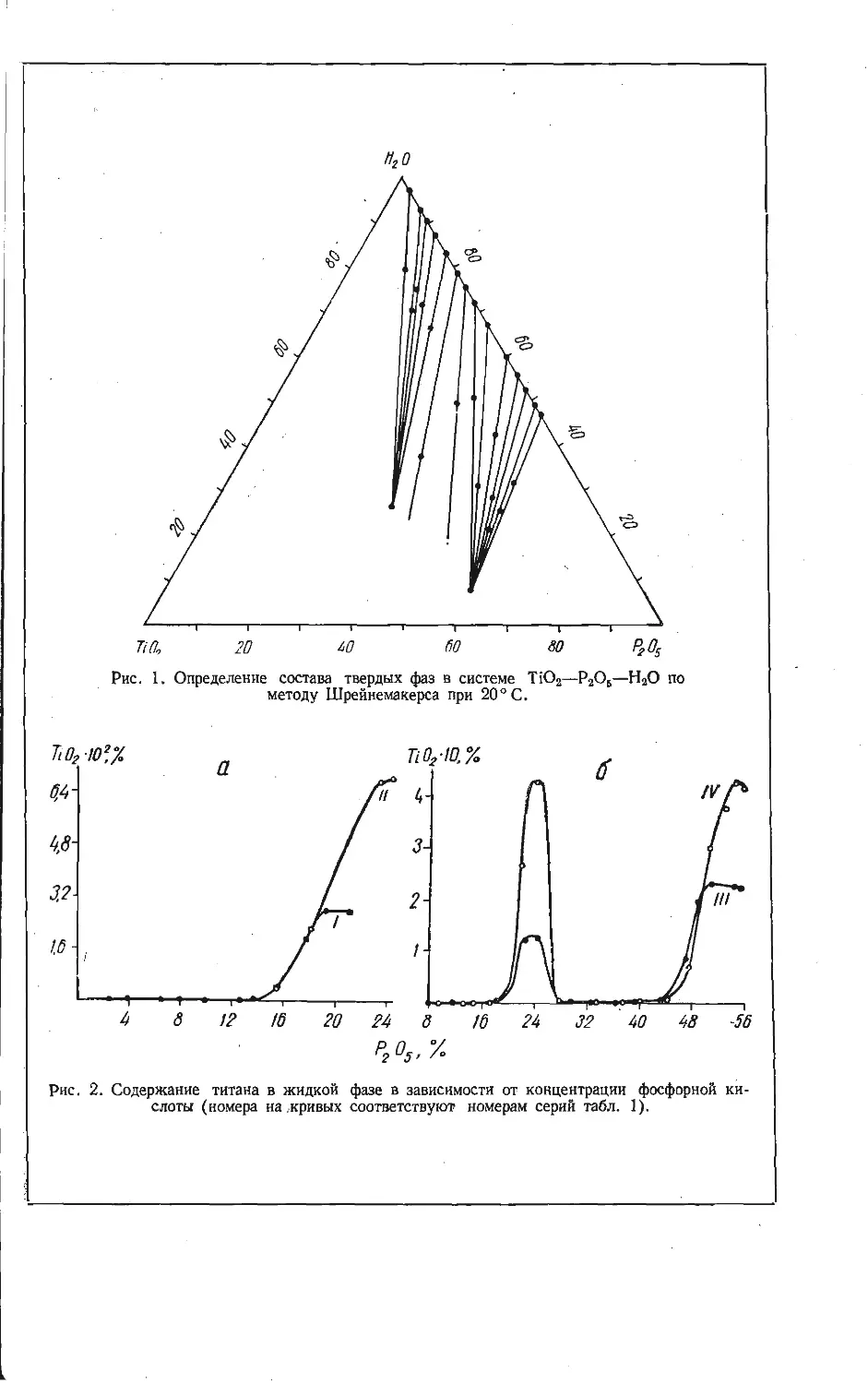
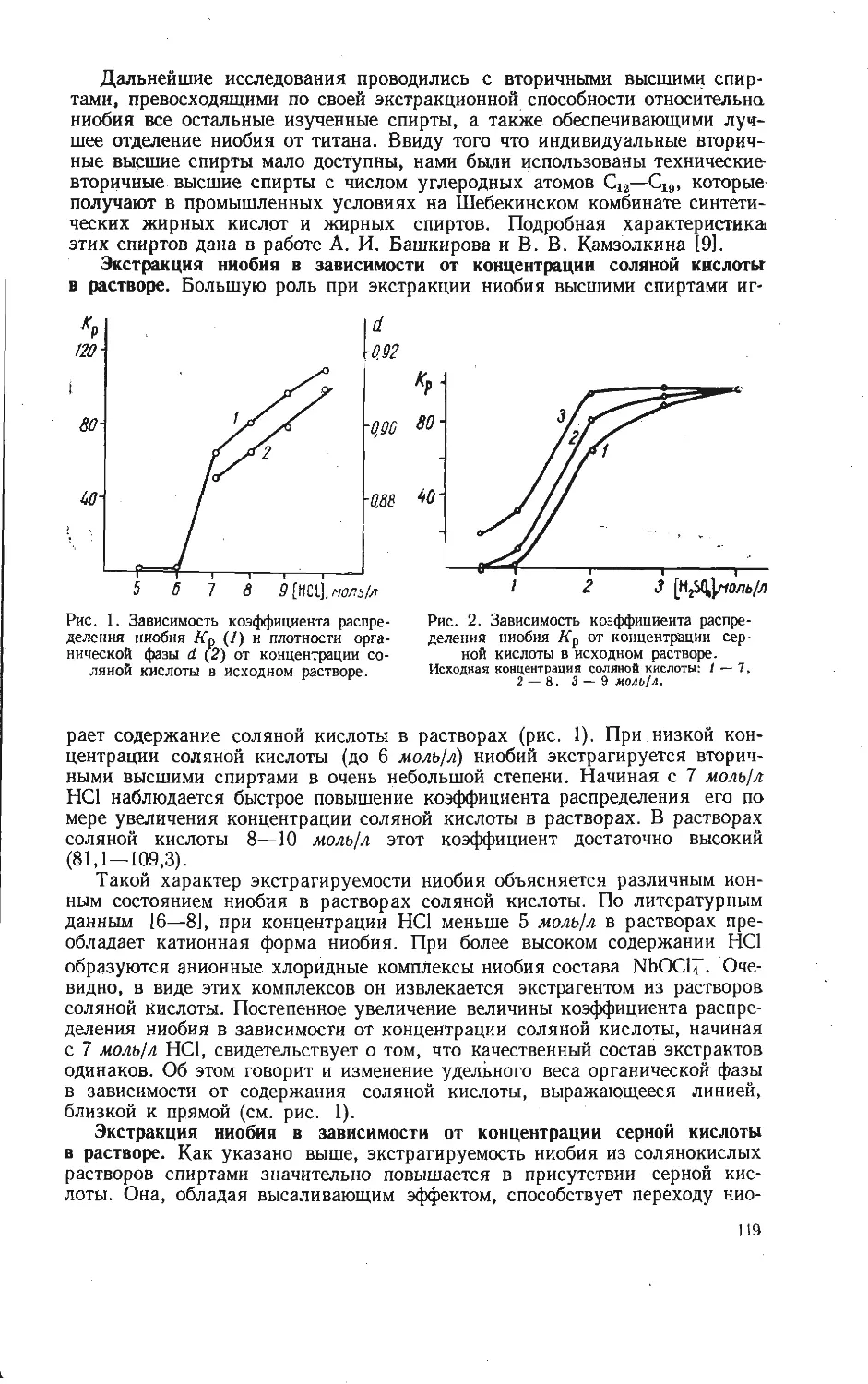
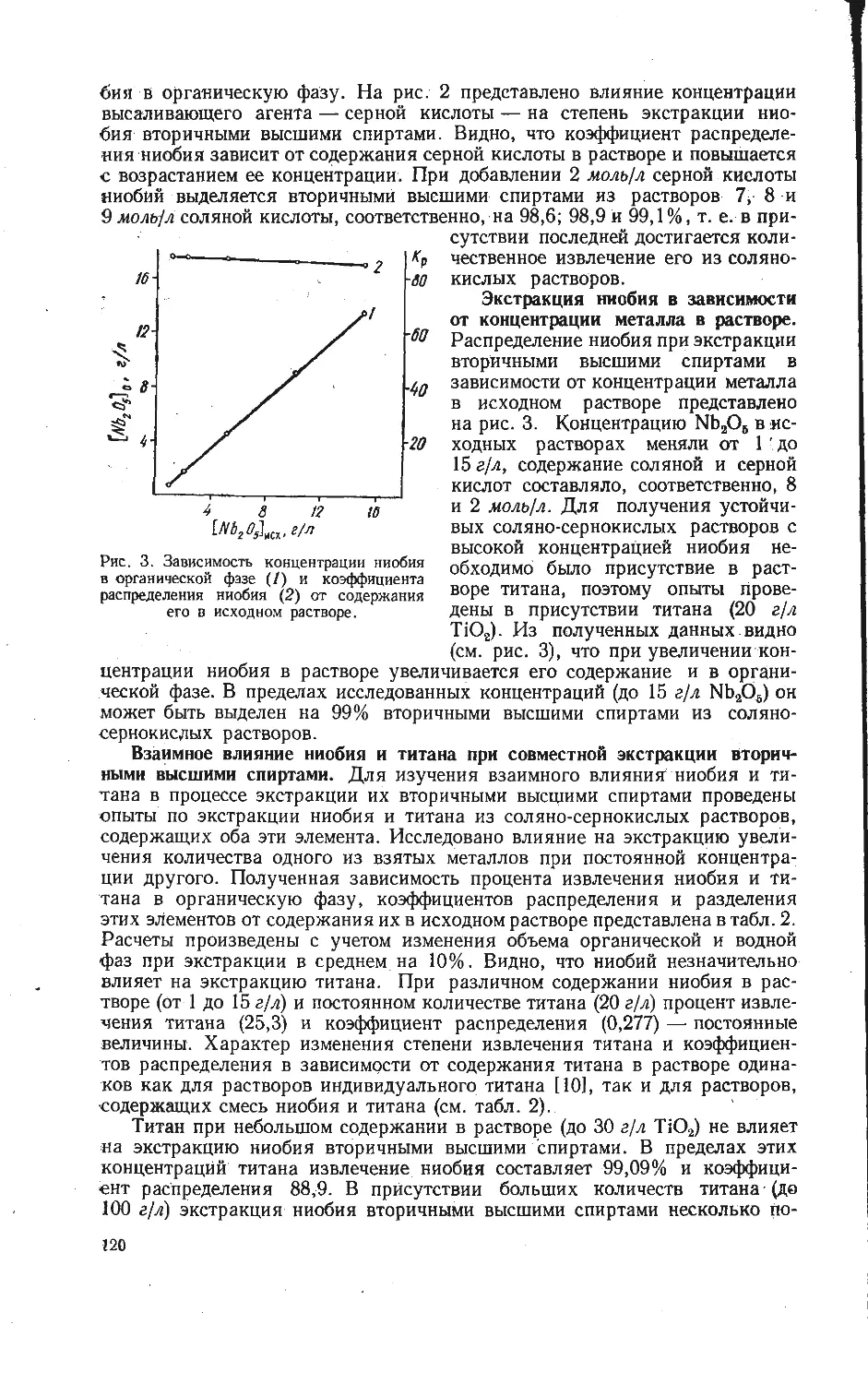
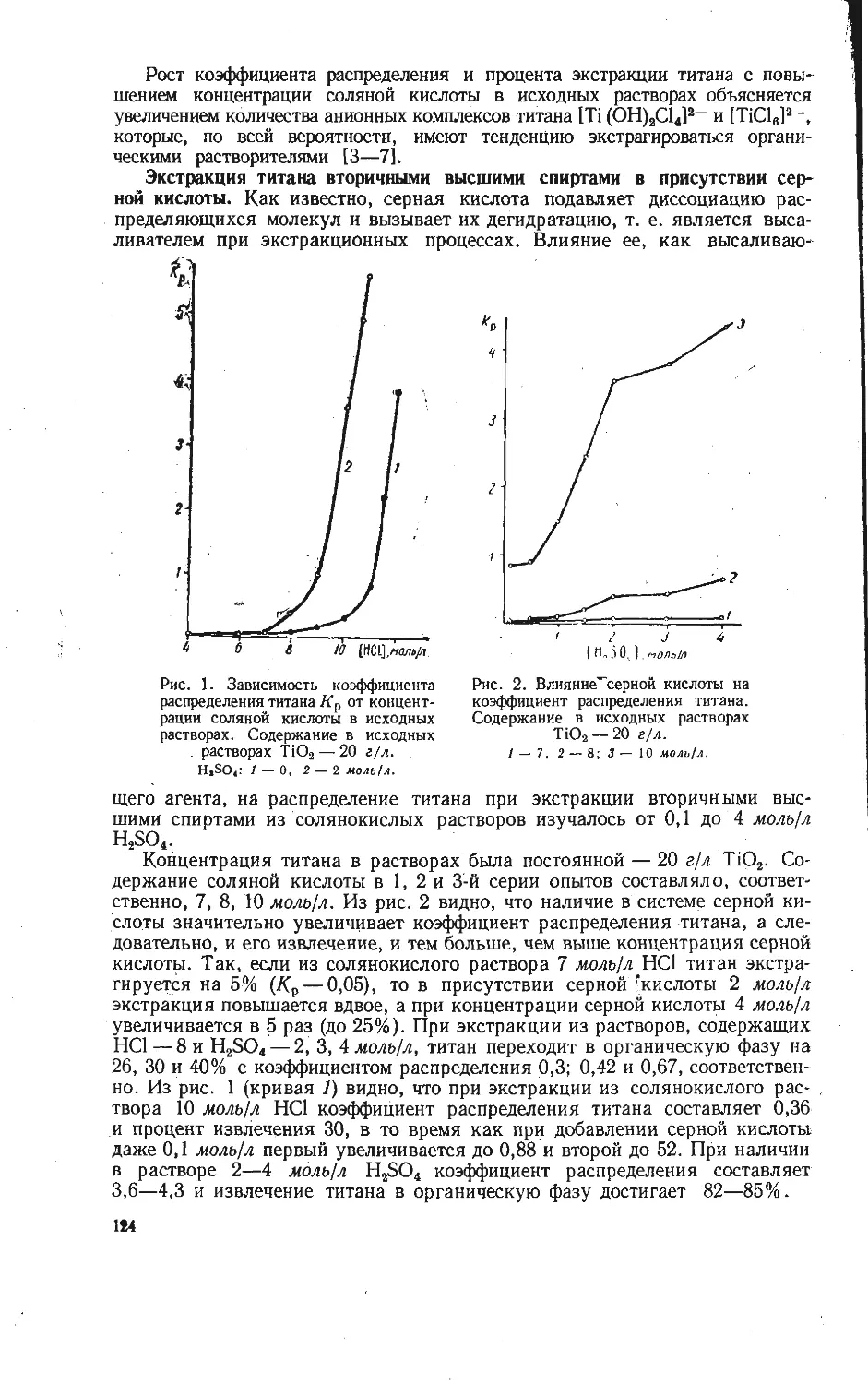
/






































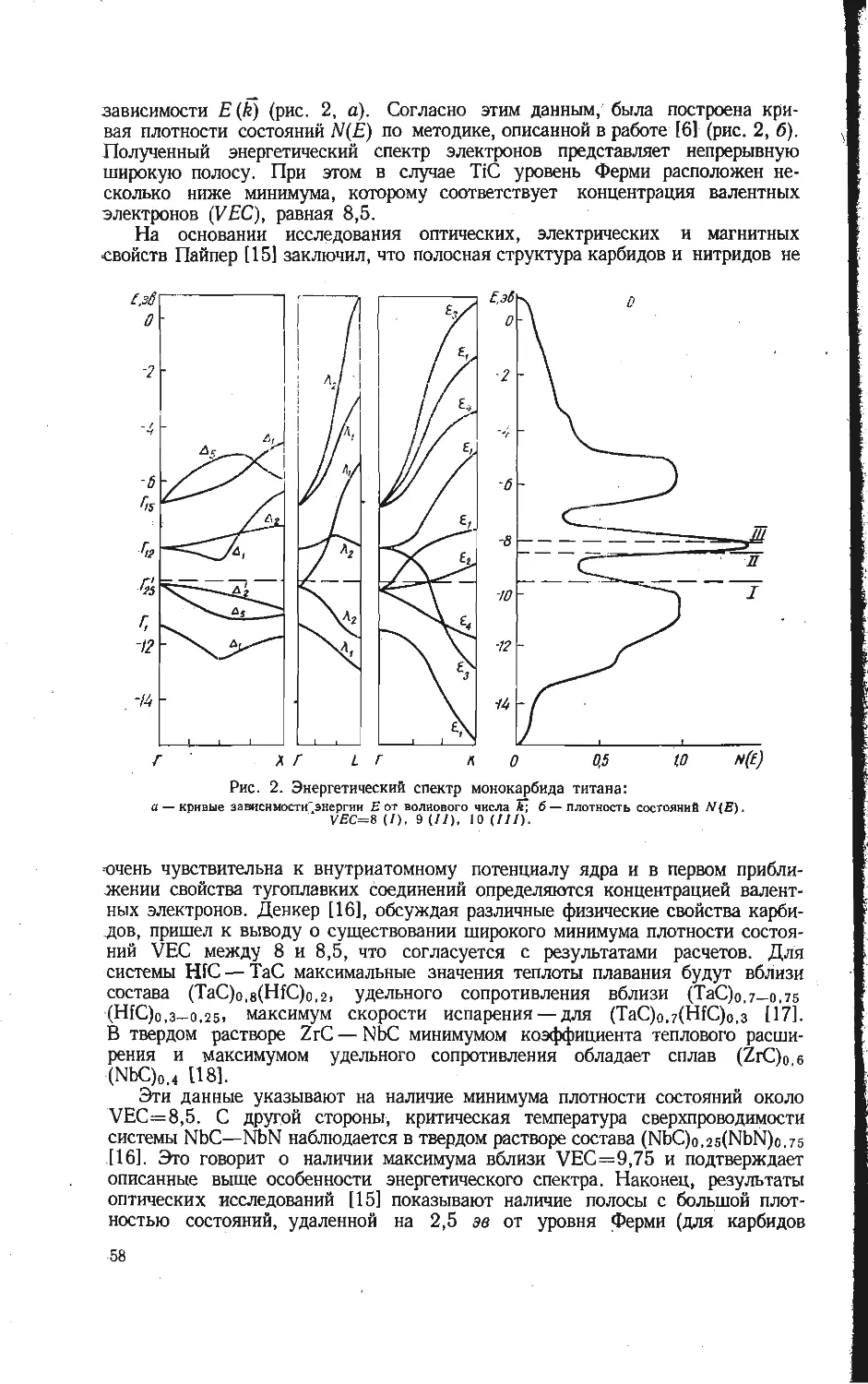




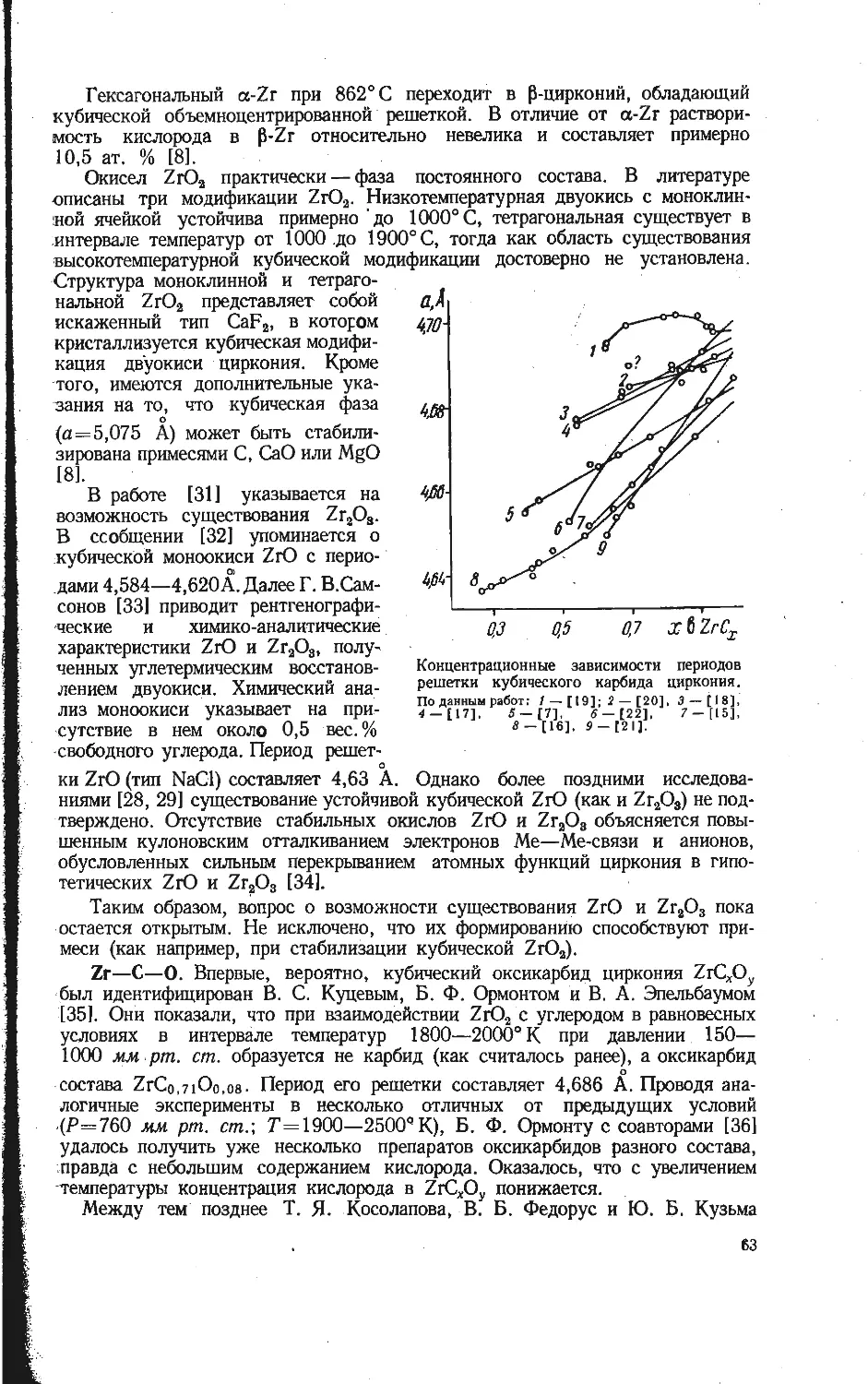







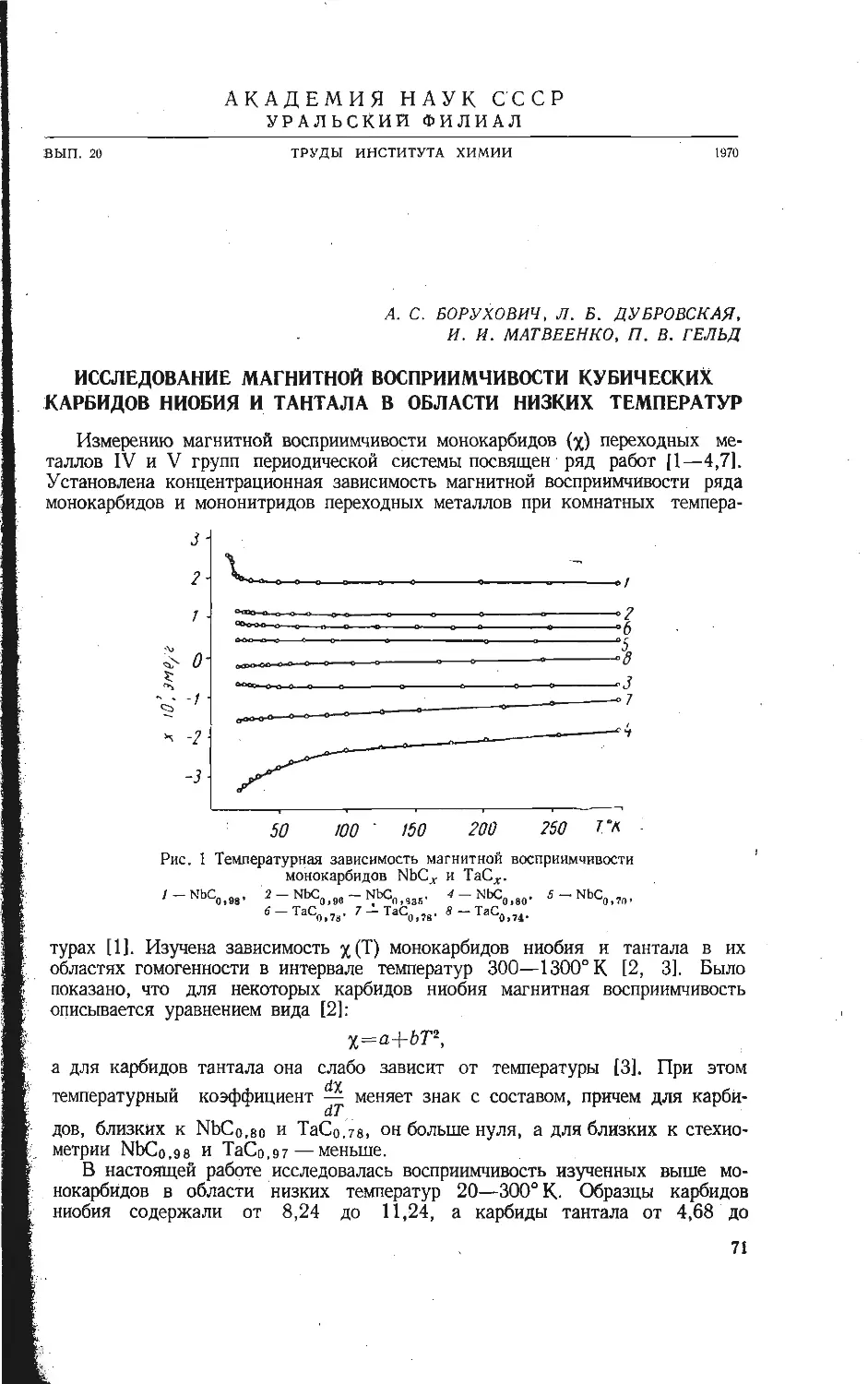





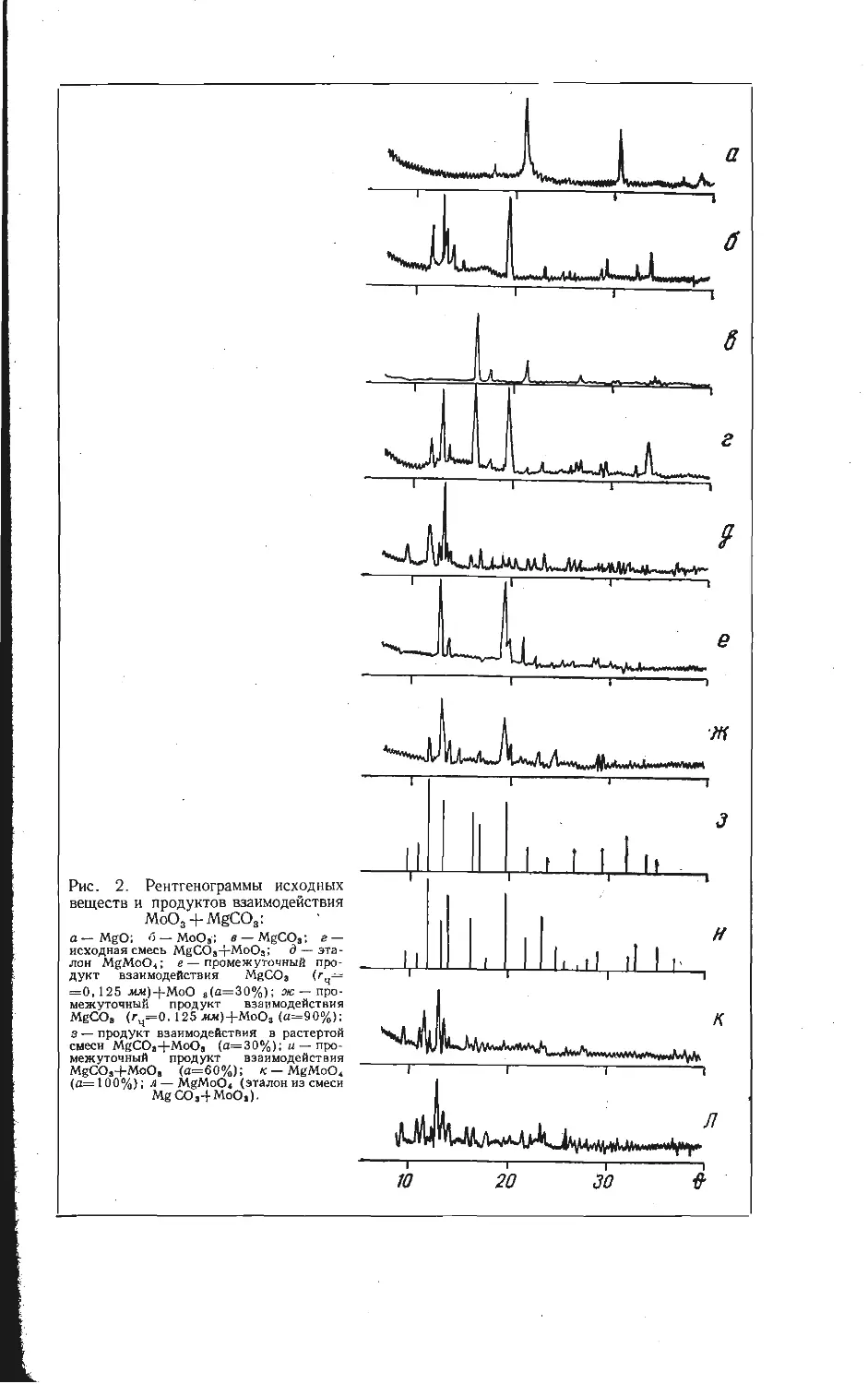









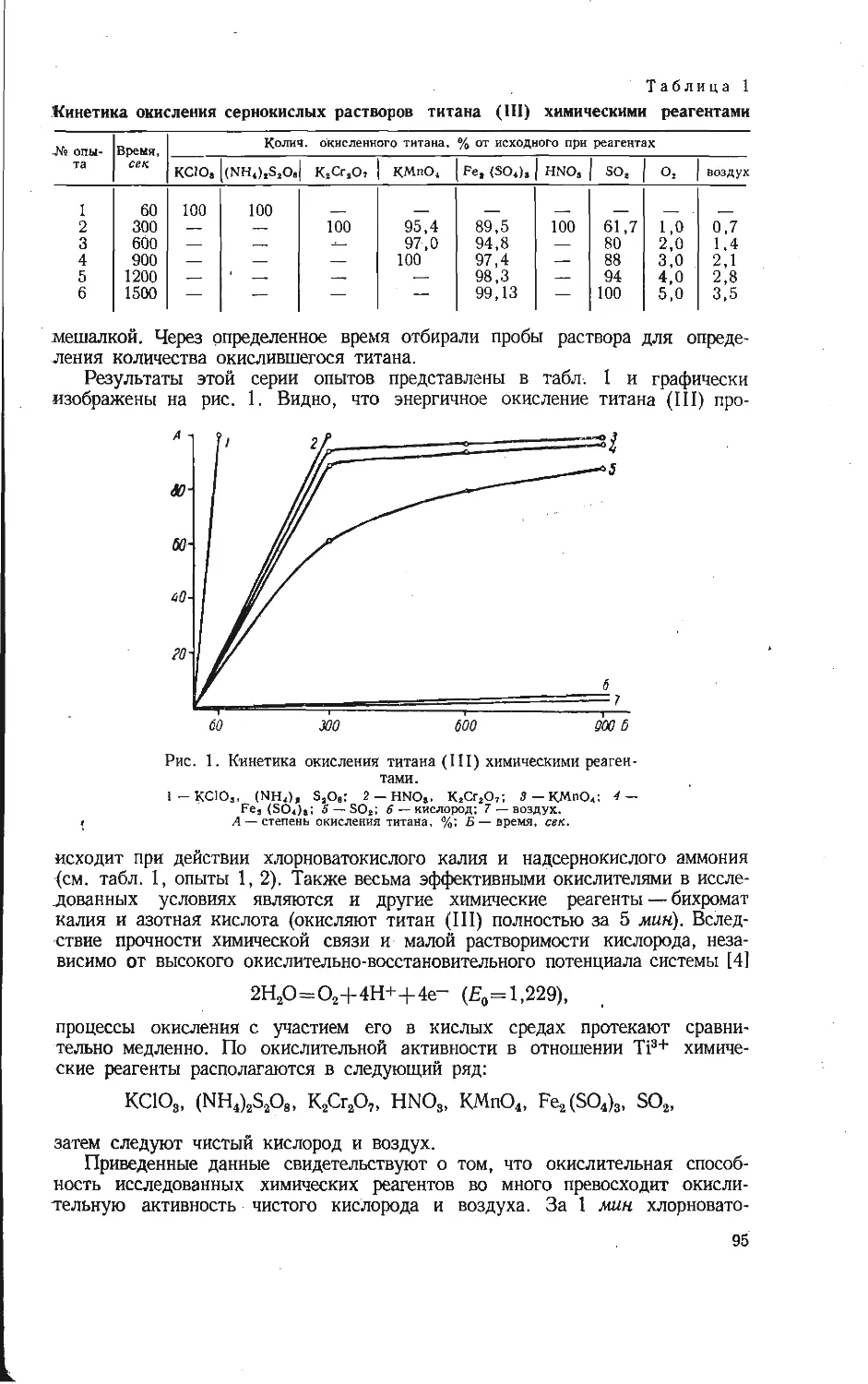














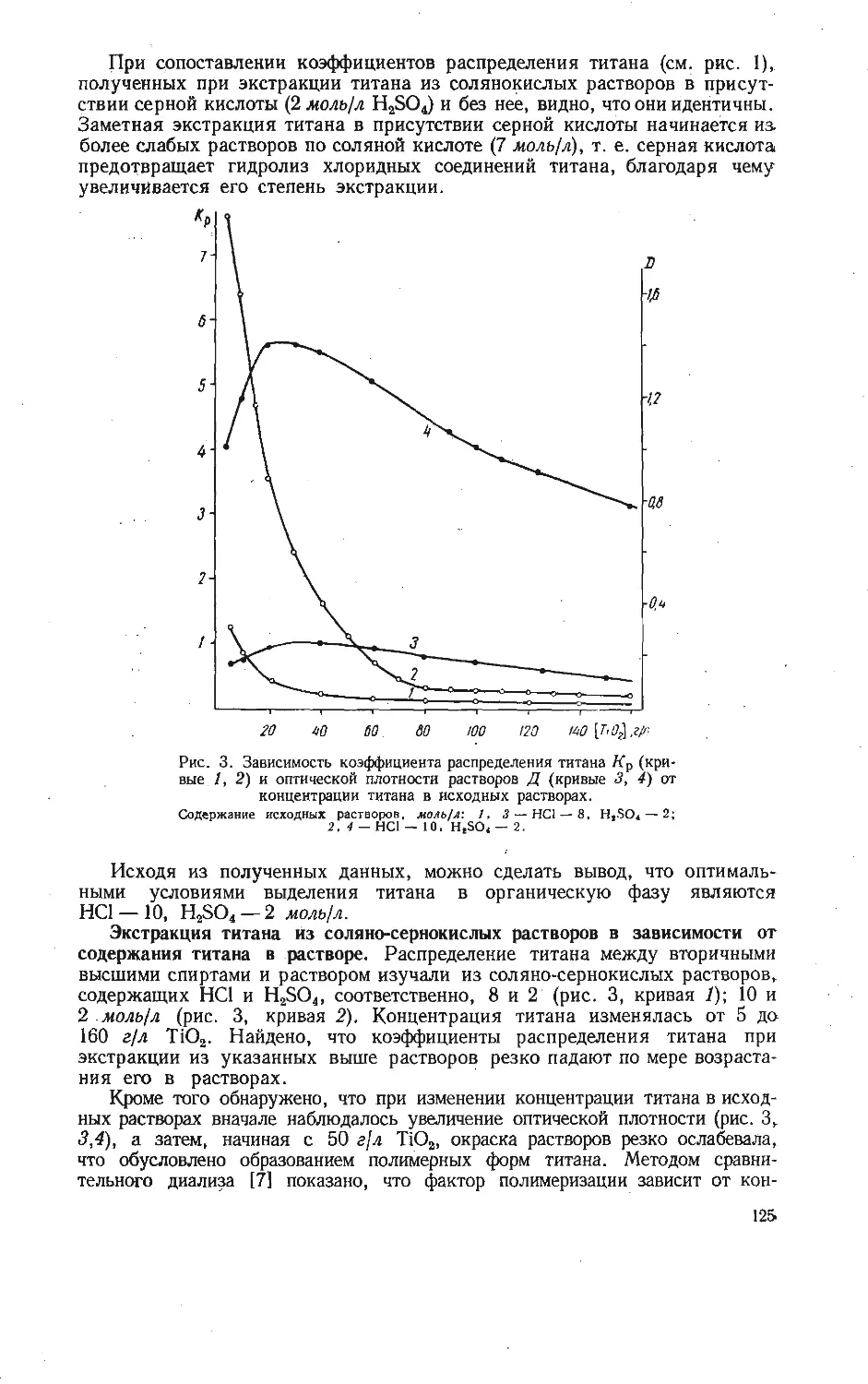


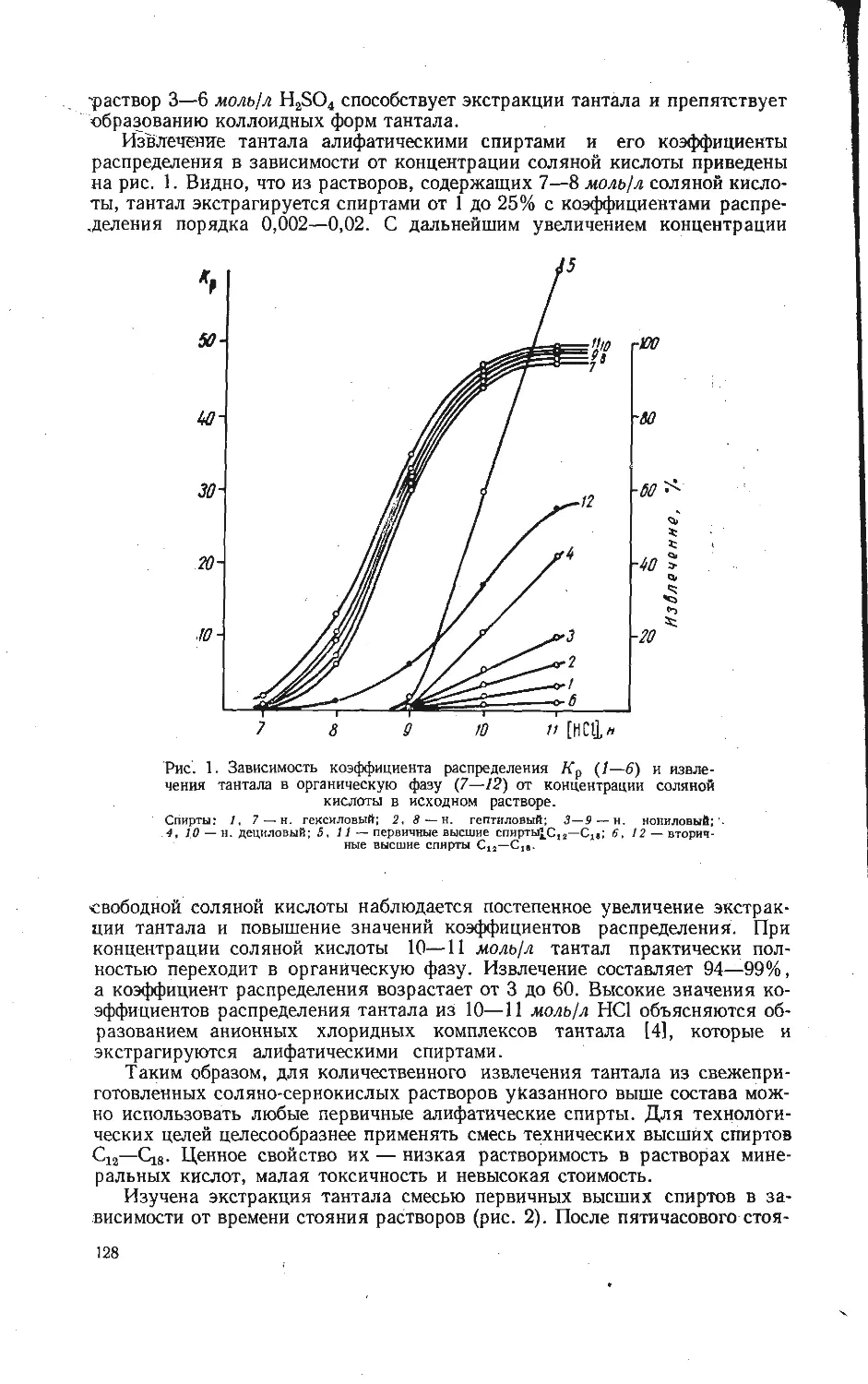
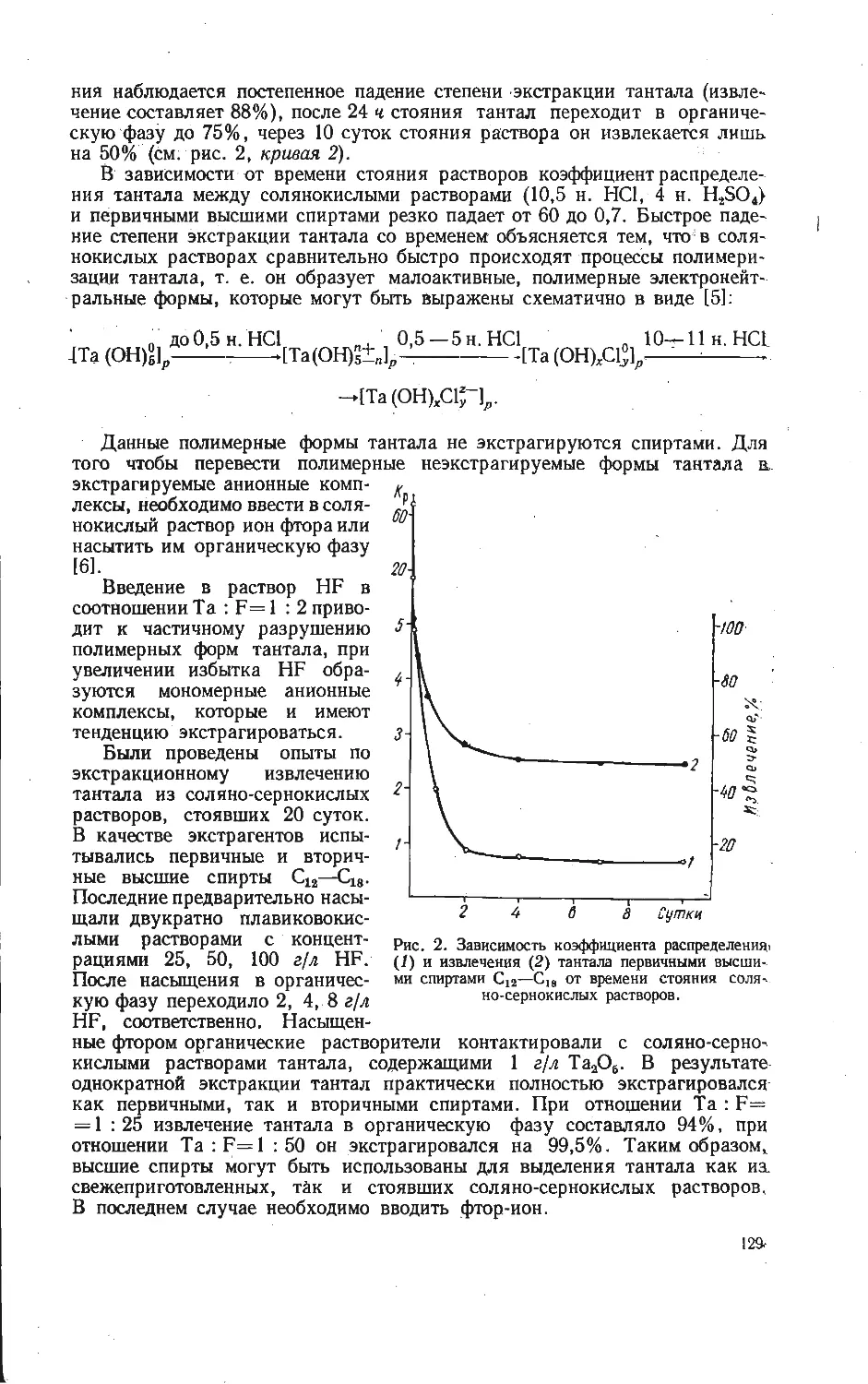



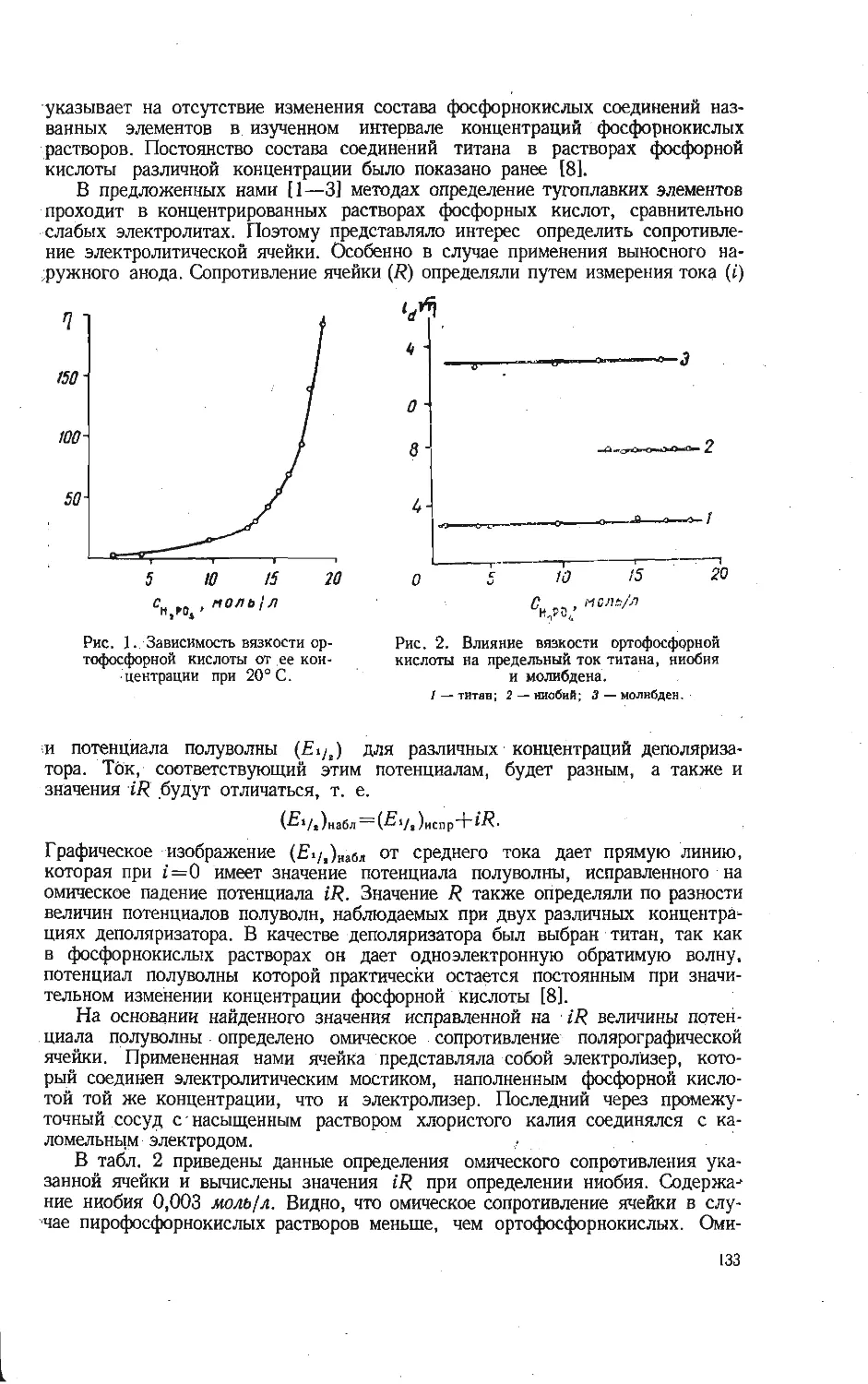




























Автор: Бамбуров В.Г.
Теги: неорганическая химия термодинамика энергетика химия редкие элементы
Год: 1970




Текст
АКАДЕМИЯ НАУК СССР
уральский филиал
ИССЛЕДОВАНИЕ
СОЕДИНЕНИИ
V:0s
РЕДКИХ
ЭЛЕМЕНТОВ
5
з
V, ^4
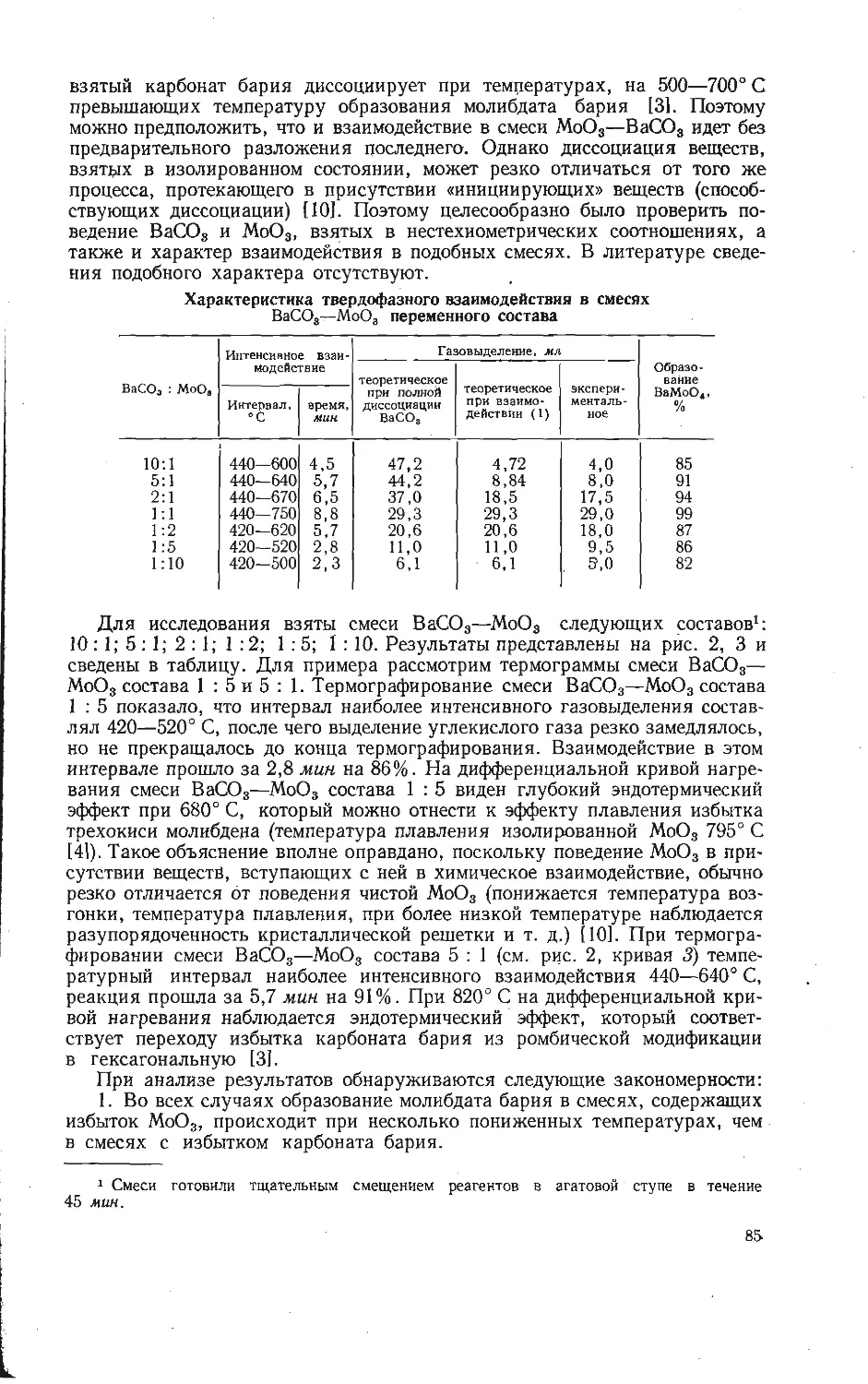
AtaV. Off +CO2^<h
СВЕРДЛОВСК • I Q7O
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970.
ИССЛЕДОВАНИЕ СОЕДИНЕНИЙ
РЕДКИХ ЭЛЕМЕНТОВ
(V, Nb, Та, Ti, Zr, Mo, Ga)
СВЕРДЛОВСК
Печатается по постановлению
Редакционно-издательского совета
Уральского филиала АН СССР
Ответственный редактор В. Г. Бамбуров
ИССЛЕДОВАНИЕ СОЕДИНЕНИЙ РЕДКИХ ЭЛЕМЕНТОВ
(V, Nb, Та, Ti, Zr, Mo, Ga)
(Труды Института химии, вып. 20)
РИСО УФАН СССР
СВЕРДЛОВСК, К-49
ПЕРВОМАЙСКАЯ, 91
Редактор изд-ва Т. И. Слесарева
Художник Я- И. Чернихов
Техн, редактор Н. Р. Рабинович Корректоры И. Д. Махнева, М. И. Зубринская
РИСО УФАН СССР 454—39 (69). НС 15359. Подписано к печати 20/XI 1970 г.
Печ. л. 10,5. Уч.-изд. л. 13,9. Формат 70x108/16.
Заказ 632. Тираж 1200. Цена 1 руб. 41 коп.
Типография изд-ва «Уральский рабочий», г. Свердловск, проспект Ленина, 49.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
А. А. ФОТИЕВ, Б. Г. ГОЛОВКИН
ОПРЕДЕЛЕНИЕ ТЕПЛОТ ОБРАЗОВАНИЯ ВАНАДАТОВ
ЩЕЛОЧНЫХ МЕТАЛЛОВ МЕТОДАМИ 'СРАВНИТЕЛЬНОГО РАСЧЕТА
Для термодинамического анализа процессов, лежащих в основе техноло-
гии извлечения ванадия, необходимо знать теплоты образования ванадиевых
соединений, экспериментальные значения которых определены только для
ванадатов натрия [1]. В настоящей работе методами сравнительного расчета
сделана оценка теплот образования ванадатов щелочных металлов. Эти зна-
чения можно найти по циклу Борна — Габера из уравнения, приведенного
в работе [2]
t/K=-Atf°+A/7°+Atf°, . (1)
где UR — энергия кристаллической решетки; —А№— теплота образования
ванадата при 298° К; —Д77к и —А/7° — соответственно, теплоты образова-
ния газообразных катиона и аниона при 298° К. Энергию кристаллических
решеток ванадатов щелочных металлов находили по уравнению А. Ф. Капу-
стинского [3]
U =287,2 (1 _ 2д345 \ (2)
Га+Гк \ ''a+'’к '
где — число ионов, образующих молекулу данной соли; zK иг, — заряды
ионов; гк и га — их радиусы. Для определения условного термохимического
радиуса иона ортованадата гуоз- использовали параметры кристаллических
решеток ортованадатов, ортофосфатов и ортоарсенатов редких элементов,
приведенные Шварцем [4]. Радиусы ортофосфата гроз-=2,38 А и ортоарсе-
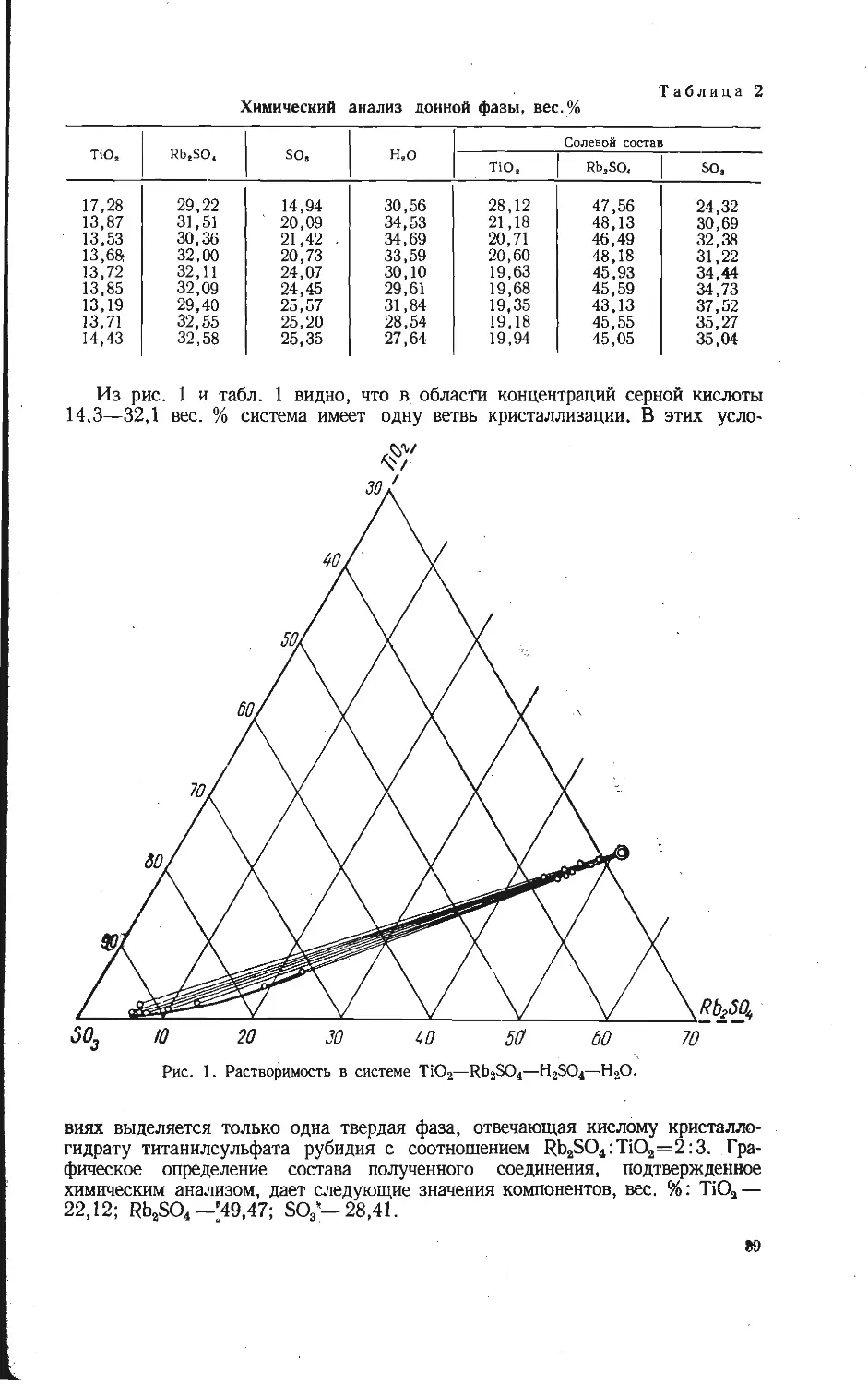
ната rAsQ3-=2,48 А взяты из работы [2]. Из табл. 1 видно, что половина
отношения суммы двух параметров кристаллических решеток диск сумме
радиусов катиона и аниона близка к величине
-----—------= 1,99. (3)
2 ^K+rvo3—)
Ортованадаты редких элементов изоструктурны с ортофосфатами и ортоар-
сенатами редких элементов, поэтому следует ожидать, что у ортованадатов
редких элементов будет аналогичное соотношение, которое даст возможность
определить радиус иона ортованадата, если известны радиусы катионов,
взятые из работ [2, 5] (табл. 2). Например, ортованадат четырехвалентного
тория имеет параметры кристаллической решетки а—7,26 и с=6,47 А [6].
а
Таблица 1
Отношение параметров кристаллических решеток фосфатов и арсенатов
к удвоенной сумме радиусов катиона и аниона
Соединение о Параметр, A A a c a-\-c
a c 2 ('x+ra) 2 ('x+ra) 2 ('’x+'a)
ТЬРО4 6,941 6,070 3,27 1,063 0,93 1,99
DyPO4 6,917 6,053 3,26 1,061 0,93 1,99
НоРО4 6,891 6,031 3,24 1,063 0,93 1,99
ЕгРО4’ 6,864 6,007 3,23 1,063 0,93 1,99
ТиРО4 6,847 5,994 3,23 1,060 0,93 1,99
LuPO4 6,798 5,961 3,18 / 1,068 0,93 2,00
TbAsO4 7,102 6,335 3,37 1,054 0,94 1,99
DyAsO4 7,074 6,307 3,36 1,053 0,94 1,99
HoAsO4 7,048 6,292 3,34 1,054 0,94 1,99
ErAsO4 7,021 6,272 ’ 3,33 1,054 0,94 1,99
TuAsO4 7,000 6,256 3,33 1,051 0,94 1,99
LuAsO4 6,952 6,230 3,28 1,063 0,95 2,01
Среднее ... 1,99
Таблица 2
Определение термохимического радиуса иона ортованадата
Параметр, A, 'k+'vo3- 4 rK [2] rV°3- 4
Соединение a c a A
CeVO4 7,398 6,498 3,474 1,02 2,454
NdVO4 7,335 6,434 3,442 0,99 2,452
EuVO4 7,239 6,370 3,402 0,97 2,432
TbVO4 7,179 6,324 3,378 0,89 2,488
HoVO4 7,124 6,292 3,354 0,86 2,494
TuVO4 7,071 6,263 3,334 0,85 2,484
LuVO; 7,026 6,231 3,314 0,80 2,514
Среднее . . , 2,48
Принимая, что радиус Th4+ равен 0,95 А, можно рассчитать по уравнению (3\
радиус иона ортованадата, который составляет 2,48 А.
Параметр элементарной ячейки кристаллической решетки содержит оди-
наковое число катионов и анионов, поэтому значение длины ребра ячейки
пропорционально величине суммы радиусов катиона и аниона. Однако не
все ионы, на данном ребре ячейки расположены на одной прямой. Это
является причиной неточной кратности отношения параметра кристаллической
решетки к сумме радиусов катиона и аниона. Приблизительно сумма всех
параметров решетки в результате частичной компенсации кратна сумме ради-
усов катиона и аниона. Так, если сумму параметров ортованадата лантана
(а=7,03; Ь—7,34 и с=6,69 А) разделить на 6 и вычесть из частного значе-
ние гьа3+ = 1,04 А, то радиус иона ортованадата окажется равным 2,47 А.
Параметры кубических решеток ортованадатов бария и стронция равны 7,859
,0 . ...
и 7,458 А, соответственно. Разделив эти значения пополам и вычтя гВа2+=1>43
4
и rs(2+ = l,27 А, получим для радиуса иона ортованадата значения, соответ-
ственно, 2,50 и 2,46 А.
Рассматривая сумму параметров кристаллической решетки как кратное
суммы радиусов катиона и аниона, найдем радиус иона метаванадата. При-
веденные в табл. 3 величины радиуса rvo- = l,78 А иона метаванадата опре-
делены по параметрам кристаллических решеток, взятых из работ [7—9].
Таблица 3
Определение условного термохимического радиуса иона метаванадата
Соединение о Параметр, A Степень кратности rK 12] 'vor
a b c A
nh4vo3 5,85 11,82 4,92 22,59 7 1,43 1,80
NaVO3 5,85 10,12 9,43 25,40 8 0,98 1,84
KVO, 5,70 10,82 5,22 21,74 7 1,33 1,83
RbVO3 6,654 11,98 4,99 22,624 7 1,49 1,74
CsVO3 5,788 12,20 5,385 23,373 7 1,65 1,69
Среднее . . . 1,78
Если неизвестно число ионов, приходящихся на каждое ребро ячейки, то
степень кратности суммы параметров кристаллической решетки к сумме
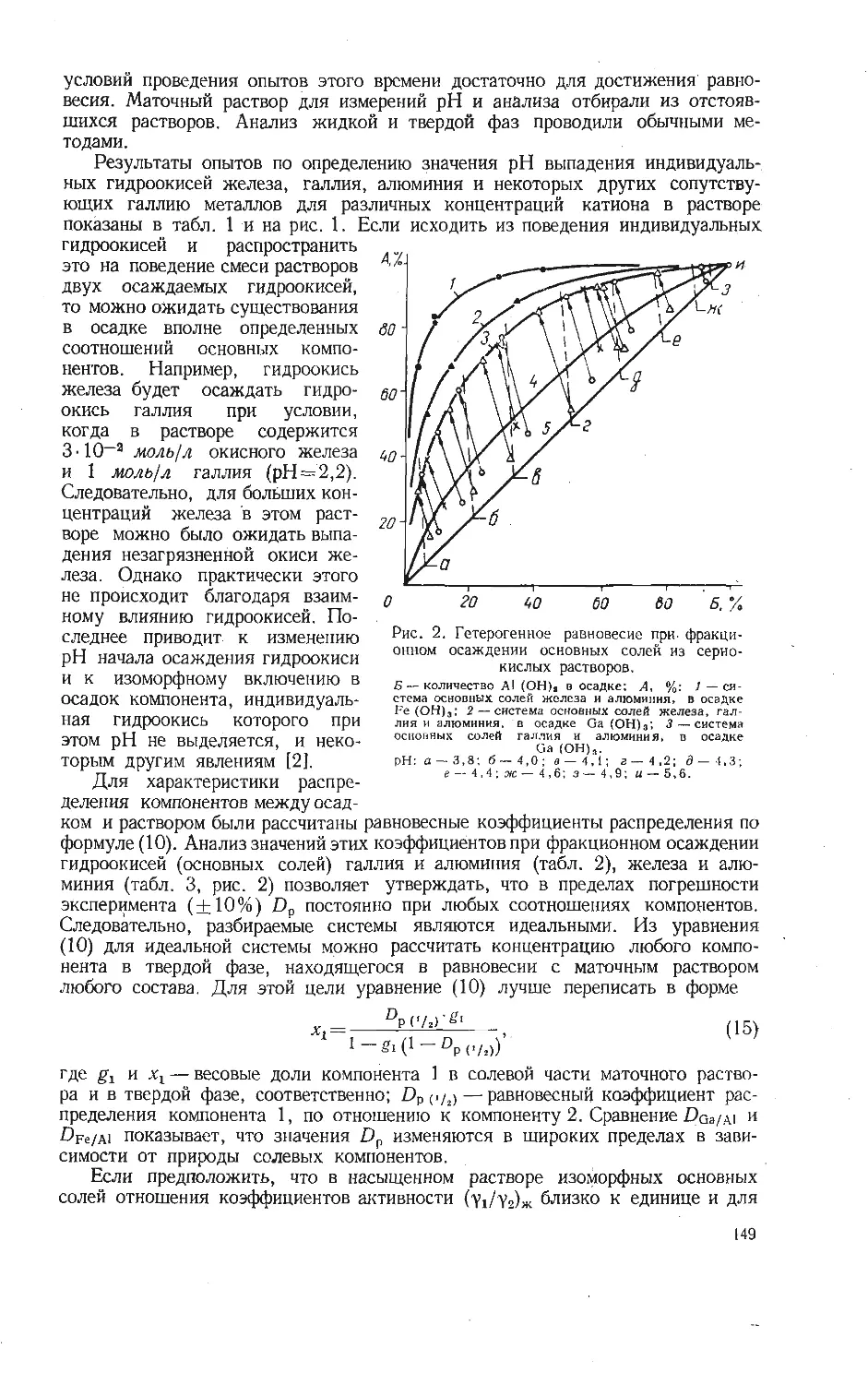
радиусов катиона и аниона можно найти следующим образом. Строится
графическая зависимость радиусов кислородсодержащих комплексных ионов,
значения которых имеются в исследовании [2], от радиуса иона основного
элемента, образующего кислородсодержащий ион. По полученной зависимости
находится предполагаемый интервал, в котором должен быть расположен
радиус искомого иона. Для иона ортованадата таким интервалом является
2,4—2,6 А, для иона метаванадата 1,7—1,8 А.. Следовательно, степень крат-
ности равна ближайшему целому числу, взятому из отношения сумм пара-
метров к радиусу катиона и приблизительного радиуса аниона. Подставив
уравнение (2) для энергии кристаллической решетки в формулу (1), записав
полученное выражение для метаванадатов натрия и кальция, теплоты образо-
вания ДЯ298 которых равны, соответственно, 274,9 и 556,7 ккал/моль [1,10],
и учтя, что теплоты образования катионов натрия и кальция равны, соот-
ветственно, 143,9 и 452,6 ккал/моль [2], получим два уравнения с двумя
неизвестными. В результате решения этой системы найдены радиус иона
метаванадата, оказавшийся равным 1,76 А, и теплота образования иона мета-
ванадата в газообразном состоянии (237,5 ккал/моль). При использовании
значений условных термохимических радиусов из работы [5] теплота образо-
вания иона метаванадата получается равной 233,0 ккал/моль.
В кристаллической решетке метаванадата ванадий окружен четырьмя
атомами кислорода, а не тремя, как это следует из химической формулы
MVO3, причем атомы ванадия соединены в виде цепочек через атомы кисло-
рода. Можно предположить, что условный термохимический радиус иона
метаванадата будет соответствовать расстояниям от атома ванадия до центров
атомов общих кислородов. По данным Эванса [11], расстояния V—О в ме-
таванадате калия равны 1,81; 1,81; 1,66; 1,65 А, в метаванадате аммония
1,80; 1,80; 1,65; 1,67 А. Усреднение этих расстояний дает близкое значение
1,73 А.
5
С помощью радиусов ионов мета- и ортованадатов по формуле (2) найдены
энергии кристаллических решеток мета- и ортованадатов щелочных металлов.
По значениям энергии кристаллической решетки ортованадата натрия, равной
500 ккал/моль, теплоты его образования (421,0 ±2,5 ккал!моль) [10] и
теплоты образования газообразного иона натрия (—143,9 ккал/г-ион) [2}
с помощью уравнения (1) определили теплоту образования газообразного иона
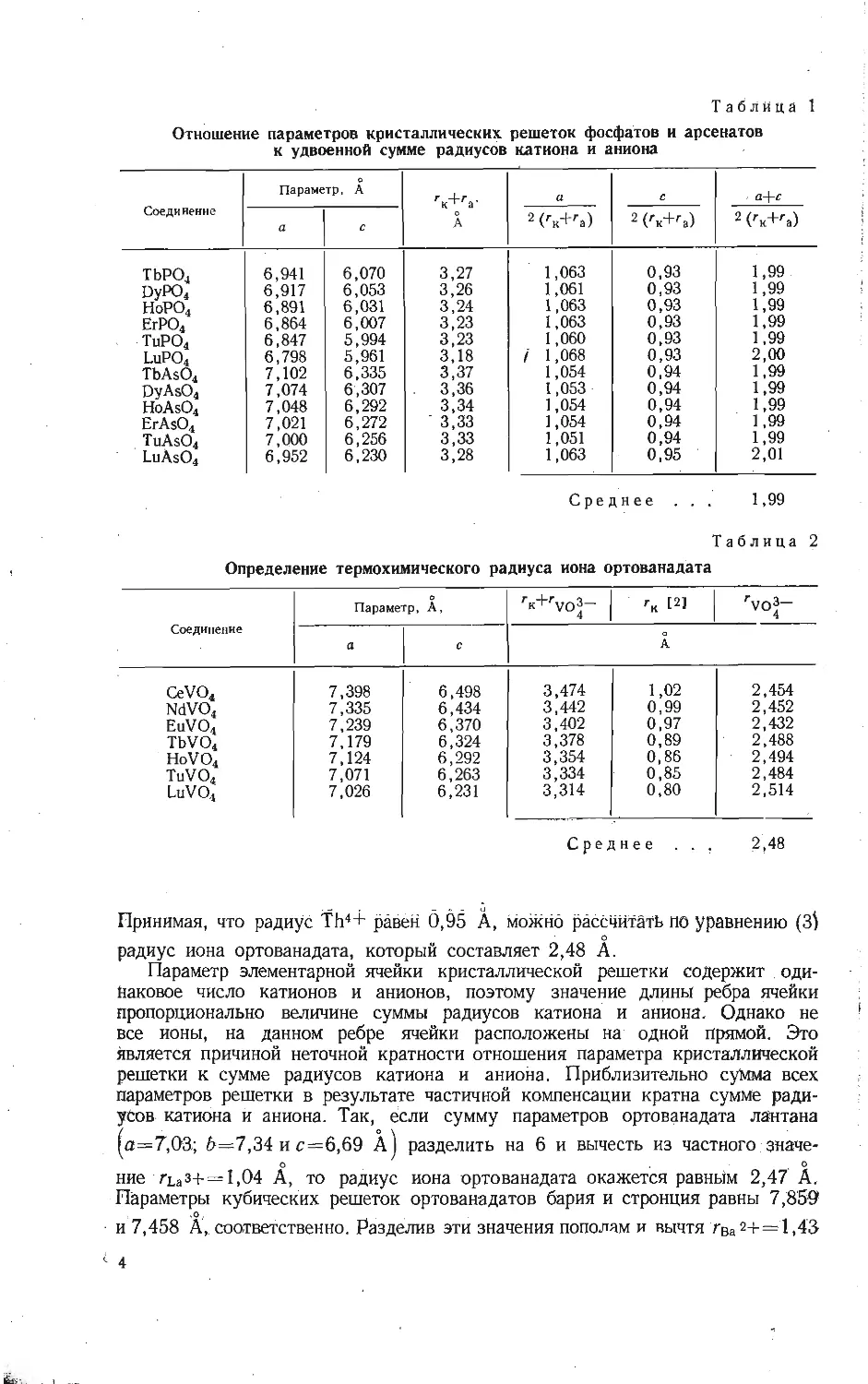
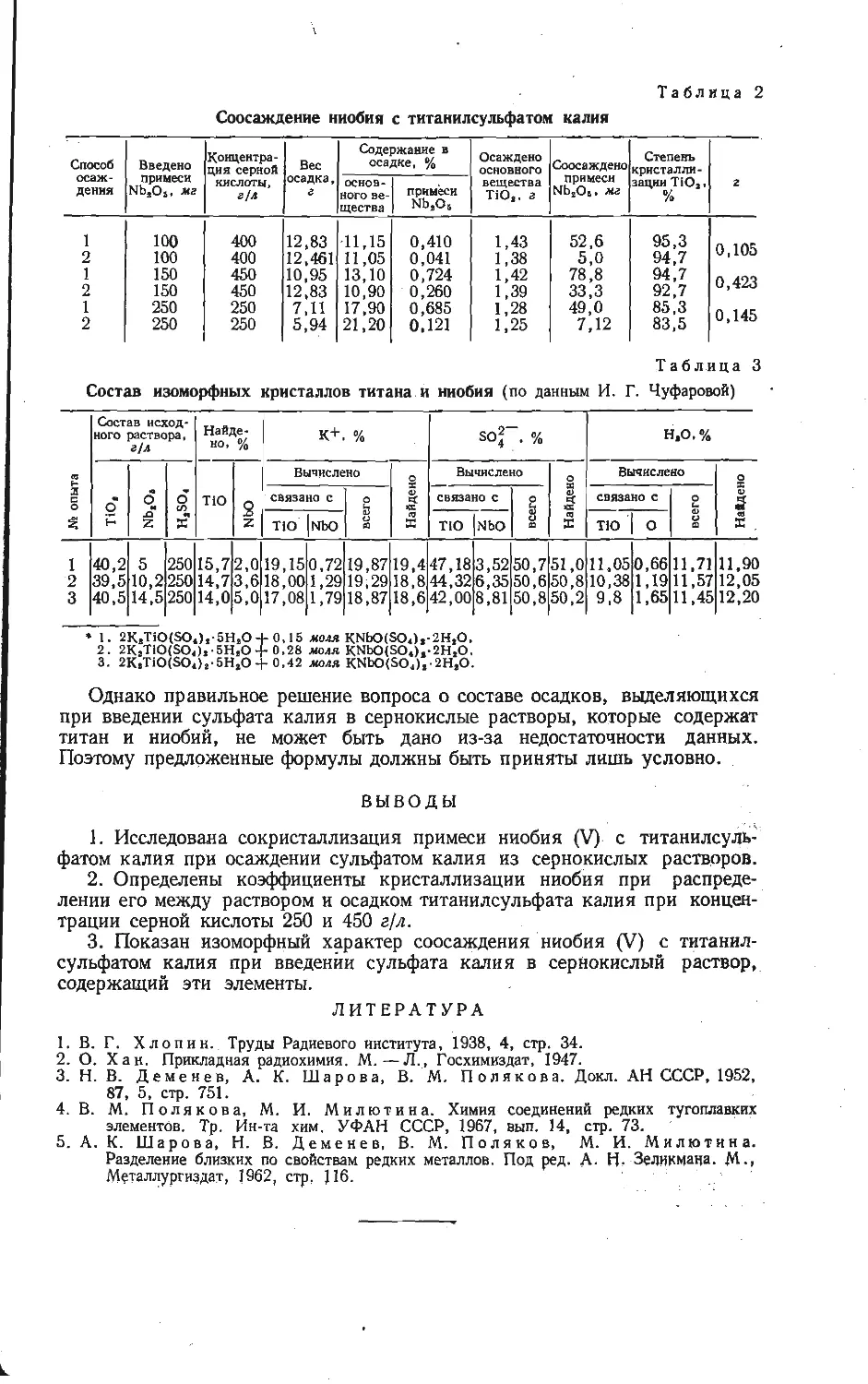
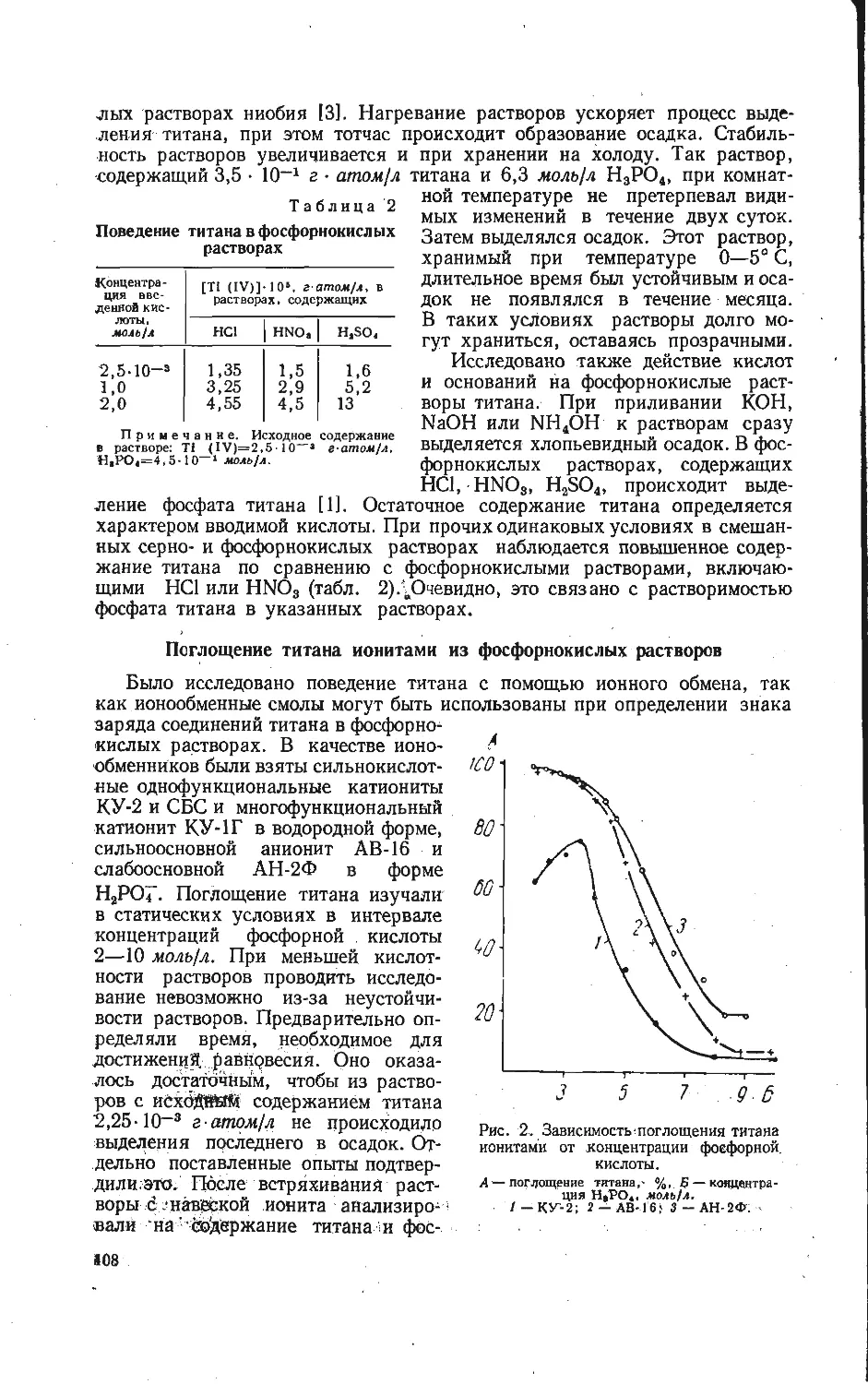
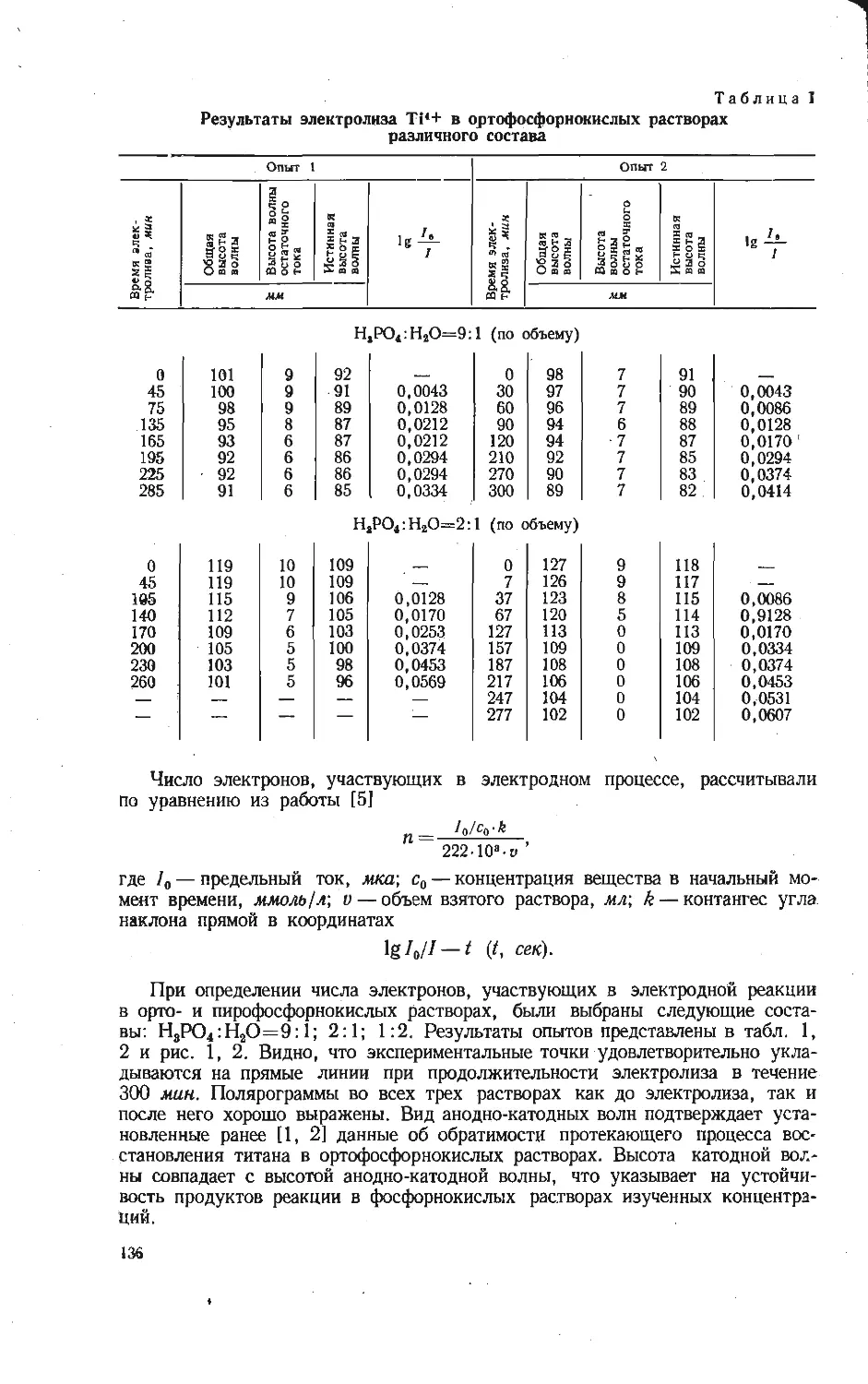
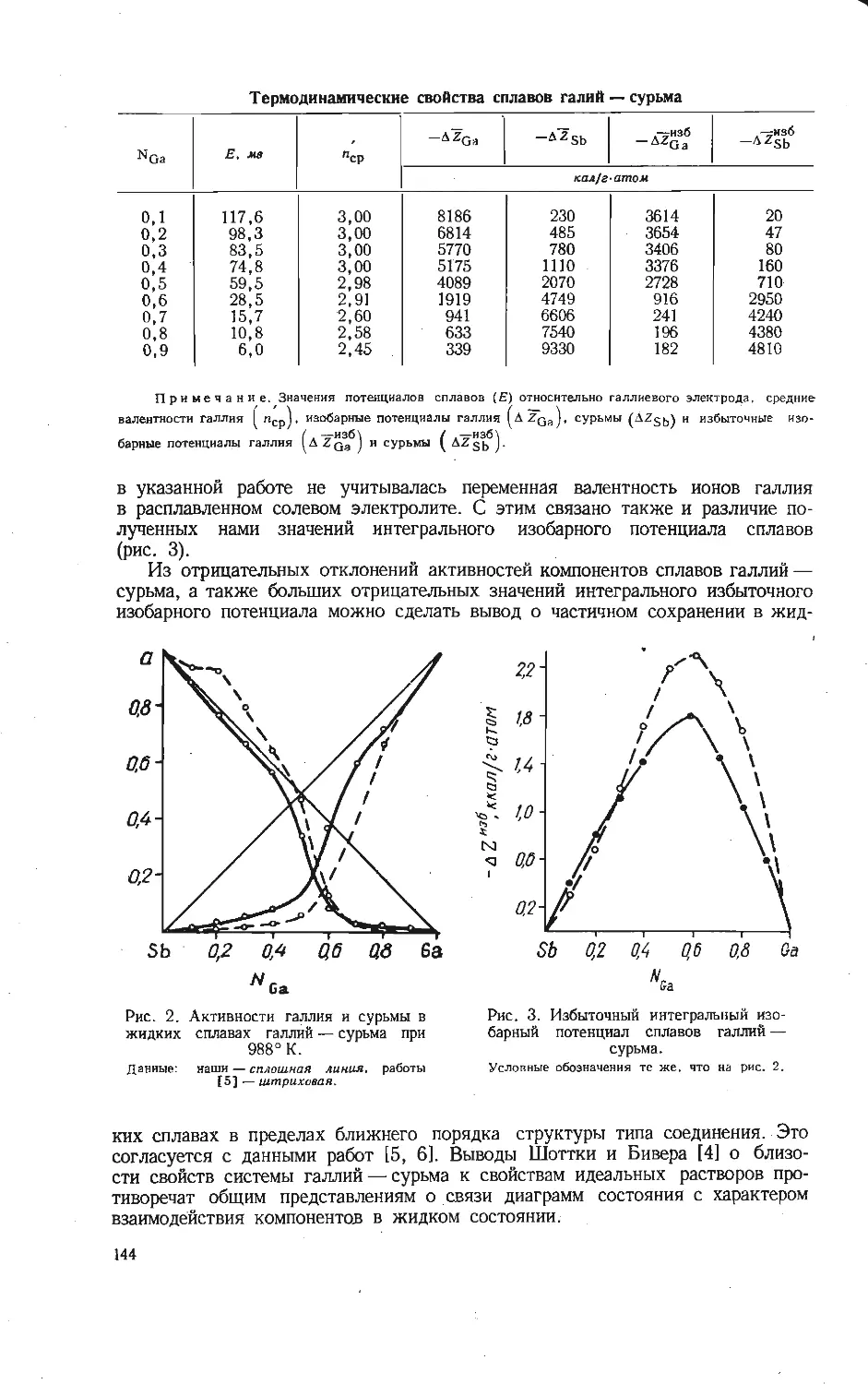
Зависимость приведенных теплот образования из элементов вана-
датов щелочных металлов от отношения V2O5: Ме2О при исполь-
зовании условных термохимических радиусов из работы [5].
/ — Na; 2 —К: 3 — Rb; 4 — Cs.
ортованадата, которая равна —48,5 ккал! г-ион. Это уравнение помогло
оценить стандартные теплоты образования из элементов мета- и ортованада-
тов, представленные в табл. 4.
По циклу Борна — Габера:
2NaVO3 (т) V2O5 (T)+Na2OT
| 2[/ыаУО, t — UNa,O
2Na++ 2VOT(r) -A^Or2“+2Nar++V2O6 (t)
найдена энергия присоединения иона кислорода к пятиокиси ванадия, состав-
ляющая, принимая условные термохимические радиусы ионов, 346,4 ккал/моль,
по данным [2], и 321,4 ккал/моль, по данным [5]. Соответствующий цикл
для ортованадата натрия позволяет найти энергию присоединения трех ионов
кислорода к пятиокиси ванадия, равной 293,8 или 191,8 ккал/моль при ис-
пользовании радиусов ионов по [2] или [5]. С помощью приведенного цикла
Борна — Габера вычислены теплоты образования из окислов мета- и орто-
ванадатов щелочных металлов (см. табл. 4).
Г. М. Матвеевым [12] показано, что приведенные значения термодинами-
ческих характеристик соединений ряда MeO-pRO2 (или MeO-pR2O3) являются
линейной функцией отношения (или Прямая в координатах при-
веденная теплота образования — отношение окислов пересекает ось ординат
в значении, равном теплоте образования окиси, соответствующего металла.
Экспериментальные значения теплот образования для ванадатов натрия ли-
нейно зависят от отношения окислов (см. рисунок). В связи с этим с помощью
графической зависимости оценены теплоты образования из элементов всех
остальных известных ванадатов щелочных металлов. При использовании
6
Таблица 4
Энергия кристаллических решеток и теплоты образования ванадатов
щелочных металлов
Соединение ЛЯ°298ОК
rK [2] rx [5] из окислов из элементов
гк 12] ГК [5] Гк [2] 'к [5]
LiVO3 193,5 206,7 32,2 29,9 270,5 279,5
NaVO, 182,0 181,5 38,7+0,5* 274,9 ±2,5*
KVO, 164,0 164,0 54,7 53,7 280,3 275,8
RbVOa 157,0 153,2 58,7 54,9 281,0 272,7
CsVO, 150,0 149,6 66,7 54,3 281,0 275,8
Li3VOa 943 973 59,9 56,4 425,8 443,8
Na3VO4 9000 900 85,4+1,4* 421,0 + 2,5*
K.VO4 807 825 105,9 107,9 <407,0 413,0
Rb8VO4 775 795 106,9 113,9 398,0 406,0
Cs3VO4 745 765 121,9 124,4 388,1 396,1
Na4V2O7 — — 127,7±2,1* 699,5±5,0*
* Из работы [I].
условных термохимических радиусов ионов по данным [5] получены следу-
ющие значения для: K4VaO7 660; Rb4V2O7 и Cs4V2O7 st 640; KV3O8,
RbV8O8 и CsV3O8^770; K3V6O14 и Rb3V5O14 1340; ~ 5470;
Rb32V18Oei и Cs32V18O81« 5280 ккал/моль.
ВЫВОДЫ
1. Определены несколькими методами сравнительного расчета условные тер-
мохимические радиусы ионов ортованадата (2,48 А] и метаванадата (1,78 А) .
Вычислены теплоты образования ионов орто- и метаванадатов, энергия при-
соединения одного и трех ионов кислорода к пятиокиси ванадия.
2. Определены энергии кристаллических решеток мета- и ортованадатов
щелочных металлов. Вычислены теплоты образования из элементов и окислов
для мета- и ортованадатов щелочных металлов. Приближенно оценены зна-
чения теплот образования из элементов для остальных известных ванадатов
щелочных металлов.
ЛИТЕРАТУРА
1. М. F. Koehler. Bureau of Mines. Report, of Ind., I960, 5700.
2. К. Б. Яцимирский. Термохимия комплексных соединений. М., Изд-во АН СССР, 1951.
3. А. Ф. Капустинский. Ж. общ. хим., 1943, 13, 7-8, 497.
4. Н. Schwarz. Z. anorg. allg. Chem., 1963, 323, 44.
5. Краткий справочник химика. Сост. В. Н. Перельман. М., Госхимиздат, 1963.
6. G. Le Flem. Theses presentees a la faculte des Scientces de L’Universite de Bordeaux
pour obtenir Le grade de Docteur es sciences physiques. L’Universite de Bordeaux, 1967.
7. M. Petrasova, J. Madar. F, Hanic. Chem. zvesti., 1958, 12, 7, 410.
8. C. Calvo. Amer Cryst. Assoc., 1958, 18, 23.
9. H. Sorum. Det Kongelige Norske videnskabers selskab Forhandlinger, 1943, 16, 11, 39.
10. E. G. King, M. F. Koehler a. L. H. Adami. Heats and free energies of forma-
tion of calcium and magnesium vanadates. Washington, U. S. Dept, of the Interior,
Bureau of Mines, 1962.
11. H. T. Evans. Z. Kristallographie, 1960, 114, 3-4, 257.
12. Г. M. Матвеев. Изв. высш, учебн.завед., Химия и хим. технология, 1958, №2,135.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА химии
1970
Л. Л. СУРАТ, Б. Г. ГОЛОВКИН
ТЕРМОДИНАМИЧЕСКИЙ РАСЧЕТ РЕАКЦИЙ, ВОЗМОЖНЫХ В СИСТЕМЕ
KC1-V2O5
Одним из компонентов, добавляемых к шихте с целью извлечения пяти-
окиси ванадия из ванадийсодержащих концентратов, является хлористый
калий (галит-сильвиновая порода). Однако в литературе нет термодинамиче-
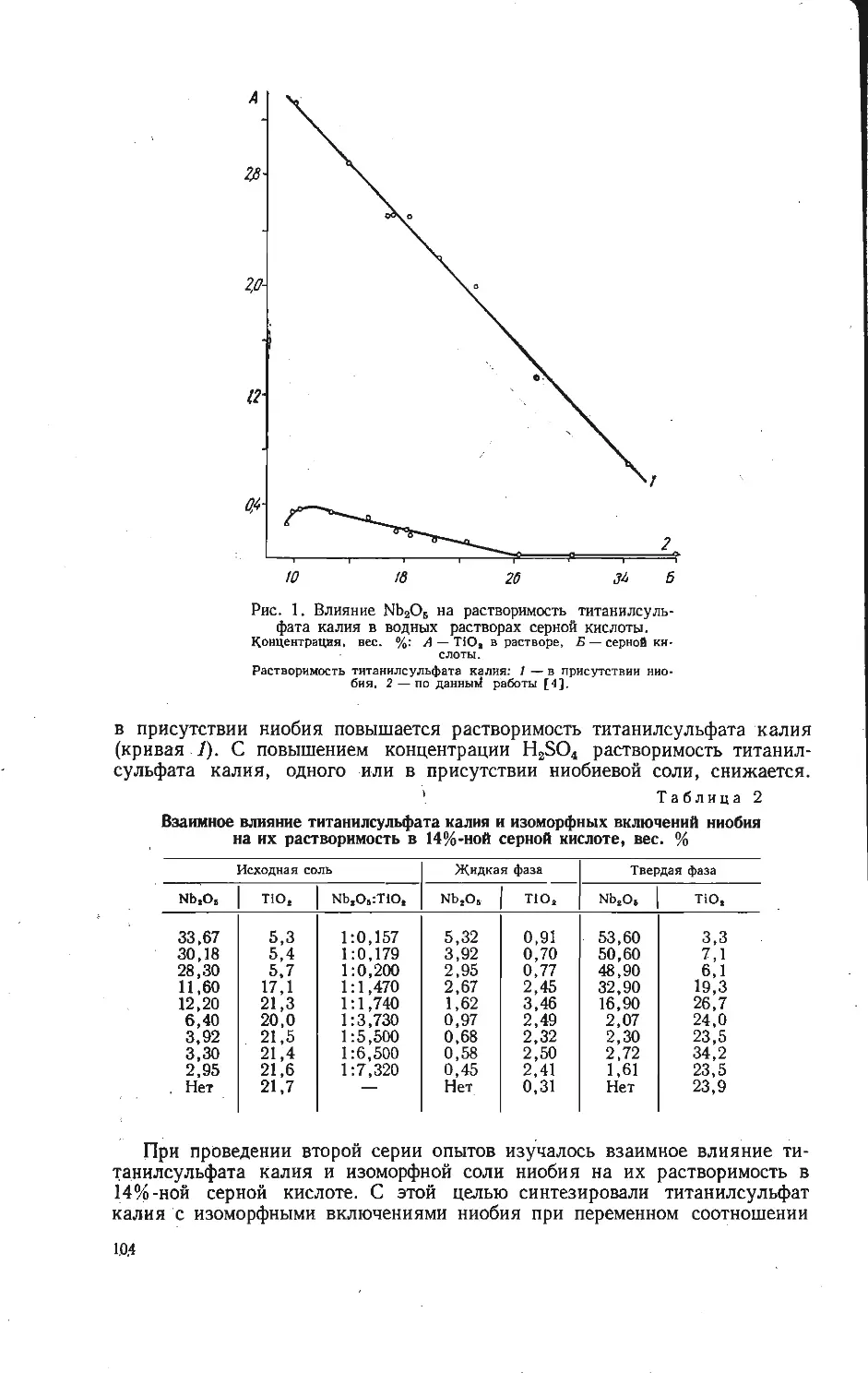
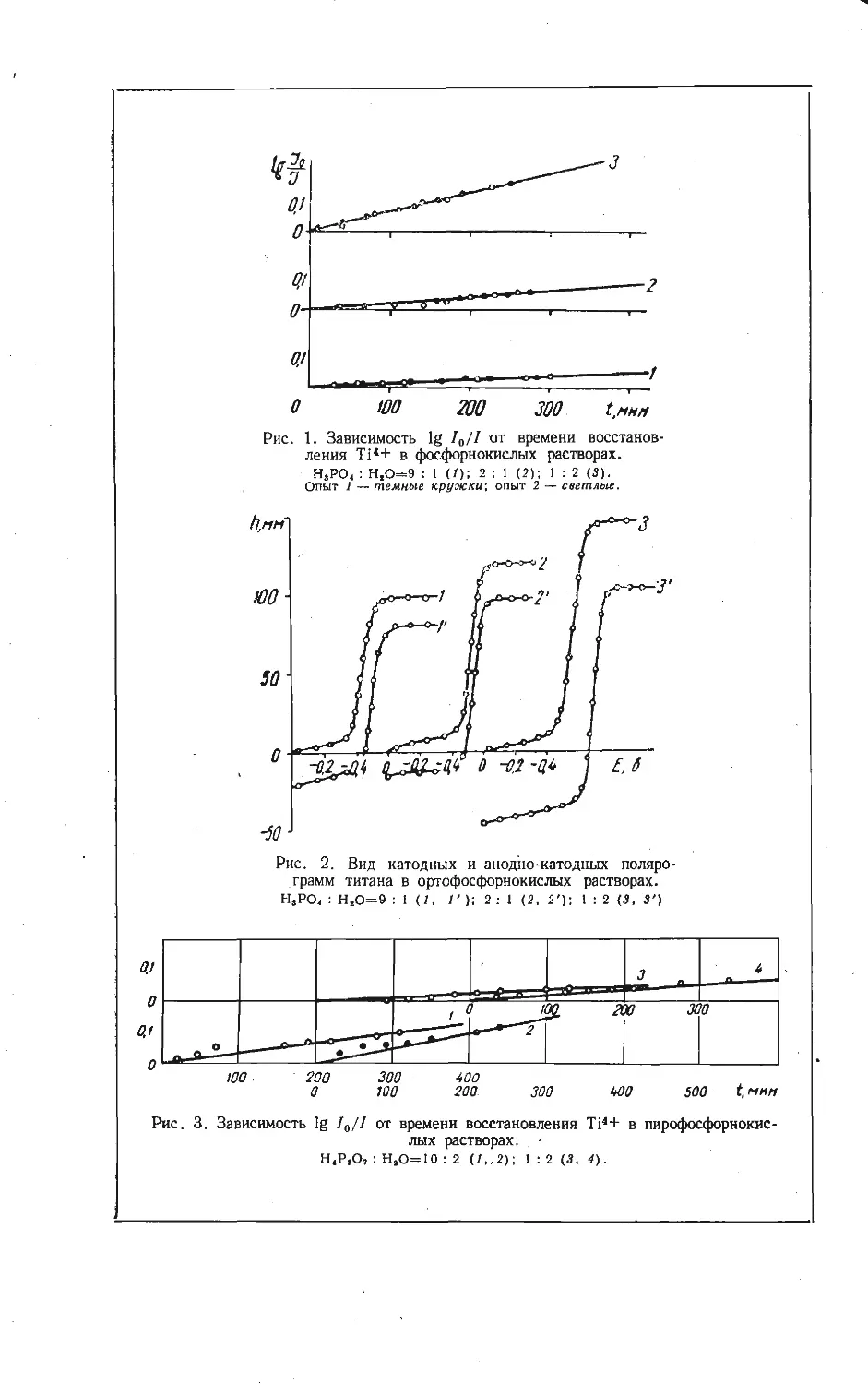
Зависимость изменения изотерм-
но-изобарных потенциалов ре-
акций (1) — (13).
Номера на кривых соответствуют но-
мерам реакций.
ской характеристики возможных реакций
между хлористым калием и пятиокисью
ванадия. Установлено [1], что в системе
NaCl — V2O5 осуществляются реакции с
образованием соединений, характерных для
системы Na2O — У2ОВ. Большое число фазо-
вых составляющих системы К2О — V2O5
определяет сложность химизма взаимодей-
ствия пятиокиси ванадия с хлористым
калием. Трудность термодинамического рас-
чета заключается в отсутствии каких-либо
экспериментальных величин. Применение же
сравнительных методов оценки термодинами-
ческих характеристик дает возможность про-
вести анализ в достаточно хорошем прибли-
жении.
В данной работе рассмотрены реакции
образования ванадатов калия из исходных
продуктов, а также реакции последователь-
ного превращения ванадатов калия при доба-
влении пятиокиси ванадия. Предполагается,
что первым продуктом взаимодействия хло-
ристого калия и пятиокиси ванадия было
соединение KV4Oio,s
2 КО + 4 V2O5+72 О2 - 2KV4O,0.5 +С12. (1)
При других соотношениях исходных ком-
понентов протекают следующие реакции:
2KO + 3V206+1/202=2KV308+02, (2)
6KCI+5V2O5+3/2O2=2K3VsOm+3C12, (3)
2KO+V206+1/202=2KV03+O2, (4)
4KC1+V2O5+O2=K4V2O7+2C12, (5)
6KC1 + V2O6-p/2 O2=2K3VO4+3C12. (6)
8
При добавлении к ортованадату калия пятиокиси ванадия образуется пиро-
ванадат калия, который, взаимодействуя с пятиокисью ванадия, претерпевает
ряд превращений по следующей схеме:
2K3vo4+VA=K4V2O7+2KVO3, (7)
K4V2O7+V2O5=4KVO3, (8)
3KVO3+V2O5=K3V5O14, (9)
K3V5O14+2V2O6 = 3KV3O8, (10)
2KV308+V205=2KV4Oio,5, (H)
KVO3+V2O6=KV3O8, (12)
2KVO3+3V2O5=2KV4O10,5. . (13)
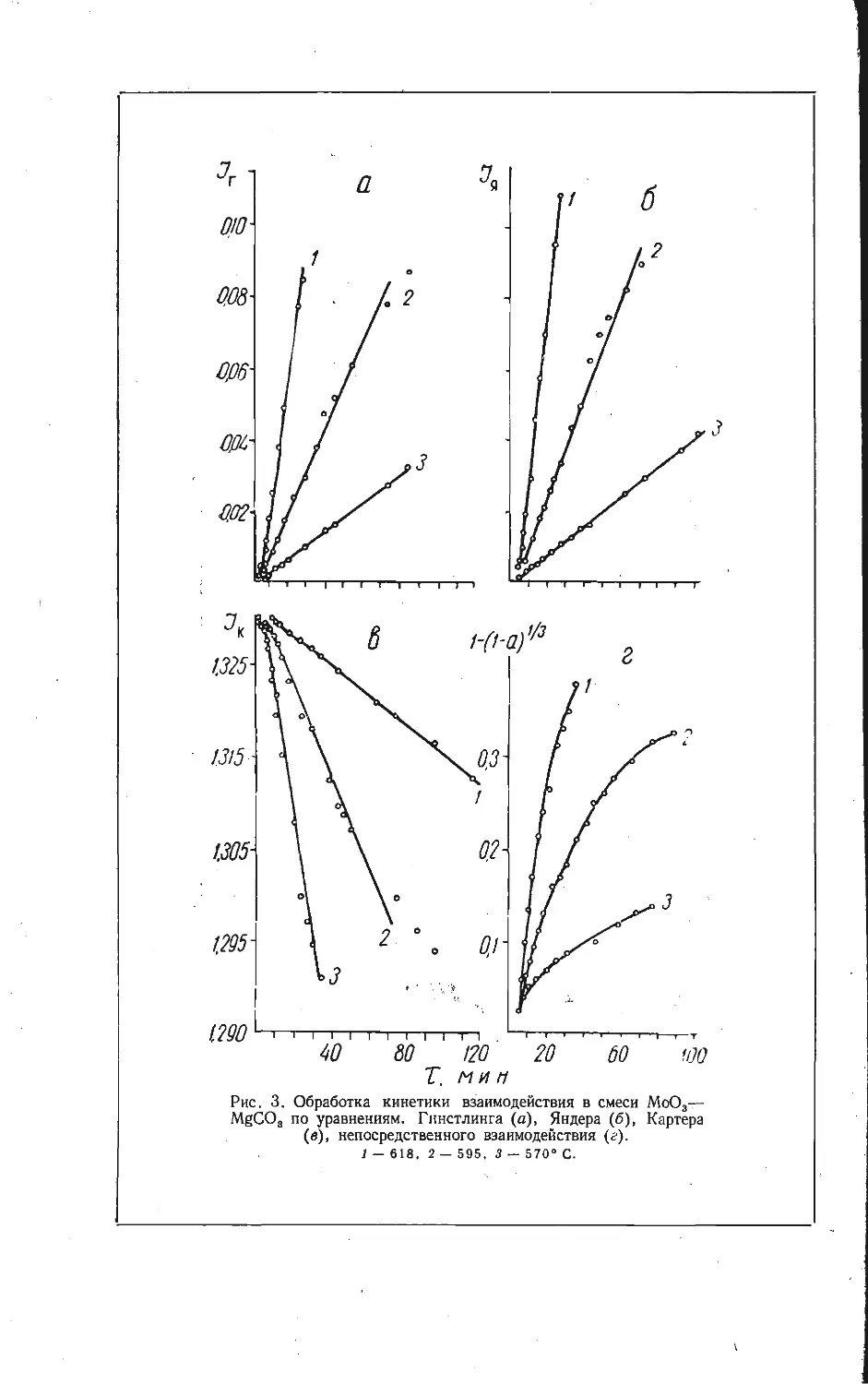
Температурная зависимость теплоемкостей ванадатов вычислена по методу
Н. А. Ландия [2] (табл. 1) с применением формулы
Ср=(С0+аат.ТЗ/2)л.
При расчетах использованы температуры плавления ванадатов калия, по-
лученные экспериментально при изучении системы К2О— V2O5 [3]. Стандарт-
ные энтропии ванадатов калия рассчитаны по методу аддитивности из окис-
лов, примененному Келли [4] для вычисления энтропий силикатов. В рабо-
те [5] сделана оценка значений энтальпий образования ванадатов калия
(мета- и орто-), с использованием величин энергии кристаллической решетки.
Энтальпии образования остальных ванадатов калия определены по методу
Г. М. Матвеева [6]. Температурная зависимость изменения изотермно-изобар-
ных потенциалов образования ванадатов калия рассчитана по уравнению
Г иббса — Гельмгольца.
Таблица 1
Термодинамические характеристики, использованные для расчета
Соединение или элемент Агрегатное состо- яние с у '-е 00 СП О 04 £ <] ' 1 0 кал 5298* град моль С р=а+Ь- 10->Т+ Р+с- 10“«Т‘ Температур- ный интервал, ° К Величина, харак- теризующая фазовое превра- щение
а ь с ^пер» °К Д/Уу . ккал^моль
о; к* к* V* KV4O10)5 KV„Oe K3V5O14 KVO3 K4v2o7 K3vo4 Газ Твердое Жидкое Твердое » » » » » » 1010 770 1340 275,8 660 413 49,02 18,2 7,0 74,4 58,6 113,5 27,4 78,3 50,9 6,15 5,24 7,70 5,57 74,70 44,87 87,72 14,91 61,54 39,93 3,10 5,55 0,97 24,80 54,39 92,64 33,34 23,53 8,12 —0,92 7,70 —17,67 —29,20 —14,80 —1,28 1,73 298—1500 273—336,5 336,5—373 273—2053 298—801 298—759 298—678 298—793 298-1183 298—1573 336,5 1043 2053 801 759 678 793 1183 1573 -0,571 19,0
* Взяты из работы [7].
В табл. 2 сведены уравнения зависимости изотермно-изобарных потенциа-
лов образования ванадатов калия из элементов от температуры. Для пяти-
окиси ванадия и хлорида калия аналогичные характеристики взяты из
работы [7]. По этим данным найдены значения изотермно-изобарных потен-
9
Т аблица 2
Значение коэффициентов в уравнениях температурных изменений
изотермно-изобарных потенциалов образования некоторых соединений
Соединение Температурный интервал, ° К Д2°=Д-|-В7’In 7’4-С7Ч-Д7'!4-Е7Ч, кал/моль
—А В с D- Ю-« D-1 о—•
v2o5 298—943 381960 —17,84 228,50 5,20
КС1 298—1043 104900 .—. 24,0 — —
KV4O10>5 298—336,5 1014507 —14,89 326,06 0,45 —2,оа
KV4O101S 336,5—801 1015165 —12,43 312,8 —2,32 —2,09
KV8o8 298—336,5 778865 1,68 171,87 —16,73 2,33
KV„Os 386,5—759 778913 4,14 158,61 —19,53 2,33
KaVaO14 298—336,5 1340325 —1,11 331,48 —24,71 13,34
KaVaO14 336,5—678 1340480 6,27 291,90 —33,03 13,34
KVO, 298—336,5 275141 5,12 40,13 —11,09 2,24
KVOa 336,5—793 275189 7,58 26,87 —13,86 2,24
K4V2O, 298—336,5 661877 —7,95 217,73 5,73 —0,32
K4V2O, 336,5—1183 662118 1,89 159,95 —11,42 —0,32
Kavo4 298—336,5 413731 —6,34 134,27 13,4 —0,6
KaVO4 336,5—1573 413886 1,04 94,69 5,08 —0,6
циалов реакций (1) — (13) образования ванадатов калия. Как видно и»
рисунка, в области низких температур возможно протекание реакций (1) — (3).
Экстраполирование кривой 4 до оси абсцисс показывает, что реакция обра-
зования метаванадата калия протекает при температурах выше 500° С. Это
подтверждается экспериментально при сплавлении смеси хлористого калия и
пятиокиси ванадия с соотношением 1:1 при 700° С. Также возможны реакции
(7) — (13), причем термодинамически самая вероятная из них реакция (13) —
образование соединения КУ40ю,5- Из этого можно заключить, что это со-
единение в области низких температур наиболее устойчивый продукт, оно
образуется первым при любом стехиометрическом соотношении.
Полученные выводы находятся в хорошем согласии с экспериментальными
результатами изучения кинетики взаимодействия в системе КС1 — V2O5 [8].
ЛИТЕРАТУРА
1. бУв. Слободин. Физико-химические исследования соединений редких тугоплавких
элементов (Ti, V, Nb, Та). Труды Ин-та хим. УФАН СССР, 1966, вып. 9, стр. 51.
2. Н. А. Ландия. Ж- физ. хим., 1951, 25, 8, 927.
3. А. А. Фотиев, М. П. Глазырин, С. И. Алямовский. Ж. неорг. хим., 1967,
12 5 1325.
4. К. К.’Kelley. Bull. U. S. Bur. Min., 1932, N 350.
5. А. А. Фотиев, Б. Г. Головкин. Ст. в наст, сб., стр. 3.
6. Г. М. Матвеев. Изв. высш, учебн. завед., Химия и хим. технология, 1958, № 2,135-
7. О. Кубашевский, Э. Эванс. Термохимия в металлургии. М., ИЛ, 1954.
8. А. А. Фотиев, Л. Л. Сурат. Ст. в наст, сб., стр. И.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
А. А. ФОТИЕВ, Л. Л. СУРАТ
ИССЛЕДОВАНИЕ ВЗАИМОДЕЙСТВИЯ В СИСТЕМЕ
ПЯТИОКИСЬ ВАНАДИЯ — ХЛОРИСТЫЙ КАЛИЙ
Для извлечения ванадия из ванадийсодержащих полупродуктов применяют
окислительный обжиг с добавками солей щелочных металлов. Наряду с суль-
фатом, карбонатом и хлоридом натрия, в качестве добавок используют хлорид
калия, содержащийся вместе с хлоридом натрия в сильвините. Хлорид калия,
-как и хлорид натрия [1], взаимодействует с пятиокисью ванадия с образо-
ванием ряда окисных соединений, в виде которых ванадий отделяется от
-примесей при дальнейшей переработке. Несмотря на практическую важность
процесса взаимодействия пятиокиси ванадия с хлористым калием, в литера-
туре отсутствуют какие-либо данные относительно фазового состава и усло-
вий образования соединений в системе V2Og— КС1. »
Цель настоящей работы — изучение кинетики взаимодействия хлористого
калия с пятиокисью ванадия при молярном соотношении KCl:V2Og=l:3 и
2:1 и построение диаграммы плавкости неравновесной системы V2Og— КС1
в атмосфере воздуха. В качестве исходных компонентов использованы пяти-
окись ванадия квалификации «о. с. ч.» и хлористый калий квалификации
<х. ч.>, предварительно прокаленные при 400° С. Исследование кинетики
взаимодействия проводили в вертикальной печи в атмосфере воздуха при
различных температурах. Температуру в печи контролировали платина-плати-
нородиевой термопарой, вставленной в кварцевую трубку и подключенной
к потенциометру типа ПП. Платиновый тигелек с навеской при помощи
тонкой платиновой проволоки подвешивали на кварцевой пружине. Изменение
массы навески контролировали по отсчетному микроскопу МИР-1. Фазовый
•состав конечных продуктов определяли оптическим и рентгеновским методами.
Согласно данным, имеющимся в литературе [2, 3], пятиокись ванадия в об-
ласти температур до 700° С существенно не изменяет массы в результате
диссоциации или испарения.
Изучению поведения хлористого калия при нагревании посвящена работа
В. И. Спицына, В. Н. Шостака и М. А. Меерова [4], которые установили,
что при 700° С навеска хлористого калия (0,25 а) на воздухе в течение 1 ч
изменила массу на 0,49%. Действительно, при 700° С происходит заметная
сублимация хлористого калия. Прямолинейная зависимость степени сублима-
ции и испарения от продолжительности эксперимента при различных темпе-
ратурах указывает на протекание реакции нулевого порядка. Отсутствие
процесса образования окиси калия и хлора подтверждено химическим анали-
зом остатка на щелочь и термодинамическим расчетом. Вычисленные энергии
активации сублимации и испарения, соответственно, равны 49,9 и
39,3 ккал/моль. Эти данные в пределах ошибки опыта совпадают с соответ-
ствующими теплотами процессов Д//°убл=49,7 ккал/моль и Д/7нсп=
11
=39,0 ккал/моль [5, 6]. Как уже отмечалось ранее [7], близость этих
величин можно объяснить тем, что в эндотермических процессах, к которым
относятся испарение и сублимация хлористого калия, повышение энергии при
разупорядочении системы обусловлено ослаблением и разрывом определенных
межмолекулярных связей, а не образованием промежуточных активированных
комплексов.
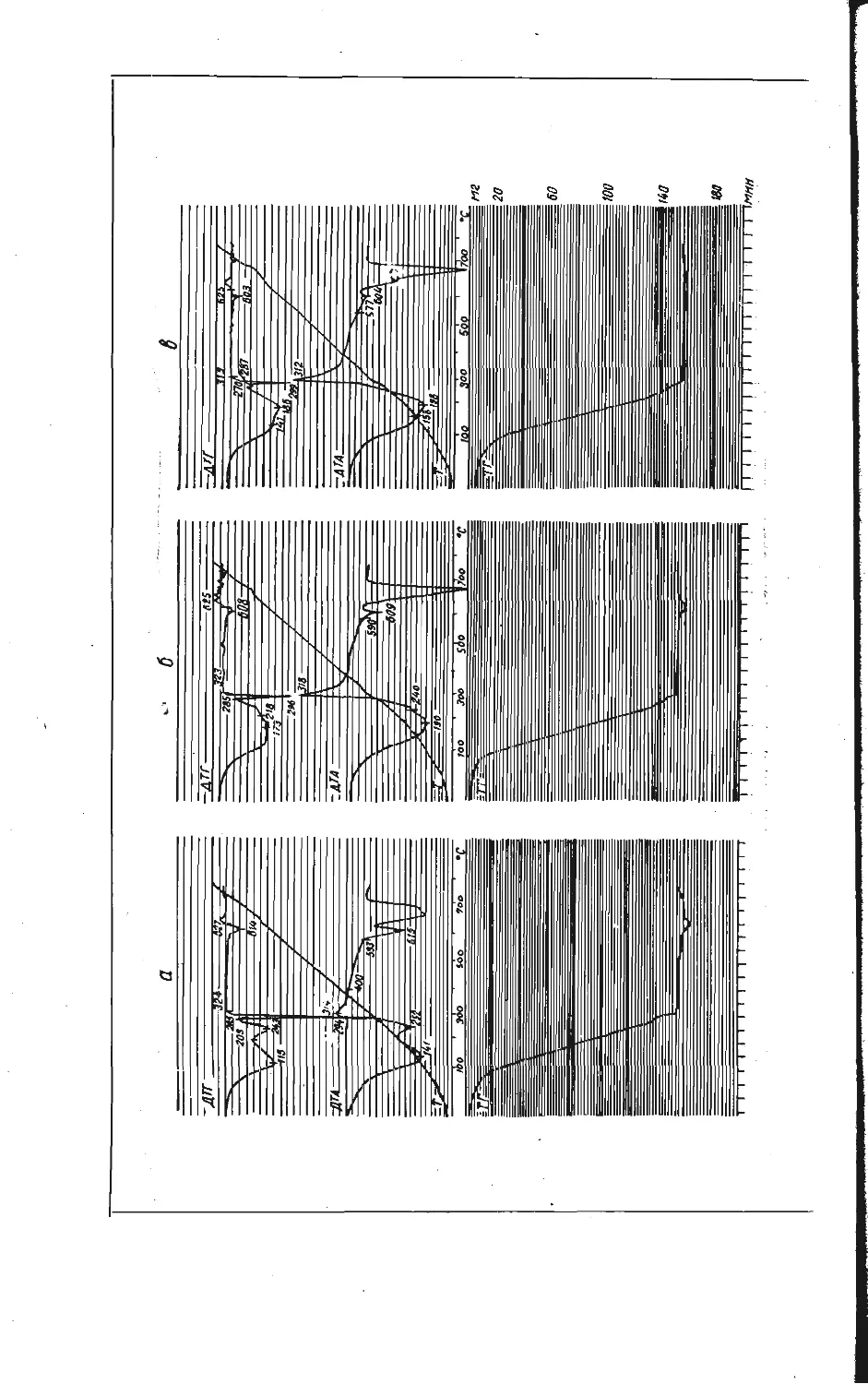
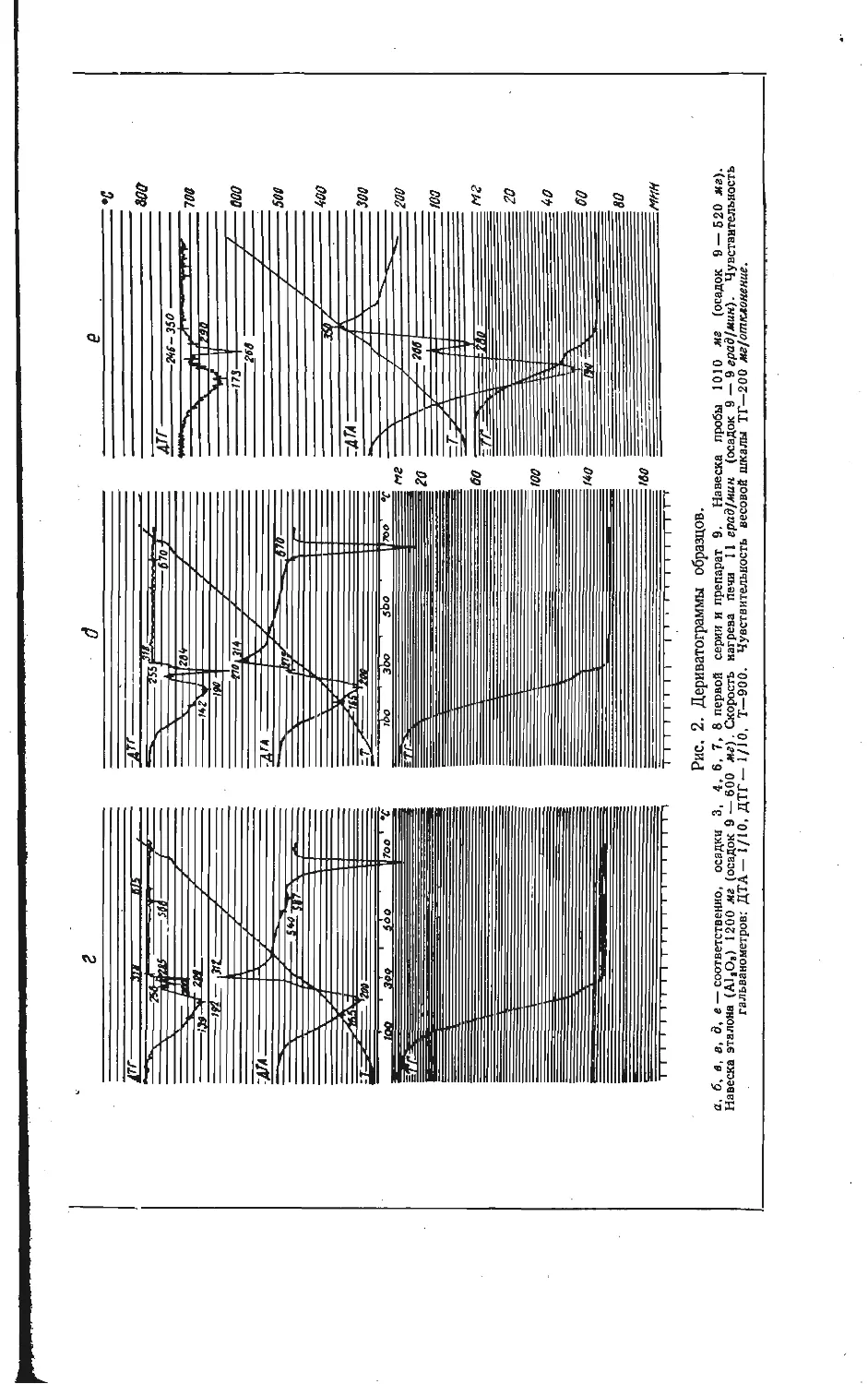
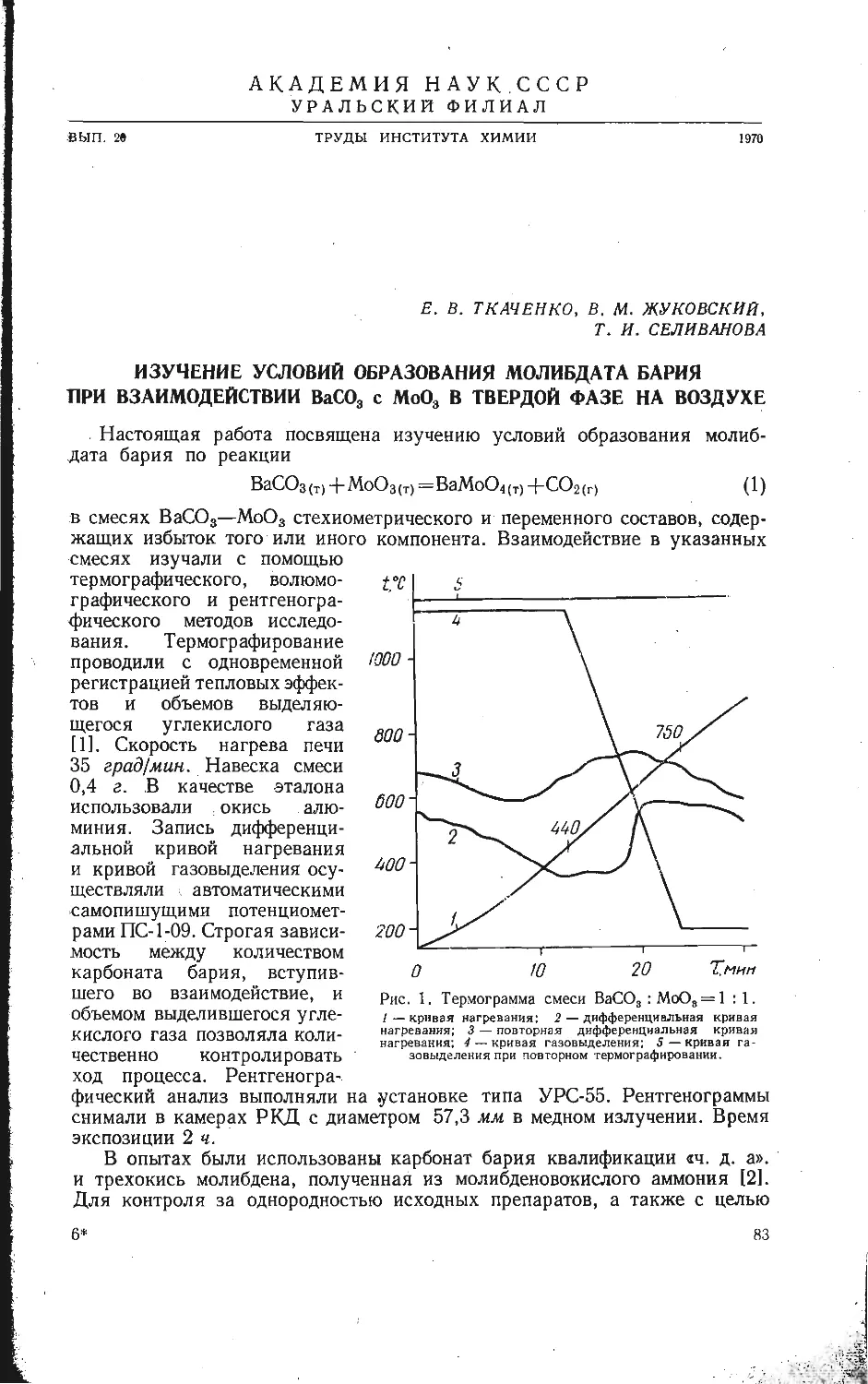
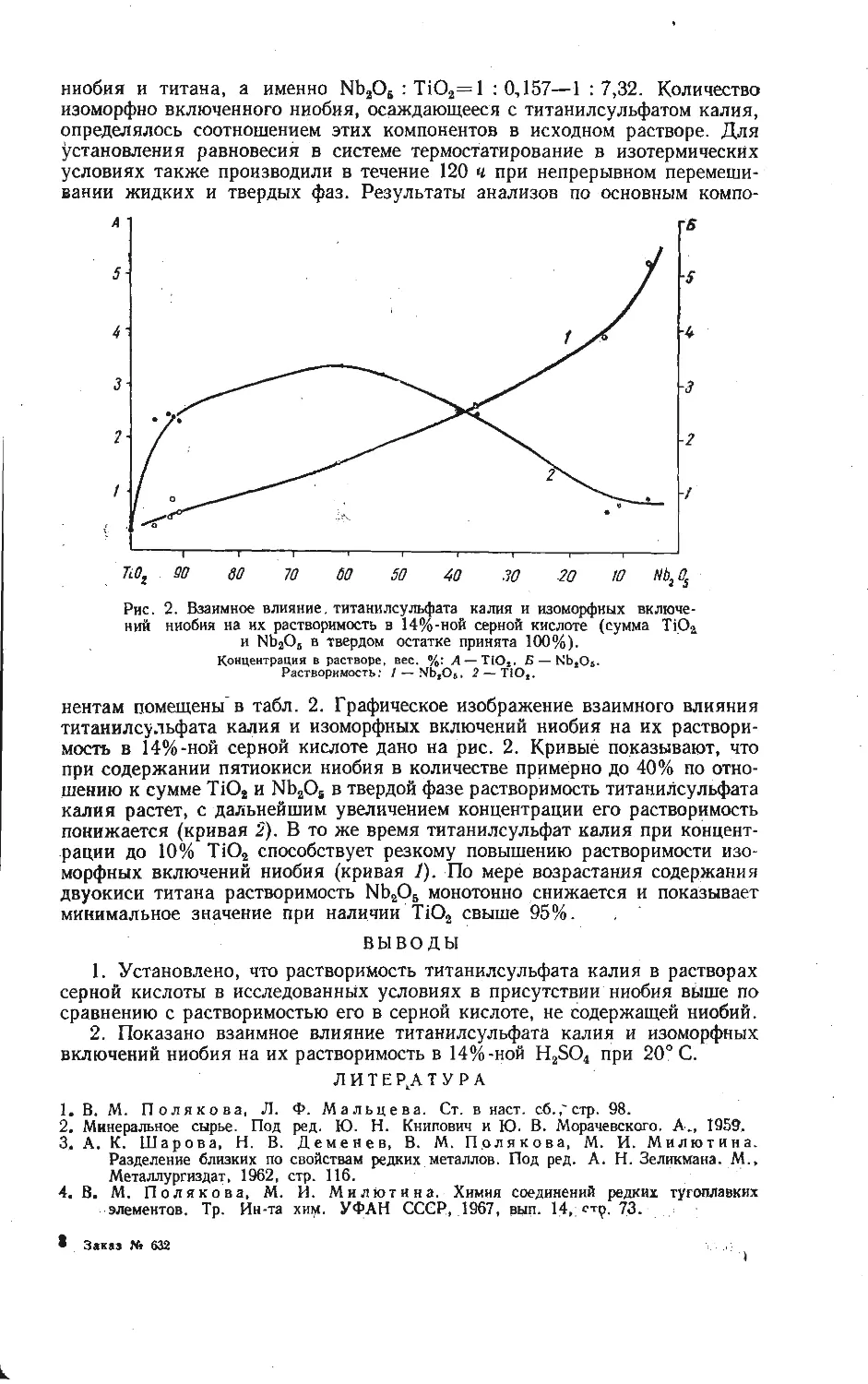
Зависимость образования калий-ванадиевой бронзы KV6O15 от температуры
изучена на смеси, составленной из
Рис. 1. Зависимость образования калий-
ванадиевой бронзы KV6OJ5 в расчете на
V2O6 от температуры при скорости на-
* гревания смеси 10 град/мин.
Из рис. 1 видно, что образование
порошкообразных компонентов КС1 и
V2OB, взятых в молярном отношении
1:3. Навески смеси (около 1 а)
нагревали до различных температур
со скоростью 10 град/мин, после
чего определяли убыль массы в
результате реакции. Для определе-
ния количества образовавшейся
бронзы KVeO1B полученные составы
обрабатывали 5 % -ным раствором
аммиака, чтобы перевести в раст-
вор непрореагировавшие КС1 и V2O5,
затем промывали 5 %-ным раствором
соляной кислоты, в которой про-
исходил гидролиз соединений
KV4Oio,4 и K2VbOi3,3, и снова
раствором аммиака до полного раст-
ворения примесных фаз. Таким об-
разом, в нерастворимом осадке по-
лучали чистую фазу KV6O1B и весо-
вым методом определяли ее массу.
бронзы KVeO15 в выбранном режиме
нагревания фиксируется в заметных количествах при температурах выше
470° С. В области более низких температур в результате взаимодействия
хлористого калия и пятиокиси ванадия найдена в основном фаза КУ40ю,4,
образующаяся по реакции
2KC1+4V2O5+| O2=2KV4O10.4+Cl2.
(1)
Таким образом, бронза KVeO1B является продуктом последовательного взаи-
модействия по реакции
KV4O! о, 4+V2O8=KVeO15+4 О2- (2)
О
Максимальное количество бронзы KVeO1B обнаружено при температуре около
530°С, соответствующей перитектическому плавлению KV4Oio,4 [8]. После
появления жидкой фазы количество бронзы уменьшается за счет ее окисле-
ния, т. е. реакция (2) сдвигается справа налево.
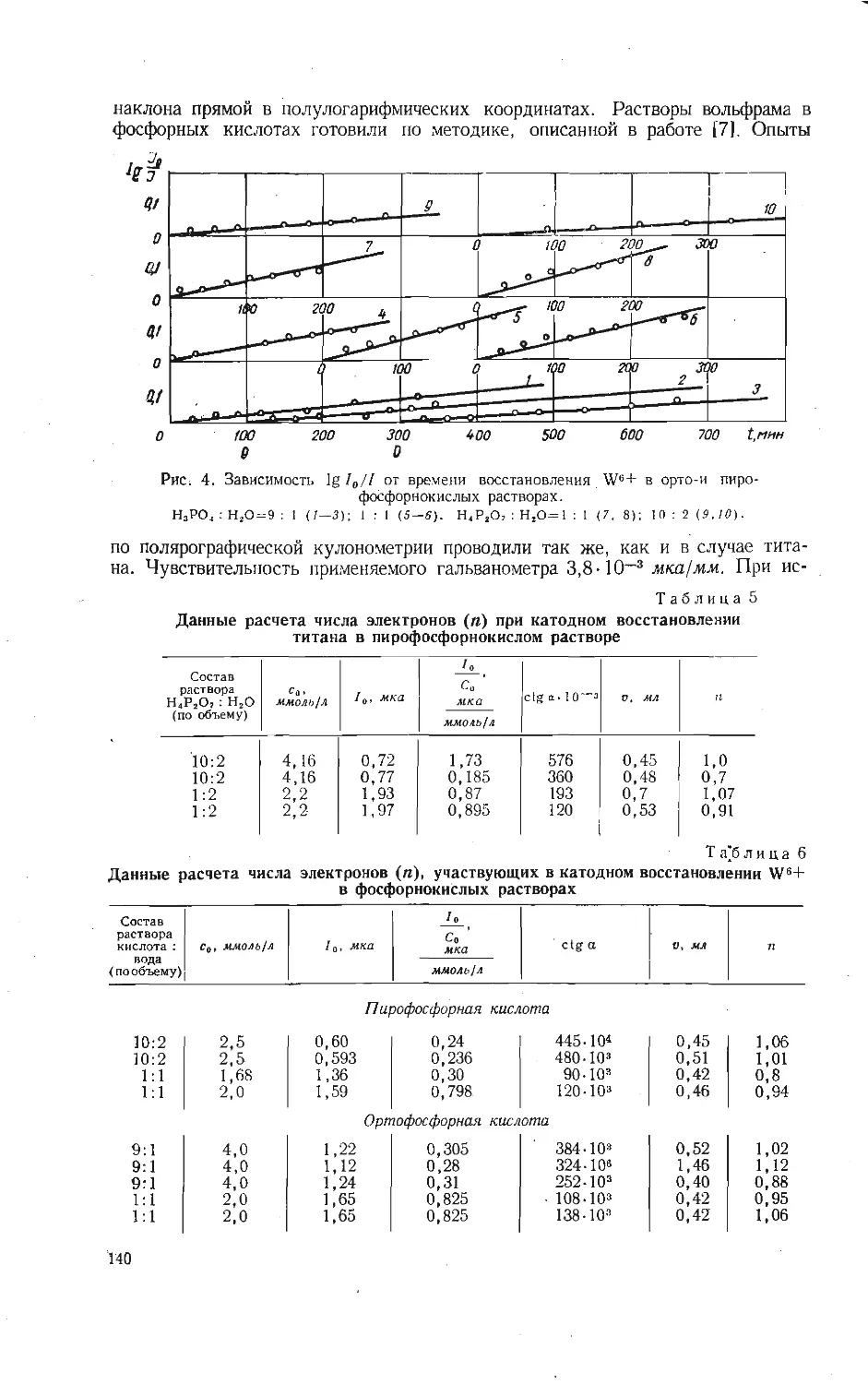
Кинетика взаимодействия КС1 с V2OB в молярном отношении 1:3 и 2:1
представлена на рис. 2. Убыль массы смеси, взятой в соотношении
KC1:V2OB=1:3, происходит за счет выделения хлора по реакции (1). Замет-
ная скорость последней наблюдается при температуре 355° С; при 450° С
процесс протекает полностью в течение 1 ч. Константы скорости реакции
при различных температурах вычислены по уравнению [9]:
/==1 — ЗД(Х)_(1 _Х)2/3=^Т.
На основании этих данных построена зависимость lg k-----— и найдена ка-
12
жущаяся энергия активации образования KV4O10,4 по реакции (1), равная
34,0 ккал/моль, что значительно меньше энергии активации сублимации
хлористого калия. Следовательно, механизм массопередачи вещества в газовой
фазе при взаимодействии КС1(г) и У2О6(т), по-видимому, маловероятен.
На рис. 26 показана кинетика взаимодействия КС1 с V2O5 в молярном
отношении 2:1. Штриховые линии при 25; 33,3; 42,3 и 60 мол. % КО
указывают на области теоретического образования соответственных фаз:
KV4Oio,4, KV3O8, K2V50i3,3 и K3V5O14. На основании рентгенофазового
анализа продуктов взаимодействия установлено, что вначале образуется фаза
Рис. 2. Кинетика взаимодействия смесей КС1 и V2OS, взятых в соотношениях 1 : 3(a) и
2 :1 (б).
а) : I — 355, 2 — 370, 3 — 400, 4 — 410, 5 — 4i0, 6 — 450° С; б) : 1 — 360, 2 — 420, 3 — 500,
4 — 600, 5 — 700, 5 — 750° С.
KV4Oio,4 по реакции (1). При повышении температуры образование этого
соединения идет с большой скоростью, после чего последовательно возника-
ют соединения KV3O8 и K2V60i3,3. При температуре выше 490°С фаза
KV3O8 не обнаруживается вследствие ее перитектического распада. Дальней-
шее превращение приводит к образованию фаз K3V5O14 и KVO3 согласно
следующему ряду превращений:
V2O6->KV4O10,4-4KV8O8-^K2V6O13.3-4K3V5O14^KVO3.
Как было отмечено выше, ванадиевая бронза KV6O15 найдена при 470—530° С.
Начиная с температуры 700° С происходит сублимация хлористого калия
в заметных количествах, что обнаруживается по конденсации паров на хо-
лодных стенках кварцевой трубки. В результате этого параллельно протека-
ющего процесса нарушается стехиометрический состав смеси и после окон-
чания взаимодействия в продукте обнаруживается смесь K3V5O14 и KVO(.
Построенные по кинетическим кривым зависимости скорости взаимодей-
ствия пятиокиси ванадия с различным количеством хлористого калия позво-
ляют сделать вывод об уменьшении скорости образования ванадиевых соеди-
нений с увеличением концентрации в них щелочного элемента (рис. 3).
По-видимому, наиболее вероятный механизм взаимодействия хлористого калия
с пятиокисью ванадия — диффузия иона калия в решетку пятиокиси ванадия.
Состояние неравновесной системы V2O5 — КС1 в условиях атмосферы
воздуха иллюстрирует диаграмма плавкости (рис. 4), построенная по термо-
13
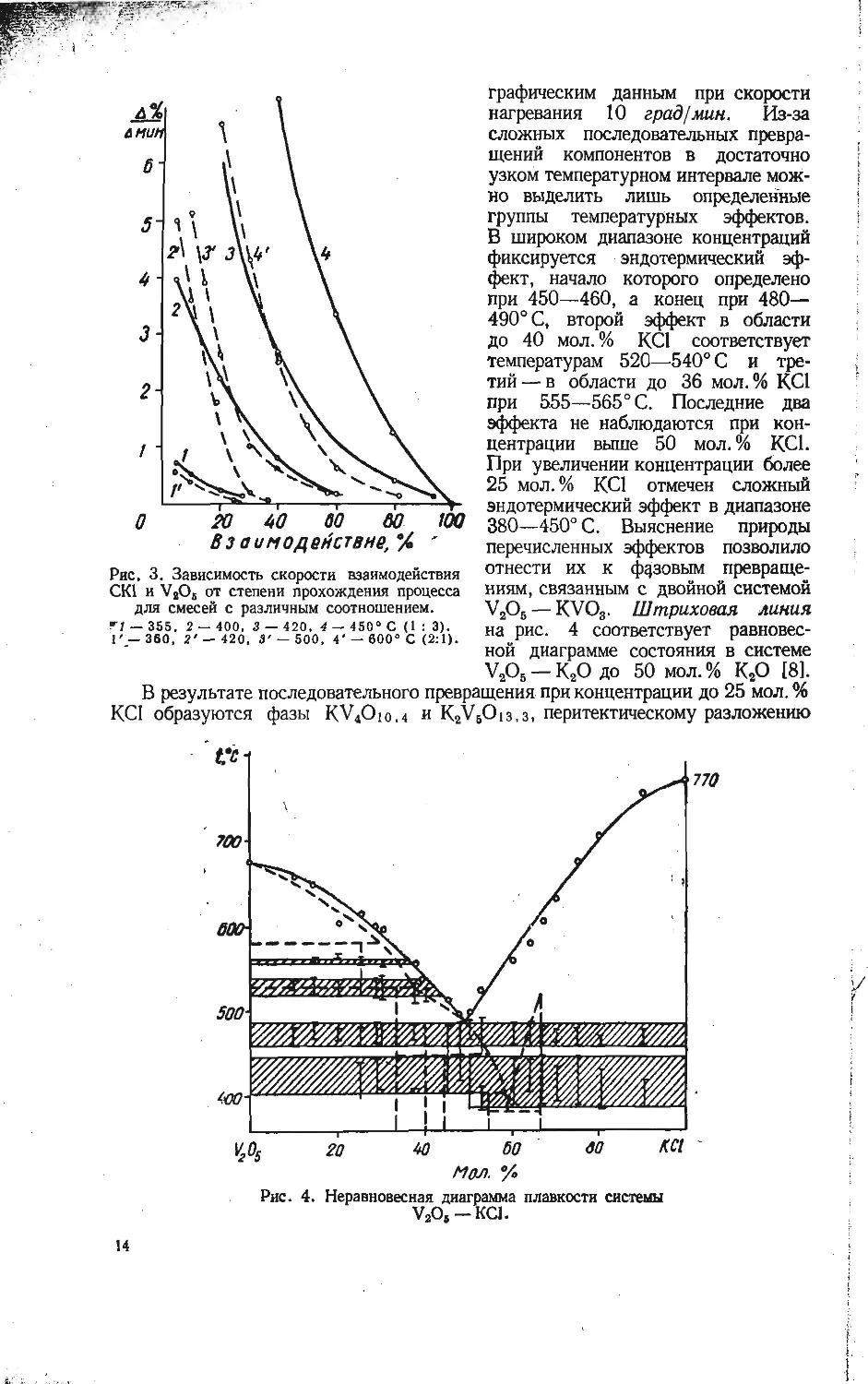
Рис. 3. Зависимость скорости взаимодействия
СК1 и V2O5 от степени прохождения процесса
для смесей с различным соотношением.
fl — 355, 2 — 400. 3 — 420, 4 — 450° С (1 : 3).
1'— 360, 2' — 420, 3' — 500, 4' — 600° С (2:1).
графическим данным при скорости
нагревания 10 град/мин. Из-за
сложных последовательных превра-
щений компонентов в достаточно
узком температурном интервале мож-
но выделить лишь определенные
группы температурных эффектов.
В широком диапазоне концентраций
фиксируется эндотермический эф-
фект, начало которого определено
при 450—460, а конец при 480—
490° С, второй эффект в области
до 40 мол. % КС1 соответствует
температурам 520—540° С и тре-
тий— в области до 36 мол. % КС1
при 555—565° С. Последние два
эффекта не наблюдаются при кон-
центрации выше 50 мол. % КС1.
При увеличении концентрации более
25 мол. % КС1 отмечен сложный
эндотермический эффект в диапазоне
380—450° С. Выяснение природы
перечисленных эффектов позволило
отнести их к фазовым превраще-
ниям, связанным с двойной системой
V2O6 — KVO3. Штриховая линия
на рис. 4 соответствует равновес-
ной диаграмме состояния в системе
V2O5 — К2О до 50 мол. % К2О [8].
В результате последовательного превращения при концентрации до 25 мол. %
КС1 образуются
фазы КУ40ю,4 и K2V5O 1з.з, перитектическому разложению
которых и отвечают первые два эффекта. Третий эффект при 560° С, по-ви-
димому, соответствует полному окислительному разложению бронзы KVeO15,
возникающей при высокотемпературном взаимодействии по реакции (2).
В области высоких концентраций хлористого калия (бойее 50 мол. %) первый
эндотермический эффект соответствует появлению легкоплавкой эвтектики
между K8VSO14 и KVO3 около 380° С и перитектическому разложению
KgVsO14 при 404°С (более 25 мол.%). Второй эффект аналогичен первому,
описанному для низкой концентрации хлористого. калия. Самая низкая тем-
пература ликвидуса равна 500° С при 50 мол. % КС1.
ВЫВОДЫ
1. Исследована кинетика сублимации и испарения хлористого калия.
Процессы описываются уравнением нулевого порядка и имеют энергии акти-
вации, соответственно, 49,9 и 39,3 ккал/моль.
2. Показано, что образование калий-ванадиевой бронзы KV8O15 происходит
при взаимодействии соединений КУ40ю,4 и V2O5 в области температур
470—550° С.
3. На основании изучения кинетики взаимодействия смеси КС1 и V2OB
установлено последовательное образование соединений
V2O6 KV4O, о, 4 -> KV3O8 -4 K2V6O, з. з -+ K8V5O14 -> KVO3.
4. Термографическим методом построена диаграмма плавкости неравно-
весной системы V2O6 — КС1.
ЛИТЕРАТУРА
1. Б. В. Слободин, А. А. Фотиев. Изв. АН СССР, Неорганические материалы
1967, 3, 1, 146.
2. Е. Р. Milan. J. Phys. Chem., 1929, 33, 498.
3. А. Д. Поляков. Ж- физ. хим., 1946, 20, 1021.
4. В. Н. Спицын, В. Н. Шостак, М. А. Мееров. Ж. общ. хим., 1952, 22, 5, 758.
” 5. К. К. Kelly. Bull. U. S. Bur. Min., 1935, № 3835.
6. К. Niwa. J. Fac. Sci., 1938, 2, 201; 1940, 3, 17 и 75.
7. А. А. Фотиев, Б. В. Слободин. Ж- физ. хим., 1965, 39, 12, 3099.
8. А. А. Фотиев, М. П. Глазырин, С. И. Алямовский. Ж. неорг. хим., 1967,
12, 5, 1325.
9. А. М. Ги нет л инг, Б. И. Броунштейн. Ж. прикл. хим., 1950, 23, 12.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ТРУДЫ ИНСТИТУТА ХИМИИ 1970
ВЫП. 20
А. А. ФОТИЕВ, Б. Г. ГОЛОВКИН
ОПРЕДЕЛЕНИЕ СООТНОШЕНИЯ СКОРОСТЕЙ ИСПАРЕНИЯ,
СУБЛИМАЦИИ ХЛОРИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
И ВЗАИМОДЕЙСТВИЯ ИХ С ПЯТИОКИСЬЮ ВАНАДИЯ
Основа существующего технологического метода извлечения ванадия в на-
шей стране — окислительный обжиг ванадийсодержащих полупродуктов с
добавками солей щелочных металлов, в частности галит-сильвиновой породы
(К, Na) С12. Несмотря на значительное количество работ по технологии из-
влечения ванадия, механизм высокотемпературного взаимодействия пятиокись
ванадия — хлорид щелочного металла выяснен недостаточно. При образова-
нии ванадиевых соединений в результате реакции пятиокиси ванадия с хло-
ридами щелочных металлов в зависимости от условий могут быть следую-
щие взаимодействия: твердое — газ, твердое — жидкое, твердое — твердое.
Механизм твердое — твердое может быть при значительной скорости диффу-
зии атомов. Лимитирующей стадией в этом случае была бы скорость диффу-
зии ионов в кристаллической решетке. Механизм твердое — жидкое при взаи-
модействии пятиокиси ванадия с хлоридами щелочных металлов наблюдается
либо в условиях образования легкоплавковой эвтектической жидкой фазы,
либо при более высоких температурах в результате плавления одного из ре-
агентов. Однако в рассматриваемом случае самые низкие температуры эвтек-
тик в системах V2O5—MCI лежат выше температур фиксируемого образова-
ния ванадиевых соединений. В механизме твердое — газ в качестве основной
лимитирующей стадии предполагается процесс сублимации хлоридов щелоч-
ных металлов. Рассмотрению этого возможного механизма и посвящена дан-
ная статья.
В табл. 1. приводятся литературные сведения по теплотам испарения и
сублимации хлоридов щелочных металлов [1 —18].
Давление насыщенного пара определялось в большинстве работ эффузи-
онным методом Кнудсена [19]. По данным В. И. Спицына и В. И. Шоста-
ка [20], имеются следующие соотношения количеств испарившихся хлоридов
за 1 ч в потоке воздуха при 800° С, г\ CsCl: RbCl: LiCl: КС1: NaCl =
=25,5:12,0:10,9:8,2:3,2; в потоке паров воды, соответственно:
34,5:22,3:20,2:10,0:4,4 и в потоке аммиака: 33,0:19,3:18,2:7,7:2,7. Тредвелл
и Вернер [21] исследовали скорости сублимации хлоридов калия, рубидия и
цезия при 440° С и нашли, что эти скорости относятся друг к другу, как
CsCl: RbCl:KCl = 84:2,5:1. В этой же работе показано, что изменение массы
хлоридов прямо пропорционально времени сублимации, т. е. скорость субли-
мации относится к реакции нулевого порядка. Аналогичный вывод следует
и из исследований сублимации хлористого лития [18], испарения и сублима-
ции хлористого натрия [22]. Как показали Фишбек и Шнайдт [23], для про-
цессов испарения и диссоциации в пределах ошибки опыта наблюдается сов-
падение энергии активации с теплотами превращения. Позднее Кнаке, Шмоль-
10
Таблица 1
Теплоты сублимации и испарения хлоридов щелочных металлов
по данным различных авторов, ккал/моль
Теплота По данным Теплота По
сублимации | испарения сублимации | испарения данным
Хлорид лития Хлорид калия
46,7 46,8 47,0 37,2 36,87 32,89 36,0 39,6 [2 4 5 6 7 8] ] 50,6 51,15 48,8 51,72 49,9 49,7 51,6 39,0 0] 3 [Г 6 [3 7 7
Хлорид натрия 50,2 51,72 43,3 [15] 18
51,8 -—• 1 — 33,6 [6
53,0 —- [9 -— 22,7 [11]
50,5 -— [ 0 -— 40,5 2
52,2 —- 2 — 43,1 [5
52,5 40,7 7 — 41,7 1 3
— 39,7 [6 —. 42,2 8
— 36,0 [U
-— 44,3 [ 2
—. 46,7 5
— 43,1 [13 Хлорид цезия
Хлорид рубидия 44,1 40,3 [15]
49,7 —- [14 — 39,4 [61
47,5 —- 1 — 37,4 4
49,3 — [Ю — 39,8 5
46,4 41,6 [15 — 38,3 8
45,5 — [2 49,8 —. [ 4
50,3 39,6 [7, 0] 48,6 38,2 7
—- 36,3 [6
— 37,8 4
— 46,6 5
ке и Странский [24], изучая кинетику сублимации хлористого и йодистого
калия, нашли их энергии активации (46,3—48,459 и 42,732—44,106 ккал/моль,
соответственно). Эти значения также хорошо совпадают с теплотами процес-
сов [7]. А. А. Фотиев и Б. В. Слободин [22, 25] на основании изучения
кинетики определили энергию активации испарения сульфата натрия
77 ккал/моль (теплота испарения 79) и энергии активации испарения и суб-
лимации хлористого натрия 39,2 и 53 ккал/моль, соответственно (см. табл. 1).
Ан. Н. Несмеянов и Л. А. Сазонов [18] интегральным вариантом метода
Кнудсена [19] определили скорость сублимации хлористого лития при
808—889° К. Рассчитанная нами по этим данным энергия активации субли-
мации равна 45,9 ккал/моль. В настоящей работе в единых методических
условиях изучена кинетика испарения и сублимации хлоридов щелочных ме-
таллов и установлено соотношение скоростей этих процессов в ряду от лития
до цезия со скоростями взаимодействия хлоридов щелочных металлов с пяти-
окисью ванадия в твердой фазе.
Изучение кинетики испарения и сублимации хлоридов щелочных металлов
проведено следующим образом. В электрическую печь, установленную в вер-
тикальном положении, помещали кварцевую трубку, через нижний конец
которой была введена платино-платинородиевая термопара. Над спаем термо-
пары помещали тигилек с навеской хлорида, крепившийся с помощью пла-
тиновой нити к контуру, фиксирующему изменение массы. Для того чтобы
испарившаяся соль не конденсировалась на платиновой нити, через систему
2
Заказ № 632
17
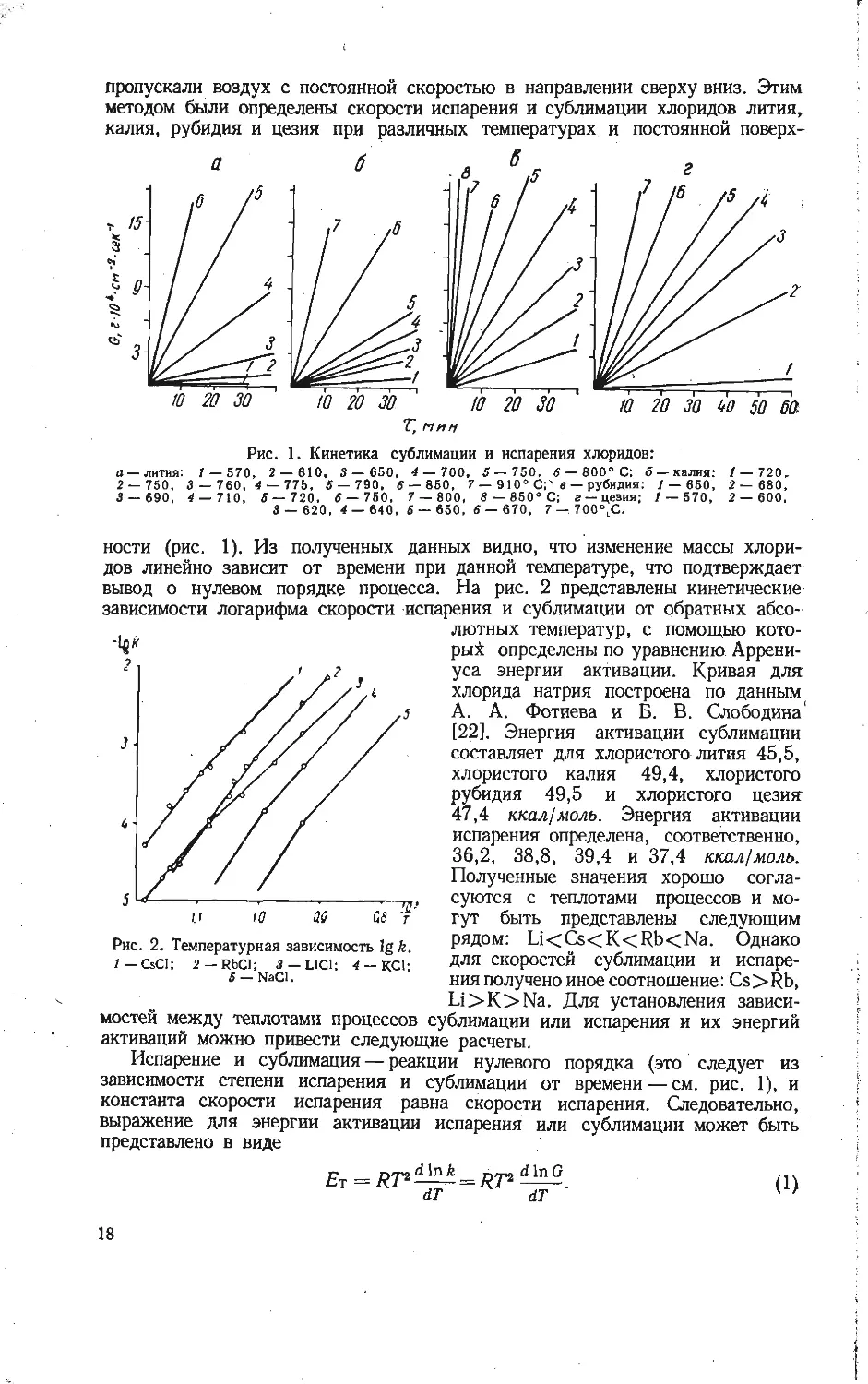
пропускали воздух с постоянной скоростью в направлении сверху вниз. Этим
методом были определены скорости испарения и сублимации хлоридов лития,
калия, рубидия и цезия при различных температурах и постоянной поверх-
V, мин
Рис. 1. Кинетика сублимации и испарения хлоридов:
а —лития: / — 570. 2 — 610, 3— 650, 4 — 700, 5— 750, в — 800° С: б —калия: / — 720.
2 — 750, 3 — 760. 4 — 775, 5 — 790, 6 — 850, 7 — 9 10° С; в — рубидия: / — 650, 2 — 680,
у — 690, 4 — 710, 5 — 720. 6 — 750, 7 — 800, 8 — 850° С; г —цезия; / — 570, 2 — 600,
3 — 620, 4 — 640, 5 — 650, 6 — 670, 7 — 700°LC.
Рис. 2. Температурная зависимость lg k.
/ —CsCI; 2 —RbCl; 3 — LIC1; 4 — KCl;
5 — NaCl.
ности (рис. 1). Из полученных данных видно, что изменение массы хлори-
дов линейно зависит от времени при данной температуре, что подтверждает
вывод о нулевом порядке процесса. На рис. 2 представлены кинетические
зависимости логарифма скорости испарения и сублимации от обратных абсо-
лютных температур, с помощью кото-
рый определены по уравнению Аррени-
уса энергии активации. Кривая для
хлорида натрия построена по данным
А. А. Фотиева и Б. В. Слободина'
[22]. Энергия активации сублимации
составляет для хлористого лития 45,5,
хлористого калия 49,4, хлористого
рубидия 49,5 и хлористого цезия
47,4 ккал/моль. Энергия активации
испарения определена, соответственно,
36,2, 38,8, 39,4 и 37,4 ккал/моль.
Полученные значения хорошо согла-
суются с теплотами процессов и мо-
гут быть представлены следующим
рядом: Li<Cs<K<Rb<Na. Однако
для скоростей сублимации и испаре-
ния получено иное соотношение: Cs>Rb,
Li>K>Na. Для установления зависи-
мостей между теплотами процессов сублимации или испарения и их энергий
активаций можно привести следующие расчеты.
Испарение и сублимация — реакции нулевого порядка (это следует из
зависимости степени испарения и сублимации от времени — см. рис. 1), и
константа скорости испарения равна скорости испарения. Следовательно,
выражение для энергии активации испарения или сублимации может быть
представлено в виде
— руг G
T dT ~~ dT
(1)
18
Подставив в формулу (1) выражение для скорости испарения, взятое из
работ [15, 18, 19, 23, 24, 26],
G=ap
(2)
М , 2
------, г см*-сек,
InRT
где G — скорость испарения в данной среде; р — давление насыщенного пара
испаряющегося вещества при температуре Т°К, мм pm. ст.-, М — молеку-
лярный вес; R — газовая постоянная, а — коэффициент испарения, равный
отношению степеней испарения вещества в данной среде к испарению в ва-
кууме, получим для энергии активации испарения и сублимации выражение
f М dln —-
/ » z б? In Ct
' 2л/?Т п'гг_____' 7 I пуа
dT dT
Второе слагаемое в формуле (3) не зависит от температуры и поэтому
равно нулю, следовательно
1 / —
(3)
dT v 1
din ар
ET=RT*-----
din —
Ет = RT2
dT
Для практического расчета энергии активации преобразуем формулу (4)
4,575Т1Т2
Ет
(4)
(5)
Теплота испарения или
Клаузиуса—Клапейрона
сублимации вещества находится по уравнению
L^.R'T^
Вычтем из выражения для энергии активации (4) теплоту испарения суб-
лимации (6)
(6)
din ?
ET — Lr = RTi---KL —=
dT dT
=RT2 — (inp — In Vt — Inp)= — RT2
dT r dT 2
J F -RT
(7)
Как известно, термохимическая теплота превращения равна разности энер-
гий активации прямой и обратной реакций:
Ь=Ег — Е2. (8)
Фишбек и Шнайдт [23] полагают, что скорость конденсации газов почти
всегда определяется количеством столкновений молекул газа с поверхностью
конденсированной фазы. Из этого следует, что энергия активации скорости
испарения равна теплоте испарения. Однако при высоких температурах одного
столкновения молекул газа с поверхностью конденсированной фазы недоста-
точно. Требуется, чтобы молекула газа, столкнувшаяся с поверхностью кон-
денсированной фазы, несколько уменьшила свою энергию, чтобы удержаться
в поверхностном слое. Значение этого эффекта лежит в пределах ошибок
опыта. Следует отметить, что энергия активации конденсации не равна нулю
и при низких температурах.
2*
19
Так как молекула газа обладает тремя поступательными степенями сво-
боды, то энергия каждой степени свободы равна Сконденсированная мо-
лекула может перемещаться только в горизонтальной плоскости, но не в пер-
пендикулярном направлении, и поэтому ее энергия меньше на одну степень
свободы. Отсюда становится понятным, почему в равенстве (7) теплота испа-
RT
рения больше энергии активации испарения на величину —.
Согласно данным М. Борна и М. Гепперт-Мейера [27], находим зависи-
мость логарифма давления насыщенного пара от температуры
lnp=—
RT ,J
0
3
где С — константа Нернста; С=С0+у1пЛ4.
равна
У(СП-СЖ.Т) dT
0 dT+C,
RT
О)
Универсальная постоянная Co
с 1п (2л)^-7?Д 10 17
CGSE ед.,
(10>
где Lo — теплота превращения при 0°К; Сп, Ст,ж — мольные теплоемкости
пара, твердого тела или жидкости; N — число Авогадро, h — постоянная
Планка. Обозначим интеграл как <р(Т) и спотенцируем выражение (9):
р=мУМеСа~"^+^(Т>. (И)
Зависимость скорости испарения от температуры определим по уравнению
Аррениуса:
Ет
G=A(T)e Rr , (12)
где Er — энергия активации испарения при Т° К. Решая уравнения (2) и
(12), находим
.----Ет
4(П-»Р1/ (13)'
Подставим уравнение (13) в выражение (11)
. cCo+~Rf~~
р/2л/?7'
Затем выражение (14) вставим в формулу (12), учтя, что по уравнению (7)
^-о=^о>
Ео :
аМ* 10,17-^ + фСП
G(/)=— е ЕГ . (15)
У 2nRT
Для температуры кипения Тк при нормальном давлении по формуле (9)
можно найти выражение для энергии активации при 0°К:
£о=£о=/?Тк=[1пр-ф(7’к)-|1пЛ4-С0]. (16)
Если в первом приближении, согласно (26), считать, что
т
(Сп~СЖ)Т)Л = (Сп-Сж,т)Т,
о
20
тогда
т
In Г,
и формула (15) примет вид
г аМ2 ж ~'Л ‘°. 17~-. 2
GT ——— Т * е кт, г/см2-сек.
V 2лЛ>
(17)
(18)
В выражении (18) первый множитель, зависящий от температуры, растет
значительно медленнее, чем второй. Например, при высоких температурах
разность теплоемкостей для двухатомных молекул близка к единице:
СП-СЖ1Т^1; [Cn^R, CT^3R, СТ^СЖ и
*~ж. т
7" R
2 ~ Т° ~ 1
(19)
Учтя это и подставив числовые значения констант, входящих в равенство (18),
получим
G(T)=6700aM2e RT , г/см2-сек. (20)
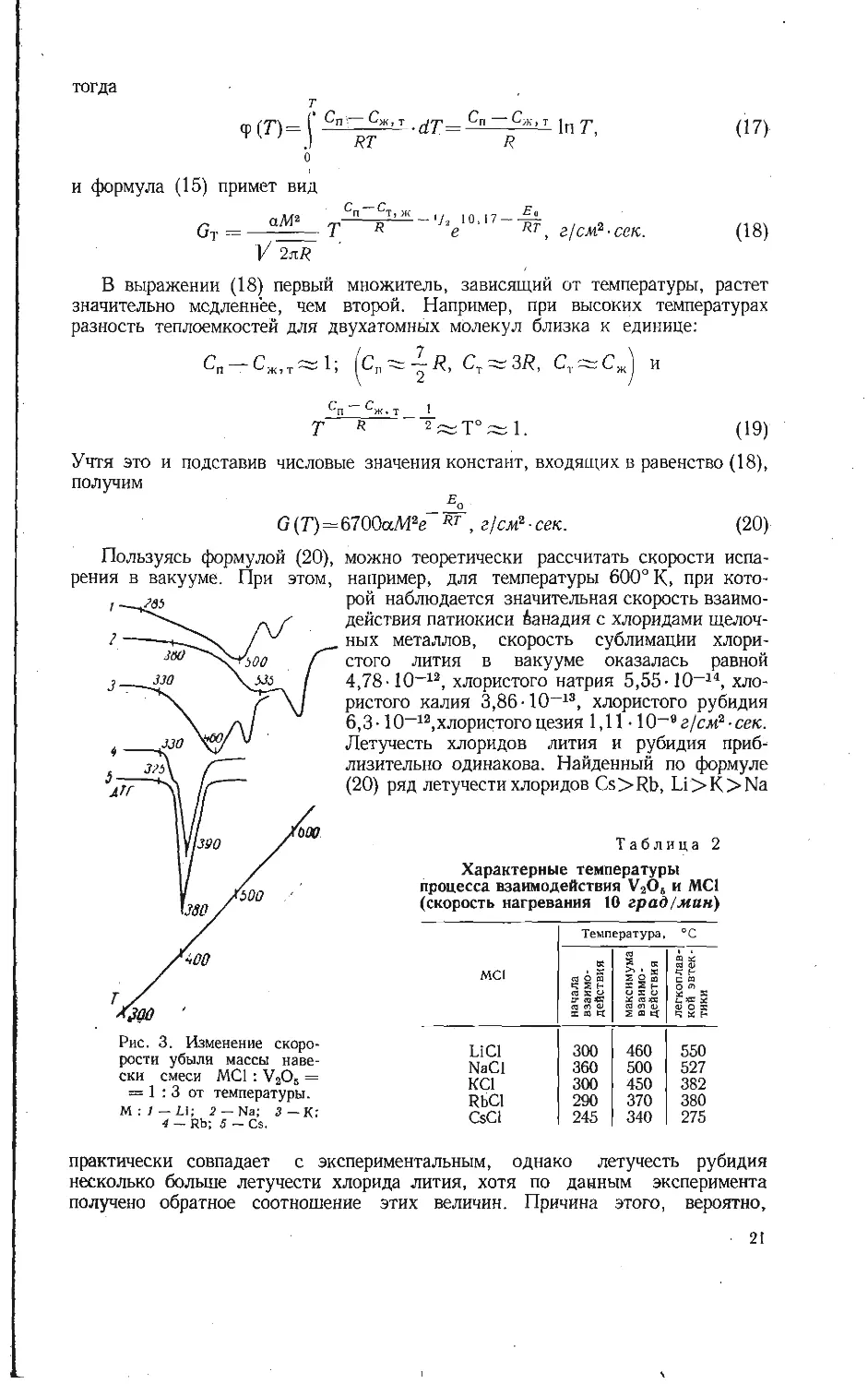
Пользуясь формулой (20),
рения в вакууме. При этом,
Рис. 3. Изменение скоро-
рости убыли массы наве-
ски смеси MCI : V2O5 =
= 1:3 от температуры.
1Л : 1 — Ы; 2 — Na; 3—К:
4 — Rb; 5 — Cs.
можно теоретически рассчитать скорости испа-
например, для температуры 600° К, при кото-
рой наблюдается значительная скорость взаимо-
действия патиокиси Аанадия с хлоридами щелоч-
ных металлов, скорость сублимации хлори-
стого лития в вакууме оказалась равной
4,78-10-12, хлористого натрия 5,55-10-14, хло-
ристого калия 3,86-10-13, хлористого рубидия
6,3-10-12,хлористого цезия 1,11 • 10-вг/сл12-сек.
Летучесть хлоридов лития и рубидия приб-
лизительно одинакова. Найденный по формуле
(20) ряд летучести хлоридов Cs>Rb, Li>K>Na
Таблица 2
Характерные температуры
процесса взаимодействия V2O6 и MCI
(скорость нагревания 10 град/мин)
Температура, °C
<4 К 2 к ffl £ 03 о
MCI g g « | g « «3 5 0 OXO a* a <5 bC « <s ca to <u a cn о X « tt 2 ® Ef f- u И Q to s u « x 4 X H
LiCl 300 460 550
NaCl 360 500 527
KCl 300 450 382
RbCl 290 370 380
CsCl 245 340 275
практически совпадает с экспериментальным, однако летучесть рубидия
несколько больше летучести хлорида лития, хотя по данным эксперимента
получено обратное соотношение этих величин. Причина этого, вероятно,
21
-заключается в том, что эксперимент выполнен недостаточно точно:
помимо колебаний температуры в печи ±7° и скорости эффузии воздуха,
по-видимому, значительную роль играет ползучесть жидких хлоридов щелоч-
ных металлов. Так, хлорид рубидия значительно более ползуч, чем хлорид
.лития, вследствие чего у первого поверхность испарения получалась большей.
Все это, а также сильная гигроскопичность хлорида лития, затрудняли про-
:ведение эксперимента.
О скорости взаимодействия пятиокиси ванадия с хлоридами щелочных
металлов можно судить по кривым ДТГ, снятым на дериватографе (рис. 3).
Видно, что во всех случаях температуры фиксируемого начала взаимодействия
пятиокиси ванадия с хлоридами щелочных металлов лежат ниже температуры
самой легкоплавкой эвтектики системы V2O6—М2О. Температуры фиксируемого
начала максимальной скорости взаимодействия пятиокиси ванадия с хлорида-
ми щелочных металлов, представленные в табл. 2, могут служить косвенной
характеристикой взаимодействия V2O6 и MCI. Можно составить следующее
соотношение скоростей взаимодействия пятиокиси ванадия с хлоридами ще-
лочных металлов: Cs>Rb>K>Li>Na. Сравнение соотношений скоростей
сублимации и испарения хлоридов щелочных металлов с рядом скоростей их
взаимодействия и оценка количеств сублимирующей массы хлоридов позво-
ляют сделать заключение, что сублимация и испарение, по-видимому, не ме-
тут лимитировать процесс взаимодействия пятиокиси ванадия с хлоридами
щелочных металлов и, следовательно, механизм твердое—газ не может быть
преобладающим.
ВЫВОДЫ
1. Скорости испарения и сублимации и хлоридов щелочных металлов,
•определенные в единых методических условиях, сопоставлены в ряд Cs>Rb,
Li>K>Na. По кинетическим данным определены энергии активации испаре-
ния и сублимации хлоридов лития, калия, рубидия и цезия, которые в пре-
делах ошибок опыта близки к значениям теплот испарения и сублимации.
2. Найдена формула для скорости испарения и сублимации и вычислена
летучесть хлоридов при 600° К. Полученный ряд летучести практически сов-
ладает с определенным экспериментально. Показано, что теплоты процессов
испарения и сублимации больше энергий активации этих процессов на RT/2,
что позволяет, зная теплоты испарения и сублимации, находить энергии
активации испарения и сублимации и наоборот.
3. Сняты на дериватографе кривые ДТГ взаимодействия пятиокиси вана-
дия с хлоридами щелочных металлов, по которым определены фиксируемые
температуры начала и максимума взаимодействия. Сопоставлены скорости
взаимодействия пятиокиси ванадия со скоростями испарения и сублимации
хлоридов щелочных металлов. Испарение и сублимация не могут быть ли-
митирующими стадиями процесса взаимодействия пятиокиси ванадия с хло-
ридами щелочных металлов.
ЛИТЕРАТУРА
1. К. Niwa, J. Chem. Soc. Japan, 1938 , 59, 637.
2. Ан. H. Несмеянов, Л. А. Сазонов. Ж- неорг. хим., 1957, 2, 946.
3. Ан. Н. Несмеянов, Л. А. Сазонов. Ж. неорг. хим., 1957, 2, 1183.
4. Н. Wartenberg, Н. Schulz. Z. Elektrochem., 1921, 27, 568.
5. О. Ruff, S. Mugdan. Z. anorg. Chem., 1921, 117, 147.
-6. W. Kangro, H. W. Wiking. Z. phys. Chem., 1938, A183, 199.
7. О. Кубашевский, Э. Эванс. Термохимия в металлургии. М., ИЛ, 1954.
8. К. К. Kelly. Bull. U. S. Bur. Min., 1935, № 3835.
9. В. Н. Zimm, J. Е. Mayer. J. Chem. phys., 1944, 12, 361.
10. J. E. Mayer, I. H. Wintner. J. Chem. Phys., 1938, 6, 301.
11. S. Horiba, H. Baba. Bull. Chem. Soc. Japan., 1928, 3, 11.
22
12. H. Warteberg, Т. Albrecht. Z. Electrochem., 1921, 27, 162.
13. E. T. Tjock, H. Rodebusch. J. Amer. Chem. Soc., 1926, 48, 2522.
14. К. С. Краснов, Г. А. Крестов. Радиохимия, 1960, 2, 6, 671.
15. W. D. T red dwell, W. Werner. Helv. chim. acta, 1953, 36, 6, 1436.
16. R. S. Bradley, P. Volans. Proc. Roy. Soc., 1953, A217, 508, 1131.
17. V. Deitz. J. Chem. Phys., 1936, 4, 578.
18. Ан. H. Несмеянов, Л. А. Сазонов. Ж- неорг. хим., 1959, 4, 231.
19. М. Knudsen. Ann. Phys., 1915, 47, 697.
20. В. И. Спицын, В. И. Шостак. Ж. общ. хим., 1952, 22, 1063.
21. W. D. Treadwell, W. Werner. Helv. chim. acta, 1953, 36, 6, 1445.
22. А. А. Фотиев, Б. В. Слободин. Ж. физ. хим., 1965, 39, 12, 3099.
23. К- Fischbeck, К- Schneidt. Z. Elektrochim., 1932, 38.
24. A. Nacke, О. Rchmolke, G. N. Stranski. Z. Kristall, 1957, 109, 184.
25. А. А. Фотиев, Б. В. Слободин. Ж. неорг. хим., 1965, 10, 3, 569.
26. И. В. Радченко. Молекулярная физика. М., «Наука», 1965.
27. М. Борн, М. Гепперт-Мейер. Теория твердого тела. М.—Л., ОНТИ, 1938^
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА ХИМИИ
1970
Б. В. СЛОБОДИН
О ХИМИЧЕСКОМ СОСТАВЕ СОЕДИНЕНИЙ, ОБРАЗУЮЩИХСЯ
В СИСТЕМЕ У2ОБ—AgVO3
Большое внимание в последнее время уделяется исследованию систем,
в которых образуются бронзы — неорганические соединения со специфически-
ми свойствами. К ним относятся системы, включающие пятиокись ванадия и
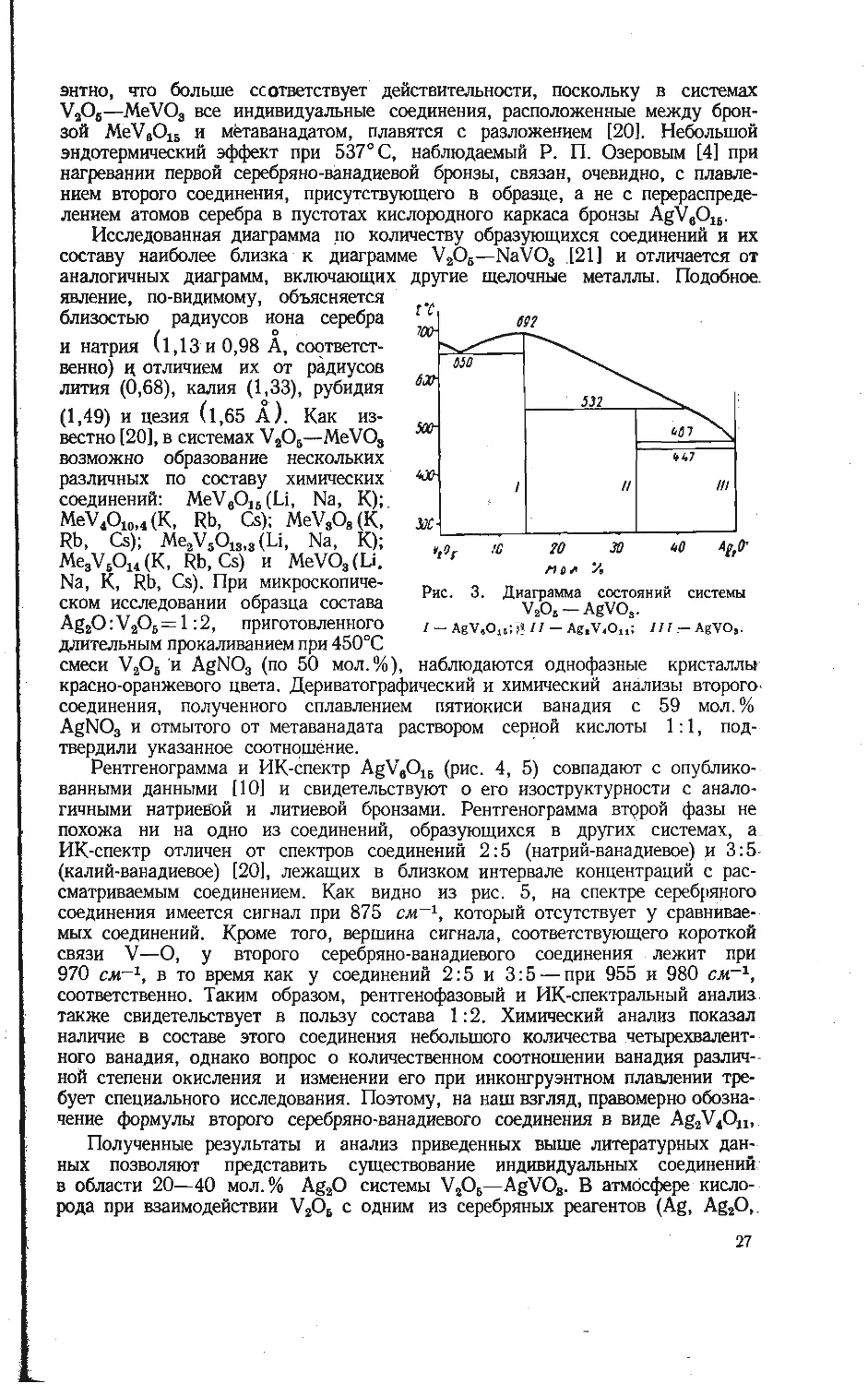
Рис. 1. Диаграмма состояний системы
V2O6-Ag2O [9].
/ - AgV,O12; 11 - Ag2V ,О,,; 777-AgVO,;
IV- Ag,V2O,; V- AgsVO,.
кислородные соединения серебра. Впер-
вые образование серебряно-ванадиевой
бронзы при нагревании смесей AgNO3
и V2O5 с молярным отношением
Ag2O:V2O5 от 1:20 до 1:1 описали
Прандтль и Муршхаузер [1]. Они на-
звали это соединение серебряным вана-
дил-ванадатом и обозначили его хими-
ческий состав в виде Ag2O- V2O4-5V2O5.
Система V2O5—AgVO3 изучалась
Флодом с сотрудниками [2] термоанали-
тическим, химическим и рентгено-
фазовым методами. При соотношении
Ag2O:V2O5 меньше 0,6 происходит
образование P-фазы AgaO • V2O4 • 5V2Oa:
Ag2O+6 V 2О5=Ag2O • V 2О4 • 5 V 2О6+
+7А- (1)
При соотношении 0,2—0,6 и темпера-
туре 536° С, по сведению этих авторов,
наблюдается Образование второго сое-
динения по реакции:
сс-фаза+жидкость -> у-фаза. (2)
Кислород, выделенный при кристаллизации первичной P-фазы, в ходе реак-
ции (1) вновь поглощается слишком медленно. В результате образуется не-
равновесное по кислороду соединение 5Ag2O-0,9V2O4-llV2O5(yi). Соответ-
ствующие ему стабильные у-кристаллы, образующиеся при соотношении
Ag2O:V2O5 от 0,6 до 0,9, имеют состав 5Ag2O- 12V2O5 (у2). При измельче-
нии и нагревании до 450° у4-фаза медленно адсорбирует кислород.
Уодсли [3] относит ванадиевые бронзы к нестехиометрическим окислам
ванадия бертолидного типа. Для p-фазы он ' приводит переменный состав
AgxV0215(0,08<x<0,17), для у-фазы AgyVO2)6, (у=0,33). Р. П. Озеров [4]
подтвердил образование p-фазы состава Ag2O:V2O5=l :6 и провел кристал-
лохимическое изучение подобных соединений.
24
4 Б в
‘ 1 0* 1 т 1 1 а [2]
— —।— П_ J, б [3]
WL 1 6 [5]
I
ГТ 1 г [5]
1 1 1
ь % з; <-j Г i । 9 [5]
Гт к г 51, тт —1 е [2 в]
1 “ |LT —1— I 1—1 J—1 [С]
. . . .' ! J ‘ГТ' £ ; ПА ЛА^Ц? 3 1/0]
Дешанвер и Ревау [5] нашли соединения переменного состава AgxV2O5
0-фаза (х=0,17—0,45) и 6-фаза (х=0,60—0,80). Между этими областями
гомогенности находится двухфазная область. На воздухе образуется 0-фаза,
подобная описанной выше, и е-фаза, область гомогенности которой лежит
в пределах х=1—1,5. Дальнейшее увеличение содержания серебра приво-
дит к образованию мета-
ванадата AgVO3. Фазы 0
и 6 содержат четырехва-
лентную форму ванадия,
е-фаза термически разла-
гается при 500° С, давая
новые фазы. Соединение
6 выше 500° С окисляется
с образованием смеси 0-
и е-фаз:
4Ag0,7V2O5+1/2O2 —>
—► Ag2V 4ОИ 2Ag0(4V 2О5.
(3)
Эта реакция не дает
возможности в присутст-
вии воздуха выделить в
чистом виде 6-фазу. Три
области существов|ания
однофазных соединений
приведены в работе
[6]: a=Ag0_01V2O5, 0 =
= Ago>29—0,41V2Os и 6 —
—Ago,67—о,86^205. При
увеличении содержа-
ния серебра сосуществуют
6-фаза и металлическое
серебро. 0-фаза, изотроп-
ная аналогичным фазам
других систем, отнесена
к моноклинной синго-
нии, а 6-фаза — к псев-
доорторомбической. Тер-
мическая прочность бронз убывает от 0- к 6-фазе. Андерсон [7, 8] приводит
соединение, которое аналогично 6-фазе, описанной Харди и др. [6]. Им же
подробно исследована кристаллическая структура.
В 1966 г. Флури и Кохмюллер [9] с помощью термического анализа
в токе кислорода построили диаграмму состояний системы V2OS—Ag2O до
70 мол. % Ag2O (рис. 1). Они считают, что условия проведения исследования
исключают образование бронз. Установлено образование пяти индивидуаль-
ных соединений: AgV7O18; Ag2V4Ou; AgVO3; Ag4V2O7 и Ag3VO4. Метавана-
дат серебра обладает, по данным этих авторов, тремя полиморфными форма-
ми, температурные пределы существования которых соответствуют следующей
схеме:
i'o , 1 20 1 30 1 >4) 50
/7/6 /3 25 >'2 35
0222 0330 0.667 /,333
Рис. 2. Химический состав и области гомогенности сое-
динений, образующихся в системе V2O5—AgVO3.
А — состав и область гомогенности. Б — метод и условия
синтеза: а — Ag2O-|-VsO6. воздух; б — AgVOa4*V2O5;
в — Ag+ViO6; г —Ай+УгОБ> вакуум; д — Ag+V2OS1 аргон;
е — Ag+VaO64-HaO, давление; ж — Ag2V+V2O6, кислород;
з—AgVO8+V2O5, воздух. В — литературные данные, ♦ — сое-
динения, в которых ванадий находится в частично восстановленной
форме (бронза).
<?<
342-122 4 78
а —* 0 <— у расплав,
t t
Они привели рентгенограммы всех обнаруженных соединений и модифи-
каций.
25
При термической обработке смесей AgVO3 и V2OB образуются, по мнению
авторов исследования [10], две фазы с общей формулой AgxV^4V^’5O„.
Фаза I (х=0,29—0,43; /п=0,32—0,44; /1=1,56—1,68; у=4,96—5,05) изо-
морфна натрий-ванадиевой 0-бронзе. Фаза ПА (х=0,80—0,99; т—0,06—
0,20; /г = 1,80—1,94; у=5,32—5,46) отличается от натриевой, у-фазы.
Однако, будучи обработанной в горячей разбавленной азотной кислоте, она
переходит в фазу 11 В (х=0,67—0,77; /п=0,05—0,09; /г=1,91—1,95;
у=5,22—5,36), ИК-спектр и рентгенограмма которой аналогичны натриевой
у-фазе.
На рис. 2 представлены литературные данные о составе и областях гомо-
генности кислородных серебряно-ванадиевых соединений, образующихся в
системе V2OS—AgVOs. Они свидетельствуют о противоречивости полученных
результатов и различии в обозначениях, используемых исследователями для
одних и тех же объектов. Однако, несмотря на это, специфичность образую-
щихся соединений и возможность использования бронзы Ag—V в качестве
катализатора [11, 12] обусловили подробное изучение кристаллохимии [3, 8,
13, 14], электрических и других свойств [15—17] кислородных серебряно-
ванадиевых соединений.
Настоящая работа посвящена изучению диаграммы состояния системы
V2OB—AgVOs и определению состава образующихся в ней соединений. Мето-
ды исследования — дериватографический, термооптический, рентгенофазовый,
ИК-спектрометрия и химический. Система изучалась в атмосфере воздуха.
Методика исследования
' Образцы для исследования готовили перемешиванием пятиокиси ванадия
и нитрата серебра квалификации «х. ч.», взятых в определенном соотноше-
нии. Смесь нагревали и охлаждали в дериватографе со скоростью 10 град/мин
до 800—900° С. Дериватограммы первоначального нагревания фиксировали
неравновесные процессы, происходящие в сложной системе, при повторном
нагревании образцы находились в равновесном состоянии. Расплав из тигель-
ка выливали на фарфоровую пластинку, охлаждали, измельчали и исследовали
на термоприставке к микроскопу МИН-8 [18] со скоростью изменения темпе-
ратуры 1—2 град/мин.
Экспериментальная часть и обсуждение результатов
Диаграмма состояний системы V2OB—AgVO3, построенная по результатам
дериватографического и термооптического анализа, приведена на рис. 3.
В такой системе возможно существование двух соединений. Первое из них
с температурой плавления 692° С образуется при соотношении Ag2O:V2OB=
= 1:6. Судя по данным химического анализа и дериватографии, шестая часть
ванадия находится в этом соединении в четырехвалентном состоянии, что
позволяет представить его состав в виде AgV6O1B.
Эвтектическая точка между этим соединением и V2OB расположена при
650°С и 3,8% Ag2O. В интервале 0—14,3% AgaO находится область двух-
фазных смесей (V2O5-|-AgVeO16). При повышении содержания серебра обра-
зуется второе серебряно-ванадиевое соединение, плавящееся с разложением
при 532° С. Эвтектическая горизонталь между ним и метаванадатом соответ-
ствует 467° С. Метаванадат серебра AgVO3 плавится конгруэнтно в промежу-
точной точке [19], т. е. при температуре солидуса.
Построенная диаграмма состояний системы V2O6—AgVO3 отличается по
конфигурации от диаграммы Флури и Кохмюллера [9]. Как и следовало
ожидать, соединением с наименьшим содержанием серебра является бронза
состава Ag2O:V2OB=l :6, а не 1:7. Второе соединение плавится инконгру-
26
Рис. 3. Диаграмма состояний системы
V2O5- AgVO3.
/ - AgV,0.6;Я II- Ag,V4Otl; III - AgVO,.
энтно, что больше ссответствует действительности, поскольку в системах
V2O8—MeVO3 все индивидуальные соединения, расположенные между брон-
зой MeVeO1B и мётаванадатом, плавятся с разложением [20]. Небольшой
эндотермический эффект при 537° С, наблюдаемый Р. П. Озеровым [4] при
нагревании первой серебряно-ванадиевой бронзы, связан, очевидно, с плавле-
нием второго соединения, присутствующего в образце, а не с перераспреде-
лением атомов серебра в пустотах кислородного каркаса бронзы AgVeO1B.
Исследованная диаграмма по количеству образующихся соединений и их
составу наиболее близка к диаграмме V2OB—NaVO3 [21] и отличается от
аналогичных диаграмм, включающих
явление, по-видимому, объясняется
близостью радиусов иона серебра
и натрия (1,13 и 0,98 А, соответст-
венно) ц отличием их от радиусов
лития (0,68), калия (1,33), рубидия
(1,49) и цезия (1,65 А). Как из-
вестно [20], в системах V2OB—MeVO8
возможно образование нескольких
различных по составу химических
соединений: MeVeOj5(Li, Na, К);
MeV4O10,4(K, Rb, Cs); MeV8O8(K,
Rb, Cs); Me2VBO1313(Li, Na, K);
Me3VBO14(K, Rb, Cs) и MeVO3(U.
Na, K, Rb, Cs). При микроскопиче-
ском исследовании образца состава
Ag2O: V2OB = 1:2, приготовленного
длительным прокаливанием при 450°С
смеси V2OB и AgNO3 (по 50 мол. %), наблюдаются однофазные кристаллы
красно-оранжевого цвета. Дериватографический и химический анализы второго,
соединения, полученного сплавлением пятиокиси ванадия с 59 мол.%
AgNO3 и отмытого от метаванадата раствором серной кислоты 1:1, под-
твердили указанное соотношение.
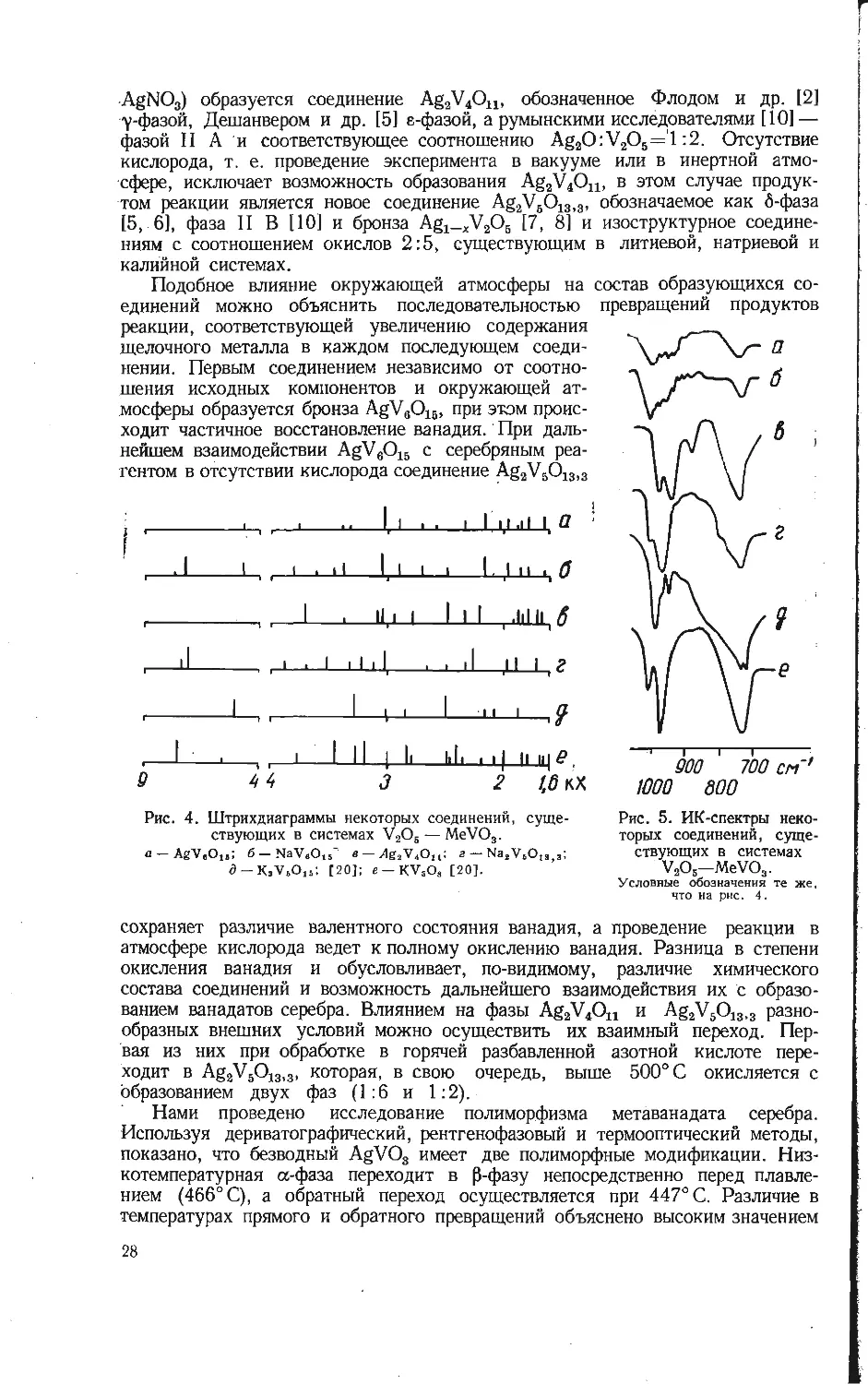
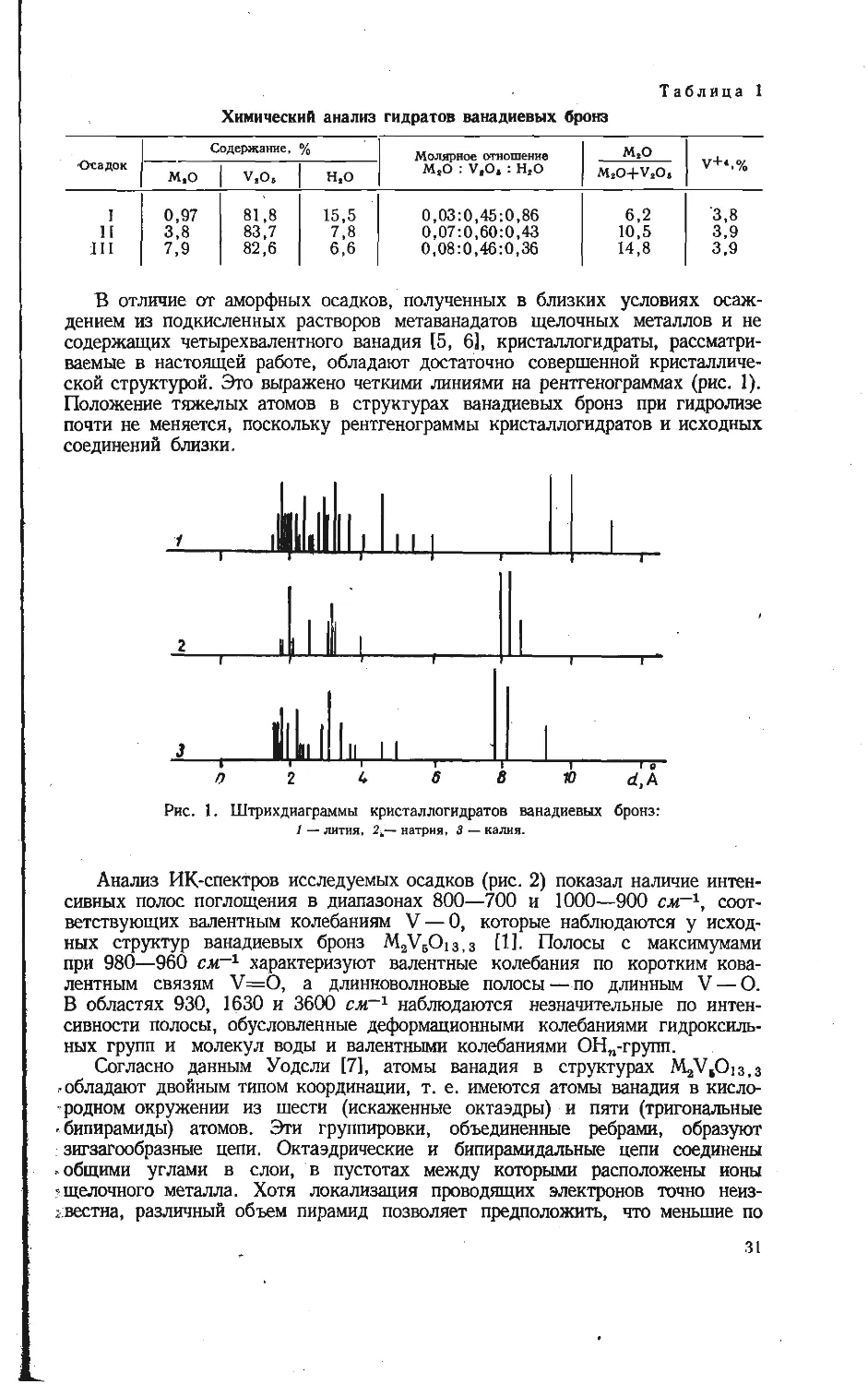
Рентгенограмма и ИК-спектр AgVeO1B (рис. 4, 5) совпадают с опублико-
ванными данными [10] и свидетельствуют о его изоструктурности с анало-
гичными натриевой и литиевой бронзами. Рентгенограмма второй фазы не
похожа ни на одно из соединений, образующихся в других системах, а
ИК-спектр отличен от спектров соединений 2:5 (натрий-ванадиевое) и 3:5-
(калий-ванадиевое) [20], лежащих в близком интервале концентраций с рас-
сматриваемым соединением. Как видно из рис. 5, на спектре серебряного
соединения имеется сигнал при 875 см-1, который отсутствует у сравнивае-
мых соединений. Кроме того, вершина сигнала, соответствующего короткой
связи V—О, у второго серебряно-ванадиевого соединения лежит при
970 см-1, в то время как у соединений 2:5 и 3:5 — при 955 и 980 см-1,
соответственно. Таким образом, рентгенофазовый и ИК-спектральный анализ
также свидетельствует в пользу состава 1:2. Химический анализ показал
наличие в составе этого соединения небольшого количества четырехвалент-
ного ванадия, однако вопрос о количественном соотношении ванадия различ-
ной степени окисления и изменении его при инконгруэнтном плавлении тре-
бует специального исследования. Поэтому, на наш взгляд, правомерно обозна-
чение формулы второго серебряно-ванадиевого соединения в виде Ag2V4On,
Полученные результаты и анализ приведенных выше литературных дан-
ных позволяют представить существование индивидуальных соединений
в области 20—40 мол. % Ag2O системы V2OB—AgVO8. В атмосфере кисло-
рода при взаимодействии V2OB с одним из серебряных реагентов (Ag, Ag2O,.
27
AgNO3) образуется соединение Ag2V4Ou, обозначенное Флодом и др. [2]
у-фазой, Дешанвером и др. [5] е-фазой, а румынскими исследователями [10] —
фазой II А и соответствующее соотношению Ag2O:V2OS=1:2. Отсутствие
кислорода, т. е. проведение эксперимента в вакууме или в инертной атмо-
сфере, исключает возможность образования Ag2V4On, в этом случае продук-
том реакции является новое соединение Ag2V8O13)3, обозначаемое как 6-фаза
[5, 6], фаза II В [10] и бронза Ag1_^V2O6 [7, 8] и изоструктурное соедине-
ниям с соотношением окислов 2:5, существующим в литиевой, натриевой и
калийной системах.
Подобное влияние окружающей атмосферы на состав образующихся со-
единений можно объяснить последовательностью превращений продуктов
реакции, соответствующей увеличению содержания
щелочного металла в каждом последующем соеди-
нении. Первым соединением независимо от соотно-
шения исходных компонентов и окружающей ат-
мосферы образуется бронза AgV6O18, при этом проис-
ходит частичное восстановление ванадия. При даль-
нейшем взаимодействии AgV6O15 с серебряным реа-
гентом в отсутствии кислорода соединение Ag2V5O1313
,________I—, 1____ы_lrj । I и .11 I, Q
,—J------1_ ,—I—i—_1—1-1д—I—1-----1, ,1 и I, б
,----------, ,__I____._Ид_1____1_1_1 ,.|||||/
,—J--------, J 1111,_________J______,н ।,г
,________I—,,________I___1_1___I__ ।
,—I——।—,,—i__l 11—|_Ь__________iili i| и u|g
0 4 4 3 2
Рис. 5. ИК-спектры неко-
торых соединений, суще-
ствующих в системах
V2O5—MeVO3.
Условные обозначения те же,
что на рис. 4.
Рис. 4. Штрихдиаграммы некоторых соединений, суще-
ствующих в системах V2O5 — MeVO3.
a- AgV«Olt; 6-NaV„Ols’ e-Ag2V4Olt: г - Na,VsO1S)3;
a-K3V6O15; [20]; e-KVsO„ [20].
сохраняет различие валентного состояния ванадия, а проведение реакции в
атмосфере кислорода ведет к полному окислению ванадия. Разница в степени
окисления ванадия и обусловливает, по-видимому, различие химического
состава соединений и возможность дальнейшего взаимодействия их с образо-
ванием ванадатов серебра. Влиянием на фазы Ag2V4Ou и Ag2V5O13,3 разно-
образных внешних условий можно осуществить их взаимный переход. Пер-
вая из них при обработке в горячей разбавленной азотной кислоте пере-
ходит в AgaV5O13>3, которая, в свою очередь, выше 500° С окисляется с
образованием двух фаз (1:6 и 1:2).
Нами проведено исследование полиморфизма метаванадата серебра.
Используя дериватографический, рентгенофазовый и термооптический методы,
показано, что безводный AgVO3 имеет две полиморфные модификации. Низ-
котемпературная a-фаза переходит в 0-фазу непосредственно перед плавле-
нием (466° С), а обратный переход осуществляется при 447° С. Различие в
температурах прямого и обратного превращений объяснено высоким значением
28
энергии активации процесса перестройки кристаллической решетки. Это при-
водит к перегреву и переохлаждению при полиморфных превращениях. По-
добные явления часто наблюдаются при фазовых превращениях многих вана-
диевых соединений. Судя по рентгенограммам и ИК-спектрам, p-фаза изо-
структурна метаванадатам калия, рубидия и цезия и относится к ромбической
сингонии.
ВЫВОДЫ
1. Построена диаграмма состояний системы V2O8—AgVO3 в атмосфере
воздуха. Показано образование в ней двух соединений AgVeO18 и Ag2V4Ou,
лервое из которых относится к кислородным ванадиевым бронзам.
2. На основании экспериментальных и литературных данных рассмотрены
'особенности взаимодействия пятиокиси ванадия с серебряными реагентами в
зависимости от окружающей атмосферы.
ЛИТЕРАТУРА
1. U. Prandtl, Н. М ц г sc h h a use г. Z. anorg. Chem., 1908 , 60,441.
2. H. Flood, Th. Krog, H. So rum. Tids. Kjemi Bergvesen Met., 1946, 6, 32.
Цит. no Chem., Abstr., 1946, 40, 6358.
3. A. D. Wads ley. Acta cryst., 1957, 10, 261.
4. P. П. Озеров. Кристаллография, 1959, 4, 201.
5. A. Deschan vers, B. Reveau. C. R. Acad. Sci., 1964, 259, 3553.
6. A. Hardy, 1. Galy, A. Casa lot, M. Pouchard. Bull. Soc. Chim. Fran-
ce, 1965, № 4, 1056.
7. S. Andersson Acta Chem., Scand., 1965, 19, 269.
8. S. Andersson. Acta Chem., Scand., 1965, 19, 1371.
'9. P. Fleury, R. К о h 1 m u 11 e г. C. R. Acad. Set., 1966 , 262.
10. I. Lukacs, C. Strusievici, C. L i t e a n u. Z. anorg. allg. Chem., 1967,
349 92
11. Пат. США № 3012043, 19.06.58.
12. Пат. Чехословакии № 106791, 15.03.63.
13. Р. П. Озеров. Ж- неорг. хим., 1959 , 4, 1047.
14. Р. П. Озеров. Кристаллография, 1957, 2, 226.
15. 1. Lukacs, С. Strusievici, Е. Weissmann, С. L i t е a n u. Rev. Rou-
main Chim., 1966, 11. 335.
16. Д. С. Волженский, М. В. Пашковский, Л. Г. Свеколкина. Ж-, неорг.
• хим., 1963, 8, 255.
17. 3. И. Ори атская. Физика твердого тела, 1964, 6, 1254.
18. М. П. Глазырин, А. А. Фотиев. Ж. физ. хим, 1967, 41, 479.
19. И. И. Новиков. Докл. АН СССР, 1955, 100, 1119.
20. А. А. Фотиев, С. И. Алямовский, М. П. Глазырин, Н. В. Баусова.
Химия соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН СССР,
1967, вып. 14, стр. 29.
21. М. П. Глазырин, А. А. Фотиев. Изв. АН СССР, Неорганические мате-
риалы, 1968, 4, 76.
АКАДЕМИЯ НАУК СССР
уральский филиал
вып. 20
ТРУДЫ ИНСТИТУТА химии
1970
А. А. ФОТИЕВ, Р. Н. ПЛЕТНЕВ
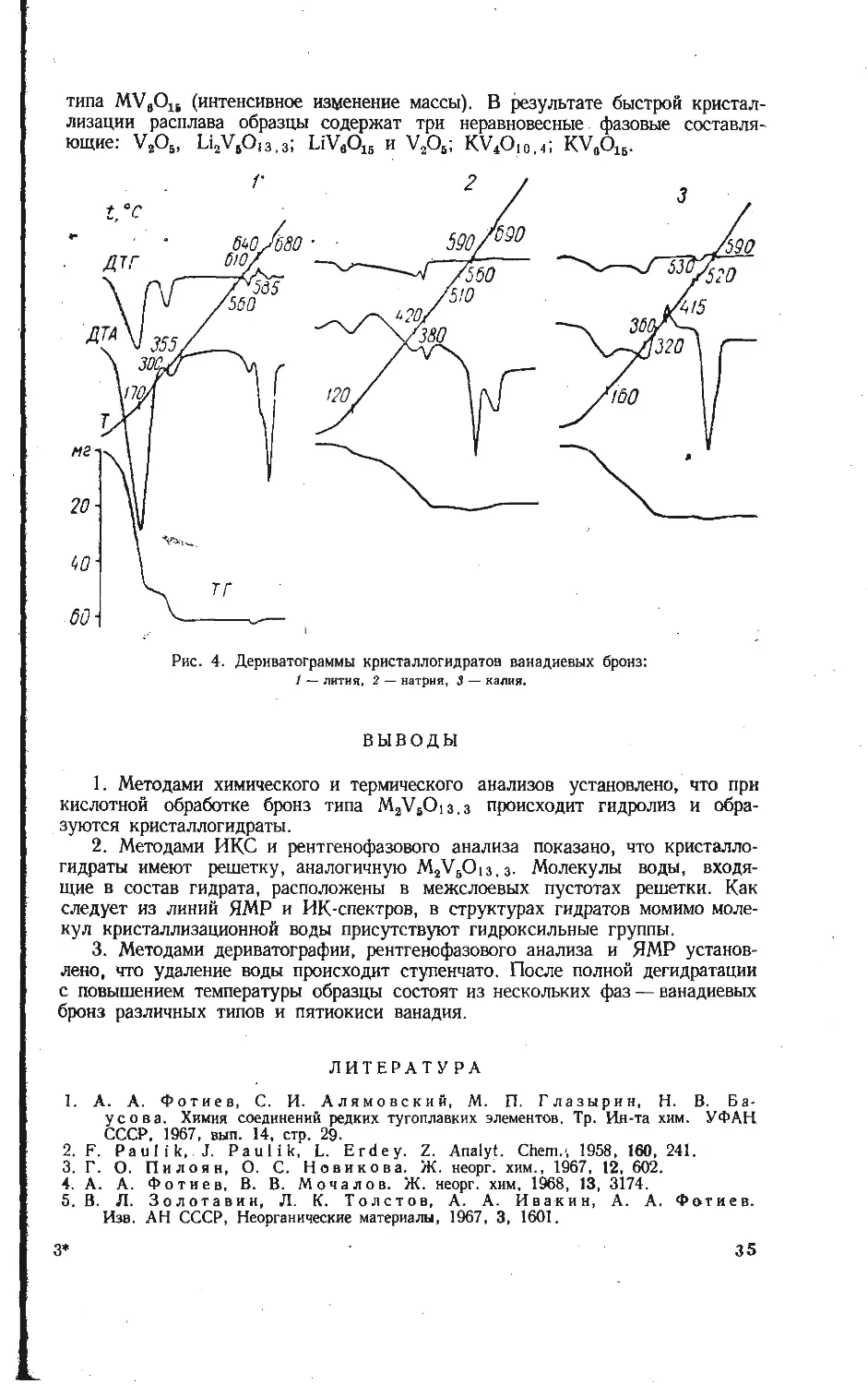
ИССЛЕДОВАНИЕ ГИДРАТОВ ВАНАДИЕВЫХ БРОНЗ
В процессе получения некоторых растворимых соединений ванадия при
обжиге ванадийсодержащих концентратов с добавками солей щелочных эле-
ментов, например сильвинита, в качестве промежуточных фаз могут возни-
кать ванадиевые бронзы типа М2У6О|з,з> где M = Na, К [1]. Эти соедине-
ния взаимодействуют с небольшой скоростью с водой, а при кислотной
обработке гидролизуются с образованием кристаллогидратов. Аналогичные
замечания относятся к бронзе Li2V60i3,3.
Учитывая возможность возникновения гидратов в технологическом про-
цессе ванадиевого производства и отсутствие каких-либо литературных дан-
ных относительно их природы, состава и свойств, нами проведено изучение
гидратированных ванадиевых бронз лития, натрия и калия различными физико-
химическими методами.
Методика исследования
Кристаллогидраты ванадиевых соединений лития (I), натрия (II) и калия
(III) получены в результате обработки бронз типа MaV60i3,s_x раствором:
20%-ной серной кислоты при рН=0,9— 1,1. Осадки после промывки водой,
подкисленной до соответствующего значения pH, высушивали на воздухе
и анализировали: на ванадий — объемным методом, на щелочной металл — мето-
дом пламенной фотометрии. Термический анализ проводили на деривато-
графе системы Ф. Паулика, И. Паулика и Эрдей [2]. Энергию активации
дегидратации воды рассчитывали способами, описанными в работах [3, 4].
Порошковые рентгенограммы получены в установке УРС-55 в камере ВРС-3
с кассетой диаметром 143,3 мм на KaCr-излучении. Интенсивность линий
оценивали визуально по пятибалльной системе. ИК-спектры снимали на спект-
рометре UR-10 в области 3800—400 см-1; образцы готовили в виде суспен-
зий в вазелиновом масле. Спектры ядерного магнитного резонанса (ЯМР)
записывали на спектрометре JNM-3, BL-1 в полях около 2650, 6580 и
7500 э при комнатной температуре или при—196° С.
Экспериментальная часть
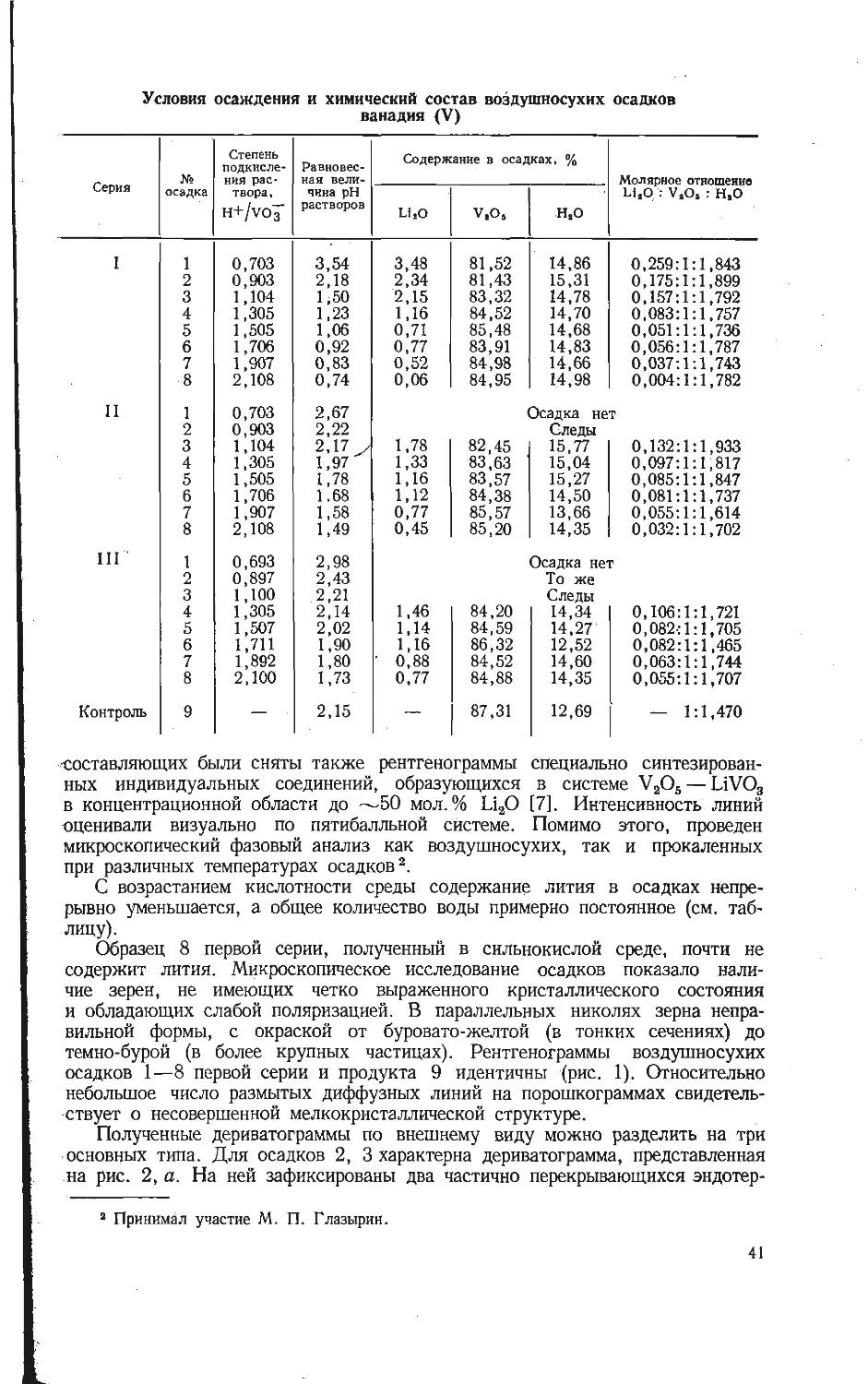
Из результатов химического анализа следует, что несмотря на единые
условия приготовления осадков, концентрация лития значительно меньше
концентации натрия или калия, в то время как содержание воды умень-
шается от осадка I к осадку III (табл. 1). Так как в исследуемых соеди-
нениях присутствует около 4% четырехвалентного ванадия от общего коли-
чества, можно считать, что при образовании кристаллогидратов восстановлен-
ная форма ванадия, содержащегося в бронзах, не окисляется.
30
Таблица 1
Химический анализ гидратов ванадиевых бронз
Осадок Содержание, % Молярное отношение М£О : V.O. : НгО м,о v+‘.%
М,0 v,O, н.о MjO-f-VaOj
I 0,97 81,8 15,5 0,03:0,45:0,86 6,2 3,8
11 3,8 83,7 7,8 0,07:0,60:0,43 10,5 3,9
ill 7,9 82,6 6,6 0,08:0,46:0,36 14,8 3,9
В отличие от аморфных осадков, полученных в близких условиях осаж-
дением из подкисленных растворов метаванадатов щелочных металлов и не
содержащих четырехвалентного ванадия [5, 6], кристаллогидраты, рассматри-
ваемые в настоящей работе, обладают достаточно совершенной кристалличе-
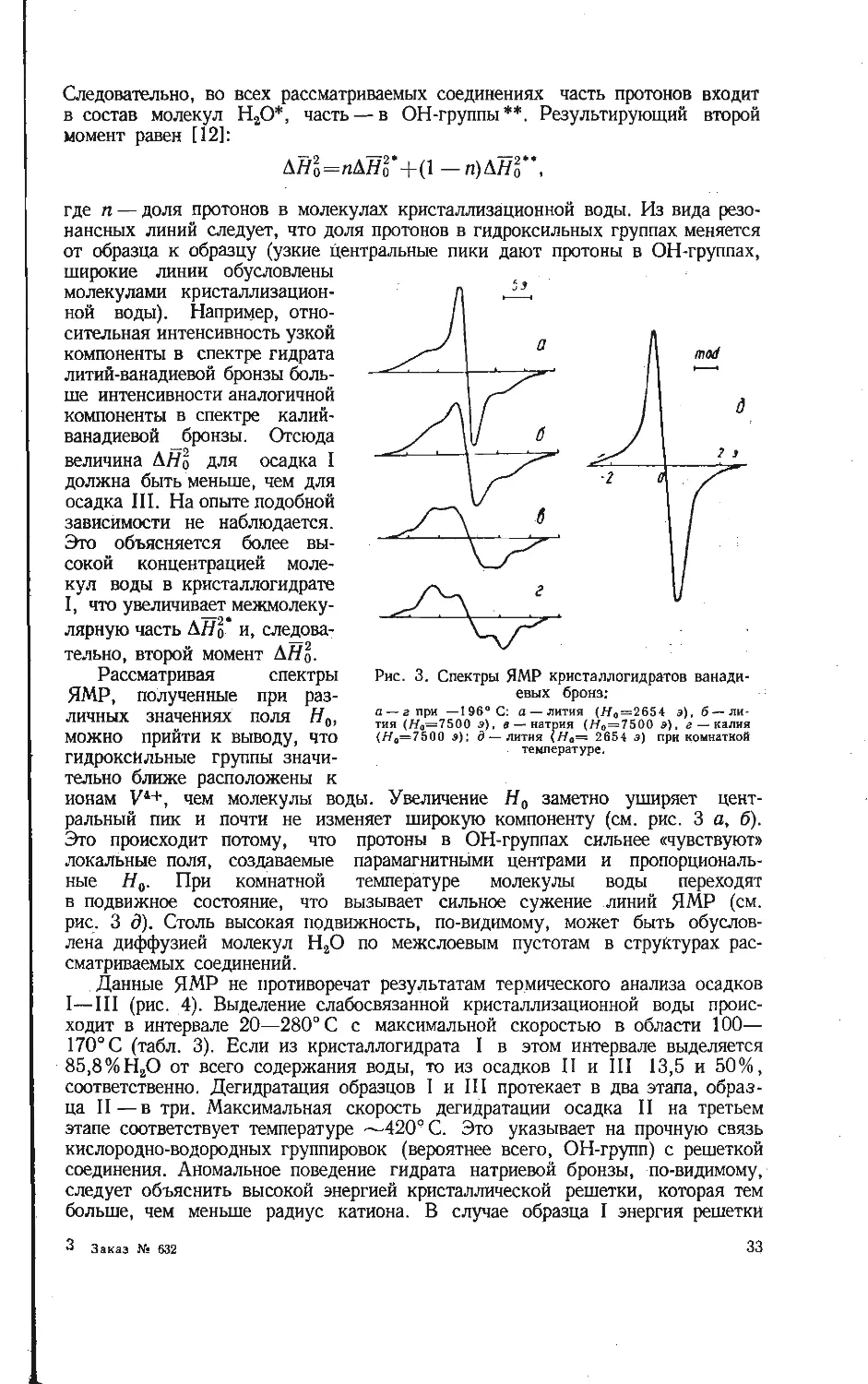
ской структурой. Это выражено четкими линиями на рентгенограммах (рис. 1).
Положение тяжелых атомов в структурах ванадиевых бронз при гидролизе
почти не меняется, поскольку рентгенограммы кристаллогидратов и исходных
соединений близки.
Анализ ИК-спектров исследуемых осадков (рис. 2) показал наличие интен-
сивных полос поглощения в диапазонах 800—700 и 1000—900 см-1, соот-
ветствующих валентным колебаниям V — 0, которые наблюдаются у исход-
ных структур ванадиевых бронз M2V50i3,3 [1]. Полосы с максимумами
при 980—960 см-1 характеризуют валентные колебания по коротким кова-
лентным связям V=O, а длинноволновые полосы — по длинным V — О.
В областях 930, 1630 и 3600 см~г наблюдаются незначительные по интен-
сивности полосы, обусловленные деформационными колебаниями гидроксиль-
ных групп и молекул воды и валентными колебаниями ОН„-групп.
Согласно данным Уодсли [7], атомы ванадия в структурах М^О^.э
обладают двойным типом координации, т. е. имеются атомы ванадия в кисло-
родном окружении из шести (искаженные октаэдры) и пяти (тригональные
- бипирамиды) атомов. Эти группировки, объединенные ребрами, образуют
зигзагообразные цепи. Октаэдрические и бипирамидальные цепи соединены
- общими углами в слои, в пустотах между которыми расположены ионы
^щелочного металла. Хотя локализация проводящих электронов точно неиз-
;. вестна, различный объем пирамид позволяет предположить, что меньшие по
31
объему пирамиды содержат атомы V4+. Пустотами и слабой 'связью щелоч-
ного металла с ванадийкислородным каркасом объясняется, по-видимому,
значительная подвижность катионов, которые, переходя при гидролизе из
поверхностных слоев кристалла в раствор, создают условия для перемещения
ионов из внутренних слоев. С другой стороны Пх-^г,т
3600 3400 1800 14001200 1000 800
Частота, см''
Рис. 2. ИК-спектры кристаллогидратов ванадиевых
бронз М2_ vVBOJ3)3-nH2O.
М: 1 - Li; 2 - Na; S - Ki - Ы,УБО
Спектры ЯМР (резонанс на протонах)
, размеры межслоевых пустот
позволяют диффундировать
внутрь структуры молекулам
воды с образованием различ-
ных связей. Это подтверж-
дается, в частности, тем,
что образование кристалло-
гидратов в момент подкис-
ления ванадиевых бронз
происходит с большой ско-
ростью. Ионы лития, обла-
дая меньшим по сравнению
с атомами натрия и калия
радиусом, могут, вероятно,
легко и до значительных
степеней замещаться кисло-
родно-водородными группи-
ровками.
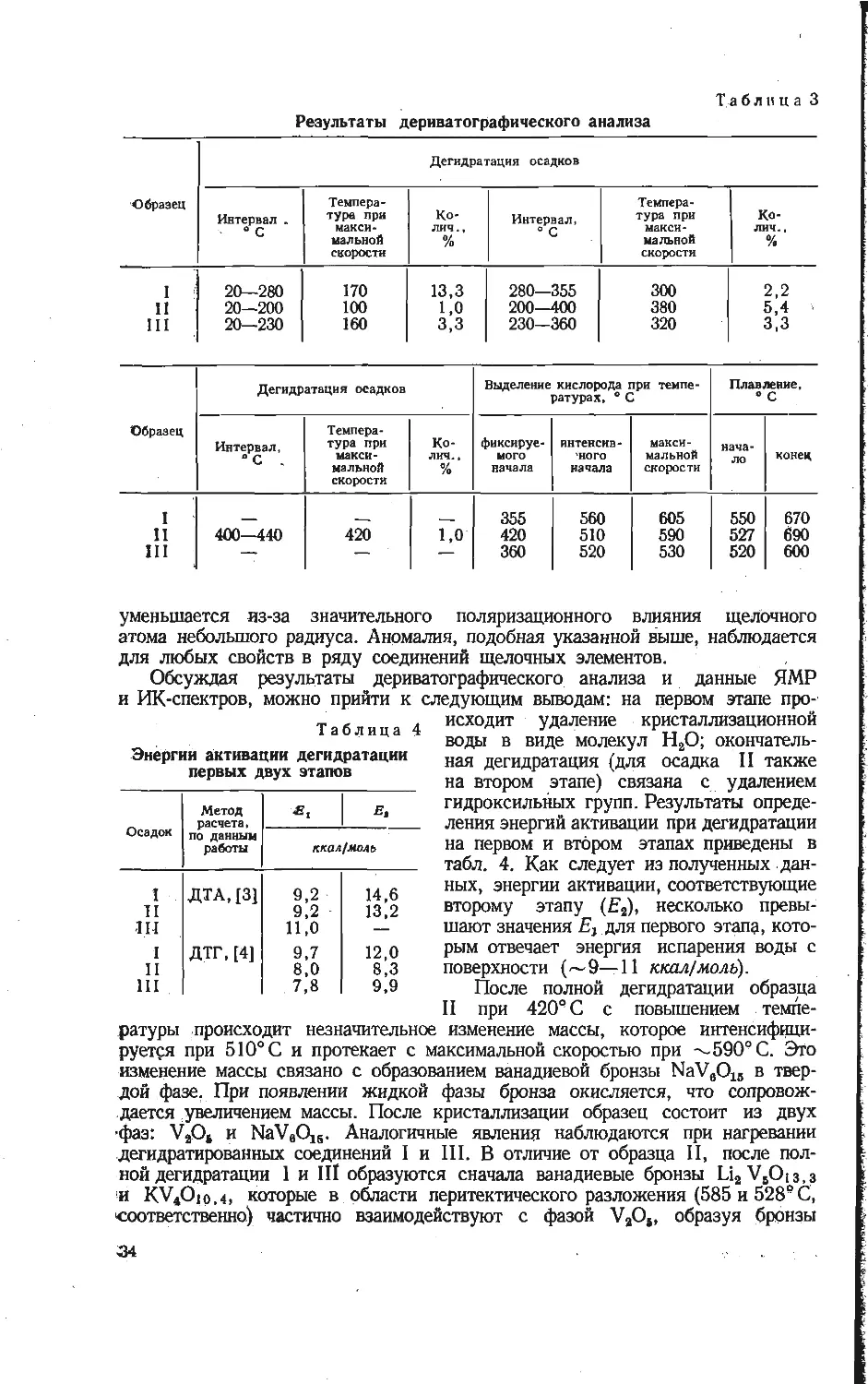
осадка I при —196° С заметно
отличаются в разных полях (рис. 3): при уменьшении поля относительная
интенсивность центральной компоненты возрастает. Аналогичный эффект, но
менее значительный, наблюдается и для гидратов II и III. Это обстоятель-
ство следует объяснить парамагнетизмом ионов V4+, присутствующих
в исследуемых соединениях. Наличие электронного парамагнетизма должно
уширить линию ЯМР и привести к асим-
метрии и сдвигу максимума поглощения
в сторону сильного поля [8]. Уширение,
асимметрия и сдвиг зависят от концент-
рации парамагнитных центров, поскольку
чем выше концентрация, тем большее
число ядер находится в поле парамагнит-
ных ионов. В нашем случае указанные
эффекты незначительны, так как содер-
жание ионов V4+ не очень велико (мень-
ше 4%).
Если парамагнитное вещество поме-
щено во внешнее постоянное магнитное
поле величины Но, то второй момент
А№=А/7о+(^о)2 [9], где k — коэффициент, определяемый величиной эффек-
тивного момента парамагнитного иона и расстоянием ядро — парамагнитный
центр; А//о — не зависящая от поля часть второго момента, обусловленная
лишь ядерным диполь-дипольным взаимодействием.
В табл. 2 приведены значения А//о Для различных величин внешнего
поля и значения A/7q (экстраполяция к /7о=О). Рассмотрение формы резо-
нансных кривых и значений вторых моментов АЯо позволяет сделать заклю-
чение о том, что не все протоны входят в состав молекул кристаллизацион-
Таблица 2
Результаты измерений ЯМР
при —196° С
Осадок Второй момент, э2
/7о=7500 э Н„ = 2654 э но=о (экстрапо- ляция)
I 21,2+1,0 18,9±0,9 18,5+1,2
II 17,8±0,9 17,4 + 0,9 17,2± 1,2
III 18,4±0,9 17,6±0,8 17,3±1,2
АН2
линии ЯМР будет равен:
ной воды. Если бы это было не так, то величины АЯо составляли бы
—24 э2 [10]. Кроме того, наблюдаемые значения вторых моментов намного
превосходят величины А//о (1—3 э2) для протонов в ОН-группах [10, 11]-
32
Рис. 3. Спектры ЯМР кристаллогидратов ванади-
евых бронз:
а — г при —196° С: а — лития (Яо=2654 э), б — ли-
тия (Яо=7500 з), в — натрия 7500 э), г — калия
(Яо=7500 5); д — лития (Но— 2654 а) при комнатной
температуре.
Следовательно, во всех рассматриваемых соединениях часть протонов входит
в состав молекул Н2О*, часть — в ОН-группы**. Результирующий второй
момент равен [12]:
ДЙ2о=пД/7о2‘+(1 — п)ЬН20*',
где п — доля протонов в молекулах кристаллизационной воды. Из вида резо-
нансных линий следует, что доля протонов в гидроксильных группах меняется
от образца к образцу (узкие центральные пики дают протоны в ОН-группах,
широкие линии обусловлены
молекулами кристаллизацион-
ной воды). Например, отно-
сительная интенсивность узкой
компоненты в спектре гидрата
литий-ванадиевой бронзы боль-
ше интенсивности аналогичной
компоненты в спектре калий-
ванадиевой бронзы. Отсюда
величина Л/7б для осадка I
должна быть меньше, чем для
осадка III. На опыте подобной
зависимости не наблюдается.
Это объясняется более вы-
сокой концентрацией моле-
кул воды в кристаллогидрате
I, что увеличивает межмолеку-
лярную часть Д/Уо* и, следова-
тельно, второй момент А До-
Рассматривая спектры
ЯМР, полученные при раз-
личных значениях поля Но,
можно прийти к выводу, что
гидроксильные группы значи-
тельно ближе расположены к
ионам У4+, чем молекулы воды. Увеличение Но заметно уширяет цент-
ральный пик и почти не изменяет широкую компоненту (см. рис. 3 а, б).
Это происходит потому, что протоны в ОН-группах сильнее «чувствуют»
локальные поля, создаваемые парамагнитными центрами и пропорциональ-
ные Но. При комнатной температуре молекулы воды переходят
в подвижное состояние, что вызывает сильное сужение линий ЯМР (см.
рис. 3 д). Столь высокая подвижность, по-видимому, может быть обуслов-
лена диффузией молекул Н2О по межслоевым пустотам в структурах рас-
сматриваемых соединений.
Данные ЯМР не противоречат результатам термического анализа осадков
I—III (рис. 4). Выделение слабосвязанной кристаллизационной воды проис-
ходит в интервале 20—280° С с максимальной скоростью в области 100—
170° С (табл. 3). Если из кристаллогидрата I в этом интервале выделяется
85,8% Н2О от всего содержания воды, то из осадков II и III 13,5 и 50%,
соответственно. Дегидратация образцов I и III протекает в два этапа, образ-
ца II — в три. Максимальная скорость дегидратации осадка II на третьем
этапе соответствует температуре ~420°С. Это указывает на прочную связь
кислородно-водородных группировок (вероятнее всего, ОН-групп) с решеткой
соединения. Аномальное поведение гидрата натриевой бронзы, по-видимому,
следует объяснить высокой энергией кристаллической решетки, которая тем
больше, чем меньше радиус катиона. В случае образца I энергия решетки
3
Заказ № 632
33
Результаты дериватографического анализа
Та блица 3
Образец Дегидратация осадков
Интервал . ° С Темпера- тура при макси- мальной скорости Ко- лич., % Интервал, ° с Темпера- тура при макси- мальной скорости Ко- лич. , %
I 20—280 170 13,3 280—355 300 2,2
II 20—200 100 1.0 200—400 380 5,4
III 20—230 160 з.з 230—360 320 3,3
Образец Дегидратация осадков Выделение кислорода при темпе- ратурах, ° С Плавление, ° с
Интервал, °C . Темпера- тура при макси- мальной скорости Ко- лич. , % фиксируе- мого начала интенсив- 'НО го начала макси- мальной скорости нача- ло конец
I 355 560 605 550 670
11 400—440 420 1,0 420 510 590 527 690
III — — — 360 520 530 520 600
уменьшается из-за значительного поляризационного влияния щелочного
атома небольшого радиуса. Аномалия, подобная указанной выше, наблюдается
для любых свойств в ряду соединений щелочных элементов.
Обсуждая результаты дериватографического анализа и данные ЯМР
и ИК-спектров, можно прийти к следующим выводам: на первом этапе про-
Таблица 4
Энергии активации дегидратации
первых двух этапов
Осадок Метод расчета, Е,
по данным работы ккал/моль
I ДТА.131 9,2 14,6
II 9,2 13,2
III 11,0 —
I ДТГ, [4] 9,7 12,0
II 8,0 8,3
III 7,8 9,9
исходит удаление кристаллизационной
воды в виде молекул Н2О; окончатель-
ная дегидратация (для осадка II также
на втором этапе) связана с удалением
гидроксильных групп. Результаты опреде-
ления энергий активации при дегидратации
на первом и втором этапах приведены в
табл. 4. Как следует из полученных дан-
ных, энергии активации, соответствующие
второму этапу (Е2), несколько превы-
шают значения Ег для первого этапа, кото-
рым отвечает энергия испарения воды с
поверхности (~9—11 ккал!моль).
После полной дегидратации образца
II при 420° С с повышением темпе-
происходит незначительное изменение массы, которое интенсифици-
--------- 590° С. Эго
ратуры
руется при 510° С и протекает с максимальной скоростью при
изменение массы связано с образованием ванадиевой бронзы NaVeOle в твер-
дой фазе. При появлении жидкой фазы бронза окисляется, что сопровож-
дается увеличением массы. После кристаллизации образец состоит из двух
•фаз: VjOg и NaVeOls. Аналогичные явления наблюдаются при нагревании
дегидратированных соединений I и III. В отличие от образца II, после пол-
ной дегидратации 1 и III образуются сначала ванадиевые бронзы Li2V60i3,3
и KV4Oio.4) которые в области перитектического разложения (585 и 528°С,
соответственно) частично взаимодействуют с фазой VaO4, образуя бронзы
34
типа MVeOls (интенсивное изменение массы). В результате быстрой кристал-
лизации расплава образцы содержат три неравновесные фазовые составля-
ющие: V2O5, Li2VBOi3,3; LiVeO16 и V2O5; KV4Oi0,4; KVeO15.
Рис. 4. Дериватограммы кристаллогидратов ванадиевых бронз:
1 — лития, 2 — натрия, 3 — калия.
ВЫВОДЫ
1. Методами химического и термического анализов установлено, что при
кислотной обработке бронз типа М2У6О1з,з происходит гидролиз и обра-
зуются кристаллогидраты.
2. Методами ИКС и рентгенофазового анализа показано, что кристалло-
гидраты имеют решетку, аналогичную M2V50i3.3. Молекулы воды, входя-
щие в состав гидрата, расположены в межслоевых пустотах решетки. Как
следует из линий ЯМР и ИК-спектров, в структурах гидратов момимо моле-
кул кристаллизационной воды присутствуют гидроксильные группы.
3. Методами дериватографии, рентгенофазового анализа и ЯМР установ-
лено, что удаление воды происходит ступенчато. После полной дегидратации
с повышением температуры образцы состоят из нескольких фаз — ванадиевых
бронз различных типов и пятиокиси ванадия.
ЛИТЕРАТУРА
1. А. А. Фотиев, С. И. Алямовский, М. П. Глазырин, Н. В. Ба-
усова. Химия соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН
СССР, 1967, вып. 14, стр. 29.
2. F. Paulik, J. Paulik, L. Erdey. Z. Analyt. Chem.1, 1958, 160, 241.
3. Г. О. Пилоян, О. С. Новикова. Ж. неорг. хим., 1967, 12, 602.
4. А. А. Фотиев, В. В. Мочалов. Ж. неорг. хим, 1968, 13, 3174.
5. В. Л. Золотавин, Л. К. Толстов, А. А. Ивакин, А. А. Фотиев.
Изв. АН СССР, Неорганические материалы, 1967 , 3, 1601.
3*
35
6. В. Л- Золотавин, Л. К. Толстов, А. А. Фотиев, Ю. Ф. Букреев.
Докл. АН СССР, 1967, 175, 595.
7. A. D. Wadsley. Acta cryst., 1957, 10, 261.
8. N. Bloembergen. Physica, 1950, 16, 95.
9. S. Yano, S. Sasaki. J. Phys. Soc. Japan, 1958 , 2, 227.
10. I. Ebert, G. Seifert. Kernresonans im Festkorper. Leipzig, 1966, S. 216.
11. A. L. Porte, H. S. Gutowsky, J. E. Boggs. J. Chem. Phys., 1962, 36,
1965.
12. С. П. Габуда, Ю. В. Гагаринский, С. А. Дурасова, А. Г. Лун-
дин. Ж. структ. хим., 1964, 5, 303.
АКАДЕМИЯ НАУК СССР
уральский филиал
®ЫП. 20
труды института химии
1970
А. А. ФОТИЕВ, Е. Д. СТОЛЕР,
Р. Н. ПЛЕТНЕВ, Ю. М. БЕЛЯКОВ
ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС В ЩЕЛОЧНЫХ
ВАНАДИЕВЫХ БРОНЗАХ
Состав и структура щелочных ванадиевых бронз типа MVeO15 хорошо
известны [1—4], вместе с тем их электронное строение изучено мало. До
сих пор вызывает споры вопрос о природе парамагнитных центров, представ-
ляющий интерес для понимания ряда физических свойств.
Неспаренные электроны в ванадиевых бронзах могут располагаться вблизи
атомов ванадия (с образованием ионов V4+) или щелочного металла или
могут делокализоваться в зоне проводимости, образующейся за счет перекры-
тия ЗсЬорбиталей атомов ванадия. Проведенное изучение бронз состава
LiV6O15 и NaVeO15 методами магнитного резонанса показало [5], Что цент-
рами локализации электронов являются атомы ванадия. Представляет интерес
исследование других типов ванадиевых бронз. Это, в частности, позволило
бы установить влияние состава и структуры бронз на природу парамагнитных
центров.
В настоящем сообщении приведены результаты изучения спектров ЭПР
щелочных ванадиевых бронз различного состава: MVeO15, М2У5О1з.з(М=1л,
Na, К); МУ4ОН ,4(М=К, Rb), гидратов M2_xV5Oi3,3(M=Li, Na, К). Образ-
цы бронз получены путем сплавления на воздухе стехиометрических количеств
пятиокиси ванадия (о. ч.) и соли М2СО3 (х. ч.). Контроль фазовой чистоты
осуществляли рентгенографическим и микроскопическим методами. Получение
гидратов бронз описано в работе [6]. Спектры ЭПР записывали на спектро-
метре РЭ-1301 при комнатной температуре. Наблюдаемую ширину линии
А//?ксп определяли как расстояние между максимумом и минимумом произ-
водной. Истинную ширину АДИСТ и фактор анизотропии 6 оценивали спосо-
бом [7], используемым при анализе асимметричных линий в поликристаллах.
Небольшая асимметрия большинства резонансных кривых (б < 2) позволила
произвести оценку частоты обмена ve с дипольной шириной линии АНДИП [8].
Результаты измерений ЭПР приведены в таблице.
Полученные спектры соединений представляют собой асимметричные син-
глеты с центром резонанса при g 1,96. Наблюдаемые величины отвечают
значениям ^-фактора для ионов V4+ в других соединениях [9, 10], в том
числе в бронзах LiVeO16 и NaV6O15 [5]. Это обстоятельство, а также малость
ширины Зй-зоны (проводимость) позволяют предполагать, что в рассматрива-
емых объектах неспаренные электроны (как и в случае бронз LiV8O15
и NaVeO16) локализуются в основном вблизи атомов ванадия с образованием
У4+-центров. Для объяснения экспериментальных значений удельной электро-
проводимости необходимо допустить некоторую подвижность электронов [5L
Сравнением интегральных интенсивностей спектров бронз различных соста-
вов и MfiSO4-H2O была произведена оценка числа спинов, дающих вклад
37
Результаты ЭПР ванадиевых бронз щелочных элементов
Вещество А^ЭКСП’ э дяист. э & . £ мгц Д"дип- » N
LiV.O13 145 60 2,0 1,96 49 180 0,6
NaV.O13 225 135 1,0 1,96 60 250 0,9
KV,O15 170 60 2,5 1,97 40 180 0,8
90 24 3,0 1,96 .—• .— 0,7
125 75 1,0 1,96 55 180 0,6
30 11 7,5 1,97 —- •— 0,8
70 22 4,0 1,97 —• — 0,3
RbV4O1014 95 41 2,5 1,97 — — 0,6
105 38 2,5 1,96 40 140 0,1
140 45 2,5 1,96 45 190 0,2
К2-Л^13>83’8пН2О 80 25 4,0 1,97 — — 0,2
Rbg—б^1з»з' яНгО 50 16 6,0 1,97 — — 0,4
в резонансе. В таблице приведены числа неспаренных электронов на одну
формульную единицу N. Сопоставляя значения N для бронз различных
составов, можно заметить, что для бронз с отношением М:У > 1:6, по-ви-
димому, не реализуется описанная в [5] модель, в которой атомы щелочного
Кристаллическая структура V2O3 (1), MVeOls (2) и M2V5O33
(3) по данным [2].
металла полностью ионизированы и дают электроны и ионы щелочного
металла (один атом щелочного металла дает один У4+-центр). Так как кон-
центрация парамагнитных центров несколько уменьшается при переходе
к бронзам с большим содержанием щелочного металла, то можно предполо-
жить, что неспаренные электроны локализованы в основном на атомах вана-
дия. С другой стороны, не исключается участие в создании У4+-центров
дефектности по кислороду.
Непосредственно из формы резонансных линий, а также из значений
фактора анизотропии 6 следует, что симметрия окружения парамагнитных
центров (V4+) зависит от размеров и количества атомов щелочного металла.
Матрицей для структуры ванадиевых бронз являются тригональные бипира-
миды пятиокиси ванадия. Атомы щелочного металла, внедренные в пустоты
кристаллической структуры, искажают формирующий ее кислородный каркас
и понижают симметрию окружения ионов ванадия (см. рисунок). Изменение
симметрии тем значительнее, чем больше размеры щелочных атомов. Однако
если считать, что степень искажения кристаллической структуры связана
лишь с размерами щелочных атомов, то у натриевых бронз фактор анизо-
тропии должен быть больше, чем у литиевых; на самом деле, как показы-
вает опыт, асймметрия линии ЭПР натриевых бронз типа MVeOlg и
MgVgOi3.3 незначительна. Видимо, необходимо учитывать еще фактор поля-
ризации катиона, влияющий на структуру и физико-химические свойства
бронз. Увеличение концентрации щелочного металла в бронзах M2V5Oi3,3 по
сравнению с MVeOls еще больше понижает симметрию окружения ионов
четырехвалентного ванадия, о чем свидетельствует рост фактора анизотропии.
Для бронз щелочных металлов состава M8V50i3,3 наблюдаемая ширина
линии заметно меньше, чем для бронз MVeO15, легированных теми же ато-
мами. Если считать, что величины обменного взаимодействия между пара-
магнитными центрами в обоих типах бронз примерно одинаковы, то умень-
шение наблюдаемой ширины линии в бронзах с большей концентрацией
щелочного металла указывает еще раз на то, что основную роль в создании
парамагнитных центров играют электроны, локализованные около атомов
ванадия.
Обменные взаимодействия между парамагнитными ионами, возникающие
благодаря ковалентному характеру связей, приводят к сужению резонансных
кривых и близости их формы к функции распределения Лоренца. Значитель-
ным обменным взаимодействием объясняется и отсутствие сверхтонкой струк-
туры. Спектры, обладающие СТС, можно получить при магнитном разбавле-
нии образцов. У гидратов бронз обменное сужение линии менее заметно,
так как наличие в кристаллической структуре молекул воды ослабляет
обменное взаимодействие между ионами четырехвалентного ванадия.
ЛИТЕРАТУРА
1. Р. П. Озеров. Усп. хим., 1955 , 8, 951.
2. A. D. Wadsley. Acta cryst., 1955, 8, 695; 1957, 10, 261.
3. 1. Galy, M. Pouchard, A. Casalot, P. Hagenmuller. Bull. Soc. Mine-
ral. Crist., 1967, 90, 544.
4. А. А. Фотиев, С. И. Алямовский, M. П. Глазырин, H. В. Bayco-
ва. Химия соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН СССР,
1967, 14, стр. 29.
5. J. Gen del, R. М. Cotts, М. Sienko. J. Chem. Phys., 1967, 37, 220.
6. А. А. Фотиев, P. H. Плетнев. Ст. в наст, сб., стр. 30.
7. Я. С. Лебедев. Ж. структ. хим., 1962, 3, 2.
8. Л. А. Блюменфельд, В. В. Воеводский, А. Г. Семенов. Применения
ЭПР в химии. Новосибирск, Изд-во СО АН СССР, 1962.
9. К. D. Bowers, J. Owen. Repts. Progr. Phys., 1955, 18, 335.
10. C. A. Hutchinson, L. S. Singer. Phys. Pev., 1953, 89, 256.
АКАДЕМИЯ НАУК СССР
уральский филиал
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
Л. К. ТОЛСТОВ, А. А. ИВАКИН, А. А. ФОТИЕВ
ИЗУЧЕНИЕ ПРОДУКТОВ ГИДРОЛИТИЧЕСКОГО ОСАЖДЕНИЯ
ВАНАДИЯ (V) В СИСТЕМЕ LiVO3 — HNO3 — Н20
Настоящая работа является продолжением серии исследований, посвящен-
ных изучению природы и свойств продуктов гидролитического осаждения
ванадия (V). Ранее нами описаны особенности термического поведения гидра-
тированных осадков, полученных подкислением растворов метаванадатов
натрия и калия [1—3]. В данном сообщении приведены результаты исследо-
вания системы LiVO3 — HNO3 — Н2О.
Экспериментальная часть
Гидратированные осадки ванадия (V) получали при комнатной темпера-
туре подкислением азотной кислотой растворов метаванадата лития. Было
проведено три серии осаждений. Начальная концентрация ванадия в раство-
рах первой, второй и третьей серий составляла, соответственно, 0,1915;
0,0383 и 0,0236 г-атом)л. Степень подкисления характеризовали величиной
отношения H+/VO7 и изменяли в каждой серии в пределах 0,7—2,1. По
достижении состояния равновесия осадки отфильтровывали, промывали охлаж-
денной и подкисленной до соответствующего .pH дистиллированной водой,
высушивали до воздушно-сухого состояния и анализировали. Осадок 9 гото-
вили следующим образом. Пятиокись ванадия квалификации «х. ч.» плавили
в платиновой чашке и тонкой струей при перемешивании вливали в холод-
ную воду (см. таблицу). После некоторого отстаивания содержимое отфиль-
тровывали для отделения от нерастворившегося осадка. Полученные таким
путем коллоидные растворы темно-малинового цвета содержали до 17 г/л
V2O5 и имели кислую реакцию (pH=2,15). Растворы выпаривали до получения
слизистой массы. Последнюю высушивали при температуре 150° С до сухого
состояния и измельчали.
Ванадий определяли объемным методом (индикатор — фенипантраниловая
кислота) [4], литий — эмиссионной фотометрией пламени на установке, опи-
санной ранее [5]. В качестве аналитической была избрана линия 670,8 нм,.
в связи с этим фотоумножитель ФЭУ-19м был заменен на ФЭУ-221. Терми-
ческий анализ проводили на дериватографе системы Ф. Паулика, И. Паулика
и Эрдеи [6], позволявшем из одной навески образца одновременно фиксиро-
вать: температуру (Т), изменение веса (ТГ), скорость изменения веса (ДТГ)
и тепловые эффекты (ДТА). Рентгенограммы препаратов получали на уста-
новке УРС-55 в порошковых камерах ВРС-3 на СгКа-излучении. Режим
съемки: £7=35 кв, Г—14 ма, экспозиция 9 ч. Для идентификации фазовых.
1 Методика определения лития разработана Ю. Ф. Букреевым.
40
Условия осаждения и химический состав воздушносухих осадков
ванадия (V)
Серия № осадка Степень подкисле- ния рас- твора, H+/VO? Равновес- ная вели- чина pH растворов Содержание в осадках, % Молярное отношение LifO : VSO6 : HtO
ЫЕО V,os нао
I 1 0,703 3,54 3,48 81,52 14,86 0,259:1:1,843
2 0,903 2,18 2,34 81,43 15,31 0,175:1:1,899
3 1,104 1,50 2,15 83,32 14,78 0,157:1:1,792
4 1,305 1,23 1,16 84,52 14,70 0,083:1:1,757
5 1,505 1,06 0,71 85,48 14,68 0,051:1:1,736
6 1,706 0,92 0,77 83,91 14,83 0,056:1:1,787
7 1,907 0,83 0,52 84,98 14,66 0,037:1:1,743
8 2,108 0,74 0,06 84,95 14,98 0,004:1:1,782
II 1 0,703 2,67 Осадка нет
2 0,903 2,22 Следы
3 1,104 2,17 / 1,78 82,45 15,77 0,132:1:1,933
4 1,305 1,97 1,33 83,63 15,04 0,097:1:1,817
5 1,505 1,78 1,16 83,57 15,27 0,085:1:1,847
6 1,706 1.68 1,12 84,38 14,50 0,081:1:1,737
7 1,907 1,58 0,77 85,57 13,66 0,055:1:1,614
8 2,108 1,49 0,45 85,20 14,35 0,032:1:1,702
III 1 0,693 2,98 Осадка нет
2 0,897 2,43 То же
3 1,100 2,21 Следы
4 1,305 2,14 1,46 84,20 14,34 0,106:1:1,721
5 1,507 2,02 1,14 84,59 14,27 0,082:1:1,705
6 1,711 1,90 1,16 86,32 12,52 0,082:1:1,465
7 1,892 1,80 ' 0,88 84,52 14,60 0,063:1:1,744
8 2,100 1,73 0,77 84,88 14,35 0,055:1:1,707
Контроль 9 — 2,15 — 87,31 12,69 — 1:1,470
•составляющих были сняты также рентгенограммы специально синтезирован-
ных индивидуальных соединений, образующихся в системе V2O5— LiVO3
в концентрационной области до ~50 мол. % Li2O [7]. Интенсивность линий
оценивали визуально по пятибалльной системе. Помимо этого, проведен
микроскопический фазовый анализ как воздушносухих, так и прокаленных
при различных температурах осадков2.
С возрастанием кислотности среды содержание лития в осадках непре-
рывно уменьшается, а общее количество воды примерно постоянное (см. таб-
лицу).
Образец 8 первой серии, полученный в сильнокислой среде, почти не
содержит лития. Микроскопическое исследование осадков показало нали-
чие зерен, не имеющих четко выраженного кристаллического состояния
и обладающих слабой поляризацией. В параллельных николях зерна непра-
вильной формы, с окраской от буровато-желтой (в тонких сечениях) до
темно-бурой (в более крупных частицах). Рентгенограммы воздушносухих
осадков 1—8 первой серии и продукта 9 идентичны (рис. 1). Относительно
небольшое число размытых диффузных линий на порошкограммах свидетель-
ствует о несовершенной мелкокристаллической структуре.
Полученные дериватограммы по внешнему виду можно разделить на три
основных типа. Для осадков 2, 3 характерна дериватограмма, представленная
на рис. 2, а. На ней зафиксированы два частично перекрывающихся эндотер-
а Принимал участие М. П. Глазырин.
41
вода удалялась с энергией активации
Рис.
а, б,
в.
1. Штрихдиаграммы воздушно-
сухих образцов.
г — соответственно, осадки 1, 5,
8, 9.
мических эффекта (кривая ДТА) с экстремумами при 141 и 252° С, сопро-
вождающиеся интенсивной убылью веса (кривая ТГ) и отвечающие, вероятно,
удалению сорбированной и кристаллизационной воды. Выделение последней,
судя по кривой ДТГ, заканчивается при ~285°С. Расчет энергии активации
процесса дегидратации образцов По кривым ДТГ и Т (в координатах
lg V — 1 /Т, где V — скорость процесса) показал следующее. Для осадков 2
и 3 в интервале температур от комнатной до 115—119° С сорбированная
£=10—11 ккал/моль, а кристаллиза-
с Е=8—10 ккал/моль. Полученные
величины энергии активации хорошо
согласуются с температурной зависи-
мостью значений скрытой теплоты паро-
образования воды (Q) [8]. Это позво-
ляет рассматривать выделение сорби-
рованной и кристаллизационной воды
с точки зрения процесса испарения с
постоянной поверхности (в этих усло-
виях E=Q), а кинетика дегидратации
подчиняется уравнению нулевой ско-
рости (V=const). В интервале темпера-
туры ~285—324° С наблюдается рез-
кое уменьшение веса образца (кри-
вая ТГ), связанное с окончательным обезвоживанием, которое протекает со зна-
чительной скоростью (максимальная скорость на кривой ДТГ отвечает 294° С).
Рентгенографическое и микроскопическое исследование осадков, прокален-
ных при температурах ниже и выше 294° С, показало, что при удалении
сорбированной и кристаллизационной воды структура осадков не нарушается,
а отщепление последней, наиболее прочно связанной воды, сопровождается
перестройкой кристаллической решетки образцов. Это сопровождается выде-
лением тепла, но так как в нашем случае этот процесс и дегидратация
осадков протекают одновременно, то взаимообратные тепловые эффекты почти
компенсируют друг друга и на кривой ДТА для осадка 3 заметен лишь
небольшой экзотермический эффект с максимумом при 314° С. Таким обра-
зом, наиболее прочно связанная последняя вода определяет структуру гидра-
тированных осадков, и ее следует считать конституционной. Эффекты на
дериватограммах, зафиксированные выше температуры окончательного обезво-
живания, обсуждаются ниже.
Дериватограмма образца 6 (см. рис. 2, в) отвечает второму типу и отли-
чается от таковой образца 3 формой и положением эндотермических эффек-
тов, соответствующих удалению сорбированной и кристаллизационной воды,,
а также ясно выраженным экзотермическим эффектом с максимумом при
температуре 312° С, который на термограмме осадка 3 только намечается.
Кроме того, на кривой ДТГ зафиксированы два пика — при 280 и 299° С,,
соответствующие максимальной скорости удаления конституционной воды.
Дериватограмму образца 4 (см. рис. 2, б) можно рассматривать как резуль-
тат совмещения дериватограмм осадков 3 и 6.
И, наконец, осадку 8, почти не содержащему лития, соответствует тре-
тий тип дериватограмм. Его дериватограмма (см. рис. 2, б) почти не отличается
от дериватограммы осадка 6; однако на ней ясно выражен эндотермический
эффект, отвечающий удалению конституционной воды. Максимальная ско-
рость выделения этого типа воды наблюдается при более низкой температуре,
чем в случае осадков 2—6, содержащих литий, а именно — при 270° С.
Дериватограмма образца 9 (см. рис. 2, е) почти соответствует таковой для
осадка 8. На ней хорошо разрешен эндотермический эффект с минимумом
при 280° С, соответствующий удалению конституционной воды. Однако выде-
42
ление воды, связанной менее прочно (сорбированной и кристаллизационной),
протекает практически в одну стадию и общее содержание ее существенно
меньше, чем в образцах 2—8. Это и понятно, так как указанный осадок
получен высушиванием при температуре ~150°С. Кроме того, экзотермиче-
ский эффект, соответствующий структурной перестройке осадка 9, несколько
сдвинут в сторону более высоких температур.
Дериватограмма осадка 7 (см. рис. 2, г) — промежуточная между таковыми
для образцов 6 и 8. Это следует, например, из сравнения кривых ДТГ.
На них отчетливо видны пики, соответствующие максимальной скорости вы-
деления конституционной воды при температурах ~270 и 299° С. При этом
вместе с уменьшением содержания лития вполне закономерно величина пер-
вого пика возрастает, а второго — уменьшается,
Как видно, осадки, образующиеся при подкислении растворов метавана-
дата лития, несмотря на различное содержание в них лития и судя по
данным рентгенографических и дериватографических исследований, имеют,
по-видимому, одну и ту же структуру и близкие термические свойства.
Отличие в термических свойствах заключается лишь в том, что максимумы
скорости удаления кристаллизационной и конституционной воды из осадков
с повышенным содержанием лития (осадки 2—4) расположены в области
более высоких температур. Аналогичной структурой и термическими свой-
ствами обладает гидратированная пятиокись ванадия (осадок 9). Она, однако,
содержит по сравнению с наиболее близким к ней по составу осадком 8
относительно много конституционной воды. Соотношение Н2Оконст: V2O5 в гидра-
тированной пятиокиси ванадия равно 0,337, а в осадке 8—0,224 моль/моль.
Это отличие, по-видимому, связано с разными условиями получения рассма-
триваемых препаратов. Если содержание лития может колебаться в широких
пределах и это почти не сказывается на свойствах осадков, то конституци-
онная вода играет весьма важную роль — ее удаление приводит к необрати-
мой перестройке структуры осадков.
В результате кристаллохимической перестройки решетки обезвоженных
осадков образуются новые фазы, состав и количество которых зависят от
содержания лития в осадках. Так, в осадке 8 (Li20=0,06%) примерно при
290° С, согласно рентгенографическому и микроскопическому анализам, обна-
руживается, в основном, фаза пятиокиси ванадия, сохраняющаяся и при
более высоких температурах. Плавление этого образца по данным деривато-
граммы (см. рис. 2, д) начинается при 670° С и близко к температуре плав-
ления пятиокиси ванадия (673° С). Перестройка структуры осадков 1—7
(содержание Li2O=3,48—0,52%) сопровождается образованием нескольких
фаз.
Так, например, при нагревании осадка 2 приблизительно при 290° С,
судя по рентгенографическим данным, появляются три фазы: литий-ванади-
евые бронзы LiVeO15 и Li2V5Oi3,5_x [9] и пятиокись ванадия. При дальней-
шем повышении температуры прокаливания соотношение фазовых составля-
ющих, согласно изменению интенсивности на рентгенограммах, меняется:
количество LiVeO15 уменьшается, a Li2VBOi3,5_x и V2O6 увеличивается.
По-видимому, метастабильная фаза LiVeO16 распадается согласно следу-
ющему уравнению
2LiVeO16 4-1=^ O2^3,5V2O6+Li2V60,3,5-х. (1)
В момент появления эвтектической жидкости фазы реакция (1) протекает
справа налево, что согласуется с диаграммой состояния V2O5 — LiVO3 [7].
Как видно, например, из дериватограммы осадка 3 (см. рис. 2, а), выделение
кислорода при образовании бронзы LiVeO1B (а соответственно, и уменьшение
43
t I I » I I I ( । t > ,t I I I I I t I i (
9
9
v
г
д
е
Рис. 2. Дериватограммы образцов.
а, б, в, е, д, е — соответственно, осадки 3, 4, 6, 7, 8 первой серии и препарат 9. Навеска пробы 1010 яг (осадок 9 — 620 яг).
Навеска эталона (А1,О,) 1200 яг (осадок 9 — 600 мг). Скорость нагрева печи 11 град/мин (осадок 9 — 9 град/мин). Чувствительность
гальванометров: ДТА — 1/10, ДТГ — 1/10, Т—900. Чувствительность весовой шкалы ТГ—200 яг/отклонение.
веса на кривой ТГ) происходит в интервале 593—627QC. При последующем
нагревании бронза LiVeO16 окисляется (увеличение веса на кривой ТГ)
с одновременным полным расплавлением системы
2LiVeOi5(T)+1/2O2(r)[1л2У12О31]ж. - (2)
При медленном охлаждении (—10—12 ч) расплавленных образцов 1—7
конечными продуктами являются V2OS и Li2V6O|3,5_x, а при достаточно
быстром — неравновесные фазовые составы, содержащие, помимо упомянутых,
также некоторое количество фазы LiVeO16. Это подтверждается рентгеногра-
фическим и микроскопическим исследованиями, причем оптические свойства
этих соединений и их рентгенограммы соответствуют описанным в литера-
туре [7, 9—11].
ВЫВОДЫ
1. Проведено исследование продуктов гидролитического осаждения вана-
дия в системе LiVO3 — HNO3—Н2О в интервале H+/VO? от 0,7 до 2,1.
2. На основании изучения состава и термических свойств осадков в зави-
симости от степени подкисления растворов установлено образование продук-
тов, близких по своей природе к гидратированной пятиокиси ванадия.
3. Отмечено присутствие во всех образцах конституционной воды, удале-
ние которой ведет к необратимой структурной перестройке осадков и сопро-
вождается образованием в них в зависимости от содержания лития различных
фазовых составляющих, присущих системе V2O6 — LiVO3 (в том числе
Li — V-бронз).
ЛИТЕРАТУРА
1. А. А. Ивакин. Физико-химические исследования соединений редких тугоплавких
элементов.—Тр. Ин-та хим. УФАН СССР, 1966, вып. 9, стр. 107.
2. В. Л. Золотавин, Л. К. Толстов, В. В. Мочалов, Ю. Ф. Букреев.
Журнал ВХО им. Д. И. Менделеева, 1967, 12, 216.
3. В. Л. Золотавин, Л. К. Толстов, А. А. Фотиев, Ю. X. Букреев.
Докл. АН СССР, 1967, 175, 595.
4. В. С, Сырокомский, В. В. Степин. Заводская лаборатория, 1936, 5 , 263.
5. В. Л. Золотавин, Ю. Ф. Букреев, Л. К. Толстов, И. Я, Безруков.
Ж. прикл. спектр., 1965 , 2, 461.
6. F. Paulik, I. Paul i k, L. Erdey. Z. Anal. Chem., 1958, 160, 241.
7. А. А. Фотиев, M. П. Глазырин, H. В. Баусова. Ж. неорг. хим., 1968,
13, 1936.
8. Краткий справочник химика. Под ред. Б. В. Некрасова. М., Госхимиздат, 1954,
стр. 310.
9. А. А. Фотиев, С. И. Алямовский, М. П. Глазырин, Н. В. Баусо-
ва. Химия соединений редких тугоплавких элементов (V, Nb, Та, Ti, Zr). Тр.
Ин-та хим. УФАН СССР, 1967, вып. 14, стр. 29.
10. М. П. Глазырин, А. А. Фотиев. Кристаллография, 1964, 9, 506.
11. A. Hardy, I. Galy, A. Casalot, М. Pouchard. Bull. Soc. Chim. France,
1964, № 11, 2808; 1965, № 4, 1056.
АКАДЕМИЯ НАУК.СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА ХИМИИ
1970
В. А. ПЕРЕЛЯЕВ, Г. П. ШВЕЙКИН
ВЗАИМНАЯ РАСТВОРИМОСТЬ НЕКОТОРЫХ ПРОМЕЖУТОЧНЫХ
ОКИСЛОВ ПЕРЕХОДНЫХ МЕТАЛЛОВ IVa И Va ПОДГРУПП
В последние годы тугоплавкие окислы переходных металлов IVa и Va
подгрупп периодической системы все больше применяются в производстве
новых технических материалов, к которым предъявляются жесткие требова-
ния. Практически в большинстве случаев, когда нужен материал, способный
одновременно выдерживать окислительную атмосферу и высокую температуру,
останавливаются на тугоплавких окислах. Для последних характерны высо-
кие температуры плавления, твердость, коррозионная стойкость, износоустой-
чивость, а также специфические термохимические, электрические и магнитные
свойства. Эти свойства тугоплавких окислов связаны с особенностями элек-
тронного строения переходных металлов и объясняются тем, что если
у простых металлов (например, натрий, олово и т. д.) в формировании
физико-химических свойств активное участие принимают только внешние
валентные электроны, то у переходных металлов существенную роль играют,
кроме валентных S-электронов, электроны незаполненных d-оболочек атома.
Такая’особенность электронного строения определяет высокий уровень меж-
атомных сил связи и высокую прочность . кристаллической решетки.
Кислород с электронной конфигурацией ls22s®2p4 резко отличается
по свойствам от металлов переходных групп и является одним из наиболее
электроотрицательных элементов. Так как электроотрицательность элемента —
мера способности его атомов принимать валентные электроны, то кислород
в большей степени склонен к захвату электронов и достройке р-оболочки
до электронной конфигурации неона. Переменная валентность кислорода,
а также возможность отдачи своих электронов при реакциях взаимодействия
открывают возможность образования различных кислородных соединений
и твердых растворов кислорода в металлах.
Для промежуточных окислов переходных металлов IVa и Va подгрупп
необходимо иметь в виду следующие особенности их кристаллического и
электронного строения:
1. Промежуточные окислы в основном реализуются в виде моноокисей
со структурой типа NaCl, полуторных окислов со структурой типа а-А12О3
и двуокисей со структурой искаженного рутила (TiO2).
2. Во всех этих структурах имеется октаэдрическое окружение атома
металла атомами кислорода, связанное, по-видимому, с тем что в атомах
переходных металлов IVa и Va подгрупп d-орбиты могут принимать макси-
мальное участие в образовании связи [1].
3. Согласно валентной схеме Рандля—Бильца [2—5], в результате обра-
зования в окислах Me—О-связей 2s- и 2р-оболочки неметаллических атомов
полностью заполнены.
47
4. Совокупность электрических и магнитных свойств указывает на то,
что в промежуточных окислах, в отличие от большинства окислов других
переходных металлов, наблюдаются сильные Me—Ме-обменные взаимодейст-
вия, обусловленные значительными перекрываниями d-орбит [6, 7].
5. Анализ структурно-дефектных свойств показывает, что в промежуточ-
ных окислах, особенно в моноокислах, имеется существенное анионное экра-
нирование Me—Ме-связей, это оказывает большое влияние на многие их
свойства [8, 9].
6. Рассматриваемые промежуточные окислы переменного состава и
в большинстве случаев представляют собой насыщенные растворы разнова-
лентных окислов с собственной структурой [10].
Для выяснения и уточнения этих особенностей промежуточных окислов
определенный интерес представляет вопрос о их взаимной растворимости.
Твердые растворы получают все большее значение в науке и технике,
поскольку при их образовании расширяется диапазон полезных свойств раз-
личных материалов, которые можно регулировать изменением состава и элек-
тронной концентрации, что особенно важно при создании, например, новых
полупроводников. Твердые растворы окислов играют существенную роль
и в качестве защитных пленок при окислении сплавов. В настоящее время
различают ряд эмпирических правил и критериев взаимной растворимости
соединений [11, 12], которые определяются изоморфизмом соединений; оди-
наковым типом химической связи; непрерывной взаимной растворимостью
металлов, входящих в рассматриваемое соединение; близостью размеров за-
мещающих атомов и т. д. Однако эти критерии нуждаются в уточнении.
Все это послужило причиной для рассмотрения вопросов, связанных с взаим-
ной растворимостью промежуточных окислов IVa и Va подгрупп.
Взаимная растворимость моноокисей
Титан и ванадий образуют кубические моноокислы со структурой NaCl.
Этим окислам ТЮХ и УСД присущи широкие области гомогенности
(0,85<х< 1,25) и статистическое распределение вакансий в обеих 'подре-
шетках (15—16%). Моноокись ниобия, в отличие от моноокисей титана
и ванадия, обладает особой кубической структурой, которая близка к NaCl.
Если в решетке типа NaCl существует октаэдрическое окружение атомов
металла атомами кислорода, то в решетке NbO — квадратичная координация
для каждого атома [13]. Кроме того, NbO отличается постоянным составом
и также обладает высокой дефектностью (~25%). .
В моноокисях титана, ванадия и ниобия заметно проявляется эффект
экранирования Me — Ме-связей, что приводит в условиях структуры NaCl
к образованию вакансии как в кислородной, так и металлической подрешет-
ках и частичному или полному переходу экранированных Me—Ме-связей
в неэкранированные [8]. В связи с этим, учитывая близкие структуроразмер-
ные факторы и сходную природу межчастичного взаимодействия, можно
было бы ожидать заметную взаимную растворимость всех моноокисей. Однако
согласно [13], растворимость ТЮ и NbO при 1600—1700° С оказалась не-
большой (<5 мол. %), тогда как VO и NbO обладают достаточно большей
взаимной растворимостью (NbO в VO около 17, VO в NbO около 30 мол.%).
При этом происходит формирование фаз переменного состава путем замеще-
ния металлических атомов одного сорта другим при сохранении типа решетки,
т. е. налицо образование твердых растворов замещения. Эти выводы хорошо
согласуются с данными [14], согласно которым стехиометрические ТЮ и VO
взаимно малорастворимы. Однако фазы с повышенным содержанием кислорода
(TiOi.ao и VOj.25) обладают заметной взаимной растворимостью (до 25%),
причем формирование подобных твердых растворов замещения со структурой
48
NaCl происходит при длительном спекании окислов. Природа образующихся
твердых растворов замещения тесно связана с особенностями электронного
строения TiO^ и VOX. Так как изменение их состава в пределах области
гомогенности сопровождается уменьшением комплектности одной из подре-
шеток и увеличением другой при сохранении средней плотности валентных
электронов, то это приводит к реализации в этих окислах насыщенных и
локализованных Me—О- и Me—Ме-связей [9, 14].
Следовательно, если образование твердого раствора связано с заметным
изменением средней плотности валентных электронов, как, например, для
системы ТЮ—NbO, VO, то в этом случае часть Me—Ме-связей окажется
ненасыщенной, а раствор термодинамически не устойчивым. Увеличение взаим-
ной растворимости для этих систем с ростом содержания кислорода можно
объяснить тем, что в этом случае электрические характеристики определяются
в основном Me—О-связями, в то время как Me—Me-связи имеют второстепенное
значение. Для объяснения достаточно большой взаимной растворимости VO и NbO
нужно учитывать, что замещающие друг друга атомы металла находятся в одной
подгруппе, т. е. у них одинаковое число внешних электронов. Это приводит
к тому, что образование твердого раствора не связано с заметным изменением
средней плотности валентных электронов. Но несмотря на заметную взаимную
растворимость этих окислов, она все же остается ограниченной из-за суще-
ственного различия перекрывания d-волновых функций атомов ванадия и ни-
обия [9, 14]. В работе [13] рассмотрен вопрос о возможности образования
твердого раствора между NbO и гипотетической окисью ТаО. Авторы пока-
зали, что в результате нагрева соответствующих смесей при 1700—2000° С
в вакууме во всех случаях образуется твердый раствор не типа (Nb, Та) О,
a (Nb, Та) О2 и металлическая фаза. Действительно, если бы окись ТаО
существовалах, то NbO и ТаО обладали бы достаточно большой взаимной
растворимостью по аналогии с NbO и VO.
Взаимная растворимость полуторных окислов
Полуторные окислы титана и ванадия (Ti2O3 и V2O3) кристаллизуются
в изоморфной структуре корунда (а-А12О3). Согласно данным [15], V2O3
обладает узкой областью гомогенности, тогда как Ti2O3 присуща заметная
концентрационная область [16, 17]. При рассмотрении. окислов Ti2O3nV2O3
необходимо учитывать не только Me—О-, но и достаточно сильные Me—Ме-
взаимодействия, так как в этих соединениях существует значительное пере-
крывание d-орбит [6, 18]. Кроме того, в структуре корунда экранирование
Me—Me-связи приводит к увеличению взаимного расстояния между атомами
кислорода на общей грани двух занятых октаэдров [7]. Это дает возможность
заключить, что эффект экранирования Ме-Ме-связи в этих окислах суще-
ственно меньше, чем в моноокислах со структурой NaCl. Поэтому в Ti2O3
и V2O3 не образуются дефекты, и они имеют устойчивую кристаллическую
структуру [9]. Сведения о взаимной растворимости Ti2Os и V2O3 ограничены.
На основании данных [19], при 1600° С в высоком вакууме в системе
Ti2O3 — V2O3 образуется непрерывный ряд твердых растворов, периоды реше-
ток которых плавно изменяются с составом. Из сопоставления рентгеногра-
фических, подсчитанных в предположении взаимного замещения Ti3+ и V3+,
и пикнометрических плотностей делается вывод, что в данном случае обра-
зуются твердые растворы замещения. Это находится в полном согласии
с данными работы [20], в которой авторы, наряду с определением электро-
проводности, изучали при 1450° С концентрационную зависимость параметров
гексагональной решетки твердых растворов системы Ti2O3—V2O3 с содержа-
нием V2O3 лишь до 10 мол. %.
1 Причины неустойчивости ТаО обсуждены в работе [9].
4 Заказ № 632
49
Как отмечено выше, в системе Ti2O3 и V2O3 в отличие от системы
ТЮ—VO образуется непрерывный ряд твердых растворов. Это и понятно,
так как рассматриваемые окислы имеют устойчивую, без дефектов, кристал-
лическую структуру, близкие размеры замещающихся атомов и сходную
по своим энергетическим характеристикам природу межчастичного взаимодей-
ствия.
Взаимная растворимость двуокисей
Двуокись титана (ТЮ2) встречается в природе в виде трех модификаций:
анатаза, брукита и рутила. Наиболее устойчивой модификацией двуокиси
титана является рутил. Пределу его устойчивости отвечает состав от TiOi
до TiO2 [21]. Кристаллическая структура рутила имеет тетрагональную цеп-
ную решетку и обычно описывается в виде ионной модели, основанной,
на ионах Ti4+ и О2~ [22, 23].
Двуокись ванадия (VO2), согласно [24], относится к гомологическому
ряду окислов ванадия VnO2n_1( где п — целое число. Этот ряд существует
в пределах от VOi.e до VO2. Окисел VO2 предел этого ряда (при п = со).
Кристаллическую структуру VO2 можно рассматривать как идеализированную
структуру типа Мо02 [25], в которой атомы металла связаны с шестью ато-
мами кислорода октаэдром типа МеО6. Такие октаэдры образуют цепи МеОв,
в направлении оси С. Если в нормальной тетрагональной структуре рутила
атомы металла внутри цепочки расположены на одинаковом расстоянии друг
от друга, то в структуре типа МоО2 они находятся поочередно то на более
близком, то на более далеком расстоянии. В результате этого образуются
пары Me—Me, а октаэдры из атомов кислорода деформируются. Кроме того,
между катионами с короткими расстояниями существуют направленные
Me—Ме-взаимодействия, которые осуществляются благодаря перекрыванию
электронных облаков свободных d-электронов. Выигрыш энергии, связанный
с этим, способствует стабилизации деформированной структуры.
Двуокись ниобия (NbO2) кристаллизуется в структуре, близкой к типу
рутила с узкой областью гомогенности: NbO>i,g4— NbO<2,o9- Структура по-
строена также из октаэдров NbOe, которые соединены ребрами и углами,
как и в случае рутила; однако в ряду повторяющихся вдоль оси октаэдров
чередуются короткие и длинные Me—Ме-расстояния [26].
В рассматриваемых двуокисях, за исключением ТЮ2, наряду с О—О-
связями, реализуются сильные Me—Me-связи и наблюдается их экранирова-
ние. В структуре искаженного рутила экранирование Me—Me-связи приводит
к увеличению взаимного расстояния между атомами кислорода на общем
ребре двух занятых октаэдров [7]. Поэтому, учитывая сказанное выше,
можно ожидать, что окислы состава МеО2 обладают достаточной взаимной
растворимостью. Действительно, в системе TiO2—VO2 при 1000° С в широком,
интервале составов от ТЮ2 до Tio.25Vo.75O2 образуются неограниченные
твердые растворы без образования промежуточных соединений [27]. Это со-
гласуется с данными [28], согласно которым в упомянутой системе образуются
непрерывные твердые растворы, причем значения параметров решетки откло-
няются от закона Вегарда незначительно. И только на рентгенограммах для
твердых растворов, бедных по TiO2 (с 20 мол. % и менее), обнаруживаются
характерные для моноклинной решетки VO2 сверхструктурные линии, которые
не соответствуют типу рутила. В системе Ti02NbO2, согласно [29], при 1000° С.
образуется непрерывный ряд твердых растворов со структурой рутила;
структура NbO2 при внедрении ионов Ti вначале сохраняется, рентгенограммы
твердых растворов вплоть до состава Ti0,2—Nbo,a02 содержат все сверх-
структурные линии, характерные для решетки NbO2, которые в дальнейшем:
с понижением содержания ниобия (до состава Tio.25Nbo.75O2) совершенно
исчезают. При этом переход от структуры NbO2 к структуре рутила (TiO2)
50
и обратно происходит непрерывно. Эти данные хорошо согласуются с рабо-
тами [27, 30]. Для системы VO2—NbO2 при 1000° С также наблюдается
непрерывный ряд твердых растворов на основе фазы рутила. Согласно [27, 31],
тетрагональная область твердого раствора (VxNb1_x)O2 простирается от
х=0,1 до х=0,9. И это удивительно, так как оба окисла не обладают
простой рутиловой структурой. VO2 кристаллизуется в моноклинной иска-
женной структуре рутила, a NbO2 — в тетрагональной сверхструктуре.
Заключение
Из приведенного выше обзора видно, что, несмотря на однотипность
в структурном отношении окисных фаз, в двойных системах, например
ТЮХ—VOX, TiOx—NbOx, VOX—NbOx, не всегда образуется непрерывный
ряд твердых растворов.
Для системы TiO2—VO2 область существования твердых растворов
хотя и протяженна, но ограниченна (при х=0,25), а для системы
Ti2O3—V2O3 наблюдается полная взаимная растворимость. Интересно поведе-
ние твердых растворов на основе кубических решеток моноокисей титана
и ванадия. Оказалось, что стехиометрические TiOx и VOX (х= 1) не растворяют-
ся друг в друге, в то время как составьте предельным содержанием кисло-
рода (х^=; 1,25) образуют твердые растворы (Т1ХУ2_ХО) 1,16-1,25. Причина этого,
как отмечено выше, различие электронно-валентной структуры TiOx и VOX,
а также постоянство средней плотности валентных электронов и насыщае-
мость Me—О- и Me—Ме-связей в растворах, образующихся на их основе.
Согласно [10], в большинстве случаев промежуточные окислы переходных
металлов — это насыщенные твердые растворы разновалентных окислов
с собственной структурой. В пользу этого представления указывают данные
по теплотам образования, теплоемкостям, электрическим и магнитным свой-
ствам, которые характеризуют такие окислы, как субнеоднородные механиче-
ские смеси. Косвенным доказательством этого является также электрохими-
ческое поведение анодов из низших окислов титана, нитридов и карбидов
тугоплавких металлов. В этом процессе при одинаковой анодной плотности
тока в электролит переходят катионы не одной валентности, а нескольких
(например, при электролизе ТЮ в электролит переходят катионы Ti2+ и Ti3+,
а при электролизе Ti2O3 — катионы Ti3+ и Ti4+) [32]. Это согласуется и
с данными [33] по изменению средних атомных объемов окислов в индиви-
дуальных системах (V, Ti, Nb)Ox в областях 0,5 < х < 2, где наблюдается
их постоянство, хотя средняя электронная плотность, приходящаяся на атом,
значительно изменяется. Это подтверждают работы Рюдорфа [29, 31]. На
основании полученных им структурных, магнитных и электрических свойств
твердых растворов (\\_Х1ЧЬХ)О2 было установлено, что рутиловая фаза со-
стоит из двух рядов твердых растворов:
(Kl±2xV3+Nb^j О2 при 0 < х < 0,5;
(V?±xNbf±xNb£Li)O2 при 0,5 < х < 1,0,
при этом при х=0,5 предполагается электронный переход V4+ + Nb4+—>
—>V3++Nb6+, так как NbO2, по сравнению с VO2, более сильный восстано-
витель. Измерения магнитной восприимчивости и электропроводности рутило-
вой фазы (V!_xNbx)O2 подтвердили, что превращение между VO2 и NbO2
связано с процессом восстановления. При составе (Vo,5Nbo,s)02 ванадий
имеет степень окисления +3, а ниобий 4-5. Этот двойной оксид имеет ре-
шетку рутила и образует с VO2 и NbO3 твердые растворы.
По данным, полученным для рутиловой фазы (Tio,5Nbo,5)02, оба ка-
тиона находятся в состояниях 4-4, а не 4-3 (Ti) и 4-5 (Nb), как в случае
4*
51
(V0,6Nb0>5) 02. Можно допустить, что при реакциях между TiO2 и. NbO2>.
благодаря электронному переходу Ti4+ + Nb4+ —> Ti8+ -f- Nb5+ при составе-
(Ti0,5Nb01B) О2 появляется оксид TiNbO4. Однако против этого говорят воз-
никающие взаимодействия между магнитными катионами при большом диа-
магнитном разбавлении и различия устойчивости соответствующих окис-
лов [29]. Интересно поведение твердого раствора (TixTax_z) О2. Оказалось,,
что область его существования простирается лишь до состава (Ti0>BTa01B) О2.
В связи с этим предполагается [29], что между TiO2 и ТаО2 в отношении
1:1 существует ряд твердых растворов (Ti]i2xТ^+Та®+)02.
Исходя из этих данных можно предположить, что поведение твердого
раствора в системе Ti—V—О также обусловлено различным валентным со-
ставом как отдельных фаз в системах Ti—О и V—О, так и возможным
частичным изменением его в процессе образования твердых раство-
ров. Отсутствие непосредственных измерений валентных состояний за-
трудняет анализ поведения твердых растворов. Однако, если исходить,
из предположения, что в решетках окислов обменное взаимодействие главным
образом наблюдается между теми атомами металла, которые находятся
в одном и том же валентном состоянии, то становятся понятными некоторые
особенности изученных твердых растворов.
Прежде всего в TiO2 титан находится преимущественно в виде Ti4+.
В окисле VO2, наряду с V4+, как уже отмечено, могут присутствовать
катионы V3+ и V5+. Присутствие V6+ в VO2, по-видимому, основная причина
образования ограниченных твердых растворов (TixVx_z) О2. Электронный пере-
ход VB+—>V4+ в этом случае невозможен из-за насыщенности валентной связи
между катионами Ti4+. Что касается V3+, то здесь, очевидно, возможен
частичный переход Ti4++V3+—>Ti3++V4+, что также будет способствовать-
образованию твердых растворов. В окислах типа корунда (Ti2O3 и V2O3),
по-видимому, существуют такие сочетания валентности титана и ванадия,
которые обеспечивают полное взаимодействие между ними.
Рассматриваемая здесь точка зрения относительно образования твердых,
растворов нуждается в дальнейшем экспериментальном обосновании.
ВЫВОДЫ
1. Дан литературный обзор по вопросу взаимной растворимости некото-
рых промежуточных окислов переходных металлов IVa и Va подгрупп.
2. Показано, что для систем TiO2—VO2, NbO2 область существования
твердых растворов хотя и протяженна, но и ограниченна, а для системы
Ti2O3—V2O3 наблюдается полная взаимная растворимость. Наряду с этим:
твердые растворы на основе кубических решеток стехиометрических монооки-
сей титана и ванадия не образуются, в то время как составы с предельным
содержанием кислорода обладают заметной взаимной растворимостью (до 25%).
3. Поведение твердых растворов в данных системах рассмотрено с точки
зрения различного валентного состава как отдельных фаз, так и возможного
частичного изменения его в процессе образования твердых растворов.
ЛИТЕРАТУРА
1. S. L. Altman, С. A. Coulson, W. Hume-Ro the г у. Proc. Roy. Soc , • 1957 ь
A, 240, 145.
2. R. E. Rundle. Acta Cryst., 1948, 1, 180.
3. W. Hume-Rothery. Philos. Mag., 1954, 44, 357.
4. H. Krebs. Acta Cryst., 1956, 9 , 95.
5. H. Bulz. Z. Phys., 1958, 153, 338.
6. J. B. Goodenough. Phys. Rev., 1960, 117, 1442.
7. В. А. Цхай, П. В. Гельд, Г. П. Швейкин, В. А. Пере л яе в. Химия вы-
сокотемпературных материалов. Тр. Второго всесоюз. совещ. по химии окислов.
при высоких тем-pax, г. Ленинград, 1965. Л., «Наука», 1967, стр. 19.
52
8. П. В. Гельд, В. А. Цхай. Доклад на Постоянном межинститутском коллоквиуме-
по твердым фазам переменного состава. Л., 1962, вып. 32 (2).
9. П. В. Гельд, В. А. Цхай, Г. П. Швейкин, В. А. Переляев. Изв.
АН СССР, Неорганические материалы, 1967, 3, 1835.
10. С. М. Ария. Доклад на Постоянном межинститутском коллоквиуме по твердым,
фазам переменного состава. Л., 1963, вып. 41 (11).
11. И. И. Корнилов. Изв. АН СССР, Металлургия и горное дело, 1964, 6, 19.
12. Г. С. С а м с он о в, Я. С. Уманский. Твердые соединения тугоплавких металлов,
М., Металлургиздат, 1957.
13. G. В г а ие г*, N. М ora v i t z. Z. anorg. allg. Chem., 1962, 317, 13.
14. П. В. Гельд, Г. П. Швейкин, С. И. Алямовский, В. А. Цхай. Изв..
АН СССР, Неорганические материалы, 1967, 12, 2001.
15. П. В. Гельд, С. И. Алямовский, И. И. Матвеенко. Ж- структ. хим.,
1961, 2, 301.
16. S. A n d е гss on, В. С о 11 е n, U. К uy 1 en s t i г n а, А. М a gn е 1 i. Acta Chem.
Scand., 1957, И, 1641.
17. M. Е. Straumanis, Т. Egima. Acta Cryst, 1962, 15, 404.
18. F. J. Morin. Phys. Rev. Letters, 1959, 3, 34.
19. В. А. Переляев, Г. П. Швейкин, С. И. Алямовский. Изв. АН СССР,
Неорганические материалы, 1968, 4, 1372.
20. Т. Tawakubo, Т. Janagi, S. Nomura. Phys. Soc. Japan, 1960, 15, 2102.
21. M. Девис, С. Бирченол. Химическая технология и металлургия титана, ч. I.
М., ИЛ, 1954, стр. 171.
22. Т. Hurlen. Acta Chem. Scand., 1959, 13, 365.
23. Б. Ф. Ормонт. Структура неорганических веществ. М.—Л., Гостехтеориздат,
1950.
24. J. Stringer. J. Less-Common Metals., 1965, 8, 1.
25. G. Andersson. Acta Chem. Scand., 1956, 10, 623.
26. В. O. Marinder. Acta Chem. Scand., 1961, 15, 707.
27. В. O. Marinder, A. Magneli. Acta Chem. Scand., 1958, 12, 1945.
28. W. Riidorf, G. Walter, G. Stadler. Z. anorg. allg. Chem., 1958 , 297,1.
29. W. Rfldorf, H.-H. Luginsland. Z. anorg. allg. Chem., 1964, 334, 125.
30. H. J. Goldschmidt. Metallurgia, 1960, 62, 216.
31. W. Riidorf, J. Marklin. Z. anorg. allg. Chem., 1965, 334, 142.
32. Л. E. Ивановский, M. В. Смирнов. Изв. Вост. фил. АН СССР, 1967, 10, 68 _
33. Г. П. Швейкин, П. В. Гельд, С. И. Алямовский, В. А. Цхай. Ж. физ.,
хим., 1968, 42, 2178.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. И. ПОТОРОЧА, В. А. ЦХАЙ, П. В. ГЕЛЬД
К РАСЧЕТУ ЭНЕРГЕТИЧЕСКОГО СПЕКТРА МОНОКАРБИДА ТИТАНА
Одной из важнейших задач физики и физической химии твердого тела является
проблема его энергетического спектра. Создание количественной или полу-
количественной теории открывает широкие возможности для установления
корреляции между атомными характеристиками и строением твердого тела,
с одной стороны, и физическими их свойствами, с другой. Достигнуты боль-
шие успехи при анализе лишь простых систем, в частности непереходных
металлов.
В последнее время возрос интерес к теоретическим исследованиям элек-
тронных энергетических спектров некоторых тугоплавких карбидов, нитридов
и окислов [1—41. При этом используются различные приближенные расчет-
ные методы, опробованные на металлах. Развитие этого направления в кван-
товой химии диктуется необходимостью уточнения природы связей, зависи-
мости электронного спектра от состава тугоплавких соединений и строения
атомов-компонентов. Накопленный обширный экспериментальный материал
по физико-химическим свойствам упомянутых соединений, по-видимому, может
быть эффективно использован для этой цели лишь с привлечением резуль-
татов квантово-механических расчетов.
Нельзя считать выясненным также вопрос об условиях применимости
и точности различных приближенных методов в передаче тех или иных па-
раметров зонной структуры. Они нуждаются в дальнейшем усовершенствова-
нии и детализации в каждом конкретном случае и особенно при использовании
их в полуэмпирическом варианте. Таким образом проведение расчетов зонной
структуры с использованием аппарата квантовой химии имеет большое зна-
чение и в методическом отношении.
В данной работе приводится краткое изложение основных этапов расчета
электронного спектра монокарбида титана методом сильной связи. Этот ме-
тод в приложении к металлам был развит Флетчером [5], Слэтером и Косте-
ром [6] и подробно освещен в обзоре Месбаума [7]. Различные его варианты
использованы авторами работ [2—4] и для расчета зонной структуры неко-
торых соединений тугоплавких металлов.
Как известно, установление энергетического спектра электронов сводится
к решению уравнения Шредингера:
^r(r)=£;Vr(r), Я=--^А+У(г). (1)
A k k 8n‘m
54
В методе сильной связи решение находят с помощью блоховской функции
гРг(7) = 2арФр(7,Т). (2)
р
фр(7, Ъ~=- Se-,rMP(7-X). (3)
р У «i
—►
Здесь фр(г — Rt) — волновая функция свободного атома, расположенного
в точке Rf, р — обозначение совокупности квантовых чисел п, I и т. Нор-
мировка функции (3) проводится в пределах одной области, включающей Я
элементарных ячеек. Между областями предполагаются периодические гра-
ничные условия. При этом блоховская сумма (Фр) включает одну атомную
орбиталь на элементарную ячейку. Если последняя содержит несколько
одинаковых атомов (например, решетка алмаза), то для каждой из позиций
составляется своя блоховская сумма.
* —> —>
Умножая уравнение (1) на Фр( г, k ) и интегрируя по всему простран-
ству, получаем
^ap{Hpq — ESpq)=G. (4)
Hpq=JФ; 7 k) НФ, (7, k)d т, [Sp,=]фрФ4 d т. (5)
Таким образом, задача сводится
ний (4). Для того чтобы она имела
чтобы детерминант, составлен-
ный из коэффициентов при ар,
обращался в нуль:
I Яр, (7)-Е (7) Sp? 1=0. (6)
В приведенном расчете учи-
тываются лишь шесть валент-
ных орбиталей атомов металла
(3d — 4s) и три — атомов угле-
рода (2р). В этом случае сис-
тема (4) состоит из девяти линей-
ных уравнений и сводится к
решению секулярного уравнения
(6) 9-го порядка, что сопряжено
с большими математическими
трудностями. Расчеты могут быть
упрощены с помощью теории
групп. Для некоторых симметрич-
к решению системы линейных уравне-
не нулевые решения ар=г=0, необходимо
Рис. 1. Зона Бриллюэна для гранецентрирован-
ной кубической решетки.
ных направлений вектора k (100), (ПО) и (111) она позволяет привести
детерминант (6) к квазидиагональному виду. Секулярное уравнение распа-
дается на ряд уравнений более низшего порядка.
Как известно, кубической гранецентрированной структуре соответствует
объемноцентрированная в ^-пространстве. При этом первая зона Бриллюэна
имеет вид усеченного октаэдра. Направление (ПО) в ^-пространстве харак-
теризуется (рис. 1) симметрией С2г, с элементами Е, С^, 1С2г и /С2<—
Действием последних на угловые части перечисленных выше атомных функ-
ций можно найти характеры приводимых представлений Х(Е) = 9; Х(С2Х)|) = 1;
Х(С22)=3; X (С2 (_Xy)) = 3. Используя таблицы характеров непроводимых
55
представлений из работы [8] и формулу av —— у g‘iX?’ Xf (см. например [9])t
находим разложение
2==4S1+S2+2b+2S4-
Оно показывает, что секулярное уравнение 9-го порядка для фиксированных
значений волнового числа k в направлении (110) распадается на одно урав-
нение 4-го порядка, два 2-го и одно 1-го порядка.
Направление (111) характеризуется группой симметрии С3о с элемента-
ми Е, 2С3, 3/С2, для которой аналогичным образом было найдено разложе-
ние на неприводимые представления: Л=3%1-|-613. Уравнение (6) в этом
случае распадается на три кубических, два из которых эквивалентны.
Наконец, для направления (001), обладающего группой симметрии Ctv
с элементами Е, Сг, 2С4, 2/С4 и 27С2, разложение имеет вид
A = 3A1+A2+A2+4As.
В этом случае исходное уравнение 9-го порядка распадается на кубическое,
два идентичных квадратных и два линейных.
Базисные функции, соответствующие неприводимым представлениям, для
рассмотренных выше направлений, приведены в работе [10]. С их помощью
для заданного значения вектора k в одном из рассмотренных направлений
автоматически могут быть получены уравнения более низких порядков.
•Общую запись этих уравнений можно передать выражением (6). Однако
индексы р и q будут пробегать не через индексы девяти исходных атомных
функций, а через обозначения базисных функций данного неприводимого
представления. Число базисных функций определяет порядок уравнения
(детерминанта).
Базисные функции при расчете матричных элементов были взаимно орто-
гонализированы по методу Левдина [11]. Рассмотрим процедуру расчета Hpq.
Согласно формулам1 (5) и (3),
(7)
Ri RJ J
Суммы интегралов по одному из индексов (например RJ) при различных Рд-
совпадают между собой. Вследствие этого суммирование по Ri в формуле (7)
может быть заменено простым умножением на N. При этом R: будет опреде-
лять положение центрального атома, которое удобно совместить с началом
координат. Тогда, согласно (7), имеем:
Нм=^ еik *J f (Л H^q & - Rj) 2 е Epq (I, т, п). (8)
я/ J
Выражения матричных элементов Hpq через Epq приводятся в работе [6]
и в настоящей статье дополнены расчетами. При вычислении Epq потен-
циальная энергия V (7) в гамильтониане (1) бралась приближенно в виде
суммы внутриатомных сферических потенциалов, рассчитанных по методу
Хартри — Фока и локализованных в различных узлах решётки. При этом
учитывались лишь двухцентровые интегралы и пренебрегались трехцентровые.
Последние представляют слагаемые, которые появляются при учете потен-
циалов атомов, расположенных в узлах. Эти узлы не совпадают с местом
1 В формуле (7) фр и ф9 — атомные функции. Матричный элемент, построенный
.на базисных функциях, определяется через Hpq.
.56
локализации атомных орбиталей Wp и Чг?. Задача таким образом сводится к вы-
числениям двухцентровых интегралов. Атомные волновые функции, фигури-
рующие в выражении Epq, должны быть представлены как линейные ком-
бинации функций пространственно квантованных по отношению к оси, связы-
вающей два узла решетки. Иными словами, орбитали рх, ру и pz должны
быть выражены через линейную комбинацию ра, рп, a dxy, dyZ через da,
d„, db -орбитали, где о, л и 6 — компоненты углового момента на ось,
связывающую два атома [7]. Интегралы, включающие угловые части волно-
вых функций в прямоугольной системе Epq, будут выражаться через моле-
кулярные интегралы (рр0), (рря), (dda) и т. д. Выражения для Epq, через
интегралы для двухатомных молекул приведены в работе [6] и дополнены
расчетами в настоящей статье.
При нахождении молекулярных интегралов численные значения потенциа-
лов внутри свободных атомов [12] аппроксимировались аналитической функ-
цией вида
V(r)=_J£±₽Cr(
(9>
где а, р и у — постоянные, соответственно, равные для атомов углерода
и титана: a^l; рх=5,0066; ух=1,7587 и сх2=Л; 02=2О,8О6О; у2=2,0211.
В интервале значений г от 0,00974 до 2,91359 радиуса Бора ошибка аппрок-
симации не превосходила 11% для С и 20% для Ti. (В области перекры-
вания волновых функций средняя ошибка будет меньше, поэтому величина
обменного интеграла изменится незначительно).
Необходимые для целей настоящей работы двухцентровые интегралы
имеют вид8
(10).
Радиальная часть волновой функции взята в форме Слэтера
Двухцентровые интегралы для решетки
карбида титана
Обозначение Обменный интеграл, эв Обозначение Интеграл перекры- вания
(4s4sa) —6,065 5(4s4sa) 0,505
(4«2р<т) 3,334 S (4s2pa) —0,083
(4s3do) —4,405 S (4s3da) 0,310
(2p3da) —3,870 S(2p3do) 0,175
(2p3dn) 1,784 S(2p3dn) —0,093
(2p2pa) 1,150 S (2p2pa) —0,050
(3d3dit) ‘ 0,459 S (3<Шл) —0,026
(3d3do) —1,096 S(3d3da) 0,043
(2р2рл) —0,166 S(2p2pn) 0,011
двухцентро'вых молекулярных
интегралов
/(г)=Аг'1*-1е-«\ (11).
Z*
где А — нормировочный множитель; 6=—, Z*=Z— у — эффективный заряд
п*
ядра; у — постоянная экранирования, которая определялась по Бацанову;
п* — эффективное главное
квантовое число по Слэтеру.
Сравнение выражений (9) —
(11) показывает, что действие
содержащегося под интегралом
(10) потенциала У(г) сводится
к изменению показателя г и
коэффициента в экспоненте
радиальной части волновой
функции (11). В связи с этим
нетрудно показать, что J мо-
жет быть выражено через инте-
гралы перекрывания [13]. Вычи-
сление последних проводилось
по методу, описанному в ра-
боте [14]. Результаты вычислений
сведены в таблицу.
Секулярные уравнения были решены для каждого выбранного направле-
ния k в четырех-пяти точках. Результаты представлены графически в виде
’ Запись интеграла приведена по Дираку.
57’
зависимости Е (k) (рис. 2, а). Согласно этим данным, была построена кри-
вая плотности состояний N(E) по методике, описанной в работе [6] (рис. 2, б).
Полученный энергетический спектр электронов представляет непрерывную
широкую полосу. При этом в случае TiC уровень Ферми расположен не-
сколько ниже минимума, которому соответствует концентрация валентных
электронов (VEC), равная 8,5.
На основании исследования оптических, электрических и магнитных
•свойств Пайпер [15] заключил, что полосная структура карбидов и нитридов не
Рис. 2. Энергетический спектр монокарбида титана:
а — кривые зависимости’энергии Е от волнового числа k\ б — плотность состояний #(Е).
VEC=8 (/), 9 (II), 10 (III).
очень чувствительна к внутриатомному потенциалу ядра и в первом прибли-
жении свойства тугоплавких соединений определяются концентрацией валент-
ных электронов. Денкер [16], обсуждая различные физические свойства карби-
дов, пришел к выводу о существовании широкого минимума плотности состоя-
ний VEC между 8 и 8,5, что согласуется с результатами расчетов. Для
системы HfC — ТаС максимальные значения теплоты плавания будут вблизи
состава (TaC)o,8(HfC)o,2, удельного сопротивления вблизи (ТаС)о,7-о,75
(HfC)o,з-0,25. максимум скорости испарения — для (TaC)o.7(HfC)0,3 [17].
В твердом растворе ZrC — NbC минимумом коэффициента теплового расши-
рения и максимумом удельного сопротивления обладает сплав (ZrC)o 6
(NbC)0,4 [18].
Эти данные указывают на наличие минимума плотности состояний около
VEC=8,5. С другой стороны, критическая температура сверхпроводимости
системы NbC—NbN наблюдается в твердом растворе состава (NbC)o,25(NbN)o,75
[16]. Это говорит о наличии максимума вблизи VEC=9,75 и подтверждает
описанные выше особенности энергетического спектра. Наконец, результаты
оптических исследований [15] показывают наличие полосы с большой плот-
ностью состояний, удаленной на 2,5 эв от уровня Ферми (для карбидов
58
V группы и нитридов IV группы), что подтверждает наличие третьего макси-
мума в зависимости плотности состояний от энергии (см. рис. 2, б).
Резкое уменьшение коэффициента Холла [19], а также термо-э.д.с.
[19, 20] при незначительном увеличении концентрации вакансий в подрешетке
углерода говорит о том, что N (Е) ниже VEC=8 должна увеличиваться,,
напротив рост термо-э. д. с. с повышением температуры [20, 21] указывает
на уменьшение N (Е) при VEC>8, т. е. в области больших энергий. Это
также совпадает с результатами нашего расчета.
Все эти, а также многие другие экспериментальные данные находятся
в хорошем согласии с установленным энергетическим спектром электронов,
в тугоплавких соединениях со структурой типа NaCl.
ЛИТЕРАТУРА
1. Е. V. Ern, А. С. Switendick. Phys. Rev., 1965, 137А, 1927.
2. R. G. Lye a. E. M. Lo go t h e t i s. Phys. Rev., 1966, 147, 682.
3. H. Bilz. Z. Phys., 1958, 153, 338.
4. P. Costa a. R. R. Conte. Nucl. Met. AIME, 1964, 10, 29.
5. G. C. Fletcher. Proc. Phys. Soc. London, 1952, A65, 192.
6. I. C. Slater u. G. F. Koster. Phys. Rev., 1954, 94, 1498.
7. A. Muss baum. Solid. State. Phys., 1966, 165, 18.
8. L. P. Bouckaert, R. Smoluchowski a. Ei. Wigner. Phys. Rev., 1936,.
50, 58.
9. К. Бальхаузен. Введение в теорию поля лигандов. М., «Мир», 1964.
10. D. G. Bell. Rev. mod. Phys., 1954, 26, 31.
11. P. О. Low din. Advances in Phys., 1956, 5, 67.
12. F. Herman. Atomic Structure Calculation, Preutice Hall, Inc. Englewood Cliffs,.
New Jersey, 1963.
13. A. Lof th us. Mol. Phys., 1963, 6, 115.
14. A. Lof thus. Mol. Phys., 1962, 5, 105.
15. J. Piper. Nuclear Metallurgy, AIME., 1964, 4, 24.
16. S. P. Denker.' J. Less-Common Metals., 1968. 14, 1.
17. Г. В. Самсонов, И. Г. Баранцева, В. Н. Па дерн о. Теплофизика высо-
ких температур, 1968, 6, 2, 248.
18. И. Г. Баранцева, В. Н. Падерно. Изв. АН СССР, Неорганические материалы,
1968, 4, 8, 1364.
19. А. И. Августиник, О. А. Голикова, Г. М. Климашин, Л. В. Коз-
ловский, В. С. Нешпор. Исследование в области химии силикатов и окислов.
Под ред. А. И. Августиника. М. -Л., «Наука», 1965, стр. 241.
20. О. А. Голикова, А. И. Августиник, Г. М. Климашин, Л. В. Коз-
ловский, С. С. Орданьян, В. А. Снеткова. Физика твердого тела,,
1965, 7, 12, 3698.
21. R. G. Lye. J. Phys. Chem. Soc., 1965, 26, 407.
ВЫП. 20
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
труды института химии
197®
С. И. АЛДМОВСКИЙ, Ю. Г. ЗАЙНУЛИН, Г. П. ШВЕЙКИН,
П. В. ГЕЛЬД, | Е. Н. ЩЕТНИКОВ |
СИСТЕМА Zr—С—О
Переходные металлы IV и V групп могут образовывать оксикарбидные
фазы МеС^Оу с кубической (типа NaCl) и с гексагональной структурой,
близкой к типу анти-Сс112. К настоящему времени относительно хорошо изу-
чены и достаточно подробно, описаны в литературе оксикарбиды Ti V, Nb,
Та, (например, [1—51). Этого нельзя сказать об оксикарбидах циркония.
Сведения, касающиеся условий их синтеза, термической устойчивости, обла-
стей гомогенности, структурных особенностей, комплектности решетки, либо
крайне скудны и отрывочны, либо вовсе отсутствуют. В настоящем сообще-
нии обобщены литературные данные по некоторым характеристикам
2гСхОу-фазы.
Однако прежде чем говорить о работах, посвященных тройной системе
Zr—С—О, целесообразно коротко остановиться на бинарных системах Zr—С
и Zr—О.
Zr—-С. Образуется лишь один карбид ZrCx. Относительно полный и
обстоятельный обзор исследований (опубликованных до 1955—1957 гг.),
касающихся изучения условий синтеза, структурных и других характеристик
ZrCx, приведен в работах [6—10]. Из ранних исследований прежде всего
следует отметить работы [11 —13], в которых был впервые получен и иден-
тифицирован карбит ZrCx. Так по данным [13], он характеризуется струк-
турой типа NaCl, а величина его периода решетки составляет 4,710А.
Позднее Бургере и Базарт [14], получившие ZrCx прикаливанием угольной
нити в атмосфере четыреххлористого циркония, нашли, что периоды решетки
синтезированных ими образцов карбидов изменяются с составом от 4,687
до 4,670А.
Уманский, Шукст и Петрусевич (цит. по [7]) впервые провели системати-
ческое исследование системы Zr—С. Они рентгенографически изучали кар-
биды циркония в области составов ZrCo.35— ZrCo.go(4,4—10,6 вес. % С).
Образцы готовили карбонизацией при 1700° С порошкообразного металла
сажей в вакууме. По их данным, препарат состава ZrCo.ss оказался двух-
фазным, он состоял из ZrCx и твердого раствора углерода в a-Zr. Осталь-
ные образцы содержали лишь кубический карбид ZrCz, область однофазности
которого отвечала составам ZrC0,37 — ZrCo.ss- Период решетки с увеличением
содержания углерода возрастал при этом практически линейно от 4,6553 до
4,6795 А. Упомянутым авторам не удалось получить эквиатомного карбида
ZrCi.oo- Следует отметить, что в исходном металлическом цирконии, исполь-
зованном в работе [7], присутствовало около 1 % титана.
Несколько отличные данные были получены А. Е. Ковальским и
•60
Т. Г. Макаренко [15], изучавшими препараты, приготовленные спеканием
порошка циркония с сажей. Согласно результатам химического и рентгено-
графического анализа, область существования ограничивается пределами
ZrCo,56—ZrCi.oo (36—50 ат. %С). При этом периоды решетки карбида,
также возрастающие с увеличением концентрации углерода, изменялись от
4,652 до 4,687 А.
Г. В. Самсоновым и Н. С. Резиновой [16] были приготовлены образцы
в области брутто-составов ZrCo.oi—ZrCjjo (0,22—15,4 вес.% С). Синтез
осуществлялся в вакууме (10-5—10~4 мм рт. ст.) в интервале температур
1500—1900°С. Авторы показали, что препараты состава ZrCo.os — Zito,25
(0,64—3,12 вес. % С) двухфазные и содержат твердый раствор углерода
в цирконии и ZrCx. Образцы, соответствующие концентрационному интервалу
ZrCo,27 — ZrCi.oo, оказались однофазными, а периоды решетки ZrCx меня-
лись от 4,581 до 4,673 А.
Бенезовский и Руди [17] также исследовали сплавы циркония с угле-
родом, но на этот раз приготовленные из гидрида циркония и сажи методом
горячего прессования. По их данным, область однофазного существования
Zitx заключена в пределы ZrCo.54 — ZrCi.oo (35—50 ат.% С). Позднее эти
результаты подтверждены ими в совместной работе с Новотным [18]. Кон-
центрационные зависимости периода решетки ZrCx в обеих работах практи-
чески совпадают (4,677—4,696 [17] и 4,675—4,694 А [18]).
Наконец, недавно Сара [19] привел подробные сведения по системе
Zr—С, которые получены методами дифференциально-термического, рентгено-
графического и металлографического анализов. Область однофазного суще-
ствования карбида циркония находится в пределах ZrC0,63 — ZrCi.oo и практи-
чески не зависит от температуры. Периоды решетки 2гСх-фазы изменяются
от 4,6941 до 4,6983 А, достигая в отличие от всех предыдущих данных
(О
максимума (4,7021 А) для образца состава ZrCo.835 . Такая концентрацион-
ная зависимость межплоскостных расстояний встречается довольно редко.
Во всяком случае она отсутствует для других кубических карбидов металлов
IVa—Va подгрупп.
Следует упомянуть работы [20—22], в которых синтезированы однофазные
препараты ZrCx. Так, Ю. И. Вильк с соавторами [21] получали образцы
карбида циркония в интервале составов ZrCo,54— ZrCo,75 (а=4,6524 —
О
4,6981 А), а Битнер и Горецкий [20] — в пределах ZrCo,67 — ZrCo.g?
О
(а=4,689—4,691 А). Наконец, согласно В. С. Куцеву [22], параметры ре-
шеток кубического карбида увеличиваются от 4,653 (при х=0,518) до
4,693 А (при х=0,910).
Как видно из приведенного выше материала, данные, касающиеся системы
Zr—С, не совпадают и требуют дальнейшего уточнения (см. рисунок и табл. 1).
Это относится как к пределам области гомогенности ZrCx, так и к концен-
трационной зависимости периодов его решетки. Достаточно указать на то,
что богатая по металлу концентрационная граница области однофазного
существования карбида по данным различных авторов колеблется от ZrCo.2?
до ZrCo.63 (см. рисунок). Эти результаты не могут быть связаны с темпе-
ратурной зависимостью пределов области гомогенности, тем более что таковая
для ZrCx практически отсутствует [19].
Большие расхождения наблюдаются также и при сопоставлении концентра-
ционных зависимостей периодов решетки ZrCx (см. рисунок). Так, например,
для препарата состава ZrCo,7 значение периода решетки, по данным различ-
О.
ных авторов, колеблется от 4,66 до 4,70 А. Возможно, это обстоятельство
61
Таблица 1
Условия синтеза, концентрационные области гомогенности (х, — х2)
и параметры решетки (а) кубического карбида циркония
Исходный материал Темпера- тура син- теза, ° С Х1 — xt а, А По дан- ным работ Химический анализ
Zr, сажа Zr, сажа Zr, сажа ZrH2, сажа ZrH2, сажа ZrH2, сажа ZrC, Zr, сажа 1700 2000 1900 1830 1830 3300 1820 0,37—1,00 0,58—1,00 0,27—1,00 0,54—1,09 0,54—1,00 0,63—1,0 0,54—0,76 4,6553—4,6795 4,652—4,687 4,582—4,673 4,677—4,696 4,675—4,694 4,6941—4,6983 4,6524—4,6981 [7] [15] 16 18 17] 19] [21]* Собщ» ^СВОб» ^СВЯЗ* ^гобщ- Собщ» Ссвоб> 2гоощ Данных нет То же » Собщ> 2гобщ Ссвоб> Ссвяз» 2гобщ Данных нет
Zr, ZrH2, сажа 2200 0,67—0,96 4,684-4,691 [20]*
♦ В указанных работах область гомогенности не определялась.
связано с наличием в них неконтролируемых примесей (О, N, Н, и др.). Вероят-
ность этого видна, в частности, хотя бы из того, что удовлетворительное
согласие в величинах периодов решеток наблюдается в тех исследованиях,
в которых использованы одинаковые исходные препараты (цирконий и сажа
[7, 15, 16, 21]; гидрид циркония и сажа [17—20]). Однофазность кубиче-
ского карбида циркония в широкой концентрационной области обусловли-
вает наличие вакансий по крайней мере в подрешетке углерода (по аналогии
с VCX, NbCx, ТаСх). Однако детальный характер их распределения остается
пока невыясненным. Во всяком случае, не исключена склонность к упорядо-
ченному распределению вакансий в карбиде циркония некоторых составов.
Об этом, в частности, Говорят результаты нейтронографического исследова-
ния [23], согласно которому обнаружено упорядочение атомов углерода в
препаратах, близких по составу к ZrCo.cs- Только в последнее время удалось
выявить упорядочение в ячейках карбидов TiCx[23], VCo,87s[24], V2C [25],
Nb2C[25], Та2С[26]. Ранее экспериментальные данные по этому вопросу
отсутствовали, чаще всего предполагалось, что в углеродной подрешетке
указанных фаз существует статистически беспорядочное распределение вакан-
сий.
Zr—О. Большинство исследователей указывает на существование в этой
системе лишь двух полиморфных фаз твердого раствора кислорода в металле
и двуокиси ZrO2. По данным [27—29], низкотемпературная модификация
металла (a-Zr) растворяет около 29—31 ат. % О с образованием гексаго-
нального раствора внедрения a-Zr(O). В противоположность указаниям [27]
и согласно данным [28] концентрационная зависимость периода а носит
экстремальный характер. Период с увеличением содержания кислорода воз-
растает монотонно. Отметим, что, согласно [27], в области существования
a=Zr(O) следует различать две фазы: a(ZrO0+x) и a' (Zr3O1+y). В ячейке
первой из них атомы кислорода распределены статистически беспорядочно,
а во второй они упорядочены. Последнее подтверждается на рентгенограм-
мах этой фазы сверхструктурными линиями. Позднее экстремальный ход
периода а от х был подтвержден в работе [29]. Наконец, И. И. Корнилов и
В. В. Глазова [30], изучая систему Zr—О методами физико-химического ана-
лиза, обнаружили при 600° С сингулярные точки, ссответствующие соеди-
нениям ZreO и Zr,O, индивидуальность и природа которых требует дальней-
шего изучения.
68
Гексагональный a-Zr при 862° С переходит в 0-цирконий, обладающий
кубической объемноцентрированной решеткой. В отличие от a-Zr раствори-
мость кислорода в p-Zr относительно невелика и составляет примерно
10,5 ат. % [8].
Окисел ZrO2 практически — фаза постоянного состава. В литературе
описаны три модификации ZrO2. Низкотемпературная двуокись с моноклин-
ной ячейкой устойчива примерно ’до 1000° С, тетрагональная существует в
интервале температур от 1000 .до 1900° С, тогда как область существования
высокотемпературной кубической модификации достоверно не установлена.
Структура моноклинной и тетраго-
нальной ZrO2 представляет собой
искаженный тип CaF2, в котором
кристаллизуется кубическая модифи-
кация двуокиси циркония. Кроме
того, имеются дополнительные ука-
зания на то, что кубическая фаза
О
(а=5,075 А) может быть стабили-
зирована примесями С, СаО или MgO
18].
В работе [31] указывается на
возможность существования Zr2Os.
В сообщении [32] упоминается о
кубической моноокиси ZrO с перио-
дами 4,584—4,620А. Далее Г. В.Сам-
сонов [33] приводит рентгенографи-
ческие и химико-аналитические
характеристики ZrO и Zr2O3, полу-
ченных углетермическим восстанов-
лением двуокиси. Химический ана-
лиз моноокиси указывает на при-
сутствие в нем около 0,5 вес. %
свободного углерода. Период решет-
47Z7-
Ш
W
4Д4-
Концентрационные зависимости периодов
решетки кубического карбида циркония.
По данным работ: 1 — [191; 2 — [20], 3— [18],
4 —[17], 5 — [7], 6-[22], 7 —[15],
8 — [16]. 9 - [21].
ки ZrO (тип NaCl) составляет 4,63 А. Однако более поздними исследова-
ниями [28, 29] существование устойчивой кубической ZrO (как и Zr2O3) не под-
тверждено. Отсутствие стабильных окислов ZrO и Zr2O3 объясняется повы-
шенным кулоновским отталкиванием электронов Me—Me-связи и анионов,
обусловленных сильным перекрыванием атомных функций циркония в гипо-
тетических ZrO и Zr2O3 [34].
Таким образом, вопрос о возможности существования ZrO и Zr2O3 пока
остается открытым. Не исключено, что их формированию способствуют при-
меси (как например, при стабилизации кубической ZrO2).
Zr—С—О. Впервые, вероятно, кубический оксикарбид циркония ZrCxOy
был идентифицирован В. С. Куцевым, Б. Ф. Ормонтом и В. А. Эпельбаумом
[35]. Они показали, что при взаимодействии ZrO2 с углеродом в равновесных
условиях в интервале температур 1800—2000° К при давлении 150—
1000 мм рт. ст. образуется не карбид (как считалось ранее), а оксикарбид
состава ZrCo,7iOo,o8- Период его решетки составляет 4,686 А. Проводя ана-
логичные эксперименты в несколько отличных от предыдущих условий
(Р=760 мм рт. ст.-, Т=1900—2500” К), Б. Ф. Ормонту с соавторами [36]
удалось получить уже несколько препаратов оксикарбидов разного состава,
правда с небольшим содержанием кислорода. Оказалось, что с увеличением
температуры концентрация кислорода в ZrCxOy понижается.
Между тем позднее Т. Я. Косолапова, В. Б. Федорус и Ю. Б. Кузьма
63
[37], изучая взаимодействие ZrO2 с ZrCo.gg в вакууме (Р=5-10~3 мм рт. ст.)
при температурах от 1800 до 21009С, обнаружили ZrCxOy с большим содер-
жанием кислорода (табл. 2). Е. Е. Котляр, Т. Я. Косолапова и Г. Н. На-
зарчук [38] указывают на образование в системе Zr—С—О непрерывного ряда
твердых растворов. Объектами исследования служили продукты взаимодействия
ZrOa с карбидом циркония, которые получали нагреванием соответствующих
шихт в вакууме (Р=10-2 лш рт. ст.) в течение 1 ч и в интервале температур
от 1000 до 2000° С.
В работе [39] указывается на возможность образования оксикарбида
циркония ZrO2C при взаимодействии циркония с окисью углерода. Он обла-
дает, по-видимому, тетрагональной ячейкой, которая представляет собой ис-
каженный тип CaF2. При этом атомы углерода занимают октаузлы исходной
ячейки тетрагональной ZrO2. Эти авторы указывают температурную область
(900—1100° С), в которой возможно формирование этого оксикарбида.
Таблица 2
Условия синтеза, составы й параметры решетки (а) оксикарбидов циркония
Исходный материал Условия синтеза Состав оксикарбида о а, А По дан- ным работ Химический анализ *
Р. мм рт. ст. Г’, к
ZrO2, сажа 11—935 1814—2020 ZrC0)71Oo,0e 4,686 [31] Собщ» Ссвоб»
^1*общ>
2гО2, сажа 760 1930 2гСо,влОо >ов 4,691
2080 2080 ^Г^0»70^0>05 ZrC0,e7Oo >04 4,696 4,694 [35] СО61Ц- Ссвоб> р
2100 ZrCo>6sOo,o2 — ^связ
ZrC, ZrO2 ю-» 2073 ZfCo>7bOo>17 4,5671
2173 2гС0,72О0,2в 4,577
2273 ^^0» 70^0,23 4,561
2373 ZrC0,eo00,57 4,583 [36] Ссвяз» Ссвоб» Собщ» 2гобщ
2473 4,583
2573 «—. 4,594
2673 — 4,605]
* Кислород определялся по разности.
Опубликованные сведения о характеристиках оксикарбидов в циркония
исчерпываются этими немногими данными (см. табл. 2).
Отметим следующее. Согласно Г. П. Швейкину с соавторами [40], кон-
центрационная область однофазного существования кубических оксикарбидов
циркония должна быть относительно узкой и располагаться вблизи стороны
Zr—С треугольника Zr—С—О. Последнее связано, в частности, с тем, что
в отличие, например, от системы Ti—С—О и V—С—О, для которых харак-
терно формирование устойчивых и изоструктурных кубическим карбидам
(TiCx и VCX) моноокисей (VOX, TiOx), в системах Me—С—О (где Me=Zr,
Hf, Та) стабильные фазы МеО не образуются. Все это обусловливает широ-
кие области гомогенности оксикарбидов титана, ванадия [1—3], отчасти
ниобия [4] и заметно более узкие для ТаСхОу [5] и, по-видимому, для
ZrCxOy, HfCxOy. В этой связи данные, полученные Е. Е. Котляр с соавто-
рами [39], представляют большой интерес и нуждаются в дальнейшем под-
тверждении.
Совсем не исследованы препараты с малыми концентрациями углерода.
Не исключена возможность, что на основе твердого раствора a-Zr(O) также
могут формироваться гексагональные оксикарбиды, близкие к типу Ме2(С, О),
64
характерному для систем V—С—О, Nb—С—О, Та—С—О [3—5]. Правда,
здесь происходит внедрение меньших по размеру атомов кислорода (0,66 А)
в октаэдрические пустоты ячеек V2C, Nb2C, Та2С, частично занятых крупными
О
атомами углерода (0,77 А). В случае фазы a-Zr(O) дело обстоит как раз
наоборот. Однако следует учесть, что размер октаэдрических пустот в эле-
ментарной ячейке a-Zr (О) заметно больше (0,66), чем V2C (0,55), Nb2C
(0,60) и Та2С (0,60 А).
В связи с упомянутым в работе [39] оксикарбидом ZrO2C вызывает инте-
рес участок диаграммы ZrO2—С. О стабилизации кубической (тип CaF2)
двуокиси циркония примесями углерода уже указывалось. Учитывая, что
в структуре (идеальной или искаженной СаР2) все октаэдрические пустоты
остаются свободными, можно предположить постепенное их заполнение ато-
мами углерода вплоть до состава ZrO2C. Нечто подобное, в частности,
наблюдается в случае оксинитридов циркония, образующихся на основе
ZrO2 [41].
Несколько слов о концентрационной зависимости межплоскостных рас-
стояний в оксикарбидах циркония. Данных о кубической ZrQtOy-фазе очень
мало, поэтому сопоставляем характеристики лишь трех образцов (с х=const
и у=const):
о;
а, А
ZrCo,7O........................................ 4,6941
ZrCo,7oOo,o5 ....................................4,696
ZrCo.7iOo,o8 ....................................4,686
ZrCo,7oOo,23 ........... ........................ 4,561
Видно, что с увеличением у периоды решетки ZrCxOy убывают. То же самое,
исключая многие другие аналогичные кубические фазы внедрения, имеет
место и в оксикарбидах титана. Однако в последнем случае такая концен-
трационная зависимость периодов решетки объясняется тем обстоятельством,
что с увеличением содержания кислорода в ТЮх-фазе параметры ее решетки
убывают. Причина уменьшения а в случае ZrCxOy пока неясна. Возможно,
что это обусловлено ростом концентрации вакансий в подрешетки металла
с увеличением содержания кислорода. Во всяком случае этот вопрос требует
дальнейшего экспериментального разрешения.
К настоящему времени сведения о комплексности элементарной ячейки
ZrCxOy отсутствуют. Однако можно полагать, что у оксикарбидов циркония,
богатых по кислороду, должна быть дефектность не только в металлоидной
(по крайней мере для составов, где х+у<1), но и в металлической подре-
шетке. Действительно, кубические фазы VCxOy; TiCxOy, NbCxOy, NbOyN2
характеризуются вакантностью в обеих подрешетках. Это объясняется боль-
шими перекрываниями атомных функций соответствующих металлов, в част-
ности, обусловливающих двойную дефектность их моноокисей. Ранее уже
отмечалось [34], что в силу этих причин в случае формирования кубической
моноокиси циркония, последняя обладала бы весьма значительной концентра-
цией вакансий. Это, по-видимому, должно сказываться и на комплектности
решеток оксикарбидов циркония, особенно богатых по кислороду. Рост дефект-
ности решетки оксикарбида циркония с увеличением содержания в нем кисло-
рода можно предполагать также из следующих соображений. В работе [42]
указывается, что увеличение комплектности ячейки кубической УСхОу-фазы
с ростом концентрации углерода обусловлено тенденцией к сохранению по-
стоянства числа внешних электронов на пару узлов элементарной ячейки
(Ne/4) в области существования оксикарбида ванадия. То же самое, по-ви-
димому, можно ожидать и в случае оксикарбидов циркония. Так, например,
если в ZrCxOy заменить один атом углерода (четыре внешних электрона)
5
Заказ № 632
65
атомом кислорода (шесть электронов), то для сохранения постоянства Ne/4
(при x+y=const) необходимо удалить из решетки часть атомов Zr.
О наличии вакансий в подрешетке циркония, очевидно, говорит суще-
ствование оксикарбида состава ZrCo,eoOo,57 [37]. Здесь х+у=1,17, а его
период решетки 4,583 А. Согласно последним данным [19], это значение
периода заметно меньше amin для ZrCx (4,6941 А).
Таким образом, из сказанного выше следует, что подробных сведений,
касающихся условий синтеза, термической устойчивости, структурных и дру-
гих характеристик оксикарбидов циркония пока не имеется, поэтому целесо-
образны систематические их исследования. Не исключена возможность, что
могут быть получены новые материалы с ценными и интересными свойствами.
ЛИТЕРАТУРА
1. Н. Nischimura, Н. Kimura. J. Japan Inst. Met., 1956, 20, 524.
2. Л. X. Пивоваров, Е. Я. Вржец, Г. С. Креймер, Ф. Г. Закиров.
Ж. неорг. хим., 1967, 12, 1743.
3. М. А. Гуревич, Б. Ф. Орм о нт. Ж. неорг. хим., 1958, 3, 403.
4. С. И. А л я м о вс к и й, П. В. Г е л ь д, Г. П. Швейкин. Изв. АН СССР, Ме-
таллургия и горное дело, 1963, 6, 139.
5. Л. Б. Дубровская, С. И. Алямовский, Г. П. Швейкин, П. В. Г ельд.
Ж. неорг. хим., 1968, 13, 233.
6. Г. В. Самсонов, Я. С. Уманский. Твердые соединения тугоплавких метал-
лов. М., Металлургиздат, 1957.
7. Я. С. Уманский. Карбиды твердых сплавов. М., Металлургиздат, 1947.
8. У. Б. Блюменталь. Химия циркония. М., ИЛ, 1963.
9. Г. В. Самсонов, К. И. Портной. Сплавы на основе тугоплавких соединений.
М., Оборонгиз, 1961.
10. Г. В. Самсонов. Тугоплавкие соединения, М., Металлургиздат, 1963.
11. Т roost. С. R. Acad. Sci., 1893, 116, 1228.
12. A. van Ark el. Phisyca, 1924, 4, 286.
13. A. van Arkel, J. Boer. Z. anorg. allgem. Chem., 1925, 148, 347.
14. W. Burgers, J. Basart. Z. anorg. allgem. Chem., 1934, 219, 209.
15. A. E. Ковальский и T. Г. Макаренко. Микротвердость. М., Изд-во
АН СССР, 1951, стр. 187.
16. Г. В. Самсонов, Н. С. Рози нова. Изв. сект. физ.-хим. анализа АН СССР,
1956, 27, 126.
17. F. Benesovsky, Е. Rudy. Planseeber. Pulvermet., 1960, 8, 66.
18. Н. Nowotny, F. Benesovsky, E. Rudy. Monatsheft. Chem., 1960, 91, 348.
19. R. V. Sara. J. Amer. Ceram. Soc., 1965, 48, 243.
20. H. Bittner, H. Goretzki. Monatsheft. Chem., 1962, 93, 1000.
21. JO. И. Вильк, С. С. Орданьян, P. Г. Аварбе, А. И. Августиник,
Г. П. Рыжова, Ю. ГА. Омельченко. Высокотемпературные неорганические
соединения. Киев, «Наукова думка», 1965.
22. В. С. Куцев. Исследование равновесия в системе Ti—С—О. (Канд, дисс., ру-
копись). М., 1958.
23. Н. G о г е t z k i. Phys. Status Solidi, 1967, 20, K141.
24. С. H. Noviori, R. Lorenzelli, P. Costa. Compt. Rend., 1966, 263, B775.
25. K. Yvo on, H. Nowotny, R. Kieffer. Monatsheft. Chem., 1967, 98, 34.
26. A. L. Bowman, T. C. Wallace, J. L. Jarnell, R. J. Wenzel
E. K- Storms. Acta Cryst., 1965, 19, 6.
27. R. F. Domagala, D. J. Me Pheson. U.S.A.E.C. Publ. COO, 1953, 181;
J. Metals, 1954, 6, 238.
28. B. Hol mb erg, T. Dagerhamm. Acta Chem. Scand., 1961, 15, 919.
29. Г. П. Швейкин, С. И. Алямовский, П. В. Гельд, H. В. Баусова.
Химия соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН СССР,
1967, вып. 14, стр. 23.
30. И. И. К о р н и л о в, В. В. Глазова. Изв. АН СССР, Неорганические материалы,
1965, 1, 1778 и 1834.
31. Г. А. Меерсон, Г. В. Самсонов. Ж. прикл. хим., 1952, 25, 744.
32. N. Shonberg. Acta Chem. Scand., 1954, 8, 627.
33. Г. В. Самсонов. Укр. хим, ж. 1957, 23, 287.
34. В. А. Цхай, П. В. Гельд, Г. П. Швейкин, В. А. Переляев. Химия высоко-
температурных материалов. Л., «Наука», 1965, стр. 19.
66
35. В. С. Куцев, Б. Ф. Ормонт, В. А. Эпельбаум. Докл. АН СССР, 1955,
104, 567.
36. В. И. Желанкин, В. С. Куцев, Б. Ф. Ормонт. Ж. неорг. хим., 1958 , 3,
1237.
37. Т. Я. Косолапова, В. Б. Федорус, Ю. Б. Кузьма. Изв. АН СССР,
Неорганические материалы, 1966, 2, 1516.
38. Е. Е. Котляр, Т. Я. Косолапова, Т. Н. Назарчук. Изв. АН СССР,
Металлы, 1967, 1, 200.
39. J. Р. Guerlet, Р. Lehr. С. R. Acad. Sci., ser. С., 1967 , 264, 1833.
40. Г. П. Швейкин, С. И. Алямовский, П. В. Гельд. Физико-химические
исследования соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН
СССР, 1966, вып. 9, стр. 12.
41. J.-C. Gilles. Rev. Hautes Temper, et Refract, 1965, 2, 238.
42. E. H. Щетников, П. В. Гельд, Г. П. Швейкин, С. И. Алямовский.
Изв. АН СССР, Неорганические материалы, 1966, 2, 91.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
Ю. Г. ЗАЙНУЛИН, С. И. АЛЯМОВСКИЙ,
Г. П. ШВЕЙКИН, | Е. Н. ЩЕТНИКОВ, | П. В. ГЕЛЬД
РАСТВОРИМОСТЬ БЕРИЛЛИЯ В КУБИЧЕСКИХ КАРБИДАХ
Ti, V, Nb, Та
Одним из основных путей создания высокопрочных материалов, необхо-
димых для новой техники, является их синтез из тугоплавких «металлидов»
(карбидов, нитридов) и металлов [1]. В литературе описан ряд систем с уча-
стием карбидов IVa — Va подгрупп и некоторых металлов [2]. Однако работ,
посвященных изучению взаимодействия указанных карбидов с бериллием, не
имеется. В настоящем сообщении приводятся результаты исследований о
растворимости бериллия в кубических (типа NaCl) карбидах Ti, V, Nb, Та.
Исходными препаратами служили карбиды состава TiCo.65, TiCo.91, VCo.eo,
NbCo.72, NbCo,77, ТаСо,7б, а также металлический бериллий чистоты 99,5%.
Брикетированные (предварительно тщательно перемешанные и истертые) смеси
исходных компонентов, содержащие от 2,9 до 30 ат. % Be (через каждые
3 ат.%), вместе с соответствующим карбидом, взятым в качестве эталона,
обжигали в вакууме (10-4—10~5 мм рт. ст.) вначале при 1100° С в тече-
ние 10 ч, а затем при 1500° С — 40 ч. Каждый раз после 10-часового спека-
ния образцы перебрикетировали и подвергали рентгенофазовому анализу.
Рентгеносъемки производили в СиКа -излучении на порошковых камерах диа-
метром 114,6 мм. Полученные образцы выборочно анализировали на Весво6,
Весвяэ, Ссмз и переходный металл. Плотности определяли вакуумно-пикномет-
рическим методом с использованием в качестве рабочей жидкости декалина.
Как показали рентгенографические исследования образцов, спеченных при
1100° С в течение 10 ч, заметной растворимости Be в рассматриваемых кар-
бидах практически не наблюдается. Она достигается для некоторых образцов
лишь после дополнительного 10-часового обжига при 1500° С. Коротко оста-
новимся на особенностях взаимодействия металлического бериллия с отдель-
ными карбидами Nb, Та, V и Ti при 1500° С.
NbCo.72 (NbCo,77)+Be. Повышение температуры до 1500° С приводит к тому,
что после 10 ч обжига образцов на их рентгенограммах четко фиксируется
лишь одна кубическая фаза (типа NaCl), изменение периодов решетки кото-
рой свидетельствует об образовании твердого раствора Be в исходном карби-
де ниобия. С увеличением содержания Be период ячейки NbCxBe -фазы рас-
тет от 4,436 (для NbCo,72) до 4,460 А (для NbC0.74Be0.209)-В дальнейшем с
увеличением содержания Be параметры решетки кубической фазы остаются
постоянными. Последующий 30-часовой обжиг указанных препаратов не вно-
сит никаких изменений в рентгенографические характеристики образцов.
То же самое наблюдается и в случае исходного карбида NbCo,77- По резуль-
татам рентгенографирования как NbCo.72, так и NbCo,7 7 в данных условиях
растворяют около 11 ат. % Be.
68
ТаС0,7бЧ-Ве. В случае ТаС0,7б отмечена такая же картина, как и при
взаимодействии NbCx с Be. Периоды кубической решетки твердого раствора
с увеличением содержания Be возрастают от 4,423 до 4,438 А. Однако в
отличие от системы МЬСЛ— Be в образце состава ТаС0,7бВе0,4з помимо куби-
ческой фазы обнаруживаются дополнительные интерференции (не принадле-
жащие ни к Be, ни к его карбидам), интенсивность которых возрастает с
увеличением содержания Be. Они также, очевидно, принадлежат к трехком-
понентной ТаС,.Веу-фазе, которую пока не удалось идентифицировать. Раст-
воримость Be в ТаСо,7б достигает при этом приблизительно 12 ат. %.
VCo.eo + Be. Взаимодействие карбида ванадия с Be по сравнению с NbCx
и ТаСЛ протекает значительно медленнее. Так, лишь 30-часовое спекание
приводит к образованию кубического твердого раствора VCJJe^, периоды ре-
шетки которого также растут с увеличением содержания Be. Однако и 40-ча-
совой обжиг при 1500° С не приводит к полной гомогенизации образцов, со-
держащих V, С, Be. В твердых растворах NbCvBey и ТаС_,.Веу, то их рент-
генограммы уже после 10 ч прокалки при 1500° С характеризуются четкими
и разрешенными на ах и а2-линиями, начиная с углов 2'ft примерно 40°.
TiCo.gi (TiC0,65)4-Be. Обжиг препаратов TiC0,9i+yBeB течение 10 ч при
1100° С и 40 ч при 1500° С не привел к образованию кубического твердого
раствора TiCJBey. Действительно, во всех случаях на рентгенограммах на-
блюдается интерференционная картина кубической фазы с периодом решетки,
отвечающим исходному TiCo.gi. При съемке механической смеси. TiC0,9i,+
-f-15 ат. % Be на рентгенограмме не обнаруживается интерференций металли-
ческого бериллия вследствие его малой рассеивающей способности. Это сви-
детельствует, очевидно, об исключении вероятности образования твердых
растворов TiCxBey без изменения периодов решетки с составом. Вначале от-
сутствие твердых растворов TiCxBey связывалось нами с тем, что карбид
состава TiC0>91 в отличие от остальных использованных здесь карбидов об-
ладает максимальной комплектностью элементарной ячейки [3]. Это казалось
и обеспечивает «жесткость» ячейки TiCo.gi• Для проверки дополнительно был
использован карбид состава TiCo.65, содержащий наибольшее число вакансий
из всех исходных карбидов. Однако и в этом случае твердый раствор также
не образовывался.
В заключение несколько слов о типе кубического твердого раствора
NbCrBey. Элементарная ячейка исходного карбида ниобия составов NbCx,
где х< 1 (как и VCX, ТаСх, TiCj, характеризуется наличием вакансий в
подрешетке углерода, атомы которого занимают октаэдрические пустоты
в практически плотнейшей упаковке крупных атомов металла. Как уже ука-
зывалось, период решетки NbCxBey, а также TaCtBey, VCxBey с увеличени-
ем содержания Be возрастает. Очевидно, что при учете размерных соотноше-
ний такая концентрационная зависимость периодов решетки 1МЬСхВеу-фазы
может быть объяснена либо внедрением атомов Be (гВе=1,13, Гыь=1,45,
rv = 1,34, гт1 = 1,46, гТа=1,46 А) в междоузлия Ме-подрешетки, либо их
вхождением в свободные октаэдрические пустоты углеродной подрешетки.
Действительно, вследствие высокой вероятности s — р-переходов [4] Be мо-
жет вести себя и как металлоид. Анализ экспериментальных плотностей (р)
и рассчитанных на их основе чисел частиц Nb(ziNb), Ве(иВе) и C(zic) (см.
таблицу), приходящихся на элементарную ячейку, с одной стороны, указы-
вает на вероятность образования твердого раствора по типу замещения — внед-
рения. Иначе говоря, часть атомов Nb замещается меньшими атомами Be,
а рост периодов решетки обеспечивается частичным внедрением атомов бе-
риллия в междоузлия Ме-подрешетки. Во всех случаях пмь+«ве>4 и рас-
тет с увеличением содержания Be. С другой стороны, не исключена воз-
можность внедрения бериллия в металлоидную подрешетку. Правда, при
69
Характеристика некоторых образцов твердого раствора Be в NbCv
Состав по данным химического ана- лиза о a, A p, г/см* Число частиц в элементарной ячейке
nNb п Be "с
NbC0>72 4,436 7,68 3,98 .— 2,87
NbC0>77 4,445 7,70 4,01 .—. 3,09
NbC0t74Be0 jQg 4,449 7,46 3,88 0,19 2,86
NbC0,74Beo, Ю5 4,456 7,30 3,79 0,40 2,99
NbC017eBe0,05 4,448 7,45 3,84 0,19 3,03
NbC0,79Be0>105 4,451 7,30 3,75 0,39 2,96
NbCO}79Beo ,14 4,456 7,23 3,72 0,52 2,94
Примеча ни e. Да = 0 001 A; Aa = = 0,01— 0,02 г/см.
этом будут образовываться более крупные по размерам вакансии в Nb-под-
решетке, способствующие уменьшению периодов ячейки. Сопоставление экспе-
риментальных и рассчитанных интенсивностей (из предположения образова-
ния либо одного, либо другого типа твердого раствора) не приводит к одно-
значным выводам. Из-за малой рассеивающей способности атомов бериллия
теоретические интенсивности для обеих моделей твердого раствора практиче-
ски не различаются. Однако, на наш взгляд, следует отдать предпочтение
твердому раствору замещения — внедрения.
В этом случае трудность образования кубических твердых растворов Be с
VC, и TiCx может быть объяснена в последних более сильными, чем в NbCx
и ТаСЛ, Me — С- и Me — Ме-взаимодействиями, которые предварительно необ-
ходимо разорвать для замещения V или Ti бериллием. В пользу этого гово-
рит, в частности, сопоставление характеристических температур (0). У кар-
бидов титана, ванадия, ниобия и тантала эта величина, по литературным
данным, характеризуется следующими числами:
[5] [6]
TiC . ........................... 841 741
VC ............................. 750 531
NbC.............................. 660 470
ТаС ............................. 454 318
[7]
910, 980
940
780, 805
630
Хотя 0мес, по данным [5—7], и отличаются друг от друга, однако после-
довательность 0т1с>0ус>9кьс>0тас всюду сохраняется.
выводы
Исследовано взаимодействие высших карбидов Ti, V, Nb, Та с металли-
ческим бериллием при 1500° С в вакууме. За исключением карбида Ti взаи-
модействие остальных при указанной температуре приводит к образованию
кубических (типа NaCl) твердых растворов, построенных, по-видимому, по
типу замещения — внедрения. Трудность образования кубических твердых
растворов Be в VCX и их отсутствие в TiCx объясняются в последних более
сильными, чем в NbCx и ТаСЛ, Me — С- и Me — Ме-взаимодействиями.
ЛИТЕРАТУРА
1. И. И. Корнилов. Физико-химические основы жаропрочности сплавов. М., Изд-во
АН СССР, 1961.
2. Б. К. Вульф. Тройные металлические фазы в сплавах. М., Металлургиздат, 1964.
3. Г. В. Самсонов, Я. С. Уманский. Твердые соединения тугоплавких метал-
лов. М., Металлургиздат, 1957.
4. Г. В. С а м с о н о в. Бериллиды. Киев, «Наукова думка», 1966.
5. И. Н. Францевич. Вопросы порошковой металлургии и прочности материалов,
1956, вып. 3, стр. 14.
6. Г. В. Самсонов, В. С. Нешпор. Там, же, 1958, вып. 5, стр. 3.
7. Б. Н. Ощерин. Порошковая металлургия, 1962, № 1, 11.
АКАДЕМИЯ НАУК СССР
уральский филиал
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
А. С. БОРУХОВИЧ, Л. Б. ДУБРОВСКАЯ,
И. И. МАТВЕЕНКО, П. В. ГЕЛЬД
ИССЛЕДОВАНИЕ МАГНИТНОЙ ВОСПРИИМЧИВОСТИ КУБИЧЕСКИХ
КАРБИДОВ НИОБИЯ И ТАНТАЛА В ОБЛАСТИ НИЗКИХ ТЕМПЕРАТУР
Измерению магнитной восприимчивости монокарбидов (%) переходных ме-
таллов IV и V групп периодической системы посвящен ряд работ [1—4,7].
Установлена концентрационная зависимость магнитной восприимчивости ряда
монокарбидов и мононитридов переходных металлов при комнатных темпера-
50 100 ' 150 200 250 Гл
Рис. 1 Температурная зависимость магнитной восприимчивости
монокарбидов NbCr и ТаСх.
'-*'ЬСО>И. 2 - NbC0j9„ - NbCn„3S. '-NbC0>,0. 5-NbC0)7n,
в-ТаС0,78- 7~ТаС0,^ *~ТаС0„4.
турах [1]. Изучена зависимость %(Т) монокарбидов ниобия и тантала в их
областях гомогенности в интервале температур 300—1300° К 12, 3]. Было
показано, что для некоторых карбидов ниобия магнитная восприимчивость
описывается уравнением вида [2]:
Х=а+ЬТ2,
а для карбидов тантала она слабо зависит от температуры [3]. При этом
dY
температурный коэффициент — меняет знак с составом, причем для карби-
dT"
дов, близких к NbCo.eo и ТаСо.78, он больше нуля, а для близких к стехио-
метрии NbCo,98 и ТаСо.97 — меньше.
В настоящей работе исследовалась восприимчивость изученных выше мо-
нокарбйдов в области низких температур 20—300° К. Образцы карбидов
ниобия содержали от 8,24 до 11,24, а карбиды тантала от 4,68 до
71
6,05 вес. % связанного углерода. Методики приготовления и результаты хими-
ческого и рентгенографического анализов образцов изложены ранее [2—4].
Все образцы контролировали на ферромагнитные примеси химическим анали-
зом и по зависимости %(Н). Загрязнение их не обнаружено. Измерения про-
водили на магнитных весах, описанных в работе [5], и на торзионных весах
системы Свечкарева [6].
На рис. 1 представлены результаты измерения магнитной восприимчивости
монокарбидов NbCt и ТаСЛ указанных выше составов. Видно, что в исследо-
ванной области температур магнитная восприимчивость NbCx незначительно
меняется с температурой, а для ТаСх эта зависимость еще слабее. Для всех
изученных нами образцов карбидов (за исключением NbCo.eo ниже 50°К)
магнитная восприимчивость вплоть до 30° К описывается уравнением
Х=Хо+аЛ
где %0 — значение % при 7=0° К, получаемое экстраполяцией прямолинейной
зависимости % (Т); а=— — тангенс угля наклона прямой к оси абсцисс.
dT
Определенные таким образом значения Хо и а для разных составов об-
разцов следующие:
Хо -1 О’. эме/г
а-1010, эме]г-град
NbC0,,0.................... 0,40 +0,26
NbC0(80 ........................ -2,35 +2,00
NbC0(835 ....................... -0,74 - 0,23
NbC0>ee.................... 1,10 —0,43
NbCo,oe................... 1,85 -0,33
TaC0,74.................... 0,17 +0,40
TaC0,78 ................... —1,45 +1,13
TaC0183 ........................ —1,00 +0,60
TaC9t97 ................... 0,745 —0,33
Как видно, и в области низких температур наблюдается минимум х для
составов NbCo.eo и ТаСо,78» а также закономерное изменение а при переходе
Рис. 2. Распределение
плотности электронных
состояний в Me—Ме-
полосе карбидов со
структурой типа NaCl.
штриховая линия —
граница Ферми.
NbCo,7o- г-ТаСв.,а>
г-ТаСо,78> <- ИЬСо.зо.
5~NbCo,M- *-ТаС0>в„
что приводит к резкому
через этот минимум. Это подтверждается и схемой
распределения плотности электронных состояний,
построенной по результатам [2, 3] и настоящей ра-
боты (рис. 2). Полученные результаты согласуются
с моделью энергетического спектра электронов,
предложенной Бильцем [7] для фаз внедрения со
структурой типа NaCl, если предполагать, что основ-
ной вклад в восприимчивость дают электроны прово-
димости. Справедливость такого предположения
была ранее обоснована [2, 3].
Появление «хвостов» и резкое изменение темпе-
ратурного хода х в области Т^20°К у монокар-
бидов Nb и Та, близких к стехиометрии, можно
объяснить близостью критических температур пере-
хода в сверхпроводящее состояние, которое соп-
ровождается появлением пика в зависимости х (Т).
Особенно это видно на образце1 состава NbCo,g8,
имеющего Тк=11,7°К [8]. Аналогичная зависимость
наблюдалась в работе [9]. Резкое уменьшение маг-
нитной восприимчивости у NbCo.eo в области водо-
родных температур, возможно, связано с особен-
ностью полосы проводимости карбида этого состава,
увеличению собственного магнитного момента элек-
тронов проводимости при этих температурах. На это, в частности, указывает
появление кюри-вейссовской зависимости % У NbCo.eo ниже 30° К.
1 Этот образец измеряли вплоть до 13° К.
72
выводы
1. Измерена магнитная восприимчивость монокарбидов ниобия и тантала
в областях гомогенности при температурах 20—300° К. Резкое возрастание
X вблизи температур перехода в сверхпроводящее состояние наблюдается у
близких к стехиометрии карбидов.
2. Обнаружено появление кюри-вейссовской зависимости восприимчи-
вости у NbC0,80 ниже 30° К.
3. Приведены значения коэффициентов уравнения, описывающего зависи-
мость х = х(7', х) для всех измеренных образцов.
ЛИТЕРАТУРА
1. Н. Bittner, Н. Goretzki. Monatsh. Chem., 1962 , 93, 1000.
2. И. И. Матвеенко, Л. Б. Дубровская, П. В. Гельд, М. Г. Третни-
ков а. Изв. АН СССР, Неорганические материалы, 1965, 1, 7, 1062.
3. Л. Б. Дубровская, И. И. Матвеенко. Физ. металлов и металловед., 1965,
18, 2, 199.
4. Л. Б. Дубровская, Г. П. Швейкин, П. В. Гельд. Физ. металлов и ме-
талловед., 1964, 17, 1, 83.
5. Л. Б. Дубровская, И. И. Матвеенко, Р. А. Климов. Физические свой-
ства сплавов. Свердловск, 1965, стр. 62.
6. В. И. Чеч ерников. Магнитные измерения. М., Изд-во МГУ, 1963.
7. Н. В i 1 z. Z. Phys., 1958, 153, 338.
8. A. L. Giorgi, Е. G. Sklarz, Е. К. Storms, A. J. Bowman, В. Т. Matt-
hias. Phys. Rev., 1962, 125 , 3 , 837.
9. Т. Н. Geballe, В. Т. Mattias, J. Р. Remeika, А. М. Clogston,
V. В.'Compton, J. Р. М a i t а, М. I. Williams. Phys., 1966, 2, 293.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА ХИМИИ
1970
В. Г. ЗУБКОВ, И. И. МАТВЕЕНКО, Л. Б. ДУБРОВСКАЯ,
Г. Д. БОГОМОЛОВ, П. В. ГЕЛЬД
ОБ УПОРЯДОЧЕНИИ В СТРУКТУРЕ ОКСИКАРБИДОВ ТИТАНА
Ранее1 было установлено, что изоморфные моноокись ТЮ и монокарбид
TiC образуют непрерывный ряд взаимных твердых растворов так называемых
оксикарбидов со структурой типа NaCl, в которой атомы металла образуют
ГЦК-упаковку. Вопро'с о характере расположения атомов металлоидов не мог
быть решен на основании рентгеноструктурных исследований ввиду малых
амплитуд рассеяния рентгеновских лучей атомами углерода и кислорода.
Между тем экспериментальные данные (устойчивость образцов против окис-
ления на воздухе, в минеральных кислотах) обнаруживали особенности вбли-
зи состава TiCo.sOo.s-
Мы измерили электросопротивление образцов разного состава в ряду
твердых растворов TiOi.o— TiCi.o и получили зависимость, характерную для
упорядочения систем, с широким минимумом вблизи состава TiCo.sOo.s и
двумя максимумами вблизи составов TiC0.12O0.93 и TiCo.erOo, 12- Для выясне-
ния характера упорядочения в структуре оксикарбидов титана провели ней-
тронографическое исследование образца TiC0.43O0.58, электросопротивление
которого близко к минимуму.
На нейтронограмме наряду с линиями, разрешенными для структуры типа
NaCl, обнаружены сверхструктурные рефлексы, которые удалось проиндици-
ровать в кубической примитивной ячейке с параметром а=4,280 А.
Были предложены две возможные модели упорядоченного расположения
атомов в структуре оксикарбида TiCo.sOo.s:
4Ti 2C 20
a) (0, 0, 0) (0, V2, 0) е/2,о, о)
(V2, V2, 0) (0, V2, V2) (V2, 0, V2) (V2, V2. V2) (0, 0, Va)
6) (0, 0, 0) (Va, 0, 0) (Va, Va, Va)
(Va, V2. 0) (0, V2, V2) (V2, 0, 1/2) (V2, ’/г, Va) (0, Va, 0)
Все другие способы расположения атомов Ti, С, О приводят либо к куби-
ческой решетке с удвоенным параметром, либо к понижению сингонии.
В случае отклонения от состава TiCo.sOo.s в ту или иную сторону атомы из-
быточного металлоида комплектно заполняют собственные позиции и стати-
стически занимают позиции недостающего металлоида.
К сожалению, вопрос об однозначности в определении типа упорядоче-
ния в случае поликристалла не может быть решен из-за близости амплитуд
когерентного рассеяния нейтронов для атомов углерода и кислорода и из-за
высокой симметрии исследуемого объекта.
1 См. Nishimura, Kimura. Japan. Inst. Met., 1956, 20 , 524.
74
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА химии
1970
В. М. ЖУКОВСКИЙ, А. С. ВИНОГРАДОВА,
Е. В. ТКАЧЕНКО
\
КИНЕТИКА ТВЕРДОФАЗНОГО СИНТЕЗА МОЛИБДАТА МАГНИЯ
В СТЕХИОМЕТРИЧЕСКИХ СМЕСЯХ МоО3 И MgCO3
Интерес к твердофазному синтезу разного рода молибдатов за последнее
время заметно возрос, что обусловлено рядом ценных свойств [1—4]. Одна-
ко условия их получения, кинетика и механизм синтеза пока изучены слабо.
Об особенностях синтеза молибдата магния при взаимодействии трехокиси
молибдена с карбонатом магния имеется пока единственная работа [5], в
какой-то степени характеризующая условия получения молибдата магния, но
совершенно не освещающая кинетических закономерностей и механизма про-
цесса.
Настоящая работа посвящена изучению влияния ряда факторов (темпера-
туры, степени контакта реагентов, давления и природы газовой фазы, круп-
ности частиц карбоната магния) на кинетику синтеза молибдата магния в
твердой фазе по реакции
МоОз (Tj+MgCOa (Т) =MgMoO4 (?) +СО2 (Г>. (1)
В опытах использовали кристаллический карбонат магния (магнезит Саткин-
ского месторождения) и трехокись молибдена, полученную прокаливанием
молибденовокислого аммония квалификации «х. ч.» [6]. Чистоту исходных
веществ оценивали методом спектрального анализа. Примерное содержание
примесей, %:
МоО3: Mg — 0,01; Si —0,01; Са -- 0,01; Al<0,01; Fe — следы; Со и
Zn —0,01.
MgCO3: Fe —0,3; Si —0,003; Al<0,001; Cu>0,001 <0,01; Mn>0,001 <
<0,01; Ca —0,001.
Исследование кинетики образования MgMoO4 проводили при 550—620° С
на порошкообразных и брикетированных смесях MgCO3: МоО3=1 :1 в атмо-
сфере воздуха и углекислого газа в интервале давлений 10—760 мм рт. ст.
Для изучения влияния крупности частиц магнезита на кинетику взаимо-
действия (1) использованы четыре фракции MgCO3:
1) тонкорастертая, с размером частиц менее 0,020 мм; характеризующаяся
удельной поверхностью —0,3 м2/г;
2) гч=0,020—0,0225 мм;
3) гч=0,040—0,045 мм;
4) гч=0,100—0,125 мм.
Во всех случаях применяли тонкорастертую трехокись молибдена с удельной
поверхностью — 1 м2/г. Смеси трехокиси молибдена с карбонатом магния
определенной крупности готовили тщательным смешением компонентов.
Кинетические исследования проводили на установке, принципиально не от-
75
личающейся от описанной ранее [7]. Чувствительность пружинных весов
Мак-Бэна составляла 0,256 мг/деление шкалы отсчетного микроскопа МИР-2,
навеска образца во всех опытах была примерно одинаковой (120 мг). Смеси
разогревали в заранее нагретой до нужной температуры печи, в которую помещали
кварцевую трубку. Температуру контролировали потенциометром типа КП-59
в комплекте с платино-платинородиевой термопарой, укрепленной на наруж-
ной стенке трубки. Разница температур между наружной частью трубы и
навеской составляла не более 5° С; ее учитывали.
Рентгенограммы исходных веществ, промежуточных, конечных продуктов
реакции и эталонного образца молибдата магния снимали на аппарате УРС-50 им
Рис. 1. Зависимость степени превраще-
ния (а) от времени и дисперсности пре-
паратов при 595°С.
Разложение: 1 — MgCO3 (тонкорастертый),
2 — MgCO3 (rq—0,045 мм); взаимодействие:
3 — в смесях MgCO3—МоО3 (тонкорастертая),
4 — МоО3—MgCO3 (гч=0,045 мм).
в СиКа -излучении при скорости враще-
ния счетчика 4 град/мин. Запись диф-
рактограмм проводили в интервале уг-
лов 9—40°. Эталонный образец MgMoO4
готовили прокаливанием эквимолярной
смеси MgO (квалификации «х. ч.»)
с МоО3 при 600° С в течение 6 ч.
Твердофазное взаимодействие в сис-
теме МоО3 — MgCO3 представляет собой
сложный физико-химический процесс,
складывающийся из ряда сопряженных
между собой последовательных и парал-
лельных стадий. Так, в изученном
нами температурном интервале наряду
с взаимодействием между МоО3 и
MgCO3 по реакции (1) образование
MgMoO4 возможно представить сово-
купностью таких реакций:
MgCO3(T)ZtMgO(T)+CO2 (г), (2)
MgO(T)+MoO3 (т) =MgMoO4 (т), (3}
т. е. через предварительную диссоциацию карбоната магния.
В свою очередь реакции (1) — (3) йогут осуществляться за счет:
а) непосредственного взаимодействия частиц реакционной смеси в местах
их тесного контакта;
б) возгонки трехокиси молибдена и транспорта ее частиц к поверхности
карбоната или окиси магния;
в) диффузионного переноса атомов или ионов реагирующих веществ через
кристаллическую решетку образовавшегося продукта реакции — MgMoO4.
Из-за большой сложности процесса вначале была изучена диссоциация
магнезита, взятого в чистом виде. Оказалось, что заметное разложение MgCO»
на воздухе происходит уже при 520° С, причем с повышением температуры
скорость диссоциации быстро возрастает. Полученные результаты согласуются
с данными других авторов по условиям разложения саткинских магнезитов
[8—10]. Заметно влияет на разложение степень дисперсности магнезита: чем
меньше частицы карбоната, тем больше скорость разложения. Кажущаяся
энергия активации процесса диссоциации MgCO3) рассчитанная по уравнению
Аррёниуса, составляла 50 ±5 ккал/моль.
Далее гравиметрическим методом была изучена кинетика взаимодействия
(1) в зависимости от температуры и крупности частиц магнезита. Было по-
казано, что с ростом температуры и увеличением степени дисперсности час-
тиц MgCO3 процесс протекает с большей скоростью и более полно.
На рис. 1 приведены сравнительные результаты кинетики взаимодействия
MgCO3 с МоО3 и разложения магнезита при температуре 595° С. Видно, что.
76
Рис. 2. Рентгенограммы исходных
веществ и продуктов взаимодействия
MoO3 + MgCO3:
a— MgO. б —МоО,: в —MgCO3; г —
исходная смесь MgCO3+MoO3; д — эта-
лон MgMoO(; е—промежуточный про-
дукт взаимодействия MgCO3 (rq=
= 0,125 jkjh)4-MoO „(а=30%); ж — про-
межуточный продукт взаимодействия
MgCO3 (гч=0, 125 л.«)-|-МоО, (а=90%);
3 — продукт взаимодействия в растертой
смеси MgCO3-)-MoO3 (а=30%); и — про-
межуточный продукт взаимодействия
MgCOs4-MoO, (а=60%); к—MgMoO.
(а= 100%); л — MgMoO, (эталон из смеси
MgCOj-f-MoO,).
10 20 30
1200 ' ............... । । । i । . L~............... т
40 80 120 20 60 Ю0
T. мин
Рис. 3. Обработка кинетики взаимодействия в смеси МоОэ—
MgCO3 по уравнениям. Гинстлинга (а), Яндера (б), Картера
(в), непосредственного взаимодействия (г).
1 - 618, 2 - 595, 3 - 570° С.
собственно процесс диссоциации магнезита осуществляется быстрее, чем твер-
дофазное взаимодействие (1). Поэтому можно было бы предположить, что
взаимодействие в смеси МоО3— MgCO3 непременно должно идти через ста-
дию предварительного разложения последнего. Однако диссоциация веществ,
Рис. 4. Зависимость скорости взаимодействия
в смеси МоО3—MgCO8 от давления прессова-
ния для разных степеней превращения.
1 — 30; 2 — 40; 3 — 50; 4 — 60; 5 — 70; 6 — 80%.
взятых изолированно, может резко отличаться от того же процесса, проис-
ходящего в присутствии «инициирующих» (способствующих диссоциации) или
«ингибирующих» (затрудняющих диссоциацию) веществ [11].
Меньшая скорость газовыделения из смесей МоО3 — MgCO3 по сравнению
с таковой из чистого препарата MgCO3 показывает, что присутствие трехоки-
си молибдена затрудняет разложение карбоната магния. Скорее всего это
обусловлено тем, что на поверхности частиц MgCO3 образуется плотный слой
молибдата магния, который пре-
пятствует удалению продукта
разложения — углекислого газа,
резко повышая его давление в
зоне реакции. Следовательно,
полученные экспериментальные
результаты можно истолковать
и таким образом, что в присут-
ствии МоО3 диссоциация MgCO3
подавлена и решающая роль при-
надлежит взаимодействию (1), в
свою очередь развивающемуся
через ряд последовательных ста-
дий. Это вполне обосновано еще
и потому, что средняя величина
кажущейся энергии активации
процесса (1), рассчитанная по
уравнению Аррениуса, состав-
ляет 62 + 5 ккал/моль, что на
12 к.кал/моль больше по сравне-
нию с величиной энергии акти-
вации процесса разложения карбо-
ната магния.
Для того, чтобы выяснить значимость каждого из возможных процессов
при образовании молибдата магния в смеси MgCO3 — Мо03 — непосредствен-
ное взаимодействие (1) или синтез MgMoO4 через предварительное разложе-
ние карбоната — [реакции (2) и (3)], — необходимо установить, возможно
ли в реакционной смеси присутствие окиси магния как продукта разложения
MgCO3. Был проведен рентгенографический анализ ряда образцов (рис. 2).
Из сопоставления рентгенограмм видно, что окись магния на промежуточных
этапах взаимодействия не обнаружена. Единственным продуктом взаимодей-
ствия между МоО3 и MgCO3 был молибдат магния.
Полученный результат может быть объяснен двояким образом. С одной
стороны, в присутствии трехокиси молибдена диссоциация карбоната магния
действительно в значительной степени подавлена, с другой — если диссоциа-
ция MgCO3 и имеет место, то образующаяся окись магния немедленно всту-
пает во взаимодействие с трехокисью молибдена, что приводит к образова-
нию молибдата. Из литературы известно [12, 13], что окислы, получающиеся
при разложении карбонатов, в момент своего образования очень активны
вследствие большой разупорядоченности их кристаллической решетки.
Таким образом, результаты рентгенографического анализа не дают одно-
значного ответа о роли отдельных элементарных стадий. Однако они пока-
зывают, что метод' непрерывного взвешивания, используемый нами для изу-
чения кинетики образования молибдата магния при взаимодействии в смесях
79
Рис. 5. Взаимодействие в порошкообразной (4)
и брикетированной (Б) смесях МоО3—MgCO3
в атмосфере углекислого газа и воздуха.
Р> мм pm. ст.: 1— 10 (воздух), 2—10 (СО2),
3 — 600 (воздух, 4 — 600 (СО2>-
MoOs—MgCO3, вполне правомерен. Убыль навески, обусловленная удалени-
ем углекислого газа, связана с единственным процессом — синтезом
MgMoO4 по суммарному уравнению (1).
В пользу того, что стадия разложения карбоната не играет решающей
роли, говорит ряд других опытных фактов. Во-первых, обработка эксперимен-
тальных результатов по известным кинетическим уравнениям показала, что
на начальных этапах (степень
превращения не более 20%)
результаты кинетических ис-
следований удовлетворительно
описывались уравнением
1—(1—(а)
Это уравнение выведено [14]
в предположении, что лими-
тирующей стадией процесса
является собственно твердо-
фазное взаимодействие (рис. 3).
На последующих этапах (сте-
пень превращения реагентов
более 20 %) эксперименталь-
ные результаты подчиняются
диффузионным кинетическим
уравнениям [14, 15]:
Яндера —
[1—(1—a)'^]2=k2 • т, (б)
Г инстлинга —
1 — 2/3fl — О — a)‘/*=k3-r, (в)
Картера —
[1+(г— l)a]V. +
+ (г-1)(1-'а)г/«=г+
+2(1-2)^, (г)
го
где klt k2, k3, kt — постоян-
ные; a — степень превращения
реагентов; т — время; г0 —
начальный радиус частицы;
2 — объем продукта реакции,
образованного на единицу из-
расходованного компонента,
т. е. отношение эквивалент-
ных объемов. Во-вторых, ис-
следование влияния предвари-
тельного брикетирования исходной смеси на процесс синтеза MgMoO4 в твер-
дой фазе вплоть до давлений 11100 кг/см2 показало (рис. 4), что повы-
шение давления прессования (Рпрс) увеличивает скорость процесса (1), которая
имеет максимальное значение при Рпрс~8000 кг!см2. Дальнейшее увели-
чение давления прессования приводило к уменьшению скорости взаимодей-
ствия. Максимум скорости особенно отчетливо проявляется на начальных
этапах взаимодействия (см. рис. 4, /^5).
Брикетирование реакционной смеси может влиять на скорость процесса
в силу двух обстоятельств: 1) улучшается контакт между реагирующими
80
веществами, что способствует ускорению твердофазной реакции; 2) при
прессовании уменьшается пористость брикета, вследствие чего может быть
затруднен отвод газообразных продуктов реакции и скорость суммарного
процесса в целом уменьшается. Следовательно, увеличение до определен-
ного предела скорости реакции с увеличением давления прессования сви-
детельствует о заметной роли непосредственного взаимодействия, особенно
на начальных этапах процесса, и сравнительно небольшой значимости ста-
дии диссоциации карбоната магния, которую брикетирование должно было
бы затруднять. Замедление реакции при значительных величинах давле-
ния прессования скорее всего обусловлено тем, что наиболее медленной
стадией становится стадия удаления углекислого газа из зоны реакции.
Это предположение кажется вполне обоснованным еще и потому, что наи-
большая скорость взаимодействия зафиксирована в порошкообразных сме-
сях, полученных разрушением брикетов. При этом степень контакта меж-
ду частицами компонентов оставалась высокой, а отвод газообразных
продуктов не был затруднен.
На кинетику синтеза молибдата магния при взаимодействии (1) оказы-
вает влияние давление и природа внешней газовой среды (рис. 5). Опыты
проводили на порошкообразной и брикетированной (Рпрс=8500 кг/см2)
смесях МоО3 с MgCO3 (гч=0,022—0,020 мм) в атмосфере углекислого газа
и воздуха при 595° С. Видно, что уменьшение давления приводит к увели-
чению скорости и полноты протекания процесса, причем реакция более
чувствительна к среде углекислого газа, чем воздуха. Ускорение процесса
при уменьшении давления газовой фазы может быть объяснено облегче-
нием десорбции углекислого газа, выделяющегося в результате реакции
(1). При этом в поры навески наряду с молекулами продукта реакции мо-
гут попадать и молекулы внешней газовой атмосферы. Таким образом, при
одинаковом значении внешнего давления парциальное давление СО2 внутри
образца больше в том случае, когда внешняя газовая атмосфера состоит из
<Х)2, а не из воздуха. Поэтому и скорость процесса в этом случае за-
медляется больше.
Анализ всей совокупности полученных нами экспериментальных дан-
ных позволяет высказать некоторые суждения об особенностях твердофаз-
ного взаимодействия в системе МоО3—MgCO3 при 595—620° С, единствен-
ный продукт которого MgMoO4.
Образование молибдена магния по реакции (1) представляет собой слож-
ный процесс, при котором варьируется механизм с изменением относитель-
ного значения отдельных его стадий как по ходу эксперимента, так и в за-
висимости от его условий. Так, на начальных этапах происходит главным
образом твердофазное взаимодействие, в котором по ходу процесса (с воз-
никновением продукта реакции) изменяется лимитирующая стадия, хотя
в какой-то мере нельзя исключить возможности предварительной диссо-
циации карбоната магния. По мере протекания процесса с образованием
слоя молибдата магния, разделяющего реагенты, взаимодействие стано-
вится возможным благодаря направленной диффузии реагирующих частиц
через слой MgMoO4, и диффузионные стадии начинают играть решающую
роль. Подчинение экспериментальных результатов диффузионным уравне-
ниям Яндера, Картера и Гинстлинга подтверждает это. Заключитель-
ная стадия взаимодействия — отвод газообразного продукта реакции (угле-
кислого газа) из зоны реакции. Поэтому при определенных условиях (до-
статочно малая пористость исходных смесей, достигнутая сильным сжа-
тием, увеличение давления внешней газовой среды) отвод его может лими-
тировать процесс образования MgMoO4.
Наконец, исходя из литературных данных, нельзя полностью исклю-
чить возможность образования молибдата магния при пространственном
6 Заказ № 632
81
разобщении реагентов, так как в исследованном нами температурном ин-
тервале трехокись молибдена способна возгоняться. Скорость ее возгонки,
в присутствии других веществ заметно возрастает [И].
ВЫВОДЫ
1. Изучено влияние температуры, крупности частиц реагентов, давле-
ния прессования реакционных смесей, природы и давления газовой фазы
на кинетику синтеза MgMoO4 в стехиометрических смесях МоО3—MgCO3.
2. Показано, что синтез молибдата магния представляет собой слож-
ный физико-химический процесс, протекающий через ряд параллельных
и последовательных стадий, относительная значимость которых меняется
как в зависимости от условий ведения процесса, так и по ходу последнего.
ЛИТЕРАТУРА
1. А. Н. Зобнина, И. П. Кисляков, Изв. АН. СССР, Неорганические мате-
риалы, 1966, 12, 2199.
2. А. Н. Зобнина, И. П. Кисляков, Н. В. Стрелина. Изв. АН СССР,
Неорганические материалы, 1965, 3, 507.
3. Н. А. Батраков. Синтез вольфраматов и молибдатов с целью получения новых
материалов для радиоэлектроники. Автореф. канд. дисс. Свердловск, 1965.
4. А. М. Гинстлинг, Т. П. Фрадкина. Ж. прикл. хим., 1952, 12, 1268,
6. Б. М. Рейнгольд, В. Н. С м а г у н о в. Сборник научных трудов ИРГИРЕДМЕТа,
1965, 12, стр. 330.
6. Ю. В. Карякин, И. И. Ангелов. Чистые химические реактивы М., Госхим-
издат, 1955.
7. В. Г. Власов, В. М. Жуковский. Кинетика и катализ, 1962, 3, 6, 882.
8. В. В. Печковский, А. Г. Звездин. Уч. зап. Перм. ун-та, 1960 , 7, 1, 33.
9. А. Г. Звездин. Исследование кинетики диссоциации некоторых сульфатов и карбо-
натов. Автореф. канд. дисс. Пермь, 1965.
10. А. А. Байков. Собрание трудов, т. 5. М., Изд. АН СССР, 1948.
11. С. Б. Шеболдаев. Исследование механизма взаимодействия твердых окислов.
металлов с углеродом. Автореф. канд. дисс. М., 1967.
12. Гетерогенные химические реакции. Под ред. М. М. Павлюченко, Минск, 1965.
13. Химия твердого состояния. Под ред. В. Гарнера. М., ИЛ, 1966.
14. П. П. Будников, А. М. Гинстлинг. Реакция в смесях твердых веществ.
М., Стройиздат, 1965.
15. Н. А. Торопов, В. П. Б а р з а к о в с к и й. Высокотемпературная химия силикат-
ных и других окисных систем. М., Изд-во АН СССР, 1963.
АКАДЕМИЯ НАУК,СССР
УРАЛЬСКИЙ ФИЛИАЛ
вып. 2е
ТРУДЫ ИНСТИТУТА химии
1970
Е. В. ТКАЧЕНКО, В. М. ЖУКОВСКИЙ,
Т. И. СЕЛИВАНОВА
ИЗУЧЕНИЕ УСЛОВИЙ ОБРАЗОВАНИЯ МОЛИБДАТА БАРИЯ
ПРИ ВЗАИМОДЕЙСТВИИ ВаСО3 с МоО3 В ТВЕРДОЙ ФАЗЕ НА ВОЗДУХЕ
Настоящая работа посвящена изучению условий образования молиб-
дата бария по реакции
ВаСОз(Т)+МоОз(Т) =ВаМоО4(Т) -|-СО2(г) (1)
Рис. 1. Термограмма смеси ВаСО3 :Мо08 = 1 : 1.
/ — кривая нагревания: 2 — дифференциальная кривая
нагревания; 3 — повторная дифференциальная кривая
нагревания; 4 — кривая газовыделения; 5 — кривая га-
зовыделения при повторном термографировании.
в смесях ВаСО8—МоО3 стехиометрического и переменного составов, содер-
жащих избыток того или иного компонента. Взаимодействие в указанных
смесях изучали с помощью
термографического, волюмо-
графического и рентгеногра-
фического методов исследо-
вания. Термографирование
проводили с одновременной
регистрацией тепловых эффек-
тов и объемов выделяю-
щегося углекислого газа
[1]. Скорость нагрева печи
35 град/мин. Навеска смеси
0,4 а. В качестве эталона
использовали ;окись алю-
миния. Запись дифференци-
альной кривой нагревания
и кривой газовыделения осу-
ществляли автоматическими
самопишущими потенциомет-
рами ПС-1-09. Строгая зависи-
мость между количеством
карбоната бария, вступив-
шего во взаимодействие, и
объемом выделившегося угле-
кислого газа позволяла коли-
чественно контролировать
ход процесса. Рентгеногра-
фический анализ выполняли на установке типа УРС-55. Рентгенограммы
снимали в камерах РКД с диаметром 57,3 мм в медном излучении. Время
экспозиции 2 ч.
В опытах были использованы карбонат бария квалификации «ч. д. а»,
и трехокись молибдена, полученная из молибденовокислого аммония [2].
Для контроля за однородностью исходных препаратов, а также с целью
6*
83
определения формы и размера их частиц порошки ВаСО3 и МоО3 просмат-
ривали в электронном микроскопе марки «Карл Цейсс Д-2» с разрешаю-
щей способностью 100 А. Частицы карбоната бария имели палочкообраз-
ную форму, с ярко выраженной кристаллической огранкой. Их примерный
размер 0,2—5 мк в длину, 0,2—1,5 мк в поперечнике. Образец трехокиси
молибдена полидисперсный, ча-
стицы неправильной формы,,
огранка кристаллическая. Раз-
мер частиц МоО3 колеблется в
пределах от 0,2 до 2 мк. Удель-
ная поверхность исходных пре-
паратов, определенная на при-
боре ПСХ-2, оказалась равной
~ 0,3 м2/г для ВаСО3 и 0,2 м2/г
для МоО3. Термографический и
рентгенографический анализы
показали полное совпадение
характеристик ВаСО3 и МоО3
с имеющимися в литературе
[3-51.
Известно [6], что теплота
образования молибдата бария по-
реакции (1) мала ккал/моль).
Поэтому в пределах чувстви-
тельности термографического
метода можно ожидать, что
эффект, связанный с образова-
нием ВаМоО4, не будет улавли-
ваться и на дифференциальной
кривой нагревания интервал
взаимодействия не определить.
Действительно, на термограмме
нагревания эквимолярной смеси
ВаСО3—МоО3 отчетливо выра-
женные эффекты отсутствуют
(рис. 1). Однако температурный
интервал взаимодействия можно
с большой точностью определять
газоволюметрически, регистри-
руя объем выделившегося в
Рис. 2. Термограммы нагревания (d) и кривые
газовыделения (б) смесей ВаСО3—Мо03 пере-
менного состава.
А: 1 — BaCOs; 2 — 10 : 1; 5 — 5:1; 4 — 2: I;
5 — 1:1.
Б: 1 — МоО3; 2 — 1:10;3 — 1:5;-#— 1:2; 5—1:1.
результате реакции углекислого газа. Видно, что по кривой газо-
выделения — образование молибдата бария прошло в интервале
440—750° С. Отсутствие тепловых эффектов на повторной кривой нагре-
вания той же смеси (рис. 1, кривая 3) указывает на два обстоятельства:
а) взаимодействие по реакции (1) прошло практически нацело; б) образо-
вавшийся молибдат бария устойчив, по крайней мере, до 1000° С. Сопо-
ставление полученных данных с литературными [7] показывает довольно-
хорошее совпадение их между собой. Так, интервал взаимодействия между
ВаСО3 и МоО3 на воздухе, определенный Н. А. Батраковым [7], составляет
400—750° С (по Тамману 460—700° С). °
Образование молибдата бария может проходить как при предваритель-
ном разложении карбоната бария [7,8], так и за счет возгонки МоО3 [9].
В ряде случаев подчеркивается большое значение диффузионных процес-
сов, обеспечивающих твердофазное течение реакции [9]. Индивидуально
84
взятый карбонат бария диссоциирует при температурах, на 500—700° С
превышающих температуру образования молибдата бария [3]. Поэтому
можно предположить, что и взаимодействие в смеси МоО3—ВаСО8 идет без
предварительного разложения последнего. Однако диссоциация веществ,
взятых в изолированном состоянии, может резко отличаться от того же
процесса, протекающего в присутствии «инициирующих» веществ (способ-
ствующих диссоциации) [10]. Поэтому целесообразно было проверить по-
ведение ВаСО3 и МоО3, взятых в нестехиометрических соотношениях, а
также и характер взаимодействия в подобных смесях. В литературе сведе-
ния подобного характера отсутствуют.
Характеристика твердофазного взаимодействия в смесях
ВаСО8—МоО3 переменного состава
ВаСОэ : МоО, Интенсивное взаи- модействие Газовыделение, мл Образо- вание ВаМоОд, %
теоретическое при полной диссоциации ВаСО3 теоретическое при взаимо- действии (1) экспери- менталь- ное
Интервал, °C время, мин
10:1 440—600 4,5 47,2 4,72 4,0 85
5:1 440—640 5,7 44,2 8,84 8,0 91
2:1 440—670 6,5 37,0 18,5 17,5 94
1:1 440—750 8,8 29,3 29,3 29,0 99
1:2 420—620 5,7 20,6 20,6 18,0 87
1:5 420—520 2,8 11,0 11,0 9,5 86
1:10 420-500 2,3 6,1 6,1 Э,0 82
Для исследования взяты смеси ВаСО3—МоО3 следующих составов1:
10 :1; 5 : 1; 2 : 1; 1 :2; 1 :5; 1 : 10. Результаты представлены на рис. 2, 3 и
сведены в таблицу. Для примера рассмотрим термограммы смеси ВаСО3—
МоО3 состава 1 : 5 и 5 : 1. Термографирование смеси ВаСО3—МоО3 состава
1 : 5 показало, что интервал наиболее интенсивного газовыделения состав-
лял 420—520° С, после чего выделение углекислого газа резко замедлялось,
но не прекращалось до конца термографирования. Взаимодействие в этом
интервале прошло за 2,8 мин на 86%. На дифференциальной кривой нагре-
вания смеси ВаСО3—МоО3 состава 1 : 5 виден глубокий эндотермический
эффект при 680° С, который можно отнести к эффекту плавления избытка
трехокиси молибдена (температура плавления изолированной МоО3 795° С
[41). Такое объяснение вполне оправдано, поскольку поведение МоО3 в при-
сутствии веществ, вступающих с ней в химическое взаимодействие, обычно
резко отличается от поведения чистой МоО3 (понижается температура воз-
гонки, температура плавления, при более низкой температуре наблюдается
разупорядоченность кристаллической решетки и т. д.) [10]. При термогра-
фировании смеси ВаСО3—МоО3 состава 5 : 1 (см. рис. 2, кривая 3) темпе-
ратурный интервал наиболее интенсивного взаимодействия 440—640° С,
реакция прошла за 5,7 мин на 91%. При 820° С на дифференциальной кри-
вой нагревания наблюдается эндотермический эффект, который соответ-
ствует переходу избытка карбоната бария из ромбической модификации
в гексагональную [3].
При анализе результатов обнаруживаются следующие закономерности:
1. Во всех случаях образование молибдата бария в смесях, содержащих
избыток МоО3, происходит при несколько пониженных температурах, чем
в смесях с избытком карбоната бария.
1 Смеси готовили тщательным смещением реагентов в агатовой ступе в течение
45 мин.
85
Рис. 3. Зависимость количества выделившегося угле-
кислого газа от состава реакционной смеси.
У—2 — теоретическое газовыделение при полной диссоциации
ВаСО, (У), при осуществлении взаимодействия (2); 3 — экс-
периментальная кривая.
2. В смесях переменного состава интенсивное взаимодействие идет в
более узком интервале температур, чем в смесях эквимолярного состава.
Температурный интервал взаимодействия сужается тем больше, чем
больше избыток одного из компонентов (см. рис. 2, 3).
3. Не только скорость, но и, в заметной мере, полнота процесса является
функцией состава смеси, ее «нестехиометричности». Наиболее полно взаи-
модействие завершалось в смесях эквимолярного состава.
4. Термограммы смесей переменного состава отличаются от таковых
стехиометрического состава еще и наличием глубоких эндотермических
эффектов, которые мож-
но отнести к превра-
щениям компонента,
находящегося в из-
бытке.
5. Наконец, было
замечено, что интен-
сивность взаимодействия
тем выше, чем совер-
шеннее контакт между
исходными реагентами.
Результаты, получен-
ные в нашей работе,
позволяют полагать, что
образование молибдата
бария происходит за
счет прямого взаимо-
действия исходных фаз
по реакции (1). В то
же время из данных
Н. А. Батракова [7],
Е. С. Жмудь и Е. П.
Остапченко [8], следует,
что взаимодействие идет
через предварительное
разложение карбоната
бария. Вывод о харак-
тере взаимодействия в этой смеси проверен специальным анализом резуль-
татов, полученных на смесях стехиометрического и переменного составов.
Количество углекислого газа, которое должно выделиться из смесей раз-
ного состава при навеске 0,4 г, было подсчитано, исходя из двух моделей:
а) взаимодействие в смеси ВаСО3—МоО3 идет без предварительного разло-
жения карбоната бария; б) только после предварительной диссоциации
ВаСО3.
Расчетные данные для смесей различного состава сравнили с экспери-
ментальными. Как оказалось, последние (рис. 4, кривая <3) полностью сов-
пали с расчетными, выполненными для случая непосредственного взаимо-
действия реагентов (рис. 4, кривая 2) без предварительной диссоциации кар-
боната бария. Об этом говорят характерные только для ВаСО3 пики на
термограммах смесей ВаСО3—МоО3, содержащих избыток карбоната бария
(см. рис. 2).
Наконец, рентгенографический анализ образцов, полученных в изотер-
мических условиях, показал отсутствие линий окиси бария на различных
этапах взаимодействия ВаСО3 с МоО3. Рентгенограммы показывают, что
с увеличением степени взаимодействия интенсивность линий исходных
компонентов убывает, а линии образующегося молибдата бария становятся
86
более яркими. Рентгенографирование конечного продукта реакции (1),
когда образование ВаМоО4 прошло практически полностью, показало, что
линии фаз Мо03 и ВаСО3 исчезли. Фиксировались только линии молибдата
бария. Рентгенографический анализ смеси ВаСО3—МоО3 состава 5 : 1
после термографирования показал, что конечный продукт взаимодействия—
молибдат бария. Наряду с линиями ВаМо04 обнаружены линии фазы ВаСО31
находившегося в избытке в исходной смеси компонентов. Линий ВаО не
обнаружено.
Таким образом, совокупность полученных данных свидетельствует о
том, что образование ВаМоО4 в смесях ВаСО3—МоО3 происходит без пред-
варительной диссоциации ВаСО3.
ВЫВОДЫ
1. С помощью термографического, волюмометрического и рентгеногра-
фического методов исследования в твердой фазе на воздухе изучены усло-
вия образования молибдата бария в смесях ВаСО3—МоО3 стехиометри-
ческого и переменного составов.
2. Показано, что образование ВаМоО4 в условиях непрерывного разо-
грева смеси ВаСО3—МоО3 наблюдается в интервале температур' 440—
750° С. Скорость процесса и его полнота зависят от состава и состоя-
ния навески.
3. Установлено, что взаимодействие в смеси ВаСО3—МоО3 происходит
без предварительного разложения карбоната бария.
ЛИТЕРАТУРА
1. Л. Г. Берг. Введение в термографию. М., Изд-во АН СССР, 1961.
2. Ю. В. Карякин, И. LH. Ангелов. Чистые химические реактивы. М., Госхим-
издат, 1955.
3. Г. Реми. Курс неорганической химии, т. 1. М., ИЛ, 1963.
4. О. А. С он г ин а. Редкие металлы. М., «Металлургия», 1964.
5. Л. И. Миркин. Справочник по рентгеноструктурному анализу поликристаллов. ЛА.,
Изд-во физ.-хим. лит., 1961.
6. Э. В. Брицке. А. Ф. Капустинский и др. Термические константы неоргани-
ческих веществ. М. —Л., Изд-во АН СССР, 1949.
7. Н. А. Батраков. Синтез вольфраматов и молибдатов с целью получения новых мате-
риалов для радиоэлектроники. Автореф. канд. дисс. Свердловск, 1964.
8. Е. С. Жмудь, Е. П. Остапченко. Ж. струк. хим., 1961, 2, 1, 33.
9. П. П. Будников, А. М. Гинстлинг. Реакции в смесях твердых веществ.
М., Стройиздат, 1965.
10. С. Б. Шеболдаев. Исследование механизма взаимодействия твердых окислов
металлов с углеродом. Автореф. канд. дисс. М., 1967.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. М. ПОЛЯКОВА, Л. Ф. МАЛЬЦЕВА, С. И. АЛЯМОВСКИЙ
ИССЛЕДОВАНИЕ СИСТЕМЫ ТЮ2—Rb2SO4—H2SO4—Н2О ПРИ 20° С
Настоящая работа является продолжением исследований по взаимодей-
ствию сернокислых растворов двуокиси титана с сульфатами щелочных
металлов. Одним из авторов этой статьи методом растворимости установ-
лено образование двойной сернокислой соли титана с калием в системе
TiO2—K2SO4—H2SO4—Н2О при 20° С [11. Данное исследование посвящено
изучению условий образования двойных сульфатов титана с другим аналогом
калия по периодической системе, а именно — с рубидием.
В литературе имеются сведения только о сульфатных соединениях руби-
дия с титаном (III) [2]. Из сернокислых растворов титана, восстановленного
до трехвалентного состояния, выделен двойной сульфат рубидия с титаном,
отвечающий формуле Rb2SO4 • 3Ti2 (SO4)3 • 24Н2О. Имеются указания [2] на обра-
зование рубидий-титановых квасцов RbTi(SO4)2-12Н2О. Двойные сульфаты
рубидия с титаном (IV), выделяемые из кислых сред, нам неизвестны.
Изучение растворимости в системе ТЮ2—Rb2SO4—H2SO4—Н2О прово-
дили изотермическим методом по общепринятой методике, использованной
в работе [1]. Состав полученных соединений определяли методом «остатков»
Скрейнемакерса и прямым химическим анализом их после промывки спиртом
или отжатия под прессом. В качестве исходных веществ при изучении
системы использовали сульфат рубидия квалификации «ч», сернокислый
раствор титана (26,5 г/л TiO2, 127,0 г/л H2SO4), приготовленный по мето-
дике [3], и серная кислота (уд. вес 1,82). Методика эксперимента, отбор
и анализ проб и построение диаграмм сообщены ранее [1]. Пробы переме-
шивали в термостате в изотермических условиях до наступления равновесия,
которое достигалось через четверо суток. Полученные результаты приведены
в табл. 1, 2 и на рис. 1.
Таблица 1
Растворимость в системе TiO2— Rb2SO4—H2SO4—Н2О при 20° С
Состав жидкой фазы, вес.% Солевой состав, вес. % Твер- дая фаза
тюг RbjSO, H2SO< H2O тю, Rb2SO2 SO,
1,08 0,79 4,34 3,03 7,36 11,77 87,22 84,41 9.44 5,88 38,00 22,56 52,56 71,56
0,65 2,82 14,29 82,24 4,28 18,90 76,82
0,32 2,15 18,65 78,88 1,81 12,17 86,02
0,19 1,98 21,94 75,89 0,95 9,87 89,91 C/j d от н *
Ю.13 1,69 26,29 71,29 0,56 7,24 92,20
0,08 1,95 29,53 68,44 0,31 7,46 92,23 СО
0,06 2,06 30,52 67,36 0,22 7,62 92,16
0,05 2,03 32,10 65,82 0,18 7,22 92,50 2Rb2S( •Н2
«8
Таблица 2
Химический анализ донной фазы, вес. %
TiO2 RbgSO, so, H2O Солевой состав
TiO, RbgSO, so3
17,28 29,22 14,94 30,56 28,12 47,56 24,32
13,87 31,51 20,09 34,53 21,18 48,13 30,69
13,53 30,36 21,42 . 34,69 20,71 46,49 32,38
13,68s 32,00 20,73 33,59 20,60 48,18 31,22
13,72 32,11 24,07 30,10 19,63 45,93 34,44
13,85 32,09 24,45 29,61 19,68 45,59 34,73
13,19 29,40 25,57 31,84 19,35 43,13 37,52
13,71 32,55 25,20 28,54 19,18 45,55 35,27
14,43 32,58 25,35 27,64 19,94 45,05 35,04
Из рис. 1 и табл. 1 видно, что в области концентраций серной кислоты
14,3—32,1 вес. % система имеет одну ветвь кристаллизации. В этих усло-
Рис. I. Растворимость в системе TiO2—Rb2SO4—H2SO4—Н2О.
виях выделяется только одна твердая фаза, отвечающая кислому кристалло-
гидрату титанилсульфата рубидия с соотношением Rb2SO4:TiO2=2:3. Гра-
фическое определение состава полученного соединения, подтвержденное
химическим анализом, дает следующие значения компонентов, вес. %: ТЮ2 —
22,12; Rb2SO4—’49,47; SO3'—28,41.
89
Вычислено для безводной соли состава
2Rb2SO4• 3TiOSO4• SOs вес. %: TiO2 —
21,91, Rb2SO4 — 48,81, SO3 —29,26.
Сопоставление расчетных и графических
Таблица 3
Растворимость Т О2 в зависимости
от TiO2:Rb2SO4 в исходном ра-
створе
Исходный раствор Состав жидкой фазы, вес. %
TiO,:Rb,SO, ТЮ RbjSO, HaSO, Н,О
1:3 1,00 2,18 14,74 82,08
1:4 0,49 2,84 14,51 82,16
1:5 0,16 3,92 13,26 82,66
1:6 0,095 5,85 13,38 80,22
1:7 0,065 6,81 14,14 78,99
Примечание. В исходном растворе
содержалось ТЮ2— 24, H2SO,—170 г/л.
данных по составляющим компонентам
дает удовлетворительное совпадение.
Химический анализ образцов твердых
фаз, промытых спиртом и высушенных
до постоянного веса или хорошо отжа-
тых под прессом, показал, что в иссле-
дуемой системе образуется кислый кристал-
логидрат титанилсульфата рубидия,
имеющий состав 2Rb2SO4 • 3TiOSO4 •
• H2SO4-nH2O. Количество кристалли-
зационной воды (п) зависит от ряда фак-
торов: от кислотности раствора, условий
высушиваний соли и др. В условиях нашего исследования п менялось от
3 до 9 молей Н2О на одну молекулу титанилсульфата рубидия.
Для 2Rb2SO4-3TiOSO4-H2SO4-7H2O:
Найдено, %:
Вычислено, %:
ТЮ, Rb,SO4 so, н„о
19,37 43,14 25,86 11,63
19,40 42,90 25,75 11,95
В табл. 3 показана зависимость растворимости двуокиси титана от кон-
центрации сульфата рубидия.
Видно, что с увеличением в растворе концентрации сульфата рубидия,
при прочих равных условиях, растворимость двуокиси титана уменьшается.
Согласно химическому анализу, состав выпадающих соединений в зависи-
мости от TiO2:Rb2SO4 аналогичен приведенному в табл. 1. Соль кислая
и’содержит кристаллизационную воду. О возможности получения кислых
солей рубидия из сернокислых растворов сообщается в работах [4, 5.] Кроме
того, с изменением условий кристаллизации при введении в сернокислый
раствор титана (IV) сульфата рубидия в твердом виде нами получена соль
с отношением TiOSO4:Rb2SO4=2:l в следующих условиях: исходный раствор
400 г/л H2SO4, 27 г/л ТЮ2; отношение TiO2:Rb2SO4=l :3,5; продолжитель-
ность кристаллизации 20 ч без перемешивания.
Соль с таким же отношением компонентов получается при сливании серно-
кислых растворов с концентрацией H2SO4—140—200, TiO2 — 25 г/л и вод-
ного 40%-ного раствора сульфата рубидия в количестве TiO2:Rb2SO4=l :4,5.
Соли имели состав Rb2SO4 • 2TiOSO4 • 6НаО. Кристаллизация этих солей про-
текала без перемешивания, в отличие от кислого кристаллогидрата, который
выделялся в равновесных условиях, достигающихся после длительного пере-
мешивания.
Термическое исследование типичного образца титанилсульфата рубидия
проводили на дериватографе системы братьев Паулик и Эрдей [5] в сле-
дующих условиях: скорость нагрева 10 град/мин, чувствительность ТГ
100 мг, ДТГ 1/10, Т 900° С, ДТА 1/10, навеска 100 мг. Термограмма
ДТА, термогравитограмма ДТГ и потери веса ТГ при нагревании титанил-
сульфата рубидия в атмосфере воздуха приведены на рис. 2. В интервале
100—400° С происходит выделение кристаллизационной воды (6Н2О), состав-
ляющей по ТГ 8,5% веса образца. Теоретический расчет для потери солью
6 молей ъоцы равняется 8,85%, что подтверждают экспериментальные дан-
ные. Потере кристаллизационной воды соответствуют слабо выраженные
тепловые эффекты на ДТА при 200, 285 и 400° С.
90
Рис. 2. Кривые нагревания двойной сернокислой
соли рубидия с титаном.
Рис. 3. Микрофотография кристаллов 2RbiS0<’3Ti0S04-H2SO4-6H2O, х4000.
Таблица 4
Межплоскостные расстояния (d)
и относительные интенсивности
линии (/) 2Rb2SO4-3TiOSO4-H2SO4-
•6Н2О (СиКа -излучение)
I d, А 1 G d, А
О. сл. 4,77 Сл. 2,540
Сл. 4,57 О. сл. 2,473
Сл. 4,30 Сл. 2,390
Ср. 4,14 Сл. 2,313
О. сл. 3,95 О. сл. 2,252
С. 3,78 Сл. 2,196
О. с. 3,62 О. сл. 2,111
С. 3,38 О. сл. 2,065
О. с. 3,30 Сл. 2,038
С. 3,18 О. сл. 1,982
О. с. 3,04 О. сл. 1,909
Ср. 2,88 Сл. 1,865
Ср. 2,84 О. сл. 1,835
Ср. 2,761 О. сл, 1,789
О. сл. 2,712 О. сл. 1,763
О. сл. 2,642
Примечание. О. ел. — очень слабо,
сл. —слабо, ср. — средце, о. с. —очень си-
льно, с. — сильно.
Это указывает на ступенчатый харак-
тер обезвоживания соли, причем потеря
основной массы воды, как видно по кри-
вой ТГ, протекает до 285° С.
Тепловой эффект при 500° С соответст-
вует началу распада соли на составляю-
щие компоненты1. В районе температур
500—700° С происходит удаление SO3
и Н2О, связанной с RbHSO4, суммар-
ные потери которых составляют 14,35%
веса по ТГ и сопровождаются неболь-
шими тепловыми эффектами на ДТА,
совпадающими с эффектами на ДТГ при
580, 618 и 678° С. Это также свидетель-
ствует о ступенчатом характере разложе-
ния соединения.
Эндотермический эффект при 705° С
отвечает формированию конечных про-
дуктов разложения титанилсульфата руби-
дия с появлением новых фаз — двуокиси
титана и персульфата рубидия, образую-
щегося при взаимодействии среднего суль-
фата рубидия с SO3 в момент его выде-
ления, что согласуется с литературными
данными [4]. Общая потеря веса об-
разца по ТГ (22,85%) близко отвечает
разложения по суммарной реакции
протеканию процесса термического
2Rb2SO4 • 3TiOSO4 • H2SO4 • 6Н2О+О2 2Rb2S2O8+
+ 3TiO2+7H2O|+2SO3t,
в соответствии с которой вычисленные потери веса составляют
Ниже приводятся результаты химического анализа остатка от
ния титанилсульфата рубидия и теоретически рассчитанный состав
вышеприведенной реакции, %:
23,45%.
нагрева-
согласно
Найдено...............
Вычислено для ЗТЮ2 +
+2Rb2S2O8..........
тю2 Rb2S2Oa STiO 2+ 4-Rb2S 2О2
26,10 72,83 98,93
, 24,83 75,17 100,00
Экспериментальные результаты удовлетворительно согласуются с вычис-
ленными; небольшое расхождение можно объяснить погрешностью химиче-
ского анализа. Таким образом, при нагревании кислой соли титанилсульфата
рубидия конечные продукты разложения — персульфат рубидия и двуокись
титана.
Электронно-микроскопический анализ титанилсульфата рубидия (х4000)
показал, что практически вся масса образца состоит из однотипных кристал-
лов ярко выраженной прямоугольной формы размером 5—10 мк (рис. 3),
которые лишь в некоторых случаях объединены-^ крупные агрегаты. Тита-
нилсульфат рубидия состава 2Rb2SO4 • 3TiOSO4 • H2SO4 6Н2О обладает со-
вершенным кристаллическим строением и характеризуется сложной струк-
турой. Действительно, на его рентгенограммах (СиКа -излучение) в отно-
сительно небольшом интервале углов (10—25°) обнаруживается около 30
1 TiOSO4+Rb2SO4+2RbHSO4.
92
линий. В табл. 4 даны межплоскостные расстояния и относительные интен-
сивности линий для типичной рентгенограммы—2Rb2SO4 3TiOSO4 • H.2SO4 •
6Н2О.
ВЫВОДЫ
1. Изучена растворимость в системе ТЮ2—Rb2SO4—H2SO4—Н2О при 20° С.
Устойчивой твердой фазой в системе является ранее неизвестный кислый
кристаллогидрат титанилсульфата рубидия — 2Rb2SO4 • 3TiOSO4 • H2SO4 • nH2O.
2. Изучена термическая устойчивость титанилсульфата рубидия. Дегидра-
тация и химические превращения, протекающие при нагревании соли, носят
ступенчатый характер.
ЛИТЕРАТУРА
1. В. М. Полякова, М. И. Милютина. Химия соединений редких тугоплавких
элементов (V, Nb, Та, Ti, Zr). Тр. Ин-та хим. УФАН СССР, 1967, вып. 14,
стр. 73.
2. Gmelins. Handbuch anorg. Chem. Titan, 1951, S. 41, 413.
3. В. M. Полякова, E. И. Чернявская. Ст. в наст, сб., стр. 94.
4. Ф. М. Перельман. Рубидий и цезий. М., Изд-во АН СССР, 1960.
5. А. Н. Киргинцев, В. А. Леонова. Изв. АН СССР, серия хим., 1968, 6, 1166.
6. F. Paulik, I. Paulik, Z.Erdey. Z. analyt. Chem., 1958, 160,141.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. а* ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. М. ПОЛЯКОВА, Е. И. ЧЕРНЯВСКАЯ
ОКИСЛЕНИЕ СЕРНОКИСЛЫХ РАСТВОРОВ ТРЕХВАЛЕНТНОГО
ТИТАНА ХИМИЧЕСКИМИ РЕАГЕНТАМИ
В предыдущих сообщениях [1, 2] были изложены результаты исследо-
вания окисления ионов титана (III) кислородом в сернокислых средах.
В настоящей статье приведены данные по окислению ионов трехвалент-
ного титана химическими реагентами, являющимися окислителями в кислых
растворах. Для проведения опытов сернокислый раствор титана приготавли-
вали следующим образом. Двуокись титана марки «х. ч.» обрабатывалась
концентрированной серной кислотой (из расчета две весовые части H2SO4
на одну весовую часть двуокиси титана) в течение 2—3 ч при 150” С при
частом перемешивании, а затем 2 ч при 300° С с обильным выделением
густых паров серного ангидрида.
Полученную сметанообразную массу сернокислой соли титана охлаждали,,
разбавляли водой в десятикратном количестве по отношению к взятой дву-
окиси титана и перемешивали при 60—70° С в течение 1 ч. После фильтра-
ции оставшуюся неразложенной часть двуокиси титана вновь обрабатывали
серной кислотой аналогичным образом. Сульфатную массу выщелачивали
раствором после первой фильтрации и перемешивали в течение 1 ч при
60—70° С. Полученный сернокислый раствор титана содержал 90—100 г/л
двуокиси титана и 180—250 г/л серной кислоты. Восстановление сернокис-
лых растворов титана производили электролитически в электролизере со свин-
цовым катодом и платиновым анодом при плотности тока на катоде
0,014 а/см1 [3].
Опыты по окислению ионов трехвалентного титана проводили при 20° С,
при концентрации его 30 г/л, кислотность растворов составляла 200 г/л,
за исключением тех опытов, когда изучали зависимость степени окисления
Ti3+ от концентрации серной кислоты в растворе. Для окисления серно-
кислых растворов титана (III) были выбраны следующие окислители: суль-
фат трехвалентного железа, хлорноватокислый калий, марганцевокислый
калий, надсернокислый аммоний, бихромат калия, азотная кислота и сер-
нистый газ. Расход окислителей рассчитывался в соответствии с реакцией (/),
аналогично которой составляли уравнения реакций и для других окисли-
телей:
Ti2 (SO4)3+К2Сг2О7+3H2SO4=2TiOSO4+K2SO4 +
+Cr2(SO4)3+3H2O+O2. (1)
Восстановленный сернокислый раствор титана помещали в сосуд, закры-
тый резиновой пробкой , с клапаном Бунзена. Затем в раствор вводили рас-
считанное количество заданного окислителя и перемешивали магнитной
94
Таблица 1
Кинетика окисления сернокислых растворов титана (III) химическими реагентами
-Ns опы- та Время, сек Колич. окисленного титана, % от исходного при реагентах
ксюа (NHa)2S2Oa| KaCr2O, КМпО, Fe, (SO,)a HNOa so2 O2 воздух
1 60 100 100 , „
2 300 — — 100 95,4 89,5 100 61,7 1,0 0,7
3 600 .—- 97,0 94,8 — 80 2,0 1,4
4 900 — — — 100 97,4 — 88 3,0 2,1
5 1200 -—- -—- —— — 98,3 — 94 4,0 2,8
6 1500 — — — — 99,13 — 100 5,0 3,5
мешалкой. Через определенное время отбирали пробы раствора для опреде-
ления количества окислившегося титана.
Результаты этой серии опытов представлены в табл. 1 и графически
изображены на рис. 1. Видно, что энергичное окисление титана (III) про-
Рис. 1. Кинетика окисления титана (III) химическими реаген-
тами.
1-KCIOS, (NH4), SaOa; 2 —HNOa, K2Cr2O7; 3 - KMnO4; 4 —
Fe2 (SO,)a; 5 — SO2; 6 — кислород; 7 — воздух.
Л — степень окисления титана, %; Б — время, сек.
исходит при действии хлорноватокислого калия и надсернокислого аммония
(см. табл. 1, опыты 1, 2). Также весьма эффективными окислителями в иссле-
дованных условиях являются и другие химические реагенты — бихромат
калия и азотная кислота (окисляют титан (III) полностью за 5 мин). Вслед-
ствие прочности химической связи и малой растворимости кислорода, неза-
висимо от высокого окислительно-восстановительного потенциала системы [4]
2Н2О=О2+4Н++4е- (£0= 1,229),
процессы окисления с участием его в кислых средах протекают сравни-
тельно медленно. По окислительной активности в отношении Ti3+ химиче-
ские реагенты располагаются в следующий ряд:
КС1О3, (NH4)2S2O8, К2Сг2О7, HNO3, KMnO4, Fe2(SO4)3, SO2,
затем следуют чистый кислород и воздух.
Приведенные данные свидетельствуют о том, что окислительная способ-
ность исследованных химических реагентов во много превосходит окисли-
тельную активность чистого кислорода и воздуха. За 1 мин хлорновато-
95
кислый калий и надсернокислый аммонии полностью окисляют ионы титана
(III) в сернокислом растворе (200 г/л H2SO4 и 30 г)л Ti3+). Между тем
за 25 мин, кислород окисляет ионы титана (III) в этих же условиях
только на 5 %, а воздух — на
Рис. 2. Окисление титана (III) химическими
реагентами в зависимости от кислотности
(продолжительность 5 мин).
J-K,Cr»O,'. 2- КСЮ,; 3 - Fe, (SO,), 4 —SO,;
5 — воздух.
А— степень окисления титана, %; Б— концентрация
серной кислоты г/л.
На степень окисления титана (III)
серной кислоты в растворе не оказывает
3,5% от исходного содержания
титана в растворе.
Для выяснения влияния кис-
лотности на кинетику окисления
титана (III) различными окисли-
телями проведена серия опытов
при изменении концентрации сер-
ной кислоты в растворе от 100
до 400 г!л. Результаты этих опы-
тов представлены в табл. 2 и на
рис. 2.
При окислении сернокислых
растворов титана (III) сульфа-
том трехвалентного железа и
сернистым газом наиболее благо-
приятными условиями за первые
5 мин окисления является кон-
центрация серной кислоты, рав-
ная 200 г/л (см. табл. 2). С повы-
шением кислотности до 300—
400 г/л H2SO4 окисляемость рас-
творов замедляется, т. е. кислота
в данном случае депрессивно дей-
ствует на процесс окисления
титана (III).
Хлорноватокислый калий пол-
ностью окисляет ионы титана (III)
при концентрации серной кис-
лоты 150—300 г/л и при повы-
шении кислотности до 400 г/л
окисление замедляется.
Окисление Ti3+ воздухом на-
чинает затормаживаться уже при:
концентрации серной кислоты
200 г/л и монотонно падает с
повышением кислотности,
бихроматом калия концентрация
тормозящего влияния в исследо-
Таблица 2‘
Окисление титана (III) химическими реагентами в зависимости от концентрации
серной кислоты, %
Кис- Fe (SO <). К,Сг,О ксю, so, Воздух
лот- иость, Время, мин
г/л 5 10 30 5 10 30 5 10 30 5 10 30 во
100 68 80 81 95 98 100 87 91 94 30 84 95
150 81 87 92 96 100 — 100 — — 60 84 89 19
200 89 94 97 100 — — 100 — -—. 62 80 88 14
300 79 82 85 100 -—• .—- 100 — .— 61 77 81 9
400 61 67 84 100 — — 89 89 89 31 55 67 6.
96
Влияние
окислителя
сернокислых растворов" титана (III)
кислотности раствора.
Таблица 3
количества вводимого
на кинетику окисления
№ опыта Время, мин Колич. окисленного титана, % от исходного, при вве- дении окислителя, % от теор. необходимого
25 50
1 5 24 48
2 10 25 50
3 20 25 50
4 60 25 50
5 120 25 50
6 Сутки 25 50
Условия опытов: кон-
Примечание. ------------------
центрация Tia+ 30» серной кислоты 200 г/л;
химический реагент КС1О8 •
ванных условиях. Наиболее полное окисление в течение первых 5 мин про-
текает при 200—400 г/л H2SO4.
Таким образом, кривые (см. рис. 2) наглядно показывают изменение
степени окисления сернокислых растворов титана (III) различными химиче-
скими реагентами от концентрации серной кислоты в растворе. Для всех
реагентов окисление затормаживается с повышением
Исключением является лишь бихромат
калия: на его окислительную актив-
ность концентрация серной кислоты в
растворе в указанном интервале не влияет.
Как уже указывалось, расход хими-
ческого реагента, потребный для окис-
ления титана (III), рассчитывался в соот-
ветствии с уравнением (1). В связи с тем,
что количество вводимого реагента, тео-
ретически необходимого для окисления
по реакции (1), довольно значительно,
исследована кинетика окисления Ti®+
хлорноватокислым калием с введением
25—50% реагента от рассчитанного.
Результаты опытов с меньшим количест-
вом химического реагента, вводимого в
сернокислый раствор титана (III), приве-
дены в табл. 3.
Видно, что степень окисления титана
находится в прямой связи с количеством
Следовательно, для полного окисления сернокислых растворов титана (III)
необходимо вводить количество окислителя в соответствии с теоретически
рассчитанным по реакции (1).
(III) хлорноватокислым калием
вводимого в раствор окислителя.
вывод
Установлено, что окисление сернокислых растворов титана (III) химиче-
скими реагентами — КС1О8, (NH4)2S2O8, К2Сг2О7, КМпО4, HNOS, Fe2 (SO4)3—
протекает много быстрее, чем кислородом. Повышение концентрации сер-
ной кислоты в окисляемых растворах оказывает замедляющее действие
на процесс окисления титана (III) исследованными химическими реагентами
за исключением бихромата калия.
ЛИТЕРАТУРА
1. В. М. Полякова, Е. И. Ч4е р н^в с'к а я. Исследование физико-химических свойств
соединений редких тугоплавких элементов (V, Nb, Та, Ti, Hf, Mo, Be). Tp. Ин-та
хим. УФАН СССР, 1970, вып. 17, стр. 148.
2. В. М. Полякова, Е. И. Чернявская. Там же, стр. 155.
3. Н. В. Деменев, М. И. Милютина, А. К. Шарова, А. П. Штин. Ж- неорг.
хим., 1957, 2, 467.
4. В. Латимер. Окислительные состояния элементов и их потенциалы в водных раство-
рах. М., ИЛ, 1954.
7 Заказ № 632
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. М. ПОЛЯКОВА, Л. Ф. МАЛЬЦЕВА
СОКРИСТАЛЛИЗАЦИЯ НИОБИЯ С ТИТАНИЛСУЛЬФАТОМ КАЛИЯ
В СЕРНОКИСЛЫХ РАСТВОРАХ
В литературе ограничены данные по поведению ниобия (V) в процессе
выделения титана сульфатом калия из сернокислых растворов, содержащих
ниобий (V) и титан (IV).
В настоящую работу включены исследования по изучению сокристал-
лизации примеси ниобия с титанилсульфатом калия из кислых сред. Для
выяснения характера соосаждения изучено влияние концентрации осади-
теля, микрокомпонента, кислотности и других факторов на количество со-
осажденного ниобия. Зная масштабы и характер соосаждения, можно пред-
видеть влияние некоторых факторов на количество сокристаллизующихся
примесей, что позволяет сделать более надежными методы концентрирова-
ния или выделения элементов из растворов с малым их содержанием.
Теоретические и практические вопросы соосаждения изучены академи-
ком В. Г. Хлопиным и его учениками. По данным [1, 2] процесс сокристал-
лизации лучше всего характеризует значение равновесного коэффициента
кристаллизации. Последний рассчитывается в соответствии с уравнением
В. Г. Хлопина:
х С1 —у)
У (1-*)’
где х и у — микро- и макрокомпоненты в твердой фазе; (1—х) и (1—у) —
микро- и макрокомпоненты в жидкой фазе; D — коэффициент кристал-
лизации.
Методика исследования сводилась к определению коэффициента кри-
сталлизации (О) в зависимости от различных факторов. Сокристаллизация
ниобия с титанилсульфатом калия изучалась при концентрации серной
кислоты 250 и 450 г/л. Эксперименты проводили следующим образом;
К взятому объему сернокислого раствора титана, содержащему примесь
ниобия, нагретому до температуры 60—70° С, добавляли определенное ко-
личество сульфата калия, заданное условиями опыта. Осаждение прово-
дили в специальных пробирках для определения растворимости. После
растворения сульфата калия нагревание прекращали, пробирки с раство-
рами закрепляли в термостате и проводилось перемешивание. Равновесие
между раствором и осадком достигали путем изотермической кристаллиза-
ции из пересыщенного раствора. Предполагалось, что при введении суль-
фата калия в сернокислый раствор, содержащий титан и примесь ниобия,
сокристаллизация последнего будет обусловлена образованием изоморф-
ных смешанных кристаллов двойных сернокислых солей титана и ниобия.
98
Результаты опытов по сокристаллизации ниобия (V) с титанилсульфа-
том калия из растворов с концентраци ей серной кислоты 250 и 450 г/л в за-
висимости от содержания микрокомпонента в растворе представлены в
табл. 1. Из ее данных можно заключить, что в пределах погрешности опыта
Таблица 1
Зависимость коэффициента кристаллизации (D) от содержания микрокомпонента
в растворе
Введено Найдено в осадке Степень кристаллизации, %
основное ве- щество TiO,. г примесь NbtOs, мг ТЮ„ г Nb,Os. мг основного ве- щества примеси
450 г/л H2SO4
1,4 63 1,370 18,6 97,86 29,52 0,0092
1,4 91 1,375 32,0 98,21 35,16 0,0098
1,4 109 1,370 35,1 97,86 32,20 0,0104
1,4 127 1,373 39,1 98,07 30,78 0,0088
1,4 445 1,370 51,0 97,86 35,17 0,0118
С р е д н ее . . 0,01
250 г/л H2SO4
1,5 25 1,36 4,0 90,5 16,0 0,020
1,5 50 1,33 8,4 88,9 16,7 0,025
1,5 100 1,33 19,2 88,9 19,2 0,029
1,5 150 1,34 32,0 89,4 21,4 0,032
1,5 200 \ 1,35 46,4 90,0 23,2 0,032
Среднее. . 0,028
величина D остается постоянной, не зависящей ни от концентрации серной
кислоты, ни от содержания микрокомпонента в растворе. Некоторые ко-
лебания в ее значении можно объяснить тем, что исследуемая система
K2TiO(SO4)2xH2O—Nb(V) сложна и работа с ней сопряжена с определен-
ными трудностями. Видно также, что с увеличением концентрации серной
кислоты от 250 до 450 г/л одновременно возрастает степень кристаллизации
основного вещества (титана) и степень сокристаллизации примеси (ниобия).
Таким образом, распределение микрокомпонента ниобия (V) между твердой
и жидкой фазами подчиняется линейному закону В. Г. Хлопина и свиде-
тельствует об изоморфном характере образующихся осадков. В этом убеж-
дают и изотермы сокристаллизации ниобия (V) с титанилсульфатом калия,
полученные при концентрациях серной кислоты 250 и 450 г/л (см. рисунок).
С помощью метода В. Г. Хлопина также возможно однозначно решить
вопрос, является ли захват примеси адсорбционным или изоморфным.
Были проведены две серии опытов. В первой титанилсульфат калйя выделяли
из раствора, содержащего приместь ниобия (V), во второй — без примеси.
Затем к пульпе с заранее образованным осадком титанилсульфата калия
добавляли количество примеси ниобия (V), аналогичное в первой серии опы-
тов; Время перемешивания в обоих случаях было одинаковое. Степень
кристаллизации основного вещества (титанилсульфата калия) в обеих се-
риях также примерно одинакова. По данным В. Г. Хлопина [1], если имеет
место изоморфный захват, то количество примеси, соосажденное в первой
7*
99
и второй серии опытов, должно значительно отличаться. В случае адсорбции
количество соосажденной примеси в том и другом случаях должно быть
одинаково.
Для сопоставления результатов этих экспериментов рассчитывали от-
ношение (z) количества примеси, соосажденной с титанилсульфатом калия
в первой (х4) и второй (х2) серии опытов в соот-
ветствии с выражением
Изотерма сокристаллизации
ниобия (V) с титанилсуль-
фатом калия.
Количество ниобия, мг: со-
осажденного (Д), оставшегося
в растворе (Б).
Концентрация серной кислоты,
г/л: 1 — 250, 2 — 450.
Очевидно, что в случае изоморфизма величина z
должна значительно отличаться от единицы и
наоборот, если имеет место адсорбция, то коли-
чество соосажденной примеси должно быть близ-
кой к единице. Как следует из табл. 2, при
концентрациях серной кислоты 250, 400 и 450 г/л
значения z для ниобия (V) составляют соответ-
ственно 0,145; 0,105; 0,423, т. е. значительно
отличаются от единицы и являются доказа-
тельством того, что примесь ниобия образует
с титанилсульфатом калия смешанные крис-
таллы.
Поскольку исследуемая система подчиняется
закону Хлопина, микрокомпонент как в жидкой,
так и в твердой фазах должен находиться в оди-
наковом молекулярном состоянии. Можно пред-
положить, что смешанные кристаллы образуются
одновременно замещением ионов TiCXSOJ^-
ионами NbO(SO4)7 и внедрением последних в
межузловое пространство для компенсации заряда.
Исследованиями одного из авторов настоя-
щей статьи [3—5] показано, что при взаимо-
действии сернокислых растворов титана (IV)
с сульфатом калия в области концентраций
серной кислоты 12,6—22,7 вес. % образуется
титанилсульфат калия. В нем отношение двуокиси
титана к сульфату калия равно 1 : 1 и состав
соли выражается формулой K2TiO(SO4)2xH2O.
Изучен состав осадков (табл. 3), образующихся при взаимодействии суль-
фата калия с сернокислыми растворами титана, содержащими ниобий
TiO2:Nb2OB==l :3-i-1:4. Установлено, что ниобий образует изоморфные
кристаллы с титанилсульфатом калия указанного состава. Рассчитанный со-
став осадков выражается формулой
K2TiO(SO4)2 • 2,5Н2О; KNbO(SO4)2 • 2Н2О.
Для установления состава осадков, образующихся при взаимодействии
сульфата калия с сернокислыми растворами титана, более концентрирован-
ными по ниобию, осаждение производили из растворов, содержащих
Ti : Nb=l : 0,9, и кислотности раствора 230 г/л. Расчет состава образую-
щихся осадков, проведенный на основании химического анализа, показал,
что в наших условиях наряду с двойными сульфатными соединениями ти-
тана и ниобия образуется гель ниобиевой кислоты. Состав осадка ориенти-
ровочно может быть выражен формулой
K2TiO(SO4)2-xH2O; KNbO(SO4)2 • уН2О; Nb2O6.
100
Таблица 2
Соосаждение ниобия с титанилсульфатом калия
Способ осаж- дения Введено примеси Nb2Oa, мг Концентра- ция серной кислоты, г /л Вес осадка, & Содержание в осадке, % Осаждено основного вещества TiOa, г Соосаждено примеси Nb2O6, мг Степень кристалли- зации TiOa, % 2
основ- ного ве- щества примеси МЬ2Ой
1 100 400 12,83 •11,15 0,410 1,43 52,6 95,3 0,105
2 100 400 12,461 11,05 0,041 1,38 5,0 94,7
1 150 450 10,95 13,10 0,724 1,42 78,8 94,7 0,423
2 150 450 12,83 10,90 0,260 1,39 33,3 92,7
1 250 250 7,11 17,90 0,685 1,28 49,0 85,3 0,145
2 250 250 5,94 21,20 0.121 1,25 7,12 83,5
Таблица 3
Состав изоморфных кристаллов титана и ниобия (по данным И. Г. Чуфаровой)
№ опыта Состав исход- ного раствора, г]л Найде- но, % к+. % so2 . % Н,О,%
о О Z I ’OS'H ТЮ NbO Вычислено Найдено Вычислено Найдено Вычислено Найдено
связано с всего связано с всего связано с всего 1
ТЮ NbO ТЮ NbO ТЮ о
1 40,2 5 250 15,7 2,0 19,15 0,72 19,87 19,4 47,18 3,52 50,7 51,0 11,05 0,66 11,71 11,90
2 39,5 10,2 250 14,7 3,6 18,00 1,29 19,29 18,8 44,32 6,35 50,6 50,8 10,38 1,19 11,57 12,05
3 40,5 14,5 250 14,0 5,0 17,08 1,79 18,87 18,6 42,00 8,81 50,8 50,2 9,8 1,65 11,45 12,20
• 1. 2K,TiO(SO4)t-5H,O4-0,15 жоля KNbOfSO.J.^H.O,
2. ZKjTiOfSOJj-SHjO-)-0,28 л<оля KNbO(SO.).-2H,O,
3. 2KtTiO(SO4)t-5H,O + 0,42 моля KNbO(SO4)j-2Н,О.
Однако правильное решение вопроса о составе осадков, выделяющихся
при введении сульфата калия в сернокислые растворы, которые содержат
титан и ниобий, не может быть дано из-за недостаточности данных.
Поэтому предложенные формулы должны быть приняты лишь условно.
ВЫВОДЫ
1. Исследована сокристаллизация примеси ниобия (V) с титанилсуль-
фатом калия при осаждении сульфатом калия из сернокислых растворов.
2. Определены коэффициенты кристаллизации ниобия при распреде-
лении его между раствором и осадком титанилсульфата калия при концен-
трации серной кислоты 250 и 450 г/л.
3. Показан изоморфный характер соосаждения ниобия (V) с титанил-
сульфатом калия при введенйи сульфата калия в сернокислый раствор,
содержащий эти элементы.
ЛИТЕРАТУРА
1. В. Г. Xлопин. Труды Радиевого института, 1938, 4, стр. 34.
2. О. Хан. Прикладная радиохимия. М.—Л., Госхимиздат, 1947.
3. Н. В. Де мен ев, А. К. Шарова, В. М. Полякова. Докл. АН СССР, 1952,
87, 5, стр. 751.
4. В. М. Полякова, М. И. Милютина. Химия соединений редких тугоплавких
элементов. Тр. Ин-та хим. УФАН СССР, 1967, вып. 14, стр. 73.
5. А. К. Шарова, Н. В. Деменев, В. М. Поляков, М. И. Милютина.
Разделение близких по свойствам редких металлов. Под ред. А- Н, Зеликмана. М.,
Металлургиздат, 1962, стр, 116.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП^Ж ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. М. ПОЛЯКОВА, Л. Ф. МАЛЬЦЕВА
ВЗАИМНОЕ ВЛИЯНИЕ ТИТАНИЛСУЛЬФАТА КАЛИЯ
И ИЗОМОРФНЫХ ВКЛЮЧЕНИЙ НИОБИЯ НА ИХ РАСТВОРИМОСТЬ
В РАСТВОРАХ СЕРНОЙ КИСЛОТЫ
В настоящем сообщении приведены данные по изучению взаимного
влияния титанилсульфата калия и изоморфных солей ниобия на их раство-
римость в серной кислоте различной концентрации х. Растворимость изу-
чалась изотермическим методом при 20° С в термостате с механическим пе-
ремешиванием. Постоянство температуры поддерживали автоматически
е точностью ± 0,1 ° С. Установление состояния равновесия в системе конт-
ролировали отбором проб жидкой фазы через определенные промежутки
времени. При постоянном содержании титана и ниобйя в повторных ана-
лизах равновесие считалось достигнутым. В данной Системе оно устанав-
ливалось через 120 ч. После этого твердую фазу отделяли от раствора фильт-
рованием через тигель Шотта № 2, промывали охлажденным спиртом и вы-
сушивали в сушильном шкафу при 60° С или на воздухе при комнатной тем-
пературе до постоянного веса.
Пробы твердой и жидкой фаз анализировали на содержание титана,
ниобия, калия и сульфат-иона. Ниобий определяли танниновым методом,
титан —- колориметрически на ФЭКН-57, сульфат-ион — путем осажде-
ния хлористым барием, калий — весовым методом или методом пламенной
фотометрии. Кислотность раствора устанавливали титрованием щелочью
с индикатором метиловым оранжевым. Содержание воды в твердой фазе
рассчитывали по разности. На основании данных анализа вычислялись
концентрации компонентов системы в жидкой и твердой фазах.
Для приготовления сернокислых растворов ниобия и титана с после-
дующим получением из них титанилсульфата калия с изоморфными вклю-
чениями ниобия были взяты двуокись титана марки «специальный», пяти-
окись ниобия марки «ч», серная кислота с уд. весом 1,82 и дважды пере-
кристаллизованный сульфат калия. Сернокислый раствор титана и ниобия
приготовляли следующим образом. К навеске двуокиси титана и пяти-
окиси ниобия, взятых в определенном соотношении и помещенных в фар-
форовую или платиновую чашку, приливали двукратное (по объему) ко-
личество концентрированной серной кислоты. Смесь медленно нагревали
на песчаной бане. В начале разложения смесь довольно часто перемеши-
вали во избежание «сцементирования» неразложившихся частиц окислов
образующимися сульфатами. Затем разложение продолжалось при обиль-
ном выделении паров серного ангидрида в течение 1,5—2 ч при ~ 300° С.
7 Полученные нами изоморфные сернокислые соли титана и ниобия содержали и гидра-
тированную пятиокись ниобия [1]. Поэтому данные, приведенные в статье, по раствори-
мости ниобия являются суммарными, относящимися к изоморфным включениям и гидро-
окиси ниобия.
102
После охлаждения полученные сульфаты титана и ниобия выщелачивали
водой, взятой в отношении твердого к жидкому 1 : 10, при 80—85® С с пе-
ремешиванием в течение 1 ч. Отфильтрованный раствор (Ne 1) имел кислот-
ность 180—250 г/л серной кислоты, концентрацию двуокиси титана 60—
80 г/л и пятиокиси ниобия 5—6 г/л.
Получение сернокислых растворов с более высоким содержанием Nb2O,
и относительно низкими концентрациями двуокиси титана и серной кислоты
описанной выше методикой представляет определенную трудность, и в ли-
тературе не описано. Поэтому в данной статье подробно приводим метод,
примененный нами. Навеску пятиокиси ниобия помещали в стеклянный
стакан и вводили десятикратное количество концентрированной серной
кислоты и пятикратное количество сульфата аммония [2]. Смесь нагревали
на песчаной бане до исчезновения неразложившихся частиц (—1,5—2 ч).
Из полученного прозрачного; раствора аммиаком осаждали гидроокись
ниобия. После промывки декантацией она растворялась й концентриро-
ванной серной кислоте, содержащей титан в количестве 10% к ниобию.
Затем стакан с раствором помещали в воду со льдом и медленно добавляли
дистиллированную воду. При этом возможно выпадение осадка, но обычно
на второй день раствор осветлялся и становился прозрачным. Таким путем
удается получать раствор (№ 2), содержащий серной кислоты до 400, пяти-
окиси ниобия до 20 и двуокиси титана до 2 г/л.
Сернокислые растворы № 1 и № 2 служили исходными. Из них приго-
тавляли расчетным путем растворы с заданными концентрациями компо-
нентов: пятиокиси ниобия, двуокиси титана и серной кислоты. Необходи-
мые для исследования соли титанилсульфата калия с изоморфными вклю-
чениями ниобия синтезировали следующим путем. В нагретый до темпе-
ратуры 60—70° С сернокислый раствор, содержащий ниобий и титан, при
включенной мешалке вводили небольшими порциями мелко растертый суль-
фат калия из расчета 150—200 г/л. После растворения соли нагревание
прекращали. При охлаждении раствора осаждался титанилсульфат калия
с изоморфными включениями ниобия [1, 3].
Таблица 1
Растворимость титанилсульфата калия с изоморфными включениями ниобия
в водных растворах серной кислоты, вес. %
Жидкая фаза Твердая фаза
H,SO4 Nb,Os TiO, K,so4 SO4 H,O K.so4 Nb,O, TIO, so4 | H,O
7,75 1,06 3,84 7,3 . 13,6 78,23 34,6 8,7 26,8 44,7 4,35
10,20 1,12 3,34 6,9 15,7 76,72 37,6 8,1 24,5 46,1 0,50
13,10 0,97 3,37 6,3 19,2 73,63 34,0 6,1 20,8 50,7 7,20
14,13 0,94 2,92 5,1 19,2 74,65 49,2 0,6 20,8 51,4 5,20
17,22 0,95 2,52 4,5 22,3 72,20 41,9 0,9 21,5 53,6 5,10
17,05 0,89 2,57 4,4 22,3 72,22 39,6 1,4 22,2 52,0 7,20
18,50 0,94 2,56 4,2 23,7 71,91 43,0 0,9 21,3 52,6 5,85
20,60 0,84 2,20 3,7 25,9 69,39 45,7 1,5 22,1 53,2 2,66
23,30 0,85 1,92 3,4 28,3 67,31 46,0 1,1' 22,3 52,0 4,00
27,80 0,85 1,35 2,5 32,8 63,83 39,8 2,0 22,4 55,6 2,20
34,40 0,60 0,71 1,9 39,9 57,95 42,4 1,8 24,0 50,6 4,55
При изучении растворимости титанилсульфата калия с изоморфными
включениями ниобия в первой серии опытов при постоянном содержании
титана и ниобия, равным 3,7 : 1, изменяли концентрацию серной кислоты
в пределах от 7 до 34 вес.%. Результаты этой серии опытов представлены
в табл. 1 и графически изображены на рис. 1. На последнем для сравнения
нанесены данные, взятые из работы [4] по растворимости титанилсульфата
калия в водных растворах серной кислоты (кривая 2). Видно, что при 20* С
1»Э
Рис. 1. Влияние Nb2O5 на растворимость титанилсуль-
фата калия в водных растворах серной кислоты.
Концентрация, вес. %: А — TiOt в растворе, Б — серной ки-
слоты.
Растворимость титанилсульфата калия: 1 — в присутствии нио-
бия, 2 — по данный работы [4].
в присутствии ниобия повышается растворимость титанилсульфата калия
(кривая 7). С повышением концентрации H2SO4 растворимость титанил-
сульфата калия, одного или в присутствии ниобиевой соли, снижается.
' Таблица 2
Взаимное влияние титанилсульфата калия и изоморфных включений ниобия
на их растворимость в 14%-ной серной кислоте, вес. %
Исходная соль Жидкая фаза Твердая фаза
Nb,O, тю, NbsO,:TiO, NbtO„ тю, NbBOB TiO,
33,67 5,3 1:0,157 5,32 0,91 53,60 3,3
30,18 5,4 1:0,179 3,92 0,70 50,60 7,1
28,30 5,7 1:0,200 2,95 0,77 48,90 6,1
11,60 17,1 1:1,470 2,67 2,45 32,90 19,3
12,20 21,3 1:1,740 1,62 3,46 16,90 26,7
6,40 20,0 1:3,730 0,97 2,49 2,07 24,0
3,92 21,5 1:5,500 0,68 2,32 2,30 23,5
3,30 21,4 1:6,500 0,58 2,50 2,72 34,2
2,95 21,6 1:7,320 0,45 2,41 1,61 23,5
. Нет 21,7 — Нет 0,31 Нет 23,9
При проведении второй серии опытов изучалось взаимное влияние ти-
танилсульфата калия и изоморфной соли ниобия на их растворимость в
14%-ной серной кислоте. С этой целью синтезировали титан ил сульфат
калия с изоморфными включениями ниобия при переменном соотношении
1,0.4
ниобия и титана, а именно Nb2O& : TiO2= 1 : 0,157—1 : 7,32. Количество
изоморфно включенного ниобия, осаждающееся с титанилсульфатом калия,
определялось соотношением этих компонентов в исходном растворе. Для
установления равновесия в системе термостатирование в изотермических
условиях также производили в течение 120 ч при непрерывном перемеши-
вании жидких и твердых фаз. Результаты анализов по основным компо-
Рис. 2. Взаимное влияние. титанилсульфата калия и изоморфных включе-
ний ниобия на их растворимость в 14%-ной серной кислоте (сумма TiO2
и Nb2O6 в твердом остатке принята 100%).
Концентрация в растворе, вес. %: Л —TiOt, Б — NbtO6.
Растворимость: / — NbfO6, 2 — TlOt.
нентам помещены в табл. 2. Графическое изображение взаимного влияния
титанилсульфата калия и изоморфных включений ниобия на их раствори-
мость в 14%-ной серной кислоте дано на рис. 2. Кривые показывают, что
при содержании пятиокиси ниобия в количестве примерно до 40% по отно-
шению к сумме ТЮ2 и Nb2Og в твердой фазе растворимость титанилсульфата
калия растет, с дальнейшим увеличением концентрации его растворимость
понижается (кривая 2). В то же время титанилсульфат калия при концент-
рации до 10% TiO2 способствует резкому повышению растворимости изо-
морфных включений ниобия (кривая 1). По мере возрастания содержания
двуокиси титана растворимость Nb2O5 монотонно снижается и показывает
минимальное значение при наличии Т1О2 свыше 95%.
ВЫВОДЫ
1. Установлено, что растворимость титанилсульфата калия в растворах
серной кислоты в исследованных условиях в присутствии ниобия выше по
сравнению с растворимостью его в серной кислоте, не содержащей ниобий.
2. Показано взаимное влияние титанилсульфата калия и изоморфных
включений ниобия на их растворимость в 14%-ной H2SO4 при 20° С.
ЛИТЕ PtA ТУРА
1. В. М. Полякова, Л. Ф. Мальцева. Ст. в наст. сб.,'стр. 98.
2. Минеральное сырье. Под ред. Ю. Н. Книпович и Ю. В. Морачевского. А., 1959.
3. А. К. Шарова, Н. В. Деменев, В. М. Полякова, М. И. Милютина.
Разделение близких по свойствам редких металлов. Под ред. А. Н. Зелйкмана. М.(
Металлургиздат, 1962, стр. 116.
4. В. М. Полякова, М. И. Милютина. Химия соединений редких тугоплавких
элементов. Тр. Ин-та хим. УФАН СССР, 1967, вып. 14,; стр. 73.
• Заказ № 632
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 197»
А. М. ГОРЕЛОВ, А. П. ШТИН
ИССЛЕДОВАНИЕ СВОЙСТВ ФОСФОРНОКИСЛЫХ РАСТВОРОВ ТИТАНА
Фосфорная кислота осаждает из сернокислых растворов фосфаты титана
[1,2], которые растворяются в избытке фосфорной кислоты вследствие ком-
плексообразования титана. В литературе нет сведений о природе и свой-
ствах фосфорнокислых растворов Ti (IV). Цель настоящей работы — опре-
делить стабильность фосфорнокислых растворов титана. Для выяснения
знака заряда соединений титана в указанных растворах изучено поглоще-
ние его некоторыми ионообменными смолами.
Устойчивость фосфорнокислых растворов титана
Для исследования использованы фосфорнокислые растворы титана.
Наблюдения над ними показали, что в широкой области концентраций фос-
форной кислоты при комнатной температуре происходит выделение титана
в осадок. Устойчивость фосфорнокислых растворов в зависимости от кон-
центрации титана и фосфорной кислоты устанавливали следующим образом.
К рассчитанному объему исходного раствора добавляли возрастающее от
опыта к опыту количество 85%-ной Н3РО4 и содержимое разбавляли водой
до определенного объема. Исследование проводили при постоянном содер-
жании в серии растворов Ti(IV), переменной концентрации фосфорной ки-
слоты и при комнатной температуре. При стоянии растворов происходит
выделение осадков. Изменения, протекающие в растворах, контролировали
путем периодического определения в растворе титана. По достижении
равновесного содержания растворы анализировали на содержание в них
титана и фосфорной кислоты. Результаты опытных данных представлены
в табл. 1 и на рис. 1.
Видно, что максимальное выделение титана в осадок происходит из рас-
творов с концентрацией фосфорной кислоты 0,1—2 моль1л. Независимо от
исходного содержания в растворах титана при стоянии наблюдается появление
опалесценции растворов с последующим выделением осадков. При концентра-
ции фосфорной кислоты 3,5—4 моль1л и содержании титана 2,5-10—8 —
—1-10~а г-атом/л растворы устойчивы. С повышением в них исходного
содержания титана до 4,55-Ю-2 и 8,125-10—2 г-атом!л при указанной
выше концентрации фосфорной кислоты он также выделяется. Количество
титана/в осадке возрастает с увеличением фосфорной кислоты до 5 моль]л.
С дальнейшим повышением концентрации фосфорной кислоты содержание его
106
Таблица 1
Зависимость выделения титана в осадок от концентрации в исходном растворе
титана и фосфорной кислоты
Исходное содержание титана, г- атом/л
2,5-10—* 5-10—* 1 • 1 о-1 4,55- 10,—г 8,125-10—=
а 6 а 6 а б а б ° б
1,2 1,5 2,0 2,5 2,75 3,0 to Ю ю Сл Сл “-J 4л. W О О О О О О О 1 1 1 1 1 1 W W to Й. «Л 0,1 0,2 0,5 0,8 1,0 1,2 1,5 1,8 2,0 2,5 2,75 3,0 3,5 Нет Нет 3,3-10—6 8,1-10-“ 1,3-10—6 2,0-Ю-6 4,4-10—5 6,6-10-5 1,4-10-5 1,7-10—3 3,7-10-3 5,0-10-3 5,0-10-а 0,1 0,2 0,5 0,8 1,0 1.2 1,5 1,8 2,0 2,2 2,5 2,75 3,0 3,2 3,5 3,6 Нет Нет 3,4-10-5 8,2-10-« 1,4-10-5 2,1-10-5 4,3-10-5 6,7-10-6 1,41-10-5 2,2-10-5 2,8-10-3 3,8-10-з 6,0-Ю-з 8,0-10-з 1,0-10-2 1,0-10—2 1.0 1,7 2,3 2,9 3,7 4,1 4,3 4,9 5,4 6,4 6,9 7,7 8,4 8,6 9,4 10 10,4 10,5 10,9 1,3-10-5 4,0-10-5 7,2-10-5 3,0-10-5 1,8-Ю-з 1,9-10—2 3,2-10-5 2,0-10-5 3,7-10-5 5,5-10-5 7,2-10-5 1,2-Ю-з 2,0-10-з 4,6-Ю-з 1,6-Ю-з 3,8-10-2 4,4-10-2 4,55-10-2 4,55-Ю-з 2,2 2,6 3,3 3,9 4,4 4,7 5,7 6,0 6,5 7,3 7,7 8,2 9,3 9,9 10,6 10,7 11,6 12,1 6,5-10-5 2,1-10-5 3,25-Ю-з 6,5-10-2 7,2-10-5 4,1-10-5 5,9-10-5 3,9-10-5 5,9-10-5 9,1-10-5 1,2-10-з 1,8-10-з 1,3-10-2 3,6-10-2 6,2-10-2 6,8-10-2 8,125-Ю-з 8,125-10-»
Примечание. Равновесная концентрация в растворе: а — НаРО4, леоль/л; б — титана, г-атом/л^
в растворе увеличивается, и при [Н3РО4], равной 11,5—12 моль!л, растворы
становятся стабильными.
А 10'2
Рис. 1. Влияние концентрации фосфорной кислоты и исходного содерЖаяййя титана на
устойчивость растворов.
А — содержание в растворе титана, моль/л. Б — концентрация Н8РО4,
Исходная lnIV]: / — 2,5-10~«; 2 — 5-10—*; 3— 1- 10“*; 4— 4,55- 10—'; 5—8,125- 10“' моль!л.
Проведенные исследования показали, что устойчивость фосфорнокис-
лых растворов титана определяется концентрацией в растборах фосфорной
кислоты и титана. Аналогичное поведение наблюдается и в фосфорнокис-
8*
107
Таблица 2
Поведение титана в фосфорнокислых
растворах
Концентра- ция вве- денной кис- лоты, моль/а [Ti (IV)]-10», г-атом/л, в растворах, содержащих
НС1 HNO, HjSO,
2,5-Ю-3 1,35 1,5 1,6
1,0 3,25 2,9 5,2
2,0 4,55 4,5 13
Примечание. Исходное содержание
в растворе: Ti (IV)=2,5 10"1 е атом/л,
Н.РО<=4,5-1 0—1 моль!л.
лых растворах ниобия [3]. Нагревание растворов ускоряет процесс выде-
ления титана, при этом тотчас происходит образование осадка. Стабиль-
ность растворов увеличивается и при хранении на холоду. Так раствор,
содержащий 3,5 • 10-1 г атом/л титана и 6,3 модь/л Н3РО4, при комнат-
ной температуре не претерпевал види-
мых изменений в течение двух суток.
Затем выделялся осадок. Этот раствор,
хранимый при температуре 0—5° С,
длительное время был устойчивым и оса-
док не появлялся в течение месяца.
В таких условиях растворы долго мо-
гут храниться, оставаясь прозрачными.
Исследовано также действие кислот
и оснований на фосфорнокислые раст-
воры титана. При приливании КОН,
NaOH или NH4OH к растворам сразу
выделяется хлопьевидный осадок. В фос-
форнокислых растворах, содержащих
НС1, HNOS, H2SO4, происходит выде-
ление фосфата титана [1]. Остаточное содержание титана определяется
характером вводимой кислоты. При прочих одинаковых условиях в смешан-
ных серно- и фосфорнокислых растворах наблюдается повышенное содер-
жание титана по сравнению с фосфорнокислыми растворами, включаю-
щими НС1 или HNO3 (табл. 2).^Очевидно, это связано с растворимостью
фосфата титана в указанных растворах.
Поглощение титана ионитами из фосфорнокислых растворов
Было исследовано поведение титана с помощью ионного обмена, так
как ионообменные смолы могут быть использованы при определении знака
заряда соединений титана в фосфорно-
кислых растворах. В качестве ионо-
юбменнйков были взяты сильнокислот-
ные однофункциональные катиониты
КУ-2 и СБС и многофункциональный
катионит КУ-1Г в водородной форме,
сильноосновной анионит АВ-16 и
слабоосновной АН-2Ф в форме
НаРО7. Поглощение титана изучали
в статических условиях в интервале
концентраций фосфорной . кислоты
2—10 модь/д. При меньшей кислот-
ности растворов проводить исследо-
вание невозможно из-за неустойчи-
вости растворов. Предварительно оп-
ределяли время, необходимое для
достижения равновесия. Оно оказа-
лось достаточным, чтобы из раство-
ров с исхдДйШя содержанием титана
2,25• 10-3 г-атом/д не происходило
выделения последнего в осадок. От-
дельно поставленные опыты подтвер-
дили.это. После встряхивания раст-
воры с .'навеской ионита анализиро-
вали на'со^ржание титана и фос-
Рис. 2. Зависимость поглощения титана
ионитами от концентрации фосфорной,
кислоты.
А — поглощение титана,- %, S — концентра-
ция Н,РО4, моль/л.
1— КУ-2; 2 — АВ-16; 3 — АН-2Ф.
108
формой кислоты. На основании опытных данных рассчитывали про-
цент поглощения (рис. 2) и коэффициент распределения <р общепринятым
методом [4] по формуле
Мг v
ф=------1------,
М — Мг т
где М и Л4Х— количество титана в исходном растворе и сорбированное
ионитом, соответственно; и — объем раствора, взятого для исследования, мл;
т — масса ионита, г. В наших опытах v=50 мл, т=0,5 г. Результаты
определения <р представлены в табл. 3 и 4.
Таблица 3
Поглощение титана катионитами в фосфорнокислых растворах
Концентрация KV-2 KV-1Г СБС
HjPO4. моль/л I п I II I и
2,0 8,45 169,1 22,5 0 22,5 0
2,5 7,12 219,3 — — — —
3,0 6,50 250,0 — — — —.
3,5 5,52 312,0 — — -— —.
4,0 9,64 136,3 — — — —
5,0 14,9 52,0 — — — —
6,0 18,8 20,6 22,5 0 22,5 0
8,0 21,4 '6,06 — — — —
10,0 21,4 6,06 — — — —
Примечание. / —равновесная [Ti (IV)-104 г-атом/л- II — коэффициент рас-
пределения (<р).
Титан из фосфорнокислых растворов сорбируется на катионите КУ-2
и не сорбируется на КУ-1Г и СБС (см. рис. 2 и табл. 3). При увеличении
кислотности растворов ионообменная сорбция титана на катионите КУ-2
возрастает, а затем падает. Известно, что поглощение катионов катионо-
Таблица 4
Поглощение титана анионитами в фосфорнокислых растворах
АЕ Мб АН-2Ф
Концентрация Н3РО4, моль/л I II Концентрация HSPO4, моль/л I
2,0 3,25 6900 1,87 1,95 1.158.000
2,5 5,2 3150 2,3 3,90 5553
3,0 9,12 2400 2,8 5,85 4254
3,5 15,5 1367 3,4 10,8 2087
4,0 24,6 853 4,3 24,7 821
4,5 41,5 448 4,7 40,3 464
5,0 71,5 218 5,5 78 191
5,9 117 75 6,6 137 67
7,0 169 35 7,6 163 40
8,0 202 13 8,3 182 25
8,8 208 9 9,0 182 25
9,8 208 9 — — —
Примечание. I — равновесная [Т! (IV)] -1 О5 г-атом,/л\ II — коэффициент рас-
пределения (<р).
обменной смолой уменьшается по мере повышения кислотности среды [5].
Снижение сорбируемости титана с увеличением концентрации фосфорной
кислоты не может быть объяснено одним изменением кислотности раство-
ров, так как имеет значение образование фосфатных анионных или нейт-
ральных комплексов. В изученном интервале концентраций фосфорной
109
кислоты наблюдается достаточно четко выраженное поглощение титана
анионитами (см. табл. 4). С ростом концентрации фосфорной кислоты сорб-
ция титана уменьшается. Анионное поглощение титана в 2—4 моль Н3РО4
составляет 90% и более (см. рис. 2). Это показывает, что в фосфорнокис-
лых растворах титан находится преимущественно в анионной форме. Сорб-
ция титана катионитом КУ-2 указывает и на присутствие в растворе кати-
онных форм титана. Это может быть объяснено малой устойчивостью фос-
фатных анионных комплексов титана. А. А. Гринберг [6] указал в качестве
общих положений, что способность к комплексообразованию минимальна
по краям в больших периодах и максимальна в центре: наиболее склонны
к комплексообразованию элементы группы VIII, имеющие лабильные
электронные оболочки с незаполненными d-ячейками. Результаты опреде-
ления устойчивости некоторых ацидокомплексов элементов четвертого пе-
риода показали, что стабильность анионных комплексов титана выражена
слабее, чем у элементов, относящихся к группе VIII [7]. Следует учитывать
также возможность нарушения равновесия между различными ионными
формами, существующими в растворе, в процессе их поглощения катиони-
том [8].
Полученные данные по ионообменному поведению титана в фосфорно-
кислых растворах показывают, что он, очевидно, находится в растворе
преимущественно в виде анионных фосфатных комплексов. Проведенные
ранее исследования по поглощению ниобия и,тантала ионитами говорят
о том, что ниобий также присутствует в фосфорнокислом растворе в виде
анионных комплексов [3].
ВЫВОДЫ
1. Исследована устойчивость фосфорнокислых растворов титана. Их
стабильность зависит от концентрации в растворе титана, фосфорной кис-
лоты и температуры.
2. Получены кривые поглощения титана ионитами. Характер кривых
поглощения титана из фосфорнокислых растворов различной концентра-
ции показывает, что титан в этих растворах находится преимущественно
в виде анионных фосфатных комплексов.
ЛИТЕРАТУРА
1. В. И. Спицын, Е. А. Ипполитова. Ж- анал. хим., 1951, 6, 5.
2. А. М. Горелов, А. П. Штин, С. В. Золота вин а. Исследование физико-хими-
ческих свойств соединений редких тугоплавких элементов. Тр. Ин-та хим. УФАН
СССР, 1970, вып. 17, стр. 115.
3. А. П. Штин, А. К. Шарова. Химия и технология редких металлов. Тр. Ин-та хим.
УФАН СССР, 1963, вып. 7, стр. 91.
4. Д. И. Рябчиков, И. К. Цитович. Ионообменные смолы и их применение. М.,
Изд-во АН СССР, 1962.
5. Л. С. Александрова, Т. Б. Гапон, Н. В. Филатова, К. В. Чмутов. Тру-
ды комиссии по аналитической химии, 1955, 6 (9), 235.
6. А. А. Гринберг. Введение в химию комплексных соединений. М.—Л., «Химия»,
1966.
7. И. К. Цитович. Докл. АН СССР, 1961, 136, 114.
8. Б. И. Набиванец. Усп. хим., 1965, 34, 949.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА ХИМИИ
1970
А. П. ШТИН, А. М. ГОРЕЛОВ, С. В. 30Л0ТАВИНА
ИЗОТЕРМА 20° С СИСТЕМЫ TiO2 — Р2О6 — Н2О
В литературе вопросы изучения фосфатов титана освещены недостаточно.
Существующие указания относятся в основном к описанию способов приго-
товления той или иной соли и к выяснению ее химического состава. Соглас-
но исследованиям Мерца [1], осадок фосфата титана после длительной
промывки приближается к составу 2ТЮа • РаО5 ЗН2О. Гош [2] указывает,
что состав свежеосажденного фосфата титана отвечает формуле 2ТЮ2Р2ОБ-
•ЗН2О, а прокаленного — Ti2P2Og. Эриксон [3] для прокаленной соли при-
нимает формулу Ti2PaO8. Другие исследователи [4] отмечают, что предло-
женные формулы фосфатов титана неверны и не могут быть установлены.
Викт. И. Спицын и Е. А. Ипполитова [5] при осаждении фосфата титана из
растворов показали, что высушенный при комнатной температуре осадок пред-
ставляет собою гидрофосфат «титанила» — ТЮНРО4-2,5Н2О. По данным
[6, 7], из фосфорнокислых растворов при pH, равном 3—4, выделяется осадок,
приближающийся к составу TiO (Н2РО4)2 • TiOHPO4 ЗНаО. В концентрирован-
ной фосфорной кислоте при удалении нагреванием фтористоводородной ки-
слоты образуется фосфат — Ti(HPO4)2.
Литературные данные о фосфатах титана противоречивы. Это свидетель-
ствует о необходимости проведения дальнейших исследований. Цель настоя-
щей работы — изучение системы TiO2 — Р2О5 — Н2О при 20° С методом рас-
творимости.
Ранее было показано,’'-что в широкой области концентраций фосфорной
кислоты при комнатной температуре происходит выделение титана в осадок
[8]. Вследствие этой особенности изучение равновесия в системе проводили
кристаллизацией твердой фазы. Для этого готовили фосфорнокислый раствор
титана. Исходными материалами служили свежеосажденная гидроокись тита-
на и концентрированная фосфорная кислота. Было замечено, что растворе-
ние гидроокиси титана в фосфорной кислоте происходит эффективнее при
комнатной температуре, чем при нагревании.
Методика исследования системы следующая: к серии проб фосфорнокис-
лого раствора титана добавляли возрастающее от опыта к опыту количество
85 %-ной Н3РО4. Для сохранения постоянной концентрации титана растворы
доливали до определенного объема водой. Содержание титана в смесях с
фосфорной кислотой в сериях проб составляло: 4-10-3; 1-10-2; 4,5- 1СН2 и
8,1 -10-2 (соответственно, серии I — IV в табл. 1). Выбор небольших кон-
центраций титана в исследуемой системе определялся условиями получения
исходного раствора титана и необходимостью изучения системы с небольши-
ми содержаниями фосфорной кислоты. Растворы в стеклянных ампулах по-
мещали в термостат при температуре 20+0,5° С и периодически встряхива-
ли. Для каждой серии ставили параллельные опыты. Причем каждая из
111
Таблица 1
Состав растворов и твердых фаз в системе TiO2—P2Ot—Н 2О
Жидкая фаза, вес. % «Остаток», вес.% Твердая фаза
TiO, P,OS ТЮ, р.о. н,о
I серия
Следы 0,88 8,8 8,9 82,30
1,2-10-5 2,52 9,62 10,9 79,48
4,2-10-5 4,02 10,22 12,3 77,48
1,1-10—* 6,41 9,11 12,96 77,93
2ТЮ2-Р205-6Н2О
1,з-ю-« 8,07 11,23 15,71 73,06
1,8-10—4 10,06 12,20 19,0 66,80
3,9-10—4 12,57 25,67 27,03 47,30
1,1-10-’ 13,95 13,77 21,68 64,55
4,5-10-’ 15,42 8,88 19,90 71,22
1,9-10-* 17,70 — — —
2,8-10-* 19,22 Осадка нет
II се р и я
Следы 1,8 8,80 9,13 82,07
4,0-10-8 4,53 9,55 12,00 78,45
5,1-10-в 5,74 10,92 13,95 73,13
1,3.10-* 7,54 12,05 16,49 71,46 ЗТЮд • PjOj • 6Й2О*
2,2-10—4 9,64 10,36 16,50 73,14
4,6-10-* 13,81 11,41 19,50 69,09
4,0-10—’ 15,45 24,8 27,80 47,40
2,2-10-* 18,25 36,95 32,89 30,18
4,8-10-* 20,93 14,09 29,45 56,46
6,7-10-* 23,24 —. — —
6,8-10-* 24,38 Осадка нет
III серия
2,5-10-* 8,08 9,74 14,98 75,28
4,1-10-* 11,60 9,69 17,05 73,26
5,3-10-* 14,72 10,59 19,33 70,03
2,3-10-’ 18,30 29,96 33,41 36,63
1,2-10—1 22,60 31,77 36,6 31,63
1,27-10—1 24,83 25,48 35,02 39,50
1,3-10-’ 29,68 19,65 47,5 32,75
2,3-10-’ 32,75 15,89 44,93 39,18
3,4-10-’ 35,52 16,43 47,06 36,51
4,8-10-’ 39,54 16,02 48,53 35,45
1,1-10-* 43,75 20,87 53,5 25,63
8,9-10-’ 47,10 16,40 53,1 30,41
2,0-10—1 49,23 6,0 51,02 .42,98
2,34-10—1 51,05 .—- — —
2,31-10—1 54,64 Осадка нет
2,30-10—1 56,68 То же
ТЮ2-Р205-6Н2О
2TiO2P2O5H2O
112
Продолжение табл. 1
Жидкая фаза вес, % «Остаток», вес. % Твердая фаза ’
ТЮ, . P.O, тю, P.O, н,о
2ТЮ2-Р2О5-6Н2О
IV серия
3,4-10—4 9,45 9,62 • 15,1 75,28
4,0-10-4 13,27 10,18 18,01- 71,20
5,0-10—4 14,72 9,46 19,25 71,29
1,4-10—8 17,22 11,22 22,25 66,53
2,7-10—1 21,26 27,33 34,12 38,55
4,3-10—1 24,51 15,12 35,29 49,59
4,6-10—3 28,02 10,95 38,40 50,65
3,7-10—3 33,37 20,02 48,90 31,08
3,6-10-3 37,62 13,36 46,22 40,42
5,3-10—» 40,02 10,85 46,50 42,65
1,1-10—» 44,39 18,09 52,89 29,02
7,3-10—3 47,59 22,72 52,60 24,68
3,0-10—1 50,79 18,24 55,54 26,22
3,8-10—1 52,86 12,59 55,26 32,15
4,3-10—1 54,30 — —. —
4,2-Ю-1 55,40 Осадка нет
TiO2-P2O5-H2O
ампул служила для отбора одной пробы жидкой и твердой фаз. При стоя-
нии растворов появляются осадки. Скорость выделения последних возрастает
с уменьшением в системе концентрации фосфорной кислоты. В растворах,
содержащих до 10—12% Р2ОБ, образование твердых фаз проходит через
стадию коллоидного раствора с последующей коагуляцией осадка, а при
большем содержании фосфорного ангидрида образование осадка идет без ви-
димой опалесценции растворов. Пробы выдерживали в термостате до достиже-
ния равновесного состояния, которое определяли постоянством состава жид-
кой фазы. Далее осадки отделяли от жидкости фильтрованием через стек-
лянный фильтр №4 и отжимали насколько возможно. Часть осадка промы-
вали ацетоном или спиртом для удаления маточника. Жидкую фазу, отжа-
тый остаток, а также отмытую твердую фазу анализировали на содержание
в них Т1О2 и Р2ОБ. Воду рассчитывали по разности.
Данные по определению состава растворов и твердых фаз сведены в
табл. 1 и отложены на плоскости концентрационного треугольника (рис. 1).
Из-за небольшого содержания титана в жидкой фазе кривая насыщенных
растворов на концентрационном треугольнике практически совпадает со сто-
роной Р2ОБ— Н2О. Поэтому отдельно в системе прямоугольных координат
(рис. 2) представлена зависимость содержания титана в растворах от кон-
центрации фосфорной кислоты.
Для выяснения состава твердых фаз в исследуемой системе были исполь-
зованы два метода:
1. Химический анализ отмытых ацетоном осадков. Он показал, что для
растворов ниже 18% Р2ОБ отношение ТЮ2:Р2О5 очень близко к 2:1, что
отвечает брутто-составу 2ТЮ2-Р2ОБ-хН2О. Выделенной соли с учетом воды,
входящей в состав осадка, можно приписать формулу 2ТЮ2 • Р2ОБ • 6Н2О. Ре-
зультаты химического анализа отмытых ацетоном воздушносухих осадков
представлены в табл. 2.
Для растворов, содержащих 28% Р2ОБ и более отношение TiO2:P2O5 в
осадках соответствует 1:1. Химический состав соединений, полученных пос-
ле удаления избытка фосфорной кислоты спиртом и высушенных при 100Q С,
приведен в табл. 3.
Видно, что выделенное соединение можно представить в виде фосфата
титана — Ti(HPO4)2. Согласно [7], фосфат титана указанного состава был
113
Таблица 2'
Химический анализ осадков в системе ТЮ2—Р2ОБ—Н2О при содержании
до Г8% Р2ОБ
Содержание Р3О6 в жид- кой фазе, % Найдено, % Рассчитано по формуле 2ТЮ2-Р2О6-6Н2О
TiO, н2о (по раз- ности) молярные отношения Т1О2 : Р2О5 : Н2О
ТЮ, Р,о, н,о
2,52 39,06 34,53 26,41 2,01:1,0:6,05
4,53 10,06 39,14 38,60 34,85 35,11 26,01 26,29 1,98:1,0:5,93 1,96:1,0:5,92 39,02 34,63 26,34
16,49 38,31 35,17 25,98 1,90:1,0:5,72
Таблица 3
Химический анализ осадков в системе TiO2—Р20Б—Н2О
при содержании свыше 28% Р2ОБ
Содержа - нне Р2О6 в жидкой фазе, % Найдено, % Рассчитано по формуле TiO2P2O6-H2O
ТЮ, Р,О, Н2О (по раз- ности) молярные отношения TiO2 : Р2ОБ:Н2О
ТЮ, Р,О6 Н,0
30,72 36,36 39,64 43,06 50,52 33,35 33,28 33,32 33,40 33,02 59,20 59,08 59,29 59,05 59,68 7,45 7,64 7,39 7,55 7,30 1:1,01:1,01 1:1:1,04 1:1:1,03 1,04:1:1,05 1:1,06:0,99 33,33 59,16 7,50
получен при нагревании раствора Ti (IV) в фтористоводородной и фосфорной;
кислотах. По мере удаления HF из раствора выделялся кристаллический
осадок фосфата титана. Выше уже было указано, что при нагревании смеси
гидроокиси титана и концентрированной фосфорной кислоты не удается по-
лучить необходимый по концентрации титана раствор, вследствие образова-
ния малорастворимого в этих условиях фосфата титана. Вероятно, при ком-
натной температуре титан в фосфорной кислоте склонен образовывать пере-
сыщенные растворы.
2. Метод «остатков» Шрейнемакерса дал аналогичную картину (см. рис. 1).
Для исследованных точек системы ниже 18% Р2ОБ лучи, соединяющие со-
став равновесных растворов и отжатых «остатков», удовлетворительно пере-
секают фигуративную точку состава фосфата титана — 2TiO2 • Р2ОБ • 6Н2О.
При концентрации фосфорной кислоты 28% и выше лучи кристаллизации
указывают на образование нового соединения, отвечающего составу фосфата
титана — TiO2 • Р2ОБ • Н2О. Для растворов, содержащих более 18% Р2ОБ, в
выделяющейся твердой фазе количество TiO2 ниже, а Р2ОБ несколько превы-
шает отвечающее составу 2TiO2 Р2ОБ • 6Н2О. Вероятно, это связано с нали-
чием в осадках двузамещенного фосфата — Ti(HPO4)2. Аналогичное явление
наблюдается и для ряда других тройных систем [9]. Графическая интерпре-
тация полученных данных позволяет судить о том, что изотерма TiO2 —
Р2ОБ — Н2О в области концентрации 1—56% Р2ОБ характеризуется дву-
мя твердыми фазами. В интервале 1—18% Р2ОБ образуется фосфат титана
ТЮНРО4-2,5Н2О и при 28—53% Р2ОБ — фосфат Ti(HPO4)2. Данные хими-
ческого анализа состава солей, расчета по формулам и графического опреде-
ления согласуются. Известные по данным [5, 6] фосфорнокислые соединения
титана — TiOHPO4-2,5H2O и Ti(HPO4)2 — были получены препаративным пу-
тем из сернокислых растворов титана при действии на них кислых фосфа-
114
Рис. 1. Определение состава твердых фаз в системе ТЮ2—Р2О6—Н2О по
методу Шрейнемакерса при 20° С.
Рис. 2. Содержание титана в жидкой фазе в зависимости от концентрации фосфорной ки-
слоты (номера на кривых соответствуют номерам серий табл. 1).
тов натрия или аммония, или из фосфорнокислых растворов при нагревании
и удалении фтористоводородной кислоты.
При растворении гидроокиси титана в фосфорной кислоте, по аналогии
с растворением гидроокисей ниобия или тантала [10, 11], возможно комплек-
сообразование по схеме:
TiO2-xH2O+6H3PO4=H2 [Ti (Н2РО4)в]+(х+2) Н2О.
Образование прозрачных растворов при взаимодействии ТЮ2-хН2О с концент-
рированной фосфорной кислотой говорит в пользу указанной схемы. При
определенном содержании в растворе фосфорной кислоты происходит, очевидно,
разрушение комплексного соединения и выделение фосфата титана
Н2 [Ti (H2PO4)e]=Ti (НРО4)2+4Н8РО4
или при разбавлении растворов водой образуется фосфат «титанила»
Н2[Ti(H2PO4)J+xH2O=TiOHPO4-2,5H2O+5H3PO4+(x — 3,5) Н2О.
Процесс образования ортофосфатов титана в растворах Н8РО4 различной кон-
центрации можно представить также по реакциям:
TiO2-xH2O+nH3PO4-TiOHPO4-2,5H2O+(x — 1,5) Н2О+
+(п - 1) Н3РО4 -> Ti (НРО4)2+(х+2) Н2О+(п - 2) Н8РО4.
При значительных избытках фосфорной кислоты или упаривании таких сме-
сей равновесие будет все более смещаться вправо до образования фосфата
титана. Наличие достаточного количества воды приводит к образованию фос-
фата оксокатиона титана.
ВЫВОДЫ
1. Методом изотермической кристаллизации найдено, что в системе
TiO2 — Р2О6 — Н2О при 20° С происходит образование двух равновесных,
твердых фаз: TiOHPO4-2,5H2O и Ti(HPO4)2. Определены области выделе-
ния указанных фосфатов.
2. Высказаны некоторые соображения о возможном ходе процесса обра-
зования ортофосфатов титана в растворах фосфорной кислоты различной кон-
центрации.
ЛИТЕРАТУРА
1. Merz. J. Pract. Chem., 1866, 1, 99, 162.
2. J. C. Ghosch. J. Ind. Chem. Soc., 1931, 8, 695.
3. Ericson. Iron. Age, 1903, 27, 4.
4. Tcheng Da-Tchang, Li-Hououg. С. ч., 1935, 200, 2173.
5. Викт. И. Спицын, Е. А. Ипполитова. Ж- анал. хим., 1951, 6, 1,5.
6. Д. И. Курбатов, С. А. Павлова. Физико-химические исследования соединений;
редких тугоплавких элементов (Ti, V, Nb, Та). Тр. Ин-та хим. УФАН СССР, 1966,
вып. 10, стр. 65.
7. Д. И. Курбатов, С. А. Павлова. Там же, стр. 73.
8. А. М. Горелов, А. П. Штин. Ст. в наст, сб., стр. 106.
9. Е. Б. Бру цк у с. Исследования по прикладной химии. М.—Л., Изд-во АН СССР,.
1955.
10. И. В. Тананаев и Г. Б. Сейфер. Докл. АН СССР, 1962, 144, 6, 1314.
11. Г. Б. Сейфер, И. В. Тананаев. Ж. неорг. хим., 1963, 8, 1, 63.
АКАДЕМИЯ НАУК СССР
уральский филиал
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
И. Г. ЧУФАРОВА, Н. И. ПЕТУНИНА
ЭКСТРАКЦИЯ НИОБИЯ ИЗ СОЛЯНО-СЕРНОКИСЛЫХ РАСТВОРОВ
СПИРТАМИ
В литературе описана экстракция ниобия из солянокислых растворов
отдельными представителями различных классов органических соедине-
ний. Так, Хикс и Джильберт [11 показали возможность извлечения ниобия
из солянокислых растворов диизопропилкетоном. Элленбург, Лэддикот,
Мур [2] предложили метод выделения оксихлоридных комплексов ниобия
трибензиламином. В. Н. Старцев и Е. И. Крылов [3,41 изучили экстрак-
цию ниобия из солянокислых растворов трибутилфосфатом. И. М. Гибало,
И. П. Алимарин и др. [5] установили, что ниобий из солянокислых раство-
ров может быть выделен ацетофеноном, бензальдегидом, трибутилфосфа-
том. Онц также показали, что использование смеси растворителей (напри-
мер, трибутилфосфата и хлороформа) приводит к значительному увеличе-
нию степени экстракции ниобия.
Известно, что в солянокислых растворах при концентрации НС1 выше
5 моль!л ниобий находится в растворе в виде анионных комплексов [6—8].
В связи с этим целесообразно было испытать в качестве экстрагентов нио-
бия из солянокислых растворов спирты — органические растворители^
способные к экстракции анионов. В литературе вопросы экстракционной
способности класса спиртов освещены недостаточно. Не установлено, как
влияет на экстракцию ниобия длина цепи, наличие изомерии в молекуле
спирта.
Цель настоящей работы — изучить и сопоставить основные характе-
ристики процесса экстракции ниобия из солянокислых растворов различ-
ными спиртами. Степень экстракции ниобия спиртами значительно повы-
шается в присутствии высаливающих веществ, таких, как серная кислота,
поэтому экстракция ниобия исследовалась в соляно-сернокислых раство-
рах. Проведение этой работы позволило выявить наиболее эффективные
экстрагенты для извлечения ниобия из соляно-сернокислой среды.
Экспериментальная часть1
Методика работы. Исходные солянокислые растворы ниобия готовили
растворением свежеосажденной гидроокиси ниобия в соляной кислоте.
Растворы ниобия с высокой концентрацией металла получали путем пере-
вода ниобия в раствор совместно с титаном. Для приготовления соляно-
сернокислых растворов в солянокислые растворы нйобия вводили опреде-
ленное количество серной кислоты. Экстракцию ниобия изучали при
комнатной температуре. Равные объемы органической и водной фаз переме-
1 Принимали участие Л. И. Санталова и И. И. Тюфякова.
117
Таблица 1
Экстракция ниобия и титана
из соляно-сернокислых растворов
различными спиртами
Спирт ^pNb KpTi Кразд
Н. гексиловый 8,7 0,4 21,7
Н. гептиловый 13,1 0,6 21,8
Н. октиловый Вторичный окти- 19,2 0,9 21,3
ловый . , . 27,8 0,8 34,7
Н. нониловый 25,6 1,3 19,7
Н. дециловый Первичные спирты 29,2 1,7 17,1
С12—С18 . . Вторичные спирты 45,5 1,6 28,4
Cj2—С19 . . 81,1 0,3 270,3
Примечание. Исходная концентра-
ция: НС1—8, H2SO4—2 моль/л.
шивали на магнитной мешалке в течение 5 мин, затем после 10-минутного
отстаивания фазы разделяли. Аналитическое определение ниобия прово-
дили весовым танниновым методом. Малые количества его анализировали
колориметрически с формазаном. Плотность органической фазы измеряли
пикнометрическим методом.
Сопоставление экстракции ниобия из соляно-сернокислых растворов
различными спиртами. В качестве растворителей для экстракции ниобия
из соляно-сернокислых растворов использовали алифатические спирты
с различным числом углеродных атомов в молекуле от С4 до С19. Экстракция
ниобия низшими спиртами (н. бутило-
вым, н. амиловым, изоамиловым) не была
изучена из-за высокой растворимости их
в воде. С увеличением молекулярного
веса растворимость спирта в воде пони-
жается и появляется возможность экст-
ракции соляно-сернокислых растворов
ниобия. Результаты, полученные при
экстракции ниобия из соляно-серно-
кислых растворов спиртами в зависи-
мости от длины углеводородного ради-
кала, приведены в табл. 1. Исходная
концентрация Nb2O5 и ТЮ2 в раство-
рах составляла, соответственно, 1 и
20 г/л. Видно, что экстракция ниобия
зависит от количества атомов углерода
в спирте: удлинение углеродной цепи
приводит при одной и той же кислот-
ности к увеличению коэффициента рас-
пределения ниобия. Эта закономерность,
по-видимому, связана с тем, что высшие
спирты образуют с ниобием более проч-
ные комплексы.
Сравнение экстракционной способности первичных и вторичных спир-
тов указывает на то, что экстракция ниобия из соляно-сернокислых раство
ров зависит от положения гидроксильной группы в углеродном скелете
спирта при одинаковом числе углеродных атомов. Это влияет на прочность
комплексного соединения ниобия, образованного со спиртами. Эффектив-
ность вторичных спиртов в качестве экстрагентов ниобия выше, чем первич-
ных. Наиболее пригодны для количественного извлечения ниобия из соля-
нокислых растворов вторичные высшие спирты с числом углеродных атомов
С12—С19, для них коэффициент распределения ниобия достигает высоких
значений (Кр—81,1). Низкий коэффициент распределения ниобия и высо-
кая растворимость низших спиртов не позволяют использовать их для це-
лей экстракции. Коэффициенты распределения титана также зависят от
длины углеводородного радикала спирта (см. табл. 1). Экстрагируемость ти-
тана спиртами при указанной кислотности растворов сравнительно низкая.
Коэффициент распределения титана несколько возрастает с увеличением
молекулярного веса спирта. Вторичные спирты экстрагируют титан в мень-
шей степени, чем первичные.
Помимо характеристики экстрагентов по степени извлечения элемен-
тов очень важную роль играет их разделительная способность. Наиболь-
ший коэффициент разделения ниобия и титана достигается для вторичных
высших спиртов (Кразд=270). При экстракции ниобия из соляно-сернокис-
лых растворов вторичными высшими спиртами происходит отделение его
от значительных количеств титана.
118
Дальнейшие исследования проводились с вторичными высшими спир-
тами, превосходящими по своей экстракционной способности относительна
ниобия все остальные изученные спирты, а также обеспечивающими луч-
шее отделение ниобия от титана. Ввиду того что индивидуальные вторич-
ные высшие спирты мало доступны, нами были использованы технические-
вторичные высшие спирты с числом углеродных атомов С12—С19, которые
получают в промышленных условиях на Шебекинском комбинате синтети-
ческих жирных кислот и жирных спиртов. Подробная характеристика
этих спиртов дана в работе А. И. Башкирова и В. В. Камзолкина [9].
Экстракция ниобия в зависимости от концентрации соляной кислоты
в растворе. Большую роль при экстракции ниобия высшими спиртами иг-
Рис, 1. Зависимость коэффициента распре-
деления ниобия Кр (/) и плотности орга-
нической фазы d (2) от концентрации со-
ляной кислоты в исходном растворе.
Рис. 2. Зависимость коэффициента распре-
деления ниобия Кр от концентрации сер-
ной кислоты в исходном растворе.
Исходная концентрация соляной кислоты: / — 7»
2 — 8, <3 — 9 моль!л.
рает содержание соляной кислоты в растворах (рис. 1). При низкой кон-
центрации соляной кислоты (до 6 моль/л) ниобий экстрагируется вторич-
ными высшими спиртами в очень небольшой степени. Начиная с 7 моль/л
НО наблюдается быстрое повышение коэффициента распределения его по
мере увеличения концентрации соляной кислоты в растворах. В растворах
соляной кислоты 8—10 моль/л этот коэффициент достаточно высокий
(81,1—109,3).
Такой характер экстрагируемое™ ниобия объясняется различным ион-
ным состоянием ниобия в растворах соляной кислоты. По литературным
данным [6—8], при концентрации НС1 меньше 5 моль/л в растворах пре-
обладает катионная форма ниобия. При более высоком содержании НС1
образуются анионные хлоридные комплексы ниобия состава NbOClT. Оче-
видно, в виде этих комплексов он извлекается экстрагентом из растворов
соляной кислоты. Постепенное увеличение величины коэффициента распре-
деления ниобия в зависимости от концентрации соляной кислоты, начиная
с 7 моль/л НС1, свидетельствует о том, что качественный состав экстрактов
одинаков. Об этом говорит и изменение удельного веса органической фазы
в зависимости от содержания соляной кислоты, выражающееся линией,
близкой к прямой (см. рис. 1).
Экстракция ниобия в зависимости от концентрации серной кислоты
в растворе. Как указано выше, экстрагируемость ниобия из солянокислых
растворов спиртами значительно повышается в присутствии серной кис-
лоты. Она, обладая высаливающим эффектом, способствует переходу нио-
119
бия в органическую фазу. На рис. 2 представлено влияние концентрации
высаливающего агента — серной кислоты — на степень экстракции нио-
бия вторичными высшими спиртами. Видно, что коэффициент распределе-
ния ниобия зависит от содержания серной кислоты в растворе и повышается
с возрастанием ее концентрации. При добавлении 2 моль/л серной кислоты
ниобий выделяется вторичными высшими спиртами из растворов 7, 8 и
9 моль]л соляной кислоты, соответственно, на 98,6; 98,9 и 99,1%, т. е. в при-
Рис. 3. Зависимость концентрации ниобия
в органической фазе (/) и коэффициента
распределения ниобия (2) от содержания
его в исходном растворе.
сутствии последней достигается коли-
чественное извлечение его из соляно-
кислых растворов.
Экстракция ниобия в зависимости
от концентрации металла в растворе.
Распределение ниобия при экстракции
вторичными высшими спиртами в
зависимости от концентрации металла
в исходном растворе представлено
на рис. 3. Концентрацию Nb2Os в ис-
ходных растворах меняли от 1 'до
15г/л, содержание соляной и серной
кислот составляло, соответственно, 8
и 2 моль/л. Для получения устойчи-
вых соляно-сернокислых растворов с
высокой концентрацией ниобия не-
обходимо было присутствие в раст-
воре титана, поэтому опыты прове-
дены в присутствии титана (20 г/л
ТЮ2). Из полученных данных видно
(см. рис. 3), что при увеличении кон-
центрации ниобия в растворе увеличивается его содержание и в органи-
ческой фазе. В пределах исследованных концентраций (до 15 г/л Nb2O6) он
может быть выделен на 99% вторичными высшими спиртами из соляно-
сернокислых растворов.
Взаимное влияние ниобия и титана при совместной экстракции вторич-
ными высшими спиртами. Для изучения взаимного влияния ниобия и ти-
тана в процессе экстракции их вторичными высшими спиртами проведены
опыты по экстракции ниобия и титана из соляно-сернокислых растворов,
содержащих оба эти элемента. Исследовано влияние на экстракцию увели-
чения количества одного из взятых металлов при постоянной концентра-
ции другого. Полученная зависимость процента извлечения ниобия и ти-
тана в органическую фазу, коэффициентов распределения и разделения
этих элементов от содержания их в исходном растворе представлена в табл. 2.
Расчеты произведены с учетом изменения объема органической и водной
фаз при экстракции в среднем на 10%. Видно, что ниобий незначительно
влияет на экстракцию титана. При различном содержании ниобия в рас-
творе (от 1 до 15 г/л) и постоянном количестве титана (20 г/л) процент извле-
чения титана (25,3) и коэффициент распределения (0,277) — постоянные
величины. Характер изменения степени извлечения титана и коэффициен-
тов распределения в зависимости от содержания титана в растворе одина-
ков как для растворов индивидуального титана [10], так и для растворов,
содержащих смесь ниобия и титана (см. табл. 2). '
Титан при небольшом содержании в растворе (до 30 г/л TiO2) не влияет
на экстракцию ниобия вторичными высшими спиртами. В пределах этих
концентраций титана извлечение ниобия составляет 99,09% и коэффици-
ент распределения 88,9. В присутствии больших количеств титана (до
100 г/л) экстракция ниобия вторичными высшими спиртами несколько по-
120
Таблица 2
Экстракция ниобия и титана вторичными высшими спиртами при совместном
присутствии в растворе
Концентрация в исходном растворе, г/л Извлечено в органическую фазу, % *pNb •KpTi Кразд
Nb,O, тю, Nb,O, | тю,
1 20 99,08 25,3 88,13 0,277 318,1
2 20 99,08 25,2 88,13 0,275 320,4
5 20 99,07 25,3 87,35 0,277 315,3
10 20 99,06 25,3 86,25 0,277 311,3
15 20 99,05 25,4 85,37 0,278 307,1
5 5 99,09 61,0 88,91 1,280 69,46
5 10 99,08 53,5 88,12 0,942 93,54
5 30 99,09 24,0 88,91 0,258 344,2
5 40 97,00 22,1 26,46 0,232 114,00
5 60 92,05 17,2 9,47 0,170 55,74
5 80 84,20 15,3 4,36 0,148 29,52
5 100 75,09 12,1 2,47 0,113 21,94
П р н,м ечание. Исходная концентрация: НС1—8, H2SO4—2 моль}л.
давляется. В этих условиях в органическую фазу за однократную экстрак-
цию извлекается 75% ниобия. На основании определенных коэффициентов
распределения ниобия и титана рассчитаны коэффициенты их разделения,
которые достигают высоких значений (до 344).
Следовательно, при совместном присутствии этих элементов в растворе
и содержании TiO2 до 30 г/л экстракция ниобия вторичными высшими спир-
тами протекает достаточно полно (до 99%) и происходит отделение значи-
тельных количеств титана.
ВЫВОДЫ
1. Изучена и сопоставлена экстракция ниобия из соляно-сернокислых
растворов различными спиртами. С увеличением количества атомов угле-
рода в спирте экстракция ниобия возрастает.
2. Замена первичного спирта на вторичный при одном и том же коли-
честве атомов углерода в спирте приводит к повышению коэффициента рас-
пределения ниобия.
3. Установлено, что вторичные высшие спирты наиболее эффективные
экстрагенты для извлечения ниобия из соляно-сернокислых растворов.
4. Экстракция ниобия спиртами определяется как концентрацией соля-
ной кислоты, так и содержанием серной кислоты в исходном растворе. Для
количественного извлечения ниобия в органическую фазу экстракцию сле-
дует проводить в растворах соляной кислоты 8 моль/л, содержащих сер-
ной кислоты 2 моль/л.
5. Изучено взаимное влияние ниобия и титана в процессе экстракции
вторичными высшими спиртами. Ниобий незначительно влияет на экстрак-
цию титана. Титан при содержании до 30 г/л также не оказывает влияния
на экстракцию ниобия. В присутствии значительных количеств титана
экстракция ниобия вторичными высшими спиртами несколько подавляется.
ЛИТЕРАТУРА
1. Н. G. Hicks, R. S. Gilbert. Anal. Chem., 1954, 26, 1205.
2. I. J. Eilenburg, G. W. Leddi cotte, F. L. Moore. Anal. Chem., 1954,
26, 1045.
3. В. H. Старцев, E. И. Крылов. Ж. неорг. хим., 1966, 11, 12, 2820.
4. В. Н. Старцев, Е. И. Крылов. Ж. неорг. хим., 1967, 12, 2, 526.
9 Заказ № 632
121
5. И. М. Гибало, И. П. Алимарин, Д. С. Альбадри, Г. В. Еремина.
Радиохимия, 1967, 9, 1, 118.
6. Е. А. Субботина, Д. М. Чижиков, О. В. Альтшулер. Разделение
близких по свойствам редких металлов. М., Металлургиздат, 1962, стр. 98.
7. Б. И. Набиванец. Ж. неорг. хим., 1964, 9, 5, 1079.
8. 1. Н. Kanzelmeyer, I. Ryah, Н. Freund. J. Amer. Chem. Soc., 1956, 78,
3020.
9. А. И. Башкиров, В. В. Кам зол к ин. Химическая наука и промышленность,
1959, 4, 5, 606.
10. Н. И. Пе ту и и на, И. Г. Чуфарова. Ст. в наст, сборнике, стр. 123.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
Н. И. ПЕТУНИНА, И. Г. ЧУФАРОВА
ЭКСТРАКЦИЯ ТИТАНА ИЗ СОЛЯНОКИСЛЫХ РАСТВОРОВ ВТОРИЧНЫМИ
ВЫСШИМИ СПИРТАМИ
За последнее время большое внимание уделяется исследованию свойств
хлоридов ниобия и, в частности, их экстракции органическими раствори-
телями [1]. Но так как титан ближайший спутник ниобия, то интересно
было изучить его поведение при экстракции из солянокислых сред. В ли-
тературе об этом имеются лишь немногочисленные сведения. В работе [2]
приводятся результаты по изучению экстракции титана кислыми алкил-
фосфатами и в [3—5] изучена система TiCl4—НС1—ТБ.Ф. Помимо этого нам
не удалось встретить каких-либо работ, посвященных экстракции хлори-
дов титана. Целесообразно было провести исследование по экстракции
титана алифатическими спиртами. Была изучена экстракция титана спир-
тами в зависимости от количества атомов углерода в спирте [ 11.
Предметом настоящего исследования является экстракция титана вто-
ричными высшими спиртами из солянокислых растворов в зависимости от
кислотности, от содержания металла в исходных растворах и изучение
влияния высаливающего действия серной кислоты на распределение титана.
Экспериментальная часть1
Методика работы. Исходные солянокислые растворы титана готовили
растворением четыреххлористого титана квалификации «ч» в соляной ки-
слоте соответствующей концентрации.
Методика проведения исследований описана в предыдущей статье [1].
Определение оптической плотности исходных растворов проводили на фо-
токолориметре ФЭКН-57 (светофильтр № 2, кювета 20 мм).
Экстракция титана в зависимости от концентрации соляной кислоты.
Изучение экстракции титана вторичными высшими спиртами из соляно-
кислых растворов проводилось в зависимости от кислотности растворов
(2—12 моль/л НС1). Концентрация титана в пересчете на двуокись титана
во всех опытах одна и та же (20 г/л TiO2). Рассчитанные коэффициенты
распределения титана от концентрации соляной кислоты приведены на рис. 1.
Видно, что из слабосолянокислых растворов (до 6 моль/л НС1) титан прак-
тически не экстрагируется. Из солянокислых растворов 7 моль)л экстраги-
руется лишь на 5%, с коэффициентом распределения 0,05. С дальнейшим
увеличением концентрации свободной соляной кислоты постепенно повышается
экстракция титана и значение коэффициентов распределения: при 9 моль/л
титан переходит в органическую фазу до 14% (Кр— 0,2), 10 моль/л — до
30% (Кр —0,36) и 12 моль/л — до 83,5% (Кр —3,9).
1 Принимали участие Г. А. Кушкина и Л. И. Санталова.
9*
123
Рост коэффициента распределения и процента экстракции титана с повы-
шением концентрации соляной кислоты в исходных растворах объясняется
увеличением количества анионных комплексов титана [Ti (ОН)2С1412- и [TiCle]2~,
которые, по всей вероятности, имеют тенденцию экстрагироваться органи-
ческими растворителями [3—7].
Экстракция титана вторичными высшими спиртами в присутствии сер-
ной кислоты. Как известно, серная кислота подавляет диссоциацию рас-
пределяющихся молекул и вызывает их дегидратацию, т. е. является выса-
ливателем при экстракционных процессах. Влияние ее, как высаливаю-
Рис. 1. Зависимость коэффициента
распределения титана Яр от концент-
рации соляной кислоты в исходных
растворах. Содержание в исходных
. растворах ТЮ2 — 20 г/л.
HtSO<: 1 — 0, 2—2 моль/л.
Рис. 2. Влияние"серной кислоты на
коэффициент распределения титана.
Содержание в исходных растворах
ТЮ2 — 20 г/л.
1 — 7, 2 — 8;3 — 10 моль/л.
щего агента, на распределение титана при экстракции вторичными выс-
шими спиртами из солянокислых растворов изучалось от 0,1 до 4 моль/л
H2SO4.
Концентрация титана в растворах была постоянной — 20 г/л TiO2. Со-
держание соляной кислоты в 1, 2 и 3-й серии опытов составляло, соответ-
ственно, 7, 8, 10 моль/л. Из рис. 2 видно, что наличие в системе серной ки-
слоты значительно увеличивает коэффициент распределения титана, а сле-
довательно, и его извлечение, и тем больше, чем выше концентрация серной
кислоты. Так, если из солянокислого раствора 7 моль/л НС1 титан экстра-
гируется на 5% (/Ср — 0,05), то в присутствии серной "кислоты 2 моль/л
экстракция повышается вдвое, а при концентрации серной кислоты 4 моль/л
увеличивается в 5 раз (до 25%). При экстракции из растворов, содержащих
НС1 —8 и H2SO4— 2, 3, 4 моль/л, титан переходит в органическую фазу на
26, 30 и 40% с коэффициентом распределения 0,3; 0,42 и 0,67, соответствен-
но. Из рис. 1 (кривая 1) видно, что при экстракции из солянокислого рас-
твора 10 моль/л НС1 коэффициент распределения титана составляет 0,36
и процент извлечения 30, в то время как при добавлении серной кислоты
даже 0,1 моль/л первый увеличивается до 0,88 и второй до 52. При наличии
в растворе 2—4 моль/л H2SO4 коэффициент распределения составляет
3,6—4,3 и извлечение титана в органическую фазу достигает 82—85%.
124
При сопоставлении коэффициентов распределения титана (см. рис. I),
полученных при экстракции титана из солянокислых растворов в присут-
ствии серной кислоты (2 моль/л H2SO4) и без нее, видно, что они идентичны.
Заметная экстракция титана в присутствии серной кислоты начинается из.
более слабых растворов по соляной кислоте (7 моль/л}, т. е. серная кислота
предотвращает гидролиз хлоридных соединений титана, благодаря чему
увеличивается его степень экстракции.
Рис. 3. Зависимость коэффициента распределения титана /Ср (кри-
вые 1, 2) и оптической плотности растворов Д (кривые 3, 4) от
концентрации титана в исходных растворах.
Содержание исходных растворов, моль!л\ 1, З — НСА — 8, Н,8О, — 2;
2,4 — НС1 — 10, HtSO4 — 2.
Исходя из полученных данных, можно сделать вывод, что оптималь-
ными условиями выделения титана в органическую фазу являются
НО — 10, H2SO4 — 2 моль/л.
Экстракция титана из соляно-сернокислых растворов в зависимости от
содержания титана в растворе. Распределение титана между вторичными
высшими спиртами и раствором изучали из соляно-сернокислых растворов,
содержащих НС1 и H2SO4, соответственно, 8 и 2 (рис. 3, кривая /); 10 и
2 моль/л (рис. 3, кривая 2). Концентрация титана изменялась от 5 до
160 г/л TiO2. Найдено, что коэффициенты распределения титана при
экстракции из указанных выше растворов резко падают по мере возраста-
ния его в растворах.
Кроме того обнаружено, что при изменении концентрации титана в исход-
ных растворах вначале наблюдалось увеличение оптической плотности (рис. 3,
3,4), а затем, начиная с 50 г/л TiO2, окраска растворов резко ослабевала,
что обусловлено образованием полимерных форм титана. Методом сравни-
тельного диализа [71 показано, что фактор полимеризации зависит от кон-
125
центрации титана и ионов С1— и почти не зависит от концентрации водород-
ных ионов. Количество полимеров титана возрастает с увеличением концен-
трации Ti4+ и С1~ и достигает максимального значения при концентрации
титана 50 г/л и соляной кислоты 10 моль!л. В работе [7] высказано предпо-
ложение, что при НС1 больше 8—9 моль/л в состав полимерных форм входит
гидратированное звено Ti(H2O)n ... Cl — Ti(H2O)n .... По всей вероятности,
эти гидратированные формы титана не экстрагируются вторичными высшими
спиртами. Таким образом, экстракция титана последними зависит от содер-
жания его в растворе. С увеличением концентрации титана коэффициент
распределения резко падает.
ВЫВОДЫ
1. Изучена экстракция титана вторичными высшими спиртами в зави-
симости от концентрации соляной кислоты. Максимальное извлечение ти-
тана достигается при экстракции из растворов, содержащих 10—12 моль/л
НС1.
2. Показано влияние серной кислоты на степень экстракции титана
вторичными высшими спиртами из солянокислых растворов. Присутствие
серной кислоты повышает извлечение титана в органическую фазу.
3. Изучена экстракция титана из соляно-сернокислых растворов в за-
висимости от его содержания в растворах. С изменением концентрации
титана в исходном растворе в сторону концентрирования происходит за-
метное уменьшение извлечения титана в органическую фазу.
ЛИТЕРАТУРА
1. И. Г. Чуфарова, Н. И. Петунина. Ст. в наст, сб., стр. 117.
2. М. Л. Н а фта н о ви ч, А. С. Черняк. Сборник научных трудов Иркутского на-
учно-исследовательского института редких металлов, 1961, вып. 9, стр. 140.
3. В. Н. Старцев, Е. И. Крылов, Ю. А. Козьмин. Ж. неорг. хим., 1965,
10, 10, 2367.
4. В. Н. Старцев, Е. И. Крылов, Ю. А. Козьмин. Ж. неорг. хим., 1966,
11, 10, 2312.
5. В. Н. Старцев, Е. И. Крылов, Ю. А. Козьмин, Ж. неорг. хим., 1967,
12 3 762.
6. Б. И. Набиванец. Ж. неорг. хим., 1962, 7, 3, 417.
7. Б. И. Набиванец, Л. Н. Кудрицкая. Ж. неорг. хим., 1967, 12, 5, 1165.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ филиал
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970'
Н. И. ПЕТУНИНА, И. Г. ЧУФАРОВА
К ВОПРОСУ ЭКСТРАКЦИИ ТАНТАЛА ИЗ СОЛЯНО-СЕРНОКИСЛЫХ
РАСТВОРОВ СПИРТАМИ
Экстракция тантала из соляно-сернокислых растворов изучена недо-
статочно. В литературе опубликовано сравнительно немного работ, посвя-
щенных исследованию закономерностей распределения тантала в системе
органический растворитель — соляная кислота. Имеются лишь указания
на возможность экстракции тантала из Юн. HCI трибутилфосфатом (извле-
чение 82%) и ацетофеноном (извлечение 50%) [11, смесью трибутилфосфата
и хлороформа (извлечение 71%) [2].
В настоящей работе ставилась задача изучить процесс экстракции тан-
тала различными алифатическими спиртами из соляно-сернокислых рас-
творов в зависимости от молекулярного веса спирта, концентрации соляной
кислоты, времени стояния исходных растворов, а также исследовать влия-
ние иона фтора на экстракционный процесс. Полученные данные позволят
выявить наиболее эффективные экстрагенты для извлечения тантала из
соляно-сернокислых растворов и рассмотреть некоторые аспекты состоя-
ния тантала в этих растворах в отсутствии и в присутствии иона фтора.
Экспериментальная часть1
Исходные соляно-сернокислые растворы тантала готовили разведением
сернокислого раствора тантала соляной кислотой соответствующей кон-
центрации.
Методика проведения экстракции описана в сообщении [3].
Аналитическое определение тантала в макроколичествах проводили
весовым танниновым методом. Микроколичества его анализировали коло-
риметрически с бутилродамином. Для опытов использовали первичные
индивидуальные спирты: н. гексиловый, н. гептиловый, н. нониловый,
н. дециловый, а также технические смеси первичных и вторичных спиртов
С12—С18, любезно предоставленные Шебекинским комбинатом жирных
спиртов и кислот.
Предварительные опыты показали, что тантал из свежеприготовлен-
ных соляно-сернокислых растворов удовлетворительно экстрагируется
спиртами. Поэтому была изучена экстракционная способность спиртов в
отношении тантала из названных растворов в зависимости от концентра-
ции соляной кислоты (7—11 моль/л) и длины углеродного радикала спирта.
Во всех опытах исходная концентрация тантала и серной кислоты состав-
ляла, соответственно, 1 г/л Та2О5, 4 моль/л H2SO4. Введение в солянокислый
Принимали участие И. И. Тюфякова, Л. И. Санталова.
127
раствор 3—6 моль/л H2SO4 способствует экстракции тантала и препятствует
образованию коллоидных форм тантала.
Извлечение тантала алифатическими спиртами и его коэффициенты
распределения в зависимости от концентрации соляной кислоты приведены
на рис. 1. Видно, что из растворов, содержащих 7—8 моль/л соляной кисло-
ты, тантал экстрагируется спиртами от 1 до 25% с коэффициентами распре-
.деления порядка 0,002—0,02. С дальнейшим увеличением концентрации
гЮО
-80
-60
О?
*
-4/7 >
а
ъ
-20
Рис. 1. Зависимость коэффициента распределения (/—6) и извле-
чения тантала в органическую фазу (7—/2) от концентрации соляной
кислоты в исходном растворе.
Спирты: /, 7 — н. гексиловый; 2, 8 — н. гептиловый; 3—9— н. нониловый;’.
4, 10 — в. дециловый; 5,11 — первичные высшие спирты^С12—С1в; 6, 12 — вторич-
ные высшие спирты С12—С18.
свободной соляной кислоты наблюдается постепенное увеличение экстрак-
ции тантала и повышение значений коэффициентов распределения. При
концентрации соляной кислоты 10—11 моль/л тантал практически пол-
ностью переходит в органическую фазу. Извлечение составляет 94—99%,
а коэффициент распределения возрастает от 3 до 60. Высокие значения ко-
эффициентов распределения тантала из 10—11 моль/л НС1 объясняются об-
разованием анионных хлоридных комплексов тантала [4], которые и
экстрагируются алифатическими спиртами.
Таким образом, для количественного извлечения тантала из свежепри-
готовленных соляно-сернокислых растворов указанного выше состава мож-
но использовать любые первичные алифатические спирты. Для технологи-
ческих целей целесообразнее применять смесь технических высших спиртов
С12—Cj8. Ценное свойство их — низкая растворимость в растворах мине-
ральных кислот, малая токсичность и невысокая стоимость.
Изучена экстракция тантала смесью первичных высших спиртов в за-
висимости от времени стояния растворов (рис. 2). После пятичасового стоя-
128
ния наблюдается постепенное падение степени экстракции тантала (извле-
чение составляет 88%), после 24 ч стояния тантал переходит в органиче-
скую фазу до 75%, через 10 суток стояния раствора он извлекается лишь
на 50% (см. рис. 2, кривая 2).
В зависимости от времени стояния растворов коэффициент распределе-
ния тантала между солянокислыми растворами (10,5 н. НС1, 4 н. H2SO4)
и первичными высшими спиртами резко падает от 60 до 0,7. Быстрое паде-
ние степени экстракции тантала со временем объясняется тем, что в соля-
нокислых растворах сравнительно быстро происходят процессы полимери-
зации тантала, т. е. он образует малоактивные, полимерные электронейт-
ральные формы, которые могут быть выражены схематично в виде [51:
п до 0,5
ДТа (ОН)°6]р------
н.НС1
0,5 —5н.НС1 . 10—Пн. на
[Ta(OH)S+„lp---------[Та (ОН)£$р—----------
-ЧТа(ОН)хаГ1,.
Данные полимерные формы тантала не экстрагируются спиртами. Для
того чтобы перевести полимерные неэкстрагируемые формы тантала а.
экстрагируемые анионные комп-
лексы, необходимо ввести в соля-
нокислый раствор ион фтора или
насытить им органическую фазу
[61.
Введение в раствор HF в
соотношении Та : F= 1 : 2 приво-
дит к частичному разрушению
полимерных форм тантала, при
увеличении избытка HF обра-
зуются мономерные анионные
комплексы, которые и имеют
тенденцию экстрагироваться.
Были проведены опыты по
экстракционному извлечению
тантала из соляно-сернокислых
растворов, стоявших 20 суток.
В качестве экстрагентов испы-
тывались первичные и вторич-
ные высшие спирты С12—-С18.
Последние предварительно насы-
щали двукратно плавиковокис-
Рис. 2. Зависимость коэффициента распределения,
(1) и извлечения (2) тантала первичными высши-
ми спиртами С12—CJS от времени стояния соля-
но-сернокислых растворов.
лыми растворами с концент-
рациями 25, 50, 100 г/л HF.
После насыщения в органичес-
кую фазу переходило 2, 4, 8 г/л
HF, соответственно. Насыщен-
ные фтором органические растворители контактировали с соляно-серно-
кислыми растворами тантала, содержащими 1 г/л Та2О6. В результате
однократной экстракции тантал практически полностью экстрагировался
как первичными, так и вторичными спиртами. При отношении Та : F=
= 1 : 25 извлечение тантала в органическую фазу составляло 94%, при
отношении Ta:F=l : 50 он экстрагировался на 99,5%. Таким образом^
высшие спирты могут быть использованы для выделения тантала как из.
свежеприготовленных, так и стоявших соляно-сернокислых растворов,
В последнем случае необходимо вводить фтор-ион.
129<
выводы
1. Изучено распределение тантала между соляно-сернокислыми рас-
творами и алифатическими спиртами от Св до Cj8 из свежеприготовленных
и стоявших растворов. Все спирты являются эффективными экстрагентами
.для тантала.
2. Установлено, что тантал практически полностью извлекается из
стоявших соляно-сернокислых растворов в присутствии фтор-иона при от-
ношении Та : F=1 : 25ч-1 : 50.
ЛИТЕРАТУРА
1. И. М. Гибало, Дж. С. Альбадри, Г. А. Еремина. Ж. анал. хим., 1967, 22,
5, 816.
2. И. М. Гибало, И. П. Алимар и^н, Дж.'С. Альбадри, Г. А. Еремина. Радио-
химия, 1967, 9, 1, 118.
3. И. Г. Чуфарова, Н. И. Петунина. Ст. в наст, сб., стр. 117.
4. Б. И. Набиванец. Ж. неорг. хим., 1962, 7, 12, 2739.
5. А. К. Бабко, Б. И. Набиванец. Проблемы современной химии координационных
соединений, вып. 2. Изд-во ЛГУ, 1968, стр. 111.
6. А. К. Бабко, Г. И. Гр ид чин а. Ж. неорг. хим., 1963, 8, 1, 52.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970-
Д. И. КУРБАТОВ, С. А. ГОВОРКО, 3. П. КОРТЕВА
ЗАВИСИМОСТЬ ДИФФУЗИОННОГО ТОКА ТУГОПЛАВКИХ ЭЛЕМЕНТОВ
(Ti, Nb, Мо) ОТ КОНЦЕНТРАЦИИ ФОСФОРНОЙ КИСЛОТЫ
И ОПРЕДЕЛЕНИЕ СОПРОТИВЛЕНИЯ ЭЛЕКТРОЛИТИЧЕСКОЙ ЯЧЕЙКИ
Для полярографического определения тугоплавких элементов (Nb, Ti, W
и др.) ранее [1—3] были предложены в качестве основного электролита кон-
центрированные растворы фосфорных кислот с повышенной вязкостью. Изме-
нение состава раствора влияет на величины, входящие в уравнение Илькови-
ча. Наибольшее влияние на диффузионный ток с изменением состава раство-
ра оказывает коэффициент диффузии (О), входящий в упомянутое уравнение
в степени 1/2. Так как коэффициент диффузии на основании закона Стокса —
Эйнштейна обратно пропорционален вязкости (л), то зависимость диффузион-
ного тока от вязкости может быть выражена соотношением
idV г) = const.
В литературе относительно выполнения указанной зависимости данные
противоречивы. Так, Вавруха [4], изменяя вязкость среды путем добавления
до 50% сахарозы, нашел, что величина для ряда катионов (РЬ, Си,
Zn, Со и др.) не постоянна. В то время как Мак-Кензи [5] указывает для
этих катионов постоянство id^r], сохраняющееся до 47% содержания саха-
розы. Шоландер [6] на многочисленных примерах подтвердил постоянство
величины idV^л: путем использования электролитов и неэлектролитов вяз-
кость растворов в некоторых его опытах изменялась в 16 раз. Этот автор
указал на сольватацию и комплексообразование, как на причины, вызыва-
ющие отклонение данной величины от постоянного значения. Он подтвердил
также ранее высказанное соображение [5], что изменение вязкости путем до-
бавления лиофильного коллоида значительно меньше сказывается на умень-
шении диффузионного тока, чем это имеет место при таком же изменении
вязкости истинного раствора. Очевидно, частицы деполяризатора сравнитель-
но легко перемещаются в промежутках между коллоидными частицами. На-
блюдаемое уменьшение тока Zn2+, Pb2+ в присутствии желатины, как ука-
зывает Кольтгоф [7], связано с образованием комплексов этих катионов с же-
латиной. Таким образом, изучая зависимость величины id V\| от состава рас-
твора, можно качественно судить о комплексообразовании иона деполяризато-
ра с молекулами фона.
Методика приготовления растворов описана ранее [1—3]. При измерении
применен полярограф ПА-1; растворы термостатированы. Температуру под-
держивали 25±0,1°С.
Изменение концентрации фосфорной кислоты сильно меняет ее вязкость.
Интересно было изучить влияние вязкости раствора на предельный ток. Это
135
Мо 05 —< 1 1 b 1 ь см см 1 I bbbbb CM CM CM CM •—*
Nb b- b- CD to CD CD CD CD CD ।।।।
н CM CO b 1 1 CM CM CM O co LQ 05 CD | | 00 CD тН Ю Ю CM* CM* CM* CM* CM*
т о Д а 36 No b- CO co CD tH TH CD CM CM CD* o' CD* O* 2?111-
н 0,196 0,26 0,46 . . b- CO Ю 1 | ЮСООЮО о’о’^’г-Г см*
1 ₽ 00 t-< OO OO CO tH CO ~ О 00 b- r—< »—< T—1 to CD CM Ю CD b- b’ to CO 05 CM CM CD CM CD lO tH CO CM <
с приоора, ccmfceK, 0,1314 0,1314 0,1218 0,1218 0,1218 0,1314 0,1218 0,1218 0,1218 0,1314 0,1218 0,1218
Il s с тН О 00 tO 05 CM* o’ CO* CO* tH 05 tH CD CD Ю »—< I—1 T—< 00 05 co to CD OO CD — 00* th CD* Ю* CM* tH CM CM
tig и а b- b- CO CM 05 CM тН О oo CO ссГ ОС? 05 •—Г CD b- Ю CO CO 05 b- Ю 05 CM 05 CO CD 1—< »—• 00 CO tH Th b-*lD b-*CO*CM*rw CM »—<»—t
Время истечения раствора, сек. 00 О CD О b- TH CM* CD i-** 00 OO b- CM CD b^tQ tH co CM Ю О Ю О CD CD CM 05 Ю TH oT.oT o? cm OO TH CM Ю CM >—'
п С 3 а С V/<1VOW ‘Edoaiocd 18,95 17,9 17,2 16,2 15,3 14,4 13,4 12,8 9,65 4,92 4,11 1,9
с Е П раствора, г/см* 1,866 1,831 1,787 1,751 1,720 о ю to to to 00 tH COCO О CD < О 05 о CD CD TH co CM О r-1 < 1—< »—< »—< »—<<
Состав раствора Н8РО4 : Н2О (по объему) Йех. H3PO4 9,5:0,5 9:1 8,5:1,5 8:2 to ci co co CM TH 05 Ю b- CD —< r-<
имеет значение для проверки указан-
ной выше зависимости предельного
тока от вязкости при изменении ее
в широких пределах, а также для
выяснения влияния концентрации
фосфорной кислоты на состав сое-
динений изучаемых элементов. В
наших опытах вязкость варьировали
путем изменения концентрации од-
ного компонента (Н8РО4), в отличие
от [6], где для изменения вязкости
использовано большое количество
веществ (сахароза, глюкоза и др.).
Кроме того, в наших исследованиях
фосфорная кислота одновременно
являлась комплексообразователем
для изучаемых ионов. Вязкость раст-
воров фосфорной кислоты опреде-
ляли методом капиллярного истече-
ния, используя капиллярный виз-
козиметр Пинкёвича. Диаметр капил-
ляра 1,2 мм. Кинематическую вяз-
кость жидкости определяли по урав-
нению
v=x-c,
где v — кинематическая вязкость
жидкости, сст\ т — время истече-
ния жидкости, сек-, с —постоянная
вискозиметра.
Для двух вискозиметров она сос-
тавляла 0,12187 и 0,131’40 сст/сек,
соответственно. Динамическая вяз-
кость или коэффициент вязкости (т])
находили согласно выражению
T] = VQ,
где q—-удельный вес жидкости.
Определение проводили в термо-
стате, при температуре 20 ±0,05° С.
На рис. 1 представлена зависимость
вязкости растворов фосфорной кис-
лоты от ее концентрации.
Данные по определению влия-
ния вязкости на предельный ток
титана, ниобия и молибдена приве-
дены в табл. 1 и рис. 2. Из них
видно, что соотношение idYt) = const
сохраняется для диффузионных то-
ков титана, ниобия и молибдена
в широких интервалах концентраций
фосфорной кислоты. Особенно это
относится к титану: вязкость раст-
вора изменялась более чем в 1Q0 раз.
Постоянство значений произведения
132
указывает на отсутствие изменения состава фосфорнокислых соединений наз-
ванных элементов в изученном интервале концентраций фосфорнокислых
растворов. Постоянство состава соединений титана в растворах фосфорной
кислоты различной концентрации было показано ранее [8].
В предложенных нами [1—3] методах определение тугоплавких элементов
проходит в концентрированных растворах фосфорных кислот, сравнительно
слабых электролитах. Поэтому представляло интерес определить сопротивле-
ние электролитической ячейки. Особенно в случае применения выносного на-
ружного анода. Сопротивление ячейки (7?) определяли путем измерения тока (О
Рис. 1 . Зависимость вязкости ор-
тофосфорной кислоты от ее кон-
центрации при 20° С.
О 5 /5 ' 20
Рис. 2. Влияние вязкости ортофосфорной
кислоты на предельный ток титана, ниобия
и молибдена.
1 — титан; 2 — ниобий; 3 — молибден.
и потенциала полуволны для различных концентраций деполяриза-
тора. Тбк, соответствующий этим потенциалам, будет разным, а также и
значения iR будут отличаться, т. е.
(^*/«)набл = )испрН-*'^-
Графическое изображение (Е1/,)набл от среднего тока дает прямую линию,
которая при i=0 имеет значение потенциала полуволны, исправленного на
омическое падение потенциала iR. Значение R также определяли по разности
величин потенциалов полуволн, наблюдаемых при двух различных концентра-
циях деполяризатора. В качестве деполяризатора был выбран титан, так как
в фосфорнокислых растворах он дает одноэлектронную обратимую волну,
потенциал полуволны которой практически остается постоянным при значи-
тельном изменении концентрации фосфорной кислоты [8].
На основании найденного значения исправленной на iR величины потен-
циала полуволны определено омическое сопротивление полярографической
ячейки. Примененная нами ячейка представляла собой электролизер, кото-
рый соединен электролитическим мостиком, наполненным фосфорной кисло-
той той же концентрации, что и электролизер. Последний через промежу-
точный сосуд с • насыщенным раствором хлористого калия соединялся с ка-
ломельным электродом. ;
В табл. 2 приведены данные определения омического сопротивления ука-
занной ячейки и вычислены значения iR при определении ниобия. Содержа-'
ние ниобия 0,003 моль/л. Видно, что омическое сопротивление ячейки в слу-
чае пирофосфорнокислых растворов меньше, чем ортофосфорнокислых. Оми-
133
Таблица 2
Сопротивление электролитической ячейки и омическое падение напряжения iR
при полярографировакии ниобия в орто- и пирофосфорнокислых растворах
различной концентрации
Ортофосфорнокислые растворы Пирофосфорнокислые растворы
Н,РО4: НеО (объемы) Др 1 0е, а ДЕ. в ом iR, в Н4Р2О7:Н2О (объемы) Д/- 10е, а дв, в ом iR, в
Н3РО4(уд. 2,55 0,015 5,8 0,0016 Н4Р207 (уд. 2,4 0,007 2,8 0,0005
вес 1,86) вес 1,91)
9:1 — — 5,6 0,0021 9,5:0,5 3,5 0,008 2,2 0,0005
8:2 1,54 0,007 4.6 0,0023 9:1 4,2 0,009 2,1 0,0006
7,5:2,5 1,54 0,007 4,5 0,0025 8,5:1,5 3,3 0,008 2,4 0,0009
1:1 3,33 0,015 4,5 — 7,5:2,5 5,1 0,011 2,2 0,0011
1:2 5,47 0,019 3,5 — 7:3 4,7 0,017 2,7 0,0016.
1:4 5,49 0,013 2,4 —
1:9 7,98 0,018 2,3 —
П римечание. Концентрацию деполяризатора изменяли от 2 • 1 0 8 до 2* 1 0 8 моль/л.
ческое падение напряжения iR с увеличением концентрации кислоты умень-
шается, и в кислоте 9:1 (оптимальные условия определения ниобия)
составляет 0,0006 и 0,002 b для пиро- и ортофосфорнокислого растворов, со-
ответственно (концентрация ниобия 3-10-3 моль/л). В случае использования
в качестве анода донной ртути сопротивление ячейки уменьшается.
ВЫВОДЫ
1. Изучено влияние вязкости растворов ортофосфорной кислоты на пре-
дельный ток тугоплавких элементов. Соотношение т| = const сохраняется
для диффузионных токов титана, ниобия и молибдена в широких пределах
концентраций кислоты.
2. Постоянство произведения itf=V<r] для названных элементов указывает
на отсутствие изменения состава фосфорнокислых соединений этих элементов
в изученном интервале концентраций ортофосфорной кислоты. Особенно это
относится к титану, где вязкость растворов фосфорной кислоты изменялась
более чем в 100 раз.
3. Найдено, что омическое падение напряжения iR в ячейке в случае
применения выносного анода (фон Н4Р2О7 или HSPO4:H2O=9:1—по объе-
му) в оптимальных условиях определения ниобия (CNb=0,003 моль/л) со-
ставляет 0,0006—0,002 Ь. Применение внутреннего анода значительно сни-
жает эту величину.
ЛИТЕРАТУРА
1. Д. И. Куратов. Ж- анал. хим., 1959, 14, 63.
2. Д. И. Курбатов. Ж. общ. хим., 1959, 14, 743.
3. Д. И. Курбатов, И. С. Скорый ина. Ж. анал. хим., 1962, 17, 711.
4. I. Vavruch. Collecktion czechoslov. Chem. Commims, 1947, 12, 429.
5. H. A. Me Kenzie. J. Council sci. Ind. Research, 1948, 1, 210.
6. A. Scholander. Sbornik I. Mezinar. polarograf. sjezdu, I. dil, stz. 260, Prirodoved.
vydavatelstvi, Praha, 1951.
7. I. M. Kolthoff, J. J. Lingane. Polarography, p. 54, Interscience, N. Y., 1941.
8. Д. И. Курбатов, С. А. Павлов. Физико-химические исследования соединений ред-
ких тугоплавких элементов (Ti, V, Nb, Та) Тр. Ин-та хим. УФАН СССР, 1966,
вып. 10, стр. 73.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20
ТРУДЫ ИНСТИТУТА ХИМИИ
1970
Д. И. КУРБАТОВ, 3. П. КОРТЕВА
ОПРЕДЕЛЕНИЕ ЧИСЛА ЭЛЕКТРОНОВ, УЧАСТВУЮЩИХ
ПРИ ЭЛЕКТРОХИМИЧЕСКОМ ВОССТАНОВЛЕНИИ Ti*+ И W«+
В КОНЦЕНТРИРОВАННЫХ ОРТО- И ПИРОФОСФОРНОКИСЛЫХ
РАСТВОРАХ С ПОВЫШЕННОЙ ВЯЗКОСТЬЮ
Ранее Д. И. Курбатовым [1—3] показано, что восстановление титана как
в орто-, так и пирофосфорнокислых растворах в широких областях измене-
ния концентрации протекает обратимо с участием одного электрона. Число
электронов, участвующих в электродной реакции, было определено из накло-
на прямой в полулогарифмических координатах. Однако в случае необрати-
мых процессов, например восстановление Nb(V) в фосфорнокислых растворах
[3, 4], такой метод определения числа электронов, участвующих в элект-
родной реакции, непригоден. Обычно в этом случае используют метод поля-
рографической кулонометрии, предложенный А. Г. Стромбергом и Т. М. Мар-
качевой [5]. В фосфорнокислых растворах при увеличении их концентрации
до 100% содержания кислоты коэффициент вязкости (т]) растворов значи-
тельно увеличивается в 100 и более раз [6]. Метод полярографической куло-
нометрии применяется для растворов средней концентрации, где т] имеет не
очень высокое значение.
Представляло большой практический интерес проверить применимость по-
лярографической кулонометрии [5] для определения числа электронов, участ-
вующих в электродном процессе в растворах с повышенной вязкостью. Для
проверки были взяты обратимые системы восстановления Ti4+-|-e=i=tTi3+ и
\V6+4-e^Ws+, где число электронов, участвующих в катодном восстановле-
нии, известно. Оно определено из угла наклона прямой в полулогарифмиче-
ских координатах [1—2] в широких областях концентрации фона (Н3РО4 и
Н4Р2О7), а следовательно, и изменения коэффициента вязкости (т]). Растворы
готовили по прописи условий полярографического определения титана [1—3].
Применяемый электролизер описан в работе [7]. Измерения проводили на ви-
зуальном полярографе типа ПА-1. К исходному раствору титана, приготов-
ленному на концентрированной орто- или пирофосфорной кислоте, прибавля-
ли воду и кислоту и получали рабочие растворы фосфорной кислоты различ-
ной концентрации.
Для опытов брали небольшой объем испытуемого раствора, помещали его
в электролизер специальной конструкции [7] и в течение 20—30 мин проду-
вали очищенный водород для удаления из раствора кислорода. Затем пред-
варительно определяли потенциал электрода, при котором устанавливался
предельный ток. Этот потенциал задавался на ртутный капельный электрод
в дальнейших опытах по микрокулонометрическому определению числа элект-
ронов, участвующих в электродной, реакции. Силу тока определяли по зер-
кальному гальванометру, чувствительность которого составляла
3,8-10_3 мка/мм.
13S
Таблица I
Результаты электролиза Ti‘+ в ортофосфорнокислых растворах
различного состава
Опыт 1 Опыт 2
z 3 Л « 3 н и- о « я и к я я о О и я Высота волны остаточного тока Истинная высота волны lg±- I емя элек- элиза, мин 5 н 3 s з з О и ш Высота волны остаточного тока Истинная высота волны "т
ММ £2. СО ь ММ
Н,РО4:Н2О=9:1 (по объему)
0 101 9 92 0 98 7 91
45 100 9 91 0,0043 30 97 7 90 0,0043
75 98 9 89 0,0128 60 96 7 89 0,0086
135 95 8 87 0,0212 90 94 6 88 0,0128
165 93 6 87 0,0212 120 94 7 87 0,0170'
195 92 6 86 0,0294 210 92 7 85 0,0294
225 92 6 86 0,0294 270 90 7 83 0,0374
285 91 6 85 0,0334 300 89 7 82 0,0414
н 1РО4:Н2О=2:1 (по объему)
0 119 10 109 — 0 127 9 118
45 119 10 109 — 7 126 9 117 —
105 115 9 106 0,0128 37 123 8 115 0,0086
140 112 7 105 0,0170 67 120 5 114 0,9128
170 109 6 103 0,0253 127 113 0 113 0,0170
200 105 5 100 0,0374 157 109 0 109 0,0334
230 103 5 98 0,0453 187 108 0 108 0,0374
260 101 5 96 0,0569 217 106 0 106 0,0453
.— ,—- -— -—. — 247 104 0 104 0,0531
— — — — — 277 102 0 102 0,0607
Число электронов, участвующих в электродном процессе, рассчитывали
по уравнению из работы [5]
fi
222-103-п ’
где 10 — предельный ток, мка; с0 — концентрация вещества в начальный мо-
мент времени, ммоль/л; v — объем взятого раствора, мл; k — контангес угла
наклона прямой в координатах
lg h/I — t У, сек}.
При определении числа электронов, участвующих в электродной реакции
в орто- и пирофосфорнокислых растворах, были выбраны следующие соста-
вы: Н8РО4:Н2О=9:1; 2:1; 1:2. Результаты опытов представлены в табл. 1,
2 и рис. 1, 2. Видно, что экспериментальные точки удовлетворительно укла-
дываются на прямые линии при продолжительности электролиза в течение
300 мин. Полярограммы во всех трех растворах как до электролиза, так и
после него хорошо выражены. Вид анодно-катодных волн подтверждает уста-
новленные ранее [1, 2] данные об обратимости протекающего процесса вос-
становления титана в ортофосфорнокислых растворах. Высота катодной вол-
ны совпадает с высотой анодно-катодной волны, что указывает на устойчи-
вость продуктов реакции в 4юсфорнокислых растворах изученных концентра-
ций.
136
В табл. 3 приведены резуль-
тэты расчета числа электронов 2}
в этих условиях. Видно, что вое- s
становление Ti4+ протекает во ю
всех изученных растворах с уча- ”
стием одного электрона. Это сог-
ласуется с ранее полученными дан-
ными о числе электронов [1, 2]
по наклону прямых в полулогариф-
мических координатах. Следо- £
вательно, метод полярографиче- £
ской кулонометрии, предложенной 'g
А. Г. Стромбергом и Т. М. Мар- ©
качевой [4], применим к раство- ~~
рам высокой вязкости.
Ниже приводим результаты II
определения числа электронов 9.
при катодном восстановлении ти- ®
тана на ртутном капельном элект- о
роде в растворах пирофосфорной ="
кислоты, предложенных Д. И. Кур- х
батовым [3] в качестве фона при ©,
полярографическом определении S
титана. Методика приготовления £
растворов титана и используемая °"
аппаратура те же, что и в слу- з
чае ортофосфорнокислых раст- 5
воров титана. Чувствительность §
гальванометра 4 10~3 мка/мм.
Нами исследовано восстановле-
ние титана в двух растворах ©
состава Н4Р2О7: Н2О= 10:2 и 1:2 ’©'
(по объему), представляющих
практический интерес при опре- о
делении титана в присутствии +
некоторых других элементов. Из р
табл. 4 и рис. 3 видно, что в л
случае пирофосфорнокислого рас- |
твора, так же как и ортофос- 2
форнокислого раствора, экспери- В"
ментальные точки хорошо уклады- |
ваются на прямых в координатах -
lg 10/1 — t. Обратимость процесса £
подтверждает совпадение анодно- |
катодной волны, снятой в вое- £
становленном растворе после £
электролиза. Данные расчета
числа электронов показывают,
что при восстановлении титана
(IV) в пирофосфорнокислом раст-
воре на ртутном капельном элект-
роде участвует один электрон
(табл. 5).
Рассмотрим пример примене-
ния микрокулонометрического
10 Заказ № 632
Рис. 1. Зависимость 1g Io/I от времени восстанов-
ления Т14+ в фосфорнокислых растворах.
НаРО4 : Н,О=9 : 1 (/); 2 : 1 (2); 1 : 2 (2).
Опыт 1 — темные кружки-, опыт 2 — светлые.
Рис. 2. Вид катодных и анодно-катодных поляро-
грамм титана в ортофосфорнокислых растворах.
НаРО. : НаО=9 : I (/, /' ); 2 : 1 (2, 2'); 1 : 2 (3, 3')
О 100 200 300 Ы)0 500 t.MHrf
Рис. 3. Зависимость 1g /0// от времени восстановления Ti4+ в пирофосфорнокис-
лых растворах.
Н4Р,О, : НаО=Ю : 2 (/,,2); 1 : 2 (3, 4).
Таблица 3
Данные расчета числа электронов (л), участвующих в катодном
восстановлении Ti4+ в ортофосфорнокислых растворах различной
концентрации
Состав раствора Н,РО4: Н2О (по объему) ^0- ммоль/л /0, мка Л)., с0 мка ctg а - 10—а V, мл Л
ммоль/л
9:1 5,1 1,049 0,205 552 0,57 0,9
5,1 1,037 0,205 492 0,47 0,96
2:1 10,0 4,14 0,414 300 0,463 1,2
10,0 4,48 0,448 288 0,456 1,2
1:2 9,6 9,84 1,021 96 0,455 0,98
10,0 10,49 1,049 96 0,446 1,01
10,0 10,26 1,026 96 0,447 1,0
Таблица 4
Результаты электролиза Т1*+
в пирофосфорнокислых
растворах различного состава
Опыт 1 Опыт 2
Время элек- тролиза, мин Общая высота волны Высота волны остаточ- ного тока Истинная I высота волны 1g / емя элек- элиза, мин Общая высота волны Высота волны остаточ- ного тока к со а н з s о 2 3 3 о X ю а lg^ /
зил* £ аз н мм
Н4Р207:Н20=10:2 (по объему)
0 30 118 118 5 5 113 113 — 0 35 126 123 5 5 121 118 0,0086
90 116 5 111 0,0086 65 121 5 116 0,0170
120 115 5 110 0,0128 125 118 5 113 0,0294
150 114 5 109 0,0170 155 117 5 112 0,0334
180 112 5 107 0,0212 185 116 5 111 0,0374
210 111 5 106 0,253 215 115 5 110 0,0414
240 НО 5 105 0,0334 275 112 5 107 0,0531
300 108 4 104 0,03741 335 108 5 103 0,0682
330 107 4 103 0,0414 — — — — —
Н4РО2:Н2О=1:2 (по объему)
0 170 9 161 — 0 174 10 164 —
20 164 9 155 0,0128 30 161 9 152 0,0294
45 161 9 152 0,0253 60 150 8 142 0,0607
70 156 9 145 0,0453 90 149 8 141 0,0645
160 150 9 141 0,0569 120 148 8 140 0,0682
190 147 9 138 0,0682 150 145 8 137 0,0792
220 145 9 136 0,0719 210 137 8 129 0,1038
310 133 5 128 0,1004 —- — — — —
—
метода к определению числа электронов, участвующих при катодном восстанов-
лении в фосфорнокислых растворах повышенной вязкости. Д. И. Курбатовым
и И. С. Скорыниной [8] было показано, что восстановление вольфрама на. ртут-
ном капельном катоде в пирофосфорнокислых растворах протекает обратимо
с участием одного электрона. Число электронов также определено по углу
10*
130
наклона прямой в полулогарифмических координатах. Растворы вольфрама в
фосфорных кислотах готовили по методике, описанной в работе [7]. Опыты
Рис. 4. Зависимость 1g /„// от времени восстановления W®+ в орто-и пиро-
фосфорнокислых растворах.
Н3РО4 : Н2О = 9 : 1 (/ -3); 1 : 1 (5—5). Н4Р2О, : Н2О=1 : 1 <7, 8); 10 : 2 (9,10).
по полярографической кулонометрии проводили так же, как и в случае тита-
на. Чувствительность применяемого гальванометра 3,8-10~3 мка/мм. При ис-
Таблица 5
Данные расчета числа электронов (и) при катодном восстановлении
титана в пирофосфорнокислом растворе
Состав раствора Н4Р2О7 : Н2О (по объему) са, ммолъ/л 10, мка Со ' мка ммолъ/л ctg а • 1 0 3 V. МА п
10:2 4,16 0,72 1,73 576 0,45 1.0
10:2 4,16 0,77 0,185 360 0,48 0,7
1:2 2,2 1,93 0,87 193 0,7 1,07
1:2 2,2 1,97 0,895 120 0,53 0,91
Т а'б л и ц а 6
Данные расчета числа электронов (и), участвующих в катодном восстановлении W®+
в фосфорнокислых растворах
Состав раствора кислота : вода (пообъему) с0, ммоль/л 10, мка 1а Со ’ мка ммоль/л ctg а V, МА П
Пирофосфорная кислота
10:2 2,5 0,60 0,24 445-10* 0,45 1,06
10:2 2,5 0,593 0,236 480-103 0,51 1,01
1:1 1,68 1,36 0,30 90-Ю3 0,42 0,8
1:1 2,0 1,59 0,798 120-Ю3 0,46 0,94
Ортофосфорная кислота
9:1 4,0 1,22 0,305 384-Ю3 0,52 1,02
9:1 4,0 1,12 0,28 324-10® 1,46 1,12
9:1 4,0 1,24 0,31 252-103 0,40 0,88
1:1 2,0 1,65 0,825 108-Ю3 0,42 0,95
1:1 2,0 1,65 0,825 138-Ю3 0,42 1,06
140
следовании взяты растворы состава Н4Р2О7:Н2О=10:2; 1:1 и Н3РО4:Н2О =
=9:1; 1:1 (по объему). Результаты электролиза приведены в табл. 6 и на
рис. 4.
Из рис. 4 видно, что опытные данные хорошо укладываются на прямые
линии в координатах 1g 70/7 — t. Вычисленное значение числа электронов,
участвующих в реакции, близко к единице. Это подтверждает данные [8],
полученные ранее, а также применимость полярографической кулонометрии к
растворам повышенной вязкости.
ВЫВОДЫ
1. Показано, что метод полярографической кулонометрии пригоден для
определения числа электронов, участвующих при катодном восстановлении на
ртутном капельном катоде в растворах повышенной вязкости (концентриро-
ванные фосфорные кислоты).
2. Методом полярографической кулонометрии определено число электро-
нов при катодном восстановлении титана (IV) и вольфрама (VI) в орто- и
пирофосфорнокислых растворах в широких пределах концентрации. Во всех
изученных растворах фосфорных кислот при катодном восстановлении титана
и вольфрама участвует один электрон, т. е. протекают реакции Ti4++e Ti3+
и W6+4-e W5+.
ЛИТЕРАТУРА
1. Д. И. Курбатов. Изв. СО АН СССР, 1958, 10, 35.
2. Д. И. К у р б а т о в, С. А. Павлова. Физико-химические исследования соединений
редких тугоплавких элементов (Ti, V, Nb, Та). Тр. Ин-та хим. УФАН СССР, 1966,
вып. 10, стр. 73.
3. Д. И. Курбатов. Ж. анал. хим., 1959, 14, 6, 743.
4. Д. И. Курбатов. Ж. анал. хим., 1959, 14, 1, 63.
5. А. Г. Стромберг, Т. М. Маркачева. Ж. физ. хим., 1954, 28, 4, 671.
6. Д. И. Курбатов, С. А. Говорко, 3. П. К о р т е в а. Статья в наст, об., стр. 131.
7. Д. И. Курбатов, С. А. Говорко, 3. П. Кортева. Исследование физико-хими-
ческих свойств соединений редких тугоплавких элементов (V, Nb, Та, Ti, Hf, Mo,
Be). Тр. Ин-та хим. УФАН СССР, 1970, вып. 17, стр. 162.
8. Д. И. Курбатов, И. С. Скорынина. Ж. анал. хим., 1962, 17, 6, 711.
АКАДЕМИЯ НАУК СССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
В. Н. ДАНИЛИН, С. П. ЯЦЕНКО
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ЖИДКИХ СПЛАВОВ
СИСТЕМЫ ГАЛЛИЙ — СУРЬМА
Сплавы галлия с сурьмой характеризуются образованием металлида экви-
атомного состава. Полупроводниковые свойства этого соединения в значи-
тельной мере объясняют большое внимание, уделяемое системе галлий —
сурьма. В литературе имеются существенные расхождения в данных термо-
графических [1—3], термодинамических исследований [4, 5] и измерений вяз-
кости сплавов этой системы [6]. Для успешного проведения синтеза метал-
лида из жидкой фазы наряду с данными по диаграмме состояния важно
знание свойств сплавов в жидком состоянии. В связи с этим интересно
было изучить термодинамические свойства жидких сплавов галлия с сурьмой.
Экспериментальная часть
Термодинамические свойства бинарной системы галлий — сурьма были
найдены из измерений ЭДС концентрационных цепей типа:
Оаж | (KCl+LiCl)+GaCltGaCl | GawSbi _ N)x. (1)
Электродом сравнения служил жидкий металлический галлий. Сплавы гал-
лий— сурьма (Ga^Sbi -n), где N— атомно-долевое содержание галлия, были
жидкими. Поскольку менее благородным компонентом сплавов галлий — сурьма
является галлий, то в качестве электролита была использована эвтектическая
смесь хлоридов лития и калия, содержащая 8,9-10-3 мольных долей хло-
ридов галлия. Постоянство концентрации хлоридов галлия достигали раство-
рением одинаковых количеств металлического галлия в токе хлористого во-
дорода под расплавом электролита. Схема измерительной ячейки, использо-
ванной нами при исследовании термодинамических свойств системы галлий —
сурьма, представлена на рис. 1. Полу элементами ячейки служили 17-образные
кварцевые трубки. Отверстие в нижнем колене полуэлемента диаметром
1,5—2 мм было на 2—4 мм выше уровня электролита в пробирке, что на-
дежно устраняло перенос галлия в сплав. Контакт между полуэлементами
обеспечивался тонкой пленкой электролита.
Измерения ЭДС проводили в инертной атмосфере, для создания которой
использовали гелий. Инертный газ очищали от влаги многократным пропу-
сканием через склянки с пятиокисью фосфора. Для очистки от кислорода
служила титановая губка, нагретая до 850—900° С. Хорошая герметичность
аппаратуры позволяла получать в реакционном пространстве при комнатной
температуре разрежение порядка 5-10-3 мм рт. ст. Для создания инерт-
ной атмосферы производили вакуумирование и последующее заполнение инерт-
ным газом не менее трех раз. Затем создавали небольшое избыточное давле-
142
ние инертного газа, чтобы избежать попадания атмосферного воздуха в про-
цессе длительных опытов. Заданную температуру поддерживали тигельной
печью сопротивления. Для исключения помех внешнего электрического поля.
а также для лучшего термостатирования
реакционную пробирку помещали в метал-
лический термоблок. Автоматическое регу-
лирующее устройство поддерживало задан-
ные значения температуры с точностью
+ 1 — 2°. Контроль температуры осущест-
вляли с помощью хромель-алюмелевой
термопары, кварцевый чехол которой был
всегда опущен в электролит измеритель-
ной ячейки.
Для приготовления сплавов были взяты
высокочистые металлы, содержащие не
более 1-10-3 вес. % примесей. Сплавы
готовили переплавкой необходимых коли-
честв компонентов над расплавом электро-
лита в инертной атмосфере. При поме-
щении их в электролит герметичность
измерительной ячейки не нарушалась. Для
измерения ЭДС гальванических элементов
(1) применена обычная компенсационная
схема с использованием высокоомного
потенциометра постоянного тока, зеркаль-
ного гальванометра и элемента Вестона.
Результаты опытов и обсуждение
Неоднократно проведенными опытами
установлено, что использованная мето-
дика не позволяет получить надежные
значения температурных градиентов ЭДС.
Это связано, вероятно, со значительной
упругостью паров хлорида галлия при
высоких температурах, приводящей к кон-
центрационным изменениям в электро-
лите. В связи с этим термодинамические
Рис. 1. Схема измерительной ячейки.
1 — вольфрамовые токоотводы, 2 — крепле-
ние U-образных трубок, 3 — U-образные
кварцевые трубки, 4 — отверстия 0
1,5—2 леи. 5 — электролит. 6 — кварцевый
чехо/т термопары, 7 — сплав галлий — сурьма
или чистый галлий, 8 — кварцевая пробирка,
9 — экраны из фторопласта, 10 — резиновая
пробка. 11— хромель-алюмелевая термо-
пара.
свойства галлия (активность и изобарный потенциал) в жидких сплавах с
сурьмой были рассчитаны для температуры 988° К по значениям ЭДС кон-
центрационных элементов, представленных в таблице.
В работе [7] нами сообщалось о наличии переменной валентности хлори-
дов галлия в эвтектической смеси хлоридов лития и калия, находящихся
в равновесии с чистым жидким галлием. Это учтено при расчете термодина-
мических свойств галлия введением средних валентностей хлоридов галлия,
находящихся в равновесии со сплавом, которые были рассчитаны по методи-
ке работы [8]. Вычисление термодинамических свойств второго компонента
проведено, как описано в работе [9]. Средние валентности ионов галлия, на-
ходящихся в равновесии со сплавом галлий — сурьма, парциальные изобар-
ные и парциальные избыточные изобарные потенциалы сурьмы и галлия были
вычислены для температуры 988° К (см. таблицу). Для активностей (а) гал-
лия и сурьмы при температуре 988° К в жидких сплавах галлий — сурьма
характерны отрицательные отклонения от закона Рауля (рис. 2). Отличие
значений активностей, полученных нами, от данных работы [5] можно объяс-
нить как разными температурами проведения исследования, так и тем, что
143
Термодинамические свойства сплавов галий — сурьма
NGa Е. мв пср -AZSb А7И36 - AZGa .-уизб - AZSb
кал/г-атом
0,1 117,6 3,00 8186 230 3614 20
0,2 98,3 3,00 6814 485 3654 47
0,3 83,5 3,00 5770 780 3406 80
0,4 74,8 3,00 5175 1110 3376 160
0,5 59,5 2,98 4089 2070 2728 710
0,6 28,5 2,91 1919 4749 916 2950
0,7 15,7 2,60 941 6606 241 4240
0,8 10,8 2,58 633 7540 196 4380
0,9 6,0 2,45 339 9330 182 4810
Примечание. Значения потенциалов сплавов (Е) относительно таллиевого электрода, средние
валентности галлия ( пср)’ изобарные потенциалы галлия IaZq^I. сурьмы (AZgb) и избыточные изо-
барные потенциалы галлия (A Z
/ .-=-изб\
и сурьмы AZsb J.
в указанной работе не учитывалась переменная валентность ионов галлия
в расплавленном солевом электролите. С этим связано также и различие по-
лученных нами значений интегрального изобарного потенциала сплавов
(рис. 3).
Из отрицательных отклонений активностей компонентов сплавов галлий —
сурьма, а также больших отрицательных значений интегрального избыточного
изобарного потенциала можно сделать вывод о частичном сохранении в жид-
Рис. 2. Активности галлия и сурьмы в
жидких сплавах галлий — сурьма при
988° К.
Данные: наши — сплошная линия, работы
[5] — штриховая.
Рис. 3. Избыточный интегральный изо-
барный потенциал сплавов галлий —
сурьма.
Условные обозначения те же, что на рис. 2.
ких сплавах в пределах ближнего порядка структуры типа соединения. Это
согласуется с данными работ [5, 6]. Выводы Шоттки и Бивера [4] о близо-
сти свойств системы галлий — сурьма к свойствам идеальных растворов про-
тиворечат общим представлениям о связи диаграмм состояния с характером
взаимодействия компонентов в жидком состоянии.
144
выводы
1. Методом ЭДС изучены термодинамические свойства жидких сплавов
системы галлий — сурьма при температуре 988° К.
2. Установлены значительные отрицательные отклонения активностей ком-
понентов сплавов галлий — сурьма от закона Рауля и отрицательные значе-
ния избыточного изобарного потенциала компонентов и сплава.
3. Полученные нами термодинамические данные так же, как и данные
Л. Н. Герасименко, А. Г. Морачевского и др. [5], позволяют сделать заклю-
чение об ошибочности мнения Шоттки и Бивера [4] о близости свойств жид-
ких сплавов галлий — сурьма к свойствам идеальных растворов.
ЛИТЕРАТУРА
1. I. Greenfield, R. Smith. J. Metals., 1955, 7, 2, 357.
2. К. Koster u. В. Thoma. Z. Metallkunde, 1955, 46, 4, 291.
3. В. M. Глазов, Д. А. Петрова. Изв. АН СССР, ОТН, 1958, 4, 125.
4. W. F. Schottky а. М. В. Be ver. Acta Met., 1958 , 6, 320.
5. Л. H. Герасименко, А. Г. Морачевский, И. Б. Сладков и Л. Н. Лож-
кин. Защитные металлические и оксидные покрытия, коррозия металлов и исследо-
вания в области электрохимии. М.—Л., «Наука», 1965, стр. 241.
6. В. М. Глазов. Изв. АН СССР, Металлургия и топливо, 1960, № 5, 190.
7. В. Н. Данилин, С. П. Яценко. Теория и практика амальгамных процессов. Алма-
Ата, «Наука», 1966, стр. 20.
8. Н. Г. Илющенко, Н. И. Корнилов, А. И. Анфиногенов, Электрохимия
расплавленных солевых и твердых электролитов. Тр. Ин-та электрохимии УФАН
СССР, 1966, вып. 8, стр. 65.
9. В. Н. Данилин, С. П. Яценко. Ж. физ. хим., 1967, 41, 879.
АКАДЕМИЯ НАУК СССР
уральский филиал
вып. 20
труды института химии
1970
С. П. ЯЦЕНКО, Г. М. РУБИНШТЕЙН
ОСАЖДЕНИЕ И РАЗДЕЛЕНИЕ ГИДРООКИСЕЙ (ОСНОВНЫХ
СУЛЬФАТОВ) АЛЮМИНИЯ, ГАЛЛИЯ И ЖЕЛЕЗА
Во многих случаях в технологических процессах и аналитической прак-
тике взаимное влияние гидроокисей при их осаждении из раствора, а также
закономерность распределения компонентов между раствором и твердой фазой
не учитывается должным образом [1—3]. Предполагается, что гидроокиси
ведут себя в смеси так, как они вели себя в индивидуальных растворах,
т. е. их осаждение идет поочередно в соответствии с изменением pH раст-
вора. Однако уже многократно отмечалось взаимное влияние гидроокисей
при их осаждении [4—6]. При практическом проведении гидроксидного раз-
деления благодаря резкому местному изменению pH происходит совместное
выделение разделяемых ионов металлов, присутствующих в растворе. Уста-
новление фазового равновесия в многокомпонентных водно-солевых системах
и закономерности распределения компонентов между раствором и твердой
фазой — решающее в рассматриваемых процессах осаждения и разделения
гидроокисей. Основы термодинамической теории распределения изоморфных
солевых компонентов между раствором (ж) и твердой фазой (т) были созда-
ны работами Венслоу [7], Б. Н. Никольского, В. И. Парамоновой [8] и дру-
гими [9—11], а для многокомпонентных систем впоследствии были развиты
в статьях Г. Н. Горштейна [12], Ю. Д. Третьякова и др. [13, 14]. Рас-
смотрим некоторые элементы этой теории в применении к случаю нахожде-
ния в равновесии между раствором и осадком соединений галлия и алю-
миния.
Химический потенциал любого компонента (i) системы равен
(Р<)ж=(Р?)ж+^7’1п(а,)ж; (1)
(Р'<)т==(Р/°)т+^7’1п(а1)т. (2)
В этих уравнениях (а£)ж и (а,)т — активность t-компонента в жидкой и твер-
дой фазах, соответственно, а (р°)ж и (ц?)т— химические потенциалы i-ro
компонента в стандартном состоянии.
Изменение свободной энергии (при постоянном давлении) при равновесии,
как известно, равно нулю, а химические потенциалы в жидкой и твердой
фазах равны, т. е.
AF=0, (ц,-)ж=(н()т. (3)
Следовательно, в общем случае можно записать
(PL - 14) - (Р?ж - и?т)+^ 1п ' п ;ж =о, (4)
(а2т) *-а1ж
146
где п± и п2 — валентность ионов первого и второго сорта. Из формулы (4)
получаем (5) (^т)л‘
или после возведения в степень (nj^)-1 l/nt Л/п, (6)
В уравнениях (5) и (6) значения постоянных связаны: -
К=К?‘п'=ехр(-^), (7)
где 1
- Н°т) - пг (|Х°Ж - ц«т). (8)
В интересующем нас частном случае nj=n2 (поскольку валентность алюми-
ния, галлия и окисного железа равна 3), и, учитывая, что а1=\1т1, можно
записать:
В этом уравнении Yi и у2 — коэффициенты активности ионов 1 и 2 в жид-
кой и твердой фазах.
Величина, стоящая в левой части уравнения (9), характеризует распреде-
ление между твердой фазой и раствором компонента 1 относительно компо-
нента 2. Эта величина называется равновесным коэффициентом распределе-
ния (Di/J:
jjf (т1/т2)т (gi/galr (Ю)
(т1/т2)ж
rpfi. (gi)T — вес. % i-ro компонента в твердой фазе и (gt)x — вес. % i-ro ком-
понента в равновесном маточном растворе. Отсутствие данных о коэффици-
ентах активности электролитов в насыщенных растворах затрудняет расчет
величины Dy, по формуле (9). Однако для идеальных систем равновесные
коэффициенты распределения являются постоянными во всем интервале кон-
центраций. В таких системах коэффициенты активности компонентов твердо-
го раствора при любых соотношениях равны единице и отношения коэффи-
циентов активности катионов в равновесном маточном растворе при любых
соотношениях представляют собой постоянную величину [12, 15]. Верно
и обратное положение, что постоянство равновесных коэффициентов распре-
деления — необходимый и достаточный критерий идеальности системы. Дей-
ствительно, постоянное значение Oi/2 соответствует постоянному отношению
<V1W(V‘/ Vl)T. Коэффициенты активности солевых компонентов, например
<3а (ОН)3 и А1 (ОН)3, могут быть выражены, как
Yi = Уоа>+ • Yoh—> (П)
Y2=Yai«<--Yoh-> (12)
и, учитывая, что согласно условиям идеальности (YGa>+/YAi3 f-)jK=const и
(Yoa>+/YAi»+)T=const, следует, что Yi=Y2=:Const или
din у1=й(1пу2. (13)
147
Согласно уравнению Гиббса — Дюгема, для бинарного раствора компонентов
1 и 2 в твердой фазе имеем
dlnY1= — ^-dlny2. (14)
Уравнения (13), (14) одновременно справедливы лишь при условии, когда
Y1=l, у2=1. Однако в этом случае твердые фазы представляют собой иде-
альный раствор изоморфных компонентов.
Выведенные соотношения для обмена ионов жидкой и твердой фаз не
зависят от механизма обмена (поверхностный, объемный). Можно было пред-
положить, что указанные закономерности должны сохраняться и при осаж-
дении основных солей или гидроокисей железа, галлия и алюминия. Для
подтверждения этого и проведены излагаемые ниже исследования.
Экспериментальная часть
Осаждение основных сульфатов трехвалентного железа, галлия и алюми-
ния проводилось прибавлением по каплям 0,5 н. щелочного раствора при
интенсивном перемешивании. Состав исходного сернокислого раствора: галлия
Таблица 1
Значения pH осаждения гидроокисей
некоторых металлов (с учетом
образования гидроксокомплексов)
pH
Гидро-
ОКИСЬ
начала осаж-
дения при
исходной кон-
центрации
осаждаемого
иона
Рис. 1. Влияние концентрации катиона в рас-
творе на pH выпадения основной соли.
1— Fes+; 2 —Ga’-!’; 3 — Al’+; 4 — Сг’ + ; 5 — Zn*+;
6 — Fe2 + ; 7 —Ni2 + ; 8 — Mn2+.
t I 0.01
1 МОЛЬ МОЛЬ
Fe (ОН), ' 1,45 2,35 3,8 14,0
Ga (ОН), 2,20 3,05 4,0 6,9
А1 (ОН), 3,25 4,05 5,2 7,8
Сг(ОН), 4,05 4,95 6,8 12,0
Zn (ОН)2 5,45 6,40 8,0 10,5
Fe (ОН)2 6,50 7,50 9,7 13,5
Ni (ОН)2 6,70 7,70 9,5 14,0
Мп (ОН)2 7,80 8,80 10,4 14,0
* Остаточная концентрация осаждаемого
иона -с 10”6 маль/л.
0,7; 2; 5 г/л и алюминия — 30 г/л. Отношение первого ко второму при пол-
ном осаждении осадка составляло, вес.%: 2,3; 6,7 и 16,6. (Состав осадка 1
соответствует существующему отношению галлия к алюминию в таллиевом
концентрате, получаемом при карбонизации глиноземных растворов алюмини-
евого завода [16]). Соосаждение галлия и алюминия с гидроокисью железа
изучалось на растворах, содержащих железа 7, галлия 0,7 и алюминия
30 г/л. Высокая ионная сила исследуемых растворов (J>7) и повышенная
температура способствовали высокой растворимости осадка, что благоприят-
ствовало быстрому установлению равновесия. Приготовленные смеси интен-
сивно перемешивались в термостате при 80° С в течение трех суток, а затем
отстаивались несколько часов при 20° С. Установлено, что для выбранных
148
условий проведения опытов этого времени достаточно для достижения равно-
весия. Маточный раствор для измерений pH и анализа отбирали из отстояв-
шихся растворов. Анализ жидкой и твердой фаз проводили обычными ме-
тодами.
Результаты опытов по определению значения pH выпадения индивидуаль-
ных гидроокисей железа, галлия, алюминия и некоторых других сопутству-
ющих галлию металлов для различных концентраций катиона в растворе
показаны в табл. 1 и на рис. 1. Если исходить из поведения индивидуальных
гидроокисей и распространить
это на поведение смеси растворов
двух осаждаемых гидроокисей,
то можно ожидать существования
в осадке вполне определенных
соотношений
нентов.
железа
окись
когда
зло-2
Рис. 2. Гетерогенное равновесие при- фракци-
онном осаждении основных солей из серно-
кислых растворов.
Б — количество А! (ОН)8 в осадке; А, %: /—си-
стема основных солей железа и алюминия» в осадке
Fe (ОН)Э; 2 — система основных солей железа, гал-
лия и алюминия, в осадке Ga (ОН)3; 3 — система
основных солей галлия и алюминия, в осадке
Ga (ОН)а.
pH: а— 3,8; 6—4,0; в— 4,1; г — 4,2; 5—4,3;
е — 4,4 ; ж — 4,6; з — 4,9; и — 5,6.
основных компо-
Например, гидроокись
будет осаждать гидро-
галлия при условии,
в растворе содержится
моль/л окисного железа
и 1 моль/л галлия (pH = 2,2).
Следовательно, для больших кон-
центраций железа в этом раст-
воре можно было ожидать выпа-
дения незагрязненной окиси же-
леза. Однако практически этого
не происходит благодаря взаим-
ному влиянию гидроокисей. По-
следнее приводит к изменению
pH начала осаждения гидроокиси
и к изоморфному включению в
осадок компонента, индивидуаль-
ная гидроокись которого при
этом pH не выделяется, и неко-
торым другим явлениям [2].
Для характеристики распре-
деления компонентов между осад-
ком и раствором были рассчитаны равновесные коэффициенты распределения по
формуле (10). Анализ значений этих коэффициентов при фракционном осаждении
гидроокисей (основных солей) галлия и алюминия (табл. 2), железа и алю-
миния (табл. 3, рис. 2) позволяет утверждать, что в пределах погрешности
эксперимента (±10%) £)р постоянно при любых соотношениях компонентов.
Следовательно, разбираемые системы являются идеальными. Из уравнения
(10) для идеальной системы можно рассчитать концентрацию любого компо-
нента в твердой фазе, находящегося в равновесии с маточным раствором
любого состава. Для этой цели уравнение (10) лучше переписать в форме
—Dp(-/±g' (15)
(/,))
где gi и хг — весовые доли компонента 1 в солевой части маточного раство-
ра и в твердой фазе, соответственно; ПР(>/2) — равновесный коэффициент рас-
пределения компонента 1, по отношению к компоненту 2. Сравнение DQa/Ai и
E>Fe/Ai показывает, что значения Dp изменяются в широких пределах в зави-
симости от природы солевых компонентов.
Если предположить, что в насыщенном растворе изоморфных основных
солей отношения коэффициентов активности (У1/у2)ж близко к единице и для
149
Таблица 2
Гетерогенное равновесие при фракционном осаждении гидроокисей (основных солей) галлия и алюминия из сернокислых растворов
[Ga* - % Ga (ОН)3, Al - % Al (ОН)3, D Oa/A1=(Ga*/AI*)T/(Ga*/AI*)llt]______________________________________________________________
Исходный раствор, еион/л pH при рав- новесии Результаты анализа В осадке, % от исходного Коэффициент распределения
твердая фаза жидкая фаза
АР+ Ga«+- 10s Ga* Al* Ga* Al* Ga* Al* ДСа/А1 Дм/Ga
1,1 1,0 3,75 0,035 0,43 0,085 8,23 29 5 7,9 0,13
1,1 1,0 3,95 0,0665 1,187 0,0535 7,47 55,4 13,7 7,8 0,13
1,1 1,0 4,1 0,0842 2,04 0,0358 6,62 70,2 23,5 7,6 0,13
1,1 1,0 4,2 0,1013 3,55 0,0187 5,11 84,4 41 7,8 0,13
1,1 l.o 4,3 0,11 . 5,13 0,0101 3,53 91,6 59,2 7,5 0,13
1,1 1,0 4,3 0,11 5,03 0,0084 3,63 93 58 9,6 0,10
1,1 1,0 4,4 0,1158 6,67 0,0042 1,99 96,5 77 8,2 0,12
1,1 2,9 3,8 0,1186 0,463 0,2284 8,2 34,2 5,35 9,1 0,11
1,1 2,9 3,9 0,164 0,839 0,183 7,82 47,4 9,7 8,3 0,12
1,1 2,9 4,0 0,209 1,36 0,138 7,3 60,4 15,7 8,1 0,12
1,1 2,9 4,1 0,271 2,565 0,0761 6,095 78,1 29,6 8,5 0,12
1,1 2,9 4,2 0,3085 4,25 0,0385 4,41 89,3 49,1 8,3 0,12
1,1 2,9 4,9 0,3435 8,01 3,5-10-3 0,65 99,1 92,4 7,9 0,13
1,1 7- ,2 3,8 0,336 0,588 0,534 8,07 38,7 6,8 8,6 0,12
1,1 7,2 4,1 0.649 2,44 0,221 6,22 74,6 28,2 7,6 0,13
1,1 7,2 4,25 0,793 4,74 0,077 3,92 91,2 54,8 8,5 0,12
Среднее. . . 8,2 0,12
Таблица 3
Гетерогенное равновесие при фракционном осаждении гидроокисей (основных солей) железа и алюминия из сернокислых растворов
[ Fe* - % Fe (ОН)3, Al* - % Al (0Н)3, PFe/A1 = (Fe*/Аl*)T/(Fe*/А1*)ж]____________________________________
Исходный раствор, г -ион/л Ионная сила J Результаты анализа В осадке. % от исходного Коэффициент распределения
Fe>+ АР-Ь твердая фаза жидкая фаза Fe* Al* ^Fe/Al ДА1/Ре
Fe* 1 Al* Fe* Al*
0,125 1,1 8,8 0,913 0,329 0,427 8,331 68,1 3,8 54,0 0,018
0,125 1,1 8,6 1,118 0,736 0,222 7,924 83,5 8,5 54,2 0,018
0,125 1,1 8,5 1,090 0,762 0,250 7,90 81,3 8,8 45,3 0,022
0,125 1,1 7,9 1,284 2,42 0,056 6,24 95,8 28,0 59,2 0,017
0,125 1,1 7,4 1,310 3,64 0,030 5,02 97,7 42,0 60,2 0,017
Среднее, . . 55 1,8-10—«
Таблица 4
Равновесие при фракционном осаждении гидроокисей (основных солей) железа,
галлия и алюминия из сернокислых растворов .
[Fe* - % Fe (ОН),, Al* - % Al (ОН),, Оа* - % Ga (ОН),,
DGa/Fe+Ai = (6а*/Ре*+А1*)т/(0а*/Ре+А1*)ж]
Исходный раствор, г ион1л Результаты анализа
Fe»+ Ga»+ Al’+ твердая фаза жидкая фаза .
Fe* | Ga* Al* Fe* Ga* I Al*
0,125 0,125 0,125 0,01 0,01 0,01 1,1 1,1 1,1 1,065 1,170 1,288 0,0646 0,0841 0,106 0,606 1,230 2,970 0,275 0,170 0,052 0,0564 0,0369 0,015 8,05 7,43 5,69
Исходный раствор, г.ион/л В осадке, % от исходного Коэффициент распределения
Fe»+ Ga«+ Al«+ Fe Ga Al ^Ga/Fl-J-Al ^Ga/Al •
0,125 0,125 0,125 0,01 0,01 0,01 1,1 1,1 1,1 79,5 87,5 96,2 53,4 69,5 87,7 7,0 14,2 34,3 5,72 7,22 9,55 15,2 13,8 13,5
Сред нее . . . 14,2
идеальных систем
следует, что
(Yi/Y2)T тоже равно единице, то из уравнений (9) и (10)
(16)
где и Л2— растворимости, выраженные в моль/л. Приняв значения раст-
воримости гидроокиси галлия, равным 5,5-10-® (определено нами для зна-
чения ионной силы J —>0), гидроокиси железа 1,41-10-® [17] и гидроокиси
алюминия 1,84-10-5 [18], получим
Dp (Ga/Al) =
'1,84-10-° у/,
, 5,5-10-® /
(17)
Dp (Fe/Al) =
1,84-10-°
1,41-10-®
(18)
Полученные результаты согласуются с экспериментом (см. табл. 2 и 3).
В табл. 4 приведены коэффициенты распределения, вычисленные для рав-
новесия при фракционном осаждении гидроокисей (основных солей) железа,
галлия и алюминия из сернокислых растворов. Видно, что в отличие от «би-
нарных» (Di/,) коэффициенты распределения одного компонента к сумме
двух других и в идеальных системах являются не постоянной величиной,
а функцией отношения (£1/£2+£3)ж, что согласуется с данными работ
Г. Н. Горштейна [12, 15] и Ю. Д. Третьякова [13, 14].
ВЫВОД
Исследованы закономерности распределения компонентов между раствором
и твердой фазой. Показана применимость термодинамической теории при
осаждении основных солей (гидроокисей) железа, галлия и алюминия из ра-
створов с высокой ионной силой. Характер распределения компонентов меж-
151
ду сернокислым раствором и твердой фазой основных солей перечисленных
металлов подтверждает, что в пределах погрешности эксперимента эти си-
стемы являются идеальными.
ЛИТЕРАТУРА
1. Т. Moeller, Н. F. Kremers. Chem. Rev., 1945, 37, 97.
2. Н. Н. Миронов, А. И. Односевцев. Ж. общ. хим., 1956, 26, 960.
3. Б. В. Громов. Цветные металлы, 1948 , 21, 3 , 29.
4. Н. В. Аксельруд, В. Б. Спиваковский. Ж. неорг. хим., 1958 , 3, 269.
5. Н. В Аксельруд, В. Б. Спиваковский. Ж. неорг. хим., 1959, 4, 989.
6. Н. В. Аксельруд, В. Б. Спиваковский. Ж. неорг. хим., 1958, 3, 1748.
7. Vanselow. J. Amer. Chem. Soc., 1932, 54, 1307.
8. Б. П. Никольский, В. И. Парамонов. Усп. хим., 1939, 8, 1535.
9. А. Р. Ratner. J. Chem. Phys., 1933, 1, 789.
10. A. Hill, G. Durham, J. Ricci. J. Amer. Chem. Soc., 1940, 62, 2723.
11. H. В. Шишкин, E. А. Крогус. Ж. общ. хим., 1950 , 20, 986.
12. Г. H. Горштейн. Ж. неорг. хим., 1963, 8, 1461.
13. Ю. Д. Третьяков. Ж. неорг. хим., 1961, 6, 985.
14. Ю. Д. Третьяков, Л. К. Силаева. Ж. неорг. хим., 1961, 6, 2203.
15. Г. Н. Горштейн. Ж. неорг. хим., 1958, 3, 51.
16. Н. И. Еремин. Галлий. М., Металлургиздат, 1964.
17. G. Almkvist. Z. anorg. Chem., 1918, 103, 240.
18. Н. Remy, A. Kuhlmann. Z. anal. Chem., 1924, 65, 161.
' АКАДЕМИЯНАУКСССР
УРАЛЬСКИЙ ФИЛИАЛ
ВЫП. 20 ТРУДЫ ИНСТИТУТА ХИМИИ 1970
С. П. ЯЦЕНКО
КОНСТАНТЫ РАВНОВЕСИЯ РЕАКЦИЙ ВЗАИМОДЕЙСТВИЯ
ГИДРООКИСИ ГАЛЛИЯ С РАСТВОРОМ ЕДКОГО НАТРИЯ
Кислотные свойства у гидроокиси галлия выражены сильнее, чем у гид-
роокиси алюминия. Наши данные о большей устойчивости растворов галлата
натрия [1, 2] по сравнению с алюминатными [3] впоследствии были подтвер-
ждены работами Б. Н. Иванова-Эмина и Л. А. Нисельсона [4]. Т. В. Пер-
мякова и И. С. Лилеев [5] методом диализа получили устойчивые галлатные
растворы с молярным отношением Na2O/Ga2O3, равным 0,1—0,2, и содер-
жанием Ga2O3 до 4,3 г/л. Растворы были прозрачны и не разлагались в те-
чение длительного времени. Эти авторы придерживаются мнения о существо-
вании в указанном случае истинного раствора таллиевой кислоты, что отли-
чает галлий от алюминия.
Расчет константы равновесия реакции —
Ga(OH)34-OH-^2Ga(OH)“, (а)
при отсутствующих значениях коэффициентов активности ионов Ga (ОН)~
mGa(OH)7
можно провести построением кривых зависимости К =-------- от ионнои
тон-
силы растворов с последующим экстраполированием на значение ji., равное
нулю. При этом коэффициенты активности ионов равнялись единице.
С. И. Кузнецов [6] и Рассел [7] указанным способом вычислили константы
равновесия Кй для алюминатных растворов, Ренделл и Спенсер [8] — для
моноокиси свинца в растворах едкого натрия и калия. Значения молярных
концентраций /Поаон^ и тон“ для температур 18 и 60° С взяты из работ
{9, 10] по изучению диаграммы растворимости системы Ga2O3 — Na2O—Н2О
(табл. 1).
Отсутствие данных о растворимости в разбавленных щелочных растворах
(для 60° С минимальная концентрация 5,5% Na2O) затрудняет графическое
экстраполирование при определении Ка. Более того, для низких значений
/Пон- равновесие достигается чрезвычайно медленно, и разброс значений
равновесной растворимости в исследованиях [9, 11] очень велик. С. И. Куз-
нецов [6, 12] проверил правильность экстраполирования согласно гипотезы
Мак-Иннеса [13], по которой сходные по свойствам ионы в разбавленных
растворах обладают одинаковыми коэффициентами активности. Значения для
алюминат-иона сопоставлены с известными данными для плюмбит-иона [8].
Нами для экстраполирования при 20° С использованы полученные значения
П Заказ № 632 1 53
Таблица 1
Растворимость в системе Ga2O3—Na2O—Н2О по данным [9, 10]
/°, с . Na,О GafOa Л Хс = - 'lGa(OH)7 /
т (ОН)-
вес. % г-ион 1000 г вес. % г ион 1000 г
-ig кс
18 2,41 0,819 2,84 0,32 0,391 0,41 1,07
18 4,00 1,43 5,67 0,67 0,468 0,33 1,45
18 4,81 1,74 6,28 0,754 0,433 0,36 1,58
18 9,00' 3,68 12,05 1,63 0,443 0,36 2,30
60 8,86 3,67 13,43 1,84 0,525 0,28 2,35
60 9,80 4,30 16,45 2,38 0,555 0,26 2,58
60 12,23 5,76 19,20 2,99 0,520 0,29 2,96
по электропроводности растворов галлата натрия (табл. 2). Эквивалентная
электропроводность равна, по [14],
Х=а(%°— (1)>
где а — степень диссоциации; — предельная эквивалентная электропровод-
ность электролита при бесконечном разведении; — предельный коэффици-
ент наклона для эквивалентной электропроводности; с — молярная концент-
рация.
Фуосс и Шидловский [15] предложили следующее решение этого урав-
нения для определения величины степени электролитической диссоциации:
“=vs(4 (2)
Для малых значений
S(z) = l+zH—(3)'
при этом
г=6х/мХ°)-’/г- (4)
Харнед и Оуэн [14] полагают, что это уравнение наиболее пригодно для
определения константы диссоциации из одной серии измерений электропро-
водности.
Для нахождения %0 строили график X от V с и кривую экстраполиро-
вали до нулевой концентрации. Значение 1° равно 96,20 ом.-1-см2. Предель-
ная ионная электропроводность, согласно закону Кольрауша [16], равна
%0—XNa+4-A,0a(OH
4
(5)
откуда (ОН)_=96,2 — 50,1 =46,1.
(Для сравнения напомним, что по-
движность алюминатного аниона при 25° С равна 38 [17]).
Значение константы диссоциации галлата натрия вычислено по уравнению
^днс
са2-/ +
1 — а ’
(6)
154
где для разбавленных раст-
воров электролита 1,1 значе-
ние коэффициента актив-
ности рассчитано по фор-
муле
0,5046с
1+V с
В пределах ошибки изме-
рений, как видно из табл. 2,
значение константы остает-
ся постоянным при изме-
нении концентрации в 100
раз. Величина среднего зна-
чения Кдис взята нами в
качестве Ка для 20° С. Ка
для 60° определена экст-
раполированием Кс по кри-
вой, которая симбатна анало-
гичной кривой для 20° С.
В результате Ка равно 0,05
и 0,29, соответственно, при
20 и 60° С. Из этих зна-
чений произведения актив-
ности ионов, Н* и Ga (ОН)-
вычисленные как произве-
дения Ka-Kw, при тех же
температурах, соответствен-
но, равны 3,4-10~16 и
2,78 -10~14. Уравнение изо-
бары реакции взаимодейст-
вия гидроокиси галлия с
едким натром от темпера-
туры может быть легко
получено из найденных
значений константы равно-
весия Ga (OH)S 4- ОН- X
X Ga (ОН)- при 20 и 60° С:
1g К а
— 8400
4,57Т
ф5,0.
(8)
В уравнении (8) —8400 —
тепловой эффект реакции,
кал/моль Ga(OH)3. Тепло-
вой эффект для реакции
взаимодействия гидроаргил-
лита с едким натром, оп-
ределенный по зависимости
констант равновесия от тем-
пературы, в работе С. И. Куз-
нецова [6] установлен рав-
ным7000,а по данным Рас-
села1 [7] —7340 кал/моль
А1(ОН)3.
Примечание. Средняя Хдис
11*
155
Стандартные изменения изобарно-изотермического потенциала реакции (а),,
вычисленные по формуле
bZ°=-RT\nKa, (9)
для 20 и 60° С, соответственно, равны 1,73 и 0,82 ккал. По значению Ка
при 25° С (получено интерполированием для 20 и 60° С) и величин измене-
ния изобарно-изотермического потенциала реакций образования из элементов
ГИДрОКСИЛИОНа AZoH— И ГИДРООКИСИ ГаЛЛИЯ AZaa(oH)3 можно вычислить
AZfla (он)- по уравнению
AZGa(OH)- = AZoafoH), + AZqh--RT In Ka. (10)
Подставив соответствующие значения из работ [18, 19, 20], получим
AZGa(OH)- = — 199,0 —37,5 — 1,985-298,16-In0,08 = —235 ккал.
4
выводы
Подвижность галлат-аниона при 20°С определена равной 46,1 ом~1-см2.
Уравнение изобары реакции взаимодействия гидроокиси галлия с едким
натром от температуры имеет вид
Стандартные изменения изобарно-изотермического потенциала этой же
реакции для 20 и 60° С, соответственно, равны 1,73 и 0,82 ккал. Измене-
ние изобарно-изотермического потенциала реакции образования иона галлата
из элементов следующее:
AZoa(OH)- =—235 ккал!моль.
ЛИТЕРАТУРА
1. С. П. Яценко, Н. В. Деменев. Ж- неорг. хим., 1959 , 4, 6, 1437.
2. С. П. Яценко, Н. В. Деменев. Ж. неорг. хим., 1959, 4, 4, 869.
3. С. И. Кузнецов, В. А. Д е ре в я н к и н. Физическая химия производства глинозема
по способу Байера. М., Металлургиздат, 1964.
4. Б. Н. Иванов-Эмин, Л. А. Нисельсон, Л. Е. Ларионов. Ж. неорг. хим.,
1962, 7, 3, 522.
5. Т. В. Пермякова, И. С. Лилеев. Ж. неорг. хим., 1960, 5, 1999.
6. С. И. Кузнецов. Ж- прикл. хим., 1952, 25, 7, 748.
7. A. S. Russell. J. Metals., 1955, 7, 1123.
8. М. Rendall, Н. М. Spenser. J. Amer. Chem. Soc., 1928, 50, 6, 1572.
9. С. В. Геворкян, Н. А. Гуревич. Изв. АН Арм. ССР, Химические науки, 1957,
10, 6, 387.
10. С. В. Геворкян. Автореф. канд. дисс. М., 1958.
И. Б. Н. Иванов-Эмин, Я. А. Нисельсон, Н. Н. Гвоздева. Ж- неорг. хим.,
1962, 7, 5, 1150.
12. С. И. Кузнецов. Производство глинозема. М., Металлургиздат, 1956.
13. Г. Н. Льюис, М. Рен да л л. Химическая термодинамика. М., ОНТИ, 1936.
14. Г. Харнед, Б. Оуэн. Физическая химия растворов электролитов. М., ИЛ, 1952.
15. R. М. Fuoss, Т. Shedlovsky. J. Amer. Chem. Soc., 1949, 71, 1496.
16. F. К о h 1 г a u s c h, L. Hol born. Leitvermogen derElektrolyte. Teubner. Leipzig, 1916.
17. I. Heyrovsky. J. Chem. Soc., London, 1920, 1013.
18. H. И. Еремин. Галлий. M., Металлургиздат, 1964.
19. W. М. Salt man, N. H. Nachtrieb. J. Electrochem. Soc., 1953, 100. 126.
20. В. M. Латимер. Окислительные состояния элементов и их потенциалы в водных
растворах. М., ИЛ, 1954.
АКАДЕМИЯ НАУК СССР
уральский филиал
ВЫП. 20
труды института химии
1970
Б. И. ЗИЛЬБЕРГЛЕЙТ, С. П. ЯЦЕНКО
ОБ ИЗУЧЕНИИ РЕАКТИВНОГО ЭЛЕКТРОПЕРЕНОСА
В ЖИДКИХ МЕТАЛЛИЧЕСКИХ СПЛАВАХ
В работах [1, 2] был предложен метод изучения реактивной диффузии
в жидких сплавах. Он основан на измерении расстояния, проходимого гра-
ницей твердой и жидкой фаз внутри капилляра вследствие взаимной диффу-
зии компонентов в жидкой фазе в тече-
ние определенного времени (смДрису-
нок). Для расчета коэффициента гете-
родиффузии использованы формулы
Вагнера [3].
Описанный метод может быть при-
менен для исследования явлений элект-
ропереноса в жидких сплавах. Если на
концы диффузионного образца нало-
жить разность потенциалов, то компо-
ненты будут перемещаться в резуль-
тате как диффузии, так и электро-
переноса. Направление и скорость
перемещения границы фаз будут зави-
сеть от соотношения коэффициентов
диффузии, величин и знаков подвиж-
Схема опыта для изучения реактивного
электропереноса в жидких металличе-
ских сплавах.
AV — разность потенциалов; £ — вытота стол-
бика жидкой фазы; Т^п — температура опы-
та; Л, Б — компоненты системы. Остальные
обозначения приведены в тексте.
ностей перемещающихся частиц компо-
нентов сплава. Уравнение, описываю-
щее процесс диффузии в электриче-
ском поле, имеет вид
D-^ — UE — = — , (1)
дх2 дх dt ' '
где U — электрическая подвижность. Второе слагаемое в левой части отри-
цательно, если диффузия и электроперенос направлены в разные стороны,
и положительно — в противном случае. Это уравнение можно достаточно
просто обобщить для бинарной системы. При этом вид его остается неиз-
менным, а под D и U следует понимать, соответственно, коэффициент
гетеродиффузии и гетероподвижность, определяемые выражениями:
Е>=ад+Д2А1, (2)
t/=t/1A2+672A1,
(3>
где А\ и N., — мольные доли компонентов.
157
Гетероподвижность из уравнения (3) связана с обычно определяемой
в опытах по электропереносу относительной подвижностью простым соотно-
шением
U=Ui — UikNi, (4)
где Ui — подвижность i-ro компонента, a Uik=Ui — Uk.
Решение уравнения (1) найдено в ряде работ, например в [4]. Однако
в нашем случае граничные и начальные условия отличны от рассмотренного
в упомянутой выше работе.
Поскольку при диффузии в электрическом поле, как и в случае простой
диффузии, верхняя граница жидкой фазы — изоконцентрационная поверх-
ность C=CS, определяемая линией ликвидуса, а концентрация источника
С=Сг — величина постоянная, то можно записать условия в виде
С (0, 0)=С, '
С (0, t)==C[
C(S, t)=Cs
(5)
При этом предполагается, что исходная концентрация в диффузионном
пространстве Со=О.
Легко показать, что с учетом (5) уравнение (1) решается следующим
образом:
IUE
C,=CrexpJ—£ —
(6)
для разнонаправленных диффузии 'и электропереноса и
С5=С,ехр
' £ U2E2
2D 4D
(7)
если их направления совпадают.
Поскольку направление электропереноса нельзя предсказать априори, то
правильный выбор из (6) и (7) может быть сделан лишь на основании ре-
зультатов двух опытов — при Е>0 и при Е=0. Если окажется, что в дан-
ной системе £e>o>g£=o, то диффузия и электроперенос направлены в одну
и ту же сторону; при £,е>о<Ле=о направления противоположны.
Решение (6), (7) получено в предположении, что силы электрического
поля и электронного ветра действуют аналогично и вызывают движение
компонентов сплава по механизму, отличному от диффузионного [5]. Гетеро-
подвижность, определяемая таким образом, является линейной комбинацией
эффективных подвижностей компонентов. Для определения зарядов ионов и
эффективных сечений рассеяния электронов на ионах необходимо привлечь
дополнительные данные, например по электросопротивлению сплава.
Необходимое условие применимости предлагаемого метода — знание
диаграммы состояния сплава. Для бинарных систем эти диаграммы, как
правило, известны достаточно подробно, что может обеспечить методу опре-
деленную универсальность. Основным преимуществом его по сравнению
с описанными в литературе являются простота и большая скорость выполне-
ния эксперимента, так как здесь нет необходимости делать выдержку для
достижения стационарного состояния. .
, ВЫВОДЫ
1. Предложен способ изучения реактивного электропереноса в жидких
металлических сплавах.
158
2. Получено решение уравнения диффузии, в электрическом поле, соот-
ветствующее условиям реактивной диффузии и реактивного электропереноса
в полубесконечном капилляре из бесконечно мощного источника.
ЛИТЕРАТУРА
1. П. М. Шурыгин, Б. И. Зильберглейт. Изв. высш. учеб, завед., Черная метал-
лургия, 1963, 8, 13.
2. П. М. Шурыгин, Б. И. Зильберглейт. Экспериментальная техника и методы
высокотемпературных измерений. Труды II Всесоюзного совещания. М., «Наука»,
1966, сто. 192.
3. В. Зайт. Диффузия в металлах. М., ИЛ, 1958.
4. Л. П. Холпанов, С. И. Дракин. Изв. высш. учеб, завед., Химия и химическая
технология, 1960, 1, 14.
5. Д. К. Бела щен к о. Усп. хим., 1965, 34, 3.
СОДЕРЖАНИЕ
А. А. Фотиев, Б. Г. Головкин. Определение теплот образования ванадатов
щелочных металлов методами сравнительного расчета ......................... 3
Л. Л. Сурат, Б. Г. Головкин. Термодинамический расчет реакций, возможных
в системе КС1 — V2OB...................................................... 8
А. А. Фотиев, Л. Л. Сурат. Исследование взаимодействия в системе пяти-
окись ванадия — хлористый калий........................................... 11
А. А. Фотиев, Б. Г. Головкин. Определение соотношения скоростей испа-
рения, сублимации хлоридов щелочных металлов и взаимодействия их с пяти-
окисью ванадия............................................................ 16
Б. В. Слободин. О химическом составе соединений, образующихся в системе
V2O5 —AgVO3 . . .......................................................... 24
А. А. Фотиев, Р. Н. Плетнев. Исследование гидратов ванадиевых бронз . . 30
А. А. Фотиев, Е. Д. Столер, Р. Н. Плетнев, Ю. М. Беляков. Элект-
ронный парамагнитный резонанс в щелочных ванадиевых бронзах............... 37
Л. К. Толстов, А. А. Ивакин, А. А. Фотиев. Изучение продуктов гидро-
литического осаждения ванадия (V) в системе LiVO3 — HNO3 — Н2О ... 40
В. А. Переляев, Г. П. Швейкин. Взаимная растворимость некоторых проме-
жуточных окислов переходных металлов IVa и Va подгрупп.......................... 47
В. И. Потороча, В. А. Цхай, П. В. Гельд. К расчету энергетического
спектра монокарбида титана...................................................... 54
С. И. Алямовский, Ю. Г. Зайнулин, Г. П. Швейкин, П. В. Гельд,
I Е. Н. Щетников |. Система Zr—С—О.............................................. 60
JO. Г. Зайнулин, С. И. Алямовский, Г. П. Швейкин,
| Е. Н; Щетников |, П. В. Гельд. Растворимость бериллия в кубических
карбидах Ti, V, Nb, Та.................................................... 68
А. С. Борухович, Л. Б. Дубровская, И. И. Матвеенко, П.В. Гельд.
Исследование магнитной восприимчивости кубических карбидов ниобия и тан-
тала в области низких температур.............................................. 71
В. Г. Зубков, И. И. Матвеенко, Л. А. Дубровская, Г. Д. Богомо-
лов, П. В. Ге ль д. Об упорядочении в структуре оксикарбидов титана . . 74
В. М. Жуковский, А. С. Виноградова, Е. В. Ткаченко. Кинетика
твердофазного синтеза молибдата магния в стехиометрических смесях МоО3
и MgCO3................................................................... 75
•Е. В. Ткаченко, В. М. Жуковский, Т. И. Селиванова. Изучение ус-
ловий образования молибдата бария при взаимодействии ВаСО3 с МоОа
в твердой фазе на воздухе......................'.......................... 83
В. М. Полякова, Л. Ф. Мальцева, С. И. Алямовский. Исследование
системы TiO2 — Rb2SO4— H2SO4— Н2О при 20° С............................... 88
В. М. Полякова, Е. И. Чернявская. Окисление сернокислых растворов
трехвалентного титана химическими реагентами...............................94
В. М. Полякова, Л. Ф. Мальцева. Сокристаллизация ниобия с титанил-
сульфатом калия в сернокислых растворах................................... 98
В. М. Полякова, Л. Ф. Мальцева. Взаимное влияние титанилсульфата калия
и изоморфных включений ниобия на их растворимость в растворах серной
кислоты.................................................................. 102
А. М. Горелов, А. П. Штин. Исследование свойств фосфорнокислых растворов
титана.................................................................. 106
А. П. Штин, А. М. Горелов, С. В. 3 о л о т а в и н а. Изотерма 20°С систе-
мы TiO2 — Р2ОВ— Н2О............................................................ Ill
•И. Г. Чуфарова, Н. И. Петунина. Экстракция ниобия из соляно-сернокис-
лых растворов спиртами................................................... 117
160
Н. И. Петунина, И. Г. Чуфарова. Экстракция, титана из солянокислых
растворов вторичными высшими спиртами.................................... 123
Н. И. Петунина, И. Г. Чуфарова. К вопросу экстракции тантала из соля-
но-сернокислых растворов спиртами........................................ 127
Д. И. Курбатов, С. А. Говорко, 3. П. К о рте в а. Зависимость диффузи-
онного тока тугоплавких элементов (Ti, Nb, Мо) от концентрации фосфор-
ной кислоты и определение сопротивления электролитической ячейки .... 131
Д. И. Курбатов, 3. П. Кортева. Определение числа электронов, участвую-
щих при электрохимическом восстановлении Ti4+ и We+ в концентрирован-
ных орто- и пирофосфорнокислых растворах с повышенной вязкостью .... 135
В. Н. Данилин, С. П. Яценко. Термодинамические свойства жидких сплавов
системы галлий — сурьма.................................................. 142
С. П. Яценко, Г. М. Рубинштейн. Осаждение и разделение гидроокисей
(основных сульфатов) алюминия, галлия и железа........................... 146
С. П. Яценко. Константы равновесия реакции взаимодействия гидроокиси галлия
с раствором едкого натрия........................................... 153
Б. И. Зильберглейт, С. П. Яценко. Об изучении реактивного электропере-
носа в жидких металлических сплавах................................. 157
УДК 546.881:536.7
Определение теплот образования ванадатов щелочных металлов методами сравни-
тельного расчета. Фотиев А. А., Головкин Б. Г. «Исследование соединений ред-
ких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Сравнением параметров кристаллических решеток ортофосфатов и ортоарсенатов редко-
земельных элементов с изоструктурными им ортованадатами найден условный радиус орто-
О
ванадата, равный 2,48А. При рассмотрении суммы параметров кристаллической решетки
как кратного суммы радиусов катиона и аниона определен условный радиус иона метава-
надата. С помощью условных термохимических радиусов по формуле Капустинского вычис-
лены энергии кристаллических решеток ванадатов щелочных металлов. Из равенства энергии
кристаллической решетки, суммы теплот образования соединения и составляющих его ионов
определены теплоты образования ванадатов щелочных металлов.
Таблиц 4. Иллюстраций 1. Библиогр. 12 назв.
УДК 546.881'32:536.7
Термодинамический расчет реакций, возможных в системе КС1—V2O5. Сурат Л. Л.,
Головкин Б. Г. «Исследование соединений редких элементов (V, Nb, Та, Ti, Zr,
Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Проведен термодинамический расчет реакций, возможных в системе КС1—V2O5. Уста-
новлено, что ванадиевое соединение KV4O10 5 в области низких температур наиболее ус-
тойчивый продукт; оно образуется первым при любом стехиометрическом соотношении.
Таблиц 2. Иллюстраций 1. Библиогр. 8 назв.
УДК 546.881'32:541.123
Исследование взаимодействия в системе пятиокись ванадия — хлористый калий.
Фотиев А. А., Сурат Л. Л. «Исследование соединений редких элементов (V, Nb,
Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Рассмотрено взаимодействие пятиокиси ванадия с хлористым калием в разных соотно-
шениях. Установлено, что образование калий-ванадиевой бронзы KVeOI6 происходит при
взаимодействии соединений KV4O10 4 и V2OB. Найдена последовательность образования
соединений в изучаемой системе от V2O. до KVO3. На основании термографических данных
построена диаграмма плавкости системы V2O6—КС1.
Иллюстраций 4. Библиогр. 9 назв.
УДК 546.881:543.226
Определение соотношения скоростей испарения, сублимации хлоридов щелочных ме-
таллов и взаимодействия их с пятиокисью ванадия. Фотиев А.А. иГоловкинБ.Г.
«Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та
хим. УФАН СССР, 1969, вып. 20.
Сравнением теплот с энергиями активации испарения и сублимации показано, что они
практически равны. Теоретическая разность между теплотой испарения или сублимации,
представляемой из уравнения Клаузиуса—Клапейрона, и энергией активации испарения и
сублимации равна RT/2. Так как эта величина не превышает ошибку в определении энер-
гии активации, то равенство теплот испарения и сублимации их энергиям активации нахо-
дит свое объяснение. Выведена формула для зависимости скорости испарения галогенидов
щелочных металлов в вакууме в зависимости от температуры, молекулярного веса и энер-
гии активации. Исследована кинетика испарения и сублимации хлоридов щелочных метал-
лов. Из сопоставления соотношений скоростей сублимации и испарения хлоридов щелочных
металлов со скоростями процессов их взаимодействия с пятиокисью ванадия сделан вы-
вод, что сублимация и испарение, по-видимому, не являются лимитирующими стадиями
162
этих процессов и, следовательно, механизм твердое — газ не может быть преобладающим.
Таблиц 2. Иллюстраций 3. Библиогр. 27 назв.
УДК 546.881'57:543.226
О химическом составе соединений, образующихся в системе iV2O5—AgVOa. Слобо-
дин Б. В. «Исследование соединений редких элементов (V, Nb. Та, Ti, Zr, Mo, Ga)».
Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Построена диаграмма состояний системы V2OB—AgVO3 в атмосфере воздуха. Показано
образование в ней двух соединений AgVeO1B и Ag2V4On, из которых первое относится
к кислородным ванадиевым бронзам. На основании экспериментальных и литературных
данных рассмотрены особенности взаимодействия пятиокиси ванадия с серебряными реаген-
тами в зависимости от атмосферы.
Иллюстраций 5. Библиогр. 23 назв.
УДК 546.881
Исследование гидратов ванадиевых бронз. Фотиев А. А., Плетнев Р. Н. «Иссле-
дование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим.
УФАН СССР, 1970, вып. 20.
Ванадиевые бронзы M2VBO13 3, где M=Li, Na, К, при кислотной обработке гидроли-
зуются с образованием кристаллогидратов. Методами рентгеновского анализа и ИКС уста-
новлено, что эти соединения имеют кристаллическую решетку, аналогичную безводному
исходному составу. Молекулы воды, входящие в состав гидрата, расположены в межслое-
вых пустотах решетки. В структуре гидратов, как установлено ЯМР, помимо молекул
кристаллизационной воды, имеются гидроксильные группы. В работе обсуждены результаты
термического анализа кристаллогидратов.
Таблиц 4. Иллюстраций 4. Библиогр. 12 назв.
УДК 546.881
Электронный парамагнитный резонанс в щелочных ванадиевых бронзах. Фоти-
ев А. А., Столер Е. Д., Плетнев Р. Н., Беляков Ю. М. «Исследование соеди-
нений редких элементов (V,Nb, Та, Ti, Zr, Mo, Ga)». Тр. Йн-та хим. УФАН СССР,
1970, вып. 20.
Получены спектры ЭПР щелочных ванадиевых бронз: MVeOjB и M2VBO1313, где
M=Li, Na, К; MV4O104, где М=К, Rb и гидратов М2— tVEO13 3-пН2О. Результаты ис-
следования позволяют предполагать, что центрами локализации неспаренных электронов
являются атомы ванадия. Симметрия окружения атомов ванадия зависит от степени иска-
жения кристаллической решетки. Показано наличие обменных взаимодействий, которые
ослабевают в гидратах ванадиевых бронз.
Таблиц 1. Иллюстраций 1. Библиогр. 10 назв.
УДК 546.881'34:543.226
Изучение продуктов гидролитического осаждения ванадия (V) в системе
LiVO3—HNO3—п2О. Толстов Л. К., Ивакин А. А., Фотиев А. А. «Исследо-
вание соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН
СССР, 1969, вып. 20.
Проведено комплексное физико-химическое исследование продуктов гидролитического
осаждения ванадия (V), выпадающих в системе LiVO3—HNO3—Н2О в интервале H+/VO^“
от 0,7 до 2,1 и при различных исходных концентрациях ванадия в растворе. На основании
изучения состава и термических свойств осадков установлено наличие соединений, близких
по своей природе к гидратированной пятиокиси ванадия. Отмечено присутствие во всех
образцах «конституционной» воды, удаление которой ведет к необратимой структурной
перестройке осадков и сопровождается образованием в них в зависимости от содержания
лития различных фазовых составляющих, присущих системе V2OB—Li2O (в том числе
литий-ванадиевых бронз LiVeO1B и Li2VBO( 315_ж).
Таблиц 1. Иллюстраций 2. Библиогр. 11 назв.
УДК 541.123.2—165+546.821—31
Взаимная растворимость некоторых промежуточных окислов переходных металлов
IVa и Va подгрупп. ПереляевВ. А., Швейкин Г. П. «Исследование соединений
редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)», Тр. Ин-та хим. УФАН СССР, 1970,
вып. 20.
На основании Литературных данных рассмотрен вопрос о взаимной растворимости не-
которых промежуточных окислов переходных металлов. Установлено, что твердые растворы
на основе кубических решеток стехиометрических моноокисей титана и ванадия TiOj 0—V! 0
163
не образуются, в то время как для систем Ti2O3—V2O3 и ТЮ2—VO2, NbO2 наблюдается
образование твердых растворов замещения на основе фаз типа корунда и рутила, соответ-
ственно. Поведение твердых растворов в данных системах рассматривается с точки зрения
различного валентного состава как отдельных фаз, так и взаимного частичного изменения
его в процессе образования твердых растворов.
Библиогр. 33 назв.
УДК 546.821'261:539,2
К расчету энергетического спектра монокарбида титана. Потороча В. И., Ц х а й В. А.,
Гельд 11. В. «Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)».
Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Отражены основные этапы расчета энергетического спектра монокарбида титана методом
сильной связи, в котором учитывались атомные орбитали 3d и 4s титана и 2р углерода.
Секулярные уравнения были решены для четырех-пяти значений вектора К в каждом из
направлений (100); (110) и (111). Кривая плотности состояний изображала непрерывную
полосу и имела три четко выраженных максимума. Уровень Ферми для TiC проходит
немного ниже минимума. Следуемый за ним максимум соответствует заполнению полосы
9,7—9,8 внешними электронами. Дано краткое обсуждение результатов расчета и их со-
поставление с экспериментальными данными.
Таблиц 1. Иллюстраций 2. Библиогр. 21 назв.
УДК 546.831'26'21:541.12.013.2
'Система Zr—С—О. Алямовский С. И., Зайнулин Ю. Г., Швейкин Г. П.,
Гельд П. В., (Щетников Е. Н. | «Исследование соединений редких элементов (V,Nb,
Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Статья посвящена литературному обзору систем Zr—С, Zr—О и Zr—С—О. Затраги-
ваются вопросы условий образования и структурных характеристик указанных систем. При-
веденный материал позволяет наиболее целесообразно выбрать направление дальнейших ис-
следований системы Zr—С—О и подчеркивает их важность. Авторы, анализируя как ли-
тературные, так и собственные данные, делают некоторые предположения относительно
структурных характеристик оксикарбидов циркония.
Таблиц 2. Иллюстраций 1. Библиогр. 42 назв.
УДК 546.261'45
Растворимость бериллия в кубических карбидах Ti, V, Nb и Та. 3 а й н у л л и н Ю. Г.,
Алямовский С. И., Швейкин Г. П., (Щетников Е. Н. |, Гельд П. В.
«Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та
хим. УФАН СССР, 1969, вып. 20.
Приведены сведения о взаимодействии карбидов титана, ванадия, . ниобия, тантала
(составов TiC0 65, TiC0.9|, VC0.80, NbC0 77, ТаС0 76) со структурой типа каменной
соли при температурах 1100 и 1500° С в вакууме (5-10—8 мм рт. ст.) с бериллием. За
исключением карбида титана в данных условиях образуются кубические твердые растворы
(типа NaCl), периоды решетки которых возрастают с увеличением содержания бериллия.
Предполагается, что указанные твердые растворы построены по типу замещения — внедрения.
Таблиц 1. Библиогр. 7 назв.
УДК 538.546.261
Исследование магнитной восприимчивости кубических карбидов ниобия и тантала
в области низких температур. Борухович А. С., Дубровская Л. Б., Матве-
енко И. И., Гельд П. В. «Исследование соединений редких элементов (V, Nb, Та,
Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Изложены результаты измерений магнитной восприимчивости кубических карбидов нио-
бия и тантала в их областях гомогенности при низких температурах. Обнаружено появле-
ние кюри-вейссовской зависимости % у карбида состава NbC0 80 ниже 30° К. Получен-
ные результаты обсуждены исходя из имеющихся представлений об электронной структуре
этих соединений.
Иллюстраций 2. Библиогр. 9 назв.
УДК 546.852'261:539.106
Об упорядочении в структуре оксикарбида титана. Зубков В. Г., Матвеен-
ко И. И., Дубровская Л. Б., Богомолов Г. Д., Гельд П. В. «Исследование
соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН
СССР, 1969, вып. 20.
164
Методом рассеяния нейтронов обнаружено упорядочение атомов углерода и кислорода
в оксикарбиде титана. Предложены возможные модели упорядочения. Измерено электро-
сопротивление образцов в ряду твердых растворов TiOj 0—TiC|_0. Получена зависимость,
характерная для упорядоченных твердых растворов, с минимумом вблизи состава TiC0 5О0 5.
Библиогр. 1 назв.
УДК 541 124.16—31—38
Кинетика твердофазного синтеза молибдата магния в стехиометрических смесях
МоО3 и MgCO3. Жуковский В. М., Виноградов А. С., Ткаченко Е. В.
«Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та
хим. УФАН СССР, 1970, вып. 20.
Исследовано влияние различных факторов (температура, крупность частиц исходных
реагентов, давление прессования реакционных смесей, природы и давление газовой фазы)
на кинетику твердофазного синтеза нормального молибдата магния MgMoO4 в стехиомет-
рических смесях МоО3—MgCO3. Высказаны соображения о возможном механизме синтеза.
Показано, что синтез молибдата магния представляет собой сложный физико-химический
процесс, протекающий через ряд параллельных и последовательных стадий, относительная
значимость которых меняется как в зависимости от условий ведения процесса, так и по
ходу последнего.
Иллюстраций 5. Библиогр, 15 назв.
УДК 541.124.16—31—38
Изучение условий образования молибдата бария при взаимодействии ВаСО, с МоО3
в твердой фазе на воздухе. Ткаченко Е. В., Жуковский В. М., Селива-
нова Т. И. «Изучение соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)».
Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Изложены результаты исследования условий образования нормального молибдата окиси
бария — ВаМоО4 при его твердофазном синтезе в смесях ВаСО3—МоО3 стехиометрического
и переменного составов, полученные с помощью термографического, волюмометрического и
рентгенографического методов. Образование ВаМоО4 в условиях непрерывного разогрева
смеси ВаСО3—МоО3 наблюдается в интервале температур 440—750° С. Скорость процесса
и его полнота зависят от состава и состояния навески. Взаимодействие в смеси происходит
-без предварительного разложения карбоната бария.
Таблиц 1. Иллюстраций 3. Библиогр. 10 назв.
УДК 541.123.6
Исследование системы TiO2— Rb2SO4—H2SO4—Н2О при 20° С. Поляков В. М.,
Мальцев Л. Ф., Алямовский С. И. «Исследование соединений редких элементов
(V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1969, вып. 20.
Настоящая работа является продолжением исследований взаимодействия сернокислых
растворов двуокиси титана с сульфатами щелочных металлов. При изучении системы
ТЮ2—RbSO4—H2SO4—Н2О установлено, что устойчивой твердой фазой является не опи-
санный в литературе титанилсульфат рубидия—2Rb2SO4-3TiOSO4-H2SO4-nH2O. Определены
-концентрационные границы существования этой соли по серной кислоте и исследованы
некоторые ее свойства. Препаративно в условиях кристаллизации без перемешивания получен
титанилсульфат рубидия с отношением TiOSO4:Rb2SO4 = 2:1. Согласно химическому ана-
лизу эта соль имеет состав Rb2SO4-2TiOSO4-6H2O.
Таблиц 4. Иллюстраций 3. Библиогр. 6 назв.
УДК 546.823—145.15:542.943
Окисление сернокислых растворов трехвалентного титана химическими реагентами.
Полякова В. М., Чернявская Е. И. «Исследование соединений редких элементов
(V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Приведены данные по окислению ионов титана (III) химическими реагентами, явля-
ющимися окислителями в кислых растворах. Окисление сернокислых растворов титана (III)
химическими реагентами — SO2, К2Сг2О7, КС1О3, Fe2(SO4)3, KMnO4, HNO3 — протекает во
много раз быстрее, чем кислородом. Повышение концентрации серной кислоты в окисля-
емых растворах оказывает замедляющее действие на процесс окисления титана (III) иссле-
дованными химическими реагентами за исключением хромовокислого калия.
Таблиц 3. Иллюстраций 2. Библиогр. 4 назв.
УДК 546.824+546.822.5:542.65—145.15
Сокристаллизации ниобия с титанилсульфатом калия в сернокислых растворах.
Полякова В. М., Мальцева Л. Ф. «Исследование соединений редких элементов
4V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
165
Исследована сокристаллизация примеси Nb5+ с титанилсульфатом калия при осаждении
его сульфатом калия из сернокислых растворов. Определены коэффициенты кристаллизации,
ниобия при распределении его между раствором и осадком титанилсульфата калия. Показан
изоморфный характер соосаждения ниобия (V) с титанилсульфатом калия при введении
сульфата калия в сернокислый раствор, содержащий эти элементы.
Таблиц 3. Иллюстраций 1. Библиогр. 5 назв.
УДК 661.882.53+661.888.53:062.122
Взаимное влияние титанилсульфата калия и изоморфных включений ниобия на их
растворимость в растворах серной кислоты. Полякова В. М.. Мальцева Л. Ф.
«Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo,.Ga)». Тр. Ин-та
хим. УФАН СССР, 1970, вып. 20.
' Приведены данные по изучению взаимного влияния титанилсульфата калия и изоморф-
ных включений ниобия на их растворимость в серной кислоте различной концентрации.
В присутствии ниобия растворимость титанилсульфата калия в исследованных условиях
повышается в сравнении с растворимостью его в серной кислоте, не содержащей ниобий.
При содержании титана в растворе в количестве 8—10 вес. % по отношению к ниобию-
растворимость изоморфных кристаллов последнего достигает максимального значения.
Таблиц 2. Иллюстраций 2. Библиогр. 4 назв.
УДК 546.824'18—145-15
Исследование свойств фосфорнокислых растворов титана. Г ореловА. М.,Штин А. П.
«Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та
хим. УФАН СССР, 1970, вып. 20.
Определена стабильность фосфорнокислых растворов титана в зависимости от исходной
концентрации титана и фосфорной кислоты. Методом ионного обмена показано, что в этих
растворах доминирующими являются анионные формы титана.
Таблиц 4. Иллюстраций 2. Библиогр. 8 назв.
УДК 546.824'18.013
Изотерма 20° С системы TiO2—Р2О5—Н2О. Штин А. П., Горелов А. М., Золо-
та вин а С. В. «Исследование соединений редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)».
Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Исследована изотерма 20° С TiO2—P2OS—Н2О. Методом «остатков» Шрейнемакерса
и непосредственным анализом промытых твердых фаз установлено образование в зависимо-
сти от концентрации фосфорной кислоты двух фосфатов титана.
Таблиц 3. Иллюстраций 2. Библиогр. 11 назв.
УДК 546.882+66.061-5
Экстракция ниобия из соляно-сернокислых растворов спиртами. Чуфарова И. Г.,
Петунина Н. И. «Исследование соединений редких элементов (V, Nb, Та, Ti, Zr,
Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Изучены и сопоставлены основные характеристики процесса экстракции ниобия из
соляно-сернокислых растворов различными спиртами, содержащими от 6 до 18 атомов
углерода. С увеличением молекулярного веса спирта коэффициент распределения ниобия
повышается. Степень экстракции ниобия возрастает также при замене первичного спирта на
вторичный при одном и том же количестве атомов углерода в спирте. Вторичные высшие
спирты — наиболее эффективные экстрагенты для извлечения ниобия из соляно-сернокислых
растворов. Изучено взаимное влияние ниобия и титана в процессе экстракции вторичными
высшими спиртами.
Таблиц 2. Иллюстраций 3. Библиогр. 10 назв.
УДК 546.824+66.061.5
Экстракция титана из солянокислых растворов вторичными высшими спиртами.
Петунина Н. И., Чуфарова И. Г. «Исследование соединений редких элементов
(V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Йн-та хим. УФАН СССР, 1970, вып. 20.
Приведены результаты исследования экстракции титана вторичными высшими спиртами
из солянокислых растворов в зависимости от содержания соляной кислоты (2—12 моль/л)
и металла (5—160 г/л TiO2) в исходных растворах и изучено влияние высаливающего
действия серной кислоты на распределение титана. Наибольшее извлечение титана дости-
гается при экстракции из растворов, содержащих 10—12 моль/л НС1. Присутствие серной
кислоты повышает извлечение хлорида титана в органическую фазу. С изменением кон-
центрации титана в исходном растворе извлечение в органическую фазу уменьшается в ре-
зультате образования неэкстрагируемых полимерных форм его.
Иллюстраций 3. Библиогр. 7 назв.
166
УДК 546.83.66.061.5
К вопросу экстракции тантала из соляно-сернокислых растворов спиртами. П е т у-
нина Н. И., Чуфарова И. Г. «Исследование соединений редких элементов (V, Nb,
Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Изучен процесс экстракции тантала различными алифатическими спиртами из соляно-
-сернокислых растворов в зависимости от молекулярного веса спирта, концентрации соляной
кислоты, времени стояния исходных растворов. Установлено, что все спирты (от С9 до
'C1S) — эффективные экстрагенты для тантала. Изучено влияние иона фтора на экстракцию
тантала из стоявших соляно-сернокислых растворов. Введение иона фтора при отношении
Ta:F = 1:25ч-1:50 способствует переходу полимерных неэкстрагируемых форм тантала
в экстрагируемые мономерные анионные комплексы.
Иллюстраций 2. Библиогр. 6 назв.
УДК 543.253
Зависимость диффузионного тока тугоплавких элементов (Ti, Nb, Мо) от'Концентра-
ции фосфорной кислоты и определение сопротивления электролитической ячейки.
Курбатов Д. И., Говорко С. А., КортеваЗ. П. «Исследование соединений ред-
ких элементов (Nb, V, Та, Ti, Zr, Mo, Ga)». Тр. Ин.та хим. УФАН СССР, 1970, вып. 20.
Зависимость ij = const сохраняется для диффузионных токов титана, ниобия и
молибдена в широких пределах концентраций фосфорной кислоты, что указывает на отсут-
ствие изменения состава фосфорнокислых соединений этих элементов в изученном интервале
концентрации кислоты. Особенна это относится к титану, где коэффициент вязкости ра-
-створа фосфорной кислоты изменяли более чем в сто раз. Омическое падение напряжения
iR с выносным анодом в концентрированных орто- и пирофосфорных кислотах составляет
при концентрации диполяризатора 0,003 моль/л 0,002 и 0,0006 в соответственно.
Таблиц 2. Иллюстраций 2. Библиогр. 8 назв.
УДК 543.253
Определение числа электронов, участвующих при электрохимическом восстановлении
ТН+ и We+ в концентрированных орто- и пирофосфорнокислых растворах с повы-
шенной вязкостью. Курбатов Д. И., Кортева 3. П. «Исследование соединений
редких элементов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970,
вып. 20.
На примере изучения обратимого восстановления Ti4+ и We+ на ртутном капельном
катоде в концентрированных растворах фосфорной кислоты показано, что метод поляро-
графической кулонометрии применим для растворов, имеющих повышенную вязкость
’(т| > 100 ест/сек).
Таблиц 6. Иллюстраций 4. Библиогр. 8 назв.
УДК 541.123.2:536.77
Термодинамические свойства жидких сплавов системы галлий — сурьма. Дани-
лин В. Н., Яценко С. П. «Исследование соединений редких элементов (V, Nb, Та,
Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Из измерений ЭДС концентрационных цепей
Ga» I (KCl+LiCl)+GaCl3+GaCl | (Ga^Sb j ,
где N — атомно-долевое содержание галлия, получены термодинамические свойства жидких
сплавов галлий — сурьма при 988° К. Из отрицательных отклонений активностей сплавов
галлий —сурьма, а также больших отрицательных значений интегрального избыточного изо-
барного потенциала сплавов сделан вывод о частичном сохранении в жидких сплавах в
пределах ближнего порядка структуры типа соединения.
Таблиц 1. Иллюстраций 3. Библиогр. 9 назв.
УДК 541.123.7:536.7
Осаждение и разделение гидроокисей (основных сульфатов) алюминия, галлия и
железа. Яценко С. П., Рубинштейн Г. М. «Исследование соединений редких эле-
ментов (V, Nb, Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Исследованы закономерности распределения компонентов в «двойных» и «тройных»
системах между раствором и твердой фазой при осаждении основных сульфосолей железа,
галлия и алюминия из растворов с высокой ионной силой при повышенных температурах.
Показана применимость термодинамической теории к осадкам, полученным после длитель-
ного выдерживания при условиях их образования. Значение коэффициента распределения
для «бинарной» системы постоянно при любых соотношениях компонентов и не постоянно
для отношения коэффициента распределения одного компонента к сумме двух других в
«тройной» системе.
Таблиц 4. Иллюстраций 2. Библиогр. 18 назв.
167
УДК 541.123.31:536.7
Константы равновесия реакций взаимодействия гидроокиси галлия с раствором ед-
кого натрия. Яценко С. П. «Исследование соединений редких элементов (V, Nb, Та,
Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
По экспериментальным значениям электропроводности галлата натрия определена кон-
станта его диссоциации. Величина среднего значения взята в качестве константы равновесия
реакции
Ga (ОН)3+ОН- Ga (ОН)^.
Из найденных значений константы при 20 и 60° С получено уравнение изобары реакций
8400
1g Ка = —-------+5,0.
ё а 4,577"
Таблиц 2. Библиогр. 20 назв.
УДК 541.11
Об изучении реактивного электропереноса в жидких металлических сплавах. Зиль-
берглейт Б. И., Яценко С. П. «Исследование соединений редких элементов (V, Nb,
Та, Ti, Zr, Mo, Ga)». Тр. Ин-та хим. УФАН СССР, 1970, вып. 20.
Предложен способ изучения реактивного электропереноса в жидких металлических
сплавах. Решено уравнение диффузии в электрическом поле, соответствующее условиям
реактивной диффузии и реактивного электропереноса в полубесконечном капилляре из бес-
конечномощного источника.
Иллюстраций 1. Библиогр. 5 назв.
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр. Строка Напечатано —г Следует читать
50 4-я снизу Tif),2 — NbO0,sO2 Т1о,2^Ьо,802
124 Подпись 1 — 1, 2—8; 3 — 10 НС1: 7 — 7; 2—8;
к рис . 2 моль/л 3 —10 моль/л
134 10-я снизу Ж. общ. хим. Ж. анал. хим.
Заказ 632