/
















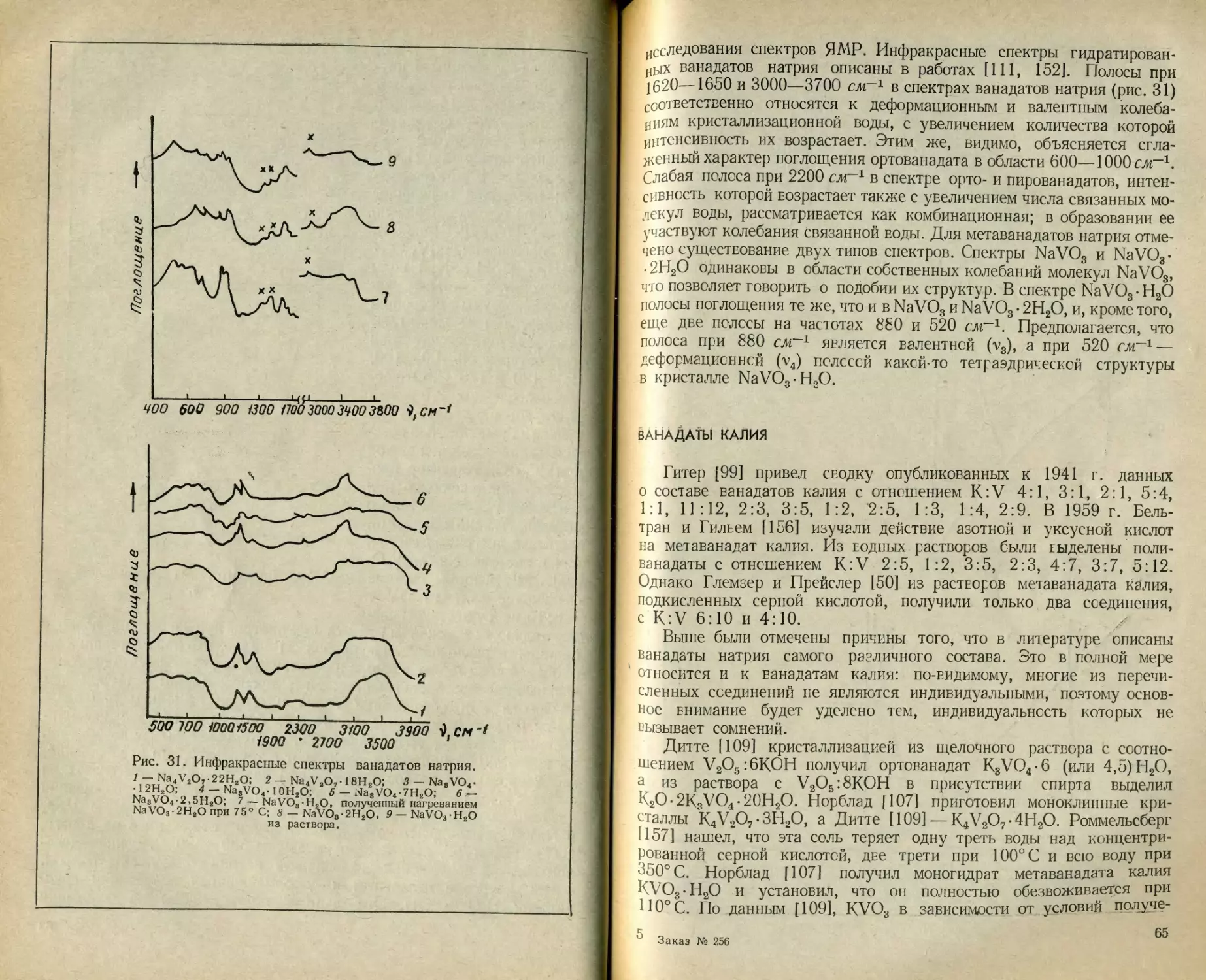

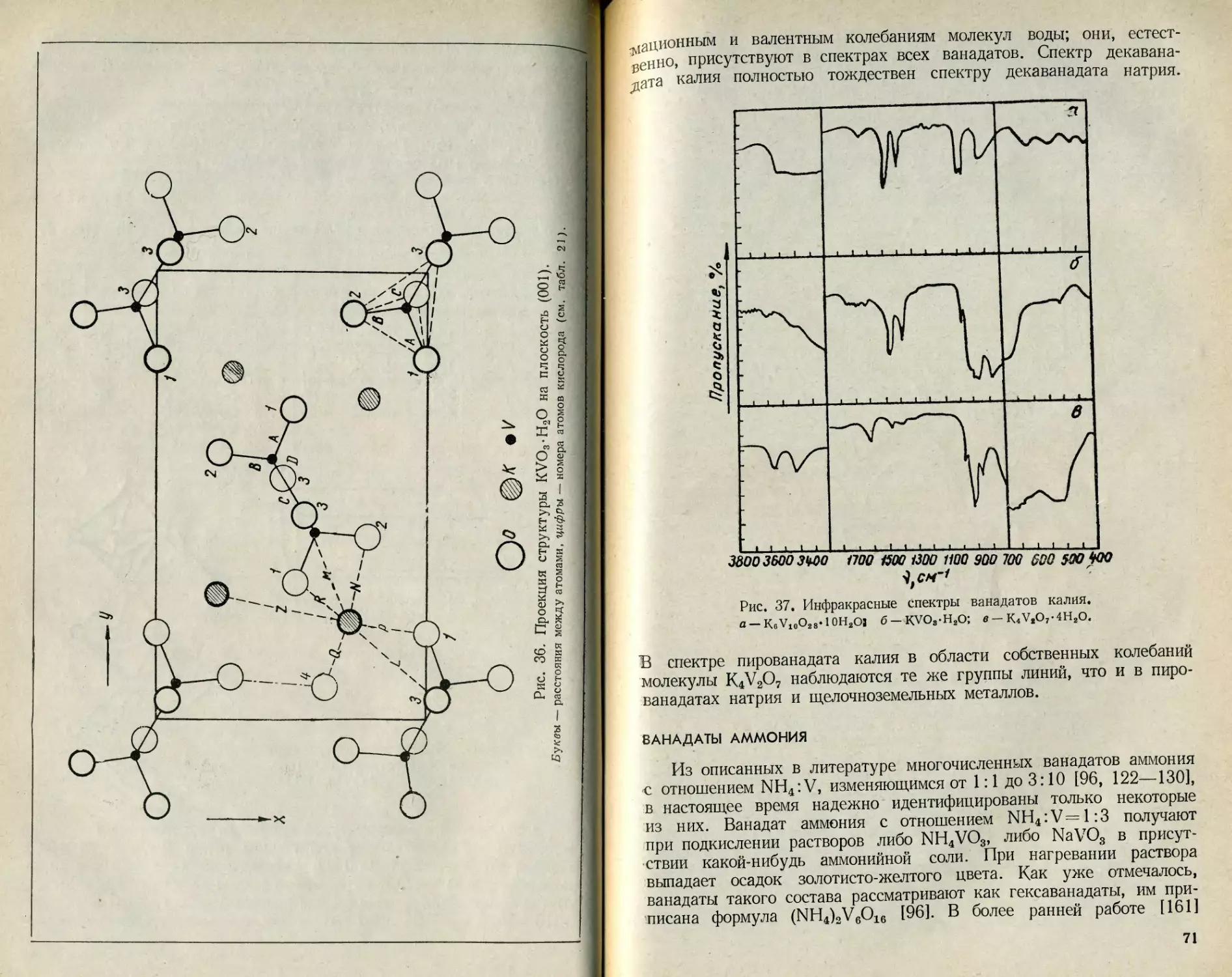


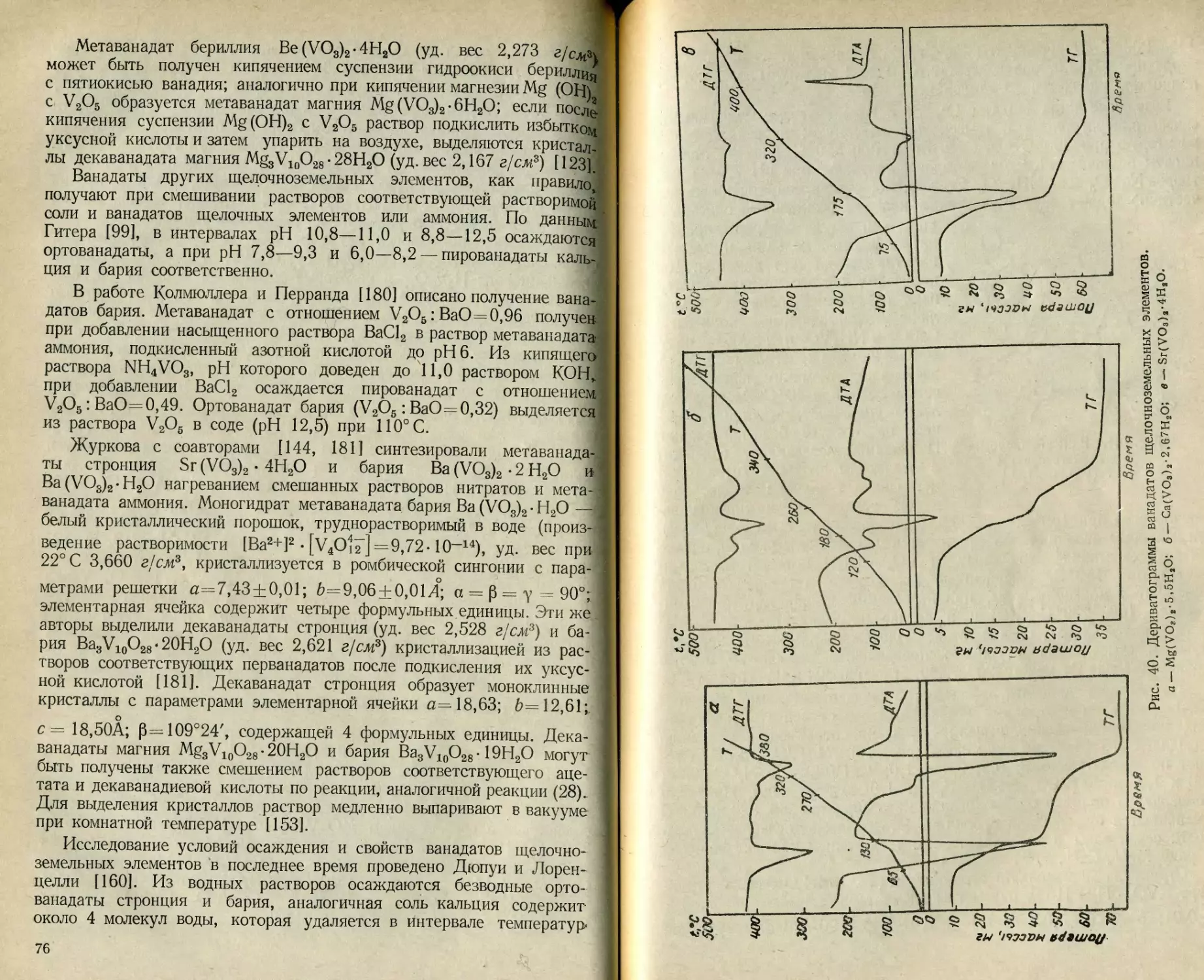

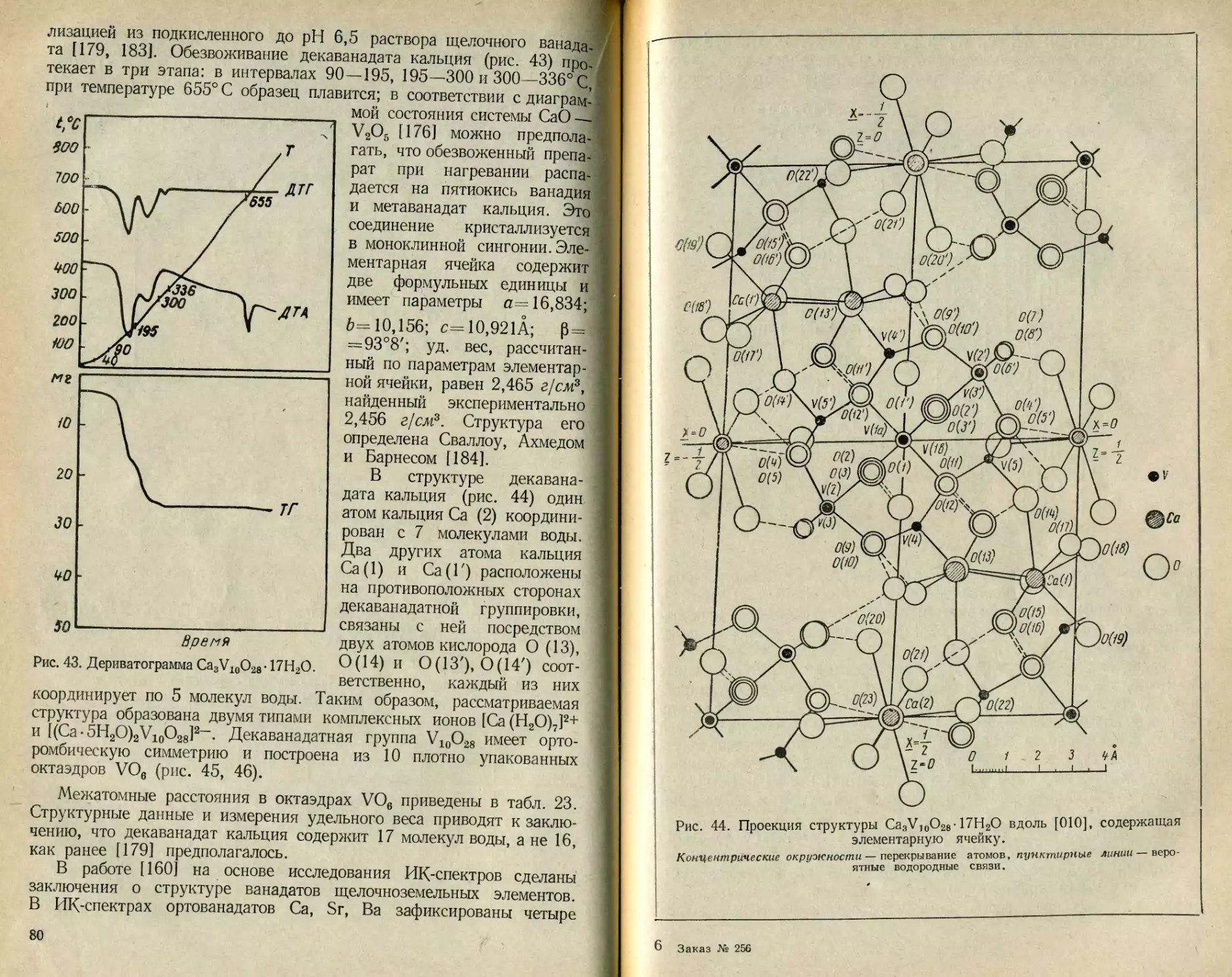
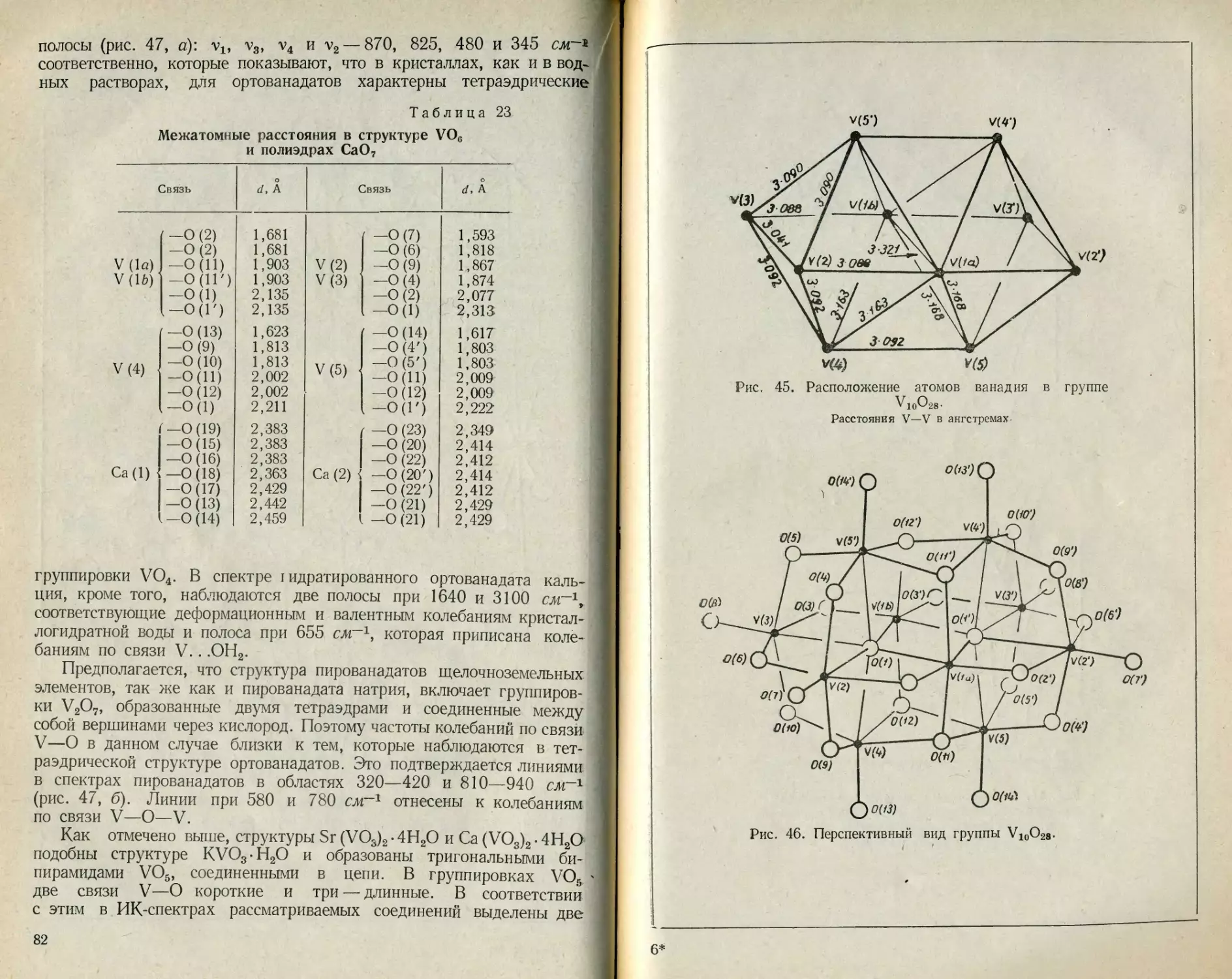
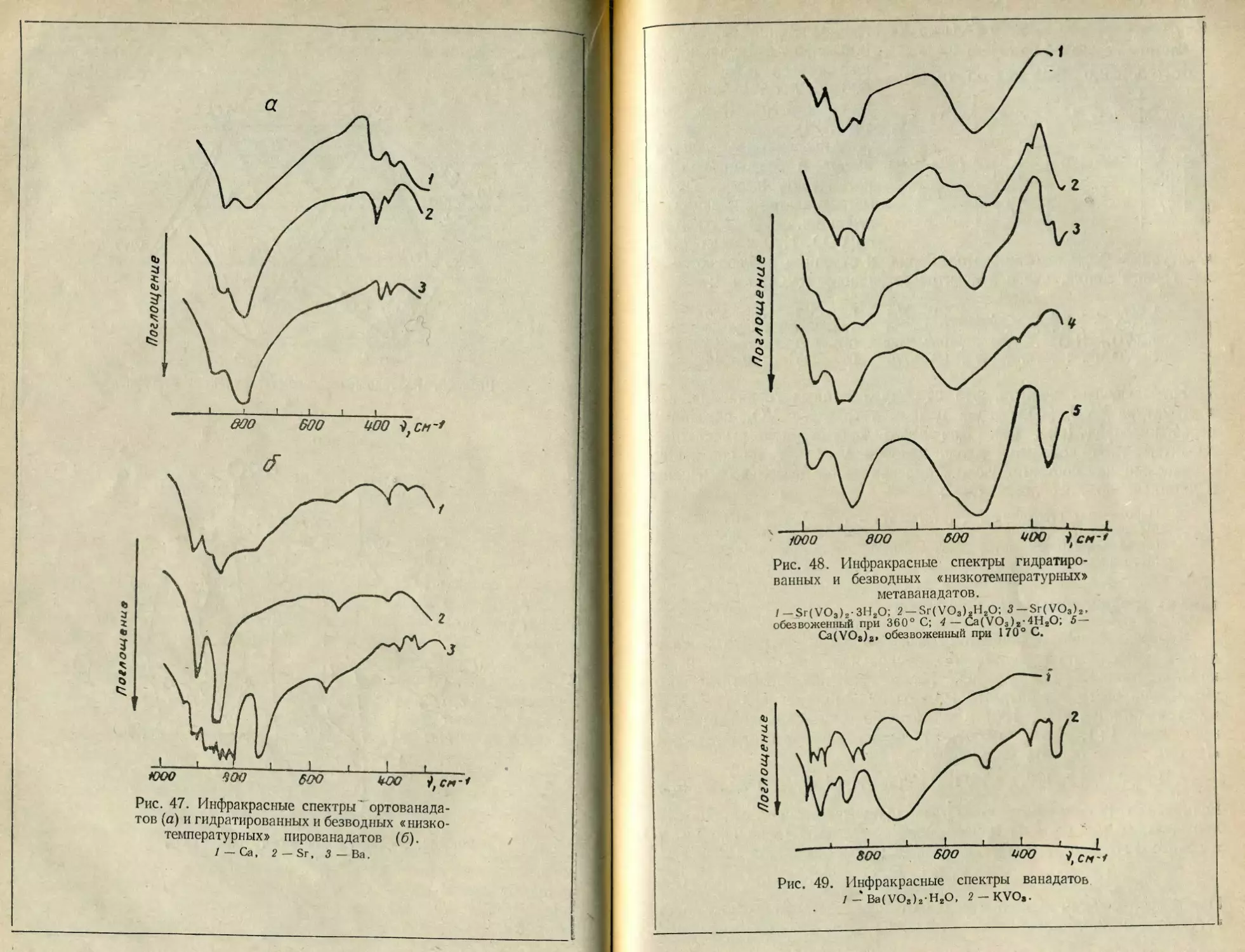

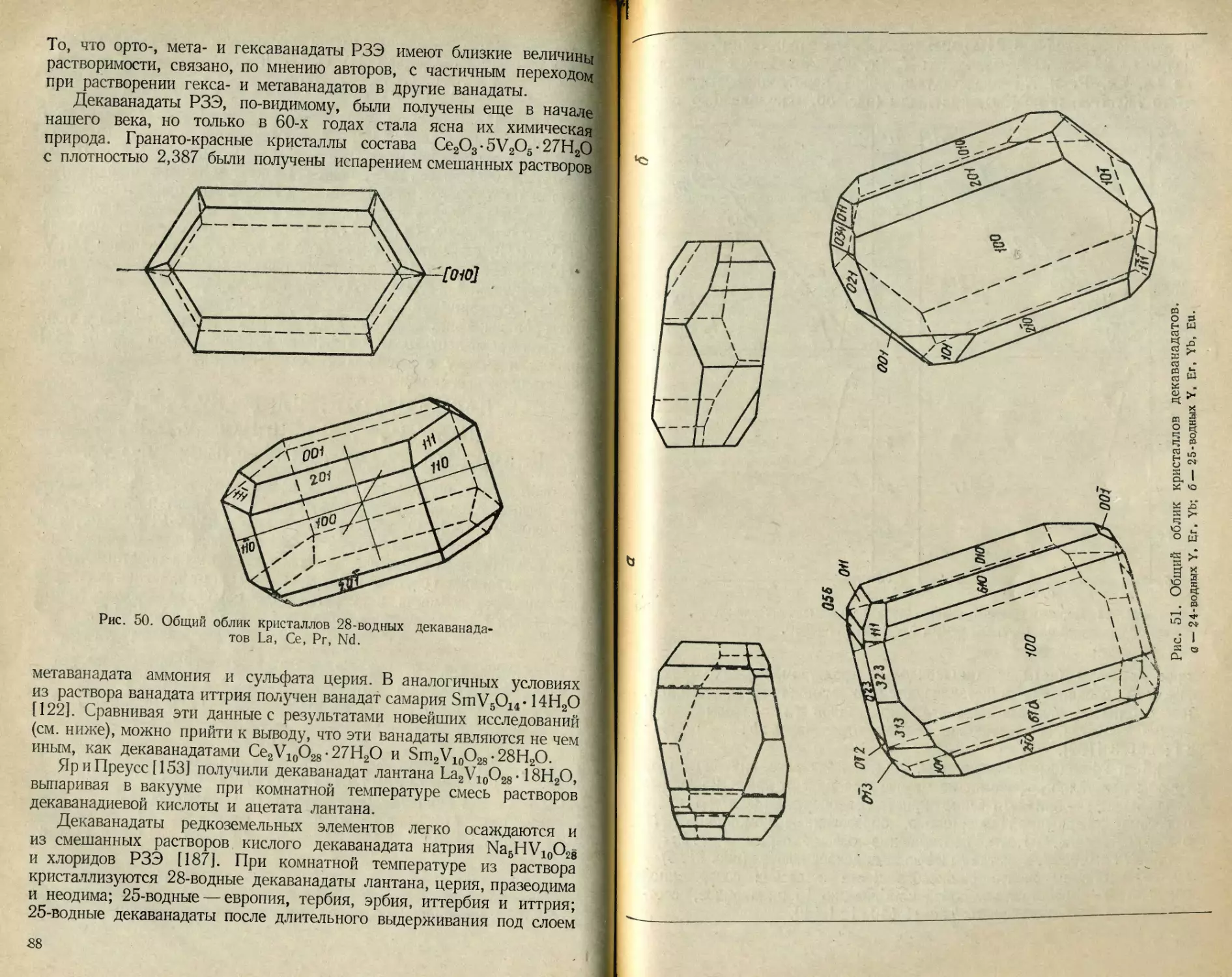
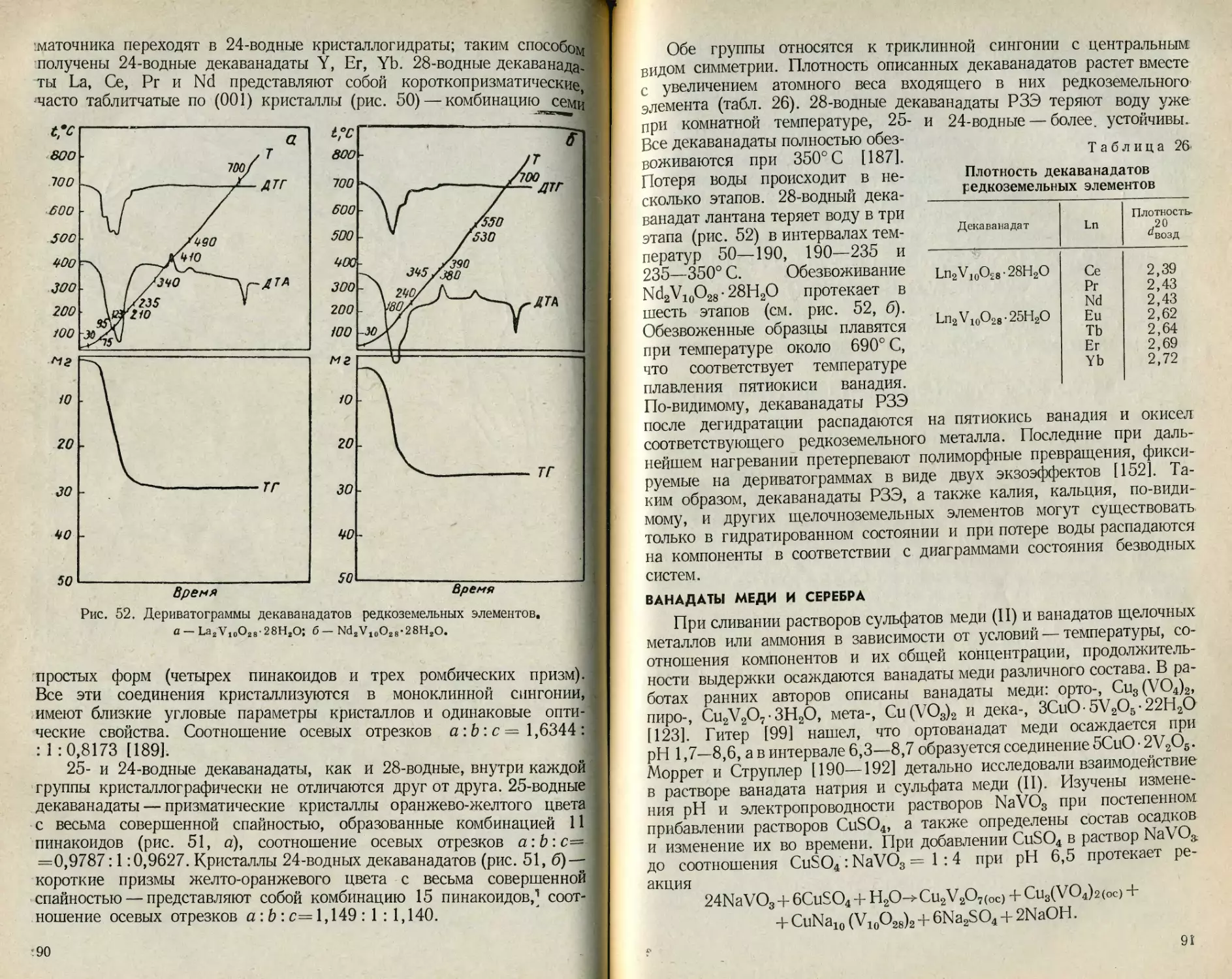



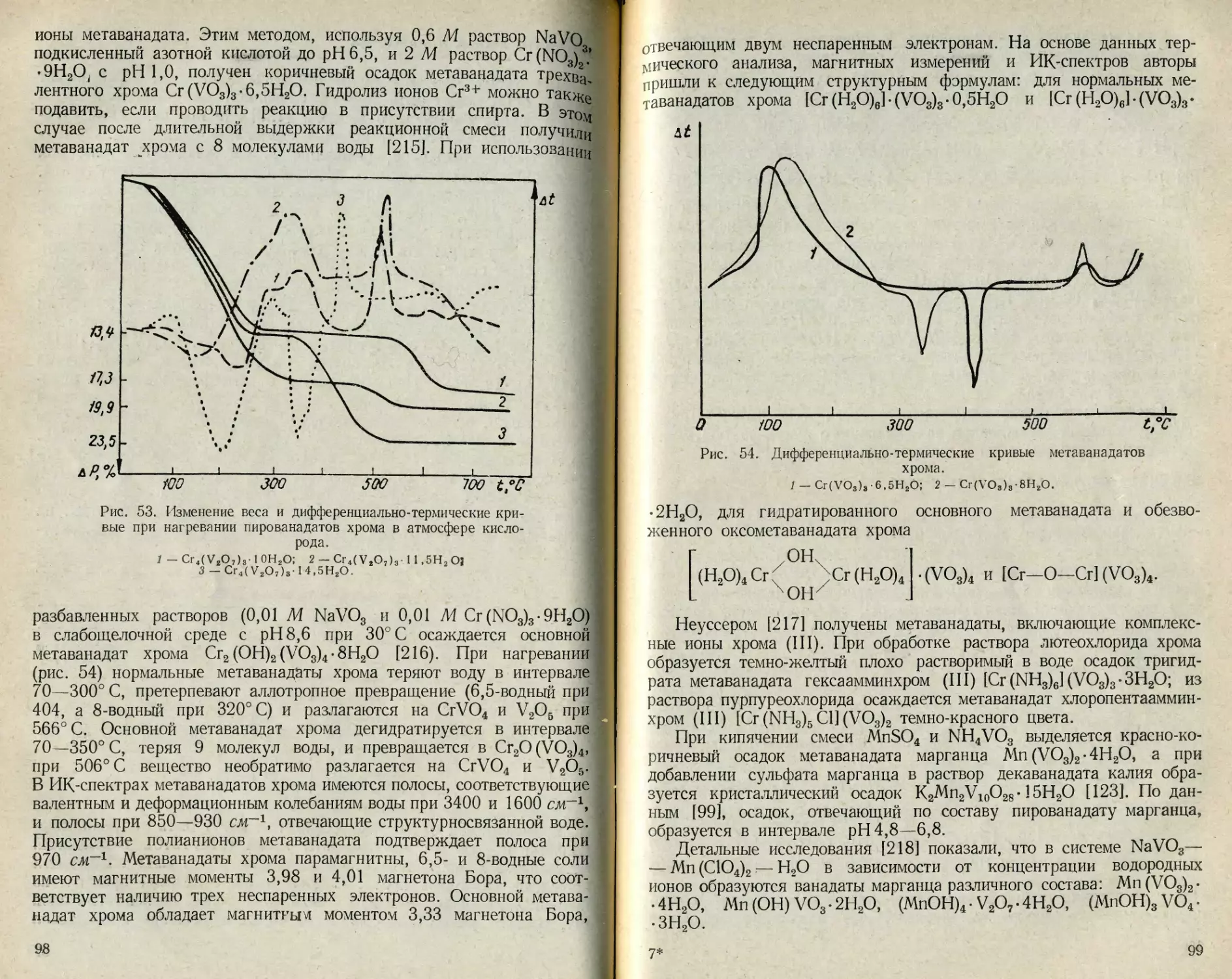

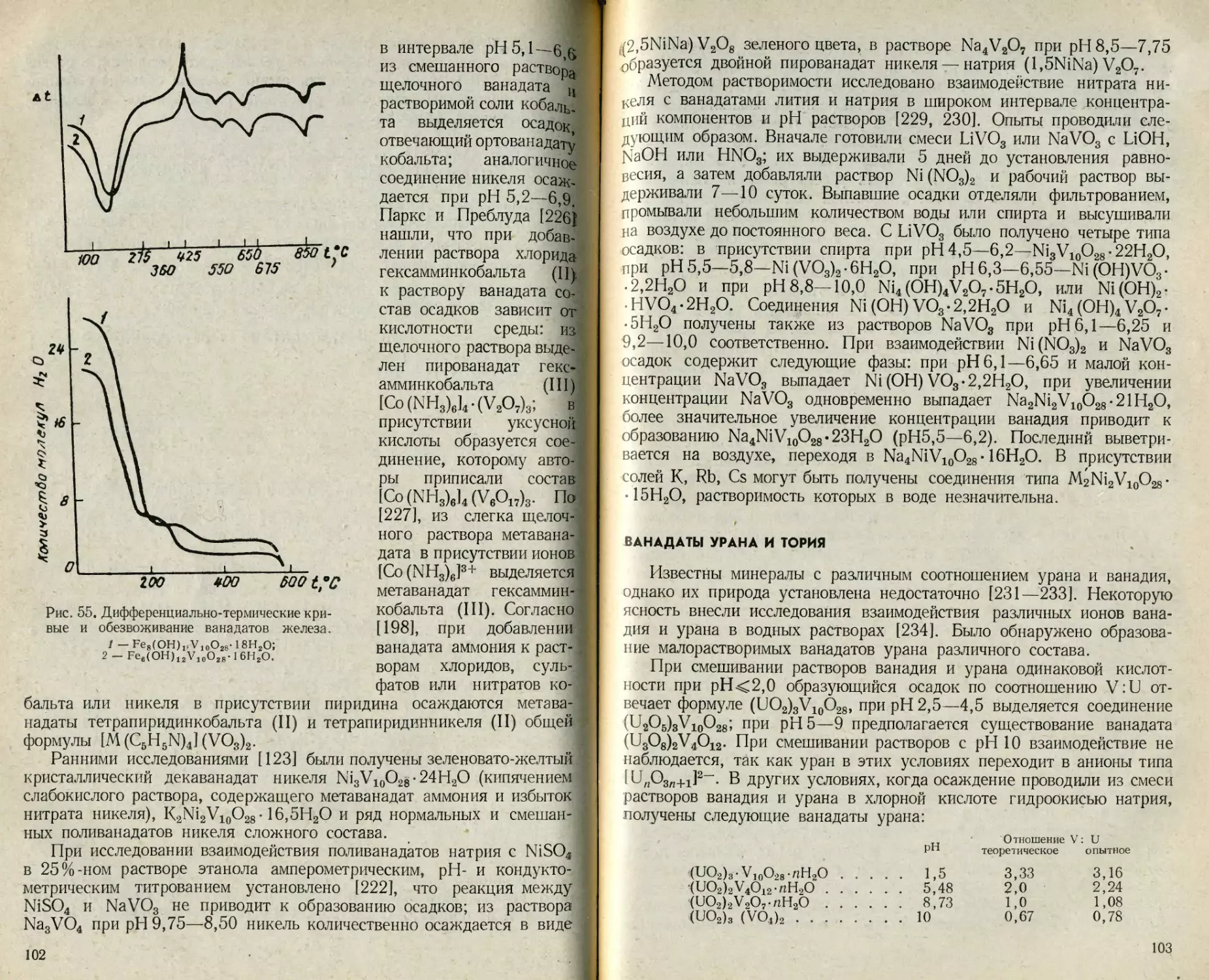


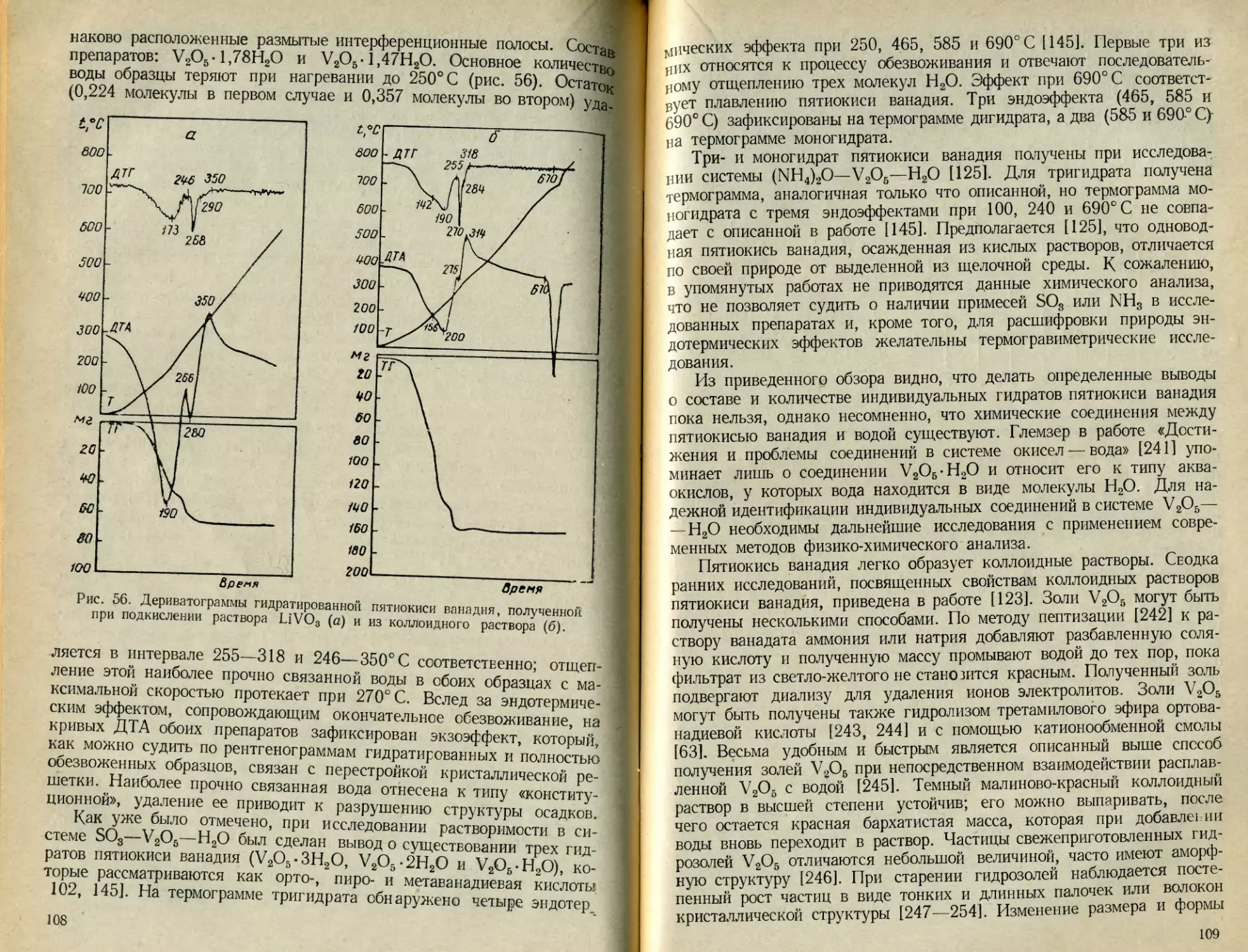



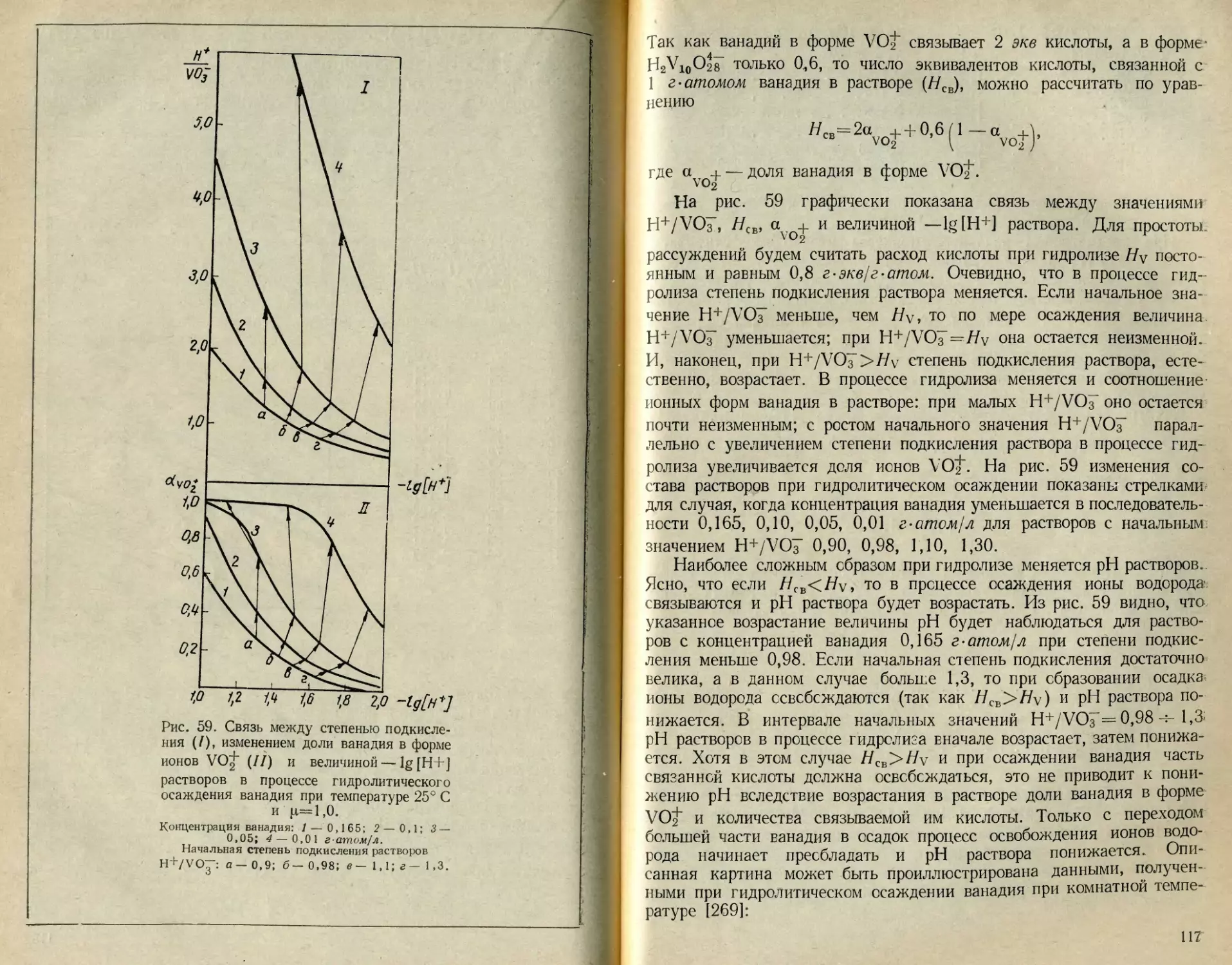
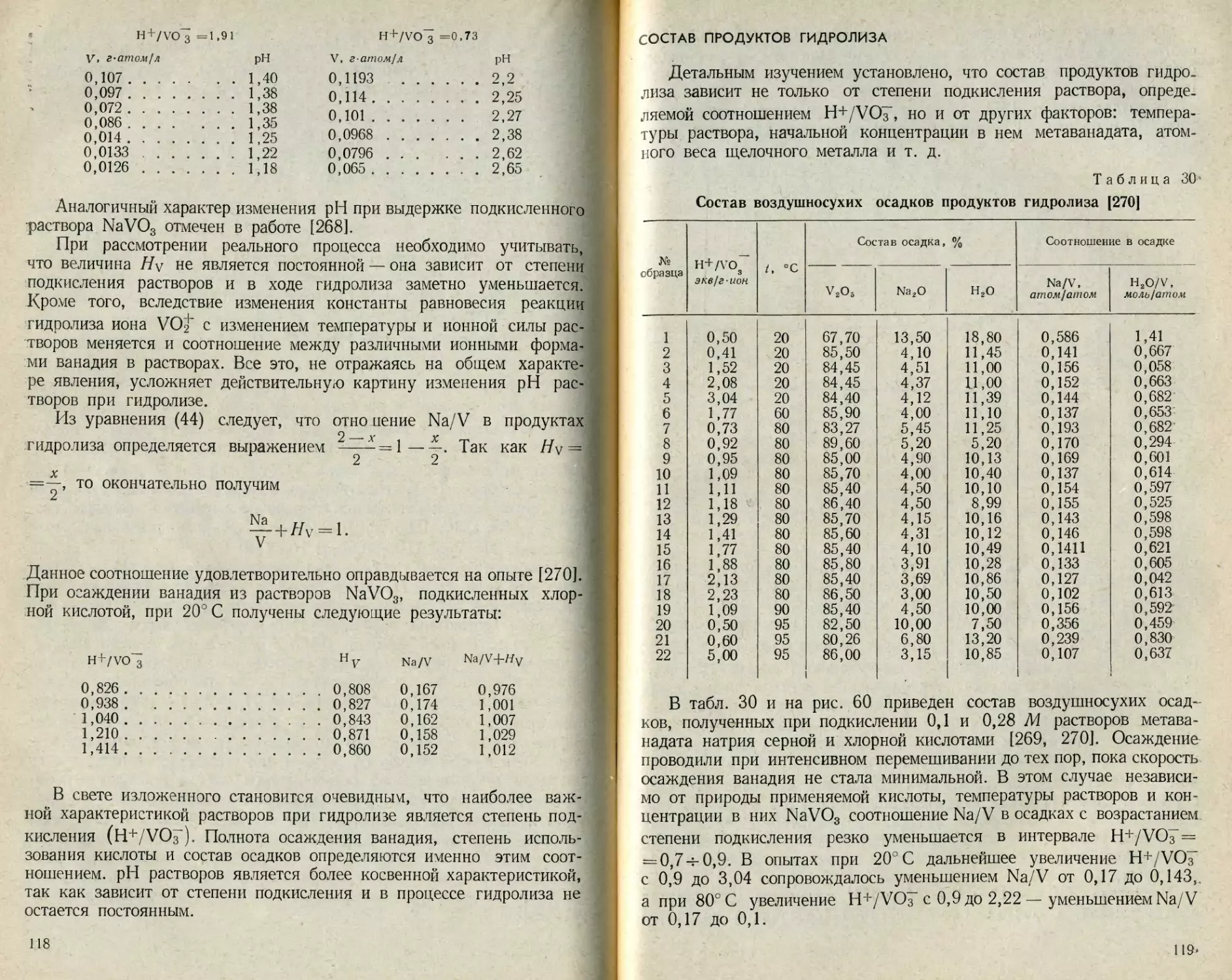
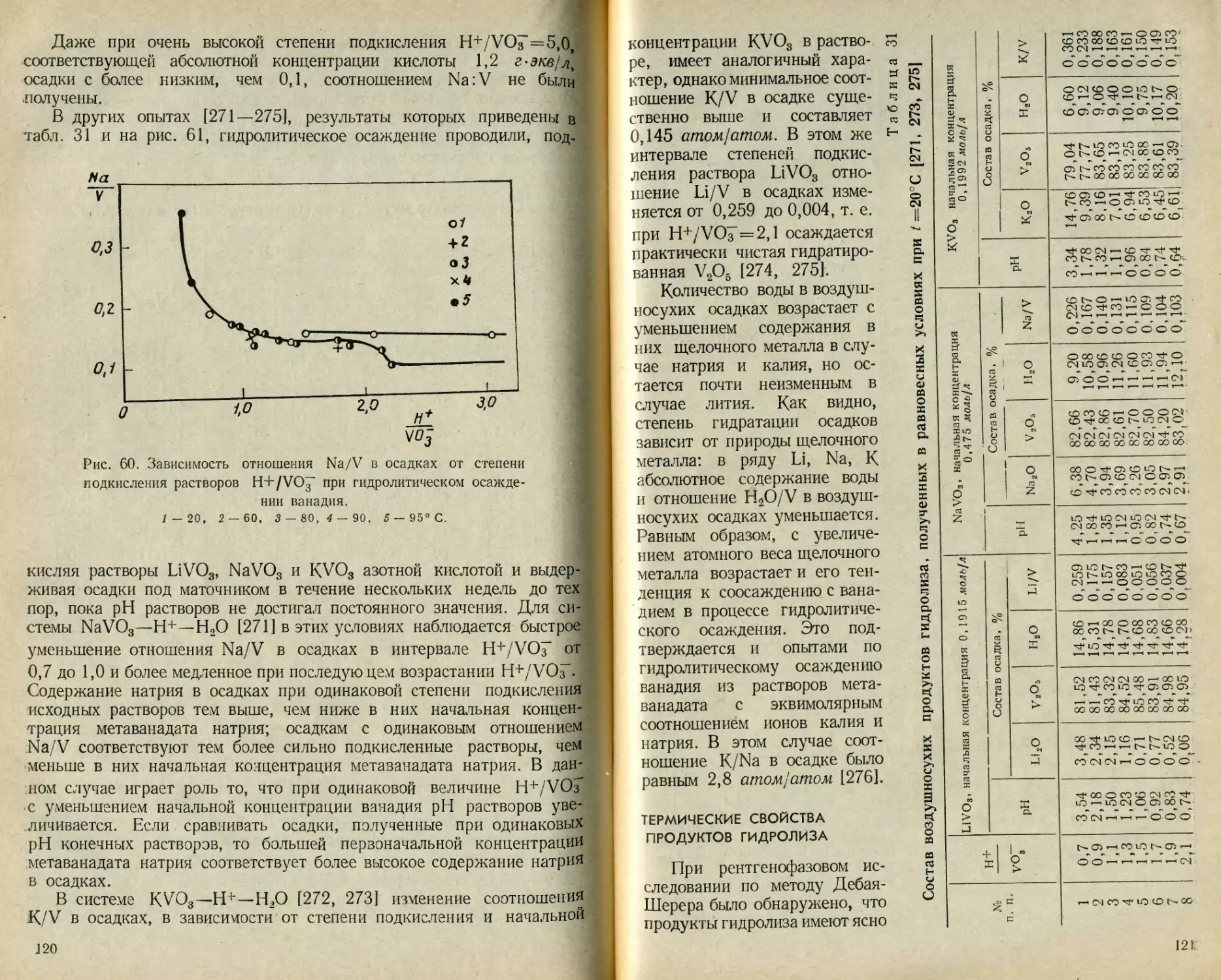
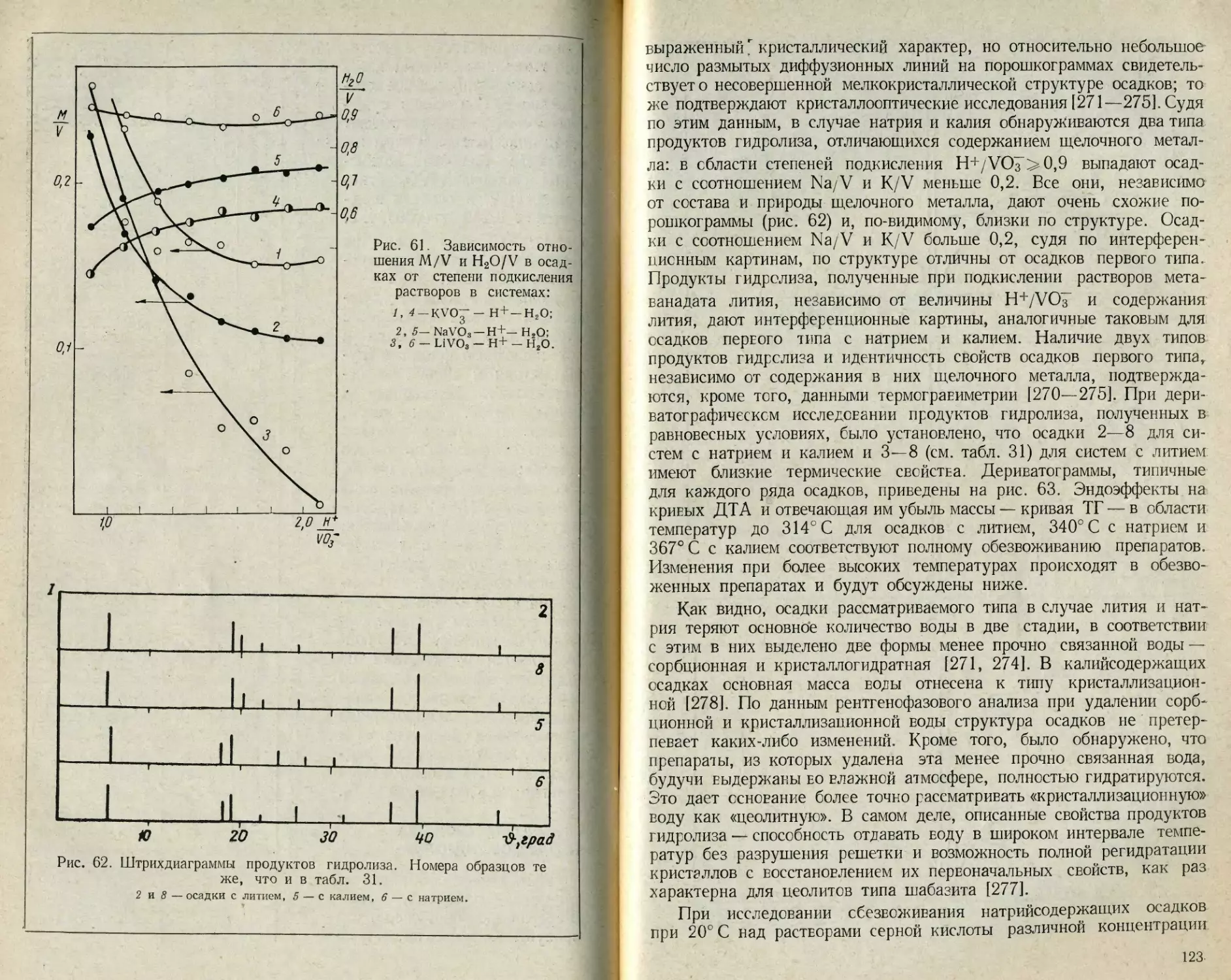
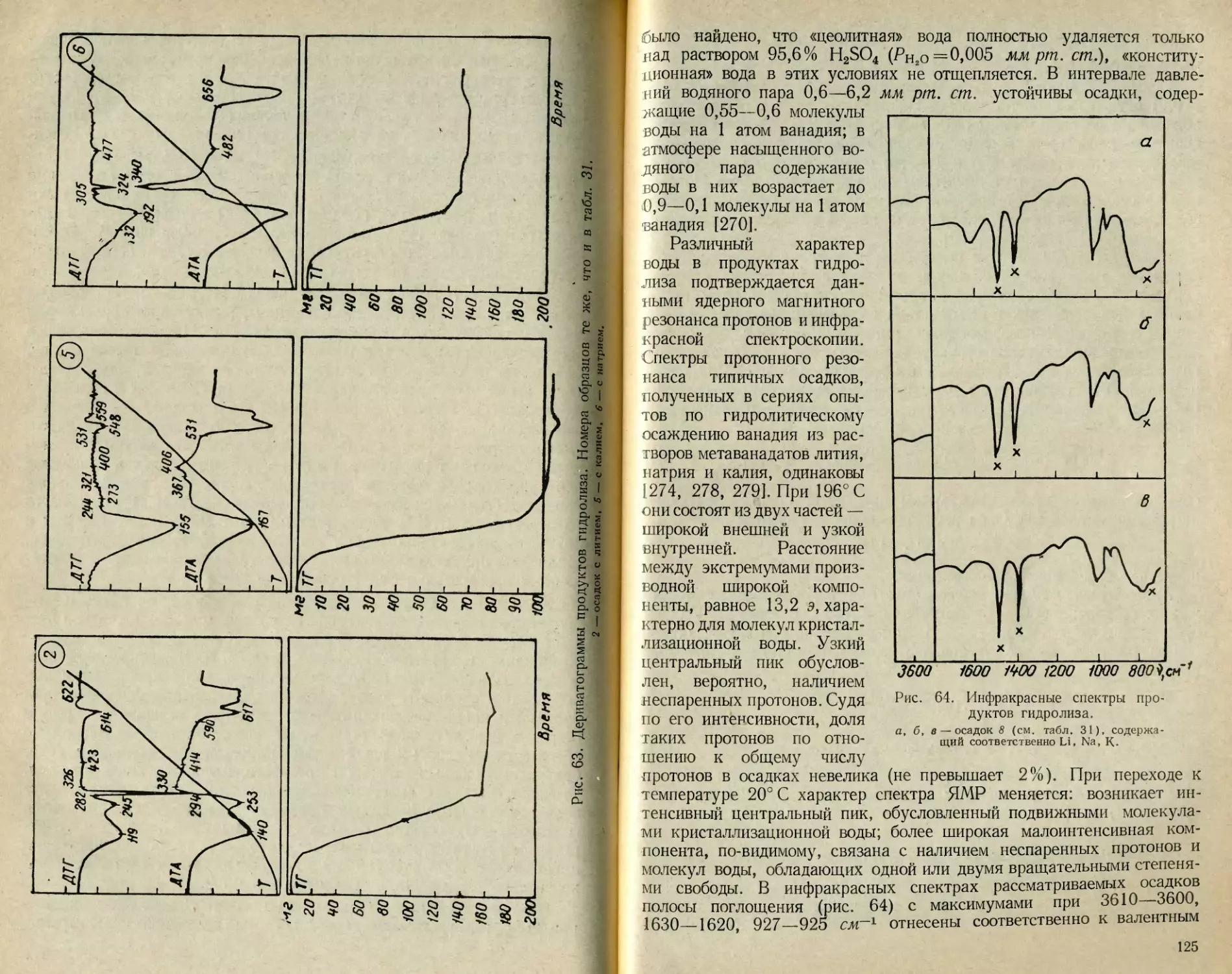

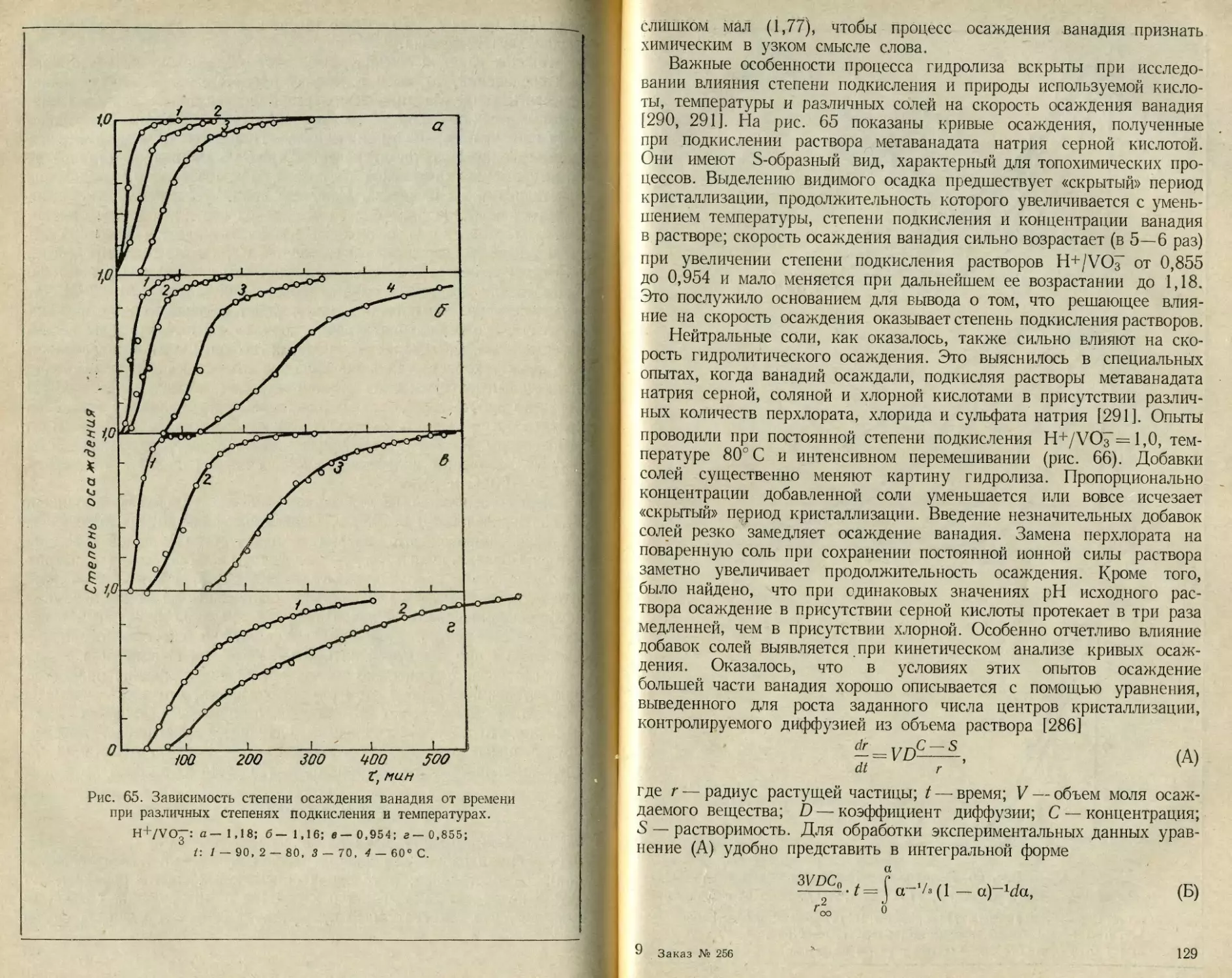
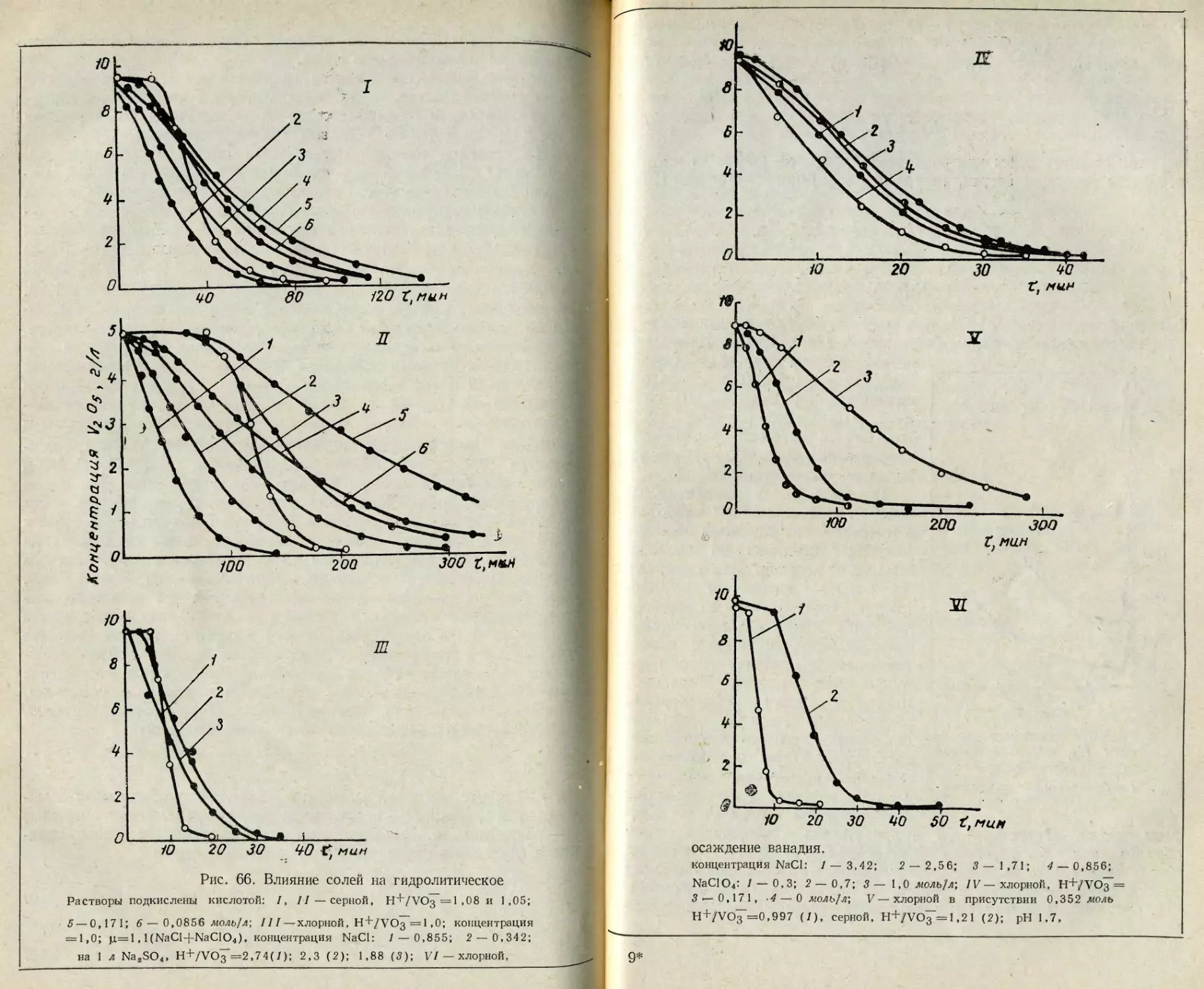








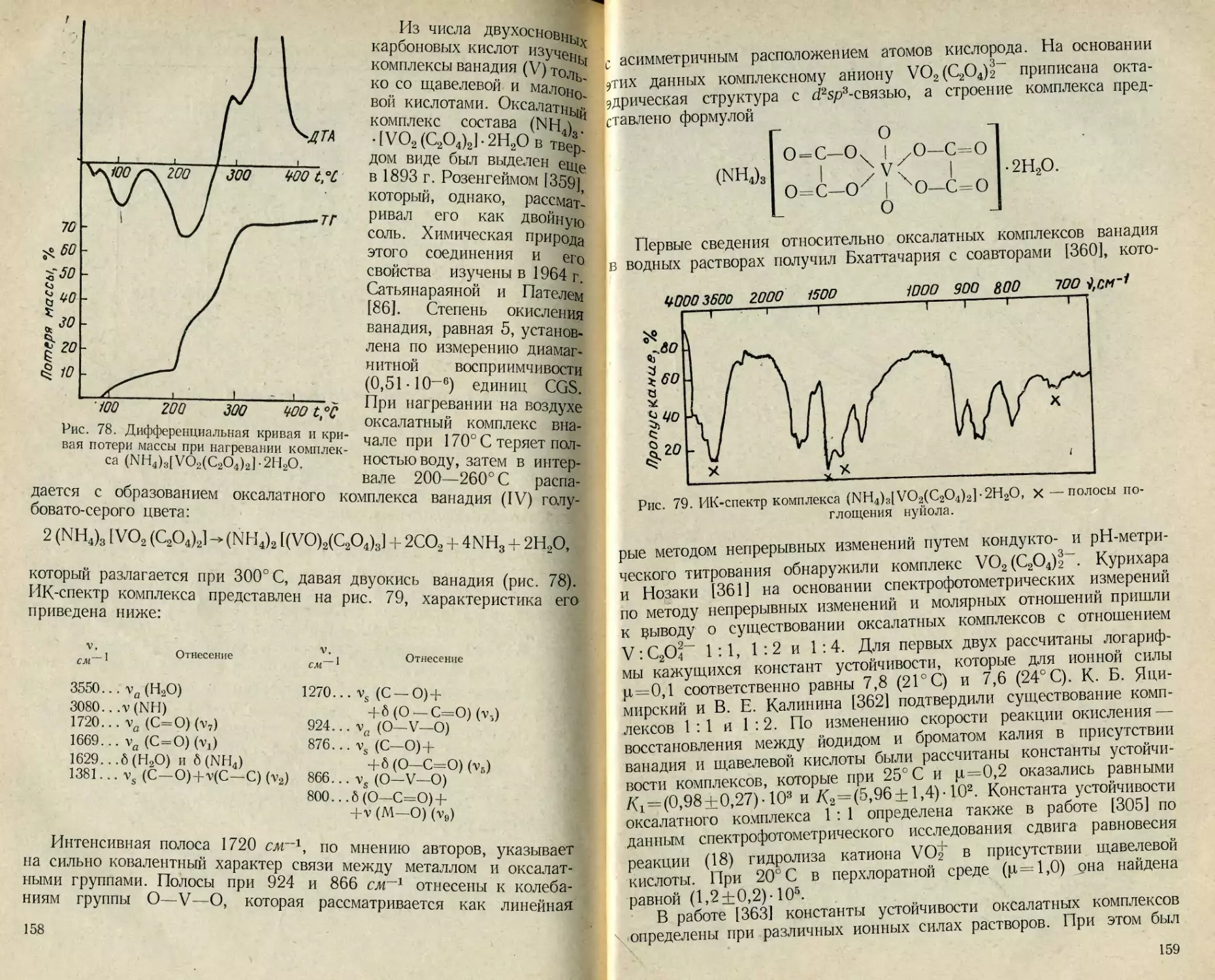
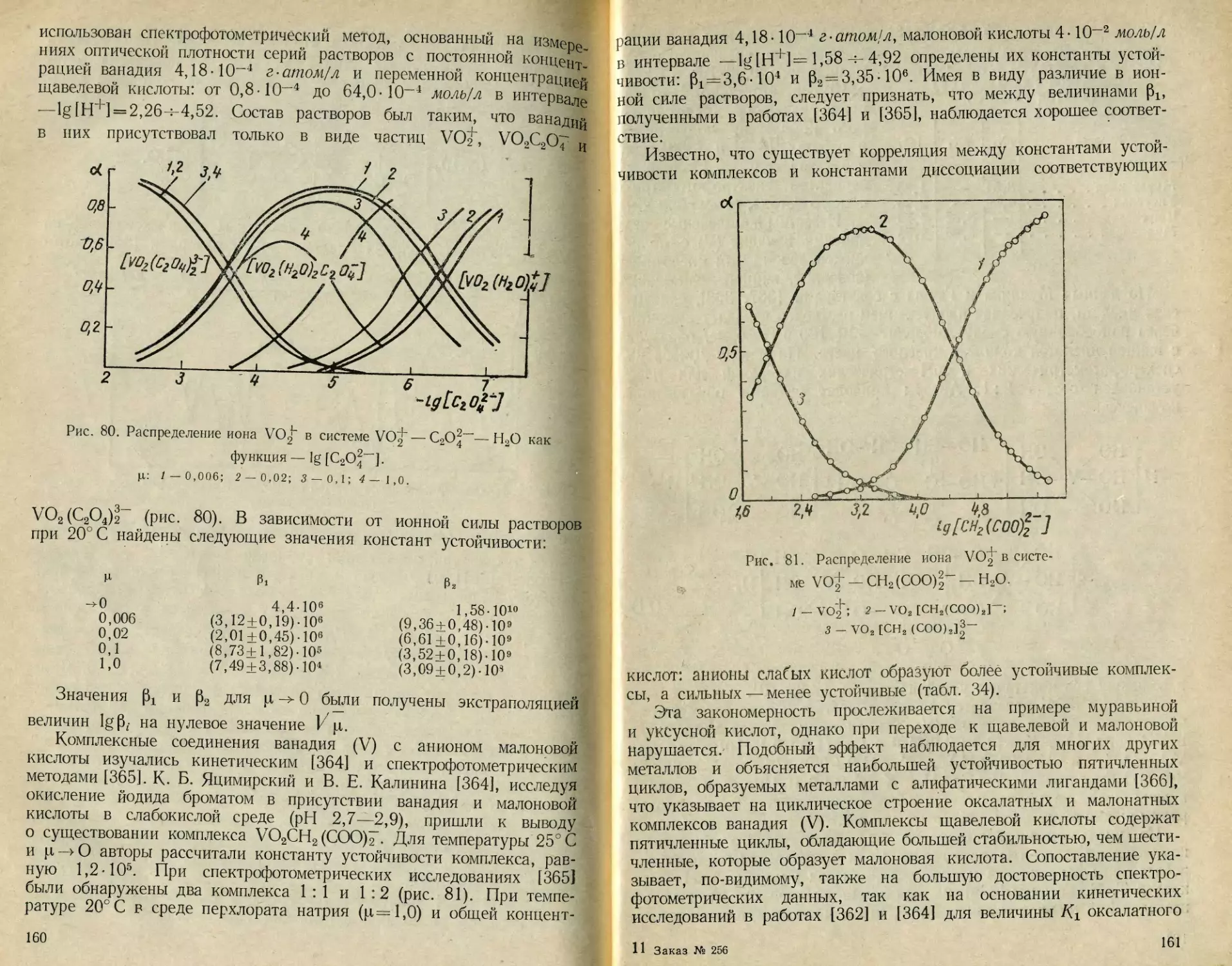

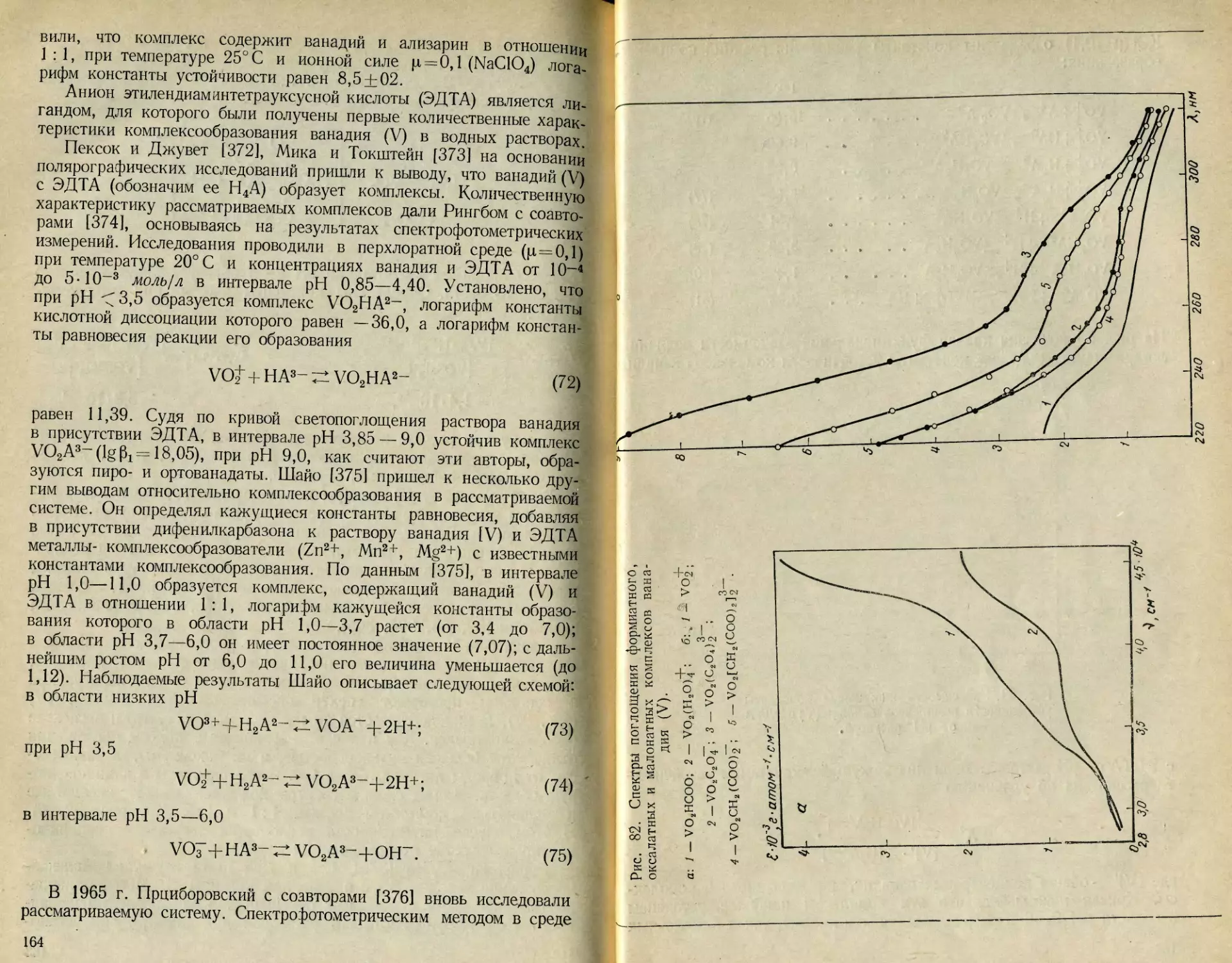















Текст
* АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ НАУЧНЫЙ ЦЕНТР
хими
ПЯТИВАЛЕНТНОГО
ВАНАДИЯ В ВОДНЫХ
РАСТВОРАХ
СВЕРДЛОВСК, 1971
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ НАУЧНЫЙ ЦЕНТР
труды института химии
1971
химия
ПЯТИВАЛЕНТНОГО
ВАНАДИЯ В ВОДНЫХ
РАСТВОРАХ
СВЕРДЛОВСК
УДК 546.881.5—145.2
Настоящая работа представляет собой первую попытку си-
стематизации обширного материала по химии пятивалентного
ванадия в водных растворах. В ней освещены: ионное состоя-
ние ванадия (V) в водных растворах; гетерогенные рав-
новесия в водно-солевых системах, включающих соединения
ванадия; получение ванадатов различных металлов методами
кристаллизации и осаждения. Значительное внимание уделено
рассмотрению закономерностей гидролитического осаждения
ванадия и природе продуктов гидролиза. Детально описаны
комплексные соединения ванадия (V).
В основу работы положены результаты оригинальных ис-
следований по химии пятивалентного ванадия, выполненных в
лаборатории окисных систем Института химии Уральского
научного центра АН СССР под руководством авторов данной
книги А. А. Ивакина и А. А. Фотиева.
Книга рассчитана на работников вузов, научно-исследова-
тельских учреждений и промышленных предприятий.
Печатается по постановлению
Редакционно-издательского совета
Уральского научного центра АН СССР
Ответственный редактор Г. П. Швейкин
ВВЕДЕНИЕ
Химия ванадия богата и разнообразна: она включает соединения
всех степеней окисления от —1 до +5. Настоящая работа
Касается только соединений пятивалентного ванадия в водных рас-
творах, которые играют особенно важную роль среди большого числа
образуемых им соединений. Все наиболее широко известные
ванадийсодержащие реактивы, такие как пятиокись ванадия, вана-
даты аммония, щелочноземельных и других элементов, являются
соединениями пятивалентного ванадия. Легкость получения, хорошая
растворимость, удобство в обращении делают их незаменимыми при
изучении химии ванадия. Значительную роль соединения пятива-
лентного ванадия играют в технологии: независимо от исходного
сырья и конечного продукта практически все методы переработки,
очистки и получения соединений ванадия включают стадии, связан-
ные с переводом ванадия в пятивалентном состоянии в раствор
и последующим его осаждением.
Процессы получения технической пятиокиси ванадия очень похожи
независимо от исходной руды [1, 2]. Измельченную руду смешивают
с различными натрийсодержащими солями — NaCl, Na2SO4, Na2CO3—
и обжигают при температурах 850—1100° С. Во время обжига
окислы ванадия превращаются в растворимые в воде соединения.
Обожженную шихту последовательно обрабатывают водой и
5—6%-ным раствором серной кислоты. Полученные растворы смеши-
вают и при pH 1,5—2,0 гидролизом выделяют техническую пяти-
окись ванадия, основная масса которой после плавления использу-
ется для производства феррованадия, а остальная часть — для полу-
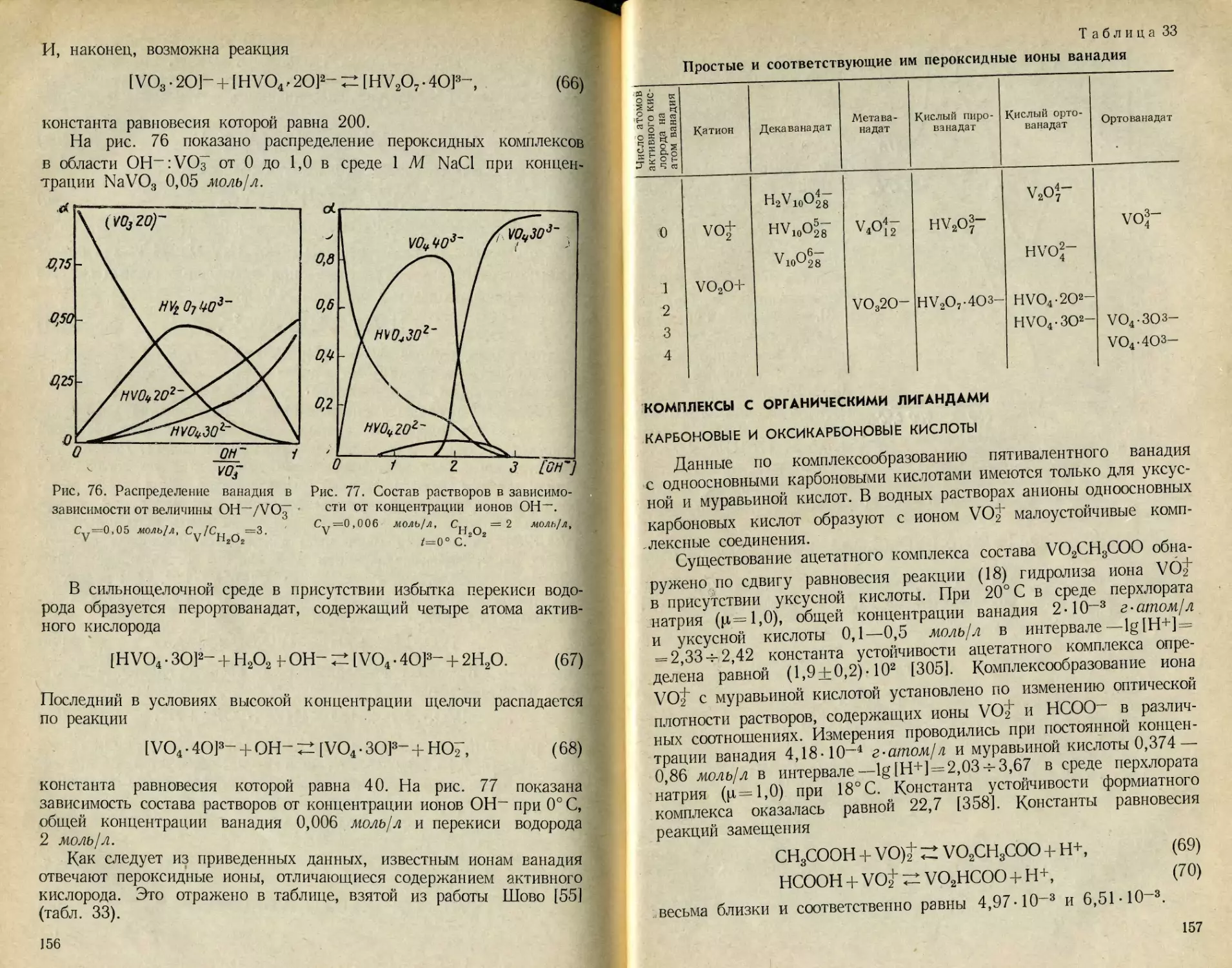
чения чистой пятиокиси ванадия и других соединений.
При получении чистых соединений ванадия техническую пяти-
окись обрабатывают крепким раствором едкого натра и по реакции
обменного разложения
NaVO3 + NH4C1^NH4VO3+NaCl
из раствора выделяют ванадат аммония, который может быть допол-
нительно очищен перекристаллизацией [3, 4]. Прокаливанием на
3
воздухе из метаванадата аммония получают чистую пятиокись вана^
дия. Образование ванадатов натрия и кальция имеет важное значение
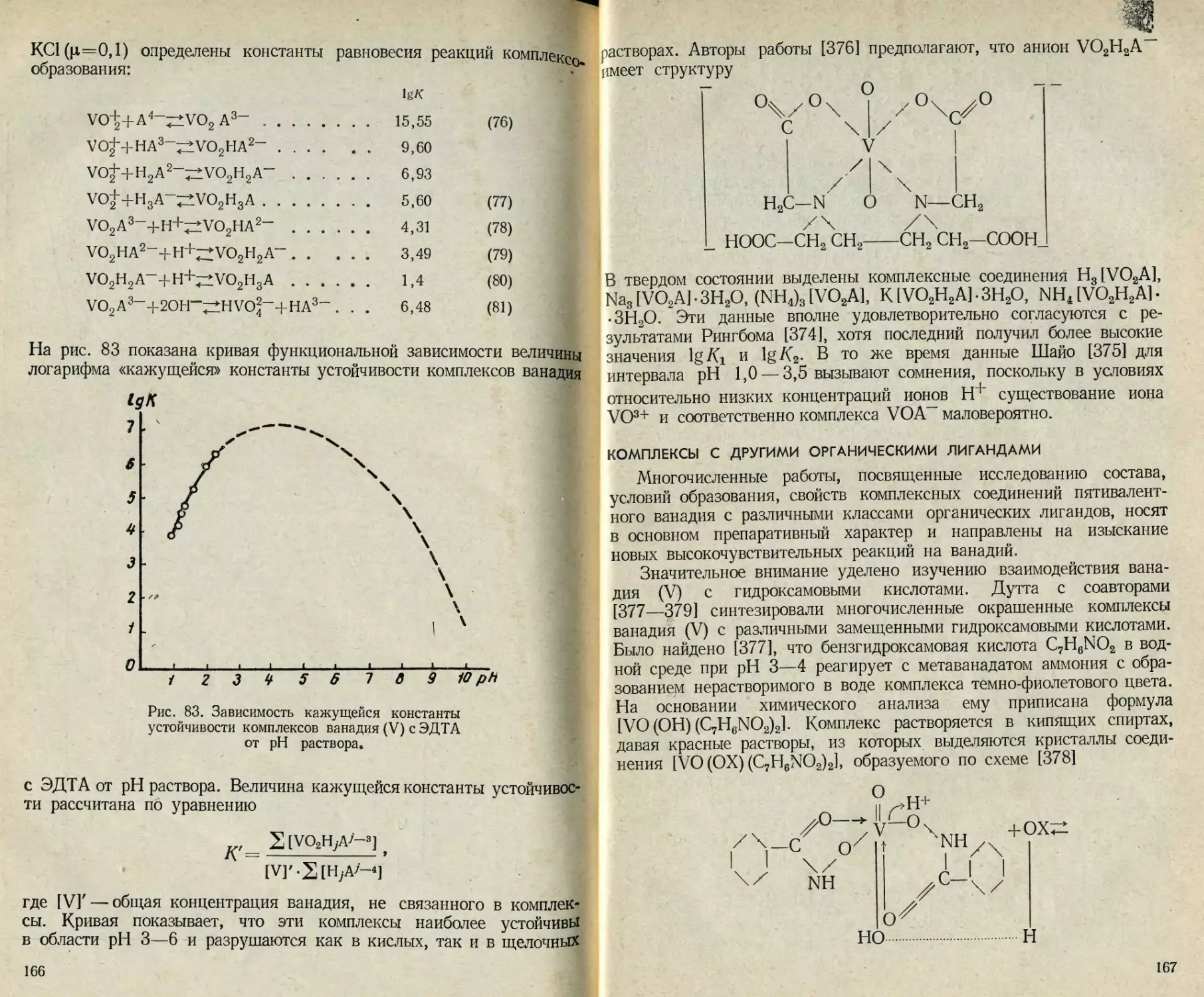
в процессах попутного извлечения ванадия из полупродуктов глино-
земного производства [5, 6].
Некоторые ванадаты могут использоваться в качестве рабочих
веществ в оптических квантовых генераторах, как люминофоры
и т. д. [7, 8]. В связи с этим к соединениям ванадия предъявляются
высокие требования по чистоте, стехиометрическому и фазовому
составу и кристаллической структуре. Между тем имеющиеся мето-
ды твердофазного синтеза ванадатов несовершенны и в ряде случаев
не удовлетворяют предъявляемым требованиям. В частности, этим
путем не удается получить ванадаты, свободные от примесей вана-
дия низших степеней окисления. Использование методов осаждения
из растворов позволяет легко решить эту задачу.
Проблема извлечения ванадия и получения его чистых соедине-
ний решается не только путем применения обычных методов хими-
ческой технологии (осаждение труднорастворимых соединений или
кристаллизация), но и благодаря все более широкому использованию
ионнообменных и экстракционных процессов [9—И].
На первых этапах развития химии ванадия (V) главное внимание
уделялось синтезу ванадатов различных элементов и выяснению-
форм нахождения ванадия в растворах. Затем необходимость созда-
ния эффективных методов анализа, выделения и очистки потребовала
тщательного исследования реакций комплексообразования, опреде-
ления состава и устойчивости образующихся соединений. Этот этап
развития химии ванадия (V) продолжается и сейчас. В самые по-
следние годы определены структуры ванадатов некоторых металлов.
Все более широкое привлечение физических методов (магнитные,
спектры поглощения в ультрафиолетовой, видимой и инфракрасной
областях, радиоспектроскопия и т. д.) к исследованию соединений
ванадия (V) позволяет глубже понять их химическую природу.
Важной лгобемялгтью пятивалентного ванадия является то, что
его состояние в водных растворах зависит от pH среды. В сильна
щелочных растворах он существует в виде моноядерной частицы —
ортованадат-иона; с уменьшением концентрации щелочи возникают
полиядерные анионные частицы; максимальная степень конденсации,
равная 10, характерна для слабокислых растворов. В достаточно
кислых растворах ванадий (V) существует в виде моноядерного
оксокатиона. Выяснению форм состояния ванадия в водных растворах
посвящены многочисленные исследования, однако до сих пор этот
вопрос далек от полного разрешения. Многообразие ионных форм
ванадия (V) в растворах приводит к образованию многочисленных
соединений с различным соотношением окислов металлов и ванадия.
И во многих случаях до сих пор не ясно, имеем ли мы дело с ин-
дивидуальными соединениями или со смесью нескольких веществ.
Ванадий в форме оксокатиона образует ряд стабильных комплексов
с разнообразными лигандами, многие из которых давно нашли прак-
тическое применение.
4
Значительный самостоятельный интерес представляет химия
оксокатиона ванадия (V). Ванадий в кислой среде является сильным
окислителем, это его свойство используется в аналитической хи-
мии [12]. Большое число работ посвящено исследованию каталити-
ческой активности ванадия (V); пятиокись ванадия — основной ком-
понент многих катализаторов, широко применяемых в промышлен-
ности. В кислой среде ванадий (V) является катализатором процессов
окисления многочисленных органических соединений [13].
Все указанное послужило основанием для написания книги,
посвященной химии пятивалентного ванадия в водных растворах.
На необходимость специального рассмотрения соединений пятивалент-
ного ванадия указывает также Селбин [14] в обзоре по химии
оксованадия (IV). Здесь не рассматриваются электрохимические
свойства системы V (V)—V (IV). Эти вопросы с достаточной полно-
той изложены в недавно вышедшей монографии А. С. Гончаренко [15].
Глава I
ИОННОЕ СОСТОЯНИЕ ВАНАДИЯ (V) В РАСТВОРАХ
Изоморфизм минералов Pb5 (VO4)3 Cl — ванадинита и Ca5(PO4)3F —
апатита, установленный Раммельсбергом [16], и синтез кристалли-
ческих ванадатов, по составу подобных соответствующим фосфатам,
послужили основанием для Раскоу [17] присвоить трем группам
ванадатов наименования ортованадата (МзУО4), пированадата
(m5v2o7) и метаванадата (M'VOg).
Эти формулы в дальнейшем использованы также для описа-
ния состава соединений ванадия в щелочных и нейтральных рас-
творах.
Первые попытки установить ионное состояние ванадия в рас-
творах были предприняты Дюльбергом [18], который использовал
метод кондуктометрического титрования. Ввиду сложности и раз-
нообразия ионных форм ванадия в растворах для решения этой
задачи в более поздних исследованиях применялись разнообразные
физико-химические методы. Наибольшее число результатов, харак-
теризующих количественные закономерности сосуществования раз-
личных ионных форм пятивалентного ванадия в растворах, получено
потенциометрическим методом, особенно с тех пор, когда для обра-
ботки экспериментальных данных стали использовать метод Силле-
на [19], основанный на гипотезе «ядро — звенья». Однако до сих
пор вопрос об ионных формах ванадия в растворах не вполне ясен.
Наибольшие разногласия вызывает состав ионов в нейтральной
области, где ванадий существует в форме метаванадата. Речь идет
о степени конденсации метаванадата, которому приписывают форму
тримера V3Og“ и тетрамера V40t7- Выводы о структуре метавана-
дат-иона во многих случаях сделаны различными авторами на осно-
ве данных, полученных при использовании одного и того же мето-
да исследования. Что касается вопроса о составе ионов ванадия
в слабокислых и кислых растворах, где долгое время господство-
вала концепция гексаванадиевой кислоты H4V6O17, то начиная
с работ Россотти [20] все исследователи единодушно пришли к вы-
воду о существовании ванадия в этих условиях в форме декавана-
дат-иона.
6
Пятивалентный ванадий, обладая амфотерными свойствами,
Б водных растворах может присутствовать и в анионной, и в кати-
онной форме; кроме того, в зависимости от pH среды и общей
концентрации ванадия в растворе меняется и степень конденсации
ионов. В сильнощелочных и сильнокислых средах, а также при
низкой концентрации ванадия в растворе (меньше 10~4 г-атом/л)
существуют мономерные формы. Максимальная степень конденсации
наблюдается в слабокислой среде, где ион ванадата содержит 10 ато-
мов ванадия.
В результате многочисленных исследований с использованием раз-
нообразных физико-химических методов получены достаточно надеж-
ные и убедительные данные, касающиеся ионного состояния пяти-
валентного ванадия в растворах, что позволяет составить цельную
картину гидролиза ионов ванадия в широком диапазоне pH при
температурах 20—25° С.
Ниже изложены основные результаты работ по исследованию
ионных форм ванадия и соответствующих равновесий, характеризу-
ющих отдельные стадии гидролиза, в порядке убывающей щелочно-
сти среды.
РЕАКЦИИ ГИДРОЛИЗА ИОНОВ ВАНАДИЯ
Равновесия ортованадат-иона (табл. 1). На основании кондукто-
метрического титрования Дюльберг [18] установил, что в воде орто-
ванадат натрия гидролизуется по реакции
VO1“+H2OZ±HVO^ + OH“. (1)
Аналогичное заключение на основании потенциометрических иссле-
дований сделали Бриттон и Робинсон [21], Дюкре [22]. В других
работах Бриттон и Робинсон [23, 24] установили, что в растворе
с общей концентрацией ванадия 0,0844 М. гидролизуется только
49% ортованадат-ионов, а растворимость серебряной соли в раство-
рах ортованадата хорошо описывается выражением
[Ag+P [HVO1“] [ОН-]= 1024.
Первые количественные данные получили Суше и Шолл [25],
которые определили константу равновесия реакции (1) криоскопи-
ческим методом в среде расплавленной глауберовой соли. Близкое
значение величины К получили Ньюмен с сотрудниками [26] на
основании результатов спектрофотометрических измерений. Ингри
и Брито [27], Ю. И. Санников, В. Л. Золотавин, И. Я- Безруков
128] изучили реакцию протонирования иона VO^-
VO1- + H+Z±HVC>4_ (2)
потенциометрическим методом. Шварценбах и Гейер [29] путем
потенциометрического титрования в струйном аппарате, обеспечива-
7
Рассчитано нами по данным работы [26].
еМ измерение в контролируемые малые промежутки времени
азличных средах, определили равновесия последовательных ре-
акций
HVOi~ + Н+ H2vor, (3)
H2VO7 + Н+ H3VO4. (4)
Авторы отмечают, что ход изменения величин Ig К в зависимости
оТ степени протонирования хорошо согласуется с аналогичными
величинами для ортофосфорной кислоты, логарифмы констант про-
тонирования которой соответственно равны 11,8; 6,7; 2,10.
Равновесия между орто- и пированадат-ионами (табл. 1). Сте-
пень конденсации пированадат-иона изучалась многими авторами.
Еще Дюльберг [18], основываясь на результатах криоскопических
исследований, пришел к выводу, что в растворах, содержащих
гидроксил-ион и ванадий в отношении 2:1, ванадий существует
в виде димера. Аналогичные данные получены Лидером и Яром
[30, 31] на основании исследования коэффициента диффузии; Суше
[32] пришел к заключению, что в достаточно концентрированных по
ванадию растворах ванадата с ОН“: V=2 :1 произведение раствори-
мости солей серебра может быть описано уравнением
[ Ag+]4 [V2O47-]=const.
Изучение пированадатных растворов Русселем и Салманом [33]
с помощью ионного обмена показало, что растворы с ОН~ : V=2 : 1
содержат ванадий в форме димера с зарядом —3. На основании
криоскопических исследований раствора Na4V2O7 в расплавленной
Na2SO4-10H2O Шварценбах и Париссакис [34] установили, что
реакция димеризации
2Н vor V2C>7~ + Н2О (5)
зависит от концентрации ванадия в растворе.
Ньюмен с сотрудниками [26] спектрофотометрическим методом
определили константу равновесия реакции димеризации (5) в обла-
сти pH 11 —14. Комбинируя данные авторов для реакций (2)
и (5), можно рассчитать константу равновесия реакции гидролиза
иона VO4~
2VO4~+H2O zt V2O47~+2OH“. (6)
По данным потенциометрических исследований Ю. И. Санников
с соавторами [28] рассчитали константу равновесия реакции
2VO4“" + 2Н+ ИV2O47- + Н2О. (7)
Ингри и Брито [35], использовав для обработки результатов
потенциометрического титрования метод Силлена [19], пришли
к выводу о том, что димеризация иона HVO4~ протекает по ре-
акции
2HVO4~ 7± HV2O7~ + ОН“. (8)
9
Эти данные хорошо согласуются с приведенными выше результатами
исследований Русселя и Салмона [33].
Равновесия между пиро- и метаванадат-ионами. Вопрос о сте-
пени конденсации метаванадат-иона, как уже отмечалось, до сих
пор остается открытым. Одна группа исследователей, начиная с
Дюльберга [18], считает ион метаванадата тримером V3Oq ^. Заклю-
чение о тетрамерности метаванадата сделано в более позднее время,
однако расхождение во мнениях сохраняется до сих пор и в ряде
новейших работ концентрация тримера остается.
При решении данного вопроса были использованы разнообразные
экспериментальные методы. Основываясь на криоскопических дан-
ных, Дюльберг [18] пришел к выводу о тримерности метаванадата,
аналогичное заключение сделали Яндер и Аден [36] по результатам
изучения диффузии. Робинзон и Синклайр [37] из определения
упругости водяных паров над растворами с ОН-: V = 1 вычислили
среднюю степень конденсации, которую они нашли равной трем.
К такому же заключению пришли Суше и Карпени [38, 39],
исходя из титриметрического определения растворимости солей
серебра в растворах метаванадата, Руссель и Салмон [33] — на
основании изучения сорбции ванадия ионообменными смолами, а
также Шиллер и Тило [40], Шварценбах и Гейер [29] — в резуль-
тате спектрофотометрических и потенциометрических измерений.
Недавно получены данные из спектров ЯМР о структуре соедине-
ний ванадия [41, 42]. Говард и Ричардс [41] нашли, что их резуль-
таты находятся в лучшем согласии с представлением о тримерности
ванадата, однако и концепция тетрамера полностью не может
быть отвергнута.
Другая многочисленная группа исследователей приписывает мета-
вана дат-иону структуру тетрамера. Среди них едва ли не первыми
эту точку зрения высказали Яндер и Яр [30, 31], основываясь на
результатах диффузионных исследований.
Этот вывод подтвержден опытами по диализу [43], результатами
изучения растворимости метаванадата аммония [44], а также кри-
оскопическими исследованиями [34, 45—49]. Аналогичные данные
получены Глемзером и Прейслером [50] при использовании спектро-
фотометрического и криоскопического методов исследования. К вы-
воду о тетрамерности метаванадат-иона пришли также Симон и Яр
[51] на основании измерений коэффициента диффузии, Сток и Яр
[52] в результате изучения седиментационного равновесия на ультра-
центрифуге. Хазель, Макнаб и Сантини [53], Лефевр [54], Шово
[55], Ю. И. Санников с соавторами [28], основываясь на результа-
тах потенциометрических исследований, также склоняются в пользу
существования тетрамера.
В то же время Ингри и Брито [35] потенциометрически опреде-
лили степень конденсации равной трем. Однако возвращаясь к этим
результатам и анализируя их с помощью электронносчетной маши-
ны [56], они пришли к заключению о возможности существования
10
меТаванадат-иона в обеих формах — V3Og~ и V4O1? — в зависимо-
сти от общей концентрации ванадия в растворе.
Переходя к количественной характеристике равновесий с участи-
ем метаванадат-иона, остановимся вначале на работах, основанных
на представлениях о тримерности строения последнего (табл. 2).
Суше и Карпени [38], исходя из результатов потенциометриче-
ского исследования, пришли к заключению о наличии равновесия
между орто- и метаванадат-ионами:
V3O9~+ЗН2О Zt ЗН vol- + ЗН+. (9)
Шиллер и Тило [40] изучили эту реакцию спектрофотометрически
и для ее константы равновесия получили близкое значение. Для
реакции
3HVO1-+ЗН2О Zi VgOg- + ЗОН- (9а)
Ингри и Брито [35] по данным потенциометрического исследования
рассчитали константу равновесия 1g—10,42. Имея в виду, что
в условиях эксперимента, описанного в работе [57] (pi=0,5, NaCl,
/=25° С), —1g [Н+] [ОН~]= 13,70, можно рассчитать 1g К для реак-
ции (9а) по формуле
lg/C(9) = 3 lg [Н+] [ОН-] - lg^9a).
Как видно, значение lg/C(9a), полученное таким путем, хорошо
согласуется с результатами предыдущих авторов. С другой стороны,
в работах Лефевра [54] и Ю. И. Санникова с соавторами [28] при-
ведены количественные характеристики равновесия с участием мета-
ванадат-иона в форме тетрамера.
Результаты потенциометрического титрования в интервале pH
7—11, обработанные на основе метода потенциометрических поверх-
ностей, привели Лефевра к заключению о наличии системы равно-
весий:
i/4V401rziH+ + HV01- (10)
(Н)
y2V40tr 72 Н+ + HV2O?- (12)
Ш+Н2О- 2H+ + V4O?r. (13)
Для реакций (10) и (11) Ю. И. Санников с соавторами [28] нашли
близкие значения 1g К. Что касается реакции (13), то существова-
ние иона не отмечено другими исследователями.
Приведенные данные свидетельствуют о необходимости дальней-
ших прецезионных исследований для решения вопроса о степени
конденсации метаванадат-иона.
Наряду с усовершенствованием методик эксперимента, по-види-
мому, требуются и дальнейшие усовершенствования способов мате-
матической обработки экспериментальных данных с использованием
11
Рассчитано по величине 1g К для реакции (9а).
12
счетно-решающих устройств. Как показано в сообщении Брито, Ин-
j рИ и Силлена [56], в некоторых случаях экспериментальные данные
потенциометрических измерений можно одинаково хорошо интерпре-
тировать, допуская существование различных групп ионных форм
ванадия.
Равновесия с участием декаванадат-ионов. В промежуточной
области pH от 2 до 6 ванадий существует в форме оранжево окра-
шенного иона с наибольшей степенью конденсации. Еще со времени
Дюльберга [18], который впервые провел титрование растворов ва-
надия, считали, что в слабокислых растворах ванадий существует
в форме гексаванадиевой кислоты H4V6O17. Эта точка зрения нахо-
дила подтверждение в работах Карпени и Суше [38,58], Труилло
[59] и долгое время оставалась господствующей. Однако уже в
1933 г. Яндер и Яр [30] рассматривали это соединение как пента-
ванадат Na2H2V5O16. Бриттон и Робинсон [23], Бриттон и Вильфорд
[60], Хазель, Макнаб и Сантини [53] на основании данных потен-
циометрического титрования пришли к выводу о существовании
ванадия в форме декаванадат-иона ¥10О2?, а Дюкре [22] сделал
вывод, что оранжевый цвет слабокислых растворов ванадия обуслов-
лен наличием иона H3V2O7.
И, наконец, в 1956 г. появилась работа супругов Россотти [20],
которые после обработки экспериментальных данных потенциометри-
ческого титрования по методу Силлена [19] пришли к выводу о су-
ществовании шестиосновной декаванадиевой кислоты H6V10O28. С тех
пор данные Россотти подтверждены многими исследователями и бы-
ло показано, что прежние измерения могут быть также интерпре-
тированы на основе концепции декаванадата. Наличие декавана-
дат-иона подтверждено измерением коэффициентов диффузии [51],
кондуктометрическим титрованием [61—63], криоскопическими изме-
рениями [46, 50], методом ЯМР [41], изучением седиментационного
равновесия в ультрацентрифуге [64] и спектрофотометрическим
методом [55, 65, 66].
Декаванадиевая кислота в водных растворах легко отщепляет
четыре протона, последние два протона присоединены более прочно
и константы диссоциации иона H2V10O2? легко определяются обыч-
ным титрованием. Количественная характеристика равновесий
HV10O52r^V10O|r+H+ (14)
H2V10O24r HV10Ofr+H+ (15)
изучена потенциометрическим методом в работах Россотти [20],
Шово [55], Шварценбаха и Гейера [29], Ю. И. Санникова с соавто-
рами [28] (табл. 3). В области pH 5—7 декаванадат-ионы сосущест-
вуют с метаванадат-ионами. На основании потенциометрических
измерений Ю. И. Санников с соавторами [28] пришли к заключе-
нию, что в этом случае имеет место равновесие между дека- и тет-
рамером
HV10O2r + 5ОН~ 2,5V401r + ЗН2О, (16)
13
сл
Реакция
14. HV10O^
16. HV10O|7+5OH_7±2,5V4O^7+3H2O
17. 3HV10O^+6H2Oz±10V3o3-+15H+
18. 10VO++8H2O-H2V10O427+14H+
Равновесия декаванадат-иона
Таблица 3
Л Концентрация ванадия, z, °C 1
г-атом [л » а Среда Iff К Литература
2-IO-2 — 2-10—3 I 25 ! 1 ,0 NaClO4 —5,80+0,1 [20]
10-1—2,5.10-2 — 1 ,0 NaClO4 —6,12 [55]
1 —- - 20 0 ,1 NaClO4 —6,94 [29]
20 i ,0 NaClO4 —6,06 [29]
1 20 0, 1 N(CH3)4C1 —7,52 [29]
•* 1 20 1, 0 N(CH3)4C1 —7,60 [29]
20 0, 1 LiCI —6,90 [29]
— 1 20 1, 0 LiCI —6,00 [29]
— 1 20 0, 1 KC1 —6,40 [29]
20 1, 0 KC1 —5,55 [29]
“““ 20 0, 1 RbCl —6,20 [29]
1 20 0,. 1 CsCl —6,00 [29]
2-Ю-2 — 2-Ю-3 2-10-2 —2-Ю-3 25 1, 25 3,( 0 NaClO4 0 NaCiO. —3,6±0,3 —6,5 + 0,3 [20] [20]
10-2 — 10—4 20 0,! 2 NaClO4 —3,65±0,35 [28]
10-1 —2,5-10-2 - | 1,< ) NaClO4 —3,70 [55]
20 • 0/ I NaClO. —4,34 [29]
20 1 ,0 NaC104 —3,60 [29]
20 0 , 1 N (NH3)4C1 —4,45 [29]
20 0 , 1 N (CH3)4C1 —4,50 [29]
20 0 , 1 LiCI -4,4 [29]
20 1 ,0 LiCI -3,7 [29]
20 0 ,1 KC1 —3,9 [29]
20 1 ,0 KC1 -3,1 [29]
20 0 , 1 RbCl -3,7 [29]
___ 20 0 51 CsCl —3,3 [29]
10-2 —10—1 20 0 , 2 NaClO4 —35,55 [28]
10-2—5-10-3 20 1 ,0 NaClO4 —108,0 [29]
2-10-2 — 2-Ю-з 25 1 ,0 NaClO4 —6,75±0,15 [20]
2-10-2 — 2-Ю-з 25 < 5,0 NaClO4 —5,50±0,15 [20]
io-3—10—4 25 ; 5,0 NaClO4 —5,80* [65]
Ю-2 — 10-4 20 ( 3,2 NaClO4 —7,50±0,3 [28]
» Определение произведено спектрофотометрическим методом, во всех
остальных случаях - потенциометрическим.
а по мнению Шварценбаха и Гейера [29] — между дека- и триме-
ром
3HV10Ot8”+6H2Ozi 10V3C>9_ + 15H+. (17)
При достаточной концентрации ионов водорода степень полимери-
зации ванадат-ионов резко уменьшается и декаванадат-ион H2V10O2?
переходит в оксикатион VO^. Соответствующая этому процессу
реакция
10VOt+8H2Oz± H2V10O^+ 14Н+ (18)
изучена впервые Россотти [20] потенциометрически, а также Нью-
меном [65], Шово [55] — спектрофотометрически и Ю. И. Саннико-
вым с соавторами [28] — потенциометрически.
Равновесия моноядерных форм (табл. 4). В пределах концентра-
ций 10-1—10~4 г-атом/л, как видно из описанного, ванадий
в виде моноядерных частиц существует только в кислых (pH < 2)
или сильнощелочных (pH 13) растворах. Однако многочисленные
исследования [35, 40, 67, 68] показывают, что при малых концент-
рациях (менее 1-10~4 г-атом/л) ванадий при всех значениях pH
существует только в форме моноядерных частиц.
Ннгри и Брито [35] потенциометрически изучили равновесие
реакции гидролиза ортованадат-иона
HVO*-^ VOT + OH-. (19)
Спектрофотометрически Шиллер и Тило [40] определили констан-
ты равновесия реакций
HVO^+H+ZtVOr + H2O, (20)
V3O?r^3VOr. (21)
По распределению ванадия между водным раствором и изобу-
тил-карбинолом в зависимости от pH Дирссен и Сикайн [67, 68]
рассчитали 1g/С реакций
HVO3ZtVOr+H+, (22)
VOt + H2OZtHVO3+H+. (23)
Близкое значение для реакции (22) получили К. Б. Яцимирский
и В. Е. Калинина [69] кинетическим методом.
Для реакции
vot + H3ozt VO7+2H+ (24)
константу равновесия можно рассчитать, поскольку известны кон-
станты равновесия реакций (22) и (23). Расчет и экспериментальное
определение электрохимическим методом [70] дали совпадающие
результаты (см. табл. 4).
Ион VO^. Исследованию состояния ванадия (V) в кислых водных
растворах посвящены многочисленные работы. Несмотря на это,
16
Равновесий моноядерных форм
бЗ
&
31
я
ю
«J
Рассчитано по [35] при lg [1ТГ] [ОН ]=—13,7.
Рассчитано по величинам 1g К для реакций (22) и (23).
2
Заказ № 256
17
вопрос о формах существования ванадия (V) в таких растворах все
еще является дискуссионным. Первоначально считали, что в сильно-
кислых растворах ванадий находится в форме метаванадиевой кисло-
ты HVO3 [71, 73], однако авторы работ [72, 74—76] на основании:
изучения окислительно-восстановительного потенциала пятивалент-
ного ванадия указали на существование в растворах катиона VO^.
Некоторые авторы обсуждали возможность существования в
кислых средах ионов VO3+ и V (ОН)з+ [75—78].
Дюкре [22] при определении растворимости пятиокиси ванадия
в хлорной, серной и соляной кислотах и измерении pH нашел, что
в указанных растворах отношение [V]: [Н+] остается постоянным
и, следовательно, пятиокись ванадия переходит в раствор по реак-
ции
V2O5+2H+^2VOt+H2O,
(25)
vo+]
для температуры 100 е С
константа равновесия которой /С=
и ионной силы р,->0 определена равной 1,36-10~х.
Ла Салль и Коббл [79] провели аналогичное исследование, под-
держивая в растворах постоянство ионной силы, и нашли, что вели-
чина [V]: [Н+] для хлорной кислоты сохраняет удовлетворительное
постоянство и на основании этих и спектрофотометрических данных
подтвердили протекание реакции (25). Ими определены: теплота
растворения V2O5 в 0,16—0,63/ИНС1О4 по реакции (25) (А/У°=
=—5,78 + 0,09 ккал)моль)^ изменение энтропии (AS0——25,7 +
+ 0,4 кал!град • моль) и рассчитана энтропия иона VO2-(aq) (S°=
= 5,5 + 0,5 калIград-моль). Хотя по данным изучения равновесий
нельзя сделать выбора между альтернативными формулами VO^
и V (OH)f, но на основе структурно-энтропийных расчетов авторы
[79] пришли к заключению, что ион VO^ подобен иону NpO^, а
структура v (OH)t маловероятна. Позднее Бертранд с соавторами
[80] вновь измерили теплоту растворения пятиокиси ванадия
в 0,25 — 0,50А1 НС1О4. Для реакции (25) найдены А//0——8,4 +
+ 0,3 ккал/моль V2O5 и AS° =—34,5 кал! град-моль V2O5, а для
иона VO^, кроме того, рассчитаны термохимические константы
AG°= —140,3 кал/моль, —155,3 кал/моль и AS°=
=—9,9 кал/град -моль. Авторы работы [80], не указывая на при-
чины существенных расхождений в величинах термодинамических
констант в сравнении с результатами Ла Салля и Коббла [79],
отмечают что их данные получены на нескольких образцах V2O5,
приготовленных различными способами. Из величин Д//° и AS0 для
реакции (25) по уравнению
AS0 _ дя°
2,303/?Г
18
жн0 рассчитать величину К- По данным работ [79, 80], она ока-
залась соответственно равной 2,06-10-1 и 2,04-10-1.
Ряд исследователей [14, 29, 72, 81—83] допускает возможность
рисоединения протонов к иону VO?. Миштра и Саймонс [81]
редполагают существование таких ионных форм, как [V (ОН)4(Н2О)]+
ли [VO(OH)2(H2O)2]+. Для объяснения зависимости констант ско-
ости от кислотности при исследовании кинетики окисления вана-
дием (V) спиртов и фенолов допускается существование ионов
[VO(OH)P+ и [VO (ОН)3Р+[82, 84].
Шварценбах и Гейер [29], исследуя процесс нейтрализации кис-
лых растворов ванадия в струйном аппарате, высказали мнение, что
ион VOt не может быть просто продуктом протонирования ортова-
Г ОН "I-
надат-иона со структурой
и состав его либо
[V (ОН)4 (Н2О)]+, либо [V (ОН)4 (Н2О)2]+. В работе [85] ион VOt
в водных растворах рассматривается как [VO2(H2O)4]+.
Составить цельную картину гидролиза ванадия на основании
литературных данных чрезвычайно трудно, так как большинство
исследований касалось состава лишь отдельных ионов и было выпол-
нено различными методами в самых различных условиях в отноше-
нии ионной силы растворов, природы «нейтральных» ионов, общей
концентрации ванадия и т. д. Все же большая часть количествен-
ных характеристик получена потенциометрическим методом с исполь-
зованием предложенного Силленом [19] нового метода обработки
данных потенциометрического титрования, что позволило получить
весьма надежные результаты. Для обработки результатов потенцио-
метрического титрования по методу Силлена строят график зависи-
мости среднего числа гидроксильных ионов (Z), связанных с одним
атомом металла, как функцию логарифма равновесной концентрации
водородных ионов — 1g [Н+] при разных концентрациях металла.
(олученные кривые подобны кривым титрования и наглядно отра-
жают области существования тех или иных ионов.
Если исходить из иона VO]!~, как это принято в работах Рос-
сотти [20] и Ю. И. Санникова [28], каждой ионной форме ванадия
будет соответствовать определенное число Z. Для VO]!" оно равно
нулю, для нейтральной частицы HVO3 — единице, для иона VO7—
двум, для VOi- — четырем.
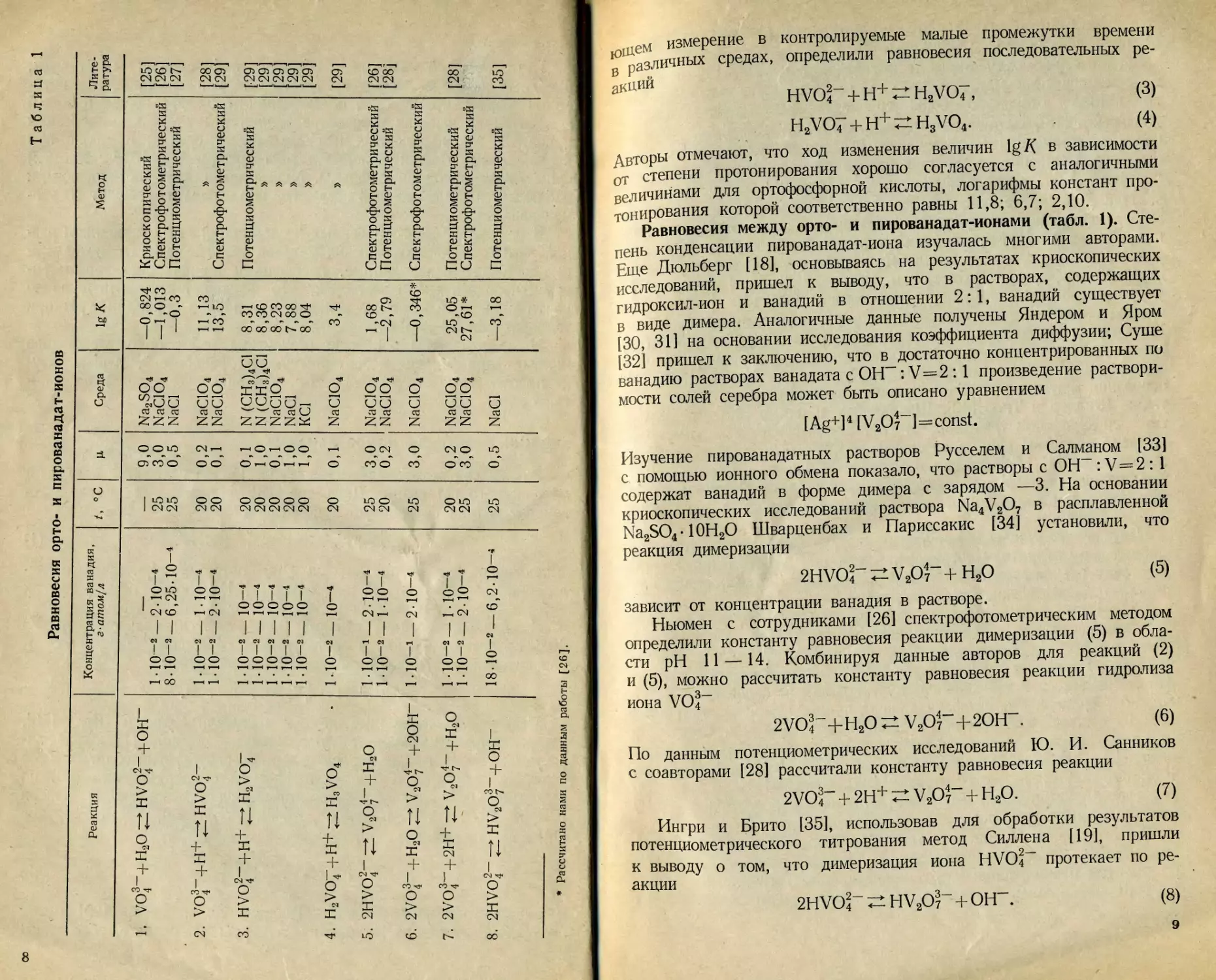
На рис. 1 приведена диаграмма зависимости числа гидроксиль-
ных ионов, связанных с одним атомом ванадия, от — 1g [Н+], со-
ставленная по данным различных авторов. Как видно, отчетливые
ступени на кривых'характерны только для ионов VO?, V3O9-"(V4Ot2),
HVO4“ и VO^. Широкая ступень в области —lg [Н+]-^3~н6 соот-
ветствует всем трем декаванадат-ионам. Кривые, полученные при
2* 19
различных концентрациях ванадия, проходят параллельно друг дру-
гу. Это означает, что ионный состав ванадия в данном случае не
меняется. Изменение формы кривой свидетельствует о появлении
других ионов. Это хорошо видно в области —lg [Н+}= 1 н-5, где
при [V] > 10-4 г-атом! л в равновесии с ионами VO^ сосуществуют
декаванадат-ионы, а при [V] < 1 • 10“4 — моноядерная молекула
HVO3. Наглядное представление о составе растворов пятивалентного
ванадия дают диаграммы распределения, построенные в координатах
а/—lg [Н+], где а — доля данного иона.
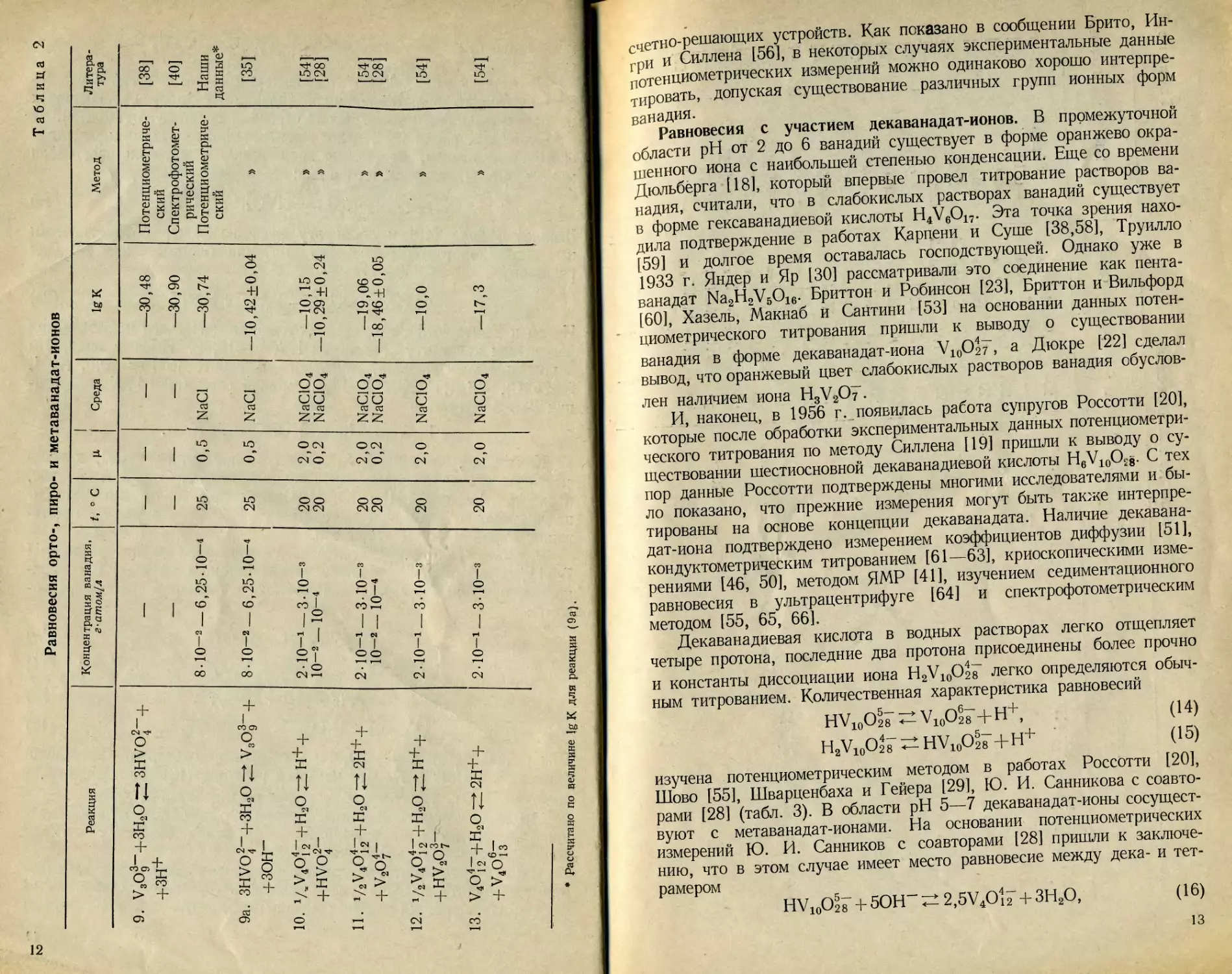
На рис. 2 приведены диаграммы распределения, полученные
различными авторами в равновесных условиях при концентрациях
ванадия 10-1—10-2 а-атом/л. Необходимо иметь в виду, что
данные Россотти [20] получены при быстром титровании и правая
часть их диаграммы не отражает равновесного состава в области
—lg [Н+] от 3—4 и выше, в то же время левая описывает истин-
ные равновесия. Это связано с тем, что реакции (16) или (17) про-
текают слева направо весьма медленно, а реакция (18) — достаточно
быстро. В кислой области данные Россотти и Ю. И. Санникова
с соавторами аналогичны, несущественная разница, по-видимому,
20
-ig[n*]
Рис. 2. Зависимость состава растворов ^ванадия (V) от — 1g [Н+].
и t,° c [V], моль/л Данные
а—0,2 (NaClO4) 20 10“2 [28]
6—1,0 (NaClOJ 25 2 -10—2 [20]
в—0,5 (NaCl) 25 4- IO”2 [25]
г—1,0 (NaClOJ 20 10“1 [55]
6—3,0 (NaClO4) 20 10~2 [26] ₽
обусловлена различием в ионной силе растворов. В области
—lg[H+] = 5 :-6, по данным Шово [55], при [VJ^IO-1 г-атом/л
в заметных количествах присутствуют ионы V10O28~, а при более
низкой концентрации ванадия ~ 10-2, они не существуют. В области
—1g [Н+] = 6-н8 преобладают ионы метаванадата, вопрос о степени
конденсации которого остается открытым. Возможно, как отмечалось
выше, в этом случае справедливо мнение Брито с соавторами [56],
что степень конденсации метаванадата возрастает вместе с увеличе-
нием концентрации ванадия в растворе. В области —1g [Н+] = 9н-11,
по данным работ [26] и [28], ванадий существует в форме HVO1~
и V2O7"", однако, по мнению Брито с соавторами [35], в этих усло-
виях пированадат имеет состав HVgO?--, хотя позднее [56] они уже
не отрицают и возможности существования ионов V2O7~. И, нако-
нец, как видно по [28], в области —1g [Н+]=9ч-11 в растворе
преобладают ионы V2O7~", а согласно [26] — ионы HVO*-. .
Состав растворов при концентрации ванадия 10~4 г-атом!л
и ниже приведен на рис. 3. В этих условиях область существова-
ния иона VO2' расширяется, а ионов декаванадата и метаванада-
та— сужается. При концентрации менее 10~4 г-атом/л существуют
только моноядерные ионы, хотя по данным [35] мономер VO3" в за-
метных количествах присутствует уже в растворах с [V] = 6,25-10—4
и даже [V]=4-10““2.
Сведения об ионных формах пятивалентного ванадия, несмотря
на их противоречивость и неполноту, могут быть представлены
в форме диаграммы состояния [40], на которой зафиксированы
области существования различных ионов в зависимости от концент-
рации ванадия и pH растворов.
На рис. 4 приведена диаграмма состояния ванадия в растворах
с учетом работ Ингри и Брито [351 и Дирссена и Сикайна [68].
Как и на диаграмме Шиллера и Тило [40], сплошные линии прохо-
дят по участкам, где равновесные формы находятся в соотношении
1:1, пунктирные означают, что их ход только предположителен,
на заштрихованных участках равновесные формы существуют в раз-
личных соотношениях и белые поля — области преимущественного
существования отдельных ионов.
В заключение необходимо еще раз отметить недостаточную изу-
ченность реакций гидролиза пятивалентного ванадия в водных рас-
творах. Об этом свидетельствуют появившиеся в ряде последних
работ предположения о существовании новых форм ванадия: поли-
ванадата— промежуточной формы между моно- и декамером [40]
и нестойкого полииона V12O|7 [50].
Весьма интересные результаты, по-видимому, могут быть полу-
чены при детальном исследовании влияния природы и концентрации
«нейтральных» ионов на условия равновесия и состав ионных форм
ванадия. Сопоставляя потенциометрические данные работ [35]
22
-lg[ti+]
Рис. 3. Зависимость состава растворов ванадия
(V) от — 1g [Н+].
Ц t,° с
а— 0,2 (NaC104) 20
6-0,5 (NaClO4) 25
в—0,5 (NaCl) 25
г-3,0 (NaClO4) 25
[V], моль]л Данные
10~4 [28]
10~6 [67]
6.25-10—4 [35]
10“4 [26]
Кристаллические соли
14 « 12 11 10 9 8 1 6 5 4 U 2 1 рн
Рис. 4. Диаграмма состояния ванадия (V) в водных растворах при комнатной
температуре,
((pi=0,5; NaCl) и [28] (p,=0,2; NaCl), можно заметить, что в интер-
вале Z=2h-3 в соответствии с различным ходом кривых образова-
ния картины гидролиза существенно различны.
СТРУКТУРА И СПЕКТРЫ ПОГЛОЩЕНИЯ ИОНОВ ВАНАДИЯ
Изучение равновесий ионных форм не позволяет сделать опреде-
ленные выводы о их структуре в растворах. В случае ванадия
дело осложняется образованием полимерных частиц. В твердых
соединениях пятивалентный ванадий имеет координационное число
4, 5 и 6.
Наличие группировок VO^ в кристаллах установлено методом
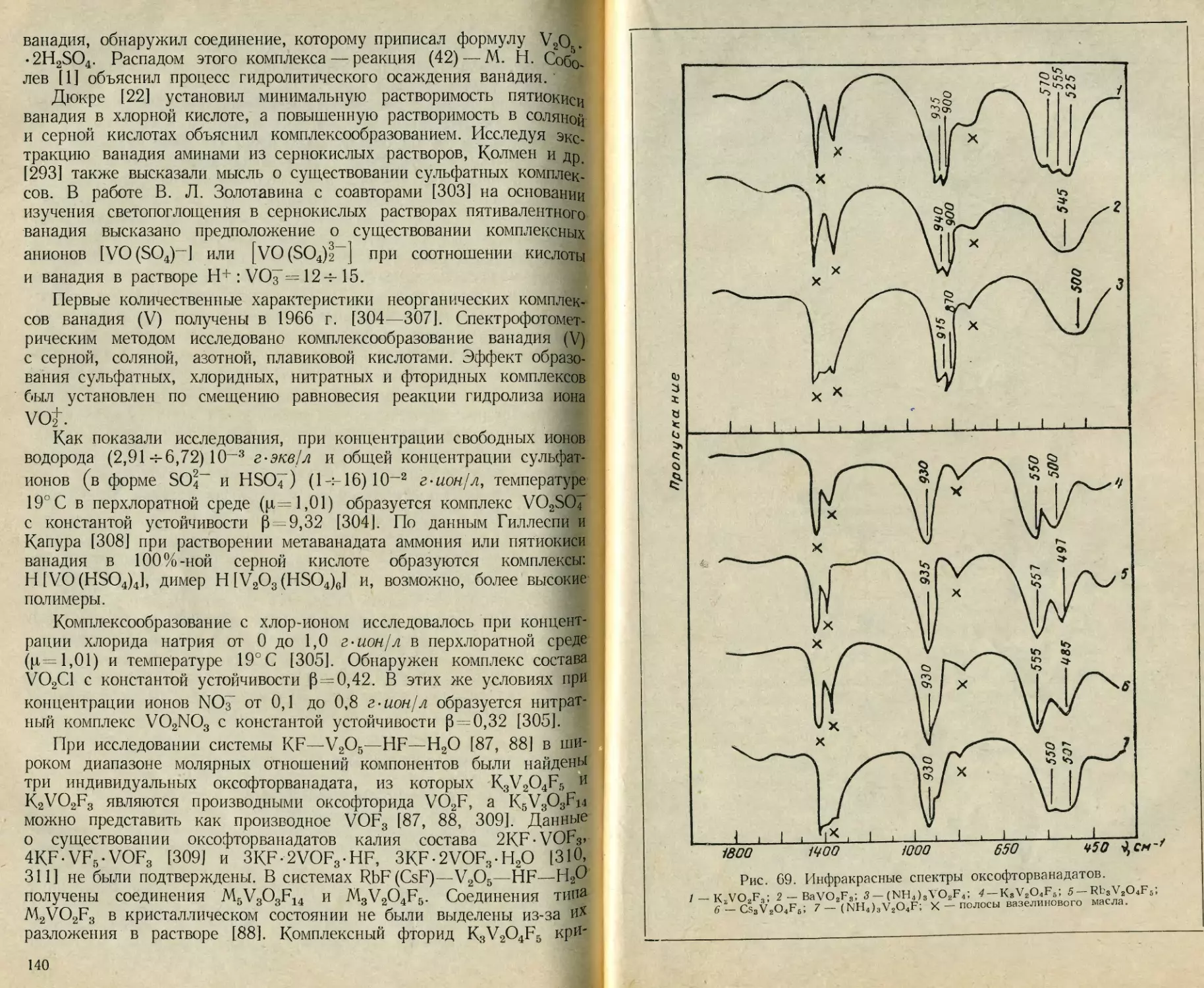
ИК-спектроскопии. По данным [86], оксалатный комплекс
(NH4)3 [VO2(C2O4)2]-2H2O
имеет октаэдрическую структуру, включающую линейную группи-
ровку О—V—О с асимметричным расположением атомов кислорода.
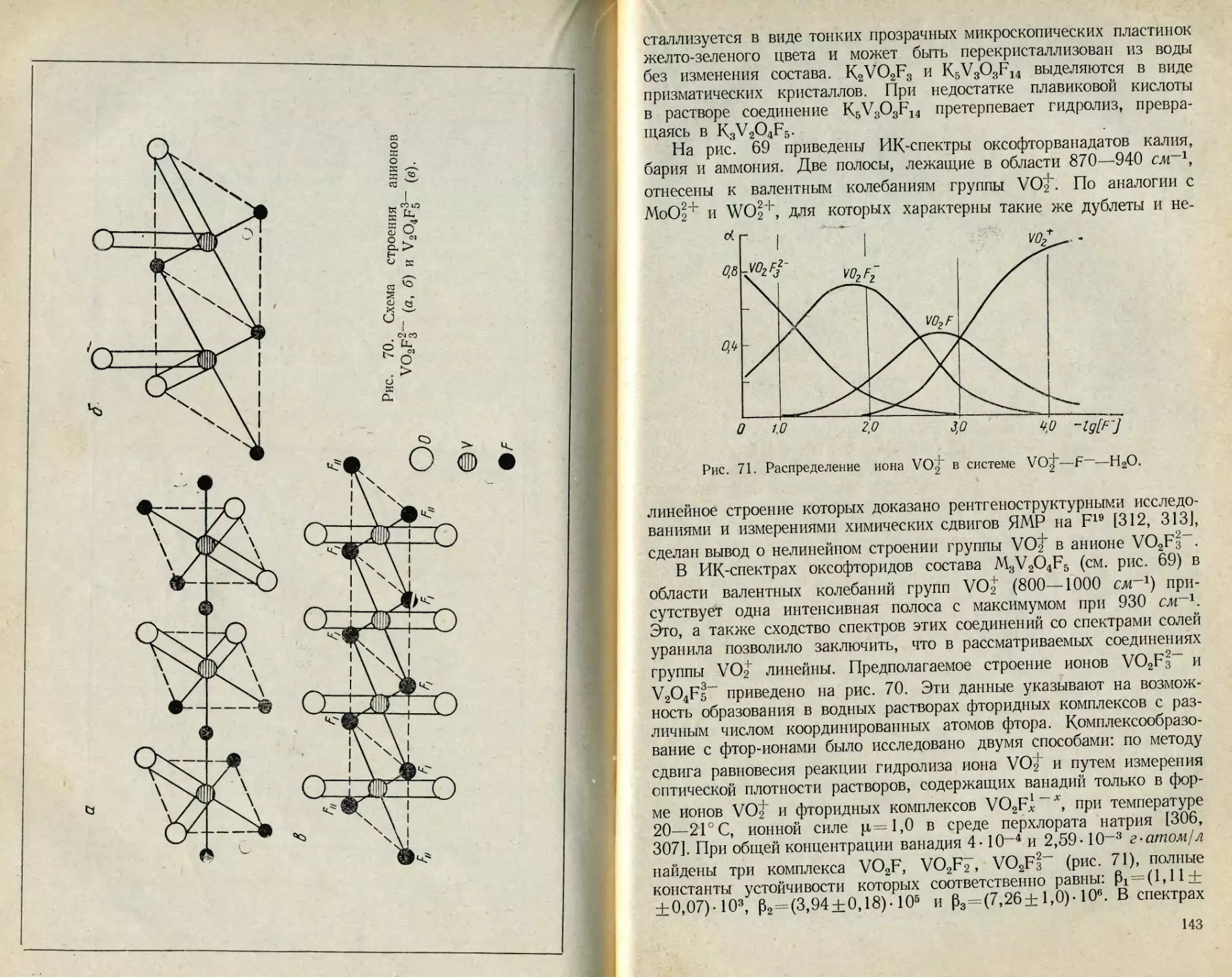
При исследовании комплексных фторидов найдено, что группы VOj’
в анионе V2O4Fs- линейны, а в анионах VO2F4- и VO2Fl“ не
линейны, при этом предполагается октаэдрическая или бипирами-
дальная конфигурация аниона VO2Ff- [87, 88]. В соединениях
VO2C1, VO2F и VO2SbF6 также обнаружены дискретные группиров-
ки VOt, характеризуемые симметрией Cs и C2v [89].
Таким образом, для пятивалентного ванадия в соединениях, где
присутствуют группировки VO^, по-видимому, характерно координа-
ционное число 6, а сам ион VO^ часто имеет линейное строение.
Для комплексов ванадия (V) в водных растворах, так же как
и для комплексов ванадия (IV), Бончев и Николов [85] принимают
симметрию С4у. Спектр поглощения ванадия (V) в кислых растворах
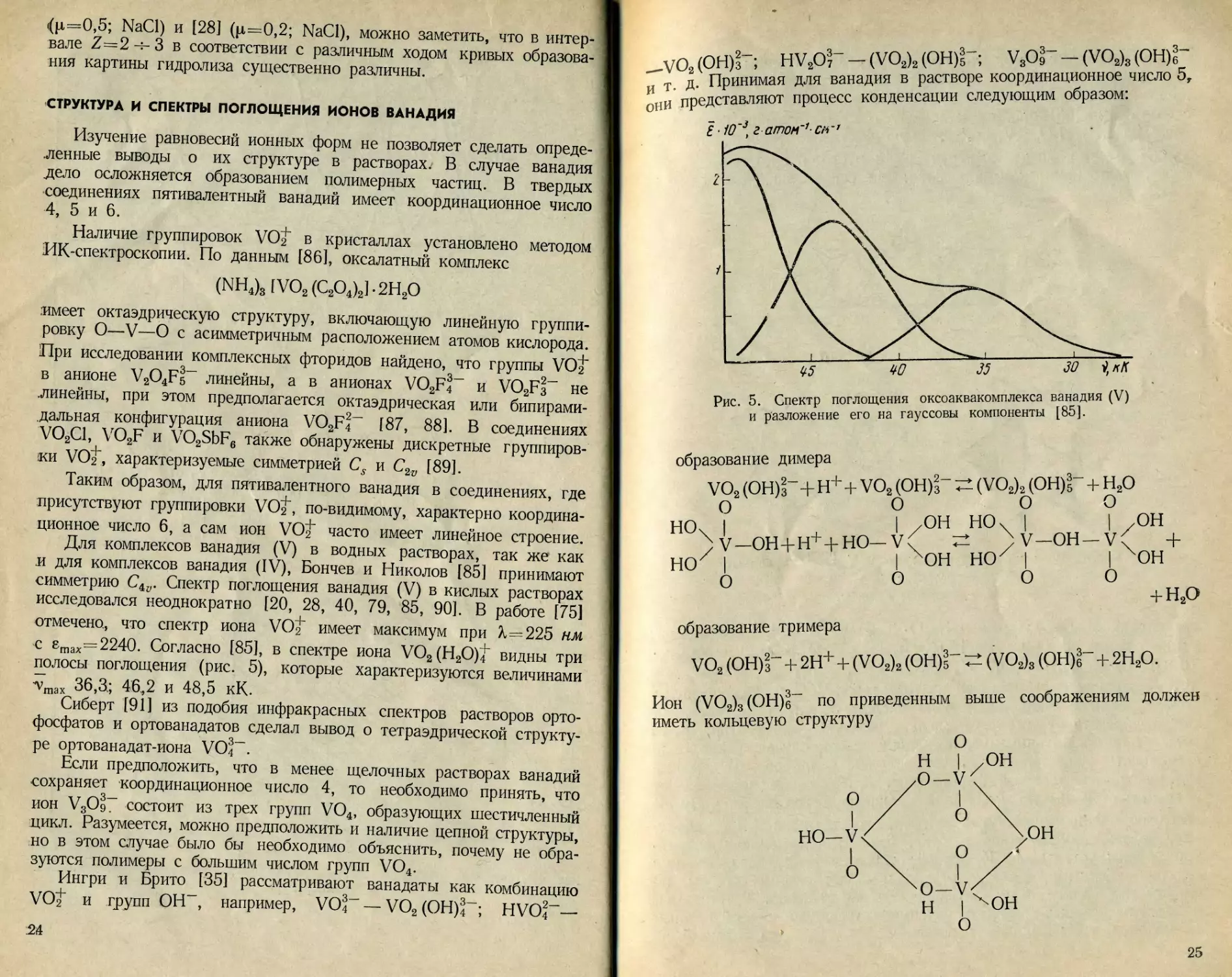
исследовался неоднократно [20, 28, 40, 79, 85, 90]. В работе [75]
отмечено, что спектр иона VO^ имеет максимум при Х=225 нм
с 8тах=2240. Согласно [85], в спектре иона VO2(H2O)t видны три
полосы поглощения (рис. 5), которые характеризуются величинами
•vmax 36,3; 46^2 и 48,5 кК.
Сиберт [91] из подобия инфракрасных спектров растворов орто-
фосфатов и ортованадатов сделал вывод о тетраэдрической структу-
ре ортованадат-иона VO4”.
Если предположить, что в менее щелочных растворах ванадий
-сохраняет координационное число 4, то необходимо принять, что
ион V3O|“ состоит из трех групп VO4, образующих шестичленный
цикл. Разумеется, можно предположить и наличие цепной структуры,
но в этом случае было бы необходимо объяснить, почему не обра-
зуются полимеры с большим числом групп VO4.
Ингри и Брито [35] рассматривают ванадаты как комбинацию
VOt и групп ОН-, например, VO4- — VO2(OH)4~; HVO4-—
.24
_VO2(OH)I-; HV2or - (VO2)2 (ОН)Г; V3O93--(VO2)3(OH)r
и т. Д- Принимая для ванадия в растворе координационное число 5У
они представляют процесс конденсации следующим образом:
Рис. 5. Спектр поглощения оксоаквакомплекса ванадия (V)
и разложение его на гауссовы компоненты [85].
образование димера
VO2 (ОН)Г+н++VO2 (OH)23- (VO2)2 (OH)|- + Н2О
О ООО
НОх I I /ОН НО/ I I /ОН
) V—ОН+Н++НО—v( )V—ОН —V/ +
НОХ I I Хон HOZ I I хон
о ООО
+ Н2О
образование тримера
VO2 (ОН)Г + 2Н+ + (VO2)2 (ОН)Г (VO2)3 (ОН)63- + 2Н2О.
Ион (УО2)3(ОН)Г по приведенным выше соображениям должен
иметь кольцевую структуру
25
На основании того, что при кондуктометрическом титровании
декаванадиевой кислоты в присутствии ионов Ва2+, La3+ расход
щелочи составляет более 6 эквивалентов на 1 моль H6V10O2g, Яр
с соавторами [63] пришли к выводу о существовании комплексов
типа [Ba (ОН)2 H2On_2V10O2g]6“. Было высказано мнение, что ион
V10Olr имеет компактную структуру с пустотой внутри и способен
образовывать комплексы включения. На основе данной модели для
Рис. 6. Возможные структуры иона V10O2g в водных
растворах.
а, б, в — описание в тексте.
соединений 4BaO*5V2O5*aq и Na2O-4CuO-5V2O5-2H2O предложены
рациональные формулы Ва3[Ва(ОН)2 H2OxV10O28] • 4Н2О и Na3Cu3-
• [Cu(OH)4-V10O28]-4H2O. Предполагается, что в растворе ион
Vi0O2? должен быть построен из тетраэдров VO4, поскольку другие
конфигурации с координационным числом 5 или 6 не позволяют
сконструировать пустотелую модель. Двойную цепочку (рис. 6, а)
нельзя принять во внимание, поскольку в этом случае можно было
бы ожидать существование более высоких полимеров. Высокосим-
метричное тело шаровой формы (рис. 6, б) также не позволяет
объяснить ступенчатую диссоциацию декаванадиевой кислоты. Лучше
всего, по мнению упомянутых авторов, свойства иона объяс-
няет модель, изображенная на рис. 6, в. В ней можно различить
три группы тетраэдров. К первой относятся 4 тетраэдра, которые
связаны с другими через три угла и имеют, таким образом, по
одному свободному атому кислорода. Вторая включает 2 тетраэдра,
каждый из которых связан с другими тетраэдрами первой группы.
И, наконец, третья включает оставшиеся 4 тетраэдра, каждый из
.26
которых также связан только с двумя тетраэдрами: с одним из
первой группы и с одним из собственной.
Рентгеноструктурные исследования, однако, показали, что в кри-
сталлических декаванадатах анион У10О2? обладает компактной
структурой, построенной из октаэдров VO6 (см. рис. 45). Чтобы
устранить это противоречие, упомянутые авторы позднее [153] вы-
нуждены были предположить, что в растворе существует равновесие
между двумя описанными модификациями декаванадат-иона — пусто-
телой (Л) и компактной (В):
V10olr И)+м" (Н2О)Г [Мп (h2o)4v10o28]4“
1 ^fMn(OH)(H2O)„_1V10O28P-+H+
V10O2? (В) + зм" (Н2О)*+ MVv10O28. (Зп - х) Н2О + хН2О.
Весьма интересные заключения о структуре ванадат-ионов сде-
ланы Говардом и Ричардсом [41] на основе результатов исследований
равновесий поливанадатов в водных растворах методом ЯМР на ядрах
V51. Подтверждено существование ионов VO^-, HVO4“, V2O7~,
HV2O|~, VOT, V10O^, HV10O258“, H2V10O^, vot. Как уже отме-
чалось, полученные результаты находятся в согласии с предположе-
нием о существовании иона V3O9~, но не исключается и У4О|Г-
Поскольку сдвиг и ширина резонансной линии ванадия в широком:
интервале не зависят от концентрации кислоты, существование
иона VO3+ не подтверждается. Для иона УО4~ характерна малая
ширина резонансной линии, что указывает на его высокую симмет-
ричность и тетраэдрическую структуру. Сделан вывод о том, что
другие бесцветные ионы ванадия построены из тетраэдров VO4,
Окрашенные ионы имеют более широкие резонансные полосы, сви-
детельствующие о менее симметричном электрическом окружении
атомов ванадия в них. Установлено, что в ионе У10О2? можно
выделить три группы атомов ванадия (2, 2 и 6), отличающиеся
различной симметричностью их электрического окружения. Для
иона VOt характерно сильно асимметричное электрическое окру-
жение, которое авторы объясняют различным положением двух
атомов кислорода.
Различие в строении и составе ионов ванадия (V) в водных
растворах в зависимости от pH и концентрации ванадия находит
отражение в качественном изменении вида полярографических кри-
вых [92, 93]. При pH 1,0 ванадий дает при восстановлении на
ртутном капельном электроде две волны, одна из которых, возни-
кающая при потенциале окисления ртути, соответствует восстанов-
лению VOt, а вторая — V (V) до V(IV). При pH 3,0 наблюдается
три волны: первая соответствует восстановлению декаванадатов
и частично иона VO^, вторая — неконденсированных форм ванадия.
В интервале pH 5,5 — 9,2 на полярограммах зафиксированы две
волны, причем с ростом pH высота первой снижается, а второй
27
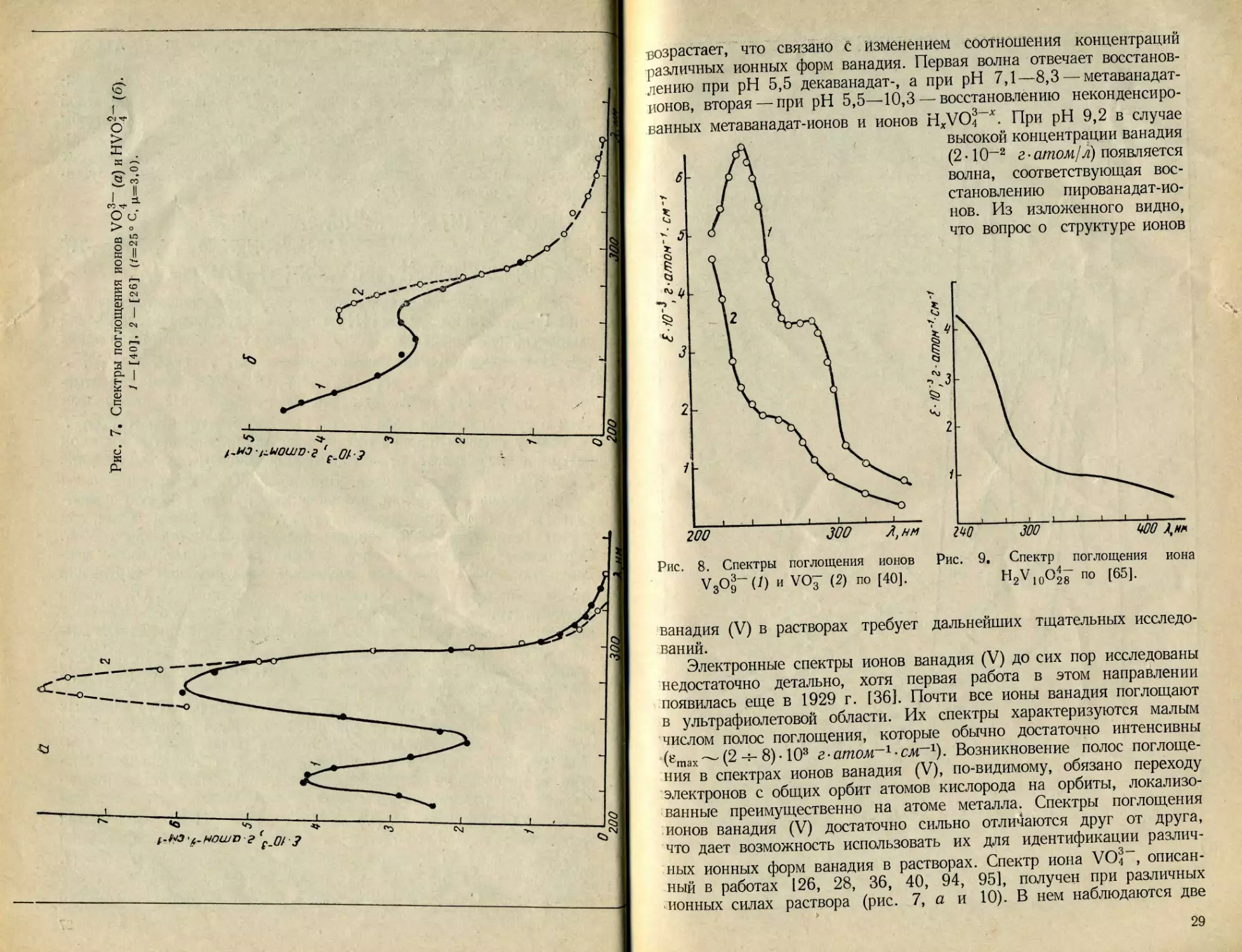
Рис. 7. Спектры поглощения ионов VO4 (aJnHVO^ (б).
1 — [40], 2 — [26] (/=25° С, |Л=3,О).
возрастает, что связано с изменением соотношения концентрации
азличных ионных форм ванадия. Первая волна отвечает восстанов-
лению при pH 5,5 декаванадат-, а при pH 7,1—8,3 — метаванадат-
ионов, вторая — при pH 5,5—10,3
ванных метаванадат-ионов и ионов
V3O®- (/) и VO7 (2) по [40].
— восстановлению неконденсиро-
HXVO4-X. При pH 9,2 в случае
высокой концентрации ванадия
(2 • 10-2 г • атом!л) появляется
волна, соответствующая вос-
становлению пированадат-ио-
нов. Из изложенного видно,
что вопрос о структуре ионов
ио зоо иоо
Рис. 9, Спектр поглощения иона
^2^10^28 П0
ванадия (V) в растворах требует дальнейших тщательных исследо-
ваний.
Электронные спектры ионов ванадия (V) до сих пор исследованы
недостаточно детально, хотя первая работа в этом направлении
появилась еще в 1929 г. [36]. Почти все ионы ванадия поглощают
в ультрафиолетовой области. Их спектры характеризуются малым
числом полос поглощения, которые обычно достаточно интенсивны
(етах — (2 8) • 103 г• атом-1 • см-1). Возникновение полос поглоще-
ния в спектрах ионов ванадия (V), по-видимому, обязано переходу
электронов с общих орбит атомов кислорода на орбиты, локализо-
ванные преимущественно на атоме металла. Спектры поглощения
ионов ванадия (V) достаточно сильно отличаются друг от друга,
что дает возможность использовать их для идентификации различ-
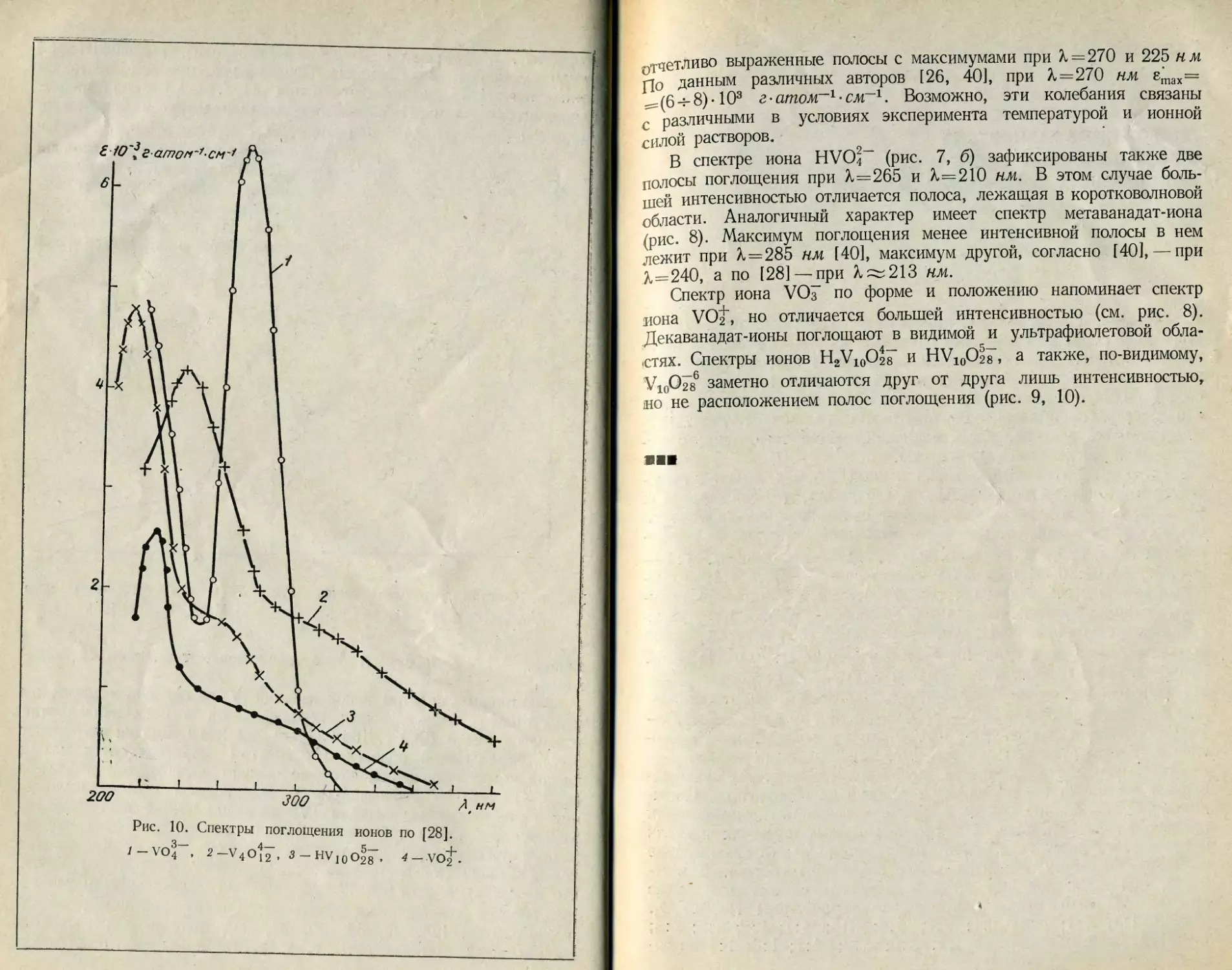
ных ионных форм ванадия в растворах. Спектр иона VO4~, описан-
ный в работах [26, 28, 36, 40, 94, 95], получен при различных
шонных силах раствора (рис. 7, а и 10). В нем наблюдаются две
29
£10~з гатом^-см'1
200 JOO Л, нм
Рис. 10. Спектры поглощения ионов по [28].
3— 4 5 _l
i — VO 4 , 2 —V 4 О J 2 > 5 — НV j q О2 8 » 4 — VО2 •
отчетливо выраженные полосы с максимумами при л=270 и 225 нм
данным различных авторов [26, 40], при Л=270 нм етах=
(6 4- 8) • 103 г • атом-1 • см-1. Возможно, эти колебания связаны
различными в условиях эксперимента температурой и ионной
силой растворов.
В спектре иона НУО|~ (рис. 7, б) зафиксированы также две
олосы поглощения при Х=265 и Х=210 нм. В этом случае боль-
шей интенсивностью отличается полоса, лежащая в коротковолновой
области. Аналогичный характер имеет спектр метаванадат-иона
рис. 8). Максимум поглощения менее интенсивной полосы в нем
лежит при X —285 нм [40], максимум другой, согласно [40], — при
X=240, а по [28] — при Z^213 нм.
Спектр иона VO? по форме и положению напоминает спектр
дана vot, но отличается большей интенсивностью (см. рис. 8).
Декаванадат-ионы поглощают в видимой и ультрафиолетовой обла-
стях. Спектры ионов Н2У10О2Г и НУ10О1Г, а также, по-видимому,
УкРГ/ заметно отличаются друг от друга лишь интенсивностью^
да не расположением полос поглощения (рис. 9, 10).
1
Глава II
ГЕТЕРОГЕННЫЕ РАВНОВЕСИЯ
В ВОДНО-СОЛЕВЫХ СИСТЕМАХ,
ВКЛЮЧАЮЩИХ СОЕДИНЕНИЯ ВАНАДИЯ
Растворимость соединений ванадия изучена недостаточно. Больше
всего данных по растворимости ванадатов натрия, аммония и пяти-
окиси ванадия. Это объясняется в основном потребностями про-
мышленности, так как ванадаты натрия являются промежуточными
соединениями при извлечении ванадия почти из всех видов сырья,
а из перекристаллизованного метаванадата аммония получают чис-
тую пятиокись ванадия. Эти соединения, кроме того, широко ис-
пользуются как химические реактивы.
В зависимости от того, в каких ионных формах ванадий суще-
ствует в водных растворах, можно предполагать, что при различных
pH в равновесии с раствором должны существовать твердые кри-
сталлогидраты ванадатов, например щелочных металлов, с соотно-
шением М2О: V2O5=3:1 — ортованадат, 2:1— пированадат, 1:1 —
метаванадат, 6:10 — нормальный декаванадат, 5:10 и 4:10 — кис-
лые декаванадаты. Соединения такого состава действительно полу-
чены кристаллизацией из водных растворов. Кроме того, было
обнаружено, что при определенных условиях из растворов можно
выделить безводное соединение с отношением M2O:V2O5=1:3 [96].
Это соединение описывается формулой K2V6O16, хотя соответствую-
щий анион в растворах не существует. С другой стороны, тщатель-
ные исследования систем М2О—V2O5 показали, что в системе Na2O—
V2O5 соединение состава 3Na2O-5V2O5 не существует, но в анало-
гичной системе с калием оно есть [97, 98].
Как видно, состав ванадатов щелочных и других металлов за-
висит и от природы катиона, и от условий их образования. По-ви-
димому, эти обстоятельства, а также сложность идентификации
индивидуальных соединений в рассматриваемых системах привели
к тому, что в литературе можно найти указания на существование
ванадатов щелочных элементов самого различного состава. В част-
ности, только Битером [99] описаны ванадаты натрия, получаемые
кристаллизацией из водных растворов, с соотношением Na2O:V2O5,
равным 4:1; 3:1; 2:1; 1:1; 2:3; 5:8; 3:5; 4:7; 1:2; 3:7; 2:5;
1:3; 1:4. Состав соединений 4:1; 2:3; 5:8; 4:7; 1:3; 1:4 не со-
32
тствует стехиометрии простых ванадатов, предсказания о суще-
°Т 'овании которых могут быть сделаны на основании представлений
СТ ионном состоянии ванадия в водных растворах или по данным
° ового состава в системе Na2O—V2O5. Во многих случаях, однако,
д шствование ванадатов необычного состава можно объяснить не
?дя за рамки современных воззрений на природу соединений
в аДИЯ, хотя при этом приходится выдвигать дополнительные пред-
ожения, которые не имеют надежного теоретического обоснова-
ния. Необходимо признать, что вопрос о природе и составе ванада-
а еще не нашел достаточно полного разрешения. Это следует
иметь в виду при изучении химии пятивалентного ванадия.
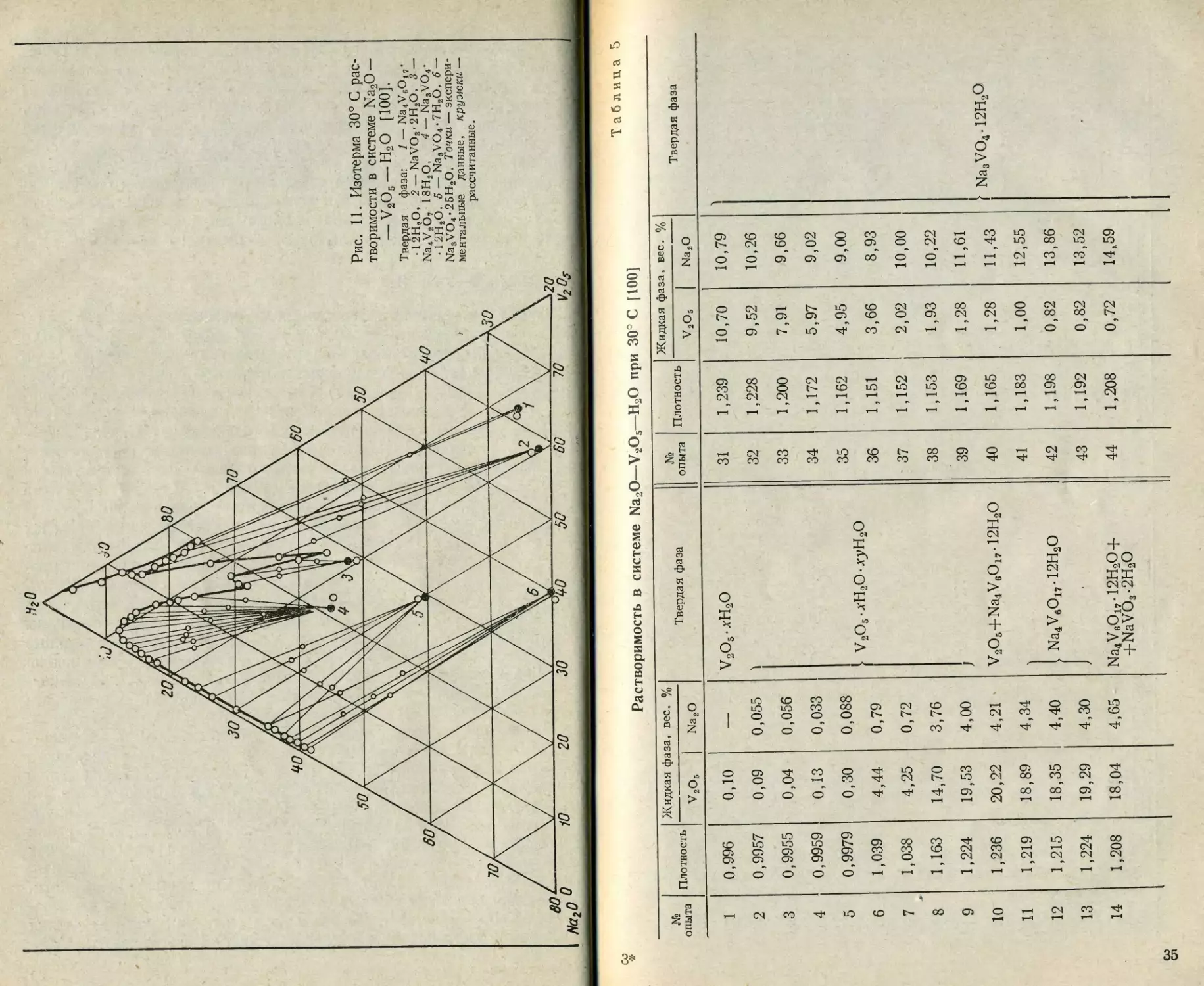
СИСТЕМА Na2O—V9O5—Н2О
Детальное изучение гетерогенных равновесий с участием вана-
датов натрия началось с 1937 г., когда Кил и Манфредо [100]
опубликовали результаты весьма тщательно выполненной работы по
исследованию растворимости в рассматриваемой системе при темпе-
ратуре 30° С (табл. 5, рис. 11). Согласующиеся результаты получи-
ли в 1938 г. Робинсон с соавторами [101], изучившие эту систему
при 25° С. В 1940 г. аналогичная работа проведена С. С. Макаро-
вым и А. Т. Репой [102], которые получили изотермы растворимости
при 25 и 75° С. В этих работах состав твердых фаз устанавливали
методом Скрейнемакерса. Наконец, в 1953 г. Менцель и Мюллер
[103] исследовали растворимость ванадатов натрия при 20° С, когда
соотношение Na2O:V2O5 в растворах менялось в широких пределах
от 1:1 до 252,2:1 (табл. 6). Состав твердой фазы (отношение
Na2O:V2O5) определяли непосредственным анализом тщательно про-
мытых ледяной водой и высушенных на воздухе осадков. Иденти-
фикацию фаз в осадках осуществляли рентгенографически по мето-
ду Дебая-Шерера. Данные работ [100] и [103] с учетом разницы
в температурах хорошо согласуются между собой (рис. 12). Данные
С. С. Макарова и А. Т. Репы сильно отличаются от них как по
величине растворимости, так и по составу твердых фаз. А. П. Яцен-
ко и С. П. Яценко [104] исследовали растворимость ортованадата
натрия при 20° С в щелочных алюминатных и галлатных растворах.
Установлено, что равновесные концентрации ванадия в алюминат-
ных растворах практически не отличаются от таковых для чисто
щелочных растворов [103]. В галлатных растворах наблюдаются
значительные отклонения, зависящие от концентрации галлата
(рис. 13). Растворимость пятиокиси ванадия в щелочных алюминат-
ных растворах при температурах 20, 30, 45 и 75° С, концентрации
Щелочи 8,4—36,5% (в процентах Na2O) и мольных отношениях
окиси натрия и глинозема 4,5; 17,0; 32,0 исследовали В. Д. По-
номарев с соавторами [105]. Установлено, что растворимость пяти-
окиси ванадия сильно зависит от концентрации щелочи и темпера-
тУры растворов (рис. 14). Изменение концентрации окиси алюминия
исходных растворах практически не влияет на растворимость V2O5.
Заказ № 256
33
3
Таблица 5
Растворимость в системе Na2O—V2O6—Н2О при 30° С [ 100]
№ опыта Плотность Жидкая фаза, вес. % Твердая фаза № опыта Плотность Жидкая фаза, вес. % Твердая фаза
V2O5 Na2O V2O5 Na2O
1 0,996 0,10 — V2O5-xH2O 31 1,239 10,70 10,79
2 0,9957 0,09 0,055 32 1,228 9,52 10,26
3 0,9955 0,04 0,056 33 1,200 7,91 9,66
4 0,9959 0,13 0,033 34 1,172 5,97 9,02
5 0,9979 0,30 0,088 35 1,162 4,95 9,00
V2O5-xH2O-xyH2O
6 1,039 4,44 0,79 36 1,151 3,66 8,93
7 1,038 4,25 0,72 - 37 1,152 2,02 10,00
8 1,163 14,70 3,76 38 1,153 1,93 10,22
9 1,224 19,53 4,00 39 1,169 1,28 11,61
10 1,236 20,22 4,21 * V2O6+Na4V6O17-12H2O 40 1,165 1,28 11,43 Na3VO4-12H2O
И 1,219 18,89 4,34 41 1,183 1,00 12,55
12 1,215 18,35 4,40 I Na4VeO17-12H2O 42 1,198 0,82 13,86
13 1,224 19,29 4,30 J 43 1,192 0,82 13,52 •
14 1,208 18,04 4,65 Na4V6O17-12H2O+ 44 1,208 0,72 14,59
+NaVO3-2H2O
Со сл
36
Таблица 6
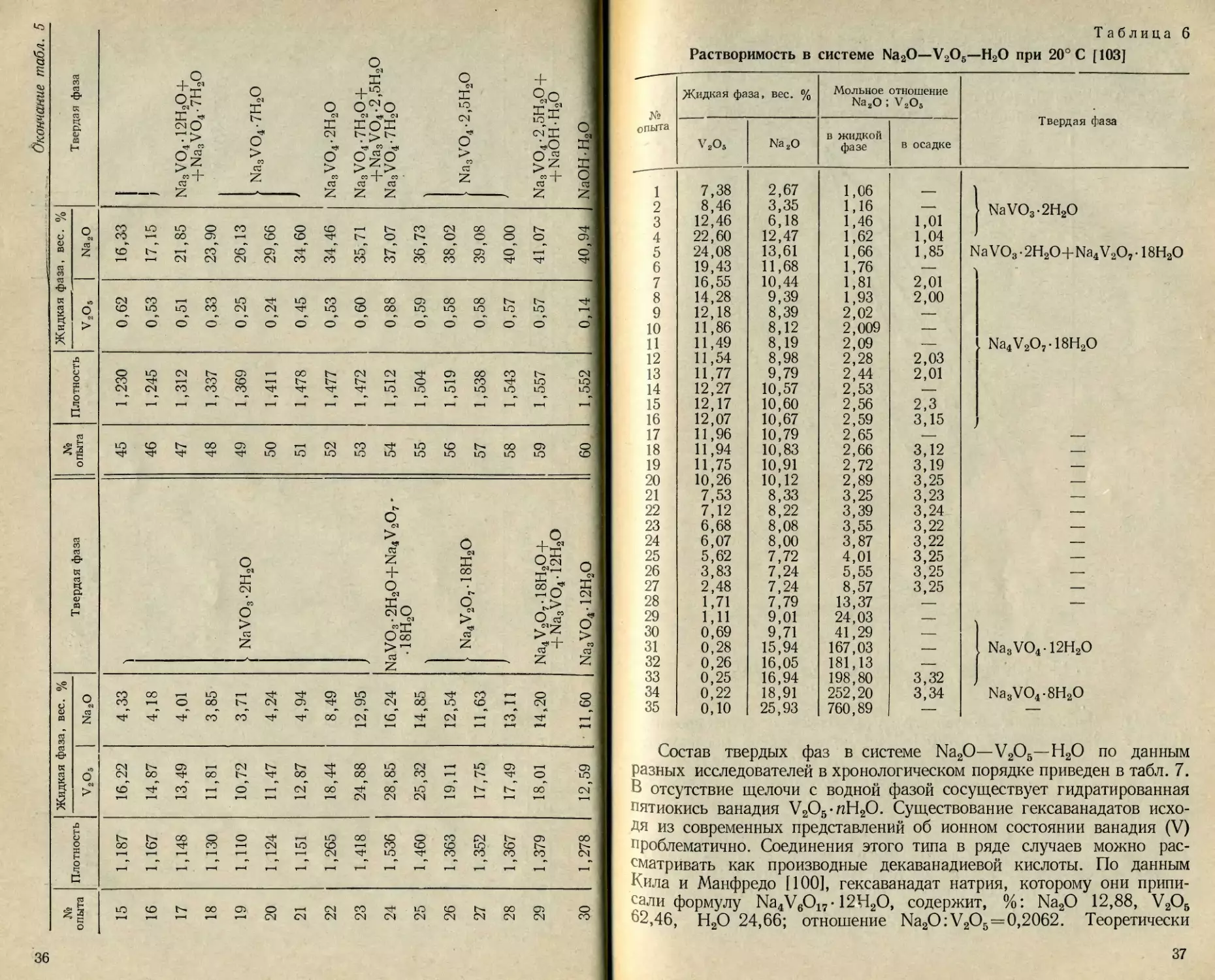
Растворимость в системе Na2O—V2O5—Н2О при 20° С [103]
№ опыта Жидкая фаза, вес. % Мольное отношение Na2O ; V2O5 Твердая фаза
v2o5 Na2O В жидкой фазе в осадке
1 7,38 2,67 1,06
2 8,46 3,35 1,16 —* } NaVO3-2H2O
3 12,46 6,18 1,46 1,01
4 22,60 12,47 1,62 1,04
5 24,08 13,61 1,66 1,85 NaVO3-2H2O+Na4V2O7- 18Н2О
6 19,43 11,68 1,76 —
7 16,55 10,44 1,81 2,01
8 14,28 9,39 1,93 2,00
9 12,18 8,39 2,02 —
10 11,86 8,12 2,009 —
11 11,49 8,19 2,09 — Na4V2O7-18H2O
12 11,54 8,98 2,28 2,03
13 11,77 9,79 2,44 2,01
14 12,27 10,57 2,53 —
15 12,17 10,60 2,56 2,3
16 12,07 10,67 2,59 3,15
17 11,96 10,79 2,65 — —
18 11,94 10,83 2,66 3,12 —
19 11,75 10,91 2,72 3,19 —
20 10,26 10,12 2,89 3,25 —
21 7,53 8,33 3,25 3,23 —
22 7,12 8,22 3,39 3,24 —
23 6,68 8,08 3,55 3,22 —
24 6,07 8,00 3,87 3,22 —
25 5,62 7,72 4,01 3,25 —
26 3,83 7,24 5,55 3,25 —
27 2,48 7,24 8,57 3,25 —
28 1,71 7,79 13,37 — —
29 1,11 9,01 24,03 —
30 0,69 9,71 41,29 —
31 0,28 15,94 167,03 — Na3VO4-12H2O
32 0,26 16,05 181,13 —
33 0,25 16,94 198,80 3,32
34 0,22 18,91 252,20 3,34 Na3VO4-8H2O
35 0,10 25,93 760,89 — —
остав твердых фаз в системе Na2O—V2O5— Н2О по данным
разных исследователей в хронологическом порядке приведен в табл. 7.
В отсутствие щелочи с водной фазой сосуществует гидратированная
пятиокись ванадия V2O5-nH2O. Существование гексаванадатов исхо-
дя из современных представлений об ионном состоянии ванадия (V)
проблематично. Соединения этого типа в ряде случаев можно рас-
сматривать как производные декаванадиевой кислоты. По данным
ила и Манфредо [100], гексаванадат натрия, которому они припи-
сали формулу Na4V6O17-12Н2О, содержит, %: Na2O 12,88, V2O5
62,46, Н2О 24,66; отношение Na2O:V2O5=0,2062. Теоретически
37
гексаванадат указанного состава должен содержать Na2O ‘14,00,
V2O5 61,6%; Na2O:V2O5=0,2275. Для декаванадата натрия
Na6V10O28 • пН2О соотношение Na2O:V2O5 = 0,2080. Таким образом]
состав соединения, полученного в рассматриваемой работе, лучше
всего отвечает декаванадату и соответствует формуле Na6V10O28.
Матвее. %
*- /
Рис. ]2. Растворимость в системе Na2O-V2O5— Н2О.
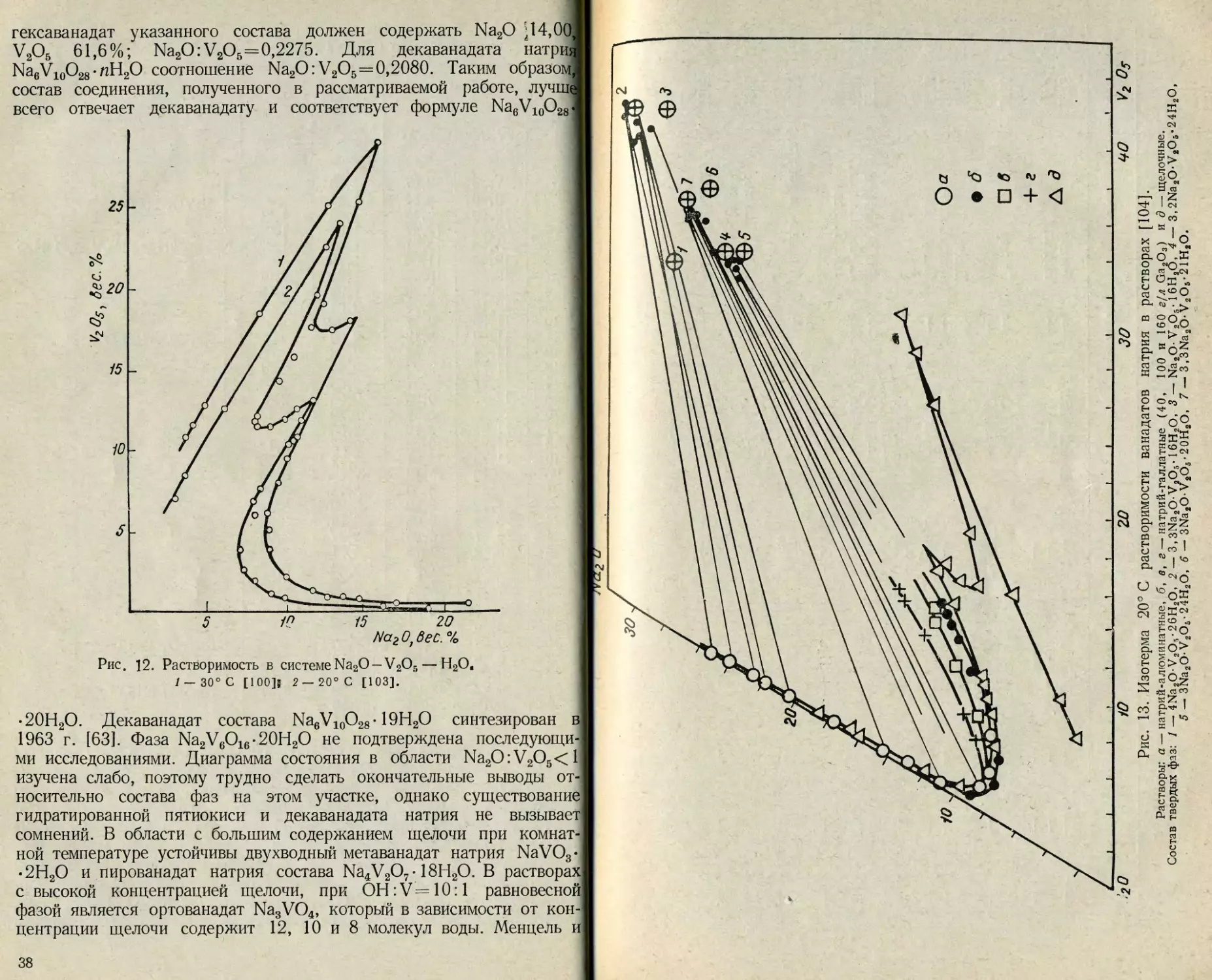
/ — 30° С [100]| 2 —20° С [103].
5
•20Н2О. Декаванадат состава Na6V10O28-19Н2О синтезирован в
1963 г. [63]. Фаза Na2V6O16-20H2O не подтверждена последующи-
ми исследованиями. Диаграмма состояния в области Na2O:V2O
изучена слабо, поэтому трудно сделать окончательные выводы от-
носительно состава фаз на этом участке, однако существование
гидратированной пятиокиси и декаванадата натрия не вызывает
сомнений. В области с большим содержанием щелочи при комнат-
ной температуре устойчивы двухводный метаванадат натрия NaVO3«
• 2Н2О и пированадат натрия состава Na4V2O7- 18Н2О. В растворах
с высокой концентрацией щелочи, при ОН :V= 10:1 равновесной
фазой является ортованадат Na3VO4, который в зависимости от кон-
центрации щелочи содержит 12, 10 и 8 молекул воды. Менцель и
38
Рис. 13. Изотерма 20° С растворимости ванадатов натрия в растворах [104].
Растворы: а — натрий-алюминатные, б, в, г — натрий-галлатные (40, 100 и 160 г/л Ga2O3) и д — щелочные.
Состав твердых фаз: 1 — 4Na2O-V2O5 • 26Н2О, 2 — 3,3Na2O-V2O5-1 6Н2О, 3 — Na2O- V2O5-1 6Н2О, 4 — 3,2Na2O-V2O6-24H2O,
5 — 3Na2O-V2O5-24H2O, 6 - 3Na2OV2O6• 20H2O, 7 — 3,3Na2OV2O6-21H2O.
Примечание. Б. в, — безводное соединение, -4-лН2О — то же, плюс п молекул воды.
40
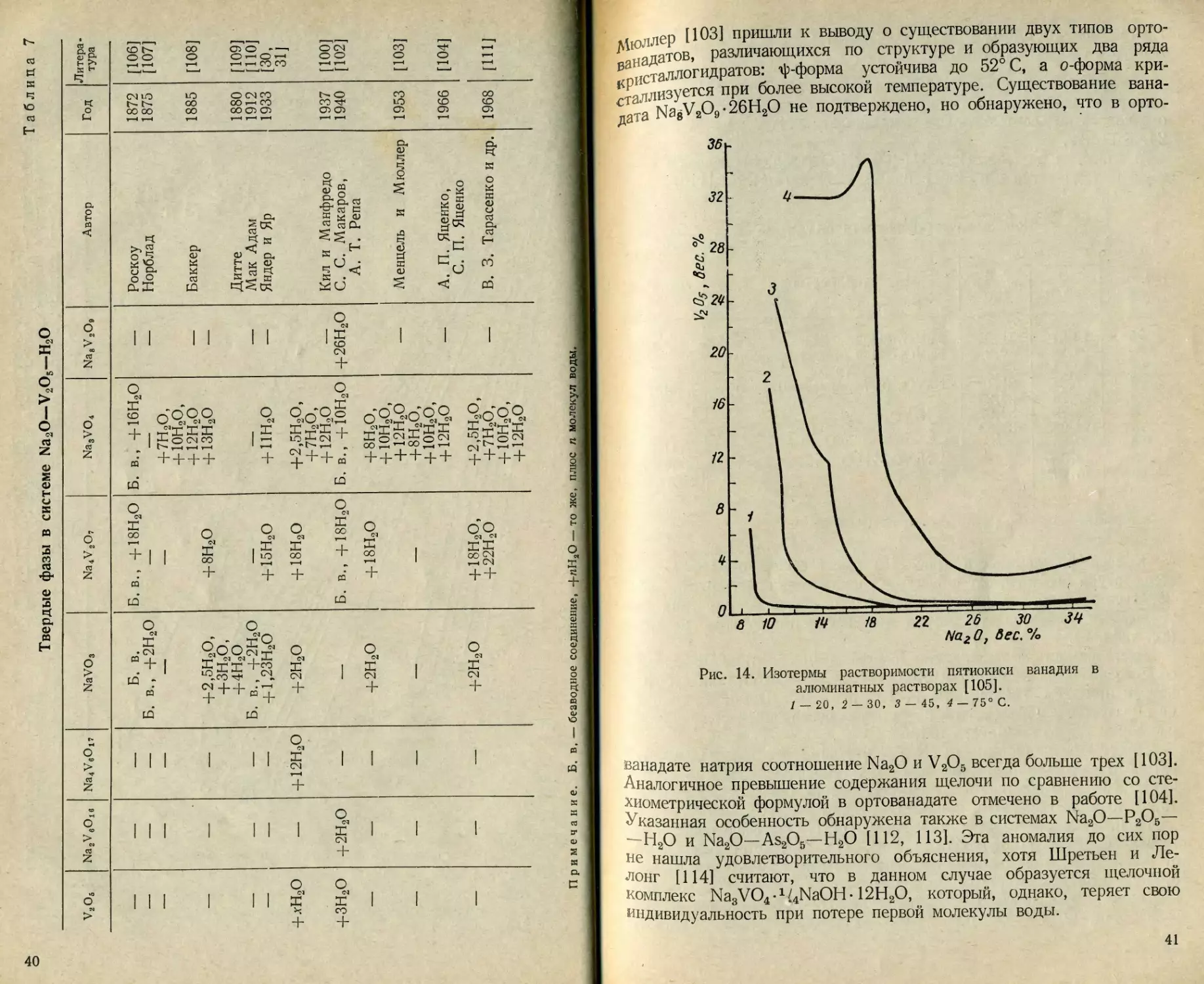
мюллер [ЮЗ] пришли к выводу о существовании двух типов орто-
™ патов, различающихся по структуре и образующих два ряда
₽писталлогидратов: ф-форма устойчива до 52° С, а о-форма кри-
сталлизуется при более высокой температуре. Существование -------
С a NagV2O9 • 26Н2О не подтверждено, но обнаружено, что в
вана-
орто-
Рис. 14. Изотермы растворимости пятиокиси ванадия в
алюминатных растворах [105].
/ — 20, 2 — 30, 3 — 45, 4 — 75° С.
ванадате натрия соотношение Na2O и V2O5 всегда больше трех [103].
Аналогичное превышение содержания щелочи по сравнению со сте-
хиометрической формулой в ортованадате отмечено в работе [104].
^казанная особенность обнаружена также в системах Na2O—Р2О5—
^Н2О и Na2O—As2O5—Н2О [112, 113]. Эта аномалия до сих пор
не нашла удовлетворительного объяснения, хотя Шретьен и Ле-
лонг [114] считают, что в данном случае образуется щелочной
Комплекс Na3VO4 •1 /4NaOH • 12Н2О, который, однако, теряет свою
индивидуальность при потере первой молекулы воды.
41
РАСТВОРИМОСТЬ ВАНАДАТОВ НАТРИЯ В ВОДЕ
И ВОДНО-СОЛЕВЫХ РАСТВОРАХ
Величина растворимости декаванадата натрия при 30° С, рассчи
тайная из данных табл. 5, для опыта 10, когда состав раствора i
осадка точно соответствует декаванадату Na6V10O28, составляе'
24,43 вес. %. 1
Политерма растворимости NaVO3-2H2O (табл. 8) изучена в ин
тервале температур 25—75° С Мак Адамом и Пайрлом [110].
Таблица
Политерма растворимости NaVO3-2H2O
t, °C NaVO3 Твердая фаза Литература
вес. % г/100 г Н2О
25 13,27 15,3 NaVO3-2H2O
40 23,20 30,2 NaVO3-2H2O (метастабильная)
60 40,61 68,4 То же
25 17,42 21,10 NaVO3 (метастабильная) [ПО]
40 20,78 26,23
60 24,79 32,97 1 NaVO3
75 27,98 38,83 1 t
20 9,40 — [103]
25 12,80 " — NaVO3-2H2O [115]
30 14,43* — - Наши данные
* Рассчитано по данным табл. 5 для опыта 19.
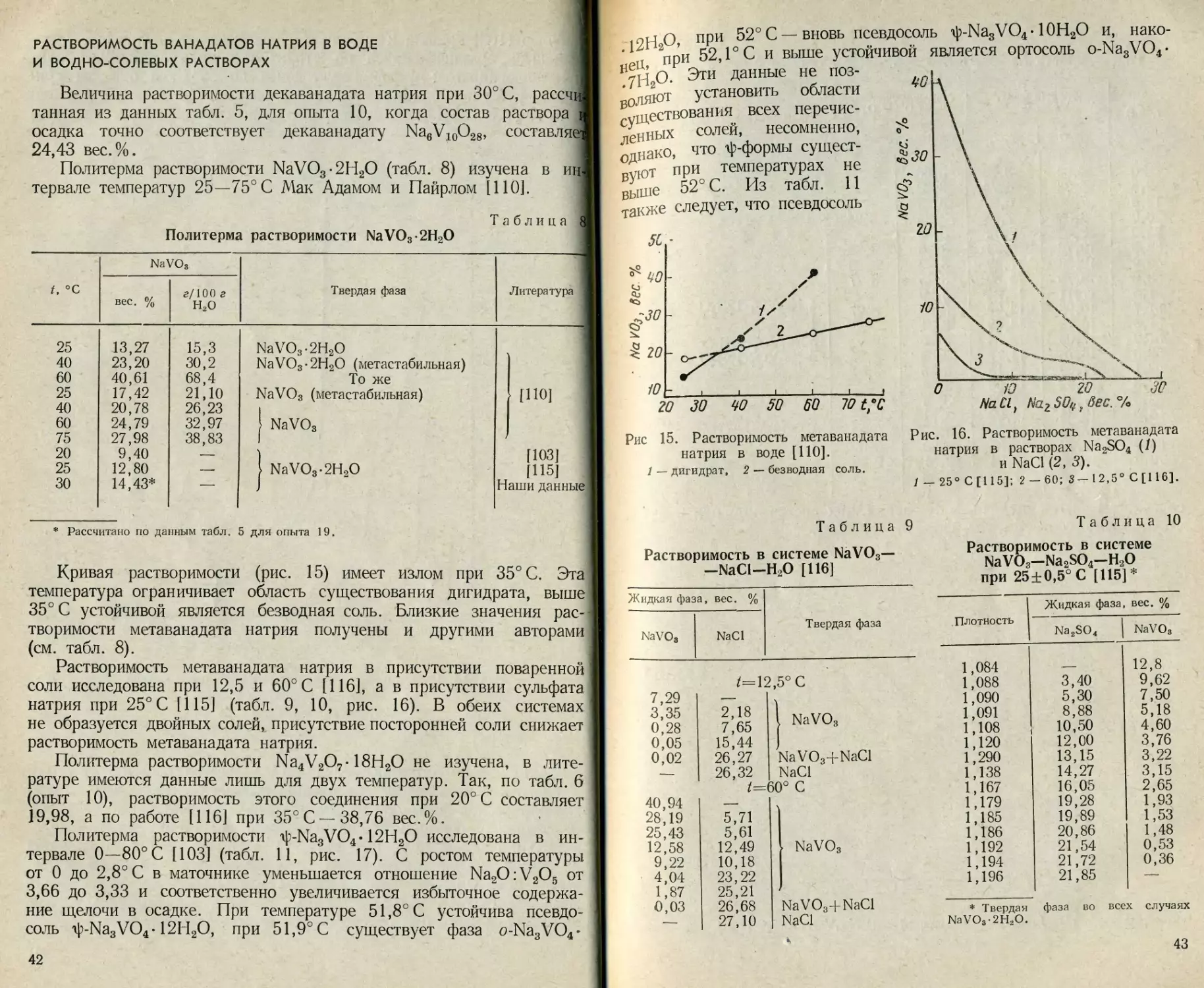
Кривая растворимости (рис. 15) имеет излом при 35° С. Эта
температура ограничивает область существования дигидрата, выше
35° С устойчивой является безводная соль. Близкие значения рас-
творимости метаванадата натрия получены и другими авторами
(см. табл. 8).
Растворимость метаванадата натрия в присутствии поваренной
соли исследована при 12,5 и 60° С [116], а в присутствии сульфата
натрия при 25° С [115] (табл. 9, 10, рис. 16). В обеих системах
не образуется двойных солей, присутствие посторонней соли снижает
растворимость метаванадата натрия.
Политерма растворимости Na4V2O7-18Н2О не изучена, в лите-
ратуре имеются данные лишь для двух температур. Так, по табл. 6
(опыт 10), растворимость этого соединения при 20° С составляет
19,98, а по работе [116] при 35° С —38,76 вес.%.
Политерма растворимости ф-Ыа3УО4-12Н2О исследована в ин-
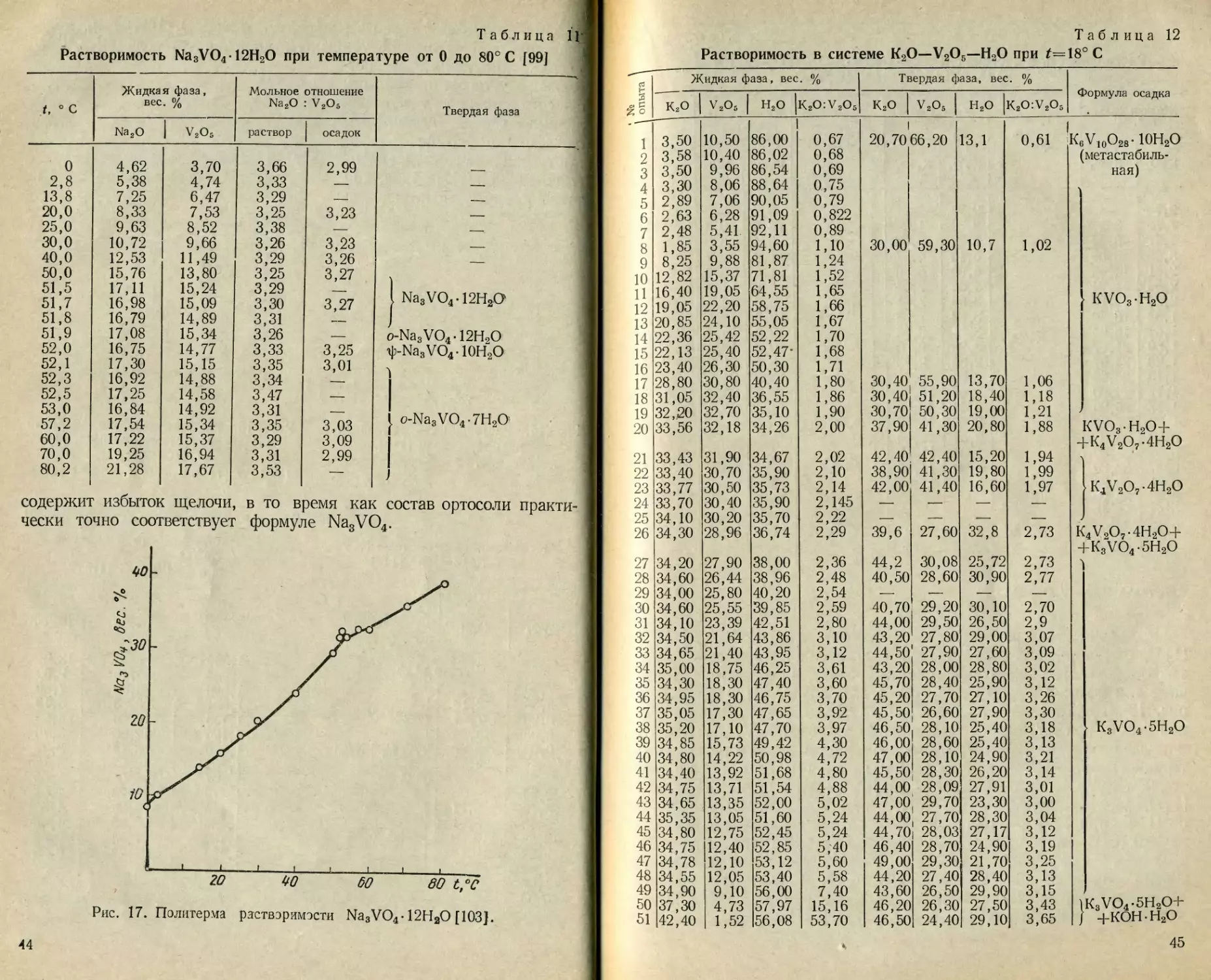
тервале 0—80 С [103] (табл. И, рис. 17). С ростом температуры
от 0 до 2,8° С в маточнике уменьшается отношение Na2O:V2O5 от
3,66 до 3,33 и соответственно увеличивается избыточное содержа-
ние щелочи в осадке. При температуре 51,8° С устойчива псевдо-
соль ф-Ыа3УО4-12Н2О, при 51,9°С существует фаза o-Na3VO4-
42
12Н2°> пРи 52оС —вновь псевдосоль ip-NasVO4-ЮН2О и,
*ец, при 52, Г С и выше устойчивой является ортосоль o-Na3VO4*
НуН2О. Эти данные не поз-
воляют установить области
существования всех перечис-
ленных солей, несомненно,
однако, что ф-формы сущест-
вуют при температурах не
выше 52° С. Из табл. 11
также следует, что псевдосоль
нако-
Рис. 16. Растворимость метаванадата
натрия в растворах Na9S04 (/)
и NaCl (2, 3).
1 — 25° С [115]; 2 — 60; 3— 12,5° С [116].
Рис 15. Растворимость метаванадата
натрия в воде [110].
1 — дигидрат, 2 — безводная соль.
Таблица 10
Таблица 9
Растворимость в системе NaVO3—
—NaCl—Н2О [116]
Жидкая фаза, вес. % Твердая фаза
NaVO, NaCl
/=12,5° С
7,29
3,35 2,18
0,28 7,65 1 NaVO3
0,05 15,44
0,02 26,27 NaVO34-NaCl
26,32 NaCl
50° С
40,94
28,19 5,71
25,43 5,61
12,58 12,49 NaVO3
9,22 10,18
4,04 23,22
1,87 25,21
0,03 26,68 NaVO3+NaCl
— 27,10 NaCl
Растворимость в системе
NaVOg—Na2SO4—Н2О
при 25±0,5°С [115]*
Плотность Жидкая фаза, вес. %
Na2SO4 NaVOs
1,084 12,8
1,088 3,40 9,62
1,090 5,30 7,50
1,091 8,88 5,18
1,108 10,50 4,60
1,120 12,00 3,76
1,290 13,15 3,22
1,138 14,27 3,15
1,167 16,05 2,65
1,179 19,28 1,93
1,185 19,89 1,53
1,186 20,86 1,48
1,192 21,54 0,53
1,194 21,72 0,36
1,196 21,85 - —
* Твердая
NaVO3-2H2O.
фаза во всех случаях
43
Таблица 11
Растворимость Na3VO412H2O при температуре от 0 до 80° С [99]
t, ° с Жидкая фаза, вес. % Мольное отношение Na2O : V 2O5 —— ' 1 Твердая фаза
Na2O v2o5 раствор осадок
0 2,8 13,8 20,0 25,0 30,0 40,0 50,0 51,5 51,7 51,8 51,9 52,0 52,1 52,3 52,5 53,0 57,2 60,0 70,0 80,2 4,62 5,38 7,25 8,33 9,63 10,72 12,53 15,76 17,11 16,98 16,79 17,08 16,75 17,30 16,92 17,25 16,84 17,54 17,22 19,25 21,28 3,70 4,74 6,47 7,53 8,52 9,66 11,49 13,80 15,24 15,09 14,89 15,34 14,77 15,15 14,88 14,58 14,92 15,34 15,37 16,94 17,67 3,66 3,33 3,29 3,25 3,38 3,26 3,29 3,25 3,29 3,30 3,31 3,26 3,33 3,35 3,34 3,47 3,31 3,35 3,29 3,31 3,53 2,99 3,23 3,23 3,26 3,27 3,27 3,25 3,01 3,03 3,09 2,99 " . — О V ' о V Z М и3 03 со w . со со < £> g 11 11111 Л; to ъ О° Q
содержит избыток щелочи, в то время как состав ортосоли практи
чески точно соответствует формуле Na3VO4.
-----1-----1... - i—-j । - i- .... । t _
20 40 00 80 t,°C
Рис. 17. Политерма раствэримэсти Na3VO4-12H2O [103].
44
Таблица 12
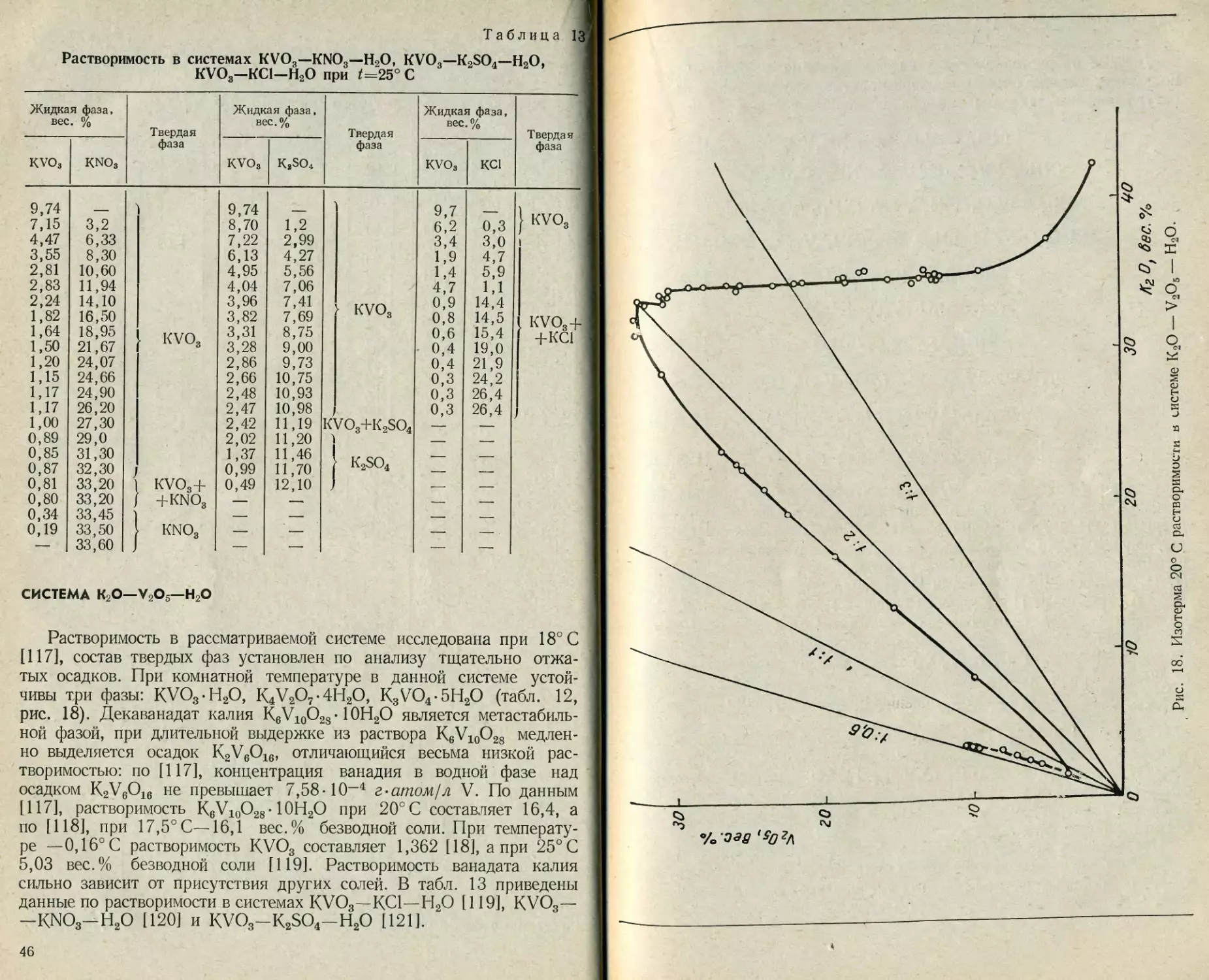
Растворимость в системе К2О—V2O5—Н2О при £=18° С
№ ) опыта Жидкая фаза, вес. % Твердая фаза, вес. % Формула осадка
к2о V2os н2о K2O:V2O5 к2о v2o5 н2о K2O:V2O5
1 3,50 10,50 86,00 0,67 1 20,70 66,20 13,1 1 0,61 1 C6V]0O28. ЮН2О
2 3,58 10,40 86,02 0,68 (метастабиль-
3 3,50 9,96 86,54 0,69 ная)
4 3,30 8,06 88,64 0,75
5 2,89 7,06 90,05 0,79
6 2,63 6,28 91,09 0,822
7 2,48 5,41 92,11 0,89
8 1,85 3,55 94,60 1,10 30,00 59,30 10,7 1,02
9 8,25 9,88 81,87 1,24
10 12,82 15,37 71,81 1,52
11 12 16,40 19,05 19,05 22,20 64,55 58,75 1,65 1,66 - KVO3-H2O
13 20,85 24,10 55,05 1,67
14 22,36 25,42 52,22 1,70
15 22,13 25,40 52,47- 1,68
16 23,40 26,30 50,30 1,71
17 28,80 30,80 40,40 1,80 30,40 55,90 13,70 1,06
18 31,05 32,40 36,55 1,86 30,40 51,20 18,40 1,18
19 32,20 32,70 35,10 1,90 30,70 50,30 19,00 1,21 >
20 33,56 32,18 34,26 2,00 37,90 41,30 20,80 1,88 KVO3-H2O+ 4-K4V2O7 -4Н2О
21 33,43 31,90 34,67 2,02 42,40 42,40 15,20 1,94 ч
22 33,40 30,70 35,90 2,10 38,90 41,30 19,80 1,99
23 33,77 30,50 35,73 2,14 42,00 41,40 16,60 1,97 • K4V2Oj-4H2O
24 33,70 30,40 35,90 2,145 — —
25 34,10 30,20 35,70 2,22 — — — ——
26 34,30 28,96 36,74 2,29 39,6 27,60 32,8 2,73 K4V,O7-4H2O+ +K3VO4-5H2O
27 34,20 27,90 38,00 2,36 44,2 30,08 25,72 2,73 ч
28 34,60 26,44 38,96 2,48 40,50 28,60 30,90 2,77
29 34,00 25,80 40,20 2,54 — —• - — " —
30 34,60 25,55 39,85 2,59 40,70 29,20 30,10 2,70
31 34,10 23,39 42,51 2,80 44,00 29,50 26,50 2,9
32 34,50 21,64 43,86 3,10 43,20 27,80 29,00 3,07
33 34,65 21,40 43,95 3,12 44,50 27,90 27,60 3,09
34 35,00 18,75 46,25 3,61 43,20 28,00 28,80 3,02
35 34,30 18,30 47,40 3,60 45,70 28,40 25,90 3,12
36 34,95 18,30 46,75 3,70 45,20 27,70 27,10 3,26
37 35,05 17,30 47,65 3,92 45,50 26,60 27,90 3,30
38 35,20 17,10 47,70 3,97 46,50 28,10 25,40 3,18 . K3VO4-5H2O
39 34,85 15,73 49,42 4,30 46,00 28,60 25,40 3,13
40 34,80 14,22 50,98 4,72 47,00 28,10 24,90 3,21
41 34,40 13,92 51,68 4,80 45,50 28,30 26,20 3,14
42 34,75 13,71 51,54 4,88 44,00 28,09 27,91 3,01
43 34,65 13,35 52,00 5,02 47,00 29,70 23,30 3,00
44 35,35 13,05 51,60 5,24 44,00 27,70 28,30 3,04
45 34,80 12,75 52,45 5,24 44,70 28,03 27,17 3,12
46 34,75 12,40 52,85 5,40 46,40 28,70 24,90 3,19
47 34,78 12,10 53,12 5,60 49,00 29,30 21,70 3,25
48 34,55 12,05 53,40 5,58 44,20 27,40 28,40 3,13
49 34,90 9,10 56,00 7,40 43,60 26,50 29,90 3,15 1K3VO4-5H2O+
50 37,30 4,73 57,97 15,16 46,20 26,30 27,50 3,43
51 42,40 1,52 56,08 53,70 46,50 24,40 29,10 3,65 / +KOH-H2O
45
Таблица 13
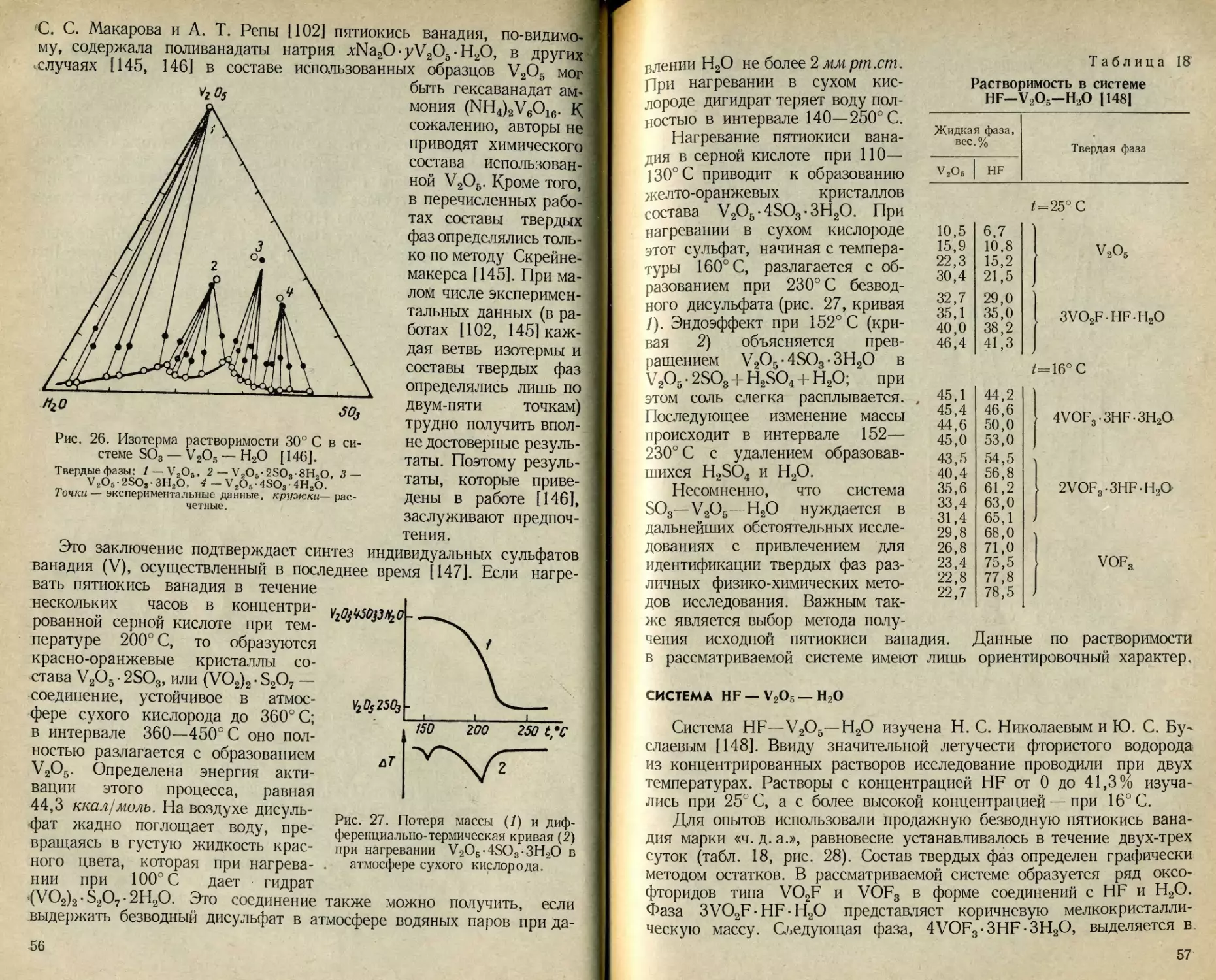
Растворимость в системах KVO3—KNO3—НоО, KVO3—K2SO4-—H2O,
KVO3-KC1-H2O при f=25° С
Жидкая фаза, вес. % Твердая фаза Жидкая фаза, вес. % Твердая фаза Жидкая фаза, вес. % Твердая фаза
KVO3 KNO3 KVO, K,so4 KVO3 КС1
9,74 7,15 4,47 3,55 2,81 2,83 2,24 1,82 1,64 1,50 1,20 1,15 1,17 1,17 1,00 0,89 0,85 0,87 0,81 0,80 0,34 0,19 3,2 6,33 8,30 10,60 11,94 14,10 16,50 18,95 21,67 24,07 24,66 24,90 26,20 27,30 29,0 31,30 32,30 33,20 33,20 33,45 33,50 33,60 KVO3 KVO3+ / 4-KNO3 1 KNO3 1 9,74 8,70 7,22 6,13 4,95 4,04 3,96 3,82 3,31 3,28 2,86 2,66 2,48 2,47 2,42 2,02 1,37 0,99 0,49 1,2 2,99 4,27 5,56 7,06 7,41 7,69 8,75 9,00 9,73 10,75 10,93 10,98 11,19 11,20 11,46 11,70 12,10 KVO3 KVO3+K,SO4 1 f K2SO4 9,7 6,2 3,4 1,9 1,4 4,7 0,9 0,8 0,6 0,4 0,4 0,3 0,3 0,3 0,3 3,0 4,7 5,9 1,1 14,4 14,5 15,4 19,0 21,9 24,2 26,4 26,4 KVO3 KVO3 + +КС1 1
СИСТЕМА К2О—V2O5—Н2О
Растворимость в рассматриваемой системе исследована при 18 С
[117], состав твердых фаз установлен по анализу тщательно отжа-
тых осадков. При комнатной температуре в данной системе устой-
чивы три фазы: KVO3-H2O, КЛ72О7-4Н2О, K3VO4-5H2O (табл. 12,
рис. 18). Декаванадат калия K6V10O28 • 10Н2О является метастабиль-
ной фазой, при длительной выдержке из раствора K6V10O28 медлен-
но выделяется осадок K2V6O16, отличающийся весьма низкой рас-
творимостью: по [117], концентрация ванадия в водной фазе над
осадком K2V6O16 не превышает 7,58-10-4 г-атом 1л V. По данным
[117], растворимость K6V10O28- ЮН2О при 20° С составляет 16,4, а
по [118], при 17,5° С—16,1 вес. % безводной соли. При температу-
ре —0,16° С растворимость KVO3 составляет 1,362 [18], а при 25° С
5,03 вес. % безводной соли [119]. Растворимость ванадата калия
сильно зависит от присутствия других солей. В табл. 13 приведены
данные по растворимости в системах KVO3—КС1—Н2О [119], KVO3—
—KNO3-H2O [120] и KVO3-K2SO4-H2O [121].
46
Рис. 18. Изотерма 20° С растворимости в системе К2О— V2O
СИСТЕМА NH3—V2O5—Н2О
Данные по растворимости ванадатов аммония в различных вод
но-солевых системах имеют важное практическое значение. В лите
ратуре описаны ванадаты аммония различного состава:
(NH4)2O«V2O5, или NH4VO3 [122-124]
(NH4)2O-V2O5-H2O, или NH4VO3.H2O [125]
2(NH4)2O• 3V2O5• 14H2O, или (NH4)4V6O17.14H2O [125]
3(NH4)2O-5V2O5-6H2O, или (NH4)6V10O28 • 6H2O [126, 127]
(NH4)2O • 2 V2O5 • 5H2O [126, 128]
3 (NH4)2O-7V2O5-4H2O [123]
2 (NH4)2O-5V2O5-5H2O [126, 128]
(NH4)2O.3V2O5, или (NH4)2V6O16 [122, 123,126, 129]
4(NH4)2O-13У2О5-18(или 22)H2O [126, 130]
3 (NH4)2O40V2O5-15 (или 18)H2O [126, 130]
Однако в большинстве случаев идентификация соединений была
основана только на химических анализах. Система NH3—V2O5—Н2О
впервые была изучена Кельмерсом [129]. Состав твердых фаз (рис. 19)
устанавливался по Скрейнемакерсу методом алгебраической экстра-
поляции и рентгенографически. Позднее эта система изучалась
П. И. Федоровым с соавторами [125] (рис. 20), В. Г. Бамбуровым
и А. А. Ивакиным [131] (табл. 14, рис. 21). В работах [129, 131]
получены вполне согласующиеся результаты: при 20° С устойчивы
только две твердые фазы: безводные метаванадат и гексаванадат
аммония (NH4VO3 и (NH4)2V6O16). Гексаванадат аммония, по-види-
мому, отличается низкой растворимостью, а метаванадат — умерен-
ной (достигает максимального значения при концентрации 5,5 вес. %
NH3 в растворе).
По данным работы [125], в рассматриваемой системе (см. рис. 20)
устойчивы одноводный метаванадат аммония NH4VO3-H2O и гекса-
ванадат аммония состава (NH4)4V6O17-14Н2О. Авторы объясняют
эти расхождения в составе твердых фаз в сравнении с работой [129]
влиянием температуры и использованием разных методов определения
состава твердой фазы.
Что касается других поливанадатов аммония, описанных в лите-
ратуре, то все они получены при выпаривании подкисленных рас-
творов и, по-видимому, при обычных температурах либо нестабильны,
либо отличаются высокой растворимостью. Кроме того, нет доста-
48
очных доказательств, что все описанные поливанадаты являются
ндивидуальными фазами, хотя соединение состава 3(NH4)2O-
• 5V2O5-6H2O соответствует нормальному декаванадату (NH4)6V10O28-
«6Н2О, возможность существования которого не вызывает сомне-
ния.
HZQ
Рис. 19. Изотерма 30° С растворимости в системе NH3— V2O5 — H2'J,
область низких концентраций NH3 [129].
Растворимость NH4VO3 в воде изучалась неоднократно [45,
131 — 135] и основные результаты представлены на рис. 22. Боль-
шая часть данных, полученных различными исследователями, удов-
летворительно согласуется между собой, хотя в некоторых случаях
различие достигает 30% (относительных). Указанное различие не
случайно, и его причиной является медленность достижения равно-
весия растворения NH4VO3 в воде, вероятно, вследствие влияния
на этот процесс медленно устанавливающихся гидролитических рав-
новесий с участием метаванадат-ионов [28] и значительных отличий
в структуре твердой фазы — метаванадата аммония и тетрамерных
(или тримерных) метаванадат-ионов в растворе. На рис. 22 через
экспериментальные точки проведена плавная кривая. Полученные
таким образом при различных температурах величины растворимости
NH4VO3, по-видимому, будут лучше всего соответствовать действи-
тельным значениям, особенно в области температур 10—60° С, где
имеется наибольшее число данных:
4 Заказ № 256 49
I, °C NH4VO3.
вес. %
t, °C
nh4vo3,
вес. %
10...............0,37
20...............0,63
30...............0,95
40...............1,30
50...............1,75
60...............2,50
70...............3,45
80...............4,50
90...............5,50
Растворимость в системе NH3—V2O6—H2O
Таблица 14
f=30° с [129] t= =20°С [131]*
Жидкая фаза, г/л Плотность Жидкая фаза,, вес. %
NH3 v2o5 Твердая фаза раствора, г/см? NH3 v2o5
0,4 0,06 0,12 0,12 0,13 0,12 0,13 0,12 0,14 0,12 0,17 0,18 0,23 0,29 0,31 0,38 0,50 0,63 0,84 1,20 1,23 1,26 1,54 1,55 1,88 3,4 4,6 5,4 5,9 6,2 6,6 7,7 8,1 9,4 9,9 10,3 12,2 12,7 0,34 0,49 0,68 0,69 0,70 0,68 0,84 0,68 0,79 0,66 0,63 0,63 0,63 0,64 0,68 0,68 0,71 0,85 0,87 1,15 1,00 1,04 1,11 1,25 1,25 1,75 1,76 1,78 1,70 1,80 1,80 1,70 1,63 1,56 1,55 1,38 1,25 1,20 7 > (NH4)2V6OJ6 (NH4)2V6O16 (NH4)2VeOl6+NH4VO3 (NH4)2VeO16+NH4VO3 NH4VO3 > NH4VO3 1,0029 0,9953 0,9947 0,9938 0,9929 0,9925 0,9912 0,9898 0,9882 0,9862 0,9899 0,9847 0,9841 0,9841 0,9795 0,9787 0,9747 0,9734 0,9730 0,9728 0,9700 0,9672 0,9606 0,9599 0,9592 0,9574 0,9555 0,9441 0,8891 0,10 0,88 1,12 1,53 1,77 2,16 2,52 3,10 3,38 3,60 3,84 4,48 4,71 5,53 5,64 6,40 6,56 6,95 7,46 7,60 7,90 8,87 9,29 9,30 9,51 9,88 10,30 12,50 20,61 0,54 0,79 0,93 1,13 1,26 1,38 1,45 1,40 1,63 1,66 1,71 1,78 1,79 1,81 1,79 1,79 1,78. 1,77 1,75- 1,73 1,71 1,67 1,59 1,60 1,54 1,54 1,50 1,32 0,65
* Твердая фаза во всех опытах NH4VO3.
50
4*
nh^ vo3, Вес %
/г
Рис. 24. Растворимость в системе NH4VO3 — NH4C1 — Н2О
при 25° С.
Поданным: / — [135], 2 —[134].
система
для рас-
Высаливающее действие хлорида
метаванадата аммония применяется i
надия из растворов, а также при
получении метаванадата аммония
и очистке его перекристаллиза-
цией из водных растворов [4,
136—139], поэтому
NH4VO3—NH4C1-H2O исследо-
валась неоднократно. Первые
систематические данные
сматриваемой системы были полу-
чены В. В. Андреевым [133], а
затем П. И. Федоровым с соав-
торами [134] и П. И. Пинаевым
[135] (табл. 15, рис. 23, 24).
В работах [134] и [135] за исклю-
чением расхождения в величине
растворимости метаванадата аммо-
ния в чистой воде получены впол-
не согласующиеся результаты.
Вследствие сильного высалива-
ния метаванадата аммония хло-
ристым аммонием на диаграм-
мах растворимости отсутствует
поле кристаллизации последнего
и полное высаливание метава-
надата аммония происходит в
растворах, не насыщенных хло-
ристым аммонием.
С повышением температуры*
увеличивается расход хлористого
аммония на водные растворы
практике при выделении ва-
Та блица 15
Растворимость метаванадата
аммония в водных растворах
хлористого аммония *
Жидкая фаза, Жидкая фаза,
вес. % вес 0/ /0
NH4VO3 iNH4C1 NH4VO3 NH4C1
<=12,5±2 °C /=35° С
0,440 1,140
0,085 0,261 0,497 0,296
0,018 0,550 0,171 0,721
Не обн. 2,270 0,149 1,067
0,080 1,574
/=25° С 0,026 2,540
0,006 4,470
0,770 — Следы 8,980
0,454 0,125
0,164 0,087 0,417 0,590 /=60° с
0,030 1,153 2,490 0,551 0,210 Следы
0,017 0,010 0,005 1,536 4,385 6,492 1,340 2,490 20,060
nh4vo3.
* Во всех случаях твердая фаза
Данные для температур 12,5; 35 и 60° С.
взяты из работы [133], для 25° С—[134].
как видно, при высаливании
аммония.
53
РАСТВОРИМОСТЬ ВАНАДАТОВ ДРУГИХ МЕТАЛЛОВ
Сведения по растворимости простых и двойных ванадатов лития,
калия, бериллия, серебра и других металлов немногочисленны. Эти
данные сгруппированы в табл. 16 и 17.
Ванадаты с соотношением М1 :V=3:5 в настоящее время рас-
сматриваются как декаванадаты, поэтому приведенным в табл. 17
соединениям KZnV5O14-8H2O и KMnV5O14-8H2O нужно приписать
формулы K2Zn2V10O28- 16Н2О и K2Mn2V10O28- 16Н2О соответственно.
Таблица 16
Растворимость ортованадата лития в воде [140]
/, ° С L13VO4, вес. % Твердая фаза z.° с Li з V O4, вес. % Твердая фаза
0 20,8 28,6 30,2 35,2 2,4 4,6 5,25 5,91 6,25 Li3VO4-9H2O / 38,4 40,0 45,0 50 60 5,09 4,2 3,70 2,8 2,6 Li3VO4-H2O
Таблица 17
Растворимость ванадатов в воде
Ванадат t, ° c Растворимость Твердая фаза Лите- ратура
KZnV5O|4 KMnV5O14 Be(VO3), AgVO3 " HgVO3 Ba(VO3), 18 25 20 20 22 0,41 вес.% 1,70 вес. % 0,1 вес.% ПР=5-10~7 ПР=26-10~8 ПР=9,72-10-14 KZnV6O|4-8H2O KMnV5O14 • 8Н2О Be(VO3),-4H,O [141] [141 (142 [143 [132 [144
СИСТЕМА SO3—V2O5—Н2О
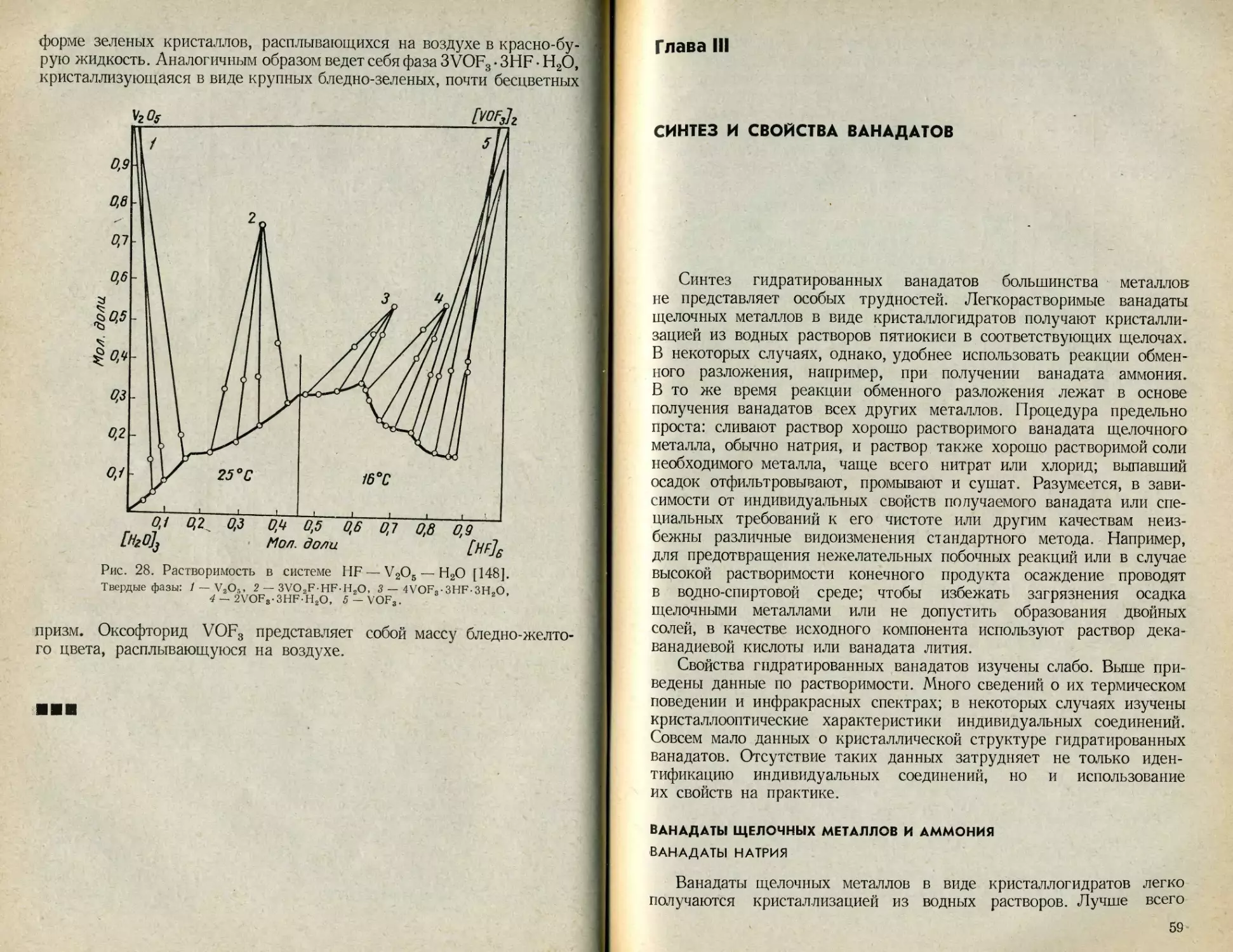
Система SO3—V2O5—Н2О изучалась неоднократно. В 1940 г.
С. С. Макаров и А. Т. Репа [102] исследовали при 25 и 75° С
растворимость пятиокиси ванадия, полученной путем приливания к
раствору Na3VO4 50 %-ной H2SO4 по реакции
2Na3 VO4 + 3H2SO4 - V2O5 + 3Na2SO4 + H2O. (26)
Они пришли к заключению, что изотерма растворимости состоит из
четырех ветвей, отвечающих существованию трех гидратных форм:
V2O5-3H2O; V2O5-2H2O; V2O5-H2O и безводной пятиокиси ванадия
(рис. 25).
Аналогичное исследование провели в 1963 г. П. И. Федоров с
соавторами [145]. Пятиокись ванадия они получали действием серной
кислоты на раствор метаванадата аммония. Осадок промывали до
исчезновения реакций на аммоний и сульфат-ион, высушивали
54
при 105° С. Данные по растворимости при 25° С получены на об-
разцах пятиокиси ванадия, предварительно прокаленной при 200,
450 и 700° С. Во всех трех случаях кривые растворимости качествен-
но подобны, но различаются количественно. Установленную зависи-
мость — чем выше температура прокаливания пятиокиси ванадия,
тем меньше ее растворимость в серной кислоте — авторы объясняют
различной величиной зерна твердых фаз, которая зависит от темпе-
ратуры предварительного прокаливания исходной пятиокиси вана-
дия. Их выводы о составе твердых фаз совпадают с выводами
С. С. Макарова и А. Т. Репы [102].
Иные результаты получены Ланфордом и Килом [146], которые
исследовали растворимость пятиокиси ванадия в серной кислоте при
30° С (рис. 26). В опытах использовали пятиокись ванадия, полу-
ченную путем осаждения из раствора метаванадата аммония при
VzOs
Рис. 25. Изотермы растворимости в системе H2SO4 — V2O5 — Н2О.
а — 75° С [102], б — 25° С [145].
Состав твердых фаз: 1— V2O5, 2—V2O5-H2O, 3— V2O5-2H2O, 4 — V2O5-3H2O.
действии серной кислотой с последующей промывкой и высушиванием
осадка при 100е С. По данным этих авторов, состав твердых фаз
отвечает формулам: V2O5-2SO3-8H2O; V2O5 • 2SO3 • ЗН2О; V2O5-
•4SO3-4H2O и безводной V2O5, которая существует в области кон-
центраций серной кислоты от 7,90 до 37,69 вес.%. В то же время,,
по данным работ [102] и [145], безводная V2O5 при 25° С устойчи-
ва при высоких концентрациях H2SO4 (60—65 вес.%). Во всех
трех указанных работах, кроме того, для одинаковых концентраций
серной кислоты получены существенно различные величины раство-
римости пятиокиси ванадия (см. рис. 26). Эти противоречия можно
объяснить двумя причинами. Во-первых, исследовалась, вероятно,
не чистая пятиокись ванадия, а смесь поливанадатов натрия или
аммония и гидратированной пятиокиси ванадия. Например, в опытах
55
С. С. Макарова и А. Т. Репы [102] пятиокись ванадия, по-видимо-
му, содержала поливанадаты натрия xNa2O-yV2O5-H2O, в других
.случаях [145, 146] в составе использованных образцов V2O5 мог
Рис. 26. Изотерма растворимости 30° С в си-
стеме SO3 — V2O5 — Н2О [146].
Твердые фазы: 1— V2O5, 2 — V2O5- 2SO3-8H2O, 3 —
V2O5-2SO3-3H2O, 4 - V2O5-4SO3 -4H2O.
Точки— экспериментальные данные, кружки— рас-
четные .
быть гексаванадат ам-
мония (NH4)2V6O16. к
сожалению, авторы не
приводят химического
состава использован-
ной V2O5. Кроме того,
в перечисленных рабо-
тах составы твердых
фаз определялись толь-
ко по методу Скрейне-
макерса [145]. При ма-
лом числе эксперимен-
тальных данных (в ра-
ботах [102, 145] каж-
дая ветвь изотермы и
составы твердых фаз
определялись лишь по
двум-пяти точкам)
трудно получить впол-
не достоверные резуль-
таты. Поэтому резуль-
таты, которые приве-
дены в работе [146],
заслуживают предпоч-
тения.
Это заключение подтверждает синтез индивидуальных сульфатов
ванадия (V), осуществленный в последнее время [147]. Если нагре-
вать пятиокись ванадия в течение
нескольких часов в концентри-
рованной серной кислоте при тем-
пературе 200° С, то образуются
красно-оранжевые кристаллы со-
става V2O5 • 2SO3, или (VO2)2 • S2O7 —
соединение, устойчивое в атмос-
фере сухого кислорода до 360° С;
в интервале 360—450° С оно пол-
ностью разлагается с образованием
V2O5. Определена энергия акти-
вации этого процесса, равная
44,3 ккал/моль. На воздухе дисуль-
фат жадно поглощает воду, пре-
вращаясь в густую жидкость крас-
ного цвета, которая при нагрева- .
нии при 100° С дает гидрат
<VO2)2-S2O7-2H2O. Это
Рис. 27. Потеря массы (1) и диф-
ференциально-термическая кривая(2)
при нагревании V2O5-4SO3-3H2O в
атмосфере сухого кислорода.
если
соединение также можно получить,
выдержать безводный дисульфат в атмосфере водяных паров при да-
56
рлении Н20 не более 2 мм рт.ст.
При нагревании в сухом кис-
лороде дигидрат теряет воду пол-
ностью в интервале 140—250° С.
Нагревание пятиокиси вана-
дия в серной кислоте при НО—
130° С приводит к образованию
Таблица IS’
Растворимость в системе
HF—V2O5—Н2О [148]
Жидкая фаза,
вес. %
Твердая фаза
V2O5 HF
желто-оранжевых кристаллов
состава V2O5-4SO3-3H2O. При
нагревании в сухом кислороде
этот сульфат, начиная с темпера-
туры 160° С, разлагается с об-
разованием при 230° С безвод-
ного дисульфата (рис. 27, кривая
/). Эндоэффект при 152° С (кри-
вая 2) объясняется прев-
ращением V2O5-4SO3-3H2O в
V2O5 • 2SO3 + H2SO4 + Н2О; при
этом соль слегка расплывается. ,
Последующее изменение массы
происходит в интервале 152—
230° С с удалением образовав-
шихся H2SO4 и Н2О.
Несомненно, что система
SO3—V2O5—Н2О нуждается в
дальнейших обстоятельных иссле-
дованиях с привлечением для
идентификации твердых фаз раз-
личных физико-химических мето-
дов исследования. Важным так-
же является выбор метода полу-
/=25° С
10,5
15,9
22,3
30,4
32,7
35,1
40,0
46,4
45,1
45,4
44,6
45,0
43,5
40,4
35,6
33,4
31,4
29,8
26,8
23,4
22,8
22,7
чения исходной пятиокиси ванадия.
6,7
10,8
15,2 ’
21,5
29,0 '
35,0
38,2 >
41,3
V2O5
3VO2F-HF.H2O
/=16° с
44,2
46,6
50,0
53,0
54,5
56,8
61,2
63,0
65,1
68,0
71,0
75,5
77,8
78,5
4VOF3.3HF-3H2O
2V0F3-3HF-H2O
VOF3
Л
Данные по растворимости
в рассматриваемой системе имеют лишь ориентировочный характер.
СИСТЕМА HF —V2O5—Н2О
Система HF—V2O5—Н2О изучена Н. С. Николаевым и Ю. С. Бу-
слаевым [148]. Ввиду значительной летучести фтористого водорода
из концентрированных растворов исследование проводили при двух
температурах. Растворы с концентрацией HF от 0 до 41,3% изуча-
лись при 25° С, а с более высокой концентрацией — при 16° С.
Для опытов использовали продажную безводную пятиокись вана-
дия марки «ч.д. а.», равновесие устанавливалось в течение двух-трех
суток (табл. 18, рис. 28). Состав твердых фаз определен графически
методом остатков. В рассматриваемой системе образуется ряд оксо-
фторидов типа VO2F и VOF3 в форме соединений с HF и Н2О.
Фаза 3VO2F • HF • Н2О представляет коричневую мелкокристалли-
ческую массу. Следующая фаза, 4VOF3-3HF-3H2O, выделяется в
форме зеленых кристаллов, расплывающихся на воздухе в красно-бу-
рую жидкость. Аналогичным образом ведет себя фаза 3VOF3 • 3HF • Н2О,
кристаллизующаяся в виде крупных бледно-зеленых, почти бесцветных
Рис. 28. Растворимость в системе HF — V2O5 — Н2О [148].
Твердые фазы: 1 — V2O5, 2 — 3VO2FHF-H2O, 3 — 4VOF3- 3HF- 3H2O,
4 — 2VOF3-3HF-H2O, 5 — VOF3.
призм. Оксофторид VOF3 представляет собой массу бледно-желто-
го цвета, расплывающуюся на воздухе.
Глава III
СИНТЕЗ И СВОЙСТВА ванадатов
Синтез гидратированных ванадатов большинства металлов
не представляет особых трудностей. Легкорастворимые ванадаты
щелочных металлов в виде кристаллогидратов получают кристалли-
зацией из водных растворов пятиокиси в соответствующих щелочах.
В некоторых случаях, однако, удобнее использовать реакции обмен-
ного разложения, например, при получении ванадата аммония.
В то же время реакции обменного разложения лежат в основе
получения ванадатов всех других металлов. Процедура предельно
проста: сливают раствор хорошо растворимого ванадата щелочного
металла, обычно натрия, и раствор также хорошо растворимой соли
необходимого металла, чаще всего нитрат или хлорид; выпавший
осадок отфильтровывают, промывают и сушат. Разумеется, в зави-
симости от индивидуальных свойств получаемого ванадата или спе-
циальных требований к его чистоте или другим качествам неиз-
бежны различные видоизменения стандартного метода. Например,
для предотвращения нежелательных побочных реакций или в случае
высокой растворимости конечного продукта осаждение проводят
в водно-спиртовой среде; чтобы избежать загрязнения осадка
щелочными металлами или не допустить образования двойных
солей, в качестве исходного компонента используют раствор дека-
вана диевой кислоты или ванадата лития.
Свойства гидратированных ванадатов изучены слабо. Выше при-
ведены данные по растворимости. Много сведений о их термическом
поведении и инфракрасных спектрах; в некоторых случаях изучены
кристаллооптические характеристики индивидуальных соединений.
Совсем мало данных о кристаллической структуре гидратированных
ванадатов. Отсутствие таких данных затрудняет не только иден-
тификацию индивидуальных соединений, но и использование
их свойств на практике.
ВАНАДАТЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ И АММОНИЯ
ванадаты натрия
Ванадаты щелочных металлов в виде кристаллогидратов легко
получаются кристаллизацией из водных растворов. Лучше всего
59
Таблица 19
Температуры дегидратации
и теплоты гидратации орто-
и пированадатов натрия [103]
Ступень дегидратации /, ° с Qo > кал! моль
.Ортованадат натрия
oNa3V04-12H20
12-10 18,2 12276
10—7 20,5 12361
7-2 52,2 13713
2—1/2 60,7 13910
i/a-0 — 14565
<0 р т о в а н адат н атрия
ip-Na3 VO4-12H2O
12-10 19,9 12351
10->8 23,1 12459
8—31/2 43,9 13300
^1/2-1/2 63,5 14180
1/2-0 — 14937
JI и р о в а н адат н атрия
Na4V2O7-18Н2О
18->12 18,9 12282
Й2->10 30,9 12819
ДО—>2 34,0 12921
2-»0 13108
ф-соли на первой и второй
по мнению авторо] в [103],
изучены свойства ванадатов натрия,
условия их выделения очевидны из диа-
граммы состояния системы Na2O—
V2O5—Н2О. Как правило, исходят из
безводной пятиокиси ванадия или
метаванадата аммония, которые раст-
воряют в едком натре или соде и по-
лучают раствор NaVO3. Добавляя в
последний необходимое количество
щелочи или подкисляя его, получают
соответствующие ванадаты [99, 139].
Ортованадат натрия Na3VO4-
• 12Н2О кристаллизуется в виде бес-
цветных игольчатых кристаллов: при
обыкновенных условиях, как описано
выше, он существует в двух фор-
мах— псевдосоль (ф-фаза) и ортосоль
(о-фаза). Мензель и Мюллер [103],
установившие этот факт, нашли, что
ф- и о-формы дают различные дебае-
граммы и образуют два ряда кристал-
логидратов. При постоянном давлении
водяных паров 10 мм рт. ст. были
определены температуры устойчивости
различных кристаллогидратов, а по
зависимости давления водяных паров
над ними от температуры рассчитаны
теплоты их гидратации Qo (табл. 19).
Различие величин теплоты гидратации
ступени всего на 108 кал объясняет,
появление при 25° С наряду с
10-воД’
ной также 12-водной ф-соли. Аналогичные данные получены этими
же авторами для пированадата натрия (см. табл. 19).
Состав твердых фаз в системе Na2O—А12О3—V2O5—Н2О при
различных температурах и концентрациях щелочи по данным [111]
приведен в табл. 20. Описанный впервые 22-водный пированадат
натрия быстро выветривается на воздухе, переходя в Na4V2O7- 18Н2О.
По данным [205], при нагревании на воздухе (рис. 29) дегидра-
тация ванадатов натрия Na3VO4-12Н2О, Na4V2O7 • 22Н2О и NaVO3-
• 2Н2О протекает ступенчато по следующим схемам:
48° 108° 140° 294° 444°
Na3VO4- 12Н2О — ЮН2О — 7Н2О Ш 2,5Н2О — 0,5Н2О — Na,VO4;
26° 48° 90° QR° 114° 494°
Na4V2O7-22H2O —18Н2О —12Н2О-»10Н2О—2Н,0 -* 0,5Н2О —
85° 9 3 ° 280°
->Na4V2O~; NaVO,-2H,O^ 1Н2О-> 0,5Н2О — NaVO3.
Лукач с соавторами [149] синтезировали ванадаты натрия состава
NaVO3(l,34-3)H2O; Na4V2O7(15-16)Н2О; Na3VO4(10-11)Н2О и ис-
60
Таблица 20
Твердые фазы в системе Na2O—А12О3—V2O5—Н2О (111J
Соединение Условия образования Форма кристаллов, показатели .преломления
f, ° С Na2O в растворе, %
Na3VO4-12H2O 20 23,82—8,46 21,92—11,09 Вытянутые шестигранные
30 призмы
45 26,36—13,2 No= 1,500
Na3VO4.10H2O, 75 26,41—18,14 Ne = 1,529
20 23,82—27,45 Ромбододекаэдры
Обычно встречается 30 21,17—23,4 N=1,539
с Na3VO4-12H2O или 45 25,14—32,00
с Na3VO4-7H2O 75 24,9—28,4
Na3VO4-7H2O 20 28,1—36,0 Трапецеидальные пластины
30 25,4—36,0 Ng= 1,579
45 25,14—36,0 Nm= 1,562
75 24,9—36,0 Np= 1,542
Na3VO4-2,5H2O 75 34,0—36,0 Короткие, плоские таблички Ne= 1,696 No=1,662
Na4V2O7-22H2O 20—25 11,0—9,0 Тонкие пластинки No=1,539 Ne= 1,531
Na4V2O,- 18H2O 30 11,0—8,0 Шестиугольные пластинки
45 14,81—11,33 Ng= 1,551 Nm= 1,546 Np= 1,540
NaVO3-2H2O 45 12,2—10,27 Тонкие иглы Ng= 1,767 Np=l ,710
NaVO3-H2O *4. 76 17,0—12,4 Тонкие иглы Ng= 1,830 Nm= 1,734 Nd=1,714
следовали термогравиметрически. По их данным, при нагревании
на воздухе ортованадат Na3VO4-10H2O теряет воду в четыре этапа:
максимумы соответствующих эндотермических эффектов лежат
при температурах 58, 70, 150 и 190° С. Пированадат состава
Na4V2O7- 16,ЗЗН2О и метаванадат NaVO3-3H2O обезвоживаются
в три этапа. Пированадат последовательно теряет 8,33; 7,0 и 1,0люлей
воды и максимумы скоростей дегидратации соответствуют темпера-
турам 50, 130 и 180° С, а для метаванадата 50, 90 и 110° С.
По данным Б. В. Слободина с соавторами [150], синтезированный
ими метаванадат натрия состава NaVO3-l,5H2O при 20—100°С
теряет 0,45 моля Н2О, при 100—140° С отщепляет еще около
моля Н2О и последние 0,01 моля Н2О медленно удаляются
в интервале 140—300° С. Как видно, данные различных авторов
ио дегидратации ванадатов натрия сильно различаются. Причина
этого, по-видимому, — различия в условиях нагревания: величине
навески, скорости подъема температуры и т. д./Ja также то, что
61
Рис. 29. Дифференциально-термические
кривые нагревания ванадатов натрия.
1 — NaVO3-H2O, 2 — NaVOs-2H2O; 3 —
Na4V2N7-1 8Н2О; 4 — Na3VO4-25H2O; б —
Na3VO4-7H2O; 6 — Na8VO4-1 0H2O; 7 —
Na3VO412H2O.
для исследования были взяты
образцы различного состава —
в некоторых случаях они могли
быть не монофазными.
Ванадаты натрия с соот-
ношением Na:V=6:10 впер-
вые синтезированы еще в
1901 г. [151], но интерес к ним
возродился только в 50-х го-
дах, когда была выяснена при-
рода этих соединений и их
стали рассматривать как дека-
ванадаты Na6V10O28 • иН2О.
Декаванадат натрия легко*
получить кристаллизацией из
водного раствора метаванадата*
натрия, подкисленного в соот-
ветствии с уравнением реакции
10VO? + 4H+=iV10O^+
+ 2Н2О. (27>
Для подкисления обычно-
используют минеральные кис-
лоты [152], но удобнее всего
уксусная кислота [63]. Не-
большое количество образо-
вавшегося в момент подкисле-
ния бурого аморфного осад-
ка гидратированной пятиокисгг
ванадия удаляют фильтрова-
нием. При стоянии на воз-
духе из раствора медленно*
выпадают крупные, хорошо
сформированные кристаллы рубиново-красного цвета состава
Na6V10O28(20-7-24)H2O [152]. Хорошие результаты получают, если
вместо кислоты использовать катионообменную смолу в Н-форме [142].
Преимущество этого способа заключается в том, что раствор не со-
держит посторонних ионов, так как ионы щелочного металла обме-
ниваются на ионы Н+ смолы и удаляются из раствора. И, наконец,
предложен способ, основанный на реакции между декава надиевой:
кислотой и ацетатом соответствующего металла (153]:
H6v10o28+6СН3СООМ M6V10O28+6СН3СООН.
(28)»
В литературе описаны декаванадаты натрия, содержащие 23 [151],
19 [63], 24 [152], 5,9 [46], 18,2 [154], 18 и 14 [155] молекул воды.
24-водный декаванадат натрия теряет некоторое количество воды:
уже при комнатной температуре, интенсивно выветриваясь на воз-
62
духе [152]. По [155],
18-водный декаванадат нат-
рия на воздухе переходит
в устойчивый 14-водный.
Тот и другой обезвожи-
ваются в эксикаторе над
концентрированной H2SO4
и переходят в четырехвод-
ный; последний поглощает
влагу из воздуха. Судя по
кривым ДТА и ДТГ (рис.30),
при нагревании Na6V10O28 •
• 24Н2О [152] потеря воды
происходит в четыре эта-
па — максимумы скоростей
дегидратации и минимумы
эндоэффектов лежат при
температурах 40—60, 100,
120—130 и 170° С. Отщеп-
ление последних 3,0 молей
Н2О хорошо заметно на
кривой ТГ; полностью обез-
воживание заканчивается
при 255° С. Характер про-
цессов, протекающих при
нагревании обезвоженного
образца, можно объяснить,
учитывая диаграмму состоя-
ния в системе Na2O—V2O5
[97]. В данной системе
соединение состава Na:V=
Рис. 30. Дериватограмма декаванадата
натрия.
=6:10 не существует; сле-
довательно, обезвоженный декаванадат натрия при нагревании должен
претерпеть разложение. Из кривой ТГ видно, что при нагревании до
530° С образец теряет в весе, а в области второго эндоэффекта первона-
чальный вес восстанавливается. Этот процесс связан с выделением
кислорода при образовании бронз и его поглощением при плав-
лении [97]. Максимальное количество бронзы в образце накапли-
вается к моменту его плавления, а температуры эндоэффектов
на кривой ДТА полностью совпадают с температурами превращений,
зафиксированных на соответствующем участке диаграммы состояния
системы Na2O—V2O5. Таким образом, в момент обезвоживания
декаванадат натрия, по-видимому, распадается на свободные V2O5
и NaVOs, взаимодействие между которыми, отраженное на кривых
ДТГ, ДТА и ТГ, протекает в интервале 300—530° С. В инфракрас-
ном спектре поглощения декаванадата натрия обнаружены полосы,
соответствующие валентным и деформационным колебаниям груп-
пировок Н2О и ОН. Эти данные подтверждены на основании
63
Поглосце нае —► По&лоще
Рис. 31. Инфракрасные спектры ванадатов натрия.
1 — Na4V2O7 • 22Н2О; 2 — Na4V2O7 • 18Н2О; 3 — Na8VO4-
•12Н2О; 4 — Na3VO4-10H2O; 5 — Na8VO4-7H2O; 6 -
Na3VO4-2,5Н2О; 7 — NaVO3-H2O, полученный нагреванием
NaVO3-2Н2О при 75° С; 8 — NaVO3-2H2O, 9 — NaVO3 H2O
из раствора.
исследования спектров ЯМР. Инфракрасные спектры гидратирован-
иях ванадатов натрия описаны в работах [111, 152]. Полосы при
1620—1650 и 3000—3700 т-1 в спектрах ванадатов натрия (рис. 31)
соответственно относятся к деформационным и валентным колеба-
ниям кристаллизационной воды, с увеличением количества которой
интенсивность их возрастает. Этим же, видимо, объясняется сгла-
женный характер поглощения ортованадата в области 600—1000 m”1.
Слабая полоса при 2200 смт1 в спектре орто- и пированадатов, интен-
сивность которой возрастает также с увеличением числа связанных мо-
лекул воды, рассматривается как комбинационная; в образовании ее
участвуют колебания связанной воды. Для метаванадатов натрия отме-
чено существование двух типов спектров. Спектры NaVO3 и NaVO3-
• 2Н2О одинаковы в области собственных колебаний молекул NaVO3,
что позволяет говорить о подобии их структур. В спектре NaVO3-H2O
полосы поглощения те же, что и в NaVO3 и NaVO3 • 2Н2О, и, кроме того,
еще две полосы на частотах 880 и 520 т”1. Предполагается, что
полоса при 880 смг1 является валентной (v3), а при 520 —
деформационной (v4) полосой какой-то тетраэдрической структуры
в кристалле NaVO3-H2O.
ВАНАДАТЫ КАЛИЯ
Гитер [99] привел сводку опубликованных к 1941 г. данных
о составе ванадатов калия с отношением K:V 4:1, 3:1, 2:1, 5:4,
1:1, 11:12, 2:3, 3:5, 1:2, 2:5, 1:3, 1:4, 2:9. В 1959 г. Бель-
тран и Гильем [156] изучали действие азотной и уксусной кислот
на метаванадат калия. Из водных растворов были выделены поли-
ванадаты с отношением K:V 2:5, 1:2, 3:5, 2:3, 4:7, 3:7, 5:12.
Однако Глемзер и Прейслер [50] из растворов метаванадата калия,
подкисленных серной кислотой, получили только два соединения,
с K:V 6:10 и 4:10.
Выше были отмечены причины того, что в литературе описаны
ванадаты натрия самого различного состава. Это в полной мере
относится и к ванадатам калия: по-видимому, многие из перечи-
сленных соединений не являются индивидуальными, поэтому основ-
ное внимание будет уделено тем, индивидуальность которых не
вызывает сомнений.
Дитте [109] кристаллизацией из щелочного раствора с соотно-
шением V2O5:6KOH получил ортованадат K3VO4-6 (или 4,5) Н2О,
а из раствора с V2O5:8KOH в присутствии спирта выделил
K2O-2K3VO4-20H2O. Норблад [107] приготовил моноклинные кри-
сталлы K4V2O7-3H2O, а Дитте [109] — K4V2O7-4H2O. Роммельсберг
1157] нашел, что эта соль теряет одну треть воды над концентри-
рованной серной кислотой, две трети при 100° С и всю воду при
350° С. Норблад [107] получил моногидрат метаванадата калия
KVO3-H2O и установил, что он полностью обезвоживается при
110° С. По данным [109], KVO3 в зависимости от условий получе-
ч 65
° Заказ № 256
Рис. 32. Дериватограммы
ванадатов калия.
а — KVO8-H2O; б — K4V2O, •
•4Н4О.
70
Рис. 33. Дериватограммы вана-
датов калия.
а — K3VO4-5H2O; б KeVioOae’
• 1 0Н2О.
Время
ния может содержать 1,25; 1,5; 2 и 3, а по [157]—7 молекул
воды.
Радау [118] выделил декаванадат калия К5 V10O28 • 10Н2О путем
упаривания раствора KVO3, подкисленного уксусной кислотой.
Это моноклинные кристаллы с а:6:с=0,5902:1:0,6040; [3—117°40'.
Эго соединение позднее было получено подкислением раствора
метаванадата калия смолой Дауэкс-50 в Н-форме [142], а подки-
слением азотной кислотой до pH 5,5 получен 8-водный декаванадат
калия [152]. Пэ данным Кельмерса [95], декаванадат калия не явля-
ется стабильным соединением. При обычных условиях из раствора
KeV10O2S медленно выделяется осадок K2VGO16. Эго подтверждено
в работе [117] при исследовании системы К2О—V2O5—Н2О. ]
Ранее были описаны гексаванадат калия безводный и кристал-
логидраты, содержащие 1,5 и 6 молекул воды [123]. Детально
условия получения гексаванадата калия описаны в работе [96].
Раствор метаванадата калия нагревают до 85° С и подкисляют
серной, хлорной или соляной кислотами по реакции
6KVOS + 4Н+ K2V6O16 + 4К+ + 2Н2О. (29)
Выпавший осадок оранжевого цвета состоит из мелких кристаллов
K2VGO16 моноклинной системы. 1
Термические свойства ванадатов калия описаны в работах
[107, 117]. Вода в молекуле KVO3-H2O связана относительно слабо
и полностью удаляется при 110° С (рис. 32). Четырехводный пиро-
ванадат калия полностью обезвоживается в интервале 120—300° С
в три стадии; при этом последние 1,5 молекулы Н2О удаляются
с высокой скоростью и, судя по кривой ДТГ, этот процесс в свою
очередь протекает многоступенчато, путем последовательного бур-
ного выделения малых порций воды. Ортованадат калия K3VO4-5H2O
теряет воду в четыре стадии (рис. 33): от 70 до 110° С удаляются
1,5 молекулы в интервале 110—170° С — в три ступени — после-
дующие 1,5, далее при 170—320° С еще 1,5 и, наконец, от 320
до 400° С отщепляются последние 0,5 молекулы Н2О; обезвоженный
препарат плавится при 420° С в полном соответствии с диаграммой
состояния системы К2О—V2O5 [98].
Кристаллическая структура KVO3-H2O описана в работах
[158, 159]. Он образует короткопризматические кристаллы (рис. 34),
которые относятся к ромбической системе [158]. Элементарная
ячейка содержит четыре формульных единицы. Параметры ее:
о
а=8,15; Ь= 13,58; с—3,69 А. Расчетная плотность 2,53, опреде-
ленная экспериментально 2,52 г)см3. Проекция структуры KVO3-H2O
на плоскость (001) и общий вид ее представлены на рис. 35 и 36,
а в табл. 21 приведены межатомные расстояния и углы между
связями. Видно, что каждый атом ванадия окружен 5 атомами
кислорода, которые образуют искаженную тригональную бипира-
миду, а тригональные дипирамидальные полиэдры образуют непре-
рывные цепи.
68
Каждый атом калия шестью атомами кислорода связан с вана-
дий-кислородными цепями и, кроме того, координирует 2 молеку-
jjbi воды, включающие О4 и О4.
В инфракрасных спектрах (рис. 37) гидратированных ванадатов
калия можно выделить несколько основных групп полос поглоще-
ния [И7, 152, 160]. Полосы в области 500—1000 см-1 характери-
зуют колебания по связям ванадий — кислород.
Таблица 21
Межатомные расстояния и углы между связями в KVO3H2O [159]
Межатомные расстояния Углы между связями
Связь Вектор о d, А Связь Вектор О d, к Связи Векторы Угол, град
V—Ох А 1,67 К—03 L 3,43 Oj-V-Oa А, В 106
v—оа В 1,65 К—О3 М 3,56 O,-V-O3 Д, С 128,9
V—03 С 1,97 К—Оо А/(2) 3,09 O2-V-O3 В, С 124,8
V—О3 0(2) 1,93 К—О! Р(2) 2,27 O,-\-O3 A, D 98,6
V—V 3,116 к-о4 Q (2) 2,78 O2—V—O3 В, D 100,7
К—к Z 4,168 к—ох Я (2) 2,97 О3—V—03 С, D 73,8
O3—V—O3 D, D 147,5
Рис* 34. Типичный кристалл
KVO3.H2O,
Рис. 35. Тригонально-бипирамидаль-
ные цепи в структурах KVO3-H<>O,
Ca(VO3)2-4H2O, Sr(VO3)2.4H2O.
В соответствии со структурой в области собственных колебаний
молекулы KVO3 выделены две группы полос поглощения [160].
Полосы в области 850—950 см—1 характеризуют колебания вдоль
коротких связей V—О, а интенсивные полосы в области 540—600 см-1
отвечают колебаниям по длинным связям V—О. Полосы в области
1610—1670 и 3510—3590 см—1 относятся соответственно к дефор-
69
I
Рис. 36. Проекция структуры KVO3-H2O на плоскость (001).
Буквы — расстояния между атомами, цифры — номера атомов кислорода (см. табл. 21).
рационным и валентным колебаниям молекул воды; они, естест-
венно, присутствуют в спектрах всех ванадатов. Спектр декавана-
«дата калия
полностью тождествен спектру декаванадата натрия.
Рис. 37. Инфракрасные спектры ванадатов калия.
а — KgV10O28. 1 OH2OJ б — KVO3-H2O; в - K4V8O7- 4Н2О.
В спектре пированадата калия в области собственных колебаний
молекулы K4V2O7 наблюдаются те же группы линий, что и в пиро-
ванадатах натрия и щелочноземельных металлов.
ВАНАДАТЫ АММОНИЯ
Из описанных в литературе многочисленных ванадатов аммония
с отношением NH4: V, изменяющимся от 1:1 до 3:10 [96, 122—130],
в настоящее время надежно идентифицированы только некоторые
из них. Ванадат аммония с отношением NH4:V—1:3 получают
при подкислении растворов либо NH4VO3, либо NaVO3 в присут-
ствии какой-нибудь аммонийной соли. При нагревании раствора
выпадает осадок золотисто-желтого цвета. Как уже отмечалось,
ванадаты такого состава рассматривают как гексаванадаты, им при-
писана формула (NH4)2V6O16 [96]. В более ранней работе [161]
71
для этой соли дана формула (NH4)2VO2V5O14. Бельтран и Гильем [162)
действием уксусной кислоты на раствор ванадата аммония получили
осадок состава 3 (NH4)2O-5V2O5-6H2O, который, по-видимому, следует
рассматривать как декаванадат аммония (NH4)6V10O28-6H2O. ]
Метаванадат аммония NH4VO3 — наиболее известное и хорошо
изученное соединение — является одним из главных исходных про-
Рис. 38. Термическое разложение метаванадата ам-
мония.
а — потеря массы; б — дифференциально-термические кривые
(/) и кривые газовыделения (2) при нагревании NH4VO8 в
атмосфере кислорода и азота.
дуктов при получении чистых соединений ванадия. Это — белые
или слабо-желтые мелкие кристаллы уд. веса 2,326 г/см?. NH4VCU
имеет орторомбическую структуру с параметрами элементарной
ячейки «=4,96; 6= 11,82; «=5,63 А; «:/?:«= 0,420:1:0,476. Эле-
ментарная ячейка содержит четыре формульных единицы. Это озна-
чает, что данное вещество не является гидратом [163]. При нагре-
вании оно разлагается по реакции
2NH4VO3= V2O5 + 2NH3 + Н2О, (30)
72
которая лежит в основе промышленных методов получения чистой
пятиокиси ванадия. Реакция протекает в несколько стадий с обра-
зованием промежуточных продуктов; к их числу относят (NH4)2O-
.(V2O5)3, или (NH4)2V6O16 [164, 165, 167, 170], HVO3 [168],
NH3-H2O(V2O5)2 [166]. На основании термографических, и рентге-
нографических исследований Сатава [167] пришел к выводу, что
на воздухе при разложении NH4VO3 образуется сначала NH4V3Og,
я затем V2O5. В инертной атмосфере (N2, СО2) при температуре
ниже 400° С, по этим же данным, образуется (NH4)2O-2VO2-5V2O5
(или NH4V6O15), который постепенно восстанавливается с образо-
ванием промежуточных окислов V6O13, VO2, VO1j67 и V2O3.
Несколько позже Танигухи и Инграхем [170] исследовали опи-
сываемый процесс в атмосфере кислорода и азота, используя термо-
гравиметрию, рентгенографию и химический анализ твердых и газо-
образных продуктов разложения. По этим данным (рис. 38), неза-
висимо от состава газовой среды на первой стадии разложения,
протекающей в две ступени, происходит реакция
6NH4VO3= (NH4)2O • (V2O5)3 + 4NH3 + H2O, (31)
максимум скорости которой лежит при 225° С. На второй стадии
разложение сопровождается вначале эндотермическим эффектом
(/=310, 325° С), связанным с освобождением воды, а затем экзо-
термическим процессом каталитического разложения аммиака
на поверхности пятиокиси ванадия (325, 375° С). При разложении
в азоте ванадий частично восстанавливается, о чем свидетельствует
темный цвет продуктов разложения. Найдено, что константа равно-
весия реакции (31) и изменение свободной энергии описываются
уравнениями:
1g К-10,148 — 5,052 • 10-3/Т,
АС° = 23,115 — 46,47.
СМЕШАННЫЕ И КИСЛЫЕ ДЕКАВАНАДАТЫ
ЩЕЛОЧНЫХ МЕТАЛЛОВ И АММОНИЯ
Ионам HV10O2? и H2V10O2s" должны соответствовать кислые
декаванадаты M5HV10O28 и M4H2V10O28 с отношением M:V, равным
1:2 и 2:5. Ванадаты натрия, калия и аммония с таким соотноше-
нием М: V и различным количеством кристаллизационной воды
описаны в работах ранних исследователей [123]. Сейчас трудно
решить, являются ли описанные соединения декаванадатами или
некоторые их них представляют собой смеси других индивидуаль-
ных соединений. Однако существование кислых декаванадатов уста-
новлено вполне надежно, и индивидуальные соединения этого типа
синтезированы Яром и Преуссом [153] и Шварценбахом и Гей-
ером [142]. Кислый четырехзамещенный декаванадат цезия получен
следующим способом [153]. Карбонат цезия обработали уксусной
кислотой, к полученному раствору ацетата цезия добавили необхо-
димое количество раствора декаванадиевой кислоты и смесь упарили
73
в вакууме при комнатной температуре. Выделившиеся кристаллы^
имели состав Cs4H2V10O28-5H2O.
Весьма просто получаются пятизамещенные двойные декавана-
даты натрия и калия, рубидия, цезия [142]. Если в раствор, содерГ
жащий пятизамещенный декаванадат калия K5HV10O28 (или рубидия,,
или цезия) добавить какую-либо»
соль натрия, например NaCl, то
выпадают желтые, сверкающие-
золотом листочки состава
K4NaHV10O28- ЮН2О, отличаю-
бремя
Рис. 39. Дериватограмма
K4NaHV10O28-10H2O.
щиеся весьма низкой раствори-
мостью. Это свойство, по мне-
нию авторов [142], можно ис-
пользовать для количественного*
определения натрия. Соедине-
ние K4NaH V10O28 • 10Н2О от-
носится к триклинной син-
гонии, параметры элементарной
ячейки а=8,59; Ь—10,36; с=
= 11,01 А; а=110°35'; [3=
= 86°45', у=114°. Вычислен-
ная плотность 2,63 г/см\
экспериментально найденная
2,66 г/см?.
При нагревании на воздухе,,
судя по кривой ДТГ, это сое-
динение обезвоживается в три
этапа (рис. 39); в интервале
50—150° С отщепляется 9 мо-
лекул Н2О, от 150 до 190° —
еще одна и, наконец, при 190—
210° образец полностью обез-
воживается, что соответствует
выделению половины молекулы
воды, образующейся за счет
структурных гидроокислов
в соединении K4NaHV10O28.
В ряду аналогичных соединений калия, рубидия и цезия раство-
римость понижается. В работе [142] получены нормальные двойные
декаванадаты состава M5NaV10O28- 10Н2О, где М—К, Rb, Cs. Внешне-
эти соединения не отличаются от кислых двойных декаванадатов
и образуются при добавлении в раствор нормального декаванадата
калия (рубидия, цезия) соли натрия. В этом случае точно так же
понижается растворимость в ряду декаванадатов от калия к цезию.
Описаны двойные декаванадаты натрия и аммония. Декаванадат
Na2(NH4)4- V10O28-11Н2О получен при выдерживании при комнат-
ной температуре раствора метаванадата натрия 0,355 моль/л, под-
74
численного соляной кислотой в соотношении H+:VO?=0,52 в при-
сутствии 0,855 моль/л NaCl и 0,374 моль/л NH4C1 [171].
Г. Ф. Пинаев [172], изучая растворимость в системе Na+,
NHt/Cl“", VO?, в определенных областях поля растворимости
обнаружил декаванадат аналогичного состава, отличающийся только
содержанием кристаллизационной воды (10 молекул Н2О вместо 11).
g работе Фридериксона и Хаузена [173], посвященной исследова-
нию инфракрасных спектров соединений ванадия, в числе прочих
упомянуто соединение Na4 (NH4)2V10O28 • 16Н2О, но, к сожалению,
отсутствуют какие-либо указания на способ его получения.
На протяжении многих лет считалось твердо установленным,
что в слабокислой среде ванадий существует в форме гексавана-
диевой кислоты H4V6O17 и некоторые соединения ванадия рассмат-
ривались как соль этой кислоты. Позже, однако, более точные
эксперименты заставили отказаться от этой точки зрения и на основе
концепции декаванадат-иона появилась возможность выяснить хими-
ческую природу многочисленных природных и искусственных соеди-
нений ванадия. В то же время в литературе появляются сообщения
о синтезе твердых ванадатов, которым приписывается формула
M4V6O17-nH2O. Впервые соединения с соотношением M:V=2:3
описаны еще в конце прошлого века [123]. Позже Глемзер и Прей-
слер [50] подкислением растворов метаванадатов увеличивающимся
количеством серной или азотной кислот получили только ванадат
натрия с Na:V 6:10, 4:10 и ванадаты калия с I<:V 6:10, 5:10,
4:10, которые, очевидно, соответствуют K(Na)6V10O28, K5HV10O28
и K(Na)4H2V10O28. В аналогичных условиях Бельтран и Гильем [156],
однако, получили ванадат калия с отношением M:V=2:3. И, на-
конец, в 1967 г. П. И. Федоров с соавторами [125] пришли к вы-
воду о существовании гексаванадата (NH4)4V6O17-14Н2О. Такие
отрывочные сведения не позволяют сделать однозначного заклю-
чения о существовании гексаванадатов этого типа. Необходимы
дальнейшие тщательные исследования соответствующих безводных
и водных систем с широким использованием комплекса физико-хими-
ческих методов для идентификации индивидуальных соединений.
ВАНАДАТЫ ЩЕЛОЧНОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
Гидратированные ванадаты щелочноземельных металлов с соотно-
шением окислов A1O:V2O5 10:3, 3:1, 2:1, 6:7, 3:4, 2:3, 4:7,
1:2, 3:7, 1:3 описаны в работах [99, 123]. Некоторые из этих
ванадатов могут и не быть индивидуальными соединениями. В без-
водных системах МО—V2O5 для элементов от Mg до Ва обнаружены
соединения с соотношением MO:V2O5=4:1 [174], 3:1, 2:1, 16:9,
3:2, 1:1 [175—177]. Гидратированные ванадаты кальция и магния
найдены в природе в виде минералов: Ca2V2O7-9H2O — пинтадоит,
Са (VO3)2 • 4Н2О — россит, Са (VO3)2 • 2Н2О — метароссит, СаО •
• 3 V2O5 • 9Н2О — хеветит [178], Ca3V10O28 • 17Н2О — паскоит
и K2Mg2V10O28 • 16Н2О — гуммерит [179].
75
Метаванадат бериллия Be(VO3)2-4H2O (уд. вес 2,273 г/см?\
может быть получен кипячением суспензии гидроокиси бериллия
с пятиокисью ванадия; аналогично при кипячении магнезии Mg (ОН)
с V2O5 образуется метаванадат магния Mg(VO3)2-6H2O; если после
кипячения суспензии Mg(OH)2 с V2O5 раствор подкислить избытком
уксусной кислоты и затем упарить на воздухе, выделяются кристал-
лы декаванадата магния Mg3V10O28-28H2O (уд. вес 2,167 г/см?) [123].
Ванадаты других щелочноземельных элементов, как правило,
получают при смешивании растворов соответствующей растворимой
соли и ванадатов щелочных элементов или аммония. По данным
Гитера [99], в интервалах pH 10,8—11,0 и 8,8—12,5 осаждаются
ортованадаты, а при pH 7,8—9,3 и 6,0—8,2 — пированадаты каль-
ция и бария соответственно. I
В работе Колмюллера и Перранда [180] описано получение вана-
датов бария. Метаванадат с отношением V2O5: ВаО=0,96 получен
при добавлении насыщенного раствора ВаС12 в раствор метаванадата
аммония, подкисленный азотной кислотой до pH 6. Из кипящего
раствора NH4VO3, pH которого доведен до 11,0 раствором КОН„
при добавлении ВаС12 осаждается пированадат с отношением
V2O5: ВаО=0,49. Ортованадат бария (V2O5: ВаО=0,32) выделяется
из раствора V2O5 в соде (pH 12,5) при 110° С. 1
Журкова с соавторами [144, 181] синтезировали метаванада-
ты стронция Sr (VO3)2 • 4Н2О и бария Ba (VO3)2 • 2 Н2О и
Ba(VO3)2-H2O нагреванием смешанных растворов нитратов и мета- \
ванадата аммония. Моногидрат метаванадата бария Ba(VO3)2-H2O— I
белый кристаллический порошок, труднорастворимый в воде (произ-
ведение растворимости [Ва2+]2 • [V40fr] =9,72-10-14), уд. вес при <
22° С 3,660 г/см?, кристаллизуется в ромбической сингонии с пара-
метрами решетки <2=7,43 + 0,01; 6=9,06 + 0,01+1; а = р = у = 90°;
элементарная ячейка содержит четыре формульных единицы. Эти же
авторы выделили декаванадаты стронция (уд. вес 2,528 г!см?) и ба- 1
рия Ba3V10O28 * 20Н2О (уд. вес 2,621 г/см?) кристаллизацией из рас- ;
творов соответствующих перванадатов после подкисления их уксус-
ной кислотой [181]. Декаванадат стронция образует моноклинные
кристаллы с параметрами элементарной ячейки <7=18,63; 6= 12,61;
с = 18,50А; |3=109о24', содержащей 4 формульных единицы. Дека-
ванадаты магния Mg3V10O28 • 20Н2О и бария Ba3V10O28-19Н2О могут
быть получены также смешением растворов соответствующего аце- !
тата и декаванадиевой кислоты по реакции, аналогичной реакции (28)..
Для выделения кристаллов раствор медленно выпаривают в вакууме
при комнатной температуре [153].
Исследование условий осаждения и свойств ванадатов щелочно- 1
земельных элементов в последнее время проведено Дюпуи и Лорен-
целли [160]. Из водных растворов осаждаются безводные орто-
ванадаты стронция и бария, аналогичная соль кальция содержит
около 4 молекул воды, которая удаляется в интервале температура
76
145—200° С. Пированадаты Са, Sr, Ва осаждаются из водного рас-
твора при pH 9,0, они содержат от 0 до 1 молекулы воды, которая
полностью удаляется до температуры 250° С.
Безводные пированадаты Са, Sr и Ва имеют две модификации:
«низкотемпературную» и «высокотемпературную»; первая переходит
во вторую при 300° С для Са и Sr и при 320° С для Ва. Метава-
надаты щелочноземельных металлов получены при pH 7,5 из рас-
творов NaVO3 или NH4VO3. Свежеосажденные препараты на воздухе
частично теряют воду. Метаванадат кальция, осажденный описанным
Рис. 41. Ванадий-кислород-
ные цепи в структуре
Sr(VO3)2-4H2O.
способом, всегда содержит примеси
пированадата кальция и метава-
надатов натрия или аммония. Для
получения чистого препарата посту-
пают следующим образом. Сплав-
ляют эквимолярные количества
СаСО3 и NH4VO3 при 800° С, полу-
ченные желтые кристаллы Са (VO3)2
растворяют в минимальном коли-
честве воды при 40° С* Раствор мед-
ленно упаривают, заканчивая про-
цесс на воздухе без нагревания.
Полученный метаванадат содержит
4 молекулы воды. Тетрагидрат
метаванадата кальция теряет воду
в две стадии, обезвоживание за-
канчивается при 180° С; при 90° С соль содержит 1 молекулу воды.
Метаванадат стронция кристаллизуется с 5 молекулами воды, однако
на воздухе стабилен тригидрат Sr (VO3)2-3H2O, который обезвожи-
вается при 160° С. Высушенный на воздухе метаванадат бария
содержит 1 молекулу воды, при нагревании дегидратируется в ин-
тервале 160—250° С. Как и в случае пированадатов, безводные
метаванадаты Са, Sr, Ва имеют две модификации: «низкотемператур-
ную» и «высокотемпературную»; превращение первой во вторую
происходит для Са между 200 и 350, Sr — при 394 и Ва —при 440° С.
По данным дериватографического исследования, метаванадаты
магния, кальция и стронция обезвоживаются ступенчато и на кри-
вых нагревания можно выделить участки, отвечающие удалению
определенного числа молекул воды (рис. 40): Mg(VO3)2-5,5H2O те-
ряет последовательно 3; 0,5; 0,5 и 1,0; Са (VO3)2-2,67H2O— 0,67;
1,0 и 1,0 и Sr (VO3)2-4H2O — 3,0 и 1,0 молекул воды. При этом
температура полного обезвоживания метаванадатов 360, 320 и 300° С
уменьшается от магния к стронцию. Это отражает ослабление проч-
ности связи воды в метаванадатах с увеличением ионных радиусов
в ряду Mg2+, Са2+, Sr2+.
Структура метаванадата стронция Sr (VO3)2-4H2O близка струк-
туре KVO3 • Н2О [182], параметры элементарной ячейки а = 7,38 +
+ 0,01; £>=33,6 + 0,01; с= 7,16 + 0,01 А; р = 90°. В его структуре
78
каждый атом ванадия координирован с 5 атомами кислорода, обра-
зующими тригональные бипирамиды. Последние образуют непрерыв-
ные цепи и связаны друг с другом общими ребрами (рис. 41, 42).
дтомы стронция обладают девятерной координацией. Три атома
кислорода 0(1), 0(2), 0(2') принадлежат одновременно ванадиевой
цепочке и связывают с ней атомы стронция, остальные О (4'), О (5'), О (5)
о © И
О ^0
Рис. 42. Схематическое изображение структуры Sr(VO3)2-4H2O.
Пунктирные линии — координации атомов кислорода, принадлежащих молекулам
воды.
принадлежат молекулам воды. Межатомные расстояния и углы между
связями в структуре Sr (VO3)2 • 4Н2О приведены в табл. 22. Анало-
гичную структуру имеет и метаванадат кальция Са (VOS)2-4H2O [159].
Декаванадат кальция Ca3V10O28-17Н2О может быть получен кристал -
Т а б л и ц а 22
Межатомные расстояния и углы между связями в структуре
Sr(VO3)2-4H2O
Межатомные расстояния Углы между связями
Связь rf+0,02, о А Связь J±0,02, О A Связи Угол, град
V—0(1) 1,65 Sr'—0(1) 2,54 0(2')—V—0(1) . 107
V—0 (2") 1,66 Sr'—0 (4') 2,62 0(1)—V—0 (3) 100
V—0 (3) 1,93 Sr'—0 (2) 2,74 0(1)— v—0 (3'") 123
V—0 (3") 1,97 Sr'—0(5) 2,73 0(2')—V—0 (3) 100
V—V'" 3,12 Sr'—0(2') 2,68 0 (2")—V—0 (3"') 131
Sr'—0 (5) 2,73 0(3)—V—0 (3') 146
Sr'—Sr' 3,99
79
лизацией из подкисленного до pH 6,5 раствора щелочного ванада,
та [179, 183]. Обезвоживание декаванадата кальция (рис. 43) про,
текает в три этапа: в интервалах 90—195, 195—300 и 300—336° С,
при температуре 655° С образец плавится; в соответствии с диаграм-
Рис. 43. Дериватограмма Ca3V10O28-17H2O.
мой состояния системы СаО —
V2O5 [176] можно предпола-
гать, что обезвоженный препа-
рат при нагревании распа-
дается на пятиокись ванадия
и метаванадат кальция. Это
соединение кристаллизуется
в моноклинной сингонии. Эле-
ментарная ячейка содержит
две формульных единицы и
имеет параметры 16,834;
6=10,156; б?=10,921А;
~93°8'; уд. вес, рассчитан-
ный по параметрам элементар-
ной ячейки, равен 2,465 г) см3,
найденный экспериментально
2,456 г) см3. Структура его
определена Сваллоу, Ахмедом
и Барнесом [184].
В структуре декавана-
дата кальция (рис. 44) один
атом кальция Са (2) координи-
рован с 7 молекулами воды.
Два других атома кальция
Са(1) и Са(1') расположены
на противоположных сторонах
декаванадатной группировки,
связаны с ней посредством
двух атомов кислорода О (13),
0(14) и 0(13'), 0(14') соот-
ветственно, каждый из них
координирует по 5 молекул воды. Таким образом, рассматриваемая
структура образована двумя типами комплексных ионов [Са (Н2О)7]2+
и [(Ca-5H2O)2V10O28]2-_. Декаванадатная группа V10O28 имеет орто-
ромбическую симметрию и построена из 10 плотно упакованных
октаэдров VO6 (рис. 45, 46).
Межатомные расстояния в октаэдрах VO6 приведены в табл. 23.
Структурные данные и измерения удельного веса приводят к заклю-
чению, что декаванадат кальция содержит 17 молекул воды, а не 16,
как ранее [179] предполагалось.
В работе [160] на основе исследования ИК-спектров сделаны
заключения о структуре ванадатов щелочноземельных элементов.
В ИК-спектрах ортованадатов Са, Sr, Ва зафиксированы четыре
80
4XJ
рис. 44. Проекция структуры Ca3V)0O28- 17Н2О вдоль [010], содержащая
элементарную ячейку.
к.™.,.™.,..™ «,₽— - ™=“ ....."'°
6 Заказ № 256
полосы (рис. 47, a): vx, v3, v4 и v2— 870, 825, 480 и 345 см~~*
соответственно, которые показывают, что в кристаллах, как и в вод-
ных растворах, для ортованадатов характерны тетраэдрические
Таблица 23
Межатомные расстояния в структуре VO6
и полиэдрах СаО7
Связь О (1, А Связь о d, А
-О (2) 1,681 -0 (7) 1,593
-О (2) 1,681 -0(6) 1,818
v (1а), -0(11) —0(11') 1,903 V (2) -0(9) 1,867
V(lfe) 1,903 V(3) -0 (4) 1,874
-0(1) l-O(l') 2,135 -0(2) 2,077
2,135 -0(1) 2,313
—0(13) 1,623 ( —0 (14) -О (4') 1,617
-о (9) 1,813 1,803
V(4) • -0(10) 1,813 V(5) • -0 (5') 1,803
-0(11) 2,002 -0(11) 2,009
-0(12) 2,002 -0(12) -O(l') 2,009
1-0(1) 2,211 2,222
1—0(19) 2,383 ( —О (23) 2,349
-0(15) 2,383 —0 (20) 2,414
-0 (16) 2,383 —О (22) 2,412
Са(1) 1 —0(18) 2,363 Са (2) i —О (20') —0 (22') 2,414
-0(17) 2,429 2,412
-0(13) 2,442 -0 (21) 2,429
1-0(14) 2,459 1 —0(21) 2,429
группировки VO4. В спектре i идратированного ортованадата каль-
ция, кроме того, наблюдаются две полосы при 1640 и 3100 см-1,
соответствующие деформационным и валентным колебаниям кристал-
логидратной воды и полоса при 655 см-1, которая приписана коле-
баниям по связи V.. .ОН2.
Предполагается, что структура пированадатов щелочноземельных
элементов, так же как и пированадата натрия, включает группиров-
ки V2O7, образованные двумя тетраэдрами и соединенные между
собой вершинами через кислород. Поэтому частоты колебаний по связи
V—О в данном случае близки к тем, которые наблюдаются в тет-
раэдрической структуре ортованадатов. Это подтверждается линиями
в спектрах пированадатов в областях 320—420 и 810—940 см-1
(рис. 47, б). Линии при 580 и 780 см-1 отнесены к колебаниям
по связи V—О—V.
Как отмечено выше, структуры Sr (VO3)2-4H2O и Са (VO3)2 • 4Н2О
подобны структуре KVO3-H2O и образованы тригональными би-
пирамидами VO5, соединенными в цепи. В группировках VO5
две связи V—О короткие и три — длинные. В соответствии
с этим в ПК-спектрах рассматриваемых соединений выделены две
82
Расстояния V—V в ангстремах
6*
а
/7ое/чощ ени&
Рис. 47. Инфракрасные спектры ортованада-
тов (а) и гидратированных и безводных «низко-
температурных» пированадатов (б).
1 — Са, 2 — Sr, 3 — Ва.
Рис. 48. Инфракрасные спектры гидратиро-
ванных и безводных «низкотемпературных»
метаванадатов.
/-Sr(VO3)2-3H2O; 2 — Sr(VO3)2H2O; 3-Sr(VO3)2,
обезвоженный при 360° С; 4 — Ca(VO3)2-4Н2О; 5—
Ca(VOs)2, обезвоженный при 170° С.
*_J1----L-—J1----I____I___i |
800 600 UOO5^-/
Рис. 49. Инфракрасные спектры ванадатов.
1 - Ba(VOs)2-H2O, 2 — KVOe.
Таблица 24 группы линий в области
Частоты колебаний по связям V-0 900—850 см-1 для ко рот -
Са (VO3)2-4H2O Sr (VOS) 2*4H2O ких связей и в области 600—540 см~1 для длин- ных связей (рис. 48, табл. 24).
0 d, А V, см 1 0 d, А V, см~1
1,63 1,67 1,93 1,97 845—900 605 1,65 1,66 1,93 1,97 850 -900 540 Аналогичные спектры имеют и безводные «низ- котемпературные» формы метаванадатов кальция и стронция. В спектрах Ba(VO3)2 • Н2О и высокотем-
пературных форм метаванадатов бария и стронция наблюдаются те
же группы линий, что и в спектре безводного KVO3, см—1:
KVO3 ..................... 915—853 680—502 389—350
Sr(VO3)2 ................. 950—830 700—530 358
Ba(VO3)2-H2O.............. 950—800 676—580 350—330
Ba(VO3)2 ................. 960—770 680—480 360—316
Это позволяет считать, что структура данных соединений, как
и структура KVO3, образована цепями тетраэдров VO4, связанных
через кислород (рис. 49). Для этого типа структуры частотные
характеристики колебаний вдоль цепочки V—О—V по сравнению
с таковыми для соответствующих пированадатов несколько смещены
в длинноволновую область, см—1:
Пированадат стронция........................ 738 580
Метаванадат стронция........................ 700 530
Пированадат бария .......................... 724 564
Метаванадат бария . ........................ 668 504
ВАНАДАТЫ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
Кристаллогидраты ванадатов редкоземельных элементов осаж-
даются при сливании растворов хлоридов или нитратов РЗЭ и ва-
надатов аммония или натрия. На кривых потенциометрического
титрования растворов нитратов РЗЭ раствором метаванадата аммония
имеется минимум [185, 186]. Соотношение РЗЭ и ванадия в мини-
муме равно 1:1, что соответствует выпадению ортованадата РЗЭ.
Процесс можно описать уравнением
La (NO3)3+NH4 VO3+H2O=La VO4+NH4NO3+2HNO3. (32)
Если же титровать раствор метаванадата аммония азотнокислым
.лантаном, то минимум на кривой соответствует соотношению La: V
в осадке 1 :3, это указывает на выпадение метаванадата РЗЭ:
La (NO3)3+3NH4VO3-La (VO3)3+3NH4VO3. (33)
При выдерживании в контакте с маточным раствором ортованадат
вступает в дальнейшие реакции и в конце концов соотношение La:V
86
0 осадке становится равным 1 :2; предполагается, что конечным про-
дуктом реакции является LaHV2O7 [187]:
2LaVO4+3HNO3=LaHV2O7+H2O4-La(NO3)3. (34)
В работе [187] осаждение ванадатов редкоземельных элементов
изучали, смешивая растворы LnCl3 и подкисленный до определенной
степени раствор метаванадата натрия. Смесь выдерживали при тем-
пературе 20° С до тех пор, пока pH раствора над осадком не при-
обретал постоянного значения. Было найдено, что при pH исходного
раствора метаванадата натрия от 8 до 6,55 осадки в случае лантана
имеют соотношение La: V = 1 : 2, в случае празеодима и неодима
Ln:V=2:3, а в интервале pH от 5,26 до 4,75 соотношение Ln:V
в осадках составляет 1:5. Состав полученных соединений был опи-
сан формулами LaHV2O7, Pr4(V2O7)3 и Ln2V10O28.
На основании результатов кондуктометрического титрования
растворов декаванадиевой кислоты раствором едкого натрия в при-
сутствии трехвалентных ионов лантана и церия авторы работы [63]’
пришли к выводу о возможности существования в водных раство-
рах комплексных ионов состава
[Ln (H2O)nH2V10O28]-; [Ln (H2O)nHV10O28l2-;
[Ln (H2O)„V10O2e]3-; [Ln (OH) (ЦОК-MoOJ4-;
[Ln (OH)2 (H2O)„_2V10O28p-; [Ln (OH)3 (H2O)„_8V10O28]e-.
Условия получения и некоторые свойства орто-, мета- и гексаванада-
тов редкоземельных элементов изучены Е. А. Арбит и В. В. Сереб-
ренниковым [188]. Ортованадаты состава LnVO4-2H2O получены
приливанием в растворы нитратов РЗЭ раствора метаванадата аммо-
ния. Если порядок сливания растворов сменить на обратный, выпа-
дают метаванадаты состава LnV3O9-4H2O. При смешивании растворов
метаванадатов аммония и нитратов РЗЭ с равным объемом спирта
образуются осадки гексаванадатов состава Ln4(V6O17)3-48H2O.
Таблица 25
pH осаждения и растворимость ванадатов редкоземельных элементов
Ванадат La Рг Nd Sm Eu Gd Dy Ho Lu V
pH осаждения
Орто . . 5,15 -и - 5,00 4,80 4,50 4,50 4,90 5,20 5,90 5,40
Мета . 5,40 5,10 4,90 4,70 4,45 4,40 4,80 5,30 5,70 4,80
Р ’астворимость при 25° С, 1 • 105 моль/л
Орто . . 31 — 6,1 5,6 4,2 1,9 4,7 6,о 10,5 •
Мета 17,2 5,3 5,1 4,4 3,9 3,2 3,7 4,6 9,3 3,6’
Величина pH растворов при образовании ванадатов и их раство-
римость изменяются в ряду редкоземельных элементов: от лантана
до гадолиния уменьшаются, далее увеличиваются до лютеция (табл. 2 ).
87
и- „
То, что орто-, мета- и гексаванадаты РЗЭ имеют близкие величины
растворимости, связано, по мнению авторов, с частичным переходом
при растворении гекса- и метаванадатов в другие ванадаты.
Декаванадаты РЗЭ, по-видимому, были получены еще в начале
нашего века, но только в 60-х годах стала ясна их химическая
природа. Гранато-красные кристаллы состава Се2О3 • 5V2O5 • 27Н2О
с плотностью 2,387 были получены испарением смешанных растворов
Рис. 50. Общий облик кристаллов 28-водных декаванада-
тов La, Се, Pr, Nd.
метаванадата аммония и сульфата церия. В аналогичных условиях
из раствора ванадата иттрия получен ванадат самария SmV5O14« 14Н2О
[122]. Сравнивая эти данные с результатами новейших исследований
(см. ниже), можно прийти к выводу, что эти ванадаты являются не чем
иным, как декаванадатами Ce2V10O28 • 27Н2О и Sm2V10O28-28H2O.
Яр иПреусс [153] получили декаванадат лантана La2V10O28- 18Н2О,
выпаривая в вакууме при комнатной температуре смесь растворов
декаванадиевой кислоты и ацетата лантана.
Декаванадаты редкоземельных элементов легко осаждаются и
из смешанных растворов кислого декаванадата натрия Na6HV10O2g
и хлоридов РЗЭ [187]. При комнатной температуре из раствора
кристаллизуются 28-водные декаванадаты лантана, церия, празеодима
и неодима; 25-водные — европия, тербия, эрбия, иттербия и иттрия;
25-водные декаванадаты после длительного выдерживания под слоем
S8
:маточника переходят в 24-водные кристаллогидраты; таким способом
получены 24-водные декаванадаты Y, Er, Yb. 28-водные декаванада,
ты La, Се, Рг и Nd представляют собой короткопризматические
-'часто таблитчатые по (001) кристаллы (рис. 50) — комбинацию семи
Рис. 52. Дериватограммы декаванадатов редкоземельных элементов,
о La2V10О28’28Н20; б — Nd2Vю^28"28Н2О.
простых форм (четырех пинакоидов и трех ромбических призм).
Все эти соединения кристаллизуются в моноклинной сингонии,
имеют близкие угловые параметры кристаллов и одинаковые опти-
ческие свойства. Соотношение осевых отрезков а : b : с = 1,6344 :
: 1 : 0,8173 [189].
25- и 24-водные декаванадаты, как и 28-водные, внутри каждой
группы кристаллографически не отличаются друг от друга. 25-водные
декаванадаты — призматические кристаллы оранжево-желтого цвета
с весьма совершенной спайностью, образованные комбинацией 11
пинакоидов (рис. 51, п), соотношение осевых отрезков а:Ь:с=
=0,9787:1:0,9627. Кристаллы 24-водных декаванадатов (рис. 51, б) —
короткие призмы желто-оранжевого цвета с весьма совершенной
спайностью — представляют собой комбинацию 15 пинакоидов,] соот-
ношение осевых отрезков а : b : с= 1,149 : 1 : 1,140.
:90
Обе группы относятся к триклинной сингонии с центральным
видом симметрии. Плотность описанных декаванадатов растет вместе
с увеличением атомного веса входящего в них
элемента (табл. 26). 28-водные декаванадаты РЗЭ теряют
при комнатной температуре, 25-
Все декаванадаты полностью обез-
редкоземельного
воду уже
24-водные — более, устойчивы.
Т а б л и ц а 26
Плотность декаванадатов
редкоземельных элементов
воживаются при 350° С [187].
Потеря воды происходит в не-
сколько этапов. 28-водный дека-
ванадат лантана теряет воду в три
этапа (рис. 52) в интервалах тем-
ператур 50—190, 190—235 и
235—350° С. Обезвоживание
Nd2V10O28 • 28Н2О протекает в
шесть этапов (см. рис. 52, б).
Обезвоженные образцы плавятся
при температуре около 690° С,
что соответствует температуре
плавления пятиокиси ванадия.
По-видимому, декаванадаты РЗЭ
Декаванадат Ln Плотность- /У20 “возд
Ln2V10O28 • 28H2O Ce 2,39
Pr 2,43
Nd 2,43
Ln2 V юО28 • 25H2O Eu 2,62
Tb 2,64
Er 2,69
Yb 2,72
и
после дегидратации распадаются на пятиокись ванадия и окисел
соответствующего редкоземельного металла. Последние при даль-
нейшем нагревании претерпевают полиморфные превращения, фикси-
руемые на дериватограммах в виде двух экзоэффектов [152]. Та-
ким образом, декаванадаты РЗЭ, а также калия, кальция, по-види-
мому, и других щелочноземельных элементов могут существовать
только в гидратированном состоянии и при потере воды распадаются
на компоненты в соответствии с диаграммами состояния безводных
систем.
ВАНАДАТЫ МЕДИ И СЕРЕБРА
При сливании растворов сульфатов меди (II) и ванадатов щелочных
металлов или аммония в зависимости от условий — температуры, со-
отношения компонентов и их общей концентрации, продолжитель-
ности выдержки осаждаются ванадаты меди различного состава. В ра-
ботах ранних авторов описаны ванадаты меди: орто-, Cu3(VO4)2,
пиро-, Cu2V2O7 • ЗН2О, мета-, Cu(VO3)2 и дека-, 3CuO-5V2O5-22H2O
[123]. Гитер [99] нашел, что ортованадат меди осаждается при
pH 1,7—8,6, а в интервале 6,3—8,7 образуется соединение 5СиО-2V2O5.
Моррет и Струплер [190—192] детально исследовали взаимодействие
в растворе ванадата натрия и сульфата меди (II). Изучены измене-
ния pH и электропроводности растворов NaVO3 при постепенном
прибавлении растворов CuSO4, а также определены состав осадков
и изменение их во времени. При добавлении CuSO4 в раствор NaVOa
до соотношения CuSO4: NaVO3 =1:4 при pH 6,5 протекает ре-
акция
24NaVO3 + 6CuSO4 + H2O->Cu2V2O7(OC) + Cu3(VO4)2(Oc) +
4- CuNa10 (V10O28)2 + 6Na2SO4 + 2NaOH.
? 91
При выдерживании реакционной массы состав осадка приближается
к составу ортованадата за счет дальнейшего взаимодействия:
CuNa10 (V10O28)2+Cu2V2O7 + H2O^2Na5HV10O28+Cu3 (VO4)2.
Образование ортованадата меди протекает полностью, если соотно-
шение CuSO4:NaVO3 в смешанном растворе составляет 1:2, а
исходный раствор NaVO3 содержит избыток щелочи, так что
NaOH : NaVO3 — 0,666; при этом в конечном растворе pH 8,65.
В растворах с соотношением CuSO4:NaVO3 = 1:2 первоначально
образовавшийся ортованадат меди, избыток щелочи и кислый дека-
ванадат натрия взаимодействуют по реакции
10Na5HV10O28+ 10Cu3(VO4)2+ 18CuSO4+ lONaOH-p 14H2O^
->24Cu2Na (V5O16H2) 4- 18Na2SO4,
при этом pH раствора понижается до 3—3,5. В конечном счете
взаимодействие между CuSO4 и NaVO3 можно описать суммарной
реакцией 1
10NaVO3 + 4CuSO4+2H2O^2Cu2NaH2V5O16 + 4Na2SO4, (35)
которая при нагревании растворов протекает очень быстро. В ре-
зультате авторы пришли к заключению о существовании 5-зарядного
аниона пентаванадата H2V50i^.
Эту точку зрения не разделяют Яр и Преусс [193], которые,
основываясь на представлении о том, что ион V10O2? существует
в виде пустотелого шара и способен образовывать комплексы вклю-
чения типа [М11 (ОН)/2(Н2О)ш_лУ10О28]~(4+п), рассматривают пента-
ванадат как соль декаванадиевой кислоты Na2Cu-[Cu(OH)4V10O28l-aq.
Химизм процессов при взаимодействии CuSO4 и NaVO3 они описы-
вают последовательностью таких реакций:
120NaVO3 + 30CuSO4 4- 5H2O->5Cu2V2O7 + 5Cu3 (VO4)2 4-
+ 5Na6[Cu (OH)2V10O281 + 5Na6V10O28 + 30Na2SO4;
5Cu2V2O7 + 5Na6[Cu(OH)2V10O281->5Cu3(VO4)2+5Na6V10O28 + 5H2O;
1 OCif 3( VO4)2 + 10Na6V10O28 +18CuSO4 4- 24H2O ->
->12Cu3Na2 [Cu(OH)4V10O28]+ 18Na2SO4.
На основании имеющихся данных трудно отдать предпочтение той
или иной схеме, однако вопрос о природе двойного поливанадата
меди и натрия можно, по-видимому, решить, изучая его струк-
туру.
В работах [191, 192, 194—196] описаны условия получения и
некоторые свойства двойного поливанадата меди и натрия и ортова-
надата меди.
Высушенный при 105° С поливанадат меди отвечает формуле
Cu2NaH2V5O16 • 2Н2О, при нагревании на воздухе он полностью обезво-
живается в интервале температур 100—300° С [192].
92
Ортованадат меди Cu3(VO4)2-3H2O осаждается после сливания
эквимолярных количеств 1 Л4 растворов CuSO4, NaVO3 и NaOH
и перемешивания в течение 48 ч при 40°, в маточном растворе
Б этих условиях pH 5,4 [192]. При нагревании на воздухе препарат
полностью обезвоживается в интервале 256—400° С, при 720° С
начинает терять кислород, а после длительной выдержки при 800° С
получен продукт состава Cu3V2O7, или 3CuO-V2O4 [196].
По данным Яра и Преусса [153], при взаимодействии уксусно-
кислой меди (II) и декаванадиевой кислоты из раствора выпадает
декаванадат меди состава Cu3 [Си (ОН)2 V10O28] • 18Н2О. Позднее
эти же авторы [193] синтезировали нормальный декаванадат калия
и меди состава K4(CuV10O28)-7H2O, а также сложные купрованадаты
калия и цезия, смешивая 200 мл 0,1 М раствора KVO3 или CsVO3
(pH 6,3) и 20 мл 1 М раствора CuSO4 (pH 3,8) при 60° С. Полу-
чены соединения состава 8CuO-5/2K2O- 10V2O5-9H2O, которым при-
писана формула
Cu3K2[Cu(OH)4V10O28]
КОН
Cu3K2[Cu(OH)4V10O28]
•9Н2О.
При сливании эквимолярных количеств растворов NaVO3 и
u(NH3)4SO4 в аммиачной среде в присутствии ацетона при 0° об-
разуются темно-фиолетовые игольчатые кристаллы метаванадата куп-
ритетраммина [Cu(NH3)4](VO3)2-2H2O [197]. В инфракрасном спектре
этого соединения обнаружены полосы, отвечающие группировке
Cu(NH3)4]2+ и ванадат-иону. Значительное смещение полос погло-
Таблица 27
Идентификация полос ИК-спектра (ел-1) метаванадата купритетраммина
[197]
[Cu(NH3)4] (VO8)2-2H2o Си (NH3)2+ Na4V4O12- 12Н2О Отнесение
3500 1 * 3500 3400 } ”S И (Н2°)
3360 3200 3270 • v (NHg)
1620 1595 1620 (NH3)+6H2O
1250 1245 970 6S(NH3)
925 860 880 ' vs и MW)
810 — —
710 709 1 er (nh3)
650 — 620 +va (V-O-V)
— — 550 +rs (V-O-V)
ч 420 — v (Cu—N)
330 280 6 (V-O-V)
93
щения в ванадате купритетраммина по сравнению с соответствуй
щими частотами для NaVO3 (табл. 27) связано, по мнению авторов
с сильным возмущающим действием на решетку (VO3)rt катиона
[Cu(NH3)4]2+. В присутствии пиридина из растворов CuCl2, CuSO
или Си (NO3)2 при комнатной температуре выпадает комплексное
соединение [Cu(C5H5N)4 ](VO3)2 [198]. :
Ванадаты серебра, как правило, получают из смешанных раствор
ров ванадатов щелочных металлов соответствующего состава или
ванадата аммония и азотнокислого серебра [99, 123, 143, 160,
199—201]. По [99], орто-, пиро- и метаванадаты серебра осаждают-
ся в интервалах pH 4,8—7,0; 3,8—4,1 и 3,2—3,5 соответственно,,
а при pH 1,0—1,9 образуется соединение состава Ag2O-2V2O5’
Шивахаре [202] нашел, что после осаждения ортованадата серебра
маточный раствор имеет pH 3,7. По результатам кондуктометри-
ческого титрования раствора Na3VO4, предварительно подкисленного
до pH 3,7 раствором азотнокислого серебра, он пришел к выводу
об образовании в этих условиях декаванадата серебра Ag6V10O28 • хН2О.
По [153], при взаимодействии уксуснокислого серебра с декавана-
диевой кислотой выпадает декаванадат серебра состава Ag6V10O28 • 4Н2О.
Орто-, пиро- и метаванадаты серебра, полученные осаждением при
взаимодействии метаванадата аммония и азотнокислого серебра,
не содержат воды [160, 195]. По другим данным, однако, метава-
надат серебра, полученный в этих условиях, содержит 0,1—0,25 моле-
кулы воды, которая полностью удаляется при 130—330° С [150, 201L
ВАНАДАТЫ ЦИНКА, КАДМИЯ И РТУТИ
*
Ванадаты этой группы изучены недостаточно, сведения о них
немногочисленны и имеют отрывочный характер. 5
Метаванадат цинка Zn (VO3)2 • 2Н2О осаждается из смешанного
раствора NH4VO3 и Zn(NO3)2 [123]. Из подкисленного раствора
KVO3 в присутствии солей цинка кристаллизуется двойной декава-
надат K2Zn2V10O28-16Н2О, изоструктурный аналогичному соединению
магния, в виде триклинных призм оранжево-красного цвета, с пара-
метрами элементарной ячейки а= 10,76; b— 11,17; с^ 8,77; а=104°50',
Р=109°29'; y=65°05z; Z—1 [179]. В структуре этого соединения
10 октаэдров VO6 образуют такую же компактную группу, как и в
структуре декаванадата кальция. Ионы цинка координируют по шесть
молекул Н2О, а ионы калия связаны через атсмы кислорода с ионами
V10O2F; оставшиеся две молекулы воды заполняют пустоты в струк-
туре [183]. I
Взаимодействием ацетатов цинка и кадмия с декаванадиевой
кислотой синтезированы декаванадаты Zn3V10O2s-16Н2О и Cd4V1nO98-
•15Н2О [153].
В работе [123] описано получение Cd(VO3)2 кристаллизацией
из концентрированного раствора метаванадата щелочного металла
и кадмиевой соли; декаванадата (KCdo,5)3V10028-21H20 — кристалли-
зацией из смешанного раствора KVO3 и CdSO4 в присутствии уксус-
94
цой кислоты, а также ванадатов CdV6O16-2H2O, 2CdO-3V2O5- 15Н2О
51 аналогичного последнему ванадата цинка.
Из ванадатов ртути (II) в этой работе описан только метавана-
дат, который можно получить из смешанного раствора NH4VO3 и
Hg(NO3)2. Согласно [203], из растворов щелочных ванадатов полу-
чены орто- и пированадаты ртути, которые, по [99], осаждаются
при pH 5,4 — 6,0 и 4,4 — 5,0 соответственно. Взаимодействие между
Hg(NO3)2 и NaVO3 в 20 %-ном растворе этанола изучено методами
потенцио- и кондуктометрического титрования [204]. В интервале
pH 6,4 — 6,9 образуется смешанный ванадат состава Na2O-HgO‘
^2V2O5, а при pH 4,5 —5,0 — метаванадат HgO-V2O5. Смешанный
метаванадат отличается низкой устойчивостью и при повышении
концентрации Hg24' превращается в HgO-V2O5.
При добавлении к растворам МС12, MSO4 или M(NO3)2, содер-
жащим пиридин (М — Zn2+, Cd2+, Hg2+), раствора NH4VO3 все три
металла образуют комплексы типа [М (C5H5N)2] (VO3)2 [198].
Гитером [99] приведены также и некоторые данные по ванада-
там одновалентной ртути. Метаванадат ртути (I) содержит две мо-
лекулы воды; орто-, пиро- и метаванадаты ртути (I) осаждаются при
pH 2,3 — 5,0; 1,0—1,2 и 0,4 — 0,8 соответственно.
ВАНАДАТЫ АЛЮМИНИЯ, ИНДИЯ И ТАЛЛИЯ
Из ванадатов этой группы описаны только отдельные соединения.
Метаванадат алюминия осаждается из раствора NH4VO3 при добав-
лении растворимой соли алюминия [123]. При взаимодействии аце-
тата алюминия и декаванадиевой кислоты образуется декаванадат
алюминия состава Al2 [Al (OH)3V10O28b 15,5Н2О [153].
Метаванадат индия In(VO3)3-2H2O получен при добавлении 1пС13
в раствор NaVO3 [123]. Известны ванадаты одновалентного таллия:
T1VO3 — соединение плохо растворимое в воде; T14V2O7, который
можно получить при смешивании растворов Na3VO4 и T12SO4, также
слаборастворим: в 100 частях воды при 14° С растворяется 0,02
части соли, а при 100° С — 0,026 части [123].
ВАНАДАТЫ ЦИРКОНИЯ, СВИНЦА И ОЛОВА
Литература по ванадатам элементов IV группы весьма ограни-
чена, данные имеются лишь по соединениям циркония, свинца и
отчасти олова.
С. М. Танатер и Е. К. Куровский [205] получили оксохлорвана-
дат mZrCl4-nZr3(VO4)4-pZrO2 смешиванием щелочного ванадата и
ZrCl4. Пейронель [206] выделил цирконилванадат 3ZrO2-2V2O5-
•9Н2О нагреванием смешанного раствора NH4VO3 и ZrO(NO3)2.
В бинарной системе ZrO2 — V2O5 обнаружен пированадат циркония
ZrV2O7 [207]. Саксена и Джайн [208] по кривым прямого и обрат-
ного потенциометрического титрования раствора Zr(SO4)2 растворами
ванадатов натрия определили состав поливанадатов циркония. При
добавлении в раствор ортованадата натрия раствора Zr(SO4)2 в ин-
тервале pH 9,0 — 9,5 образуется промежуточное соединение 3ZrO2-
9
• 2Na2O-4V2O5, при дальнейшем приливании выпадает ортованадат
3ZrO2-2V2O5. При обратном титровании образуется только ортова.
нанят циркония. В случае растворов пиро- и метаванадата натрИя
образуются соответственно пиро- и метаванадат цирконила ZrO2.
•V2O5 и ZrO2-2V2O5. Образование и осаждение орто-, пиро- и
метаванадатов цирконила происходит при pH 5,8 — 7,5; 4,0—5,5;
3,0 — 4,0 соответственно.
Для свинца (II) известны почти все основные типы ванадатов.
Ортованадат свинца выпадает в виде белого нерастворимого осадка
Pb3(VO4)2 при сливании растворов ацетата свинца и ортованадата
натрия; из раствора метаванадата аммония и азотнокислого свинца
в присутствии уксусной кислоты в зависимости от соотношения
реагентов при кипячении могут быть выделены пиро- и метаванадат
свинца [24,'123, 209]. Согласно [99], орто- и пированадат свинца
выпадают при pH4,0—11,6 и 1,8 —2,4, а метаванадат свинца ме-
тодом осаждения получить невозможно, однако более поздние иссле-
дования [210] показали, что в растворах орто-, пиро- и метаванада-
тов натрия соответствующие ванадаты свинца образуются при
pH 7,5 — 9,0; 5,75 — 7,25; 4,25 — 5,25. По данным В. Л. Золота-
вина ]132], орто- и пированадат свинца по сравнению с метавана-
датом отличаются меньшей растворимостью. Е. Г. Тимофеева с со-
авторами [211] в результате подробного изучения условий осаждения
метаванадата свинца при взаимодействии растворимых солей свинца
и метаванадата натрия или аммония отметили важность величины pH
исходных растворов, концентрации и соотношения реагентов для
получения монофазных препаратов. По их данным, при использовании
ацетата свинца, независимо от условий опытов в осадок выпадает
смесь мета-, пиро- и ортованадата свинца. Метаванадат свинца почти
стехиометрического состава (PbO: V2O5=1 :0,99) был осажден при
сливании эквимолярных количеств 0,1 М растворов NaVO3 или
NH4VO3 и Pb(NO3)2, подкисленных азотной кислотой до pH 3,0.
Лучшие результаты получаются при использовании разбавленных
растворов Pb(NO3)2 и NaVO3; избыток свинца в растворе почти не
влияет на осаждение. Температура исходных растворов не сказы-
вается на составе осадков, однако из нагретых до 40—50° С рас-
творов выделяются хорошо отстаивающиеся и легко фильтруемые
осадки. Согласно [195], при сливании растворов NaVO3 и Pb(NO3)2,
предварительно подкисленных до pH 3,0—3,5 при 60° С осаждается
ванадат свинца Pb4VeO18 • Н2О, который при нагревании отщепляет
воду в. интервале ПО—205° С.
Для олова (IV) известен ванадат состава SnV2O7-2,5H2O-0,5NH3
[194, 195]. При нагревании это соединение претерпевает изменения,.,
которые могут быть описаны схемой
SnV2O7 • 2,5Н2О • 0,5NH3 SnV2O7 • Н2О • 0,5NH3
—0,5NH3
— Н2О
360°
Sn V 2О7 3 SnO2+V2O5.
96
6дНАДАТЬ! ХРОМА И МАРГАНЦА
Для хрома известны почти все типы ванадатов. Ортованадат
хрома CrVO4 может быть получен сливанием растворов соли хрома
(III) и ванадата щелочного металла [123] или при окислении суль-
фата ванадила бихроматом калия в аммиачной среде [212]. Ортова-
надат хрома — нестойкое соединение и, согласно [212], уже при
лромывании свежеосажденного препарата водой осадок меняет состав,
частично превращаясь в пированадат. По данным Оливье с соавто-
рами [213, 214], осаждением из водной среды не удается получить
чистый пированадат хрома из-за образования полимерных комплексов
Cr (III). Эту трудность можно обойти, если синтез осуществлять
в спиртовой среде. Осадки пированадатов щелочных или щелочно-
земельных элементов обрабатывают избытком 0,26 714 раствора
Cr (NO3)3-9H2O в этиловом спирте. После восьмидневной выдержки
образуется пированадат хрома в виде мелкозернистого зеленого по-
рошка. Из Na4V2O7 было получено два препарата Сг4 (V2O7)3-ЮН2О
и Cr4(V2O7)3-11,5Н2О. В случае Ca2V2O7 осадок имел состав
Cr4(V2O7)3- 14,5Н2О. При нагревании в атмосфере кислорода пиро-
ванадаты хрома претерпевают сложные изменения: вода удаляется
в две стадии, одновременно препараты переходят в кристаллическое
состояние и затем разлагаются на CrVO4 и V2O5 (табл. 28, рис. 53).
По мнению авторов, отличие температур термических эффектов пи-
рованадата хрома с 14,5 молекулы воды обусловлено различием в
энергиях связей ванадий — кислород, присущим исходным соедине-
ниям Na4V2O7 и Ca2V2O7. Пированадаты хрома парамагнитны, их
магнитный момент соответствует наличию трех неспаренных элект-
ронов.
Таблица 28
Термические эффекты пированадатов хрома
Соединение ° с Потеря массы, % Температура,' °C
кристаллиза- ции разложения на CrV04+V2O5 т
Cr4(V2O,)3-10H2O 70—240 588—610 13,0 4,4 238—420 490
Cr4(V2O7)3-ll,5H2O 80—320 490—510 17,7 2,2 238—420 490
Cr4 (V2O,)3-14,5Н2О 70—240 325—500 13,4 10,1 238—315 641
В зависимости от условий осаждения могут быть получены нор-
мальный и основной метаванадаты хрома [215, 216]. Нормальный
метаванадат хрома получают, предотвращая образование полимерных
продуктов гидролиза хрома. С этой целью осаждение проводят из
концентрированных растворов с возможно более низкой величиной
pH, однако достаточной для того, чтобы в растворе преобладали
7
Заказ № 256
97
ионы метаванадата. Этим методом, используя 0,6 М раствор NaVO
подкисленный азотной кислотой до pH 6,5, и 2 М раствор Cr(NO3)3.*
• 9Н2С\ с pH 1,0, получен коричневый осадок метаванадата трехвц.
лентного хрома Cr (VO3)3-6,5H2O. Гидролиз ионов Сг3+ можно также
подавить, если проводить реакцию в присутствии спирта. В этом
случае после длительной выдержки реакционной смеси получили
метаванадат хрома с 8 молекулами воды [215]. При использовании
Рис. 53. Изменение веса и дифференциально-термические кри-
вые при нагревании пированадатов хрома в атмосфере кисло-
рода.
2 - Cr4(V2O7)3-1 0Н2О; 2 - Cr4(V2O7)3-11,5Н2 Оз
<3 — Cr4( V2O7)3-1 4,5Н2О.
разбавленных растворов (0,01 М NaVO3 и 0,01 7И Cr (NO3)3 • 9Н2О)
в слабощелочной среде с pH 8,6 при 30’С осаждается основной
метаванадат хрома Сг2 (ОН)2 (VO3)4 • 8Н2О [216). При нагревании
(рис. 54) нормальные метаванадаты хрома теряют воду в интервале
70—300° С, претерпевают аллотропное превращение (6,5-водный при
404, а 8-водный при 320° С) и разлагаются на CrVO4 и V2O5 при
566° С. Основной метаванадат хрома дегидратируется в интервале
70—350° С, теряя 9 молекул воды, и превращается в Cr2O(VO3)4,
при 506° С вещество необратимо разлагается на CrVO4 и V2O5.
В ПК-спектрах метаванадатов хрома имеются полосы, соответствующие
валентным и деформационным колебаниям воды при 3400 и 1600 см~г,
и полосы при 850—930 см”1, отвечающие структурносвязанной воде.
Присутствие полианионов метаванадата подтверждает полоса при
970 смг1. Метаванадаты хрома парамагнитны, 6,5- и 8-водные соли
имеют магнитные моменты 3,98 и 4,01 магнетона Бора, что соот-
ветствует наличию трех неспаренных электронов. Основной метава-
надат хрома обладает магнитный моментом 3,33 магнетона Бора,
98
отвечающим двум неспаренным электронам. На основе данных тер-
мического анализа, магнитных измерений и ПК-спектров авторы
пришли к следующим структурным формулам: для нормальных ме-
таванадатов хрома [Cr (H20)J-(V03)3-0,5H20 и [Cr(H2O)6]-(VO3)3*
Рис. 54. Дифференциально-термические кривые метаванадатов
хрома.
1 — Cr(VO3)8-6,5Н2О; 2 — Cr(VO3)3-8H2O.
• 2Н2О, для гидратированного основного метаванадата и обезво-
женного оксометаванадата хрома
онч
(Н2О)4 Сг Г )Сг (Н2О)4
ХОНХ
• (VO3)4 и lCr-0-Cr] (VO3)4.
Неуссером [217] получены метаванадаты, включающие комплекс-
ные ионы хрома (III). При обработке раствора лютеохлорида хрома
образуется темно-желтый плохо растворимый в воде осадок тригид-
рата метаванадата гексаамминхром (III) [Cr(NH3)6I (VO3)3-3H2O; из
раствора пурпуреохлорида осаждается метаванадат хлоропентааммин-
хром (III) [Cr(NHS)6Cl](VO3)2 темно-красного цвета.
При кипячении смеси MnSO4 и NH4VO3 выделяется красно-ко-
ричневый осадок метаванадата марганца Мп (VO3)2-4H2O, а при
добавлении сульфата марганца в раствор декаванадата калия обра-
зуется кристаллический осадок K2Mn2V10O28- !5Н2О [123]. Подан-
ным [99], осадок, отвечающий по составу пированадату марганца,
образуется в интервале pH 4,8—6,8.
Детальные исследования [218] показали, что в системе NaVO3—
— Мп(С1О4)2— Н2О в зависимости от концентрации водородных
ионов образуются ванадаты марганца различного состава: Mn(VO3)2-
• 4Н2О, Мп (ОН) VO3 • 2Н2О, (МпОН)4 • V2O7 • 4Н2О, (МпОН)3 VO4 •
•ЗН9О.
у*
99
Химизм процессов, приводящих к образованию перечисленных
соединений, описывается следующими реакциями: |
при pH 5—6 2Mn2++V40jr = Mn,V4019; ]
pH 6-7 Mn2++OH-=Mn (ОН)+ ' I
и 4Mn (ОН)+ + V4Otr=(MnOH)4 V4O12;
pH 7—10 2Mn2V4O12+4OH-=(MnOH)4V2O7+V2O?“+2H2O;
pH 10—12 3(MnOH)4V2O7+6OH~=4(MnOH)3VO4+2VOi_+3H2O.
В слабокислых растворах (H+:VOr=0,5 и pH > 4,5) при p.=
= 1,0 в среде перхлората натрия кристаллизуется двойной декава-
надат Na4MnV10O£8-25H2O. В присутствии этанола осадки образу-
ются быстро и в зависимости от состава исходных растворов, кроме
упомянутого Na4MnV10O28 • 25Н2О выпадают Mn3V10O28 • 15Н2О,
Na2Mn2V10O2g-19Н2О. При замене ванадата натрия на ванадат ка-
лия в слабокислых растворах в зависимости от состава исходных
растворов образуются только K2Mn2V10O28 • 11Н2О или K4MnV10O28 •
•ЮН2О, отличающиеся меньшей растворимостью, чем аналогичные
соединения натрия. В системе с литием образуется только простой
декаванадат марганца. I
Известен метаванадат дипиридинмарганец (II) [Мп (C5H5N)2] •
•(VO3)2, который осаждается из смешанного раствора NH4VO3 и
растворимой соли марганца в присутствии пиридина [198].
ВАНАДАТЫ ЖЕЛЕЗА, КОБАЛЬТА И НИКЕЛЯ
По данным [99], ванадаты железа состава Fe2O3-V2O5 и 2Fe2O3-
• 3V2O5 осаждаются из растворов при pH 1,0—1,8. |
Систематическое исследование состава, условий образования и
свойств ванадатов железа провели В. Л. Золотавин и Ю. И. Сан-
ников с соавторами [219—225]. При исследовании методами физико-
химического анализа (растворимость, pH, светопогашение, скорость
фильтрации и т. д.) систем Fe(C104)3 — Na5HV10O28—Н2О; Fe(C104)3—
— NaVO3 — Н2О; Fe (С1О4)3 — Na4V2O7 — Н2О и Fe (С1О4)3 —
— Na3VO4 — Н2О найдено, что осадки постоянного состава образу-
ются только в первых двух. В системах с декаванадатом и метава-
надатом натрия осаждаются два типа соединений с отношением
V:Fe 1,25 и 1,65 : Fe8 (OH)18V10O28 • 18Н2О и Fe6 (OH)12V10O£8 • 16Н2О
[220, 221]. Осаждение сопровождается понижением pH растворов
и может быть описано уравнениями:
8Fe3+ +18Н2О + HV10O^ Fe8 (OH)18V10O28 + 19Н+; (36)
6Fe3+ + 12H2O+HV10Oir -> Fe6 (ОН)12 V10O28 + 13H+; (37)
8Fe3++ 16H2O+ lOVO? -> Fe8(OH)18V10O28+ 14H+; (38)
6Fe3++ 10H2O+10VO? Fee (OH)12V10O28 + 8H+. (39)
При исследовании процесса замещения железа в осажденных соеди-
нениях ионами серебра установлено, что в состав соединения
100
fe8 (OH)18V]0O.8 входят различно гидратированные атомы железа,
лрочность связи которых с анионами декаванадата различна; в соеди-
нении Fe6 (OH)12V10O28 все шесть атомов железа связаны с одина-
ковым числом ОН-групп. Это позволило рассматриваемым соедине-
ниям приписать формулы
Fe2(OH)t]2 г . п ,, г
Гс znuA+1 • [Ую°2Г] И [Fe (OH)t] 6 • [V10Ofr] .
[Fe
Предполагаемые формулы подтверждены результатами измерения
магнитной восприимчивости ванадатов железа [225]. Найдено, что
в состав октаферридекаванадата входят два диамагнитных Fe2(OH)5~
я четыре парамагнитных иона Fe(OH)2~, а внешняя сфера гексафер-
ридекаванадата образована шестью парамагнитными ионами Fe (ОН)^.
Магнитные свойства ванадатов приведены ниже:
Fe8 (ОН)18у10О28-18Н2О Fee(OH)12Vio028‘16Н2О
Молярная восприим-
чивость х-106 при тем-
пературах:
77° К 13,26 26,36
194° К 12,94 21,15
295° К 7,15 10,75
Магнитный момент, рБ . 1,28 1,81
Константа Вейсса, ° К . 65 68
Температуры максимальной скорости процессов при нагревании, °C:
Удаление воды.......... 127 (—22Н?О)
320 (— 2Н2О)
Удаление воды и образо-
вание FeVO4 ............. 542 (— ЗН2О)
123 (—16Н2О)
265 (— 4Н2О)
558 (— 2Н2О)
На рис. 55 показаны кривые обезвоживания ванадатов железа.
Предполагается, что при этом Fe8 (OH)18V10O28 • 18Н2О последова-
тельно превращается в Fe8O4 (ОН)10У10О28 и Fe8O10 • (ОН)6 • У10О9§;
a Fe6 (OH)12V10O28 • 16Н2О в Fe6 (OH)12V10O28 и Fe6O4(OH)4V10O28.
В системах с Na4V2O7 и Na3VO4 ванадаты железа определенного
состава, по-видимому, не образуются [222, 223].
При нагревании смешанного раствора NH4VO3 и Co(NO3)2, слегка
подкисленного азотной кислотой, выделяются красноватые кристаллы
метаванадата кобальта Co(VO3)2-H2O [123]. Выделен [196] метава-
надат кобальта состава 2Co(VO3)2*5H2O, или Co(VO3)2-2,5H2O, про-
цесс дегидратации которого можно описать схемой
2Со (VO3)2• 5Н2О 2Со (VO3)2 • 3,5Н2О 2Со (VO3)2 -
• 2,5Н20—2Co(VO8)2.
Из смешанного раствора KVO3 и CoSO4 выделяются кристаллы двои
кого декаванадата калия и кобальта K2Co2V10O28- 16,5Н2О;_в 100
частях воды при 17,5° С растворяется 0,48 частей соли [123]. 11о[9У1,
101
в интервале pH 5,1—-6 g
из смешанного раствора
щелочного ванадата 3
растворимой соли кобал J
та выделяется осадок
отвечающий ортованадату
кобальта; аналогичное
соединение никеля осаж-
дается при pH 5,2—6,9.
Паркс и Преблуда [226J
нашли, что при добав-
лении раствора хлорида
гексамминкобальта (II),
к раствору ванадата со-
став осадков зависит от
кислотности среды: из
щелочного раствора выде-
лен пированадат гекс-
амминкобальта (III)
[Co(NH3)e]4-(V2O7)3; в
присутствии уксусной
кислоты образуется сое-
динение, которому авто-
эы приписали состав
Со (NH3)6]4 (VeO17)3. По
[227], из слегка щелоч-
ного раствора метавана-
дата в присутствии ионов
[Со (NH3)6]3+ выделяется
метаванадат гексаммин-
кобальта (III). Согласно
[198], при добавлении
ванадата аммония к раст-
ворам хлоридов, суль-
фатов или нитратов ко-
Рис. 55. Дифференциально-термические кри-
вые и обезвоживание ванадатов железа.
1 -Fe8(OH)rV10O28-18H2O;
2 Fee(OH)I2V10O28-1 6Н2О.
бальта или никеля в присутствии пиридина осаждаются метава-
надаты тетрапиридинкобальта (II) и тетрапиридинникеля (II) общей
формулы [M(C5H5N)4](VO3)2.
Ранними исследованиями [123] были получены зеленовато-желтый
кристаллический декаванадат никеля Ni3V10O2g-24H2O (кипячением
слабокислого раствора, содержащего метаванадат аммония и избыток
нитрата никеля), K2Ni2V10O28-16,5Н2О и ряд нормальных и смешан-
ных поливанадатов никеля сложного состава.
При исследовании взаимодействия поливанадатов натрия с NiSO4.
в 25 %-ном растворе этанола амперометрическим, pH- и кондукто-
метрическим титрованием установлено [222], что реакция между
NiSO4 и NaVO3 не приводит к образованию осадков; из раствора
Na3VO4 при pH 9,75—8,50 никель количественно осаждается в виде
102
(2,5NiNa) V2O8 зеленого цвета, в растворе Na4V2O7 при pH 8,5—7,75
□бракуется двойной пированадат никеля — натрия (l,5NiNa) V2O7.
Методом растворимости исследовано взаимодействие нитрата ни-
келя с ванадатами лития и натрия в широком интервале концентра-
ций компонентов и pH растворов [229, 230]. Опыты проводили сле-
дующим образом. Вначале готовили смеси LiVO3 или NaVO3 с LiOH,
NaOH или HNO3; их выдерживали 5 дней до установления равно-
весия, а затем добавляли раствор Ni (NO3)2 и рабочий раствор вы-
держивали 7—10 суток. Выпавшие осадки отделяли фильтрованием,
промывали небольшим количеством воды или спирта и высушивали
на воздухе до постоянного веса. С LiVO3 было получено четыре типа
осадков: в присутствии спирта при pH 4,5—6,2—Ni3V10O28-22H2O,
при pH 5,5—5,8—Ni (VO3)2 • 6Н2О, при pH 6,3—6,55—Ni (OH)VO3 •
• 2,2Н2О и при pH 8,8—10,0 Ni4(OH)4V2O7-5H2O, или Ni(OH)2-
• HVO4-2H2O. Соединения Ni (ОН) VO3-2,2H2O и Ni4 (ОН)4 V2O7 •
• 5Н2О получены также из растворов NaVO3 при pH 6,1—6,25 и
9,2—10,0 соответственно. При взаимодействии Ni(NO3)2 и NaVO3
осадок содержит следующие фазы: при pH 6,1—6,65 и малой кон-
центрации NaVO3 выпадает Ni (ОН) VO3-2,2H2O, при увеличении
концентрации NaVO3 одновременно выпадает Na2Ni2V10O28-21H2O,
более значительное увеличение концентрации ванадия приводит к
образованию Na4NiV10O28• 23Н2О (рН5,5—6,2). Последний выветри-
вается на воздухе, переходя в Na4NiV10O28-16Н2О. В присутствии
солей К, Rb, Cs могут быть получены соединения типа M2Ni2V10O28-
•15Н2О, растворимость которых в воде незначительна.
ВАНАДАТЫ УРАНА И ТОРИЯ
Известны минералы с различным соотношением урана и ванадия,
однако их природа установлена недостаточно [231—233]. Некоторую
ясность внесли исследования взаимодействия различных ионов вана-
дия и урана в водных растворах [234]. Было обнаружено образова-
ние малорастворимых ванадатов урана различного состава.
При смешивании растворов ванадия и урана одинаковой кислот-
ности при рН<2,0 образующийся осадок по соотношению V:U от-
вечает формуле (UO2)3V10O28, при pH 2,5—4,5 выделяется соединение
(U2O5)3V10O28; при pH 5—9 предполагается существование ванадата
(U3O8)2V4O12. При смешивании растворов с pH 10 взаимодействие не
наблюдается, так как уран в этих условиях переходит в анионы типа
I илО3л+1]2“. В других условиях, когда осаждение проводили из смеси
растворов ванадия и урана в хлорной кислоте гидроокисью натрия,
получены следующие ванадаты урана:
Отношение V: U
pH теоретическое опытное
(UO2)3 • V10O28 • пН2О . . . . . 1,5 3,33 3,16
(UO2)2V4O12-«H2O . . . . . . 5,48 2,0 2,24
(UO2)2V2O,-nH2O . . . . . . 8,73 1,0 1,08
(UO2)3 (VO4)2 .... .... 10 0,67 0,78
103
Заключение о их индивидуальности было сделано на основании тер.
мографических исследований; все они теряют воду в интервале тем.
ператур 100—200° С. Гидратированные препараты рентгеноаморфны
кристаллическая структура появляется после прокаливания при 200° с
у дека- и пированадата и при 520° С у мета- и ортованадата урана.
Давно было известно, что при сливании растворов солей тория
и ванадатов щелочных металлов образуются ванадаты тория. Пиро-
ванадат тория ThV2O7 • 6Н2О, который рассматривался также как
Th(HVO4)2-5H2O, был получен обработкой разбавленного раствора
хлорида тория раствором метаванадата аммония; выделены метавана-
дат Th(VO3)4-nH2O и гексаванадат Th (V6O16)2 • 8Н2О [123]. Стехио-
метрия соединений, образующихся при взаимодействии солей тория
и ванадатов натрия, исследована методами амперо-, pH- и кондукто-
метрического титрования [235, 236]. При титровании раствора нит-
рата тория растворами орто-, пиро- и метаванадата натрия образу-
ются осадки. Их состав установлен по изломам на кривых титрова-
ния. В областях pH 7,5—6,0; 6,0—4,8; 4,8—3,5 осаждаются орто-
ванадат 3ThO2-2V2O5, пированадат ThO2-2V2O5 и метаванадат
ThO2-2V2O5 соответственно. В случае ортованадата осаждение тория
протекает количественно.
Среди большого числа сведений относительно синтеза того или
иного класса соединений наиболее важна теория, или те общие
принципы, которые лежат в основе методов их синтеза. К настоя-
щему времени получен значительный экспериментальный материал,
касающийся условий получения ванадатов почти всех элементов пе-
риодической системы. Данные по ванадатам натрия, калия и аммония
получены путем исследования гетерогенных равновесий, имеют стро-
гий количественный характер и поэтому являются научной основой
при разработке любых методов получения ванадатов заданного со-
става. О ванадатах большинства других металлов сведения получены
препаративным методом и во многих случаях имеют характер кон-
кретных прописей применительно к синтезу того или иного соедине-
ния. Практическая ценность этих данных несомненна, но они дают
мало общих и теоретических сведений, относящихся к рассматрива-
емому классу соединений в целом. Первая и практически пока един-
ственная попытка выявить некие общие закономерности образования
ванадатов в водных растворах была предпринята Гитером [99], кото-
рый определил pH осаждения около 60 ванадатов различных метал-
лов. Выводы, к которым пришел автор, сводятся к следующему.
В ряду ванадатов, содержащих металлы одной группы, осаждение
ванадатов определенного состава протекает при различных pH в за-
висимости от природы присутствующих в растворе посторонних
ионов; число ванадатов, образуемых различными металлами, уменьша-
ется с возрастанием атомного веса металлов; pH образования и ра-
створимость ванадата данного металла тем ниже, чем выше его атом-
ный вес. Эти выводы, полученные на ограниченном количестве экс- I
периментального материала при использовании весьма несовершенных
методов идентификации индивидуальных соединений, не выходят за>
104
}дмки простых качественных представлений; они имеют характер ра-
бочей гипотезы, которая нуждается в усовершенствованиях и видо-
изменениях. Со времен работ Гитера накоплен значительный экспе-
иментальный и теоретический материал, касающийся ионного состоя-
ния ванадия в водных растворах, состава, структуры и свойств ва-
надатов, однако отсутствие количественных характеристик равновесий
в водно-солевых системах, включающих пятивалентный ванадий и
другие металлы, не позволяет наметить рациональные пути синтеза
и критически оценить всю сумму имеющихся в литературе сведений
о составе и условиях образования ванадатов. Эта важная область
химии пятивалентного ванадия ждет своих исследователей.
Глава IV
ГИДРОЛИТИЧЕСКОЕ ОСАЖДЕНИЕ ВАНАДИЯ
ГИДРАТИРОВАННАЯ ПЯТИОКИСЬ ВАНАДИЯ
И ДЕКАВАНАДИЕВАЯ КИСЛОТА
Если выдержать пятиокись ванадия в контакте с водой, то ра-
створ приобретает кислую реакцию и в нем обнаруживается некото-
рое количество ванадия. Величины растворимости пятиокиси ванадия
в воде, по литературным данным (табл. 29), сильно различаются, осо-
бенно при высоких температурах. Это, по всей вероятности, связано
с медленностью достижения равновесия и с тем, что в некоторых
случаях исходные препараты не являлись чистой пятиокисью вана-
дия, а содержали некоторые количества щелочных металлов, имели
различную структуру и физико-химические свойства. 1
Таблица 29
Растворимость пятиокиси ванадия в воде
i, °C Растворимость, вес. % Твердая фаза Метод получения исходной пятиокиси ванадия Лите- ратура
~20 0,125 Прокаливанием NH4VO3 [237}
25 0,07 — То же [238]
100 0,07 » [238]
18 0,18—0,188 — Осаждена из раствора ванадата деист-
вием кислоты; прокаливанием [1Г
NH4VO3
100 0,20—0,221 -— То же [1}
30 0,101 V2O5-xH2O Прокаливанием NH4VO3 [100}
25 0,04 VoO5-3H2O Осаждена из раствора Na3VO4 деист-
75 0,95 V,O6-3H,O вием серной кислоты [102}
30 0,11 V2O5.a-H2O Прокаливанием NH4VO3 [146]
По-видимому, наиболее достоверные значения растворимости в во-
де приведены в работах [100, 146, 237]: они близки по величине
и получены в условиях достаточно длительной выдержки. Однако
неясным остается вопрос, устойчивы ли безводная пятиокись ванадия
или какой-либо гидрат в качестве донной фазы в водных растворах.
В литературе описаны гидраты пятиокиси ванадия различного состава:
V2O5-3H2O(H3VO4) и V2O5-2H2O(H4V2O7) [102, 125, 145], V2O5.
• H2O(HVO3) [79, 102, 125, 145, 239], 3V2O5.2H2O(H4V6O17) U23],
106
V2P6-0,5H2O(H2V4O11) [239], но в большинстве случаев заключение
об их индивидуальности основано только на данных химического
анализа.
Хюттиг и Кёниг [239] исследовали процесс обезвоживания гид-
ратированной пятиокиси ванадия. Исходный препарат был получен
подкислением раствора NH4VO3 азотной кислотой. Осадок после дли-
тельной выдержки под слоем маточника многократно промывали во-
дой и, как указывают авторы, не содержал солей аммония. Его со-
став отвечал формуле V2O5-2H2O. Препарат был рентгеноаморфным
и при нагревании медленно терял воду в широком интервале темпе-
ратур. Другие препараты гидратированной пятиокиси ванадия гото-
вили, подвергая исходный осадок длительному воздействию повышен-
ной температуры, давления, встряхивания и т. д. Таким путем были
получены препараты V2O5-nH2O, где п=3,34-ь0,44. Как правило,
характер обезвоживания подвергнутых старению образцов изменялся,
они постепенно теряли основную массу воды, но последние 0,5 мо-
лекулы отдавали ступенчато при 260 или 275 С. Один из образцов,
полученный при длительном взбалтывании исходного препарата под
слоем маточника, имел состав V2O5-H2O, терял воду в две ступени;
0,5 молекулы при 258 и 0,5 молекулы при 275° С. На рентгенограм-
ме наблюдались четкие интерференционные линии. Авторы рассмат-
ривают его как индивидуальное химическое соединение — метавана-
диевую кислоту HVO3. Рентгенограмма препарата V2O5-0,5H2O была
похожа на рентгенограмму безводной пятиокиси ванадия, но интер-
ференционные линии смещены; его также рассматривали как инди-
видуальное химическое соединение H2V4On. В работе [79] отмеча-
ется существование индивидуального соединения HVO3. В природе
гидратированная пятиокись ванадия встречается в виде минерала
алоита V2O5-H2O [178].
Детальное исследование гидратированной пятиокиси ванадия про-
ведено в работе [240]. Препараты для исследования были получены
двумя способами. В первом случае раствор метаванадата лития под-
кисляли азотной кислотой (Н+:УОГ= 2,1) и образовавшуюся суспен-
зию выдерживали в течение длительного времени при комнатной тем-
пературе до установления постоянной величины pH. Осадок отфильт-
ровывали, промывали слабым раствором азотной кислоты и высуши-
вали на воздухе. Для приготовления другого образца в платиновой
пашке расплавляли чистую пятиокись ванадия и тонкой струей вы-
ливали в воду. После кратковременного перемешивания и отделения
фильтрованием (через бумажный фильтр) нерастворившегося остатка
получили кислый раствор (pH2,15) ярко-малинового цвета, содержа-
щий в коллоидном состоянии 17,67 г)л V2O5. Об этом свидетель-
ствовала легкая коагуляция раствора под действием солей и кислот.
Коллоидный раствор медленно упаривали до пастообразного состоя-
ния, а затем высушивали при 100° С до получения твердой массы.
Оба препарата, по-видимому, обладали одной и той же кристалличе-
ской структурой, так как на их рентгенограммах зафиксированы оди-
107
наково расположенные размытые интерференционные полосы. Состав
препаратов: V2O5-1,78H2O и V2O5- 1,47Н2О. Основное количество
воды образцы теряют при нагревании до 250° С (рис. 56). Остаток
(0,224 молекулы в первом случае и 0,357 молекулы во втором) уда.
Рис. 56. Дериватограммы гидратированной пятиокиси ванадия, полученной
при подкислении раствора LiVO3 (а) и из коллоидного раствора (б).
ляется в интервале 255—318 и 246—350° С соответственно; отщеп-
ление этой наиболее прочно связанной воды в обоих образцах с ма-
ксимальной скоростью протекает при 270° С. Вслед за эндотермиче-
ским эффектом, сопровождающим окончательное обезвоживание, на
кривых ДТА обоих препаратов зафиксирован экзоэффект, который,
как можно судить по рентгенограммам гидратированных и полностью
обезвоженных образцов, связан с перестройкой кристаллической ре-
шетки. Наиболее прочно связанная вода отнесена к типу «конститу-
ционной», удаление ее приводит к разрушению структуры осадков.
Как уже было отмечено, при исследовании растворимости в си-
стеме SO3—V2O5—Н2О был сделан вывод о существовании трех гид-
ратов пятиокиси ванадия (V2O5-3H2O, V2O5-2H2O и V2O5-H2O), ко-
торые рассматриваются как орто-, пиро- и метаванадиевая кислоты
102, 145]. На термограмме тригидрата обнаружено четыре эндотер_
108
^ических эффекта при 250, 465, 585 и 690° С [145]. Первые три из
лих относятся к процессу обезвоживания и отвечают последователь-
ному отщеплению трех молекул Н2О. Эффект при 690° С соответст-
вует плавлению пятиокиси ванадия. Три эндоэффекта (465, 585 и
690° С) зафиксированы на термограмме дигидрата, а два (585 и 69&°Q
на термограмме моногидрата.
Три- и моногидрат пятиокиси ванадия получены при исследова-
нии системы (NH4)2O—V2O5—Н2О [125]. Для тригидрата получена
термограмма, аналогичная только что описанной, но термограмма мо-
ногидрата с тремя эндоэффектами при 100, 240 и 690° С не совпа-
дает с описанной в работе [145]. Предполагается [125], что одновод-
ная пятиокись ванадия, осажденная из кислых растворов, отличается
по своей природе от выделенной из щелочной среды. К сожалению,
в упомянутых работах не приводятся данные химического анализа,
что не позволяет судить о наличии примесей SOs или NH3 в иссле-
дованных препаратах и, кроме того, для расшифровки природы эн-
дотермических эффектов желательны термогравиметрические иссле-
дования.
Из приведенного обзора видно, что делать определенные выводы
о составе и количестве индивидуальных гидратов пятиокиси ванадия
пока нельзя, однако несомненно, что химические соединения между
пятиокисью ванадия и водой существуют. Глемзер в работе «Дости-
жения и проблемы соединений в системе окисел — вода» [241] упо-
минает лишь о соединении V2O5-H2O и относит его к типу аква-
окислов, у которых вода находится в виде молекулы Н2О. Для на-
дежной идентификации индивидуальных соединений в системе V2O5—
— Н2О необходимы дальнейшие исследования с применением совре-
менных методов физико-химического анализа.
Пятиокись ванадия легко образует коллоидные растворы. Сводка
ранних исследований, посвященных свойствам коллоидных растворов
пятиокиси ванадия, приведена в работе [123]. Золи V2O5 могут быть
получены несколькими способами. По методу пептизации [242] к ра-
створу ванадата аммония или натрия добавляют разбавленную соля-
ную кислоту и полученную массу промывают водой до тех пор, пока
фильтрат из светло-желтого не становится красным. Полученный золь
подвергают диализу для удаления ионов электролитов. Золи V2O5
могут быть получены также гидролизом третамилового эфира ортова-
надиевой кислоты [243, 244] и с помощью катионообменной смолы
[63]. Весьма удобным и быстрым является описанный выше способ
получения золей V2O5 при непосредственном взаимодействии расплав-
ленной V2O5 с водой [245]. Темный малиново-красный коллоидный
раствор в высшей степени устойчив; его можно выпаривать, после
чего остается красная бархатистая масса, которая при добавлении
воды вновь переходит в раствор. Частицы свежеприготовленных гид-
розолей V2O5 отличаются небольшой величиной, часто имеют аморф-
ную структуру [246]. При старении гидрозолей наблюдается посте-
пенный рост частиц в виде тонких и длинных палочек или волокон
кристаллической структуры [247—254]. Изменение размера и формы
109
частиц коллоидной V2O5 в процессе старения приводит также к увс.
личению оптической плотности и светорассеяния золями пятиокиси
ванадия [244]. После коагуляции под действием электролитов (ВаС12
КС1, NaCl) наблюдается падение pH раствора [255]. Под действием
ультразвука увеличивается устойчивость золя V2O5, что находит от-
ражение в увеличении коагулирующей концентрации электролита [256].
Растворы пятиокиси ванадия в воде окрашены в желтый цвет
и имеют кислую реакцию (pH 3,5—3,6) [12]. Это указывает на
^6 Vю
Рис. 57. Кондуктометрическое титрова-
ние декаванадиевой кислоты.
существовав ие достаточ г о
сильной ванадиевой кис-
лоты, относительно состава
которой имелось несколько
точек зрения. Вначале ее
рассматривали как комплекс-
ную изополикислоту состава
H2[O(V2O5)2,5] [60], как
кислоты: H3VO4, HVO3 [12]
и H4V10O27 [53]. После
работы супругов Россотти
[20], которые установили
существование декаванадат-
ионов, ее стали рассматри-
вать как декаванадиевую
кислоту H6V10O28. Эта последняя точка зрения была подтверждена экс-
периментально и в настоящее время является общепринятой. Растворы
декаванадиевой кислоты получают при пропускании раствора метава-
надата щелочного металла, чаще всего натрия, через катионообмен-
ную смолу в Н-форме [62]. По другому методу пятиокись ванадия
обрабатывают раствором перекиси водорода, свободной от фосфорной
кислоты. Образовавшаяся первоначально пероксованадиевая кислота
довольно быстро распадается и в растворе остается свободная дека-
ванадиевая кислота [55, 63]. При кондуктометрическом [61, 63] или
рН-метрическом [62, 257] титровании декаванадиевой кислоты раст-
вором щелочи наблюдается три точки перегиба, соответствующие ис-
пользованию 4, 5 и 6 эквивалентов ОН~ (рис. 57). Четыре из шести
протонов декаванадиевой кислоты в растворах отщеплены, а анион
H2V10O28 диссоциирует ступенчато. Декаванадиевая кислота плохо
растворима: уже 0,002 М растворы ее неустойчивы, и вследствие
реакции
^6^10^28 5V2O5 • aq + хН2О
(40)
в них возникают коллоидные частицы гидрата пятиокиси ванадия.
Состояние равновесия в таком растворе устанавливается приблизи-
тельно через 800 ч [63]. Моуллик и Муллик [258] рассматривают
это явление как процесс полимеризации, описываемый уравнением
2ROH^ROR+H2O,
но
где ROH — ванадиевая кислота. Используя кондуктометрический и
спектрофотометрический методы, они нашли, что кинетика процесса
полимеризации описывается уравнением второго порядка.
В присутствии щелочи ион У10О2Г довольно медленно распада-
ется, переходя, в зависимости от концентрации ионов ОН"’, в бес-
цветные мета-, пиро- или ортованадат-ионы. Кинетика этого процесса
исследована спектрофотометрическим методом при ионной силе ра-
створа [i=l,5 (NaC104) и комнатной температуре [551. Оказалось,
что деполимеризация декаванадат-иона под действием щелочи является
реакцией первого порядка в отношении ванадия и ионов ОН~; ско-
рость ее описывается уравнением
d ГVjoO^I 1 г 4
-----!_2°- 28 J =0,0162 [QH^] V10O^ .
dt
При подкислении раствора метаванадата натрия образуются ионы
HgVjoO^r, HV10O52F и VOt в соотношении, определяемом степенью
подкисления и общей концентрацией ванадия в соответствии с рав-
новесием реакции (18) гидролиза ионов VO^. При спектрофотомет-
рическом определении константы равновесия реакции гидролиза [65]
было найдено, что смешение растворов при комнатной температуре
не дает устойчивых результатов; если же смешанный раствор нагреть
почти до кипения, а затем охладить, то равновесие устанавливается
быстро и получаются вполне воспроизводимые результаты. Аналогич-
ное явление было отмечено при спектрофотометрическом исследовании
кинетики деполимеризации иона H2V10O2? под действием кислоты
[259]. Скорость распада декаванадат-ионов при избытке кислоты опи-
сывается уравнением
d [H2V10O^rl г 4
-------С/ [H2V10O24r]n [Н+]-,
а величины п и т зависят от условий опыта. Если растворы
Na4H2V10O28, полученные подкислением раствора NaVO3, быстро на-
греть и охладить, а затем добавить избыток кислоты, то они приоб-
ретают оптическую плотность, соответствующую переходу всего ва-
надия в форму ионов VO2 за 10—50 мин (в зависимости от избытка
кислоты). Величины пит в этом случае равны единице, а 7<=2,51.
В опытах без нагревания п изменяется от 1,3 в начале опыта до
4,0—4,5 в конце, а для достижения равновесия требуется около
суток. К сожалению, природа этого интересного явления до сих пор
не выяснена.
Из сравнения коэффициентов скорости видно, что распад декава-
надата под действием кислоты протекает значительно быстрее, чем
в щелочной среде. Это и понятно, так как в первом случае взаимо-
действие происходит между разноименно заряженными частицами.
Опыты показывают также, что из всех известных ионных форм ва-
надия (V) декаванадат-ион кинетически наиболее стабилен, его рас-
111
пад протекает достаточно медленно и в некоторых случаях с этим
необходимо считаться. В то же время образование его при нейтра-
лизации сильнокислых растворов щелочью или щелочных растворов
ванадия кислотой протекает практически мгновенно.
Можно предполагать, что высокозарядные анионные формы вана-
дия, например, анионы декаванадиевой кислоты способны выступать
в качестве лигандов, образуя в растворе с ионами противоположного
знака комплексные частицы. Качественно этот процесс обнаружен
при кондуктометрическом титровании декаванадиевой кислоты ще-
лочью в присутствии двух- и трехзарядных катионов [63]. Шварцен-
бах и Гейер [29] методом потенциометрического титрования в струй-
ном аппарате установили существование комплексов декаванадиевой
кислоты с катионами щелочных металлов и определили их константы
устойчивости при /=20° С, 1,0:
m+ + hv10o5 дДМНУ10О4 .
28 28
м + + v10o6- zi м v10os“. .
28 28
м + + mv10o‘~ m2vwo‘"
2 о 2 8
Li+ Na+ K+ Rb+ Cs+
4 5 25 60 160
40 40 250 600 1500
4 4 10 — —
Как видно, способность к комплексообразованию с декаванадат-
ионами в ряду Li+, Na+, К+, Rb4", Cs+ возрастает, в то время как
с обычными лигандами наблюдается обратная последовательность.
Причина аномалии пока не ясна. Эти же авторы нашли, что ион
HVO4- образует комплексные частицы NaHVO? и KHVO?, констан-
ты устойчивости которых при £—20° С, р—1,0 соответственно равны
2,0 и 1,1.
ГИДРОЛИТИЧЕСКОЕ ОСАЖДЕНИЕ ВАНАДИЯ
При подкислении растворов ванадатов щелочных металлов силь-
ными минеральными кислотами выпадает красно-коричневый осадок.
Дюльберг [18] описал образование этого осадка при осаждении из
растворов ортованадата натрия и назвал его гидратированной пяти-
окисью ванадия V2O5-zzH2O. Он считал, что данное соединение в
своем составе не содержит натрия, доказывая это получением осадка
гидратированной пятиокиси при взаимодействии V2O5 с перекисью
водорода.
Описанный процесс гидролитического осаждения лежит в основе
промышленных методов извлечения ванадия из растворов и в зави-
симости от характера исходного сырья и требований, предъявляемых
к готовой продукции, может довольно широко варьироваться. Оса-
док, получаемый в заводских условиях, помимо обычных примесей
Са, Mg, Fe, Сг, Si, Р, Ti содержит всегда 6—10% Na2O и известен
как техническая пятиокись ванадия [1, 2, 260]. При осаждении ва-
надия из нейтральной среды растворы подкисляют серной, соляной
или азотной кислотами [1, 2, 260—-263]. Кислые растворы нейтра-
112
дизуют, как правило, содой [262]. Для получения более чистой пя-
тиокиси ванадия гидролиз проводят в присутствии аммонийных со-
лей [264]. Все эти и другие варианты практического осуществления
гидролиза выработаны в результате долгой практики на основе эмпи-
рического подхода. Химизм и механизм процессов гидролиза изучены
недостаточно.
РАСХОД КИСЛОТЫ И ИЗМЕНЕНИЕ pH РАСТВОРОВ
В ПРОЦЕССЕ ГИДРОЛИЗА
В общих чертах условия гидролитического осаждения ванадия
сформулированы Э. Ф. Краузе с соавторами [263] и сводятся к сле-
дующему:
1) осаждение ванадия возможно из растворов различной концен-
трации с использованием любой минеральной кислоты;
2) необходимая кислотность раствора определяется концентрацией
ванадия;
3) вне пределов оптимальной кислотности растворов ванадий вы-
падает не полностью;
4) с ростом температуры скорость осаждения увеличивается.
Процесс осаждения рассматривается как результат образования
плохорастворимой ванадиевой кислоты
6VOr+3H+->HVeO?r+H2O, (41)
а величина pH раствора — как основной фактор, определяющий вы-
падение ванадиевой кислоты в осадок.
Н. П. Слотвинский-Сидак с соавторами [262] установили, что при
осаждении из промышленных сернокислых растворов, нейтрализован-
ных содой, едкой щелочью, аммиаком и карбонатом аммония, опти-
мальная величина pH растворов лежит в узком интервале значений
(1,1 ч-1,9). М. Н. Соболев [1] указывает на зависимость продолжи-
тельности осаждения от концентрации ванадия в растворе и приводит
данные, согласно которым pH растворов после осаждения увеличи-
вается. Он считал, что в растворе образуется комплексная сернова-
надиевая кислота примерного состава V2O5-2H2SO4. После нейтрализа-
ции свободной кислоты начинается гидролиз и кислотность раствора
возрастает вследствие освобождения кислоты
V2O5 • 2H2SO4 + хН2О -> V2O5 • ^H2O + 2H2SO4. (42)
По данным Равитца и др. [265], растворы ванадия с рН<2 об-
ладают буферными свойствами и при осаждении ванадия pH таких
растворов не меняется; если pH растворов выше указанной величи-
ны, то для поддержания ее постоянства во время осаждения требу-
ется добавлять кислоту. Авторы рассматривают процесс осаждения
как образование кислого гексаванадата натрия Na2H2V6O17. Эта же
точка зрения изложена в работах [266, 267].
А. Б. Бектуров с сотрудниками [268] изучали гидролиз, подкис-
ляя растворы NaVO3 и KVO3 серной или соляной кислотой при ком-
8 Заказ № 256
113
натной температуре и выдерживая смеси в течение двух-трех недель
до установления равновесия. По их мнению, при степени подкисле-
ния1, равной 0,55—1,2 для NaVO3 и 0,5—1,5 для KVO3, выпадают
осадки, которые по своему составу являются не чистой пятиокисью
ванадия, а солями пентаванадиевой или декаванадиевой кислот, со-
держащими ванадий в катионной и анионной форме: NaHVO2V5O14.
•3,5Н2О и K2(VO2)3V5O15-4H2O. Влияние степени подкисления!
растворов на результаты гидролитического осаждения ванадия и из-
менение pH в ходе гидролиза детально исследованы в работе 1269].
Ванадий осаждали подкислением раствора NaVO3 серной или со-
ляной кислотой при 60—80< С. В ходе осаждения измеряли кон-
центрацию ванадия в растворе (У) и содержание кислоты в растворе
за вычетом связанной с ванадием (Н). При этом условно принима-
лось, что весь ванадий, находящийся в растворе, присутствует в
форме Н2¥10О2Г и количество связанной с ним кислоты определя-
лось по результатам потенциометрического титрования, исходя из
уравнения реакции
10VOT+6H+= H2V10O27+2H2O. (43у
Если отношением характеризовать изменение содержания кис-
лоты в растворе при осаждении 1 г-атома ванадия, то выражение-
0,6 Ну будет определять расход кислоты на осаждение
1 г-атома ванадия. Таким образом, зная величину Ну, мы опреде-
лим стехиометрические коэффициенты в уравнении, описывающем,
процесс осаждения
2NaVO3 + хН+Na2O • V2O5 • aq + xNa+ + - H2O. (44)
2 2
Из рис. 58 видно, что в процессе осаждения величина Н уменьша-
ется почти пропорционально уменьшению концентрации ванадия в
растворе. Отношение при малых степенях подкисления раствора’
(H+/VO3~<0,8) падает с 0,2 до 0,16, при больших (H+/VO?> 1,0)-
к концу осаждения, напротив, возрастает до 0,23—0,24. Иными
словами, расход кислоты при осаждении зависит от степени подкис-
ления раствора: чем выше отношение H+/VOT, тем выше расход ки-
слоты. Меняется он и в процессе осаждения, возрастая при
H+/VO?>1,0 и падая при H+/VO?<0,8. В условиях не очень вы-
сокой кислотности на осаждение 1 г-атома ванадия в среднем за-
трачивается 0,8 г-же кислоты; при недостатке кислоты в растворе
осаждение протекает не полностью. Следовательно, в уравнении (44)
Ц Степень подкисления растворов метаванадатов удобно характеризовать ве-
личиной H+/VO^, т. е. отношением числа эквивалентов кислоты, добавленных,
на 1 моль NaVO3.
114
2__х
х>1,6 (так^как x=2Hv), а коэффициент------
и отношение Na:V
атом!атом в осадке равно или меньше 0,2.
Зная расход * кислоты на осаждение ванадия, можно выяснить,
каким образом в процессе гидролиза изменяется pH раствора. Для
10
Рис. 58. Н—[V] при осажде-
нии ванадия из подкисленных
растворов NaVO3 (1 — 11 —
серной, 12 —17 — соляной
кислотой).
Цифры на кривых — номера опы-
тов
Номер опыта 1, ° С h+/vo3
1 80 0,683
2 80 0,740
3 80 0,806
4 80 1 ,000
5 70 0,866
6 70 1 ,022
7 70 1,181
8 70 1,330
9 60 2,532
10 80 2,532
1 1 80 2,730
12 80 0,767
13 80 0,700
14 80 0,890
15 80 1,092
16 80 1,331
17 60 2,220
У Ю мюк/л
этого необходимо знать состав растворов в зависимости от степени,
подкисления. Последний легко рассчитать, если известны ионные
формы, в которых ванадий существует в растворе при данных усло-
виях, константы равновесия реакций перехода их друг в друга и
концентрация ванадия. К сожалению, константы равновесия известны
только для температуры 25° С и р = 0,2 и 1,0 (см. главу I). Мы
провели расчет для р.==1,0 в интервале pH 1,0—2,0 при концентра-
циях ванадия 0,165—0,01 г-атом!л. В этих условиях ванадий в ра-
створе находится только в форме ионов VO^ и H2V10O28 , которые
образуются при подкислении раствора NaVO7 по реакциям (43) и
VO? + 2Н+ = VOt + Н2О.
(45)
115
В*
Рис. 59. Связь между степенью подкисле-
ния (/), изменением доли ванадия в форме
ионов VO+ (//) и величиной — 1g [Н+]
растворов в процессе гидролитического
осаждения ванадия при температуре 25° С
и р=1,0.
Концентрация ванадия: 1 — 0,165; 2 — 0,1; 3 —
0,05; 4— 0,01 г-атом.!л.
Начальная степень подкисления растворов
H+/VOT: а — 0,9; б — 0,98; в — 1,1; г — 1,3.
О
ак как ванадий в форме VO^- связывает 2 экв кислоты, а в форме-
H2Vi0O2V только 0,6, то число эквивалентов кислоты, связанной с
1 г • атомом ванадия в растворе (Ясв), можно рассчитать по урав-
нению
Нс = 2а + +0,6(1—а
vo2 vojJ
где а —доля ванадия в форме VO^.
vo2
На рис. 59 графически показана связь между значениями
H+/VO¥, Ясв, и величиной —lg[H+] раствора. Для простоты,
рассуждений будем считать расход кислоты при гидролизе //v посто-
янным и равным 0,8 г-же! г-атом. Очевидно, что в процессе гид-
ролиза степень подкисления раствора меняется. Если начальное зна-
чение H+/VO? меньше, чем //у, то по мере осаждения величина
H+/VO7 уменьшается; при Н+/VO? =//v она остается неизменной.
И, наконец, при Н+/¥О?>Яу степень подкисления раствора, есте-
ственно, возрастает. В процессе гидролиза меняется и соотношение-
ионных форм ванадия в растворе: при малых H+/VO7 оно остается
почти неизменным; с ростом начального значения H+/VO7 парал-
лельно с увеличением степени подкисления раствора в процессе гид-
ролиза увеличивается доля ионов vot. На рис. 59 изменения со-
става растворов при гидролитическом осаждении показаны стрелками
для случая, когда концентрация ванадия уменьшается в последователь-
ности 0,165, 0,10, 0,05, 0,01 г-атом/л для растворов с начальным
значением H+/VO7 0,90, 0,98, 1,10, 1,30.
Наиболее сложным образом при гидролизе меняется pH растворов.
Ясно, что если //св</7у, то в процессе осаждения ионы водородам
связываются и pH раствора будет возрастать. Из рис. 59 видно, что
указанное возрастание величины pH будет наблюдаться для раство-
ров с концентрацией ванадия 0,165 г-атом] л при степени подкис-
ления меньше 0,98. Если начальная степень подкисления достаточно
велика, а в данном случае больше 1,3, то при образовании осадка
ионы водорода освобождаются (так как //св>//у) и pH раствора по-
нижается. В интервале начальных значений Н+/¥Оз'= 0,98-ь 1,3;
pH растворов в процессе гидролиза вначале возрастает, затем понижа-
ется. Хотя в этом случае //св>Яу и при осаждении ванадия часть
связанной кислоты должна освобождаться, это не приводит к пони-
жению pH вследствие возрастания в растворе доли ванадия в форме
VOt и количества связываемой им кислоты. Только с переходом
большей части ванадия в осадок процесс освобождения ионов водо-
рода начинает преобладать и pH раствора понижается. Опи-
санная картина может быть проиллюстрирована данными, получен
ными при гидролитическом осаждении ванадия при комнатной темпе
ратуре [269]:
117
H+/VO з =1,91
V, S'атом [л pH
0,107.................1,40
0,097 .............. 1,38
0,072 ............... 1,38
0,086 ............... 1,35
0,014.................1,25
0,0133................1,22
0,0126................1,18
H+/VO з =0,73
V, г-атом/л pH
0,1193 ..........2,2
0,114................2,25
0,101............... 2,27
0,0968 ............. 2,38
0,0796 ............. 2,62
0,065 .............. 2,65
Аналогичный характер изменения pH при выдержке подкисленного
•раствора NaVO3 отмечен в работе [268].
При рассмотрении реального процесса необходимо учитывать,
что величина Ну не является постоянной — она зависит от степени
подкисления растворов и в ходе гидролиза заметно уменьшается.
Кроме того, вследствие изменения константы равновесия реакции
гидролиза иона VOt" с изменением температуры и ионной силы рас-
творов меняется и соотношение между различными ионными форма-
ми ванадия в растворах. Все это, не отражаясь на общем характе-
ре явления, усложняет действительную картину изменения pH рас-
творов при гидролизе.
Из уравнения (44) следует, что отно пение Na/V в продуктах
гидролиза определяется выражением -------=1------. Так как Ну =
х
то окончательно получим
Na
Данное соотношение удовлетворительно оправдывается на опыте [270].
При осаждении ванадия из растворов NaVO3, подкисленных хлор-
ной кислотой, при 20е С получены следующие результаты:
H+/VO з
0,826 ........................
0,938 ..........................
1,040 ..........................
1,210 ..........................
1,414 ..........................
ну Na/V Na/V-pHy
0,808 0,167 0,976
0,827 0,174 1,001
0,843 0,162 1,007
0,871 0,158 1,029
0,860 0,152 1,012
В свете изложенного становится очевидным, что наиболее важ-
ной характеристикой растворов при гидролизе является степень под-
кисления (H+/VO3). Полнота осаждения ванадия, степень исполь-
зования кислоты и состав осадков определяются именно этим соот-
ношением. pH растворов является более косвенной характеристикой,
так как зависит от степени подкисления и в процессе гидролиза не
остается постоянным.
118
СОСТАВ ПРОДУКТОВ ГИДРОЛИЗА
Детальным изучением установлено, что состав продуктов гидро-
лиза зависит не только от степени подкисления раствора, опреде-
ляемой соотношением H+/V07, но и от других факторов: темпера-
туры раствора, начальной концентрации в нем метаванадата, атом-
ного веса щелочного металла и т. д.
Таблица 30’
Состав воздушносухих осадков продуктов гидролиза [270]
Состав осадка, % Соотношение в осадке
№ h+/vos t, °C
образца v2o5 Na2O н2о Na/V, атом] атом H2O/V, мяль/атом
экв/г -ион
1 0,50 20 67,70 13,50 18,80 0,586 1,41
2 0,41 20 85,50 4,10 11,45 0,141 0,667
3 1,52 20 84,45 4,51 11,00 0,156 0,058
4 2,08 20 84,45 4,37 11,00 0,152 0,663
5 3,04 20 84,40 4,12 11,39 0,144 0,682
6 1,77 60 85,90 4,00 11,10 0,137 0,653
7 0,73 80 83,27 5,45 11,25 0,193 0,682
8 0,92 80 89,60 5,20 5,20 0,170 0,294
9 0,95 80 85,00 4,90 10,13 0,169 0,601
10 1,09 80 85,70 4,00 10,40 0,137 0,614
11 1,11 80 85,40 4,50 10,10 0,154 0,597
12 1,18 80 86,40 4,50 8,99 0,155 0,525
13 1,29 80 85,70 4,15 10,16 0,143 0,598
14 1,41 80 85,60 4,31 10,12 0,146 0,598
15 1,77 80 85,40 4,10 10,49 0,1411 0,621
16 1,88 80 85,80 3,91 10,28 0,133 0,605
17 2,13 80 85,40 3,69 10,86 0,127 0,042
18 2,23 80 86,50 3,00 10,50 0,102 0,613
19 1,09 90 85,40 4,50 10,00 0,156 0,592
20 0,50 95 82,50 10,00 7,50 0,356 0,459
21 0,60 95 80,26 6,80 13,20 0,239 0,830
22 5,00 95 86,00 3,15 10,85 0,107 0,637
в табл. 30 и на рис. 60 приведе! н состав воздушносухих осад-
ков, полученных при подкислении 0,1 и 0,28 М растворов метава-
надат; а натрия серной и хлорной кислотами [! 269, 270J. Осаждение
проводили при интенсивном перемешивании до тех пор, пока скорость
осаждения ванадия не стала минимальной. В этом случае независи-
МО от природы применяемой кислоты, температуры растворов и кон-
центрации в них NaVO3 соотношение Na/V в осадках с возрастанием
степени подкисления резко уменьшается в интервале H+/VO?=
= 0,7 ч-0,9. В опытах при 20° С дальнейшее увеличение H+/VO3
с 0,9 до 3,04 сопровождалось уменьшением Na/V от 0,17 до 0,143,
а при 80° С увеличение H+/VO? с 0,9 до 2,22— уменьшениемNa/V
от 0,17 до 0,1.
119>
Даже при очень высокой степени подкисления Н+/УОз =5,0,
соответствующей абсолютной концентрации кислоты 1,2 г-экв]^
осадки с более низким, чем 0,1, соотношением Na:V не были
получены.
В других опытах [271—275], результаты которых приведены в
табл. 31 и на рис. 61, гидролитическое осаждение проводили, под-
Рис. 60. Зависимость отношения Na/V в осадках от степени
подкисления растворов H+/VO^" при гидролитическом осажде-
нии ванадия.
1 — 20, 2 — 60, 3 — 80,4 — 90, 5 — 95° С.
кисляя растворы LiVO3, NaVO3 и KVO3 азотной кислотой и выдер-
живая осадки под маточником в течение нескольких недель до тех
пор, пока pH растворов не достигал постоянного значения. Для си-
стемы NaVO3—Н+—Н2О [271 ] в этих условиях наблюдается быстрое
уменьшение отношения Na/V в осадках в интервале H+/VOV от
0,7 до 1,0 и более медленное при последующем возрастании H+/VOF.
Содержание натрия в осадках при одинаковой степени подкисления
исходных растворов тем выше, чем ниже в них начальная концен-
трация метаванадата натрия; осадкам с одинаковым отношением
Na/V соответствуют тем более сильно подкисленные растворы, чем
меньше в них начальная концентрация метаванадата натрия. В дан-
ном случае играет роль то, что при одинаковой величине H+/VO7
с уменьшением начальной концентрации ванадия pH растворов уве-
личивается. Если сравнивать осадки, полученные при одинаковых
pH конечных растворов, то большей первоначальной концентрации
метаванадата натрия соответствует более высокое содержание натрия
в осадках.
В системе KVO3—Н+—Н2О [272, 273] изменение соотношения
K/V в осадках, в зависимости от степени подкисления и начальной
J20
концентрации KVO3 в раство- Й
ре, имеет аналогичный хара- а
ктер, однако минимальное соот- *
ношение K/V в осадке суще- ч
ственно выше и составляет
0,145 атом/атом. В этом же ь-
интервале степеней подкис-
ления раствора LiVO3 отно-
шение Li/V в осадках изме-
няется от 0,259 до 0,004, т. е.
при H+/VO7=2,1 осаждается
практически чистая гидратиро-
ванная V2O5 [274, 275].
Количество воды в воздуш-
носухих осадках возрастает с
уменьшением содержания в
них щелочного металла в слу-
чае натрия и калия, но ос-
тается почти неизменным в
случае лития. Как видно,
степень гидратации осадков
зависит от природы щелочного
металла: в ряду Li, Na, К
абсолютное содержание воды
и отношение H^O/V в воздуш-
носухих осадках уменьшается.
Равным образом, с увеличе-
нием атомного веса щелочного
металла возрастает и его тен-
денция к соосаждению с вана-
дием в процессе гидролитиче-
ского осаждения. Это под-
тверждается и опытами по
гидролитическому осаждению
ванадия из растворов мета-
ванадата с эквимолярным
соотношением ионов калия и
натрия. В этом случае соот-
ношение K/Na в осадке было
равным 2,8 атом!атом [276].
ТЕРМИЧЕСКИЕ СВОЙСТВА
ПРОДУКТОВ ГИДРОЛИЗА
При рентгенофазовом ис-
следовании по методу Дебая-
Шерера было обнаружено, что
продукты гидролиза имеют ясно
3 X X ф га Я О О 74 сз Z СО О 0 О LO Ь 1—»- со ь- о> со сч о о О) CO^COCOCOCOtNOH
v § ‘ Ю Tf LO СЧ Ю О1
го ОЧ СО СО ' Oi ОО Ь- СО
о а
X гфг-чг-чгчСООО
св OiLQr-cO’—lONTf
СО cOr-uOOOtQuOCOO
X о CMr-tr-iOOOOO
(=5 а?
о 1_О oooooooo
Хи
о чо
X т—ч CO r-ICO ococooco
о ОС CO b~ b- CO OO CD OH
о га С4
и к Я М-4
о Я га 1 T < T 11 < 4 •
X о
X га СО. О И Л 0400040400»—<ООЮ
К га о LQ co LQ СП о о
о Ef О о v—। r—। CO LQ CO
Рм X с и CO CO 00 OO OO OO GO CO
X к га Г) OOTfLOCO-—< b- 04 CD
X к ^t<CO,—<b-(^LQC)
X t-Q
>~4 COOIOIt-i о О О o -
3*
о
X я
а -ФОООООСОСЧОО^
О X LO^LQCMOOlOOb-
со Ом CO 04 'r~> r—• О О О
о
X
ь О CO 'O N о гч
X 4 W
св О О г-н О)
CJ
О
е?' t- r-’tMCO'xFLQCDb-GC
С
121
Рис. 61. Зависимость отно-
шения M/V и H2O/V в осад-
ках от степени подкисления
растворов в системах:
1, 4-KVO-- Н+-Н2О;
2, 5— NaVO3 —Н+— Н2О;
3, 6 — LiVO3 — Н+-Н2О.
Рис. 62. Штрих диаграммы продуктов гидролиза. Номера образцов те
же, что и в табл. 31.
2 и 8 — осадки с литием, 5 — с калием, 6 — с натрием.
выраженный' кристаллический характер, но относительно небольшое
число размытых диффузионных линий на порошкограммах свидетель-
ствует о несовершенной мелкокристаллической структуре осадков; то
же подтверждают кристаллооптические исследования [271—275]. Судя
по этим данным, в случае натрия и калия обнаруживаются два типа
продуктов гидролиза, отличающихся содержанием щелочного метал-
ла: в области степеней подкисления H+/VO?>0,9 выпадают осад-
ки с соотношением Na/V и K/V меньше 0,2. Все они, независимо
от состава и природы щелочного металла, дают очень схожие по-
рошкограммы (рис. 62) и, по-видимому, близки по структуре. Осад-
ки с соотношением Na/V и K/V больше 0,2, судя по интерферен-
ционным картинам, по структуре отличны от осадков первого типа.
Продукты гидролиза, полученные при подкислении растворов мета-
ванадата лития, независимо от величины H+/VO? и содержания
лития, дают интерференционные картины, аналогичные таковым для
осадков первого типа с натрием и калием. Наличие двух типов
продуктов гидролиза и идентичность свойств осадков первого типа,
независимо от содержания в них щелочного металла, подтвержда-
ются, кроме того, данными термогравиметрии [270—275]. При дери-
ватографическсм исследовании продуктов гидролиза, полученных в
равновесных условиях, было установлено, что осадки 2-—8 для си-
стем с натрием и калием и 3—8 (см. табл. 31) для систем с литием
имеют близкие термические свойства. Дериватограммы, типичные
для каждого ряда осадков, приведены на рис. 63. Эндоэффекты на
кривых ДТА и отвечающая им убыль массы — кривая ТГ — в области
температур до 314° С для осадков с литием, 340° С с натрием и
367° С с калием соответствуют полному обезвоживанию препаратов.
Изменения при более высоких температурах происходят в обезво-
женных препаратах и будут обсуждены ниже.
Как видно, осадки рассматриваемого типа в случае лития и нат-
рия теряют основное количество воды в две стадии, в соответствии
с этим в них выделено две формы менее прочно связанной воды —
сорбционная и кристаллогидратная [271, 274]. В калийсодержащих
осадках основная масса воды отнесена к типу кристаллизацион-
ной [278]. По данным рентгенофазового анализа при удалении сорб-
ционной и кристаллизационной воды структура осадков не претер-
певает каких-либо изменений. Кроме того, было обнаружено, что
препараты, из которых удалена эта менее прочно связанная вода,
будучи выдержаны ео влажной атмосфере, полностью гидратируются.
Это дает основание более точно рассматривать «кристаллизационную»
воду как «цеолитную». В самом деле, описанные свойства продуктов
гидролиза — способность отдавать воду в широком интервале темпе-
ратур без разрушения решетки и возможность полной регидратации
кристаллов с восстановлением их первоначальных свойств, как раз
характерна для цеолитов типа шабазита [277].
При исследовании обезвоживания натрийсодержащих осадков
при 20е С над растворами серной кислоты различной концентрации
123
х
J____I____I_____।____1
было найдено, что «цеолитная» вода полностью удаляется только
над раствором 95,6% H2SO4 (Рн2о =0,005 мм рт. ст.), «конститу-
ционная» вода в этих условиях не отщепляется. В интервале давле-
ний водяного пара 0,6—6,2 мм рт. ст. устойчивы осадки, содер-
жащие 0,55—0,6 молекулы
воды на 1 атом ванадия; в
атмосфере насыщенного во-
дяного пара содержание
воды в них возрастает до
(0,9—0,1 молекулы на 1 атом
ванадия [270].
Различный характер
воды в продуктах гидро-
лиза подтверждается дан-
ными ядерного магнитного
резонанса протонов и инфра-
красной спектроскопии.
пектры протонного резо-
нанса типичных осадков,
полученных в сериях опы-
тов по гидролитическому
осаждению ванадия из рас-
творов метаванадатов лития,
натрия и калия, одинаковы
[274, 278, 279]. При 196° С
они состоят из двух частей —
широкой внешней и узкой
внутренней. Расстояние
между экстремумами произ-
водной широкой компо-
ненты, равное 13,2 э, хара-
ктерно для молекул кристал-
лизационной воды. Узкий
центральный пик обуслов-
лен, вероятно, наличием
неспаренных протонов. Судя Рис. 64. Инфракрасные спектры про-
ПО его интенсивности, доля дуктов гидролиза.
а, б, в — осадок 8 (см. табл. 31), содержа-
таких протонов по отно- щий соответственно Li, Na, К.
шению к общему числу
протонов в осадках невелика (не превышает 2%). При переходе к
температуре 20° С характер спектра ЯМР меняется: возникает ин-
тенсивный центральный пик, обусловленный подвижными молекула-
ми кристаллизационной воды; более широкая малоинтенсивная ком-
понента, по-видимому, связана с наличием неспаренных протонов и
молекул воды, обладающих одной или двумя вращательными степеня-
ми свободы. В инфракрасных спектрах рассматриваемых осадков
полосы поглощения (рис. 64) с максимумами при 3610 о ,
1630—1620, 927—925 смг1 отнесены соответственно к валентным
в
к
* I I_____]____I___।___I__ .
3600 1600 1400 1200 1000 8001ы*
125
колебаниям ОНл-группировок (vOh/7), деформационным колебаниям
молекул воды (дОн2) и ОН-групп (ёонп) [280].
Осадки с повышенным содержанием щелочных металлов (М'/V >
>0,2) изучены менее тщательно. Их дериватограммы, оставаясь по-
хожими на дериватограммы осадков первого типа, отличаются более
сложным характером эндотермических эффектов и расположением
максимумов на кривых ДТА и ДТТ, а структура, судя по рентге-
нограммам, отличается от структуры осадков первого типа. По дан-
ным кристаллооптических исследований и ИК-спектроскопии, осадкам,
содержащим повышенные количества натрия и калия, приписаны
структуры гексаванадатов [280].
Как отмечено выше, при полном обезвоживании происходит не-
обратимая перестройка структуры продуктов гидролиза, которая,
как можно ожидать, должна сопровождаться образованием новых
фазовых составляющих в соответствии с диаграммой состояния си-
стемы окись щелочного металла — пятиокись ванадия. В случае
литийсодержащих осадков структурная перестройка приводит к об-
разованию трех фаз: литийванадиевых бронз LiV6O15 и Li2V60i3,5
и V2O5 [274], соотношение между которыми зависит от содержания
Li2O. При температурах, близких к 585е С, вначале протекает ре-
акция - 1
Li2VeO 1 з. 5 - z + 3,5V2O5 = 2Li V6O15+О2, (46)
обусловливающая уменьшение массы препарата. При последующем
нагревании бронза LiV6O15 окисляется, образец плавится
2LiV6O15 + V2O2=Li2V12O31 (47)
и масса образца вновь увеличивается.
Аналогичная картина наблюдается при нагревании калийсодер-
жащих осадков: после обезвоживания в них образуются V2O5 и
бронзы KVeO15 и KV4Oio,5-%- В этом случае потери массы образ-
цов и последующее ее увеличение можно объяснить с помощью
реакций:
VА+KV4010,5- A-^KV6О15+^^ О2; (48)
2KVeO15+1/2O2=K2Vi2O13. (49)
Подобные изменения веса при нагревании обезвоженных натрий-
содержащих образцов связаны с образованием натрийванадиевых
бронз [271, 273].
КИНЕТИКА ГИДРОЛИТИЧЕСКОГО ОСАЖДЕНИЯ
Гидролитическое осаждение ванадия представляет собой весьма
сложный процесс, результаты которого — полнота и скорость выде-
ления ванадия, состав осадков и т. п.—зависят от многих факто-
126
ров. Для практического осуществления весьма важна его кинети-
ческая характеристика.
Так как ванадий осаждается за счет образования малораствори-
мых соединений, то рассматриваемый процесс в общем случае можно
отнести к типу топохимических реакций, связанных с осаждением
кристаллических осадков за счет химической реакции в жидкой
фазе или на поверхности кристалла. При этом возможны все ва-
рианты от полного лимитирования кристаллизационной стадией до
лимитирования химической реакцией, предшествующей выделению
кристаллического вещества. Наиболее полно теоретически и экспе-
риментально исследован случай осаждения вещества из пересыщен-
ных растворов, когда осаждение не сопровождается новообразова-
нием объемных зародышей кристаллов и лимитируется процессами
диффузии вещества к поверхности растущего зародыша из объема
раствора или диффузией на поверхности кристаллов [281 — 287].
В литературе ограниченное число работ посвящено кинетике гид-
ролитического осаждения ванадия, однако в практике давно было
известно, что нагревание доведенных до оптимальной кислотности
растворов ускоряет осаждение ванадия; отмечалась зависимость про-
должительности осаждения от концентрации ванадия в растворе [1].
Согласно работе [288], время для осаждения ванадия можно со-
кратить путем точного контроля химического состава раствора и его
температуры. Рекомендуется поддерживать в растворе высокое сум-
марное содержание сульфата и хлорида натрия 5—20%, температу-
ру 75-100°С и рН-2ч-3.
Первое серьезное исследование кинетики гидролиза выполнено
Е. И. Буровой [289] в 1936 г. Осаждение ванадия проводилось из
растворов пированадата натрия в присутствии соляной кислоты,
которую брали в количестве, эквивалентном всему количеству натрия,
находящемуся в растворе, плюс еще некоторый избыток. Выявлено,
что на процесс осаждения ванадия ускоряющее влияние оказывает
перемешивание, высокая начальная концентрация ванадия и повы-
шенная температура. При этом максимальная скорость осаждения
достигается при некоторой оптимальной степени подкисления исход-
ного раствора. Установлено также ускорение осаждения при введе-
нии затравки, однако в этом случае оказалось, что очень малые ее
количества даже несколько замедляют процесс осаждения. Было
найдено, что кинетика гидролиза подчиняется уравнению, выведен-
ному в предположении, что кристаллизация идет через стадии по-
следовательных процессов образования и роста зародышей
(а —л) (£>+*),
at
где х — концентрация ванадия в момент времени /; а=С0 — эо
(Со — концентрация ванадия начальная и Соо—в момент равнове-
сия); b — постоянная, в условиях описываемых опытов равная нулю.
Автор не делает заключений о характере лимитирующих стадий, но
отмечает, что температурный коэффициент скорости в данном случае
127
Рис. 65. Зависимость степени осаждения ванадия от времени
при различных степенях подкисления и температурах.
H+/VO—: а — 1,18; 6—1,16; в— 0,954; г— 0,855;
t: 1 — 90, 2 — 80, 3 — 70, 4 — 60° С.
слишком мал (1,77), чтобы процесс осаждения ванадия признать
химическим в узком смысле слова.
Важные особенности процесса гидролиза вскрыты при исследо-
вании влияния степени подкисления и природы используемой кисло-
ты, температуры и различных солей на скорость осаждения ванадия
[290, 291]. На рис. 65 показаны кривые осаждения, полученные
при подкислении раствора метаванадата натрия серной кислотой.
Они имеют S-образный вид, характерный для топохимических про-
цессов. Выделению видимого осадка предшествует «скрытый» период
кристаллизации, продолжительность которого увеличивается с умень-
шением температуры, степени подкисления и концентрации ванадия
в растворе; скорость осаждения ванадия сильно возрастает (в 5—6 раз)
при увеличении степени подкисления растворов H+/VO7 от 0,855
до 0,954 и мало меняется при дальнейшем ее возрастании до 1,18.
Это послужило основанием для вывода о том, что решающее влия-
ние на скорость осаждения оказывает степень подкисления растворов.
Нейтральные соли, как оказалось, также сильно влияют на ско-
рость гидролитического осаждения. Это выяснилось в специальных
опытах, когда ванадий осаждали, подкисляя растворы метаванадата
натрия серной, соляной и хлорной кислотами в присутствии различ-
ных количеств перхлората, хлорида и сульфата натрия [291]. Опыты
проводили при постоянной степени подкисления H+/VO7=l,0, тем-
пературе 80° С и интенсивном перемешивании (рис. 66). Добавки
солей существенно меняют картину гидролиза. Пропорционально
концентрации добавленной соли уменьшается или вовсе исчезает
«скрытый» период кристаллизации. Введение незначительных добавок
солей резко замедляет осаждение ванадия. Замена перхлората на
поваренную соль при сохранении постоянной ионной силы раствора
заметно увеличивает продолжительность осаждения. Кроме того,
было найдено, что при одинаковых значениях pH исходного рас-
твора осаждение в присутствии серной кислоты протекает в три раза
медленней, чем в присутствии хлорной. Особенно отчетливо влияние
добавок солей выявляется при кинетическом анализе кривых осаж-
дения. Оказалось, что в условиях этих опытов осаждение
большей части ванадия хорошо описывается с помощью уравнения,
выведенного для роста заданного числа центров кристаллизации,
контролируемого диффузией из объема раствора [286]
= (А)
dt г
где г — радиус растущей частицы; t—-время; V — объем моля осаж-
даемого вещества; D — коэффициент диффузии; С — концентрация;
S — растворимость. Для обработки экспериментальных данных урав-
нение (А) удобно представить в интегральной форме
а
3VDC-°. / — j V3 (1 — а)“Ма, < Б)
9
Заказ № 256
129
Концентрация
Рис. 66. Влияние солей на гидролитическое
Растворы подкислены кислотой: I, // — серной, н+/УОз"=1 ,08 и 1,05;
5 — 0,171; 6 — 0,0856 моль]л\ III — хлорной, H+/VO3 = 1,0; концентрация
= 1,0; ц=1,1 (NaCl+NaClO4), концентрация NaCl: 7 — 0,855; 2 — 0,342;
на 1 л Na2SO4, Н+/УО^=2,74(/); 2,3 (2); 1,88 (5); VI — хлорной,
t, мин
ЮО 200 JOO
Z,muh
осаждение ванадия.
концентрация NaCl: / — 3,42; 2 — 2,56; 5—1,71; 4 — 0,856;
NaClO4: 1 — 0,3; 2— 0,7; 3— 1,0 моль'/л', IV—хлорной, H+/VO3 =
5 — 0,171, -4— 0 молъ'/л\ V—хлорной в присутствии 0,352 моль
H+/VC\f=0,997 (/), серной, H+/VO^=1,21 (2); pH 1,7,
9*
где а —степень осаждения; г^=г при /=6о; £0 —начальная кощ
а
центрация. При и \ сН/*(1 — a)“Ma=/D уравнение (g\
d о I
переходит в
/\е>/—Id .
(В)
Если скорость роста контролируется реакцией р-порядка на по-
верхности растущих частиц, то уравнение (Б) приобретает вид [286]
а
I\pt = а~2/з (1 — а)_рб/а
б
(Г)
или
(Д)
Величины интегралов 1D и I при р=1, 2, 3, 4, в зависимости от
степени осаждения а, приведены в работе [286]. Для описания ки-
Рис. 67. Зависимость вели-
чины KD от концентрации
электролитов.
/ — IV — серии опытов, приведен-
ных на рис. 66.
усом частиц. Изменение 1(D
образом, может быть связано с
Как показали непосредственные
нетики гидролиза в конце осажде-
ния (при а>0,8~:-0,9) оказалось
применимым уравнение (Д) при
р=1 и был сделан вывод, что в
этом случае рост частиц осадка кон-
тролировался реакцией первого по-
рядка на поверхности частиц.
Величина Kd в уравнении (В)
имеет смысл константы скорости,
и изменение ее отражает влияние
различных факторов на скорость
осаждения ванадия. Оказалось, что
I\d сильно зависит от концен-
трации электролитов в растворе
(рис. 67): при увеличении ионной
силы раствора она вначале сильно
уменьшается, проходит через мини-
мум и затем медленно возрастает.
Вместе с ростом концентрации
солей уменьшается и продолжи-
тельность «скрытого» периода кри-
сталлизации. При неизменной на-
чальной концентрации ванадия в
растворе величина I\d определяется
коэффициентом диффузии и ради-
в присутствии электролитов, таким
изменением этих обоих параметров,
измерения, с ростом концентрации
электролитов увеличивается размер частиц осадка и его агрегация,
что, очевидно, и приводит к уменьшению величины /<ф. Увеличение
132
размера частиц при постоянной концентрации ванадия может быть
вязано либо с уменьшением количества первоначально образовав-
шихся зародышей, либо с быстрой агрегацией. В обоих случаях это
должно привести к уменьшению общей поверхности осаждения и,
соответственно, к снижению скорости осаждения, что и наблюдается
а действительности. Последующее увеличение /<D при высоких кон-
центрациях электролитов до некоторой степени можно объяснить
возрастанием коэффициента диффузии с ростом ионной силы раство-
ра и высаливающим действием электролитов.
Тот факт, что процесс гидролитического осаждения (при не очень
высоких концентрациях ванадия в растворе) лимитируется диффу-
зией вещества к поверхности растущих частиц и не сопровождается
новообразованием объемных зародышей, указывает на незначитель-
ное влияние температуры на скорость осаждения ванадия на дан-
ной стадии гидролиза. Можно предполагать, что температура в боль-
шей степени влияет на скорость образования зародышей, поэтому
температурный коэффициент скорости осаждения 1,7—2,0 обусловлен
в значительной мере именно процессом образования устойчивых
объемных зародышей в начальный момент осаждения.
Более сильное тормозящее действие ионов SO4~" и С1~ по срав-
нению с перхлоратом, по-видимому, обусловлено связыванием части
ванадия в сульфатные и хлоридные комплексы.
О СТРУКТУРЕ ПРОДУКТОВ ГИДРОЛИЗА
И МЕХАНИЗМЕ ГИДРОЛИТИЧЕСКОГО ОСАЖДЕНИЯ
Результаты исследования количественных характеристик реакций
гидролиза и свойств получаемых осадков позволяют на более широ-
кой основе обсудить химизм этого важного процесса. Как уже от-
мечалось, одни исследователи рассматривают продукты гидролиза
Таблица 32
Возможные реакции гидролиза
Реакция
Расход
кислоты
экв/г-атом V
Соотношение
в осадке Na/V
атом/атом
2NaVO3+2H+=V2O5 • aq+2Na++H2O
6NaVO,+3H4-=Na3HV6O]7+3Na+ +
H-H2O
6NaV03+4H+=Na2HoV6017+4Nai~-|-
+H2O
6NaVO3+6H+=H4V6Oi74-6Na++H2O
1,0
0,5
0,667
1,0
0 [18]
0,5 [263]
0,333 [265]
0 [292]
как гидратированную пятиокись ванадия [1, 18], другие — как ва-
надиевые кислоты и их соли [263, 265—267, 292]. Из рассмотрения
возможных реакций гидролиза, уравнения которых приведены в
табл. 32, с точки зрения состава продуктов и расхода кислоты на
133
их образование видно, что количественные характеристики
реакций отличаются от полученных опытным путем. I „
Лучше всего по расходу кислоты (0,833 экв/г-атом V) и состав гидратации. Еще одной характерной особенностью цеолитов
продуктов (Na/V=0,167) экспериментальным данным соответствхл?^ 1^-гяется Т0, что с°ДеРжание воды в их элементарных ячейках зави-
уравнение, приведенное в работе [268]: И от числа и размеров обменных катионов, находящихся во внут-
6NaVO3+5H+ - NaH VO2V5O14+5Na++2H2O.
цкристаллических пустотах. В частности, количество молекул воды
кристаллах, рассчитанное на элементарную ячейку, в калийных
^абазитах всегда меньше, чем в натриевых, а в натриевых меньше,
в литиевых. При изменении катионного состава цеолитов, т. е.
ори замещении, например, одного щелочного металла на другой,
структура решетки может не меняться, но изменяются параметры
I элементарной ячейки, плотность цеолитов [277]. Точно так же ха-
[рактер структуры продуктов гидролиза не зависит от природы и
концентрации (в определенных пределах) щелочного металла, но со-
держание в них цеолитной воды определяется именно этими факто-
рами. Все описанные свойства цеолитов связаны с особенностями
1пх структуры. Цеолиты представляют собой алюмосиликаты, в кото-
рых чередующиеся алюминий- и кремний-кислородные тетраэдры
соединены в трехмерные каркасы, пронизанные системой регулярных
каналов и сообщающихся полостей минимальных размеров, сравни-
мых с размерами молекул. Щелочные и щелочноземельные катионы,
компенсирующие отрицательные заряды алюминий-кислородных те-
траэдров каркаса, располагаются в пустотах кристаллической решет-
| ки, занимая лишь сравнительно малую часть их объема. Не занятая
катионами большая часть пустот в кристаллической решетке цео-
литов заполнена молекулами воды [277].
Можно предполагать, что отмеченная аналогия свойств цеолитов
[и продуктов гидролиза обусловлена сходством их структур. В таком
(50}
•H2O [270]. Позднее было най-
что определенное методом ЯМР отношение числа протонов
Однако, как показано выше, содержание щелочных металлов в про,
дуктах гидролиза не постоянно. Учитывая это, а также то, ЧТо
наиболее высококонденсированные поливанадаты рассматриваются
как декаванадаты (см. главу I), продуктам гидролиза была припи-
сана формула Nax(VO2)4 _xH2V10O28-H2O [270]. Позднее было най-
дено, 1
в. молекулах Н2О к числу неспаренных протонов в осадках лучше
всего соответствует формуле Мх (VO2)5 _ ¥HV10O28 • пН2О, где M=Li
Na, К [274, 278, 279].
Наконец, можно предполагать, что продукты гидролиза являются
кислыми солями декаванадиевой кислоты типа Na6 _ xHxV10O28 • nH2O.
В таком случае осадкам, исходя из содержания в них ванадия и
натрия, следует приписать состав от Na2H4V10O28 • пН2О до
NaH5V10O28-nH2O. Такие соли должны содержать значительное
количество неспаренных протонов, но это не наблюдается. Продукты
гидролиза содержат относительно мало неспаренных протонов, но в
то же время процесс осаждения сопровождается связыванием кис-
лоты и протекает в условиях, когда в растворах ванадий находится
в форме катиона VO^ и аниона H2V10O
жение в формуле Mx(VO2)s _xHV10O28-nH2O. Вместе с тем трудно [случае продукты гидролиза необходимо отнести к структурам кар-
касного типа и представить состоящими из ванадий-кислородных
полиэдров, объединенных в пространственный каркас с каналами
28 • Все это находит отра-
5_
2 8 являются элементами струк-
допустить, что ионы VO^ и HV10O
туры осадков, поэтому данная формула в определенной степени
носит формальный характер, хотя и очень хорошо отражает химизм и пустотами, в которых размещены катионы щелочных металлов,
образования продуктов гидролиза. W Г™ ....л „
В литературе нет сведений о структуре продуктов гидролиза, и
все суждения о ее характере и особенностях могут быть основаны
лишь на рассмотрении свойств осадков. К числу наиболее важных
свойств продуктов гидролиза относятся:
1) наличие кристаллогидратной воды и обратимое ее выделение
без разрушения решетки кристаллов, что позволяет рассматривать
эту менее прочно связанную воду как цеолитную;
2) зависимость содержания цеолитной воды от природы и коли-
чества щелочного металла;
3) постоянство характера кристаллической структуры независимо
от природы и содержания щелочного металла;
4) наличие конституционной воды, удаление которой сопровож-
дается необратимой перестройкой структуры кристаллов.
Эти особенности продуктов гидролиза делают их похожими на
цеолиты, одно из важнейших свойств которых — способность к обра-
неитрализующие отрицательный заряд ванадии-кислородного каркаса,
|и молекулы воды. Предположение о существовании каналов в струк-
туре калийсодержащих продуктов гидролиза было высказано в ра-
боте [279] на основании данных протонного магнитного резонанса.
В связи с изложенным встает вопрос о состоянии конституцион-
ной воды в продуктах гидролиза. Исследование Н-форм цеолитов
[Химическими, термогравиметрическими и ИКС-методами показало,
что при замене различными способами щелочных металлов, компен-
сирующих отрицательный заряд алюминий-кремний-кислородного кар-
каса, на ион водорода, свободный протон или протон, ассоциирован-
ный с молекулой цеолитной воды (протон иона Н3О+) вследствие их
большой реакционной способности, легко взаимодействует с алюми-
Ний-кислородным каркасом с образованием структурных гидроксиль-
ных групп, связанных с атомами кремния, по схеме
М+ Н
134
135
Вода, образующаяся за счет структурных гидроксилов, выдел
ется при нагревании после полного удаления цеолитной воды [277]
По-видимому, и в случае продуктов гидролиза, наличие воды в Ко‘
торых в форме неспаренных протонов установлено методом проток
ного магнитного резонанса [274, 278, 279], можно говорить о при'
сутствии структурных гидроксилов, связанных с атомами ванадий
Данные инфракрасной спектроскопии [280] подтверждают присущ^
вие гидроксилов в продуктах гидролиза, что соответствует так^е
результатам термогравиметрических исследований. Вероятно
конституционная вода, которая при нагревании удаляется в по-
следнюю очередь, образуется именно за счет структурных гидр0.
ксилов. jg
Таким образом, по своей химической природе продукты гидро,
лиза можно рассматривать как кислую соль некой поливанадиевой
кислоты. Ванадий-кислородный каркас представляет собой бесконеч^
ный полимерный анион этой кислоты, суммарный отрицательный
заряд которого нейтрализован отчасти ионами щелочного металла,
отчасти за счет протонов структурных гидроксилов. В таком случае
гидратированную пятиокись ванадия можно рассматривать как поли-
ва на диевую кислоту.
Выше было показано, что, судя по расходу кислоты на гидро-
литическое осаждение ванадия из подкисленных растворов метавана-
дата натрия, состав продуктов гидролиза лучше всего отвечает
формуле Na2 (VO2)2H2V10O28, или Na2H2V12O32. Тогда состав предпо-
лагаемой поливанадиевой кислоты можно описать формулой
(H4V12O32)/I, или (У2О5-0,ЗЗН2О)л. Препараты гидратированной пяти-
окиси ванадия, описанные в работе [240], по содержанию консти-
туционной воды (0,224 и 0,357 молекулы Н2О на молекулу V2O5)
близко соответствуют этой формуле. В работе [240] также отмечена
близость свойств и природы гидратированной пятиокиси и продуктов
гидролитического осаждения ванадия.
Исходя из каркасной модели и имея в виду, что продукты гид-
ролиза содержат относительно мало гидроксилов, но в то же время
процесс осаждения сопровождается связыванием значительных коли-
честв кислоты, можно предположить, что образование каркасной
структуры происходит в результате реакций поликонденсации либо
между простейшими ионами VOf" и более сложными декаванадат-
ионами, либо между декаванадат-ионами. Так как ионы H2V10O28
обладают высоким отрицательным зарядом, то непосредственное
взаимодействие между ними затруднено. Однако положительно за-
ряженные ионы VO2H могут служить мостиками, которые как бы
сшивают крупные анионы H2V10O2T. Последние, являясь остатками
шестиосновной кислоты, содержат четыре свободных группы
—V—О—, которые могут участвовать в образовании связей по
схеме
Р этом случае для образования осадка требуется определенное соот-
ношение в растворе между концентрациями ионов VOt и H2V10O2C.
реакция поликонденсации может протекать и при непосредственном
взаимодействии декаванадат-ионов при участии ионов водорода,
например, по схеме
Скорость этого процесса зависит от концентрации ионов водорода
и H2V10O24r. Образование связей при конденсации может происхо-
дить и за счет участия в реакции части ОН-групп ионов H2V10O28
В работе [63] описан предполагаемый механизм образования золей
! гидрата пятиокиси ванадия при старении растворов свободной дека-
ванадиевой кислоты: агрегаты V10O2? (см. рис. 6, в) в процессе
конденсации объединяются в тела
’трубчатой формы (рис. 68), стенки
которых образованы бесконечной
Цепочкой тетраэдров, намотанной
на цилиндр по винтовой линии.
•При этом продукт конденсации
соответствует формуле (У2О5)Л-
I -ЗН2О. Согласно [63], описанный
Механизм объясняет полочкообраз-
ную форму частичек золей пяти-
окиси ванадия.
В последнее время сделано
Немало для выяснения механизма
гидролитического осаждения ва-
надия. Однако сложность поведения ванадия в растворах и недо-
статок экспериментальных данных по структуре и свойствам про-
дуктов гидролиза еще не позволяют дать сколько-нибудь детальную
и строго обоснованную картину процесса гидролиза.
136
137
Глава V
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ ВАНАДИЯ
На различных стадиях извлечения ванадия из руд и концентра-
тов или при получении чистых соединений почти всегда приходится
иметь дело с водными растворами пятивалентного ванадия. Успешное
применение обычных приемов химической технологии (растворения,
осаждения в виде малорастворимых соединений, кристаллизации
и т. д.) немыслимо без учета ионного состояния ванадия в растворах.
Поэтому химия пятивалентного ванадия в водных растворах до сих
пор в основном сводилась к решению этого сложного вопроса. Бла-
годаря усилиям многочисленных исследователей в течение последних
пятидесяти лет были установлены ионные формы, в которых вана-
дий (V) существует при различных pH в водных растворах.
Расширение объема производства, повышенные требования к ка-
честву и все увеличивающееся разнообразие производимых соедине-
ний, а также вовлечение в промышленную переработку новых слож-
ных по составу видов сырья, требуют совершенствования существу-
ющих и разработки новых способов извлечения ванадия. Поэтому
наряду с традиционными приемами химической технологии все чаще
используются такие способы, как ионный обмен, экстракция [293—-
295]. Применение этих методов, как правило, основано на реакциях
комплексообразования. Создание новых эффективных способов кон-
центрирования, извлечения, очистки и определения ванадия требует
тщательного исследования реакций комплексообразования, опреде-
ления состава и устойчивости образующихся в растворах соедине-
ний, их взаимных превращений. Прогресс, в значительной мере
связанный с бурным развитием химии комплексных соединений, так-
же способствовал пробуждению в последнее время интереса к химии
комплексных соединений пятивалентного ванадия, хотя его комплек-
сообразующая способность была замечена уже давно. Так, М. Н. Со-
болев [1] считал, что процесс гидролитического осаждения ванадия
связан с распадом сернокислого комплекса. Дюкре [22], основываясь
на повышенной растворимости пятиокиси ванадия в серной и соля-
ной кислотах, высказал предположение об образовании соответству-
ющих комплексов. Однако эти и другие подобные заключения не
выходили за рамки предположений и фактически не учитывались
138
^ри разработке и осуществлении способов извлечения ванадия, хотя
некоторые реакции комплексообразования нашли применение в ана-
литической химии ванадия [296, 297].
Основная часть публикаций, посвященных комплексным соеди-
нениям пятивалентного ванадия, появилась за последние десять
дет. В большинстве опубликованных работ основное внимание уде-
ляется синтезу и определению состава образующихся комплексов
Я лишь в некоторых из них даны количественные характеристики
реакций комплексообразования пятивалентного ванадия в растворах.
Таким образом, химия комплексных соединений пятивалентного ва-
надия только «рождается». Исследования в этой области важны с
точки зрения совершенствования технологии и аналитической химии
ванадия и представляют самостоятельный интерес, так как химия
комплексных соединений ванадия (V) характеризуется рядом особен-
ностей. В отличие от обычных металлов-комплексообразователей,
ионы которых в воде существуют в виде аквакомплексов, пятива-
лентный ванадий не образует аквакомплексов, способных к существо-
ванию даже в сильнокислых растворах, и его простейшим известным
ионом является диоксо-ион VO^. Равновесия с участием таких ионов
исследованы гораздо меньше, чем равновесия с участием аква-ионов.
Однозарядные типичные катионы, такие как ионы щелочных метал-
лов, отличаются весьма малой склонностью к комплексообразованию
и вначале ожидали, что это свойственно также однозарядным катио-
нам типа МО^. Однако детальные исследования ионов пятивалент-
ного нептуния и плутония (NpO^ и PuOt) показали, что для них
характерна достаточно сильная тенденция к ассоциации с органи-
ческими и неорганическими лигандами и по своей комплексообразу-
ющей способности они близки к таким типичным комплексообразо-
вателям, как двухвалентные ионы Zn2+, Mn2+, Cd2+ [298—301].
Экспериментальный материал позволяет утверждать, что ион VO1"
отличается еще большей комплексообразовательной способностью.
Комплексные соединения пятивалентного ванадия рассматрива-
ются в таком порядке: 1) комплексы с анионами неорганических
кислот, 2) гетерополисоединения, 3) соединения с перекисью водо-
рода, 4) комплексы с органическими лигандами.
Комплексы с неорганическими лигандами
г
КОМПЛЕКСЫ С АНИОНАМИ НЕОРГАНИЧЕСКИХ КИСЛОТ
Предположения о комплексообразовании ванадия (V) с неорга-
ническими лигандами высказывались многочисленными исследовате-
лями, но работы по изучению состава комплексов и определению
Констант устойчивости соответствующих комплексов появились лишь
В последнее время.
Берцелиус [302], исследуя осадки, полученные при медленном
выпаривании на воздухе концентрированных сернокислых растворов
139
ванадия, обнаружил соединение, которому приписал формулу V2O .
• 2H2SO4. Распадом этого комплекса — реакция (42) — М. Н. Соб’о.
лев [1] объяснил процесс гидролитического осаждения ванадия.’
Дюкре [22] установил минимальную растворимость пятиокиси
ванадия в хлорной кислоте, а повышенную растворимость в соляной
и серной кислотах объяснил комплексообразованием. Исследуя экс-
тракцию ванадия аминами из сернокислых растворов, Колмен и др,
[293] также высказали мысль о существовании сульфатных комплек-
сов. В работе В. Л. Золотавина с соавторами [303] на основании
изучения светопоглощения в сернокислых растворах пятивалентного
ванадия высказано предположение о существовании комплексных
2_
VO(SO4)2 при соотношении кислоты
анионов [VO (SO4)~] или
и ванадия в растворе Н+ : VO3 = 12 — 15.
Первые количественные характеристики неорганических комплек-
сов ванадия (V) получены в 1966 г. [304—307]. Спектрофотомет-
рическим методом исследовано комплексообразование ванадия (V)
с серной, соляной, азотной, плавиковой кислотами. Эффект образо-
вания сульфатных, хлоридных, нитратных и фторидных комплексов
был установлен по смещению равновесия реакции гидролиза иона
vot. Я
Как показали исследования, при концентрации свободных ионов
водорода (2,91 4-6,72) 10~3 г‘Экв1л и общей концентрации сульфат-
ионов (в форме SO и HSO7) (116) 10~2 г-ион/л, температуре
19 С в перхлоратной среде (р = 1,01) образуется комплекс VO2SO?
с константой устойчивости (3 = 9,32 [304]. По данным Гиллеспи и
Капура [308] при растворении метаванадата аммония или пятиокиси
ванадия в 100%-ной серной кислоте образуются комплексы:
Н [VO (HSO4)4], димер Н [V2O3 (HSO4)6] и, возможно, более высокие
полимеры.
Комплексообразование с хлор-ионом исследовалось при концент-
рации хлорида натрия от 0 до 1,0 г-ион/л в перхлоратной среде
(р=1,01) и температуре 19° С [305]. Обнаружен комплекс состава
VO2C1 с константой устойчивости (3=0,42. В этих же условиях при
концентрации ионов NOF от 0,1 до 0,8 г-ион! л образуется нитрат-
ный комплекс VO2NO3 с константой устойчивости (3 = 0,32 [305].
При исследовании системы KF—V2O5—HF—Н2О [87, 88] в ши-
роком диапазоне молярных отношений компонентов были найдены
три индивидуальных оксофторванадата, из которых K3V2O4F5 и
K2VO2F3 являются производными оксофторида VO2F, a K5V3O3Fi4
можно представить как производное VOF3 [87, 88, 309]. Данные
о существовании оксофторваиадатов калия состава 2KF-VOF3,
4KF-VF5.VOF3 [309] и 3KF-2VOF3.HF, 3KF.2VOF3.H2O [ЗЮ,
311] не были подтверждены. В системах RbF(CsF)—V2O5—HF—Н2О
получены соединения M5V3O3F14 и M3V2O4F5. Соединения типа
M2VO2F3 в кристаллическом состоянии не были выделены из-за их
разложения в растворе [88]. Комплексный фторид K3V2O4F5 крИ'
140
Пропуска ние
Рис. 69. Инфракрасные спектры оксофторванадатов.
/ —KzVO2F3; 2 —BaVO2F3; 3 — (NH4)8VO2F4; 4 — KSV2O4F5; 5 — Rb3V2O4F5;
6 _ Cs3V2O4F5; 7 — (NH4)3V2O4F; X — полосы вазелинового масла.
сталлизуется в виде тонких прозрачных микроскопических пластинок
желто-зеленого цвета и может быть перекристаллизован из воды
без изменения состава. K2VO2F3 и K5V3O3F14 выделяются в виде
призматических кристаллов. При недостатке плавиковой кислоты
в растворе соединение K5V3O3F14 претерпевает гидролиз, превра-
щаясь в K3V2O4F5.
На рис. 69 приведены ИК-спектры оксофторванадатов калия,
бария и аммония. Две полосы, лежащие в области 870—940 т-1,
отнесены к валентным колебаниям группы VO^. По аналогии с
МоО|+ и WO|+, для которых характерны такие же дублеты и не-
Рис. 71. Распределение
иона УОф в системе УОф—F-—Н2О.
линейное строение которых доказано рентгеноструктурными исследо-
ваниями и измерениями химических сдвигов ЯМР на F19 [312, 313],
сделан вывод о нелинейном строении группы УОф в анионе VO2F|“.
В ИК-спектрах оксофторидов состава M3V2O4F5 (см. рис. 69) в
области валентных колебаний групп VO2 (800—1000 сыт1) при-
сутствует одна интенсивная полоса с максимумом при 930 см~±
Это, а также сходство спектров этих соединений со спектрами солей
уранила позволило заключить, что в рассматриваемых соединениях
группы VOJ линейны. Предполагаемое строение ионов VO2F1“ и
V2O4F53" приведено на рис. 70. Эти данные указывают на возмож-
ность образования в водных растворах фторидных комплексов с раз-
личным числом координированных атомов фтора. Комплексообразо-
вание с фтор-ионами было исследовано двумя способами: по методу
сдвига равновесия реакции гидролиза иона VO2 и путем измерения
оптической плотности растворов, содержащих ванадий только в фор-
ме ионов УОф и фторидных комплексов VO2F^~X, при температуре
20—2ГС, ионной силе р=Я,0 в среде перхлората натрия [306,
307]. При общей концентрации ванадия 4-10-4 и 2,59-10~3 г-атом/л
найдены три комплекса VO2F, VO2FF, УО2Рз~ (рис. 71), полные
константы устойчивости которых соответственно равны: р4=(1,11 ±
±0,07). 103, р2-(3,94±О,18).1О5 и р3-(7,26± 1,0). 106. В спектрах
143
б ’Ю"3, г атом-i см-1
1.5
0,9
0.1
0,5
Л/Г
39 9i
Рис. 72. Спектры поглощения ком-
плексных оксофторидов ванадия.
1 — VO^~; 2 — VO2F; 3 — VO2f7;
2__
4 — VO2F
г з
фторидных комплексов (рис. 72) с
увеличением числа присоединенных
атомов фтора интенсивность погло-
щения в областях X > 270 и Z <
< 230 нм уменьшается, а в области
Х--230:270 нм, напротив, воз-
растает, и в спектре иона VO2F3"
наблюдается отчетливо выраженная
полоса поглощения с максимумом
при Л--240 нм и ътах= 1,7-103.
Кроме того, видно, что в спектрах
всех фторидных комплексов полоса
поглощения с максимумом, лежа-
щим в области 7 — 220 нм, сохра-
няется, но ее интенсивность, по-
видимому, падает с увеличением
числа присоединенных атомов фтора.
ГЕТЕРОПОЛИСОЕДИНЕНИЯ
Г етерополисоединения состав-
ляют обширную группу многооснов-
ных кислот и их солей общей фор-
мулы Н3 [ЭО (МеОт)2],
центральный комплексообразователь,
Me — металл шестой или пятой
системы. Чаще всего в состав лиган-
где Э —
побочных групп периодической
дов комплексных гетерополианионов входят молекулы окислов одного
лишь металла, но существуют гетерополианионы, которые содержат
различные лиганды, включающие окисли двух и даже трех метал-
лов — смешанные гетерополикислоты.
Наибольшее количество гетерополисоединений образуют элементы
четвертой и пятой групп периодической системы, особенно много —
кремний и фосфор. Наиболее хорошо изучены гетерополисоединения,
где в качестве лигандов выступают молибден и вольфрам. Ванадий
(V) может играть роль центрального атома, служить в качестве
лиганда, а вместе с молибденом и вольфрамом образует многочис-
ленные смешанные гетерополисоединения. Сводка ранних исследо-
ваний по гетерополисоединениям ванадия приведена в работах [123,
314]. Им же посвящена монография Е. А. Никитиной [315], вышед-
шая в 1962 г., в которой рассмотрены весьма подробно основные
аспекты химии гетерополисоединений. Поэтому в данной работе де-
тально будут освещены только фосфорнованадиевые и смешанные
фосфорномолибденованадневые гетерополисоединения, главным обра-
зом, по результатам исследований, выполненных после 1960 г. Эти
классы гетерополисоединений ванадия имеют большое практическое
значение и наиболее хорошо изучены. Все другие будут рассмотрены
в самых общих чертах на основе упомянутой монографии [315].
144
Известны гетерополисоединения, центральным атомом в которых
является ванадий; этим соединениям приписаны формулы:
М3 [V (С2О4) (МоН4)3]-хН2О и M2H[VO2(C2O4)(MoH4)].xH2O.
Для 3-молибдооксалатованадатов известны натриевая, калиевая, ам-
мониевая и бариевая соли, для 1-молибдооксалатованадатов получены
натриевая и аммониевая соли [315].
Более многочисленны гетерополисоединения, в которых ванадий
выступает в качестве лиганда, особенно много получено смешанных
гетерополисоединений.
Среди гетерополисоединений йода известны ванадиевые соедине-
ния. Получена йодноватованадиевая кислота V2O5- J2O5-4H2O и не-
которые ее соли, но основность кислоты не установлена. Синтези-
рованы ванадойодаты типа ^MO2-nJ2O5-pV2O5-xH2O с различным
соотношением окислов [315].
Описаны ванадотеллураты натрия состава Na2O • 2V2O5 • 5ТеО • 2Н2О,
Na2O • 2V2O5 • 6ТеО2 • 12Н2О и Na2O • 2V2O5 • 10ТеО2 • ЗН2О, которые
могут быть получены при обработке ванадата натрия в нейтральном
или кислом растворе тетрахлоридом теллура [123]. Ванадотеллу-
раты другого типа получили Прасад и Патхак [316] путем взаимо-
действия теллуровой кислоты и пятиокиси ванадия в аммиачном
растворе. После упаривания и кристаллизации было получено веще-
ство состава 3 (NH4)2O-2TeO3-3V2O5- ЮН2О. Это желтые кристаллы,
плохо растворимые в холодной и хорошо в горячей воде, легко раз-
лагаемые кислотами и щелочами. По реакции обменного разложения
в растворе выделены соли состава ЗМО-2ТеО3-ЗУ2О5-иН2О, где для
М=Ва, Sr, Са, Ag, Pb, Ni п=2, 8, 8, 6, 5, 8 соответственно. Ани-
онная природа ванадотеллурата аммония была доказана путем сорб-
ции на анионите. При нагревании аммониевая соль теряет 4 молекулы
Н2О при 110° С и 6 молекул Н2О при 260—300° С. Удаление ам-
миака при 360° С сопровождается экзотермическим эффектом. Свин-
цовая соль теряет воду в два этапа при 105 и 280е С. ИК~спектр
аммониевой соли содержит полосы поглощения воды и связей V—О
и Те—О.
Смешанные гетерополисоединения ванадия в качестве центральных
атомов содержат водород, бор, кремний, германий, фосфор, мышьяк,
а в состав лигандов, кроме ванадия, входят молибден или вольфрам
или тот и другой одновременно.
Описано много смешанных гетерополисоединений с водородом в
качестве комплексообразователя, но, по-видимому, большая часть из
них—двойные соли или просто смеси [123].
Существует несколько рядов аквамолибдованадатов типа М3Нр-
TH2(MoO3)„(V2O5)J-xH2O с различными п и т [315]. Среди ак-
вавольфрамванадиевых гетерополисоединений различают три ряда
солей [317]. Состав солей первого ряда, называемого желто-красным,
2М2О- V2O5-4WO3-xH2O, второго, оранжевого, 5M2O-3V2O5-6WO3-
• хН2О, третьего, пурпурно-красного, 4M2O-3V2O5- 12WO3-xH2O.
Соотношение V/W в этих рядах соответственно равно 1:2; 1:1; 1:2.
10 Заказ № 256
145
Как было установлено на основании спектрофотометрических и
pH-метрических измерений [318, 319], в водных растворах могут
существовать два ряда изополианионов, включающих ванадий и
вольфрам или молибден. Соединения первого ряда относятся к типу
гексавольфрамата, второго ряда — к типу декаванадата.. В области
pH 6,5—8,5 изополианион содержит V и W или Мо в отношении 1: 2
2VO?+4WO4_+6H+^
4—
+зн20
(51)
W4 '
. V2^19 .
при pH < 3,0 образуется Н
при pH 2—3 протекает реакция
W4
. V2Oi9
з—
. В более кислых растворах
VOr + 5WO4~ + 8H+
WB у-
_VO19.
(52)
И, наконец,
Мо3
к типу гексавольфраматов
При pH < 4,5 один атом
отнесен ванадатомолибдат
ванадия в декаванадатионе
4 —
может быть замещен атомом вольфрама или молибдена; образующийся
изополианион имеет структуру декаванадата [V9W (Mo)O2gl5-. Кроме
того, выделено соединение состава 5 (NH4)2O- 14WO3-3V2O5-37H2O,
которое отнесено к ряду декаванадата и рассматривается как про-
rW7 Не-
изводное аниона v п
Из соединений бора получена боровольфрамванадиевая кислота
и ее таллиевая соль 5Tl2O-B2O3-20WO3-2V2O5 [315]. Известны
вольфрамванадосиликаты с отношением SiO2: WO3: V2O5 = 1:10:1;
1:9:1; выделена кремневольфрамванадиевая кислота Н8 [Si (W2O7)5 •
•(V2O6)]-xH2O; аналогичные соединения получены с молибденом
[315].
смешанные гетерополикислоты с германием в качестве централь-
ного атома и лигандами, содержащими молибден или вольфрам
и ванадий, описываются общей формулой Н8 [Ge (М2О7)5 (V2O6)] -хН2О,.
где М—-Мо или W. Для вольфрама получены кристаллогидраты
с х=20, 25, 28, 30. Получены соли мышьякововольфрам (молиб-
ден) ванадиевых кислот, например, (NH4)6H [As (W2O7)4 (V2O6)2] •
•xH2O, (NH4)6H[As(W2O7)3(V2O6)3].%H2O [315].
Из числа смешанных гетерополисоединений, содержащих вана-
дий, лучше всего изучены соединения фосфора. На реакциях обра-
зования окрашенных фосфорномолибденванадиевых и фосфорно-
вольфрамванадиевых соединений основаны колориметрические-
методы определения фосфора и ванадия [296, 297]. Фосфорно-
вольфрамванадиевые кислоты и их соли содержат меняющееся коли-
чество радикалов У20б~, частично заменяющих радикалы W2Oy~.
146
Эти соединения принадлежат к насыщенному ряду и являются
производными кислоты H7[P(W2O,)n(V2Oe)m-xH2O, ' где п-\-т—6.
Выделены три типа аммониевых солей (NH4)€ Н [Р (W2O7)rt •
• (V2O6),Z/• хН2О, в которых п и т равны 4 и 2, 3 и 3, 2 и 4,
и свободная кислота Н7 [P(W2O7)5(V2O6)]-%H2O [315]. Более детально
изучены свойства фосфорномолибденванадиевых соединений. Среди
них наиболее известна кислота Н7 [Р (Мо2О7)5 • (V2O6)] • хН2О [315].
Шово с сотрудниками [320—322] нашли, что помимо кислоты
с соотношением Р:Мо: V= 1:10:2, в зависимости от условий, обра-
зуются кислоты с Р:Мо: V= 1:11:1 и 1:9:3. Криви и Кртыл [323]
выделили цезиевую соль фосфорномолибденванадиевой кислоты
и приписали ей формулу Cs3H4 [Р (Мо2О7)5 (V2O6)-xH2O.
Ю. Ф. Шкаровский [324] на основании измерений оптической
плотности изомолярных серий растворов пришел к несколько иным
выводам относительно состава фосфорномолибденванадиевых комп-
лексов. При pH 5,8—3,0 и 0 соотношение фосфат: молибдат (ni)
близко 1:6 независимо от концентрации ванадата; при pH2,0
/71=1:6 и 1:3 для УО3] <^0,018 и 0,024—0,048 моль/л соот-
ветственно; при pH 1,0 т= 1:6 и 1:4 для [УОУ] <0,025 и 0,05
соответственно. Соотношение фосфат — ванадат равно 1:3, 1:2
и 1:1 при pH 5,8; 3,0 и 1,0 (и 0) соответственно и сохраняется
постоянным в интервале концентраций молибдата от 0,05 до
0,4 моль/л.
По мнению автора, существование от 1 до 3 атомов ванадия
в молекуле фосфорномолибденванадиевого комплекса, которые
не могут быть заменены молибденом как лигандом, свидетельствует
о неравноценной роли обоих металлов в структуре тройного ком-
плекса. Шигдинос и Халлада [325] синтезировали все три типа
фосфорнрмолибденовых кислот и на основании результатов химиче-
ского анализа и потенциометрического титрования предложили для
них формулы Н4 [РМо14УО40] • 34Н2О, Н5 [PMo10V2O40] • 32Н2О,
Н6 [РМо9У3О40]-34Н2О. На кривых титрования указанных кислот
имеются две ступени. Первая из них (pH —3,0) соответствует ней-
трализации водородов, способных к замещению; на второй стадии
(pH —8,5) происходит распад гетерополианиона, например,
IPMo11VO4014- + 22OH-^HPO4-t 11МоО4' + Н УС)‘Г + ЮН2О.
Авторами, кроме того, получены натриевая и аммониевая соли
фосфор-11 -молибденванадиевой кислоты Na4 [PMonVO40] • 8Н2О
и (NH4)3H[PMonVO40].75H2O. В свете приведенных данных цезие-
вой соли, описанной в работе [323], по мнению авторов, следует
приписать состав Cs3H2 [PMo10V2O40]-%H2O. Получены ИК-спектры
поглощения синтезированных соединений. В ИК-спектрах найдены
полосы поглощения воды, связей Р—О, Мо—О; полоса поглощения
связи V—О маскируется полосами поглощения Мо—О. ^^Ф-спектре
при Х = 310 нм обнаружена полоса поглощения (е
характерная для фосфор-12-молнбденовой кислоты. Авторы рас
сматривают описанные соединения на основе модели Кеггина для
10* 147
17000—18100),
фосфор-12-молибденовой кислоты, в которой часть молибдена заме-
щена на атомы ванадия.
Начиная с ранних работ Гиббса, Дитте и Фридгейма с сотрут
никами [326—330], которые выделили два типа фосфорнованадие-
вых солей: пурпурсоли с отношением V:P=12:1 и лютеосоли
с V:P=1:1, фосфорнованадиевые гетерополисоединения исследова-
лись неоднократно.
Из пурпурсоединений Фридгеймом были получены семизаме-
щенные аммонийные и калиевые соли фосфор-12-ванадиевой кислоты
состава 7 (NH4)2O • Р2О5 • 12V2O5 • 26Н2О, 7К2О • Р2О5 • 12V2O5 • 26Н2О,
которые, как полагал автор, являются солями кислоты Н7 [Р (V2O6)6].
• 13Н2О. Изучение процесса дегидратации аммониевой соли [3311
показало, что она не содержит внутрисферной воды и ей следует
приписать координационную формулу (NH4)7 [Р (V2O6)6b 13Н2О.
В работе [331] также выделен ванадофосфат калия 5К2О-Р2О5-
• 10V2O5-5H2O, которому на основании исследования процесса
дегидратации приписана формула К5Н2 [РО (V2O6)5] • 24Н2О. При
обменном разложении ванадофосфата аммония с солями цезия
и рубидия получены пятизамещенные соли Cs5H2[PO(V2O6)5]« 12,5Н2ОГ
Rb5H2 [РО (V2O6)5] • 22Н2О [331]. Суше и Дюбуа [332j, повторив
работу [331], получили натриевые, калиевые, рубидиевые, бариевые
и стронциевые соли фосфор-12-ванадиевой кислоты Н7 [PV12O36]..
По их мнению, существуют только пурпурованадофосфаты, которые
представляют собой соединения насыщенного ряда. По мнению
Яндера с сотрудниками [333], ванадофосфаты не являются гетеро-
полисоединениями и должны рассматриваться как продукты взаи-
модействия октаванадиевой кислоты [H4V8O28]6- и фосфатов, а
отношение V:P в ванадофосфорной кислоте кратно 8. Однако эта
точка зрения не подтверждена другими исследователями. Рассел
с соавторами [334], исследуя состав ионов, поглощенных на анионо-
обменной смоле из фосфорнованадиевых растворов, пришли к за-
ключению о существовании гетероиопов [HPV10O30]4-" при pH 4,9—
5,9 и [H2PV10O30]3~ при pH 1,8—2,9. А. Б. Бектуров и А. К. Иль-
ясова [335] изучали взаимодействие растворов метаванадата натрия
и калия с различными кислотами в присутствии ортофосфорной
кислоты и ее солей. По их данным, фосфорнованадиевые гетеропо-
лиионы темно-вишневого цвета образуются в растворах, характери-
зуемых степенью подкисления Н+:У07=0,65-ь0,18, после того
как метаванадат полностью перейдет в оранжевые пента (дека)-
ванадат-ионы. Авторы отмечают, что фосфорнованадиевые соедине-
ния, в зависимости от природы катиона, имеют различную степень
замещенности и различное отношение V:P. Полученные соединения^
охарактеризованные составами Na3 (H2V6O16)2PO4-nH2O, K5(V7O18V
• РО4 • мН2О, (CN3H6)7 (V6O16)2. пН2О, (NH4)7 (V6O16)2PO4 • nH2O,
рассматриваются как продукты конденсации двух молекул изопо-
ливанадатов и фосфат-иона Н2РОГ- В частности, образование вапа-
дофосфата калия представлено схемой, согласно которой взаимодей-
148
ствие Н2РО4 с ионом H3V50is сопровождается замещением двух
ионов Н+ на катионы VO2 :
Рис. 73. Термограмма фосфорно-
ванадиевой кислоты.
ОК О—К
| НО—(V5O14) н2к2 , , |
0=Р-ОН + + 4 VOt-> 0=Р—О-V5O14 (VO2)2K2 +
1 НО—(V5O14) H2K2 |
OH O-V5O14 (VO2)2K2
+ 2H2O + 4H+
Данная схема встречает возражение хотя бы в том отношении,
что в настоящее время нет доказательств существования пентава-
надат-иона H3V5O^-
Лютеофосфорнованадиевые соединения, открытые Дитте [327],
неоднократно описывались в литературе и различные авторы при-
писывают им различное строение:
VOPO4-xH2O [336], H2[VO3PO4P
• 4Н2О [337], V2O5 • Р2О5 • 4Н2О
[338], H4VPO7 [339] и VOPO4-
•2Н2О [340]. Рассел с соавто-
рами [334], изучая равновесие
в системе V2O5—Р2О5—Н20 при
25° С, установили образование
малорастворимых соединений
V2O5-PoO5-6H2O и 2V2O53P2O5.
• ЗН2О, которым они припи-
сывают строение VOPO4-3H2O
или VO2H2PO4-2H9O и (V02).
• (НР04)о*5Н20 или (V02)«
•н2ро4]2.н3ро4.зн2о.
В работе [339] фосфорно-
ванадиевую кислоту синтезиро-
вали растворением V2O5 в расплав-
ленной ортофосфорной кислоте.
Выделенный золотисто-зеленый осадок приблизительно отвечал фор-
муле VOPO4-2Н2О• 0,1Н3РО4. При нагревании он теряет 2,05 моля
Н2О при 120—180° С и 0,1 моля Н2О при 240° С; при 830° С осадок
теряет избыточную Р2О5 (рис. 73). Полученный VOPO4 растворим
в воде с образованием 0,06 моля оранжево-красного раствора
с pH 3. При добавлении солей Ва2+- РЬ2+ и Hg+ образуются осадки.
Состав бариевой соли близок формуле BaVPO7, в связи с чем
водные растворы ванаданфосфата рассматриваются как пирована-
диевофосфорная кислота H4VPO7, строение которой выражается
формулой
ном °;он
V—О—Р
HOZ хон
149
Иной точки зрения на природу ванаданфосфата придерживаются
Л. А. Кадушкина и Л. К. Курмангужина [340], которые иссле-
довали желто-зеленые соединения состава V2O5-P2O5-4H2O и темно-
зеленые V2O5-P2O5-2,5H2O, полученные кристаллизацией в системе
V2O5—Н3РО4—Н2О. На основании термогравиметрических иссле-
дований было сделано заключение, что вода, входящая в состав
первого соединения, имеет кристаллогидратный характер и наличие
обменных водородов исключается. Второе соединение рассматривается
как продукт присоединения к молекуле ванаданфосфата фосфорной
кислоты с образованием двойного ванаданфосфата (VOPO4)2-H3PO4.
При длительном выдерживании водных растворов, содержащих
от 5 до 20 г/л VOPO4-2H2O при 70° С ванаданфосфат подвергается
гидролизу с образованием H6V10O28 и Н3РО4, и в осадок выделяется
.либо декаванадиевая кислота, либо ее смеси с VOPO4 или
VO2H2PO4.
Розенгейм и Янг [341] установили, что при кристаллизации
лютеофосфорнованадиевая кислота захватывает фосфорную кислоту.
Ими получена аммониевая и калиевая соли состава M2O-2V2O5-
•PgOs’SHgO. На основании определения прочности связи воды
предполагается, что фосфорнованадиевая кислота одноосновна и ее
соли имеют формулу iM [РО2 (VO3)2]-3H2O, а лютеосоли с отноше-
нием V: Р = 1:1 — производные аниона [РО3 (VO3)]2~[315]. Однако в ра-
боте [321] лютеофосфорнованадиевые соединения рассматриваются
как фосфаты ванадила VO2" и им приписаны формулы M(VO2)«
• НРО4 и M(VO2)2PO4.
Комплексы ванадия (V) с фосфорной кислотой существуют в вод-
ных растворах. Моулик с соавторами [258, 342] нашли, что вана-
диевая кислота, полученная пропусканием раствора ванадата аммо-
ния через катионообменную смолу в Н-форме, в растворе взаимо-
действует с ортофосфорной кислотой, образуя два комплекса: один
красного цвета с отношением V:P=2:1 и другой, бесцветный,
с отношением V:P=1:3. Состав комплексов установлен методом
Джоба на основе спектрофотометрических измерений. Комплекс 2:1
имеет максимум поглощения при 500 нм, а на кривой поглощения
комплекса 1:3 в интервале 320—350 нм имеется плато. Реакции
образования комплексов представлены уравнениями
Н3РО4 4- 2 (HVO3) Н [РО2 (VO3)2] + 2Н2О, (53)
ЗН3РО4 + HVO3 Н [V (НРО4)3] + ЗН2О. (54)
По мнению авторов [258, 342], в анионе [PO2(VO3)2] два из четы-
рех атомов кислорода, находящихся в тетраэдрической координации
по отношению к фосфору, замещены на две группы VO3, а комп-
лекс 1:3 рассматривается как производное HVO3, в которой три
атома кислорода замещены на группы НРО4. Определены константы
устойчивости изученных комплексов [321-^6,162 -101, [З13 1,809 -109.
Следует отметить, что интерпретация результатов в рассматриваемой
работе сделана без учета ионного состояния ванадия и фосфора
150
в растворах. В частности, ванадиевой кислоте приписана фор-
мула HVO3. Однако известно, что ванадиевая кислота существует
как HVO3 только при весьма малых концентрациях ванадия (~10~5
моль/л) [67—69], при более высоких ее рассматривают как H6V10O28
[20, 55, 62, 63, 257]; кроме того, в смесях с фосфорной кислотой
ванадий может переходить хотя бы частично в катионную форму.
Ортофосфорная кислота в растворах частично ионизирована,
и в реакциях может участвовать весь набор частиц от Н3РО4
до РО4. Эти обстоятельства не позволяют считать выводы авторов
работ [258, 342] о составе и константах устойчивости фосфатных ком-
плексов ванадия достаточно обоснованными.
А. А. Ивакин и Э. М. Воронова [343], основываясь на том, что
в присутствии фосфорной кислоты оптическая плотность растворов,
содержащих ванадий в форме декаванадатов, понижается, пришли
к заключению об образовании фосфатных комплексов. При темпера-
туре 25 С, ионной силе растворов ц=1,0 в среде перхлората
натрия, общей концентрации ванадия 2-10“3 г -атом/л и ортофос-
форной кислоты 0,01 моль/л в интервале — 1g [Н+] = 3,25-?-4,47
данные спектрофотометрических измерений интерпретированы, исходя
из предположения о существовании комплексов VO2HPO? и VO2-
’(HPO4)f~ с константами- устойчивости 4,1-104и 2,05-109. Ранее
[344] сообщалось о существовании ванадофосфата с отношением
V:P=1:2.
Одной из важных с точки зрения технологии является система
ванадия (V) с фосфорной кислотой. Однако, как видно из приве-
денного в данном разделе материала, фосфорнованадиевые соеди-
нения изучены еще недостаточно. Особенно много необходимо
сделать, чтобы выяснить ионные формы ванадофосфатов в водных
растворах.
ПЕРОКСОВАНАДАТЫ
9
Давно известно, что пятиокись ванадия и ванадаты реагируют
с перекисью водорода, образуя перекисные соединения типа над-
кислот и пероксосолей. Однако, несмотря на многообразие работ,
посвященных изучению пероксосоединений ванадия, выполненных
на протяжении более столетия, до сих пор нет ясности в понимании
их химической природы, точного состава, строения, условий обра-
зования. В связи с применением этих соединений в аналитической
химии, одни исследователи изучали их свойства в растворе, другие
стремились их выделить в твердом состоянии, изучить их состав, стро-
ение, условия образования и свойства.
Шейер [345] при действии перекиси водорода на метаванадаты
в присутствии спирта выделил соединения состава MVO4, где
M=Li, Na, К, NH4, Ag, Меликов и Писаржевский [346] получили
перпированадаты, соответствующие гипотетической кислоте H4V2On,
действием щелочного раствора перекиси водорода на перметава-
15R
па даты. П. Г. Меликов и Е. Елчанинов [347] выделили голубые
перортованадаты аммония и калия (NH4)3VOG-2,5H2O, K3VOG-2,5H2O
и (NH4)2HVO7, обрабатывая растворы ванадатов аммония и калия
на холоду перекисью водорода в присутствии аммиака и поташа
и осаждая спиртом. Яр [348] получил соединения, которым припи-
сал формулы КН2 [VO2(O2)2]-H2O, (NH4)2-H [VO2(O2)2]-aq. Из силь-
нощелочных растворов в присутствии большого количества Н2О2 при
0°С он получил сине-фиолетовые кристаллы состава М3 [V (O2)4]-aq,
где M=Li, Na, NH4, К. Соединения аналогичного состава M3VO8
(M=NH4, К) получили Белтран и Риос [349]. Белтран [350]
выкристаллизовал при обычной температуре желтую соль, содер-
жащую 2,5 атома активного кислорода на 1 атом ванадия. Предпо-
лагается [351], что это продукт разложения приведенного выше
соединения.
Н. Г. Григорьева и К. Н. Дергачева [351], пытаясь изучить
систему Na3VO4—Н2О2—Н2О при 0°С методом растворимости
(рис. 74), нашли, что равновесие в системе устанавливается плохо,
тю-видимому, из-за разложения как твердых остатков, так и жидкой
фазы. Было получено черно-фиолетовое кристаллическое соединение,
которому соответствовала валовая формула Na3VO8-7H2O. Это со-
единение, кроме того, было синтезировано иным путем: сплав 1 г V2O5
с 3 г NaOH растворяли сначала в 4—5%, затем в 40% Н2О2
352
при охлаждении льдом. Раствор отфильтровывали и перекисное
соединение осаждали спиртом. Выделенное и промытое спиртом
вещество анализировали в «сыром» виде, а также после подсуши-
вания над СаС12. Из кривых нагревания (рис. 75) сырого и сухого
продуктов видно, что разложение первого начинается при 70° С
при одновременном эндотермическом эффекте, связанном с удалением
воды; при 110е С начинается бурное выделение кислорода с разло-
Рис. 75. Кривые нагревания пероксованадатов натрия.
а — «сырой» (пунктир — выделение активного кислорода), б—«сухой».
жением вещества и экзотермическим эффектом. Сухое вещество
разлагается при 115 С со вспышкой. При комнатной температуре
за 60 суток Na3VO8 потерял 3,53 % активного кислорода. Анало-
гичным способом был синтезирован перпированадат калия, состав
которого отвечал формуле K4V2On с 4 атомами активного кисло-
рода. «Сырой» перпированадат калия при 105° С дегидратируется,
а при 150° С начинается разложение продукта с бурным выделением
кислорода. K4V2O11 достаточно устойчив при комнатной температуре:
за 60 суток потеря активного кислорода составила 1,59%.
Появление, а затем обесцвечивание красно-коричневой окраски,
когда избыток перекиси водорода медленно добавляется к кислому
эаствору ванадия (V), известно более ста лет. Майер и Павлетта
352] объяснили это образованием в сернокислом растворе вначале
красного комплекса
(VO)2 (SO4)3 + 2H2O2->[V (О2)]2 (SO4)3 + 2Н2О,
(55)
который в присутствии избытка перекиси водорода переходит в жел-
тую пероксокислоту,
[V (О2)]2 (SO4)3 + 6Н2ОНт?2 2V (О2)(ОН)3 + 3H2SO4.
h2so4
(56)
153;
Однако эта схема неудовлетворительна, хотя бы потому, что в сер-
нокислом растворе предполагается наличие иона VO3+, а не VOJ" .
Согласно Яру [348], в щелочных или слабокислых растворах обра-
зуются желтые дипероксоортованадат-ионы [VO2 (О2)2]3~, а в силь-
нокислых— красно-коричневые катионы пероксованадия [V (О2]3+,
соотношение между которыми определяется равновесием
[ VO2 (О2)2]3- + 6Н+ [V (О2)]3+ + Н2О2 + 2Н2О. (57)
По мнению Румпфа [353], в концентрированной соляной кислоте
пероксованадат имеет состав VO2C13. А. К. Бабко и А. И. Волкова
[354], исследовавшие взаимодействие перекиси водорода и ванадия
(V) в растворах колориметрическим методом, нашли, что в слабо-
щелочной среде (pH ~ 8) комплекс содержит V и Н2О2 в соотно-
шении 1:2, а в кислой (lA'HCl) — отношение V:H2O2=1:1.
Основываясь на том, что изменение кислотности в широких пре-
делах не приводит к изменению оптической плотности растворов,
они сделали вывод, что комплекс с соотношением V:H2O2=1:1
представляет собой продукт присоединения молекулы перекиси
водорода [VOx(H2O2)]5~2x; найдена величина его константы диссо-
циации в Ш НС1:
ГуО5-2х1[Н о2]
Л = J—х_------- 2J =3 • 10-5,
[vox (Н2О2)5“2х1
откуда константа устойчивости равна 3,33-104. Ранее Шеппи
и Тредвелл [355] в среде 7 М H2SO4 колориметрическим методом
определили константу устойчивости комплекса 1:1, равную 8,2-104.
На основании спектрофотометрических исследований Дин [356]
нашел, что в кислых растворах существуют два комплекса; крас-
ный — состава 1:1 в форме катиона — хорошо сорбируется катио-
нитом и желтый-—1:2 в форме аниона — сорбируется анионооб-
менной смолой. Определены константы устойчивости комплекса 1:1
в Ш H2SO4, НС1О4, равные соответственно 3,5-104 и 3,4-104,
а также константа равновесия реакции перехода красного комплекса
в желтый
«красный» + Н2О2 Zt «желтый» + 3/2 H2SO4,
при 22°С равная 2,2±0,2. Найдено, что максимум поглощения
красного комплекса в W H2SO4 лежит при 454 нм, а в 1N НС1О4 —
при 460 нм. Дин, исходя из близости величин констант устойчи-
вости комплекса 1:1, полученных им и в работе [355], несмотря
на большое отличие от концентрации кислоты, пришел к выводу,
что ион Н+ в заметной степени в реакцию не вступает и процесс
образования комплекса 1:1 можно упрощенно описать уравнением
VOt + H2O2-VO(O2)+ + H2O, (58)
где (О2) означает пероксогруппу.
154
Флуд с соавторами [357] на основании pH-метрических и спект-
рофотометрических исследований пришли к выводу, что при раство-
рении пятиокиси ванадия в растворах перекиси водорода pH раст-
вора понижается вследствие диссоциации образующейся пероксова-
надиевой кислоты
Н3УО5
H+ + H2vo?.
(59)
В сильнокислых растворах наблюдается равновесие
H4VOtzl2H+ + H2VOr.
(60)
По мнению авторов, поскольку добавки кислоты не меняют опти-
ческие свойства раствора, спектры ионов Н4УО^и Н2УО5" анало-
гичны; обесцвечивание раствора при добавлении щелочи связано
с дальнейшей ионизацией частицы Н2УОГ
H2VO? Н+ + HVO^“.
(61)
Как видно, данные работ [354, 355] относительно состава пер-
оксокомплексов ванадия и константы устойчивости комплекса 1:1
хорошо совпадают, но заключения о природе его различны.
Шово [55] проведены систематические исследования взаимодей-
ствия перекиси водорода с ванадием в кислых и щелочных растворах
криоскопическим и спектрофотометрическим методами. Для кислых
и нейтральных растворов полученные данные хорошо согласуются
с результатами работ указанных выше авторов. В растворах мета-
ванадата в присутствии перекиси водорода образуется ион, содер-
жащий на один атом ванадия два атома активного кислорода
[УО3-2О]~. Поскольку один атом «активного» кислорода соответ-
ствует одной молекуле перекиси водорода, то состав приведенного
комплекса будет отвечать отношению У:Н2О2 И:2. При подкисле-
нии раствора перметаванадата отщепляется одна молекула Н2О2
и образуется катион перванадила:
[VO3 ."20]- + 2Н+ 72 [VO2O]+ + Н2О2. (62)
Ион [УО2*2О]- может гидролизоваться, процесс гидролиза проте-
кает по уравнению
[УО3• 20]- + Н2О Z2 [Н УО4 • 2OF- + Н+ (63)
Для [L1= 1,0 (NaCl) логарифм константы равновесия реакции гидро-
лиза равен—7,15. В слабощелочной среде образуется перпирова-
надат, содержащий на один атом ванадия три атома активного
кислорода:
[УО3 • 20]- + ОН- + Н2О2 72 [НУО4 • ЗО]2- + Н2О. (64)
Ион [НУО4-ЗО]2- также может гидролитически расщепляться,
освобождая одну молекулу Н2О2:
[НУО4 • ЗО]2- + Н2О 72 [НУО4 • 2О]2- + Н2О2. (65)
155
И, наконец, возможна реакция
[ VO3 • 20]- + [HVO4,2О]2~ [HV2O7 • 4О]3~
(66)
константа равновесия которой равна 200.
На рис. 76 показано распределение пероксидных комплексов
в области OH~:VO? от 0 до 1,0 в среде 1 M NaCl при концен-
трации NaVO3 0,05 моль!л.
0,15
450-
Z?25
О
ОН'' 1
Рис, 76. Распределение ванадия в
зависимости от величины OH~/VO^~ *
С_ =0,05 моль/л, с.т/с„ =з.
V V н2о2
VOu
О,в
0,6
4*
42
0 12 3 [ОН']
Рис. 77. Состав растворов в зависимо-
сти от концентрации ионов ОН~.
С =0,006 моль/л, Стт =2 моль/л,
V Н2О2
/=о° с.
НУ0п20г"
В сильнощелочной среде в присутствии избытка перекиси водо-
рода образуется перортованадат, содержащий четыре атома актив-
ного кислорода
(67)
Последний в условиях высокой концентрации щелочи распадается
по реакции
[ VO4 • 4OF- + ОН- И [ VO4 • ЗОР- + НОГ,
(68)
константа равновесия которой равна 40. На рис. 77 показана
зависимость состава растворов от концентрации ионов ОН~ при 0° С,
общей концентрации ванадия 0,006 моль I л и перекиси водорода
2 моль/л.
Как следует из приведенных данных, известным ионам ванадия
отвечают пероксидные ионы, отличающиеся содержанием активного
кислорода. Это отражено в таблице, взятой из работы Шово [55]
(табл. 33).
156
Таблица 33
Простые и соответствующие им пероксидные ионы ванадия
Число атомов 1 активного кис- лорода на атом ванадия Катион Декаванадат Мета ва- надат Кислый пиро- ванадат Кислый орто- ванадат Ортованадат
h2v10o427 V2O4-
0 vo+ hv10o^ v4o47 HV2O3- VO»"
V10O28 HVO'S-
1 VO2O+
2 VO32O- HV2O7-4O3- HVO4-2O2~
3 HVO4.3O2- VO4-3O3-
4 VO4-4O3-
КОМПЛЕКСЫ С ОРГАНИЧЕСКИМИ ЛИГАНДАМИ
КАРБОНОВЫЕ И ОКСИКАРБОНОВЫЕ КИСЛОТЫ
Данные по комплексообразованию пятивалентного ванадия
с одноосновными карбоновыми кислотами имеются только для уксус-
ной и муравьиной кислот. В водных растворах анионы одноосновных
карбоновых кислот образуют с ионом VO^ малоустойчивые комп-
.лексные соединения.
Существование ацетатного комплекса состава VO2CH3COO обна-
ружено по сдвигу равновесия реакции (18) гидролиза иона VOt
в присутствии уксусной кислоты. При 20° С в среде перхлората
натрия (р—1,0), общей концентрации ванадия 2-10-3 г-атом/л
и уксусной кислоты 0,1—0,5 моль/л в интервале—lg[H+] =
= 2,ЗЗч-2,42 константа устойчивости ацетатного комплекса опре-
делена равной (1,9 ±0,2)-102 [305]. Комплексообразование иона
VOt с муравьиной кислотой установлено по изменению оптической
плотности растворов, содержащих ионы VOt и НСОО~ в различ-
ных соотношениях. Измерения проводились при постоянной концен-
трации ванадия 4,18-10~4 г-атом/л и муравьиной кислоты 0,374 —
0,86 моль/л в интервале—1g [Н+]=2,03 ч- 3,67 в среде перхлората
натрия (ц=1,0) при 18°С. Константа устойчивости формиатного
комплекса оказалась равной 22,7 [358]. Константы равновесия
реакций замещения
СН3СООН + VO)t 7Z VO2CH3COO 4- Н+, (69)
НСООН + vot VO2HCOO + Н+, (70)
весьма близки и соответственно равны 4,97-10~3 и 6,51 -10-3.
157
Рис. 78. Дифференциальная кривая и кри-
вая потери массы при нагревании комплек-
са (NH4)3[VO2(C2O4)2]-2H2O.
Из числа двухосновны
карбоновых кислот изучеНь
комплексы ванадия (V) толь
ко со щавелевой и малоно
вой кислотами. Оксалатные
комплекс состава (NH )
•[УО2(С2О4)21-2Н2Овтвер;
дом виде был выделен
в 1893 г. Розенгеймом [359]
который, однако, рассмат-
ривал его как двойную
соль. Химическая природа
этого соединения и его
свойства изучены в 1964 г.
Сатьянараяной и Пателем
[86]. Степень окисления
ванадия, равная 5, установ-
лена по измерению диамаг-
нитной восприимчивости
(0,51-10-6) единиц CGS.
При нагревании на воздухе
оксалатный комплекс вна-
чале при 170° С теряет пол-
ностью воду, затем в интер-
вале 200—260° С распа-
дается с образованием оксалатного комплекса ванадия (IV) голу-
бовато-серого цвета:
2 (NH4)3 [VO2 (С2О4)21 - (NH4)2 l(VO)2(C2O4)3] + 2СО2 + 4NH3 + 2Н2О,
который разлагается при 300° С, давая двуокись ванадия (рис. 78).
ИК-спектр комплекса представлен на рис. 79, характеристика его
приведена ниже:
V_L 1 Отнесение
см 1
3550... va (Н2О)
3080.. .v(NH)
1720... vQ(C=O) (v7)
1669... va (C=O) (vj
1629... б (H,O) и 6(NH4)
1381... vs (C—0)4-v(C—C)
v ’ i Отнесение
CM 1
1270... vs (C —O) +
+6(0 —C=O) (v5)
924... va (О—V—O)
876... vs (C—O) +
+ 6 (O—C=O) (v5)
866... vs (О—V—O)
800... d(O—C=O) +
+v (M—O) (v9)
Интенсивная полоса 1720 им1, по мнению авторов, указывает
на сильно ковалентный характер связи между металлом и оксалат-
ными группами. Полосы при 924 и 866 см-1 отнесены к колеба-
ниям группы О—V—О, которая рассматривается как линейная
158
с асимметричным расположением атомов кислорода. На основании
этих данных комплексному аниону VO2(C2O4)2~ приписана окта-
эдрическая структура с с!25р3-связью, а строение комплекса пред-
ставлено формулой
Первые сведения относительно оксалатных комплексов ванадия
в водных растворах получил Бхаттачария с соавторами [360], кото-
4000 3600 2000 1500
1DOO 900 800 700 1СМ-1
Рис. 79. ИК-спектр комплекса (NH4)3[VO2(C2O4)2]-2H2O, X —полосы по-
глощения нуйола.
рые методом непрерывных изменений путем кондукто- и рН-метри-
ческого титрования обнаружили комплекс VO2 (C2O4)f~. Курихара
и Нозаки [361] на основании спектрофотометрических измерений
по методу непрерывных изменений и молярных отношений пришли
к выводу о существовании оксалатных комплексов с отношением
V: С2С)Г 1:1, 1:2 и 1:4. Для первых двух рассчитаны логариф-
мы кажущихся констант устойчивости, которые для ионной силы
р,—0,1 соответственно равны 7,8 (21° С) и 7,6 (24° С). К. Б. Яци-
мирский и В. Е. Калинина [362] подтвердили существование комп-
лексов 1:1 и 1:2. По изменению скорости реакции окисления —
восстановления между йодидом и броматом калия в присутствии
ванадия и щавелевой кислоты были рассчитаны константы устойчи-
вости комплексов, которые при 25° С и ц=0,2 оказались равными
Л1 = (0,98+ 0,27)-103 и /<2=(5,96 +1,4) -1()2. Константа устойчивости
оксалатного комплекса 1 : 1 определена также в работе [305] по
данным спектрофотометрического исследования сдвига равновесия
реакции (18) гидролиза катиона VO^ в присутствии щавелевой
кислоты. При 20° С в перхлоратной среде Qx=l,O) она найдена
равной (1,2±0,2)-105.
В работе [363] константы устойчивости оксалатных комплексов
определены при различных ионных силах растворов. При этом был
159
использован спектрофотометрический метод, основанный на измерь
ниях оптической плотности серий растворов с постоянной концент-
рацией ванадия 4,18-10~4 г-атом/л и переменной концентрацией
щавелевой кислоты: от 0,8-10~4 до 64,0-10~4 моль! л в интервале
—lg [Н] = 2,26-^4,52. Состав растворов был таким, что ванадий
в них присутствовал только в виде частиц VOjt, VO2C2C)7 и
Рис. 80. Распределение иона VOj“ в системе VOj — С2О4 — Н2О как
функция —1g [С2О4~].
ц: 1 — 0,006; 2 — 0,02; 3 — 0,1; 4—1,0.
VO2(C2O4)2 (рис. 80). В зависимости от ионной силы растворов
при 20° С найдены следующие значения констант устойчивости:
->0
0,006
0,02
0,1
1,0
4,4-106
(3,12 + 0,19) • 106
(2,01+0,45). 106
(8,73± 1,82) • 105
(7,49 + 3,88) • 104
1,58-Ю10
(9,36 + 0,48). 109
(6,61+0,16).Ю9
(3,52 + 0,18)-109
(3,09 + 0,2)40*
Значения и р2 для р 0 были получены экстраполяцией
величин 1g р; на нулевое значение V р.
Комплексные соединения ванадия (V) с анионом малоновой
кислоты изучались кинетическим [364] и спектрофотометрическим
методами [365]. К. Б. Янимирский и В. Е. Калинина [364], исследуя
окисление йодида броматом в присутствии ванадия и малоновой
кислоты в слабокислой среде (pH 2,7—2,9), пришли к выводу
о существовании комплекса УО2СН2(СОО)Г. Для температуры 25° С
и р—>0 авторы рассчитали константу устойчивости комплекса, рав-
ную 1,2 ДО5. При спектрофотометрических исследованиях [365]
были обнаружены два комплекса 1:1 и 1:2 (рис. 81). При темпе-
ратуре 20° С в среде перхлората натрия (р = 1,0) и общей концент-
160
ации ванадия 4,18-10~4 г-атом! л, малоновой кислоты 4-10~2 моль! л
в интервале —1g [Н+]= 1,58-н 4,92 определены их константы устой-
чивости: р! = 3,6-101 и 3,35-106. Имея в виду различие в ион-
ной силе растворов, следует признать, что между величинами рь
полученными в работах [364] и [365], наблюдается хорошее соответ-
ствие.
Известно, что существует корреляция между константами устой-
чивости комплексов и константами диссоциации соответствующих
Рис. 81. Распределение иона УОф в систе-
ме УО+ — СН2 (СОО)2“ — Н2О.
1 _ vot; 2 -- VO2 [СН2(СОО)2]~;
3 — VO2 [СН2 (СОО)2]3-
4W
кислот: анионы слабых кислот образуют более устойчивые комплек-
сы, а сильных — менее устойчивые (табл. 34).
Эта закономерность прослеживается на примере муравьиной
и уксусной кислот, однако при переходе к щавелевой и малоновой
Нарушается. Подобный эффект наблюдается для многих других
металлов и объясняется наибольшей устойчивостью пятичленных
циклов, образуемых металлами с алифатическими лигандами [366],
что указывает на циклическое строение оксалатных и малонатных
комплексов ванадия (V). Комплексы щавелевой кислоты содержат
пятичленные циклы, обладающие большей стабильностью, чем шести-
членные, которые образует малоновая кислота. Сопоставление ука-
зывает, по-видимому, также на большую достоверность спектро-
фотометрических данных, так как на основании кинетических
исследований в работах [362] и [364] для величины Ki оксалатного
11 Заказ № 256
161
Таблица 34
Константы кислотной диссоциации
кислот и константы устойчивости
комплексов с ванадием (V)
(р, =1,0; t =20°С)
1— . - —- Кислота pKi РК2 IgPi
Муравьиная 3,54 * 1,36
Уксусная 4,59 2,28
Щавелевая — 3,32 4,88
Малоновая — 4,96 4,56
По данным Муллика и Гхоша с
комплекса получено меньшее
значение по сравнению с Д
малонатного (0,98-103 и 1,2\
X105 соответственно). В то же
время значения рх для оксалат-
ного комплекса, полученные
в работах [305] и [363] при
использовании различных
вариантов спектрофотометри-
ческого метода, весьма близки.
Известны комплексные сое-
динения ванадия (V) с поли-
диоксикарбоновыми кислотами
(яблочной, винной, слизевой),
соавторами [367, 368], ванади-
евая кислота, полученная пропусканием растворов ванадата аммония
через ионообменную смолу Амберлит — IR 120 в Н-форме, образует
с винной кислотой комплекс красного цвета. По методу Джоба из
спектрофотометрических и pH-метрических измерений для него
установлен состав 2:1. Авторы приводят реакцию образования
комплекса:
-НО\ /ОН-
НО—V— он
_HOZ хон_
НО—СН—СН—он
+но— С С—ОН+
о о
"НО/ /ОН'
но—V—он
_нох хон_
-НОХ /О—СН—СН—/ОН-
НО—V—ОН НО—V—он
_НОУ 4 О—с с—о7 хон_
2
(71)
и величину его константы устойчивости, 7,35-104.
В работе [364] приводятся данные о составе и константах устой-
чивости комплексов ванадия (V) с кислотами: винной VO2C4O6H5
и VO2(С4ОбН5)Г, ^=(2,3±0,2). 102 и tf2=(l,6±0,l). 102; яблочной
VO2C4O5H5 и VO2(C4O5H5)T, 7С1=(4,9±0,9)-102 и /<2-(3,3±
±0,7). 102; слизевой VO2C6O8H9, К= (1,9±0,4)-103. Как видно,
в данном случае предполагается образование комплекса с одноза-
рядными анионами оксикарбоновых кислот.
Из числа оксикарбоновых кислот ароматического ряда как
комплексующие агенты по отношению к пятивалентному ванадию
исследованы салициловая и сульфосалициловая кислоты. А. И. Вол-
кова и Т. Е. Гетьман [369] на основании спектрофотометрических
исследований пришли к заключению, что в водно-спиртовых раство-
162
pax при pH 2 с салициловой кислотой образуется розовый комплекс
состава 1 : 1, а в водных в присутствии большого избытка салици-
лата при pH 2—4 — синий состава 1 :2. Строение этих комплексов
изображено следующим образом:
Сульфосалициловая кислота при pH 2—4 образует также комплекс
1 :2 синего цвета. Определены константы диссоциации этих комп-
лексов:
_ [Усвоб] [Sal2-]2
vo:Sa*2 |vosai7
^VOSSal2 =
[Усвоб] [SSaP~]2
[VOSSal^]
= 1,4-10-22,
= 5,8-10-22.
Поскольку при расчете констант диссоциации не учитывалось ион-
ное состояние ванадия, не связанного в комплекс, то эти данные
имеют весьма приближенный характер.
Выше (см. главу II) спектры ионов ванадия (V) были интерпре-
тированы как системы полос переноса заряда с общих орбит атомов
кислорода на орбиты, локализованные преимущественно на атоме
металла. Аналогично происхождение спектров поглощения неокра-
шенных комплексов пятивалентного ванадия. В литературе описаны
электронные спектры комплексов ванадия (V) различного состава
с муравьиной, щавелевой, малоновой, лимонной, сульфосалициловой
кислотами [82, 85, 305, 365].
Характерной особенностью спектров поглощения комплексов
ванадия (V) с лигандами, координированными через кислород,
является то, что спектры этих комплексов по своей конфигурации
в основном повторяют спектр оксоаквакомплекса ванадия (V)
(рис. 82). В них наблюдается та же система полос, что и в спектре
оксоаквакомплекса. Как правило, наблюдаемые полосы поглощения
отличаются большей интенсивностью, а их максимумы, в зависимо-
сти от природы лиганда, более или менее сдвинуты в длинноволно-
вую область. Весьма схожие спектры поглощения имеют оксалатные
и малонатные комплексы состава 1:1 и 1:2 (см. рис. 82, б).
В этом случае интенсивность поглощения возрастает пропорцио-
нально количеству присоединенных лигандов.
Банерджи и Дей [370], Сез и Дей [371] изучили комплексо-
образование ванадия (V) с ализарином красным S. Методом непре-
рывных изменений и молярных отношений, измеряя электропровод-
ность и оптическую плотность растворов при pH 3,5, они устано-
11*
163
вили, что комплекс содержит ванадий и ализарин в отношении
1:1, при температуре 25° С и ионной силе р = 0,1 (NaC104) лога-
рифм константы устойчивости равен 8,5 + 02.
Анион этилендиаминтетрауксусной кислоты (ЭДТА) является ли-
гандом, для которого были получены первые количественные харак-
теристики комплексообразования ванадия (V) в водных растворах.
Пексок и Джувет [372], Мика и Токштейн [373] на основании
полярографических исследований пришли к выводу, что ванадий (V)
с ЭДТА (обозначим ее Н4А) образует комплексы. Количественную
характеристику рассматриваемых комплексов дали Рингбом с соавто-
рами [374], основываясь на результатах спектрофотометрических
измерений. Исследования проводили в перхлоратной среде (pt==O, 1)
при температуре 20° С и концентрациях ванадия и ЭДТА от 10~4
до 5-10~3 моль/л в интервале pH 0,85—4,40. Установлено, что
при pH <3,5 образуется комплекс VO2HA2-, логарифм константы
кислотной диссоциации которого равен —36,0, а логарифм констан-
ты равновесия реакции его образования
VOt + НА3- 71VO2H А2- (72)
равен 11,39. Судя по кривой светопоглощения раствора ванадия
в присутствии ЭДТА, в интервале pH 3,85 — 9,0 устойчив комплекс
VO2A3- (1g 18,05), при pH 9,0, как считают эти авторы, обра-
зуются пиро- и ортованадаты. Шайо [375] пришел к несколько дру-
гим выводам относительно комплексообразования в рассматриваемой
системе. Он определял кажущиеся константы равновесия, добавляя
в присутствии дифенилкарбазона к раствору ванадия [V) и ЭДТА
металлы- комплексообразователи (Zn2+, Mn2+, Mg2+) с известными
константами комплексообразования. По данным [375], в интервале
pH 1,0—11,0 образуется комплекс, содержащий ванадий (V) и
ЭДТА в отношении 1:1, логарифм кажущейся константы образо-
вания которого в области pH 1,0—3,7 растет (от 3,4 до 7,0);
в области pH 3,7—6,0 он имеет постоянное значение (7,07); с даль-
нейшим ростом pH от 6,0 до 11,0 его величина уменьшается (до
1,12). Наблюдаемые результаты Шайо описывает следующей схемой:
в области низких pH
VO3++H2A2- 71 VOA~+2H+; (73)
при pH 3,5
VOt+H2A2~Zi VO2A3~+2H+; (74)
в интервале pH 3,5—6,0
VO74-HA3- 71 VO2A3-+OH“.
(75)
В 1965 г. Прциборовский с соавторами [376] вновь исследовали
рассматриваемую систему. Спектрофотометрическим методом в среде
164
Рис. 82. Спектры поглощения формиатного,
оксалатных и малонатных комплексов вана-
дия (V).
КС101=0,1) определены константы равновесия реакций комплекс^
образования: .
IgK
VO++A4-ztVO2 А3-............ 15,55 (76)
VO++HA3~72VO2HA2~........... 9,60
VO^+H2A2-t2VO2H2A~.......... 6,93
VO++H3A-ztVO2H3A............. 5,60 (77)
VO2A3-+h+^±VO2HA2-........... 4,31 (78)
VO2HA2-+h+^VO2H2A_........... 3,49 (79)
VO2H2A_+H+7±VO2H3A........... 1,4 (80)
VO2A3“4-2OH_^HVO2-+HA3-. . . 6,48 (81)
На рис. 83 показана кривая функциональной зависимости величины
логарифма «кажущейся» константы устойчивости комплексов ванадия
Рис. 83. Зависимость кажущейся константы
устойчивости комплексов ванадия (V) с ЭД ТА
от pH раствора.
с ЭДТА от pH раствора. Величина кажущейся константы устойчивос-
ти рассчитана по уравнению
S [УО2Н^-3] >
[V]'.S[H;A>-4
где [V]' — общая концентрация ванадия, не связанного в комплек-
сы. Кривая показывает, что эти комплексы наиболее устойчивы
в области pH 3—6 и разрушаются как в кислых, так и в щелочных
Z
166
•астворах. Авторы работы [376] предполагают, что анион VO2H2A
имеет структуру
НООС—СН2 СН2---СН2 СН2—СООН-
В твердом состоянии выделены комплексные соединения H3[VO2A],
Na3[VO2A]-3H2O, (NH4)3[VO2A], К [ VO2H2A] • ЗН2О, NH4[VO2H2A]-
• ЗН2О. Эти данные вполне удовлетворительно согласуются с ре-
зультатами Рингбома [374], хотя последний получил более высокие
значения 1g и 1g К2- В то же время данные Шайо [375] для
интервала pH 1,0 — 3,5 вызывают сомнения, поскольку в условиях
относительно низких концентраций ионов Н+ существование иона
VO3+ и соответственно комплекса VOA- маловероятно.
КОМПЛЕКСЫ С ДРУГИМИ ОРГАНИЧЕСКИМИ ЛИГАНДАМИ
Многочисленные работы, посвященные исследованию состава,
условий образования, свойств комплексных соединений пятивалент-
ного ванадия с различными классами органических лигандов, носят
в основном препаративный характер и направлены на изыскание
новых высокочувствительных реакций на ванадий.
Значительное внимание уделено изучению взаимодействия вана-
дия (V) с гидроксамовыми кислотами. Дутта с соавторами
[377—379] синтезировали многочисленные окрашенные комплексы
ванадия (V) с различными замещенными гидроксамовыми кислотами.
Было найдено [377], что бензгидроксамовая кислота C7H6NO2 в вод-
ной среде при pH 3—4 реагирует с метаванадатом аммония с обра-
зованием нерастворимого в воде комплекса темно-фиолетового цвета.
На основании химического анализа ему приписана формула
[VO(OH)(C7H6NO2)2]. Комплекс растворяется в кипящих спиртах,
давая красные растворы, из которых выделяются кристаллы соеди-
нения [VO (OX) (C7H6NO2)2], образуемого по схеме [378]
167
где НОХ — метиловый, этиловый или пропиловый спирты. Методом
Джоба установлено, что в водно-спиртовой среде при pH 3—5,
устойчив комплекс состава 1 :3. Эти же авторы [378, 379] с нитро-
хлор-, фтор- и различными алкилзамещенными гидроксамовыми
кислотами получили многочисленные соединения приведенных выше
типов: оксогидроксокомплексы VO (ОН) (RH)2 и алоксокомплексы
VO(OX)(RH)2. Электронные спектры всех синтезированных комп-
лексов в этаноле имеют максимум поглощения в области 420—
480 нм и коэффициент экстинкции еМах— 1000—2000.
По данным Минчевского и Скорко-Трибула [380], тиофено-
гидроксамовая кислота при pH 3 образует с ионами VO^ комплекс
состава V:R=1:3, а при pH 7,7 в состав комплекса входит ион
H2VO7, а отношение V:R^>3. А. Л. Маркман с соавторами [381]
установили, что в водно-метанольных растворах (1:4) 2-окси-З-
нафтогидроксамовая кислота с ванадием (V) образует два комплекса:
темно-синий 1:1с Х,маХ=540юи и коричневый 1:2 с ХМах=450юи,
величины ступенчатых констант устойчивости которых соответствуют
рД\=^3,58 и р/С2—3,20. По их мнению, эти комплексы имеют сле-
дующую структуру:
Реакцию образования оранжевого комплекса ванадия (V) с капро-
гидроксамовой кислотой при pH 1—5 в водно-ацетоновой среде пред-
ложено использовать для спектрофотометрического определения ва-
надия [382]. Позднее на основании спектрофотометрических измере-
ний [383] высказано предположение, что при pH 1,2 и ниже
комплексы ванадия (V) с капрогидроксамовой кислотой в зависимо-
сти от соотношения реагентов имеют состав 1 : 1 или 1 :2 и струк-
туры:
(ОН)2,
168
3
(ОН) (Н2О).
2 J
Интенсивно окрашенные соединения образуются при взаимодей-
ствии в водной среде ионов ванадия (V) с ортофенолкарбоновыми
кислотами и ортофенолами [384—392]. Свойства этих соединений
наиболее подробно исследованы в работах С. Я. Шнайдермана с со-
авторами [385—392]. Наибольшего развития процесс комплексо-
образования в системах ванадий (V) — ортофенолкарбоновые кислоты
или ортофенолы достигает в слабокислых и нейтральных средах.
Предполагается, что за счет нейтрализации водорода спиртовых
групп образуются устойчивые хелатные соединения, в которых ион
VOt связан с электроотрицательными атомами кислорода спиртовых
групп.
Протокатеховая (3,4-диоксибензойная) кислота [386] образует
синие комплексы состава 1:1 и 1:2 анионного характера. Предпо-
ложительно формула комплекса 1:1
сосг
-I
о—vo2
Аналогичного состава соединения образуются в присутствии
галловой кислоты и пирогаллола. При взаимодействии ванадия (V)
с пирогаллокарбоновой (2, 4, 3-триоксибензойной) кислотой в рас-
творе наряду с комплексообразованием протекает окислительно-вос-
становительная реакция и образуется смесь комплексов ванадия (V)
и (IV) [387, 388].
Ванадий (V) с пирокатехином образует несколько окрашенных
комплексов [389]. Комплекс состава 1 : 1 розового цвета (^х^бОО нм)
образуется при pH 1—3 за счет вытеснения водорода одной спир-
товой группы
В интервале pH 3—10 устойчивы синие комплексы 1:1 и 1:2,
в спектрах поглощения которых имеется два максимума при X—380
и 580 нм. Установлено, что образование комплексов протекает
с выделением двух ионов водорода, а опыты по электромиграции
169
и сорбции на ионообменных смолах показали у комплексов отрица^
тельный заряд. Им приписано следующее строение:
Рассчитаны их константы диссоциации, соответственно
[vo+] [Сен4р2-
1 [vo2c6h4O7‘
=2,2-10-12
С хромотроповой кислотой
плексы состава 1 Г1 и 1 : 2 с м;
[390]. Предполагается, что оС
идет вследствие замещения во;
в пери-положении
[VOH-][C6H4O2-]2
[VO (С6Н4О2)-]
= 4,58-10~24.
ванадий (V) образует желтые ком-
ссимумом поглощения при Х=370/ш
•азование комплексов в этом случае
уродов спиртовых групп, находящихся
экстракт которого окрашен в розовый цвет [391]. Предполагаемая
структура комплекса представлена схемой
—О . .Антипирин
HQ—VO2
В^интервале pH 3—7 при большом избытке пирокатехина протекает
ступенчатое образование тройных комплексов 1:1:1 и 1:2:1, вод-
ные растворы и дихлорэтановые экстракты которых окрашены в си-
ний цвет. При этом предполагается, что в дихлорэтановых экстрактах
тройные комплексы являются продуктами ионной ассоциации катиона
антипириния с существующими в водных растворах двойными пиро-
катехинатными комплексами ванадия:
Н+«Антипирин
VO2
Определены константы диссоциации этих комплексов
Vo+] [HChps-]
[VO2HChp2—]
=5,4-10-6,
[VO3+] [HChp3-]2
[VO (HChp)®-]
= 1,65-ю-10.
Фенольные комплексы ванадия (V) реагируют с органическими
основаниями, в частности, с антипирином и его производными, давая
интенсивно окрашенные смешанные комплексы. Такие соединения
отличаются малой растворимостью в воде и хорошей в органических
растворителях.
В системе ванадий (V) — пиpv/XYU. 1 V-Anil-Clil 1 И11Ир'
2—4 образуется тройной комплекс состава 1:1:1
В системе ванадий (V) — пирокатехин-антипирин в области pH
гтт^л 1 1 . 1, дихлорэтановый
и
Н+- Антипирин.
Комплексные соединения ванадия (V) с галловой, пирогаллокар-
боновой и 3,4-диоксибензойной кислотами реагируют с антипирином
в узком интервале кислотности (pH 3,5), образующиеся соединения
содержат ванадий, фенолкарбоновую кислоту, антипирин в соотно-
шении 1:2:3 [392].
Дутта и Лайри [393] синтезировали комплексы ванадия (V)
с 3-гидрокси-1,3-дифенил-триазеном
Ими получены два изомера пиридин-диоксо-моно-3-гидрокси-1,3-ди-
фенил-триазендиоксованадия [VO2 (C12H10N3O) (C5H5N)] золотисто-
желтого и темно-зеленого цвета; а-пиколин-диоксо-моно-3-гидрокси-
1,3-дифенил-триазендиоксованадия [VO2 (C12H10N3O) (CH3C5H4N)] и
анил ин-д иоксо-моно-3-гидрокси-1,3-дифенил-триа зенд и оксованадия
[VO2(C12H10N3O) (C6H5NH2)], оба золотисто-желтого цвета. В данном
случае координация лигандов с ионом VOj" осуществляется через
кислород гидроксильной группы и азот.
170
171
В работе [394] Дутта и Симал описали получение оксо-этокСск
бцг-салициламидоксимата ванадия (V) [VO (ОС2Н5) (C7H7O2N2)2] жед
того цвета и диоксо-а-нитрозо-(3-нафталата и диоксо-р-нитрозо-ц
нафталата ванадия (V) [VO2 (C10HgO2N)], окрашенных в темно-крас
* ный цвет. I
А. К. Бабко с соавторами [395] спектрофотометрическим мето-
де м изучили комплексы манганона с ванадием (V). Установлено
что при Су = 4 • 10-5 моль/л и концентрации манганона 4 • 10"5
— 5,2-10~4 моль!л при pH 3 образуются комплексы 1:1, а при
pH 5 комплексы 1 :2. При изучении экстракции их хлороформом
найдено, что комплекс 1:1 катионного характера, а 1:2 — анион-
ного. Приведены реакции образования комплексов
Установлено образование комплекса ванадия (V) с 4-(2-пиридилазо)-
резорцинолом
ОН
172
красного цвета, состава 1:1 в буферном фосфатном растворе при
pH 5,25—6,20 [396]. Данную реакцию предложено использовать
для спектрофотометрического определения ванадия [396, 397]. Ана-
логичного состава комплекс нашли А. К. Бабко с соавторами [398]
в системе ванадий (V) — 4-(2-пиридилазо)-резорцинолгуанидин.
В. Г. Жарковский и А. Г. Пилипенко [399] изучили условия
экстрагирования хлороформом комплекса ванадия с N-бензоилфенил-
гидроксиламином и предположили, что комплекс имеет состав
^О3 (C13H10O2N)4.
По данным Монтегю [400], N-бензоил-гидроксиламин образует
хелатные комплексы с псевдокислотными свойствами состава
(C13H10O2N)2.VO2H, (C13H10O2N)2.VO2CH3 и (C13H10O2N)2-VO2H-HC1.
Подобные комплексы образуют и салицил альдоксим, ацетилацетон,
бензоилацетон, изохинолин, гидразид и салицилизен-1,3-диамино-
пропан. Комплексообразование ванадия (V) с тетрабром-пирокате-
хин-дифенилгуанидином исследовали А. И. Бусев и 3. П. Карякина
[401].
Для спектрофотометрического определения ванадия предложено г
также использовать его комплексы с метилтимоловым синим состава [
1 :2. Комплекс устойчив при pH 2,6—3,8 и характеризуется
^niax"580 — 585 нм, е=1,9-104 [402]. Известно также об образо-
вании ванадием (V) комплексов с углеводами: сахаром, глюкозой,
декстрином в присутствии NaOH с соотношением ванадий: углевод-
1 : 1,8 [403].
Для фотоколориметрического и спектрофотометрического методов
определения ванадия в различных материалах и условиях предло-
жены другие многочисленные реакции комплексообразования (V)
с разнообразными органическими реагентами. К ним можно отнести
N-бензоилфенилгидроксиламин [404], И-2-тиофенкарбонил-И-п-толил-
гидроксиламин и Ы-2-тиофенкар5онил-П-фенилгидроксиламин [405],
а-нафтиламин [406], оксихинолин [407], диантипирилвинилбензоме-
тан и диантипирил-3,4-диметоксифенилметан [408], морин [409],
N-фу роилфенил гидроксиламин [410], 8 — гидроксихинолин [411 ],
протокатехиновая кислота [412], N-бензоил-фенил-гидроксиламин
[4133, 1-(0-каРбоксифенил)-3-гидрокси-3-фенилтриазен [414] и др.
ОСОБЕННОСТИ КОМПЛЕКСООБРАЗОВАНИЯ ВАНАДИЯ
Для пятивалентного ванадия характерно образование связей
с кислородом, и комплексные соединения с органическими и неор-
ганическими лигандами, координированными через кислород, весьма
распространены. В то же время известно лишь небольшое число
комплексов с лигандами, координирующимися с ванадием посредством
атомов азота и кислорода. Из соединений этого класса комплексы
с этилендиаминотетрауксусной кислотой отличаются большой устой-
чивостью. Из галоидосодержащих комплексов, по-видимому, только
фторидные обладают достаточной прочностью и могут быть надежно
обнаружены в водных растворах. Что касается соединений с лиган-
173
дами, содержащими в качестве донорных атомов только атомы азота
то сведения о них полностью отсутствуют и, естественно, нельзя
судить об устойчивости таких соединений.
В табл. 35 приведены значения констант устойчивости комплек-
сов ванадия (V) в водных растворах. Как видно, количественные
характеристики реакций комплексообразования имеются только для
лигандов, координированных через кислород, не считая ЭДТА,
а также для фтора и хлора. Но несмотря на ограниченность сведе-
ний, сейчас можно выявить некоторые общие закономерности комп-
лексообразования ванадия (V) и оценить его комплексообразующую
способность.
Ступенчатые константы устойчивости для ряда одноядерных
комплексов, образуемых ионами металла с одинаковыми лигандами,
обычно убывают по величине. Это можно проследить на примере
фторидных, малонатных и оксалатных комплексов, однако у фос-
фатных комплексов первая и вторая ступенчатые константы устойчи-
вости весьма близки.
Обычно сопряженные основания сильных кислот образуют с ио-
нами металлов слабые комплексы, а сопряженные основания слабых
кислот — прочные комплексы [366]. Из числа исследованных с вана-
дием анионы одноосновных кислот, расположенные в порядке убыва-
ния силы этих кислот, образуют ряд NOF~C1~>F~>НСОО~>
>СН3СОО~, а относительная устойчивость комплексов ванадия (V)
с теми же лигандами увеличивается в направлении NO7<CI~<
< HCOO~<CH3COO~<F~. Несоответствие между устойчивостью
фторидного комплекса и силой плавиковой кислоты, по-видимому,
объясняется различием в природе лигандов, взятых для сравнения;
в то же время в ряду F~>C1~ устойчивость комплексов падает.
Аналогичные ряды для многоосновных кислот выглядят следующим
образом:
SO*- > С2О*- > СН2(СОО)|- > НРО*- > ЭДТА4-,
SO* < НРО*-<СН2(СОО)2-< С2О1~<ЭДТА4-.
Причины, обусловливающие несоответствие между силой щавелевой
и малоновой кислот и устойчивостью соответствующих комплексов,
обсуждены выше. Положение фосфорной кислоты в ряду многоос-
новных кислот можно объяснить, например, тем, что если она и
образует циклы, то они могут быть только четырехчленными, т. е.
менее устойчивыми, чем пяти- или шестичленные.
Другое доказательство циклического строения оксалатных и ма-
лонатных комплексов связано с явлением хелатного эффекта, заклю-
чающегося в том, что, как правило, хелатные комплексы более
устойчивы, чем их нехелатные аналоги [366]. В нашем случае под-
ходящей парой для сравнения могут служить комплексы формиат-
ный 1:2 и оксалатный 1:1. Константа устойчивости формиатного
комплекса 1 : 2 неизвестна, но малая устойчивость формиатного комп-
лекса 1 : 1 (Рх=23,8) позволяет предполагать, что она также неве-
174
175
СП
12 Заказ № 256
Окончание табл. 35
Кислота, соответствующая лиганду Комплексный ион Метод t, °C ц (среда) Igp Литература
Щавелевая 1 vo2c2oy Спектрофотометри- ческий 20 l,0(NaC!O4) 5,08 [305]
vo2c2oy To же 20 l,0(NaC104) 4,87 [363]
VO2(C2O4)^ » 20 ->0 10,2 [363]
VO2(C2O4)3- Кинетический 25 0,2 4,76 [362]
V O2(C2O4)2 Спектрофотомет- 20 !,0(NaClO4) 8,49 [363]
Малоновая рический
VO2CH2(COO)y Кинетический 25 ->0 5,08 [364]
vo2ch2(coo)7 Спектрофотометри- 20 1,0 (NaClOJ 4,55 [365]
ческий
VO2[CH2(COO)2]3- То же 20 l,0(NaC104) 6,52 [365]
Винная H2[V2(OH)8C4O6H4] То же — — - 4,86* [368]
VO2C4O6H5 Кинетический — -0 2,36* [364]
V O2(C4O6H5)2 То же - - ' -^0 4,57* [364]
Яблочная vo2c4o5h5 » — ->0 2,69* [364]
vo2(c4o5h5)7 » — ->0 5,21* [364]
Слизевая VO2C6O8H9 » - - ->0 3,28* [364]
Салициловая VOSalC Спектрофотометри- 23 0,2(KNO3) 21,85 [369]
ческий
Сульфосалициловая VOSSaly То же 23 0,2(KNO3) 21,23 i [369]
Ализарин красный S 1 : 1 » 0,l(NaClO4) 1 8,5 [371]
"4
3,58 [382]
2-окси - 3 - нафтогидроксамовая кислота VO2C11OsNH8 Спектрофотомет- рический 6,78 [382]
VO2(CuO8NH8)7 То же
20 0,1 (NaClOJ 18,05 [374]
vo2a° »
vo2a3- » 20 0,1 (KC1) 15,55 [376]
vo2ha2~ » 20 0,1 (NaClO4) 11,39* [374]
vo2ha2- » 20 0,1 (KC1) 9,6* [376]
• vo2h2a~ » 20 0,1 (KC1) 6,93* [376]
vo2h3a“ » 20 0,1 (KC1) 5,6* [376]
11,65 [389]
VO2C6H4Oy » ZU
Пирокатехин 90 23,34 [389]
vo(c6h4o2)7 » ZU
90 - —— 5,27* [390]
Хромотроновая кислота . . • VO,HChp2- » 90 9,78* [390]
VO(HChp)|— » 1 zu
* Лигандом является протонированный ион кислоты.
лика. В то же время оксалатный комплекс 1 : 1 отличается значи
тельной устойчивостью (|3Х = 1,2 • 105).
Чтобы оценить склонность пятивалентного ванадия к комплексе
образованию, можно сравнить константы устойчивости некоторых
изученных комплексов его с константами устойчивости аналогичных
комплексов других металлов [299]. В первую очередь это сопостав-
ление необходимо провести по отношению к ближайшим аналогам
----tz , выполнить эту задачу
эти элементы в кислых
' и Ta(OH)t и
либо с образованием гидроксоацидокомплек-
тельной устойчивостью = 1,2 • 105).
образованию, можно сравнить константы устойчивости некоторых
изученных комплексов его с константами устойчивости аналогичных
комплексов других металлов [299]. В первую очередь это сопостав-
ванадия — ниобию и танталу. К сожалению,
не представляется возможным, так как г"
растворах образуют гидроксокомплексы NbO(OH)t и Та(ОН)^ и
реакции комплексообразования, как правило, происходят либо с вы-
теснением ионов ОН“, .
сов МОл(ОН)тАГ [415, 416].
Таблица 36
Константы устойчивости комплексов ванадия (V) и нептуния (V)
Центральная группа " —• —
Кислота Соста в комплек- сов М:А в уо+ lg₽/ Литература в NpO^ Литература
Соляная . . . Уксусная . . . Щавелевая . . ЭДТА .... * Для РиО^~ 1:1 1:1 1:1 1:2 1:1 1:1 • 1,01 1,0 0 0 0,1 0,1 —0,377 2,28 6,64 10.20 18,05 15,55 [305] 305 363 363 374 376] 0 0 0,05 —0,17* 1,08 3,92 7,36 9,70 [417] 301 301 301 [301
Более всего данных для сравнения имеется по комплексам неп-
туния (V) который также существует в виде диоксокатиона NpO2+
Из табл. 36 и рис. 84 следует, что ванадий образует более устой-
чивые комплексы, чем нептуний, и между логарифмами констант
устойчивости обоих металлов наблюдается линейная корреляция.
Ион NpO2, подобно другим ионам типа MO^h, характеризуется
симметричной линейной (или близкой к линейной) структурой
О М О и, подобно уранилу, способен образовывать прочные эк-
ваториальные связи перпендикулярно М—О. По мнению Э. Т. Чу-
динова и И. К. Швецова [299], на прочность комплексов, кроме
того, влияет распределение зарядов в оксокатионе. Последнее та-
ково, что при образовании связей на каждый атом лиганда в эква-
ториальной плоскости катиона действует эффективное поле от всего
сложного иона, по величине приблизительно такое как будто на
нептунии сосредоточен заряд +2. ’
178
Аналогичные соображения справедливы при рассмотрении комп-
лексов ванадия (V). Ион VOj", как и ион NpOf, склонен к обра-
зованию прочных ковалентных связей в экваториальной плоскости,
перпендикулярной оси О—V—О [86]. Это обстоятельство, а также
тот факт, что радиус иона ванадия (V), равный 0,57 А [418], мень-
12
Рис. 84. Зависимость lg₽n(MAn) от lgpn(VO2A„)
для комплексов с различными лигандами при р=1,0:
1 — нитрат; 2 — хлорид; 3 — сульфат; 4 — ацетат; 5 —фто-
рид; 6 — малонат; 7 — оксалат (ц=1,0), 8 — оксалат (ц->
->0); 9 — оксалат (1g р2, ц,= 1,0); 10 — ализарин S (1g |32);
11—оксалат (lg р2, ц->0), M=Z — NpO^, II — La3+,
III — Lu3 + , IV—прямая с наклоном, равным единице.
те радиуса иона нептуния (V), 0,88 А [419], определяет повышенную
устойчивость комплексов пятивалентного ванадия.
Поскольку в водных растворах ванадий (V) существует в виде
оксоакваиона VO2(H2O)t, то комплексообразование следует рассмат-
ривать, как процесс замещения лигандами молекул воды, располо-
женных, во внутренней сфере иона УО2(Н2О)^:
VO2(H2O)t] +пкт-^ [VO2(H2O)4_„An-"m] +«н2о.
По своей комплексообразующей способности ванадий (V) ближе
всего к ионам тяжелых трехвалентных лантанидов. При срав-
нении комплексообразующей способности ванадия (V) и трехва-
лентных ионов лантана и лютеция на графике в координатах
lgPn(VO2AJ/lgP^(MA/2) (см. рис. 84) получаются две прямые. Наклон
12*
179
прямой для лантана меньше единицы (0,85), а для лютеция несколько
больше (1,06). На основании этих данных, разумеется, нельзя ут^
верждать, что между пятивалентным ванадием и лантанидами есть
полная аналогия, так как сравнение проводилось для ограниченного
числа типов лигандов, а именно для лигандов, координированных
через кислород, и для галогенидов. Надо иметь в виду и различие-
в характере химической связи в комплексах ванадия (V) и редко-
земельных элементов. Высказанные здесь соображения по поводу
комплексообразующей способности носят предварительный характер
из-за недостатка данных по константам устойчивости комплексов
пятивалентного ванадия. Однако такая оценка является полезной,,
она позволит предсказать свойства еще не исследованных комплек-
сов и более широко использовать их на практике. И
Из приведенного обзора видно, что в области комплексообразо-
вания пятивалентного ванадия предстоит еще многое сделать, чтобы
выяснить природу даже простейших соединений.
ЛИТЕРАТУРА
1. М. II. Соболев. Извлечение ванадия и тигана из уральских титано-
магнетитов. М.— Л., ОНТИ, 1936.
2. У. Ростоке р. Металлургия ванадия. М.,- Изд-во иностр, лит., 1959»
3. J. Meyer. Z. Electrochem., 1909, 15, 26.
4. L. Moser, О. Brandl. Monatsh, 1929, 51, 169.
5. В. Д. Пономарев, В. 3. Тарасенко, А. И. 3 а з у б и н, В. IL
Беспалов. Авт. свид. № 198306 от 29/IV 1967.
6. Д. И. А б и hi е в, В. Д. Пономарев, В. С. Мальце в, И. И. С и -
р о к о. Тр. Ин-та металлургии и обогащения АН Каз. ССР, 1964, 11,85.
7. Н. Gobrecht, G. И е i n s о 1 а г. Z. Phys., 1957, 147, 350.
8. Z. FI. В г i х n е г, Р. Q. Flournoy. J. Electrochem. Soc., 1965. 112, 303..
9. R. A. G u s t i s о n, F. W. Hurd, R. M. Fowler. Пат. США 3131993.C.A.,
1956, 50.
10. E. H. G r a b t г e с, V. E. Padilla. J. Less-Common Metals, 1961, 3,437.
11. R. R. Swanson H. N. Punning, J. E. FI a u s e. Engng and Mining*
1961, 10, 162.
12. В. С. Сырокомский, Ю. В. Клименко. Ванадатометрия. М.,
Металлургиздат, 1950.
13. У. А. Уотерс. Механизм окисления органических соединений. М.,
«Мир», 1966.
14. J. S е 1 b i n. Chem. Rev., 1965, 65, 153.
15. А. С. Гончаренко. Электрохимия ванадия и его соединений. М.*
«Металлургия», 1969.
16. С. F. R a m m е 1 s b е г g. Pogg. Ann., 1856, 98, 249.
17. FI. E. Roscoe. Phil. Trans., 1868, 158, 1.
18. P. Dullberg. Z. phys. Chem.. 1903, 45, 129.
19. L. G. S i 11 e n. Acta Chem. Scand., 1954, 8, 299.
20. T. J. C. R о s s о 11 i, H. R о s s о 11 i. Там же, 1956, 10, 957.
21. H. T. S. Britton, R. A. Robinson. J. Chem. Soc., 1930, 1261.
22. L. P. D u c r e t. Ann. Chim. France, 1951, 6, 705.
23. И. T. S. Britton, R. A. Robinson. J. Chem. Soc., 1932, 1955.
24. И. T. S. Britton, R. A. Robinson. Там же, 1930, 2328.
25. P. Sou ch ay, R. Scholl. Bull. Soc. Chim. France, 1950, 17, 824.
26. L. N e w m e n, W. J. La Fleur, F. J. В г о u s a i d e s, A. M. Ross;
J. Amer. Chem. Soc., 1958, 80, 4491.
27. N. Ingri, F. Brito. Anal. Real. Soc. Espan. fis. у quim., 1960, 56B, 165.
28. Ю. И. Санников, В. Л. Золота вин, И. Я. Безруков. Ж- пе-
орг. хим., 1963, 8, 923.
29. G. S с h w а г z е n b а с h, G. Geier. Helv. Chim. Acta, 1963, 46, 906.
30. G. J a n d e r, K. F. J a h r. Z. anorg. Chem., 1933, 211, 49.
31. G. J a n d e r, K. F. J a h г. Там же., 1933, 212, 1.
32. P. S о u c h a у. Bull. Soc. Chim. France, 1951, 18, 932.
33. R. U. R u s s e 1, J. E. Salmon. J. Chem. Soc., 1958, 4708.
34. G. Schwarzenbach, G. Parissakis. Helv. Chim. Acta., 1958, 41*
2425.
35. N. I n g r i, F. Brito. Acta Chem. Scand., 1959, 13, 1971.
36. G. J a n d e r, T. Aden. Z. phys. Chem., 1933, 144, 197.
37. R. A. Robinson, D. A. Sinclair. J. Chem. Soc., 1934, 642.
38. P. S о u ch a y, G. Carpeni. Bull. Soc. Chim. France, 1946, 13, 1601
39. P. S о u c h а у. Там же, 1947, 14, 914.
40. К. Schiller, E. Thilo. Z. anorg. Chem., 1961, 310, 261
41. O. W. Howarth, R. E. Richards. J. Chem. Soc., 1965, 864.
181
42. J. V. Hatton, Y. Saito, W. G. Schneider. Canad. J. Chem., 196S
43, 47.
43. H. Brinzinger, J. Wallach. Z. anorg. Chem., 1935, 244, 193.
44. R. Trujillo, E. Tejera. Anal. Real. Soc. Espan. fis. у quim., 1954
50B, 399.
45. L. S h о e p p. Dissertation, Techn, Univ. Berlin, 1959. Цит. no [46].
46. K. F. J a h r, L. S h 0 e p p. Z. Naturforsch., 1959, 14b, 467.
47. A. W. Nauman, C. J. H a 11 a d a. Inorg. Chem., 1964, 3, 70.
48. K. F. Jahr, J. Fuchs. Z. Naturforsch., 1959, 14b, 468.
49. K. F. Jahr, A. Schroth, J. Fuchs. Там же, 1963, 18b, 1133.
50. О. G 1 e m s e r, E. P r e i s 1 e r. Z. anorg. Chem., 1960, 303, 303.
51. J. Simon, K. F. Jahr. Z. Naturforsch., 1964, 19b, 165.
52. U. P. Stock, K. F. Jahr. Там же, 1963, 18b, 1134.
53. J. F. Hazel, W. M. McNabb, J. R. Santini. J. Phys. Chem., 1953
57, 681.
54. J. Lefebvre. J. Chim. Phys., 1957, 54, 567.
55. F. Ch auveau. Bull. Soc. Chim. Erance, 1960, 5, 810.
56. F. Brito, N. J n g r i, L. G. S i 11 e n. Acta Chem. Scand., 1964, 18, 1557.
57. N. Jngri. Там же, 1959, 13, 758.
58. G. C a r p e n i, P. Souchay. J. Chim. Phys., 1945, 42, 149.
59. R. Trujillo. Anal. Real. Soc. Espan, fis у quim., 1954, 50B, 533.
60. H. T. S. Britton, G. W e 1 f 0 r d. J. Chem. Soc., 1940, 764.
61. K. F. Jahr, L. Schoepp, J. Fuchs. Z. Naturforsch., 1959, 14b, 469.
62. J. M a g e 1, E. Richardson. J. Inorg. Nucl. Chem., 1960, 15, 272.
63. K. F. Jahr, J. Fuchs, F. Preuss. Chem. Ber., 1963, 96, 556.
64. O. G 1 e m s e r, E. P r e i s 1 e r. Naturwiss, 1959, 46, 474.
65. L. Newman, R. Quinlan. J. Amer. Chem. Soc., 1959, 81, 547.
66. I. Lukacs, C. L i t e a n u, C. S t г u s i e v i c i. Studii sicercetari chim. Acad.
RPR. Fil. Cluj, 1963, 14, 265.
67. D. Dyrssen, T. Sekine. Acta Chem. Scand., 1961, 15, 1399.
68. D. Dyrssen, T. Sekine. J. Inorg. Nucl. Chem., 1964, 26, 981.
69. К. Б. Я Ц и м и p с к и й, В. E. Калинина. Ж. неорг. хим., 1964, 9, 1117.
70. Ф. 3. Д ж а б а р о в, С. В. Горбачев. Там же, 1964, 9, 2399.
71. Т. F. Rutter. Z. anorg. Chem., 1907, 52, 368.
72. С. D. Coryell, D. M. Yost. J. Amer. Chem. Soc., 1933, 55, 1909.
73. H. T. S. Britton. J. Chem. Soc., 1934, 1842.
74. F. Foerster, F. Bottcher. Z. phys. Chem., 1930, 151, 321.
75. J. E. Carpenter. J. Amer. Chem. Soc., 1934, 56, 1847.
76. A. B. Hart, J. R. Partington. J. Chem. Soc., 1940, 1532.
77. J. Meyer, A. Pawletta. Z. phys. Chem., 1927, 125, 49.
78. J. B. Ramsay, E. L. Colichman, L. Pack. J. Amer. Chem. Soc.,
1946, 68, 1695.
79. M. J. La Salle, G. W. Cobble. J. Phys. Chem., 1955, 59, 519.
80. G. L. Bertrand, G. W. S t a p 1 e t о n, C. A. Wulff, L. G. Hepler.
Inorg. Chem., 1966, 5, 1283.
81. H. C. Myshra, M. C. R. Symons. J. Chem. Soc., 1962, 4411.
82. J. S. Littler, W. A. Waters. Там же, 1959, 1299 и. 4046.
83. R. S h a n k e r, S. N. Swami. J. Indian. Chem., 1963, 40, 105.
84. К. Б. Я ци мирский, Г. С. Николов. Ж. Физ. хим., 1970, 44.
1129 и 1400.
85. Р. R. В о n t с h е v, G. S. N i к о 1 о v. J. Inorg. Nucl. Chem., 1966, 28,
2609.
86. D. N. S athy anar ay ana, С. C. Patel. Bull. Chem. Soc. Japan, 1964,
37, 1736.
87. Ю. Я. Харитонов, Ю. А. Б у с л a e в. Изв. АН СССР, сер. хим.,
1964, № 5, 808.
88. Р. Л. Давидович, В. II. Сергиенко, Л. М. М у р з а х а н о в а.
Изв. СО АН СССР, сер. хим., 1968, вып. 1, № 2, 58.
89. К. Deh nick е, G. W е i d 1 е i n. Angew. Chem., 1966, 23, 1065.
90. S. C. Turman, C. S. Garner. J. Amer. Chem. Soc., 1950, 72, 2422.
182
91. H. Siebert. Z. anorg. Chem., 1954, 275, 225.
92. I. Fi 1 ip о v i c, Z. H a h 1, Z. G a sp a г а с, V. К 1 em e n ci c. J. Amer.-
Chem. Soc., 1954, 76, 2074.
93. В. А. Миркин, M. T. Козловский Изв. высш. учеб, завед. СССР,
Хим. и хим. технол., 1963, 6, 901.
94. К. F. Jahr, Н. W i t z m a n n. Z. anorg. Chem., 1933, 212, 13.
95. K. F. Jahr, H. W i t z m a n n. Z. phys. Chem., 1934, A168, 283.
96. A. D. Keim er s. J. Inorg. Nucl. Chem., 1961, 21, 45.
97. M. П. Глазырин, А. А. Фотиев. Неорг. мат-лы, 1968, 4, 82.
98. M. П. Глазырин, А. А. Фотиев. Ж. физ. хим., 1968, 42, 2435.
99. Р. М. Guiter. Ann. Chim., 1941, 15, 5.
100. S. J. Ki ehl, E. J. M a n f r e d o. J. Amer. Chem. Soc., 1937, 59, 2118.
101. R. A. Robinson, G. B. Jones, A. W. W у 1 i a, J. E. В r u n d e 11.
Trans. Proc. Roy. Soc. New. Zealand, 1938, 68, 39. Цит. по C.A. 1939,
33 3245
102. С. С. Макаров, A. T. Репа. Изв. АН СССР, OXH, 1940, 3, 349.
103. H. Menzel, G. Muller. Z. anorg. Chem., 1953, 272, 81.
104. . А. И. Яценко, С. П. Яценко. Ж- прикл. хим., 1966, 39, 76.
105. В. Д. Пономарев, В. 3. Тарасенко, Г. П. Шибанов. Тр. Ин-та
металлургии и обогащения АН Каз. ССР, 1966, 16, 70.
106. М. Ros koe. Liebigs Ann., 1872, 8, 103. Цит. по [102].
107. I. A. N о г b 1 a d. Bull. Soc. Chim., 1875, 23, 64. Цит. по [102].
108. Н. Backer. Liebigs Ann., 1885, 229, 286. Цит. по [102].
109. A. Ditte. Ann. Chim. Phys., 1880, 13, 190. Цит. по [102].
110. D. J. McAdam, С. A. P i e r 1 e. J. Amer. Chem. Soc., 1912, 34, 604.
111. В. 3. Тарасенко, В. Д. Пономарев, А. И. Зарубин,.
Б. А. Бочкарев. Тр. Ин-та металлургии и обогащения АН Каз.
ССР, 1968, 27, 58.
112. Н. Menzel, Е. V. Sa hr. Z. Electrochem., 1937, 43, 104.
113. H. Menzel, W. Hagen. Z. anorg. Chem., 1937, 233, 49.
114. A. C h r e t i e n, M. L e 1 о n g. C.r, 1966, C262, 478.
115. R. Trujillo, E. T e j e r a. J. Amer. Chem. Soc., 1953, 75, 741.
116. В. В. Андреев. Ж- прикл. хим., 1957, 30, 299.
117. А. А. Ивакин, А. П. Яценко. Ж. неорг. хим., 1971, 16, 1689.
118. С. R a d a u. Liebigs Ann., 1889, 251. 129.
119. R. Trujillo, E. F. Machado. Anal. Real. Soc. Espan. fis. у quim.,,
1955 51B, 751
120. R. Trujillo, E. F. T e j e г а. Там же, 1951, 47В, 496.
121. R. Trujillo, L. Pastor. Там же, стр. 501.
122. M. Lachartre. Bull. Soc. Chim. France, 1924, 35, 321.
123. J. W. Mellor. Inorganic and theoretical Chemistry. London, Green, 1928..
124. J. S. Lukesh. Acta Cryst., 1950, 3, 476.
125. П. И. Федоров, Л. M. Акулкина, E. С. Разгон. Ж- неорг. хим.,
1967, 12, 195.
126. J. Beltran, C. Guillem. Anal. Real. Soc. Espan. fis. у quim., 1957,
53B, 745.
127. I. Lindquist. Ци.т. no [20].
128. J. Beltran, C. Guillem. Anal. Real. Soc. Espan. fis. у quim., 1953,
52B 215
129. A D. Kelmers. J. Inorg. Nucl. Chem., 1961, 17, 168.
130. J. Beltran, C. Guillem. Anal. Real. Soc. Espan. fis. у quim., 1957,
53 В, 735.
131. В. Г. Б а м б у p о в, А. А. Ивакин. Тр. Ин-та хим. УФАН СССР,
1966, вып. 10, 25.
132. В. Л. 3 о л о т а в и н. Ж- анал. хим., 1947, 2, 364.
133. В. В. Андреев. Ж. общ. хим., 1954, 24, 1730.
134. П. И. Федоров, Л. М. Акулкина, Е. С. Разгон. Ж. неорг. хим.,.
1963, 8, 258.
135. П. И. П и н а е в. Там же, 1965, 10, 965.
136. И. К. Кр х. Уч. зап. Сарат. ун-та, 1938, 1, 54.
18Х
137. J. J. Lingake, L. M e i t s. J. Amer. Chem. Soc., 1946, 68, 2443.
138. O. A. Cook. Там же, 1947, 69, 331.
139. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы М.,
Госхимиздат, 1955.
140. A. Rosenheim, W. R е g 1 i n. Z. anorg. Chem., 1921, 120, 103.
141. H. T. S. Britton, K. A. Robinson. J. Chem. Soc., 1930, 2334.
142. G. Schwar zenbach, G. Geier. Helv. Chem. Acta, 1961, 44, 859.
143. P. Britton. J. Amer. Chem. Soc., 1916, 38, 2365.
-.144. L. Zurkova, J. Cor ba, V. Sucka. Chem. zvesti, 1968, 22, 73.
145. П. И. Федоров, H. П. С л о т в и н с к и й - С и д а к, Г. Н. Цимба-
лист. Ж. неорг. хим., 1963, 8, 2593.
146. О. Е. Land ford, S. J. Ki ehl. J. Amer. Chem. Soc., 1940, 62, 1660.
147. J. T u d о, B. J о 1 i b о i s, G. Laplace. C. r., 1969, cep. C, 17, 978.
148. H. С. Николаев, Ю. А. Буслаев. Химия редких элементов. М.,
Изд-во АН СССР, 1955, вып. 2, 57.
149. I. Lukacs, С. Strusievici, С. L i t е a n u. Stadii si certetori de
Chimie, 1962, 13, 171.
150. Б. В. Слободин, P. H. Плетнев, Л. К. Толстов. Ж. физ. хим.,
1969, 43, 1566.
151. Von Rex. Dissert. Berne, 1901. Цит. по [98].
152. Л. К. Толстов, А. А. Ивакин, Р. Н. Плетнев, Б. В. Сло-
бодин. Тр. Ин-та хим. УНЦ АН СССР. В печати.
153. К. F. Jahr, F. Preuss. Chem. Вег., 1965, 98, 3297.
154. С. Liteanu, I. Lukacs, C. Strusievici. Omagin Acad. Prof. Raluca
Ripan Editure, Academici Republicii Socialiste, 1966, 333.
155. А. К. Ильясова, А. Б. Бектуров. Ж. неорг. хим., 1962, 7, 2149.
156. J. Beltran, C. Guillem. Anal. Real. Soc. Espan. fis. у quim., 1959,
55B, 801.
157. C. F. Rammelsberg. Wied Ann., 1883, 20, 938. Цит. no [123].
158. C. L. Christ, J. R. Clark, H. T. Evans. Acta Cryst., 1954, 7, #01.
159. T. Howard, H. T. Evans. Z. Krist, 1960, 114, 257.
160. T. Dupuis, V, Lorenzelli. Thermal Anal., 1969, 1, 15.
161. Гун Дун, Сюн Ж у - ж э н ь. Науч, вестник, КЭСЮЭ ТУНБАО, 19^8,
№ 7, 215. Цит. по Реф. ж., Хим., 1959, 24, 85595.
162. J. Beltran, С. Guillem. Anal. Real. Soc. Espan. fis. у quim., 1957,
53B, 217, 223. Цит. по. Реф. ж., Хим., 1957, 24, 76716.
163. V. Synecek, F. H a n i c. Physic, j. Czech., 1954, 4, 120.
164. P. Dubois, P. Breton. C.r., 1938, 206, 1969.
165. К. T a r a m a. S. Ter ahis hi. J. Chem Soc. Japan, 1952, 55, 68.
166. T. A. S e s b e s. Istanbul Univ Teh. Fak. Mecmuasi Ser. C, 1955, 20, 2?2.
Цит. no [170].
167. V. Satava. Collect. Czechosl., Chem. Commun., 1959, 24, 2172. Цит. no
Реф. ж., Хим., 1960, 14, 56510.
168. J. T r a u. Roczniki Chem., 1962, 36, 1365.
169. C. Duval. Inorganic, thermogravimetric analysis, Elsevier Publ. Co Inc-
Amsterdam, 1963, Цит. no [170].
170. M. Taniguchi, T. R. Ingraham. Canad. J. Chem., 1964, 42, 24^7.
171. А. А. Ивакин. Тр. Ин-та хим. УФАН СССР, 1967, вып. 14, 119.
172. Г. Ф. П и н а е в. Автореф. дисс. Свердловск, 1965.
173. L. D. Frederickson, D. М. Hausen. Anal. Chem., 1963, 35, 818.
174. Н. В a u er, W. Balz. Z. anorg. Chem., 1965, 340, 225.
175. А. А. Фотиев, В. А. Макаров. Кристаллография, 1969, 14, 723.
176. A. H. Морозов. Металлург, 1938, 12, 21.
177. А. А. Фотиев, В. А. Макаров, В. Л. Волков, Л. Л. С у р а т,
Ж. неорг. хим., 1969, 14, 277.
178. Н. Е. Dunn, D. L. Е d 1 u n d, Т. G. Griffin. Rare Metals Handbook,
Reinhold, Pablishing Corporation, N. Y., 1954.
179. H. T. Evans, M. E. M г о s e, R. Marvin. Amer. Mineral, 1955, 40, 314.
180. R. Kohlmuller, J. Perran d. Bull. Soc. Chim. France, 1964, 3, 642.
181. F. H a n i c, L. Zurkova. Chem. Zvesti, 1961, 15, 486.
184
182. P. Sedlacek, К. Dorenberger-Schiff. Acta Cryst., 1965, 18, 407.
183. H. T. Evans, A. G. Swallow, W. H. Barnes. J. Amer. Chem. Soc.,
1964, 86, 4209.
184. A. G. Swallow, F. R. Ahmed, W. H. Barnes. Acta Crystal., 1966,
21, 397.
185. В. Г. Гулиа, О. Г. Немкова, В. К. Д е й н а к о в. Ж- неорг. хим.,
1962, 7, 84.
186. Е. А. А р б и т, В. В. Серебренников. Там же, 1965, 10, 410.
187. А. А. Ивакин, Н. И. Игнатьева, М. И. Глазырин. Там же,-
1967, 12, 50.
188. Е. А. А р б и т, В. В. Серебренников. Тр. Томск, ун-та, 1963,
157, 311.
189. М. П. Глазырин, А. А. Ивакин. Кристаллография, 1964, 9, 927.
190. A. Morette, N. Strupler. С. г., 1959, 249, 2326.
191. A. Morette, N. Strupler. Bull. Soc. Chim. France, 1961, 1, 154.
192. N. Strupler. Ann. Chim., 1965, 10, 345.
193. K. F. J a hr, F. Preuss. Chem. Ber.. 1966, 99, 1602.
194. S. К. Bhattacharyya, G. S. De, N. C. D a 11 a. Proc. Second. Int.
Conrg. Thermal anal., Worcester, 1968.
195. S. K. Bhattacharyya, J. Ghosh. Там же.
196. S. К. Bhattacharyya, N. C. D a 11 a. J. Thermal Anal., 1969, 1, 75.
197. M. D e с о t-A 1 b e r t, M. Dartiguenave, Y. Dartiguenave. C.r.,
1968, C266, 117.
198. P. G. S p a c u. Bull. Sect. Sci. Acad, roumaine, 1939, 22, 42, Цит. по C.A.
1940, 34, 5774.
199. L. Malaprade. Bull. Soc. Chim. France, 1950, 9-10, 765.
200. P. Fleury, R. К о h 1 m u 11 e r. C.r., 1966, C262, 475.
201. Б. В. Слободин. Ж- неорг. хим. 1969, 14, 2027.
202. G. С. S h i v a h а г е. J. Inorg. Nucl. Chem., 1966, 28, 657.
203. M. B. R a n e, К. К о n d i a n. J. Indian Chem. Soc., 1931, 8, 289. Цит.
no [99].
204. R. S. Saxena, С. P. Sharma, M. L. Mittal. J. pract. Chem., 1967^
36, 225.
205. С. M. T а н а т e p, E. К. Кур овский. Ж- Рус. физ.-хим. о-ва, 1909,
41, 813.
206. G. Peyronel. Gazz. Chim. ital., 1942, 72, 77. Цит. по [208].
207. А. В u г d е s е, М. В о г 1 v г a*. Ann. Chim., 1960, 50, 1570.
208. R. S. Saxena, M. C. Jain. J. Inorg. Nucl. Chem., 1968, 30, 3130.
209. A. Morette. Bull. Soc. Chim. France, 1950, 5, 527.
210. R. S. Saxena, M. C. Jain. Z. Naturforsch., 1969, 24b, 173.
211. E. Г. Тимофеева, И. И. Калиниченко, В. Д. Никитин,
А. И. И у р т о в. Ж. неорг. хим., 1960, 5, 1168.
212. F. R е v е n g. Bull. Soc. Chim. France, 1946, 11-12, 677.
213. D. О 1 i v i e r, P. R a b e 11 e. C.r., 1967, C265, 1451.
214. D. Olivier, B. Combe. Там же, 1968, C267, 877.
215. J. A m i e 1, D. Olivier, M. D e s s о 1 i n. Там же, 1967, C264, 1045.
216. D. Olivier. Там же, 1967, C264, 1176.
217. E. N e u s s e r. Z. anorg. Chem., 1938, 239, 240.
218. В. Л. Золота вин, В. H. Булыгина, И. Я- Безруков.
Ж. неорг. хим., 1970, 15, 429, 435.
219. В. Л. Зол отав ин, Ю. И. Санников. Тр. Ин-та хим. УФАН
СССР, 1963, вып. 7, 73.
220. Ю. И. Санников, В. Л. 3 о л о т а в и н. Ж. неорг. хим., 1963, 8, 418.
221. Ю. И. Санников, В. Л. 3 о л о т а в и н. Там же, стр. 428.
222. В. Л. 3 о л о т а в и н, Ю. И. Санников. Там же, стр. 969.
223. В. Л. 3 о л о т а в и и, Ю. И. С а п н и к о в. Там же, стр. 973.
224. Ю. И. Санников, В. Л. 3 о л о т а в и н. Там же, стр. 1151.
225. Ю. И. Санников, Е. И. Крылов, В. Л. Зол отав ин. Там же,
стр. 1157.
226. W. G. Parks, Н. J. Р г е b 1 u d a. J. Amer. Chem. Soc., 1935, 57, 1676.
185
227. J. G. Murgulescu, V. Alexa. Z. anal. Chem., 1942, 123, 341.
228. R. S. Saxena, О. P. Sharma. Indian J. Chem., 1947, 5, 9.
229. В. И. Елфимов, И. Я. Безруков, В. Л. 3 о л о т а в и н. Изв. АН
СССР, 1970, 6, 607.
230. В. И. Елфимов, И. Я- Безруков, В. Л. Зол отав ин. Тр. Ин-та
хим. УФАН СССР, 1971, вып. 23, 124.
231. М. В. Соболева, И. Л. Пудовкина. Минералы урана. М., Гос-
геолтехиздат, 1957.
232. И. И. Л ип ил ин а. Уранил и его соединения. М., Изд-во АН СССР, 1959.
233. Ю. В. Морачевский, Л. И. Беляева. Геохимия, 1956, 7, 20.
234. И. Я- Безруков, В. Л. 3 о л о т а в и н, А. Б. Аскеров, В. В. Про-
коп ч у к. Там же, 1965, 9, 1120.
235. R. S. Saxena, О. Р. Sharma. J. Inorg. Nucl. Chem., 1966, 28, 195.
236. R. S. Saxena, О. P. Sharma. J. Indian Chem. Soc., 1966, 43, 209.
237. H. Gessner. Kolloidchem. Beiheft, 1924, 19, 213. Цит. no [129].
238. J. Meyer, N. A u 1 i c h. Z. anorg. Chem., 1928, 172, 321.
239. G. F. Hiittig, A. Konig. Там же, 1930, 193, 81.
240. Л. К. Толстов, А. А. Ивакин, А. А. Фотиев. Тр. Ин-та хим.
УФАН СССР, 1970, вып. 20, 40.
241. О. Glemser. Angew. Chem., 1961, 73, 785.
242. W. Biltz. Ber., 1904, 37, 1095.
243. W. P r a n d 11, L. Hess. Z. anorg. Chem., 1913, 82, 103.
244. M. Kerker, G. Jones, I. B. Read, N. P. Yang, M. D. Melvin.
J. Phys. Chem., 1954, 58, 1147.
245. E. Muller. Z. Roll., 1911 8, 302.
246. 3. Я. Берестнева, Г. А. Корецкая, В. А. Каргин. Коллоид,
ж., 1952, 24, 73.
247. Е. Suite, К. Т a k i у a m a. Proc. Japan Akad., 1954, 30, 752.
248. 3. Я. Берестнева, Г. А. Корецкая, В. А. Каргин. Докл.
АН СССР, 1948, 59, 1121.
249. А. В. Бромберг, В. М. Лукьянович, В. В. Немцова, Л. В.
Радушкевич, К. В. Г л ушков. Ж. физ. хим., 1953, 27, 379.
250. Н. Thielle. Disc. Faraday Soc., 1954, 8, 294.
251. В. Tamamushi. Sci. Papers Coll. Gen. Univ. Tokyo, 1956, 6, 37. Цит.
по Реф. ж., Хим., 1957, 30, 234.
252. N. H. Bhuiyan. Pakistan. J. Sci. Res., 1957, 9, 5. Цит. по Реф. ж.,
Хим., 1958, 1, 642.
253. К. Takiyama. Bull. Chem.- Soc. Japan, 1958, 31, 329, 535.
254. И. Ф. Ефремов. Коллоид, ж., 1954, 26, 264.
255. R. S h a n k e r Rai. Kolloid Z., 1961, 179, 160.
256. S. Prakash, A. P a n d e y. Kolloid. Z., 1959, 167, 39.
257. R. P. Mitra, A. K. Banerjie. J. Sci. Ind. Res. (Indian), 1960,19b, 492.
258. S. P. M о u 1 i k, D. К- M u 1 1 i k. Z. phys. Chem., Neue Folge, 1966, 48, 196.
259. А. А. Ивакин. Тр. Ин-та хим. УФАН СССР, 1967, вып. 14, 113.
260. М. Н. Соболев. Получение ванадия из керченских железных руд.
М., ОНТИ, 1935.
261. Н. П. Слотвинский-Сидак, В. И. Потапов, П. И. Аверин.
Цвет, мет., 1965, 5, 67.
262. Н. П. Слотвинский-Сидак, В. И. Потапов, Л. Е. Колпа-
ков, П. И. Федоров, Г. А. Ловецкая, Ф. А. Хомяков,
В. И. С а н д л е р. Там же, 1966, 10, 64.
263. Э. Ф. Краузе, Е. И. Бурова, О. И. Воробьева, В. И. Во-
робьева.. Уч. зап. МГУ, 1934, вып. 2, 201.
264. В. Burwell. J. Metals, 1961, 18, 562.
265. S. F. Ravitz, L. W. Nicholson, C. J. C h i n d g r e n, L. С. В a u e r 1 e,
F. P. Willi as, M. T. Martinson. Metal technology, 1948, 14, 2178.
266. H. E. Dunn, A. D. Wallace, B. Mauer. Пат. США 2551733. C.A.
1951, 45, 8216.
267. L. C. Bauerle, L. W. Nicholson. Пат. США 2784075. C.A. 1957,
51, 8400.
186
268 А. Б. Бектуров, А. К. Ильясова, Р. А. Г е с к и н а. Ж- неорг.
хим. 1962, 7, 2134.
269. А. А. Ивакин. Ж. прикл. хим. 1963, 36, 2121.
270. А. А. Ивакин. Тр. Р1н-та хим. УФАН СССР, 1966, вып. 9, 107.
271. В. Л. 3 о л о т а в и н, Л. К. Толстов, А. А. Ивакин, А. А. Фо-
тиев, В. В. Мочалов, Ю. Ф. Букреев. Неорг. мат-лы, 1967^
3, 1601.
272. В. Л. Золотавин, Л. К. Толстов, А. А. Фотиев, Ю. Ф. Бук-
реев. Докл. АН СССР, 1967, 175, 595.
273. Л. К. Толстов, В. Л. Золотавин, А. А. Ивакин, Ю. Ф. Бук-
реев, М. П. Глазырин, В. В. Мочалов, С. И. А л я м о в с к и й.
Тр. Ин-та хим. УФАН СССР, 1970, вып. 17, 96.
274. Л. К. Толстов, В. Л. Золотавин, А. А. Фотиев, Ю. Ф. Бук-
реев, Р. Н. Плетнев. Неорг. мат-лы, 1968, 4, 1754.
275. Р. Н. Плетнев, В. Л., Золотавин, Л. К. Толстов. Тр. Ин-та
хим. УФАН СССР, 1970, вып. 17, 104.
276. А. А Ивакин. Там же, 1967, вып. 14, 119.
277. С. П. Жданов, Е. И. Егорова. Химия цеолитов. Л., «Наука», 1968.
278. В. Л. Золотавин, Р. Н. Плетнев, Л. К. Толстов. Тр. Урал,
политехи, ин-та, 1967, сб. 163, 106, ПО.
279. В. Л. Золотавин, Р. Н. Плетнев, Л. К- Толстов, А. П. Сте-
панов, Ж. Всесоюз. хим. о-ва им. Д. И. Менделеева, 1968, 13, 355-
280. А. А. Фотиев, Л. К. Толстов, Ж. неорг. хим., 1970, 15, 1011.
281. С. 3. Рогинский. Ж. физ. хим., 1940, 12, 427.
282. С 3. Рогинский, О. М. Тодес. Изв. АН СССР, ОХИ, 1940, 3, 331-
283. О. М. Тодес. Изв. АН СССР, ОХН, 1942, 2-3, 106.
284. О. М. Тодес, Л. Н. Сосновский, Ж. физ. хим., 1946, 1, 51.
285. A. Nilsen. Acta Chem. Scand., 1958, 12, 951.
286. A. Nilsen. Там же, стр. 784.
287. A. Nilsen. Там же, стр. 1680.
288. A. Q. Lundquist. Пат. США 2733980. С.А., 1956, 50, 8980.
289. Е. И. Бурова. Уч. зап. МГУ, 1936, вып. 6, 15.
290. А. А. Ивакин. Ж. прикл. хим., 1963, 36, 1894.
291. А. А. Ивакин, В. Л. Волков. Исследования в области химии и
технологии минеральных солей и окислов. Под ред. М. Е. Позина.-
М.— Л., «Наука», 1965, стр. 66.
292. Т. S. Perrin. Пат. США 2614905. С.А., 1953, 47, 2443.
293. К. Колмен, К. Броди, Э. Мур, Д. Кроуз. Экстракция в анали-
тической химии. М., Изд-во иностр, лит., 1961.
294. А. А. Ивакин, Н. И. П е т у н и н а. Исследования в области химии
и технологии минеральных солей и окислов. Под ред. М. Е. Позина.
М.— Л., «Наука», 1965, стр. 180.
295. Klemme г, Rosenbaum. Mining Rev., 1955, 58, 6.
296. А. И. Пономарев. Методы химического анализа минералов и гор-
ных пород. М., Изд-во АН СССР, 1955.
297. Г. Ш а р л о. Методы аналитической химии. М.— Л., «Химия», 1966.
298. Г. Т. С и б о р г. Дж. Дж. К а ц. Химия актинидных элементов. М.,.
Атомиздат, 1960.
299. Э. Т. Чудинов, И К. Швецов. Радиохимия, 1965, 7, 188.
300. Ю. А. Золотов, И. М. Маров, А. И. Москвин. Ж. неорг. хим.,.
1961, 6, 1055.
301. А. И. Москвин, И. Гэлэцеану, А. В. Лапицкий. Докл. АН
СССР 1Q63 14Q 011
302. J. J. Berzelius’ Pogg. Ann., 1831, 22, 1. Ци.т. по [123].
303. В. Л. Золотавин, И. Я. Безруков, Ю. И. Санников. Ж- неорг.
хим., 1961, 6, 581.
304. А. А. Ивакин. Ж. прикл. хим., 1966, 39, 277<
305. А. А. Ивакин. Там же, стр. 2406.
306. А. А. Ивакин. Ж. неорг. хим., 1967, 12, 1787.
307. А. А. Ивакин, Э. М. Воронова. Там же, 1969, 14, 1557.
187
308. R. J. G e 11 e s p i e, R. Kapoor. Canad. J. Chem., 1966, 44, 1203.
-309. A. K. S. Gupta. Z. anorg. Chem., 1960, 304, 328.
310. E. Petersen. J. pract. Chem., 1888, 40, 271. Цит. no [87].
311. A. P i с c i n i, G. Giorgis. Gazz. Chim. Ital., 1892, 22, 55. Цит. no [87].
312. D. G г a n d j e a n, R. Weiss. C.r., 1966, 262, 1864.
313. Ю. H. Буслаев, В. А. Щербаков, M. E. Д я т к и и а. Ж. структ.
хим., 1965, 6, 16.
314. Gmelin — Kraut. Hb. anorg. Chem., Vanadium, Abt. A, 1908, 3, 125.
315. E. А. Никитина. Гетерополисоединения. M., Госхимиздат, 1962.
•316. S. Prasad, К- С. Р a t h a k. Indian J. Chem., 1967, 5, 14.
317. A. Rosenheim, M. Pieck. Z. anorg. Chem., 1916, 98, 225.
318. F. Ch a uve a u. Bull. Soc. Chim. France, 1960, 5, 834.
319. F. Chauveau, P. Souchay. Там же, 1963, 3, 561.
320. M. P. Courtin, F. Chauveau, P. Souchay. C.r., 1964, 258, 1246.
321, B. Charreton, G. Berth о. Там же, 1965, 261, 2903.
322. В. Charreton, F. Chauveau, G. Berth о, P. Courtin. Chim
anal. (Fr.), 1965, 47, 17.
•323. I. Krivy, J. Krtil. Collection Czech. Chem. commun., 1964, 29, 587.
324. Ю. Ф. Ш к a p о в с к и й. Ж- неорг. хим., 1967, 12, 2363.
-325. G. А. Т s i g d i n о s, C. J. H a 11 a d a. Inorg. Chem. (Amer.), 1968, 7, 437.
326. W. Gibbs. Amer. Chem. J., 1885, 7, 214. Цит. no [315].
327. A. Ditte. C.r., 1885, 102, 1019. Цит. no [315].
328. C. Friedheim. Ber., 1890, 23, 1530; 24, 1505. Цит. no [315].
329. C. Friedheim, W. Handerson. Ber., 1902, 35, 2245. Цит. no [315].
330. C. Friedheim, A. M u c h a e 1 i s. Z. anorg. Chem., 1894, 5, 437. Цит.
no [315].
331. A. Rosenheim, M. Pieck. Там же, 1946, 98, 225.
•332. P. Souchay. S. Dubois. Ann. Chim., 1948, 3, 88.
333. G. Jander. Z. anorg. Chem., 1933, 211, 49; 212, 1; 214, 143; 215, 310.
Z. phys. Chem., 1934, 168, 283.
334. R. U. Russel, I. E. Salmon, H. R. T i e t z e. J. Chem. Soc., 1961, 3211.
335. А. Б. Б e к т у p о в, А. К. Ильясова. Тр. Ин-та хим. наук АН Каз.
ССР, 1964, 10, 130.
336. G. Jander, К- F. Jahr, Н. W i t z m a n n. Z. anorg. Chem., 1934, 217,78.
337. F. Ephraim. Inorg. Chem., 1954, 534.
338. Л. H. Ш e л у д я к о в, Л. С. Любимова. Изв. АН Каз. ССР, сер.
хим., 1961, 1(19), 63.
339. Г. Б. С е й ф е р, И. В. Т а н а п е в. Ж- неорг. хим., 1963, 8, 1011.
340. Л. А. Кадушкина, Л. К- К у р м а н г у ж и н а. Хим. и хим. технол.,
1964, 2, 59, 67.
341. A. Rosenheim, К. Н. Jang. Z. anogr. Chem., 1923, 129, 181.
342. S. P. Moulik, B. N. Ghosh, D. К. Mui lick. J. Indian Chem. Soc.,
1963, 40, 743.
343. А. А. Ивакин, Э. M. Воронова. Тр. Ин-та хим. УФАН СССР, 1969,
вып. 17, 144.
344. W. В i 11 z. Z. anorg. Chem., 1942, 249. 1.
345. A. Scheuer. Там же, 1898, 16, 284.
346. П. Г. Меликов, Л. В. П и с а р ж е в с к и й. Исследования над переки-
сями, С.-Пб., 1900.
347. П. Г. Меликов, Е. Е л ч а и и и о в. Вег., 1909, 42, 2291.
348. К. F. Jahr. Z. Elektrochem., 1941, 47, 810.
349. J. Beltran, R. Rios. Anal. Real. Soc. Espan. fis. у quim., 1952, 48B, 388.
350. J. Beltran, A. Castano, F. Castano. Там же, 1956, 52В, 105.
351. H. К. Григорьева, К- Н. Дергачева. Изв. АН СССР. ОХН,
1962, 6, 943.
352. J. Ale у е г, А. Р a w 1 е 11 a, Z. angew. Chem., 1926, 39, 1284.
353. М. Е. Rumpf. Ann. Chim., 1937, 8, 456.
354. А. К. Бабко, А. И. Волкова. Ж. общ. хим., 1952, 22 (84), 1108.
355. Y. Shaeppi, W. D. Treadwell. Helv. Chim. Acta, 1948, 31, 577.
356. G. A. Dean. Canad. J. Chem., 1961, 39, 1174.
188
3&7. P. Flood, Т. J. Lewis, D. H. Richards. J. Chem. Soc., 1963, 5024.
358. А. А. И в а к и н, Э. М. Воронов а. Тр. Ин-та хим. УФАН СССР,
1969, вып. 17, 134.
359. A. Rosenheim. Z. anorg. Chem., 1893, 4, 352.
360. Р. К. В h a 11 а с h а г у а, М. С. Savena, S. N. Banerji. Proc. Nat.
Acad. Sci., India, 1961, A30, 75.
361. H. Kurihara, T. Nozaki. J. Chem. Soc. Japan, Pure Chem. Sec.,
1962, 83, 708.
362. К. Б. Я ци мирский, В. E. Калинина. Ж. неорг. хим., 1964, 2,
1328.
363. Э. М. Воронова, А. А. Ивакин. Там же, 1969, 14, 1564.
364. К. Б. Яцимирский, В. Е. Калинина. Изв. высш. учеб, завед.
СССР, Хим. и хим. технол., 1965, 8, 385.
-365. А. А. Ивакин, Э. М. Воронова. Тр. Ин-та хим. УФАН СССР,
1969, вып. 17, 139.
-366. Ф. Р о с с о т т и. В кн. «Современная химия координационных соедине-
ний». Под. ред. Дж. Льюиса и Р. Уилкинса. М., Изд-во иностр, лит.,
1963.
367. D. К. Mullick, S. Р. Moulik, В. N. Ghosh. J. Indian. Chem. Soc.,
1963, 40, 137.
368. В. N. Ghosh, S. P. Moulik, K. N. S e n g u p t a, P. R. Pal. Там же,
стр. 509.
369. А. И. Волкова, Т. Е. Г е т ь м а н. Укр. хим. ж., 1963, 29, 1240.
370. S. N. Banerji, А. К. Dey. Z. anorg. Chem., 1961, 309, 226.
371. R. L. Seth, A. K. Dey. J. Indian Chem. Soc., 1962, 39, 724.
372. R. L. P e c s о k, R. S. Ju vet. J. Amer. Chem. Soc., 1953, 75, 1202.
373. K. Mick a, A. To ck st ein. Chem. Listy, 1954, 48, 648.
374. A. Ringbom, S. Siitonen, B. Skrifvars. Acta Chem. Scand., 1957,
11, 351.
375. J. S a j o. Acta Chim. Acad. Sci Hung., 1958, 16, 115.
376. L. Przyborowski, G. Schwarzenbach, T. Zimmerman. Helv.
Chim. Acta, 1965, 48, 1556.
377. R. L. D u 11 a, S. Lahiry. J. Indian Chem. Soc., 1962, 39, 860.
378. R. L. D u 11 a, S. Lahiry. Там же, 1963, 40, 53.
379. R. L. Dutt a, S. Ghosh. Там же, 1967, 44, 369.
380. I. Minczewski, Z. Skorko-Trybula. Taianta, 1963, 10, 1063.
381. А. Л. Маркман, Б. T. О с т p о б p о д, Э. К. А б д у с а л а м о в а. Изв.
высш. учеб, завед. СССР, Хим. и хим. технол., 1969, 12, 97.
382. В. Ф. Попова, А. Л. Маркман. Узб. хим. ж., 1966, № 4, 18.
383. В. Ф. Попова, А. Л. Маркман. Изв. высш. учеб, завед. СССР,
хим. и хим. технол., 1970, 13, 332.
384. С. Y. Nester, М. N о b 1 s. Z. anal. Chem., 1959, 167, 81.
385. С. Я. Шнайдерман, Г. И. Хрусталев. Ж. общ. хим., 1959,29,20.
386. С. Я. Шнайдерман, Е. П. Клименко, Г. П. Прокопьева. Ж.
неорг. хим., 1969, 14, 1552.
387. С. Я. Шнайдерман, Е. П. Клименко. Там же, 1967, 12, 2132.
388. С. Я. Шнайдерман, Е. П. Клименко. Там же, стр. 127.
389. С. Я. Шнайдерман, Е. П. Клименко, Е. Н. Князева,
Н. В. Черная. Там же, 1969, 14, 1238.
390. С. Я. Шнайдерман, Е. П. Клименко, Г. Н. Прокопьева.
Изв. высш. учеб, завед. СССР, Хим. и хим. технология., 1969, 12,
1811.
391. С. Я. Шнайдерман, Е. П. Клименко, А. Н. Демидовская.
Ж- неорг. хим., 1969, 14, 731.
392. Е. П. Клименко, С. Я. Шнайдерман. Укр. хим. ж., 1969, 35, 580.
393. R. L. Dutt a, S. Lahiry. J. Indian Chem. Soc., 1963, 40, 67.
394. R. L. Dutt a, A. S у a m a 1. Там же, 1967, 44, 381.
395. А. К. Б а б к о, А. И. В о л к о в а. Т. Е. Г е т ь м а н. Ж неорг. хим.,
1967. 12, 3365.
396. Y. Shi jo, Т. Takeuchi. Jaoan Analyst, 1965, 14, 115.
18
397. К. Н. Богдасаров, X. А. Ахмедова, О. А. Татаев
лабор., 1969, 35, 12. * Во^
398. А. К- Б а б к о, А. И. Волкова, Т. Е. Г е т ь м а н. Ж. неопг vn
1966, 11, 374. 1 ’ Хим-
399. Ф. Г. Жаровский, А. Т. Пилипенко. Укр. хим. ж., 1964, 25, 2чп
400. R. Montegui. Anal. Real. Soc. Espan. fis. у guim., 1964, B60, 277
401. А. И. Бу сев, 3. П. Карякина. Ж. неорг. хим., 1967, 12, 1561.
402. В. Н. Тихонов. Ж- анал. хим., 1966, 21, 1172.
403. Pakistan J. Sci. and Ind. Res., 1962, 5, Цит. по Реф. ж., Хим., 1963 N о
404. Н. Е i n a g а, Н. Ishii. Japan Analyst, 1968, 17, 836.
405. S. С. T о n d о n. Anal. Chem., 1961, 33, 1267.
406 Л П Санецкая, M. M. Тепляков. Ж- анал. хим., 1961, 16, 731.
407 М Тосихаоу, И. Йоси о, У. Кацу я. Химиэдзу коге дайгану кэикю
хокону. Repts Himeji Inst: Technol., 1965, A № 18,99. Цит. по Реф.
ж., Хим., 1967, 6Г146.
408. В. Н. П о д ч а й н о в а, А.
лабор., 1965, 31, 790.
В. Долгарев, В. Я- Дергачев. Завод.
409. Н. Kohara, N. Yshibashi, Y. Hakamura, К. Ueno. Japan Analyst
1966, 15, 938.
410. A. T. Пилипенко, И. П. Середа, Э. А. Шпак. Завод, лабор
1966, 32, 660.
411. N. Kurmaian, D. Satyanarayana, U. Pandu, R. Rao. Taianta
1967, 14. 495.
412. T. Matsumoto, M. Satake, T. Yonekubo. Nippon Kagaku Zasshi
1968, 89, 944.
413. H. E i n a g a, H. Ishii. Japan Analyst, 1968, 17, 836.
414. A. K. Majundar, S. C. Saha. Anal. Chim. Acta, 1969, 44, 85.
415. Б. И. H а б и в а н e ц. Ж. неорг. хим., 1963, 8, 2302.
416. Б. И. H а б и в а н e ц. Там же, 1966, 11, 2732.
417. S. W. R a b i d е a u. J. Amer. Chem. Soc., 1956, 78, 2705.
418. С. С. Б а ц а н о в. Ж- структ. хим., 1962, 3, 616.
419. В. 3 а х а р и а з е н. В кн. «Актиниды». Под. ред. Г. Сиборга и Дж.
Каца. М., Изд-во иностр, лит., 1955, стр. 626.
ОГЛАВЛЕНИЕ
Введение .......................................................... 3
Глава I. Ионное состояние ванадия (V) в растворах . . . ’ 6
Реакции гидролиза ионов ванадия.............................. 7
Структура и спектры поглощения ионов ванадия.................24
Глава II. "етерогенные равновесия в водно-солевых системах, вклю-
чающих соединения ванадия.......................................32
Система Na2O—V2O5—Н2О........................................34
Растворимость ванадатов натрия в воде и водно-солевых растворах 42
Система К2О—V2Ot5—Н2О........................................46
Система NH3—V2O6—Н2О .......................................*48
Растворимость ванадатов других металлов .................... 54
Система SO3—V2O6—Н2О.........................................54
Система HF—V2O6—Н2О..........................................57
Глава III. Синтез и свойства ванадатов.............................59
Ванадаты щелочных металлов и аммония............................
Ванадаты натрия................................................
Ванадаты калия..............................................65
Ванадаты аммония............................................71
Смешанные и кислые декаванадаты щелочных металлов и аммония . 73
Ванадаты щелочноземельных элементов..........................75
Ванадаты редкоземельных элементов............................86
Ванадаты меди и серебра......................................91
Ванадаты цинка, кадмия и ртути...............................94
Ванадаты алюминия, индия и таллия............................95
Ванадаты циркония, свинца и олова.............................—
Ванадаты хрома и марганца ...................................97
Ванадаты железа, кобальта и никеля................,.........100
Ванадаты урана и тория.......................................ЮЗ
Глава IV. Гидролитическое осаждение ванадия.......................106
Гидратированная пятиокись ванадия и декаванадиевая кислота ... —
Гидролитическое осаждение ванадия ..........................112
Расход кислоты и изменение pH растворов в процессе гидролиза . 113
Состав продуктов гидролиза.................................119
Термические свойства продуктов гидролиза...................121
Кинетика гидролитического осаждения........................126
О структуре продуктов гидролиза и механизме гидролитического
осаждения...............................................133
Глава V. Комплексные соединения ванадия...........................138
Комплексы с неорганическими лигандами.......................139
Комплексы с анионами неорганических кислот...................—
Гетерополисоединения.......................................144
Пероксованадаты.........................................-..151
Комплексы с органическими лигандами........................ 157
Карбоновые и оксикарбоновые кислоты .........................—
Комплексы с другими органическими лигандами...............167.
Особенности комплексообразования ванадия....................173
Литература ...................................................... 181
1
9
ХИМИЯ ПЯТИВАЛЕНТНОГО ВАНАДИЯ
В ВОДНЫХ РАСТВОРАХ
Труды Института химии УНЦ АН СССР, вып. 24
РИСО УНЦ АН СССР
СВЕРДЛОВСК, К-49
ул. ПЕРВОМАЙСКАЯ, 91
Редактор изд-ва Г. Е. Никитюк Обложка художника А. А. Лебедева
Техн, редактор Н. Р. Рабинович Корректоры Н. Д. Махнева, М. И. Зубринская
РИСО УНЦ АН СССР № 519—54 (70). Сдано в набор 28.IV.71 г. НС 11021.
Формат 60Х901/1б. Печ. л. 12. Уч.-изд. л. 13,18. Подписано к печати 6.1.1972 г.
Тираж 1200. Цена 1 р. 31 к. Заказ 256.
Типография изд-ва «Уральский рабочий», г. Свердловск, проспект Ленина, 49.
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр. Строка Напечатано Следует читать *
68 14-я снизу 420° С 1300°с
113 15-я снизу увеличивается уменьшается
116 1-я снизу а—0,9; б—0,98; в—1,1; г—1,3 й — 1,3; 6—1,1; в—0,98 г—0,9
149 5-я снизу BaVPO7 Ba2VPO7
154 5-я снизу отличие от различие в
164 16-я сверху —36,0 —3,60
184 9-я сверху Sucka Sucha
Заказ № 256