при-
бор с фиксированным углом («Autosizer»), а фирма Nicomp
Instruments (сейчас фирма является филиалом HIAC/Royco
Instruments) также выпускает много моделей приборов, напри-
мер лазерный измеритель частиц — модель 220. Все эти приборы
дают «средний» диаметр частиц и показатель распределения по
'размерам.
Ультрамикроскопия. Классический метод [80] определения
размера коллоидных частиц за пределами разрешения оптиче-
ского микроскопа — это использование затемненного поля наблю-
дения (ультрамикроскоп) с целью подсчета числа частиц в еди-
нице объема. Зная массу частиц на единицу объема, легко опре-
делить их средний диаметр.
Дерягин и сотр. [81] улучшили этот метод путем введения
жидкостной техники для упрощения подсчетов. С тех пор и дру-
гие авторы ввели усовершенствование методики [82].
Уолш и др. [83] сконструировали жидкостной ультрамикро-
!С'коп с диапазоном измерения от 0,1 до 2,0 мкм. Используя детек-
тирование и измерение интенсивности лазерного излучения (с при-
менением фотоумножителя), они получили возможность измерять
’также распределение по размерам. Их детекторное устройство
'Пригодно для материалов с пониженными показателями прелом-
ления, таких как глина, полимеры или частицы масла. Однако,
оно непригодно для измерения размеров частиц материалов
'с высоким показателем преломления, например диоксида титана.
6.3.6. Методы определения площади поверхности
Газовая адсорбция. Зная площадь поверхности частицы на
единицу веса и ее плотность, можно найти средний размер час-
тицы. Так, если частица рассматривается как правильная сфера,
то отношение объема к площади равно г/3, где г — радиус экви-
валентной сферы.
Газовая адсорбция [84] — наиболее частый метод опреде-
ления площади поверхности твердого тела. В принципе методика
газовой адсорбции может применяться к любой системе газ —
твердое тело, но на практике метод ограничен теми типами адсорб-
ции, с которыми мы сталкиваемся.
Для определения площади поверхности необходимо иметь
соответствующие значения площади поверхности адсорбирую-
щейся молекулы и знать число молекул, образующих монослой.
Уравнения для определения этих параметров зависят от природы
сил взаимодействия между газом и твердым веществом. Если эти
силы совершенно неспецифические, т. е. наблюдается физическая
адсорбция, то монослойное покрытие можно рассмотреть с исполь-
зованием полуэмпирического уравнения ВЕТ. Если, с другой
стороны, наблюдается химическая адсорбция, то также получа-
ется монослой, но занятая адсорбированными молекулами пло-
щадь будет зависеть от строения кристаллической решетки ато-
мов субстрата. Многие системы газ — твердое тело по типу ад-
сорбции являются промежуточными, и им нелегко дать опреде-
ление. Это справедливо, например, для активированных углей
или глин. w
В классическом оборудовании для измерения площади по-
верхности используется вакуумная стеклянная посуда. Современ-
ное доступное промышленное оборудование обычно изготовля-
ется из металла и использует электронные приборы вместо тради-
ционных ртутных манометров и прибора Маклеода.
Площадь поверхности сорбции газов определяется из гра-
фика полной изотермы адсорбции с помощью таких автомати-
ческих приборов, как Carlo Erba’s Sorptomatic или Micromeritics
Digisorb 2600. Хотя автоматика может уменьшить время обра-
ботки результатов, она не может изменить время достижения
равновесного состояния. Для ускорения измерений приборы скон-
струированы таким образом, что в них заложены некоторые до-
пущения относительно природы уравнения ВЕТ, что позволяет
определять площадь поверхности по одной экспериментальной
точке. Это относится к прибору Micromeritics 2200, использую-
щему статистический метод, и прибору Perkin — Elmer’s Sorpto-
meter, использующему динамический адсорбционный метод. Оба
прибора способны определить площадь поверхности менее чем
за час после подготовки образца.
Подготовка образца имеет большое влияние на результат
измерений, и следует позаботиться о том, чтобы природа поверх-
ности не изменялась в результате предварительной обработки.
Одно из преимуществ измерения площади поверхности путем
газовой адсорбции заключается в возможности определить нали-
чие пористости. При температуре жидкого азота последний кон-
денсируется в порах согласно уравнению Кельвина; форма изо-
термы [85] показывает размер пор от 1,5 до 30 нм, причем наи-
более удачные результаты получаются для IV типа изотермы.
(Размеры пор выше 8 нм обычно определяются с использова-
нием ртутной порометрии. Типовые промышленные приборы для
определения пористости могут работать в пределах до 7,5 нм,
что соответствует давлению порядка 2000 атм.)
Адсорбция растворенных веществ. Альтернативный путь изме-
рения площади поверхности — использование адсорбции раство-
ренных веществ, например жирных кислот, из растворов [86,
87] Несмотря на то, что площадь поперечного сечения молекулы
жирной кислоты в вертикальной ориентации составляет 20,4 А2
на молекулу, последняя необязательно примет это положение при
адсорбции на всех поверхностях и во всех растворителях. Поэтому
необходимо вначале, перед проведением измерений площади по-
верхности, установить природу адсорбции и эффективную пло-
щадь, занимаемую молекулой.
Адсорбция красителей также часто использовалась для изме-
рений, поскольку их концентрацию можно легко и точно опреде-
лить колориметрически [88]: В этом случае также требуется тща-
тельность в определении площади, занимаемой молекулой, о чем
пишут Киплинг и Уилсон [89], определившие, что площадь, при-
ходящаяся на одну молекулу метиленового голубого, равна 102—
108 А2. Лайнж и др. [90, 91], однако, нашли что при адсорбции
на одну молекулу этого красителя приходится 69,6—76,0 А2, что
примерно соответствует образованию димера.
При определении площади поверхности путем адсорбции кра-
сителей считают [92], что необходимо предварительно провести
-калибровку по более надежному методу газовой адсорбции.
г Грегг и Синг [93] полагают, что при использовании адсорб-
ции красителей для измерения площади поверхности должны
’соблюдаться следующие условия:
• 1) следует ограничиваться случаями, когда краситель доста-
-точно растворим и на изотерме появляется четкое плато;
т-г 2) должна быть известна ориентация молекул;
г > 3) должно быть известно число молекулярных слоев.
-о, Однако, если применяется эта методика, площадь поверхно-
сти может быть легко определена даже без спектрофотометра,
который используется обычно для определения концентрации
красителя в растворе. Например, для определения концентрации
красителя в маточном растворе после адсорбции необходимо лишь
разбавить раствор, чтобы цвет стал слабее, чем цвет раствора
известной концентрации. Затем раствор помещают в испытатель-
ный сосуд в качестве эталона цвета. Путем прибавления извест-
ного объема цветного раствора во второй аналогичный сосуд и
разбавления его чистым растворителем до тех пор, пока цвет
в обоих сосудах станет идентичным, находят полученную концен-
трацию. Здесь глаз выступает в роли компаратора цвета.
«Поверхностные» методы. Для измерения площадей поверх-
ности также предлагается калориметрическое определение теплот
погружения [94]. Однако, преимущества метода часто перевеши-
ваются малым количеством выделяемой теплоты, вследствие чего
Для проведения измерений требуется тщательность и хорошее
приборное оснащение [95].
Еще в 1934 г. предложен метод [94], который был использо-
ван для определения размеров чешуек алюминия [96].
На приборе типа весов Лангмюра производится разрыхление
и уплотнение распределенных на поверхности’ воды чешуек до
тех пор, пока не получится постоянная, компактная, плоская
поверхность. Зная вес образца и размеры поверхности, занимае-
мой слоем, можно вычислить размеры частиц. В работе [98]
таким образом определен размер чешуек слюды в диапазоне 2—
30 мкм и частиц песка в диапазоне 50—200 мкм. Аналогичные
вычисления проведены для субмикронных частиц латексов, распре-
деленных на поверхности раздела воздух — вода [99] и масло —
вода [100]
6.3.7. Какой из методов является наилучшим!
На этот вопрос нет однозначного ответа. Так, два независи-
мых метода лучше одного, повторенного, так как в сочетании они
более информативны. Например, если средний размер частиц ТЮ2
определить из удельных площадей поверхности, используя адсорб-
Рис. 6.18. Числовое распределение размеров частиц
Рис. 6.19. Массовое распределение размеров частиц
SO 100 200 SOO 1000 3000 (нм)
Рис. 6.20. Сравнение распределений частиц по размерам на одном и том же образце
латекса с использованием:
а) дисковой центрифуги Джойса—Лобеля; б) электронной микроскопии (подсчитано ок.
4000 частиц); в) PCS, Brookhaven BI-90; г) RCS, Nanosizer N4; d) HOC, Micrometric
НРС 5600; e) PCS, Nicomp 200; ж) RCS, Malvern Autosizer
* Для в, e, г, ж оси ординат нелинейны; для г максимум шкалы «у» 40%, для осталь-
ных — 100%
цию азота, а также по методу седиментации (или электронной
микроскопии), то найденные средние значения не обязательно
будут коррелировать, даже если сделаны все поправки. Это свя-
зано с наличием пленки на поверхности частиц. Использование
лишь одного метода увеличивает возможность ошибки.
Стоит также сравнить распределения по размерам, получен-
ные численным (рис. 6.18) и массовым (6.19) методами на одном
и том же приборе. Различия очевидны.
На рис. 6.20 показаны результаты измерений распределения
по размерам частиц одного и.того же образца латекса; исполь-
зованы электронная микроскопия (подсчитано свыше 4000 час-
тиц), дисковая центрифуга, четыре различных прибора PCS и
гидродинамическая хроматография.
Невозможно рассмотреть в одной главе все 400 методов из-
мерения частиц [101]. Авторская классификация методов в «ти-
повом» виде может не сочетаться с другими классификациями.
Однако при таком изложении можно дать некоторые идеи о воз-
можных методиках, не называя все видоизменения.
Описанные выше методы можно разделить на две категории.
К первой относятся методы, основанные на новейших дорого-
стоящих приборах с микрокомпьютерами. В этой главе рассмот-
рены только основные принципы и лишь некоторые приборы.
Ко второй категории относятся методы, которые возможно
осуществить без больших затрат. В целом эта группа имеет тяго-
тение к классическим методам и, конечно, не менее правомочна,
чем современные автоматизированные методики.
ПРОМЫШЛЕННОЕ ПРОИЗВОДСТВО
ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Ф. К. Фаркас
7.1. ВВЕДЕНИЕ
Теория смачивания и стабилизации частиц пигмента в плен-
кообразующей системе обсуждалась в гл. 5. Важно подчеркнуть,
что целью процесса диспергирования пигмента является смачи-
вание и отделение первичных пигментных частиц от агрегатов
и агломератов с их последующей стабилизацией в соответствую-
щей пленкообразующей системе, т. е. в смоле или растворах
пленкообразователей.
Все стадии этого процесса важны и существенно влияют на
использование пигмента, производительность оборудования и
свойства конечного продукта. Схема его представлена в виде
диаграммы на рис. 7.1. Чтобы воспрепятствовать реагрегации
в течение и после диспергирования, важно выбрать правильные
соотношения пигментов, смол и растворителей. Кроме того, вто-
рая стадия введения растворов смол или растворителей должна
выполняться в специальном оборудовании для диспергирова-
ния, чтобы исключить возможность «коллоидного слипания»
(флокуляции) в конце процесса приготовления лакокрасочного
материала. Процесс выполняется в различного типа дисперги-
рующем оборудовании, где силы сдвига воздействуют на пигмент-
ные агрегаты и разделяют первичные пигментные частицы. Эта
стадия часто называется перетиром. Межмолекулярные силы
также оказывают существенное влияние на смачивание поверх-
ности пигмента и на процессы самопроизвольного диспергиро-
вания. Весьма желательно обеспечить максимальное проявление
межмолекулярных сил любой лакокрасочной системы для быстрого
Рис. 7.1. Схема технологической линии по изготовлению краски
получения устойчивой дисперсии пигментов при минимальных
усилиях сдвига. Процесс смачивания влияет на чистоту и насы-
щенность цвета, долговечность ряда пигментов чувствительных
к сдвиговым усилиям, время и энергию, затрачиваемые на диспер-
гирование. Условия, в которых проявляется действие физических
сил сдвига в различном диспергирующем оборудовании, также
влияют на процесс.
7.2. ПРИМЕНЕНИЕ ДИСПЕРГАТОРОВ
Смачивание играет важную роль в диспергировании пигмен-
тов и, следовательно, в производстве лакокрасочных материалов.
Все пигменты содержат такие загрязнения, как воздух, влага и
газы, адсорбированные на их поверхности. Для смачивания час-
тиц эти загрязнения должны в большинстве случаев быть вытес-
нены диспергирующей средой. Поэтому весьма важно, чтобы
смачивание пигмента диспергатором было достаточно эффектив-
ным для преодоления или, по крайней мере, для снижения сил
когезии в жидкости и поверхностного натяжения на границе
раздела фаз жидкость — твердое тело, обеспечивая адгезию функ-
циональных групп диспергатора к поверхности пигмента. Боль-
шинство связующих, применяемых в производстве лакокрасоч-
ных материалов, могут считаться диспергаторами; их смачиваю-
щая эффективность зависит от молекулярной массы, структуры
и присутствия функциональных групп, таких как карбокси-, гид-
рокси-, амино- и сложноэфирных групп. В настоящее время полу-
чены диспергаторы определенного состава, которые более эффек-
тивны, чем большинство пленкообразующих в красках; они явля-
ются многоцелевыми и совместимы с большим числом красок.
Так как электрические заряды в молекулах жидкости и пиг-
мента ответственны за процесс смачивания, полярность ингре-
диентов, смол, растворителей и твердых частиц играет важную
роль при диспергировании. Полярность определяется строением
молекул и расположением электрических зарядов, т. е. поляр-
ность зависит от того, симметричны молекулы или асимметричны.
Диспергаторы и пленкообразователи адсорбируются на по-
верхности пигмента посредством полярных групп (якорных групп),
а неполярные радикалы растворяются в жидкой фазе. Это явле-
ние называется «пространственной стабилизацией». Количество
растворителя, совместимость ингредиентов и порядок их введения
имеют большое значение для обеспечения стабильности. Если
стабилизирующие радикалы полностью или частично выделяются
из раствора в результате изменения растворяющей способности,
может наступить реагрегация и флокуляция с ухудшением каче-
ства конечного продукта; этот недостаток в большинстве случаев
невозможно устранить или восстановление качества обходится
слишком дорого. Широкий ряд смачивающих вешеств (поверх-
ностно-активных веществ используется в лакокрасочной про-
мышленности для уменьшения поверхностного натяжения на гра-
нице раздела твердое тело — жидкость. В результате обработки
ПАВ создаются новые поверхности, на которых облегчается ад-
сорбция пленкообразователя при диспергировании. ПАВ могут
быть катионными, если адсорбируемый ион положительно заря-
жен, анионными, если адсорбируемый ион отрицательно заряжен,
или неионными, активность которых может быть обусловлена
наличием полярных и неполярных групп в молекуле.
Смачивающими агентами являются соли органических ами-
нов, щелочные мыла, сульфированные масла, простые эфиры
гликолей и т. п. Имеется очень много таких продуктов с различной
эффективностью;. Некоторые из них или их комбинации являются
стандартными компонентами водных красок (в частности, эмуль-
сионных) обеспечивающими ионную стабилизацию. Пленкообра-
зователи требуемого строения — акриловые и гидролитически
стабильные алкиды — после нейтрализации амином или неоргани-
ческим основанием также могут использоваться для достижения
пространственной и ионной стабилизации, в особенности при про-
изводстве колеровочных паст, для которых важна текучесть.
В связи с тем, что избыточные количества поверхностно-
активных веществ ухудшают многие свойства красок, важное
значение приобретает определение их оптимального количества
для обеспечения нужной степени диспергирования и стабилиза-
ции. Однако прежде чем заниматься стадией диспергирования,
необходимо определить эффективность смачивания различными
смолами пигментов и наполнителей, присутствующих в лакокра-
сочном материале. Это позволит максимально использовать
сырье, сократить время диспергирования, получить высокое каче-
ство конечного продукта.
7.3. МЕТОДЫ ОПТИМИЗАЦИИ СОСТАВОВ ДЛЯ ДИСПЕРГИРОВАНИЯ
Принципы составления рецептур, применяемые в лакокрасоч-
ной промышленности для диспергирования пигментов, основы-
ваются главным образом на эмпирических данных, многолетнем
опыте и лабораторных оценках временных и материальных затрат
с целью установления рабочих (не обязательно оптимальных)
условий для процесса образования дисперсии.
Можно с уверенностью сказать, что необходимо сделать по
крайней мере шесть лабораторных проб в шаровых мельницах
на системе с данными пигментом, пленкообразователем и раство-
рителем, варьируя объемное содержание пигмента (ОКП) и су-
хой остаток раствора пленкообразователя, чтобы получить пред-
ставление о приемлемой технологичности. То же справедливо
для бисерной мельницы и для аппаратов высокоскоростного типа
для диспергирования (HSD).
Для проведения таких испытаний требуется по крайней мере
три недели, а в случае красок, содержащих несколько пигментов
и пленкообразователей, обычно необходимо несколько недель.
Последующие опытные и промышленные испытания часто тре-
буют дальнейшей корректировки рецептуры.
Прежде всего, для приготовления пигментной пасты в емко-
сти смешивают определенные количества пигмента и раствора
смолы до получения необходимой консистенции, затем отвеши-
вают остальные компоненты и загружают в аппарат для диспер-
гирования, доводя объем до требуемой степень заполнения.
Этот технический прием может обеспечить или ис обеспечить
хорошее качество диспергирования. Давно общепризнано боль-
шое значение и существенное влияние процесса' на экономиче-
ские показатели и качество конечного продукта. Невозможно
добиться удовлетворительного диспергирования пигментов и на-
полнителей при произвольном составлении рецептур пигментных
паст. Ф. К Дэниэл [1] разработал способ, получивший назва-
ние «точка текучести», при помощи которого были получены со-
ставы пигментной пасты улучшенного качества. Этот способ за-
ключается в титровании навесок пигмента растворами смолы пере-
менной концентрации при перемешивании шпателем. Конечная
точка достигается, когда смесь начинает стекать со шпателя
порциями с интервалами в 1 с. Таким образом устанавливается
объем раствора смолы, израсходованного на взятую навеску
пигмента. Концентрация раствора смолы, при которой расхо-
дуется наименьший объем, считается оптимальной для смачива-
ния. Пигмент и раствор смолы в установленном соотношении
загружают в шаровую или бисерную мельницу, поскольку для
этих типов машин требуются пасты с практически одинаковой
вязкостью. Сложность состоит в том, что не все предварительно
смешанные пигментные пасты будут обладать удовлетворитель-
ной текучестью для уверенного определения конечности точки,
как, например, в случае органических пигментов и тонкодисперс-
ных наполнителей. Для этих материалов рекомендуется серия
испытаний в шаровой мельнице.
Работа Гугенгейма [2] была основана на ряде промышлен-
ных экспериментов с использованием высокоскоростных машин
для диспергирования. В результате этой работы было получено
эмпирическое выражение для определения оптимальных соста-
вов пигментных паст для данного типа машин:
-~^= (0,9 + 0,69СР.„с + о,025л)
где Л4р.„„ — масса раствора пленкообразователя, г; А4„ — масса пигмента, г;
С|>.„и—сухой остаток раствора пленкообразователя, %; i] - вязкость раствора
пленкообразователя, пуаз; Р—маслоемкость по Гарднеру-Когеману, (г/100 г).
На практике состав пигментных паст для высокоскоростного
диспергирования можно рассчитать по эмпирическому методу
Гугенгейма. Например, если сухой остаток раствора пленкообра-
зователя равен 40%, его вязкость — 5 пуаз, а маслоемкость
пигмента—25 (для заданной степени пигментирования), то
M|I I]G = (0,9 + 0,69 • 0,40 + 0,025 • 5,0) -^-=0,325.
М„ 100
Таким образом, получаем следующие соотношения ингре-
диентов пасты*
В масс. ч. В %
Пленкбобразователь (0,40-0,325) =0,0130= 9,81
Растворитель (0,60-0,325) =0,195= 1-4,72
ПигмеЦт = 1,000 = 75,47
Сумма 1,325 100,00
1 11 I
Нет сомнения^ что эти способы определения состава пигмент-
ных паст полезнь) в практической работе. Однако им не достает
точности для оцейки эффективности смачивания пигмента плен-
кОобразующей системой — важного фактора, обеспечивающего
стабильность краски. Кроме того, получаемый результат зависит
От опыта оператора и точность получаемых субъективных ре-
зультатов составляет +25%. Поэтому соотношение пигмент/
пленкообразователь, полученное с исходным (недиспергирован-
ным) пигментом, не является надежной основой для выбора
рецептуры, которая была бы оптимальной для всего процесса
Диспергирования. Дальнейшее отделение от агрегатов и стабили-
ш
Рис. 7.2. Влияние объемной концентрации связующего на скорость и эффективность
диспергирования
зация первичных частиц пигмента все более затрудняется. Про-
цесс диспергирования прекращается независимо от продолжи-
тельности работы оборудования. Увеличение сухого остатка
выше оптимального для улучшения текучести приведет только
к возрастанию времени диспергирования (рис. 7.2).
7.4. ИСПОЛЬЗОВАНИЕ ИНСТРУМЕНТАЛЬНЫХ МЕТОДОВ
ДЛЯ ВЫБОРА СОСТАВА ПАСТ
Виршинг, Хауг и Хамман исследовали адсорбцию масла на
пигментах, используя пластограф Брабепдера [3], и получили
хорошо воспроизводимые результаты.
Это побудило автора изучить возможность применения инст-
рументальных методов для определенных составов паст. В резуль-
тате двадцатилетних исследований, проводимых отделением кра-
сок фирмы ICI, этот способ был разработан и нашел применение
для определения оптимальных составов паст для различных
методов диспергирования. Из результатов боле^ чем 11 000 испы-
таний, выполненных на огромном числе пигментов, наполнителей,
пленкообразующих сред, растворителей и добавок, найдено, что
количественные соотношения этих ингредиентов весьма специ-
фичны и могут быть точно измерены.
Соотношения ингредиентов сильно влияют на вязкость и ско-
рость диспергирования, определяют применимость данного пиг-
мента для получения дисперсии и определяют порядок добавле-
ния ингредиентов для получения стабильных продуктов с наи-
высшим качеством, которое может быть достигнуто для данной
системы. Каждый пигмент в среде, содержащей несколько ПАВ,
имеет большее сродство к одному из них и, следовательно,
для достижения максимальной стабилизации требуется опти-
мальное и пропорциональное количество этого предпочтитель-
ного ПАВ.
Диспергируемость пигмента или, соответственно, эффектив-
ность смачивания пленкообразователем илр раствором поверх-
ностно-активного вещества определяется На пластографе Бра-
бендера. На этом приборе можно достичь необходимой степени
диспергирования пигментной пасты при стандартных условиях
и скоростях сдвига. I
На процесс диспергирования и конечной результат не ока-
зывает влияние субъективный фактор. Разработаны и приме-
няются специальные методы, которые поддерживают темпера-
туру пигментных паст близкую к температуре диспергирования
в промышленных условиях. I
Конечные результаты ряда этих испытаний можно непосред-
ственно использовать для выбора оптимального состава пигмент-
ных паст для различных способов диспергирования. С другой
стороны, состав пигментных паст можно^' рассчитать, исходя из
предварительно полученных значений ОКП и сухого остатка
раствора пленкообразователя. Такие данные могут быть введены
в компьютерную систему и использованы для вычисления опти-
мальных составов, исходя из накопленных результатов для боль-
шинства пигментов, наполнителей, пленкообразователей, рас-
творителей и прочих добавок, которые могут потребоваться для
получения конечного продукта.
Затем можно с помощью спектрофотометра проанализиро-
вать качество пигментирования путем накрасок на стеклянных
пластинках и, используя полученные соотношения, рассчитать
наилучший возможный состав и способ получения для выбран-
ной пленкообразующей системы. Очевидно, что это экономит
большое количество времени и средств по сравнению с методом
проб и ошибок, который все еще применяется в промышленно-
сти для получения пригодного, но не обязательно оптимального
состава. Кроме того, данная методика обеспечивает точный каче-
ственный контроль пигментов, наполнителей, пленкообразевате-
лей, смачивателей и прочих добавок при условии, что были уста-
новлены соответствующие стандарты сравнения.
Конечные значения ОКП и градуировочная кривая, получен-
ная для стандартных условий, позволяют быстро определить
разницу между различными составами, которую нельзя устано-
вить другими методами, но которая весьма важна для производ-
ства. Этот метод может также использоваться для контроля
производства пленкообразователей и диспергаторов путем изме-
рения влияния скорости перемешивания, загрузки и темпера-
туры на эффективность смачивания. Этот метод исследования
неоценим при разработке новых диспергаторов, пленкообразо-
вателей и их пигментирования с целью получения новых про-
дуктов.
7.4.1. Выбор раствора пленкообразователя
с наилучшей смачивающей способностью
и оптимальным содержанием в пигментной пасте
Для получения кривой эффективности смачивания прово-
дится ряд измерений с растворами пленкообразователя в раз-
личных растворителях и различной концентрацией, разными
диспергаторами и пигментами. На графике откладывают объем-
ное содержание пигмента в конечной точке при испытании на
пластографе и содержание пленкообразователя в пигментной
пасте. Эта кривая является характеристической для данной
системы и позволяет выбрать пленкообразователь или дисперги-
рующий раствор с наилучшей смачивающей способностью, кото-
рые необходимо использовать для диспергирования данного
пигмента. Она также позволяет классифицировать различные
пленкообразователи по эффективности смачивания, что необхо-
димо' для последующей оптимизации составов. При оценке плен-
кообразователей желательно исключить из стандартной пигмент-
ной пасты различные смачиватели и добавки.
Однако их необходимо вводить в рецептуру и требуется оце-
нивать их влияние при выборе оптимального состава пигмент-
ной пасты. В большинстве случаев они не оказывают сущест-
венного влияния на выбранный состав.
По результатам испытаний на пластографе для различных
концентраций растворов пленкообразователя рассчитывают объ-
емную концентрацию пигмента (ОКП). В рассмотренном ниже
примере в качестве пигмента был испытан технический углерод,
пленкообразователи — алкидная смола (а) и азотсодержащая
смола (м). Полученные результаты представлены ниже:
Сухой остаток
раствора плеико-
образователя, %
10
15
20
30
Соответствующие зна-
чения ОКП
Общий объем раствора
пленкообразователя, мл
Смола (а) Смола (п)
50 43
45 38
43,6 42
57 48
Смола (а)
14,25
15,47
16,04
12,75
Смола (п)
16,23
17,98
16,55
15,00
Приведенные данные, а также кривые эффективности сма-
чивания (рис. 7.3), показывают, что смола (п) характеризуется
лучшей смачивающей способностью и поэтому ёе следует исполь-
зовать для диспергирования технического углерода. С другой
стороны, поскольку смола (а) хуже смачивает технический угле-
род, она не будет ухудшать стабильность пигментной пасты,
если ее добавлять после введения смолы (м).
Рис. 7.3. Кривые эффективности смачивания технического углерода алкидной (а)
и азотсодержащей (п) смолами
оке, %
Рис. 7.4, а. Кривые эффективности смачивания для некоторых пигментов:
/__Темный лимонный хром (Хром желтый); 2 — Монастраль голубой FBN (Фталоциа-
ниновый голубой); 3— Монастраль зеленый GN (Фталоцианиновый зелеиый); 4— Ру-
на RH472 (Диоксид титана белый); 5 — Фиблэк AN550 (Уголь черный); 6 — Перманент-
ный красный F3RK70 (Нафтол красный)
Рис. 7.4, б. Кривые эффективности смачивания для Монолита красного У в некото-
рых растворах пленкообразователей:
1 — средний алкид в алифатическом углеводороде; 2 — полиметилметакрилат в смеси
эфира и ароматического углеводорода; 3 — акриловый сополимер в ароматическом угле-
водороде; 4 — жирный алкид в смеси алифатического и ароматического углеводородов;
5— другой жирный алкид в смеси алифатического и ароматического углеводородов; 6 —
другой акриловый сополимер в смеси алифатического и ароматического углеводородов
При 15%-м сухом остатке наибольшее количество пигмента
приходится на единицу объема пигментной пасты и время дис-
пергирования минимально.
Формы кривых эффективности смачивания для различных
пигментов и растворов пленкообразователей различны и спе-
цифичны (рис. 7.4).
Причины использования объемного показателя (ОКП) вме-
сто массового становятся понятными, когда по результатам
испытаний начинают рассчитывать рецептуры пигментных паст,
применяемые на практике. С помощью ОКП очень просто можно
рассчитать оптимальные составы пигментных паст для различ-
ных способов диспергирования в промышленных условиях.
Данные лабораторных испытаний можно непосредственно
использовать при переходе на промышленное оборудование. При
этом в лабораторных условиях только для оптимального состава
проверяются скорость и степень диспергирования.
Вообще (см. рис. 7.5, а и б) при значениях сухого остатка
ниже оптимального (нестабильная область) в ходе процесса
Диспергирования может произойти частичная реагрегация из-за
недостаточного количества стабилизирующего пленкообразова-
теля в пасте. С другой стороны, при значениях сухого остатка
выше оптимального перестабилизированная область будет непро-
Рис. 7.5, а. Расположение частиц:
/ — нестабильная область (низкий сухой остаток); // — оптимальный сухой остаток; /// —
перестабнлизнрованная область (высокий сухой остаток); IV — рефлокулированиый
агрегат; V — устойчивый; VI — стабильный, но воздействие процессов смачивания и
диспергирования уменьшено.
I — частица пигмента; 2 — якорная группа: 3 — стабилизирующая полимерная цепь
Рис. 7.5, б. Влияние сухого остатка раствора пленкообразователя на ОКП:
/ — ведостабилизированная область; И — оптимум; III — перестабилизированная
область.
порционально увеличивать время диспергирования для достиже-
ния требуемой степени перетира. В случае необходимости можно
принимать компромиссное решение (чувствительные пигменты,
составы с низким содержанием растворителей) и использовать
значения сухого остатка и, соответственно, ОКП выше оптималь-
ных, определенных по кривой эффективности смачивания.
Для классификации диспергаторов и растворов пленкообра-
зователей независимо от их смачивающей или диспергирующей
эффективности в качестве дополнительного и важного пара-
Рис. 7.6. Пластографическая кривая
метра можно использовать работу, затраченную на смешение
при диспергировании (площадь, ограниченная пластографиче-
ской кривой под точкой когерентности). Чем меньше работа,
тем выше эффективность смачивания диспергатором или раство-
ром пленкообразователя. Типичная пластографическая кривая
представлена на рис. 7.6.
Так как для испытываемой системы условия стандартизи-
рованы, использование этого показателя в сомнительных слу-
чаях имеет большое значение.
Эффективность оптимизации составов пигментных паст также
очевидна, когда интенсивность окраски покрытий сравнивается
с окраской неоптимизированных стандартных образцов. На
рис. 7.7 показано, как может быть получена экономия в сырье
и времени диспергирования. На графиках приведены зависи-
мости интенсивности окраски от времени перетира для трех
пигментов, диспергированных в растворах: 1) алкида средней
жирности в ароматическом углеводороде и 2) жирного алкида
в алифатическом углеводороде до и после оптимизации.
Ниже приведены правила для диспергирования пигментных
паст и их последующего использования для красок:
1. Смачивающая и диспергирующая способность диспергато-
ров, пленкообразователей и добавок по отношению к разным
пигментам и наполнителям различна.
Время перетира, ч
Время перетира, ч
Рис. 7.7, а. Дисперсия Монастраля голубого FBN (Фталоцианинового голубого)
в растворе среднего алкида в ароматическом углеводороде:
Ст.— до оптимизации: ОКП=10%, ОКС = 15%
Опт.— После оптимизации: ОКП = 13%, ОЦС = 20%
Рис. 7.7, б. Дисперсия Монастраля голубого FBN (Фталоцианинового голубого)
в растворе жирного алкида в алифатическом углеводороде:
Ст.— До оптимизации: ОКП = 14%, ОКС = 20%
Опт.— После оптимизации: ОКП = 21%, ОКС=15%
Рис. 7.7, в. Дисперсия Монастраля зеленого GN (Фталоцианинового зеленого)
в растворе среднего алкида в ароматическом углеводороде:
Ст.—До оптимизации: ОКП=12%, ОКС=16%
Опт.—После оптимизации: ОКП = 16%, ОКС = 25%
Рис. 7.7, г. Дисперсия Монастраля зеленого GN (Фталоцианинового зеленого)
в растворе жирного алкида в алифатическом углеводороде:
Ст.— До оптимизации; ОКП = 11,5%, ОКС = 30%
Опт.— После оптимизации: ОКП —15%, ОКС=25%
Время перетира, ч Время перегара, ч
Рис. 7,7, д. Дисперсия Перманентного красного F3RK70 (Нафтола красного) в
растворе среднего алкида в ароматическом углеводороде:
Ст.— До оптимизации: ОКП = 20%, ОКС = 20%
Опт.—После оптимизации.- ОКП = 22%, ОКС = 20%
Рис. 7.7, е. Дисперсия Перманентного красного F3RK70 (Нафтола красного) в
растворе жирного алкида в алифатическом углеводороде:
Ст.—До оптимизации: ОКП = 14%, ОКС = 20%
Опт.— После оптимизации: ОКП = 23%, ОКС = 20%
2. Поэтому в средах, содержащих несколько ПАВ, каждый
пигмент будет взаимодействовать предпочтительно с одним ПАВ.
Следовательно, на стадии диспергирования необходимо приме-
нять оптимальные и пропорциональные количества предпочти-
тельного ПАВ для каждого пигмента с целью достижения макси-
мальной стабилизации.
3. Эти характеристики могут быть измерены инструменталь-
ными методами с использованием пластографа Брабендера.
Диспергирующая способность или эффективность смачивания
иленкообразующсго материала и других компонентов опреде-
ляют стадии диспергирования, загрузки и переработки и позво-
ляют выбрать оптимальные время диспергирования и наилуч-
шие условия использования исходного сырья.
4. Даже небольшие различия в эффективности смачивания
компонентов пленкообразователей, которые вводятся на стадии
диспергирования и конечных стадиях, могут снизить стабиль-
ность системы (например, изменение цвета со временем). Вза-
имодействие добавок, облегчающих диспергирование, и прочих
добавок имеет аналогичный эффект и применять их в оптимизи-
рованных системах необязательно.
5. Значения сухого остатка выше оптимального могут быть
использованы в следующих случаях:
а) чтобы защитить поверхности определенных пигментов от
разрушения под. действием компонентов пигментной пасты при
оптимальных значениях сухого остатка;
б) чтобы обеспечить необходимую вязкость (содержание
растворителя) пасты;
в) чтобы обеспечить большую гибкость процессу при диспер-
гировании на промышленном оборудовании.
6. Не следует диспергировать никакой пигмент любым мето-
дом при значении сухого остатка или содержании диспергатора
меньше оптимального на 5 %. Например, если сухой остаток
равен 20%, то предельно допустимый низший уровень равен
15%. Иначе в процессе диспергирования в пигментной пасте
возникнет недостаток стабилизатора, будет происходить реагре-
гация и окажется невозможным достижение необходимой сте-
пени диспергирования независимо от времени перетира.
В общем случае при значении сухого остатка на 10% выше
оптимального время перетира увеличивается непропорционально.
7. Не следует разбавлять на второй стадии диспергирован-
ную пасту растворителями или растворами пленкообразователей
с очень высоким сухим остатком. Это может привести к частич-
ной или полной флокуляции, которую трудно устранить. Необ-
ходимо увеличить концентрацию пленкообразователя на 8—10%
сверх оптимального сухого остатка для второй стадии за счет
добавления порциями раствора соответствующего пленкообра-
зователя. Это гарантирует необходимую стабильность пасты
при кратковременном хранении и предохраняет от коагуляции
при последующей переработке.
8. Необходимо соблюдать осторожность, если колеровочная
паста содержит более эффективный диспергатор, чем готовая
эмаль. Некоторые пигменты в эмали могут быть дестабилизи-
рованы и/или флокулированы, если более чем половинное коли-
чество необходимого для них стабилизатора вводится с коле-
ровочной пастой. Однако, если колеровочная паста содержит
менее эффективный диспергатор, чем эмаль, то произойдет
частичная или полная флокуляция. Степень ее будет зависеть
от различий в смачивающей способности пленкообразователя
колеровочной пасты и наиболее эффективного пленкообразова-
теля или смеси пленкообразователей эмали. Предполагается,
что в обоих случаях пленкообразователи колеровочных паст и
эмалей совместимы.
9. Важно на всех стадиях производства поддерживать неиз-
менным состав растворителя, чтобы воспрепятствовать выделе-
нию пленкообразователей из раствора. Даже частичная нерас-
творимость или несовместимость может привести к необратимой
флокуляции. Особенно чувствительны прозрачные дисперсии.
10. Для того, чтобы извлечь максимальную пользу от опти-
мизации пигментных составов, необходимо обеспечить оптималь-
ные условия работы диспергирующего оборудования.
Эмали и соответствующие пигментные пасты можно клас-
сифицировать по составу следующим образом:
1) один пигмент—один пленкообразователь;
2) два или более пигментов — один пленкообразователь;
3) один пигмент — два или более пленкообразователей;
4) два или более пигментов — два или более пленкообразова-
телей.
-< В случаях 1 и 2 для пигментов нет вариантов и они будут
Диспергироваться до определенной степени при оптимальном
значении сухого остатка. В случаях 4 и 3 для пигментов имеется
возможность выбора и поэтому их необходимо диспергировать
в наилучшем пленкообразователе или в их смеси при концентра-
циях, которые определяются и рассчитываются из оптимальных
значений для каждого компонента.
Ниже приведен пример 1, который показывает, как рассчи-
тать состав пигментной пасты на основе значений ОКП и сухого
остатка. Он применим к случаю 1. Если значение сухого остатка
пленкообразователя одно и то же для двух пигментов в системе
с одним пленкообразователей (случай 2), следует использовать
тот же самый расчет. Предполагается, что ОКП также одина-
ковы. Если, однако, сухой остаток и ОКП различны для двух
и более пигментов в одном пленкообразователе, следует рассчи-
тывать по приведенному примеру 2, в разных пленкообразова-
телях — по примеру 3.
7.4.2. Как применять результаты испытания
для определения составов пигментных паст!
В идеальном случае необходимо хранить данные, полученные
при оптимизации пигментных паст, и использовать их для состав-
ления рецептур из различных комбинаций пигментов и пленко-
образователей без дальнейших испытаний.
Установленные значения ОКП и сухого остатка раствора
пленкообразователя, используемого для диспергирования, опре-
деляют состав пигментной пасты, который используется для
последующих расчетов количества пигментной пасты, загру-
жаемой в различное диспергирующее оборудование.
Основные уравнения для расчетов:
М, / МИсх. р. ПО " бцсх. р. по
= Р V; Мпо ----Е;
Мд.р. ПО“ Л4ИСХ.р. поЧ-Л4р.
где: М. — масса; р — плотность; V — объем; 7ИПо — масса пленкообразователя, г;
Мнх.р.по — мас<;а исходного раствора пленкообразователя, г; /Ил.р.по—масса
раствора пленкообразователя, используемого для диспергирования, г; Л4Р — мас-
са растворителя, г; С,|сх.р.по — сухой остаток исходного раствора пленкообразо-
вателя, %; Сд.р.по — сухой остаток раствора пленкообразователя, используемого
для диспергирования, %.
Практический пример 1
ТЮг’, Крон RN45. Этот пигмент необходимо диспергировать
в растворе жирного алкида с сухим остатком 15%. Соответ-
ствующее значение ОКП—43,5 %. Сухой остаток исходного
раствора алкида равен 40 %
Пигмент
15%-ный раствор алкида
Объемные Плотность, Массовые
единицы г/см3 единицы
43,5 X 4,1 = 178,35
56,5 X 0,788 44,52
100,0 222,87
Далее рассчитаем массу алкида в 44,52 г 15%-ного раствора:
.. 44,52-15 сс_о
Мю =---------=6,678 г.
100
Рассчитаем массу раствора смолы с сухим остатком 40%,
требуемую для получения 44,52 г 15%-ного раствора алкида:
-,678 • 100= 16,695 г.
40
Необходимое количество растворителя составит:
44,520 г—16,695 г=27,825 г.
Следовательно, пигментная паста содержит:
Пигмент
Алкид с сухим остатком 40 %
Растворитель
178,35 г
16,695 г
27,825 г
Практический пример 2
Требуется рассчитать состав смесевой пигментной пасты
исходя из данных для пигмента ТЮг и наполнителя BaSO4, взя-
тых в весовом соотношении 20/80. В качестве диспергатора
используется раствор алкидной смолы в уайт-спирите (сухой
остаток 40%).
Этап 1. Напишем оптимальные значения ОКП и сухого
остатка, используемого для диспергирования пленкообразователя
для каждого из компонентов пигментной пасты. Приведенные
значения были получены с помощью пластографа Брабендера:
Для TiO-2 ОКП = 43,5% при Сл.рПо = 15%;
Для BaSO« ОКП = 38,35% при Сл.р.„о = 20%.
Этап 2. Переведем указанные величины в объемные и весо-
вые единицы:
к р м
тю2 43,5 4,1 178,35
15%-ный раствор алки- 56,5 0,788 44,52
да 100,0
BaSOj 38,35 4,1 157,24
20%-ный раствор алки- 61,65 0,798 49,20
да 100,0
Этап 3. Запишем и определим объем г соотношения пигментов в пигментной пасте шгментной смеси:
м Р V
ТЮ2 20 4,1 4,88
BaSC>4 80 4,1 19,51
Too 24,39
Этап 4. Найдем пропорциональные количества диспергирую-
щего раствора и его объемы для каждого пигмента:
ТЮ2 : 178,35 : 44,52 : Л4л р.„„; мл р ро= 44-52'20 = 4,99 г;
Р' 178,35
Кд. р. ПО -
4,99
0,788
= 6,34 см3.
BaSO.>: 157,24 : 49,20=80 : Мд.р.„о;
.. 49,20-80 25,05 О1 3
Л4д.р.пО— ]5724 —25,05 г, I4 p.no— 0 7д8 —31,37 см .
Этап 5. Определим объемную концентрацию смеси пигментов
окпсм, %:
Объем пигментов 24,39 см3;
Объем диспергирующих растворов 31,37 + 6,34 = 37,79 см2,
ОКПсм =------------100 = 39,28 %.
24,39 + 37,71
Этап 6. Рассчитаем сухой остаток смеси пленкообразовате-
леи Осм.по, /о •
Для 15%-ного
lp.uo —
4,99-15 п_с
Л4По=-------—=0,75 г.
100
раствора алкида Л4Р.,1О 25,03 г;
Для 20%-ного
-----------------------— 0,01 Г, Орм по —
100--------------------25,03 + 4,99
25,03-20
5,01 +0,75
Этап 7. Рассчитаем массу исходного раствора пленкообра-
зователя (AfHCX.p.„o) и массу растворителя (Л1Р), требуемые для
конечной рецептуры:
0 75
Для 15%-ного раствора алкида Л4ИСХ Р по=—:-100=1,86 г;
40
Л4Р = 4,99 — 1,86 = 3,13 г.
5 01
Для 20%-ного раствора алкида Л4„сх р п0=—: 100=12,52 г;
40
Мр = 25,03—12.52=12,51 г.
Общая масса растворителя = 15,64 г.
Для пересчета на 100 единиц объема пигментной пасты полу-
, , ЮО
ченные значения умножим на коэффициент ——=1,61.
Этап 8. Запишем окончательный состав пигментной пасты:
ОКП = 39,28 %; Сухой остаток = 19,15%
V р м
ТЮ2 7,86 4,10 32,2
BaSO.i 31,42 4,10 128,8
Раствор алкида (40%-ный) 28,44 0,84 23,83
Уайт-спирит 32,28 0,78 25,18
100,00
Практический пример 3
Требуется найти сухой остаток смеси пленкообразователей
Ссм по и ОКПсм, исходя из значений Сд.р.п0 и ОКП, чтобы устано-
вить необходимые количества стабилизатора для пигмента в
объемных единицах пигментной пасты.
Этап 1. Запишем индивидуальные оптимальные значения ОКП
и Сд р.по для пигментов, взятых для образования пигментной
пасты для автомобильного верхнего покрытия:
Пигмент ОКП. % Од р. „о. % Пленкообразователь
Монолит желтый 8 20 Меламиновая смола 1 в ксилоле
TiOs 42 15 Акриловая смола в кси- лоле
Титан желтый 36 25 Меламиновая смола 2 в ксилоле
Красный железо- окисный пигмент 35 20 Алкидная смола в ксилоле
Этап 2. Переведем указанные значения в объемные и весовые
единицы:
V Р М
Монолит желтый 8 1,6 12,8
20%-ный раствор меламиновой смо- 92 0,91 83,72
лы 1 100
TiO2 42 4,1 168
15%-ный раствор акриловой смолы 58 0,89 51,62
100
Титан желтый 36 4,3 154,8
25%-ный раствор меламиновой смо- 64 0,92 58,88
лы 2 100
Красный железоокисный пигмент 35 4,3 150,5
Алкидная смола 65 0,9 58,5
100
Этап 3. Определим массовые отношения для четырех пигмен-
тов в пигментной пасте для получения требуемого цвета покры-
тия и найдем общий объем пигментной смеси. Предположим,
что требуются следующие массовые отношения:
М р V
Монолит желтый 2,85 1,6 1,78
тю2 18,36 4,1 4,58
Титан желтый 46,72 4,3 10,87
Красный железоокисный пигмент 32,07 4,3 7,46
100,00 24,70
Этап 4. Определим соответствующие массы и объемы раство-
ров для диспергирования для каждого пигмента:
Монолит желтый:
ТЮ2:
.. 83,72-2,85 _ 18,64 оп/о ,
Мд.р.по— 12 8 —18,64 г, Ид.р.по— q —20,48 см3.
КА 51,62-18,36 _сл ,, 5,64 с 3
Л4д.р.по=-----—------—5,64 г; гд.р „о— —6,34 см .
1 об и,оУ
Титан желтый:
58,88-46,72
ДР"°- 154,8
Красный железоокисный пигмент:
17 77
= 17,77 г; 1/д.,„о=-==19,32 см3.
Л4д. р. по —
58,5-32,07
= 12,47 г;
150,5
12 47
Удрпо=_090-= 13,86 С“3
Суммарная масса = 54,22 г;
Суммарный объем = 60,00 см3.
Этап 5. ОКПсм = о J4'76 -100 = 29,16 %.
24,70 + 60,00
Коэффициент для пересчета на 100 единиц объема
24,70+60,00
*
Этап 6. Вычислим сухой остаток смеси пленкообразователей,
используемых для диспергирования:
Для 20%-иого раствора меламиновой смолы 1: .. 18,64-20 ,„о Л4ПО = — =3,728 г;
Для 15%-ного раствора акриловой смолы: Л4ПО= 5'64'15 =0,846 г; 100
Для 25%-ного раствора меламиновой смолы 2: ^17.77-25 100
Для 20%-ного раствора алкидной смолы: 12 47-20 Мпо=- -- =2,494 г. 100
Суммарная масса пленкообразователей = 11,510 г.
Ссм.по= ^’-51° -100 = 21,11 %.
54,22
Этап 7. Рассчитаем массу исходных растворов пленкообразо-
жателей и массу растворителей:
Для меламиновой смолы 1:
Для акриловой смолы:
.. _ 3,728 . лп_--с
А4исх. р. по— *100—5,56 г,
Мр= 18,64 — 5,56= 13,08 г.
Л4„сх.р.по=-5^-100= 1,29 г;
65,5
Мр = 5,64—1,29=4,35 г;
Для меламиновой смолы 2:
4 442
Л411сх.р.по=—~——100 = 7,40 г;
60
Мр= 17,77 — 7,40= 10,37 г;
Для алкидной смолы:
2 404
М„сх.р.„о=-^—100= 4,99 г;
50
Мр= 12,47 — 4,99 = 7,48 г.
Суммарная масса растворителя = 35,28 г.
Для пересчета на 100 объемных единиц пигментной пасты
умножим полученные значения на коэффициент 1,181.
Этап 8. Запишем окончательную рецептуру пигментной пасты:
V р М
(ОКП = 29,82 % сухой оста-
ток= 12,15%)
Монолит желтый 2,10 1,6 3,37
ТЮ2 5,41 4,1 21,68
Титан желтый 12,84 4,3 55,68
Fe2O3 8,81 4,3 37,87
Меламиновая смола 1 6,32 1,04 6,57
Акриловая смола 1,52 1,00 1,52
Алкидная смола 6,20 0,95 5,89
Меламиновая смола 2 8,74 1,00 8,74
Ксилол 48,06 0,868 41,67
100,00
Глава 8
ПОКРЫТИЯ ДЛЯ ЗДАНИИ
Д. А. Грейстоун
8.1. ВВЕДЕНИЕ
Важной и широко применяемой группой покрытий являются
материалы для отделки зданий. Эта глава посвящена вопросам
отделки любых типов жилища, начиная с частных домов до
школ, госпиталей, офисов и облегченных промышленных по-
строек. К покрытиям для химических заводов, нефтяных вышек
и морских сооружений предъявляются более жесткие требова-
ния (см. гл. 11).
Характерные особенности предлагаемых здесь покрытий со-
стоят в том, что они предназначены для улучшения качества по-
верхности по цвету, текстуре и другим характеристикам внешнего
вида. Действительно, данный вид покрытий наиболее известен
как «декоративные покрытия», хотя их можно также называть
«архитектурные покрытия», «краски для дома» или «краски ши-
рокого потребления».
Первые наскальные рисунки носили не просто утилитарный
характер, а являлись произведениями искусства и представляли
собой простейший пример отделки жилища. Краски для зданий
различаются не только по цветовым характеристикам. Подчер-
кивая важность эстетического фактора, не следует забывать
важную защитную роль, которую играют покрытия. Необходимо
отметить, что использование черных металлов без защитного
покрытия крайне ограничено. Существует еще более тонкий
аспект в применении защитных покрытий — иногда покрытие
и подложка не должны соприкасаться друг с другом. Покрытия
на инертных подложках сохраняют свои свойства дольше, чем
при нанесении на простейший строительный материал. Дефор-
мация деревянных частей, например, может вызывать отслаива-
ние покрытия. С другой стороны, непроницаемые пленки при-
водят к гниению дерева, если влага не имеет выхода.
Другой важный аспект применения покрытий, особенно при
внутренней отделке зданий,— это эффект освещенности. Белые
и пастельные тона играют важную роль в увеличении естест-
венной и искусственной освещенности, в то же время выбор цве-
та может влиять на настроение и самочувствие человека.
Абсолютно точной классификации покрытий не существует.
Поэтому классификационные признаки, используемые при мар-
кетинге, разработке или производстве красок, могут быть совер-
шенно различными; существует также различие при описании
признаков, используемых при патентовании и продаже на
рынке.
Описание покрытий может быть связано только с ограничен-
ным количеством признаков и их необходимо выбирать таким обра-
зом, чтобы подчеркнуть область применения различных групп
материалов.
Ниже представлены некоторые признаки, которые исполь-
зуются при описании продукции, отражая ее наиболее важные
характеристики.
Характеристики материала
Тип
Внешний вид покрытия
Рыночная характеристика
Назначение
Область применения
Состав
Свойства покрытия
Вид подложки
Пример
Краска, эмаль, лак
Блеск (матовое, шелковистое, полуматовое,
глянцевое)
Кроющее или прозрачное
Цвет, структура
Оптовая, розничная продажа
Грунтовка, промежуточный слой, отделка
Потолки, стены, окна, полы
Акриловый, алкидный, на основе раствори-
теля, водоразбавляемый
Проницаемость, эластичность, фунгицидность,
стойкость к коррозии
Дерево, металл, кирпич
Широкий выбор классификационных признаков труден при
логической классификации покрытий для зданий, которые долж-
ны быть рассмотрены с различных позиций.
В этой главе рассмотрены факторы, которые влияют на выбор
или улучшение качества покрытий для зданий. Обсуждены неко-
торые общие закономерности влияния типа связующего и объем-
ной концентрации пигмента (ОКП), но приведение рецептур
не предусматривается, так как в литературе ||], а также у по-
ставщиков сырья достаточно информации по данному вопросу.
8.2. РЕЦЕПТУРНЫЕ ОСОБЕННОСТИ И ОГРАНИЧЕНИЯ
Роль лакокрасочника обычно сводится к созданию покрытий
или красочных систем, отвечающих специфическим требованиям
рынка, хотя иногда особенности рецептуры могут подсказать
путь к выбору соответствующих исходных материалов. Некото-
рые примеры взаимосвязей такого типа схематически показаны
на схеме:
Цвет, структура, блеск,
укрывистость,
прозрачность,
текстура и т. д.
Реология
Время высыхания
*
Тип связующего
Пигментный состав
Растворитель
Технический
Производство
Распределение
Продажа
Вид подложки,
внутренняя
или наружная окраска,
погодные условия,
климат
Стены, потолок, окна
и т. д.
Физико-механические
свойства
Адгезия
Проницаемость
Химические
и биологические
свойства
Соотношение
компонентов
Способ
диспергирования
обычно рыночные ситуации выявляют путем тщательного ана-
лиза рыночного спроса и связывают эту ситуацию с областями
применения соответствующих материалов. В большинстве слу-
чаев задаются требования по внешнему виду покрытий, которые
в сочетании с другими требованиями обусловливают необходи-
мость выбора того или иного материала.
Изучив необходимые требования к покрытиям, включая ме-
тоды нанесения и характеристики внешнего вида, разработчик
рецептур намечает программу работ и выбирает соответствую-
щие исходные сырьевые материалы. Прежде, чем разрабатывать
какой-либо состав, необходимо обратить внимание на некоторые
ограничения, имеющие важное значение для декоративных лако-
красочных материалов.
8.2.1. Стабильность при хранении
Важной характеристикой лакокрасочного декоративного ма-
териала является его стабильность при хранении. Материал
должен сохранять свои свойства в течение нескольких лет после
его изготовления. Особое значение имеет сохранение стабиль-
ности малярных характеристик и скорости высыхания покрытия.
В процессе хранения материал не должен образовывать твер-
дого осадка и -в нем не должны протекать необратимые измене-
ния, приводящие к образованию поверхностных пленок или
к коагуляции. Если возможно использование материала при
низких температурах, необходимо обеспечить сохранность
свойств материала при замерзании и оттаивании. В водных
красках может протекать гидролиз эфирных связей, что исклю-
чает употребление некоторых видов связующих. Во многих кра-
сочных композициях при хранении происходят реологические
изменения, которые сказываются на малярных характеристиках.
В водных и, реже, в неводных красках с течением времени
возможно увеличение вязкости. Это обстоятельство может быть
учтено при разработке рецептуры, так как экспериментально
подтверждено, что в большинстве случаев происходит стаби-
лизация вязкости во времени. Можно с полным основанием
утверждать, что обеспечение высокой стабильности при хранении
является наиболее важным этапом разработки рецептур и пред-
ставляет собой лимитирующую стадию в цикле работ по созда-
нию нового продукта. Возможно несколько ускорить получение
данных по стабильности материала при хранении путем выдерж-
ки его при повышенной температуре (60 °C) и встряхивании на
вибрационном приборе для имитации эффекта транспортных
перевозок. Однако в целом можно утверждать, что неразумно
разрабатывать новые материалы со сроком хранения менее
12 мес.
8.2.2. Окружающая среда и здоровье
Декоративные краски применяются в различных областях
как профессиональными малярами, так и неопытными нович-
ками. Краски для внутренних работ обычно используются в поме-
щениях с ограниченной вентиляцией, поэтому необходимо на
банках с красками помещать инструкции с описанием необхо-
димых мер предосторожности. Это не исключает приложения
отдельного описания правил по технике безопасности. Декора-
тивные краски обычно содержат около 50% по объему летучих
растворителей, которые в результате испарения создают высокие
концентрации паров в воздухе после нанесения краски. По этой
причине ассортимент растворителей, применяемых в красках для
внутренней отделки зданий, весьма ограничен. Преимущество
должно быть отдано двум группам красок, в которых раство-
рителями в основном являются вода или алифатические угле-
водороды типа уайт-спирита.
Требования безопасности для здоровья предъявляются также
и к высохшей пленке; использование в красках токсичных ком-
поненгов, таких как, например, свинцовые пигменты, необходимо
существенно ограничить.
В то время как водные краски можно считать наиболее бла-
гоприятными с точки зрения окружающей среды, имеется ряд
совместимых с водой растворителей, например эфиры гликолей,
применение которых связано с описанной выше проблемой ток-
сичности [2].
8.2.3. Образование пленки
В процессе высыхания покрытия происходит превращение
жидкой пленки в твердое состояние. Это положение является
общим, однако факторы, благодаря которым происходит этот
процесс, оказывают влияние на формирование пленки. В отличие
от эмалевых красок для автомобилей и некоторых промышлен-
ных красок, в случае декоративных покрытий использование
высокой температуры для формирования пленки невозможно.
Также невозможно осуществить многие химические реакции,
происходящие при использовании двухупаковочных продуктов,
так как это либо слишком дорого, либо слишком токсично.
В целом, механизм образования декоративного покрытия бази-
руется на сочетании процессов коалесценции и окисления, кото-
рые описаны в разд. 8.6.
8.2.4, Нанесение
Метод нанесения является еще одним важным показателем
при работе с декоративными красками. Хотя иногда возможно
применение специальных аппликаторов для нанесения некото-
рых красок, но наиболее часто применяют известные инстру-
менты: кисти, валики, ролики и, реже, распылители. Это обстоя-
тельство вызывает необходимость использования красок с
хорошим розливом, не оставляющих следов от кисти и не обра-
зующих брызг при движении валиков или роликов, т. е. требует
применения материалов с хорошими реологическими характери-
стиками. Многие из этих показателей трудно замерить, но при
субъективной оценке внешнего вида покрытия необходимо учи-
тывать дефекты, которые вызываются образованием неисчезаю-
щих следов от кисти, плохой очисткой поверхности и повышенной
вязкостью. Все это не представляется чем-то необыкновенным
для профессиональных маляров, однако для широкого потре-
бителя необходимо дать инструкцию, содержащую практические
рекомендации по применению. В конечном итоге многообразие
требований, возникающих в каждой конкретной ситуации, нельзя
удовлетворить путем создания единственной рецептуры.
Разработка красок не является строго лимитированным про-
цессом; различные разработчики рецептур могут создавать аль-
тернативные составы, которые должны проверяться по основным
показателям, выдвигаемым требованиями рынка.
8.3. СООТНОШЕНИЕ ПИГМЕНТ — СВЯЗУЮЩЕЕ — РАСТВОРИТЕЛЬ
Если принять во внимание наиболее существенные характе-
ристики, декоративные краски можно рассматривать как дис-
персии пигментов в растворах связующих при различных соот-
ношениях компонентов. После превращения в сухую пленку
в покрытии преобладающим компонентом является пигмент,
однако следует иметь в виду, что в некоторых случаях в пленке
высохшего покрытия может оставаться воздух, что имеет особое
значение для декоративных материалов. Связующее может
также претерпевать физические и химические превращения,
в результате чего возможно возникновение внутренних напря-
жений, которые следует принимать во внимание.
Если принять общее количество жидкой рецептуры за 100%,
удобно представить различные соотношения компонентов в виде
треугольной диаграммы, как это показано на рис. 8.1. Эта диа-
грамма является полезной иллюстрацией для описания соотно-
шений между компонентами покрытия. В случае покрытий со
специальными свойствами диаграмма может быть использована
как основа для создания композиции с требуемыми характе-
ристиками.
На рис. 8.1 композиция «X» содержит 20% пигмента, 40%
связующего и 40% растворителя. Прямая, проходящая через
точки S и X, соответствует композиции, имеющей постоянное
Рис 8.1. Соотношения пигмент — связующее — растворитель
соотношение пигмент — связующее (которое можно определить
по оси РВ) и переменный состав твердой фазы, в то время
как линия АА' определяет композицию с постоянным значе-
нием твердой части, но с различным соотношением пигмент —
связующее. Так как линия, характеризующая содержание рас-
творителя, расположена горизонтально, то линия, относящаяся
к общему содержанию сухого остатка, расположена на некото-
ром расстоянии от основания диаграммы и соответствует 100%.
Прямая, проходящая от точки Р через точку X — это постоян-
ное содержание сухого связующего, количество которого можно
определить по оси SB. Практическая ценность диаграммы со-
стоит в том, что она позволяет по геометрическим соотношениям
решить проблему соотношения компонентов рецептуры без про-
ведения комплекса соответствующих экспериментов [3].
Из диаграммы, представленной на рис. 8.1, не видно, выбраны
ли для расчетов объемные или весовые соотношения, однако
важно учитывать и то и другое. Если краска изготавливается
в лаборатории, компоненты взвешивают и химический состав
обычно выражают в массовых процентах, которые необходимо
разделить на плотности каждого компонента, чтобы получить
необходимые значения объемных соотношений. Во многих слу-
чаях свойства как краски, так и сухого покрытия более четко
можно установить по объемным соотношениям, причем краска
обычно продается и используется с учетом ее объема. Так как
большинство пигментов имеют плотность больше единицы, диа-
грамма для объемных соотношений будет отличаться от диа-
граммы для массовых соотношений.
8.3.1. Соотношение пигмент — связующее
Одним из наиболее важных параметров красочных компози-
ций является объемная концентрация пигмента (ОКП), которая
представляет собой часть от общего объема сухого остатка в су-
хой красочной пленке.
где У„ — объем пигмента; — объем связующего.
Этому кажущемуся простым соотношению уже около 50 лет;
обзор его исторического развития сделан Стигом [4], который
не только обосновал концепцию ОКП, но и широко исследовал
роль соотношения пигмент — связующее в красочных системах
[5]. Исследования в этой области нельзя считать законченными;
в настоящее время продолжаются дебаты вокруг расшифровки
понятия ОКП применительно к пластинчатым пигментам, а также
цветным вспомогательным добавкам непигментного происхожде-
ния (см. ниже).
Для более правильного понимания роли соотношения пиг-
мент__связующее целесообразно ввести геометрические пред-
ставления, которые можно легко развить на базе простой си-
стемы. Эти соотношения могут быть использованы как качест-
венная основа для интерпретации свойств краски, однако не
следует забывать о вводимых при этом элементарных допуще-
ниях. Свойства реальных систем могут значительно отклоняться
от модели, особенно при воспроизведении количественных харак-
теристик. Например, расчетная ОКП может существенно отли-
чаться от реальной, если связующее обладает склонностью
к сморщиванию при высыхании или отверждении.
Наиболее просто значение ОКП можно определить в том
случае, если предположить, что в состав сухой красочной пленки
входят только пигмент и связующее; в действительности же
различные добавки могут влиять на этот показатель. Трудно-
стей можно избежать путем определения значения Кс+Кп, т. е.
общего объема сухой пленки (исключая воздух).
Объем пигмента (К„) обычно проще определить, однако лако-
красочники чаще включают в рецептуру такую общую пигмент-
ную часть, которую считают наиболее предпочтительной. Су-
ществует множество способов определения базовых значений
ОКП, которые могут быть использованы в красках для спе-
циальных целей. Если это не оговорено особо, можно применить
простейшее определение (т. е. процент или долю объема пигмента
в сухой пленке).
8.3.2. Критическая объемная концентрация пигмента (КОКП)
С точки зрения развития концепции наиболее показательно
рассмотреть изменения, которые происходят при последователь-
ном добавлении порций связующего к основному сухому пиг-
менту до тех пор, пока связующее не окажется в избытке.
Опыты можно проводить также путем уменьшения количества
пигмента до тех пор, пока в системе не останется только одно
связующее. Начальные и конечные условия соответствуют крае-
вым значениям 100% и 0% на шкале ОКП.
При добавлении связующего к основному сухому пигменту
достигается экспериментальная точка, в которой количество
добавленного связующего соответствует полному заполнению
всех пустот между частичками пигмента. Объемная концентра-
ция пигмента в этой точке называется критической или КОКП
и представляет собой важнейшую характеристическую точку
при определении свойств пленки.
Декоративные краски содержат растворители, которые улету-
чиваются при высыхании. Это необходимо учитывать при опре-
делении ОКП, которая может быть выше, ниже или рав-
ной кокп.
В сухих пленках, где содержание пигмента выше КОКП,
количество связующего недостаточно для полного смачивания
всей поверхности пигментных частиц, в результате п 'енка ста-
новится пористой. В дезагрегированных, хорошо диспергирован-
ных красочных системах при содержании пигмента ниже КОКП
пигментные частички не вступают в контакт друг с другом.
Процесс, описанный выше, в значительной мере условный.
В реальных системах проявляется влияние многих дополнитель-
ных факторов, например адсорбция связующего поверхностью
частиц пигмента.
Имеются существенные различия между растворами и дис-
персиями связующих с точки зрения проницаемости их в пустоты
между частицами пигментов, а также взаимодействия с поверх-
ностью частиц. Существует богатая практика использования
ОКП как одного из параметров для описания свойств покрытий.
Обычно глянцевые краски имеют низкий ОКП (15—25%), в то
время как в грунтовках, полуматовых отделочных красках для
стен и красках для внутренних работ ОКП прогрессивно воз-
растает. Зная ОКП в краске, можно определить другие свой-
ства, такие как укрывистость, оттенок и т. д.
При графическом описании взаимосвязи свойств и ОКП зна-
чения ОКП целесообразно откладывать на абсциссе. Но в целом
более полезную информацию дает отношение ОКП к потен-
циальному значению КОКП, несмотря на то, что в реальной
композиции содержание пигмента ниже КОКП. Это отношение
играет роль внутреннего ориентира, компенсируя возможные
отклонения при выборе пигментной части для альтернативных
составов. Для описания этого отношения вводится термин «отно-
сительная ОКП» (X):
РКП
КОКП '
«Относительная ОКП» во многих случаях является более
предпочтительным параметром, чем ОКП, определенная графи-
ческим способом. Значения X будут больше 1, если содержание
пигментов в покрытиях превышают критическую величину [6].
8.З.2.1. Факторы, влияющие на характер упаковки пигментов
В связи с тем, что в сухой пленке часть объема занимает
воздух, вводится понятие упаковочного фактора пигмента, кото-
рый представляет собой отношение объема пигмента к суммар-
ному объему смеси пигмента, связующего и воздуха в сухой
пленке:
гю И» — обьсм пустот в сухой пленке.
Если в пленке нет пустот, а содержание пигмента ниже
КОКП, в этом случае ОКП = Р; если содержание пигмента выше
КОКП, Р будет оставаться постоянным и равным КОКП (так
как Ис заменяется на Ив). В реальных системах возможны
отклонения в связи с тем, что упаковочный фактор пигмента
может меняться в результате флокуляции. ОКП при этом может
увеличиваться до конечной точки 1,0 (100%).
8.3.2.2. Расстояние между частицами
Расстояние между частицами зависит от множества факто-
ров, в том числе от распределения частиц по размерам. Важно
также учитывать геометрические формы частиц В идеальной
системе принимается упрощение, допускающее, что частицы
имеют сферическую форму, равные размеры и равноудалены
друг от друга. В этом случае для ромбической упаковки объем
пустот равен 26%, и при полном его заполнении связующим
ОКП будет равна 0,74. Отсюда можно вывести уравнение для
расчета диаметра частиц:
d = \/0,74/О КП — 1. (8.3)
Сближение частиц при увеличении ОКП будет влиять на
эффективность рассеивания света частицами пигмента (разд.
8.4.3).
Частицы пигментов имеют очень большую поверхность. На-
пример, один килограмм ТЮг может иметь поверхность около
5000 м2. В результате этого значительное количество связую-
щего может адсорбироваться на поверхности пигмента. Частицы
пигмента, покрытые оболочкой связующего, влияют на упаковоч-
ный фактор пигмента как более крупные частицы.
Рассмотренные закономерности упаковки частиц становятся
более сложными, если их относить к реальной смеси, в которой
присутствуют частицы различных размеров. Предположим, на-
прймер, что два лабораторных стакана наполнены бисером
с диаметром частиц 1 см и 1 мм. Объем пустот будет приблизи-
тельно равен 26%. Но если бисер перемешать в отношении
3:1, маленькие частицы будут заполнять промежутки между
большими и общий объем пустот окажется значительно меньше.
Этот пример иллюстрирует эффект увеличения КОКП. Реальные
пигменты и смеси пигментов с наполнителями являются еще
более сложными системами, чем описанная выше. Поэтому
расчет КОКП становится малореальным и требует эксперимен-
тального определения. КОКП двухкомпонентной смеси с части-
цами различного размера, как правило, выше, чем для каждого
компонента в отдельности.
8.3.2.3. КОКП и маслоемкость
Простейший способ определения приблизительного значения
КОКП для определенного пигмента состоит в добавлении льня-
ного масла к сухому пигменту до получения однородной массы.
Для проведения испытания используется резиновый шпатель,
с помощью которого осуществляется тщательное механическое
смешение компонентов. С давних времен результат, выражен-
ный как масса льняного масла, поглощенного 100 г пигмента,
обозначается термином «маслоемкость». Если полученный ре-
зультат выразить в объемных, а не в массовых единицах, то
это и будет эквивалентом КОКИ- Высокое значение КОКП под-
разумевает низкую маслоемкость и наоборот. По причинам, рас-
смотренным выше, невозможно предсказать значение КОКП для
смеси, исходя из известных значений для отдельных пигментов,
и поэтому необходимо провести определение маслоемкости смеси
с помощью резинового шпателя. Для многих комбинаций важ-
нейших пигментов и наполнителей известны условия для обес-
печения максимальной упаковки, что достигается использова-
нием наполнителя с различными размерами частиц.
Использование маслоемкости как общего метода определе-
ния КОКП может быть допустимо при условии, что пигмент
будет вести себя аналогично во всех жидких связующих, что
не всегда имеет место. Поэтому не удивительно, что значения
маслоемкости, определенные в различных жидких средах, раз-
личаются между собой. Химики должны иметь информацию об
установленной величине маслоемкости и способе ее измерения;
при этом значение маслоемкости, полученное для льняного масла,
также может быть полезной качественной характеристикой для
правильного выбора пигмента, хотя этот показатель в редких
случаях будет точно соответствовать КОКП для других систем.
Связующее всегда следует рассматривать совместно с дан-
ным пигментом; при определении КОКП связующее играет двой-
ную роль: часть его, |/ал, адсорбируется на поверхности пигмен-
та, а другая часть, заполняет пустоты между частицами.
Это положение можно проиллюстрировать следующим уравне-
нием [5]:
КОКП =
И,
K.+ (V„„+V'e„)
(8.4)
В принципе, значение К-в может быть рассчитано исходя из
модели упаковки частиц. Однако при таком расчете оказывается,
что |/ад будет иметь большую величину, чем следует из теории
адсорбции. Хьюсман [7] особо подчеркивает, что формула (8.4)
может быть использована для расчета Еад только при условии,
что паста пигмента в конечной точке определения маслоемкости
будет состоять из индивидуальных диспергированных частиц.
На практике в системе обычно присутствуют агрегаты, величина
поверхности которых неизвестна. Поэтому окончательное зна-
чение маслоемкости зависит от эффективной плотности агрега-
тов, их упаковки и смачиваемости связующим. Хьюсман пред-
ложил модель, основанную на определении эффективной плот-
ности частиц, которая учитывает присутствие агрегатов. Асбек
предложил термин «предельная критическая объемная кон-
центрация пигмента» (ПКОКП) для обозначения монодисперс-
ного состояния системы и вывел соотношение между КОКП
и ПКОКП исходя из размеров индивидуальных частиц пигмента
(d) и пигментных агломератов (О) [8]:
КОКП = ПКОКП-(ПКОКП)2 (8.5)
Если ОКП прогрессивно возрастает до значений выше КОКП,
вторая точка перехода может быть достигнута только в том
случае, если имеется достаточно связующего для покрытия всех
частиц, что соответствует значению Еал в уравнении 8.4 и все
«свободное» связующее заменено воздухом. Кастелз и др. [9]
опубликовали экспериментальные данные, которые подтверж-
дают существование этой точки и представляют значительную
ценность' для установления оптимальных соотношений между
пигментом и связующим, что, в конечном итоге, влияет на опти-
мальные характеристики покрытий. Кремер использовал реоло-
гический метод для определения толщины адсорбционных слоев
на частицах пигментов и наполнителей [10].
8.3.2.4. Денсиметрический метод определения КОКП
В покрытиях, разработанных с учетом упомянутых выше кри-
тических точек, содержится воздух, который заполняет пустоты
после испарения растворителя. Четкое определение точки, при
которой под влиянием воздуха в пленке начинает возрастать
ОКП, является наглядным методом определения КОКП. При-
сутствие воздуха в сухой пленке уменьшает ее плотность, поэтому
объем воздуха может быть вычислен как разность между заме-
ренным значением Оа и теоретическим значением плотности От
Плотность может быть измерена многими способами, вклю-
чая гидростатическое взвешивание [11] и ртутную поромет-
рию [12].
На кривой зависимости плотности от ОКП должен присут-
ствовать пик, соответствующий КОКП. Соотношение между ко-
личеством воздуха, пигмента и связующего в сухой пленке также
A
100 % воздуха
Рис. 8.2. Соотношения между КОКП и содержанием воздуха
может быть представлено в виде треугольной диаграммы, кото-
рая дает обширную информацию. Стейн показал, как с помощью
диаграммы можно определить ОКП без проведения экспери-
ментов [13].
На рис. 8.2 состав любой сухой пленки, в рецептуре которой
содержание пигмента ниже критической точки, отложен на базо-
вой линии Р — В. Любая композиция, содержащая пигмент
выше критического количества, включает воздух (это соответ-
ствует прямой А — В) и располагается внутри треугольника.
Любой отрезок, выходящий из точки А, откладывает на Р — В
состав, соответствующий композиции с постоянным ОКП и умень-
шенным содержанием воздуха.
Если в каком-то покрытии заданное значение ОКП выше
критического, а содержание воздуха в пленке определено экспе-
риментально, состав сухой пленки будет соответствовать точке
пересечения линий ОКП и процентного содержания воздуха,
на рисунке 8.2 это проиллюстрировано для пленки, содержа-
щей 10% воздуха, с ОКП = 0,4. Если допустить, что пустоты
между частицами пигмента не изменяются по объему при умень-
шении количества связующего, то процент содержания пигмента
можно определить экстраполяцией к базовой линии, соответ-
ствующей нулевому содержанию воздуха при КОКП = 0,36. Уве-
личение значения ОКП в данной системе может происходить
при соответствующем увеличении содержания воздуха, что
можно отобразить на соответствующей линии ОКП, причем эта
линия будет такой же, которая соответствует КОКП = 0,36.
\Цля реальных композиций линия, соответствующая КОКП,
может не быть абсолютно прямой. Например, при изготовлении
красок с сильно различающимися значениями ОКП могут воз-
никнуть новые факторы, которые будут влиять на упаковку пиг-
мента. Но для большинства практических целей достаточная
точность определения КОКП может быть обеспечена одним про-
стым измерением.
8.3.2.5. Пористость
Из рис. 8.2 следует, что часть воздуха, присутствующего
в системе, можно рассматривать как общую пористость (это
обозначено на горизонтальной оси). Другими словами, пори-
стость соответствует проценту объема, исключая объем пигмента.
Второй способ обозначения пористости обычно называют индек-
сом пористости, который может быть определен на графике
методом экстраполяции отрезка, проведенного из точки Р на ось
«воздух — связующее» через точку, соответствующую определен-
ному составу. В примере, показанном на рис. 9.2, композиция
имеет общую пористость 0,1 и индекс пористости 0,16. Если
необходимо изменить свойства данной рецептуры, это возможно
сделать путем изменения одного или обоих этих параметров.
Поскольку они оказывают различное влияние на свойства кра-
сок, важно знать степень отклонений, допустимых при состав-
лении рецептур.
Пористость и индекс пористости могут быть выражены сле-
дующими уравнениями [14]:
। КОКП ,с
Общая пористость = 1-—---. (8.7)
, КОКП (1-ОКП)
Индекс пористости = 1----2--------.
1 ОКП (1-КОКП)
(8-8)
8.3.2.6. Критическая ОКП для дисперсионных связующих
До настоящего момента при обсуждении вопроса о КОКП
мы полагали, что связующее может легко проникать и пол-
ностью заполнять промежутки между частицами пигмента; счи-
тали также, что связующее — это раствор. Но для многих кра-
сочных систем, особенно для декоративных водных красок, свя-
зующее представляет собой дисперсию, содержащую частицы,
которые могут претерпевать различные деформации при форми-
ровании пленок. При этом возможны ситуации, когда дефор-
мационные процессы в пленках окажутся не двумерными, что
вызывает необходимость некоторой модификации приведенных
выше уравнений. Можно предположить, что на характер ура-
ковки пигмента и дисперсии связующего будут влиять размеры
частиц связующего (также как и пигмента) и температура стек-
лования (Тс) дисперсии полимера. Тс является полезной, но не
абсолютной характеристикой для определения способности дис-
персии связующего к коалесценции и образованию пленки. Эта
способность может быть в дальнейшем улучшена путем введения
специальных пластификаторов, обычно известных как коалесци-
рующие агенты, которые временно повышают текучесть и дефор-
мацию полимерных частиц.
Однако даже при условии использования коалесцирующих
добавок проникновение частиц связующего и их контакт с части-
цами пигмента в значительной мере затруднен, в результате
для связывания определенного количества пигмента необходимо
большее количество дисперсии полимера, чем раствора поли-
мера, т. е. КОКП для краски на основе дисперсии будет меньше,
чем КОКП для краски на основе раствора эквивалентного коли-
чества полимера.
Отношение количества раствора к количеству дисперсии
полимера, необходимых для связывания данной смеси пигмен-
тов, может быть названо «эффективностью связующего» или
«индексом связующего материала» «е» [15], который можно вы-
разить уравнением:
объемная маслоемкость
е=--------------------------.
объемная дисперсионная емкость
Если индекс связующего известен, индекс пористости для
краски на основе дисперсионного связующего получают исходя
из известной маслоемкости путем умножения уравнения (8.6)
на е. Затем изменяют действительную объемную адсорбцию
латекса и переводят ее в КОКП латекса (ЛКОКП) [17, с. 201]:
„ , ЛКОКП (1-ОКП)
I юристость латекса = 1----5.
ОКП (1-ЛКОКП)
Графические методы определения е или ЛКОКП описаны
в работе [16].
Использование некоторых дисперсий полимеров (латексов)
с заранее известным низким индексом связующего открывает
путь для регулирования пористости покрытий (см. разд. 8.5.3).
8.4. СЛЕДСТВИЯ ПОРИСТОСТИ
Увеличение объемной концентрации пигмента в любом связую-
щем приводит к существенному изменению свойств покрытий.
Понятно, что скорость изменения свойств связана с появлением
пористости, экстремальное значение которой соответствует КОКИ.
Свойства, которые заметно реагируют на изменение ОКП и свя-
' разделены на три основ-
занной с ней
ныв группы:
Механические
I свойства
Плотность
Прочность
Модуль упругости
Адге;1
ИЯ
пористостью, могут быть
Проницаемость
Коррозионная стойкость
Склонность к пузырению
Загрязняемость
Выпотевание компонентов
Оптические свойства
Рассеяние света
Контрастность цвета
Интенсивность цвета
Блеск
Обобщенные графические зависимости, характеризующие из-
менение свойств при изменении ОКП, опубликованы во многих
источниках [17]. В принципе точка перегиба или излома на гра-
фике, соответствующая значению КОКП, открывает возмож-
ность для определения собственно КОКП. Однако на практике
различные свойства по-разному реагируют на объем содержаще-
гося в пустотах воздуха и это отражается на значениях КОКП.
8.4.1. Механические свойства
Плотность пленки достигает максимального значения при
объемной концентрации пигментов, соответствующей критиче-
ской, и, как было отмечено ранее, это наиболее чувствительный
и точный метод определения КОКП. Известно [17], что механи-
ческие свойства и адгезия достигают максимального значения
при КОКП, но для практического использования этих характе-
ристик требуется дополнительная обработка данных.
Влияние количества наполнителя на механические свойства
полимеров широко исследовано. В общем случае модуль эластич-
ности при увеличении наполнения будет постепенно возрастать;
одновременно должно снижаться удлинение при разрыве. В боль-
шинстве случаев увеличивается прочность при разрыве, но эта
характеристика зависит от многих факторов, например эффек-
тивности взаимодействия между пигментом и связующим. В тех
случаях, когда достигается высокая прочность, удлинение имеет
низкое значение. Непрерывное возрастание прочности при при-
ближении к КОКП не является, однако, строго обязательным.
Не всегда также соблюдается известное положение о том, что
адгезия максимальна при наполнении, соответствующем КОКП.
Необходимо четко представлять различия между адгезией и
адгезионным состоянием (или практической адгезией). Явление
«адгезия» представляет собой сочетание взаимодействия раз-
личных факторов, включая механическую адсорбцию, диффу-
зию и электростатическое взаимодействие; при этом результаты
измерения адгезии могут существенно различаться в зависимо-
сги от выбранного метода проведения испытаний, а также состоя-
ния подложки (чистота, пористость и т. д.). 7
Бикерман обратил внимание на важную роль в оценке адге-
зии слабых пограничных слоев. В работе [18] приведены харак-
терные примеры использования метода адгезии для определе-
ния кокп.
8.4.2. Свойства, связанные с проницаемостью |
Проницаемость для жидкостей, газов и паров — важнейшее
свойство покрытий. Проницаемость водяных паров представляет
особую важность с точки зрения воздействия на процесс корро-
зии металлов и регулирования влажности древесины.
Поскольку пары проникают через связующее, а не через пиг-
мент, можно предположить, что увеличение ОКП должно умень-
шать проницаемость. Это обычно наблюдается в тех случаях,
когда пигмент недостаточно смочен связующим. Отклонения при
изменении проницаемости могут служить основой достаточно
тонкого метода определения смачиваемости пигментов [19].
Независимо от того, хорошо или плохо смачивается пигмент,
проницаемость резко увеличивается при достижении КОКП, что
объясняется появлением в пленке большого количества микро-
пор. Это сказывается на изменении коррозионной стойкости по-
крытий и склонности к образованию пузырей. Изменяется также
склонность к миграции некоторых компонентов эмалей в поверх-
ностный слой покрытия, что обычно связано с потерей блеска.
8.4.3. Оптические свойства
В белых красках укрывистость и белизна обеспечивается
их высокой светорассеивающей способностью (в цветных крас-
ках более важной характеристикой является поглощение света).
По своему назначению диоксид титана является главным источ-
ником рассеяния света. Для большей наглядности удобно
рассмотреть три отдельных явления: поверхностное отражение,
рефракцию и дифракцию, которые являются составными ча-
стями явления «светорассеяние».
8.4.3.1. Дифракция
Волновая теория света позволяет объяснить различные явле-
ния, приводящие к повышению рассеяния. В соответствии
с механизмом светорассеяния, рассеивающая способность уве-
личивается при уменьшении размера частиц до оптимального
значения, которое равно приблизительно половине длины волны
света.
\ Частица оптимального размера может отразить света при-
близительно в 4 раза больше, чем на нее падает, так как ее
рассеивающая поверхность в 4 раза больше геометриче-
ской.
\В случае диоксида титана для достижения максимальной
рассеивающей способности агломераты частиц ТЮг должны
быть разрушены. Это затрудняет
кра
ния
све'
ь разрушены. Это затрудняет повышение укрывистости
сок с TiOs, которые находят широкое применение. С точки зре-
оптических требований размер частиц для оптимального
‘Торассеяния можно рассчитать; при этом надо учитывать
зависимость оптических характеристик от длины волны. Практи-
ческие результаты также зависят от распределения частиц по
размерам и форме. В красках, содержащих наполнители, ча-
стицы титанового пигмента заполняют пустоты между более
крупными частицами наполнителя, поэтому в данном случае
достигается такой же эффект, как при более высокой ОКП
диоксида титана. Однако Т1О2 не может заполнить пустоты в ком-
позициях с наполнителями, если ее количество превосходит
собственную КОКП. По этой причине важнейшим требованием,
предъявляемым к пигментам для высоконаполненных рецептур,
является максимально высокая маслоемкость. Адсорбционные
слои на частицах пигмента обеспечивают сохранение промежут-
ков между ними, что необходимо для проявления оптической
эффективности при плотной упаковке.
В противоположность сказанному, частицы более мелких раз-
меров могут быть использованы для заполнения пустот между
частицами TiOs с целью повышения ее эффективности. Два
подхода к составлению рецептуры в конечном итоге приводят
к получению покрытий с различными свойствами, что позволяет
использовать различные варианты при составлении конкретных
рецептур.
Пористость пленки является функцией упаковки и масло-
емкости всей пигментной части, в то время как свойства, опре-
деляемые поверхностью покрытия, зависят от маслоемкости
основного пигмента.
8.4.3.2. Рефракция
Свет может преломляться частицами и, вследствие этого,
рассеиваться. Направление рассеяния зависит от коэффициента
рефракции. Чем меньше радиус частиц пигмента, тем больше
общая поверхность, доступная для взаимодействия со светом.
Рассеяние за счет рефракции увеличивается до критической
точки, при которой длина волны падающего света соизмерима
с размером частиц и луч света проходит через раствор без пре-
ломления.
При небольших углах падающего света коэффициент отра-
жения описывается уравнением закона Френеля: /
W"2""' Y I
\ п-г + п, / j
для части светового потока, которая проходит через материал,
имеющий показатель преломления щ, и для части, которая отра-
жается от поверхности материала с показателем преломления п2-
Так как воздух имеет более низкий показатель преломления,
чем любое связующее, его попадание в пленку увеличивает све-
торассеяние в результате появления границы раздела пиг-
мент/воздух. Таким образом, рецептуры, в которых наполнение
выше КОК.П,„имеют более высокую укрывистость, как, например,
в случае краски, известной под названием «сухая корка». Это
следует учитывать, если последствия увеличения пористости не
создают каких-либо проблем. Такие краски часто используют
для окраски потолков и стен. Оптические свойства открывают
другую возможность определения КОКП: так как на графике
зависимости ^светорассеяния от КОКП имеется довольно чет-
кий пик. Значение красящей способности вещества также имеет
экстремальное значение в области КОКП.
Если краска, разработанная для стен, будет применяться
для окраски поверхностей с различной абсорбционной способ-
ностью к свету, могут возникнуть трудности с получением оди-
наковых оттенков цвета. Для алкидных красок установлено, что
при ОКП значительно ниже КОКП может быть обеспечена опти-
мальная однородность по цветовым характеристикам покрытий.
8.5. КРАСКИ, СОДЕРЖАЩИЕ ВОЗДУХ И ПОЛИМЕРНЫЕ НАПОЛНИТЕЛИ
Белый цвет, встречающийся в природе, например белый снег,
лепесток цветка или морская пена, не требует для своего появле-
ния присутствия TiOa- Белизна возникает в результате взаимо-
действия света с поверхностью раздела и связана с наличием
пустот, как это было написано выше. Вполне понятно, что это
явление может служить не требующим больших затрат потен-
циальным источником для повышения укрывистости лакокра-
сочных покрытий, и поэтому привлекает внимание разработчи-
ков и производителей декоративных красок.
Представления о повышении укрывистости за счет микро-
полостей по аналогии с покрытием типа «сухой корки», описаны
ранее. В принципе, в зависимости от применяемого способа,
возможно повысить укрывистость без ухудшения других свойств
краски (таких, например, как загрязняемость), которые непо-
средственно связаны с увеличением пористости.
\ Когда микропустоты, содержащие воздух, полностью капсу-
лированы, можно получить покрытие с низким содержанием
ТПЭг, но с высокой укрывистостью и высоким блеском. Однако
до\ настоящего времени в большинстве случаев отдается пред-
почтение использованию базовых пигментов и наполнителей,
особенно для получения матовых и шелковистых покрытий.
Использование полимерных наполнителей и капсулирован-
ного воздуха представляет большой интерес при интерпретации
понятия КОКП и ее простейших расчетах. Не всегда оказывается
ясным, являются ли полимерные частицы пигментом или состав-
ной частью связующего. По всей видимости, они занимают про-
межуточное положение между двумя важнейшими компонен-
тами красок, а это открывает новые возможности в области
разработки рецептур.
Исследование этого вопроса важно не только для объектив-
ной оценки свойств новых пигментов, но и для более рациональ-
ного использования старых. Часто оказывается, что требова-
ния, предъявляемые к новым органическим пигментам, не могут
быть реализованы в стандартных рецептурах в связи с возни-
кающими трудностями введения пигментов. Взвесив все за и
против, можно создать рецептуры, каждая из которых будет
оптимальной в зависимости от выбранного способа пигменти-
рования. В дальнейшем каждая рецептура может быть оптими-
зирована с учетом различных потребительских свойств. Следует,
однако, отметить, что интерес к новым подходам, связанным
с пигментированием красок, будет возрастать, если окажется,
что стоимость диоксида титана будет непрерывно увеличиваться.
Использование микропустот в созданных человеком мате-
риалах подробно описано Сейнером в 1978 г. [20]. Он также
дал интерпретацию теоретического анализа Керкера и др. при-
менительно к покрытиям [21]. После этого появилось много
практических разработок, которые были запатентованы, а также
возросла коммерческая активность. Чальмерс и Вудбридж по-
дробно исследовали коммерческую конъюнктуру в 1983 г. [22].
Они описали четыре метода предварительной обработки поли-
мерной крошки, которые приведены в следующих разделах.
8.5.1. Измельчение
Delux Australia Ltd описали использование как плотной, так
и вспененной полиэфир-стирольной крошки. Плотная крошка
имеет сравнительно крупные размеры (порядка 40 мкм) и может
поставляться как в чистом виде, так и окрашенная. Основной
областью применения этой крошки являются рецептуры матовых
красок с хорошей устойчивостью к истиранию. Неокрашенная
крошка используется лишь для интенсивных цветов. Глубоко
матовые отделочные материалы с хорошей стойкостью к исти-
ранию имеют широкий спрос в Австралии и Южной Африке,
несмотря на то, что они дороже материалов с традиционными
наполнителями. Для снижения стоимости материала при усло-
вии сохранения укрывистости используется вспененная крошка.
Вспененная крошка изготавливается методом двойного эмуль-
гирования в воде при использовании в качестве исходного сырья
ненасыщенного полиэфира, отвержденного стиролом. Для уско-
рения свободнорадиального отверждения применяется окисли-
тельно-восстановительная система. При проведении процесса
необходимо соблюдать предосторожности. Образующиеся кру-
пинки имеют размеры от 25 до 0,7 мкм, хотя более типичными
являются размеры 11—14 мкм. При этом может быть получена
крошка, содержащая включения ТЮг. На практике применение
крошки с включениями ТЮ2 эффективно в тех случаях, когда
требуется обеспечить хорошую плотность пленки и при этом
используется латекс, обеспечивающий высокое значение ОКП
(крошка рассматривается как пигмент).
В современных исследованиях рассматривается возможность
применения более глубокой степени сшивки, что может привести
к расширению области применения красок. Так как рецептуры
красок имекЗт некоторую специфику, их техническая пригод-
ность зависит от относительной стоимости и особых требований
потребителя. Крошка, которая пригодна для одних потребите-
лей, может быть непригодной для других. Трудности, возникаю-
щие при выборе между оптимальной рецептурой, содержащей
крошку, и традиционными материалами, хорошо проанализиро-
ваны в работах [24, 25]. Опубликовано руководство по исполь-
зованию вспененной крошки [26].
8.5.2. «Микроблоки» (Фирма Berger Jenson & Nicholson}
«Микроблоки» обычно представляют собой агрегаты мелких
частиц, образующихся при проведении реакции полимеризации
в водной среде Они используются в разных системах по анало-
гии с применением полимерной крошки. Предполагают, что их
неправильная геометрическая форма обеспечивает получение
более прочных пленок, чем в случае сферической формы крошки.
В отличие от пористой крошки, где включения Т1О2 обеспечивают
значительный цветовой эффект, применение «Микроблоков» тре-
бует довольно высокого наполнения ТЮг, в результате чего
получаются глубоко матовые покрытия с высоким ОКП.
8.5.3. Полимерные наполнители
(Glidden Coatings & Resings Division)
Ремиг и др [27, 28] выдвинули предположение, что очень
тонкая крошка полистирола или полистирольный латекс с раз-
мером ячейки примерно 0,1—0,6 мкм вызывает образование
микропор в красочной пленке и, тем самым, повышает укрыви-
стость. Размеры этой так называемой крошки имеют тот же поря-
док, что и частицы латекса в эмульсионных красках, и поэтому
крошка обычно смешивается с латексом в соотношении 1:1.
Такие композиции могут быть использованы в шелковистых и
матовых красках для повышения ОКП выше критической, при
этом обеспечивается высокая укрывистость при естественной
сушке покрытия и очень незначительное уменьшение сплошности
пленки.
Использование полистирольных пигментов является нагляд-
ным примером тех трудностей, которые встречаются при приме-
нении традиционных методов расчета ОКП- С одной стороны,
твердые частицы латекса с высокой Тс будут вести себя как
очень тонкие частицы наполнителя (т. е. должны входить в урав-
нение для ОКП), а с другой стороны, их можно рассматривать
как латекс с нулевой или низкой связующей активностью. Пра-
вильное решение, очевидно, находится между этими двумя экстре-
мальными положениями. Соответствующим подбором коалесци-
рующих добавок можно добиться более равномерного слияния
полимерных частиц по сравнению с неорганическими наполни-
телями, но полной коалесценции частиц добиться невозможно.
Для получения оптимальных свойств покрытия необходим
тщательный выбор коалесцирующих добавок и других компо-
нентов краски, что не исключает возможность создания мате-
риалов с помощью других рецептурных решений [29].
8.5.4. «Ropague»
Фирма Rohm & Haas [31] разработала добавки для повыше-
ния укрывистости, представляющие собой крошку вспененного
сополимера стирола с акрилатами, суспендированную в воде
(обычно 37% по весу или 52% по объему).
В процессе высыхания покрытий, содержащих такой «укры-
вающий полимер», вода, испаряющаяся с поверхности частиц,
должна быть замещена воздухом. Образовавшиеся воздушные
пустоты выполняют роль источника рассеяния и непосред-
ственно влияют на повышёние укрывистости. Установлено, что
четыре части (по объему) «укрывающего полимера» дают при-
мерно такой же эффект, как одна часть ТЮг [30]. Коммерчески
доступный материал [«Ropaque» ОР-62] содержит однородные
мелкие частицы размером примерно 0,4—0,5 мкм. Он может
быть использован для повышения кроющей способности; кроме
того, равномерным распределением между частицами ТЮг этот
продукт предотвращает образование крупных агрегатов. По-
скольку частицы продукта имеют меньшую поверхность, чем
соответствующий объем диоксида титана, то для их связывания
тре уется меньшее количество пленкообразователя. Другими
словами, может быть увеличена КОКП, что открывает возмож-
ности для разработки композиций с более высокой ОКП. Из
сказанного следует, что «укрывающий полимер» правильнее
рассматривать как пигмент, нежели как составную часть свя-
зующего.
При введении в рецептуры уже существующих красок новых
добавок, повышающих укрывистость, следует производить кор-
ректировку рецептур исходя из объемных, а не весовых соотно-
шений компонентов. Изготовители красок должны вносить соот-
ветствующие коррективы в техническую документацию.
8.6. ПРИРОДА КРАСОЧНОГО СВЯЗУЮЩЕГО
8.6.1. Общие положения
-В предыдущих разделах основное внимание уделялось влия-
нию изменения соотношений пигментов и связующих на свойства
рецептур красок. Не менее важную роль для лакокрасочника
играет правильное понимание химической и физической природы
пигментов и связующих. Эта весьма сложная проблема более
или менее детально рассматривается в настоящей книге. Пред-
принимается попытка связать некоторые общие положения тео-
рии с их влиянием на свойства декоративных красок строитель-
ного назначения, а также наметить пути разработки красок
специального назначения.
Ассортимент строительных красок имеет специальные под-
разделы: краски для пола, для защиты бетона, для плаватель-
ных бассейнов и т. д. Эти краски изготавливаются на основе
различных связующих, однако основу ассортимента крупното-
нажных декоративных красок строительного назначения состав-
ляют две группы красок: «водные» и «краски на растворителях»
(более точно: «водные» и «неводные»). В состав первой группы
входит небольшое количество полярных растворителей, напри-
мер спиртов или производных этиленгликоля, тогда как невод-
ные группы красок обычно содержат алифатические раствори-
тели и иногда также некоторое количество ароматических угле-
водородов.
Уместно задать вопрос: почему соотношения между водными
красками и красками на растворителях различны в различных
областях применения и разных странах?
В целом существует общая тенденция перехода от красок
на растворителях к водным краскам во всех областях приме-
нения. Объем применения водных красок зависит от условий
эксплуатации (например от климата), специальных требований,
от экологических и экономических требований и иногда от исто-
рических традиций. В Англии приблизительно 60% декоратив-
ных красок выпускается на водной основе, а в США свыше
70% [32]
Разработчик рецептур должен знать, каким образом природа
связующего влияет на свойства лакокрасочного покрытия и
каковы будут практические результаты от применения новых
материалов, выбранных для разработки красок. Прежде всего
необходимо определить природу пригодного растворителя и уста-
новить, какие физические и химические процессы будут происхо-
дить при высыхании пленки.
Декоративные краски на растворителях обычно являются
пожароопасными, имеют сильный, неприятный запах, но не за-
мерзают при отрицательных температурах. С другой стороны,
водные краски не пожароопасны, но могут вызывать коррозию
и замерзают. Эти выводы очень важны, но этого не достаточно
для полной характеристики свойств краски. Существенно иметь
четкое представление о физической природе полимерного свя-
зующего. Необходимо также ясно понимать различие между
«раствором» и «дисперсией» полимера. Одним из наиболее оче-
видных различий растворов и дисперсий является их реологи-
ческое поведение. Благодаря взаимодействию между частицами
дисперсии имеют более низкую текучесть, что сказывается на
малярных характеристиках красок.
Растворы и дисперсии различаются также по степени воз-
растания вязкости при испарении растворителя. Вязкость дис-
персии растет быстрее, чем вязкость раствора. Многие недо-
статки водных красок, например, неудовлетворительный розлив
иди сохранение следов от кисти, связаны с быстрым изменением
вязкости при испарении воды [33]. С другой стороны, более
высокая текучесть растворов создает проблему получения по-
крытий без натеков на вертикальных поверхностях и затрудняет
окраску острых кромок. Максимальные различия в текучести
декоративных красок можно проиллюстрировать путем сравне-
ния вязкости матовых эмульсионных и жидких глянцевых кра-
сок. В целом следует отметить, что для решения проблемы теку-
чести содержание твердой фазы в вододисперсионных красках
обычно бывает более низким, чем в красках на основе раство-
рителей.
Следующим этапом в построении модели, описывающей неко-
торые аспекты создания красок, является рассмотрение хими-
ческой природы связующего. Для того, чтобы полимер мог быть
использован в качестве связующего для красок, он должен быть
жестким и прочным. Экспериментальные исследования и тео-
ретические данные показывают, что полимеры с большой моле-
кулярной массой в значительной мере отвечают этим требова-
ниям, но недостаточно хорошо растворяются в растворителях,
пригодных для использования в декоративных красках. Поэтому
необходимо использовать низкомолекулярные полимеры, кото-
рые хорошо растворяются, но затем повышают молекулярную
массу в результате сшивки в процессе высыхания.
Хотя существуют различные способы отверждения покры-
тий, на практике приемлемы только некоторые из них. С учетом
влияния атмосферы наиболее распространенным способом
сшивки является процесс, протекающий через стадию окисле-
ния. Процесс самоокисления протекает при формировании по-
крытий из растворов масел или полимеров, модифицированных
маслом в уайт-спирите, которые в течение многих лет являются
основой лакокрасочной промышленности.
Изучение механизма окисления явилось предметом многих
серьезных исследований, однако основные характеристики про-
цесса развиты в гидроперокисдной теории, выдвинутой Фарме-
ром в 1942 г. В начальной стадии окисления происходит незна-
чительное увеличение вязкости и образование гидроперекисей
на активных метиленовых группах. Эта стадия сопровождается
также ростом количества сопряженных связей, образование
которых протекает по радикальному механизму и сопровож-
дается отщеплением водорода от активных метиленовых групп
с последующей перегруппировкой. Вторичный процесс окисле-
ния, который катализируется сиккативами, протекает на поверх-
ности покрытия и сопровождается распадом перекисей с после-
дующей рекомбинацией и образованием связей С—С, С—О—С
и С—О—О—С [34].
К сожалению, самоокисление масел ведет к нежелательным
эффектам, связанным с появлением запаха и пожелтением
пленки. Процесс самоокисления трудно остановить. Поэтому,
вследствие очень глубокой сшивки, покрытие становится хруп-
ким и заметно снижаются его прочностные характеристики.
Сшивку можно предотвратить путем использования предва-
рительно синтезированных высокомолекулярных продуктов, при-
чем из-за различия в растворимости, о чем говорилось выше,
этот процесс легче протекает в дисперсии, чем в растворе. Про-
цесс пленкообразования протекает по механизму коалесценции.
Следует иметь в виду, что не во всех дисперсиях полимеров про-
исходит коалесценция, но для того, чтобы получить пленку с нуж-
ными характеристиками, следует выбирать связующее с опреде-
ленным набором свойств (Тс и др.). Образование пленок дости-
гается для большого количества полимеров. Такие пленки не
требуют отверждения и сохраняют термопрочность и жесткость.
Согласно Диллону и др. [35], движущей силой коалесценции
является поверхностное натяжение и вязкое течение полимер-
ных молекул. Браун отмечает важную роль давления в капилля-
рах [36]. В последующих работах рассматривается влияние
тех и других явлений на процесс пленкообразования [37].
Несмотря на то, что образованные из дисперсий пленки мо-
гут выглядеть вполне однородными для невооруженного глаза,
микроскопические исследования показывают, что такие пленки
обычно имеют гетерогенную структуру, построенную по типу
взаимопроникающей сетки. Такая структура существенно влияет
на их свойства. Их проницаемость, например, как правило,
выше, чем проницаемость пленок, полученных из растворов.
Кроме того, проницаемость во всех случаях у несшитых пленок
выше, чем у сшитых.
Ниже приведены общие характеристики четырех основных
групп рецептур декоративных
Полимерная дисперсия;
(пленка формируется только в ре-
зультате коалесценции)
Композиция нолимер/олигомер;
(пленка формируется в результате
самоокисления)
красок:
Водные (с добавкой
растворителей)
I группа
Эмульсионные или
латексные краски
IV группа
Водоразбавляемые
алкиды
Неводные (в основ-
ном на уайт-спирите)
II группа
Неводные диспер-
сии «НВД»
III группа
Масляные или ал-
кидные краски
8.6.2. Некоторые особенности применения красок
Наибольший объем выпускаемых промышленностью декора-
тивных красок приходится на I группу рецептур — эмульсион-
ные или латексные краски, а также на III группу — алкидные
краски, однако во многом их свойства диаметрально противо-
положны. Основные различия между водными и неводными
красками даны в табл. 8.1.
Как следует из приведенных в таблице данных, ни один из
типов красок не имеет явного преимущества и во многих слу-
чаях могут применяться и водные, и неводные краски. Выбор
того или иного типа покрытия будет зависеть от конкретной
области применения и от требований по внешнему виду, а также
От торговой и экономической ситуаций и законодательных огра-
ничений.
Рассмотрим для примера, как осуществляется окраска внут-
ренних помещений с помощью матовых или глянцевых красок
в Англии. Матовые краски используются при окраске больших
поверхностей стен и потолков; в этих случаях важнейшими тре-
бованиями являются легкость нанесения, быстрое высыхание и
отсутствие запаха. С точки зрения этих требований водоэмуль-
сионные краски имеют несомненные преимущества. Недостаточно
хороший розлив и неисчезающие следы от кисти не очень за-
метны на матовых покрытиях и не являются серьезными недо-
статками этих красок. На глянцевых покрытиях последствия
плохого розлива более заметны, но в соответствии с сущест-
вующим стандартом такие покрытия можно применять для стен.
Для наклонных поверхностей, например плинтусов и дверей,
существует более жесткий стандарт, требования которого могут
быть обеспечены при применении красок на растворителях. То
Таблица 8.1. Основные различия между покрытиями
Основные причины свойств Покрытия на основе водных красок (эмульсионные краски)
различия Положительные характеристики Отрицательные характеристики
Природа растворите- ля: вода или органи- ческий растворитель Негорючесть Легкость отмывки от за- грязнения Быстрое высыхание Незначительный исходный запах Низкая токсичность Плохая стойкость свежего покрытия к обрызгиванию водой Проблемы при заморажи- вании и оттаивании Медленное высыхание на холоде, во влажных усло- виях Коррозия железного крепе- жа Ускорение коррозии Склонность к вспучиванию
* Дешевый, легко доступный растворитель Возможность повторного нанесения в тот же день Плохое смачивание масля- ных шпатлевок Склонность к биоразруше- нию
Физическое состояние: дисперсия или раствор Относительно высокая проницаемость Простота нанесения Неисчезающие слезы от кисти Хорошая кроющая способ- Недостаточный розлив ность Пониженный сухой остаток Т рудность достижения блеска Образует корку иа крышке тары
Химическая природа связующего: термо- пластичное или термо- активное Отсутствие пожелтения Незначительное повышение хрупкости при старении Длительное сохранение эла- стичности- Сохранение исходного от- тенка Отсутствие запаха при ста- рении Способность адсорбировать пыль Склеивание свежсокрашен- Прозрачность для ультра- фиолетовых лучей
же самое можно сказать об окраске кухонных помещений и
ванных комнат, где вследствие конденсации влаги краски на
растворителях также являются более пригодными.
Полуглянцевые краски содержат значительные количества
компонентов, растворимых в органических растворителях; по-
крытия на их основе целесообразно использовать в тех случаях
основе водных красок и красок на растворителях
Основные причины различия свойств Покрытия на основе неводных красок (масляные или аклидные краски) Положительные Отрицательные характеристики характеристики
Природа растворите- ля: вода или органи- ческий растворитель Водостойкость пленки на Пожароопасность ранних стадиях высыхания Потребность в специальных разбавителях Отсутствие проблем хране- Сильный исходный запах ния при низких температу- рах и коррозионных про- блем
Физическое состоя- ние: дисперсия или раствор Отсутствие склонности к Сравнительно низкая ПДК вспучиванию Возможность длительного Возможность нанесения сле- пребывания в открытой та- дующего слоя как правило ре через сутки Совместимость с маслины- ми шпатлевками Относительно низкая проницаемость Хороший розлив Длительный отлип после на- Легкость нанесения несения Высокий сухой остаток Возможность получения высокого глянца Хорошая впитываемость
Химическая природа связующего: термо- пластичное или термо- активное Образование более жесткой Склонность к пожелтению пленки Повышение хрупкости при Легкая отмываемость от за- старении грязнений Появление запаха при ста- рении Образование поверхностной пленки при храпении в таре
когда требуется высокая износостойкость и стойкость к мытью:
в коридорах школ, госпиталях, в заводских помещениях и т. д.
Общий баланс использования полуглянцевых красок в Англии
делится между водными красками и красками на растворителях,
с некоторым преимуществом в пользу водных красок с улучшен-
ными технологическими характеристиками.
В случае глянцевых покрытий для наружных работ основу
ассортимента составляют краски на растворителях. При этом
высокий блеск и поэтому хороший розлив более существенны,
чем недостатки, обусловленные сильным запахом при высыхании
и пожелтении. Для окраски окон высокоглянцевые краски на
растворителях находят более широкое применение, поскольку
они имеют ряд преимуществ перед водными красками. К числу
преимуществ относятся возможность окраски при неблагоприят-
ных погодных условиях, а также возможность нанесения по
шпатлевке на льняном масле. Эти преимущества особенно ярко
проявляются при окраске наружных деревянных поверхностей,
причем в странах с различными климатическими условиями и
различной технологией строительства эти преимущества прояв-
ляются по-разному. Страны, в которых производится большое
количество деревянных конструкций, чаще используют водные
краски для внутренних работ, так как они позволяют получить
прочные покрытия, не требующие особого ухода при эксплуа-
тации.
8.6.3. Пути совершенствования рецептур
На основе приведенных выше данных можно сделать вывод,
что выбор того или иного материала, отвечающего определен-
ному комплексу потребительских свойств, является отнюдь не
простой задачей. При разработке новых рецептур всегда нужно
хорошо представлять, какие свойства по сравнению со свой-
ствами известной рецептуры должны быть улучшены. Чаще
всего могут изменяться физические и химические характери-
стики материала, на что следует обратить внимание.
В разд. 8.6.1 приведены обобщенные данные, позволяющие
реализовать разнообразные возможности при разработке новых
рецептур. Для иллюстрации рассмотрим материалы, относя-
щиеся ко II и IV группам рецептур. Возникает вопрос, почему
материалы этих групп сравнительно мало используются на прак-
тике и обладают ли они какими-либо преимуществами по сравне-
нию с материалами, относящимися к другим группам рецептур?
Рецептуры, объединенные в группу II, обычно относятся к невод-
ным дисперсиям полимеров (НВД) [38]. В первом приближе-
нии краски на основе полимерных дисперсий, представленные
в этой группе, могут быть сопоставлены с красками на водных
дисперсиях (латексах), входящими в I группу рецептур, хотя
имеется много различий в технологии производства и в стабиль-
ности этих материалов. Технология красок на НВД была раз-
работана позднее, чем технология красок на водных дисперсиях.
Если сравнить два близких но составу полимера, приме-
няемых в водной и неводной средах, очевидные преимущества
будет иметь водная система. Обычно при разработке новых
материалов используют растворители с минимальной горючестью
и низкой стоимостью. Однако следует отметить, что материалы
II группы имеют ряд достоинств, как, например, быстрое высы-
хание, а применение растворителей улучшает совместимость
связующих с различными пластификаторами и смолами, что
позволяет повышать атмосферостойкость и коррозионную стой-
кость. В целом, рецептуры, относящиеся ко II группе, по своим
свойствам занимают промежуточное положение между рецепту-
рами I и III групп.
Наиболее перспективной областью применения материалов
этой группы является наружная окраска, так как в этом случае
необходимо использовать быстросохнущие эластичные покры-
тия, стойкие к омыванию дождем сразу же после нанесения.
Такие покрытия могут наноситься как по кирпичной кладке,
так и по дереву; возможно использование этих материалов и
в других областях [39].
Четвертая группа рецептур включает растворимые в воде
масляные и алкидные материалы. Для придания водораствори-
мости в состав пленкообразователей алкидного или другого типа
вводят кислотные или основные функциональные группы, кото-
рые затем с помощью нейтрализующих добавок переводятся
в водорастворимые «мыла». Одним из примеров материалов
этого типа являются малеиновые аддукты, нейтрализуемые
аммиаком или аминами. Для придания водорастворимости ши-
роко применяется также тримеллитовый ангидрид. Дозировка
нейтрализатора должна очень строго соблюдаться, поскольку
она может влиять на вязкость краски, равномерность высыха-
ния и некоторые свойства покрытия [40].
При использовании водорастворимых полимеров важное зна-
чение имеет их гидролитическая стабильность. Неустойчивость
сложноэфирной связи к гидролитическому воздействию ограни-
чивает применимость соответствующих материалов, особенно
там, где требуется длительный срок службы. Кроме того, при
использовании аммиака в качестве нейтрализатора появляется
неприятный запах.
В целом, при переходе от группы I к группе IV наблюдается
улучшение свойств, например розлива, но появляется и ряд
недостатков: пожелтение и запах для группы III. Кроме того,
материалы всех групп имеют некоторые собственные особен-
ности.
Подводя итог всему сказанному, можно утверждать, что для
разработки декоративных красок общего назначения наиболее
широкое распространение получили водные дисперсии полиме-
ров. Однако дальнейшие исследования могут изменить эту си-
туацию, так как в последнее время возрастает интерес к при-
менению смесей растворов полимеров с дисперсиями как в вод-
ных, так и неводных системах. Можно примерно представить,
какое место будет занимать смесевой продукт такого типа
в классификационных группах. Точные характеристики свойств
предсказать трудно; они могут быть установлены лишь в резуль-
тате эксперимента. Для некоторых водорастворимых полимеров
наблюдается аномальное изменение вязкости растворов при раз-
бавлении, что связано с набуханием полимерных агрегатов,
однако это явление можно использовать для получения ряда
положительных свойств.
Применение комбинаций водных дисперсий и растворов по-
зволяет добиться блеска покрытий. Такие покрытия находят
широкое применение для отделки наружных деревянных поверх-
ностей. Роль алкидной составляющей в смесевых красках со-
стоит в улучшении адгезии к побеленным поверхностям, сниже-
нии проницаемости и улучшении впитываемости в необработан-
ную древесину. Алкидная составляющая может использоваться
либо в виде раствора, либо в виде истинной эмульсии. Мате-
риалы на основе эмульсии алкидов по своим свойствам зани-
мают промежуточное положение между материалами I и IV групп
рецептур.
8.7. ПРОБЛЕМЫ КОЛЕРОВАНИЯ КРАСОК
8.7.1. Общие положения
В большинстве стран основу ассортимента составляют деко-
ративные краски белого цвета; за ними в порядке уменьшения
объема выпуска следуют краски пастельных тонов, а затем ин-
тенсивных темных цветов. Конечно, имеются национальные тра-
диции применения различных цветовых оттенков покрытий;
существуют также различные требования по их отражающей
способности. В Англии наибольшей популярностью для наруж-
ной окраски дверей, гаражей и т. п., пользуются черный и тем-
ные тона.
Все страны столкнулись с противоречием, обусловленным
ограничениями по допустимым с точки зрения сервиса характе-
ристикам материалов и экономическими затратами на их произ-
водство. Проблема усугубляется тем, что для выпуска красок
разных цветов необходимо иметь на производстве. несколько
технологических линий. При решении этой проблемы необходимо
учитывать соотношение между количеством красок, готовых
к употреблению (т. е. красок светлых тонов, или колерных кра-
сок, изготовленных непосредственно на заводе) и количеством
красок, колеруемых перед применением. При этом необходимо
учитывать, что для колеровки красок требуется специальное
оборудование, предназначенное для дозировки колеровочных
паст, их расфасовки в мелкую тару и для смешения их с красоч-
ной основой.
Колерные краски широко распространены в мире, но в разных
странах характер их производства не одинаков. Так, в Северной
Америке, Австралии и Скандинавских странах наиболее широко
применяются готовые к употреблению краски, в то же время
в Англии практикуется использование, готовых к употреблению
красок лишь нескольких основных тонов и одновременно по-
ставляются колеровочные пасты, объем которых составляет
около 10% от общего объема выпускаемых красок.
Каких-то простых причин, которые объясняют эти различия
между странами исходя из их географических, экономических
и социальных особенностей, не существует. Например в Англии,
где плотность населения очень высока, поставщикам красок
выгодно организовывать централизованные склады для снабже-
ния потребителей. В то же время, такая организация не очень
практична там, где потребитель разбросан на большой терри-
тории. Можно также отметить, что в Англии нет потребности
в разработке большого разнообразия цветов. Кроме того, спрос
на высокоглянцевые краски в Англии находится в противоречии
с- техническими ограничениями на производство колеровочных
паст для водных красок и красок на растворителях, обеспечи-
вающих сохранение исходного глянца.
Что бы ни служило основанием для существовавших ранее
различий, совершенно очевидно, что теперь наступил период
внедрения эффективных достижений, базирующихся на возмож-
ности широкого использования микропроцессоров и компьютер-
ной техники. Существенные преимущества внедрения новой
технологии могут быть достигнуты при хорошо организованной
работе по маркетингу и поиску рынков сбыта, причем эта работа
должна предшествовать постановке новых исследований. Произ-
водство красок требует больших капитальных вложений и по-
этому какие-либо изменения технологии обходятся очень дорого.
При организации производства новых материалов трудно бывает
показать преимущества, которые могут быть достигнуты при
использовании более дорогих колерных красок, а изменения
качества красок неизбежно отражаются на их продажной
цене.
Производство колерных красок требует участия квалифици-
рованных рабочих, так как продолжительность производствен-
ного цикла составляет, всего 5—10 мин, в течение которых эта
работа должна быть выполнена. Производственный цикл можно
сократить при использовании различных автоматизированных
систем колерования красок, и, несмотря на то, что это скажется
на увеличении стоимости, определенная степень автоматизации
необходима для дополнительного контроля, поскольку высокое
качество окупает все дополнительные затраты. Заключительные
стадии производства некоторых колерных красок могут быть
достаточно сложными, что отражается на их стоимости, поэтому
требуется четкое сопоставление преимуществ, которые дают
новые разработки, с затратами на их создание.
Декоративные колерные краски значительно отличаются от
материалов, используемых при ремонтной окраске автомобилей.
Последние обычно включают более широкую гамму цветов, в том
числе глубокие черные тона, а также краски с металлическим
оттенком. Существенные различия имеются и в технологии
окраски, так как для окраски автомобилей могут использоваться
как нитроцеллюлозные материалы, так и двухупаковочные поли-
уретаны с изоцианатными отвердителями. Колерование авто-
мобильных красок проводят в емкости, в которую по весу добав-
ляется пигментированная основа и необходимые добавки пигмен-
тов, после чего производится тщательное перемешивание. Деко-
ративные строительные краски колеруются путем добавления
к краске данного цвета определенного объема колеровочной
пасты, после чего определяется общий объем готовой краски.
Количествен добавляемой колерной пасты обычно не превышает
7% объема самой краски. Декоративные краски поставляются
в емкостях, заполненных примерно на 95% от допустимого
объема; на емкостях имеется соответствующая маркировка. Го-
товая краска после колерования может занимать объем от 96%
до 102% от допустимого объема, что создает некоторые труд-
ности при определении ее стоимости и затрудняет сравнение
с готовыми для потребления красками. Наиболее простым реше-
нием этой проблемы могло бы быть заполнение емкости 95%
основы и 5% пасты, однако в этом случае возникнут сложности
с производством ряда цветов, и если ввести это соотношение, то
потребуется расширение ассортимента рецептур красочных основ
и колеровочных паст.
8.7.2. Дозирующие машины
За некоторым исключением, основой любого процесса коле-
рования является дозирующая машина, которая должна обеспе-
чить подачу заданного объема колеровочной пасты. Такие ма-
шины могут быть разделены на три группы: ручные, полуавто-
матические и автоматические. Как ни странно, автоматические
машины были разработаны раньше, чем полуавтоматические,
хотя различия между ними весьма условны [41]
Любая машина должна иметь емкость для колеровочной
пасты и механизм (ручной или автоматический) для ее периоди-
ческого перемешивания. Обычно колеровочная паста подается
с помощью поршневого насоса, а объем пасты контролируется
по длине хода поршня. В ручных машинах непосредственно
с валом поршня связана градуированная шкала, деления кото-
рой соответствуют определенному объему подаваемого мате-
риала (в разных странах цена деления может выражаться в раз-
них единицах), в Англии шкала для измерения ооъема жидко-
сти имеет цену деления 1 /96 унции. Сохранение этих архаичных
единиц измерения и связанные с ними трудности обусловлены
большими расходами на переоснащение и переделку машин.
Рассмотренный метод дозирования пигментных паст ограни-
чивает количество цветов, которые могут быть получены путем
колерования основного тона.
При использовании автоматических машин работа двигателя
контролируется электронным устройством, что обеспечивает не-
прерывную подачу колеровочных паст. В этом случае практиче-
ские ограничения связаны только с точностью определения объема
подаваемого материала.
Расположение емкостей для колеровочных паст у различных
машин может быть различным. Наиболее распространенной кон-
струкцией является карусель, при вращении которой емкости
с краской поочередно попадают под общую точку дозирования
и подачи колеровочной пасты. В другой модификации карусель-
ной машины имеются неподвижные емкости с основной краской,
к которым при вращении карусели последовательно переме-
щается дозатор колеровочной пасты. С помощью карусельного
механизма можно одновременно осуществлять подачу в опреде-
ленной последовательности нескольких колеровочных паст. Это
ускоряет этап колеровки в общем производственном цикле,
однако при большой длине трубопроводов могут образовываться
застойные зоны, которые ухудшают качество краски.
При работе на ручных колеровочных машинах оператор дол-
жен строго следить за соблюдением рецептуры и последователь-
ностью загрузки; эти данные должны иметься в технологической
карте или должны быть нанесены на микрофиши. После выбора
красочной основы оператор распаковывает ее, проверяет каче-
ство и направляет на участок колерования, где выполняются
соответствующие операции и тщательное перемешивание. Во
многих случаях при колеровании возникает необходимость в
добавлении трех или более различных колеровочных паст, по-
этому оператор может допустить довольно много ошибок.
После добавления колеровочной пасты емкость должна быть
закрыта и передана на соответствующую перемешивающую ма-
шину. Существует несколько типов полностью автоматизиро-
ванных машин, которые имеют электронную рецептурную базу
данных, поэтому после пуска электродвигателя операция коле-
рования может быть выполнена автоматически.
Ряд операций, которые обычно ускоряются при использова-
нии машин, позволяют выбрать нужную красочную основу. Кро-
ме того, с помощью машин проверяют правильность выбора
соотношения компонентов для получения требуемой рецептуры.
Часто колеровочная паста в начале процесса подается через
отверстие в крышке емкости, которая закрывается пластмассе-
вой пробкой. С помощью системы компьютеров одновременно
с подбором колера можно печатать этикетки и другие данные
о краске. Несмотря на целый ряд преимуществ, автоматиче-
ские -машины более чем на порядок дороже ручных машин и
поэтому их применение на средних производствах не экономично.
Полуавтоматические машины представляют собой нечто
среднее между двумя упомянутыми видами колеровочных машин.
Эти машины могут существенно различаться деталями, однако
у всех имеется поршень, приводимый в движение электродвига-
телем, и ручная подача колеровочной пасты. Такие машины
обладают многими преимуществами при производстве неболь-
ших партий материалов.
Что касается уменьшения стоимости в будущем, то это свя-
зано с удешевлением отдельных элементов технологии, разра-
боткой более надежных методов контроля, позволяющих отка-
заться от многих ограничений, которые закладываются в рецеп-
туры. . Сюда же относятся и мероприятия по повышению
сохранности продукции при хранении на складе, а также созда-
ние банка данных, содержащего информацию о ее качестве.
Уже сегодня имеется в продаже система [42], обеспечиваю-
щая проведение колеровочных работ на базе спектрофотомера,
связанного с автоматической колеровочной машиной. Многие
потребители предпочитают применять не стандартные цвета,
а различные промежуточные оттенки, поэтому изготовитель
должен иметь возможность наглядно продемонстрировать полу-
чаемые при колеровании оттенки цвета. Для того, чтобы выпус-
кать краски таких оттенков, необходимо иметь интенсивные
колеровочные пасты и очень точные дозирующие машины. Все
это, безусловно, будет увеличивать общую стоимость красок
и поэтому неприемлемо для производства небольших партий.
Достижения в области технологии дозирования должны, безус-
ловно, оказать влияние на совершенствование технологии пере-
мешивания красок, что уже сегодня является лимитирующей
стадией в автоматических и полуавтоматических машинах.
В настоящее время возникают трудности при сбыте красок,
требующих обычного перемешивания с помощью мешалок. Для
этих целей применяют машины, действие которых основано либо
на встряхивании, либо на вращении емкости с краской в двух
плоскостях. Магнитные, ультразвуковые или .вибрационные ме-
тоды перемешивания оказались практически непригодными. Для
усовершенствования процесса перемешивания разрабатываются
бесшумные, высокоскоростные машины. Создаются машины с
повышенной пропускной способностью, выдерживающие боль-
шие нагрузки, снабженные автоматическим гидравлическим за-
твором. Энергетические особенности процесса перемешивания
создают определенные трудности для совмещения этого про-
цесса с процессом розлива красок.
В заключение следует отметить, что хотя дозирующие ма-
шИНы являются неотъемлемой частью всего процесса колерова-
ния все же имеются возможности колерования без использо-
вания машин. Примером является применение градуированных
шприцев, заполняемых колеровочной пастой, которую потреби-
тель может сам добавить в краску для получения необходимого
ему оттенка. Конечно, этот метод ограничивает выбор цвета
и затрудняет его воспроизведение. В США применяют другой
вариант колерования, заключающийся в том, что к продаваемой
краске прилагаются маленькие пластмассовые коробочки с по-
рошком определенного колера, который может добавляться в
краску непосредственно при продаже [43]. Менее удобным яв-
ляется способ колерования, применяемый в Германии. Здесь пред-
варительно взвешенная колерная добавка заворачивается в во-
дорастворимую пленку, которая помещается в защищающую от
повреждения упаковку. Хотя при использовании таких методов
колерования, в принципе, можно получить большое разнообра-
зие цветов, на практике ассортимент их ограничен. Поэтому труд-
но конкурировать с тем многообразием цветов, которое может
обеспечить машинный метод колерования [44].
8.7.3. Колеровочные пасты и красочные основы
8.7.З.1. Цвет и совместимость
Требования, рассмотренные в предыдущих разделах, во мно-
гом являются общими для всех колеровочных паст, хотя в ряде
случаев имеются некоторые различия. Наиболее важным и слож-
ным вопросом является совместимость. Промышленные колеро-
вочные пасты обычно предназначаются для одного вида про-
дукции; универсальная колеровочная паста может использо-
ваться лишь для таких материалов, в состав которых входят
одинаковые растворители. Для декоративных красок наиболее
целесообразно использовать универсальные колеровочные пасты,
как для водоразбавляемых систем, так и для систем, разбав-
ляемых углеводородными растворителями. Это противоречивое
требование может быть удовлетворено в результате применения
продукта с компромиссными свойствами, что не может не отра-
зиться на качестве красок, особенно при больших масштабах
их использования. В этом заключается существенное различие
между готовыми к употреблению и колерованными красками
одного и того же цвета.
Соотношение объемов выпуска красок на растворителях и
водных красок постоянно меняется в сторону увеличения вы-
пуска последних, и, вероятно, наступит такой момент, когда
будет необходимо выпускать колеровочные пасты лишь для водо-
разбавляемых красок. Пока же необходимо четко представлять
требования, которые предъявляются к соответствующим про-
дуктам для водных и неводных красок.
Наиболее простым видом колеровочных паст являются ком-
позиции, содержащие разнообразные красящие вещества, дис-
пергированные в этиленгликоле с добавкой различных поверх-
ностноактивных веществ. Такие композиции могут замедлять
высыхание (особенно в случае алкидных красок) и увеличивать
чувствительность к воде покрытий на основе водных красок.
В таких странах, как Англия, где в основном используются
краски, колеруемые на месте потребления, существует еще одна
проблема, обусловленная изменением вязкости красок при вве-
дении колеровочных паст и необходимостью их тщательного
перемешивания. Для того, чтобы уменьшить влияние измене-
ния вязкости, целесообразно ввести ограничения по максималь-
ному количеству добавляемых колеровочных паст. Некоторые из-
готовители решают эту проблему путем создания колеровочных
паст нового поколения [45], содержащих минимальное количе-
ство гликоля. Несмотря на ряд преимуществ, такие пасты склон-
ны к комкованию и часто возникает необходимость в подборе
соответствующего сопла для краскораспылителя, чтобы избежать
его засорения при проведении окраски.
При проведении колеровочных работ необходимо правильно
выбрать колеровочную пасту и основной тон, так как они могут
существенно различаться по оттенку и интенсивности. В идеаль-
ном случае при проведении колеровочных работ хорошо было бы
иметь одну бесцветную основу и пять очень интенсивных колеро-
вочных паст белого, черного и трех основных цветов. Однако
на практике, в силу экономических и технических причин, такой
путь не находит применения. Влияние пигментов на оттенок краски
зависит от многих факторов; кроме того, цвета некоторых пигмен-
тов являются недостаточно чистыми.
Очень большое количество цветов может быть получено на
основе голубого фталоцианинового пигмента, красного азокра-
сителя и ариламидного желтого красителя при смешении их
с белым и черным цветами, но существует еще много других
цветов, которые невозможно получить с помощью названных
выше пигментов. Применение новых колеровочных паст увеличи-
вает разнообразие цветов, однако если новая колеровочная
паста позволяет создать мало новых оттенков, то увеличение
стоимости может оказаться неоправданным.
При выборе пигментов для колеровочных паст необходимо
учитывать их экономичность, цвет, укрывистость и прочность;
в этом отношении требования, предъявляемые различным про-
изводителям, являются общими.
Типичный ассортимент колеровочных паст выпускается на
основе следующих пигментов: черный, белый, органический крас-
ный, органический желтый, неорганический красный, неоргани-
ческий желтый и органический голубой. Ио многообразию оттен-
ков ассортимент у различных производителей может разли-
чаться и зависит от вида применяемой основы, веяний моды и
от опыта по выпуску материалов с учетом затрат, которые необ-
ходимы для их модификации.
Помимо упомянутых колеровочных паст находят применение
пасты фиолетового, оранжевого, зеленого, коричневого и других
цветов, полученные на основе желтого и красного органических
пигментов (см. гл. 3, где рассматриваются типы пигментов, ис-
пользуемые в декоративных красках).
Важной характеристикой колеровочных паст является их ин-
тенсивность, которая прямо зависит от концентрации пигмента.
Для многих целей будет справедливым утверждение, что более
высокая концентрация пигмента предпочтительнее. При допу-
стимости введения колеровочной пасты в количестве, максимум
5—10%, целесообразно использовать пасту с высокой концен-
трацией пигмента, так как в этом случае можно получить макси-
мальное количество глубоких тонов.
Трудность применения очень интенсивных колеровочных паст,
а иногда и паст с нормальной интенсивностью, заключается
в том, что их необходимо точно дозировать, особенно при необ-
ходимости получения пастельных тонов. Такая точность не может
быть обеспечена большинством дозирующих машин, и поэтому
интенсивные колеровочные пасты (например, паста на фтало-
цианиновом голубом пигменте) используются только для полу-
чения интенсивных тонов.
При существующем уровне точности добавление интенсив-
ной колеровочной пасты производится с недогрузкой, а затем
осуществляется дополнительная подколеровка небольшими пор-
циями пасты. Для рационального использования рассмотренных
типов колеровочных паст необходимо иметь соответствующие
типы красочных основ. Обычно используют смеси красочных
основ, эффективность которых зависит от ОКП диоксида титана.
В этих основах содержание ТЮг меняется от нуля до макси-
мального верхнего предела, характерного для готовых к употреб-
лению белых красок, поэтому имеется возможность получать
глубокие, средние и светлые тона. Для дальнейшего расшире-
ния ассортимента цветов необходимо использовать цветные
основы, содержащие в простейшем случае красные и желтые
пигменты; можно использовать и другие цвета, получаемые при
применении смесевых пигментов. Каждая новая основа должна
обеспечивать соответствие требованиям, предъявляемым к крас-
кам различного типа и назначения, поэтому не очень выгодно
иметь большое разнообразие основ.
В последнее время наблюдается тенденция отказа от созда-
ния окрашенных основ, так как применение интенсивных коле-
ровочных паст снижает потребность в таких основах.
8.7.3.2. Консистенция и воспроизводимость цвета
Обычно после проведения операции колерования нет необхо-
димости в дальнейшей подгонке цвета, так как краска должна
соответствовать эталону цвета. Это, в свою очередь, требует
высокого качества как колеровочных паст, так и красочных
основ, которые должны по своим свойствам точно соответство-
вать требуемым параметрам. Так как колеровочная паста обычно
добавляется по объему, то необходимо знать ее плотность и
стабильность; обычно при производстве используют вакуумное
оборудование.
При производстве колеровочных паст должна соблюдаться
воспроизводимость интенсивности от замеса к замесу; точно так
же при производстве красочных основ должна соблюдаться
воспроизводимость оттенков, поскольку при использовании основ
и колеровочных паст с невоспроизводимыми цветовыми харак-
теристиками невозможно сохранить свойства соответствующей
-рецептуры. Если воспроизводимость не будет соблюдаться, банк
рецептур может сильно увеличиться, а это может стать источ-
ником появления ошибок, особенно при ручном колеровании.
На практике очень сложно получить одинаковые оттенки
в разных средах, и поэтому нет ничего странного, что для водных
красок и красок на растворителях рецептуры различны. В разра-
ботанной рецептуре колеровочной пасты очень сложно заменить
какой-либо пигмент на другой, поскольку в случае замены бу-
дут изменяться цветовые характеристики, т. е. расширяться ас-
сортимент рецептур.
В заключение следует отметить, что колеровочные пасты дол-
жны иметь удовлетворительные реологические характеристики,
обладать достаточной текучестью для перекачивания по трубо-
проводам и не создавать трудностей при нанесении краски мето-
дом распыления. Кроме того, они не должны загустевать и оседать
при хранении, хотя большинство дозирующих машин обеспечи-
вает их перемешивание. Необходимо также учитывать влияние
компонентов колеровочной пасты на состояние окрашиваемой
конструкции. Алюминий, например, подвергается коррозии под
действием гликоля.
8.8. РОЛЬ ПОДЛОЖКИ
Свойства декоративных покрытий лишь в незначительной сте-
пени зависят от природы подложки, однако в большинстве слу?
чаев свойства подложки имеют важное значение при выборе
того или иного вида декоративного покрытия. Выбор декоратив-
ного покрытия может быть продиктован необходимостью обеспе-
чения комплекса специальных свойств; в других случаях при-
нимается во внимание потенциальная возможность взаимодей-
ствия покрытия с подложкой. В некоторых случаях необходимые
свойства будут достигаться при нанесении выбранной системы
покрытий непосредственно на подложку; в других случаях на
подложку наносится специальный грунтовочный слой, который
перекрывается другими слоями, соответствующими по своим
свойствам требованиям условий эксплуатации.
Целесообразно разделить подложки для строительных красок
на три большие группы, включающие дерево, металлы и камен-
ную кладку. Небольшую, но постоянно растущую группу под-
ложек составляют органические пластики. Эти материалы тре-
буют специального изучения для определения их долговечности,
так как составляющие компоненты пластиков подвержены ста-
рению и обесцвечиванию. При возобновлении окраски наличие
сохранившегося покрытия можно не принимать во внимание,
однако в ряде случаев следует учитывать состав и характери-
стику старого покрытия и условия, в которых оно эксплуати-
ровалось. Так, например, в случае битумных покрытий и неко-
торых защитных составов для обработки дерева при перекраске
возникают проблемы в связи с миграцией битума в наружные
слои при проникновении активных растворителей из наносимого
материала в нижний слой.
Увеличение толщины пленки изменяет многие важные свой-
ства покрытий и, в первую очередь, проницаемость.
Как уже отмечалось, покрытия должны быть стойкими к
действию климатических и погодных факторов. Эти свойства
могут зависеть от взаимодействия покрытия с подложкой. В боль-
шинстве случаев требуется разрабатывать специальные рецеп-
туры красок для наружных и внутренних работ, что связано
ро спецификой воздействия факторов окружающей среды на
поверхностный слой покрытия, а также с необходимостью защиты
.самой подложки от воздействия этих факторов. Различные усло-
вия эксплуатации создают характерные проблемы, которые оп-
ределяют выбор того или иного покрытия в зависимости от вида
подложки. Например, дерево должно быть стойким к биоразру-
шению и защищено от проникновения влаги; цемент имеет сильно
щелочную поверхность, а металлы подвергаются коррозии. В каж-
дом из этих явлений активная роль отводится воде, и поэтому
справедливо будет сказать, что’ важнейшим назначением покры-
тий является торможение тех процессов, в которых вода прини-
мает активное участие. К таким процессам, в первую очередь,
следует отнести проницаемость. Однако проницаемость, допусти-
мая для разных типов покрытий, может существенно различаться.
> В заключение еще раз отметим, что характеру подложки
следует уделять серьезное внимание, так как от вида подложки
во многом зависят причины разрушения покрытий со всеми
вытекающими отсюда последствиями; от характера подложки
зависят также требования, предъявляемые к покрытиям декора-
тивного назначения.
Глава 9
АВТОМОБИЛЬНЫЕ КРАСКИ
Д. А. Ансделл
9.1. ВВЕДЕНИЕ
Требования, предъявляемые к качеству автомобильных по-
крытий, весьма разнообразны и определяются их назначением.
В ассортимент этих материалов входят краски для защиты
днища, обеспечивающие получение покрытий, стойких к корро-
зии и ударам мелких камней, а также декоративных поверхност-
ных покрытий с высокой эластичностью. Все эти материалы
должны отвечать требованиям массового производства, т. е
должны обеспечивать получение прочных, эластичных покрытий,
экономичных в эксплуатации.
Конструкция корпуса автомобиля претерпела значительные
изменения в течение этого века. В настоящее время основным
конструкционным материалом для корпуса является, главным
образом, мягкая сталь, однако могут быть использованы и другие
сплавы, а также детали из пластика. Очертания корпуса очень
сложны и отдельные его части труднодоступны для окраски.
Производительность современных заводов весьма высока и
составляет примерно 45 автомобилей в час, что выдвигает опре-
деленные требования к материалам и технологии окраски, кото-
рые должны соответствовать производственному циклу. Произ-
водство легковых автомобилей во всем мире (за исключением
Рис. 9.1 Производство легковых автомобилей в 1983 г.
СССР И стран СЭВ) составило примерно 28,1 млн. единиц, для
чего потребовалось около 570 млн. л. красок. В Великобритании,
для сравнения, произведено 1,044 млн. автомобилей и потребо-
валось 20,0 млн. л. краски. На диаграммах, приведенных ниже
(рис. 9.1 и 9.2), представлено распределение этих данных для
разных стран.
Используемые лакокрасочные материалы подразделяются
на две принципиально различающиеся группы: грунтовки и вы-
равнивающие слои (шпатлевки), обозначаемые как нижележа-
щий слой (система покрытия), и верхний (отделочный) слой.
Для современных покрытий соотношение между различными
группами грунтовки : шпатлевки : материалы для отделочного
слоя составляет (в %%) 30:20:50. В разных частях света
имеются различия в технологии окраски, особенно в нанесении
отделочного слоя, а это в значительной мере определяет требова-
ния к свойствам красок, режимам проведения окраски и неко-
торым деталям процесса. Основным назначением покрытий яв-
ляется обеспечение защитных и декоративных свойств. С этой
целью процесс окраски подразделяется на ряд отдельных этапов.
Эти этапы (сочетания отдельных операций) выполняются в опре-
деленном порядке, и, хотя функции каждой операции специфичны,
можно оценить вклад каждой из них в обеспечение требуемых
свойств покрытия.
Рис. 9.2. Объем выпуска красок в 1983 г.
Процесс окраски включает следующие этапы: подготовка по-'
верхности металла, грунтование, шпатлевание, нанесение поверх-
ностного слоя. Первые два этапа обеспечивают защитные свой-
ства, последующие — декоративные свойства.
В основу современной технологии окраски положены три
метода нанесения грунтовочных слоев: метод распыления, метод
окунания и метод электроокраски (наиболее широко приме-
няемый в настоящее время).
Основой для такой классификации являются методы нанесе-
ния грунтовки, так как это является основным отличительным
признаком; все остальные стадии процесса практически одина-
ковы. Все три метода нанесения грунтовки предусматривают
соответствующую подготовку поверхности. Шпатлевки и поверх-
ностные слои обычно наносят распылителем. Могут быть исполь-
зованы различные методы распыления, например пневматиче-
ское (ручное или автоматическое), либо электростатическое.
Более подробно эти методы будут описаны ниже.
9.1.1. Система окраски с грунтовкой, наносимой распылителем
Система включает следующие материалы:
Цинксодержащая грунтовка — только для внутренних поверхностей;
Грунт-шпатлевка — 40—50 мкм;
Отделочный слой — 45—55 мкм.
Цинксодержащую грунтовку наносят на внутренние поверх-
ности деталей корпуса до сварных работ. Это обеспечивает за-
щиту от коррозии поверхностей, которые в дальнейшем стано-
вятся недоступными для окраски.
Этот метод применялся до разработки грунтовок, наносимых
окунанием. В настоящее время он применяется только в тех слу-
чаях, когда малый объем производства делает невыгодным или
неприемлемым применение окунания или электроокраски.
9.1.2. Системы окраски с грунтовкой, наносимой окунанием
Система включает следующие материалы:
Противокоррозионная грунтовка, наносимая окунанием — 12—18 мкм;
• Шпатлевка — 40—50 мкм;
Отделочный слой — 45—55 мкм.
Эта система широко применялась на линиях массового про-
изводства до внедрения в начале 60-х годов метода электро-
окраски. Она и сейчас еще используется в очень ограниченном
масштабе в тех случаях, когда превалирует малый объем произ-
водства или другие факторы, например стоимость.
9 13. Система с грунтовкой, наносимой методом электроокраски
Система включает следующие материалы:
Грунтовка, наносимая методом электроокраски
(анодным или катодным осаждением) — 20—25 мкм;
Шпатлевка — 23—35 мкм;
Отделочный слой — 45—55 мкм.
В настоящее время электронанесение наиболее широко ис-
пользуется в массовом производстве. Оно представляет собой
эффективную, относительно простую операцию с высокой сте-
пенью автоматизации. Если в начале 60-х гг. широко применялся
метод анодного осаждения, то в настоящее время быстро рас-
пространяющийся метод катодного осаждения стал основным
в процессе окраски.
В описанных выше технологических процессах все покрытия
требуют горячей сушки; это необходимо для того, чтобы достиг-
нуть максимально высоких показателей качества и обеспечить
возможность организации процесса в рамках конвейера. Темпе-
ратура сушки может варьировать от 180 °C для грунтовок, нано-
симых методом катодного осаждения, до 80 °C при восстанов-
лении отделочного слоя. Типичная схема процесса показана на
рис. 9.3.
9.1.4. Почазатепи качества покрытия
. На различных производствах могут существовать собствен-
ные спецификации качества покрытий. Однако эти спецификации
могут меняться, например в зависимости от вида отделочного
слоя покрытия, а также от локальных заводских условий про-
изводства. Обычно качество покрытия определяется следующими
показателями: 1) внешний вид (блеск и внешние различия);
2) декоративные свойства (цвет и уровень блеска); 3) механи-
ческие характеристики и стойкость к действию ударов мелких
камней; 4) адгезия; 5) коррозионная стойкость и влагостой-
кость; 6) стойкость к действию бензина и растворителей; 7) об-
щая химическая стойкость, в том числе стойкость к действию
кислот; 8) твердость и стойкость к повреждениям; 9) ремонто-
способность.
Материалы, обеспечивающие достижение показателей, опи-
саны ниже. Технология применения чрезвычайно сложна и дли-
тельна; она включает нанесение материалов и горячую сушку.
Объемы энергетических затрат, трудозатрат и капиталовложе-
ний являются значительными. При дальнейшем усовершенство-
вании материалов основное внимание будет уделяться не только
улучшению качества покрытий, но и обеспечению экологических
требований.
Предварительная промывка Сушка
Рис. 9.3. Процесс окраски в условиях завода
9.2. ПОДГОТОВКА ПОВЕРХНОСТИ
\ Подготовка поверхности преследует три цели:
\ 1) Удаление окалины и жировых компонентов, находящихся
на поверхности стали, а также временных консервационных за-
щитных покрытий.
| 2) Улучшение адгезии краски путем создания фосфатного
слря, который лучше всего совместим с последующими слоями
грунтовки.
3) Создание барьера, препятствующего проникновению кор-
розионноактивных элементов через пленку покрытия.
Обычно подготовка поверхности включает в себя следующие
операции: 1) удаление ржавчины; 2) щелочное обезжиривание;
3) промывка водой; 4) фосфатирование металла; 5) промывка во-
дой с целью деминерализации фосфатного слоя.
В зависимости от индивидуальных требований и условий
производства могут применяться методы распыления, распыле-
ния — окунания или окунания; последние два метода более пред-
почтительны в современных крупногабаритных установках.
Удаление ржавчины. Наиболее пригодным методом является
применение минеральных кислот, особенно при большой толщине
слоя ржавчины или при наличии слоя окалины; предпочтение
обычно отдается использованию составов на основе фосфорной
кислоты. Хотя фосфорная кислота действует медленнее, чем
соляная или серная кислоты, опасность загрязнения поверхно-
сти растворимыми солями значительно меньше, поскольку боль-
шинство фосфатов металлов малорастворимы.
Щелочное обезжиривание. Щелочные агенты широко приме-
няются в установках с распылением или окунанием, в которых
масла и жировые загрязнения частично смываются и эмуль-
гируются в щелочном растворе. Физическое воздействие, воз-
никающее при впрыскивании щелочного агента, в существенной
мере способствует удалению твердых частиц, присутствующих
на поверхности изделия.
Составы для обезжиривания изменяются в зависимости от
множества факторов, таких как тип подлежащих удалению ма-
сел или жировых загрязнений, вид подвергающегося обработке
металла и т. д. Однако необходимо контролировать силу щелочи,
чтобы избежать снижения активности поверхности металла.
Типичными щелочными реагентами являются каустическая сода,
тринатрийфосфат и карбонат натрия; они применяются с детер-
гентами различного типа для эмульгирования масел и жировых
смазок.
Фосфатирование металла (преобразующие покрытия). Ис-
пользуются два основных типа фосфатов:
фосфат железа — вес покрытия 0,2—0,8 г/м2;
фосфат цинка — вес покрытия 0,5—4,5 г/м'-’.
При увеличении веса фосфатного покрытия повышается его
стойкость к коррозии, но снижается механическая прочность
и адгезия последующих слоев покрытия. Таким образом, вер
фосфатного покрытия является его важной характеристикой, й
на практике всегда необходимо подбирать такой вес, который
обеспечивает требуемый уровень других свойств.
Фосфат железа аморфен, а его пленка имеет малый вес.
Фосфат железа используется, в основном, при окраске рефриже-
раторов, моечных машин и металлических деталей, которые
эксплуатируются в не слишком агрессивных коррозионных усло-
виях. Исходным требованием для таких покрытий является соче-
тание высокой механической прочности и адгезии; фосфат же-
леза, образующий покрытие с низким весом, идеален для дан-
ных целей.
Фосфат цинка является более универсальным покрытием,
особенно при автоматизированной окраске; его получение —
важная составная часть всего процесса окраски. Особенность
покрытия на основе фосфата цинка состоит в том, что оно
может быть получено путем кристаллизации из' раствора непо-
средственно на металлической поверхности, так как в кислых
растворах возможно существование фосфатов различного хими-
ческого состава, находящихся в равновесии.
Химическое равновесие можно представить в виде следую-
щей схемы:
3Zn(H2PO4)2 < > Zn3(PO.i)2 -р 4Н3РО4
Растворимая Нерастворимая Свободная
соль соль кислота
Это равновесие сохраняется до тех пор, пока остается неиз-
менной концентрация исходных компонентов. Исходной реакцией
процесса фосфатирования является взаимодействие свободной
фосфорной кислоты с металлом:
Fe + 2Н3РО„---------> Ре(Н2РО.1)з + Н2.
Подложка Свободная Растворимый
кислота фосфат железа
В результате этой реакции равновесие, установившееся в
соответствии с первым уравнением, сдвигается вправо и раство-
римый фосфат цинка переходит в нерастворимый фосфат цинка
с выделением свободной кислоты. Пленка кристаллического
фосфата начинает расти, и этот процесс продолжается до тех.
пор,- пока вся подложка не будет полностью покрыта фосфат-
ной пленкой. Образование в качестве побочного продукта раство-
римого фосфата железа замедляет процесс и отравляет фосфат-
ный раствор, поэтому в раствор вводится добавка окислителя,
который переводит растворимый фосфат железа в нераствори-
мый осадок.
С подготовка поверхности обработкой ингибиторами коррозии.
(Стальная поверхность или подложка включает кристаллическую
Хшетку железа, углерод, растворенный в железе, карбид же-
леза и нерастворенные частицы углерода.
\ Коррозия представляет собой окислительный процесс, проте-
кающий, в основном, в среде электролитов. На влажной поверх-
ности стали за счет примесей в металле (карбиды, углерод
и V. д.) могут образовываться микрогальванические элементы
с ярко выраженными катодными (отрицательными) и анодными
(положительными) участками. В присутствии воды и солей (элек-
тролитов) эти участки становятся электродами микрогальвапи-
ческих элементов, при работе которых развивается процесс
коррозии. Ионы двухвалентного железа, образующиеся на ка-
тодных участках, реагируют с кислородом воздуха и гидроксиль-
ными ионами электролита, в результате чего образуются оксиды
и гидроксиды железа. Эти процессы свидетельствуют о начале
коррозии и для предотвращения ее дальнейшего распростране-
ния необходимо изолировать поверхность металла от воздействия
внешней среды.
Обычная краска — плохой изоляционный материал, ибо не
существует покрытий, непроницаемых для влаги. Кроме того,
любое незначительное повреждение поверхности покрытия до
металла, например царапина, полученная при ударе' камнем,
неизбежно приводит к появлению коррозии, которая будет рас-
пространяться под пленкой из-за недостаточной адгезии, свой-
ственной почти всем лакокрасочным пленкам на необработан-
ных металлических поверхностях.
Фосфатные пленки являются прекрасным изоляционным ма-
териалом. Рост кристаллов фосфатов начинается с активных
участков поверхности металла и продолжается до тех пор, пока
не покрывает всю поверхность. Такая кристаллическая струк-
тура по своему действию ведет себя как «керамическая» изо-
ляция, не подвергающаяся воздействию влаги, особенно в тех
случаях, когда она перекрыта красочной пленкой. Появление
царапин или других видов местных повреждений не представ-
ляет серьезной опасности при эксплуатации фосфатированных
изделий.
9.3. ГРУНТОВАНИЕ
Назначение грунтовки состоит в усилении противокоррозион-
ной защиты. Это достигается введением в состав рецептур грун-
товок антикоррозионных пигментов; связующее при этом взби-
рается так, чтобы обеспечить не только антикоррозионные, но
и необходимые механические свойства.
За последние 50 лет конструкция автомобилей настолько
изменилась, что для нанесения грунтовочных слоев на различные
Таблица 9.1. Эволюция методов грунтования корпусов автомобилей /
Период Тип корпуса Тип грунтовок Особенности метода J
До войны Корпус/шасси Грунтовки, нано- симые распыле- нием Пригодность для всех пр- верхностей
После войны до 1960 г. Сварные конст- рукции (цельно- сварной корпус) Грунтовки на ос- нове растворите- лей для нанесе- ния окунанием Трудоемкость Неэкономичность Неудовлетворительная защита внутренних по- верхностей Пожароопасность Вредность для здоровья
1960—1966 гг. Сварные конст- рукции (цельно- сварной корпус) Водорастворимые грунтовки для на- несения окунани- ем Пониженная вредность Остальные показатели как у предыдущего типа
1966—1977 гг. Сварные конст- рукции (цельно- сварной корпус) Анодная электро- окраска Автоматизация Высокая производитель- ность Пониженная вредность Эффективность Однородность пленки
1977 - Сварные конст- рукции (цельно- сварной корпус) Катодная элект- роокраска Высокие защитные свой- ства Хорошая стабильность Более высокая проницае- мость
участки корпуса необходимо использовать различные методы
окраски. В табл. 9.1 представлена эволюция методов грунтова-
ния корпусных конструкций автомобилей.
Совершенствование технологии нанесения грунтовок на кон-
струкции корпуса автомобилей непосредственно связано с раз-
витием техники сварки. Прежде всего, внедрение автоматизи-
рованной технологии сварки во время Второй мировой войны
и применение точечной сварки привело к появлению цельно-
сварных конструкций корпуса. В конструкциях такого типа мно-
гие участки поверхности оказываются фактически недоступны
для окраски, например, различные крепежные элементы каркаса
и дверные подпорки. Нанесение грунтовок на эти поверхности
методом распыления осуществить невозможно. Для этого был
предложен метод окунания, позволяющий нанести грунтовку на
все поверхности подобно тому, как это делалось при окраске
внутренних поверхностей летательных аппаратов.
Первоначально грунтовки для нанесения методом окунания
изготавливались на основе растворителей, однако в 60-х гг. для
этой цели были предложены водорастворимые грунтовки, обла-
дающие пониженной пожароопасностью и менее вредные для
здоровья. Водорастворимые грунтовки имеют более низкую эф-
фективность и менее экономичны, чем грунтовки на растворите-
L тем не менее их внедрение явилось еще одним важным до-
Дрижением в технологии грунтования автомобильных корпусов,
так как эти грунтовки позволили применить метод электроосаж-
двНИЯ.
\ 9.3.1. Нанесение грунтовки методом распыления
\В этом методе грунтования используют цинконаполненные
грунтовки, а иногда другие противокоррозионные материалы,
которые наносят на внутренние поверхности корпусных конструк-
ций до сварки корпуса. Таким способом производится защита
от коррозии таких поверхностей, которые становятся недоступ-
ными для окраски в последующих операциях.
Метод распыления очень широко использовался до тех пор,
пока не был разработан метод нанесения грунтовки окунанием.
В настоящее время распыление используется только тогда, когда
малые объемы продукции не оправдывают затрат на создание
установки для электроокраски, а также в тех случаях, когда
для содержания более современных установок требуются боль-
шие средства.
9.3.2. Нанесение грунтовки окунанием
Исходные материалы. Эти материалы используются для обес-
печения защиты внутренних частей корпуса автомобиля. Так
как на эти поверхности в дальнейшем другие покрытия не нано-
сят, применяемые грунтовки должны обеспечивать высокую стой-
кость к коррозии и хорошую адгезию как к предварительно
обработанной фосфатирующим составом поверхности, так и
к необработанному металлу.
Из-за сложной конфигурации и больших размеров корпуса
автомобиля грунтовки для нанесения окунанием должны иметь
хороший розлив. Кроме того, грунтовки должны обладать до-
статочной стабильностью, поскольку ванны, в которых произво-
дится грунтование, по величине должны быть соизмеримы с кор-
пусами автомобилей.
В настоящее время используется четыре основных типа грун-
товок для нанесения методом окунания: алкидные, алкидно-
эпоксидные, эпоксиэфирные и эпоксидные.
В большинстве случаев в состав грунтовок, наносимых оку-
нанием, входят растворители, хотя используются и водораство-
римые материалы. Наиболее широко применяются в промыш-
ленности алкидные грунтовки, так как они сочетают хороший
внешний вид и сравнительно низкую стоимость. Эпоксидные
грунтовки разработаны позднее с целью улучшения стойкости
к коррозии, однако они более дорогие и их применение в ваннах
Для окраски окунанием сопряжено с некоторыми сложностями.
Пигментная часть. Традиционно в рецептурах грунтово^
использовался красный железоокисный пигмент, однако в на/
стоящее время более предпочтительны черные и серые цвета.
В качестве основных пигментов используют красный железб-
окисный пигмент и сажу. Для усиления антикоррозионных
свойств часто применяют хромат цинка.
Типичные наполнители — бариты (сульфат бария) и блан-
фикс (осажденный сульфат бария).
Первый отличается твердостью, придает хорошую устойчи-
вость к вспузыриванию, второй способствует сохранению ста-
бильности грунтовки в окрасочной ванне. Объемная концентра-
ция пигмента (ОКП) обычно составляет 20—30%.
Технологический процесс. Этот процесс применяется в очень
ограниченных масштабах и в тех случаях, когда определяющим
фактором является малый объем продукции или другие эконо-
мические показатели.
Погружение корпуса автомобиля в ванну с краской может
быть неполным («частичное окунание»); в этом случае корпус
погружается лишь до окон. При частичном окунании за счет
неполного погружения и малой площади окрашиваемых участ-
ков удается избежать подтеков и наплывов. После стекания
избытка краски и кратковременной выдержки на воздухе, сле-
дующий слой может наноситься «мокрым по мокрому», после
чего система покрытия подвергается воздушной сушке.
При полном окунании корпуса автомобиля сложной формы
могут образовываться подтеки и наплывы, которые перед нане-
сением следующего слоя должны быть удалены растворителем.
Иногда исправление дефектов производится перед нанесением
следующего слоя краски на высохшем слое грунтовки.
Интересный способ нанесения грунтовки окунанием был пред-
ложен в середине 60-х гг. и получил название «ротационное оку-
нание».
При этом способе окрашиваемый корпус закрепляется на
поперечной оси специального конвейера, при вращении которого
последовательно осуществляются различные стадии окраски:
предварительная обработка (окунание в раствор фосфата цин-
ка), грунтование и затем сушка. Преимуществом этого метода
является совмещение операции предварительной обработки кор-
пуса с равномерным нанесением слоя грунтовки по всей поверх-
ности. Однако такая установка оказалась весьма дорогой и
сложной в эксплуатации, а скорость выполнения операций была
относительно низкой. Это обстоятельство, а также многие дру-
гие факторы привели к замене метода окунания на метод электро-
осаждения грунтовочных материалов.
В целом метод окунания весьма грязен, трудоемок и непро-
изводителен. Метод электроосаждения имеет много преимуществ
(см. 9.3.3) благодаря тому, что он обеспечивает лучшую защиту
внутренних поверхностей и позволяет возвращать растворитель
Л окрасочный цикл при использовании грунтовок как на водной
основе, так и на органических растворителях.
I 9.3.3. Электроокраска
'Метод электроокраски был разработан в 1930-х гг., однако
электроокраска автомобилей получила распространение начиная
с 1963 г. после того, как основные стадии процесса были отра-
ботаны в других отраслях промышленности. Метод электро-
окраски заключается в осаждении под действием электрического
тока на окрашиваемую поверхность водорастворимого пленко-
образователя, являющегося полиэлектролитом.
В настоящее время при массовом производстве практически
все выпускаемые автомобили грунтуют этим способом. При этом
корпус автомобиля погружают в соответствующую водную
краску, которая представляет собой композицию с очень низкой
вязкостью и плотностью, способную осаждаться при действии
тока на аноде или на катоде.
9.3.3.1. Анодная электроокраска
Анодная электроокраска применялась в автомобильной про-
мышленности до 1977 г. Технология получения необходимых
связующих была относительно простой, легко доступной, а сами
связующие отвечали требованиям автомобильной промышлен-
ности.
Типы связующих. Имеется четыре основных типа связующих:
а) малеинизированные масла, б) алкидно-фенольные, в) эфиры
многоатомных спиртов (эпоксиэфиры), г) малеинизированный
полибутадиен.
Первоначально лакокрасочные материалы для анодной элек-
троокраски базировались на связующих групп «а» и «б». Однако,
в дальнейшем они были заменены материалами на основе свя-
зующих групп «в» и «г», так как в этих случаях повышаются
противокоррозионные свойства покрытий.
Пигментная часть. Пигменты, применяемые в лакокрасочных
материалах для анодной и катодной электроокраски, должны
иметь следующие характеристики:
а) простое строение;
б) стабильность (например при сушке); чистота, отсутствие
водорастворимых примесей (например хлоридов и сульфатов);
в) во всех случаях придавать или улучшать коррозионную
стойкость. •
Объемная концентрация пигмента обычно составляет око-
ло 6%.
Основные пигменты — диоксид титана рутильной формы выс-
шего качества в смеси с минимальным количеством сажи для
материалов серого цвета, а также красный железоокисный пиг-
мент высшего качества для материалов красного цвета.
Все наполнители должны быть высшего качества, не содер-
жать растворимых ионизируемых примесей, таких как, напри-
мер, хлориды или сульфаты. В качестве антикоррозионных при-
годно значительное количество пигментов; типичным примером
является силикохромат свинца.
Механизм осаждения. Под действием тока в системе происхо-
дят три основные реакции: электролиз, электрофорез и электро-
эндоосмос.
Схема перевода связующего в растворимую форму:
RCOOH +КОН------* RCOOK +Н2О,
нерастворима растворима
например, R — эпоксиэфир
(В качестве основания в данном примере использован гидроксид
калия, однако можно применять также амины).
I. Электролиз
а) диссоциация связующего:
RCOOK RCOO- + K+,
б) электролиз воды:
4Н2О -f==t ЗН+ + ЗОН~ + Н2О.
Восстановление на катоде:
Н.2О + ЗН++4е->- 2Н24-ОН~.
Накапливающиеся гидроксильные группы ОН“ могут быть
удалены электродиализом.
Окисление на аноде:
ЗОН - 4ё---Н2О4-О2 + Н+.
Происходит подкисление в результате выделения Н+, одновре-
менно протекает реакция:
RCOO +Н+-----> RCOOH.
нерастворимое осажденное покрытие,
(образующееся в результате электро-
коагуляции) .
Растворение металла (побочная реакция происходит при на-
личии загрязняющих примесей):
' М-лё —->- М" +
II. Электрофорез. При подаче напряжения на элек-
троды, погруженные в водную дисперсию, заряженные частицы
начинают перемещаться к соответствующим электродам. Это
явление называется электрофорезом.
При электроокраске происходит движение отрицательно за-
яженых ионизированных частиц краски в направлении анода
(Положительного электрода). Электрофорез определяет меха-
низм движения заряженных частиц, но он не обусловливает
происходящее осаждение краски на аноде.
। III- Электроэндоосмос. Слой электроосажденной на
аноде краски содержит окклюдированную воду. Эта вода уда-
ляется из покрытия путем проникновения через пористую мем-
брану, образуемую красочной пленкой, в результате процесса,
называемого «электроэндоосмос».
Практическая реализация процесса. На практике основной
стадией процесса является подача напряжения между рабочей
поверхностью (анодом) и прогивоэлектродом (катодом). В ре-
зультате на рабочей поверхности осаждается нерастворимое
в воде покрытие, которое не может быть также растворено
повторно.
При этом образуется практически сухая, плотная пленка,
имеющая очень высокую твердость и хорошую адгезию к пред-
варительно обработанной (раствором фосфата цинка) металли-
ческой подложке. Пленка покрыта очень тонким слоем жидкой
краски, которая легко удаляется при промывке водой. В про-
цессе сушки (165 °C) происходит сшивка связующего и полу-
чается жесткая, прочная полимерная пленка.
Вес осажденной краски существенно зависит от количества
электричества, пропускаемого через раствор. По мере осажде-
ния пленки возрастает ее электрическое сопротивление и пленка
поляризуется, в результате значительно возрастает сопротивле-
ние и процесс осаждения прекращается. Таким образом, при
осаждении краски на каком-то участке поверхности возрастает
местное сопротивление этого участка и электроосаждение начи-
нается на соседних или более отдаленных участках. Этот эффект
обеспечивает сплошное укрывание всех участков окрашиваемой
поверхности.
В результате получается исключительно равномерная по тол-
щине пленка без подтеков, наплывов и других обычных дефектов.
Процесс очень эффективен в условиях массового производ-
ства, т. к. он позволяет без больших трудностей нанести грун-
товку на все поверхности сварной конструкции корпуса авто-
мобиля. Этот процесс может быть полностью автоматизирован
и характеризуется высоким процентом использования лакокра-
сочных материалов.
Основные требования к установке:
а) Установка должна иметь мощную циркуляционную си-
стему, обеспечивающую интенсивное перемешивание материала
и предотвращающую оседание пигмента, т. к. краска имеет низ-
кий сухой остаток (12%).
б) Требуется хорошее охлаждение и тщательная фильтрация
краски с целью обеспечения ее стабильности и удаления при-
месей.
в) Установка должна обеспечивать равномерное, плавное
перемещение деталей, подлежащих окраске.
г) Должно быть предусмотрено промывочное устройство для
удаления поверхностного слоя жидкой краски, а также устрой-
ство для ультрафильтрации (см. 9.3.3.2) для очистки и после-
дующей регенерации воды.
Регулирование pH, Накопление щелочи, образующейся па
катоде (смотри механизм электроосаждения, описанный выше)
при электролизе в процессе электроосаждения, повышает pH
окрасочной ванны и поэтому для сохранения исходных свойств
краски избыток щелочи должен быть удален. Это можно сделать
двумя путями: электродиализом и дробным подкислением.
При электродиализе вокруг катода устанавливается селек-
тивная мембрана Хионный обмен}. Ионы основания могут сво-
бодно проходить через мембрану, в то время как анионы (кол-
лоидные частицы краски) задерживаются ею.
Метод дробного повышения кислотности (снижение щелоч-
ности) вполне понятен- При проведении процесса электроосаж-
дения добавляются небольшие порции кислоты, которые нейтра-
лизуют образующуюся щелочь.
Выбор метода регулирования pH зависит от природы связую-
щего, способа нейтрализации и типа самой установки. Наиболее
предпочтительным методом является электродиализ, так как он
может быть полностью автоматизирован и обеспечивает непре-
рывную подачу материала в соответствующую емкость. Кроме
того, ионообменная мембрана может использоваться в течение
длительного времени, если, конечно, она не будет повреждена
механически.
Недостатки метода анодного электроосаждения грунтовок.
а) Разложение фосфатов. При анодном электро-
осаждении возникают очень сильные электрические поля, кото-
рые разрушают связи металла с ионами фосфатов (разложение
фосфатов). Это приводит к ухудшению адгезии фосфатного
покрытия к стальной подложке. При нанесении последующих
слоев покрытия по всей толщине пленки будут развиваться
внутренние напряжения, которые могут привести к местному
отслоению покрытия до металла и разрушению покрытия вокруг
места его повреждения.
Первоначально при нанесении грунтовок методом анодного
осаждения часто наблюдались случаи, когда при небольшом
местном нарушении целостности фосфатного слоя происходило
его полное разрушение. Это приводило к отслаиванию покры-
тия до чистого металла и развитию интенсивного коррозионного
процесса («подпленочной коррозии»).
। Недостаток, связанный с разрушением фосфатного покрытия,
был устранен путем повышения интенсивности его взаимодей-
ствия с подложкой, т. е. за счет увеличения плотности упаковки
тонких кристаллов фосфатов. При использований таких фосфат-
ных покрытий при небольшом весе (1,8 г/м2) удалось снизить
Опасность возникновения «подпленочнбй коррозии».
шб) Низкая стойкост'ь к омылен и.ю. Химизм анод-
ной электроокраски основан на том, что для формирования по-
крытий применяются пленкообразующие в кислотной форме.
Поэтому, если осажденную сухую пленку поместить в щелочную
среду, то будут образовываться ' водорастворимые полимер-
ные мыла.
При разрушении покрытия до металла щелочь одновременно
воздействует и на стальную подложку 'и на плёнку осажденного
покрытия, в результате чего начинается коррозия металла и
одновременно образуется натриевая соль ‘(мыло) полимерного
связующего. Растворение' грунтовочного слоя приводит к ухуд-
шению адгезии всей системы покрытия и еще в большей Степени
осложняет коррозионные проблемы.
loot,
• ;!1 '. I. ..
;;г
9.3.3.2. Нанесение грунтовки методом катодного осаждения
(Катодная электроокраска)
Катодная электроокраска теоретически всегда считалась наи-
более предпочтительным методом окраски по следующим причи-
нам:' 1) покрытия не подвержены воздействиям, отрывающим
их от подложки; 2) смолы в катионной форме дают щелочную
реакцию, т. е. по своей природе являются ингибиторами корро-
з‘йи; кроме того они устойчивы к омылению. '
Однако сложности, возникающие при выборе полимерной
сист'емк, препятствовали быстрому внедрению этого метода.
В начале 1970 г. в автомобильной промышленности Северной
Америки наблюдалась' вспышка коррозионного разрушения ме-
таллов, особенно стальных корпусов. ПоследЬвавшиё' за этим
законодательные акты предусматривали ужесточение требова-
ний по борьбе с коррозией, что послужило импульсом для раз-
работки метода катодной электроокраски, который был внедрен
вЬ всей автомобильной промышленности Северной Америки
в 1978 г.
Пленкообразующая основа. Все современные материалы для
катодной электроокраски базируются на' связующих эпоксид-
ного типа, отверждаемых аминами, которые стабилизируются
в водной среде нейтрализацией различными кислотами. Для
достижения оптимальных защитных свойств в' краски необхо-
димо вводить отвердители, а формирование покрытий проводить
пРи повышенной температуре (165—180°C).
Пигментная часть. Основные требования к пигментам такие
же, как и в системах для анодной электроокраски: простота
строения, высокая степень чистоты, способность увеличивать
коррозионную стойкость и ускорять процесс отверждения.
Объемная концентрация пигмента обычно составляет при-
близительно 10%. Наиболее распространенные цвета — от се-
рого до черного, причем основные пигменты и наполнители прак-
тически не отличаются от аналогичных материалов, применяе-
мых в композициях для анодной окраски.
Механизм осаждения. В основе механизма осаждения лежат
те же законы электрохимии, которые использовались при анод-
ном осаждении. Основные различия можно проиллюстрировать
следующими схемами:
Переход полимера в раствор:
R R
R—N; 4-R'COOH R—N—H+ + R'COO-
"t, R R
растворяющая
кислота
-jJ’
а) Диссоциация воды:
4H2O ч=ь 3H+-R3OH~ 4-H2O
б) Восстановление на катоде:
Н2О + ЗН+4-4ё--+ 2Н2 + ОН~
R R
R— N—Н++ОН- > R—N: +Н2О
I I
R R
нерастворимое осажденное
покрытие
в) Окисление на аноде:
ЗОН~ —4е----> Н2О+О2 + Н +
Н + удаляется при электродиализе
R'COO-+H+'----->- R'COOH
Эксплуатационные характеристики. В Северной Америке тех-
нология катодной электроокраски быстро вытеснила технологию
анодной электроокраски. Экспериментально было установлено,
что катодная электроокраска обеспечивает получение покрытий
с более высокими противокоррозионными свойствами и другими
преимуществами, причем необходимый комплекс свойств обеспе-
чивается даже при нанесении тонких пленок примерно 12 мкм
непосредственно по металлу без его предварительной обработки.
Это очень существенно для изделии, имеющих короочатое сече-
,.мр так как в этом случае покрытия должны обладать высокой
НИС» 1
прочностью.
V Автомобильная промышленность Японии также быстро пере-
строилась, за ней последовала Западная Европа, несмотря на
тб что в это время в Европе технология анодной электроокраски
достигла вполне удовлетворительных результатов. Установки
катодной электроокраски теперь можно встретить в любой части
света, например на Тайване, в Малайзии, Индонезии, на Фи-
липпинах и т. д„ причем все установки обеспечивают высокое
качество окраски; соответствующее наивысшим стандартам.
На рис. 9.4 представлена диаграмма, характеризующая уро-
вень совершенствования противокоррозионной защиты металла
с помощью покрытий, полученных катодной или анодной электро-
окраской. Противокоррозионные свойства определялись путем
испытания покрытия в камере солевого тумана по методу ASTM.
Требования к установке. Установка для катодной электро-
окраски (рис. 9.5) в основном аналогична установке для анод-
ной электроокраски в части систем, обеспечивающих циркуля-
цию, охлаждение и фильтрацию краски и равномерную и ста-
бильную работу источника тока. Основное различие состоит
в том, что в данном случае необходимо использовать материалы,
стойкие к кислотам, либо защищать поверхности из конструк-
ционной стали хорошими кислотостойкими покрытиями.
Наиболее подходящим материалом является нержавеющая
сталь, из которой изготавливаются насосы, вентили и трубопро-
воды. Однако высокая стоимость нержавеющей стали часто яв-
ляется препятствием для ее широкого использования, и там, где
Рис. 9.4. Усовершенствование метода противокоррозионной защиты (Сравнение
покрытий, полученных методами катодного и анодного осаждения, дается при
одинаковой толщине пленки (18—22 мкм), нанесенной по слою фосфата цинка)
Промывная вода
Конвейер
Ванна для промывки
Ванна для промывки
Слив
Ванна для
электроосаждения
(футер ованная)
Подача краски
Возврат краски
Тепло
обменник
—| Фильтр j
Ультрафильтрация ультрафильтрат
Ванна для электро-
лита (анолита)
Промывная вода
Слив
Слив
Сборник
Rmcs. 9.5. Участок жатодной электроокраски
возможно, применяют различные заменители, например поли-
винилхлорид или другие полимерные материалы.
Ванны для электроокраски и камеры для промывки также
должны быть изолированы кислотостойким материалом, хотя
камеры для промывки чаще выполняются из нержавеющей стали.
Промывка окунанием. Лакокрасочные материалы для катод-
ной электроокраски имеют относительно высокий сухой остаток
(20—22%) и поэтому наиболее эффективным методом про-
мывки в этом случае является метод окунания.
При подъеме изделия из камеры для промывки лакокрасоч-
ный материал стекает с поверхности более полно, чем при обыч-
ной промывке методом распыления. Понятно, что чем более
эффективна промывка, тем более экономичен процесс.
Для эффективного удаления поверхностного слоя краски
с окрашенного изделия в установке для промывки окунанием
должна быть обеспечена турбулизация промывочного раствора.
Ультрафильтрация. Ультрафильтрация представляет собой
процесс прохождения материала через мембрану с очень малыми
порами под действием давления. В случае электроокраски под
ультрафильтрацией следует понимать процесс разделения дис-
персионной среды (вода, растворитель и водорастворимые соли)
и дисперсной фазы (смолы и пигменты).
Необходимая скорость движения краски составляет при-
мерно 4—4,5 м/с или 150, л/мин. Для предотвращения осажде-
ния пигмента в трубопроводах, что может привести к наруше-
нию нормального потока краски, эту скорость необходимо под-
держивать на требуемом уровне.
Назначение ультрафильтрации:
а) Экономия краски. Это является наиболее важным вопро-
сом. При повторном использовании фильтрата после промывки
окрашенного изделия и снятия поверхностного слоя эффектив-
ность использования краски может возрасти на 15—20%.
б) Очистка. Растворимые соли, которые попадают в камеру
промывки с окрашенных изделий и которые ухудшают внешний
вид и эксплуатационные свойства покрытий, могут быть уда-
лены посредством ультрафильтрации.
в) Сохранение качества краски. Вообще при катодной элек-
трбокраске очистка является обязательным процессом, обеспе-
чивающим поддержание свойств краски в некоторых определен-
ных пределах. Это необходимо для того, чтобы избежать ухуд-
шения эксплуатационных характеристик покрытия и обеспечить
качественное нанесение краски.
г) Обезвоживание. В тех случаях, когда сухой остаток краски
в ванне для окраски становится слишком низким, а свободный
объем ванны достаточно велик, сухой остаток может быть уве-
ичен простым сливом ультрафильтрата и заменой его на све-
жую краску.
Методы регулирования pH. Регулирование pH осущест-
вляется с помощью установки электродиализа. Вокруг анода
устанавливается селективная ионообменная мембрана, которая
пропускает ионы кислоты, образующиеся в процессе электро-
лиза. Контроль производится автоматически с помощью анали-
тической аналоговой системы. Количество подаваемой из источ-
ника неионизированной воды возвратной жидкости контроли-
руется с помощью счетчика проводимости.
Предварительная обработка поверхности. Получение фос-
фатной пленки методом распыления, которое осуществляется
при анодной электроокраске, непригодно в случае катодной
электроокраски, так как приводит к снижению адгезии во влаж-
ных условиях этого метода. Причиной ухудшения адгезии, как
было установлено, является отслаивание вторичного слоя фос-
фата цинка; (химическое название «гопеит», Zn:i(РО4)ИКС)).
Этот вторичный фосфат называют словом «трава», из-за его
внешнего вида, а также из-за того, что он «растет» на поверх-
ности фосфата цинка (химическое название «фосфофиллит»,
Zn2Fe(PO4)2-4H2O). При анодной электроокраске эта «трава»
в значительной мере отщепляется благодаря разрушению под-
ложки, при катодном осаждении она практически сохраняется.
Высокие когезионные силы в эпоксидной пленке, получен-
ной методом катодной электроокраски, приводят к увеличению
напряжения в слабом слое вторичных фосфатов при набухании
в результате поглощения воды. В результате на границе раздела
слоев появляются «сколы», что само по себе свидетельствует
о снижении адгезии.
Для решения этой проблемы были разработаны методы по-
лучения гомогенных фосфатных покрытий, наполненных желе-
зом, с высокоразвитой тонкой кристаллической структурой и
минимальной толщиной слоя вторичного фосфата цинка. Для
большей гарантии можно обработать полученный фосфатный
слой слабым раствором какого-либо хромата. Получаемый при
этом эффект называют «эффектом закрепления», в результате
кристаллы скрепляются и повышается гомогенность поверхности.
В Европе и Северной Америке вторичная обработка фос-
фатного слоя считается ведущим направлением в области улуч-
шения качества и эксплуатационных характеристик покрытий.
Однако из-за различных ограничений этот метод не нашел при-
менения в Японии.
В этой стране более широкое распространение получил метод
фосфатирования путем окунания изделий в ванну, в которой
обеспечивается равномерное перемешивание раствора для фос-
фатирования, вследствие чего достигается более высокая сте-
пень однородности фосфатного покрытия и образуются в основ-
ном первичные фосфаты.
Общая оценка состояния проблемы и пути её усовершен-
ствования. Основным преимуществом метода катодной электро-
окраски является усиление противокоррозионных свойств
покрытий. Степень улучшения этих свойств весьма значительна,
причем в сочетании с использованием парафиновых смазок,
которые часто применяют для защиты изделий, имеющих короб-
чатое сечение, можно с гарантией обеспечить шестилетний срок
противокоррозионной защиты и отсутствие каких-либо серьез-
ных повреждений на защищенных поверхностях. Технологиче-
ский процесс и качество обработки отличаются стабильностью
и обеспечивают высокие эксплуатационные характеристики
защищаемых изделий, что очень существенно при массовом
производстве.
На рынках появились и некоторые новые разработки. К ним
относится, например, толстослойное покрытие (толщина около
35 мкм), которое сводит до минимума операцию грунтования
и имеет низкую температуру отверждения. Применение этого
материала не дает преимуществ с точки зрения снижения энер-
гозатрат, однако он может применяться в тех случаях, когда
на корпусе автомобиля имеются детали из пластмассы.
9.4. СИСТЕМА ПОКРЫТИЯ, СОДЕРЖАЩАЯ ГРУНТОВКУ,
ШПАТЛЕВКУ И ПРОМЕЖУТОЧНЫЙ СЛОИ
При окраске собранного корпуса автомобиля независимо от
того, наносится защитное или отделочное покрытие, обязатель-
ной операцией является шпатлевание. Назначение этой опера-
ции состоит в устранении любых даже незначительных дефектов
поверхности и следов, возникающих при прессовании и сборке.
При фосфатировании не происходит устранения указанных
дефектов, поскольку широко применяемые составы# наносимые
окунанием или электроосаждением, вследствие их малой кон-
центрации не обеспечивают получение таких же эффектов, как
при нанесении шпатлевки. Более того, природа процесса элек-
троосаждения такова, что она еще в большей мере выявляет
дефекты металла.
Кроме способности к устранению всех дефектов шпатлевка
должна обладать и некоторыми другими свойствами. Важными
свойствами являются механические показатели, эластичность,
влагостойкость и легкость обработки при шлифовании, так как
это необходимо для отделки поверхности перед окончательной
окраской.
На разработку новых материалов для шпатлевки в какой-то
степени оказало влияние изменение технологии сборки и приме-
нение новой технологии окраски, например электроокраски. Пер-
воначально разрабатывались рецептуры шпатлевок, которые на-
носились толстым слоем (40—50 мкм), имели высокую пористость
и требовали значительных усилий для получения гладкой поверх-
ности при шлифовании. По этой причине часто по слою такой
шпатлевки наносят промежуточный слой (с низким содержанием
пигментов) и лишь затем наносят отделочные покрытия. Кроме
того шпатлевки, наносимые по слою грунтовки, полученной оку-
нанием или распылением, а также по предварительно обработан-
ному металлу, должны обладать хорошей адгезией к указанным
поверхностям.
Первоначально в качестве связующего в шпатлевках’ исполь-
зовались алкидные смолы, которые затем были заменены сначала
эпоксиэфирами, а затем более современными полиэфирными
смолами. Основу ассортимента составляли шпатлевки на раст-
ворителях Однако материалы на водной основе теперь привле-
кают все большее внимание благодаря хорошему розливу и
легкости шлифования.
Материалы на эпоксидной основе наиболее широко приме-
няются при анодной электроокраске благодаря их прекрасной
адгезии й водостойкости.- Эти материалы использовали также
на разных стадиях разработки метода катодной электроокраски,
однако практически не была разработана технология, обеспе-
чивающая качественное выполнение очень трудоемкой операции
шлифования. За последние годы в связи с развитием техноло-
гии, обеспечивающей получение покрытий с металлическим эф-
фектом, стало возможным применение эпоксидных материалов
в качестве основы для шпатлевок.
Низкая устойчивость алкидных шпатлевок к действию УФ-
облучения (наблюдается меление на границе раздела покровного
слоя и шпатлевки) приводит к отслаиванию отделочного покры-
тия, обладающего металлическим эффектом. Для устранения
этого недостатка были разработаны полиэфирные шпатлевки,
обладающие более высокой устойчивостью к действию УФ лучей.
Полиэфирные шпатлевки в настоящее время считаются луч-
шими по всем показателям для катодной электроокраски. Эти
материалы отвечают основным требованиям по химическим и
физическим свойствам и могут применяться либо вообще без
шлифования, либо для придания необходимого внешнего вида
эта операция проводится в минимальном объеме.
В настоящее время имеются предпосылки для разработки
шпатлевок, не требующих шлифования и обеспечивающих хоро-
шую адгезию покрывных слоев.
9.4.1. Состав связующего
В качестве связующего используют растворы алкидов, эпо-
ксиэфиров или полиэфиров, которые реагируют с соответствую-
щей меламино- или мочевиноформальдегидной смолой.
Покрытия выдерживают в течение 20 мин при 150—165 °C
(наиболее эффективно) для получения сшитой нерастворимой
пленки.
В алкидных и эпоксиэфирных материалах механизм отвержде-
ния часто является смешанным и представляет собой сочетание
самоокисления и термосшивки. Эфиры обычно являются произ-
водными льняного, таллового или дегидратированного касторо-
вого масел. Они используются для придания пленке необходи-
мой эластичности, твердости и нерастворимости при определен-
ных условиях сушки.
Не содержащие масла полиэфирные материалы являются тер-
мореактивными и отверждаются только под действием темпе-
ратуры. Однако необходимая эластичность может быть обеспе-
чена введением в структуру полимера длинноцепных синтети-
ческих жирных кислот.
9.4.2. Пигментирование
Первые шпатлевки представляли собой высокопигментиро-
ванные системы с объемной концентрацией пигмента (ОКП)
30—35% в зависимости от физических и химических показате-
лей и характеристик верхнего покрытия. В состав пигментной
части обычно входят основной пигмент (для придания укры-
вистости и цвета) и наполнители (для экономичности, если позво-
ляют эксплуатационные требования).
Обычно шпатлевку наносят распылением в два слоя «мокрым
,по мокрому» по слою грунтовки, нанесенной окунанием, либо
по фосфатированному металлу (толщина пленки примерно
50 мкм). В шпатлевках для первого слоя применяется красный
железоокисный пигмент, а для второго слоя — обычно серый пиг-
мент. В зависимости от качества второго слоя определяется
необходимость шлифования. При внедрении метода электро-
окраски применение материалов на основе железоокисных пиг-
ментов уменьшилось и в настоящее время используют в основном
шпатлевки с обычной толщиной пленки 30—35 мкм.
Основные пигменты: красный железоокисный пигмент
или высококачественный диоксид титана рутильной формы (ана-
таз имеет низкую стойкость к УФ-лучам, что препятствует его
применению).
Пигменты-наполнители значительно дешевле, чем
основные пигменты, однако при их выборе стремятся не только
не ухудшить свойства исходного материала или готового покры-
тия, но и улучшить их показатели. При правильном подборе
наполнителей могут быть улучшены такие свойства, как кон-
систенция, розлив, стойкость к оседанию пигмента в краске.
Одни наполнители могут улучшить механические свойства су-
хого покрытия, другие повысить водостойкость и препятство-
вать о разованию вздутий. В качестве наполнителей наиболее
широко применяются следующие материалы:
Бариты (сульфат бария): основной наполнитель, наиболее
пригоден для повышения устойчивости к образованию вздутий.
Тяжелый, ухудшает полируемость покрытий.
Каолин (силикат алюминия): обеспечивает хорошую поли-
руемость. Обладает высокой маслоемкостью, что приводит к сни-
жению блеска.
Виннофил (карбонат кальция, покрытый стеаратной обо-
лочкой) : структурирующая добавка, препятствующая оседанию
пигмента.
Тальк (силикат магния): раньше тальк обладал значи-
тельной растворимостью в воде; в настоящее время этот пока-
затель существенно улучшен. Хороший наполнитель для улучше-
ния полируемое™ и для снижения стоимости.
Объемная концентрация пигмента может изме-
няться в пределах 30—55% в зависимости от типа отделочного
покрытйя (например-под акриловые~лаки необходимо использо-
вать шпатлевки с ОКП 55%) и от технических требований,
например механических свойств, внешнего вида и легкости поли-
рования. ,
9.5. УСОВЕРШЕНСТВОВАННЫЙ ПРОЦЕСС
После освоения метода анодной электроокраски подготовку
поверхностей корпусов автомобилей стали производить по опре-
деленной схеме, включающей предварительную обработку ме-
талла, нанесение грунтовки методом электроосаждения, шпатле-
вание или нанесение промежуточного слоя. Позднее был разра-
ботан усовершенствованный процесс, целью которого являлось
устранение серьезных недостатков анодной электроокраски и
улучшение технологичности отдельных этапов окраски.
Основные стадии усовершенствованного процесса: 1) предва-
рительная обработка (нанесение фосфата цинка методом рас-
пыления); 2) нанесение шпатлевки методом распыления на листы
для наружной обшивки корпуса автомобиля (полная или ча-
стичная сушка); 3) грунтование методом электроокраски
(стандартный процесс); 4) окончательная сушка.
Основные преимущества этого процесса заключаются в сле-
дующем:
1. Большая толщина пленки и, как следствие, усиление про-
тивокоррозионной защиты внутренних поверхностей корпуса
(благодаря повышению изолирующих свойств покрытия и на
внутренних и на наружных поверхностях).
2. Значительное повышение стойкости к появлению подпле-
ночной коррозии и образованию пузырей (не происходит раз-
рушения фосфатного покрытия на наружных поверхностях).
3. Более высокая адгезия и устойчивость системы покрытия
к повреждениям при ударе камнями после полного завершения
окраски — процесс позволяет использовать такие рецептуры
шпатлевок, которые обеспечивают более высокую сохранность
фосфатного слоя по сравнению с грунтовками, наносимыми
методом электроокраски.
4. Более легкая окраска корпуса, выполненного из разно-
родных материалов, например из стали, цинка и алюминия. Эти
металлы имеют различные электрические параметры, что может
создавать определенные трудности при электроокраске.
Некоторые производители уже опробовали этот процесс,
используя шпатлевки на растворителях или порошковые по-
крытия.
Широкое применение этого метода сдерживается рядом фак-
торов:
1. Неудовлетворительный внешний вид и качество покрытия
на границе раздела шпатлевки и грунтовочного слоя, наносимого
методом электроокраски. Эта проблема, однако, может быть
решена при использовании шпатлевок на водной основе, имею-
щих соответствующие электрические характеристики и хорошо
совместимых с другими слоями покрытия.
2. Для модернизации установки требуются капитальные
затраты.
3. Разработка имеющего ряд преимуществ метода катодной
электроокраски и одновременное создание усовершенствованной
технологии предварительной обработки металла перед окраской
и методов контроля.
Порошковые покрытия, наносимые методом электроосажде-
ния (ЭПП). Электроосаждение порошковых покрытий является
еще одним примером усовершенствованной технологии, хотя этот
метод предусматривает нанесение грунтовки обычным методом
катодной электроокраски.
Появившийся в Японии метод получения ЭПП предусматри-
вает применение порошковой шпатлевки, которая в виде водной
суспензии путем электроосаждения наносится вторым слоем на
фосфатированный корпус автомобиля. ЭПП применяют в основ-
ном для наружных поверхностей. После подъема корпуса авто-
мобиля из ванны и сушки покрытия производят катодную элек-
троокраску.
Методом катодной электроокраски невозможно нанести по-
крытие на поверхности, изолированные ЭПП. Наилучшие резуль-
таты могут быть получены при окраске поверхностей пола,
деталей коробчатого сечения и т. д. Для этих целей выбирают
рецептуры, отвечающие предъявляемым требованиям.
Этот процесс очень легко поддается автоматизации благо-
даря соответствующей консистенции материала; из технологиче-
ской схемы исключена распылительная камера, материалы не
содержат органических растворителей. В целом метод Э1111
в современном виде хотя и обеспечивает получение шпатлевоч-
ных покрытий с различной толщиной пленки, но не позволяет
полностью скрыть дефекты или текстуру поверхности, поэтому
перед нанесением покрывных слоев покрытия требуется тща-
тельное шлифование поверхностей.
9.6. НЕСКАЛЫВАЮЩИЕСЯ (ПРОТИВОУДАРНЫЕ) ПОКРЫТИЯ
Основное назначение применяемых в настоящее время про-
тивоударных покрытий состоит в повышении стойкости наиболее
уязвимых поверхностей корпуса автомобиля к разрушению при
ударах камнями.. В первую очередь это касается дверных поро-
гов', передней и задней частей корпуса ниже бампера, днища.
В зависимости от назначения автомобиля, эти покрытия могут
наноситься на различные уровни по высоте корпуса. Противо-
ударные ' покрытия являются толстослойными (50—100 мкм) и
наносятся^ огокръш по мокрому» по слоюсоответствующей
шпатлевки.
Связующее представляет собой одноупаковочную компози-
цию на основе блокированного ароматического изоцианата,
отверждаемого циклоалифатическим диамином.
Пленка высушивается вместе со слоем шпатлевки (например,
20 мин при 150 °C) и имеет резиноподобную структуру и хоро-
шие механические свойства. Помимо основных, пигментов в со-
став красок могут входить и наполнители, хотя от количества
наполнителей будут зависеть механические свойства покрытий.
В состав красок вводят также тиксотропные добавки, например
аэросил, которые препятствуют оседанию пигментов и позволяют
получать пленки большой толщины.
9.7. ОТДЕЛОЧНЫЕ ПОКРЫТИЯ ДЛЯ АВТОМОБИЛЕЙ
Различные требования и ограничения, выдвигаемые промыш-
ленностью и отдельными покупателями, обусловили многообра-
зие видов отделочных покрытий для автомобилей. Все виды
покрытий могут отличаться комплексом свойств; качество их
определяется особенностями технологии и контролируется с по-
мощью- различных методов испытаний.
• Основным требованием, предъявляемым к покрытиям, яв-
ляется высокая долговечность, т. е. очень хорошая стабильность
цвета и устойчивость к разрушению под действием УФ-облуче-
ния. Эти требования в сочетании с возможностью организации
массового производства позволяют отнести автомобильные по-
крытия к разряду особых материалов.
Существуют два основных вида отделочных покрытий: покры-
тия с насыщенным (или неразбавленным) цветом и покрытия
с металлическим оттенком. Эти покрытия могут быть разделены
на следующие группы:
1. Алкидные или полиэфирные покрытия с насыщенным
цветом.
2. Термоотверждаемые акрилатные покрытия на основе не-
водных дисперсий (НВД) с насыщенным цветом и с металличе-
ским оттенком.
3. Термопластичные акрилатные покрытия с насыщенным
цветом и с металлическим оттенком.
4. Покрытия с металлическим оттенком, состоящие из основ-
ного и прозрачного слоев.
Для получения покрытий с насыщенным цветом во всем
мире наиболее широко используются алкидные материалы, так
как они имеют низкую стоимость и не вызывают затруднений
при производстве. Покрытия с металлическим оттенком обычно
изготавливаются на основе термореактивных акрилатных связую-
щих (или НВД), поскольку алкиды не обеспечивают необходи-
мую долговечность таких покрытий. Термопластичные акрилатные
лаки широко используются кампанией Дженерал Моторе как
для покрытий с насыщенным цветом, так и с металлическим от-
тенком.
Более поздняя технология, связанная с применением системы
покрытий с прозрачным слоем, очень выигрышна, так как покры-
тия обладают повышенным блеском, хорошим внешним видом
и прекрасной долговечностью. В качестве связующего для про-
зрачного слоя применяют термореактивные акрилатные смолы
с добавками соответствующих адсорбентов УФ-лучей.
9.7.1. Алкидные или полиэфирные покрытия
Химизм процесса. Алкидные покрытия изготавливаются на
основе смол, получаемых при взаимодействии многоатомных
спиртов (глицерин, гликоль и т. д.) и двухосновных кислот (фта-
левый ангидрид), модифицированных маслами или синтетиче-
скими жирными кислотами для обеспечения необходимой проч-
ности, эластичности, твердости и т. д. Масла или жирные кислоты
используются для обеспечения сохранности цвета и стойкости
к пожелтению (стойкости к окислению). Наиболее пригодными
являются жирные кислоты масла из кокосового ореха или
3,5,5-триметилгексановая кислота, которая одновременно играет
роль реактивного пластификатора. В типичных алкидах содер-
жание масла составляет примерно 35%.
Процесс сшивки, необходимый для образования нераство-
римой пленки, происходит при взаимодействии алкида (или
полиэфира) и меламиноформальдегидного конденсата, содержа-
щего N-метилольные группы или их эфиры:
Алкидная смола
СООН ОН
1~
Алкидная смола
СООН О
I
СН2' + ROH
NH
Меламиноформальдегидная смола
Меламиноформальдегидная смола
NHCHjOR
"“Л
Алкидная смола
СО ОН
I
О +ROH
СН2
Г
NH
Меламиноформальдегидная смола
(R - Н или алкил)
Для достижения необходимой степени отверждения сушку
проводят в течение 20 мин при температуре 130 °C.
Общая характеристика. Общие характеристики алкидов со-
стоят в следующем:. возможность широкого использования для
получения насыщенных цветов, относительно высокий сухой
остаток (~50%), низкая стоимость, легкость нанесения, высо-
кая долговечность, возможность использования в качестве под-
слоя материалов разной природы, склонность к грязеудержанию
(повышенная гидрофильность пленки), сравнительно плохая по-
лируемость.
Высокий сухой остаток, низкая стоимость и легкость нане-
сения (двухслойное нанесение «мокрым по мокрому» обеспечи-
вает толщину пленки 40—50 мкм) явились причиной широкого
распространения этих материалов. Благодаря указанным свой-
ствам покрытия хорошо укрывают дефекты поверхности и имеют
очень хороший внешний вид после сушки. По прочностным ха-
рактеристикам эти покрытия не уступают термореактивным акри-
латным покрытиям, что было установлено в результате двухлет-
них испытаний во Флориде (5° южной широты).
Однако, с другой стороны, они имеют повышенную склонность
к удержанию растворителя по сравнению с акрилатными мате-
риалами и поэтому пленка отличается повышенной загрязняе-
мостью. Следствием этого является необходимость частого мытья
окрашенных поверхностей; кроме того, недостаточно хорошая
шлифуемость затрудняет устранение дефектов поверхности в
условиях промышленного производства.
9 7 2- Материалы на основе неводных акрилатных дисперсии,,
отверждаемых при повышенной температуре
Химизм процесса. Термореактивные акрилатные композиции
изготавливают на основе различных акриловых сополимеров.
Для синтеза этих сополимеров в зависимости от предъявляемых
к композициям требований используются смеси разных моно-
меров. Сшивка осуществляется путем взаимодействия акрило-
вого сополимера с меламино-формальдегидной смолой, в струк-
туре которой содержатся свободные гидроксильные группы.
Полимеры, содержащие гидроксильные группы, могут быть по-
лучены также на основе гидроксилсодержащих акриловых моно-
меров. Отверждение при 130 °C в течение 20 мин приводит к
образованию нерастворимой в . воде сшитой полимерной пленки:
ROH
На основе таких полимеров были разработаны термореак-
тивные покрытия интенсивных цветов, а также покрытия с ме-
таллическим оттенком. Для получения высококачественных
покрытий с металлическим оттенком в состав красок вводится
невсплывающая алюминиевая пудра (в виде пасты). Чешуйки
невсплывающей алюминиевой пудры (длина чешуек 15—45 мкм)
распределяются в покрытии беспорядочно, что обусловливает
зеркальное отражение света при наблюдении с различных ра-
курсов. При введении в состав красок цветных пигментов можно
значительно расширить цветовую гамму получаемых покрытий.
Этот вид отделочных покрытий был разработан в конце
1950-х — начале 1960-х гг. и получил широкое распространение
во всем мире, однако и сегодня еще ведутся работы по модифи-
кации рассматриваемых покрытий. К недостаткам этого вида
покрытий можно отнести необходимость нанесения трех слоев
из-за низкого сухого остатка красок, а также сложности, свя-
занные с получением металлического оттенка. Разработка отде-
лочных покрытий на основе неводных дисперсий (НВД) в конце
1960 г. позволила решить эти проблемы.
В наиболее распространенных термореактивных акрилатных
композициях связующее используется в виде растворов, а в ма-
териалах на основе НВД те же полимеры применяются в виде
дисперсий в смеси органических растворителей.
Несмотря на то, что значительная часть полимера остается
нерастворенной, невысокая вязкость краски позволяет осущест-
вить ее нормальное нанесение.
Стабилизация дисперсий обеспечивается введением в состав
полимеров длинных алифатических цепей (т. е. применение
графт-сополимеров), которые химически прививаются к молеку-
лам основного полимера в процессе сополимеризации. Такие
дисперсии, которые можно назвать «сверхстабильными», зна-
чительно превосходят системы, в которых стабилизатор распре-
деляется на поверхности частиц в результате физического или
полярного взаимодействия.
Когда значительная часть алифатических углеводородных
цепей располагается в непрерывной фазе дисперсии, они запол-
няют весь свободный объем и создают стабилизирующую обо-
лочку вокруг каждой частицы. Для предотвращения возможной
коалесценции частиц в непрерывной фазе в состав НВД вво-
дится некоторое количество растворителя для самого полимера.
Количество растворителя^должно быть таким, чтобы стабили-
зирующее действие алифатических цепей не нарушилось при
хранении материала в таре или при перекачке по трубопрово-
дам в производственных условиях. Этот растворитель как бы
создает перегородку между полимером (вызывая размягчение
и набухание частиц полимера) и непрерывной фазой дисперсии.
После нанесения покрытия происходит испарение алифатического
углеводорода, являющегося нерастворителем для полимера, и
набухшие частицы полимера коалесцируют, образуя однородную
пленку. Завершение процесса формирования покрытия произ-
водится при горячей сушке. Структура полученной пленки в
основном аналогична структуре пленки, получаемой из раствора
соответствующего полимера.
Основной стадией технологического процесса получения
этих материалов является стадия приготовления дисперсий, в
которых практически весь полимер находится в дисперсной
фазе.
Реологические характеристики таких материалов не позво-
ляют широко их использовать на действующих промышленных
поточных линиях. Однако, определенные положительные резуль-
таты в этой области могут быть получены при переводе части
полимера в раствор. Поэтому ведутся довольно интенсивные
исследования, направленные на выбор оптимальных соотноше-
ний между диспергированным и растворенным полимером для
каждой конкретной полимерной системы.
Реологические характеристики таких термореактивных акри-
латных композиций своеобразны и от них зависят качество вос-
произведения металлического оттенка, полнота использования
сухого вещества краски и качественное нанесение покрытия без
подтеков и неровностей.
Общие характеристики. Для систем на основе НВД харак-
терны: высокая прочность покрытий насыщенных тонов и покры-
тий с металлическим оттенком (однослойное покрытие); более
низкий сухой остаток, чем у алкидных материалов (30%); необ-
ходимость нанесения в два или три слоя; хорошая полируемость;
полное удаление растворителя; необходимость применения высо-
кокачественного подслоя, например, эпоксиэфирных покрытий,
-г. Термореактивные акрилатные покрытия на основе НВД мо-
гут иметь как насыщенные цвета, так и металлический оттенок,
причем может быть получен большой ассортимент цветов. Для
этих материалов характерно быстрое, полное испарение раство-
рителя, что снижает загрязняемость покрытий, а легкая поли-
руемость позволяет быстро устранять местные дефекты, которые
могут- возникать при окраске на поточных линиях.
;"н В качестве нижнего слоя под эти отделочные покрытия необ-
ходимо применять материалы с высокими эксплуатационными
характеристиками, например эпоксиэфирные или полиэфирные.
Достоинством термореактивных акрилатов является возмож-
ность их применения непосредственно по грунтовке, нанесенной
методом электроосаждения, так как они имеют к этой грунтовке
высокую адгезию.
* Алкидные материалы из-за их плохой адгезии применять
в этих условиях нельзя. В связи с тем, что акрилатные мате-
риалы применяются всегда в виде двухслойной системы, состоя-
щей из грунтовки, наносимой методом электроосаждения, и
покрывного слоя, эти материалы нашли широкое применение
в-' автомобильной промышленности.
а Покрытия с металлическим оттенком. Основная особенность
покрытий с металлическим оттенком состоит в том, что они
производят определенный «зрительный эффект», т. е. создают
впечатление монохроматической поверхности независимо от угла
наблюдения. Этот простейший оптический эффект зависит от ори-
ентации металлических чешуек параллельно поверхности, вслед-
ствие чего количество отраженного от поверхности света будет
определяться углом наблюдения. Таким1 образом, при взгляде
на такую поверхность под любым углом будет создаваться впе-
чатление, что эта поверхность имеет интенсивную окраску. В дей-
ствительности алюминиевые чешуйки выполняют роль маленьких
плоских зеркал.
Для достижения оптимальной ориентации чешуек должны
соблюдаться следующие условия:
а) покрытия должны наноситься методом распыления для
достижения максимальной равномерности;
б) пленка покрытия должна обладать максимальной усадкой
после нанесения, поскольку в этом случае невсплывающие алю-
миниевые чешуйки способны сориентироваться параллельно
поверхности;
в) чешуйки должны обладать минимальной склонностью к
случайной переориентации после нанесения покрытия.
Обычные термореактивные композиции растворного типа в
значительной степени отвечают предъявляемым требованиям, од-
нако необходим правильный подбор смеси растворителей, вида
и вязкости связующего и соответствующей марки алюминиевой
пудры. Работа по нанесению покрытий должна выполняться
операторами высокой квалификации, так как довольно сложно
получить покрытие с равномерными цветовыми характери-
стиками.
Слишком высокая влажность приводит к тому, что алюминие-
вые чешуйки осаждаются неравномерно и на поверхности появ-
ляются более светлые и более темные участки. Темные полосы
(черная кайма) могут также образовываться вокруг отверстий
в корпусе или на сгибах. Для устранения этих дефектов при-
меняют низковязкие эмали, в результате чего получаются тонкие
покрытия, которые быстро высыхают и имеют хороший розлив
тг блеск.------------
Использование композиций дисперсионного типа в сочетании
с составами на основе растворителей позволяет решить многие
проблемы, поскольку открывается возможность для улучшения
реологических характеристик красок.
Как было отмечено выше, дисперсии полимера не оказывают
такого сильного влияния на вязкость покрытия, как раствор
того же самого полимера. Однако при переходе от дисперсии
к раствору после испарения из жидкой фазы осадителя наблю-
дается резкое увеличение вязкости. Если это происходит в мок-
рой пленке после ее нанесения на поверхность, то будет иметь
место более быстрое нарастание вязкости, чем это произошло бы
просто в результате испарения жидкости из емкости. Вследствие
этого резко снижается подвижность алюминиевых чешуек, огра-
ничивается возможность их переориентации и легче достигается
желаемый внешний вид покрытия.
Расслаивание. Те же процессы, которые меняют реологиче-
ские характеристики композиций при переходе от дисперсии к
раствору, будут оказывать влияние и на расслаивание покрытий
дисперсионного типа. В более толстых покрытиях дефекты,
связанные с расслаиванием, проявляются более интенсивно.
Улучшение стойкости к расслаиванию особенно важно для
высоко наполненных пигментированных систем, особенно для
белых и оранжевых цветов.
Устойчивость к «образованию кратеров». Образование кра-
теров (иногда это называется «вскипанием») объясняется быст-
рым удалением при горячей сушке содержащегося в пленке
избыточного количества воздуха, удерживаемого растворителем.
Обычно это происходит при чрезмерно большой толщине мокрой
пленки. Растворы термореактивных акрилатных материалов осо-
бенно склонны к этому, так как пленка их очень быстро «схваты-
вается» сразу после нанесения покрытия.
Можно было предположить, что для дисперсионных систем
возникнут те же самые проблемы, если состав полимерного
связующего будет один и тот же. Как показывает опыт, диспер-
сионные системы имеют определенные преимущества. Это, ве-
роятно, можно объяснить двумя причинами:
а) более тонкое измельчение дисперсии в процессе распы-
ления;
б) применение в композициях осадителя, не взаимодействую-
щего с полимером, в результате чего происходит быстрое и пол-
ное удаление растворителя в процессе сушки.
9.7.3. Термопластичные акрилатные лаки
Основы химических процессов. Высыхание акрилатных лаков,
также как и любых лаков вообще, происходит за счет испарения
растворителя. Основой акрилатных лаков чаще всего является
твердый пластифицированный полиметилметакрилат. В качестве
пластификаторов обычно применяют бутилбензилфталат, а также
полимерные фталаты линейной структуры, представляющие
собой производные жирных кислот кокосового масла. Поверх-
ностная пластификация приводит к улучшению ряда свойств:
повышается стойкость к растрескиванию, улучшается адгезия
к нижележащим слоям, повышается эластичность, снижается
склонность к удержанию растворителя.
В основном акрилатные полимеры, применяемые для данного
вида отделочных материалов, имеют среднюю молекулярную
массу приблизительно 90 тыс. Покрытия на основе этих полиме-
ров обладают превосходным блеском, длительно сохраняю-
щимся при эксплуатации в атмосферных условиях. Композиции
на основе полимеров со средней молекулярной массой свыше
105 тыс. и сухим остатком, соответствующим обычным промыш-
ленным материалам, при нанесении распылением склонны к обра-
зованию паутины или длинных нитей. Низкомолекулярные
полимеры образуют пленки с неудовлетворительными эксплуа-
тационными свойствами и низкой долговечностью. Кроме того,
сохранность блеска покрытия при повышении молекулярной
массы свыше 105 тыс. увеличивается очень незначительно, так
как при повышении молекулярной массы необходимо разбав-
лять материалы для обеспечения возможности его нанесения
методом распыления и, соответственно, снижать сухой остаток.
Смеси растворителей для лаков представляют собой сложные
композиции и относительно дороги, состав растворителей под-
бирается таким образом, чтобы обеспечить необходимую вяз-
кость, скорость испарения растворителя и хороший розлив.
Чтобы снизить до минимума содержание в пленке остаточного
растворителя, необходимо использовать растворители, не содер-
жащие высококипящих фракций, и строго контролировать тем-
пературу испарения растворителя, уходящего из пленки в послед-
нюю очередь. Применение пластификаторов способствует более
полному испарению растворителя, поскольку пленка сохраняется
в «жидком» состоянии более продолжительное время. Это позво-
ляет также снизить усадочные напряжения, возникающие при
высыхании или отверждении пленки (особенно в акрилатных
лаках).
Хотя акрилатные лаки обычно высыхают на воздухе, на прак-
тике процесс сушки может быть ускорен нагреванием в течение
30 мин при температуре 90 °C. Для достижения необходимого
блеска в этом случае требуется шлифование поверхности, кото-
рое производится по схеме: сушка — шлифование — сушка.
В этом случае не до конца высушенная пленка (15 мин при
82 °C) шлифуется и затем досушивается (20 мин при 154 °C),
в результате чего получается бездефектная. блестящая пленка.
Общие характеристики. Для термопластичных акрилатных
материалов характерны: очень хорошая долговечность, как в слу-
чае покрытий насыщенных цветов, так и покрытий с металли-
ческим оттенком; .стабильность и воспроизводимость процесса
производства; хорошая полируемость и ремонтоспособность;
ярко выраженный металлический эффект; низкий сухой остаток
1(15—20%) и связанная с этим необходимость нанесения мио-'
послойного покрытия; необходимость применения больших коли-
честв дорогих растворителей; высокая стоимость сырья; необхо-
димость нанесения- специального подслоя под покрытие.
Рекомендуется ступенчатый процесс обработки: сушка в тече-
ние 15 мин при 82 °C, шлифование и затем окончательная сушка
|в течение 20 мин при 154 °C (температура нагрева металла)?
Отделочные покрытия на основе акрилатных лаков широко
[используются во всем мире, в том числе фирмой Дженерал
Моторе, а также производителями престижных автомобилей
[типа «Ягуар». Высокое качество внешней отделки, особенно
в случае покрытий с металлическим оттенком, стабильные тех-
нологические характеристики (полируемость, легкость ремонта)
остигаются только при строгом контроле производственного
процесса. Покрытия с металлическим оттенком отличаются пре-
восходным внешним видом, для их получения используют мате-
риалы с низким сухим остатком и высокой вязкостью на основе
высокомолекулярных акриловых полимеров. Проявление метал-
лического эффекта обусловлено быстрым «схватыванием» пленки,
полным испарением растворителя и малой толщиной пленки,
эграничивающей геометрическую переориентацию и всплывание
рлюминиевых чешуек.
Акрилатные лаки имеют ряд недостатков, которые сводятся
[с следующему:
а) Низкий сухой остаток вызывает необходимость нанесения
четырехслойного покрытия для достижения требуемой толщины
пленки (55—60 мкм) и при этом сушку покрытия требуется
проводить по ступенчатому режиму. Нанесение многослойного
покрытия усложняет и удлиняет процесс, что сказывается на
его стоимости.
б) Внешний вид покрытия, высушенного для ускорения даже
при сравнительно низких температурах (80—90 °C), неудовлет-
ворительный, что связано с необходимостью дополнительного
полирования.
в) Высокая стоимость сырья, особенно растворителей.
г) Недостаточная межслойная адгезия, обусловленная видом
применяемого подслоя (обычно эпоксиэфирное покрытие с высо-
ким ОКП, равным 55%).
От качества подслоя во многом зависит внешний вид покрыв-
ного материала. Для повышения адгезии наносится специаль-
ный промежуточный слой, что еще больше увеличивает стои-
мость окраски.
9.7.4. Технология окраски с применением основного слоя
и прозрачного наружного слоя
Как было указано выше, при производстве автомобилей
в настоящее время в основном используются отделочные покры-
тия с металлическим оттенком. Таким покрытиям благодаря при-
влекательному внешнему виду, вероятно, и в дальнейшем обес-
печен большой спрос, т. к. и по цветовым характеристикам и по
свойствам они отвечают требованиям потребителей. Однослой-
ные отделочные покрытия с металлическим оттенком в том виде,
в каком они сейчас существуют, готовятся на основе термо-
пластичных или термореактивных акрилатных связующих.
Однако они имеют ряд недостатков по сравнению с покры-
тиями насыщенных цветов:
а) пониженный блеск, особенно для покрытий светлых тонов;
б) ограниченное применение некоторых видов пигментов,
например, органических;
в) пониженная устойчивость в кислых средах;
г) трудности нанесения (для улучшения этого показателя
целесообразно использовать покрытия на основе НВД термо-
реактивных акрилатов).
Несколько десятилетий назад был предложен способ введе-
ния алюминиевых чешуек в самостоятельный слой покрытия
(в основной слой), который перекрывается слоем прозрачного
связующего. Такая технология используется при нанесении мно-
гоцветных покрытий на велосипедные рамы.
Многие европейские производители автомобилей отдают
предпочтение этой технологии, поэтому с конца 1960-х гг. при
окраске широко применяется система с основным слоем, содер-
жащим алюминиевые чешуйки, и прозрачным наружным слоем.
Основной слой обеспечивает укрывистость и металлический
оттенок, а прозрачный слой придает блеск, чистоту тона и повы-
шенную долговечность. Такая технология получила широкое рас-
пространение и в настоящее время используется в Западной
Европе, Японии и интенсивно развивается в США.
Химизм процесса.
Свойства основного слоя: а) высокая укрывистость,
которая достигается при нанесении тонкой пленки (10 мкм);
б) быстрое удаление растворителя — продолжительность сушки
перед нанесением прозрачного покрытия составляет 2—3 мин;
в) максимальный металлический эффект достигается при низ-
ком сухом остатке (20%); г) «совместимость» с прозрачным
покрытием, ’г. е. хорошая адгезия и отсутствие стекания про-
зрачного покрытия.
В- качест-ве—связующего для основного слоя используется
термореактивное полимерное связующее, модифицированное дру-
гими полимерами типа ацетобутирата целлюлозы, вводимыми
в состав композиции для улучшения условия формирования
пленки и ускорения испарения растворителя. В качестве связую-
щего применяют либо не содержащий масел полиэфир, либо
термореактивный акрилатный полимер, активно взаимодействую-
щий с азотсодержащими смолами (например меламиновыми).
Характеристики этих двух типов связующих приведены ниже:
Полиэфирное
связующее
Акрилатное
связующее
Позволяет получить продукт с очень низким сухим остатком
(10—12%). обеспечивающий прекрасный металлический оттенок
и легкость нанесения (в основном используется в Западной Ев-
ропе)
Более высокий сухой остаток, чем у полиэфирного связующего
(15—20%), менее выраженный металлический эффект, но более
высокая гладкость покрытия и меньшая продолжительность про-
цесса окраски. (В основном используется в Японии и США)
Свойства прозрачного слоя: а) высокая прозрач-
ность и «совместимость» с основным слоем; б) обеспечивает
повышенную стойкость к действию УФ-лучей, выдерживает более
3-х лет в условиях экспозиции в штате Флорида.
В качестве связующего для прозрачного слоя используют
термореактивный акрилатный полимер, отверждаемый мелами-
новой смолой с добавками поглотителей ультрафиолетовых лу-
чей и светостабилизаторов.
Для получения максимального блеска и защиты от ультра-
фиолетовых лучей толщина пленки наносимого материала долж-
на составлять 35—50 мкм.
Известны два типа прозрачных слоев.
Прозрачный слой на осно-
ве растворов акрилатных
связующих
Прозрачный слой невод-
ных дисперсий акрилатов
Характеристики
Обеспечивает получение покрытий с высо-
ким блеском и прекрасной прозрачностью;
обычно наносится два слоя.
Обеспечивает получение однослойного по-
крытия с меньшей прозрачностью, чем при
использовании растворов
Процесс нанесения. Основной слой наносят в виде двухслой-
ного покрытия «мокрым по мокрому» с непродолжительной про-
межуточной сушкой на воздухе. Это необходимо для получения
высокой укрывистости и хорошего внешнего вида. После нане-
сения второго слоя покрытие выдерживается на воздухе (2—
3 мин); иногда сушка ускоряется подачей теплого воздуха, после
чего наносится 1 или 2 слоя прозрачного покрытия. Обычно
толщина пленки основного слоя составляет 15 мкм, прозрач-
ного слоя — 35—50 мкм. Окончательное отверждение покрытия
производится при 130 °C в течение 30 мин.
Цвет и пигментирование. Для пигментирования покрытий
с металлическим оттенком используются такие пигменты, кото-
рые при соответствующих методах диспергирования и стабили-
зации обеспечивают прозрачность покрытия. Полная прозрач-
ность -цветных пигментов позволяет с помощью металлических
чешуек получить яркое, сверкающее покрытие с металлическим
эффектом.
При обеспечении максимальной прозрачности металлический
оттенок будет зависеть от ориентации металлических чешуек.
Как было описано выше, максимальный металлический эффект до-
стигается тогда, когда каждая чешуйка ориентируется параллель-
но поверхности. Отражающую способность покрытия с металли-
ческим оттенком можно измерить с помощью гониофотометра,
который часто используется для замеров отражения непигментй-
рованных серебристых покрытий. В таких покрытиях можно не
учитывать отражение или поглощение света за счет цветных
пигментов.
На рис. 9.6 представлены сравнительные данные для различ-
ных покрытий с металлическим оттенком. Из рисунка видно яв-
ное превосходство полиэфирных покрытий с низким содержанием
сухого остатка. Отражение измеряли при стандартном угле паде-
ния (45°) и различных углах наблюдения и полученные данные
откладывали на графике. Более высокое положение на кривой
соответствует лучшему отражению, что характеризует степень
ориентации металлических чешуек параллельно поверхности.
Ориентация металлических чешуек. Как уже отмечалось ранее,
важнейшим фактором, влияющим на ориентацию алюминиевых
чешуек, является «схватывание» или усадка пленки в процессе
высыхания. Испарение растворителя из пленки в процессе высы-
хания ограничивает свободу передвижения чешуек (типичный
Рис. 9.6. Гомиофотометрические кривые (серебристый цвет).
Сухой остаток
при нанесении
Кривая А: основной слой на полиэфирном связующем 12%
Кривая В: термопластичный акрилатный лак 15%
Кривая С: основной слой на акрилатном связующем 18%
Кривая D: термореактивное акрилатное связующее 25%
размер 10—25 мкм). Силы поверхностного натяжения, а также
довольно большие размеры чешуек способствуют ориентации
чешуек параллельно поверхности. Однако, в случае каких-либо
отклонений от параллельного расположения чешуек получаются
покрытия с различным внешним видом, иными словами, покры-
тия, в которых все чешуйки расположены абсолютно параллельно
подложке, обеспечивают более высокий блеск по сравнению с
покрытиями, где чешуики ориентированы под разными углами,
например до 20°. В первую очередь это относится к основным
слоям с очень яркими тонами и низким сухим остатком. Кроме
того, толщина сухой пленки приблизительно равна или часто
может быть меньше, чем длина чешуек. Это очень ограничивает
движение чешуек, особенно если происходит усадка тонкой плен-
ки на завершающей стадии процесса сушки покрытия. Все эти
факторы обуславливают преимущества применения для основного
слоя материалов с низким сухим остатком.
Состав подслоя. Можно утверждать, что наиболее высокое
качество системы покрытия, включающей основной слой и про-
зрачный слой, может быть обеспечено при применении полиэфир-
ной шпатлевки (не содержащей масла), так как в этом случае по-
вышается стойкость системы покрытия к расслаиванию. Как было
отмечено ранее, эпоксидные материалы склонны к расслаиванию
на границе раздела подслой — основной слой, особенно при воз-
действии УФ-излучения. Следствием этого является преждевре-
менное разрушение системы покрытия, состоящей из основного
и прозрачного слоев. .
Применяемые в настоящее время полиэфирные шпатлевки,
а также прозрачные слои, содержащие поглотители УФ-излуче-
ния и светостабилизаторы, позволили решить эту проблему.
Практически внешний вид покрытий с основным слоем как
при испытаниях во Флориде, так и в эксплуатационных условиях,
был совершенно одинаков (см. ниже).
Внешний вид покрытий и долговечность. Испытания долговеч-
ности во Флориде (5° южной широты) представляют собой тради-
ционный способ определения долговечности в натурных условиях,
используемый в автомобильной промышленности-; Флорида явля-
ется весьма подходящим районом для таких испытаний благодаря
высокому уровню УФ-излучения и повышенной влажности.
Ранее в качестве основы для прозрачных слоев применялись
алкиды, однако такие покрытия разрушались под действием
УФ-лучей в течение 12 мес.
Современные прозрачные слои на основе термореактивных ак-
рилатных связующих имеют гарантированную долговечность от
3 до 5 лет. Такой высокий уровень долговечности обеспечивается
современной технологией, связанной с применением, основного и
прозрачного слоев.
Следует отметить, что в соответствии с существующими
стандартами высококачественное покрытие должно выдерживать
без разрушения испытания во Флориде в течение двух лет. Для
данного вида покрытий возможно использовать более широкий-ас-
сортимент пигментов, чем для термореактивных и термопластич-
ных акрилатов, т. к. при использовании органических пигментов
практически не происходит изменения цветовых характеристик
при экспозиции в атмосферных условиях.
Органические пигменты имеют высокую прозрачность и низкую
укрывистость и иногда обладают недостаточной стойкостью. Од-
нако, поскольку к основному покрытию не предъявляются требо-
вания по блеску, можно использовать высоконаполненные пиг-
ментированные тонкие пленки (толщина 15 мкм). В этом случае
прозрачный слой будет обеспечивать необходимый блеск и защиту
от ультрафиолетового излучения.
9.7.5. Пигментирование отделочных покрытий для автомобилей
Выбор пигментной части для получения покрытия нужного
цвета должен производиться с учетом требований рынка. В связи
с этим автомобильная промышленность всегда предъявляла наи-
более высокие требования к качеству сырья и использовала ма-
териалы только сорта «А», обеспечивающие наивысшую долговеч-
ность покрытий при эксплуатации в жестких условиях и хорошую
укрывистость__при невысокой стоимости. Поэтому для большин-
ства отделочных покрытий непригодны многие дешевые пигменты
с ограниченной светостойкостью; и вообще ассортимент применяе-
мых пигментов значительно уже, чем в других областях, где ис-
пользуются декоративные покрытия.
9.7.5.1. Насыщенные цвета
Для различных видов отделочных покрытий для автомобилей
вопросы пигментирования очень похожи. Как правило (не считая
некоторых незначительных различий во внешнем виде), алкиды
и полиэфиры, а также термореактивные акрилаты пигментируют-
ся одинаково. Однако белые акрилатные лаки склонны к мелению,
красные и фиолетовые — к обесцвечиванию до более светлых
оттенков; сохранность цвета фталоцианинового голубого хуже,
чем в других видах связующих. Существует ряд общих требований
к применяемым пигментам.
Долговечность. Применение многих органических пигмен-
тов либо полностью исключается, либо они могут быть использо-
ваны в ограниченных концентрациях как в чистом виде, так и в
смеси с другими пигментами, обеспечивающими высокую долго-
вечность.
Укрывистость и блеск. Очень чистые, яркие цвета
часто невозможно получить, так как избыток пигмента, необходи-
мого для получения укрывистого покрытия, значительно снижает
блеск. Чаще используют неорганические пигменты; благодаря их
природной укрывистости находят применение такие интенсивные
органические пигменты, как, например, синий, зеленый и фиоле-
товый.
Цена. Большинство органических пигментов очень дороги
и во многих случаях имеют низкую красящую способность. Исклю-
чение составляют фталоцианиновый голубой и зеленый, которые
значительно дешевле других и благодаря их высокой кроющей
способности могут использоваться при небольших концентрациях.
Выцветание. Многие органические красные и желтые пиг-
менты не могут быть использованы, поскольку они обладают склон-
ностью к выцветанию.
Метамерия. Метамерия представляет собой явление, за-
ключающееся в том, что два одинаковых по цвету образца при
различных условиях освещения становятся отличными друг от
друга по цвету (например при дневном свете и под вольфрамо-
вой лампой). Это явление часто наблюдается в тех случаях,
когда используются различные образцы пигментов, имеющих от-
личия от «эталонов». Наиболее характерно это явление для же-
лезоокисных и фталоцианиновых пигментов.
Применение хроматов свинца. В состав автомобиль-
ных отделочных покрытий часто входят пигменты на основе хро-
мата, сульфата и молибдата свинца, т. к. эти пигменты обладают
высокой яркостью и интенсивностью цвета и низкой стоимостью.
Однако применение этих пигментов имеет ряд ограничений:
1. Под действием солнечного света пигменты на основе хрома-
та свинца темнеют. Это особенно заметно в светло-желтых тонах
и зависит от сезонных изменений погоды и содержания загряз-
нений в промышленной атмосфере.
2. Хроматы свинца также чувствительны к действию разбав-
ленных кислот, под влиянием которых они обесцвечиваются. Дей-
ствие кислот может проявляться в появлении белых пятен или
полной потере цвета.
3. Пленки покрытий кислотного отверждения, применяемые
при ремонте, также будут быстро темнеть в результате взаимо-
действия кислоты с пигментом в поверхностном слое покрытия.
Разнообразные проблемы, связанные с загрязнением окружаю-
щей среды, заставляют отказываться от применения хромата
свинца в красках для автомобилей. Однако многие чистые и
яркие пигменты, которые применяют вместо хромата свинца, очень
дороги и затрудняют получение покрытий с требуемым блеском
и укрывистостью. Пигменты желтых цветов обладают особенно
низкой долговечностью.
В настоящее время выпускается довольно большое количество
красных пигментов, не содержащих соединений свинца, однако
для полного отказа от соединений свинца необходимо разрешить
целый ряд проблем как в области цветовых характеристик, так
и промышленного производства красок.
9.7.5.2. Однослойное покрытие с металлическим оттенком
Для термопластичных и термореактивных акрилатных покры-
тий вопросы пигментирования решаются практически одинаково,
хотя в ряде случаев органические желтые и фталоцианиновые
пигменты имеют некоторые преимущества при использовании в
термопластичных акрилатных покрытиях.
Долговечность. К органическим пигментам предъявляют-
ся более жесткие требования, чем к пигментам насыщенных цве-
тов, особенно при получении светлых тонов. Эти требования
ограничивают ассортимент пригодных пигментов, которые можно
применять лишь в тех случаях, если их свойства могут быть улуч-
шены добавками, поглощающими ультрафиолетовое излучение,
или введением в состав покрытия прозрачного железооксидного
пигмента. В покрытиях с металлическим оттенком используют
в основном неорганические пигменты. При этом могут быть полу-
чены красные и желтые тона с высокой прозрачностью покрытий,
с прекрасной долговечностью (конечно, при добавлении поглоти-
теля УФ-лучей) и достаточно дешевые. Единственным недостат-
ком таких пигментов является недостаточно насыщенный цвет.
Укрывистость и блеск. В любом отделочном покрытии
с металлическим оттенком укрывистость обеспечивается за счет
алюминиевых чешуек, тогда как введение прозрачной колеровоч-
ной пасты придает покрытию определенный тон. Если не использо-
вать черный пигмент, то глубокие темные цвета с металлическим
оттенком (при, низком содержании алюминия) получить гораздо
сложнее, чем светлые тона. Сложнее всего добиться чистого, яр-
кого красного цвета, так как большинство допустимых пигментов
имеет плохую красящую способность; при этом количество алю-
миния должно быть небольшим, чтобы не получить серо-розовый
оттенок.
Стоимость. Наиболее дешевым и прочным пигментом, при-
годным для покрытий с металлическим оттенком, является про-
зрачная окись железа, на основе которой могут быть получены
наиболее популярные оттенки золотистого, бежевого, коричневого
и бронзового цветов. Также широко используются голубой и зе-
леный фталоцианиновые пигменты благодаря их высокой крася-
щей способности и сравнительно низкой стоимости.
Стабильность цвета. Покрытие с металлическим оттен-
ком должно соответствовать эталону цвета при различных ракур-
сах осмотра. На цвет покрытия с металлическим оттенком оказы-
вают большое влияние условия формирования пленки. Краска
должна наноситься таким образом, чтобы при сушке алюминиевые
чешуйки ориентировались параллельно поверхности подложки
для достижения максимальной яркости и требуемого оттенка.
Очевидно, что при замене одного вида распылителя на другой
может изменяться цвет краски, поэтому там, где возможно, для
получения равномерного цвета используют автоматические распы-
лительные устройства.
Выбор алюминиевой пудры. Существуют несколько
сортов алюминиевой пудры, отличающихся размером чешуек. Как
правило, при увеличении размеров частиц возрастает яркость,
цвет становится более интенсивным (менее серым) и усиливается
металлический оттенок -покрытия. С другой стороны, блеск, чи-
стота цвета, укрывистость и красящая способность ухудшаются.
В связи с этим необходимо найти компромисс между укрывисто-
стью, блеском и яркостью. Наиболее пригодными для решения
этой проблемы являются чешуйки среднего и мелкого размеров.
Очень грубые частицы нужно обязательно удалять.
9.7.5.3. Система покрытия, состоящая из основного
и прозрачного слоев
Проблемы пигментирования для обоих типов покрытия практи-
чески одинаковы. К покрытиям предъявляются следующие тре-
бования:
Укрывистость. Укрывистость обычно достигается при тол-
щине пленки 10—20 мкм. Как и в случае однослойного покрытия,
требуемая укрывистость обеспечивается за счет алюминиевых
чешуек, поэтому к полиэфирным покрытиям с низким сухим остат-
ком предъявляется ряд требований, обеспечивающих четкое про-
явление металлического оттенка. Как и в предыдущем случае,
наибольшую трудность вызывает получение ярких красных тонов.
Важным преимуществом данного типа покрытий по сравнению с
однослойными покрытиями является отсутствие необходимости
получения высокого блеска, поэтому при введении большого ко-
личества алюминиевой пудры сохраняется возможность получе-
ния разнообразных светлых тонов.
Стоимость. Требования по стоимости аналогичны требова-
ниям для однослойного покрытия.
Равномерность окраски и д о л г о в е ч н о с т ь. В ос-
новном слое покрытия может использоваться очень широкий ас-
сортимент пигментов, так как в состав прозрачного верхнего
слоя вводится поглотитель УФ-лучей и светостабилизатор (про-
зрачный слой выполняет две функции: предотвращает разруше-
ние покрытия под действием УФ-лучей и защищает от разрушения
пигменты в основном слое).
Многие органические пигменты склонны к изменению цвета
при экспозиции в атмосферных условиях, но они могут применять-
ся в системе с основным и прозрачным слоями, что позволяет
таким образом значительно расширить ассортимент цветов. Это
в первую очередь относится к покрытиям светлых пастельных
тонов, в которых за счет применения разнообразных пигментов
и оптимальной ориентации алюминиевых чешуек можно получить
очень интересные привлекательные тона.
При нанесении основного слоя необходимо учитывать те же
требования, что и для однослойного покрытия.
Выбор алюминиевых чешуек. Как и в случае одно-
слойного покрытия, наиболее пригодными являются алюминиевые
чешуйки среднего и малого размеров. Грубые частицы использо-
Таблица 9.2. Отделочные покрытия для автомобилей: сравнение основных свойств
Свойства Алкидно-меламиновое Термореактивное акрилатное НВД TeF мопластичное 1Крилатное Система с основным и прозрачным слоями
Покрытие насыщен- Насыщенные цвета Насыщенные цвета и по- Насыщ енные цвета и по- Покрытие с металличе-
ных цветов или с ме- крытия с металлическим крытия с металлическим ским оттенком
таял и ческим оттенком ... оттенком оттен к<! !>м
Внешний вид Высокий блеск, 85% при Несколько меныпий Высокг й блеск, 85% при Высокий блеск 90% при
Сухой остаток при на- угле 20° блеск 80% при угле 20° угле 2( О угле 20°
40—45% 30—35% 12—18J ^0 Основной слой:
несении распылением Количество слоев при Два Два-три Три-nei ыре 12% (полиэфирное по- крытие); 18% (акрилат- ное покрытие), прозрач- ный слой 35% Два — для основного
нанесении распыле- нием Совместимость с под- Удовлетворительная (не- Вполне удовлетвори- Очень высокая ОКП слоя, один-два —для прозрач- ного слоя Рекомендуется полиэфир-
слоем достаточная адгезия к тельная (хорошая адге- (55%), необходимо на- ное покрытие
Режим сушки электропокрытшо) 20 мин при 130 °C зия к электропокрытшо) 20 мин при 130 °C несение подслоя, тре- буется нанесение проме- жуточного слоя (плохая адгезия к электропокры- тию) 30 мин при 155 °C (сту- 20 мин при 130 °C
Полируемость Неудовлетворительная Хорошая пенчатый режим) Отличная Хорошая
Ремонтоспособность Перекраска листов Перекраска листов Местная подкраска Незначительная местная
(краска с кислотным ка- тализатором) (краска с кислотным ка- тализатором) подкраска Для прозрачного слоя — лак с кислотным катали- затором
вать не рекомендуется. Крупные частицы (до 30 мкм) не только
снижают укрывистость, но и ухудшают качество покрытия, по-
скольку они могут проступать через очень тонкую пленку (10—
20 мкм) основного слоя (напоминают по внешнему виду рассы-
панную крупу) и способствуют разрушению пленки.
В дополнение к сказанному, при использовании крупных
частиц возникают проблемы при электростатическом нанесении
покрытий (разряд через заземление трубопроводов). В табл. 9.2
приведено сравнение основных типов автомобильных покрытий.
9.8. РЕМОНТНАЯ ОКРАСКА В ЗАВОДСКИХ УСЛОВИЯХ
После предварительной отделки и окраски корпуса автомобиля
окончательную окраску производят после сборки. На завершаю-
щей стадии отделки могут произойти повреждения красочной
пленки. Если возникает необходимость ремонта покрытия, то при-
меняемая для этих целей краска должна обладать теми же свой-
ствами, что и исходная краска.
Ремонтная окраска может включать перекраску листов или
больших поверхностей (дверей, капота и т. д.), либо местную под-
краску.
Выбор ремонтного покрытия зависит от вида и технологии на-
несения основного покрытия.
Термореактивные отделочные материалы (перекраска листов
или больших поверхностей). Для этих целей используют алкид-
ные и термореактивные акрилатные материалы, а также систему
покрытия, состоящую из основного и прозрачного слоев. Сушка
эмалей или лака для прозрачного слоя ускоряется введением
в состав материалов небольших количеств кислотного катализа-
тора с последующей выдержкой в течение 30 мин при 90 °C или
10 мин при 100 °C. Применение более высоких температур не-
допустимо, поскольку приводит к разрушению находящихся внут-
ри корпуса пластмассовых деталей.
В настоящее время для ремонта листов или больших поверх-
ностей в заводских условиях используют двухупаковочные системы
(на основе полиуретанов или акрилатов). Эти материалы имеют
высокую долговечность, хороший блеск и для их сушки необходима
сравнительно низкая температура (15 мин при 80 °C). Однако
они имеют два недостатка: высокую токсичность из-за присут-
ствия в системе в качестве катализатора изоцианата, работа
с которым требует особой предосторожности, а также необходи-
мость полного соответствия по цвету исходным материалам.
Термопластичные акрилатные лаки (местная подкраска). По
своей природе эти материалы при ремонтных работах не вызывают
затруднений. В основном термопластичные акрилатные лаки при-
меняют для местной подкраски, по их можно использовать и для
ремонта листов или больших поверхностей. Они прекрасно поли-
руются, что позволяет легко удалять дефекты, вызываемые за-
грязнениями или некачественным распылением краски.
Использование немодифицированного продукта значительно
облегчает процесс и уменьшает трудности равномерного нанесе-
ния. Минимальное время сушки составляет всего 15 мин при
80 °C, после чего производится полирование для получения
максимального блеска.
9.9. ОКРАСКА ПЛАСТМАССОВЫХ ДЕТАЛЕЙ КОРПУСА
В настоящее время изделия из пластмасс находят широкое
применение в автомобильных конструкциях. Эти материалы все
более широко используются для деталей внешней отделки, на-
пример для бамперов, щеток и вентиляционных решеток и т. д.
Пластмассовые детали используют не только благодаря их при-
влекательному внешнему виду, но также из-за высокой стойкости
к механическим разруш-ения-М-И—кофрозии. ____
Дальнейшее развитие в этой области должно привести к ис-
пользованию пластмасс для других деталей корпуса, например
обшивки дверей или капота.
При окрасйе пластмассовых деталей, однако, возникает целый
ряд проблем, связанных с адгезией, режимами сушки, устойчиво-
стью пластмасс к растворителям, равномерностью нанесения по-
крытия и т. д. В настоящее время технология окраски автомоби-
лей разработана для цельносварных конструкций из мягкой стали.
Процесс предусматривает сушку при температуре 165 °C для
грунтовочных покрытий и 130 °C — для отделочных слоев. При
использовании пластмассовых деталей нужно либо усовершенство-
вать для них уже существующую технологию, либо разрабаты-
вать новую.
Существует много различных видов пластмасс, свойства кото-
рых влияют на процесс их окрашивания. Основные различия
между разными видами пластмасс приводятся ниже.
9.9.1. Листовой материал, получаемый горячим формованием
Важными преимуществами данного вида пластмасс являются:
а) высокий модуль упругости (сохранение жесткости в горизон-
тальном положении); б) высокая температура деформации (ма-
териал выдерживает воздействие температуры до 180 °C, кото-
рая требуется для проведения электроокраски); в) высокая
стойкость к действию растворителей.
К недостаткам относятся волнистый рельеф поверхности, а
также выделение газов при отверждении, в результате чего на
поверхности образуются пузыри. Обычно поверхность изолиру-
ется путем нанесения полиуретановой пленки либо из расплава,
либо методом распыления.
9.9.2. Полиуретаны, получаемые методом экструзии
•• > К достоинствам этих материалов относится широкий'диапазон
значений жесткости (от очень жесткого до резиноподобного)
и высокая прочность. К недостаткам можно отнести наличие по-
верхностных или подповерхностных пор, отсутствие стабильности
размеров (армированный стекловолокном материал при пересуш-
ке увеличивается в размерах) и недостаточную стойкость к
растворителям., особенно при армировании стекловолокном.
9.9.3. Пластики, получаемые методом экструзии из расплава
К материалам этого типа относятся поликарбонаты-, АБС-плас-
тики, полиамиды (в' основном армированные стекловолокном) и
полипропилен. Важнейшими преимуществами .материалов этого
типа являются прочность и жесткость, хорошее качество поверх-
ности и широкий диапазон изменения эластичности — от жесткого
до пластичного. К наиболее существенным недостаткам относятся
высокая хрупкость при низких температурах, во многих случаях
низкая температура температурной деформации (ТТД), а также
возможность образования «задиров» при формовании вследствие
аморфной структуры материалов.
9.9.4. Проблемы окраски
Адгезия. Обеспечить хорошую адгезию отделочного покры-
тия к модифицированному полипропилену очень сложно, поэтому
необходимо использовать специальную грунтовку, улучшующую
адгезию. При окраске других видов пластмасс проблемы, связан-
ные с адгезией, практически не возникают.
Температурная деформация. Пластмассовые детали
при повышении температуры могут коробиться или прогибаться.
Температура, при которой пластмасса будет деформироваться,
зависит от состава полимера, от вида наполнителя и армирующего
материала,- а также величины и направленияк-меднют-йй^ой на-
грузки. Таким образом, хотя для каждого полимера известен
верхний температурный предел, при котором полимер плавится или
размягчается, на практике допустимая темперадурамфижонтаже
и сборке пластмассовых деталей должна быть знаимтельп® ниже.
Так как пластмассы под действием температурыэдеформиру-
ются, окраску пластмассовых деталей можно производить вне
основного конвейера (например деталей из полиуретанов) и
осуществлять их монтаж либо после проведения,электроокраски
(например при использовании полипропилена, -армированного
стекловолокном и большинства полиамидов), либо монтировать
после окончательной отделки корпуса (например в случае некото-
рых полиамидов, или пластиков, полученных.горячим формова-
нием и т. д.). Если окраска производится вне основного конвейера,
получить цвет, полностью совпадающий с цветом корпуса, весьма
трудно.
Текстура поверхности. Для того, чтобы получить ка-
чественное равномерное покрытие при использовании различных
партий одного и того же материала, необходимо использовать
один и тот же подслой.
Устойчивость к действию р а с т в о р и те л е й. Мно-
гие стандартные растворители, входящие в состав красок, чрезвы-
чайно сильно разрушают поверхность некоторых пластмасс, вызы-
вая растрескивание и ухудшение механических свойств изделия.
Однако при незначительном воздействии растворителя может
улучшиться адгезия покрытия.
Влияние механических свойств. Если на окрашен-
ной поверхности при изгибе или ударе на покрытии появляются
трещины, они могут явиться причиной разрушения самой пласт-
-м-а-ссовой осчшвы. Иными-словами, неудовлетворительная схема
окраски может служить источником снижения механических ха-
рактеристик пластмассовых деталей.
9.9.5. Процесс окраски и используемые материалы
. Окраска на к о н в е й е р е. Типовой процесс окраски цель-
носварных корпусов был описан выше. Под этот процесс могут
быть приспособлены только пластмассы, полученные горячим фор-
мованием, а также некоторые виды полиамидов, так как эти ма-
териалы выдерживают температурный режим электроокраски
(165—180 °C) без деформации и их окраска может быть выпол-
нена стандартными отделочными материалами.
Процесс окраски горячеформованных деталей включает нане-
сение либо слоя расплавленного связующего, либо полиуретано-
вого покрытия. Эта операция производится для выравнивания
и изоляции поверхности. После этого пластмассовые детали мон-
тируются на корпусе и вся конструкция поступает на дальнейшую
обработку, где подвергается очистке, фосфатированию и электро-
окрашиванию. Затем производится окончательная окраска по
стандартной схеме, включающей нанесение шпатлевки и отделоч-
ного покрытия.
Окраска вне основного конвейера. В этом случае
окраска пластмассовых деталей может производиться на спе-
циальном участке, либо на другом предприятии. Окрашенные
детали затем монтируются на полностью окрашенном корпусе ав-
томобиля. При таком способе выполнения работ необходимо при-
менять специальные лакокрасочные материалы и соответствую-
щую технологию окраски.
Цвет окрашенных пластмассовых деталей должен соответ-
ствовать цвету корпуса автомобиля, поэтому возникают опреде-
ленные трудности при проведении окраски, особенно в случае
покрытий с металлическим эффектом.
Детали из полиуретанов, полученные методом экструзии из
расплавов, обычно окрашиваются вне основного конвейера. Если
детали не нуждаются в дополнительной сборке, на их поверхность
может быть нанесено эластичное покрытие на основе одноупако-
вочного полиуретанового материала, отверждаемого меламино-
формальдегидной смолой при 120 °C.
В США этот метод окраски является стандартным, однако в
Европе для окраски такого вида деталей, а также деталей из гиб-
ких термопластов чаще используют двухкомпонентные материалы,
на основе которых получают прозрачные покрытия и покрытия
без металлического оттенка.
Применение двухупаковочной композиции позволяет снизить
температуру сушки ниже 100 °C, расширяет рецептурные воз-
можности и ускоряет в целом процесс окраски. Так как процесс
полностью автоматизирован, то при такой технологии окраски не
возникают проблемы токсичности.
Подгонка цвета при окраске вне основного конвейера явля-
ется сложным процессом, который зависит от формы и расположе-
ния деталей. Кроме того, при поломке или замене деталей разли-
чия по цвету должны быть минимальными, независимо от того,
окрашены ли детали на основном конвейере или вне его. В покры-
тиях с металлическим оттенком в качестве основного слоя может
быть использован тот же материал, который применяется при
окраске металлических поверхностей, а в качестве прозрачного
слоя может применяться либо стандартный, либо двухкомпонент-
ный лак. Во многих случаях стандартный материал дает доста-
точно хорошие результаты.
9.9.6. Окрасочная линия для местной окраски
На ряде пластмассовых деталей между стадиями электроокрас-
ки и нанесения отделочного слоя производится местная окраска
соответствующими лакокрасочными материалами. Эта операция
выполняется в основном для небольших деталей, таких, как, на-
пример, вентиляционные решетки из полиамидов.
Большинство других армированных пластмасс допускает обыч-
ный режим окраски, что позволяет получать покрытия с доста-
точно хорошим внешним видом.
9.10. ОКРАШИВАНИЕ РАСПЫЛЕНИЕМ
Внедрение массового поточного производства автомобилей
в сочетании с разработкой новых синтетических лакокрасочных
материалов явилось основным фактором, определившим приме-
нение технологии распыления. Принцип окраски распылением за-
ключается в раздроблении жидкой краски на тонкодисперсные
частицы и перенос их на поверхность корпуса автомобиля. Перво-
начально для распыления применялся сжатый воздух, но сейчас
находят применение новые способы, такие как электростатиче-
ское распыление, характеризующееся более эффективным перено-
сом материала на подложку.
Этот метод, однако, приводит к образованию пылевого об-
лака, поэтому необходимо принимать меры для удаления этой пы-
ли и выброса ее в атмосферу или, в ряде случаев, пропускания
ее через систему очистки. В настоящее время для этих целей при-
меняются высокоэффективные и пожаробезопасные распылитель-
ные камеры с водяной завесой. В камерах этого типа выходящий
воздух пропускают через воду, которая смывает пыль в цистерну
для хранения промывных вод..
Основные^требования, предъявляемые к методу распыления,
состоят в следующем;
1. Обеспечить надежный, гибкий, удобный и безвредный ме-
тод нанесения, тгри поторомддостигается необходимая толщина
пленки за ограниченный срок.
' 2. Обеспечить получение тонкой пленки с хорошим розливом,
лишенную таких дефектов, как «апельсиновая корка», потеки,
кратеры. Часто для того, чтобы избежать кратеров, в краску вво-
дят поверхностно-активные вещества.
3. Получать «влажную» пленку покрытия, при нанесении кото-
рой не образуется'пыль.
4. При нанесении материалов с металлическими пигментами
предотвратить грубую шероховатость покрытия.
5. Обеспечить максимальное использование материала и эф-
фективность переноса без ухудшения внешнего вида покрытия и
его характеристик!
9.10.1. Воздушное распыление
Основным методом, применяемым в массовом производстве,
является медод-«подачи под давлением», хотя для окраски очень
малых участков в лабораторной практике и при ремонте использу-
ется такжё':нодачй под вакуумом.
По первому методу разбавленная краска (хорошо перемешан-
ная) подартся под давлением по линии подачи к пистолету-рас-
пылителю. Из распылителя краска подается через игольчатый
клапан; при этом количество краски определяется степенью на-
жима на курок и давлением. Тонкая струя краски, выходящая
из распылителя, измельчается током сжатого воздуха, поступаю-
щего через отверстия в головке распылителя. Отверстия сопел
можно направлять так, чтобы обеспечить равномерное распыле-
ние частиц. .
Благодаря многообразию конструкций и скорости окрашива-
ния воздушный краскораспылитель до сих пор доминирует над
другими методами нанесения. Несмотря на значительные потери
краски в результате того, что не вся краска достигает окрашивае-
мой поверхности, он все еще широко применяется при нанесении
шпатлевок, промежуточных и декоративных слоев, включая по-
крытия с металлическим оттенком.
Когда поток частиц выбрасывается из сопла распылителя,
часть краски попадает на уже окрашенную поверхность детали.
Эти потери неизбежны, поскольку необходимо тщательно окраши-
вать также углы и грани; но часть краски уносится за пределы
детали потоком сжатого воздуха, как бы вследствие «отбивки» от
поверхности.
Частицы краски подвергаются действию двух факторов. Одним
из них является поступательное движение частиц, которое удер-
живает их при движении в направлении к окрашиваемому объек-
ту; другим — поток воздуха, который стремится отклонить траек-
торию частиц и направить их по различным направлениям. Оче-
видно, действие потока воздуха проявляется тем сильнее, чем
меньше размер частиц и чем мощнее поток. Грубое распыление
требует меньшего давления воздуха и отбивка краски может быть
снижена, но в результате ухудшается качество покрытия.
Необходимо установить такое давление воздуха, при котором
обеспечивается достижение требуемого качества покрытия и при-
емлемая степень отбивки краски и пылеобразования. Другими
факторами, которые влияют на выбор давления воздуха, явля-
ются вязкость и поверхностное натяжение. Чем выше эти показа-
тели, тем больше энергия, необходимая для распыления краски,
и тем мощнее должен быть поток воздуха.
Типичные значения вязкости для нанесения распылением при-
ведены ниже:
Тип покрытий Вязкость при 25 °C Количество слоев (мок- рый по мокрому)
Шпатлевки 25 с, вискозиметр BSB4 Два
Алкидные покрывные эма- 25 с, вискозиметр BSB4 (25 с Два
ли Термореактивные акри- для интенсивных цветов) 33 с для покрытий с метал- Два — три
латные, неводные диспер- сии лическим оттенком, вискози- метр BSB3
Термопластичные акри- 35 с, вискозиметр BSB3 Три — четыре
латные Основной слой (полиэфир- 25 с, вискозиметр BSB3 Два
ный) Прозрачный слой 35 с, вискозиметр BSB4 Два
Примечание. BSB — Британский стандартный вискозиметр типа «В»
Истинная эффективность воздушного распылителя при нор-
мальных условиях окраски низка: только 40% краски попадает
на корпус автомобиля, 60% составляют потери (20% — за счет
отбивки, 40% —за счет пылеобразования).
Необходимость повышения эффективности распыления очевид-
на. Более экономичны автоматические распылительные установки,
однако в последние годы разработан ряд новых методов окраски,
открывающих перспективу существенной экономии красок, в част-
ности электростатическое и воздушно-электростатическое распы-
ление, распыление под колоколом, дисковое распыление.
Для воздушного распыления к способам снижения потерь
краски помимо описанного ранее снижения давления относится
горячее распыление с пониженным давлением.
Новейшая технология окраски, которая далее будет рассмот-
рена, может быть полностью автоматизирована. Новые методы
обладают рядом преимуществ, к числу которых относятся эконо-
мия рабочей силы и стабильность качества, не говоря о заметном
увеличении эффективности окраски.
9.10.2. Распыление с подогревом при-пониженном давлении
Этот метод нашел применение для нанесения шпатлевок, но
он редко используется в современной технологии. Нагрев краски
до 60—80 °C снижает вязкость и поверхностное натяжение, об-
легчая автоматизацию процесса. Соответственно, для распыления
необходимо более низкое давление; более высокий сухой остаток
снижает потери за счет отбивки. Однако потери за счет пылеобра-
зования увеличиваются из-за увеличения сухого остатка, а также
возникают трудности при выборе растворителя. Представляют
проблему и потери за счет испарения растворителя.
9.10.3. Электростатическое распыление
Принцип электростатического распыления прост. Когда части-
цы краски распыляются в электрическом поле, они приобретают
заряд и притягиваются к поверхности изделия, которое обычно
заземлено. Существуют различные конструкции установок для
электростатического распыления; в автомобилестроении наиболее
широко применяются установки с вращающимися колоколами
или дисками, которые обеспечивают возможность нанесения всего
ассортимента материалов и для подслоя и для декоративных по-
крытий.
На рис. 9.7 представлена типичная схема установки. Краска
подается насосом из напорного бачка через промежуточную
емкость. Вращение распылителя отбрасывает краску к периферии,
где происходит ее распыление за счет центробежных сил, но глав-
ным образом за счет действия электростатических сил. Под дей-
ствием электростатического поля частицы краски притягиваются
к окрашиваемой поверхности равномерно, что делает электроста-
Рис. 9.7. Установка электростатического распыления
тическое распыление высокоэффективным в отношении использо-
вания краски. Основные параметры процесса, обеспечивающие
максимальную эффективность: скорость вращения — 30—
40 тыс. об/мин; напряжение 90—110 кВ. Уменьшенное пылеобра-
зование устраняет необходимость применения крупногабаритных
окрасочных камер, а также интенсивного воздухообмена. Этот
метод позволяет полностью автоматизировать процесс со всеми
вытекающими из этого преимуществами.
Однако некоторые закрытые и другие поверхности трудно
окрасить этим методом, поскольку частицы краски притягиваются
более близко расположенными частями сложной конструкции кор-
пуса автомобиля, и скорость движения частиц недостаточна для
более глубокого их проникновения. Этот недостаток преодоле-
вается с помощью воздушно-электростатического распыления,
основанного на совместном использовании сжатого воздуха и
электростатического поля.
Установка снабжена изолированным бачком, имеющим зазем-
ленную штангу с высоковольтным электродом в воздушной распы-
лительной головке, напряжение в которой обычно колеблется в
пределах 30—60 кВ. Ток разряда, стекающий с высоковольтного
электрода, создает вокруг распылителя область, обогащенную
ионами с одинаковым зарядом, которые зарядсают,,частицы рас-
пыляемой краски. ।
За пределами головки распылителя преобладает инерция аэро-
динамического потока, но по мере приближения частиц краски к
окрашиваемой поверхности заземленного корпуса электростатиче-
ские силы (благодаря электростатическому полю между электро-
дом распылителя и изделием) становятся более значимыми и
способствуют притяжению распыленных частиц к окрашиваемой
поверхности.
Максимальная экономия краски обычно достигается за счет
поддержания наибольшего допустимого напряжения, что создает
максимальную разность потенциалов между распылителем и окра-
шиваемой деталью. За счет того, что скорость распыленных частиц
вблизи окрашиваемого изделия поддерживается практически на
минимальном уровне, обеспечивается хорошее распыление и расте-
кание «осаждающейся» пленки.
Воздушно-электростатические распылители могут управляться
вручную либо автоматически. Этот метод занимает промежуточ-
ное положение между менее эффективным воздушным распыле-
нием и высокоэффективным электростатическим распылением, и
в сравнении с последним обеспечивает лучшую однородность
и гладкость пленки.
Электростатическое распыление применяется также для
получения покрытий с металлическим оттенком. Основные огра-
ничения, допускаемые при получении таких покрытий, описаны
в предыдущих разделах. Fla практике электростатическое распы-
ление заметно ухудшает условия всплывания металлических пиг-
ментов (тональный контраст) и затрудняет получение покрытий
с металлическим оттенком.
В результате многочисленных исследований этого явления был
предложен следующий механизм.
При сравнении скорости нанесения краски и степени распы-
ления наблюдаются значительные различия этих характеристик
для разных материалов. Например, типичная скорость нанесения
покрытия для основного слоя ’с низким сухим остатком состав-
ляет:
Электростатическое распыление
(высокоскоростной колокол)
Воздушно-электростатическое
распыление
Воздушное распыление
300 500 см3/мин
450—500 см3/мин
800 см3/мин
Распыление
удовлетвори-
тельное
Хорошее
Отличное
При ручном методе распыления основной слой наносится с
очень большой скоростью и распыляется исключительно хорошо.
Есть основания полагать, что в момент ударения распыленной кап-
ли о поверхность подложки алюминиевые чешуйки распределя-
ются параллельно поверхности в результате действия значитель-
ных усадочных сил. Во влажном слое нанесенной краски происхо-
дит дополнительная ориентация некоторого количества частиц,
но быстрая усадка и поверхностное натяжение быстро приводят к
«поворачиванию» частиц в идеальное положение (параллельно
поверхности), что дает оптимальный эффект.
Меньшая скорость и относительно меньшая степень распыле-
ния при электростатическом распылении приводят к тому, что при
ударе усадочные силы ниже и чешуйки не стремятся к быстрой
и полной ориентации относительно поверхности и располагаются
более беспорядочно.
В дальнейшем усадка и поверхностное натяжение улучшают
ориентацию, но, принимая во внимание начальное положение,
образующееся покрытие уступает по внешнему виду и качеству
покрытию, нанесенному ручным способом. Именно по этой причине
основной слой с металлическим оттенком наносят «смешанным»
методом с целью оптимизации внешнего вида и обеспечения
эффективности окраски, а также снижения до минимума необхо-
димости подправлять покрытие после нанесения.
9.10.4. Эффективность окраски (практические аспекты)
С точки зрения абсолютной эффективности какого-либо опре-
деленного типа оборудования трудно выделить специфические по-
казатели. Существуют значительные различия между заводами,
зависящие от того, наносится покрытие вручную или автоматиче-
ски, от размеров распылительной камеры и от различия в разме-
рах и форме окрашиваемых деталей. Фактические показатели мо-
гут значительно отличаться от идеала.
Все же некоторые основные значения эффективности, основан-
ные на результатах практической эксплуатации различных уста-
новок, приводятся ниже. Они предназначены только для того,
чтобы дать некоторое сравнительное представление относительно
эффективности реально используемых методов:
Высоковольтное электростатическое распыле- 75 ±5%
ние (высокоскоростной колокол)
Воздушно-электростатическое распыление 60 + 5%
Воздушное распыление 40 + 5%
На практике широко используется в массовом производстве
«совмещение» или «комбинирование» различных методов. Усред-
ненный типовой процесс приведен в табл. 9.3. Подобные системы
сочетают достоинства и недостатки отдельных методов распыле-
ния, реализуя следующие преимущества: 1) максимальное соот-
ветствие требованиям по внешнему виду при толщине пленки
(45—55 мкм); 2) оптимальный уровень эффективности; 3) воз-
можность окраски сложных цельносварных конструкций, вклю-
чающих наружные и внутренние поверхности; 4) реализация
п. п. 1), 2) и 3) в пределах приемлемых интервалО’вчвремени и
пространства; 5) минимальные затраты труда.:
9.11. ГОРЯЧАЯ СУШКА
(: ‘
Ранее были описаны различные типы материалов, применяе-
мых при окраске корпусов автомобилей. К ним относятся водо-
разбавляемые эпоксидные материалы для электроокраски и много-
компонентные покрывные краски, содержащие растворители, та-
кие как алкидно-полиэфирные и акрилатные эмали. Органические
растворители представляют собой сложные смеси алифатических
и ароматических углеводородов, спиртов, эфиров и кетонов. Крас-
Таблица 9.3. Типичный технологический процесс окраски (упрощенная схема)
Тип покрытия 1 участок 2 и 3 участки 4 участок Обдув! воздух( а •м 5 участок 6 участок 7 участок
Электростатиче- ское распыление (в ысокос корсет- ной колокол) Ручное распыле- ние (воздушное) Ручное распыле- ние (воздушное)* Электростатиче- ское (высоко- скоростной колокол) Воздушио- электроста- тическое Воздушно- эл ектроста - тическое
Автоматиче- Ручное Ручное Автоматиче- Ручное Ручное
ское Основной слой Основной слой Основной слой ское Прозрачный Прозрач- Прозрачный
Покрытие с металли- Наружная по- Внутренняя Наружная по- слой Наружная по- ный слой Внутренняя слой Наружная
ческим оттенком, верхность поверхность верхность верхность поверх- поверхность
включающее основ- ной и прозрачный слой 1 слой 2 слоя 1 слой 1 слой ность 1 слой 1 слой
Алкидное покрытие с Алкидное по- интенсивным цветом крытие Наружная по- верхность 1 слой Алкидное по- крытие Наружная по- верхность Ручная под- краска Алкидное по- крытие Внутренняя поверхность 1 слой
Примечания: I. Ручное распыление применяется для получения максимального металлического эффекта.
2. Полностью электростатическое распыление применяется для нанесения 1-го слоя только для лучшей эффективности.
3. Воздушно-электростатическое распыление применяется для обеспечения требуемого внешнего вида и эффективности.
и на растворителях наносятся либо методами окунания, либо
етодами распыления. В соответствии с принятой технологией
-и материалы требуют горячей сушки по следующим причинам:
1 Для того, чтобы достигнуть высокого уровня качества, отве-
чающего требованиям автомобильной промышленности, т. е. полу-
чить прочное защитное покрытие.
2 Для того, чтобы уложиться в ограниченный промежуток
времени, определяемый скоростью конвейера.
н 3. Для того, чтобы контролировать или ускорить испарение
растворителя и свести к минимуму загрязнения и выбросы.
Иными словами, операция горячей сушки приводит к отвержде-
нию пленки, оказывая влияние на различные химические пре-
вращения или реакции поперечной сшивки, в результате чего
образуется прочное защитное покрытие. В процессе пленкообразо-
вания выделяются растворители и побочные продукты реакции,
что может создать проблему загрязнения окружающей среды.
Температура горячей сушки колеблется в широких пределах.
Если для современных катодных покрытий, получаемых методом
электроокраски, требуется сушка в течение 20 мин при 180 °C,
то в других случаях условия отверждения могут быть значитель-
но мягче— 15 мин при 80 °C. На первой стадии горячей сушки
температуру необходимо поднимать с контролируемой скоростью,
чтобы избежать дефектов пленки, таких как образование крате-
ров или «вскипание» (вследствие выброса растворителя или вклю-
чений воздушных пузырьков). Как только температура достигает
требуемой величины, ее обычно поддерживают на этом уровне в те-
чение всего периода, необходимого для полного отверждения.
Типичный график включает подъем температуры в течение 8—
10 мин и выдержку при данной температуре 20 мин.
9.11.1. Технология горячей сушки
Существуют два типа печей, которые могут применяться для
сушки автомобильных покрытий: печи с конвективным обогревом,
применяемые более широко, и печи с радиационным или инфра-
красным обогревом.
В печах с конвективным обогревом происходит непрерывное
омывание потоком воздуха поверхности изделия — идеальный
случай для корпусов сложной формы. Радиационный обогрев
более пригоден для изделий регулярной формы, но часто он при-
меняется совместно с конвективным обогревом, например, чтобы
обеспечить подогрев на первой стадии с целью уменьшения вред-
ных выбросов или обеспечить нагрев утолщенных частей корпуса.
9.11.2. Конструкция конвективных печей
Выбор оптимальной конструкции печей производится с учетом
многих факторов, которые должны учитывать требования техно-
логии, расположение завода, расход энергии, экологические
требования и капитальные затраты.
Конфигурация печи. При печной сушке процесс вклю-
чает две стадии: контролируемый подъем температуры и после-
дующую выдержку, в течение которой смолы либо вступают в
реакцию, либо, как в случае акриловых лаков, равномерно расте-
каются на поверхности. Для этого необходимы две отдельных
зоны, каждая со своей системой нагрева и контроля. Конвектируе-
мый воздух поступает с высокой скоростью в пространство печи,
обычно через распределительную систему, смонтированную на
крыше печи. Зоны подогрева и выдержки могут быть далее под-
разделены на две или более зон каждая.
Вентиляция печи. При горячей сушке автомобильных
эмалей в атмосферу печи выделяются горючие соединения, что
создает взрывоопасную среду. Чтобы избежать этого, в простран-
ство печи необходимо подавать свежий воздух и, соответственно,
.отводить избыточ-ны-й- воздух-из- печи. Очевидно, этот воздух со-
держит разбавленные горючие соединения, выделяющиеся с по-
верхности краски, и не должен поступать в рабочую зону.
Нагрев печи. Каждая зона печи снабжена приточно-
вытяжной системой, которая отсасывает определенный объем воз-
духа из печи, смешивает его со свежим воздухом для вентиляции,
подводит тепло к этой смеси для поддержания теплового режима
печи и затем нагнетает воздух в печь через распределительные
отверстия или щели.
Требования к подаче воздуха. Количество свежего
воздуха, подаваемого в печь, рассчитывается в соответствии с
количеством растворителя, выделяющегося из покрытия: счита-
ется необходимым, чтобы концентрация растворителя в воздухе
не превышала 25% от нижнего предела взрываемости. Этот
объем свежего воздуха может подаваться в печь либо через фильтр
в зону нагрева, либо через уплотнения печи, либо в зону прямого
сгорания.
Топливо и метод нагрева. В Великобритании и боль-
шинстве стран Европы в качестве горючего используется природ-
ный газ. Поскольку этот газ не содержит серу, он применяется
в системах прямого пламенного нагрева всех окрасочных уста-
новок, так как опыт и испытания показали, что при этом не наблю-
дается снижение качества покрытий. Там, где природный газ не-
доступен, применяется бутан-пропановая смесь, которая также
содержит ничтожную примесь серы.
Другие системы предусматривают сжигание дистиллирован-
ного масла; это приводит к необходимости применять менее эффек-
тивный непрямой нагрев (его эффективность составляет около
70% по сравнению с прямым пламенным нагревом).
9.11.3. Выделение паров и запах
Как сказано ранее, при печной сушке автомобильных покры-
тий выделяются различные продукты, включающие смесь органи-
ческих растворителей, продукты химических реакций и деструк-
ции. Это может привести к образованию значительного количе-
ства паров и ощутимого запаха, а также к конденсации указан-
ных продуктов в трубах вытяжной системы. Однако наиболее
серьезного внимания требует проблема выделения значительного
количества растворителей. В США существует законодательство
по этому вопросу, значительные меры по обеспечению безопас-
ности предпринимаются и в Европе.
Контроль уровня выделений промышленных предприятий, в
том числе и из печей для горячей сушки, впервые был установлен
в США в 60-х гг., поскольку большинство лакокрасочных мате-
риалов подвергается горячей сушке и содержит органические
растворители. Последние рассматриваются как источник загряз-
нения ввиду токсичности продуктов их фотохимического разложе-
ния в окружающей атмосфере. В конце 60-х гг. власти Лос-Анже-
леса и Сан-Франциско ввели правила, согласно которым ограни-
чивалось количество различных растворителей, используемых в
композициях. Эти правила в дальнейшем получили название
«Закон 66» или «Правило 3» основного раздела этого закона.
В соответствии с этими документами предусматривается, что пе-
ред выбросом в окружающую атмосферу 90% углеводородов
должны быть окислены до двуокиси углерода.
Недавно введенные федеральные правила предусматривают
нормы по выделению углеводородов. Действительные уровни вы-
бросов зависят от местоположения предприятия. Они могут коле-
баться в пределах 1,4—2,3 кг/ч или 100—300 т/год. Этот уровень
может быть достигнут либо путем контроля выбросов из печей,
либо подбором рецептуры лакокрасочного материала, либо тем
и другим одновременно.
Во Франции и Англии приняты положения «Закона 66», но в
других странах Европы более широко используется норма, уста-
новленная в ФРГ. Это норма определяет точные количественные
пределы содержания растворителей любых типов.
Кроме новой технологии окраски, в настоящее время исполь-
зуются различные способы предотвращения загрязнения окру-
жающей среды. К ним относятся термическое разложение, ката-
литическое сжигание, адсорбция углем, жидкостные скрубберы,
дезодорация. Из этих способов наиболее широко применяется тер-
мическое разложение; но в последнее время вполне удовлетво-
рительные результаты были получены при использовании метода
каталитического сжигания.
Термическое разложение представляет собой про-
пускание загрязненного воздуха через зону интенсивного горения,
в которой сжигается первичное топливо — газ или дистиллиро-
ванное масло для подъема температуры до необходимой величи-
ны и поддержания ее на этом уровне в течение требуемого
времени. Таким способом сложные углеводороды окисляются до
двуокиси углерода и воды.
Метод каталитического сжигания с успехом приме-
няется в различных областях техники для очистки газов. Метод
дает существенное снижение расхода топлива по сравнению с
термическим разложением без необходимости использования
(либо в ограниченном масштабе) теплообменного оборудования.
Извлекаемые газы поступают в камеру с катализатором, на кото-
ром реагируют с кислородом воздуха.
Применение каталитического сжигания получило развитие в
середине 70-х гг. Катализатор не может действовать бесконечно
долго, но, если исключить возможность его загрязнения или отрав-
ления, то срок службы катализатора может достигать 5 лет.
9.11.4. Перспективы дальнейшего развития
печной сушки
*
Развитие и разработка других систем сушки покрытий тесно
связаны с развитием технологии окраски вообще. В основе но-
вых направлений работ в этой области выдвигаются требования
по снижению энергозатрат и решению проблемы удаления воз-
духа.
Любые изменения технологии сушки должны быть связаны с
использованием новых методов радиационного нагрева, поскольку
существующие методы конвективного нагрева весьма близки к
пределу возможностей.
В настоящее время радиационные методы нашли применение
в различных областях; для сушки плоских поверхностей, напри-
мер, стен, листового материала, тканей, бумаги и пластика. По-
видимому, эти методы могут быть использованы в самом общем
виде для окраски металла. К числу новых методов относятся элек-
тронно-лучевая сушка, ультрафиолетовое облучение, индукцион-
ный нагрев.
Пока еще не ясно, какой именно метод окажется наиболее
приемлемым. Преимущество электронно-лучевого и УФ-методов
заключается в том, что они наиболее пригодны для сушки деталей
из пластмасс, применение которых в автомобилестроении непре-
рывно возрастает из-за снижения веса корпуса. Инфракрасный
нагрев имеет преимущество по причине сокращения времени суш-
ки, уменьшения длины конвейерной линии, минимального загряз-
нения и гибкости технологического процесса (например возмож-
ности использования в сочетании с конвективным обогревом).
Ниже приведены типичные режимы печной сушки:
Грунтовки, наносимые окунанием 30 мин 160 °C при 150 °C; 20 мин при
Грунтовки, наносимые электро- осаждением: анодного типа 20 мин при 165 °C; 20 МИН при
катодного типа 175 °C 20 мин при 165 °C; 20 МИН при
Шпатлевки 180 °C 30 мин при 150 °C; 20 мин при
Покрывные слои: алкиды, термореактивные ак- рилаты тер мо пл астичные акрил атные лаки Ремонтные покрытия: термореактивные эмали с катализатором 165 °C 20 мин 30 мин 30 мин при при при 130 °C 155 °C 90 °C; 10 мин при
двухупаковочные эмали термопластичные самовосстапа вливающиеся 100 °C 15 мин при 80 °C минимальная сушка 15 мин при
80 °C
Примечание: Приведены температуры металлической подложки, а не
температуры воздуха.
9.12. ВНЕШНИЙ ВИД И МЕТОДЫ ЕГО ОЦЕНКИ
Требования к качеству, предъявляемые автомобильной про-
мышленностью, наиболее высоки. Методы испытания систем по-
крытия в целом и его отдельных «слоев» разрабатываются таким
образом, чтобы воспроизвести условия, которые встречаются на
практике, и чтобы иметь представление о качестве покрытия в
натурных условиях. Очевидно, методы испытания нижних слоев
покрытия должны существенно отличаться от методов испытания
покрывных слоев; однако, испытания всей системы покрытия пред-
ставляют такой же, если не еще больший интерес.
Стандарты качества покрытия за последние годы значительно
возросли. Например, если ранее стандартное испытание грунтовок
в течение 10 дней (240 ч) в камере солевого тумана считалось
вполне достаточным, то теперь время испытания увеличено до
20 дней (480 ч). В соответствии с существующим стандартом испы-
тания стойкости покрытия по внешнему виду ранее проводились
при экспозиции во Флориде в течение 12 мес, однако для современ-
ных систем, включающих основной и прозрачные слои, этот срок
увеличен до 3—5 лет. Сейчас особое внимание уделяется опреде-
лению химической стойкости, например к действию кислоты.
Объект испытания и его стойкость составляют единую комп-
лексную систему, которая рассмотрена в гл. 16. Однако основные
свойства, показатели качества и методы испытания автомобильных
покрытий обобщены и сопоставлены ниже. Оценке подлежат внеш-
ний вид и защитные свойства.
9.12.1. Внешний вид
Учитываемые показатели: цвет; прозрачность; гладкость, т. е.
отсутствие дефектов в виде кратеров и апельсиновой корки; блеск
и индивидуальное впечатление (важно для привлечения потре-
бителя) .Примечание: Помимо состава и качества покрывного
слоя, на внешний вид покрытия в целом существенное влияние
оказывает система нижележащих слоев.
9.12.2. Защитные свойства
Физические свойства. Учитываемые показатели: твердость;
гибкость; стойкость к удару; адгезия; стойкость к ударам камней;
стойкость к растрескиванию на холоде, т. е. устойчивость к дей-
ствию экстремальной температуры и влажности.
Химическая стойкость: стойкость к действию бензина, кислот,
щелочей, воды, влажности, сопротивление коррозии и подпленоч-
НОЙ. коррозии; стойкость к действию факторов _вне.шлей среды,
т. е. ультрафиолетовому излучению и влажности.
9.12.3. Методы испытаний
Типичные методы испытаний и показатели качества представ-
лены в табл. 9.4.
Ниже приводится краткое описание некоторых видов типовых
испытаний.
Различные фирмы используют свои собственные методы испы-
таний и делают упор на различные показатели качества. Эти ме-
тоды применяются для испытания систем покрытий, нанесенных
по полной схеме, либо, если необходимо, только для испытания
грунтовок.
1. Степень отверждения (испытание для отверждае-
мых материалов). 20 двойных протираний чистой белой тканью,
смоченной метилизобутилкетоном (МИБК). «Выдерживает» озна-
чает отсутствие смывания или нарушения пленки краски.
2. Адгезия: крестообразный надрез (на расстоянии 1,5 или
2,0 мм). Испытание образцов покрытий проводится методом над-
реза до и после выдержки в воде (см. п. 9 ниже) в течение 120,
240 и 480 ч. «Выдерживает» означает отсутствие отслаивания
или его наличие не более 5%.
3. Твердость («Такой», вдавливание на приборе Такона;
твердометр Кнупа). Определение твердости по Такону представ-
ляет общепринятый метод в автомобилестроении, особенно при
испытании покровных слоев. Могут использоваться другие методы
определения твердости: карандашный твердомер, роликовый нож,
маятниковый прибор (Кениг, Персов и др.).
Стойкость к действию ударов к а м н я м и. Имеется
множество приборов, но самым простым и эффективным являет-
Таблица 9.4. Типичные показатели качества различных систем покрытий
Испытание Алкндно- меламиновые Термореактивные акрилатные неводные днсперсни Термопла- стичные акрилатные лаки Основной И прозрачный слон с металлическим оттенком
Блеск (20°) 85% 85% 85% 90%
Твердость (метод «Та- кон — Кнуп») 5—7 8—14 14—18 8—12
Адгезия (крестообраз- Отслоение Отслоение Отслоение Отслоение
ный надрез) <5% <5% <5% <5%
Бензостойкость (бен- зин фирмы Шелл — медленное капание на поверхность) Превосход- ная Очень хоро- шая Прекрасная Очень хорошая
Кислотостойкость (раствор Ih-HoSO^; 48 ч) Выдержи- вает Выдерживает Выдержи- вает Выдерживает
Деформация поверх- ности — — 55—65 °C —
Стойкость к удару камнями Превосход- ная Очень хорошая Хорошая Превосходная
Стойкость при ударе Выдержи- вает Выдерживает Выдержи- вает (в за- висимости от цвета) Выдерживает
Водопроницаемость (40 °C) Без пузы- рей Без пузырей Без пузы- рей Без пузырей
> 240ч > 240 ч > 240 ч > 240 ч
Влагостойкость (100% отн. влажн. Без пузы- рей Без пузырей Без пузы- рей Без пузырей
40 °C) > 96 ч > 96 ч >96 ч > 96 ч
Коррозионная стой- кость (солевая каме- ра) > 480 ч >480 ч > 480 ч > 480 ч
Стойкость к подпле- ночной коррозии Выдержи- вает Выдерживает Выдержи- вает Выдерживает
Испытание во Флори- де: блеск при 20° (после промывки) че- рез 2 года 70% 65% 75% 80%
Грунтовка Катодная Катодная Катодная Катодная
электроок- раска электроокраска электроок- раска электроокраска
Шпатлевка Эпокси- эфирная или поли- эфирная Полиэфирная Эпоксиэфир- ная с высо- ким ОКП (~55%) Полиэфирная
Обработка поверхно- сти металла перед окраской Фосфат цинка Фосфат цинка Фосфат цинка Фосфат цинка
ся следующий метод: 100 штук гаек Витворта диаметром 1 /4 дюйма
высыпают через трубку длиной 15 футов (457 см) и диаметром
2,5 дюйма (6 см) на окрашенную пластину, установленную под
углом 45°. Результат оценивается по степени разрушения пленки
краски. «Выдерживает» означает разрушение не более 5%.
Стойкость при ударе. Г руз 2 фунта (1 кг) сбрасывается
с высоты 10 дюймов (25 см) и 20 дюймов (50 см) на окрашенную
пластину (удар с обратной стороны). «Выдерживает» означает
отсутствие разрушения пленки краски.
Кислотостойкост ь. Окрашенную пластину погружают
на 96 ч при комнатной температуре в 0,1 н. серную кислоту. «Вы-
держивает» означает, что покрытие сохранилось без изменений.
Щелочестойкост ь. Окрашенную пластину погружают на
240 ч при комнатной температуре в 0,1 н. раствор едкого натра.
«Выдерживает» означает, что покрытие сохранилось без изме-
нений.
Примечание: испытания по последним двум пунктам до-
полняют испытания, приведенные в табл. 9.4.
Влагостойкость (длительное испытание). Пластины по-
мещают в камеру (100% отн. влажн., 40 °C). «Выдерживает»
означает-огсу,11сгвтге~1тотертщадт’езщщ пузырей, изменения цвета
после испытания не менее 96 ч.
Водостойкость при полном погружении (дли-
тельное испытание). Пластины погружают в ванну с перемешивае-
мой деминерализованной водой при 40 °C. «Выдерживает» озна-
чает отсутствие потери адгезии, пузырей, изменения цвета после,
минимум, 240 ч. В отдельных случаях наблюдение проводят че-
рез 500 ч.
Камера солевого тумана ASTM В117 (5% раствор
соли при 35 °C). Испытываемые пластины помещают в камеру
при описанных выше условиях. Перед испытанием на пластинах
делают Х-образный надрез. Осмотр пластин проводится через
240, 480, 840 ч (свыше 240 ч обычно проводится испытание толь-
ко для грунтовок, наносимых методом катодного электроосажде-
ния) . «Выдерживает» означает появление коррозии не более, чем
в 2 мм от надреза. Этот метод является главным испытанием для
грунтовок и проводится либо только на загрунтованных пласти-
нах, либо на пластинах, окрашенных по полной схеме.
Стойкость к коррозии при отслаивании по-
крытия. Этот метод предназначен для воспроизведения условий
коррозионного воздействия в местах отслаивания после испыта-
ния падающими осколками камней и погружения в воду.
Вначале окрашенную по полной схеме пластину погружают в
деминерализованную воду при 40 °C. Затем пластину подвергают
испытанию падающими камнями и далее испытанию в солевой
камере ASTM В117. Кроме того, в дальнейшем может быть прове-
дено испытание в атмосферных условиях. «Выдерживает» означа-
ет отсутствие коррозии в месте отслаивания.
Испытания (на стенде) во Флориде (5° южной
широты). Флорида — район, который подавляющее большинство
автомобилестроителей предпочитает для стендовых испытаний
автоэмалей. Он характеризуется высокой влажностью в сочетании
с высокой интенсивностью ультрафиолетового излучения; это со-
здает особенно жесткий режим для испытания отделочных слоев
покрытий, стабильности цвета пигментов.
Хотя эти испытания можно рассматривать как хороший абсо-
лютный метод определения качества покрытий, тем не менее всегда
необходимо учитывать ряд факторов. Оценка стойкости покрытия
часто зависит от времени года, когда начинается испытание, и
от конкретных погодных условий в течение года. Другой источник
погрешности при оценке качества, проявляющийся в форме обра-
зования микропузырей, часто бывает связан с ростом грибов (пле-
сени) в виде маленьких углублений и идущих от них нитей или
полос. Следовательно, испытание обычно дает «относительные»
результаты по отношению к известным стандартным показателям,
включенным в какую-либо программу испытаний, что позволяет
избежать ошибочной оценки результатов. «Выдерживает» подра-
зумевает то, что указано в табл. 9.4.
Показателем качества служат хороший блеск и сохранность
цвета, отсутствие разрушений после 2 лет (например, для алки-
дов, термоотвержденных акрилатных эмалей и лаков). Системы
покрытий, включающие основной и прозрачный слои, выдержи-
вают испытания во Флориде до 5 лет.
Ускоренные испытания. При разработке рецептур но-
вых красок необходимо уже на первых этапах работы иметь пред-
ставление относительно атмосферостойкости покрытий, поэтому
нет времени ждать 2—3 года для окончания испытаний на стенде
во Флориде, чтобы получить эти сведения. В связи с этим были
предприняты усилия для разработки методики ускоренных испы-
таний защитных свойств покрытий.
Имеются различные аппараты, в которых покрытие подверга-
ется совместному воздействию УФ-излучения, высокой влажности
и температуры. Ни один из этих методов не позволяет предсказать
результаты испытаний во Флориде, поскольку очень трудно искус-
ственным путем воспроизвести спектр УФ-излучения, идентичный
естественному солнечному освещению. Тем не менее, если покры-
тие выдерживает в течение 2000 ч один или несколько циклов уско-
ренных испытаний, это позволяет достаточно уверенно предска-
зать стойкость покрытия в условиях Флориды.
Типичные аппараты поставляются фирмой «Atlas» (источ-
ник УФ-излучения — лампы с угольной нитью или ксеноновые
лампы), фирмой «Q-Panel» Со. (аппарат УФ-излучения с ксеноно-
вой лампой QUV), Ксенотест (ксеноновая лампа). Испытания
атмосферостойкости покрытий при повышенной солнечной радиа-
ции проводятся на испытательной станции в Аризоне (циклы
EMMA и EMMAQUA) путем концентрирования естественного
солнечного света с помощью системы зеркал.
9.13. ПЕРСПЕКТИВЫ РАЗВИТИЯ
Перспективы дальнейшего развития технологии окраски авто-
мобилей неразрывно связаны с вопросами экономики, энерго-
затрат, с экологическими проблемами и требованиями потреби-
телей. При этом основные усилия должны быть направлены
на улучшение защитных свойств и внешнего вида. Существуют
различные пути решения этих вопросов, каждый из которых имеет
свои достоинства и недостатки; к сожалению, компромиссное
решение, удовлетворяющее всем противоречивым требованиям,
практически невозможно.
С точки зрения экологии был достигнут определенный про-
гресс в отношении выбросов в атмосферу отработанного воздуха
из печей. Как было сказано ранее, в настоящее время для этой
цели разработаны механические, термические и химические ме-
тоды очистки, к которым относится сжигание газов, применение
скрубберов. и-фильтров с-шсг-ивирх^в-ан-ны-м углем.
Однако, все еще остается огромное количество загрязненного
растворителями воздуха, выделяющегося из окрасочных камер, и
потому требуется большой расход энергии при окраске (на прак-
тике 50% эйергии, потребляемой в окрасочной камере, расходу-
ется только на перемещение и нагревание воды и воздуха). Во-
прос о снижении температуры при отверждении покрытий также
представляет собой проблему, заслуживающую серьезного внима-
ния. Особое значение эта проблема приобретает в связи с увели-
чением объема применения пластмасс в автомобилестроении.
В экономическом аспекте продолжается рост стоимости орга-
нических растворителей одновременно с ростом цен на раститель-
ные масла. Если принять во внимание, что краски, наносимые
распылением, имеют сухой остаток около 12%, то, несмотря на
многие достоинства этих материалов, их применение становится
неприемлемым не только по экологическим, но и по экономическим
соображениям.
Существуют три направления разработки новых материалов
с пониженным содержанием растворителей, либо не содержащих
растворителей: покрытия с высоким содержанием сухого остатка,
порошковые покрытия и водоразбавляемые материалы. Ниже
приводится обсуждение этих направлений.
9.13.1. Покрытия с высоким содержанием сухого остатка
Преимущества данных материалов по сравнению с существую-
щими, а также возникающие при их использовании проблемы
можно обобщить в следующем виде:
Достоинства Проблемы и недостатки
Допускают применение суще- Труднее получить покрытия с такими же
|ствующей технологии окраски свойствами
Допускают применение суще-
ствующего оборудования для
нанесения
Имеют одинаковые параметры
процесса
Труднее обеспечить необходимый внешний вид
покрытий с металлическим оттенком
Затруднен контроль реологических свойств.
Необходимо использовать оборудование для
автоматического распыления с высоким расхо-
дом материала, обеспечивающим экономич-
ность
Содержание сухого вещества: для пигментирован-
ных материалов — до 70%, для покрытий с металлическим оттен-
ком (основной слой) —20—40%, прозрачный слой — до 55%.
Тип связующих: алкидные, полиэфирные, акрилатные.
Тип материалов: грунтовки, шпатлевки, противокорро-
зионные материалы, отделочные покрытия с интенсивным цветом
и с металлическим оттенком.
Очевидные преимущества заключаются в том, что для нанесе-
ния материалов с высоким сухим остатком может быть использо-
вана та же самая технология, что и для материалов обычного
типа. Некоторые недостатки этих материалов обусловлены тем, чго
в их составе необходимо использовать смолы с меньшей молеку-
лярной массой и вязкостью. Это приводит к повышению текуче-
сти эмали при нанесении и увеличивает опасность образования
подтеков. Отсюда следует, что необходимо строго контролиро-
вать реологические свойства материалов, поскольку от этих
свойств зависит нормальное, качественное нанесение покрытия.
Кроме того, при меньшей молекулярной массе смолы должны
обладать более высокой реакционной способностью, так как толь-
ко при этих условиях можно получить покрытие с требуемыми
физическими и химическими свойствами.
Изменение и регулирование реологических свойств материалов
может быть достигнуто путем применения микрогелей. Микро-
гели, по сути дела, представляют собой органические наполни-
тели и вводятся в очень малом количестве; они также способны
образовывать сшитые структуры, и после завершения процесса
сушки эти наполнители могут входить в состав структуры пленки.
Покрытия с металлическим оттенком, включающие основной и
прозрачные слои, представляют собой пример того, какие превос-
ходные результаты может дать применение микрогелей в компози-
циях с высоким сухим остатком. Применяемые в настоящее время
в Европе композиции имеют очень низкий сухой остаток (12%),
что обеспечивает требуемый уровень ориентации металлических
чешуек относительно поверхности подложки. Это обусловливает
необходимую привлекательность внешнего вида покрытия.
Увеличение содержания сухого вещества в краске снижает
усадку при формировании пленки, что ухудшает цветовые характе-
ристики покрытия. Однако введение небольшого количества соот-
ветствующего микрогеля устраняет возможность какой-либо пере-
ориентации частиц алюминиевой пудры после нанесения, и поэто-
му уменьшается влияние эффекта всплывания на внешний вид
покрытия. Таким образом, содержание сухого вещества может
быть увеличено до 20—40% без ухудшения эстетических качеств
покрытия.
9.13.2. Покрытия со сверхвысоким содержанием сухого вещества
К таким покрытиям относятся материалы, содержащие более
70% сухого вещества. Однако для их широкого внедрения суще-
ствуют некоторые препятствия. Например, не может быть и речи
о покрытиях с металлическим оттенком, поскольку ухудшение их
декоративных свойств настолько заметно, что такие покрытия
совершенно неприемлемы.
Пигментированные материалы интенсивных цветов должны
использоваться в виде двухкомпонентных композиций, в состав
которых могут вводиться реакционноспособные мономеры или ак-
тиваторы с высоким содержанием сухого вещества, ввиду ограни-
ченной жизнеспособности композиций. На предприятиях, занятых
выпуском массовой продукции, при применении таких материалов
используется оборудование для нанесения двухкомпонентных кра-
сок со сложной системой дозировки. В разработке оборудования
для нанесения наблюдается определенный прогресс, однако соот-
ветствующий рост цен и сложность применения значительно огра-
ничивают использование этих материалов, в результате чего в на-
стоящее время интерес к ним невелик.
9.13.3. Водоразбавляемые материалы
Преимущества
Экологически безопасны
Дешевый и доступный разбави-
тель
Допускают применение обычной
технологии изготовления
Допускают применение обычной
аппаратуры для нанесения
Имеют одинаковые параметры
процесса
Проблемы и недостатки
Выделение паров воды при печной сушке
Необходимость более жесткого контроля тем-
пературы и влажности при нанесении
Повышенная текучесть; необходимость конт-
роля реологических свойств
Ограниченный выбор связующих (стабиль-
ность)
Склонность к образованию дефектов на по-
верхности вследствие загрязнения
Предел содержания сухого вещества не имеет никаких ограни-
чений с точки зрения экологии.
Тип смолы: алкиды, полиэфиры, эпоксиды, акрилаты.
Тип материала: грунтовки, шпатлевки, промежуточные
слои, покрытия, стойкие к удару, покрытия интенсивных цветов
и с металлическим оттенком
Применение воды в качестве растворителя для органических
покрытий всегда рассматривалось как весьма заманчивая цель.
Однако имеются некоторые трудности, ограничивающие широкое
применение этих материалов, например образование кратеров при
печной сушке, повышенная текучесть и чувствительность к за-
грязнению маслоподобными веществами. Водоразбавляемые ком-
позиции были разработаны в первую очередь для подслоя, так
как отмеченные выше проблемы сами собой привели к необходи-
мости более тщательного выбора состава таких композиций и
оказалось, что получить отделочные материалы на водной основе
чрезвычайно трудно. Только использование окрасочных камер с
кондиционированным воздухом может сделать эту работу доста-
точно успешной, но высокая стоимость оборудования и обслужи-
вания ограничивает внедрение таких материалов.
Более ранние разработки основывались на водорастворимых
смолах и.водных пигментных пастах. Однако недавно за короткое
время был достигнут успех в области стабилизации частиц поли-
меров в воде. Оказалось, что дисперсии многих полимеров в воде
получать достаточно легко, причем некоторые из этих дисперсий
имеют необычные реологические характеристики. Позднее были
разработаны методы регулирования текучести водных полимерных
систем путем «структурирования».
Водные полимерные дисперсии позволяют более полно удалять
воду при печной сушке, расширяют ассортимент применяемых
смол и образуют более гладкие пленки вследствие более тонкого
измельчения частиц. Кроме того, по причине более высокой теку-
чести на покрытиях из водных дисперсий менее видны дефекты
поверхности, например кратеры от попадания грязи при окраске.
Несмотря на то, что на основе водных дисперсий разработано
значительное количество рецептур, до сих пор не удается полу-
чить вододисперсионные покрытия толщиной около 50 мкм (т. е.
толщиной покрывного слоя автомобильного покрытия с интенсив-
ным цветом). Однако водные дисперсии полимеров могут быть эф-
фективно использованы при разработке рецептур композиций для
основного слоя, поскольку его толщина должна быть в пределах
10—15 мкм. В этом случае общая толщина покрытия обеспечива-
ется за счет толщины прозрачного слоя (35—50 мкм).
Дальнейшее усовершенствование реологических свойств вод-
ных дисперсий может быть достигнуто путем более тонкого диспер-
гирования и хорошего распыления при нанесении основного слоя
обычным пистолетом. На водной основе могут быть разработаны
рецептуры основного слоя для покрытий с металлическим оттен-
ком с такой же яркостью цвета и набором оттенков, как лучшие
составы на основе растворителей с низким содержанием сухого
остатка (~ 12%), которые при эквивалентном количестве сухого
вещества содержат 55—60% растворителей. Свойства таких ос-
новных слоев гораздо меньше зависят от температуры и влаж-
ности, чем свойства других материалов. Они легко могут сочетать-
ся с композициями на основе органических растворителей. Однако
перед нанесением прозрачного слоя должна быть произведена
обдувка поверхности воздухом.
9.13.4. Порошковые покрытия и водные пасты
Преимущества порошковых
покрытий
100% содержания сухого
вещества (отсутствие рас-
творителей)
Высокая эффективность
при нанесении
Хорошие свойства пленки
(при большой толщине)
Проблемы и недостатки
Необходимость специального оборудования для из-
готовления
Необходимость специального оборудования для
нанесения
Ограниченный набор цветов по соображениям эко-
номики
Трудность подбора цвета
Металлические пигменты не просвечивают
Трудность перекраски
Необходимость большой толщины для достижения
надлежащего внешнего вида
Шпатлевки требуют тщательной шлифовки для до-
стижения надлежащего внешнего вида покрывного
слоя
Неэкономичность при окраске деталей сложной
формы
Более высокая температура для обеспечения розлива
и отверждения
Взрывоопасность пыли
Тип смол: эпоксидные, полиэфирные, акрилатные.
Существует множество проблем, связанных с порошковыми
покрытиями, поэтому считают, что возможности их применения
ограничены. Важнейшим преимуществом этих покрытий является
полное отсутствие загрязнения среды выделяющимися раствори-
телями. Однако трудно представить себе, что области их примене-
ния в ближайшее время могут быть расширены, если не считать
возможность применения грунтовки и однослойных покрытий в ма-
шиностроении. Недостаточно хороший внешний вид покрытий де-
лает проблематичным их применение в автомобилестроении, если
не считать возможности их использования в составе прозрачного
слоя для покрытий с металлическим оттенком. Однако и в этом
случае более высокая температура отверждения создает препят-
ствия для их внедрения.
Водные порошковые пасты позволяют решить неко-
торые проблемы нанесения порошковых покрытий, однако остают-
ся вопросы, связанные с высокими затратами энергии и средств,
трудностью восстановления покрытий и ухудшением внешнего ви-
да вследствие грубых размеров частиц порошка.
9.13.5. Основные слои с интенсивным цветом
Такие композиции могут быть разработаны либо на основе
систем с растворителями и высоким сухим остатком, либо на осно-
ве водных дисперсий. Они могут обеспечивать лучшую укрыви-
стость в тонкой пленке основного слоя и менее связаны требова-
ниями по степени глянца, поскольку главными критериями явля-
ются гладкость и свойства пленки. Однако не все пигменты можно
использовать без ухудшения качества пленки; в ряде случаев мо-
жет происходить растрескивание покрытия при большом наполне-
нии некоторыми пигментами.
Основное преимущество таких покрытий состоит в том, что по
внешнему виду они не отличаются от обычных покрытий с метал-
лическим оттенком, но позволяют использовать более широкий ас-
сортимент пигментов, которые в обычных покрытиях с металли-
ческим оттенком не могут применяться. Последнее в первую оче-
редь относится к пигментам, не содержащим свинца.
9.13.6. Прозрачные слои
Современные материалы для прозрачных слоев содержат около
40% (масс.) сухого вещества. Для снижения содержания органи-
ческих растворителей целесообразно применение низкомолекуляр-
ных смол в сочетании с соответствующим микрогелем для контро-
ля текучести. Может возникнуть потребность в разработке микро-
геля с определенными оптическими характеристиками, обеспечи-
вающими требуемый глянец и прозрачность для улучшения
внешнего вида покрытия.
Совершенствование водных дисперсий также может привести
к их использованию для разработки прозрачных слоев. Однако
необходимость строгого контроля температуры и влажности, а
также более высокая толщина прозрачного слоя на водной основе
создает значительное количество ограничений.
Порошковые краски и водные пасты также рассматривались
в качестве возможных материалов для прозрачных слоев, хотя их
недостатком является высокая температура отверждения'. Другой
разновидностью являются прозрачные слои на основе двухупако-
вочных материалов; достоинства и недостатки материалов этого
типа были рассмотрены выше.
9.13.7. Пигментирование
Наблюдается тенденция роста потребления чистых и ярких
тонов для покрытий с интенсивным цветом и металлическим оттен-
ком. По соображениям токсичности весьма желательно прекра-
тить использование свинцовых и хроматных пигментов. Это озна-
чает, что возникает определенная потребность в органических
пигментах, которые сочетали бы чистоту и яркость цвета хромат-
ных пигментов с высокой прочностью. Если для красок с интенсив-
ными цветами требуется высокая укрывистость, то для пигментов,
применяемых в покрытиях с металлическим оттенком, требуется
прозрачность.
Кроме того, новая очень сложная технология выдвигает воз-
росшие требования к пигментам и их функции в схеме покрытия.
Пигменты должны быть использованы в сочетании с другими свя-
зующими, для которых они первоначально не предназначались;
новые системы покрытий подвергаются воздействию таких условий
и нагрузок, которые трудно было предсказать несколько лет тому
назад.
9.13.8. Окраска пластмасс
Проблема окраски композитных корпусов, включающих раз-
личные металлы и пластики, была рассмотрена в разд. 9.9. Особые
требования к окраске подобных сборных конструкций следует
принимать во внимание при разработке новых систем покрытий,
поскольку применение пластмасс продолжает расти.
Основным фактором являются температурные ограничения, и
-нринциииальным решеиием-проблемы окраски различных пласт-
масс является разработка новых систем покрытий с низкой тем-
пературой отверждения.
9.13.9. Технология, оборудование и нанесение
покрытий электроосаждением
В технологии окраски методом распыления, который применя-
ется при нанесении шпатлевок и покрывных слоев, возросло
использование нового оборудования — робототехники и турбо-
эчектростатических (высокоскоростных) установок. Это обеспе-
чивает более высокую эффективность окраски и меньшее выделе-
ние растворителей. При внедрении таких методов окраски исполь-
зование ручного труда составляет 40—50%, тогда как при автома-
тическом распылении — 60—70%.
При нанесении грунтовок в общем объеме окрасочных работ
большую долю составляют электроосаждаемые покрытия. Метод
электроосаждения представляет собой чрезвычайно эффективный
метод окраски сложных изделий фактически без применения
растворителей и является наиболее эффективным и экономичным
из всех используемых методов. Его достоинства так велики, что
трудно предвидеть масштабы его внедрения.
Внедрение недавно разработанных толстослойных грунтовок,
наносимых методом электроосаждения, в автомобилестроении
открывает возможность снизить толщину последующих слоев
(шпатлевки и промежуточного слоя), наносимых распылением,
вследствие чего снижается объем выделяющихся органических
растворителей. Можно ожидать, что когда всю систему подслоя
можно будет наносить методом электроосаждения, органические
растворители будут полностью исключены из этой стадии окраски
корпусов автомобилей.
9.13.10. Заключение
Разработка материалов с высоким сухим остатком привела к
появлению композиций, содержащих микрогели для регулирова-
ния реологических характеристик, в результате чего была разра-
ботана новая технология нанесения систем покрытий, включаю-
щих основной слой с высоким сухим остатком и прозрачный слой.
В дальнейшем, по-видимому, будет отдано предпочтение системе
покрытий, включающей основной слой с интенсивным цветом и
прозрачный слой с использованием той же самой технологии
окраски. Эти материалы рассматривались как наиболее перспек-
тивные вплоть до широкого внедрения водоразбавляемых компо-
зиций.
Разработка водоразбавляемых материалов предопределила
возможность их применения для покрытий всех типов. Толсто-
слойные грунтовки, наносимые методом электроосаждения, по-
зволили снизить, а иногда и полностью исключить необходимость
нанесения подслоя, который сам также может быть водоразбав-
ляемым.
В области декоративных покрытий появление водных поли-
мерных дисперсий с контролируемой реологией уже дало ряд но-
вых материалов, например композиций на водной основе для
основного слоя системы покрытий с металлическим эффектом.
Для основного слоя интенсивного цвета пригодна та же техноло-
гия, которая применяется при нанесении прозрачных слоев по
основным слоям обоих типов.
Наиболее ясной перспективой разработок в рассматриваемой
области является создание покрытий с низкой загрязняемостью
в сочетании с новым оборудованием, например электроосажде-
нием и автоматическим высокоскоростным электростатическим
распылением. Такое сочетание сможет удовлетворить возросшие
требования в части снижения затрат энергии, экологии и необхо-
димости внедрения более экономичных и менее длительных про-
цессов.
Г л а в а 10
ЭМАЛИ ДЛЯ РЕМОНТНОЙ ОКРАСКИ АВТОМОБИЛЕЙ
А. Г. Моуби
10.1. ВВЕДЕНИЕ
Название «Эмали для ремонтной окраски автомобилей» отно-
сится к лакокрасочным материалам, которые применяются для
восстановления окраски автомобилей после их выпуска с завода.
В заводских условиях корпус автомобиля окрашивается как обыч-
ная металлоконструкция, а применяемые материалы могут от-
верждаться- при повышенной температуре (чаще всего 150—
160 °C). Но автомобиль, у которого кроме корпуса есть еще и ре-
зиновые шины, пластмассовые детали, обивка, запас, бензина в
баке, уже нельзя окрашивать эмалями, отверждающимися при
такой температуре. Следовательно, для ремонта должны быть
использованы материалы другого типа, и поставщики должны
стремиться -к тому, чтобы ремонтные эмали обеспечивали полу-
чение покрытий такого же качества, что и исходные (заводские)
лакокрасочные материалы.
Объем эмалей для ремонта, поставляемых на рынок, сопоста-
вим с объемом заводских эмалей. Во всем мире в 1984 г. было
произведено приблизительно 250 млн. л декоративных эмалей и
примерно такое же количество грунтовок, растворителей и вспо-
могательных материалов.
Коммерческие автомобили — грузовики и автобусы — обычно
окрашивают материалами, которые более близки к авторемонтным
эмалям, чем к материалам для заводской окраски автомобилей
или промышленным эмалям общего назначения. Объем производ-
ства эмалей для коммерческих автомобилей составляет 40% об-
щего производства ремонтных эмалей. Требования к качеству этих
эмалей обычно несколько ниже, чем к качеству эмалей для ремон-
та легковых автомобилей, хотя в ряде случаев выдвигаются
требования по стойкости к действию химических веществ и окру-
жающей атмосферы.
Ремонтные автоэмали применяются, главным образом, в сле-
дующих случаях:
— восстановление или перекраска после случайной аварии;
— исправление повреждений или дефектов при изготовлении
на заводе или после транспортировки перед продажей;
— перекраска по собственной инициативе владельца с целью
улучшения внешнего вида, либо изменения цвета.
В развитых странах преобладают первые две причины. Полная
перекраска связана, главным образом, с перепродажей подержан-
ных автомобилей, где новая краска может оказать влияние на
то, будет или не будет продан автомобиль. Необходимость изме-
нения цвета краски возникает очень редко. Подавляющая часть
работ по ремонту покрытия обусловлена необходимостью восста-
новления покрытия после аварии.
В развивающихся странах имеется тенденция к старению пар-
ка автомобилей, служба ремонта после аварий менее развита и
полная перекраска автомобилей представляет собой обычную
практику.
Даже в развитых странах заметны различия в культуре отно-
шения к автомобилю. Это проявляется в уровне требований к
внешнему виду автомобилей, что, в свою очередь, накладывает
определенный отпечаток на отрасль, занимающуюся ремонтом
покрытий. Различия в требованиях к качеству окраски автомоби-
лей, допустим, в Лиссабоне и Цюрихе, весьма существенны, и это
отражается на уровне сложности технологии ремонта и применяе-
мого оборудования.
Целью большинства авторемонтных предприятий является
восстановление внешнего вида автомобиля до того состояния, в
котором он сошел с конвейера.
В большинстве стран Запада авария автомобиля воспринима-
ется как удар по личности, оскорбление морали и образа жизни
владельца, поэтому восстановление автомобиля до «первоначаль-
ного» состояния, чрезвычайно существенно. При возвращении
автомобиля из ремонта единственным стандартом, по которому
владелец может оценить качество работы, является отделочный
слой покрытия. Каждый владелец прежде всего осматривает
окраску и оценивает работу по наличию дефектов окраски. Сохра-
нение цвета и аккуратность являются наиболее важными отличи-
тельными характеристиками ремонтных покрытий.
На практике при восстановлении окраски необходимо ориен-
тироваться на заводскую окраску и, следовательно, на тех по-
ставщиков, которые выпускают материалы для заводской окраски.
Однако, автоэмали часто выбирают исходя не из их ремонто-
способности, а только по внешнему виду покрытияшНоэтому по-
ставщики красок для ремонта, которые часто также являются
поставщиками автоэмалей для заводов, должны принимать во
внимание это обстоятельство.
Автомобильные эмали для ремонта применяются в самых раз-
нообразных условиях. Во многих развивающихся странах счита-
ется вполне нормальной перекраска автомобиля на открытом воз-
духе, часто прямо на улице. В противоположность этому многие
большие авторемонтные мастерские имеют для этой цели очень
сложное промышленное оборудование. Такие различия, которые
часто сочетаются с низким уровнем квалификации рабочих, вы-
двигают самые разнообразные требования при- разработке рецеп-
тур красок. Это вносит существенные отличия по сравнению
TcFокраской на заводах, где конвейерные линии обслуживаются
высококвалифицированными рабочими и окраска проводится в
строго контролируемых условиях. Главной особенностью прове-
дения работ в авторемонтных мастерских, от которой зависят
свойства авторемонтных эмалей, является различие между сушкой
на воздухе (при температуре окружающего воздуха) и печной
сушкой. При авторемонте температура печной сушки должна
быть такой, чтобы температура металла не превышала 80 °C,
хотя современные авторемонтные эмали обычно имеют оптималь-
ную температуру при печной сушке до 60 °C.
Отсутствие оборудования для печной сушки на авторемонт-
ных предприятиях обычно создает неблагоприятные условия при
окраске, которая часто проводится в условиях запыленности и
колебаний температуры и влажности. Для материалов воздушной
сушки, применяемых в таких условиях, существенно важно, чтобы
время высыхания «от пыли» было предельно малым, порядка не-
скольких минут. Это предполагает использование быстросохну-
щих лакокрасочных материалов физического высыхания. Исклю-
чается применение эмалей, высыхающих за счет окисления кисло-
родом воздуха, либо двухупаковочных материалов, медленно
отверждающихся при комнатной температуре (в противном слу-
чае они становятся нестабильными при хранении или имеют корот-
кий срок жизнеспособности). Однако использование таких мате-
риалов для ремонта возможно, если создать условия, в которых
исключено пылеобразование. В надлежащих условиях такие си-
стемы имеют большие преимущества перед быстросохнущими
материалами как в технологии нанесения, так и в отношении ка-
чества покрытия.
Вследствие этого, практически во всех развитых странах и в
.очень немногих развивающихся странах ремонтная окраска авто-
|мобилей производится в распылительных камерах, непосредствен-
но смонтированных с печами для низкотемпературной горячей
|сушки, либо сам автомобиль поступает в печь для сушки (комби-
нированная окрасочная камера—печь).
10.2. ПОКРЫВНЫЕ СИСТЕМЫ
10.2.1. Введение
Ограничения в отношении температуры исключают применение
|обычно используемых на заводах материалов и технологий и де-
лают непригодными для авторемонтных предприятий такие виды
материалов, как термоотверждаемые акрилаты, термопластичные
акрилаты со ступенчатым режимом сушки, алкидно-меламиновые
материалы высокотемпературной сушки.
Кроме того, следствием характерной структуры ремонтных
предприятий с большим количеством установок для мелкого ре-
монта является медленное усовершенствование технологии, и по-
этому все основные технологические приемы, применявшиеся в
промышленности на протяжении всей истории ее существования,
используются и в настоящее время.
Существуют две побудительные причины для изменения техно-
логии: необходимость увеличения производительности авторе-
монтных предприятий и требования многих поставщиков автомо-
билей относительно применения материалов, способных длитель-
но сохранять противокоррозионные свойства.
В результате появилась тенденция к применению материалов
с высоким сухим остатком и большей толщиной слоя, наносимых
при меньшем количестве слоев, обеспечивающих необходимый
блеск после нанесения, т. е. не требующих полирования. Перспек-
тивно также применение более прочных и стойких систем покры-
тий, в частности, акрилатных.
10.2.2. Нитроцеллюлозные эмали
На ранней стадии развития автомобилестроения почти все
эмали для заводских условий и для ремонта изготавливались на
нитроцеллюлозной основе. На конвейерных линиях нитроцеллю-
лозные эмали давно уже были заменены акрилатными и алкид-
ными эмалями.
В авторемонте нитроцеллюлозные эмали используются до на-
стоящего времени, хотя в большинстве развитых стран они посте-
пенно вытесняются с рынка. Основное преимущество материалов
физического высыхания типа нитроцеллюлозных эмалей (и термо-
пластичных акрилатов) в авторемонте заключается в высокой
скорости воздушной сушки, что в сочетании с легкостью полирова-
ния позволяет получить хорошие результаты в условиях слабо
оснащенных и запыленных авторемонтных мастерских. Дополни-
тельные достоинства этих материалов — легкость нанесения и воз-
можность перекраски через любые сроки. Основными недостатка-
ми нитроцеллюлозных эмалей для авторемонта являются понижен-
ная прочность, хрупкость, плохая устойчивость при УФ-облучении
К другим недостаткам относятся необходимость полирования
из-за низкого глянца, слабая сохранность глянца, низкий сухой
остаток, низкая толщина пленки, низкая стойкость к действию
растворителей (бензина).
Нитроцеллюлозные эмали как правило пластифицируют обыч-
ными пластификаторами, полимерными пластификаторами и не-
высыхающими алкидами.
10.2.3. Термопластичные акрилатные эмали (ТПА)
ТПА эмали для ремонта, в основном, имеют примерно те же
характеристики, что и нитроцеллюлозные, и за исключением тех
случаев, когда требуется более высокая прочность и стойкость
к УФ-излучению, считаются взаимозаменяемыми с нитроцеллю-
лозными эмалями.
Подобно нитроцеллюлозным эмалям, ТПА высыхают в резуль-
тате испарения растворителя. В отличие от заводских эмалей,
ремонтные эмали не подвергают печной сушки для достижения
лучшего розлива. В состав полимеров, составляющих основу ТПА
лаков, входит значительное количество метилметакрилата, сополи-
меризованного с различными другими акриловыми мономерами,
что обеспечивает сочетание требуемой твердости, гибкости и ад-
гезии; в состав лаков входят также низкомолекулярные пласти-
фикаторы.
Достоинства ТПА для ремонта заключаются в очень быстром
высыхании на воздухе, хорошей прочности, стойкости к УФ-излу-
чению, легкости очистки от загрязнений, хорошей полируемости,
возможности получения прекрасного металлического эффекта.
К недостаткам следует отнести плохую стойкость к растворителям
(.бюнзину).,.низкий сухой остаток,...малую толщину пленки, хруп-
кость, низкий глянец (необходимость полировки).
10.2.4. Алкидные материалы
Алкидные покрытия, используемые для ремонта, являются
быстросохнущими материалами с низким содержанием масел. Они
могут высыхать на воздухе при добавке обычных кобальтовых и
свинцовых сиккативов или применяться в виде двухкомпонент-
ных составов в сочетании с меламиновыми смолами или изо-
цианатами. При использовании меламиновых смол алкиды могут
высыхать при температуре металла 70—80 °C; добавка изоциана-
тов ускоряет отверждение при любой температуре, но обычно
«ускоренную сушку» проводят при 40-—60 °C.
Алкиды были первыми «эмалями» (в отличие от физически
высыхающих «лаков»), нашедшими применение для ремонта авто-
мобилей. Эти материалы имеют ряд технологических преиму-
ществ, да.кдф дающихся в более высоком сухом остатке эмалей и,
следовдтрльцо, большей толщине и глянце покрытия, что исключи-
ло необходимость трудоемкой ручной операции полирования.
Однако алкиды медленнее высыхают «от пыли», чем материалы
на основе нитроцеллюлозы или ТПА, и, следовательно, их можно
наносить, только при отсутствии пыли в окружающей среде. Их
внедрение' в авторемонте сопровождалось разработкой соответ-
ствующих распылительных камер.
Основным недостатком алкидных покрытий'является трудность
исправления дефектов, обусловленная тенденцией не полностью
отвержденных пленок к размягчению при нанесении следующего
слоя. В результате любую ошибку при окраске либо случайное
повреждение невозможно устранить до тех пор, пока пленка не
высохнет полностью. Для алкидов, высыхающих за счет окисления,
этот период составляет несколько дней, но пленки', сшйтыё за
счет добавки меламиновых смол или изоцианатов, можно пере-
крывать следующим слоем значительно быстрее.
В дополнение к сказанному следует отметить, что алкидно-
смоляные'системы не предназначаются для введения алюминий
вой пудры, т. ё. непригодны для получения покрытий с металли-
ческим оттенком.
Алкидные смолы при введении металлических пигментов'!нё
обеспечивают нужный эффект, поскольку ориентация частиц алю-
миния происходит слишком медленно. • ’ :
10.2.5. Акрилатные эмали
Акрилатные эмали представляют собой материалы на основе
сополимеров'алкидов и ТПА, являясь материалами"гибридного
типа, разработанными с целью увеличения толщины и глянца эма-
лей на основе ТПА при сохранении быстрого высыхания «от пыли»,
свойственного быстросохнущим покрытиям. Свойства этих мате-
риалов оказались промежуточными по отношению к обоим типам
эмалей. Как и обычные алкиды,..они .могут быть использованы
в виде двухкомпонентных составов, в которых вторым компонен-
том является изоцианат.
Это приводит к более интенсивной сшивке, Повышению твер-
дости и прочности, но несколько уменьшает в*ремя высыхания и
требует в связи с этим нанесения в условиях* отсутствия пыли.
’ d • . ’
10.2.6. Акрилуретэновые эмали ’
Недавно внедренный в практику авторемонта'очень-ценный
материал разработан на основе гидроксилсодержайтих-акрилатов,
применяемых исключительно в сочетаний- с‘изоцйййатами. Не-
смотря на сравнительно медленное высыхание «от пыл'й»', во всех
остальных отношениях акрил-уретановые эмали прево’сходят дру-
гие эмали, применяемые для авторемонта и, такиМ^бВ^Уазом, ис-
пользованные ранее материалы можно считатй'^ст'ё^ёв'ййми,' за
исключением узкоспециализированных областей’Ргф'ййёйеЙия.
Акрил-уретановые эмали особенно пригоднь! для’-ййтбрёмонта
благодаря многочисленным достоинствам: прочйийй^Фохран-
ности блеска, твердости, гибкости, легкости п'ИгМёкйй'ройания и
высокому глянцу. ' '' ' '
При использовании сравнительно низкомолекулярных акрило-
вых смол толщйна покрьлтия может быть'увеличена по сравне-
нию с обычной толщиной ремонтных покрытий; при этом реакция
сшивки изоцианатами протекает с такой скоростью, которая по-
зволяет использовать этй эмали в широком интервале температур,
требуемых на практике. Отвержденное с достаточной скоростью
происходит даже ниже 5 °C. ’ '
При применении ускорителей горячей сушки для оптимизации
процесса на авторемонтных предприятиях разработан типовой
режим отверждения акрил-уретановых эмалей, предусматриваю-
щий выдержку в течение 30—40 мин при температуре 80—100 °C,
при этом температура металла поднимается максимум до 60 °C.
10.2.7. Системы, включающие основной и прозрачный слои
Разработка покрытий с металлическим оттенком, включающих
основной и прозрачный слои, применяемых при окраске автомо-
билей на заводах (см. 9.7.4) неизбежно привела к необходимости
внедрения подобных материалов и в авторемонте, поскольку эти
материалы позволяют осуществлять перекраску с сохранением
соответствующего внешнего вида.
Основными требованиями, предъявляемыми к основному слою,
являются высок-ая-у-крьгв-и-етоеть и-хереш-ая адеезия-к-грунтовке и
к верхнему прозрачному слою. Требования глянца и прочности
обеспечиваются этим прозрачным слоем и, следовательно, не
предъявляются к основному слою. Обычные ремонтные системы
разработаны таким образом, чтобы покрытие имело глянец и проч-
ность, и не могут быть удовлетворительно использованы для
восстановления заводских покрытий.
С другой стороны, основной компонент пленкообразующего
основного слоя отверждается за счет испарения растворителя и
поэтому в равной мере пригоден и для ремонта.
После внедрения покрытий с металлическим оттенком была
разработана технология ремонта основного слоя, предусматри-
вающая применение обычных автоэмалей для этого слоя, модифи-
цированных полиэфирами, акриловыми смолами или нитроцеллю-
лозой. В состав многих ремонтных эмалей кроме металлического
пигмента входят полиэтиленовый воск или другие добавки,
предназначенные для регулирования всплывания алюминиевых
частиц.
Таким 'образом, автоэмали для основного слоя обычно исполь-
зуются для ремонта покрытия почти без модификации. Хотя неко-
торые автоэмали, применяемые для основного слоя, содержат в
своем составе меламиновую смолу, которая почти не влияет на
процесс отверждения в условиях ремонта, твердость, прочность
и междуслойная адгезия основного слоя под используемым при
ремонте акрилуретановым прозрачным слоем вполне достаточны.
В тех случаях, когда ремонтный основной слой не идентичен
заводскому, технология окраски, тем не менее, подобна той, ко-
торая применяется при нанесении обычной автоэмали для основ-
ного слоя на базе полиэфиров или акрилатных смол.
10.3. ЦВЕТ
10.3.1. Введение
Цвет является основной проблемой при ремонте покрытия.
Количество и разнообразие цветов окраски автомобилей возраста-
ло очень быстро, начиная с 70-х гг., когда цвет превратился в
весьма важный фактор при продаже автомобилей, фактор, кото-
рый был использован поставщиками при столкновении с чрезвы-
чайно требовательным рынком. Это особенно четко проявилось на
европейском рынке. Установлено, что в основных европейских
странах количество колеров в 1985 г. превысило 10 000, увеличив-
шись в 5 раз по сравнению с 1965 г.
Ассортимент колеров, которые потребовалось исправлять при
ремонте, расширялся за счет вариаций колеров на автостроитель-
ных заводах. Эти вариации могут быть вызваны как отклонениями
от стандарта по времени хранения и технологии изготовления
краски, состояния оборудования на конвейерной линии, так и раз-
личиями между красками, оборудованием или технологией, при-
меняемой на двух или более автозаводах, производящих автомо-
били, номинально окрашенные одним и тем же цветом.
Работники авторемонта ориентируются на цвет автомобиля,
поступающего на ремонт, а не на простое восстановление завод-
ской окраски.
10.3.2. Исправление колера в заводских условиях
и смешанные схемы колеровки
Подгонка цвета, нарушенного при заводской окраске, под
требуемый согласно спецификации колер представляет собой ти-
пичную и постоянно встречающуюся проблему.
Эта проблема будет существовать до тех пор, пока на заводах
применяется весь большой набор колеров, требуемых рынком.
В настоящее время становится все более очевидным непрак-
тичность такого подхода и более приемлемым представляется
способ подгонки цвета путем смешения различных колеров. Опе-
рация смешения колеров может выполняться как в заводских
условиях, так и непосредственно у потребителя.
Первоначально использование смешанных схем колеровки осу-
ществлялось лишь применительно к наименее часто встречаю-
щимся колерам. Однако теперь такие схемы почти полностью
охватили все кодеры, применяемые для устранения заводского
брака. Это произошло потому, что при огромном разнообразии
колеров и низкой стоимости материалов для устранения индиви-
дуального брака их производство становится мало рентабельным.
Колера для исправления заводского брака все еще составляют
заметную статью расходов в тех странах, где недостаточно разви-
ты рынки сбыта и где имеется большое количество малых авто-
ремонтных предприятий. То же относится к странам, где имеется
относительно малое количество поставщиков автомобилей, и по-
тому каждый индивидуальный колер заметно влияет на цену.
Самый большой рынок в мире — рынок США — также придержи-
.вается этих .позиций, хотя быстрый рост импорта автомобилей
возбудил интерес к применению смешанных схем, которые в на-
стоящее время почти полностью преобладают в Европе. Реальные
размеры рынка США позволяют поставщикам красок установить
экономически оправданные объемы поставки индивидуальных ко-
леров с учетом запаса, который комплектуется при выпуске гото-
вых автомобилей.
Опыт наиболее развитых стран подтверждает, что 700—
1000 наиболее распространенных колеров могут обеспечить менее
60—75% потребности, вследствие чего тенденция к применению
смешанных схем становится необходимой и оправданной.
10.3.3. Ремонтные смешанные схемы
- Типичная- ремонтнаясмешанттая- схема- основана на использо-
вании ряда однопигментных красок, тщательно контролируемых
(каждая партия) по оттенку и'красящей способности. Эти краски
смешивают в соответствии с инструкцией, составленной разра-
ботчиком краски,, с подгонкой под колер окраски автомобиля.
Инструкции обычно изготавливают в форме таблицы с указанием
количества (по весу, или объему) смешиваемых ингредиентов,
необходимых дляполучения определенного объема точно по-
добранного колера.
Оборудование для составления смешанной ремонтной компози-
ции состоит из мешалки для перемешивания всех компонентов
с целью получение однородной по цвету во всем объеме смеси
и приспособления для дозировки по весу или по объему. При ра-
боте используются таблицы с расшифровкой состава колеров.
Количрст^р,.пигментов, которые могут быть использованы в
смешанной схеме колеровки, обычно ограничено; в основном при-
меняется, J30—,40 пигментов, в том числе несколько составов с
рцзличэд^ррдаржанием алюминиевой пудры для покрытий с ме-
талличе^эд^рд-тецком. В некоторых составах количество пигмен-
тов мод^т.фыть.удвоено, чем достигается возможность колеровки
очень малцм,и;.дрбавками составов, например для колеровки свет-
лых цветрВ|.;|Хмпичный набор пигментов, включаемых в смешанные
схемы ре^рру^фй. окраски, а также смеси, имеющие максималь-
ную укрывистость, приведены ниже:
Цвет Пигмент Пример
Белый Двуокись титана ' SCM Tiona RH 472
Синий и,фталоцианин меди BASF Гелиоген толубой В 6975 F
Фталоцианин меди Hoechst Хостаперн голубой BFL
Индантрон , ICI Монолит голубой 3R
Калий ферро/феррицианд Манокс голубой 10.SF
Зеленый Хлорбромфталоцианин меди Хлорфталоцианин меди ICI Монастраль зеленый GY ICI Монастраль зеленый GN
Желтый Хромат свинца Хромат-сульфат свинца Флавантрон Тетрахлоризоиндолин Азометнновын комплекс меди Гидратированная окись же- леза (HI) Ciba Geigy Иргазин желтый 2 GTLN Bayer Байферрокс 3910
Красно- оранжевый Окись железа (III) Молибдат-хромат свинца Моноазонафтол AS Хинакридон Дибромантрон Нафтол AS Перилен Диметил хинакридон Производное тиоиндиго Транс-хинакридон линейный Диоксазин Bayer Байферрокс НО—160 BASF Сикомин L 3135S Hoechst Новоперн красный F 3 RK —70 Ciba Geigy Цинкуазиа красный Y ICI Монолит красный 2 Y Hoechst Хостаперн коричневый HFR Bayer R64I8/R 6436 Hoechst Хостаперн ярко-розо- вый Е Sandoz Сандорин Бордо 2 RL Ciba Geigy Цинкуазиа фиоле- товый RT-887-D Hoechst Хостаперн фиолетовый RL специальный
Темно-синий Газовая сажа/печная сажа Degussa Сажа специальная 10
Глубоко-чер- Газовая сажа/канальная са- Degussa Сажа специальная
ный жа FW 200
Алюминие- Тонкодисперсный Eckert 601
вый Среднедисперсный Грубодисперсный Тоуо 8160 Silberline 3166
Большую помощь при разработке смесей оказывает использо-
вание данных по спектрам отражения, обрабатываемых с по-
мощью компьютера. При сопоставлении на фотоколориметре
спектра отражения покрытия на автомобиле и смесевых схем,
компьютер выдает ряд возможных составов пигментных смесей,
из которых можно выбрать наиболее приемлемый вариант, ру-
ководствуясь определенными критериями, например пределами
отклонения цвета, стоимостью пигмента или коли-.чеСТвй1Л 'пигмен-
тов в смеси. При разработке рецептур покрытий с металлическим
оттенком компьютер все же не может дать удовлетворительных
результатов по той причине, что коэффициент отражения зависит
от угла отражения; но, тем не менее, компьютер чрезвычайно поле-
зен для ускорения времени подбора состава пигментной смеси
даже и в этом случае.
10.4. ПЕРСПЕКТИВЫ РАЗВИТИЯ
10.4.1. Отношение к технологии автомобилестроения
Основное различие между заводской и ремонтной окраской
обусловлено ограничением температуры сушки автомобиля в пол-
костью собранном виде при ремонте и, по-видимому, будет про-
должать оставаться основным фактором, разделяющим две тех-
нологии окраски, несмотря на стремление снизить температуру
печной сушки в условиях автозавода. Снижение температуры
сушки обусловлено, с одной стороны, стремлением уменьшить
энергозатраты, с другой — тенденцией к использованию пласт-
масс в конструкции корпуса. Лишь немногие пластмассы способ-
ны выдержать условия обычной печной сушки в автомобилестрое-
нии без разрушения. Следовательно, до тех пор, пока детали из
пластмасс не будут окрашиваться отдельно и высушиваться в
специальных печах низкотемпературной сушки (с большой вероят-
ностью нарушения колера), необходима разработка новой техно-
логии окраски и сушки автомобильных покрытий при низкой
температуре. Печная сушка при температуре порядка 100—
130 °C может быть применена без разрушения конструкционных
пластмасс однако и эга температура все еще достаточно высока
для авторемонта.
Как мы уже говорили, при внедрении схемы, включающей
основной и прозрачный слой, снижается различие в технологии
окраски на заводе и в авторемонте только применительно к про-
зрачному слою. По-видимому, в ближайшие несколько лет значи-
тельная доля колеров для новых машин как в случае интенсивных
тонов, так и в покрытиях с металлическим оттенком, будет про-
изводиться по технологии с применением основного и прозрач-
ного слоев. Возможно, только пастельные тона и белые краски бу-
дут производить по технологии обычных однослойных покрытий,
так что технология подбора колеров станет в автомобилестроении
и в ремонте очень близкой, если не одинаковой.
В действительности набор пигментов, применяемых разработ-
чиками автоэмалей для создания новых колеров, значительно
превосходит возможности смешанных схем ремонтных покрытий.
В результате колер ремонтной эмали часто получается несколько
отличным от оригинального цвета автомобиля. Удовлетворитель-
ный по качеству ремонт может потребовать перекраски значи-
тельной поверхности автомобиля; в крайнем случае может потре-
боваться полная перекраска автомобиля, поскольку имеющимися
пигментами не удается точно воспроизвести достаточно близкий
колер.
Поставщики ремонтных эмалей прилагают большие усилия
для разработки составов смесей всех колеров, которые можно
получить смешением, для распространения этих сведений и выдачи
рекомендаций ремонтным предприятиям по способам идентифи-
кации колеров.
Стремление к большему разнообразию цветов автомобилей,
что стало возможным после разделения защитных и декоративных
функций в схеме, состоящей из основного и прозрачного слоев,
может явиться стимулом к подбору колера на месте, т. е. при про-
ведении ремонта становится реальным получить колер, идентич-
ный заводскому. Имеются ограничения в отношении ряда пигмен-
тов, которые можно добавлять к смеси, чтобы достигнуть требуе-
мого оттенка; эти ограничения можно преодолеть при введении
пигментов, покрытых оболочкой из слюды. Тем не менее, не сле-
дует недооценивать общепринятый принцип использования сме-
шанных схем для окраски корпусов на заводе; по-видимому,
будут предприняты попытки дальнейшего расширения применения
ремонтных смешанных схем.
10.4.2. Новые системы связующих
Разработка акрилуретановых материалов привела в резуль-
тате к появлению связующих, наиболее отвечающих требованиям
авторемонта. Но у этих связующих имеется один крупный недоста-
ток, который препятствует фундаментальному изменению техноло-
гии окраски с использованием этих материалов. В то же время
очевидно, что дальнейшее их усовершенствование может привести
к получению покрытий с выдающимися характеристиками.
Основным недостатком этих материалов является присущая
изоцианатам токсичность. Существует постоянная опасность для
рабочих приобрести чувствительность к действию изоцианатов в
результате вдыхания паров (аэрозолей) при распылении и вслед-
ствие неаккуратной работы, либо дефектов оборудования. Воз-
действие изоцианатов даже при очень низкой концентрации может
вызвать астматические симптомы, и вследствие высокой чувстви-
тельности работники бывают вынуждены уйти из авторемонтной
промышленности.
Исследования в области связующих для ремонтных эмалей
направлены на разработку материалов такого же высокого ка-
чества, как акрилуретановые, но без изоцианатов.
Высокая гибкость и прекрасная атмосферостойкость акрилат-
ных систем дает основания полагать, что эмали нового поколения
для авторемонта будут создаваться на основе акрилатов.
В патентной литературе встречается ряд мадефиадрр., посвя-
щенных трехмерной сшивке гидроксилсодер.жащих акриловых
смол, однако трудно превзойти уретановые материалы, образо-
вание которых может протекать даже при очень низкой темпера-
туре. К числу запатентованных материалов сшитого типа отно-
сятся композиции на основе меламиновых смол, полиэпоксидов
и полиангидридов.
Все они должны быть пластифицированы путем введения
внутреннего или внешнего пластификатора.
Трудно предполагать, какой из этих материалов может вы-
теснить из области авторемонта применяющиеся сейчас акрилуре-
тановые эмали, и сможет ли вытеснить вообще.
Глава 11
ОКРАШИВАНИЕ СУДОВ
Р. Ламбурн
11.1. ВВЕДЕНИЕ
Окрашивание судов и хранилищ для нефти и газа ставит перед
технологом целый ряд труднопреодолимых проблем в области
защиты от коррозии. Полное или частичное погружение в соленую
воду, обрызгивание с последующим высушиванием ветрами явля-
ются крайне агрессивными условиями. Все это требует совершен-
ствования лакокрасочных покрытий, защищающих сталь, из кото-
рой выполнен корпус судна. В добавление к проблемам коррозии
следует отметить, что об астание подводной части судов морскими
организмами приводит к замедлению скорости движения судна
и, отсюда, к увеличению расхода топлива.
Гитлиц [1] приводит пример экономического ущерба от об-
растания в с-лучае эксплуатации очень крупных грузовых транс-
портов с рабочей скоростью 15 узлов. Расход топлива при этом
составляет около 170 тонн в день.
Допуская, что судно находится в море в течение 300 дней в
году, стоимость топлива достигает 4 млн. долл, (при цене 80 долл,
за тонну в 1981 г.). Автор указывает, что даже умеренное об-
растание может увеличить потребность в топливе на 30% с целью
поддержания оптимальной рабочей скорости, т. е. расходы могут
возрасти более чем на 1 млн. долл, в год. При постоянном увели-
чении стоимости топлива представляется очевидным, что наличие
эффективных компЬзиций, препятствующих обрастанию, составля-
ет главный интерес судовладельца. Следует отметить, что при от-
сутствии эффективного средства против обрастания на 1 м2 по-
верхности за 6 мес. может поселиться около 50 кг «ракушек и
водорослей» в зависимости от окружающих условий.
Обрастание также способствует разрушению защитного по-
крытия, что приводит к более быстрому появлению коррозии.
Судовладелец стремится избежать частых и дорогостоящих поста-
новок судов в сухие доки и, следовательно, он заинтересован в
лакокрасочных материалах с улучшенными противокоррозионны-
ми и противообрастающими свойствами.
Поскольку только часть корпуса судна, погруженная в воду,
подвергается обрастанию, важно ясно представлять особенности
поверхностей, подлежащих окрашиванию, так как для каждой из
поверхностей необходим свой тип краски. Зона между ватерли-
нией при загрузке судна балластом и ватерлинией при полной
загрузке судна известна как переменная ватерлиния. Иногда она
располагается ниже уровня воды, а иногда подвергается воз-
действию морской атмосферы. К тому же эта зона может подвер-
гаться разрушениям при трении о пирсы, причал и другие суда,
например о буксиры, несмотря на то, что для предотвращения
трения применяются кранцы.
Выше переменной ватерлинии находятся надводный борт и
надстройки.
Кроме того, существуют специальные краски для палуб, внут-
ренних помещений, двигателей и т. д. Для военно-морских судов
были разработаны вспучивающиеся покрытия, предотвращаю-
щие распространение пламени, однако в процессе старения или
последующего перекрашивания их эффективность снижается. Из
опыта, полученного во время фолклендской войны, сомнительно,
чтобы эти покрытия оказали какой-либо значительный эффект на
предотвращение пожара при прямом попадании ракеты. Однако
такие покрытия могут быть полезны для уменьшения распростра-
нения пламени от слабых источников возгорания.
Процесс окрашивания судов можно рассматривать в двух ас-
пектах: окрашивание во время сборки судна и перекрашивание
при эксплуатации. Трудности окрашивания судов в процессе их
эксплуатации возникают из-за ограниченных возможностей со-
здать благоприятные условия для окрашивания. Следовательно,
применяемые краски должны быть достаточно надежными, чтобы
их можно было наносить даже при неблагоприятных условиях,
например в случае недостаточно хорошо подготовленной поверх-
ности. Определенные зоны, такие как надводный борт и над-
стройка, могут часто перекрашиваться, причем последняя даже
во время нахождения судна в море.
В этой главе мы коротко рассмотрим требования, предъявляе-
мые к основным типам судовых красок. Однако сначала рассмот-
рим упрощенный механизм коррозии стали и методы ее устранения
или, по крайней мере, замедления.
11.2. КОРРОЗИЯ
о я •?. .
Коррозия — это электрохимический пронесу, основу которого
составляет реакция между металлическим железом, кислородом
и водой, приводящая к образованию гидратированного оксида
железа, или ржавчины.
Суммарная реакция выглядит следующим образом:
4Fe + 3O2+2H2O = 2Fe2O3-H2O. (11.1)
Общепринятый механизм данной реакции включает анодный и ка-
тодный процессы. На поверхности железа или стали при контакте
с водой образуются локализованные аноды и катоды, на которых
эти процессы происходят. Поток электронов, составляющих «кор-
розионный ток», проходит через металл и одновременно происхо-
Коррозионный ток
Морская вода
Рис. 11.1. Процесс коррозии
дит эквивалентный перенос заряда через воду или электролит
гидроксид-ионами (рис. 11.1). На электродах протекают следую-
щие процессы:
на аноде — образование ионов двухвалентного железа при
потере электронов:
4Fe-^4Fet++8e (11.2)
на катоде — образование гидроксид-ионов:
4Н2О+2О2+8ё-^8ОН-. (Hj3)
Первоначал,ьным продуктом окисления является, таким образом,
гидроксид железа(II):
4Fe+++8OH~ 4Fe(OH)2. (11.4)
В присутствии избытка кислорода гидроксид железа(II) окисля-
ется до гидратированного оксида железа(III), который и явля-
ется ржавчиной. Анодные и катодные области на поверхности
металла образуются вследствие неоднородности структуры этой
поверхности. Неоднородность может быть обусловлена целым ря-
дом факторов, включающих наличие границы раздела с зернами
примесей, наличие вмятин и микроскопических дефектов, которые
вызывают локальные градиенты концентраций электролита или
кислорода в растворе. Любой из этих факторов может послужить
причиной образования разности потенциалов между соседними
областями на поверхности металла, достаточной для возникно-
вения гальванического процесса.
При строительстве судов необходимо окрашивать сталь, на
поверхности которой имеется слой оксидов толщиной до 60 мкм,
образующийся при ее производстве. Сталь подвергается горячей
прокатке до требуемой толщины, причем эта операция выполня-
ется в температурной области 800—900 °C. Окисление стали про-
исходит при охлаждении. Образующийся оксидный слой называют
«прокатной окалиной» Окалина, если ее не удалить, может вы-
звать коррозию вследствие того, что образуется значительная
разность потенциалов между ней и неокрашенной сталью (около
300 мВ) при погружении стали в такой электролит, как морская
вода. При появлении трещины или разрыва, проходящего через
слой окалины до металла, образуется гальваническая ячейка и
начинается процесс коррозии. Ржавчина образуется у границы
анода, который находится в контакте с соленой водой, но не не-
посредственно на анодном участке. Таким образом, анод медлен-
но растворяется и происходит точечная коррозия. Катодный
участок не подвергается подобному разрушению.
В настоящее время окалина удаляется путем дробеструйной
обработки и очищенная сталь загрунтовывается еще до сборки
изделий, что не гарантирует полного предотвращения коррозии.
Как же ослабить или устранить коррозию? Возможны два
подхода: а) устранение или ингибирование электрохимических
процессов и б) удаление воды или кислорода из потенциального
очага коррозии.
Катодный процесс требует присутствия воды и кислорода на
поверхности металла, так что два фактора, указанные выше,
взаимосвязаны. На практике очень трудно предохранить поверх-
ность металла от контакта с водой и кислородом вследствие
того, что большинство связующих, входящих в состав красок, име-
ют весьма высокую проницаемость для этих веществ. К материа-
лам с низкой проницаемостью для воды и кислорода относятся, на-
пример, воски, что объясняется их кристаллической природой.
Низкой проницаемостью обладают также кристаллические поли-
меры, но их трудно ввести в состав композиций, высыхающих
на воздухе. Вообще же, традиционные судовые покрытия не пред-
отвращают процессы на катодных участках. Для того, чтобы
коррозия происходила на анодных участках, ионы железа должны
перейти в электролит. Последнее может быть подавлено исполь-
зованием лакокрасочных систем двух типов.
В системах первого типа в качестве пигмента применяется
металлический цинк в достаточно большом количестве, чтобы осу-
ществлялся контакт между частицами цинка и поверхностью
железа. Вследствие того, что цинк более электроотрицателен по
сравнению с железом, в гальванической паре, возникающей меж-
ду этими двумя металлами, железо является катодом, а цинк —
анодом. В результате этого подавляется разрушение железа, а
коррозии подвергается цинк.
В системы второго типа входят краски, содержащие ингиби-
рующие пигменты, такие, как свинцовый сурик, свинцовые белила
или оксид цинка. Эти пигменты используются вместе со связую-
щими на основе высыхающих масел, которые подвергаются авто-
окислению. При этом наряду с формированием сшитых пленок про-
исходит частичное расщепление триглицеридов высыхающего
масла с образованием продуктов окисления, например азелаино-
вой кислоты. Эта кислота реагирует с пигментами, образуя свин-
цовые или цинковые мыла. Мыла реагируют с поверхностной
оксидной пленкой на металле и предотвращают доступ воды,
приводящей к образованию ржавчины. Другая группа пигментов
характеризуется определенной растворимостью в воде. К ней отно-
сятся хроматы цинка, бария,стронция и свинца. В случае их при-
менения на стали образуется оксидная пленка, защитные свой-
ства которой усиливаются образующимися железо-хромо-оксид-
ными комплексами. Однако в связи с тем, что в последнее время
возросли экологические требования и учитывая токсичность этих
пигментов, проводятся работы с целью исключения их из всех кра-
сок. Для судов были использованы два других пути подавления
электрохимических процессов, вызывающих коррозию:
1. Применение «жертвенных» анодов. Здесь аноды помещают-
ся прямо на корпус судна и для этих целей используются менее
электроотрицательные, чем сталь корпуса, сплавы. Такие аноды не
красятся, а их назначение — подвергаться коррозии вместо стали.
При необходимости изношенные «аноды» заменяют на новые.
2. Применение регулируемого внешнего тока, подаваемого на
корпус, с целью подавления растворения ионов железа. Этот ме-
тод защиты от коррозии называется методом «наложенного» тока.
Однако в любом случае требуется окрашивание корпуса судна.
11.3. ПОДГОТОВКА ПОВЕРХНОСТИ
Для того чтобы получить лакокрасочные покрытия с удовлет-
ворительными эксплуатационными свойствами, необходимо уда-
лить прокатную окалину. Окалина не только непосредственно
участвует в процессе коррозии, но еще и отслаивается со време-
нем от поверхности стали й, следовательно, не может служить под-
ходящим субстратом для окрашивания. Таким образом, подготов-
ка поверхности особенно важна в тех случаях, когда стремятся
получить наилучший эффект от лакокрасочного покрытия.
Экономически эффективная поверхностная обработка сталей,
покрытых прокатной окалиной, была разработана только в начале
шестидесятых годов. До второй мировой войны, когда сборка су-
дов осуществлялась с помощью заклепочных соединений деталей
киля и каркаса, окалина обычно удалялась выдерживанием в
атмосферных условиях в течение года и более; стальные листы
подвергались атмосферным воздействиям и ржавели. Ржавчина
давала возможность счистить окалину проволочной щеткой и при
этом сама удалялась вместе с окалиной. После второй мировой
войны в связи с развитием производства сварных судов короткое
время, отводимое на строительство, не давало возможности при-
менить этот способ. Не так давно стал использоваться целый ряд
альтернативных методов, включая кислотное травление, пламен-
ную очистку и дробеструйную обработку. Последний из них и по
сей день является наиболее удовлетворительным и общеисполь-
зуемым способом подготовки поверхности.
Обычно применяют две разновидности способа: механическую
дробеметную и дробеструйную очистку. В первом случае в качестве
абразива широко используют стальную дробь или крупный песок,
которые после отделения от частиц окалины снова возвращают в
процессе очистки. Поскольку абразив грубее, чем пыль, в которую
превращается окалина, его можно отделить с помощью циклон-
ного сепаратора. Некоторое истирание абразивных частиц приво-
дит, тем не менее, к постепенному расходу абразива.
В машинах, осуществляющих данный способ, применяется
ротационная мешалка, которая выбрасывает частицы абразива на
поверхность с высокой скоростью.
При дробеструйной очистке используется сжатый воздух,
подающий абразив к очищаемой поверхности. Этот способ более
универсален, так как с его помощью можно счищать окалину
непосредственно на месте на собранных конструкциях, сварных
швах и т. д., но обладает недостатком, связанным с образованием
пыли. Модифицированный или вакуумноструйный способ позво-
ляет удалять абразив и отводить пыль с помощью создания ва-
куума вокруг чистящего сопла. Однако этот способ намного мед-
леннее и используется только на малых поверхностях в тех слу-
чаях, когда пылеобразование недопустимо или не может контроли-
роваться. После дробеструйной очистки стали очень важно как
можно быстрее нанести покрытие, чтобы предотвратить образова-
ние ржавчины на свежей поверхности. Для этой цели используют
грунтовки.
11.4. ГРУНТОВКИ, НАНОСИМЫЕ ПОСЛЕ ДРОБЕСТРУЙНОЙ
ОБРАБОТКИ ПОВЕРХНОСТИ
Грунтовки наносятся на поверхность методом распыления не-
посредственно после дробеструйной очистки. Они должны очень
быстро сохнуть (в пределах нескольких минут), чтобы в дальней-
шем не подвергаться повреждениям при последующих операциях
на корпусе судна. Грунтовки не должны оказывать отрицатель-
ного влияния на последующие производственные процессы. На-
пример, они не должны препятствовать сварочным работам. Необ-
ходимо также, чтобы свойства грунтовки обеспечивали возмож-
ность нанесения последующего покрытия.
Широко используют 4 типа грунтовок:
1. Одно- и двухупаковочные композиции на основе поливинил-
бутираля и фенольной смолы, отверждаемые кислотами.
2. Двухупаковочные эпоксидные композиции холодного от-
верждения, пигментированные красным оксидом железа и, иногда,
ингибирующими пигментами, например хроматами.
3. Двухупаковочные эпоксидные композиции холодного от-
верждения, пигментированные цинковой пылью.
4. Одно- и двухупаковочные цинк-силикатные композиции.
Жизнеспособность грунтовки для практического использова-
ния должна составлять примерно 8 ч. Эпоксидные смолы обычно
отверждаются полиамидами, такими как версамид 140 или 115.
В двухупаковочную цинк-силикатную композицию порошко-
образный цинк вводится при перемешивании непосредственно пе-
ред использованием.
Типичные рецептуры таких грунтовок, описанные Банфилдом
[2], приведены ниже:
Одноупаковочная фенольно-поливинилбутиральная грунтовка
Содержание, %
Поливинилбутираль (например, Mowital В60Н) 4,7
Phenodur PR263 (фенольная смола) 4,7
Красный оксид железа (синтетический) 8,6
Асбестин (силикат магния) 1,4
Фосфорная кислота (d= 1,7) 1,0
Мет илэтил кетон 30,0
Бутанол 19,6
Изопропанол 30,0
.3 100,0
Двухупаковочная эпоксидная грунтовка
Основа Содержание, %
Эпикот 1004 12,0
Фосфат цинка (BS 5193: 1975) 8,0
Красный оксид железа (синтетический) 10,0
Асбестин 1,5
Тальк (размер зерна 5 мкм) 15,3
Бентон 27 (препятствует оседанию пигмента) 0,4
Толуол 23,0
Изопропанол 11,5
Отвердитель
Версамид 140 (полиамид) 1,8
Толуол 11,0
Изопропанол 5,5
100,0
Двухупаковочная эпоксидная цинкнаполненная грунтовка
Основа Содержание. %
Цинковая пыль (Zincoli 620 или Durham Chemi- 80,0
cals «ультрафайн»)
Бентон 27 1 ,о
Оксид кальция 0,3
Эпикот 1001 4,0
Толуол 6,6
Изопропанол 3,3
Отвердитель
Версамид 115 1,8
Толуол 2,0
Изопропанол 1,0
100,0
Примечание: Оксид кальция поглощает воду, реагируя с влагой,
содержащейся в растворителях. В противном случае влага будет взаимо-
действовать с цинком, образуя водород.
Стилсиликатная цинкнаполненная грунтовка
Этилсиликат (Dynasil Н500)
Бентон 38 (препятствует осаждению пигмента)
Тальк (5 мкм)
Толуол
Изопропанол
Целлозольв
Цинковая пыль (Zincoli 615)
Содержание, %
20,0
1,4
4,0
5,3
5,3
4,0
60,0
100,0
Цинковая пыль смешивается с основой перед применением. Гото-
вый продукт имеет жизнеспособность около 8 часов.
Фенольно-поливинилбутиральные грунтовки используются наи-
более широко и на них можно наносить большинство материалов,
за исключением случаев воздействия на покрытия сильных рас-
творителей. Практически любая краска может наноситься по
грунтовкам на основе двухупаковочной эпоксидной композиции,
пигментированной красным оксидом железа или цинковой пылью.
Недостатком этих типов грунтовок, однако, является то, что они
двухупаковочные и более дорогостоящие, нежели грунтовки фе-
нольно-поливинилбутирального типа.
Цинк-силикатные грунтовки используются, главным образом,
в тех случаях, когда они могут быть перекрыты цинк-силикат-
ными покрывными материалами.
Грунтовки, содержащие металлический цинк, обеспечивают
самую лучшую общую защиту от коррозии, но они обладают рядом
практических недостатков, которые иногда вызывают необходи-
мость применения других типов грунтовок. Так, продукты корро-
зии цинка чувствительны к воде и до того', как поверхность покры-
вается другой краской, они должны быть удалены промывкой
водой из шланга под давлением. Другой проблемой, связанной
с цинксодержащими грунтовками, является образование паров
оксида цинка при сваривании покрытого ими металла. Это очень
часто вызывает недовольство сварщиков, хотя не установлено,
чтобы пары оксида цинка представляли угрозу для здоровья *.
Вдыхание паров цинка может вызвать так называемую «цинково-
паровую лихорадку». Это явление известно в гальванической
промышленности уже свыше ста лет.
11.$. СИСТЕМЫ ОКРАСКИ ДЛЯ СУДОВ
О защите листовой стали после ее производства уже кратко
говорилось выше. Последующее окрашивание судов во время и
* Последнее утверждение нельзя признать справедливым: установлено, что
оксид цинка вызывает как острые, так и хронические отравления. См. Вредные
химические вещества. Неорганические соединения элементов I—IV групп. Л.:
Химия, 1988, с. 146. П р и м. редактора.
после сборки представляет собой очень сложную технологию.
Как уже указывалось, различные поверхности нуждаются в
различной обработке. Для каждой поверхности необходима це-
лая лакокрасочная система, так как невозможно удовлетворить
все требования, предъявляемые к каждой отдельной поверхности,
с помощью только одной краски.
Таким образом, основное значение приобрели многослойные
системы, в которых каждый отдельный вид краски наносят в не-
сколько слоев до достижения толщины пленки, обеспечивающей
максимальное снижение скорости коррозии, протекающей, напри-
мер, по механизму питтинговой коррозии.
Типичные системы для окрашивания надводного борта и над-
строек, переменной ватерлинии и подводной части, являющиеся
традиционными материалами, приведены в табл. 11.1.
В последние годы большинство изменений, происходящих в
технологии окрашивания и разработке лакокрасочных рецептур,
были связаны с потребностями, вызванными развитием строитель-
_ства. супертанкеров. Интеноивное развитие супертанкеров нача-
лось после открытия Суэцкого канала в 1967 г. по окончании
ближневосточной войны. Супертанкеры потребовались для того,
чтобы сделать транспортировку нефти с Ближнего Востока эко-
номически йыгодной. Это было недостижимо при эксплуатации
обычных танкеров начала 50-х гг., имеющих брутто-тоннаж
около 30 тыс. Средний же супертанкер 80-х гг. имеет брутто-тон-
наж около 300 тыс., причем он управляется меньшим экипажем,
чем его предшественник. Это означает, что меньшее число людей
должно выполнять техническое обслуживание и ремонтную окрас-
ку гораздо более крупного судна. Общим итогом этих изменений
явилось требование к разработке таких красок, использование
которых позволило бы сократить время пребывания судна в сухом
доке. Для достижения этого необходимы краски с улучшенными
эксплуатационными свойствами, обеспечивающие и достаточную
толщину при их нанесении меньшим количеством слоев.
Таким образом, было разработано новое поколение красок,
использование которых позволило увеличить междоковый период
с 9—12 мес. до 24—30 мес. Но даже это не считается пределом
современной технологии и некоторые изготовители красок стре-
мятся увеличить «междоковый» период до 5 лет.
Эти требования были удовлетворены в результате разработ-
ки систем толстослойных покрытий, которые могут применяться
при толщине каждого слоя около 100 мкм. Данные материалы
как правило наносят методом безвоздушного распыления. Один
краскораспылитель установки безвоздушного распыления спо-
собен подать от 50 до 80 л краски за час, окрашивая при этом
150—400 м2 поверхности при требуемой толщине пленки.
Во избежание образования наплывов при окрашивании верти-
кальных поверхностей применяемые краски должны обладать
Таблица 11.1. Традиционные типы судовых красок
Поверхность Лакокрасочная система Тип связующего Число слоев Толщина сухой пленки, (мкм)
Подводная часть Г рунтовка Ф е н о л ф о р м альдегидная смола, модифицированная маслом; битум 3—4 40-60
Барьерный противокорро- зионный слой № 1 Битум/резинат кальция 1 50—60
Противообра- стаютее по- крытие Растворимая матрица на основе уплотненного льняного масла и резината кальция с медным или оловоорганическим биоцидом * 1 50—75
Переменная ватерлиния Грунтовка 655 Фенолформальдегидная смола, модифицированная тунговым и льняным маслом 2—3 40—60
11окрыиной слой Фенолформальдегидная смола, модифицированная тунговым мас- лом 2 40—60
Надводный борт и над- стройка Быстровысы- хающая грун- товка Алкидная смола средней «жирно- сти», модифицированная льняным маслом (55—60% масла) 2 50
Промежуточ- ный слой Алкидная смола средней «жирно- сти», модифицированная льняным маслом (50—60% масла) 1 50
Эмаль Алкидная смола высокой «жир- ности», модифицированная сое- вым маслом (65% масла) 1—2 35—50
* Существует 3 разновидности: «Атлантическая», «Тропическая» н «Свсрхтрони-
ческая», которые различаются выбором ядохимиката.
реологическими свойствами неньютоновской жидкости, и при этом
хорошей текучестью при перемешивании, что облегчает их распы-
ление. С другой стороны, они должны быстро структурироваться
в жидкой пленке. Так, например, они могут иметь вязкость поряд-
ка 0,5—1 Па-с при скорости сдвига 10 000 с_|, а при низкой ско-
рости сдвига порядка 10~2 с“', развиваемой под действием силы
тяжести на вертикальной поверхности, вязкость будет ~ 100 Па-с.
Это достигается введением в краски тиксотропных добавок или
загустителей, таких как монтмориллонитовые глины или поли-
амиды. Многие высокоэффективные системы являются двухупа-
ковочными.
Противокоррозионная защита, обеспечиваемая при примене-
нии этих систем, достигается в большей степени за счет подавле-
ния электрохимических процессов толстым барьерным слоем по-
крытия, и в меньшей степени за счет специфического действия
ингибирующих пигментов, хотя используемые грунтовки часто
содержат их. Токсичность свинца и хроматов требует их замены.
В связи с этим возрастает использование фосфата цинка.
11.5.1. Краски для надводного борта
и надстроек
Для удовлетворения требований, предъявляемых к окраске
надводного борта и надстроек, можно использовать существую-
щие промышленные лакокрасочные материалы, включая грунтов-
ки, краски для промежуточных и наружных слоев, которые высы-
хают при температуре окружающей среды.
Эти материалы долгое время выпускали и на масляносмоля-
ных, и алкидных связующих. Так, грунтовки как правило содержа-
ли масляносмоляное связующее (например фенолформальдегидную
смолу, модифицированную маслом) с ингибирующим пигментом,
-обычно свитщовым суриком. Промежуточный слой обычно изго-
тавливался на основе алкида средней «жирности» на льняном
масле. Верхний слой покрытия готовился на основе жирного
алкида, или алкида, модифицированного сс-метилстиролом. Пиг-
ментирование в последнем случае давало возможность достигнуть
нужной укрывистости и других декоративных свойств. Винилиро-
ванная алкидная смола обладает более высокой скоростью вы-
сыхания, но при нанесении последующего слоя необходимо
соблюдать осторожность, поскольку первоначальное высыхание
обусловлено испарением растворителя и может произойти вспу-
чивание, если последующий слой наносится после сушки в про-
межутке от 12 до 16 ч, особенно при низкой температуре
окружающего воздуха. Данный эффект объясняется относительно
низкой скоростью образования поперечных сшивок в связующем
этого типа. В современных высокоэффективных системах исполь-
зуется широкий ряд связующих, в том числе эпоксидные смолы,
полиуретаны и хлорированный каучук. Последний часто приме-
няется в смесях с акриловыми или алкидными смолами. Так,
например, типичная система эпоксидного покрытия для надвод-
ного борта обычно предполагает нанесение двух достаточно тол-
стых слоев грунтовки и промежуточного слоя и последующего
одного слоя эпоксидной эмали. Общая толщина сухой пленки при
этом составляет примерно 300 мкм. В случае надстройки, где тре-
буется обеспечение блеска покрытия, используют один слой грун-
товки, перекрываемой эпоксидным промежуточным слоем и поли-
уретановым верхним слоем. В этом случае толщина сухой пленки
составляет приблизительно 200 мкм.
Примеры рецептур для толстослойного эпоксидного покрытия
с красным оксидом железа и белой эпоксидной эмали приведены
ниже:
Состав толстослойного эпоксидного покрытия
на основе красного оксида железа
Основа Содержание, %
Синтетический красный оксид железа 5,0
Асбестин (силикат магния) 22,0
р-Кристобаллит (кремнезем), проходящий через сито 325 меш 18,0
Эпикот 1001 16,0
Бентон 27 2,0
Ксилол 14,0
Бутанол 5,0
Отвердитель
Версамид 115 (полиамид) 8,0
Целлозольв 10,0
100,0
Типичный состав белой эпоксидной эмали
Основа Содержание, %
Диоксид титана рутильной формы (например, Tioxide R-CR2) 30,0
Аэросил 380 (препятствует расслаиванию) 0,5
Эпикот 1001 23,0
Ароматические фракции нефти. Solvesso 100 (нафта) 16,5
Метилизобутилкетон 8,0
н-Бутанол 3,0
Отвердитель
Версамид (полиамид) 11,0
Катализатор (например Anchor К54) 1,0
Ксилол 7,0
100,0
Эпоксидные смолы могут сшиваться аминами, причем обычно
для этих целей используются диэтилентриамин и триэтилентетр-
амин. Часто осуществляют предварительное взаимодействие ами-
нов с эпоксидными смолами с образованием аддуктов, которые
имеют некоторые преимущества по сравнению с чистыми аминами.
Обычно аддукты получают с частью той эпоксидной смолы, кото-
рая используется в качестве пленкообразующего в краске. За
счет применения такого приема можно ликвидировать или осла-
бить неприятный запах свободного амина и добиться более удоб-
ного соотношения основа:отвердитель, равного примерно 2:1 или
3:1 вместо 10:1.
Двухупаковочные полиуретаны также используются в эмалях
для верхнего покрытия. В основу эмали входит гидроксилсодер-
жащая насыщенная полиэфирная смола (например Desmo-
phen 650); отвердителем служит раствор изоцианата (например
Desmodur N) в смеси ксилола и целлозольвацетата (1:1), при
концентрации 75%.
Полиуретаны используются в тех случаях, когда требуется
хороший и устойчивый блеск. Толстослойные высокоэффективные
покрытия также могут быть изготовлены на основе одноупаковоч-
ных композиций, в частности на хлоркаучуке или виниловых
смолах. Хлор каучуковые краски получили более широкое распро-
странение, чем виниловые, хотя последние были давно разрабо-
таны для использования в военно-морском флоте США и Канады.
Типичные составы грунтовки, промежуточного и верхнего слоев,
на основе хлоркаучука, приведены ниже:
Хлоркаучуковая грунтовка
Содержание, %
Невсплывающая алюминиевая пудра — 65% в бензиновом растворителе (Нафта) 20,0
Оксид цинка 1,0
Тиксатрол ST (тиксотропный агент) 0,5
Бланфикс 10,0
Аллопрен R10 (хлоркаучук) 15,0
Церехлор 70 (пластификатор на основе хлорированного парафина) 7,5
Церехлор 42 (пластификатор на основе хлорированного парафина) 7,5
Оксид пропилена (стабилизатор) 0,1
Ксилол -28,4
Solvesso 100 (Нафта) 10,0 100,0
Для приведенного выше состава требуется тщательный подбор
рецептуры и' контроль производства. В этом составе пластифика-
тором служит предварительно приготовленная смесь твердого
(воскообразного) хлорированного парафина с жидким хлориро-
ванным парафином. Тиксотропный агент (загуститель) основан
на гидрированном касторовом масле. Его тиксотропное действие
проявляется только при введении в процессе диспергирования,
когда температура поднимается выше 40 °C. Однако температура
не должна достигать 55 °C во избежание потери тиксотропного
эффекта. Алюминиевая пудра не перетирается с другими пигмен-
тами, а вводится после охлаждения замеса.
Оксид пропилена является стабилизатором краски при ее хра-
нении, который «поглощает» ионы С1—, медленно выделяющиеся
из хлоркаучука. Для повышения блеска покрытия в рецептуре
белого верхнего слоя используются пониженные количества на-
полнителя (по сравнению с промежуточным слоем), применяется
Бентон 38 вместо Тиксатрола ST и более высокое содержание
Церехлора 70. Однако получаемый блеск все равно ниже по срав-
нению с блеском на основе эпоксидных или полиуретановых по-
крытий.
Все эти краски наносят безвоздушным распылением. Типич-
ной системой является следующая:
Толщина сухой
пленки (мкм)
Грунтовка (один слой) 50
Промежуточный слой (два слоя) 200
Верхнее покрытие (один слой) 50
Всего: ”300
11.5.2. Краски для переменной ватерпинии
Для окрашивания переменной ватерлинии используются высо-
кокачественные композиции на основе тех же связующих, что и
в красках для надводного борта. Для этих целей хлоркаучуки
представляются наилучшими пленкообразователями вследствие
их исключительной межслойной адгезии. Это особенно важно,
если иметь в виду повреждения лакокрасочных покрытий в ре-
зультате истирания и ударов о кранцы и причалы, а также необ-
ходимость повторных окрашиваний.
При эксплуатации очень больших грузовых транспортов об-
растание также может быть важной проблемой при окраске зоны
переменной ватерлинии, и на практике обычно используют проти-
вообрастающую композицию в качестве верхнего слоя лакокра-
сочного покрытия.
При этом обычно применяют те же самые противообрастаю-
щие покрытия, что и для подводной части судов.
Хлоркаучуковое промежуточное покрытие (белое)
Содержание, %
Диоксид титана (например Tioxide R-CR2) 16,0
Бланфикс 14,0
Оксид цинка 1,0
Тиксатрол ST (тиксотропный агент) 1,0
Аллопрен R10 (хлоркаучук) 14,0
Церехлор 70 (хлорированный пластификатор) 7,0
Церехлор 42 (хлорированный пластификатор) 7,0
Оксид пропилена (стабилизатор) 0,1
Solvesso 100 (ароматическая фракция нефти) 10,0
Ксилол 29,9
100,0
Хлоркаучуковый порывный слой (белый)
Содержание, %
Диоксид титана (например Tioxide R-CR2) Бланфикс Оксид цинка Бентон 38 (тиксотропный агент) н-Бутанол (вызывает набухание) Аллопрен R10 Церехлор 70 Церехлор 42 Оксид пропилена Ксилол Solvesso 100 18,0 2,0 1,0 1,0 0,5 16,0 12,0 4,0 0,1 30,4 15,0 100,0
11.5.3. Краски для подводной части судов
Высокоэффективные толстослойные системы покрытий, описан-
ные применительно к надводному борту и переменной ватерлинии,
также используются и для окрашивания подводной части, однако,
поскольку эстетический аспект в данном случае не является глав-
ным, представляется возможным составлять более дешевые ком-
позиции, вводя в них каменноугольный пек, без ущерба для их
качества. Краски, полученные таким путем, обычно шоколадно-
коричневого или черного цвета, но это не имеет никакого значе-
ния. Содержание каменноугольного пека в эпоксидном толсто-
слойном покрытии можно варьировать в широких пределах. Чем
больше каменноугольного пека, тем ниже маслостойкость и
химическая стойкость, однако данный факт менее важен для под-
водных композиций. В рецептуре, удовлетворяющей требованиям
Министерства обороны Великобритании (ВМС) около 60—65%
общего количества связующего составляет каменноугольный пек.
Обычно выбирают каменноугольный пек в соответствии с совме-
стимостью е эпоксидной смолой, и хотя он считается инертным,
тем не менее может реагировать с эпоксидными группами вслед-
ствие содержания акаменноугольном пеке-фенольных гидроксиль-
ных групп. В связи с этим каменноугольный пек часто вводится
в отвердитель двухупаковочной композиции ибо, в противном
случае, снижается гарантийный срок хранения композиции.
*
Типичный состав двухупаковочной краски для подводной части судна
Основа Содержание, %
Бариты 20,0
Асбестин 20,0
Эпикот 1001 11,0
Бентон 27 1,0
Ксилол 10,0
Solvesso 100 5,0
н-Бутанол 3,0
Отвердитель
BSC Norsip 5 (75% раствор ароматического 25,0
каменноугольного пека в ксилоле)
Версамид 140 2,5
Synolide 968 0,3
Ксилол 2,2
100,0
После смешения компонентов краска наносится методом без-
воздушного распыления с образованием сухого покрытия толщи-
ной 125 мкм. Жизнеспособность при 15 °C составляет около 4 ч.
Скорость реакции удваивается при увеличении температуры на
10 °C и сокращается наполовину при аналогичном понижении
температуры. Таким образом, краска может иметь жизнеспособ-
ность в пределах от 8 ч при 5 °C до 2 ч при 25 °C. Скорость отверж-
дения также влияет на возможность нанесения последующих
слоев краски.
Для достижения удовлетворительной межслойной адгезии и во
избежание вспучивания точно определяются максимальное и ми-
нймальное время нанесения последующего слоя в зависимости от
температуры нанесения.
Данный тип композиции может быть использован как для
судов на стадии их сборки, так и при их ремонтной окраске.
В первом случае возникает проблема, заключающаяся в том,
что шротивообрастающее покрытие наносится непосредственно
перед сдачей судна в эксплуатацию, а это может произойти зна-
чительно позже оптимального времени, необходимого для нане-
сения наружного слоя по эпоксидной краске с каменноугольным
пеком. Тогда используют так называемое «адгезивное покрытие».
Требования к адгезивным покрытиям также точно определены
Министерством обороны Великобритании. Состав, удовлетворяю-
щий ранному требованию, приведен ниже:
Алюминиевая пудра (не всплывающая)
Основной сульфат свинца
Гильсонит (битум)
Льняное полимеризованное масло, 5,0 Па-с
Нефтяной пек 90/160
Содержание, %
14,0
14,0
22,4
5,6
44,0
100,0
Адгезивное покрытие наносится методом безвоздушного рас-
пыления, в результате чего высохшая пленка имеет толщину
порядка 50 мкм. Благодаря этому противообрастающее покры-
тие можно наносить непосредственно перед спуском судна на
воду.
В других красках для подводной части судов могут также
использоваться хлоркаучук и виниловые смолы (см., например,
монографию Банфилда [2] по этому вопросу).
11.5.4. Противообрастающие покрытия
11.5.4.1. Проблема обрастания
Морские организмы, как растения, так и животные, способны
прикрепляться к корпусам судов. Для жизнедеятельности расте-
ний требуется дневной свет, так что обрастание происходит на
верхней зоне погруженного в воду корпуса судна. Наиболее рас-
пространенным видом водорослей является энтероморфа, имею-
щая длинные зеленые трубкообразные нити, которые очень быстро
растут при благоприятных условиях. Коричневые и красные
водоросли требуют меньше света и поэтому могут расти на ниж-
них зонах корпуса, или даже на днище судна. Прочное прикреп-
ление данных растений может произойти всего за несколько
часов, если не приняты меры, препятствующие этому.
Среди морских животных организмов способностью при-
крепляться к судам обладают балянусы, мидии, полихеты, мшан-
ки, гидроиды, асцидии и губки. Наиболее известными (достав-
ляющими самые большие неприятности) из них являются баг/я-
нусы. Поскольку они не требуют света для своего роста, их мо^но
обнаружить на всех погруженных в воду частях корпуса судна.
Их личинкам необходимо всего около 48 ч для того, чтобы прочно
прикрепиться к поверхности. О последствиях обрастания 'уже
было упомянуто, но заслуживает повторения то, что самым важ-
ным последствием для больших современных судов является! воз-
растание стоимости передвижения судна по воде в результате
увеличения торможения, вызванного обрастанием. /
Первые «противообрастающие» краски, основанные на ирполь-
зовании биоцидов, были разработаны в середине XIX в. Это'были
масляносмоляные композиции, содержащие оксиды ртути и
мышьяка. Впоследствии ввиду высокой токсичности и всё воз-
растающей стоимости оксида ртути были разработаны к|эа-ски,
содержащие оксид меди(1). Оксид меди(1) и сегодня остается
наиболее широко применяемым биоцидом в традиционных типах
композиций.
11.5.4.2. Традиционные типы противообрастающих покрытий
Существует два типа традиционных противообрастающих
красок, классифицированных в соответствии с их принципом
действия. Они подразделяются на краски с растворимой матри-
цей (краски на полностью растворимой основе) и на краски кон-
тактного типа.
Краски с растворимой матрицей. В касках данного типа
биоцид диспергирован в связующем, которое слабо растворимо
в морской воде. Биоцид медленно высвобождается по мере раство-
рения связующего. Скорость растворения должна контролиро-
ваться очень тщательно, так как недостаточная концентрация
биоцида у поверхности не сможет предотвратить прикрепление
морских организмов, тогда как слишком быстрое растворение
будет означать, что эффективность противообрастающего покры-
тия будет очень кратковременной. Связующим, используемым
в этом типе противообрастающих красок, может служить смесь
резината кальция и полимеризованного льняного масла (в соот-
ношении 3:1). Наилучшая рецептура для влажных тропиков
может иметь следующий состав:
Оксид мсди(1)
Оксид цинка
Оксид трибутилолова
Красный железоокисный пигмент
Парижские белила
Тальк (10 мкм)
Бентон 38
н-Бутаиол
Смесь 3:1 резината кальция и уплотненного льняного масла
в виде 60% раствора в ароматических растворителях
Уайт-спирит
366
Содержание, %
24,0
5,0
2,0
8,0
9,0
6,0
0,3
0,1
41.0
4.6
100,0
ВОрил
играе
ваниг
зом, 1
\ В красках для «Атлантического» или «Тропического» клима-
та\обычно не требуется использование оксида трибутилолова.
^Покрытия контактного типа. Противообрастающие покрытия
контактного типа названы так потому, что в их состав вводится
большое количество пигмента, так что диспергированные частицы
пигмента (биоцида) контактируют друг с другом во всем объеме
сухой пленки.
Связующее в основном не растворяется в морской воде, поэто-
му при растворении биоцида на поверхности остается пористая
пленка связующего. А поскольку связующее все же частично раст-
" о, то баланс между его растворимостью и нерастворимостью
г определенную роль в регулировании скорости выгцелачи,-
биоцида. Хотя состав этих красок подбирают таким обра-
1тобы получить плотноупакованную структуру пигментов в
пленке, представляется сомнительным, достигается ли это на
практике и нужно ли этого достигать. Однако это не является глав-
ной проблемой. Типичный состав для противообрастающего по-
крытия контактного типа приведен ниже:
Содержание, %
Оксид меди(1) 57 4
Асбестин 2,4
Канифоль 16,1
Смесь модифицированной фенолформальдегидной смолы и по- 9,0
димеризованного льняного масла 1:2; 60% раствор в Сольвен-
те Нафта 90/190
Хлорированный пластификатор 5,4
Сольвент-нафта 90/190 9,7
100,0
Одной из наиболее важных особенностей противообрастаю-
щих композиций является скорость выщелачивания биоцидов.
Различные организмы обладают различной степенью чувстви-
тельности к биоцидам. В случае балянусов для предотвращения
их прикрепления к поверхности судна оказывается достаточно
выщелачивание около 10 мкг меди на 1 см2 в день. Имеются дан-
ные, подтверждающие существование синергизма действия раз-
личных биоцидов. Это является одной из причин, по которой оксид
меди(1) и оксид трибутилолова используются вместе в противо-
обрастаюших красках, которые применяют в более жестких
условиях, например в «супертропических» водах.
11.5.4.3. Современные противообрастающие покрытия
Строительство все более крупных судов (например, супертан-
керов) требует лакокрасочных покрытий с более длительным сро-
ком службы между ремонтной окраской, вызванной разрушением
покрытий и коррозией, защищающих на более длительный пе-
риод. В 1954 г. Ван-дер-Керк и Луйтен [3] сообщили об изучении
биоцидных свойств соединений трибутилолова. Эти исследовате-
ли пришли к выводу, что данные материалы характеризуются
широким спектром действия, а именно, что они являются токсич-
ными для большинства морских организмов. Впоследствии оикид
трибутилолова начали использовать в комбинации с оксидом
меди(1). Однако, совершенно очевидно, что при использовании
оксида меди(1) и при достигаемых на практике толщинах пленки
невозможно обеспечить эффективный срок действия противооб-
растающего покрытия более 18 мес. В результате возросло приме-
нение оловоорганических производных, и сейчас в распоряжении
разработчиков красок имеется целый набор этих веществ. Лабо-
раторные эксперименты показали, что производные трибутилолова
в 100 раз эффективнее оксида меди [4]. При практических испы-
таниях оказалось, что количества оловоорганического соеди гения,
равные 1/10—1/20 от количества оксида меди(1), будут обеспе-
чивать тот же эффект действия против обрастания водорослями
и балянусами [5]. Очень важно, чтобы оловоорганические |соеди-
нения были доступными, иначе не будут более эффективными,
чем соединения меди. Таким образом, необходимо внимательно
относиться к составу краски для того, чтобы сделать использо-
вание биоцида максимально эффективным.
На рынке в настоящее время укоренились в качестве биоцидов
несколько оловоорганических производных. Главными из них яв-
ляются оксид трибутилолова, фторид трибутилолова и фторид
трифенилолова.
Оксид трибутилолова — это жидкость, смешиваемая с основ-
ными растворителями для красок. Данное вещество имеет отно-
сительно высокую растворимость в морской воде (25%о), что
дает возможность использовать его, когда требуется высокая
скорость выщелачивания. Оно оказывает пластифицирующее дей-
ствие на пленку, и это накладывает ограничение на его количество
(около 13% по массе) в виниловой системе.
В современных промышленных составах оксид трибутилолова
обычно используется совместно с оксидом меди(1) при содержа-
нии в сухой пленке в количестве 2—5%.
Фторид трибутилолова — белое, высокоплавкое воскообразное
вещество, которое нерастворимо в большинстве растворителей
для красок. Поэтому он вводится как пигмент в количестве до
30% от массы сухой пленки. Данное соединение менее растворимо
в морской воде, чем оксид (10%о) и часто используется как един-
ственный биоцид в красках на основе таких пленкообразующих
систем, как виниловый полимер — канифоль или хлоркаучук —
канифоль. Фторид трибутилолова выпускается в виде порошка
или пасты. Паста более удобна в обращении и легко может быть
введена в краску при высокоскоростном смешивании.
Производные трифенилолова также являются полезными
биоцидами. Фторид трифенилолова свыше 10 лет использовался
в качестве фунгицида. Это белый порошок с растворимостью
в Дорской воде менёе чем 1%о. Краски, содержащие этот биоцид,
могут обеспечить эффективную защиту от морского обрастания
на арок до двух лет. Оба продукта гидролиза фторида трифенил-
олова в морской воде — хлорид трифенилолова и гидроксид
трифенилолова — являются твердыми и не диффундируют сквозь
пленку покрытия. Следовательно, фторид трифенилолова наибо-
лее эффективен при использовании в композициях, которые мо-
гут растворяться или подвергаться эрозии.
Традиционные противообрастающие системы, основанные на
выщелачивании биоцида, характеризуются логарифмическим сни-
жением концентрации биоцида на границе раздела покрытие —
вода. Таким образом, для обеспечения длительного срока службы
покрытия оно должно вначале выделять более высокое количество
биоцида, чем требуется для эффективного предотвращения об-
растания. Когда концентрация доступного биоцида падает ниже
значения, необходимого для предотвращения обрастания, в плен-
ке все еще остается некоторое остаточное его количество. Приме-
нение таких композиций становится неэффективным по двум
причинам: во-первых, вследствие использования дорогостоящего
компонента краски, во-вторых, удваивание толщины пленки уве-
личивает эффективное время жизни покрытия всего на 13%, если
высвобождение биоцида протекает только по механизму диффу-
зии. Следовательно, если было бы возможно достигнуть постоян-
ной и оптимальной скорости выщелачивания, то это было бы зна-
чительным шагом вперед. Крупное достижение такого рода было
сделано в начале 1970-х гг. в Великобритании благодаря работам
компании International Paint в области оловоорганических поли-
мерных композиций.
Полимеры триалкилоловоакрилатов впервые были описаны
Монтермосо, Эндрюсом и Маринелли [6] в 1958 г. Эти полимеры
были получены в связи с разработкой термически стабильных
полимеров и их биоцидная активность не определялась. Либ-
рик [7] в 1965 г. первым заявил о возможности использования
оловоорганических полимеров в качестве биоцидов, однако только
в 1978 г. эти соединения получили одобрение со стороны амери-
канского агентства по защите окружающей среды. К тому време-
ни они уже прочно укоренились в Европе и Японии. Полимеры,
которые содержат оловоорганические группы, являются простыми
статистическими со- или терполимерами, имеющими, как правило,
следующую структуру:
Bu3Sn—О
С—О
СНз
сн2—сн
с—сн2
с=о
о—СНз
У
369
Введение биоцида в полимер гораздо эффективнее по срав'не-
нию с использованием полимерных производных, которые обычно
добавляются в краску в форме пигмента. Содержание биоцида
в полимере можно изменять в широких пределах, также как и
температуру стеклования полимера. Вероятно, самым большим
преимуществом является возможность обеспечить контролируемое
высвобождение биоцида не только посредством физическим про-
цессов, а главным образом за счет химического процесса гидро-
лиза. Преимущества новых противообрастающих оловооргани-
ческих полимеров заключаются в постоянной скорости высвобож-
дения биоцида и контролируемой скорости эрозии, самоочищении,
а при высоких скоростях эрозии — самополировании.
Составление рецептур противообрастающих красок на основе
оловоорганических полимеров представляет, однако, ряд йесьма
специфических проблем. Использование оловоорганических биоци-
дов в быстроразрушающихся вследствие эрозии системах увели-
чивает первоначальную стоимость судна, но это в значительной
мере оправдывается снижением времени пребывания судна в сухом
доке. Основной разработчик противообрастающих полимерных
оловоорганических красок — International Paint (Великобрита-
ния), и Nippon Oils and Fats Company (Япония) — ведут работы
в различных направлениях. International Paint видит преиму-
щества этого вида материалов в сочетании противообрастаю-
щего действия с самополированием и предлагает продукты, кото-
рые подвергаются более быстрой эрозии, чем продукты японской
компании. Так, для противообрастающих покрытий самополи-
рующегося типа обычно скорость эрозии составляет 10—12 мкм
в мес. В этом случае для достижения 2—3 лет эффективной служ-
бы потребуется нанесение до четырех слоев краски толщиной
по 100 мкэд.,,Японская система подвергается эрозии со скоростью
3—6 мкм в мес., так что адекватную защиту обеспечивают два
слоя по 100 мкм. Однако, как мы уже отмечали ранее, шерохова-
тость поверхности влияет на энергозатраты, связанные с поддер-
жанием эксплуатационной скорости крупных судов. При исполь-
зовании композиций самополирующегося типа можно получить
добавочную экономию, которая не осуществима с медленно раз-
рушаемыми вследствие эрозии покрытиями. Харпур и Милн [8]
указывают, что на традиционно окрашенных судах, если они хо-
рошо окрашены, шероховатость корпуса возрастает на 25 мкм
в год при первоначальном среднем значении НО—125 мкм.
Гидродинамики установили, что расход энергии для движения
судна зависит от шероховатости корпуса судна в степени 1/3,
так что увеличение шероховатости на 10 мкм может привести к
дополнительному увеличению расхода топлива на 1%. Однако
маловероятно, чтобы можно было реализовать преимущества эф-
фекта самополирования на старых судах, которые уже под-
верглись значительной коррозии.
РЕОЛОГИЯ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
\ Т. А. Страйвенс
I 12.1. ВВЕДЕНИЕ
В настоящей главе рассмотрены реологические характеристики
(текучесть) исходных лакокрасочных материалов и жидких пле-
нок по'сле их нанесения, а также методы измерения реологических
свойств красок для обоих состояний. Вкратце уделено внимание
также тем областям лакокрасочной технологии, где роль реологии
очень важна: при диспергировании (смачивании) пигментов,
транспортировке красок по трубопроводам и операции смешения.
Из-за многообразия практических ситуаций, связанных с те-
чением красок, как теория, так и экспериментальные данные,
приводимые здесь для понимания таких процессов, оказываются
отрывочными или во многих отношениях неполными. Однако,
правильное понимание и умение контролировать такие ситуации
позволяет успешно использовать реологические особенности при
практическом применении красок. Законодательство относительно
контроля выделений в атмосферу (загрязнение воздуха) и по
вопросам техники безопасности ужесточается во всем мире. След-
ствием этого является ограничение ассортимента лакокрасочных
материалов как у их разработчиков, так и у потребителей. Из-за
этих ограничений возникло две тенденции предусматривающие
создание красок с максимально высоким содержанием нелетучих
веществ (чтобы выделялось меньше растворителя при пленко-
образовании) и водных красок (менее токсичных по своей при-
роде) . Неизбежным результатом в том и другом случаях являют-
ся краски с более сложными реологическими свойствами, как
в объеме, так и в пленке.
12.2. ОБЩИЕ ЗАМЕЧАНИЯ О РЕОЛОГИИ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Утверждение, что контроль реологии красок является сущест-
венным для успешного использования последних можно под-
твердить рядом примеров. Наиболее характерным является на-
несение красок на поверхность. Независимо от использованного
метода нанесения, процесс складывается из трех стадий: I) по-
дачи краски из емкости в аппарат для нанесения; 2) подачи крас-
ки из этого аппарата к поверхности для образования тонкой, ров-
ной пленки; 3) растекание по поверхности, коалесценция частиц
полимера (для эмульсионных красок) и испарение растворителя.
На каждую из этих стадий значительное влияние оказывает
реология.
12.2.1. Стадия транспортировки
При хранении в емкости краска обычно имеет низкую вязкость,
поэтому ее легко можно наносить любым выбранным методом.
Краска может легко проникнуть в пространство между щетиной
кисти или в поры ручного валика, где будет удерживаться капил-
лярными силами или силами поверхностного натяжения во время
подачи к окрашиваемой поверхности. Конечно, если общий вес
краски, переносимой на кисти после погружения последней в ем-
кость с краской, достаточен для преодоления капиллярных сил,
то краска будет капать или стекать с кисти. Если же это количе-
ство краски, не считая потерь от капания, слишком мало, то при
окраске кистью получится либо чересчур тонкая пленка, либо
при большей толщине будет окрашена малая площадь поверх-
ности. В первом случае в результате слишком быстрого испаре-
ния вязкость пленки будет значительно возрастать, что приведет
^ ухудшению растекания краски после нанесения (см. ниже).
Во втором случае операция окраски становится слишком трудо-
емкой и продолжительной. 1
Что определяет оптимальную толщину пленки? Кроме свойств
растекания (см. ниже), важными показателями являются требуе-
мая интенсивность цвета, кроющая способность, а также защит-
ные свойства получаемой пленки.
Увеличение количества краски, удерживаемой на кисти, мо-
жет быть достигнуто либо за счет увеличения вязкости краски,
либо путем введения добавок, регулирующих реологические
свойства. При обоих подходах одновременно достигается сниже-
ние или предотвращение оседания пигментов при хранении лако-
красочного материала в таре. Однако, одновременно растут меха-
нические усилия, потребные для распределения наносимой краски
в виде пленки (важное соображение для ручных методов окраски).
Для того, чтобы удовлетворить требованиям потребителя, реоло-
гическая структура должна быстро разрушаться при относительно
низких напряжениях сдвига, возникающих при погружении кисти
в краску, или при более высоких напряжениях, возникающих при
воздействии ручного валика. Разрушение структуры облегчает
процессы переноса краски и ее проникновения в поры, но затем
структура должна быстро восстановиться, чтобы не допустить
капание, стекание и т. п.
Для промышленных методов окраски, включая распыление
или валиковое нанесение, соображения аналогичны. Так, при
распылении вязкость лакокрасочного материала должна быть
настолько низкой, чтобы обеспечивалось перекачивание краски
через тонкое сопло распылителя под минимальным давлением.
Обычно такие краски готовят разбавлением материалов с более
высоким содержанием нелетучих веществ непосредственно перед
использованием, так что оседание не является проблемой. Меха-
низм каплеобразования можно легко понять для простых жид-
костей [1], но влияние таких факторов, как присутствие частиц
пигментов или полимеров в суспензии или дисперсии или присут-
ствие полимера в растворе, на размер капель и распределение
частиц по размерам при распылении большей частью неизвестно.
Однако можно ожидать, что снижение вязкости исходного лако-
красочного материала понизит размер капель, что приведет к уве-
личению потерь краски в виде малых капель, уносимых от окра-
шиваемой поверхности потоком воздуха. В равной степени размер
капель и, соответственно, общая площадь поверхности капель
влияют на испарение растворителя с поверхности и, значит, будут
влиять на реологические свойства пленки, полученной в резуль-
тате соударения и коалесценции капель на поверхности окраши-
ваемого объекта. В случае промышленного валикового окрашива-
ния краска должна быть гораздо более вязкой, чтобы легко на-
текать на поверхность валика, где она может быть распределена
в ровный слой под действием вспомогательного лезвия, другого
валика и др. В этой ситуации механическая работа, необходимая
для распределения краски, гораздо менее важна. Однако краска
должна быть вязкой настолько, чтобы предотвратить ее стекание
или «сбрасывание» под действием центробежной силы.
Важной особенностью этих процессов является очень высокая
скорость течения жидкой краски и, соответственно, скорость окрас-
ки, в результате чего к краске прилагаются высокие напряжения
и усилия деформации. Следует, однако, заметить, что краска на-
ходится в струе при распылении (или в зазоре между валиками)
такое короткое время, что устойчивое состояние никогда не дости-
гается, и, следовательно, только скоростные методы измерения,
вероятно, дадут удовлетворительные реологические параметры.
Такие методики требуют сложного оборудования и приборов,
особенно при высоких напряжениях и скоростях деформаций,
достигаемых при нанесении. Шурц [2] ссылается на скорость
сдвига 105 с”1, достигаемую за 1 мс в высокоскоростной валковой
машине. Такие высокие значения с еще большей вероятностью
могут быть получены в том случае, если в рецептуре краски имеет-
ся полимер в виде раствора. При этом присутствие полимера в
концентрациях, характерных для типичных лакокрасочных мате-
риалов, и при молекулярной массе около 10 тыс., может привести
к появлению структурированных систем как при истечении краски
из сопла распылителя, так и при нанесении пленки, выходящей
из зазора валковой машины. Гласс [3] показал, что структурная
вязкость «загущенной» водоэмульсионной краски влияет на такие
свойства последней при нанесении валиком, как образование
полос, разбрызгивание и т. д. Можно предположить, что возник-
новение структурной вязкости может воспрепятствовать разрыву
струй, в результате чего при распылении образуются капли.
По закону Троутона структурная вязкость жидкости втрое больше
вязкости при сдвиге. Однако присутствие десятых долей процента
высокополимера с большой молекулярной массой в растворе
может увеличить структурную вязкость настолько, что она станет
больше вязкости при сдвиге в 10000 раз.
12.2.2. Пленкообразование
После подачи краски из емкости к окрашиваемой поверхности,
например с помощью кисти, следуют движения руки маляра над
поверхностью, в результате чего краска переходит с кисти на эту
поверхность и распределяется на ней ровным слоем. При этом
воздействие руки на средство окрашивания вызывает сдвиг и
сжатие щетины кисти или пенорезины или войлока — типичных
материалов для изготовления ручного валика. Такое усилие «вы-
талкивает» краску из пустот кисти (валика). Возникающие про-
цессы истечения очень сложны и, возможно, не могут быть описаны
-количественно Однако п ред пр ин имались. поп ытки-п риблизительно
измерить соответствующие скорости сдвига для кисти (прямо
или путем корреляции с оценкой укрывистости ньютоновских
красок). В литературе [4] упоминаются значения 15—30 с-1 для
окунания кисти и 2500—10000 с_| для распределения краски
кистью. Кьюг [5] попытался измерить усилия, возникающие при
окраске кистью, и соотнести их с реологическими свойствами
красок, используя при этом простые теоретические уравнения.
12.2.3. Растекание пленки краски
Свыше двадцати лет назад было сделано предположение, что
характерные неровности, получающиеся на поверхности пленки
краски при окраске кистью, поверхностные дефекты, образующие-
ся при нанесении краски с помощью аппликатора, и дефекты,
получаемые при нанесении валиком, имеют общее происхождение,
связанное с гидродинамической неоднородностью и нестабильным
растеканием краски, попадающей в зазор между подложкой и
выступающей кромкой приспособления для нанесения [6]. Хотя
физический процесс понятен, результат не всегда возможно опре-
делить количественно. Теоретические работы по этому эффекту
опубликованы [7, 8].
Выше обращалось внимание на роль вязкости в процессе пере-
носа краски с валика на поверхность при окрашивании. Гласс [3]
измерил вязкость' для водоэмульсионных красок, загущенных
водорастворимыми полимерами, такими, как различные производ-
ные целлюлозы, сополимеры акриловой кислоты с акриламидом,
полиэтиленоксид, и пытался соотнести явления разбрызгивания
краски и образования поверхностных штрихов с результатами
своих измерений. К сожалению, из-за ограничений, накладывае-
мых методикой, которую автор использовал в своих эксперимен-
т.ах, он был вынужден применять такие концентрации и молекуляр-
ные массы загущающих полимеров, которые приводят к образо-
ванию паутины и нитей при нанесении красок, т. el создают явные
дефекты поверхности при нанесении краски валиком. Он также
измерил остаточное удлинение [9] и нормальное напряжение
при постоянном усилии сдвига., Первое измерение было выполнено
с использованием методики Доджа [10], в которой образец вна-
чале подвергался высокоскоростному усилию сдвига (скорость
сдвига 2600 с-1), а затем сразу же измерялась эластичность как
функция времени снятия усилия сдвига с замером величины низко-
частотной вращательной деформации сдвига (частота 0,3 Гц,
максимальная скорость сдвига 0,07 с~‘). Такие измерения легко
провести, например, на реогониометре Вайсенберга. Далее, при-
знав важность восстановления эластичности, Гласс пытался
описать влияние большинства эффектов, которые он изучил, на
внутреннюю вязкость. Однако, когда значение этой вязкости было
низким или умеренным, он нашел, что скорость восстановления
эластичности хорошо коррелирует с исчезновением следов от ва-
лика и растеканием. । .. .
. При исследовании процессов нанесения красок можно придти
к заключению, что, во-первых, течение краски происходит при
очень высокой скорости сдвига, а во-вторых, краска подвергается
когезионному измельчению или на выходе из сопла распылителя
или на подложке между выступающей кромкой оборудования для
нанесения и слоем, прилежащим к субстрату. Оба процесса за-
нимают доли секунды. Показано [ф1], что высокая скорость сдви-
га при нанесении краски полностью разрушает любую структуру,
имеющуюся в краске до нанесения, и что пленка'.краски становит-
ся слишком жесткой, так как два фактора препятствуют ^нормаль-
ному когезионному растеканию пленки в результатедвндкого те-
чения: I) быстрое удаление оборудования для нанесения от суб-
страта ведет к появлению напряжений, которые нс могут быть
сняты путем поперечного течения жидкости; 2) болишинстео лако-
красочных составов содержит в растворе некоторое ^количество
полимера, что придает раствору заметную эластичноотвош
Итак, пока имеет место обычный распад жидкости на нити
в результате кавитации, присутствие эластичного компонента
в краске и соответствующая скорость распада приводят только
к уменьшению поперечного сечения нитей и к увеличению времени
их сохранения по сравнению с тем, что можно было бы ожидать
в случае обычной вязкой жидкости. Присутствие пигментных или
полимерных частиц в красочной суспензии может быть причиной
зарождения процесса кавитации. [12]. Такое объяснение пол-
ностью исключает значение структурной вязкости,.хотя она может
быть столь же важна, как и эластичность при некоторых условиях
нанесения; и, как указывает Уолтерс [13] , эта вязкость не являет-
ся априори функцией сдвиговой вязкости. В самом деле, некоторые
недавние работы с разбавленными растворами высокополимеров
I[14а] показали, что резкое увеличение структурной вязкости
при определенных скоростях деформации связано с полным рас-
прямлением полимерных молекул в направлении приложенной
деформации, что находится в резком противоречии с глобулярной
теорией; в соответствии с ней молекулы принимают форму клубка
и в таком виде сохраняются в поле напряжений сдвига, приме-
няемом для измерения модуля эластичности при сдвиге. Уолтерс
приводит пример для водного раствора полиакриламида с
концентрацией 100 ч. на миллион, который имеет сдвиговую вяз-
кость 1,4-10~3 Па-с, а структурная вязкость при этом равна
9 Па-с [146].
Таким образом, хотя некоторые представления о физическом
смысле процессов, при котором на поверхности возникают де-
фекты при "пленкообразовании, являются понятными, но нет еди-
ного мнения насчет того, какие реологические свойства краски
-ответственны -з-а -образование лзаких дефектов.
Если рассмотреть процесс растекания жидкой пленки на по-
верхности, от которого зависят такие важные свойства, как равно-
мерность цвета, кроющая способность и т. д., то окажется, что
не существует ясного понимания влияния на эти показатели
реологических свойств краски. Хотя в работах [9, 10] ясно пока-
зано наличие связи между скоростью восстановления эластич-
ности и неравномерностью поверхностного растекания, опубли-
кованные теоретические и экспериментальные работы на эту тему
не внесли ничего существенно нового в пионерские работы Орчар-
да [15], выполненные около 25 лет назад. Действительно, недав-
нее издание авторитетного сборника [4] рассматривает течение
краски и диспергирование пигментов даже без учета эластичности
при сдв,йге й структурной вязкости [16]. Однако, существуют
бесспорные доказательства проявления вязкоэластических свойств
красок и дисперсий пигментов [17, 18].
Ситуация, .еще более усложняется эффектами испарения рас-
творителя, Это не только влияет на реологию красок, но и на по-
верхностное натяжение на границе раздела «мокрая» пленка/
воздух. Поверхностное натяжение и сила тяжести приводят к воз-
никновению сдвиговых напряжений, что улучшает процесс расте-
кания. В результате испарения возрастает концентрация раствора
полимера и происходит охлаждение поверхности пленки. Оба эти
эффекта приводят к возникновению тангенциальных сил сдвига
на поверхности (силы Левича-Ариса). В работе [19] недавно
доказано, что градиент гидростатического давления в пленке
краски, обусловленный поверхностным натяжением, несуществе-
нен для объяснения результатов выравнивания поверхности плен-
ки, что утверждают Смит и др. [6]. В этой работе [6] сделана
попытка продемонстрировать как теоретически, так и эксперимен-
тально с использованием алкидных красок растворного типа, что
тогда как поверхностное натяжение стремится создать плоскую
поверхность пленки независимо от профиля поверхности субстра-
та, градиент поверхностного натяжения, возникающий над по-
верхностью жидкой пленки краски, стремится создать пленку
единой толщины, т. е. профиль поверхности пленки точно отра-
жает профиль поверхности субстрата.
Вдобавок, испарение растворителя приводит к возникновению
градиента концентрации растворителя по толщине пленки и, сле-
довательно, к градиенту плотности. Как градиент плотности, так
и градиент поверхностного натяжения, могут вызвать образова-
ние в жидкой пленке циркулирующих потоков. Последние могут
привести к дезориентации (рандомизации) алюминиевой пудры
в автомобильных верхних покрытиях с металлическим оттенком,
хотя в большинстве публикаций по этому вопросу рассматривает-
ся в качестве контролирующего фактора только вязкость пленки
[20]. В крайних случаях они могут привести к образованию ячеи-
стых структур Бенарда, которые часто можно наблюдать на по-
верхности кипящих или быстро испаряющихся жидкостей. По-
верхностные дефекты лакокрасочных или полимерных покрытий,
вызываемые этими причинами, рассмотрены в работах [21, 22]
и других.
' Таким образом, представленные соображения позволяют оце-
нить роль сил, связанных с реологией наносимой краски, однако
важно понять также, каким образом можно реализовать реологи-
ческие особенности пленок, обусловленные сложностью их физи-
ческого и химического состава.
Можно ожидать, что пленка будет не только вязкоэластичным
материалом, но при низких значениях приложенных напряжений
будет проявлять упругие свойства. Эти свойства нелинейны и
зависят от времени приложения и величины напряжения сдвига
при нанесении. Некоторые сведения о сложности реологического
поведения объемных систем аналогичного состава можно узнать
в опубликованных много лет назад работах Оноги с сбтр. по дис-
персиям полимерных частиц [23]. В свете этого попь/тки смодели-
ровать поведение пленок при растекании путем рассмотрения их
как ньютоновских или псевдопластических жидкостей могут по-
казаться слишком простыми, так как на основе этих концепций
можно выполнить лишь простейшие реологические измерения.
Кроме того, наличие градиента концентраций по толщине пленки
свидетельствует о том, что реология будет изменяться по толщи-
не пленки. В то же время влияние градиента плотности в пленке,
вероятно, должно сказываться в меньшей степени.
Во всестороннем обзоре [24] рассмотрена роль различных
факторов, влияющих на растекание и другие процессы течения
жидких пленок, особенно с учетом дефектов поверхности. Указы-
вается, что силы, влияющие на растекание, находятся в пределах
3—5 Па, а на стекание с поверхности — около 0,8 Па. Скорости
сдвига при ’-растекании находятся в пределах 0,001—0,5 с-1.
Поскольку именно усилие сдвига, возникающее от действия сил
тяжести и поверхностного натяжения, контролирует растекание,
стекание й т. п., ‘оценивать скорость сдвига неуместно; иначе го-
воря, при измерении реологических свойств лучше использовать
приборы для измерения усилия, чем более распространенные при-
боры для измерения скорости сдвига.
12.2.4. Необходимые реологические характеристики
для нанесения красок
В первую очередь, структура краски должна разрушаться и
краска должна стать низковязкбй для облегчения переноса с
помощью (или через) приспособления для нанесения. Из-за вы-
соких скоростей сдвига и коротких промежутков времени, харак-
терных для процессов переноса краски на поверхность, как элас-
тичность, так и вязкое течение,- могут изменить характер поверх1-
ностных дефектов плёнкш Последние","как можно ожидать, воз-
растают из-за нестабильного гидродинамического течения, что
связано с когезионным -разрушением потока краски на выходе
сопла распылителя или на поверхности раздела между пленкой,
прилегающей к субстрату; и выступающей кромкой, например
валика, движущегося вдоль окрашиваемой подложки.
Краска должна оставаться низковязкой достаточное время
для того, чтобы растечься по дефектам поверхности в достаточ-
ной степенй1-,(зависящей от того, что требуется от пленки — блеск
или- защитны©’: свойства, связанные с толщиной). Однако при
низкой вязкости краска будет стекать с вертикальной поверхности
под действием силы тяжести. Если толщина слишком велика,
может стЙъ заметным эффект стекания, и на поверхности про-
явятся дефекты в-чвиде «наплывов». Для проявления таких эффек-
тов требу^зя некоторое время после окраски.
Такимкй&в^^м, за исходной низкой вязкостью должно после-
довать резкбк^-ее увеличение, связанное с испарением раствори-
теля или с быстрым восстановлением реологической структуры,
разрушенной при . воздействии напряжений сдвига в процессе
переноса краски на поверхность. В обоих случаях эффект одина-
ков; высыхающая пленка фактически неподвижна, и процесс
стекания прекращается до того, как он станет заметным. На по-
терю растворителя могут влиять различная летучесть раствори-
телей и растворимость компонентов в смеси растворителей, обра-
зующих жидкую фазу. Эти эффекты растворимости, в свою оче-
редь, будут контролировать рост вязкости высыхающей пленки
по мере испарения растворителя. При испарении наблюдается
охлаждение поверхности пленки, особенно в случае быстроиспа-
ряющихся растворителей, что также может повлиять на вязкость
пленки.
12.3. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИЗМЕРЕНИЯ РЕОЛОГИЧЕСКИХ
СВОЙСТВ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ ДО И ПОСЛЕ НАНЕСЕНИЯ
12.3.1. Реологические свойства до нанесения
Для изучения свойств красок при нанесении и растекании
можно использовать много приборов. Однако в пределах ограни-
чений, отмеченных ранее, многие краски можно рассматривать
как квазиньютоновские жидкости. Также есть необходимость в
простых приборах для контроля качества и измерения вязкости
красок потребителем перед нанесением. Эта необходимость в ос-
новном удовлетворяется специальными приборами, выпускаемыми
лакокрасочной промышленностью.
Эта группа простых приборов состоит в основном из разно-
образных сосудов для истечения жидкостей различных конструк-
ций и упрощенных вращательных приборов, например воронки
ICL Сосуды для истечения применяются и в других отраслях, на-
пример в нефтехимической промышленности. Принцип их устрой-
ства показан на рис. 12.1. Известный объем краски помещается
в вертикально расположенный цилиндрический сосуд, дно кото-
рого имеет короткий капилляр известной длины и диаметра. Крас-
ка вытекает через отверстие в дне сосуда (обычно после удаления
пальца испытателя); секундомером замеряется время истечения.
Концом истечения обычно считается момент, когда непрерывная
струя краски распадается на капли. Возникает несколько вопро-
сов для обсуждения. Во-первых, поскольку масса жидкости изме-
няется при испытании, движущая сила течения (обусловленная
силой тяжести) через капилляр также меняется. Если краска
Рис. 12.1. Сосуд для истечения (примерно в масштабе 1:1)
является неньютоновской жидкостью, то результат измерения
вязкости может быть сильно искаженным. Во-вторых, капилляр
обычно короткий, следовательно, стабильных условий течения
внутри него не получается, и это, а также ошибки в начале и
конце определения, может повлиять на результат, особенно если
материал слегка структурирован. В-третьих, присутствие абразив-
ных частиц в краске может привести к износу металлического
капилляра, и, значит, такие приборы должны поверяться ньюто-
новскими жидкостями с известной вязкостью. Дно сосуда может
быть коническим или плоским; первое, очевидно, уменьшает ошиб-
ки в начале опыта. Наконец, испытания проходят нормально при
температуре окружающей среды, но предпочтительно тщательно
контролировать температуру краски до и во время испытаний,
чтобы получить точные и сравнимые результаты. Этот тип испы-
таний должен применяться только для красок, близких к ньюто-
новским жидкостям. Различные виды таких приборов приведены
в работе [25].
- Примером другого тина приборов может служить-вискозиметр
фирмы ICI, впервые описанный Манком [26, 27]. Вискозиметр
состоит из фиксированной пластины, термостатируемой при темпе-
ратуре 25,0±0,1 °C, и расположенной над ней воронки, приводи-
мой во вращение со скоростью 900 об/мин с помощью двигателя.
Скорость сдвига — 10000 с-1, предел измерений 0—0,5 Па-с при
точности измерений выше 0,01 Па-с (цена деления шкалы при-
бора). Воронка усечена, чтобы уменьшить износ и предупредить
«застревание» частиц в зазоре. Для проведения измерений тре-
буются малые количества образца (менее 1 мл), а сам процесс
осуществляется легко и быстро. Очевидно, этот прибор имеет пре-
имущества по сравнению с воронками, заключающиеся в том,
что температура образца тщательно контролируется, а вязкость
измеряется-в условиях (высокая скорость сдвига), характерных
для условий нанесения краски. Однако прибор дает только одну
точку при измерениях и, следовательно, не показывает реологи-
ческих свойств краски в покое и в процессе растекания. Для оцен-
ки последнего необходимо производить измерения при низких
скоростях (напряжениях) сдвига. Прибор для таких измерений,
релаксационный вискозиметр ICI с низким усилием сдвига, будет
описан ниже.
Известны и другие методы измерения исходной вязкости крас-
ки, приведенные в работе [28]. Особую проблему с точки зрения
реологии представляют тиксотропные (высокоструктурирован-
ные) краски. Найти простые способы контроля реологических
свойств здесь очень трудно.
Проводить измерения для красок в состоянии покоя относи-
тельно несложно — можно использовать разновидность вибра-
ционного метода, который рассматривается ниже, в разделе, по-
священном реологии при хранении красок,— но исключительно
трудно точно измерить разрушение и восстановление структуры.
Прекрасный обзор имеющихся методов дан Уолтоном [28]. Со-
гласно опыту автора, существует только один удовлетворитель-
ный метод полного разрушения структуры, заключающийся в
воздействии на образец высокоскоростного усилия сдвига в те-
чение продолжительного времени (это также устраняет ошибки,
связанные с реологической «предысторией» образца, проистекаю-
щей из способа подготовки этого образца и загрузки его в рео-
метр), с последующим определением времени восстановления
структуры. Это может быть сделано путем уменьшения скорости
сдвига до очень малых значений и установления зависимости
вязкости от времени; или, еще лучше, путем измерения динами-
ческой вязкости и модуля эластичности как функции времени
при малых вращательных напряжениях [10]. Такие измерения
можно сделать с использованием вискозиметров с низким уси-
лием сдвига (LSV фирмы ICI).
Стандартные методы измерения вязкости красок, разработан-
ных официальными организациями, например Британским инсти-
тутом стандартов (BSI), основаны на применении различных
воронок [25]. Однако, увеличивается число методов, основанных
на использовании ротационных вискозиметров, например ворон-
ки ICI (ASTM D4287-83) или вискозиметра Штурмера (ASTM
D562-81). Германский институт нормирования описывает метод
определения «реограмм и вязкостей» лаков и красок, с использо-
ванием ротационных вискозиметров DIN53214 (1982). Такие же
приборы упоминаются во французском и чехословацком стандар-
тах [30, 31 ].
12.3.2. Растекание пленки краски
12.3.2.1. Прямые измерения реологических свойств при растекании
)
Основные проблемы при таких измерениях заключаются в
следующем: ;
1. Реология красочной пленки исключительно сложна не толь-
ко в связи с вязкоэластическими свойствами, но и с нелиней-
ностью.
2. Реология может быстро изменяться во времени вследствие
изменений в составе (в процессе восстановления реологической
структуры изменяется соотношение растворителей, меняется со-
держание нелетучих веществ и т. д. по мере испарения раствори-
теля) .
3. Вследствие малых объемов возможна также неоднородность
состава по толщине пленки.
Особая роль, следовательно, должна отводиться быстрым ме-
тодам анализа (короткое время определения и высокая скорость
воспроизведения) и максимизации информации, получаемой из
каждого определения. По этой причине мы начинаем обзор с рас
смотрения ударных и высокочастотных колебательных методов
измерения вязкости.
Ударный метод (метод «отскакивающего» шарика). Ударные
методы широко использовались в различных модификациях для
испытания полимеров в виде цилиндрических или дисковых об-
разцов и для испытания механических свойств отвержденных пле-
нок в процессе их эксплуатации (см. Главу 13). Однако их исполь
зованию для изучения растекания пленок и процессов отвержде
ния не придавалось значения, за исключением короткой работы
Сноу [32], посвяшенной методу «отскакивающего» шарика. Для
объемных образцов полимера аналогичный метод кратко проана-
лизирован в работах [33—36].
Автор работы [37] подтвердил оригинальные результаты Сноу.
Типичные кривые показаны на рис. 12.2, где высота отскока та
рика является функцией времени высыхания краски. Данные
получены для стального шарика диаметром 0,5 см (вес 0,5 г), ко-
торый сбрасывали на стеклянную плитку толщтптой-около 1,25 см,
окрашенную испытуемой краской. По мере сушки пленки в связи
с испарением растворителя вязкость нарастает, следовательно,
60г
Рис. 12.2. Зависимость высоты отскока шарика от времени сушки покрытия (тол
щина пленки 200 мкм, подложка — стекло):
/ — средний размер шариков; 2 — малый размер шариков
энергия, диссипированная пленкой во время удара шарика по
стеклу, также растет, в результате чего высота отскока шарика
в первые моменты времени уменьшается. Однако, когда начинает-
ся отверждение (по механизму автоокисления или по механизму
«лакового типа», как показано в примере), эластичность пленки
несколько увеличивается, и высота отскока снова растет. Можно
построить простую теорию, связывающую высоту отскока с вяз-
костью пленки, исходя из момента количества движения шарика
и потери энергии, однако много важных факторов в этой простой
теории не может быть учтено. Для получения такого соотношения
лучше построить калибровочную кривую для стандартных ньюто-
новских масел и использовать эмпирическую формулу. К факто-
рам, которые следует учитывать, относится гидродинамическая
сила, предотвращающая соприкосновение шарика с поверхностью
субстрата, а также мгновенная упругость жидкости в момент
удара (десятые доли миллисекунды).
Несмотря на эти недостатки, метод прост для реализации.
Единственными необходимыми приборами являются стеклянные,
металлические или даже деревянные пластинки, а также набор
шариков различного диаметра или плотности и градуированная
трубка для определения высоты отскока шарика. При регулируе-
мом нагреве металлической плитки можно использовать эту ме-
тодику для изучения процесса отверждения термореактивных
систем, аналогично тому, как описано Гордоном и Гривсоном [38]
для полимеров. В отличие от метода катящегося шарика (будет
описан ниже), эта методика дает возможность измерять увели-
чение пластичности пленки при отверждении.
Высокочастотные методы (методы импеданса). Механический
импеданс волн эластического сдвига, распространяющихся в
среде, изменяется ввиду присутствия вязкоэластичного слоя на
поверхности среды. Если волны, проходящие через эластичный
слой, полностью поглощаются этим слоем, изменения характери-
стического импеданса могут быть соотнесены с реологическими
параметрами материала слоя. Это трудно получить для пленок
многих красок, но, несмотря на это, метод можно использовать
для измерения изменений, происходящих в пленке при высуши-
вании и отверждении.
На практике импульсы высокочастотных колебаний генери-
руются пьезоэлектрическим кристаллом, присоединенным к под-
ложке. После прохождения через подложку затухающие колеба-
ния вновь преобразуются в электрический сигнал. Измеряются
фазовый угол и затухание колебаний, а также изменение зна-
чений каждого из этих параметров, которые используются для
сравнительной оценки скоростей изменений, протекающих в вы-
сыхающей и отвержденной пленках различных красок. Каждый
из импульсов направляется на подложку под малым углом, а от-
раженные импульсы детектируются приемником-кристаллом, как
4
Рис. 12.3. Импедансный прибор Майерса:
I — передающий кристалл; 2 — пленка; 3 — принимающий кристалл; 4 — кварцевый
брусок
это сделано в приборе Майерса (рис. 12.3) [11, 39—42]; в другом
варианте ряд импульсов отражается перпендикулярно от поверх-
ности подложки вдоль того же направления, по которому они
были генерированы, таким образом, чтобы их можно было детек-
тировать тем же кристаллом, как в методе Мьюиса [43]. В обоих
случаях изучаемая краска наносится на подложку.
Хотя это точно не установлено, складывается четкое впечат-
ление, что эти методы ограничены чувствительностью приборов
и могут использоваться для измерений в ограниченном диапазоне
значений вязкоэластических параметров. Следовательно, для
многих лакокрасочных материалов методики эти могут приме-
няться для- измерения свойств пленок только для ограниченной
области общего процесса сушки — отверждения пленок. Более
того, как можно ожидать, адгезия высыхающей пленки к мате-
риалу подложки оказывает сильное (и, по-видимому, неизвестное)
влияние на результаты [42]. В работе [11] использованы часто-
ты 2—100 МГц, и измеренное затухание сигналов представляется
набором экспоненциально затухающих эхо. Мьюис использовал
в своем методе [43] несколько меньшие частоты — около 100 кГц.
Метод катящегося шарика. Этот метод, как и метод «отскаки-
вающего» шарика, прост по замыслу и исполнению. Окрашенная
панель наклоняется под определенным углом, и время, необходи-
мое для того, чтобы маленький стальной шарик прокатился на
определенное расстояние по окрашенной поверхности, измеряется
как функция времени сушки (отверждения). В другом варианте
расстояние, на которое прокатится шарик, измеряется как функ-
ция времени. Метод введен Вольфом и Зайдлером [44], которые
использовали его для изучения влияния различных растворителей
и пластификаторов на вязкость нитроцеллюлозных покрытий
при сушке. В работе [45] метод использован для корреляции
вязкости и поверхностного растекания эмульсионных красок;
Тэйлор и Фостер [46] применяли его для изучения реакционной
способности эмалей горячей сушки в температурном диапазоне
140—160° С, а Геринг [47] —для изучения изменений вязкости
в высоконаполненных эмалях, наносимых методом электрофореза.
В этой работе выводится простая теория, основанная на рассмот-
рении баланса действующих сил, которую, однако, мы считаем
неадекватной объясняемым результатам.
Недавно в работе [48] описан остроумный автоматизирован-
ный вариант этого метода. Окрашенная панель помещается на
наклоненный вращающийся стол, находящийся в камере с контро-
лируемой температурой. Шарик располагается на краю панели
и освещается источником света, который также освещает ряд
фотоэлементов. Выход этих фотоэлементов используется для
точного контроля скорости вращения стола, которая такова, что
шарик остается неподвижным относительно пучка света. При
развитии этой теории авторы сделали допущение, что такие фак-
торы, как поверхностное натяжение и, соответственно, смачива-
ние шарика, ускорение слоя краски при вращении и течение краски
вокруг шарика, влияния не оказывают.
Работы, посвященные исследованию трения качения по поверх-
ности твердых полимеров, а также методы механического испы-
тания твердости пленок красок (см. гл. 13), могут создать впе-
чатление, что величины эластичности могут быть получены так
же, как и в методе «отскакивающего» шарика, но этого пока
что не было сделано. В отличие от метода «отскакивающего»
шарика, это может потребовать модификации экспериментальной
методики.
Релаксационный вискозиметр ICI с низким усилием сдвига.
Этот прибор разработан для того, чтобы имитировать условия
высокой скорости сдвига, которые возникают при течении под
действием низких (уменьшающихся) усилий, например при окра-
шивании кистью. Теория и описание этого прибора даны в рабо-
тах [48, 49].
Принцип устройства показан на рис. 12.4. Он основан на
применении торзионного маятника. Если маятник закрутить под
малым углом и отпустить, то кольцевая пружина будет раскру-
чиваться, а подвеска маятника будет возвращаться в исходное
(равновесное) положение. При этом инерция подвески заставит
ее пройти далее, за равновесную точку, настолько же закручивая
пружину в противоположную сторону. Затем движение подвески
пойдет в противоположную сторону, и колебания будут продол-
жаться неопределенное время (рис. 12.4,6) с амплитудой и ча-
стотой, определяемыми массой (инерцией) подвески и модулем
упругости материала пружины (и его физическими значе-
ниями) .
В реальной ситуации потери энергии есть всегда, в результате
колебания затухают во времени (рис. 12.4, в). Если противовес
во время колебаний соприкасается с вязкой жидкостью, колебания
будут еще более затухать. Если вязкость этой жидкости возрас-
тает, затухание колебаний усиливается вплоть до прекращения
колебаний, и отклонение плавно переходит во времени от первого
максимума отклонения к равновесному состоянию. Система, как
говорят, является самозатухающей (рис. 12.4, г), и это условие
используется в приборе.
б)
Рис. 12.4. Принцип действия вискозиметра с низким усилием сдвига:
а) схематическое изображение; б) синусоидальная кривая колебаний; в) затухающая
кривая колебаний г) сверхзатухающая кривая колебаний
В приборе, изображенном на рис. 12.5, жидкость (образец
краски) помещается между параллельными пластинами (S), при-
чем нижняя фиксирована и термостатирована, а верхняя вместе
с опорным стержнем (F) и измерительной лопастью (L) является
противовесом маятниковой системы. Опорный стержень опирает-
ся на подшипник (D), который препятствует любым движениям,
кроме вызываемых торзионных перемещений, с минимальными
потерями от трения. Измерительная лопасть является подвижной
частью отрегулированного кварцевого колебательного контура,
так что малейшее движение вызывает разбаланс напряжения,
что используется для измерения отклонения. Наверху стержня
находится кольцевая пружина (К) Вся система может опускать-
Рис. 12.5. Релаксационный вискозиметр
ся и подниматься вдоль вертикального столбика (С) с помощью
пружинного рычага.
При измерениях в начальном состоянии верхняя пластина от-
клоняется на угол 5°, вводится образец и прибор запускается.
При этом пластина освобождается и начинает колебаться (вплоть
до нулевого угла отклонения). Результирующая кривая затухания
во времени может быть получена приблизительно за 30 с, когда
плита автоматически возвратится к исходному положению (и бу-
дет готова к следующему опыту).
Прибор автоматически измеряет и записывает три значения
отклонения (6) через 3, .10 и 25 с после начала измерений (время
t), а также два значения вязкости (путем измерения среднего
наклона dQ/dt в диапазоне отклонений 80—60% при начальном
отклонении 40—30%). Эти значения, а также значения мгновен-
ных отклонений во время испытаний могут регистрироваться на
дисплее прибора.
С помощью зубчатой передачи верхняя плита может вращать-
ся с высокой скоростью (900 об/мин, что соответствует максималь-
Рис. 12.6, а. Типичные результаты, полученные на вискозиметре с низким усилием
сдвига:
1 — исходное состояние краски до приложения усилия сдвига; 2 — скорость сдвига
2500 с"’ в течение 4 с; измерение — через 5 мин после прекращения сдвига; 3— условия
сдвига те же, измерение — через I мин после прекращения сдвига; 4 — условия сдвига
те же, измерение — немедленно после прекращения сдвига
ной (на краю) скорости сдвига 2500 с ') в течение нескольких
секунд, чтобы добиться имитации высокой скорости сдвига, воз-
никающей при кистевой окраске. Таким образом, можно получить
семейства кривых для находящегося в покое образца немедленно
после приложения усилия сдвига и через некоторое время после
этого (чтобы изучить восстановление структуры в тиксотропных
красках). Как видно из рис. 12.6, эти кривые прекрасно коррели-
руют с результатами субъективных оценок растекания краски,
основанными на крупномасштабных окрасках кистью, произве-
денных опытными малярами. Это, вероятно, связано с восстанов-
лением тиксотропной структуры, что имеет более сильное влияние
на начальные стадии растекания краски при окраске кистью,
чем испарение растворителя, по крайней мере, для испытанных
красок.
Используя теорию Орчарда и уравнение движения торзион-
ного осциллятора, в работе [48] авторы смогли выбрать подходя-
щие кольцевые пружины, чтобы получить оптимальные значения
исходного напряжения (3—5 Па). Далее, по мере раскручивания
пружины и движения верхней плиты к нулевому отклонению,
движущая сила уменьшается, т. е. напряжение, приложенное к
образцу, уменьшается, как это и бывает на практике.
Рис. 12.6, б. Типичные кривые для красок с различными характеристиками при
кистевом окрашивании и растекании:
1 — хорошее течение; умеренная скорость восстановления структуры; хорошая выравни-
ваемость рисок от кисти; хорошая стабильность к «стеканию»; 2 — прекрасное течение;
малая скорость восстановления структуры; прекрасная выравннваемость рисок от кисти;
плохая стабильность к «стеканию»; 3 — хорошее разрушение структуры с быстрым вос-
становлением; примерно через 30 с риски от кисти прекращают выравниваться, т. е. это
свойство представлено умеренно, но хорошая стабильность к «стеканию»; 4 — малое раз-
рушение; плохое течение; слабое образование рисок от кисти; прекрасная стабильность
к «стеканию»
Предложено еще два типа приборов [49]. В одном из них коль-
чатая пружина закручивается с контролируемой скоростью
на угол 180°, в результате чего, к образцу передается напряже-
ние, линейно возрастающее во времени. Во время закручивания
мгновенные отклонения верхней плиты записываются как функция
времени. Ньютоновские жидкости дают отклонение, пропорцио-
нальное квадрату времени (считая от начала испытаний). Однако,
если в краске имеется слабоэластичная структура, исходный гра-
фик «отклонение — время» будет скорее линейным, чем парабо-
лическим, с наклоном, пропорциональным эластичности; затем,
при некотором критическом напряжении, начинается разрушение
структуры, и этот график приближается к графику, характер-
ному для ньютоновских жидкостей. Таким образом, этим прибо-
ром можно изучать кинетику разрушения структуры.
Во втором варианте пружина колеблется взад и вперед с раз-
личной частотой и скоростью. Таким способом к образцу можно
приложить синусоидальную нагрузку с различной частотой и
амплитудой. Путем измерения фазового угла и амплитуды дви-
жения верхней плиты можно получить модули динамической вяз-
кости и эластичности образца, как функцию частоты (диапазон
0,02—3 Гц) и амплитуды напряжений (4 значения0,6; 1,5;
5 и 10 Па).
Изучение вязкоэластических свойств суспензий и детальное
описание прибора приведены в работах [50—52]. Поскольку
прибор оборудован приспособлениями для получения высоких
скоростей сдвига, как описано выше для прибора LSV, можно
изучить кинетику структурирования более детально, и, поскольку
в разных частотных интервалах можно измерить вязкость и элас-
тичность как функцию времени, эта информация более полезна,
чем в случае прибора LSV. Последний проще и доступнее, тогда
как колебательные вискозиметры пока в основном являются ис
следовательскими приборами.
Салазковый метод. Если поместить пленку между субстратом
. и тонкой-пдастинкой- (скажем^-маленьким-нредмет-ным стеклом),
то можно получить простейшую установку для изучения сдвига.
Присоединяя через шкив гирю к этому предметному стеклу и запи-
сывая перемещение его как функцию времени, можно определить
вязкость пленки краски. Такая простая методика описана в ра-
боте [53]. В этой работе обсуждены практические трудности, свя-
занные с геометрией сдвига и краевыми эффектами.
Торзионный маятник (шнур). Эти методы можно использовать
для изучения ранних стадий отверждения, пока краска еще жид-
кая или полужидкая. Они более подробно описаны в следующей
главе.
Другие методы. В рамках этой главы возможно описать только
несколько из огромного числа возможных методик. Автор выбрал
некоторые из них, где он имеет личный опыт работы, причем такие,
которые дают абсолютные значения реологических параметров,
и, кроме того, широко распространенные в промышленности.
Более подробный обзор вискозиметров всех типов, применяе-
мых в лакокрасочной промышленности, дан в работе [28].
12.3.2.2. Прямое измерение растекания пленки
В дополнение к измерению реологических характеристик пле-
нок при их растекании были также предприняты попытки прямого
измерения растекания или других явлений течения.
Если лакокрасочный материал способен распространяться
по поверхности субстрата, и можно нанести на поверхность покры-
тия ряд регулярно расположенных полосок, то в этом случае воз-
можно прямо наблюдать направление растекания при высыхании
и измерить амплитуду и длину волны как функцию времени. Такие
измерения были сделаны с использованием методов интерферен-
ции света Уаплером [54], а также Кхешги [55], который исполь-
зовал метод топографии Мойре. Уаплер сравнил свои результаты
с теоретическими представлениями Бирмана [56]. Авторы ра-
боты [55] помимо изучения выравнивания поверхности при расте-
кании также использовали свою методику для изучения профилей
поверхности в жидкости, стекающей под наклоном, в жидких за-
весах, стекающих вертикально через щель, и для изучения воз-
мущений в профилях поверхности, возникающих из-за столкно-
вения с поверхностно-активными частицами. Хотя методика Мойре
менее чувствительная, чем интерференционные методы, она более
гибка и охватывает широкий диапазон отклонений (от нескольких
мкм до нескольких см), при хорошей чувствительности.
Существует много технологических методов для определения
растекания краски на поверхности, а также для выявления круп-
ных дефектов течения. Они приведены в обзоре [28]. Обычный
образец испытательного устройства для измерения стекания пред-
ставляет собой емкость, в торце которой вырезан ряд щелей раз-
личной ширины. Передвигая устройство вдоль стеклянной пла-
стинки, можно получить ряд параллельных полосок краски раз-
личной толщины. Испытательная пластинка затем помещается
на подставку под углом обычно 60° и визуально наблюдается
состояние кромки каждой полоски при высыхании. Стойкость к
стеканию оценивается по минимальной толщине пленки, при ко-
торой кромка за время наблюдения значительно не деформируется.
Такой прибор описан в работе [57]. В работе [56] сделана попыт-
ка теоретически описать процесс стекания и другие подобные
явления.
12.4. РЕОЛОГИЯ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
ПРИ ПРОИЗВОДСТВЕ И ХРАНЕНИИ
Можно назвать четыре области, где контроль или измерение
реологических свойств могут быть полезны: а) измельчение пиг-
ментов; б) транспортировка красок или промежуточых материа-
лов по трубопроводам и т. д.; в) операции смешения; г) хранение.
12.4.1. Измельчение пигментов
Пигменты поставляются изготовителю лакокрасочных мате-
риалов в большей или меньшей степени в форме сухих порошков,
которые по различным причинам содержат множество пигментных
агломератов и агрегатов. Цель диспергирования пигментов со-
стоит в разрушении этих агломератов и агрегатов и в обеспечении
полного смачивания поверхности пигмента жидкой средой лако-
красочного материала, что предотвращает последующую флоку-
ляцию пигмента.
Достижение требуемых оптических свойств пленки сильно
зависит от качества диспергирования пигментов, поэтому важно
оптимизировать условия этого процесса.
Хотя основные принципы диспергирования пигментов хоро-
шо известны, применить их для оптимизации условий дисперги-
рования на практике часто бывает трудно. В работах [58, 59] дан
обзор этих принципов и причин, влияющих на флокуляцию пиг-
ментов в лакокрасочных материалах. В японской работе [60]
рассматриваются методы определения размеров частиц и их рас-
пределения, а также степени диспергирования. Паттон [61] рас-
сматривает практические аспекты этих процессов.
Основываясь на реологии дисперсий, можно сформулировать
ряд принципов. Во-первых, эффективность помола зависит от ко-
личества диссипированной механической энергии. Для ее макси-
мизации необходимо, чтобы средняя вязкость в смеси пигмент —
среда должна быть как можно более высокой, т. е. содержание
пигмента в лакокрасочном материале должно быть высоким.
Пределом Является эффективная объемная доля около 0,64, когда
достигается критическая упаковка пигментных частиц и смесь
-быстро- статювится -подобной твердому--делу. -Н-а практике этот
предел снижается настолько, насколько велика доля анизотроп-
ных пигментных частиц, агломератов, агрегатов.
Начальный эффективный объем, занятый пигментными части-
цами, будеТ больше, чем общий эффективный объем отдельных
частиц из-за объема, занимаемого пигментными агрегатами и
агломератами. По мере разрушения последних при диспергирова-
нии эффективный пигментный объем уменьшается, и вязкость
системы падает. Кроме того, часть механической энергии рас-
сеивается в виде тепла, и, если его не отводить, уменьшение вяз-
кости будет прогрессировать. Общим результатом этого снижения
вязкости является уменьшение диссипирования механической
энергии при диспергировании, что приводит к уменьшению эффек-
тивности диспергирования. Таким образом, время достижения
постоянной вязкости при диспергировании может быть исполь-
зовано для оценки эффективности процесса диспергирования.
Однако, нужно отметить, что изменение размеров частиц и рас-
пределения частиц по размерам может наблюдаться без значи-
тельного изменения эффективного объема и, следовательно, рео-
логических свойств дисперсии. Для измерения этих характеристик
более подходят нереологические методы [60].
Реологические методы для исследования процесса дисперги-
рования использованы многими авторами. Остерл [62] применил
|их для изучения характера диспергирования и эффективности
различных видов диспергирующего оборудования. Маккей [63]
изучил эффективность различных диспергаторов в процессах
диспергирования органических пигментов; Хиртджис и Смите
[64] использовали реологические измерения для изучения диспер-
гирования двуокиси титана в алкидном связующем, обратив ос-
новное внимание на роль молекулярной массы алкида и присут-
ствие жирных кислот. В работах [17, 65, 66] рассмотрены вязко-
эластические свойства пигментных дисперсий, и с помощью этих
свойств изучены направления процессов диспергирования в шаро-
вых мельницах, проведена оценка природы взаимодействия пиг-
ментных частиц, а также эффективность диспергаторов.
12.4.2. Течение по трубопроводам
Обзор ранней литературы по этому вопросу сделан Вельтма-
ном [67]. Паттон [68] также посвятил главу этой проблеме.
В общем, суть проблемы заключается в том, чтобы вычислить
давление, требуемое для перекачивания взятой краски по трубо-
проводу с требуемой скоростью течения. Хотя для ньютоновских
жидкостей (как для ламинарного, так и для турбулентного ре-
жимов) это сделано, неньютоновские жидкости составляют более
серьезную проблему. Измерив кажущуюся вязкость как функцию
скорости сдвига в заданном диапазоне значений (на ротационном
вискозиметре) и применив эмпирические уравнения, например
Кассона или Бингама, можно получить приблизительные данные
о необходимом давлении, пригодные для инженерных расчетов.
Однако временные эффекты (тиксотропия) могут сделать эти
расчеты неверными, особенно при низких скоростях течения. Кроме
того, сильные взаимодействия в материале увеличивают его упру-
гость, что может привести к неприемлемо высокому исходному
давлению, необходимому для начала течения материала. В этом
случае более полезны измерения с помощью трубопроводного
реометра (аналогичного капиллярному вискозиметру, но с более
широким отверстием).
12.4.3. Смешение
Реологические характеристики смешиваемых материалов
имеют важное значение при проектировании оборудования и изу-
чении эффективности процессов смешения. По этому вопросу вы-
пущен ряд книг, например книга Олдшу [69].
12.4.4. Хранение
Определение изменений реологической структуры при хране-
нии представляет собой особенно сложную проблему, что видно
на примере тиксотропных красок. Если бидон с краской открыть,
взять образец и поместить в реометр, структура по крайней мере
частично окажется разрушенной (так же как и при погружении
кисти в бидон). Следовательно, такие измерения желательно
производить прямо в бидоне.
рибор для таких измерений, известный как USCAK *, раз-
работан фирмой ICI несколько лет назад [70]. Его принцип дей-
ствия, кратко описанный в работе [49], аналогичен рассмотрен-
ному выше вискозиметру LSV. Круглый столик в верхней части
прибора колеблется вокруг центра благодаря возвратно-поступа-
тельному движению кольчатой пружины с частотой около 10 Гц.
Амплитуда и фазовый угол движения измеряются в соответствии
с амплитудой и углом привода (аналогично вискозиметру LSV).
Если цилиндрический бидон (контейнер) расположен на столике,
то разница фазовых углов столика и привода равна нулю, а от-
ношение амплитуд имеет конечное значение, зависящее от массы
бидона. Оно принимается как единица шкалы прибора и все по-
следующие измерения приводятся к этой единице для взятой
массы. Когда бидон, содержащий вязкоэластический материал
(например, тиксотропную краску), помещается на столик, отно-
шение амплитуд и разница фазовых углов меняются, поскольку
энергия в этом случае диссипируется из-за вязкоэластических
свойств содержимого билона. Эти_и.гм.гшгш-ия-м4>гу-г-бь1ть исполь-
зованы для вычисления динамической вязкости и эластичности
краски. На практике, путем сложных математических расчетов
строится градуировочный график, с помощью которого можно
получать результаты прямо из показаний прибора. Изменения
веса бидона можно учесть с помощью цифрового потенциометра.
Поскольку сдвиговые волны быстро рассеиваются и т. к. макси-
мальный угол отклонения равен 5° для контейнера емкостью 250 мл
и диаметром 9 см, максимальное усилие сдвига настолько мало,
что массу образца фактически можно считать неизменной.
Измерения можно сделать очень быстро. Образцы, хранящие-
ся в бидонах, взвешиваются и помещаются на столик, вес высве-
чивается на шкале потенциометра, и обычно через 30 с дисплей
показывает значения разницы фазовых углов и отношение ампли-
туд. Отсюда легко вычисляются значения динамической вязкости
и эластичности. Изменения реологической структуры проявляются
в основном в величине упругости и, в меньшей степени, в вязкости.
Прибор также можно использовать для определения оседания
и кинетики структурирования при гелеобразовании.
* Аббревиатура от английского oscillating can rheometer (осциллирующий
реометр для бидона) — Примем, переводчика.
Глава 13
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОКРЫТИИ
Т. А. Страйвенс
13.1. ВВЕДЕНИЕ
Механические свойства покрытий во многом определяют уро-
вень их защитных свойств, а также в определенной степени влияют
на декоративные функции покрытий в течение срока их эксплуа-
тации. Покрытия подвергаются большому числу разнообразных
механических воздействий и деформаций. Они могут подвергать-
ся воздействию больших сил, действующих на малой площади в
течение очень коротких промежутков времени, например при уда-
рах камней, гравия и т. п. в случае автомобильных покрытий, или
последовательному воздействию медленной циклической деформа-
ции, что имеет место в случае декоративных покрытий по дереву
(например для оконных рам), так как древесина расширяется
и сжимается соответственно изменению температуры и атмосфер-
ной влажности. Такие силы и деформации могут быть велики:
порядка гигапаскалей на единицу площади при ударе или 10—15%
растяжения при деформациях древесины (такие деформации
анизотропны в силу характерной структуры древесины). Эти ос-
новные механические свойства покрытий имеют наибольшее прак-
тическое значение, поскольку напряжение или растяжение могут
привести к пластическому течению (необратимая деформация),
или разрушению пленки в результате растрескивания.
Пленки красок в процессе эксплуатации могут испытывать
разнообразные механические деформации. Кроме того, физико-
механические свойства покрытий сами по себе изменяются при
старении. От уровня механических показателей зависит долговеч-
ность покрытия в процессе эксплуатации, т. е. сохранность физи-
ческой однородности пленки определяет время, в течение которого
покрытие может выполнять свои защитные функции. Постоянное
воздействие воздуха и влаги (росы) на поверхность покрытия
приводит к постоянному выщелачиванию низкомолекулярных
веществ, таких как остатки растворителя, пластификатор или
низкомолекулярные полимерные фракции, а также продуктов
деструкции, которые могут размягчать покрытие и увеличивать
его стойкость к хрупкому разрушению (растрескиванию). Кроме
того, воздействие кислорода воздуха и света (особенно его УФ-со-
ставляющей) может активировать различные фотолитические
реакции с образованием свободных радикалов и пероксидов,
вследствие чего может увеличиться степень сшивки пленки
и, соответственно, ее хрупкость. В конечном итоге происходит
разрушение пленки путем растрескивания или на ее поверхности,
или, чаще, внутри пленки. Проникновение воды в пленку может
оказывать благоприятное воздействие, поскольку вода часто
«смягчает», пластифицирует пленку. Однако, если последнее со-
четается с проникновением кислорода или анионов, это может
привести к механическому разрушению вследствие увеличения
хрупкости или накопления твердых продуктов коррозии на поверх-
ности раздела покрытие/металл.
Большинство красок и покрытий основано на органических
полимерах. Они поэтому вязкоэластичны и их реакция на меха-
ническую нагрузку заметно отклоняется от линейной. Кроме того,
механические свойства матричного полимера могут существенно
изменяться благодаря присутствию мелких полимерных частиц
(выделяющихся при смешении компонентов и отверждении),
частиц пигментов, наполнителей и т. д. Роль этих частиц в моди-
фицировании свойств покрытия аналогична роли, которую играют
-наполнители при определении механических свойств полимерных
композитов в массе. Предприняты попытки применить идеи и
теории, выдвинутые для полимерных композитов, для интерпрета-
ции механических свойств лакокрасочных пленок.
В дополнение следует отметить, что большинство систем по-
крытий многослойны. Хотя пленкообразователь может быть один
и тот же, природа и состав пигментов могут изменяться от слоя
к слою. К неоднородности механических свойств, как по толщине,
так и вдоль поверхности пленки, могут привести процессы удале-
ния растворителя и пространственно-неоднородного сшивания
при отверждении. Эти процессы обычно приводят к усадке поли-
мерной матрицы и возникновению внутренних напряжений, ко-
торые изменяют ее механические свойства на подложке. В этой
связи часто возникает вопрос, соответствуют ли результаты
испытаний покрытий исходным свойствам материала, и в ряде
случаев результаты испытаний вообще утрачивают свою цен-
ность.
Достаточно сказано о сложности и важности механических
свойств покрытий, а также о важности их изучения при натурных
или искусственных условиях испытаний.
Настоящая глава построена следующим образом: После крат-
кого обзора вязкоэластичности и механических свойств полимеров
рассмотрены методы определения этих свойств, особенно для
окрашенных образцов, т. е. для адгезированных пленок. После
обзора практически используемых методов механических испыта-
ний покрытий и их интерпретации на более фундаментальной
основе глава завершается разделом о применении акустической
эмиссии для записи изменений механических свойств при искус-
ственном старении.
13.2. ВЯЗКОЭЛАСТИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
Выше были рассмотрены концепции и экспериментальные
методики для измерения вязкоэластических свойств. Эти вопросы
более подробно обсуждены в книгах [1—3] и обзорных статьях
[4, 5].
Характерным свойством большинства полимеров с достаточно
высокой молекулярной массой или степенью сшивки является
то, что они представляют собой эластичные твердые вещества
при комнатной температуре. Если к образцу вязкоэластического
твердого полимера приложить постоянную механическую нагрузку
(эксперимент по изучению ползучести) или усилие растяжения
(эксперимент определения релаксации напряжения), то отклик
будет преимущественно эластическим в том случае, если времени
для перемещения макромолекул или их сегментов относительно
друг друга недостаточно. В ответ на механическое воздействие
они могут передвигаться путем изменения конфигурации, вытяги-
ваясь и изменяя начальные длины связей и углы между ними.
Когда нагрузка снимается, макромолекула возвращается в ис-
ходное состояние. Так запасается и освобождается механическая
энергия (эластический отклик). Аналогичный процесс запасания
и выделения механической колебательной энергии имеет место,
если колебательное (синусоидальное) механическое напряжение
(динамический эксперимент) прилагается к образцу, причем ча-
стота достаточно высока.
Если это время увеличится (или, что то же самое, частота
колебаний уменьшится), то оно может стать достаточным, чтобы
значительное число полимерных макромолекул смогло перегруп-
пироваться друг относительно друга, а их сегменты заняли новые
состояния с равновесными величинами длин связей и углов между
ними. Таким образом, при удалении механической нагрузки не
будет движущей силы, которая могла бы вернуть полимерные
макромолекулы в начальные состояния, следовательно, значи-
тельное количество энергии рассеивается. Хотя на вид нельзя
обнаружить изменений формы образца, но на микроуровне проис-
ходят необратимые деформации. Продолжение этого процесса
приведет к необратимым изменениям формы. Так, даже стеклян-
ная пластина или стекловолокно будут необратимо изгибаться
или вытягиваться под нагрузкой, приложенной достаточно про-
должительное время.
Если температура образца повышается, эти необратимые пере-
группировки облегчаются за счет избыточной поступательной
вращательной и колебательной энергии, которой обладают моле-
кулы полимеров. Иначе говоря, образец по мере роста темпера-
туры переходит от стеклообразного состояния через каучукопо-
добный промежуточный продукт (особенно, если полимерные
макромолекулы сшиты химически) к вязкой жидкости. Соответ-
ствующие изменения, связанные с накоплением энергии и ее рас-
сеянием наблюдаются и в том случае, если уменьшается часто-
та или возрастает время эксперимента. Наблюдаемая в этих слу-
чаях эквивалентность названа принципом суперпозиции вре-
мени — температуры или принципом Уильямса-Ландела-Ферри
(WLF) [6]. Этот принцип имеет огромную экспериментальную
важность. Большинство полимеров имеет широкое молекулярно-
массовое распределение и, как результат, широкий диапазон
временных промежутков для перегруппировок. Изменение меха-
нических свойств, таким образом, происходит в широких пределах
частот или промежутков времени. Поскольку многие из приборов
для определения механических свойств работают только в сравни-
тельно узких диапазонах частот или временных интервалов, точное
определение таких переходов механических свойств («температура
стеклования» или Тс) является проблемой и вызывает необходи-
мость иметыв распоряжении много приборов с различными частот-
ными или временными пределами. Однако, если один прибор
используется для исследования „ойрдзиа при -нескольких различ-
ных температурах, то с помощью принципа WLF результат можно
свести к одной стандартной температуре, т. е. охватить гораздо
более широкий диапазон частот, чем позволяет прибор. Конечно,
в случае покрытий при таком эксперименте следует учесть влия-
ние температуры только на сам образец (с учетом испарения рас-
творителя и пластификатора, дополнительной сшивки или термо-
деструкции) прежде чем применять методику для определения
7\. Значение Тс и особенно ее изменение при воздействии окру-
жающей среды имеет огромное практическое значение для оценки
и предсказания свойств покрытия.
Если приложить достаточную механическую нагрузку в/тече-
ние времени, при котором проявляются эластические свойства,
можно вызвать протекание необратимых перегруппировок. По-
ведение образца становится нелинейным, измеряемая реакция
становится непропорциональной подведенной механической энер-
гии, и при достаточно большом значении последней наблюдается
механическая текучесть и разрушение на макроуровне. Таким
образом, свойства текучести и хрупкого излома также зависят
от времени (частоты) и температуры. В другом случае, когда
при деформации образца с постоянной скоростью растяжения
измеряется мгновенное усилие как функция растяжения, можно
установить, что результаты таких измерений зависят как от ско-
рости деформации, так и от температуры.
Подверженность необратимым изменениям определяется ря-
дом факторов. Важное значение имеет гибкость цепей. Так, гибкий
углеводородный полимер полиизобутилен перегруппировывается
гораздо легче, чем жесткие полимеры с громоздкими боковыми
цепями или жесткими кольчатыми структурами в основной или
боковых цепях (полистирол, целлюлоза и т. п.).
Другой важной характеристикой для оценки механических
свойств является наличие поперечных сшивок между молекулами,
их число, а также соотношение гибкости молекул и длины фраг-
мента цепи между поперечными сшивками (так, вулканизованный
каучук является более жестким, чем невулканизованный). Упоря-
доченность полимерных цепей, наличие межмолекулярных взаимо-
действий, таких как водородные связи, ионные взаимодействия
и т. п., увеличивают Тс.
Присутствие низкомолекулярных фракций, громоздких гибких
боковых цепей, как, например, в случае алкилметакрилатов, увели-
чивает пространство между макромолекулами, облегчает пере-
группировки и снижает 7\. Грубо говоря, чем длиннее полимерные
цепи, тем более они запутаны и тем выше Тс. Для большинства
полимеров Тс не растет заметно после молекулярной массы 20 тыс.
Многие полимерные покрытия готовятся на основе полимеров с
диапазоном молекулярных масс от 5 тыс. до 20 тыс., что необхо-
димо для получения низковязких растворов, обеспечивающих
легкость нанесения красок. Вследствие этого рост Тс по мере суш-
ки или отверждения покрытия оказывает решающее влияние
на его свойства.
Представления о вязкоэластических свойствах твердых ве-
ществ аналогичны представлениям о вязкоэластических свойствах
жидкостей, о чем уже говорилось в гл. 14. Так, кажущийся модуль
(или деформируемость), графически представленный как функция
времени при разных температурах, может быть использован как
параметр для экспериментов с контролируемой нагрузкой. Анало-
гично, истинная и кажущаяся составляющие модуля (обычно
называемые, соответственно, эластичностью и потерей модуля)
графически представляют как функцию частоты в динамических
экспериментах также при различных температурах. Однако мно-
гие промышленные приборы работают при единственной фикси-
рованной или приблизительно постоянной частоте; в этом случае
обычно строят графики зависимости истинной составляющей мо-
дуля и отношения кажущейся и истинной составляющих («тан-
генс потерь», обозначаемый tg б) от температуры. Большинство
этих приборов работает в режиме постепенного подъема темпе-
ратуры. Тс в этом случае определяется при максимальном значе-
нии tg6. Следует помнить, что на величину 7\. влияет скорость
подъема температуры.
В других видах динамических экспериментов, например при
диэлектрических измерениях, широко используются диаграммы
Арганда, в которых строятся графики зависимости кажущейся
составляющей от истинной. Получаются плавные, искаженные
полукруглые или круглые дуговые графики, в которых точки вдоль
графика соответствуют каждому использованному значению ча-
стоты. Искажение дуги или ее ограничение на угле, меньшем
180° (часть полукруга), связаны с наличием более одного харак-
co
Рис. 13.1, а. Идеализированное представление динамических механических свойств
твердого (сшитого) полимера:
1 — стеклообразный; 2 — вязкоэластический; 3 — резиноподобный
Рис. 13.1, б. Идеализированная диаграмма Арганда для механических свойств
вязкоэластического твердого вещества:
/ — высокая частота; 2 — низкая частота
Рис. 13.2. График Арганда динамического механического анализа (ДМА) для
полиэфирной пленки (ср. с рис. 13.5):
1 — наблюдается; 2 — вычислено
теристического времени или с распределением характеристиче-
ских времен. Представить эти параметры на диаграмме Арганда
легче, чем другими способами. Существенно ограниченный диа-
пазон частот, пригодных для динамических механических изме-
рений, ограничивает применение последних. Однако в работах
[7, 8] показано, что метод может быть вполне приемлем, особенно
когда требуются сравнения с данными других динамических ис-
пытаний, например диэлектрических. Недавно в работах [9—11]
использованы диаграммы Арганда для исследования результатов
динамических механических испытаний высыхающих алкидных
и эмульсионных красок. В этих работах выведено простое урав-
нение, и путем сравнения с результатами электронной микроско-
пии и других физических методов анализа найдены параметры
этого уравнения, связанные со степенью сшивки и целостностью
пленки. В работе [12] также дан теоретический обзор по исполь-
зованию диаграмм Арганда и связь последних с динамическими
механическими измерениями свойств пленок.
Примеры для иллюстрации этих данных даны на рис. 13.1, а
и 13.1, б в виде идеализированных кривых. На рис. 13.2 приведены
некоторые экспериментальные результаты, полученные из неопуб-
ликованных данных автора; для полиэфирных пленок 6" и <У ана-
логичны, но не идентичны с G" и G'.
13.3. МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
Так же, как и в случае вязкоэластических свойств полимеров,
ниже дано только очень краткое введение; более подробные
Рис. 13.3. Идеализированные кривые напряжение — удлинение для хрупкого (а)
и пластичного (б) разрушения твердых веществ
сведения можно найти в специальных книгах и обзорах. При
рассмотрении основных свойств полимеров можно выделить два
основных вида разрушения: гибкое (ползучесть) и хрупкое (рас-
трескивание). На рис. 13.3, а и 13.3, б приведены идеализирован-
ные кривые напряжение — удлинение для этих двух типов раз-
рушения.
Если сила приложена к образцу в течение короткого времени,
например в случае удара, то этого времени недостаточно, чтобы
вызвать пластическую деформацию, т. е. чтобы полимерные моле-
кулы или звенья успели переместиться друг относительно друга
и занять новые равновесные положения. Следовательно, полимер
реагирует на воздействие подобно гибкому стеклу (рис. 13.3, а),
и при условии, что напряжение не превысит определенного пре-
дела, хрупкого разрушения не происходит. В принципе, возможно
вычислить предел прочности при хрупком разрушении, зная энер-
гии связей и межмолекулярные силы (см., например 117]). На
практике, однако, значения этих величин на несколько порядков
ниже найденных теоретически. Это обычно связано со структур-
ными неоднородностями образца (щели, трещины, включения
инородных частиц, присутствие деструктированного полимера),
которые приводят к локальной концентрации напряжений. Такой
вывод делает Гриффит [18] на основании теории хрупкого разру-
шения твердых веществ, которая опирается на предположение,
что именно трещины ответственны за снижение прочности по
сравнению с теоретической. Хотя эта простая теория имеет много-
численные существенные недостатки и многократно дополнялась
(см., например [19]), ес основные положения, несомненно, пра-
вильны и неоднократно подтверждены [20].
Если, однако, приложенное усилие действует значительное
время и происходит вязкая деформация, то напряжение прохо-
дит через максимум (напряжение ползучести) и дальнейшая де-
формация образца приводит к его окончательному разрушению.
Этот тип разрушения называют гибким. Сделаны попытки пред-
сказать напряжение ползучести с использованием теории, осно-
ванной на уравнениях, изложенных в работе [21].
Важность времени и температуры при определении характера
разрушения (хрупкое или гибкое) очевидна. В работе [22] рас-
смотрена применимость принципа WLF к исследуемым свойствам.
В работе [23] обсуждались как теория, так и экспериментальные
данные о влиянии содержания пигментов на механические свой-
ства красочных пленок.
13.4. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
МЕХАНИЧЕСКИХ СВОЙСТВ ПОКРЫТИЙ
13.4.1 . Введение
В предыдущем разделе дан общий обзор вязкоэластических
свойств полимеров и их интерпретация. Имеется много работ по
экспериментальным методам определения вязкоэластических
свойств пленок и интерпретации результатов. Поскольку многие
из них представлены в журнале «Progress in Organic Coatings»
или других легко доступных источниках, в этом разделе предла-
гается сжатое рассмотрение методик и результатов. Упомянутые
обзоры можно использовать для более детального и полного
изучения проблемы.
13.4.2 . Экспериментальные экспресс-методы
Обзор таких методов дан в работе [24]. В большинстве из
них используются свободные пленки, которые подвергаются рас-
тяжению с применением промышленных приборов. В испытании
на ползучесть к образцу прилагается нагрузка и возникающая
деформация измеряется как функция времени. Если нагрузку
резко снять, можно исследовать также эластическую релаксацию.
Специальный прибор для таких измерений описан в работах [25,
26].
В экспериментах по релаксации напряжения образец быстро
растягивается до заранее определенной величины, и напряжение
записывается как функция времени. Типичный прибор для этого
описан в работе [27].
Во всех этих испытаниях используются свободные пленки.
Из-за наличия внутренних напряжений часто дебатируется вопрос
о том, насколько такие результаты применимы для оценки меха-
нтгческих “свойств “системы гтокрытие —-подложка. Но этой при-
чине и для получения сопоставимых результатов испытаний по-
крытий при старении и при воздействии окружающей среды,
удобно использовать относительно малые образцы покрытий, на-
несенных непосредственно на подложку.
Для этих целей фирмой ICI сконструирован пневматический
микроиндентор [28]. При перемещении нагруженной иглы внутрь
пленки покрытия изменяется расстояние между поверхностью
пластины и маленьким соплом, через которое подается воздух.
Изменения давления, получающиеся вследствие этих перемеще-
ний, пневматически усиливаются и записываются на диаграм-
мном запи'сываюш.ем устройстве. Образец вырезается из окрашен-
ной пластины. На кончике иглы закреплен стальной или сапфиро-
вый шарик (радиус 0,001—0,025 см). Образец помещается на
обогреваемую плиту под иглой. Температура плиты контролирует-
ся устройством Фригистора, которое позволяет выдерживать
образец в пределах от —20 до -|- 90° С в течение нескольких
минут. Максимальное отклонение записывающего устройства
соответствует перемещению иглы при вдавливании на 6 мкм.
С диаграммного записывающего устройства можно снимать по-
казания с точностью около 0,1 мкм, а точность измерения почти
вдвое выше. Примеры кривых, полученных с помощью этого при-
бора для алкидных пленок в зависимости от температуры и вре-
мени климатических испытаний, приведены в статье [28]. По
форме кривых определена характеристика механических свойств
пленок, т. е. дан ответ на вопрос, проявляют ли пленки при испы-
таниях пластические, каучукоподобные, вязкоэластические или
стеклообразные свойства. Теоретическая интерпретация несколько
сложнее. Ее можно сделать с применением теории Герца [29] с
дополнением [30,31] для вязкоэластических материалов. Следует,
однако, иметь в виду, что силовое поле под вдавливающей иглои
исключительно нелинейно; оно изменяется с глубиной вдавлива-
ния и зависит от нагрузки. Так, если толщина пленки слишком
мала или глубина вдавливания слишком велика, это силовое поле
может взаимодействовать с подложкой и изменять характер за-
висимости глубины вдавливания от времени. Моррис [32] оценил
эти аспекты критически. Несмотря на это, прибор очень полезен;
из дискового образца диаметром около 25 мм можно вырезать
ряд малых образцов для исследований в течение всего времени
климатических испытаний или для опытов с изменением темпера-
туры при вдавливании. Этим способом можно оценить температуру
стеклования. Во всех случаях можно точно оценить изменения
механических свойств при климатических испытаниях.
13.4.3 . Динамические методы
Вопрос о динамических механических испытаниях красочных
пленок детально обсужден в обзоре [33], в котором рассматри-
ваются не только экспериментальные методики, но и приводятся
методики обработки результатов и расчеты характеристик свойств
покрытий и т. п.
Существует четыре основных метода: а) методы свободно
колеблющегося торзионного маятника; б) резонансные методы
при вынужденных колебаниях; в) нерезонансные методы при
вынужденных колебаниях; г) методы ультразвукового импеданса.
Метод а) обычно низкочастотный, и осуществляется при един-
ственном значении частоты около 1 Гц. Методы б). и в) обычно
применяют при частотах от 1 Гц до 10 кГц, причем единственное
значение частоты применяется только для резонансных методов.
Метод г), как следует из его названия, использует высокочастот-
ный диапазон (от сотен кГц до 10 МГц).
13.4.3.1 . Метод свободно колеблющегося торзионного маятника
Устройство напоминает вращающийся часовой маятник; окра-
шенный образец (обычно в виде узкой полоски) образует под-
веску, на которую навешивается груз. Грузу придается легкое
горизонтальное вращение, и он колеблется взад-вперед, вращая
испытываемый образец вокруг вертикальной оси также в обоих
направлениях. Рассеивание энергии в образце и подвеске при-
водит к уменьшению амплитуды вращений, и, таким образом,
масса груза (точнее момент инерции маятника) и эластичность
образца (или подвески) определяют частоту колебаний. Измеряя
частоту и амплитуду затухания колебаний, можно найти эластич-
ность при воздействии усилия сдвига и уменьшение модуля (G'
и G").
Рис. 13.4. Схема перевернутого
торзионного маятника:
/ — гиря; 2 — шкив; 3 — противовес;
4 — образец; 5 — опорная- плита;
6 — зажим
Хрупкость или пластичность многих образцов, например сво-
бодных пленок, затрудняет использование одного этого измере-
ния в достаточном широком температурном диапазоне, т. к. растя-
жение пленки под грузом может влиять на результаты. Сущест-
вуют два решения этой проблемы: или маятник переворачивается
и вес гирь уравновешивается, чтобы минимизировать силу растя-
жения, приложенную к образцу (показано схематически на
рис. 13.4), шш покрытие наносится на металлическую фольгу
(или стеклоткань), которая образует подвеску простого маятни-
кового устройства и выдерживает вес гири. Если использована
металлическая фольга, то для определения модуля покрытия
необходимо предварительно определить его для фольги. Однако
для точного определения модулей серьезной проблемой является
геометрический фактор, поскольку уравнение для модуля содер-
жит разность кубов толщины покрытия с фольгой и собственно
фольги [33]. Поскольку толщину часто бывает трудно определить
достаточно точно и она может изменяться с температурой (в ре-
зультате отверждения, потери растворителя и т. п.), это может
привести к серьезным ошибкам. Менее очевидный недостаток за-
ключается в том, что рабочая частота также изменяется с темпе-
ратурой (из-за изменений эластичности покрытий, которые могут
быть значительными даже в армированных пленках). Эти труд-
ности могут быть преодолены, если использовать методы, рас-
смотренные ниже.
Несмотря на эти недостатки, использование стеклонитей для
армирования пленки весьма распространено, особенно для изуче-
ния реакций отверждения. Этот метод получил название «тор-
зионный анализ», причем имеется промышленное оборудование
для его проведения [34, 35]. При неправильной геометрии нитей
невозможно получить модуль покрытия, но для изучения реакций
отверждения метод вполне удовлетворителен. Если производится
постепенное изменение температуры, результаты могут зависеть
от скорости ее изменения. Гораздо более серьезным, с нашей
точки зрения, является вопрос о смачивании и адгезии (соответ-
ственно жидких и твердых покрытий) к волокнам стеклоткани,
что может влиять на результаты. Имеются работы, в которых
показано, что существенные отклонения может внести склеивание
волокон стеклоткани.
13.4.3.2 . Резонансные методы при вынужденных колебаниях
Резонансная техника заключается в приложении к окрашен-
ному образцу синусоидальной силы и измерении амплитуды про-
исходящих изменений как функции частоты. Резонансная частота
определяется как частота, соответствующая максимальной («пи-
ковой») амплитуде. Путем измерения этого параметра и «шири-
ны пика» можно вычислить модуль. «Ширина пика» определяется
как диапазон частот, за пределами которого амплитуда имеет
значение, равное пиковой амплитуде, деленной на 2. Иногда воз-
можно определить более высокочастотные резонансные пики,
возникающие ввиду протекания более сложного типа деформа-
ции, но они обычно меньше по пиковой амплитуде, чем при нор-
мальной деформации. Метод прост для использования. Образец
свободной или' адгезированной пленки прочно закрепляется в
вибраторе'одним концом и подвергается вибрации. Амплитуда ко-
лебаний свободного конца может быть изменена микроскопически.
Необходимо прочное закрепление образца,, иначе может возник-
нуть псевдор.езонанс. В другой разновидности метода может быть
использована окрашенная тонкая полоска стали (например .брит-
венное лезвие), которая подвешивается на тонкой нити, а колеба-
ния возбуждаются с помощью электромагнита.
Все эти методы имеют общий недостаток. Если покрытйе не-
достаточно твердое (диссипация энергии мала), трудно иденти-
фицировать пик резонансной частоты с точностью,, соответствую-
щей свободной пленке (эта частота, не будет сильно облегчаться
от ее значений для чистого субстрата). rj .. •
Так же как и для маятникового метода, опиу|ЗДн<рш выше,
данный метод основан на использовании одной ч^рдо^ь^и любой
фактор, изменяющий модуль эластичности пшррыгтия,,: например
температура или степень отверждения, будет сдву^ть,,резонанс-
ную частоту. Толщина оказывает несколько .ме^Ц|е^г,влияние
для свободных пленок, так как она входит в ура-внени.е в виде
квадратного члена; однако для армированных образцов уравне-
ния более сложны и включают линейные, квадратичные и куби-
ческие члены в соотношениях толщин покрытия и субстрата [36,
37J. Для большей точности теория также должна учитывать
адгезию покрытия к подложке.
13.4.3.3 . Нерезонансные методы при вынужденных'колебаниях '
Более предпочтительным по сравнению с вышеописанными
методами является измерение амплитуды, как функции частоты
(как и в резонансном методе), и нахождение разности фаз между
приложенной силой и волновым откликом. Этот метод для твер-
дых пленок является точным аналогом метода, используемого
для определения вязкоэластических свойств красок, описанного
в гл. 11 и 12. Он позволяет использовать набор частот .при постоян-
ной температуре, с применением принципа WLF или набор темпе-
ратур при постоянной частоте. Первый вариант является более
точным методом определения Тс, а второй предпочтителен для
изучения процессов отвержденйя. Совокупность таких методов
называют «динамическим механическим анализом» (ДМА).
Важно, что одна из основных конструкций приборов ДМА кон-
тролирует и частоту, и температуру. Геринг [38] опубликовал
результаты, полученные на таком приборе, для образцов красок
(использовалась деформация изгиба).
Диапазон частот может быть в принципе широким (от 10~5
до 104 Гц), однако ряд факторов ограничивает этот диапазон.
Характерным ограничителем является чувствительность измери-
тельного оборудования. Главный лимитирующий фактор верхнего
предела частоты состоит в том, что выше резонансной частоты
на результат все более и более сильно влияет инерция движущих-
ся частей аппарата, а вклад инерции образца также растет с ча-
стотой^_Таким образом, предел частот может быть расширен
только путем снижения до минимума усилий, необходимых для
перемещения движущихся частей прибора. При использовании
образцов в виде полосок толщина покрытия при определении
растяжения становится не столь существенной, так как она входит
в уравнение как линейный член.
Несколько лет назад мы изготовили аппарат для динамиче-
ских испытаний, в котором пытались применить эти принципы [39].
Полоска окрашенного образца зажималась в маленьких вин-
товых алюминиевых зажимах, каждый из которых жестко при-
соединялся к прбчному стальному каркасу. Подвижная под-
веска состоит из спирали, присоединенной с помощью легкой
скобы к зажиму. Специальное устройство позволяет скобке дви-
гаться толькб'ёертикально. Магнит, окружающий спираль, жестко
присоединён" к' основанию прибора. Другой зажим выведен на
крестбобразнуф> пружинную систему, в центре которой располо-
жен сердечник преобразователя линейного смещения (LVDT).
Спираль LVDT и пружинная система жестко закреплены на ос-
новании прибора, которое может передвигаться вверх и вниз с
помощью винта, расположенного вверху прибора. Прибор нахо-
дится в среде (воздух, инертный газ) с контролируемой темпера-
турой. Были изучены модельные алкидные покрытия с целью уста-
новления влияния их химической природы и климатических ис-
пытаний на динамические характеристики (рис. 13.5). Даже для
очень мягких пленок на алюминиевой фольге чувствительность
прибора достаточна для определения значения модулей. Анало-
гичные устройства описаны в обзоре [40].
Для нерезонансных измерений можно использовать динамиче-
ский микроиндентор [41]. Однако могут возникнуть проблемы
в том случае, если поверхность пленки недостаточно эластична
или слишком мягка, чтобы обеспечить достаточную адгезию или
IO10
Угловая скорость, рад/сек
Рис. 13.5. Результаты испытаний фталевого полиэфира (1 неделя выдержки, испы-
тание при 44° С)
постоянный контакт с поверхностью во время колебательного
цикла. Тем не менее, этот перспективный путь измерения вязкоэла-
стических свойств пленок непосредственно на подложке. Ампли-
туда колебаний должна быть малой, чтобы свести ".к минимуму
влияние подложки. Более современная и интересная модификация
приборов для термомеханического анализа (ТМА) описана в
работе [42].
13.4.3.4 . Методы ультразвукового импеданса
Эти методы применительно к изучению отверждения покрытий
развивались Майерсом [43]; он же описал технику измерений и
используемые приборы.
Принцип устройства приборов показан схематически в преды-
дущей главе на рис. 13.3. Волны сдвига, возникшие в поверхност-
ном слое, проходят сквозь поверхность раздела с другим слоем
пропорционально тому, насколько близки импедансы механиче-
ского сдвига. Если применить датчик для детектирования отражен-
ной энергии, можно установить, что отражение ослабляется прямо
пропорционально импедансу субстрата. Применяются подложки
трапециедального сечения. Найдено, что подходящим материалом
подложки для распространения сдвиговых волн является оплав-
ленный кварц, причем оптимальный угол наклона—11° [44];
можно также применять стальные полоски [45].
С помощью пьезоэлектрического преобразователя, присоеди-
ненного к наклоненной поверхности пластины, при частотах 2—
100 МГц ' генерируются импульсы продолжительностью 4 мкс.
Они распространяются внутрь окрашенной поверхности и отра-
жаются от нее, собираясь в ряд экспоненциально затухающих
импульсов, и это затухание записывается. Однако, из этих изме-
рений можно получить только действительную составляющую
сложного модуля сдвига (G'). Мнимую составляющую модуля
сдвига (G") можно найти, измерив фазовые соотношения на
поверхности раздела. Однако G' относительно нечувствительна
к любому измеряемому параметру, кроме затухания [46], следо-
вательно фазовый угол при отражении имеет предел.
На практике при изучении затухания в процессе перехода
от жидкого к твердому состоянию во время отверждения строится
график зависимости этого затухания от времени отверждения,
причем получается сигмаподобная кривая [47].
Используя, аналогичный принцип, но направляя ряд крутиль-
ных колебательных импульсов на поверхность оплавленного квар-
цевого стержня, Мьюис [48] создал прибор, работающий при
частоте 100—150 кГц. Покрытие наносится на поверхность стерж-
ня, и извернётся угол фазового отражения и затухание, поэтому
можно’ вычислить' обе составляющие модуля сдйига. Как указы-
валось ” [49], диапазон частот позволяет с помощью
этого прибора изучить различные стадии процесса высыхания.
Прибор ;МлЛбйёа'более подходит к изучению начальных стадий
высыхаЙия^ЙЯЙ' Утверждения, тогда как более высокочастотное
оборудование предпочтительнее для исследования заключитель-
ных стай.й'й^п'роцесса.
Однако в описанных методах требуется сложное электронное
оборудование, и поэтому обсуждается вопрос, не может ли гораздо
более простая техника (метод отскока шарика, крутильный шну-
ровой анализ) быть настолько же эффективной при изучении
отверждения или высыхания.
13.4.4 . Измерение конечных свойств
Эти свойства 'покрытий можно измерить так же, как и для
полимеров, т. е. путем растяжения прямоугольного образца сво-
бодной пленки при контролируемой скорости и построения графи-
ка напряжение — деформация при нескольких скоростях дефор-
мации. Температура и влажность при испытаниях должны контро-
лироваться. Использование этих измерений описайО Эвансом [50].
Методы исследования адгезированных пленок описываются реже,
за исключением технологических тестов, описания которых обыч-
но кратки. Симпсон [51] предложил циклическую разрывную
машину, в которой можно испытать покрытия на металлических
пластинах до начала и во время ускоренных климатических ис-
пытаний. Разрушение покрытия наблюдается визуально по появ-
лению трещин на поверхности. Такие испытания можно исполь-
зовать для предсказания долговечности или для регистрации
изменения механических свойств покрытий при атмосферном
старении. Гораздо более чувствительными приборами для опре-
деления разрушения в результате растрескивания (которые так-
же определяют вид подповерхностного разрушения — когезион-
ный или адгезионный) являются приборы акустической эмиссии,
которые будут рассмотрены в последнем разделе данной главы.
Основная проблема при использовании свободных пленок
заключается в трудности получения качественных образцов, т. к.
например, наличие краевых дефектов будет серьезно влиять на
точность и воспроизводимость измеренных параметров. Вообще,
лучше применять методы испытаний покрытий, нанесенных непо-
средственно на субстраты.
13.5. ОБСУЖДЕНИЕ ЭКСПЕРИМЕНТАЛЬНЫХ МЕТОДОВ
Выбор наиболее пригодного экспериментального метода из
их разнообразия вызывает определенные вопросы и должен учи-
тывать многие факторы (время, необходимое оборудование, цена),
однако наиважнейшим определяющим фактором является цель
испытаний. Можно перечислить ряд общих принципов выбора
метода испытаний:
1. Если это экспериментально возможно, испытание покры-
тия на подложке всегда предпочтительнее испытания свободных
пленок.
2. Механические свойства всегда зависят от температуры и
временных параметров (периодичности, времени, скорости рас-
тяжения и т. п.) и часто от влажности. Совершенно необходимо
строго контролировать первые два параметра, а также влажность
воздуха при испытаниях. Здесь кроется причина ошибок, неадек-
ватности в большинстве технологических тестов, применяемых
в промышленности для оценки механических свойств (будут опи-
саны ниже).
3. Если требуется оценка или предсказание долговечности
покрытия, то следует измерять и конечные свойства.
Однако, если целью испытаний является изучение процессов
высыхания и отверждения или влияние химических (физических)
изменений в структуре покрытия (изменение рецептуры, измене-
ния в процессе старения и др.), то динамические испытания при
малых нагрузках (деформации) могут дать наибольшую инфор-
мацию и являются наиболее легкими для интерпретации резуль-
татов, исходя из физико-химии покрытии, ни также являются
более надежными, так как случайные дефекты покрытий менее
сказываются в условиях воздействия малых сил или деформаций.
13.6. ТЕХНОЛОГИЧЕСКИЕ ИСПЫТАНИЯ МЕХАНИЧЕСКИХ СВОЙСТВ
Существует обширный ряд методов испытаний твердости,
гибкости и других свойств, используемых в лакокрасочной про-
мышленности для испытания механических свойств покрытий.
Все они достаточно полно описаны в обзорах [52—54], и ниже
дается только краткий комментарий с более детальным рассмотре-
нием некоторых хорошо отработанных тестов, которые дают воз-
можность соотнести результаты с такими фундаментальными
характеристиками, как вязкоэластические свойства.
13.6.1. Испытания твердости
Как указывал Сато [54], термин твердость неточен. Его смысл
характеризует жесткость вещества, т. е. сопротивляемость внешне
приложенной силе. Это свойство зависит как от величины прило-
женной силы, так и от скорости ее приложения. Существуют три
основных типа измерения твердости: методом вдавливания, ме-
тодом царапания и маятниковым методом.
13.6.1.1. Измерения твердости методом вдавливания
Этот метод разработан на базе методик Бринеля или Рокуэла,
применяемых для резин и пластмасс. Для уменьшения влияния
подложки глубина вдавливания должна быть малой. ASTM D1474
предусматривает применение прибора Тукона для измерения твер-
дости по Кноопу Алмазный индентор имеет наконечник узкой
ромбоэдрической формы, и покрытие, нанесенное на массивную
металлическую или стеклянную подложку, деформируется путем
вдавливания индентора под грузом 25 г в течение 18 + 2 с. После
удаления груза измеряется длинная диагональ отпечатка, полу-
чившегося при вдавливании, и вычисляется число Кноопа (KHN) *.
Меркурио [55] построил графики зависимости KHN и модуля
Юнга от температуры для покрытий полиметилметакрилата и
показал, что оба графика имеют одинаковую форму и значения
Д идентичны (110°С). В работе [56] с использованием прибора
автора показано, что твердость, определенная методом вдавли-
вания для ряда термопластичных покрытий, пропорциональна
модулю Юнга. Возможно, наиболее чувствительным прибором
этого типа является микроиндентор ICI, описанный выше. Недо-
статок использования точечных конических или призматических
* KHN—аббревиатура от английского «Knoop Hardness Number» — число
твердости по Кноопу (Прим, переводчика).
инденторов состоит в том, что они приводят к прерывистым полям
напряжений в покрытии. Сферический индентор ICI не имеет
этого недостатка, так как максимальная глубина вдавливания,
составляющая 6 мкм, сводит влияние подложки к минимуму.
Большинство исследователей, работающих с этим прибором,
определяют глубину вдавливания за фиксированное время с мо-
мента начала испытания. Следует также иметь в виду, что с по-
мощью сферического индентора можно определить зависимость
деформации при сдвиге от времени при разных температурах
путем построения графика в координатах глубина вдавливания —
время.
13.6.1.2. Измерение твердости методом царапания
Эти испытания разнообразны и включают качественное испы-
тание царапающим карандашом, обычно применяемым в про-
мышленности, а также точечные нагруженные инденторы, кото-
рыми проводят по пленке с постоянной скоростью, с последующим
измерением ширины получившейся царапины [57]. Меркурио [55]
утверждает, что твердость пленки по карандашу связана с удли-
нением при разрыве, т. е. покрытие разрушается только когда
максимальное напряжение, характеризующее твердость, превы-
сит прочность пленки при разрыве.
13.6.1.3. Маятниковый метод
В этом методе используется вращательное движение шарико-
вых опор маятника взад-вперед по горизонтальной поверхности
покрытия. Такое движение обусловливает свободное колебание
маятника с амплитудой, затухающей во времени. .Исходная дви-
жущая сила определяется начальным отклонением и равнодей-
ствующим вращательным моментом, возникающим :из-за сил гра-
витации, когда маятник отпущен. Энергия движения.рассеивается
в покрытии, следовательно, частота и декремент колебаний свя-
заны с разностью вязкоэластических свойств покрытия и неокра-
шенной стеклянной или металлической массивной подложки.
Твердость по маятнику может быть выражена различными спо-
собами: временем в секундах, временем, необходимым для умень-
шения амплитуды колебаний вдвое (или в другое число раз) от
исходного значения, числом колебаний, а также в относительных
единицах как процент от времени, измеренного на стандартной
стеклянной пластинке. Существующее представление о том, что
измеренная твердость обратно пропорциональна способности по-
крытия гасить колебания маятника, неверно [58]. Поэтому недо-
статочно корректно сравнивать покрытия с различными вязко-
эластическими свойствами только по их твердости, хотя такое
сравнение правомерно при одинаковых вязкоэластических свой-
ствах.
Имеются два основных типа маятников: распространенные
в Европе маятники Кенига и Персожа, в которых используются
инденторы в качестве опоры вращения маятника, и маятники
типа балансира Шварда, популярные в Японии и в США, где
круглая опора с закрепленным маятником вращается взад-вперед
по окрашенной подложке. Первый тип маятников превосходит
второй в точности и в воспроизводимости и имеет меньшую пло-
щадь контакта с пленкой [59, 60]. Балансир Шварда анализиро-
вался в работах [61—63].
Для меламиналкидных покрытий было произведено сравне-
ние температурной зависимости декремента свободных вращатель-
ных колебаний и декремента маятника Кенига [64, 65]. Эти два
метода хорошо коррелируют. Следует, однако, помнить, что сма-
чивание влияет на площадь контакта маятника с поверхностью
в точке враще'ния, и что эта площадь будет возрастать по мере
роста температуры и размягчения пленки, в результате чего ша-
-рдковые опоры индентора будут шэ-рр-у-ж-ат-ьоя—в—покрытие. Не-
смотря на это, для вязкоэластических свойств можно получить
хорошие результаты, а простота методики делает ее привлека-
тельной. Это детально проанализировано Сато [54].
13.6.2. Испытания гибкости
Существует два основных типа испытаний: испытания на из-
гиб, при которых окрашенные пластинки огибаются вокруг опра-
вок, и тест Эриксена, в котором подложка деформируется боль-
шим индентором, оканчивающимся полусферой.
В испытании на изгиб тонкая окрашенная с одной стороны
металлическая пластинка огибается неокрашенной стороной во-
круг оправок различного диаметра. Суть испытания состоит в
том, чтобы определить наименьший диаметр оправки, при кото-
ром наступит растрескивание. Очевидно, что для получения срав-
нимых результатов важно контролировать температуру и скорость
огибания.
В тесте Эриксена полусферический индентор вдавливается в
подложку с неокрашенной стороны. Измеряется глубина вдавли-
вания, при которой начинается растрескивание пленки. В этом
случае также необходимо контролировать температуру и скорость
деформации.
Оба теста могут быть более чувствительными, если использо-
вать специальный прибор для контроля скорости деформации,
а также акустический эмиссионный детектор — более точный
прибор для определения растрескивания, чем визуальное наблю-
дение. То, что это возможно при испытании на гибкость, было
показано в работе [66]. Использованный в этой работе прибор
не так чувствителен, как прибор акустической эмиссии, его можно
применять для различных покрытий.
13.6.3. Испытания на удар
Трудно предсказать ударную стойкость на основе вязкоэласти-
ческих свойств покрытия из-за сложного характера профилей
деформации и напряжений и отсутствия достоверных данных о
механических свойствах при очень коротком времени воздействия.
Тимошенко и Гудиер [67] разработали теорию удара, которая
позволяет оценить величину его наиболее важных параметров.
Используя эту теорию, Зорл [68] установил время удара, которое
составляет десятки микросекунд. Это согласуется с эксперимен-
тальными данными. Например, Кирби [69] нашел, что это время
составляет 18±3 мкс при комнатной температуре (8-мм сталь-
ной шарик, удар по толстой стеклянной пластине со скоростью
1,7 м/с), а Калвит [70] установил значение 100 мкс (монолит
ПММА, температура ниже его температуры стеклования, 5-мм
стальной шарик, скорость удара 0,7 м/с).
Обычным требованием, предъявляемым к автомобильным
эмалям, является стойкость к удару гравием. В работе [71] описан
простой прибор для проведения таких испытаний. Здесь лег-
кий стальной сферический индентор вдавливается в покрытие
со скоростями до 25 м/с. Измеряются минимальная скорость, при
которой повреждение покрытия становится видимым визуально,
и площадь поврежденной поверхности. В результате этих измере-
ний было установлено, что существует температурный интервал
в районе 7’с покрытия, в котором стойкость к удару является опти-
мальной. Аналогичные выводы следуют из работ [72, 73], в ко-
торых сравниваются результаты измерений стойкости покрытий
к ударам гравия и вязкоэластических свойств, покрытий.
Априори можно ожидать, что поведение покрытия при ударе
будет также сильно зависеть от формы ударяющего тела и угла,
под которым наносится удар. Это было подтверждено многими
авторами, в частности в работе [74], где произведено сравнение
конических и сферических ударяющих тел. Также были подтвер-
ждены выводы относительно взаимосвязи Тс покрытия и оптималь-
ной ударной стойкости.
При определении ударной стойкости важными являются меха-
нические свойства отдельных слоев многослойного покрытия и
их соотношение. Технологические испытания основаны на падении
стадартных тел через трубку на покрытие (например британский
стандарт BS AU 148). По другому способу тщательно отсортиро-
ванный гравий или стальная дробь сильным потоком воздуха
направляются на окрашенную пластину (как в приборе для испы-
тания стойкости к ударам гравия).
13.7. АКУСТИЧЕСКАЯ ЭМИССИЯ
Использование акустической эмиссии для обнаружения тре-
щин, возникающих при воздействии напряжений в инженерных
конструкциях, например в нефтяных платформах в Северном море,
в сосудах высокого давления, на самолетных крыльях и т. д.,
стало обычным делом в последние годы. Однако использование
этого метода для изучения покрытий, подвергающихся воздей-
ствию напряжений, является новинкой. Методика, предложенная
впервые фирмой ICI, находит значительное применение при по-
стоянном контроле за состоянием покрытий и даже, в некоторых
случаях, в предсказании их долговечности при натурных испыта-
ниях, как естественных, так и ускоренных, испытаниях коррозион-
ной стойкости путем разбрызгивания соли и т. п. Кроме того,
метод очень полезен для оценки влияния изменений рецептуры
лакокрасочного материала на механические свойства покрытия
и для оценки этих свойств для отдельных слоев системы покрытия,
а также для уяснения того, как эти свойства «накладываются»
друг на друга, формируя общее свойство всей системы. Методика
в принципе очень проста. Любое внезапное микроскопическое
перемещение-В-деле, .например -обрадование и развитие трещины,
может вызвать акустическую эмиссию. Например, напряжение
концентрируется на растущем конце трещины. По мере распро-
странения последней энергия напряжения выделяется в двух
главных фо'рмах: термической и акустической. Последняя излу-
чается как деформационная волна из источника и преломляется
и отражается твердыми включениями и поверхностями раздела,
пока не достигнет поверхности тела. Здесь волны можно воспри-
нимать чувствительными датчиками, обычно пьезоэлектрическими
или мощными преобразователями. Затем усиленный сигнал от
преобразователя анализируется. Простыми примерами акустиче-
ской эмиссии при частотах и интенсивностях, воспринимаемых
человеческим ухом, являются растрескивание льда на водоеме
или скрип деревянных ступеней лестницы под весом человека.
Покрытие наносится на одну сторону полоски фольги, послед-
няя помещается в зажимы разрывной машины, присоединяется
преобразователь, и образец растягивается. Появляющийся «шум»
анализируется и строится график зависимости некоторых «шумо-
вых» характеристик от общего напряжения. Хотя испытание на
растяжение наиболее распространено, однако можно использо-
вать изгиб или любой другой вид деформации. Единственное
существенное ограничение состоит в том, что не должно быть
постороннего шума от скольжения образца в зажимах. Это един-
ственный источник шума, от которого следует защищать прибор.
Поэтому используют малые скорости деформации.
В работах [75—77] установлено, что для этих целей следует
применять пьезоэлектрические устройства с узким диапазоном
частот (около 150 кГц). Для увеличения чувствительности детек-
тора существенна хорошая акустическая связь между поверх-
ностью преобразователя и образцом; этого легко достичь, исполь-
зуя тонкий слой силиконовой смазки.
Число акустических сигналов
Рис. 13.6. Влияние влажности и ускоренное™ испытаний на звуковую эмиссию
при деформации (алкидная краска для наружных работ):
/ — влажное испытание; 11 — сухое испытание; 1 - образование трещин и шелушение;
2 — трещины; 3 — исходная краска
Методы анализа, пригодные для характеристики акустической
эмиссии, многочисленны. Из-за одновременного существования
многих источников шума, а также из-за изменения вида волн,
как при прохождении через образец, так и в детекторе, по акусти-
ческой эмиссии образцов покрытий очень трудно проанализиро-
вать сложные сигналы, чтобы получить информацию об исходном
источнике сигнала. Существует слишком мало теоретических
или экспериментальных работ с модельными системами. Сложная
техника частотного или амплитудного анализа обычно мало при-
емлема, хотя последняя может дать информацию о резком изме-
нении механизма разрушения покрытия, например, если наблю-
дается переход от микро- к макрорастрескиванию при обычных
величинах напряжения. Для характеристики покрытий предла-
гается также использовать простые методики анализа, такие как
построение графиков зависимости числа колебаний от общего
значения напряжения. На основе этих графиков можно проводить
анализ изменения свойств покрытия при натурных испытаниях,
изучение влияния изменений рецептуры лакокрасочного материала
на механические свойства и т. п. Пример такого использования
приведен на рис. 13.6. Видно, что на алкидные пленки сильное
влияние оказывает влага и в большинстве случаев происходит
ухудшение адгезии.
Глава 14
ДЕКОРАТИВНЫЕ СВОЙСТВА ПОКРЫТИЙ.
ОСНОВНЫЕ ПОЛОЖЕНИЯ
Т. Р. Буллетт
14.1. ВВЕДЕНИЕ
Одним из наиболее важных свойств красок является возмож-
ность с их помощью изменять внешний вид подложки практически
без ограничений. Маскировка и украшение всегда были двумя
основными направлениями использования красок. Ремесленники
использовали их для имитации внешнего вида природных мате-
риалов, причем основными объектами имитации были древесина
и полированный мрамор. Термин «эмаль» пришел в лакокрасоч-
ную промышленность в то время, когда люди научились изготав-
лингать краски, воспроизводящие внешний Вид и твердость стекло-
видных эмалированных драгоценностей; успехи в этой области
были столь велики, что появилась необходимость квалифициро-
вать такие лакокрасочные материалы как «эмалевые краски».
Однако краски по-прежнему остаются самостоятельным видом
композиций, которые могут после нанесения образовывать глад-
кую поверхность (с высоким блеском или полностью матовую)
с различными типами структуры и широким диапазоном цветов.
Настоящая глава в основном посвящена рассмотрению физи-
ческих эффектов, определяющих основные декоративные свой-
ства — блеск, укрывистость, цвет. В то время как отражение,
поглощение, рассеяние света подчиняются объективным законам
физики, внешний вид пленки во многом зависит от наблюдателей;
их физиологических особенностей, и часто от психологии.
14.2. ФИЗИКА ОТРАЖЕНИЯ НА ГРАНИЦЕ РАЗДЕЛА ПОКРЫТИЕ/ВОЗДУХ
14.2.1. Плоские поверхности
Когда луч света достигает поверхности раздела двух материа-
лов с различной оптической плотностью, часть света отражается,
а остаток распространяется во втором материале с изменением
направления (рефракция) (рис. 14.1). Количество отраженного
света зависит от показателей преломления двух сред и от угла па-
дения пучка света. Количественное определение отражения ослож-
няется тем, что поляризованный в плоскости поверхности свет
отражается легче, чем свет, поляризованный перпендикулярно. Это
в какой-то степени аналогично тому, что плоский камейь, брошен-
ный горизонтально над водой, будет отскакивать от воды, но будет
тонуть, если его длинная ось расположена вертикально. Матема-
Рис. 14.1. Отражение и преломление
на поверхности раздела
воздух/покрытие:
/__падающий свет; 2 — отраженный,
3 — преломленный
тически [1] коэффициент отражения для света, поляризованного
в плоскости поверхности, выражается следующим образом:
Г sin (/—<) I2
I sin (т-Н) J ’
(14.1)
и для света, поляризованного под углом к поверхности:
где i — угол отражения, г — угол преломления.
На рис. 14.2 показана зависимость Rp и Rsot угла падения света
при показателе преломления второго материала п2 = 1,5 (значение,
типичное для красок).
Для неполяризованного света коэффициент отражения имеет
среднее значение между Rs и Rv и возрастает линейно, при п2 = 1,5
от 0,04 (4%) при перпендикулярном падении света до 1 (100%)
при скольжении света по поверхности (/ = 90°).
Из уравнений (14.1) и (14.2) представляют интерес два след-
ствия. Во-первых, при г4-/ = 90°, tg(r-H) становится бесконечным
и /?р = 0, а это означает, что отраженный свет полностью поляризо-
ван в плоскости поверхности. При г -Н = 90°, sin г — cos I и из урав-
нения Снелла n = sin i/sin r=tg i. Это основа углового метода
Брюстера для определения показателя преломления, в котором
определяется угол падения света, при котором отраженный луч
может полностью отсекаться путем использования поворотного
поляризующего фильтра. Методика требует высокоточной аппара-
туры и может использоваться для определения показателя пре-
ломления черного стекла (используемого как стандарт при изме-
рении блеска) или лаковых пленок на черном стекле, а также (с не-
которым снижением точности) для пленок с высоким блеском.
Рис. 14.2. Зависимость /?, и Rp от угла падения света (п2=1,5)
Рис. 14.3. Увеличение зеркального отражения с ростом отношения показателей
преломления
Во-вторых, при i = 0 коэффициент отражения уменьшается по
формуле:
/?=(П'~Пг), (14.3)
\П| +«2 /
где /11 и п-2 — показатели преломления первого и второго материалов.
На рис. 14.3 показано, как возрастает R с увеличением отноше-
ния nt/tix в диапазоне от 1,2 до 2,0. Как будет показано в дальней-
шем, интенсивность зеркального отражения резко возрастает
с увеличением показателя преломления, т. е. более интенсивное
отражение происходит от покрытий на основе смол с высоким по-
казателем преломления (например фенольных смол), и менее ин-
тенсивное — в случае смол с низким п (например ПВА). Эти заме-
чания очень важны как для практической работы с блескомера-
ми, так и для составления рецептур красок для покрытий с высоким
блеском на их основе.
14.2.2. Эффекты структуры поверхности
Выше рассматривалась оптически плоская поверхность. Если
поверхность по тем или иным причинам неоднородна, луч света па-
дает на ее различные участки под различными углами. Отражен-
ный свет в результате распространяется под разными углами
и чисто зеркальное отражение нарушается. Величина структурных
неоднородностей поверхности, необходимых для нарушения зер-
кального отражения, зависит от длины волны и угла падения света.
При углах падения от 0 до 45° шероховатости поверхности, экви-
валентной длине волны света (0,4—0,7 мкм), достаточно для того,
чтобы завуалировать зеркальное отражение; для скользящего
луча требуется более сильно выраженная текстура для нарушения
отражения. Таким образом, при появлении повреждений на бле-
стящей поверхности покрытия под воздействием окружающей сре-
ды первым эффектом является потеря блеска, наблюдаемая под
большими углами к поверхности. Полностью матовую пленку
можно получить путем введения более крупных по сравнению
с длиной волны частиц. Для этого достаточен диаметр частиц 10—
15 мкм (в толстых пленках).
14.3. СВЕТОРАССЕЯНИЕ И СВЕТОПОГЛОЩЕНИЕ ПОКРЫТИИ
14.3.1. Общие положения
Свет, преломившийся в покрытии, частично поглощается, а час-
тично рассеивается. Светорассеяние на пигментных частицах зави-
сит от их размера, длины волны света и соотношения показателей
преломления пигмента и полимерного связующего. Поглощение
в пигментных частицах зависит от длины пути света через частицу
и оптической плотности пигментированной пленки. Большинство
теоретических работ по светоотражению пленками покрытий (на
пример работы Кубелки и Мунка [17]) предполагают, что много-
кратное рассеяние на большом числе частиц приводит к полному
рассеянию потока света, подобно явлениям, наблюдаемым в обла-
ках или густом тумане. Важно представлять себе, что такой подход
несовершенен в случае тонких или слабопигментированных пленок,
где не достигается необходимая степень светорассеяния.
14.3.2. Отражение на поверхностях раздела
Свет, прошедший через пленку, частично поглощается подлож-
кой, а частично ею отражается; часть отраженного света вновь
проходит через пленку, так что пленка тем светлее, чем выше
светоотражающие свойства подложки.
Полное математическое описание этих явлений сложно даже
даш упрощенного случая, когда глянцевая пленка постоянной тол-
щины нанесена на матовую подложку. Это связано с тем, что зна-
чительная часть (обычно более половины) света, рассеянного на
пигментных частицах, подвергается отражению на границе разде-
ла пленка/воздух (рис. 14.4) и, таким образом, ослабляется вновь
ввиду поглощения пигментами или подложкой прежде, чем вто-
рично достигнет поверхности раздела. Аналогичная ситуация воз-
никает на границе раздела пленка/подложка, когда свет подвер-
гается отражению между подложкой и пигментированным слоем
[3]. На рис. 14.5 показан последовательный ряд актов внутрен-
него отражения, которые происходят до тех пор, пока весь свет не
поглотится или окончательно не отразится от пленки.
Влияние поверхности раздела воздух/плеика можно весьма
просто вычислить суммированием геометрических прогрессий внут-
ренних отражений:
(14.4)
(1 —/?Е) (!—>?,) Rp
1 — R\Rp
Рис. 14.4 Внутреннее отражение на поверхности раздела воздух/покрытие:
I/ — чистое связующее; 2 — воздух, 3 — поток света; 4 — полное внутреннее отражение;
5 — пигмент
Таблица 14.1. Влияние отражений на поверхности раздела воздух/пленка на коэффициент отражения глянцевых пленок (/?Е=0,05; Rl=0,55)
ЯР Наружное отражение Яр Наружное отражение
Коэффициент отражения суммарный Коэффициент диффузного отражения Коэффициент отражения суммарный Коэффициент диффузного отражения
Г. 0,05 0,072 0,022 0,60 0,433 0,383
0,10 0,095 0,045 0,70 0,537 0,487
0,20 0,148 0,098 0,80 0,661 0,611
0,30 0,204 0,154 0,90 0,812 0,762
0,40 0,269 0,219 0,95 0,901 0,851
0,50 0,345 0,295 1,00 1,000 0,950
Где /?т — коэффициент отражения от глянцевой пленки; /?Е —
коэффициент наружного отражения от поверхности раздела;
— коэффициент внутреннего отражения от поверхности разде-
ла; /?Р — коэффициент отражения от пигментированной пленки
на подложке.
! Табл. 14.1 иллюстрирует изменение /?т в зависимости от /?Р для
7?е=О,О5 и Ri = 0,55, причем типичные значения даны для угла
освещения 45° и показателя преломления среды 1,5. Можно отме-
тить, что влияние поверхности раздела должно значительно умень-
шить отражение. Вследствие этого большинство неглянцевых мате-
риалов (включая матовые покрытия) заметно темнеют, если их
Ьокрыть лаком. Для очень темных поверхностей эффект лакирова-
ния зависит от того, будет ли наблюдатель видеть зеркально отра-
Рис. 14.5. Внутренние отражения на поверхности и подложке:
1 — падающий свет; 2 — воздух; 3 — чистое связующее; 4 — пигмент; 5 — подложка
женный от поверхности свет; если это происходит (например,
поверхность освещается рассеянным светом, отраженным от об-
лачного неба или стен), исходный цвет кажется светлее. Это кажу-
щееся посветление темных цветов часто воспринимается сильнее,
чем можно предположить на основании табл. 14.1, так как для со-
вершенно однородного освещения со всех направлений /?Е возрас-
тает почти до 0,1.
Одно существенное следствие отражения на поверхности разде-
ла важно в рассмотрении оптических свойств пленок. Свободная
пленка (или пленка, наложенная на прозрачную фольгу) имеет по-
верхность раздела воздух/пленка с обратной стороны такую же,
как и с освещенной. Эта вторая поверхность раздела также имеет
коэффициент внутреннего отражения около 0,55 для рассеянного
света. Таким образом, свободная пленка (неукрывающей толщи-
ны) будет иметь такую же отражательную способность, как если
бы она была нанесена на светло-серый субстрат, если же эту пленку
свободно положить на черно-белые клетки шахматной доски, кон-
трает будет ш нотоште ньше, чем для той же пленки, нанесенной на
доску.
14.3.3. Светорассеяние белых пигментов
Белые пигменты изготавливают из прозрачных, почти бесцвет-
ных материалов, используемых в красках в виде мелких частиц.
Соотношение между размерами частиц и светорассеянием изуча-
лось в 1908 г. Маем [4], который показал, что максимальное
светорассеяние на единицу количества материала имеет место для
частиц с диаметром несколько меньшим, чем длина волны света.
Рис. 14.6 показывает изменение рассеяния в зависимости от диа-
Рис. 14.6. Зависимость рассеяния от размера одиночных сферических частиц
(вычислено для рутильной двуокиси титана в льняном масле)
метра сферических частиц. Строго говоря, эта кривая относится
к рассеянию на единственной частице, т. е. когда свет рассеива-
ется каждой частицей. В пленке оптимальный для светорассеяния
размер частиц не сильно отличается, за исключением случаев очень
сильного пигментирования, где светорассеяние значительно мень-
ше из-за того, что частицы расположены гораздо ближе друг к дру-
гу. Промышленные белые пигменты выпускаются с диаметром
частиц, который обеспечивает наилучшее рассеяние зеленого света
(для максимальной кроющей способности лакокрасочного мате-
риала); это составляет для рутильного диоксида титана около
0,25 мкм. Частицы этого размера менее эффективны при рассеянии
желтого или красного света, так что тонкие белые пленки прони-
цаемы для оранжевого света.
В работах по рутильному диоксиду титана оценена степень,
в которой коэффициент светорассеяния уменьшается при повыше-
нии концентрации пигментов. Для частиц оптимального для свето-
рассеяния размера при низких концентрациях рассеяние на части-
цу уменьшается примерно вдвое при ОКП = 30%. Для частиц та-
кого размера увеличение ОКП выше 30% не дает дальнейшего
прироста кроющей способности; последняя может даже упасть
в диапазоне концентраций, где прирост рассеяния от увеличения
числа частиц меньше, чем уменьшение рассеяния от более плотной
упаковки. При очень высоких ОКП связующего недостаточно,
чтобы заполнить промежутки между пигментными частицами,
и кроющая способность растет. Имеются данные, что при ОКП =
= 30%, несколько более крупные частицы (например 0,4 мкм вме-
сто 0,25'мкм) дают лучшую укрывистость. Также показано [5],
что изменение содержания рутильных частиц путем добавки мел-
ких частиц наполнителя с низким показателем преломления (таких
как тонкоизмельченный диоксид кремния или карбонат кальция)
значительно улучшает укрывистость при высокой ОКП.
Эти выводы совпадают с предположением, что при высоких кон-
центрациях пигментов рассеяние происходит на гранях пустот
между частицами в большей степени, чем на отдельных частицах.
Теория светорассеивающих систем, которая базируется на пред-
ставлениях о наличии пустот с низким показателем преломления,
образующихся в непрерывной среде с высоким показателем пре-
ломления, может быть столь же эффективной, как и другие теории.
Предпринято много попыток для использования пустот в пленке
с целью улучшения ее укрывистости. Простой метод заключается
в эмульгировании несмешивающейся жидкости, (например уайт-
спирита) в водном растворе желатина; при высыхании образуется
непрозрачная белая пленка, наполненная мелкими сферическими
пустотами. Другие, более сложные композиции позволяют полу-
чить такие же результаты при лучших свойствах пленки [6—8].
Можно представить, что наиболее удачным промышленным спо-
собом использования этого эффекта является применение компо-
зиции из пигментных частиц и мелких частиц смолы, распределен-
ных в системе с мелкими пустотами и содержащих мелкие частицы
пигментов с высоким показателем преломления [9]. Теория также
говорит, что если частицы материала с высоким показателем пре-
ломления оптимального размера покрыты оболочкой с низким по-
казателем преломления, то концентрация кроющего пигмента и ка-
чество дисперсии не очень существенно влияют на конечный ре-
зультат [10].
14.3.4. Светопоглощение пигментами
Все пигменты поглощают излучение определенных длин волн,,
но белые пигменты интенсивно поглощают только в УФ-области.
Черные пигменты поглощают свет всех видимых длин волн, но мо-
гут быть прозрачны в ИК-области, что важно для создания маски-
рующих красок. Большинство цветных пигментов сильно погло-
щают в определенных областях видимого спектра, но прозрачны
вдругих областях. В п.ленках.—гд-с—пъетиые- пигменты смешаны
с рассеивающими белыми частицами, общее поглощение и, следо-
вательно, глубина цвета зависят от размера частиц цветных пиг-
ментов. Если частицы полностью диспергированы, поглощение
возрастает обратно пропорционально размеру частиц. Это объ-
ясняется тем, что поперечное сечение каждой частицы пропорцио-
нально квадрату ее диаметра d2, а число частиц в единице объема
пропорционально 1/d3. Следовательно, общее поперечное сечение
частиц пропорционально \/d. Поскольку путь падающего света
через частицы достаточно велик и большая часть его успевает
поглотиться, интенсивность цвета приблизительно обратно про-
порциональна d; если поглощение на частицу гораздо меньше, то
и уменьшение интенсивности цвета меньше. На рис. 14.7 показаны
данные Карра [11] для красок, содержащих органические пигмен-
ты, при различном времени перетира. Для сильно поглощающего
Рис. 14.7. Зависимость коэффициен-
та поглощения от обратного размера
частиц для органических пигментов
[11J:
/ — Фталоцианиновый голубой; 2 —
Пигмент зеленый В (оба смешаны в
соотношении I : 12,5 с диоксидом ти-
тана)
фталоцианинового голубого пропорциональность между К и 1 /d.
наблюдается для размера частиц ниже 0,15 мкм, тогда как для
менее интенсивного Пигмента зеленого В эта зависимость не
соблюдается.
14.4. ЦВЕТ СМЕСЕЙ ПИГМЕНТОВ И ПИГМЕНТИРОВАННЫХ ПЛЕНОК
14.4.1. Восприятие цвета
Полное рассмотрение цветового восприятия в этой главе не
предусмотрено. Читатель отсылается к работам [12—16]. Для об-
щего представления достаточно знать, что в дневном свете челове-
ческий глаз различает цвета с длинами волн 0,40—0,75 мкм
в виде трех первичных составляющих (приблизительно, синих,
зеленых и красных) и что различаемый цвет представляет собой
определенное сочетание этих составляющих. Сопоставление ре-
зультатов анализа цветовых различий для света с известным рас-
пределением спектральной энергии показывает, что любое вос-
приятие какого-либо цвета вызвано сочетанием разных цветов
в широком диапазоне длин волн. Рис. 14.8 иллюстрирует распре-
деление спектральной чувствительности в системе трех координат
X, Y и Z, в соответствии с международной системой CIE. Следует
отметить, что ощущение красного цвета вызывается светом с дли-
нами волн 0,43—0,45 мкм в фиолетовой части спектра и 0,55—
0,65 мкм в желтой, оранжевой и красной частях спектра. Приве-
денные «усредненные кривые» получены на основе данных несколь-
ких наблюдателей (все с достоверно нормальным цветоощуще-
нием), производивших измерения в стандартизованных условиях.
Цветовосприятие любого конкретного наблюдателя, вероятно,
несколько отличается от усредненного, и на него, безусловно,
влияют окружающие цвета, возбудители, воздействующие на гла-
за, и другие факторы. Однако большинство практических цветовых
измерений укладывается на среднюю кривую как показано на
рис. 14.8; эти кривые получены с помощью колориметров.
Из набора соответствующих кривых для любого распределения
энергии падающего света (£;.) и значений коэффициентов отраже-
ния (/?;.) можно рассчитать значения соответствующих координат
цвета Ма-тематическиэто ъьтражается ’уравнениями:
(14.5)
На практике обычно представляют в виде таблицы_величины
распределения энергии источника освещения £>. и х-,_, у-,,, z>.
при интервале длин волн, например, 10 нм и вычисляют X, У, Z пу-
тем суммирования. Если интервал длин волн велик, то несколько
снижается точность, особенно для материалов с резкими пиками
поглощения.
Относительные величины X, У, Z соответствуют глубине цвето-
вого восприятия, и в системе CIE называются координатами цвет-
ности и обозначаются как х, у и z, при этом
х=Х/(Х+У+2); y=r/(A'4-r + Z); z=Z/(X+ Y+Z). (14.6)
Абсолютные значения А', У, Z соответствуют яркости цвета.
Основные цвета в системе CIE выбраны так, что значение У соот-
ветствует количеству отраженного света, х и у показывают степень
отклонения цвета от нейтрального белого или серого. Таким обра-
зом, У, X и У полностью характеризуют цвет поверхности при опре-
деленном освещении. Если освещение меняется, например, от днев-
ного тусклого света к свету вольфрамовой лампы, У, х и у изменя-
ются. Поскольку все кривые на рис. 14.8 широкие, возможно для
двух различных кривых спектрального отражения получить одина-
ковые интегральные величины X, У и Z при одинаковом освещении.
Такие цветовые пары разрушаются путем изменения источника
освещения. Два цвета такого типа называются «метамерными»,
а явление образования цветовых пар только при одном освеще-
нии и не соблюдающееся при другом — «метамерией». Цвет, кото-
Рис. 14.9. Кривые отражения при метамерическом смешении цветов
рый воспринимается как изменение оттенка при изменении осве-
щения называется «дихроическим». На рис. 14.9 показаны кривые
отражения для трех образцов. Для образцов А и В освещение осу-
ществлялось лампой дневного света Макбета с цветовой темпе-
ратурой 7500 К; для В и С — холодной флуоресцентной лампой
белого света. При освещении вольфрамовой лампой подобные цве-
товые пары получить не удалось.
14.4.2. Цвет пигментированных пленок
Объективной величиной, определяющей цвет пленки, является
количество света, отраженного при каждой длине волны видимого
спектра, или, точнее, количество такого света, воспринятое наблю-
дателем. Первая успешная попытка соотнести отражение со свето-
рассеянием и светопоглощением в пленке сделана Кубелкой и
Мунком [17, 18]. Они сделали допущения, что тонкий горизонталь-
ный срез пленки (рис. 14.10) будет, во-первых, одинаково рассеи-
вать свет в прямом и обратном направлениях пропорционально
коэффициенту светорассеяния S и объему среза, и, во-вторых,
Рис. 14.10. Поглощение и рассеивание в
слоях покрытий по Кубелке-Муику:
1 — падающий свет (/); 2 - свет, рассеянный
в обратном направлении (ISdh); 3 — слой
толщиной dh-. 4 — поглощено IKdh; 5 — свет,
рассеянный в прямом направлении (ISdh)
поглощать свет пропорционально коэффициенту абсорбции К и
объему среза.
Из этих простых предпосылок можно вычислить поток пропу-i
щенного и отраженного света. Для пленки определенной толщи-
ны h конечное выражение для R сложное. I
£ г J —ЗЛд/Х (K + 2S) ।
RP=------------- ------------ J __________-_____ (14.7?)
K+S+^(K+2S)-K+S-^K(K~+‘2S) e-^^IK(K+2S)
Для пленок неопределенной толщины выражение упрощается:
Rv = S/{K+S+y'K(K+2S) ], (14.8)
и его можно преобразовать:
K/S=(1-/?p)2/2/?p. (14.9)
Отношение К/S, таким образом, определяет отражение от пиг-
ментированного слоя некоторой толщины, которая обеспечивает
полную укрывистость. Ввиду того, что отражение на поверхности
раздела воздух/покрытие не рассматривается в исходных уравне-
ниях Кубелки-Мунка, /?Р не является таким же, как и отражение
от пленки краски (см. 14.3.2 выше). Однако, имея дело с цветом
пигментированных пленок, удобно вычислить Rp при каждой длине
волны и затем, сделав соответствующие поправки, получить R^.
Дальнейшим развитием теории цвета пигментированных пле-
нок явилось предположение Дункана [19] о том, что коэффициен-
ты поглощения и рассеяния различных пигментов должны склады-
ваться пропорционально концентрации пигментов:
7</S=(C17<1 + C27<2 + ...)/(C1S1 + C2S2 + ...). (14.10)
Предположение о том, что присуствие других пигментов в плен-
ке не влияет на степень светорассеяния или светопоглощения каж-
дой пигментной частицей, подобно ряду аналогичных законов
в физике, например закону Дальтона для парциальных давлений,
является полезным рабочим правилом. Оно позволяет вычислить
RP и затем R для любой смеси пигментов, если известны соответ-
ствующие коэффициенты поглощения и светорассеяния для инди-
видуальных пигментов.
14.4.3. Предсказание цвета
Составление смеси пигментов, дающей желаемый цвет пленки,
путем расчетов в значительной степени вытеснило составление
смесей методом проб и ошибок. Первые программы предсказания
цвета были составлены для упрощенного случая (для толщин пле-
нок, обеспечивающих укрывистость), но мощные современные ком-
пьютеры сделали возможным распространить такие вычисления
и на пленки с меньшей толщиной. В таких более сложных вычисле-
ниях применяются абсолютные концентрации пигментов, а не отно-
сительные, использовавшиеся в более ранних расчетах. Точность
предсказания будет зависеть от того, с какой степенью точности
лежащая в основе расчетов теория Кубелки-Мунка позволяет оп-
ределить светорассеяние и светопоглощение в пленке. Недостатки
(теории могут быть частично нивелированы-путем использования
(значений К и S, определенных для каждого пигмента, дисперги-
рованного в связующем, при концентрациях, близких к той, кото-
рая применяется в конечной смесевой системе.
\ Известны многие различные программы предсказания цвета.
Они могут основываться на вычислении количества выбранных
пигментов, которые по координатам цвета X, У, Z близки к пара-
метрам желаемого цвета, а также на других вычислениях, напри-
мер на подборе пар пигментов с близкими величинами R}_.
(( Полезным является изучение влияния на цвет малых измене-
ний концентрации каждого пигмента. Это влияние можно легко
вычислить, так как в основе расчетов лежит обычный прием,
основанный на методе аппроксимаций, применяемом для решения
многих задач. Зная вклад каждого из оттенков, легко вычислить
количества добавок, необходимых для корректировки цвета; эти
корректировки будут точны только тогда, когда поведение лако-
красочного материала подчиняется зависимостям Кубелки-Мунка
и Дункана. Основные возмущения в системе, такие как флотация
одного из пигментов или флокуляция в смесевой системе, не могут
быть учтены.
14.5. ИЗМЕНЕНИЯ В ПИГМЕНТИРОВАННЫХ ПОКРЫТИЯХ
г 14.5.1. Введение
Обсуждение факторов, влияющих на внешний вид покрытия,
будет неполным без учета изменений, происходящих при пленко-
образовании и после его завершения. Для красок растворного'
типа основное изменение — это быстрое сжатие пленки при высы-
хании и затем продолжающееся медленное сжатие по мере удале-
ния остатков растворителя в течение многих дней и даже месяцев.
Окислительная среда также способствует усадке пленки (до 10%
за 3 мес.) благодаря удалению разрушающихся летучих продук-
тов; эти процессы приводят к возрастанию показателя преломле-
ния пленкообразователя, уменьшая таким образом светорассея-
ние на частицах белых пигментов. Некоторые основные пигменты,
такие как цинковые и свинцовые белила реагируют с кислотными
продуктами окисления, образуя металлические соли на поверх-
ности пигментных частиц. Этот процесс в сочетании с общей усад-
кой приводит к огрублению поверхности со значительной потерей
глянца, если частицы (их конгломераты) велики; примером явля-
ется помутнение пленок с оксидом цинка.
Латексные краски образуют пленки путем агрегации и частич-
ной коалесценции полимерных глобул по мере удаления воды из
Рис. 14.11. Схематическая структура низкокачественной пленки латексной краски:,
1 — частично скоалесцировавшие полимерные частицы; 2 — группы пигментных частиц
/
пленки. Этот процесс сопровождается уменьшением светорассея-
ния на латексных частицах и частичным образованием агрегатов
из пигментных частиц. В результате наблюдается дальнейшее
уменьшение светорассеяния, приводящее к падению кроющей спо-
собности сухой пленки и увеличению глубины цвета, поскольку
колеровочные пигменты менее подвержены изменениям. Для ла-
тексной пленки трудно достичь высокого глянца, поскольку пиг-
менты имеют тенденцию к образованию конгломератов между
группами коалесцировавших полимерных глобул, что приводит
к увеличению шероховатости поверхности (рис. 14.11). Одним из
методов предотвращения этого является введение части водораст-
воримого пленкообразователя, который может предотвратить сли-
пание пигментных частиц и ослабить процесс усадки.
14.5.2. Изменение укрывистости пленок
По мере высыхания белые или слегка окрашенные пленки по
причинам, приведенным в 14.5.1, а также по ряду других причин,
могут сильно изменять укрывистость. Основные факторы, влияю-
щие на это явление — изменение показателя преломления и изме-
нение концентрации.
Изменение показателя преломления. Светорассеяние пропор-
ционально (п.\—по)2/(И1 + п2)2, где Hi и иг — показатели прелом-
ления пигмента и среды, в которой он диспергирован. При высыха-
нии и окислении пленки алкидной смолы на основе сильнополиме-
ризованного растительного масла, полученной из раствора в уайт-
спирите, по может возрасти от 1,46 до 1,53. Для пигмента (рутиль-
ный диоксид титана) уменьшение светорассеяния составляет
14% (Hi =2,7). Для латексной краски, где среда разбавлена по
меньшей мере на 60% водой (и =1,34), уменьшение светорассея-
ния может быть даже больше. Важно, что обратный эффект мож-
но получить для слегка сшитого латекса, если после пленкообра-
зования часть пигмента находится на границе с воздухом. Рутиль-
ная частица на воздухе теоретически рассеивает на 158 % больше
света, чем в среде с пг = 1,5, однако на практике это полностью
не достигается, так как в сухой пленке частицы не полностью изо-
лированы друг от друга. Однако это обстоятельство является глав-
ным резервом улучшения укрывистости. Описанное явление иногда
называют «сухой укрывистостью»; оно объясняет, почему деше-
s
-----1----J----1----1_____I____I_______
5 10 15 20 25 30
ОКП,
Рис. 14.12. Зависимость коэффициента светорассеяния (вычислено по объему
пигментов) от ОКП.
1 — размер частиц 0,34 мкм; 2 — размер частиц 0,44 мкм
вая, неотвержденная пленка водоэмульсионной краски может
иметь лучшую укрывистость, чем более дорогая, полностью
отвержденная и содержащая больше основного пигмента.
Изменение концентрации. Пигменты рассеивают свет наиболее
эффективно, когда каждая частица находится на достаточном рас
стоянии от соседней. Эксперименты показали, что коэффициенты
рассеяния остаются практически постоянными до ОКП = 5%,
а затем быстро падают (рис. 14.12) [20—22]. Из этого следует,
что увеличение концентрации по мере испарения растворителя из
пленки выше 5% приводит к ухудшению укрывистости. Этот эф-
фект усугубляется некоторой флокуляцией пигментов В конечном
результате происходит большее падение кроющей способности, чем
можно ожидать вследствие изменения показателя преломле-
ния [23].
14.5.3. Изменение цвета
Уменьшение светорассеяния (по причинам, обсуждавшимся
выше) обычно приводит к значительному увеличению глубины цве-
та по мере высыхания пленки; исключениями являются случаи
сильного пигментирования, при которых проявляется «сухая укры
вистость», а также применение пористых подложек, которые могут
впитывать пленкообразователь. Для большинства пленок измене-
ние цвета при высыхании обратимо, так что возможно контроли-
ровать цвет сухой пленки путем регулирования цвета жидкой крас-
ки. Это дает возможность полностью автоматизировать цветовой
контроль при производстве красок добавлением жидких колеро-
вочных паст для получения требуемого цвета.
14.6. ФЛУОРЕСЦЕНЦИЯ И ФОСФОРЕСЦЕНЦИЯ
До сих пор в данной главе обсуждались лишь явления отраже-
ния или поглощения падающего света пленкой или подложкой.
Рассмотрение оптических свойств покрытий было бы неполным без
упоминания об изменениях отраженного света, которые имеют мес-
то для некоторых материалов, и сопровождаются возбуждением
эмиссии света другими видами энергии.
Флуоресценция — это явление поглощения излучения материа-г
лом, с последующим выделением поглощенной энергии в виде излу-
чения с большей длиной волны (т. е. в виде квантов с более низкой
энергией). Выделение энергии может быть мгновенным или проис-
ходить в течение значительного промежутка времени, что зависит
от возможности возвращения атомов из возбужденного состояния
в нормальное. Замедленное выделение энергии называется фосфо-
ресценцией. Последний термин часто употребляется для опреде-
ления световой эмиссии, связанной с химическими процессами
(хемолюминесценция^ и с радиоактивностью.—
Флуоресценция весьма распространена в покрытиях, ибо неко-
торые материалы преобразуют часть УФ-излучения, присутствую-
щего в дневцом свете, в видимое излучение, либо синий цвет,
например, превращают в зеленый. В результате свет определен-
ных длин волн, отражающийся от поверхности, может по интен-
сивности превосходить падающий свет тех же длин волн. Флуо-
ресцирующие краски и красители для дневного света используют
для нанесения предупредительных надписей. Флуоресценция зна
чительно усложняет цветовые измерения по двум причинам. Во-
первых, при измерениях следует принимать в расчет только часть
излучения в близкой к ультрафиолетовой области, присутствую-
щей в источнике излучения, под освещением которого рассматри-
вается покрытие. Во-вторых, нельзя предполагать, что свет, отра-
женный от покрытия, будет той же длины волны, что и падающий,
так что необходимо производить освещение в полном спектре (а не
последовательно, пучками с узким диапазоном длин волн), и затем
анализировать отраженный свет. Фосфоресценция дает еще боль
ше проблем при измерении, но, к счастью, ограничивается малым
числом материалов, применяемых только для очень специальных
целей.
14.7. ОЦЕНКА ЦВЕТА
14.7.1. Введение
Общеизвестно, что цвет существует только в воображении
наблюдателя; никто не может знать, как другой человек ощущает
цвет — так же или иначе. Из опыта, однако, известно, что есть
общие моменты в восприятии. Также возможно вывести некоторые
соотношения между цветовосприятием и физическими факто-
рами — распределением длин волн и интенсивностью (см. 14.4.1).
Некоторые из этих соотношений и различий восприятия обсуж-
даются ниже.
14.7.2. Аномальное цветовосприятие
По крайней мере 10% мужчин, и значительно меньшее число
женщин имеют цветовосприятие, значительно отличающееся от
нормального, описанного в 14.4.1. Аномалии могут быть различных
типов, наиболее часто встречаются протоаномалии (уменьшенная
чувствительность красных рецепторов) и дейтероаномалии (умень-
шенная способность отличать красный и зеленый цвета). Это лег-
ко обнаруживается на аномалиоскопе, в котором смешиваются
красный и зеленый цвета с образованием спектрально желтого
цвета. Для наблюдателя с протоаномалией требуется большая
добавка красного цвета, а с дейтероаномалией — большая добав-
ка зеленого цвета, чем наблюдателю с нормальным цветовосприя-
тием для получения одинакового желтого цвета. Быструю проверку
можно также сделать по тесту Ишихара [24], в котором применя-
ются пластинки с нанесенными на них различными цветными пят-
нами. Гораздо реже встречаются случаи отсутствия цветовосприя-
тия (в основном воспринимаются только оттенки серого цвета)
или явление тританопия. В последнем случае уменьшенное
восприятие синего цвета приводит к тому, что путаются синие
и зеленые цвета, а также некоторые розовые и желтые цвета.
Даже наблюдатель с нормальным цветовосприятием инди-
видуально дифференцирует свет, поступающий на различные
участки сетчатки глаза. Это связано с анатомическими особен-
ностями; около центральной, наиболее чувствительной части сет-
чатки конические рецепторы расположены близко и дают наиболь-
шее пространственное различие, но дальше к периферии они пере-
межаются с рецепторами в виде палочек, которые большей частью
функционируют в тусклом свете и отвечают за характеристики
зрения в сумерках (когда желтый цвет темнеет, а синий относи-
тельно светлеет).
14.7.3. Усталость и контрастность
Фотохимические изменения в сетчатке, происходящие под воз-
действием света, связаны с мозгом электрическими нервными им-
пульсами. Этот сложный механизм исключительно важен в пере-
даче информации об изменении интенсивности, но в определенные
моменты при экстремальных условиях или при наличии резких
контрастов наступает усталость. Это приводит к ослаблению цве-
товосприятия в тех случаях, когда в поле зрения присутствуют
очень яркие и насыщенные цвета, и к аномалиям цветовосприя-
тия, особенно если цвета контрастные. Эти факторы имеют некото-
рое отношение к визуальной оценке цвета. Так, образцы лучше
всего сравнивать со стандартом на фоне, аналогичном по яркости
фону стандарта.
14.7.4. Оценка цветовых различий
Для цветовых измерений можно применить систему CIE. Раз-
ница между двумя цветами, определяемыми параметрами Х|( У|,
2| и Х%, Y2, Zi (где параметры X, У, Z измерены вдоль прямолиней-
ных осей), равна -^(Xf —Лг)2-|- (У| — Е2) + (Zi —ZY)' К сожа-
лению, равные расстояния между цветами в такой системе не соот-
ветствуют одинаковой разнице цветовосприятия. Это наблюдал
Мак Адам [25]. На рис. 14.13. показаны эллипсы Мак Адама для
цветов с постоянной яркостью, но разными х в у. Действительно,
на графике yi=f(x) разница цвета в зеленой области сильно пре-
увеличена и уменьшена в синей и оранжево-коричневой областях.
Было.пред1финято_мног.о по.иыток трансформировать цветовые ха-
рактеристики X, Y, Z в другие характеристики, .дающие большее
соответствие с визуальной оценкой. Точное решение проблемы
Рис. 14.13. Эллипсы Мак-Адама. Размер эллипса соответствует 10-кратному стан-
дартному отклонению цветовой пары по отношению к цвету, представленному
центральной точкой
невозможно из-за того, что визуальная оценка изменяется с величи-
ной поля зрения и цвета окружающего пространства и в некоторой
степени — с уровнем освещения, однако найдены наилучшие при-
ближения. Так, восприимчивость глаза к изменению яркости
(представленная координатой Y в системе CIE) нелинейна; т. е.
если для белого цвета У = 100, то серый, находящийся точно посре-
дине между белым и черным, будет значительно темнее, чем цвет,
для которого У=50. С другой стороны, х и у можно преобразовать
в новые координаты а и р, которые преобразуют эллипсы Мак
Адама в фигуры, близкие к кругам более однородного размера. До-
казано, что из многих преобразований наиболее полезным явля-
ется система Т*, а*, Ь*, предложенная в 1976 г. [16, 26]; ее- при-
менение стандартизовано. Эта система и использует функции
кубического корня координат X, У, Z для всех светлых цветов и линей-
ные функции — для темных. Цветовое различие вычисляется как
-д/Д7Д-|-Аа (для цветов, близких к белому). Для цветных
образцов вводится также различие АС* (характеризует глубину
цвета) и А//* (характеризует оттенок).
14.7.5. Влияние освещения
Влияние изменения распределения энергии источника света на
восприятие цвета было показано ранее. Эти эффекты очень важны
во всех испытаниях; даже укрывистость белой краски может быть
значительно лучше, если измерять ее на свету, содержащем боль-
шую часть синего излучения, чем в случае доминирования красного
излучения. Международные классификации источников освещения
обычно соотносятся с солнечным светом или светом вольфрамовой
лампы накаливания. В системе CIE используют стандартные ис-
Таблица 14.2 Распределение энергии стандартных источников (нормализовано
при £.здо=100)
Длина волны, им Относительная спектральная энергия Длина волны, нм Относительная спектральная энергия
Источник А. 5 Источник С (днев- ной свет) Источник А (воль- фрам) Источиик О.,5 Источник С (днев- ной свет) ИстОЧник А (воль- фрам)
320 20,2 0,01 560 100,0 100,0 100,0
340 30,0 2,6 580 95,8 92,9 114,4
360 46,6 12,3 600 90,0 85,2 129,0
380 50,0 31,3 9,8 620 87,7 83,7 143,6
400 82,8 60.1 14,7 640 83,7 83,4 158,0
420 93,4 93,2 21,0 660 80,2 83,5 172,0
440 104,9 115,4 28,7 680 78,3 79,8 185,4
460 117,8 116.9 37,8 700 71,6 72,5 198,3
480 115,9 117,7 48,2 720 61,6 64,9 210,4
500 109,4 106,5 59,9 740 75,1 58,4 221,7
520 104.8 92,0 72,5 760 46,4 55,2 232,1
540 104.4 97.0 86,0 780 63,4 56,1 241,7
точники:А (вольфрамовая лампа с цветовой температурой
2854 К); В (солнечный свет); С (дневной свет). Источники В и С
обычно использовали для колориметрии до тех пор, пока не выяс-
нилось, что измерения на красках, содержащих рутильный диоксид
титана, не соответствуют визуальному опыту. Это потребовало
более тщательного анализа спектральных кривых и разработки
новых стандартных источников с большим содержанием коротко-
волнового излучения. Сейчас наиболее широко используется стан-
дартный источник Z>65, основанный на естественном дневном свете
с цветовой температурой 6500 К; его энергетическое распределение
находится в диапазоне 320—780 нм (см. табл. 14.2) и сравнимо
с распределением источников А и С.
14.8. ДРУГИЕ ДАННЫЕ
В работе [13] дан, возможно, лучший обзор для технолога-
- лакок-р асоч-н и ка.
Работа [14] дает ясное представление о современном состоянии
проблемы и содержит перечень современной литературы. Мак Ла-
рен [16] подробно анализирует цветовые свойства пигментов
и красителей. Работа [12], написанная лучшими специалистами
по физике цвета, является, вероятно, лучшей книгой в этой облас-
ти. В работе [15] наиболее исчерпывающе рассмотрены вопросы
цветовосприятия, математические методы представления цвето-
вого поля и вычисления цветовых различий, но она предназначена
скорее для узких специалистов, чем для технологов общего про-
филя. Книга [2] рекомендуется для получения общих представле-
ний о вопросах, затронутых в этой главе.
Глава 15
ХАРАКТЕРИСТИКА И КОНТРОЛЬ ВНЕШНЕГО ВИДА
Т. Р. Буллетт
15.1. БЛЕСК
15.1.1. Классификация
Покрытия можно классифицировать на следующие типы в за-
висимости от степени зеркального отражения пленки:
Высокоглянце- вое Пленка проявляет яркое, зеркальное отражение при любом угле наблюде- ния
Пол у глянце вое Пленка проявляет зеркальное отра- жение при малых углах наблюдения, но кажется матовой при больших углах
Полуматовое Пленка кажется матовой при любом угле наблюдения; при углах, близких к 90°, возможно зеркальное отраже- ние
Матовое Пленка не дает зеркального отраже- ния ни при каких углах
Для оценки уровня блеска высокоглянцевого и полуглянцевого
покрытий иногда используют термин «масляный глянец», который
характеризует внешний вид свеженанесенных покрытий на основе
старинных масляных красок [1].
Глянец не является простым свойством, величина которого мо-
жет быть оценена на линейной шкале. Существует несколько
различных характеристик, которые должны быть рассмотрены:
а) интенсивность зеркального отражения, или яркость, либо
отражение при углах, равных или близких к зеркальному;
б) четкость изображения, позволяющая рассмотреть все де-
тали строения отражающей поверхности;
в) глянец при направлении потока света по касательной к по-
верхности; отражение в этом случае происходит под углами, близ-
кими к углу зеркального отражения.
Даже этих трех типов характеристик недостаточно для уста-
новления всех различий между наблюдаемыми визуально различ-
ными поверхностями, особенно в случае полуглянцевых поверхно-
стей, где необходимо анализировать распределение отраженного
света при широком наборе углов, а также в случае матовых покры-
Рис. 15.1. Распределение отраженного света:
1 — падающий свет; 2 — полное зеркальное отражение; 3 — полное рассеяние
тий, где неровности поверхности могут вызвать преимущественно
обратное отражение, в результате чего некоторые пленки могут
вьгглядетБгораздобояее-бл-ест-яищ-ми-при наблюдении в направле-
нии падающего света, чем при обычном наблюдении в направле-
нии зеркального отражения.
Чтобы порять причины различий во внешнем виде, можно рас-
смотреть распределение света, отраженного от некоторых типич-
ных пленок. На рис. 15.1 и 15.2 иллюстрируются данные, получен-
ные при освещении очень узким пучком света под углом 60°. Для
идеальной зеркальной поверхности весь свет отражается зеркаль-
но. В других случаях интенсивность отраженного света зависит от
косинуса угла отражения, как показано на рис. 15.1; для обшир-
ной поверхности при любом направлении наблюдения яркость оди-
накова. Для полуглянцевых белых покрытий характерен круговой
график отражения с пиком, соответствующим углу зеркального
отражения; узость пика и его высота в точке зеркального отраже-
ния являются мерой глянца покрытия. Для полуглянцевых покры-
тий форма пика меняется с величиной глянца (рис. 15.2).
Рис. 15.2. Распределение отраженного света от полуглянцевой черной и белой
пленки:
/ — белая; 2 — черная
15.1.2. Измерение глянца при зеркальном отражении
Многие годы в Великобритании использовался стандартный
метод измерения глянца (британский стандарт BS 3900: Part D2:
1967), в котором зеркальное отражение под углом 45° сравнива-
ется с зеркальным отражением от черного глянцевого стеклянного
стандарта. Этот метод положен в основу стандарта BS 3900: Part
D5: 1980 [3], который соответствует международному стандарту
ISO 2813-1978; последний, в свою очередь, основан на гораздо бо-
лее старом стандарте ASTM [2]. Раздел D5 включает метод изме-
рения, основанный на сравнении с отражением от черного стеклян-
ного стандарта, но используются углы падения света 20, 60 или
85°, в соответствии с измеряемым уровнем глянца и целями изме-
рений. Угол 20° обеспечивает чувствительность измерения для
высокоглянцевых покрытий, а 85° — используется только для из-
мерений глянца покрытий, близких к матовым.
Из рис. 15.2 ясно, что количество отраженного света, зарегист-
рированное как зеркальное отражение от полуглянцевого покры-
тия, зависит от отклонения углов от угла, соответствующего зер-
кальному отображению, воспринимаемому блескомером. Рис. 15.3
воспроизводит данные стандарта BS 3900: Part D5 и показывает,
как производится определение по данной методике. Подлежит
контролю как диапазон углов падающего света, который зависит
от размера лампы накаливания и соотношений фокальной длины
коллиматорных линз, так и диапазон воспринимаемых углов, кото-
рый зависит от углового размера в приемном устройстве. Увеличе-
Рис. 15.3. Оптическая схема блескомера под углом 60° (BS 39Q0: Part D5):
G—лампа; L] и L2—линзы; В — воспринимающее устройство; Р—пленка покрытия;
£1 = 82 = 60 + 0,2°; ов = 4,4 + 0,1°; о2 = 0.75 + 0,25°; / — отражение нити накаливания
Табли-на 15.1. Показатели преломления связующих и сравнительные данные
интенсивностей зеркального отражения материалов
Материал nD Зеркальное отраже- ние (стандарт из BS 3900:DS:1980 принят на 100)
Стеклянный стандарт (BS 3900: D2) 1,523 88,1
Стеклянный стандарт (BS 3900: D5) 1,567 100,0
Политетрафторэтилен 1,350 45,5
Иолививдлиденхлорид 1,420 61,7 .
Полибутил акрил ат 1,466 73,2
Полмви нил ацетат 1,466 73,2
Полиэфирные смолы 1,523—1,540 88,1—92,6
Алкидные смолы на основе сильно полимери- 1,530—1,550 89,9—94,7
зова иных масел??
Эпоксидные смолы 1,550—1,600 94,7—109,1
Хлоркаучук 1,550 94,7
1 600 1,630 109 I 117 6
Фенольные смелы (немодифицированные) Ц660—Ц700 126,1 —137^8
ние любого из этих углов уменьшает различимость при высоком
глянце [4].
Следует обратить внимание на ряд других важных моментов:
1. Интенсивность отраженного света зависит от показателя
преломления, а также от гладкости поверхности (см. уравнение
14.3). По этой причине показатель преломления черного стеклян-
ного стандарта должен быть определен точно. Вдобавок, краска
на основе связующего с высоким показателем преломления будет
иметь более высокое значение зеркального отражения, чем краска
на основе связующего с более низким показателем преломления.
В табл. 15.1 приведены показатели преломления некоторых типич-
ных связующих и величины зеркального отражения от совершенно
идентичных поверхностей (относительно черного стеклянного стан-
дарта, глянец которого принят за 100 единиц). Человеческий глаз
также имеет склонность к большему восприятию блеска, отражен-
ного от поверхности с более высоким показателем преломления
(бриллианты отличаются от стекла!), но поверхности, имеющие
дефекты, блескомерами могут быть оценены неверно из-за раз-
ности показателей преломления.
2. Направленные дефекты поверхности, например остаточные
штрихи от кисти, сильно влияют на глянец, измеренный поперек
направления дефектов, но гораздо меньше — на глянец, измерен-
ный вдоль направления дефектов.
3. Подложки должны быть совершенно гладкими, чтобы разни-
ца углов падения и отражения была минимальной. Обычно наносят
пленки на стеклянные пластины. Если измеряются прозрачные
пленки, стекло должно быть или непрозрачным или окрашенным
с нижней стороны, чтобы предотвратить второе зеркальное отра-
жение от нижней поверхности раздела воздух/стекло; простого
помещения стеклянной пластины на черную поверхность недоста-
точно.
15.1.3. Отчетливость восприятия глянца
Существует ряд недостатков метода определения зеркального
отражения для оценки глянца высокоглянцевых поверхностей.
Аномалии связаны с разницей показателей преломления, о которой
уже упоминалось; это может привести к возрастанию зеркального
отражения со временем эксплуатации для алкидных глянцевых
покрытий, что не коррелируется ни с каким улучшением глянца.
Вторая проблема заключается в том, что точные измерения зер-
кального отражения возможны только на плоской поверхности;
метод неприемлем, например, для кривых поверхностей корпусов
автомобилей. Еще более важно, что блескомеры зеркального отра-
жения не способны определить значительные различия в блеске,
полученные за счет более тонкого диспергирования частиц пигмен-
тов; эти различия проявляются в повышении ясности зеркального
отражения без значительного изменения количества света, отра-
женного в конусе 1—2° вокруг зеркального угла. Многих из этих
недостатков можно избежать, если оценивать блеск по четкости
изображения.
Прямые визуальные методы оценки четкости изображения
основаны на определении четких границ линий или фигур в контро-
лируемых условиях [5]. Методика измерений достаточно чувстви-
тельна, но является субъективной, как и все подобные методы.
Несмотря на это, последняя методика наиболее часто употребля-
ется для оценки глянца покрытий корпусов автомобилей. Также
предпринимались многочисленные попытки установить физический
Рис- 15.4. Различие отображения от точечных источников:
1 — зеркальный угол для А; 2 — зеркальный угол для В; 5 — угловое сканирование
эквивалент четкости изображения Его можно представить каквы-~
соту пика на кривой зеркального отражения (рис. 15.2). На
рис. 15.4 представлено зеркальное отражение от двух ярких точек
объекта под несколькими разными углами по отношению к поверх-
ности. Эти две точки будут давать четкое изображение, если
минимум кривой /?Л + /?В будет значительно ниже высот пиков,
соответствующих отражению из точек А и В. Прямое измерение
этого различия требует очень точных приборов. Гораздо более пря-
мым методом является сканирование фотометром одного отражен-
ного луча, проходящего через линзу на плоскость, на которой запи-
сывается ширина угла между высотой пика и полупика. Новые
экспериментальные приборы, внедряемые ASTM, базируются на
сравнении значений зеркального отражения, полученных при ис-
пользовании линз с очень малыми и большими размерами отвер-
стий, через которые идет луч. Эта методика устраняет аномалии,
связанные с показателем преломления для среднего диапазона
1_нели.чин глянпа, но является недостаточно чувствительной для
высоких значений глянца.
15.1.4. Оценка глянца
Зеркальное отражение под низкими углами к поверхности явля-
ется источником ослепительного блеска, например в случае наруж-
ных поверхностей столов под ярким светом. Часто оно является
нежелательным при внутренней отделке помещений, так как может
выявить неровность стен и потолков. Имеется метод оценки так
называемого «эффективного блеска», который заключается в изме-
рении зеркального отражения под углом 85° к нормали [3]. Ре-
зультаты, полученные этим методом, очень зависят от гладкости
Рис. 15.5. Оптическая схема блескомера под углом 85° (BS 3900: Part 05):
G — лампа; 1., и L,—линзы; В — воспринимающее устройство; Р — пленка покрытия;
ei = 62 = 85 + 0,1"; = 4,0 + 0,3"; 02 = 0,75 + 0,25; 7 — отражение нити накаливания
испытываемых образцов и от точности центровки блескомера. Ви-
зуальные методы выглядят проще и, следовательно, более привле-
кательны. С 1940 по 1980 гг. применялся стандартный визуальный
британский метод, описанный в конечном виде в BS 3900: Part
D3:1975 [7]. Методика давала воспроизводимые результаты при
работе одного и того же наблюдателя, однако неизбежно имели
место расхождения в случае различных наблюдений. В конце кон-
цов, несмотря на многочисленные попытки улучшить испытатель-
ный прибор и стандартизировать условия наблюдения, метод был
заменен упоминавшимся выше методом по измерению зеркального
отражения по углом 85° (рис. 15.5).
15.2. УКРЫВИСТОСТЬ
15.2.1. Общие положения
Одной из практически важных характеристик при нанесении
пленки краски является однородность цвета поверхности. Если
пленка нанесена при толщине, недостаточной для обеспечения
полной укрывистости, что редко встречается в случае очень темных
или «металлических» покрытий, неравномерность цвета может
быть вызвана различиями в толщине пленки или цвете подложки.
В большинстве случаев измерения укрывистости (кроющей способ-
ности) основаны на способности пленки укрывать различные цве-
та подложки. Укрывистость выражается в виде отношения конт-
растностей (в виде частей или процентов), т. е. отражение пленки
на черном фоне делится на отражение той же пленки на белом. Для
очень ярких красок изменения цвета, обусловленные изменением
толщины пленки на однородной подложке, могут на практике быть
более значительными. Примерами могут служить влияния рисок
от кисти и т. п., которые часто видны на окрашенных поверхностях.
Такие эффекты связаны с реологическими свойствами красок. Их
значение может быть уменьшено на практике путем соответствую-
щего выбора нижних слоев покрытия. Один из практических мето-
дов оценки влияния толщины пленки для водоэмульсионных кра-
сок заключается в окраске большой школьной доски: первый слой
наносится на всю поверхность, второй — на нижнюю половину
и третий — на правую четверть; затем сравнивается сплошность
в областях, окрашенных одним, двумя и тремя слоями. Испытания
такого рода, проведенные опытными мастерами, дают возможность
сравнить относительные характеристики укрывистости для различ-
ных красок в условиях, имитирующих практическую окраску.
15.2.2. Определение отношения контрастностей
Наиболее широко распространенным методом оценки укры-
вистости является нанесение пленки контролируемой толщины на
подложку, содержащую черные и белые участки. Вначале исполь-
зовалась шахматная доска, и контраст между черными и елыми
участками оценивался визуально. Такая оценка субъективна и не-
воспроизводима; современные методы основаны на фотометриче-
ских измерениях. Существует ряд проблем, связанных с определе-
нием отношения контрастностей, которые заслуживают внимания
и обсуждения; некоторые из них — практические, другие — теоре-
тические, а некоторые представляют собой интерпретацию резуль-
татов, полученных при практической окраске.
15.2.2.1. Измерение отношения контрастностей
Достоверные результаты можно получить только на однород-
ных пленках известной толщины. Может быть эффективным налив
краски на плоские черно-белые стеклянные пластинки с помощью
аппликатора. Другим методом определения является нанесение
краски на прозрачную синтетическую пленку равномерной толщи-
ны (например на полиэфирную) и проведение измерений при опти-
ческом контакте окрашенной синтетической пленки со стеклянной
пластиной (оптический контакт дает жидкость с показателем пре-
ломления, идентичным показателю преломления синтетической
пленки) [8]. В обоих методах толщина покрытия определяется
исходя из веса краски на единицу площади.
Белые покрытия гораздо более проницаемы для красного света,
чем для синего, так что для достижения соответствия с визуаль-
ной оценкой сочетание светового источника и записывающей си-
стемы должно по спектральному отклику приближаться к челове-
ческому глазу при дневном свете. Отклонения от этого условия
являются одной из главных причин ошибок в измерениях отноше-
ния контрастов.
15.2.2.2. Теоретические основы контрастных соотношений
Если предположить, что пленка имеет коэффициент отраже-
ния До на неотражающей поверхности и Д на поверхности с ко-
эффициентом отражения До, и светопропускание пленки Т, то мож-
но показать, что
R = R<> + T2-RC/(1-RORC). (15.1)
Это выражение неточно, поскольку не учитывает зависи-
мость Т от изменения угла падающего света, а также возможность
измерения До путем нанесения слоя краски, что устраняет поверх-
ность раздела стекло/воздух на стандартной стеклянной поверх-
ности. Однако это уравнение полезно при рассмотрении влияния
изменения До на контрастность. В табл. 15.2 показано изменение Д
в зависимости от До для двух типичных случаев: белая краска
(До = О,7; 7 = 0,25) и серая краска с хорошей укрывистостью
(До = 0,5; 7 = 0,1). В обоих случаях видно, что для светлых суб-
стратов Д гораздо более чувствительна к изменению До, чем для
Таблица 15.2. Влияние яркости субстрата на коэффициент отражения белых
и светло-серых покрытий
Коэффициент ' отражения субстрата Белая краска (/?n = 0,7; Т = 0,25) Серая краска (/?<> = (),5; 7 = 0,1)
R Отношение контрастностей («../«) R Отношение контрастностей (/?..//?)
0 0,700 1,00 0,500 1,000
0,1 0,707 0,99 0,501 0,998
0,2 0,715 0,98 0,502 0,996
0,4 0,735 0,95 0,505 0,990
0,6 0,772 0,91 0,509 0,982
0,8 0,814 0,86 0,513 0,975
0,9 0,852 0,82 0,516 0,969
1,0 0,908 0,77 0,520 0,961
темных. Из этих вычислений следует вывод, что для получения
воспроизводимых результатов при измерении контрастных соот-
ношений точная характеристика черного субстрата не является
обязательной, но отражательная способность белого субстрата
должна очень тщательно контролироваться.
Вычисления, приведенные в табл. 15.2, также показывают, на-
сколько поглощение света пленкой влияет на укрывистость; в слу-
чае белой краски приблизительно только 1 /20 часть падающего
света поглощается при прохождении через пленку, а в случае серой
краски поглощается около 40%.
15.2.2.3. Связь с практической окраской
Из сказанного выше может показаться, что гораздо легче
«укрыть» неоднородность темного субстрата, чем светлого. Одна-
ко, если принять во внимание изменения значений R, которые воз-
растают с увеличением толщины пленки, окраска темных субстра-
тов становится не таким простым делом. Оптимальный цвет суб-
страта (или подслоя) должен быть близким к цвету толстой пленки
покрытия, которое необходимо нанести.
Для красок темного цвета, которые обычно обладают лучшей
укрывистостыо, чем светлые краски, определение величины отно-
шения контрастностей не очень существенно; для них недостатки
укрывания поверхности обычно связаны с появлением тонких
штрихов на пленке, обусловленных недостаточной текучестью и де-
фектами нанесения покрытия.
Для белых красок, вероятно, наиболее полезной характеристи-
кой может служить степень укрывания, которую можно получить
при окрашивании поверхности со средней скоростью нанесения.
Международный стандарт ISO 3906—1980, на основе которого
британским институтом стандартов разработан британский стан-
дарт BS3900: Part D6:1982 [9], описывает метод определения
отношения контрастностей, соответствующий средней скорости
окраски 20 м2/л. При этом толщина «мокрого» слоя составляет
около 50 мкм, что является типичным при кистевом нанесении. Ме-
тод предусматривает измерения на шести пленках, различающихся
скоростью нанесения краски, и последующую интерполяцию.
Другой подход заключается в определении веса или толщины
пленки, которая обеспечивает требуемую укрывистость путем
построения графика зависимости отношения контрастностей от
веса пленки с последующей интерполяцией или экстраполяцией
полученных данных. Когда этот метод был впервые разработан,
требуемый уровень укрывистости характеризовался отношением
контрастностей 0,98. Было выбрано именно это значение, посколь-
ку 2% разницы в факторе яркости, как было принято, является
наименьшим контрастом, который человеческий глаз может ясно
различить.-; Такое высокое значение отношения контрастностей,
по всей видимости, слишком велико, так как на практике окраска
редко производится по черной и белой поверхностям. Существуют
также серьезные экспериментальные трудности точного опреде-
ления высоких значений отношения контрастностей из-за неизбеж-
ных ошибок в измерении параметров яркости. Поэтому предпоч-
тительнее работать с отношением контрастностей 0,95 по черному
и белому, что на практике соответствует получению удовлетвори-
тельной укрывистости белой краски Применительно к белой крас-
ке эта цифра должна достигаться при скорости нанесения не ни-
же 10 м2/л или 20 м2/л для двухслойного покрытия. Вычисления
показывают, что для белого покрытия с почти нулевым светопогло-
щением отношение контрастностей 0,95 (в расчете на 2 слоя соот-
ветствуют примерно 0,85 для одного слоя) Таким образом, значе-
ние 0,85, достигаемое в вышеупомянутом британском стандарте,
предполагает достижение удовлетворительной укрывистости при
нанесении двух равномерных слоев.
15.2.3. Укрывистость и коэффициенты светорассеяния
Укрывистость можно соотнести со светопоглощением и свето-
рассеянием по теории Кубелки-Мунка. Это основа метода оценки
укрывистости, предложенного в США и стандартизованного в этой
стране (ASTM D2805), в ФРГ (D1N 53162), а также в Англии
(BS 3900: Part: 1983 (10] ив международном стандарте ISO
6504/1 —1983. Рабочая формула соотношений Кубелки-Мунка,
используемая в этом методе, такова:
^l-^Ca-fecosfe-SQ (]52)
a + b cos b-St — Rq
где й = 0,5(/?„ +!)//?«, ; fc = a—/?то ; R — коэффициент отражения пленки тол-
щиной /, нанесенной на подложку с отражательной способностью /?с; S — коэффи-
циент светорассеяния на 1 мкм; I — толщина пленки, мкм; R™ — отражательная
способность пленки такой толщины, при дальнейшем увеличении которой R не уве-
личивается.
Очевидно, что уравнение для определения коэффициента отра-
жения на черной подложке RB (До = 0) упростится:
/?в = I/(a + b cos b-St), (15.3)
или иначе:
/ 1 — aRr, \
St = \/b • arcos ——-J. (15.4)
Для упрощения вычислений произведение St обычно определяется
из графиков зависимости St от Дв и Д^. Определив St, возможно
затем вычислить t или скорость нанесения, соответствующую отно-
шению контрастностей 0,98.
Метод требует определения только Д^, что гораздо легче сде-
лать для светлых красок путем их нанесения на матовое стекло или
белую керамическую плитку и измерения Дв для пленки известной
толщины. Для определения Дв пленку следует нанести на черное
стекло. Ее можно нанести также на полиэфирную пленку и нало-
жить на черное стекло, предварительно слегка смочив его несколь-
кими каплями уайт-спирита для получения оптического контакта.
Рис. (5.6. Коэффициенты рассеяния из графика зависимости /?/(!—R) от веса
пленки (окись цинка при ОКП = 20% в льняном масле)
Несмотря на сложность теории и явно громоздкую форму урав-
нений 15.2—15.4, метод весьма полезен для вычислений.
Для простого случая, когда светопоглощением в пленке можно
пренебречь благодаря светорассеянию, получается упрощенное
выражение для Дв:
RB=St/(St+l) St=R/(l-R). (I5.5)
График зависимости R/ (1 —R) от t представляет собой прямую
с наклоном, характеризующим величину S. На рис. 15.6 показаны
результаты измерений при различных длинах волн для пленок
белых красок, нанесенных на черную стеклянную подложку. Вид-
но, что при более длинных волнах сохраняется линейная зависи-
мость и S растет с уменьшением длины волны. При длине волны
450 нм, где нельзя пренебречь светопоглощением пигментов, ли-
нейность теряется.
15.3. ХАРАКТЕРИСТИКА И КОНТРОЛЬ ЦВЕТА
15.3.1. Введение
Цвет можно контролировать путем визуального сравнения об-
разца со стандартными цветными эталонами. Л4ожно доказывать,
что в конечном счете глаз является единственным справедливым
арбитром, но возникает много вопросов, из которых самым труд-
ным, вероятно, является: «Чей глаз?» Помимо генетических ано-
малий, которые рассматривались выше, известно, что цветовос-
приятие изменяется с возрастом из-за возникновения в глазах
желтой пятноподобной пигментации, и что стандартный наблюда-
тель — это статистическая абстракция. Все это — сильные аргу-
менты в пользу контроля цвета путем физических измерений. Эти
измерения и их интерпретация должны быть тесно связаны с визу-
альным восприятием наблюдателя. На практике имеется два вида
цветового контроля — визуальный и инструментальный.
15.3.2. Визуальный цветовой контроль
15.3.2.1. Цветовые картотеки и стандарты
Было предпринято много попыток для создания исчерпываю-
щих визуальных цветовых картотек. Наиболее универсальной яв-
ляется картотека «Munsell book of colour», впервые опубликован-
ная в 1929 г. [II]. Вся картотека насчитывает 40 листов, каждый
для отдельного оттенка, и охватывает весь спектр от красного че-
рез пурпурный к фиолетовому (РВ по номенклатуре Мансела).
Цвета на каждом листе сгруппированы в ряды с одинаковым зна-
чением «уровня», (соответствует значению У) и в колонки с рав-
ным значением «хроматичности» (соответствует насыщенности
1
4
Рис. 15.7. Расположение в цветовой книге Мансела:
1 — белый; 2— величина; 3 — черный; 4 — оттенок; 5 — цвет; 6 — нейтральные серые;
7 - - желтый; 8 — фиолетово-синий
цвета) (рис. 15.7). Каждый цвет имеет три характеристики: отте-
нок, уровень и насыщенность, например 5 YR/5/10 — это насыщен-
ный оранжевый цвет. Широкий диапазон цветов в картотеке Ман-
села представлен в виде самостоятельных цветов (каждый может
быть глянцевым или матовым), но картотека позволяет интерполи-
ровать или экстраполировать цвета. Картотека была стандартизо-
вана путем замера отражения для каждого цвета.
Принимая во внимание, что человеческий глаз способен разли-
чить как минимум полмиллиона цветов при оптимальных условиях
наблюдения, неудивительно, что, хотя картотека Мансела способ-
на характеризовать цвета, она не исключает использование инди-
видуальных цветовых карточек для целей точной характеристики
цвета. Другая причина применения таких индивидуальных карто-
чек для промышленных целей состоит в том, что визуальное цве-
товое сравнение трудно стандартизовать, особенно в тех случаях,
когда глянец и текстура сравниваемых поверхностей неидентичны.
Таким образом, для разработки метода контроля цвета прежде
всего необходимо тщательное изготовление эталонов с последую-
щим использованием полученных таким образом цветовых харак-
теристик в качестве рабочих стандартов.
15.3.2.2. Визуальное цветовое сравнение
Помимо выбора наблюдателя с нормальным или средним цве-
товосприятиём, важнейшим фактором для правильной оценки при
визуальном сравнении цвета является освещение.
— Доетаточноувилетьиз-м-енение цвета-си них-ил и пурпурных цве-
тов в лучах солнечного света, чтобы понять, насколько сильно
может зависеть цвет от изменения освещения. Старый метод срав-
нения цветов предполагал в качестве источника освещения рас-
сеянный дневной свет, вероятно, наиболее воспроизводимый есте-
ственный источник. Большинство сравнений сейчас производят
в камерах с тщательно подобранными флуоресцентными лампами
и контролируемыми условиями наблюдения, приведенными в стан-
дартах: международном ISO 3668—1976 и британском BS 3900:
Part D1 : 1978 [12]. Эти стандарты предусматривают как естест-
венный рассеянный дневной свет интенсивностью не менее 2000 люкс,
так и искусственное освещение с интенсивностью 1000—4000 люкс.
Для общего употребления рекомендуется нейтральный серый фон
с уровнем освещенности 15% (Мансел N4—N5), а для белых цве-
тов предпочтительнее более яркий фон с уровнем освещенности
30% (Мансел N6). Образцы лучше всего располагать так, чтобы
их кромки частично перекрывались; рассматривать их следует
с расстояния не менее 500 мм. Метамеризма можно избежать, на-
пример, путем переключения на вольфрамовую лампу или на дру-
гой источник с радикально отличающимся спектральным распреде-
лением. Метамеризм, действительно, часто является причиной
ошибок при визуальном цветовом сравнении. В крайних ситуа-
циях, если сравниваются сильно метамерические цвета, наблюда-
тель может видеть красноватую область с одной стороны линии
соприкосновения образца и стандарта и сине-зеленую область —
с другой стороны.
Проблемой является возможное выцветание или другое изме-
нение цвета стандартов. Рабочие стандарты, которые показывают
значительное изменение цвета при регулярных инструментальных
проверках, должны быть заменены.
15.3.3. Инструментальный контроль цвета
-15.3.3.1. Введение
Существует два основных метода измерения цвета поверхно-
стей. Первый, называемый трехстимульной колориметрией, напо-
минает визуальный метод, и заключается в прямом определении
параметров X, Y, Z (см. 14.4.1). Второй метод основан на опреде-
лении коэффициента отражения (/?) в каждом из диапазонов длин
волн, в котором чувствителен глаз, с последующим вычислением
визуальных характеристик путем суммирования произведений R
на стандартные величины распределения чувствительности для
трех цветовых характеристик (х, у, z). Трехстимульная колоримет-
рия теоретически имеет преимущества, особенно для флуоресцент-
ных материалов.
15.3.3.2. Трехстимульная колориметрия
Трехстимульный колориметр имеет три основных части:
1) источник освещения, обычно лампа при строго фиксирован-
ном напряжении;
2) набор трех комбинаций фильтров, используемых для регу-
лирования распределения энергии падающего или, лучше, отра-
женного света;
3) фотоэлектрический детектор, преобразующий интенсив-
ность отраженного света в электрический сигнал.
Общее требование заключается в том, чтобы произведение рас-
пределения энергии источника (Ек), пропускания фильтра (/ч),
и чувствительности детектора (S?.) соответствовало бы произве-
дению спектрального распределения чувствительности глаза и рас-
пределения энергии источника (например D65), к которому долж-
ны относиться трехстимульные значения. Абсолютное соответствие
невозможно, но в лучших образцах колориметров достигается не-
обходимый уровень точности. Обычно не стремятся сравнивать два
пика распределения х; полагают, что высота коротковолнового
пика близка к высоте максимума распределения параметра z и из-
меренная величина х, обозначаемая хм, увеличивается пропорцио-
нально увеличению измеренной величины г, т. е. трехстимульные
параметры х, у, z могут быть выражены в следующем виде:
x=xM+0,l8zM; у=ум; 2 = 2м-
Измерения, проведенные на трехстимульном колориметре,
обычно относительны, причем прибор калибруется по стеклянному
или керамическому стандартам. Поскольку точное соответствие не
всегда достигается при работе прибора, для большей точности
стандартизация должна проводиться с использованием калибро-
вочных стандартов тех же цветов, что и у испытываемых материа-
лов. Наиболее употребительны трехстимульные колориметры для
экспресс-сравнения близких цветов. Как и для всех современных
систем цифровой записи, необходимо помнить, что точность ре-
зультатов больше зависит от вводимого сигнала, чем от компью-
тера; если спектральная чувствительность системы лампа —
фильтр — фотодетектор неточна, то и результат обработки сигнала
скорее всего будет ошибочен, несмотря на точность вычислений
и количество обработанных величин.
15.3.3.3. Спектрофотометрия
Для точных измерений цвета в абсолютных величинах можно
использовать спектрофотометр, т. е. измерять отражение для
каждой из длин волн по очереди и затем вычислять трехстимуль-
ные величины. Преимущества метода перед трехстимульной коло-
риметрией заключаются в том, что полученной информации доста-
точтгсг для вычисления цветовых величин при любом источнике
света, и в том, что автоматически обнаруживается метамеризм.
К недостаткам относится дороговизна высококачественных спект-
рофотометров и длительность измерений. Как и для колориметров,
для всех видов вычислений, связанных с этими измерениями, мож-
но применять встроенные и приставные компьютеры.
В спектрофотометре свет обычно расщепляется в спектр с по-
мощью призмы или дифракционной решетки, и каждая из полос
соответствующих длин волн отбирается по очереди для измерений.
Разработаны приборы, в которых узкие полосы отбираются путем
интерференционных фильтров. Если необходимо изучать флуорес-
центные материалы, образец должен освещаться полным спектром,
а отраженный свет — разлагаться для анализа [13]. Спектральное
разрешение прибора зависит от узости полос, применяемых для из-
мерений. Для большинства работ с красками ширина полосы
в 10 нм дает чаще всего достаточное разрешение. Теоретически
спектрофотометр способен прямо сравнивать отраженный свет
с падающим, но его обычно калибруют по матовому стеклянному
стандарту, предварительно откалиброванному в международно
зарегистрированной лаборатории. Должна быть сделана проверка
оптического нуля путем измерений с черной ловушкой света, так
как пыль и другие помехи могут привести к неправильным пока-
заниям.
15.3.3.4. Условия освещения и наблюдения
Для всех типов цветоизмеряющего оборудования полученные
результаты и их корреляция с визуальными определениями зависят
от углов освещения и наблюдения. Эта зависимость в максималь-
ной степени важна для черных глянцевых образцов.Колориметрия
Таблица 15.3. Стандартные условия освещения и наблюдения
Условия измерений
Освещен ие Наблюдение
Обозначение (краткое)
Прямое (45±5) °
Прямое (0±10)°
Полусферическое с фоку-
сирующей сферой
Полусферическое с фоку-
сирующей сферой и по-
глощением глянца
Прямое (8±2)°
То же
Прямое (0±10)°
Прямое (45±5)°
Прямое (8±2)°
То же
Полусферическое с фокуси-
рующей сферой
Полусферическое с фоку-
сирующей сферой и с погло-
щением глянца
45°/нормальное (45/0)
Нормальное/45° (0/45)
Диффузпое/8° (d/8)
Диффузное/8° (cZ/8);
исключено зеркальное от-
ражение
8°/диффузное (8/d)
8°/диффузное (8/d); ис-
ключено зеркальное от-
ражение
Примечание. В последних четырех условиях выбран угол 8°, а ие 0°, так как при
нормальном освещении (наблюдении) могут возникнуть сложности с отражениями между
глянцевыми образцами и оптикой для освещения (наблюдения); при угле 8° зеркальное
отражение большей частью устраняется.
лаков и красок описывается в международном стандарте I.SO 7724,
который воспроизводится в британском стандарте BS 3900; в нем
предусматривается шесть разновидностей условий освещения и на-
блюдения (табл. 15.3). Из них первое (45/0) и четвертое (d/8)
с исключенным зеркальным отражением, вероятно, в наибольшей
степени соответствует визуальному испытанию в специальном бок-
се для цветовых измерений. Однако, третье и пятое условия, где
имеется зеркальное отражение, обладают преимуществом с точки
зрения снижения влияния различий глянца на цветовые измере-
ния; при этом получаются результаты, соответствующие цвету,
видимому под облачным небом или в комнате при рассеянном осве-
щении.
Многие цветоизмерительные приборы сейчас включают сфери-
ческие светильники для освещения или наблюдения. Сфера внутри
покрыта матовой белой краской, которая должна иметь однород-
ную отражательную способность во всем видимом спектре (обыч-
ным пигментом для верхнего покрытия является чистый сульфат
бария). Имеются специальные гнезда для образца, которые долж-
ны занимать небольшую часть общей площади поверхности,
поскольку иначе вторичные отражения от образца могут увеличить
насыщение цвета внутри сферы. При тщательной разработке гео-
метрии сферы и применении светоулавливающих конусов можно
добиться устранения компонентов зеркального отражения и, та-
ким образом, измерять только диффузное отражение.
1S.3.3.S. Допустимые отклонения цвета
При оптимальных условиях глаз может различать исключи-
тельно малые цветовые различия, порядка 1% значения Y или по-
казателя яркости и около 0,1АЕ в системе CIE L, а, b [14]. На
практике нет смысла контролировать цвет в таких пределах, даже
если возможно применить точные методы измерений. Для повсед-
невного контроля, следовательно, необходимо установить приемле-
мые пределы допустимых отклонений цвета. Наилучшая корреля-
ция из визуальных оценок, вероятно, может быть дана по разни-
цам психометрической яркости L*, насыщенности С"пь и оттенка
Hfr,, как определено в формулах CIELAB, 1976. Эти пределы мож-
но вычислить из допустимых изменений параметров X, Y, Z для
любого цвета, но с помощью компьютера их легко можно выразить
прямо как Д, С и Н.
15.4. ЦВЕТОВОЙ КОНТРОЛЬ ПРИ ПРОИЗВОДСТВЕ КРАСОК
Ранее упоминались работы по вычислению цвета смесей пиг-
ментов и компьютеризованное составление рецептур и корректи-
ровка цвета. Такие системы обеспечивают максимальные преиму-
щества, если производится тщательный качественный контроль
всех пигментов и связующих [15]. Метамеризма можно избежать,
если число применяемых для изменения оттенка пигментов будет
минимально (обычно не более трех) и если они имеют воспроизво-
димые цветовые характеристики. При тщательном контроле боль-
шинство красок может быть приготовлено из одноцветной основы
и цветных пигментов путем объемного смешивания. Такой ло-
гичный подход целесообразен при поставках красок для розничной
продажи, когда требуется большое разнообразие оттенков, напри-
мер в материалах для ремонта автомобилей. Смесевые системы
широко используются для розничной продажи на рынках Сканди-
навии и Северной Америки, но они не получили распространения
в Великобритании.
Глава 16
ОЦЕНКА ДОЛГОВЕЧНОСТИ ПОКРЫТИИ
Р. Ламбурн
16.1. ВВЕДЕНИЕ
Долговечность можно определить как способность покрытия
противостоять внешним воздействиям; т. е. оставаться неизмен-
ным при воздействии окружающей среды и различных неблаго-
приятных факторов. К неблагоприятным относятся факторы, вызы-
вающие напряжения, которые могут быть непродолжительными,
например удар, или продолжительными, например медленное рас-
тяжение или сжатие пленки и подложки. Окружающие условия
оказывают огромное влияние на долговечность покрытия, поэтому
методы испытаний систем покрытий всегда разрабатываются та-
ким образом, чтобы воспроизвести практические условия приме-
нения последних. Они обычно разрабатываются, чтобы ускорить
процессы разрушения испытываемых покрытий. Задачей ускорен-
ного испытания является предсказание момента разрушения по-
крытия. Другой аспект определения долговечности состоит в том,
чтобы определить способность покрытия противостоять наруше-
ниям условий эксплуатации; и в этом случае используются также
различные методы испытаний. Для всех видов покрытий суще-
ствует два типа испытаний: тесты на химическую стойкость и тесты
физические или механические. Следует принять во внимание, что
долговечность покрытия часто зависит от природы подложки.
Разнообразие тестов объясняется многими обстоятельствами.
На рынках промышленных красок изготовитель сталкивается
с множеством методик, выдвигаемых потребителем. Среди разра-
ботчиков методик испытаний следует выделить Американское об-
щество испытания материалов (ASTM), Британский институт
стандартов (BSI), Германский институт нормативов (DIN), Меж-
дународную организацию стандартов (ISO), британские органи-
зации — Общество изготовителей и торговцев двигателями
(SMMT), Министерство обороны, Ассоциацию исследователей
красок и др.
16.1.1. Причины разрушения покрытий
Из множества причин разрушения покрытий некоторые уста-
новлены, и можно предпринимать попытки бороться с ними. Строи-
тельные краски с автоокисляющимися связующими имеют внут-
ренние причины деструкции. Процесс окисления не прекращается
с высыханием пленки, в результате образуется сильно сшитая
пленка. Продукты окисления выделяются из пленки, процесс сши-
вания приводит к повышению хрупкости пленки, и, в конце концов,
наблюдается ее растрескивание и отслаивание от подложки. До-
статочная долговечность современных глянцевых наружных по-
крытий связана с тщательным выбором пленкообразователя, что
сводит к минимуму окисляемость пленки, увеличивает прочность
пленки при растяжении, твердость и др.
Разрушение промышленных покрытий, например на стираль-
ных машинах, не составляет проблемы в течение срока службы
самой защищаемой машины, если покрытие обладает удовлетвори-
тельной стойкостью к стиральным порошкам или щелочам. Для
автотранспортных средств покрытие не только должно сохранять
соответствующий внешний вид при воздействии УФ-лучей и погод-
ных условий вообще, но и должно быть стойким к ударам мелких
камней, а также предотвращать распространение ржавчины в ме-
стах, подвергаемых различным воздействиям. Можно привести
и другие примеры. Очевидно, что покрытие должно обладать и хи-
мической стойкостью, и оптимальными механическими свойст-
вами, так как разрушение покрытий может иметь место как вслед-
ствие воздействия каждого из этих факторов, так и вследствие
совместного их воздействия.
*
16.1.2. Методология
Испытания долговечности проводятся как для контроля каче-
ства стандартных продуктов, так и при разработке новых мате-
риалов. Из-за большого числа образцов, подвергаемых испыта-
ниям, часто невозможно проводить испытания со статистически
достоверным числом образцов в каждой из сравниваемых серий.
В лучшем случае испытания можно провести на двух параллель-
ных образцах; важно при этом включить в каждую из испытывае-
мых серий стандартный (контрольный) образец с установленными
свойствами — базис для сравнения. Если стандартная композиция
ведет себя хуже, чем ожидалось, вся серия должна быть признана
сомнительной и испытания следует повторить. Это может произой-
ти, если возникнут необычные обстоятельства, например перегрев
или загрязнение тестового раствора, что могло бы в других случаях
пройти незамеченно.
16.1.3. Подготовка образцов для испытаний
Тип пластин часто специфичен. Часто используют пластины
из мягкой стали 150X100 мм. Пластина должна быть очищена от
поверхностных загрязнений и коррозии, тщательно обезжирена
трихлорэтиленом и высушена, затем отшлифована тонкой наж-
дачной бумагой и обработана уайт-спиритом до полного удаления
каких-либо поверхностных загрязнений. После этого наносят
испытываемый материал в соответствии с его свойствами и типом
.испытаний, включая любую предварительную обработку пласти-
\ны и нанесение грунтовки и/или промежуточного покрытия. Сле-
дует позаботиться о том, чтобы обезжиренные перед окрашивани-
ем пластины не были загрязнены руками или иным путем.
' Окрашенные пластины высушивают на воздухе или, при необ-
ходимости, отверждают при нагревании. В некоторых случаях
окрашивают одну сторону, а вторую защищают (например от
коррозии) другим материалом. Этот последний может быть быст-
росохнущей краской или, в некоторых случаях, восковым покры-
тием. Обычно способ защиты обратной стороны пластины указы-
вается в методике испытаний.
При испытании продуктов в процессе их разработки на прак-
тике имеют дело только с одним экспериментальным лакокрасоч-
ным материалом как частью всей системы. Например, если испы-
тывается краска для верхнего покрытия, то ее следует применять
в композиции со стандартными грунтовкой и/или промежуточным
покрытием. Аналогично, при испытании экспериментальной грун-
товки все остальные компоненты общей системы покрытия должны
быть стандартными.
Применяются не только стальные, а в некоторых испытаниях
также алюминиевые или жестяные пластины; в ряде случаев может
использоваться дерево, твердый картон, асбоцемент и стекло, осо-
бенно при механических и климатических испытаниях.
16.2. ЙСП1 ТАНИЯ ХИМИЧЕСКОЙ СТОЙКОСТИ
Здесь не предполагается приводить детальных методик. Те из
них, которые являются модификацией стандартных методик (или
идентичны им), указаны в списке литературы. Эти методики
описаны в той степени, чтобы читатель мог их оценить; при жела-
нии применить их неизбежно обращение к специальной литературе.
Если не придерживаться строго установленных методик, трудно
получить воспроизводимые результаты, поэтому исследователь
должен критически относиться к данным, полученным не в соответ-
ствии с утвержденными методиками. Во многих случаях тесты
разработаны изготовителями красок для собственных нужд или
по согласованию с потребителем.
16.2.1. Специальные испытания
16.2.1.1. Водостойкость
Целью этого испытания является оценка стойкости покрытия
к проникновению дистиллированной воды. Метод отражен в бри-
танском стандарте для автомобильной промышленности SMMT
[57]. Он применяется для широкого круга промышленных лако-
красочных материалов.
Испытание проводится в термостатируемой водяной бане (тем-
пература воды 38 + 0,35 °C), снабженной механической мешалкой.
Пластинки, подготовленные как описано выше в 16.1.3, поме-
щаются в держатели, изготовленные из инертного материала, вер-
тикально, попарно, повернутые друг к другу обратными поверх-
ностями. Через 24 ч пластинки вынимаются, аккуратно обтираются
сухой, мягкой тканью и немедленно осматриваются на предмет
появления пузырей и потери глянца. Пузыри, образовавшиеся на
расстоянии ближе 12 мм от края пластины, не принимаются во
внимание. Затем пластины вновь опускаются в воду, и испытание
продолжается (как правило, не менее 7 дней). Пластины осматри-
ваются каждые 24 ч вплоть до окончания испытаний, после чего
их высушивают при комнатной температуре; пластины можно за-
тем использовать для других испытаний. Чаще всего пластины за-
тем испытывают на потерю адгезии; обычно есть возможность
установить, имело ли место падение адгезии между покрытием
и подложкой лли межпу слоями л системе л о крытия.
Образование пузырей обычно оценивается с использованием
фотографий, опубликованных в ASTM 714-56. Пузыри классифи-
цируют по размеру, числу или концентрации. Концентрация пузы-
рей имеет 4 категории: «малая», «средняя», «средней плотности»
и «плотная». Также оценивается потеря глянца. Если имеет место
значительное образование пузырей, то нет смысла оценивать поте-
рю глянца приборами, так что это делается по числовой шкале
в сравнении с образцом, не подвергавшимся испытаниям. Исполь-
зуется шкала от 1 до 10, где 10 — «отлично» (нет потери глянца),
а 1 — полная потеря глянца.
16.2.1.2. Влагостойкость: BS3900: Part F2
Это испытание отличается от предыдущего тем, что оно отно-
сится к поведению покрытий при условиях циклических воздей-
ствий температуры и влажности, как это бывает для автомобилей
и других окрашенных металлических поверхностей в различных
климатических условиях. Тест часто упоминается в правительст-
венных контрактах. Испытание проводится в камере влажности,
где поддерживается влажность около 100%, а температура цикли-
чески изменяется между 42 и 48° с тем, чтобы обеспечить конден-
сацию на пластинках. Нагревание производится с помощью по-
гружного нагревателя воды, помещаемого на дне камеры. Темпе-
ратура воздуха непрерывно изменяется циклически между двумя
экстремумами за 60 + 5 мин. Циркуляция воздуха поддерживается
с помощью вентилятора, чтобы в любой точке воздушного прост-
ранства камеры различие температур в любой момент времени
не превышало 1 °C.
Пластинки для испытаний, подготовленные так же, как и для
испытаний на водостойкость, визуально осматриваются каждые
$4 ч на предмет появления признаков ухудшения внешнего вида.
При их возвращении в камеру они помещаются в другую точку
камеры, чтобы свести к минимуму любые влияния, которые могли
бы быть вызваны местоположением образца. После окончания
испытаний образцы выдерживают 24 ч при комнатной темпера-
туре и затем проверяют на потерю глянца, адгезии, изменение цве-
та или появление хрупкости. В некоторых случах можно снять по-
лоску краски с пластинки и исследовать появление подпленочной
коррозии.
16.2.1.3. Стойкость к солевым брызгам
Эти испытания являются, возможно, самыми распространен-
ными испытаниями коррозионной стойкости и, вместе с тем, наи-
более спорными. Хорошо известно, что соли, такие как хлористый
натрий, могут вызвать быструю коррозию железных субстратов,
поэтому полезно иметь информацию о поведении конкретных
систем при защите таких подложек от коррозии как для неповреж-
денных, так и для поврежденных пленок. При интерпретации
данных ввиду плохой воспроизводимости результатов испытаний
возникают сомнения. Однако эти испытания признаны и, несмот-
ря на проблему воспроизводимости, являются весьма полезными
при отсутствии более точных данных по коррозии. Они, таким
образом, вряд ли могут быть отброшены. Лишь некоторые авторы
полагают эти испытания далекими от практики вследствие ускоре-
ния коррозионного процесса и невоспроизводимости результатов
по определению степени повреждения пленки, получаемой в раз-
ных тестах.
Обычно используют два теста: непрерывного и прерывистого
разбрызгивания солевого раствора.
Метод непрерывного разбрызгивания. Тест описан в британ-
ском стандарте BS 3900: Part F4—1968. Покрытие подвергается
обработке в течение определенного времени и затем проверяется
степень его повреждения. Следует критически относится к интер-
претации результатов теста; он не предназначен для ускоренных
испытаний, его назначение — воспроизведение воздействия погод-
ных условий.
Пластины подготавливают как указано в BS 3900: Part АЗ и А4
(в основном соответствует описанию в разделе 16.1.3). Обратную
сторону и края покрывают хорошим защитным материалом есте-
ственной сушки. Пластины выдерживают 24 ч перед началом испы-
таний. Если выдержка больше, это следует отметить, так как это
может повлиять на результаты испытаний. С помощью скальпеля
покрытие прорезается до металла на расстоянии 25 мм от верха
и от низа пластины. Разрез должен идти параллельно длинной
стороне пластины, причем важно, чтобы на поверхности металла
появилась царапина. Испытание проводится в химически инертном
(т. е. стеклянном или пластиковом), плотно закрытом крышкой
сосуде. Солевой туман получается при разбрызгивании искусствен^
ной морской воды с помощью пульверизатора. Пластины нахо<-
дятся в специальных гнездах, их лицевые стороны наклонены при-
мерно на 15° к вертикали Распыление проводится таким образом,
чтобы брызги не попадали непосредственно на поверхность плас-
тин. Раствор, стекающий с испытываемых пластин, не подается
вновь для распыления. Пластины проверяют через 48 ч, 1, 2 и 3 не-
дели. Их ополаскивают в проточной воде, сушат фильтровальной
бумагой и сразу же проверяют на появление пузырей, коррозии
вблизи надреза и адгезию. Образование пузырей оценивают так
же, как описано выше. В этом случае следует обратить особое вни-
мание на образование пузырей па расстоянии 3, 6 и 12 мм от надре-
за. Потеря адгезии оценивается по легкости удаления покрытия
ногтем и по тому, насколько далеко от надреза распространяется
ухудшение адгезии. Наблюдения оценивают: «без изменений»,
«легкое снижение адгезии» и «сильное снижение адгезии». С целью
получения воспроизводимых результатов, готовят стандартный
солевой раствор, причем используют только соли аналитической
чистоты. Состав испытательного раствора таков: NaCl — 26,5 г;
MgCl —2,4<г; MgSO4 —3,3 г; КО — 0,73 г; NaHCO3 — 0,20 г;
NaBr — 0,28 г; СаСК— 1,1 г; дистиллированная вода—до
1000 мл Хлористый кальций должен добавляться последним. Тем-
пература раствора при испытаниях — 20±2 °C.
Прерывистое разбрызгивание. Тест аналогичен предыдущему
за исключением того, что солевой туман подают ежедневно 8 раз
по 10 минут с интервалом 50 минут. Испытания проводят 5 дней
подряд, а затем — перерыв на 2 дня. Испытание не такое жесткое,
как непрерывное разбрызгивание.
Существует множество вариантов таких тестов. В некоторых
из них на пластины наносят крестообразный надрез по диагоналям
Указывается, что потрею адгезии легче обнаружить (и что она,
возможно, проявляется на более ранней стадии) в центре пересе-
чения. Обычно указывается тип скальпеля и прилагаемое усилие
при царапании. Последнее условие выполняется путем располо
жения пластины на лабораторных весах и нагружения ее весом
1 кг при царапании.
16.2.1.4. Стойкость к щелочам и моющим средствам
Применяется как минимум три теста на щелочестойкость. Они
основаны на использовании тринатрийортофосфата, безводного
карбоната натрия и гидроксида натрия, соответственно. Эти тесты
обычно используются при испытаниях промышленных верхних
покрытий, которые могут применяться для бытовых приборов, та-
ких как стиральные машины. Кроме того, можно также использо-
вать тест на стойкость к моющим средствам. Щелочные составы
имеют различную степень агрессивности. Пластинки для всех тес-
тов подготавливают как указано в 16.1.3. Обычно окрашивают обе
^стороны пластины и торцы, но можно окрашивать и одну сторону,
обрабатывая другую и торцы расплавленным парафиновым вос-
Дом (т. пл. 49—60 °C). Если необходимо окрашивать обе стороны
Пластины, удобно иметь у края ее маленькое отверстие для подве-
шивания при сушке (в печи или на воздухе). Фирма 1CI предлагает
такие концентрации и температуры растворов:
Тринатрийортофосфат, 1% (масс.) в дистиллированной воде, 75 °C;
Безводный карбонат натрия, 10% (масс.) в дистиллированной воде, 65 °C;
Гидроксид натрия, 5% (масс.) в дистиллированной воде, 25 °C.
В этих испытаниях пластины частично погружаются в раство-
ры на глубину 7—10 см. Как и в вышерасмотренных тестах, они
поддерживаются неметаллическими держателями в положении,
близком к вертикальному. (Влияние частичного погружения дает
удобную возможность сделать прямые сравнения влияния раство-
ра на систему покрытия.) Существенно, чтобы сравнения с приня-
тым стандартом проводились с включением контрольного образца
в испытываемую серию. Образцы вынимаются для проверки после
четырехчасового погружения и затем последовательно каждые 24 ч.
После удаления из раствора пластины промывают проточной во-
дой, сушат замшей и проверяют на образование пузырей, размяг-
чение, потерю глянца и разъедание. Затем сушат 1 ч при комнат-
ной температуре и проверяют на растрескивание и шелушение. За-
тем вновь погружают в раствор на 24 ч и всю процедуру проверки
образца повторяют. Образование пузырей оценивается по методу
ASTM, как указывалось выше в тесте на водостойкость. Твердость
или степень размягчения можно определять по сравнению показа-
телей с помощью карандаша твердости (см. 16.3.2) для погружен-
ной и непогруженной в раствор частей покрытия. Потеря адгезии
обычно видна по отслоению или «сморщиванию» покрытия. Пленка
при этом может легко отслаиваться ногтем. Более точно адгезию
можно оценить по методу решетчатого надреза (см. 16.3.5). Поте-
рю глянца, обесцвечивание или появление пятен оценивают каче-
ственно: «нет», «слабое», «сильное».
Испытание на стойкость к моющим средствам проводится так
же, как и на щелочестойкость. В нашей лаборатории использова-
ли 5% раствор средства «Deepio» при 74 + 0,5 °C. Время ис-
пытаний меньше, чем на щелочестойкость; пластины проверяют
через 1, 2, 4, 6, 24 и 48 ч после начала испытаний. Критерии
ухудшения свойств — те же, что и для тестов на щелочестойкость.
Эти тесты являются произвольными, но несмотря на это могут
дать важную практическую информацию.
В большинстве случаев используемые тесты разрабатываются
совместно изготовителями и потребителями красок.
16.2.1.5. Стойкость к растворителям
Стойкость к растворителям может испытываться по разным
причинам. Испытания на стойкость к действию бензина или ди-
зельного топлива проводятся для композиций, которые могут кон-
тактировать или подвергаться периодическому действию брызг
этих жидкостей, например, в случае покрытий для автомобиле^,
топливных емкостей и т. д. Для оценки степени отверждения
или сшивки композиций часто применяются полярные раствори-
тели, например кетоны.
В случае испытаний па бепзостойкость предпочтительнее
использовать синтетический бензин известного состава из-за
широкого диапазона составов промышленного бензина и возмож-
ного присутствия антидетонационных присадок. При испытаниях
на стойкость к дизельному топливу рекомендуется топливо с ани-
линовой точкой от 60 до 70 °C.
При испытаниях на стойкость к действию растворителей ре-
комендуется метилэтилкетон. Подготовленные (как описано выше)
пластины погружаются в соответствующую жидкость на глубину
10 см в положении, близком к вертикальному. Все испытания
такого рода* проводят при комнатной температуре. Пластины
осматривают через 1, 2, 4, 6, 24 и 48 ч. В случае испытаний
с летучими растворителями определяют образование пузырей,
твердость, адгезию и обесцвечивание сразу же и через 5 минут,
необходимых для улетучивания растворителя с поверхности по-
крытия. Смысл этого испытания состоит в том, чтобы определить
исходную степень размягчения и оценить восстановление твердо-
сти после испарения растворителя, если это имеет место. В случае
дизельного топлива значительного испарения за это время не
происходит, и допускается выдержка пластины в течение 1 мин.
перед испытаниями.
Этот тип испытаний, разумеется, допускает использование
любого типа растворителя в зависимости от того, какую инфор-
мацию желательно получить. Его можно использовать для опреде-
ления степени отверждения; в этом случае обычно применяют
растворитель или смесь растворителей, присутствующих в исход-
ной краске.
Кроме испытаний при погружении в растворитель, стойкость
к его действию можно оценивать путем протирки покрытия раство-
рителем.
В этом случае окрашенная пластина протирается кусочком
мягкой ткани, смоченной растворителем. Стойкость к раствори-
телю при этом можно оценивать по числу «протирающих» движе-
ний, необходимому для разрушения пленки или «протирания»
ее до следующего слоя. По-видимому, можно выполнить опреде-
ленное число таких движений и оценить внешний вид пленки
по пятибалльной шкале, где 0 показывает, что нет действия
^растворителя, а 5 — полное разрушение (растворение) пленки;
между этими экстремумами будут наблюдаться различные
Степени потери глянца и разрушения пленки.
\ 16.2.1.6. Стойкость к образованию пятен
Важно, чтобы кухонное оборудование (холодильники, моечные
млшины и т. д.) окрашивались материалами, стойкими к пятно-
орразованию для широкого диапазона используемых домашних
продуктов. Следовательно, необходимо испытывать новые покры-
тия и сравнивать с существующими стандартами. Пластинки
в этом случае используют больших размеров (300X100 мм)
из-за значительного числа используемых загрязнителей.
Типичные применяемые загрязнители — губная помада, крас-
ный восковой мелок, черный и коричневый обувной крем, горчич-
ная паста, томатный кетчуп и ломтик лимона. Они наносятся
на поверхность и каждый растирается часовым стеклом. Через
24 ч поверхность очищается мягкой тканью. Степень пятнообразо-
вания оценивается визуально. Важно одновременно проводить
сравнительные испытания со стандартным покрытием.
16.3. ИСПЫТАНИЯ МЕХАНИЧЕСКИХ СВОЙСТВ ПОКРЫТИЙ
Значение механических ствойств пленок и методы их измерения
обсуждены детально в гл. 13. Общие методы определения меха-
нических свойств во многих случаях оказываются пригодными
применительно к краскам, на их основе разработаны стандартные
тесты. Здесь будет обсуждено примененение некоторых часто
используемых тестов.
Мы рассмотрим испытания на стойкость к удару, изгибу
и истиранию; методы измерения адгезии и отслаивания. Хотя
эти тесты типа «выдерживает — не выдерживает испытания»
согласно принятому стандарту применяются к новым (не под-
вергавшимся климатическому воздействию) пленкам, их также
можно применять к пленкам, подвергшимся такому воздействию.
В ,этом случае они могут быть использованы как дополнение
к ускоренным или естественным климатическим испытаниям.
16.3.1. Стойкость к удару
Имеется три типа таких испытаний: маятниковый тест, испы-
тание ударом падающего тела и тест Эриксена.
16.3.1.1. Маятниковый тест
Прибор состоит из двух вращающихся рычагов, каждый из
которых опирается на образец (стальные трубки, окрашенные
испытываемой краской). Один из испытываемых образцов вра7
щается на горизонтальной оси, другой расположен горизонтально
неподвижно. Механизм устроен таким образом, что рычаги вра-
щаются один относительно другого, и вращающийся образец
скользит по неподвижному. Повреждение покрытия оценивается
визуально. Это одни из немногих тестов, не использующих плоские
стальные пластины. В качестве подложек используются стальн ле
трубки с внутренним диаметром 2,8 см и наружным 3,7 см и длиной
12,5 см. Методика подготовки образцов—обычная. Хотя тест
прост для проведения, требуется некоторый опыт для интерпре-
тации результатов. Степень повреждения покрытия оценивается
через потерю адгезии.
16.3.1.2. Тест с помощью падающего груза
Этот тест разработан для того, чтобы определить, насколько
покрытие на металлической подложке может выдерживать быст-
рую деформацию^ Пластинка с нанесенным покрытием подверга-
ется воздействию падающего груза с определенной высоты. Удар
может наноситься по окрашенной поверхности или с обратной
стороны пластины, согласно требованиям на материал (тест
BS 3900. Part ЕЗ (Impact). Прибор состоит из стального бруска,
скользящего вертикально вдоль двух направляющих, и являюще-
гося держателем рабочего тела. Брусок и рабочее тело падают
под собственным весом на испытываемую пластинку, зажатую
двумя держателями. Пластины подготавливают как описано выше.
В этом тесте важна толщина пленки; обычно, если не делается
никаких оговорок, она составляет 25 мкм. Толщина подложки
должна быть постоянной. В этом случае размеры ее несколько
меньше, чем в испытаниях на химстойкость и обычно составляют
100X50X1,257 мм Разрушение покрытия оценивается по появле-
нию растрескивания или потере адгезии на деформированном
участке.
16.3.1.3. Тест Эриксена
Это испытание позволяет оценить, насколько покрытие способ-
но выдержать медленную деформацию подложки, которая посте-
пенно деформируется с неокрашенной стороны путем вдавливания
шарообразного индентора. Глубина вдавливания, при которой на-
блюдается растрескивание, является мерой деформационных характе-
ристик. Имеется два типа аппаратов для таких испытаний: The
Erichsen Lacquer Testing Mashines, Модель 229/E (ручная)
и Модель 225/Е (механическая).
Для обеих моделей требуется пластинка шириной 70 мм.
Пластинка закрепляется окрашенной стороной вверх. Полусфери-
ческий индентор диаметром 20 мм через специальное отверстие
вдавливается в обратную сторону пластинки. Микроскоп, распо-
Сложенный по центру, позволяет оператору наблюдать за деформа-
цией покрытия и остановить индентор с появлением первых
разрушений. Цифровой микрометр непрерывно показывает глу-
бину вдавливания. В модели 229/Е привод индентора ручной,
а в модели 225/Е — механический, причем скорость вдавливания
составляет 0,2 мм/с.
Адгезионное разрушение наблюдается в виде кольцевых тре-
щин в верхней части деформированного покрытия и часто сочетаю-
щихся с ними радиальных трещин. В этом случае делается вывод,
что покрытие не выдерживает испытания. Этот тип разрушений
часто возникает внезапно. Когезионное разрушение проявляется
в виде четких отдельных трещин. В этом случае адгезия может
быть хорошей, но когезионная прочность недостаточна для увели-
чения деформации. Этот
адгезионный, и для его
наблюдения.
тип разрушения не так очевиден, как
обнаружения необходимы тщательные
16.3.2. Испытания твердости
Может показаться, что испытания твердости не связаны с
определением долговечности покрытий, так как общие механиче-
ские свойства пленок рассмотрены в гл. 13. Однако здесь мы рас-
смотрим только некоторые из стандартных тестов. Во-первых,
их можно применять для оценки степени отверждения покрытия,
что оказывает сильное влияние на долговечность. В некоторых
случаях изменение твердости в результате внешних воздействий
дает возможность установить механизм процессов, протекающих
при климатических испытаниях.
Разработано множество тестов для получения информации
о твердости покрытий. Очень трудно определить абсолютное значе-
ние твердости; можно лишь утверждать, что это сложная функция
механических свойств материала, связанная с сопротивлением
его деформации. Это определение, однако, слишком простое,
поскольку материалы бывают хрупкими, пластичными, эластич-
ными и т. д., и понятно, что два материала, подвергающиеся
под нагрузкой деформации в равной степени, могут отличаться
в своем поведении после снятия нагрузки. Например, один мате-
риал может деформироваться непрерывно, а другой нет, т. е.
первый подвергается пластической, а второй—эластической
деформации. Технолог, как правило, имеет более прагматический
взгляд на твердость, и поэтому он предпочитает простые, стандарт-
ные методы измерения твердости. Так, метод, связанный с измере-
нием свойств тонкой пленки на различных подложках, важен для
установления влияния подложки на твердость пленки. Принято
поэтому измерение твердости производить на твердых подложках
типа стекла или стали путем действия давления на испытываемое
покрытие.
Может возникнуть необходимость измерений твердости прямо
на производстве, так что в некоторых случаях требуются простые
тесты и портативное оборудование. Если это возможно, то всегда
предпочтительнее лабораторное проведение испытаний при станг
дартных условиях и тщательной подготовке образцов. (
Из многочисленных методов испытаний твердости с целыб
изучения долговечности покрытий наиболее распространенными
методами являются измерения с помощью карандаша твердости,
маятникового прибора и индентора. Последние два метода опи-
саны в гл. 13, причем индентор 1CI может дать ценную информа-
цию почти по любому типу пленок.
Метод измерения твердости с помощью карандаша исполь-
зуется чаще для свежих пленок не подвергшихся старению. Это
один из простейших тестов на твердость. При испытаниях исполь-
зуют набор карандашей с различной твердостью как базис для
сравнения. Так же, как геологи используют шкалу твердости
Мора с четко определенным набором стандартных материалов,
возможно использовать аналогичный путь определения твердо-
сти пленок с помощью условных значений твердости по карандашу,
используя ряд специально разработанных реперных точек для
карандашей различной твердости.
Карандаши используются как показано на рис. 16.1; высту-
пающая часть грифеля имеет длину примерно 6 мм. Последний
имеет цилиндрическую форму, а его кончик сточен тонкой абразив-
ной бумагой. Угол нажима на поверхность — 45°, сила нажима
должна быть максимальной, но чуть меньше требующейся для
излома грифеля. Твердость покрытия соответствует твердости
карандаша, который еще не повреждает покрытие. Метод хорошо
воспроизводится. Карандаши используются с твердостями в диа-
пазоне от 6В до 9Н Если примерные характеристики краски
известны, то работать со всем диапазоном карандашей нет не-
обходимости. На твердость могут повлиять атмосферные условия
(температура и влажность), поэтому все пленки должны подготав-
ливаться, подвергаться старению и испытываться в одинаковых
условиях.
Оригинальный метод Вольфа-Виборна, основанный на том же
принципе, отличается только тем, что твердость определяется
45°
Рис. 16.1. Карандаш для опреде-
ления твердости
до самому мягкому карандашу, который уже оставляет след
на поверхности.
Чуть лучшей воспроизводимости можно добиться с использова-
нием специального держателя карандаша («Erichsen Scratch
Pencil Hardness Tester Model 291»).
16.3.3. Стойкость к царапанию
Цель этого теста — оценить способность покрытия выдержи-
вать царапание. Стальная игла определенного веса со сферическим
кончиком опускается на поверхность пленки и протаскивается
по поверхности с постоянной скоростью.
Метод применяется в лакокрасочной промышленности много
лет и стандартизован (BS 3900: Part Е32 (Scratch)). Оценка
результатов испытания дается по принципу: «выдерживает» или
«не выдерживает». Испытания проводятся с иглой определенного
веса или с увеличением веса до тех пор, пока не наступит
повреждение покрытия.
Прибор состоит из горизонтальной движущейся плиты, на
которой закреплена окрашенная пластинка покрытием вверх. Эта
плита движется под иглой со скоростью 3—4 см/с. Сама «игла»
состоит из стержня, к которому припаян стальной шарик диа-
метром 1 мм. Она зафиксирована в держателе на конце противо-
веса. Держатель нагружается гирями, и пластина приводится
в движение. Игла аккуратно опускается на поверхность. Для
испытания требуется длина царапания не менее 6 см.
Хотя для этого типа тестов возможны как ручные, так и элек-
тромеханические аппараты, последние предпочтительнее, так как
дают более воспроизводимые результаты.
Пластины готовят как описано ранее. Они могут быть сталь-
ными, оловянными или алюминиевыми, размерами 50X125 мм.
Результаты оцениваются путем визуального осмотра.
16.3.4. Испытания на изгиб
Этот тип испытаний, применяющихся к покрытиям, которые
могут подвергаться изгибу в результате соответствующего воз-
действия на подложку, очень широко распространен в промышлен-
ности.
Наиболее общим способом испытаний является изгибание
подложки, на которую нанесено покрытие, вокруг цилиндрической
оправки известного диаметра. Покрытие затем проверяется на
растрескивание или потерю адгезии. Хотя обычно этот тест назы-
вают испытанием на гибкость, он является тестом комбинирован-
ным и включает испытание адгезии, гибкости и растяжимости.
Одна из форм теста (британский стандарт'BS 3900 * Part El: 1966
(Bend)) использует специальное шарнирное устройство, к кото-
рому присоединена цилиндрическая оправка, так что она может
свободно вращаться вокруг специальной цапфы. Испытываемая
пластинка с покрытием вставляется в зазор между оправкой
и имеющимся клапаном окрашенной стороной против направления
изгибания. Шарнир приводит оправку в движение и пластинка
огибается вокруг нее. Время изгиба — 1 — 1,5 с. Покрытие осматри-
вается сразу же после изгибания с целью выявления растрескива-
ния и потери адгезии. Трещины ближе 6 мм к краю пластинки
во внимание не принимаются. Тест проводится после двухчасовой
выдержки покрытия при комнатной температуре. Применяется
набор оправок диаметром 25, 19, 12,5, 10, 6,3, 5 и 4,2 мм.
Измерения производят с постепенным ужесточением условий ис-
пытания, т. е. последовательно уменьшая диаметр оправки. Вели-
чина максимального диаметра, при которой наступит разрушение
покрытия, является показателем прочности на изгиб. Размер
пластинки строго определен — 100X50 мм. Толщина — не выше
0,3 мм. Материал пластины — олово или алюминий.
Этот исключительно простой тест может быть очень инфор-
мативен, так как он соответствует условиям эксплуатации многих
покрытий. Он может дать информацию о применимости материа-
лов, которые будут подвергаться воздействиям соответствую-
щих внешних условий после их нанесения на окрашиваемое из-
делие.
Видоизменением этого метода является использование оправок,
закрепленных на стальной опоре. Метод применяется для более
толстых подложек, причем чем последняя толще, тем меньший
диапазон диаметров оправок можно успешно использовать в этом
тесте. Так, стальную пластину толщиной 0,45 мм можно успешно
изгибать вокруг всех оправок, тогда как при толщине подложки
0,975 можно использовать только оправки диаметром более 8 мм.
16.3.5. Адгезия (метод решетчатого надреза)
Адгезия является очень трудноизмеряемым, но в то же время
важнейшим свойством покрытия. Используя метод решетчатого
надреза, можно получить некоторые полезные эмпирические дан-
ные. В этом тесте матрица с рядом близко расположенных
лезвий вдавливается в покрытие в двух взаимно перпендикуляр-
ных направлениях. Второй раз вдавливание производится поверх
первого вдавливания, в результате получается решетчатый надрез.
Поверх него приклеивается кусочек липкой ленты, который затем
резко снимается. Адгезия пленки оценивается по количеству
удаленного при этом покрытия.
В тесте фирмы ICI матрица, изготовленная из закаленной
углеродистой стали, содержит 9 параллельных лезвий шириной
1,5 и длиной 25 мм. Острие каждого лезвия имеет радиус 0,075—
0,1 мм. Матрица вдавливается в покрытие гидравлическим прессом
при усилии 140 атм. После получения отпечатка приклеивается
лента (с использованием мягкого ластика). Лента выдерживается
Ю с и затем резко отрывается под углом 120°. Адгезия считается
хорошей, если отслоение покрытия невелико или отсутствует;
умеренной — если имеет место некоторое отслоение, но маленькие
частицы покрытия остаются на подложке в центре всех или боль-
шинства квадратов, получившихся при надрезе; плохой —
если наблюдается почти полное отслоение покрытия.
Если нет подходящей матрицы и пресса, можно применить
разновидность этого испытания с использованием скальпеля: про-
резая им пленку до подложки получают аналогичную решетку.
В некоторых случаях, при плохой адгезии, может наблюдаться
некоторое отслоение пленки при надрезании до приклеивания
липкой ленты. В этих случаях следует повторить надрез более
тщательно в другом месте.
16.4. УСКОРЕННЫЕ КЛИМАТИЧЕСКИЕ ИСПЫТАНИЯ
Долговечность современных покрытий такова, что при воз-
действии естественных погодных условий они могут слабо разру-
шаться за время, превышающее 1 год (в некоторых случаях срок
службы гарантируется до 10 лет). Чем больше долговечность
покрытия, тем сложнее его испытать. Средства ускорения этих
процессов искались многие годы, в результате разработан ряд
стандартных методов ускоренных испытаний. Из этого не следует,
что хорошая корреляция между результатами естественных
и ускоренных испытаний достигается всегда, поэтому результа-
тами последних всегда следует пользоваться с крайней осторож-
ностью.
Разрушение органических покрытий при внешних воздействиях
связано с влиянием излучения (особенно УФ-излучения), влаж-
ности и температуры. Процессы деструкции являются результа-
тами химических изменений (окисление) и механических нагрузок.
В ускоренных климатических испытаниях используют методы
интенсификации этих процессов таким образом, чтобы разруше-
ние пленки происходило в такой же степени, как и при естественных
испытаниях, но за более короткое время. Для этих целей разрабо-
тан ряд аппаратов искусственной погоды (везерометров), в кото-
рых при циклическом воздействии излучения и влажности дости-
гаются заметные изменения покрытия при сроке выдержки
до 2000 ч.
Тип везерометра и цикл испытаний может указываться потре-
бителем лакокрасочной продукции, особенно в случае крупных
потребителей, например автомобильных фирм. Часто это уста-
навливается по согласованию между изготовителем и потребите-
лем; потребитель также может разработать и собственные тесты,
которым должен соответствовать продукт.
Ряд таких везерометров находит широкое применение, напри-
мер аппараты Марра, Атласа и др. Каждый из них ускоряет
деструкцию покрытия, но результаты не всегда можно сравнивать.
Прибор Марра соответствует британскому стандарту BS 3900:
Part F3 и включает следующие циклы испытаний:
4 ч — влажные условия — распыление включено, вентиляция выключена;
2 ч — сухие условия — распыление выключено, вентиляция включена;
10 ч — влажные условия — распыление включено, вентиляция выключена;
2 ч — сухие условия — распыление выключено, вентиляция включена;
5 ч — влажные условия — распыление включено, вентиляция выключена;
1 ч — остановка — для обслуживания аппарата и осмотра образцов.
Прибор состоит из круглой емкости с расположенным в центре
источником излучения (1,6 кВт углеродная дуга). Окрашенные
пластинки (размер 100X150 мм, подготовленные как и ранее)
укреплены'по периферии внутренних опорных концентрических
колец, которые медленно вращаются в процессе испытаний. Цикл
-можх'Л-.изменятьсяв-зависшмости-от различных щелей, но обычно
включает чередующиеся периоды влажных и сухих условий ис-
пытания.
Везерометр Атласа XW соответствует ASTM Е42-57. В нем
используются несколько меньшие пластины (75 X 150 мм). Прибор
сконструирован в виде прямоугольной камеры. Источник излуче-
ния — углеродная дуговая УФ-лампа. Здесь пластины облучаются
непрерывно с периодическим трехминутным опрыскиванием водой
через каждые 20 мин.
Прибор Атласа XWR предусматривает более жесткие испыта-
ния. Здесь применяется не содержащая светофильтра углеродная
дуговая лампа с высоким процентом УФ-излучения. За циклом
УФ-облучения (1 ч) следует орошение в темноте (1ч). Продолжи-
тельность теста определяет разработчик материала (обычно не
более 1000 ч).
Везерометр QUV отличается от двух описанных выше тремя
особенностями. В нем в качестве источника излучения используют
флуоресцентные трубки, пластины закреплены в камере стацио-
нарно, и конденсация воды на пластинах происходит не от
распыления, а от разности температур. В аппарате обеспечивается
вращение флуоресцентных трубок, чтобы исключить различия
в результатах испытаний в связи с малыми, но важными разли-
чиями мощности облучения.
Приборы типа QUV завоевали популярность для испытаний
автомобильных эмалей и используются чаще, чем приборы Марра
и Атласа XW, XWR. Воспроизводимость опытов требует обсужде-
ния, однако результаты, которые дает прибор QUV, лучше согла-
суются с данными естественных испытаний. Как бы то ни было,
результаты испытаний в аппарате типа QUV быстро становятся
одним из критериев, которому должны соответствовать автомо-
бильные эмали, особенно в Северной Америке.
16.4.1. Методы оценки: определение критериев
Покрытие, подвергшееся ускоренному или естественному ста-
рению, оценивается по изменению ряда свойств, таких как глянец
и цвет, и по специфическим видам повреждений: а) меление;
б) бронзирование; в) растрескивание; г) образование пузырей;
д) отслаивание; е) коррозия подложки; ж) коррозия при нанесе-
нии надреза; з) эрозионное разрушение; и) образование пятен
от воды; к) загрязняемость и грязеудержание; л) стойкость
к заплесневению; м) меление пленки; н) выцветание. Эти терми-
ны определены в британском стандарте BS 2015 (1965).
В большинстве случаев делается визуальная оценка изменений
и разрушений покрытия с использованием в некоторых случаях
увеличительного стекла или микроскопа. Иногда измерения
глянца или цвета проводятся инструментальными методами, но
во многих случаях это несущественно. Сравнение, как правило,
проводят с покрытиями, не подвергнутыми испытаниям. В боль-
шинстве случаев принимается оценочная шкала от 0 до 10. Так,
при оценке глянца балл 0 показывает полное сохранение глянца,
а балл 10 соответствует полностью матовой поверхности, т. е.
показывает полную потерю глянца. При проведении оценок глянца
обычно учитывается интенсивность отражения от пленки [1].
Изменение цвета может быть многообразно, и часто характер
изменения так же важен, как и степень изменения цвета. Цвет
может становиться темнее или блекнуть, становиться более мутным
или более чистым. Выцветание может быть истинным или кажу-
щимся. Например, меление создает впечатление выцветания, но
в этом случае исходный цвет может быть восстановлен путем
очистки поверхностного слоя и удалением покрытия, подвергше-
гося мелению. Помутнение приводит к увеличению серого оттенка,
т. е. к потере чистоты цвета, что не обязательно связано с выцвета-
нием. Специфические изменения цвета можно зарегистрировать
путем сравнения со стандартом.
Оценку глянца и цвета можно проводить до и после протирания
покрытия влажной тряпкой с последующей сушкой.
Меление определяется как «образование рыхлого порошко-
образного слоя на поверхности пленки, вызванное нарушением
целостности связующего и обусловленное разрушающими факто-
рами при климатических испытаниях». Мелению могут под-
вергаться покрытия любого цвета, но чаще встречается у покрытий
пастельных тонов. В случае покрытий глубоких тонов, особенно
синих и каштановых, меление дает эффект сильного блеска и на-
зывается бронзированием. Меление может быть легко вызвано,
например, при воздействии на покрытие указательным пальцем.
Кончиком пальца слегка надавливают на покрытие, и количество
удаленного «мелящего» слоя сравнивают со стандартами. То же
относится и к бронзированию. Растрескивание — это специфи-
ческий случай разрушения пленки, в котором трещины пронизы-
вают по меньшей мере один слой покрытия и могут распростра-
няться сквозь всю систему покрытия. Существует три типа рас-
трескивания: а) микрорастрескивание, когда трещины видны в ма-
ломощный оптический микроскоп с увеличением в 16 или 32 раза;
б) незначительное растрескивание, когда трещины мелкие, но ясно
видны невооруженным глазом; в) значительное растрескивание,
когда эффект больше и сразу очевиден. Вид образца после
растрескивания может быть различным: а) мелкие линейные,
почти параллельные трещины; б) угол между трещинами неопре-
деленный, а трещины могут объединиться в сплошную сетку;
в) «гусиные лапы», где трещины расходятся лучами, напоминая
по форме птичьи лапы; г) волосяные и структурные трещины
(только для покрытий по древесине).
Все эти-дефекты оцениваются согласно фотостандартам, где
перечислено большинство из таких дефектов. Использование фото-
стандартов для оценки образования пузырей уже упоминалось
в связи с испытаниями химической стойкости.
Дефекты отслаивания, ржавления и коррозии также оценива-
ются относительно фотостандартов. Эрозия подобна мелению,
однако является более серьезным дефектом, так как может затро-
нуть и нижележащие слои покрытия. Она может быть вызвана
действием капель дождя, частиц гравия, принесенных ветром, или
одновременным действием обоих этих факторов. Образование
пятен от воды вызывается воздействием капель, в результате чего
на поверхности покрытия остаются пятна более светлого тона,
чем остальное покрытие. Пятна могут быть сплошными и отдель-
ными. Этот эффект может оцениваться согласно фотостандартам.
Загрязнение и плесневение поверхности покрытия чаще имеют
место в естественных условиях, но не при ускоренных испытаниях.
Грязь и плесень на поверхности покрытия могут присутствовать
одновременно и чтобы различить их часто требуется использовать
микроскоп с 16-кратным увеличением. Пластинки осматривают
после протирания половины их длины мягкой влажной тканью;
при этом не следует удалять грязь со второй половины. Плесень
характеризуется паутиноподобными отростками, исходящими
из темного центра. Остатки их видны даже после протирки, и
обычно можно наблюдать повторный рост плесени на вымытом
участке покрытия при дальнейших климатических испытаниях.
«Внутреннее меление пленки» — один из редких дефектов. Он
выглядит подобно обычному мелению, но не удаляется при про-
мывке. Эфлоресценция — это появление на поверхности покрытия
по цементу или кирпичу порошкообразных отложений, вызванных
миграцией водорастворимых солей через пленку и последующим
испарением воды. В некоторых случаях осадок может сформиро-
ваться на верхней части любого покрытия, но обычно пленка
отслаивается и разрушается из-за эфлоресценции под покрытием.
16.5. ЕСТЕСТВЕННЫЕ КЛИМАТИЧЕСКИЕ ИСПЫТАНИЯ
При воздействии на систему покрытия природной среды, ве-
роятно, можно получить наиболее надежные данные по свойствам
покрытия. Это верно в том случае, если будут приняты во внимание
всевозможные изменяющиеся погодные факторы, которые могут
оказать значительное влияние на поведение покрытия при испыта-
ниях. Поскольку естественные испытания приводят к более
медленной деструкции, чем ускоренные, особенно важно разрабо-
тать такие экспериментальные методики, которые бы исключили
ошибки при проведении испытаний в течение нескольких лет.
Требуется четкое ведение документации, так как испытания
могут занять ряд лет, и за это время могут произойти изменения
в составе обслуживающего персонала, т. е. персонал, начинавший
испытания, и персонал, делающий выводы по результатам послед-
них, может отличаться. Важным является выбор географической
точки для испытаний. Если краска предназначается для исполь-
зования в промышленном районе, например на металлургическом
заводе, возможно лучше испытывать пластины именно в таком
районе, а не в сельском. С другой стороны, может быть полезным
проводить испытания в таких местах, о которых известно, что в них
происходит более быстрое разрушение покрытия, чем в местах его
предполагаемого использования. Так, многие технологи считают,
что районы Техаса и Флориды предпочтительнее в этом смысле,
и что в этих районах старение покрытия происходит быстрее,
чем в Англии. Это связано с более высокими уровнями УФ-излуче-
ния в этих местах, а также с большими перепадами температуры
и влажности.
16.5.1. Выбор образцов для испытаний
Требования к пластинам для испытаний определяются не
только их природой, но также и физической формой и методом
изготовления. Наиболее часто используемые материалы для опыт-
ных пластин — это древесина, мягкая сталь, алюминий и неко-
торые виды каменных материалов, например асбоцементные
плиты. Выбор зависит от назначения покрытия. Так, строительны;
краски могут испытываться на деревянных или каменных подлож-
ках, тогда как автомобильные эмали — только на стальных. В не-
которых случаях возможны исключения, например, для компози-
ций, специально разработанных для защиты пластиковых упако-
вочных материалов, важно использовать соответствующую под-
ложку. Древесина, вероятно, наиболее изменчивый материал среди
всех подложек. Она анизотропна, подвержена изменениям в связи
с изменениями температуры и влажности, особенно чувствительна
к воде.
Большинство материалов испытывается на плоских образцах
определенного размера. Время испытаний обычно от 3 -6 мес.
до 5 лет. Некоторые композиции с повышенной защитной спо-
собностью могут испытываться дольше. Методы оценки анало-
гичны методам, описанным для ускоренных испытаний (см. 16.4).
Многие годы для естественных испытаний декоративных по-
крытий применяли плоские пластины (размером 150X300 мм)
из сосны высшего качества из Британской Колумбии, так как эти
пластины дают хорошую воспроизводимость результатов и менее
требовательны к условиям испытаний по сравнению с другими
подложками. Однако в последнее время сделан ряд попыток
по разработке других методов испытаний, более приближенных
к реальным условиям эксплуатации покрытий. Например, разрабо-
тан метод, основанный на испытании глянцевых покрытий на
оконных рамах в домах. Дом ориентирован таким образом, что
достигается' максимальное облучение прямым солнечным светом.
Температура и влажность внутри дома изменяются циклически,
чем достигается конденсация воды на ловерхнюгти стекол и ее
стекание на рамы. Метод имитирует наихудшие условия при ис-
пользовании декоративных покрытий в доме, например в кухнях
и ванных комнатах. Этот метод получения данных довольно
дорог, поэтому многие работы посвящены созданию аналогичных
методов испытаний, но другими путями. Так, Paint Research
Association разработала камеры для естественных испытаний,
аналогичные описанным выше, с той разницей, что стекло заме-
нено на прямоугольные куски фанеры, но так же используются
деревянные рамы.
Оба этих метода предназначены для изучения проблемы со-
хранения целостности пленки на стыках различных частей древе-
сины, в которых волокна расходятся в разные стороны. Различия
в величинах адсорбции или десорбции воды в зависимости от
направления волокон значительны. Поперек волокон древесины
водопоглощение может вызвать увеличение размеров подложки
до 8%, а водопоглощение вдоль волокон —только до 1,0%. Эти
пространственные изменения могут вызвать сближение или отда-
ление мест стыка между упомянутыми различными частями под-
ложки. Таким образом, покрытие подвергается значительным
механическим нагрузкам в местах стыка, и это — одна из главных
причин разрушения пленки в таких участках.
Более простой метод для исследования таких эффектов пред-
ложен фирмой 1CI. Ввиду его простоты и дешевизны мы рас-
смотрим его несколько подробнее.
16.5.2. Метод 1CI для испытания столярных секций
В основе метода используется подход, предусматривающий
создание таких же жестких условий испытаний, с которыми можно
столкнуться в реальных условиях. Учитываются все факторы,
которые с наибольшей вероятностью вносят вклад в быстрое
разрушение покрытия. Наиболее важными факторами являются
проникновение воды через торцы и сочленения деревянных частей
рамы, а также продолжительность контакта с водой, особенно
когда она собирается в местах сочленения или между рамой
и подоконником. Поэтому испытываемый образец соединяется
с емкостью для воды, которая примыкает к столярной секции.
Торец последней, примыкающий к воде, неокрашен. Испытывае-
мые образцы выставляются под углом 45° к югу, чтобы они
поглощали максимум УФ-энергии. Тест эффективен как по стоимо-
сти, так и по достоверности результатов. Следует подчеркнуть,
что природа деревянной подложки сильно влияет на качество
покрытия, причем ее влияние во многих случаях сказывается
сильнее, чем свойства самой краски. Это должно быть учтено
при постановке эксперимента. Поскольку климатические испы-
тания приводят к существенному разбросу результатов, необхо-
димо проводить ряд повторных испытаний, чтобы получить верные
сравнительные данные разных систем. Количество повторных
испытаний зависит от разброса результатов и стандартного
отклонения внутри серий, и для независимых систем желательно
проводить не менее четырех повторных испытаний. Под независи-
мыми системами понимаются две системы, не имеющие сущест-
венного сходства друг с другом, т. е. не имеющие общих свойств.
Если такое сходство есть, то можно использовать статистические
методы для анализа результатов, при этом количество повторных
испытаний может быть уменьшено.
В малых сериях возможно вырезать испытываемые образцы
из одного окрашенного деревянного участка изделия. Это сводит
к минимуму разброс результатов, если позаботиться, чтобы образ-
цы были взяты в случайном порядке. Следует, однако, помнить,
что результаты испытаний относительны, и делая окончательные
выводы, следует соблюдать осторожность. Общая достоверность
результатов повышается при повторных испытаниях на образцах,
вырезанных из другой части столярной секции. Для больших
серий число столярных секций должно быть две или более. Испы-
тываемые образцы, вырезанные из этих секций, объединяются
и используются в случайном порядке.
16.5.2.1. Подготовка образцов
Испытываемые образцы (длиной 187,5 мм) вырезаются из
столярных секций и нумеруются. Вырезанные образцы не должны
иметь серьезных дефектов (плохо обработанные торцы, сучки
и т. д.). Верхний торец покрывается одним слоем алюминий-
содержащей грунтовки, а затем — испытываемой системой. По-
следняя наносится на всю поверхность образца, за исключением
нижней части, примыкающей к емкости с водой. Время межслой-
ной сушки должно быть не меньше рекомендуемого для данной
системы.
16.5.2.2. Процедура испытаний и оценки
При оценке рассматриваются две части испытываемого образ-
ца: ниже 50 мм, примыкающая к воде, на предмет определения
растрескивания и отслаивания (так как именно в этой области
можно ожидать наиболее быстрого появления таких дефектов)
и выше 137,5 мм на предмет появления всех остальных дефектов,
описанных в 16.4. Критерии оценки обсуждены в 16.4.1.
16.5.3. Регистрация данных испытаний
Запись данных климатических испытаний производится столь
продолжительные периоды времени и количество данных может
быть-^тюя-ь-юбшир-ньш и р-азнеебр-азным,-что- записи требуется
производить систематически Систематичность необходима для
стандартизации методов испытаний. Если данные записаны в опре-
деленном порядке, их обработка может быть легко компьютеризо-
вана. Важно, однако, чтобы принятая система записи была гибкой
и принимала во внимание такие изменения, которые не были зара-
нее определены, но могут приобрести значительную важность
через некоторое время. Система должна также включать деталь-
ную запись об испытываемой композиции. Типичная запись данных
должна, помимо результатов испытаний, содержать, как минимум,
следующию информацию: наименование испытываемой серии; из-
готовитель краски; цель испытаний; данные об условиях при
начале испытаний; место испытаний; тип краски; подложка; си-
стема нумерации слоев краски; связующее; пигментирование;
различные варианты состава (если это важно); условия нанесения
(температура, влажность, толщина пленки, время межслойной
сушки).
Также полезно записывать погодные условия, чтобы можно
было учесть любые аномальные условия, которые могут повлиять
на испытываемые образны.
16.5.4. Сравнение долговечности
при естественных и ускоренных испытаниях
Целью испытаний долговечности является установление со-
ответствия свойств продукта определенным требованиям. В боль-
шинстве случаев покрытия достаточно долговечны в течение
многих месяцев, даже лет, при выдержке в естественных условиях,
.длительно сохраняя основные свойства до появления первых
признаков разрушения. Этот период времени может существенно
сдерживать (в отсутствие других данных) сбыт продукта до тех
пор, пока не будет достигнута уверенность в свойствах краски.
Если этот период можно сократить, то соответствующие решения
разработчика материала ускорятся и многие проблемы будут
быстрее решены. Преимущества ускоренных испытаний заклю-
чаются в уменьшении стоимости разработки и в выигрыше време-
ни, необходимого для этой разработки.
Ускорения испытаний долговечности можно достичь различны^
ми путями. Если главными причинами деструкции или ухудшения
свойств покрытия являются облучение, тепло и влага, можно
передать продукт для испытания в ту часть мира, где наблюда-
ется более высокая температура и более интенсивное солнечное
облучение, чем в Англии. Если исследуется рост плесени, то
имеются регионы, более благоприятные для проведения испытаний,
т. е. такие регионы, где наблюдается высокая температура и влаж-
ность, как, например, в Малайзии. Часто, однако, исследователь
стремится ускорить разрушение покрытия в большей степени, чем
этого можно достичь в естественных тропических условиях, и тогда
он прибегает к оборудованию, описанному в 16.4. При этом есть
риск, что поведение покрытия в более жестких условиях испыта-
ний может сильно отличаться от поведения в реальных условиях.
В этом случае предполагается с некоторым допущением, что если
исследуемое покрытие показывает себя хуже стандартного с из-
вестными свойствами в принятых условиях испытаний, то и на
практике оно будет хуже. Если же экспериментальное покрытие
обнаруживает лучшие свойства по сравнению со стандартным
при ускоренном испытании, то нет гарантии, что то же будет
наблюдаться и на практике в реальных условиях. В целом, условия
испытаний должны быть составлены таким образом, чтобы как
можно ближе воспроизвести тип воздействия на покрытие, который
может иметь место на практике. Сравнительное распределение
излучения для солнечного света и различных искусственных источ-
ников приведено в табл. 16.1. [2]. Ксеноновая лампа мощностью
6500 Вт с внутренним боросиликатным покрытием и внешним
фильтром дает излучение, наиболее близкое к солнечному. Следует
ожидать, что интенсивное УФ-излучение будет гораздо агрессив-
нее, что и случается реально. В результате данные везеро-
метрии с УФ-источником гораздо труднее подлежат интерпретации
по сравнению с данными, полученными при испытаниях в везеро-
метрах с менее агрессивными источниками излучения. Несмотря на
это, некоторые основные потребители красок, например автомо-
билестроители, могут требовать проведения испытаний в этих
особо агрессивных условиях, хотя полученные данные могут не
коррелировать с условиями эксплуатации покрытий.
Скотт [2] утверждает, что лабораторные испытания могут
разрабатываться для ускорения процессов деструкции или для
имитации широкого диапазона условий окружающей среды.
Таблица 16.1 Сравнительное распределение излучения
Длина волны, НМ Солнечный свет, % 6500 Вт ксеноновая лампа, % Углеродная дуга (открытое пламя), % Флуоресцент- ный источ- ник, %
300 0,01 0,01 0,5 14,0
300—340 1,6 1,5 2,5 70,0
340—400 4,5 5,0 11,0 13,0
Всего ниже 400 6,1 6,5 14,0 97,0
400—750 48,0 51,5 34,0 3,0
750 46,0 42,0 52,0 0,0
Всего от 400 до 750 94,0 93,5 86,0 3,0
Первое является традиционным путем использования везеро-
метров при стандартных режимах испытаний. Второе позволяет
воспроизвести в лабораторных условиях естественные природные
условия в любой точке м-ира. Современные везерометры могут
быть запрограммированы на создание естественных циклов излу-
чения, температуры, сушки-смачивания, содержания атмосферных
загрязнителей, например оксидов азота, диоксида серы и озона.
Этот тип оборудования дает возможность более строгого подхода
к изучению влияния окружающей среды на деструкцию покрытия.
ЛИТЕРАТУРА
К главе 2
1. Kraft W. М., American Paint J. 41 (28) 96 (1957)
2. Heydon Newport Chemical Corporation, United States Patent 2 973 331 (1961).
3. Rooney J. F., Official Digest 36 (475 Part 2) 32 (1964).
4. Shell Chemicals, Cardura ЕЮ, Technical Manual CA 1.1.
5. Herzberg S„ 1. Paint Technol. 41 222 (1969).
6. Shell Chemicals, The manufacture of ’Cardura’ resins. Technical Manual
CA 2.1; Shell Chemicals, A review of technical information of Cardura E10,
Technical Note TN-R.83.01.
7. Holmberg K. & Johansson J., Proc. 8th International Conference Organic
Coatings Science and Technology, Athens, 255 (1982).
8. Baker A. S., Paint and Resin 52 (2) 37 (1982); Imperial Chemical Industries,
British Patent 1 370 914 (1972).
9. Bobalek E. G., Moore E. R., Levy S. S., & Lee С. C., J. App. Polym. Sci. 8 625
(1964).
10. Imperial Chemical Industries, British Patent, 1 242 054 (1971).
11. Imperial Chemical Industries, British Patent, 1 594 123 (1978).
12. Pond P. S. & Monk C. J. H., J. Oil Col. Chem. Assoc. 53 876 (1970).
13: Tysall L. A., Calculation techniques in the formulation of alkyd and related
resins, Paint Research Association, 1982.
14. Patton T. C. Alkyd resin technology (formulaing techniques and allied calcu-
lations), Interscience, 1962.
15. Bernardo J. J. & Bruins P., J. Paint Technol. 40 558 (1968).
16. Weiderhorn N. M., American Paint J. 41 (2) 106 (1956).
17. Nelen P. J. C., 17th FATIPEC congress 283 (1984).
18. Shackleford W. E. & Glaser D. UK, J. Paint Technol. 38 293 (1966).
19. Turpin E. T., J. Paint Technol. 47 (602) 40 (1975).
20. AMOCO Chemicals Corporation, How to process better coating resins with
AMOCO IPA and TMA.
21. AMOCO Chemicals Corporation, Processing unsaturated polyesters based on
AMOCO isophthalic acid (IPA).
22. Imperial Chemical Industries, United States Patent, 3 050 533 (1962).
23. Fradet A. & Marechai E., Advances in Polymer Science 43 51 (1982).
24. Price J. G., Proc. 3rd International Conference Organic Coatings Science
and Technology, Athens 464 (1977).
25. Price J. G., Silicone resins as modifiers for waterborne coatings, Dow Corning,
England, 1978.
26. California Research Corp., Unites States Patent, 2 904 533 (1959).
27. Standart Oil, United States Patent, 3 252 941.
28. Imperial Chemical Industries, British Patent, 784, 611 (1957).
30. Brown W. H. & Miranda T. J., Official Digest 36 (475 Part 2), 92 (1964).
31. Klein D. H„ J. Paint Technol. 42, 335 (1970).
32. DuPont de Nemours E. 1., British Patent 763 158 (1956).
33. DuPont de Nemours E. I., British Patent 807 895 (1959).
34. Zimrnt UK S., Ind. Chem. Prod. Res. Dev. 18 91 (1979).
35. Taylor J. R. & Price T. I., J. Oil Col. Chem. Assoc. 50 139 (1967).
36. Chrictenson R. M. & Hart D. P., Official Digest 33 684 (1961).
37. Vogel H. A. & Bittie H. G„ Official Digest 33 699 (1961).
38. PPG Industries, British Patent 1 556 025 (1975).
39. PPG Industries, British Patent 1 559 284 (1976).
40. Murdock J. D. & Segall G. M„ Official Digest 33 709 (1961).
41. Yanagihara T., Progress in Organic Coatings 11 205 (1983).
42. Thompson M. W., In. Polymer Colloids, Buscall R., Corner T., & Stageman J. F.
(eds), Elsevier Applied Science, 1985.
43. Imperial Chemical Industries/Dulux Australia, United States Patent 4 322
328 (1982); Bromley C. W. A., Colloids & Surfaces 17 (1) (1986).
44. Blackley D. C., Emulsion polymerisation, Applied Science, 1975.
45. Llewellyn I. & Pearce M. F., J. Oil Chem. Assoc. 49 1032 (1966).
46. Bondy C., J. Oil. Chem. Asooc. 49 1045 (1966).
47. Various papers in Science and technology of polymer colloids. Volume 1 and 2,
Poehlein С. IV., Ottewill R. H., & Goodwin J. W. (eds) Martinus Nijhoff; 1983.
48. Imperial Chemical Industries, United States Patent 4 403 003 (1983).
49. Backhousr A. J., J. Coatings Technol. 54 (693) 83 (1982).
50. Westrenen IV. J. van & Nieuwenhuis IV. H. M., J. Oil Col. Chem. Assoc. 54 747
(1971).
51. Gordon P. G., Davies M. A. S., & Waters J. A., J. Oil. Col. Chem. Assoc. 67 197
(1984).
52. Hoechst, British Patent 1 541 891 (1976).
53. Rohm and Haas, British Patent 1 072 894 (1967).
54. Walbridge D. J., In: Science and technology of polymer colloids Volume 1,
Poehlein G. IV., Ottewill R. H., & Goodwin J. IV. (eds). Martinus Nijhoff.
1983.
55. Walbridge D. J., In: Dispersion polymerisation in organic media, Barrell К. E. J.
______„ (ed), Wilpy, 1975. —____________
56. F. A., J. Oil. Col. Chem. Assoc. 54 342 (1971).
57. Rempp P. F. & Franta E., Advances in polymer Science 58 1 (1984).
58. Barrett К. E. J. & Thompson M. W., In: Dispersion polymerisation in organic
media, Barrett К. E. J. (ed.), Wiley, 1975.
59. Imperial Cfiemical Industries, British Patent 1 157 630 (1969).
60. Blank IV. J. & Hensley IV. L., J. Paint Technol. 46 (593) 46 (1974).
61. Krishnamurti K., Progress in Organic Coatings 11 167 (1983).
62. Lomax J. & Swift G„ J. Coatings Technol. 50 (643) 49 (1978).
63. Moss N. S. & Demmer C. G., J. Oil Col. Chem. Assoc. 65 249 (1982).
64. Robinson P. V., J. Coatings Technol. 53 ( 674) 23 (1981).
65. Hill L. IV. & Wicks Z. IV., Progress in Organic Coatings 8 161 (1980).
66. Grant P. M., J. Coatings Technol. 53 (677) 33 (1981)
67. Schenck H. IL, Spoor H., & Marx M., Progress in Organic Coatings 7 1 (1979).
68. Cook B. A., Ness N. M., & Palluel A. L. L. In: Industrial electrochemical processes,
Khun A. T. (ed), Elsevier, 1971.
69. Wismer M., Pierce R. E., Bosso J. F., Chricterson R. M., Jerabek R. D.,
Zwack R. R., J. Coatings Tech. 54 (688) 35 (1982). ,1
70.. Kordomenos P. I. & Nordstrom J. D., J. Coatings Technol. 54 (686) 33 (1982).
71. Imperial Chemical Industries, European Patent 15655; Imperial Chemical
Industries, European Patent 109760; Doroszkowski A., Colloids & Surfaces
17 (1),. 13 (1986).
72. PPG Industries, United States Patent 3 947 339 (1976).
73. BASF., United States Patent 3 455 806 (1969).
74. SCM., United States Patent 3 975 251 (1976).
75. Imperial Chemical Industries, British Patent 2 050 381 (1981.). -
76;. PPG Industries, United States Patent 4 017 438 (1973). , ,
77. PPG Industries, United States Patent 3 799 854 (1974).
78. PPG Industries. United States Patent 4 101 486 (1978).
79. Gibson D. & Leary B., J. Coatings Technol. 49 53 (1977); Noren G. K., Polymer
News 10 39 (1984).
80. Athey R. D. Progress in Organic Coatings 7 289 (1979).
81. Hill L. IV. & Wicks Z. IV., Progress in Organic Coatings 10 55 (1982).
82. Blank IV. /., J. Coatings Technol. 54 ( 687) 26 (1982).
83. Potter T. A., Schmelzer H. G., & Baker R. D., Progress in Organic Coatings
12 321 (1984).
84. Roffey C. G., Protopolimerisation of surface coatings, Wiley, 1962; O’Hara K-,
Polymers Paint Colour J. 175 254 (1985).
85. Dowbenko R., Friedlander C., Gruber G., Pruenal P., & VFismer M., Progress
in Organic Coatings 11 71 (1983).
86. Green P. X'.. Polymers Paint Colour J. 175 246 (1985); Hageman H. J., Progress
in Organic Coatings 13 123 (1985).
87. Harris S. T., The technology of powder coatings. Portcullis Press, 1976.
К главе 3
1. Colour Index — pigments and solvent dyes, 3rd edition, Society of Dyers and
Colourists (1982).
2. Sappok R. JOCCA 61 299 308 (1978).
3. Honigtnann B., Lenne H. U., & Schrddel R., Zeit. fur Kristallographie 122
Nr 314 (1965).
4. Kaluza U., Physical/chemical fundamentals of pigment processing for paints
and printing inks, Lack u. Chemie, Edition (1981).
5. Merkle K., & Herbst IV., Farbe u. Lack 74 Nr 12 (1968).
6. Hafner O., JOCCA 57 268 273 (1974).
7. Schafer H., Farbe u. Lack 77 1081—1090 (1971).
К главе 4
1. Acima Chemical Industries Ltd, PO Box CH9470, Buchs SG (Switzerland).
2. Albright & Wilson Ltd, PO Box 3, Oldbury, Warlcy, W. Midlands B68 ONN.
3. Allied Colloids Ltd, PO Box 38, Low Moor, Bradford.
4. Bayer (UK) Ltd, Bayer House, Strawberry Hill, Newbury, Berks.
5. Bevaloid Ltd, Flemingate, Beverley, Yorkshire.
6. Borchers (Gerb) AG, Elsabethstrasse 14, Postfach 1812, D-4000, Dusseldorf 1,
W. Germany.
7. Bush Boake Allen Ltd, Blackhorse Lane, London E17 5QP.
8. Byk Chemie GmbH, Abelstrasse 14, D-4230 Wesel, W. Germany.
9. Capricorn Chemicals Ltd, 1 Sugar House Lane, Stratford, London E15 2QN.
10. Central Soya, 1825 North Lawrence Avenue, Chicago 39, Ill, USA.
11. Chemitrade Ltd, Berkeley Square House, Berkeley Square, London W1X 5LA.
12. Chemacord Ltd, 43 Upper Wickham Lane, Welling Kent DA16 3AJ.
13. Ciba-Geigy. Industrial Chemicals Division, Trafford Park, Manchester Ml7
1WT.
14. Cray Valley Products Ltd, Farnborough, Kent BR6 7EA.
15. Croxton & Garry Ltd, Curtis Road, Dorking, Surrey RH4 1XA.
16. Cuprinol Ltd, Adderwell, Frome, Somerset BAH 1NL.
17. Degussa Ltd, Stanley Green Trading Estate, Cheadle Hulme, Cheshire SK8 6RW.
18. Diamond Shamrock Process Chemicals Ltd, 147 Kirkstall Road, Leeds LS3 1JN.
19. Dow Corning Ltd, Reading Bridge House, Reading, Berks, RG1 8PN.
20. Drew Ameroid International, PO Box 6, 4930 AA Geertrudenberg, Netherlands.
21. Durham Chemicals Ltd, Birtley, Chcster-le-Street, Durham DH3 1QX.
22. EFTA Chemicals BV, PO Box 358, 2180 AJ Hillegom, Netherlands.
23. Esso Chemical Ltd, Arundel Towers, Portland Terrance, Southampton SO9 2GW.
24. Fritzsche Dodge & Alcott, Fincdon Road, Lindhurst Estate, Links Road,
Wellingborough, Northants.
25. W. R. Grace Ltd, Northdale House, North Circular Road, London NW10 7UH.
26. Haeffner, H. & Co. Ltd, Nursery Industrial Estate, Station Road, Chepstow,
Gwent NP6 5PB.
27. Henkel Chemicals GmbH, Dusseldorf, W. Germany, (UK) Merit House. The
Hyde, Edgware Road, London NW 9.
28. Hercules Ltd, 20 Red Lion Street, London WC1R 4PB.
29. Hoechst (UK) Ltd, Hoechst House, 50 Salisbury Road, Hounslow, Middx.
30. ICI pic, Mond Division, The Heath, Runcorn, Cheshire.
31. ICI pic. Organics Division, Hexagon House, Blackley, Manchester.
32. ICI pic, Speciality Chemicals, Cleeve Road, Leatherhead, Surrey.
33. Industrial Perfumes Ltd, Commerce House, Commerce Road, Brentford, Middx.
34 K&K Greeff Industrial Chemicals Ltd, Suffolk House, George Street, Croydon
CR9 3QL.
35. KMZ Chemicals, Claire House, Bridge Street, Leatherhead, Surrey K22 8BZ.
36. Fratelli Lamberti, S. p. A., 21041 Albizzate, Italy.
37. Langley Smith & Co Ltd, Langley House, 19—21 Christopher Street. Finsbury
Square, London EC2.
38. Lankro Chemicals Ltd, PO Box 1, Eccles, Manchester M30 OBH.
39. Manchem Ltd, Ashton New Road, Clayton, Manchester МН 4AT.
40. ML Chemicals (UK) Ltd. 2 National Trading Estate, Norman Avenue, Hazlegro-
ve, Stockport, Cheshire.
41. NL Chemicals (UK) Ltd, St Ann’s House, Wilmslow, Chechire SK9 111G.
42. Occidental Chemical Corporation, 10889 Wiltshire Boulevard, Los Angeles
California 90024, USA.
43. Perchem Ltd, West Road, Temple Fields, Harlow, Essex.
44. Pluss Stauffer, CH-L665 Oftrigen, Switzerland.
45. PRB SA, Avenue de Brogneville 12, Bl 150, Brussels, Belgium.
46. Priem Wilhelm GmbH & Co. D-4800 Bielefeld 1, Osningstrasse 12, W. Germany.
47. . Proprietary Perfumes International Ltd, Wilesborough Road, Ashford, Kent
TN24 OLT.
48. Ransburg (UK) Ltd, Ransburg House, Hamm Moor Lane, Weybridge, Surrey
K15 2RH.
^“Rfroffe^outerrc~(UK) Ltd, Tfutrond+orrse, Г6Т/+66 Fleet Street, London EC4.
50. Rohm & Haas (UK) Ltd, Lennig House, 2 Masons Avenue, Croyden, CR9 3NB.
51. Sandoz Products Ltd, PO Box Horsforth No. 4, Calverley Lane, Horsforth,
Leeds LSI8 4RP.
52. Schwegmann, Bernd, KG, Buchenwag 1, D5300 Bonn 3, PO Box 300 860,
W. Germany.
53. Servo BV, PO Box 1, 7490 AA Delden, Netherlands.
54. Shell Chemicals (UK) Ltd, J Northumberland Avenue, Trafalgar Square,
London WC2 5LA'.
55. Streetlev Minerals Ltd, PO Box, Gateford Hill, Worksop, Nottinghamshire
S81 8AF.
56. Sterling Industrial, Capeltown, Sheffield S30 4YP.
57. John E. Sturge Ltd, Lifford Chemical Works, Lifford Lane, Birmingham
B30 3JW.
58. Thor Chemicals Ltd, Ramsgate Road, Margate, Kent CT9 4JY.
59. (The) White Sea & Baltic CoLtd, Concordia Works, Parkinson Approach.
Garforth, Leeds, LS25 2HR.
60. Whitfield Chemicals & Polymers Ltd. 23 Albert Street, Newcastle-under-Lyme,
Staffs ST5 UP.
61. Wilson & Mansfield, 48 Gresham Street, London EC2.
62. Wymouth Lehr, 158 City Road, London EC1V 2PA.
К главе 5
1. Balfour /. G„ OCCA Conference 39—49 (1977).
2. Asbeck W. K.. Off. Digest 33 65—83 (1961).
3. Balfour L G., JOCCA 60 No 9 365—76 (1977).
4. Peacock J., XI Fatipec Congress 193—9 (1972).
5. Elm A. C., Off. Digest 33 (433) 163.
6. Patton T. C., p. 146 In Paint flow and pigment dispersion Interscience Publ.
(1964).
7. Heertjes P. M. & Witvoet W. C., Powder Tech. 3 339 (1970).
8. Washburn E. 0., Phys. Rev. 17 374 (1921).
9. Good R. J., Chem. and Ind. 600 (1971).
10. Doroszkowski A., Lambourne R., & Walton J., Colloid & Polymer Sci. 255
869—901 (1977).
11. Hare E. F. & Zisman W. A., J. Phys. Chem. 59 33t> (1955).
12. Crowl V. T„ JOCCA 55 (5) 388—420 (1972).
13. Parfitt G. D., XIV Fatipec Congress 107 (1978).
14. Hughes W., Fatipec Congress, 67—82 (1970).
15 Howard P. B. & Parfitt G. D., Croatica Chemica Acta 45 189—194 (1973);
ibid 50 15—30 (1977) see also JOCCA 54 356—362 (1971).
16. Keesom W. H., Phys. Zeit 22 129 (1921).
17. Debye P., Phys. Zeit 21 178 (1920).
18. London F., Zeit Phys. 63 245 (1930).
19. Hamaker H. C., Rec. trav chim 55 1015 (1936) ibid 56 3, 727 (1937).
20. Hamaker H. C., Physica 4 1058 (1937).
21. Visser J., Adv. in Colloid and Interface Sci. 3, 331 (1972).
22. Gregory J., Adv. in Colloid and Interface Sci. 2 396 (1970).
23. Void M., J. Coll. Sci. 16 1 (1961).
24. Casimir H. B. 0'., and Polder D., Phys. Rev. 73 360—372 (1948).
25. Schenkel J. H. & Kitchener J. A. (see also Tabor D.) Trans. Farad. Soc. 56 161
(1960) (see also Tabor D.) in p. 23—45 in: Colloid dispersions Ed. J. W. Goodwin
Special Publ./Royal Soc. of Chem.
26. Osmond D. W. J. & Vincent B., & Waite F. A., J. Colloid Interface Sci. 42 (2)
262—269 (1973) also ibid 42 (2) 270—285 (1973).
27. Visser J. Adv. in Colloid and Interface Sci 15 157—69 (1981).
28. La Мег V. J. Colloid & Interface Sci. 19 291 (1969).
29. Verwey E. J. W., & Overbeek J. Th, GTheory of lyophobic colloids Elsevier
Publ. Co. Inc. (1948).
30. Lyklema H. In: Colloidal dispersions Chapter 3 J. W. Goodwin (ed.) Special
Publ./Royal Soc of Chem.
31. Overbeck Th. G., Adv. Coll, and Interface Sci. 16 17—30 (1982).
32. Smith A. L. In: Dispersion of powders in liquids p. 127, Chapter 3 3rd ed. by
G. D. Parfitt, Applied Sci. Publ. (1981).
33. O’Brien R. W. & White L. R., J. Chem. Soc. Farad 11 2 1607 (1978).
34. Wiersema P. H., Loeb A. L., & Overbeek Th. G. J. Coll. Sci. 22 78 (1966).
35. Van Oss C. J. J. Coll. Interface Sci. 21 117 (1966).
36. Franklin M. /. B. JOCCA 51 499— 523 (1968).
37. Beck F. Progress in Organic Coatings 4 1—60 (1976).
38. Schenkel J. H., Kitchener J. A., Experientia 14 425 (1958) Mackor E. L., Rev.
Trav. Chim. 70 747—62 (1951).
39. Rank Bros. Bottisham,. Cambridge UK.
40. James A- M., In: Surface and colloid science R. J. Good & R. R. Stromberg
(eds). Plenum Press Chapter 4, vol. 11 121 —185 (1979).
41. Hunter R., In: Zeta potential in colloid Science Chapter 4, p. 125—178 Academic
Press (1981).
42. Hogg R., Healy T. W., & Fuerstenan D. IV., Trans. Faraday Soc. 62 1638 (1966).
43. Overbeek Th. G., Disc. Farad. Soc. 42 10 (1966).
44. McGowan D. N. L. & Parfitt G. D. Disc. Farad. Soc. 42 225 (1966).
45. Osmond D. J. W. ibid p. 247.
46. Lyklema H., Adv. Coll, and Interface Sci. 2 94 (1968).
47. Overbeek Th. G., Disc. Farad. Soc. 42 10 (1966).
48. Vincent B. Adv. Colloid and Interface Sci. 4 193—277 (1974).
49. Parfitt G. D. & Peacock J. In: Surface and colloid Science Vol. 10 Chapter 4
Matijevic (ed.) Plenum Press 0978).
50. Osmond D. J. W. Sci. and Tech, of Polymer Colloids Vol. 2 369 (1983) NATO
ASI series Poehlein, Otteweill, & Goodwin (eds).
51. Napper D. H. Polymeric stabilisation of colloidal dispersions Academic Press
(1983).
52. Doroszkowski A. & Lambourne R. J. Coll, and Interface Sci. 26 214—221 (1968).
53. Asbeck W. E. & Van Loo M., Ind. Eng. Chem. 46 1291 (1954).
54. Doroszkowski A. & Lambourne R. Disc. Farad. Soc. 65 253—262 (1978).
55. Garvey M. J., J. Coll, and Interface Sci. 61 (1) 194 —196 (1972).
56. Doroszkowski A. & Lambourne R. CID Congress Chemie Physique et Applications
Protiques Vol. 2 part 1 73—81 Barcelona (1968).
57. Walbridge D. J In: Dispersion polymerisation in organic liquids Chapter 3
Barrett (ed.): Wiley-Interscience Publ. (1974).
58. Zigmondy R. Z. Anal. Chem. 40 697 (1901).
59. Mackor E. L. J. Coll. Sci. 6 492 (1951).
60. Fischer E. W. Kolloid-Z 160 120 (1958).
61. Clayfield E. J. & Lumb E. C. J. Coll. Sci. 22 269 (1966).
62. Meier D. 1. J. Phys. Chem. 71 1861 (1967).
63. Hesselink F. Th. j. Phys. Chem. 73 3488 (1969).
64. Hesselink F. Th. J. Phys. Chem. 75 65 (1971).
65. Evans R. & Napper D. H., Kolloid Z-Z Polymere 251 409—414 (1973) ibid 329.
66. Ottewill R. H. & Barclay L„ & Harrington A., Kolloid-Zuz Polymers 250 655
(1972).
67. Ottewill R. H.. Cairns R. J. R., Osmond D. W. J., & Wagstaff I. J. Coll, and
Interface Sci. 54 45 (1976).
68. Hotnola A. M. & Robertson A A. J. Coll. Interface Sci. 54 286 (1976).
69. Doroszkowski A. & Lambourne R. J. Polymer Sci. C34 253 (1971).
70. Doroszkowski A. & Lambourne R. J. Coll, and Interface Sci. 43 97 (1973).
71. Klein J. Nature 288 248 (1980) see Adv. Coll and Interface Sci. 16 101 (1982).
72. Napper D.H., Trans. Farad. Soc. 64 1701 (1968).
72—73. Napper D. H., J. Coll, and Interface Sci. 32 106—114 (1970).
74. Evend R. & Davison J. B., Napper D. H. J. Polymer Sci. B10 449— 453 (1972).
ZJu Flory, p__/.. In. JRtinciples—at pcilyrncr—chemistry,-p, 523 Cornell Uni. Press.
76. Ottewill R. H. & Walker T. Kolloid Z 227 (1/2) 108—116 (1968).
77. Napper D. H., Kolloid Z u Z Polymere 234 1149 (1969).
78. Waite F. A. & Osmond D. J. W., Vincent B., Kolloid Z u Z Polymere 253 676—682
(1975).
79. Napper D’H. Trans. Farad. Soc. 64 1701 (1968).
80. Napper D. H. & Feigin R. I. J. Coll, and Interface Sci. 75 525 (1980).
81. Asakara S. & Oosawa F. J. Chem. Phys. 22 1255 (1954). —
82. Asakara S. & Oosawa F. J. Polymer Sci. 33 183 (1958).
83. Li-in-on F. K., Vincent B., & Waite F. A. ACS Symposium Series 9 165 (1975).
84. Vrij A., Pure Appl. Chem. 48 471 (1976)
85. Joanny J. F. & De Gennes P. G., J. Polymer Sci. Polymer Phys. 17 1073 (1979).
86. Pathmamanoharan C., Hek Th. de. & Vrij A.. Colloid Polym. Sci. 259 769
(1981).
87. Vincent B., J. Coll, and Interface Sci. 73 No 2 (1980).
88. Clarke J., Vincent B., J. Chem. Soc. Farad. Trans. 177 1831—43 (1981).
89. Scheuijens J. M. H. M. & Fleer G. J., Adv. Coll. Interface Sci. 16 361 (1982).
90. Fleer G., Scheutjens J. H. M. H., & Vincent B., In Polymer adsorption and
dispersion stability pp. 245—263 B. Vincent & E. D. Goddard (eds.) A. C. S.
Symp. Ser. (1984) 240.
91. Sperry P. R , J. Coll, and Interface Sci. 99 97—108 (1984).
92. Thompson M. W., Graetz C., Waite F. A., Waters !. A., EP13478.
93. Crowl V. T., JOCCA 46 169—205 (1961).
94. Glasstone S. Textbook of physical chemistry 2nd ed. p. 822 MacMillan & Co.
(1956)
95. Koral L. & Ullman R., Eirich F., J. Phys. Chem. 62 541 (1958).
96. Gilliland E. R. & Gutoff E. B., J. Applied Polymer Sci. 3 (7) 26—42 (1960).
97. Ellerstcin S. & Ullman R., J. Polymer Sci. 55 161, 129 (1961).
98. Mizuhara К., Hara K.. & Imoto T., Koll.-Z u Z Polymers 229 17—21 (1969).
99. Howard G. J. & McConnell P., J. Phys. Chem. 71 2974 , 2981—2991 (1967).
100. Perkel R. & Ullman R„ J. Poly. Sci. 54 127—148 (1961).
101. Kiselev A. V., Lygin V. I. et al. Colloid J USSR 30 291—295 (1968).
102. Thies C„ J. Phys. Chem. 70 No 12 3783—3789 (1966).
103. Eirich F. R., Effects of polymers on dispersion properties Symposium 125—143
(1983).
104. Lipatov Yu. S. & Sergeeva L. U., Adsorption of polymers, Halsted Press
(1974) ISBN 0—470— 54040—0.
|IO5. Hobden J. F. & Jellinek H. H. G. J. Poly. Sci. II 365 (1953).
106. Patat Von, Franz & Schliebeuer C„ Macromol Chem. 44—6 643—668 (1961)..
107. Fetter R. E., Moyer E. S., & Ray L. N., J. Polymer Sci. B7.529—533 (1969).
108. Baker A. S., Jones G. M., & Nicks P. F., BP 1370914 (1974).
109- Miyamoto T., Tomoshige S., & Inagaki H., Poly J. 6 No. 6, 564—570 (1974).
110. Butter R., Tan Y., & Challa Cr., Polymer 14 (4) 171—2 (1973).
111. Walbridge D. J., Scott E. I., & Young С. H. Vth Conference in Organic Coatings
Science and Technology 526—546 (1979) Athens. Coatings Science and Techno-
logy 526—546 (1979) Athens.
112. Solomon D. H., Swift J. D., & Murphy A. J.. J. Macromol. Sci.— Chem. A5
(3) 587—601 (1971).
113. Smoluchowski M. Z Phys. 17 557 (.1916); Z Phys. Chem. 92 129 (1917).
114. Lichtenfeld J. W., Th. Pathmamanoharan, & Wiersema P. H., J. Coll and
; Interface Sci. 49 281 (1974).
115. Lips A. & Wills E„ Trans. Farad. Soc. 69 1226 (1973).
116. Overbeek J. Th. G., In:- Colloid science Vol. 1 p. 282 Kruyt (ed.), Elsevier
(1952).
1.17. Spielman L. A. J. Coll, and Interface Sci. 33 562 (1970).
118. Honig E. P., Roebersen G. J., & Wiersema P. H., J. Coll, and Interface Sci.
36 97 (1971).
119. Gedan H., Lichtenfeld H., & Sonntag H., Coll, and Polymer Sci. 260 1151
(1982).
120. Void M. J., J. Coll. Sci. 14 168—174 (1959).
121. Smith A. E. & Pemberton E. W., Pig. and Resin Tech. 9 Nos. 11, 8 (1980).
122. Strivens T. A. J. Coll. Interface Sci. 57 (3 ) 476—87 (1976).
123. Albers 117. & Overbeek J., Th., G., J. Coll. Sci. 15 480—502 (1960).
124. Doroszkowski A. & Lambourne R., J. Coll. Sci. 26 128—130 (1968).
125. Kalwza U., Pig. Resin Tech. 9 Nos. 4—7 (1980).
126. Hornby M. R. &Murley D. R., Progress in Organic Coatings 3 261 (1975).
127. Menold R., Luttge B., & Kaiser W., Adv. Coll, and Interface Sci. 5 281—335
(1976).
.128. Balfour J. G. & Hird M. J., JOCCA 58 331 (1975).
129. Doroszkowski A. & Armitage В. H. XVII Fatipec Congress 2, 311—330 (1984).
К главе 6
I. Kaye В. H. Chem. Eng. 73 239 (1966).
2. Deity J., The particle analyst Reticles and Graticules, Vol. 1 Nos. 77 — Ann
Arhor Sci. Publ. (1965).
"3. ASTM; E20—68 (1974) section 14.02.
4. Sichel H. Sin., Silverman L., Billings C., & First M., Particle size analysis
in industrial hygiene, New York, NY, Academic Press (1971) p. 248.
5. Hoel P. G., Introduction to mathematics and statistics 3rd ed. J. Wiley & Sons
Inc. New York (1962).-
6. Hatch T. & Choate S. P., J. Franklin Inst. 207 369—387 (1929).
'7. Jelinek Z. K„ In: Particle size analysis p. 14 Ellis Horwood Ltd (1974).
ISBN 85 312 0021.
8. Jackson M. R.,Iglarsh H. & Salkowski M. Powder Tech. 3 317—322 (1969/70).
9. Swenson R. A. & Attle J. R.l Counting, measuring and classifying with image
analysis. Am. Lab. 11 4 (1978).
10. Attle J. R., Oneg D., & Swenson R. A., International Lab. Oct. 1980 pp. 35—48.
11. Quantimet 920, Cambridge Instruments Ltd Rustat Rd. Cambridge CB1 3QH.
12. Magiscan 2, Joyce—Loebl, Gateshead NE11 OQW Tyne & Wear.
13. Menold R., Luttge B., & Kaiser W., Adv. Coll. & Interface Sci. 5 281—335
(1976).
14. Menold R., et al. ibid. p. 306.
15. Cruz-Orive L. M., J. of Microscopy 131 (3) 265—290 (1983) also Schwartz H. A.
& Saltykov (1958) see Saltykov (.1958) Stereometric metalography (2nd Ed.)
(Moscow: Metallurgizdat).
16. Andreassen Pipette, Technico Andreassen pipette apparatus PBW—200—W
technical brochure ex Gallenkamp,
17. BS3406: Part 2: 1963.
18. Jelinek Z. K., p. 77 loc. cit.
19. Svorovsky L. & Allen C J., Particle size analysis 442—450 M. J. Groves (ed.)
Heyden & Son Ltd (1978) ISBN 085501 1580.
20 ASTM D3360- 06.02.
21. Berg S„ ASTM Special Tech. Publ. No. 234, p. 143—171 (1958).
22. Jarrett B. A. & Heywood H.. In: A comparison of methods for particle size
analysis British J. Applied Sci. Supplement Vol. 3, p. 21 (1954).
23. Oden S., Alexanders Colloid chemistry Vol. 1 p. 877—882 Chem. Catalogue Co.
NY (1926).
24. Siebert P. C., In: Particle size analysis Ann Arbor Sci. (1977) Stockham J. D.
& Fochtman C. G., (eds) p. 52.
25. Scott K. J. & Mumford D., Powder Tech. 5 321—328 1971/2.
26. Murley R. D. XII FATIPEC Congress 377—383 (1974).'
27. Svedburg T„ Ind. Eng. Chem (Anal, ed.) 10 113—127 (1938).
28. Joyce—loebl Centrifuge, Vickers Company, Adv. Information brochure 8 :83.
29. Oppenheimer L. E., J Coll, and Interface Sci. 92 (2 ) 350—357 (1983)
30. Hornby M R. & Tunstall D F BP 1 387, 442.
31 Silverman L., Billings С. E., First M IV, Particle size analysis in industrial
hygiene p. 137—146 Academic Press (197T). ISBN 0—12—643750—5.
32. Dallavalle J. H., Micromeritics, 2nd ed., Isaac Pitman & Son Ltd (1948), p. 20.
33. Herdan G., Small particle statistics Butterworth 2nd revised ed. (1960) p. 360.
34. Carmen P. C., J. Soc. Chem. Ind 57 225 239 (1938).
35. Lea F M. & Nurse R. W7., J. Soc. Chem Ind. London 58 277 (1939).
36. Gooden E. C. & Smith С. M., Ind. Eng. Chem. (Anal ed.) 12 497 (1940).
37. Rigden P. J., J. Soc. Chem. Ind. 66 130—136 (1947).
38. Pechukas A. & Gage F. W., Ind. Eng. Chem. (Anal, ed.) 18 370—373 (1946)
also J. App. Chem. (London) 1 105 (1951).
39. Carmen P. C. & Malherbe P. le R., J. Soc. Chem. Ind. 69 139—193 (1950)
and J. Appl. Chem. I 105—108 (1951).
40. Hutto F. B. & Davies D. W., Off. Dig. 31 429 (1959).
41. Gel permeation chromatography, Altgelt К. H. & Segal L., (eds) Marcel
Dekker Inc. ISBN 0—8247—1006—1 (1971).
42. Yau W. W., Kirkland J. J., & Bly D. D., Modern size exclusion liquid chromato-
graphy J. Wiley & Sons (1979).
43. Otocka E. P., Hellman M. Y., & Muglia P. M., Macromolecules 5 1273 (1972).
44. Belenku В G. & Gankina E. S., J. Chromatog. 141 13—90 (1977)
45. Small H., Saunders F. L., & Sole J., Adv. Coll. Interface Sci. 6 237—266 (1976).
46. Small H Anal. Chem. 54 (8) 892A—8a (1982).
47. McGowan G. R., J. Coll. Interface Sci. 89 (1) 94—106 (1982), also Nagy D. J..
Silebi C. A., & McHugh A. J., J. Appl. Poly Sci. 26 (5) 1555—1578 (1981).
48. Giddings J. C. & Caldwell K. D., Anal. Chem. 56 (12) 2093—9 (1984).
49. Rudin A. & Frick C. D., Polymer Latex 11, preprints of Conference (item 12),
London, May 1985.
50. Thornton T. & Maley R., idid. (item 14)
51. Giddings J. C„ J. Chem. Phys. 49 No. 1 81 85 (1968).
52. Hovingh M. E., Thompson G. H., & Giddings J. C., Anal. Chem. 42 195—203
(1970).
53. Kirkland J. J., Rementer S. W„ & Yau W. IV., Anal. Chem. 53 1730—6 (1981).
54. Kirkland J J. & Yau W. №.. Anal Chem. 55 (13) 2165—70 (1983).
55. Heidenreich E., In: Particle size analysis Groves (ed.) Heyden (1978).
ISBN 035501 1580.
56. Leschowski K-, Powder Tech. 24 115—124 (1979).
57. Daeschner H. IV., Powder Tech. 2 349 (1968/9).
58. Crawley D. F. C., J. Sci. Instruments Ser. 2 1 576 (1968).
59. Whitby К. T„ ASTM Special Tech. Publ. No. 234 pp. 3—25 (1958).
60 Daeschner H. IF., Seibert E. E., & Peters E. D., ASTM Special Tech Publ.
No. 234 26—50.
61 Arietti R., Tenca F., & Scarpone A., XVI FATIPEC Congress (1978) 277—300.
62. Coulter W. H., USP 2, 656, 508 (1953).
63. Coulter W. H., Proc, of National Electronic Conf. 12 1034 (1956).
64. Morgan В. B., Research, London 10 271 (1957).
65. Kubitschek H. E., Nature 182 234—5 (1958). See also Research 13 128 (1960).
66. Industrial Bibliography Oct. 1982, produced by Coulter Electronics Ltd.
Luton UK.
67. Allen T., Chapter 13 in Particle size measurement 2nd ed. Chapman & Hall,
London (1974). SBN 412 13490X.
68. Particle Data Ltd Cheltenham, Gloucestershire GL50 3AT.
69. Kerker M., The scattering of light and other electromagnetic radiation Academic
Press (1969).
70. Collins E. A., Davidson J. A. & Daniels C. A., J. Paint Tech. 47 No. 604, 35—56
(1975).
71. Melik D. H. & Fogler H. S., J. Coll. & Interface Sci. 92 (1) 1983.
72. Tunstall D. F., UKP 2, 046, 898.
73. Nobbs J. H., Paint Research Assoc. Progress Report No. 6 33—37 (1984).
74. Meeten G., J. Colloid & Interface Sci. 72 471 (1979).
75. Weiner В. B., Ch. 5. Modern methods of particle size analysis. Barth H. G.
(ed.) Wiley Interscience PubL (1984) also Planiz P. E., ibid. Chapter 6.
76. Johnston P. R. & Swanson R., Powder Tech. 32 119—124 (1982).
77. Lee S. P., Tscharnuter W. & Chu B., J. Polym. Sci. 10 2453 (1972).
78. Weiner В. B., Chapter 3 in Modern methods of particle size analysis Barth H. G.
(ed.) Wiley Interscience Publ. (1984).
79. Lines R. W. & Miller В. V., Powder Tech. 24 91—96 (1979).
80. Siedentopf H. & Zsigmondy R., Ann. Physic 10 1 (1903).
81. Derjaguin В. V., Vlasenko G. Ja., Storozhilova A. I., & Kudrjavteeva N. M.,
J. Coll. Sci. 17 605—627 (1962).
82. McFadyen P. & Smith A. L., J. Col. Interface Sci. 45 573 (1973).
83. Walsh D. J., Anderson J., Parker A.. & Dix M. J., Coll. & Polym. Sci. 259
1003—9 (1981).
84. Gregg S. J. & Allen K. S. W., Adsorption, surface area and porosity Academic
Press (1967).
85. Gregg S. J. & Sing K. S. W., ibid. p. 7.
86. de Boer J. H., Houben G. M. M., Lippens В. C., & Meijs, J. Catalysis 1 1 (1962).
87. Kipling J. J. & Wright E. H. M„ J. Chem. Soc. 855—860 (1962).
88. Herz A. H., Danuer R. P., & Janusonis G. A. In: Adsorption from agueous
solution Webb W. W. & Matijevic E. (eds.) Advances in Chemistry Series
79 (1968) pp. 173—197.
89. Kipling J. J., Wilson R. B., J. AppL Chem. 10 109—113 (1960).
90. Linge H. G. & Tyler R. S., Proc. Australas Inst. Min. Metall 271 27—33 (1979).
91. Barker N. W. & Linge H. G., Hydrometallurgy 6 (3—4), 311—326 (1981).
92. Padday J. F.. Surface Area Determination Proc. hit. Symp. 1969, 331—340
Everett D. H. (ed.), Butterworth.
93. Gregg S. J. & Sing K- S., Adsorption surface area and porosity Academic
Press p. 294 (1967).
94. Harkins W. O. & Jura G., J. Am. Chem. Soc. 66 1362 (1944).
95. Gregg S. J., The surface chemistry of solids 2nd ed. Chapman & Hall (1961)
p. 280.
96. Edwards J. D. & Wray R. I., Aluminium paint and powder 3rd ed. (1955)
Rheinhold Publ. Corp. p. 18.
97. Edwards J. D. & Mason R. B., Ind. Eng. Chem. Anal Ed. 6 1951 (1934).
98. Capes С. E. & Coleman R. D., Ind. Eng. Chem. Fundamentals 12 No. 1 1246
(1973).
99. Shepard E. & Tcheurekedjian N., J. Colloid Interface Sci. 28 481 (1968).
100. Doroszkowski A. and Lambourne R., J. Polym. Sci. Part C34 253 (1971).
101. Scarlet B., In Particle size analysis Stanley—Wood N. C. & Allen T (eds ),
Wiley Heyden (1982), p. 219.
К главе 7
1- Daniel F К. Natl. Paint, Varn, Lacquer Assoc. Sci. Sect. Citjc. No. 744 (1950).
2. Guggenheim S. Off. Dig. 30 No. 402, 729 (1958).
3. Wirsching F., Haug R., & Hamann K. Deutsche Farben Zeits Oct. (1957).
4. Farkas F. K. Unpublished work.
К главе 8
1. Flick E. W. Waterborne paint formulations (1975), Solvent based paint formula-
tion's (1977), Industrial water borne trade paint formulations (1980), Handbook
of paint, raw ma terials (1982), Noyes Data Corporatin, New Jersey,1 USA.
2. Dawoodi 7. Ethoxy propanol: a substitute for ethyl glicol ether. Polimer'Paint
Colour Journal 176, No. 4159, pp. 41—45, Jan. 22 (1986).
3. Hesler К. K- & Loftsrom J. R., Applications of simplex lattice design experimen-
tation to coatings research. Journal of coatings Technology 53, No. 674, 33
(1981).
4. Steig F. B., The influence of PVC on paint properties. Progr. Organic Coatings,
June 1 (4), 3512 73 (1973).
5. Steig F. B., Pigment/bindcr geometry. Pigment handbook, Vol. Ill (T. Patton,
ed.), Wiley—Interscience, New .York, _pp. 203=2-17—(1-973).
6. Bierwagen 0. P. & Hay T. K. The reduced pigment volume concentration as
an important parametr in interpreting and predicting the properties of organic
coatings. Prog. Org. Coatings 3, 281 —303 (1975).
7. Huisman H. F. Pigment volume concentration of the oil absorption of pigments.
Journal of coatings Technology 56, No. 712, pp. 65—79 (1984).
8. Asbeck V7. K. Dispersion and agglomeration effects on coatings performance.
JCT 49, No. 635, 59—70 (1977).
9. Castels R. et al. Particle packing analysis of coatings above the critical pigment
volume concentration. Journal of coatings Technology 55, No. 707, pp. 53—59
(1983).
10. Cremer M. The measurement and importance of adsorption layers in paints.
European Suttiement to Polymers Paint Colours Journal Oct. 5, pp. 85—93
(1983).
11. Cole R. J. Determination of critical PVC in dry surface coating films. Journal
of Oil and Colour Chemists’ Association, pp. 776—780 (1962).
12. Rasenberg C. J. F. M. & Huisman H. F. Measurement of the critical pigment
volume concentration by means of mercury porosimetry. Progr. Org. Coatings,
Sept. 13 (3/4) 223—235 (1985).
13. Steig F. R. Density method for determining the CPVK of. flat latex paints.
Journal of coatings Technology 55, No. 696, pp. Ill —114 (1983).
14. Steig F. R. Numerical expressions of film porosity, American Paint and Coatings
Journal, Oct. 9, pp. 46—50 (1978).
15. Bernardi P. Parametrs affecting the CPVC of resins in aqueous dispersion,
Paint Technol. 27, 24 July (1963),
16. Rowland R. H. & Steig F. B. Grafical solution to CPVC problems in latex
paints. Journal of coatings Technology 54, No. 686, pp.. 51—56 (1982),
17. Patton T. C, Paint flow and pigment dispersion 2nd ed., John Wiley- Interscience,
New York, Ch. 7, p. J72 (1979).
18. Reddy J. N. et al. Studies of adhesion: role of pigments. Journal of Paint
Technology 44, No. 566, pp. 70—75 (1972).
19. Funke W. On the relation between the pigment vehicle interaction and liquid
water absorpsion of paint films. J. Oil Col. Chem. Assoc. 50, 942 975 (1967).
20. Seiner J. A. Microvoids as pigments: a review, Ind., End. Chem. Prod. Res. Dev.
17, No. 4, pp. 302—317 (1978).
21. Kerker M. et-al. Pigmented microvoid coatings. Journal of Paint Technology
47, No. 603, pp. 33—42 (1975).
22. Chalmers J. R. & Woodbridge R. J. Air and polymer—extended paints. European
Suttiement to Polymers Paint Color Journal, Oct. 5, pp. 94—101 (1983).
23. Dulux Australia Ltd., PO Box 60, Clayton, Victoria 3168, Australia.
24. Hislop R. W. & McGinley P. L. Microvoid coatings. Journal of Coatings
Technology 50, No. 642, pp. 69—77 (1978)
25. Letters to the Editor. Journal of Coatings Technology 50, No. 645, pp. 112 —113
(1978).
26. Goldsbrough et al. Prog, in Org. Coat., 10, 35 (1982).
27. Ranting A. & Floyd F. L. Plastic Pigment: A novel approach to microvoid
hiding. Journal of Coatings Technology 51, No. 658, p. 63 (1979).
28. Ranting A. & Ranting P. F. Plastic Pigment, J. Oil Col. Chem. Assoc. 64,
pp. 439—447 (1981).'
29. Letters to the Editor. Journal of Coatings Technology 52, No. 663, April and
August (1980).
30. Hill W. H. et al. A more efficient additive for lower cost hiding. Resin Review,
XXXV, No. 3, pp. 3—10 (1985).
31. Rohm and Haas Co., Philadelphia, PA 19105, Ropacue OP—62 Trade sales
Data Sheets.
32. Water holds Healthier future of coatings. Polymers Paint Colour Journal,
175, No. 4156, p. 825 (1985).
33. Smith N. D. P., Orchard S. E. & Rhind Tutt A. J. The phisics of brushmarks,
J. Oil Colour Chemists Assoc. Sept. 44 (9) 618—633 (1961).
34. Solomon D. H. The chemistry of organic film formers, 2nd ed. R. E. Krieger
Publishing Co., Inc. (1977).
35. Dillon R. E. et al. Sintering of synthetic latex particles. J. Colloid Science,
6, p. 108 (1951).
36. Brown G. L. Formation of films from polymer dispersions, J. Polymer Science,
22. p. 423 (1956).
37. Sheetz D. P. Formation of films bv drying of latex, J. AppL Polymer Sci.,
9.(11), 3759—73 (1965).
38. Barrett K, E. J. (ed.) Dispersion polymerisation in organic media, John Wiley
& Sons (1975).
39. Bromley C. W. A. & Gr-.iystone J. A. Coating composition, BP, 2, 164, 050 A.
40. Surfase Coatings, Vol. I, Ch. 22 — Water reducible resins, OCCAA, Chapmen
& Hall Ltd. 2nd ed. (1983).
41. Wide E. G. Advances in paint dispensing Polymers Paint Colour Journal,
175, No. 4146, pp. 454—461 (1985).
42. Atkinson D. Developments in colour matching and dispensing of paints. Paint
and Resin, Dec. pp. 29—30 (1985).
43. Colony powder colorants, Color Corporation of America, 200 Sayre Street,
Rockford, Illinois 61 101.
44. Herbol colour pill. Hoechst A. G., Frankfurt/Main, 80.
45. Wintermix colourant pastes. Winter OY, 33270 Tampere 27 puh. 440300.
К главе 9
Payne H. F., Organic coatings technology. Volume I (Oils, resins, varnichcs, and
polymers). New York, John Wiley, (1954).
Payne H. F., Organic coatings technology, Volume 11 (Pigments and pigmented
coatings), New York, John Wiley, (1961).
Solomon D. H., The chemistry of organic film formers, Robert E. Krieger Publishing,
(1977).
Waring D. J., Automobile paint systems (paper for Institute of Production Engineers).
February 1984.
Baylis R. L., Trans. Inst. Metal Finishing 50 80—86 (1972).
Waghorn M. J., Non-aqueous dispersion finisches. IMF/ОССА Symposium Warwick
University pp. 228—233 (1973).
Ansdell D. A., Painting of plastic body components for cars Industrial and Production
Engineering 3 30—35 (1980).
Strong E. R., The reduction of lacquer waste in spraying, FIRA Technical Report
21. (November 1965).
Eaton M. J., Product finishing, July 1983.
Inshaw J. L., Jour. Oil and Colour Association, July 1983, 183—185
К главе 10
The refinishers handbook: published by Imperial Chemical Industries in association
with the Vehicle Builders Repairers Association (1983).
К главе 11
1. Gitlitz M. H. J. Coatings Tech. 53 46 (1981).
2. Banfield T. A. (1) «Protective Painting of Ships and Structural Steel». SITA
Ltd, Manchester (1978). (2) JOCCA 63 53 (1980); JOCCA 63 93 (1980).
3. Van der Kerk G. J. M. & Luijten J. G. A. J. Appl. Chem. 4 314 (1954).
4. Evans C. J. & Smith P. J. JOCCA 58 160 (1975).
5. Chromy L. & Uhacz K. JOCCA 61 39 (1978).
6. Montermoso J. C., Andrews T. M. & Marinelli L. P. J. Polym. Sci. 32 523 (1958).
7. Leebrick /.US Patent 3167473 (1965).
8. Harpur W. & Milne A. Proc. 5th PRA Int. Conf., May 1983, 45—50.
К главе“12
I. Richardson E. G., in: Hermans J. J. (ed.) Flow properties of disperse systems.
North-Holland Ch. VI pp. 266—98 (1953), Green H. L. Loc. cit. Ill Ch. VII
pp. 299—322.
2. Schurz J. EUCEPA 20th Conf. Budapest, Paper 11/13 (1982) World Surface
Coatings Abstr., 83/8306.
3. Glass J. E. J. Coatings Technol. 50 (641) 56 (1978).
4. Patton T. C. Paint flow and pigment dispersion: A rheological approach to
coating and ink technology. 2nd edn. Willey—Interscience pp. 365—7 (1979).
5. Huge Y. J. Coatings Technol. 55 (701) 59 (1983).
6. Smith N. D. P., Orchard S. E., & Phind—Tutt A. 1. J. Oil & Col. Chem. Assocn.
44 618—33, part. pp. 621—3 (1961) Patton T. C. Loc. cit. [4], pp. 553.
7. Pearson J. R. A. J. Fluid Meeh., 7 481 (I960).
8. Savage M. D. J. Fluid Mechanics 80 743 (1977).
9. Glass J. E. J. Oil Col. Chem. Assoch. 58 169 (1975).
10. Dodge J. S. J. Paint Technol. 44 (564) 72 (1972).
IL Myers R. R. J. Polymer Sci., Pt. C, (35), 3 (1971).
12. Trevenna D. H. J. Phys., D: Appl. Phys., 17 2139 (1984).
13. Walters K. Rheometry Chapman & Hall, pp. 219 (1975).
14. (a) Keller (b) Loc. cit. [13J p. 233.
15. Orchard S. E. J. Appl. Sci. Res., All, 451 (1962).
16. Wu S. ACS. Division of Org. Coatings & Plastics Chem., papers. 37 (2) 315,
323 (1977).
17. Strivens T. A. (Unpublished results).
18. Amari T. & Watanabe K. Polymer Engineering Rev. 3 (2—4), 277 (1983).
19. Overdiep W. S., in: Spalding D. B. (ed.) Physicochemical hydrodynamics.
V. G. Levich Festschrift. Vol. IL Advance Publications Ltd, 683 (1985).
20. See, for example, Wojtkowiak J. J. J. Coatings Technol. 51 (658), Ill (1980).
21. Anand J. N. & Balwinski R. Z. .1. Coll. & Interfac. Sci., 31 (2), 196 (1969).
Anand J. N. Ibid., 203, Anand /. N. & Karam, H. J. Ibid., 208.
22. Higgins B. G. & Scriven L. E. Ind. Eng. Chem., Fundamentals 18 (3), 208 (1979).
23. Matsumoto T., Segawa Y., Warashina Y., & Onogi S. Trans. Soc. Rheol 17
(1), 47 (1970).
24. Kornum L. O. & Raaschou Nielsen H. K. Progress in Organic Coatings 8 275
(1980).
25. McElvie A. N. Progress in Org. Coatings 6 49 (1978).
26. Monk C. J. H. J. Oil & Col. Chem. Assocn. 49 543 (1965).
27. Manufactured by Research Equipment (London) Ltd, 64 Wellington Rd. Hampton
Hill, Middlesex, UK.
28. McGuigan J. P. Viscosity and Consistency Ch. 3.2. pp. 181—212. in: Sward G. G.
(ed.) Paint testing manual (ASTM Special Technical Publication No. 500).
13th ed. American Society for Testing of Materials (1972).
29. Walton A. J. Paint Manufacture, 47 (5) 13—7 (1977) Ibid. [6], 16—22, 24.
30. Association Francaise de Normalisation, NFT 30—029 (1980).
31. Urad pro Normalised CSN67 3016 (1981).
32. Snow С. I. Official Digest Г392) 907 (1957).
33. Flom D. G. ,1. Appld. Physics 31 306 (I960).
34. Tillett J P. A. Proc. Phys. Soc. B67 677 (1954).
35. lenckel E. & Klein E. Z. Naturf. 7a 619 (1952).
36. Рао Y. H. J. Appld. Phys., 26 1082 (1955).
37. Strivens T. A. (Unpublished results).
38. Gordon M. & Grieveson В. M. J. Polymer Sci. 29 9 (1958).
39. Myers R. R. Official Digest 33 (439) 940 (1961).
40. Myers R R. & Schultz R. K. Official Digest 34 (451), 801 (1962).
41. Myers R R. & Schultz R. K. J Appld. Polymer Sci. 8 755 (1964)
42. Myers R R , Klimek J., & Knauss C. J. J. Paint Technol., 38 (500) 479 (1966).
43. Mewis J. FATIPEC IX Congr. Brussels. Proc. pt. 3 pp. 120—4 (1968).
44. Wolff H. & Zeidler G. Paint & Varnish Production Manager 15 (8) 7 (1936).
45. Quach A. & Hansen С. M. J. Paint Technol., 46 (592) 40 (1974).
46. Taylor R. & Foster H. J. J. Oil Col. Chem. Assoc" 54 1030 (1971).
47. Goring W.. Dingerdissen N., & Hartmann C Farbe und Lack 83 (4) 270 (1977).
48. Cplclough M L., Smith N D P.. & Wright T. A. J. Oil Col. Chem. Assocn.
63 183 (1980).
49. Strivens T. A. Quarterly Report of the Paint Research Assocn. (79/4), II (1979).
50. Strivens T. A. Colloid and Polymer Sci. 261, 74 (1983).
51. Strivens T. A. Colloids and Surfaces 18 395 (1986).
52. Strivens T. A. (In preparation).
53. Kornum L. O. FATIPEC Congr. XIV Budapest Proc. pp. 329—36 (1978).
54. Wapfer D. Farbe und Lack 81 (8) 717; (9) 822; (10) 924 (1975).
55. Kheshgi H. S. & Striven L. E Chem. Eng. Sci 38 (4) 525 (1983).
56. Biermann M. Rheol. Acta 7 (2) 138 (1968).
57. Schaeffer B. Official Digest 34 (453) 1110 (1962)
58. Parfitt G. D FATIPEC Congr. XIV Budapest Proc. pp. 107—117 (1978).
59. Kaluza U. Progress in Org. Coatings 10 289 (1982).
60. Tsutsui K- & Ikeda S. Progress in Org. Coatings 10 235 (1982).
61. Patton T. C. Loc. cit. ref. [4], ch. 7—12.
62. Oesterle К- M. FATIPEC Cong. XIV Budapest Proc. pp. 329—36 (1978).
63. McKay R. B. FATIPEC Congr. XIII Cannes, Proc. pp. 428—34 (1976).
64. Heertjes P. ,1'1 & Smits С I. Powder Technology 17 197 (1977)
65. Mewis I & Strivens T. A. Paper given at 8th lot Congr. of Chem. Eng., Chem.
Equipment & Apparatus (CHISA), Prague (1984)
66. Zosel A. Rheol. Acta 21 72 (1982).
67. Weltmann R. N. In: Eirich F. R. (ed.) Rheology: theory and applications.
Vol. Ill Ch. 6. Rheology of pastes and paints, pp. 236—40 (I960).
68. Patton T. C. Loc. cit. ref. [4]. Ch. 6. pp. 156—183.
69. Oldshue J. Y. Fluid mixing technology. Chem. Eng McGraw—Hili Publications
Co. (1983)
70. Tins instrument is currently being commercially developed by Revenhead
Designs Ltd., Gregge St, Heywood, Lancs., UK.
К главе 13
1. Ferry 1. D. Viscoelastic properties of polymers. J. Wiley, New York, 2nd cd.
(1970).
2. Nielsen L. E. Mechanical properties of polymers. Reinhold Publishing Corpn.,
New York (1962).
3. McCruni N. G., Read В. E. & Williams G. Anelaslic and dielectric effects in
polymeric solids, J. Wiley, New York (1967).
4. Graessley W. W. Adv. Polymer Sci. (1974) (16), 1.
5. Hedvig P. J. Polymer Sci., Macromol. Rev. 15 375 (1980).
6. Williams M. L., Landel R. F. & Ferry J. D. J. Amer. Chem. Soc. 77 3701 (1955).
7. Havriliak S. & Negami S. Brit. J. Appld. Physics Ser. 2 2 1301 —15 (1969).
8. Havriliak S. & Negami S. Polymer 8 161—210 (1967).
9. Laout J. C. & Duperray B. XlVe Congres AFTPV, Aix-les-Bains 143 (1981).
10. Laout J. C. XVIth. FATIPEC Congress, Liege I, 165 (1982).
II. Duperray B., Laout J. C. & Mansol J. L. Double liaison (330). 61 (1983).
12. Toussaint A. & Dhoni L. Prog, in Org. Coatings 11 (2) 139 (1983).
13. Ward I. M. Mechanical properties of solid polymers, Wiley—Interscience
(1981).
14. Andrews E. H. Fracture in polymers Oliver & Boyd (1968).
15. Kinloch A. J. & Young R. J. Fracture behaviour of polymers Applied Science
Publishers (1983).
16. Kinloch A. J. Adv. in Polymer Sci. (1985) 72, 46.
17. Cuthrell R. E. In: Chompff A. J. & Newman S. Polymer networks: structural
and mechanical properties Plenum Press, p. 121.
18. Griffith A. A. Phil. Trans. Roy. Soc. (London) A221 163 (1920/1).
19. Loc. cit. ref. [14], pp. 123—31.
20. Kerry T. E. -j. Pblymcr ScC 50 313 П96Т)".
21. Loc. cit. ref. [13].
22. Smith T. L. & Stedry P. J. J. Appld. Physics 31 1982 (I960).
23. Toussaint A. Proc. 5th Int. Conf, on Organic Coatings Sci. and Tech., Athens
(1979) 489.
24. Zosel A. Prog. Org. Coatings 8 (I) 47 (1980).
25. de Jong J. & van Westen G. C. J. Oil Colour Chem. Assoc. 55 989 (1972).
26. Zosel A. Farbe Lack 82 115 (1976).
27. Pierce P. E. In: Myers R. R. & Long J. S.(eds.), Treatise on coatings. Vol. 2,
Pt. 1, Ch. 4, Marcel Dekker, New York, p. 112 (1969).
28. Monk C. J. H. & Wright T. A. Oil Col. Chemists Assocn. 48 520 (1965).
29. Hertz H. J. Reine Angew. Math. 92 156 (1881).
30. Lee E. H. & Radok J. R. M. Proc. 9th Internal. Congr. Appld. Maths. 5 321
(1957).
31. Radok J. R. M. Quart. Appld. Maths 15 198 (1957).
32. Morris R. L. J. J. Oil Col. Chem. Assocn. 56 t>55 (1973).
33. Inoue Y. & Kobatake Y. Kolloid-Z 160, 44 (1958)
34. Roller M. B. & Gillam J. K. J. Coatings Tech. 50 ( 636), 57 (1978).
35. Roller M. B. Metal Finishing 78 ( 3) 53; (4), 28 (1980).
36. van Hoorn H. & Bruin P. Paint Varnish Prodn. 49 (9) 47 (1959).
37. Yamasaki R. S. Official Digest 35 992 (1963).
38. Gearing J. W. E. Polymer Paint СоГ. J. 172 (4081) 687 (1982).
39. Strivens T. A. (Unpublished results).
40. Whorlow R. W. In: rheological techniques, Chap. 5, Ellis Horwood (1980)
41. Smith N. D. P. & Wright T. A. (Unpublished results).
42. Reichert K-H. W. & Donnebrink G. Farbe u Lack 86 (7) 591 (1980)
43. Myers R. R. J. Polymer Sci., C, (35), 3, (1971).
44. Mason W. P., Baker W. O., McSkitnmin H. J. & Heiss J. H. Phys. Rev. 75 936
(1949).
45. Peck G. (Private Communication).
46. Myers R. R. & Knauss С. I. Polymer colloids, Plenum Press, New York (1971).
47. Myers R. R., Klimek J. & Knauss C. J. J. Paint Technol. 38 479 (1966).
48. Mewis J. FATIPEC IX, Brussels (1968) 3—120.
49. Newis J. In: Watters K. (ed.) Rheometry: industrial applications, John Wiley
Ch. 6, p. 323 (1980).
50. Evans R. M. In: Myers A. R. & Long J. S. (eds.) Treatise on coatings Vol. 2,
Pt. I, Ch. 5. Marcel Dekker, New York (1969).
51. Simpson L. A. FATIPEC XVI, Liege 1 33 (1982).
52. Corcoran E. M. In: Sward G. G (ed.) Paint testing manual (13th edn.) ASTM
Special Publication No. 500, Ch I, Hardness p 281 (1972)
53. Schurr G. G. In: loc. cit. ref. [52], Pt. 5, Ch. 4, Flexibility, p. 333.
54. Sato K. Prog. Org. Coatings 8 (I) 1, (1980).
55. Mercurio A. Official Digest 33 987 (1961).
56. Inone Y. Seni Gakkaishi 6 147 (1950)
57 Inone Y. Loc. cit. ref. [56] p. 150.
58. Inone Y. & Ito Y. Shikizai Kyokaishi 27 37 (1954)
59 Persoz B. Reint Pigm. Vernis 21 194 (1945).
60. Inone Y. & Ikeda K. Kobunshi Kagaku 11 409 (1954).
61. Baker D. J., Elleman A. J. and McKelvie A. N. Official Digest 22 1048 (1950).
62. Roberts J. & Steels M. A. J. Appld. Polymer Sci., 10 1343 (1966).
63. Pierce P. E., Holsworth R. M. & Boerio F. J. J. Paint Technol 39 593 (1967).
64 Sto K. & Inone Y. Kogyo Kagaku Zasshi 60 1179 (1957)
65. Sato К & Inone Y. Kobunshi Kagaku 15 421 (1958).
66 Strivens T. A & Bahra M. S. (Unpublished results).
67. Timoshenko S. & Goodier J. N. Theory of elasticity, McGraw-Hill, New York,
p. 383 (1951).
68. Zorll U. FATIPEC IX Congr. Brussels (1968) p. 3.
69. Kirby P. L. Brit. J. Appld. Physics 7 227 (1956).
70. Calvit H. H. J. Meeh. Phys. Solids 15 141—150 (1967).
71 Zosel A. Farbe Lack 83 9 (1977)
72. Bender H. S J. Appl. Polymer Sci. 13 1253 (1969).
73. Bender H. S. .1. Paint Technol. 43 (552) 51 (1971).
74. Breinsberger J. & Koppelmann J. Farbe u Lack 88 (11) 916 (1982).
75. Strivens T. A. & Rawlings R. D. J. Oil CoL Chem. Assocn. 63 412 (1980).
76. Strivens T. A. & Bahra M. S. J. Oil CoL Chem. Assocn. 66 (II) 341 (1983).
77. Strivens T. A., Bahra M. S. & Williams—Wynn D E. A J. Oil Col. Chem.
Assocn. 67 (5) 113; (6) 143 (1984).
К главе 14
1. Heavens О. S. Optical properties of thin solid films, Butterworth (1955).
2. Hunter R. S. The measurement of appearance, John Wiley & Sons (1975).
3. Ross IR. D. Theoretical computation of light scattering power, J. Paint Tech.
43 50 (1971).
4 Mie G. Ann. Physik 25 377 (1908).
5. Steig F. B. Effect of extenders on hiding power of titanium pigments, Off. Dig.
31 52 (1959)
6. Siener I. A. & Gerhart H. L. XI FATIPEC Congress 1972, Proceedings 127.
7. PPG Industries US patent 3 669 729.
8. Chalmers J. R. & Woodbridge R. I. Air and polymer extended paints. Paint R. A.
5th International Conference, P. R. A.. Progress Report 3 16 (1983).
9 Kershaw R. W. A new class of pigments, Australian OCCA Proceedings and
, News,8 No. 84 (1971).
10 Loney S. T. Scattering of light by white pigment particles. Paint R. A. Technical
Paper No. 213 (1960).
11. Carr W. Dispersion—the neglected parameter, J. Oil Col. Chem. Assoc. 65
373 (1982).
12. Wright W. D. The measurement of colour 4th ed. Adam Hilger (1969).
13. Judd D. B. & Wyszecki G. Color in business, science and industry 3rd ed. John
Wiley (1975).
14. Billmeyer F. W. & Saltzman M Principles of color technology 2nd ed. John
Wiley (1981).
15. Wyszecki G. & Stiles W. S. Color sciences — concepts and methods 2nd ed.
John Wiley (1982).
16. McLaren K- The colour science of dyes and pigments, Adam Hilger (1983).
17. Kubelka P. & Munk F. Z. Tech. Physik 12 593 (1931).
18 Kubelka P. J Opt. Soc Amer 38 448 (1948)
19. Duncan D. R. The colour of pigment mixtures, J. Oil Col. Chem. Assoc. 32
296 (1949).
20. Vial F. On the dependency of scattering coefficients on PVC, IX FATIPEC
Congress 1968 1—40.
21. Tioxide Pic Opacity with tioxide pigments, BTP Leaflet 107.
22. Steig F. B. Hiding power of titanium pigmants, Off. Dig. 29 439 (1957).
23. Bullett T. R. & Holbrow C. L. Wet and dry opacity of paints, J. Oil Col. Chem.
Assoc. 40, 991 (1957).
24. Ishihara S. Test for colour blindness, Tokyo, Kanehara Shuppan (1973).
25. MacAdam D. Visual sensitivity to color differences in daylight, J. Opt. Soc.
Amer. 32 247 (1942).
26. Robertson A. R. The CIE color difference formulae, Color Res. Appl. 2 7 (1977).
К главе 15
I. BS 2015 : 1965 : Glossary of Paint Terms.
2. ASTM D523— 1939 revised as D523 — 67.
3. BS 3900 : Part D5 : 1980 Measurement of spesular gloss of non-metallic paint
films at-20°, 60° and 85°.
4. Bullett T. R. & Tilleatd D. L. Optical properties of paints — the basis of instru-
mental measurements, J. Oil. Col. Chem. Assoc. 36 545 (1953).
bv-H-tm-ter-R- S. Gloss eva-krat-fen-ef ffi-aJer+ate -ASTM Bulletin No. 186 48 (1952).
6. ASTM D1471 Test for 2-parameter 60° specular gloss.
7. BS 3900: Part D3 : 1975 Assessment of sheen (withdrawn June 1983).
8. BS 3900 : Part D4 : 1974 Comparison of contrast radio (hiding power) of paints
of the sajne type and colour.
9. BS 3900 : Part D6 : 1982 Determination of contrast radio (opacity) of lightcolou-
red paints at a fixed spreading rate, Using polyester foil.
10. BS 3900 : Part D7 : 1983 Determination of hiding power of white and lightcolou-
red paints by the Kubelka-Munk method.
11. Munsell A. H. Munsell book of color, Munsell Color Co., Baltimore, Maryland,
USA.
12. BS 3900 : Part DI : 1978 Visual comparison of the colour of paints.
13. Billmeyer F. W. Colorimetry of fluorescent specimens. A state of the art review.
Technical Report NBS—GCR 79 — 185. National Bureau of Standards
Washington DC (1979).
14. Tilleard D. L. Tolerances in colour matching J. Oil Col. Chem. Assoc. 40 952
(1957).
15. Johnston R. M. Color control in the small paint plant, J. Paint Tech. 41 415
(1969).
К главе 16
I. A visual method of gloss estimation JOCCA 47 (II) 867 (1964).
2. Scott J. L. JOCCA 65 182 (1982).
дополнительная литература на русском языке
Общая
1. Сорокин М. Ф., Кочново. 3. А., Шодэ Л. Г. Химия и технология пленкообра-
зующих веществ. М.: Химия, 1989. 480 с.
2. Охрименко И. С., Верхоланцев В. В. Химия и технология пленкообразующих
веществ. Л.: Химия, 1978. 392 с.
3. Яковлев Л. Д. Химия и технология лакокрасочных покрытий. Л.: Химия,
1989. 384 с.
4. Лившиц М. Л., Пшиялковский Б. Н. Лакокрасочные материалы: Справочное
пособие. М.: Химия, 1982. 360 с.
5. Дринберг С. А., Ицко Э. Ф. Растворители для лакокрасочных материалов:
Справочное пособие. Л.: Химия, 1986. 208 с.
6. Рейбман А. И. Защитные лакокрасочные покрытия. Л.: Химия, 1982. 320 с.
7. Зимон А. Д. Адгезия пленок и покрытий. М.: Химия, 1977. 352 с.
8. Вакула В. Л., Притыкин Л. М. Физическая химия адгезии полимеров. М.:
Химия, 1984. 224 с.
9. Адгезивы и адгезионные соединения/Под ред. Л. X. Ли: Пер. с англ. М.: Мир,
1988. 226 с.
К главам 1 и 2
I. Паттон Т. Технология алкидных смол. Составление рецептур и расчеты:
Пер. с англ./Под ред. К. П. Беляевой. М.: Химия, 1970. 127 с.
2. Седов Л. Н., Михайлова В. В. Ненасыщенные полиэфиры. М.: Химия, 1977.
241 с.
3. Могилевич М. М„ Плисе Е. М. Окисление и окислительная полимеризация
непредельных соединений. М.: Химия, 1990. 240 с.
4. Олигоорганосилоксаны/Под ред. М. В. Соболевского. М.: Химия, 1985. 269 с.
5. Соломон Д. Г. Химия органических пленкообразователей: Пер. с англ. М.:
Химия, 1971. 320 с.
6. Сырье и полупродукты для лакокрасочных материалов. Справочное пособие/
Гольденберг М. М., Ермолаева Т. А., Лившиц М. Л. и др. М.: Химия, 1978.
512 с.
7. Лившиц Р. М., Добровинский Л. А. Заменители растительных масел. М.:
Химия. 160 с.
8. Яковлев А. Д. Порошковые краски. Л.: Химия, 1987. 216 с.
9. Чернин И. 3., Смехов Ф. М., Жердев Ю. В. Эпоксидные полимеры и компо-
зиции. М.: Химия, 1982. 232 с.
10. Финкельштейн М. И. Промышленное применение эпоксидных лакокрасочных
материалов. Л.: Химия, 1983. 120 с.
II. Молотова В. А. Промышленное применение кремнийорганических лакокра-
сочных покрытий. М.: Химия, 1978. 112 с.
12. Лившиц Р. М., Семина Р. А. Лакокрасочные материалы с пониженным
содержанием органических растворителей. М.г Химия, 1989. 80 с.
К главе 3
I. Ермилов П. И., Индейкин Е. А., Толмачев И. А. Пигменты и пигментиро-
ванные лакокрасочные материалы. Л.: Химия, 1987. 200 с.
2. Индейкин Е. А., Лейбзон Л. И., Толмачев И. А. Пигментирование лакокра-
сочных материалов. Л.: Химия, 1986. 160 с.
3. Беленький Е. Ф.. Рискин И. В. Химия и технология пигментов. Л.: Химия,
1974 . 656 с.
4. Липатов Ю. С. Физическая химия наполненных полимеров. М.: Химия, 1977.
304 с.
К главе 4
1. Козлов П. В., Папков С. П. Физико-химические основы пластификации
полимеров. М.: Химия, 1982. 224 с.
2. Сырье и полупродукты для лакокрасочных материалов. Справочное пособие/
Гольдберг М.М., Ермолаева Т. А., Лившиц М. Л. и др. М.: Химия 1978. 512 с.
3. Розенфельд И. Л. Ингибиторы коррозии. М.: Химия, 1977. 352 с.
4. Толстая С. И., Шабанова С. А. Применение поверхностно-активных веществ
в лакокрасочной промышленности. М.: Химия, 1976. 176 с.
5. Химические добавки к полимерам: Справочник/Под ред. И. П. Масловой.
М.: Химия, 1981. 264 с. ,
К главам 5 и 6
I. Липатов Ю. С. Межфазные явления в полимерах. Киев: Наукова думка,
1980. 260 с.
2. Ребиндер П. А. Избранные труды. Поверхностные явления в дисперсных
системах. М.: Наука, 1978. 368 с.
3. Яминский Б. В., Пчелин В. А., Амелина Е. А., Щукин Е. Д. Коагуляционные
контакты в дисперсных системах. М.: Химия, 1982. 186 с.
А. -Коузов- Д—Д,—Основы—анализа дисперсного сэстава—промышленных пылей
и измельченных материалов. Л.: Химия, 1971. 280 с.
5. Коузов П. А., Скрябина Л. Я. Методы определения физико-химических свойств
промышленных пылей. Л.: Химия, 1983. 143 с.
6. Дринберг С. А., Верхоланцев В. В. Органодисперсные лакокрасочные мате-
риалы и по'крытия. М.: Химия, 1976. 144 с.
7. Елисеева В. И. Полимерные дисперсии. М.: Химия, 1980. 216 с.
8. Андрианов Е. И. Методы определения структурно-механических характеристик
порошкообразных материалов. Л.: Химия, 1982. 256 с.
К главе?
I. Кочнова 3 А., Фомилева Т. Н., Сорокин Н. Д. Аппаратурно-технологические
схемы производства пленкообразующих веществ. М.: Химия, 1978 . 92 с.
2. Горловский И. А., Козулин Н. А. Оборудование заводов лакокрасочной
промышленности. Л.: Химия, 1980. 376 с.
3. Индейкин Е. А., Лейбзон Л Н., Толмачев И. А. Пигментирование лакокра-
сочных материалов. Л.: Химия, 1986. 160 с.
4. Манусов Е. Б. Контроль и регулирование технологических процессов лако-
красочных производств. М.: Химия, 1977. 116 с.
К главе 8
I. Гарин В. Н.. Долгополов Н. Н. Полимерные защитные и декоративные покрытия
строительных материалов. Л.: Стройиздат, 1975. 192 с.
2. Депкер И. И., Кулешова И. Д. Защита изделий из алюминия и его сплавов
лакокрасочными покрытиями. М.: Химия, 1985. 144 с.
3. Гольдберг М. М., Корюкин А. В., Кондрашев Э. К Покрытия для полимерных
материалов. М.: Химия, 1980. 288 с.
4. Буглай Б. М. Технология отделки древесины. М.: Лесная промышленность,
1973. 304 с.
5. Грицевич Е. П. Малярные работы. М.: Стройиздат, 1981. 160 с.
К главам 9 и 10
I. Чеботаревский В. В., Кондрашов Э. К- Технология лакокрасочных покрытий
в машиностроении. М.: Машиностроение, 1978. 295 с.
2. Крылова И. А., Коган Н. Д., Ратников В. И. Окраска электроосаждением.
М.: Химия 1982. 248 с.
3. Карякина М И. Лакокрасочные материалы для защиты сельскохозяйственной
техники. М.: Химия, 1985. 112 с.
4. Розенфельд И. Л., Рубинштейн Ф. И., Жигалова К Л- Защита металлов от
коррозии лакокрасочными покрытиями. М.: Химия, 1987. 224 с.
5. Шангин Ю. А. Восстановление лакокрасочного покрытия легкового автомо-
биля. М.: Транспорт, 1989. 205 с.
6. Рейхельт В. Антикоррозионная защита автомобилей/Пер. с нем. М.: Транспорт,
1977. 104 с.
7. Окрасочные работы в машиностроении. Справочник/Под ред. Е. В. Искры.
Л.: Машиностроение, 1984. 256 с.
8. Вадас Э. Изготовление и ремонт деталей машин с пластмассовым покры-
тием/Пер. с венгерского. М.: Машиностроение, 1986. 320 с.
• К главе 11
I. Дринберг С. А., Калаус Э. Э., Левит Н. И., Рожков Ю. И., Фрост А. М.
Судовые покрытия. Л.: Судостроение, 1982. 201 с.
2. Гуревич Е. С., Рухадзе Е. Г., Фрост А. М. и др. Защита от обрастания. М.:
Наука, 1989. 560 с.
3. Гуревич Е. С., Искра Е. В., Куцевалова Е. И. Защита морских судов от
обрастания. Л.: Судостроение, 1978. 200 с.
4. Искра Е. В. Лакокрасочные материалы и покрытия в судостроении. Л.:
Судостроение, 1984. 367 с.
К главам 12 — 16
1. Карякина М. И. Испытание лакокрасочных материалов и покрытий. М.:
Химия, 1988. 272 с.
2. Зубов И. И., Сухарева Л. А. Структура и свойства полимерных покрытий.
М.: Химия, 1982. 256 с.
3. Карякина М. И. Физико-химические основы процессов формирования и ста-
рения покрытий. М.: Химия, 1980. 216 с.
4. Гуревич М. М., Ицко Э. Ф., Середенко М. М. Оптические свойства лако-
красочных покрытий. Л.: Химия, 1984. 120 с.
5. Санжаровский А. Г. Физико-механические свойства полимерных и лакокра-
сочных покрытий. М.: Химия, 1978. 184 с.
6. Цветовой ассортимент лакокрасочных материалов и его нормирование. М.:
ВНИИТЭ, 1986. 124 с.
7. Андрющенко Е. А. Светостойкость лакокрасочных покрытий. М.: Химия,
1986. 185 с.
ОГЛАВЛЕНИЕ
Предисловие к русскому изданию . . . . 5
Предисловие к английскому изданию 7
Глава 1 Состав лакокрасочных материалов и их применение — общее
введение (Р. Ламбурн)............................................... 9
1.1. Краткий исторический обзор . .9
1.2. Лакокрасочные покрытия . .... 12
1.3. Компоненты красок . . .15
1.3.1. Полимерные или олигомерные пленкообразователи . . 1G
1.3.2. Пигменты 22
1.3.3. Растворители 25
1.3.4. Добавки . . 27
1.4. Методы нанесения . . . . . ________—.—.—,—,—,.............. 29
Глава 2 Органические пленкообразователи (Дж. Бентли) . . . . 30
2.1. Введение . . ...... 30
2.2. Природные полимеры 35
2.3. Алкиды t . .35
2.3.1. Состав алкидов . . 38
2.3.2. Реакции и строение алкидов ... .41
2.3.3. Контроль процесса синтеза алкидов . 42
2.3.4. Описание алкидов........................................ 42
2.3.5. Составление рецептур алкидов . 43
2.3.6. Модифицированные алкиды . ... 44
2.4. Полиэфиры 47
2.4.1. Состав . ... ... .......... 48
2.4.2. Получение. 49
2.4.3. Модификация .... 50
2.4.4. Ненасыщенные полиэфиры . 50
2.5. Акриловые полимеры 52
2.5.1. Состав и синтез 53
2.5.2 Термопластичные акриловые полимеры . . 56
2.5.3. Термореактивпые акриловые материалы . . . 57
2.6. Эмульсионные полимеры ... .... .......... 60
2.7. Дисперсионная полимеризация в неводных средах .64
2.8. Водоразбавляемые материалы .... .......... 68
2.9. Материалы для электроосаждения . 72
2.10. Материалы с высоким сухим остатком . ... 74
2.11. Радиациоиио-отверждаемые пленкообразователи . 76
2.12. Порошковые составы 78
Глава 3. Пигменты для красок (Дж. Ф. Ролинсон) . 80
3.1. Введение ... 80
3-2. Определения................................................... 80
3.3. Требования к качеству пигментов 81
3.3.1. Внешний вид....................................... . ; 81
3.3.2. Стойкость . 81
3.3.3. Долговечность . 81
3.4. Классификация пигментов . . 84
3.5. Номенклатура пигментов . 84
3.6. Группы пигментов 86
3.6.1. Тонеры .86
3.6.2. Литоли .... 87
3.6.3. Красочные лаки........................................... 87
3.6.4. Пигменты, модифицированные канифолью ... . 88
3.6.5. Пигменты на основе нафтола, Нафтола AS* и нафтанилидов 88
3.6.6. BON-пигменты . . ......... ... 89
3.6.7. Кубовые пигменты...................................... 89
3.6.8. Керамические пигменты (синтетические смешанные фазовые
оксиды) ... 90
3.6.9 Пигменты, полученные флашинг-процессом 90
3.7. Токсичность ... 91
3.8. Свойства пигментов 91
3.8.1. Химическая природа........... . . . 92
3.8.2. Кристаллическая структура 93
3.8.3. Форма частиц......................... 94
( 3.8.4. Размер и распределение частиц по размерам ... 96
3.8.5. Показатель преломления... ...... 97
3.8.6. Природа поверхности пигментов и покрытий на пигментных ча-
стицах .................. . . . .98
3.8.7. Диспергируемость ... ......... 102
3.8.8. Цвет и другие свойства пленкообразователя . . 103
3.9. Выбор пигментов 104
,. 3.9.1. Тип лакокрасочного материала . . ... 105
г 3.9.2. Свойства красок.......................................... ПО
3.9.3. Укрывистость и прозрачность ПО
3.9.4. Цвет и смешивание пигментов . 111
3.9.5. Количество пигментов в краске........................... 113
О
Гл а в а 4. Добавки в краски (Р. А. Джеффе, В. Джонс) . 114
4.1. Введение...................................................... 114
4.2. Усилители противокоррозионных свойств 115
4.3. Пеногасители . . ............. 115
4.4. Добавки, препятствующие осаждению.................. . 116
4.5. Добавки, препятствующие образованию поверхностной пленки при
хранении красок .... 118
4.6. Ингибиторы коррозии тары ........... 118
4,7. Добавки, поглощающие влагу и предотвращающие газовыделение 119
4.8. Диспергаторы ... . .119
4.9. Сиккативы...................... . ... 120
4.10. Регуляторы электрических свойств . 122
4.11. Ингибиторы коррозии . . . 123
4.12. Добавки, предотвращающие флотацию и флокуляцию . . 123
4.13. Консерванты для хранения краски в таре . .... 124
4.14. Биоциды ..... .... 125
4.15. Инсектициды .................. . . 125
4.16. Добавки, повышающие белизну покрытий . ... 126
4.17. Дезодоранты................................................. 126
4.18. Добавки, поглощающие УФ-излучение............................ 126
Глава 5. Физическая химия дисперсий (Л Дорожковский) 128
5.1. Введение...................................................... 128
5.2. Погружение и смачивание пигмента............................. 129
5.3. Деагломерация (механическое разрушение агломератов) 132
5.4 Диспергирование — коллоидная стабилизация 134
5.4.1. Силы, действующие между макроскопическими телами . 134
5.4.2. Стабилизация дисперсий с помощью заряда . 137
5 4.3. Зета-потенциалы . ............. 138
-5’4.4. Измерение электрофоретической подвижности . 139
5.4.5. Объяснение коллоидной стабильности 141
5.5. Стерическая (или полимерная) стабилизация 149
5.6. Полная флокуляция и полная стабилизация . 152
5.7. Адсорбция ... 156
5 7 1. Изотермы адсорбции...................................... 157
5.7.2. Свободная энергия адсорбции и изотермы адсорбции 158
5.7.3. Адсорбция и температура . . . 162
5.7.4. Скорость адсорбции и равновесие 162
5.7.5. Специфическая адсорбция 164
5.8. Скорость флокуляции . . . I ? Г 164
Глава 6. Размер частиц и его измерение (А. Дорожковский) . 170
6 1. Введение . 170
6 2. Определения . 171
6.2.1. Требования к счету частиц................................ 172
6.2.2. Средний размер частиц и способы его выражения 174
6.2.3. Кривые размер — частота его наблюдения ... 175
6.3. Методы измерения частиц . 179
6.3.1. Прямые измерения . 180
6.3.2. Седиментометрические методы 181
6.3.3. Хроматографические методы 186
6.3.4. Методы ситового анализа .... 188
6.3.5. Оптические методы (светорассеяние) . . 191
6.3.6. Методы определения площади поверхности . ... 197
6.3.7. Какой из методов является наилучшим? . 200
Глава 7. Промышленное производство лакокрасочных материалов
(Ф. К Фаркас) .... 203
7 1. Введение....................................................... 203
7 2 Применение диспергаторов............................... 204
7.3. Методы оптимизации составов для диспергирования................ 205
7.4. Использование инструментальных методов для выбора состава паст 208
7.4.1. Выбор раствора пленкообразователя с наилучшей смачивающей
способностью и оптимальным содержанием в пигментной пасте 209
7.4.2. Как применять результаты испытаний для определения составов
пигментных паст?......................... . . . 218
Г л а в а 8. Покрытия для зданий (Д А Грейстоун) 224
8 1 Введение........................................................ 224
8.2. Рецептурные особенности и ограничения 22о
8.2.1. Стабильность при хранении . . . ............... . 226
8.2.2. Окружающая среда и здоровье 227
8.2.3. Образование пленки . 228
8.2.4. Нанесение................................................ 228
8.3- Соотношение пигмент — связующее — растворитель .
8.3.1. Соотношение пигмент — связующее...............
8.3.2. Критическая объемная концентрация пигмента (КОКП) .
^.4. Следствия пористости
, . 8.4.1. Механические свойства ............
- . 8.4.2. Свойства, связанные с проницаемостью
' 8.4.3. Оптические свойства
О
8'.5. Краски, содержащие-воздух1 и пол и мер н Ле наполните*! и . л:.
8.5.1. Измельчение............................
8.5.2. «Микроблокн» (Фирма Berger Tenson & Nicholson).'. . .
8.5.3. Полимерные наполнители (Glidden Coatings' & Resings 'Divizion)
J 8.5.4. «Ropaque» . . . .
.8,6. Природа красочного связующего
J,‘ 8.6.1. Общие положения .
8.6.2. Некоторые особенности применения красок .
8.6.3. Пути совершенствования рецептур
6‘7. Проблемы колерования красок .
1 8.7.1. Общие положения .
8.7.2. Дозирующие машины .....
'1'' 8.7.3. Колеровочные пасты и красочные основы
8.8. Роль подложки . . . . .
Глава 9. Автомобильные краски (Д. А. А неделя)
9:1. Введение
< \ о w .
9.1.1. Система окраски с грунтовкой, наносимой распылителем .
' ' 9.1.2. Система окраски с грунтовкой, наносимой'окунанием . . .
9.1.3. Система окраски с грунтовкой, наносимой методом электро-
]и,1 окраски................................. . ,
9.1.4. Показатели качества покрытия
9.2. Подготовка поверхности
9;.3. Грунтование . .
Of)t. 9.3.1. Нанесение грунтовки методом распыления .
9.3.2. Нанесение грунтовки окунанием
9.3.3. Электроокраска . .
9.4'. Система покрытия, содержащая грунтовку, шпатлевку и промежуточный
I ()< слой .
1 9.4.1. Состав связующего
9.4.2. Пигментирование .' Л!1 ' .
9.5. Усовершенствованный процесс...............
9.6. Нескалывающиеся (противоударные)"п6кр'ытйяг.
•9.7. Отделочные покрытия , для автомобилей . /...
9.7.1. Алкидные или полиэфирные покрытия . . .......
‘ 9.7.2. Материалы на основе неводных акрилатных дйсперсий, отверж-
- даемых при повышенной температуре . .
9.7.3. Термопластичные акрилатные лаки . .......... . .
9.7.4. Технология окраски с применением основного слоя и прозрач-
ного наружного слоя............................................
” 9.7.5. Пигментирование отделочных покрытий для автомобилей
9.8. Ремонтная окраска в заводских условиях.............. . . . .
9.9: Окраска пластмассовых деталей корпуса................, ... .
9.9.1. Листовой материал, получаемый горячим формованием . 310
9.9.2. Полиуретаны, получаемые методом экструзии 311
9.9.3. Эластики, получаемые методом экструзии из расплава 311
9.9.4. Проблемы окраски....................................... 311
9.9.5. Процесс окраски и используемые материалы . 312
9.9.6. Окрасочная линия для местной окраски . 313
9.10. Окрашивание распылением 313
9.10.1. Воздушное распыление.................................. 315
9.10.2. Распыление с подогревом при пониженном давлении........316
9.10.3. Электростатическое распыление . . . . 316
9.10.4. Эффективность окраски (практические аспекты) . 319
9.11. Горячая сушка . 319
9.11.1. Технология горячей сушки . . 321
9.11.2. Конструкция конвективных печей 321
9.11.3. Выделение паров и запах............................... 323
9.11.4. Перспективы дальнейшего развития печной сушки 324
9.12. Внешний вид и методы его оценки . 325
9 12 1_Внешний вид._. .. -_. __. . .___*_____ . 326
9.12.2. Защитные свойства . 326
9.12.3. Методы испытаний . . 326
9.13. Перспективы развития 330
9.13.1. Покрытия с высоким содержанием сухого остатка..........330
9.13.2. Покрытия со сверхвысоким содержанием сухого вещества . 332
9.13.3. Водоразбавляемые материалы ... 332
9.13.4. Порошковые покрытия и водные пасты 334
9.13.5. Основные слои с интенсивным цветом 334
9.13.6. Прозрачные слои 335
9.13.7. Пигментирование . 335
9.13.8. Окраска пластмасс......................................336
9.13.9. Технология, оборудование и нанесение покрытий электро-
осаждением ........... ........................... 336
9.13.10. Заключение 337
Глава 10. Эмали для ремонтной окраски автомобилей (21. Г. Моуби). 338
10.1. Введение .... 338
10.2. Покрывные системы 340
10.2.1. Введение . . . 340
10.2.2. Нитроцеллюлозные эмали . ... 341
10.2.3. Термопластичные акрилатные эмали (ТПА) 341
10.2.4. Алкидные материалы 373
10.2.5. Акрилатные эмали . . 373
10.2.6. Акрилуретановые эмали ... . 373
10.2.7. Системы, включающие основной и прозрачные слои . . 344
10.3. Цвет ' . ... 345
10.3.1. Введение . .... 345
10.3.2. Исправление колера в заводских условиях и смешанные
схемы колеровки............................................... 345
10.3.3. Ремонтные смешанные схемы 346
10.4. Перспективы развития 347
10.4.1. Отношение к технологии автомобилестроения . 347
10.4.2. Новые системы связующих................................349
Глава 11. Окрашивание судов (Р. Ламбурн) 350
11.1. Введение . 350
11.2. Коррозия..................................................... 351
11.3. Подготовка поверхности........................................354
11.4. Грунтовки, наносимые после дробеструйной обработки поверхности 355
11.5. Системы окраски для судов . . 357
11.5.1. Краски для надводного борта и надстроек . 360
11.5.2. Краски для переменной ватерлинии 363
11.5.3. Краски для подводной части судов . 363
11.5.4. Противообрастающие покрытия . 365
Глава 12. Реология лакокрасочных материалов (Т. А. Страйвенс) 371
12.1. Введение . . 371
12.2. Общие замечания о реологии лакокрасочных материалов 371
12.2.1. Стадия транспортировки 372
12.2.2. Пленкообразование ... 374
12.2.3. Растекание'пленки краски . 374
12.2.4. Необходимые реологические характеристики для нанесения
красок . . . ......................... 378
12.3. Экспериментальные методы измерения реологических свойств лако-
красочных материалов до и после нанесения.......................... 379
12.3.1. Реологические свойства до нанесения 379
12.3.2. Растекание пленки краски 381
12.4. Реология лакокрасочных материалов при производстве и хранении 391
12.4.1. Измельчение пигментов . 391
12.4.2. Течение по трубопроводам 393
12.4.3. Смешение 393
12.4.4. Хранение 393
Глава 13. Механические свойства покрытий (Т. А. Страйвенс) 395
13.1. Введение................................................... 395
13.2. Вязкоэластические свойства полимеров 397
13.3. Механические свойства полимеров...............................401
13.4. Экспериментальные методы определения механических свойств по-
крытий . 403
13.4.1. Введение.............................................. 403
13.4.2. Экспериментальные экспресс-методы . 404
13.4.3. Динамические методы ... 405
13.4.4. Измерение конечных свойств . 410
13.5. Обсуждение экспериментальных методов . . . 411
13.6. Технологические испытания механических свойств . 412
13.6.1. Испытания твердости 412
13.6.2. Испытания гибкости 414
13.6.3. Испытания на удар . . 415
13.7. Акустическая эмиссия................................... .... 415
Глава 14. Декоративные свойства покрытий. Основные положения
(Т. Р Буллетт) .
14 1 Введение ... ......... .
14.2. Физика отражения на Границе раздела покрытие/воздух
14.2.1. Плоские поверхности ....
14.2.2. Эффекты структуры поверхности ... . . . .
14.3. Светорассеяние и светопоглощение покрытий . ?'".• ? '.
14.3.1. Общие положения . .
14.3.2. Отражение на поверхностях раздела
14.3 3. Светорассеяние белых пигментов
14.3.4. Светопоглощение пигментами .
sir.».' И
14.4. Цвет смесей пигментов и пигментированных пленок .
14.4.1. Восприятие цвета .....
14.4.2. Цвет пигментированных пленок ... ; j-.-Vi’l •. .
14 4 3. Предсказание цвета .
14.5. Изменения в пигментированных покрытиях .
14.5.1. Введение .....................
14.5.2. Изменение укрывистости пленок
14.5.3. Изменение цвета . . .__».....................
14.6. Флуоресценция и фосфоресценция
14.7. Оценка цвета .
14 7.1. Введение..............
14.7.2. Аномальное цветовосприятие
14.7.3. Усталость и констрастность
14.7 4. Оценка цветовых различий
14.7.5. Влияние освещения .
14.8. Другие данные .
Глава 15. Характеристика и контроль внешнего вида (Т. Р. Буллетт)
15.1 Блеск
15.1.1. Классификация
15.1.2. Измерение глянца при зеркальном отражении .
15 1 3. Отчетливость восприятия глянца
15 14. Оценка глянца .
15.2. Укрывистость
15.2.1. Общие положения
15.2.2. Определение отношения контрастностей .
15.2.3. Укрывистость и коэффициенты светорассеяния .
15.3. Характеристика и контроль цвета
15.3.1. Введение
15.3.2. Визуальный цветовой контроль . .
15.3.3. Инструментальный контроль цвета .
15.4. Цветовой контроль при производстве красок .
Глава 16. Оценка долговечности покрытий (Р. Ламбурн.) . .
|16.1 Введение
16.1.1. Причины разрушения покрытий
16.1.2. Методология...................
16.1 3. Подготовка образцов для испытаний..................
16.2. Испытания химической стойкости
16.2.1. Специальные испытания .
16.3. Испытания механических свойств покрытий
16.3.1. Стойкость к удару............
16.3.2. Испытания твердости..........
16.3.3. Стойкость к царапанию .
16.3.4. Испытания на изгиб..............
16.3.5. Адгезия (метод решетчатого надреза)
16.4. Ускоренные климатические испытания .
16.4.1. Методы оценки: определение критериев .
16.5. Естественные климатические испытания . . . . .
Л 16.5.1. Выбор образцов для испытаний....................
16.5.2. Метод 1CI для испытания столярных секций .
16.5.3. Регистрация данных испытаний .
16.5.4. Сравнение долговечности при естественных и ускоренных ис-
пытаниях . . . . .
Литература...................’.....................................
ИЗДАТЕЛЬСТВО «ХИМИЯ»
ГОТОВИТСЯ К ВЫПУСКУ
Корсунский Л. Ф., Калинская Т. В., Степин С. Н. Неоргани-
ческие пигменты: Справ, изд.— СПб.: Химия, 1992.— 25 л. (в
пер.): 4 р. 50 к., 8000 экз.
Приведены свойства неорганических пигментов и области их приме-
нения. Рассмотрено пигментирование различных систем: лакокрасочных
материалов различного назначения, пластмасс, синтетических волокон,
искусственных кож, бумаги и др.
Для инженерно-технических и научных работников лакокрасочной и
других отраслей промышленности, связанных с применением пигментов.
Может быть полезна студентам химико-технологических вузов.
Кочкин В. Ф., Коваленко Д. Г., Сакар А. Г. Защитно-декора-
тивные покрытия радиоэлектронных средств.— СПб.: Химия,
1992 — 12 л. 1 р. 60 к.
Рассмотрены особенности защиты и отделки радиоэлектронных средств.
Приведена технология получения лакокрасочных и гальванических по-
крытий. Проанализирована зависимость свойств покрытий от структуры и
свойств применяемых для них материалов. Описано оборудование для по-
лучения покрытий. Дана экономическая и экологическая оценка исполь-
зуемых процессов и материалов. Рассмотрены новые материалы для по-
лучения защитно-декоративных покрытий.
Для инженерно-технических работников, связанных с применением
лакокрасочных и гальванических покрытий в радиоэлектронной и других
отраслях промышленности. Может быть полезна специалистам, занимаю-
щимся созданием материалов для покрытий, а также преподавателям и
студентам химико-технологических вузов.
Горловский И. А., Козулин Н. А., Евтюков Н. 3. Оборудова-
ние заводов лакокрасочной промышленности: Учеб, пособие
для вузов.— 4-е изд., перераб. и доп.— СПб.: Химия, 1992.—
25 л. (в пер.): 4 р. 20 к.
Описано основное технологическое оборудование, применяемое на за-
водах лакокрасочной промышленности при производстве пленкообразова-
телей, пигментов и пигментированных лакокрасочных материалов с обос-
нованием выбора оптимальных типов машин и аппаратов. Четвертое из-
дание (3-е изд.— 1980 г.) переработано и дополнено сведениями о новых
видах оборудования. Рассмотрены пути интенсификации работы машин и
аппаратов.
Для студентов химико-технологических вузов. Может быть полезна
инженерно-техническим работникам проектных организаций и предприя-
тий лакокрасочной и других отраслей промышленности.
ИЗДАТЕЛЬСТВО «ХИМИЯ»
ГОТОВИТСЯ К ВЫПУСКУ
Исидоров В. А. Органическая химия атмосферы.— 2-е изд.,
ререраб: и доп.— СПб.: Химия, 1992,—-20 л. (в пер.): 4 р. 30 к.
Во втором издании (1-е изд.— 1985 г.) обобщены новейшие’сведения
по источникам и составу органических компонентов земной атмосферы, их
химическим и фотохимическим превращениям. Введены данные о фотости-
мулированных реакциях с участием аэрозольных частиц. 'Более детально
, изложены современные методы количественного анализа органических
соединений, присутствующих в атмосфере й следовых количествах.
Для специалистов по экологической и санитарной химии, химиков-
аналитиков и геофизиков. Может быть полезна студентам и аспирантам
вузов.
Вредные химические вещества. Кислород- и галогенсодержа-
щие органические соединения: Справ. изд./А. Л. Бандман,
Ю. С. Каган, Н. В. Кокшарева и др.; Под ред. В. А. Филова,
Л. А. Тиунова и А. А. Потехина.— СПб.:- Химия, 1992.— 42 л.
(в пер.): 6 р. 90 к.
В продолжающемся справочном издании содержатся сведения о га-
логенпроизводных ароматических углеводородов (материал дополняет
вышедший в 1990 г. справочник «Углеводороды. Галогенпроизводные уг-
леводородов») и о кислородсодержащих органических соединениях, за-
грязняющих окружающую и производственную среду. Приведены данные
об их свойствах, получении и применении, источниках' их поступления в
окружающую среду, содержании в почве, воздухе, воде, продуктах пита-
ния и т. д. Даны предельно допустимые уровни в разных средах, санитарно-
гигиеническая и токсикологическая характеристики, меры профилактики
и защиты.
Для химиков разного профиля, токсикологов, врачей, работников са-
нитарно-гигиенических служб и лиц, ответственных за технику безопасно-
сти и охрану окружающей среды.
Вредные химические вещества. Азотсодержащие органиче-
ские соединения: Справ. изд./Л. А. Базарова, Э. Л. Балабанова,
А. Л. Бандман и др.; Под ред. Б. А. Курляндского, В. А. Филова,
Б. А. Ивина.— СПб.: Химия,. 1992.— 40 л. (в пер.): 2 р. 30 к.
ИЗДАТЕЛЬСТВО «ХИМИЯ»
ГОТОВИТСЯ К ВЫПУСКУ
Справочник инженера-химика. (Справочник Перри): Справ.
изд./Под ред. Р. Перри и Д. Грина; Пер. с англ, под ред. В. С. Бес-
кова.— СПб.: Химия, 1992.— 100 л.— Пер. изд.: США, 1984 (в
пер.) :15 р.
В первой книге перевода 6-го американского издания фундаменталь-
ного справочника представлены необходимый математический аппарат в
применении к химической технологии и данные по физико-химическим
свойствам вешеств. Впервые в справочной химической литературе приво-
дится обширный раздел по кинетике химических реакций и проектирова-
нию химических реакторов. На русском языке справочник, обобщающий
достижения в области химической технологии за последние двадцать лет,
будет издан в 1992—1994 гг. в четырех книгах. В них рассмотрены типовые
процессы химической технологии, проблемы энергоресурсов, экологии,
экономики, биотехнологии; приведены примеры расчетов и проектирова-
ния технологических систем.
Для широкого круга инженерно-технических и научных работников
химической и смежных с ней отраслей промышленности. Может быть по-
лезен преподавателям, аспирантам и студентам вузов.
Рабинович В. А., Хавин 3. Я. Краткий химический справоч-
ник: Справ. изд./Под ред. А. А. Потехина и А. И. Ефимова.—
3-е изд. перераб. и доп.— СПб.: Химия, 1992.— 33 л. (в пер.):
5 р. 100 000 экз.
Приведены данные о физических и термодинамических свойствах
свыше 1800 веществ, сведения по химическому равновесию, свойствам
растворов, электрохимии, строению веществ и т. д. В 3-е издании (2-е изд.—
1978 г.) включены новые материалы, в частности основы современной но-
менклатуры, характеристика природных полимеров, сведения по лабора-
торной технике и др. Ряд данных уточнен и обновлен.
Может служить настольным справочным пособием для научных и
инженерно-технических работников, преподавателей и студентов вузов,
учащихся техникумов и старших классов средней школы.
Книги можно приобрести в местных магазинах, распространяющих научно-
техническую литературу.
В случае отказа обращайтесь по адресам:
103031, Москва, ул. Петровка, 15, магазин № 8 «Техническая книга»;
198147, Санкт-Петербург, Московский пр., 54, магазин № 21 «Книги по химии».
Заказ будет выслан наложенным платежом.