/










































































































Автор: Музгин В.Н. Хамзина Л.Б. Золотавин В.Л. Безруков И.Я.
Теги: аналитическая химия неорганическая химия химия
Год: 1981




Текст
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ*
АНАЛИТИЧЕСКАЯ ХИМИЯ
ВАНАДИЯ
В.'Н. Музгин, Л. Б. Хамзина, В. Л. Золотавин, И. Я. Безруков
гей]
ИЗДАТЕЛЬСТВО «НАУКА»
МОСК В А 1981
Серия: «Аналитическая химия элементов»
Главный редактор
член-корреспондент АН СССР Ю. А. Золотов
Редакционная коллегия:
И. П. Алимарин, Ю. И. Беляев, А.И.Бусев, М.П. Волынец, А. Н. Ермаков, В. М. Иванов, А. В, Карякин, Н.М. Кузьмин, С. Б. Саввин, И. М. Ростоцкая (ученый секретарь)
Редактор тома «Аналитическая химия ванадия»
Н. С. Полуэктов
Адрес редколлегии: 117334, Москва, Воробьевское шоссе, 47а Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
205С6-180
М (55(02)-81
Б.3-82-125-1980 18С4ССССС0
© Издател*е.твр.«Нау'ка», 1981 г.
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов» — составит около 50 томов. Потребность в подобного рода издании назрела давно. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом возникло настоящее издание — серия «Аналитическая химия элементов», которое осуществляется впервые. Издание серии было начато по инициативе академика А. П. Виноградова, который с 1958 по 1975 г. был ее главным редактором.
Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий пла'н как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы, как физические, так и физико-химические, излагаются применительно для количественного определения данного элемента начиная с анализа сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
Монографии содержат обширную библиографию, доведенную до последних лет; они рассчитаны на широкий круг химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также
3
на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» выходят самостоятельно по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, бериллию, редкоземельным элементам и иттрию, никелю, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, селену и теллуру, алюминию, нептунию, трансплутониевым элементам, платиновым металлам, радию, кремнию, германию, магнию, рению, марганцу, кадмию, ртути, кальцию, фосфору, литию, олову, серебру, цинку, золоту, рубидию и цезию, вольфраму, мышьяку, сере, плутонию, азоту, стронцию, сурьме, барию, хрому и брому.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Ванадий в промышленном масштабе стали применять лишь с начала XX в. До настоящего времени его основным потребителем (до 90%) является черная металлургия. Ванадий используют в производстве быстрорежущих, инструментальных и конструкционных сталей и чугунов. Благодаря его легирующим, раскисляющим и карбидообразующим свойствам повышается качество и эксплуатационные характеристики материалов. Ванадий применяют для получения сплавов на нежелезной основе (медно-вана-диевые, титано-ванадиевые сплавы, ванадиевые бронзы и др.). Большой интерес представляют ванадиевые сплавы с добавками вольфрама, ниобия, циркония и некоторых элементов, сверхпроводящие сплавы ванадия с галлием, кремнием, титаном и т. д. Перспективно использование чистого ванадия и его сплавов в ядерной энергетике, в ракето- и самолетостроении.
Соединения ванадия, обладающие ценными каталитическими, люминесцентными, полупроводниковыми и другими свойствами, находят все большее применение в химической, радиоэлектронной, лакокрасочной, керамической, текстильной промышленности, а также в других отраслях народного хозяйства.
Многообразие ионных форм и степеней окисления ванадия определяют его высокую подвижность в земной коре и значительную рассеянность в природе. Ванадий редко образует крупные скопления руд, но легко концентрируется в почвах, наземных и водных растениях. В связи с высокой токсичностью соединений ванадия необходим падежный контроль за его содержанием в окружающей среде, отходах производства, биологических объектах и т. д. Это особенно важно из-за быстрого роста добычи и широкого потребления нефти с высоким содержанием ванадия.
В монографии рассмотрено состояние ионов ванадия в растворах, имеющее важное значение в понимании химии процессов отделения и определения ванадия, а также при анализе ванадия и его соединений. Большое внимание уделено наиболее перспективным физическим и физико-химическим методам контроля промышленных и природных объектов, и в частности методам определения ультрамалых количеств ванадия. Подробно изложены титриметри-ческие, фотометрические, спектральные, атомно-абсорбционные, рентгенофлуоресцентные, активационные и некоторые другие методы определения ванадия в различных объектах. Из всех перечисленных методов в настоящее время наибольшее распространение получили фотометрические и титриметрические методы определения ванадия.
5
[ Большое значение в аналитической химии ванадия имеют методы концентрирования и выделения. Широкое распространение получили методы экстракции комплексных соединений ванадия, в частности р-дикетонатов. Хорошие результаты получают при использовании ионообменной хроматографии, где изменением степени окисления ванадия или ионного состояния можно легко отделить ванадий практически от всех элементов. Для отделения микрограммовых количеств ванадия применяют распределительную хроматографию па бумаге или в тонком слое сорбента.
В список литературы к монографии включены оригинальные работы главным образом последних двух десятилетий, подробно не" рассмотренные в руководствах и обзорах по аналитической химии ванадия.
В работе над монографией обобщен длительный опыт работы авторов в области аналитической химии ванадия.
Авторы выражают благодарность и признательность редактору монографии академику АН УССР Н. С. Полуэктову, рецензентам кандидатам химических наук Т. М. Малютиной и А. В. Дол-гореву за ценные критические замечания и пожелания.
Глава I
ФИЗИКО-ХИМИЧЕСКАЯ И ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА ВАНАДИЯ И ЕГО СОЕДИНЕНИЙ
ОБЩИЕ СВЕДЕНИЯ
Андре Мануэль дель Рио в 1801 г. обнаружил в свинцовом блеске неизвестный элемент и назвал его эритронием, а в 1805 г. Кол-лет Декостиль показал, что полученное вещество является загрязненным хромом. Дель Рио согласился с этим и таким образом отказался от своего открытия. В 1830 г. Н. Сефстрём вновь обнаружил этот элемент и назвал его ванадием, а в 1831 г. Ф. Вёллер доказал тождество ванадия и эритрония. Хрупкий металлический порошок ванадия впервые получил Г. Роско в 1869 г. [172]. Ковкий ванадий удалось получить Д. Мардену и М. Ричу только в 1927 г. [440].
Ванадий — переходный d-элемент 3(/-ряда. Он расположен в четвертом периоде и возглавляет подгруппу ванадия в пятой группе периодической системы Д. И. Менделеева. Порядковый номер ванадия 23. Электронная структура его атомов ls22s22p63s23p63(/34s2. Атомная масса ванадия по углеродной шкале 50,9415. В природе существует два стабильных ^изотопа ванадия: 60V (0,24%) и 5IV (99,76%). Свойства изотопов ванадия приведены в табл. 1.
Распространенность ванадия в природе. По распространенности в земной коре ванадий относится к типичным редким элементам. Среднее содержание его в земной коре, по А. П. Виноградову (1962 г.), равно 0,015%.
Высокая химическая активность, переменная валентность, способность к образованию комплексных соединений объясняют обилие ванадиевых минералов в природе (их около 70 [68]) и химических соединений (особенно искусственных), по числу которых ванадий уступает только углероду.
Из всех известных минералов ванадия 40 являются ванадатами. Промышленное значение имеют только шесть минералов (табл. 2).
Ванадий — очень рассеянный элемент, в природе встречается во многих объектах, но содержание его в них невелико и близко к среднему содержанию в земной коре (табл. 3).
7
Таблица 1
Изэтопы ванадия [361, 455, 884а]
Массовое число Характер излучения Период полураспада Энергия излучения, Мэв
Э-частиц V-лучей
46 Р+ 0,426 сек. 6,3 +0,4 0,16; 0,8
47 Р+(97%) ^(3%) 31,1+0,1 мин. 1,89 +0,01 0,1609; 1,3914; 1,5523; 1,7949; 2,000; 2,163
48 К Р+ ЛГ/Р+ = О,69 15,99+0,08 дней 0,695+0,003 0,9289; 0,9443; 0,9833; 1,2181; 1,3114; 1,3381; 1,438; 2,2401; 2,255; 2,650
49 К 330 дней 0,610+0,01 Пет
50 К 6-Ю15 лет 0,780; 1,580
51 Стабильный
52m р+, ИП 3,76+0,02 мин. 2,470+0,03 0,201; 0,397; 0,500—0,900; 1,4329; 1,800- 4,000
53 ₽- 2,0 мин. 2,530 1,000
54 р- 55 сек. 3,300 0,835; 0,990; 2,210
Примечаете: g- — электрон; g+ — позитрон; К — захват орбитального электрона К, L- и М-слоя с испусканием характеристического рентгеновского излучения; ИП — изомерный переход; т — возбужденное состояние ядерных изомеров.
Получение и применение ванадия. Из-за малого содержания ванадия в рудах и концентратах производство ванадия становится рентабельным только при одновременном извлечении большинства компонентов. Поэтому ванадий извлекают попутно при производстве алюминия, титана, урана и др. Основное количество производимого в СССР ванадия получают из уральских титаномагнетитов. Ванадийсодержащие железорудные концентраты вначале проходят доменную плавку, а затем в конверторах ванадий из чугуна переводят в шлак, из которого гидрометаллургическим путем получают пятиокись ванадия [240].
Пятиокись ванадия служит сырьем для производства всех соединений ванадия, феррованадия, ванадиевых катализаторов и люминофоров и металлического ванадия. Основной потребитель ванадия — черная металлургия, где его используют как легирующий элемент при выплавке специальных сортов сталей. Далее небольшие добавки его существенно повышают прочность стали, уменьшают размер зерна, снижают склонность стали к перегреву, улучшают свариваемость и многие другие физико-механические, технологические и эксплуатационные свойства.
Для создания разнообразных марок высоколегированных углеродистых сталей ванадий комбинируют с хромом, никелем, мар-»
8
Таблица 2
Минералы ванадия, имеющие промышленное значение [68, 403, 549]
Минерал . Формула минерала Содержание ванадия, %
Фольбортит HCu3(VO4)2-3H2O 7,62—7,66
Минасрагит V2(OH)2(SO4)3- 15Н2О 7,94
Роскоэлит KV2(OH)2AlSi3O10 10-12
Карнотит K2(UO2)2(VO4)2-3H2O 10,1—10,4
Тюямунит Ca(UO2)2(VO4)2-8H2O 10,3—12,4
Патронит VS4(V2S5+nSe) 19—24
ганцем, бором, вольфрамом и другими элементами. Его используют также в качестве заменителя дефицитных и дорогостоящих легирующих элементов; так, например, в ФРГ при производстве твердых сплавов ванадием заменяют часть вольфрама. Металлический ванадий и его сплавы представляют интерес для развития таких отраслей, как ракетостроение, атомная промышленность. Ванадаты применяют при производстве люминофоров, в керамическом деле и в качестве катализаторов.
Токсичность ванадия и его различных соединений. Многие химические соединения ванадия являются ядами с самым разнообразным и часто весьма сильным действием на организм. Ванадий токсичен как в виде катионов, так и в виде анионов. Степень токсичности зависит от валентности ванадия, дисперсности частиц аэрозоля и растворимости соединений в биологических средах. Наиболее токсичны соединения пятивалентного ванадия, предельно допустимые концентрации которых в виде аэрозолей равны 0,1 —0,5 мг/м? воздуха [172]. Токсичность соединений ванадия обусловливает необходимость контроля иногда очень малых содержаний ванадия в объектах самЪй разной сложности.
Таблица 3
Среднее содержание ванадия в некоторых объектах (%) [549]
Объект Содержание Объект Содержание
Породы изверженные карбонатные Глины, сланцы Песчаники 0,015 0,0003 0,012 0,002—0,005 Почвы Животные и растения Морская вода Донные морские отложения 0,01 Ю-4 5-Ю-8 0,02
9
ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
Ванадий — мономорфный голубовато-серый металл. Полиморфные превращения его при 1550 и —28 н---38° С не подтвержде-
ны более поздними исследованиями и связаны, по-видимому, с наличием примесей [440]. Плотность ванадия чистотой 99,8—99,9% при 20° С равна 6,11 г/см3 [440], а температура плавления его 1919° С [923]. Загрязненный ванадий в зависимости от степени чистоты и способа получения металла может плавиться при температурах от 1900 до 1700° С. Его удельное сопротивление при комнатной температуре колеблется в пределах от 22,6 до 35,8 мком-см [963]. Магнитная восприимчивость ванадия чистотой 99,8%, отнесенная к единице массы, равна 5,00-10~6 э.м.е./а при 25° С [440].
Ванадий обладает высокой коррозионной стойкостью в органических и некоторых неорганических агрессивных средах. По стойкости к действию хлористоводородной и серной кислот он значительно превосходит титан и нержавеющую сталь. Ванадий в меньшей степени, чем ниобий и тантал, окисляется в атмосфере СО2,‘ обладает высокой стойкостью в расплавленных металлах, применяющихся в качестве теплоносителей [172]. Среди тугоплавких металлов ванадий имеет самую низкую сопротивляемость окислению на воздухе. Окисление начинается при 300° С и становится очень быстрым при 600—700° С, так как образующаяся при этом пятиокись ванадия расплавляется (%л = 675° С) и стекает с поверхности металла [172]. Хлористоводородная, бромистоводородная и холодная серная кислоты не действуют на ванадий, но азотная, фтористоводородная и горячая серная кислоты, смеси азотной и хлористоводородной кислот (1:4), а также растворы гидроокисей натрия и калия растворяют его [172, 549].
Высокая реакционная способность ванадия обусловливает сложность его химического поведения, большую рассеянцость в природе, включая растительный и животный мир, трудности отделения ванадия от сопутствующих элементов.
СОЕДИНЕНИЯ ВАНАДИЯ
В соединениях ванадий проявляет переменную валентность от 2-[- до 54-. Ионные радиусы (в А) равны: V(II) 0,72, V(III) 0,67, V(IV) 0,61 и V(V) около 0,4 [172]. Потенциалы ионизации атома ванадия равны (эв): I 6,74, II 15,13, III 30, 31, IV 48,35 и V 68,7 [172]. Атомный радиус ванадия равен 1,35 А для координационного числа (к.ч.) 8 и 1,36 А для к.ч. 12 [440].
Неорганические соединения
Соединения с кислородом. В системе ванадий—кислород получено большое количество окислов ванадия, а точнее, фаз, так как многие из них имеют области гомогенности. В настоящее врб-
10
мя с различной степенью достоверности можно говорить о существовании кислородных соединений (фаз), которые приведены в табл. 4. Исчерпывающие сведения о фазовой диаграмме состояния в системе ванадий—кислород можно получить из справочника [515]. Химические свойства окислов ванадия приведены в работах [172, 467, 544, 549].
VO нерастворим в воде, имеет основной характер и растворяется в разбавленных кислотах с образованием солей катиона V2+, водные растворы которых окрашены в фиолетовый цвет. Представляя уравнения гидролиза катионов металла в виде
7мп+ + рн2о = м„ (он)^"^ + рн+
и обозначая константы реакций гидролиза 0да, гидролиз катиона V2+ можно охарактеризовать следующими данными: в интервале концентраций ионов ванадия 1-10-2—5-10“5 М 1g |3П = —6,49 [139]. Кроме этого, имеются сведения [422] о существовании димера V2Q2+ при концентрации ванадия выше 2,5-10“3 М. Добавление гидроокисей к растворам солей двухвалентного ванадия вызывает образование буро-коричневого гидрата закиси V(OH)2, который быстро окисляется на воздухе в серо-зеленый V(OH)3.
V2O3 имеет основной характер, в воде, растворах гидроокисей и кислот, кроме азотной и фтористоводородной, не растворяется. Водные растворы солей трехвалентного ванадия окрашены в зеленый цвет. При добавлении к ним раствора аммиака (до pH 4—5) осаждается V(OH)3, переходящий под действием кислорода воздуха в коричневый продукт. Ион V3+ в водных растворах гидролизуется с образованием гидроксокомплексов: V(OH)2+, V2(OH)i+, V(OH)| и V2(OH)|+. Константы реакций гидролиза определены потенциометрическим методом в среде 37И КС1 при 25° С и равны: 1g Зи = —3,07, 1g р22 = —3,96, 1g р12 ~ —7,5 и 1g р23 ~ —8,7 [669]. Согласно другим данным [422], при 25° С в 3 и 17И NaCl 1g =-3,15 ± 0,05 и -2,85^+0,05; 1g ₽22 = -4,10 + 0,05 и —3,90 + 0,05, 1g р12 = —7,30 и —6,70 соответственно. Для реакции
V (ОН)2+ VO+ + Н+
при 25° С 1g р — —3,53, а для реакции димеризации иона V(OH)2+ 1g ₽20 = 2,2 [422].
V2O4 амфотерна. При растворении ее в неокисляющих кислотах образуются ионы ванадила VO2+ или диванадила V2O2+, имеющие синий цвет. Окраска их заметна при концентрации >0,1 мг/мл. Гидролиз иона ванадила сопровождается образованием только двух ионов: VO(OH)+ и (VO)2-(ОН)2+. Более высокие полимеры в растворе не обнаружены. Константы гидролиза в 3 М перхлоратном растворе при концентрации ванадия от 0,25 до 0,005 М равны: 1g рп = —6,0 + 0,1 и 1g р22 = —6,88 + 0,04 [1037], а по данным [918], 1g |3П = —5,36.
Из 0,01 М растворов солей ванадила при pH ~ 4 осаждается серовато-бурая гидроокись ванадила, произведение растворимости
11
Таблица 4
Некоторые характеристики фазовых составляющих систем ванадий—кислород [515, 530, 544]
Фаза Сингония и тип решетки Параметры решетки, А Магнетизм фазы Магнитный момент, цв Константа Кюри, "К Константа Вейсса, °К ( пл’°C Цвет
а-Фаза VOci—0,01 Р-Фаза VOo,18-0,33 6-Фаза VOo,455—0,54 (V2O) VOo,75—1,24 VO1.27 V2O3 (VO 1,5) Кубическая объемноцеи-трированная Тетрагональная объемно-центрированная Моноклинная Кубическая, типа NaCl Тетрагональная или кубическая объемноцентри-рованные Ромбоэдрическая а = 3,0240-г-3,0495 а = 2,990; с = 3,264 для VOo, 185 а = 2,940; с = 3,523 для VOo,28 а = 14,710; 6 = 14,630; с = 17,925; р = 90,633° для VOo,530 а = 4,062 = 4,125 а = 16,623; с = 16,515 или а = 2,90 а = 4,952; с = 14,002 Антифер- 2,83 . 168 — 72 2000 1977 Фиолетовый Черный
VO 1,67 (V3O5) VO 1,75 (V4O7) Моноклинная Типа рутила а = 9,983; 6 = 5,031; с = 9,835; а = 138,80е ромаг-нитная » » 2,77 2,82 235 + 3 130 + 3 — 302 — 142
VO1.801 (V5O9) * Триклинная а = 5,465; 6 = 7,001; с = 7,825; а = 94,Г; Р = 97,65°; 7= 109,03° 2,60 162 — 142
Таблица 4 (окончание)
Фаза Сингония и тип решетки Параметры решетки, А Магнетизм фазы Магнитный момент, Цц Константа Кюри, “К Константа Вейсса, •К tпл-°C Цвет
VO1,84 (Veon) VO 1,86 (V,o13) VO1.87 vo2 (V2O4) Типа рутила То же » Моноклинная (при <70° С) Тетрагональная (при > 70° С) а = 5,77; 6 = 4,50; с = 5,39; р = 122,16° а = 4,54; с = 2,85 Парамагнитная » Антиферромагнитная » 2,14 2,30 2,96 345±3 — 47 —107 —292 1545 Голубой Индиговый
Veo13 (V12O26) VO2.33 (V3O7) (V9O21) v205 Моноклинная » Ромбическая а =11,90; 6 = 3,671 с = 10,122; р= 100,87° аГ= 21,93; 6 = 3,68; с = 18,51; р = 96,45° а =11,519; 6 = 3,564; с = 4,373 » Диамагнитная 1,95 (2,13) 1,32 0 165 -237 —30 710 675 Коричневый (за висит от-способа получения)
которой равно 7,4-10-23 [298]. При pH 8—10 VO(OH)2 растворяется. Как V2O4, так и VO(OH)2 при растворении в щелочах образуют соли поливанадистой кислоты H2V4O9 или H2V2O5, называемые ванадисто-кислыми солями, ванадитами или гипованадатами.
V2O5 имеет отчетливо выраженный кислотный характер. При растворении в растворах щелочей или взаимодействии при нагревании с окислами основного характера она образует ванадаты, состав которых зависит от соотношения реагирующих компонентов. Термическим путем получены мета-, пиро- и ортованадаты многих металлов (щелочных, щелочноземельных, некоторых переходных <7-, а также и />-металлов) [512, 531]. Фазовые диаграммы состояния систем окисел металла—пятиокись ванадия описаны в работах [516, 530]. В некоторых системах наряду с ванадатами отмечается образование кислородных ванадиевых бронз, содержащих некоторое количество ванадия в степени окисления 4+ [516, 530, 531].
При растворении пятиокиси ванадия в кислотах образуется катион VO2, а в сильнокислой среде обнаружен катион VO3+ [511].
Многочисленные сведения о составе ванадатов, получаемых из водных растворов, часто противоречивы, а формулы их не всегда достаточно обоснованы. Противоречия возникают из-за неправильного учета ионного состояния как ионов пятивалентного ванадия, так и катионов металла, образующих ванадат.
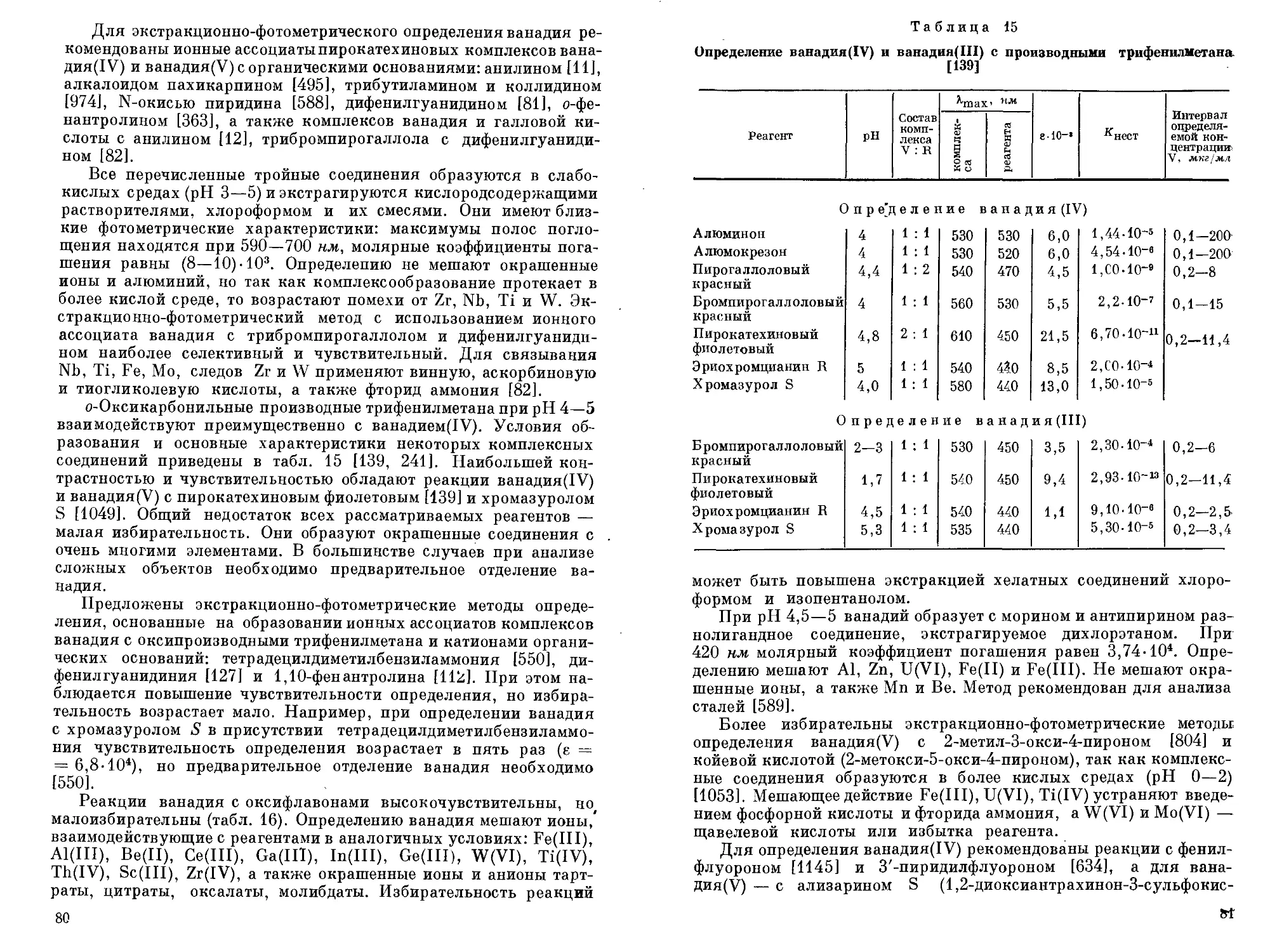
В таблице 5 приведены сведения о константах равновесия ионов пятивалентного ванадия в водных растворах. Все уравнения равновесных! реакций расположены в таблице в порядке возрастания величины В = H+/VOf“, характеризующей затраты эквивалентов кислоты на превращение ортованадат-иона в ион, получаемый по соответствующей равновесной реакции. Для каждого уравнения дана расчетная формула, которая позволяет наглядно представить линейную зависимость pH от логарифма концентрации ванадия в растворе (с) для различных степеней превращения (а) исходных форм.
На рисунке 1 представлена номограмма для определения состояния ионов пятивалентного ванадия в водных растворах, построенная на основании данных табл. 5. Линии номограммы разделяют области преобладания ионов. На номограмму нанесены не все линии, обозначенные индексами в табл. 5, а только те, для которых константы равновесия найдены в перхлоратной среде.
Кроме указанных в табл. 5 значений, имеются сведения о константах, найденных в средах: NaCI, КС1, CsCl, LiCl, N(CH3)4C1 [1069], Na2SO4 [1103] и NaClO4 [688, 945, 994, 1037].
Из растворов, подкисленных до отношения В 2,6, ванадий осаждается. Скорость осаждения значительно увеличивается при нагревании растворов. Поскольку природа осадков до конца не была выяснена, то называли их по-разному: гидратированная пяти; окись ванадия [468, 1180], декаванадаты ванадия [54], гекса- и 14
Таблица 5
Константы равновесия ионов пятивалентного ванадия в водных растворах
IgK [Среда t, °C c Метод pH1/2 Индекс Литература
При В = 1
1. VOr+H+?tHVOr •I 1 4
[HVOf] 1—<х
pH:=Ig^_Ig—-^-=Ig^ + Jg —
+11,131 0,2 I NaClOi I 20 I tO'2-Rr4 I ПТ | 11,131 la I[452]
+13,5 | 0,1 | NaC104 | 20 | НН-Ю'4 I СП | 13,5 | 16 J [1069]
2. VOf + HaO HVOf + OH-[VOf] fl _ a)
ph = - ig kw + ig к + ig [Hv012_] = -Ig Kw + Ig К + Ig
—1,013| 3,0 I NaClOj I 25 | lO^-lO1 | СП |12,887| 2 | [945]
3. 2HVOf j±V2O4" + H4O
lgC = -lgK + lg2-(-1^-)a
—2,79| 0,2 I NaCIO4 I 20 I 10'2-10'4 I ПТ I I За I [452]
1,68] 3,0 I NaCIO4 I 25 | 102— IO'4 J СП J | 36 |[945]
4. 2VO|~ + 2H+ V2O4- + H,0
[VOf]2 2(1—a)2
PH = Va Ig К + 4/2 lg-f 4 = V2 Ig К + Va Ig--~ + V* !g c
1'/4и7 J
25,5 | 0,2 | NaClO4 | 20 | IO'2—IO'4 I ПТ |12,525| 4 |[452]
При В = 1,5
5. 2HVOf Л HV2C>3" + OH-
[HVOfP 2(1— а)2
pH = -Ig Kw + IgX + Ig tHy-pa-j = -ig K» + ig К +lg-—- + ig c
—3,18 | 0,5 | NaCl | 25 | 1,8• IO'3—6,2• 1Q-4| ПТ | 10,72] 5 | [803]
6. V4O4“ + H2O jt V4O6' + 2H+
[V4O«-] а
pH -= -i/t Ig К + i/2 Ig = -4/2 Ig К + V2lg
—17,3 I 2,0 | NaCIO4 I 20 [ г.Ю^-З.Ю'3 [ ПТ | 8,65 I 6 f[886]
15
Таблица 5 [продолжение)
lg к Среда t, *c c Метод pHi/2 Индекс Литература
При В = 2
7. nvof+ h+^h2vo;
[HVOf] 1 —а
pH = lg* + Ignv^=lg*+Ig“
8,23 | 0,1 | NaC104 | 20 | IQ^-IO'4 | ПТ | 8,23 | 7 | [1069]
8. V3O39- + 3H2O 3HVOf + 3H+
, , [HVOfp 3a3
pH = -1/, 1g К + 1/, 1g - = -i/з lg К + i/з 1g r— + 3/3 1g c
L'3 J9 J
-30,481 III I nT |10,16l 8a I [1102],
—30,90| III I СП |10,30| 86 | [1061]
9. 3HVOf + 3H2O it V3O®- + 3OH-
[HVOf]3 Заз
pH — •~Ig^H,+7з +Jg [y3Q3_] ~ *W + Vs lg *4-V3 lg + + 2/з lg c
—10,421 0,5 | NaCl | 25 | 8.10~2—6,25.10*4 | ПТ |10,215| 9 | [803|
10. V4 V4O£ + H2O HVO f + H+ [HVOf] 4*Ла
pH = -lg К + lg = -lg K-V Ig^^T + Vilgc
—10,291 0,2 I NaClO4 I 20 I 10'2-10'4 I ПТ I Ю,29| 10a I[452]
— 10,15] 2,0 ) NaClO4 | 20 | 2-Ю'1—3-10~3 | ПТ | 10,15| 106 | [886]
и. V» v4o*; 4- H2O it V2Of 4- 2H+
[V2Of] а
pH-----1/2 lg #4-1/»lg Qi-,»/; = ~V» lg К 4-1/2 lg —4- i/4 lg с ч
lv4ul2J. I1 —a)
— 18,461 0,2 I NaClOi I 20 I 10~2-10'4 I ПТ I 9,23 I Ha 1 [452]
— 19,06] 2,0 ) NaClO* | 20 | 2.10'1—З-Ю'® | ПТ | 9,48 | 116 | [886]
12. Vss V4On + H2O HV2O3"+ H+ [HV20n a
pH = -lg К 4- lg [ViO4-]l/2 = -lg* + lg (1_a)1/2 4- V» lg c
—10,0 2,0 NaClO4 20 2-10'1—3-10'3 ПТ 10,0 12a [886]
—10,29 0,2 NaClO4 20 Ю-a—IO'4 ПТ 10,29 126 [452]
16
Таблица 5 (продолжение)
IgK Среда t, ’C c Метод рн1/г Индекс Литература
13. HVO2 ?iVO3+OH-
[HVOf] pH = -lg Kw + lg К + lg j -ivu3j
—6,0 I 0,5 I NaCl I 25 | 6,25-104 | ПТ | 7,9
14. HVOf + H+ VO3 + H2O
[HVOf] 1—а
pH = lg 7C + lg^Ig^+lg —
8,44
= — lg^w + lg^+lg^-^
13
|[803]
8,44
—5,55
Ацетатный буферный
Ацетатный буферный
[vo;j
5-Ю-6
СП
15. V3O® 3VO3 1 —• а lg с = Чг 1g К + 4i 1g -35г
3,6.10“2—2,8-10-4
СП
14
15
[1061
>[1061]
16
|[209]
16. 4VO; v4o412
lg с = -1/з 1g К + 1/3 1^-(11а)1
10,65| 3,0 | SrCl„NaCl| 20 | I I I
При В = 2,5
17. 3HV10O5~ +6 Н2О 10V3O|- + 15Н+
[V:iO®"]10 10заю
pH = —’/15 1g К + 1/15 lg п6-,8 = —1/15 lg К + 1g 31° 11 — CtV + 7/1® 1g С [ПУ10и 28-1 ' '
—108,0| 1,0 | NaC104 | 20 | 10~2-5-10~3 [ ПТ [ 6,94 | 17 |[1069]
18. HV10O52; + 5OH- = 2,5V4O4- + 3H2O
[V4O«-]2’6 10a2,5
pH = -lg Kw - i/5 lg К -h 1/5 lg - 5 = -lg Kw-i/5lg A+Vs lg42->5 (1_аЛ
LnviQU28J ' '
+ 3/ю 1g с
—35,55| 0,2 1 NaClO4 1 20 | 10“2—10-4 | ПТ | 6,68 | 18 |[452]
19. HV10O5--V10O62-+H+
[HVi0O5-] a
pH = -lg К + lg —= -lg К + lg
L V10V 28-1
—6,94 0,1 NaClO4 20 ПТ 6,94 19a [1069]
—6,C6 1,0 NaClOt 20 ПТ 6,C6 196 [1069]
17
Таблица 5 (продолжение)
lg К IJ. Среда t, °C c Метод ₽H1/2 Индекс Литература
При В = 2,6
20. h2v10o£ hv10o|; + нт [HV10O|7] а
—3,65 | 0,2 I NaCIO4 I 20 I IO-2—IO-4 I ПТ I 3,65 I 20a I[452]
—3,60 | 1,0 | NaCIO4 | 25 | 2-10~2— 2-10~3 | ПТ | 3,60 ] 206 |[1O37]
При В = 3 2i. hvo, vo; 4-н+ о о 1
[VO“] а
pH = -lg К 4- 1g = -lg К +.Ig
—3,78 | 0,5 | NaC104 | 25 | 1 • 10'“-1 • 10~7 | ЭК | 3,78 | 21 | [726]
22. n2vo; + H+ H3VO4 [H2vo;] . 1-a
pH = ]g к + lg = h к + lg -v-
3,4 | 0,1 |NaCIO4 | 20 [ 10~2—IO'4 | ПТ | 3,4 [ 22 |[1069]
При В = 4
23. 10VO2 + 8H2O H2V10O42; + 14H+ [H2V10O“r] а
PH = — Vu lg K + Vu lg ~i-Vz7'll0~ = - Vi4 lg К 4-1/14 lg ]o(1T,auo -—9/u lg c
—7,5 I 0,2 I NaC104 I 20 I IO'2—IO-4 I ПТ I 0,656l 23a I [452]
-6,75 J 1,0 I NaC104 | 25 | 2-10~2-2.10-3 | ПТ | 0,603| 236 | [1037]
24. VO2 4- H2O HVO3 4- H+ [HVO3] а
pH = -lg- К 4- lg [VQ+j =—lgK4-lgi_a
—3,2161 0,061 NaC104 I 25 I 6,1.10-“ I КН I 3,2161 24a I [616]
-3,2 | 0,5 | NaClO4 | 25 | IO-1—Hr7 | ЭК | 3,2 | 246 | [726]
25. VO2 4- H2O VO; 4- 2H+
[VO3] а
pH = -1/2 lg к 4- 1/2'lg ~~ = - v2 lg к 4-1/2
L ’ O2]
-5,26
ЭХ I 2,63 I 25 I [154]
18
Таблица 5 (окончание)
1g К
м- Среда f, °C С Метод рн1/г Индекс
Литература
При В = 6
26. V0* + 2Н+ V03+ + Н20
[V0+2] 1-а
Рн = -V21g к + v2 lg = -1/2 lg к + V2 lg
2,79 || || 4,58-10-3- | СП | 1,395| 26 Ц5И]
II || 6,09-10-3 I I | I
Примечание. К — константа равновесия реакции; р — ионная сила, М; с — концентрация ванадия, г-атом/л-, pH,/, — значение pH в расчетном уравненйи при с = 1 о—
иа — Чг; а —степень превращения исходной формы; В = H+/VO4 ; Kw — ионное произведение воды при указанной температуре; ПТ — потенциометрический, СП — спектрофотометрический, ЭК — экстракционный, КН — кинетический и ЭХ — электрохимический методы определения [констант; индексы уравнений расчета введены для обозначения соответствующих им линий на номограмме (см. рис. 1).
додекаванадаты щелочных металлов [210]. Доказать, что они являются гидратами пятиокисиванадия, никому не удалось [1180]. ИК-спектры этих осадков схожи с ИК-спектрами гекса- и додекаванадатов щелочных металлов [210]. Считая образование этих соединений промежуточной стадией между анионными и катионными формами, по нашему мнению [1177], их следует рассматривать как двойные декаванадаты ванадия и щелочных металлов, а состав представить равноценными гекса- и додекаванадатам формулами M4(VO2)2V10O28 и M2(VO2)2V10O27.
В сильнокислой среде катионы щелочного металла в соединениях могут частично или полностью замещаться катионами водорода. Изучение процесса гидролитического осаждения ванадия(У) при кипячении и длительном выдерживании растворов, подкисленных до отношения В от 2,3 до 4,0, показало, что графическая зависимость pH от логарифма остаточной концентрации ванадия (су) представляет собой две пересекающиеся прямые, одна из которых характеризует процесс осаждения, а вторая — растворения. В табл. 6 представлены рассчитанные методом наименьших квадратов параметры уравнения pH = а + Ъ lg cv и состав осадков, для которых приняты следующие обозначения: I - M4_xHx(VO2)2V10O28-nH2O, II-M2_xHx(VO2)2V10O27-nH2O (М = = Li+, Na+, К+, NHt); III - Na2VeO15(NO3)2; IV - NH4VO3 [1177].
Параметр а в табл. 6 характеризует растворимость осадков, а параметр Ъ — наклон линий осаждения и растворения, который связан с природой процессов. Состав осадков установлен следующими методами: дериватографии и ИК-спектроскопии, полного химического и рентгенофазового анализов.
Обширность области существования твердых фаз (см. рис. 1) свидетельствует о метастабильности декаванадат-ионов в растворе.
19
-igc
6 9
1Z pH
Рис. 1. Номограмма для определения состояния ионов V(V) в среде NaC104 Обозначения линий соответствуют индексам табл. 5. Заштрихована гетерофазная область растворов NaVOs (время осаждения 1,5 часа, температура 100° С)
Многочисленные исследования взаимодействия ванадат-ионов с катионами металлов в водных растворах [52, 53] и их совместного поведения в области pH 0—14 [23, 50, 51, 76, 100, 168] позволили сформулировать основные положения теории образования и син-' теза ванадатов в водных растворах, хорошо согласующиеся с современными представлениями о составе ионов ванадия(У) и взаимодействующих с ними катионов металлов.
На рисунке 2 приведены в качестве примера номограммы для определения состояния ионов щелочноземельных металлов, аммония и алюминия, построенные на основании данных о константах реакций гидролиза и произведений растворимости гидроокисей [53, 298]. Подобные номограммы можно построить для большинства катионов металлов, гидролиз которых хорошо изучен. На основании номограмм (см. рис. 1, 2) можно сделать следующие заключения.
20
Таблица 6
Результаты исследования процесса гидролитического осаждения ванадия (V)
Состав растворов Условия осаж-• дения Параметры уравнений Состав осадков на стадии
время t, °C осаждения растворения осажде-НИЯ растворения
а ъ а ъ
0,3 м 1,5 час. 100 3,63 0,66 0,15 -0,64 И-и II
LiVO3+ +H2SO4 55 сут. 20 4,00 0,48 0,24 -0,79 I-HI II
0,3 м 1,5 час. 100 5,19 0,49 -1,31 —1,13 I II
NaVO3-1- +HNO3 55 сут. 20 5,41 0,38 0,095 -0,82 14-Ш II+II1
0,3 М 1,5 час. 100 7,54 0,43 0,12 —0,68 I I
KVO3+ + HNO3 25 сут. 20 6,18 0,14 -0,16 —0,72 I I4-H
0,15 М 1,5 час. 1С0 8,09 0,8 -0,22 —0,84 H-IV I+II
nh4vo3+ + H2SO4 25 сут. 20 6,04 0,87 —0,28 —0,68 I 1+П
Ванадаты в виде кристаллогидратов образуются лишь в том случае, когда взаимодействующие ванадат-ион и катион металла могут сосуществовать. Поскольку многие катионы существуют в негидролизованном виде только в кислой среде, где ванадий(У) находится в форме декаванадат-ионов, то большинство кристаллогидратов являются декаванадатами. Например, известны кристаллогидраты типа М3У10О28-геН2О, где М = Са, Zn, Мп, Cd, Со, Ni, а п = 17, 19, 15, 15, 22, 22 [52, 76, 100]. Кроме того, из рис. 2 следует, что пиро- и ортованадаты аммония не существуют, а катионы щелочноземельных металлов способны образовать кристаллогидраты дека-, мета-, пиро-, а иногда и ортованадатов.
В присутствии избытка катионов щелочных металлов или аммония наряду с нормальными ванадатами образуются двойные, например, МзМ^У^Огз-пНгО или M4MIIV10O28-nH2O, где М1 = Na+, К+, NHJ, Rb+, Cs+; = Са2+, Zn2+, Cd2+, Mn2+, Со2+, Ni2+ [23, 52, 76,100]. Растворимость двойных ванадатов, как правило, меньше растворимости нормальных ванадатов и убывает с ростом радиуса катиона щелочного металла. Поэтому выделение нормальных ванадатов проводят в присутствии ионов лития или натрия, а для повышения полноты осаждения нормальной соли в раствор добавляют ацетон или этанол.
При взаимодействии любых ванадат-ионов с легко гидролизующимися катионами металлов (см. рис. 1 и 2) одновременно протекают реакции сопряженного гидролиза и образования основных, преимущественно рентгеноаморфных, ванадатов. Поскольку ванадаты щелочных металлов подобны карбонатам или фосфатам и яв-
21
Рис. 2. Номограммы для Определения состояния ионов алюминия, аммония и щелочноземельных элементов
а — рассчитано по данным [53]; б, в — по [298, 628]; пунктирные линии рассчитаны из значений nPjfe(OH)n
' 10 12 pH
ляются неявными основаниями, то добавление их к раствору соли металла вызывает сдвиг равновесия
Ме”+ + zH2O Me (ОН)”-* + zH+
(х = р/<? в гидроксокомплексах типаМед(ОН)р”9~р)) в правую сторону и изменение pH в область гидролизованного состояния катионов металла. Сопряженность* процесса гидролиза обусловлена двумя причинами: 1) связыванием освобождающихся по указанной реакции ионов водорода вводимыми ванадат-ионами и образованием ванадат-ионов состава, соответствующего области pH гид-
22
ревизованного состояния катионов металла (см. рис. 1 и 2); 2) связыванием гидроксокомплексов образующимися ванадат-иона-ми в малорастворимые основные ванадаты. Например, гидродекаванадат гидроксоалюминия [Al(OH)x]6^,/3_xIIy V)(IO28 образуется независимо от того, с каким ванадат-ионом взаимодействует соль алюминия [53]. Степень гидролиза (величина х) зависит от природы взаимодействующих ионов и их отношения в исходной смеси. Она симбатна гр а мм-атомному отношению V/Ме и возрастает в ряду ванадат-ионов: дека- < мета- <; пиро- < ортованадат.
Избыток ванадата щелочного металла не оказывает влияния на состав образовавшегося соединения, если ванадат-ионы осадка и добавляемого раствора могут находиться в равновесии (см. рис. 1). В противном случае pH раствора смеси будет повышаться сначала до pH существования промежуточных ванадат-ионов, а затем до pH раствора добавляемого ванадата. Состав соединения при этом изменяется как за счет увеличения содержания гидроксильных ионов в гидрооксокатионе, так и за счет одновременного образования промежуточных менее полимеризованных ванадат-ионов. При деполимеризации ванадат-ионов возникают избыточные слабо связанные или не связанные с осадком ванадат-ионы, которые диффундируют в раствор. Малая подвижность этих полимерных ионов в твердой фазе способствует образованию двойных основных ванадатов со щелочными металлами. По этой же причине часто возникают коллоидные состояния осадков. Подобные явления наблюдаются и при взаимодействии осадков основных ванадатов с гидроокисями щелочных металлов.
Поведение ванадат-ионов совместно с катионами металлов при постепенном повышении pH от 2 до 13 характеризуется медленным последовательным превращением декаванадатов в мета-, пиро-,а иногда и в ортованадаты. Состав образующихся при этом ванадатов также зависит от природы катионов металлов, исходных концентраций, pH, температуры и времени выдержки. При pH <2 образуются так называемые продукты гидролитического осаждения ванадия. Состав продуктов аналогичен приведенному в табл. 6.
Существование гексаванадат- (УеОД) и тетраванадат-ионов (V4O13), а также соответствующих им ванадатов металлов следует считать пока недостаточно обоснованным.
Термическое поведение ванадатов, полученных из растворов, зависит от их состава. Как кристаллогидраты, так и основные ванадаты дегидратируются. Дегидратация их протекает в несколько стадий, что свидетельствует о разном характере связи молекул воды в соединениях. Полная дегидратация основных солей наступает при температуре выше 400° С, а кристаллогидратов — до 350* С. В результате дегидратации большинство ванадатов распадается с образованием рентгеноаморфных продуктов. При содержании в соединениях щелочных металлов образуются легкоплавкие кислородно-ванадиевые бронзы и ванадаты щелочных металлов.
23
Растворимость в ряду ванадатов уменьшается от дека- к ортованадатам. По характеру растворения в воде ванадаты можно разделить на две группы. К первой группе относятся конгруэнтно растворяющиеся ванадаты, состав которых при растворении не изменяется. Вторую группу составляют ванадаты, растворение которых сопровождается гидролизом (инкопгруэнтное растворение). Их большинство. Гидролизуются не только ванадат-ионы, но и катионы некоторых металлов. В результате образуются либо основные ванадаты, либо менее растворимые нормальные ванадаты.
Пероксосоединения ванадия. При обработке разбавленных растворов ванадатов щелочных металлов перекисью водорода в слабокислой среде образуются желтые растворы, содержащие моно- и дипероксованадат-ионы [VO3(O2)]3-, [VO2(O2)2]3- или фиолетово-синие растворы с анионами [У(О2)4]3", [VO2(O2)2]3', [V2O2(O2)5]4~. В сильнокислой среде образуются коричнево-красные растворы, содержащие пероксосоли радикала, [V(O2)]3]+. Известны монопероксометаванадаты MI[VO2(O2)], монопероксоортованадаты M|[VO3(O2)]-гаН2О, дипероксоортованадаты М3[УО2(О2)2] • гаН2О, тетрапероксоортованадаты М3[У(О2)4], тетрапероксодиванадаты М4[У2О3(О2)4], пентапероксодиванадаты M4[V2O2(O2)6] (М = К+, Na+, NH4). Пероксосоединения ванадия плохо растворимы в спирте, в воде подвергаются гидролизу с образованием перекиси водорода, которая в свою очередь разлагается с выделением кислорода.
Соединения с галогенами [439]. Со фтором ванадий образует фториды VF2, VF3, VF4 и VF5, оксифториды VOF2 и VOF3, двойные фториды MI[VF6] (М1 = К+, Na+, Ag+), Ba[VF6]2 и др. и двойные оксифториды K2[VOF4], M2[VOF4(H2O)] (М1 = Na+,K+, NH4), [МП(Н2О)6] [VOF4(H2O)] (Мп = Ni2+, Zn2+), (NH4)3[VOFJ, M4VOFJ и M1[VOF6] (M1 = K+, Na+, NH4). Хотя оксифторид ванадия VO2F не выделен, оксифторсоли типа M2[VO2F3] и Ml[VO2F4] известны, например K2[VO2F3], (NH4)2[VO2F3] и
(NH4)3[VO2FJ.
С хлором ванадий образует хлориды VC12, VC13 и VC14, оксихлориды VOC1, VOC12 и VOC13, а также и двойные хлориды K[VC14], K2[VC15]. Кроме того, известны гексамин- и гексакватри-хлориды ванадия [V(NH3)6]C13 и [V(H2O)6]C13.
С иодом и бромом ванадий образует ди- и тригалогениды VHal2 и VHal3, оксибромиды VOBr2, VOBr3 и V2O3Br4. Из водных растворов получены VBr4SbBr3-7H2O и VOJ2-4,5H2O. При взаимодействии с водой дигалогениды разлагают ее с выделением водорода, а тригалогениды легко гидролизуются. Низкие температуры кипения некоторых галогенидов ванадия (VF5 111,2° С, УСЦ 148,5* С и VOC13 126,7° С) позволяют отделять ванадий и проводить его очистку.
В качестве растворителей галогенидов и оксигалогенидов вана-
24
дня часто применяют спирты, эфиры, хлороформ, бензол, сероуглерод и др.
Соединения с водородом. При комнатной температуре в ванадии растворяется до 2 ат. % водорода. Фазовая диаграмма системы ванадий—водород изучена в области от 2 до 50 ат. % водорода (172]. В области от 2 до 30 ат. % водорода существуют а- и р-фазы, а гидрид ванадия VII с кубической решеткой гомогенен в области 38,6—50 ат. % водорода (а — 4,147 4,159 А).
Соединения с углеродом. Диаграмма состояния системы ванадий—углерод известна в двух вариантах [172]. Область гомогенности V2C лежит при 30—33,3 ат. % углерода, a VC — при 42,5—47,5 ат. % углерода (при 1650° С). Кроме того, имеются сведения о существовании карбидов ванадия V3C, V3C2 и V4C3 [439, 450].
В тройной системе ванадий—углерод—кислород обнаружены две оксикарбидные фазы следующего состава: б-фаза VC0!28O0i8 — VC0,e2O0il7 и е-фаза VCoi630 VCoj880 0 0 8 [172].
Нитриды. В системе ванадий—азот установлено существование V3N и VN с областью гомогенности первого при 25—33 ат. % азота, а второго — 42,9—50 ат. % азота [439, 443].
Фосфиды. В системе ванадий—фосфор имеются четыре фазы: V3P, V2P, VP и VP2. Фаза V2P имеет переменный состав V2P—Vj 6Р [439].
Арсениды. В системе ванадий—мышьяк достоверно установлено существование Двух соединений: V3As и VAs. Существование фазы V2As предположительно [439].
Соединения с серой. Сульфиды. В системе ванадий—сера найдены четыре фазы: V5S4, V2S3, VS4 и V3S [439]. Кроме указанных сульфидов, полученыVS(или V2S2), VS2 и V2SB. Последний встречается в природе в виде минерала патронита.
Сульфаты. Из растворов серной кислоты получены сульфаты ванадия [V(H2O)6]SO4-Н2О, (NH4)2SO4-VSO4-6H2O, V2(SO4)3-reH2O (п = 0, 3, 4, 9, 10 и И), H2SO4-V2(SO4)3- 12Н2О, квасцы MV(SO4)2-12H2O (М = К+, Rb+, Cs+, NH|, Т1+) и сульфат ванадила VOSO4-7iH2O (п = 2, 3, 3,5, 5 и 6,5) [439].
Из растворов азотной и хлорной кислот выделены VO(Nt)3)2 и VO(C1O4)2.
Комплексные соединения
При образовании комплексных соединений ванадий участвует в любой из известных степеней окисления. Заряд его катионов в растворах не превышает трех. Ванадий в степенях окисления 4 + и 5+ образует устойчивые оксикатионы VO2+ и VO|+, а иногда VO3+. По способности преимущественно координироваться в комплексных соединениях с донорными атомами лигандов N,0 и S катионы ванадия (IV) и ванадия(У) относятся к ряду металлов, которые образуют с кислородом более прочную связь, чем с азотом, а катионы ванадия(Ш) — к ряду металлов, у которых связь
25
с азотом прочнее, чем с кислородом. При образовании комплексных соединений, как правило, возникают гетероциклы, в которых катионы ванадия являются акцепторами электронных пар кислорода и азота. Наиболее прочными оказываются комплексные соединения, в которых возникает один или несколько пяти- или (с двойной связью) шестичленных циклов.
Для ванадия в разных степенях окисления характерны следующие функциональные аналитические группировки атомов (ФАГА) [78, 274, 348]:
V(V)
-C-N-
II I
О он
=N(O)-C
''sNa(K)
V (IV) -ОН -соон 1>он >-он /N
он ^соон ^-OH(SH)
V(IV) и V(V) =N—ОН
\n-ch2-cooh
ОН НО
Необходимо отметить также, что исследования комплексных соединений ванадия с органическими реагентами выполнены в основном спектрофотометрическим методом, возможности которого в ряде случаев использованы не полностью. Многие исследователи ограничиваются изучением спектрофотометрических характеристик исходных компонентов и соединений, установлением соотношения компонентов в соединении и даже не всегда определяют константы их устойчивости. Вопрос о состоянии ионов ванадия в соединениях чаще всего остается невыясненным. Особенно это касается ванадия(М), ионный состав которого весьма сложен. Во многих случаях не решается даже такой вопрос, в какой — катионной или анионной — форме находится ванадий(У) в соедине-1 нии. Это видно хотя бы из того, что при образовании большинства комплексных соединений с органическими реагентами при оптимальном pH (2—5) ванадий в соединениях представляют либо в катионной форме VO3+, VO (ОН)2+, VOJ, либо в виде анионов V40i7 и VeOt,, существование которых, однако, не доказано. Кроме того, не всегда учитывается изменение характера спектра, обусловленное переходом бесцветных ионов ванадия в оранжевоокра-шенные декаванадат-ионы. Найденные константы устойчивости соединений в таких случаях не отражают в полной мере химизм процессов и носят условный характер. »
26
Основные сведения о соединениях ванадия с органическими реагентами, используемых в анализе, даны при изложении методик в соответствующих разделах.
Ацидокомплексы. Катионы ванадия, взаимодействуя с анионами кислот, образуют комплексные соединения. В кристаллическом виде выделены следующие комплексные соединения:
K4[V(CN)e]-3H2O
K3[V(CN)6]
K2[VO(CN)4]-5H2O
M3[V(C2O4)3]-3H2O
(NH4)2[VO(C2O4)2]-2H2O (NH4)2[V2O2(C2O4)3].54-8H»O
K2[V2O2(C2O4)3]-4H2O
K3[V(SCN)„].4H2O (NH4)3[V(SCN)8]-4H2O Na3[V(SCN)e]-12H2O K2[VO(SCN)4]-5H2O (NH4)2[VO(SCN)4].5H2O M3[V(C3O4H2)3] (M = Na+, K+, NH4) (NH4)2[VO(C3O4H2)2].4H2O (NH4)2[VO(CsO4H2)2]-3H2O
Гетерокомплексы. Известны гетерополисоединения, в которых ванадий присутствует как центральный элемент-комплексообра-зователь или как кислый радикал в форме координированном группы. Большинство гетерополисоединений ванадия додержит в качестве координированных групп кислотные радикалы V2O8~, \У2ОГ, Мо2ОГ, С2ОГ и др.
р* Формулы фосфорцомолибденованадиевых и фосфорновольфра-мованадиевых кислот представляют в виде H7[P(Mo2O7)m (У2О8)ПЬ •zH2O и H7[P(W2O7)m (V2O8)J -а:Н2О (где п = 2, 3, 4, а т = 4, 3, 2, поскольку п + т = 6). Они являются производными фосфорно-ванадиевой кислоты Н7[РУ12О38] или Н7[Р(У2О6)8]. В свободном состоянии получены кислоты Н7[Р(Мо2О7)5(У2О8)Ьа:Н2О, H7[P(W3O7)6(V3O8)bzH2O и H7[P(V2O6)8bl3H2O и их соли CssH4[P(Mo307)5(V206)bzH20, (NH4)eH[P(W2O7)m(P2O8)nbzH2O, K8H[P(W2O7)5(V2O8)bzH2O и Na3H4[PV12O38b
Известны фосфорнованадовольфрамомолибденовые кислоты H7[P(V208)(W207)4(Mo207)bzH20, H7[P(V2O8)(W2O7)3(Mo2O7)2b
• zH2O, H7[P(V20e)(W307)2(Mo207)3bzH30, H7[P(V2O8)(W2O7).
• (Mo2O7)4bzH2O и их соли.
Существование фосфорномолибденованадиевых комплексов с нечетным числом атомов ванадия поставило под сомнение возможность эквивалентной замены ванадия молибденом как лигандом. В результате была предложена формула фосфор-12-молибде-новой кислоты, в которой часть молибдена может замещаться на атомы ванадия. Препараты выделенных в свободном состоянии кислот соответствуют формулам Н4[РМопУО40Ь34Н2О, Н6[РМо10У2О40Ь32Н2О и Н8[РМо9У3О40Ь34Н2О, а соли—формулам Na4[PMO11VO40b8H2O и (NH4)3H[PMO11V0wb75H20.u На кривых титрования этих кислот наблюдаются две ступени: нейтрализации при pH 3 и распада соединения при pH 8,5.
Мышьяк образует мышьякованадовольфрамовые кислоты,
27
Таблица 7
Константы устойчивости комплексных соединений ванадии
Лиганд Комплексный Метод ъпре- igP
ИОН деления константы д (среда) ратура
Нитрат-ион [VO2NO3] Спектрофо-тометричес- 20 1,0 (NaC104) —0,49* [545]
кий
Сульфат- [VO2SO4]~ » 19 l,Cl(NaC104) 0,97 [545]
ион
Фосфат-иои [PO2(VO3)2]~ » 4,79 [545]
[V(HPO4)S]- » 9,26* [545[
[VO2HPO4]~ » 25 1,0 (NaClO4) 4,60* [545 [
[VO2(HPOJ2p- » 25 1,0 (NaC104) 9,31* [545]
Фторид-!ЮН [VO.2F] » 20 1,0 (NaClO,) 3,04 [545]
|V02f2]~ » 20 1,0 (NaClO4) 5,60 [545],,
[VO2F3]2~ » 20 1,0 (NaC104) 6,86 [545]
Хлорид-иоп [VO2C1] » 19 l,Cl(NaC104) -0,38 [545]
Перекись V(V):H2O2=1:1 » 20 (1/V HC1) 4,52 [545]
водорода » — (7/W H,SO4) 4,91 [545]
» — (1/V H2SO4) 4,54 [545]
» — (1А HC1O4) 4,53 [545]
Этиленди- [VO2A]*~ » 20 0,1 (NaClOJ 18,05 [545]
аминтетра- [VO2A]4- » 20 0,1 (KCI) 15,55 [545]
уксусная [VO2HA]2- » 20 0,1 (NaClO4) 11,39* [545]
кислота (Н4А) [VO2HA]2~ [VO2H2A]~ » » 20 20 0,1 (KCI) 0,1 (KCI) 9,6* 6,93* [545] [545]
[VO2H3A] » 20 0,1 (KCI) 5,6* [545]
[VH2A] Потенцио-мстриче- 20 0,1 (KCI) 12,7* [78]
ский
[VHA] » 20 0,1 (KCI) 25,9* [78]
[VHA]~ » 20 0,1 (KCI) 3,50* [165a]
[VOHA]~ 20 0,1 (KCI) 4,4* [165a]
[VOH»A] Потенциометрический, полярогра- 20 0,1 (KCI) 18,77* [78]
фический
Алюмион [VOC22H16O8N] Спектрофо-тометриче- 3,041 [348]
ский
Салицилат- [VOSal,]- )> 23 0,2 (KNO3) 21,85 Г545]
ИОН
Сульфосали- [VOSSal]- 23 0,2 (KNO3) 21,23 [545]
цилат-ион Пирокате- [VO2C6HtO2r 20 11,65 [545]
хин [VO(C6H4O2)2]- » 20 23,34 1545] f
28
Таблица 7 (окончание)
Лиганд Комплексный ион Метод определения константы t, «с ц (среда) igP Литература
Оксалат- [VO2C2O4]- Спектрофо-тометричес- 20 ц—>0 6,64 [545]
ион [vo.ao4]~ кий » 20 1,0 (NaClOJ 4,87 [545]
[VO2(CA)2]3- » 20 ц—>0 10,2 [545]
[VO.2(C2O4)2]3~ » 20 1,0 (NaC104) 8,49 [545]
* Протонированный ион.
аммонийные соли которых имеют состав (NH4)6H[As(V2Oe)2-•(W2O7)4]-zH2O и (NH4)6H[As(V2O6)3(W2O7)3bzH2O.
Кремний и германий, так же как мышьяк и фосфор, способны образовывать гетерополисоединения с вольфрамом, молибденом и ванадием. Известны кремневанадовольфрамовая кислота H8[Si(V2O6)(W2O7)5]-zH2O и ее полностью замещенные соли калия и натрия, кремневанадомолибденовая кислота H8[Si(VsO6) • • (Мо2О7)5]-2:Н2О, германованадовольфрамовая кислота H8[Ge-• (V2O6)(W2O7)5] -а:Н2О, германованадомолибденовая кислота H8[Ge(V2O6)(Mo2O7)5]-zH2O и их соли K5H3[Ge(V2O6)(W2O7)5]• •zH2O, K5H3[Ge(V2O0)(Mo2O7)5bzH2O.
Имеются гетерополисоединения, полученные на основе ванадиевых кислот: желтые модификации ванадовольфраматов
(Mi = Na+, К+, Ag+), Ba2[V2W4O18]-13H2O, оранжевые формы типа M5[V3W3O18]-а:Н2О (A/1 = Na+, К+, NH£), пурпурные соединения M5[V3W7O3I]-а:Н2О и M5[V3W6O28]-a:H2O, ванадомолибдаты M4[V2Mo4O18]-а:Н2О и M4[V2Mo3Oi6]-а:Н2О, молибдооксалатованадаты M3[V(C204)(Mo04)3] -а:Н2О и M2H[VO2(C2O4)(MoO4)] -zH2O.
Восстановление гетерополисоединений сопровождается образованием яркоокрашенных гетерополисиней, которые так же широко используются в анализе, как и сами гетерополисоединения.
Сведения о константах устойчивости некоторых комплексных соединений ванадия в разных степенях окисления с органическими и неорганическими лигандами приведены в табл. 7, в последующих разделах книги и в монографиях [615,1181 —1183].
29
Глава II
МЕТОДЫ ОБНАРУЖЕНИЯ ВАНАДИЯ
Обнаружение ванадия проводят различными химическими, физико-химическими и физическими методами. Чаще всего используют реакции ионов ванадия(1У) и ванадия(У), устойчивых при обычных условиях.
ХИМИЧЕСКИЕ МЕТОДЫ
Для обнаружения ванадия применяют реакции образования* окрашенных соединений с неорганическими и органическими реагентами, реакции окисления ванадием(У) различных органических реагентов с образованием окрашенных продуктов, а также реакции осаждения малорастворимых соединений ванадия [9, 15, 243, 274, 275, 434, 498, 499, 523].
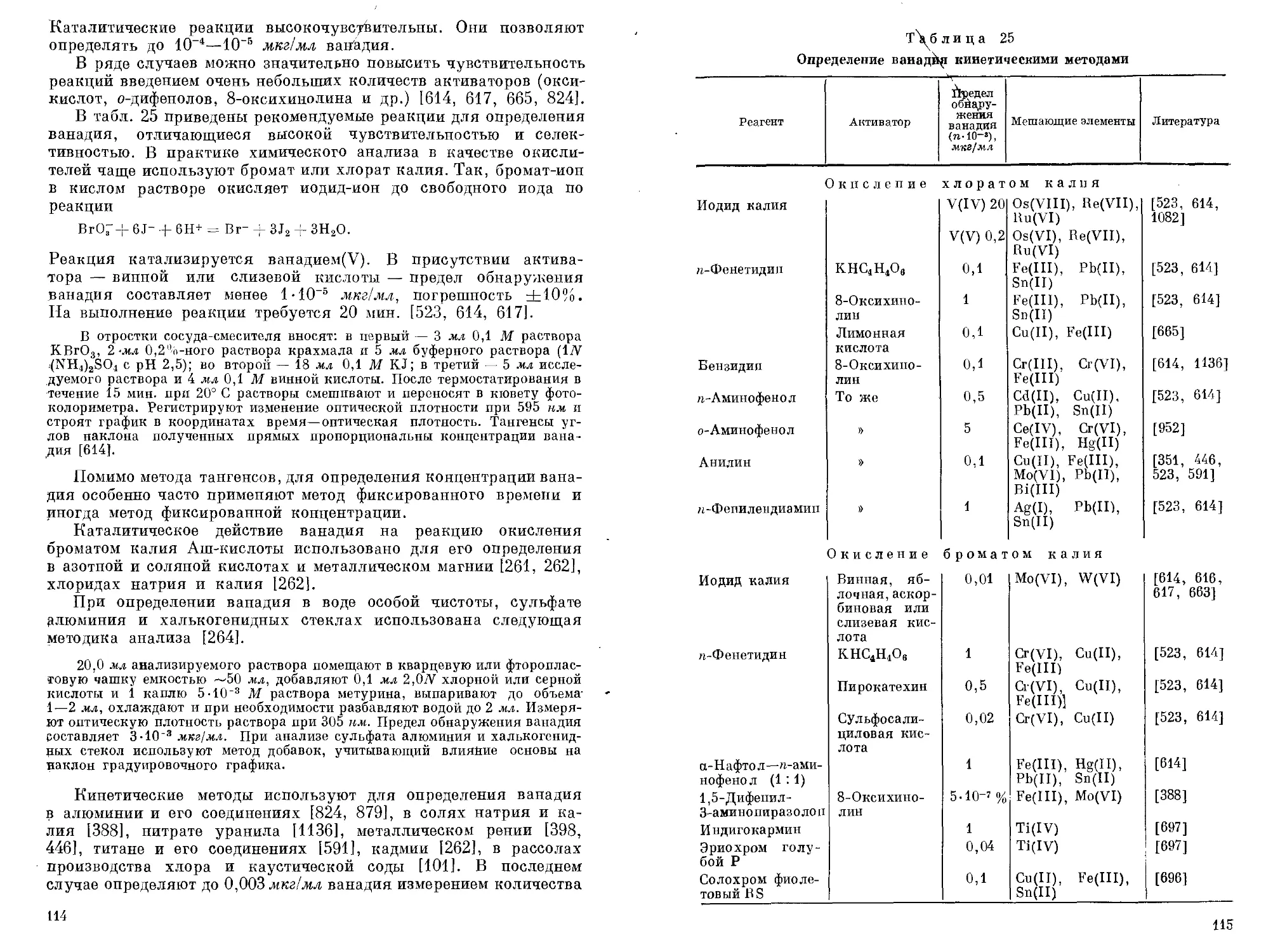
Реакции с неорганическими реагентами. Для определения ванадия(У) широко применяют реакцию с перекисью водорода [9, 15, 57, 434, 457, 523, 572]. Реакция возможна как в кислой, так и в щелочной среде; образующиеся при этом перекисные соединения имеют различный состав и окрашены соответственно в красный и желтый цвета. Максимальная интенсивность окраски перекисного соединения, обеспечивающая наибольшую чувствительность реакции, возникает при соотношении компонентов 1 : 1 и при кислотности раствора не менее 9 молей серной кислоты на 1 моль ванадия [35]. Предел обнаружения составляет 2,5 мкг ванадия [523].
Обнаружению ванадия мешают титан, молибден и хром, перекисные соединения которых окрашены соответственно в желтооранжевый, карминно-красный и синий цвета; вольфрам, образующий с ванадием желтое гетерополисоединение, а также железо и медь — своей окраской. Титан и железо связывают в бесцветные фторидные и фосфатные комплексы, а перекисное соединение хрома экстрагируют диэтиловым эфиром [57, 243, 434, 523].
В микротигле или на фарфоровой пластинке смешивают 2—3 капли исследуемого раствора с 1 каплей 20%-ного раствора H2SO4 и через 2—3 мин. добавляют 1 каплю 1 %-ного раствора Н2О2. В зависимости от концентрации ванадия появляется красное или розовое окрашивание. При малых содержаниях ванадия рекомендуется проводить холостой опыт [243].
Описан [15, 572] метод качественного обнаружения ванадия в рудах, сталях и шлаках. Он позволяет обнаружить ванадий 30
в присутствии мышьяка, молибдена, фосфора, платины и вольфрама.
Реакции с органическими реагентами- 8-Оксихинолин в уксуснокислом растворе с ванадием(У) образует желтый осадок внутрикомплексного соединения, который при нагревании переходит в черно-синий, вероятно, вследствие восстановления ванадия [215, 243]. При обработке желтого осадка хлороформом образуется устойчивый красно-фиолетовый раствор [434, 523]. Наиболее полно хелатное соединение экстрагируется из уксуснокислой среды с pH 3,3—4,5 [457, 598]. В качестве экстрагента, кроме хлороформа, используют пропанол, бутанол и изоамиловый спирт. Окрашенные соединения с 8-оксихинолином образуют также А1(Ш), Fe(III), Mo(VI), Ti(IV), W(VI) и др. [243, 457, 523]. Железо и титан перед определением осаждают щелочами, а молибден и вольфрам — хлоридом бария [215]. Хром(Ш) определению не мешает, так как его оксихинолинат не образуется в этих условиях [270].
В микропробирку помещают 1 каплю исследуемого раствора, 2 капли 2 М СН3СООН, каплю реактива (2,5%-ного раствора 8-оксихинолина в 60%-ном растворе уксусной кислоты), несколько капель хлороформа (или изоамилового спирта) и взбалтывают. В присутствии ванадия слой экстрагента окрашивается в цвета от темно-красного до фиолетового. Предел обнаружения составляет 0,1 мкг, предельное разбавление 1:5- 10? [243, 434].
Реакцию рекомендуют для обнаружения ванадия в силикатах [141] и феррованадии [18].
Показано, что добавление азида натрия к хлороформному экстракту изменяет окраску раствора из красноватой в зеленую и снижает предел обнаружения ванадия до 0,05 мкг в 0,15 мл экстракта. Метод применен для обнаружения ванадия в горных породах [1141].
Для обнаружения ванадия применяют метод осадочной хроматографии на колонке, заполненной растертым в порошок 8-окси-хннолином или его смесью с А12О3 в отношении 1 : 1 [733]. Окрашенные осадки оксихинолинатов металлов располагаются в определенном порядке, зависящем от растворимости их в воде, величины pH раствора и характера среды. Метод позволяет обнаружить ванадий(У) в присутствии висмута, вольфрама, железа, кобальта, меди, никеля, урана и цинка по образующемуся темносерому окрашиванию.
Ксиленоловый оранжевый в ацетатном буферном растворе (pH 4,5) образует с ванадием(У) окрашенное в красный цвет соединение. Предел обнаружения составляет 0,25 мкг/мл. При использовании в качестве маскирующего реагента 1,2-диамин-циклогексан—Ь1,Ь1,ЬГ,ЬГ-тетраацетата натрия (комплексон IV) определению мешают кратные количества Mo(VI) (18), U(VI) (10), In (40), Y (150), Cu(II) (250), Bi(III) (350), Hg(II) (400), Tli(IV) (500), Al и Pb(103), Fe(HI) (2-103), Zn (2,5-103), Cr(III) и Zr(IV) (3-103), Ni(II), Co(II), Mn(II) и др. (>103) [7].
31
Кверцетин-З-рутинозид в присутствии персульфата аммония в фосфорнокислой среде образует с ванадием(У) окрашенное в оранжево-красный цвет соединение. Обнаружению ванадия мешают кратные количества Fe(II) и Fe(III) (100), Ti(IV) (15), Ce(IV) (40), Mn(VII) (240), Cr(III) (180) [523].
KI.».! испытуемого раствора приливают 1 мл 10%-ного раствора персульфата аммония, 1 мл 0,5%-ного раствора реактива в метаноле и 3 мл 85%-ного раствора Н3РО4. Железо маскируют добавлением 2 мл 5%-ного раствора комплексона III, титан — 2 мл 3%-ного раствора^ фторида натрия, церий и марганец восстанавливают 1—2 каплями Н2О2. Предел обнаружения ванадия в присутствии персульфата аммония составляет 0,3 мкг/мл [523].
Резацетофепоксим в уксуснокислой среде (pH 2) взаимодействует с ванадием(У) с образованием коричневого окрашивания. Предел обнаружения ванадия равен 0,5 мкг. Определению ванадия мешают ионы Cu(II) за счет образования осадка с реагентом, ионы Cr(VI) и Mn(VII) — своей окраской. Ионы Ni(II) повышают предел обнаружения. Мешающее действие титана(1У) устраняют фторид-ионами, а железо(Ш) — фосфат-ионами [825>].
3,3',3"-Т риоксиаурин образует с метаванадат-ионами сине-черный осадок или голубой раствор при pH 5—10, с ортова-надат-ионами он не взаимодействует. Максимальная интенсивность окраски достигается через 25 мин. [523]. Предел обнаружения составляет 0,007 мкг ванадия. Реакции мешают ионы серебра(1), меди(П), висмута(Ш), железа(Ш), свинца(П), сурь-мы(Ш), олова(П) и олова(ГУ) [523].
Ванадий(У) окисляет анилин до анилинового черного [9, 275, 364, 498,499, 523]. Реакцию проводят в присутствии конц. HNO3 или НС1 в микропробирке или на фильтровальной бумаге. Определению мешают окислители К2Сг2О7, КС1О8, КСЮ и др., которые предварительно восстанавливают кипячением раствора' с хлористоводородной кислотой.
На полоску фильтровальной бумаги наносят 1 каплю спиртового раствора анилина, а затем каплю испытуемого раствора, предварительно выпаренного досуха с азотной кислотой и разбавленного водой. Избыток азотной кислоты не мешает. В присутствии ванадия в зависимости от количества его в испытуемом растворе появляется сине-зеленое или зеленоватое кольцо. Предел обнаружения составляет 5 лкг/0,01 лм. [499].
Реакцию можно использовать для обнаружения ванадия в минералах, сталях и шлаках [275, 364, 498].
Для обнаружения ванадия(У) применяют реакцию окисления бензидина и его производных (диаминбензидин и дифенилбензидин) [14, 15, 243, 274,275,353, 434], а также 3,3'-диметилнафтидина [848]. При использовании бензидина предел обнаружения ванадия равен 0,03 мкг.
Обнаружение ванадия(1У) проводят косвенным методом, используя реакцию окисления его ионами Fe(III) в нейтральном или слабоаммиачном растворе. Образующееся по реакции количество железа(П), эквивалентное ванадию(1У), определяют по реакции с диметилглиоксимом [353, 434, 523] или с 2,2'-дипиридилом [248, 32
523]. Предел обнаружения ванадия по этим реакциям составляет 0,1—1,0 мкг.
Мйкрокристаллоскопические реакции. Для обнаружения более чем 0,07 мкг ванадия(У) используют реакции образования кристаллических осадков Ag4V2O7 и T1IVOC1J [248]. 0,025 мкг ва-надия(1У) обнаруживают по образованию красных ромбических кристаллов при взаимодействии с дипиридиловым комплексом железа [412].
КИНЕТИЧЕСКИЕ МЕТОДЫ
Свойство ионов ванадия катализировать многие окислительно-восстановительные реакции используют в аналитической химии для его качественного обнаружения и количественного определения. Кинетические методы анализа высокочувствительны и специфичны [614]. Для обнаружения ванадия применяют реакции окисления некоторых органических веществ (анилина, бензидина, гидрохинона, ге-фенетидина и др.) хлоратом, броматом или перйодатом щелочных металлов, а также перекисью водорода.
Реакция окисления ге-фенетидина броматом калия протекает медленно с образованием бесцветного или окрашенного в слабофиолетовый цвет продукта. В присутствии ванадия(У) реакция идет быстро и продукт ее окрашен в интенсивный фиолетовый цвет. Обнаружение ванадия(У) с помощью этой реакции проводят следующим образом [614].
К 1 мл исследуемого раствора прибавляют 1 мл 0,1%-ного раствора га-фепетидипа, 1 мл насыщенного раствора бромата калия и доводят объем до 5 мл водой. Нагревают на водяной бане в течение нескольких минут и наблюдают за окраской, которая появляется в присутствии 1 мкг ванадия. Одновременно проводят холостой опыт. Выполнению реакции мешают ионы олова, ртути, свинца. В присутствии 1 мл 0,1%-ного раствора 8-оксихино-лина или 1мл 1%-ного раствора пирокатехина предел обнаружения ванадия снижается до 0,01 или 0,0006 мкг соответственно. Мешающими в этом случае являются ионы Fe(III), Cu(II), Cr(VI).
Реакция окисления бензидипа броматом калия использована для обнаружения ванадия капельным методом в сталях и сплавах [523].
На зачищенную поверхность образца помещают 1 каплю 70%-ного (по объему) раствора HNO3 и через 1—2 мин. 5 капель насыщенного раствора фосфата натрия и тартрата натрия-калия и несколько крупинок карбоната натрия. Перемешивают и погружают в раствор полоску фильтровальной бумаги, предварительно пропитанную 1%-ным раствором бромата калия и высушенную. Когда влажная зона поднимается на 3—5 см, помещают над ней 1 каплю 1%-ного раствора бензидина в уксусной кислоте, содержащего 5% ацетата натрия. При содержании 0,2% ванадия в стали в месте соприкосновения растворов образуется сине-зеленая полоска. Если содержание ванадия меньше 0,2%, окрашивание появляется только через 0,5—1 мин [523].
Мешающее действие некоторых ионов может быть устранено введением маскирующих реагентов. Так, ионы Fe(III) при окислении галловой кислоты персульфатом аммония могут быть связаны фосфат-ионами в неактивный комплекс, а при окислении п-фене-тидина броматом калия — сульфосалициловой кислотой. В последнем случае сульфосалициловая кислота пе только устраняет
2 В. Н. Музгин и др.
33
мешающее действие ионов железа, но и является активатором реакции, что снижает предел обнаружения ванадия [614]. Для качественного обнаружения ванадия могут быть использованы практически все каталитические реакции, приведенные в главе IV.
ЛЮМИНЕСЦЕНТНЫЕ МЕТОДЫ
Известны всего две качественные флуоресцентные реакции на ванадий с резорцином и церулеином.
Резорцин образует с ванадием(У) комплексное соединение, при облучении которого УФ-светом возникает красное свечение, исчезающее примерно через 5 мин. [64, 484, 596].
Каплю анализируемого раствора помещают в микропробирку или на фарфоровую пластинку, прибавляют 1 каплю 1%-ного раствора резорцина, 4 капли 20;V HjSOa и после перемешивания рассматривают при УФ-освещении. Предел обнаружения ванадия составляет 1 мкг, предельное разбавление 1 : 50С00 [484].
Церулеин при взаимодействии с ванадием в кислом растворе после восстановления его цинком в присутствии этанола позвц-ляет обнаружить'до 16 мкгЧ!мл по появлению желтой флуоресценции раствора. Аналогичную реакцию с церулеином дают ионы вольфрама(У1), молибдена(У1), олова(П), титана(ГУ) и урана(У1) [64, 484, 596].
СПЕКТРАЛЬНЫЕ МЕТОДЫ
В самых разнообразных объектах (металлы, порошковые пробы, растворы) легко обнаружить до 1-10“3% ванадия методами эмиссионной оптической спектроскопии. В качестве источника возбуждения спектра можно использовать дугу переменного пли постоянного тока либо высоковольтный искровой разряд. Ванадий определяют по аналитическим линиям V II 309,31; V II 310,23; V II 311,07; V I 318,54; V I 438,00; V I 438,47 нм. Наиболее характерными и чувствительными линиями ванадия являются три линии: 318,34; 318,40 и 318,54 нм [183, 785].
Методами пламенной фотометрии по линиям 528, 550 и 578 нм можно обнаружить до 1 мкг!мл ванадия [72]. В пламени закись азота—ацетилен по линии 437,9 нм обнаруживают до 0,01 мг!мл ванадия [1138]. Такой же предел обнаружения ванадия наблюдается и в кислородно-ацетиленовом пламени [739, 843].
Методом атомно-абсорбционной спектрофотометрии по поглощению света линией ванадия 318,4 нм в пламени закись азота— ацетилен обнаруживают 0,02 мг/мл ванадия, а при использовании графитового трубчатого атомизатора — 3-10-6 мг!мл [420, 843, 844, 1138]. Применение рентгенофлуоресцентного анализа позволяет обнаружить до 2-10“4%Упо линии Ка^ (0,25 нм) [296].
ДРУГИЕ|МЕТОДЫ
Для качественного обнаружения ванадия возможно применение твердофазных реакций (так называемый «метод растирания»), реакций сплавления, «пленочных» реакций (образование окрашенных пленок при непосредственном нанесении реагента .на 34
исследуемый образец) и методов бумажной и тонкослойной хроматографии.
При растирании испытуемого образца с несколькими кристалликами 8-оксихинолина . в присутствии ванадия в пробе смесь окрашивается в желто-оранжевый цвет, который при нагревании порошка переходит в густо-синий [18, 215]. При растирании анализируемого образца с сульфатом железа(П) порошок окрашивается в красно-коричневый цвет, переходящий в темно-синий при нагревании [215].
Сплавление ванадийсодержащих материалов с 8-оксихиноли-ном позволяет обнаружить несколько микрограммов ванадия по окрашиванию плава в черно-коричневый цвет. Обнаружению ванадия мешают ионы урана(У1) и железа(Ш), 8-оксихинолинаты которых окрашивают пробу в черно-зеленый и красно-коричневый цвет соответственно. Обработка пробы раствором аммиака позволяет перевести ванадий в виде ванадата аммония в раствор и отделить его от мешающих элементов [523, 741].
Несколько капель раствора упаривают досуха, добавляют 0,3 г 8-оксихинолина и повышают температуру до 250° С. Образование черно-коричневого плава указывает на присутствие ванадия в пробе. Данным методом можно обнаружить до 0,05 мкг ванадия [741].
Установлено, что 2 мкг ванадия могут быть обнаружены в присутствии 75 мг трехокиси молибдена и 14 мг трехокиси вольфрама. Метод рекомендован для обнаружения ванадия в минералах [523, 741].
Для обнаружения ванадия используют также сплавление пробы с карбонатом натрия или калия в присутствии азида натрия [141, 572], с пиросульфатом калия или гипофосфитом аммония [141, 572, 794, 944].
При нанесении на исследуемый образец насыщенного раствора солянокислого анилина в конц. НС1 в присутствии ванадия образуется пленка зеленого цвета, переходящего в черный. Наилучшие результаты получают при использовании разбавленного раствора в отношении от 1 : 4 до 1 :10. При уменьшении концент-трации кислоты скорость образования пленки замедляется, а в более концентрированных растворах образуется черная пленка и образец сильно разрушается [372, 572]. Данная реакция дает хорошие результаты на минералах ванадините, тюямуните, карнотите. Несколько худшие результаты отмечены на деклуазите и фольбортите [372].
Для обнаружения ванадия широко применяют методы хроматографии на бумаге и в тонком слое. В этих методах после разделения элементов их легко обнаруживают обработкой хроматограмм соответствующими реагентами-проявителями. Практически все методы хроматографического разделения элементов на бумаге и в тонких слоях сорбентов, описанные в главе III, могут быть использованы для обнаружения ванадия в разнообразных объектах. Следует особенно отметить возможность обнаружения ванадия различной степени окисления методами хроматографии на бумаге 1564, 1051, 1109].
2*
Глава III
МЕТОДЫ ОТДЕЛЕНИЯ ВАНАДИЯ ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
МЕТОДЫ ОСАЖДЕНИЯ
Методы отделения ванадия осаждением его труднорастворимых соединений применяют в аналитической химии довольно редко.
Лучшим способом отделения ванадия на стадии разложения пробы от многих элементов (в частности, от железа, титана и циркония) является сплавление пробы с карбонатом натрия или калия (при необходимости с небольшим количеством нитрата натрия) или с перекисью натрия. При последующем выщелачивании плава водой ванадий в виде хорошо растворимых ванадатов щелочных элементов переходит в фильтрат вместе с молибденом, вольфрамом, марганцем, мышьяком, ураном, фосфором и хромом [115, 480, 544, 607]. Следует отметить, что осадок всегда захватывает небольшие количества ванадия и указанный способ можно применять только для отделения сравнительно больших количеств ванадия.
Осаждение ванадия неорганическими реагентами
Ванадаты кальция, бария, свинца, железа и других элементов — труднорастворимые соединения, и их образование может быть использовано для выделения ванадия.
Из сульфидно-щелочных растворов ванадий осаждают окисью кальция. При pH 10,8—11 выпадает ортованадат кальция Ca3(VO4)2, при pH 7,8—9,3 — пированадат Ca2V2O7, а при pH 5,1 — 6,5 — метаванадат Ca(VO3)2 [492]. Ванадий вместе с ураном(У1) может быть осажден из уксуснокислых растворов в виде уранил-аммонийванадата или уранованадата кальция. В дальнейшем уран отделяют от ванадия осаждением уранилфосфата [385].
Осаждение ванадата бария из щелочного раствора использовано для отделения его отбора, который остается в растворе в виде бората бария [1156].
Нитрат серебра количественно осаждает ванадий при pH 6,2— 6,8 в виде AgVO3, а при pH 8—8,5 в виде Ag3VO4, что использовано для отделения ванадия от большого числа элементов [894].
Осаждение ванадата свинца применяют для предварительного группового отделения его от железа, бора, калия, таллия и неко
36
торых других элементов. Совместно с ванадием осаждаются молибден] VI), вольфрам(У1), хром(У1) [115, 786]. Для отделения ванадия от вольфрама осадок ванадата и вольфрамата свинца обрабатывают раствором карбоната калия при кипячении. При этом вольфрам переходит в раствор, а ванадат свинца остается без изменения.
Сульфиды. Оксисульфид ванадия VOS может быть осажден сульфидом аммония после восстановления ванадия(У) сернистой кислотой и удаления ее избытка кипячением. В фильтрате остаются калий и другие щелочные элементы [115].
Пентасульфид ванадия V2S8 осаждается совместно с вольфрамом, марганцем и молибденом при групповом разделении элементов тиоацетамидом из кислых растворов, а из щелочных растворов — еще и с никелем и свинцом. Осаждение ванадия тиоацетамидом из щелочных растворов при pH 10,1—10,2 может быть использовано для его отделения от вольфрама [908].
Гетерополисоединениц. Осаждение ванадия в виде молибдепо-ванадиевофосфорной [810, 811] или ванадиевофосфорной [581] гетерополикислот применяют для его отделения от многих элементов и последующего определения спектрофотометрическим [810] или атомно-абсорбциоппым [811] методом.
Для выделения и концентрирования ванадия используют со-осаждение его на гидроокиси железа, алюминия, магния, титана, цинка или циркония [115, 310, 371, 379, 488, 503, 573, 988, 1170], на пероксиде тория [229], фосфоромолибдате аммония [5С>5а].
Осаждение ванадия органическими реагентами
Купфброн из кислых растворов (~1 TV по НС1 или H2SO4) осаждает ванадий. Метод применяют для отделения ванадия от алюминия, бериллия, бора, урана(У1), фосфора, хрома (III) и некоторых других элементов [115, 521, 577, 607, 657, 1065]. Совместно с ванадием осаждаются железо, ниобий, олово, тантал, титан, цирконий. В присутствии фтористоводородной кислоты происходит отделение от вольфрама. Для выделения малых количеств ванадия в качестве коллектора вводят в раствор железо(Ш) и избыток осадителя. При анализе нержавеющей стали предложено отделять титан от ванадия осаждением титана из солянокислого раствора (~4 N) небольшим количеством осадителя (1 — 2 мл 6%-ного раствора купферона на 100 мл раствора пробы). Ванадий в этих условиях не осаждается [303].
8-Оксихинолин и его производные из слабокислых растворов (pH 2,7—6,1) осаждают ванадий вместе с большой группой элементов. При pH «С 4 хром(У1) не осаждается и может быть отделен от ванадия [16, 115, 457, 947, 988]. В присутствии перекиси водорода в уксусно-ацетатной среде ванадпй(У) не осаждается 8-оксихинолином и может быть отделен от алюминия [115].
Бензоат аммония количественно осаждает ванадий(1У) в виде бензоата ванадила УО(С6Н6СО2)2. Реагент используют для от
37
деления ванадия от алюминия, магния, меди, молибдена, титана и вольфрама. Железо и хром осаждаются вместе с ванадием [115, 521].
1Ч-Бензоил-№-фенилгидроксиламин применяют для группового разделения элементов. При pH ~6 осаждается бериллий, при pH 4,0 — алюминий, железо, в интервале pH 3—6 осаждаются торий, скандий и РЗЭ, в более кислой среде — ванадий, гафний, ниобий, олово, сурьма, тантал, титан и цирконий [115, 10421. Ванадий осаждается и в присутствии комплексона III [115, 607].
Для осаждения ванадия применяют диэтилдитиокарбаминат натрия [254, 269], таннин [115, 211, 269] и многие другие органические реагенты [115, 146, 147, 149, 269, 939, 1085]. При групповом концентрировании элементов и их последующем спектральном определении осаждение проводят совместным действием 8-оксихинолина, таннина и тионалида [701а, 1085, 1171].
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
При электролизе исследуемых сернокислых растворов (—-0,1 М) ванадий вместе с ураном, алюминием и фосфором остается в растворе, а железо, хром, висмут, кадмий, кобальт, медь, никель, олово, цинк и другие элементы осаждаются на ртутном катоде и могут быть отделены от ванадия [34, 115, 457, 821, 1062]. После электроосаждения часть элементов (кадмий, медь, свинец, цинк) может быть извлечена из амальгамы методом анодного растворения, а оставшиеся элементы выделяют отгонкой ртути.
При отделении ванадия от железа и других элементов на ртутном катоде поддерживают плотность тока 0,15—0,3 а.!смг\ Если ванадия в растворе много, то он затрудняет электроосаждение отделяемых элементов и необходимо предварительное разбавление раствора. При уменьшении кислотности раствора на аноде наблюдается выделение окислов ванадия [115, 457].
МЕТОДЫ ЭКСТРАКЦИИ
Экстракционные методы разделения элементов получили широкое распространение в аналитической практике благодаря простоте выполнения операций, экспрессности и достаточно высокой избирательности [202—204, 337, 478, 598]. Эти методы наиболее часто используют для концентрирования и отделения ванадия от сопутствующих и мешающих элементов перед определением его титриметрическими, фотометрическими, спектральными и другими методами.
В связи с тем, что ванадий в водных растворах может существовать в различных степенях окисления, при проведении процесса экстракции необходимо принимать специальные меры к стабилизации данного состояния ванадия. Наибольшее значение при экстракционном отделении имеет ванадий(У), который вполне устойчив в азотнокислых, сернокислых и фторидных растворах.
38
Рис. 3. Зависимость степени экстракции ванадия и некоторых других элементов 0,01 М раствором 8-оксихинолина в хлороформе от pH [478]
1 — молибден(УГ); 2 — ванадий(У); г — никель(П); 4 — уран(У1); 5 — кобальт(П); в — марганец(П)
В солянокислых растворах необходимо считаться с частичным восстановлением ванадия(У) хлорид-иопом до ванадия(1У). Следует отметить, что некоторые реагенты, используемые для экстракции (]У-бензоил-]>[-фенилгидроксиламин, купферон, 5-хлор-8-меркаптохинолин и другие), восстанавливают ванадий(У) до ванадия(1У), который, как правило, экстрагируется хуже [40а; 202, с. 163]. В качестве стабилизаторов ванадия(У) обычно используют небольшие добавки хлората калия, перманганата калия и некоторых других окислителей. В растворах бромистоводородной и иодистоводородной кислот ванадий может находиться в состоянии окисления (IV) и (III).
Ванадий экстрагируют обычно либо в виде комплексов с анионами неорганических кислот, либо чаще всего в виде комплексных и внутрикомплексных соединений с различными органическими реагентами [202-204, 337, 478, 598, 998, 1118, 1138].
8-Оксихинолин образует с ванадием(У) соединение, хорошо растворимое в хлороформе и других органических растворителях (бензол, толуол, амиловый спирт, бутанол, амилацетат и др.). При pH 2—6 ванадий(У) количественно экстрагируется 0,1 М растворов 8-оксихинолина в хлороформе в виде комплексного соединения VO2C9HeON. Использование мепее концентрированных растворов 8-оксихинолина (0,003—0,07 М) сужает интервал pH до 2,3—3,3, при котором происходит количественная экстракция ванадия. В области pH ]> 9 ванадий практически не экстрагируется. Зависимость степени экстракции ванадия и некоторых других элементов от pH раствора представлена на рис. 3.
Совместно с ванадием могут экстрагироваться оксихинолинаты палладия, молибдена, железа(Ш), таллия, никеля, циркония и некоторых других элементов. Селективность разделения элементов можно повысить введением в раствор маскирующих агентов (ацетат-, фторид-, циапид-ионов, этилендиаминтетрауксусной кислоты и т. д.). Так, при отделении ванадия от урана используют уксуснокислые растворы [202], а при отделении от железа, алюминия, хрома, марганца, кобальта и никеля экстракцию ванадия проводят 0,3%-ным раствором 8-оксихинолина в изобутаноле из растворов с pH 3,5—4,5, содержащих фторид натрия [598].
Ванадий(ГУ) в присутствии гидразина экстрагируется хлороформом в виде оксихинолината ванадила VO(C9HeON)2, который на воздухе медленно переходит в комплекс ванадия(У) [478]. Эти-лендиаминтетрауксусная кислота образует с ванадием(1У) прочный комплекс, который остается в водной фазе, что было использовано
39
Рис. 4. Зависимость степени экстракции ванадия и некоторых других элементов 0,005 М раствором купферона в хлороформе от pH [478] 1 — вольфрам(У1); 2 — торий(1У)| 3 — иттрий(Ш); 4 — молибден(У1); 5 — ванадий(У); в — титан(ГУ);
7 — цирконий(1У)
для отделения ванадия от урана [577] и вольфрама [202,482].
Отделение ванадия(У) рекомендуют проводить следующим образом.
К анализируемому раствору прибавляют 2 мл 0,1 М раствора виннокислого натрия и разбавленным раствором NaOH устанавливают pH 9-—10. Разбавляют водой до 20 мл и экстрагируют мешающие элементы несколькими порциями 0,1 М раствора 8-оксихинолина в хлороформе (до постоянства цвета органической фазы). Отделяют водную фазу, устанавливают pH 3—5 с помощью НС1 и экстрагируют ванадий двумя порциями по 20 мл раствора 8-оксихинолина в хлороформе. Вместе с ванадием в экстракт переходят молибден^!), вольфрам(УТ) и цирконий [478, с. 291]. -
Отделение ванадия от сопутствующих элементов с помощью 8-оксихинолина используют достаточно широко [271, 311, 478, 591, 702а, 752, 769, 889]. Реже применяют экстракцию ванадия(У) некоторыми производными 8-оксихинолина, такими, как 8-окси-хинальдин(2-метил-8-оксихинолин) [478], 5,7-дихлор-8-оксихино-лин [843], 5,7-дибром-8-оксихинолин [561].
Купферон используют для экстракционного отделения ванадия [702, 821, 1043, 1110] и особенно часто при групповом выделении примесей [420, 702а, 829, 975, 984]. В интервале pH 0—2,5 купфе-ронат ванадия(У) количественно экстрагируют хлороформом, ацетоном, зтилацетатом и другими растворителями из серно- или солянокислых растворов. В этих условиях не экстрагируются и могут быть отделены от ванадия уран, алюминий, никель, цинк [202, 478, 597]. Зависимость степени экстракции ванадия и некоторых других элементов от pH представлена на рис. 4. Ванадий(ТУ) количественно экстрагируется этилацетатом из 0,5 N по НС1 растворов, содержащих 3—6% купферона [478].
Гидроксамовые кислоты — достаточно селективные экстрагенты на ванадий. Бенз-, салицил-, хинальдин-, тиофено-гидроксамовые кислоты, Н-бензоил-Ы-о-толил-, Ы-2-теноил-Ы-фенил-‘, Ы-фуроил-Ы-фенил-, Ы-циннамоил-Ы-фенилгидроксиламины и другие производные образуют с ванадием(У) окрашенные хелатные соединения, которые количественно экстрагируются различными органическими растворителями [202, 478, 560, 598, 777].
Особенно широко применяют экстракцию ванадия(У) раствором Ы-бензоил-Ы-фенилгидроксиламина. (БФГА) в хлороформе из солянокислых сред (2,8—4,3 N по НС1). Метод использован для отделения и определения ванадия в металлах, их окислах и сплавах [128, 202, 513, 598, 718, 732, 824, 1042], горных породах [718, 972, 1042] и других объектах [202, 399, 478, 488, 984]. Бензол 40
экстрагирует комплекс ванадия с БФГА несколько лучше, чем хлороформ. Зависимость степени экстракции ванадия и титана растворами БФГА в бензоле от pH и при различном содержании в анализируемом растворе фторида натрия представлена па рис. 5.
Выделение радиоизотопа 48У без носителя из облученного протонами титана проводят с помощью БФГА следующим образом [40а].
Анализируемый образец (100 мг) растворяют в небольшом количестве конц. HF, прибавляя по каплям конц. HNO3. Добавляют 1 мл конц. H2SO4 и упаривают раствор до появления паров SO3. Остаток охлаждают, растворяют в 34 мл воды, прибавляют 115 мл холодного 0,6 М раствора фторида натрия и 4 мл 7 N NaOH. Вносят 3—4 капли водного раствора бромфенолового синего и добавляют по каплям 1 N NaOH до перехода зеленой окраски в синюю (pH — 4,5). Экстрагируют ванадий трижды порциями по 10 мл 0,05 М раствором БФГА в бензоле. Экстракты объединяют, промывают 10 мл 0,6 М раствора фторида натрия с pH 4—5 и реэкстрагируют ванадий 6 N НС1. Время выделения ванадия < 40 мин. [40а].
Трибутилфосфат количественно извлекает ванадий(У) из солянокислых растворов О 6У НС1) в виде комплексной кислоты состава Н(Н2О)т(ТБФ)2УО2С12 [203]. Снижение кислотности исходного раствора, а также разбавление ТБФ керосином или хлороформом заметно уменьшают степень экстракции ванадия. Применение в качестве высаливателей хлорида аммония или магния резко увеличивает степень экстракции ванадия. Отделение ванадия от мешающих элементов с помощью ТБФ проводят следующим образом.
К 5 мл анализируемого раствора добавляют конц. НС1 и дистиллированную воду до общего объема 25 мл так, чтобы полученный раствор был — 67V по НС1. Экстрагируют 10 мл 100%-ным ТБФ, отделяют органическую фазу и реэкстрагируют ванадий двумя порциями (по 20 мл) воды [375].
Метод использован для экстракционного отделения основы (ванадия), концентрирования примесей (алюминия, кобальта, меди, скандия, хрома, цинка и щелочноземельных элементов) с их последующим спектральным определением [375].
Хорошие результаты получены при экстракции ванадия(У) растворами три-н-бутилфосфинокиси в толуоле. Триизобутилфосфат обладает худшими экстракционными свойствами [203, 775].
1-(2-Пиридилазо)-2-нафтбл (ПАН) образует в области pH 3,5—4,5 с ванадием(У) синее хелатное соединение, которое количественно экстрагируется хлороформом, амиловым спиртом, диэтиловым эфиром, бензолом и четыреххлористым углеродом [478, 603]. При определении ванадия в нефтепродуктах методом атомноабсорбционной спектрофотометрии его извлекают из пробы с помощью 2 М НС1, а затем экстрагируют раствором ПАН в хлороформе [734].
Р-Дикетоны, такие, как ацетилацетон, теноилтрифторацетон, а также 1-фенил-3-метил-4-бензоилпиразолоп-5, широко используют для экстракционного выделения ванадия [204, 337, 459, 478, 594, 801, 1054].
41
Рис. 5. Зависимость от pH среды степени экстракции ванадия (7—3) и титана (4—6) 0,05 М раствором 1\-бензоил-?<-фенилгидроксиламина в бензоле при различных концентрациях фторида натрия в водной фазе [40а]
Концентрация NaF, М: 1 и 4 — 0,08; 2 и 5 — 0,2; 3 и в — 0,332
Рис. 6. Зависимость степени экстракции ванадия(Ш) (7), ванадия(1У) (2) и ванадия(У) (3) смесью ацетилацетона и хлороформа (1 ; 1) от pH среды [915]
А цетилацето и в хлороформе (1 : 1) при pH 2,0—3,0 количественно экстрагирует ванадий(Ш). В этих условиях ва-надий(1У) экстрагируется на 80%, а ванадий(У) < 70% (рис. 6). Ванадий(У) более эффективно экстрагируется раствором ацетилацетона в бутаноле или бензоле [478, 1054].
Разделение ванадия и железа может быть осуществлено экстрагированием ацетилацетоната ванадия(Ш). Этот метод рекомендован для анализа железосодержащих материалов [337, 915].
1 г образца растворяют в 50 мл НС1 (1 : 1) и раствор выпаривают до 25 мл. Добавляют 2 мл 30%-ного раствора Н2О2 и 10 мл H2SO4 (1 : 1) и упаривают до паров SO3; если появился осадок карбидов, его растворяют несколькими каплями HF и Н2О2. Раствор охлаждают, разбавляют до 25 мл, переносят в колбу на 300 мл, добавляют 1,5 г чистого порошка цинка и восстанавливают ванадий (железо, молибден) в атмосфере N2 или СО2. После охлаждения раствор декантируют, охлаждают на льду, добавляют конц. раствор аммиака (— 10 мл) до pH 2 и переносят в делительную воронку (общий объем раствора не должен превышать 80 мл). Экстрагируют ванадий вначале 80 мл смеси ацетилацетона в хлороформе (1 : 1), затем 10 мл хлороформа. Объединенную органическую фазу выпаривают досуха, добавляют 5 мл конц. HaSOi и 25 мл конц. HNO3. Выпаривают до паров SO3, добавляя по каплям конц. HNO3 до разложения всех органических веществ. Охлаждают, разбавляют водой до 25 мл и определяют содержание ванадия тем или иным методом, чаще спектрофотометрическим [915].
0,3 М раствор теноилтрифторацетона в «-бутаноле количественно извлекает ванадий(У) при pH 2,5—4,1. Максимум экстракции (~70%) ванадия(1У) 0,25 М раствором теноилтрифторацетона в бензоле наблюдается при pH 4,0 [478, с. 115].
1-Ф енил-3-метил-4-бензоилпиразолон-5 (ФМБП) при концентрации в водной фазе, >7 • 10-3 М образует с ванадием(1У) и ванадием(У) внутрикомплексное соединение, количественно экстрагируемое хлороформом в присутствии этанола при pH 2—4 [204]. При использовании бензольных растворов ФМБП с концентрацией >10~3 М вападий(У) восстанавливается
42
им до ванадия(1У), что подтверждено спектрами ЭПР органической фазы. Экстракция ванадия растворами ФМБП использована для концентрирования ванадия и других примесей при химикоспектральном анализе соединений щелочных металлов [204, 205, 4591.
Дитиокарбаминаты очень часто используют для выделения ванадия и других элементов [478, 568, 929, 992]. Количественно экстрагируют ванадий(У) 0,01—0,03 М растворы диэтилдитиокар-бамината натрия в четыреххлористом углероде, этилацетате или хлороформе при pH 3—6. Более устойчивы, особенно в кислых средах, диэтилдитиокарбаминат диэтиламмония, 0,04%-ный раствор которого в СС14 экстрагирует ванадий(У) при рН1 4,0—5,5, и пирролидиидитиокарбаминат аммония, 0,2%-пый раствор которого в хлороформе экстрагирует ванадий(У) при pH ~1,0 [478].
Дитиокарбаминаты реагируют с большим числом элементов и имеют сравнительно ограниченный интервал pH существования комплексов, поэтому их чаще всего используют для группового выделения элементов при их спектральном [170, 173, 272, 273, 525, 676, 851, 976, 992, 1060, 1138] или атомно-абсорбционном [702а, 843] определении. Для увеличения числа экстрагируемых элементов используют смеси дитиокарбамипатов с купфероном [170, 843], 8-оксихинолином [170, 173, 272,976, 992] или дитизоном [676, 976]. Избирательность экстракционного разделения элементов может быть повышена применением маскирующих реагентов [352, 478].
При химико-спектральном определении примесей (висмут, кадмий, медь, свинец, цинк и другие элементы) в ванадии предложено концентрировать их экстракцией хлороформом в виде дитио-карбаминатов из растворов с pH 7,5—8,0. В этих условиях вана-дий(У) остается в водной фазе [375].
Гетерополикислоты ванадия(У) хорошо растворимы в кислородсодержащих органических растворителях, таких, как простые и сложные эфиры, кетоны, альдегиды, что используют для экстракционного отделения ванадия от кобальта, марганца, никеля, хрома, циркония и других элементов [337].
Фосфорновольфрамованадиевая кислота из сернокислых (2,5— 3,5 N) или азотнокислых (17V) растворов хорошо экстрагируется метилизобутилкетоном [1032], изобутанолом [190, 593], /г-гекса-полом [655]. Метод использован для выделения и определения ванадия в самых разнообразных объектах [13, 190, 568, 593, 607, 655, 1032].
Фосфорномолибдепованадиевая кислота экстрагируется обычно из 0,5—1N растворов H2SO4, HNO3 или НС1О4 метилизобутилкетоном [461, 941], З-метил-1-бутанолом, диэтиловым эфиром [337], смесью диэтилового эфира с бутилацетатом, бутанолом [337] или с /i-пентанолом [265, 811]. Экстракция этой гетерополикислоты смесью бутанола и хлороформа (1 : 4) использована для отделения ванадия и его последующего определения атомно-абсорбционным методом [843, с. 727].
43
Таблица 8
Методы экстракции ванадия и отделение его от некттэрых элемента»
Отделяемые элементы Водная фаза Органическая фаза Литература
Al, Be, Сн,Сг, Fe, Ti, Mo, Mg, Zr Mo, Ti, Zr, Cr, Cu. Be, Ba, Sr Co, Al, Fe, Cr, Zr, Ti, W, Ni, Mn Fe, Al, Mn, Cr, U u Ti, Cr Al, Sr, Ca и другие элементы и Fe, Al, Cr, Mn, Co, Ni Ti, Al, Zr, Nb, Pb, Co, Ni и другие элементы Fe P, F, As Nb, Ti, Sc, Ta, Cr, Fe, Al, Se Ti, Fe Ti, Zr, Al, Zn, Mg, Mn, Ni 6—8 M HC1 pH 3,5 (ацетатный буферный раствор) Растворы сульфосалициловой кислоты, pH 3—4 ЗА растворы кислот НС1, H2SO4, HNO3 или СН3СООН pH 6,2,—0,5 М NaCl pH 3,0 цитратные растворы, д иэт и л д и тио ка р -баминат натрия 0,1-0,3 N НС1, диэтилдитиокар-баминат натрия pH 0,4-0,5, азотнокислые или сернокислые растворы pH 3,5—4,5, фторид натрия 6А' НС1 5-7 М НС1, роданид калия pH 2,25-2,93 8,5 М НС1 pH 4,5—6,0, 4-(2-пиридилазо)-резорцин 1-6 N H2SO4, трихлорацетат натрия (50%) 0,2%-ный раствор N-ж-толил-о-метоксибенз-гидроксамовой кислоты в хлороформе 0,5%-ный раствор 2-окси-3-нафтогидрокса-мовой кислоты в н-окта-ноле 0,1%-пый раствор тиофено-гидроксамовой кислоты в бутаноле 0,1%-ный раствор N-циннамоил-Х-фенилгнд-роксиламина в хлороформе 4%-ный раствор н-про-пил-2,3,4-триоксибензо-ата в н-пентаноле Хлороформ » 2%-ный раствор ди-этилдитиокарбамината натрия в этилацетате 0,3%-ный раствор 8-оксихинолина в изобутаноле Смесь метилпропилке-тона с четыреххлористым углеродом (3:2) Три-н-бутилфосфат в керосине (1:1) 0,2 М раствор ди- (2-этилгексил) фосфорной кислоты в керосине 2-Этилгексанол с петро-лсйным эфиром (83:17) 0,С5%-ный раствор нитрона в хлороформе 0,25%-ный раствор ацетонгидразида антраниловой кислоты в ацетоне с хлороформом (8:5) [624, 6261 [379] [202, с.115] [143, 174] [816] [506] [569] [421] [598] [756] [124] [853] [203 , 964] [405] [160]
44
Таблица 8 (окончание)
Отделяемые элементы Водная фаза Органическая фаза Литература
и HCl (1:1), 0.1%-ный раствор KC1O4 Амилацетат с толуолом (1:1) [270]
As, F, Р pH 2,5-5,0 0,1 М раствор Амберлита SA-2 в керосине с 5% 1-деканола [1001]
Al, Са, Cr, Mg, Мп, pH 3—4, 0,1 M раствор БФГА в этаноле Хлороформ [128]
Al, Cr, Fe, Ti, и pH 5,2—5,5, пиридин, роданид аммония » [1165]
и pH 2 Ацетилацетон с хлороформом (1:1) [598]
Al, Be, La, U, Мп, Ni, Zn, Ag, Са pH 0—2,5, купферон Смесь метилизобутилкетона и масляной кислоты (8:2) Бутилацетат [1043] [975]
Sn, Zr, W, Al, pH 4—6, пирро- Метилизобутилкстон [420]
Mn, Ca, Ba, Cr Si’ лидиндитиокарба-минат аммония Хлороформ [702а, 821,1060]
Другие случаи. Для экстракционного отделения ванадия от мешающих при анализе элементов или его концентрирования используют также простые и сложные эфиры [268, 791], высшие вторичные спирты [964], кетоны [268, 756, 1155], 8-меркаптохино-лип и его аналоги [43, 478], а-бензоиноксим и другие оксимы [662, 891], окись мезитила и многие другие экстрагенты [27, 149, 202— 204, 268, 326, 337, 478, 598, 625, 702а, 853, 891, 964, 1053]. В табл. 8 приведены некоторые методы экстракционного отделения ванадия от сопутствующих элементов.
В аналитической практике часто используют экстракционное отделение мешающих элементов от ванадия [306, 412, 418, 605, 708, 787]. Так, железо при определении ванадия в стали экстрагируют из НС1 смесью метилизобутилкетона и четырех-
хлористого углерода (3 : 2) [1063], а при определении ванадия в рудах — изопропанолом из 87V НС1 [766]. Титан и цирконий отделяют от ванадия экстракцией их из 0,5—4%-ных растворов серной кислоты, содержащих 2% перекиси водорода, раствором алкиламина Амберлит LA-2 в хлороформе [772]. При химикоспектральном [342, 375] и полярографическом анализах ванадия [1117] примеси концентрировали и отделяли от основы экстракцией их дитиокарбаминатов хлороформом при pH 7,5—8,0 [375] и дитизонатов четыреххлористым углеродом при pH 9,0 [342, 1117]. В табл. 9 приведены примеры экстракционного отделения различных элементов от ванадия.
45
Таблица 9
Методы экстракции элементов и отделение их от ванадия
Экстрагируемые элементы Водная фаза Органическая фаза Литература
Cd,Си,Bi,РЬ, Fe,Ni,Co,Mn pH 7,5 — 12, дцэтилди-тиокарбаминат натрия Хлороформ [375,598]
Cu,Bi,Pb,Zn pH 9, винная кислота 0,3%-ный раствор дитизона в СС14 [342,598, 1117]
Cr (VI) pH 1 — 3 (при pH 3,5 — 5,0 экстрагируется V (V); 5%-ный раствор трп-н-октил-н-дециламнна в керосине [1033]
Си (И) Пиридин, гпдрокспл-амин, КВг пли KJ Хлороформ [813]
Mo(Vl) pH 1,5 (тиогликолевая кислота, хлорид трибу-тиламина) Дихлорметан [1173]
и (VI) 0,1 -0,4 М H2SO4 0,1217 раствор пзо-октил-бензиламина в бензоле или четыреххлористом углероде [557]
и (VI) pH 1 (при pH 2 экстрагируется ванадий) 0,1217 раствор три-н-ок-тиламина в керосине в присутствии 3% трибутилфосфата [1039]
Fe (III), Mo (VI), W(VI) pH 6 (аскорбиновая и этилендиаминтетраук-сусная кислоты) 0,05М раствор 8-оксихинолина в хлороформе [202,482]
Re (VII) 0,3-1,51V NaOH Ацетилацетон или хинолин [1123,1166]
Re (VII) ЗА' H2SO4 Амиловый спирт, метилциклогексанон или циклогексанон [1168]
Re (VII) IN H3PO4 (перекись водорода) Изоамиловый спирт [1167]
Fe (III) ~ IN HC1 с добавлением ~ 5% роданида калия 35%-ный раствор трибу-тпл фосфата в хлороформе [411]
Fe(III) 10- 111V HC1 Изопропиловый или диизопропиловый эфир [223,708]
Fe (II) pH 5, 1,10-фенантролин, гидроксиламин Нитробензол [906] ‘
Та (V) 12V HF и 12V HNO3 Т р и б у ти лф о сфат [132]
Mo (VI) 4,5-6 M HC1 0,05Л/ раствор три-н-ок-тиламина в толуоле [418]
Mo (VI) 6Ar HC1 Диэтиловый эфир, дп-изоироппловый эфир, н-амиловый спирт или «-амилацетат [848]
Mo (V) Mo (VI) 11V HC1 (10% лимонной кислоты, 2% роданида калия) Диизопропиловый эфир, н-амиловый спирт и н-амилацетат (1:1:1) [848]
0,3 - 0,41V ИС1 Изоамиловый спирт с хлороформом (1:1) [203]
46
Таблица 10
Разделение ионов ванадия различной степени окисления методами экстракции
Экстрагируемый ион Отделяемый ион Водная фаза Органическая фаза Литература
V(V) V(IV) 6-7 M HC1 Трибутилфосфат или 20— —1С0%-ный раствор триизобутилфосфата в бензине [203, 775]
V(V) V(IV) 1A HC1 .0,4%-иый раствор 5,7-ди-бром-8-оксихинолииа в хлороформе [202, 561]
V(V) V(IV) pH 1,5—2, азотнокислый раствор 0,6М раствор три-н-ок-тилфосфинокиси в керосине [337, с. 154]
V(V) V(IV) 8 A HC1 0,1М раствор трополона в хлороформе [202]
V(V) V(IV) 4A H2SO4, БФГА Хлороформ [128]
V(V) V(IV) pH 3,5-3,8 0,1 М раствор трп-н-октил-амина в керосине [564]
V(V) V(IV) 0,2A H2SO4 н-Бутанол [564]
V(IV) V(III) pH 2-3,0,2%-ный раствор сульфосалициловой и лимонной кислот 40%-ный раствор ди-2-этил-гексилфосфорной кислоты в керосине [562]
Экстракция — один из наиболее универсальных методов разделения и выделения ионов различных степеней окисления одного и того же элемента, он успешно используется в аналитической практике. В табл. 10 приведены методы экстракционного разделения ионов ванадия в различной степени окисления. Следует отметить, что изменение степени окисления ванадия или мешающих элементов применяют для повышения селективности экстракционных методов разделения [203, 204, 478].
МЕТОДЫ ХРОМАТОГРАФИИ
Ионообменная хроматография
Для отделения ванадия от сопутствующих элементов или примесей широко применяют различные ионообменные методы, теория и практика которых изложена в монографиях [214, 438, 451, 519, 542, 582]. В зависимости от того, в какой степени окисления находится ванадий в растворе и стоящих перед исследователем задач, применяют различные анионо- и катионообменные смолы.
В качестве примера можно привести разделение ванадия(1У) и титана(1У) из кислых растворов, содержащих 0,1 М НС1 и 1 М HF [451]. Если необходимо отделить малые количества титана от больших количеств ванадия, то целесообразно сорбировать
47
титан па анионите, при этом ванадий не сорбируется и будет находиться в фильтрате. В другом случае для отделения малых количеств ванадия от больших количеств титана целесообразнее использовать катионит, на котором при данных условиях сорбируется только ванадий, который после промывки ионообменной колонки можно десорбировать раствором 1—3 М HCI.
Разделение на катионитах. Ванадий(1У) в водных растворах находится в основном в виде ванадил-иопа VO2+, который в кислых растворах вполне устойчив и количественно сорбируется катионитами как из хлоридпых, так и из сульфатных растворов. В разбавленных растворах ванадий(У) в щелочной среде катионитами в Н-форме не поглощается, однако в кислой среде может происходить его частичное восстановление или осаждение [177, 451, 542]. Подробное исследование поведения ванадат-иона показало [17], что уже при pH 6,0 на катионите СБС поглощается -~20% ванадия.
С повышением кислотности раствора сорбция ванадия возрастает, достигая ~85% при концентрации серной кислоты ~0,17V. Это связано с тем, что в кислых растворах возможно существование ионов VO2 и даже VO3+. При дальнейшем повышении концентрации кислоты поглощение ванадия катионообменной смолой резко уменьшается, а при 1,27V H2SO4 практически весь ванадий оказывается в фильтрате, что связано как с сильным регенерирующим действием раствора серной кислоты такой концентрации, так и с частичным образованием гексавападатов по реакции
6VO,- + 2Н3О+ [V„O,,]4" + ЗН2О.
Введение в слабокислые растворы ванадия(У) перекиси водорода в концентрации 0,25—1,0% предотвращает его сорбцию катионитом, так как образуется достаточно устойчивая надкислота H[VO2(O2)]. Это было использовано для отделения ванадия от железа при анализе стали, чугуна, феррованадия [17, 480], от титана, титана и железа, от кобальта, меди, никеля и частично хрома [480, 582].
Германий(1У) отделяют от ванадия(У) сорбцией этих элементов на катионите КУ-2 в Н-форме и вымыванием германия посред-ством 17V HCI [587]. Вместе с ванадием(У) остается на смоле и ти-тан(1У). Разделение последних осуществляют вымыванием ванадия 10%-ным раствором NaOH, а титана — 4—57V НС1 [582, 587]. Ванадий в элюате определяют фотометрическим методом.
На катионите Дауэкс-50\УХ8 отделяют ванадий(У) от большого числа элементов (кадмий, кобальт, медь, марганец, никель, свинец, титан и т. д.) [582, 705, 710, 748, 750, 856]. После поглощения всех катионов на смоле проводят селективное вымывание ванадия в виде перекисного комплексного соединения 0,3—1%-ным раствором Н2О2 [582, 748], 0,57V НС1 [582, 710] или ацетатной буферной смесью (pH 6—7) 0,026 М по сульфосалициловой кислоте и 0,05 М по CH3COONa [750].
48
' Ванадий отделяют от вольфрама и молибдена из слабокислых растворов (pH 1,0) в присутствии лимонной кислоты. В этих условиях ванадий сорбируется катионитами в виде УО2+, а молибден^!) и вольфрам(У1) образуют прочные цитратные комплексы и не сорбируются [451, 582, 999].
Ванадий с помощью катионитов отделяют от хрома из солянокислых растворов с pH 1,0. В этих условиях сорбируется ванадий^), а хром в виде иона СгО4- остается в фильтрате [582]. Более надежным методом является введение в анализируемый раствор роданид-иона, который восстанавливает ванадий(У) до ванадия(1У) и образует прочный комплекс с хромом, который катионитом не сорбируется [1063].
Навеску стали растворяют в 40 мл H2SO4 (1 : 4), прибавляют несколько капель HNO3 (1 : 2), выпаривают до появления паров SO3, охлаждают, прибавляют 1 мл H2SO4 (1 : 4), 20—25 мл 4 N KSCN, кипятят несколько минут и по охлаждении сорбируют ванадий на катионите СБС в аммонийной форме. Колонку отмывают водой до отрицательной реакции на роданид-ион. Хром определяют в фильтрате, а ванадий элюируют с колонки раствором H2SO4 (1 : 8) и определяют потенциометрическим методом [582].
При отделении ванадия от фосфора, а также серы и кремния предварительно восстанавливают ванадий(У) до VO2+ с помощью солянокислого гидроксиламина ийи сернистого ангидрида и сорбируют на катионите КУ-2, СБС-Р, СДВ-3, КС-22 или Вофатит-Р [92, 582]. Метод применяют при анализе металлического ванадия, пятиокиси ванадия, феррованадия, хромового концентрата [92, 480, 784, 999]. Следует отметить, что высокая кислотность раствора в присутствии цитратов, оксалатов или тартратов заметно снижает сорбцию ванадия [582, 746].
Вымывание ванадия(1У) 2М раствором NH4SCN с катионита Амберлит IR-120 использовано для его отделения от ртути(П) и железа(П) [900]. Отделение ванадия(1У) от никеля на смоле Амберлит-200 описано в работе [832].
Разделение рения(УП) и ванадия(У) возможно из раствора с pH 3 на смоле СБС в Н-форме. Рений вымывают раствором с pH 3—4 в форме ReO^, а ванадий — раствором аммиака (1 : 4) [582].
Ванадий(У), цирконий(1У), хром(Ш) с тайроном (натриевая соль пирокатехин-3,5-дисульфокислоты) образуют комплексные соединения при pH 5—6, которые не сорбируются на смоле КУ-2. Метод предложен для отделения ванадия(У) от марганца(П), который в этих условиях сорбируется [118].
Для отделения урана проводят сорбцию урана и ванадия парафенолсульфокатионитом ПФСК, а затем вымывают ванадий (совместно с молибденом) 5%-ным раствором Н2О2 в виде перекисных соединений. Уран вымывают 5%-ным раствором H2SO4 [447]. Б присутствии комплексона III ванадий(У), а также медь(П), же-лезо(Ш), вольфрам(У1), молибден(У1) и свинец совершенно не сорбируются катионитами КУ-2 и Амберлитом IRC-50 в широком интервале pH (2—8) исследуемого раствора, в то время как уран прочно сорбируется катионитом [447], что и было использовано
49
для его отделения. Совместное отделение ванадия(У) и алюминия от молибдена(У1) и урана(У1) на смолах Вофатит F, Амберлит IR-120, Дауэкс-50 описано в работе [846].
Разработаны методы отделения ванадия(У) от титана(1У) и ниобия(У) на смоле flay3KC-50WX8 с использованием органических кислот [1007]. Возможно разделение ванадия(У) и молибдена^!) на сульфоугле, который из кислого раствора поглощает преимущественно ионы МоО2+ и практически не сорбирует ванадий [582]. Ванадий(У) можно отделить от вольфрама(У1) после их сорбции на А12О3 и последующей десорбции ванадия солянокислым раствором с pH 1, содержащим 0,4% Н2О2. Ванадий в элюате определяют титриметрическим методом [582]. Вымыванием перекисных соединений ванадия с А12О3 отделяют его от железа, молибдена и вольфрама [582].
Электродиализом с применением катионитовых мембран отделяют POj от ванадия(1У). Ванадий в виде VO2+ практически полностью переходит в католит [802].
Групповое ионообменное концентрирование применяют для определения ванадия и других элементов методами атомно-абсорбционной спектроскопии в сталях [789], силикатных горных породах [1111], рентгеноспектральным — в морской воде [929], керосине и минеральных маслах [646, 890], горных породах [770], активационным — в сталях [1063] и природных водах [889], эмиссионным спектральным — в горных породах [770] и природных водах [422, 6761, спектрофотометрическим — в морской воде [1029].
Разделение на анионитах. Ванадий(1У) в солянокислой среде заметно сорбируется слабоосновными анионитами (типа ЭДЭ-10П, АН-2Ф, АВ-16Г), и тем больше, чем выше кислотность раствора. Сильноосповной анионит АВ-17 практически совсем не сорбирует четырехвалентный ванадий [389, 451]. Сорбируемость ванадия(У) из солянокислых растворов анионитами невелика, кроме того, некоторые аниониты восстанавливают ванадий(У) до ванадия(ТУ) [391].
Отделение ванадия от молибдена основано на слабой сорбции VO3 анионитами] ЭДЭ-10П, Амберлит-400, Амберлит-410, Вофатит L-150 в Cl-форме из кислых растворов (pH 1). Ванадий полностью вымывается ОДУ HCI, в то время как молибден элюируется лишь 4У НС1 [582]. Ванадий в элюате определяют методом фотометрии.
Ванадий(У) отделяют от рения(УП) на анионите ММГ-1 в ОН-форме из ОДУ раствора по НС1. Частично сорбированный ванадий вымывают ОДУ HCI, рений — 2,5У NaOH [582]. На анионитах АВ-27 и АВ-17 в Cl-форме сорбируют ванадий(У) и рений(УП) из растворов 1—2У НС1 или 3—5У NaOH. Ванадий элюируют 1У НС1 и определяют гравиметрическим методом, а рений вымывают 2,5У NaOH или 8У НС1 [151, 582].
Отделение висмута, свинца и цинка от макроколичеств ванадия
50
основано на избирательном поглощении примесей из 2,52V HG1 анионитами ТМ или ЭДЭ-1ОП в Gl-форме и последующей десорбции цинка и свинца O,O22V HG1 и висмута 22VIH2SO4. Ванадий(У) в этих условиях не сорбируется [479, 582].
Из солянокислых растворов на анионитах возможно отделение ванадия(У) от хрома(У1), а также от титана(1У) и железа(Ш), что использовано при анализе стали [451, 582]. На анионите АВ-17 в РО4-форме из раствора 0,5Ar по Na3PO4 отделяют ванадий(У) от хрома(У1) и в фильтрате определяют ванадий фотоколоримет-рическим методом [582]. G использованием хлорида лития в качестве комплексообразующего реагента отделяют ванадий(У) от алюминия, галлия, железа(Ш) и титапа(1У) на анионитах ЭДЭ-10П и АВ-17 [38, 451].
При ионообменном отделении ванадия(1У) весьма эффективно использование смесей HG1 или H2SO4 с HF ([389-—391, 451]. Ва-надийДУ) из таких растворов практически не сорбируется, что дает возможность отделить его от элементов, образующих прочные фторидные комплексные анионы (ниобий,! титан, цирконий, уран(У1) и др.). В этих же условиях ванадий(У) обладает высокой сорбируемостью. Разработаны методы отделения ванадия от скандия и титана(1У) [451], вольфрама и молибдена [582], кадмия, кобальта, меди, магния, марганца, никеля, свинца, серебра, цинка [705].
При определении ванадия, молибдена и вольфрама в сталях и жаропрочных сплавах 1 г образца растворяют в HCI (1 : 1), добавляют 10 мл 65%-ного раствора HNO3, выпаривают досуха, приливают 10 мл конц. HNO3 и снова выпаривают досуха (эту операцию повторяют 2—3 раза). Сухой остаток растворяют в 50 мл раствора 1 JV по HF и 0,1 N по НС1, пропускают через колонку с анионитом АВ-17, предварительно промытым таким же раствором. В фильтрате, содержащем железо, никель и хром, определяют ванадий фотометрическим методом с формазаном [582].
Ванадий(У), молибдеп(У1), вольфрам(У1) и некоторые другие элементы образуют в растворе аскорбиновой кислоты при pH 4 отрицательно заряженные комплексы, которые неодинаково прочно сорбируются сильноосновным анионитом Амберлит IRA-400 (в аскорбатной форме). При промывании колонки 0,12V HG1 количественно элюируется только ванадий(У), что использовано для отделения его от вольфрама, железа, молибдена и других элементов [451, 582].
Ванадий(У) в слабокислом растворе восстанавливается тиогли-колевой кислотой до солей ванадила, образующих с избытком кислоты комплексные соединения, хорошо сорбирующиеся анионитами Дауэкс-1 и Дауэкс-2 при pH 2—6. При pH < 2 ванадий практически не сорбируется. Желтое комплексное соединение молибдена(У1) с тиогликолевой кислотой прочно сорбируется этими анионитами в ЭО4-форме при pH 1—6, что использовано для отделения ванадия от молибдена. Молибден элюируют раствором NH4OH (1 : 7), содержащим 3% (NH4)2S2O8. При отделении 10— 20 мг молибдена от 40—200 мг ванадия получены хорошие результаты [451, 582].
51
Разделение скандия(Ш), ванадия(1У) и титана(1У) основано на сорбируемости комплексов этих элементов из 10-4 М НСООН анионитом Дауэкс-1 в формиатной форме и последующем селективном вымывании указанных элементов [451]. Скандий вымывают раствором 0,1 М по HCI и 0,1 Мпо Н2С2О4, а затем элюируют на-надий раствором 0,4 М по НС1 и 1 М по Н2С2О4 и лишь затем десорбируют титан 0,1 М НС1.
При определении ванадия в сталях его предварительно выделяют сорбцией на смоле Де-ацидит FF из ацетатного буферного раствора (рН2,5—3,0), содержащего 0,5% маннитола, добавленного для устранения осаждения железа. Железо(Ш) в этих условиях не сорбируется и остается в элюате. Ванадий элюируют в виде VO3 0,6М NaOH, затем хром(Ш) 8 М НС1 и, наконец, молибден (VI) 1 М HCI [451].
Для разделения бария(П), скандия(Ш), хрома(Ш) и ванадия (IV) применяют последовательное элюирование растворами, со-, держащими 0,01 М этилендиаминтетраацетата и переменное количество хлорида аммония. Элементы вымываются в указанном порядке при содержании в растворах хлорида аммония соответственно 0,01 М (pH 4,6), 0,1 М (pH 4,6), 0,3 М (pH 9,0) и 2М (pH 4,5) [451].
Для отделения ванадия от алюминия использована хорошая сорбируемость ванадия(1У) сильноосновным анионитом Амберлит IRA-400 из 0,1—1,0 М HCI с добавкой 0,5—2,0 М NH4SCN [451].
Отделение ванадия(У) совместно с бериллием, галлием, магнием от урана(У1) проводилось на анионите Дауэкс-1 Х8 в NO3-форме, предварительно промытом смесью 5 М HNO3 и метанола (1 : 19). Пробу растворяют в 1 мл 5 М HNO3, добавляют 19 мл метанола и сорбируют уран и все примеси на анионите. Ионы ванадия^), бериллия, галлия, магния вымывают раствором смеси 5 М HNO3 и СН3ОН (1 : 19), затем элюируют уран(У1) раствором смеси ЬМ HNO3, СН3ОН и Н2О (1 : 10 : 9) [855].
Растворы кислот, содержащие метанол, применяют также для отделения ванадия от никеля [451] и рения [570]. На анионите АВ-17 отделяют ванадий(У) от молибдена(У1), вольфрама(У1) и рения (VII) из водно-этанольных растворов HCI и НСЮ4 [109].
В присутствии 5—6% перекиси водорода из сернокислых или солянокислых растворов ванадий(1У) и ванадий(У) в виде перекисных соединений полностью сорбируются анионитом Дауэкс-1 Х8 или Вофатит L-150, что было использовано для его отделения от хрома(Ш), хрома(У1) [276] и алюминия(Ш) при анализе феррованадия [938].
Хлоридные комплексные анионы урана(У1) из солянокислых растворов (>8V) полностью поглощаются анионитами ПЭ-9 и ЭДЭ-10П [329], Дауэкс-1 и Дауэкс-2 [451] в Cl-форме; ванадий(У) в этих условиях не образует сколько-нибудь устойчивых комплексов, практически полностью переходит в элюат и может быть определен фотометрическим методом. Из растворов НС1 с pH 3 на смоле ЭДЭ-10П в Cl-форме количественно сорбируются ванадий^), 52
вольфрам(У1), молибден(У1) и хром(У1) и могут быть отделены от урана(У1), который полностью переходит в фильтрат [586]. Анионит Амберлит IRA-400 из растворов с pH 1 количественно сорбирует ион Сг2О?-, а ванадий(У) в форме катиона остается в фильтрате [1153].
Сильноосновной анионит Дауэкс-2 в ЭО4-форме избирательно сорбирует сульфатные комплексы урана из сернокислого раствора с pH 2, что было использовано для отделения урана(У1) от больших количеств ванадия(У), железа(Ш), хрома(У1) и других элементов. Во избежание частичной сорбции ванадия и железа перед пропусканием их через анионит рекомендуют в исследуемые растворы добавлять сернистую кислоту для восстановления же-леза(Ш) и ванадия(У) [529а].
Ванадий(У) и карбонатный комплекс урана(У1) сорбируют на смоле Амберлит IRA-400 в СО3-форме. Ионы УОД поглощаясь на смоле, вытесняют эквивалентное количество карбонат-ионов, что используется для косвенного определения ванадия [1115]. На анионитах АВ-16 и АВ-17 в СО3-форме возможно разделение ванадия(У), урана (VI), вольфрама(У1) и молибдена(У1) из растворов с pH 2,5 [602]. Молибден и - вольфрам вымывают 0,5 А (NH4)2CO3, уран — IN (NH4)2CO3, а ванадий — ЗА NaOH.
Из 10%-ных растворов Na2CO3 отделяют ванадий(У) от ура-на(У1) и стронция сорбцией на анионите Амберлит IRA-400 в Cl-форме. Ванадий вымывают 10%-ным раствором Na2CO3 [935]. Предложен метод отделения ванадия(У) от вольфрама(У1), молибдена^!), ниобия(У), титана(1У) и циркония(1У) из фторидных растворов на сильноосновном анионите Дауэкс-1. Ванадий вымывают 10,1 М HF [570].
На анионите Амберлит А-21 в SCN-форме проводят отделение ванадия(1У) от никеля [749]. Используя аниониты АВ-17 или ЭДЭ-10П в роданидной форме, можно разделить алюминий (III), ванадий(У), галлий(Ш) и железо(Ш) [39]. После сорбции из 0,5Л7 NH4SCN с pH 0,3 последовательно вымывают со смолы алюминий раствором 2М NH4SCN с pH 1,0, галлий — 0,1 М HCI, ванадий 1А NaOH и железо — 1А НС1.
Отделение ванадия(ГУ) от мышьяка(Ш) проводят из 0,5 М HN3 на сильноосновном анионите Биорад AG1 [958]. Разделение ванадия(У) и молибдена(У1) осуществлено на смоле Дауэкс-1 Х8 из азотнокислых водноорганических растворов [1152] и на смоле Дайон SA-100 в ОН-форме из щелочных растворов [570, 9121. Ванадий вымывают 2М NaOH, а молибден — смесью 0,1М NaOH и IM NaCI (1 : 1).
Анионообменное выделение ванадия использовано для его атомно-абсорбционного определения в горных породах [858] и водах [857], при эмиссионном спектральном анализе геологических материалов [627] и плутония [632], при спектрофотометрическом определении ванадия(У) и, ванадия(1У) в морской и природной водах [841].
53
Хроматография на бумаге
Достаточно простым и удобным способом отделения ванадия от целого ряда сопутствующих элементов является хроматография на бумаге.
В качестве специфических проявляющих реагентов для обнаружения ванадия после его отделения обычно используют 1%-ные водные растворы роданида или хромата калия, образующие в присутствии ванадия окрашенные соответственно в синий или красно-оранжевый цвета зоны, либо применяют смесь равных объемов 2N H2SO4 и 20%-ного раствора Н2О2 (коричнево-красное окрашивание) [551]. Некоторые другие проявляющие реагенты приведены ниже, а также в монографии [102].
Основы метода, техника работы и практическое применение рассмотрены в монографиях [551, 580]. В табл. И приведены сведения о методах отделения ванадия от других элементов и разделения и обнаружения ионов ванадия разных степеней окислепия.-Проведено подробное исследование поведения ванадия в ходе систематического анализа методом хроматографии на бумаге [660, 985], при использовании в качестве подвижных растворителей растворов органических кислот [874], этилендиаминтетрауксус-ной кислоты [1119], 2-метилтетрагидрофурана [633], салициловой, сульфосалициловой или тиосалицилов ой кислот [108], различных систем органических растворителей, содержащих хлороформ [651, 652].
В качестве одного из примеров отделения ванадия от большой группы элементов можно привести следующую методику [551].
К анализируемому раствору добавляют 2 капли 2,5%-ного раствора 8-оксихинолина в 2N СН3СООН, образовавшийся осадок экстрагируют небольшим количеством (— 2 мл) хлороформа, упаривают и наносят на полоску хроматографической бумаги. После высушивания на воздухе полоску подвешивают в камере (или цилиндре с крышкой) так, чтобы нижний край полоски, куда нанесена проба, касался подвижного растворителя (смесь равных объемов изоамилового спирта и 2 TV СН3СООН). Rf для ванадия составляет 0,90 и мало зависит от его концентрации. Метод позволяет обнаружить до 2—5 мкг ванадия при большом избытке таких элементов, как хром, молибден, вольфрам и многих других [551].
Рекомендовано применение техники кольцевой бани для отделения ванадия(У) от титана(ТУ) и циркония(ТУ) [658], вольфрама^!) и молибдена(У1) [932], хрома(У1), железа(Ш), кобаль-та(П), висмута(Ш) и серы(У1) [778].
Предложен ряд методов количественного определения ванадия после предварительного отделения его от других элементов методом хроматографии на бумаге.
Так, при использовании в качестве подвижного растворителя смеси ацетона, хлороформа и конц. НС1 (71 : 20 : 9) ванадий(У) (Rf = 0,28) отделяется от большой группы элементов. После проявления хроматограммы 2,5%-ным раствором 8-оксихинолина в 60%-ном растворе уксусной кислоты пятно ванадия вырезают, выдерживают в течение часа в атмосфере NH3, опускают в стаканчик, прибавляют 4,5 мл бидистиллята, 0,5 мл 14%-ного раствбра 54
Таблица И
Отделение ванадия от других элементов и разделение ионов ванадия различной степени окисления методами хроматографии на бумаге
Разделяемые ионы Подвижный растворитель Проявляющий раствор Rf Литература
Re(VII), Амиловый спирт— 0,5 г таинипа Зависит от кои- [760]
Fe(III), Mo(VI), конц. НС1—30%- и 0,5 г уксус- центрации вана-
W(VI), V(V) ный раствор Н,О2 (19:6:2) нокислого нат- рия в воды дия
V(V), Mo(VI), \V(VI) Бутанол — анизол—6ЛГ НС1 (4:1:1) Пентанол— диоксан—2/V НС1 (25 : 5 ; 2) Пентанол—ацетон—2/V НС1 (25 : 5 : 2) 5 %-ный раст-ворТ1С14вкоиц. НС1, затем 25%-ный раствор K.SCN 0,18; 0,65; 0,00 0,07; 0,40; 0,15 0,05; 0,42; 0,25 [1140] *
Re(VII'), 0,1— 0,5%-ные 1 % -ный раст- 0,94; 0,70; 0,36; [108]
Mo(VI), V(V), растворы салици- вор SnCl2 в 6.V 0,18 для раство-
W(VI) ловой, сульфоса-ЛИцИЛОВОЙ или тиосалициловой кислот в 96%-ном этаноле НС1, затем 50%-ный раствор NH4SCN ров сульфосалициловой кислоты
Fe(III), V(V), U(VI) Конц. СНзСООН-конц. HNO3 (19 : 1) 5%-ный раствор K4[Fe(CN)e] 0,18; 0,54,- 0,84 (330]
V(V), Mo(VI) W(V1), Cr(VI), Pb(II), Cd(II), Mg(II), Zn(II), Al(III), Mn(II), Cu(I) Изоамиловый спирт—2N СН3СООН (1 :1) Для V(V} равен 0,90 и практически не зависит от концентрации ванадия [862]
Более 40 ионов Этанол—конц. НС1-Н.0 (15:4:1) 0,2-ный раствор морина в 96%-ном эта- ноле Определению мешает Ti(lV) [1064]
Fe(III), V(V), U(VI) Ацетон—30% -ный раствор Н20а — конц. НС1 (18:1:1) 0,97; 0,18; 0,64 [551, c. 694]
Fe(IH), Gr(VI), Этанол—5Y НС1 0,56; 0,47; 0,27; [551,
Al(III), V(V), U(VI) (9:1) 0,38; 0,57 c. 694]
14 ионов н-Бутапол, насыщенный 1%-ным раствором НС1 TI(III)>Zn(lI)> >Ве(П)>1п(Ш)> >V(IV)>Mg(II)> >Ti(IV), Al(III), Cr(VI)> Ег(Ш), Ce(III), Y(III), Th(IV), Dy(III) [551, c.694]
Ti(lV), V(V), Mo(VI) н-Бутанол—конц. НС1—30%-ный раствор Н2О2— Н2О (10 :1 : 1 : 8) 0,09; 0,27-; 0,79 [551, c.694]
55
Т аблица И (прсдолжение)
Разделяемые ионы Подвижный растворитель Проявляющий раствор Rf Литература
U(VI), Cu(II), Co(II), V(V), Mn(VII), Ni(II) V(V), Mo(vl), W(VI) Fe(III), U(VI), Be(II), V(V), A1(III), Th(IV), Zr(IV) V(V), Cr(VI) V(V), Cl(VI), U(V1) Ci (VI), V(V), W(vl), Mo(VI) V(V), Ag(D, Pb(II), Hg(II), Cu(II), Cd(II), Sb(III), Bi(III), Tl(III), Fe(III), Al(III),Cr(III), Sn(II), Sn(IV), Ni(II), Co(II), Mn(II), U(1V), Ce(III),Th(IV), Zr(IV), Zn(II), Ca(II), Ba(II), Sr(II), Te(lV), La(III), Mg(II) Fe(III),Mo(VI), Co(II), Cu(II), Tl(III), Mn(VlI), V(V), Ni(II), Cr(Vl), Al(III) V(IV), V(V), Mo(VI), W(VI), Zr(lV), U(VI) Тетрагидрофуран—конц. HCI (10 : 3) 3—7 1N HNO3 Ацетон—конц. НС1-ЩО (8:1:2) Этанол—метанол— конц. НС1—Н2О (50 : 30 : 4 : 15) Ацетон—конц. СН3СООН или конц. НС1—Н2О (различные соотношения) силл-Ко л ли дин— «зо-бутанол— конц. НС1 (10 : : 38:52), Ацетоуксусный эфир— хлоргидрат метиламина— конц. НС1 (20:2:7) Этилацетат—н-бутапол — конц. НС1 (9:6:5) X лорофорМ—этанол (1 : 1), хлороформ—метанол (1:3), хлороформ— бутанол (3:1), хлороформ—изопропанол (1 : 1) 1%-пый раствор таннина в NH4OH 0,5 0 таннина и 1 г CH3COONa в 10 мл 60%-ного этанола 5%-ный раствор K4[Fe(CN)e], затем НС1 5%-ный раствор пирогаллола в этаноле 1%-пый раствор ализарина в этаноле 1 % -ный раствор рубеановодо-родной кислоты в этаноле, содержащий 0,5% салицилальдо-ксима, выдержанный над конц. NH4OH 10%-ный раствор таннина в 2N СН3СООН, пары NH3 0,98; 0,91; 0,78; 0,56; 0,5£); 0,27 0,96; 0,69; 0,00 Fe>U>Be> >V>Al>Th> >Zr 0,50; 0,80 0,20; 0,53; 0,69 Cr>V>W>Mo 1,00; 0,85; 0,60; 0,55; 0,35; 0,25; 0,20; 0,07; 0,07; 0,00 [551, с. 696] [552] [896] [993] [1083] [1161] [1004] [619] [652]
56
Таблица 11 (окончание)
Разделяемые ионы Подвижный растворитель Проявляющий раствор Ну Литература
V(V), V(IV) Этанол—копц. СН3СООН—20%-ный раствор CH3COONa(5:2:3) 1%-ный раствор 8-оксихинолина в диэти-ловом эфире 0,5; 0,8 [1109]
V(IV), V(III) Этанол—конц. СН3СООН— 40%-пый раствор CH3COONa (10 : 1 : 2) То же 0,68; 0,97 [1109]
пиридина в 0,2 N HNO3, 0,5 мл 1,5%-ного раствора дифенилкарбазида в этаноле, 3,0 мл ацетона и титруют 0,1%-ным раствором нитрата свинца до обесцвечивания малиново-розовой окраски раствора. Относительная погрешность Определения ванадия не хуже 5% [551, с. 703].
Описан метод полярографического определения ванадия после его отделения на бумаге 5%-ным раствором НС1 в бутаноле. Хроматограмму разрезают па две части по пятну молибдена так, что на нижней части остаются железо, висмут, кадмий и сурьма вместе с молибденом, а п?а верхней — ванадий, медь, уран, свинец и титан. В этих группах нет мешающих элементов для их последующего полярографического определения при использовании салициловой кислоты в качестве фона [551, с. 705, 794]. Ванадий после его отделения от других элементов методом хроматографии на бумаге определяют спектрофотометрическим методом в виде 8-оксихинолината [257, 1109] или фосфорновольфрамованадиевой гетерополикислоты [671, 1161], а также применяют активационный метод анализа бумажных хроматограмм [674].
Для повышения специфичности и скорости процессов разделения применяют хроматографическую бумагу, импрегнированную различными реагентами [551, 885, 1118]. На бумаге, пропитанной гидроксиламином, можно отделить ванадий(1У) от хрома(Ш) и обнаружить его в виде голубого пятна [257]. В табл. 12 приведены некоторые условия отделения ванадия от сопутствующих элементов на импрегнированной бумаге.
Методы хроматографии на бумаге применяют для количественного определения ванадия в сталях [619, 671, 685, 993], ферровольфраме и феррованадии [583], органических соединениях [700], рудах [685].
Хроматография в тонком слое
Хроматография в топком слое сорбента характеризуется большей экспрессностью и четкостью разделения элементов по сравнению с методами хроматографии на бумаге. Однако для получения строго постоянных и воспроизводимых значений Rf необхо-
57
Таблица 12
Отделение ванадия от других элементов методом хроматографии на импрегнированной бумаге
Разделяемые ионы Пропиты вающий бумагу реактив Подвижный растворитель я/ Литература
26 ионов, в Амберлит 1R-2, 5 различных сис- Для V(V) [1078]
том числе содержа- тем растворите- равен 0,0 для
V(V), Т1(1), Cd(II) щий ~45% лей, содержащих всех систем
Амберлита малоновую кис- растворителей
Mo(VI), W(VI) IRA-400 в мало-патпой форме; аннонообмеппая бумага SB-2 0,05 М раствор лоту 0,50^-ный раст- 0,00; 0,03; 0,30 [751]
V(V) V(V), Fe(III) анионита Аликват 336 в толуоле, ЯО4-форма 0,1 М раствор вор Н.,О.. в 0,15М H2SO4 0,25 М НС1 0,54; 0,09; 0,39 [1052]
Ti(IV) V(IV), V(V) дипонилнафта-ленсульфоновой кислоты в гептапе 10%-пый раст- Фосфатиоцптрах- 0,06; 0,83 [1051]
V(IV), Cr(III), вор ZrOCl2 в НС1, затем 12%-пый раствор H-jPO j в 47V НС1 Гпдроксиламин, пый буферный раствор с pH 7 Сернокислый V > Ст > Мо [257]
Mo(VI) V(V), W(VI), пары NO2 0,1 М раствор 2- раствор (pH 1,6 —1,7) Ацетон — 12.V V > W > Fe > [709]
Fe(III), Cr(VT) тепоплтрпфтор- • НС1 (8:2) >Сг
V(V), Nb(V), Ta(V) V(V), Bi(IlI), ацетона в бензоле Катпонообмеппая 1%-ный раст- V > Nb > Та Для V(V) [1077]
Cd(II), Co(II), бумага Амберлит вор Н2О2 в 0,01 М равен 0,36,
Cu(II), Fe(III), SA-2 в H2SO4 для остальных
Ni(II), Pb(II) V(IV), Ga(III), Н-форме 0.1 М раствор 0,001 .1/ HNO3 элементов 0,06 0,11; 0,78; 0,91 [1006]
Mo (VI) V(IV), U(VI), SnCl?, затем 0,25 М раствор арсената натрия То же 0,1 .1/ HNO3 0,69; 0,48; [1006]
Th(IV), Ag(I), Sn(II), Bi(III) Zr(lV) Более 40 Вольфрамат или Изучено 0 систем 0,40; 0,22; 0,22; 0,16; 0,08 [1005]
попов, в том числе V(IV), Mo(VI), U(VI) молибдат титана (сначала пропитывают бумагу 0,25 М раствором TiOCl2, затем 0,25М раствором Na»MoO4 или Na.,WO4) различных растворителей (конц. HCI, HCOONH4, КС], н-бутапол)
58
Таблица 12 (окончание)
Разделяемые ионы Пропитывающий бумагу реактив Подвижный растворитель я/ Литература
V(V), Fe(III), Ti(IV), Mo(VI), W(VI) 0,5 %-пый раствор рутила в метаполе Этанол или метанол — IN НС1 (10:1) 0,90; 0,80; 0,70; 0,51; 0,32 для метанольных и 0,28; 0,72; 0,60; 0,48; 0,32 для этапольных растворов [1120]
Димо очень тщательное соблюдение всех условий проведения процесса разделения [30, 102, 103, 579]. Техника работы при использовании метода хроматографии в тонком слое во многом сходна с работой на хроматографической бумаге. В качестве сорбентов при отделении ванадия от других элементов обычно используют силикагель или окись алюминия; толщина сухого сорбционного слоя 0,1—0,3 мм. В табл. 13 приведены некоторые условия отделения ванадия от сопутствующих элементов методом хроматографии в тонком слое.
Метод хроматографии в тонком слое применяют для количественного определения вападия(У) методом диффузной отражательной спектроскопии после его отделения от Bi(III), Cd(II), Co(II), Cu(II), Fe(III), Mn(II), Ni(II), Pb(II), U(VI) [920].
Электрофорез
Электрофорез неорганических ионов на бумаге и в топком слое сорбента-носителя позволяет существенно уменьшить время разделения, использовать значительно большие количества вещества [102, 579]. Разделение элементов методом электрофореза основано на различии подвижности образующихся ионов этих элементов с комплексообразующим реагентом (электролитом) под действием электрического поля.
Разделение более 30 элементов методом электрофореза на слоях целлюлозы проводили из 0,1 М раствора а-оксиизомасляной кислоты. Градиент потенциала 50 в!см, сила тока 5—15 а, продолжительность электрофореза 20 мин., проявитель—1%-ный раствор 8-оксихинолина в этаноле. Установлено, что ванадий мигрирует к аноду и за 20 мин. перемещается па 40 мм от линии старта (pH электролита от 2,72 до 6,0). При pH 8,8 скорость миграции ванадия возрастает до 60 мм. В этих условиях ванадий наиболее надежно может быть отделен от всех исследованных элементов, в том числе от молибдена, марганца, хрома, железа, никеля, урана, циркония и многих других [102, с. 128].
59
Таблица 13
Отделение ванадия от других элементов методами хроматографии в тонком слое
Разделяемые ионы Сорбент Подвижный растворитель Проявляющий раствор Rf Литература
Cr(VI),V(V),Ti(IV) Силикагель G А ми лацетат—эти л ацетат— конц. НС1—конц. СНдСООН (20:20:30:11) Раствор NaBrO, затем подкисленный раствор Н2О2 Сг > V > Ti [102, c. 54]
V(V), Mo(VI), Se(VI), Te(VI) » Диэтилоксалат—конц. HCI (60:1) 0,1 М раствор тпокарбо-ната калия 0,65; 0,60; 0,77; 0,42 [102, c. 57]
н-Бутилацетат—конц. FICI (40: :0,6) 0,43; 0,67; 0,55; 0,23
V(V), W(VI), Mo(VI), Be(VII) Al2O3, незакрепленный слой Растворы серной или азотной кислоты различной концентрации, смеси серной кислоты с ацетоном и метанолом 10%-ный раствор SnCI2 в HCI, затем 50?/»-ный раствор KSCN Для 1,5—4,5 N HNO3h0,3 — — 0,6TVH2SO4 Re > Mo >• > v>w [104]
Ag, Cd,Co, Cu, Fe(III), Mn, Mo, Ni, Pd, Rh, Ru, Sn, V(V), Zn, Zr(IV), Ti(IV) Целлюлоза или силикагель, пропитанные 5%-ным раствором трибу-тплфосфата в хлороформе Растворы НС1 (0,1 — 9 М) 2%-ный раствор 8-оксихинолина в хлороформе [102, c. 66]
V(V), Au(III), TI(III), U(VI) Силикагель пли целлюлоза, закрепленная крахмалом Гексан—трибутилфосфат— 4 ,7A- HNO3 (100:1:10) 510%-ный раствор 8-ок-(сихинолинав этаноле, пары NH3 0,10; 0,85; 0,9; 0,9 [102, c. 85]
V(V), V(IV), V(III) Силикагель MNS-HS н-Бутанол, насыщенный 1 М НС1 0,2%-ный раствор кверцетина в этаноле или пары NH3 V(V) >V(IV)> > V(III) [102, c. 90]
Bi(III), Co(II), Cu(II), Cr(III), Fe(II), Mo(VI), Ni(II), U(VI) ,V(V) Силикагель Бензол или смесь циклогексана с хлороформом (3:7) Диэтил дитиокарбамино-вая кислота Cu > Mo>Ni> >Co>Bi>Fe> >V>Cr,U [102, c. 119]
Таблица 13 (окончание)
Разделяемые ионы Сорбент Подвижный растворитель Проявляющий раствор Rf Литература
Более 30 ионов, в том числе V(V) Силикагель 5%-ный раствор HCI в смеси метилизобутилкетона с амилацетатом (2:1) 0,05%-ный раствор дитизона в четыреххлористом углероде [1080]
Более 50 ионов, в том числе V(V) Диэтил амин этилце л лю-лоза (ДЕАЕ) Смеси метанола, бутанола, тетрагидрофурана или циклогексана с растворами HCI 2 %-ный раствор AgNO3—конц. NH4OH (3:1) Даны зависимости Rf от pH [872]
39 ионов, в том числе V(V), Fe(III), U(VI) Силикагель HF254 17 систем водно-органических растворов (ацетон, метилацетат, 10%-ный раствор HCI в различных соотношениях) 1%-ный раствор K4[Fe(CN)6] [1149]
V(V), W(VI), Re(VII), Mo(VI) А1ХО3 Растворы салициловой, сульфосалициловой или тиосалициловой кислоты в этаноле 1%-ный раствор SnCI2 в 6М НС1, затем 50%-ный раствор NH4SCN Re > Мо > > V> W [Ю5, 108]
33 иона, в том числе V(V) Силикагель Конц. HCI—метанол—амиловый спирт — амилацетат — ме-тилпзобутилкетои (1:1:1:1:1) 0,05%-ный раствор дитизона в четыреххлористом углероде Для V(V) равен 0,42 [Ю80а]
Fe(III), Mo(VI), Sb, Au, Sn, Те, Hg, As, Cd, Zn, Co, Pt, Cu, Pd, Bi, Mn, Pb, V(V) Силикагель Конц. HCI—амилацетат—ме-тилизобутилкетон (1 : 16 ; 8) 0,С5%-ный расгвор дитизона в четыреххлористом углероде 0,75; 0,68; 0,68; 0,67; 0,55; 0,45; 0,41; 0,37; 0,33; 0,27; 0,22; 0,21; 0,17; 0,13; 0,07; 0,07; 0,05; 0,05 [1080а]
Метод электрофореза в тонком слое окиси алюминия применен для разделения ванадия(У), вольфрама(У1), молибдена(У1) и рения (VII). В качестве электролита использован раствор 0,1 N H2SO4. Для обнаружения элементов хроматограмму обрабатывали 35 % -ным раствором двухлорида олова в 6 N HCI и затем 50%-ным раствором цианида калия [106].
Электрофорез на бумаге предложен для разделения ванадия(У) и таллия(Ш) [1079] и ванадия(У), молибдена(У1) и рения(УП) [107]. В качестве электролита применяют 1%-ный раствор Н2О2 в 0,01 М H2SO4 [1079] или в 0,05 N NaOH [107]. В 0,05 М растворе У-(2-оксиэтил)-иминоуксусной кислоты отделяют ванадий(У) от большой группы элементов, в том числе от мышьяка(У), тантала^), ниобия(У), хрома(У1) [820]. В 0,5У НС1 разделяют перекисные соединения ванадия(У), молибдена(У1) и титана(1У). Зоны ванадия и титана перемещаются к катоду (соответственно красное и оранжевое окрашивание), а зона молибдена — к аноду (желтая полоса) [551, с. 694]. Проведено систематическое . исследование подвижности ионов ванадия(У), урана(У1), то-рия(1У), титана(1У), молибдена(У1) и многих других элементов в различных смесях хлористоводородной и винной кислот и показана возможность их разделения [592].
Метод электрофореза на бумаге применен для определения ванадия, никеля, кобальта и молибдена в стали с использованием в качестве электролита 10%-ного раствора HNO3 [859].
ДРУГИЕ МЕТОДЫ
Ванадий с некоторыми хелатообразующими реагентами, такими, как трифторацетилацетон, теноилтрифторацетилацетон, бис-(триметилсилил)-трифторацетамид, салицилальдимин, образует соединения, имеющие достаточную летучесть для их использования в газовой хроматографии [809, 1118]. Обычно после конденсации разделяемых компонентов ванадий определяют спектрофотометрическим методом [1118]. Использование газового хроматографа Микротек 2500 с пламенно-ионизационным детектором позволяет обнаружить до 10~10 г ванадия [809].
При пропускании сухого хлористого водорода над анализируемой пробой можно отделить ванадий в виде его летучего оксихлорида, который может быть количественно поглощен водой или другим подходящим раствором [115]. В разработанном химикоспектральном методе анализа пятиокиси ванадия операцию концентрирования примесей проводят следующим образом.
2 г пробы помещают в кварцевую лодочку и в кварцевой печи при температуре 300° С отгоняют в анадий в токе сухого хлористого водорода в течение ЙО мин. Затем остаток ох лаждают, добавляют несколько капель HNO3 и продолжают отгонку еще в течение 15 мин. В концентрате примесей остается не более 1 • 10 5 г ванадия. Вместе с ванадием отгоняются молибден, мышьяк, вольфрам, германий и олово [338].
При атомно-абсорбционном определении примесей в тетрахлориде ванадия применяют предварительное концентрирование цх путем частичной отгонки (94—95%) УС14 в потоке аргона (1 л!мин при 60° С) из навески пробы, равной 4 г [442].
Глава IV
МЕТОДЫ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ВАНАДИЯ
ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ
Гравиметрическими методами ванадий определяют в виде У2ОБ> V2S5, ванадатов и соединений с органическими реагентами [57> 115, 162, 193, 194, 320, 833, 928, 943].
Для определения ванадия рекомендованы малорастворимые ванадаты свинца, ртути, серебра и т. д. [57, 193, 194, 724, 894]. По современным представлениям о ионном состоянии ванадия(У) в водном растворе осадки ванадатов определенного состава можно получить только в узком интервале pH (см. рис. 1). В состав осадка, несмотря на принимаемые меры предосторожности, часто входят щелочные металлы, которые нельзя удалить при промывании осадка. Прокаливание осадков иногда сопровождается их плавлением, в процессе которого самопроизвольно образуются кислородные ванадиевые бронзы и часть ванадия переходит в другую степень окисления. Кроме того, возможно взаимодействие расплава с керамикой тигля.
Непостоянство состава осадков ванадатов и их весовой формы, а также необходимость предварительного отделения ванадия от большинства катионов не позволяют рекомендовать эти методы для гравиметрического определения ванадия.
Для осаждения ванадия рекомендовано много органических реагентов (табл. 14). Из них наиболее известны купферон и 8-окси-хинолин. Эти реагенты образуют осадки со многими элементами, поэтому ванадий предварительно отделяют. Получаемые осадки мелкодисперсны и плохо фильтруются. При их прокаливании до V2O5 происходит частичное восстановление ванадия, поэтому осадок обрабатывают несколькими каплями азотной кислоты и вновь прокаливают [115]. Температура прокаливания осадков (800° С) выше температуры плавления пятиокиси ванадия, поэтому во избежание взаимодействия плава с керамикой тигля работают с платиновыми тиглями. Методики трудоемки и длительны, хотя считаются достаточно точными [194, 928].
Предложенные в последнее время новые органические реагенты образуют с ванадием кристаллические осадки с большой молекулярной массой, что позволяет определять 10—50 мг ванадия с достаточной точностью, но большинство реагентов не опробовано
63
Таблица 14
Определение ванадия гравиметрическими методами
Реагент Условия осаждения Температура обработки осадка,°C Весовая форма Примечание Литература
Определение ванадия(1У)
Бензоат аммония, 0,37%-иый водный раствор O,67V НС1 при кипячении 800 V2O5 He мешают Cu, Al, Mg, Mo(VI), VV(VI), Ti(IV) Мешают Fe(III), Cr(III) [57]
5,7-Дибром- 8-оксихино-лин, 1%-ный ацетоновый раствор pH 4-5 150-200 VO(C9H4ONBr2)2 Мешают Al, Zn, Co, Ni, Mn(II) [16]
5,7-Дииод-8-оксихино-лин, 1%-ный ацетоновый раствор pH 4—5 170-180 VO(C9H4ONJ2)2 Мешают Al, Zn,Co,Ni, Mn(II) [16]
2-Оксинафтил-1-мети-ленэтиламин, 2%-ный спиртовый раствор pH ~3, при нагревании в присутствии аскорбиновой кислоты 110 VO(C13H12ON)2 Мешают Pb и Cu He мешают Fe(III) [147]
2-Оксинафтил-альд оксим, 2%-пый спиртовый раствор pH ~3, при нагревании в присутствии аскорбиновой кислоты 110 VO(CnH8O2N)2 Мешают Ni, Cu и Ti (1 мг/мл) He мешают Fe(III) [147]
Бензоил-гс-нитроацетат-аналид pH 4,0—5,5, в присутствии 5%-ного раствора CHgCOONHa, при нагревании НО \ VO(Ci5HnO4N2)2 He мешают Ag, Tl, а в присутствии сульфосалициловой кислоты и фтор-иона — Со, Мп, Zn, Cr(III), Fe(III), [9С4]
Музгин и др.
ы Таблица 14 (окончание)
Реагент Условия осаждения Температура обработки осадка,°C Весовая форма Примечание Литература
Циклопентанон-2-кар-боксианалид, 0,3%-ный водно-спиртовый раствор pH 5—6 в присутствии 5%-ного раствора CH3COONH4 при нагревании 110 V O(C12H12O2N)2 Mo(VI), Ti(IV), W(VI), U(VI); а иод-иона — Hg и Cd He мешают Cu, Co, Ni, Al, Zn, Fe(III), Ce(III), Zr(IV), в присутствии иод-иона— Hg [904]
Определение ванадия (V)
Диантипирилфенилме-тан, 1%-ный спиртовый раствор Слабокислый на холоду раствор, 100-105 [(C11H11ON2)2CHCeH5]i. h4v6o17 Мешают многие ионы [146]
о-Оксиацетофеноксим, 2о/„-пый спиртовый раствор pH 2,2—4,8 при нагревании до 35—40° С (2,5-кратный избыток реагента) 110 VO(OH)(C8H8O2N)2 Мешают Cu, Pd, Ti He мешает Fe(III) в присутствии F", C2O42~ и тартрат ионов [989]
Купферон, 4%-ный раствор ~1А HCI или холоду Н2ЗО4‘на 800 v205 Мешают многие ионы [21, 22, 194, 320]
8-Оксихинолин, 4%-ный спиртовый раствор 05 pH ^4,5, при НИИ нагрева- 120—140 800 VO(OH)(C9HeNO), v208 То же [577, 666, 947]
на реальных объектах, не указаны мешающие элементы. Иэ перечисленных в табл. 14 органических веществ следует отметить производные 2-оксинафтойного альдегида, а также бензоил-п-нит-роацетатаналид, с помощью которых можно определять содержание ванадия в феррованадии [147] и в сталях [904] без предварительного отделения железа.
Гравиметрические методы для определения ванадия в настоящее время используются значительно реже, чем все другие методы.
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Определение ванадия проводят оксидиметрическими и комплексонометрическими методами. Наибольшее распространение получили оксидиметрические методы.
Оксидиметрическое титрование
Ванадий в водных растворах может быть получен в различных степенях окисления (II, III, IV, V), поэтому в качестве титрующих растворов применяют как окислители, так и восстановители.
Титрование восстановителями. Ванадий(У) является сильным окислителем и в кислых растворах взаимодействует с большинством восстановителей, переходя в ванадий(1У), поэтому для титрования рекомендованы растворы солей железа(П), олова(П), титана(Ш), аскорбиновой кислоты и др.
Для получения точных результатов ванадий предварительно полностью переводят в пятивалентное состояние действием различных окислителей. Для этой цели рекомендованы перманганат калия, хлорная кислота, персульфат аммония в присутствии ионов серебра или Co-Cu-катализатора, озон, бромат калия. Наибольшее применение получил перманганат калия. Он удобен тем, что в присутствии Н3РО4 на холоду взаимодействует с ванадием и не окисляет хром(Ш). Для полноты окисления необходим достаточный избыток перманганата калия. Считается, что розовая окраска раствора от избытка перманганата калия должна сохраняться 2—3 мин., затем избыток его устраняют различными способами.
При наличии в растворе хрома добавляют восстановитель (азид натрия, щавелевую кислоту или нитрит натрия). Из перечисленных реагентов чаще всего применяют нитрит натрия или калия, избыток которого перед титрованием разрушают мочевиной [245] или сульфаминовой кислотой [639]. Применение щавелевой кислоты нежелательно, так как она может частично восстанавливать ванадий из-за индуцирования этой реакции ионами перманганата [522]. Нитрит-ион может также восстанавливать ванадий(У), если в растворе присутствует в большом количестве фосфорная кислота [356].
66
Широко распространенным и наиболее точным методом определения ванадия является титрование его раствором соли желе-за(П) [414]. В кислой среде железо(П) восстанавливает ванадий(У) до ванадия(1У). Методом амперометрического титрования показано, что эта реакция в 1—8W H2SCh протекает стехеометрично и не зависит от форм существования ионов ванадия(У) в растворе [475].
Для определения точки эквивалентности в зависимости от кислотности среды используют различные индикаторы [535]:
Индикатор Концентрация H.SOi, N
Дифениламин 1—3
Дифениламинсульфоновая кислота 1—5
Фенилантраниловая кислота 4—11
о-Фенантролинат железа (ферроин] 5—15
2,2'-Дикарбоксидифениламин 20—24
4-Сульфофенилантраниловая кислота 18—23
Из перечисленных индикаторов чаще применяют фенилантраниловую кислоту. Следует иметь в виду, что изменение концентрации H2SOa от 1 до 25 N приводит к уменьшению окислительного потенциала этого индикатора от 0,97 до 0,75 в и соответственно к изменению полноты взаимодействия ванадия и железа с индикатором [3].
Точка эквивалентности может быть определена потенциометрически [93, 414, 1011] и фотометрически [373].
При титровании ванадия раствором соли железа(П) следует обращать внимание на присутствие в растворах фосфат- и фторид-ионов. Наличие в растворах этих ионов в количествах, необходимых для связывания железа, благоприятствует титрованию. Увеличение концентрации фосфорной кислоты от 0,5 до 12 М приводит к возрастанию не только потенциала V(V)/V(IV), но и потенциала У(1У)/У(П1). Одновременно падает значение потенциала Fe(III)/Fe(II) [93, 1011]. Так, в 6,5 М Н3РОа первый потенциал равен 0,95, второй — 0,50, а третий — 0,28 в [93], следовательно, в присутствии больших количеств фосфорной кислоты ванадий может быть оттитрован частично до ванадия(Ш), что повлечет за собой большую ошибку определения.
Титрованию мешают бромат-, иодат- и нитрат-ионы, а также все окислители, взаимодействующие с железом(П). Хром, часто сопутствующий ванадию, должен быть переведен перед определением в хром(Ш).
К 20—30 мл раствора, полученного после растворения навески образца, добавляют 3—5 мл Н3РО4 (уд. в. 1,70). Если необходимо, то для восстановления хрома добавляют несколько миллилитров соли железа(П) (40 г/л). Частично восстановленный ванадий окисляют 0,5—3%-ным раствором перманганата калия до появления устойчивой в течение 2—3 мин. розовой окраски. Избыток перманганата калия восстанавливают несколькими каплями 0,5%-ного раствора нитрита натрия, вводя 2—3 капли избытка. Раствор перемешивают и сразу же вносят 2—3 г сухой мочевины или 4—5 мл 10%-ного раст
3*
67
вора сульфаминовой кислоты. Приливают равный объем серной кислоты (1 : 1), при этом общая кислотность раствора должна быть ~ 5—9 N. Охлаждают, добавляют 2—4 капли фенилантраниловой кислоты, окрашивающей раствор в ярко-вишневый цвет. Титруют 0,1—0,02 N раствором железа(П) до перехода окраски раствора в светло-зеленую [165].
Концентрацию титрующего раствора устанавливают по бихромату калия, приготовленному из перекристаллизованной соли 1245].
При определении содержания ванадия в рудах, шлаках и сталях титр раствора соли железа(П) устанавливают по стандартным образцам, содержащим хром и вольфрам в количествах, близких к анализируемым объектам [453]. В некоторых случаях, например при определении содержания ванадия в феррованадии и высоколегированных сталях, содержащих вольфрам, предпочтительнее методы обратного титрования. При этом избыток железа(П) титруют раствором бихромата калия с индикатором дифениламин-сульфонатом бария [1143] или раствором сульфата церия с о-фе-нантролином в присутствии фторида натрия или фосфорной кислоты, которые способствуют понижению потенциала железа [245].
Раствором соли железа(П) можно титровать и ванадий(1У) в присутствии не менее чем 10,5 М Н3РО4 в инертной атмосфере с индикаторами метиловым синим, тионином или точку эквивалентности установить потенциометрически. Определению не мешают ионы железа(П), хрома(Ш), марганца(П) и вольфрама(У1) [1011].
В качестве титранта применяют раствор фотохимически генерируемого вольфрама(У) [1100] и растворы титана(Ш) [1041], ниобия(Ш) [1073], аскорбиновой [374] и тиомалоновой [1075] кислот, растворы сульфата гидразина [1014], гидрохинона [931] и гидразида изоникотиновой кислоты [1015] с визуальными и инструментальными способами индикации точки эквивалентности.
При анализе бинарных смесей ванадия(У) с железом(П I), церием(1У), марганцем(УП), хромом(У1), ванадием(1У) в качестве титрантов используют растворы олова(П) [1040, 1121], меди(1) [925], хрома(П) [1127] и ванадия(П) [690].
Титрование окислителями. В водных растворах ванадий(У) легко восстанавливается до низших степеней окисления (II, III, IV) под действием различных восстановителей. Чаще всего ва-надий(У) восстанавливают до ванадия(1У) и титруют растворами окислителей. Ванадий(У) восстанавливают сернистым ангидридом, сульфитом или бисульфитом натрия. Избыток восстановителя после подкисления раствора удаляют в виде сернистого газа при кипячении и пропускании углекислого газа. Железо в растворе должно отсутствовать, так как оно восстанавливается одновременно с ванадием.
Сероводород для восстановления ванадия применяют только в том случае, если необходимо одновременно осадить тяжелые металлы [21, 580]. При его использовании могут быть получены завышенные результаты [115]. Соли железа(П) в кислых раство-
68
pax быстро восстанавливают ванадий] V). Избыток восстановителя окисляют на холоду персульфатом аммония, который в отсутствие ионов серебра лишь очень медленно взаимодействует с ванадием] IV) и хромом(Ш) [45, 165, 518, 847]. Рекомендуют также использовать для восстановления ванадия 8—15 %-ный раствор нитрита натрия, избыток которого восстанавливают мочевиной [1067] или сульфаминовой кислотой [355].
Перманганатометрия. Соли ванадия(1У)' окисляются перманганатом калия с заметной скоростью только в слабокислой среде (~0,1У H2SO4) и при нагревании до 50—80° С. При меньшей кислотности раствора (pH 2) ванадий(1У) начинает окисляться кислородом воздуха, а при увеличении концентрации кислоты реакция замедляется [245]. Однако при добавлении к раствору фосфорной кислоты реакция, наоборот, настолько ускоряется, что ее можно проводить при комнатной температуре в широком интервале кислотности раствора (0,1—1,0Лг) [716а].
Титрование обычно проводят без индикатора до появления слабо-розовой окраски раствора, устойчивой в течение 1—2 мин. Считают, что при введении эмпирической поправки на окрашивание раствора перманганатом калия (индикаторная ошибка) результаты определения вполне надежны.
При наличии в растворе ионов хрома(Ш) титрование следует проводить на холоду в присутствии фосфорной кислоты и индикатора. При титровании 0,01—0,05.У раствором перманганата калия применяют индикаторы ферроин [716а, 1016] или фенилантраниловую кислоту [165]. Точку эквивалентности можно установить потенциометрически [847] или фотометрически [355]. Титрованию мешают все восстановители и вольфрам. Не мешают хром, никель, кобальт и уран. Присутствие ионов хлора нежелательно.
В качестве восстановителей ванадия(У) обычно применяют сернистый газ [115], иногда нитрит натрия [355, Юб?!. При наличии в анализируемом растворе железа, ниобия, титана, тантала, хрома, марганца применяют соли железа(П) [45, 165, 518, 847].
К 200—250’ мл раствора ванадия прибавляют 20 мл смеси (320 мл H2SO4 (1 : 1), 80 мл Н3РО4 (уд. в. 1,70) и 600 мл воды). Затем добавляют 0,1 N раствор сульфата железа(П) до перехода цвета раствора в бледно-желтый или зеленый (в присутствии хрома) и 5—10 мл избытка. Хорошо перемешивают, выдерживают 1—2 мин., добавляют 20 мл 7,5-%ного раствора персульфата аммония. Раствор тщательно перемешивают 1 мин. для полного окисления избытка железа и сразу же титруют 0,05—0,1 N раствором перманганата калия до появления розовой окраски, не исчезающей 30 сек. [165].
При восстановлении ванадия(У) в цинковом, висмутовом или никелевом редукторе получают ванадий(П) или ванадий(Ш), которые титруют раствором перманганата калия с индикаторами феносафранином до ванадия(111), дифениламинсульфонатом бария до ванадия]V) [245].
Определение ванадия и урана при совместном присутствии проводят потенциометрическим титрованием после предварительного восстановления их в висмутовом редукторе [458].
69
Хроматометрия. В сернокислой среде бихромат калия взаимодействует только с ванадием(1П), но в присутствии следов марганца(П) [548}, а также в 3—12 М растворе фосфорной кислоты [1013] он может окислять и ванадий(ГУ).
Определение ванадия(Ш) рекомендуют проводить в присутствии катализаторов ионов меди(П) в сернокислой среде [689а] и железа(Ш) в фосфорнокислой среде [1012]. В качестве индикаторов используют фенилантраниловую кислоту или дифениламин [689а],'а также дифениламинсульфонат бария и ферроин [1012]. Точку эквивалентности устанавливают потенциометрически [689а, 1012] и фотометрически [1013].
Цериметрия. Сульфат церия(1У) окисляет ванадий(П), ванадий(Ш) и ванадий(1У) до ванадия(У). В качестве индикаторов применяют ферроин в растворе ~2У по H2SO4 или 7,0—8,5У ио СН3СООН [1105], фенилантраниловую кислоту и дифениламинсульфонат бария в 10 У H2SO4 в присутствии Н3РО4 и солей меди [689а], трифенилметановые красители (эриозеленый Б и др.) в 0,1—0,2JV H2SO4 [1016, 1017].
Описан метод определения ванадия(1У) с применением флуоресцирующего индикатора родамина 6Ж. В 1,5—2,5У H2SO4 раствор церия(ГУ) окисляет вначале ванадий(1У) и только затем родамин 6Ж [64, 716, 1010].
К 50 мл анализируемого раствора 1,5—2,5 У по H2SO4 добавляют 1,0— 1,5 мл конц.’ Н3РО4 и 0,2 мл 0,035%-ного водного раствора родамина 6Ж. Титруют 0,05 N раствором сульфата церия в 1 У H2SO4 при комнатной температуре на дневном свету или при электрической подсветке до исчезновения желто-зеленой флуоресценции раствора. При титровании 5—20 мл 0,05 У раствора ванадия(ТУ) погрешность определения не превышает 2% [1010].
Необходимо соблюдать указанную кислотность раствора, так как при более высокой кислотности ванадий окисляет родамин 6Ж, а при меньшей — осаждается сульфат церия [1010]. Варьируя кислотность раствора и индикаторы, можно одновременно определять ванадий с железом [763, 1105] или с молибденом [716, 1009].
Иодометрия. В кислых растворах ванадий(У) восстанавливается иодидом калия до ванадия(1У). В щелочных растворах иод окисляет ванадий(1У) до ванадия(У). На стехиометрич-ность обеих реакций существенное влияние оказывают условия их протекания, поэтому разработаны лишь косвенные методы йодометрического определения ванадия.
Восстановление ванадия(У) иодидом калия проводят в 0,4—1 У растворах по H2SOi или HG1 в инертной атмосфере. Большой избыток иодида калия и высокая кислотность раствора способствуют дальнейшему частичному восстановлению ванадия до V(III) [245, 580]. Быстрому протеканию реакции способствует введение катализаторов, например маннита [970]. Выделившийся иод титруют раствором тиосульфата натрия и рассчитывают содержание ванадия. Этим методом можно определить ~10—50 мкг ванадия с погрешностью 1,5% [970].
70
В щелочной среде (pH 8—10) ванадий(1У) окисляется до ва-надия(У) не только иодом, но и кислородом воздуха. Поэтому анализируемый раствор вливают в титрованный раствор иода. Избыток иода титруют раствором арсенита натрия [245, 1146]. Этим способом определяют ванадий в присутствии меди [1146]. Титрованию мешают ионы, восстанавливающие иод в щелочной среде (мышьяк(Ш), сурьма(Ш), железо(П) и др.).
Другие методы. В последнее время все большее значение приобретают методы определения ванадия в низших степенях окисления (II и III). Для титрования ванадия(П) рекомендованы растворы соли железа(Ш) с индикаторами сафранин Т, феносафранин и нейтральный красный [690, 926]. При титровании необходимо учитывать, что в 8—10 М H3POi ванадий(П) окисляется до вана-дия(Ш), а в 1У H2SO4 — до ванадия(1У) [690]. Точку эквивалентности можно определить и потенциометрически.
Для титрования ванадия(Ш) применяют растворы окислителей ванадата аммония, бихромата калия, сульфата церия(1У), соли железа(Ш). На кривых потенциометрического титрования раствором соли церия(1У) наблюдают два скачка потенциала, соответствующие окислению сначала ванадия(Ш) до ванадия(1У), а затем ванадия(1У) до ванадия(У). При использовании индикаторов фенилантраниловой кислоты, дифениламинсульфоната бария и дифениламина к растворам ванадия в качестве катализатора рекомендуют добавлять соли меди(П) [689а].
Для определения ванадия(1У) в качестве титрующих растворов рекомендованы в кислой среде бромат калия [753] и трикарбо-натокобальтиат гексаминокобальта(Ш) [645], а в щелочной среде — феррицианид калия в присутствии тартрат-ионов [534, 642] или комплексона III [541].
Комплексонометрическое титрование
Ванадий(У) в водных растворах существует в виде сложных ионов, поэтому его взаимодействие с комплексоном III замедленно и в большой степени зависит от pH. При pH ^1,8 ванадий титруют с индикаторами ксиленоловым оранжевым [830] или вариами-новым синим в присутствии железа(П) [1125], а при pH 6,7—6,9 — с индикатором дифенилкарбазоном [1045]. Однако эти методы нельзя считать надежными. Более распространены методы титрования ванадия(1У), который взаимодействует с комплексонодг III в широком интервале pH [424, 578]. Для восстановления ванадия используют сульфит натрия или сернистую кислоту [828], аскорбиновую кислоту [997], гидроксиламин [309]. Титрование ванадия(1У) раствором комплексона III можно проводить в присутствии различных индикаторов. При pH 3—3,5 используют Н-бензоил-И-фенилгидроксиламин [84, 126, 828], пирокатехин [653], галлеин [833], 1-(2-пиридилазо)-2-нафтол в присутствии комплексоната меди [745], дитизон в присутствии дитизоната цинка в 60%-ном этаноле [537]. При pHJj—5,5 применяют феррон
71
(7-иод-8-оксихинолин) [902], ксиленоловый оранжевый [309] и хромоксановые красители [347]. Ни один из перечисленных индикаторов не имеет особых преимуществ.
К раствору, содержащему 2—30 мг ванадия(1У), добавляют 10 мл ацетатного буферного раствора и по каплям 6 М NaOH до pH 3.’ Разбавляют раствор этанолом до 100 мл и приливают по каплям, интенсивно перемешивая, 2 мл 2%-ного раствора Н-бензоил-И-фенилгидроксиламина в 95%-ном этаноле. Титруют 0,05 М раствором комплексона III до перехода красной окраски раствора в голубую. Титрованию не мешает присутствие кратных количеств AI и V(V) (200), Мп(П) (100) и Сг(Ш) (50). Мешают равные количества Fe(III) и Ti(IV) [126].
Ванадий может быть надежно определен методами обратного титрования. К анализируемому раствору приливают комплексон III и оттитровывают непрореагировавший избыток его растворами солей различных металлов.
Ванадий(У) определяют при pH 3,5—6. Избыток комплексона III титруют раствором соли цинка в присутствии индикатора ди-фенилкарбазона [277] или растворами солей цинка, свинца или кадмия с пирокатехиновым фиолетовым [1045].
При определении ванадия(1У) избыток комплексона III титруют растворами солей свинца [864], цинка [528, 997], тория [996] и висмута [8401 с ксиленоловым оранжевым, меди — с кальцеином [1157] или 4,4-диаминостильбен-Й,Н,Н',Н'-тетрауксусной кислотой [842], висмута и свинца с пирогаллоловым красным [9011, марганца с эриохром черным Т [424], кобальта с экстракционным определением конечной точки [678].
Раствор, содержащий 2—50 л«г’ванадия(1У), подкисляют до pH 2—3 НС1 (1 : 1). (Если необходимо, то для связывания алюминия добавляют 0,5 г фторида аммония.) Добавляют избыток 0,05 М раствора комплексона III, 1 г аскорбиновой кислоты, тщательно перемешивают, разбавляют до 100 мл водой, добавляют раствор уротропина до pH 5—5, 5, приливают 1—2 капли индикатора ксиленолового оранжевого и титруют избыток комплексона III 0,05 М раствором хлорида цинка [997].
Комплексонометрические методы не имеют преимуществ перед методами оксидиметрического титрования. Их используют при анализе ванадатов или сплавов на основе ванадия в том случае, если другие компоненты определяют также комплексонометрически.
Описаны методы совместного определения ванадия с алюминием, никелем, кобальтом [175], свинцом [2771, железом и алюминием [309]. Во всех случаях вначале определяют сумму ванадия(У) и элемента методом обратного титрования при pH 5—6 и затем содержание элемента. Последний титруют в кислой среде, маскируя ванадий перекисью водорода [528], или в щелочной среде при pH > 10, где ванадий не дает прочных комплексонатов [175, 277].
Другие методы титрования
Для определения малых количеств ванадия предложены органические титранты — водные растворы красителей, которые являются одновременно индикаторами. Ванадий(У) титруют раст
72
вором метилового оранжевого' в ~60 %-пом растворе серной кислоты [176]. Метод позволяет определять ~40 мкг ванадия в присутствии равных количеств титана, молибдена, вольфрама и значительно больших количеств марганца(П), алюминия, цинка, железа(Ш), кобальта, хрома(Ш). Мешают окислители, а также железо(П) и висмут(Ш). Для спектрофотометрического и амперометрического титрования используют растворы ферроцена [471], для потенциометрического — тиооксина [91].
Ванадий(У) можно определять методами осаждения, используя в качестве титрующих растворов нитрат серебра [951] или раствор 5,7-дибром-8-оксихинолина в ацетоне [661].
Опробованы косвенные методы определения, основанные на том, что при взаимодействии ванадия(1У) с тартратом натрия-калия, маннитом, глицерином, тростниковым сахаром выделяется кислота в количествах, эквивалентных ванадию, которую титруют раствором щелочи с индикатором метиловым оранжевым. Ошибки определения большие [206].
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Для определения ванадия применяют методы, основанные на измерении оптической плотности растворов, содержащих ионы ванадия(У), ванадия(ГУ), ванадия(1П) или окрашенные продукты взаимодействия ванадия с неорганическими и органическими реагентами, а также продукты окислительно-восстановительных реакций ванадия с органическими реагентами.
Методы определения, основанные на измерении оптической плотности растворов, содержащих ионы ванадия(У), ванадия(ГУ) и ванадия(Ш), малочувствительны, но позволяют быстро определять ванадий в простых по составу объектах: окислах, растворах, ванадатах.
Методы, основанные на цветных реакциях ванадия с различными реагентами, чрезвычайно многочисленны, но в практике химического анализа используют лишь некоторые из них. Наибольшее распространение получил фосфорновольфраматный и экстракционно-фотометрический метод с использованием N-бензоил-N-фенилгидроксиламина. Исследованиями последних лет показано, что более высокая чувствительность и специфичность определения ванадия могут быть достигнуты при использовании других органических производных гидроксиламина, а также производных гидразина. Перспективно использование азопроизводных гетероциклических аминов и нафтолов.
Интенсивно изучаются ионные ассоциаты и смешанно-лигандные соединения, применение которых позволяет определять ванадий в сложных по составу объектах без предварительного разделения.
В качестве маскирующего реагента для большинства элементов, сопутствующих ванадию, следует рекомендовать комплексон IV [423], применение которого позволяет определять вана-
73
дий без предварительного отделения с такими реагентами, как ксиленоловый оранжевый, 4-(2-пиридилазо)-резорцин и др.
Методы, основанные на способности ванадия(У) окислять различные органические реагенты с образованием окрашенных продуктов, широко используют при анализе металлов и сплавов.
Обзор фотометрических методов определения ванадия приведен в работах [161, 162, 185, 194, 1118].
Определение ванадия по! окраске ионов ванадия (V), ванадия (IV)[Jh ванадияJ(III)
Ионы ванадия в степенях окисления II, III, IV и V различно окрашены и имеют характерные абсорбционные спектры. Ярко выраженные максимумы полос поглощения могут быть использованы для фотометрического определения ванадия [907].
Ванадий(У) в растворе в зависимости от его концентрации и кислотности среды может находиться в различных ионных состояниях, которые характеризуются определенными полосами поглощения [195]. Однако сложные ионные равновесия затрудняют использование абсорбционных спектров ванадия(У). Для его определения рекомендуют только две полосы поглощения с максимумами при 330 нм в конц. H2SO< и при 270 нм в 1 М NaOH (рис. 7). Молярные коэффициенты погашения соответственно равны 4,6• 103 и 8,4-103 [538]. Определению мешают многие элементы.
Окрашенные ионы Co(II), Ni(II), Gu(II) и др. предварительно осаждают в виде гидроокисей. Хром отделяют в виде оксихино-лината или хромата свинца [1050]. Большие количества желе-за(Ш) отделяют электролизом на ртутном катоде. Титан маскируют фторид-ионами [1068]. Для уменьшения влияния больших количеств железа и титана их вводят в растворы сравнения [190]. Описан метод определения ванадия с предварительным отделением его в виде купфероната [165, 539]. Определению ванадия при 270 нм в щелочной среде не мешают W, Ti, Zr, а также равные количества Re, Мо и Nb [281, 538]. Для повышения избирательности определения ванадия в кислой среде оптические плотности растворов измеряют при 450—480 нм. В этом случае определению не мешают любые количества Nb и Та, а также <J2%Mo, ^6%Fe и Мн [1068]. Этим методом определяют ванадий в рудах [165], сталях и железе [1175], сплавах [539], карбидах и окислах вольфрама, ниобия, титана, тантала [1068], тетрахлориде титана [190, 565].
Синие растворы ванадия(1У) в 1 М H2SO< имеют две полосы поглощения при 360 и 760 нм (рис. 8). Увеличение кислотности раствора приводит к гипсохромному сдвигу второй полосы [538], Определение ванадия(1У) чаще проводят при 760 нм. Чувствительность при этом невелика (е = 19), а избирательность выше. В сочетании с дифференциальным способом измерения оптической плотности раствора можно получить при определении больших количеств ванадия достаточно точные результаты [538]. Таким 74
£ М~3
Рис. 7. Спектры поглощения растворов ванадия(У) IM NaOH (7) и 18MH2SOj (2) [538]
Рис. 8. Спектры поглощения растворов V(III) (7 и 2) и V(IV) (3 и 4) (СФ-4, I = 20 мм) [200]
Концентрация солей ванадия, N: 1 — 5,4-10~2;2 — 2,0-10“3; 3 — 1,8-10-’; 4 — 4,2-10-
1,4 — в 1 М H2SOa; 2,3 — в 1 М HtSO<, 1 М NH<SCN, 60% ацетона
образом ванадий определяют в окислах [882], катализаторах [861], феррованадии [538].
Количественное определение ванадия(Ш) проводят путем измерения оптической плотности растворов при 400'—420 нм (см. рис. 8). Для замедления окисления ванадия кислородом воздуха создают кислую среду (6—7 N H2SOi). Определению не мешают медь (до 0,5 М) и ванадий(ГУ) (до 0,1 М) [238]. Этот метод используют для определения ванадия(Ш) в хлоридных расплавах [386].
Характерная окраска ионов ванадия сохраняется в стеклах, что было использовано для качественной оценки содержания ванадия(Ш), ванадия(1У) и ванадия(У) в минералах, синтетическом корунде и александрите [142].
Определение ванадия по образованию окрашенных соединении с неорганическими реагентами
Перевиснсе' соединение^ ванадия. Перекись водорода взаимодействует с ванадием в кислой среде с образованием устойчивого комплексного иона [V(O2)]3+ красно-оранжевого цвета [350, 457, 577, 580]. Его молярный коэффициент погашения при 460 нм равен '—200 [311]. Перекисное соединение образуется в широком интервале концентраций кислоты (0,6—6 N H2SO4) и при содержании перекиси водорода 0,03—0,09%. Замечено, что чем больше кислотность анализируемого раствора, тем больше должна быть концентрация перекиси'водорода'[457]. Допустима замена серной
кислоты на хлористоводородную или азотную. Окраска растворов устойчива в течение 1—2 суток и подчиняется закону Бера в широком интервале концентраций ванадия.
45 мл раствора, содержащего от 0,2 до 2 мг ванадия(У), 1—2 N по минеральной кислоте, помещают в мерную колбу на 50 мл, добавляют 0,5—1 мл 3%-ного раствора перекиси водорода, перемешивают, разбавляют водой до метки и измеряют оптическую плотность раствора при 450 им [457].
Определению ванадия мешают титан(1У), железо(Ш), хром, вольфрам и молибден, а также восстановители J~ и Вт-. Влияние титана 1%) устраняют добавлением фторид-ионов. Железо маскируют фосфат-ионами. Малые количества хрома добавляют в раствор сравнения для устранения его влияния [457, 518, 743]. Мешающее действие вольфрама ослабляют введением перекиси водорода в щелочной раствор и только затем его подкисляют. Это приводит к превращению желтого соединения Na2[B7(O2)4] в бесцветное [HWOJ.
В виде перекисного соединения ванадий определяют в сталях [350, 457], шлаках [320], рудах и минералах [21, 457], феррохроме, металлических хроме и марганце [320], ферровольфраме, ферротитане [743], сплавах на основе ниобия [518], золе нефтей [90], органических веществах [378]. При совместном присутствии молибдена, титана и ванадия их определяют в виде перекисных соединений путем измерения оптической плотности соответственно при 300, 390 и 500 нм [485, 508].
Применяют также разно лигандные соединения ванадия с перекисью водорода и 4-(2-пиридилазо)-резорцином [40], комплексоном III [397, 483], дифениламином [368] или 2-(3,5-дибром-4-метил-2-пиридилазо)-5-диэтиламинофенолом [845].
Гетерополикислоты ванадия- Чаще всего определение ванадия проводят по окраске фосфорновольфрамованадиевой кислоты. Соединение образуется в растворе любой минеральной кислоты с концентрацией до 2,4 N при соотношении Н3РО4 : Na2WO4 от 3 : 1 до 20 : 1 и содержании вольфрамата натрия, равном 0,01—0,1 М. Молярный коэффициент погашения при 400 нм равен 1,4-10®. Оптимальная концентрация фосфорной кислоты 0,5 М, а .вольфрамата натрия — 0,025 М. Последний рекомендуют вводить после фосфорной кислоты. Для быстрого развития окраски растворы кипятят. При малом содержании ванадия (менее 10 мкг/мл), кислотности растворов, близкой к 0,5 N, и в отсутствие железа кипячение раствора не обязательно.
К анализируемому раствору (> 1,0 мг V) добавляют H2SO4 (1 : 4) ( в таком количестве, чтобы конечная концентрация ее была 0,5 N), 5 мл Н3РО4 (1 : 2), 5 мл 10%-ного раствора вольфрамата натрия, разбавляют водой до' ~45 мл и нагревают до кипения. Охлажденный раствор переносят в мерную колбу на 50 мл и водой доводят до метки. Перемешивают и измеряют оптическую плотность раствора относительно раствора холостого опыта при 400 нм [311].
Определению мешают окрашенные ионы Сг(Ш), Со(П), Си(П) и др., большие количества Ti(IV), Zr(IV), Bi(III), Sb(V), Sn(IV), 76
образующие осадки фосфатов или основных солей, ионы аммония и калия, дающие малорастворимые осадки фосфорновольфрама-тов, и восстановители. Молибден(У1) образует с реагентами окрашенные соединения и мешает при более чем. 200-кратном избытке. Железо(Ш) не мешает до 20 Л4г/5О мл. Небольшие количества Mo, Ti, Fe, Zr связывают фторид- и тартрат-ионами. Окрашенные ионы вводят в стандартный раствор. Большие количества мешающих элементов осаждают щелочами [117, 121, 122] или отделяют от ванадия электролизом на ртутном катоде [406]. Иногда ванадий осаждают купфероном [191, 406], экстрагируют в виде оксихино-лината [141, 457] или диэтилдитиокарбамината [568]. При анализе руд ванадий отделяют в процессе вскрытия пробы сплавлением со щелочами [141, 406, 417, 509, 529].
Описаны методы определения ванадия в сталях [117], шлаках и черных металлах [121, 122], цирконии [568], алюминии [191]. Ванадий без отделения определяют в золе растений, почв [155] и нефтей [677]. Фосфорновольфраматным методом определяют 0,001 — 3% ванадия.
Описаны методы определения ванадия, основанные на экстракции гетерополикислоты метилизобутилкетоном [1032], изобутанолом [190, 593] или гексанолом [655] из 2,5—3,5 N H2SO4 или 1 N HNO3. При этом определению не мешают большие количества хрома, алюминия, титана, кобальта, железа и молибдена. Этим методом ванадий определяют в минералах [1032], сталях, шлаках, соединениях вольфрама, феррохроме, ферротитане, феррофосфоре [13, 593], алюминии [655], титане [190].
Аликвотную часть раствора 5 —10 л<л(0—0,2л«гУ) нейтрализуют 20%-ным раствором NaOH до pH 4 и приливают 3 мл 5 N HNO3, добавляют 0,5 мл 1%-ного раствора Н2О2 для восстановления хрома, 5 мл Н3РО4 (1 : 2). Затем окисляют ванадий) 0,1 N раствором КМпО4. Спустя 3 мин. избыток окислителя восстанавливают 1%-ным раствором NaNO2. Добавляют 1 мл 10%-ного раствора вольфрамата натрия и нагревают до кипения. После охлаждения раствор переносят в делительную воронку и разбавляют до 25 мл. Добавляют 10 мл изобутанола и экстрагируют в течение 2 мин. Органическую фазу фильтруют через сухой фильтр и измеряют оптическую плотность при 400 нм относительно раствора холостого опыта. При определении 0,02—0,1% ванадия в феррофосфоре погрешность составляет 15% [13].
При восстановлении гетерополикислоты хлоридом олова(П) [84, 150], аскорбиновой кислотой [410] или тиомочевиной [436] в 0,2—0,75 N fl2SO4 образуется окрашенное в красно-фиолетовый цвет соединение.
Анализируемый раствор (0,2—1 мг V) помещают в мерную колбу на 50 мл и нагревают до кипения, прибавляют4 мл 5%-ного раствора Na2WO4, 3 мл Н3РО4 (уд. в. 1,7) и нагревают до 90—95° С (контроль термометром). Выдерживают 2 мин. и охлаждают.’ Добавляют 2 мл 0,5%-ного раствора SnCl2, разбавляют до метки водой и измеряют оптическую плотность при 510—530 нм [150].
Разработаны ускоренные методы определения ванадия в чугунах, сталях и рудах, в которых для исключения влияния ме-
тающих элементов применяют стандартные растворы сталей и руд, не содержащих ванадий [150], или раствор той же стали используют в качестве раствора сравнения [436].
Фосфорномолибденованадиевая кислота образуется в 0,5—1 7V H2SCh при большом избытке молибдена. Окраска раствора достигает максимума через 10 мин. и начинает ослабляться через ~80 мин. Молярный коэффициент погашения при 323 нм равен 1,2-104. Определению мешают те же ионы, что и в фосфорноволь-фраматном методе, а также Nb, Ge, As, W [810]. Предел обнаружения может быть снижен вдвое, если гетерополикислоту восстановить оловом(П) и экстрагировать продукт реакции метилизобу-тилкетоном [941]. Метод рекомендован для одновременного определения ванадия с титаном [942] или с ниобием [668]. При этом используют различие в скорости разложения соответствующих гетерополикислот.
Для определения ванадия рекомендована также двойная мо-либденованадиевая кислота [1151].
Роданидные комплексные соединения ванадия. Ванадий(Ш) и ванадий(1У) в кислых (0,1—1 N) растворах взаимодействуют с роданид-ионами, давая окрашенные соединения переменного состава. В присутствии органических растворителей (этанол, метанол, ацетон и др.) и при большом избытке лиганда (4 М) образуются координационно-насыщенные комплексные ионы [V(SCN)6]3-и [VO(SCN)4]2~, молярные коэффициенты погашения которых равны соответственно ~7-103 при 400 нм и ~130 при 760 нм. Определению ванадия мешают элементы, взаимодействующие с роданид-ионами в аналогичных условиях (Fe(III), Си, Bi, Со, Ti(III) и др.). Их предварительно отделяют в виде гидроокисей или электролизом на ртутном катоде.
Роданидный комплекс ванадия(Ш) используют для определения 0,001—0,5% ванадия в сталях [982], а роданидный комплекс ванадия(ГУ)— для определения ванадия в растворах [742]. Так как максимумы полос поглощения в спектрах роданидных комплексов ванадия не перекрываются (см. рис. 8), то их применяют для определения валентного состояния ванадия в растворах, окислах, ванадиевых стеклах и шпинелях [138, 200].
Предложен экстракционно-фотометрический метод определения ванадия(Ш), основанный на экстракции ионного ассоциата роданидного комплекса с дифенилгуанидином из слабокислых растворов с pH 1 [163].
Определение ванадия по образованию окрашенных соединений с органическими реагентами
Методы определения ванадия с органическими реагентами очень многочисленны и разнообразны.
Определение ванадия с кислородсодержащими реагентами. Реагенты, содержащие окси- и карбокси- или две окиси-группы
78
в о^шо-положении друг к другу, взаимодействуют преимущественно с ванадием(1У) в слабокислых или нейтральных средах с образованием окрашенных комплексных соединений.
Пирокатехин (о-диоксибензол) образует с ванадием при pH 4,7—7,0 окрашенное комплексное соединение с отношением компонентов 2:1. Молярный коэффициент погашения при 590 нм равен 6800 [973].
Определению не мешают Се, U, La, As, Pb, Sn, Th, Zr, Cd, Zn (20%), Sb (<10%), Ga (< 10%), а также щелочные и щелочноземельные металлы. Мешают окрашенные ионы Cu(II), Ni(II), Co(II), Cr(III) и ионы, взаимодействующие с реагентом: Fe(III), Al(III), Ti(IV), W(VI), Mo(VI), Nb(V), Bi(III). Большие количества многих элементов отделяют в виде гидроокисей [944] или хроматографически [797]. Менее чем 100-кратные количества Fe, Со, Ni, Cr, Mo, W и Си маскируют аскорбиновой кислотой, цитрат- и цианид-ионами и комплексоном III [84], 104-кратные количества алюминия — фторид-ионами [973].
К раствору, содержащему 50—250 мкг ванадия, предварительно нейтрализованному щелочью до pH 3—5, добавляют 2—5 мл 10%-ного раствора сульфита натрия, 0,5 мл 10%-ного раствора фторида натрия (для связывания алюминия), 5 мл 20%-ного раствора пирокатехина, 10 мл 20%-ного раствора ацетата аммония и разбавляют до 50 мл. Окраска развивается через 25—30 мин. и остается постоянной в течение часа. Измеряют оптическую плотность при 600 нм [973].
Измерением оптической плотности при 800 нм полностью исключают влияние Nb, W, Ti, Bi, образующих желтые комплексы с реагентом. При выполнении этих условий метод высокоселективен и позволяет определять ^>10~4% ванадия в минералах [84, 417], сталях [943], металлическом уране и плутонии и их сплавах [797], а также топливе [431].
Описан экстракционно-фотометрический метод определения ванадия (V), молибдена(У1) и хрома(У1) отдельно и в смесях на основе их комплексных соединений с т^ет-бутил-пирокатехином [684].
Тайрон (Na-соль пирокатехин-3,5-дисульфокислоты) образует различно окрашенные соединения с ванадием(Ш) при pH 8—9, с ванадием(1У) при pH 5,8—6,4 и ванадием(У) при pH ~1 [561]. Его используют для определения ванадия(Ш) [561] и ва-надия(У) [369]. Протокатеховая (3,4-диоксибензойная) кислота, протокатеховый альдегид, пирогаллол, пирогаллол-4-карбо-новая кислота, галловая кислота, гваякол, 2,3-диоксинафталин, 2,3-диоксинафталин-6-сульфокислота взаимодействуют' с ванадием (IV) в аналогичных условиях. Реакции существенно не от-личаются'друг от друга по чувствительности и избирательности и мало используются для определения ванадия [1101]. Более селективными считаются методы, основанные на экстракции ионных ассоциатов комплексных соединений ванадия(1У) и ванадия (V) с этими реагентами и катионами органических оснований.
79
Для экстракционно-фотометрического определения ванадия рекомендованы ионные ассоциаты пирокатехиновых комплексов вана-дия(1У) и ванадия(У) с органическими основаниями: анилином [И], алкалоидом пахикарпином [495], трибутиламином и коллидином [974], N-окисью пиридина [588], дифенилгуанидином [81], о-фе-нантролином [363], а также комплексов ванадия и галловой кислоты с анилином [12], трибромпирогаллола с дифенилгуанидином [82].
Все перечисленные тройные соединения образуются в слабокислых средах (pH 3—5) и экстрагируются кислородсодержащими растворителями, хлороформом и их смесями. Они имеют близкие фотометрические характеристики: максимумы полос поглощения находятся при 590—700 нм, молярные коэффициенты погашения равны (8—10)-103. Определению не мешают окрашенные ионы и алюминий, но так как комплексообразование протекает в более кислой среде, то возрастают помехи от Zr, Nb, Ti и W. Экстракционно-фотометрический метод с использованием ионного ассоциата ванадия с трибромпирогаллолом и дифенилгуанидином наиболее селективный и чувствительный. Для связывания Nb, Ti, Fe, Мо, следов Zr и W применяют винную, аскорбиновую и тиогликолевую кислоты, а также фторид аммония [82].
о-Оксикарбонильные производные трифенилметана при pH 4—5 взаимодействуют преимущественно с ванадием(1У). Условия образования и основные характеристики некоторых комплексных соединений приведены в табл. 15 [139, 241]. Наибольшей контрастностью и чувствительностью обладают реакции ванадия(ТУ) и ванадия(У) с пирокатехиновым фиолетовым [139] и хромазуролом S [1049]. Общий недостаток всех рассматриваемых реагентов — малая избирательность. Они образуют окрашенные соединения с очень многими элементами. В большинстве случаев при анализе сложных объектов необходимо предварительное отделение ванадия.
Предложены экстракционно-фотометрические методы определения, основанные на образовании ионных ассоциатов комплексов ванадия с оксипроизводными трифенилметана и катионами органических оснований: тетрадецилдиметилбензиламмония [550], дифенил гуанидиния [127] и 1,10-фенантролина [112]. При этом наблюдается повышение чувствительности определения, но избирательность возрастает мало. Например, при определении ванадия с хромазуролом S в присутствии тетрадецилдиметилбензиламмония чувствительность определения возрастает в пять раз (е = = 6,8-104), но предварительное отделение ванадия необходимо [550].
Реакции ванадия с оксифлавонами высокочувствительны, но, малоизбирательны (табл. 16). Определению ванадия мешают ионы, взаимодействующие с реагентами в аналогичных условиях: Fe(III), А1(Ш), Ве(П), Ce(III), Ga(lH), In(III), Ge(III), W(VI), Ti(IV), Th(IV), Sc(III), Zr(IV), а также окрашенные ионы и анионы тартраты, цитраты, оксалаты, молибдаты. Избирательность реакций 80
Таблица 15
Определение ванадия(1У) и ванадия(Ш) с производными трифенилметана [139]
Реагент рн Состав комплекса V : R ^тах’ н<м е-10-» ^нест Интервал определяемой концентрации^ V, мкг/мл
комплекса реагента
Определение ванадия (IV)
Алюминон 4 1 : 1 530 530 6,0 1,44-10~5 0,1—200
Алюмокрезон 4 1 : 1 530 520 6,0 4,54.10-в 0,1—200
Пирогаллоловый красный 4,4 1 : 2 540 470 4,5 1,С0-10~9 0,2-8
Бромпирогаллоловый красный 4 1 : 1 560 530 5,5 2,2-10-7 0,1-15
Пирокатехиновый фиолетовый 4,8 2 ; 1 610 450 21,5 6,70-Ю-11 0,2-11,4
Эриохромцианин R 5 1 : 1 540 4?0 8,5 2,00.10-»
Хромазуро л S 4,0 1 : 1 580 440 13,0 1,50-10-5
Определение в а надия (III)
Бромпирогаллоловый красный 2-3 1 : 1 530 450 3,5 2,30-10-* 0,2-6
Пирокатехиновый фиолетовый 1,7 1 : 1 540 450 9,4 2,93.1g--13 0,2-11,4
Эриохромцианин R 4,5 1 : 1 540 440 1,1 9,10.10-е 0,2—2,5-
Хромазурол S 5,3 1 : 1 535 440 5,30-10-5 0,2-3,4
может быть повышена экстракцией хелатных соединений хлороформом и изопентанолом.
При pH 4,5—5 ванадий образует с морином и антипирином разнолигандное соединение, экстрагируемое дихлорэтаном. При 420 нм молярный коэффициент погашения равен 3,74-104. Определению мешают Al, Zn, U(VI), Fe(II) и Fe(III). Не мешают окрашенные ионы, а также Мп и Be. Метод рекомендован для анализа сталей [589].
Более избирательны экстракционно-фотометрические методы определения ванадия(У) с 2-метил-3-окси-4-пироном [804] и койевой кислотой (2-метокси-5-окси-4-пироном), так как комплексные соединения образуются в более кислых средах (pH 0—2) [1053]. Мешающее действие Fe(III), U(VI), Ti(IV) устраняют введением фосфорной кислоты и фторида аммония, a W(VI) и Mo(VI) — щавелевой кислоты или избытка реагента.
Для определения ванадия(1У) рекомендованы реакции с фенил-флуороном [1145] и З'-пиридилфлуороном [634], а для вана-дия(У) — с ализарином S (1,2-диоксиантрахинон-З-сульфокис-
Таблица 16
Определение ванадия(У) с оксифлавонами
Реагент Условия определения Состав комплекса V : R ^тах, нм е • 10“« Интервал определяемой концентрации V, мкг/мл Литература
рн Органический растворитель
3,5,7-Триоксиф ла-вон 3-4 Хлороформ 1 : 2 560 4,9 <5 [255]
3,5,7,4'-Те.траок-сифлавон * 3-7 Этанол 1 : 1 425 13,8 <2,3 [759]
Морин (3,5,7,2',4'-пентаоксифлавон) 3-4,5 Изопентанол 1 : 2 530 — 5-20 [256]
Кверцетинсульфокислота (3,5,7, 3',4'-лентаокси-флавон-6-сульфо-кислота) К верцетинрутино-зпд 4,4-4,7 3,5—6,0 1 : 1 1 : 2 430 435 20,3 0,2—1,0 0,5-14,3 [219] [715]
* Реагент применяют для определения ванадия(ТУ). Реакция с ванадием(У) менее чувствительна.
лота) [1048, 1089], трополоном [1030], производными окси-4-пи-ридона [1124], салициловым альдегидом [871], основанием Шиффа, полученным сочетанием 1-амино-8-нафтол-3,6-дисульфокислоты с салициловым альдегидом [990], госсиполом (2,2'-ди(1,6,7-триокси-3-метил-5-изопропил)-8-альдегидонафтол) [536] и его производными [497], а-нафти л амином [629], производными нафтола [622] и таннина [839].
Наиболее избирательным и чувствительным реагентом является 1-фенил-2-метил-3-окси-4-пиридон. Экстракционно-фотометрическому определению ванадия(У) в 4,0 М НС1 мешает только Та, допустим 10-кратный избыток U(VI), Ga(III), Nb(V), Zr(IV) и Ti(IV), а также 100-кратный избыток Fe(III) [1148а].
Производные гидроксил амин а с одним или двумя органическими радикалами взаимодействуют с ванадием(У). Присоединение металла по окси- и карбокси-группам приводит к образованию прочных хелатных соединений, в большинстве случаев нерастворимых в воде, но хорошо растворимых в органических растворителях.
Гидроксамовые кислоты (производные гидроксиламина с одним органическим радикалом) в слабокислых растворах pH 2—5 образуют с ванадием(У) комплексные соединения, свойства которых приведены в табл. 17 [137].
Бенз-, салицил- и никотингидроксамовые кислоты образуют соединения, растворимые в водно-спиртовых растворах. Все осталь-82
/Таблица 17
Определение ванадия(V) с гидроксамовыми кислотами [137]
Реагент ipH Состав комплекса V : R ^тах’ нм £ • 10—3 Предел обнаружения V, мкг/см2 Интервал определяемой концентрации V, мкг/мл
Бензгидроксгмовая 1-5,5 1 : 2 450 3,5 0,012 0,2—10
Салицилгидроксамо-вая 3-3,5 1 : 1 470—480 0,017 0,05—5
Никотингидроксамо-вая 2-4,5 1 : 3 470 4,0 0,014 0,5-10
Изоникотипгидрок-самовая Хинальдингидро кса-ыовая 2,5-3,9 3-4,8 1 : 3 440 450 0,012 0,013 1-8
2-Нафто гидроксамовая 2-4 1 : 2 450 0,009 1—10
2- О кси-3-нафтогид ро -ксамовая 2,5-4 1 : 1 540 3,75 — 0,02-8
2-Тиофенгид роксамо -вая 2-Бром-З-тиофенгид- роксамовая 3,5-7 2 1 : 3 1 : 2 550 465 2,1 0,01 0,016 0,5-6
п-Метоксибензотио-гидроксамовая * 5 1 : 3 450 10,1 0,0025 0,5-3
Капрогидроксамовая 2 1 : 2 460 3,2 0,017 0,4-30
* Определение ванадия(1У).
ные дают осадки, растворимые в кислородсодержащих растворителях (высокомолекулярных спиртах, кетонах, альдегидах и эфирах).
Для фотометрического определения используют водно-спиртовые растворы и экстракты хелатов. Экстрагируемость комплексных соединений зависит от кислотности среды, природы растворителя, а также характера органического радикала. По степени увеличения экстрагируемости хелатов ванадия изобутиловым эфиром может быть составлен следующий ряд: изоникотин- никотин-^> пиколин- хинальдингидроксамовая кислоты [723]. Реагенты малоизбирательны. При различных условиях с ними образуют комплексные соединения ионы многих элементов.
Чаще всего для определения ванадия применяют б е н з г и-дроксамовую кислоту [311, 327, 799, 868, 1074, 1160], которая в слабокислой среде образует с ванадием(У) окрашенный осадок, растворимый в водно-спиртовом растворе. Окраска раствора устойчива в течение дня [311]. При pH 2—4 окрашенное соединение экстрагируют 2-октанолом [799], гексанолом [311, 868], 3-гептанолом [327], метилизобутилкетоном [1074]. Определению
83-
мешают Fe(III), Ti(IV), Mo(VI), Nb(V), Hg(II), Co(II) [799]. Железо удаляют электролизом [1160] или предварительно экстрагируют совместно с титаном в виде бензгидроксамата при pH 8,5 бутанолом [799]. Железо совместно с ураном отделяют перед измерением оптической плотности реэкстракцией 20%-ным раствором (NH4)2HPO4 при pH — 1,5 из гексанольного экстракта [868].
Большие количества молибдена, ниобия, а также титана и железа можно предварительно экстрагировать в виде хлоридных комплексов из 6 М НС1. Небольшие количества железа и титана маскируют фторид-, пирофосфат- и малонат-ионами [327]. Непосредственно определяют 0,6 мкг/мл, а с экстракцией — 0,05 мкг/мл ванадия [799]. Этим методом определяют ванадий в водных растворах [327], биологических материалах [799], тетрафториде и окислах урана [868], сталях и нефти [1160], минералах, рудах, горных породах [1074].
К 20 мл (0,2 мг V) раствора добавляют 2 мл хлорацетатного буферного раствора (pH 2,5). С помощью pH-метра устанавливают pH 2. Вводят 1 мл 1 %-ного раствора бензгидроксамовой кислоты, 10 мл метилизобутилкетона и экстрагируют окрашенное соединение. Экстракты высушивают безводным сульфатом натрия и измеряют оптическую плотность при 450 нм относительно раствора холостого опыта [1074].
Большинство из синтезированных в последнее время гидроксамовых кислот не имеют особых преимуществ перед бензгидроксамовой, за исключением капро- и метоксибензтиогидроксамовой, которые образуют соединения с ванадием в более кислых средах (0,3—0,8 N H2SO4 и 6 М НС1 соответственно) [419, 638, 648, 723, 1093, 1094]. Это существенно снижает число мешающих элементов При анализе почв, нефтей, соединений урана, сталей мешают практически только Fe(III), Mo(VI), Ti(IV) и Nb(V), которые должны быть предварительно удалены [1093—1095].
Производные гидроксиламина с двумя органическими радикалами (двузамещенные гидроксамовые кислоты) — высоко избирательные реагенты на ванадий(У). Наиболее изучен и широко используется Н-бензоил-Н-фенилгидроксиламин(Н-бензоил -N-фе-нилгидроксамовая кислота) [19, 311, 348].
N - б е в з о и л - N-ф е в и л гидр о к с и л а м и в (БФГА) взаимодействует с ванадием(У) в кислой среде (9 N — pH 6) с образованием окрашенного хелатного соединения. Молярный коэффициент погашения в водно-этанольном растворе с pH 1,9—2,8 при 480 нм равен 2670. Окраска растворов при pH 2,6 устойчива в течение 5 час. БФГА взаимодействует с большинством металлов, однако окрашенные хелаты образуют немногие элементы и при различной кислотности растворов: Ti(IV) (< 9,6 N), Nb(V) (3—62V), Ce(IV) (3 V), U (VI) (> 3,52V), Mo(VI) (IN - pH 6); Co(II) (pH - 10), Ni(II) (pH - 5), Fe(II) (pH - 5), Fe(III) (0,5 N), Cu(II) (pH 3—11) [478]. Определению ванадия мешают также Mn(II), Th(IV), W(VI), Cr(III) [19].
Образующийся хелат ванадия можно экстрагировать хлороформом при pH 5,1—6,0. Молярный коэффициент погашения органи-84
Рис. 9. Спектры поглощения раствора комплекса вана-дия(У) с 1\-бензоил-Н-фенил-гидроксиламином в хлороформе (СФ-4а, раствор сравнения — хлороформ) [348]
1 — pH 5; 2-6 м на
ческой фазы равен 3700, максимум полосы поглощения смещается до 445 нм. При исследовании пригодности различных растворителей для экстракции комплекса ванадия с БФГА был получен следующий ряд: бутанол > пентанол хлороформ амилацетат бутилацетатбензол гексанол толуол метилизобу-тилкетон четыреххлористый углерод [513]. Для работы рекомендован хлороформ [19].
Показано [311, 1042], что большая избирательность достигается, если проводить экстракцию хлороформом из очень кислых растворов (18—22 N по H2SO4 или 3—6 N по НС1) и при 10-кратном избытке реагента. При этом экстрагируется пурпурно-красное соединение с широкой полосой поглощения (490—630 нм) и более высоким молярным коэффициентом погашения (е — 4500 -4- 5100) (рис. 9). Найдено, что в этом соединении отношение ванадия к БФГА равно 1 : 2 [19, 311, 599]. Определению ванадия мешают W(VI), Nb(V), Hf [409]. Ионы Zr(IV), Mo(VI), Ti(IV) должны быть удалены, если содержание их в пробе больше, чем ванадия, в 200, 400 и 10 раз соответственно [488]. Хром восстанавливают до Сг(Ш). Концентрация азотной кислоты не должна быть выше 1 М. Присутствие окислителей и восстановителей нежелательно.
Аликвотную часть раствора (~ 25 мл^, содержащую 20— 200 мкг V, помещают в делительную воронку, добавляют по каплям 0,1 N КМпО4 до слаборозового окрашивания, НС1 (уд. в. 1,19) до 5—9 М концентрации. Добавляют 0,5%-ный раствор М-бензоил-М-фенилгидроксиламина в хлороформе (по 2 мл на каждый мг V), затем 10 мл хлороформа и перемешивают 2 мин. Разделяют фазы и водный раствор промывают 10 мл хлороформа, собирают экстракты, фильтруют через сухой фильтр, разбавляют до 50 мл в мерной колбе хлороформом и измеряют оптическую плотность при 530 нм относительно хлороформа [348].
Если экстракцию комплексного соединения проводить из 0,5— 5 М H2S04 в присутствии 0,05—6,5 М HF или 0,05—2 М KF, то определению не мешает 500 мг Ti(IV), Ta(V), Nb(V), Zr(IV) и 100 мг Mo(VI) и W(VI) [718]. Возможно одновременное определение ванадия и титана [1148]. Изучены условия селективной экстракции микроколичеств элементов Ti, Zr, Hf, Nb, Та и V с последующим фотометрическим определением каждого с БФГА [878]. Показано [128], что ванадий(У) можно определять в присутствии ванадия(1У).
Х-Бензоил-Н-фенилгидроксиламин используют для определения ванадия в железе и сталях [348, 513], горных породах и рудах
85
[348, 796, 972], солевых рассолах [488], морской воде [382], цементе [599], хлориде титана [348], урановых соединениях [24, 878, 1148], алюминии [24], ниобии и тантале [718], нефтяных продуктах [513, 798].
Определение ванадия с БФГА предпочтительнее, чем с перекисью водорода, пирокатехином и в виде фосфорновольфрамована-диевой кислоты [783].
Хлор- [362, 897, 899], сульфо- [649], нитро- [761] и азо-производные [683] Х-бензоил-Х-фенилгидроксиламина не имеют особых преимуществ перед БФГА.
В последние годы синтезировано большое число новых органических производных гидроксиламина. Проведено сравнительное изучение цветных реакций ванадия(У) с реагентами, содержащими ароматические или гетероциклические радикалы (бензоил-, метоксифенил-, фенилацетил-, о-, м~, n-толил-, 2-фуроил-, 2-те-ноил-, циннамоил- и др.) в различных комбинациях [777, 897, 1129], а также с ненасыщенными гидроксамовыми кислотами [654].
В результате для определения ванадия(У)рекомендованы N-2-теноил-Х-тг-толил- и Х-2-теноил-Х-фенил- [1128], Х-.ч-толил-Х-о-метоксибензоил- [624, 626, 777], Х-толил-Х-циннамоил- [1129], Х-га-толил-Х-2-фуроил- [625], Х-2-фуроил-Х-фенил- [399, 401], Х-о-метоксифенил-Х-2-теноил- [618], Х-бензоил-Х-о-толил- [898], Х-циннамоил-Х-фенилгидроксиламины [143, 174]. Эти реагенты по чувствительности и избирательности превосходят БФГА. Их сравнительная характеристика приведена в табл. 18. Все реагенты в 2,5—8 М НС1 образуют с ванадием хелатные соединения в отношении 2 : 1 при достаточном избытке реагента. Соединения экстрагируются органическими растворителями, преимущественно хлороформом. Молярные коэффициенты погашения при 510—555 нм равны (3-?7)-103. Они отличаются в основном по избирательности. Определению ванадия с большинством реагентов мешают титан(1У), молибден(У1), ниобий(У), цирконий(1У) и гафний(1У).
Одним из наиболее избирательных реагентов является Х-л-толил-Х-о-метоксибензоилгидроксамовая кислота. Определению ванадия не мешают щелочные и щелочноземельные металлы, Ti(IV), Zr(IV), Cr(III), Mn(II), Fe(III), Co, Xi, Cu(ll), Zn, Cd, Hg(II), Pb, Al, As(III), редкоземельные элементы и анионы СГ, F~, РО^”, С2О1~, цитраты и тартраты [624, 626].
Аликвотную часть раствора, содержащую до 5 мкг/мл ванадия, помещают в делительную воронку. Устанавливают кислотность раствора 6—8 М НС1 в объеме 10 мл, добавляют 4 мл 0,5%-ного раствора реагента в хлороформе и 6 мл хлороформа, экстрагируют и переносят органическую фазу в мерную колбу на 25 мл.[Водный раствор дважды промывают 5 мл растворителя, объединяют экстракты, доливают до метки хлороформом и измеряют оптическую плотность относительно хлороформа [624].
Аликвотную часть раствора, содержащую не более 0,1 мг V (V), 0,1 мг Fe(III) и 1 мг U(VI), помещают в делительную воронку. Устанавливают кислотность раствора 4 М НС1 в объеме 15—20 мл, добавляют 7—10 мл
86
Таблица 18
F» Определение ванадия(У) с производными гидроксиламина
реагент сНС1, м Д'тах, нм а. 10 ’ Кратные количества ионов, метающих определению Ванадия Литература
N-EeH3onn-N-фенилгидрок-силамин 5-9 530 4,5-5,1 Zr(IV) 200; Mo(VI) 400; Ti(IV), Cr(VI) 10; Fe(IH); Юз; Hf [19, 143]
N-2-®ypona-N-фенилгидрок- -силамин 3-7 530 5,6 Ti(IV), Nb(V), Mo(VI), W(VI) >100—1000 [399—401]
N-Циннамоил-N-фенилгидро-ксиламин 2—10 540 6,3 Ti(IV) 10; Mo(VI) 20; Zr(IV)200; NO2~, Cr(VI) [143, 174]
N-2-TeHonn-N-феиилгидрок-•силамин 5 530 5,7 Mo(VI), Ti(IV), Zr(IV) [1128]
N-о-Метокси-фенил-№-2-те-ноилгидрок-силамин 3-6 545 7,2 30—100 мг Ti(IV), Zr(IV), Fe(III), Mo(VI), U(VI), PO43-, F- [618]
N-Eenaoii.T-N- о-тОлилгидро-ксиламин 4—8 510 Ti(IV), который маскируют NaF [898]
М-л-Толил-М-2-фуроилгид-роксиламин 3-4 540 3,0 Ti(IV) и Zr(IV) 300 [625]
Т4-,и-Толил-М-о-метоксибен-зоилгидроксил-амин 6-8 550 6,5 [624, 626]
0,1%-ного раствора ЦФГА в хлороформе и экстрагируют комплекс V(V). Водный раствор нейтрализуют аммиаком до pH 1, добавляют 2 мл 0,5% -ного раствора ЦФГА в этаноле и экстрагируют комплекс Fe(III) 5 мл пентанола. Оставшийся водный раствор доводят до pH 6—7 и аналогично извлекают комплекс U(VI). Экстракты разбавляют до 25 мл соответствующими растворителями и измеряют оптическую плотность при 536, 453 и 413 нм соответственно [174].
Проведено сравнительное изучение свойств И-бензоил-М-фе-нилгидроксиламина и N-2-фуроил-К’-фенилгидроксиламина [401], а также И-циннамоил-Н-фенилгидроксиламина [143] и показано, что^их применение повышает селективность определения ванадия в легированных сталях [143, 400] и рудах [400]. Возможно последовательное определение ванадия, железа и урана [174].
Для определения ванадия рекомендованы формальдоксим [311], салицилальдоксим [520], о-оксиацетофеноксим [989а]. Реакции малоизбирательны. Необходимо предварительное отделение всех элементов, взаимодействующих с реагентами Ni, Со, Cu(II), Mn(II), а также Fe(III) [311], W [520], Pd(II), Ti(IV) [989а].
87
Определение ванадия с азотсодержащими органическими реагентами. Самое большое число цветных реакций, применяемых для определения ванадия, дают реагенты, содержащие азот. При взаимодействии с ванадием они образуют прочные хелатные соединения.
Одним из первых реагентов, предложенных для определения ванадия, был 8-оксихинолин. При pH 2,0—5,5 с ваиади-ем(У) он образует окрашенный осадок, растворимый в органических растворителях. Как видно из рис. 10, хлороформный раствор данного соединения имеет два максимума поглощения при 380 и 550 нм. Молярные коэффициенты погашения соответственно равны 5,4-103 и 3,0-103 [311, 457, 478]. Определение ванадия более избирательно при 410—420 нм [768]. Оптическая плотность экстрактов возрастает при увеличении концентрации оксихинолина в растворе [1126], а также в ряду растворителей: ксилол толуол бензол дихлорэтан •< и-бутанол хлороформ изобутанол. Чаще используют хлороформ, так как устойчивость экстракта выше. Для повышения чувствительности определения к экстракту добавляют бутанол [481].
С 8-оксихинолином реагируют многие металлы (Al, Со, Zn, Ni, Fe, Mo, W, U, Cu, Ti, Bi). Их предварительно отделяют чаще всего, экстракционными методами. Все элементы, кроме Mo, Ti, W, Zr, экстрагируют в виде их оксихинолинатов [478, 637]. Железо(Ш) и молибден(У1) экстрагируют в виде хлоридных комплексов диэтиловым эфиром из 6 N НС1 [482], уран(У1) — раствором ТБФ в хлороформе из 5 N HNO3 [637].
Если экстракцию оксихинолината ванадия проводить смесью бутанола с бензолом из 0,05 М сернокислого раствора, содержащего фосфаты, то определению мешают лишь Си, Мо и W [115].
Описаны довольно селективные методы определения ванадия, основанные на различии pH образования и экстракции оксихинолинатов ванадия и примесей [311, 478].
Анализируемый раствор (~ 250 мкг ванадия) с pH 2,8 ± 0,2 помещают в делительную воронку и экстрагируют ванадий вместе с примесями двумя порциями 0,5%-ного раствора 8-оксихинолина в хлороформе в течение 2 мин. Экстракт промывают водой, подкисляют до pH ~ 3 и реэкстрагируют ванадий в течение 5 мин. двумя порциями буферного раствора с pH 9,4 (к 800 мл воды добавляют 40 мл раствора аммиака (уд. в. 0,92) и 20 мл HN03 (уд. в. 1,4), проверяют pH на потенциометре и разбавляют до 1 л). Затем раствором 4 7V НС1 устанавливают pH 2,8 + 0,2 и снова экстрагируют ванадий двумя порциями 0,1 %-ного раствора 8-оксихинолина в течение 2 мин. Экстракты объединяют, разбавляют хлороформом до 50 мл и измеряют оптическую плотность при 380 нм относительно раствора холостого опыта [311].
г
Описаны методы определения ванадия в природных водах [345, 457], рудах [580], металлическом уране и его соединениях [637], бихромате калия [481] и других материалах. Дана методика одновременного определения ванадия и вольфрама [482].
Для повышения избирательности и чувствительности определения ванадия рекомендуют использовать тройные соединения на 88
Рис. 10. Спектр поглощения раствора комплекса ванадия(У) с 8-оксихино-лином в хлороформе (Z = 20 мм) [348]
Рис. 11. Спектры поглощения растворов ксиленолового оранжевого (7) и его комплекса с ванадием(ТУ) (2, 3) (pH 3, Z = 10 мм, СФ-4а) [32а]
Растворы сравнения: 1,3 — вода; г — раствор реагента
основе 8-оксихинолина и азида натрия [1019] или о-дифенола [1081].
Исследовано взаимодействие ванадия с 5,7-дибром- и 5,7-дииод-производными 8-оксихинолина [357, 792]. Образуемые комплексные соединения экстрагируют высокомолекулярными спиртами. Молярные коэффициенты погашения равны (6-;- 7) • 103. Основным мешающим элементом является железо(Ш), малые количества которого могут быть замаскированы ионами РО’” и F-.
Ванадий(Ш), ванадий(1У) и ванадий(У) взаимодействуют с ферроном (7-иод-8-оксихинолин-5-сульфокислота) с образованием хелатных соединений, пригодных для фотометрического определения [870, 902а, 1164].
Экстракционно-фотометрическое определение ванадия(У) проводят при pH 2,5—3,5 по образованию окрашенного соединения с 6-окси-1,7-фенантролином в присутствии 1,2-пропандиола [719].
Фталексоны — красители трифенилметанового ряда, включающие в свой состав группы—СН2К(СН2СООН)П в орто-поло-жении к карбоксигруппе. В кислой среде они взаимодействуют как с ванадием(У), так и с вацадием(1У) с образованием интенсивно окрашенных хелатных соединений различного состава (табл. 19).
Ксиленоловый оранжевый — наиболее перспективный реагент для определения ванадия. Устойчивость его соединений с ванадием сочетается с высокими значениями молярных коэффициентов погашения, достаточной контрастностью реакций и доступностью реагента.
Для фотометрического определения используют комплексные соединения, образуемые ванадием(1У) и ванадием(У). При избытке реагента и оптимальном значении pH 3—5 окраска развивается через 30 сек. и устойчива более суток [673]. Спектры поглощения реагента и его комплекса с ванадием(1У) приведены на рис. 11. Оптическую плотность растворов измеряют при 530 и
89
Таблица 19
Определение нанадия(1У) и ванадия(У) с фталексонами [125]
Реагент pH Состав ком-। плекса V : В ^тах’ ил< Е-10-з ^нест Интервал определяемой концентрации V, мкг/мл
комплекса реагента
О п ределе пие в ан а дня (IV)
Глицинкрезоло- 5 1 : 1 560 430 18,2 1,60-10-’ 0,2—1,2
вый красный 1 : 2 530 30,5 1,СО-10-м
Глицинтимоловый 5 2 : 1 590 430 9,5 3,0 .10-6
синий 1 : 1 580 13,4 1,3 -10-9 0,1-1,5
1 •. 2 540 21,5 8,0 .10~1в
Ксиленоловый 3 1 : 1 560 440 36,0 3,31.10-и
оранжевый 1 : 2 520 24,0 2,14.10-ie 0,С4—2,0
Метилтимоловый 3-4 1 : 1 590 435 16,0 5,0—20
синий
О п ределение ванадия (V)
Ксиленоловый 4 2 : 1 580 440
оранжевый 1 : 1 590 20,0
1 : 2 520 13,0 0,1—4,5
Фталексон S 3 1 : 1 540 434 21,0 2,12-10-6 0,25-1,25
Тимолфтал ексон 4 2 : 1 580 420 25,3 2,40.10-12 1-3,5
Крезолфталексон 3 1 : 1 580 6,0
М етилтимо л овый 3-4 1 : 2 580 440 19,0 4,30.10-13
СИНИЙ 5-7 1 : 1 510 9,8 6,30.10-’ 0,15-1,6
550 нм в зависимости от состава раствора сравнения. В этих условиях предел обнаружения составляет 0,0026 мкг/см2 [222, 815]. Определению мешают все элементы, взаимодействующие с реагентом в этих условиях, а также дающие окрашенные ионы в растворах (Fe(III), Al, Cu, Zn, Cd, Ga, Bi(III), Zr, Th, Ti(IV), Ni, Co, Mn, Sc(III)). Их предварительно отделяют от ванадия различными способами. Описан, например, метод ионнообмён-ного разделения [815]. Выбор маскирующих средств ограничен, так как определению ванадия мешают тартрат- и цитрат-ионы, а также перекись водорода и комплексон III [815]. В качестве маскирующих реагентов рекомендованы фторид калия [966] и комплексон IV (1,2-диами11ииклогексап-Ат,А,К',А'-тетраук-сусная кислота) [222, 673]. В их присутствии окраска развивается медленно (через 10 мин.) и необходим достаточный избыток реактива.
5—10 мл] анализируемого раствора (5—50 мкг V) нейтрализуют 2 7V NaOH до pH 3—5, добавляют 5 мл 0,01 М раствора комплексона IV, 5 мл ацетатного буферного раствора с pH 4,5, 5 мл 0,001 М раствора ксиленоло
90
вого оранжевого и разбавляют до 50 мл. Через 10 мин. измеряют оптическую плотность раствора при 550 нм относительно раствора холостого опыта, к которому добавлен реагент и комплексон III [673], или относительно раство ра реагента [222].
При использовании в качестве маскирующего реагента комплексона IV ванадий может быть определен в золе нефтей без предварительного отделения.
Реакция ванадия с ксиленоловым оранжевым может быть использована для его определения в растворах [276, 673], нефти [222], жидком топливе [815], металлическом уране и его соединениях [672].
Для определения ванадия(1У) и ванадия(У) рекомендован также метилтимоловый синий [504, 507]. Окрашенные соединения образуются при pH 3—4 и при достаточном избытке реагента. Оптическую плотность раствора измеряют при 580 нм. Мешают многие элементы, но выбор маскирующих средств шире, чем при использовании ксиленолового оранжевого. Определению не мешает 20-кратный избыток фтор-, тартрат-, пирофосфат-ионов [504, 1106]. Основную массу мешающих элементов отделяют при вскрытии образцов щелочами. Метод рекомендован для определения ванадия в титановых шлаках и концентратах [507].
Хелатные соединения ванадия(1У) с фталексонами в присутствии дифенилгуанидиния экстрагируют бутанолом [32, 90]. Обзор по экстракции ионных ассоциатов приведен в работе [125].
Разнолигандное соединение ванадия(У) с тимолфталексоном и фтор-ионами образуется при pH 2,3—3 и 125-кратном избытке ионов фтора в растворе, содержащем 40% ацетона. При 610 нм молярный коэффициент погашения равен 5,7-103. Реакция достаточно избирательна, так как определению не мешают кратные количества следующих элементов: W 0,5, Fe(III) 10, Cu(II) 10, Mo(VI) 20, Cr(VI) 50, Cr(III) и Al 100, Co 500, Mn(II) и Ni 2000. Мешают аскорбиновая и щавелевая кислоты. Метод рекомендован для анализа сталей. Ванадий определяют после экстракции железа(Ш) в виде хлоридных комплексов из 6 М НС1 [567].
ГидразидьГорганических кислот образуют с ванадием(У) хелатные соединения за счет присоединения металла по карбокси- и амино-группам [156]. Соединения ванадия с гидразидами бензойной [394], никотиновой [865], салициловой [1072], а также анисовой, антраниловой, капроновой и др. [156] кислот неустойчивы и начинают разлагаться через ~10 мин. после образования. Комплексные соединения состава V : R = 1 : 1 имеют максимумы полос поглощения в ближней УФ-области спектра. Реакции высокоизбирательны. В кислой среде, кроме ванадия, с реагентами взаимодействуют только молибден и ртуть. Для практического использования рекомендован гидразид 2-оксибензойной кислоты [156].
Изучены соединения ванадия с ацетонгидразидом [159] и бен-зоилгидразидом [234] антраниловой кислоты, а также с бензоил-фенилгидразидом [937]. Ванадий(У) реагирует с ацетонгидра-
91
Рис. 12. Влияние концентрации БГАК на оптическую плотность раствора Комплекса VORA^-S (1 • 10-4 М ванадия(У), I = 10 мм, 540 нм, Specord UV VIS) [234]
Рис. 13. Зависимость оптической плотности ацетоновых растворов комплексов ванадия(У) с гидразидами антраниловой кислоты от кислотности среды [234] Концентрация растворов, М: ванадия — 2-10~4; АГАК или БГАК — 2-Ю-3; 1 — АГАК, 2,3 — БГАК; 1,2 — НС1; 3 — H2SO4 (540 н.и, I = 10 леи Specord UV-VIS)
зидом антраниловой кислоты (АГАК) в 0,2—0,9 М HCI (H2SO4) с бензоилгидразидом антраниловой кислоты (БГАК) в 1,5— 3,5 М НС1 и 1—4 М H2SO4 при большом избытке реактивов в присутствии 60—80% ацетона. Предполагаемый состав соединений VORA2S, где R — реагент (АГАК или БГАК), А — анион (Cl~, HSO4), S — ацетон. На рис. 12 показано влияние концентрации БГАК на оптическую плотность раствора. Максимумы полос поглощения расположены при 540 нм и молярные коэффициенты погашения равны 5100 + 100. Комплекс ванадия с БГАК более устойчив к действию кислот, а также ионов NOJ (рис. 13) [234].
5 мл раствора (2—200 мкг V) переносят в мерную колбу на 25 мл, добавляют 1 мл H2SO4 (уд. в. 1,84), 13 мл 0,02 М раствора БГАК в ацетоне и разбавляют до метки нодой. Оптическую плотность раствора измеряют при 540 нм относительно раствора холостого опыта [234].
Определению ванадия с обоими реагентами мешают сильные окислители (СЮ2, JО2, СггО?”, Н2О2), а определению с АГАК — еще Fe(III) и Mo(VI). Для устранения мешающего действия железа и молибдена применяют фосфорную кислоту и комплексон III. Не мешают определению ванадия 104-кратные количества щелочных и щелочноземельных металлов, РЗЭ, Al, In, TI, Sc, U, Ni, Re, Rh, Pt, Ru, Pd и 1500-кратные — Co, Th. Разработаны селективные методы определения ^8-IO-4 % ванадия в сталях, металлах, силикатных породах [159, 234].
Окрашенное соединение ванадия с АГАК извлекается смесью ацетона с хлороформом в присутствии 50%-ного раствора трихлорацетата натрия. При этом наблюдается смещение максимума
92
Таблица 20
Определение вападия(У) с гетероциклическими оксиазосоединениями
Реагент pH Состав комплекса V : R ^тахт нм ^нест Литература
6-(2-Пиридилазо)-тимол 6-(2-Пиридилазо)-3,4-ди метилфенол 3,1-4,6 2,6-5,4 640 630 0,37 0,70 2,37.10-и [326] [433]
4-(2-Пиридилазо)- резорцин 5-6 1 : 1 545 3,7 3,4-10—15 [31, 621]
5-(2- Пиридилазо )-2-мо -ноэтиламин-4-га-крезол 2,0-5,0 1 : 1 560 3,8 1,88-10-’ [148]
2-(2-Пиридилазо)-5-ди-этил -л4-а минфе ио л 3-4 1 : 1,5 600 5,25 1,36-io-6 [149]
2-(2-Тиазолилазо)- 5-ди-этиламинфенол 3,2-5,0 1 : 1 580 4,2 1,46-10-* [553]
Н-Метиланабазин-2'-азо-5-диэтиламинфенол 5,0 1 ; 1 580* 5,2* 2,5-10-13 [496]
* Данные приведены для водных растворов, остальные — для хлороформных.
полосы поглощения (лтах = 530 нм) и в шесть раз возрастает чувствительность определения (е = (9,57-н—0,3) • 103) [160].
Для определения ванадия используют гетероциклические оксиазосоединения — азопроизводные ароматических аминов (пиридина, тиазолина, анабазина) и фенолов с оксигруппой в. орто-положении к азогруппе (табл. 20).
4-(2-П иридилазо)-резорцин (ПАР) образует с ванадием(Ш), ванадием(1У) и ванадием(У) при pH 5 — 6 соединения одинакового состава V : ПАР = 1:1. Несмотря на то что все они имеют одинаковые фотометрические характеристики (Zmax = =545 нм, е = (3,2-н 3,7)-103), изучение ЭПР- и ИК-спектров показало, что ванадий сохраняет в них свою степень окисления [31]. Для определения ванадия обычно используют комплексные соединения ванадия(У) [621, 755, 841, 1169]. При этом мешают многие элементы (Fe(III), Со, Ni, Ti(IV), Cr(III), U(VI), Nb(V), Ta(V), Al, Mn, Zn, Cu, Hg(II), РЗЭ, Bi, Ga, Sc). При использовании в качестве маскирующего реагента комплексона IV избирательность определения возрастает и допустимо присутствие следующих кратных количеств элементов: Ag 10; Al, Со, Cr, Си, Fe(III), Zn, Hg(II),PbnNi 25; Mn, Mo(Vl). W(VI) и Sn 50. Мешают определению Nb(V), U(VI), Ti(IV) [423, 1169].
К анализируемому раствору (2—40 мкг V) добавляют 4 мл 0,01 М раствора комплексона IV, 5 мл фосфатного буферного раствора pH 6,5, 2 мл 0,1 % -ного раствора ПАР (0,1 г реактива растворяют в 0,01 N NaOH), воды до 50 мл и через 5 мин. измеряют оптическую плотность при 545 нм относительно раствора холостого опыта [1169].
Метод применяют для определения ванадия в морской воде [841] и золе нефтей [1169].
93:
Рис. 14. Зависимость оптической плотности хлороформных экстрактов ионных ассоциатов ванадия(У) от природы катиона партнера [405]
Концентрация растворов, М. ванадия — 4-10-8; 4-(2-пиридилазо)-резорцина— 2-10-3; 1 — диантипирилметана — 0,12; 2 — антипирина — 0,26; 3 — пиридина — 2,5; 4 — хинина — 1,6-10-’; 5 — нитрона — 2-Ю-’, (! = 30 м, 540 нм, СФ-4а)
Рис. 15. Спектры поглощения хлороформных растворов 4-(2-пиридилазо)-резорцина и нитрона (7), их ассоциата с ванадием(У) (2) и водного раствора комплекса ванадия с 4-(2-пиридилазо)-резорцином (3) (pH 6,0, I = 30 мм, СФ-4а) [405]
Концентрация растворов, М: ванадия — 1,1-10-’; ПАР — 3-10-’; нитрона — 3-10-4
Так как комплексное соединение ванадия с ПАР имеет анионный характер, то его можно экстрагировать хлороформом из водных растворов с pH 4—6 в виде ионного ассоциата с крупными органическими катионами: тетрафенилфосфония и тетрафенилар-сония [1090], тетрадецилдиметилбензиламмония [950], а также с хинином [36], пиридином, антипирином, диантипирилметаном и нитроном [405]. Как видно иэ рис. 14, применение нитрона обеспечивает, наибольшую чувствительность определения ванадия. Максимум оптической плотности наблюдается при 540 нм (рис. 15). Определению не мешают Al, Mn, Cr, Zr, W, а также ионы ON", F". Допустимо присутствие кратных количеств следующих элементов: Nb, Та и Sn 25, Мо 50. Влияние 1000-кратных количеств железа устраняют введением тиомочевины, а любых количеств титана — фторида аммония. Метод позволяет определять 10-4% ванадия в тетрахлориде титана без отделения основы [405].
Для повышения избирательности в координационную сферу окрашенного комплекса ванадия(1У) и ванадия(У) с ПАР вводят гидроксиламин [297] или перекись водорода [40]. Тройное комплексное соединение ванадия(У) с ПАР и перекисью водорода с отношением компонентов 1:1:1 образуется при pH 0,5—3. Молярный коэффициент погашения при 540 нм равен 16 400 [404]. На рис. 16 приведены спектры поглощения растворов ПАР и его комплексного соединения с ванадием(У) в присутствии перекиси водорода. Реакция отличается высокой избирательностью. Из более шестидесяти проверенных элементов определению не ме
94
шают кратные количества следующих: Cr(III), Mn(II), Mo(VI), Ni(II), Fe(III) 5; Zr(IV), Zn, T1(I), Ga, Sc, V(IV), Hf,rAl, La, Ti(IV) 20, а также соизмеримые количества Gu(II), Bi, Go. В растворе могут находиться анионы WO^-, РО®-, РгО?-, а также цитраты, тартраты, оксалаты, тиомочевина и комплексон III. Железо
Рис. 16. Спектры поглощения растворов 4-(2-пиридилазо-ре-зорцина) (7) и его комплексного соединения с ванадием(У) в присутствии перекиси водорода (2) (СФ-10) [40]
(600-кратный избыток) и титан (105-кратный избыток) маскируют фторидом аммония. Этим методом рекомендуют определять ванадий в рудах, сталях и сплавах [40], тетрахлориде титана и его сплавах [280, 386, 387, 404].
К аликвотной части раствора (10—60 мкг V) добавляют 5—10 мл 10%-но-го раствора фторида аммония для связывания железа и титана, 20 мл 1 N H2SO4, 2 мл 1,96-10“4 М раствора ПАР, 3 мл 3%-ного раствора Н2О2, разбавляют до 100 мл раствором той же серной кислоты и нагревают на кипящей водяной бане 2 мин. После охлаждения измеряют оптическую плотность раствора при 540 нм относительно раствора холостого опыта [40].
Состав тройного комплексного соединения ванадия(1У) или ванадия(У) с ПАР и гидроксиламином зависит от pH раствора. Показано, что во всех комплексных соединениях степень окисления ванадия равна 5+ [297]. Реакция высокочувствительна и селективна как в слабощелочной, так и в сильнокислой среде. Определение ванадия в щелочной среде (pH 7—9) основано на образовании довольно устойчивого комплексного соединения с отношением компонентов 1:1:1. При 500 нм молярный коэффициент погашения равен 3,9-104. Определению мешают Мп, Мо, Zn, Al, Fe, Ni, Go, Gu. Их мешающее действие можно устранить введением маскирующих средств: фосфата, пирофосфата, цитрата, тартрата или комплексона III. В присутствии последнего ~20-кратные количества железа почти не мешают определению. Вольфрам(У1) с ПАР в этих условиях не взаимодействует [297].
К нейтральному раствору, содержащему (2 -? 8) -10“5 М ванадия, добавляют 2 мл боратного буферного раствора с pH 9,2, 0,5 мл 1 10“ 3 М раствора ПАР, 1 мл 1 М раствора солянокислого гидроксиламина, доводят объем до 10 мл и нагревают раствор в течение 5—10 мин. на кипящей водяной бане. Раствор охлаждают и измеряют оптическую плотность при 500—520 нм [297].
В 0,4 N H2SO4 комплекс ванадия(У) с ПАР и гидроксиламином не разрушается, что позволяет увеличить селективность определения ванадия без введения маскирующих средств. Определению 2 мкг ванадия не мешают кратные количества следующих элементов: Мо 3, Ti 75, Gr 15, Re 100, Th и Zr 200, Bi и Sn 6C0,
95
U, Мп, Al, La, Be более 1000. Мешают Cu, Fe, W. Метод рекомендован для определения ванадия в урановых сплавах [305].
Все другие реагенты, приведенные в табл. 20, синтезированы в последние годы. Из них наиболее перспективными следует считать азопроизводные диэтиламинфенола, отличающиеся высокими молярными коэффициентами погашения ((4,2 -4- 5,2) • 104). Соединения экстрагируются хлороформом и дихлорэтаном. Так как они образуются в кислых средах, то определению мешают в основном Fe(III), Cu(II), Ti(IV), Cr(III) [149], а также для некоторых реагентов U(VI) [553], Ni, Со, Bi, In, Ga [496]. Мешающее действие других элементов значительно меньше, и допустимые их количества индивидуальны для каждого реагента. В качестве маскирующих средств рекомендованы для урана(У1) фосфат-1553], для железа(Ш) и титана(1У) фторид- [148, 496], для алюминия, молибдена, вольфрама и хрома(Ш) тартрат-ионы [496]. Ни в одном случае не изучалось маскирующее действие комплексона IV. Только два реагента использованы для анализа реальных образцов: 5-(2-пиридилазо)-2-моноэтиламин-4-п-кре-зол для шлаков [148] и 2-(2'-пиридилазо)-5-диэтил-л{-аминфе-нол для сталей и шлаков [149].
Изучено тройное комплексное соединение ванадия(У) с 2-(3,5-дибром-4-метилпиридилазо)-5-диэтиламинфенолом и перекисью водорода. Соединение экстрагируется хлороформом и характеризуется высоким молярным коэффициентом погашения (e6i5=5-104). Определению ванадия не мешает 15 мг Сг(Ш), 12 мг Fe(III) и 13 мг Ti(IV) [845].
1-(2-П иридилазо )-2-н а ф т о л (ПАН) реагирует с ванадием(1У) и ванадием(У) при pH 3,0—4,5 с образованием окрашенного в синий цвет хелатного соединения (лтах =560 нм) с отношением компонентов 1:1 [936, 1150]. В водном растворе окраска комплекса неустойчива, но может быть стабилизирована добавлением персульфата аммония и ацетона [1150].
Комплексное соединение ванадия с ПАН хорошо экстрагируется хлороформом. Молярный коэффициент погашения при 615 нм равен 1,69-104 [1107]. Определению ванадия с ПАН мешают многие элементы: Cu, Fe(III), Ti(IV), Hf, Со, Ni, Cr(III), Bi, Mn, U(VI), Zr и др. [478].
о-0ксиазосоединения, синтезированные на основе хромотроповой кислоты, а-нафтола, фенола и их производных, взаимодействуют с ванадием(1У) и ванадием]V). Все эти соединения содержат оксигруппы в о/шго-положении к азогруппе.
Для определения ванадия предложены о-оксиазопроизводные, такие, как арсеназо I [295], хромотроп 2В и 2-(4-сульфофенил~ азо)-хромотроповая кислота [1144], а также о,о'-диоксиазопроиз-водные: пикраминазо Н [179], галлион [29, 179], люмогаллион [325], магнезон [334], эриохром зеленый В [505], кислотный хром темно-синий К [247, 332], сульфонитразо [44], кальцихром [216], невазолон НС (2-окси-3-сульфо-5-нитробензол-(1-азо-2')-1/-окси~ нафталин-4'-сульфокислота) [46], солохром фиолетовый [675],
.96
солохром черный RN [812] и солохром прочный серый RS [837], сульфоназо [73], сульфохлорфенолы С и М и сульфонитрофенолы М, С и К [449].
Ванадий взаимодействует с этими реагентами в слабокислой среде (pH 3—7) и образует комплексные соединения с отношением компонентов 1:1. Молярные коэффициенты погашения равны (1,6-н 4,6)-10% что обеспечивает хорошую чувствительность определения. Все предложенные реагенты в основном групповые. Так, с арсеназо I можно определять U(VI) и U(IV), Zr, Th, Sc, La, Се, РЗЭ, Al, Be, Ti, Nb, Ta, In и др., с сульфохлорфенолом С - Nb, Zr, Sc, Mo, Al, Ga, In, Cu.
В большинстве случаев при определении ванадия необходимо предварительное отделение мешающих элементов. Маскирующие средства специфичны для каждого реагента. Среди предложенных азосоединений наиболее перспективными считаются сульфонитразо [44] и сульфоназо [73].
Сульфоназо [сульфон-бис-4-оксифенил-(3-азо-2')-1'-окси-8'-аминонафталин-3',6'-дисульфокислота] в слабокислой среде (pH 4—5) образует с ванадием(У) комплексное соединение с отношением компонентов 1:1. При 620—640 нм молярный коэффициент погашения равен 1,68-104. Окраска соединения развивается через 15 мин. при нагревании на водяной бане и устойчива длительное время.
В условиях определения ванадия (pH 4—5) сульфоназо дает чувствительные реакции с Ga, Al, Си, Sc, Со, менее чувствительные — с In, Th, Bi, U. Мешают определению ванадия все перечисленные элементы, а также гидролизующиеся в условиях определения Fe, Ti, W, Zr, Sn, восстановители, фосфат- и цитрат-ионы. Не образуют окрашенных соединений и не мешают определению ванадия 500—1000-кратные количества щелочных, щелочноземельных металлов, РЗЭ, Zn, Pb, Ge, Мп, 100—200-кратные количества As, Se, Mo, Fe, Re, Ni. В качестве маскирующего реагента может быть использован фторид натрия. В этом случае 0,2—5 мкг ванадия в 5 мл раствора можно определять в присутствии равного количества железа и кратных количеств элементов: Ti, Zr, Th, Bi, Sn 50—100, As, U, W, Ga, Al, Sc —'1000. Однако большой избыток фторида натрия снижает интенсивность окраски. Перед определением железо(Ш) восстанавливают аскорбиновой кислотой, Сг и Со удаляют, ~25 мкг Си связывают тиомочевиной [73, 85, 119, 120].
К 1—5 мл анализируемого раствора, если необходимо для связывания мешающих элементов, добавляют 1,5 мл 4%-ного раствора NaF, 2 мл 5%-ного раствора Н3ВО3, 0,5 мл 10%-ного раствора аскорбиновой кислоты. Нейтрализуют раствором аммиака (по индикаторной бумаге конго), добавляют 15 мл ацетатного буферного раствора с pH 4,4, вводят 2 мл 0,02%-ного раствора сульфоназо, нагревают на водяной бане 10 мин.; раствор охлаждают, разбавляют до 25 мл и измеряют оптическую плотность при 620 нм относительно раствора холостого опыта [119].
4 В. Н. Музгин и др.
97
Рис. 17. Спектры поглощения растворов сульфонитразо (7) и его комплексного соединения с ванадием(1¥) (2) (СФ-10, раствор сравнения — вода) [44]
Разработаны методы^ определения ванадия в металлическом галлии после предварительного отделения основы экстракцией, в очищенной воде, а также в окиси кальция [73]. Предложены методы анализа стали [85], анодных порошков, выделенных из сплавов на основе ниобия и тантала [119, 120].
Сульфонитразо [2-окси-3-сульфо-5-нитробензол-(1-азо-2')-1'-оксинафталин-3',8'-дисульфокислота] реагирует с ва-надием(1У) в кислой среде (pH 2) с образованием окрашенного комплексного соединения с отношением компонентов 1:1. Молярный коэффициент погашения при 574 нм равен 2,7-104. Спектры поглощения реагента и его комплекса с ванадием даны на рис. 17. При использовании таких активных маскирующих средств, как тиомочевина, тиогликолевая, винная и фосфорная кислоты, этот метод становится достаточно избирательным и позволяет определять ванадий в стали [44] и в сплавах на титановой и железной основе [413] без предварительного его выделения. Синтез реагента описан в работе [44].
Аликвотную часть раствора (0,4—25 мкг V) переносят в мерную колбу на 50 мл, добавляют 10 мл 10%-ного раствора аскорбиновой кислоты, 2 мл 10%-ного раствора NaBF4, 1 мл 5%-ного раствора тиогликолевой кислоты (в присутствии Мо), 5 мл 4%-ного раствора пирокатсхин-3,5-дисульфокислоты (в присутствии Ti) и перемешивают, через 5 мин. приливают 2,5 мл 0,2%-ного раствора сульфонитразо. Устанавливают pH 2,3 4-0,3, разбавляют водой до метки, перемешивают и через 10 мин. измеряют оптическую плотность раствора при 582 нм. Относительная погрешность Ну 2—5% при определении 0,14—0,49% ванадия [44].
Для повышения избирательности и чувствительности определения ванадия с азокрасителями предложены тройные соединения: ванадий(1У) — арсеназо I — хинин [295], а также ванадий(У) — сульфонитрофенол К — гидроксиламин [448] у Первое соединение экстрагируется хлороформом, что позволяет увеличить чувствительность определения в ~5 раз. Применение второго комплекса позволяет определять ванадий в более кислой среде (pH 1), что также существенно увеличивает чувствительность и избирательность реакции.
Для определения ваиадия(У) используют также дифенилкарба-зон [122, 469] и производные 3-окси-1,3-дифенилтриазена [686].
Формазан [N, И'-ди(2-окси-5-сульфофенил)-С-цианформа-зан] с ванадием(1У) и ванадием (V) образует окрашенные ком-
98
илексные соединения различного состава в зависимости от кислотности среды. Практический интерес представляют комплексные соединения с отношением компонентов 1:2, образующиеся при pH 3,5—5,5. Молярный коэффициент погашения при 610 нм равен 2,5-104. Определению ванадия мешают многие элементы: Со, Ga, Mn(II), Fe(III), Ti(IV), U(IV) и U(VI) и др. Часть из них отделяют от ванадия сорбцией на катионите, другие — при щелочном сплавлении образцов. Более чем 60-кратные количества хрома отгоняют в виде хлористого хромила [201].
Определение ванадия с серусодержащими органическими реагентами. Для определения ванадия(1У) и ванадия(У) рекомендованы пиразолон-дитиокарбаминат натрия [79], 8-меркаптохинолин [91], 6-хлор-8-меркаптохинолин [43], 2-меркаптопиридин-Н-ок-сид [80], производные фентиазина [641] и ионный ассоциат дитио-гликолевого комплекса ванадия(1У) с первичными аминами [807].
Определение ванадия по продуктам окисления органических реагентов
Характерной особенностью ванадия(У) является его способность в сильнокислой среде (H2SO4 или Н3РО4) окислять органические соединения с образованием интенсивно окрашенных продуктов. Окраска прямо пропорциональна содержанию ванадия в растворе и может быть использована для его фотометрического определения. Для этой цели рекомендованы производные анилина, бензидина, дифениламина и диантипирилметана, лей-кооснования красителей (табл. 21).
Достоинством реакций данного типа является быстрота определения, хорошая чувствительность и избирательность. Определению ванадия в большинстве случаев мешают сильные окислители (Cr(VI), Ce(IV), Mn(VII), Ан(Ш) и С12), а также окрашенные ионы. Ванадий перед определением окисляют перманганатом калия в умереннокислых растворах. Избыток окислителя восстанавливают нитритом натрия и мочевиной или щавелевой кислотой.
Среди производных дифениламина наиболее известны дифенил-аминсульфонат бария и фенилантраниловая кислота [61, 429, 1129]. Устойчивая окраска продуктов окисления фенилантраниловой кислоты образуется в >9 М H2SO4 при двойном избытке реагента. Определению мешают большие количества железа, которые связывают в бесцветный фосфатный комплекс.
К 10 мл раствора, содержащего 10—100 мкг V, добавляют по каплям 0,1 N КМпСЦ до не исчезающей в течение 5 мин. розовой окраски, которую устраняют 0,05 М NaNO2 и дают 1 каплю в избыток, сразу добавляют 0,5 г мочевины. Раствор переносят в мерную колбу на 50 мл, добавляют 5 мл Н3РО4 (4 : 1) и доливают H2SO4 (1 : 1) почти до метки. Приливают 8—10 капель 0,1%-ного раствора фенилантраниловой кислоты, доводят до метки кислотой. Через 5 мин. измеряют оптическую плотность при 570 нм относительно раствора сравнения, не содержащего реагента [429].
4*
99
Таблица 21
Определение Еанадия(У) по продуктам окисления органических реагентов
Реагент Условия определения ^тах » нм Предел обнаружения V, мК2,'мл Литература
концентрация H2SO4, М время развития окраски, мин.
о-Амивфенол i.,8*1 14 340 <20 [1116]
п-Амин~К,№'-диметил-анилин pH 4-6 40 512, 552 0,01 [848] [923]
Бензидин -9 25 430 0,02 [84, 691,
3,3'-Д иаминбепзидип 1—2t2 15 470 -0,1 1028, 1097]
о-То ЛИДИИ 3 -4 20 5 555 445 2,1 -0,02 [848] [506, 923]
о-Диапнзидин 3-6 25 440 0,1 [848 , 923]
Дифепиламипсульфояат бария 5 5 570 0,3 [429, 1122]
Фенплаятраяплевая кислота 5-12 5 570 0,2 [61, 429]
2,2'-Дикарбоксидифениламин 12 5 610 0,08 [171, 348, 367]
N-Метилдифениламип -4-сульфокислота 3 5 540 0,2 [171]
Вариаминовый синий pH 2-5 5 -400 0,5 [423, 731]
3,3'-Диметплпафтиди1£ Разбавленная*2 15 550 <0,08 [640 , 848]'
Диаитипирилвипилбен-золметан 6—7*М2 60 550 0,05 [407]
Диаитипирил-3,4-диме -токсифепилметан 6—7*1>”2 15 470 0,03 [407]
Лейкооспование метиленовой синей Слабокислая среда 10 650 0,2 - [764]
ч-Анизидин 2,2 25 440 2 [1116]
« В среде НС1. *2 В среде Н3РО».
Метод рекомендован для анализа сталей [429], титановых • сплавов [61], графита и неочищенной каменной соли [1122].,
Исследована возможность применения других дифениламин-карбоновых кислот для фотометрического определения окислителей [533]. Для определения ванадия(У) рекомендован ванадокс-2,2'-дикарбоксидифениламин как наиболее избирательный и чувствительный реагент [171, 348, 367]. Спектры поглощения реагента и продукта его окисления приведены на рис. 18. Определению не мешают <'50мг Fe(III), Mn(II), Ni, Со, Cu, Mo(VI), W(V1),
3 мг Nb(V), Сг(Ш), а также Ce(IV), если его в два раза больше, чем ванадия.
100
Рис. 18. Спектры поглощения растворов 2,2'-дикарбоксиди-фениламина (7) и продукта его окисления ванадием(У) (2) (12 М H2SO4, СФ-4а) [348]
К аликвотной части раствора (3—20 мкг V), охлажденной до 5—10° С, добавляют по каплям 0,1 N КМпО4 до не исчезающей в течение 3 мин. окраски. Избыток окислителя восстанавливают 0,05 М NaNO2 и 0,3—0,5 г мочевины. Раствор переносят в колбу на 25 мл, прибавляют 12 М H2SO4 почти до метки, перемешивают, приливают 0.2 мл 0,1%-ного раствора ванадокса в 0,1 %-ном растворе Na2CO3 и доводят кислотой до метки. Раствор перемешивают и через 5 мин. измеряют оптическую плотность при 610 им. [171, 348].
Реагент рекомендован для определения ванадия в сложнолеги» рованных сталях и титаномагнетитовых рудах [171, 348, 367].
Среди производных бензидина наибольшее применение нашли 3,3'-диаминбензидин [691, 1028, 1097] и о-толидин [506, 923, 1097]. Проведено сравнительное изучение и найдены оптимальные условия определения ванадия с п-амин-Ь^ЬГ-диметиланилипом, 3,3'-диметилнафтидином, о-дианизидином и 3,3'-диаминбензиди-ном. Показано, что наименее устойчивая окраска продуктов окисления наблюдается у последнего реагента, а наиболее чувствительный — первый из перечисленных [848]. При использовании этих реагентов определению ванадия мешают не только окислители, но и другие ионы Fe(IlI), Сг(Ш), Ti(IV), которые предварительно отделяют различными методами.
Описано определение ванадия с 3,3'-диаминбензидином в продуктах алюминиевой промышленности [848], морской воде [1028], каучуке [1097], с о-толидином в топливе [923] и титане [506].
Очень высокая чувствительность и избирательность достигнуты при определении ванадия(У) с производными диантипирил-метана. Молярный коэффициент погашения продуктов окисления диантипирил-3,4-диметоксифенилметана при 470 нм равен 9,2-104 [407]. Определению не мешают большинство сопутствующих ванадию элементов: Fe(III), Ti(lV), Ni, Со, Cu(II), Cr(III), W(VI), Mo(VI), Nb(V), Ta(V) и др. Метод рекомендован для определения ванадия в сталях [407].
Окисленные формы реагентов диантипирилвинилбензолметана и диантипирил-3,4-диметоксифенилметана имеют катионный характер и в присутствии анионов SCN-, СЮ4, CdJg, J~, FeCl|~, SnClg и др. экстрагируются хлороформом и дихлорэтаном [157, 407].
101
Определение ванадия косвенными методами
Определение ванадия(1У) проводят по реакции
V(IV) + Fe(III) V(V) + Fe(II),
Которая протекает при pH 3—9. Образующееся в результате реакции железо(П) эквивалентно количеству ванадия и может быть точно определено по известным реакциям с 2,2'-дипиридилом [194, 556] или о-фенантролином [83, 606]. Определению мешают более 200-кратные количества железа. Не мешают W, Mn(VH), Cr(VI), Mo, Pb, Zn, Sn(IV), Ti(IV) и др. [556]. Метод рекомендован для анализа руд, минералов, сплавов, металлов [33, 194, 556, 606, 607].
Определение ванадия(П) проводят по реакции
ТЮ Y2~ + VY2~ + 2Н+ TiY- + VY" + Н2О,
где Y1' — ион ЭДТА. Содержание ванадия устанавливают путем измерения оптических плотностей при 450 и 530 нм. Метод рекомендован для определения ванадия в катализаторах [765].
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Значения окислительно-восстановительных потенциалов ванадия в водных растворах зависят не только от соотношения и концентраций его ионных форм, но и от pH среды. Нормальные окислительно-восстановительные потенциалы ванадия (табл. 22) рассчитаны из термодинамических данных с точностью ±0,001—0,1 в 1123].
Таблица 22
Нормальные окислительнэ-вэсетанэвительчыз потенциалы ванадия [123]
Высшая степень окисления Низшая степень окисления Нормальный электродный потенциал, в Высшая степень окисления Низшая степень окисления Нормальный электродный потенциал, в
V2+ V ТВ —1,2 VO2 + 2Н+ VO2+ + Н2О +1,00
V:i+ \Г2+ -0,25 VO* +4Н+ Vtb+2H2O -0,38
VO2+ ± 2Н+ V3+ + Н2О +0,36 vo; + 4H+ V(OH)+4 + С13,
По изменению свободной энергии различных окислительно-восстановительных реакций рассчитаны зависимости <р—pH для системы ванадий—вода при 25° С и построены соответствующие диаграммы [477а, 995]. Существенным недостатком диаграмм является то, что в них нашли отражение не все современные представления о состоянии ионов ванадия в водных растворах. В этом смысле более точна диаграмма <р—pH, изображенная на рис. 19,
102
Рис. 19. Равновесные системы между растворимыми и нерастворимыми соединениями ванадия [995]
Таблица 23
Окислительно-восстановительные потенциалы систем
Система Среда t, °c ф, в Литература
HVO3 + 3H+/VO2+ 4- 2Н2О +0,92 [695a]
VOt + 2H+/VO2+ + Н,0 1 М НС1 25 +1,0216 [491a]
4,715 М Н3РО4 25 +1,080 [491a]
0,836 М Н4Р2О7 25 +1,113 [491a]
3,08 М NaHSO4 25 +1,033 [491a]
2,37A/(NH1)2SO1 25 +0,864 [491a]
5,4 М NH4F 25 +0,805 [491a]
VOf /V(IV) 1 N NaOH -0,74 [491a]
2 N NaOH -0,85 [491a]
5 N NaOH —0,85 [491a]
VO2+ +2H+/VS+ -'г Н2О 1 M H2SO4 +0,33 [1150a]
3 M H2SO4 +0,29 [1150a]
5 M H2SO4 +0,20 [1150a]
уЗ+/у2+ H2SO4 -0,255 [821a]
103
но и она не учитывает влияния ионной силы растворов и процессов комплексообразования. Поэтому, безусловно, заслуживают внимания сведения об экспериментальных измерениях реальных потенциалов ванадийсодержащих систем в разных средах (табл. 23).
Потенциометрические методы
Метод основан на резком изменении потенциала индикаторного электрода вблизи точки эквивалентности при титровании. В методе используют реакции окисления—восстановления, комплексообразования и осаждения. Растворы, содержащие ванадий в низших степенях окисления, титруют растворами окислителей. Для определения двухвалентного ванадия рекомендованы растворы хлората, йодата, бромата и бихромата калия, сульфата четырехвалентного церия и трэхвалентного железа [730], для двух-и трехвалентного ванадия в присутствии гидроксиламина при 80° С — сульфата четырехвалентного церия [68, 71], а для трех-и четырехвалентного ванадия в концентрированной ортофосфор-ной кислоте — растворы бихромата калия [934, 1012].
Ванадий(У) титруют растворами соли железа(П) в кислой среде [93, 480, 571, 693, 847, 860, 954, 983, 1084] и в щелочном растворе глицерина [659], сульфата гидразина в присутствии катализатора OsO4 [720], тиосульфата натрия в присутствии катализатора ионов Gu2+ [1020], растворами молибдена(Ш) [921], хрома(П) [266, 1099], титана(Ш) [722] и аскорбиновой кислоты [1087].
Методом комплексонометрического титрования ванадий определяют в виде четырехвалентного, для чего ванадий(У) восстанавливают сульфитом натрия при кипячении. Затем добавляют избыток комплексона III, уротропиновый буферный раствор (pH 8) и титруют раствором двухвалентной ртути с индикаторным электродом из амальгамы серебра [836]. Метод пригоден для титрования смесей двух, трех и четырех катионов в присутствии ионов щелочноземельных или тяжелых металлов. Подобным образом определяют ванадий, хром, алюминий, медь в ильмените [838].
Осаждение ванадия в виде ортованадата серебра проводят при pH 11,5, используя серебряный индикаторный электрод [838, 1057]. K2(VO)3[Fe(GN)(;]2 осаждают из водных растворов сульфата ванадила ферроцианидом калия [1023].
Полярографические методы
Полярографическое поведение пятивалентного ванадия сильно зависит от pH среды. В кислых растворах он дает две волны, первая соответствует восстановлению ионов Hg22+, получаемых в результате окисления металлической ртути ионами пятивалентного ванадия, а вторая — восстановлению четырехвалентного ванадия до двухвалентного (срг, = —0,83 в отн. нас. к.э.) [246, 267]. На последнюю волну дополнительно накладывается волна 104
водорода, и поэтому затруднено количественное определение ванадия .
Подробно изучено поведение ванадия(У) в фосфатных, боратных и карбонатных буферных растворах при pH от 2 до 12,5. В фосфатном буферном растворе при pH 3 и ^9 наблюдаются только по две волны, между pH 3 и 5 — три волны и при pH между 5 и 9 — четыре волны. Полагают, что первая волна в растворах с pH 2—5,5 обусловлена процессом адсорбции полива-надат-ионов на ртутном электроде, так как поливанадат-иопы обладают поверхностно-активными свойствами, а при потенциалах, более положительных, чем —0,5 в (отн. нас. к.э.), поверхность ртути имеет положительный заряд. К тому же отмечено, что добавление поверхностно-активных веществ приводит к сильному уменьшению высоты этой волны. Аналогичное влияние оказывают и хлорид-ионы, которые начинают адсорбироваться при потенциале —0,2 в (отн. нас. к.э.). Вблизи электрокапиллярного максимума (около —0,5 в отн. нас. к.э.) начинается’десорбция поли-ванадат-ионов, чему соответствует вторая волна. Одновременно с десорбцией происходит восстановление ванадия(У) до вана-дия(1У), которое протекает необратимо [437].
Благодаря десорбции ванадата при потенциалах, более отрицательных, чем —0,5 в (отн. нас. к.э.), в растворах с pH 5 появляется минимум на полярограмме. Потенциал полуволны необратимого восстановления ванадия(У) до ванадия(1У) с возрастанием pH сдвигается в сторону более отрицательных значений. Это в конечном счете приводит к разделению первой и второй волн в интервале pH от 5 до 9. Адсорбционная волна сильно уменьшается с ростом pH выше 5,5 и исчезает совсем при pH 9, в то время как высота волны восстановления ванадия не зависит от pH п остается постоянной.
Необратимость процесса восстановления обусловлена еще и тем, что образующийся ванадил-ион может вступать во взаимодействие с гидроксил- или ванадат-ионами, давая мало растворимые гидроокись ванадила или ванадилванадаты. Последние, вероятно, образуют мономолекулярный адсорбционный слой на поверхности ртути.
При pH 5,5 вторая волна перерождается в третью, которая обусловлена только процессом восстановления ванадия(У) до ва-надия(ГУ) (она исчезает при пропускании SO2 через раствор). Потенциал полуволны ее равен —0,8 в (отн. нас. к.э.) и сдвигается в сторону более отрицательных значений при повышении pH. На расстоянии чуть более —0,5 в (отн. нас.к.э.) от этой волны наблюдается четвертая волна, обусловленная восстановлением ванадия(ГУ) до ванадия(П). При pH )> 7,5 наклон четвертой волны уменьшается и расстояние между третьей и четвертой волнами сокращается. Повышение pH приводит к сближению волн за счет сдвига потенциалов полуволн (третьей волны в сторону более отрицательных, а четвертой — в сторону более положительных значений). Одновременно уменьшается высота третьей
105
волны с соответствующим возрастанием высоты четвертой волны, а в растворах с pH 12,5 наблюдается только одна волна, отвечающая восстановлению ванадпя(У) до ванадия(П). Все процессы восстановления необратимы.
В присутствии многих реагентов, например комплексонов, на полярограммах ванадия(У) наблюдаются две или три одноэлектронные волны. Число волн, их высота и положение зависят от pH раствора. Поэтому в практическом анализе целесообразно предварительно ванадий(У) перевести в какую-нибудь низшую степень окисления. Полярографических волн, соответствующих восстановлению ванадия ниже двух, не обнаружено [267].
Перекись водорода, ионы NO3 и Mo(VI) оказывают каталитическое действие на процессы восстановления ванадия(У) [644, 834, 854]. Присутствие перекиси водорода при pH < 5 вызывает увеличение высоты пика восстановления ванадия(У) до вана-дия(1У) [854]. Нитрат-ионы обусловливают трехкратное увеличение высоты волны восстановления ванадия(1У) до ванадия(П) при полярографировании растворов ванадия(У) на фоне 1 М NHjCl + 1 М NH4OH [644[. В присутствии ионов Mo(Vl) облегчается восстановление ванадия(У) до ванадия(Ш) на фоне 0,1 М этилендиаминтетрауксусной кислоты при pH 4,5—6. По этой причине из трех одноэлектронных полярографических волн ва-надия(У) остается только две волны [834].
В уксуснокислой среде (2 М GH3GOONa -ф 2 М GH3GOOH -ф -ф 0,01 % желатины) волны восстановления ванадия(У) до вана-дия(1У) (ф‘/г > 0в отн. нас. к.э.) и до ванадия(Ш) (ф1/г = —1,2 в отн. нас. к.э.) плохо развиты и на последнюю накладывается ток фона [267].
Электрохимическое поведение четырех - и пятивалентного ва- надия было изучено на платиновом микроэлектроде и ртутном капающем электроде на фоне 0,5 N Nad с добавлением HG1 и NaOH [123]. Найдено, что вид вольтамперных кривых сложен и определяется прежде всего значением pH фона. Это обусловлено состоянием ионов как пяти-, так и четырехвалентного ванадия. Полярографические волны отвечают необратимому восстановлению определенных ионных форм ванадия в области pH от 3 до 10,3, осложненному различиями в их адсорбционных свойствах и вторичными процессами, в частности образованием на поверхности электродов пленок из ванадилванадатов и гидроокиси ваца-дила [123].
Такое поведение ванадия затрудняет его определение в слабо-кислых, нейтральных и слабощелочных растворах в отсутствие комплексообразователей и мешает определению других элементов в его присутствии.
В табл. 24 приведены потенциалы полуволн и составы рекомендуемых фонов для определения ванадия.
Полярографическим методом ванадий определяют в самых разнообразных объектах: сталях [26, 334, 692], рудах [227, 1114], 106 :
Таблица 24
Потенциалы полуволн и состав фона при определении ванадия
Деполяризатор Состав раствора Реакция <Р./г> о Литература
V(V) 0,05 M H2SO4, 0,CC5% желатина 5-»4 4-2 0 1 —0,98J [267]
0,1 M H2SO4, комплексон I 5 — 4 0 1
4 — 3 -0,2 [267]
3 — 2 -1,2 J
0,1 M комплексон I, pH 6,82 5 — 4 4-2 0 1 -1,2 / [267]
0,1 M комплексон I, pH 9,55 5-2 —1,22 [267]
0,1Л/ комплексон III, 0,02 М 5-2 -1,3 [349]
NagCgHgOv
0,С2 М комплексон III, 0,2 М 5 — 2 —1,37 [267]
KCN
5 мл копц. Н4Р2О7 па 50 .ил про- 5 — 4 -1,03 [6Ю]
бы, 0,С04% полиакриламид, pH
10-11,5
1 М К2С2О4, pH 4,6 5 — 4 4-2 —1,33 / [267]
1М К2С2О4, pH 7,6 5 — 4 4 — 2 -0,05 ) -1,32 f [267]
0,1 М L1OH 5 — 2 -1,7 [267]
IM NH4C1, 1 м nh4oh 5 — 4 4 — 2 —0,97 ) -1,26 / [267]
0,5 М NaOH, 0,5 М бпс-(2-окси- 5-2 -1,65 [717]
пропил)-2-оксиэтилепаМин или
бпс-(2-окспбутил)-2-окспэтилен-
амип
V(IV) 1 М NaOH 4 — 5 —0,432 [267]
1 М NaOH, 0,01% желатина 4-5 -0,35 [267]
2 М NaOH 4-5 —0,438 [267]
0,5 М NaOH 4-5 -0,426 [267]
0,25 М NaOH 4 — 5 —0,419 [267]
v4o2~ (?) 0,122 М NaOH 4-5 -0,5 )
S [267]
v2o53- (?) -0,4 J
v4o2- (?) 0,061 М NaOH 4 — 5 -0,49)
[267]
v3or (?) -0,39]
v4o2- (?) 0,03 М NaOH 4 — 5 -0,47)
> [267]
v2o52- (?) -0,Зб]
0,5 М Na2CO3, 0,5 М К2СО3, pH 4 — 5 -0,342 [267]
9,4
IM KSCN, 0,2М NaOH 4 — 5 -0,30 [267]
107
Таблица 24 (продолжение)
Деполяризатор Состав раствора Реакция Ф1/,. в Литература
V(IV) 1 M КНСОз, насыщ. СО2, pH 7,6 4 5 -0,22 [2671
НМ КС1, IM NaOH 4 —> 5 -0,39 [267]
1MKF, IM NaOH 4 _»5 -0,44 [267]
IM Na3CeH3O7, IM NaOH 4 5 —0,432 [267]
Na3PO4, насыщ. р-р, 1 М NaOH 4- -»5 —0,37 [267]
К2С2О4, насыщ. р-р, IM NaOHj 4 ->5 —0,43 [267]
0,5 М H2SO4 4 -»5 -0,52 я [267]
0,05 М H2S04 4 -2 —0,85 [267]
IM K2SO4, pH 6,1, не буферир. 4 ^2 —1,290 [267]
Р-Р 1 М К2С2О4, pH 5,71 4 ^2 —1,309 [267]
1 М К2С2О4, pH 9,4J 4 —»2 —1,500 [267]
IM KSCN, pH 1,5, 0,005% желатина 4 3 3 -2 -0,21 [ -0,54 / [714]
1 М НС1О4 4 -н>2 -0,93 [924] ;
Аскорбиновая к-та, pH 10,98 7 4 ^2 -1,56 [924]
0,5 М KSCN, 1М СН3СООН, 4 ->3 —0,2381 [267]
0,004% агар-агара 3 —»2 —0,563/
V(III) 0,5 М H2SO4 3 —>2 -0,508 [267]
0,5MH2SO4, 0,005% J-лкелати- 3 —>2 —0,55 [267]
на
1JM К2С2О4, pH 3,5—6,5 3 —>2 -1,136 *’ [267]
V(OH)2+ 1М СНзСООН, pH 4,1 3 — 2 -0,97 1 [267]
VO* -1,232/
1М СНзСООН, pH 5,4 3 — 2 —0,9781 -1,245/ [267]
1М СНзСООН, pH 6,3 3 —>2 —1,0081 —1,300/ [267]
1 М КВг 3 — 2 -0,43 1 -0,87 / [267]
V(III) IM KJ, pH 2,5 3 —>2 -0,46-1 —0,71 } —0,98J [267]
Ацетатный буферный р-р, pH 5,0 3 —♦2 -0,981 —1,24/ [267]
0,5 М NaClO4, 0,2 М СН3СООН 3 —»2 НН о © ОО [1104]
Ьмл конц. Н4Р2О7 на 50 .ил пробы, 0,004% полиакриламида, pH 5,5—9,0, 0,06 М комплексона III 3 -»2 -1,28 [6Ю]
V(II) 0,5 М H2SO4 2 -> 3 -0,5 [267]
108
Т а б л й\ц а 24 (окончание)
Деполяризатор \ Состав раствора Реакция ф1/2. в Литература
\(П) 1 М К2С2О4, pH 4,5 2 3 — 3 — 4 —1,136] -0,05 / [267]
1 М К2С2О4, pH 6,5 2 ->3 —1,091 [267]
1М КС2О4, pH 10 2 3 1 14 *1 [267]
Ацетатный буферный р-р, pH 5,4 2 — 3 -0,886 [267]
3 _5 —0,106
IM KJ 2 3 —0,49 *2 [267]
Комплексон Ш, pH 5,0—8,3 2 3 —1,270 *3 [267]
Смесь НС1О4 и NaClO4, ц = 0,2 2 -^3 Зависит от [656]
состава смеси *3
*1 Необратимая волна. *2 Волна плохо выражена. *3 Обратимая волна.
металлах [244], нефти [217, 762], растительных материалах [821] и др.
На фоне 0,1 N HG1 по катодно-анодной волне (<р4 г = — 0,5 в отн. нас. к.э.) ванадий определяют в быстрорежущей стали.
0,2 г образца, содержащего более 5% ванадия, растворяют в 10 мл НС1 (1 : 1) при нагревании, добавляют 15 мл 60%-ного раствора НС1О4 для окисления хрома (если присутствует более 1 % вольфрама, осадок отфильтровывают). К остатку несколькими порциями при нагревании прибавляют 0,5 г NaCl для удаления олова и большей части хрома. После охлаждения соли растворяют в 180 мл горячей воды и осаждают гидроокись железа прибавлением NH4OH (1 : 1). Раствор кипятят несколько минут, осадок отстаивают, фильтруют, промывают несколько раз горячей водой (до получения фильтрата, не окрашенного хроматом), растворяют в 20 мл горячей HCI (1 : 3) и пе-реосаждают железо для полного отделения от молибдена. Затем осадок на фильтре растворяют в 20 мл HCI (1 : 1), фильтр промывают 15 мл горячей воды, к полученному раствору прибавляют несколькими порциями 0,3 г алюминия для восстановления ванадия(У), осторожно нагревают, защищая от окисления воздухом. После охлаждения разбавляют водой до 50 мл и по-лярографируют [26].
Описан быстрый полярографический метод определения ванадия в сталях, основанный на измерении высоты катодной волны, образующейся при восстановлении ванадия(У) до ванадия(1У) в кислой среде. Метод не требует предварительного отделения ванадия от других элементов, за исключением вольфрама, который осаждают в виде WO3 [692].
В минеральном сырье ванадий определяют полярографически на фоне 0,2 М KGN и 0,02 М комплексона III (<pi/2 = —1,37 в отн. нас. к.э.).
Навеску 0,5—1 г обрабатывают в платиновом тигле смесью 5—10 мл 40%-ного раствора HF и 0,5 мл H2SO4 (1 : 1). Сухой остаток прокаливают, сплавляют с 6 г Na2CO3 и 0,5—1 г буры. Плав охлаждают, добавляют твердую Na2O2 (для восстановления марганца), нагревают до кипения, фильтруют, промывают теплым 0,5%-ным раствором Na2CO3, подкисляют НС1, упа-
109
ривают до 70 мл и доводят водой до 100 мл. К 25 мл полученного раствора прибавляют 5 мл 0,2 М комплексона III, 0,2 мл 0,5%-ного раствора даела-тина, разбавляют водой до 50 мл и полярографируют. Определению мешают ионы СгО^~ и Сг3+, искажая волну [1114].]
Для определения ванадия в рудах (карнотите, ванадините) используют осциллополярографию на фоне ацетатного буферного раствора с pH 6 (1 М GH3COONH4 + 1 М СН3СООН) [927]. В металлическом висмуте полярографическое определение ванадия проводят на фоне NaNO3 и NaF [244]. Содержание ванадия в нефти определяют переменно-токовой полярографией на фоне 2 М K.SCN и 0,45 М Н28О4.
К 10 мл нефти добавляют 20 мл раствора 2 М по KSCN и 0,45 М по H2SO4 и экстрагируют 10 мин. Экстракт разбавляют до 25 мл и полярографируют от 0 до 0,8 в (отн. нас. к. э.) [217].
В нефтяных продуктах ванадий и никель определяют с помощью пульсполярографии.
Пробу, содержащую 5—10 мкг никеля и ванадия, выпаривают досуха, сжигают в муфеле при 600° С, остаток обрабатывают смесью конц. HNO3 и конц. HCI (1 : 3), снова выпаривают досуха, растворяют в 10 мл 0,1 М НС1, прибавляют воду до 20 мл и полярографируют. В этих условиях ванадий и никель образуют четкие пики соответственно при —0,78 и —1,20 в (отн. ртутного анода) [762].
Предложен метод полярографического определения ванадия в растительных материалах. Ванадий предварительно отделяют от железа и других элементов электролизом на ртутном катоде, от марганца — экстракцией хлороформом купфероната ванадия,-а от титана — экстракцией хлороформом диэтилдитиокарбами-ната ванадия. Полярографирование ведут на фоне буферного раствора 9 М по NHaOH и 2 М по NHiCl.
Навеску (5 г) обрабатывают конц. HNO3 и НСЮ4, а затем конц. H2SO4, разбавляют водой до 50 мл, фильтруют и проводят электролиз на ртутном катоде при 6 в в течение 30 мин. К раствору прибавляют бромную воду для окисления ванадия, а затем кипятят для удаления избытка брома, фильтруют, прибавляют 1%-ный водный раствор купферона и экстрагируют купфе-ронаты V, Ti, Мо и W хлороформом. Выпаривают хлороформ, остаток обрабатывают HNO3 и 5 М H2SO4, переливают в делительную воронку вместе с 3 мл 10%-ного раствора CH3COONH4 (предварительно очищенного экстракцией хлороформом в присутствии 8-оксихинолина) и добавляют 2 капли 0,1%-ного спиртового раствора бромкрезолового зеленого, нейтрализуют раствором NH4OH до pH 4,5—5,0, добавляют 2%-ный водный раствор диэтил-дитиокарбамината натрия и экстрагируют карбаминаты V, Мо и W хлороформом. Органическую фазу выпаривают, остаток разлагают кислотами, упаривают до белых паров SO3, прибавляют 2 капли Н2О„ (для окисления ванадия), 2,5 мл буферного раствора (9 М NH4OH и 2 MNH4C1), 0,05 г безводного Na2SO3 и через 10 мин. полярографируют от —0,6 до —1,3 в. При очень малом содержании ванадия объем 6 М HCI уменьшают до 0,1 мл, а буферного раствора — до 0,5 мл. Количество ванадия определяют по градуировочному графику, используя стандартные растворы NH4VO3 в аналогичных условиях. Чувствительность метода 0,006 мкг ванадия [821].
110
Амперометрические методы
Для определения ванадия\применяют реакции окисления— восстановления, осаждения или комплексообразования. За процессом титрования следят по изменник тока восстановления или окисления одного из участников реакций или «иона-свидетеля». В качестве индикаторных электродов используют ртутный капельный либо платиновый микроэлектрод. Контроль за наступлением точки эквивалентности довольно прост и надежен, а методически амперометрическое титрование не отличается от обычных титри-метрических методов.
В качестве титрантов применяют растворы восстановителей: соли железа(П) [20, 360, 370, 474, 707, 708, 1126, ИЗО], аскорбиновой кислоты [110, 1086], 8-меркаптохинолина [383, 384], 2,4-дитиобиурета [491]; окислителей: соли железа(Ш) [111, 1087], иода [1088]; осадителей для ванадат-ионов: соли серебра [1058, 1059], таллия [220, 1056], меди [778], 8-меркаптохинолина [383], а для катионов ванадия в разных степенях окисления — гекса-цианоферроата калия [472], селенистой кислоты [713]и комплек-сообразователей — тайрона [123].
Раствор соли железа(П) является наиболее распространенным титрантом при определении ванадия в минералах, рудах и горных породах [20, 360], сталях, жаропрочных сплавах и шлаках [370, 487, 527], чистых солях ванадия [474] и хромите [707]. Анализируемый материал вскрывают смесью кислот [487, 527], сплавлением с пиросульфатом калия [20, 360, 370] или смесью буры и соды (2 : 3) [707]. Раствором КМпОа окисляют ванадий до вана-дия(У). Избыток КМпОг восстанавливают нитритом натрия, а мочевиной устраняют избыток нитрит-ионов. Титрование раствором соли железа(П) обычно проводят в сернокислых средах (1—6 N H2SOa). Иногда для понижения окислительно-восстановительного потенциала системы Fe(II)/Fe(III) добавляют фосфорную кислоту [360, 527, 708].
Ионы железа(П) восстанавливают не только ванадий(У) до ва-надия(РА), но и xpoM(VI) до хрома(Ш), а марганец(УП) до мар-ганца(П). Поэтому при определении ванадия часто одновременно с ним определяют содержание хрома и марганца после предварительного окисления их до высших степеней персульфатом аммония в присутствии катализатора — нитрата серебра при кипячении.
0,2—1 г минерала или горной породы сплавляют с минимальным количеством пиросульфата калия. Большие количества SiO2 отгоняют обработкой фтористоводородной кислотой при кипячении в платиновой посуде. Кислоторастворимые минералы разлагают хлористоводородной кислотой или смесью последней с азотной кислотой (3 : 1). К влажным солям прибавляют 3 мл конц. H2SO4 и нагревают до белых паров SO3.
Сплав или остаток растворяют в 30—40 мл 1А H2SO4, прибавляют 1 — 1,5 мл конц. Н3РО4, 1 мл 1%-ного раствора AgNO3 и 1—2 г (NH4)2S2O8, нагревают и кипятят до полного разложения персульфата аммония. После охлаждения разбавляют 1;V H2SO4 до 50 мл. Аликвотную часть (5—20 мл) испытуемого раствора титруют 0,01—0,03 N раствором соли железа(П) и определяют сумму содержаний ванадия, хрома и марганца. К другой порции (10—15 мл)
111
по каплям прибавляют 0,2" -ный раствор нитрита натрия до исчезновения розовой окраски. Затем быстро прибавляют 0,2—0,5 г мочевины и через одну минуту титруют раствором соли железа(П) и находят сумму вападия и хрома. К этой же аликвотной части на холоду (при температуре ниже 20° С) по каплям добавляют 0,17V КМпО4 до розовой окраски, которую обесцвечивают каплями 0,2*!,-ного раствора нитрита натрия, добавляют 0,2—0,5 г мочевины п титруют ванадий(У) раствором соли желсза(П) [360].
Продолжительность анализа 20 мин. Относительная погрешность определения сотых долей процента ванадия составляет ±15%, а десятых долей — ±8—10%.
При определении ванадия в сталях, чугунах и сплавах 163], феррованадии [383, 384] в качестве титранта используют раствор 8-меркаптохинолина (тиооксин), который в интервале от 2N Н280з до pH 2 восстанавливает ванадий(У) до ванадия(1У), а при титровании на фоне фосфатного буферного раствора (pH 5,4 и 6,8) в присутствии NH4F осаждает его без восстановления. Точка эквивалентности в обоих случаях фиксируется при мольном отношении ванадий : тиооксин, равном 1:1, а на фоне 8 N и 16 N H2SO4 — при отношении 2:1. Титрование проводят по току окисления тиооксина на платиновом электроде при +1,1 в отн. меркур-иодного электрода [383, 384] или при +0,9 в отн. нас. к.э. [63]. На фоне 2 М GH3GOONa можно титровать ванадий(1У), точка эквивалентности для которого соответствует мольному отношению ванадий(1У) : тиооксин, равному 1 : 2 [522].
При использовании аскорбиновой кислоты в качестве титранта растворы ее стабилизируют добавками муравьиной кислоты и комплексона III (0,05 М аскорбиновая кислота с добавками 4 мл 75%-ного раствора муравьиной кислоты и 0,1 г комплексона III) [110]. Титр раствора устанавливают по KJO3. При анализе стали титрование ванадия(У) проводят на фоне 0,1 N H2SO4.
2,4-Дитиобиурет — более сильный восстановитель, чем аскорбиновая кислота. Его используют в амперометрическом титровании ванадия(У) при анализе сталей, шлаков и феррованадия [491]. Для ускорения реакции и получения стабильных результатов необходимо присутствие ионов меди(П).
1 г стали растворяют при нагревании в 40 мл H2SO4 (1 : 4), к охлажденному раствору добавляют 5 мл HNO3 (уд. в. 1,4) и нагревают до белых паров SO3. Затем по каплям добавляют 2°/-ный раствор КМпО“, 10 мл З”, -ной Н2О2 для восстановления МпО~ и Сг2О,~) [и кипятят до полного разложения Н2О2. (Объем раствора после охлаждения доводят водой до 100 мл. К аликвотной части добавляют 15 мл 6 М HCI, 1 мл 10~3 М сульфата меди, воду до 30 мл и титруют 10 3 М раствором 2,4-дитиобиуретг при +0,7 в (отн. пас. к. э.) [491].
Осаждение ванадат-ионов растворами нитрата серебра [1058, 1059], нитрата таллия [220, 1056] и сульфата меди [778] требует строгого соблюдения рекомендованных условий для обеспечения стехиометрических соотношений компонентов в осадках. Эти методы пригодны для определения ванадия только в чистых солях и не представляют практического интереса при анализе сложных объектов.
112
Кулонометрические методы
В этом весьма перспективном аналитическом методе ванадий определяют титрованием окислителями или восстановителями, генерированными электролитически внутри ячейки. Точку эквивалентности устанавливают спектрофотометрическим, потенциометрическим, полярографическим или амперометрическим методом, а также визуально — по изменению окраски индикаторов. Содержание ванадия находят по закону Фарадея, по затратам количества электричества на процесс окисления или восстановления ванадия.
Микрограммовые количества ванадия определяют электроге-нерируемыми ионами Fe(II) с амперометрическим установлением точки эквивалентности [123, 221, 427, 695, 721, 754, 917, 956]. Наряду с ионами Fe(II) в качестве титрантов-восстановителей применяют ионы Mo(V) [178, 611], W(V) [612], U(V) [981], Ti(III) [703, 835, 888], Cu(I) [916] и V(III) [4], а в качестве титрантов-окислителей — Cr(Vl) [253], Fe(III) [1096], хинон [555].
Простым кулонометрическим титрованием определяют ванадий в стали, используя установку, в которой для восстановления V(V) в присутствии ионов Fe(III) применяют метод внутреннего электролиза с Pt- и Zn-электродами [957]. Точку эквивалентности контролируют потенциометрически. Методика позволяет определять не только ванадий, но и хром, марганец, серу и кислоту.
Определение ванадия методом кулонометрии при контролируемом потенциале применяют крайне редко из-за необходимости отделения его от многих мешающих элементов и длительности подготовки проб при анализе сложных объектов. Принципиальная возможность такого определения очевидна, но только для простых объектов [192, 437, 1027].
Другие электрохимические методы
Для определения ванадия иногда применяют кондуктометрический метод и метод высокочастотного титрования. Кондуктометрический метод применяют только с использованием очень простых реакций [286, 554, 717, 830]. Высокочастотное титрование ванадия проводят 1,5%-ным раствором 8-оксихинолина в водноспиртовой среде (1 : 1) [186].
КИНЕТИЧЕСКИЕ МЕТОДЫ
Ванадий оказывает сильное каталитическое действие на многие окислительно-восстановительные реакции. Скорость реакций в большинстве случаев прямо пропорциональна концентрации ванадия [614, 1118].
Каталитическое действие проявляют соединения как вана-дия(1У), так и ванадия(У), причем активность анионной формы последнего значительно ниже, чем катионной или нейтральной.
ИЗ
Каталитические реакции высокочувствительны. Они позволяют определять до 10-4—10“5 мкг! мл ван'адия.
В ряде случаев можно значительно повысить чувствительность реакций введением очень небольших количеств активаторов (оксикислот, о-дифенолов, 8-оксихинолина и др.) [614, 617, 665, 824].
В табл. 25 приведены рекомендуемые реакции для определения ванадия, отличающиеся высокой чувствительностью и селективностью. В практике химического анализа в качестве окислителей чаще используют бромат или хлорат калия. Так, бромат-ион в кислом растворе окисляет иодид-ион до свободного иода по реакции
ВгОГ+ 6J- + 6Н+ = Вг- + 3J2 4- ЗН2О.
Реакция катализируется ванадием(У). В присутствии активатора — винной или слизевой кислоты — предел обнаружения ванадия составляет менее 1 -10-5 мкг!мл, погрешность + 10%. На выполнение реакции требуется 20 мин. [523, 614, 617].
В отростки сосуда-смесителя вносят: в первый — 3 мл 0,1 М раствора КВгО3, 2-мл 0,2%-ного раствора крахмала и 5 мл буферного раствора (12V (NH4)2SO4 с pH 2,5); во второй — 18 мл 0,1 М KJ; в третий — 5 мл исследуемого раствора и 4 мл 0,1 М винной кислоты. После термостатирования в течение 15 мин. при 20° С растворы смешивают и переносят в кювету фотоколориметра. Регистрируют изменение оптической плотности при 595 нм и строят график в координатах время—оптическая плотность. Тангенсы углов наклона полученных прямых пропорциональны концентрации ванадия [614].
Помимо метода тангенсов, для определения концентрации ванадия особенно часто применяют метод фиксированного времени и цногда метод фиксированной концентрации.
Каталитическое действие ванадия на реакцию окисления броматом калия Аш-кислоты использовано для его определения в азотной и соляной кислотах и металлическом магнии [261, 262], хлоридах натрия и калия [262].
При определении ванадия в воде особой чистоты, сульфате длюминия и халькогенидных стеклах использована следующая методика анализа [264].
20,0 мл анализируемого раствора помещают в кварцевую или фторопластовую чашку емкостью ~50 мл, добавляют 0,1 мл 2,0N хлорной или серной кислоты и 1 каплю 5-10-3 М раствора метурина, выпаривают до объема-1—2 мл, охлаждают и при необходимости разбавляют водой до 2 мл. Измеряют оптическую плотность раствора при 305 нм. Предел обнаружения ванадия составляет 3-10’3 мкг!мл. При анализе сульфата алюминия и халькогенидных стекол используют метод добавок, учитывающий влияние основы на наклон градуировочного графика.
Кинетические методы используют для определения ванадия в алюминии и его соединениях [824, 879], в солях натрия и калия [388], нитрате уранила [1136], металлическом рении [398, 446], титане и его соединениях [591], кадмии [262], в рассолах производства хлора и каустической соды [101]. В последнем случае определяют до 0,003 мкг!мл ванадия измерением количества 114
Т\б лица 25
Определение ваяадйр кинетическими методами
Реагент Активатор Предел обнаружения ванадия (п-10-3), лркз/мл Мешающие элементы Литература
Иодид калия Окисление хлорат V(IV) 20 ом к а л и я Os(VIII), Re(VTI), [523, 614,
/г-Фенетидип КНС4Н4О8 V(V) 0,2 0,1 Ru(VI) Os(VI), Re(VII), Ru(VI) Fe(III), Pb(II), 1082] [523, 614]
8-Оксихино- 1 Sn(II) Fe(III), Pb(II), [523, 614]
ЛИН Лимонная 0,1 Sn(II) Cu(II), Fe(III) [665]
Бензидин кислота 8-Оксихипо- 0,1 Cr(III), Cr(VI), [614, 1136]
п-Аминофенол ЛИН То же 0,5 Fe(III) Cd(II), Cu(II), [523, 614]
о-Аминофенол » 5 Pb(II), Sn(II) Ce(IV), Cr(VI), [952]
Анилин » 0,1 Fe(III), Hg(II) Cu(II), Fe(III), [351, 446,
гг-Фепилен диамин » 1 Mo(VI), Pb(II), Bi(III) Ag(I), Pb(II), 523, 591] [523, 614]
Иодид калия Окисление Винная, яб- бромат 0,01 Sn(II) о м калия Mo(VI), W(VI) [614, 616,
п-Фенетидин лочная,аскорбиновая или слизевая кислота КНС4ЩО6 1 Cr(VI), Cu(II), 617, 663] [523, 614]
Пирокатехин 0,5 Fe(III) Cr(VI), Cu(II), [523, 614]
Сульфосали- 0,02 Fe(III)] Cr(Vl), Cu(II) [523, 614]
а-Нафтол—п-ами- циловая кислота 1 Fe(III), Hg(II), [614]
нофенол (1 : 1) 1,5-Дифепил- 8-Оксихино- 5-10-’% Pb(II), Sn(II) Fe(III), Mo(VI) [388]
З-аминопиразолоп Индигокармин лин 1 Ti(IV) [697]
Эриохром голу- 0,04 Ti(IV) [697]
бой Р Солохром фиоле- 0,1 Cu(II), Fe(III), [696]
товый RS Sn(II)
115
/
Таблица 25 (окончание)
Реагент Активатор Предел обнаружения ванадия, (n-10-з), мка/мл Мешающие элементы Литература
Краситель бордо R 5 Cu(II), Ru(III) [753a]
Метурпн (N-фе-нил-ЬГ-окси-Ы’-метилмочевипа) 3 Cr(III), Cr(VI) [263, 264]
Аш-кислота(1-амино-8-нафтол-3,6-сульфокислота) Оки с пение пе] 0,5 с у л ь ф Bi(III), Mo(Vl), Sb(III), Se(VI), W(VI) itom аммония [262, 614]
Галловая кислота Пирокатехин Ок Иодид калия Анилин л-Хлоранилин Бензидин Тиосульфат натрия и с л о и и е п £ 2,5 2 рекип 7- 10-е % 20 0,05 1 5 Ag(b, Fe(III), Ni(II), Mo(VI), U(VI) Cr(III), Cr(VI), Mo(VI), Ti(IV) ю водорода Mo(VI), W(VI) Cu(II), Fe(III) Cu(II), Fe(III) Cu(II), Fe(III), Nb(V), Ta(V), W(VI), Zr(IV) [614, 729] [41] [290] [948] [949] [14] [523]
Люминол (гидразид о-аминофта-левой кислоты) Люминол Вос Соли, Сг(Ш) Окислен становлени 20 и е к и с V(IV) 1 е с о л я Co(II), Cu(II), Fe(HI) i о p о д о м Cu(II), Mn(VII), NH^ м и титан а(Ш) [614] [398]
Краситель «Виктория голубой В» (СззНзаКзС!) V(V)1,6 V(IV) 16 Mo(VI), Os(VIII), W(VI), U(VI) [523, 614] '
водорода, выделившегося за 30 мин. контакта пробы с амальгамой натрия. Кроме ванадия, процесс разряда водорода и разложения амальгамы катализируют хром и молибден [101].
ЛЮМИНЕСЦЕНТНЫЕ МЕТОДЫ
Для ванадия известно всего несколько люминесцентных реакций. G церулеином ванадий дает желтую флуоресценцию после восстановления его цинком до V(II) и добавления спирта. Метод
116
позволяет обнаружить до 16 ккг/мл ванадия. Проведению реакции мешают вольфрам(У1), молибден(У1), олово(П), титан(ГУ) и уран(У1) [64; 484, с. 315; 596]. '
Ванадий дает с резорцином в 20 N H2SO4 или конц. H3POj красную флуоресценцию, которая исчезает через 5 мин. Предел обнаружения ванадия составляет 2,5 мкг/мл. Проведению реакции не мешают ионы вольфрамата, железа(Ш), перманганата, титана(1У), урана(У1), 100-кратные количества молибдена(У1), 25-кратные — хрома(У1) и 10-кратные — церия(ТУ) [64; 484, с. 314; 596; 1018].
Описан метод люминесцентного определения 0,1—0,8 мкг ванадия(У) в водно-спиртовом растворе’с 1,4-диамино-5-нитроантра-хиноном [1035].
К анализируемому раствору, содержащему 0,1—0,8 мкг ванадия(У), добавляют 25 мл этанола, 9 мл 2N HCI, 1 мл 10'1 М спиртового раствора 1,4-диамино-5-нитроантрахинона (дважды перекристаллизованный пз спиртового раствора) и разбавляют до 50 мл водой. Раствор нагревают в течение 2 час. при 80—85° С (уменьшение объема восполняется спиртом), охлаждают и при комнатной температуре измеряют интенсивность люминесценции при 550 нм. Источник возбуждения — ксеноновая лампа (X == 475 нм). Интенсивность люминесценции не изменяется в течение двух дней, погрешность определения ванадия не превышает 2%.
Предложен метод определения ванадия (до 0,5 нг/мл), основанный на измерении интенсивности флуоресценции, возникающей при взаимодействии ванадия с растворами ароматических карбоновых кислот (бензойной, орто-, мета- и терефталевой, 1,2,3- и 1,2,4-бензолтрикарбоновых кислот) в присутствии амальгамы цинка [852].
К 1 мл анализируемого раствора с] pH 4—5 прибавляют 1 мл 2-10-3 М раствора бензойной кислоты в 0,5 %-пом растворе аммиака с pH 4,0, затем добавляют 0,5 мл ацетатного буферногораствора с pH 5,2 и 0,5 мл амальгамы цинка. Смесь встряхивают в течение 2 мин., разбавляют 2%-ным раствором сульфата натрия в 5%-ном метаноле до 10 мл и измеряют флуоресценцию раствора по линии 410 нм при облучении УФ-светом с /. = 300 нм. Градуировочная зависимость линейна в интервале 0,5—100 нг/мл ванадия. Определению ванадия мешают титан и железо при их содержании более 20 нг’мл [852].
Показана возможность определения ванадия в интервале 2,5-1Сг4—1-101 мг!мл по измерению люминесценции, возникающей при взаимодействии ванадия(У) с силоксеном в 6 М растворе •серной кислоты. Сильные окислители, а также ионы железа(Ш), меди(П), ртути(П), титана(1У), тория(1У), серебра(Т), молибде-Ha(VI) и вольфрама(УТ) мешают определению ванадия [133].
Соединения ванадия(У) в ряде случаев тушат люминесценцию.
По тушению люминесценции люминола с перекисью водорода в присутствии катализаторов (меди, кобальта или гемина) определяют до 2-10“5 мг!мл ванадия(У) [37, с. 208—214]. В кремнии высокой чистоты ванадий определяют по тушению люминесценции комплексного соединения галлия с люмогаллионом ИРЕ А [396]. Предел обнаружения 0,2 мкг ванадия.
117
СПЕКТРАЛЬНЫЕ МЕТОДЫ
Спектральные методы анализа, основанные на регистрации спектров излучения атомов в оптической области спектра, благодаря своей универсальности и экспрессности нашли широкое применение для контроля состава разнообразных технологических материалов и природного сырья.
Потенциал ионизации ванадия сравнительно невысок и составляет 6,74 эв. В связи с этим в спектре (даже в маломощных дуговых разрядах) одновременно появляются как дуговые (V I), так и искровые (V II, V III) линии, а общее количество линий ванадия в интервале длин волн 200—1000 нм превышает несколько тысяч (-5000).
Наиболее чувствительные и характерные линии для эмиссионного спектрального определения ванадия приведены в табл. 26.
Таблица 26
Аналитические линии ванадия [ 183, 444, 785]
Длина ВОЛНЫ, НЛ* Интенсивность линий в Энергия возбуждения, эв Предел обнаружения, % Длина волны, нм Интенсивность линий в Энергия возбуждения, эв Предел обнаружения. %
дуге искре дуге искре
11 268,80 150 500R 4,65 0,03 I 318,40 500R 400 R 3,90 0,003
11309,31 100R 400R 4,40 0,003 I 318,54 500R 400R 3,96 0,003
II 310,23 70 300 R 4,36 0,001 I 411,18 100R 100R 3,31 0,01
II311,07 70 300 R 4,33 0,001 I 437,92 200R 200R 3,13 0,001
11311,84 70 200R 4,31 0,003 1 438,47 125R 125 R 3,11 0,01
11312,53 80 200 R 4,29 0,01 I 438,99 80 R 60R 3,10 0,01
1318,34 200R 100R 3,91 0,001 I 440,85 30R 20 R 3,08 0,01
Примечание. R — данные линии испытывают сильное самообращение.
Для обнаружения и определения ванадия чаще всего используют линии V II 311,07; V I 318,34; V I 318,40 и V I 318,54 нм. При спектральном анализе различных по составу проб и при использовании спектральных приборов средней дисперсии или широких щелей квантометров необходимо учитывать возможность наложения линий некоторых элементов, присутствующих в пробе, на аналитические линии ванадия (табл. 27).
При определении ванадия по линии V I 437,92 нм необходимо учитывать мешающее влияние меди и циркония при их содержании в пробе около 3%, а также кобальта, никеля и тантала, если их концентрация превышает 10%.
Описания используемых в промышленности спектральных методов определения ванадия в монолитных образцах (стали, чугуны, сплавы и т. д.) содержатся в монографиях [56, 77, 180, 393, 454, 493, 514, 544, 5761. В качестве источника возбуждения при
118
меняют в основном высоковольтный искровой разряд или дугу переменного тока. Анализируемая проба включается одним из электродов, вторым (подставным) электродом обычно являются стержни из спектрально чистого углерода, меди, железа, алюминия и т. Д. либо из анализируемого материала. Выбор и стандартизация формы и массы электродов, качество подготовки рабочей поверхности анализируемой пробы и стандартных образцов влияют на точность анализа.
При использовании высоковольтной конденсированной^ искры генератор ИГ-2, ИГ-3 или ИВС-23 включают по сложной схеме
Таблица 27
Элементы, мешающие определению ванадия спектральными методами [183, 239, 444, 785]
V II 311,071 ни V I 318,398 нм V I 318,540 ни
элемент длина волны, нм элемент длина волны, "НМ элемент длина волглы, НМ
Fe 311,028 Fe 318,357 Fe 318,490
311,084 Ti 318,396 318,531
Ti 311,062 318,402 Zr 318,507
311,067 W 318,404 Ag 318,509
Mo 311,064 Ag 318,415 Mo 318,510
311,085 Nb 318,422 W 318,520
Mn 311,068 Ni 318,437 Os 318,533
311,085 Mo 318,440 Rh 318,533
Co 311,083 Cr 318,434 Re 318,557
Cr 314,087 Ta 318,455 Mo 318,571
Be 311,086 Fe 318,462 Co 318,595
{С = 0,01 мкф, L = 0,01 мгн, задающий искровой промежуток 3 мм, аналитический промежуток 2 мм), обеспечивающей один разряд за полупериод питающего тока. Подставной электрод — угольный, медный или алюминиевый стержень, заточенный на конус, время предварительного обыскривания 60—80 сек. Экспозиция определяется условиями освещения щели, светосилой применяемого спектрального прибора и характеристикой приемника излучения (фотопластинки или фотоумножителя).
В дуговом режиме (генераторы ДГ-1, ДГ-2, ГЭУ-1 или ИВС-26) систему поджига регулируют на один разряд за полупериод питающего тока, аналитический промежуток 2 мм, ток дуги 3—5 а. Подставной электрод — угольный или медный стержень, заточенный на конус. Предварительный обжиг — 5—10 сек.
Следует отметить, что при использовании дугового возбуждения погрешность определений, как правило, больше, чем в случае высоковольтного искрового разряда. Наложение магнитного поля (40—1000 гс) конусообразной формы на разряд дуги перемен-
119
ного тока позволяет существенно повысить воспроизводимость, результатов единичных определений. Объясняется это повышением стабильности разряда и более регулярным поступлением вещества в разрядный промежуток [87, 88].
Разработаны методы спектрального определения ванадия в титановых [42, 74, 140, 145, 323, 576, 965], алюминиевых [289 , 576, 911], кобальтовых [544], урановых [712, 1110], никелевых [145, 328, 544], хром-молибден-никелевых сплавах [145, 432], сплавах некоторых тугоплавких металлов [145, 884, 965], ферросплавах 1514, 576].
При анализе крупногабаритных изделий и отливок часто применяют методы с предварительным отбором пробы, основанные на переносе вещества пробы в электрических разрядах па вспомогательный электрод (металлический или графитовый). Последующий спектральный анализ перенесенного вещества осуществляют при наиболее благоприятных условиях возбуждения [493, 566, 584]. Включения в сталях и сплавах и ликвационную неоднородность отливок определяют с применением микроспектраль-пых методов анализа [251, 319].
Спектральные методы определения ванадия в порошковых пробах (горные породы, руды, минералы и другие геологические объекты) описаны в монографиях и обзорах [208, 224, 239, 444]. Наиболее часто анализируемую пробу помещают в кратер угольного электрода и возбуждают спектр в дуге переменного [98, 336, 428, 445, 922] или постоянного [282, 366, 462, 590, 613, 636] тока. Для уменьшения влияния химического и минералогического состава проб на результаты спектрального анализа применяют различные разбавители и буферные смеси. Наиболее простой и эффективной буферной смесью можно считать угольный порошок, содержащий 10% ВаСО3 [282, 428, 462]. При возбуждении спектра в дуге переменного тока проба разбавляется этой смесью в отношении 1 : 2 и помещается в кратер угольного электрода; ток дуги 20 а. Определяют до 10~3% ванадия в железных рудах и силикатных горных породах с относительной погрешностью 9—16% [428]. В случае применения дуги постоянного тока рекомендуют смешивать пробу с указанной буферной смесью в отношении 1:1, ток дуги поддерживают равным 13 а. Метод использован для определения до 5-10~5% ванадия в основных и ультраосновных породах, а также в других геологических материалах [282, 462].
Аналогичную технику спектрального анализа применяют и при определении ванадия в ряде чистых металлов и сплавов после их перевода в окислы. Это объясняется тем, что применение окис-лов, как правило, снижает предел обнаружения ванадия, а также и других примесных элементов и, кроме того, существенно облегчается приготовление стандартных образцов. Анализ чистых металлов подробно описан в главе V.
Следует отметить, что в процессе разогрева угольного электрода соединения ванадия, присутствующие в пробе, легко восстанавливаются с образованием труднолетучих оксикарбидов вана-120
дня, при этом время испарения ванадия из кратера электрода возрастает. Скорость испарения ванадия из пробы обычно повышают добавлением хлорида аммония, хлорида натрия и т. д., что способствует образованию легколетучего окситрихлорида ванадия.
При спектральном анализе нефти [6, 58, 252, 279, 735, 781, 979], нефтепродуктов [252, 279, 933, 979], углей, сланцев и лигнитов [962, 1174], крекинг-катализаторов [905], растительных материалов [208, 1110], обычно применяют предварительное -озоление проб, смешение с какой-либо буферной смесью и испарение определенной навески полученной смеси из кратера угольного электрода в дуге переменного или постоянного тока. Возможно применение и металлических (алюминиевых) электродов [1174].
Для повышения точности определений и унификации методик спектрального анализа пробы сплавляют с различными плавнями [288, 770, 818, 819, 850, 1110] или переводят в раствор [144, 701а, 850, 910]. Дальнейшее спектральное определение ванадия проводят обычно испарением плава или сухого остатка пробы из кратера угольного электрода в дуговом разряде [288, 701а, 770, 818], либо плав брикетируют с угольным порошком и брикет анализируют в искровом разряде, так же как и в случае анализа монолитных проб [850], либо анализируемый раствор вводят непосредственно в плазму источника возбуждения тем или иным способом [604, 850, 910].
Принудительная подача вещества пробы в облако разряда уменьшает роль и влияние различных физико-химических процессов, протекающих в объеме пробы в кратере угольного электрода. Наиболее просто это может быть реализовано с помощью вращающихся тарельчатых или ленточных электродов, на которые нанесен слой пробы, непрерывной подачей пробы из кратера электрода (трубчатый поршневой электрод) или при просыпке и вдувании анализируемого порошка в разрядное облако.
При анализе шлаков [86] и агломерата [299] использована техника вращающегося горизонтального дискового электрода, по окружности которого насыпается тонким слоем измельченная проба. Спектр возбуждают в дуге переменного тока (8 а), скорость вращения диска 0,5 об!мин, аналитический промежуток 2,5 ми. Используют медные [299] или никелевые [86] электроды. Определяют содержание ванадия в интервале 0,3—15% с относительной погрешностью 1,2—7% [86], а в агломерате в интервале 0,3— 0,8% с погрешностью 0,2—0,4% [299].
Разработан метод определения ванадия в концентрате и агломерате титаномагнетитовых руд с использованием техники поршневого электрода [3441.
Пробу смешивают с угольным порошком в отношении 1:3, помещают в нижний полый графитовый электрод,-и со скоростью 2—3 мм/мин она выталкивается в разрядный промежуток дуги переменного тока. Сила тока дуги 4 а, аналитический промежуток 2,5 мм, предварительный обжиг 90 сек. Относительная погрешность определения 0,3—1% ванадия не превышает 5%.
121
Различные модификации метода «просыпки-вдуванил» порошковых проб в плазму дугового разряда обеспечивают равномерное введение пробы в облако разряда, стабилизируют разряд, повышают воспроизводимость определений по сравнению с методом испарения вещества из канала угольного электрода и повышают интенсивность спектральных линий элементов, расположенных в начале и середине рядов летучести [444, 704а].
Описан оригинальный метод определения ванадия в сливных шлаках феррованадиевого производства [11.
Проба жидкого шлака непосредственно из ложки всасывается в стальные трубки (внешний диаметр 8 мм, толщина стенки 1 мм). После охлаждении трубка с застывшим шлаком стачивается на плоскость и служит одним и» электродов;^противоэлектрод — угольный стержень, заточенный на конус. Спектр возбуждают в дуге переменного тока (11—12 а) (обжиг 15—20 сек., экспозиция 30 сек.) и регистрируют на приборе ФЭС-1. При определении ванадия в интервале концентраций 0,3—1,5% относительная погрешность не превышает 7,5%.
В аналитической практике очень часто объектом анализа являются технологические растворы или естественные воды. Кроме того, некоторые твердофазные объекты иногда специально переводят в раствор для исключения влияния химического, минералогического и гранулометрического состава пробы на результаты определений или для снижения предела обнаружения путем использования методов предварительного химического обогащения. В большинстве случаев после упаривания и высушивания раствора на каком-либо коллекторе применяют метод испарения пробы из кратера угольного электрода.
Так, после концентрирования ванадия методами ионного обмена [170, 422, 627, 676, 770], экстракции [170, 173, 273, 459, 976, 1110] или осаждения [701а, 1174] концентрат примесей па угольном порошке после высушивания помещают в кратер угольного электрода и возбуждают спектр в дуговом разряде. В растительных материалах определяют до 3-10~5% ванадия после озоления и концентрирования примесей экстракцией хлороформом комплексных соединений с дитизоном, купфероном или пирролидиндитио-карбамипатом и при возбуждении спектра концентрата в дуге постоянного тока 10 а [1110]. . ,
Для уменьшения трудоемкости анализа, повышения точности, а в ряде случаев и чувствительности определений анализируемый раствор высушивают на торце электрода или пропитывают им угольный электрод и возбуждают спектр в дуговом разряде. Этот метод использован для определения до 2-10~5% ванадия в сталях [631], нефтепродуктах и биологических материалах [279, 314, 933].
Непосредственный анализ жидких проб проводят с использованием вертикального вращающегося дискового электрода, частично погруженного в анализируемую жидкость [279]. Для определения до 10~4% ванадия в известняках и близких им по составу материалах применен капиллярный электрод. Пробу растворяют и возбуждают спектр в дуге переменного тока 11—12 а [910]. При 122
определении ванадия в нефтях и мазуте пробы после разбавления хлороформом вводят в трубчатый электрод и возбуждают спектр в дуговом разряде [601].
Более перспективным следует считать введение анализируемого раствора в виде аэрозоля непосредственно в плазму разряда. В качестве источника возбуждения спектров используют дуговые плазмотроны [144, 279, 604], факельный высокочастотный разряд [144, с. 127] и высоковольтный искровой разряд. Аэрозоль вводят через осевое отверстие в нижнем электроде [144, с. 59] или между горизонтально расположенными графитовыми электродами [850]. Методы нашли применение при определении ванадия в титановых сплавах [144, с. 59,127], нефтепродуктах [279, 601], образцах сложного состава [604].
Значительное повышение экспрессности анализа порошковых проб можно получить при использовании аэрозольно-искрового метода анализа суспензий [343]. При определении ванадия в титаномагнетитовых рудах и агломерате применение этого метода позволяет использовать наиболее эффективный способ мокрого измельчения, устранить агрегатирование частиц пробы соответствующим выбором состава жидкой фазы и влияние минералогического и химического состава проб на результаты анализа. Относительная погрешность определения ванадия не превышает 3,2% [291].
МЕТОДЫ ЭМИССИОННОЙ ПЛАМЕННОЙ СПЕКТРОФОТОМЕТРИИ
В низкотемпературных пламенах (типа пропан—бутан—воздух и ацетилен—воздух) определение ванадия возможно лишь по измерению интенсивности молекулярных полос моноокиси ванадия с максимумами при 528,0; 550,0; 573,7 и 578,0 нм [72]. Это обусловлено высокой энергией диссоциации моноокиси ванадия VO, равной 5,5 эв. Использование высокотемпературных пламен, таких как кислород—ацетилен и особенно закись азота—ацетилен, дает возможность определять до 3 мкг/мл ванадия по атомным линиям V I 318,5 и V I 437,9 нм при существенном уменьшении различного рода влияний [630, 739, 740, 795, 800].
При пламенно-фотометрическом определении до 0,01% ванадия в низко-и высоколегированных сталях 1 г пробы растворяют в смеси соляной и азотной кислот, раствор упаривают, остаток сушат при 200° С в течение 5 мин., растворяют в 10 мл конц. НС1; объем доводят водой до 200 мл и вдувают аэрозоль раствора в пламя закись азота—ацетилен. Относительная погрешность определения не превышает 4% [740].
После экстракции ванадия из пробы в виде его оксихинолина-та каким-либо органическим растворителем (этилацетатом, метил-изобутилкетоном, амилацетатом или хлороформом) экстракт непосредственно распыляют в водородно-кислородное пламя. Этим методом определяют до 50 мкг/мл ванадия по молекулярной полосе 528,0 нм [769]. Следует отметить, что выбор пламени для определения ванадия в данном случае нельзя признать удачным. Повы
123
шение восстановительных свойств этого пламени может быть достигнуто применением органических растворителей, в частности этанола [1092]. При этом предел обнаружения ванадия снижается лишь до 2—8 мкг/мл.
Значительного снижения предела обнаружения ванадия, а также и других элементов, образующих труднодиссоциируемые моноокиси, можно достичь применением разделенных пламен. Так, если в пламени закись азота—ацетилен предел обнаружения ванадия составляет 0,2 мкг/мл, то при использовании этого жп пламени, но защищенного аргоном или азотом, можно снизить предел обнаружения ванадия до 0,05 мкг/мл. Это сравнимо с результатами, получаемыми в методе атомно-абсорбционной спектрофотометрии [420, с. 60; 795, 844].
МЕТОДЫ АТОМЫ О-АБСОРБЦИОННОЙ И АТОМНО-ФЛУОРЕСЦЕНТНОЙ СПЕКТРОФОТОМЕТРИИ
Методы атомно-абсорбционной спектрофотометрии, основан ные на резонансном поглощении излучения свободными атомами элементов, обладают высокой селективностью, точностью, низкими пределами обнаружения, простотой аппаратурного оформления и т. д. Они нашли широкое применение при анализе самых разнообразных объектов [300, 420, 466, 808, 843, 1038, 1138, 1158]. Наиболее часто для атомизации ванадия используют пламена кислород—ацетилен и особенно закись азота—ацетилен. В качестве источника первичного излучения обычно применяют лампы с полым катодом или безэлектродпые ВЧ-лампы. Спектральные ли-нии'ванадия, рекомендуемые при атомно-абсорбционном анализе, а также чувствительность его определения приведены в табл. 28.
Таблица 28
Определение ванадия атомио-абсорбциокным спектрофотометрическим методсм [420, 466, 1038, 1138]
X, им Сила осциллятора Чувствительность определения ванадия в пламенах, мкг!мл (на 1% поглощения)
C2H2-N2O С2Н2-О2
305,63 3,3
306,05 2,8
306,64 0,5-2,9
318,34 318,40 0,5 1 0,66 J 1,2—1,3 25
318,54 0,40 1,0 25
370,36 0,5-5,2 50
385,54 385,58 0,98 } 2-6,2 50
437,92 0,20 4—4,4 50
124
Предел обнаружения ванадия в пламени закись азота—ацетилен составляет 0,01 мкг!мл [843, с. 7051. Возможно применение в качестве окислителя окиси азота, что позволяет несколько снизить предел обнаружения ванадия [1038], однако из-за высокой токсичности этот газ в аналитической практике применяется крайне редко.
В пламени закись азота—ацетилен эффективность атомизации ванадия превышает 0,9, а 10% его находится в ионизованном состоянии. Поэтому для повышения поглощения излучения линиями ванадия в анализируемые и стандартные растворы вводят соли щелочных элементов в качестве ионизационных буферов. Усиление поглощения ванадия наблюдается в присутствии алюминия, титана, железа и фосфорной кислоты [766, 789, 843], а серная кислота несколько уменьшает абсорбцию ванадия [1038]. Введение в анализируемые растворы некоторых органических растворителей снижает предел обнаружения ванадия. Так, добавление к водным растворам ~8% диэтиленгликоля приводит к увеличению величины регистрируемого сигнала почти на 50% [1043].
Описаны методы пламенного атомно-абсорбционного определения ванадия в сталях [694, 800, 849, 1098, 1133], чугуне [694], карбидных включениях в стали [1055], соединениях и сплавах титана [130, 662, 869], в алюминии и его соединениях [1170], никелевых сплавах [1155], рудах, шлаках и горных породах [600, 647, 766, 858,880,975, 987, 1131], природных водах [780, 857, 998], в катализаторах синтеза аммиака [494], в нефти и нефтепродуктах [667, 1071, 1147], во взвешенных в воздухе частицах [1135] и других объектах. В качестве примера приведен атомно-абсорбционный метод определения ванадия в низко- и высоколегированных сталях [420 , 849].|
1 г стали растворяют в 10 мл НС1 (уд. в. 1,18) и 5 мл HNO3 (уд. в. 1,42). По окончании реакции к раствору добавляют 10 мл НС1О4 (уд. в. 1,54). Раствор медленно упаривают до начала выделения паров хлорной кислоты и продолжают упаривание еще в течение 5 мин. Затем раствор охлаждают, добавляют 50 мл воды, фильтруют и разбавляют водой до 100 мл. Раствор распыляют в стехиометрическое пламя закись азота—ацетилен и определяют поглощение ванадия по самой чувствительной линии V I 318,4 нм. Для устранения влияния железа на результаты анализа его вводят в стандартные растворы в постоянной концентрации, равной 10 мг!мл [849].
Для снижения предела обнаружения ванадия в различных объектах и уменьшения влияния концентрации посторонних веществ в распыляемом растворе применяют методы предварительного концентрирования. В связи с высокой селективностью самого метода атомно-абсорбционного анализа к специфичности методов концентрирования не предъявляются жесткие требования; как правило, одновременно выделяют целую группу определяемых элементов. Наибольшее распространение ввиду простоты техники исполнения нашли экстракционные методы. Особенно часто концентрирование ванадия проводят экстракцией метилизобутилке-тоном комплекса с купфероном или пирролидиндитиокарбамина-том аммония [420, с. 168; 702, 1043, 1155], а также смесью дихлор-
125
метана с трифениларсином, 5,7-дихлор-8-оксихинолином и другими экстрагентами [662, 734, 811, 843J.
Предложено использовать ионообменное концентрирование ванадия на смоле Цеокарб 225 при анализе стали [789], на сульфированном полистирольном катионите AG50-X8 при анализе силикатных горных пород [1111] и в некоторых других случаях [420, с. 172; 857, 858].
Ванадий и другие примеси при анализе алюминия и его соединений концентрируют соосаждением на гидроокисях циркония и алюминия. После растворения осадка на фильтре раствором HCI (1 : 1) определяют до 5 мкг ванадия [1170].
Интересен метод определения ванадия, который основан на образовании молибденованадиевофосфорной кислоты, экстракции ее смесью w-пентанол—диэтиловый эфир (1 : 4 по объему), реэкстракции водой и определения молибдена по линии 313,2 нм. Содержание молибдена пропорционально и в несколько раз больше концентрации ванадия. При использовании воздушно-ацетиленового пламени относительная погрешность метода не превышает 3,4%, а чувствительность определения в пересчете на ванадий составляет 0,05 мкг/мл (на 1% поглощения) [811, 843, с. 726]. Усовершенствование методики и применение пламени закись азота—ацетилен позволило получить чувствительность определения ванадия 0,011 мкг/мл [843, с. 727].
В последнее время все больше внимания уделяется непламенным методам атомизации. Применение плазменных атомизаторов (высокочастотный и дуговые разряды) позволяет определять ванадий примерно с такой же чувствительностью, что и в пламени закись азота—ацетилен [466, 11181. Существенное снижение предела обнаружения ванадия в различных объектах наблюдается при использовании в качестве атомизаторов графитовых, вольфрамовых или танталовых лодочек, графитовых кювет и т. д. [420, 466, 734, 913, 961, 1112, 1118]. Стандартная графитовая кювета позволяет при температуре атомизации 2700° К определять до 20 нг ванадия в 1 мг пробы [961]. При анализе нефтепродуктов с использованием угольной нити как атомизатора определяют до 0,3 нг ванадия, а с предварительной экстракцией определяют 7-10"7% ванадия (по отношению к исходной пробе) [734].
Методом атомно-флуоресцентной спектрофотометрии с приме-' пением пламени закись азота—ацетилен, защищенного аргоном, и безэлектродной разрядной лампы с микроволновым возбуждением в качестве источника света определяют до 0,07 мкг!мл ванадия по линии V I 318,4 нм [843, с. 705]. Установлено, что из 23 исследованных ионов только алюминий и вольфрам увеличивают регистрируемый сигнал, а присутствие кобальта ведет к его некоторому снижению.
426
РЕНТГЕНОСПЕКТРАЛЬНЫЕ МЕТОДЫ
Рентгеноспектральные методы, особенно его флуоресцентный вариант, прочно вошли в практику работы промышленных предприятий и научно-исследовательских институтов при анализе самых разнообразных материалов. Теоретические основы и различные практические приложения рентгенофлуоресцентпого анализа изложены в монографиях [296, 393].
Определение ванадия рентгеноспектральным методом проводят по самой интенсивной линии 2 (0,25 нм) с пределом обнаружения до 2-10-4% и относительной’погрешностью 0,1—2%. Вследствие сильного поглощения линии V воздухом необходимо применять вакуумные рентгеновские спектрометры.
Метод применен для определения ванадия в сталях [296, с. 213; 623, 875, 1172], бериллии и его окислах [681], ванадий-галлиевых сплавах [682], металлическом алюминии и его окисле [879], титане и титановых сплавах [315], ванадий-кремниевых пленках [49], шлаках [819], ильмените [546], магнетите [426], глиноземе [1044], горных породах и минералах [819, 1159], морской воде [929], коррозионных средах [1065], катализаторах крекинга нефти [1022], нефтепродуктах [646], битуминозных породах [967], смесях окислов, получаемых из металлов, сплавов и горных пород [296, с. 207]. В ряде случаев для снижения предела обнаружения ванадия применяют методы предварительного концентрирования: экстракции [890, 929, 967], ионного обмена [646, 770, 890, 929], осаждения купфероном в присутствии коллектора [1065].
Методом рентгеноспектрального анализа определяют валентное состояние ванадия. С использованием рентгеновского спектрометра высокого разрешения измеряют соотношения менаду химическим сдвигом линии ванадия и шириной линии Ка. По полученным результатам строят диаграмму в координатах сдвиг линии — ширина линии Ка, где выделяют области, соответствующие соединениям ванадия(У), ванадия(1У) и ванадия(Ш). Метод применен для анализа перовскита и катализаторов, содержащих ванадий [767].
В последнее время для экспрессного определения ванадия, особенно в геологической практике, применяют портативные рентгенорадиометрические анализаторы [307, 500, 1026]. В этих приборах для возбуждения характеристического рентгеновского спектра пробы используют радиоизотопные источники. В частности,, для определения ванадия рекомендуют 55Fe, 109Cd и 147Рш. Метод анализа применен для определения ванадия в сталях [500, 10261, горных породах, рудах и сплавах [1026].
Абсорбционным рентгеноспектральным методом по А-скачку поглощения с применением в качестве источника излучения радиоактивного изотопа 55Fe определяют ванадий в полимерных пленках [547].
12Т
АКТИВАЦИОННЫЕ МЕТОДЫ
Активационные методы анализа являются одними из самых чувствительных методов определения большого числа элементов и применимы к самым разнообразным объектам. Теоретические основы, техника и практическое применение активационных методов приведены в монографиях [70, 608] и обзорах [490, 1118, 1138]. Некоторые характеристики методов определения ванадия приведены в табл. 29.
Таблица 29j
Определение ванадия методами активационного анализа [70, 258, 490, 608, 1138]
Условия активации Ядерные реакции кэв Мешающие элементы Предел обнаружения ванадия, мг
Тепловые нейтро- 51V(/i, T)52V 3,77 мин. 320 1 «•Ti, 62Сг, 0,002
иы, 1013 нейтрон) 1434 / Б5МП
/см2-сек 3191
Быстрые нейтро- 61V(/i, p)51Ti 5,80мин. 605 1 50Ti, иСг 0,3
иы, 109 нейтрон! 980]
/ см2 • сек 986 1
51V(ra, a)I8Sc 1,83 дня 1040 «8Ti, «Ст 0,2
1314 J
5iV(ra,ara)«Sc 3,45 дня 180 40Ca,47Ti, 48Ti 0,2
Протоны, Е = ==11,5 Мяв 61V(p, «)51Cr 27,8 дней 323 54Fe 0,3
Наибольшее распространение из всех активационных методов нашел нейтронно-активационный, позволяющий определять до 10-9 г ванадия по ядерной реакции Б1У (га, у)Б2У. Практически 100%-ная распространенность изотопа B1V и достаточно высокое сечение реакции активации на тепловых нейтронах (4,5 барн) благоприятствуют применению метода.
При количественном определении ванадия необходимо учитывать влияние первичных интерферирующих реакций Б2Сг (га, р)Б2У и ББМп (га, а)Б2У. Кроме того, определению мешают 27А1, 6БСи, 107Ag и 103Rh, которые в результате ядерных реакций образуют радиоактивные изотопы с близкими к Б2У периодами полураспада и энергиями излучения.
Разработаны нейтронно-активационные методы определения ванадия в стали [70, 774, 1063], железе высокой чистоты [11081, вольфраме [1046], ниобииД8631, титане, его сплавах и соединениях [70, 732], графите [70, 292], рудах и горных породах [608, 1162, 1139], природных водах [752, 889], биологических материалах [70, 829], сырой нефти, ее фракциях и золах [10, 60, 293, 294, 744], воздухе [986, 1176]. Большинство методов определения ванадия 128
128
основано на кратковременном (1—10мин.) облучении пробы в ядер-ном реакторе потоком нейтронов 1013 нейтрон1см2-сек, выдерживании («охлаждении») пробы в течение 1—6 мин. и измерении (2 мин.) активности B2V по у-пику 1,43 Мэе на сцинтилляционном многоканальном у-спектрометре с кристаллом NaJ(Tl) или Ое(Ы)-де-тектором.
В ряде случаев ванадий определяют активационным методом после его предварительного концентрирования методами ионного обмена [889, 1063], экстракции [662, 752, 1063, 11181, а также после отделения ванадия методами хроматографии на бумаге [674].
Для повышения точности и чувствительности активационного метода анализа применяют радиохимическое выделение радиоактивного изотопа 82V. С этой целью обычно применяют методы экстракции [70, с. 271; 732, 829, 889].
Концентрат природной воды (3—4 мл) после облучения в течение 5 мин. в канале ядерного реактора переносят в раствор 8 N NaOH, содержащий радиоактивный индикатор (4eV, = 330 дней) и устанавливают pH 4 с по-
мощью разбавленной НС1. Ванадий экстрагируют 1%-ным раствором 8-оксихинолина в хлороформе и реэкстрагируют нитратно-аммиачной буферной смесью с pH 9,4. Реэкстракт промывают хлороформом, измеряют активность 62V, а после его полного раепада (20—30 мин.) — активность 49V для определения химического выхода ванадия. Предел обнаружения 0,1 мкг!л [889].
Применение быстрых нейтронов с энергией ~14 Мэе предложено для определения ванадия по реакции B1V(rc, p)B1Ti в сталях [1063] и фосфатах [89]. Ампульные источники нейтронов 2B2Cf [977] и Рн—Be [2351 позволяют определять ванадий в полевых условиях и контролировать различные технологические процессы. Предел обнаружения ванадия при потоке нейтронов 107 нейтрон! !смг-сек составляет 10-2%.
Активацию пробы протонами предложено использовать для определения ванадия в алюминии [711], ниобии [863], титане [258] и других материалах [346]. При энергии протонов 11,5—15 Мэв и интенсивности пучка 10 мка определяют до 2,8-10-4% ванадия в титане [258] и до 3,6- 10-в% в алюминии [711]. Следует отметить, что сечения реакций ванадия на у-квантах высокой энергии и быстрых нейтронах существенно меньше, чем на тепловых нейтронах, поэтому применение таких методов анализа оправданно лишь в некоторых специальных задачах [70, 608, 1138].
МАСС-СПЕКТРАЛЬНЫЕ МЕТОДЫ
Аналитическое применение масс-спектральные методы получили лишь в последнее десятилетие, что связано в основном с освоением производства масс-спектрометров с двойной фокусировкой и с искровыми источниками ионов [312, 526,>574]. Главным достоинством методов искровой масс-спектрометрии является низкий абсолютный (до 10~12 г) и относительный (до 10"8%) предел обнаружения элементов. 5
5 В Н. Музгин и др.
129
В монографии [312J и обзоре [1118] приведены литературные данные по масс-спектральному определению ванадия в металлах (алюминии, меди, натрии, никеле, цирконии), сплавах, биологических объектах, геологических пробах, образцах лунного грунта и других материалах.
При анализе двуокиси титана пробу смешивают с пятиокисью ниобия (внутренний стандарт) и угольным порошком в отношении 1:1:2 или с порошком металлического серебра в отношении 1:1: 10. Смесь брикетируют под давлением 300 т/см2 и полученные брикеты используют в качестве электродов. Масс-спектры получают при напряжении искрового источника до 100 кв. Предел обнаружения ванадия составляет 1-10~7%, относительная погрешность ~15% [806].
Ванадий в железе особой чистоты, полученном методом зонной плавки, определяют следующим образом. Из соответствующих зон слитка вырезают электроды длиной 8—12 мм и поперечным сечением 1—2 мм2 и после химической полировки устанавливают в ионном источнике. Спектры возбуждают при длительности импульсов 20 мксек, и частоте следования импульсов 1000 гц. Предел обнаружения ванадия составляет 1 • 1О~5/о, относительная погрешность ~25% [914].
Масс-спектральным методом анализа определяют ванадий с пределом обнаружения 1,9 мг!м3 в воздухе и менее 0,02 мг/мл в природных водах [1118].
МЕТОДЫ ЭЛЕКТРОННОГО И ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
У ионов ванадия, соответствующих низшим степеням окисления, происходит застройка Зй-оболочки (для иона V(IV) конфигурация 3d1, для иона У(Ш) — 3d2, для иона V(II) — 3d3), приводящая к парамагнетизму и, следовательно, к возможности наблюдения электронного парамагнитного резонанса (ЭПР).
Поскольку площадь под линией ЭПР пропорциональна числу парамагнитных частиц, измерение ее величины может быть использовано для определения доли ванадия, находящегося в низших степенях окисления. Этот метод был применен при исследовании неорганических и органических соединений ванадия различной степени окисления [99, 204], для определения степени восстановления ванадия(У) в ванадий(1У) [27], для определения содержания ванадия(1У) в пятиокиси ванадия [826] и других объектах [470], для исследования процесса соосаждения ванадия(1У) с гидроокисями различных металлов [310]. Методом ЭПР находят тип соединения, в виде которого находится ванадий в нефти [758], его количественное содержание (предел обнаружения до 1-10-в?6) в ней [1031], а также определяют содержание ванадия в фосфатных стеклах [940].
Ванадий] V), находящийся в растворе, восстанавливают до ванадия(1У) действием 2,5-кратного избытка сернокислого гидроксиламина при pH 4. К 200 мл полученного раствора добавляют 0,5 г катионита КУ-2, перемеши-130
вают в течение 20 мин., смолу отфильтровывают, промывают водой, сушат и переносят в ампулу, которую помещают в резонатор ЭПР-спектрометра, и регистрируют интенсивность сигнала ЭПР. Погрешность определения ванадия не превышает 3% (без предварительного концентрирования ванадия методом ионного обмена погрешность достигает 5,6%) [470].
В тех случаях, когда удается получить сигнал магнитного резонанса ядер ванадия, возможно его количественное определение путем измерения] площади под резонансной кривой. Метод ядер-ного магнитного резонанса (ЯМР) был использован при изучении состояния воды в гидратированных соединениях ванадия [402а, 1070], для установления степени чистоты пятиокиси ванадия [893], при изучении механизма образования комплексных соединений ванадия(1У) с арсеназо [1118].
Отмечается, что спектроскопия ЯМР на высоких частотах — один из наиболее чувствительных методов анализа чистоты и стехиометрии VO2 [893].
5*
Глава V
ОПРЕДЕЛЕНИЕ ВАНАДИЯ В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
Содержание ванадия в природных и промышленных объектах колеблется в очень широких пределах. Природные вещества, воды, нефти, руды характеризуются низким содержанием ванадия — от 1СГ8 до 2%. Ванадий в этих объектах определяют фотометрическими, кинетическими и спектральными методами. При анализе промышленных концентратов, ванадиевых шлаков, сплавов, минеральных отложений (на стенках котлов и лопатках турбин) наибольшее применение нашли титриметрические методы.
Определению ванадия химическими методами часто предшествует отделение его от мешающих элементов. Многие примеси отделяют в момент разложения пробы сплавлением с окислительно-щелочными плавнями с последующим выщелачиванием ванадия водой в виде ванадата. Фотометрическое определение ванадия сочетают с экстракционным отделением его в виде различных соединений с органическими реагентами.
Нитридные включения сталей (нитриды ванадия, алюминия и хрома) не переходят в раствор при электролитическом растворении. Это используют для отделения нитридных включений от основы и для определения содержания азота, нитридов ванадия, алюминия и хрома в низко- [3331 и среднелегированных [5711 сталях.
ГОРНЫЕ ПОРОДЫ, РУДЫ И ПРОДУКТЫ ИХ ПЕРЕРАБОТКИ •
Основными промышленными концентратами ванадия являются шлаки металлургического производства, полученные при выплавке стали и чугуна, а также отходы титанового и алюминиевого производства. Чаще используют конверторные шлаки, содержащие 12—24% ванадия, который входит в состав шпинелида (твердого раствора окислов двух- и трехвалентных металлов). На долю шпинелида приходится 45—50% шлака, остальная часть представлена орто- и метасиликатами.
Руды и основные шлаки растворяют в соляной, хлорной, азотной, смесях серной и фосфорной кислот или обрабатывают последовательно азотной и фтористоводородной кислотами. Неразло-жившийся остаток сплавляют с содой и растворяют плав в соляной кислоте или в смеси фтористоводородной и азотной кислот
132
и присоединяют к основному раствору. Определению ванадия в большинстве случаев мешают нитрат- и фторид-ионы, поэтому анализируемые растворы упаривают с серной кислотой до паров серного ангидрида.
Силикатные породы и кислые шлаки разлагают сплавлением. Выбор плавня определяется минералогическим составом образцов. Ванадиевые шлаки и большинство горных пород спекают или сплавляют с содой и небольшим количеством нитрата или перекиси натрия. Окисленные руды сплавляют с пиросульфатом калия. Руды, содержащие тяжелые металлы, а также титаномагнетиты и хромиты сплавляют с перекисью натрия или Na2CO3. Щелочные плавы обычно растворяют в воде. При этом ванадий переходит в раствор в виде ванадата, а элементы, образующие нерастворимые гидраты и основные соли, выделяются в осадок. Таким образом отделяют основную массу железа, титана, циркония и т. д. перед фотометрическим определением. Перед титриметрическим определением плавы лучше растворять в кислотах.
Навеску титаномагнетитовой руды, концентрата или агломерата помещают в железный или фарфоровый тигель, в который вносят 6—8 г смеси для сплавления (Na2CO3 и Na2O2 в отношении 1 : 1), тщательно перемешивают, закрывают крышкой и сплавляют при 650—700° С в течение 1—2 мин. с момента расплавления. Тигель охлаждают, помещают в стакан и выщелачивают ванадий 50—70 мл горячей воды. Тигель и крышку извлекают и обмывают над стаканом водой. Далее поступают различными способами: перед фотометрическим определением отбирают аликвотную часть пробы и отфильтровывают осадок, а перед титриметрическим определением добавляют серпую кислоту и растворяют основную массу осадка [115].
При определении ^>0,1% ванадия используют оксидпметриче-ские методы с визуальной или инструментальной регистрацией точки эквивалентности [20, 21, 45, 84, 115, 425, 5801 (см. главу IV).
Ввиду малого содержания ванадия в минеральном сырье для его определения чаще используют различные фотометрические методы. Наибольшее распространение получил фосфорновольфра-матный метод [13, 21, 84, 121, 141, 150, 406, 417, 457, 529, 544, 580]. Ванадий (0,005—0,5%) определяют в растворе, полученном после обработки щелочного расплава водой. Этот метод является стандартным для анализа железных, хромовых и титаномаг-петитовых руд, их концентратов и агломератов (ГОСТ 15848. 17—70 и 18262.9—72). Чувствительность определения может быть повышена, если гетерополикислоту экстрагировать органическими растворителями [13, 1032].
Для определения ванадия в горных породах широко применяют М-бензоил-]У-фенилгидроксиламин (БФГА). Метод достаточно избирателен и позволяет определять до 0,001 % вападия при использовании маскирующих средств и экстракции окрашенного комплекса из силыюкислых растворов [718, 972].
В платиновый тигель вносят 0,5—1 г растертой горной породы, прибавляют 0,5 мл H2SO4 (1 : 1), 5—10 мл HF (уд. в. 1,13) и нагревают до паров серного ангидрида. Остаток растворяют в 10 мл НС1 (уд. в. 1,26) и фильтруют.
133
К фильтрату добавляют 12—18 .ил той же НС1, 25 мл воды, несколько капель 5%-ного раствора КМпО4 до розовой окраски и нагревают на водяной бане до полного просветления раствора. После охлаждения добавляют 0,1 г сульфосалициловой кислоты и 0,1—0,3 г NH4F, если необходимо маскировать примеси. Экстрагируют ванадий 10 мл 0,1%-ного раствора БФГА в хлороформе в течение 30—60 сек. Экстракт фильтруют через сухой фильтр и измеряют оптическую плотность при 530 нм относительно хлороформа [972]-.
Аналогично определяют ванадий в железных рудах [7961, магнетите, ильмените, бокситах, сульфидных конкрециях [972]. Метод принят в качестве стандартного при определении ванадия в глиноземе (ГОСТ 13583-8—71). Показано, что этот метод лучше, чем методы с применением перекиси водорода и пирокатехина, а также фосфорновольфрамовой гетерополикислоты [783]. Вместо БФГА можно использовать 1У-фуроил-1У-фепилгидроксиламин [400].
Рекомендован [40] избирательный метод, основанный на образовании тройного комплексного соединения ванадия с 4-(2-пириди-лазо)-резорцином (ПАР) и перекисью водорода.
Растворяют 0,1 г руды при умеренном нагревании в смеси 10 мл H2SO1 (1 : 3) и 5 мл HNO3 (1 : 1). Раствор упаривают до паров серного ангидрида» переносят в мерную колбу на 100 мл и разбавляют водой. Отбирают аликвотную часть (10—100 мкг ванадия) раствора в мерную колбу на 50 мл, добавляют 5—10 мл 10%-ного раствора NH4F, 20 мл 1 У H2SO4, 2 мл 1,96-• 10 4 М водного раствора ПАР, 3 мл 3%,-ного раствора Н2О2 и доводят до метки 1 У H2S04. Нагревают на водяной бане 2 мин. После охлаждения до комнатной температуры измеряют оптическую плотность при 540 нм [40].
Для определения ванадия в глиноземе, бокситах и глинах используют реакцию окисления диметилнафтидина, диаминбензиди-на, о-дианизидина [848], а в титапомагнетитовых рудах — реакцию окисления ванадокса [3671.
2,5 г пробы глинозема сплавляют 2 часа в платиновом тигле с 5 г NaHC03 и 3 г Н3ВО3 при 1000° С. Плав растворяют в 100 мл воды, добавляют 50 мл 5%-ного раствора H2S04 и разбавляют водой до 250 мл. Отбирают аликвотную часть, прибавляют несколько капель 2%-ного раствора о-дианизидина в 0,5 М растворе уксусной кислоты. Разбавляют до 100 мл и через 10 мин. измеряют оптическую плотность при 470 нм. Определению ^0,1 мкг ванадия не мешают 20-кратные количества Al, Ti и Zn. Возможно одновременное определение хрома [848].
В вольфрамовых рудах ванадий определяют с 2,2'-дипиридилом [5561. Для анализа минерального сырья рекомендуют также реакции ванадия с дифенилкарбазоном [122], пирокатехином [11, 84], кислотным хромсиним [332] и другими реагентами.
При определении ванадия в горных породах, рудах и шлаках широко используют физические методы, особенно методы эмиссионной спектроскопии [1, 86, 98, 224, 239, 282, 288, 291, 299, 336, 366, 428, 444, 445, 462, 502, 575, 604, 613, 636, 701а, 770, 818, 850, 922, 980].
Для анализа титаномагнетитовых руд и агломератов используют, например, аэрозольно-искровой метод.
300 мг пробы смачивают 0,5 мл насыщенного водного раствора изоамилового спирта и измельчают в вибромельнице 5—10 мин., добавляют 4,5 мл 134
тсго же раствора и полученную взвесь количественно переносят в распылительное устройство [343]. Суспензию распыляют потоком воздуха (рабочее давление в системе распылителя 0,4 атм) между горизонтально расположенными медными электродами диаметром 6 мм, заточенными на конус 120°, с площадкой диаметром 1 мм. Генератор высоковольтного искрового разряда включают по сложной схеме (С = 0,005 мк/, индуктивность отключена, ток 2,4 а), межэлектродный промежуток 2,0 мм, расстояние распылитель—ось электродов 3,5 мм. Непосредственно перед экспозицией (30 сек.) проводят обыскривание в течение 1 мин. без введения пробы. Расход пробы не превышает 1 мл/мин. При использовании аналитической пары линий V II 311,1—Са II 317,9 нм относительная погрешность не превышает 3,2% [291].
При атомно-абсорбционном определении ванадия в силикатных породах, руДах, шлаках и т. д. обычно используют кислотное разложение проб [600, 647, 766. 880, 11311, реже применяют сплавление с карбонатом натрия [819, 975] или с пиросульфатом калия [600].
300 мг пробы смачивают 0,5 мл воды и 3 мл конц. HF (добавляют 2,8 г борной кислоты) [647] или к 200 мг пробы добавляют 0,75 мл конц. НС1, 0,25 мл конц. HN03 и 5 лмконц. HF [880] и нагревают в течение 30—40 мин. при 110° С в герметически закрытом сосуде из фторопласта. После охлаждения реакционную смесь разбавляют водой до 100 мл. Раствор распыляют в пламя закись азота—ацетилен и измеряют оптическое поглощение линии V I 318,4 нм. Градуировочный график линеен от 0,3 до 10—20 мкг ванадия в 1 мл. При более высоком содержании ванадия необходимо дополнительное разбавление. Относительная погрешность определения составляет 1,2—4% [647].
В горных породах и рудах ванадий определяют методами рентгеноспектрального [426, 546, 770, 819, 1159], рентгепорадиомет-рического [1026] и активационного [490, 977, 1139, 1162, 1163] анализа.,
НЕФТЕПРОДУКТЫ, БИОЛОГИЧЕСКИЕ МАТЕРИАЛЫ И ЗОЛЫ
Содержание ванадия в нефти достигает 0,01%, а в золах — до 30% [279]. Для определения ванадия широко используют фотометрические и спектральные методы, которые применяют в основном после озоления образцов. Используют различные способы озоления: сжигание образцов с последующим прокаливанием остатка при 500° С, или разложение образцов концентрированной серной кислотой с добавлением азотной кислоты, или сочетают оба способа [1178].
Фотометрическое определение ванадия проводят фосфорно-вольфраматным методом [524, 677], по реакции окисления бензидина и других веществ [923], по реакциям образования окрашенных соединений ванадия с ксиленоловым оранжевым [815], N-бензоил-N-фенилгидроксиламином [513, 798], 4-(2-пиридилазо)-резорци-ном [1169], м-метокси-И-бензтиогидроксамовой [1094] и бензгид-роксамовой [799] кислотами. В качестве стандартного для определения ванадия в золе нефтей принят фосфорновольфраматвый метод (ГОСТ 10364—63). Наиболее избирательным является метод
135
с 4-(2-пиридилазо)-резорцином в присутствии комплексона IV (циклогексапдиаминтетрауксуспая кислота) [11691.
Пробу нефти минерализуют конц. H2SO4. К слабокислому раствору (2— 40 мкг ванадия) прибавляют 4 мл 0,01 М раствора комплексона IV, 5 мл фосфатной буферной смеси с pH 6,5, 2 мл 0,1%-ного раствора ПАР в 0,01 М NaOH. Разбавляют до 50 мл и измеряют оптическую плотность через 5 мин. при 545 нм относительно раствора холостого опыта. Этим методом можно Определить (1 + 5)-10 4% ванадия с относительной погрешностью 8—40% 11169].
Определение ванадия в нефтях и нефтепродуктах осуществляют методами эмиссионной [6, 58, 59, 252, 279, 316, 601, 735, 773, 781, 933, 979], атомно-абсорбционной [667, 734, 913, 961, 1071, 1147], рентгеновской [646, 890] спектрометрии и активационного анализа [10, 60, 294, 744].
Метод эмиссионной спектроскопии используют для определения ванадия в растительных материалах [208, 1110, 1178], лигнитовых [962] и угольных [Ц74] золах.
10 г анализируемого растительного материала озоляют при 450° С, остаток сплавляют с карбонатом натрия и плав растворяют в соляной кислоте. Ванадий (вместе с молибденом и вольфрамом) экстрагируют 6%-ным раствором купферона в хлороформе при pH 1,5—2,0. Экстракцию повторяют при pH 0,5. Объединенный экстракт упаривают с 90 мг угольного порошка, содержащего 1% SnO2, высушивают, прокаливают при 450° С и добавляют 30 мг ЫгСО3. Спектр пробы возбуждают в дуге постоянного тока (10 а). Предел обнаружения ванадия составляет 3-10~6% [1110].
При спектральном анализе нефтепродуктов и биологических материалов хорошие результаты получены при использовании метода пропитывания угольного электрода [316].
Раскаленный угольный электрод с плоским торцом опускают в нефтепродукт или жидкий биоматериал и через 5—10 мин. контакта охлажденный до комнатной температуры электрод извлекают из пробы и высушивают в течение 20—40 мин. при температуре не выше 500° С. Спектр возбуждают в дуге переменного тока. Для стабилизации разряда применяют пропитку угольногопротивоэлектрода 1,4%-ным раствором уксуснокислого лития. При содержании ванадия от 3-10*5 до 1 • 10*2% относительная погрешность определения не превышает 12%. Предел обнаружения ванадия может быть спи-» жен до 1-Ю5 % при замене торцевого электрода угольным диском диаметром 20 лл [316].
В золе некоторых мазутов и минеральных отложений на рабочих поверхностях котлов и турбин ванадий можно определять комплексонометрическим титрованием [309] или полярографическим методом [116].
ПРИРОДНЫЕ И СТОЧНЫЕ ВОДЫ
Для определения ванадия и других элементов в водах широко применяют методы эмиссионной [170, 173, 205, 237, 273, 422, 676, 976, 1085], атомно-абсорбционной [857, 998] спектроскопии, рентгеноспектрального [929, 1065] и активационного [752, 889] анализа. Определяемые элементы предварительно концентрируют экст-136
ракцией [170, 173, 202, 205,676, 752, 929, 976], ионообменной хроматографией [170, 422, 676, 889, 998] или осаждением [1065,1085].
К анализируемому раствору добавляют 10 мл ацетатно-хлоридного буферного раствора с pH 2,0, 5 мл 1%-ного этанольного раствора 1-фенил-З-метил-4-бензоилпиразолона-5 (ФМБП) и разбавляют водой до 25 мл. Через 15 мин. дважды экстрагируют образовавшееся соединение хлороформом по 5 мл. Экстракты переносят на 100 мг угольного порошка в кварцевом тигле. В водную фазу после нейтрализации до pH 7,0—7,5 вновь прибавляют порцию ФМБП и дважды экстрагируют примеси, как указано выше. К объединенному экстракту в тигле добавляют 1—2 мл 2,5%-ного раствора щавелевой кислоты и упаривают под ИК-лампой досуха, перемешивают 15—20 мин. и 25 мг порошка помещают в кратер угольного электрода (диаметром и глубиной 4,5 мм). В канал верхнего угольного противоэлектрода набивают угольный порошок с 5% хлорида натрия. Спектр возбуждают в дуге переменного тока (16 а). При использовании спектрографа ИСП-28 предел обнаружения составляет 0,3 мкг ванадия [205],
Фотометрическое определение ванадия в морских и природных водах в основном проводят после предварительного концентрирования ванадия соосаждением с гидроокисью железа [345, 488, 595] или сорбцией из кислых растворов [841, 1028]. Вападий определяют по реакции с 4-(2-пиридилазо)-резорцином [841], диамин-бензидином [1028], Х-бензоил-Х-фенилгидроксиламином [488], перекисью водорода [595], 8-оксихинолином [345]. Для анализа рассолов и сточных вод предложены также экстракционно-фотометрические методы с Х-фуроил-^Х-фенилгидроксиламипом [399]. и Х-бензоил-Х-фепилгидроксиламипом [409].
Пробу 50 .мл (-0,04л1г V/л) подкисляют 3—6 каплями9 М НС1, добавляют 1—2 капли 0,1 N КМпО4, выдерживают на водяной бане при 60° С ~1 мин., и удаляют избыток окислителя 1—2 каплями 1%-ного раствора нитрита натрия, затем добавляют 2 мл 30%-ного раствора мочевины. Добавляют 10 мл насыщенного раствора хлорида натрия и 9 М НС1 до ~3 М общей кислотности раствора, добавляют 20 мл 0,2%-ного раствора N-бензоил-Х-фенилгидрок-силамина в хлороформе, экстрагируют 1 мин. и разделяют фазы. Органический слой отделяют, фильтруют через вату if измеряют оптическую плотность раствора относительно хлороформа. Продолжительность анализа 10 мип.(бе» выпаривания). При малом содержании ванадия («С 0,02 м.'/л) его соосаж-дают с 50 мг железа(Ш) при pH 6—7. Железо в дальнейшем ходе анализа отделять не требуется [409].
В воде особой чистоты ванадий (до 10-7 %) определяют с сульфоназо [73] или по каталитической реакции окисления Х-фепил-Х-окси-Х'-метилмочевипы броматом калия [263, 264].
ВОЗДУХ
Методом эмиссионной спектроскопии определяют до 10“ €% ванадия в атмосферной пыли [8, 314, 1113].
Воздух просасывают через фильтр ФПП-15 без подложки, фильтр озо-ляют, смешивают золу в отношении 2:1с буферной смесью (1 часть угольного порошка на 9 частей хлорида серебра) и навеску 3 мг сжигают в дуге переменного тока (12 а). Относительная погрешность определения ванадия <10% [8].
137
Описано применение методов атомно-абсорбционной пламенной спектрофотометрии [1135] и активационного анализа [986, 1176] для определения до 0,01 мкг/м3 ванадия в городской пыли.
Пробу пыли, полученную прокачиванием воздуха через фильтр, облучают в реакторе потоком нейтронов (2-ь5)-1012 нейтрон/см2-сек и снимают сразу же после облучения у-спектр пробы. Отмечено аномально высокое содержание ванадия (до 2 мкг/м3) в воздушном бассейне некоторых городов [1176].
ПОЧВЫ
Ванадий в почвах в основном определяют методами эмиссионного спектрального [26, 207, 208, 701, 728, 992, 1060,1134], активационного [293] и атомно-абсорбционного [987] анализа. В зависимости от содержания ванадия и чувствительности метода его определяют либо непосредственно [207, 293, 728, 1134], либо после предварительного экстракционного концентрирования [992, 1060].
Спектральное определение ванадия в почвах обычно проводят путем испарения пробы из кратера угольного электрода в дуге переменного тока. В связи с тем, что колебания состава почв сравнительно малы и практически не оказывают влияния на результаты определения ванадия и других микроэлементов, подавляющее большинство типов почв анализируют используя один комплект стандартных образцов [207, 208].
Образец почвы прокаливают при 450° С, измельчают и помещают в кольцевой кратер (глубина 2 мм, ширина 1 мм, внутренний диаметр 2,5 лы»), выточенный в торцевой части стандартного угольного электрода. Верхний электрод — угольный стержень, заточенный на конус с площадкой диаметром 1 мм. Ток дуги 18—20 а, время экспозиции 1 —1.5 мин. Определяют до 104% ванадия с относительной погрешностью 8—15% [208].
При активационном определении ванадия анализируемую пробу облучают в канале реактора потоком нейтронов 1,8-1013 нейтрон/см2-сек в течение 2 мин. и через 5 мин. проводят идентификацию 62V по периоду полураспада и энергии у-излучения. Предел обнаружения 10 4?о ванадия, погрешность анализа 7% [293].
При фотометрическом определении ванадия пробы почвы спекают и затем ванадий извлекают водой. Перевод ванадия в раствор осуществляют также многократной обработкой почвы смесями различных кислот. Для определения ванадия (^>3-10 3%) используют фосфорновольфраматпый метод [155] или реакцию вапа-дия(У) с капрогидроксамовой кислотой [419]. Для определения ванадия в почве применяют также осциллополярографический метод [302].
СОЕДИНЕНИЯ И СПЛАВЫ ВАНАДИЯ
Ванадий в неорганических соединениях, сплавах и окислах чаще всего определяют титриметрическими методами. Ванадаты растворяют в воде, сплавы и окислы — в различных кислотах. Феррованадий растворяют в разбавленных кислотах — азотной или смеси серной и азотной. При содержании в сплаве более 4 % кремния и более 1 % углерода добавляют еще конц. HF, которую затем удаляют выпаривая раствор с серной кислотой до паров 138
серного ангидрида [350]. Сплавы, содержащие хром и вольфрам, сплавляют со щелочами [860].
При оксидиметрическом определении ванадия в феррованадии [350, 639, 805], сплавах [278, 860, 909] и окислах [242, 313] в качестве титрующего раствора используют соли железа(П) или КМпО4 (см. главу IV). Для получения более точных результатов рекомендуют метод обратного титрования, когда избыток железа(П) титруют раствором бихромата калия [350]. Применяют как визуальный метод регистрации точки эквивалентности, так и потенциометрический с биметаллическими электродами (Pt—) [860]. Так, в феррованадии содержание ванадия определяют потен циометрически согласно ГОСТ 132117.1—67.
Методы комплексонометрического титрования применяют для определения ванадия в двухкомпонентных сплавах и ванадатах [175, 277, 883]. Эти методы удобны в том случае, если одновременно определяют оба компонента: ванадий и свинец [277], ванадий и алюминйй (или никель, кобальт, железо) [175]. Иногда труднорастворимое соединение растворяют непосредственно в титрованном растворе комплексона III. Таким образом определяют содержание ванадия в ванадате свинца [277] и в гексацианоферроате ванадила [864].
Навеску 0,1 г ванадата свинца растворяют при кипячении в смеси 40 мл 0,05 М комплексона III и 30 мл ацетатного буферного раствора с pH 6. Раствор охлаждают и переносят в мерную колбу на 250 мл. К аликвотной части (50 мл) добавляют 5 мл ацетатного буферного раствора с pH 6, 40 мл воды, 0,5 мл дифенплкарбазида (1%-ный раствор в ледяной уксусной кислоте), 15 мл стандартного раствора 0,02 .V соли цинка и титруют избыток цинка 0,05 М раствором комплексона III до перехода окраски от красной до отчетливой светло-желтой. Ко второй аликвотной части (50 мл) добавляют 10 мл раствора аммиака (1 : 1), 40 мл воды, нагревают до 80—90° С и к охлажденному раствору добавляют 7—8 капель раствора кислотного хром темно-синего и титруют раствором соли цинка комплексон III как избыток, введенный при растворении навески, так и освободившийся при разложении комплексоната ванадия. Переход окраски от синей к малиновой отчетлив. Из сопоставления результатов титрования рассчитывают содержание обоих компонентов— ванадия и свинца [277].
В феррованадии после отделения железа щелочью ванадий может быть определен гравиметрическим методом с купфероном [320] или без отделения железа с производными р-нафтойпого альдегида [147]. Рекомендованы также фотометрические методы, основанные на собственной окраске ионов ванадия(У) или вана-дия(1У). В сочетании с дифференциальным способом измерения оптической плотности эти методы дают достаточно точные результаты [538, 539].
Определение ванадия в феррованадии спектральным методом проводят при помощи спектрографа средней дисперсии типа И СП-28 и искровом возбуждении спектра [514, 576].
Генератор высоковольтной конденсированной искры включают по сложной схеме: С = 0,01 миф, L = 0,05 мгн, один разряд за полупериод питающего тока, вспомогательный промежуток 3 мм, аналитический промежуток 2 мм. В качестве подставного электрода используют алюминиевый стержень
139
диаметром 6—10 мм, заточенный на полусферу. Применение угольных или . медных электродов снижает точность определения. Рабочая поверхность кусковых проб или специальных отливок феррованадия должна быть тщательно зачищена, а при необходимости отполирована. Предварительное -обыскривание 80 сек., экспозиция 15—20 сек. Аналитическая пара линий V II 322,69 Fe II 322,78 нм, градуировочный график строят в координатах 'AApe-v — 1g (сге/су), содержание ванадия рассчитывают по формуле
_ 100 - А
V i_|_ /с } , где А — сумма содержания всех элементов примесей (обычно 5—7%), определяемая одновременно с ванадием спектральным методом (данные по углероду вводят по результатам химического анализа). Относительная погрешность определения ванадия —0,6% [514].
Для определения ванадия в фосфатах ванадия и титана применяют активационные методы [89].
КАТАЛИЗАТОРЫ
Общее содержание ванадия в катализаторах на основе пяти-окиси или трехокиси ванадия определяют обычными титриметри-ческими методами. Определение степени окисления ванадия в отработанных катализаторах описано в главе VI. Малые количества ванадия в катализаторах на основе алюминия и кремния определяют фотометрическими методами, основанными на реакциях с перекисью водорода [485] и 1-фенил-3-метил-4-бензоилпиразоло-ном-5 [594]. При анализе катализаторов крекинга нефти и синтеза аммиака типа СА-Р и СА-3 применяют различные физические методы.
Описан [905] метод спектрального анализа катализаторов крекинга нефти.
Навеску катализатора (силикат алюминия) смешивают в отношении 10 : 1 с буферной смесью, состоящей из 90 частей угольного порошка, 10 частей карбоната лития и 0,37 частей окиси германия. Спектр пробы возбуждают в угольной дуге переменного тока (6 а). Метод позволяет определить до 0,001% ванадия с относительной погрешностью 10—15%.
Предложено два варианта рентгепоспектрального метода опре^ деления ванадия и других элементов в катализаторах крекинга нефти [1022]. В первом случае пробу сплавляют с Ы2В4О7 в отношении 1 : 3, во втором готовят плав с отношением компонентов проба : La2O3 : Li2B4O7 = 1 : 1 : 8. Второй способ приготовления пробы позволяет получить более правильные результаты.
1 г полученного плава, проплавленного 10 мин. при 1100° С, измельчают, добавляют 20 мг Н3ВО3 и прессуют таблетку на подложке из 8 г борной кислоты (диаметр таблетки 32 мм, давление 4,5 т/см2'). Измеряют интенсив-
ность линии VKa с помощью проточного пропорционального счетчика и вво-. дят поправку на возможное наложение линии TiK^ [1022].
Рекомендуют определять ванадий в катализаторах синтеза аммиака атомно-абсорбционным методом.
Навеску пробы (—2 г) растворяют в смеси конц. HNO3 и HCI (1 : 3), прибавляют 30 мл этанола (для повышения оптической плотности) и разбав
140
ляют водой до 100 мл. Анализ проводят в пламени закись азота—ацетилен методом добавок. Оптическое поглощение измеряют по линии V 318,4 им. Относительная погрешность определения 1,5—2,5% V не превышает 1% [494].
ЖЕЛЕЗО, ЕГО СПЛАВЫ, СТАЛИ И ЧУГУНЫ
Для определения ванадия в сталях и чугунах широко используют методы эмиссионного спектрального анализа [56, 77, 88, 180, 393, 454, 514, 526, 543, 566, 576, 584, 631]. Определение проводят, как правило, с применением спектральных приборов средней дисперсии с кварцевой оптикой и фотографической регистрацией спектра. Использование квантометров позволяет в течение нескольких минут одновременно определять в пробе содержание целой группы элементов. В качестве источника возбуждения спектра обычно используют искровой и дуговой разряды.
При искровом возбуждении генератор высоковольтной конденсированной искры включают по сложной схеме: С = 0,01 мкф, L — 0,05 мгн, один разряд за полупериод питающего тока, задающий искровой промежуток 3 мм, аналитический 2 мм, подставной электрод — медный или угольный стержень, заточенный на конус или полусферу. Предварительное обыскривание 60 сек. Аналитическая пара линий V II 311,07—Fe II 308,31 нм. В случае применения дуги переменного тока активизатор регулируют на получение одного разряда за полупериод питающего тока, аналитический промежуток 2 мм, ток дуги может меняться от 1,5 до 8 а (обычно 5 а), подставной электрод — медный или угольный, предварительный обжиг 10 сек. Аналитическая пара линий V I 318,34—Fe I 318,49 нм. Дуговой режим обеспечивает большую экспрессность анализа, однако погрешность определения оказывается больше, особенно при повышении содержания углерода в стали.
При спектральном анализе конструкционных и инструментальных сталей, а также сталей специального назначения предъявляются более жесткие требования к точности определения химического состава, но одновременно увеличивается влияние состава и структуры сталей на результаты анализа. Все это вынуждает прибегать к детализации сталей по отдельным группам марок. Каждую группу или даже марку стали анализируют по отдельному градуировочному графику, используя стандартные образцы соответствующего химического состава и термомеханической обработки. Предпочтительно использование жесткого режима высоковольтной конденсированной искры, обеспечивающего большую точность и универсальность анализа. Условия возбуждения спектра аналогичны приведенным выше, однако в случае искрового разряда индуктивность уменьшается до 0,01 мгн, а емкость увеличивается до 0,02 мкф. При искровом возбуждении в качестве аналитической пары используют линии V II 311,07—Fe II 315,42 нм для сталей, содержащих хром, никель, марганец, ниобий. При анализе быстрорежущих и легированных конструкционных сталей в качестве линий сравнения используют Fe II 311,66 и Fe II 308,31 нм соот
141
ветственно, а также аналитические пары линий V II 306,33— Fe II 306,22 нм и V II 310,23—Fe II 307,57 нм.
В случае дугового возбуждения в зависимости от состава сталей используют следующие аналитические пары линий: V II 311,07— Fe II 315,42 и Fe I 311,66 нм; V II 313,03-Fe II 313,54 нм; V I 318,54—Fe I 318,49 нм.
При фотоэлектрической регистрации спектра и применении генераторов с электронным управлением измеряют интенсивность линий V I 411,18 или V I 437,92 нм; линией сравнения служит Fe I 440,77 нм или фон. Условия возбуждения: ток дуги 1,5—3 а, фаза поджига 90°, обжиг 7—10 сек., экспозиция 30—45 сек. Относительная погрешность определения ванадия составляет 1—2%, что в несколько раз лучше, чем при фотографической регистрации спектра.
В высокочистом железе ванадий определяют после отделения основы экстракцией диэтиловым эфиром [225, 791] или изопропиловым спиртом [223] из 6 N НС1.
1 г высокочистого железа растворяют в 20 мл 6 N НС1 и окисляют добавлением 1 мл конц. НА'О3. Раствор упаривают до появления паров А'О2, разбавляют до 40 мл посредством 6 N НС! и экстрагируют железо (!!!) диэтиловым эфиром. В водный раствор добавляют в качестве внутреннего стандарта висмут (0,1 мг) и 100 мг чистого графитового порошка и выпаривают досуха. 30 мг пробы помещают в канал графитового электрода и возбуждают спектр в дуге постоянного тока. Определяют до 1 -10 4% ванадия [791].
Широко применяют атомно-абсорбционные методы определения ванадия в чугунах [694], сталях [420, 694, 789, 800, 843, 849, 1055, 1098, 1133] и различных сплавах [130, 420, 662, 843, 869, 1155], а также рентгеноспектральные методы [393, 500, 623, 875, 1172].
При определении ванадия в низкоуглеродистых легированных сталях и чугунах 0,5 г пробы растворяют в 10 мл HCI (1 : 1), добавляют по каплям 1 мл HNO3 (уд. в. 1,41) и кипятят до появления окислов азота. После охлаждения добавляют 2 мл раствора А1С13 (25 мг Al/мл) и разбавляют водой до 50 мл. (Введение до 1 мг Al/мл в стандартные и анализируемые растворы устраняет влияние состава проб и кислотности растворов на результаты анализа.) Раствор распыляют в пламя закись азота—ацетилен и измеряют оптическое поглощение линии V I 318,54 нм. Воспроизводимость результатов опре-' делений при содержании ванадия 0—0,1; 0,1—0,25; 0,25—2,5% составляет соответственно + 0,002, + 0,01 и +0,07%. Чувствительность метода равна 1,5 мкг \'/мл (на 1% поглощения) [694].
Перед титриметрическим и фотометрическим определением сплавы, чугуны и стали растворяют при нагревании в растворах кислот. Нелегированные стали растворяют в серной, а стали, содержащие вольфрам,— в смеси серной и фосфорной кислот, увеличивая количество последней по мере роста содержания вольфрама. Высокохромистые стали (при последующем отделении хрома) растворяют в соляной кислоте. Для растворения карбидов добавляют азотную или хлорную кислоту, перекись водорода или персульфат аммония. В дальнейшем ходе анализа, как правило, избыток окислителя нежелателен, поэтому растворы после разложения карбидов упаривают с серной кислотой до паров серного ангидрида.
142
Для определения ванадия используют полярографические [25], амперометрические [63, 527] и другие инструментальные методы, но чаще применяют обычное визуальное титрование растворами соли железа(П) или перманганата калия [320, 350, 501, 1076].
Так как большинство легированных сталей содержит хром и вольфрам в количествах, больших, чем ванадий,,то необходимо предварительно их отделить. Согласно ГОСТ 12351—66, при содержании вольфрама более 3% его осаждают в виде вольфрамовой кислоты, а при содержании хрома более 3% предварительно осаждают ванадий в виде купфероната или отгоняют хром в виде хлористого хромила. Влияние меньших количеств вольфрама устраняют фосфорной кислотой или фторидом натрия. Хром(У1) перед титрованием восстанавливают до хрома(Ш).
Чаще всего в качестве титрующего раствора используют сульфат железа(И). Навеску стали растворяют в кислотах и ванадий окисляют на холоду перманганатом калия. После восстановления избытка, окислителя ванадий(У) титруют раствором соли желе-за(П) с любым индикатором, чаще с фенилантраниловой кислотой (ГОСТ 12351-66).
0,05—0,1 г стали растворяют при умеренном нагревании в 40—50 мл смеси кислот (1 л раствора смеси содержит 160 мл H2SO4 (уд. в. 1,84) и 120 мл Н3РО4 (уд. в. 1,7)). По окончании растворения пробы приливают 0,5—1,0 мл HNO3 (уд. в. 1,4) и продолжают нагревание до растворения карбидов и затем до появления паров SO3. Если карбиды не растворились, то добавляют малыми порциями (по 0,1—0,2 г) твердую соль KNO3 или (NH4)2S2O8 и продолжают нагревание еще 2—3 мин. Охлаждают раствор, обмывают стенки колбы водой и вновь упаривают до паров 8О3.'Охлаждают и растворяют соли в 200 мл воды при нагревании. Раствор охлаждают до 10—15° С и по каплям приливают 0,5%-ный раствор КМпО4 до появления устойчивой в течение 3—5 мин. розовой окраски. Избыток окислителя восстанавливают несколькими каплями 2%-ного раствора нитрита натрия, вводя 2—3 капли избытка. Добавляют 2 г сухой мочевины (или 4—5 мл 10%-ного раствора сульфаминовой кислоты); перемешивают, добавляют 80 мл H2SO4 (1 : 1), 2—3 капли 0,2%-ного раствора феиилантраниловой кислоты и титруют 0,02—0,1 N раствором соли железа(П) до перехода окраски индикатора из малиновой в чисто-зеленую.
Титр соли железа устанавливают по стандартным образцам сталей, содержащим хром и вольфрам в количествах, близких к анализируемым. При содержании ванадия в стали более 0,05 % погрешность определения составляет ±0,01% [350]. После определения ванадия в том же растворе может быть определен хром [165].
Точку эквивалентности определяют потенциометрическим [301, 692, 1034] или амперометрическим методом с двумя платиновыми электродами. С амперометрической индикацией точки эквивалентности можно определять 0,02—0,1% ванадия в сталях (ГОСТ 22536.12—77) и 0,05—0,5% ванадия в феррохроме (ГОСТ 21600.15—76) без предварительного отделения хрома.
Перед титрованием ванадия раствором перманганата калия его совместно с хромом восстанавливают сульфатом железа(П), затем избыток восстановителя окисляют персульфатом аммония на холоду (без серебра). Титрование проводят при комнатной температуре.
143
Навеску стали растворяют описанным'выше способом. Раствор упаривают до паров серного ангидрида. Охлаждают, разбавляют водой до 200—250 мл. Приливают титрованный 0,05 N раствор соли железа(П) до перехода окраски раствора в зеленый или бледно-желтый цвет, затем еще избыток 15—20 мл и выдерживают 1—2 мин.^ Приливают 20мл 7,5%-ного раствора персульфата аммония, хорошо перемешивают и через 1—2 мин. медленно титруют 0,05 N раствором КМпО4 на холоду до устойчивой в течение 2 мин. розовой окраски. Индикаторную ошибку определяют в параллельной пробе. Оттитрованный раствор нагревают до исчезновения окраски, охлаждают и вновь добавляют КМпО4 до такого розового окрашивания, как в первой пробе. Индикаторная ошибка для каждой марки стали постоянна, так как обусловлена содержанием элементов, дающих окрашенные ионы [350].
При содержании ванадия в стали более 0,1% погрешность определения с учетом индикаторной ошибки составляет ±0,03% [350, 847]. Предложено несколько вариантов этого метода, позволяющих одновременно с ванадием определять хром [165, 350, 650, 847].
В качестве титрующих растворов при определении ванадия в сталях рекомендованы также феррицианид [534], метиловый оранжевый [176]. Описано комплексонометрическое определение ванадия в мягких магнитных сплавах [1157], но эти методы не имеют особых преимуществ перед описанными ранее.
Для определения ванадия в низколегированных сталях и сплавах широко используют фотометрические методы. В большинстве случаев перед определением основную массу железа отделяют действием щелочи [117, 122, 357, 944, 1107, 1175[. Применяют также хроматографические [201, 638], электролитические [544] и экстракционные [482, 567] методы отделения. В некоторых случаях железо предварительно восстанавливают тиомочевиной [436], аскорбиновой кислотой или маскируют фторид-, фосфат-ионами [40, 194], тиогликолевой кислотой [436].
Ванадий в нелегированных сталях чаще всего определяют фосфорновольфраматным методом [117, 122, 150, 165, 350, 436, 501, 544, 593]. Определение ванадия проводят как с предварительным отделением основной массы железа [117, 122, 457], так и без, отделения. В последнем случае для ослабления мешающего действия железа и других элементов в качестве раствора сравнения используют раствор той же стали без добавления вольфрамата натрия (ГОСТ 22536.12—77). Железо с титаном и молибденом маскируют фторидом натрия. Этим методом определяют от 10“3 до 5% ванадия [544]. Применяют экстракционно-фотометрический вариант этого метода [544, 593], а также используют продукты восстановления гетерополикислоты хлоридом олова [150, 501] или тиомочевиной [436].
Фосфорновольфраматный метод широко используют при анализе ферросплавов. Он рекомендован в качестве стандартного для определения ванадия в ферротитане (ГОСТ 14250—69). При этом основную массу железа и титана предварительно отделяют щелочью. Экстракционно-фотометрический вариант этого метода применяют при анализе феррохрома [13].
144
0,4—0,2 г феррохрома растворяют при нагревании в 20—30 мл H2SO4 (1 : 4), приливают 3—4 мл HNO3 (уд. в. 1,4) и продолжают нагревание до начала выделения паров SO3. Охлаждают, добавляют 20 мл H2SO4 (1 : 4), фильтруют в мерную колбу на 100 мл и разбавляют той же кислотой. Отбирают 5—10 мл раствора, прибавляют 4 мл Н3РО4 (уд. в. 1,7) и 6 мл 5%-ного раствора Na2WO4, добавляют 0,1 N КМпО4 до устойчивой розовой окраски, перемешивают в течение 2—3 мин., восстанавливают избыток окислителя 4%-ным раствором щавелевой кислоты. Через 5 мин. прибавляют 1 мл HNO3 (уд. в. 1,4) и 5—8 мл изобутилового спирта, экстрагируют гетерополикислоту. Определяют содержание ванадия методом стандартных серий или измеряют оптическую плотность раствора органической фазы при 400 нм [13].
Аналогично определяют ванадий в феррониобии после растворения его в 40%-ном растворе фтористоводородной кислоты [607] и феррофосфоре [13].
В виде перекисного соединения определяют ванадий в стеллите, нихроме, ферромарганце, феррохроме, ферровольфраме [350], ферротитане [743].
Для определения ванадия(У) в жаропрочных сплавах, сложнолегированных сталях и чугунах рекомендуют использовать реакции окисления фенилантраниловой кислоты и дифениламинсуль-фоната бария [429], бензидина [350], 2,2'-дикарбоксидифепиламипа [171, 367], диантипирилвинилбензолметана и других производных антипирина [407]. Применение этих реагентов не требует отделения основы, но окислители должны быть предварительно восстановлены, например хром(У1) переведен в хром(Ш). Определения проводят в растворах кислот: 12 М H2SO4 [171], 3,5 М Н3РО4 [407] или их смесях [429]. Перед определением ванадий окисляют перманганатом калия на холоду.
Навеску стали, содержащей 0,1—1,0 мг ванадия, растворяют в 30 мл смеси конц. кислот HNO3 и НС1 в отношении 3:1. Раствор выпаривают с 20 мл H,SO4 (1 : 1) до паров SO3. Прибавляют 20—30 мл воды, 1—2 капли 30%-ного раствора Н2О2 для восстановления хрома в хром(Ш) и нагревают до кипения. Отфильтровывают нерастворившийся осадок. Фильтрат и промывные воды собирают в мерную колбу на 100 мл, отбирают 10 мл, окисляют ванадий 0,1 N КМпО4, добавляя его по каплям до неисчезающего розового окрашивания. Избыток окислителя через 5 мин. восстанавливают 0,05 N NaNO2 и 0,5 г мочевины. Раствор переносят в мерную колбу на 50 мл, приливают 5 мл Н3РО4 (4 : 1) и H2SO4 (1 : 1) почти до метки. Добавляют 8— 10 капель 0,2%-ного раствора фенилантраииловой кислоты и той же серной кислотой доводят до метки. Перемешивают раствор и через 5 мин. измеряют оптическую плотность раствора при 540 нм [429].
Для экстракционно-фотометрического определения ванадия в стали используют М-бензоил-И-фенилгидроксиламин и его производные. Экстракцию окрашенных комплексных соединений проводят из сильнокислых растворов. В присутствии фторида натрия определению мешают только восстановители и окислители, которые разрушают реагенты [400, 513, 618, 625, 718].
Определение ванадия в виде тройного комплексного соединения на основе 4-(2-пиридилазо)-резорцина и перекиси водорода достаточно избирательно в присутствии фторида натрия [40].
Среди новых реагентов, рекомендованных для определения ванадия в сталях, следует отметить Невазол НС [46], сульфонитра-
145
зо [44, 413], гидразиды антраниловой кислоты [159, 234]. Комплексные соединения ванадия с этими реагентами устойчивы к действию многих маскирующих средств, что позволяет определять ванадий без отделения от основной массы железа и легирующих элементов.
Малые количества ванадия в образцах жаропрочных сплавов можно определять с цианформазаном [201] и тимолфталексоном [567]. Так как сплавы содержат значительное количество хрома, вольфрама, титана и молибдена, то определение возможно только после предварительного отделения ванадия от этих элементов.
Для определения ванадия в сталях рекомендованы также реагенты: дифенилкарбазон [122], пирокатехин [588], 8-оксихинолин и его производные [357, 482], 1-(2-пиридилазо)-2-нафтол [1107], кислотный хром темно-синий К. [332], сульфоназо [85], арсеназо [295] и др.
ТИТАН И ПРОДУКТЫ ЕГО ПЕРЕРАБОТКИ
Содержание ванадия постоянно контролируют при производстве титана и его соединений. В титановой губке содержание ванадия колеблется от 0,001 до 0,05%, в титановых сплавах достигает 3,5%, а в тетрахлориде титана по техническим условиям не должно быть больше 0,001%. Для его определения используют спектральные, фотометрические и значительно реже другие методы [1179].
Титан и его сплавы растворяют в разбавленной серной кислоте или в растворе фторида аммония, приготовленном на серной кислоте. Карбиды окисляют азотной кислотой или персульфатом аммония. Ферротитан растворяют в смеси фтористоводородной и азотной кислот. Пробы тетрахлорида титана отбирают пинеткой и медленно, по каплям, вводят при охлаждении в раствор серной кислоты (1 : 1). Затем в большинстве случаев растворы упаривают с серной кислотой до паров SO3.
В титановых сплавах ванадий определяют титриметрическими методами, используя растворы перманганата калия [540] или соли железа(П) [953]. Разработаны стандартные методы определения ванадия (ГОСТ 19863.2—74),’а также ванадия и хрома при совместном присутствии (ГОСТ 19863.3—74), основанные на титровании их раствором соли Fe(II) в сернокислой среде с фецилантраниловой кислотой.
Для анализа титановых материалов применяют фотометрические методы. Определение ванадия (до 5-10~4%) в металлическом титане и тетрахлориде титана осуществляют экстракционнофотометрическим методом в виде фосфорновольфраматного комплекса ванадия. Титан при этом связывают фторидом натрия [190]. Такое же количество ванадия можно определять по окраске его ионов [190, 540, 565].
2 г тетрахлорида титана отбирают градуированной пипеткой и жидкость вводят по каплям в стакан, содержащий 35 мл H2SO4 (1 : 1) и покрытый стеклом. Стакан охлаждают водой. Затем раствор выпаривают до паров SO3. К горячему раствору прибавляют в три приема 0,03 г NaNO3 для окисления
146
ванадия. Раствор охлаждают до ~50° С и доводят объем H2SO4 (уд. в. 1,84)» до 20 мл. Оптическую плотность раствора измеряют при 445 нм относительно раствора тетрахлорида титана, не содержащего ванадий и приготовленного аналогично. Можно использовать и метод стандартных серий [190].
Применяют также методы, основанные на реакции окисления ванадием(У) дифенилбензидина [1008], фенилантраниловой кислоты [61], о-толуидина [506]. Последний реагент используют послн отделения ванадия в виде диэтилдитиокарбамината.
Разработан весьма избирательный метод определения ванадия О 10~4%) в очищенном тетрахлориде титана, возгонах и расплавах титанового производства с ацетонгидразидом антраниловой кислоты (АГАК) [160].
Растворяют 1,2 мл (2 г) тстрахлорида титана в 10 мл 6 A H2SO4 при слабом нагревании в течение 2—3 мин. В охлажденный раствор добавляют 6 мл воды, 1 каплю 0,1 N КМпО4, выдерживают 1—2 мин., добавляют 1 каплю 0,1 N Н2С2О4, 8 мл 0,25%-ного раствора АГАК, 15 мл 50%-ного раствора трихлорацетата натрия и 5 мл хлороформа. Экстрагируют 1 мин. и фазы разделяют в делительной воронке. Фильтруют органический слой через вату и измеряют оптическую плотность при 530 нм [160].
Для определения ванадия в тетрахлориде титана рекомендованы цветные реакции с У-бензоил-К-фенилгидроксиламином [348] и 4-(2-пиридилазо)-резорцином в присутствии перекиси водорода [280, 387, 404] или нитрона [405].
Описаны методы спектрального определения ванадия в металлическом титане и его двуокиси [28, 42, 74, 140, 145, 152, 228, 323, 359, 532, 851, с. 151; 965], а также в тетрахлориде титана [903, 955].
Металлический титан переводят в двуокись, разбавляют угольным порошком в отношении 9:1, добавляют 6% серебра (по отношению к окиси титана в виде 6,6%-ного раствора AgCl в 18%-ном растворе аммиака). Навеску смеси (~33 мг) вносят в кратер угольного электрода типа «рюмки» и спектр возбуждают в дуге переменного тока (6,5 а), экспозиция 45 сек.. Ванадий определяют в интервале концентраций 5-10~3—3-10 х% [152].
До 2-10’5% ванадия определяют в титане после концентрирования примесей и ванадия экстракцией хлороформом в виде пирро-лидиндитиокарбаминатов и дитизонатов [851, с. 151].
Метод атомно-абсорбционной спектрофотометрии используют для определения ванадия в соединениях и сплавах титана [130, 662, 868]. Образцы сплавов растворяют в смесях концентрированных кислот (H2SO4 4- HF, HCI + HF, HCI + HBF4), разбавляют водой и распыляют в пламя закись азота—ацетилен. Определяют от 0,5 до 6% ванадия с относительной погрешностью 2% по линии V I 318,5 нм. Метод использован для аттестации стандартных образцов титановых сплавов [130].
Предложено определять ванадий в виде его оксихлорида в тетрахлориде титана методом ИК-спектроскопии [971].
147
УРАН, ТОРИЙ, ПЛУТОНИЙ И ИХ СОЕДИНЕНИЯ
Для определения ванадия в урановых материалах чаще применяют фотометрические и спектральные методы. Иногда используют титриметрические методы [887].
Перед фотометрическим определением ванадия уран, его сплавы и окислы растворяют в азотной кцслоте или в смесях ее с другими кислотами. Для определения ванадия в урановых соединениях, металлическом уране, плутонии и их сплавах используют экстракционно-фотометрический метод с ]У-бензоил-Ь7-фенилгид-роксиламином. Комплексное соединение экстрагируют из 4—8 М ПС1 хлороформом и измеряют оптическую плотность при 530 нм [24, 878, 1148].
Пробу урана или его сплава растворяют в 15 мл HNO3 (1 : 1) и 5 мл НС1О4 (уд. в. 1,67), раствор упаривают до интенсивных белых паров. Остаток растворяют в воде, добавляют 3—4 капли 0,1 N КМпО4 и кипятят 10 мин., прибавляя НС1 до конечной концентрации раствора 4 М. Аликвотную часть раствора, содержащую 10—200 мкг ванадия, экстрагируют 10 мл 0,1 % -него раствора Х-бензоил-М-фенилгидроксиламина в хлороформе и измеряют оптическую плотность экстракта при 530 км. Калибровочный график строят в присутствии 0,05 г!мл урана. Предел обнаружения составляет <~2-10_?% ванадия [24].
При наличии в пробе более 0,1% железа его маскируют Н3РО4. Одновременно в экстракте можно определить и титан при 420 нм [1148]. Рекомендованы также бенз- и п-метоксигидроксамовая кислоты [868, 1095]. В уране, плутонии и их сплавах, конструкционных материалах ванадий можно определить с пирокатехином после предварительного ионообменного разделения в 6 М HG1. Определению не мешают <3% Mo, 5-102% Со, Cu, Fe, Мп, Th, W, Zr [797].
В металлическом уране, его окислах и солях ванадий определяют без предварительного отделения с ксиленоловым оранжевым [672] и 4-(2-пиридилазо)-резорцином в присутствии Н2О2 [305].
0,25 г урана обрабатывают 1 мл 65 %-него раствора азотной кислоты, выпаривают досуха при слабом нагревании, растворяют в 10 .ил воды и 6 Ял 0,5 М НС1, затем вводят 2,5 мл 1 М NaOH и 5 мл 0,004 М раствора ксиленолового оранжевого, разбавляют до 25 мл и через 2 часа измеряют оптическую плотность при 530 нм относительно раствора контрольного опыта [672].
Для определения ванадия в уране используют фосфорноволь-фраматный метод после экстракционного отделения ванадия в виде купфероната [457].
Подробно описаны методы спектрального определения примесей, в том числе и ванадия, в атомных материалах [182, 225]. Метод фракционной возгонки примесей в дуге постоянного тока широко используют при определении до 10-4% ванадия в металлическом уране и его соединениях [182, 635, 679, 699, 704, 712, 814, 1002, 1021] и до 2-10-3% ванадия в плутонии [182]. В качестве «носителей» применяют Ga2O3, AgCl, NaGl, NaF, AgF и их смеси. Такое же количество ванадия в тетрафториде урана определяют при использовании дугового разряда в аргон-кислородной атмо
148
сфере [991]. Предварительное физическое обогащение с применением испарителя позволяет определять лишь до 10-3% ванадия [308]. В плутонии ванадий и другие элементы определяют методом медной искры после предварительного ионнообменного концентрирования. Предел обнаружения ванадия равен 1-1СГ4% [632].
В ураноферритах ванадий определяют методом фракционной дистилляции, применяя в качестве «носителя» окись галлия или смесь хлорида серебра с фторидом стронция (7 : 3), или методом непосредственного введения раствора примесей в искровой разряд после их экстрагирования дифенилгидроксиламином и купфероном [984]. В последнем случае определяют до 10-Б% ванадия.
Определение ванадия в тории и его соединениях спектральным методом описано в работах [182, 229]. Ввиду сложности спектра тория хорошие результаты можно получить только при использовании приборов высокого разрешения или при отделении основы в виде оксалата. Фильтрат после осаждения тория выпаривают досуха, добавляют окись бериллия и спектр примесей возбуждают в дуге постоянного тока. Предел обнаружения составляет 5-1СГ5% ванадия [229].
АЛЮМИНИЙ, ЕГО СПЛАВЫ И СОЕДИНЕНИЯ
В алюминии, сплавах на его основе и продуктах алюминиевой промышленности ванадий определяют фотометрическими, спектральными, кинетическими методами [824 , 879] и кулонометрическим титрованием [322].
Для определения ванадия рекомендованы различные варианты фосфорновольфраматного метода. В алюминиевых сплавах 0,005— 0,5% ванадия определяют без предварительного его отделения восстановлением гетерополикислоты хлоридом олова(П) согласно ГОСТ 11740-66. В алюминии высокой чистоты 5-10“5% ванадия определяют после предварительного отделения его купфероном совместно с железом [191] или экстрагируют фосфорновольфрамо-ванадиевую гетерополикислоту гексанолом [655].
В щелочных алюминатных растворах ванадий удобно определять косвенным методом с применением о-фенантролината железа [33]. В алюминии, его окислах, сплавах 0,02—5% ванадия определяют экстракционно-фотометрическим методом с К-бензоил-К-фенилгидроксиламином [24, 824].
0,5 г алюминия или его сплава (<5% ванадия) растворяют в смеси 40 мл HNO3 (1: 1), 6 мл НСЮ4 (уд. в. 1,64), добавляют несколько капель конц. HF. Раствор упаривают до интенсивных белых паров, остаток растворяют в воде и далее поступают как при анализе сплавов урана (см. с. 148). Более чем 0,1% железа маскируют фосфорной кислотой. Калибровочный график строят в присутствии 0,0125 г!мл алюминия. Погрешность определения составляет ^3,7% ванадия [24].
В техническом алюминии ванадий(У) определяют по собственной окраске, возникающей в концентрированной серной кислоте, методом стандартных серий согласно ГОСТ 12697—67. Рекомендо
149
ваны также цветные реакции ванадия(У) с о-дианизидином, 3,3'-диаминбензидином и другими реагентами [848], а также вариами-новым синим [423, 731].
При определении ванадия непосредственно в металлическом алюминии и его сплавах спектральным методом в качестве источника возбуждения спектра пробы используют высоковольтный искровой разряд. Предел обнаружения ванадия составляет 0,01 — 0,001% [289, 576], а с применением защитной атмосферы (азот) — 2-10-4% [911]. Анализ соединений алюминия методом сухого остатка [236, 969] или методом фракционной дистилляции примесей из А12О3 в дуге постоянного тока [153, 236] позволяет снизить предел обнаружения ванадия до 10'4—7 ИГ5 %. При анализе окиси алюминия рекомендуют использовать тонкостепные электроды [366а].
Пробу окиси алюминия смешивают с угольным порошком в отношении 2 : 1 п помещают в тонкостенный анод (диаметр канала 3,2—3,5 мм, толщина стенки 0,25—0,3 .v.w). Спектр пробы возбуждают в дуге постоянного тока 12 а. Предел обнаружения ванадия составляет 5-10 5% [366а].
Дальнейшего снижения предела обнаружения ванадия в алюминии и его соединениях достигают использованием предварительного химического концентрирования элементов-примесей экстракцией [851] или осаждением основы в виде хлорида газообразным НС1 [225] и последующим спектральным анализом концентрата примесей.
МЕТАЛЛЫ ОСОБОЙ ЧИСТОТЫ И ИХ СОЕДИНЕНИЯ
Для определения ванадия в веществах особой чистоты .используют главным образом методы спектрального, атомно-абсорбционного, активационного и фотометрического анализов. В некоторых случаях применяют и другие методы определения ванадия.
Спектральные методы анализа позволяют одновременно с ванадием определять и другие элементы-примеси [182, 225, 477, 526, 11381.
Бериллий и его соединения предварительно переводят в окисел.
0,25 г окисла бериллия смешивают с 0,192 г буферной смеси (двуокись олова, гидроокись бария и угольный порошок (0,1 : 10 : 33,0)) и помещают в канал графитового электрода глубиной 7 и диаметром 4 мм. Спектр возбуждают в дуге постоянного тока (проба является анодом, ток дуги 12 а, межэлектродный промежуток 4 мм, экспозиция 35 сек.). Предел обнаружения ванадия составляет 1-10 3%, погрешность определения 10% [892].
Экстрагирование основного ацетата бериллия хлороформом с последующим возбуждением спектра концентрата примесей в дуге постоянного тока снижает предел обнаружения до 5-10~5% (при коэффициенте обогащения, равном 20—25). Погрешность метода не хуже 20% [227].
Висмут гредварнтельно гереводят в Bi2O3.
15С
3—5 г висмута растворяют в 10—15 мл! N HNO3, выпаривают досуха, прокаливают полчаса при 550° С и смешивают со стекловидной формой окиси бериллия в отношении 5:1. 120 мг полученной смеси помещают в кратер угольного электрода и возбуждают спектр в дуге постоянного тока (12 а). Проба служит анодом, экспозиция 2 мин., аналитическая линия V 318,5 нм. Предел обнаружения ванадия составляет 2-10-4%. Он может быть снижен до 2-10 6%, если предварительно отделить висмут осаждением в виде иоди-да [260].
Вольфрам окисляют до WO3, смешивают с равным количеством угольного порошка, содержащего 0,3% хлорида натрия. 50 мг смеси помещают в кратер угольного электрода и возбуждают спектр в дуге постоянного тока (7 а). Определяют до 1 -10-4% ванадия с погрешностью ~10% [725, 866J. Аналогичные результаты получены и при использовании в качестве буферной смеси иодида калия и фторида натрия в отношении 4 : 1 [392].
В металлическом галлии и его арсениде ванадий совместно с другими элементами определяют в концентрате примесей после отделения основы экстракцией изопропиловым эфиром из 8 N НС1. Концентрат примесей анализируют методом сухого остатка [9601 или испарением из кратера угольного электрода [959]. Предел обнаружения ванадия в обоих случаях не хуже 1 -10'5%.
Германий или двуокись германия анализируют следующим образом [225, 226].
6 г металлического германия растворяют в 100 мл смеси конц. НС1 и конц. HNO3 (4 : 1) и при постепенном нагревании отгоняют тетрахлорид германия. В случае анализа двуокиси германия соответствующую навеску пробы обрабатывают 90 мл смеси конц. НС1 и конц. HNO3 (8 : 1). К 5—10 мл оставшегося после дистилляции германия раствора приливают 5 мл конц. HNO3, вводят 173 л/г двуокиси германия (полученной из исходной пробы), дважды выпаривают при добавлении азотной кислоты (по 5 мл), высушивают и остаток прокаливают при 500° С. 40 мг пробы помещают в кратер угольного электрода и возбуждают шГектр в дуге переменного тока (12 а), экспозиция 2 мин. Определяют по линии V I 268,8 нм до 2-Ю 5% ванадия. Предел обнаружения ванадия может быть снижен до 2-10~6% при использовании полной отгонки тетрахлорида германия и возбуждения спектра сухого остатка концентрата примесей на торце угольного электрода в искровом разряде [226].
Иттрий, лантан и редкоземельные элемент ы анализируют в виде окислов. В окиси лантана, а также других РЗЭ определяют до 2-10“4% ванадия, возбуждая спектр пробы (смешанной в отношении 1:1с угольным порошком) в дуге постоянного тока (15 а) [477, 771]. Предел обнаружения ванадия может быть снижен до 10-5% при использовании метода фракционной дистилляции примесных элементов в дуге переменного тока (15 а) и добавок полимерных фторуглеродов к пробе в качестве термохимического буфера. Оптимальное соотношение компонентов анализируемых смесей (окиси РЗЭ, угольного порошка, фторопласта-3) при анализе окислов иттрия и лантана составляет 1 : : 0,1 : 1, а при анализе окислов церия и европия — 1 : 0,5 :1 [187].
Кальций предварительно переводят в окисел. Окись кальция разбавляют в отношении 1 : 9 угольным порошком и помещают
151
в кратер угольного электрода (анода). Спектр пробы возбуждают в дуге постоянного тока (10—12 а). Определяют до 10~3% ванадия с погрешностью не хуже 5% [747].
Литий и другие щелочные элементы. При анализе лития и его сплавов образцы растворяют в специальном сосуде в этаноле или воде и фильтруют. Осадок па фильтре растворяют в небольшом количестве конц. HNO3 и методом пористого электрода определяют до 1СГ3% ванадия [968]. Снижение предела обнаружения ванадия до 10-6% достигается предварительным концентрированием примесей экстракцией, выпариванием экстракта на угольном порошке (коллектор) и возбуждении спектра полученного концентрата в дуге постоянного или переменного тока. Используют экстракцию (при pH 5,5—6,0) диэтилдитиокар-баминатов и 8-оксихинолинатов примесей хлороформом при анализе щелочей особой чистоты [272] и солей редких щелочных металлов [525]. Применяют также в качестве групповых экстрагентов 1-феиил-3-метил-4-бензоилпиразолон-5 [204, 205, 459], 1-(2-пириди-лазо)-2-нафтол [203, 603].
Марганец. 20 мг МпО2 помещают в кратер графитового анода. Возбуждают спектр в дуге постоянного тока с графитовым катодом при 10 а. Предел обнаружения ванадия ~0,01% [9191. Более высокая чувствительность достигается при испарении из кратера анода смеси окисла марганца с угольным порошком [817].
Медь. При анализе электролитически чистой бескислородной меди ванадий в количествах 1-10~Б—1,5- 1СГ4О/6 определяют по линии VII 268,8 нм, а при больших содержаниях (10-4—1,5-10~3%)—-по линии V II 268,9 нм, в обоих случаях линией сравнения служит линия Си I 287,1 нм.
Пробу меди 2 г прессуют в брикет определенной формы, помещают в углубление угольного электрода, закрепленного в охлаждаемом водой держателе, и анализируют в дуге постоянного тока с графитовым противоэлектро-дом при 15 а в течение 1,5 мин. [1003]. Возможно химическое концентрирование элементов-примесей. При этом основное количество меди выделяют электролизом на платиновом катоде в азотнокислой среде при токе 2,5—3 а и напряжении 2,5—3 в. Затем электролит выпаривают, остаток прокаливают при 550° С и анализируют в дуге постоянного тока с угольными электродами [34].
Молибден. При определении ванадия в молибдене применяют следующую методику.
0,5 г молибдена растворяют в 15 мл смеси конц. HNO3 и конц. НС1 (1 : 3) при нагревании, разбавляют водой до 50 мл, помещают в делительную воронку и приливают 10 мл насыщенного спиртового раствора а-бензоинокси-ма. Образующийся осадок флотируют 25 мл хлороформа. Операции осаждения и флотации повторяют. Кислый раствор примесей упаривают с добавлением нескольких порций (по 1 мл) азотной кислоты, добавляют раствор нитрата бериллия, содержащий 100 мг бериллия, упаривают, высушивают и прокаливают вначале при 250° С, затем 30 мин. при 500° С. Относительное обогащение примесей 5—10 раз. 60 мг приготовленной пробы помещают в кратер угольного электрода (анода) и возбуждают спектр в дуге постоянного тока (12 а). Экспозиция 2 мин. Предел обнаружения ванадия составляет 2-10 [230].
152
Ванадий определяют в молибдене и его трехокиси при использовании слаботочной дуги постоянного тока в аргоновой атмосфере [930].
При определении ванадия в молибдате аммония особой чистоты его предварительно концентрируют соосаждением с фосфорномо-либденовой гетерополикислотой.
50 г (NH4)2Mo04 растворяют в 200 мл воды, подкисляют 3 ТУ НС1 до pH 2 и добавляют 0,2—0,25 ммоля дигидрофосфата аммония (в виде 0,5%-ного раствора NH4H2PO4). Встряхивают 3 мин., центрифугируют в течение 5 мин., осадок высушивают при 100° С и взвешивают. Концентрат разбавляют угольным порошком в отношении 1:1, помещают в кратер угольного электрода типа «рюмка» и возбуждают спектр в дуге переменного тока (10 а). Предел обнаружения ванадия по линии 318,40 нм составляет 1-10-5% [565а].
Отмечается, что с повышением содержания элементов-примесей (ухудшением степени очистки препарата) степень соосаждения ванадия уменьшается до 70%.
Никель. Пробу окиси никеля помещают в графитовый анод типа «рюмка» и сверху покрывают слоем (~1 мм) угольного порошка. Спектр пробы возбуждают в дуге постоянного тока (9 а), межэлектродный промежуток 2 мм. Предел обнаружения ванадия 10~3% при относительной погрешности 18—20% [304]. Уменьшение погрешности определения до 8—16% достигается при использовании буферной смеси окиси индия и угольного порошка (1 : 2), которой разбавляется проба (1 : 1). Спектр возбуждается в дуге постоянного тока (10 а) [831]. Предел обнаружения ванадия может быть снижен до 3-10~4% путем предварительного отделения никеля флотацией его диметилглиоксимата хлороформом [225].
Ниобий. Ванадий в ниобии определяют после перевода его в пятиокись, которую смешивают с угольным порошком в отношении 1 : 1 и анализируют двумя методами: непосредственным испарением из угольного анода или просыпкой пробы в разрядное облако дуги переменного тока. В первом случае достигается предел обнаружения ~3-10~3%, во втором ~1-10-2% [358]. Снижение предела обнаружения до 3-1СГ4% достигается применением носителей — хлорида натрия [189] или смеси хлорида и фторида серебра (1 : 1) [877], а также при возбуждении спектра смеси пробы с угольным порошком (4 : 3) в дуговом разряде в контролируемой атмосфере (70% аргона -% 30% кислорода) [335].
Олово. Для определения ванадия в олове применяют следующую методику.
0,5 г SnO2 сплавляют с 1 г Na2CO3 и 1 г NagBO^, плав растворяют в 25 мл HCI (1 : 4), добавляют 5 мл 1-10 2%-ного раствора соли молибдена, служащего внутренним стандартом, и разбавляют водой до 50 мл. Одну каплю полученного раствора наносят на торец угольного электрода диаметром 6,4 мм. Спектр возбуждают в угольной дуге переменного тока (4 я). На каждую экспозицию используют два таких электрода. В интервале концентраций 0,001—0,05% погрешность определения ванадия 8—20% [969].
Аналогичная чувствительность определения ванадия достигается и при непосредственном анализе двуокиси олова, а концентрирование примесей путем отгонки хлорида олова из солянокисло
153
го раствора в присутствии перекиси водорода позволяет снизить предел обнаружения до 5-10-5% ванадия [225].
Ртуть. Для определения ванадия и других элементов применяют метод, описанный ниже.
Через 25 .и.г ртути при 150° С в течение 8 час. пропускают воздух и растворяют получившиеся окисли в 40 мл 0,8 TV HNO3 двукратным распылением ртути. К полученному раствору добавляют 2 мл 18 М H2SO4, 10 мл раствора, содержащего 1 мг/мл Fe2O3, выпаривают досуха, высушивают и прокаливают при 600° С. Остаток смешивают с (NH4)2SO4 в соотношении 1 : 4, измельчают и прессуют четыре таблетки. Спектр концентрата возбуждают в дуге постоянного тока (7 а) при использовании медных электродов [698].
Разработан метод анализа ртути с предварительным концентрированием примесей, основанный на растворении ртути в HNO3 и последующем селективном восстановлении ее [376].
50 г ртути растворяют в конц. HNO3, затем восстанавливают ее до металла добавлением 55 мл конц. муравьиной кислоты. Водный раствор примесей упаривают с 49 мг угольного порошка и высушивают под ИК-лампой. Полученный концентрат помещают в кратер угольного электрода и возбуждают спектр в дуге переменного тока 16 а. В канал верхнего угольного электрода помещают угольный порошок, содержащий 8% хлорида натрия. Предел обнаружения ванадия составляет 1-10 7% при средней квадратичной погрешности <20% [376].
Св и н е ц. Большую группу элементов-примесей, в том числе и ванадий, определяют в свинце и его соединениях с применением предварительного химического обогащения.
Свинец растворяют в HNO3 (1 : 1), осаждают в виде сульфата действием H2SO4 (1:1) при нагревании (~100° С). Раствор декантируют, а осадок кипятят еще 2 час. с HN03 (1 : 1). Растворы объединяют, добавляют 3—4 мл суспендированного осадка и выпаривают до полного удаления паров H2SO4. Остаток прокаливают 30 мин. при 550° С, смешивают с угольным порошком в соотношении 5:1, навеску полученной пробы (100 мг) помещают в кратер анода и возбуждают спектр в дуге постоянного тока 5а. Предел обнаружения ванадия без обогащения (основа в виде PbSO4) 2-10*4, а после обогащения (при коэффициенте обогащения 40) 2-10'6% [65, 225].
Т а н т а л | анализируют в виде пятиокиси, которую получают прокаливанием металлической стружки при 800° С.
Полученную пятиокись тантала смешивают в отношении 2:1с угольным порошком, содержащим 2,5% хлорида натрия, и тщательно растирают в течение 5—10 мин. Пробу помещают в кратер угольного электрода (диаметр 1,3, глубина 3 мм) и возбуждают спектр в дуге переменного тока 10 а. Предел обнаружения ванадия 4-10 4% [189]. Применение в качестве спектроскопического буфера смеси, состоящей из 30% порошкообразного серебра, 20% фторида бария и 50% хлорида серебра, позволяет снизить предел обнаружения ванадия лишь до 10 4% [876]. Использование метода просыпки-вдуванпя [358, 359] или закрепление (приклеивание) небольших количеств пятиокпсп тантала к плоским торцам электродов с последующим возбуждением спектра в дуге переменного тока [687] позволяет определить только (2 -е 3) • 10‘ s% ванадия.
Предложено концентрировать примеси путем экстракционно-хроматографического отделения тантала трибутилфосфатом, закрепленном на пористом фторопласте-4, из растворов 1ЛГ по HF и HNO3.
154
Элюат, содержащий ванадий и другие примеси, упаривают на 50 мг угольного порошка, содержащего 2 мг NaF. Спектр концентрата пробы возбуждают в дуге постоянного тока 15 а. Предел обнаружения ванадия составляет 1-10'^% [132].
Хром растворяют в 6 X НО, добавляют 60 %-ный раствор НСЮ4 и отгоняют основу в виде хлористого хромила СгО2С12 [225, 790]. При концентрировании примесей на коллекторе из сульфата стронция с последующим возбуждением спектра в дуге постоянного тока (12 а) предел обнаружения ванадия составляет 5-Ю"4% [225]. Аналогичная чувствительность получена и при анализе раствора после отгонки хрома методом пористого электрода или методом вращающегося дискового электрода с использованием высоковольтного искрового разряда [790].
Цинк перед анализом растворяют в концентрированных кислотах HNO3 или НС1.
1 г пробы растворяют в 4 мл конц. HNO3, вводят 1 мг алюминия в виде нитрата и осаждают раствором аммиака гидроокись алюминия. Осадок отфильтровывают, растворяют в минимальном количестве азотной кислоты и повторяют осаждение раствором аммиака. Концентрат примесей на гидроокиси алюминия растворяют в нескольких каплях азотной кислоты и выпаривают на 20 мг угольного порошка, содержащего 7% хлорида натрия. Полученную пробу помещают в кратер угольного электрода и возбуждают спектр в дуге переменного тока 9 а. Предел обнаружения ванадия составляет 1 -10 5% [503].
Анализ раствора, полученного растворением цинка в конц. ИС1 с последующим упариванием до удельного веса 1,95 г/см9, методом сухого остатка позволяет обнаружить до 10~4% ванадия [377].
Металлический цирконий анализируют с использованием графитового электрода в дуговом [736] или искровом [727] разряде. Предел обнаружения ванадия в обоих случаях составляет 2-10“3%. Предел обнаружения ванадия можно снизить до 1-Ю"3—5-10“4% путем перевода металлического циркония в двуокись (прокаливание порошка металла при 1000° С), применения метода фракционной дистилляции с носителем (25% хлорида серебра или 20% фторида бария) и возбуждения спектра в дуге постоянного тока 12 а [182, 392, 476, 965]. При возбуждении спектра двуокиси циркония в газовом разряде с полым катодом определяют до 1-10“3% ванадия [182]. Предварительное отделение основы флотацией осадка миндальнокислого циркония изоамиловым спиртом с последующим концентрированием примесей на коллекторе из сульфата свинца и возбуждением спектра в дуге постоянного тока (12 а) позволяет снизить предел обнаружения ванадия до 3-10"4% [182, 476]. При экстракции примесей хлороформом в виде пирролидиндитиокарбаминатов и дитизоиатов и возбуждении спектра сухого остатка концентрата на торце электрода в искровом разряде определяют до 2-10-5% ванадия [182, 851].
Разработаны методы спектрального определения ванадия и других примесей в молибдатах натрия и редкоземельных элементов [4301, в титанате бария [28], титано-цирконатах свинца [590], марганцево-цинковых ферритах [67]. Во всех случаях используется
155
дуга постоянного тока (10—12 а), проба смешивается с угольным порошком (обычно в отношении 1 : 1)ииспаряетсяиз кратера угольного электрода. Определяют до 1-10"3—5-10“4% ванадия с погрешностью 10—20%.
Активационные методы анализа использованы для определения ванадия в алюминии [711], вольфраме [1046], графите [70, 292], железе высокой чистоты [1108], ниобии [863], титане и его сплавах [70, 258, 732], фосфатах ванадия и титана [89].
При определении ванадия в фосфатах ванадия и титана навеску пробы 0,5 г помещают в полиэтиленовую ампулу, которую затем завертывают в медную фольгу. Для облучения проб и стандартных образцов использован генератор быстрых нейтронов (Еп 14 Мэе, поток нейтронов ~107 нейтрон! см2-сек). Через 1 мин. после конца облучения (время облучения 15 мин.) измеряют в течение 200 сек. активность 61Ti по у-пику 320,1 кэв на у-спектро-метре с 1024-канальным анализатором и Се(Ы)-детектором. Для устранения погрешности определения за счет непостоянства нейтронного потока используют способ мониторирования, который проводят по измерению наведенной удельной активности медной фольги. Учет мешающего влияния реакции 54Сг (и, a)51Ti проводят по у-пикам с энергией излучения 990,0 и 835,0 кэв. Погрешность определения ванадия <4% [89].
Ме'год атомно-абсорбционной спектрофотометрии использован для определения ванадия в никелевых сплавах [1155], соединениях и сплавах титана [130, 662, 869].
При анализе никелевых сплавов навеску пробы 0,5 г растворяют в 5 мл конц. HNO3 и 3 мл конц. НС1, упаривают, растворяют остаток в 5 мл конц. НС1 и 2 мл конц. HF и разбавляют водой до 100 мл. Раствор распыляют в пламя закись азота—ацетилен и измеряют оптическую плотность по линии V 318,5 нм. Оптимальная концентрация ванадия в анализируемом растворе составляет 100 мкг/мл. При меньших содержаниях ванадия используют предварительную экстракцию примесей метилизобутилкето-ном [1155].
Для определения ванадия в различных металлах, сплавах и соединениях используют спектрофотометрические методы с предварительным концентрированием ванадия или отделением основы.
В карбидах вольфрама, титана и кобальта, окислах вольфрама определяют до 1-10“4% ванадия фосфорновольфраматным методом, экстрагируя гетерополикислоту изобутанолом [593]. Этим же методом определяют ванадий в цирконии после экстракционного отделения ванадия в виде диэтилдитиокарбамината из фторидных растворов при pH 1 [568].
Определение ванадия в металлах, карбидах и окислах вольфрама, ниобия, тантала, титана [1068], сплавах на основе железа, кобальта никеля, хрома [539] проводят по собственной окраске ионов ванадия(У) после предварительного отделения основы. Отделение проводят либо осаждением купферонатов с последующим озолением и сплавлением осадка со щелочами и выщелачиванием ванадия водой [539], либо сразу сплавляют анализируемые пробы с окислительным щелочным плавнем и растворяют его в воде в присутствии перекиси водорода [1068].
156
Т'
В ниобии и тантале ванадий определяют экстракционно-фотометрическим методом с К-бензоил-Г^-фенилгидроксиламином [718].
0,1—0,5 г металла растворяют в 8 мл 12,5 М H2SO4, 2 мл 2,5 М НС1 и 4 мл конц. HNO3 при нагревании. Раствор упаривают до паров SO3. Операцию упаривания повторяют цри добавлении 1 мл конц. H2SO4. Остаток растворяют в 5 мл воды и 8 мл 2,5 М HF, приливают 4 мл 10%-ного раствора (NH4)2Fe(SO4)2, разбавляют до 40 мл, вносят 5 мл 10%-ного раствора (NH4)2S2O8, доводят общий объем до 50 мл и экстрагируют ванадий 10 мл 0,1 %-него раствора БФГА в хлороформе в течение 2 мин. Экстракцию повторяют еще три раза, применяя 5, 3 и 3 мл реагента соответственно. Объединенный экстракт разбавляют до 25 мл хлороформом и измеряют оптическую плотность при 475 нм [718].
В металлическом галлии определяют до 10“3% ванадия с сульфоназо после предварительного отделения основы экстракцией диэтиловым эфиром [73]. Этот же реагент применяют для определения ванадия в анодных порошках, выделяемых из сплавов на основе титана и никеля [120].
Для определения ванадия используют также косвенные методы. Так, в хроме определяют 0,01—0,001 % ванадия с применением ферроортофенантролина после отделения ванадия совместно с железом в виде купферонатов [606, 607]. В меди и цинке ванадий определяют косвенным молибденороданидным методом после предварительного отделения избытка фосфорномолибденовой кислоты экстракцией диэтиловым эфиром [265].
Предложены кинетические методы определения ванадия в металлах особой чистоты: магнии и его соединениях [261], рении и его препаратах [446], титане [591], карбонате кадмия [262], окислах алюминия [824, 879], нитрате уранила [1136], галогенидах щелочных металлов [41, 262], минеральных кислотах [261, 262], воде особой чистоты [263].
Ванадий в сплавах на основе ниобия [518] и бора [909] можно определить титриметрическим методом с перманганатом калия.
ДРУГИЕ ОБЪЕКТЫ
Ванадий в элементном боре, борном ангидриде ибо р ной кислоте определяют спектральным методом анализа.
5 г бора растворяют в 60 мл конц. HNO3 с добавлением 10 мл конц. HF при осторожном нагревании. В случае анализа борной кислоты или окиси бора соответствующую навеску пробы смачивают только водой. К охлажденному раствору добавляют 200 мг угольного порошка, 10 мг нитрата натрия, 10 мл 30%-ного раствора HF и 10 мл конц. HNO3. Упаривают до полного удаления паров кислот и повторяют 4—5 раз обработку пробы смесью-HN03 и HF. Остаток прокаливают при 350° С в течение 10 мин. и 35 мг пробы загружают в кратер угольного электрода. Спектр возбуждают в дуге постоянного тока (12 а), экспозиция 1 мин. Предел обнаружения ванадия составляет 1-10-в% [66].
В карбиде кремния прямым спектральным методом определяют до 1 • 1СГ5% ванадия [402]. В кварце и оптической двуокиси кремния определяют до 10“4% ванадия, возбуждая спектр
157
пробы в дуге постоянного тока (10 а) [365]. Ванадий в тетрахлориде кремния и трихлорсилане определяют спектральным методом в концентрате, полученном после отгонки кремния с трихлорметаном. Предел обнаружения составляет 1-10“8% [1142]. При анализе элементного кремния основу отгоняют в виде тетрафторида в токе HF и HNO3. Примеси концентрируют на сульфате стронция и определяют в дуге постоянного тока до 2-10“5% ванадия [225, 477].
В элементном иоде ванадий определяют спектральным методом после отгонки основы.
2—12 г измельченного иода смешивают с 20 мг угольного порошка, содержащего 4% хлорида натрия, и возгоняют иод при 60—65° С. После окончания возгонки пробу выдерживают при 80° С в течение 30 мин. 20 мг полученного концентрата помещают в кратер угольного электрода и возбуждают спектр в дуге переменного тока 10 а. При исходной навеске 12 г предел обнаружения ванадия составляет 3• 10~7% [408].
Аналогичные результаты получены при анализе иодистово-.дородной кислоты. Примеси предварительно концентрировали экстракцией в виде 8-оксихинолинатов хлороформом из навески 50 г. Экстракт озоляют на угольном порошке и возбуждают спектр концентрата в дуге переменного тока 16 а [271].
При анализе селена определяемые элементы, в том числе и ванадий, экстрагируют в виде 8-оксихинолинатов и дитизонатов хлороформом последовательно при pH 3, 5, 7 и 9. Объединенные экстракты примесей упаривают до минимального объема, остаток наносят на торец угольного электрода и спектр возбуждают в высоковольтном искровом разряде. Определяют до 10-5% ванадия [851]. Такой же предел обнаружения ванадия достигнут и при предварительной отгонке селена из сернокислого раствора с последующим возбуждением спектра концентрата примесей в дуге постоянного тока 13 а [823].
Веере ванадий определяют спектральным методом после отгонки основы.
Навеску пробы (~1 г) нагревают на воздухе при 300° С, остаток растворяют в небольшом количестве 6 М НС1 и добавляют НУлсг смеси графитового порошка с 10% хлорида натрия. После высушивания пробу помещают в кратер угольного электрода и возбуждают спектр в дуге постоянного тока 13 а. Предел обнаружения ванадия составляет 5 • 10~7% [822].
Прямым спектральным методом определяют в графите или уг о л ь ны х э ле кт р о д а х до 3-10"3% ванадия [895]. Использование метода прикатодного слоя снижает предел обнаружения ванадия до1 • 1О"3°о ,что было использовано при анализе радиоактивного графита [1154]. Указывается, что добавление свинца или его солей к графитовому порошку обеспечивает быстрое испарение ванадия и титана и предел обнаружения ванадия снижается до 10“6% [1066]. Разработаны методы анализа углерода с предварительным концентрированием примесей (сжигание основы в токе воздуха при 900° С) и спектральным анализом концентрата [1091], я также при наложении магнитного поля на дуговой разряд [946].
Глава VI
ОПРЕДЕЛЕНИЕ ВАЛЕНТНОГО СОСТОЯНИЯ ВАНАДИЯ
Очень сложной и важной задачей является определение степени окисления и форм соединений ванадия в промышленных объектах. Эта задача в основном решена только для простых объектов: окис-лов, катализаторов и стекол. Обзор методов определения дан в [158].
Степень окисления ванадия определяют физическими методами без разрушения образцов. Стекла анализируют методами абсорбционной и ИК-спектроскопии [142, 456], aj окисли — методами рентгеновской спектроскопии высокого разрешения [767].
Чаще всего для определения валентного состояния ванадия используют титриметрические и фотометрические методы, применение которых возможно только после растворения образцов. При переведении их в раствор необходимо применять особые меры предосторожности для сохранения истинного соотношения степеней окисления ванадия. Растворы защищают от действия кислорода воздуха и предотвращают возможные^ процессы межионного взаимодействия, например
V(V) + V(III)-*V(IV).
Окислы ванадия(1¥) и ванадия(У) избирательно растворяют в щелочи или аммиаке и только после этого остальные окислы растворяют в кислотах в инертной атмосфере [242, 313, 380, 585, 867].
Для переведения в раствор анализируемых образцов используют также другие растворители, например раствор комплексона III в ЗМ Н3РО4 [134]. Продукты восстановления окситрихлорида ванадия последовательно извлекают горячими растворителями: VClg — водой, VC12 — насыщенным раствором хлорида калия,. VOC1 — 10%-ным раствором оксалата аммония в 5 %-ной щавелевой кислоте, V2O3 — 5%-ным раствором азотной кислоты [460].
Для растворения различных окислов ванадия и катализаторов используют также титрованные растворы восстановителей [473] или окислителей [199]. При всех способах растворения анализируемых образцов в водных растворах возможно одновременное существование только следующих комбинаций ионов: V(V) и V(IV); V(IV) и V(III); V(III) и V(II).
159
Таким образом, анализ растворов сводится к определению двух различных ионов ванадия при их совместном присутствии или после их предварительного разделения. Обзор методов разделения приведен в работе [564] и в главе III.
ОПРЕДЕЛЕНИЕ ВАНАДПЯ(1У) И ВАНАДИЯ(У)
Для титриметрического определения чаще всего используют растворы соли железа(П) и перманганата калия с визуальной или инструментальной индикацией точки эквивалентности.
Прямым титрованием определяют только содержание ванадия одной степени окисления. Затем находят общее содержание ванадия после дополнительной операции окисления или восстановления всего ванадия в зависимости от избранного титрующего раствора. Эти методы опробованы при анализе солей [926, 1011], ванадиевых катализаторов [670], окислов [313], ванадиевых стекол [242, 259], технического тетрахлорида титана [510].
2—3 г измельченного катализатора кипятят 30—40 мин. в H2SO4 (1 : 1), охлаждают, разбавляют водой до 200—250 мл. Аликвотную часть полученного раствора (50—100 мл) разбавляют водой до 400 мл, подкисляют 5 мл Н3РО4 (уд. в. 1,70) и через 1 мин. титруют ванадийДУ) 0,1 N раствором К.МпО4 до розового окрашивания. Вводят 20 мл раствора железа(П) [смесь 183 г FeSO4-7H2O, 100 мл H2SO4 (уд. в. 1,84) и 2000 мл воды], окисляют избыток железа 10 мл 10%-ного раствора персульфата аммония и через 2 мин. титруют весь ванадий 0,1 ТУ раствором КМпО4. Содержание ванадия(У) находят по разности [670].
Для определения ванадияДУ) и ванадия(У) в их смесях и раздельно предложен один титрующий раствор — сульфат ванадия(П), который в зависимости от кислотности среды и индикатора может быть использован для титрования как ванадия(У), так и ванадияДУ) [689]. ВанадийДУ) можно титровать раствором соли железа(П) в 10,5 М Н3РО4 [1011]. Предложены методы амперометрического титрования с двумя индикаторными электродами [167, 474]. В качестве титрантов рекомендованы сульфат железа(П) [167] и ферроцен [471]. Разработаны методы потенциометрического и фотометрического определения V(V) и УДУ), основанные на их взаимодействии с 8-меркаптохинолином 191].
Описан метод, сочетающий фотометрическое определение общего содержания ванадия в щелочной среде при 270 нм с титриметрическим определением ванадияДУ) [1036].
Для совместного определения ванадияДУ) и ванадия(У) предложены различные фотометрические методы. При pH 1 ванадий^) взаимодействует с тайроном и может быть определен в присутствии 10-кратного количества ванадияДУ) [561]. Содержание ванадияДУ) в пятиокиси определяют косвенным путем: в щелочной среде окисляют ванадийДУ) с помощью соли железаДП) и эквивалентное ванадиюДУ) количество железа(П) определяют с о-фенантролином [97]. Описаны экстракционно-фотометрические методы определения ванадия(У) в присутствии ^>500-кратного количества ванадияДУ) с 1-фенил-3-метил-4-бензоилпиразоло-160
ном-5 [594]. Определению не мешают большие количества многих элементов.
Оба иона ванадия определяют после их предварительного разделения экстракционными, электролитическими и хроматографическими методами [560, 563, 564]. После экстракции раствором трибутилфосфата в керосине ванадий(У) непосредственно определяют в органической фазе с бензгидроксамовой кислотой, а ва-надий(1У) — в водном слое с кислотным хром темно-синим К [560].
Для определения ванадияДУ) и вападия(У) рекомендован экстракционно-фотометрический метод с N—бензоил-N-фенилгидроксиламином [126, 128[.
Для определения ванадия(У) 0,1—0,5 г материала растворяют в 20 мл 4 N H2SO4 в течение 20—30 мин. в атмосфере СО2. Охлаждают, разбавляют 4А' H2SO4 до 100 мл. К 5—10 мл раствора в делительной воронке приливают 5 мл 4 N H2SO4, 1 мл 0,1 М спиртового раствора БФГА, 4 мл этанола и разбавляют водой до 20 мл. Прибавляют 5 мл хлороформа, встряхивают 1 мин., отделяют органический слой и повторяют экстракцию. Объединяют экстракты, фильтруют через сухой фильтр и измеряют оптическую плотность при 510 нм относительно раствора хлороформа.
Для определения ванадия(1У) к водному раствору добавляют 5 М NaOH до pH 3—4, 10 мл ацетатного буферного раствора, 1 мл 0,1 М раствора БФГА и дважды экстрагируют 5 мл хлороформа. Через 1 час измеряют оптическую плотность так же, как указано ранее [128]. Метод может быть использован для анализа стекол, окислов, шпинелей и других материалов [128].
Экстракционно-фотометрическое определение ванадия(1У) можно заменить комплексонометрическим титрованием [126]. Определению не мешают 100-кратные количества Mii(Il), Al, Са, Mg и большие количества фосфат-ионов. Мешают титан, желе-зо(Ш) и хром(Ш). Их влияние может быть устранено применением соответствующих маскирующих средств. Определению ванадия(У) не мешает 200-кратное количество ванадия(1У).
ОПРЕДЕЛЕНИЕ ВАНАДИЯ(Ш) И ВАНАДИЯ(1У)
Титриметрические методы определения этих валентных форм ванадия могут быть разделены на две группы в зависимости от того, в виде какого соединения находится ванадий в образце.
Если соединение растворяется в неокисляющих кислотах, то ванадий(Ш) в инертной атмосфере титруют растворами перманганата калия [55], соли железа(Ш) [926], ванадата аммония [242] или бихромата калия [380, 1012] с визуальной и амперометрической индикацией точки эквивалентности. В другой порции этого раствора определяют общее содержание ванадия. Если анализируемые образцы медленно и плохо растворяются в неокисляющих кислотах, то для их растворения используют кислые растворы окислителей, по расходу которых определяют содержание ванадия(Ш) [199, 510, 1025]. Затем определяют общее содержание ванадия. Ванадий(1У) находят по разности.
Навеску окисла растворяют при нагревании в 4—5-кратном избытке железо-аммонийных квасцов при кислотности раствора 6—9 N по H2SO4
i/26 F. Н. Музгин и др. 161
в атмосфере СО2. Охлаждают, добавляют 2—3 капли индикатора фенилантраниловой кислоты и титруют железо(П), выделившееся в количестве, эквивалентном содержанию ванадия(Ш), 0,1 N раствором КМпО4. Затем по каплям добавляют 1%-ный раствор КМпО4 для окисления всего ванадия. Раствор подогревают для разложения индикатора. Избыток КМпО4 разрушают нитритом натрия и мочевиной и титруют весь ванадий раствором соли железа(П) с тем же индикатором. Содержание ванадия(ГУ) рассчитывают по разности [199].
Таким же образом анализируют технический тетрахлорид титана [510].
Растворы, содержащие ванадий(Ш) и ванадий (IV), титруют амперометрически комплексоном III [136], тайроном [558, 561], пирокатехином [559], солями хрома(У1) [135].
Для фотометрического определения ванадия(Ш) и ванадия(1У) предложены различные реагенты. Пирокатехиновый фиолетовый взаимодействует с обоими ионами ванадия в кислой среде. При pH 1,25—2,75 ванадий(Ш) образует комплексное соединение с полосой поглощения при 540 нм, а ванадий(1У) при pH 4—5 дает интенсивно окрашенное соединение с двумя полосами поглощения при 562 и 610 нм [557]. Последнее соединение рекомендовано для определения ванадия(1У) в присутствии 500-кратных количеств ванадия(Ш). Предел обнаружения равен 0,2 мкг/мл ванадия(1У) [561].
Ванадий(Ш) образует с тайроном интенсивно окрашенное комплексное соединение при pH 8—9 (е670 = 9900), которое используют для его определения в присутствии 33-кратных количеств ва-надия(1У). Предел обнаружения составляет 0,02 мкг/мл [561).
В одних и тех же условиях ванадий(Ш) и вападий(1У) образуют с роданидом калия или аммония различно окрашенные комплексные соединения (Атах для У(Ш) = 400 нм,-для V(IV) = 760 нм), которые могут быть использованы для их определения в растворах, ванадиевых стеклах и шпинелях [138, 200].
5—10 мл анализируемого раствора, содержащего смесь сульфатов ва-надия(Ш) и ванадияПУ), помещают в мерную колбу на 50 мл, добавляют 7 мл H2SO4 (1 : 1) и 9,7 г KSCN. После растворения соли добавляют 30 мл ацетона и доводят до метки водой. Оптическую плотность раствора измеряют при 400 и 760 нм относительно фонового раствора [138].
Таким образом можно определить >0,01 мкг ванадия(Ш) в присутствии 100 мг ванадия(IV) и >1,1 мкг ванадия(1У) в присутствии 125 мкг ванадия(Ш) [200].
Исследовано взаимодействие ванадия(Ш) и ванадия(1У) с эриохром красным Б [129]. Ванадий(1У) определяют в присутствии ванадия(Ш) или ванадия(У) в виде ионного ассоциата с эриохром красным Б и дифенилгуанидинием.
Для определения ванадия(Ш) можно использовать собственную окраску иона в 3—3,5 М H2SO4. Таким образом можно определить более чем 0,7 мкг!мл ванадия(П1) в присутствии 10 мг ванадия(ГУ) [238].
162
ОПРЕДЕЛЕНИЕ ВАНАДИЯ(П) И ВАНАДИЯ(Ш)
Из-за низких значений окислительных потенциалов ванадия(П) и ванадия(Ш) анализируемые объекты растворяют в растворах окислителей — железо-аммонийных квасцах или бихромате калия. Степень окисления ванадия определяют из сопоставления расхода окислителя на растворение образца и общего содержания ванадия [199].
В присутствии большого количества фосфорной кислоты желе-зо(Ш) окисляет только ванадий(П) и не взаимодействует с вана-дием(Ш). Разработаны методы прямого и обратного определения ва-надия(П) в присутствии ванадия(Ш). Ванадий(П) непосредственно титруют раствором соли железа(1П) с индикатором нейтральным красным [926] или титруют непрореагировавший с ванадием(П) избыток железа(Ш) раствором соли хрома(П) с потенциометрической индикацией точки эквивалентности [94]. Ванадий(Ш) в обоих методах рассчитывают по разности после определения общего содержания ванадия.
Для определения ванадия(П) предложен косвенный фотометрический метод по реакции восстановления комплексоната титана(1У) [765].
Ванадий(П) можно определить с 2,9-диметил-1,10-фенантро-лином (е = 1,14-103) в присутствии равного количества ванадия (Ш) и 20-кратного количества железа(П) [62].
6*
Глава VII
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В ВАНАДИИ И ЕГО СОЕДИНЕНИЯХ
Современная паука и техника предъявляют весьма жесткие требования к чистоте и постоянству состава используемых материалов. Это в полной мере относится к ванадию и его соединениям. Особенно необходимо отметить важность определения таких примесей, как кислород, азот, водород, углерод, ухудшающих свойства металлического ванадия.
Основными методами анализа ванадия на примеси до сих пор являются химические, хотя в последнее время значительное внимание уделяется развитию физических методов, таких, как спектральные, радиоактивационные и др.
ХИМИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
При определении элементов-примесей в ванадии и его соединениях необходимо учитывать сильное мешающее действие ванадия на результаты анализа. Лишь в небольшом числе случаев возможно непосредственное определение примесей в присутствии ванадия. Так, разработаны спектрофотометрические методы определения хрома с дифепилкарбазидом [218], циркония — с арсе-назо(Ш) [517], европия — по окраске Еп(Ш) [881], железа — с о-фенаптролином [285], титана — с диаптппирилметаном [324]. В большинстве случаев мешающее влияние ванадия устраняют введением маскирующих реагентов или применяют методы предварительного отделения.
Для маскирования ванадия используют аскорбиновую [47, 48], щавелевую [164], лимонную [164, 1132], винную [47, 164, 381, 415, 517] кислоты, гндроксиламин [284, 395], сернокислый или солянокислый гидразин [69, 267], перекись водорода [249, 517], а также различные смеси этих реагентов.
Более универсальными являются методы предварительного отделения определяемых элементов от ванадия и других мешающих элементов. Особенно часто используют экстракцию хлороформом или четыреххлористым углеродом 8-оксихинолинатов [441, 787], дитизонатов [381, 1117] или карбаминатов [318, 352, 354, 375, 381] элементов-примесей. Широко применяют и другие экстрагенты [96, 306, 411, 461, 813, 848, 906]. Используют методы ионного обмена [381, 480, 582, 848, 935, 999] и осаждения [381, 415, 786, 860, 873].
164
Описано определение Cd, Со, Cu, Fe, Ni, Pb и Zn-в соединениях ванадия с предварительным концентрированием их на ртутном катоде из разбавленного раствора серной кислоты [1062]. После отделения элементов от ванадия их определяют спектрофотометрическими пли полярографическими методами.
Разработаны методы химического анализа ванадиевого концентрата [860] и ванадиевой контактной массы [1024]. В концентрате, помимо ванадия, определяют алюминий, железо, кальций, магний, марганец, мышьяк, никель, серебро, серу, титан, углерод, фосфор, хром, а в контактной массе — алюминий, калий, кальций, железо, натрий.
В табл. 30 приведены методы определения основных элементов-примесей в ванадии и ванадиевых материалах.
Определение газов в ванадии является важной аналитической задачей. Азот в ванадии может быть определен с высокой чувствительностью классическим методом Кьельдаля (обработка конц. H2SO4 при температуре кипения). Однако высокая устойчивость нитрида ванадия к действию серной кислоты значительно увеличивает время анализа. Поэтому для разложения пробы обычно используют различные смеси с серной кислотой, имеющие более высокую температуру кипения [577, с. 545], хлорную кислоту [381] или проводят сплавление со щелочью [457, 788].
Навеску металлического ванадия растворяют в хлорной кислоте, азот при этом восстанавливается до аммиака, прибавляют избыточное количество щелочи, аммиак отгоняют под вакуумом и поглощают водой. Содержание азота определяют титрованием серной кислотой. Предел обнаружения 4-10-3% [381].
При определении азота в нитриде ванадия хорошие результаты получены при разложении пробы нагреванием с конц. HF в специальной бомбе при 140° С в отсутствие окислителей [664].
Сплавление ванадия со щелочью КОН проводят при 500—600° С в течение 15—20 мин. в токе инертного газа. Выделившийся NH3 поглощают разбавленной НС1. В полученном растворе устанавливают pH 9,8—10,3 и вводят NaClO. Образующийся мопохлорамин взаимодействует с раствором тимола при pH 11,5—11,9. Через час раствор фотометрируют при Л = 660 нм [788].
В последнее время азот, кислород и водород определяют методами вакуум-плавления [231, 544, 577, 643, 706, 738].
Кислород в ванадиевых сплавах определяют методом вакуум-плавления в графитовых тиглях с применением вспомогательной ванны из сплава 75% Ni Ц- 25% Fe (разбавление 1: 10 с добавкой 10—12% олова от веса ванны, при 1650° С) либо из платины (разбавление 1 : 10, при 1920° С). Время экстракции в обоих случаях 7 мин. Предел обнаружения составляет 0,007 % [231].
При определении газов в ванадии и его сплавах применяют также спектральные [166, 181, 184, 737, 738] и активационные [233] методы анализа.
6* В Н. Музгин и др. 165
Таблица 30
Методы определения элементов в ванадии и ванадиевых материалах
Определяемый элемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
А1 Экстракционнофотометрический с 8-оксихинолином, К = 390 нм V(V) маскируют Н2О2, А1 экстрагируют в виде 8-окоихиполината хлороформом из аммиачного раствора с pH 8—10,7 V, V2OS, VO2C1 2-10-э [131, 381, 441, 787]
Фотометрический с алюминоном, X, = 520 нм, pH 5,4—5,5 V (V) в присутствии Н2О2 сорбируют на анионите Вофатит L-150 Феррованадий 0,01 [938]
Фотометрический со стильбазо, ^=496 нм, pH 4,3-6 Экстракция купферо-натов Fe и V хлороформом из среды 3,5 N НС1 То же 0,01 [317]
Фотометрический с пирокатехиновым фиолетовым, \ = 578 нм, pH 5,9-6,3 Экстракция ацетил-ацетоната А1 хлороформом в присутствии Н2О2, реэкстракция 12 A НС1 » 0,СС1 [718а]
As Экстракционно-фотометрический с (NH4)2MoO4, Л = 840 нм Отгонка AsH3 [и экстракция мышьяко-вомолибденовой гетерополикислоты изоамиловым спиртом V, v206 5-Ю-5 [381]
Bi Фотометрический с KJ, Л = 460 нм Bi сорбируют на анионите ТМ или ЭДЭ-10П из 2,5 N НС1 и элюируют 2 N H2SO4 Экстракция диэтил-дитиокарбамината Bi хлороформом и реэкстракция 6 N НС1 V, ферро-ванадий V 2-10-1 «10%) 1-10-4 [479, 480] [352]
Экстракционнофотометрический с дитизоном, 7.— 40) и 620 нм Экстракция дитизо-ната Bi из цианиднощелочного раствора с pH 8—10 четыреххлористым углеродом V, V2O5 1 • 10-“ [131,381]
Полярографический на фоне ацетата натрия, pH 6,0 Экстракция дитизо-вата Bi четыреххлористым углеродом из среды ~ 2,5 N NaOH V «16%) [1117]
С Кондуктометрический (измерение электропроводности раствора) Пробу сжигают в токе О2, СО2 поглощают 0,01 N раствором Ва(ОН)2 V 5-10-4 (10—20%) [1137]
166
Таблица 30 {продолжение)
Определяемый влемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
С Г азохроматогра-фическое' определение СО2 Образец сжигают в токе О2 V 5-105 [435]
Са Колориметрический с мурекси-дом, А = 506 нм Ванадий отгоняют в токе СС14 и С12 V, V2O6 bio-4 [381]
Cd Колориметрический с дитизоном, А = 518 нм Экстракция дитизона-та Cd четыреххлористым углеродом (pH 9-10) Экстракция диэтпл-дитиокарбамината Cd хлороформом, реэк-стракция 0,2 N НС1 v, V2O6 V, V2O5 2-10-5 5-10-5 [131, 381] [352, 381]
Полярографический па фоне NH4C1 Соосаждение Cd на Fe(OH)3 V 2О6 1•10-з [415]
Полярографический на фоне ацетата натрия, pH 6,0 Экстракция дитизо-ната Cd четыреххлористым углеродом из среды ~2,54VNaOH V «2%) [1117]
Со Экстракционно-фотометрический с питрозо-П-солыо, А = 420 (520) нм Экстракция комплекса Со с а-нитрозо-р-нафтолом хлороформом при pH 3—4, V(V) маскируют лимонной кислотой v205 1-10-5 [96]
Сг Фотометрический с дифевилкарбази-дом в 2 N H2SO4, V(V) в отсутствие Н3РО4 не мешает определению Сг Магнитные сплавы 1С0 мкг [218]
А = 540 нм V (V) осаждают купфероном и доизвле-кают экстракцией хлороформом Экстракция купферо-ната V(V) хлороформом Сорбция V(IV) и V(V) на анионите Дауэкс-1Х8 из 0,1 М НС1, содержащей 5—6% Н2О2 V, V2O5 Иттрий-ванадиевые фосфоры Соединения V 5.10-* «20%) (0,7%) [381] [597] [276]
Си Колориметрический с дитизоном, А = 535 нм Экстракция дитизо-ната Си четыреххлористым углеродом при pH 2,5 V, V2O6 1-Ю-4 [131, 381]
6**
167
Таблица 30 [продолжение)
Определяемый элемент .Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
Си Фотометрический с 2,2'-бицинхопи-новой кислотой, pH 5,0, X = 560 нм V(V) маскируют гидроксиламином при pH 3 Феррованадий, V2O5 0,02 «2,5%) [284]
Полярографический на фойе пири-дива и NH4C1, ср , = —0,54 в V(IV) маскируют сернокислым гидразином Феррованадий 1.10~3 [267, с. 196]
Полярографический на фоне NH4C1 Соосаждение Си на Fe(OH)3 v205 1 • IO-3 «10%) [415]
Полярографический на фоне ацетата натрия, pH 6,0 Экстракция дитизо-ната Си четыреххлористым углеродом из раствора ~2,5 7V NaOH V (15%) [1117]
F Фотометрический с ализарин-цирко-ниевым комплексом, X = 525 нм Сплавление пробы с Na2CO3 и ВаСО3 (1 : : 1 : 5) и выщелачивание F” водой V2O5 При определении 10— 20 мкг ошибка <3 мкг [96]
Fe Титриметрический с Ce(SO4)2, индикатор ферроин Осаждение ванадата свинца Ванадиевые шлаки [786] '
К олориметриче -ский с сульфоса- V(V) маскируют винной кислотой V, V2O5 5-10*4 [131, 381]
лициловой кислотой, Л = 430 нм Осаждение Fe(III) на Cd(OH)2 v206 1-10~4 [415]
Экстракционнофотометрический с KSCN, X = 518 нм Экстракция Fe(III) 35РА-ПЫМ раствором ТБФ в хлороформе Ванадаты РЗЭ 5-IO*6 [411]
Экстракционно-фотометрический с метилизоб утилке-топом, NH4SCN, А = 490 нм Экстракция Fe(III) амилацетатом из 8 N НС1 Хлориды ванадия 1-10-8 [306]
Фотометрический V(IV) маскируют со- Металл, 0,01- [97, 285,
с 1,10-фенантроли-ном, А = 500-s--1-510 нм лянокислым гидро-ксиламипом сплавы, V2O5 4 104 609]
Экстракционно-фотометрический с 1,10-фенантроли-ном, X = 515 нм Экстракция Fe(II) в виде перхлората нитробензолом при pH 5,0 Металл 3-10~4 [906]
168
Таблица 30 (продолжение)
Определяемый элемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
Fe Экстракционно-фотометрический с батофенантроли-цом (4,7-дифенил-1,1-фепантроли-иом) в присутствии С1О~ при pH. 3, А, = 533 нм Fe(III) восстанавливают Na2S2O4 п экстрагируют хлороформом при pH 4—6 V, сплавы, V2O5, NH4VO3 1 • Ю-* (~2°;>) [757]
Мп Колориметрический по марганцевой кислоте (с KJO3), X = 525 нм Осаждение Ми на Fe(OH)3 V 5 -10* [381]
Мо Экстракцпопно-фотометрический с о-нитрофенилфлу-ороном, X =- 540 нм Экстракция диэтил-дитиокарбамипата Мо хлороформом V, V2O5 3.10-- [318, 354]
Экстракционно-фотометрический с KSCN, Z =490 нм Экстракция Мо три-«-октиламшюм в толуоле или 20%-ным раствором ТБФ в бензоле Экстракция Mo(VI) диэтиловым эфиром или Mo(IV) смесью диизопропилового эфира, «-амилово- го спирта и «-амилацетата (1:1: 1) V, V2O5 V, ванадаты 1.10-(15%) «10%) [283, 418] [818]
Фотометрический с KSCN, X = 490 нм Сорбция Mo(VI) из 4 .V НС! анионитами А В-17, Варной AD или Дауэкс-2Х8 и вымывание 1 N НС1 To же 0,05 [848]
Фотометрический с кислотным хром фиолетовым К, 1 TV НС1, Х=620 «л Г равпметрический с а-бепзоинокси- мом V(IV) маскируют винной кислотой V(IV) сорбируют на катионите AG50W-X8 Сплавы Фосфор-номолиб-денова-падиевая кислота и ее соли (0,5%) [517] [999]
Nb Экстракцпошю-фотометрический с пирокатехином и трибеп з иламино м, А, = 370 нм V(I V) маскируют тио-гликолевой и винной кислотами, Nb экстрагируют 0,1 М раствором трибепзилами-на в хлороформе V, V2O5 5.10-5 (7-13%) [605]
Таблица 30 (продолжение)
Определяемый элемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
Nb Фотометрический с тиохромином, 2,5 W H2SO4, Л = = 410 нм V(IV) маскируют аскорбиновой и винной кислотами Сплавы (3—8%) [47]
Фотометрический с кислотным хром фиолетовым К V(IV) маскируют винной кислотой Сплавы (0,3%) [518]
Ni Гравиметрический с диацетилдиоксимом или с диме-тилглиоксимом V(V) не мешает определению никеля Ванадаты, катализаторы (0,7%) [486]
Титриметрический с комплексоном III V(V):Ni<20: 1 не мешает, pH 4,0 Ванадаты (С1%) [169]
Экстракционнофотометрический с диметилглиокси-мом, Л = 460 нм Экстракция диметил-глиоксимата никеля хлороформом при pH 8,5—9,5; V(V) маскируют лимонной кислотой v2o5 1104 [96]
Р Гравиметрический в виде (NH4)3PO4-•12МоО3 Г равиметрический в виде NH4MgPO4 V(IV) маскируют лимонной кислотой V(IV) сорбируют на катионите К У-2 [из 0,3 N НС1 V(IV) сорбируют на катионите AG50W-X8 Феррованадий Феррованадий, v2o5 Фосфор-номолиб-денова-надиевая кислота и ее соли [1132]- [92, 480] [999]
Титриметрический косвенный с комплексоном III, 0,05 MMgSO4 V(IV) маскируют комплексоном III Соединения V (0,9%) [779]
Фотометрический V(V) сорбируют на V, V2O6 10-2— [582, с.
c(NH4)2MoO4 или с аскорбиновой кислотой катионите КУ-2, Дау-экс-50 или Вофа-тит F Фосфор осаждают с Fe(OH)3 ванадаты Феррованадий Ю-4 0,01 274; 746, 784] [873]
Экстракционно-фотометрический с (NH4)2MoO4, Л = — 420 нм Экстракция фосфор-номолибденовой ге-терополикислогы ме-тилизобутилкетоном V, v2os, феррованадий 1-10-3 [131, 381, 461, 517]
170
Таблица 30 [продолжение)
Определяем мый элемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) Литература
РЬ S Sb Si Sn Ti Колориметрический с дитизоном, X = 420 нм Полярографический на фоне ацетата натрия, pH 6,0 Полярографический на фоне 10%-ного раствора NaOH, Ф1/2 = = —0,80 в Гравиметрический в виде BaSO4 К ондуктометриче-ский (измерение электропроводности раствора) Колориметрический с фенилфлуо-роном, А — 530 нм Экстракционнофотометрический с (NH4)2MoO4, А = = 660 нм К олориметриче-ский с л-нитрофе-нилфлуороном, А = 530 нм Фотометрический с Н2О2) А = 410 нм Экстракция РЬ в виде дитизбната четыреххлористым углеродом или диэтилдитиокар-бамината хлороформом Сорбция РЬ на анионите ТМ или Э ДЭ-10П из 2,5 N НС1 и элюирование 0,02 N НС1 Экстракция дитизо-ната РЬ четыреххлористым углеродом из —2,5 N NaOH V(V) не мешает, Cr(VI) маскируют NaF V(IV) и Fe(II) сорбируют на К У-2 из 0,3 N HCI Образец сжигают в токе О2 и SO2, поглощают 0,001 N раство-ром'К2Сг2О7 'в 0,001 N H2SO4 Экстракция Sb в виде пиридилроданидного комплексного соединения диэтиловым эфиром Экстракция кремнемолибденовой гетерополикислоты изОами-ловым спиртом Сорбция V(V) на катионите КУ-2 Экстракция диэтил-дитиокарбамината олова хлороформом Осаждение Ti(OH)4 совместно с Fe(OH)3 и А1(ОН)3 V, V2O5 V V v206 Феррованадий V V, V2O6 V, V2O6 V V, V2O6 Концентрат V 4-105 2-10i «10%) «16%) 1•10-з (Ю-16%) 5-10'3 1 • IO-3 l-10-з l-10-з [131,352, 381] [479, 582] [1117] [415] [92] [1137] [381] [381] [582] [381] [860]
171
Табл и ц а 30 (окончание)
Определяемый элемент Метод и условия определения Метод отделения ванадия или определяемых элементов Объект анализа Предел обнаружения, % (погрешность) 1 ,1 Гптера-тура
Ti Фотометрический V(IV) маскируют гид- V.v2o5, 5-10’5 [48. 324]
и с диантипирилме-таиом, К = 390 нм Фотометрический с тимолом в 13 Л/ H2SO4, Х=470»л« Полярографический на фоне ацетатного буферного раствора, pH 5,0 Полярографиче- ро ксилампиоы или аскорбиновой кислотой Соосаждение Ti па А1(ОН)., V(V) восстанавливают N2H4-H2SO4 V(V) восстаиавлпва- хлориды ванадия V2O5. растворы Paci во- 2-10~4 1 • IO5 (2,5%) [381] [69, 267] [331. 163]
W ский на фоне молочной кислоты (3-7%), <pt/2-= — 0,2 в Фотометрический ют до \ (IV) Осаждение W (и ча- ры. руды Сплавы (0,3%,) [517]
Zn с KSCN, К--:: — 410 нм Фотометрический стпчио V) танинном и р-лафтохпнолииом V(V) маскируют вин- \;о3 2 • 10-* [317, 415]
Zr, с дитизоном, X = = 538 нм, pH 9—11 Полярографический на фоне ацетата натрия и винной кислоты Тптриметричес кий пой кислотой Zn сорбируют па анионитах ТЛ1 или ЭДЭ-10П из 2,5 .V НС1 и элюируют 0.02 N HCI Экстракция дитпзо-пата Zn четыреххлористым углеродом из раствора ~ 2,5 Л' NaOH V(V) маскируют Н2О2 V V Силе, вы 2-10 1 «10°;) 110’1 (13,5?.) (0,5%) [479. 480] [1117] [249, 517]
HI с комплексоном Ш в 0,2 N H2SO4 Фотометрический с арсепазо III в 2 N НОД = 665/4.4 V(V) не мешает Сплавы 0,2 [517]
Примечание, Методы анализа ванадия, (го сплавов и соединений изложены в изданиях ГПРЕДМВТа (ЦМТУ 05-162—69, ТУ 18-05-106—70, ОСТ *7-70—73 и др.).
172
ФИЗИЧЕСКИЕ МЕТОДЫ
Спектральные методы
Спектральные методы определения примесей имеют существенные преимущества перед химическими: возможность одновременного определения большого числа элементов, быстрота выполнения анализа, использование в большинстве случаев малых проб и сравнительно несложная предварительная подготовка вещества к анализу. Однако при спектральном определении примесей в чистом ванадии и его соединениях возникают дополнительные трудности, обусловленные сложным спектром ванадия и возможностью наложения его линий на аналитические линии элементов-примесей.
В работе [785] приведены полуколпчественные методы анализа ванадия, чувствительность определений и учтены возможные наложения линий для 42 элементов.
При анализе ванадия в виде его пятиокиси в угольной дуге на спектрографах средней дисперсии (ИСП-22 и ИСП-51) показана возможность определения' большого числа элементов с чувствительностью 1 • 10~2—1 • 10"3 [381, 416]. Для 13 наиболее часто встречающихся элементов (Al, As, Cd, Си, Bi, Fe, Mg, Мп, P, Pb, Sb, Si, Zn) разработаны условия проведения спектрального анализа. Применение приборов высокой дисперсии (ДФС-13) и использование NaCl и AgCl в качестве «носителя» позволили снизить предел обнаружения до 3 • 10 3—1-10~б% для 24 элементов-примесей [187, 188].
30 мг пятиокиси ванадия смешивают с 10 мг угольного порошка и 10 мг хлорида серебра и помещают в кратер (3,8 х 7 Л1Л1) угольного электрода с шейкой (2,6 X 5 мм). Угольный иротивоэлектрод, заточенный на конус, имеет кратер (1,8 X 15 мм), заполненный угольным порошком, содержащим 6% хлорида натрия. Спектр пробы возбуждают в дуге переменного тока при 10 а в течение 90 сек. (время экспозиции) [188].
Аналогичная чувствительность была получена для 22 элементов при анализе пятиокиси ванадия в газоразрядной трубке с полым катодом [339, 341]. Применяя униполярную дугу с искровым активпзатором, определяют в ванадии и его соединениях 18 элементов с пределом обнаружения 10“2—10-4%. Образцы металлического ванадия переводят в пятиокнсь ванадия, смешивают с графитовым порошком, Li2CO3 и Ga2O3. Относительная погрешность <^4,4% [680].
Описан анализ пятиокиси ванадия методом сухого остатка с использованием высокоинтенсивной дуги постоянного тока (23 а) [489]. При массе пробы 0,3 мг одновременно определяют 16 элементов в интервале концентраций 0,1—0,0001% при относительной погрешности менее 1096.
В люминофорной пятиокиси ванадия определяют до 3-10'6 % кадмия и 2-10”4?6 цинка при возбуждении спектра в дуге переменного тока (9 а) [95]. Анализируемую пробу смешивают с уголь-
173
ним порошком и иодидом серебра в отношении 3 : 1 : 1 и 75 мг смеси помещают в кратер угольного электрода.
Разработан аэрозольно-искровой метод анализа растворов ванадатов алюминия, гадолиния, иттрия и лантана при их непосредственном введении в искровой разряд [232]. Предел обнаружения редкоземельных элементов (от лантана до лютеция) и иттрия составляет 0,1—0,0001 мг/мл при коэффициенте вариации 8—10%.
В лигатурах на основе ванадия определяют 1,5—3% алюминия в искровом и 0,1—1% кремния в дуговом [250] разрядах.
В первом случае лигатуру ванадий-алюминий растворяют вразб. НЖ). (1 : 1), упаривают, прокаливают при. 450° С; полученные окислы смешивают с угольным порошком (1 : 1), содержащим 20% K,SO4, и навеску (100 мг) прессуют в таблетки диаметром 4 мм. Спектр пробы возбуждают в высоковольтном искровом разряде. Относительное стандартное отклонение 6%.
Во втором случае (при определении кремния) лигатуру ванадий-молибден-алюминий-хром-железо растирают в порошок, смешивают с угольным порошком (1 : 1), содержащим 20% окиси кобальта, и смесь помещают в кратер угольного электрода. Спектр пробы возбуждают в дуге переменного тока (12 а). Относительное стандартное отклонение 15%.
Прямым спектральным методом определяют 0,01—1% магния в металлическом ванадии. 2,5 г образца растворяют в 70 мл смеси конц. НС1, конц. HNO3 и Н2О (3 : 2 : 10) и разбавляют до 100 мл. Раствор помещают в пористый электрод и возбуждают спектр в искровом разряде [1000].
При использовании дуги постоянного тока в атмосфере аргона возможно определение кислорода в ванадии в интервале концентраций 0,004—0,5% по линиям О I 777,2—Ar I 789,1 и О Т 777,5 и Аг I 789,1 нм с коэффициентом вариации 5—6% [737, 738]. Описан метод определения азота в целом ряде металлов, в том числе и в ванадии [464, 465]. Методом спектрально-изотопного анализа определяют водород, азот и кислород в чистом ванадии и феррованадии [166, 181, 184]. При определении водорода время изотопического уравновешивания при 1000° С и диаметре образцов 8—10 мм составляет для ванадия 10 и феррованадия 15 мин. Предел обнаружения 0,5 слАИОО г, а относительная погрешность определения ^10% [181].
Эффективным способом снижения предела обнаружения элементов является применение предварительного концентрирования элементов-примесей как химическими, так и физическими методами.
В отечественной практике спектрального анализа широко используется метод физического обогащения — испарения примесей из основы [182, 308]. Метод применим к тугоплавким основам. Хотя пятиокись ванадия является легколетучим окислом, легкость образования труднолетучих карбидов ванадия позволила разработать анализ ванадия методом испарения [340]. При предварительном прогреве пробы в испарителе при температуре 800—1400° С ванадий восстанавливается углеродом, образуя трудно летучие оксикарбиды [339]. Летучесть ванадия резко уменьшается и в спектре конденсата примесей, полученного даже при
174
1900° С, наблюдаются только отдельные, наиболее интенсивные линии ванадия. Элементы-примеси, такие, как висмут, таллий, кадмий, возгоняются из пробы почти полностью уже при 1500° С. Дальнейшее повышение температуры пробы приводит к испарению и более труднолетучих элементов, таких, как хром, никель и др.
Порошки эталонов и проб (40 мг), тщательно смешанные с угольным порошком в весовом отношении 5:1, помещают в угольные стаканчики с отверстием диаметром 4 и глубиной 8 мм (внешний диаметр 6 мм). Стаканчик зажимают между массивными охлаждаемыми водой графитовыми щечками испарителя и над ним на расстоянии 1,5—2,0 мм ‘устанавливают приемник, представляющий собой угольный электрод диаметром 6 мм и длиной 4—5 см, заточенный на плоскость и обожженный в дуге переменного тока (10 а) в течение 30 сек. Температуру стаканчика контролируют пирометром ОППИР-017. При проведении процесса испарения пробу медленно^ нагревают до рабочей температуры (1500—1900° С) в течение 1,5—2 мин. и выдерживают в течение 2 мин. Конденсат примесей на приемнике сжигают в угольной дуге переменного тока (ток 6 а, экспозиция 35 сек). Спектры фотографируют на спектрографе ИСП-28. Предел обнаружения составляет 10ч—103%; при введении в пробу 1% натрия (в виде хлорида или нитрата) он снижается почти на порядок [340].
Применение газового разряда в полом катоде к анализу конденсата примесей, полученного методом испарения, позволяет снизить предел обнаружения до ИГ5 % для большого числа элементов [341].
Разработан метод концентрирования примесей при анализе пятиокиси ванадия, основанный на образовании и отгонке легколетучих соединений ванадия.
Пробу пятиокиси ванадия при 300° С обрабатывают газообразным хлористым водородом, при этом ванадий отгоняют в виде окситрихлорида и в полученном концентрате определяют 19 элементов в дуге переменного тока методом сухого остатка с пределом обнаружения 10“6—-10~4% [338].
При определении малых количеств урана в сложных растворах, содержащих большие количества ванадия, предложено сорбировать уран на синтетических ионообменных смолах с последующим озолением сорбента и спектральным анализом полученного концентрата [422].
Разработан метод спектрального анализа ванадия высокой чистоты с предварительным отделением элементов-примесей или основы методами экстракции [375]. В первом случае предложено концентрировать примеси пятикратной экстракцией их диэтил-дитиокарбаминатов хлороформом при pH 7,5—8,0; ванадий при этом должен находиться в пятивалентном состоянии.
1 г ванадия разлагают в 30 мл HNO3 (1 : 3), упаривают досуха, прокаливают, растворяют в 50 лм NH4OH (1 : 10) и добавляют 50 мл Н2О. Из полученного раствора экстрагируют примеси раствором диэтилдитиокарбамино-вой кислоты в хлороформе [375].]
Предел обнаружения висмута, кадмия, меди, свинца, таллия, цинка и некоторых других элементов составляет 10~4—10"8%.
175
Таблица 31
Сравнение результатов анализа пятиэкнеи ванадия различными методами спектрального анализа
При определении алюминия проводят двукратную экстракцию 5 мл 2%-ного раствора 8-оксихинолина в хлороформе при pH 10,0.
Во втором случае основу (ванадий) экстрагируют из 6 Л НС1 трибутилфосфатом при соотношении фаз 1:1. После повторной экстракции в водной фазе остается до 1—3 мг ванадия и практически полностью щелочноземельные элементы, -алюминий, галлий, кобальт, медь, скандий, хром и цинк. Висмут и свинец экстрагируются вместе с ванадием. В дальнейшем концентрат примесей упаривают на 50 мг угольного порошка, помещают в кратер угольного электрода и возбуждают спектр в дуге постоянного тока 10 а. Предел обнаружения составляет п-10~5% при относительной погрешности определения 20% [3751.
Предложено [342] при определении примесей в пятиокиси ванадия экстрагировать их в виде дитизонатов хлороформом при pH 9,0. Элементы-примеси реэкстрагируют несколькими миллилитрами конц. HNO3, реэкстракт собирают во фторопластовый тигель и упаривают до нескольких капель. Показано, что последующее спектральное определение предпочтительнее проводить с использованием алюминиевых электродов, на торце одного из которых высушивают концентрат примесей. Спектр возбуждают в дуге переменного тока 6 а. Метод использован для определения 10 4—10 висмута, меди, свинца и цинка. Коэффициент ва-
риации 9—12%. Сравнительные результаты определения примесей в пятиокиси ванадия представлены в табл. 31.
При определении щелочных и щелочноземельных элементов широкое применение нашли методы эмиссионной и атомно-абсорбционной спектрофотометрии, которые характеризуются высокой чувствительностью (10“6—10~3 мг!мл) и хорошей точностью (относительная погрешность составляет 2—3%) [75, 196—198, 287, 1024]. В качестве горючих газов используют смеси пропан—бутан или ацетилен—воздух. Методом атомно-абсорбционной спектрофотометрии рекомендуют определять в ванадии и его различных соединениях алюминий [782], магний [212], свинец [213], железо, кобальт и никель [212] с относительной погрешностью 5—6%. При определении малых концентраций (5-10“6—3-10'5%) железа, кадмия, меди и цинка в тетрахлориде ванадия из 4 г пробы отгоняют в потоке аргона (1 л/мин) при 60° С основную часть (94— 95%) основы. Остаток растворяют в водном растворе изопропилового спирта, подкисленного 1 — 2 каплями конц. НС1, раствор разбавляют до концентрации хлорида ванадия <4% и распыляют в пламя ацетилен—воздух [442].
Актива {ионные методы
Методом нейтронной активации и спектрометрии у,у-совпа-дений определяют в пятиокиси ванадия мышьяк и вольфрам [620], углерод [793], золото, медь, марганец и цинк [ИЗ], рутений, осмий и иридий [1047].
175
177
Пробу весом 20 мг и соответствующие стандартные образцы пятиокиси ванадия, содержащие по 1 10 5 г определяемых элементов, облучают в течение 8 час. в канале ядерного реактора потоком нейтронов 1,8 • 1013 нейтрон/ /см2-сек, затем снимают спектры на у-спектрометре со 100 канальным анализатором и рассчитывают активность 198Ап, 56Мп, 60Со по у-пикам 0,41, 0,84, 1,33 Мэе соответственно. Для определения мышьяка, вольфрама, сурьмы и молибдена пробу через 15 час. после облучения растворяют в присутствии носителей (по 10 мг соответствующих элементов) и проводят их химическое выделение. Мышьяк отгоняют в виде AsBr3 с последующим осаждением действием NH4H2PO4. Сурьму, молибден и вольфрам осаждают в виде соответствующих кислот конц. HNO3, из этого осадка вольфрам и молибден переводят в раствор, обрабатывая его NH4OH. Затем отделяют молибден от вольфрама экстракцией его амилацетатом и осаждают Н2МоО4 и H2W04 соответственно из органической и водной фаз. Полученные осадки промывают водой и эфиром, сушат при 105° С, взвешивают и измеряют активность 187W, 122Sb, 76As и 88Мо на торцовом счетчике СИ-2Б. Радиохимическую чистоту выделенных изотопов контролируют по периоду полураспада и по энергии у-спектра. Средняя относительная погрешность анализа 10—20% при определении до 10“7—10 8| г золота, марганца, кобальта и до 10-8—10“’г вольфрама, сурьмы, мышьяка и молибдена [114].
Активация образцов быстрыми нейтронами (14 Мэв) использована для определения стехиометрии состава окислов ванадия и титана [978].
Фотоактивационный метод использован для определения в ванадии титана [2] и кислорода по реакции 16О (у, тг)15О с последующим выделением изотопа 15О восстановительным плавлением образца в вакууме [233]. При использовании линейного ускорителя электронов ЛЭУ-25 в ванадии определяют до 5-10‘6% кислорода [233].
Разработан метод послойного определения углерода (до 10"4%) и циркония (10'3%) в ванадии активацией заряженными частицами (дейтронами) по реакциям I2C {d, n)13N и .90Zr (d, 2n)90Nb [321].
Масс-спектральные методы
Разработана методика определения большого числа элементов-примесей в пятиокиси ванадия с помощью искрового масс-спектрографа Маттауха—Герцога с двойной фокусировкой [827]. Анализируемую пробу смешивают с 9% порошкообразного графита, смачивают глицерином, прессуют при давлении 4 т/см2 и помещают в искровую камеру в качестве электродов.
Описана аналитическая процедура контроля состава тетрахлорида ванадия в процессе получения ванадия особой чистоты [5].
Система напуска и ионизационная камера масс-спектрометра МИ-1305 подогреваются до температуры 25 и 165° С соответственно. Ионизирующее напряжение в камере поддерживается на уровне 60 в, ток эмиссии 1,5 ма. В техническом тетрахлориде ванадия найдено до 50% окситрихлорида ванадия и обнаружены хлориды германия, железа, мышьяка, олова, сурьмы, титана на уровне ю*2—ю_®%.
ЛИТЕРАТУРА
1. Абрамович В. И., Корсунская М. К., Ткачева С- И., Шубина С. В.— В кн.: Химия и технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 458.
2. Абэ С.— Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 87, 714 (1966); РЖХим, 1967, 8Г166.
3. Авилов В. Б., Золотарь Р. В.— Ж. аналит. хим., 25, 1418 (1970).
4. Агасян П. К., Басов В. В., Костромин А. И.— Там же, с. 1933.
5. Агафонов И. Л., Фаерман В. И.— Тр. по химии и хим. техно л. (Горький), вып. 3 (38), 99 (1974).
6. Азизов В. А., Вометун Е. А.— Докл. АН ТаджССР, 11, 25 (1968).
7. Александров А., Будаевски О.— Науч. тр. Висш. ин-т хранит, и вкус, пром-ст (Пловдив), 12, 297 (1965); РЖХим, 1966, 23Г83.
8. Александров В. В., Гуния Г. С-, Гунченко А. И., Туркин Ю. И.— Тр. Гл. геофиз. обсерв., вып. 314, 104 (1974).
9. Алексеев В. В. Курс качественного химического полумикроанализа. М.: Химия, 1973.
10. Алешин Г. В., Смольянинов С. В., Мещеряков Р. В., Глухов Г.Р. — В кн.‘ Химия и технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 468.
11. Али-Заде Т.Д., Гамид-Заде Г. А., Абдуллаева Р. Ю., Агамирова О. М., Ганиева Э. М.— Изв. вузов. Химия и хим. технол. , 19, 330 (1976).
12. Али-Заде Т.Д., Гамид-Заде Г. А., Агамирова О.М.— Азерб. хим. ж., № 1, 97 (1973).
13. Аликеева Э. А., Козловская В. М.— Тр. Ленингр. н.-и. и проект, ин-та основн. хим. пром-сти, вып. 4, 264 (1971).
14. Алимарин И. П$— Ж. прикл. хим., 17, 83 (1944).
15. Алимарин В. В., Архангельская В,Н. Качественный полумикроанализ. М.; Л.: Госхимиздат, 1952.
16. Алимарин В. В., Крюков В. Г-— Ж. аналит. хим., 10, 56 (1955).
17. Алимарин В. В., Медведева А. М.— Зав. лаб., 20, 911 (1954).
18. Алимарин В. В., Ветрикова М. В. Качественный и количественный ультрамикрохимический анализ. М.: Химия, 1974.
19. Алимарин В. В., Судаков Ф. В., Головкин Б. Г-— Усп. хим., 31, 989 (1962).
20. Алимарин В. В., Фрид Б. В.— Зав. лаб., 18, 1300 (1952).
21. Анализ минерального сырья: Сборник методов химического анализа/ Под ред. Ю. Н. Книпович, Ю. В. Морачевского. Л.: Госхимиздат, 1956.
22. Анализ черных металлов/ Под ред. С. В. Липина. Свердловск; Москва: Металлургиздат, 1951.
23. Антипова Ж- А., Безруков И. Я., Глазырин М. В., Золота-вин В. Л.— Ж. неорг. хим., 21, 437 (1976).
24. AnmoHujeeuh В.— Гласник Хем. друшт. (Београд), 31, 305 (1966).
25. Асаока X.-- Бунсэки кагаку (Japan Analyst), 10, 255 (1961); РЖХим, 1961, 24Д70.
26. Астафьев В. В., Рубикович Р. С., Яковлева С. А.— Изв. АН СССР. Сер. физ., 19, 192 (1955).
27. Астахов А. В., Клименко Е. В., Князева Е. В., Грачевский В. В.— Ж. общ. хим., 45, 7 (1975).
1'79'
28. Атрошенко М. П., Егорова Т. Н.— Научно-техн. сб. «Радиодетали», вып. 2 (23), 41 (1971).
29. Ахмедов С. А., Мирзаева Х.А., Абдуллаев М.Ш.— В кн.: Методы определения неметаллических и других вредных примесей в промышленных материалах. М.: МДНТП им. Ф. Э. Дзержинского, 1977, с. 67.
30. Ахрем Л. А.. Кузнецова А. И. Тонкослойная хроматография. М.: Наука, 1964.
31. Бабенко Н. Л., Бусев А. И., Симакова Л. К.—- Ж. аналит. хим., 25, 1539 (1970).
32. Бабенко Н. Л., Бусев А. И., Чистяченко И. Н. —Ж. неорг. хим., 19, 1587 (1974); 21, 1282 (1976).
32а. Бабенко Н. Л., Бусев А. И., Чистяченко И. И-— Там же, 17, 1865 (1972).
33. Бабенко Н.Л., Сухорукова II. В.— Тр. Ин-та металлург, и обогащ. АН КазССР, 23, 85 (1967).
34. Бабина Ф. Л-, Карабаш А. Г., Пейзулаев III. И., Семенова Е. Ф.— Ж. аналит. хим., 20, 501 (1965).
35. Бабко А. К. Физико-химический анализ комплексных соединений в растворах. Киев: Изд-во АН УССР, 1955.
36. Бабко А. К., Волкова А. И., Гетьман Т. Е.— Ж. неорг. хим., 11, 374 (1966).
37. Бабко А. К., Дубовенко Л. И., Луковская Н.М. Хемилюминесцентный анализ. Киев: Техника, 1966.
38. Багбанлъг И. Л., Гусейнов И. К., Аллахвердиева Э. Г.— Азерб. хим. ж., Л«2, 116 (1969).
39. Багбанлъг И. Л., Гусейнов И. К., Посадовская А. Н., Багбан-лы С. И.— В кн.: Исследования в области неорганической и физической химии. Баку: Элм, 1971, с. 218.
40. Багдасаров К.Н., Ахмедова Х.А., Татаев О. А.—-Зав. лаб., 35, 12 (1969).
40а. Багреев В. В., Петрухин О. М., Раковский Э. Е., Золотов Ю. А.— В кн.: Химические основы экстракционного метода разделения элементов. М.: Наука, 1966, с. 69.
41. Бакал Г. Ф., Лисецкая Г. С.— Укр. хим. ж., 37, 594 (1971).
42. Баканов Д. Г.— В кн.: Фотоэлектрические методы спектрального анализа. М.: Оборонгиз, 1961, с. 66.
43. Банковский Ю. А., Лобанова Е. Г.— Изв. АН ЛатвССР. Сер. хим. наук, № 3, 113 (1960).
'А. Баренбаум М. Е., Дедков Ю. М., Орлова Е. С.— Ж. аналит. хим., 27, 1967 (1972).
45. Барков Б. Я.— Зав. лаб., 12, 627 (1946).
46. Басаргин Н. И., Яковлев П. Я., Бусев А. И., Занина И. А.— Там же, 35, 411 (1969).
47. Басаргин II. Н., Яковлев П. Я., Панарина Н. А., Онучина Г. В.— Там же, 37, 143 (1971).
48. Баусова Н. В., Решетникова Г. А.— Тр. Ин-та химии УФ АН СССР, вып. 10, 107 (1966).
49. Башкиров Ю.А., Денкер В. И.— Изв. АН СССР. Неорг. матер., 6, 20 (1970).
50. Безруков И. Я., Золотавин В. Л., Аскеров А. Б., Прокопчук В. В.— Геохимия, № 9, 1120 (1965).
51. Безруков И. Я., Золотавин В. Л., Водопьянова В. П., Ребрин О.И.—• Тр. Ин-та химии УНЦ АН СССР, вып. 31, 88 (1975).
, 52. Безруков И. Я., Калугина Н. И., Попков В. М.— В кн.: Химия и технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 372.
53. Безруков И. Я., Романцева С. Ю., Спиридонов В. А.— Ж. неорг. хим., 22, 343 (1977).
54. Бектуров А. Б., Ильясова А. К., Гескина Р.А.— Там же, 7,2134 (1962).
55. Бектурова Г. Б., Сангина О. А., Захаров В. А.— Зав. лаб., 38, 143 (1972).
180
56. Белъкевич Я. П. Руководство по спектральному анализу металлов. М.; Л.: Судпромгиз, 1950.
57. Берлъ — Лунге. Химико-технические методы исследования. Л.; М.: ГОНТИ, 1938. Т. 1, 2.
58. Берман Е. Л.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 242.
59. Биктимирова Т. Г., Байбазаров А. А.— Химия и технол. топлив и масел, № 7, 50 (1969).
60. Бланкова Т. Н., Русяев В. Г., Бочкарев Б. Н.— В кн.: Ядерно-гео-физические методы. Новосибирск: Наука, 1972, с. 181.
61. Блинова В. И., Подчайнова В. Н-, Харламов И. Я., Фридман Г. И.— Технология легких сплавов: (Научно-технический бюл. ВИЛСа), № 1, 99 (1973).
62. Блох М. Ш., Бабенко II. Л., Бусев А. И.— Ж. аналит. хим., 30, 1369 (1975).
63. Боговина В. II., Усатенко 10. И., Мальцев В. Ф.— Зав. лаб., 32, 412 (1966).
64. Божеволънов Е.А. Люминесцентный анализ неорганических веществ. М.: Химия, 1966.
65. Бондаренко Л. С., Пейзулаев III. И., Карабаш А. Г.— В кн.: Методы анализа веществ высокой чистоты. М.: Наука, 1965, с. 315.
66. Бондаренко Л. С., Савинкова И. П., Пейзулаев III. II., Каратыгина Г. Ф., Веренцова Р. М.— Там же, с. 483.
67. Бондаренко С. К., Мамот Ж. А.— В кн.: Материалы II Межотраслевого совещания по методам получения и анализа ферритовых материалов и сырья для них. М.: ИРЕА, 1969, ч. 2, с. 74.
68. Борисенко Л. Ф. Ванадий (минералогия, геохимия и типы эндогенных месторождений). М.: Недра, 1973.
69. Ботев И., Еленкова Н., Шейтанов X.— Докл. Волг. АН, 19, 1059 (1966).
70. Боуэн Г., Гиббонс Д. Радиоактивационный анализ. М.: Атомиздат, 1968.
71. Брикун И. К., Козлосский М. Т.— Изв. АН КазССР. Сор. хим. наук, 2, 8 (1965),
72. Брицке М. Э. Анализ металлургических продуктов методом эмиссионной фотометрии пламени. М.: Металлургия, 1969.
73. Брудзъ В. Г., Шафран И. Г., Смирнова К. А., Дранкина Д. А., Зеличенок С. Л., Подольская Б. Л.— В кн.: Тр. ВНИИ хим. реактивов и особо чистых хим. веществ. М.: ИРЕА, 1963, вып. 25, с. 17.
74. Бубыръ С. II., Вовк В. II., Васильева Т. Г.— Зав. лаб., 33, 706 (1967).
75. Букреев IO. Ф>., Золотавин В. Л., Безруков И. Я., Толстов Л. К.— Там же, 32, 1085 (1966).
76. Булыгина В. Н., Безруков И. Я., Золотавин В. Л.— Ж. неорг. хим., 15, 429, 435 (1970).
77. Бураков В. С., Янковский А. А. Практическое руководство по спектральному анализу. Минск: Изд-во АН БССР, 1960.
78. Бургер К. Органические реагенты в неорганическом анализе. М.: Мир, 1975.
79. Бусев А. И., Бырько В. М., Квсситадзе А. Г.— Сообщ. АН ГССР, 60, 577 (1970).
80. Бусев А. И., Бырько В. М., Хоанг Ким Иен, Новикова Н. Н.— Вести. МГУ. Химия, 13, № 3, 319 (1972).
81. Бусев А. И., Карякина 3. П.— Там же, № 6, 77 (1966); Ж. аналит. хим., 22, 1506 (1967); Ж. неорг. хим., 12, 1561 (1967).
82. Бусев А. И., Карякина 3. П.— Ж. аналит. хим., 22, 1350 (1967).
83. Бусев А. И., Полянский Н. Г. Применение органических реактивов в неорганическом анализе. М.: Изд-во АН СССР, 1958.
84. Бусев А. И., Типцова В. Г., Иванов В. М. Руководство по аналитической химии редких элементов. М.: Химия, 1978.
85. Бутенко Г. А-, Гржегоржевский А. С., Каленченко Т-Я.— Вопр.
181
химии и хим. технол.: Респ. межвед. томат, науч.-техн, сб., вып. 28, 64 (1973).
86. Буянов Н. В.— Изв. АН СССР. Сер. физ., 12, 439 (1948).
87. Буянов Н. В., Замараев В. П., Туманов А. К. Повышение точности спектрального анализа магнитной стабилизацией. М.: Металлургия, 1971.
88. Буянов Н. В., Замараев В. П., Туманский С. С.— Зав. лаб., 32, 1354 (1966).
89. Вайвадс Я. К., Гедровиц Я. Я., Констант 3. А., Пелекис Л. Л.— Изв. АН ЛатвССР. Сер. физ. и техн, наук, № 5, 8 (1976).
90. Вакамацу И., Отомо М.— Нихон кагаку дзасси (J. Chom. Soc. Japan, Pure Chem. Sec.), 90, 595 (1969); РЖХим, 1970, 15Г134.
91. Величко В. В., Супрунович В. И., Усатенко Ю- И.— В кн.: Химия и химическая технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 432.
92. Вербицкая В. А., Степин В. В., Онорина И. А.— Тр. ВНИИ станд. обр. и спектр, эталон., 4, 136 (1968).
93. Веселаго Л. И.— Ж. аналит. хим., 23, 384 (1968).
94. Веселаго Л. И.— Там же, 20, 335 (1965). '
95. Видищева А. Я., Перфильева 3. В.— В кн.: Атомная спектроскопия и спектральный анализ. Киев: Наукова думка, 1974, с. 100.
96. Винковецкая С. Я.— Науч. тр. Н.-и. и проектн. ин-та редкомет. пром-сти, 42, 202 (1972).
97. Винковецкая С. Я., Левицкая Т. И.— Зав. лаб., 34, 278 (1968).
98. Витушкина И. Н., Гинзбург В. Л.— В кн.: Спектральный анализ в цветной металлургии. М.: Металлургия, 1960, с. 299.
99. Вишневская Г. П., Тишков П. Г.— Докл. АН СССР, 142, 841 (1962).
100. Водопьянова В. П., Безруков И. Я., Золотавин В Л.— Ж. неорг. хим., 17, 1331, 2426, 2429 (1972).
101. Волков Г. И., Мельникова С. А.— Ж. аналит. хим., 25, 2049 (1970).
102. Волынец М. П. Тонкослойная хроматография в неорганическом ана* лизе. М.: Наука, 1974.
103. Волынец М. П., Ермаков А. Н.— Усп. хим., 89, 934 (1970).
104. Гайбакян Д. С.— Арм. хим. ж., 24, 861 (1971); Уч. зап. Ереванск. ун-та. Естеств. науки, № 1 (116), 71 (1971).
105. Гайбакян Д. С.— В кн.: Металлургия рения. М.: Наука, 1970, с. 80.
106. Гайбакян Д. С., Саградян С- И.— Арм. хим. ж., 24, 668 (1971).
107. Гайбакян Д. С., Саградян С- И.— Ж. аналит. хим., 25, 2386 (1970).
108. Гайбакян Д. С., Саркисян Ж. В.— Там же, 29, 1991 (1974).
109. Гайбакян Д. С., Худавердян Д. X.— Арм. хим. ж., 28, 390 (1975).
110. Галлай 3. А., Типцова В. Г., Пешкова В. М.— Ж. аналит. хим., 12, 469 (1957).
111. Галлай 3. А., Шеина Н. М.— Там же, 16, 706 (1961).
112. Ганаго Л. И., Бухтеева Л. Н-—>,Там же, 32, 1537 (1977).
113. Ганиев А. Г., Назмитдинов М. К., Рыбное В. В., Московцева Г. А.— В кн.: Ядерная физика и ее применение. Ташкент: Фан, 1966, ч. I, с. 144.
114. Ганиев А. Г., Назмитдинов М. К., Сабиров С-— Изв. АН УзССР. Сер. физ.-мат. наук, № 1, 40 (1968).
115. Гиллебранд В. Ф., Ленделъ Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М.: Госхимиздат, 1960.
116. Голикова Н. Д., Ростовская А. А.— Химия и технол. топлив и масел, № 11, 52 (1969).
117. Голова Т. А. — Тр. Гос. н.-и. и проектн. ин-та нефт. маш., вып. 4 (14), 187 (1967).
118. Головатый Р. Н., Ощаповский В. В.— Укр. хим. ж., 28, 518 (1962).
119. Голубцова Р. В., Савватеева С. М., Ярошенко А. Д.— Зав. лаб.,
34, 1184 (1968).
120. Голубцова Р. Б., Ярошенко А. Д.— Ж. прикл. хим., 44, 1005 (1971).
121. Голъцберг И. М., Коваль Г. Л., Клемешов Г. А.— В кн.: Новые ме
182
тоды анализа на металлургических и металлообрабатывающих заводах. М.: Металлургия, 1964, с. 46.
122. Голъцберг И.М., Коваль Г. Л., Клемешов Г. А.— Тр. Укр. н.-и. ин-та металлов, вып. 11, 387 (1965).
123. Гончаренко А. С. Электрохимия ванадия и его соединений. М.: Металлургия, 1969.
124. Гончаров А. I., АринЛчева А. Т., Лукьянович II. М.— Шстп. Кшвськ. ун-та. Сер. ф!з. та х!м., 6, 90 (1966).
125. Гордеева М. Н.— В кн.: Проблемы современной аналитической химии. Л.: Изд-во ЛГУ, 1976, вып. 1, с. 131.
126. Гордеева М. Н., Рындина А. М.— Вести. ЛГУ, № 16, 147 (1972).
127. Гордеева М. Н., Рындина А. М.— Там же, № 22, 153 (1974).
128. Гордеева М. Н., Рындина А. М., Белянина Л. II.— В кн.: Инстру-
ментальные и химические методы анализа. Л.: Изд-во ЛГУ, 1973, с. 30.
129. Гордеева М. Н., Рындина А. М., Станевич Т. В.— Вести. ЛГУ, № 22, 126 (1976).
130. Горлова Н. М.— В кн.: Унификация и стандартизация методов анализа химического состава материалов. М.: МДНТП им. Ф. Э. Дзержинского, 1976, с. 17.
131. Горюшина В. Г., Бирюкова Е. Я— В кн.: Сборник научных трудов Гиредмета. М.: Металлургия, 1959, т. 2, с. 156.
132. Горянская Г. И., Каплан Б. Я., Ковалик И. В., Мерисов 10. И., Назарова М. Г.— Ж. аналит. хим., 27, 1498 (1972).
133. Григоренко Ф. Ф., Дубовеико Л. II.— Укр. хим. ж., 38, 814 (1972).
134. Григорьева М. Ф., Захаренко В. М., Церковницкая И. А.— Вести. ЛГУ, № 16, 133 (1972).
135. Григорьева М. Ф., Захаренко В. М., Церковницкая И. А.— Зав. лаб., 42, 660 (1976).
136. Григорьева М. Ф., Захарян Л. В., Церковницкая И. А.— Там же, 43, 153 (1977).
137. Григорьева М. Ф., Слесарь II. И., Церковницкая И. А.— В кн.: Проблемы современной аналитической химии. Л.: Изд-во ЛГУ, 1976, вып. 1, с. 113.
138. Григорьева М. Ф., Церковницкая И. А.— Вести. ЛГУ, № 10, 124 (1976).
139. Григорьева М. Ф., Церковницкая И. А.— В кн.: Проблемы современной аналитической химии. Л.: Изд-во ЛГУ, 1976, вып. 1, с. 25, 74.
140. Грикитп, И. А., Галушко Е. Г., Макаренко В. С., Пепгруиько М. II.— Тр. Всесоюз. н.-и. и проект, ин-та титана, 3, 283 (1969).
141. Гровс А. Анализ силикатов. М.: Изд-во иностр, лит., 1953.
142. Грум-Гржимайло С. Б., Щербина В. В.— Геохимия, № 11, 971 (1961).
143. Гусакова Н. Н., Еременко С. Н., Муштакова С. П., Фруми-на Н. С.— Ж. аналит. хим., 30, 721 (1975).
144. Гусарский В. В., Фридман Г. И. Эмиссионная спектроскопия аэрозолей в металлургии. М.: Металлургия, 1974.
145. Гусарский В. В., Фридман Г. И., Медведева М. С.— В кн.: Унификация и стандартизация методов анализа химического состава материалов. М.: МДНТП им. Ф. Э. Дзержинского, 1976, с. 134.
146. Гусев С. И., Бейлис Р. Г., Соколова Е. В.— Ж. аналит. хим., 6, 43 (1951).
147. Гусев С. И., Кумов В. И., Соколова Е. В.— Там же, 15, 180 (1960).
148. Гусев С. И., Шаламова Г. Г.— Там же, 22, 1357 (1967).
149. Гусев С. И., Шаламова Г. Г.— Там же, 23, 686 (1968).
150. Давыдов А. Л., Вайсберг 3. М.— Фотоколориметрические методы анализа черных, цветных металлов и руд. Изд-во АН УССР, 1943, с. 18.
151. Дарбинян М. В., Даниелян А. А.— Изв. АН АрмССР. Сер. хим. наук, 18, 462 (1965).
152. Дегтярева О. Ф-, Синицина Л. Г-, Негина В. Р., Чикишева Л. С.—
183
В кн.: Спектральные и химические методы анализа материалов. М.: Металлургия, 1964, с. 7.
153. Дегтярева О. Ф., Синицина Л. Г., Проскурякова А. Е.— Ж. аналит. хим., 18, 510 (1963).
154. Джабаров Ф. 3., Горбачев С. В.— Ж. неорг. хим., 9, 2399 (1964).
155. Добрицкая Ю. И.— Почвоведение, № 2, 35 (1969).
156. Долгорев А. В.— В кн.: Химия и химическая технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 438.
157. Долгорев А. В.— Там же, с. 447.
158. Долгорев А. В., Золотавин В. Л., Сериков Ю. А.— Зав. лаб., 40, 1425 (1974).
159. Долгорев А. В., Лукачина В. В., Карпова О. И.— Ж. аналит. хим., 29, 721 (1974).
160. Долгорев А. В., Пальчикова Т. И.— Там же, 31, 1168 (1976).
161. Долгорев А. В., Пальникова Т. И.— Там же, 30, 2020 (1975).
162. Долгорев А. В., Подчаймова В. Н., Дергачев В. Я.— В кн.: Современные методы анализа материалов. М.: Металлургия, 1969, с. 208.
163. Долгорев А. В., Сериков Ю. А.— Ж. неорг. хим., 21, 2065 (1976).
164. Драницкая Р. М., Гаврильченко А. И., Карпий Н. Л., Палей Л. Н,— Ж. аналит. хим., 26, 2137 (1971). •
165. Дымов А. И.— Технический анализ. М.: Металлургия, 1964.
165а. Дятлова Н. М., Темкина В. Я., Колпакова И. Д. Комплексоны. М.: Химия, 1970, с. 98.
166. Егоров В. II.— Ж. аналит. хим., 32, 82 (1977).
167. Елизарова Г. Л., Кузнецова А. С.— Там же, 23, 50 (1968).
168. Елфимов В. И., Безруков И. Я., Золотавин В. Л.— Ж. неорг. хим., 15, 2391 (1970).
169. Елфимов В.И., Безруков И. Я., Золотавин В. Л. — Тр. Ин-та химии УНЦ АН СССР, вып. 23, 122 (1971).
170. Еременко В. Я. Спектрографическое определение микроэлементов в природных водах. М.: Изд-во АН СССР, 1960.
171. Еременко С. И., Аграновская Л. А., Фрумина II. С., Будникова В. А.— Зав. лаб., 42, 780 (1976).
172. Ефимов Ю. В., Барон В. В., Савицкий Е. М. Ванадий и его сплавы. М.: Наука, 1969.
173. Жаворонкина Т. К., Скопинцев Б. А., Климов И. Т.— Океанология, 4, 205 (1964).
174. Жаровский Ф. Г., Сухомлин Р. И.— Ж. аналит. хим., 21, 59 (1966).
175. Жданов А. К., Вархударъян А. А.— Там же, 32, 92 (1977).
176. Жигалкина Т. С., Черкесов А. И.— Там же, 16, 505 (1961).
177. Жуков А. И., Гареев В. И., Маркова В. М.— Ж. неорг. хим., 7, 1724 (1962).
178. Жзнъ Хун-дз.— Кэсюэ тунбао (Kexue tongbao), № 6, 549 (1965); РЖХим, 1966, 13Г32.
179. Заду мина Э. А., Черкесов А. И.— Изв. вузов. Химия и хим. технол., 12, 1483 (1969).
180. Зайдель А. Н. Основы спектрального анализа. М.: Наука, 1965.
181. Зайдель А. Н., Иванова Т. Ф., Петров А. А., Федоров В. В., Чумакова Н. М.— Зав. лаб., 29, 693 (1963).
182. Зайдель А. И., Калитеевский И. И., ЛиписЛ. В., Чайка М. П. Эмиссионный спектральный анализ атомных материалов. М.: Физматгиз, 1960.
183. Зайдель А. Н., Прокофьев В. К., Райский С. М., Славный В. А., Шрейдер Е. Я. Таблицы спектральных линий. М.: Наука, 1977.
184. Закорина И. А., Лазеева Г. С., Петров А. А.— Ж. аналит. хим., 23, 1688 (1968).
185. Занина И. А.— Тр. Центр, н-и. ин-та черн. мет., вып. 66, 26 (1969).
186. Заринский В. А., Ермаков В. И. Высокочастотный химический анализ. М.: Наука, 1970.
187. Захария Н. Ф., Измайлова Д. Н., Стайков А. И.— Тр. по химии и хим. технол. (Горький), вып. 3 (24), 77 (1969).
184
188. Захария Н. Ф., Стайнов А. И.— Ж. аналит. хим., 26, 1348 (1971).
189. Захария Н. Ф., Стайнов А. И., Анбиндер И. С.— В кн.: Спектроскопия атомов и молекул. Киев; Наук, думка, 1969, с. 88.
190. Зинченко В. А., Баринова О. Д — В кн.: Титан и его сплавы. М.: Изд-во АН СССР, 1962, вып. 8, с. 251.
191. Зинченко В. А., Горулева А. И.— В кн.: Методы анализа веществ высокой чистоты. М.: Наука, 1965, с. 278.
192. Зозуля А. П. Кулонометрический анализ. Л.: Химия, 1968,
193. Золотавин В. Л.— Ж. аналит. хим., 2, 346 (1947).
194. Золотавин В. Л.— В кн.: Методы определения п анализа редких элементов. М.: Наука, 1961, с. 462.
195. Золотавин В. Л., Безруков И. Я., Санников Ю. И.— Ж. неорг. хим., 6, 584 (1961).
196. Золотавин В. Л., Букреев Ю. Ф., Музгин В. Н., Пророк М. М.— Тр. Ин-та химии УФ АН СССР, вып. 17, 159 (1970).
197. Золотавин В. Л., Букреев Ю. Ф., Толстов Л. К.— Ж. прикл. спек-троск., 9, 18 (1968).
198. Золотавин В. Л., Букреев Ю. Ф., Толстов Л. К., Безруков И. Я.— Там же, 2, 461 (1965).
199. Золотавин В. Л., Левашова Л. В.— Зав. лаб., 28, 161 (1962).
200. Золотавин В. Л., Левашова Л. Б., Долгорев А. В.— Ж. аналит. хим., 17, 336 (1962).
201. Золотавин В. Л., Федорова Н. Д.— Тр. ВНИИ станд. обр. и спектр, этал., 1, 31 (1964); 2, 92 (1965).
202. Золотов Ю. А. Экстракция внутрикомплексных соединений. М.: Наука, 1968.
203. Золотов Ю. А., Иофа Б. 3., Чучалин Л. К. Экстракция галогенидных комплексов металлов. М.: Наука, 1973.
204. Золотов Ю. А., Кузьмин И. М. Экстракция металлов ацилпиразоло-нами. М.: Наука, 1977.
205. Золотов Ю. А., Сизоненко А. Г., Золотовицкая Э. Я., Яковенко Е. И.— Ж. аналит. хим., 24, 20 (1969).
206. Золотухин В. К.— Ж. прикл. хим., 10, 1651 (1937).
207. Зырин Н. Г., Белицина Г. Д., Обухов А. И., Пацукевич 3. В.— Агрохимия, № 5, 119 (1964).
208. Зырин Н. Г., Обухов А. И,— В кн.: Методы определения микроэлементов в природных объектах. М.: Наука, 1976, с. 95.
209. Ивакин А. А., Гуревич В. А., Глазырин М. П.— Ж. неорг. хим., 19, 2397 (1974).
210 Ивакин А. А., Чуфарова И. Г., Петунина Н. И., Плетнев Р. И., Горшков В. В.— Там же, 22, 214 (1977).
211. Иванов Д. И., Иванова И. И., Орлова И. П.— Тр. Комис, по аналит. хим. АН СССР, 15, 306 (1965).
212. Иванов И. П., Михельсон Д. М.— В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1971, вып. 18, с. 85, 91.
213. Иванов И. П., Михельсон Д. М., Баранов С. В., Пофралиди Л. Г.— Там же, с. 100.
214. Иониты в цветной металлургии/Под ред. К. В. Лебедева. М.: Металлургия, 1975.
215. Исаков П. М. Количественный химический анализ руд и минералов методом растирания порошков. М.: Госгеолиздат, 1955.
216. Исии Т., Муся С.— Бунсэки кагаку (Japan Analyst), 14, 886 (1965); РЖХим, 1966, 15Г121.
217. Исии X., Эйнага X.— Там же, 19, 371 (1970); РЖХим, 1970, 24Г102.
218. Ито С.— Там же, 14, 15 (1965); РЖХим, 1966, 7Г135.
219. Йонэкубо Т., Сатакэ М., Мацумото Т.— Там же, 18, 640 (1969); РЖХим, 1970, 2Г105.
220. Йосимура Г., Уно С., Ногути X.— Там же, 12, 42 (1963); РЖХим, 1963, 19ГЗЗ.
221. Йосимура Т., Мива Т., Такэмура Т., Ито И.— Там же, 11, 1243 (1962); РЖХим, 1963, 17Г97.
7 В Н. Музгин и др.
185
222. Йоцуянаги Т.,Ито Д.,Аомура К.— Тамже, 18, 1498 (1969); РЖХим, 1970, 15Г179.
223. Кавамура К., Ватанабэ Т., Марата И., Фурукава Т.— Бунко кэн-кю (J. Spectroscop. Soc. Japan), 16, 225 (1968); РЖХим, 1968, 21Г100.
224. Калинин С. К., Файн Э. Е. Спектральный анализ минерального сырья. Алма-Ата: Изд-во АН КазССР, 1962.
225. Карабаш А. Г., Пейзулаев Ш. И.— В Кн.: Труды Всесоюзной научно-технической конференции по применению радиоактивных и стабильных изотопов и излучений в народном хозяйстве и науке. М.: Изд-во АН СССР, 1958, с. 36.
226. Карабаш А. Г., Пейзулаев Ш. И., Морозова Г. Г., Смиренки-на И. И.— Тр. Комис, по аналит. хим. АН СССР, 12, 25 (1960).
227. Карабаш А. Г., Пейзулаев Ш. И., Слюсарева Р. Л., Липатова В. М-— Ж. аналит. хим., 14, 94 (1959).
228. Карабаш А. Г., Пейзулаев Ш. И., Сотникова Н. П., Сазанова С. К.— Тр. Комис, по аналит. хим. АН СССР, 12, 108 (1960).
229. Карабаш А. Г., Пейзулаев Ш. И., Усачева В. П., Морозова Г. Г., Мешкова В. М-, Лобанова В. Л.— Ж. аналит. хим., 16, 217 (1961).
230. Карабаш А. Г., Самсонова 3. Н., Смирнова-Аверина Н. И., ,Пейзу-лаев III. И.— Тр. Комис, по аналит. хим. АН СССР, 12, 255 (1960).
231. Карленко Г. С., Бурцев В. Т., Ефимов Ю. В.— Ж. аналит. хим., 26, 1222 (1971).
232. Карпенко Л. И., Фадеева Л. А., Шевченко Л. Д.— Там же, 29, 454 (1974).
233. Карпов Ю. А., Кудинов Б. С., Кузьмин Л. Е., Пронман И. М., Федорченко В. Я.— В кн.: Активационный анализ в народном хозяйстве. Ташкент: Фан, 1974, с. 130.
234. Карпова О. И., Пилипенко А. Т., Лукачина В. В.— Ж. аналит. хим., 32, 1142 (1977).
235. Карташев Е. Р., Штаиь А. С.— Тр. ВНИИ радиац. техн., вып. 5, 117 (1970).
236. Карякин А. В., Павленко Л. И., Бабичева Г. Г.— Ж. аналит. хим., 26, 1344 (1971).
237. Карякин А. В., Павленко Л. И., Сафронова Н. С.— Там же, 30, 775 (1975).)
238. Карякин Ю. В., Завалъская А. В.— Там же, 23, 1742 (1968).
239. Катченков С. И. Спектральный анализ горных пород. Л.: Недра, 1964.
240. Качканарский ванадий/Под. ред. В. И. Довгопола, Н. Ф. Дуброва. Свердловск: Средне-Уральск. кн. изд-во, 1964.
241. Кашковская Е. А., Мустафин И. С.— Тр. Комис, по аналит. хим. АН СССР, И, 97 (1960).
242. Китайгородский И. И., Фролов В. К.— Зав. лаб., 26, 418 (1960).
243. Клячко Ю. А., Шапиро С. А. Курс химического качественного анализа. М.: Госхимиздат, 1960.
244. Коваленко П. Н.— В кн.: Электрохимические и оптические методы анализа. Ростов н/Д.: РГУ, 1963, с. 43.
245. Колътгоф И. М-, Белчер Р., Стенгер В. А., Матсуяма Дж. Объемный анализ. М.: Госхимиздат, 1961, т. 3, с. 719, 730.
246. Кольтгоф И. М-, Лингейн Дж. Дж. Полярография. М.; Л.: Госхимиздат, 1948.
247. Кондрашова В. П., Петрашень В. И.— Изв. вузов. Химия й хим. технол., 5, 210 (1962).
248. Коренман И. М. Микрокристаллоскопия. М.: Госхимиздат, 1955.
249. Корнилова В. И., Назарчук Т. Н.— Ж. аналит. хим., 23, 1866 (1968).
250. Корнилова О. А.— Там же, 29, 1427 (1974).
251. Королев Н. В., Рюхин В. В., Горбунов С. А. Эмиссионный спектральный микроанализ. Л.: Машиностроение, 1971.
252. Коростелева Н. А., Копрова Н. А.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 237.
253. Костромин А. И., Ахметов А. А., Орлова Л. Н.— Ж. аналит. хим., 25, 195 (1970).
186
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
275.
276.
277.
278.
Котляр Е. Е., Назарчяук Т. II.— Тамже, 16. 688 (1961).
Кохара X., Исибаси И., Ито 10.— Бунсэкп кагаку (Japan Analyst), 16, 315 (1967); РЖХпм. 1968, 23Г61.
Кохара X., Исибаси II., Ханамура И., Уэно К.— Там же, 15, 938 (1966); РЖХим, 1967, 11Г94.
Коцудзи К,— Там же, 13, 27 (1964); РЖХпм, 1964, 19Г64.
Краснов И. II., Севастьянов Ю. Г., Константинов И. О., Виноградова В. Г., Малухин В. В. — В кн.: Активационный анализ в народном хозяйстве. Ташкент: Фан, 1974, с. 133.
К раснопольская М. Б., Гаркун А. И., Залесских Т. II., Ганелина Е. III.— Зав. лаб., 43, 137 (1977).
Крауз Л. С., Карабаш А. Г., Пейзулаев Ш. II., Липатова В. М., Молева В. С.— Тр. Комис, по аналит. хим. АН СССР, 12, 175 (1960). Крейнгольд С. У., Божеволънов Е. А.— Зав. лаб., 31, 784 (1965).
Крейнгольд С. У., Божевотънов Е. А., Сосенкова Л. И. — В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1969, вып. 16, с. 175, 187, 196.
Крейнгольд С. У,, Пантелеймонова А. А., Жаворонкова 3. К.— В кн.: Методы анализа химических реактивов п препаратов. М.: ИРЕА, 1973, вып. 21, с. 183.
Крейнгольд С. У., Пантелеймонова А. А., Попонова Р. В,— Ж. аналит. хим., 28, 2179 (1973).
Кристаллев П. В., Кристаллева Л. Б.— Тр. Перн, политехи, пп-та, № 106, 3 (1971).
Круковская Е. Л., Талипов Ш. Т., Чижова Л,— Науч. тр. Ташкентск. ун-та, вып. 264, 48 (1961).
Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М.: Госхимиздат, 1959.
Кузнецов В. И, — Ж. общ. хим., 22, 2083 (1952).
Кузнецов В. И., Козырева Л. С.— Ж. аналит. хим., 8, 90 (1953).
Кузнецов В. И., Серякова И. В.— В кн.: Экстракция. Теория применения п аппаратура. М.: Атомиздат, 1962, вып. 2, с. 227.
Кузьмин II. М.— Ж. аналит. хим., 22, 451 (1967).
Кузьмин И. М., Беляев Б. II., Калиначенко В. Р., Якименко Л. М,—
Зав. лаб., 29, 691 (1963). t
Кузьмин И. М., Якименко Л. М., Калиначенко В. Р.— В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1968, вып. 15, с. 84.
279.
280.
281.
282.
283.
284.
Кульберг Л. М. Органические реактивы в аналитической химии. М.; Л.: Госхимиздат, 1950.
Кульберг Л. М., Альтерзон Г. С., Вельтман Р. И. Капельный анализ. М.; Л.: Госхимиздат, 1951.
Курода Р., Кирияма Т.— Бунсэки кагаку (Japan Analyst), 19, 1285 (1970); РЖХим, 1971, 7Г168.
Кусакина Н. П., Якимец Е. М.— Тр. Уральск, политехи, ин-та, № 121, 91 (1962).
Кучкина Е. Д., Горбенко Ф. П., Грановский И. В., Морозова Е. II., Якушевская Ф. Т.— В кн.: Материалы II Межотраслевого совещания по методам получения и анализа ферритовых материалов и сырья для них. М.: ИРЕА, 1969, ч. 2, с. 149.
Кюрегян С. К. Эмиссионный спектральный анализ нефтепродуктов. М.: Химия, 1969.
Лазарев №. М. Технология легких сплавов: (Научно-технический б юл. ВИЛСа), № 4, 76 (1977).
Лазарев А. II., Харламов И. П., Яковлев П. Я., Яковлева Е. ф. Справочник химика-аналитика. М.: Металлургия, 1976.
Лактионова И. В., Агеева Л. В., Симонова Л. В.— Ж. аналит. Хим., 26, 554 (1971).
Левашова Л. Б., Водопьянова В. П.— Тр. Уральск, политехи, ин-та, № 193, 14 (1971).
Левашова Л. В., Петрова Э. А.— Там же, № 226. 142 (1975).
7*
187
285. Левашова Л. Б., Усолкин А. Н.— Там же, с. 18.
286. Лекторская И. А.— В кн.: Теория и практика полярографического анализа. Кишинев: Штиинца, 1962, с. 261.
287. Лерман Р. В., Полонская М. С.— В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1971, вып. 18, с. 73.
288. Лесникова Е. Н.— Изв. АН СССР. Сер. физ., 18, 288 (1954).
289. Ливанов В. А., Горохов В.П., Голофаев Т.Н., Малявкина В. П.— Там же, 26, 914 (1962).
290. Лисецкая Г. С., Бакал Г. Ф.— Укр. хим. ж., 36, 709 (1970).
291. Лисиенко Д. Г., Муззин В. Н., Золотавин В. Л.— Ж. прикл. спект-роск., 15, 388 (1971).
292. Лобанов Е. М., Валиева М. Г., Усманова М. М.— В кн.: Нейтронноактивационный анализ. Ташкент: Фан, 1971, с. 73.
293. Лобанов Е. М., Минеалиев Г. Г.— В кн.: Активационный анализ элементного состава геологических объектов. Ташкент: Фан, 1967, с. 84.
294. Лобанов Е. М., Умаров М. У., Азизов Н. А., Нуманов И. У.— Докл. АН ТаджССР, 12, 33 (1969).
295. Лозановская И. И., Петрашенъ В. И.— Ж. аналит. хим., 22, 1196 (1967).
296. Лосев Н. Ф. Количественный рентгеноспектральный флуоресцентный анализ. М.: Наука, 1969.
297. Лукачина В. В., Пилипенко А. Т., Карпова О. И.— Ж. аналит. хим., 28, 86 (1973).
298. Лурье Ю. Ю. Справочник по аналитической химии. М.: Химия, 1971.
299. Лыткина Н. И., Преображенская Г. П.— В кн.: Материалы 2-го Уральского совещания по спектроскопии. Свердловск: Металлургиз-дат, 1959, с. 154.
300. Львов Б. В. Атомно-абсорбционный спектральный анализ. М.: Наука, 1966.
301. Львова О. В.— Зав. лаб., 12, 373 (1946).
302. Макаров В. А., Сопельняк Д. С.— Тр. Ставропольск с.-х. ин-та, 3, 150 (1973).
303. Мальцев В. Ф.— В кн.: Производство труб. Харьков: Металлургиздат, 1962, вып. 6, с. 151.
304. Мальцев В. Ф., Мазан Л. К., Ларченко А. Ф.— В кн.: Производство труб. М.: Металлургия, 1971, вып. 26, с. 210.
305. Мальцева Л. С., Шаламова Г. Г., Гусев С. И.— Ж. аналит. хим., 29, 2053 (1974).
306. Малютина Т. М., Орлова В. А.— Науч. тр. Н.-и. и проект, ин-та редкомет. пром-сти, 47, 66 (1975).
307. Мамиконян С. В., Воробьев Г. В., Козлов О. В., Мартищенко Л. Г.— Тр. ВНИИ радиац. техн., вып. 4, 111 (1970).
308. Мандельштам С. Л., Семенова Н. Н., Туровцева 3. М.— Ж. аналит. хим., 11, 9 (1956).
309. Маринов В-~ Химия и индустрия (НРБ), 43, 353 (1971).
310. Маров И. Н., Евтикова Г. А., Малофеева Г. И., Илларионова Е. В.— Ж. аналит. хим., 30, 1972 (1975).
311. Марченко 3. Фотометрическое определение элементов. М.: Мир, 1971, с. 128—137.
312. Масс-спектрометрический метод определения следов/Под ред. М. С. Чупахина. М.: Мир, 1975.
313. Маховка П. П.— Зав. лаб., 23, 533 (1957).
314. Мацумаэ А., Хасэзава Т.— Бунсэки кики (Anal, and Instrum.), 8, 508 (1970); РЖХим, 1971, ЗГ196.
315. Мацумура Т., Котани Н., Гото Т.— Бунсэки кагаку (Japan Analyst), 19, 1393 (1970); РЖХим, 1971, 11Г170.
316. Маширева Л. Г., Зимина К. И., Иванов Н- Г.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 195.
317. Маэкава С., Като К.— Бунсэки кагаку (Japan Analyst), 17, 70 (1968); РЖХим, 1968, 21Г106.
188
318. Мелентьев В. И., Федорова Л. Я.— В кн.: Технический анализ в металлургии. М.: ВИНИТИ АН СССР, 1966, с. 5.
319. Менке Г., Менке Л. Введение в лазерный эмиссионный микроспект-ральный анализ. М.: Мир, 1968.
320. Методы анализа металлов и электролитных ванн. М.: Оборонгиз, 1944.
321. Мещеряков Р.П., Обливанцев А.Н., Рыбасов А. Г., Тронов Г. 11., Яковлев Б. М.— Тр. Н.-и. ин-та ядерной физики, электроники и автоматики при Томск, политехи, ин-те, вып. 6, 16 (1976).
322. Миха Т., Фукуда К., Мидзуикэ А,— Бунсэки кагаку (Japan Analyst), 19, 786 (1970); РЖХим, 1970, 24Г183.
323. Мидзуно Т., Мацумура Т.— Нихон киндзоку гаккайси (J. Japan Inst. Metals), 33, 944 (1969); РЖХим, 1970, 8Г164.
324. Минин А. А., Милютина Л. ЛУч. зап. Пермск. ун-та, № 111, 130 (1964).
325. Мирзаева Х.А., Татаев О. А.— В кн.: Физико-химические методы анализа и контроля производства. Махачкала: Дагестанский ун-т, 1973, с. 66.
326. Мирзакасимов Т. М., Рахмапгуллаев К., Талипов Ш. Т.— Узб. хим. ж., № 6, 15 (1970).
327. Миямото М., Катагири МБунсэки кагаку (Japan Analyst), 14, 443 (1965); РЖХим, 1966, 1Г64.
328. Младенцева О. И., Горожанкина Н. П., Сухенко К. А., Аксенова А . В.— Тр. Комис, по аналит. хим. АН СССР, 12, 355 (1960).
329. Морачевский Ю. В., Гордеева М. Н.— Вести. ЛГУ, 10, 148 (1957).
330. Морачевский Ю. В., Гордеева М. Н., Круглова Т. Е.— Зав. лаб., 24, 790 (1958).
331. Морачевский Ю. В., Сахаров А. А.— Ж. аналит. хим., 13, 83, 467 (1958).
332. Морачевский Ю. В., Церковницкая И- А.— Там же, 16, 106 (1961).
333. Моргун И.Д., Ларина О. Д., Смирнова А. В., Домогапгских Л. А., Поплавская Н. Ф.— Зав. лаб., 35, 265 (1969).
334. Морден Э. А., Росинская Э. С., Власов Н. А., Жесько В. П.— Там же, 39, 409 (1973).
335. Морошкина Т. М., Коврижных В. М., Мельников Ю. А.— Ж. аналит. хим., 26, 1352 (1971).
336. Морошкина Т. М., Прокофьев В. К.— Там же, 11, 714 (1956).
337. Моррисон Дж., Фрейзер Г. Экстракция в аналитической химии. М.: Госхимиздат, 1961.
338. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф.— Ж. аналит. хим., 19, Ш (1964).
339. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 29.
340. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф., Балаев В. Н.— Зав. лаб., 30, 697 (1964).
341. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф., Улыбышева Л. В.— Тр. ВНИИ станд. обр. и спектр, этал., 1, 127 (1964).
342. Музгин В. Н., Курбатова И. Б.— В кн.: Химия и технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 420.
343. Музгин В. Н., Лисиенко Д. Г.— Ж. аналит. хим., 29, 666 (1969); Ж. прикл. спектроск., И, 583 (1969).
344. Музгин В. Н., Москаленко Н. И., Суриков В. Т.— В кн.: Вопросы общей и прикладной физики. Алма-Ата: Наука, 1969, с. 238.
345. Муликовская Е. П.— Тр. Всесоюз. н.-и. геол, ин-та, 117, 79 (1964).
346. Муминов В. А., Мухаммедов С., Султанов В., Хайдаров Р.А.— В кн.: Ядерно-физические методы анализа и контроля технологических процессов. Ташкент: Фан, 1974, с. 50.
347. Мустафин И. С., Кашковская Е. А,— Зав. лаб., 23, 519 (1957).|
348. Мустафин И. С., Нехаенко Т. М., Фрумина Н. С. Органические реактивы для определения неорганических ионов. Ассортимент реактивов на ванадий. М.: НИИТЭХИМ, 1969.
189
349. Муся С., Такао II.— Бунсэки кагаку (Japan Analyst), 9, 233 (1960); РЖХим, 1960, № 21, 84395.
350. Мухина 3. С., Никитина Е. И., Туранова Л. М., Володарская Р. С., Поляк Л. Я., Тихонова А. А. Методы анализа металлов и сплавов. М.: Оборонгиз, 1959.
351. Назаренко В. А., БирюЪ E.A.— IB. аналит. хим., 10, 28 (1955).
352. Назаренко В. А., Бирюк Е. А.— Зав. лаб., 25, 28 (1959).
353. Назаренко В. А., Полуэктов Н.С. Полумикрохимический анализ минералов и руд. М.; Л.: Госхимиздат, 1950'.
354. Назаренко В. Л., Шустова М. В.— В кн.: Спектральные и химические методы анализа материалов. М.: Металлургия, 1964, с. 150.
355. Намики X., Ватанабэ К.— Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 85, 371, АЗО (1964); РЖХим, 1965, 17Г98.
356. Намики X., Ватанабэ К., Навамура Ф.— Бунсэки кагаку (Japan Analyst), 13, 98 (1964); РЖХим, 1965, 4Г66.
357. Нзуэн Ши Зунъ, Рыженко В. Л., Шарое,ский Ф. Г.— Укр. хим. ж., 35, 206 (1969).
358. Педлер В. В., Иванов Н. П., Шокина II. Т. — Науч. тр. Н.-и. и проект, ин-та редкомет. иром-сти, 10, 399 (1963).
359. Педлер В- В., Шокина Н. Т., Андриканис Э. Н.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 90.
360. НепеинаЛ.А.— В кн.: Количественный анализ минералов и горных пород физическими методами/Тр. Ин-та геологии и геофизики СО АН СССР, 1961, вып. 32, с. 61.
361. Несмеянов Ан. Н., Лапицкий А. В., Руденко Н- П. Получение радиоактивных изотопов. М.: Гостехиздат, 1954.
362. Пехаенко Т. М., Фрумина II. С., Мустафин И. С., Высокосова И. А.— Тр. Комис, по аналит. хим. АН СССР, 17, 352 (1969).
363. Нигай К. Г., Турсуков А. А., Рахимов X. Р.— Тр. Ташкенток., ун-та, вып. 513, 7 (1976).
364. Никитина Е. И. Ускоренные полумикрохимические методы анализа металлов и сплавов. М.: Госхимиздат, 1956.
365. Никитина О. Н.— В кн.: Методы анализа веществ высокой чистоты. М.: Наука, 1965, с. 67.
366. Никитина О. Н.— В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1966, вып. 12, с. 46.
366а. Никитина О. Н., Зилъберштейн X. И.— Тр. по химии и хим. технол. (Горький), вып. 3 (24), 79 (1969).
367. Никурашина М. Л., Фрумина Н. С.— В кн.: Физико-химические исследования в области органических и некоторых неорганических соединений. Куйбышев: Куйбышевск. политехи, ин-т, 1974, с. 15.
368. Нисида X.— Бунсэки кагаку (Japan Analyst), 13, 1259 (1964); РЖХим, 1965, 24Г176.
369. Нисикава С., Никагава Й., Сатакэ М., МацумотоТ.— Там же, 15, 944 (1966); РЖХим, 1967, 11Г95.
370. Новак В. П., Мальцев В. Ф., Боговина В. И.— Зав. лаб., 31, 295 (1965).
371. Новиков А. И., Рузанкин В. И., Дедова В. Ф.— Радиохимия, 11, 347 । (1969).
372. Новиков В. А.— Советская геология, 8, 75 (1945).
373. Новоселова И. М., Пушкарева Т. А., Барковский В. Ф.— Тр. Уральск, н.-и. хим. ин-та, вып. 26, 62 (1971).
374. Носака X., Нагасима К., Фудзисиро М.— Титаниуму, 7, 101 (1959); РЖХим, 1960, № 11, 42450.
375. Боткина М. А., Петрова Е. И., Черкашина Т. В., Чернихов Ю. А.— Тр. Комис, по аналит. хим. АН СССР, 15, 80 (1965).
376. Овруцкий М. И., Фрезер С. В., Голенкова Л. Г., Белозуб Т. Н.— В кн.: Методы анализа химических реактивов и препаратов. М.: ИРЕА, 1971, вып. 18, с. 59.
377. Ода Н., Цуноо С., Opuma II.— Нихон киндзоку гаккайси (J. Japan Inst. Metals), 24, 355 (1960); РЖХим, 1961, 11Д176.
378. Ольховская Р. В., Капелюх М. В., Иванова В. И., Масалова И. С., Лубеницкая И. Я.— Тр. Н.-и. и проект, ин-та азотн. пром-сти и прод. орг. синт., вып. 5, ч. 2, 17 (1970).
379. Остроброд Б. Г., Маркман А. Л., Абдусаламова 9. В.— Изв. вузов. Химия и хим. технол., 16, 843 (1973).
380. Остякова А: К., Салибаев Т. О., Азербаева Р. Г.— В кн.: Науч. тр. Казахск. политехи, ин-та, № 33, 526 (1971).
381. Отраслевые технические инструкции РЭТИ, № 170—58, № 174—58, № 175—58. М.: ГИРЕДМЕТ, 1958.
382. Охта Н., Куме И., Тераи М., Араки Т-— Бунсэки кагаку (Japan Analyst), 24, 206 (1975); РЖХим, 1975, 18Г170.
383. Павлова И. М., Абетова Э. К.~ В кн.: Химия и химическая технология. Алма-Ата: Казахск. ун-т, № 3/4, 1965, с. 298.
384. Павлова И. М., Сангина О. А.— Изв. АН КазССР. Сер. хим., 2, 33 (1967).
385. Палей П. Н.— В кн.: Материалы Международной конференции по мирному использованию атомной энергии. Женева, 1955, М.: Металлург-издат, 1958, т. 8, с. 268.
386. Палъникова Т. И., Холмогоров С. Ы., Чеснокова Н. А.— В кн.: Химия и технология ванадиевых соединений. Пермь: Кн. изд-во, 1974, с. 443.
387. Пальникова Т. И., Холмогоров С. Н., Чеснокова Н. А.— Зав. лаб., 37, 1429 (1971).
388. Панталер Р. П., Черноморд Л. Д.~ В кн.: Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты. Харьков: ВНИИ монокристаллов, 1972, ч. 2, с. 92.
389. Пахолков В- С., Корбут А. Я.— Изв. вузов. Цветн. металлург., № 3, 116 (1963).
390. Пахолков В. С., Корбут А. Я.— Там же, № 5, 100 (1962).
391. Пахолков В. С., Симаков С. Е.— Там же, № 5, 82 (1964).
392. Певцов Г. А., Рагинская Л. К., Манова Т.Г., Сотникова В. С.— Тр. Всесоюз. н.-и. ин-та хим. реактивов и особо чистых хим. веществ. М.: ИРЕА, 1969, вып. 31, с. 150.
393. Петров В. И. Оптический и рентгеноспектральнып анализ. М.: Металлургия, 1968.
394. Пилипенко А. Т., Карпова О. И., Луканина В. В.—Ж. аналит. хим., 32, 1369 (1977).
395. Пилипенко А. Т., Карпова О. И., Луканина В. В., Грачевский В. В.— Там же, 27, 78 (1972).
396. Пилипенко А. Т-, Костышина А. П., Назарчук Н- М.— Укр. хим. ж., 42, 521, 633 (1976).
397. Пилипенко А. Г., Лория II. В,-Сообщ. АН ГССР, 62 , 313 (1971).
398. Пилипенко А. ТМитрополигпска Е. В., Луковская И. М.— Укр. хим. ж., 41, 1196 (1975).
399. Пилипенко А. Г., Середа И. П., Шпак Э. А.— Зав. лаб., 32, 660 (1966).
400. Пилипенко А. Т-, Шпак 9. А., Курбатова Г. Г.— Ж. аналит. хим., 22, 1014 (1967).
401. Пилипенко А. Т-, Шпак 9. А., Рубан П. П.— Укр. хим. ж., 29, 1209 (1963).
402. Пихтин А.Н.— Зав. лаб., 31, 559 (1965).
402а. Плетнев Р. Н., Степанов А. П., Музгин В. Н.— Тр. Уральск, политехи. ин-та, № 167, 149 (1968).
403. Поваренных А. С. Кристаллохимическая классификация минеральных видов. Киев: Наук, думка, 1966.
404. Пограничная Р. М., Резник Б. Е., Зезянова А. Г., Перубащенко В. В.— Зав. лаб., 39, 948 (1973).
405. Пограничная Р. М., Резник Б. Е., Перубащенко В. В., Зезянова А. Г., Цевина А. В.— Ж. аналит. хим., 30, 180 (1975).
406. Подвальная Р. Л.— В кн.: Методы химического анализа минерального сырья. М.: Недра, 1965, вып. 8, с. 23.
407. Подчайнова В.Н., Долгорев А. В., Дергачев В. Я.— Зав. лаб., 31, 790 (1965); Ж. аналит. хим., 21, 53 (1966).
191-
408. Поливанова Н.' Г., Фрадкин Э. Г.— Зав. лаб., 33, 970 (1967).
409. Полонская И. А., Мирошник Е- А.— Там же, 40, 1196 (1974).
410. Полотебнова Н. А., Крачун С. В., Данилина Л. М.— Ж. неорг. хим., 21, 3172 (1975).
411. Полуэктов Н. С., Лауэр Р. С., Огниченко С. Ф.— Зав. лаб., 37, 1050 (1971).
412. Полуэктов И. С., Назаренко В. А.— Ж. прикл. хим., 10, 2105 (1937).
413. Поляк Л. Я.— Ж. аналит. хим., 32, 278 (1977).
414. Поляк Э. А., Городенцева Т. Б.— Тр. Всесоюз. н.-и. ин-та станд. обр. и спектр, этал., 6, 41 (1970).
415. Поляк Э. А., Перкина Л. С.— Зав. лаб., 29, 161 (1963).
416. Поляков П. М„ Русанов А. К.— Там же, 21, 1070 (1955).
417. Пономарев А. И. Методы химического анализа силикатных и карбонатных горных пород. М.: Изд-во АН СССР, 1961.
418. Пономарева А. А., Агринская Н. А., Анохина Л. Г.— В кн.: Новые методы химического анализа материалов. М.: МДНТП им. Ф. Э. Дзержинского, 1971, № 1, с. 88.
419. Попова В. Ф., Маркман А. Л.— Узб. хим. ж., № 4, 18 (1966); Тр. Ташкентск. политехи, ин-та, вып. 64, 3 (1970). '
420. Прайс В. Аналитическая атомно-абсорбционная спектроскопия. М.: Мир, 1976.
421. Пржевальский Е. С., Николаева Е. Р., Климова Н. С.— Вести. МГУ. Сер. мат., мех., астр., физ., хим., 13 (3), 217 (1958).
422. Прокофьев В. К., Морошкина Т. М., Богданова И. В.— Физ. сб. Львовск. гос. ун-та, вып. 4 (9), 112 (1958).
423. Пршибил Р. Аналитическое применение этилендиаминтетрауксусной кислоты и родственных соединений. М.: Мир, 1975.
424. Пршибил Р. Комплексоны в химическом анализе. М.: Изд-во иностр, лит., 1960.
425. Пчелинцев Д. А.— В кн.: Методы химического анализа и химического состава минералов. М.: Наука, 1967, с. 39.
426. Пятков А. Г., Романенко И. М., Шушпанов А. П., ЛукашВ.И.—• Зав. лаб., 41, 431 (1975).
427. Пятницкий И. В., ЕрисовВ. Ю.— Укр. хим. ж., 29, 1088 (1963).
428. Пятова В. Н., Алексеева В. М., Русанов А. К.— В кн.: Спектральный анализ в геологии. М.: Наука, 1971, с. 185.
429. Рабовский Г. В., Кузнецова Т. Ф., Белоногова В. А.— Ж. аналит. хим., 14, 578 (1959).
430. Рагинская Л. К., Манова Т. Г., Сотникова В. Н.— Зав. лаб., 36, 1348 (1970).
431. Рапепорт Л. Г., Юткевич Р. М.— Химия и технол. топлив и масел, № 3, 57 (1975).
432. Рафф Е. Л,— Зав. лаб., 31, 184 (1965).
433. Рахматуллаев К., Рахматуллаева М. А., Шоазизов Н.— Узб. хим. ж., № 5, 25'(1970).
434. Реакции и реактивы для качественного анализа неорганических соеди-нений/Под ред. А. С. Комаровского. М.; Л.: Госхимиздат, 1950.
435. Ревин Ю. В., Клен А. А., Ларин Н. В.— Ж. аналит. хим., 30, 1007 (1975).
436. Резник В. Е., Ганзбург Г. М., Филь А. П.— В кн.: Комплексообразование, межмолекулярное взаимодействие и соосаждение в некоторых системах. Днепропетровск: Днепропетр. ун-т, 1970, с. 164.
437. РечницГ. А. Электроанализ при контролируемом потенциале. Л.: Химия, 1967.
438. Риман В., Уолтон Г. Ионообменная хроматография в аналитической химии. М.: Мир, 1973.
439. Рипан Р., Четяну И. Неорганическая химия. М.: Мир, 1972. Т. 2.
440. Ростокер У. Металлургия ванадия. М.: Изд-во иностр, лит., 1959.
441. Рубан II. Н., Виноградова К. А., Исаева С. М., Аветисян Ю. А.— Тр. Ин-та металлург, и обогащ. АН КазССР, 12, 120 (1965).
192
442. Рудневский II. К., Демарии В. Т., Туманова А. II., Молянова А. С.— Тр. по химии п хим. технол. (Горький), вып. 3 (33), 84 (1974).
443. Руководство ио препаративной неорганической химии/Под ред. Г. Брауэра. М.: Изд-во иностр, лит., 1956.
444. Русанов А. К. Основы количественного спектрального анализа руд и минералов. 2-е изд. М.: Недра, 1978.
445. Рыспекова 3. А., Дрояронова И. С.— В кн.: Исследования в области химических и физических методов анализа минерального сырья. Алма-Ата; Казахск. н.-и. ин-т минер, сырья, 1971, с. 156.
446. Рябчиков Д. И., Лазарев А. II., Лазарева В. И.— Ж. аналит. хим., 19, 1110 (1964).
447. Рябчиков Д. II., Сенявин М. М,— В кн.: Материалы Международной конференции по мирному использованию атомной энергии. Женева, 1955. М.: Металлургиздат, 1958, т. 8, с. 328.
448. Саввин С. Б., Минеева В. А., ОхаиоваЛ.А., Пачаджанов Д. Н.— Ж., аналит. хим., 26, 2364 (1971).
449. Саввин С. Б., Прописцова Р. Ф.— Там же, 23, 813 (1968).
450. Самсонов Г. В. Тугоплавкие соединения: Справочник по свойствам: п применению. М.: Металлургиздат, 1963.
451. Самуэльсон О. Ионообменное разделение в аналитической химии. М.;. Л.: Химия, 1966.
452. Санников Ю. И., Золотавин В. Л., Безруков И. Я.— Ж. неорг. хим., 8, 923 (1963).
453. Сапи.р А. Д.— Зав. лаб., 24, 949 (1960).
454. Свентицкий II. С. Визуальные методы эмиссионного спектрального анализа. М.: Физматгиз, 1961.
455. Селинов И. П. Изотопы. М.: Наука, 1970. Т. 1.
456. Сембаев Д. X., Саурамбаева Л. И., Ким С. М.— Ж. аналит. хим., 33, 391 (1978).
457. Сендел Е. Б. Колориметрические методы определения следов элементов. М.: Мир, 1964.
458. Сер^овская В. В.— Тр. Уральск, политехи, ин-та, № 121, 81 (1962).
459. Сизоненко II. Т.. Золотовицкая Э. С., Яковенко Е. И., Беленко Л. Э.— В кн.: Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты. Харьков: ВНИИ монокристаллов, 1971, ч. 2, с. 50.
460. Симакова Л. К.. Бабенко Н. А., Бусев А. И.— В кп.: Химия и химическая техполон'я ванадиевых соединений. Пермь: Кп. изд-во, 1974, с. 426.
461. Симапуки Т,— Бунсэки кагакм (Japan Analyst), 16, 1141 (1967); РЖХим, 1968, 19Г145.
462. Симонова Л. В., Павленко Л. И., Карякин А. В.— В кн.: Спектральный анализ в геологии. №.: Наука, 1971, с. 227.
463. Синякова С. И., Цветкова Л. А.— Цит. по [267].
464. Скотников С. А.— Тр. Ин-та металлург, им. А. А. Байкова АН СССР, вып. 6, 117 (1960).
465. Скотников С. А., Лазарева И. Ю.— Зав. лаб., 36, 420 (1970).
466. Славии В. Атомно-абсорбпионная спектроскопия. И.: Химия, 1971.
467. Славинский М. П. Физико-химические свойства элементов. М.: Металлургиздат, 1952.
468. Соболев М. Н. Извлечение ванадия и титана из уральских титаномаг-петитов. М.; Л.: ОНТИ, 1936.
'469 . Соколович В. Б.. ГомбаеваГ.Е.— Изв. Томск, политехи, ин-та, № 250, 16 (1975).
470. Соложенкин П. М., Сидоренко Г. Г.— Докл. АН ТаджССР, 16, 45 (1973).
471. Соломатин В. Т., Козина Г. В., Ржавичев С. П., Артемова Т. И.— Ж. аналит. хим., 32, 1742 (1977).
472. Сангина О. А., Вектурова Г. Б., Захаров В. А.— В кн.: Химия и химическая технология. Алма-Ата: Казахск. ун-т, 1968, вып. 7/8, с. 221.
473. Сангина О. А., Бектурова Г. Б., Захаров В. А., Пичугина Т. Г.—
193
В кн.: Прикладная и теоретическая химия. Алма-Ата: Казахск. ун-т, 1976, вып. 7, с. 67.
474. Сангина О. А., Савицкая И. С.— Зав. лаб., 29, 401 (1963).
475. Сангина О. А., Студенская Л. С., Вектурова Г. В., Маслова П. И., Городенцева Т. В.— Тр. ВНИИ станд. обр. и спектр, этал., 4, 112 (1968).
476. Сотникова Н. П., Романович Л. С., Пейзулаев Ш. И., Карабаш А. Г.— Тр. Комис, по аналит. хим. АН СССР, 12, 151 (1960).
477. Спектральный анализ чистых веществ/ Под ред. X. И. Зильберштейна. Л.: Химия, 1971.
477а. Справочник химика. М.; Л.: Химия, 1965, т. 3, с. 762.
478. Старый. Экстракция хелатов. М.: Мир, 1966.
479. Степин В. В., Поносов В. И., Мельчекова 3. Е.— В кн.: Ионообменная технология. М.: Наука, 1965, с. 93.
480. Степин В. В., Силаева Е. В., ПлиссА. М., Курбатова В. И., Федорова И. Д., Поносов В. И.— В кн.:’ Анализ черных металлов, сплавов и марганцевых руд. М.: Металлургия, 1971.
481. Степина И. П., Масалович В. М., Черных М. И.— Тр. Уральск, н-и. хим. ин-та, вып. 21, 31 (1971). ,
482. Столяров К. П.— Вести. ЛГУ, № 22, 140 (1963).
483. Столяров К. П., АгрестФ. В.— Ж. аналит. хим., 19, 457 (1964).
484. Столяров К. П., Григорьев Н. И. Введение в люминесцентный анализ неорганических веществ. Л.: Химия, 1967.
485. Страутиня А. К., Шиманская М. В.— Изв. АН ЛатвССР. Сер. хим. наук, № 5, 535 (1967).
486. Страутиня А. К., Шиманская М. В.— Там же, № 6, 747 (1970).
487. Студенская Л. С., Макагонова Л. И.— Тр. ВНИИ станд. обр. и спектр, этал., 1, 12 (1964).
488. Судзуки М., Такэупги Ц.— Когё кагаку дзасси (J. Chem. Soc. Jap. Ind. Chem. Sec.), 66, 191 (1963); РЖХим, 1964, 1Г168.
489. Суриков В. T., Ваусова Н. В., Музгин В. И.— Тр. Ин-та химии УНЦ АН СССР, вып. 25, 152 (1973).
490. Сухов Г. В.— В кн.: Методы определения микроэлементов в природных объектах. М.: Наука, 1976, т. 3, с. 141.
491. Сухоручкина А. С., Усатенко Ю. И.— Зав. лаб., 35, 647 (1969).
491а. Сырокомский В. С., Авилов В. В.— Зав. лаб., 17, 1163 (1951); 19, 135 (1953).
492. Сысоев Л. П., П олывянный И. Р.— Тр. Ин-та металлург, и обогащ. АН КазССР, 25, 54 (1967).
493. Таганов К. И. Спектральный анализ металлов и сплавов с предварительным отбором пробы. М.: Металлургия, 1968.
494. Талалаев В. М., Миронова О. И., Ахмед М. М.— Ж. аналит. хим., 26, 1327 (1971).
495. Талипов III. Т., Джиянбаева Р. X., Горъковая Г. П.— Науч. тр. Ташкенток. ун-та, вып. 264, 91 (1964).
496. Талипов Ш. Т., Джиянбаева Р. X., Мартиросов А. Е.— И ЗВАН КазССР. Сер. хим., № 6, 1 (1968).
497. Талипов Ш. Т., Чапрасова Л. В., Урманов Г., Ташходжаева А . Т., Иноятов А.— Докл. АН УзССР, № 4, 37 (1974).
498. Тананаев Н. А. Дробный анализ. М.; Л.: Госхимиздат, 1950.
499. Тананаев И. А. Капельный метод. М.; Л.: Госхимиздат, 1954.
500. Танзмура Т,— Бунсэки кики (Anal, and Instrum.), 7, 806 (1969); РЖХим, 1970, 14Г137.
501. Теплоухов В. И. Экспресс-анализ стали. М.: Металлургиздат, 1961.
502. Тилепова 3. С.— В кн.: Вопросы общей и прикладной физики. Алма-Ата: Наука, 1972, с. 201.
503. ТипцоваВ.Г., Дворцан А. Г., Голицына М. И.— Ж. аналит. хим., 23, 1684 (1968).
504. Тихонов В. И,— Там же, 21, 1172 (1966).
505. Тихонов В. Н— Там же, 26, 2142 (1971).
194
506. Тихонов В. И.— В кн.: Химия и химическая технология. Чебоксары: Чувашек, ун-т, 1971, вып. 2, с. 64.
507. Тихонов В. Н., Гранкина М. Я., Вернигора В. Я.— Ж. аналит. хим., 22, 359 (1967).
508. Тихонов В. И., Подчайнова В. И.— Изв. вузов. Химия и хим. технол., 8, 724 (1965).
509. Тихонова А. ИБогданченко А. И., Спесивцева Н. С.— Тр. Всесоюз. н.-и. и проектн. ин-та титана, 1, 264 (1967).
510. Тихонова А. И., Усатенко IO. И., Харькова Л. Т.— Там же, 2, 366 (1968).
511. Толмачев В. Н., Серпухов Л. Н.— Ж. физ. хим., 30, 134 (1956).
512. Толстов Л. К., Ивакин А. А., Фотиев А. А.— Тр. Ин-та химии УФ АН СССР, вып. 20, 40 (1970).
513. Томиока X.— Бунсэки кагаку (Japan Analyst), 12, 271 (1963); РЖХим, 1963, 21Г85.
514. Топалов Л. И., Шаевич А. В., Шубина С. Б. Спектральный анализ ферросплавов. Свердловск: Металлургиздат, 1962.
515. Торопов Н. А., Варзаковский В. И., Бондарь И. А., Удалов Ю. И. Диаграммы состояния силикатных систем: Справочник. Л.: Наука, 1970. Т. 2.
516. Торопов И. А., Варзаковский В. И., Лапин В. В., Бурцева Н. Н. Диаграммы состояния силикатных систем: Справочник. Л.: Наука, 1969. Т. 1.
517. Трамм Р. С.— В кн.: Аналитическая химия редких металлов и полупроводниковых материалов. М.: МДНТП им. Ф. Э. Дзержинского, 1970, с. 120.
518. Трамм Р. С., Певзнер К. С., Кушнарева Р. А.— В кн.; Химические методы анализа ниобия и сплавов на его основе. М.: ОНТИ ГИРЕДМЕТ, 1970, с. 36, 38.
519. Тремийон Б. Разделение на ионообменных смолах. М.: Мир, 1967.
520. Третьяк 3. Л.— Шсник. Харк1в шиптехн. iH-ту, № 92, вып. 6, 22 (1974).
521. Умланд Ф., Янсен А., ТиригД., ВюншГ. Комплексные соединения в аналитической химии: (Теория и практика применения). М.: Мир, 1975.
522. Усатенко Ю. И., Бондаренко М. М., Гринберг Е. И.— Зав. лаб., 19, 500 (1953).
523. Файглъ Ф., Ангер В. Капельный анализ неорганических веществ. М.г Мир., 1976. Т. 1.
524. Федоровская Н. П., Миессерова Л. В.— Тр. Ин-та горюч, ископ., 23, 3 (1967).
525. ФедяшинаА. Ф., Юделевич И. Г., Строкина Т. Г.— Изв. СО АН СССР. Сер. хим. наук, вып. 2, № 7, 71 (1965).
526. Физические методы анализа следов элементов/Под ред. Д. Мориссо-на. М.: Мир, 1967.
527. Филенка А. И.— Зав. лаб., 29, 1423 (1963).
528. Филипов Д., Кучева М.— Докл. Волг. АН, 17, 467 (1964).
529. Финкельштейн Д. Н., Борецкая В. А. Методы анализа минерального сырья. М.: Госгеолиздат, 1958, с. 183.
529а. Фишер С., Кунин Р,— В кн.: Материалы Международной конференции по мирному использованию атомной энергии. Женева, 1955. М.: Металлургиздат, 1958, т. 8, с. 343.
530. Фотиев А. А., Ивакин А. А.— Тр. Ин-та химии УФ АН СССР, вып. 19, 9 (1970).
531. Фотиев А. А., Шульгин Б. В., Москвин А. С., Гаврилов Ф.Ф. Ванадиевые кристаллофосфоры: Синтез и свойства. М.: Наука, 1976.
532. Фрйткин В. Г.— Зав. лаб., 30, 170 (1964).
533. Фрумина И. С.— Тр. Комис, по аналит. хим. АН СССР, И, 420 (1960).
534. Фрумина Н. С., Мустафин И. С.— Ж. аналит. хим., 15, 671 (i960).
535. Фрумина Н. С., Мустафин И. С., Никурашина М. Л., Милованова Г. С.— Тр. Комис, по аналит. хим. АН СССР, 17, 138 (1969).
195
536. Хадеева Л. А., Талипов Ш. Т.— Науч.тр. Ташкентск. ун-та, вып. 264, 130 (1964).
537. Хара С. Бунсэки кагаку (Japan Analyst), 10, 629 (1961); РЖХим, 1962, 8Д12.
538. Харламов И. П. Спектрофотометрический анализ сплавов. М.: Металлургия, 1969, с. 71, 170.
539. Харламов И. П., Яковлев П. Я., Лыкова М. И.— Зав. лаб., 28, 802 (1962).
540. Хасимото С., Като С.— Сумитомо Киндзоку (Sumitomo Metals), И, 147 (1959); РЖХим, 1962, 8Г150.
541. Хаяси С. Фукуи дайгаку гакугэй гакубу кие (Mem. Fac. Liber. Arts Fukui Univ.), 2, № 12, 37 (1962); РЖХим, 1963, 22Г58.
542. Херине P. Хелатообразующие ионообменники. М.: Мир, 1971.
543. Химический и спектральный анализ в металлургии: Практическое руководство. М.: Наука, 1965.
544. Химия и технология редких и рассеянных элементов. Т. 1. Химия редких и рассеянных элементов/Под ред. К. А. Большакова. М.: Высшая школа, 1965.
545. Химия пятивалентного ванадия в водных растворах.— Тр. Ин-та химии УНЦ АН СССР, вып. 21, 64 (1971).
546. Хисано К., Ояма К.— Бунсэки кагаку (Japan Analyst), 18, 1508 (1969); РЖХим, 1970, 12Г134.
547. ХиславскийА- Г., Плотников Р. И.— В кн.: Аппаратура и методы рентгеновского анализа. Л.: СКВ РА, 1971, вып. 8, с. 126.
548. Хитаров П. И.— Зав. лаб., 4, 491 (1935).
549. Холодов В. Н. Ванадий: (Геохимия, минералогия и генетические типы месторождений в осадочных породах). М.: Наука, 1968.
550. Хориути Й., Нисида X.— Бунсэки кагаку (Japan Analyst), 18, 850 (1969); РЖХим, 1970, 7Г91.
551. Хроматография на бумаге / Под ред. И. М. Хапса, К. М. Мацека. М.: Изд-во иностр, лит., 1962.
552. Ху Чжи-дэ, Се Сю-цзюнъ.— Хуасюэ тунбао (Huaxue tongbao), 15, 312 (1963); РЖХим, 1964, 22Г46.
553. Худякова Т. А., Крешков А. П. Теория и практика кондуктометрического и хронокондуктометрического методов анализа. М.: Химия, 1976.
554. Хци Шуй-цзе, Дэн-Синъ-Цзянь.— Кэсюэ тунбао (Kexue tongbao), 17, 84 (1966); РЖХим, 1966, 17Г102.
555. Цзо Цзун-цзи, Чжу Лэн-лин.— Хуасюэ сюэбао (Acta chim. sinica), 31, 299 (1965); РЖХим, 1966, 13ГЗЗ.
556. Цзян Жи-Хуэй.— Дичжи юй каньтань (Dizhi yu kantan),*24, 21 (1958); РЖХим, 1959, № 23, 81951.
557- Церковницкая И. А., Григорьева М. Ф,— В кн.: Применение органических реагентов в аналитической химии. Л.: Изд-во ЛГУ, 1969, с. 45, 56, 74, 83, 149, 165.
558. Церковницкая И. А., Григорьева М. Ф.— Ж. аналит. хим., 21, 1395 (1966).
559- Церковницкая И. А., Григорьева М. Ф. Методы Количественного определения элементов. Л.: Изд-во ЛГУ, 1964, с. 72, 101.
560. Церковницкая И. А., Григорьева М. Ф., Ковязина Л. Н.— Вести. ЛГУ, № 22, 129 (1971).
561. Церковницкая И. А., Григорьева М, Ф., Кустова И. А. Химия редких элементов. Л.: Изд-во ЛГУ, 1964, с. 153.
562. Церковницкая И. А., Каухова Л. В.— Изв. вузов. Химия и хим. технол., 14, 684 (1971).
563. Церковницкая И. И., Лугинин В- А.— Вести. ЛГУ, № 10, 162 (1969).
564. Церковницкая И. А., Лугинин В. А.— Ж. аналит. хим., 24, 54 (1969).
565. Цеховолъская Д. И., Меренкова В. М.— Зав. лаб., 31, 946 (1965).
565а. Цыганок Л. П., Чуйко В. Т., Резник В. ЕМазан Л-КСтець Т. В.— Там же, 39, 169 (1973).
566. Цюпка П. Н., Савкив С. В., Сабадах В. И.— Там же, 40, 965 (1974).
567- Черкесов А. И., Смирнов А. И., Казаков Б- И- — Там же, 41, 933 (1975).
196
568. Чернигов Ю. А., Добкина Б. М.— Там же, 22, 519, 1019 (1956).
569. Чернигов Ю. А., Добкина Б. М.— Там же, 16, 402 (1950).
570. Чернобров С. М.— Там же, 37, 1 (1971).
571. Чиркова С. Н., Шишкина В. И., -Паздникова Т. С.— Тр. Уральск, н.-и. пн-та черн. металл., 9, 23 (1970).
572. Чуева М. [I. Минералогический анализ шлихов и рудных концентратов. М.: Госгеолиздат, 1950.
573. Чуйко В. Т., Тодоров И. А.— Изв. вузов. Химия и хим. технол., 3, 988 (1960).
574. Чупахин М. С., Крючкова О. И., Рамендик Г. И. Аналитические возможности искровой масс-спектрометрии. М.: Атомиздат, 1972.
575. Шаврин А. М.~ Изв. АН СССР. Сер. физ., 14, 673 (1950).
576. Шаевич А. Б., Шубина С. В. Промышленные методы спектрального анализа. М.: Металлургия, 1965.
577. Шарло Г. Методы аналитической химии. Количественный анализ неорганических соединений. М.; Л.: Химия, 1965.
578. Шварценбах Г., Флашка Г. Комплексонометрическое титрование. М.: Химия, 1970.
579. Шеллард Э. Количественная хроматография на бумаге и в тонком слое. М.: Мир, 1971.
580. Шеллер В. Р., Поузл А. Р. Анализ минералов и руд редких элементов. М.: Госгеолтехиздат, 1962.
581. Шелудяков Л. Н., Любимова Л. С.— Изв. АН КазССР. Сер. хим., вып. 1 (19), 63 (1961).
582. Шемякин Ф. М., Степин В. В. Ионообменный хроматографический анализ металлов. М.: Металлургия, 1970.
583. Шемякин Ф. М., Харламов И. 1Г.~ Тр. 1-го Моск. мед. ин-та, 17, 241 (1962).
584. Шерстюк А. И., Вовк В. Н., Власова Л. И.— Зав. лаб., 40, 147 (1974).
585. Шестов А. П., Полотнюк О. В., Шерман Ю. Г., Карева А . М.— А. с. 186754 (СССР); опубл. 14.11.66, РЖХим, 1968, 2Г68П.
586. Шишков Д. А.— Годишник Минно-геол, ин-та, 7, 203 (1961); 9, 393 (1964).
587. Шишков Д. А., Петева-Горданова С.— Докл. Волг. АН, 17, 1027 (1964).
588. Шнайдерман С. Я., Демидовская А. Н.— Хим. технология: Науч.-произв. сб., № 2 (56), 37 (1971); РЖХим, 1972, 5Г151.
589. Шнайдерман С. Я., Прокофьева Г. И.— Ж. аналит. хим., 25, 2368 (1970).
590. Штепа Л. П.— В кн.: Материалы III Межотраслевого совещания по методам получения и анализа ферритовых, сегнето- и пьезоэлектрических материалов и сырья для них. Донецк; ВНИИ реактив-электрон, 1970, ч. 1, с. 282.
591. Шустова М. Б., Назаренко В. А,— Зав. лаб., 26, 1339 (1960).
592. Щербак О. В.— В кн.: Минеральное сырье. М.: ВИМС, 1961, вып. 3, с. 35.
593- Щербаков В. Г., Вейцман Р. М.— Зав, лаб., 20, 663 (1954).
594. Щербакова С. А., Мельникова Н. В., Пешкова В. М.— Ж. аналит. хим., 31, 318 (1976).
595. Щербинина С. Д., Петрова С. К).— Энергетик, № 8, 21 (1972).
596. Щербов Д. П. Флуориметрия в химическом анализе минерального сырья. М.: Недра, 1965.
597. Эдзава М,— Бунсэки кагаку (Japan Analyst), 19, 1026 (1970); РЖХим, 1971, 7Г169.
598. Экстракция в аналитической химии и радиохимии / Под ред. Ю. А. Золотова. М.: Мир, 1961.
599. Энага X., Исии X,— Вунсэки кагаку (Japan Analyst), 17, 836 (1968); РЖХим, 1969, 7Г159.
600. Эндо И., Хата Т., Накахара Ю.— Там же, 18, 833 (1969); РЖХим, 1970, ЗГ156.
601. Эрдеи Л.— Ж. аналит. хим., 24, 48 (1969).
197
602. Эристави Д. И., Эристави В. Д., Куциава Н. А., Данелия А. Г.— Тр. Груз, политехи, пн-та, № 3 (167), 22 (1974).
603. Юделевич И. Г., Федяшина А. Ф.— Изв. СО АН СССР. Сер. хим. наук, № 7, вып. 3, 82 (1971).
604. Юделевич И. Г., Черевко А. С., Богданова Е. С.— Там же, Л’ 9, вып. 4, 83 (1972).
605. Ягнятинская Т. Я.— Зав. лаб., 36, 158 (1970).
606. Яковлев П. Я., Разумова Г. П.— Там же, 24, 1450 (1958).
607. Яковлев П. Я., Федоров А. А., Буянов Н. В. Анализ материалов металлургического производства. М.: Металлургиздат, 1961.
608. Якубович А. Л., Зайцев Е. И., Пржиялговский С. М. Ядерно-физические методы анализа минерального сырья. М.: Атомиздат, 1969.
609. Ямамото Я., Соэдзима С., Харада М.— Бунсэки кагаку (Japan Analyst), 19, 1356 (1970); РЖХим, 1971, 11Г168.
610. Ямаути С.— Тамже, 18, 129 (1969); РЖХим, 1969, 21Г192.
611. Янъ Хуэй-нии, Сюй Шао-лин.— Кэсюэ тунбао (Kexue tongbao), 6, 548 (1965); РЖХим, 1966, 13Г31.
612. Янъ Хуэй-юй, Жэнъ Хун-дэ,— Хуасюэ сюэбао (Acta chim. sinica), 32, 191 (1966); РЖХим, 1967, 4Г56.
613. Ярош Н. А.— В кн.: Спектральный анализ в геологии и геохимии. М.: Наука, 1967, с. 175.
614. Яцимирский К. Б. Кинетические методы анализа. М.: Химия, 1967.
615. Яцимирский К. Б., Васильев В. И. Константы нестойкости комплексных соединений. М.: Изд-во АН СССР, 1959.
616. Яцимирский К. Б., Калинина В. Е.— Ж. неорг. хим., 9, 1117, 1328 (1964).
617. Яцимирский К. БКалинина В. Е. — Изв. вузов. Химия и хим. техно л., 8, 378, 385 (1965).
618. Abbasi S. А. - Anal. Lett., 9, ИЗ (1976).
619. Ackerman G., Schlosser G.— Z. Chem., 3, 471 (1963).
620. Adams F., Hoste J.— Acta chim. Acad. sci. hung., 52, 115 (1967).
621. Agarwala В. V., Dey A. K.— Microchem. J., 12, 162 (1967).
622. Agarwala В. V., Sangal S. P., Dey A. K.— Mikrochim. acta, N 2, 442 (1968).
623. Agrawal R. M., Khanna- P. P.— Govt India Atom. Energy Commis. Rept., N 699 (1973).
624. Agrawal Y. AT.—Anal. Lett., 5, 863 (1972).
625. Agrawal Y. K.— Ibid., 7, 729 (1974).
626. Agrawal Y. K., Chattapadhyaya M. C., Abbasi S. A ., Bodas M. G.— Separ. Sci., 8, 613 (1973).
627. Ahrens L. H., Edge R. A., Brooks R. R.— S. Afric. Industr. Chem.. 15, 102 (1961).
628. Аке О,— Svensk kem. Tidskr., 73, 482 (1961).
629. Albert F. M., Stoia M.~ Z. anal. Chem., 202, 420 (1964).
630. Alder J. F., ThompsonK. C., West T. S.— Anal. chim. acta, 50, 383 (1970).
631. Alvarez H. C., Burriel M. F.— Inform, quim. anal., 27, 235 (1973).
632. Andersson L.-H., J ohansson I., Soderlund R.— FOA (Foersvarets Fors-kningsanst) Repts., 4, N 8 (1970).
633. Arden T. V., Burstall T. H., Davies G. R., Lewis J. A., Linstead R. P.— Nature, 162, 691 (1948).
634. Asmus E., Jahny J.— Z. anal. Chem., 259, 269 (1972).
635. Avni R.— Spectrochim. acta, B23, 619 (1968).
636. Avni R., Boukobza A.— Appl. Spectrosc., 23, 483 (1969).
637. Awasthi S. P., Khasqiwale K. A.— Indian J. Chem., 2, 102 (1964).
638. Baas V. C., Yoe J. H.— Anal. chim. acta, 35, 337 (1966).
639. Bagshawe B.— J. Iron and Steel Inst., 170, 343 (1952).
640. Bannard L. G., Burton J. D.— Analyst, 93, 142 (1968).
641. Basinska H., Tarasiewicz M., Puzanowska-Tarasiewicz H.— Chem. anal. (Polska), 15, 317 (1970).
642. Basinska H., Wisniewski W.— Ibid., 10, 1007 (1965).
643. Baudin G., Destreumaux J., Fontaine R.— Anal. chim. acta. 38, 537 (1967).
198
644. Baumann. F.— J. Electroanal. Chem,, 6, 245 (1963).
645. Baur J. A., Bricher С. E.— Anal. Chem., 37, 1461 (1965).
646. Bergmann j. G., Ehrhardt С. H., Granatelli L., Janik J. L.— Ibid., 39, 1258, 1331 (1967).
647. Bernas B.— Ibid., 40, 1682 (1968).
648. Bhaduri A. S., Bay P.— Z. anal. Chem., 154, 103 (1957).
649. Bhargava S. P., Sogani N. C.— Bull. Acad. pol. sci. Str. sci. chim., 20, 741 (1972).
650. Bhashara В. K., Davaray N. K.— Rec. trav. chim., 84, 65 (1965).
651. Bhatnagar В. P., Poonia N. S.— Anal. chim. acta, 30, 211 (1964).
652. Bhatnagar В. P., Sharma K. D.— J. Indian Appl. Chem., 29, 113 (1966).
653. Bhattacharya P. K.— J. Indian Chem. Soc., 40, 797 (1963).
654. BhuraD. C., Tandon S. D.— Indian J. Chem., 8, 1036 (1970); Anal. chim. acta, 53, 379 (1971).
655. Biechler D. G., JordanD. E., Leslie W. D.— Anal. Chem., 35, 1685 (1963).
656. Biermann W. J-, Wong Wen-Kuei.— Can. J. Chem., 41, 2510 (1963).
657. Bishop E., Crawford A. B.— Analyst, 74, 364 (1949).
658. Biswas S. D., Dey A. K.— Mikrochim. acta, N 1, 10 (1963).
659. Blanicky P., Dolezal J.,Zyka J.— Z. anal. Chem., 240, 233 (1968).
660. Blasius E., Gottling W.— Ibid., 162, 423 (1958).
661. Bobtelsky M., Ganz M.— Israel J. Chem., 3 (1), 29, 57 (1965).
662. Bock R., Jost B.— Z. anal. Chem., 250, 358, 366 (1970).
663. Bognar J., Jellinek 0.— Mikrochim. acta, N 2, 318, 366 (1969).
664. Bollman D. H., Mortimore D. M.— Anal. Chem., 43, 154 (1971).
665. Bontschev P. R.— Mikrochim. acta, N 1, 79 (1964).
666. Barrel M., Paris K.— Anal. chim. acta, 4, 267, 279 (1950).
667. Bowman J. A., Willis J. B.~ Anal. Chem., 39, 1210 (1967).
668. Bradshaw W. S.— Chemist-Analyst, 54, 76 (1965).
669. Brito F.~ An. Real soc. esp. fis. у quim., B62, 193 (1966).
670. Brotdnkovii-Zvolskd K.— Chem. priim., 10, 303 (1960).
671. Bruch J., Watschel A— Arch. Eisenhiittenw., 34, 583 (1963).
672. Budesinsky B.— Coll. Czech. Chem. Communs, 28, 1858 (1963).
673. Budevsky O., Pribil R.— Taianta, 11, 1313 (1964).
674. Budzynski A. Z., Beer J. Z.~ Anal. chim. acta, 46, 281 (1969).
675. Burney J., Pleniceanu M., Burnea L.— Rev. roum. chim., 19, 501 (1974).
676. Burriel M. F., Alvarez H. C.— Inform, quim. anal., 15, 151 (1961); 16, 68 (1962).
677. Buzds L.— Magyar kc-m. lapja, 23, 716 (1968).
678. Cameron A. J., Gibson N. A.—Anal. chim. acta, 25, 429 (1961).
679. Capdevila C.— An. Real soc. esp. fis. у quim., B63, 155 (1967).
680. Carpenter L., Hazen K.— U.S. Bull. Mines. Rept. Invest., N 6182, 16 (1963).
681. Carpenter L., Nishi J. M., FehlerR. H.— Appl. Spectrosc., 20, 359 (1966).
682. Carton F. W. J., Parker A., Walting J.— Res. Group. U.K. Atom. Energy Auth., N AERE-R5452 (1967).
683. Cassidy R. M., Ryan D. E — Can. J. Chem., 46, 327 (1968).
684. Caton L., Mitoserin O., Popescu M., Mincea M.— Chim. anal. (Bucharest), 1, 114 (1971).
685. Cerny P-— Chem. listy, 50, 2026 (1956); Coll. Czech. Chem. Communs, 22, 613 (1957).
686. Chakraborti D.— Anal. chim. acta, 71, 196 (1974).
687. Chandola L. C., Venkatasubramanian R.— Z. anal. Chem., 266, 127 (1973).
688. Chauveau F.— Bull. Soc. chim. France, 810 (1960).
689. Chawla K. L., Tandon J. P.— Indian J. Chem., 5, 566 (1967).
689a. Chawla K. L., Tandon J. P.— Ibid., 6, 332 (1968).
690. Chawla K. L., Tandon J. P.— Taianta, 13, 545 (1966).
691. Cheng K. L.— Ibid., 8, 658 (1961).
692. Ciantelli G., Raspi G.— Chim. e ind., 47, 303 (1965).
693. Claassen A., Bastings L.— Z. anal. Chem., 202, 241 (1964).
694. Cobb W. D., Harrison T. S.— Metallurgia and Metal Forming, 39, 167 (1972).
199
695. Coke W. D., Reilley C. NFurman N. H.— Anal. Chem., 24, 205 (1952). 695a. Corjella C. D., Jost D. M.— J. Amer. Chem. Soc., 55, 1909 (1933).
696. Costache D.— Chim. anal. (Bucharest), 1, 241 (1971); Rev. roum. chim., 16, 849 (1971).
697. Costache D., SasuR.— Rev. roum. chim., 16, 1211, 1861 (1971).
698. Cotterill J. C.— Ind. Group. U.K. Atom. Energy Auth., N 196 (1959).
699. Cotterill J. C.— Res. Group. U.K. Atom. Energy Auth., N AERE-AM, 94 (1963).
700. Crouzolon P.— Chim. anal., 50, 483 (1968).
701. Cruft E. F.— Econ. Geol., 59, 458 (1964).
701a. Cruft E. F., Husler J.— Anal. Chem., 41, 175 (1969).
702. Crump-Wiesner H. J., Feltz H. R.— Anal. chim. acta, 55, 29 (1971).
702a. Crump-Wiesner H. J., Purdy W. C.— Taianta, 16, 124 (1969).
703. Cata F., Chui Jii Yen.— Rev. Chim. Acad. (Polska), 7, 127 (1962).
704. Czakow J.— Techn. Repts Ser. Intern. Atom. Energy Agency, 62, 6 (1966).
704a. Czakow J., Minczewski J.— Chem. anal. (Polska), 5, 863 (1960).
705. Danielson L.— Acta chem. scand., 19, 607, 1889 (1965).
706. De'Aeth L. A. Determination of Gaseous Elements in Metals. New York: Wiley, 1974, p. 474.
707. Dean G. A.— Analyst, 87, 754 (1962).
708. Dean G. A., Herringshaw J. F.— Taianta, 10, 793 (II 63).
709. De'Anil K., Bhattacharyya C. R.— Separ. Sci., 6, 621 (1971).
710. De'Anil K., Majumdar S. K.— Z. anal. Chem., 191, 40 (1962).
711. Debrun J. L., Barrandon J. N., Albert Ph.— Ann. chim. (France), 5, 357 (1970 — 1971).
712. Delespaul J., Lievens F.— Spectrochim. acta., 16, 1486 (1960).
713. Deshmukh G. S., Asthana О. P., Pillai V. S. N.— Z. anal. Chem., 185, 429 (1962).
714. Deshmukh G. S., Srivastava J. P.—Bull. Chem. Soc. Japan, 34, 325 (1961).
715. Dev B., Jain B. D.— Proc. Indian Acad. Sci., A57. 142 (1963).
716. Dikshitulu L. S. A.— Z. anal. Chem., 202, 344 (1964).
716a. Dikshitulu L. S. A., Rao G. G.— Z. anal. Chem., 189, 421; 191, 254 (1962).
717. Dolezal J., Petrus V., Zyka J.— J. Electroanal. Chem., 3, 169 (1962).
718. Donaldson E. M.— Taianta, 17, 583 (1970).
718a. Donaldson E. M.— Ibid., 18, 905 (1971).
719. Donghetry J. A., Mellon M. G.— Anal. Chem., 37, 1096 (1965).
720. Dragulesky C., Mitranesky M.— Studio si cercetari stiinto chim., Acad. RPR, Baza Timisoara, 9, 21 (1962).
721. Drew H. D., Fitzgerald J. M.— Anal. Chem., 41, 974 (1969).
722. Dufek О., Tuma B.— Hutn. listy, 14, 246 (1959).
723. Dutta R. L.— J. Indian Chem. Soc., 35, 243 (1958); 36, 285, 339 (1959).
724. Duval C., Morette A.— Compt. rend. Acad, sci., 230, 545 (1950).
725. Dyck R.— Anal. Chem., 37, 1046 (1965).
726. DyrssenD., Sekine T.— J. Inorg. Nucl. Chem., 26, 981 (1964).
727. Easterday C. L.— Anal. Chem., 31, 1867 (1959).
728. Ecrement F.— Meth. phys. anal., 7, 128 (1971).
729. Eishman M. J., Skougstag M. W.— Anal. Chem., 36, 1643 (1964).
730. Erdey L., Mdzor L.— Acta. chim. Acad. sci. hung., 3, 469 (1953).
731. Erdey L., Szabadvary F.— Z. anal. Chem., 159, 429 (1958).
732. Erdtmann G., Aboulwafa O.— Ber. Kernforschungsanlage Julich, N 1170, '60 (1975).
733. Erlenmeyer H., Dahn H.— Helv. chim. acta, 22, 1369 (1939).
734. Everett G. L., West T. S., Williams R. W.-— Anal. chim. acta, 66, 301 (1973)
735. Farhan F. M., Pazandeh B.— Analusis, 3, 201 (1975).
736. Farrel R. F., Harter G. I., Jakobs R. M.— Anal. Chem., 31, 1550 (1959).
737. Fassel V. A., Altpeter L. L.— Spectrochim. acta, 16, 443 (1960).
738. Fassel V. A., Dallman W. E., Skogerboc R., Harrigan V. M.— Anal. Chem., 34, 1364 (1962).
200
739. Fassel F. A., Myers R.B., Kniseley R. TV.—Spectrochim. acta, 19, 1187 (1963).
740. Fassel F. A., Slack R. IF., Kniseley R. NAnal. Chem., 43, 186 (1971).
741. Feigl F., Baumjeld L.— Anal. chim. acta, 3, 15 (1949).
742. Feinstein H. G.— Ibid., 15, 141 (1956).
743. Finch J.— N. Z. J. Sci., 14, 36 (1971); РЖХим, 1971, 23Г123.
744. Flaherty J. P., Eldridge H. B.~ Appl. Spectrosc., 24, 534 (1970).
745. Flaschka H., Abdine H.— Chemist-Analyst, 45, 58 (1956).
746. Fleps I'., Inczedy JMagyar kcm. folyoirat, 62, 322 (1956).
747. Florian K., Lavrin A., Matherny M., Petrickova-Florianova K,— Spectrosc. Lett., 2, 377 (1969).
748. Fritz J. S., James S., Rettig T. A.— Anal. Chem., 34, 1562 (1962).
749. Fritz J. S., Kaminski E. E.— Taianta, 18, 541 (1971).
750. Fritz J. S., Palmer T. A.— Ibid., 9, 393 (1962).
751. Fritz J. A., Topping J. J.- - Ibid., 18, 865 (1971).
752. Fujinaga T., Kusaka Y., Koyama M. et al.— J. Radioanal. Chem., 13, 301 (1973).
753. Fuller C. IF., Oitaway J. M.— Analyst, 94, 32 (1969).
753a. Fuller C. W-, Otlaway J. M.— Ibid., 95, 28, 34, 41, 139 (1970).
754. Furman N. H., Reilley C. N., Cooke W. D.— Anal. Chem., 23. 1665 (1951).
755. Gagliardi E., llmaier В.— Mikrochim. acta, N 1, 180 (1967)..
756. Gagliardi E., PFoss H. Р,— Anal. chim. acta, 48, 107 (1969).
757. Gahler A. R., Hammer R. N-, Shubert R. C.— Anal. Chem., 33, 1937 (1961).
758. Galambos G., Korosi G., Sikles P., Tiidbs F.— Magyar kirn. folyoirat, 79, 364 (1973).
759. Garg B. S., Singh R. P.— J. Indian Chem. Soc., 48, 1119 (1971).
760. Geyer R., Herrmann W., Bogatzki B.— Wiss. Z. Techn. Hochschule Chem. Leuna-Merseburg, 9, 1 (1967).
761. Ghosh N. N., Siddhanta G.— Z. anal. Chem., 253, 207 (1971).
762. Gilbert D.D.— Anal. Chem., 37, 1102 (1965).
763. Gill J. S., Rao G. N.— Z. anal. Chem., 256, 201 (1971).
764. GiufjreL., CapizziF. M.— Ann. chim. (Itai.), 50, 1547 (1960).
765. Giuffre L., Cassani F.— Chim. e ind., 47, 512 (1965).
766. Goecke R.~ Taianta, 15, 871 (1968).
767. Gohshi Y., Nakamura T., Yoshimura M.— X-Ray Spectrom., 4, 117 (1975).
768. Gonzalez G. F., Catalina L.— An. edafol. у agrobiol., 27, 75 (1968); РЖХим, 1968, 23Г60.
769. Goto H., Sudo E.— Japan Analyst, 10, 1213 (1961).
770. Govindaraju K.— Analysis, 3, 116 (1975).
771. Gramporohit S. V., Swamy S. K.— Govt India Atom. Energy Comniis. Rept, N B. A. R. C.- 473‘, 1970.
772. Green H.— Metallurgia, 66, 52 (1962); 70, 299 (1964).
773. Gregorowicz Z.— Chem. anal. (Polska), 3, 19 (1958).
774. Gruber E., Sorautin H.— Mikrochim. acta, N 2, 262 (1971).
775. Gunzler G.~ Z. Chem., 4, 233 (1964).
776. Gupta С. M., Joshi M. P.— J. Pract. Chem., 38, 358 (1968).
777. Gupta V. K., Tandon S. G.— Anal. chim. acta, 66, 39 (1973).
778. Haba F. R., Wilson C. L.— Mikrochim. acta, N 1, 196 (1963).
779. Habersberger K.— Chem. prum., 12, 547 (1962).
780. Hagadone, M., Zeitlin H.— Anal. chim. acta, 86, 289 (1976).
781. Hansen J., Hodkins C. R.— Anal. Chem., 30, 368 (1958).
782. Hansen R. K., Bachman R. Z., O' Laughlin J. W-, Banks С. V.— Anal, chim. acta, 46, 217 (1969).
783. Hartman V.— Acta geol. et geogr. Univ, comen. GeoL, N 15, 147 (1968).
784. Hartmann SZ. anal. Chem., 151, 332; 153, 112 (1956).
785. Harvey С. E. A Method of Semi-Quantitative Spectrographic Analysis. Clendal (California), Applied research laboratories, 1947.
786. Hasek Z.— Hutn. listy, 25, 815 (1970).
201
787. Hashitani H., Motojima К.— Japan Analyst, 7, 478 (1958).
788. Hashitani H., Yoshida H., Adachi T.— Anal. chim. acta, 76, 85 (1975).
789. Headridge J. B., Somerbutts M.— Lab. Pract., 23, 99 (1977).
790. Heffelfinger R. E., Blosser E. R., Perkins О. E., Henry W. M.— Anal. Chem., 34, 621 (1962).
791. Heffelfinger R. E., Chase D. L., Rengstorff G. W- P., Henri/ W. M.— Ibid., 30, 112 (1958).
792. Heitner-Wirguin C., Ganez M.— Taianta, 14, 671 (1967).
793. Hislop J. S., Wood D. A.— Res. Group. U. K. Atom. Energy Auth., N AERE-R6165 (1969).
794. Hitoshi T., Yoshisida S.— Sci. Repts. Res. Inst. Tohoku Univ., set. A, 3, 640 (1951); C. A., 47, 2081i (1953).
795. Hobbs R. б1., Kirkbright G. F., West T. S.— Analyst, 94, 554 (1969).
796. Hofer A., Heidinger R.— Z. anal. Chem., 246, 125 (1969).
797. Huart A., Chaussabel L., Corgier J., Deron S., Detwiller J., Disant C.— Note CEA, N 1341, 55 (1970).
798. Hulanicki A., Karwowska R.— Chem. anal. (Polska), 18, 709 (1973).
799. Hulcher F. H.— Anal. Chem., 32, 1183 (1960). •
800. Husler J.— Atom.' Absorpt. Newslett., 10, 60 (1971).
801. Ikehata A., Shimizu T.— Bull. Cheni.j Soc. Japan, 38, 1385 (1965); РЖХим, 1966, ЗГ88.
802. Inczedy J., Erdey L.— Koloriszt. ert., 6, 10 (1964).
803. Ingri NBrito F.— Acta chem. scand., 13, 1971 (1959).
804. lunglickel H. E., Klinger N.— Z. anal. Chem., 206, 275 (1964).
805. Jaboulay В. E.— Chim. anal., 41, 185 (1959).
806. Jackson P. F. S., Whitehead J.— Analyst, 91, 418 (1966).
807. Jacobsen E., Strom P.— Anal. chim. acta, 53, 309 (1971).
808. Jacocca D., Goldwasser A., Chiglione M.— Acta cient. venezol., 19, 99 . (1968).
809. Jacquelot P., Thomas G.— Bull. Soc. chim. France, 3167 (1970); J. Chro-matogr., 66, 121 (1972).
810. Jakubiec R., Boltz D. F.— Anal. chim. acta, 43, 137 (1968).
811. Jakubiec} R., Boltz D. E.~ Anal. Lett., 1, 347 (1968).
812. Janauer G. E., Tera F.— Mikrochim. acta, N 4, 599 (1961).
813. JancikF., KorblJ.— Taianta, 1, 55 (1958).
814. Janda L., Schausberger J., Schroll E.— Mikrochim. acta, N 1, 122 (1963).
815. Janousek J. J.— Coll. Czech. Chem. Communs, 27, 2972 (1962).
816. Jasim F.— Taianta, 16, 752 (1969).
817. Job V. A., Kartha S. B., Krishnamurty G., Gopal S.— 1,. anal. Chem., 271, 26 (1974).
818. Joensuu O. J., Suhr N. H.— Appl. Spectrosc., 16, 101 (1962).
819. Johnson W. Analysis Calcareous of the Materials. London: Soc. Chem. Ind., 1964, p. 306.
820. Jokl V., Majer J.— Acta geol. et. geogr. Univ, comen. Geol., N 15, 77 (1968).
821. Jones G. B.— Anal. chim. acta, 17, 254 (1957).
821a. Jones G. B., Colvin J. H.— J. Amer. Chem. Soc., 66, 1573 (1944).
822. Joshi B. D., Bangia T. R., Dalvi A. G. I.— Mikrochim. acta, N 5, 829 (1974).
823. Joshi B. D., Bangia T. R., Dalvi A. G. I.— Z. anal. Chem., 260, 107 (1972).
824. Julietti R.— J. Brit. Ceram. Soc., 5, 47 (1968).
825. Kadarmandalgi S. G.— Indian J. Chem., 2, 291 (1964).
826. Kahn A., Livage J., Collongues R.— Phys, status solidi (a), 26, 175 (1974).
827. Kai J., Ogata Y., Ohi K., Watanabe M.~ In: Recent Developments Mass Spectroscopy: Proceedings of the International Conference on Mass Spectroscopy, Kyoto, 1969, Baltimore: Univ. Park press, 1970, p. 314.
828. Kaimal W. R. M., Shome S. C.— Anal. chim. acta, 27, 594 (1962).
829. Kaiser D. G., Meinke W. W.~ Ibid., 29, 211 (1963).
830. Kakabadse G., Wilson H. J.— Analyst, 86, 402 (1961).
202
831. Karanjikar N. P., Sakseni M. D.— Taianta, 21, 652 (1974).
832. Kawazu K.— J. Chromatogr., 120, 171 (1976).
833. Kekedy L., Balogh J.— Stud. Univ. Babes-Bolya, Ser. chem., 9, 101 (1964).
834. Kennedy J. IL, Jensen КAnal. Chem., 37, 310 (1965).
835. Kennedy J. II., Lingane J. J.— Anal. chim. acta, 18, 240 (1958).
836. Khalija H., El-Sirajy A Z. anal. Chem., 227, 109 (1967).
837. Khalija II., Farar A .— Ibid., 158, 109 (1957).
838. Khalija IL, Ismail I. A.— Microchim. J., 14, 464 (1969).
839. Kiboku AL, Yoshimura C.— Japan Analyst, 7, 488 (1958); C. A., 54.
16284 (1960).
840. Kinnunen J., Wen.nerstand B.— Chemist-Analyst, 46, 92 (1957).
841. Kiriyama T., Kuroda B.— Anal. chim. acta, 62, 464 (1972).
842. Kirkbright G. F., Hees D. J., Stephen TU. J.— Ibid., 27, 558 (1962).
843. Kirkbright G. F., Sargent Al. Atomic Absorption and Fluorescence Spectroscopy. London; New York; San Francisco: Acad. Press, 1974.
844. Kirkbright G. F., Sargent Al., West T. S.— Taianta, 16, 1467 (1969).
845. Kiss E.— Anal. chim. acta, 77, 205 (1975).
846. Klement R.— Z. anal. Chem., 136, 17 (1952).
847. Klinkmann A.— Metall, 14, 1190 (1960).
848. Klug O.N., Metlenko A. J.— Chem. anal. (Polska), 10, 819 (1965);
13, 7 (1968). I
849. К night D. Al., Pyzyna AL K.— Atom. Absorpt. Newslett., 8, 129 (1969).
850. Koch К. H., Ohls K.— Z. anal. Chem., 247, 239 (1969).
851. Koch O. G.— Mikrochim. acta, 92, 151, 347, 402 (1958).
852. Koh K. L., RyanD. E.— Anal. chim. acta, 52, 503 (1970); 57, 295 (1971).
853. Koloini T., Modic R.— Vestn. Slov. kem. drustva, 13, 69 (1966).
854. Kolthojj J. M., Parry E. P.~ J. Amer. Chem. Soc., 73, 3718, 5315 (1951).
855. Korkisch J., Ahluwalia S. S.— Taianta, 11, 1623 (1964).
856. Korkisch J., Feik F., Ahluwalia S. S.— Ibid., 14, 1069 (1967).
857. Korkisch J., Krivance H.— Anal. chim. acta, 83, 111 (1976).
858. Korkisch J., Stejjan I., Graft H.— Mikrochim. acta, N 4—5, 503 (1976).
859. Krajoran-Marjanovic V., Hlavaty AL, Habenovic M., Cukovic V. —Kem. u indistr., 7, 33 (1958).
860. Kral S.~ Huth, listy, 15, 727 (1960); 22, 699 (1967).
861. Kranz AL., Krzyzaniak J.— Chem. anal. (Polska), 5, 243 (1960).
862. Krausz I.— Magyar kem. folyoirat, 66, 218, 296 (1960).
863. Krivan V.— Anal. Chem., 47, 469 (1975).
864. Krtil J.— Z. anal. Chem., 210, 412 (1966).
865. KrychZ., Lipiec T.-— Chem. anal. (Polska), 12, 535 (1967).
866. Kucharzewski B.— Ibid., 7, 349 (1962).
867. Kuehn P. R., Howard О. H., Weber C. W.— Anal. Chem., 33, 740 (1961).
868. Kuczynski W., Nowinska K., Fiedorow R., Sczaniecki B., Katarska-Kor-nowska K.— Chem. analit. (Polska). 17, 1247 (1972).
869. Kudo Y., Hasegawa N., Yamashita T.— Japan Analyst, 20, 1319 (1971).
870. Kurmaiah N., Satyanarayana D., Rao V. P. R.— Anal. chim. acta, 35, 484 (1966).
871. Kurmaiah N., Satyanarayana D., Rao V. P. R.— Z. anal. Chem., 230, 199, 840 (1967); Taianta, 14, 493 (1967).
872. Kuroda R., Voshikuni N., Kawabuchi K.— J. Chromatogr., 47, 453 (1970).
873. Kvapil M.— Rudy, 10, N 6; Prace vyzkumnych ustavu, Priloha, N 5, 33 (1962).
874. Lacourt A ., Sommereyns G., Geyndt de E., Baruch J., Gillard J.— Nature,
163, 999 (1949).
875. Lajjolie de H.— Arch. Eisenhiittenw., 38, 535 (1967).
876. Laib R. D.— Appl. Spectrosc., 17, 160 (1963).
877. Laib R. D., Lykins J. D.— Ibid., 22, 539 (1968).
878. Lamarque G.— Rapp. CEA, N 4215, 63 (1971).
879. Landi M. F., Battaglia A.— Mettallurgia Ital., 59, 650 (1967).
880. Langmyhr F. J., Pans P. E.— Anal. chim. acta, 45, 157, 173 (1969).
203
881. Lanning E. W.— Anal. Chem., 41, 182 (1969).
882. Lanning E. IF., Weberling R. P.— Ibid., 40, 626 (1968).
883. Lassner E., Scharf R.— Planseeber. Pulvermetall., 9, 51 (1961).
884. Leao E. C., Hobart E. W., Fornwalt D. E.— Appl. Spoctrosc , 20, 400 (1966).
884a. Lederer С. M., Hollander J. M., Perlman I. Table Isotopes. New York; London; Sydney: J. Wiley, 1968.
885. Lederer M.— Anal. chim. acta, 7, 458 (1952).
886. Lefebvre J.— J. Chem. Phys., 54, 567 (1957).
887. Lievens F., Lecocq A., Constant M.— Z. anal. Chem., 247, 171 (1969).
888. Lingane J. J., Kennedy J. H.— Anal. chim. acta, 15, 465 (1956).
889. Linstedt K. D., Kruger P.— Anal. Chem., 42, 113 (1970).
890. Louis R.— Erdol und Kohle-Erdgas-Petrochem., 23, 347 (1970).'
891. Luke C. L.— Anal. chim. acta, 37, 267 (1967).
892. Lund M. A., Smith D. G. L.— Atom. Energy Res. Establ., N AM73 (1961).
893. Lynch G. F., Segel S. L., Sayer M.— J. Magnet. Reson., 15, 8 (1974).
894. Maggio F., Cavasino P.— Ann. chim., 50, 1280 (1960).
895. Maillard P., Ades C.— Can. Spectrosc., 14, 17 (1969). *
896. Majumdar A.K., Chakrabartty M.M.— Anal. chim. acta, 17, 415 (1957).
897. Majumdar A. K., Das G.— Ibid., 36, 454 (1966).
898. Majumdar A. К., Das G.— Ibid., 31, 147 (1964).
899. Majumdar A. K., Das G.— J. Indian Chim. Soc., 42, 189 (1965).
900. Majumdar A. K., Mitra В. K.— Z. anal. Chem., 208, 1 (1965).
901. Malat M., Suk F., Tenorova M.— Chem. listy, 52, 2408 (1958).
902. Malik A. U.— Indian J. Chem., 3, 316 (1965).
902a. Malik A. U.— Ibid., p. 446.
903. Malmstadt H. F., Scholz R. G.— Anal. Chem., 27, 881 (1955).
904. Mandal S. K., Das J.— J. Indian Chem. Soc., 52, 1146 (1975); 53, 134 (1976).
905. Manoliu C., Balasa T., Bulac E.— Rev. chim. (RSR), 21, 166 (1970).
906. Margerum D. W., Banke C. F.— Anal. Chem., 26, 200 (1954).
907. Martin E. L., Bentley К. E.— Ibid., 34, 354 (1962).
908. Martinez L. M. J., Maceira V- A., Burriel-Marti F.— An. quim. Real soc. exp. fis. у quim., 70, 223 (1974).
909. Matejicek F.— Chemie (Prague), N 5, 87 (1952).
910. Matherny M., Pliesovska N.— Chem. zvesti, 27, 327 (1973).
911. Matocha С. K., Petit J.— Appl. Spectrosc., 22, 562 (1968).
912. Matsui H.— Japan Analyst, 21, 96 (1972).
913. May L. A., Presley B. J.— Atom. Absorpt. Newslett., 13, 144 (1974).
914. McCrea J. MAppl. Spectrosc., 23, 55 (1969).
915. McKaveney J. P., Freiser H.— Anal. Chem., 30, 526, 1965 (1958).
916. Meier D. J., Myers R. J., Swift E. H.— J. Amer. Chem. Soc., 71, 2340 (1949).
917. Meites L.— Anal. Chem., 24, 1057 (1952).
918. Meites L.— J. Amer. Chem. Soc., 75, 6059 (1953).
919. Mellichamp J. W.— Anal. Chem., 26, 977 (1954).
920. Miketurkova F., Frei R.— J. Chromatogr., 47, 427, 435 (1970).
921. Mikhail S. Z., Stewart J. J.— Z. anal. Chem., 178, 335 (1961).
922. Miksovsky M.— Acta geol. et geogr. Univ, comen. Geol., N 15, 253 (1968).
923. Milazzo G., Verga C., Marilli NRiv. combust., 12, 684, 690 (1958).
924. Minczewsky J., Sobkowska A.— Acta chim. Acad. sci. Hung., 27, 143 (1961).
925. Misra G. J., Tandon J. P.— J. Inst. Chem., 41, 94 (1969).
926. Mittal R. K., Mehrotra R. G.— Z. anal. Chem., 209, 405 (1965).
927. Morales A., Gonzales F., Diaz C.— Chemist-Analyst, 56, 89 (1967).
928. MoretteA.— Bull. Soc. chim. France, 526 (1950).
929. Morris A. W.— Anal. chim. acta, 42, 397 (1968).
930. Morris W. F., Worden E. F.~ Appl. Spectrosc., 25, 305 (1971).
204
931. Mraz L., Simon V., Zyka J.— Coll. Czech. Chem. Commons, 24, 1487 (1959).
932. Munshi K. N., Dey A. K.— Mikrochim. acta, N 5, 874 (1962).
933. Muntean J., Toma C., Pisculescu M.— Chim. anal. (Bucharest), N 2 (4), 255 (1972).
934. Muralikrishna UЛао G. G.— Taianta, 15, 143 (1968).
935. Murthy T- K. S.— Anal. chim. acta, 16, 25 (1957).
936. Mushran S. P., Prakash O., Verma J. R.— Bull. Chem. Soc. Japan, 45,
1709 (1972); РЖХим, 1973, 2Г87.
937. Musil A., Haas W.— Mikrochim. acta, 6, 803 (1957).
938. Musil J- Hutn. listy, 23, 649 (1968).
939. Muzzarelli R. A. A.— Anal. chim. acta, 54, 132 (1971).
940. Nador B.— Acta chim. Acad. sci. hung., 40, 1 (1964).
941. Nagaosa Y., Seto R., Satake M., Yonekubo T.— J. Chem. Soc. Japan, Pure Chem. Sec., 92, 1216 (1971); РЖХим, 1972, 10Г76.
942. Nagaosa Y., Yonekubo T., Satake M., Seto R.— Japan Analyst, 21,
215 (1972); РЖХим, 1972, 17Г66.
943. Nestler C. G.,l Nobis MZ. anal. Chem., 167, 81 (1959).
944. Neunhoeffer O.— Z. anorg. und allg. Chem., 296, 208 (1958).
945. Newmen L., Quinlan R.— J. Amer. Chem. Soc., 81, 547 (1959).
946. Nickel H., Leushacke D. F., Lummerzhein D.— Z. anal. Chem., 247, 196 (1969).
947. Niericker R-, Treadwell W. D — Helv. chim. acta, 29, 1472 (1946).
948. Nishida H.— Japan Analyst, 14, 1009 (1965).
949. Nishida H.— Technol. Repts Iwate Univ., 2, 61 (1966); РЖХим, 1968, 6Г77.
950. Nishimura M., Matsunaga K., Kudo T., Obara F.— Anal. chim. acta, 65, 466 (1973).
951. Nivoli M.— Ann. chim. (Ital.), 42, 370 (1952).
952. Nomura T., Nakagawa G-— Rev. Polarogr., 21, 68 (1975).
953. Norwitz G., Codell M. Metallurgia, 57, 261, 269, 343 (1958).
954. Nouyrigat F.— Bull. Soc. chim. France, 781 (1958); Chim. anal., 10, 515 (1963).
955. Oda N., Tsunoo S., Hashimoto T.— J. Japan Inst. Metals, 23, 86 (1959); РЖХим, 1960, 26367.
956. Oelsen W., Gobbels P.— Stahl und Eisen, 69, 33 (1949).
957. Oelsen W-, Haase H., Graue G.— Angew. Chem., 64, 76 (1952).
958. Oguma K., Maruuama T., Kuroda R.— Anal. chim. acta, 74, 339 (1974).
959. Oldfield J. H., Bridge E. P.— Analyst, 86, 267 (1961).
960. Oldfield J. H., Mack D. L.— Ibid., 87, 778 (1962).
961. Omang S. H.— Anal. chim. acta, 56, 470 (1971).
962. O'Neil R. L., Suhr N. H.— Appl. Spectrosc., 14, 45 (1960).
963. Oriani P.A., Jones T. SRev. sci. instrum., 25, 248 (1954).
964. Orlandini K. A., Wahlgren M. A., Barclay J.— Anal. Chem., 37, 1148 (1965).
965. Orsag J.— Rev. univ. mines, 15, 296 (1959).
966. Otomo M.— Bull. Chem. Soc. Japan, 36, 137 (1963).
967. OtteTmann J., Friese G.— Geochim. et cosmochim. acta, 26, 599 (1962).
968. Owen L. E., Eilenburg J. Y.— Anal. Chem., 23, 1823 (1951).
969. Owens E. B.— Appl. Spectrosc., 16, 86 (1962).
970. Pais J— Magyar kern. folyoirat, 62, 252 (1956).
971. Parizkova H., Sedlacek I.— Chem. listy, 62, 975 (1968).
972. Patrovsky V,— Ibid., 60, 1545 (1966).
973. Patrowsky V,— Chem. listy, 49, 854 (1955); Coll. Czech. Chem. Communs, 31, 3392 (1966).
974. Patrowsky V.— Taianta, 16, 456 (1969).
975. Pearton D. C. G., Taylor J. D., Faure P. K., Steele T. W.— Anal, chim. acta, 44, 353 (1969).
976. Pepin D., Gardes A., Petit J., Berger J. A., Gaillard G-— Analusis, 2, 549 (1973).
205
977. Perkins R. W., Raucitelli L. A., Cooper J. A., Brown R. E.— Nucl. Appl. and Technol., 9, 861 (1970).
978. Persiani C., Cosgrove J. F.— Anal. Chem., 40, 1350 (1968).
979. Pethii A.— Acta chim. Acad. sci. hung., 51, 151 (1967).
980. Pethii A.— Bol. geol. у minero, 80, 62 (1969).
981. Phillips S. L., Kern D. M.— Anal. chim. acta, 20, 295 (1959).
982. Picasso G.— Metallurgia Ital., 50, 367 (1958).
983. Piccardi G.— Ann. chim. (Ital.), 48, 188 (1958).
984. Pichat R.~ Note CEA, N 1241, 44 (1970); N 1419, 42 (1971).
985. Pickering W. F.— J. Chromatogr., 1, 274 (1958).
986. Pillay К. K. S., Thomas С. C. Jr.— J. Radioanal. Chem., 7, 107 (1971).
987. Pinta M., Riandey C.— Meth. phys. anal., 5, 76 (1969).
988. Pittwell L. R.— Appl. Spectrosc., 12, 54 (1958).
989. Poddar S. N.— J. Indian Chem. Soc., 40, 706 (1963).
989a. Poddar S. AL—Indian J. Chem., 1, 537 (1963); Indian J. Appl. Chem., 27, 132 (1964).
990. Poddar S. N., Dey K., Sarkar S. C. N.— Indian J. Chem., 4, 371 (1966).
991. Podobnik B., Spenko M.— Anal. chim. acta, 34, 294 (1966).
992. Pohl F.A.— Z. anal. Chem., 141, 81 (1954).
993. Pollard F. H., Nickless G., Banister A. J.— Analyst, 81, 577 (1956).
994. Pope M. T., Dale В. M.— Quart. Revs. Chem. Soc. (London), 22, 527 (1968).
995. Pourbaix M. Atlas d’equilibres electrochimique. Paris: Ganthier — Villars, 1963.
996. Pribil R.— Taianta, 12, 934 (1965).
997. Pribil R., Vesely V,— Hutn. listy, 25, 813 (1970).
998. Price W. J. Effluent and Water Treatment Manual. London: Thunderbird Enterprises, 1968, p. 258.
999. Price W. J-, Maurer R. H.— Anal. Chem., 35, 595 (1963).
1000. Product. Group. U. K. Atom. Energy Auth., N 11 (S), 1959.
1001. Prosen T., Modik R.— Vestn. SIov. kem. drustva, 16, 23 (1969).
1002. Pszonicki L., Minczewski J.— Spectrochim. acta, 18, 1325 (1962).
1003. Publicover W.— Anal. Chem., 37, 1680 (1965).
1004. Qureshi M., Akhtar J.— Z. anal. Chem., 227, 89 (1967).
1005. Qureshi M., Husain W., Kahn F.— Experientia, 27, 607 (1971).
1006. Qureshi M., Sharma S. D-— Taianta, 22, 129 (1975).
1007. Qureshi M., Varshney K. G., Kaushik R. C.— Anal. Chem., 45. 2433 (1973).
1008. Radcliffe N. C., Parker J. R.— Anal. chim. acta, 52, 9 (1970).
1009. Rao В. K.— Chemist-Analyst, 54, 52 (1965); 55, 103 (1966).
1010. Rao G. G., Dikshitulu L. S. A.— Taianta, 9, 289 (1962).
1011. Rao G. G., Dikshitulu L. S. A.— Ibid., 10, 295, 1023 (1963).
1012. Rao G. G., Rao P. К.— Ibid., 13, 1335 (1966).
1013. Rao G. G., Rao P. K.— Ibid., 14, 33 (1967).
1014. Rao G. G., Rao P. V- K.— Anal. chim. acta, 65, 347 (1973); Taianta, 20, 907 (1973).
1015. Rao P. V. K., Rao G. В. B., Rao R. S.— Z. anal. Chem., 266, 367 (1973).
1016. Rao V. N., Dutt E. V. V. S.— Anal. chim. acta, 51, 553 (1970).
1017. Rao V. N., Dutt E. V. V. S.— Z. anal. Chem., 250, 192 (1970); 258, 32 (1972).
1018. Rao V. P., Rao G. G.— Ibid., 161, 406 (1958).
1019. Rao V. P. R., Anfaneyulu Y.— Mikrochim. acta, N 4, 481 (1973).
1020. Rao V. P. R., Sarma В. V. A.— Chemist-Analyst, 54, 107 (1965).
1021. Rasmussen S. W., Rodden C. J, Analytical Chemistry of the Manhatten Project. New York; Toronto; London: McGraw-Hill Book Co., 1950, p. 459.
1022. Rayburn K. A.— Appl. Spectrosc., 22, 726 (1968).
1023. Reddy D. V., Rao S. B.— Chemist-Analyst, 56, 17 (1967).
1024. Resac Z., Dvorak J.— Chem. prum., 10, 576 (1960).
206
1025. Reuter G., Jaskowsky J., Laqua W.— Z. anal. Chem., 214, 185 (1965).
1026. Rhodes J. R.— Chem. Eng. Progr. Symp. Ser., 66, 48 (1970).
1027. Rigdon L. P., Harras J. E.— Anal. Chem., 41, 1673 (1969).
1028. Riley J. P., Taylor D.— Anal. chim. acta, 41, 175 (1968).
1029. Riley J. P., Taylor D.— Ibid., 40, 479 (1968).
1030. Rizvi G. H., Singh R. P.— Taianta, 19, 1198 (1972).
1031. Roberts E. M., Rutledge R. L., Wehner A. P.— Anal. Chem., 33, 1879 (1961).
1032. Roberts J. L.~ Taianta, 18, 1070 (1971).
1033. Rohtmann H., Bauer G. Заявка ФРГ, кл. 12, n 37/14, N 224029; РЖХим, 1975, 11Л127П.
1034. Roland G. — Anal. chim. acta, 28, 93 (1963).
1035. Roman M., Garcia-Sanches F., Gomez H.A.— Quim. anal, (pura V apl.), 28, 191 (1974).
1036. Root C.— Anal. Chem., 37, 1600 (1965).
1037. Rossotti F. J. C., Rossotti H. S.— Acta chem. scand., 9, 1177 (1955);
10, 957 (1956).
1038. Rubeska J., Moldan B. Atomic Absorption Spectrophotometry. London: Iliff Books Ltd., 1969.
1039. Rusheed A., Qazi A. H., Ahmad S., Ashraj M.— Separ. Sci., 10, 507 (1975).
1040. Ruzicka E.— Chem. listy, 51, 1814 (1957).
1041. Ruzicka E., Adamek J.— Z. anal. Chem., 195, 411 (1963).
1042. RyanD. E.— Analyst, 85, 569 (1960).
1043. Sachdev S. L., Robinson J. W., West P. W.— Anal. chim. acta, 37, 12, 156 (1967).
1044. Sajo J. Femipari kutato intezet kozlmenyei, Budapest, N 10, 247 (1971).
1045. Sajo J.— Z. anal. Chem., 188, 168 (1962).
1046. Salamon A.— Acta techn. Acad. sci. hung., 78, 427 (1974).
1047' . Samadi A. A., Fedoroff MCompt. rend/Acad, sci., C282, 667 (1976).
1048. Sanual P., Mushran S. P.— Anal. chim. acta, 35, 400 (1966).
1049. Sanual P., Mushran S. P.— Mikrochim. acta, N 5—6, 959 (1965).
1050. Sarma P. L.— Anal. Chem., 36, 1076 (1964).
1051. Sastri M. N.,Rao A . P. — J. Chromatogr., 9, 250 (1962); 19, 630 (1965).
1052. Sastri M. N., Rao A. P.— Z. anal. Chem., 196, 166 (1963).
1053. Satyanarajana D., Kurmaiah N., Rao V. P. R.— Chemist-Analyst, 54, 4 (1965).
1054. Satyanarajana D., Rao V. P. R.— Indian J. Chem., 2, 164 (1964); 3, 40 (1965).
1055. Sauer К.-H., Nitsche M.— Arch. Eisenhiittenw., 40, 891 (1969).
1056. Saxena R. S., Mittal M. L.— J. Indian Chem. Soc., 41, 125 (1964).
1057. Saxena R. S., Sharma О. P.— Indian J. Chem., 2, 502 (1964).
1058. Saxena R. S., Sharma О. P.— Taianta, 11, 863 (1964).
1059. Syxena R. S., Sharma О. P.— Z. anal. Chem., 212, 286 (1965).
1060. Scharrer K., Judel G. K.— Ibid., 156, 340 (1957).
1061. Schiller R., Thilo E.—-7.. anorg. und allg. Chem., 310, 261 (1961).
1062. Schmidt W. E., Bricker С. E.— J. Electrochem. Soc., 102, 623 (1955).
1063. Schmitt B. R., Segebade C.— Z. anal. Chem., 270, 193 (1974).
1064. Schneer-Erdey A.— Taianta, 10, 591 (1963).
1065. Schneider H., Schumann H.— Z. anal. Chem., 235, 160 (1968).
1066. Schroll E., Huber-Schausberger I., Janda I., Spatzek H.— Mikrochim. acta, bJ 3, 649 (1968).
1067. Schulek E., Pais Z., Pataki L.— Ann. Univ. sci. budapest. Sec. chim., 2, 573 (1960).
1068. Schwarz H.— Z. anal. Chem., 176, 241 (1960).
1069. Schwarzenbach G., Geier G.— Helv. chim. acta, 46, 906 (1963).
1070. Schwarzmann E., Glemser O.— Angew. Chem., 73, 33 (1961).
1071. Sebor G., Lang I., Vavrecka P., Sychra V-, Weisser O.— Anal. chim. acta, 78, 99 (1975).
1072. Sekhon B. S., Pannu B. S., Chopra S. L.— Ann. chim. (Franco), 1, 69 (1976).
207
1073. Sen В. К., Maity R. К., Gupta R. N-, Bandyopadhyay P.— Anal, chim. acta, 81, 173 (1976).
1074. Sen G.N.R., Sen G.D. B.— J. Indian Chem. Soc., 50, 112 (1973).
1075. Sen S. R. N.~ Sci. and Cult., 23, 434 (1958).
1076. Seroneit A., Lelic H.— Chem. Techn., 8, 295 (1956).
1077. Sherma JAnal. Chem., 39, 1497 (1967).
1078. Sherma J., Cline C. W.— Anal. chim. acta, 30, 139 (1964).
1079. Sherma J., Evans G. H., Frame H. D., Straine H. H.— Anal. Chem., 35, 224 (1963).
1080. Sherwood A. E.— Metallurgia, 80, 209 (1969).
1080a. Sherwood A.E.— Metallurgia and Metal Forming, 39, 216 (1972).
1081. Shigematsu T., Matsui M., Munakata M., Naemura T.— Bull. Inst. Chem. Res. Kyoto Univ., 46, 262 (1968); РЖХим, 1969, 22Г103.
1082. Shiokawa T.— Sci. Res. Inst. Tohoku, Univ. Sci. Chem., 2, 613 (1950); C. A., 46, 4950 (1952).
1083. Shiro H., Takashi S.-— J. Chem. Soc. Japan, 74, 285 (1953); C. A., 47, 9855 (1953).
1084. Silvestre J, Hurth M. F.— Chim. anal. (France), 38, 253 (1956y.
1085. Silvey W- D., Brennan R.— Anal. Chem., 34, 784 (1962).
1086. Singh D., Bhatnagar U.— Israel J. Chem., 5, 29 (1967).
1087. Singh V. B., Prasad B. N.— J. Sci. Res. Banaras Hindu Univ., 12, 116 (1961 -1962).
1088. Singh V.B., Varma A. — Ibid., 15, 131 (1964—1965).
1089. Sinha S. N., Dey A. K.— J. Pract. Chem., 20, 225 (1963).
1090. Siroki M., Djordjevic C.— Anal. chim. acta, 57, 301 (1971).
1091. Skalska S., Baranska G.— Acta geol. etgeogr. Univ, comen. Geol., N 6, 401 (1961).
1092. Skogerboe R. K., Heybey A. T., Morrison G. II.— Anal. Chem., 38, 1821 (1966).
1093. Skorko-Trybula Z.— Chem. anal. (Polska), 10, 831 (1965).
1094. Skorko-Trybula Z.— Nafta (PRL), 22, 141 (1966).
1095. Skorko-Trybula Z.— Nukleonika, 10, 559 (1965).
1096. Slovak Z.— Z. anal. Chem., 220, 401 (1966).
1097. Smith A. J.— Anal. Chem., 36, 944 (1964).
1098. Smith D. C., Johnson J. R., Soth G. CAppl. Spectrosc., 24, 576 (1970).
1099. Smith D. P., Pope M. T.— Anal. Chem., 40, 1906 (1968).
1100. Smith E. J., Fitzgerald J. M.— Anal. chim. acta, 60, 367 (1972).
1101. Sommer L.—Z. anal. Chem., 187, 7, 263 (1962).
1102. Souchay P.— Bull. Soc. chim. France, 160 (1946).
1103. Souchay P., Gandebocuf J.— Compt. rend. Acad. Sci., 249, 94 (1959).
1104. Souchay P., Scholl R.— Bull. Soc. chim. France, 824 (1950).
1105. Sriramen K., Rao G. G.— Taianta, 13, 1468 (1966).
1106. Srivastava К. C., BanerjiS. K.— Chim. anal. (France), 52, 973 (1970).
1107. Staten I. W., Huffmann E. W.~ Anal Chem., 31, 2003 (1959).
1108. Steinnes E.— Scand. J. Met., 1, 137 (1972).
1109. Stevens H. M.— Anal. chim. acta, 15, 51 (1956).
1110. Strasheim A., Eve D. J.— J. S. Afric. Chem. Inst., 14, 1 (1961).
1111. Strelov; F. IP., Liebenberg C. J., Victor A.— Anal. Chem., 46, 1409 (1974).
1112. Sturgeon R. E., Chakrabarti C. L.— Ibid., 49, 90 (1977);
1113. Sugimal A.— Anal. Chem., 46, 1123 (1974).
1114. Sulcek Z. — Chem. listy, 51, 1453 (1957).
1115. Sundaram A. K., Sundaresan M., Vartak D. G.— J. Sci. and Ind. Res., BC16, B25 (1957).
1116. Suleu A., Buzura V-— Stud. Univ.. Babes-Bolyai. Ser. chem., 12, 35, 93 (1967).
1117. Suth A.— Chem. anal. (Polska), 10, 1093, (1965).
1118. Svehla G., Tolg G.— Taianta, 23, 755 (1976).
1119. Sykora J., Eybl V.— Coll. Czech. Chem. Communs, 32, 352 (1967).
208
1120. Szarvas P., Baloghne K. J-, Kelemenl., larabin Z.— Acta Univ, deb-recen, 7, 119 (1961 — 1962).
1121. Szanas P., Lantos J.- - Magyar, kem. folyoirat, 65, 145 (1959).
1122. Takenaka T.— Asahi Garasu Kenkyu Hokoku, 15, 145 (1965): C. A., 64, 14960d (1966).
1123. Talwar U. B.. Haidar В. C.— Indian J. Chem., 3, 452 (1965).
1124. Tamhina B., Vojkovic V., Herak M. J.— Croat, chem. acta, 48. 183 (1976); Mikrochim. acta, 2, 45 (1975).
1125. Tanaka M., Ishida A.— Anal. chim. acta, 36. 515 (1966).
1126. Tanaka M., Kojima I.— Ibid., 36, 522 (1966).
1127. TandonJ. P., Mehrotra R. C.— Z. anal. Chem., 164, 314 (1958).
1128. Tandon S. G., Bhattacharya S. C.— Anal. Chem., 33, 1267 (1961);
36, 1378 (1964).
1129. Tandon S. G., Bhattacharya S. C.— J. Indian Chem. Soc.. 47, 583 (1970).
1130. Tanos F.~ Tavkozlesi kut. int'z. kozl., 10, 93 (1965).
1131. Terasima A.— Japan Analyst, 22, 1317 (1973).
1132. Thomas G.— Z. anal. Chem., 230, 99 (1967).
1133. Thomerson D. R., Price IP. J.— Analyst, 96, 825 (1971).
1134. Thompson G., Bankston D- C.— Spectrochim. acta, B24, 335 (1969).
1135. Thompson R. J., Morgan G. B., Purdue L. J.— Atom. Absorpt. New-slctt., 9, 53 (1970).
1136. Tien Li-ching, Chang Wen-bing.— Sci. Sinica, 13, 1625 (1964); РЖХим, 1965, 18Г51.
1137. Tomasevic M.~ Metalurgija (Yugoslavia), 9, 15, 25 (1970).
1138. Trace Analysis. Spectroscopic Methods for Elements/Ed. J. D. Wi-nefordner. New York: J. Wiley, 1976.
1139. Turkstra J., Smith H. J., Wet de IP. J.— J. S. Afr. Chem. Inst., 24, 113 (1971).
1140. Tzou Scih-fu, Liang Shu-chuan.— Sci. Sinica, 2, 207 (1962); РЖХим, 1962, 23Д32.
1141. Vanossi R.— An. Soc. cient. argent., 135, 97 (1943); C. A., 39, 473 (1945).
1142. Vecsernyes L.— Magyar ki'm. folyoirat, 72, 377 (1966).
1143. Venkatasubramanian N., Ganapathysundraram B.— Current Sci. (India), 37, 225 (1968).
1144. Verma J. R., Mushran S. P — Rev. chim. miner., 8, 339 (1971).'
1145. Verma J. R., Prakash O., Mushran S. P.— Anal. chim. acta, 52, 357 (1970).
1146. Verma M. R., Bhuchar V- M.—~ J. Sci. and Industr. Res., 14, 19 (1955); 15, 437 (1956).
1147. Vigl’r M. 5., Gaylor U. F.— Appl. Spectrosc., 28, 342 (1974).
1148. Vita O. A., Mullins L. R., Trivisonno C. 7,— U. S. Atom. Energy Comm., N YAT-T1085 (1963).
1148a. Vojkovic U., Tamhina B., Herak M. J.— Z. anal. Chem., 276, 377 (1975).
1149. Wagif H. A.. Bayburdi A. — Analusis, 2, 746 (1974).
1150. Wakamatsu S.— Japan Analyst, 9, 284 (1960); РЖХим, 1960, № 21, 844 51.
1150a. Walden G., Hammett L., Edmons S.— J. Amer. Chem. Soc., 56, 57 (1934).
1151. Wallace G. W-, Mellon M- G.— Anal. chim. acta, 23, 355 (1960).
1152. Walter С. IP., Korkisch J.— Mikrochim. acta, N 1, 181 (1971).
1153. Warren R. J., Hazel J. F., McNabb W. M.— Anal. chim. acta, 21, 224 (1959).
1154. Webb M. S. IP., Cotterill J. C.— Ibid., 26 548 (1962).
1155. Weiss G., Blum P.~ Bull. Soc. chim. France, 1077 (1947).
1156. Welcher G. G-, Kriege H.— Atom. Absorpt. Newslett., 8, 97 (1969).
1157. Wilkins D. H., Hibbs J. E.— Anal. chim. acta, 20, 427 (1959).
1158. Willis J. B.— Nature, 207, 715 (1965).
1159. Willis J. P.— Bol. geol. у minero, 80, 74 (1969).
1160. Wise IP. M., Brendt IP. IP.— Anal. Chem., 27, 1392 (1955).
209
1161. Witwit A. S., Magee R. J., Wilson C. L.— Taianta, 9, 495 (1962).
1162. Wyttenbach A.— Helv. chim. acta, 52, 2458 (1969).
1163. Wyttenbach A., Dulakas H.— Chimia, 22, 484 (1968).
1164. Yatirajam 7., Arya S. P.— Anal. chim. acta, 86, 209 (1976).
1165. Yatirajam V., Arya $. P.— Taianta, 22, 861 (1975).
1166. Yatirajam V., Kakkar L. P.— Anal. chim. acta, 44, 468 (1969,.
1167. Yatirajam V., Kakkar L. P.— Ibid., 47, 568 (1969).
1168. Yatirajam V., Prosad R.— Z. anal. Chem., 220, 340 (1966).
1169. Yotsuyanagi T., Itoh J., Aomura K.— Mem. Fac. Eng. Hokkaido Univ.,
13, 39 (1971); РЖХим, 1972, 1Г123.
1170. Young P. N. W.— Analyst, 99, 588 (1974).
1171. Zaremba J.— Chem. anal. (Polska), 12, 305 (1967).
1172. Zemany P. D.— Spectrochim. acta, 16, 736 (1960).
1173. Ziegler M., Horn H.-G.— Z. anal. Chem., 166, 362 (1959).
1174. Zimmer K., Haypal E.— Spectrochim. acta, B24, 655 (1969).
1175. Zindel E., Zeiher R.— Z. anal. Chem., 195, 27 (1963).
1176. Zoller W. H., Gordon G. E.— Anal. Chem., 42, 257 (1970).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1177. Безруков И. Я., Романцева С. Ю., Волостное В. Г., Овсянников Б. В.— В кн.: Химия, технология и применение ванадиевых соединений: Тез. докл. III Всесоюз. совещания в г. Качканаре. Свердловск: УНЦ АН СССР, 1979, ч. 2, с. 101.
1178. Карякин А. В., Грибовская И. Ф. Эмиссионный спектральный анализ объектов биосферы. М.: Химия, 1979.
1179. Пономарев А. И., Быковская Ю. И., Веселаго Л. И. Анализ сплавов на основе ниобия, титана, хрома. М.: Наука, 1979.
1180. Theobald F.— Bull. Soc. chim. France, 1607 (1975).
1181. Hogfeldt E. Stability constants of metal-ion complexes. Part A. Inorganic ligands. ШРАС chemical data, series N 21. Oxford; New York; Toronto; Sydney; Paris; Frankfurt: Pergamon Press, 1980.
1182. Perrin D. D. Stability constants of metal-ion complexes. Part B. Organic ligands. IUPAC chemical data, series N 22. Oxford; New York; Toronto; Sydney; Paris; Frankfurt: Pergamon Press, 1979.
1183. Sillen L. G., Martell A. Stability constants of metal-ion complexes. London: The Chemical society, 1964.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Активационное определение ванадия в алюминии 129, 156 биологических объектах 128 бумаге хроматографической 129 воздухе 128 вольфраме 128, 156 горных породах 128 графите 128, 156 железе 128, 156 нефти, ее фракциях и золах 128 ниобии 128» 129, 156 природных водах 129 сталях 128 титане, его сплавах и соединениях 128, 129, 156
Амперометрическое титрование 111 аскорбиновой кислотой 111 гексацианоферроатом калия 111 2,4-дитиобиуретом 111 иодом 111 8-меркаптохинолином 111 селенистой кислотой 111 солями железа(П) 111 железа(Ш) 111 меди, серебра или талия 111 тайроном 111
Атомно-абсорбционное определение ванадия в алюминии 125 воздухе 125, 138 горных породах 125, 135 карбидных включениях 125
катализаторах синтеза аммиака 125
нефти и нефтепродуктах 125, 136
никелевых сплавах 125 природных водах 125, 136 рудах 125 сталях 125, 142 чугунах 125, 142 шлаках 125
Атомно-флуоресцентное определение ванадия 124
Ванадий
атомная масса 7
изотопы 8
история открытия 7
минералы 9
нахождение в природе 7 положение в периодической- системе элементов 7
получение 8
потенциалы электродные 102, 103
применение 5, 6
распространение в природе 7
свойства
физические 10
химические 10
соединения неорганические 10 органические 25
состояния окисления 10
спектральные линии в оптическом диапазоне 118
рентгеновском диапазоне 127
токсичность 9
Гравиметрическое определение
ванадия с
бензоатом аммония 64 бензоил-)г-нитроацетани-лидом 64
диантипирилфенилмста-ном 65
5,7-Дибром-8-оксихино-лином 64
5,7-дн иод-8-ок сихино липом 64
купфероном 65 о-оксиацетофенонокси-мом 65
2-оксинафтил-альдокси-мом 64
2-оксипафтил-1-метилен-этиламином 64
8-оксихинолином 65
Качественные методы обнаружения ванадия 30 кинетические 33 люминесцентные 34 спектральные 34 химические
микрокристаллоскопи-ческие 33
по реакции с анилином 32 бензидином и его производными 32
кверцетин-3-рутинози-дом 32
ксиленоловым оранжевым 31
8-оксихинолином 31 перекисью водорода 30
резацето феноксимом 32
3,3',3"-триокспаури-ном 32
хроматографические
в тонких слоях сорбента 35
на бумаге 35
Кинетические методы определения ванадия по реакциям восстановления
солями титана (III) красителя «Виктория
голубой В» 116 окисления
броматом калия 1-амнно-8-яафтол-3,6-сульфокислоты (Аш-кислогы) 116
п-ампнофепола 115 1,5-дифенил-З-ами-
ногшразолона 115 индигокармина 115 иодида калия 115 красителя бордо R 116
метурипа 116 а-пафтола 115 солохром фиолетового
RS 115
п-фенетидипа 33, 115 эрпохром голубого Р 115
кислородом люминола 116
перекисью водорода анилина 116 бензидина 116 иодида калия 116 люминола 116 тиосульфата натрия
116 п-хлоранилина 116 персульфатом аммония галловой кислоты 116 пирокатехина 116 хлоратом калия о-ампнофенола 115 п-аминофенола 115 анилина 115 бензидина 115 иодида калия 115 п-фенетидина 115
Кондуктометрическое титрование 113
Кулонометрическое определение ванадия ИЗ
Люминесцентное определение ванадия с
1,4-диамино-5-нитроантра-хиноном 117
211
карбоновыми кислотами 117
резорцином 117 силоксеном 117 церулеином 117
Масс-епектральнос определение ванадия в воздухе 130
железе особой чистоты 130 оксиде титана 130
Методы отделения ванадия от сопутствующих элементов дистилляция 62 осаждение 36, 37 бензоатом аммония 37 N-6eH30Hjr-N-ij>eHiTn гидроксиламином 38 ванадатов 36 гетерополикислот 37 диэтилдитиокарбамина-том натрия 38 купфероном 37 оксисульфида 37 8-оксихинолином 37 пентасульфида 37 таннином 38 фосформолибдатом аммония 37 хроматография на анионитах 50 бумаге 54 катионитах 48 тонких слоях сорбентов 57 экстракция 38
Амберлитом SA-2 45 амилацетатом 42 ацетилацетоном 46 ацетонгидразидом антраниловой кислоты 44
М-бензоил-М-фенилгид-роксиламином 39. 40 бутилацетатом 45 гетерополикислот 43 гидроксамовыми кислотами 40 в-дикетонами 41 ди-2-этилгексилфосфор-ной кислотой 47 диэтилдитиокарбамина-тов 43 купфероном 39, 40 метилизобутил кето ном 45 метилпропилкетоном 44 нитроном 44 2-окси-З-нафтогидрокса-мовой кислотой 44 8-оксихинолином 39 1-(2-пиридилазо)-2-наф-толом 41 м-пропил-2,3,4-триоксибензоатом 44 теноилтрифторацетоном тиофен-2-гидроксамовой кислотой 40, 44
М-м-толил-о-метокси-бензогидроксамовой кислотой 44 трибутилфосфатом 41 1-фенил-З-метил-4-бсн-зоилпиразолоном-5 42
Л'-циннамоил-Л'-фенил-гидроксилампном 40, 44
2-этилгсксанолом 44 электролиз на ртутном ка-
тоде 38
электрофорез на
бумаге 59, 62
тонких слоях сорбен-
тов 5"—61
Окислительно-восстановительные потенциалы ванадия 102
в различных средах ЮЗ
зависимость от pH 103
нормальные 102
Определение валентного со-• стояния ванадия ванадия(1у) и ванадия(У) 160
ванадия(Ш) и ванадия(1У) 161
ванадия(П) и вана-Дия(Ш) 163
Определение ванадия в алюминии, его сплавах и соединениях 149, 156 биологических объектах 135
боре и его соединениях 157
водах 136
воздухе 137
горных породах 132, сл.
графите 156
железе 141, 156
золе 135
иоде 158
катализаторах 140
кремнии 157
металлах особой чистоты бериллии 150 висмуте 150 вольфраме 151 галлии 151
германии 151
иттрии 151 лантане 151 литии и других щелочных металлах 152
марганце 152
меди 152
молибдене 152
никеле 153 ниобии 153 олове 153 редкоземельных элементах 151
ртути 154
свинце 154
тантале 154
хроме 155
цинке 155
цирконии 155
нефтепродуктах 135
плутонии 148
почвах 138
рудах и продуктах их переработки 132
селене 158
сере 158
солях щелочных металлов 152
сплавах и соединениях ванадия 138
сталях 141—144
титане, его сплавах и соединениях 146, 147 156
тории 148, 149
углероде 158
уране 148, 149
ферросплавах 139, 144,
14 5
чугунах 141, 142 шлаках 134, 135 Определение примесей в ванадии и его соединениях 164
физическими методами 173 активационными 177 масс-спектральными 17"8 спектральными 173
химическими и физико-химическими методами 164
азота 165 алюминия 166 висмута 166 водорода 165 вольфрама 172 газов 165 железа 168 кадмия 167 1
кальция 167 кислорода 165 кобальта 167 кремния 171 марганца 169 меди 167, 168 молибдена 169 мышьяка 166 никеля 170 ниобия 169 олова 171 свинца 171 серы 171 сурьмы 171 титана 171 углерода 166 урана 172 фосфора 170 фтора 168 хрома 167 цинка 172
циркония и гафния 172
Пламенно-фотометрические методы определения ванадия 123 атомно-абсорбционные " 124 атомно-флуоресцентные 124 эмиссионные 123
Полярографическое] определение ванадия в металлах 109, 143 минералах 109 нефти и нефтепродуктах 110, 136 растительных материалах 110
рудах 106, 109, 110 Сталях 106, 109, 143
Полярографическое поведение ионов ванадия 104— 106
Потенциометрическое титрование ванадия(У)
аскопбиновой кислотой 104
солями железа(П) 104 молибдена(Ш) 104 серебра(1) 104 титана(1П) 104 хрома(П) 104 сульфатом гидразина 104
тиосульфатом натрия 104
ванадия(1У)
бихроматом калия 104 ванадия(Ш) солями
212
ванадия(У) 104 xpOMa(VI) 104 церия(1У) 104 ванадия(П) бихроматом калия 104 броматом калия 104 йодатом калия 104 сульфатом железа(Ш) 104 сульфатом церия(1У) 104 хлоратом калия 104
Разделение ионов ванадия различных степеней окисления хроматогра фией на бумаге 56, 57 тонких слоях сорбентов 58 экстракцией 39
Растворимость ванадатов 24 гидроокиси ванадила 14
Рентгенорадио метрический метод определения ванадия 127, 135
Рентгеноспектральное определение ванадия в агломератах 127 алюминии 127 бериллии 127 битуминозных породах 127 ванадий-галлиевых сплавах 127 ванадий-кремниевых пленках 127 глиноземе 127 горных породах 127, 135 ильмените 127 катализаторах крекинга нефти 127 коррозионных средах 127 магнетите 127 минералах 127 морской воде 127 органических материалах 127 рудах 135 сталях 127 титане и титановых сплавах 127 шлаках 127
Соединения ванадия с неорганическими реагентами арсениды 25 ацидокомплексы 27 галогениды 24 гетерополисоединени я 27 гидриды 25 карбиды и оксикарбиды 25 нитриды 25
гидроокиси перекисные соединения 24 сульфаты 25 сульфиды 25 фосфиды 25
органическими реагентами 26, 27
Спектральное определение ванадия в металлических монолитных образцах 119, 120 органических объектах 121
порошковых пробах 121 технологических растворах и водах 122
Титрование ванадия 68, .104, 111 восстановителями аскорбиновой кислотой 68, 104, 111, 112 гидразидом изоникотиновой кислоты 68 гидразином 68, 104 гидрохиноном 68 метиловым оранжевым 144
солями ванадия(П) 68
вольфрама(У) 68 железа(П) 66, 67, 104, 111 меди(1) 68 ниобия(П) 68 олова(П) 66 титана(Ш) 66, 68, 104 хрома(П) 68, 104 тиомалояовой кислотой 68 комплексоном Ш 71, 104 обратное 72 прямое 71 совместно с алюминием 72 железом 72 кобальтом 72 свинцом 72 окислителями бихроматом калия 70 броматом калия 71 перманганатом калия 69
солями железа(Ш) 71, 111| церия(1У) 70
трикарбонатокобаль-тиатом гексааминокобальта 71 феррицианидом калия 71, 111
Токсичность соединений ванадия 9
Фотометрическое определение ванадия 73, 74 косвенными методами 102 по образованию окрашенных соединений с неорганическими реагентами
гетерополисоединени я-ми 76
перекисью водорода 75
роданидами 78 органическими реагентами беязоилгидразидом антраниловой кислоты 91
К-бензоил-М-фенил-гидроксилами-ном 84
гетероциклическими оксиазосоединс-ниями 93
гидразидами органических кислот 91
гидроксамовыми кислотами 83
ксиленоловым оранжевым 89
o-оксиазосоединения-ми 96 8-оксихинолином 88 4-(2-ниридилазо)-рс-зорцином 93 пирокатехином 79 серусодержащими соединениями 99 сульфаназо 97 сульфонитразо 98 формазаном 98 фталексонами 90 по поглощению света ионами
ванадия(У) 74 ванадия(ТУ) 74 ванадия(ТП) 75
Электронный парамагнитный резонанс 130
Электрофорез 59 в тонких слоях сорбента 59—61
на бумаге 59, 62
Электрохимические методы определения ванадия 102
амперометрические 111 высокочастотного титро-
вания 113
кондуктометрические ИЗ кулонометрические 113 полярографические 104
ЯДерный магнитный резонанс 130
ОГЛАВЛЕНИЕ
От редколлегии................................................ 3
Предисловие................................................... 5
»
Глава I
Физико-химическая и химико-аналитическая характеристика ванадия и его соединений...................................... 7
Общие сведения................................................ 7
Физические и физико-хпмическпе свойства...................... 10
Соединения ванадия .......................................... 10
Неорганические соединения............................... 10
Комплексные соединения.................................. 25
Глава II
Методы обнаружения ванадия................................... 30
Химические методы............................................ 30
Кинетические методы.......................................... 33
Люминесцентные методы........................................ 34
Спектральные методы.......................................... 34
Другие методы................................................ 34
Глава III
Методы отделения ванадия от сопутствующих элементов ... 36
Методы осаждения............................................. 36
Осаждение ванадия неорганическими реагентами............ 36
Осаждение ванадия органическими реагентами.............. 37
Электрохимические методы..................................... 38
Методы экстракции............................................ 38
Методы хроматографии......................................... 47
Ионообменная хроматография.............................. 47
Хроматография на бумаге................................. 54
Хроматография в тонком слое............................. 57
Электрофорез............................................ 59
Другие методы............................................... 62'
Глава IV
Методы количественного определения ванадия................... 63
Гравиметрические методы...................................... 63
Титриметрические методы...................................... 66
Оксидиметрическое титрование............................ 66
Комплексонометрическое титрование....................... 71
Другие методы титрования................................ 72
Фотометрические методы....................................... 73
Определение ванадия по окраске ионов ванадпя(У), ванадия(1У) и ванадия(Ш)................................ 74
Определение ванадия по образованию окрашенных соединений с неорганическими реагентами............................ 75
214
Определение ванадия по образованию окрашенных соединений с органическими, реагентами.........................
Определение ванадия по продуктам окисления органических реагентов ..............................................
Определение ванадия косвенными методами................
Электрохимические методы..........'.........................
Потенциометрические методы..............................
Полярографические методы................................
Амперометрические методы................................
Кулонометрические методы................................
Другие электрохимические методы.........................
Кинетические методы.........................................
Люминесцентные методы.......................................
Спектральные методы..............................-..........
Методы эмиссионной пламенной спектрофотометрии..............
Методы атомно-абсорбциоппой п атомно-флуоресцентной спектрофотометрии .......................................... . .
Рентгеноспектральные методы.................................
Активационные методы........................................
Масс-спектральные методы....................................
Методы электронного и ядерного магнитного резонанса . .
78
09 102 102 104 101 111 113 113 из 116 118 123
124 127 128 129 130
Глава V
Определение ванадия в прпродвых и промышленных объектах . . 132
Горные породы, руды и продукты их переработки.............. 132
Нефтепродукты, биологические материалы и золы.............. 135
Природные и сточные воды................................... 136
Воздух..................................................... 137
Почвы...................................................... 138
Соединения и сплавы ванадия............................. . 138
Катализаторы............................................... 140
Железо, его сплавы, стали и чугуны......................... 141
Титан и продукты его переработки........................... 146
Уран, торий, плутоний п их соединения...................... 148
Алюминий, его сплавы и соединения.......................... 149
Металлы особой чистоты и их соединения..................... 150
Другие объекты............................................ !>?
Глава VI Определение валентного состояния ванадия.................... 159
Определение ванадия(ТУ) и ванадия(У)........................ 160
Определение ванадия(Ш) п ванадия(1У)........................ 161
Определение ванадия(П) и ванадия(Ш)........................' 163
Глава VII
Определение примесей в ванадии п его соединениях 164
Химические и физико-химпческпе методы...................... 164
Физические методы........................................... 173
Спектральные методы..................................... 173
Активационные методы.................................... 177
Масс-спектральные методы................................ 178
Литература................................................. 179
Предметный указатель..................................
211
215
УДК 543:546.881
МузгивВ. Н., Хамзина Л. Б., Золотавин В. Л.,'Б езруковИ. Я. Апа-литическая химия ваиадия.’Серия: «Аналитическая химия элементов». М.: Наука, 1981. 216 с.
В монографии рассматриваются основные химические свойства ванадия и его соединений, методы обнаружения и отделения ванадия от сопутствующих элементов. Подробно рассмотрены титриметрические, фотометрические, электрохимические, спектральные, атомно-абсорбционные, рентгенофлуоресцентные, активационные и другие методы анализа»
Книга рассчитана на широкий круг научных работников, преподавателей, аспирантов и студентов вузов.
Табл. 30. Ил. 19. Библиогр. 1183 назв.
Владимир Николаевич Музгин* Людмила Борисовна
Хамзина,
^Валерии Леонидович Золотавин\ ,
Иван Яковлевич Безруков
АНАЛИТИЧЕСКАЯ ХИМИЯ ВАНАДИЯ Серия: «Аналитическая химия элементов»
Утверждено к печати ордена Лепина 11 нститутом геохимии и аналитической химии им. В. И. Вернадского Академии -наук СССР
Редактор Л. В. Юкина. Редактор издательства Р. А. Баранова Художественный редактор С. А. Литвак. Технический редактор О. М. Гуськова Корректоры В. С. Федечкина, В.'А. Шварцер
ИВ № 17321
Сдано в набор 07.07.80. Подписано к печати 10.04.81. Т-03190. Формат 60X90*'к-Бумага типографская № 1. Гарнитура обыкновенная. Печать высокая. Усл. псч. л. 13,5
Уч.-изд. л. 16,0. Тираж 2100 экз. Тип. зак. 3626. Цена 2 р.
Издательство «Наука» 117864 ГСП-7, Москва, В-485, Профсоюзная ул., 90 2-я типография издательства «Наука» 121099, Москва, Г-99, Шубинский пер., 10
СПИСОК ОПЕЧАТОК
Страница Строка Напечатано Должно быта
9 4 св. НСи3( ГОО.-ЗШО Cih(V0,)...3H,0
24 18 св. [V(O.)FJ- [V(O_,)P+
28 9 сн. Алгоме он Алюминон
169 15 св. кто3 КЮ4 '
169 26 св. Mo(IV) -W)
.',ак 3<2и В. Н. Музпш и др.