/










































































































































Автор: Годовский Ю.К.
Теги: химия тепло термодинамика физика математическая физика теплофизика полимеры издательство химия
Год: 1982




Текст
Ю. К. Годовский
ТЕПЛОФИЗИКА
ПОЛИМЕРОВ
ИЗДАТЕЛЬСТВО «ХИМИЯ»
МОСКВА, 1982
УДК 541.6:536:
Годовский Ю. К.
Теплофизика полимеров. — М.: Химия, 1982.—
280 с., ил.
В монографии обобщены теоретические представления н результаты
экспериментальных исследований в области теплофизики полимеров. Основ-
ное вмнманне обращено на связь теплофнзическнх свойств с молекулярной
и надмолекулярной структурой полимеров. Рассмотрены также теплофнзи-
ческие явления при изменении температуры и при действии механических
напряжений.
Книга предназначена для исследователей, работающих в области физи-
ки, механики и физической химнн полимеров, а также для инженерно-тех-
нических работников, занимающихся получением, переработкой и примене-
нием полимерных материалов.
280 с., 101 рис., 18 табл., список литературы 392 ссылки.
Рецензент: доктор физ.-мат. наук, профессор
А. Я. МАЛКИН.
1807000000—025
Г 050(01)—82 2582
© Издательство «Химия», 1982 1
СОДЕРЖАНИЕ
Предисловие.......................................4 5
Глава 1
ТЕПЛОЕМКОСТЬ............................................. 8
Колебательные спектры изолированных цепей. Учет взаимодейст-
вия ...................................................... 9
Колебательный спектр и теплоемкость решетки, образованной по-
лимерными цепями...........................................14
Модель Стокмейера — Хента...............................14
Континуальные модели....................................18
Низкотемпературная теплоемкость и ее связь со строением и
структурой полимеров ..................................... 26
Изменение теплоемкости при стекловании.....................40
Аморфные полимеры.......................................40
Кристаллические полимеры................................51
Литература.................................................54
Глава 2
ТЕПЛОПРОВОДНОСТЬ........................................ 56
Аморфные полимеры........................................60
Кристаллические полимеры.................................70
Анизотропия теплопроводности ориентированных полимеров 79
Литература...............................................84
Глава 3
ТЕПЛОВОЕ РАСШИРЕНИЕ...................................85
Уравнение состояния твердых полимеров. Параметр Грюиайзена 85
Тепловое расширение полимерных кристаллов............101
Литература...............................................,113
Глава 4
ТЕПЛОФИЗИКА УПРУГОСТИ ПОЛИМЕРОВ.............................114
Феноменологическая термодинамика деформирования конденсиро-
ванных тел.....................•............................115
Теплофизика упругости твердых полимеров....................118
Всестороннее расширение (сжатие).........................118
Одноосное растяжение (сжатие)............................124
Сдвиг. Кручение......................................... 125
Теплофизика упругости молекулярных сеток....................126
Термоэластичность гауссовых сеток........................127
Термоэластичность иегауссовых сеток......................136
3
Энтропийные и энергетические эффекты при деформации сеток и
их молекулярная интерпретация................................139
Внутримолекулярные эффекты.................................139
Межмолекуляриые эффекты....................................146
Большие деформации и проблема ограниченной растяжимости
цепей......................................................152
Термоэластичиость гетерофазных эластомеров...................155
Тепловые явления при обратимом деформировании твердых поли-
меров и их структурно-молекулярная интерпретация ... 165
Феноменологический анализ. Роль межмолекулярного взаимо-
действия в аморфных областях кристаллических полимеров 165
Теплофизический анализ структурных моделей ориентирован-
ных кристаллических полимеров. Сравнение теплофизических и
структурных данных.........................................179
Анизотропия термоэластичности ориентированных полимеров 194
Литература...................................................199
Глава 5
ТЕПЛОФИЗИКА ОРИЕНТАЦИОННЫХ ЯВЛЕНИЙ В ТВЕРДЫХ
ПОЛИМЕРАХ..................................................203
Термодинамика ориентации кристаллических и стеклообразных по-
лимеров ...................................................205
Температурные эффекты при ориентации твердых полимеров 223
Равномерное распространение шейки в твердых полимерах 225
Автоколебательный режим распространения шейки в твердых
полимерах...............................................233
Литература.................................................239
Глава 6
ТЕПЛОФИЗИКА РАЗРУШЕНИЯ ПОЛИМЕРОВ................241
Кинетический подход к проблеме прочности твердых тел. Энерге-
тические эффекты, связанные с разрывом макромолекул
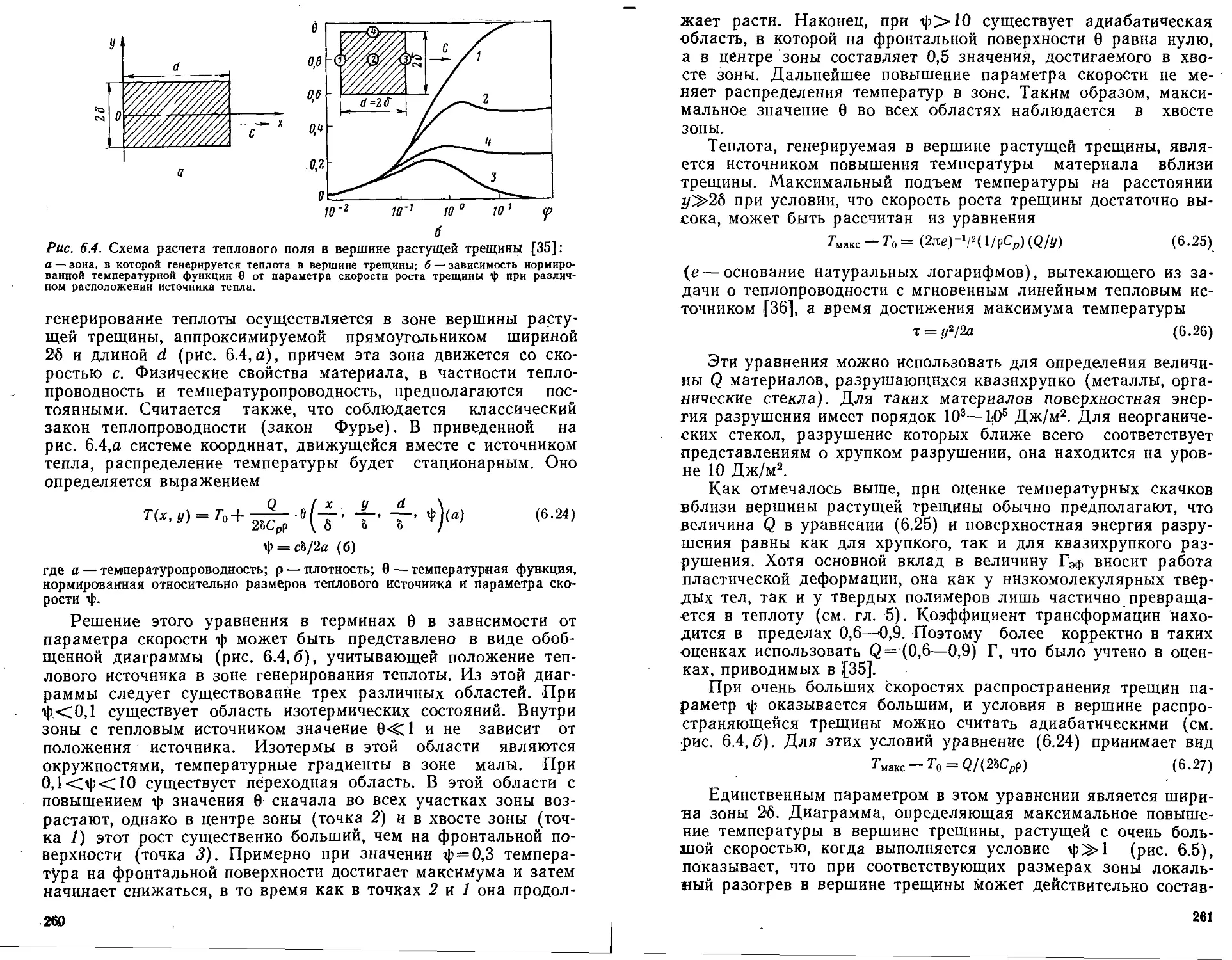
Тепловые явления при образовании и росте трещин в твердых те-
лах .......................................................
Тепловые явления при разрушении твердых полимеров
Макроскопические тепловые явления при разрушении полиме-
ров ....................................................
Микроскопические тепловые явления при медленном разруше-
нии полимеров ..........................................
Локальные экзопроцессы при росте трещин в твердых полиме-
рах ....................................................
Литература.................................................
Предметный указатель.......................................
242
255
262
263
266
272
275
277
ПРЕДИСЛОВИЕ
Среди различных разделов бурно развивающейся физики по-
лимеров важное место принадлежит обширной области, изу-
чающей тепловые явления и тепловые свойства, которую при-
нято называть теплофизикой полимеров. Исторически сложи-
лось так, что именно необычные тепловые явления, сопровож-
дающие деформацию каучука, обнаруженные и исследованные
экспериментально Джоулем и теоретически (с позиции возник-
шей в то время термодинамики) Кельвином в прошлом веке в
Англии, позволили уже тогда выделить полимеры из числа
других объектов физического исследования и привели к зарож-
дению физики полимеров. Эти исследования заложили основу
решения ключевой проблемы физики полимеров — высокой эла-
стичности полимеров, — которая была решена в середине теку-
щего столетия методами статистической термодинамики, при-
менными к цепным макромолекулам. В это же время с ис-
пользованием теоретических методов физики твердого тела бы-
ла показана возможность существования для цепных струк-
тур закономерностей изменения теплоемкости и теплового рас-
ширения, принципиально отличающихся от закономерностей
для неполимерных систем. Наконец, появление в конце 50-х
годов новых представлений о структуре твердых полимеров
стимулировало развитие новых теплофизических подходов, ко-
торые в настоящее время являются мощным инструментом изу-
чения природы твердого состояния полимеров и их структурной
гетерогенности. Таким образом, развитие физики полимеров не-
разрывно связано с развитием представлений о тепловых яв-
лениях и тепловых свойствах полимеров.
Предлагаемая вниманию читателя книга обобщает теорети-
ческие представления и результаты экспериментальных иссле-
дований в области теплофизики полимеров. Книга логически
распадается на две части. В первой части, включающей трш
первые главы, рассмотрены тепловые явления в полимерах при
изменении температуры, а вторая часть посвящена изложению
тепловых явлений при деформировании и разрушении полиме-
ров. Рассмотрение тепловых явлений в полимерах ведется с
позиций теплофизики твердого тела и молекулярной физики,
причем обращается внимание как на общность этих явлений в
полимерных и неполимерных системах, так и на различия,
обусловленные особенностями молекулярного строения и струк-
туры полимеров.
Основная особенность строения полимеров заключена в на-
личии высокой локальной анизотропии силового поля, обуслов-
ленной резким различием сил внутримолекулярного и межмо-
лекулярного взаимодействия. Межатомные силы в макромоле-
5
кулах примерно на два порядка больше, чем силы межмолеку-
лярные, и это различие является причиной сохранения доста-
точной индивидуальности макромолекул в любой полимерной
системе, а также источником высокой локальной анизотропии
физических свойств. Под действием ориентирующих полей ло-
кальная анизотропия и индивидуальность макромолекул слу-
жат источником макроскопической анизотропии физических
свойств полимеров, что особенно характерно для ориентиро-
ванных полимеров. Теории тепловых свойств полимеров приво-
дят к выводу, что наличие такой анизотропии и сохранение
макромолекулами их индивидуальности в полимерных систе-
мах должно приводить к появлению для них специфических за-
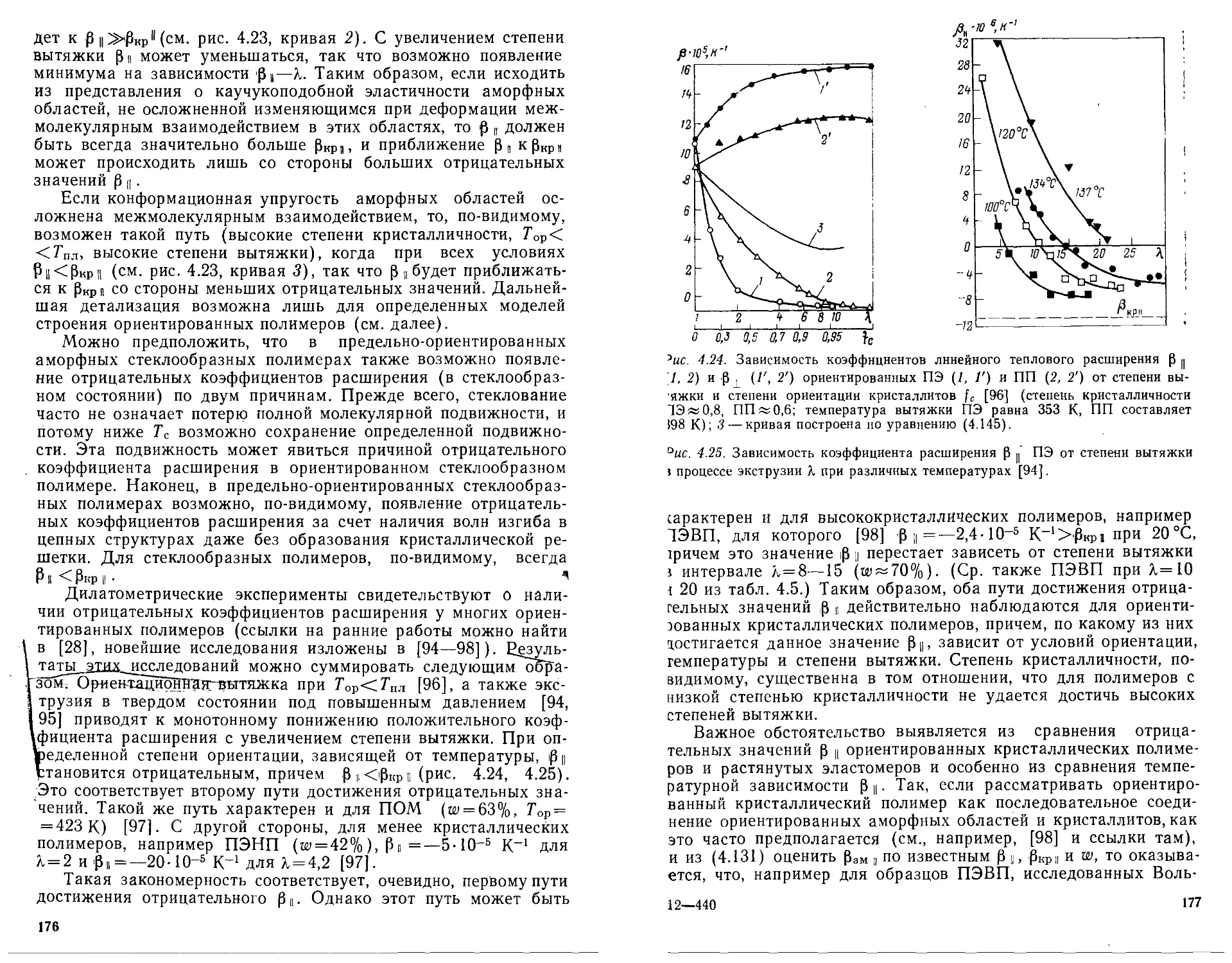
кономерностей теплоемкости, отрицательных коэффициентов
теплового расширения и других особенностей. Проанализиро-
ванные с этих позиций температурные зависимости теплоемко-
сти, теплопроводности и теплового расширения в широком
температурном интервале, включая самые низкие температу-
ры, где существенны квантовые эффекты, свидетельствуют о
наличии таких закономерностей в тех температурных интерва-
лах, где решающим оказывается колебательный спектр скелет
та макромолекулы.
Деформирование является термодинамическим процессом и
обычно сопровождается тепловыми явлениями. Однако при
рассмотрении деформаций конденсированных тел, как правило,
используют систему сугубо механических понятий, хотя с фи-
зической точки зрения термодинамический подход к описанию
деформаций является более глубоким. Характер тепловых яв-
лений, сопровождающих обратимые деформации, может быть
установлен методами теории термоупругости. Однако, оста-
ваясь в рамках феноменологической термодинамики, нельзя
выявить молекулярную природу изменений энтропии и внут-
ренней энергии при деформации и установить соотношение
между ними. Ответ на эти вопросы может быть получен лишь
в результате анализа уравнений состояния. На основе простых
уравнений состояния твердых тел и молекулярных сеток в
книге дана обобщенная термодинамическая трактовка высоко-
эластической деформации молекулярных сеток и обратимых де-
формаций твердых полимеров и с этих позиций проведена ин-
терпретация тепловых явлений при обратимых деформациях
полимеров и предпринята попытка связать эти тепловые явле-
ния с молекулярными и структурными изменениями при де-
формации.
Одна из отличительных особенностей твердых полимеров
по сравнению с низкомолекулярными веществами связана с их
способностью переходить в ориентированное состояние. В отли-
чие от обратимых деформаций ориентация твердых полимеров,
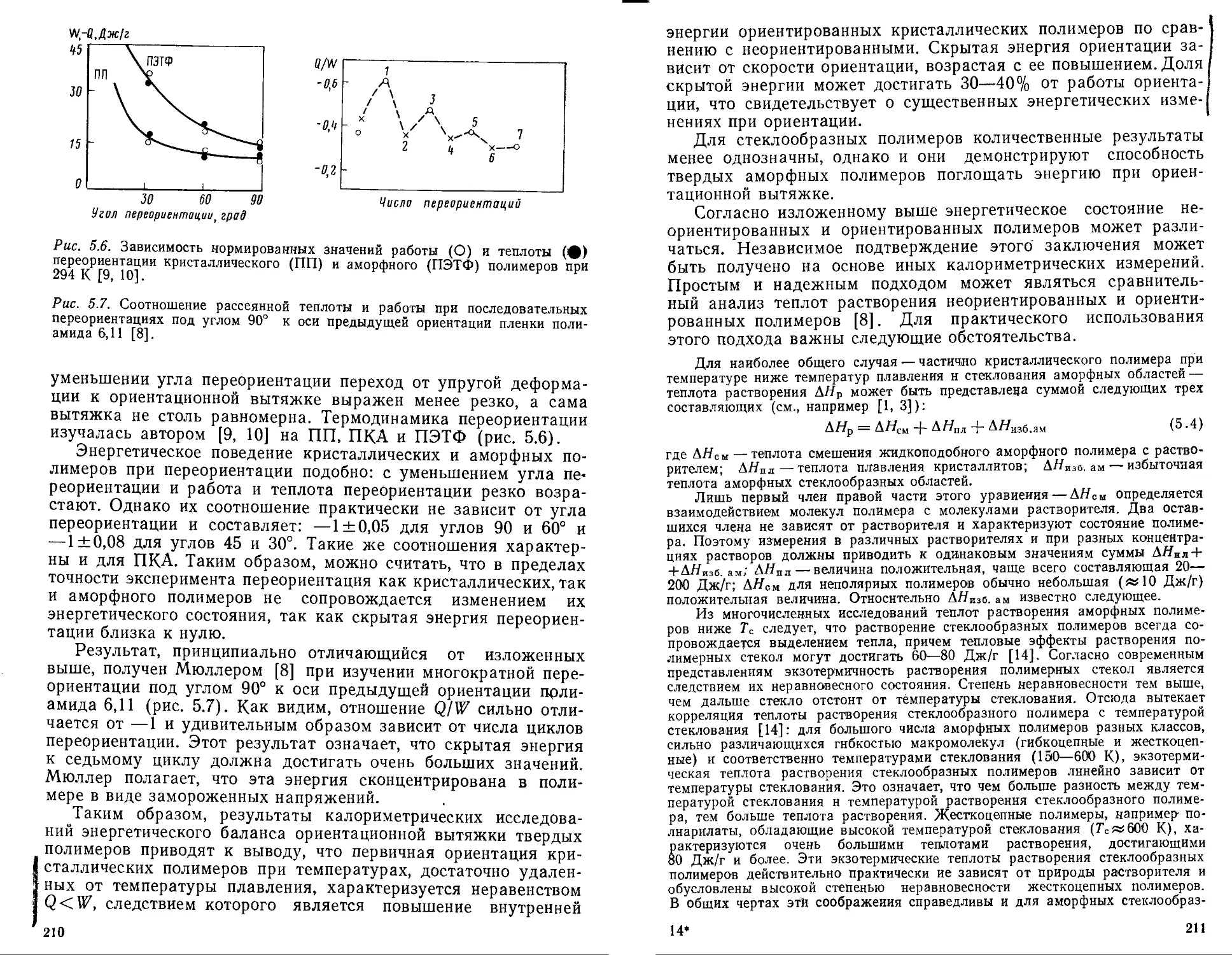
как и пластическая деформация низкомолекулярных веществ,
всегда связана с рассеянием энергии. Центральный вопрос
здесь — соотношение рассеянной и поглощенной при пластиче-
ской деформации (ориентации) энергии и связь этой поглощен-
ной энергии с молекулярными и структурными изменениями
яри ориентации.
Разрушение твердых тел неизбежно сопровождается тепло-
выми явлениями, так как идеально хрупкое разрушение редко
реализуется практически. Основными источниками тепловых
эффектов при разрушении твердых полимеров являются пла-
стические деформации и разрывы макромолекул. Оба этих
источника локализованы в очагах разрушения — трещинах, и
эта локализация может приводить к огромным локальным эк-
зоэффектам вблизи вершины растущих трещин.
По своему содержанию книга представляет собой первую
попытку обобщения основных сведений в области теплофизики
полимеров по состоянию на 1981 г. Ограниченный объем книги
не позволил, к сожалению, рассмотреть, как это первоначально
предполагалось, теплофизические аспекты фазовых переходов
в полимерах. Автор надеется восполнить этот пробел в буду-
щем.
Эта монография логически связана с предыдущей моногра-
фией автора (Ю. К. Годовский. Теплофизические методы иссле-
дования полимеров. М., Химия, 1976), посвященной рассмотре-
нию современных методов исследования тепловых явлений в
полимерах, а также анализу информации, которая может быть
получена при помощи этих методов. В отличие от предыдущей
в данной книге рассматриваются только основные принципы
теплофизики полимеров:
Автор будет искренне признателен всем коллегам за указа-
ние любых недостатков и неточностей в книге, а также за те
или иные замечания и пожелания.
Ю. К. Годовский
Глава 1
ТЕПЛОЕМКОСТЬ
Теплоемкость полимеров, так же как и других конденсирован-
ных тел, определяется их колебательным спектром. В соответ-
ствии с цепным строением макромолекул теплоемкость твер-
дых линейных полимеров в первом приближении может быть
представлена аддитивной функцией двух основных вкладов:
решеточных колебаний и характеристических колебаний от-
дельных групп атомов в повторяющемся звене или их более
или менее изолированного вращения и изомеризации. Решеточ-
ные (скелетные) колебания являются низкочастотными коле-
баниями и вносят основной вклад в теплоемкость полимеров
при низких температурах. Анализ решеточных колебаний мо-
жет быть проведен на основе скелетного приближения, в кото-
ром макромолекула рассматривается как бесструктурная це-
почка точечных масс, равных массе повторяющегося звена.
Поскольку для многих карбоцепных полимеров геометрия по-
вторяющегося звена и- силовые константы близки, единствен-
ным переменным параметром при переходе от одного полиме-
ра к другому является масса повторяющегося звена.
Характеристические колебания боковых радикалов являют-
ся оптическими колебаниями, их частоты существенно выше
частот скелетных колебаний, и потому их вклад в теплоемкость
становится ощутимым, начиная с умеренно низких температур,
и зависит от соотношения масс атомов основной цепи и боко-
вого заместителя. Характерное для полимеров существование
поворотных изомеров, различающихся конформационными
энергиями, приводит к возможности появления составляющей
теплоемкости, обусловленной различными энергетическими со-
стояниями.
Согласно квантовой теории, теплоемкость твердого тела
в гармоническом приближении может быть рассчитана на ос-
новании уравнения
где feg —постоянная Больцмана; A=ft/2rt— постоянная Планка; <о — круго-
вая частота (o>=2itv (здесь v — частота); Т—абсолютная температура;
я
s? “макс
р(,ш) —функция распределения частот, причем j p(<o)dco=3jV, где W— число
О
атомов в решетке; (Омане —максимальная частота.
Для вычисления температурной зависимости теплоемкости
достаточно знать функцию распределения частот, т. е. колеба-
тельный спектр полимера. Проще всего определить вклад в
теплоемкость характеристических колебаний, для которых мож-
но использовать функцию Эйнштейна. Если усредненная харак-
теристическая частота данного колебания данного радикала
равна но, то «спектр» такого колебания имеет вид
Р(<о) = 3№(« — шЕ) (1.2)
(где (йе — эйнштейновская частота; б — функция Дирака),
а его теплоемкость
(1-3)
или в символической форме
Cv = ЗМБ£(0Е/7) (1.4)
ЛМ/М2 ехР(еЕ/П ,
\ Т ) \Т / [екР(0Е/Т) - ip ’ J
где 0е=АшеДб — температура Эйнштейна.
Расчет низкочастотной части колебательного спектра и ре-
шеточной теплоемкости бездефектных полимеров представляет
значительно более сложную задачу. Наряду с численными ме-
тодами расчетов для этого используют ряд аналитических мо-
делей. Еще более сложной теоретической проблемой является
определение низкочастотного колебательного спектра Дефект-
ных полимеров. Наиболее интересны закономерности измене-
ния теплоемкости полимеров при низких тепературах, где су-
щественны квантовые эффекты.
КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ ИЗОЛИРОВАННЫХ ЦЕПЕЙ.
УЧЕТ ВЗАИМОДЕЙСТВИЯ
Колебательный спектр бездефектной изолированной полимер-
ной цепи можно получить, рассмотрев динамику цепи обычны-
ми методами теории гармонических колебаний идеальнух
кристаллических решеток [1—3]. Простейшим примером одно-
мерной решетки является линейная цепь, образованная N-
равными массами М, расположенными на одной прямой и на
^равном расстоянии а друг от друга (рис. 1.1). В реальной по-
I лимерной цепи (в отличие от обычно рассматриваемой матема-
ктической абстракции) возможны как продольные, так и попе-
9
М М а М
О----------о----------о
. а
м
Рис. 1.1. Отрезок линейной цепи, состоящий из одинаковых масс М, располо-
женных на одинаковых расстояниях а (0У — константа продольной упругости,.
ху — константа поперечной упругости).
речные колебания. Продольная жесткость такой цепи харак-
теризуется константой упругости ру (упругость валентных свя-
зей), а поперечная—константой упругости ху (упругости
валентных углов). Колебательный спектр цепи с закрепленны-
ми концами, в том случае, если она обладает вращательной
симметрией относительно своей оси, состоит из одной ветви
продольных колебаний и двух взаимно вырожденных попереч-
ных ветвей. Законы дисперсии этих колебательных ветвей име-
ют вид [3]
О)
46v \1/2 ka
-1 sin — —продольные колебания
(1.6а>
... [ 16ху \1/2 . ka
wt(k) — I----- I sin2-----поперечные колебания (1.об>
\ М J 2
Здесь k — волновое число — находится в интервале
В длинноволновом пределе (k — мало, ak^l) sin -g~y
и законы дисперсии принимают вид
/ Pv Х1/2
“/W =а (-§-) k (h7a>
(Xv \V2
_Z) k2 (1.76)
Таким образом, для длинных волн законы дисперсии про-
дольных и поперечных колебаний цепи оказываются различны-
ми (рис. 1.2). На основании (1.7) можно получить функции
распределения частот (плотность спектрального распределе-
ния) для обеих колебательных ветвей. Согласно обычному оп-
ределению функции распределения частот с учетом нормирова-
ния числа колебаний [2]
(1.8)
аы /ак
имеем [3]:
2N 1
Pl(“) = ' , 2 1/2 (1-9)
" (“/.макс ~
~ • --------Г--ZT"7--------<о.7 11/2 (1 ’ 10>
ш/,макс (1 ) I
L^/jMaKc' w/,MaKc/J
10
Рис. 1.2. Зависимость частоты от волно-
вого вектора (а) и (в) и плотность спек-
трального распределения (б) и (г) для
продольных (а, б) и поперечных (в, г)
колебаний модели, показанной на
рис. 1.1.
Графический вид этих рас-
пределений показан на рис. 1.2.
Их рассмотрение позволяет от-
метить, что почти во всей обла-
сти частота продольных колеба-
ний постоянна и лишь на правой
границе спектра имеется резкий
максимум. Поперечные колебания имеют максимум как при
<о—>-0, так и при то—номакс, в середине зоны плотность спект-
ра также постоянна. Так как Ху-Сфу, то максмакс- Под-
ставив распределения (1.9) и (1.10) в выражение теплоемко-
сти (1.1), можно показать, что при низких температурах
(Т<С0 = (ко/Аб) продольные колебания дают вклад в теплоем-
кость CV~7’1, а поперечные — CV~7’1/2. Таким образом, выра-
жение для теплоемкости изолированной цепи при низких темпе-
ратурах имеет вид
Cv = аТ1 + а1/2Т1/2
(1.Н)
При высоких температурах Cv принимает постоянное зна-
чение в соответствии с законом Дюлонга — Пти. Если колеб-
лющиеся массы не лежат на одной оси как, например, в ти-
пичной для полимеров зигзагообразной цепи, то такая цепь ха-
рактеризуется крутильными колебаниями с законом дисперсии
(1.66).
Приведенная на рис. 1.1 модель цепи содержит лишь один
колеблющийся элемент в элементарной ячейке. Если элемен-
тарная ячейка цепи содержит п колеблющихся элементов раз-
личной массы, то колебательный спектр такой цепи кроме
акустических ветвей содержит и оптические ветви. Согласно
теории колебаний [1, 2], число дисперсионных кривых равно
утроенному числу атомов в повторяющемся звене (ячейке).
Таким образом, даже такой полимер, как ПЭ, должен иметь
девять колебательных ветвей. Задача, как и в простейшем
случае, сводится к получению и решению секулярного уравне-
ния движения цепи. Основная сложность расчетов состоит в
том, что секулярное уравнение в этом случае имеет большой
порядок. Дисперсионные кривые и колебательный спектр изо-
лированной цепи ПЭ (рис. 1.3), рассчитанный [4] таким обра-
зом с учетом упругих постоянных, определенных по ИК- и Ра-
ман-спектрам, позволяют отметить тот факт, что частоты опти-
ческих и акустических колебаний не перекрываются и разде-
11
Рис. 1.3. Дисперсионные кривые (а) и плотность спектрального распределения
(б) изолированной цепи ПЭ [4] :
/ — крутильные (скелетные); 2 — деформационные (скелетные); 3 — СН2-крутильиые; 4 —
С—С-валентные; 5 — СН2-маятииковые; 6 — СН2-веерные; 7 — СН2-ножиичные; 8, 9 —
СНа-валентные.
лены значительной щелью, что обусловлено малой массой ко-
леблющихся атомов. Из этого вытекает важное следствие: низ-
котемпературная теплоемкость изолированной цепи ПЭ долж-
на определяться лишь акустической частью колебательного
спектра. Оценки показывают, что низкочастотные оптические
колебания (750—1500 см-1) дают вклад «2% в теплоемкость
при 120 К, а высокочастотные (2500 см-1) не дают практиче-
ски никакого вклада даже при комнатной температуре [3].
Представленная на рис. 1.3 картина дисперсионных кривых
в' общих чертах типична для всех изолированных полимерных
цепей. Изменение химического строения влияет лишь на число
дисперсионных кривых и интервалы частот для различных ча-
стотных ветвей. Общей чертой структуры колебательного
спектра изолированных цепей является наличие 2 акустических
и п—2 оптических ветвей.
Как изменится колебательный спектр цепи, если учесть
межмолекулярное взаимодействие? Можно полагать (и экспе-
римент полностью подтверждает это [3]), что учет межмоле-
кулярного взаимодействия не скажется на высокочастотных
оптических колебательных ветвях и лишь незначительно бу-
дет влиять на низкочастотные оптические колебания. В то же
время спектр низкочастотных акустических колебаний, несмот-
ря иа малость взаимодействия между цепями по сравнению с
внутрицепным взаимодействием, радикальным образом изменя-
ется при учете межцепного взаимодействия.
На рис. 1.4 приведен акустический спектр ПЭ, рассчитанный
'е учетом силового поля межмолекулярного взаимодействия по
аддитивной схеме |[3, 4]. Константы упругости межмолекуляр-
ного взаимодействия выбирали таким образом, чтобы добить-
ся соответствия с низкотемпературной теплоемкостью. Основ-
ные изменения в спектре при «включении» межмолекулярного
взаимодействия происходят в низкочастотной области, где без
учета взаимодействия р(со)—>оо при со—>0, в то время как с
учетом взаимодействия р(со)—>0 при и—>0. Низкочастотная
область спектра ПЭ, определенная [3, 4] по данным неупругого
рассеяния тепловых нейтронов, хорошо согласуется со спект-
ром ПЭ, полученным расчетным путем. Таким образом, аку-
стический колебательный спектр цепей с учетом межцепного
взаимодействия характеризуется двумя областями: низкоча-
стотной, в которой плотность колебаний монотонно падает до
0 при р(со)—>0, и высокочастотной, в которой спектр имеет
тонкую структуру, определяемую внутримолекулярными взаи-
модействиями. В области частот до 100 см-1 вид спектра оп-
ределяется межцепным взаимодействием, а в области от 100
до 500 см-1 он почти целиком определяется химическим строе-
нием цепи. Отсюда следует, что низкотемпературная теплоем-
кость (до 50—70 К) должна зависеть' как от внутримолекуляр-
ного, так и межмолекулярного взаимодействия. Выше этой об-
ласти всегда можно выделить такой интервал температур, в
котором теплоемкость определяется главным образом внутри-
молекулярными взаимодействиями. В то время как в этой по-
следней области теплоемкость должна определяться уравнени-
ем (1.П), при более низких температурах закономерность из-
менения Cv с температурой должна быть иной.
Прежде чем переходить к рассмотрению теории теплоемко-
сти полимеров в конденсированном состоянии, необходимо сде-
лать одно замечание, связанное с определением числа акусти-
а>,см-’
Рис. 1.4. Плотность спектрального распределения кристаллического ПЭ [4].
13
ческих колебательных ветвей в изолированной молекуле и в
твердом полимере, образованном макромолекулами [5]. Нуж-
но иметь в виду, что для кристаллического полимера сущест-
вует два различных элемента регулярности — повторяющееся
звено и элементарная кристаллическая ячейка. В кристалле*
должны существовать три акустических ветви (две поперечные
и одна продольная), в то время как в изолированной цепи та-
ких ветвей лишь две.
КОЛЕБАТЕЛЬНЫЙ СПЕКТР И ТЕПЛОЕМКОСТЬ РЕШЕТКИ,
ОБРАЗОВАННОЙ ПОЛИМЕРНЫМИ ЦЕПЯМИ
Модель Стокмейера — Хечта
Расчет полных колебательных спектров полимерных кристал-
лов, рассмотренный выше на примере полиэтилена, в принципе
может быть выполнен для полимера любого строения, однако
он настолько трудоемок даже при современном уровне ма-
шинных расчетов, что до настоящего времени такие расчеты
выполнены лишь для полимеров простейшего строения — поли-
этилена, полиоксиметилена и полиоксиэтилена [4].
Как уже отмечалось выше, для исследования низкочастот-
ной области колебательного спектра, определяющей низкотем-
пературную теплоемкость, нет необходимости рассматривать
все колебательные степени свободы повторяющегося звена
макромолекулы, поскольку характеристические частоты коле-
баний боковых групп и колебаний скелета лежат в разных об-
ластях спектра. Вообще говоря, это заключение, впервые сде-
ланное при анализе колебательного спектра предельных угле-
водородов Кирквудом [6] и Питцером [7], достаточно обосно-
вано лишь для макромолекул с «легкими» боковыми группа-
ми. Для других полимеров, содержащих «тяжелые» боковые
радикалы, этот метод требует дополнительного обоснования,
поскольку замена точечной массой всего повторяющегося зве-
на становится неочевидной, так как тяжелые боковые группы
могут иметь низкие характеристические частоты.
В последнее время Чебан [8, 9] теоретически рассмотрел
вопрос о скелетном приближении и показал, что простое ске-
летное приближение, при котором массу повторяющегося зве-
на можно считать сосредоточенной в точке, строго выполняет-
ся лишь в узкой области частот вблизи нулевой частоты. Если
же рассматривать всю область низких частот, то замена повто-
* Представления о нормальных колебаниях применимы и для аморфных
твердых тел, поскольку единственным требованием для существования колеба-
тельного спектра является стабильное положение каждого атома. Если при
этом существует регулярность в их расположении (кристалл), то расчет коле-
бательного спектра сильно упрощается.
14
Рис. 1.5. Модель кристалла, образован-
ного полимерными цепями (модель Сток-
мейера—Хечта); силовые постоянные:
Ру — для главной валентной связи вдоль
цепи; cty — для взаимодействия ближай-
ших звеньев двух соседних цепей; бу,
уу — для взаимодействия звена одной це-
пи со вторым ближайшим звеном сосед-
ней цепи; Ху — для деформации валент-
ных углов цепи [10].
ряющегося звена точечной массой возможна лишь при условии,
что эта точечная масса считается зависящей от частоты. Эта
модель, названная в отличие от обычного скелетного прибли-
жения обобщенным скелетным приближением, позволяет уста-
новить границы применимости обычного скелетного приближе-
ния.Так, оценки показывают, что для ПЭ простое скелетное при-
ближение оправданно в области частот 0—50 см-1 для кру-
тильных колебаний и 0—200 см-1 для деформационных коле-
баний. Обобщенное скелетное приближение является особенно
ценным при анализе теплоемкости в том случае, когда боко-
вые группы взаимодействуют только с атомами в своем звене,
образующем скелет. В этом случае реальная макромолекула
заменяется эквивалентной линейной цепочкой с точечными
массами (И(ко) и силовыми постоянными /Дсо); колебательный
спектр такой цепи может быть получен из анализа уравнений
ее движения по формулам (1.6) — (1.8).
Для теоретического расчета низкотемпературной теплоем-
кости очень полезными оказываются упрощенные модели, до-
пускающие аналитические решения и достаточно точно опи-
сывающие низкочастотную область спектра. Первая такая мо-
дель была предложена Стокмейером и Хечтом [10], а ее коле-
бательный спектр и низкотемпературная теплоемкость деталь-
но проанализированы Гененски и Ньюэлом [11] методом Бор-
на—Кармана (модель СХ). В этой модели (рис. 1.5) парал-
лельно расположенные цепи образуют простую тетрагональную
решетку, в узлах которой находятся повторяющиеся звенья
одинаковой массы М. Расстояния между цепями принимаются
одинаковыми и отличными от расстояния между звеньями
вдоль цепи. Межмолекулярное взаимодействие учитывается в
приближении ближайших соседей. Константы взаимодействия
выбирают таким образом, чтобы отразить две важнейшие осо-
бенности решеток, образованных полимерными цепями: силь-
ную анизотропию решетки и гибкость цепей. Учет этих осо-
бенностей достигается следующим выбором соотношения кон-
стант взаимодействия (рис. 1.5): ру>ху>юу»’уу=6у. Сильная
15
Рис. 1.6. Функция распределения частот валентных (а) и деформационных (б)
колебаний и полный колебательный спектр (в) для модели Стокмейера — Хеч-
та по данным работы [11].
анизотропия выражается условием py^>iay, а учет гибкости це-
пей достигается введением упругой постоянной ху.
Анализ динамики движения этой модели показал, что, по-
скольку эта решетка является простой, в ней существуют лишь
три акустические ветви колебаний, две из них являются взаи-
мовырожденными поперечными (деформационными) колеба-
ниями, а третья ветвь представляет собой продольные (валент-
ные) колебания, при которых смещения звеньев происходят
вдоль цепи. Функция распределения частот продольных и попе-
речных колебаний при соотношении упругих постоянных ху =
= 0,1 ру; ау=0,04 ру и у = 0,002 ру, а также полный колебатель-
ный спектр такого кристалла показаны на рис. 1.6.
В начальной области спектров при со—>-0 как для продоль-
ных, так и для поперечных колебаний функция распределения
частот р(о))~б)2. Однако интервал частот, в котором соблюда-
ется эта пропорциональность, для поперечных колебаний зна-
чительно уже. При и^>0,2 <йг,Макс плотность продольных коле-
баний постоянна. Это означает, что в этой области pz(co) иден-
тична р(со) изолированной цепи, т. е. в этой области спектра
межмолекулярное взаимодействие не оказывает влияния на
распределение продольных колебаний цепи.
Для поперечных колебаний за областью p(«)~w2 сначала
следует область, в которой р;(со) ~<о3/2, а затем область, где
р;(<й) аппроксимируется функцией ta>-1/2. Спектр продольных
колебаний имеет пять особенностей Ван Хова, а спектр попе-
речных колебаний — девять, из которых отчетливо проявляют-
ся лишь семь.
В соответствии с этими функциями распределения частот
согласно уравнению (1.1) температурная зависимость тепло-
емкости продольных колебаний имеет вид
м
CVl~ Т3 при 0<Т < —— (1.12а)
«Б
16
ft
CVl ~ T1 при
16yy\l/2
~лГ)
kb
<T«
/4₽y \‘/2
\ M I
kb
(1.126)
Температурная зависимость теплоемкости поперечных коле-
баний выражается зависимостями
СИ/~РприО<Т«—Д- (1.13а)
*бЫ;
Cv ~ Г5,2 при - - « Т « —(1 • 136)
<, /4ху\1/2 *Б
,40^/2 ,1бхуу/2
Су. ~ Т1/2 при - ' - « Т« 'М > (1.1Зв)
* Ьь kb
Обращает на себя внимание принципиально отличная зави-
симость теплоемкости, обусловленной продольными и попереч-
ными колебаниями в той области температур, где межцепным
взаимодействием можно пренебречь. Поперечные колебания
дают Cv^T1/2, а продольные — Cv~Tl. Появление зависимо-
сти Су~Тл/2 является следствием гибкости полимерной цепи,
обусловленной деформацией валентных углов.
Суммарная теплоемкость, определяемая акустическими ко-
лебаниями рассматриваемой модельной решетки, определяется
зависимостями
СГак = а3Л при 0 < Т « — — (1.14а)
й(^У/2
М \ М /
Суак = аз’ТЗ + аъ^тър п₽и 77—777 « Т «--------7----- И • 14б>
, / ’Ху «Б
ftf^Y/2 ft&Y/2
\М. X м
СУак = а1Р при----------- «Г < ------------ (1 • 14в)
Коэффициенты пропорциональности at в этих уравнениях
определяются константами взаимодействия ру, ху, ау и уу и
массой М.
Таким образом, характерной особенностью модели СХ яв-
ляется наличие «дебаевской» области, в которой Су~Тъ, пере-
2—440
17
-1 Л£
ходной области, в которой Су^ Т3-±-Т5/2, и достаточно широкой
области температур, в которой Cv~ 7’,4-7’1/2’. Детальный ана-
лиз показывает [11, 3], что полученные закономерности изме-
нения теплоемкости должны выполняться для типичных силь-
ноанизотропных кристаллов, образованных цепными молекула-
ми в вытянутом состоянии, в том числе и для макромолекул,
представляющих собой плоский зигзаг (типа ПЭ). К решеткам,
образованным спиральными макромолекулами, рассмотренная
теория прямо неприменима. Таким образом, хотя модель СХ
является предельно упрощенной моделью полимерного крис-
талла, однако она учитывает гибкость цепей и сильную ани-
зотропию сил взаимодействия в решетке, и потому можно по-
лагать, что она качественно правильно предсказывает наличие
характерных интервалов изменения теплоемкости полимерных
кристаллов.
Континуальные модели
Наличие сильной локальной анизотропии сил в твердых поли-
мерах, обусловленной различием связей в цепях и между ни-
ми, приводит к тому, что при низких температурах всегда су-
ществует такая область, в которой межмолекулярным взаимо-
действием можно пренебречь и в которой теплоемкость
определяется плотностью колебательных состояний изолиро-
ванной цепи. При достаточно большом числе элементов в цепи
ее можно считать упругой одномерной средой (континуумом)
и использовать для анализа ее колебаний континуальный под-
ход, введенный в физику твердого тела Дебаем. Впервые про-
блема теплоемкости цепных структур, типичными представите-
лями которых являются органические полимеры, на основе кон-
тинуального подхода была рассмотрена Тарасовым [12—15].
Для понимания существа теории теплоемкости Тарасова и ее ограниченности
целесообразно напомнить основные положения подхода Дебая [3, 16]. В отли-
чие от метода Бориа — Кармана, применение которого к анализу колебатель-
ного спектра решетки, образованной полимерными цепями, рассмотрено выше,
Дебай рассмотрел распространение упругих волн в твердом теле, считая его
упругой изотропной средой и пренебрегая атомной структурой тела.
Из теории упругости известно [17], что в такой изотропной среде каждому
волновому вектору соответствует одна ветвь продольных и две взаимно вырож-
денные ветви поперечных упругих колебаний. Эти колебания являются звуко-
выми волнами, и для них характерен простой закон дисперсии w~A. Скорости
продольных ci и поперечных ct волн определяются упругими постоянными
среды:
^]A.+'>7-U> "=]/Ж "',5>
где Е — модуль упругости; ц — коэффициент Пуассона; р — плотность среды.
Функция распределения частот континуума имеет вид
V Г 1 2 1
рб») "= "оТТ Т5" + 0)2 О-16)
zn- l J
где V — объем.
18
Внутренняя энергия такого континуума определяется суммой энергий стоя-
чих продольных и поперечных волн. Такой континуум обладает бесконечно
«большим набором собственных частот, поскольку у него отсутствует верхняя
граница частотного спектра. Реальное твердое тело содержит хотя и огромное,
яо все же конечное число частиц и потому должно иметь конечное число соб-
ственных колебаний. Исходя из этих соображений, Дебай ограничил функцию
распределения частот максимальной частотой шо и нормировал ее таким обра-
зом, чтобы суммарное число колебаний решетки было равно
'"D
p(<»)do> ==3N (1.17)
о
(где N — число структурных элементов твердого тела). Это позволяет опреде-
лить ограничивающую частицу:
о 18n2N
(Вз=__
(1-18)
4 +2с?
Если, исходя из типичных значений упругих постоянных кристаллической
решетки полимера £=240 ГПА и ц=0,3, оценить для полимеров значения с;
и ct, а затем рассчитать coD, то оказывается, что <Оо«3-1013 с-1, а соответствую-
щие такой частоте длины волн 2nci/a>D и 2яс//шо находятся на уровне пара-
метров кристаллической решетки («0,5 нм). Таким образом, введение ограни-
чивающей частоты формально соответствует тому, что в отличие от упруго-
го континуума в реальной кристаллической решетке волны длиной меньше
удвоенного расстояния между структурными элементами решетки не могут
существовать. Итак, функция спектрального распределения в теории Дебая
имеет вид
t 9Х
I —со2, 0 < СО < Шп (1.19)
р(«>)=| “Ь " D ' ’
I 0, coD
Вклад акустических колебаний в теплоемкость упругоизотропной среды,
согласно теории Дебая определяется уравнением
функция Дебая, в которой 0в=Л<опДб—температура Дебая.
Формула Дебая упрощается в предельных случаях высоких (0о<СГ) и низ-
ких температур (QdS-T). При высоких температурах Cv=3Wkp , что соответ-
ствует закону Дюлонга — Пти, а при низких температурах (Г<0в/12) из
уравнения (1.22) следует известный Т^-закон Дебая:
Cv = — nWfcr,(770D)3
о
(1-23)
19
Несмотря на простоту исходных постулатов, теория Дебая хорошо согла-
суется с экспериментальными результатами для многих твердых тел простой
структуры. Однако границы выполнения Т3-закоиа для многих тел находятся
при значительно более низких температурах (часто при T»0D/1OO), чем это
предсказывается теорией. Теория Дебая построена для изотропной упругой
среды. При наличии ярко выраженной анизотропии скорости распространения
упругих волн в различных направлениях могут сильно различаться и колеба-
ния могут не обладать определенной поляризацией. Поэтому каждому направ-
лению в пространстве в этом случае будут соответствовать три различные
функции распределения частот с тремя различными ограничивающими часто-
тами и тремя различными температурами Дебая. Предельные законы и в этом
случае должны, естественно, выполняться (Cv~3^) при высоких температу-
рах и (Су-~Т3) вблизи О К, однако в промежуточной температурной области
температурная зависимость теплоемкости может существенно отличаться от
той, которая предсказывается формулой Дебая. Твердые тела, для которых
характерно цепное и слоистое строение, являются представителями той группы
тел, для которых наблюдаются сильные отклонения от формулы Дебая. Этот
экспериментальный факт побудил Тарасова поставить вопрос о теплоемкости
таких твердых тел и он, использовав континуальный подход Дебая, рассмотрел
эту проблему.
Главная идея, положенная в основу теории теплоемкости цепных и слои-
стых структур [12—15], состоит в том, что основная доля собственных колеба-
ний в них приписывается цепям или слоям, а не всему твердому телу. Это
означает, что при 7"<С0 внутренняя энергия тела определяется функцией рас-
пределения частот для цепей или слоев. Анализ показал, что функции распре-
деления частот для линейного и двумерного континуумов различаются прин-
ципиально: для линейного континуума частоты равномерно распределены по
всему диапазону частот от 0 до (Омаке, а для двумерного они линейно возраста-
ют от 0 до (Омаке. По аналогии с приемом Дебая функции распределения частот
нормальных колебаний нормируются согласно
“макс
pm(o>)d<o = 3A (1.24)
о
Тогда обобщенная функция распределения частот имеет вид
Рт (<»)d<o = (1.25)
(щ = 3 для трехмерного континуума, т=2 для слоев и т=1 для цепей), а обоб-
щенное выражение теплоемкости можно получить, подставив это выражение
в уравнение (1.1). Сделав это, получаем выражение
в котором 0т=й(Омакс/Й£—характеристическая температура цепи 0Ь слоя 02
или трехмерного тела 03. Из этого уравнения следуют все три возможных част-
ных случая. Когда т=3, получаем трехмерный континуум Дебая [уравнение
(1.22)]. Если т=2 — двумерный континуум (слои), то
Cv = 3NkBD2(Q2/T) (1.27)
где D2(Q2/T) — двумерная функция Дебая, равная
е2/г
„ (* (д/Т¥>ехр(д/Т)
J (Е28>
о
20
Для одномерного континуума (т = 1) имеем:
Cv = 3NkBD1(QlIT) (1.29)
где Di — одномерная функция Дебая, равная
е,/г
DifOi/n = (W1 ( <L30>
J (ехр(0/Т) — I]2
о
Уравнение для двумерного и одномерного континуумов получены в пред-
положении, что взаимодействие между слоями и цепями отсутствует. При вы-
соких температурах уравнения (1.27) и (1.28), так же как и теория Дебая,,
приводят к закону Дюлонга — Пти, а при низких температурах для теплоемко-
сти цепей, слоев и трехмерного континуума можно получить следующие выра-
жения:
^ = ^(7/0!)! (I.3I)-,
С2 = 43,272/?(Т/Эя)2 (1.32)
12
С3 = — л2Я(7703)3 (1.33)
о
Из этих выражений следует, что при Ттеплоемкость веществ, состав-
ленных из невзаимодействующих слоев, следует 72-зависнмости, а из невзаимо-
действующих цепей — Т’-зависимости. Для трехмерного континуума тепло-
емкость следует Т3-закону Дебая.
Прн выводе соотношений (1.27) и (1.30) предполагалось отсутствие взаи-
модействия между слоями и цепями, что является идеальным (модельным)
случаем. В реальных твердых телах между цепями и слоями существует взаи-
модействие, которое, как мы уже убедились выше, может существенным обра-
зом изменить колебательный спектр твердого тела при низких температурах.
Для учета взаимодействия между цепями и слоями Тарасов использовал сум-
мирование дебаевской функции распределения частот для трехмерного конти-
нуума с функциями распределения для одно- или двумерного континуумов.
С физической точки зрения этот прием означает, что при очень низких темпе-
ратурах, когда длины упругих волн велики, анизотропия сил взаимодействия
не должна сказываться на длинноволновых колебаниях и в этой области темпе-
ратур (вблизи 0 К) Т3-закон Дебая должен выполняться для тел любой струк-
туры. Поэтому можно предположить, что часть собственных колебаний от 0 до
<0з распределена по закону трехмерного континуума. При повышении темпера-
туры все большую роль начинают играть собственные колебания в цепях и
слоях, и общее распределение частот начинает трансформироваться в распреде-
ление, характерное для цепей или слоев. Часть частот от а>3 д.о <вМакс распреде-
лена по закону одно- или двумерного континуума. Таким образом, с учетом
нормировки функция распределения частот для одномерного континуума с
взаимодействием цепей имеет вид
Pi(") =
9№.-'
—-------, 0 < “ “3
^З^макс
(1-34)
шмакс
сйз 5^ со < <оМакс
Функция распределения частот
дующим образом:
для двумерного континуума выражается еле-
0 < СО (Од
р2(ш) =
6N(D
.,.2 > 0) ^макс
макс
(1.35)
21
Подставляя (1.20) и (1.21) в уравнение (1.1), получаем следующие формулы
для теплоемкости взаимодействующих цепей
С113 = О1(01/Т) - (03/01)[О1(03/Т)-£>3(93/Т)] (1.36)
и взаимодействующих слоев:
С2,з = D2(Q2/T) - (03/02)[О2(03/7’) - Рв(03/Г)] (1.37)
В отличие от формулы Дебая и формул (1.13) и (1.15) формулы для взаи-
модействующих цепей и слоев являются двухпараметрическими. Этими пара-
метрами в них являются характеристические температуры 03 и 0] или 03 и 02.
При низких температурах (Т—»-0) уравнения (1.36) и (1.37) переходят
в следующие уравнения:
12
^'i,3(7'-.O) ~ (03/®i) л*/?(7’ 63)1 (1.38)
12
^2,3(Г^0) = (®з/0ц)2 —л47?(7’/03)3 (1.39)
Согласно оценкам Тарасова, кубическая зависимость теплоемкости взаимо-
действующих цепей или слоев должна наблюдаться до 7"» 0,1 03. Из уравнений
(1.36) и (1.37) следует также, что при значительном различии характеристи-
ческих температур (0i»03 и 02»03) в интервале температур, определяемом
соотношениями ТгС А- 0: и 7’=С0,1 02 для цепных структур Cv~T, а для слои-
стых структур Cv~T2. Мы видим, что учет взаимодействия изменяет темпера-
турную зависимость теплоемкости слоевых и цепных структур лишь в области
очень низких температур. Таким образом, по теории Тарасова, температурная
область изменения теплоемкости цепных и слоистых структур разделяется на
характерные интервалы: при очень низких температурах теплоемкость изменя-
ется в соответствии с дебаевским Т3-законом, затем по зависимостям (1.36) и
(1.37) она переходит в область линейного (цепи) и параболического измене-
ния (слои) и наконец при высоких температурах достигает классического зна-
чения.
Согласно теории Тарасова, при низких температурах теплоемкость инди-
видуальной цепи при условии, что межцепное взаимодействие играет второ-
степенную роль в колебательном спектре, должна подчиняться Т^-зависимости.
Эта закономерность отличается от выражения для теплоемкости (1.14в), выте-
кающего из спектрального подхода. Причина различия состоит в том, что в тео-
рии Тарасова как для продольных, так и для поперечных колебательных вет-
вей принят обычный закон дисперсии <в~А. Однако, как мы убедились выше,
этот закон характерен лишь для продольных колебаний цепи. Для одномерной
цепи с валентными углами 180° этн продольные колебания являются валентны-
ми колебаниями. Валентные колебания реальных полимерных цепей лежат в
области 1000 см-1, и такие колебания не дают практически никакого вклада в
теплоемкость не только при низких, но и при комнатных температурах.
В отличие от изотропных твердых тел, для которых не существует низко-
частотных акустических колебаний с дисперсионным законом отличным от
обычного, в реальных цепных структурах основной вклад в низкотемператур-
ную теплоемкость дают низкочастотные (0—200 см-1) скелетные крутильные
колебания (волны изгиба). Дисперсионное соотношение для таких колебаний
в длиноволновой части спектра имеет вид со~й2, а температурная зависимость
теплоемкости цепи — Cv~T'j2. Такое же соотношение между изгибными и про-
дольными колебаниями характерно и для слоевых структур, для которых вол-
ны изгиба дают вклад в теплоемкость Cv~T.
Переходя на язык континуальных моделей, можно заключить, что в тео-
рии Тарасова цепная молекула представляется жесткой линейной струной,
в которой возможны лишь продольные колебания. В действительности же ее
следует аппроксимировать тонкой гибкой иглой или гибким стержнем, в кото-
рых могут возбуждаться изгибные волны. Аналогично слоистые структуры ве-
22
дут себя подобно гибким мембранам, а не жестким пластинкам. Таким образом,,
при правильной в принципе постановке вопроса о роли анизотропии в тепло-
емкости и о решающей роли квазиодномерного и квазидвумерного спектров,
колебаний цепных и слоистых структур в теории Тарасова оказались не учтен-
ными основные колебательные моды, определяющие низкотемпературную теп-
лоемкость. Поэтому формулы (1.36) и (1.37) в принципе не могут дать кор-
ректную информацию о колебательном спектре полимерной цепи или слое. Хотя
на это уже неоднократно указывалось в литературе [18—20], тем не менее-
теория Тарасова широко и некритически используется, особенно в работах за-
рубежных исследователей, при анализе низкотемпературной теплоемкости по-
лимеров, и на основе такого анализа часто пытаются сделать определенные вы-
воды о структуре полимеров и о функции распределения частот. Так, для мно-
гих полимеров [3, 19, 20] в температурном интервале 60—200 К зависимость,
теплоемкости от температуры близка к линейной, что является поводом для
заключения о выполнении /"'-закона Тарасова. Однако уже неоднократно отме-
чалось [18—20], что приближенная линейная зависимость теплоемкости от
температуры для полимеров является результатом сложной суперпозиции раз-
личных колебательных мод, а не только колебаний скелета и не может служить
подтверждением физической обоснованности расчетов Тарасова. К этому во-
просу мы еще вернемся.
Как изменятся уравнения (1.36) и (1.37) н соответственно закономерно-
сти изменения теплоемкости в характерных температурных интервалах, если
наряду с продольной акустической колебательной ветвью будут учтены и из-
гибные колебания?
Впервые корректный анализ тепловых свойств цепных и
слоистых структур при низких температурах в рамках конти-
нуального подхода дан Лифшицем [16, 21]. Он обратил вни-
мание на тот факт, что в тонких иглах и пленках характер
температурной зависимости теплоемкости при низких темпе-
ратурах должен определяться волнами изгиба с законом дис-
персии щ~Л2. Их наличие приводит к своеобразным зависи-
мостям теплоемкости от температуры (при низких температу-
рах), когда существенны квантовые эффекты; теплоемкость,
тонких игл (одномерная структура) оказывается пропорцио-
нальной Т1/2, а тонких пластин (двумерные структуры) — Т1.
Этот важный результат был учтен при построении теории теп-
ловых свойств анизотропных кристаллов, образованных цепны-
ми молекулами или атомными (молекулярными) слоями. Це-
пи и слои в таких кристаллах отождествлялись с одно- или
двумерными структурами. Сначала было рассмотрено поведе-
ние невзаимодействующих цепей и слоев. Законы дисперсии
для цепи имеют вид
<в3 = ckz (продольные волны)
©12 = (поперечные волны)
(1.40)
где k? — проекция волнового вектора на ось г, являющуюся осью цепи; с —
скорость упругих волн; y'=cav'/n (а — расстояние между атомами в цепи,
v' — безразмерный параметр, характеризующий «поперечную жесткость» цепи).
С учетом этих законов дисперсии для теплоемкости имеем:
G = Л%Бт1 (770) V2[ 1 + ^Ц/О/Ч
где у; « 2,3//v'; $ ~ /v'.
(1.41).
23
При низких температурах
С1/ = №Бу;(7’/0)1'2 (1.42)
Аналогично для невзаимодействующих слоев, в плоскости
которых действует обычный закон дисперсии, а поперек слоев
частота пропорциональна квадрату волнового вектора, было
получено выражение для теплоемкости:
Cy=;^L-^7-~№B(T/e)1 (1.43)
12 hy
где (а')2— площадь элементарной ячейки в плоскости слоя; у'=са'/л; 0 =
йс
, л.
-kBa
Таким образом, низкотемпературная теплоемкость невзаи-
модействующих цепей и слоев практически целиком определя-
ется волнами изгиба, а вкладом от продольных колебаний
обычно можно пренебречь. Как видим, эти выводы принципи-
ально отличаются от выводов Тарасова.
Переход к термодинамике реальных цепных и слоистых
структур осуществляется путем учета взаимодействия между
цепями и слоями на модели сильноанизотропного кристалла
гексагональной системы, в котором силы взаимодействия в
одном направлении значительно отличаются от сил в двух дру-
гих направлениях. Учет взаимодействия цепей и слоев приво-
дит к появлению нескольких характерных температурных ин-
тервалов изменения теплоемкости. Для цепей
Cv = Д(Т/0)з при Т « 0т]2 (1.44)
Cf = B(T/0)6/2 при 7]20«Т«0т; (1.45)
где А и В — коэффициенты, выражаемые через упругие модули кристалла и
«поперечную жесткость» цепей; ц2 — отношение модуля сдвига к модулю Юнга
вдоль оси цепи.
При более высоких температурах, когда 0цТ<С0, взаимо-
действием цепей можно пренебречь, и в этой области теплоем-
кость определяется формулой (1.42).
Для теплоемкости взаимодействующих слоев
Cv = А(Т/9)3 при Т< (1.46)
Су = В(Т/ву при т]20 « Т « 0т] (1.47)
где А и В — коэффициенты, выражаемые через упругие модули кристалла и «по-
перечную жесткость» слоев; ц2 — отношение модуля сдвига к модулю Юнга в
плоскости слоя.
В области температур 0ц<С Т<С0 взаимодействием между
слоями можно пренебречь, и в этой области теплоемкость оп-
ределяется формулой (1.43).
Теперь можно сравнить результаты расчетов теплоемкости
решетки, образованной полимерными цепями, проведенных по
методу Борна—Кармана {уравнение (1.14)], с теоретическими
результатами Лифшица для взаимодействующих цепей, полу-
24
ченными на основе континуального подхода. Это сравнение
убеждает в том, что при корректном учете всех низкочастотных
вкладов в теплоемкость линейных структур закономерности ее
изменения, предсказываемые континуальной теорией и расче-
тами колебательного спектра, вполне адекватны.
Изящный анализ колебательных спектров сильноанизотроп-
ных кристаллов и их низкотемпературной теплоемкости, про-
веденный методом исследования динамики решетки Косевичем
[22], подтверждает существование характерных температур-
ных интервалов, в которых изменение теплоемкости цепных и
слоистых структур подчиняется закономерностям (1.43), (1.46)
и (1.47).
Баур [23] провел расчет вклада изгибных волн в теплоем-
кость цепных структур непосредственно в духе расчетов Тара-
сова. Он искал решение в виде Су=Су/+Су/, где Су —вклад
в теплоемкость продольных акустических колебаний, выражае-
мый уравнением (1.36), a CVi—вклад изгибных колебаний.
Цепи моделировались тонким стержнем, для поперечно-поля-
ризованных колебаний которого был сформулирован закон
дисперсии a~k2 и получена функция распределения частот
р('О)) ~to-I/2. При соответствующей нормировке выражение для
Cv имеет вид
CVi = 2R{Dll2(Qu/T) + (0зА/)1'-’[^з(0з//7’) -DihM ]) (1.48>
где йц2 — функция Дебая при значении т—\/2 [см. уравнение (1.26)].
При очень низких температурах, тогда Т<^'0з/, 0з*<0ш Sib
теплоемкость подчиняется Т^-закону Дебая:
Су = а.,Т- (1.49>
где
4л4/? / 1 2 \
“3 ~ 5 I VI/ + 0]^2 )
В температурном интервале ТСбз/, Оз«<0п, 0ы теплоемкость
изменяется в соответствии с зависимостью
Cv = а1'Т1 + а1/2Т1р (1.50}
где
од ’ ^*1/2 , о
(здесь Г — гамма-функция; £— дзета-функция Римана).
При высоких температурах (7'3>0з/,0з/, 0п, 0lf) полученное
выражение переходит в закон Дюлонга—Пти.
Суммирование продольных и поперечных колебаний с раз-
личающимися ограничивающими максимальными частотами
; является довольно распространенным приемом в исследовани-
i ях теплоемкости полимеров i[24—26]. Используемые для это-
25
то модели являются усложенными вариантами модели Тарасо-
ва. При этом, однако, как правило, не обращают внимание на
необходимость использования различных дисперсионных зако-
нов для продольных и поперечных колебаний. Хотя соответст-
вующим подбором ограничивающих частот при этом удается
достичь хорошего описания низкотемпературной теплоемкости,
однако такой прием скорее может рассматриваться как эмпи-
рический.
НИЗКОТЕМПЕРАТУРНАЯ ТЕПЛОЕМКОСТЬ И ЕЕ СВЯЗЬ
СО СТРОЕНИЕМ И СТРУКТУРОЙ ПОЛИМЕРОВ
Из изложенных выше теоретических представлений следует,
что с позиций физики наибольший интерес представляет теп-
лоемкость полимеров при низких температурах, при которых
проявляются квантовые эффекты и благодаря квазиодномер-
ному характеру структуры возможно появление новых пре-
дельных законов изменения теплоемкости, отличных от закона
Дебая. Суммированные и проанализированные Вундерлихом
[3] результаты многочисленных измерений теплоемкости, на-
копленных к концу 60-х годов для многих полимеров, явились
важным этапом в развитии представлений о теплоемкости по-
лимеров. Эти результаты свидетельствуют о существовании ха-
рактерных температурных областей изменения теплоемкости
полимеров. Первая область находится ниже 50 К. Здесь прояв-
ляется влияние на теплоемкость не только химического строе-
ния, но и физической структуры полимеров, т. е. межцепного
взаимодействия. В частности, в этой области теплоемкость ока-
зывается чувствительной к степени кристалличности.
Поведение кристаллических и аморфных полимеров в этой
области различается принципиально, так как в отличие от вы-
сококристаллических полимеров аморфные полимеры не под-
чиняются закону Дебая. Такое поведение является характерной
особенностью аморфных веществ любой природы. Выше 50 К
находится простирающаяся до температуры стеклования об-
ласть температур, в которой межцепное взаимодействие прак-
тически не оказывает влияния на теплоемкость и она опреде-
ляется колебательным спектром скелета и движениями боко-
вых групп. В этой области проявляются характерные для цеп-
ных структур предельные законы изменения теплоемкости,
однако наложение вкладов от низкочастотных составляющих
колебательного спектра скелета и боковых групп затрудняет
прямое сравнение теории с экспериментом. У аморфных поли-
меров в области стеклования всегда наблюдается резкое уве-
личение теплоемкости, связанное с размораживанием сегмен-
тальной подвижности. Исходя из этой общей картины, мы
рассмотрим экспериментальные данные о теплоемкости поли-
меров в характерных температурных интервалах и обсудим,
26
прямолинейные отрезки соответствуют
С^а3'Т3+апТп:
1 — п=3; 2 — п=Ы2\ 3 — я=3/2.
Рис. 1.7. Зависимость теплоемкости от температуры в логарифмических коор-
динатах для КПЭ (ф) и АПЭ (О) по обобщенным данным [3]; прямая 1 со-
ответствует уравнению (1.51), прямая 2 — уравнению (1.55).
в какой мере экспериментальные результаты согласуются с
теоретическими представлениями.
Полиэтилен представляет собой полимер, теплоемкость ко-
торого изучена наиболее полно. Особенно важным обстоятель-
ством является наличие данных для полностью кристалличе-
ского и полностью аморфного ПЭ, полученных на основании
экстраполяции зависимостей теплоемкости от степени кри-
сталличности. Таким образом, на примере ПЭ можно проана-
лизировать температурную зависимость Cv для кристалличе-
ской решетки и сравнить ее с теоретическими зависимостями,
а также сравнить поведение полимеров в упорядоченном и не-
упорядоченном состояниях при низких температурах.
Температурная зависимость теплоемкости полностью крис-
таллического (КПЭ) и полностью аморфного (АПЭ) ПЭ при-
ведена на рис. 1.7 в логарифмических координатах [3]. Для
КПЭ в температурном интервале 0<Т<:9 К зависимость теп-
лоемкости от температуры выражается прямой [23]:
lgCP = 3,01gT—3,95 (1.51)
причем отклонения не превышают 1%. Таким образом, в этом
температурном интервале теплоемкость КПЭ подчиняется
7'3-закону Дебая:
Скпэ = 0,1104-10-37'3 Дж/(моль-К) (1.52)
Из формулы Дебая (1.23) и уравнения (1.52) следует, что
6о = 9з = 260 К. Соответствующая этой дебаевской температуре
27
частота ®d = 3,4-1013 Гц (181 см-1), а значение Ct, рассчитанное
при условии с/ = 2с/, составляет при О К~2-105 см/с, что хоро-
ши совпадает с данными акустических измерений при гелиевых
температурах [27]. Согласно теории СХ и теории Лифшица,
возбуждение волн изгиба в цепных структурах должно приво-
дить к появлению температурной области, в которой вслед за
областью Су~Т3 должна появляться область изменения Су~
~7’5/2. В ранних работах [3, 25] при анализе температурной
зависимости Cv для ПЭ был сделан вывод, что такая зависи-
мость как будто не проявляется. Однако затем Баур [28] про-
вел более детальный анализ поведения теплоемкости КПЭ вы-
ше 10 К с позиций этих теорий.
На рис. 1.8 теплоемкость КПЭ представлена в координатах,
'соответствующих уравнению (1.146) и позволяющих оценить
значение показателя у Т во втором слагаемом этого уравнения.
В узкой области 15—25 К теплоемкость изменяется в соответ-
ствии с 7'5/2, а затем переходит в область («40 К), в которой
С Г3/2. Существование этой последней области также пред-
сказывается по чисто геометрическим соображениям теорией
•СХ. Изменение теплоемкости в соответствии с T^2 не является
следствием геометрических особенностей и должно проявлять-
ся для фибриллярных кристаллов любой природы [11]. Конеч-
но, это температурные области не очень велики, и потому зна-
чения характеристических температур, определенных на осно-
вании экспериментальных значений 05/2 и 03/2 из рис. 1.8, ха-
рактеризуются низкой точностью. Однако они правильно пере-
дают порядок величин и качественно свидетельствуют о су-
ществовании температурных интервалов, в которых Су~Т3-[-
-|-7’5/2 и Су~Т3-\-Т3/2, предсказанных теоретически.
Вместе с тем следует отметить, что ширина температурных
интервалов, в которых наблюдаются указанные зависимости,
значительно отличается от рассчитанной на основании модели
•СХ при разумно выбранных силовых постоянных. Так, соглас-
но уравнению (1.36) 7'5/2-зависимость Cv должна наблюдаться
в интервале 5^ 7]^90 К, в то время как реально наблюдае-
мый температурный интервал такого изменения теплоемкости
значительно уже. Это же справедливо и для других областей
изменения теплоемкости. Таким образом, для реальной, а не
модельной кристаллической решетки эта теория позволяет
.лишь качественно представить температурную зависимость Cv.
Несмотря на наличие экспериментальных данных о темпе-
ратурной зависимости теплоемкости ниже 50 К для многих
кристаллических полимеров [3], особенно полученных в по-
следние годы Лебедевым с сотр. [29], их анализ с позиции из-
ложенных выше теоретических представлений затруднен двумя
принципиальными обстоятельствами. Все кристаллические по-
лимеры в действительности являются лишь частично-кристал-
лическими и наряду с кристаллической частью содержат и
аморфную. Раньше казалось, что закономерности изменения
теплоемкости кристаллических и аморфных веществ при низ-
ких температурах должны быть подобны, как это, например,
предполагается в теории Дебая. В таком случае нет большой
разницы в том, анализируется ли теплоемкость полностью или
частично-кристаллического полимера. Однако эксперименталь-
ные исследования последних двух десятилетий убедительно по-
казали, что эти закономерности различаются принципиально.
Поэтому перенос представлений, развитых для кристалличе-
ского состояния, на частично-кристаллическое может привести
лишь к эмпирическому описанию закономерностей. В связи с
этим далее сначала будут рассмотрены результаты для аморф-
ных полимеров, а затем проанализирована роль кристаллич-
ности в теплоемкости частично-кристаллических полимеров.
Как отмечено выше, сейчас можно считать надежно уста-
новленным экспериментальным фактом, что аморфные вещест-
ва, как органические, так и неорганические, даже при очень
низких температурах не подчиняются теории Дебая [30]. Это
является довольно неожиданным, так как при очень низких
температурах длина звуковых волн велика, и потому каза-
лось, что различий в поведении теплоемкости кристаллических
и аморфных диэлектриков не должно существовать. Тем не ме-
нее экспериментально установлено, что для многих аморфных
веществ характер изменения теплоемкости при низких темпера-
турах имеет вид, показанный на рис. 1.9. Видны три характер-
ные особенности изменения низкотемпературной теплоемкости
аморфных веществ: во-первых, существенно более высокие по
сравнению с дебаевскими значениями теплоемкости, т. е. су-
ществование избыточной теплоемкости во всей низкотемпера-
турной области; во-вторых, наличие широкого максимума; на-
конец, наличие систематической дополнительной составляющей
к дебаевскому вкладу при Т<1 К. Этот линейный вклад не-
значительно зависит от природы аморфного вещества и от
степени чистоты системы, однако наличие примесей все-таки
может влиять на теплоемкость, не оказывая, однако, при этом
существенного влияния на теплопроводность. Формально эту
зависимость можно описать следующим соотношением:
Су = С17’ + СзР + сЕ£(0Е/Т) (1.53)
где Ci, с3 и сЕ, 0е — константы, определяемые нз экспериментальных данных;
£(9е/7’)—функция теплоемкости Эйнштейна [см. уравнения (1.3)—(1.5)].
Физические причины появления этой зависимости в аморф-
ных веществах не вполне ясны. Существующие в настоящее
время предположения будут рассмотрены при описании экс-
периментальных данных для полимеров.
Ниже 1 К теплоемкость измерена лишь для нескольких
аморфных полимеров. В этой температурной области теплоем-
кость описывается первыми двумя членами уравнения (1.53).
В соответствии с этим экспериментальные результаты обычно
представляют в координатах С/Т от Т2. Для трех аморфных по-
29
1 — дебаевская теплоемкость; 2 — экспери-
ментально наблюдаемая теплоемкость
[30, 31}.
Рис. 1.10. Теплоемкость аморфных полимеров ниже 1 К [30, 31]:
1 — ПК; 2, 2' — ПС; 3, 3' — ПММА; 1,2, 3 — экспериментальные зависимости; 2'г 3' — де-
баевская теплоемкость, рассчитанная из акустических данных.
лимеров такие зависимости показаны на рис. 1.10. Значения
Cj находятся в пределах 4-10~6—5-10 6 Дж/(г-К2), а постоян-
ные с3 значительно превышают константу в формуле Дебая
(1.23), рассчитанную по акустическим данным (табл. 1.1).
Существуют два типа моделей, с помощью которых пыта-
ются объяснить появление линейно зависящей от температуры
избыточной теплоемкости в аморфных веществах [30, 31]. Мо-
дели первого типа предполагают наличие в дебаевской матрице
изолированных низкочастотных колебательных мод, что, в свою
очередь, связано с предположением об определенной гетероген-
ности стекла на молекулярном уровне. Примером модели та-
кого рода является модель, предполагающая существование
внутренних полостей в неупорядоченных системах [33]. Нахо-
дящиеся внутри или на поверхности этих полостей колеблю-
щиеся структурные элементы обладают большей амплитудой
(меньшей частотой) по сравнению с основной массой осцилля-
торов. Эти низкочастотные колебания вносят определенный
вклад в теплоемкость системы, но не вносят вклада в упругие
свойства матрицы, будучи в своем поведении достаточно изо-
Таблица 1.1. Параметры уравнения (1.53) для ПК, ПС и ПММА [36*]
Полимер ср10-5, см/с eD, к Ср*-10’, Дж,'(г-КО ci-10’, Дж/Ц-К2) сз-Ю’. Дж/(г-К4)
ПММА 1,79 256 177 48 292
ПС 1,67 223 262 53 457
ПК 1,53 210 284 38 410
• Средняя скорость звука в формуле Дебая (1.23).
'* Постоянная в формуле Дебая (1.23).
30
лированными от нее. Таким образом, в отличие от упорядо-
ченных кристаллических структур, в которых низкочастотные
колебания имеют только «акустическое» происхождение, в не-
упорядоченных системах вклад в низкотемпературную тепло-
емкость могут давать низкочастотные неакустические колеба-
ния, приводящие к появлению в них избыточной теплоемкости.
Идентифицировать природу таких осцилляторов нелегко.
Полагают, [33], например, что ими могут являться примесные
атомы или другие присущие неупорядоченным системам струк-
турные элементы. Расчеты показывают, что такие низкочастот-
ные неакустические колебания действительно должны давать
вклад в теплоемкость, линейно зависящий от Т, при линейных
размерах полостей на уровне 0,3—1,0 нм и при распределении
их по размерам, обратно пропорциональном кубу размеров. Та-
ким образом, эта модель по крайней мере качественно пред-
сказывает существование избыточной теплоемкости при очень
низких температурах, линейно зависящей от температуры.
К этой же группе моделей относится ячеечная модель не-
органических стекол [34], в которой предполагается существо-
вание ячеек (зерен) среднего диаметра d. Форма и размер зе-
рен описываются случайными функциями. Предполагается
также, что каждое зерно окружено тонкой оболочкой, жест-
кость которой может меняться от «мягкой» до «жесткой». При
низких температурах длина тепловых волн сравнима с d, и по-
тому необходимо рассматривать вклад в теплоемкость не толь-
ко сердцевины зерна, но и отдельно вклад оболочки. Расчеты
показывают, что теплоемкость оболочки является линейной
функцией Т, а теплоемкость зерен должна подчиняться обыч-
ному дебаевскому закону. Таким образом, и эта модель пред-
сказывает существование избыточной теплоемкости в неупоря-
доченных системах, линейно зависящей от Т ниже 1 К.
Еще одна модель этой группы предполагает наличие в стек-
лах локализованных объемов, в которых плотность отлична от
плотности матрицы [35]. Эти локализованные участки повы-
шенной плотности, объемы которых сравнимы с координацион-
ным объемом, могут мигрировать по «решетке». Появление
линейной по температуре избыточной теплоемкости ниже 1 К,
а также особенности поведения низкотемпературной теплопро-
водности объясняются существованием ловушек, в которых мо-
гут оказываться эти мигрирующие участки. Предполагается,
что доля мигрирующих уплотнений, попавших в ловушки, долж-
на зависеть от скорости охлаждения образца так, что чем мед-
ленее охлаждение, тем меньше уплотненных участков окажет-
ся в ловушках, что должно отразиться на поведении низкотем-
пературной теплоемкости. Предпринята недавно попытка [36]
обнаружить такие ловушки на основании измерения теплоемко-
сти охлажденного с различной скоростью ПЭТФ в области
0,8—5 К не привела к успеху, хотя ниже 1 К для образцов,
31
охлажденных с различной скоростью, наблюдалось появление
характерного подъема на кривой зависимости С/Т3 от Т.
Таким образом, хотя модели первой группы предсказывают
появление дополнительного линейного вклада в теплоемкость,
они содержат параметры, которые трудно поддаются экспери-
ментальному определению (природа и число низкочастотных
осцилляторов, размеры зерен и оболочки или участков повы-
шенной плотности и их распределение).
Другая группа моделей основана на предположении о су-
ществовании энергетического распределения локализованных
резонансных колебательных мод. Наиболее распространенной
моделью этой группы является модель туннельного перехода
под действием решеточных фононов [37, 38]. В этой модели,
учитывающей фундаментальную особенность стеклообразного
состояния вещества — его неравновесность, предполагается,
что морфология неупорядоченных систем обеспечивает воз-
можность нахождения отдельных атомов или групп атомов в
нескольких квазиравновесных положениях, мало различающих-
ся энергетически (см. рис. 2.4), что практически не изменяет
внутренней энергии системы. Эти квазиравновесные положения
разделены энергетическим барьером, который при низких тем-
пературах атомы преодолеть не могут, однако под действием
решеточных коелбаний возможен туннельный переход из одно-
го положения в другое. Для объяснения появления линейного
вклада в теплоемкость при низких температурах достаточно
предположения о существовании локализованных колебатель-
ных мод с несколькими дискретными энергетическими уровня-
ми, а предположение о туннелировании позволяет объяснить
некоторые особенности поведения теплопроводности и акусти-
ческие свойства неупорядоченных систем. В настоящее время
двухуровневая резонансная модель наиболее широко использу-
ется для объяснения температурного хода избыточной тепло-
емкости аморфных веществ любой природы при сверхнизких
температурах*.
* В самое последнее время (Reynolds С. L.—J. Non-Crystal. Solids, 1979,
vol. 30, N 3, p. 371—374; 1980, vol. 37, N 1, p. 125—127; Raychaudhuri A. K„
Pohl R. O. — Solid State Comm., 1981, vol. 37, N 2, p. 105—108) эксперимен-
тально установлено, что между избыточной теплоемкостью, характеризуемой
параметром С] в уравнении (1.53), и температурой стеклования существует
четкая корреляция: значение С] уменьшается с повышением Тс. Это несколько
противоречит картине, которую можно было бы ожидать из модели туннели-
рования. Действительно, большая степень разупорядоченности, характерная
для стекол с большей Тс, должна была бы приводить к большей доле атомов
или групп атомов, способных к туннельному переходу. Возможно, что темпера-
тура стеклования определяет шкалу энергий низкотемпературных возбужден-
ных состояний, так что при одном и том же числе возбужденных состояний в
стекле с более высокой Тс они распределены в более широкой области энергий.
Это должно приводить к более низкой плотности состояний. Если эта законо-
мерность окажется характерной для широкого класса стекол, она будет важ-
ным звеном в представлениях о физической природе криогенной аномалии теп-
лофизических свойств стекол.
32
Рис. 1.11. Зависимость С/Т3 от Т для ПЭТФ различной степени кристаллично-
сти [41]:
1 — 2%; 2—17%; 5 — 41%; 4 — 59%; пуктирными линиями отмечены дебаевские значения,
рассчитанные из акустических данных; сплошные линии соответствуют уравнению
-(Cvir)D + \NkEE^ 1/Тз (см. табл. 1.2).
Рис. 1.12. Зависимость С/Т3 от Т для образца ПЭТФ со степенью кристаллич-
ности 41 % [41]:
/ — дебаевский вклад (см. табл. 1.2); 2 — линейный вклад [~ ^у^.см. уравнение (1.53)1;
3 — эйнштейновский вклад; 4 — зависимость, соответствующая уравнению (1.53).
Cv 58 7,2• 103£( 15/ Т)
—? = 382 + — + ---------J— 1 [ X 10"5Дж/(г- К4)]
Наличие аморфных областей в частично-кристаллических
полимерах должно приводить к появлению линейного вклада
в теплоемкость и этих полимеров, причем величина этого вкла-
да должна зависеть от степени кристалличности. Результаты
измерений ниже 1 К опубликованы лишь для ПЭ '[39]. Изуча-
лись изотропный и экструдированный образцы сверхвысокомо-
лекулярного ПЭ (степень кристалличности ^40%). Хотя ниже
0,2 К наблюдалось некоторое различие в теплоемкости изо-
тропного и экструдированного образцов, выше 0,2 К их тепло-
емкости совпадали. Ниже 1 К Cv обоих образцов описывается
двумя первыми членами уравнения (1.53). Однако эти данные
имеют лишь качественный характер.
Важные сведения о поведении избыточной теплоемкости бы-
ли получены при изучении влияния степени кристалличности
на теплоемкость при температурах выше 1 К [25, 40, 41]. Из
этих исследований особенно ценными представляются резуль-
таты, полученные для ПЭТФ [41], так как в них в достаточно
широком интервале проварьирована степень кристалличности,
а дебаевская теплоемкость рассчитана из независимых акусти-
ческих данных. Из рис. 1.11 отчетливо видно, что для всех ис-
следованных степеней кристалличности экспериментальные
значения Cv выше рассчитанных из формул Дебая, что свиде-
тельствует о существовании избыточной теплоемкости во всех
образцах. Пик на кривой зависимости С/Т3 от Т с увеличением
> 3-440 я.
Таблица 1.2. Параметры функций, описывающих низкотемпературную
теплоемкость ПЭТФ [4/]
Степень кристал- личности образ- ца W, % Су/ГЗ)п.1О5, Дж/(г-К4) еЕ. к ЛМ01», 1—1 N .101», 1 I— UI
2 4,21 15 10,4 10,6
17 4,05 15 9,6 11,6
41 3,82 15 5,2 8,8
59 3,64 12 1,01 2,5
степени кристалличности уменьшается и смещается в область
более низких температур. На всех экспериментальных кривых
видна также тенденция к линейному возрастанию Cv при тем-
пературе ниже примерно 1,5 К. Для образцов с низкой сте-
пенью кристалличности избыточная теплоемкость хорошо ап-
проксимируется эйнштейновской функцией теплоемкости с па-
раметрами, указанными в табл. 1.2. Число осцилляторов N
и характеристическую температуру 0ц выбирали таким обра-
зом, чтобы наилучшим образом описать максимумы. Для это-
го, как видно из табл. 1.2 и рис. 1.11, достаточно одного эйн-
штейновского слагаемого с 0е=15 К- Таким образом,
максимумы на кривых образцов с низкой степенью кристал-
личности хорошо представляются последним членом в уравне-
нии (1.53).
Поведение образцов со степенями кристалличности 41 и
59% удается описать, лишь предположив, что подъем на кри-
вой при ТС 1,5 К отражает появление линейного вклада при
понижении температуры, рассмотренного выше (рис. 1.12). Од-
нако вклад линейного члена в общую теплоемкость в этой обла-
сти температур невелик, и потому точность определения ко-
эффициента Су низка. Поведение образца с самой высокой сте-
пенью кристалличности удается описать, лишь предположив
более низкое значение 0е=12 К. Число низкочастотных осцил-
ляторов пропорционально доле аморфной фазы, однако для
образца со степенью кристалличности 59% оно существенно
меньше. Это, а также более низкое значение 0е для этого об-
разца свидетельствует, по-видимому, о влиянии степени крис-
талличности на теплоемкость аморфной фазы. Такое влияние
проявляется и в области стеклования (см. далее). Примеча-
тельно, что с повышением степени кристалличности влияние
линейного вклада проявляется при более высоких температу-
рах. Как отмечалось выше, для аморфных полимеров линейный
вклад в избыточную теплоемкость проявляется ниже 1 К, а из
рис. 1.11 и 1.12 следует, что повышение степени кристаллично-
сти как будто сдвигает начало подъема кривых С/Т3 — Т в об-
ласть более высоких температур. Однако это наблюдение не
очень согласуется с данными о ПЭ, для образца которого со
34
р"
f степенью кристалличности «40% линейный вклад в теплоем-
‘ кость начинает сказываться ниже 1 К-
; Показанное на рис. 1.11 изменение теплоемкости в зависимо-
сти от степени кристалличности наблюдается также для ПЭ
'[25] и для оксациклобутена [40], что позволяет считать его
достаточно характерным для частично-кристаллических поли-
' меров.
Для объяснения появления широкого пика избыточной теп-
лоемкости выше 1 К обычно используют те же гипотезы, что и
для объяснения линейного вклада ниже 1 К- Так, поскольку
формально избыточную теплоемкость в этой температурной об-
ласти удается описать эйнштейновской функцией теплоемко-
сти со значением 0е=12—20 К, то в ранних работах (напри-
мер, [42, 43]) широко использовалась идея Розенштока о лока-
лизации вблизи пустот низкочастотных неакустических колеба-
ний, причем в качестве таких низкочастотных осцилляторов в
полимерах рассматривались боковые и концевые группы или
разветвления. Однако такая трактовка этой аномалии теплоем-
кости осложняется рядом факторов. Так, указанные выше раз-
меры пустот могут быть меньше размеров боковых групп (на-
пример, фенильной группы в ПС) или разветвлений. Получае-
мые значения 0е слишком низки для возбуждения колебаний
таких групп, как фенильная. К тому же, сам автор этой кон-
цепции полагает, что она лучше объясняет наличие линейного
вклада в теплоемкость при температурах ниже 1 К-
Исходя из представлений о пространственной сетке зацеп-
лений, предполагали также [44], что одномерными осциллято-
рами могут служить целые последовательности в несколько де-
сятков звеньев, заключенные между двумя ближайшими за-
цеплениями в линейных макромолекулах. Хотя такое представ-
ление также формально позволяет объяснить избыточную теп-
лоемкость органических и неорганических стекол как бы с
единых позиций — в полимерах она обусловлена дискретным
спектром одномерных протяженных участков цепей, а в неор-
ганических стеклах наличием небольших упорядоченных обла-
стей, — однако неясно, в какой мере эти представления соот-
ветствуют реальной физической картине. Еще одна, более
частная гипотеза основана на представлении о появлении до-
полнительной к дебаевским колебательной моды, обусловлен-
ной кооперативным вращением объемистых боковых групп ти-
па бензольного кольца в ПС или атомов фтора в ПТФЭ [45].
Более общее представление основано на результатах изу-
чения фононных спектров стекол методом рассеяния нейтронов
[32]. Оказалось, что пик на зависимости С/Г3—Т, например,
для стеклообразного SiO2 является следствием смещения пи-
ка, характеризующего поперечные акустические колебания в
колебательном спектре аморфного стекла, в область более низ-
ких частот по сравнению с кристаллическим состоянием. Для
кристаллического состояния этот пик находится в области
3‘ 35
70 см-1, а для аморфного — в области 40 см-1. Указанное сме-
щение связывают с более низкой плотностью аморфного стек-
ла. Полагают, что такое поведение должно быть вообще ха-
рактерно для стеклообразного и кристаллического состояния
одного и того же вещества. Таким образом, согласно этим дан-
ным, избыточная теплоемкость стекла по сравнению с крис-
таллом — следствие трансформации плотности распределения
низкочастотных фононов в кристаллическом состоянии в резуль-
тате потери дальнего порядка и уменьшения плотности.
В связи с этим уместно вернуться к рис. 1.8 и рассмотреть
с учетом изложенного выше характер поведения теплоемкости
КПЭ и АПЭ при более высоких температурах и переход в ту
область температур, где их теплоемкости совпадают. Если ис-
ходить из изложенной идеи, то различие между теплоемкостя-
ми кристаллического и аморфного полимера должно быть свя-
зано с межмолекулярной (разные плотности) составляющей
теплоемкости, а . скелетная (внутримолекулярная) составляю-
щая должна быть единой для аморфного и кристаллического
состояния. Действительно, как ранние оценки [42], так и бо-
лее поздние расчеты [46] показывают, что все изменения теп-
лоемкости при переходе от КПА к АПЭ можно удовлетвори-
тельно описать, лишь изменяя температуру 0з, характеризую-
щую межцепное взаимодействие. Так, в модели Тивери и др.
[26, 46] колебательный спектр имеет вид
р(о>) =pi(a>) + 2рДсв) (1.54)
где
р (св) ~ <в2 (0 < <в <в3)
1
Р (М)--Т~2 (“з < ® “1I,t)
Здесь ton, t соответствует продольным и поперечным колебани-
ям с различными значениями tou и ton, а 0з=п:<оз/&ь- Для КПЭ
при постоянных для обеих систем значениях 9ц —205 К и 9ц =
= 350 К величина 03кпэ = 90 К, а для АПЭ значение 0алпэ =
= 50 К- С помощью этих значений удается удовлетворительно
описать низкотемпературную теплоемкость не только КПЭ и
АПЭ, но и образцов с промежуточной степенью кристаллично-
сти. Такое изменение параметра 03 свидетельствует о смещении
только низкочастотной части колебательного спектра в область
более низких частот при уменьшении плотности.
По мере повышения температуры различия между колеба-
тельными спектрами КПЭ и АПЭ нивелируются, и при 50 К
оба спектра становятся практически идентичными, так как до-
минирующую роль начинает играть спектр внутримолекуляр-
ных колебаний. Таким образом, ясно, что идея о трансформа-
ции низкочастотной части фононного спектра кристаллов при
их аморфизации является достаточно универсальной для объ-
яснения закономерностей изменения теплоемкостей стекол вы-
36
Рис. 1.13. Зависимость теплоемкости ли- Дж/(мом-к')
иейных полимеров от температуры [19]: t20
1 — ПММА; 2 —ПС; 3 — ПВДХ; 4— ПТФЭ;
5 — ПВО; 6 — ПЭ.
80
ше 1 К. Однако эта идея явля-
ется весьма плодотворной и при
объяснении линейного вклада в
Cv ниже 1 К [34, 47, 48]. Так, о
Такено и Года [48], базируясь
на теории теплоемкости жидкого гелия, развитой Ландау,
предположили, что во всех системах с пространственной разу-
порядоченностью в низкочастотной области спектра плотность
возбужденных состояний выше, чем в кристалле, вследствие
появления ротонов, что, в свою очередь, приводит к появлению
аномалий в тепловых свойствах некристаллических систем при
низких температурах.
Расчеты показали, что избыточная теплоемкость может
быть представлена двумя слагаемыми, одно из которых ока-
зывается линейно зависящим от Т, а другое зависит от Т слож-
ным образом. При очень низких температурах доминирующим
может стать первый член, что приводит к линейной темпера-
турной зависимости Су*136. При более высоких температурах
вклад в Су1136 вносят оба слагаемых. Хотя поведение второго
члена детализировать сложно, его наличие создает основу для
количественного анализа проблемы избыточной теплоемкости
аморфных тел во всей температурной области. Пока же ни од-
на из существующих гипотез и моделей не имеет достаточно
универсального характера.
Как видно из рис. 1.7, выше 50 К теплоемкость постепенно
перестает зависеть от особенностей межмолекулярного взаи-
модействия, что свидетельствует о доминирующей роли в теп-
лоемкости внутримолекулярного теплового движения, обуслов-
ленного подвижностью скелета цепи и боковых радикалов.
Многочисленные экспериментальные исследования теплоемко-
сти линейных полимеров [3, 19, 29] показывают, что зависи-
мости Су—Т выше «50 К для полимеров достаточно едино-
образны (рис. 1.13) и в этой области температур всегда можно
выделить участок, на котором изменение теплоемкости близко
к линейной. Однако эта линейность не является следствием вы-
полнения теории Тарасова, как это часто предполагается в экс-
периментальных работах, хотя бы по формальному признаку:
эти линейные зависимости, как правило, не экстраполируются
в Су = 0 при 0 К, как того требует уравнение (1.31), а обычно
отсекают положительный отрезок на оси СР. Было убедитель-
но показано, что эта линейность появляется в результате нало-
жения нелинейно изменяющихся с температурой вкладов от
низкочастотных составляющих колебательного спектра скелета
и боковых групп [18, 19]. В связи с этим, естественно, что она
37
Таблица 1 3. Характеристики колебаний групп CF? и СН2 [4, 5/]
Тип колебаний Группа CF, Группа СНз
средняя ча- стота, СМ”1 характери- стическая температу- ра 6jg, К средняя ча- стота, см-1 характери- стическая температу- ра 0£, К
Крутильные 289 416 1173 1688
Ножничные 413 594 1433 2062
Маятниковые 430 619 1022 1471
Веерные 611 879 1394 2006
Валентные симметричные 975 1403 2973 4278
Валентные асимметрии- 1350 1943 2998 4314
ные С—С-Валентные 1059 1524 1059 1524
не может служить критерием правильности модельных расче-
тов, относящихся к скелету полимерной цепи. Для проведения
такого сравнения из общей теплоемкости необходимо выделить
теплоемкость, обусловленную колебательным спектром скеле-
та полимерной цепи.
В простейшем полимере — ПЭ — колебания групп СН2 до-
статочно высокочастотны (табл. 1.3) и, как уже отмечалось,
вносят небольшой вклад (несколько процентов) в теплоемкость
в области температур 100—150 К. Это дает возможность рас-
смотреть экспериментальную зависимость теплоемкости ПЭ от
температуры непосредственно с учетом подвижности лишь ске-
лета цепи. Наиболее детально этот вопрос рассматривался в
работах [18, 19, 23, 25, 28, 42].
В температурном интервале 100^7'^210 К теплоемкость
КПЭ может быть представлена прямой 2 (см. рис. 1.7) [28]:
lgCy = 0,751g Г-0,54 (1.55)
Наклон прямой 2 составляет 3/4, что близко к величине V2,
вытекающей из теории Лифшица, и необъяснимо с позиций
теории Тарасова, согласно которой значение этого параметра
вообще не может быть меньше 1. Выводы теории Лифшица на-
ходят также экспериментальное подтверждение для цепных и
слоистых структур неорганических соединений [49, 50]. Широ-
кая экспериментальная проверка этой теории для органических
полимеров до сих пор, к сожалению, не проводилась, что отча-
сти связано с отсутствием необходимых значений констант [см.
уравнения (1.41) — (1.43)]. В температурном интервале 100^
^7’^190 К зависимость Cv—Т может быть представлена так-
же уравнением
Су = а^Т -f- а1/(1.56)
со значением ai = 0,045 Дж/(моль-К2) и «1/2 =
= 0,496 Дж/(моль-К3/2). По форме уравнение (1.56) как будто
соответствует уравнениям (1.14), (1.41) и (1.50), полученным
38
Рис. 1.14. Температурная зависимость г д
теплоемкости ПТФЭ [18, 52]: fig
I Сопт; 2 Сак; 3 С=Сак+Сопт; 4 экспе-
риментальные значения С
при учете всех колебательных 60
ветвей цепной макромолекулы,
член этих уравнений со-
ответствует продольным колеба-
ниям, а второй — поперечным.)
Однако соотношение между ко- М
эффициентами свидетельствует о
том, что это соответствие лишь
формальное, так как продольные °
колебания в рассматриваемой
области температур не возбуждены, а согласно (1.56) оба чле-
на имеют одинаковый порядок.
Для цепей более сложного строения, чем ПЭ, прямое срав-
нение экспериментальной зависимости Cv—Т с теоретическими
уравнениями, характеризующими скелетную теплоемкость, не-
возможно: для этого необходимо оценить вклад нескелетных ко-
лебаний. Рассмотрим эту процедуру на примере теплоемкости
ПТФЭ [18, 51, 52]. В связи с тем что масса атома фтора боль-
ше массы атома водорода, даже при низких температурах кро-
ме скелетных крутильных и деформационных акустических ко-
лебаний необходимо учесть ряд оптических колебательных мод.
Характеристики этих оптических колебаний приведены в табл.
1.3. При расчетах теплоемкости необходимо учитывать также
влияние одних колебательных мод на другие: например, влия-
ние маятниковых колебаний на крутильные. Рис. 1.14 демонст-
рирует вклад всех оптических колебаний в общую теплоемкость
ПТФЭ. В области 60 К нх вклад составляет около 10%, а в об-
ласти 160 К — около 50%. Полагают [18], что такое соотноше-
ние характерно для большинства линейных полимеров с тяже-
лыми боковыми атомами и группами атомов. Для боковых ра-
дикалов сложного строения необходимо учитывать возможность
заторможенного внутреннего вращения [19, 53].
В температурном интервале 60—150 К приведенные на рис.
1.14 экспериментальные значения С можно удовлетворительно
аппроксимировать линейной зависимостью. Это наглядно под-
тверждает сделанное выше заключение, что такая линейность
является обычно случайной и появляется вследствие наложения
нелинейно изменяющихся с температурой отдельных вкладов.
Выделенную описанным выше методом скелетную теплоем-
кость, как правило, анализируют в терминах двухпараметриче-
ского уравнения (1.36). Процедура сводится к подбору пара-
метров 0з и 0ь значения которых наилучшим образом описыва-
ют изменение теплоемкости в определенном температурном ин-
39
тервале. Это обычно удается сделать с точностью «10%. За-
тем этим параметрам Оз и 01 придается смысл характеристиче-
ских температур, который они имеют в теории Тарасова, и де-
лается заключение, что эта теория хорошо описывает теплоем-
кость цепных молекул. Часто 03 и 01 дифференцируют (03/ и
9з/, Ок и Оп), что позволяет иногда улучшить аналитическое
описание экспериментальных результатов. Однако представля-
ется вполне очевидным, что такой прием является обычным
эмпирическим подбором двух (или четырех) переменных пара-
метров, с помощью которых удается описать эксперименталь-
ные данные. Поэтому полученные при этом формулы можно рас-
сматривать лишь как эмпирические соотношения. Однако де-
лать какие-либо заключения о физической реальности модели
Тарасова на основании такого приема нельзя.
ИЗМЕНЕНИЕ ТЕПЛОЕМКОСТИ ПРИ СТЕКЛОВАНИИ
Аморфные полимеры
Переход полимера из стеклообразного состояния в высокоэла-
стическое, так . же как переход низкомолекулярного стеклооб-
разного вещества в жидкое состояние, сопровождается резким
(скачкообразным) увеличением теплоемкости Ср. В ранних ис-
следованиях на основе дырочной теории жидкостей предпола-
галось [54], что АСр обусловлено главным образом образовани-
ем новых дырок выше Тс- Принципиальное ограничение этого
подхода связано с тем, что ускорение релаксационных процес-
сов и изменение свойств при температуре стеклования, в част-
ности возрастание теплоемкости, должно наблюдаться и в ус-
ловиях постоянного объема. Поэтому изменение теплоемкости
при стекловании обусловлено не только увеличением дырок, но
и другими эффектами. Согласно современным представлениям
изменение теплоемкости является суммой трех основных вкла-
дов в теплоемкость, появляющихся в результате перехода по-
лимера из твердого агрегатного состояния в жидкое '[55, 56]:
АСр = ДО Н- дел 4- ДС™-1 (1.57)
Здесь ДСК представляет собой конформационную теплоем-
кость, возникающую вследствие перехода замороженных ниже
Тс низкоэнергетических конформаций в конформации с более
высокой энергией; АСД отражает вклад в теплоемкость, связан-
ный с размораживанием свободного объема и возникновением
выше Тс дополнительного числа дырок. При переходе через Тс
наряду с Ср резкие изменения наблюдаются и в других термо-
динамических характеристиках; в частности, скачкообразно из-
меняются тепловое расширение и сжимаемость, что должно при-
водить к изменению колебательной теплоемкости как вследст-
вие изменения частоты колебаний так и (особенно) увеличения
ангармоничности колебаний. Последний член в уравнении
АЛ
<(1.57) отражает изменения частоты и амплитуды колебаний
структурных единиц, т. е. суммарные изменения в колебатель-
ном спектре. Корректное определение соотношений между эти-
ми вкладами в скачок теплоемкости при Тс является сложной
теоретической и экспериментальной задачей. Ниже будут рас-
смотрены основные подходы к определению отдельных слагае-
мых в уравнении (1.57).
Согласно концепции свободного объема изменение теплоем-
кости при стекловании связано с резким увеличением концент-
рации дырок при размораживании подвижности [54]. Ниже TQ
число дырок постоянно, и теплоемкость стеклообразного поли-
мера определяется лишь колебательным спектром. Дырочная
теплоемкость определяется выражением
= (158>
\ 01 I р
где Ед — мольная энергия дырок, определяемая как разность мольных энергий
тела, содержащего и несодержащего дырки; NK — число молей дырок.
Предполагается, что равновесное распределение дырок под-
чиняется уравнению Больцмана
Л\ = Л;1(К)/Уд)'-£д/?7' (1.59)
где jV0 — число молей повторяющейся структурной единицы; Р'о— их объем;
Ед — мольный объем дырок.
Совместное рассмотрение (1.58) и (1.59) дает при Тс
ЛСД = /?(Е0/Ед) (Ед//?7’с)2е--£д/етс (1.60)
Использовав приемлемые значения соотношений У0/Уд~
^Е01Е^ = Ь—6 (Ео — энергия когезии), Вундерлих получил сле-
дующее соотношение:
ДС'р = МЛСр = 12,4 Дж/(моль-К) (1.61)
где М — молекулярная масса «бусинки» — наименьшего структурного элемен-
та, способного перемещаться как единое целое.
Анализ экспериментальных значений АС'Р для нескольких
десятков полимерных стекол показывает [54, 57, 58], что ЛС'р =
= 11,5±2 Дж/(моль-К). Эта закономерность получила название
«правило постоянного скачка теплоемкости при стекловании».
Несмотря на существующую до сих пор неоднозначность опре-
деления числа бусинок в повторяющихся звеньях многих мак-
ромолекул, это правило является важным обобщением и рас-
сматривается как одна из важнейших характеристик этого ре-
лаксационного перехода.
Большинство макромолекул характеризуется наличием не-
скольких поворотных изомеров, энергии которых различаются
на 0,5—8 кДж/моль <[59, 60]. Наиболее характерно существо-
вание одного низкоэнергетического транс-изомера и двух гош-
изомеров, обладающих большей энергией. Если соотношение
41
между изомерами изменяется при повышении температуры, то
в результате этого перехода появляется дополнительный вклад
в теплоемкость, называемый «конформационной теплоем-
костью». Конформационная теплоемкость цепи с простейшими
боковыми группами может быть рассчитана из теории Гиббса—
Ди-Марцио [61, 62]. Согласно этой теории, доля гибких связей
(fT), находящихся в высокоэнергетическом гош-состоянии, оп-
ределяется выражением [59]
/г = 2 ехр( - EK/kBT)/[1 + 2ехр( - EK/kBT)F (1.62)
где Ек = Егои1 Етранс.
Теплоемкость системы, для которой характерно существова-
ние двух энергетических состояний, может быть рассчитана ив
следующего уравнения, вытекающего из статистического ана-
лиза таких систем '[3]:
Ск и R{EK/W 2ехР(.-£к/^В__ (1.63)
' к/ Б ’ [1 + 2ехр(-£кМБГ)Р 1 7
Из (1.62) и (1.63) следует
Сх = 7?(£я/АБТ)2/Г(1-/Г) (1.64)
Конформационная теплоемкость для гош-конформаций как
функция Exlkb Т показана на рис. 1.15. Появление максимума
связано с изомеризацией при повышении температуры. Темпера-
тура, при которой наблюдается максимум, зависит от Ек и с
повышением £к от 0,5 до 8 кДж/моль возрастает примерно с
25 до 200 К.
При каких температурах можно ожидать проявления кон-
формационной теплоемкости в реальных условиях? В стекло-
образном состоянии различные изомеры заморожены, и переход
между ними ниже Тс невозможен по кинетическим соображе-
ниям, и потому этот вклад в теплоемкость в твердом состоянии
обычно не реализуется. Действительно, как следует из изложен-
ного выше, на температурной зависимости теплоемкости аморф-
ных полимеров ниже Тс обычно отсутствуют аномалии, которые
можно было бы идентифициро-
вать как проявление конформа-
ционной теплоемкости. Сегмен-
тальная подвижность размора-
живается при Тс, и это размо-
раживание сопровождается кон-
Рис. 1.15. Зависимость конформационной
теплоемкости модели с двумя энергетиче-
скими состояниями (транс- и гош-) (1)
и доля связей (2) в высокоэиергетиче-
ском состоянии (гош-) от отношения
EK/k б Т (Ек — разность энергий в гош-
и rpawc-состояннях).
42
формационными переходами. Если предположить, что дыроч-
ная составляющая теплоемкости в условиях постоянного объе-
г ма отсутствует, то простейшее предположение состоит в том,
г что весь скачок теплоемкости при постоянном объеме АСу в
' области стеклования является следствием резкого изменения
i числа гош- и транс-конформаций [63—65]. Таким образом, эта
гипотеза базируется на предположении, что слагаемое АСКОЛ
в уравнении (1.57) вносит существенно меньший вклад в из-
менение теплоемкости по сравнению с АСК. Изменение теплоем-
кости при постоянном объеме АСу можно рассчитать из термо-
динамического уравнения
CP=Cv + TV(a?/Ko6) (1.65)
где а — термический коэффициент объемного расширения; хое — сжимаемость;
V — удельный объем.
При температуре стеклования имеем:
(о
«ж
= ДСрТс —Тс^сД ( —I
\ ^об /
(1.66)
Здесь индексы «ж» и «т» относятся соответственно к жидкому
и твердому состоянию выше и ниже Тс. Произведение
ТсКА/—I согласно (1.57) и (1.66) характеризует вклад сво-
^об/
бедного объема (дырок) в изменение теплоемкости в области
стеклования.
Согласно основной гипотезе
R(EK/ksTy-fr(l -/г) - ДСр(Тс) -TcVcA(a?/*o6) (1.67)
Уравнение (1.64) и, следовательно, уравнение (1.67) полу-
чены для цепей с простыми боковыми группами, такими, как Н,
СН3, С1, (СНз)г. Если боковые группы также способны нахо-
диться в нескольких энергетически неэквивалентных конфор-
мациях, то выражение для конформационной теплоемкости мож-
но представить в более общем виде [64]:
/ Е • V
Ск = да (1.68)
i W /
, gi ехр(—EKi/kbT)
где ] _|_ g. ехр^_£ -/kyr}’ ^Ki~энеРгии соответствующих конформации
связей; gt — их статистический вес.
Рое и Тонелли [55] при определении конформационной теп-
лоемкости полимеров также исходили из поворотно-изомерной
модели полимерных цепей. Их расчеты имеют более общий ха-
рактер, поскольку базируются на обобщенном определении ста-
тистической суммы полимерной цепи ZK методами статистиче-
43
ской механики цепных молекул [59, 60], На основании ZK кон-
формационная энергия может быть определена по уравнению
£к = In ZJdT (1.69)
а конформационная теплоемкость затем рассчитана по формуле
CK = d£K/dT (1.70)
По этой схеме численным методом с использованием числен-
ных значений параметров матриц статистических весов связей,
доступных для многих полимеров [60], были рассчитаны кон-
формационные энергии большого числа макромолекул при тем-
пературах на 50 °C выше и ниже Тс, на основании которых бы-
ли определены конформационные теплоемкости в области стек-
лования (табл. 1.4). Обсуждение этих результатов будет прове-
дено далее.
Рое и Тонелли в отличие от О’Рейли полагают, что разность
АСр—АСу=#ДСд, т. е. она характеризует не только вклад свобод-
ного объема. В стеклообразном состоянии при повышении тем-
пературы в условиях полного постоянного объема постоянным
сохраняется как свободный, так и занятый объем. В жидкости
при полном постоянном объеме между свободным и занятым
объемами может происходить перераспределение, минимизирую-
щее свободную энергию жидкости. Поэтому значение ДО из
экспериментального значения ДСр можно надежно получить,
лишь рассчитав ДСу.у , т. е. сохраняя постоянным одновремен-
но и полный, и свободный объемы. Экспериментально удовлет-
ворить это требование нельзя. Предполагается, что значение
АСур- может быть рассчитано по уравнению
(Да)2
ACvV4 = ACp-TV1—- (1.71)
’ АКоб
вытекающему из дырочной теории, развитой Ноузом |[66]. Та-
кое же уравнение было получено в работе [67]. Танака [68]
использовал для расчета ДСР предложенное им выражение для
конфигурационной статистической суммы, учитывающей кон-
формационную составляющую и составляющую, связанную со
свободным объемом, и получил следующее выражение:
(1-72)
где Z«(T) — конформационная статистическая сумма изолированной цепи при Т;
Ед — свободный объем, приходящийся на одну структурную единицу; п — сте-
пень полимеризации: Но — межмолекулярная энергия когезии на моль струк-
турных единиц; первый и второй члены в правой части этого уравнения харак-
теризуют соответственно вклады ДСК и ДСд, а два последних члена появляются
вследствие изменения колебательной теплоемкости, т. е. соответствуют ДСКОЛ.
<
а
В скобках указаны значения ДСр в %.
Значение ДСр, использованное в работе Рое и Тонелли [551.
44
Наиболее полная теория скачка теплоемкости при стеклова-
нии полимеров, учитывающая все три составляющих в уравне-
нии (1.57), развита недавно Ди Марцио и Даулом (ДД) [56].
Рассмотрим ее более подробно. При построении этой теории ис-
ходили из статистической теории стеклования Гиббса—Ди-Мар-
цио [61, 62]. Как в первоначальном, так и в более совершенном
варианте этой теории предполагается, что колебательный
спектр не зависит от конформационного состояния, т. е. вкла-
ды колебательных степеней свободы в термодинамические ха-
рактеристики одинаковы выше и ниже Тс- Это соответствует
предположению, что конфигурационная и колебательная стати-
стические суммы являются независимыми. Теория Гиббса—Ди-
Марцио предсказывает лишь конфигурационно зависимые свой-
ства полимерных стекол, определяемые конфигурационной ста-
тистической суммой, а свойства, определяемые колебательной
статистической суммой, в этой теории игнорируются. По этой
причине теплоемкость, тепловое расширение и сжимаемость по-
лимерных стекол становятся равными нулю ниже температуры
Т2 (температура гипотетического фазового перехода второго ро-
да), а скачок теплоемкости ДСР при стекловании включает
лишь конформационный и дырочный вклады. В действительно-
сти ниже Тс указанные характеристики определяются колеба-
тельными модами, и, кроме того, они вносят определенный
вклад в них в жидком состоянии. Учитывая эту ограниченность
теории Гиббса—Ди-Марцио, в теории ДД наряду с конфигура-
ционной статистической суммой вводят и анализируют колеба-
тельную статистическую сумму. Полную статистическую сумму
можно представить произведением
Z(T, Р, Nr) =^e^(-PV/kBT)ZK(T, fr, n0)Zm(T, fr, n0) (1-73)
/г«0
где no — число дырок (свободных мест на решетке); Z и — колебательная ста-
тистическая сумма; Nr — число макромолекул г.
Суммирование проводится по всем энергетическим уровням,
по числу дырок и гибких связей (fr), а также по объему. Пред-
полагается, что каждая конфигурация системы с одними и те-
ми же значениями fr и по характеризуется одинаковым колеба-
тельным спектром. Использование статистической суммы в виде
произведения отражает два важных момента, упоминав-
шихся выше: ниже Тс значения fr и п0 постоянны, и термодина-
мические свойства определяются лишь Za>, а выше Тс они опре-
деляются как конфигурационными, так и колебательным
вкладами. Для Zo) использовано приближение Эйнштейна:
46
Предполагается, что со в этом выражении зависит только от
объема вследствие изменения п0 и не зависит от fr. Эта зависи-
мость имеет вид соотношения Грюнайзена (см. гл. 2):
О < Т <С00
Ve(T)
(1-75)
; Ус — объем ячейки решетки;
у — постоянная Грюнайзеиа; у'=0,5.
Уравнения (1.74) и (1.75) позволяют одновременно опреде-
лить и PVT, и EVT уравнения состояния и полностью охаракте-
ризовать термодинамическое состояние системы.
Минимизация термодинамического потенциала
G = k5T In Z = G(T, Р, Nr, fT, n0, Vc)
(1-76)
по всем параметрам позволяет вычислить энтропию системы
выше и ниже Тс, а затем и теплоемкости. Искомые выражения
для теплоемкости выше и ниже Тс имеют вид
(1-77)
где
Ср = Ср0Л( 1 -f- 'pT’ojc -f- у'Т&а) Ср Т Тс
Здесь So = zn0,/[ [(г— 2) г + 2] Л\ -f- znQ}, где z— координационное
число решетки; Sr==l—So; оо=1—ог.
Ср=Сколр(1+?7’ас) 7’<ТС
(1.80)
Из этих выражений можно получить искомую величину скач-
ка теплоемкости при стекловании:
ЛСр = у'С^рТ’да + Скр
(1-81)
Полный скачок теплоемкости состоит из двух компонентов —
член, содержащий Скол₽, отражает изменения в колебательном
спектре при стекловании, а член Cvp — конфигурационные из-
47
менения (конформационные и дырочные). Уравнение (1.65) по-
зволяет определить теплоемкости при постоянном объеме:
СуЛ = С“°л (1.82)
= Ср0Лj/г(1—/г) (1.83)
\«Б-< /
ДСу = /?(^ЬУ7г(1-/г) (1-84)
\«Б-< /
Согласно расчетам окончательное выражение для изменения
теплоемкости при Тс в теории ДД можно представить в следу-
ющем виде:
ЬСР = /?( ^7 j7r (1 - А) + 4£7’Да (1 — 4,17ТДа) +
+ 0,57’ДаСр(7’-) (1.85)
где Ср(Тс~) — значение теплоемкости до начала скачка при Тс.
Два первых слагаемых в правой части (1.85) являются кон-
фигурационными вкладами от изменения числа гибких связей
и числа дырок с изменением температуры, а третье слагаемое
характеризует изменение колебательного состояния системы
вследствие изменения температурного коэффициента объема
при Тс. Из уравнения (1.84) видно, что ДСу определяет кон-
формационный вклад, а остальные два слагаемые связаны со
сжимаемостью и объемным расширением. Это совпадает со
сделанными выше заключениями.
Согласно уравнению (1.85) для расчета скачка теплоемко-
сти ДСР необходимо знать лишь химическое строение полимера,
так как это уравнение не содержит переменных параметров.
Действительно, произведение ТДа и Ср(Т~с) является измеряе-
мыми величинами, а Ек/^в Т определяется температурой стекло-
вания. Таким образом, различие в поведении теплоемкости при
стекловании для разных полимеров определяется различием в
ТДа и Ек/^б Т.
Ди Марцио и Даул оценили типичные значения отдельных
слагаемых в уравнении (1.85) и пришли к выводу, что
(f \ JT?k
Дж
4/?ТДа( 1 — 4,17ТДа) « 2-7=------ (1.87)
моль бусинок-К
Дж
0,5ТДаСп(ГГ) « 1,25----—------ (1.88)
моль бусинок-К
При оценках использовано значение Ек//гБТ = 2,25, харак-
терное для полистирола, однако отношение Ек/^б Т не сильно
меняется при переходе от полимера к полимеру, и потому ДСу
также не претерпевает очень сильных изменений. Максимальное
значение (1.86) оценивается в 6,25 Дж на моль гибких связей.
48
Составляющая (1.87) меняется в пределах от 1,8 до 1,94 при
изменении ГД а от 0,08 до 0,14 с небольшим максимумом (1,997)
при 7’Дй=0,12. Число бусинок в повторяющемся звене опреде-
лялось как отношение объема мономерного звена к объему
СН2-группы, а число гибких связей — по химическому строению
полимера. Гибкой считалась связь, вращение вокруг которой
приводит к изменению формы цепи.
Сумма всех вкладов дает 9,25 Дж/(моль-К). Это значение
отличается от среднего значения ЛСР = 11,5 Дж/(моль-К), по-
лученного Вундерлихом для многих стекол, менее чем на 20%.
Такую точность предсказания значения ДСР можно рассматри-
вать как очень высокую, учитывая, что теория не содержит пе-
ременных параметров, а точность экспериментального определе-
ния ДСР находится на уровне нескольких процентов.
Уравнение (1.85) позволяет сразу же сделать некоторые ка-
чественные выводы и о поведении теплоемкости СР полимеров
в жидком состоянии. С повышением температуры конформаци-
онный вклад в Ср должен уменьшаться. Дырочный вклад мало
изменяется с повышением температуры, а колебательный вклад,
хотя и возрастает, но не так быстро, как ниже Тс. Поэтому
можно ожидать, что температурный коэффициент изменения
теплоемкости выше Тс будет меньше, чем в стеклообразном
состоянии. Такая закономерность действительно наблюдается
[69].
Перейдем теперь к более детальному анализу отдельных
вкладов в ДСр и сравним экспериментальные результаты с тео-
ретическими. В табл. 1.4 суммированы экспериментальные зна-
чения ДСр для двенадцати самых распространенных полимеров
и результаты расчетов отдельных вкладов. Бросаются в глаза
заметные расхождения в оценках конформационного вклада.
Сравнение теории ДД с экспериментом приводит к выводу, что
вклад ДСК наибольший и может достигать половины ДСр, 30%
ДСр приходится на ДСД, а оставшиеся 20% принадлежат ДСК0Л.
Еще большая доля в ДСР принадлежит конформационному
вкладу, согласно оценкам О’Рейли и Гавличека и др. С другой
стороны, Рое и Тонелли полагают, что конформационному вкла-
ду принадлежит не самая большая часть ДСр и что ДСД со-
ставляет от '/з до */2 ДСр. В расчетах О’Рейли и Гавличека и
др. колебательный вклад вообще не принимался во внимание,
и потому можно полагать, что его учет должен уменьшить зна-
чение ДСК.
По-видимому, самым важным выводом из этих результатов
является заключение, что практически во всех случаях величи-
ны отдельных вкладов сравнимы между собой и ни одним из
них нельзя пренебрегать при расчетах ДСР. Это заключение,
с одной стороны, опровергает широко распространенное пред-
ставление концепции свободного объема, что изменение теплоем-
кости в области стеклования обусловлено главным образом раз-
мораживанием свободного объема и увеличением числа дырок
4—440
49
Рис. 1.16. Зависимость исчезающей
при охлаждении части разности эн-
тропий AS„c, от температуры [70, 71]:
t — ПВХ; 2 — ПЭМА; 3 — цас-1,4-полиизо-
прен.
при повышении температуры, и, с другой стороны, не дает ос-
нований переоценивать вклад конформационных изменений в
ЛСр. Наконец, признание важной роли ДСК0Л вклада в ДСр
свидетельствует о некоторых различиях в колебательных спект-
рах полимеров в жидком и стеклообразном состояниях.
Возможный характер этих изменений был подвергнут недав-
но детальному анализу с использованием высокоточных изме-
рений температурной зависимости теплоемкости полимерных и
неполимерных стекол {70, 71]. Если изменения в колебательном
спектре (частота колебаний) не вносят значительного вклада
в ДСр, то разность энтропий двух стекол с различной термиче-
ской предысторией будет оставаться постоянной при понижении
температуры в область 0 К. Если же в стекле, в котором замо-
рожены конформации с большей энергией, колебательный
спектр отличен (включает более мягкие колебания) от колеба-
тельного спектра стекла с большим содержанием низкоэнерге-
тических конформаций, то разность энтропий таких систем при
охлаждении будет уменьшаться. Можно предполагать, что это
снижение будет подобно дебаевской кривой теплоемкости, т. е.
сначала падение будет довольно медленным, а затем его ско-
рость резко возрастет. С другой стороны, если существенны из-
менения ангармоничности, а не частоты колебаний, то эта раз-
ность энтропий должна сначала резко уменьшаться с пониже-
нием температуры, а затем падение должно замедлиться. По
мнению Голдстейна, вклад, связанный с ангармоничностью,
трудно отличить от изменений теплоемкости, обусловленных вто-
ричными релаксационными процессами в стеклообразном со-
стоянии, которые также могут давать вклад в низкотемператур-
ную теплоемкость. Однако можно все-таки полагать, что теп-
лоемкость не очень чувствительна к этим вторичным процессам
и, если такое падение будет наблюдаться, его логичнее связать
с изменением ангармоничности колебаний.
Результаты анализа температурной зависимости разности
энтропий на примере трех полимеров (фщ-1,4-полиизопрена,
ПЭМА и ПВХ) привели к следующим выводам. Конфигураци-
онная часть теплоемкости ДСК составляет 0,79 для полиизопре-
на, 0,15 для ПЭМА и 0,58 для ПВХ. Для ПВХ это хорошо сов-
падает с оценками ДД, а для ПЭМА — с оценками Рое и То-
нелли. Для всех трех полимеров сразу же вслед з& температу-
рой стеклования на зависимости А5ИСЧ — Т наблюдается резкое
падение (рис. 1.16). Для полиизопрена небольшое падение на-
50
блюдается и ниже 50 К, а для ПЭМА — ниже 100 К, однако бо-
лее значительны изменения Д5ИСЧ сразу после стеклования. Для
ПВХ вообще наблюдается лишь этот высокотемпературный
спад. Эти результаты позволяют заключить, что более значи-
тельная часть ДСК0Л связана к изменением ангармоничности ко-
лебаний.
Кристаллические полимеры
Двухфазная модель кристаллических полимеров связывает со-
отношение аморфных и кристаллических областей со значением
скачка теплоемкости простой линейной зависимостью:
w = 1 - ДСр/ДСР>ам (1.89)
где ДСр, ам и ДСр — скачок теплоемкости при Тс соответственно в полностью
аморфном и частично-кристаллическом полимере.
Предполагается, что это соотношение выполняется при всех
значениях степени кристалличности от 0 до 1. Однако в дейст-
вительности такая зависимость в лучшем случае существует
при небольших степенях кристалличности (рис. 1.17), а при
дальнейшем возрастании кристалличности становится пробле-
матичным само отыскание области стеклования на температур-
ной зависимости С?. Действительно, если экспериментальную
зависимость, приведенную на рис. 1.17, экстраполировать на
ДСр = 0, то оказывается, что при ww 75% скачок теплоемкости
в области предполагаемого стеклования будет отсутствовать.
Это действительно наблюдается для высококристаллических по-
лимеров [73, 74]. Непропорциональное уменьшение ДС₽ с уве-
личением степени кристалличности, особенно при w>50%, при-
водит к тому, что рассчитанные по формуле значения w обычно
выше полученных другими методами, в том числе и по теплоте
плавления. Следует подчеркнуть, что явление непропорциональ-
ного уменьшения ДСр с увеличением w, а затем и исчезновение
скачка не связано с чувствительностью методов определения Ср.
Оно, несомненно, является отражением неадекватности двух-
фазной модели для высококристаллических полимеров.
Сложность однозначной идентификации стеклования высоко-
кристаллических полимеров по температурной зависимости Ср
отчетливо видна на примере наиболее широко исследованного
полимера ПЭ. Дискуссия о температуре стеклования ПЭ про-
должается уже много лет, а общего мнения до сих пор не су-
ществует. Обычно называют три интервала температур, в кото-
рых предполагается стеклование аморфных областей ПЭ: 150±
±10; 195+10; 240+10 К [74—76]. Наиболее детальный анализ
стеклования ПЭ по температурной зависимости СР проведен
Битти и Карашем [74]. Из этого анализа почти с несомнен-
ностью следует, что, согласно калориметрическим данным, стек-
лованию линейного ПЭ может соответствовать лишь область
4»
51
150±10 К. Рассмотрим основные аргументы, позволяющие сде-
лать это заключение.
Самой важной особенностью поведения СР является то, что
хотя в высококристаллических полимерах скачкообразное из-
менение СР часто не наблюдается, однако такой скачок появля-
ется лишь при стекловании и не появляется ни при каких дру--
гих релаксационных переходах. Это является определенным пре-
имуществом калориметрии по сравнению с другими методами
(например, дилатометрией, динамическим механическим анали-
зом) при идентификации природы релаксационных переходов.
Оказалось (рис. 1.18), что скачок теплоемкости в ПЭ наблюда-
ется лишь в области температур 150± 10 К и отсутствует в двух
других температурных областях. И для других высококристал-
лических полимеров, таких, как ПТФЭ и ПОМ, калориметрия
позволяет вполне определенно установить области стеклования.
Еще более показательной является зависимость абсолютных
значений теплоемкости (рис. 1.19) от степени кристалличности
при двух температурах — выше и ниже Тс- Уже отмечалось,
что ниже Тс теплоемкости полимера в аморфном и кристалли-
ческом состоянии практически тождественны. Горизонтальная
прямая на рис. 1.19 еще раз это подтверждает. Выше Тс вслед-
ствие дополнительных вкладов в теплоемкость при расстеклова-
нии аморфных областей СР начинает зависеть от степени крис-
талличности (наклонная прямая). Это является еще одним под-
тверждением того, что Тс линейного ПЭ находится ниже 180 К.
Наконец, следует упомянуть, что «холодная» кристаллизация
при разогреве сильноаморфизованного образца ПЭ наблюдает-
ся в области температур 160—180 К [78], что совместимо с
Рис. 1.17. Зависимость скачка теплоемкости при стекловании от степени крис-
талличности для линейного ПУ иа основе гексаметилендиизоцианата и диэти-
ленгликоля [72].
Рис. 1.18. Изменение теплоемкости в частично-кристаллических полимерах при
стекловании ,[74]:
ПЭ (степень кристалличности 35%); 2 — ПЭ (степень кристалличности 55%); 3 —
ПТФЭ; 4 — ПОМ.
К9
Рис. 1.19. Зависимость теплоемкости линейного ПЭ от
степени кристалличности (разные точки соответству-
ют данным различных авторов) [74].
йредставлением о Тс=150±10 К и несовме-
стимо с двумя другими приводимыми зна-
чениями Тс, так как кристаллизация ниже
Тс обычно протекать не может.
Таким образом, ЛСР при стекловании
аморфных областей кристаллических поли-
меров уменьшается быстрее с ростом сте-
пени кристалличности, чем это предпола-
гается двухфазной моделью, и при боль-
ших степенях кристалличности скачок мо-
жет вообще исчезать. Молекулярные при-
чины этого явления неясны, и потому
трудно оценить характер изменения от-
дельных вкладов в ДСР при частичной
кристаллизации полимера.
Можно предположить, что наиболее чувствительным к крис-
талличности является конформационный вклад ДСКР по следую-
щим соображениям. В отличие от статистических клубков в
аморфных полимерах аморфные области кристаллических поли-
меров включают петли различной длины, натянутые участки
цепей, закрепленные в различных кристаллитах, концы цепей.
Конформационный набор таких цепей по очевидным соображе-
ниям сильно обеднен. Такие цепи до некоторой степени можно*
отождествить с отдельными участками сильно растянутых це-
пей аморфных высокоэластических полимеров и для оценки их
конформационной теплоемкости использовать представления
Волькенштейна и Птицына [59], предсказавших теоретически
уменьшение конформационной теплоемкости растянутых поли-
мерных цепей.
Теория этого эффекта основана на следующих представле-
ниях. Растяжение полимерных цепей, обладающих различными
конформационными энергиями, сопровождается переходом одних
изомеров в другие (см. гл. 4). Теплоемкость растянутой цепи
(при постоянной длине) должна быть меньше теплоемкости ис-
ходной нерастянутой цепи, так как фиксирование длины ограни-
чивает поворотную изомерию, уменьшая тем самым конформа-
ционную теплоемкость цепи. Внутримолекулярная энергия рас-
тянутых полимерных цепей выражается формулой (4.72). Диф-
ференцирование этой формулы при постоянных V и А дает
2 \ Л /
_ d In <r2)n d-’ In <r-’>0’
d In T (d In T)-’
(1.90)
53
где Cv, i —значение Cv в нерастяиутом состоянии, т. е. при Х = 1.
Для цепей с симметричными боковыми группами и поворот-
ными изомерами (0°, ±120°)
CV,K = CV>1 - vk3 ~ (V + X - 2) + (1.91)
Для изотактических цепей типа [—СНг—CHR—]п
(Х-> + Х-2)|'^-¥ (1.92)
2 Л \ R^l /
Из этих формул действительно видно, что теплоемкость рас-
тянутых цепей ниже, чем нерастянутых. Оценки показывают
[59], что |ДСу| «4- ПУ4 Дж/(см3-К). Уже при растяжениях
порядка Х=10 уменьшение конформационной теплоемкости
сравнимо с самой величиной конформационной теплоемкости, а
при Х=20—25 конформационная теплоемкость может оказаться
полностью замороженной даже без учета негауссовых эффектов
при больших растяжениях. В связи с этим непропорциональное
уменьшение АСР при Тс частично может быть следствием рез-
кого возрастания доли сильно вытянутых цепей в аморфных
областях с ростом степени кристалличности и непропорциональ-
ного уменьшения конформационного вклада в ДСР. При очень
больших X конформационный вклад в ДСР по-видимому, вооб-
ще может исчезнуть. Оценить характер изменения двух других
вкладов с увеличением степени кристалличности трудно.
ЛИТЕРАТУРА
1. Марадудин А., Монтролл Э., Вейс Дж. Динамическая теория кристалличе-
ской решетки в гармоническом приближении. Пер. с англ./Под ред.
М. И. Петрашень. М., Мир, 1965. 383 с.
2. Лейбфрид Г. Микроскопическая теория механических и тепловых свойств
кристаллов. Пер. с нем./Под ред. Б. Я Мойжеса. М. — Л., Физматгиз, 1963.
312 с.
3. Вундерлих Б., Баур Г. Теплоемкость линейных полимеров. Пер. с англ, и
нем./Под ред. Ю. К. Годовского. М., Мир, 1972. 238 с.
4. Kitagawa Т., Miyazawa Т. — Adv. Polymer Sci., 1972, vol. 9, N 2, p. 336—
414.
5. Shen M., Reese W. — In: Progress in Solid State Chemistry/Ed. by
I. O. McColdin a. G. Somorjai, Oxford, Pergamon Press, 1975. Vol. 9,
p. 241—268.
6. Kirkwood J. G. — J. Chem. Phys., 1939, vol. 7, N 7, p. 506'—509.
7. Pitzer K. S. —J. Chem. Phys., 1940, vol. 8, N 9, p. 711—720.
8. Михайлов И. Д., Чебан Ю. В. — В кн.: Численные методы решения задач
математической физики и теории систем. М., Ун-т Дружбы народов, 1977,
с. 73—76.
9. Чебан Ю. В. Автореф. канд. ди с. М., Уи-т Дружбы народов, 1979.
10. Stockmayer W. Н„ Hecht С. Е.—J. Chem. Phys., 1953, vol. 21, N 11,
p. 1954—1959.
11. Genensky S. M., Newell C. F. — J. Chem. Phys., 1957, vol. 26, N 3, p. 486—
497.
12. Тарасов В. В. — ДАН СССР, 1945, т. 46, № 1, с. 22—25.
13. Тарасов В. В. — ЖФХ, 1950, т. 24, № 1, с. 111—128.
54
14. Тарасов В. В. — ЖФХ, 1953, т. 27, № 9, с. 1430—1436.
15. Тарасов В. В. Проблемы физики стекла. М., Стройиздат, 1979. 255 с.
16. Ландау Л. Д., Лифшиц Е. М. Статистическая физика. М., Наука, 1976.
Ч. 1, 583 с.
17. Ландау Л. Д., Лифшиц Е. М. Теория упругости. М., Наука, 1965. 202 с.
18. Готлиб Ю. Я., Сочава И. В. — ДАН СССР, 1962, т. 147, № 3, с. 580—
583.
19. Сочава И. В. —Вести. ЛГУ, 1964, т. 19, № 10, с. 56—64.
20. Годовский Ю. К. Теплофизические методы исследования полимеров. М.,
Химия, 1976. 216 с.
21. Лифшиц И. М. — ЖЭТФ, 1952, т. 22, № 4, с. 471—474, 475—486.
22. Косевич А. М. Основы механики кристаллической решетки. М., Наука,
1972. 280 с.
23. Baur Н.— Koll.-Ztschr. u. Ztschr. Polymere, 1970, Bd 241, N 1—2, S. 1057—
1070.
24. Kothari L. S., Tewari V. K. —J. Chem. Phys., 1963, vol. 38, N 2, p. 417—418.
25. Tucker J. E., Reese W.—J. Chem. Phys., 1967, vol. 46, N 4, p. 1388—1397.
26. Swaminathan K, Tewari S. P., Kothari L. S. — Phys. Lett., 1975, Ser. A,
vol. 51, N 1, p. 153—154.
27. Перепечко И. И. Свойства полимеров при низких температурах. М., Хи-
мия, 1977. 271 с.
28. Baur Н.— Koll.-Ztschr. u. Ztschr. Polymere, 1971, Bd 244, N 2, S. 293—303.
29. Лебедев Б. В. Автореф. докт. дис. М., МГУ, 1978.
30. Stephens R. В. — Phys. Rev., 1973, В, vol. 8, N 6, р. 2896—2905; 1976,..
vol. 13, N 2, p. 852—865.
31. Pohl R. O., Salinger G. L. — Ann. N. Y. Acad. Sci., 1976, vol. 279, N 4,.
p. 150—172.
32. Bottger H. — Phys. Stat. Solidi, 1974, Ser. B, Bd 62, N 1, S. 9—42.
33. Rosenstock H. B.—J. Non-Crystall. Solids, 1972, vol. 7, N 1, p. 123—'126.
34. Baltes H. P. — Solid. State Comm., 1973, vol. 13, N 2, p. 225—228.
35. Tantilla W. H. — Phys. Rev. Lett., 1977, vol. 39, N 9, p. 554—557.
36. Watkins G. M., Fenichel H.—J. Non-Crystall. Solids, 1980, vol. 37, N 3,.
p. 433—435.
37. Anderson P. W., Halperin В. I., Varma С. M. — Phil. Mag., 1972, vol. 25,
N 1, p. 1—9.
38. Phillips HZ. A. —J. Low Temp. Phys., 1972, vol. 7, N 3/4, p. 351—360.
39. Finlayson D. M., Mason P., Rogers J. N. — J. Phys., 1980, Ser. C, vol. 13,
N 9, p. L185—L188.
40. Yosida S., Suga H., Seki S. — Polymer J., 1973, vol. 5, N 1, p. 11—21.
41. Choy C. L, Hug M., Moody D. E. — Phys. Lett., 1975, Ser. A, vol. 54, N 4,
p. 375—377.
42. Reese W. —J. Macromol. Sci., 1969, pt A, vol. 3, N 7, p. 1257—1295.
43. Choy C. L„ Hunt R. G., Salinger G. L. — J. Chem. Phvs., 1970, vol. 52, N 7,
p. 3629—3633.
44. Zoller P., Fehl D. L., Dillinger J. R.—J. Polymer Sci., Polymer Phys. Ed.,
1973, vol. 11, N 7, p. 1441—1451.
45. Madding R. P., Fehl D. L., Dillinger J. R. — Ibid., p. 1435—1440.
46. Swaminathan K, Tewari S. P. — J. Polymer Sci., Polymer Phys. Ed. 1980,
vol. 18, N 8, p. 1707—1716.
47. Fulde P„ Wagner H. — Phys. Rev. Lett., 1971, vol. 27, N 9, p. 1280—1282.
48. Takeno S., Goda M. — Progr. Theor. Phys., 1972, vol. 48, N 5, p. 1468—
1473.
49. Андерс Э. E„ Сухаревский Б. Я-, Шестаченко Л. С. — Физ. низк. темп.,
1979, т. 5, № 7, с. 783—793.
50. Мамедов К- К-, Керимов И. Г., Мехтиев М. И., Алджанов М. А. — ДАН
АзССР, 1980, т. 36, № 8, с. 39—42.
51. Lee W. К, Choy С. L.—J. Polymer Sci., Polymer Phys. Ed., 1975, vol. 13,
N 3, p. 619—635.
52. Choy C. L., Leung W. Y., Chen F. C.—J. Polymer Sci., Polymer Phys. Ed.,
1979, vol. 17, N 1, p. 87—94.
55-
53. О’Рейли Дж. М., Карац Ф. Е. — В кн.: Переходы и релаксационные явле-
ния в полимерах/Сост. Р. Бойер. Пер. с англ. Под ред. А. Я- Малкина. М.,
Мир, 1968, с. 60—85.
54. Wunderlich В. —J. Phys. Chem., I960, vol. 64, N 8, p. 1052—1056.
55. Roe R.-J., Tonelli A. E. — Macromolecules, 1978, vol. 11, N 1, p. 114—117.
56. DiMarzio E. A-, Dowell F. — J. Appl. Phys., 1979, vol. 50, N 10, p. 6061.
57. Wunderlich B„ Jones L. D.— J. Macromol. Sci., 1969, pt B, vol. 3, N 1,
p. 67—79.
58. Wunderlich B., Gaur U. — Pure Appl. Chem., 1980, vol. 52, N 2, p. 445.
59. Бирштейн T. M., Птицын О. Б. Конформации макромолекул. М., Наука,
1964. 391 с.
60. Флори П. Статистическая механика цепных молекул. Пер. с англ./Под ред.
М. В. Волькеиштейна. М., Мир, 1971. 440 с.
61. Gibbs J. Н., DiMarzio Е. А. — J. Chem. Phys., 1958, vol. 28, N 3, p. 373—
383; N 8, p. 807—813.
62. DiMarzio E. A., Gibbs J. H., Fleming P. D., HI, Sanchez I. C. —Macromole-
cules, 1976, vol. 9, N 5, p. 763—771.
63. O'Reilly J. M. — J. Appl. Phys., 1977, vol. 48, N 10, p. 4043—4048.
64. O’Reilly J. M.— J. Polymer Sci., Polymer Symp., 1978, N 63, p. 165—172.
65. Havlicek I., Vojta V-, Ilavsky M., Hrouz J. — Macromolecules, 1980, vol. 13,
N 2, p. 357—362.
66. Nose T. — Polymer J., 1971, vol. 2, N 4, p. 437—456.
67. Goldstein M. — J. Chem. Phys., 1963, vol. 39, N 12, p. 3369—3374.
68. Tanaka N. — Polymer, 1978, vol. 19, N 7, p. 770—776.
69. Ван Кревелен Д. В. Свойства и химическое строение полимеров. Пер. с
англ./Под ред. А. Я. Малкина. М., Химия, 1976. 414 с.
70. Goldstein М. — J. Chem. Phys., 1976, vol. 64, N 11, p. 4767—4774; 1977,
vol. 67, N 5, p. 2246—2253.
71. Goldstein M. — Ann. N. Y. Acad. Sci., 1976, vol. 279, N 4, p. 68—77.
72. Годовский Ю. K-, Липатов Ю. С. — Высокомол. соед., 1968, сер. А, т. 10,
№ 1, с. 32—40.
73. O’Reilly J. М., Karacz F. Е. — Polymer Prepr, 1964, vol. 5, N 2, p. 351 —
353.
74. Beatty C. L., Karacz F. E. — J. Macromol. Sci., 1979, pt C, vol. 17, N 1,
p. 37—60.
75. Stehling F. C., Mandelkern L.—Macromolecules, 1970, vol. 3, N 1, p. 242—
252.
76. Boyer R. F. — Macromolecules, 1973, vol. 6, N 2, p. 288—299.
77. Boyer R. F.—J. Macromol. Sci., 1973, pt B, vol. 7, N 3, p. 487—501.
78. Hendra P. J., Jobic И. P., Holland-Moritz K- — J. Polymer Sci., Polymer Lett.
Ed., 1975, vol. 13, N 6, p. 365—368.
Глава 2
ТЕПЛОПРОВОДНОСТЬ
Полимеры являются диэлектриками, и их теплопроводность
обусловлена решеточными колебаниями. Квантовая теория твер-
дого тела позволяет представить механизм и температурную за-
висимость теплопроводности х твердых диэлектриков, исходя из
основной кинетической формулы [1—4]
х = ^-рСус/ (2.1)
где с — средняя скорость распространения фоионов; I — средняя длина свобод-
ного пробега фононов.
56
VPuc. 2.1. Схематическое изображение X
[температурной зависимости теплопровод-
ности кристаллических (/) и аморфных
г (2) диэлектриков.
Температурная зависимость и
идеальной кристаллической ре-
шетки должна иметь вид, пока-
занный на рис. 2.1, по следую-
щим соображениям. При нор-
мальных и высоких температу-
рах, когда возбуждено много фононов и взаимодействия меж-
ду ними (процессы переброса) происходят достаточно часто,
вероятность взаимодействия между фононами пропорциональ-
на их числу, т. е. абсолютной температуре. Так как при таких
температурах Cv практически постоянна, то I, а следовательно,
и х пропорциональны Т~г. С понижением температуры число фо-
нонов, способных к взаимодействию с перебросом, экспоненциа-
льно уменьшается. В связи с этим по такому же закону долж-
ны возрастать I и х. Дальнейшее снижение температуры долж-
но привести к тому, что I станет сравнимо с размером объекта
и перестанет зависеть от температуры. Однако это не должно
привести к независимости х от температуры, так как теплоем-
кость малочувствительна к размерам объекта и при низких
температурах сильно зависит от температуры. Поэтому после
того как граничное рассеяние фононов станет доминирующим,
зависимость х от температуры будет определяться температур-
ной зависимостью Су, т. е. в соответствии с теорией Дебая
х~Т3 вблизи О К-
Таким образом, в предположении существования лишь двух
механизмов рассеяния — фононов на фононах (процесс пере-
броса) и фононов на границах кристалла — температурная за-
висимость х кристаллического диэлектрика должна иметь мак-
симум. Сравнение теоретических значений х в максимуме, вы-
численных с учетом этих предположений, с экспериментальными
значениями х для наиболее совершенных кристаллов показало
[2, 3], что экспериментальное значение х намного ниже тео-
ретического. Оказалось, что причиной этого расхождения явля-
ется рассеяние фононов на различных дефектах кристаллов —
дислокациях, примесях, точечных дефектах, изотопах. Поэтому
метод исследования теплопроводности при низких температурах
является чувствительным методом исследования дефектов крис-
таллической структуры.
Теория низкотемпературной теплопроводности кристалличе-
ских диэлектриков позволяет количественно оценить и темпера-
турную зависимость, и значение х. Этого нельзя сказать о фи-
зической теории теплопроводности при средних и повышенных
температурах, для которых теория надежно предсказывает тем-
57
пературную зависимость теплопроводности (х~7-1), числовые
же значения х обычно находят на основе полуэмпирических за-
висимостей (см., например, [5]).
Изложенные представления развивались для твердых ди-
электриков, обладающих, несмотря на наличие примесей и де-
фектов, кристаллической структурой. Поведение теплопроводно-
сти аморфных твердых тел — стекол характеризуется двумя
принципиальными особенностями, отличающими их поведение
от поведения кристаллических тел: существенно более низкими
абсолютными значениями х при низких температурах и иной
температурной зависимостью х. Отсутствие трансляционной
симметрии в аморфных телах исключает рассеяние фононов по
механизму переброса*.
При обычных и высоких температурах длина свободного
пробега может считаться постоянной, не зависящей _от темпе-
ратуры величиной. Действительно, оценки величины I на осно-
вании формулы (2.1) показывают, что выше 100—200 К I со-
ставляет несколько десятых нанометра, т. е. находится на уров-
не ближнего порядка в стеклах. При таких температурах тепло-
проводности стекла и кристалла одного и того же вещества
сравнимы вследствие сравнимых значений I.
Длина свободного пробега фононов в стеклах при пониже-
нии температуры сначала меняется очень слабо, поэтому теп-
лопроводность х стекла должна уменьшаться с понижением
температуры так же, как и Су. Такая закономерность наблюда-
ется для многих стекол примерно до 20—50 К (см. рис. 2.1).
При более низких температурах эта закономерность резко на-
рушается. Для многих стекол, в том числе и полимерных, на
температурной зависимости х в области 5—20 К наблюдается
плато, а при дальнейшем понижении температуры х уменьша-
ется, но менее резко, чем Cv, т. е. менее резко, чем у кристалл
лических тел. Удивительно, что одинаковыми оказываются не
только температурные зависимости теплопроводности стекол
различной природы, но и абсолютные значения теплопроводно-
сти. Аномальное поведение теплопроводности стекол в криоген-
ной области тесно связано с аномальным поведением их тепло-
емкости, рассмотренным в предыдущей главе. Таким образом,
теплопроводность стекол немонотонно растет при повышении
температуры и не имеет максимума. Это позволяет предпола-
гать качественное различие механизмов теплопроводности в
кристаллических и аморфных телах.
Для объяснения механизма теплопроводности аморфных ве-
ществ потребовалось введение представления о структурном
рассеянии волн, возникающем в результате отсутствия даль-
* Несмотря на то, что понятие фоиона для аморфных твердых тел может
быть введено лишь для длинноволновых акустических колебаний, оно часто
используется для всего колебательного спектра аморфного тела.
58
него порядка в аморфных веществах ([4]. Отсутствие дальнего
порядка обусловливает неоднородность упругих свойств в мик-
рообъемах. Распространение волн в такой среде должно со-
провождаться их рассеянием в результате изменения их скоро-
сти. Основным параметром в механизме структурного рассея-
ния является соотношение между длиной волны и длиной кор-
реляции упругих свойств аморфной среды [1]. Последняя сопо-
ставима с линейным размером областей ближнего порядка. При
сравнимых размерах длины волны и длины корреляции длина
свободного пробега близка к длине корреляции, и в этом слу-
чае наблюдается интенсивное рассеяние фононов.
Дальнейший анализ механизма структурного рассеяния при-
вел к гипотезе о более эффективном рассеянии в стеклах попе-
речных колебаний, чем продольных. В связи с этой гипотезой
низкотемпературная теплопроводность стекол должна опреде-
ляться лишь продольными колебательными модами, так как
поперечные моды характеризуются малой длиной свободного
пробега. С повышением температуры резко возрастает вероят-
ность взаимодействия продольных и поперечных колебаний, ко-
торые интенсивно рассеиваются. На основе этой модели было
получено уравнение для теплопроводности продольных колеба-
тельных мод [4]:
оо
fel А „ (* ех х2
и, — —• — Ту*\ --------- • ------dx (2-2)<
Зл/i a J (ех— I)2 х- -|-у-
о
где А — константа, характеризующая длину корреляции; а — расстояние меж-
ду колеблющимися единицами; x=halkbT-, уг=(Т0!Ту (То— параметр, харак-
теризующий процесс превращения продольных колебаний в поперечные).
При Т<^Т0 уравнение (2.2) предсказывает линейную зави-
симость х от температуры и появление низкотемпературного
плато как результата наложения теплопроводности продольных
и поперечных фононов.
В отличие от теорий теплопроводности твердых тел в теори-
ях теплопроводности жидкости исходят из представления о не-
коррелированной передаче тепла через атомы или молекулы,
связанные со своими соседями ван-дер-ваальсовыми связями.
Основным параметром, определяющим теплопроводность, явля-
ется среднее расстояние между молекулами жидкости. При вы-
соких температурах, когда длина свободного пробега в стек-
лах имеет порядок расстояния между частицами, оба подхода
приводят к эквивалентным результатам, и полученные при
жидкостном подходе соотношения оказываются в конечном сче-
те идентичными уравнению (2.1) {4, 5].
Имея в виду эту качественную картину теплопроводности
диэлектриков, рассмотрим особенности теплопроводности поли-
меров.
59
АМОРФНЫЕ ПОЛИМЕРЫ
Температурные зависимости х аморфных полимеров довольно
•единообразны. Наиболее полно она исследована для ПММА
(рис. 2.2), на примере которого будет рассмотрено поведение
теплопроводности аморфных полимеров в характерных темпера-
турных интервалах.
Область температур выше 100 К. Для изменения теплопро-
водности аморфных полимеров в этой температурной области
характерно наличие излома зависимости при температуре стек-
лования. Ниже Тс теплопроводность имеет небольшой положи-
тельный коэффициент, а выше — отрицательный. Необходи-
мость появления излома при Тс не является очевидной. Соглас-
но уравнению (2.1) и рассмотренному в гл. 1 поведению тепло-
емкости при Тс скорее следовало бы ожидать скачкообразного
изменения х при Тс [в уравнение (2.1) входит лишь колеба-
тельная составляющая общего скачка ДСр]. Более того, при
повышении температуры кроме скачка х при статической Тс
должно было бы наблюдаться скачкообразное уменьшение х
в области динамической Тс, соответствующее скачкообразному
уменьшению скорости распространения упругих волн. Посколь-
ку скачкообразное изменение х при Тс и выше до сих пор не
обнаружено, Айерман [8] предположил, что перенос энергии
в аморфных полимерах вблизи Тс как со стороны стеклообраз-
ного, так и высокоэластического состояния осуществляется не
путем распространения упругих волн, а в результате некорре-
лированной передачи энергии путем внутри- и межмолекуляр-
ного взаимодействия, т. е. по механизму, характерному для теп-
лопроводности жидкостей.
Для объяснения экспериментальной зависимости теплопро-
водности аморфных полимеров от температуры был проведен
теоретический анализ структурной модели, базирующийся на
следующих двух предположениях <[8, 11]. При расчете межмо-
лекулярной теплопроводности значение I в уравнении (2.1)
можно считать не зависящим от температуры и равным средне-
Рис. 2.2. Температурная зависимость теплопроводности ПММА [6—10].
ФО
Рис. 2.3. Модельная решетка для расчета теп-
лопроводности аморфных полимеров (двойные
линии соответствуют ковалентным химическим
связям, одинарные — ван-дер-ваальсовым) [8].
му межмолекулярному расстоянию
между двумя соседними молекулами
и использовать для расчета межмоле-
кулярного взаимодействия соответст-
вующий потенциал. Второе предполо-
жение касается локальной анизотро-
пии теплопроводности, являющейся следствием цепного строе-
ния полимеров, и сводится к постулированию соотношения
между внутри- и межмолекулярной теплопроводностью: пред-
полагается, что внутримолекулярная теплопроводность намно-
го выше (минимум на порядок), чем межмолекулярная.
С учетом этих предположений была рассмотрена теплопро-
водность структурной модели (рис. 2.3), в которой атомы глав-
ной цепи образуют кубическую квазирешетку. Каждый такой
атом соединен двумя ковалентными связями с соседними ато-
мами и четырьмя ван-дер-ваальсовыми (концевые атомы имеют
одну химическую и пять межмолекулярных связей). Направле-
ние ковалентных связей в решетке статистическое (каждое из
пяти направлений узла решетки — равновероятно). Таким обра-
зом, в этой модели аморфный полимер моделируется сеткой,
образованной термическими мостиками двух типов. Ковалент-
ные связи характеризуются большой теплопроводностью хв
(низкое термическое сопротивление), а ван-дер-ваальсовы свя-
зи — низкой теплопроводностью х® (большим термическим со-
противлением). Расчет Хи, основан на уравнении Дебая (2.1),
в котором макроскопические величины заменены на молекуляр-
ные параметры: Cv = 3kB /а2Ь, c = a]lfw/m (fw — константа упру-
гости ван-дер-ваальсовой связи, т — масса частиц), 1 = а. Из
уравнения (2.1) имеем:
хда = fe(3feB/&)/ /та/ т
(2-3)
Теплопроводность полимера х пропорциональна элементар-
ной теплопроводности х®
x = fe'xw (2*4)
Вклад хв в общую теплопроводность учитывается путем вве-
дения неравенства k'z>\, причем k не зависит от температуры.
Для расчета fw использован 12/6-потенциал. Подстановка опре-
деленного таким образом значения fw в уравнение (2.3) приво-
дит к выражению, связывающему теплопроводность со средним
расстоянием между атомами. Это позволяет выразить относи-
тельный температурный коэффициент теплопроводности
(1/х) (du[dT} через коэффициент объемного теплового расшире-
61
ния а и соответственно связать скачкообразное изменение Асй
в области стеклования со скачком (1/х) {dtildT):
1 da 1 db
а ~2 „ • ,т + , '
а аТ о ат
(2.5)
Поскольку изменением валентных связей в зависимости от
температуры можно пренебречь по сравнению с изменением
ван-дер-ваальсовых связей, окончательное уравнение будет
иметь вид
/ 1 zfv \
Д |— • — )= — 5,8 Да (2-6)
\ х dT )
Таким образом, физической причиной изменения температур-
ного коэффициента теплопроводности при Тс является различие
в коэффициентах теплового расширения аморфных полимеров
до и после стеклования. Уравнение (2.6) удовлетворительно
описывает связь между пологим изломом х при Тс и скачком
Да для ряда аморфных полимеров различного химического
строения, в том числе и для ПММА [8, 11].
Существенное различие внутри- и межмолекулярной тепло-
проводности должно приводить к зависимости теплопроводности
от молекулярной массы, причем с повышением последней х
должна возрастать. Такая зависимость действительно существу-
ет, однако разные модели предсказывают различный характер
этой зависимости. Рассмотренная выше модель приводит к за-
висимости
где Хоо — теплопроводность полимера бесконечно большей молекулярной мас-
сы М; А — константа.
При выводе соотношения (2.7) предполагалось, что расчле-
нение макромолекулы по валентным связям не сопровождает-
ся изменением конформации ее участков, а в местах разрыва
валентные связи заменяются физическими. Проверка этого со-
отношения на ПДМС и ПЭО показала, что оно не выполняет-
ся, так как зависимость х от молекулярной массы существует
лишь при сравнительно небольшой молекулярной массе, а при
дальнейшем ее повышении наблюдается насыщение.
'Хансен и Хоу [13] развили основанную на «жидкостном»
подходе полуфеноменологическую теорию теплопроводности
аморфных полимеров с помощью которой, не вдаваясь в де-
тальную молекулярную динамику, а учитывая лишь различную
степень взаимодействия соседних звеньев, связанных между
собой химическими и физическими связями, удалось показать,
что теплопроводность должна зависеть от молекулярной массы,
как МV2 при малых молекулярных массах, и достигать постоян-
ного значения при дальнейшем увеличении М. Имеющиеся экс-
62
периментальные результаты лучше всего отвечают этой зави-
симости.
Физическая причина насыщения теплопроводности при по-
вышении молекулярной массы состоит, по-видимому, в том, что
при достаточно длинных цепях передача энергии через межмо-
лекулярные связи осуществляется чаще, так что при достаточно
большой длине общая теплопроводность становится нечувстви-
тельной к длине макромолекулы. Проведенные расчеты эффек-
тивной средней длины пробега V для одномерных модельных
цепей с межцепным взаимодействием привели к следующей за-
висимости [14]:
/ а 1 \-Т2
а (2-8)
где /ц — средняя длина пробега вдоль цепи; — степень полимеризации; а,-—
расстояние между мономерными единицами в цепи, аппроксимируемыми точеч-
ными массами.
Модель хорошо предсказывает длину цепи, при которой на-
блюдается [13] насыщение теплопроводности для ПЭ (Af>105,
Т=140°С) и ПС (Л1«105, T=100oC). Проведенный недавно
детальный теоретический анализ теплопроводности расплава
полимера, основанный на методе временных корреляционных
функций с использованием модели субцепей Рауза, привел к
выражению [15]:
, (Pov)2
К -------
1
Tv
(2-9)
(где ро — плотность мономерной единицы; v — параметр, харак-
теризующий взаимодействие; b — сегмент, v — коэффициент
трения), в котором длина цепи не фигурирует (цепи предпола-
гаются достаточно длинными). Таким образом, зависимость
теплопроводности от М1/2 можно считать теоретически обосно-
ванной.
Проведенное до сих пор рассмотрение касалось лишь харак-
тера температурной зависимости теплопроводности аморфных
полимеров. Нрряду с этим предпринимались попытки связать
абсолютные теплопроводности с молекулярными параметрами.
Полуэмпирические оценки Айермана [8], основанные на модель-
ных соображениях и на сравнениях экспериментальных значе-
ний теплопроводности полимеров с теплопроводностью органиче-
ских жидкостей и неорганических стекол, можно рассматривать
лишь как сугубо ориентировочные.
Бонди [16, с. 305] предложил уравнения для расчета теп-
лопроводности полимеров в размягченном состоянии. Схема
расчета близка к модели Айермана и рассматривает теплопро-
водность полимера как комбинацию теплопроводности вдоль це-
63
пи и между цепями. Теплопроводность хв вдоль макромолеку-
лы выражается следующим образом:
SCDPo< Ет V'2
*в = ., -1--- ) 0,5^/т (2.10)
М \ Ро /
где SCD — мольная теплоемкость акустических колебаний составляющих цепь
атомов в приближении Дебая; Ет — спектроскопический модуль макромоле-
кулы; множитель 0,5 отражает статистическую ориентацию участков макромо-
лекулы к направлению температурного градиента; <р& — часть сечения макро-
молекулы, доступная для распространения колебания (определяется геометри-
ческими характеристиками повторяющегося звена макромолекулы); средняя
длина свободного пробега принимается равной персистентной длине макромо-
лекулы (конформация макромолекулы в растворе отождествляется с ее кон-
формацией в высокоэластическом состоянии).
Теплопроводность между цепями может быть рассчитана по
уравнению
_R_{E^ A®, 8,8cmRT
NaAM ' \ ’ £°
(2.Н)
где £° — стандартная энергия испарения повторяющегося звена молекулярной
массы Л4; свк — число внешних степеней свободы повторяющегося звена.
Общее тепловое сопротивление определяется из соотношения
1
Хщ,
где
J_ _ 1 1 d
х 7 + d хв 7 d
(2. 12
(2-13)
d
(здесь А — поверхность одного моля повторяющихся звеньев).
Приведенные формулы относятся к цепи, не содержащей
длинных боковых групп. Их наличие создает дополнительное
сопротивление вдоль цепи за счет взаимодействия между собой.
Для полимеров с короткими боковыми группами типа ПП и ПС
полная теплопроводность хполн может быть выражена соотно-
шением
q(m) Г д(т)
^полн —- И 4- 1
<? I <?
(2.14)
в котором отношение поперечных сечений полиметиленовой це-
пи (индекс т) к сечению цепи с боковыми группами определя-
ется как
д(т) Уи(т)аР
q Vuap(m)
где ар — проекция связи на ось макромолекулы; Vu — мольный объем повто-
ряющегося звена.
Учет влияния боковых групп в этой модели приводит к
уменьшению теплопроводности и увеличению отрицательного
коэффициента d%ldT, что согласуется с экспериментальными
данными. Сравнение результатов расчета с экспериментальны-
64
ми показывает, что точность таких расчетов находится на уров-
не 30—50%.
Низкотемпературная область (ниже 100 К). Две проблемы
привлекли в последние годы наибольшее внимание к низкотем-
пературной теплопроводности аморфных веществ: природа низ-
котемпературного плато в области 5—20 К и характер темпера-
турной зависимости теплопроводности ниже 5 К (см. рис. 2.1
и 2.3). Предсказанная ранней теорией Клеменса [4] линейная
зависимость х от температуры при очень низких (<1 К) тем-
пературах [уравнение (2.2)] противоречит экспериментально
наблюдаемой зависимости [9, 10, 17]
х = АГ‘
в которой б=1,9±0,1 и Д = 3-10~2 Вт/(м-К) (вариация коэф-
фициента А для всех исследованных стеклообразных веществ
не превышает 3). Это расхождение между существовавшей тео-
рией и экспериментом породило ряд новых теоретических под-
ходов.
Клеменс [18] рассмотрел механизм теплопроводности сте-
кол, считая стекло упругим континуумом с характерными флук-
туациями упругих характеристик и плотности. В частности, на-
личие таких флуктуаций в скорости звука должно приводить к
рассеянию фононов. Процесс рассеяния может быть охаракте-
ризован временем превращения фонона с данным волновым век-
тором k в другие состояния (время релаксации)
2К
где с — средняя скорость звука; <с2> — средний квадрат флуктуации скорости
звука; Ф(р)—структурный фактор; р — разность волновых векторов k и k'
в двух состояниях.
Таким образом, частотная зависимость т определяется зави-
симостью Ф(р) и фактором <с2>. Сложность использования
этого теоретического построения связана с неопределенностью
вида зависимости Ф(р).
Следуя этой общей идеи о механизме флуктуационного рас-
сеяния фононов, Морган и Смит [19] предложили микроскопи-
ческую модель рассеяния фононов в стеклах любой природы.
Модель включает две основные характеристики — длину корре-
ляции флуктуаций соответствующего свойства и величину флук-
туаций. При наличии большой длины корреляции (100—300 нм)
теплопроводность в температурном интервале 0,1—1 К возра-
стает согласно расчетам, как Тв, где б лежит между 2 и 3. Если
длина корреляции мала (1,5—0,5 нм), то выше 1 К на зависи-
мости х—Т появляется плато. Таким образом, согласно этой
модели экспериментально наблюдаемая зависимость х—Т явля-
ется следствием существования в стеклах одновременно боль-
5—440
65.
Рис. 2.4. Схема двухуровневого потенциала
в модели туннельного перехода под дейст-
вием фононов [37, гл. 1].
ших и малых корреляционных
длин. Эксперименты по рассеянию
света в некоторых стеклах под-
тверждают существование больших
корреляционных длин. Вопросы о
том, является ли это общим прави-
лом и существуют ли такие же кор-
реляционные длины в полимерных стеклах, пока не выяснены.
Эта модель оказалась применимой к частично-кристаллическим
полимерам в предположении существования дополнительной
корреляционной длины, соответствующей размерам кристалли-
тов. Такая трактовка теплопроводности кристаллических поли-
меров будет изложена далее.
Оскотский и Смирнов [2, с. 81—85] провели теоретический
анализ теплопроводности аморфных тел при низких темпера-
турах, также рассматривая стекла как неоднородный упругий
континуум. Использовав представления о флуктуационном рас-
сеянии фононов, они получили выражения для длины свободно-
го пробега фононов, также включающие два параметра, пол-
ностью определяющие теплопроводность стекла: характерную
длину корреляции и средний квадрат величины флуктуации ско-
рости распространения фононов. Рассмотренная модель позво-
ляет качественно объяснить поведение теплопроводности в тем-
пературном интервале примерно 2—20 К-
Рассмотренный подход касается трактовки аномального по-
ведения в криогенной области лишь тепропроводности. Наряду
с этим существуют модели, на основе которых пытаются выяс-
нить механизм аномального поведения не только теплопровод-
ности, но и других свойств стекол при очень низких температу-
рах. Среди них наиболее широкое распространение получила
рассмотренная в гл. 1 модель туннельного перехода под дейст-
вием решеточных колебаний. Поскольку процесс туннелирова-
ния существен именно для объяснения аномального поведения
теплопроводности, рассмотрим его более подробно.
Согласно модели некоторые атомы или группы атомов в
аморфном теле могут находиться в двух положениях, мало от-
личающихся энергетически (рис. 2.4). Потенциал взаимодейст-
вия характеризуется небольшой асимметрией, являющейся след-
ствием различного окружения частиц в положениях, соответст-
вующих двум минимумам. Хотя при низких температурах час-
тицы самостоятельно не могут преодолеть разделяющий их
барьер, переход из одного положения в другое возможен по ме-
ханизму туннельного перехода под действием решеточных ко-
лебаний.
вб
Если предположить, что нулевые колебательные энергии
й<оо частиц в обоих минимумах одинаковы, то энергия в мини-
мумах E = ±[(Ei—Е2)2+а2]1/2, где энергия перекрывания а =
=/г<ооехр(—у,), а р,— пропорционально расстоянию между ми-
нимумами Ах и корню квадратному из высоты барьера V. При
низких температурах можно предположить достаточно однород-
ное распределение параметров Ei—Е% и у.
Взаимодействие этой двухуровневой системы с решеточными
колебаниями возникает в результате деформации потенциала
взаимодействия под действием упругой волны. Если энергия
колебания близка к разности энергий уровней, он поглощается,
а затем некогерентно испускается. Средняя длина пробега
f^co'coth и при достаточно низкой температуре Z-^co1.
Соответственно при таких температурах из уравнения (2.1)
Т3Т~' ~ Т2, что согласуется с наблюдаемой эксперименталь-
но зависимостью. Несмотря на широкую распространенность
этой модели (см., например, [17]) для объяснения низкотемпе-
ратурного поведения стекол (теплоемкость, теплопроводность,
акустические свойства и др.), она не позволяет выяснить при-
роду туннельных состояний и понять, почему для стекол самой
различной природы характерны столь близкие по абсолютному
значению и температурной зависимости теплопроводности.
К числу моделей, с помощью которых делаются попытки
объяснить криогенные аномалии теплоемкости и теплопроводно-
сти стекол, относится модель локализованных неакустических
колебаний, также кратко охарактеризованная в гл. 1. Предпола-
гаемые в ней локализованные неакустические колебания слабо
связанных с основной матрицей частиц могут взаимодейство-
вать с фононами, поглощая фононы соответствующих частот и
оказывая таким путем влияние на теплопроводность.
Рассмотренные представления позволяют выделить в колеба-
тельном спектре аморфных стекол три области '[20], отличаю-
щиеся зависимостью длины свободного пробега от тем-
пературы. В первой, самой низкотемпературной области асС®/?
(а>я — нижний предел полосы частот локализованных неакус-
тических колебаний), и средняя длина свободного пробега оп-
ределяется соотношением Z = £>®-1 (D — параметр, характери-
зующий данное вещество). Подстановка этого значения ~1
в наиболее общее выражение для теплопроводности
1 е
И(Л « — 2 J (2.16)
позволяет получить соотношение, определяющее теплопровод-
ность в этой области спектра [20]:
fegO х3ех
----т‘ J (ZT77
о
где 0^ = х = h&lk^T.
5
67
При этом предполагается дебаевская модель стекла, т. е.
Плотность акустических колебаний пропорциональна со2, однако
различия между продольными и поперечными волнами не дела-
ется.
Две последующие области спектра характеризуются нера-
венствами соЛ<со^(ос и сос<со^(О1, где <ос — верхний предел
полосы частот локализованных колебаний, a coi — максималь-
ная частота акустических колебаний в спектре. В обеих обла-
стях средняя длина пробега является малой величиной. В об-
ласти частот cor<;i(o^|coc можно ожидать, что I будет величиной
порядка межатомных расстояний вследствие сильного рассея-
ния фононов локализованными колебаниями. В области частот
«с<jcoсредняя длина пробега согласно теории [1] и экс-
периментальным результатам '[3] может считаться постоянной
и составляет несколько десятых долей нм. Суммарный вклад в
теплопроводность решеточных колебаний в этих обеих областях
выражается формулой
9,/Г
1 __р
Ъ=~с1 р(и) —----------—dx. (2.18)
3 J (ех — 1р
6^/г
где 0j =
Вид интегралов в уравнениях (2.17) и (2.18) различается,
хотя они и получены из одного и того же уравнения (2.16). Это
является следствием различной зависимости /(со) и р(со) от ча-
стоты.
Уравнения (2.17) и (2.18) могут быть использованы для ана-
лиза экспериментальной зависимости теплопроводности ПММА
(см. рис. 2.2). Прежде чем переходить к такому анализу, необ-
ходимо сделать одно замечание относительно вида р(а»). В ра-
ботах Риза {6, 7] было показано, что экспериментальные ре-
зультаты по теплопроводности ПММА в области 1—300 К уда-
ется описать с точностью 10% суммой трех составляющих:
х = хг + хз + ’<1 (2.19)
где X/ — вклад, обусловленный продольными волнами [выражается уравнени-
ем (2.2)]; — теплопроводность, обусловленная трехмерными колебаниями
(межцепное взаимодействие); Xi— теплопроводность, обусловленная колеба-
ниями скелета цепи.
Выражения для %з и %i имеют вид
х3 = — Cjz3c3/3 (2.20)
О
Xj = (2.21)
О
Хотя такое представление формально подобно модели Тара-
сова для теплоемкости, однако Ризу удалось достичь удовлетво-
рительного соответствия с экспериментом, лишь дифференциро-
68
вав одномерные колебания на продольные и поперечные и при-
няв соотношение скорости звука в продольном и поперечном
направлении равным 2,5. Такая модификация модели Тарасова
все-таки остается формальным приемом описания эксперимен-
тальных результатов. Если рассматривать температурную зави-
симость теплопроводности полимера в той области, где взаимо-
действием цепей можно пренебречь (>60 К), то теплопровод-
ность полностью аморфного ПЭ, полученная экстраполяцией,
в температурном интервале 100—190 К подчиняется закономер-
ности [21]:
х = const Л/гСуТ (2.22)
Эта закономерность еще раз подтверждает сделанные в гл. 1
выводы о решающем влиянии изгибных колебаний на тепловые
свойства цепных структур.
Чой [20] провел анализ теплопроводности ПММА и других
аморфных полимеров на основе уравнений (2.17) и (2.18)
(рис. 2.5 и табл. 2.1). При расчетах для ПММА использованы
следующие значения параметров: D—1,35-10® м/с, 0Н = 4,7 К
и с/ = 1,31-10-6 м2/с, с=1,79-103 м/с. Из двух последних вели-
чин следует, что / = 0,72 нм. Совпадение экспериментальной за-
висимости с расчетной вполне удовлетворительное во всей
температурной области 0,1—350 К.
Значения 6/?, использованные при расчетах %,• хорошо со-
гласуются с результатами, полученными при анализе избыточ-
ной теплоемкости (табл. 2.1, см. также табл. 1.2). Из табл. 2.1
видно также, что значения параметров D и 0R мало зависят
от строения макромолекул. Этим, по-видимому, объясняется
тот экспериментальный факт, что теплопроводность всех
аморфных полимеров лежит в пределах, отличающихся лишь
в 2 раза. Более того, выше 20 К теплопроводность при любой
температуре отличается от среднего значения для многих по-
лимеров лишь на 15%. Этот экспериментальный результат
оказывается полезным для объяснения теплопроводности ча-
стично-кристаллических полимеров.
Несмотря на тесную связь низкотемпературной аномалии
теплоемкости и теплопроводности стекол, существует ряд на-
Таблица 2.1. Параметры D и 0R, полученные при анализе низкотемпературной
теплопроводности аморфных полимеров [20]
Полимер D-10-5, м/с 8Я- К 9’а- К 9%. К
из зависни «ости Gy—T
ПММА 13,5 4,7 4,9 17,5
ПС 6,02 5,2 5,5 16,0
ПК 5,61 4,5 5,0 14,0
ПЭТФ 5,43 4,3 — 15,0
* См. ссылку [43] в гл. 1.
69
Рис. 2.5. Сравнение экспериментальной зави-
симости теплопроводности ПММА от темпе-
ратуры с теоретической зависимостью, соот-
ветствующей уравнениям (2.17) и (2.18)
[20] (точки соответствуют эксперименталь-
ным результатам, которые на рис. 2.2. пред-
ставлены в обобщенном виде).
блюдений, которые не удается объ-
яснить с единых позиций. В част-
ности, теплопроводность стекла ока-
зывается совершенно не чувстви-
тельной к химической чистоте и
термической предыстории образца,
в то время как теплоемкость ока-
зывается чувствительной к этим
параметрам [9]. Хотя между из-
быточной теплоемкостью стекла и
аномалией теплопроводности существует корреляция (см. табл.
2.1), однако предположение, что они обусловлены одними и те-
ми центрами, приводит к выводу об аномально-высокой (по
сравнению с известными дефектами в кристаллах) рассеива-
ющей способности этих центров. Таким образом, действитель-
ные молекулярные механизмы аномального поведения тепло-
физических свойств аморфных веществ оказываются намного
сложней, чем можно было предположить. Вряд ли их удастся
выяснить, изучая лишь температурную зависимость теплоемко-
сти и теплопроводности. Для этого, очевидно, необходимы ком-
плексные исследования с использованием различных физиче-
ских методов. Такие исследования в этой области сейчас ин-
тенсивно проводятся [17, 22, 23].
КРИСТАЛЛИЧЕСКИЕ ПОЛИМЕРЫ
Температурная зависимость теплопроводности кристаллических
полимеров, особенно при низких температурах, значительно от-
личается от таковой для аморфных полимеров. При низких тем-
пературах у кристаллических полимеров отсутствует плато и
температурный ход теплопроводности сильно зависит от степе-
ни кристалличности. Даже при сравнимых степенях кристаллич-
ности поведение теплопроводности разных полимеров может
существенно различаться. Так, для одной группы полимеров
(ПЭВП, ПОМ, ПОЭ) теплопроводность сначала возрастает, до-
стигает максимума, а затем начинает убывать (рис. 2,6, кри-
вая 1). Для другой группы полимеров (ПЭНП, ПЭТФ, ПП)
теплопроводность монотонно повышается вплоть до температу-
ры стеклования, а затем наблюдается пологий излом (рис. 2.6,
кривая 2), т. е. поведение теплопроводности кристаллических
полимеров второй группы и аморфных пол'имеров качественно
70
подобно. По абсолютным значениям теплопроводность полиме-
ров первой группы выше, чем у полимеров второй группы. Для
обеих групп 'выше — 30 К характерно увеличение теплопровод-
ности с ростом степени кристалличности. При более низких
температурах (<20 К) наблюдается противоположная зависи-
мость теплопроводности от степени кристалличности — с повы-
шением степени кристалличности теплопроводность резко па-
дает. Например, для ПЭТФ при 2 К % аморфного образца бо-
лее чем на порядок превышает х образца со степенью кристал-
личности 75% [24].
Это необычное соотношение между теплопроводностью крис-
таллических и аморфных диэлектриков, обнаруженное в послед-
ние годы у полимеров [24, 25], свидетельствует о существова-
нии в кристаллических полимерах дополнительного механизма
рассеяния фононов при очень низких температурах. В связи с
принципиально различным влиянием кристалличности на х при
различных температурах поведение теплопроводности в этих
областях будет рассмотрено раздельно.
Область температур выше 30 К. В этой температурной обла-
сти теплопроводность кристаллических полимеров должна опре-
деляться теплопроводностью кристаллической и аморфной фаз,
их соотношением и формой кристаллитов. Если последний па-
раметр не принимать во внимание, то теплопроводность двух-
фазной полимерной системы (как и любое другое физическое
свойство) может быть представлена простым' аддитивным соот-
ношением:
x = wxKp+ (1 — и»)хам (2.23)
Однако можно ожидать, что форма кристаллитов, а также
сильная анизотропия теплопроводности кристаллической ре-
шетки полимеров играют существенную роль в теплопроводно-
сти частично-кристаллического полимера. Если такой полимер
представить в виде аморфной матрицы со статистически распо-
ложенными в ней сферическими кристаллитами, причем обе фа-
зы считать изотропными, то для оп- х
ределения теплопроводности может
быть использована [11, 26] формула z
Максвелла:
х Хам хКР Хам Л.ч
—— = w---------------- (2.24)
х 2хам хКР + 2хам io1
При известных х, хам и w можно
определить хКр. Такие расчеты были
выполнены [11, 26] для некоторых -2
Рис. 2.6. Характер температурной зависимости
теплопроводности кристаллических полимеров ^-3
I — ПЭВП (ш=0,81); 2 — ПЭТФ (ш=0,51).
71
кристаллических полимеров (ПЭ, ПОМ) для той области тем-
ператур, где степень кристалличности можно считать постоян-
ной. Оказалось, что Хкр^Т’-1, что согласуется с поведением
кристаллических диэлектриков в температурной области, в ко-
торой теплопроводность определяется трехфононными процес-
сами рассеяния (процессы переброса).
Слабым местом такой модели теплопроводности частично-
кристаллических полимеров, даже в области небольших степе-
ней кристалличности, когда полимер можно в первом прибли-
жении рассматривать как аморфную матрицу с кристалличе-
скими включениями (при больших значениях w это предполо-
жение вряд ли вообще выполняется), является игнорирование
микроанизотропии теплопроводности кристаллических областей.
Уже отмечалось, что теплопроводность вдоль цепей по меньшей
мере на порядок выше, чем по ван-дер-ваальсовым связям по-
перек цепей (теоретические оценки дают для этого соотношения
величину 50 [27]). Такое же соотношение, очевидно, характер-
но и для теплопроводности в направлении оси цепи в решетке,
и для теплопроводности аморфной фазы. Поэтому следующим
этапом анализа теплопроводности кристаллических полимеров
на основе модели Максвелла явился учет анизотропии тепло-
проводности кристаллической фазы [28]. Были введены два па-
раметра, характеризующие соотношение теплопроводности
кристаллической и аморфной фаз
*11 = Хкр|]/Хам » 1 и \ = XKpj_/xaM ~ 1 (2.25)
которые позволяют представить основное соотношение в виде
X — ХЯм /2 kj_ 1 1 &|| — 1 \
= w [ —- • -—- + — •-- I (2.26)
и -]- 2хам--------------------------\ 3 &±-|-2 3 Ац 2 j
Так как /гц^>1, то это соотношение практически не зависит
от k ||, и, следовательно, оно не позволяет оценить ни темпера-
турную зависимость, ни абсолютное значение хкрц. Значения
теплопроводности аморфных полимеров выше 30 К различа-
ются очень мало, поэтому хам легко оценить с точностью
»15°/о Для любого полимера в неориентированном состоянии.
Таким образом, единственным параметром в уравнении (2.26)
фактически оказывается хкр± • Он может быть определен на
основании анализа зависимости х от степени кристалличности.
Такой анализ был проведен [20, 28] для тех кристалличе-
ских полимеров, степень кристалличности которых удается
изменять в достаточно широких пределах. Полученная в резуль-
тате такого анализа температурная зависимость теплопровод-
ности кристаллической решетки хкРд (рис. 2.7) наглядно демон-
стрирует упоминавшееся выше деление кристаллических поли-
меров на две группы, для одной из которых теплопроводность
заметно падает с возрастанием температуры, а для другой
очень слабо зависит от температуры, оставаясь во всей области
72
Рис. 2.7. Температурная зависимость теплопро- « Bmlfa-K')
водности кристаллитов в направлении, перпен- *'
дикулярном осям цепей [20]: jjg
I — ПЭ; 2 — ПОМ; 3 — ПЭТФ; 4 — ПП; 5 — темпера-
турная зависимость теплопроводности аморфного ПЭ,
полученная экстраполяцией данных для расплава и
типичная для всех аморфных полимеров (для сравне- 0,5
иия).
0,4
по абсолютному значению меньше,
чем у полимеров первой группы.
Если различия в абсолютных зна-
чениях теплопроводности можно объ-
яснить более слабым межмолекуляр- дг
ным взаимодействием в ПЭТФ и ПП ’
по сравнению с ПЭ и ПОМ, то удиви-
тельно слабую зависимость xKpi от ц,
температуры объяснить значительно
труднее. Единственным логичным
предположением оказывается гипоте- °
за о сильном рассеянии фононов в
кристаллитах таких полимеров на дефектах, что, возможно,
связано с более сложным строением повторяющихся звеньев в
полимерах второй группы. Длина свободного пробега фононов
в полимерах этой группы сопоставима с расстоянием между
отдельными атомами, и это позволило применить к ним мо-
дельный анализ, разработанный для аморфных полимеров [26].
Более высокая теплопроводность кристаллической фазы по
сравнению с аморфной обусловлена различием в их плотностях
что, в свою очередь, позволяет понять соотношение их тепло-
проводностей. В более плотной кристаллической фазе силовые
постоянные связей больше, а тепловое сопротивление меньше.
Это позволяет связать теплопроводность с плотностью. Из мо-
дельного анализа следует:
Икр — Иам
5 g Ркр Рам
Нам Рам
X — Хам л р — рам
--------- » 5,о----------
Нам Рам
(2.27а)
(2.276)
Коэффициент пропорциональности в этих уравнениях оказы-
вается таким же, как и в уравнении (2.6).
Имея в виду изложенные результаты, температурной зависи-
мости теплопроводности кристаллических полимеров (см.
рис. 2.6) выше 30 К может быть дано следующее объяснение
[20]. Для полимеров простого строения (ПЭ, ПОМ) вблизи
30 К хкр1 ^>хам, поэтому уравнение (2.26) можно упростить, и
оно принимает вид
/ 1 4- 2w \
х ~ хам I - I (2.28)
\ 1 — w /
73
В этой области температур х пропорционально хам, и по-
этому с повышением температуры наблюдается возрастание х.
Выше 100 К хКр1 и Хам оказываются сравнимыми по. величине
и х зависит от обоих этих параметров. При больших степенях
кристалличности (а>^=0,7) температурная зависимость х опре-
деляется главным образом температурной зависимостью хКр±,
т. е х должна понижаться с повышением температуры. Таким
образом, у полимеров с высокой степенью кристалличности,
начиная примерно с 30 К, теплопроводность сначала возраста-
ет, достигает максимума в области 80—100 К, а затем начинает
уменьшаться с температурой. С повышением степени кристал-
личности максимум выражен более четко и смещен в область
более низких температур. Теплопроводность образцов (полиме-
ров первой группы с низкой степенью кристалличности
(о>^0,4) в основном определяется хам, и для таких образцов
наблюдается непрерывное возрастание х с температурой вплоть
до появления пологого максимума выше 200 К. Для полимеров
второй группы Хкрх от температуры в этой области практиче-
ски не зависит, поэтому поведение теплопроводности образцов
с различной степенью кристалличности выше 30 К близко к по-
ведению Хам.
Низкотемпературная область. Наиболее важная особенность
низкотемпературной теплопроводности кристаллических полиме-
ров связана с необычным влиянием степени кристалличности на
абсолютное значение и температурную зависимость х. Выше
«30 К формула (2.26) предсказывает повышение х с увеличе-
нием w, что наиболее отчетливо наблюдается экспериментально
для полимеров первой группы. Поскольку можно ожидать, что
хКр1 по крайней мере не уменьшается с понижением темпера-
туры, то формула (2.26) предсказывает возрастание х с увели-
чением степени кристалличности также и при более низких
температурах. Однако экспериментальная зависимость оказыва-
ется совершенно иной (рис. 2.8). В
области около 10 К зависимость теп-
лопроводности от степени кристал-
личности меняется качественно. Ни-
же этой температуры повышение w
приводит к уменьшению х, причем
влияние это столь велико, что разли-
чие в теплопроводности аморфного
образца и высококристаллического
может достигать десяти и более раз.
Рис. 2.8. Зависимость теплопроводности от тем-
пературы для ПЭТФ различной степени крис-
талличности [сплошные линии — усредненные
экспериментальные данные, пунктирные — рас-
чет по уравнению (2.29)] [24]:
/ — О1=0; 2 — w—0,05; 3 — и>=о,14; 4 — w—Q,7b.
74
Столь же сильным оказывается и различие в температурной
зависимости теплопроводности.
Уже в ранних исследованиях [7, 29] было обращено (внима-
ние на важную роль кристаллитов в рассеянии фононов при
низких температурах. Наряду со структурным рассеянием, ха-
рактерным для аморфной фазы, рассматривался дополнитель-
ный механизм рассеяния в системе кристаллит — аморфная
фаза, и была установлена корреляция толщины кристаллитов и
температурно-независимого параметра, появляющегося в урав-
нениях типа (2.2) при учете дополнительного механизма рас-
сеяния на кристаллитах. В настоящее время основная идея
этого подхода формулируется следующим образом [19, 20, 24,
27]. Наличие кристаллитов в аморфной матрице обусловливает
длину корреляции порядка размеров кристаллитов (10—15 нм),
что приводит к дополнительному рассеянию фононов и умень-
шению теплопроводности. Такой подход правомерен, лишь ког-
да средняя длина пробега больше длины корреляции, т. е. при
температурах в несколько градусов К. В последнее время эта
идея о дополнительной длине корреляции была рассмотрена
количественно.
При анализе экспериментальных результатов, приведенных
на рис. 2.8, следуя теории Клеменса предполагалось [24], что
структурный фактор Ф(р) в уравнении (2.15) для времени ре-
лаксации пропорционален структурному фактору, характеризую-
щему флуктуацию электронной плотности Фг(.р). Это означает,
что в первом приближении длина корреляции может быть
отождествлена со средним значением большого периода. При
статистическом расположении кристаллитов (изотропное со-
стояние) Ф/(р) характеризуется сферической симметрией.
В предположении, что доминирующий вклад в низкотемпера-
турную теплопроводность вносят фононы с волновым вектором
pd ~ (4kBT/ft.c) (с — скорость звука), время релаксации в урав-
нении (2.16) оказывается т(Т) »r(.pd) « [Ф^)]-1. Поскольку
Ф(рл) ~Ф/(р), а 1=сх, то из уравнения (2.1) для теплопровод-
ности частично-кристаллического полимера можно получить
х(Т) =
1Ф? (Pd) ]ам
l$z(Pd)]
хам(7')
(2.29)
ЗнаяФ;(р) из малоугловых рентгеновских данных, х(Т)
можно определить на основании Хам(Т’). Полученные таким ме-
тодом зависимости (см. рис. 2.8) достаточно хорошо иллюстри-
руют основную тенденцию изменения теплопроводности с изме-
нением степени кристалличности.
При анализе низкотемпературной теплопроводности анизо-
тропного (экструдированного) и изотропного ПЭ также предпо-
лагалось [27], что фононы рассеиваются при прохождении че-
рез области, в которых скорость звука различна (кристаллиты
и аморфные области). Количественный анализ проводился на
основе теории Моргана — Смита [19], согласно которой общая
75
средняя длина свободного пробега фононов вдоль'направления
экструзии может быть представлена суммой
= /71 4- /71 (*) + (1 - (2.30)
где li(k) и ls(k) — средняя длина пробега для области продольной длины кор-
реляции и области малой корреляционной длины в аморфной фазе соответ-
ственно.
Величина продольной длины корреляции принималась рав-
ной 10 нм в соответствии с размером кристаллитов, а для дли-
ны корреляции в аморфной фазе принималось значение 0,8 нм.
Константа 1ь отражает граничные условия (~10~2 см). Таким
образом, член 1г1 характеризует рассеяние фононов, возникаю-
щее вследствие флуктуации скорости звука в системе последо-
вательно чередующихся кристаллитов и аморфных прослоек, а
член (1—w)ls~l отражает процесс рассеяния в аморфных обла-
стях. Упоминавшиеся при рассмотрении теплопроводности
аморфных полимеров большие длины корреляции (100—
300 нм) в частично-кристаллических полимерах вряд ли сущест-
вуют. Рассеянием в кристаллитах можно пренебречь в связи с
параллельным расположением цепей в кристаллитах.
Уравнение (2.30) можно применять и для анализа теплопро-
водности изотропных полимеров при низких температурах
(<30 К), когда тепловым сопротивлением кристаллитов, пер-
пендикулярным осям макромолекул, можно пренебречь по
сравнению с тепловым сопротивлением аморфных областей.
При более высоких температурах необходимо учитывать вклад
кристаллитов, что требует знания взаимного расположения
кристаллитов и аморфных областей. Если считать, что кристал-
литы и аморфные области соединены последовательно, причем
оси макромолекул во всех кристаллитах перпендикулярны на-
правлению теплового потока, уравнение (2.30) приобретает вид
i-Цк) = /7‘+ /Г1!*) 4- (1 — w)Z~’(fe) (2.31>
где lu(k) — средняя длина пробега фононов, рассеяние которых в кристалли-
тах перпендикулярно осям с происходит по механизму переброса.
Однако такое взаимное расположение кристаллитов и
аморфных областей в изотропных полимерах не реализуется.
Для статистического распределения осей кристаллитов расчеты
резко усложняются.
Сравнение теоретических зависимостей х—Т, построенных
по уравнению (2.30), с экспериментальными результатами для
ПЭ со степенью кристалличности w = 0,6 показано на рис. 2.9.
При расчетах принимались указанные выше значения длин
корреляции. Наилучшее совпадение расчетных и эксперимен-
тальных зависимостей достигалось при значениях флуктуаций
Д(2=0,08 и As2 = 0,25. Последнее значение оказывается харак-
терным для аморфных веществ (например, селена [19]). Ввиду
76
Рис. 2.9. Зависимости теплопроводности от темпе-
ратуры, построенные по уравнению (2.30) [27]:
/ — расчет для одной длины корреляции — 10 им (w=0);
2 — расчет для двух длин корреляции — 10 н 0,8 нм (w =
=0,6); 3 — расчет с учетом рассеяния по механизму пе-
реброса [см. уравнеине (2.31)]; О — изотропный ПЭ; Ф —
экструдированный ПЭ.
большого числа допущений столь хоро-
шее совпадение экспериментальных и
расчетных данных авторы [27] склонны
считать случайным. Тем не менее рас-
смотренные модельные расчеты нагляд-
но передают основные черты поведения
теплопроводности частично-кристалли-
ческого ПЭ, и можно полагать, что тео-
рия Моргана — Смита окажется плодотворной для количест-
венного описания теплопроводности как аморфных, так и крис-
таллических полимеров.
В рассмотренных до сих пор моделях роль границ кристал-
лит — аморфная область в рассеянии фононов не учитывалась,
хотя теоретически показано [30], что различие в свойствах, оп-
ределяющих упругие характеристики на межфазной границе
двух различных фаз (плотность, скорость звука), приводит к
сильному рассеянию фононов при низких температурах, причем
термическое сопротивление межфазной области тем выше, чем
больше разница в плотностях и скоростях звука. Поскольку
плотность и скорость звука для аморфной и кристаллической
фаз в полимерах различны, то можно предполагать проявление
этого эффекта и ib частично-кристаллических полимерах.
Вопрос о роли теплового сопротивления межфазных границ
в кристаллических полимерах был рассмотрен [25, 20] на мо-
дели кристаллического полимера, в которой тепловой поток
распространяется вдоль структуры, в которой последовательно
чередующиеся кристаллиты и аморфные прослойки соединены
параллельно с протяженной аморфной областью. Основное со-
отношение для теплопроводности такой модели имеет вид
— =----------------------- + (1 — х) (2.32)
Нам 2wxaM(rft/d) + (х — w)
где х — доля кристаллита в поперечном сечении структурной модели; d — раз-
мер кристаллита в направлении теплового потока; гь — тепловое сопротивление
межфазного слоя.
При выводе (2.32) тепловым сопротивлением кристаллитов
пренебрегали. Формула (2.32) содержит три параметра:
гь(Т) и Хам(Т’) (размер кристаллитов d определяется из рент-
геноструктурных данных), причем два из них зависят от темпе-
ратуры. Для полимера, аморфизующегося при закалке, ха(7)
может быть измерена. Из оставшихся двух параметров гь не-
посредственно измерить. нельзя, но его можно оценить по тео-
77
рии Литлла [30], согласно которой при очень низких темпера-
турах
15*3 / г, ГЛ-1
+ Г’3 (2.33)
где Г; и Г/ — трансмиссионные коэффициенты для продольных и поперечных
волн.
Зная эти соотношения, необычную зависимость теплопровод-
ности от степени кристалличности при очень низких темпера-
турах (см. рис. 2.8) можно качественно объяснить следующим
образом. При высоких температурах граничное тепловое сопро-
тивление пренебрежимо мало, и, поскольку теплопроводность
кристаллической фазы более высокая, чем аморфной, с повы-
шением степени кристалличности х возрастает. При понижении
температуры г* возрастает, и при некоторой температуре (для
ПЭТФ эта температура составляет — 15 К) граничное сопро-
тивление не только компенсирует возрастание теплопроводно-
сти кристаллитов с понижением температуры, но и приводит к
более сильному падению теплопроводности образца с большей
степенью кристалличности. Поэтому ниже этой температуры
взаимное расположение зависимостей для различных степеней
кристалличности меняется.
Наличием термического сопротивления на границе кристал-
лит — аморфная фаза можно объяснить и изменение наклона
зависимостей х—Т. Для аморфных веществ в температурном
интервале 2—10 К экспериментально наблюдается х~Т1/2, и
поскольку для полностью кристаллических материалов в этой
же температурной области х~Т3, то для частично-кристалличе-
ских полимеров можно ожидать х~Тв (0,5<б<3). С увеличе-
нием степени кристалличности доля межфазных границ возрас-
тает, что приводит к возрастанию вклада граничного рассеяния
фононов и, следовательно, к увеличению показателя б.
Использование уравнения (2.32) позволило дать количест-
венное описание экспериментальных данных для ПЭТФ [25],
для которого удается в широких пределах изменять степень
кристалличности и минимизиройать число входящих в уравне-
ние (2.32) параметров. Распространение этого подхода на дру-
гие полимеры является более сложной задачей. Существует еще
лишь одна серия систематических данных по низкотемператур-
ной теплопроводности ПЭ (1,2—20 К), в которых степень кри-
сталличности образцов менялась от 0,43 до 0,81 [29]. С по-
мощью изложенного подхода их удалось количественно (точ-
ность да 10%) описать выше 3 К, введя ряд упрощающих допу-
щений [20], которые, однако, не могут исказить общего
заключения о применимости этого подхода для частично-кри-
сталлических полимеров.
Представляет интерес на основе такого подхода оценить па-
раметр гь для полимеров разной химической природы и различ-
ной структуры. Результаты таких оценок [20] (табл. 2.2) пока-
78
Таблица 2.2. Параметры, полученные при анализе низкотемпературной (4 К)
теплопроводности кристаллических полимеров [25, 29, 32]
по уравнению (2.32) [20]
Полимер Степень кристаллич- ности W И-102, Вт/(м-К) Иа-102, Вт/(м-К) d, им _2—..юз м-К/Вт ГйГ3.105, М2-К/Вт
ПЭ 0,81 2,85 3,2 28,2 1,95 5,5
0,43 1,2 3,2 11,3 4,9 5,5
пом 0,71 2,8 3,8 28,2 1,27 3,6
ПП 0,57 1,2 3,8 17,0 5,0 8,5
Полиамид 66 0,42 1,0 3,8 14,5 7,5 10,8
ПЭТФ 0,39 0,95 3,75 14,1 8,7 12,2
ПХТФЭ 0,34 1,0 3,8 28,2 3,2 9,1
зывают, что термическое сопротивление гь мало зависит от
строения полимера и степени кристалличности.
Таким образом, в заключение можно отметить, что в настоя-
щее время существуют две физически обоснованные модели,
позволяющие качественно объяснить, почему теплопроводность
кристаллического полимера при достаточно низких температу-
рах всегда ниже, чем теплопроводность этого же полимера в
аморфном состоянии. Однако фигурирующие в этих моделях
параметры (г* или АД А;2 и длина корреляции в аморфной фа-
зе) не поддаются прямому измерению, что осложняет количест-
венный анализ и влечет за собой введение ряда допущений.
АНИЗОТРОПИЯ ТЕПЛОПРОВОДНОСТИ
ОРИЕНТИРОВАННЫХ ПОЛИМЕРОВ
В ориентированных аморфных и кристаллических полимерах
существует анизотропия теплопроводности. Степень анизотро-
пии зависит от степени ориентации, степени кристалличности
и температуры. Теплопроводность всегда оказывается выше в
направлении ориентации, чем в направлении, перпендикуляр-
ном ориентации, а также в изотропном состоянии. Наиболее
детально анизотропия теплопроводности исследована для
ПММА, а среди кристаллических полимеров — для ПЭ. На
примере этих полимеров рассмотрим основные закономерности
анизотропии теплопроводности ориентированных полимеров.
Аморфные полимеры. Ориентация аморфных полимеров при-
водит к увеличению доли сегментов макромолекул, направлен-
ных преимущественно в направлении ориентации. Поэтому
единственной физической причиной появления анизотропии теп-
лопроводности у аморфных некристаллизующихся полимеров яв-
ляется увеличение доли главных валентностей, расположенных
вдоль оси ориентации, и уменьшение доли валентностей, рас-
положенных поперек оси ориентации. Айерман [8] использовал
79
рассмотренную выше решеточную модель (см. рис. 2.3) и эти
общие соображения для расчета теплопроводности вдоль (х g )
и поперек (xj_) оси ориентации и получил простое соотноше-
ние, которое должно выполняться для любой степени ориен-
тации:
xjj"1 + 2х“' = Зх-1 (2»34)
Это уравнение следует и из агрегатной модели [31].
Хансен и Хоу [13] на основе теоретического анализа пред-
ложенной ими модели (см. с. 62) получили иное соотношение,
характеризующее анизотропию теплопроводности аморфных по-
лимеров:
х/хх = (Хц/х)1^ (2.35)
Несмотря на то что уравнения (2.34) и (2.35) сильно раз-
личаются по виду, их проверка на ПС, ПММА и ПВХ показы-
вает [И], что точность предсказания анизотропии х этими урав-
нениями примерно одинакова и достаточно высока (несколько
процентов). Это в значительной мере объясняется невысокими
степенями ориентации, которых удается достичь на практике
для аморфных полимеров с обычными молекулярными массами.
Кристаллические полимеры. Закономерности анизотропии
теплопроводности кристаллических полимеров, которые стали
предметом интенсивных количественных исследований лишь в
последнее время [28, 32—39], оказываются значительно 'более
сложными. Если для аморфных полимеров коэффициент анизо-
тропии теплопроводности xi/xj. обычно не превышает 2, то
соответствующий коэффициент для кристаллических полимеров
может достигать 50 и выше (рис. 2.10). Эти исследования по-
зволили выявить две важные закономерности. При высоких тем-
пературах х и непрерывно возрастает с увеличением степени
вытяжки X, в то время как xj_ сначала уменьшается по сравне-
нию с теплопроводностью изотропного образца, а затем дости-
гает постоянных значений и не зависит от X. Кроме приведен-
ных на рис. 2.10 данных для ПЭ.ВП, демонстрирующих эту за-
кономерность, такое поведение характерно для ,ПП, ПОМ,
ПЭТФ и ПВДФ [33—35]. Другая особенность состоит в том,
что при низких температурах (<30 К) анизотропия теплопро-
водности даже для предельно-ориентированных образцов ока-
зывается чрезвычайно малой. Так, для ПЭВП при низких тем-
пературах (<10 К) для всех степеней вытяжки х» «х«1,5 хц,.
Для объяснения сильной анизотропии теплопроводности
ориентированных кристаллических полимеров при обычных
температурах используют три модельных подхода. Если един-
ственной причиной анизотропии теплопроводности в ориентиро-
ванном состоянии считать ориентацию вдоль направления вы-
тяжки кристаллитов, коэффициент анизотропии тепло-
проводности которых может достигать 100, то ориента-
80
Рис. 2.10. Зависимость отношений х в /х и х± /х при 300 К от степени вытяжки
для ПЭВП (7) и ПММА (2) [11, 35].
Рис. 2.11. Теплопроводность (х-10) ориентированного ПЭ при 323 К в зависимо-
сти от функции ориентации кристаллитов и степени вытяжки X [20] [теорети-
ческие кривые для х jj (1) и х± (2) построены по уравнениям (2.37) и (2.38)].
ция кристаллитов может быть охарактеризована функцией
А = у [3< cos2<?> — 1] 0^/с<1 (2.36)
Из рентгеноструктурных исследований известно (см., на-
пример, ссылку [90] в гл. 4), что для многих кристаллических
полимеров различной степени кристалличности характерна еди-
нообразная зависимость fc от Л: при небольших степенях вытяж-
ки fc резко возрастает, а при 7. «4—5 достигает постоянного
значения на уровне 0,9. Аморфная же фаза при таких степенях
вытяжки ориентирована слабо. Учитывая эти соображения, а
также слабую анизотропию теплопроводности ориентированных
аморфных полимеров, для расчета х± и х ц ориентирован-
ных кристаллических полимеров можно использовать модифи-
цированную модель Максвелла. Расчеты приводят к следую-
щим соотношениям:
— иам
----------- ae W
+ 2xaM
k, -1
1 -]- (COS2<p)
2
1 + (COS2<p)
1
•----(sin2®)
йц + 2 2 4
1
2 “ т'
+ — <sin2<p>
(2.37)
йц — 1
= W
2
+2
~ 1
k^+2
X|| — Хам йц — 1
------------ = W !---
Хц -f- 2xaM-kn -|" 2
£.1-1
*11 +2
Й..-1
<sin2<p> + — <COS2<p>
й|| +2
- (sin2cp) + (cos2<p) j
(2.38)
W
6—44 0
81
В этих уравнениях, так же как и ,в уравнении (2.26), пред-
полагается, что k || = Хкр|1 1; <cos2 <р> и <sin2 <р> могут
Иам
быть рассчитаны ino fc, а оценено из данных по изотропным
полимерам (рис. 2.7), так что уравнения (2.37) и (2.38) факти-
чески не содержат переменных параметров. Сравнение данных,
предсказанных этой моделью, с экспериментальными (рис. 2.11)
показывает, что она правильно передает основную тенденцию
поведения анизотропии теплопроводности. Количественное соот-
ветствие наблюдается для Xi во всей области Хи fc, а для
х ||—лишь при Х=С4 (fc~0,9). При Х>4 экспериментальные
значения резко возрастают, что свидетельствует о важной роли
каких-то дополнительных факторов, не связанных с ориентацией
кристаллитов. Эти факторы оказываются существенными лишь
для теплопроводности вдоль оси ориентации, так как модель хо-
рошо передает поведение х± и при больших степенях вытяжки.
Кроме того, эта модель правильно передает зависимость
анизотропии теплопроводности от степени кристалличности.
Коэффициент анизотропии для образца ПЭ с большой степенью
кристалличности (w = 0,74) при Х=3 составляет (х|/Х±, «5,
а при w = 0,45 (xj[/x±) «2,5 [28, 38]. Это согласуется с уравне-
ниями (2.37) и (2.38). Из уравнения (2.38) следует, что для
предельной ориентации (fc=l) х ц =ха(1+2ш)/(1—w), т. е. х л
должна так же, как и хам, монотонно возрастать с температу-
рой выше — 30 К. С другой стороны, для изотропных полимеров
(/с=0) теплопроводность, например, ПЭ (см. рис. 2.6),умень-
шается с повышением температуры в интервале 100—300 К.
Таким образом, эта модель предсказывает монотонное измене-
ние наклона зависимости хц—Т с отрицательного на положи-
тельный при возрастании fc от 0 до 1. Это предсказание коли-
чественно не проверялось. Тенденция монотонного возрастания
Хц с температурой действительно существует. Из (2.37) сле-
дует, что Xi даже при [с=1 зависит как от хам, так и от хКр±.
Температурная зависимость x_l должна быть такой же, как и
зависимость х, что также наблюдается экспериментально.
Для объяснения резкого возрастания теплопроводности у
сильноориентированных кристаллических полимеров, особенно
у сверхвысокоориентированного высокомодульного ПЭ, исполь-
зуют модель «статистических кристаллических мостиков» (см.
гл. 4, с. 192). Резкое повышение теплопроводности х л при боль-
ших степенях вытяжки, так же как и возрастание модуля Дц.
объясняют существованием кристаллических мостиков между
кристаллитами соседних микрофибрилл. Теплопроводность та-
кой модели, состоящей из микрофибрилл с чередующимися кри-
сталлитами и аморфными областями, соединенными параллель-
но с межкристаллитными мостиками, определяется выраже-
нием [33]
1 1 — а _________а_______
*|Г= *кр|| + Чр|| + (1 —*)хам (2'39>
82
п
где а и b — геометрические характеристики модели.
Выше 30 К при &^>1% ЬхКр в > (1—&)хам, и роль межкрис-
таллитных мостиков становится определяющей. В этом случае
уравнение (2.39) переходит в
xll“xKpnW-“)+a]-1 (2.40)
Аналогичный анализ модуля упругости такой модели позво-
ляет связать теплопроводность с модулем. Для ПЭ в этой об-
ласти температур, где модуль не зависит от температуры (об-
ласть плато «200 К)
х,.(Т) £ц(200К)
Т(П= £----------- (2>41)
xkP||U; скрц
Значения х.ц(Т) и Е л (200 К) известны из эксперимента, и
поскольку £'Кр = 240 ГПа, то можно оценить хкрг.. Такие оценки
приводят к хКр в =31 Вт/(м-К) при 100 К, что примерно в
200 раз превышает хами еще раз подтверждает, что хкрц
Теплопроводность перпендикулярно оси ориентации опреде-
ляется выражением
х . = х п . (1 — а) +--------------
± KPJ-k &/хкр±+(1-6)/х,
« (! — а)хкр± +ахам
(2.42)
т. е. Xj_ практически не зависит от Ь, что наблюдается экспери-
ментально (см. рис. 2.10). Принимая хам = 0,18 Вт/(м-К), имеем
Xkp.l~0,6 Вт/(м-К), что совпадает с оценками, сделанными на
основании модифицированной модели Максвелла (см. рис. 2.7).
Для анализа анизотропии теплопроводности (и температу-
ропроводности) использована агрегатная модель [37, 38], осно-
ванная на следующих представлениях. Предполагается, что со-
седние кристаллические ламели ориентированы друг относи-
тельно друга коррелированным образом и вместе с аморфными
областями образуют кластер, в котором кристаллиты и аморф-
ные области сильно взаимосвязаны. Такой кластер является ис-
ходной сруктурной единицей агрегата. На основании простого
анализа получено выражение для анизотропии теплопровод-
ности:
О II
хкл±
Хкл||
(cos2<p)
(2.43)
где хкл ц /хкл± — анизотропия теплопроводности кластера.
Если считать, что ориентация кластеров однозначно описы-
вается функцией ориентации кристаллитов fc (что в действи-
тельности вряд ли справедливо), тогда анизотропия теплопро-
водности может быть оцределена на основании хКлц и хКЛ1-
Несмотря на хорошее соответствие 'проведенных расчетов с
экспериментальными результатами [37, 38], этот модельный
подход имеет ряд недостатков, основным из которых является
предположение о неизменности кластера при ориентации кри-
сталлического полимера.
Относительно природы другой закономерности — резкого
уменьшения анизотропии теплопроводности ниже 30 К и ее
практического исчезновения ниже 10 К — в настоящее время
можно лишь предположить, что поскольку в этой области тем-
ператур теплопроводность обусловлена механизмом рассеяния,
связанным с термическим сопротивлением межфазных границ
кристаллит — аморфная область, а также наличием длины кор-
реляции в флуктуациях скорости звука, то эти процессы рас-
сеяния оказываются малочувствительными к ориентации как
кристаллитов, так и аморфных областей.
ЛИТЕРАТУРА
1. Займан Дж. Электроны и фононы. Пер. с англ./Под ред. В. Л. Бонч-Бруевн-
ча. М., Издгтиялмт, 1962. 488 с.
2. Оскотский В. С., Смирнов И. А. Дефекты в кристаллах н теплопроводность.
М„ Наука, 1972. 160 с.
3. Берман Р. Теплопроводность твердых тел. Пер. с англ./Под ред. В. 3. Кре-
сина. М., Мнр, 1979. 286 с.
4. Клеменс П.—В кн.: Физика низких температур. Пер. с англ./Под ред.
А. И. Шальникова. М., Издатинлит, 1959, с. 224—314.
5. Миснар А. Теплопроводность твердых тел, жидкостей и газов и их ком-
позиций. Пер. с фр./Под ред. Э. Шпильрайна. М., Мир, 1968. 464 с.
6. Reese W. — J. Appl. Phys., 1966, vol. 37, N 2, p. 864—868; N 8, p. 3227—
3230.
7. Reese W.— J. Macromol. Sci., 1969, pt A, vol. 3, N 7, p. 1257—1295.
8. Eiertnann K.— Koll.-Ztschr. u. Ztschr. Polymere, 1964, Bd 198, N 1—2,.
S. 5—16.
9. Stephens R. B. — Phys. Rev., 1973, B, vol. 8, N 6, p. 2896—2905.
10. Stephens R. B. —Phys. Rev., 1976, B, vol. 13, N 2, p. 852—865.
11. Knappe W.~Mv. Polymer Sci., 1971, vol. 7, N 4, p. 477—535.
12. Lohe P. — Koll.-Ztschr., 1965, Bd 206, N 1, p. 115—119.
13. Hansen D., Но С. C.—J. Polymer Sci., 1965, pt A, vol. 3, N 3, p. 659—
670.
14. Morgan G. J., Scovell P. D.—-J. Polymer Sci., Polymer Lett. Ed., 1977,
vol. 15, N 4, p. 193—198.
15. Fesciyan S., Frisch H. L.—J. Chem. Phys., 1977, vol. 67, N 12, p. 5691 —
5694.
16. Bondi A. Physical Properties of Molecular Crystals, Liquids and Glasses.
N. Y„ J. Wiley, 1968. 500 p.
17. Pohl R. 0., Salinger G. L. — Ann. N. Y. Acad. Sci., 1976, vol. 279, N 4,
p. 150—172.
18. Klemens P. G. — In: Non Crystalline Solids/Ed. by V. C. Frechette. N. Y.,
J. Wiley, 1960, p. 508—529.
19. Morgan G. J., Smith D.—J. Phys., 1974, Ser. C, vol. 7, N 4, p. 649—664.
20. Choy C. L. — Polymer, 1977, vol. 18, N 10, p. 984—1004.
21. Baur H. — Koll.-Ztschr. u. Ztschr. Polymere, 1971, Bd 247, N 1—2, S. 753—
762.
22. Amorphous Solids: Low Temperature Properties/Ed. by W. A. Phillips. Hei-
delberg, Springer Verlag, 1980.
84
23. Smith T. L., Anthony P. J., Anderson A. C. — Phys. Rev., 1978, B, vol. 17r
N 12, p. 4997—5009.
24. Assfalg A. — J. Phys. Chem. Solids, 1975, vol. 36, N 12, p. 1389—1396.
25. Choy C. L., Greig D.—J. Phys., 1975, Ser. C, vol. 8, N 19, p. 3121—3130.
26. Eiermann K.— Koll.-Ztschr. u. Ztschr. Polymere, 1965, Bd 201, N 1—2,
S. 3—15.
27. Burgess S., Greig D.—J. Phys., 1975, Ser. C, vol. 8, N 11, p. 1637—1648.
28. Choy C. L., Young K. — Polymer, 1977, vol. 18, N 8, p. 769—776.
29. Kolouch R. J., Brown R. G.—’J. Appl. Phys., 1968, vol. 39, N 8, p. 3999—
4003.
30. Little Г. A. — Canad. J. Phys., 1959, vol. 37, N 3, p. 334—349.
31. Hannig J. — J. Polymer Sci., 1967, pt C, N 16, p. 2751—2763.
32. Choy C. L„ Greig D.—J. Phys., 1977, Ser. C, vol. 10, N 2, p. 169—179.
33. Gibson A. G. e. a.—J. Polymer Sai., Polymer Lett. Ed., 1977, vol. 15, N 4,
p. 183—192.
34. Choy C. L., Luk W. H., Chen F. C. — Polymer, 1978, vol. 19, N 2, p. 155—
162.
35. Choy C. L., Chen F. C., Luk W. H.—J. Polymer Sci., Polymer Phys. Ed.,
1980, vol. 18, N 6, p. 1187—1207.
36. Gibson A. G., Greig D., Ward I. M. — Ibid., p. 1481—1482.
37. Pietralla M. — Coll. a. Polymer Sci., 1981, vol. 259, N 1, p. 111—129.
38. Kilian H.-G., Pietralla M. — Polymer, 1978, vol.. 19, N 6, p. 664—672.
39. Choy C. L., Ong E. L., Chen F. C.—J. Appl. Polymer Sci., 1981, vol. 25,
N 7, p. 2325—2336.
Глава 3
ТЕПЛОВОЕ РАСШИРЕНИЕ
Тепловое расширение, или более широко — тепловая деформа-
ция, представляющее собой^измененле размеров тела.вследствие,
изменения температуры, обусловлено, так же как и теплопро-
водность, ангармоничностью "колебаний составляющих тело ча-
стиц. Наряду с теплоемкостью и теплопроводностью тепловое
расширение отражает силы, действующие между частицами, и
особенности тепловых колебаний связанньЦ"-мёжду~сбббй''"час-
тиц. Цепное строение макромолекул и различие в силах меж-
цепного и внутрицепного взаимодействия, приводящее к появле-
нию волн изгиба в цепных структурах, является причиной еще
одной характерной особенности тепловых свойств полимеров—
наличия отрицательных коэффициентов теплового расширения
в кристаллах и ориентированных полимерах и сильной анизо-
тропии теплового расширения в них.
УРАВНЕНИЕ СОСТОЯНИЯ ТВЕРДЫХ ПОЛИМЕРОВ.
ПАРАМЕТР ГРЮНАЙЗЕНА
Представления о тепловом расширении тесно связаны с урав-
нением состояния твердых тел. Вывод уравнения состояния
твердых тел, в том числе и полимерных, основан на гипотезе
Грюнайзена [1, 2], согласно которой внутренняя энергия твер-
дого тела может быть представлена в виде суммы двух компо-
85
нентов. Один из них является энергией решетки UL(V) и зави-
сит только от объема, а другой — характеризует зависящую от
; температуры энергию Ut(T,V), связанную с тепловыми колеба-
1 ниями атомов или молекул около их равновесных положений.
В связи с этим выражение для энергии кристаллической решет-
ки, образованной гармоническими осцилляторами, может быть
представлено в виде
= + + (3.1)
I 4
где nt — колебательные квантовые числа.
Зная полную энергию системы, можно определить статисти-
ческую сумму ZM=exp(—U/kB Т), затем свободную энергию F,
после чего уравнение состояния находится дифференцированием
•свободной энергии по объему. Таким образом, имеем [3]:
(3-2)
5= -йБТ1п2ш=^0 + йБт2)1п 1
В этих уравнениях Uo характеризует температурно-незави-
симое слагаемое внутренней энергии и является суммой энергии
решетки и нулевой энергии (энергии основного состояния).
Производные от частот со, по объему нельзя получить априори,
не имея колебательного спектра. Очевидно, что частоты с
уменьшением объема должны возрастать, а с увеличением объ-
ема— уменьшаться. Поэтому зависимость частот от объема мо-
жет быть представлена~|Т7 в виде-
’ - - —- у & \ d In со,
—[—Ч =------------= — У; (3.4)
сог(ау/г a in у v ’
где ус — положительная
Таким образом,
.ления имеет вид
константа, называемая параметром Грюнайзена.
с учетом постоянной yi выражение для дав-
। Грюнайзен предположил, что при изменении объема часто-
| ты всех колебательных мод меняются одинаковым образом.
®6
Тогда уравнение (3.3) принимает вид
(3.6)
Поскольку сумма во втором члене этого уравнения опреде-
ляет зависящую от температуры часть внутренней энергии, то
из (3.6) следует уравнение состояния Ми — Грюнайзена:
/д£7,. \ у
Р= “ДД +v (U~Uo)=P' + YPr (3-7)
Давление представляется суммой двух слагаемых. Первое I
слагаемое— (dU0)/дУ)т называют внутренним давлением, так I
как оно определяет зависимость части внутренней энергии твер- Л
дого тела, обусловленной статическим взаимодействием частиц, I]
от объема. ||
Отношение (U—Uq)[V называют термическим давлением, так у
как оно обусловлено тенденцией твердого тела к расширению I
под действием колебаний решетки. J
Исходя из уравнения состояния (3.7), найдем связь теплоем- р
кости Су, коэффициента объемного теплового расширения а и !
модуля объемного сжатия Кт- Из термодинамики следует [4]: ,
/ P2F \ / дР \ 1
= = -Г — (3-8)
\ (/V2 /7' \ uV Jт'
Дифференцирование уравнения (3.2) для свободной энергии
сначала по температуре, а затем по объему с учетом предполо-
жения Грюнайзена дает
или с использованием соотношения (1Л|: П'1 Ц
V Cv j ’ *""*
(3-и>
Это уравнение называется уравнениелгГрюнайзена. Оно обыч-
но используется для вычисления Y, WK 1Ьгк-все"Остальные вхо-
дящие в него величины можно определить экспериментально.
Из изложенного ясно, что введение постоянной Грюнайзена
позволяет выразить уравнение состояния и входящие в него
величины а и Кт через теплоемкость Су и зависящую от темпе-
ратуры часть внутренней энергии. Это в свою очередь дает воз-
можность использовать теоретические расчеты теплоемкости и
87
внутренней энергии твердых тел для получения уравнения со-
стояния. В частности, из теории Дебая на основании (3.3) на-
ходим уравнение состояния Дебая:
/ dFp \ = _/ дГр.Х / deD \ = _( ди° \ , иР
\ dV /г \ ' т\ dV /т \ dV )т У V
д In 0г>
у — —---------~
din У
(3.12а)
(3.126)
где UD — внутренняя энергия твердого тела Дебая.
Такое определение у предполагает опять, что все частоты, в
том числе и дебаевская частота cod, одинаковым образом зави-
сят от объема.
Так как причиной теплового расширения твердых тел явля-
ется энгармонизм колебаний составляющих его частиц, то .кон-_
^танта у в_ уравнении (3.11) отражает степень ангармонизма,
'шоскольку~^се^)стальные параметры, входящие в уравнение
(3.11), могут быть получены в гармоническом приближении.
Уравнение (3.11) позволяет качественно установить температур-
ную зависимость а. Так как у согласно гипотезе Грюнайзена от
температуры не зависит, а Кт и V зависят слабо, то из (3.11)
следует, что температурная зависимость а определяется темпе-
ратурной зависимостью теплоемкости. Поэтому при Т—>-0 ве-
личина а также стремится к 0, а при высоких температурах — к
постоянному значению.
Из многочисленных экспериментальных исследований тепло-
вых свойств твердых тел следует [2,5], что изменение частоты
колебаний с изменением объема неодинаково для всех колеба-
тельных мод при различных температурах, как это предпола-
гает уравнение (3.6), т. е. у не является константой*. Диффе-
ренцирование уравнения (3.5) приводит к соотношению
«Кт- = у(П-у- (3.13)
в котором
Макроскопическая постоянная Грюнайзена у(Г) является
теперь усредненным значением всех микроскопических парамет-
ров Грюнайзена, суммированных относительно вклада каждой
колебательной моды в соответствии с ее теплоемкостью. Таким
образом, следует четко представлять, что, в то время как для
* В связи с этим в литературе стал встречаться термин «функция Грюнай-
зена» вместо «константа Грюнайзена».
88
данного твердого тела при каждой температуре существует лишь
одна константа у(Г), каждая колебательная мода характеризу-
ется своим параметром Грюнайзена у,-. В принципе необходи-
мое усреднение может быть проведено в соответствии с уравне-
нием (3.14), что требует знания у; для каждой колебательной
моды.
Исходя из этих общих для всех твердых тел представлений
о связи свойств, определяемых гармонизмом и энгармонизмом,
Офассмотрим особенности поведения полимеров, обусловленные
наличием в них.ав.и.зотрюпии локальных сил взаимодействия
между цепями и внутри цепей? Уже отмечалось, что кристалли-
ческие решетки полимеров сильноанизотропны и эта анизотро-
пия обусловлена большим различием силовых констант межмо-
лекулярного ван-дер-ваальсового и внутримолекулярного кова-
лентного взаимодействия. К тому же полимерные решетки
обычно содержат большое число _атомов, способных к самостоя-
тельным движениям.| Поэтомурколебания решетки полимерного
кристалла представляют собой сумму характеристических коле-
баний отдельных атомов и скелетных колебаний. Вследствие
сильной анизотропии силового поля решеток скелетные колеба? I
ния могут быть подразделены на внутри- и межмолекулярньцд.
При низких температурах, когда возбуждены лишь длинновол-
новые колебания, роль внутри- и межмолекулярных потенциа-
лов сравнима и полимерная решетка может рассматриваться
как трехмерный кристалл. При более высоких температурах
доминирующим становится внутримолекулярный потенциал
взаимодействия, и в этой области температур полимерный кри-
сталл можно рассматривать как ансамбль невзаимодействую-
щих линейных «решеток». Граница между этими температур- |
ными областями, естественно, не является резкой. |
Сильное различие в потенциалах внутри- и межцепного '
взаимодействия в полимерах приводит к тому, что энгармонизм
для межцепных колебаний значительно, больше, чем для внутри-
цепных. ГГ соответствии с уравнением (3.4) параметр Грюнай-
зена, являющийся характеристикой энгармонизма, при межцеп-
ных колебаниях также должен иметь существенно (большие зна-
чения, чем при внутрицепныхд В связи с этим очевидно, что
тепловое расширение и сжимаемость полимерных кристаллов
также должны быть сильноанизотропны, причем расширение и
сжимаемость поперек цепей должны быть значите’льно больше,
чем вдоль цепей. Таким образом, очевидно, что простой пере-
нос на полимеры представлений о тепловом расширении и пара-
метре Грюнайзена, развитых для низкомолекулярных твердых
тел с центральными силам» взаимодействия между частицами
(металлы, ионные кристаллы), может привести к неверным вы-
водам. Действительно, если, например, для определения пара-
метра Грюнайзена воспользоваться уравнением (3.4), как это
обычно делается ® фидике твердого тела, то очевидно, что для
полимера можно получйть значения у, различающиеся в не-
89
сколько раз (вплоть до порядка) в зависимости от того, какое
значение Cv будет использовано в расчетах — полное значение
теплоемкости или же только та часть, которая соответствует
межцепному вкладу в Cv. Это может являться источником
принципиальных недоразумений.
Концепция решающего вклада межцепного взаимодействия
в свойства полимеров, определяемые энгармонизмом, которая
стала широко использоваться в физике полимеров в последние
годы [6—9], была сформулирована в наиболее отчетливом виде
Вада [10]. Чтобы количественно продемонстрировать плодо-
творность основной идеи, Вада использовал для расчетов мо-
дельную тетрагональную решетку Стокмейера—Хечта (см.
рис. 1.5), отражающую цепной характер полимеров и сильную
анизотропию потенциалов взаимодействия в полимерах. Рас-
смотрим колебательную моду, в которой определенное число п
повторяющихся единиц вдоль цепи (для ПЭ — это CH2-rpyn-
пы) движется когерентно. Эффективная константа межцепного
взаимодействия для этой моды равна а эффективная
константа внутрицепного взаимодействия равна 20у/га. Множи-
тели ’/2 и 2 появляются в результате того, что, когда один ко-
нец упругого стержня длиной па зафиксирован, а другой его
конец деформируется на величину х, средняя деформация вдоль
стержня равна Щх. Если вклад внутри- и межцепного взаимо-
действия одинаков, то число повторяющихся единиц цепи вдоль
оси с определяется как
п* =2(0у/ау)1/2 (3.15)
Если рассматривать межцепные колебания решетки, то ис-
ходную тетрагональную решетку можно заменить на модельную
кубическую, в которой повторяющимися единицами являются
последовательности «* эффективной массой т* = п*М (где М —
масса повторяющегося элемента тетрагональной решетки) и
изотропной константой взаимодействия
Л* = -у Nn*ay = 2[Jy/n* = (РуЯу)V2 (3.16)
Естественно, такая модель является лишь определенным
приближением к реальной ситуации. В гл. 1 показано, что, на-
пример, для ПЭ колебания изолированной цепи представлены
крутильной, деформационной и валентной модами и рядом ха-
рактеристических колебаний СН2-группы. Силовые константы
этих колебаний сильно различаются, и потому каждая колеба-
тельная мода будет характеризоваться своим значением п*.
Например, для наиболее низкочастотной крутильной моды
«*~3. Для валентной моды это значение будет существенно
большим. Однако в расчетах Вады определялось усредненное
по всем модам значение п*.
90
Учет усредненного межцепного потенциала взаимодействия
совместно с рассмотрением низкотемпературной теплоемкости^
позволяющей оценить п*, привел к формуле
<хКт — ?(^1п(ег/Г)
(3.17}
где Cinter — часть суммарной теплоемкости, связанная лишь с межцепными ко-
лебаниями.
При высоких (T>9d) температурах Cinter составляет лишь
незначительную часть измеренной калориметрически теплоем-
кости. Конкретные результаты расчетов будут рассмотрены
далее.
Для теоретического расчета параметров Грюнайзена крис-
таллических полимеров используют ряд моделей. Наиболее пол-
ное уравнение состояния кристаллического ПЭ получено Пэс-
тайном [11], детально проанализировавшим выражения- для
внутреннего Pi и термического Рт давления в уравнении со-
стояния Ми — Грюнайзена [уравнение (3.7)]. Решеточная
энергия UL была представлена в виде суммы
uL = u“ + urL
энергий притяжения и отталкивания; UaL представляется ван-
дер-ваальсовым взаимодействием между двумя любыми мети-
леновыми группами, a UrL является суммой энергий отталкива-
ния всех несвязанных атомов водорода в кристаллической
ячейке.
Зависящая от температуры составляющая свободной энер-
гии FT и соответственно термическое давление Рт были выра-
жены следующим образом:
В этих уравнениях п — число СН2-групп в цепи, 2[п — чис-
ло дебаевских частот (Od, (1—f)2n — число частот со/, характе-
ризующих колебание изолированных цепей; 7п—число
91
эйнштейновских частот сое, ро — плотность при Т=0 К, а d In cor, VD-- ,1пу (3.20а) d In а>/ Yr-- „J (3.206) d In cop TE-- dln, (3.20b)
параметры Грюнайзена соответствующих колебательных мод.
Так как со/ предполагается не зависящей от межцепного взаи-
модействия, то у/ = 0. Кроме того, поскольку силовые констан-
ты эйнштейновских колебательных мод намного больше, чем
дебаевских, их зависимостью от объема также можно прене-
бречь по сравнению с соответствующей зависимостью cod- Эти
предположения эквивалентны представлениям Вады об опреде-
ляющей роли межцепного взаимодействия. Считая, что yd оди-
накова для всех частот, имеем:
P7- = 2poMBT(TD/V)D(0D/T) (3.21)
Предположение о постоянстве yd приводит к следующей за-
висимости дебаевской температуры 0р от объема:
1
»D(V) =9D(l)expJ(VD/V)dy (3.22)
е/ т
Подстановка уравнения (3.22) в уравнение (3.21) позволяет
рассчитать Рт как функцию V. Полное давление легко опреде-
лить по уравнению (3.7).
Шен с сотр. [12] сравнили данные по сжимаемости кристал-
лической решетки ПЭ, полученные исследованием зависимости
частоты крутильных мод СН2-групп от давления, и пришли к
выводу, что уравнение Пэстайна хорошо описывает экспери-
ментальные результаты.
Баркер [6] и Бродхерст и Мопсик [13] для вывода уравне-
ния состояния кристаллического полимера и параметра Грю-
найзена использовали модель двумерных пачек цепей, основан-
ную на концепции Вады о решающей роли межцепного взаи-
модействия. При приложении гидростатического давления к
кристаллической решетке полимера она сжимается лишь в на-
правлениях, перпендикулярных цепям, а сжимаемостью вдоль
оси цепи можно пренебречь. Это позволяет рассматривать поли-
мерный кристалл как двумерную решетку. Для расчета энергии
решетки использовали потенциал Ми
i/(r/r0)= —a1(r/r0)-m + a2(r/r0)-" (3.23)
И экспоненциальный 'потенциал:
V(r/r0)E = — a1(r/r0)-m + a2exp( — Ег/го) (3.24)
где г0 — равновесное расстояние между частицами; щ, о2 Е, п, m — константы.
92
Двойное дифференцирование этих потенциалов и подстанов-
ка г/г0 —V/Vo позволяют получить выражение для модуля объ-
емного сжатия:
Кт = VUomn[4V*(n - т)]-1^ + 2) (У/У„)-<П+4)Л’ -
- (/яД-гДИ/Ио)-'"^)/-’] (3.25)
и потенциал Ми
Кг = VUomE [4V0(E-/n)]-1{[E(V/V0)'1 + (V/V0)-3«]X
X exp Е [ 1 - (VI Vo)1/2] - (/» + 2) (V/VP)-<m+«4} (3.26)
экспоненциальный потенциал
Индекс 0 в этих уравнениях относится к величинам при О К-
При V=V0 уравнение (3.25) переходит в
KT=Uomn/4Vo (3.27)
Параметр Грюнайзена в этой модели находят из следующих
соображений. Дебаевская температура связана со скоростью
звука соотношением
0D = (ЗЛ^з/4л)1 з (3.28)
*в
Подстановка этого выражения в уравнение (3.126) с учетом
(3.27) дает:
(3'2”
Для двумерной решетки скорость распространения звуковых
волн перпендикулярно направлению осей макромолекул опреде-
ляется соотношением [14]
г/ 1 \ фи
с~ 1КГ— — PJV (3.30)
Использование этого соотношения совместно с уравнением
(3.29) дает:
1 / 31пКг \
7 = — — ) —const (3.31)
2 \ д In V /т
Постоянная, несколько различающаяся в расчетах разных ав-
торов, имеет несущественный для практических расчетов поря-
док 0,2—0,3.
До сих пор речь шла о поведении кристаллических решеток
'полимеров. Проблема вывода уравнения состояния и определе-
ния параметров Грюнайзена. для аморфных полимеров явля-
ется более сложной по нескольким .причинам. Прежде всего? для-
аморфных полимеров и аморфной фазы кристаллических поли-
меров физический смысл у оказывается более сложным, так как
сжимаемость (в широкбм" смыслеУ ,в этом случае будет опреле-
ляться не только энгармонизмом колебаний, но и свободным
ДУбъеушм^-Кроме того, в аморфных~полймёрах“рёЗТО'' возрастают"
трудности количественных расчетов 'внутреннего и термического
давления, связанные со сложностью корректного определения
взаимодействия и расстояний между взаимодействующими ча-
93
стицами. Тем не менее попытки получить теоретически уравне-
ние состояния аморфных полимеров известны. Пэстайн [И]
пытался вывести уравнение состояния аморфного и частично-
кристаллического ПЭ, использовав для этого ряд предположе-
ний и эмпирических данных. Поэтому такой подход следует счи-
тать скорее полуэмпирическим, так как входящие в уравнение
константы находят эмпирическим путем.
Отдельное направление в этой области составляют работы,
посвященные теоретическому анализу уравнений состояния
стеклообразных и жидких полимеров на основе квазирешеточ-
ных и дырочных моделей жидкости. Это направление, берущее
свое начало с работы Пригожина с corp. [15] и особенно ши-
роко представленное в работах Симха с сотр. [16], основано
на использовании принципа соответственных состояний. Полу-
ченные таким путем уравнения состояния в приведенных пере-
менных применимы к таким температурам, при которых кванто-
вые эффекты не играют существенной роли. Они не могут пред-
сказать поведение твердых полимеров при низких температу-
рах. К тому же, хотя эти уравнения получают на основе теоре-
тических моделей, в них содержится ряд констант, которые
можно определить лишь эмпирически. Потому такие уравнения
также можно отнести к категории полуэмпирических. Часто
оказывается, что сугубо эмпирические уравнения типа уравне-
ния Тейта проще для практического использования (см., напри-
мер, [16, 17]).
Однако в одной из работ Симха с сотр. [18] сделана попыт-
ка использовать квантовые представления в уравнении состоя-
ния в приведенных переменных с тем, чтобы описать экспери-
ментальные результаты по тепловому расширению стеклообраз-
ных полимеров при низких температурах. Исходным пунктом
расчетов является выражение для свободной энергии в фор-
ме (3.2).
Суммирование проводится по 3cN колебательным модам, где
Зс—число, зависящих от объема степеней свободы на цепь.
Приведенные переменные определяются соотношениями
Т* = <7гЕ*/сйБ Р* = NqzE*/V* (3.32)
где qzB* — параметр, характеризующий межмолекулярную энергию на цепь.
Уравнение состояния, получаемое обычным путем по урав-
нению (3.3), имеет вид
где А и В — числовые коэффициенты.
94
Зависящая от температуры часть свободной энергии анали-
зировалась с использованием как модели Эйнштейна, так и Де-
бая. Мы приведем лишь уравнение в дебаевском приближении:
(3.34)
где О(0о/Т) — функция энергии Дебая.
Выражение для приведенного коэффициента расширения а
имеет вид
«od ~ СП$ъУ
тр,е а = аТ*; 6 = 9/Т*.
(3.35)
Для выполнения принципа соответственных состояний в
квантовой области необходимо введение дополнительного усло-
вия 0 = const. Сравнение с экспериментом показало, что для
ряда стеклообразных полимеров различной химической приро-
ды зависимость а — Г в области достаточно низких температур
(10—40 К) является универсальной. Выше этой области наблю-
дается систематическое превышение экспериментальных данных
по сравнению с теоретической зависимостью.
Сложности, возникающие при теоретическом анализе урав-
нения состояния аморфных полимеров, в равной мере присущи
и частично-кристаллическим полимерам. Их анализ проводят
обычно путем комбинирования уравнений состояния для кри-
сталла и аморфного полимера с учетом степени кристаллично-
сти (см., например, Пэстайн [11], Джейн и Симха [16]).
Учитывая двухфазную структуру частично-кристаллических
полимеров, Гиббонс [19] рассмотрел вопрос о связи парамет-
ров Грюнайзена частично-кристаллического полимера и анизо-
тропных кристаллитов. Несмотря на анизотропию кристалли-
тов, такой полимер является макроизотропным вследствие ста-
тистической ориентации кристаллитов и наличия аморфных
областей.
Для расчета у использовали обычные для физики кристал-
лов усреднения тензоров модулей упругости (усреднение по
Фойгту) и усреднение тензоров податливости (усреднение по
Ройссу) [20]. Для полимерных кристаллитов, характеризую-
щихся двумерной сжимаемостью, изотропное напряжение в бло-
ке достигается за счет анизотропного изменения объема кри-
сталлитов. Это соответствует усреднению по Ройссу и приводит
к соотношению
УЛТ2 + ?2^Т1
К?! +
(3.36)
где индексы 1 и >2 обозначают два направления, перпендику-
лярные осям макромолекул в кристаллите. Такое же усредне-
95
Рис. 3.1. Зависимость решеточного yL и
термодинамического ут параметров Грю-
найзена от температуры:
1 — fT ПЭ [8, 9]; 2 — ут ПММА [24]; 3 —
ПММА [24]; 4 — теоретическая кривая для
стеклообразования полимеров [21].
ние фактически используется и при анализе рассмотренной вы-
ше модели двумерных пачек цепей [6, 131.
Эти же представления использованы в работе [19] для ана-
лиза температурной зависимости у, выводы которой сводятся к
следующему. При низких температурах основной вклад в у
вносят длинноволновые акустические колебания, низкие частоты
которых целиком обусловлены слабыми силами межмолекуляр-
iHoro взаимодействия. При повышении температуры начинают
возбуждаться более короткие акустические, а также оптические
Ж постоянная Грюнайзена которых~мала по сравнению с
оволновыми колебаниями. Усреднение всех колебаний в
соответствии с их теплоемкостями по уравнению (3.14) должно
сопровождаться достаточно резким и значительным уменьше-
нием у с повышением температуры. При обычных температурах
(»300 К) у уже очень слабо зависит от температуры (рис. 3.1).
, Перейдем теперь к рассмотрению с позиции изложенных
теоретических представлений экспериментальных результатов.
Сразу же следует подчеркнуть важное обстоятельство. Пара-
метр у может быть вычислен на основе экспериментальных дан-
ных, полученных различными методами. Так, для его определе-
ния используют измерения теплового расширения, теплопро-
водности, теплоемкости, акустических характеристик, объемной
сжимаемости, ИК-спектров, Раман-спектров, рентгеновского
рассеяния. Для расчета параметров у применяют уравнения,
полученные на основе различных приближений. Такое много-
образие экспериментальных и расчетных методов, несомненно,
благоприятно с точки зрения совершенствования представлений
об особенностях твердого состояния полимеров на основе изме-
рений различных физических характеристик. Однако такая си-
туация одновременно создает и определенные трудности, по-
скольку при попытке сравнить результаты разных методов ока-
зывается, что они сильно различаются. Поэтому неудивительно,
что в литературе по параметрам Грюнайзена полимеров име-
ется много, на первый взгляд, противоречивых результатов.
Положение сейчас таково, что для одного и того же полиме-
ра значения у, полученные разными методами, могут разли-
чаться на порядок и более. В теплофизике полимеров сейчас
уже стало общепринятым представление, что для твердых поли-
ве
меров следует различать два типа параметров Грюнайзена j
[6, 7]. Один из них — термодинамический параметр Грюнайзе-
на ут отражает ангармоничность, усредненную по всем колеба- I
тельным модам. Другой — решеточный параметр Грюнайзена I
yL определяется лишь межцепными колебательными модами, '
т. е. только межмолекулярным взаимодействием. Это следует |
иметь в виду прежде всего при экспериментальном определе- !
нии у. Так, если у определяется по зависимости модуля объем-
ного сжатия от давления по уравнению
1 /дКт\
2 \ дР /г
(3.37)
вытекающему из (3.31)*, то это значение относится только к
межцепному взаимодействию и является уь. Так как уравне-
ние (3.17) также отражает лишь межцепное взаимодействие, то
1 (дКт\
2 \ дР )т
аКтУ
Winter
(3.38)
Для металлов и ионных кристаллов ут.«ут; для полимеров
yL« (54-20)ут. Таким образом, становится понятной одна из
причин противоречивых результатов.
Наличие обоих параметров ут и yL для одного и того же
полимера позволяет оценить теплоемкость, обусловленную лишь
межцепным взаимодействием, которую экспериментально изме-
рить очень трудно. Действительно, соотношение общей тепло-
емкости Cv и Cinter должно быть таким же, как и соотношение
Ут и ут, т. е.
Ут = ?L(C|nter/Clz) (3.39)
Теоретическое обоснование этой формулы дано Кюрро [21],
который, используя решеточную модель и потенциал Ленарда—
Джонса — Девоншира, рассчитал параметры Грюнайзена для
.аморфных полимеров в широком температурном интервале; ока-
залось, что lirn ут = 3,1б. Формула (3.39) хорошо передает тем-
т-л
пературную зависимость ут при обычных и повышенных темпе-
ратурах для ПС и ПММА, для которых имеются эксперимен-
тальные результаты. Широкая экспериментальная проверка для
разных полимеров уравнения (3.39) при низких температурах
пока не приводилась, за исключением ПММА (см. далее). Еще
одно интересное предсказание касается связи параметра Грю-
найзена с числом атомов в повторяющемся звене при высоких
температурах: параметр у должен быть обратно пропорциона-
лен этому числу.
* Из (3.31) следует
__L Д_(дКт\ /_L/oy_\
2 Кт \ дР )т/ V \дР /т
и с учетом того, что Kr—V(dP/dV)r, имеем:
Y=
1 (дКт\
2 I дР )т'
7—440
9?
Основным способом определения параметра Грюнайзена яв-
ляется расчет по уравнению (3.13) или (3.17) при известных
значениях остальных входящих в эти уравнения величин. Таким
методом можно, как правило, определить значение у при
какой-то определенной температуре. Определить ут в широком
температурном интервале расчетным путем ,не удается, так как
обычно отсутствуют необходимые данные: Кт (Т), Cv(T)
и а(Т).
Существуют обходные пути определения температурной зави-
симости у. Так, Шен с сотр. [22] предлагают, например, опреде-
лять у (Г) по уравнению
Вытекающему из известного термодинамического соотношения
Ср—Су — а2КтУТ, при подстановке в него уравнения (3.13).
Таблица 3.1. Параметры Грюнайзена для ПЭ при 298 К [8, 9, 12, 21]
Уравнение, использованное для определения Параметр Грюнайзена Метод определения
V или уг-
Уг = aKfCy/V 1 / д In Кт \ 5 1,1* Расчет
2 \ д In V 12 7,5 3,5* Объемная сжимаемость Рентгенография под дав- лением
/ д In с \ 1 д Р )т+ 3 4,0 7,7 Ультразвуковые измере- ния Измерение теплоемкости
( д In х\ 1 Yz. = ''Ц д р ~ 11,0 Измерение теплопровод- ности
УТ = [Ср— Су)!СусГГ у, отдельных колебательных мод / d 1п<вг \ \ Тг d In У ) 1,1* Измерение теплоемкости
72 см"1 — трансляционная оптиче- ская мода 1,28 ИК-Спектроскопия
730 см-1—маятниковая мода СНг-групп 0,07 ИК-Спектроскопия
11 см3 — аккордеонная мода 0,47 Раман-спектроскопия
1133 см-3 — валентная С—С-мода 0,087 То же
1296 см-1 — крутильная мода СН2-групп 0,027
2883 см"1 — асимметричная СНгва- лентиая мода 0,07
* Для кристаллической решетки.
88
При понижении температуры ут должна возрастать вследствие
исчезновения высокочастотных колебаний и приближаться к
значению yL. Теоретически при О К величина ут должна ста-
новиться равной yL. К такому выводу вводятся все теоретиче-
ские предсказания [19,21].
Наиболее полно параметры Грюнайзена исследовались для
ПЭ. Результаты, полученные различными методами, суммиро-
ваны и проанализированы в работах Шена (табл. 3.1). Хотя
Шен не разделяет параметры у на решеточные и термодинами-
ческие, мы это делаем, следуя занятой выше позиции. Приве-
денные в таблице результаты подтверждают наличие сильной
зависимости ул от метода исследования. Нужно иметь в виду,
что параметры Грюнайзена, полученные акустическими мето-
дами, являются адиабатическими, а полученные методом объем-
ной сжимаемости или теплопроводности, — изотермическими.
Можно ожидать, что адиабатические значения будут примерно
на 10% выше изотермических. Такое соотношение действитель-
но наблюдается для ряда полимеров [7]. Результаты, получен-
ные по второй формуле таблицы, демонстрируют также зависи-
мость yL от степени кристалличности.
В рентгеновских исследованиях определяется модуль объем-
ного сжатия только кристаллической решетки, тогда как обыч-
ный метод объемной сжимаемости суммирует вклады как кри-
сталлической, так и аморфной составляющих. Однако эти ре-
зультаты получены на разных образцах и, возможно, различие
значений yL обусловлено влиянием не только степени кристал-
личности, по и других факторов, например разного рода дефек-
тов структуры. К сожалению, даже для ПЭ до сих пор не про-
ведены исследования одного и того же хорошо структурно оха-
рактеризованного образца различными методами. Для количе-
ственного сравнения это важно, так как, например, сообщалось
[23], что параметры Грюнайзена для закаленного и отожжен-
ного образцов ПЭ различны. Поэтому до тех пор, пока такие
исследования не будут проведены, количественные сравнения
преждевременны.
Параметры Грюнайзена для отдельных колебательных мод
различаются уже на несколько порядков, демонстрируя вполне
понятную закономерность: моды более высокой частоты харак-
теризуются меньшим значением у, поскольку они в меньшей
степени подвержены действию возрастающего при сжатии меж-
молекулярного взаимодействия.
Выше отмечалось, что понижение температуры должно со-
провождаться вымораживанием высокочастотных колебатель-
ных мод и повышением вклада низкочастотных, что должно при-
водить к возрастанию термодинамического параметра Грюнай-
зена. Проведенные для кристаллической решетки ПЭ расчеты
(см. рис. 3.1) качественно подтверждают этот вывод. В проти-
воположность термодинамическому параметру Грюнайзена ут
зависит от температуры только через зависимость объема от
7'
99
температуры. Но поскольку объем слабо зависит от температу-
ры, то следует ожидать слабой температурной зависимости уь.
Исследования в ограниченном температурном интервале от —50
до 100 °C согласуются с этим заключением [7, 10].
Недавно такое подтверждение было получено при исследо-
вании yL ПММА в температурном интервале 4—300 К [24];
yL определяли по температурным зависимостям бриллюэновс-
кого рассеяния света и линейного теплового расширения. Срав-
нение полученной экспериментально температурной зависимос-
ти уь и рассчитанной по уравнению (3.11) (см. рис. 3.1) пре-
красно согласуется с представлением о стремлении ут к yL при
низких температурах вследствие вымораживания высокочастот-
ных колебательных мод, характеризующихся малыми значения-
ми параметра Грюнайзена. С другой стороны, при повышении
температуры должна возрастать роль дефектов кристаллической
решетки при тепловом расширении кристалла, что должно при-
водить к некоторому повышению уь. С этих позиций была рас-
смотрена зависимость параметра Грюнайзена от температуры в
интервале 15—90 °C и от давления (до 550 МПа) монокристал-
лов ПЭ [25]. При повышении температуры от 15 до 90 °C уь
действительно возрастала примерно от 6,5 до 9 при атмосфер-
ном давлении и оставалась на уровне 6,5 во всем интервале
температур при давлении 200 МПа. Наличие температурной
зависимости yL при обычном давлении и ее отсутствие при по-
вышенном давлении было объяснено одновременным влиянием
дефектов решетки, обусловливающих большие смещения ча-
стиц в узлах решетки, и зависимостью энгармонизма от темпе-
ратуры и давления.
Хотя для других полимеров исследования параметров Грю-
найзена не столь всесторонние, как для ПЭ, они все же позво-
ляют оценить значения уь и ут (табл. 3.2). Еще раз можно от-
метить, что ПЭВП высокой степени кристалличности («90%)
характеризуется более низкими значениями yL по сравнению с
Таблица 3.2. Решеточный и термодинамический параметры Грюнайзена
для полимеров при 298 К [7]
Полимер vr YT Cinter Yl ~ CV
Полиэтилен высокой плотности 4,1 0,52 0,126
Полиэтилен низкой плотности 6,4 0,38 0,059
Полипропилен (изотактический) 9,0 0,96 0,094
Полиэтиленоксид 5,0 1,02 0,204
Политетрафторэтилен 8,0 0,40 0,05
Полиметилметакрилат 4,0 0,82 0,205
Полистирол 4,4 0,79 0,180
100
ПЭНП (степень кристалличности «50%), Четкой корреляции
параметров Грюнайзена с молекулярным строением полимеров
пока не установлено.
ТЕПЛОВОЕ РАСШИРЕНИЕ ПОЛИМЕРНЫХ КРИСТАЛЛОВ
Для изотропных твердых тел и кристаллов с кубической решет-
кой тепловое расширение изотропно и может быть охарактери-
зовано одним коэффициентом объемного теплового расширения
а= (din V/dT)P [2]. При каждой температуре в равновесных
условиях такое твердое тело занимает объем, которому отвечает
минимум свободной энергии.
Положение минимума свободной энергии изменяется с тем-
пературой только вследствие изменения колебательной энтро-
пии с изменением объема [уравнение (3.9) ]. Знак производной
(dS/dV)r показывает, сжимается или расширяется тело при на-
гревании. Твердые тела расширяются при нагревании, если
(dS/dV)T положительно, и сжимаются, если (dS/dV)r отрица-
тельно. Тепловое расширение зависит также от крутизны мини-
мума, будучи обратно пропорционально крутизне, а следова-
тельно, модулю объемного сжатия Кт [уравнение (3.9)].
Тепловое расширение всех остальных кристаллов анизо-
тропно, и наряду с изменением объема при нагревании проис-
ходит изменение формы тела '[2б,"27]. Однако тепловое расши-
рение должно ббЛИДЭТБ"сймметрией кристалла (принцип Ней-
мана) и изменение температуры не может привести к исчезно-
вению какого-либо элемента его симметрии (в отсутствие фазо-
вых переходов). Тепловая деформация [е»>], возникающая при
нагреве кристалла на АТ, является тензором, все компоненты
которого могут быть представлены соотношением
(3.41)
где (Ji, — коэффициенты линейного теплового расширения.
Так как [е2Д —симметричный тензор второго ранга, то, по-
скольку АТ — скаляр, [0ij] также является симметричным тен-
зором второго ранга. Тензор теплового расширения можно при-
вести к главным осям:
Е1 = ^ДГ, е2 = ₽2Д7\ е3 = Р3ДТ (3.42)
где р2, Рз — главные коэффициенты теплового расширения.
Коэффициент объемного теплового расширения равен
а = Pi % + Р3 (3.43)
В общем случае для анизотропных сред соотношение Грю-
найзена, аналогичное уравнению (3.9), может быть записано в
виде [2, 27]
101
где в,, т — упругие податливости (<Зе//дО/)г (О/—напряжение, сопряженное
деформации в/).
Вводя постоянную Грюнайзена в виде
(dS/dej)T
(3.45)
(где Се—теплоемкость при постоянной деформации), полу-
чаем:
С 6
(3-46)
/=1
Компоненты тензора параметра Грюнайзена выражаются
соотношением
6
= (3.47)
/=1
где С,, т — изотермические модули упругости.
Таким образом, тензор параметров Грюнайзена является
симметричным тензором второго ранга.
Кристаллы с аксиальной симметрией (гексагональной и три-
гональной сингоний) обладают двумя независимыми коэффици-
ентами теплового расширения и двумя параметрами Грюнай-
зена [27]:
где а', с' — параметры кристаллической решетки.
Определение тензора расширения для кристаллических ре-
шеток более низких симметрий значительно сложнее. Расшире-
ние кубических решеток описывается уравнением (3.11) или
(3.13).
Для большинства кристаллов главные коэффициенты тепло-
вого расширения положительны, что приводит к появлению эл-
липсоида расширения. Однако у ряда кристаллов (кальций,
теллур, цинк, селен) [2] коэффициенты теплового расширения
в некоторых направлениях в определенных температурных ин-
тервалах отрицательны, что приводит к появлению сложных по-
верхностей теплового расширения. Таким образом, тепловое
расширение твердых тел, так же как и другие тепловые свойст-
ва, является отражением сил, действующих между частицами, и
U02
особенностей динамики соединенных между собой частиц, а
тензор теплового расширения анизотропных структур характе-
ризует анизотропию действующих в них сил. у
Цепное строение макромолекул является причиной высокой '
локальной анизотропии физических свойств, в том числе и теп-
лового расширения. Сильное различие в потенциалах внутри- и
межцепного взаимодействия в полимерах должно обусловли-
вать большое различие в тепловом расширении кристалличе-
ских решеток полимеров вдоль и поперек цепей. Более того,
если следовать представлениям о последовательности возбуж-
дения колебательных мод в цепных структурах в соответствии
с их вкладом в теплоемкость, то можно утверждать, что тепло-
вое расширение вдоль цепей должно вообще отсутствовать при
низких и комнатных температурах, так как при этом возможно
размораживание лишь крутильных и деформационных колеба-;
ний, характеризующихся невысокими значениями характерно!
тических температур.
Однако теоретический анализ теплового расширения цепных,
и слоистых структур, проведенный Лифшицем [28], показал,'
что возбуждение волн изгиба с отличным от Обычного законом!
дисперсии (со~/г2) должно приводить к появлению отрицатель-)
ных коэффициентов вдоль цепи в цепных структурах и в плос-
кости слоя в слоистых структурах вследствие проявления в них!
специфического мембранного эффекта. Суть эффекта состоит в )
том, что при растяжении частоты длинных волн изгиба возра- '
стают, т. е. колебания становятся как бы более жесткими. Это
отличается от обычного случая, когда при растяжении расстоя-
ния между частицами увеличиваются и частоты колебаний '
уменьшаются. Наличие мембранного эффекта в слоистых и цеп-
ных структурах означает, что возбуждение волн изгиба может
приводить только к уменьшению продольных размеров в плос-
кости слоя или вдоль цепи.
Эти соображения можно проиллюстрировать на примере
цепных структур, для которых анизотропный тензор коэффи-
циентов теплового расширения
Ь-------- (3.50)
(где etk — тензор тепловой деформации) характеризуется тремя
отличными от нуля компонентами: 0ZZ вдоль цепи и рхж и
поперек цепи.
Так как свободная энергия* твердого тела связана с тензо-
ром деформации простым соотношением
dF
Zik = ~^h (3‘51)
* Для твердых тел при обычном давлении термодинамический (изобарно-
изотермический) потенциал практически тождествен свободной энергии. Хотя
в работе [28] используют термодинамический потенциал, все выводы остаются
в силе и для свободной энергии.
103
(где Oik — тензор .напряжений), то
д-F
daz^T
d»F
(3.52)
Рхх + Руу-----J
0ОХх(уу)"Т
(3.53)
₽« =
При низких температурах аналогично уравнениям
(3.3) имеем:
(3.2) и
dF
ik
(3.54)
1 — exp
так как при T = 0 К величина etfe° = 0. Лифшиц теоретически по-
казал, что для невзаимодействующих слоев (цепей) ды/доХ)
вдоль цепи или в плоскости слоя, в то время как перпендику-
лярно цепи или слою да>/до<0. В соответствии с (3.52), (3.53)
и (3.54) это означает, что вдоль цепи и в плоскости слоя теп-
ловое расширение должно быть отрицательным, т. е. при нагре-
вавии таких тел в этих направлениях они должны сокращать-
ся, в то время как в перпендикулярных направлениях — расши-
ряться.
Таким образом, причиной появления отрицательных коэффи-
циентов теплового расширения в цепных и слоистых структу-
рах является наличие сильной анизотропии упругих сил, дейст-
вующих в кристаллических решетках таких структур. Это прин-
ципиально отличается от причин появления отрицательных ко-
эффициентов теплового расширения в твердых телах, не обла-
дающих столь сильной анизотропией [2], где они в основном
появляются в результате возникающих при поляризации ато-
мов дальнодействующих сил в кристалле.
Рассмотренные теоретические расчеты относятся к области
низких температур, где проявляются квантовые эффекты в од-
номерных и двумерных структурах. Температурная зависимость
главных коэффициентов расширения для температурной области,
где существенны лишь внутрислоевые колебания и где CV~T2,
имеют вид ~Л а ₽zz~T2. Температурная зависимость
тензора теплового расширения цепных структур в широком ин-
тервале температур до сих пор теоретически не рассмотрена.
Что касается очень низких температур, то учет взаимодействия
между цепями и слоями приводит к тому [28], что вблизи О К
как р, так и а~7'3, что вполне согласуется с температурной за-
висимостью Cv кристаллических решеток полимеров, как это и
должно быть согласно условию связи теплового расширения и
теплоемкости (см. выше).
Отрицательные коэффициенты теплового расширения вдоль
оси макромолекул в кристаллических решетках обнаружены
104
Таблица 3.3. Тепловое расширение полимерных кристаллов
Полимер р „ -105, К-1 кр || ₽кр1 -105- К-1 Температур- ный интер- вал, °C Литератур- ная ссылка
Полиэтилен —1,2 = 14,7 20—60 [30]
— 1,8 Р* = 6,0 20—120 [30]
От —1,1 до —1,3 От —180 до 60 [23]
Полиэтилеитере- фталат —2,2 Ра = 17,1 12,7 От —196 до 20 [29]
Полиамид 6 —4,5 ₽а = 6,2 Рь = 23,4 От —196 до 20 [29]
Целлюлоза —4,5 Ра = 6,0 ₽г = 2,8 От —196 до 20 [29]
Полихлоропрен —4,1 Ра= 13,1 От —196 до 20 [29]
Натуральный каучук —8,4 Ра= 12,5 ₽6=2О,5 От —196 до 20 [29]
экспериментально для многих полимеров (табл. 3.3). Таким об-
разом, явление сокращения в направлении оси макромолекул
при нагревании можно считать достаточно общим, не завися-
щим от молекулярной структуры цепей. Во всех случаях сжа-
тие вдоль цепей в среднем в несколько раз (до порядка) мень-
ше, чем расширение перпендикулярно цепям. Отсюда следует,
что коэффициент объемного теплового расширения полимерных
кристаллических решеток является положительным. Замеча-
тельно, что отрицательные коэффициенты вдоль цепей наблю-
даются не только при низких температурах, но и в области
комнатных температур, т. е. в классической области. Еще одно
важное обстоятельство, вытекающее из анализа данных
табл. 3.3, состоит в том, что отрицательные значения р незначи-
тельно изменяются от полимера к полимеру. Это позволяет
предполагать существование достаточно универсального меха-
низма теплового сокращения в полимерах.
Ценность теоретического предсказания Лифшица о появле-
нии отрицательных коэффициентов в Цепных и слоистых струк-
турах состоит в том, что оно не связано с особенностями моле-
кулярного строения этих структур и предполагает лишь нали-
чиевысокой анизотропии сил взаимодействия между цепями или
слоями и внутри их, приводящей к появлению в них волн из-
гиба с особым законом дисперсии в длинноволновой части спек-
тра. Так, отрицательные коэффициенты наблюдаются не толь-
ко у цепных макромолекул органических полимеров, но и у
твердых тел иной природы, например у цепных кристаллов тел-
лура, образованных спиральными макромолекулами этого эле-
мента [2].
105
Теория тепловых свойств цепных и слоистых структур, раз-
витая Лифшицем еще в 1952 г., остается практически неизвест-
ной для широкого круга физиков-полимерщиков за границей, и
потому, после того как в начале 70-х годов повысился интерес
к особенностям теплового расширения полимеров и стали ин-
тенсивно выясняться физические причины появления отрица-
тельных коэффициентов вдоль осей макромолекул в кристалли-
ческих решетках полимеров, возникли гипотезы [30, 31], связы-
вающие отрицательные коэффициенты с особенностями молеку-
лярной структуры полимеров. Они основаны на интуитивном
представлении о том, что повышение амплитуды поперечных
колебаний с повышением температуры должно сопровождаться
сокращением макромолекулы (вдоль ее оси, поскольку сильное
внутрицепное взаимодействие позволяет считать связи между
отдельными атомами скелета макромолекулы нерастяжимыми.
В частности, для ПЭ было высказано предположение, что со-
кращение цепи при нагревании обусловлено возрастанием ам-
плитуды внеплоскостных крутильных колебаний плоского зиг-
зага, которая может быть охарактеризована углом поворота
между двумя последовательными плоскостями, (проходящими
через соседние С—С-овязи. Величина постулированного перво-
начального угла вращения, равная 3° [30], при последующем
более детальном анализе [31] оказалась значительно больше.
Этот угол должен быть не менее 13°, чтобы именно крутильны-
ми колебаниями можно было объяснить экспериментально на-
блюдаемое сокращение для молекулы ПЭ в температурном ин-
тервале 20—100 °C.
Исходя из представлений о теплоемкости цепных структур,
можно полагать, что наряду с крутильными колебаниями ске-
лета в сокращение цепи при нагревании должны вносить вклад
и деформационные колебания. Попытки модельного теоретиче-
ского анализа сокращения цепей при нагревании были пред-
приняты в работах Кана [32] и Чоя с сотр. [33, 34]. Мы рас-
смотрим лишь результаты более совершенных расчетов, учиты-
вающих наличие как крутильных, так и деформационных коле-
баний [33, 34]. Расчеты проводились для модельных решеток
. двух типов: модельной решетки СХ (см.
т~О рис. 1.5) и решетки, образованной плоски-
, -т—А ми зигзагами, расположенными в парал-
а А дельных плоскостях. Первая модель по-
0 а \ад зволяет рассмотреть вклад лишь деформа-
\ ционных колебаний, в то время как вторая
£_Д к модель позволяет учесть и крутильную ко-
лебательную моду.
а Oq
__А Рис. 3.2. Схема изменений продольных размеров не-
I растяжимых цепей при поперечных колебаниях [33].
106
Основная идея расчетов проста и может быть наглядно про-
демонстрирована рис. 3.2. Рассмотрим поведение линейной це-
пи, образованной атомами, соединенными друг с другом нера-
стяжимыми связями длиной йо каждая. При нагревании такой
цепи вследствие поперечных колебаний атомов длина проекции
связей на направление оси цепи изменится и будет составлять
а = (а* — х2)1/2 « а0 — х2/2до
(3v55)
где предполагается, что |х| <Са0. Если эффективная крнстанта
упругости, определяющая величину поперечного смещения, рав-
на Яэф, то для двух поперечных колебательных мод имеем:
-у^эф<*2>=*Б7’ (3.56)
где <х2> — смещение, усредненное по всему ансамблю связей.
Тогда среднее значение проекции
(а) = а0 — (3.57)
Аэфао
а тепловое расширение вдоль цепи
- 1 d(a)
= (3-58>
Таким образом, расчеты сводятся к определению Кэф- Инте-
ресно, что в этой схеме никак не фигурирует ангармонизм, яв-
ляющийся молекулярной причиной теплового расширения. Од-
нако при расчетах Кэф ангармонизм поперечных колебаний при-
нимается во внимание, так что в конечном счете причиной со-
кращения цепей при нагревании также является ангармонизм
колебаний, хотя и не в направлении сокращения. В решетке,
образованной линейными цепями, Лэф определяется константой
упругости межцепного взаимодействия Kw и константой упруго-
сти Kg, соответствующей деформационным колебаниям цепи.
Выражение для эффективной константы упругости линейной ре-
шетки имеет вид
1 / 1 \ l/4 1
7~ = 77^(1,18-0,5«-0,59^+...) (3.59)
/'эф \AgKW// ху 1
где = Kw/ 16/(g.
При использовании значений Лг = 35 Н/м и Да, = 3,2 Н/м,
характерных для ПЭ, расчет дает ркрц=—1,3-10-5 К-1, что впол-
не согласуется с рентгеноструктурными данными табл. 3.3.
Для решетки зигзагообразных цепей выражение для коэф-
фициента расширения ркр а имеет вид
о ( 1 / S \ V2 1 3
107
х (1,18 - 0,54«2 - 0,59 v1 +-..)+ -1/4 X
loo Qq
X (1,18 — 0,81u2 — 1,8и3 — 2,21u4-{- ...)
(3.60)
где Кк — константа упругости, соответствующая крутильным колебаниям; s=
= sin(<Do/2) (Фо — равновесное значение валентного угла); v и и — определяют-
ся соотношением констант К® и Kg, Kw и Кк соответственно.
Оценки по уравнению (3.60) опять приводят к ркр и =
= — 1,31-10-5 К-1 для ПЭ.
Проведенный теоретический анализ указанных моделей по-
зволяет сделать ряд обобщений. Оказывается, что тепловое со-
' кращение не зависит от массы групп" атомов в повторяющемся
। звене макромолекулы, а из выражений (3.59) и (3.60) видно
также, что сокращение мало чувствительно к величине Kw- Это
I дает основание ожидать, что для полимеров с одинаковым ске-
летом значения ркр а не будут сильно различаться. Данные
табл. 3.3 не противоречат этому заключению. Сравнение резуль-
татов для двух моделей показывает также, что вклад крутиль-
ных и деформационных колебаний в сокращение примерно оди-
наков. Это объясняет, почему модель линейной решетки, для
которой характерны лишь деформационные моды, приводит к
такому же значению 0кр в , как и модель плоских зигзагов, в ко-
торой появляется крутильная колебательная мода.
[ Рассмотренные модели позволяют оценить среднее тепло-
вое сокращение цепей в классической температурной области.
Такие оценки показывают [32], что для ПЭ сокращение должно
слабо зависеть от температуры в области выше примерно
100 К. Судя по данным табл. 3.3, это наблюдается для всех
приведенных в ней полимеров. С точки зрения молекулярной
динамики несомненный интерес представляет поведение тепло-
вого расширения полимерных решеток при более низких темпе-
ратурах, когда возможно появление необычных законов темпе-
ратурного изменения теплового расширения, особенно для тен-
зора коэффициентов расширения. Теоретический анализ этой
проблемы отсутствует. Использовать рассмотренные модели для
расчетов температурной зависимости теплового расширения в
квантовой области чрезвычайно трудно.
Шен с сотр. [22] использовали для расчета коэффициенты
объемного теплового расширения а решетки ПЭ уравнение
Грюнайзена для теплового расширения твердых тел [1]:
_______Су
<2(1 — et/r/Q)2
(3.61)
где Q = VqKq/Уо, £ =
m-j- n-j-3
6
Для дебаевской модели твердого тела при значениях
0d = 315 К, Q=H6 кДж/моль и |=3,75 зависимость а—Т при-
108
Рис. 3.3. Температурная зависимость теп- _____________________
лового расширения решетки ПЭ [22]: j l
сплошная кривая — расчет по уравнению (3.61); Sr
О—экспериментальные значения, определен-
иые по температурной зависимости ИК-спект-
ров и рентгенографических кристаллических
рефлексов.
ведена на рис. 3.3. Хотя наблю- f
дается хорошее согласие с экс- 7" г
периментальными значениями а, /
вряд ли можно надеяться, что / |
уравнение (3.6) будет выпол-
няться для широкого круга по-
лимеров, так как согласие с экспериментом достигнуто при до-
статочно произвольном выборе констант. Рис. 3.3 тем не менее
показывает, что основные идеи теплофизики неполимерных
твердых тел могут быть плодотворно использованы для созда-
ния теории теплового расширения полимеров при учете их
структурных особенностей. В рассматриваемых расчетах ос-
новная особенность полимерных кристаллических решеток
формально учитывалась предположением о двумерном характе-
ре теплового расширения кристалла ПЭ, так как предполага-
лось, что тепловое расширение вдоль оси макромолекул равно
нулю. Теоретический анализ теплового расширения н-алканов
на основе подхода Грюнайзена для модели полужесткого
«ожерелья» в приближении квазигармонического потенциала,
проведенный Бродхарстом и Мопсиком [35], продемонстриро-
вал чувствительность теплового расширения к длине оже-
релья. Для длинных цепей зависимость а—Т очень близка к
представленной на рис. 3.3.
Для развития представлений об особенностях теплового рас-
ширения кристаллитов полимеров важными являются резуль-
таты сравнительного рентгенографического исследования тепло-
вого расширения решеток трех полимеров, различающихся сте-
пенью межмолекулярного взаимодействия, в температурном
интервале 5—400 К [36]. Рассмотрим эти результаты (рис. 3.4).
Температурная зависимость тепловой деформации поперек
и вдоль макромолекул единообразна, хотя они различаются и
по знаку, и по значению. Единообразие формы зависимостей
₽кР±—Т и рКр у—Т свидетельствует, очевидно, о том, что про-
дольное сокращение макромолекул является следствием попе-
речных колебаний. Качественно зависимость pi—Т для ПЭ по-
добна зависимости Cv—Т (количественное сравнение затрудне-
но отсутствием табулированных значений рКр±, особенно необ-
ходимых для области ниже 100 К). В соответствии с температур-
ной зависимостью предполагается '[36], что ниже 100 К основной
вклад в рКр1 всех трех полимеров вносят крутильные колеба-
ния, а возрастание ipKp± выше 100 К обусловлено повышением эн-
гармонизма крутильных колебаний и возбуждением деформаци-
онных колебаний. Простые оценки показывают, что в области
109
л 200 К Ркр 1 достигает «классических» значений. Действитель-
но согласно формуле Френкеля [37]
₽ » /гБ/Е/2 (3.62)
где I — линейный характеристический размер.
Для ПЭ £±=4 ГПа, а для ПВС £±«9 ГПа. Принимая
1 = 0.3—0,4 нм, имеем для ПЭ 0кр ± « 10-10-5, для ПЭ 0КР i =
= 5-10“5. Эти оценочные значения близки к экспериментальным
в области 200 К.
Несмотря на единообразие формы температурной зависимо-
сти [Зкр1 и ркр || для трех полимеров, различие в абсолютных
значениях тепловой деформации для ПКА и ПВС, в наибольшей
степени различающихся степенью межмолекулярного взаимо-
действия, составляет 4—5 раз. Возникает вопрос, каким из фак-
торов обусловлено это различие: разницей в амплитудах коле-
баний или же разницей в коэффициентах энгармонизма.
Для решения этой проблемы рассмотрим двучленное пред-
ставление упругосилового взаимодействия в твердом теле [37].
Сила, действующая между колеблющимися частицами, может
быть выражена уравнением
/ = Ма —^(Да)2 (3.63)
где k — коэффициент квазиупругой силы; g — коэффициент ангармоничности;
Да — смещение из равновесного положения.
Средняя относительная тепловая деформация ет равна
Да g Ла-
гг--------и —— • ------
а0 к До
(3.64)
где Да2 — средний квадрат
А-.;®?*"
отклонения частицы от положения равновесия.
Таким образом, различие в ет может
вызываться как фактором g/k, так и
фактором Да2. Симбатность поперечно-
го расширения и продольного сокраще-
ния для всех полимеров позволяет счи-
тать решающим фактором в величине
тепловой деформации различие в ампли-
тудах колебаний. Вариация же степени
ангармоничности при переходе от ПВС
к ПКА играет меньшую роль.
Вывод о решающем влиянии ампли-
туды колебаний в величине тепловой де-
формации вполне согласуется с особен-
ностями взаиморасположения и взаи-
модействия макромолекул в решетке
(рис. 3.5). Наибольшая тепловая дефор-
Рис. 3.4. Температурные зависимости поперечного
Р„Р1 и продольного (Кр л коэффициентов линейно-
го теплового расширения для ПВС (/), ПЭ (2) и
ПКА (3) [36].
ПО
Рис. 3.5. Схемы элементарных ячеек кристаллических решеток полимеров [36]:
а, б, в — проекции плоских грянг-зигзагов на плоскость, перпендикулярную осям макро-
молекул (значения межплоскостных расстояний указаны для комнатной температуры);
г — плоский транс-зигзаг скелета макромолекулы.
мация наблюдается в ПКА, в котором плоскости зигзагов па-
раллельны друг другу, а водородные связи вдоль длины мак-
ромолекул следуют через семь связей. Хотя в ПЭ водородные
связи отсутствуют, плоскости скелетов молекул расположены
крестообразно. Очевидно, что наибольшее влияние на увели-
чение межплоскостного расстояния оказывают те макромоле-
кулы, плоскости зигзагов которых близки к плоскости (НО).
Таких макромолекул — половина из всех, и, вероятно, следст-
вием этого является тот факт, что тепловая деформация ре-
шетки ПЭ оказывается примерно в 2 раза меньше, чем у ре-
шетки ПКА (см. рис. 3.4). Наконец, хотя в решетке ПВС все
плоскости зигзагов параллельны, однако высокая густота водо-
родных связей допускает лишь сравнительно малые амплиту-
ды колебаний. Эти соображения хорошо согласуются также с
обратной пропорциональностью между поперечным тепловым
расширением полимерных решеток и модулями поперечной
упругости.
Монокристаллы полимеров макроскопических размеров —
большая редкость, и их свойства, в том числе и теплофизиче-
ские, це изучены. Тем интереснее рассмотреть данные [38] по
тепловому расширению (рис. 3.6) монокристалла ПОМ макро-
скопических размеров (полуцилиндр высотой 2 см и диаметром
2,5 см). Несмотря на заметный разброс экспериментальных то-
чек ниже 10 К, авторам удалось установить, что
= (0,15 ± 0,005) -Ю-вГЦК-1) (Г< 15К)
₽ц = ( — 0,028 ± 0,001) •10-87’3(К-1) (Т< ЮК)
а = 20± + ₽ц =0,27- 10"87^(К-1)
Таким образом, эти результаты подтверждают, что вблизи
0 К как линейные, так и объемный коэффициенты теплового
расширения описываются кубической зависимостью от темпера-
туры, что хорошо согласуется с поведением теплоемкости, изме-
ренной на этом же образце [® интервале 3—12 К
С~ (13,0±0,5) Т3 мДж/(г-К)]-
При сравнении поведения макроскопического монокристалла
с кристаллическими решетками выявляется принципиальное от-
111
Рис. 3.6. Температурная зависимость теп-
лового расширения монокристаллов ПОМ
параллельно ((3 ) и перпендикулярно
(Р ц) направлению оси цепей [38].
3 личие в поведении р 9. У реше-
ток ркр и остается отрицательным
2 во всей области 4—400 К, а у
j X: монокристалла рмон в меняет
'а знак и становится положитель-
0 ным выше 100 К. Причины такого
-/ поведения не ясны. Возможно, что
это связано с тем, что плотность
образца была ниже кристалло-
графической плотности ПОМ.
Хотя авторы связывают дефицит плотности с наличием микро-
пор, однако вполне вероятно, что образец не обладал 100%-
ной степенью кристалличности. Поэтому возможно, что при
наличии дефектов в решетке или аморфных областей изменит-
ся знак ркр и . Макромолекулы ПОМ кристаллизуются в спи-
ральную конформацию, и это еще раз подтверждает тот факт,
что отрицательное расширение характерно как для плоских,
так и для спиральных конформаций макромолекул.
Возникает естественный вопрос, какие последствия могут
иметь для кристаллических полимеров отрицательные коэффи-
циенты расширения вдоль оси макромолекул в кристаллических
решетках. Для блочных неориентированных кристаллических
полимеров их влияние не может сказаться на макроскопиче-
ском коэффициенте теплового расширения по крайней мере по
двум причинам. Во-первых, отрицательные коэффициенты зна-
чительно меньше по абсолютному значению положительных
коэффициентов по другим кристаллографическим осям, что при-
водит согласно (3.43) к положительному коэффициенту объем-
ного расширения. Учитывая статистическое распределение кри-
сталлитов в блочных образцах, а значит, и осей макромолекул,
очевидно, что и коэффициенты расширения по любому из на-
правлений в образцах макроскопических размеров окажутся
положительными.
Вторая причина заключается в наличии аморфных областей
в кристаллических полимерах. Согласно аддитивной схеме
строения кристаллических полимеров
“ = аам(1 — w)+aKpw (3.65)
При T<ZTC аам~акр- Однако в отличие от кристаллитов для
аморфных областей нет оснований ожидать заметной анизотро-
пии теплового расширения в микрообъемах с линейными раз-
мерами 10—20 нм, типичными для размеров кристаллитов и
аморфных областей в частично-кристаллических полимерах с
112
обычной степенью кристалличности. При Т>ТС аам~(3—4)Х
Х.акр>0, поэтому наличие даже не очень значительной доли
аморфных областей приводит к весомому их вкладу в общий
коэффициент теплового расширения. Эти соображения позво-
ляют считать, что в макроскопических тепловых свойствах не-
ориентированных частично-кристаллических полимеров тепло-
вое сжатие макромолекул не должно проявляться. Это действи-
тельно наблюдается экспериментально.
Положение может коренным образом измениться при пере-
ходе к ориентированному состоянию кристаллических полиме-
ров. Такие полимеры действительно при нагревании могут со-
кращаться, однако природа этого явления вследствие двухфаз-
ной структуры кристаллических полимеров оказывается много
сложнее. Тем не менее в конечном счете оно также связано с
цепным строением полимеров. Ввиду важной роли растянутых
цепей в аморфных областях ориентированных кристаллических
полимеров в механизме их усадки, а также вследствие тесной
связи тепловых явлений при обратимых деформациях с тепло-
вым расширением проблема анизотропного теплового расшире-
ния ориентированных полимеров будет рассмотрена совместно с
термоэластическими свойствами полимеров в гл. 4.
ЛИТЕРАТУРА
1. Gruneisen Е.— In: Handbuch der Physik/Unter Schriftleitung, H. Geifer u.
K. Scheel. Berlin, Springer Verlag, 1926. Bd 10, S. 1—59.
2. Новикова С. И. Тепловое расширение твердых тел. М., Наука, 1974. 294 с.
3. Жирифалько Л. Статистическая физика твердого тела. Пер. с англ./Под
ред. В. 3. Кресина и Б. М. Струнина. М., Мир, 1975. 382 с.
4. Румер Ю. Б., Рывкин М. Ш. Термодинамика, статистическая физика и ки-
нетика. М„ Наука, 1977. 552 с.
5. Barron Т. Н. К- — Phil. Mag., 1955, vol. 46, N 378, p. 720—734.
6. Barker R. E., Jun.—J. Appl. Phys., 1967, vol. 38, N 11, p. 4034—4242.
7. Warfield R. W. — Makromol. Chem., 1974, Bd 175, N 11, p. 3285—3297.
8. Shen M., Reese W.— In: Progress in Solid State Chemistry./Ed. by
I. O. McColdin a. G. Somorjai. Oxford, Pergamon Press, 1975. Vol. 9,
p. 241—268.
9. Shen M. —Polymer Eng. a. Sci., 1979, vol. 19, N 14, p. 995—999.
10. Wada Ya., Hani A., Nishi T.. Nagai S. — J. Polymer Sci., 1969, pt A-2, vol. 7,
N 1, p. 201—208.
11. Pastine D. J.— J. Chem. Phys., 1968, vol. 49, N 7, p. 3012—3022; J. Appl.
Phys., 1970, vol. 41, N 13, p. 5085—5087.
12. Wu С. K., Jura G., Shen M.— J. Appl. Phys., 1972, vol. 43, N 11, p. 4348—
4354.
13. Broadhurst M. G., Mopsik F. I. — J. Chem. Phys., 1970, vol. 52, N 7, p. 3634—
3641.
14. Dugdale J. S., McDonald D. К. C. — Phys. Rev., 19153, vol. 89, N 4, p. 832—
834.
15. Prigogine I., Trappeniers N., Mathot V.—Disc. Farad. Soc., 1953, N 15,
p. 93—107; Prigogine J. The Molecular Theory of Solutions. Amsterdam, 1957.
448 p.
16. Simha R. — J. Macromol. Sci., 1980, pt B, vol. 18, N 3, p. 377—392; Simha R.,
Somcynsky T. — Macromolecules, 1969, vol. 2, N 4, p. 342—350; Olabisi 0.,
Simha R. — Macromolecules, /19715, vol. 8, N 2, p. 206—210, 211—016;
8—440
113
Jain R. К, Simha R.—J. Polymer Sei., Polymer Phys. Ed., 1979, vol. 17,
N 11, p. 1929—1946.
17. Айнбиндер С. Б., Тюнина Э. Л., Цируле К. Я.—Мех. композ. материалов,
1981, № 3, с. 387—392.
18. Simha R., Roe J. М., Nanda V. S. — J. Appl. Phys., 1972, vol. 43, N 11,
p. 4312—4317.
19. Gibbons T. G. — J. Chem. Phys., 1974, vol. 60, N 3, p. 1094—1100.
20. Шермергор T. Д. Теория упругости микронеоднородных сред. М., Наука,
1977. 400 с. „
21. Curro J. G. — J. Chem. Phys., 1973, vol. 58, N 1, p. 374—380.
22. Shen M„ Hansen W. N„ Romo P. C. — J. Chem. Phys., 1909, vol. 51, N 1,
p. 425—430.
23. Devis G. T„ Eby R. K, Colson J. P. — J. Appl. Phys., 1970, vol. 41, N 11,
p. 4316—4326.
24. Kato E. — J. Chem. Phys., 1980, vol. 73, N 3, p. 1020—1025.
25. Kijima T., Koga K, Imada K, Takayanagi M.— Polymer J., 1975. vol. 7,
N 1, p. 14—20.
26. Най Дж. Физические свойства кристаллов. Пер. с англ./Под ред. Л. А. Шу-
валова. 2-е изд. М., Мир, 1967. 385 с.
27. Barron Т. Н. К — J. Appl. Phys., 1970, vol. 41, N 13, р. 5044—5050.
28. Лифшиц И. М. —ЖЭТФ, 1952, т. 22, № 4, с. 475—486.
29. Wakelin J. Н., Sutherland A., Beck L. R.—J. Polymer Sci., 1960, vol. 42,
N 1, p. 278—280.
30. Kobajashi J., Keller A. — Polymer, 1970. vol. 11, N 1, p. 114—118.
31. Baughman R. H. — J. Chem. Phys., 1973, vol. 58, N 7, p. 2976—2983.
32. Кан К- H. Вопросы теории теплового расширения полимеров. Л., Изд-во
ЛГУ, 1975. 80 с.
33. Chen F. С., Choy С. L., Young К — J. Polymer Sci., Polymer Phys. Ed.,
1980, vol. 18, N 12, p. 2313—2322.
34. Chen F. C., Choy C. L., Wong S. P., Young К — J. Polymer Sci., Polymer
Phys. Ed., 1981, vol. 19, N 6, p. 971—981.
35. Broadhurst M. G., Mopsik F. I.— J. Chem. Phys., 1971, vol. 54, N 10,
p. 4239—4346.
36. Дадобаев Г., Слуцкер А. И. — ФТТ, 1981, т. 23, № 8, с. 1936—1942; Высо-
комол. соед., 1982, сер. А, т. 24, № 8, с. 1616—1622.
37. Френкель Я. И. Введение в теорию металлов. М. — Л., Гостехтеоретиздат,
1950. 383 с.
38. White G. К, Smith Т. F., Birch J. А. — J. Chem. Phys., 1976, vol. 65, N 2,
p. 554—558.
Глава 4
ТЕПЛОФИЗИКА УПРУГОСТИ ПОЛИМЕРОВ
При рассмотрении деформации твердых тел в рамках теории
упругости обычно используют систему сугубо механических по-
нятий. Вместе с тем деформирование является термодинамиче-
ским процессом. Переход от механического описания деформа-
ционных процессов к термодинамическому связан с переходом
от рассмотрения потенциальной энергии деформации к рассмот-
рению термодинамических потенциалов (внутренняя энергия,
свободная энергия и др.). С экспериментальной точки зрения
это означает, что определение механических характеристик (на-
пряжения, деформации) необходимо дополнить одновременным
114
измерением температурных или тепловых изменений, возникаю-
щих при адиабатическом или изотермическом проведении про-
цесса. Тепловые эффекты, сопровождающие деформацию,
обычно незначительны, и именно их измерение представляет ос-
новную трудность перехода от механического к термодинамиче-
скому описанию деформации. Вместе с тем в тепловых эффек-
тах, сопровождающих деформационные процессы конденсиро-
ванных тел, заложена глубокая информация о молекулярных
изменениях, происходящих при деформации. В связи с этим с
физической точки зрения термодинамический подход к описанию
деформации является предпочтительным. Блестящим примером
плодотворности термодинамического подхода к анализу меха-
низмов деформации являются эластомеры. В этой главе дано
термодинамическое рассмотрение обратимых деформаций не
только эластомеров, но и твердых полимеров.
ФЕНОМЕНОЛОГИЧЕСКАЯ ТЕРМОДИНАМИКА ДЕФОРМИРОВАНИЯ
КОНДЕНСИРОВАННЫХ ТЕЛ
Если на изотропную конденсированную систему действует несколько ти-
пов различных сил, то выражение для дифференциала энергии dU имеет вид
Т1.21
п
dU = dQ+^idXi .(4.1)
/ = 1
где dQ — теплота, подведенная к системе; £,• — обобщенная сила, сопряженная
с Ей обобщенной координатой х,-; п — число обобщенных сил.
Вслед за [1] будем считать положительной работу, совершаемую внешни-
ми силами над системой. Если система, например стержень, растягивается или
сжимается вдоль своей длины, то обобщенной является растягивающая или
сжимающая сила f, сопряженной ей координатой — длина стержня. Точно так
же для случая кручения жесткого стержня обобщенной силой является мо-
мент кручения, а сопряженной с ней координатой — угол закручивания. Если
на систему действует внешнее давление Р, то сопряженной ему координатой
будет объем. Ниже для простоты рассмотрены лишь такие случаи, когда на
систему действуют лишь две обобщенные силы, одной из которых является
давление. Экспериментальные исследования термодинамики деформации поли-
меров обычно проводят при таких условиях. Для этого случая основное тож-
дество для обратимого процесса удобно представить в виде
dU~TdS — PdV + idx (4.2)
(работа сжатия под действием давления положительна, так как dV<Z0, а рабо-
та расширения против давления отрицательна, так как rfV>0).
Тогда остальные термодинамические потенциалы имеют вид
свободная энергия iF = —SdT—PdV + idx (4.3)
энтальпия dH = — SdT VdP -f- idx (4.4)
термодинамический потенциал dG = —SdT-|- VdP -f- idx (4.5)
Для упругих тел вводятся еще четыре потенциала:
U* = U— Ь; dU* = TdS — PdV— xdi (4.6)
F* = U- — TS; dF* = - SdT — PdV - xd' (4.7)
H* =H—&; dH* =-- TdS -|- VdP—xd^ (4.8)
G* = H* — TS-, dG* = — SdT + VdP — xdi (4.9)
8* 115
На основании этих восьми термодинамических потенциалов обычными ме-
тодами можно получить 24 соотношения (соотношения Максвелла) между про-
изводными термодинамических величин. Многие нз них нам потребуются при
обсуждении термодинамики деформации полимеров, поэтому мы приведем эти
соотношения:
К этим термодинамическим коэффициентам необходимо добавить соответ-
ствующие теплоемкости. По определению
(4.18}
где у представляет собой Р либо V, az— £ либо х.
Приведенные соотношения позволяют установить связь между тепловыми
явлениями, связанными с возникновением напряжений и деформаций в упругих
116
телах. Так, если на жидкость или твердое изотропное тело действует гидро-
статическое давление, то тепловой эффект, сопровождающий сжатие, опреде-
ляется соотношением (4.13) или (4.17)
dV \ / dS \ I / dQ \
~дТ)Р~ ~\~дР!т = Т \ дР 't-
или для конечных изменений давления:
AQ = — apVTAP
(4.20)
где dp — изобарический коэффициент объемного теплового расширения.
Это соотношение показывает, что приложение давления к нормальным те-
лам, обладающим положительным Значением ар, должно сопровождаться вы-
делением тепла. Если давление прикладывается быстро, то при этом происхо-
дит адиабатическое изменение (повышение) температуры, величина которого»
определяется соотношением (4.12) или (4.16)
дТ \ / \ __ Т
dP)s~\ds)p~afV Ср
(4.21)
или для конечных изменений давления:
a VT
ЬТ =(b.P)s (4.22>
Ц. В качестве второго примера рассмотрим находящийся при постоянном дав-
лении упругий стержень длиной I, на который действует продольная сила f.
Тепловой эффект, сопровождающий растяжение стержня, определяется соот-
ношением (4.176)
(4.23>.
или для конечного значения силы:
(AQ)t,p= Tapfl^f
(4.24).
В этих уравнениях ар, f — изобарический коэффициент линейного теплово-
го расширения при постоянной силе f. Это уравнение показывает, что растяже-
ние стержня с положительным значением ар, / сопровождается поглощением
тепла. Так ведет себя большинство твердых тел.
Если усилие прикладывается быстро, то при этом происходит адиабатиче-
ское изменение (понижение) температуры, величина которого определяется со-
отношением (4.166)
/ дТ\ / dl \ Т
(37) = ~ 37 =~aP,fvT~ (4-25)-
\ of Js,p \ oS Jp f Cpj
или для конечных значений силы:
(^T)s,p = — ap,fV bf (4.26)
cP,f
Поскольку AQ=—СрДТ, то соотношения (4.22) и (4.26) можно получить,
и из соотношений (4.20) и (4.24).
Рассмотрим теперь соотношение между теплоемкостями. Если на систему
действует лишь внешнее давление, то соотношение между теплоемкостями С?
и Су определяется следующим образом [1, 2]:
117
Согласно (4.18) в общем случае существуют четыре теплоемкости Cv, х,
Cv, , СР, х и Ср'^. Первые две появляются в теоретических соотношениях для
конденсированных систем, в то время как две последние—в эксперименталь-
ных исследованиях. Соотношения между этими теплоемкостями следующие:
ъ = Ср>х -т| < ду_ < ОТ , дР . дТ ^V,x
! dV ' f _дР_
V,; = Cp.i - т 1 \ дТ . < дТ
' дх ) f
Р,х = Cp,i — rl JdT ) дТ , 1,.,
(4.28)
(4.29)
(4.30)
Выражения для термодинамических потенциалов свидетельствуют о том,
что обычно приложение сил к конденсированным телам сопровождается изме-
нением как внутренней энергии, так н энтропии (или температуры). Однако,
оставаясь в рамках феноменологической термодинамики, нельзя получить от-
вет на вопрос о молекулярной природе этих изменений и их соотношении. Это
связано с тем, что в рамках феноменологической термодинамики нельзя опре-
делить, как изменяется потенциальная энергия частиц конденсированного тела
при деформации и, следовательно, нельзя установить, как изменяется его тем-
пература или энтропия. Ответ на эти вопросы может быть получен лишь на
основе уравнений состояния. Такие уравнения могут быть получены либо мето-
дами статистической термодинамики, либо экспериментальным путем. При
анализе термодинамики деформации полимеров мы будем исходить из этого
фундаментального положения.
ТЕПЛОФИЗИКА УПРУГОСТИ ТВЕРДЫХ ПОЛИМЕРОВ
Всестороннее расширение (сжатие)
Анализ термодинамики деформации твердых полимеров умест-
но начать с рассмотрения простейшего случая — равномерного
всестороннего расширения (сжатия). Для этого необходимо
иметь уравнение состояния cr=o(V, Т). Такое уравнение мож-
но получить, объединив закон теплового расширения тела и за-
кон Гука [2, 3]. Зависимость объема от температуры имеет вид
У = И(Т0)[1 + аа(Т-Т0)] (4.31)
где аа — коэффициент объемного теплового расширения при постоянном на-
пряжении; V(T0) —объем при какой-то исходной температуре.
Можно, в частности, положить То = О К, и тогда уравнение
(4.31) принимает более простой вид:
у = у0(1+<хаТ) (4.32)
Зависимость изменения объема от напряжения устанавли-
вает закон Гука:
где Кт — модуль объемного расширения (сжатия).
118
Объединяя соотношения (4.32) и (4.33), получим уравнение
состояния:
о-'ф.чЛ.Л ~‘] <4-3”
где V для простоты обозначает V (Т, о).
Так как для 'большинства конденсированных тел аа = 10-4—
—10-5 К-1, то произведение а^Т для обычных случаев будет
значительно меньше 1, и потому величинами второго порядка
малости по а«Т можно пренебречь. В пределах такой точно-
сти уравнение (4.34) можно записать в виде
г v 1
а = Л> — (1-а0П-1 (4.35>
L*o J
В полученных ниже соотношениях переход от растяжения к
сжатию производится простой заменой а на —Р.
Изотермы такого тела представлены на рис. 4.1. С повыше-
нием температуры Кт несколько понижается, и потому наклон
изотерм падает. При анализе термодинамики деформации будем
считать Кт, а также а0 постоянными, поскольку учет их зави-
симости от температуры приводит к эффектам второго порядка
малости.
Исходя из уравнения состояния (4.35), определим теперь
термодинамические функции такого тела:
(dS\ll/dS\ С I do \
-av )dV~—^-dT — ( dv (4.36)'
I 01 /о \ 0V /т 1 \ 01 /у
Второе слагаемое находим из уравнения состояния:
I до \ V
Н? (4-37>'
\ дТ )у и0
откуда
dS = ^dT + Kja^dV=C-LdT+aaVdo (4.38)
1 "о 1
Адиабатическое изменение температуры тела, определяемое
из условия dS = O, выражается соотношением
a,„VTdo aDVTdP
dT = — —--------= —- - (4.39)
9
Интегрируя это равенство, находим:
(A7’)s =
aaTV0 (Aa)2
—---------- До +--------
CJA-V'T') 2хг
(4.40)
Так как вторым членом в скобках можно обычно прене-
бречь по сравнению с (До), а также поскольку ааТ^Д, то
уравнение (4.40) принимает вид
aaT <ХрТ
(AT)S « - — V0Aa = —- VW3
Ca СР
(4.41)
Знак температурных изменений определяется знаком аа.
Растяжение изотропных тел с положительным a а сопровожда-
ется их охлаждением, а сжатие — соответственно нагреванием.
Для тел с отрицательным аа эффекты имеют противополож-
ные знаки. Тепловой эффект изотермической деформации опре-
деляется формулой
Q = TSS =
-C°(AT)S =
[aa7V0
1-apT
До +
(Да)2'
2Х?
аоТИ0Дп и -ар7%ДР
(4.42)
Определим теперь изменение внутренней энергии в изотер-
мических условиях:
Г ос VTKt
dU = TdS + adV = a„TVdo + adV = 1—2-- x
I K>
г V 1) / V \
X(l-aaT)+KT — (l-a.T)-l \dVxKT — -1 ]dV (4.43)
L Го JJ \ Vo /
Интегрирование этого уравнения дает:
Кт
и = ^(и-ио)2
(4.44)
Таким образом, внутренняя энергия конденсированного тела
является квадратичной функцией деформации. Поскольку ра-
бота — также квадратичная функция деформации
= (4.45)
2 2 2V 1 ’
то интуитивно кажется, что зависимости внутренней энергии
и работы от деформации тождественны. Однако в действитель-
ности оказывается, что они различаются принципиально. Эти
зависимости представлены на рис. 4.2. Оказывается, что вер-
шины соответствующих парабол не совпадают. В отличие от ра-
боты, вершина параболы которой находится в начале коорди-
нат, зависимости для внутренней энергии смещены относитель-
но начала координат. Координаты этого смещения, определяе-
.120
Q;W;U
Рис. 4.2. Зависимость работы (/), теплоты (2), внутренней энергии (3) и отно-
шения тепла к работе (4) при объемной и одноосной деформации твердых тел
г положительным (а) и отрицательным (б) коэффициентом теплового расши-
рения.
мне уравнениями (4.43) и (4.44), выражаются соотношениями
а,Т
^мии— . . -г ~ (4.46}
1 “Г
иыт = - ХтМааП2 = - КТУ^МИИ (4.47)
Важным обстоятельством является инверсионный характер
изменения внутренней энергии. Согласно уравнению (4.44)
внутренняя энергия обращается в нуль при
2ааТ
<?ин = — "ГТ—— ~—2а3Т (4.48)
1 + ааТ
Уравнения (4.46) и (4.48) показывают, что инверсия-ВНУТ-
рендещэнергии должна наблюдаться ппи сжатии тел с положи-
тедышм-ас и при растяжении с отрйцатёЛБТГБШ аа. Положе-
ние инверсии пропорционально также абсолютной температуре.
Параметры инверсии для основных типов конденсированных
тел приведены в табл. 4.1.
Особого обсуждения заслуживают физические причины ин-
версии. Рассмотрим их на примере сжатия тел с положитель-
ным аа. Поскольку ^начальной области деформации U умень-
шается при сжатии, то это означает, что выделяющаяся тепло-
та превышает механическую работу. Этот на первый взгляд
парадоксальный факт вытекает и из сравнения зависимостей теп-
лоты и работы от деформации [уравнения (4.42) и (4.45)], со-
гласно которому их отношение выражается гиперболой (см.
рис. 4.2)
— Q/W — — 2ааТ/е (4.49)
а следовательно, при &—>-0 (Q/U7)—-»—оо. Естественно, такое
121
Таблица 4.1. Параметры инверсии внутренней энергии
при деформации конденсированных тел* (Т=300 К)
Всестороннее растяжение (сжатие ) Одностороннее растяжение (сжатие)
Материалы а,-105, к-1 I л с 4" я Я (7МИН. Дж/смЗ 3-105, к-1 Я g UJ o'4 X р> 8 « ч Я я
Металлы Неорганиче- ские стекла Полимеры стеклооб- разные кристалли- ческие каучукопо- добные «Органические ЖИДКОСТИ Вода Иодид серебра 4 2 20 30 70 80 20 (7>4 °C) —6,1 (7=0 °C) -0,4 50 30 3 3 2 1 2 40 —2 —1 — 10 -15 —40 —50 — 10 +4 +0,1 —0,5 —0,05 —0,5 —1,0 —4 —3 -0,5 —0,05 —0,01 2 1 7 10 —0,13 50 50 4 3 40 i । [ । । । । © । w ’1 1 1 Сл ЬО - >—» СП —0,1 <—0,1 —0,01 —0,1 <—0,001
* В таблице указаны типичные для данного класса материалов значения а, 0, Е,
и порядок величин в„„ и
i Ип мии
соотношение соблюдается и для 'растяжения с той лишь разни-
цей, что теперь (Q/IT)—Н-°°- Таким образом, при малых де-
формациях теплота по абсолютному значению может сколь
угодно сильно превосходить работу. Это, в свою очередь, озна-
чает, что -^-1—мх> при е—>-0. Явление это согласно уравнению
(4.49) должно исчезать при ао = 0 и при Т=0 К, а при обычных
значениях Т и аа должно наблюдаться для конденсированных
тел, подчиняющихся принятому уравнению состояния. Этот ре-
зультат наглядно показывает, что конкретные соотношения меж-
ду термодинамическими функциями могут быть найдены лишь
на основе уравнения состояния.
Согласно (4.43) возникающие при деформации напряжения
можно представить в виде (см. гл. 3)
X ( — =<Н— а,К.тТKOt— а„КтТ (4.50)
\ 01 ]у Кд
где а,— внутреннее напряжение (давление) (энергетическая составляющая);
Пкин — кинетическое напряжение (давление) (энтропийная составляющая).
122
Согласно простым оценкам кинетическое напряжение
акин=,аа КтТ в конденсированных телах очень велико — имеет
порядок 108 Па — и уравновешивается энергетической состав-
ляющей о,. Если теперь представить работу в виде
adV = O[dV — <TKHHdE (4.51)
то становится очевидным, что-изменение внутренней энергии на
нача-льццх-этапа^ деформации.. определяется изменением энтро-
пии—(или наоборот), поскольку oidVоКИн dV.
Таким образом, на начальных этапах деформации внешнее™
воздействие лишь изменяет объем тела, не участвуя практичен
ски в энергетическом балансе (см. рис. 4.2). Следует особой
подчеркнуть, что речь идет о соотношении тепла, работы m
внутренней энергии при переходе из недеформированного со-
стояния в деформированное. При возвращении в исходное неде-
формированное состояние соотношения между этими характери-
стиками останутся такими же и изменятся лишь знаки эффек-
тов, и потому свободная энергия деформированного состояния
оказывается, как того требует термодинамика, равной работе
деформации или, как принято в теории упругости, упругому
потенциалу.
Проведенный анализ ясно показывает, что упругие дефор-
мации конденсированных тел обычно сопровождаются как из-
менением внутренней энергии, так и изменением колебатель-
ной энтропии, и опровергает встречающееся часто утверждение,
что упругость твердых тел является энергетической. В конден-
сированных телах изменения энергии и энтропии являются
взаимосвязанными и обычно сравнимыми. Это — следствие ан-
гармоничности колебаний атомов, т. е. следствие асимметрии
потенциальной кривой взаимодействия атомов. Днгармонизм
колебаний атомов является причиной теплового расширения
(см. гл. 3) и, следовательно, определяет..лвеличины тепловых
эффектов при механических деформациях, поскольку а — един-
ственный микроскопический параметр в уравнениях для тепло-
ты деформации. Полимеры характеризуются высокими значе-
ниями коэффициентов теплового расширения, существенно
большими, чем у металлов и неорганических твердых тел, и
потому для них тепловые эффекты при деформации должны
быть особенно значительными.
(При приложении давления атомы в теле сближаются, меняй
ется их потенциальная и кинетическая энергия (температура)^
и, для того чтобы тело осталось в изотермических условиях, не|
обходимо либо подвести к нему тепло, либо отвести его. Изме-|
нение колебательной энтропии означает изменение амплитуды!
колебаний и, следовательно, частоты колебаний) К этому выво-'
ду можно прийти и формальным путем. Изменение колебатель-
ной энтропии при изменении частоты в гармоническом прибли-
жении для моля осцилляторов выражается соотношением [1]
AS = 3R ln(w/w') (4.52)
123
где со'— частота колебаний тела под напряжением.
Поскольку при сжатии обычно AS<0, то <в'><в; при расши-
рении А5>0и со'Ссо. Изменение колебательной энтропии явля-
ется следствием изменения объема при деформации.
Одноосное растяжение (сжатие)
Исходя из аналогичных представлений, уравнение состояния
упругого стержня, растягиваемого (сжимаемого) силой f вдоль
оси, можно представить в виде
а = 4 = ^|-г(’-₽л/7’)-11 (4-53>
А [ /0 J
где Е — модуль упругости (Юнга); Z=Zo(l+0e, fT) (1+е); ftp, f — коэффициент
линейного теплового расширения при постоянных давлении и силе; е=
/—л. л
=-----------___—относительная деформация; А—площадь поперечного се-
Ld+fipjT)
чения недеформированного стержня.
Полагая, как и раньше, Е и рР, у постоянными и пренебре-
гая изменением поперечных размеров при деформации (т. е. по-
лагая коэффициент Пуассона ц=0, учет которого дает эффект
второго порядка малости), можно получить соотношения, ана-
логичные трехмерному случаю. Адиабатическое изменение тем-
пературы стержня определяется соотношением
(4.И)
СР,1
Теплота деформации равна
<? = ₽Л/Т/о/ = ₽Лг7’£1/ое (4.55)
Поскольку различие между теплоемкостями СР, / и СР1;, оп-
ределяемое соотношением
cPif-cP>[ = (WTEV0 (4.56)
•является величиной второго порядка малости по (РТ), им в на-
шем приближении можно пренебречь. То же самое можно ска-
зать и о коэффициентах теплового расширения, т. е. на практи-
ке можно считать рР, /= рР, ;=рР. Выражение для внутренней
энергии имеет вид
dU = TdS + fdl - PdV = EA dl (4.57)
*0
При деформации стержень совершает работу против внеш-
него давления PdV. Однако этот член для атмосферного давле-
ния на несколько порядков меньше остальных членов уравнения
124
(4.57), поэтому им обычно можно пренебречь. Это означает,
что внутренняя энергия практически тождественна энтальпии:
U = ЕА(1 — 10у/2 (4.58)
Это выражение для U также характеризуется инверсией
(здесь и далее мы пишем просто 0 по причине, указанной
выше):
еин=-2рт (4.59)
емин^-РТ (4-60)
t/мин = - EV0 riL = - EV0 (4.61)
Соотношения между термодинамическими функциями пока-
заны на рис. 4.2. Так же как и при трехмерной деформации
Q/r = 2pT/e (4.62)
U/W = 1 +2&Т/е (4.63)
. U . । Q , п
причем и —>-оо при е—>0.
В заключение рассмотрим, какие тепловые явления будут
возникать при сокращении растянутого стержня без механиче-
ской работы (мгновенное прекращение действия растягивающей
силы). Очевидно, что запасенная в растянутом стержне механи-
ческая работа во всех случаях должна рассеиваться в виде теп-
ла. Однако знак общего теплового эффекта этого процесса бу-
дет определяться знаком U. При (}>0 величина QCokp<0, а при
Р<0 возможны три случая: QCOkp=0 при е=®ин; Qcokp<0 при
£>еин и QcoKp>0 при ®<еин. Особенно впечатляет послед-
ний случай, когда запасенная работа рассеивается и стержень
при этом поглощает тепло (охлаждается). При мгновенном
расширении сжатого стержня эффекты, естественно, будут
иметь противоположный знак.
Сдвиг. Кручение
Согласно классической теории упругости [4] при сдвиговых де-
формациях и при кручении изменяется лишь форма тела, а его
объем остается неизменным. В связи с этим из приведенных
выше рассуждений может быть сделан вывод, что при таких де-
формациях тепловые явления по порядку величины будут зна-
чительно меньше, чем при объемных деформациях. В этом мож-
но убедиться, рассмотрев уравнение состояния для сдвига в
форме [3]
т = G0[l — m(T — То)] У (4.64)
где т — напряжение сдвига; у—деформация сдвига; Go — модуль сдвига; m —
малая положительная константа.
Изотермы сдвиговых деформаций в отличие от изотерм ра-
стяжения (сжатия) (см. рис. 4.1) являются семейством прямых,
125
проходящих через начало координат. Физически это означает,
что одними лишь температурными изменениями осуществить
деформацию сдвига невозможно. Работа сдвиговой деформации
определяется выражением
2 2
а теплота может быть определена из соотношения (4.7)
Q = TG^- (4.66)
Сравнивая W и Q и имея в виду, что т(Т—То)*^!, находим,
что Q/W=Tm<g. 1. Таким образом, в этом случае отношение теп-
лоты к работе не зависит от деформации и является величиной
малой, что позволяет без всякого ущерба в случае сдвиговой
деформации пренебречь тепловыми эффектами. Это в полной
мере относится и к кручению.
ТЕПЛОФИЗИКА УПРУГОСТИ МОЛЕКУЛЯРНЫХ СЕТОК
В рассмотренных выше уравнениях состояния (4.35) и (4.53)
предполагалось, что К, Е, а м $ не зависят от температуры и
деформации (или напряжения). Как показывает проведенный
выше анализ, эти предположения, в первом приближении оп-
равдывающиеся для большинства твердых тел и жидкостей, в
том числе и полимеров, позволяют определить соотношения
между основными термодинамическими потенциалами при де-
формации и количественно предсказать характер тепловых явле-
ний при различных видах деформации. Однако имеется группа
тел, а именно каучукоподобные тела или эластомеры, для кото-
рых для 'большинства типов (геометрий) деформаций эти усло-
вия принципиально не выполняются. Действительно, при одно-
осном,растяжении каучукоподобные тела способны к огромным
(сотни процентов) обратимым деформациям. Модуль упругости
каучукоподобных тел на несколько порядков ниже модуля упру-
гости твердых тел.
В отличие от обычных твердых тел, у которых Е с повыше-
нием температуры несколько понижается, что, как отмечалось
выше, является следствием теплового расширения, у умеренно
растянутых эластомеров модуль упругости пропорционален аб-
солютной температуре. Не менее замечательной особенностью
термомеханического поведения эластомеров является зависи-
мость коэффициента линейного расширения от степени растя-
жения (растягивающей силы). Зависимость эта такова, что по-
ложительный коэффициент нерастянутого эластомера по мере
растяжения очень быстро падает и затем становится сильно от-
рицательным, сравнимым по величине с коэффициентом рас-
ширения газов, т. е. имеет порядок 1/Т. Уже один этот факт по-
зволяет на основе термодинамики предсказать инверсию тепло-
126
вото эффекта при деформации: поглощение тепла в начальных
областях деформации должно сменяться при растяжении выде-
лением тепла.
Высокая эластичность каучукоподобных тел проявляется
лишь в таких типах (геометриях) деформации, в которых мо-
жет проявляться упругость формы. Значения модуля К для
эластомеров типичны для органических соединений, и они обла-
дают такой же низкой сжимаемостью. Поэтому термодинамика
всестороннего растяжения и сжатия эластомеров должна опи-
сываться уравнениями (4.35) и (4.49).
Использованные выше уравнения состояния для твердых по-
лимеров получены эмпирическим путем. Для эластомеров со-
ответствующие уравнения состояния могут быть получены так-
же на основе статистических (молекулярных) теорий высоко-
эластичности.
Термоэластичность гауссовых сеток
Простое растяжение (сжатие). Современная статистическая
теория высокоэластичности полимеров, в которой предполага-
ется, что распределение расстояний между концами цепей в сет-
ке выражается нормальным (гауссовым) законом и в которой
учитывается, что при деформации изменяется не только энтро-
пия макромолекул, но и их энергия (конформационная), при-
водит к следующему уравнению состояния для одноосной де-
формации ;[5—7]:
, <г2>г Л _ И \
Lo ’ <г2>0 \ Vol2 /
(4-67)
где Lo и Vo — длина и объем нерастянутого образца при температуре Т и ну-
левом давлении; L и V — длина и объем сетки в растянутом состоянии при
температуре Т при действии на нее силы f и давления Р; }.=L/L0— удлинение
(или сокращение); v — число активных цепей в сетке; — фроит-фактор,
в котором <У2>;— среднее расстояние между концами цепей в сетке объе-
мом Vo; <г2>о — среднее расстояние между концами цепей в свободном со-
стоянии.
Молекулярные теории высокоэластичности, приводящие к
уравнению (4.67), базируются на нескольких постулатах, важ-
нейшими из которых являются: 1) полимерные цепи в недефор-
мированной эластомерной сетке принимают статистические
пространственные конформации; 2) межмолекулярные взаимо-
действия не зависят ют конфигураций цепей и, следовательно,
не зависят от деформации сетки; это, в свою очередь, означает,
что возникающие в сетке при деформации в условиях постоян-
ного объема напряжения являются внутримолекулярными по
своей природе; 3) свободная энергия деформированной сетки
является аддитивной функцией свободных энергий индивиду-
альных цепей в сетке.
127
.Центральным вопросом в термодинамике деформации эла-
j стойеров является вопрос о том, какой вклад в общую растяги-
' вающую силу вносят внутримолекулярные энтропийные измене-
i ния, а какой — внутримолекулярные энергетические. Согласно
уравнению (4.68) действующая на тело сила может быть пред-
ставлена в виде суммы энергетической и энтропийной состав-
ляющих:
= Hr + т — =/y+/s (4.68)
\ OL IVtT \ OL JvtT \ OL JvtT ' \O1 !v,L
Таким образом, для решения основной проблемы термоэла-
стичности каучукоподобного состояния необходимо определить
энергетическую и энтропийную составляющие растягивающей
силы в условиях постоянных объема и температуры. Другими
словами, нужно определить вклад в работу деформации изме-
нений внутримолекулярной энтропии и внутримолекулярной
энергии. Исторически сложилось так, что основным методом оп-
ределения энтропийной и энергетической составляющих при де-
формации эластомеров долгое время являлся метод определения
температурной зависимости силы f. Поэтому обычно термо-
динамика деформации эластомеров рассматривается в терми-
нах сил f, fu и fs [8]. В целях общности изложения с термоди-
намикой деформации твердых полимеров мы будем вести изло-
жение в обычных терминах — работы, внутренней энергии и эн-
тропии. Переход от одних величин к другим очевиден.
Условия Т, V = const. Так же как и для твердых стержней,
выражение для дифференциала энергии имеет вид
dU = TdS + fdL—Pd.V
(4.69)
Исходя из уравнения состояния, для работы, теплоты и внут-
ренней энергии имеем [9]:
Wv,t = -f- • Ц2-1 О-2 + 1 - 2) (4.70)
2 А
Qv.T = T(AS)yT = - -у (1 - Т U2 + Ь- 2) (4.71)
2 \ CL1 / А
С dln(r2>0 1-1 .
UV,T~—J — — (I'-f-A —2) (4.72)
2 CL л rv
где C = vkBTL? Г—(4.
\<r2>0 I
Для анализа составляющих эти уравнения удобно предста-
вить в относительной форме:
/ Д3\ rfln(r2>0
Т I —— I = — 1 + Т----------—— (энтропийная составляющая) (4.73)
\ W /у у dT
/ U У d In (Г2)о
( — I = Т —------------(энергетическая составляющая) (4.74)
128
Эти уравнения выражают основную идею статистической
теории высокоэластичности. Для цепей с различной энергией \
конформаций, т. е. с заторможенным внутренним вращением,
определенная доля затраченной на растяжение работы расходу-
ется на изменение внутримолекулярной энергии, связанной с пе-
реходом одних конформаций в другие. Знак температурного
коэффициента невозмущенных размеров макромолекулы
dln<r2>o/c/T определяется видом потенциала внутреннего вра-
щения, т. е. молекулярной структурой цепи и может быть как
положительным, так и отрицательным [5, 10]. Поэтому растя-
жение (сжатие) эластомеров может сопровождаться как уве-
личением, так и уменьшением внутренней энергии. Для цепей
со свободным вращением d'\n<Zr2>odT=O, и работа растяже-
’ТГйтгполностью превращается в тепло. Этот основной вывод «га-
зовых» теорий высокоэластичности, связывающих изменения
при деформации эластомера только с изменением конформаци-
онной энтропий (идеальная энтропийная упругость эластоме-
ров), является очень наглядной иллюстрацией основной идеи
высокоэластичности.
К сожалению, это представление настолько укоренилось, что
даже в современных руководствах, посвященных физике поли-
меров, встречаются заключения об идеальной энтропийной уп-
ругости эластомеров. Теоретические оценки [5, 10] и изложен-
ные ниже экспериментальные данные однозначно показывают,
что в подавляющем большинстве случаев наряду с конформа-
ционной энтропией происходит и изменение конформационной
энергии, причем эти изменения могут быть вполне сравнимы
по величине.
Условия Р, 7'=const. Хотя уравнения (4.73) и (4.74) чрез-
вычайно просты, при их экспериментальной проверке возника-
ет одна существенная сложность, связанная с необходимостью
проведения экспериментов при постоянном объеме. Как будет
видно из дальнейшего изложения, растяжение эластомеров со-
провождается изменением объема на уровне AV/VaslO-4. Для
определения работы деформации, т. е. упругого потенциала де-
формации, таким изменением объема можно без всякого ущер-
ба пренебречь, что равносильно предположению о полной не-
сжимаемости эластомера (коэффициент Пуассона р,—0,5).
Однако выше мы уже убедились, что даже малые изменения
объема чрезвычайно существенны при определении изменения
внутренней энергии и колебательной энтропии вследствие боль-
ших значений внутреннего и кинетического давлений. Поэтому
даже столь незначительные изменения объема (соответствую-
щие им коэффициенты Пуассона составляют 0,4996—0,4998) мо-
гут исказить значения внутримолекулярных изменении. В связи
с этим для корректного определения внутримолекулярных энер-
гетических и энтропийных составляющих нужно учитывать эти
изменения объема. В опытах при постоянном объеме они ком-
пенсируются внешним гидростатическим давлением, и эта ком-
9-440
12»
пенсация представляет основную экспериментальную сложность
таких измерений.
До сих пор проведена лишь одна серия таких исследований
[11, 12], и эти результаты будут рассмотрены далее. Обычно
же эксперименты проводят при постоянном давлении. Прямой
путь состоит в следующем. Изменение энтальпии и энтропии
при деформации эластомеров может быть представлено в виде
суммы изменений при постоянном объеме и в результате изме-
нений объема:
(АЯ)=* (dU)v т + PdV + (dU/dV)PtTdV =
= (dU)v,T + (P + Pt)dV ' (4.75)
T(AS) р>т = T(dS)VtT -f- (dS/dV)P>TdV = T(dS)v>T + PtdV (4.76)
В то время как внутреннее давление эластомеров нетрудно
рассчитать, зная а и Кт*, определение очень малых изменений
объема связано с большими экспериментальными трудностями.
Поэтому. такой чисто термодинамический подход определения
сложен и обычно используют косвенный подход, основанный на
анализе уравнения состояния (4.67). Так же как и в случае
твердых стержней, членом PdV в выражении (4.69) можно пре-
небречь. С учетом этого имеем [9]:
Up,t=^
С 1— 1
№р,т = ~ • — (Х2+Х-2) .
2 Л
l_Tdln <r2>'
dT
Qp,t = T(AS)p,t = —
2aT ] X — 1
X2 + X-2 ] X + '
d In <r2>0 2aT \X— I
- .T - + ^7—7------ -7- W + x - 2)
df X- X — 2 / X
(4-77)
(4.78)
(4-79)
где a — коэффициент объемного теплового расширения недеформированного
эластомера.
В относительной форме выражения для тепла и внутренней
энергии имеют вид
d In (r-Y, 2аТ
(Q/ W)p,r = - 1 + т 9 (4.80)
dT A — 2
rfln(r3)0 2aT
Эти уравнения показывают, что изменения энтропии и внут-
ренней энергии при деформации в условиях Р, 7 = const имеют
как внутри-, так и межмолекулярное происхождение. Внутри-
молекулярные изменения, определяемые d In<r2>o/dT, не зави-
* Объемные коэффициенты умеренно растянутых эластомеров пренебрежи-
мо мало отличаются от коэффициентов исходных нерастянутых эластомеров,
и потому их можно использовать при расчетах.
130
Рис. 4.3. Зависимость относительного изменения внутренней энергии молеку-
лярной сетки от деформации [34] :
1, 2, а- ; А,В, С- ; A- >0; В- Ц1п<Г'—° = 0: С —
\W )Р,Т \W JV,T dT dT
tflnO2>0 Л . л « \
--------- <0; (стрелками отмечены инверсии внутренней энергии).
Рис. 4.4. Зависимости внутренней энергии и теплоты от степени деформации
молекулярной сетки в соответствии с уравнениями (4.78) и (4.79) [34]:
А— теплота; В — внутренняя энергия '>|1 <0 j; С — внутренняя энергия
(——>0 | (стрелками отмечены иверсии теплоты и внутрененй энергии).
сят от деформации. Межмолекулярные изменения, определяе-
мые коэффициентом теплового расширения а, резко зависят от
деформации. Зависимости, выражаемые уравнением (4.81),при-
ведены на рис. 4.3. Различия между значениями при Р, Т=const
и V, T—const существенны в области малых деформаций. Они
определяются резкой зависимостью от деформации члена, со-
держащего а, который при "k—>-1 стремится к бесконечности, а
при больших X может становиться пренебрежимо малым по
dln<r2>0
сравнению с членом Т —
Из сравнения уравнений (4.73), (4.74) и (4.80), (4.81) сле-
дует:
/_Q\ __1 _0\ 2аТ
\W'av~\WJp.t \Wlv,r~^ + ^-2 (4’82>
7_t7_\ (U_\ _(U_\ ЖГ
\ W 'дк "Д w)P>T~\ wlv<T~ V-f-1-2 ( '83)
Уравнения (4.82) и (4.83) характеризуют относительные из-
менения энтропии и внутренней энергии, связанные с измене-
нием объема. Но в таком случае эти соотношения должны быть
идентичны соответствующим соотношениям для твердых тел
[уравнения (4.62), /ЮбЗ)]. Действительно', переходя в уравне-
9’ 131
е, имеем
(4.84)
ниях (4.82) и (4.83) к относительной деформации
(пренебрегая е2)
/_0\ _(LL\________________2аТ_________
' W JbV \ W )\V (1 + е)-'4-(I + е) — 2
2аТ 2рГ
Зе е
в полном согласии с (4.62) и (4.63). Иного и не должно было
быть, так как «жидкостные» свойства эластомеров отражены в
его уравнении состояния только через коэффициент теплового
расширения а. Это свидетельствует также о том, что в уравне-
нии состояния эластомеров межмолекулярное взаимодействие
по крайней мере в области малых деформаций отражено пра-
вильно. ,
Абсолютные изменения колебательной энтропии и внутрен-
ней энергии определяются соотношениями
А— 1
Т(Д5)дк= (Д(7)дг = СаТ — (4.85)
А
Мы видим, что изменение внутренней энергии в результате
изменения объема сетки гауссовых цепей полностью уравнове-
шивается изменением колебательной энтропии и, таким обра-
зом, это измененье объема не вносит вклада в свободную энер-
гию деформации. Джи [13] также пришел к такому заключе-
нию на основе иных рас-суждений. Вспоминая теперь, что
(dQ/dV)T^i (ди1дУ)т~аКтТ, на основании уравнения (4.85)
можно определить относительное изменение объема:
где — изотермическая сжимаемость.
Уравнение (4.86) было впервые получено Хаза.новичем [14]
и Флори [7] другим путем.
Приведенные формулы .и соотношения наглядно показывают
различие в механизме упругости твердых тел и эластомеров
при обычных условиях (Р, Т) при одноосной деформации. Уп-
ругость твердых стержней является объемной, поскольку их де-
•формация возможна только благодаря изменению объема и все
термодинамические свойства стержня связаны именно с изме-
нением объема. Упругость эластомерного стержня определяется
упругостью формы, и вся работа расходуется только на измене-
ние формы, связанное с конформационными характеристиками
макромолекул. Небольшое изменение объема, происходящее
при этом, является как бы сопутствующим эффектом, посколь-
ку связанные с ним энергетические изменения полностью ком-
пенсируются энтропийными изменениями.
Уравнения (4.78) и (4.79) предсказывают появление инвер-
сии теплоты и внутренней энергии (рис. 4.4.). Инверсия тепло-
132
ты должна наблюдаться при деформации:
(4.87)
Для цепей с заторможенным вращением точка инверсии
теплоты смещена в область больших (d ln<r2>o/dT>O) или в
область меньших (d ln<r2>o/dT<0) деформаций по сравнению
с. цепями со свободным вращением. Появление инверсии тепло-
ты является результатом конкурирующего увеличения колеб-а-
тельн7)й Тнтрогтии вслёдствиё увеличения объема и уменьшения
конформационной энтропии, обусловленного изменением формы.
Поскольку а всегда положительна, a dln<r2>0/dT обычно не
превышает ± 1,5-10-3К—то инверсия теплоты в обычных усло-
виях (7'<600 К) для цепей с заторможенным вращением долж-
на наблюдаться только при растяжении. Для цёпей со свобод-
ным вращением это будет происходить при любой температуре.
Максимум теплоты должен наблюдаться при деформации
аТ
1 _ Г d 1п
dT
(«Т)2
dT
а значение теплоты при Х<?макс
С
пмакс ___
- 3
(4.88)
(4.89)
Положение точки инверсии внутренней энергии определяет-
ся соотношением
аТ
dln(r2)0
dT
2 аТ
3 d In (г2)»
dT
(4.90)
Термоупругая инверсия внутренней энергии в отличие от ин-
версии теплоты возможна лишь для цепей с заторможенным
—внутрр-н4н<м^рап1ёшйём.ТГри_этом-^глй цепей с din <r2>o/dT<O
инверсия будет появляться при1Х>1, т. е. при растяжении, а для
цепей с d\n<r2>oJdT>0 при Х<1, т. е. при сжатии. Для значе-
ний а= (7-г-10) • 10~4 К-1 и dln<r2>0/^=±(l,54-0,5)-10^ КЧ
133
типичных для эластомеров, имеем Ху=1,3—2,2 (растяжение) и
Xi/>0,5 (сжатие). Следует подчеркнуть, что термоупругая ин-
версия внутренней энергии не связана с часто наблюдаемой
кристаллизацией при растяжении эластомеров. Она обусловле-
на разными знаками внутри- и межмолекулярных составляю-
щих внутренней энергии.
> Экстремум внутренней энергии должен наблюдаться при де-
формации:
аТ
т d In (rJ>„
dT
aT
Tdln(r^
(4.91)
Выражение для экстремума громоздко, и мы его приводить
не будем. Естественно, что в условиях постоянных V и Т ин-
версия как теплоты, так и внутренней энергии исчезает.
При мгновенном (без совершения работы) сокращении рас-
тянутого эластомерного стержня
\ W 'р т
О*
Q--Q
(4.92)
где Q* — теплота мгновенной разгрузки.
Здесь возможны три случая: Q* = 0, U=0; Q*>0, если U<0 и
Q*<0, если U>0.
В современном варианте статистической теории высокоэлас-
тичности, приводящей к уравнению (4.67), предполагается, что
фронт-фактор не зависит от объема (а значит, и от давления).
Тобольский и Шен [15] предположили, однако, что такая зави-
симость возникает вследствие изменения межмолекулярного
взаимодействия в сетке при деформации, и предложили полу-
эмпирическое уравнение состояния, учитывающее эту зависи-
мость. Оно имеет вид
vkBT <r*>t
Lo ‘<'2>o \ V ) \ V^l
(4-93)
dln<r2>0 \
din У /
— константа, зависящая от строения полимера.
T.L ,
При у=0 уравнение (4.93) переходит в классическое урав-
нение (4.67). С этим уравнением можно поступить так же, как
это 'было сделано с уравнением (4.67), и получить выражения
для термодинамических потенциалов. Мы приведем здесь лишь
два самых важных следствия из этого уравнения. Первое каса-
ется энергетической составляющей, которая имеет вид
! U \ / U \ 2аТ
W = V ~ 12IV 2"~V«r (4.94)
\ W ' у 7 \ W / р 7 Л — 2
Это уравнение отличается от уравнения (4.84) лишь наличи-
ем постоянной величины уаТ, т. е. и в этом случае энергетиче-
134
ская составляющая не зависит от деформации. Относительное
изменение объема при деформации выражается соотношением
ДР X— 1 Г у 1
— = хгс— 1 + (Х« +1 - 2)
г о Л _ 2 J
(4.95)
При у = 0 это выражение переходит в уравнение (4.86). Из-
менение колебательной энтропии и внутренней энергии, соответ-
ствующее этому изменению объема, выражается уравнением
X — 1 у
(Д(7)д7 = Т(Д5)дк = аСТ — l+-f- (X-’-j-X —2)
Л 2
(4.96)
Кручение. Флори [7] показал, что для сетки гауссовых це-
пей температурная зависимость силы, действующей на эласто-
мер при постоянном объеме, пропорциональна dln<r2>o/d7'
независимо от типа деформации.
Для сетки гауссовых цепей уравнение состояния для круче-
ния имеет вид [16]
.. l/2nvfeB7 <r2)i 6 4
М~ Ио '(г^'Д/0
(4.97)
где М — равновесная величина пары сил, необходимая для закручивания ци-
линдра длиной Z-o на Угол 0; До — радиус недеформированного образца, осталь-
ные величины имеют тот же смысл, что и в уравнении (4.67).
Анализ этого уравнения приводит к следующим
ниям:
соотноше-
е
f l/4n;v^B7’ (r->i <2q
r= Md9 = v-62
J (r2>0 4o
,o
(d In (r2>o
-1-aT + T
dT
I d In (r2)o \
(W)W = W - uT + T - \ --
\ al f
W I P>T<L dT
/MJ\ „ „ d In <r2>0
T(AS)ViT>l = w(1-T^^
\ dT ,
WT d 1П <r2>°
(At/)v,r,L = ---—-----
dl
IbS \ din (r-)n .
T — ) = — I + T--------- (энтропийная составляющая)
\ W Jv T l dT
_Tdln <r2)"
W]v<t,l dT
(энергетическая составляющая)
(4.98)
(4.99)
(4.100)
(4.101)
(4.102)
(4.103)
(4.104)
(4.105)
(4.106)
135
Видно, что относительное изменение энтропии и внутренней
энергии при деформации в условиях постоянного давления не
зависит от степени закручивания, что принципиально отличает-
ся от результата для простого растяжения. Энтропийная и
энергетическая составляющие для кручения, как и предсказыва-
ет теория [7], совпадают с соответствующими параметрами для
простого растяжения. Из уравнений (4.99) и (4.100) следует,
что инверсия теплоты и внутренней энергии при кручении долж-
на отсутствовать.
Различия между изменениями энтропии и внутренней энер-
гии при постоянных давлении и объеме, обусловленные измене-
нием объема, выражаются уравнением
T(AS)iK=(At/)4t,= -W (4.107)
Как видим, и в случае кручения изменение объема не вно-
сит вклада в свободную энергию деформации. Согласно урав-
dU ос rip
нению -&=—Т изменение объема при кручении равно
ДУ =
Л4хг0
2
1/4 nvk^T
Й' 4
— а&т
La
(4.108)
Таким образом, кручение сопровождается уменьшением
объема, и это уменьшение объема в отличие от простого растя-
жения (сжатия) параболически зависит от угла закручивания.
Эффекты, связанные с изменением объема при небольших де-
формациях кручения, должны быть значительно меньше, чем
при одноосных деформациях.
Термоэластичность негауссовых сеток
Уравнение (4.67) получено в предположении, что распределение
между концами цепей в сетке выражается гауссовой функцией.
Считают, что такое распределение вполне адекватно описывает
поведение сеток при деформациях, когда г мало по сравнению
С ДЛИНОЙ ПОЛНОСТЬЮ ВЫТЯНУТОЙ ЦеПИ, В ЧаСТНОСТИ ПрИ Г^0,ЗгМакс
[8, 17]. По мере приближения г к гмакс гауссова функция рас-
пределения все в возрастающей степени неадекватно выражает
вероятность распределения расстояний между концами цепей
в сетке и соответственно возникающую при растяжении силу.
Наиболее очевидный недостаток гауссовой функции распреде-
ления состоит в том, что она предсказывает нулевую вероят-
ность лишь для значений г, соответствующих бесконечности, а
Не ДЛЯ ЛЮбОГО Г, бОЛЬШеГО Гмакс.
Для значений г, сравнимых с Гмакс, зависимость растягиваю-
щей силы от степени растяжения имеет вид [8, 17]
> vk^T
/«—|-jP1'2[5?-1(A.y-1'2) — (4.109)
136
где S’-1—обратная функция Ланжевена; р— число статистических сегментов
в цепи, определяющее максимальную растяжимость цепи к=р'12.
Когда сетка достигает предела растяжимости к=Ур, сила
стремится к оо, так как S’-1 (О—Расхождение между значе-
ниями силы по уравнениям (4.67) и (4.109) при г = 0,3 гМакс со-
ставляет около 3%, а при г = 0,5гМакс — около 8%. При даль-
нейшем приближении г к гМакс расхождения резко возрастают.
Таким образом, уравнение (4.109) в отличие от уравнения
(4.67) предсказывает резкое увеличение растягивающей силы
При Г—>Гмакс-
Наряду с этим существуют теории, в которых предполагает-
ся, что даже при малых деформациях распределение расстоя-
ний между концами цепей не является гауссовым. В частности,
Кригбаум и Конеко [18], использовав представления о модель-
ной кубической решетке, получили для такого случая следую-
щее уравнение состояния:
(4ИО)
Со \ у о / \ л,- /
где
ф = (Зе” — е-2')В + (15eV — 10 + 3e-*i)C у ...
81 / V у/3
56 V? \ К J X
9 { V Vя/, 4 \
здесь В = -v(—) (V+TXj;
/ 8 8 \ Ек
X X4 + — + “ № ); j — ------ (Ек — конформационная энергия).
\ 5 5 / 2kv,T
Смит [19], использовав иные представления, получил
нее сложное уравнение:
vkBT
не ме-
(г2>п / 1 \ Г 2 4
v<r2>0 3X2+ 4 1
х СМ —:------------
</->» Ь 1
и б2 — сложные функции <г2>о и
Г2>,).
(4.1П)
В котором 61
(<г2>* несколько отличается от
Все эти уравнения состояния приводят к зависимости энерге-
тической составляющей от деформации и предсказывают появ-
ление подъемов на зависимости напряжение — деформация при
больших деформациях. Однако экспериментальная проверка
первого предсказания затруднена. Таким образом, эти, а также
другие негауссовы статистические теории [5, 17] предсказыва-
ют зависимость энергетической составляющей от деформации,
и лишь согласно классической статистической теории она не за-
висит от деформации.
Наряду со статистическими теориями высокоэластичности
для количественного описания больших деформаций эластоме-
ров широко используют чисто феноменологический подход, в ко-
тором, исходя из определенных соотношений между напряже-
ниями и деформациями, «конструируются» упругие потенциалы
137
деформации. Классическая статистическая теория приводит к
простейшему упругому потенциалу W с одной константой. Наи-
более широкое распространение для феноменологического опи-
сания растяжения эластомеров получил потенциал Муни — Рив-
лина (МР) [8]
1УМР = С1(Г1 — 3) +С.,(/2 — 3) (4.112)
в котором /] и /2 — первый и второй инварианты деформаций.
Для простого растяжения этот потенциал имеет вид (эласто-
мер предполагается абсолютно несжимаемым, т. е. Р=Р0)
П7МР = Cj - (X3 + X - 2) + С,
Л Л
2Х —
(4.113)
X
или, переходя к напряжению (на исходное сечение Л):
dW
о = —
дХ
(4.114)
Потенциал МР лучше описывает зависимость напряжение —
деформация эластомеров при умеренных растяжениях по срав-
нению с потенциалом W. Однако константа С2 в Ц7МР не имеет
молекулярной интерпретации, и потому соотношение (4.114)
обычно рассматривается [17] как полуэмпирическое, в котором
константа С2 характеризует степень отклонения зависимости
напряжения от деформации, предсказываемой уравнением
(4.67).
Потенциал МР не описывает другие типы деформаций [17].
При С2=0 IFMP переходит в W.
Шен [20] рассмотрел термоэластические особенности тел,
описываемых этим уравнением, и показал, что энергетическая
составляющая зависит от степени растяжения, уменьшаясь с
увеличением степени растяжения:
(X3- 1)^+.
— аТ
(4.115)
Если эластомер предполагается сжимаемым, то изменение объ-
ема при растяжении (сжатии) согласно уравнению Муни — Рив-
лина выражается соотношением
ДУ/К, = 2кт-(Х-1)(С;/Х 4-С2) (4.116)
Валанис и Ландел [21] предложили для простого растяже-
ния (сжатия) следующее уравнение состояния
/=слФ1пЛ+^,"|“Нг;] <4-"7)
которое приводит к такому выражению для энергетической со-
ставляющей (в предположении V=Vq):
[fu \B'l / д In С ат\ аТ I 2Хз/-> + 2 4
( — =(1 —--------—----- —----- 1 —---------Х------ (4.118)
\f \ д In Т 3 3j (2Х-у + 1) In X ] v '
138
Как видно, это уравнение также предсказывает зависимость
энергетической составляющей от деформации.
Важно подчеркнуть, что вытекающее из анализа этих по-
тенциалов уменьшение энергетической составляющей при растя-
жении является, по-видимому, отражением изменения межмо-
лекулярного взаимодействия по мере растяжения, а внутримоле-
кулярная часть энергетических изменений остается постоянной,
как того требует статистическая теория. Зависящая от дефор-
мации доля энергетической составляющей при очень больших
растяжениях достигает значений —0,68 и —0,07 соответствен-
но для потенциала МР и ВЛ.
Обширные исследования феноменологических уравнений со-
стояния эластомеров выполнены в последние годы Чоглом с
сотр. [22—24]. Было предложено новое феноменологическое
уравнение состояния эластомеров при растяжении:
2C/_£V
п \ К J
к п+2
(4.119)
где п — константа, характеризующая нелинейность зависимости напряжение —
деформация.
При п = 2 это уравнение переходит в рассмотренное выше
уравнение Тобольского — Шена (4.93), а если при этом и у = 0,
то — в основное уравнение состояния (4.67). В первоначальном
варианте [22] п полагали не зависящей от температуры. В та-
ком случае вытекающая из уравнения (4.119) энергетическая
составляющая не зависит от степени растяжения. Сделанное за-
тем предположение n = f(T) приводит к зависимости энергетиче-
ской составляющей от степени растяжения.
Предпринимаются попытки представить упругий потенци-
ал IF в виде ряда, содержащего в отличие от уравнения Муни —
Ривлина более двух членов [25—27]. Широкая эксперименталь-
ная проверка термоэластических свойств эластомеров, характе-
ризующихся такими потенциалами, до сих пор, по-видимому, не
проводилась, поэтому здесь они рассматриваться не будут.
ЭНТРОПИЙНЫЕ И ЭНЕРГЕТИЧЕСКИЕ ЭФФЕКТЫ
ПРИ ДЕФОРМАЦИИ СЕТОК И ИХ МОЛЕКУЛЯРНАЯ ИНТЕРПРЕТАЦИЯ
Внутримолекулярные эффекты
Реакция молекулярной сетки на приложенное напряжение ха-
рактеризуется двумя механизмами. Первый связан с изменени-
ем объема и обусловлен наличием межмолекулярного взаимо-
действия. Этот механизм совершенно аналогичен механизму
сжимаемости (расширения) обычных жидкостей. Второй меха-
низм связан с деформацией сетки, причем изменение свободной
энергии при деформации определяется конфигурационными из-
менениями активных цепей сетки. В ранних теориях предпола-
139
галась изоэнергетичность конформаций макромолекул, в связи
с чем свободная энергия деформации отождествлялась с кон-
формационной энтропией цепей. При таком представлении мо-
лекулярное строение цепей полностью игнорируется.
Согласно изложенным выше теплофизическим представле-
ниям о механизме упругости сеток, учитывающим молекуляр-
ное строение цепей, их деформация должна сопровождаться не
только изменением конформационной энтропии, но и в общем
случае изменением внутримолекулярной энергии, обусловлен-
ным неизоэнергетичностью конформаций макромолекул. Поэто-
му надежное экспериментальное определение внутримолекуляр-
ных эффектов при деформации эластомеров имеет фундамен-
тальное значение для физики полимеров вообще, поскольку по-
зволяет связать поведение макромолекул в блочном аморфном
состоянии с их поведением в растворе. Признание этого способ-
ствовало расширению экспериментальных исследований термо-
эластических свойств каучукоподобных материалов не только
классическим методом измерений температурных зависимостей
равновесных напряжений [8, 18J, но и с использованием пре-
цизионной калориметрии [28, 29]. Ниже рассматриваются наи-
более характерные экспериментальные результаты исследова-
ния внутримолекулярных эффектов при деформации эластоме-
ров и дается их молекулярная интерпретация на основе теории
поворотной изомерии.
Определению энергетических составляющих fu/f= (AU/W)v,r
многих аморфных эластомеров посвящено большое число работ,
результаты которых суммированы в обзорах [30—33] и обоб-
щающих статьях [29, 34]. Подавляющее, большинство таких ис-
следований выполнено при постоянном давлении, так что внут-
римолекулярные эффекты в них определялись косвенным спо-
собом, включающим введение поправок на изменение объема
при деформации. В связи с этим исключительную важность
имеют работы Аллена и Прайса с сотр. [11, 12, 29, 35], в кото-
рых определение внутримолекулярных эффектов для ряда
эластомеров проведено в условиях постоянного объема. Их
ценность определяется возможностью проверки достоверности
данных, полученных при Р —const, и общей экспериментальной
проверки статистической теории высокоэластичности. Поэтому
прежде всего на примере наиболее широко исследованных
различными методами эластомеров — натурального каучука и
полидиметилсилоксана — сравним данные, полученные различ-
ными методами.
Характерные результаты, суммированные в табл. 4.2, пока-
зывают, что надежные значения внутримолекулярных эффектов
могут быть получены как в условиях постоянного объема, так
и в условиях постоянного давления с использованием различных
методических подходов и различных типов деформаций. Эти же
результаты позволяют сделать еще одно очень важное заключе-
ние. Поскольку представленные данные получены в независи-
мо
Таблица 4.2. Значения энергетической составляющей
в натуральном каучуке и полидиметилсилоксане,
полученные различными методами
Метод и условия проведения эксперимента Постоянный параметр Энергетиче- ская состав- ляющая Литератур- ная ссылка
Натуральный каучук
f— Т (растяжение) V 0,2 [11]
f— Т (растяжение) V 0,12±0,02 [12
f— Т (растяжение) pa 0,18±0,03 [12
f— Т (растяжение) Р 0,24±0,03 [23
f— Т (растяжение — сжатие) Р 0,18±0,03 [36
Калориметрия (растяжение) Р 0,18±0,01 [37
Калориметрия (кручение) Р 0,20±0,02 [37
Калориметрия (растяжение) Р 0,28±0,03 . [9, 34]
Калориметрия (инверсия теплоты при Р 0,22±0,03 [9, 34]
растяжении)
Калориметрия (растяжение) 0,356 [38]
f—Т (растяжение в присутствии Р 0,16±0,03в [33]
различных количеств разных рас-
творителей)
Полидиметилснлоксан
f — Т (растяжение) V 0,25±0,01 [35]
f — Т (растяжение) р 0,27±0,02 [39]
Калориметрия (растяжение) р 0,30±0,05г [9, 34]
Калориметрия (инверсия теплоты при растяжении) р 0,25±0,03г [9, 34]
f—Т (растяжение в присутствии 50% растворителя) р 0,15±0,03 [33]
3 Хотя эксперименты проведены при постоянном давлении, для перехода к условиям
постоянного объема использовалось точное термодинамическое уравнение и эксперимен-
тально определялись необходимые термоэластнческне коэффициенты.
6 Температура опыта 44 °C.
в Среднее значение из четырех независимых исследований.
г Ненаполненные образцы и образцы, содержащие = 20% активного наполнителя.
мых экспериментах несколькими методами на образцах, разли-
чающихся степенью сшивания, условиями сшивания (неориен-
тированное аморфное состояние, неориентированное и ориенти-
рованное кристаллическое состояние, раствор), наличием или
отсутствием растворителя или наполнителя, они наглядно пока-
зывают практическую нечувствительность энергетической со-
ставляющей ко всем этим параметрам и условиям. Этот чрез-
вычайно важный экспериментальный результат свидетельствует
о независимости внутримолекулярных эффектов от факторов,
могущих влиять на межмолекулярное взаимодействие, и дока-
зывает внутримолекулярный характер рассматриваемых эффек-
тов. Такого рода данные свидетельствуют [33] также об отсут-
ствии какой-либо значительной упорядоченности в аморфных
полимерах, обусловленной межмолекулярным взаимодействием.
Из сказанного выше и данных, приведенных в табл. 4.2, следу-
ет, что применение гауссового уравнения состояния (4.67) для
анализа данных по термодинамике деформации эластомеров,
141
Рис. 4.5. Зависимость энергетической со-
ставляющей от деформации (относитель-
но участков А, Б и В, см. текст).
полученных при Р = const, позво-
ляет в первом приближении оп-
ределить внутримолекулярные
энергетические и энтропийные
эффекты, а значит, и рассчитать
температурный коэффициент не-
возмущенных размеров макромолекул. О том, почему это яв-
ляется лишь первым приближением, будет сказано далее.
Одно из основных следствий уравнения (4.67) — независи-
мость энергетической составляющей от степени растяжения. По-
этому для окончательного суждения о применимости уравне-
ния (4.67) и о корректности определения внутримолекулярных
эффектов на его основе важными являются результаты иссле-
дования зависимости энергетической составляющей от степени
деформации. И в данном случае этот вопрос наиболее детально
исследовался для натурального каучука. Схематически резуль-
таты многочисленных исследований можно представить в сле-
дующем виде (рис. 4.5). Как видно, кривая зависимости fu/f от
X может содержать три участка. Участок А — это теоретическая
зависимость при отсутствии ориентационной кристаллизации.
Большинство экспериментальных исследований [31—34] хоро-
шо согласуется с этим теоретическим выводом. Это свидетель-
ствует о независимости внутримолекулярных изменений от сте-
пени деформации. Появлению кристаллизации соответствует
участок В, который всегда отвечает снижению энергетической
составляющей, что вызвано уменьшением энергии при кристал-
лизации. Однако в некоторых исследованиях по термоэластич-
ности натурального каучука, а также фторированных сополиме-
ров, сополимеров этилена с пропиленом и других эластомеров
наблюдался участок Б [30]. Его интерпретация вызывает боль-
шие затруднения, поскольку он обычно появляется при Х<1,5,
когда уравнение (4.67) хорошо предсказывает не только внут-
римолекулярные эффекты, но и межмолекулярные (см. далее)
и, следовательно, зависимость растягивающей силы от дефор-
мации.
Предпринимались попытки связать зависимость энергетиче-
ской составляющей от степени деформации в начальной обла-
сти с зависимостью межмолекулярного взаимодействия от кон-
формации макромолекул (что противоречит основному постула-
ту теории высокоэластичности), однако специальные исследова-
ния этой проблемы показали [32], что эта зависимость скорее
явдяется экспериментальным артефактом, а не присуща эласто-
мерам. Дело в том, что в области малых деформаций член,
отражающий межмолекулярное взаимодействие в формуле
142
(4.81), резко возрастает (см. рис. 4.3) и для корректного опре-
деления внутримолекулярных эффектов в этой области необхо-
дима чрезвычайно высокая точность определения деформации.
Это требование относится ко всем методам исследования при
постоянном давлении. Эта проблема исчезает для условий по-
стоянного объема, и потому для окончательного заключения
очень важным является вывод о независимости энергетической
составляющей от степени растяжения, сделанный в соответст-
вующих исследованиях [11, 12, 29].
Важно подчеркнуть, что в области умеренных деформаций
(1,5<?.<3) практически во всех исследованиях эластичности
натурального каучука, сополимеров этилена с пропиленом и не-
которых других эластомеров, для которых при малых деформа-
циях иногда появляется участок Б, наблюдается независимость
внутримолекулярных эффектов от степени растяжения. Это свя-
зано с тем, что в этой области деформаций член 2аТ/Х2+Х—2
(или соответствующий ему член аТ/Х3—1 при исследовании за-
висимостей f—Т) становится несущественным, так что необходи-
мостью введения поправок на изменение объема в этой обла-
сти деформаций практически отпадает. Это и обусловливает
надежное определение внутримолекулярных энергетических эф-
фектов на основании уравнения (4.67) даже в тех областях
деформаций, где оно не описывает зависимости растягивающей
силы от деформации. Все сказанное выше наглядно показано
на рис. 4.6, который для четырех эластомеров при Х<2 иллю-
стрирует характер общих и внутримолекулярных энергетиче-
ских и энтропийных эффектов, полученных калориметрическим
методом [34].
Таким образом, отсюда следует общее заключение, что в об-
ласти малых и умеренных деформаций внутримолекулярные
изменения не зависят от деформации, как это и предполагается
в гауссовой статистической теории высокоэластичности.
Это заключение позволяет перейти к рассмотрению внутри-
молекулярных изменений, оцененных термодинамическими ме-
тодами, для широкого круга полимеров и рассмотреть их связь
с молекулярным строением цепей. Наиболее надежные данные
для ряда полимеров, полученные как при измерении темпера-
. турных зависимостей напряжений, так и прямым калориметри-
рованием, суммированы в табл. 4.3. Здесь же приведены рас-
считанные на основании энергетических составляющих темпе-
ратурные коэффициенты невозмущенных размеров макромолекул
и для сравнения представлены значения этой же характе-
ристики, полученные на основании вискозиметрических измере-
ний разбавленных растворов. Во всех случаях, когда при виско-
зиметрических измерениях принято в расчет взаимодействие
полимер — растворитель и его изменение с температурой, соответ-
ствие между значениями (Лп<г2>о^7’) полученными различны-
ми методами, достаточно хорошее. Это еще раз показывает не-
зависимость межмолекулярных взаимодействий от конформа-
143
Рис. 4.6. Зависимость относительного
изменения энтропии и внутренней
энергии от степени растяжения [34]:
/ Q \ ,'\U\ (Q \
\ u )р,т ( u )р,т V1 'И,т
(MJ\
— ; а — натуральный каучук (Л) и
\ " IV>T
статистический сополимер этилена с про-
пиленом (В); б — полидиметилснлоксан
(Л) и полихлоропрен (В) (стрелками отме-
чены инверсии теплоты и внутренней энер-
гии).
ционного состава, свидетельст-
вует о гауссовом распределе-
нии цепей в нерастянутых сет-
ках и подтверждает сделанный
вывод об отсутствии упорядо-
ченности в нерастянутых блоч-
ных аморфных полимерах. Из
табл. 4.3 видно также, что в
большинстве полимеров энер-
гетическая составляющая отлична от нуля и принимает как по-
ложительные, так и отрицательные значения, причем видимая
связь знака энергетической составляющей с молекулярной
структурой цепи отсутствует.
Согласно теории поворотной изомерии [5, 10, 40], положи-
тельная энергетическая составляющая возникает в тех полиме-
рах, в которых более вытянутые конформационные последова-
тельности обладают большей энергией, поскольку при растяже-
нии или повышении температуры их число возрастает. Напротив,
отрицательные значения энергетической составляющей ха-
рактерны для тех полимеров, у которых более вытянутые кон-
формации характеризуются более низкими конформационными
энергиями. Теория поворотной изомерии позволяет на основании
знаков и значений энергетических составляющих оценить раз-
ность энергий вытянутых и скрученных конформаций. Эти
оценки проводятся на основании соотношения [10]
d In </"->(> Ек
энергетическая составляющая = Т----- = — А----- (4.120)
dT k^T
где А — коэффициент, учитывающий симметрию цепи и соотношение чередую-
щихся валентных углов.
Для цепей с симметричными привесками и независимым вра-
щением вокруг соседних связей при наличии в них поворотных
изомеров (0±120°) X = 2/(2 + g-), где g=e~£K/kBT. При EK~k^
fu~f, причем если £к>0, то fu<0. Оценка разности энергий
гош- и транс-изомеров в полиэтилене на основании значений
энергетической составляющей (—0,47) по уравнению (4.120)
привела к значению £„ = 2,1 -103 Дж/моль, что очень хорошо со-
144
Таблица 4.3. Внутримолекулярные энергетические эффекты
при деформации молекулярных сеток и их сравнение
с данными вискозиметрических измерений
на изолированных макромолекулах [29—34]
Пол и мер Калориметрия* Температурная за- висимость напряже- ний** Вискози- метрия**
Z ‘/1 / /И \ \ ZJV / с л М к. V с л М V с с л «ч U V С
Полиэтилен Натуральный каучук и полинзопрен Полидиметилснлоксан Полннзобутилен цис-1,4-Полибутадиен Статистический сополи- мер этилена с пропи- леном 0,22 0,27 0,14 —0,42 0,75 0,91 0,44 — 1,43 —0,47 0,18 0,* 1 27 —0,05 0,13 —0,42 — 1,05 0,56 0,91 —0,17 0,40 — 1,40 —I,2 0,6 0,71 —0,28 0,30 — 1,4
* Значения, относящиеся к определенной температуре, обычно к 293 К.
** Средине значения для определенного температурного интервала, чаще всего 50—
100 аС.
гласуется со спектроскопическими и термодинамическими дан-
ними. Для цепей с симметричными привесками и коррелиро-
ванными конформациями соседних связей коэффициент А в
уравнении (4.120) равен 1.
Для цепей с чередующимися неравными валентными углами
(л—ci! и л—аг), к которым относится ПДМС, коэффициент А
имеет вид
1 + т,- — 2т
2/<2+g)-п_7
(1 + V)x' - 2-rj
где
1 — g t 1 — cos osj cos а2
1 1 -I- 2g sin 04 sin a2
В таких полимерах знак fu зависит от соотношения т] и х'.
Если т)< [х'—У(х')2—1]. то fu<0, а если т)>[х'—У(х')2—Ч то
fy>0. Так же как и в ПЭ, транс-конформация в ПДМС харак-
теризуется меньшей энергией по сравнению с гош-конформаци-
ей, однако в ПДМС из-за неравенства последовательных ва-
лентных углов транс-конформация является менее вытянутой
(более компактной). Поэтому энергетическая составляющая в
ПДМС в отличие от ПЭ оказывается положительной, поскольку
растяжение цепи ПДМС сопровождается переходом более
10—440 145
компактных и обладающих меньшей энергией транс-изомеров
в более вытянутые и характеризующиеся большей энергией
гош-изомеры.
Положительная энергетическая составляющая в цис-1,4-дие-
новых полимерах является следствием того, что изомеры, обла-
дающие меньшей энергией, являются более компактными. На-
против, отрицательная энергетическая составляющая в транс-
1,4-полихлоропрене появляется вследствие перехода при растя-
жении более компактных, но обладающих большей энергией изо-
меров в более вытянутые изомеры с меньшей энергией. Большие
„ dln<r2>0
отрицательные значения Т ----ат ' в статистических сопо-
лимерах этилена с пропиленом свидетельствуют о том, что как
транс-последовательности в ПЭ, так и 3-спиральные конформа-
ции изотактического ПП являются сильновытянутыми и обла-
дают более низкой энергией.
Таким образом, исследование внутримолекулярных эффектов
при деформации эластомеров и их анализ на основе теории по-
воротной изомерии являются очень плодотворным подходом к
изучению термодинамического и молекулярного аспектов высо-
коэластичности. Исследования термоэластичности молекуляр-
ных сеток позволяют оценить конформационные энергии макро-
молекул и, следовательно, предсказать свойства цепей, опреде-
ляемые их конформациями.
Известна попытка установить корреляцию между знаками и
значениями энергетической составляющей и чисто геометриче-
скими характеристиками макромолекулы [41]. В качестве такой
характеристики была выбрана площадь поперечного сечения
макромолекулы в кристаллическом состоянии So. Увеличение
площади поперечного сечения макромолекулы как будто приво-
дит к изменению знака энергетической составляющей с отрица-
тельного на положительный. Например, энергетическая состав-
ляющая равна: для полиэтилена (So = 0,183 нм2)—0,47; для по-
лиэтиленоксида (So=0,21 нм2) + 0,1 [31]; для политетр а мети -
леноксида (So = O,176 нм2) —опять —0,47 [31]. Физические при-
чины такой корреляции неясны и к ней вообще нужно относить-
ся с большой осторожностью. Теория поворотной изомерии объ-
ясняет такое изменение энергетической составляющей различи-
ем конформационной энергии изомеров, что подтверждается со-
гласованностью теоретических расчетов и экспериментальных
данных о поведении этих макромолекул в растворе [40]. С по-
зиций отмеченной корреляции такие изменения остаются необъ-
яснимыми.
Межмолекулярные эффекты
Проведенный выше теоретический анализ показывает, что по
крайней мере, одноосная деформация эластомеров при посто-
янном давлении должна сопровождаться межмолекулярными
146
УМ,а,^-Ю3,Дж'г
Рис. 4.7. Зависимость механической работы (Д), теплоты (В) и внутренней
энергии (С) от степени растяжения [34]:
а - натуральный каучук; б — статистический сополимер этилена с пропиленом.
Рис. 4.8. Внутримолекулярные и межмолекулярные изменения внутренней энер-
гии при растяжении эластомеров [34] :
А — (Д(/)р т; - В — (Д<7)у т; С — (Д(7) ду ; D — (Л(7)ду в соответствии с уравнением
(4.85); а — натуральный каучук; б — статистический сополимер этилена с пропиленом,
эффектами, связанными с изменением объема при деформации.
Корректное экспериментальное определение этих эффектов со-
пряжено с огромными экспериментальными трудностями, обус-
ловленными малыми абсолютными значениями этих эффектов.
Тем не менее измерение этих эффектов и их сравнение с пред-
сказаниями теории чрезвычайно важны для понимания моле-
кулярных механизмов деформации эластомеров и проверки кор-
ректности постулатов, лежащих в основе теории.
Наиболее очевидное проявление межмолекулярных эффек-
тов связано с инверсиями при малых деформациях, которые яв-
ляются результатом конкуренции внутри- и межмолекулярных
изменений. Калориметрические исследования одноосного растя-
жения эластомеров очень наглядно это демонстрируют
(рис. 4.7). Точка инверсии теплоты (табл. 4.4) в полном соот-
ветствии с уравнением (4.87) зависит от знака и величины энер-
гетической составляющей и от коэффициента теплового расши-
рения. Значения (\U/W)vt, определенные по уравнению (4.87),
хорошо согласуются с данными, полученными более общим ме-
тодом (см. рис. 4.6). Энергетическая составляющая в СЭП
отрицательна, и согласно уравнению (4.90) при растяжении
должна наблюдаться инверсия внутренней энергии, что прекрас-
10’
147
Таблица 4.4. Параметры инверсии теплоты и внутренней энергии
и определенные на их основании значения энергетической составляющей [34]
Полимер Инверсия теплоты Инверсия внутренней энергии
о о о +1 е*? о © +1 Jc \ U7 JVT
Натуральный каучук Сополимер этилена с пропиленом Полихлоропрен Полидиметилснлоксан 1,165 1,105 1,130 1,235 0,22 —0,40 —0,08 0,25 1,35 (±0,01) 2,37 —0,42 (±0,02) —0,10
но подтверждается экспериментально не только качественно, но
и количественно (см. табл. 4.4). Энергетическая составляющая
в полихлоропрене также отрицательна, но ее абсолютное зна-
чение существенно меньше, чем в СЭП, и потому инверсия внут-
ренней энергии в этом случае наблюдается при значительно
больших растяжениях.
Эквивалентным подходом является определение инверсии в
обычных экспериментах по изучению зависимости f—Т [8, 17,
36]. Результаты для НК, полученные этими двумя независимы-
ми методами, хорошо согласуются. .
Установленная выше независимость внутримолекулярных эф-
фектов от деформации позволяет разделить общие энергетиче-
ские изменения на меж- и внутримолекулярные (рис. 4.8). Наи-
более важным выводом из этих данных является заключение,
что предсказания, вытекающие из уравнения (4.85), согласуют-
ся с экспериментальными межмолекулярными изменениями
энергии и энтропии лишь при малых степенях растяжения (Х<
<1,3). При больших растяжениях экспериментальные кривые
лежат выше теоретических, и по мере увеличения деформации
расхождение между ними возрастает.
Эмпирическое уравнение, описывающее зависимости (ДU) w
и (Д£)д от деформации, имеет вид
X -1
(AU)bv = T(AS)av = СаТ " ' [1 + у'(Х2+ Х-2)] (4.121)
Л
Значения у' зависят от химического строения полимера и на-
ходятся в пределах 0,1—0,2. Это уравнение аналогично уравне-
нию (4.95) (у = 2у')-
Первопричиной межмолекулярных эффектов является изме-
нение объема при деформации, и потому судить о них можно и
по зависимости изменения объема. Существует несколько путей
экспериментальной оценки изменений объема: прямой (дилато-
148
метрический), калориметрический на основании уравнений
(4.85) и (4.86) и основанный на термодинамическом тождестве
(dV/dl)PtT^(df/dP)L>1 (4.122)
Типичные результаты, полученные двумя методами (рис. 4.9
и 4.10), показывают, что статистическая теория высокоэластич-
иости хорошо предсказывает увеличение объема при растяжении
эластомеров лишь до значений Х< 1,3, а при больших растяже-
ниях она недооценивает увеличение объема и, таким образом,
недооценивает межмолекулярные изменения, обусловленные из-
менениями объема при деформации сетки. Этот факт не являет-
ся неожиданным, поскольку жидкостные свойства сеток в этой
теории учитываются фактически лишь обычными коэффициен-
тами теплового расширения и сжимаемости. При Х<1,3 этого,
по-видимому, достаточно для отражения межмолекулярных из-
менений. Однако при больших удлинениях сетки на межмолеку-
лярные изменения могут влиять не только чисто жидкостные
свойства, но и специфические свойства самой сетки, прежде все-
го ее топология и неаффинная деформация узлов.
Хорошо известно, что при %= 1,5—2,0 зависимость напряже-
ний от % для многих эластомеров хорошо описывается уравне-
нием (4.114) Муни — Ривлина и отклоняется от уравнения
(4.67). Поэтому одним из подходов к анализу изменения объ-
ема при деформации является использование уравнения (4.116).
Хотя в литературе отмечалось [29, 32], что зависимость изме-
нения объема от X описывается уравнением (4.116), Трилор'
[42] заключил, что это не может быть общим правилом. Он
детально рассмотрел проблему изменений объема при различ-
ных функциональных представлениях W и пришел к выводу.
Рис. 4.9. Относительное изменение объема при растяжении эластомеров [34]:
4 — натуральный каучук; В — статистический сополимер этилена с пропиленом; 1 — ка-
лориметрические результаты (О и ) и дилатометрические результаты работы ( + ) [43];
2 — зависимости, построенные по уравнению (4.86).
Рис. 4.10. Зависимость относительного изменения объема при растяжении нату-
рального каучука [42]:
/ — экспериментальная зависимость (по данным Аллена и Прайса с сотр. [12, 29]); 2—
теория Огдена [25]; 3 — статистическая теория [уравнение (4.86)].
14»
что: во-первых, наибольшие отклонения от экспериментальных
данных характерны для статистической теории; во-вторых, наи-
лучшее совпадение наблюдается для выражения W в виде трех-
членного уравнения, предложенного Огденом [25], хотя и в
этом случае расхождения с экспериментом достаточно велики,
особенно при больших деформациях. Сказанное наглядно ил-
люстрирует рис. 4.10. В связи с этим встречающееся в литера-
туре хорошее совпадение экспериментальных данных с выраже-
нием (4.116) можно действительно считать случайным.
В ранних работах по изучению изменения объема при рас-
тяжении каучукоподобных полимеров было сделано предполо-
жение [13, 17], что линейная сжимаемость растянутого полиме-
ра изотропна даже для умеренных растяжений. Это означает,
что, так же как и в классической теории упругости изотропного
твердого тела, приложение растягивающего напряжения долж-
но сопровождаться увеличением гидростатической компоненты
напряжения на величину, составляющего одну треть этого на-
пряжения, для того, чтобы объем тела оставался неизменным.
Использование этого предположения приводит к следующему
выражению для изменения объема при растяжении [13]:
AV = ^L(2p\dL (4.123)
О I OL I
L о
т. е. изменение объема определяется производной растягиваю-
щей силы в любой точке. Хотя предположение об изотропии
сжимаемости при умеренных и больших деформациях гауссо-
вых сеток было впоследствии теоретически опровергнуто [7],
немногочисленные экспериментальные результаты [17, 29, 43]
лучше описываются уравнением (4.123), чем уравнением (4.86).
Однако это еще, возможно, не означает, что анизотропия сжи-
маемости у растянутого эластомера действительно отсутствует,
а лишь свидетельствует о наличии определенного вклада в AV,
не учитываемого гауссовой теорией.
Все эти соображения ясно указывают на то, что ни статисти-
ческая теория высокоэластичности, ни феноменологические
уравнения типа Муни — Ривлина или Огдена не могут количест-
венно предсказать изменения объема и, следовательно, связан-
ные с ними межмолекулярные изменения внутренней энергии
и энтропии при одноосной деформации эластомеров. Трилор
[42] считает, что физические причины такого расхождения в на-
стоящее время неясны, и потому для решения проблемы следует
идти по пути тщательных и всесторонних (различные типы де-
формации) экспериментальных исследований изменений объема
и межмолекулярных эффектов, а не модифицировать теоретиче-
ские представления.
Однако в связи с этим необходимо отметить, что анализ не-
зависимых данных по изменению объема ряда полимеров пока-
зывает [9, 32, 34], что они очень хорошо описываются уравнени-
150
ем (4.95), вытекающим из полуэмпирического уравнения состоя-
ния Тобольского — Шена [уравнение (4.93)] или родственного
ему феноменологического уравнения Чогла с сотр. [уравнение
(4.119)]. Константа у, отражающая зависимость фронт-фактора
от объема (или давления), в большинстве случаев оказывает-
ся отличной от нуля. Таким образом, существуют экспери-
ментальные основания считать фронт-фактор в уравнении
(4.67) зависящим от объема (давления), хотя строгое теорети-
ческое обоснование этого факта отсутствует. Если это действи-
тельно так, то константа у для эластомеров может рассматри-
ваться как экспериментально определяемый коэффициент, по-
добный коэффициентам теплового расширения и сжимаемости.
Таким образом, можно считать, что вопреки мнению Трило-
ра современная статистическая теория высокоэластичности
нуждается в определенной модификации с целью количествен-
ного предсказания межмолекулярных эффектов и зависимости
растягивающей силы от деформации при Х>1,3. Флори [44, 45]
в последнее время сравнил «аффинную сетку», в которой узлы
жестко закреплены и аффинно смещаются с деформацией,
«фантомную сетку» Джеймса и Гута, в которой цепи свободно
проходят друг через друга, а флуктуации узлов около средних
положений гауссовы и не зависят от деформации, и сетку [46],
в которой на флуктуации узлов наложены вызванные присут-
ствием соседних цепей ограничения, и показал, что эти последние
ограничения вносят вклад в свободную энергию, зависящий от
деформации и степени ограничения. Сравнение показывает, что
при малых деформациях поведение сетки с ограничениями
близко к аффинной, а при больших — к фантомной.
Новая теория Флори [46] предсказывает ряд эксперимен-
тально наблюдаемых явлений при деформации эластомеров,
в частности уменьшение приведенной силы с увеличением сте-
пени растяжения, и в отличие от уравнения Муни — Ривлина
правильно передает поведение при сжатии. Флори полагает, что
большинство отклонений экспериментальных закономерностей
от существовавших до его новой теории теоретических зависи-
мостей обусловлено именно тем, что не учитывались ограниче-
ния на флуктуацию узлов сетки. Однако, как подчеркивает сам
Флори, форма учета этих ограничений в новой теории имеет
скорее интуитивный и довольно произвольный характер, а не
строгий, и поэтому ожидать количественного совпадения теории
с экспериментом не приходится. Тем не менее эта новая теория
указывает физические пути модификации классических теорий
высокоэластичности, и это дает основание надеяться, что более
совершенный вариант новой теории позволит объяснить наблю-
даемую зависимость межмолекулярных эффектов и изменения
объема при деформации.
Возникает естественный вопрос: в какой мере недооценка
межмолекулярных эффектов в классической молекулярной
теории высокоэластичности сказывается на величине внутримо-
151
лекулярных эффектов, определенных при постоянном давлении
с учетом поправки на непостоянство объема, вытекающей из
этой теории? Поскольку уравнение (4.95) наилучшим образом
описывает межмолекулярные эффекты, воспользуемся экспери-
ментальными значениями у и оценим по уравнению (4.94) энер-
гетическую составляющую для НК (у = 0,3) и СЭП (у = 0,2).
Расчеты показывают, что член уаТ равен 0,04 для НК и 0,06 для
СЭП, в связи с чем положительная энергетическая составляю-
щая для НК несколько уменьшается, а отрицательная энерге-
тическая составляющая для СЭП несколько возрастает. После
учета этой поправки энергетическая составляющая для НК ока-
зывается еще ближе к значению, полученному при постоянном
объеме (см. табл. 4.2). Аналогичные оценки, проведенные на ос-
новании уравнения (4.116) в предположении С[ — С2, показыва-
ют, что в той области растяжений, где уравнение Муни — Рив-
лина описывает зависимость силы от деформации, поправочный
член аТ'/(С1/С2Х+1) в самом худшем случае имеет тот же поря-
док (0,05—0,08). В отличие от уравнения (4.94) величина это-
го поправочного члена зависит от X, и в связи с этим должна
появиться очень слабая зависимость энергетической составляю-
щей от степени растяжения. Физические причины появления в
этом случае такой зависимости неясны.
Проведенные оценки показывают, что погрешности, вносимые
во внутримолекулярные эффекты, вследствие неадекватного
описания статистической теорией межмолекулярных изменений
невелики и находятся на уровне точности определения энергети-
ческих составляющих. Это и объясняет практическую нечувст-
вительность энергетической составляющей к экспериментальным
условиям, условиям получения образцов, а также к условию
С2э^0.
Большие деформации и проблема ограниченной
растяжимости цепей
При больших деформациях расстояние между концами цепи
сравнимо с длиной полностью вытянутой цепи, гауссово распре-
деление становится неадекватным и появляются негауссовы
эффекты, обусловленные ограниченной растяжимостью цепей.
Кроме того, при очень больших деформациях более существен-
ным может стать вклад межмолекулярных эффектов вследствие
анизотропного усреднения силового поля между соседними сег-
ментами [47]. Принято считать [8, 17], что резкое увеличение
модуля эластомеров при больших деформациях, в частности
натурального каучука, является отражением этой ограниченной
растяжимости цепей. Аналогично трактуется обычно и резкий
подъем на кривой зависимости Муни — Ривлина (рис. 4.11).
Однако эти изменения, как неоднократно отмечалось в литера-
туре, несомненно, связаны и с кристаллизацией, протекающей
в натуральном каучуке при растяжении и приводящей к само-
152
Рис. 4.11. Зависимость отношения f/
от обратной степени растяжения:
1 — по уравнению (4.114); 2 — экспериментальная
зависимость для натурального каучука.
усилению образца, которое обеспе-
чивает возможность достижения
больших удлинений. Для многих
других эластомеров из-за низкой
прочности достичь высоких степеней
удлинения не удается. Таким образом, проблема проявления
ограниченной растяжимости цепей и связанные с ней негаус-
совы эффекты фактически корректно экспериментально не были
исследованы.
В середине 70-х годов Дж. Марк обратил на это внимание
и провел с сотрудниками цикл систематических исследований,
результаты которых суммированы в обзоре [48]. Было одно-
значно установлено, что для ненаполненных эластомеров, не
способных кристаллизоваться при растяжении, подъемы на кри-
вой зависимости Муни — Ривлина отсутствуют. Они исчезают
и в эластомерах, способных кристаллизоваться, при введении в
них растворителей. Эти, а также ряд других соображений [48]
привели к выводу, что часто наблюдаемое возрастание модуля
эластомеров при больших растяжениях, несомненно, связано с
кристаллизацией или с возникновением стеклообразных микро-
доменов в сополимерах при растяжении и что в отсутствие этих
эффектов разрыв обычных эластомеров наблюдается раньше,
чем может проявиться ограниченная растяжимость цепей. От-
меченные экспериментальные наблюдения побудили Флори [44]
заключить, что в общем случае гауссово распределение должно
быть вполне приемлемым для описания эластичности (в отсут-
ствие кристаллизации) сеток, содержащих в среднем 100 и бо-
лее последовательных связей, не только при малых и умерен-
ных деформациях, но и при больших. Однако не вызывает
сомнений, что при приближении длины растянутой макромоле-
кулы к полностью вытянутому состоянию негауссовы эффекты
должны проявиться.
Общие соображения дают основания предполагать, что про-
явление ограниченной растяжимости цепей вряд ли может вы-
ражаться в виде резкого эффекта, аналогичного проявлению
кристаллизации при растяжении. При больших растяжениях,
когда деформация становится неаффинной, должно наблюдать-
ся перераспределение напряжений между соседними цепями.
Поэтому, даже если макроскопическая деформация будет соот-
ветствовать максимальной вытянутой длине какой-либо цепи,
это еще не будет означать, что эта цепь в действительности
окажется полностью вытянутой. Возникающее в такой цепи на-
153
пряжение будет перераспределяться между соседними цепями в
сетке, и такое перераспределение явится серьезным препятстви-
ем растяжению цепей до их максимальной длины. Если это в
конце концов произойдет и длина цепей начнет приближаться
к максимальной, эффект должен проявляться постепенно из-за
последовательного включения различных цепей и не будет со-
провождаться в отличие от кристаллизации гистерезисными яв-
лениями.
Первая успешная экспериментальная попытка проникнуть в
область проявления ограниченной растяжимости цепей в моле-
кулярных сетках была осуществлена в самое последнее время
Марком с сотр. [49] на сетках полидиметилсилоксана, обла-
дающих бимодальным распределением цепей (короткие и отно-
сительно длинные цепи). Наличие коротких цепей в таких сет-
ках, по-видимому, существенно с точки зрения их очень малой
растяжимости, а длинные цепи, как полагают, ингибируют на-
чало разрушения и таким образом обеспечивают возможность
больших удлинений. На изотермах, полученных в температур-
ном интервале от 6 до 150 °C, наблюдались эффекты, однознач-
но свидетельствующие об ограниченной растяжимости цепей.
Теоретический анализ показал, что длина цепей г, при которой
начинают проявляться негауссовы эффекты, обусловленные ог-
раниченной растяжимостью, в частности подъем на зависимо-
сти Муни — Ривлина, составляет примерно 60—70% средней
максимальной длины полидиметилсилоксановой цепи гмаКс, ко-
торая определялась из соотношения гмакс = 0,134п нм (п— число
скелетных связей). Аналогичные расчеты показывают, что раз-
рыв цепей происходит при г= (0,8—0,9) гМакс-
Хотя эти результаты получены на сетках со специфическим
распределением длин цепей и не могут быть непосредственно
перенесены на другие эластомеры, они чрезвычайно важны для
понимания соотношения растяжимости полимерных цепей и уп-
ругих и прочностных свойств сеток. Они, в общем, наглядно
подтверждают представление о том, что в обычных условиях
ненаполненные эластомеры, не обладающие способностью к са-
моусилению при растяжении, разрушаются при деформациях
значительно меньших, чем те, при которых начинает проявлять-
ся ограниченная растяжимость цепей.
В связи с изложенным возникает задача изучения эластич-
ности очень густых молекулярных сеток, в которых расстояния
между узлами составляют лишь единицы — десятки скелетных
связей.
Шварц [50] теоретически рассмотрел поведение модельных
сеток, образованных цепями, содержащими от 4 до 14 связей.
При этом он исходил из следующих представлений. Длины всех
связей предполагались одинаковыми, фиксированные валентные
углы составляли л/2, а углы вращения связей принимались
равными Ойл (цис- и транс-положение). Конформационным
энергиям придавались значения £«=2,3 кДж/моль (аналогично
154
Рис. 4.12. Зависимость энергетической со-
ставляющей от деформации для модель-
ной сетки с 14 связями между узлами и
конформационными энергиями £к =
= 2,3 кДж/моль (отрицательные значе-
ния fu/f) и Ек=—0,7 кДж/моль [50]:
1 — с учетом флуктуаций узлов сетки; 2 —- с
учетом флуктуаций узлов сетки и жесткости
сшивок; 3 — обычная сетка с большим числом
связей между узлами.
для ПЭ), —0,7 кДж/моль и 0.
В такой модели все возможные
конформации цепей могут быть
получены графически и обычным
методом определены свободная и
внутренняя энергии. Проведен-
ные численным методом расчеты
показали, что при отсутствии флуктуаций узлов сетки и в пред-
положении, что они допускают свободное вращение последова-
тельных связей возникающая при одноосном растяжении энер-
гетическая составляющая растягивающей силы fu/f не зависит
от числа связей и деформации и, таким образом, не отличается
от теоретического значения fu/f для модельной сетки, образо-
ванной длинными цепями (рис. 4.12). При учете флуктуаций
узлов сетки, а также в предположении, что узлы являются
жесткими мостиками между двумя последовательными связями,
энергетическая составляющая зависит от деформации, причем
эта зависимость оказывается очень слабой для положительных
значений энергетической составляющей и очень сильной — для
отрицательных.
Этот вывод является довольно неожиданным. Сравнение
[50] с данными для радиационно-сшитого ПЭ, имеющимися в
работах Флори и Принса, не привело к однозначному выводу о
наличии такой сильной зависимости энергетической составляю-
щей в густосшитом ПЭ от деформации. Экспериментальные
данные о термоэластичности очень густых сеток с контролируе-
мой длиной цепей между узлами в настоящее время отсутст-
вуют.
ТЕРМОЭЛАСТИЧНОСТЬ ГЕТЕРОФАЗНЫХ ЭЛАСТОМЕРОВ
Улучшение механических, технологических и эксплуатационных свойств
эластомеров обычно достигается путем создания гетерофазных смесей эласто-
меров и наполнения эластомеров высокодисперсными наполнителями. Тради-
ционный подход состоит в использовании технического углерода, оксидов ме-
таллов, некоторых солей и жестких полимеров, обеспечивающих хорошее взаи-
модействие эластомера с развитой поверхностью наполнителя, следствием
которого является повышение прежде всего жесткости эластомера, а часто и
прочности. Новым подходом является создание термоэластопластов — блок-
сополимеров с чередующимися гибкими (эластомерными) и жесткими (поли-
мерными) блоками, в которых благодаря термодинамической несовместимости
155
компонентов происходит микрофазовое разделение. Термоэластопласт представ-
ляет собой микрогетерогенную систему, в которой эластомерная матрица на-
полнена доменами жесткого компонента, являющимися одновременно узлами
эластомерной сетки, и мелкодисперсным усиливающим наполнителем. Таким
образом, общим для таких систем является их гетерогенность, поскольку они
представляют собой дисперсные коллоидные системы.
Смеси эластомеров обычно также являются гетерогенными, однако их от-
личие с деформационной точки зрения от наполненных твердыми наполнителя-
ми систем состоит в способности всех фаз к высокоэластическим деформациям.
Механизм усиления и эластические свойства гетерофазных эластомеров, осо-
бенно наполненных неорганическими наполнителями, продолжают оставаться
предметом многочисленных исследований, однако доля работ, в которых эти
вопросы рассматриваются с термодинамических позиций, мала. Поэтому мно-
гие теплофизические аспекты деформирования гетерофазных эластомеров ос-
таются неизученными. Ниже будут рассмотрены теплофизические аспекты двух
важнейших проблем; природа явления размягчения наполненных эластомеров
и термоэластопластов под действием напряжения и соотношение энтропийных
и энергетических эффектов при деформировании.
Для наполненных эластомеров характерен эффект размягчения (эффект
Маллинза — Патрикеева), заключающийся в том, что при повторных после
первого растяжениях для достижения одного и того же значения деформации
требуется значительно меньшее усилие, в результате чего на графике в коор-
динатах напряжение — деформация появляется гистерезисная петля (рис. 4.13).
Этот эффект в значительной мере необратим, он не связан также с релакса-
ционными процессами, что указывает на изменение внутренней структуры на-
полненной резины. Его количественное изучение является одним из актуальных
вопросов физики эластомеров, поскольку оно может быть источником количе-
ственной информации о механизме усиления эластомеров. Считается общепри-
нятым, что первопричиной усиливающего действия дисперсного наполнителя
является образование физических и химических связей между молекулами кау-
чука и наполнителя и что именно прочностью этих связей и их способностью
перестраиваться под действием напряжений определяются деформационные и
прочностные свойства наполненных резин.
Многочисленные исследования эффекта размягчения, суммированные в об-
зорах [51—53] и монографиях [54—57], привели к нескольким различным
представлениям о механизме этого эффекта. Согласно одному из них считает-
ся, что это явление обусловлено необратимым разрывом связей полимер—на-
полнитель и наиболее напряженных участков цепей и соответственно уменьше-
нием числа эффективных цепей в сетке. В соответствии с этим механизмом
значительная часть работы, затраченной при первичном деформировании образ-
ца, должна поглощаться им и расходоваться на разрушение связей полимер —
наполнитель и цепей сетки. Согласно другой точке зрения, причина размягчения
заключается в скольжении физических узлов полимерная молекула — наполни-
тель по поверхности частичек наполнителя, приводящем к уменьшению числа
коротких цепей вследствие перераспределения длин цепей в сетке. В соответ-
ствии с этим механизмом дополнительная работа, затрачиваемая при первом
Рис. 4.13. Силовое размягчение наполненных резин (а) и термоэластоплас-
тов (б).
растяжении на преодоление сил трения полимерных цепей о поверхность части-
чек наполнителя, полностью рассеивается в виде тепла.
Маллинз [51] полагает, что большая часть эффекта размягчения обуслов-
лена неаффинным перемещением узлов сетки и зацеплений при деформации и
тем, что не все онн возвращаются в исходное состояние после разгрузки. Счи-
тается, что н в этом случае работа гистерезисной петли трансформируется в
тепло и рассеивается. Хотя в пределах каждой из этих точек зрения существует
ряд более тонких различий относительно конкретных механизмов эффекта раз-
мягчения, в основных выводах о судьбе работы, соответствующей гистерезис-
ной петле, первая точка зрения вполне противоположна остальным. Определен-
ную роль в эффекте размягчения может играть также разрушение первичной
структуры самого наполнителя, происходящее при малых деформациях. Изло-
женные точки зрения на явление размягчения, безусловно, являются упрощен-
ными, н истинный механизм конечно же является более сложным. Однако,
несмотря на это, можно полагать, что в основе явления размягчения лежит
один из указанных выше процессов.
Явление размягчения характерно н для каучукоподобных блок- и приви-
тых сополимеров [58, 59]. Наличие только химических связей между жестки-
ми доменами, играющими роль мелкодисперсного наполнителя, и эластомерной
матрицей, по-виднмому, исключает возможность перегруппировки цепей на по-
верхности доменов. Согласно современным представлениям, основанным на
структурно-механических исследованиях (например, [60, 61]), силовое размяг-
чение термоэластопластов в отличие от наполненных эластомеров в значитель-
ной мере обусловлено разрушением исходной доменной структуры. При доста-
точно больших содержаниях жесткого блока [>35% (об.)] первое растяжение
(см. рис. 4.13) по характеру совершенно аналогично вынужденной высокоэла-
стнчности стеклообразных полимеров (см. гл. 5). Действительно, при деформа-
ции в несколько процентов наблюдается предел вынужденной эластичности,
затем в образце появляется и развивается шейка. После полной трансформации
всего образца в шейку наблюдается ее равномерное растяжение. В отличие от
стеклообразных полимеров подавляющая часть деформации термоэластоплас-
тов обратима после снятия нагрузки, поскольку осуществляется за счет высо-
коэластической матрицы.
Во втором цикле предел вынужденной эластичности отсутствует и дефор-
мация подобна деформации усиленных эластомеров. По мере уменьшения до-
ли жесткого блока в сополимере кривая первого растяжения все в большей
степени становится похожей на обычную кривую растяжения наполненной ре-
зины. Важной особенностью размягчения термоэластопластов является возмож-
ность практически полного восстановления исходных механических свойств и
доменной структуры при температурах ниже температуры стеклования или
плавления жестких блоков.
Таким образом, теплофизический аспект проблемы размягчения гетерофаз-
ных эластомеров может быть сформулирован следующим образом: происходят
ли при первичном растяжении необратимые изменения структуры, сопровож-
дающиеся увеличением внутренней энергии системы, или образующаяся новая
структура энергетически эквивалентна исходной. С экспериментальной точки
зрения это означает, что кроме обычного анализа механического гистерезиса
(см., например, [62, 63]) необходимо калориметрически определить, рассеива-
ется ли вся соответствующая гистерезисной петле работа в виде тепла. Такие
исследования с использованием высокочувствительной калориметрической ме-
тодики были проведены в последние годы на наполненных силоксановых рези-
нах [64, 65] н бутадиен-стирольных термоэластопластах [66]. Эффект усиле-
ния от введения наполнителя в силоксановые эластомеры является самым боль-
шим нз наблюдаемых для всех типов резин, и потому именно в этих резинах
можно ожидать проявления энергетических изменений в наиболее четком виде.
Кроме того, эффект размягчения в этих резинах не осложнен ни кристаллиза-
ционными процессами при растяжении (при комнатной температуре), ни за-
медленными релаксационными процессами, характерными для многих резин.
Для определения энергетических изменений образцы растягивались в мик-
рокалориметре до заданной деформации и сокращались до начальной длины.
Изменение внутренней энергии образца за цикл Д17рс< (»— номер цикла), харак-
157
теризующее необратимое изменение его структуры, определялось суммирова-
нием тепловых эффектов и работы:
p.i + + Qe i (4.124}
где И7р+ и — работа растяжения и сокращения; Qp~ — тепло, выделяемое
при растяжении; Qc+ — тепло, поглощаемое при сокращении.
На рис. 4.14 представлены характерные зависимости механического гисте-
резиса ДВ7рс в первом цикле и изменения внутренней энергии ЛС7рс в первом
и последующих циклах для одного из типов силоксановых резни и бутадиен-
стирольных термоэластопластов. Близкие результаты получены и для других
силоксановых резин и СБС-термоэластопластов. В то время как для наполнен-
ной резины при всех деформациях практически вся гистерезисная петля рас-
сеивается в виде тепла, для термоэластопласта заметная доля (ж 25%) меха-
нической гистерезисной петли остается в образце, повышая его внутреннюю
энергию. Это дает основание полагать, что молекулярные механизмы размягче-
ния наполненных резин и термоэластопластов, по-видимому, различны.
Поскольку эффект размягчения резин не связан с энергетически необрати-
мыми изменениями структуры резины, то можно предположить, что наиболее
вероятным механизмом размягчения резин является не отрыв молекул поли-
мера от поверхности частичек наполнителя, а перегруппировка цепей на этой
поверхности вследствие нх скольжения. Таким образом, результаты энергети-
ческого исследования явления размягчения наполненных резин вполне согласу-
ются с наиболее обоснованной точкой зрения на молекулярный механизм этого
явления. Однако можно еще раз заметить, что сведение механизма размягче-
ния резины только к перераспределению длин цепей в полимерной сетке вслед-
ствие их скольжения по поверхности частичек наполнителя под действием на-
пряжения и попытка количественного описания этого эффекта только на осно-
ве статистической теории сетки являются слишком упрощенным подходом.
Легко представить, что в зависимости от формы и объема наполнителя, а так-
же от числа сорбированных на его поверхности полимерных цепей и от харак-
тера их упаковки при переходе реальной наполненной системы в квазиустой-
чивое состояние в процессе ее деформирования может происходить не только
проскальзывание цепей по поверхности частицы, но и ее вращение и необра-
тимое перемещение в полимерной матрице. Это, в свою очередь, должно при-
водить к соответствующему изменению пространственной структуры наполнен-
ной резины, что может быть причиной появления анизотропии свойств, в част-
ности механических.
Проверка этого предположения, проводившаяся на образцах, вырезанных
из предварительно размягченной резины под
различными углами к оси растяжения, дейст-
вительно показала [64, 65], что характер раз-
мягчения резин зависит от направления ее
вторичного растяжения, т. е. наблюдается ани-
зотропия размягчения резин при деформации.
Степень размягчения отчетливо зависит от
количества наполнителя в резине и соответст-
венно от степени ее усиления. Предварительное
растяжение сильнонаполненных резин приво-
дит к значительному размягчению и в попереч-
ном направлении. Сравнение кривых повтор-
ного растяжения в продольном направлении
Рис. 4.14. Зависимость механического гистере-
зиса ДИ7рс (1, 2) цикла растяжение — сокраще-
ние и изменения внутренней энергии за цикл
А(7рс (!', 2') от деформации:
1, /' — силоксановая резина [64]; 2, 2'— звездообраз-
ный блок-сополнмер СБС, содержащий 34% связанно-
го стирола [66].
158
и под различными углами к оси первичного растяжения свидетельствует о раз-
личном характере усиливающего действия наполнителя в различных направ-
лениях. Предварительное растяжение слабонаполненных и соответственно ме-
нее усиленных резин практически не сказывается на усиливающем действии
наполнителя в поперечном направлении.
Таким образом, важным результатом исследования энергетики силового
размягчения наполненных силоксановых резин является вывод, что при дейст-
вии механической нагрузки на наполненные резины происходит перестройка ее
исходной пространственной структуры в новую энергетически эквивалентную,
но отличающуюся от исходной структуры механическим поведением. Можно
полагать, что эта закономерность характерна и для других некристаллизую-
щнхся при растяжения резин, содержащих активный мелкодисперсный напол-
нитель.
Перейдем теперь к рассмотрению возможного механизма размягчения тер-
моэластопластов, вытекающего из факта возрастания внутренней энергии в
процессе размягчения. Совокупность имеющихся в настоящее время данных
об особенностях структурного, механического и теплового поведения термо-
эластопластов при последовательных растяжениях позволяет следующим обра-
зом представить механизм их размягчения [60, 61, 66—68]. Типичный термо-
эластопласт характеризуется микродоменной структурой. Размер, форма и
взаиморасположение доменов зависят от содержания жесткого компонента и
молекулярных характеристик обоих компонентов, а также от объема, который
занимает фаза жесткого блока в момент формирования структуры. Даже при
небольшом (~20%) содержании жесткого блока сферические домены, кон-
тактируя друг с другом, образуют достаточно непрерывную жесткую фазу.
Степень ее непрерывности возрастает с увеличением содержания жесткого бло-
ка. При начальных растяжениях (~5%) происходит нагружение этой жесткой
фазы, сопровождающееся поглощением тепла. Дальнейшее растяжение приво-
дит сначала к искажению формы сетки доменов, а затем и к нарушению ее
сплошности. При большом содержании жесткой фазы ее разрушение происхо-
дит очень резко и сопровождается образованием шейки.
После разрушения жесткой фазы напряжение переносится на эластомер-
ную матрицу, и дальнейшее развитие деформации идет по механизму каучуко-
подобной эластичности. При этом происходит дальнейшее разрушение жесткой
фазы и дробление ее на отдельные несвязанные домены различных формы и
размеров. Этот процесс завершает размягчение. После снятия напряжения в
размягченных образцах наблюдается небольшая остаточная деформация, свя-
занная. по-видимому, с остаточной деформацией доменов и с некоторой оста-
точной ориентацией цепей. Таким образом, основной причиной увеличения внут-
ренней энергии при размягчении термоэластопластов является, по-видимому,
возникновение искажений и неравновесных дефектов в жесткой фазе в резуль-
тате пластической деформации и разрушения.
Процесс размягчения термоэластопластов характеризуется ярко выражен-
ной тиксотропией. За восстановлением структуры и свойств можно наблюдать
по изменению параметров малоуглового рентгеновского рефлекса, соответст-
вующего определенному большому периоду в образце, возникающему вслед-
ствие упорядоченного расположения доменов, по восстановлению исходной
гистерезисной петли или соответствующего деформации жесткой фазы началь-
ного эндоэффекта [66, 68, 69]. Анализ показывает, что кинетика восстановле-
ния исходной сетки жесткой фазы и определяемых ее наличием свойств экспо-
ненциально зависит от температуры и, несмотря на то что оно протекает в
высоковязкой среде, имеет, судя по значению энергии активации, составляю-
щей около 40 кДж/моль, диффузионный характер. Можно полагать, что для
коагуляции доменов и восстановления первоначальной жесткой фазы сущест-
венное значение имеет как вынужденно-эластический характер деформации до-
менов, так и образовавшаяся при разрушении жесткой фазы новая поверх-
ность.
Диенстирольные термоэластопласты склонны к образованию в высшей сте-
пени регулярной надмолекулярной структуры, возникающей в результате
строго периодического расположения доменов в матрице. Экструзией или
другими подобными методами микрогетерогенную структуру этих систем ока-
159
залось возможным приводить в такое состояние, когда надмолекулярные
структурные элементы, образованные аморфными компонентами, во всем
объеме образца имеют общую ось ориентации, т. е. система приобретает «мо-
нокристаллический» характер. Такие системы были впервые получены и ис-
следованы Келлером с сотр. [70] и названы «надмолекулярными монокристал-
лами». В частности, в бутадиен-стирольных термоэластопластах, содержащих
30—35% связанного полистирола, экструзией и прессованием удается получить
«надмолекулярные монокристаллы», в которых практически бесконечные ци-
линдрические агрегаты полистирола образуют в полнбутадиеновой матрице
высокоупорядоченную гексагональную решетку с периодом 25,5 нм [67, 71].
Свойства таких монокристаллов, в частности термоэластическне [69, 71], силь-
ноанизотропны. На примере таких структур удается однозначно идентифици-
ровать механизм разрушения жесткой фазы при размягчении (рис. 4.15).
Растяжение вдоль цилиндров уже при деформации 10—‘20% приводит к
их разрушению на отдельные участки строго постоянной длины 65 нм. При
этом переход от начального крутого участка деформационной кривой к парал-
лельному оси деформации очень резкий. Из данных рентгеноструктурного ана-
лиза следует, что восстановление исходной цилиндрической структуры проис-
ходит довольно быстро, чему, вероятно, способствует образование микропу-
стот между отрезками цилиндров. Растяжение перпендикулярно оси цилинд-
ров протекает в несколько этапов. На первом этапе («100%) ориентация
всей массы цилиндров сохраняется и происходит лишь деформация ячейки.
На втором этапе (>100%) «кристалл» надламывается в наиболее слабых
местах по всей длине и принимает форму плоского зигзага, и лишь после это-
го может наступить его дробление. Кривая первичного растяжения перпенди-
кулярно оси цилиндров более похожа на кривую растяжения наполненного
эластомера. Восстановление структуры жесткой фазы и исходных свойств
протекает значительно дальше, чем при деформации вдоль цилиндров. Таким
образом, размягчение при растяжении вдоль цилиндров наступает при значи-
тельно меньшнх деформациях, чем при растяжении поперек цилиндров, а
восстановление структуры и свойств после снятия напряжения происходит
быстрее. Анализ показывает, что размягчение обычных «поликристаллических»
образцов, т. е. не обладающих «монокристаллической» структурой, представ-
ляет собой суперпозицию рассмотренных процессов.
Проведенный сравнительный анализ энергетики размягчения наполненных
эластомеров и каучукоподобных блок-сополимеров показывает, что природа
явления размягчения в этих эластомерных материалах различна. В наполнен-
ных системах она определяется физическими процессами, происходящими в
мягкой фазе в области ее контакта с наполнителем, а в термоэластопластах
она связана с их доменной структурой н определяется особенностями обра-
зования и распада жесткой фазы. Хотя основные изменения, связанные с
размягчением, происходят в первом цикле растяжение — сокращение, однако
свойства размягченного эластомера окончательно стабилизируются лишь пос-
ле нескольких таких циклов [51].
Естественно возникает вопрос об энтропийных и энергетических измене-
ниях при деформации таких размягченных наполненных эластомеров и их
зависимости от степени наполнения и деформации. Известно, что в присутст-
вии усиливающих наполнителей модуль упругости эластомера возрастает в
соответствии с уравнением Гута — Смолвуда:
Е' = Е(1 + 2,5? + 14,I?2) = Ех1 (4.125)
где Е — модуль упругости ненаполненного эластомера; <р — объемная доля
наполнителя.
Тогда для наполненных эластомеров выражения для W7, Q и U [уравне-
ния (4.77) — (4.79)] будут включать множитель т). Тем не менее энтропийная
и энергетическая составляющие в соответствии с уравнениями (4.73), (4.74) и
(4.80), (4.81) не должны зависеть от присутствия наполнителя. Это, конечно,
является следствием внутримолекулярного характера энергетической состав-
ляющей в эластомерах, и очевидно, что наполнитель не может изменить энер-
гетику конформационных переходов. Считается, что действие наполнителя
сводится к увеличению числа активных цепей в сетке [v в уравнеиин (4.67)],
160
Рис. 4.15. Схема разрушения жесткой фазы бутадиен-стирольного термоэлас-
топласта со структурой надмолекулярного монокристалла [67, 71].
Рис. 4.16. Зависимость энергетической составляющей н полидиметилснлоксано-
вых наполненных эластомерах, блок-сополимерах и привитых сополимерах от
объемного содержания наполнителя:
I — наполненная резина на основе ПДМС (наполнитель HiSil-233) [72]); 2 —Д — напол-
ненная раздиропрочная резина (Sil-52) [73], □ —наполненная резина Sil-4600 [73], О—.
блок-сополимер полиарнлатполнднметилсилоксан [73], X — привитой сополимер полиак-
рилонитрил — полидиметилснлоксан [73].
введенных в нее через связи каучук — наполнитель. Эти представления пере-
несены н на каучукоподобные блок-сополимеры.
Немногочисленные экспериментальные данные по термоэластичности на-
полненных эластомеров противоречивы. Это наглядно демонстрирует рис. 4.16,
где представлена зависимость энергетической составляющей в полидиметил-
силоксановых эластомерах [57, 72, 73]. Оставляя в стороне вопрос о несоот-
ветствии энергетических составляющих ненаполненного эластомера (см.
табл. 4.2 и 4.3), рассмотрим лишь тенденции изменения этой зависимости
с введением наполнителя. Одна из зависимостей (кривая 1) свидетельствует
о влиянии наполнителя, начиная с самых малых содержаний, причем рост
энергетической составляющей близок к экспоненциальному. Примечательно
также, что сравнительно небольшое содержание наполнителя (»15%) оказы-
вает исключительно сильное влияние на энергетическую составлиющую. Как
видим, эти данные совершенно не согласуются с изложенными ныше взгляда-
ми на роль наполнителя как причины увеличения числа активных цепей в
сетке. Кривая 2 обобщает данные как для наполненного полиднметилсилокса-
на, так и для его блок-и привитых сополимеров. Можно считать, что согласно
этим результатам до 15%-ного наполнения энергетическая составляющая не
зависит от присутствия наполнителя, а затем наблюдается довольно резкое ее
повышение. Заметим, что, хотя в обоих случаях энергетическая составляю-
щая зависит от присутствия наполнителя, она, так же как н для ненаполнен-
ных эластомеров, не зависит практически от степени растяжения.
Зависимость энергетической составляющей от содержания и от усиливаю-
щей способности наполнителя, наблюдавшейся кроме полидиметилсилоксана
для бутилкаучука [74], пластифицированного наполненного поливинилхлорида
[75] и наполненного сополимера этилена с пропиленом [76], свидетельствует
о том, что для наполненных эластомеров энергетическая составляющая теряет
свой очевидный смысл как доля только внутримолекулярных изменений. Га-
лантн и Сперлинг [72] полагают, что энергетическая составляющая в напол-
ненных эластомерах может включать деформацию водородных связей, дви-
жение и деформацию агломератов частиц наполнителя или кристаллизацию
полимерных молекул на поверхности наполнителя. Однако очевидно, что мно-
гие, если не все, нз этих факторов не должны оказывать влияния на измене-
ние внутренней энергии в условиях постоянного объема, в которых энергетиче-
ская составляющая имеет простой смысл. Поэтому можно полагать, что для
наполненных эластомеров член 2аТ/(Х2+Х—2) в уравнении (4.81) не отра-
11—440
161
жает достаточно полно межмолекулярные изменения и необходимо введение
соответствующей поправки, учитывающей наличие наполнителя. Галанта и
Сперлинг [72] попытались это сделать, введя в уравнение Тобольского — Ше-
на (4.93) множитель, учитывающий усиливающее действие наполнителя, и по-
лучили следующее выражение для энергетической составляющей:
г d 1п <Л>>(’ । rdlnf''
/ \ W}Vtr dT + dT
Енап
RT
(4.126)
где F’ — параметр, отражающий влияние наполнителя; Енл„ — суммарное из-
менение внутренней энергии, включающее и конформационную энергию пово-
ротных изомеров.
Однако попытка согласовать увеличение модуля упругости и прочности
при наполнении в предположении о неизменности числа активных цепей в
сетке приводит к непреодолимым трудностям [57]. Поэтому предложенные в
этих работах уравнения вряд ли могут иметь универсальное значение для
широкого класса наполненных эластомеров. С другой стороны, факт различ-
ного влияния на энергетическую составляющую активных (достаточно резкое
ее повышение) и неактивных (слабое повышение) наполнителей убеждает в
том, что в отличне от ненаполненных эластомеров межмолекулярные энерге-
тические изменения играют важную роль в механизме деформирования напол-
ненных эластомеров.
Диенстирольные термоэластопласты обладают способностью обратимо
деформироваться на многие сотни (вплоть до 2000) процентов, в связи с чем
они являются прекрасными модельными системами для выяснения механизма
больших каучукоподобных деформаций. При этом особенно важным обстоя-
тельством является то, что используемые в таких блок-сополимерах полидие-
ны не способны кристаллизоваться при растяжении, и это позволяет изучать
их эластические свойства при больших деформациях в отсутствие осложняю-
щей их анализ кристаллизации. С другой стороны, изучение термодинамиче-
ских особенностей больших деформаций таких систем должно прояснить роль
стеклообразных доменов, химически связанных с эластомерной матрицей, в
механизме их усиления.
Исследования термодинамики деформации диенстирольных термоэласто-
пластов проведены в последние годы [34, 69, 71, 77]. Замечательной особен-
ностью термодинамического поведения этих блок-сополимеров является неза-
висимость энергетической составляющей ии от степени растяжения, ни от со-
держания жесткого блока (рис. 4.17). (Причины небольшого отличия энерге-
тической составляющей в линейных и звездообразных блок-сополимерах пока
не выяснены.) Этот результат, по-вндимому, подтверждает вывод Флори
[44], что и при больших деформациях молекулярных сеток, содержащих в
среднем не менее 100 последовательных связей, гауссово распределение доста-
точно адэкватио описывает эластичность таких систем.
Поведение бутадиен-стирольных термоэластопластов заслуживает более
подробного рассмотрения в связи с проблемой ограниченной растяжимости
цепей. Хотя энергетическая составляющая остается практически постоянной в
очень широком интервале деформаций, анализ н терминах уравнения Муни —
Ривлина показывает [77], что вслед за линейным участком для большинства
термоэластопластов при Л«4—5 наблюдается резкий подъем, как на
;рис. 4.11. Как отмечалось выше, аналогичный подъем обычно наблюдается
для кристаллизующихся при растяжении эластомеров. Одиако кристаллизация
бутадиенов невозможна. Это подтверждается, в частности, и калориметриче-
скими данными. Связать этот подъем с проявлением ограниченной растяжи-
мости цепей, даже предположив, что энергетическая составляющая при таких
яебольших ее абсолютных, значениях является нечувствительной к появлению
ограниченной растяжимости цепей, не представлиется возможным хотя бы
потому, что после появления этого подъема термоэластопласты способны к
обратимым растяжениям до многих сотен процентов. Выше же мы убедились
в том, что ограниченная растяжимость коротких цепей ПДМС начинает про-
являться лишь при степенях растяжения макромолекул, сравнимых с макси-
мальной длиной цепей.
162
Рис. 4.17. Зависимость механи-
ческой работы и теплоты (а) и
энергетической составляющей
(б) от степени растяжения [34,
77]:
О — солпрен-406; 9 — ДСТ-30.
Объяснить наличие этого
подъема можно, лишь предпо-
ложив, что при больших растя-
жениях наряду с матрицей
начинают деформироваться
стеклообразные домены. При
таких деформациях электрон-
но-микроскопически [78]
удается наблюдать искажение
формы сферических доменов и
превращение нх в эллипсоиды
вращения с большей осью вдоль
направления растяжения. Иска-
жение формы доменов идет, по-
видимому, по механизму сдви-
га, возможно, с элементами
пластической деформации до-
менов, поскольку растяжение в
область очень больших дефор-
маций сопровождается появле-
нием небольших необратимых
деформаций, которые, однако,
через определенное время исчезают. Это дает основание считать, что появле-
ние подъемов на кривых зависимости Муни — Ринлина связано с деформиру-
емостью доменов при больших деформациях, а не с проявлением ограниченной
растяжимости эластомерных цепей.
Можно надеяться, что проявление ограниченной растяжимости удастся
обнаружить в термоэластопластах с относительно короткими эластомерными
цепями, например в полиуретановых термоэластопластах. В отличие от диен-
стнрольных блок-сополимеров, в которых эластомерные цепи содержат многие
сотни повторяющихся звеньев, в полиуретановых термоэластопластах поли-
эфирные эластомерные цепи обычно содержат лишь несколько десятков пов-
торяющихся звеньев. Однако н первых же исследованиях термоэластичиости
сегментированных полиуретанов [79] пришлось столкнуться с проблемой не-
устойчивости физических узлов под действием температуры н напряжений,
которая сводится к следующему. Деформационное поведение термоэластопла-
стов может зависеть не только от матрицы, но и от жесткости доменов, ко-
торая определяется межмолекулярным взаимодействием в доменах. Очевидно,
что прн приближении к температуре стеклования нли плавления жесткого
блока его жесткость будет понижаться и эффект усиления исчезать. Это дей-
ствительно наблюдается в СБС-блок-сополимерах [58, 59]. Однако физиче-
ские узлы могут быть недостаточно стабильными н прн температурах, дале-
ких от температуры перехода жесткого блока.
Показательным является поведение полиуретановых эластомеров, на при-
мере которых мы и рассмотрим эту проблему. Характерной особенностью
полиуретановых эластомеров является сильное межмолекулярное взаимодей-
ствие, обусловленное двумя факторами: высокой полярностью блоков, приво-
дящей к образованию сетки водородных связей, и способностью обоих бло-
ков к кристаллизации. Степень микрофазового разделения в системе опреде-
ляется конкурирующим взаимодействием блоков: межмолекулярное взаимо-
действие однородных блоков вследствие кристаллизации или нодородного свя-
зывания приводит к более полному фазовому разделению и доменообразо-
ванню, тогда как образование водородных связей между блоками приводит к
11’
163.
Рис. 4.18. Зависимость отношения
AZ7/H7 от степени растяжения для сег-
ментированного полиуретана, в кото-
ром мягким блоком являются полибу-
тадиен (1) и сложный полиэфир (2)
[80]:
/ДОг\
сплошные кривые — [-тру- пунктир —
\ /' р, п
; 1, 2-20 °C; Г, 2z-60°C.
\ W IV,Т
их смешению. Этн взаимодействия и
сильной мере зависят от температуры
н механических воздействий. Поэтому
для полиуретановых эластомеров осо-
бенно остро стоит вопрос о выявле-
нии происходящих при деформации
внутри- и межмолекулярных измене -
ний и выяснении условий стабильно-
сти фазовой структуры не только при изменении температуры, но и под дейст-
вием напряжений.
Характер энергетических изменений при деформации зависит от полярно-
сти гибких блоков полиуретановых эластомеров (рис. 4.18), что особенно на-
глядно проявляется в области повышенных деформаций. Такой эффект может
быть обусловлен как высокими отрицательными значениями внутримолекуляр-
ных энергетических составляющих, так и усилением межмолекулярного взаи-
модействия, вызванным, например, кристаллизацией гибких блоков при растя-
жении. К тому же в отличие от обычных эластомеров н диенстирольных тер-
моэластопластов для полиуретановых эластомеров зависимость доли суммар-
ных энергетических изменений от степени растяжения сохраняется и в обла-
сти больших деформаций, что в свое время [79] не позволило корректно рас-
считать внутримолекулярные энергетические составляющие различных поли-
уретановых эластомеров. Это удалось сделать, лишь рассмотрев термоэластн-
ческие свойства при малых деформациях (Х<1,6) [80]. У полярных сложно-
эфирных гибких блоков энергетические составляющие оказались небольшими
н положительными (0,1—0,2), а у полнбутадиенового блока — практически
такие же, как и в диенстирольных термоэластопластах, как и следовало ожи-
дать.
Положительные энергетические составляющие в сложноэфирных полиуре-
танах исключают возможность объяснения снижения внутренней энергии при
растяжении внутримолекулярными эффектами и приводят к выводу, что эф-
фект вызван усилением межмолекулярного взаимодействия. Анализ показал,
что это не связано с кристаллизацией гибких блоков, часто наблюдаемой в
полиуретанах с регулярным строением гибкого блока [81], так как этн эф-
фекты сохраняются и прн температурах, превышающих Тал мягкого блока, и
появляются при этих температурах и в бутадиеновом эластомере, не способ-
ном к кристаллизации. Наиболее вероятной причиной непрерывного снижения
внутренней энергии при растяжении полиуретановых эластомеров является
усиление межмолекулярного взаимодействия в жестких блоках, происходящее
в результате перестройки в них сетки водородных связей. Неустойчивость
физической сетки в полиуретановых эластомерах резко затрудняет выявление
эффектов ограниченной растяжимости относительно коротких эластомерных
депей в таких системах.
В заключение кратко остановимся на термоэластических свойствах сме-
сей эластомеров, двухфазную структуру которых можно рассматривать как
коллоидную систему типа эмульсия «полимер в полимере» [82] н которая
обычно представляет собой илн сочетание дисперсной и непрерывной фаз, нли
две непрерывные фазы (взаимопроникающие сетки). При однородной высоко-
эластической деформации таких смесей вклад отдельных компонентов в тер-
164
модннамику деформации должен определяться прежде всего соотношением,
компонентов и нх индивидуальными характеристиками. Это в первую очередь
касается внутримолекулярных конформационных изменений, которые, очевид-
но, должны аддитивно суммироваться вследствие гетерофазности системы.
Детальное калориметрическое исследование энтропийных и энергетических
эффектов при деформации гетерофазных смесей эластомеров проведено в
{83] на примере гетерогенных смесей НК и СКЭП различного состава. Эти
компоненты термодинамически несовместимы и характеризуются достаточно
большими значениями энергетических составляющих разного знака (см.
табл. 4.3).
Термодинамика деформации гетерогенных эластомерных смесей может
быть рассмотрена с тех же позиций, что и термоэластичность индивидуальных
эластомеров. Определенные на основании изложенной выше процедуры разде-
ления общих энергетических и энтропийных изменений на внутри- и межмо-
лекулярые и энергетические составляющие являются практически аддитивными
от состава смеси. У смеси, содержащей 60% НК, (&UIW)V, т=0, т. е. возраста-
ние конформационной энергии НК при растяжении полностью компенсируется
убылью конформационной энергии СКЭП. Смесь указанного состава является
моделью эластомера с идеальной энтропийной упругостью. Таким образом,
так же как и термоупругость исходных компонентов, термоупругость смесей
не является чисто энтропийной, причем внутримолекулярные энергетические
характеристики смесей последовательно меняются от одного компонента к
другому.
Несмотря на гетерогенную структуру смесей всех составов, прн деформа-
ции они ведут себя подобно однородным эластичным материалам, обладаю-
щим промежуточными термоупругими свойствами по отношению к исходным
компонентам, и каждый компонент, сохраняя индивидуальные особенности
термоупругих свойств, вносит вклад пропорционально своему содержанию.
Аддитивность энергетических составляющих наблюдается и для некоторых
статистических сополимеров [32].
ТЕПЛОВЫЕ ЯВЛЕНИЯ ПРИ ОБРАТИМОМ ДЕФОРМИРОВАНИИ
ТВЕРДЫХ ПОЛИМЕРОВ И ИХ СТРУКТУРНО-
МОЛЕКУЛЯРНАЯ ИНТЕРПРЕТАЦИЯ
Феноменологический анализ. Роль межмолекулярного
взаимодействия в аморфных областях кристаллических
полимеров
Микроскопическим параметром, определяющим знак и зна-
чение тепловых эффектов при деформировании твердых тел, яв-
ляется коэффициент теплового расширения. Длинноцепное
строение полимеров и их физическая структура обусловливают
большое разнообразие закономерностей изменения теплового
расширения. Особенно существенным с позиции термоупругости
является наличие у многих ориентированных полимеров отрица-
тельных коэффициентов линейного теплового расширения и
сильной анизотропии расширения. Все это дает основание ожи-
дать большого разнообразия в термоупругом поведении твердых
полимеров. Экспериментальные исследования термоупругости
твердых полимеров до сих пор малочисленны. Методики таких
теплофизических измерений и получаемая информация описаны
в монографии [28]. Здесь рассматриваются принципы, в соответ-
ствии с которыми на основании тепловых явлений, сопровож-
дающих обратимые деформации твердых полимеров, можно оха-
165
растеризовать происходящие при этом структурно-молекуляр-
ные изменения.
Прежде всего необходимо рассмотреть, в какой мере соотно-
шения термоупругости твердых тел, полученные при анализе
простейшего уравнения состояния, описывают тепловые явле-
ния в твердых полимерах. Априори можно ожидать, что они
должны хорошо выполняться для стеклообразных полимеров и
для кристаллических полимеров в условиях, когда их аморфные
области застеклованы, так как положенные в основу уравнения
состояния предпосылки в этом случае с очевидностью выполня-
ются. Приведенные на рис. 4.19 результаты для изотропных ПС
и ПЭТФ показывают, что теплота упругого растяжения линейно
возрастает с увеличением деформации, а отношение Q/W выра-
жается гиперболой. Кристаллизация ПЭТФ не изменяет харак-
тера зависимости тепловых эффектов от деформации и не влия-
ет на величину 0. Значения коэффициентов '0, определенные на
основании тепловых эффектов деформирования (как растяже-
ния, так и сокращения), хорошо согласуются с дилатометриче-
скими результатами (указаны в скобках): 0пс=6,8-1О-5 К-1
(«7-10~5), 0пэтф =5,4-10-5 К-1 (~5-10_5 Кг1). Если значение0
известно из независимых исследований, по теплотам упругой де-
формации можно определить модуль упругости. Таким образом,
закономерности термоупругости стеклообразных полимеров и
Рис. 4.19. Зависимость теплоты упру-
гого растяжения и отношения теплоты
к механической работе от деформации
при 20 °C для ПЭТФ (/) и ПС (2)
[28, 29]:
О — аморфный стеклообразный ПЭТФ; 9 —
кристаллический ПЭТФ (степень кристал-
личности w«0,50).
Рис. 4.20. Зависимость теплоты упругого растяжения (1, Г) и отношения теп-
лоты к работе (2, 2'} от деформации для ПЭНП при 20 °C:
1, 2 — неориентированный изотропный образец; 2'— ориентированный (степень вытяж*
ки«4) образец (растяжение вдоль оси ориентации).
166
кристаллических полимеров с застеклованными аморфными об-
ластями хорошо передаются уравнениями, вытекающими из
уравнения состояния (4.53).
Более сложным является положение с гетерогенными кри-
сталлическими полимерами, когда аморфные области находятся
выше температуры стеклования. Теперь их в принципе можно
рассматривать [84, 85] как гетерогенные негауссовы сетки,
в которых кристаллиты являются узлами сеток и мелкодисперс-
ным усиливающим наполнителем. В связи с этим можно пред-
полагать, что термоупругость кристаллических полимеров выше
Тс может быть качественно подобна термоупругости молекуляр-
ных сеток. Однако в действительности положение оказывается
совершенно иным. Кристаллическим полимерам ввиду замеча-
тельных особенностей их термоупругости, особенно в виде высо-
коориентированных пленок и волокон, далее уделяется главное
внимание. _
На рис. 4.20 приведены данные для неориентированного и
ориентированного ПЭ. Характер изменения теплоты деформа?
ции, а также соотношения теплоты и работы и в этом случае
соответствует формулам (4.55) и (4.62). Показательно, что ори-
ентация приводит лишь к изменению знака теплового эффекта,
не изменяя качественной картины термоупругого’поведения. Та-
кой характер изменения термодинамических величин выше Тс
наблюдается для многих неориентированных и ориентированных
кристаллических полимеров [28, 86, 87]. Таким образом, несмот-
ря на то что в кристаллических полимерах имеются аморфные
области, термоупругое поведение таких полимеров (по крайней
мере при степени кристалличности больше 30%) выше Тс этих
областей хорошо передается закономерностями, характерными
для твердых тел, а не для эластомеров. Так, знак теплового
эффекта в полном согласии с уравнением (4.55) определяется
знаком коэффициента теплового расширения вдоль оси дефор-
мирования.
Наряду со знаком и значением теплового эффекта количест-
венной характеристикой обратимых деформаций твердых поли-
меров, на основании которой можно судить о механизме дефор-
мации, является изменение внутренней энергии. Такие зависи-
мости для некоторых полимеров [88, 89] показаны на рис. 4.21.
Растяжение ориентированных полимеров сопровождается ин-
версией внутренней энергии, параметры которой суммированы
в табл. 4.5. Однако и знак инверсии, и молекулярные причины
принципиально отличны от инверсии внутренней энергии в эла-
стомерах, и это свидетельствует о различном механизме упруго-
сти. Значение коэффициента (3 с повышением модуля упругости
как неориентированных, так и ориентированных полимеров
уменьшается.
Согласно уравнению (4.58) сжатие изотропных полимеров
также должно сопровождаться инверсией внутренней энергии.
Однако простые оценки показывают, что для неориентирован-
167
О
Рис. 4.21. Зависимость изменения внутренней энергии от деформации для ори-
ентированных полимеров (деформация вдоль оси ориентации; номера кривых
соответствуют номерам полимеров в табл. 4.5) [89].
ных кристаллических полимеров выше Тс инверсия должна
наблюдаться в области деформаций 5—15%. Поэтому коррект-
ное экспериментальное определение при сжатии затруднено,
поскольку при этом начинают появляться необратимые дефор-
мации. Параметры инверсии AU изотропных полимеров прн
сжатии были рассчитаны по формулам (4.59) — (4.61) на осно-
вании значений ф и Е, полученных как при растяжении пленок,,
так и при малых деформациях цилиндрических образцов при
сжатии. Приведенные зависимости внутренней энергии и пара-
метры инверсии А(7 также свидетельствуют о соответствии экс-
периментальных результатов термодинамическим соотношениям,,
вытекающим из анализа уравнения состояния (4.53).j"Oco6o
следует подчеркнуть, что выделение тепла при растяжении ори-
ентированных полимеров никоим образом не связано с какими-
либо криталлизационными процессами в аморфных областях,
а является физическим следствием наличия отрицательного ко-
эффициента теплового расширения. Зависимости соотношения
тепла и работы от деформации и изменение U наглядно и одно-
значно это иллюстрируют (см. рис. 4.20 и 4.21).
Рассмотренные данные о термоэластичности твердых поли-
меров свидетельствуют о том, что их упругость, как и упругость
твердых тел, при одноосной деформации является объемной
упругостью,.а не упругостью формы, характерной для каучуко-
"Яодобной эластичности. Этот вывод очевиден для стеклообраз-
ных полимеров и для тех кристаллических полимеров, аморфные
области которых при температуре опыта находятся в застекло-
ванном состоянии, так как элементы каучукоподобной упруго-
сти при обратимых деформациях в таких полимерах проявиться
не могут. Замечательно, что соотношения термоупругости твер-
дых тел хорошо передают энергетические изменения при дефор-
мации и таких кристаллических полимеров, аморфные области
которых при температурах опытов находятся выше Тс и могут
в принципе проявлять каучукоподобную эластичность, посколь-
В К
В о
Is.
3 С
xS
s о
S>
gcc
См
СЕССССС
Й5 '
ее
йи
СЕ
СЧ со rf lO С© N. 00 О
со со о 00 о
о о о*сч* —? —Г
So о о о о
О СЧ 00
S Ю сч 00 сч сч
ЕЕ
Ей
сссссе
сч СО to
ку обратимые деформации в таких полимерах, как известна
[90], локализованы в этих аморфных областях.
Выше Тс обратимые деформации кристаллических полиме-
ров в неориентированном состоянии составляют несколько про-
центов (до ~ 10%), а в ориентированном состоянии они могут
достигать 20—30%. Эти достаточно большие деформации кри-
сталлических полимеров выше Тс принято объяснять каучуко-
подобной эластичностью аморфных областей, т. е. уменьшением
конформационной энтропии и изменением конформационной
энергии. Однако мы видим, что закономерности изменения
внутренней энергии с деформацией даже качественно не согла-
суются с закономерностями, характерными для эластомеров. Это
особенно наглядно видно для ориентированных кристалличе-
ских полимеров, в которых U сначала уменьшается, а затем воз-
растает. В эластомерах такой характер изменения внутренней
энергии при растяжении не может наблюдаться вообще. Важно-
подчеркнуть, что характер изменения U в кристаллических по-
лимерах определяется только величиной (3, а особенности внут-
римолекулярных (конформационных) энергетических изменений
в отличие от эластомеров при деформации никак не проявля-
ются.
Умеренно ориентированные кристаллические полимеры вы-
ше Тс обладают модулем упругости порядка нескольких ГПА
(несколько сот кгс/мм2) и способны обратимо деформироваться
на 15—25%. Поскольку деформация сосредоточена в аморфных
областях, то при обычных степенях кристалличности (30—70%)
реальная деформация этих областей может в несколько раз
превышать это значение. Столь большие обратимые деформа-
ции аморфных областей при сравнительно низком модуле упру-
гости должны быть обусловлены, несомненно, переходом свер-
нутых изомеров в вытянутые. Такие переходы для ориентиро-
ванных кристаллических полимеров (ПЭ, ПП, ПКА) зарегист-
рированы методом ИК-спектроскопии [90, 91]. Однако в отли-
чие от эластомеров эти гош-транс-переходы в аморфных обла-
стях кристаллических полимеров происходят в условиях изме-
нения межмолекулярного взаимодействия (изменение объема и
межмолекулярной энергии), которое и определяет модуль уп-
ругости, поскольку одни лишь гош-транс-переходы в условиях
практически неизменного межмолекулярного взаимодействия
(эластомеры) не могут обеспечить высокого модуля упругости.
Это справедливо не только для нерастянутых эластомеров,
но и для сильновытянутых макромолекул, какими являются про-
ходные участки цепей в аморфных областях. В условиях неиз-
менного межмолекулярного взаимодействия термодинамика
растяжения сильноориентированных цепей (модуль упругости
при этом может быть на два-три порядка выше модуля упруго-
сти нерастянутого эластомера) должна определяться внутримо-
лекулярными энергетическими и энтропийными изменениями^
обусловленными гош-транс-переходами. Совокупность данных
170
по термоупругому поведению двухфазных кристаллических по-
лимеров дает основание заключить, что в отличие от классиче-
ских эластомеров в термодинамике деформации кристалличе-
ских полимеров, как ниже так и выше температуры стеклования
аморфных областей, решающим является межмолекулярное
взаимодействие.
Эта особенность термоэластического поведения кристаллине-^
ских полимеров позволила нам предположить [28, 88], что, не-
смотря на локализацию обратимых деформаций в аморфных
областях, субмикроскопический размер кристаллитов и аморф-
ных областей, а также наличие молекулярной связанности
аморфных и кристаллических областей, обусловленной сущест-
вованием проходных цепей, создают такие условия при дефор-
мации этой микрогетерогенной системы, при которых принци-
пиальная возможность аморфных областей деформироваться
лишь за счет изменения формы (конформационная упругость)
не может быть реализована и они испытывают сложные дефор-
мации, связанные с преодолением межмолекулярного взаимо-
действия, т. е. с изменением их объема. Эти изменения объема
типичны для твердых полимеров, а не для эластомеров, и, что
более существенно, свободная энергия деформации связана с
этим изменением объема. Другими словами, работа деформации
связана с преодолением объемной упругости этих аморфных об-
ластей. Хотя при этом происходят и конформационные гош-
транс-переходы, обусловливающие геометрическую возможность
значительных обратимых деформаций, термодинамика этих де-
формаций определяется межмолекулярным взаимодействием •-
цепей в аморфных областях. Нам представляется, что именно I
эти сложные объемные деформации аморфных областей, вклю- j
чающие элементы как объемной, так и конформационной упру- \|
гости, приводят к уникальному комплексу термоэластических U
свойств кристаллических полимеров. \[
В связи с изложенным выше становится очевидно, что тра”^’
диционный подход к анализу модуля упругости кристаллических
полимеров, основанный на рассмотрении кристаллических поли-
меров как эластомеров, усиленных кристаллитами, является
слишком упрощенным. Вопрос о модуле упругости и других фи-
зических свойствах аморфных областей кристаллических поли-
меров выше Тс является наиболее сложным при изучении
свойств кристаллических полимеров. В настоящее время не су-
ществует обоснованного подхода к расчету модуля упругости,
коэффициента теплового расширения и других свойств аморф-
ных областей, и все оценки в той или иной степени базируются
на сравнении с эластомерами. Примером такого подхода и со-
ответственно иной точки зрения на механизм термоупругости
ориентированных кристаллических полимеров являются пред-
ставления, развитые Бонартом с сотр. [87] в результате кало-
риметрического исследования теплофизических процессов при
деформации ПЭТФ. На основании изучения влияния степени
171
Рис. 4.22. Зависимость изменения внутренней энергии от деформации для ПЭТФ
при 21 °C и степени вытяжки 0 (/), 100 (2), 200 (3), 300 (4), 400 и 500%
(5); справа показана термодинамическая модель, объясняющая инверсионное
изменение внутренней энергии в ориентированном ПЭТФ (см. текст) [87].
ориентации ПЭТФ на изменение внутренней энергии при малых
растяжениях ими установлен следующий характер измене-
ния U (рис. 4.22). Пленки с высокой степенью ориентации в от-
личие от неориентированных и слабоориентированных выделя-
ют тепло при растяжении, что приводит к инверсии U. Пара-
метры инверсии для этих пленок, приведенные в табл. 4.5, очень
хорошо согласуются с результатами наших исследований. Рас-
считанные по нашим формулам (4.59) — (4.61) значения £/мин
и 0 на основании экспериментальных значений вин и емин пре-
красно совпадают с приведенными в работе [87].
Для объяснения,, наблюдаемой инверсии U в ПЭТФ была
предложена термодинамическая модель '[87], согласно которой
растяжение двухфазного кристаллического полимера моделиру-
ется параллельным соединением идеальной пружины и поршня,
движущегося в наполненном идеальным газом цилиндре (см.
рис. 4.22). Авторы модели полагают, что пружина моделирует
поведение кристаллитов, а газ — поведение аморфных областей.
В исходном равновесном состоянии пружина растянута или
сжата настолько, чтобы компенсировать разность давлений
газа с противоположных сторон поршня в цилиндре. Знак теп-
лового эффекта определяется разностью (Pi—Р2): при Р1>Рг
растяжение сопровождается поглощением тепла, а при Pi<P2—
выделением; при Pi = P2 тепловой эффект отсутствует. Естест-
венно, что внутренняя энергия газа во всех случаях неизменна.
На основе этой модели было получено выражение для AU,
описывающее изотермы на рис. 4.22, при соответствующем выбо-
ре констант модели. В соответствии с этой термодинамической
моделью в работе [87] был предложен механизм деформации
ориентированного кристаллического ПЭТФ, согласно которому
выделение тепла при растяжении и инверсионное изменение
внутренней энергии являются следствием сжатия аморфных об-
172
ластей в фибриллах с пучностями (см. далее). Хотя формально
авторам удалось описать наблюдаемое поведение Д{7, однако
нам представляется, что такое термодинамическое моделирова-
ние нельзя признать физически обоснованным. В частности,
аморфные области ПЭТФ при температуре опытов (20 °C) на-
ходятся в стеклообразном состоянии, и отождествлять их пове-
дение с поведением идеального газа просто нелепо. К тому же
такой подход совершенно не в состоянии объяснить всего много-
образия термоупругих явлений в полимерах и в других твер-
дых телах, поскольку подразумевает наличие в системе двух
типов областей с идеальной упругостью. По этой причине рас-
смотренный подход представляется несостоятельным.
Данный нами термодинамический анализ термоупругости
твердых тел не связан ни с какими конкретными структурными
моделями и потому позволяет предсказать термоупругое пове- L
дение твердых тел любой природы. В частности, он показывает, V
что инверсия внутренней энергии не связана с двухфазностью t
тела, а характерна для гомогенных тел. Единственное требова-
ние для появления инверсии при растяжении твердых тел — на-
личие отрицательного коэффициента теплового расширения. До-
статочно ориентированные пленки ПЭТФ действительно имеют
отрицательное значение р.
Теперь уместно перейти к анализу отрицательных макроско-
пических коэффициентов расширения ориентированных кристал-
лических полимеров и выяснению соответствующих молекуляр-
ных механизмов. Сравнение отрицательных значений р ц для
ориентированных полимеров (см. табл. 4.5) с [Зкр и (см. табл. 3.3)
показывает, что при Т>ТС макроскопические значения ц
существенно превышают (JKpn- Это с очевидностью свидетельству-
ет о существовании по крайней мере еще одного отрицательно-
го вклада в 0 я. Этот вклад может быть обусловлен лишь стрем-^i
лением растянутых макромолекул в аморфных областях сокра-
титься при Т>ТС. Таким образом, сокращение ориентированных
кристаллических полимеров при нагревании может быть обус-
ловлено двумя молекулярными механизмами: конформационной
упругостью растянутых цепей в аморфных областях и отрица-
тельным значением фкр и. В отличие от изотропного состояния
в хорошо одноосноориентированных кристаллических полимерах
все кристаллиты оказываются ориентированными вдоль оси рас-
тяжения, так что оси макромолекул параллельны направлению
ориентации. При нагревании размеры кристаллитов вдоль оси
ориентации будут уменьшаться. Очевидно, что этот вклад дол-
жен быть отрицательным как выше, так и ниже Тс. Вклад
аморфных областей в макроскопический коэффициент будет оп-
ределяться степенью кристалличности, соотношением непроход-
ных и проходных цепей и степенью вытянутости последних. Та-
ким образом, центральным вопросом и здесь является выясне-
ние влияния условий ориентации и молекулярных параметров
полимера на строение аморфных областей.
173
’Для понимания особенностей теплового сокращения ориенти-
рованных кристаллических полимеров важными являются зако-
номерности, характерные для растянутых эластомеров. Исходя
из уравнения состояния молекулярных сеток (4.67), можно по-
казать [32, 92], что для изоэнергетических цепей*
Рц(М = — — • 1з ,2 13 9 (4.127)
11 1 Л’’ -j- 2 Л4 -{- £.
I Х.З — I а X3 + 1
В,(Х) =— • ---- +— —(4.128)
’ 2Т Х3 + 2 2 Х3 + 2
В недеформированном состоянии Рц(1) =ф± (1) =а/3>0. При
больших значениях X членами, содержащими а, можно прене-
бречь, и тогда имеем:
<4-|29>
1 X- — 1
Эти соотношения показывают, что растянутый эластомер яв-
ляется сильноанизотропным телом, обладает отрицательным
коэффициентом расширения вдоль оси ориентации, причем по
абсолютному значению коэффициенты расширения имеют по-
рядок коэффициентов расширения газов. Несмотря на сильную
анизотропию, коэффициент объемного теплового расширения
растянутого эластомера такой же, как и нерастянутого. Дейст-
вительно, из (4.127) и (4.128) видно, что рц +2р± =а. Из (4.127)
следует также, что инверсия знака р ц наблюдается для изоэнер-
3 ----------------------------------
гетических цепей при ЛИн(бИн) = + аТ в полном соответствии
с уравнением (4.88).
Зависимость рл(X), соответствующая уравнению (4.127), на-
блюдалась экспериментально для НК и других эластомеров до
1,5 [32, 93]. При больших удлинениях Рц оказывается даже
больше, чем предсказывают уравнения (4.127) и (4.129). Так,
при Х=1,5 значение р ц = —1,3-10-3 К-1; при Х=3 р и =—3,7><
х10~3 К-1; при Л.=4,5 р и =—4,2-10“3 К-1- При %>3 риуже очень
слабо зависит от степени растяжения, что соответствует Pi->-
—>const при больших %.
По мере растяжения эластомера, способного к кристаллиза-
ции, часть цепей оказывается в кристаллитах, причем направ-
ление осей цепей в них близко к направлению растяжения. Теп-
ловое сокращение макромолекул, находящихся в силовом поле
решетки, на 2—2,5 порядка меньше теплового сокращения не-
* Для цепей с различающейся конформационной энергией в этих выраже-
ниях присутствует температурный коэффициент невозмущенных размеров мак-
ромолекул, однако в данном контексте основные идеи проще продемонстриро-
вать на примере изоэнергетических цепей, выражения для 0 которых чрезвы-
чайно просты.
174
Рис. 4.23. Характер изменения ко-
эффициента линейного теплового
расширения от степени растяже-
ния:
1 — растянутый эластомер, способный к
ориентационной кристаллизации; 2, 3 —
кристаллический полимер; 4 — Экр([ •
закристаллизовавшихся макромолекул, и поэтому с увеличением
степени кристалличности по мере растяжения коэффициент со-
кращения может опять начать уменьшаться. Таким образом, на
гипотетической зависимости рц—X кристаллизующегося эласто-
мера может появиться минимум (рис. 4.23). В соответствии с
двухфазной моделью кристаллических полимеров растянутый
частично-закристаллизованный эластомер можно представить в
виде последовательного соединения кристаллитов и аморфных
областей, ориентированных в направлении растяжения. Тогда
₽|| = Рам|| О - W> +ЖР)|“- (4’13])
где fc — функция распределения осей кристаллитов.
Так как рам1»ркр?,, то при w<l 0ц ~рам и. Таким образом,
растянутый эластомер будет сокращаться благодаря стремле-
нию растянутых цепей увеличить конформационную энтропию.
Когда напряжение снимается с эластомера при той же тем-
пературе, при которой проводилось растяжение, то он сокраща-
ется до первоначального изотропного состояния, а кристаллиты
при этом плавятся. Напротив, в ориентированных кристалличе-
ских полимерах напряжения в аморфных областях уравновеши-
ваются кристаллитами. Поэтому даже при отсутствии внешних
напряжений ориентированный кристаллический полимер нахо-
дится в механически устойчивом состоянии. Еще одно важное
отличие от кристаллизующихся эластомеров состоит, по-види-
мому, в том, что растянутыми могут быть лишь некоторые
(проходные) цепи в аморфных областях. Все это создает широ-
кие возможности для вариации значения 0ц, и потому коэффи-
циент усадки ориентированного кристаллического полимера бу-
дет определяться степенью и температурой вытяжки, степенью
кристалличности и молекулярными параметрами. Детально эта
зависимость будет исследована далее, пока же рассмотрим ка-
чественно возможные пути достижения отрицательных коэффи-
циентов р и (см. рис. 4.23).
Общие соображения позволяют считать, что при небольших
степенях вытяжки полимеров с невысокой степенью кристал-
личности, особенно при Тор^Гпл, большая доля цепей в аморф-
ных областях окажется достаточно напряженной, и это приве-
175
дет к 0 ц ^>0кри (см. рис. 4.23, кривая 2). С увеличением степени
вытяжки Рп может уменьшаться, так что возможно появление
минимума на зависимости 0 в—к Таким образом, если исходить
из представления о каучукоподобной эластичности аморфных
областей, не осложненной изменяющимся при деформации меж-
молекулярным взаимодействием в этих областях, то 0 л должен
быть всегда значительно больше 0Крц, и приближение 0Я к0Кри
может происходить лишь со стороны больших отрицательных
значений 0 ц.
Если конформационная упругость аморфных областей ос-
ложнена межмолекулярным взаимодействием, то, по-видимому,
возможен такой путь (высокие степени кристалличности, Тор<
<Тия, высокие степени вытяжки), когда при всех условиях
₽11<0крц (см. рис. 4.23, кривая 3), так что 0 в будет приближать-
ся к 0Кр и со стороны меньших отрицательных значений. Дальней-
шая детализация возможна лишь для определенных моделей
строения ориентированных полимеров (см. далее).
Можно предположить, что в предельно-ориентированных
аморфных стеклообразных полимерах также возможно появле-
ние отрицательных коэффициентов расширения (в стеклообраз-
ном состоянии) по двум причинам. Прежде всего, стеклование
часто не означает потерю полной молекулярной подвижности, и
потому ниже Гс возможно сохранение определенной подвижно-
сти. Эта подвижность может явиться причиной отрицательного
коэффициента расширения в ориентированном стеклообразном
полимере. Наконец, в предельно-ориентированных стеклообраз-
ных полимерах возможно, по-видимому, появление отрицатель-
ных коэффициентов расширения за счет наличия волн изгиба в
цепных структурах даже без образования кристаллической ре-
шетки. Для стеклообразных полимеров, по-видимому, всегда
Р 11 < 0кр || •
Дилатометрические эксперименты свидетельствуют о нали-
чии отрицательных коэффициентов расширения у многих ориен-
тированных полимеров (ссылки на ранние работы можно найти
) в [28], новейшие исследования изложены в [94—98]). Еезуль-
I тэты эю_исследований можно суммировать следующим оора-
гзомГОриентадио^я-вьттажка при ТОр<7’пл [96], а также экс-
I трузия в твердом состоянии под повышенным давлением [94,
I 95] приводят к монотонному понижению положительного коэф-
фициента расширения с увеличением степени вытяжки. При оп-
ределенной степени ориентации, зависящей от температуры, 0 ц
становится отрицательным, причем 0 „ <0кр и (рис. 4.24, 4.25).
Это соответствует второму пути достижения отрицательных зна-
чений. Такой же путь характерен и для ПОМ (ш = 63%, Тор =
= 423 К) [97]. С другой стороны, для менее кристаллических
полимеров, например ПЭНП (ш = 42%), 0в =— 5-10~5 К-1 для
Х=2 и 0ц=—20-Ю-5 К-1 для А, = 4,2 [97].
Такая закономерность соответствует, очевидно, первому пути
достижения отрицательного 0Й. Однако этот путь может быть
176
Juc. 4.24. Зависимость коэффициентов линейного теплового расширения 0 ц
1, 2) и Р (Г, 2') ориентированных ПЭ (1, Г) и ПП (2, 2') от степени вы-
яжки и степени ориентации кристаллитов fc [96] (степень кристалличности
ЛЭ«0,8, ПП«0,6; температура вытяжки ПЭ равна 353 К, ПП составляет
198 К); 3 — кривая построена по уравнению (4.145).
аис. 4.25. Зависимость коэффициента расширения 0 ц ПЭ от степени вытяжки
5 процессе экструзии Л при различных температурах [94].
характерен и для высококристаллических полимеров, например
1ЭВП, для которого [98] 0 ц =—2,4-10~5 К-1>₽крв при 20°С,
1ричем это значение 0 ц перестает зависеть от степени вытяжки
5 интервале л=8—15 (w«70%). (Ср. также ПЭВП при Х=10
I 20 из табл. 4.5.) Таким образом, оба пути достижения отрица-
гельных значений ,0 в действительно наблюдаются для ориенти-
рованных кристаллических полимеров, причем, по какому из них
достигается данное значение 0ц, зависит от условий ориентации,
температуры и степени вытяжки. Степень кристалличности, по-
видимому, существенна в том отношении, что для полимеров с
низкой степенью кристалличности не удается достичь высоких
степеней вытяжки.
Важное обстоятельство выявляется из сравнения отрица-
тельных значений 0 ц ориентированных кристаллических полиме-
ров и растянутых эластомеров и особенно из сравнения темпе-
ратурной зависимости 0ц. Так, если рассматривать ориентиро-
ванный кристаллический полимер как последовательное соеди-
нение ориентированных аморфных областей и кристаллитов, как
это часто предполагается (см., например, [98] и ссылки там),
и из (4.131) оценить 0ам л по известным 0 ц, 0Крц и w, то оказыва-
ется, что, например для образцов ПЭВП, исследованных Воль-
12—440
177
фом и Карлом, Рам । =—5,2-10"5 К-1- Это значение примерно на
два порядка меньше, чем для достаточно растянутого эластоме-
ра. Такое же соотношение коэффициентов характерно и для
других сильноориентированных кристаллических полимеров.
Столь сильное различие в коэффициентах сокращения
аморфных областей ориентированных кристаллических полиме-
ров по сравнению с растянутыми эластомерами является, на
наш взгляд, отражением роли межмолекулярного взаимодейст-
вия в аморфных областях, препятствующего свободному сокра-
щению растянутых цепей. Межмолекулярное взаимодействие с
повышением температуры должно ослабевать, и одновременно
должна ослабевать степень напряженности аморфных областей
в результате релаксации объемных напряжений. Вследствие
этого ориентированных кристаллических полимеров с повы-
шением температуры должен возрастать. Такое поведение про-
тивоположно предсказываемому уравнением (4.127) для растя-
нутых эластомеров, согласно которому 0 ц (Л) должно изменяться
обратно пропорционально абсолютной температуре, т. е. в ре-
альных условиях слабо зависеть от температуры (учет того, что
Td In <r2>o/dT не изменяет этого вывода).
Приведенные на рис. 4.26 характерные для многих ориенти-
рованных кристаллических полимеров температурные зависимо-
сти р л свидетельствуют о сильном возрастании отрицательных
Pi с повышением температуры. В довольно узком температур-
ном интервале (50—60 градусов) возрастает в несколько раз.
Температурный коэффициент d$ildT мало зависит от типа поли-
мера и его термомеханической предыстории, хотя абсолютное-
значение р ц ПЭНП на порядок выше, чем у двух других поли-
меров. Заслуживает упоминания также то, что для многих сла-
боориентированных полимеров (ПОМ, Х = 6,5; ПЭВП, Х=4,2;
ПП, Х = 5,5; ПВДФ, Х = 4; ПКА, Х = 2,5) ниже Тс р л положите-
лен, возрастает в области стеклования и лишь при дальнейшем
повышении температуры начинает монотонно уменьшаться и при
определенной для каждого полимера и его термомеханической
предыстории температуре становится отрицательным [97]. Та-
кое поведение также согласуется с представлением о решающем
влиянии межмолекулярного взаимодействия в аморфных обла-
стях на термоэластические свойства ориентированных кристал-
лических полимеров, так как при отсутствии такого влияния
Р„ выше Тс должен был бы сразу стать сильноотрицательным,
в чем легко убедиться даже на слаборастянутом и заморожен-
ном ниже Тс эластомере.
Подводя итог совместного рассмотрения термоэластических
свойств и теплового расширения кристаллических полимеров,
можно окончательно сформулировать основное заключение, вы-
текающее из рассмотрения всей совокупности феноменологиче-
ских результатов. В двухфазных кристаллических полимерах,
содержащих как кристаллические, так и аморфные области,,
поведение аморфных областей выше Тс при деформации, вопре-
178
’ис. 4.26. Температурная зависи-
ость коэффициента 0 ц ориенти-
юванных ПЭВП (/; «> = 0,7; Л=
= 8—15) [98]; ПЭНП (2; ш = 0,43;
.=4,2) [97]; ПОМ (3; ш=0,63;
.=6,5) [97].
си общепринятому представ-
1ению, нельзя уподобить по-
»едению классических эла-
стомеров, так как их пове-
хение определяется не толь-
<о конформационными ха-
эактеристиками, но и меж-
молекулярным взаимодействием. Можно полагать, что основные
особенности физических, и прежде всего механических, свойств
двухфазных кристаллических полимеров обусловлены этим об-
стоятельством.
До сих пор все заключения базировались на феноменологи- ;
ческом анализе закономерностей и не были связаны со струк-
турными моделями ориентированных кристаллических полиме-
ров. Дальнейшая детализация теплофизического поведения мо-
жет быть достигнута на основе рассмотрения таких структурных
моделей. В связи с этим, имея в виду сформулированную выше
гипотезу; проведем теплофизический анализ деформирования
наиболее реальных структурных моделей ориентированных кри-
сталлических полимеров и рассмотрим возможные молекуляр-
ные механизмы деформации, основываясь на теплофизических
и структурных результатах. При этом мы практически не будем
касаться подходов, характерных для определения механических
и тепловых свойств композиционных материалов, которые позво-
ляют предсказать эти свойства, исходя из известных свойств со-
ставляющих композит компонентов. Основная предпосылка та-
ких теорий — неизменность свойств компонентов при создании
композита — для кристаллических полимеров не выполняется.
Теплофизический анализ структурных моделей
ориентированных кристаллических полимеров.
Сравнение теплофизических
и структурных данных
Схемы основных моделей ориентированных кристаллических по-
лимеров приведены на рис. 4.27. Если кристалл периодически
прерывается аморфными областями, в которых все макромоле-
кулы, выходящие из предыдущего кристаллита, попадают в по-
следующий, то возникает модель микрофибриллы Гесса — Стэт-
тона — Херла (ГСХ). Такая микрофибрилла состоит из последо-
вательного чередования кристаллических и аморфных областей.
Модуль упругости и коэффициент расширения такой модели
12’ 179
a — гладкая микрофибрилла со 100% проходных цепей (модель Гесса — Стэттоиа — Хер-
ла); б — гладкая микрофибрилла с ограниченным числом проходных цепей (модель Хо-
земана — Бонарта); в — микрофибрилляриая структура (микрофибриллы ГСХ) с межфиб-
риллярными аморфными прослойками; г — микрофибрилляриая структура (микрофибрил-
лы ХБ) с межфибриллярными аморфными прослойками (модель Петерлииа — Преворзека)
[90, 99, 105]; д — модель статистических кристаллических мостиков сверхориентированиых
сверхвысокомодульных гибкоцепиых полимеров [120, 121]; / — кристаллит; 2 — аморфиая-
виутрифибриллярная прослойка; 3 — аморфная межфибриллярная прослойка; 4— меж-
кристаллитный мостик.
определяются уравнениями:
Е =_____________£^11___________
II (l-w)+w(EaB||/EKp())
PaB||(J-^) + ^KP|f
Р||— / ISO 1 — W\ РавИ^1 +Ж'₽кр||
Е.,1---- Н-------
III г F I
'Скр|| Сав|| /
а теплота деформации, работа и их соотношение
п TR F ^ав||О — ®) + ж'Ркр’|1-^авП
Q = Т Рц£|Л =---------------------------£
II II (l-w) + w(EaB||/EKp||)
w= £пе2 =_______________________________
2 2 [(I - w) + №(Еав||/Екр|1)]
27'1₽ав||(1-И')+^кР||]
W . е
(4.132)
(4-133)
(4.134)
(4.135)
(4.136)
(индекс ав относится к аморфным внутрифибриллярным обла-
стям).
Растяжение такой модели выше Тс должно всегда сопровож-
даться выделением тепла, т. е. Q<0, так как рКрц и 0ав ц являют-
ся отрицательными. Изменение термодинамических функций при
растяжении микрофибриллы ГСХ характеризуется зависимостя-
ми, показанными на рис. 4.26. Обычно £кр и ^>Еав н, и потому
вкладом кристаллитов в термоэластические свойства можно
пренебречь. Макродеформация микрофибриллы ГСХ должна
совпадать с изменением большого периода при деформации,
180
а плотность аморфных пучностей при растяжении увеличи-
ваться.
В том случае, когда лишь часть макромолекул проходит из
одного кристаллита в другой, а часть возвращается в тот кри-
сталлит, из которого они вышли, возникает модель микрофиб-
риллы Хоземана — Бонарта (ХБ). Тепловой эффект и работа
обратимой деформации микрофибриллы ХБ будет также опре-
деляться уравнениями (4.132) — (4.136). Однако знак 0аь и, сле-
довательно, Q в этом случае может быть как положитель-
ным, так и отрицательным. Он будет определяться долей
проходных цепей, причем можно полагать, что при их малой
доле или недостаточной вытянутости 0ав>О и Q>0. Хотя в этом
случае проходные цепи будут иметь отрицательное значение (3,
общий коэффициент теплового расширения аморфной прослойки
может все же быть положительным. Здесь, очевидно, возможны
самые различные соотношения, однако особо следует подчерк-
нуть, что в моделе ХБ с малым числом проходных молекул
возможно поглощение тепла (положительный тепловой эффект)
при растяжении. При малом числе проходных цепей коэффици-
ент Пуассона (ц=—ej_/S|) будет, по-видимому, меньше 0,5, так
что плотность такой прослойки при растяжении должна па-
дать*. Макродеформация микрофибриллы ХБ должна соответ-
ствовать изменению большого периода при растяжении.
Исследования структуры и свойств ориентированных поли-
меров, проведенные в последние годы, показали [90, 99—101],.
что важную роль в деформационных и прочностных свойствах,
ориентированных кристаллических полимеров играют межфиб-
риллярные цепи. Две последующие модели представляют собой
микрофибриллы ГСХ и ХБ, объединенные в макрофибриллу~
межфибриллярными цепями, образующими аморфные прослой-
ки между микрофибриллами. Цепи в межфибриллярных про-
слойках предполагаются достаточно вытянутыми, в связи с чем
0ам<О. Термодинамика растяжения таких комбинированных
моделей определяется уравнениями
(l-x)-'£aB||
£H-£aM|lX+(l_w_X) + U7(EaB||/EKp(|) (4J 7)'
* Интуитивно кажется, что при упругой деформации конденсированных
тел выделение тепла обязательно связано с уменьшением объема, а поглоще-
ние — с его увеличением. Однако термодинамические соотношения (4.42) и
(4.55) показывают, что знак теплоты определяется только знаком коэффициен-
та теплового расширения, а на изменение объема в явном виде никакие огра-
ничения не накладываются. Всестороннее сжатие (объем уменьшается) жид-
костей, у которых а<0 (например, вода при 0<Г<;4°С), или всестороннее
сжатие и одноосное растяжение твердых тел, у которых а<0 (например,
иодид серебра) и, следовательно, тепло при сжатии поглощается, а при растя-
жении выделяется, является очень наглядным примером ошибочности этого
интуитивного представления. Это следует и из формальных соображений:
с учетом коэффициента Пуассона из (4.55) имеем Q = $TEe=$TEAV/V(l—2 ц);
ДУ и (1—2 ц) меняют знак одновременно, и, таким образом, знак Q по-преж-
нему определяется знаком ₽.
181
„ ____________Рам||^аМ||__________
" = F О-Х^авН
ам|| + (l_w_X)+w(EaB||/EKp|1)
(1-ХИав||
+ v и-ХГ-’^авП
£а«Н + (1_w-X)+w(£a3||/£Kp[i)
Рав||(1 ~ да) + Ркр||(да ~ X)
Х (®-Х)(£ав||/£кр||) -(1-«ц
Q = 7'P||£||e= РамцХ£амц +
+ (1-Х)£авц
W1 ~~w) +ркрц(да-х)
(®~Х)(£ав||/£кр||) + (1- да)
£.г-"
2 =£амУХ +
<;-Х)^£ав||
(1-w-X) +®(£аз||/£кр11)
Q/U7= (2Т/г)Рп
(4.138)
(4.139)
(4.140)
(4.141)
(здесь X — объемная доля аморфных межфибриллярных обла-
стей, «ам» относится к аморфным межфибриллярным областям).
Очевидно, что модель ГСХ с межфибриллярными прослой-
ками характеризуется только выделением тепла при растяже-
нии, и потому термоупругое поведение гладкой микрофибриллы
ГСХ и макрофибриллы ГСХ с межфибриллярными аморфными
областями оказывается качественно одинаковым и различия
должны носить лишь количественный характер. Как внутри-,
так и межфибриллярные области должны уплотняться при рас-
тяжении. Наиболее важным отличием поведения фибрилл ГСХ
с межфибриллярными областями должны быть, по-видимому,
несовпадение макродеформации с деформацией большого перио-
да и зависимость коэффициента р от деформации.
У модели ХБ с межфибриллярными аморфными областями
знак теплоты растяжения, определяемой по уравнению (4.139),
зависит от соотношения рам и рав и может являться функцией
удлинения даже при малых деформациях. При этом возможно,
что сначала р>0, а по мере деформации ф может изменить
знак, т. е. р<0. Уменьшение поперечных размеров при растяже-
нии может оказаться больше, чем увеличение длины, и это бу-
дет означать, что коэффициент Пуассона ц>0,5. Это соответ-
ствует уменьшению объема при растяжении такой микрогетеро-
генной системы. Хотя согласно классической теории упругости
изотермический коэффициент Пуассона для гомогенного твер-
дого тела не может превышать 0,5 [4], для гетерогенных анизо-
тропных тел такое ограничение снимается [102]. Прямые мик-
роскопические измерения коэффициентов Пуассона для ряда
ориентированных кристаллических полимеров (ПП, ПЭВП,
182
ПЭТФ, ПКА; см. табл. 4.5) [28, 103], а также данные, получен-
ные на основании измерения модулей упругости [104], показы-
вают, что эти значения могут существенно превышать 0,5,
в связи с чем рассматриваемый механизм деформации вполне
вероятен. Последняя из рассмотренных моделей является наи-
более разработанной и экспериментально обоснованной в на-
стоящее время моделью ориентированного кристаллического по-
лимера (модель Петерлина — Преворзека [99—101, 105, 106]).
Если межфибриллярные аморфные области отсутствуют (Х = 0),
то уравнения (4.137) —(4.141) переходят в уравнения (4.132) —
(4.136).
Несовпадение макродеформации и деформации большого
периода (еОбр>ет) в образцах с большим содержанием меж-
фибриллярных аморфных областей, являющееся, как полагают
(см., например, [90, с. 138]), следствием обратимого смещения
(проскальзывания) микрофибрилл друг относительно друга,
должно приводить к интересному термоупругому эффекту.
Взаимное проскальзывание микрофибрилл будет приводить к
дополнительному выделению тепла, так как работа трения хотя
бы частично превращается в тепло. При сокращении микрофиб-
риллы будут возвращаться в исходное взаимное положение, т. е.
проскальзывать в обратном направлении, что также должно со-
провождаться выделением тепла. Поэтому при деформации та-
ких образцов, во-первых, начало проскальзывания должно быть
отмечено резким экзоэффектом при растяжении и отклонением
от начальной линейной зависимости; во-вторых, при определен-
ных деформациях тепловой эффект сокращения может изме-
ниться с эндо на экзо, как это схематически показано на
рис. 4.28. Подобные эффекты не должны наблюдаться в образ-
цах, в которых межфибриллярные аморфные прослойки отсут-
ствуют.
Проведенное рассмотрение соответствует модельному анали-
зу модулей упругости двухфазных систем, развитому Такаянаги,
который применяется для расчета модулей упругости кристал-
лических полимеров [99] и который использован выше. Однако
феноменологические модели Такаянаги, хотя и могут описывать
достаточно хорошо модули упругости двухфазных систем, не
позволяют получать никакой информации о микроструктуре
полимера и ее поведении при деформации. На основании теп-
лофизического анализа такая информация, как будет видно из
дальнейшего изложения, может быть получена.
Посмотрим теперь с учетом рассмотренных моделей, какие
выводы можно сделать из приведенных в табл. 4.5 и на
рис. 4.21 данных о термоупругости ориентированных кристалли-
ческих полимеров. Независимо от химической природы полиме-
ра и условий ориентации ориентированные кристаллические по-
лимеры выделяют тепло при растяжении и характеризуются ин-
версионной зависимостью внутренней энергии. Ни в одном из
исследованных полимеров (кроме ПБТФ) не наблюдается по-
183
Рис, 4.28. Характер изменения тепловых эффектов растяжения и сокращения
микрофибриллярной структуры с обратимым проскальзыванием микрофибрилл:
1 — теплота растяжения без проскальзывания; 2 — теплота растяжения с проскальзыва-
нием; 3 — теплота сокращения без проскальзывания; 4 — теплота сокращения с проскаль-
зыванием.
Рис. 4.29. Зависимость теплоты растяжения (/, 3) н сокращения (2, 4) от де-
формации для ориентированных ПЭВП, вытянутого в 20 раз (/, 2), и ПП, вы-
тянутого в 30 раз (3, 4) [89] (см. табл. 4.5).
глощения тепла при растяжении вдоль оси ориентации. На ос-
новании этих данных, казалось бы, можно заключить, что строе-
ние ориентированных полимеров скорее всего представляется
моделью ГСХ. Поскольку рентгеноструктурные исследования
изменения большого периода при этом не приводились, никаких
заключений о наличии или отсутствии межфибриллярных
аморфных областей из одних лишь данных по термоупругости,
как будто, сделать не удается.
Однако проведенный в соответствии с рис. 4.28 анализ за-
висимости теплот растяжения и сокращения показал, что экс-
периментальные результаты вполне согласуются с этой схемой
(рис. 4.29). Но в таком случае эти данные можно рассматривать
как свидетельство наличия межфибриллярных аморфных про-
слоек в ПЭ и ПП. Характер изменения знака теплоты сокраще-
ния в сильной мере зависит от степени вытяжки, что вполне по-
нятно, если учесть, что разнодлинность межфибриллярных це-
пей уменьшается с увеличением степени вытяжки. На основании
разности данных линейных экстраполированных зависимостей и
экспериментальных значений можно сравнить тепловые эффек-
ты, связанные с обратимым проскальзыванием микрофибрилл.
Оценки показывают, что соответствующие значения близки.
Рентгеноструктурные исследования большого периода при этом
не проводились, и потому рассмотренные результаты являются
лишь косвенным доказательством существования значительного
числа межфибриллярных прослоек. Однако значения коэффици-
ента Пуассона (см. табл. 4.5) также подтверждают этот вывод.
Отсутствие же эндоэффектов при растяжении еще не означает
наличия большого числа проходных цепей во внутрифибрилляр-
ных прослойках, так как вполне возможно, что эти эффекты
184
перекрываются экзоэффектами от деформации межфибрилляр-
ных цепей.
Хотя это заключение выглядит вполне правдоподобным, од-
нако при этом нельзя исключить тот факт, что в этих исследо-
ваниях просто не были найдены условия, в которых решающая
роль в термоупругости принадлежит внутрифибриллярным про-
слойкам с небольшим числом проходных цепей, когда, как по-
казано выше, существует наибольшая вероятность эндотермиче-
ских эффектов при растяжении. Хорошо известно, что парамет-
ры фибриллярной структуры кристаллических полимеров опре-
деляются молекулярными характеристиками, исходной надмоле-
кулярной структурой и условиями ориентации. В частности, для
ПЭ рентгенографически было установлено [105, гл. 1; 107],
что низкомолекулярные полимеры характеризуются практиче-
ски полным отсутствием межфибриллярных аморфных обла-
стей, в то время как в высокомолекулярных доля этих прослоек
может быть значительной. В соответствии с этим можно ожи-
дать, что ориентированные образцы ПЭ низкой молекулярной
массы, структура которых, по-видимому, ближе всего к модели
микрофибрилл ХБ, будут поглощать тепло при растяжении,
а высокомолекулярные, которые в большей степени соответству-
ют модели Петерлина — Преворзека, — выделять тепло при рас-
тяжении.
Комплексное исследование влияния молекулярных парамет-
ров (молекулярной массы, молекулярно-массового распределе-
ния, степени разветвленности) и условий ориентации (степени
и температуры вытяжки) на механизм деформации ориентиро-
ванных ПЭ, которое включало калориметрическое изучение тер-
модинамики деформации и параллельный рентгенографический
анализ структурных изменений, было проведено в последнее
время [108—112]. Прежде всего оказалось, что растяжение ори-
ентированного ПЭ низкой молекулярной массы действительно
сопровождается поглощением тепла при растяжении, а высоко-
молекулярного ПЭ — выделением тепла (рис. 4.30). Совместный
анализ термоупругости и рентгеновских данных (табл. 4.6) в
рамках модели Петерлина — Преворзека привел к заключению,
что выделение тепла связано, по-видимому, с уплотнением меж-
фибриллярных аморфных областей, а поглощение — с разуплот-
нением внутрифибриллярных, содержащих небольшую долю
проходных цепей. Результирующий эффект зависит от соотно-
шения уплотнения и разуплотнения этих аморфных областей.
Важным результатом этих исследований явился метод ко-
личественного определения объемной доли межфибриллярных
аморфных областей в ПЭ по тепловому эффекту и изменению
большого периода при деформации. Этот метод основан на сум-
мировании вкладов тепловых эффектов от внутри- и межфиб-
риллярных аморфных областей [см. уравнение (4.139)], причем
в качестве «эталонных» образцов выбирали самый низкомоле-
кулярный и самый высокомолекулярный ПЭ. Предполагалось
185
Таблица 4.6. Структурные и теплофиэические характеристики ПЭ* [109—112]
Тепловой эф- фект начальной деформации о со & о • О о 2- О О О О я х а я а а а « х СП (П (D (Т) СП (D Л Л Ф
Р ,, -10S. (1 1 К-1 О 1Л — -'7 м 1 7 1 ' °
£-(1, ГПа , сососъ оо о о о о со СО О (7) N w-Г o' о Ю ’-ф" со о”
1 \~w~X, % , 1 1 i 00 Ю —• *Ф СМ СМ^—* ООСМ’ФСМ
1 X, % 1 1 СМ О СО О- L-C is. о — 00 СЧ — — СМ
L, нм СО 00 О 00 , СО СО —'О) СЧ СЧ ’Ф со | СЧ СЧ ”ф сч
1 1 1 1, нм о — со Ю СЧ о to N С) ® сч см со — сч СЧ СО л
% ‘«1 00 сч со О> оо С? 00 -т ~ *£ Е. со is. о- | о со со ’С со О)
Образец и его термическая предыстория** ПЭ-1*** Х=б ПЭ-2*** %=6 Изометрический отжиг Свободный отжиг Отжиг под давлением 0,7 ГПа при 264 °C Х=1б Изотермический отжиг Свободный отжиг ПЭ-3*** Л=5,5
S
S
S
О)
ч
и
(О
Е(
§
Is
IS .
с
о
7
§
Is
с
IS
о
sr
I*
Si
л
с
д’
8
о
©
с?
Рис. 4.30. Зависимость теплоты упругого растя-
жения от деформации (а) и зависимость мик-
родеформации фибрилл е (деформации большо-
го периода) от макродеформации е (6) [109]:
1—ПЭ-1 (М-4,2-104, Тор = 108°С>; 2 - ПЭ-2 (M=3-!0s,
Гор=108 -С); З-ПЭ-З (М«3-10®—8- 10s, Тор=108°С).
также, что при переходе от низкомоле-
кулярного к высокомолекулярному ПЭ
доля и степень вытянутости внутри-
фибриллярных проходных цепей изме-
няются мало и основные изменения
сводятся к появлению межфибрилляр-
ных аморфных областей. При таком
упрощающем предположении можно
полагать, что доля межфибриллярных
областей может оказаться несколько
завышенной. Проведенные на основе
этого метода оценки показали, что об-
разцы с низкой молекулярной массой
практически не содержат межфибрил-
лярных аморфных прослоек (см.
табл. 4.6). Увеличение молекулярной
массы приводит к монотонному уве-
личению доли межфибриллярных
аморфных прослоек при сопоставимых
кратностях и температурах вытяжки. Кроме того, их доля воз-
растает с увеличением кратности вытяжки при данной темпе-
ратуре и с ее понижением. Результаты этих расчетов неплохо
согласуются с теоретическими оценками [101] доли межфибрил-
лярных аморфных областей в ориентированном ПЭ и с прямы-
ми рентгенографическими исследованиями [90, 107]. Таким об-
разом, эти результаты позволяют заключить, что термоупругость
ориентированных ПЭ хорошо согласуется с моделью Петерли-
на — Преворзека.
Отжиг свободных или закрепленных ориентированных кри-
сталлических полимеров при обычном давлении приводит к су-
щественным изменениям их исходной надмолекулярной струк-
туры и, как следствие, к изменению их механических свойств
[90, 101, 105]. Наиболее радикальные изменения происходят с
предельно-вытянутыми межфибриллярными цепями и проход-
ными участками цепей во внутрифибриллярных аморфных про-
слойках. При повышенных температурах они приобретают по-
движность, достаточную в принципе для сближения кристалли-
ческих блоков, которые они соединяют. Возникающие в меж-
фибриллярных цепях силы стремятся сместить микрофибриллы
друг относительно друга, и, если отжиг проводится в свободном
187
гиванию тех частей цепи,
Рис. 4.31. Влияние отжига на 1еплоту упру-
гого растяжения ПЭ [112, 89]:
t — исходный ПЭ-2 (см. табл. 4.6); к=15; 2 — изо-
метрический отжиг; 3 — свободный отжиг.
состояния, следствием этого являет-
ся заметная макроусадка образца.
Фиксация концов образца предот-
вращает усадку. Внутрифибрилляр-
ные проходные цепи могут сблизить
соседние кристаллиты лишь при тем-
пературах отжига, близких к Тпл.
При более низких температурах ре-
лаксация внутрифибриллярных про-
ходных цепей может проходить без
заметной усадки благодаря выдер-
которые закреплены в соседних крис-
таллитах.
Расположенные во внутрифибриллярных аморфных облас-
тях складки, большие петли и концы цепей препятствуют усад-
ке. После охлаждения до комнатной температуры отожженных
образцов их механические свойства заметно ухудшаются из-за
потери натяжения вытянутых до отжига цепей вследствие усад-
ки (свободный отжиг) или в результате вытягивания участков
цепей из кристаллитов (изометрический отжиг). В последнем
случае свойства ухудшаются в меньшей степени. При длитель-
ном хранении (десятки часов) изометрически отожженных об-
разцов их модуль упругости может возвращаться на исходный
уровень или даже превосходить его, что связывают [101] с
медленной кристаллизацией отрелаксировавших участков це-
пей, не способных из-за ряда ограничений закристаллизоваться
при охлаждении. Отжиг обычно сопровождается переходом фиб-
риллярной структуры в слоевую.
Представляет несомненный интерес проследить, как такие
резкие изменения сказываются на термоупругом поведении.
Данные табл. 4.6 и рис. 4.31 показывают, что эти изменения за-
висят от степени вытяжки. Наиболее существенные изменения
наблюдаются для сильноориентированного образца при отжиге
в свободном состоянии. Наблюдаемые изменения при всех усло-
виях отжига можно объяснить существованием Двух типов
аморфных областей. Отжиг приводит к потере ориентации и на-
тяжения вытянутых участков цепей, в результате чего на на-
чальных участках растяжения вклад эндотермических эффек-
тов, связанных с разуплотнением внутрифибриллярных аморф-
ных областей, может стать преобладающим. Однако по мере
увеличения деформации эндоэффект переходит в экзо. Отжиг в
свободном состоянии сопровождается существенным снижением
модуля упругости. Хотя при изометрическом отжиге модуль ос-
188
тается практически на прежнем уровне, тепловой эффект дефор-
мации, а следовательно, и р। изменяются очень сильно вплоть
до изменения знака. Как видим, теплофизические явления при
деформации отожженных ориентированных образцов ПЭ хоро-
шо отражают структурные изменения, происходящие при от-
жиге.
Отжиг ориентированного ПЭ под высоким давлением приво-
дит к образованию кристаллов с выпрямленными цепями
(КВЦ) ![113, 114]. Размер кристаллов вдоль оси вытяжки в
КВЦ превышает 100 нм, а плотность и теплота плавления соот-
ветствуют степени кристалличности 96—98%. Образцы с КВЦ
характеризуются слоевой структурой, в которой жесткие кри-
сталлические слои больших поперечных и продольных размеров
чередуются с неупорядоченными мягкими прослойками, где лишь
некоторые цепи являются проходными. Такие образцы харак-
теризуются малым модулем упругости и эндотермическим эф-
фектом при растяжении (см. табл. 4.6). Эти данные позволяют
•отнести КВЦ к категории ХБ-макрофибрилл. Их отличие от
низкомолекулярного образца ПЭ-1 заключается лишь в значи-
тельно больших продольных и поперечных размерах кристалли-
тов и в очень высокой степени кристалличности. Эти результа-
ты свидетельствуют о том, что одних лишь больших размеров
кристаллитов и высокой степени кристалличности совершенно
недостаточно для обеспечения высоких механических показате-
лей ориентированных полимеров. Последовательное включение
даже очень небольшого числа неупорядоченных областей с ма-
лым числом проходных цепей в полимерный кристалл не позво-
ляет реализовать высокий модуль такого кристалла. Причина
этого состоит в том, что работа деформации связана в основном
с преодолением слабого межмолекулярного взаимодействия це-
лей в неупорядоченных областях, а деформация валентных уг-
лов и связей транс-конформеров кристаллитов остается нереали-
зованной.
Отмеченные закономерности характерны не только для ПЭ.
Изучение теплофизических процессов при деформации ориенти-
рованных ПП с повышенным модулем упругости (Е=«10—
15 ГПа, рис. 4.32) и ПКА (Е~8 ГПа) показало, что изменения
в их термоупругом поведении подобны изменениям в поведении
ПЭ [115]. В связи с этим интересно отметить, что закономерно-
сти термоупругости так называемых твердых эластичных воло-
кон [116], характеризующихся слоевой структурой, весьма по-
хожи на закономерности для отожженных ориентированных ПЭ
и ПП.
Таким образом, термоупругое поведение отожженных ориен-
тированных полимеров также удовлетворительно объясняется
с позиций структурных изменений в рамках микрофибрилляр-
ной модели с внутри- и межфибриллярными аморфными обла-
стями. Можно полагать, что эта модель будет приемлема для
эписания термоупругости тех кристаллических полимеров, кото-
189
Рис. 4.32. Зависимость теплового эф-
фекта от деформации для высокомо-
дульного ПП [115]:
I — растяжение; 2 — сокращение; а — ис-
ходный; б — после изометрического отжига.
рые в неориентированном состоянии характеризуются ламеляр-
ними сферолитами, так как их трансформация в фибриллярную
структуру обычно приводит к ламелярным микрофибриллам с
ограниченным числом проходных цепей [90, 105].
Ориентация аморфных полимеров всегда сопровождается
переходом менее вытянутых конформеров в более вытянутые.
После снятия напряжения они не могут вернуться в исходное
состояние только в том случае, если температура вытяжки за-
метно ниже Тс (вынужденная высокоэластичность; см. гл. 5).
При таких условиях в некоторых полимерах, способных к кри-
сталлизации, например в ПЭТФ, удается создать аморфную
текстуру [117], в которой основная часть цепей находится в
транс-конформариях. Эти цепи сдвинуты вдоль оси текстуры
вполне закономерно, как и в кристаллическом образце; однако
азимутальные повороты звеньев макромолекул и межцепные
расстояния сильно нарушены, и потому трехмерная кристалли-
ческая решетка в такой текстуре отсутствует. Модуль упругости
такой текстуры должен быть заметно выше, чем у неориентиро-
ванных образцов, поскольку ее растяжение связано в значи-
тельной мере с деформацией транс-конформации. Наряду с
транс-конформациями в такой текстуре существуют и аош-кон-
формеры, однако их переход в транс-состояния при деформации
резко заторможен (Т<7’с). Наличие транс-конформаций, обла-
дающих отрицательным коэффициентом расширения, должно
резко уменьшать коэффициент теплового расширения такой тек-
стуры или даже делать его отрицательным. В исходном состоя-
нии такая текстура должна быть до некоторой степени подобна
микрофибрилле ГСХ с той лишь разницей, что в ней нельзя вы-
делить собственно кристаллиты и аморфные области. Доля мо-
лекулярных складок в такой текстуре, по-видимому, невелика.
Рассмотрим теперь еще раз с учетом этих соображений дан-
190
ные по термоупругости ориентированных в различных условиях
пленок ПЭТФ. Как следует из табл. 4.5 (образец 7), модуль уп-
ругости аморфной текстуры, полученной при 20 °C, в 2,5 раза
превышает модуль упругости неориентированного образца,
и эта текстура характеризуется отрицательным значением Рц.
При Т>ТС термоупругое поведение текстуры должно соответст-
вовать поведению фибрилл ГСХ. Экспериментально термоупру-
гость текстурированного ПЭТФ при повышенных температурах
до сих пор, по-видимому, не исследовалась. При постановке та-
кого исследования придется, вероятно, столкнуться с проблемой
складывания цепей при кристаллизации текстуры выше Тс.
Данные Бонарта с сотр. (табл. 4.5, образец 8) показывают,
что повышение температуры ориентации до 85 °C приводит к
резкому увеличению модуля, который теперь составляет около
20% экспериментального значения модуля упругости кристал-
лической решетки [99, гл. 7], и к уменьшению коэффициента
ц, который, однако, остается отрицательным. Судя по значе-
нию плотности, такая текстура уже является частично-кристал-
лической. Повышение модуля связано с улучшением ориентации
транс-конформеров при кристаллизации. Согласно данным
ИК-спектроскопии общее содержание аош-конформеров в ней
составляет менее 10% [87, рис. 11], что позволяет исключить
наличие сколь-нибудь значительного числа складок в аморфных
областях и рассматривать такую текстуру в качестве примера
ориентированных систем, в которых внутрифибриллярные
аморфные области содержат преимущественно проходные цепи
в вытянутых конформациях. При кристаллизации аморфной
текстуры возможно возникновение границ между микрофибрил-
лами и появление межфибриллярных аморфных цепей. Кратко-
временный изометрический отжиг при повышенных температу-
рах (10 мин, 175 °C) несколько уменьшает модуль упругости и
приближает коэффициент р к нулю, что, вероятно, связано с
гош-транс-переходом в аморфных областях и появлением в них
определенного числа складок.
В отличие от ПЭТФ ПБТФ не удалось даже при быстром
охлаждении получить в полностью аморфизованном состоянии.
Поэтому ориентированные при комнатной или повышенной
температуре образцы являются частично-кристаллическими и
их растяжение как при комнатной температуре (у ПБТФ Тс=
= 40 °C), так и при повышенных температурах (120—140 °C)
характеризуется поглощением тепла*. Это дает основание пред-
положить, что у ПБТФ в таких условиях получения ориентиро-
* Для ориентированного ПБТФ характерно необычное явление полимор-
физма при деформации, проявляющееся в изменении типа решетки при опреде-
ленном напряжении (деформации) [118]. Этот фазовый переход является ре-
зультатом изменения ^периода решетки вдоль оси с (ось макромолекулы) вслед-
ствие перехода цепей под действием механического напряжения в наиболее
вытянутое состояние. В калориметрических экспериментах это проявляется в
виде резкого изменения наклона зависимости Q—
191
ванных образцов преобладает микрофибрилляриая структура с
небольшим числом проходных цепей во внутрифибриллярных
аморфных прослойках и малым содержанием межфибрилляр-
ных аморфных цепей.
Проведенный выше модельный анализ теплофизических и
структурных данных был ограничен ориентированными кристал-
лическими полимерами с обычным модулем упругости, состав-
ляющим несколько процентов от теоретического модуля кри-
сталлической решетки. Однако, по крайней мере, в одном слу-
чае (см. рис. 4.32)—высокоориентированный ПП [115]—мо-
дуль упругости волокна достигал 25—30% от модуля кристал-
лической решетки, и тем не менее наблюдаемые при обратимой
деформации исходных и отожженных образцов теплофизические
явления и структурные изменения и в этом случае удается не-
противоречивым образом объяснить, основываясь на модели
Петерлина — Преворзека. В связи с этим теперь уместно более
подробно остановиться на теплофизических свойствах сверх-
ориентированных сверхвысокомодульных гибкоцепных полиме-
ров. Такие волокна и пленки из ПЭ, ПП и ПОМ. были получе-
ны в последние годы [105] методами ориентационной вытяжки,
гидростатической экструзии в твердом состоянии и контроли-
руемой кристаллизацией потока раствора или расплава полиме-
ра. С позиций теплофизики деформирования наиболее важными
являются две характеристики этих сверхориентированных по-
лимеров — высокий модуль упругости, достигающий при темпе-
ратуре —150 °C значений >0,5Екр ь а при комнатной температу-
ре— (0,3—0,35) Екрк, и отрицательный коэффициент р ц (см.
рис. 4.25). Необходимо также подчеркнуть, что для сверхориен-
тированных полимеров характерны морфологические и струк-
турные особенности, присущие обычным хорошо ориентирован-
ным полимерам, а именно [119]: наличие четко выраженной
фибриллярной структуры и значительной концентрации ламе-
лярных кристаллитов. Наиболее существенным морфологиче-
ским отличием сверхориентированных полимеров является су-
ществование в них протяженных кристаллических участков,
продольная длина которых значительно превышает размер боль-
шого периода. i
Для объяснения свойств сверхориентированных полимеров
предложена модель «статистических кристаллических мостиков»
[120, 121], идеи которой можно уяснить на модели Петерлина —
Преворзека. Если считать, что все внутрифибриллярные проход-
ные молекулы и все цепи в межфибриллярных аморфных обла-
стях находятся в наиболее вытянутых кристаллических транс-
конформациях, являясь продолжением кристаллических цепей в
кристаллитах, из которых они выходят, то их можно рассмат-
ривать как статистические кристаллические мостики, и свойства
сверхориентированного полимера могут рассматриваться с по-
зиции степени непрерывности кристаллической структуры. Кон-
центрация кристаллических мостиков и соответственно модуль
192
Рис. 4.33. Термодинамические функции ш... . 3
идеального транс-кристалла ПЭ (/—3) и ™>иДж/см______________
высокомодульного ПЭ (/'—3'); при рас-
четах использованы следующие значения „ /
Ец и 3 J : ЕКр и =240 ГПа; Е । =70 ГПа; \/ л,
Зкр „ =—1,810-5 К"1; ₽ ц =_1.ю-5 К"1. / / / /
1, Г — Q\2,2' — W-,3,3'—U. / / / /
10 - / / / /
/ // У' 3
упругости определяются степенью / S'/
вытяжки. Переход от образцов с /
обычным модулем к высокомо- _______।
дульным является непрерывным 2 г,%
процессом и определяется долей
проходных цепей в аморфных об- х.
ластях и степенью их вытянуто- \\ z
сти. Поэтому модель Петерли- 7
на — Преворзека может рассмат--^- X
риваться как наиболее общая мо-
дель ориентированных криста л_
лических полимеров, поскольку она позволяет уяснить связь их
свойств со структурой как при обычных, так и при предельных
степенях ориентации. Важно подчеркнуть, что как модель кри-
сталлических мостиков, так и модель Петерлина — Преворзека
не предполагают наличия кристаллов очень больших размеров
(КВЦ), поскольку для достижения высоких механических ха-
рактеристик вполне достаточно наличия кристаллических мо-
стиков лишь между двумя-тремя ламелярными кристаллитами
[119, 121]. Ориентированный кристалл макроскопических раз-
меров является предельным случаем. Сравнение термоэластиче-
ских свойств такого кристалла ПЭ и высокомодульного образ-
ца ПЭ приведено на рис. 4.33.
Реализация высокого модуля в сверхориентированных кри-
сталлических полимерах оказывается возможной благодаря на-
личию предельно-ориентированных участков цепей между от-
дельными кристаллитами. Эти участки являются своего рода
«шунтами», позволяющими передавать напряжения не через
слабое межмолекулярное взаимодействие, а через скелет цепи
путем деформации валентных углов и связей этих вытянутых
участков. В изотропных полимерах этого не удается достичь
ввиду статистического распределения осей кристаллитов (це-
пей), а в ориентированных до обычных степеней полимерах это
не достигается ввиду недостаточной вытянутости цепей в аморф-
ных областях, что влечет за собой передачу напряжения в си-
стеме через межмолекулярное взаимодействие. Даже в образцах
с КВЦ, несмотря на очень высокую степень кристалличности
(>95%) и на очень большие размеры кристаллитов (>100нм),
сделать этого не удается из-за наличия неупорядоченных обла-
стей, в которых передача напряжения также осуществляется че-
13-440 ' 193
рез межмолекулярное взаимодействие. Наличие же сильноори-
ентированных «шунтов» позволяет реализовать высокие моду-
ли скелета полимерной цепи при обычных размерах кристалли-
тов (<50 нм) и обычных степенях кристалличности (<85%).
Если межкристаллитные внутри- и межфибриллярные мости-
ки действительно являются кристаллическими в обычном смыс-
ле, то сверхориентированные полимеры должны обладать вели-
колепной стабильностью размеров вплоть до температуры плав-
ления. Это обусловлено малым отрицательным коэффициентом
§Крц и слабой его температурной зависимостью. В температур-
ном интервале от —150 до 50 °C для сверхориентированного ПЭ
это действительно наблюдается [122, 123], однако выше 50 °C
отрицательный коэффициент 0 ц образца начинает стремительно
возрастать, что пытаются связать с высокотемпературными про-
цессами отжига (<120°С) [122]. Однако этому явлению может
быть дано и другое объяснение, если считать, что межкристал-
литные мостики не являются истинно кристаллическими тяжами,
а образованы сильноориентированными участками цепей. Что-
бы начать сокращаться при нагревании, таким участкам не
нужно «дожидаться» плавления или рекристаллизации. Для
этого достаточно, чтобы в условиях понижающегося с темпера-
турой межмолекулярного взаимодействия начали происходить
транс-гош-переходы, сопровождающиеся потерей ориентации и
усадкой. В отличие от эффектов изменения размеров в области
от —150 до 50 °C эффекты выше 50 °C должны быть необрати-
мы, что и наблюдается в действительности [122]. Подобные
эффекты наблюдаются и в высокомодульных волокнах ПП
[115, 124].
Анизотропия термоэластичности ориентированных полимеров
Растянутый эластомер и полимерный кристаллит обладают
ярко выраженной анизотропией теплового расширения. Сильная
анизотропия теплового расширения характерна и для твердых
ориентированных полимеров. Общим свойством как стеклооб-
разных, так и кристаллических полимеров является уменьшение
теплового расширения вдоль оси ориентации (₽ и)и увеличение
поперек (0д). Однако степень анизотропии стеклообразных и
кристаллических полимеров может сильно различаться при срав-
нимых степенях ориентации. Причины этого различия обуслов-
лены отрицательными значениями 0Крц и 0ам» (выше Тс). Коэф-
фициент анизотропии Рп /Рд. сильно зависит от типа полимера.
Для стеклообразивных ПС и ПММА он равен соответственно
1,1 и 2,5 для степени ориентации Х=4, а для ПВХ, обладающе-
го небольшой степенью кристалличности (<1О°/о), он равен 4,5
при Х=3 [125, 28]. Зависимость 0 ц и ;0±от степени ориентации
кристаллических полимеров показана на рис. 4.25. Такое соот-
ношение Щ и 0х типично для кристаллических полимеров.
104
Для теоретического анализа анизотропии теплового расши-
рения кристаллических полимеров используют два подхода.
Один из них основан на анализе эффективных коэффициентов
теплового расширения кристаллических полимеров методами,
развитыми в теории упругости и термоупругости микронеодно-
родных сред [126, 127]. Эти методы требуют знания тензоров
податливости обеих фаз и всего композита, которые обычно
отсутствуют. Однако более существенным ограничением при-
менимости этого подхода является, как уже отмечалось, пред-
положение, что в композиционном материале свойства компо-
нентов сохраняются неизменными. Хотя для двухфазного кри-
сталлического полимера это предположение йе выполняется,
тем не менее этот подход пытаются использовать для грубой
оценки анизотропии теплового расширения ориентированных
кристаллических полимеров, предполагая, что тензор теплового
расширения композита if}',/ может быть получен на основе ли-
нейной интерполяции тензоров расширения компонентов (фаз)
[96]. Считая, что кристаллические включения ориентированы в
определенном направлении, можно записать:
(4-142)
где рКр, ц и — тензоры теплового расширения кристаллической и аморф-
ной (изотропной) фаз (0кр,и — диагональный тензор с элементами Ркр,
Ркр 0 J ).
Это выражение выполняется для предельных случаев w = 0
и и>=1, и можно ожидать, что для кристаллических полимеров
(w = 0,5—0,8) оно также будет выполняться. Его можно исполь-
зовать и в том случае, когда матрица является жидкой (аморф-
ная фаза выше Тс).
Тензор теплового расширения ориентированного кристалли-
ческого полимера р(/- может быть получен весовым усреднением
тензора р'ц по углу с учетом распределения направления цепей
кристаллических блоков:
[ feRik(c)Rj$kldS3 (4.143)
где fc — функция распределения направления осей с кристаллитов; 7?,л(с) —
матрица вращения оси z (направление ориентационной вытяжки) к оси с.
Предполагая fc симметричной функцией относительно z и
подставляя действительные элементы Rik (с) и уравнение (4.142)
в уравнение (4.143), приходим к выводу, что р,,- является тензо-
ром с диагональными элементами (i0i, и р u ; Р х и ри — теп-
ловое расширение кристаллического полимера (композита)
перпендикулярно и параллельно оси ориентации):
=®Фкр+ (l-w)₽aM + y ®Л(Х)(0крх -₽кр||) (4.144)
2
₽ll = wPKp + (l —w)PaM—— w/c(l)(PKpx - Ркр||) (4.145)
13*
195
В этих уравнениях рКр= "з (2рКр1+'₽кр J)—усредненный ко-
эффициент линейного теплового расширения кристаллитов,
a fc(ty =-^-(3cos2<p— 1) (<р —угол между осями z и с). Для изо-
тропного образца fc = 0, и оба эти уравнения переходят в
Р = яфкр “Ь (1 — ^)Рам (4.146)
Из уравнений (4.144) и (4.145) видно, что средний коэффи-
циент линейного теплового расширения ориентированного об-
разца, равный pcp=y(2p1+ip1) или.уа, не зависит от степени
растяжения и равен р. Из этих уравнений очевидно также, что
зависимость р от степени растяжения появляется только вслед-
ствие ориентации оси с кристаллитов вдоль направления ори-
ентации. Когда все кристаллиты окажутся ориентированными
осями макромолекул вдоль оси ориентации наступит
насыщение (Pi и р ц перестанут зависеть от X).
Физическая необоснованность этих расчетов связана с отож-
дествлением коэффициентов расширения аморфной фазы в
изотропном и ориентированном состоянии. При этом последст-
вия этого предположения для р i и р и совершенно различны.
Если для р 1 такое отождествление не играет существенной
роли, так как и в изотропном, и в ориентированном состоянии
перпендикулярно оси ориентации тепловое расширение в основ-
ном определяется межмолекулярным взаимодействием, то для
Р ц эта предпосылка приводит к принципиальным расхождениям.
Это наглядно видно из сравнения экспериментальных и рассчи-
танных по уравнениям (4.144) и (4.145) зависимостей Pj_ и р,
от степени ориентации ПЭ и ПП (см. рис. 4.24). Для Pi со-
гласие эксперимента с теорией вполне удовлетворительное для
всех степеней ориентации, что, безусловно, является следствием
слабой зависимости р± от степени растяжения (Pi возрастает
в предельно-ориентированном состоянии лишь на 25% по срав-
нению с изотропным состоянием). Напротив, уравнение (4.145)
совсем не передает поведения р л при ориентации. Так, оно не
допускает появления отрицательных значений р (| даже при пре-
дельной ориентации кристаллитов (fc~l), так как предполагает
|1Рам | > I Ркр || | •
Второй подход связан с анализом моделей структуры ориен-
тированных кристаллических полимеров. Выражения для коэф-
фициентов рл основных моделей приведены выше [уравнения
(4.133) и (4.138)]. Рассматривая поведение моделей перпенди-
кулярно оси ориентации, можно получить уравнения для Pi-
Для гладкой микрофибриллы такое уравнение имеет вид
й даРкр±^крj, + (1—^)Рав 1 £ав ±
«’Якрл + и-иОЯав!
196
Здесь Рх может принимать только положительные значения,
так как и рКр 1, и р ,в l являются сугубо положительными. Со-
гласно формуле Френкеля [уравнение (3.62)], const.
К тому же теперь £ав сравнимо с £KPi, поэтому 0Х ~pKPi ~
~Рав±- Это совпадает с выводом композитного подхода.
Для макрофибриллы с аморфными межфибриллярными про-
слойками уравнение для р^ имеет вид
^рхда£крл + Рав1£ав(1-^)
£кр±да+ W)£aB
(1-Х) + Рам±Х (1.148)
И в этом случае <Рд может быть только положительным. При
•отсутствии межфибриллярных аморфных прослоек (Х=0)
уравнение (4.148) переходите (4.147).
Для модели со статистическими кристаллическими мостика-
ми, в которой межфибриллярные области считаются кристалли-
ческими, аналогичным способом можно получить [96]:
= V + (₽амх ~ Ркрх) + 1 j <4J49)
Ркр]| Н" (Рам|| ~ ^кр|р
(£
£ам||
(4.150)
где w' — степень кристалличности, учитывающая и долю X' межфибриллярных
областей.
Наряду с двумя наиболее важными направлениями в ориен-
тированных пленках и волокнах твердых полимеров — вдоль и
поперек оси ориентации — можно рассмотреть тепловую дефор-
мацию и под любым <р углом к оси ориентации. Для пленочных
материалов коэффициент линейного теплового расширения под
углом <р к оси ориентации определяется уравнением [126]
Рт = ₽ц C0S2? + COS2(90 - <р) (4.151)
Принимая во внимание отрицательные значения рл, очевид-
но, что в ориентированном кристаллическом полимере существу-
ет такое направление, в котором тепловая деформация отсут-
ствует. Хотя анизотропия механических свойств полимеров ис-
следована достаточно детально [104, 128], исследования тепло-
вого расширения в различных направлениях, а не только вдоль
и поперек направления ориентации отсутствуют. Исследования
термоэластичности ориентированных ПЭНП, ПКА, ПЭТФ пока-
зало [28], что растяжение под углом около 30° к оси ориента-
ции практически не сопровождается тепловым эффектом. Это
означает, что 0зо°~О. Зависимость коэффициента >0 от угла ори-
197
Рис. 4.34. Зависимость коэффициента
линейного теплового расширения
ПЭНП от угла к оси ориентации [28].
Рис. 4.35. Зависимость отношения теплоты к работе при растяжении от дефор-
мации для ПЭНП [28]:
1 _ ф=0; 2 — Ф=15°; 3 — ф=30°; 4 — <р=45°; 5 —<р=90°.
ентации, полученная при исследовании термоэластичности, при-
ведена на рис. 4.34.
Термодинамика одноосной деформации для любого выбран-
ного направления в ориентированном твердом полимере опре-
деляется соотношениями, полученными при анализе уравнения
(4.53). Для теоретического анализа необходимо знать коэффи-
циент теплового расширения и модуль упругости в данном на-
правлении. При известных р „ и PlP<₽ может быть определен
по уравнению (4.151), а расчет модуля упругости может быть
проведен на основании уравнения [128]
Е ‘= _________________U____________
? cos4<p 4- b sin22<p + с sin<cp
E|l Er 1 + с
где с = — ; * = —'-----------------
Е± Ei5o 4
(4.152)
На основании изложенного выше ясно, что в любом из на-
правлений в обычных ориентированных двухфазных кристал-
лических полимерах выше Тс деформирование должно быть свя-
зано с преодолением межмолекулярного взаимодействия и ка-
учукоподобная упругость аморфных областей не должна про-
являться при деформации в любом направлении. Эксперимен-
тальное исследование анизотропии термоэластичности проводи-
лось на ПЭНП, ПКА и ПЭТФ [28]. Исследовалось соотноше-
198
ние между теплотой деформации Qfp и работой деформации
Е ,2 Q 2fi Т
Q? = TP„E?e; = -^- = —Е- (4.153)
В качестве иллюстрации на рис. 4.35 приведены результаты
для ПЭНП. Полученные при этом значения р<₽ рассматривались
выше (см. рис. 4.34).
ЛИТЕРАТУРА
1. Ландау Л. Д., Лифшиц Е. М. Статистическая физика. М., Наука, 1976.
Ч. 1, 583 с.
2. Румер Ю. Б., Рывкин М. Ш. Термодинамика, статистическая физика и ки-
нетика. М., Наука, 1977. 552 с.
3. Надаи А. Пластичность и разрушение твердых тел. Пер. с англ./Под ред.
Г. С. Шапиро. М., Мир, 1969, с. 15—64.
4. Ландау Л. Д., Лифшиц Е. М. Теория упругости. М., Наука, 1965. 202 с.
5. Волькенштейн М. В. Конфигурационная статистика полимерных цепей.
М. — Л., Изд-во АН СССР, 1959. 466 с.
6. Flory Р. L, Новое С. A. I., Ciferri А. — J. Polymer Sci., 1969, vol. 34, N 127,
p. 337—347.
7. Flory P. /. — Trans. Farad. Soc., I9SI, vol. 57, N 5, p. 829—838.
8. Трелоар Л. Физика упругости каучука. Пер. с англ./Под ред. Е. В. Кув-
шинского М. — Л., Издатинлит, 1953. 240 с.
9. Годовский Ю. К.— Высокомол. соед., 1977, сер. А, т. 19, № 10, с. 2359—
2366.
10. Бирштейн Т. М., Птицын О. Б. Конформации макромолекул. М., Наука,
1964. 391 с.
11. Allen G., Bianchi U.. Price С. — Trans. Farad. Soc., 1963, vol. 59, N 11,
p. 2493—2502.
12. Allen G., Kirkham M. C., Padget J., Price C. — Trans. Farad. Soc., 1971,
vol. 67, N 5, p. 1278—1292.
13. Gee G. — Trans. Farad. Soc., 1946, vol. 42, N 8, p. 585—599.
14. Khasanovich T. N.—J. Appl. Phys., 1959, vol. 30, N 10, p. 948—949.
15. Tobolsky A. V., Shen M.—J. Appl. Phys., 1966, vol. 37, N 5, p. 1952—
1955
16. Treloar L. — Polymer, 1969, vol. 10, N 2, p. 291—305.
17. Treloar L. — In: Applied Fibre Science/Ed. by F. Happey, London, Academic
Press, 1979, p. 103—132.
18. Krigbaum W. R., Koneko M. — J. Chem. Phys., 1962, vol. 36, N 1, p. 99—
109.
19. Smith К — J. Polymer Sci., 1971, pt A-2, vol. 9, N 12, p. 2119—2130.
20. Shen M. — J. Appl. Phys., 1970, vol. 41, N 11, p. 4351—4356.
21. Valanis К. C., Landel R. F. — J. Appl. Phys., 1967, vol. 38, N 10, p. 2997—
3002.
22. Blatz P. J., Sharda S. C., Tschoegl N. M. — Trans. Soc. Rheol., 1974, vol. 18,
N 1, p. 145—161.
23. Sharda S. C., Tschoegl N. M. — Macromolecules, 1976, vol. 9, N 6, p. 910—
917.
24. Chang W. V., Bloch R., Tschoegl N. M. — Ibid., p. 917—926.
25. Ogden R. W. — Proc. Roy. Soc., 1972, Ser. A, vol. 326, p. 665—584.
26. Chadwick P. — Phil. Trans. Roy. Soc. London, 1974, Sec. A, vol. 276,
p. 371—403.
27. Gee G.—Macromolecules, 1980, vol. 13, N 3, p. 705—710.
28. Садовский Ю. К. Теплофизические методы исследования полимеров. М„
Химия, 1976. 216 с.
109
29. Price С. — Proc. Roy. Soc., 1976, Ser. A, vol. 351, p. 331—350.
30. Krigbaum W. R., Roe R.-J. — Rubb. Chem. Technol., 1965, vol. 38, N 5,
p. 1039—1069.
31. Mark J. E. — Rubb. Chem. Technol., 1973, vol. 46, N 3, p. 593—618.
32. Shen M., Kroucher M. — J. Macromol. Sci., 1975, pt C, vol. 12, N 2, p. 287—
329.
33. Mark J. E.—J. Polymer Sci., Macromol. Revs., 1976, vol. 11, p. 135—159.
34. Godovsky Yu. K.— Polymer, 1981, vol. 22, N 1, p. 75—86.
35. Price C„ Pedget J., Kirkham M. C., Allen G. — Polymer, 1969, vol. 10, N 7,
p. 495—499; N 8, p. 573—678.
36. Wolf F. P., Allen G. — Polymer, 1975, vol. 16, N 3, p. 209—217.
37. Allen G., Price C., Yoshimura N. — Trans. Farad. Soc., 1975, vol. 71, N 3,
p. 548—657.
38. Goritz D., Muller F. H. — Koll.-Ztschr. u. Ztschr. Polymere, 1973, Bd 251,
N 9, S. 679—688.
39. Mark I. E., Flory P. J.—J. Amer. Chem. Soc., 1964, vol. 86; N 2, p. 138—
141.
40. Флори П. Дж. Статистическая механика цепных молекул. Пер. с англ./
/Под ред. М. В. Волькенштейна. М„ Мир, 1971. 440 с.
41. Бойер Р. Ф„ Миллер Р. Л. Международная конференция по каучуку и ре-
зине, 1978, Киев . Препринт А3.
42. Treloar L. — Polymer, 1978, vol. 19, N 12, p. 1414—1420.
43. Christensen R. C., Hoeve C. A. J.—J. Polymer Sci., 1970, pt A-l. vol. 8,
N 6. p. 1503—1512.
44. Flory P. I. — Proc. Roy. Soc., 4976, Ser. A, vol. 351, p. 351—380.
45. Flory P. J. — Polymer, 1979, vol. 20, N 11, p. 1317—1320.
46. Flory P. J.—3. Chem. Phys., 1977, vol. 66, N 12, p. 5720—5729.
47. Guth E. — J. Polymer Sci., 1966, pt C. N 12, p. 89—109.
48. Mark J. E. — Polymer Eng. a. Sci., 1979, vol. 19, N 4, p. 254—259.
49. Andrady A. L„ Llorente M. A., Mark J. E.—J. Chem. Phys., 1980, vol. 72,
N 4, p. 2282—2290; vol. 73, N 3, p. 1439—1445.
50. Schwarz J. Habilitationsschrift, Clausthal, Technischen Universitat, 1978.
147S.; 26th International Symposium on Macromolecules, ШРАС, Mainz,
1979. Preprints. Vol. 3, p. 1370—1373; 9th Europhysical Conference on Mac-
romolecules. Abstracts of Papers, Jablonna, 1979, p. 121—122.
51. Mullins L. — Rubb. Chem. Technol., 1969, vol. 42, N 3, p. 339—362.
52. Вишняков И. И.—В кн.: Итоги науки и техники. Химия высокомол. со-
ед. М„ ВИНИТИ, 1975. Т. 6, с. 130—146.
53. Dannenberg Е. М. — Rubb. Chem. Technol., 1975, vol. 48, N 3, p. 410—
444.
54. Бартенев Г. M. Структура и релаксационные свойства эластомеров. М.,
Химия, 1979. 288 с.
55. Липатов Ю. С. Физическая химия наполненных полимеров. М., Химия,
1977. 304 с.
56. Эйрих Ф. Р., Смит Т. Л. — В кн.: Разрушение/Под ред. Г. Либовица. Пер.
с англ. Под ред. Ю. Н. Работнова. М., Мир, 1976. Т. 7, ч. 2, с. 104—390.
57. Мэнсон Дж., Сперлинг Л. Полимерные смеси и композиты. Пер. с англ./
/Под ред. Ю. К- Годовского. М., Химия, 1979. 439 с.
58. Ношей А., Мак-Грат Дж. Блок-сополимеры. Пер. с англ./Под ред.
Ю. К. Годовского. М., Мир, 1980. 480 с.
59. Estes С. М., Cooper S. L., Tobolsky А. V. — J. Macromol. Sci., 1970, pt С,
vol. 4, N 2, p. 313—360.
60. Fujimura M., Hashimoto T., Kawai H. •— Rubb. Chem. Technol., 1978, vol. 51,
N 2, p. 215—224.
61. Pedemonte E., Dondero G., Alfonso G. C., DeCandia F. — Polymer, 1975,
vol. 16, N 7, p. 531—540.
62. Payne A. R. — J. Polymer Sci., 1974, pt C, N 48, p. 169—196.
63. Gent A. N. — J. Appl. Polymer Sci., 1974, vol. 18, N 5, p. 1397—1406.
64. Папков В. С., Годовский Ю. К., Булкин А. Ф. и др.—Мех. полимеров,
1975, № 3, с. 387—392.
65. Папков В. С. Докт. дис. М., ИНЭОС АН СССР, 1980.
200
66. Годовский Ю. К., Тарасов С. Г. — Высокомол. соед., 1977, сер. А, т. 19,
№ 9, с. 2097—2103.
67. Тарасов С. Г., Цванкин Д. Я., Годовский Ю. К. — Высокомол. соед., 1978,
сер. А, т. 20, № 7, с. 1534—1542.
68. Leblank J. L. — J. Appl. Polymer Sci., 1977, vol. 21, N 9, p. 2419—2437.
69. Тарасов С. Г., Годовский Ю. К. — Высокомол. соед., 1980, сер. А, т. 22,
№ 9, с. 1879—1885.
70. Folkes Н. J., Keller А. — In: Physics of Glassy Polymers/Ed. by R. N. Ha-
ward. London, Applied Science Publishers, 1973, p. 548—598.
71. Godovsky Yu. K-, Tarasov S. G., Tsvankin D. Ya.— In: 27th International
Symposium on Macromolecules, Florence, 1980. Preprints. Vol. 3, p. 235—
238.
72. Galanti A. V., Sperling L. — Polymer Eng. a. Sci., 1970, vol. 10, p. 177—
184.
73. Якимцов В. П., Коврига О. В., Тарасов С. Г., Годовский Ю. К. — Высоко-
мол. соед., 1983, сер. А, т. 25.
74. Zapp R. L., Guth Е. — Industr. Eng. Chem., 1951, vol. 43, N 3, p. 430—
438.
75. Гузеев В. В., Шкаленко Ж- И-, Малинский Ю. М. — Высокомол. соед.,
1981, сер. А, т. 23, № 1, с. 161—170.
76. Jehlar Р„ Romanov A., Pollak V. — In: 26th International Symposium on
Macromolecules, Mainz, 1979. Preprints. Vol. 3, p. 1409—1413.
77. Годовский Ю. K-, Тарасов С. Г. — Высокомол. соед., 1980, сер. А, т. 22,
№ 7, с. 1613—1621.
78- Aggerwal S. L. —Polymer, 1976, vol. 17, N 11, p. 938—956.
79. Липатов Ю. С., Керча Ю. Ю., Сергеева Л. М. Структура и свойства поли-
уретанов. Киев, Наукова думка, 1970. 279 с.
80. Годовский Ю. К-, Бессонова Н. П., Миронова Н. Н. — Высокомол. соед.,
1983, сер. А, т. 25, № 1, с. 111—116.
81. Bonart R., Morbitzer L. — Koll.-Ztschr., 1969, Bd 232, N 2, S. 764—772.
82. Кулезнев В. H. Смеси полимеров. М., Химия, 1980. 302 с.
83. Годовский Ю. К-> Бессонова Н. П. — Высокомол. соед., 1977, сер. А, т. 19,
№ 12, с. 2731—2737; в кн.: 25-й Международный симпозиум по макро-
молекулам, Ташкент, 1978. Тезисы кратких сообщений. М., Наука, 1978.
Т. 6, с. 25.
84. Krigbaum W. R., Roe R.-J., Smith К. J-, Jun. — Polymer, 1964, vol. 5, N 10,
p. 533—542.
85. Kilian H.-G. — In: Macromolecular Kolloqium, Freiburg, 1980, S. 1, s. a. 77.
86. Muller F. H.— In: Rheology./Ed. by F. R. Eirich. N. Y.—London, Acade-
mic Press, 1969. Vol. 5, p. 417—489.
87. Morbitzer L., Hentze G., Bonart R. — KolLnZtschr. u. Ztschr. Polymere,
1967, Bd 216—217, S. 137—149.
88. Годовский Ю. K-, Слонимский Г. Л., Папков В. С., Дикарева Т. А. — Мех.
полимеров, 1970, № 5, с. 785—798.
89. Godovsky Yu. К. — Coll. a. Polymer Sci., 1982, vol. 260, N 5, p. 461—476.
90. Марихин В. А., Мясникова Л. П. Надмолекулярная структура полимеров.
Л., Химия, 1977. 238 с.
91. Пахомов П. М., Шерматов М., Корсу ков В. Е. и др. — Высокомол. соед.,
1976, сер. А, т. 18, № 1, с. 132—139.
92. Гут Э., Джемс Г., Марк Г. — В кн.: Химия больших молекул. Пер. с англ./
/Под ред. Б. А. Догадкина и А. Г. Пасынского. М., Издатинлит, 1948,
с. 72—134.
93. Thiele J. L., Cohen R. Е. — Rubb. Chem. Technol., 1980, vol. 53, N 2, p. 313—
320.
94. Mead W. T., Desper C. R., Porter R. S. — J. Polymer Sqi., Polymer Phys.
Ed., 1979, vol. 17, N 5, p. 859—892.
95. Capati N. J., Porter R. S. — J. Polymer Sci., Polymer Phys. Ed., 1977, vol. 15,
N 8, p. 1427—1433.
96. Choy C. L., Chen F. C., Ong E. L. — Polymer, 1979, vol. 20, N 10, p. 1191—
1198.
201
97. Choy C. L., Chen F. C., Young K. — J. Polymer Sci., Polymer Phys. Ed.,
1981, vol. 19, N 2, p. 336—352.
98. Wolf F.-P., Karl V.-H.— Angew. makromol. Chem., 1980, Bd 92, N 1433,
S. 89—106; Coll. a. Polymer Sci., 1981, vol. 259, N 1. p. 29—37; Macromol.
Chem. a. Phys. (Makromol. Chem.), 1981, vol. 182, N 6, p. 1787—1799.
99. Structure and Properties of Oriented Polymers/Ed. by I. M. Ward. N. Y.,
J. Wiley a. Sons, 1975. 500 p.
100. Peterlin A. —Intern. J. Fract., 1975, vol. 11, N 5, p. 761—780.
101. Peterlin A.— J. Appl. Phys., 1977, vol. 48, N 10, p. 4099—4108.
102. Рабинович. А. Л. Введение в механику армированных полимеров. М., Нау-
ка, 1970. 468 с.
103. Садыкова Ф. X. —Хим. волокна, 1971, № 9, с. 45—50.
104. Уорд И. Механические свойства твердых полимеров. Пер. с англ./Под ред.
А. Я. Малкина. М., Химия, 1975. 357 с.
105. Ultra-High Modulus Polymers/Ed. by A. Cifferi, I. M. Ward. London, Applied
Science Publishers, 1979. 362 p.
106. Prevorsek D. C., Tirpak G. A., Harged P. J. e. a. — J. Macromol. Sci., 1974,
pt B, vol. 9, N 4, p. 733—759.
107. Марихин В. А., Мясникова Л. П., Викторова H. Л. — Высокомол. соед.,
1976, сер. А, т. 18, № 6, с. 1302—1309.
108. Чвалун С. Н., Озерин А. Н„ Зубов Ю. А. и др. — Высокомол. соед., 1978,
сер. Б, т. 20, № 9, с. 672—674.
109. Чвалун С. Н„ Озерин А. Н., Зубов Ю. А. и др. — Высокомол. соед., 1980,
сер. Б, т. 22, № 5, с. 359—363.
ПО. Чвалун С. Н., Озерин А. Н., Зубов Ю. А. и др. — Высокомол. соед., 1981,
сер. А, т. 23, № 6, с. 1381—1387.
111. Чвалун С. И., Озерин А. Н., Зубов Ю. А. и др. — В кн.: III Международ-
ный симпозиум по химическим волокнам. Калинин, 1981. Препринты. Т. 1,
с. 116—121.
112. Чвалун С. И. Каид. дис. М„ МФТИ, 1981.
113. Зубов Ю. А., Селихова В. И., Константинопольская М. Ю. и др. — Высо-
комол. соед., 1972, сер. А, т. 14, № 9, с. 2090—2096.
114. Зубов Ю. А., Бакеев Н. Ф„ Озерин А. Я. —ДАН СССР, 1975, т. 221, № 1,
с. 121—122.
115. Годовский Ю. К. и др. — В кн.: III Международный симпозиум по хими-
ческим волокнам, Калинин, 1981. Препринты. Т. 1 (доп.), с. 66—72.
116. Goritz D., Muller F. Н.—Coll. a. Polymer Sci., 1975, vol. 253, p. 844—
851.
117. Казарян Л. Г., Цванкин Д. Д. —Высокомол. соед., 1965, т. 7, № 1, с. 80—
87.
118. Brereton М. G. е. а. — Polymer, 1978, vol. 19, N 1, р. 17—26.
119. Capaccio G. — Coll. a. Polymer Sci., 1981, vol. 259, N 1, p. 23—28.
120. Gibson A. G., Davies G. R., Ward I. M. — Polymer, 1978, vol. 19, N 6,
p. 683—693.
121. Уорд И. M. — Высокомол. соед., 1979, сер. А, т. 21, № 11, с. 2553—2566.
1'22 . Mead W. Т., Porter R. S. — J. Appl. Phys., 1976, vol. 47, N 10, p. 4278—
4288.
Г23. Gibson A. G., Ward I. M.—J. Mater. Sci., 1979, vol. 14, N 8, p. 1838—
1842.
124. Зубов Ю. А., Бакеев H. Ф., Кабанов В. А. и dp. — В кн.: Ill Международ-
ный симпозиум по химическим волокнам, Калинин, 1981. Препринты. Т. 1
(доп.), с. 53—65.
125. Hellwege К.-H., Hannig J., Knappe W. — Koll.-Ztschr. u. Ztschr. Polymere,
1963, Bd 188, N 3, S. 121—127.
126. Kardos I. L., Raisoni J., Piccarolo S., Halpin C. — Polymer Eng. a. Sci.,
1979, vol. 19, N 14, p. 1000—1009.
127. Шермереор T. Д. Теория упругости микронеоднородных сред. М., Наука,
1977. 400 с.
128. Рысюк Б. Д., Носов М. П. Механическая анизотропия полимеров. Киев,
Наукова думка, 1978. 230 с.
202
Глава 5
ТЕПЛОФИЗИКА ОРИЕНТАЦИОННЫХ
ЯВЛЕНИЙ В ТВЕРДЫХ ПОЛИМЕРАХ
Необратимые деформации твердых тел в отличие от обратимых
всегда связаны с рассеянием в виде тепла хотя бы части затра-
ченной на деформирование механической работы. Поэтому не-
зависимо от знака теплового эффекта упругой деформации
переход в область пластической деформации всегда сопровож-
дается экзотермическим эффектом.
Схема (рис. 5.1) механотермического поведения твердого
тела (металла) при одноосном растяжении, включающем обла-
сти упругой и пластической деформации, показывает, что вслед
за небольшим начальным охлаждением, соответствующим упру-
гой деформации, наблюдается резкое выделение тепла, соответ-
ствующее началу пластической деформации. Развитие пласти-
ческой деформации связано с выделением тепла независимо от
того, идет оно путем локализации (шейка) или достаточно рав-
номерно. Термодинамика пластической деформации широко ис-
следовалась на металлах и сплавах [1—3]. Основные представ-
ления в этой области сводятся к следующему.
Исходным пунктом термодинамического анализа пластиче-
ской деформации твердых тел является энергетический баланс
деформирования. Поскольку первое начало термодинамики при-
менимо как к обратимым, так и к необратимым процессам, его
использование в случае пластической деформации позволяет
независимо от условий проведения процесса определить изме-
нение внутренней энергии (энтальпии) в результате пластиче-
ской деформации
Д//скр(Д67скр) = 117(1 -Q/W) (5.1)
Идеальная пластическая деформация характеризуется пол-
ным превращением работы деформирования в тепло (W=Q),
так что энергетическое состояние твердого тела после дефор-
мации эквивалентно исходному недеформированному состоянию.
В реальных условиях работа пластической деформации твердых
тел обычно лишь частично рассеивается в виде тепла (W>Q),
в то время как определенная ее доля аккумулируется, повышая
внутреннюю энергию. Эту энергию принято называть скрытой
энергией деформации, поскольку она сохраняется в твердом
теле после снятия деформирующих напряжений. Этим скрытая
энергия отличается от макроскопической энергии упругой де-
формации, исчезающей после снятия напряжений.
В результате пластической деформации изменяются многие
свойства твердого тела и обычно увеличивается сопротивление
деформированию (деформационное упрочнение). Это указыва-
ет на то, что пластически деформированное твердое тело нахо-
203
Рис. 5.1. Механотермическое поведение низ-
комолекулярного твердого тела (/) и твер-
дого полимера (II) при одноосном растяже-
нии:
1 — область упругой деформации (твердое тело —
доли процента, твердый полимер — проценты); 2 —
область пластической деформации твердого тела;
и ориентационной вытяжки твердого полимера
(твердое тело — десятки процентов, твердый по-
лимер— сотни» процентов); 3 — пластическая де-
формация ориентированного полимера.
дится в метастабильном состоянии
при данной температуре, и, следо-
вательно, обладает свободной энер-
гией, для определения которой не-
обходимо знать изменение энтропии
в результате пластической деформа-
ции. Полное изменение энтропии:
Д5 = Д5Конфиг+Д5кол. Очевидно, что во всех реальных случаях
при пластической деформации увеличивается структурная раз-
упорядоченность, и потому ДЗконфиг растет. Колебательная эн-
тропия при этом может как возрастать, так и уменьшаться. Тем
не менее обычно превалирует Д5КОнфиг, так что при пластической
деформации Д5>0. Однако экспериментально определить AS
чрезвычайно сложно.
Современные представления о молекулярных механизмах
пластической деформации в твердых телах основаны главным
образом на дислокационных теориях. Количественные оценки
показывают, что при температурах, достаточно низких по отно-
шению к температуре плавления металла, энтропийный вклад
в свободную энергию деформированного металла несуществен,,
и потому в первом приближении свободную энергию можно счи-
тать равной скрытой энергии деформации.
Скрытая энергия деформации зависит от многих факторов,,
и указывать ее значение без их указания не имеет смысла.
Считается твердо экспериментально установленным, что зна-
чение скрытой энергии деформации сильно зависит от условий
деформации, степени и температуры деформации, чистоты ме-
талла, скорости и вида деформации и других факторов. Ее
значение для металлов и сплавов колеблется от долей процента
до десятков процентов от работы деформирования. Для после-
дующего сравнения особый интерес представляет зависимость
скрытой энергии деформации от температуры деформирования^
вернее, от соотношения температур деформирования и плавле-
ния Т/Тпл. При идентичных условиях деформирования абсолют-
ное значение скрытой энергии деформации при одинаковых сте-
пенях деформации резко снижается при увеличении отношения
ТПпл- При больших отношениях Т/Тпл металл практически
упрочняется, доля скрытой энергии чрезвычайно мала и пласти-
ческая деформация близка к идеальной (IF=Q). Особенно
204
большие значения скрытой энергии деформации характерны для
упрочняющего деформирования при низких температурах (ма-
лое Т/Тпл) и при динамических (ударных) режимах деформиро-
вания.
Пластически деформированный металл находится в метаста-
бильном состоянии и при благоприятных условиях стремится
возвратиться в исходное равновесное состояние. Движущей си-
лой процесса возврата является свободная (скрытая) энергия
деформирования. Выделение скрытой энергии деформирования
при возврате удается во многих случаях зафиксировать на тем-
пературной зависимости динамической теплоемкости. Харак-
терным оказывается наличие нескольких (двух-трех) темпера-
турных областей выделения скрытой энергии.
Согласно современным представлениям механизмы поглоще-
ния энергии при пластической деформации упрочняющихся ме-
таллов сводятся к возникновению при деформации дополни-
тельных скоплений дислокаций и отдельных дислокаций, затор-
мозившихся на препятствиях, и к появлению дополнительных
вакансий. Каждый из этих механизмов характеризуется лока-
лизацией определенной доли скрытой энергии. Считается, что
наибольшая доля скоростей энергии локализована в скоплениях
дислокаций. Наличием нескольких механизмов поглощения
энергии при деформации объясняется и многостадийный ха-
рактер ее выделения при возврате металла в исходное состоя-
ние. В процессе нагрева деформированного металла происходит
исчезновение возникших при пластической деформации вакан-
сий, распад скоплений дислокаций на отдельные дислокации и
уменьшение их числа. Считают, что каждая из этих стадий про-
текает в характерном для каждого металла температурном ин-
тервале и приводит к выделению соответствующей части скры-
той энергии. Если пластическое деформирование осуществляет-
ся выше температуры, при которой должна происходить одна
или нисколько стадий возврата, то в процессе деформирования
поглощение энергии и ее возврат протекают одновременно, что
и объясняет невозможность упрочнения металлов при пластиче-
ской деформации при больших значениях отношения Т/Тпл
(0,5—0,6) и практически полное отсутствие скрытой энергии в
деформированных в таких условиях металлах.
Имея в виду эту качественную теплофизическую картину
пластической деформации твердых тел, перейдем к рассмотре-
нию теплофизических явлений при ориентации твердых поли-
меров.
ТЕРМОДИНАМИКА ОРИЕНТАЦИИ КРИСТАЛЛИЧЕСКИХ
И СТЕКЛООБРАЗНЫХ ПОЛИМЕРОВ
При растяжении в определенных условиях изотропных кри-
сталлических и аморфных стеклообразных полимеров обычно
наблюдается явление холодной вытяжки [4], механотепловая
2и5
картина которого заключается в следующем (см. рис. 5.1). Если
кристаллический полимер растягивать в калориметре с опреде-
ленной постоянной скоростью ниже Тпл, а стеклообразный по-
лимер— ниже Тс, то обычно вначале наблюдается равномерное
растяжение образца, сопровождающееся его охлаждением (по-
глощением тепла). При определенной деформации, зависящей
от типа полимера и составляющей от нескольких процентов до
нескольких десятков процентов, однородность деформации на-
рушается, и в образце появляется резкое сужение — шейка, ко-
торая в отличие от металлов не сужается при дальнейшем рас-
тяжении непрерывно вплоть до разрыва, а сужается только до
определенных размеров и в таком состоянии распространяется
через весь образец. При некоторой степени вытяжки, опреде-
ляемой соотношением поперечных размеров исходного образца
и шейки, весь образец превращается в шейку. После этого
превращения шейка может пластически деформироваться на
многие десятки и сотни процентов.
В момент образования шейки на эффект охлаждения образ-
ца накладывается эффект резкого нагрева в месте образования
шейки. По мере развития шейки в образце рассеиваемый теп-
ловой поток стабилизируется в соответствии с постоянной ско-
ростью развития ориентации. После полной трансформации об-
разца в шейку на кривой растяжения наблюдается подъем на-
пряжения (область 3 на рис. 5.1), и рассеиваемое тепло также
возрастает. Напряжение называемое пределом вынужденной
эластичности (стеклообразные полимеры) и рекристаллизации
(кристаллические полимеры), а также напряжение развития
шейки Ош Для многих полимеров возрастают линейно с логариф-
мом скорости растяжения [5, 4] в достаточно широком интер-
вале скоростей (несколько порядков). Логарифмический закон
скоростей характерен и для предела текучести От металлов [6].
Феноменологические закономерности холодной вытяжки твер-
дых полимеров изложены в монографии Уорда [4]. Ниже рас-
сматриваются теплофизические аспекты явления ориентации
твердых полимеров.
Как и в термодинамике пластической деформации низко-
молекулярных твердых тел, главным здесь является вопрос о
скрытой энергии ориентации, т. е. о соотношении механической
работы холодной вытяжки и рассеиваемого тепла и об энерге-
тическом состоянии ориентированных полимеров. Систематиче-
ски этот вопрос исследовался Мюллером и сотр. [7, 8] и авто-
ром [9, 10]. В результате исследования влияния природы поли-
мера, скорости деформирования, фазового состава установлены
Гедующие количественные закономерности.
Для ряда кристаллических полимеров как работа ориента-
|ции, так и рассеиваемое при ориентации тепло линейно зависят
[от логарифма скорости ориентации (рис. 5.2). Для механиче-
Гской работы эта закономерность является прямым следствием
логарифмического закона скоростей, наблюдавшегося для всех
206
Рис. 5.2. Зависимость работы ориентации (а) и теплоты, рассеянной при ориен-
тации (б), от скорости растяжения для ПП (/), ПЭВП (2), ПКА (5), ПЭНП
(4) при 294 К [9, 10].
приведенных на рис. 5.2 полимеров. Таким образом, экспери-
ментальные закономерности работы и теплоты ориентации мо-
гут быть представлены для кристаллических полимеров соот-
ношениями
(5.2)
U7 = а + Ь lg Vp
Q-a' + &'lgVp
Вместе с тем относительная доля рассеиваемого в процессе
ориентации тепла от скорости растяжения практически не зави-
сит (рис. 5.3), и для исследованных кристаллических полиме-
ров Q/W<1, что свидетельствует о поглощении части работы в
процессе ориентации (скрытая энергия ориентации).
Согласно (5.1) и (5.2) эта скрытая энергия ориентации тоже
линейно возрастает с логарифмом скорости деформации
Д/7СКР = Г(1 - Q/ W) = const W = А + В lg Vp (5.3)
Соответствующие зависимости для кристаллических полиме-
ров приведены на рис. 5.4. Поскольку естественная степень
ориентации различна для разных полимеров, сравнение способ-
ности полимеров поглощать энергию при ориентационной вы-
тяжке возможно лишь при нормировке на величину естествен-
ной степени вытяжки (см. рис. 5.4, б).
Рис. 5.3. Зависимость доли работы,
рассеянной в виде теплоты при ориен-
тации кристаллических полимеров
[9, 10]:
I — ПП; 2 - ПЭВП; 3 — ПКА; 4 — ПЭНП.
-Q/W
7,0
0,9
0,8
0,1
-2 -1 IgV [мм/cj
-------------------------------------;
_____._______________________________2,3
-------------------------------------if
207
ah
GKP, Дж/г
АН скр , Дж !з
1 - ПП (Л=6);2 —ПЭВП (Л=5); 3 - ПКА (Л=3,5); 4 - ПЭНП (Х=4).
В отличие от кристаллических полимеров для аморфного
поликарбоната при двух температурах ориентации зависимость
W и Q от скорости растяжения несколько иная (рис. 5.5), хотя
и в этом случае Q/IF<1, что также свидетельствует о сущест-
вовании скрытой энергии ориентации и в ориентированных
стеклообразных полимерах. Энергетические характеристики
ориентационной вытяжки кристаллических и стеклообразных по-
лимеров, полученные в работах различных авторов для опреде-
ленной скорости деформирования, суммированы в табл. 5.1.
По данным различных авторов, значения отношения Q/W
для некоторых полимеров достаточно хорошо совпадают. В то
же время для таких полимеров, как ПКА и ПК, в работах
Мюллера получены существенно более низкие значения Q/W,
чем в остальных исследованиях. Кроме того, для зависимости
Q/W от скорости растяжения ПКА Мюллер [8] наблюдал зна-
чительное уменьшение отношения тепловой мощности к меха-
нической— от 0,62 до 0,25 при скорости растяжения менее
1 мм/с. Возможно, что столь низкие значения Q/W, соответст-
вующие очень большим скры-
тым энергиям ориентации в
ПКА и ПК, обусловлены ме-
тодическими особенностями
регистрации тепловых потоков
Рис. 5.5. Зависимость работы (/, 2) и
теплоты (Г, 2') ориентации ПК при
293 (1, Г) и 303 К (2, 2') от скорости
растяжения [8].
208
Таблица 5.1. Энергетические характеристики ориентационной
вытяжки полимеров
Полимер э Тр, к L э/ww ‘^д & № о» 1 j/жЦ ‘dsI3nv АНскр/Х, Дж/г Литератур- ные ссылки
ПЭВП 0,61 294 и 5 1,0 0,85±0,05 18,9 3,78 [9, 0]
296 — — 0,8—0,9 — •— [Н
ПЭНП 0,41 294 4 1,0 0,85±0,05 7,2 1,8 [9, 0]
293 —. 0,9 0,76—0,87 — [8
ПП 0,59 294 6 1,0 0,93±0,05 11,8 1,97 [9, 0]
— 296 — —. 0,9—1,0 — •— [И
ПКА 0,45 294 3,5 1,0 0,80±0,05 17,0 4,85 [9, 0]
— 293 1,2 0,60—0,62 — — [8
ПВХ <0,1 293 — 0,34 0,70—0,83 —• — [8
296 — — 0,60—0,70 —. — [П
ПБТФ <0,4 293 5,2 0,028 0,95±0,05 5,3 1,0 См.
ПК 0 294 2,3 0,015 0,95±0,05 — — [9, 0]
293**1 303 / 2,3 — 0,65 14,6 6,35 [8
296 — — 0,95±0,05 — — [И]
ПЭТФ
пленка 0 294 5,4 0,03 0,95±0,05 6,0 1,1 [9, 0]
волокно 0 294 5,6 0,03 0,95±0,05 6,75 1,2 [9, 0]
пленка 0 300
319 330 — 0,17 1,0± 0,03 = 0 «0 [12]
340 -
* Во всех работах, кроме [11], энергетические характеристики определялись прямым
калориметрированнем. В работе [11] отношение QIW оценено на основании измерения
распределения температуры в зоне образования шейки.
** См. рис. 5.3.
*** Данные, полученные в последнее время в лаборатории автора.
в газовом калориметре, который использовали в работах
Мюллера. Так или иначе, но данные, приведенные в табл. 5.1,
позволяют заключить, что наибольшей способностью погло-
щать энергию при деформации обладают ПКА и ПЭВП. На
примере полиэтиленов видно также, что при повышении степе-
ни кристалличности w увеличивается поглощение энергии при
ориентации и его зависимость от скорости деформации.
В предыдущей главе отмечалось, что ориентированные поли-
меры обладают ярко выраженной анизотропией термоупругих
свойств. Наряду с этим им свойственна способность к переори-
ентации, заключающаяся в том, что при растяжении под углом
к оси ориентации ориентированный полимер можно перевести в
новое ориентированное состояние. В общих чертах механотепло-
вая картина переориентации такая же, как и при первичной
ориентации. При переориентации под большими углами к' оси
первичной ориентации поведение полимера качественно мало
отличается от поведения его при первичной ориентации. При
14—440
209
Угол переориентации, граО
Рис. 5.6. Зависимость нормированных значений работы (О) и теплоты (ф)
переориентации кристаллического (ПП) и аморфного (ПЭТФ) полимеров при
294 К [9, 10].
Q/W
-0,2
Число переориентаций
Рис. 5.7. Соотношение рассеянной теплоты и работы при последовательных
переориентациях под углом 90° к оси предыдущей ориентации пленки поли-
амида 6,11 [8].
уменьшении угла переориентации переход от упругой деформа-
ции к ориентационной вытяжке выражен менее резко, а сама
вытяжка не столь равномерна. Термодинамика переориентации
изучалась автором [9, 10] на ПП, ПКА и ПЭТФ (рис. 5.6).
Энергетическое поведение кристаллических и аморфных по-
лимеров при переориентации подобно: с уменьшением угла пе-
реориентации и работа и теплота переориентации резко возра-
стают. Однако их соотношение практически не зависит от угла
переориентации и составляет: —1 ±0,05 для углов 90 и 60° и
—1 ±0,08 для углов 45 и 30°. Такие же соотношения характер-
ны и для ПКА. Таким образом, можно считать, что в пределах
точности эксперимента переориентация как кристаллических, так
и аморфного полимеров не сопровождается изменением их
энергетического состояния, так как скрытая энергия переориен-
тации близка к нулю.
Результат, принципиально отличающийся от изложенных
выше, получен Мюллером [8] при изучении многократной пере-
ориентации под углом 90° к оси предыдущей ориентации поли-
амида 6,11 (рис. 5.7). Как видим, отношение Q/W сильно отли-
чается от —1 и удивительным образом зависит от числа циклов
переориентации. Этот результат означает, что скрытая энергия
к седьмому циклу должна достигать очень больших значений.
Мюллер полагает, что эта энергия сконцентрирована в поли-
мере в виде замороженных напряжений.
Таким образом, результаты калориметрических исследова-
ний энергетического баланса ориентационной вытяжки твердых
полимеров приводят к выводу, что первичная ориентация кри-
сталлических полимеров при температурах, достаточно удален-
ных от температуры плавления, характеризуется неравенством
Q<W, следствием которого является повышение внутренней
210
энергии ориентированных кристаллических полимеров по срав-
нению с неориентированными. Скрытая энергия ориентации за-
висит от скорости ориентации, возрастая с ее повышением. Доля
скрытой энергии может достигать 30—40% от работы ориента-
ции, что свидетельствует о существенных энергетических изме-
нениях при ориентации.
Для стеклообразных полимеров количественные результаты
менее однозначны, однако и они демонстрируют способность
твердых аморфных полимеров поглощать энергию при ориен-
тационной вытяжке.
Согласно изложенному выше энергетическое состояние не-
ориентированных и ориентированных полимеров может разли-
чаться. Независимое подтверждение этого заключения может
быть получено на основе иных калориметрических измерений.
Простым и надежным подходом может являться сравнитель-
ный анализ теплот растворения неориентированных и ориенти-
рованных полимеров [8]. Для практического использования
этого подхода важны следующие обстоятельства.
Для наиболее общего случая — частично кристаллического полимера при
температуре ниже температур плавления н стеклования аморфных областей —
теплота растворения АЯР может быть представлена суммой следующих трех
составляющих (см., например [1, 3]):
Д//р = АЯСМ -f- А#пл 4" А^изб.ам (5*4)
где ДДсм—теплота смешения жидкоподобного аморфного полимера с раство-
рителем; ДЯпл—теплота плавления кристаллитов; ДЯизб. ам— избыточная
теплота аморфных стеклообразных областей.
Лишь первый член правой части этого уравиеиия — ДЯсм определяется
взаимодействием молекул полимера с молекулами растворителя. Два остав-
шихся члена не зависят от растворителя и характеризуют состояние полиме-
ра. Поэтому измерения в различных растворителях и при разных концентра-
циях растворов должны приводить к одинаковым значениям суммы ДЯНЛ +
+ДЯизб. ам/ ДДпл — величина положительная, чаще всего составляющая 20—
200 Дж/г; ДЯом для неполярных полимеров обычно небольшая («10 Дж/г)
положительная величина. Относительно ДЯизб. ам известно следующее.
Из многочисленных исследований теплот растворения аморфных полиме-
ров ниже Тс следует, что растворение стеклообразных полимеров всегда со-
провождается выделением тепла, причем тепловые эффекты растворения по-
лимерных стекол могут достигать 60—80 Дж/г [14]. Согласно современным
представлениям экзотермйчность растворения полимерных стекол является
следствием их неравновесного состояния. Степень неравновесности тем выше,
чем дальше стекло отстоит от температуры стеклования. Отсюда вытекает
корреляция теплоты растворения стеклообразного полимера с температурой
стеклования [14]: для большого числа аморфных полимеров разных классов,
сильно различающихся гибкостью макромолекул (гибкоцепные и жесткоцеп-
ные) и соответственно температурами стеклования (150—600 К), экзотерми-
ческая теплота растворения стеклообразных полимеров линейно зависит от
температуры стеклования. Это означает, что чем больше разность между тем-
пературой стеклования н температурой растворения стеклообразного полиме-
ра, тем больше теплота растворения. Жесткоцепные полимеры, например- по-
лнарилаты, обладающие высокой температурой стеклования (ТсЯйбОО К), ха-
рактеризуются очень большими теплотами растворения, достигающими
80 Дж/г и более. Эти экзотермические теплоты растворения стеклообразных
полимеров действительно практически ие зависят от природы растворителя и
обусловлены высокой степенью неравновесности жесткоцепных полимеров.
В общих чертах этй соображения справедливы и для аморфных стеклообраз-
14*
211
ных областей частично-кристаллических полимеров. В применении к стекло-
образным аморфным полимерам, в которых ДЯПл = 0, в правой части урав-
нения (5.4) сохраняются лишь ДЯои+АДизб. ам-
Составляющая ДЯИзб. аи оказывается чувствительной к физической струк-
туре полимера. Поэтому между параметрами, определяющими эту структуру
(термическая предыстория, способ приготовления образцов, молекулярная
масса и др.), и теплотами растворения существует прямая связь. Из сущест-
вования такой связи вытекает возможность использования теплот растворения
для определения скрытой энергии ориентации стеклообразных полимеров.
Ориентационная вытяжка кристаллических полимеров может сопровождаться
изменением степени кристалличности и, следовательно, пропорциональным из-
менением теплоты плавления &НПЛ.
Эти соображения являются физической основой рассматри-
ваемого подхода. Его идея ясна из рис. 5.8. Поскольку ДЯСМ
определяется только взаимодействием молекул полимера с мо-
лекулами растворителя, эта составляющая одинакова для не-
ориентированного и ориентированного состояния. При A//CKp>0
экзотермический эффект растворения ориентированного стекло-
образного полимера будет превышать экзотермический эффект
растворения неориентированного полимера, а эндотермический
эффект растворения ориентированного кристаллического поли-
мера будет меньше эндотермического эффекта растворения не-
ориентированного. Если в процессе ориентационной вытяжки
стеклообразного полимера произойдет кристаллизация, то
ДЯСКр<0. Если при этом Д//Скр>Д^р.нр, то растворение ориенти-
рованного полимера будет эндотермическим, в противном слу-
чае (Д//скр<Д//Р.нр)—экзотермическим. В процессе ориентации
кристаллического полимера его степень кристалличности может
возрасти за счет дополнительной кристаллизации аморфных об-
ластей (ДЯСКр<0). В этом случае эндоэффект растворения ори-
ентированного полимера будете превышать эффект растворения
неориентированного полимера.
6
метод определения скрытой энергии
а
Рис. 5.8. Диаграммы, иллюстрирующие
ориентации по теплотам растворения:
а — аморфный стеклообразный полимер; б — частично-кристаллический полимер; I — не-
ориентированное состояние; II — ориентированное состояние; III — раствор.
212
Рис. 5.9. Энергетическая схема опреде-
ления скрытой энергии ориентации ПВХ
по теплотам растворения (7’0р=313 К;
Уор=0,1'4 мм/с; теплоты растворения оп-
ределяли при 313 К в хлорбензоле) [8, 15,
16].
Возможность практического использования рассмотренного
подхода для оценки энергетического состояния ориентирован-
ных полимеров и скрытой энергии ориентации продемонстриро-
вана в работах Мюллера с сотр. [8, 15, 16]. В частности, при
исследовании ориентационной вытяжки ПВХ были получены
следующие результаты (рис. 5.9). Работа ориентационной вы-
тяжки W=29,8 Дж/г, теплота, рассеянная при вытяжке, Q=
= —14,9 Дж/г, скрытая энергия ориентации Д//Скр=14,9 Дж/г
(Q/W=—0,5). Теплота растворения неориентированного поли-
мера Д/7р.нр составляла 16,5 Дж/г, теплота растворения ориен-
тированного полимера ДЯр.ор равна —20 Дж/г. Скрытая энергиях
ориентации Д/7Скр, по данным теплот растворения, составляет]
3,5 Дж/г, что существенно меньше величины, определенной не-1
посредственно в процессе ориентации в деформационном кало-1
риметре. Это различие обусловлено процессом возврата ориен-
тированного образца, т. е. переходом его в более равновесное
состояние (по условиям экспериментов при определении теплот
растворения ориентированные образцы выдерживали перед рас-
творением в течение 12 ч при комнатной температуре). Энергия
возврата, по данным энергетического баланса, составляет:
ДЯВ =—11,4 Дж/г. Этот пример показывает, во-первых, плодо-
творность сравнительного исследования энергетического состоя-
ния ориентированных и неориентированных полимеров и, во-
вторых, необходимость учета всех возможных стадий изменения
энергетического состояния полимера.
Рассмотрим возможные источники изменения энергетическо-
го состояния полимеров при ориентационной вытяжке. Можно
полагать, что по меньшей мере следующие шесть источников
могут играть при этом определенную роль (перечислены в по-
рядке важности вклада): 1) изменение степени кристаллично-
сти; 2) конформационные (внутримолекулярные) энергетиче-
ские изменения в аморфных полимерах и в некристаллических
областях кристаллических полимеров; 3) замороженные (очень
медленно релаксирующие) внутренние напряжения; 4) возник-
новение дополнительных дефектов в кристаллических областях;
5) образование новой поверхности; 6) химические изменения
(радикалы, новые концевые группы).
213
Для кристаллических полимеров наиболее важным источни-
ком энергетических изменений может бытиТизменение степени
кристалличности при ориентации. Действительно, если, напри-
мер, при ориентации ПЭ степень кристалличности изменится на
3%, то соответствующее изменение энтальпии ориентированно-
го образца составит около 10 Дж/г (теплота плавления ПЭ рав-
на 293 Дж/г). Такого же порядка изменения могут быть харак-
терны и для других кристаллических полимеров.
Другим непременным источником энергетических изменений
при ориентация как аморфных, так и кристаллических полиме-
ров являютс^/внутримолекулярные (конформационные) измене-
ния. Ориентационная вытяжка стеклообразных полимеров явля-
ется вынужденной высокоэластической деформацией, разви-
вающейся ниже Тс под действием высоких локальных напря-
жений. В отличие от высокоэластичности, развивающейся выше
TQ, вынужденные высокоэластические по своей природе дефор-
мации стеклообразных полимеров не исчезают при Т<ТС после
снятия напряжений. Этому препятствует отсутствие сегменталь-
ной подвижности. В процессе вынужденной эластичности, так
же как и при обычной высокоэластичности, конформационная
энтропия уменьшается, и происходит изменение внутримолеку-
лярной конформационной энергии — ее увеличение или умень-
шение в зависимости от знака: T(d \n<r2>0/dT) (см. с. 129).
Таким образом, можно утверждать, что ориентированный стек-
лообразный полимер обладает, по меньшей мере, как скрытой
конформационной энергией, так и скрытой конформационной
энтропией. Эти компоненты при Т>ТС являются движущей си-
лой сокращения ориентированного полимера, который выше Тс
возвращается в исходное неориентированное состояние.
В отличие от ориентации при Т>ТС теплота, рассеиваемая
при растяжении стеклообразного полимера, состоит из двух со-
ставляющих: теплоты, обусловленной уменьшением конформа-
ционной энтропии при ориентации, и теплоты, связанной с пре-
одолением межмолекулярного трения. Большая часть работы
ориентации стеклообразного полимера расходуется именно на
преодоление межмолекулярного трения. Эта часть теплового
эффекта является полностью необратимой. Напротив, теплота,
обусловленная уменьшением конформационной энтропии, явля-
ется обратимой частью теплового эффекта ориентации. Разде-
лить суммарный тепловой эффект ориентации на эти две со-
ставляющие на основании лишь данных по энергетическому ба-
лансу ориентационной вытяжки нельзя. Однако оценить их со-
отношение позволяет совместное рассмотрение энергетического
баланса ориентации в стеклообразном и высокоэластическом
состоянии.
С целью визуализации этой возможности проведем мыслен-
но следующий эксперимент с идеальным энтропийно-упругим
эластомером. Подвергнем его ориентационной вытяжке ниже
Тс и полученный стеклообразный ориентированный полимер в
214
свободном состоянии мгновенно поместим в условия Т>ТС.
Образец, перейдя в высокоэластическое состояние, сократится
до первоначальных размеров (эффектом обычного теплового
расширения твердых тел пренебрегаем). Поскольку образец
сокращается без совершения механической работы, тепловой
эффект сокращения будет равен нулю. Свободную энергию со-
кращения можно определить, растянув образец при температу-
ре сокращения до его первоначальной длины. Очевидно, что
Wt>tq =—TAS<^Wt<tq - Поэтому в первом приближении обрати-
мая часть рассеиваемого при ориентационной вытяжке тепла
может быть приравнена тепловому эффекту растяжения до
необходимой длины при Т>ТС. Типичные значения работы
и тепловых эффектов растяжения некристаллизующихся нена-
полненных эластомеров до составляют несколько Дж/г.
Таким образом, обратимая часть составляет лишь малую долю
рассеиваемого при ориентационной вытяжке тепла.
В общих чертах эти соображения об изменении конформа-
ционной энтропии и конформационной энергии при ориентаци-
онной вытяжке справедливы и для аморфных областей кристал-
лических полимеров.
В ориентированных твердых полимерах определенная доля
работы ориентации может быть связана с возникновением внут-
ренних напряжений. Такие напряжения могут очень медленно
релаксировать, и эти релаксационные процессы будут сопрово-
ждаться рассеянием энергии внутренних напряжений и перехо-
дом ориентированного полимера в более метастабильное состоя-
ние.
Примером может служить рассмотренное выше опреде-
ление скрытой энергии ориентации ПВХ. Количественно оценить
долю скрытой энергии, связанной с внутренними напряжения-
ми, можно, по-видимому, изучая кинетику возврата по данным
теплот растворения. Непосредственно, экспериментально, опре-
делить теплоту, рассеивающуюся при релаксации внутренних
напряжений, чрезвычайно трудно.
При ориентации кристаллических полимеров в кристаллитах
наблюдаются все элементарные процессы, характерные для
пластической деформации низкомолекулярных тел: образова-
ние и движение дислокаций, скольжение по кристаллографиче-
ским плоскостям, двойникование, рекристаллизация *[’17]. Одна-
ко эти процессы вряд ли могут давать такой же вклад в скры-
тую энергию ориентации полимеров, какой они могут вносить
и вносят в скрытую энергию деформации низкомолекулярных
тел. Это связано прежде всего с тем, что ориентационная вы-
тяжка кристаллических полимеров обычно происходит при зна-
чении 77Тпл>0,6, которое тесно связано с соотношением тем-
пературы плавления и стеклования кристаллического полимера
(777’пл~0,66), поскольку ориентационная вытяжка кристалли-
ческих полимеров, как правило, возможна лишь при Т>ТС. Зна-
215
чительно ниже Тс кристаллические полимеры хрупко разруша-
ются. Из исследований энергетики пластической деформации
известно {1—3], что при высоких значениях относительных тем-
ператур ТПпл скрытая энергия деформации оказывается очень
малой или вообще равна нулю. Это обусловлено одновремен-
ным протеканием процессов упрочнения и возврата металлов
в таких условиях. Именно в связи с этим можно полагать, что
роль и доля пластической деформации кристаллитов в скрытой
энергии ориентации кристаллических полимеров несуществен-
ны, несмотря на то что соотношение АЯскр и W для кристалли-
ческих полимеров может находиться в той же области, что и
для низкомолекулярных твердых тел.
Ориентационная вытяжка твердых полимеров сопровожда-
ется образованиём новой поверхности (трещины серебра, крей-
зы, фибриллизация). На образование этой поверхности расхо-
дуется определенная часть работы ориентации. Однако этот
энергетический вклад не может быть большим. Так, энергия
фибриллизации (образование поверхности микрофибрилл) мо-
жет достигать в самом лучшем случае нескольких Дж/см3.
Образование шейки при ориентационной вытяжке твердых
полимеров сопровождается разрывом макромолекул, что фик-
сируется по появлению свободных радикалов [18]. Однако чис-
ло разрываемых в процессе холодной вытяжки макромолекул
невелико и обычно не превышает 1018 г-1. Поэтому энергия, за-
траченная на образование свободных радикалов, а также на
образование новых концевых групп в результате механохимиче-
ских процессов, ничтожно мала, и ее вообще можно не прини-
мать во внимание. Вопрос об энергетике разрыва макромолекул
механическими напряжениями будет детально обсуждаться в
гл. 6.
^ лТаким обр_азом> иа.рассмотренных шести источников, измене-
ния энергетического состояния ориентированных кристалличе-
ских полимеров существенными могут, по-видимому, являться
лишь первые три, причем наиболее важным из них является
изменение степени кристалличности при ориентации. Для стек-
лообразных полимеров двумя основными вкладами в скрытую
энергию ориентации являются конформационные изменения и
внутренние напряжения.
Прежде чем переходить к анализу количественных резуль-
татов (см. табл. 5.1), целесообразно кратко рассмотреть совре-
менные представления о структурных изменениях и механизмах
ориентации кристаллических полимеров. В гл. 4 на основе дан-
ных о термоупругости ориентированных полимеров мы пришли
к выводу, что эти данные лучше всего согласуются с микрофиб-
риллярной моделью Петерлина — Превррзека/ в которой посту-
лировано существование микрофибрилл с чередующимися
кристаллическими и внутрифибриллярными аморфными облас-
тями и межфибриллярных аморфных областей, объединяющих
микрофибриллы в единую гетерогенную ориентированную струк-
216
туру. Структурные процессы, приводящие к образованию такой
фибриллярной структуры при ориентационной вытяжке, сводят-
ся к следующему (19—21].
На первой стадии растяжения исходная сферолитная струк-
тура, состоящая из пластинчатых кристаллитов — ламелей, со-
единенных между собой незначительным числом общих участ-
ков макромолекул, сначала деформируется упруго. Эта стадия
сопровождается увеличением объема и соответственно погло-
щением тепла. По мере увеличения деформации агрегаты ламе-
лей постепенно начинают поворачиваться и скользить друг от-
носительно друга. На этом этапе возможны также двойникова-
ние и фазовое превращение (переход решетка — решетка) ламе-
лей. Благодаря всем этим элементам пластической деформации
происходит подготовка ламелярной структуры к ее разрушению,
Образец начинает деформироваться упругопластически. Все
моды пластической деформации сопровождаются выделением
тепла, и потому пропорциональность на зависимости Q—е исче-
зает и наклон этой зависимости постепенно начинает меняться
на противоположный.
Разрушение кристаллической структуры начинается с рас-
калывания ламелей на отдельные блоки, соединенные участка-
ми макромолекул, и их поворота в направлении действия силы.
На этом этапе происходит также наклон и скольжение отдель-
ных цепей и потеря складчатой конформации у некоторых це-
пей. Дальнейшее растяжение сопровождается локальным обра-
зованием микрошеек и формированием в них микрофибрилляр-
ной структуры. Образование микрошеек сопровождается ло-
кальным повышением температуры, обусловленной работой
пластического разрушения ламелярной структуры. Роль локаль-
ных нагревов при образовании шейки в формировании фибрил-
лярной структуры будет подробно рассмотрена в следующем
разделе.
Схематическое изображение начальных этапов пластической
деформации и превращения стопки ламелей в зоне микрошейки
в пучок плотноупакованных параллельных микрофибрилл при-
ведено на рис. 5.10. Очевидно, что строение микрофибрилл при
таком механизме фибриллизации соответствует модели ХБ
(см. с. 181), поскольку кристаллические блоки со сложенными
цепями в микрофибриллах оказываются связанными между со-
бой ограниченным числом проходных цепей. Наряду с внутри-
фибриллярными аморфными прослойками с небольшой долей
проходных цепей в процессе формирования микрошеек и осо-
бенно при пластической деформации микрофибриллярной струк-
туры возникают межфибриллярные аморфные прослойки. Таким
образом, рассмотренная схема структурных превращений дей-
ствительно соответствует образованию фибриллярной структуры
в ориентированном кристаллическом полимере, отвечающей
модели Петерлина — Преворзека. Она свидетельствует о глу-
боких структурных перестройках исходной сферолитной струк-
217
Рис. 5.10. Схематическое изображение
процесса превращения стопки ламеляр-
ных кристаллитов в зоне микрошейки в
пучок плотноупакованных микрофибрилл
(по Петерлину):
1 — кристаллические блоки; 2 — пучок микро-
{)ибрилл; 3 — зона микрошейки; 4 — стопка ла-
мелей.
туры на всех уровнях надмолекулярной организации. Однако
она не предполагает полного плавления исходных кристаллитов
в момент образования микрошейки и формирования фибрилляр-
ной структуры из образовавшегося расплава (рекристаллиза-
ция). Такое предположение широко распространено в литера-
туре для объяснения механизма ориентационной вытяжки крис-
таллических полимеров [[21]. Оно базируется на температурных
эффектах при ориентации (локальное повышение температуры,
понижение температуры плавления кристаллитов) и также бу-
дет рассмотрено в следующем разделе.
Рис. 5.10 позволяет представить причины изменения степени
кристалличности при ориентационной вытяжке. Вытяжка при
комнатной температуре, т. е. в достаточно жестких условиях,
скорее всего будет сопровождаться уменьшением степени крис-
талличности, так как^ определенная доля раздробленных кри-
сталлитов не сможет восстановиться из-за ограниченной под-
вижности. Это должно быть особенно характерно для полиме-
ров с высокой исходной степенью кристалличности, а также
для полимеров, температура стеклования которых близка к
температуре вытяжки. При повышенных температурах вероят-
ность восстановления кристалличности до исходного уровня
резко возрастает. Более того, в таких условиях степень кри-
сталличности после ориентаций может, вероятно, и возрасти.
Как понижение, так и повышение степени кристалличности при
ориентации наблюдалось для многих кристаллических полиме-
ров (см., например, [22, с. 444—452; 21, с. 201]).
Если предположить, что основной вклад в скрытую энергию
ориентации вносит изменение степени кристалличности, то дан-
ные табл. 5.1 дают основание для следующего заключения. Для
всех представленных в таблице кристаллических полимеров
степень кристалличности при ориентации понижается. Величи-
218
на этого понижения составляет для ПЭВП — 6,5%, ПЭНП —
2,5%, ПП—'8%, ПКА— 10%, ПБТФ— 4,0%. Эти оценки сдела-
ны на основании соотношения АНскр/ЛН*пЛ (где АН* пл — тепло-
та плавления полностью кристаллического полимера). Эти ре-
зультаты согласуются с представлением о более глубоком по-
нижении степени кристалличности полимеров с температурами
стеклования, близкими к температуре вытяжки (ПКА, ПБТФ,
ПП), и с более высокой степенью кристалличности (ПЭВП и
ПЭНП). Понижение степени кристалличности ПКА примерно
на 7% при вытяжке (60 °C) зафиксировано, например, по дан-
ным измерения плотности [23], что совпадает с приведенным
выше значением при учете различия в температурах вытяжки.
Сравнение энергетического состояния ориентированных и не-
ориентированных кристаллических полимеров методом теплот
растворения затруднено, так как они нерастворимы при обыч-
ных температурах. Существует еще один путь, связанный с
определением теплоемкости. Так как Cp = dHldT, то, поскольку
А^ор#= АЯи.р, Ср.ор=И=Ср.нр. Однако для практической реализации
этого подхода необходима чрезвычайно высокая точность опре-
деления теплоемкости, достижение которой осложнено мета-
стабильным состоянием твердых полимеров и протеканием в
них релаксационных процессов при изменении температуры.
Поэтому целесообразно также сравнить поведение Ср ориенти-
рованного и неориентированного полимеров в широкой области
температур, включая области переходов, в частности плавление
кристаллических и стеклование аморфных полимеров.
Идея анализа энергетических изменений при ориентации
кристаллических полимеров на основании сравнения энтальпий
ясна из схемы, приведенной на рис. 5.11. Она предполагает, что
расплавы ориентированного и неориентированного полимеров
идентичны и термодинамически равновесны. Изменение энталь-
пии с температурой для ориентированного полимера может,
например, идти двумя различными путями. Первый из этих
путей (кривая 2) соответствует предположению, что А/7Скр це-
ликом обусловлена уменьшением степени кристалличности и
при измерениях в ориентированном полимере не происходят ни-
какие процессы. В этом случае А/7Пл.ор+А/7скр=АНПл.ир. Если
же ДЯскр обусловлена не только уменьшением степени кристал-
личности, но и другими вкладами (внутренние напряжения,
конформационные изменения
219
Ср,Цж/(гК)
Рис. 5.12. Температурная зависимость теплоемкости неориентированного (пунк-
тир) и ориентированного (сплошная линия) ПЭВП, полученная методом ска-
нирующей калориметрии [9, 10].
(рекристаллизация и реорганизация кристаллитов, дополни-
тельная кристаллизация, релаксация внутренних напряжений),
то зависимость энтальпии от температуры может характери-
зоваться кривой, отличной от кривой 2. Один из возможных пу-
тей (кривая 3) может, по-видимому, привести к ЛЯПл.ор>
>Япл.нр, несмотря на то, что Д//Скр>0.
Сравнительное изучение температурной зависимости тепло-
емкости некоторых ориентированных и неориентированных по-
лимеров [9, 10] показало, что их поведение отличается, причем
это отличие наблюдается, начиная с температур несколько бо-
лее высоких, чем температура ориентационной вытяжки. Ти-
пичная зависимость показана на рис. 5.12. Во всей температур-
ной области, кроме области плавления, зависимости гладкие и
не содержат видимых областей аномального изменения. Это
подтверждает сделанное выше заключение об отсутствии замет-
ного вклада в скрытую энергию ориентации, связанного с созда-
нием дефектов в кристаллической решетке.
На аналогичных кривых для многих пластически деформи-
рованных металлов задолго до плавления наблюдаются экзо-
термические пики, соответствующие выделению скрытой энергии
деформации (1—3]. Точность определения теплоемкости поли-
меров в этих исследованиях не была столь высокой, как это
требуется для детального сравнения зависимости энтальпии от
температуры ориентированных и неориентированных образцов.
Поэтому сравнение было ограничено областью плавления. Тер-
модинамические характеристики плавления и значения скрытых
энергий ориентации приведены в табл. 5.2. Они показывают,
что температурная зависимость энтальпии ориентированных
полимеров действительно может иметь качественно различный
характер. Судя по теплоте плавления, степень кристалличности
•ориентированного ПП на 9% ниже, чем неориентированного,
и это уменьшение достаточно хорошо соответствует скрытой
энергии ориентации. Такое поведение отвечает кривой 2 на
рис. 5.11. С другой стороны, у полиэтиленов рассчитанная по
теплотам плавления,степень кристалличности ориентированных
образцов выше, чем у неориентированных. Это означает, что
220
Таблица 5.2. Термодинамические характеристики плавления
ориентированных и неориентированных полимеров [9, 10}
Полимер Гпл- к ДНпл- Дж/г W (определена по теплоте плавления) Д«пл. нр- Д^пл. ор’ Дж/г д^*скр’ Дж/г
пэвп
неориентирован- . 405 177,5 0,60
ный —12,6 15,9
ориентиров энный 411 190 0,65
ПЭНП
неориентирован- ный 385 118,3 0,40 —14,7 6,3
ттп ориентированный 385 133,0 0,45
неориентирован- 438 85,7 0,60
НЫЙ ориентированный 440 72,4 0,51 13,3 10,1
ПБТФ**
неориентирован- ный 224 54,5 ) Одинаковая 0 5,3
ориентированный 224 55,0 J
* Соответствует скорости, при которой проводили ориентацию
СР-
** Данные получены в последнее время в лаборатории автора.
образцов для измере-
изменение энтальпии в этом случае, по-видимому, соответствует
кривой 3.
Повышение температуры и теплоты плавления при ориента-
ции ПЭ, установленное также в ряде исследований [24—26],
объясняется пониженными энтальпией и энтропией аморфных
областей [27]. Это понижение связывают с высокой ориентацией
цепей в аморфных областях. Плотность аморфных областей
ориентированных ПЭ предполагается более высокой, чем плот-
ность, полученная экстраполяцией значений, характерных для
расплава. Поэтому аморфные области ориентированного ПЭ
могут давать существенный вклад в теплоту плавления.
Совместное рассмотрение теплот плавления и скрытой энер-
гии ориентации ПЭ приводит к выводу, что для ПЭ, по-видимо-
му, возможно существование механизмов поглощения энергии
при ориентации, не связанных с понижением степени кристал-
личности. Одним из них может быть возникновение внутренних
напряжений при вытяжке. Так, установлено [28], что уровень
внутренних напряжений в ПЭ, подвергнутом холодной вытяж-
ке, значительно выше, чем в исходной сферолитной структуре,
причем эти повышенные напряжения (20—30 МПа) локализо-
ваны в аморфных областях. Значение внутренних напряжений
хорошо коррелирует с значением усадки при термообработке
ориентированного ПЭ. Очевидно, что в процессе нагрева ориен-
тированного ПЭ в нем могут протекать процессы релаксации
221
этих внутренних напряжений, сопровождающиеся понижением
энтальпии с одновременным уплотнением аморфных областей,
что в результате может привести к зависимости 3 на рис. 5.11.
Хотя эти предположения требуют детальной эксперимен-
тальной проверки, их правдоподобность видна из данных о
ПБТФ (см. табл. 5.2). Теплоты плавления исходного и ориенти-
рованного образца совпадают, что свидетельствует о неизмен-
ности степени кристалличности при ориентации. В то же время
скрытая теплота ориентации составляет вполне ощутимую ве-
личину. Неизменность степени кристалличности позволяет от-
нести ее к внутренним напряжениям аморфных областей. В про-
цессе нагревания эти напряжения могут полностью отрелакси-
ровать, так что в области плавления энтальпии ориентирован-
ного и неориентированного образцов окажутся одинаковыми.
Скрытая энергия ориентации стеклообразных полимеррв, в
общем, невелика (см. табл. 5.1 )7"“°Т5гсидвным’ 'вкладом в нее
являются, вероятно, конформационные энергетические измене-
ния и внутренние напряжения. Последние будут с возрастаю-
щей скоростью релаксировать по мере приближения к темпера-
туре стеклования, что при измерениях теплоемкости может
выразиться в неравномерности хода Ср. Эта неравномерность
может усугубляться усадкой образца. Мюллер (29] специально
исследовал этот вопрос калориметрически на примере ориенти-
рованного ПС и показал, как на основе зависимости Ср—Т
можно проследить релаксацию скрытой энергии. При свобод-
ной усадке энергия конформационных превращений может ли-
бо суммироваться с энергетическими эффектами релаксации
напряжения, либо эти эффекты могут взаимно компенсировать-
ся. Достаточно резких изменений теплоемкости можно ожидать
в области стеклования, где усадка может протекать быстро.
Однако эти эффекты трудно отделить от скачкообразного повы-
шения теплоемкости, обусловленного появлением сегментальной
подвижности. Поэтому для стеклообразных полимеров более
простым является метод теплот растворения.
В отличие от первичной ориентации переориентация под раз-
личными углами протекает без поглощения энергии. Согласно
рентгеноструктурным исследованиям переориентации ориенти-
рованного ПЭ исходные стопки кристаллитов за счет поворотов
и скольжений с незначительными разрушениями переходят во
вновь образующуюся фибриллярную структуру переориентиро-
ванного образца {30, 31]. Возможность поворотов и согласован-
ных скольжений возникает, вероятно, благодаря наличию меж-
фибриллярных и внутрифибриллярных аморфных областей,
обусловливающих необходимую подвижность кристаллитов.
Однако относительно переориентации также существует пред-
положение о полном плавлении исходной фибриллярной струк-
туры и кристаллизации полученного расплава в новую фибрил-
лярную структуру.
222
ТЕМПЕРАТУРНЫЕ ЭФФЕКТЫ ПРИ ОРИЕНТАЦИИ
ТВЕРДЫХ ПОЛИМЕРОВ
Диссипация механической энергии при ориентации твердых по-
лимеров сопровождается температурными эффектами. В общем
случае величина этих эффектов определяется соотношением
диссипирующей энергии и характером теплообмена пластически
деформируемой системы с окружающей средой. Качественная
иллюстрация тепловых явлений при пластической деформации
твердых тел может быть проведена на простой модели (22, с.
429]. Рассмотрим энергетический баланс процесса деформиро-
вания твердого цилиндра длиной I и диаметром d, температура
боковой поверхности которого равна температуре окружающей
среды Т оо (рис. 5.13). Через боковую поверхность протекает
теплообмен с окружающей средой. Между отдельными частями
цилиндра через слои толщиной б/ с теплопроводностью % воз-
можен теплообмен вследствие теплопроводности. Исходное рас-
пределение температур в образце равномерно и соответствует
средней температуре Т. Деформация со скоростью ds/dt осу-
ществляется растягивающим напряжением о, приложенным к
торцам цилиндра.
Энергетический баланс процесса деформирования этой моде-
ли может быть записан в следующей форме:
Г (dT\(dW dU\
Р P\dtj \ dt dt J \ dt + dt I
где p —• плотность.
(5.5)
Первый член в правой части уравнения представляет дисси-
пирующую часть работы деформации, а второй — потери тепла
вследствие конвекции и теплопроводности соответственно. Та-
ким образом, изменение средней температуры образца опреде-
ляется скоростью диссипации и интенсивностью теплообмена.
Очевидны два предельных случая. Если скорость диссипации
энергии очень велика, так что скоростью теплообмена можно
пренебречь, то деформирование будет протекать в адиабатиче-
ских условиях, для которых уравнение (5.5) может быть пред-
ставлено в виде
pCpdT —
о
(5.6)
В этом случае температура Т образца зависит только от де
формации.
Рис. 5.13. Схема распределения тем-
ператур в модельной системе, подвер-
гающейся ориентации (объяснения см.
в тексте).
223
В другом предельном случае, когда скорость теплообмена
очень велика по сравнению со скоростью диссипации энергии,
уравнение (5.5) принимает вид
г (ат\ (dQa ,dQ*\ z[-
\ at / \ at at )
В установившемся режиме
Т «= const = Тж (5.8)
т. е. образец деформируется при постоянной температуре, рав-
ной температуре окружающей среды.
Условия деформации, близкие к адиабатическим, будут реа-
лизоваться при очень высоких скоростях деформирования дос-
таточно толстых образцов, особенно при малой общей поверх-
ности теплообмена. Изотермические условия могут быть достиг-
нуты при очень медленных скоростях ориентации, особенно при
равномерном растяжении (без шейки) образца в средах с вы-
сокими коэффициентами теплоотдачи. В большинстве же ре-
альных случаев условия деформирования отличаются от этих
двух предельных случаев, поскольку ни скоростью теплообмена,
ни скоростью диссипации энергии нельзя пренебречь.
Особый интерес с позиции температурных эффектов пред-
ставляет ориентация путем развития шейки. В этом случае вы-
деление энергии локализовано в малом объеме, что должно со-
провождаться значительными локальными разогревами. Имен-
но проблема локальных разогревов в области шейки при дефор-
мации твердых полимеров стимулировала поиски эксперимен-
тальных путей регистрации температур в зоне образования
шейки и привела к ряду гипотез и теорий, объясняющих ориен-
тационную вытяжку стеклообразных и кристаллических поли-
меров.
Равномерная вытяжка по всей длине образца или ее лока-
лизация в зоне шейки представляет, очевидно, два крайних
случая диссипации энергии с точки зрения соотношения меха-
низмов теплообмена. При однородной деформации теплообмен
осуществляется в основном путем конвекции на всей поверх-
ности образца. Это соответствует определяющей роли Qa в
уравнении (5.5). При ориентации путем образования и развития
шейки теплообмен (вследствие теплопроводности от шейки к не-
деформированным частям образца) может оказаться фактором,
контролирующим диссипацию энергии, а также размер зоны
шейки [22, 32, 33]. Модель, представленная на рис. 5.13, позво-
ляет оценить роль теплопроводности в общем теплообмене для
случая локализации деформации. Относительный вклад тепло-
проводности в общий теплообмен согласно уравнению (5.5)
равен [22].
Qa + @х « ,
I+ nd
224
где а* — коэффициент конвективного теплообмена (а также излучения).
Считая §l = d для типичных значений х = 0,2 Вт/(м-К) и
а* = 50 Вт/(м2-К) (теплоотдача в газовую фазу), для равномер-
ной ориентации образца длиной /=100 мм имеем -я Л . <
<2%. При тех же параметрах теплообмена для короткой зоны
ориентации (/=1 мм) доля теплопроводности в общем теплооб-
мене составляет 0,67. Это означает, что при ориентации в воз-
душной среде, отвод тепла из зоны шейки путем теплопровод-
ности по образцу является решающим механизмом, теплообмена.
При вытягивании полимеров в жидких средах |[а*«103 Вт/
/(м2-К)], даже при локализации деформации в зоне шейки, ре-
шающим оказывается конвективный теплообмен, а теплопро-
водность вдоль образца играет второстепенную роль.
Равномерное распространение шейки в твердых полимерах
Проблема локального разогрева полимеров при ориентационной
вытяжке возникла в начале 50-х годов в связи с представле-
ниями о последовательном размягчении при ориентации стекло-
образных полимеров. Пытаясь разрешить ряд противоречий в
развиваемой в то время Мюллером теории холодной вытяжки,
основанной на представлении о сосуществовании двух фаз
(изотропной и ориентированной), Мюллер и Екель [34, 35]
предположили, что выделяющегося при ориентации тепла дос-
таточно для локального разогрева стеклообразного полимера
до температуры, которая может превышать температуру стек-
лования. В соответствии с этой гипотезой в узкой области фор-
мирования шейки происходит локальный разогрев материала
выше температуры стеклования, так что в каждый данный мо-
мент ориентации подвергается фактически не твердый полимер,
а полимер, находящийся в высокоэластическом состоянии. Это
тепловыделение происходит последовательно, обеспечивая пос-
ледовательность размягчения. Подсчет, выполненный для ПВХ,
исходя из предположения адиабатичности процесса превраще-
ния работы в теплоту в зоне шейки, привел к выводу, что вы-
деляющегося тепла должно быть достаточно для нагрева образ-
ца с 20 до 90 °C, т. е. выше Тс ПВХ. Так возникла гипотеза об
адиабатическом разогреве при вытяжке стеклообразных поли-
меров с образованием шейки.
Вслед за Мюллером и Екелем идея об адиабатическом разо-
греве была использована Маршаллом и Томпсоном [32] для
объяснения закономерностей ориентации стеклообразного
ПЭТФ. Проведенные ими расчеты позволяют количественно
проиллюстрировать роль адиабатического разогрева при вы-
тяжке. Основой расчетов являлись деформационные кривые,
полученные в условиях, близких к изотермическим (рис. 5.14).
Затем предполагалось, что при высоких скоростях деформиро-
15—440 225
Рис. 5.14. Кривые растяжения ПЭТФ прн
различных температурах (а) н зависи-
мость температуры образца при адиаба-
тическом растяжении (б) [32, 4] :
1 — 20 °C; 2 — 40; з — 60; 4 — 80; 5 — 100; 6 —
140 °C; 7 — зависимость, полученная расчетом
для адиабатических условий при начальной
температуре 20 °C.
вания ориентация протекает в
адиабатических условиях. Адиа-
батическая деформационная кри-
вая рассчитывалась в предполо-
жении, что вся работа превра-
щается в теплоту. В результате
расчета была получена адиаба-
тическая кривая растяжения
(кривая 7 на рис. 5.14), наклон
которой практически от началь-
ных степеней удлинения до пол-
ного перехода в ориентированное
состояние оказывается отрица-
тельным. Это соответствует неустойчивому протеканию ориен-
тации. В действительности даже при достаточно высоких скоро-
стях процесс ориентации протекает достаточно устойчиво.
iB связи с этим было сделано предположение, что эта устой-
чивость достигается за счет передачи тепла из области шейки
в граничную с ней неориентированную область. Приведенные
выше оценки роли теплопроводности по образцу показывают,
что такая возможность при вытяжке на воздухе существует.
Рассчитанные изменения температуры в зоне шейки в зависи-
мости от кратности вытяжки позволили оценить размер обла-
сти, в которой происходит формирование шейки при холодной
вытяжке ПЭТФ.
? Представление об адиабатическом разогреве было перенесено
: и на кристаллические полимеры [33, 35, 36], и возникла идея
о локальном плавлении кристаллитов под действием адиаба-
' тического разогрева и последующей кристаллизации образую-
щегося расплава при ориентирующем влиянии напряжения.
Гипотеза адиабатического разогрева инициировала много-
численные попытки экспериментального определения распреде-
ления температур вдоль образца, подвергаемого ориентацион-
ной вытяжке. Для регистрации скачка температуры применяли
три метода: термопарный, термоиндикаторный и метод регист-
рации ИК-излучения. Характерные результаты, полученные все-
ми тремя методами на различных полимерах, приведены на
рис. 5.15. Как ранние измерения, так и новейшие с использова-
нием ИК-излучения свидетельствуют о сильной зависимости
скачка температуры от скорости деформирования. Можно выде-
лить две области. При скоростях растяжения ниже 0,1 мм/с
226
скачки температур не превышают нескольких градусов незави-
симо от природы полимера и его фазового состава. Выше
0,1 мм/с скачок резко возрастает и теперь уже зависит от при-
роды полимера, его фазового состава и степени кристаллично-
сти. При скоростях деформирования, обычных для лаборатор-
ных исследований (<10 мм/с), скачки температур достигают
нескольких десятков градусов, но не превышают 60—70 °C.
В технологических процессах вытягивания волокон, когда ско-
рости вытяжки составляют I02—103 м/мин, скачки температур
могут достигать 100 °C и более. Таким образом, судя по приве-
денным на рис. 5.15 экспериментальным данным, саморазогрев
при вытяжке твердых полимеров может оказывать влияние на
процесс вытяжки лишь при скоростях деформирования выше
0,1 мм/с.
Возможность ориентационной вытяжки с образованием шей-
ки при малых скоростях деформирования, когда наблюдаемые
экспериментально скачки температур на 1—2 порядка меньше
тех, которые необходимы для перехода стеклообразного поли-
мера в высокоэластическое состояние или для плавления крис-
таллитов в кристаллических полимерах, позволила отвергнуть
гипотезу последовательного размягчения как основного меха-
низма образования шейки в твердых полимерах (5, 10, 39]. Для
этого были и другие основания. Так, эта гипотеза не могла
объяснить возникновения шейки в исходном однородно-растя-
гиваемом образце, когда не только отсутствует саморазогрев,
но и наблюдается охлаждение. Кроме того, необъяснимым
оставался такой фундаментальный экспериментальный факт
как то, что холодная вытяжка идет тем легче (при тем мень-
шем напряжении), чем меньше скорость растяжения, т. е. чем
ближе условия вытяжки к изотермическим.
Рис. 5.15. Зависимость скачка температуры от скорости деформации:
7 —ПП (И К-из лучение) [11]; 2 — ПП (термопара, ИК-излучение) [37J; 3 — ПК (ИК-излу-
чение) [11]; 4 — ПЭНП (термопара) [88]; 5 — ПЭТФ (расчет) [4, с. 271]; 5 — ПЭВП
(ИК-излучение) [11]; 7 — ПКА (ф—термопара, □ — термоиидикатор) |[8, 35]; 8 — стекло-
образная резина СКН-40 (термопара) [39]; 9 — полиамид 6.6 (термопара) [22, с. 432];
10 — ПВХ (термопара) [8, 35]; измерения для СКН-40 выполнены при —50’С, все осталь-
ные—при обычных температурах (20—40 "С).
15*
227
Расстояние по длине пленки, мм
Рис. 5.16. Распределение температуры
вдоль плеикн полиамида, подвергаемой
холодной вытяжке со скоростью 1 мм/с
[8]; сплошная линия — эксперименталь-
но зарегистрированное распределение;
пунктирная — теоретическое распределе-
ние, полученное на основании расчетов с
использованием данных энергетического
баланса.
Зафиксированные термопарой или термоиндикатором скач-
ки температур в области образования шейки при малых ско-
ростях составляют лишь несколько процентов от той величины,
которая может быть получена на основании анализа энергети-
ческого баланса. Действительно, если рассчитать повышение
температуры при ориентации, исходя из уравнения (5.6) и дан-
ных табл. 5.1, как это было сделано в свое время Мюллером
[7, 8], то, например, для ПВХ при скорости растяжения 0,3 мм/с
АТ = 69 °C, что примерно на порядок выше реально наблюда-
емого скачка. Такой расчет основан на идеализированной ади-
абатической схеме, когда предполагается что все тепло, гене-
рируемое при стационарном развитии шейки, сначала идет иа
повышение температуры граничной изотропной части образца
и лишь затем отводится в окружающее пространство и в дру-
гие области образца.
Недостаток такого расчета вполне очевиден в свете рассмот-
ренных выше соотношений механизмов теплообмена при вытяж-
ке. Тем не менее такое расхождение дало основание [8] поста-
вить вопрос о корректности измерений температур термопарой
или термоиндикатором. Этот вопрос становится вполне законо-
мерным, если считать, что зона локализации пластической де-
формации значительно меньше, чем размер термопары или зер-
на термоиндикатора (при регистрации скачков этими методами
существуют и другие экспериментальные трудности). Данные
о распределении температур вдоль пленки полиамида, зареги-
стрированном термоиндикатором и. полученном расчетом на
основе энергетического баланса, принципиально различаются
(рис. 5.16). Если такое расхождение действительно имеет место,
то оно может оказаться важным фактором в представлениях о
холодной вытяжке. Для решения этой проблемы существенными
оказываются результаты ИК-измерений (не требующих кон-
такта с образцом). Результаты, например, для ПП (см.
рис. 5.15), полученные двумя методами в независимых исследо-
ваниях, очень хорошо совпадают. Поэтому есть основания счи-
тать, что все используемые методы регистрации дают надежные
значения скачков температур, усредненные по поверхности с
228
линейными размерами в доли миллиметра. Что касается ло-
кальных разогревов в микрообъемах с линейными размерами в
десятки нм, то этот вопрос требует специального эксперимен-
тального изучения.
Несмотря на то что локальные разогревы не могут играть-
решающей роли в образовании шейки, они могут влиять на весь
процесс растяжения. Уорд [4, с. 269] проанализировал роль
локальных разогревов при холодной вытяжке стеклообразного
ПЭТФ и установил, что, как и для других полимеров, предел
текучести от линейно возрастает с логарифмом скорости дефор-
мирования в интервале скоростей 10-3—>10 с-1. В то же время
напряжение образования шейки ош меняется симбатно от толь-
ко при малых скоростях (10-1 с-1), а при более высоких ско-
ростях ош не возрастает с увеличением скорости растяжения,
а снижается. Это снижение объясняется разогревом, который
облегчает развитие ориентации. При больших скоростях про-
цесс деформирования все в большей степени приближается к
адиабатическому, и эффективная температура, при которой
происходит развитие шейки, возрастает.
Как уже отмечалось в применении к кристаллическим поли-
мерам, гипотеза адиабатического разогрева предполагает плав-
ление исходных кристаллитов и новую кристаллизацию с обра-
зованием кристаллитов, цепи в которых расположены в на-
правлении ориентирующего напряжения. Однако расчеты пока-
зывают f 1(9, 41], что даже в самых неблагоприятных условиях
генерируемого тепла при вытяжке при обычных температурах
недостаточно для плавления не только столь высокоплавких
полимеров, как ПКА (7’пл = 230°С) и ПП (7’Пл = 170°С), но даже
ПЭ (7’йл= 140°С). При этом существенно, что процесс ориента-
ционной вытяжки и связанные с ним структурные и морфоло-
гические превращения одинаковы в широком интервале скоро-
стей и температур. Но при малых скоростях локальные разогре-
вы столь малы, что вытяжку можно считать близкой к изотер-
мической. Поэтому локальный разогрев не может быть основ-
ной причиной рекристаллизации кристаллических полимеров
при ориентационной вытяжке. Тем не менее идея о рекристал-
лизации широко распространена и обычно используется для
объяснения структурных перестроек при ориентационной вы-
тяжке кристаллических полимеров [1, 7, 21]. В связи с этим
возникает вопрос о других возможных физических причинах
рекристаллизации.
Другая идея о рекристаллизации основана на представлении
о зависимости температуры плавления анизотропных кристал-
литов от их ориентации относительно действующих на них сил
[42, 43]. Считается, что при приложении растягивающих усилий
к анизотропному кристаллиту температура плавления должна
повышаться, если направление действия силы совпадает с на-
правлением осей макромолекул. Растяжение во всех других
направлениях должно сопровождаться понижением температу-
16—440
229
ры плавления. Предполагается также, что при растяжении тем-
пература плавления может достигать температуры, при которой
проводится вытяжка. Если деформирование осуществляется вы-
ше температуры стеклования аморфных областей, образующий-
ся при плавлении расплав вновь кристаллизуется, но уже наи-
более благоприятным по отношению к напряжению действия
силы образом. Такой механизм означает фазовый переход (ре-
кристаллизацию) под действием напряжений.
Гипотеза о фазовом переходе была постулирована без ка-
ких-либо количественных оценок. Поэтому немедленно возни-
кает вопрос, достаточны ли развиваемые при образовании шей-
ки усилия для необходимого понижения температуры плавле-
ния. Эти значения АГ не являются малыми и часто должны
составлять 150—200 (ПП, ПКА) и даже 300 °C (ПТФЭ). Такое
снижение температуры может быть обусловлено лишь значи-
тельными напряжениями, появление которых при обычных зна-
чениях От и Ош (20—50 МПА) не очевидно. Такие оценки были
впоследствии сделаны [44] в предположении, что деформация
аморфно-кристаллического полимера выше Тс, локализованная
в аморфных областях, осуществляется по высокоэластическому
механизму, что приводит к развитию таких напряжений на
торцевых неверностях кристаллитов, которые, по мнению авто-
ров, эквивалентны снижению гидростатического давления в
этой области кристаллита.
Использование уравнения Клапейрона — Клаузиуса, связы-
вающего изменение температуры плавления с изменением дав-
ления, показало, что для снижения температуры плавления ПЭ
и ПП до комнатной необходимы отрицательные давления по-
рядка нескольких сот МПа, т. е. примерно на порядок выше
От или ош- В связи с этим уместно заметить, что уравнение
Клапейрона — Клаузиуса предполагает зависимость Гпл от
истинно гидростатического давления и выполняется независимо
от размеров и ориентации кристаллитов*. Кроме того, в пре-
дыдущей главе показано, что аморфные области находятся в
сложнонапряженном состоянии при растяжении кристалличе-
ского полимера. Несомненно, что одноосное растяжение приво-
дит к возникновению сложнонапряженных состояний и в кри-
сталлитах. Поэтому неориентированный полимер не является
аналогией одноосно-растянутой сетки с возникшими в ней одно-
осно-ориентированными кристаллитами, для которой было по-
лучено' «одномерное» уравнение Клапейрона—Клаузиуса 1[45]
и которое использовалось при оценках. Наконец, наличие таких
больших отрицательных давлений должно сопровождаться тем-
пературными и тепловыми эффектами, отличными от тех, кото-
рые наблюдаются экспериментально. Поэтому такой подход
является упрощенным.
* Отрицательные гидростатические давления в зоне образования шейки
могут появляться вследствие особого распределения напряжений [33] нлв
взаимодействия микрофибрилл в зоне образования микрошеек [19].
230
Недавно проведены расчеты снижения температуры плавле-
ния, основанные на модели последовательного разгибания скла-
док в кристаллитах со сложенными макромолекулами (модель
Кобаяси [21]) |[46]. При таком механизме рекристаллизации
возможно, судя по оценкам для ПЭ, значительное снижение
температуры плавления кристаллитов со сложенными цепями
при значениях напряжений, сравнимых по порядку с от. Ре-
кристаллизация путем распрямления складчатых молекул мо-
жет привести лишь к возникновению микрофибрилл типа ГСХ
(см. рис. 4.27).
Важным аспектом образования шейки по механизму рекри-
сталлизации является кинетический аспект. Обычно рассматри-
вается такой случай, при котором 7’с<<7’<<7’Пл (ПЭ, ПП), когда
Т — комнатная температура, т. е. когда Т находится достаточно
близко к максимуму колоколообразной зависимости скорости
кристаллизации от температуры [47, 48]. В этом случае ско-
рость кристаллизации таких полимеров, как ПЭ и ПП, должна
быть чрезвычайно высокой, и потому плавление кристаллитов
и новая кристаллизация кинетически могут оказаться нераз-
личимыми. Однако для ПКА вытяжка легко осуществляется
при 7’^7’с (40°С), когда кристаллизация расплава должна
протекать чрезвычайно медленно и можно надеяться обнару-
жить существование расплава вблизи шейки. Однако наличие
аморфного состояния между вытянутой и невытянутой частями
в ПКА, а также в полиамиде 6,6 [22] обнаружить не удается.
Нами этот вопрос специально исследовался на примере
ПБТФ (7’пл = 224°С, 7’с = 40°С). Исходный неориентированный
полимер со сферолитной структурой и степенью кристалличности
<0,5 подвергался ориентационной вытяжке при комнатной тем-
пературе в деформационном микрокалориметре (термодинами-
ческие параметры ориентации приведены в табл. 5.1). В рекри-
сталлизационном механизме обычно предполагается, что, если
ориентация проводится при 7’<7’с, кристаллический полимер
аморфизуется. Единственным описанным в литературе случаем
является, по-видимому, аморфизация ПЭТФ [49]*. Аморфные
области неориентированного ПБТФ при температуре опытов
находятся ниже Тс (хотя, по-видимому, выше температуры
хрупкости). Тем не менее аморфизовать ПБТФ в процессе
ориентационной вытяжки невозможно. Рентгенограмма ориен-
тированного образца свидетельствует о хорошей кристалличе-
ской текстуре (рис. 5.17).
Исследования методом ДСК показали, что в ориентирован-
ных образцах отсутствуют кристаллизационные процессы, а
теплоты плавления исходного и ориентированного полимера
одинаковы (см. табл. 5.2), что свидетельствует об одинаковой
степени кристалличности.
* Неоднократные наши попытки воспроизвести подобный эксперимент не
увенчались успехом, так как образцы хрупко разрушались.
16* 231
Рис. 5.17. Рентгенограммы ориентиро-
ванных (при комнатной температуре)
полимеров:
— ПБТФ; б — ПЭТФ, вытянутый прн мед-
ленном, равномерном распространении шей-
ки; в — ПЭТФ, вытянутый в автоколеба-
тельном режиме.
Нам представляется, что приведенные результаты свидетель-
ствуют об отсутствии рекристаллизационного механизма ориен-
тационной вытяжки в таких полимерах, как ПБТФ, ПКА,
ПЭТФ при их деформировании вблизи Тс и ввиду общности
закономерностей при всех температурах ставят под сомнение
вообще возможность полного плавления исходной структуры и
новой кристаллизации. Такие сомнения высказывались еще в
работах Лазуркина [5, 39]. Совокупность всех возможных тем-
пературных эффектов может привести |[19, 20] лишь к повыше-
нию молекулярной подвижности в зоне образования шейки,
достаточному для разрушения ламелярной структуры путем
раскалывания на отдельные микроблоки и формирования из них
микрофибрилл (см. рис. 5.10). При этом, вероятно, возможно
разрушение отдельных наиболее дефектных кристаллитов и до
молекулярного состояния, что может привести к снижению сте-
пени кристалличности. Однако основная масса микроблоков,
по-видимому, не подвергается полной рекристаллизации и в
результате скольжений и поворотов переходит в микрофибрил-
лярные образования. Важная роль при этом может принадле-
жать кооперативному перемещению дислокаций [17].
Такой механизм ориентационной вытяжки приводит к опре-
деленным трудностям трактовки структурных превращений
(температурная зависимость большого периода 1[19—21]), одна-
ко он в большей степени, чем представление о полной рекри-
232
сталлизации, соответствует теплофизической картине ориента-
ционной вытяжки кристаллических полимеров. Наконец, такой^
механизм вполне соответствует наиболее общему представлению
о переходе как стеклообразных, так и кристаллических полиме-
ров в ориентированное состояние как о релаксационном явле- г
нии, обусловленном размягчением материала под действием | ,
приложенных напряжений и его упрочнением при ориентации \
[5, 39]. и
Автоколебательный режим распространения шейки
в твердых полимерах
Рассмотренные выше результаты выявляют теплофизические
особенности стационарного режима образования и развития
шейки при больших деформациях твердых полимеров. Однако
для некоторых твердых полимеров в определенных температур-
но-скоростных условиях ориентационная вытяжка проходит в
режиме, принципиально отличающемся от равномерного рас-
пространения шейки. В этом случае через определенный про-
межуток времени после прохождения через предел текучести
и выхода на напряжение ош шейка начинает распространяться
не равномерно, а регулярными скачками, причем напряжение
меняется во времени периодически (рис. 5.18). Явление перио-
дического распространения шейки было обнаружено для ПЭТФ
[50, 51] и ПКА {50, 33] и детально исследовано в работах
[12, 51] на примере ПЭТФ. Основной физической причиной по-
явления автоколебательного режима распространения шейки,
как отмечалось во всех указанных работах, является интенсив-
ный локальный саморазогрев. Рассмотрим, следуя [12, 51], за-
кономерности и критические условия возникновения автоколеба-
тельного режима, учитывая решающую роль локальных разо-
гревов. Совместный анализ кинетики изменения напряжения,
условной* тепловой мощности и истинной скорости перехода
полимера в шейку при различных режимах растяжения ПЭТФ
(рис. 5.19) позволяет следующим образом охарактеризовать это
явление.
Растяжение стеклообразного ПЭТФ с низкой скоростью
(<0,5 мм/с) сопровождается появлением и равномерным раз-
витием шейки. Локальный разогрев в области формирования
шейки при таких скоростях невелик и не превышает нескольких
градусов (см. рис. 5.15). Однако при определенном соотноше-
нии скорости растяжения и условий теплоотвода деформиро-
вание начинает принимать скачкообразный характер. Локаль-
ные разогревы, зарегистрированные термопарой и термоинди-
* Эта зависимость не является истинной зависимостью тепловой мощности
от времени из-за инерционности калориметрической ячейки. Тем не менее она
качественно подтверждает соответствие характера изменения тепловой мощно-
сти и механического напряжения.
О 10 20 30 t.c
Рис. 5.18. Возникновение автоколеба-
ний при растяжении стеклообразного
ПЭТФ с постоянной скоростью [51].
катером, при этом достигают многих десятков градусов. Изме-
нение режима распространения шейки сопровождается изме-
нением внешнего вида образца. При медленном, равномерном
развитии шейка прозрачна, при автоколебательном режиме
шейка характеризуется периодическими чередованиями проз-
рачных и непрозрачных участков, причем эта периодичность
соответствует периодичности возрастания и спада напряжения.
При дальнейшем повышении скорости растяжения (Vp>
>10 мм/с) автоколебательный режим опять сменяется равно-
мерным распространением шейки.
. В автоколебательном режиме образование шейки происхо-
дит в области уменьшающихся напряжений, причем скорость
развития шейки примерно на 1—2 порядка превышает среднюю
скорость растяжения. Предполагается, что решающим условием
осциллирующего развития шейки являются локальные разогре-
вы, которые в данном случае оказываются особенно велики из-
за резкого снижения напряжения. Следствием локального скач-
ка температуры является локальное возрастание податливости.
В момент резкого падения напряжения условия близки к ади-
абатическим, и дальнейшая деформация образца начинает осу-
ществляться за счет растяжения существующей шейки. Это
растяжение продолжается до тех пор, пока напряжение опять
не достигнет критического значения, при котором скорость пе-
рехода в ориентированное состояние (развития шейки) резко
возрастает, и предыдущий цикл вновь повторяется. Многократ-
ное повторение этих циклов приводит к автоколебательному
механизму распространения шейки.
Роль локальных разогревов в автоколебательном режиме
наглядно проявляется при изменении соотношения механизмов
теплообмена. Локальные разогревы при деформировании на
воздухе оказываются большими из-за второстепенной роли
конвективного теплообмена при локальном развитии деформа-
ции. Переход к деформированию в жидких средах (как пока-
зывают приведенные выше оценки) должен резко повысить
роль конвективного теплообмена даже при условии локализа-
ции деформации. Действительно, растяжение ПЭТФ в воде не
приводит к возникновению автоколебательного режима ни при
каких скоростях растяжения (51]. Высокий коэффициент теп-
лоотдачи в воду препятствует появлению больших локальных
разогревов, что исключает возникновение автоколебательного
режима.
Для появления автоколебаний кроме скорости деформиро-
вания и условий теплообмена важным фактором является по-
датливость образца (или всей системы нагружения), поскольку
от жесткости деформируемой части образца зависит, будет ли
он деформироваться или разрушаться. Для существования
автоколебательного режима требуется высокая податливость.
Этим определяется тот экспериментальный факт, что система
выходит на автоколебательный режим лишь спустя некоторое
время, за которое шейка развивается на определенную длину
и в течение которого податливость быстро возрастает и дости-
гает необходимого критического значения.
Таким образом, критические условия, при которых возникает I
автоколебательный режим распространения шейки в твердых I
полимерах, зависят от скорости деформирования, условий теп-«
лообмена и жесткости системы. '
Теоретически автоколебательный механизм распространения
шейки при растяжении твердых полимеров, основанный на
представлении о том, что автоколебания развиваются в резуль-
Рис. 5.19. Изменение напряжения (а), условной тепловой мощности (б) и ис-
тинной скорости перехода полимера в шейку (в) при различных режимах
растяжения i[12, 51]:
1 — стационарный режим растяжения с формированием прозрачной однородной шейки;
2 — автоколебательный режим с образованием в шейке чередующихся прозрачных и не-
прозрачных полос; 3 — нестационарный высокоскоростной режим, характеризующийся
формированием непрозрачной шейки и резко выраженным неустановнвшимся режимом
тепловыделения.
235
тате упругого деформирования образца и локальных разогре-
вов, был рассмотрен Баренблаттом [52]. Ранее им же [53] была
развита теория равномерного распространения шейки в твердых
полимерах при ориентации, основанная на аналогии этого явле-
ния с явлениями, относящимися к классу «самоподдерживаю-
щихся режимов» (типа распространения пламени). Подобно
равномерному распространению пламени, скорость которого
определяется локальной температурой и процессами переноса
(теплопроводность, массоперенос), равномерное распростране-
ние шейки определяется соотношением скоростей ориентацион-
ного превращения неориентированного полимера и скоростью
передачи деформации к неориентированным частям полимера.
На основании анализа баланса ориентированного материала в
зоне перехода в шейку получено уравнение, описывающее прев-
ращение полимера в шейку при равномерной вытяжке и имею-
щее решение лишь при определенном напряжении, действующем
в неориентированной области.
Эти идеи использованы и при построении теории автоколе-
бательного распространения шейки, однако в отличие от равно-
мерного изотермического распространения при этом рассматри-
вается неизотермическая модель превращения. Теоретический
анализ этой модели показывает, что при определенных соотно-
шениях податливости системы и теплообмена существует интер-
вал скоростей, в котором автоколебательное распространение
шейки действительно возможно.
Недавно в модель Баренблатта введены более общие соот-
ношения, определяющие переход неориентированного материа-
ла в ориентированный, и показано, что автоколебательный ре-
жим распространения шейки возможен и в изотермических
условиях [54]. При достаточно низких температурах, если ско-
рость диффузии напряжений превышает скорость диффузии
ориентированного материала, равномерное распространение
шейки оказывается нестабильным, и должен возникнуть авто-
колебательный режим.
Наряду с представлением о решающей роли локальных разо-
гревов в возникновении автоколебательного режима распро-
странения шейки в твердых полимерах возможно и другое
объяснение этого явления, основанное на представлении об
ориентационной кристаллизации. Рассмотрим сначала ряд кос-
венных данных. Использование термопары и термоиндикатора
позволило зарегистрировать скачок температуры в ПЭТФ при
скорости растяжения 0,7 мм/с, составляющий около 70 °C, при-
чем, судя по спектру, зафиксированному термоиндикатором,
этот скачок наблюдается в побелевших участках шейки (51].
Однако согласно рис. 5.15 при таких скоростях в условиях рав-
номерного распространения шейки локальные разогревы долж-
ны быть значительно меньше и не должны превышать 20—30 °C.
Поэтому можно предположить, что существует еще какой-то
источник локального разогрева, наблюдаемого при автоколеба-
тельном режиме при данной скорости деформирования. Таким
источником может являться теплота кристаллизации. Если это
так, то высокий локальный разогрев является не причиной по-
явления скачкообразного режима распространения шейки, а
его следствием. Качественным подтверждением наличия кри-
сталличности в побелевших участках пленки является сохране-
ние ими размеров при прогреве, в то время как прозрачные-
участки пленки способны к усадке (51]. Важным обстоятельст«
вом служит также тот факт, что осцилляционный режим рас-
пространения шейки может наблюдаться лишь в кристаллизую-
щихся полимерах (ПКА, ПП, ПЭТФ). Такие режимы не воз-
никают при ориентации не способных к кристаллизации стек-
лообразных полимеров, например ПК (И, 55], даже при
больших скоростях растяжения (1—10 мм/с), когда локальные
разогревы достигают многих десятков градусов.
Термодинамическим критерием ориентационной кристалли-
зации является ДЯскр<0. Как следует из табл. 5.1, при мед-
ленном, равномерном распространении шейки в ПЭТФ ДДСкр>0.
Нами был изучен энергетический баланс ориентационной вы-
тяжки аморфного стеклообразного ПЭТФ в автоколебательном
режиме распространения шейки при скорости растяжения
2 мм/с при комнатной температуре. Формирование режима бы-
ло близким к показанному на рис. 5.18. Отношение интеграль-
ного теплового эффекта ориентации к работе ориентации со-
ставляло —Q/IT=1,3—1,35, а ДНскр = —22 Дж/г. Таким обра-
зом, энергетический баланс ориентационной вытяжки свиде-
тельствует о частичной кристаллизации.
Прямым доказательством кристаллического характера об-
разцов, полученных в автоколебательном режиме, служат рент-
геноструктурные данные (см. рис. 5.17). При медленном, рав-
номерном вытягивании аморфного ПЭТФ образующаяся шейка
рентгеноаморфна и характеризуется аморфной текстурой, не-
однократно наблюдавшейся в различных исследованиях. Шейка,
образовавшаяся в автоколебательном режиме, характеризуется
кристаллической текстурой. Исследования, проведенные мето-
дом ДСК, также показывают, что полученные в автоколеба-
тельном режиме образцы являются частично-кристаллическими.
Теплота плавления таких образцов ДДпл = 48 Дж/г, что соот-
ветствует степени кристалличности около 0,4. Различие в
ДДпл и ДДскр может быть связано с компенсирующим эффектом
внутренних напряжений и с совершенствованием кристалличе-
ской текстуры в процессе измерений теплоты плавления. В свя-
зи с проблемой энергетического состояния ориентированного
ПЭТФ можно отметить что аморфная текстура ПЭТФ является
своего рода кристаллической заготовкой (10, 56], так как по-
давляющая часть звеньев в ней находится в транс-конформации
(см. с. 190). Таким образом, кристаллический характер образ-1
цов, полученных в автоколебательном режиме распространения!
шейки, доказан двумя прямыми независимыми методами.
Рис. 5.20. Зависимость скорости распространения
шейки от приложенного напряжения [55].
Пакула и Фишер [55] исследовали
возможность появления осциллирующего
режима при холодной вытяжке ПЭТФ,
ПП, ПКА и ПК не только при постоян-
ной скорости перемещения подвижного
зажима, но и при постоянном напряже-
нии. В условиях постоянной скорости
растяжения эти авторы также наблюда-
ли появление автоколебательного режи-
ма в ПЭТФ при 20 и 40 °C в области
скоростей деформирования, совпадающих с приводимыми в
работе [51]. Образцы, полученные в автоколебательном режи-
ме, были частично-кристаллическими. При температурах выше
60 °C автоколебательный режим не наблюдался ни при каких
скоростях деформирования, и полученные в таких условиях об-
разцы были прозрачными и рентгеноаморфными.
В режиме ориентации при постоянной нагрузке скорость
распространения шейки сильно зависит от приложенного на-
пряжения (рис. 5.20). Наиболее важным в этой зависимости
является разрыв в скорости распространения шейки в очень
узком интервале напряжений. При этом различие в скоростях
в двух областях — ниже и выше разрыва — составляет более
двух порядков. Связь этой закономерности с приведенной на
рис. 5.19 очевидна. Развитие шейки при напряжениях ниже
критических, как показывают оптические и рентгеноструктур-
ные исследования, приводит к возникновению прозрачных об-
разцов с аморфной текстурой, а растяжение при напряжениях
выше критических сопровождается образованием оптически
непрозрачного кристаллического материала (рентгенограммы,
приведенные в работе [55], практически идентичны показанным
на рис. 5.17). В работе [55] подчеркивается, что наличие крити-
ческих напряжений характерно лишь для ориентационных про-
цессов в кристаллизующихся полимерах, причем они могут
наблюдаться как ниже температуры стеклования (ПЭТФ), так
и выше (ПП). Для ПК автоколебательный режим распростра-
нения шейки осуществить не удалось ни при каких скоростях.
Неустойчивое (автоколебательное) распространение шейки
должно появляться при растяжении со скоростями, находящи-
мися в интервале между двумя стабильными режимами—'выше
и ниже критического напряжения. Специальные опыты показа-
ли, что неустойчивое распространение шейки может наблюдать-
ся и в том случае, когда локальные разогревы малы.
Таким образом, появляющийся при определенных условиях
в кристаллизующихся полимерах автоколебательный режим
распространения шейки может быть отнесен к категории дина-
238
мических переходов беспорядок — порядок, и по характеру
' проявления он подобен ориентационной кристаллизации. По-
• скольку локальная кристаллизация сопровождается локальным
выделением тепла, при автоколебательном режиме должны
наблюдаться дополнительные по сравнению с равномерным
распространением шейки локальные разогревы. Резкое сниже-
ние напряжения (см. рис. 5.19) можно объяснить локальной
кристаллизацией и связанным с ней локальным удлинением.
Это означает резкое уменьшение локальной скорости деформи-
рования и переход в область «докритического» устойчивого
режима. Затем напряжения постепенно возрастают из-за воз-
растания локальной скорости ориентации. Но, как только на-
пряжение перейдет в область «сверхкритического», опять про-
исходит локальная кристаллизация, и весь цикл повторяется.
При таком подходе в автоколебательном режиме должны суще-
ствовать два механизма локальных разогревов. Один, связан-
ный с локальным превращением работы пластической деформа-
ции в теплоту, вполне аналогичен локальным разогревам при
равномерном распространении шейки; другой —обусловлен
локальной кристаллизацией. Этот второй источник разогревов
должен быть более мощным. Полученное в работе [51] распре-
деление температур по зонам шейки не противоречит такому
представлению.
Явление скачкообразного развития пластической деформации характерно
для многих твердых тел, прежде всего металлов и сплавов при деформирова-
нии в области гелиевых температур [57, 58]. Одна из гипотез, объясняющих
скачкообразный характер деформации, основана на представлении о терми-
ческой нестабильности процесса деформации вследствие локального повыше-
ния температуры в момент скачкообразного развития пластической деформа-
ции. При гелиевых температурах саморазогрев может быть следствием очень
низких значений теплоемкости и теплопроводности. На основе гипотезы о ло-
кальных разогревах введен целый ряд критериев термомеханической неустой-
чивости низкотемпературной деформации, полученных для различных условий
протекания процесса (адиабатический и различные условия теплообмена с
окружающей средой) [59—61]. Однако многочисленные экспериментальные
исследования природы скачкообразного развития пластической деформации
твердых тел при гелиевых температурах приводят к выводу, что гипотеза
локальных разогревов не является универсальной и не позволяет объяснить
многие экспериментальные данные [57, 61]. В результате этих исследований *
утвердилось представление, что локальные разогревы появляются вследствие I)
падения напряжения, а не являются их причиной. Ч
ЛИТЕРАТУРА
1. Болыианина М. А., Панин В. Е. — В кн.: Исследования по физике твердого
тела. М., Изд-во АН СССР, 1957, с. 193—233.
2. Титченер Э. Л., Бевер М. Б. — В кн.: Успехи физики металлов. М.,
ГНТИЛЧЦМ, 1961. Сб. 4, с. 290—395.
3. Bever М. В., Holt D. L., Titchener A. L. — Progr. Mater. Sci., 1973, vol. 17,
p. 1—190.
4. Уорд И. Механические свойства твердых полимеров. Пер. с англ./Под ред.
А. Я- Малкина. М., Химия, 1975. 350 с.
5. Lazurkin Yu. S. — J. Polymer Sci., 1958, vol. 30, p. 595—604.
6. Надаи А. Пластичность и разрушение твердых тел. Пер. с англ./Под ред.
Г. С. Шапиро. М., Издатинлит, 1954. Т. 1, 647 с.
7. Engelter Ad., Muller F. H. — Koll.-Ztschr., 19158, Bd 157, N 2, S. 89—111.
8. Muller F. H. — In:*Rheology/Ed. by F. R. Eirich. N. Y. — London, Academic
Press, 1969. Vol. 5, p. 417—489.
9. Годовский Ю. К. Докт. дис. М., НИФХИ им. Л. Я- Карпова, 1971.
10. Годовский Ю. К. Теплофизические методы исследования полимеров. М.,
Химия, 1976. 216 с.
11. Maher J. IF., Haward R. N., Hay J. N. — J. Polymer Sci., Polymer Phys. Ed.,
1980, vol. 18. N 11, p. 2169—2179.
12. Андрианова Г. П., Попов Ю. В., Арутюнов Б. А. — Высокомол. соед., 1976,
сер. А, т. 18, № 10, с. 2311—2317.
13. Maron S. Н., Filisko F. Е. — J. Macromol. Sci., 1972, pt В, vol. 6, N 2,
p. 413—429.
14. Волынская А. В., Годовский Ю. К-> Папков В. С. — Высокомол. соед., 1979,
сер. А, т. 21, № 5, с. 1059—1063.
15. Slotting J., Muller F. Н. — Koll.-Ztschr. u. Ztschr. Polymere, 1970, Bd 238,
N 1—2, S. 459—470; 1970, Bd 240, N 1—2, S. 790—791.
16. Muller F. H., Engelter Ad. — Koll.-Ztschr., 1960, Bd 171, N 2, S. 152—153.
17. Вундерлих Б. Физика макромолекул. Пер. с англ. М„ Мир, 1976. Т. 1.
624 с.
18. Кауш Г. Разрушение полимеров. Пер. с англ./Под ред. С. Б. Ратнера. М.,
Мир, 1981. 440 с.
19. Peterlin А. — J. Mater. Sci., 1971, vol. 6, N 6, p. 490—508.
20. Peterlin A. — In: Ultra-High Modulus Polymers/Ed. by A. Cifferi, I. Ward.
London, Applied Science Publishers, 1979, p. 279—320.
21. Марихин В. А., Мясникова Л. П. Надмолекулярная структура полимеров.
Л., Химия, 1977. 238 с.
22. Зябицкий А. Теоретические основы формования волокон. Пер. с англ. М.,
Химия, 1979. 504 с.
23. Комков Ю. А., Шишкин Н. И. — Высокомол. соед., 1972, сер. Б, т. 14, № 4,
с. 295—298.
24. Peterlin A., Meinel G. — J. Appl. Phys., 1965, vol. 36, N 10, p. 3018—3022.
25. Fischer E. W., Hinrichsen G.— Koll.-Ztschr. u. Ztschr. Polymere, 1966,
Bd 213, N 1, S. 28—38.
26. liters K.-H.—Angew. makromol. Chem., 1970, Bd 12, N 1, S. 89—104.
27. Peterlin A. —J. Polymer Sci., 1967, pt C, N 18, p. 123—132.
28. Kubat J., Peterman J., Rigdahl M. — Coll. a. Polymer Sci., 1975, vol. 253,
N 5. p. 875—880.
29. Muller F. //.-Koll.-Ztschr., 1959, Bd 165, N 1, S. 96—107.
30. Герасимов В. И., Цванкин Д. Я.— Высокомол. соед., 1970, сер. А, т. 12,
№ 11, с. 2599—2609.
31. Генин Я. В., Герасимов В. И., Цванкин Д. Я. — Высокомол. соед., 1973,
сер. А, т. 15, № 8, с. 1798—1808.
32. Marshall I., Thompson А. В. — Proc. Roy. Soc., 1954, Ser. A, vol. 221, N 1147,
p. 541—557.
33. Hookway D. C. — J. Text. Inst., 1968, vol. 49, N 7, p. 292—316.
34. Mutter F. H„ Jacket K. — Koll.-Ztschr., 1952, Bd 145, N 2, S. 145—148.
35. Jacket K. — Koll.-Ztschr., 1954, Bd 137, N 2, S. 130—164.
36. Muller F. H. — Kunststoffe, 1954, Bd 44, N 6, S. 569—572.
37. Schwarz G. — Coll. a. Polymer Sci., 1981, vol. 259, N 2, p. 149—155.
38. Nakamura M., Skinner S. M. — J. Polymer Sci., 1955, vol. 18, N 2, p. 423—
426.
39. Лазуркин Ю. С. Докт. дис. M., ИФП АН СССР, 1954.
40. Vincent Р. I. — Polymer, 4960, vol. 1, N 1, р. 7—19.
41. Meinel G., Peterlin A.—J. Polymer Sci., 1971, pt A-Й, vol. 9, N 1, p. 67-—83.
42. Horsley R. A., Nancarrow H. A. — Brit. J. Appl. Phys., 1951, vol. 2, N з'
p. 345—349.
43. Каргин В. А., Соголова T. И.—ЖФК, 1953, т. 27, № 10 с. 1039—1042-
№-11, с. 1208—1212, 1213—1216.
44. Баранов В. Г., Френкель С. Я., Волков Т. И., Гаспарян К. А. — ФТТ, 1969.
т. 11, № 5, с. 1220—1227.
45. Flory Р. J. — J. Amer. Chem. Soc., 1956, vol. 78, N 20, p. 5222—5235.
46. Беляев О. Ф., Зеленев Ю. В. — Высокомол. соед., 1980, сер. А, т. 22, № 2,
с. 471—477.
47. Годовский Ю. К. — Высокомол. соед., 1969, сер. А, т. 11, № 10, с. 2129—
2135.
48. Вундерлих Б. Физика макромолекул. Пер. с англ. М., Мир, 1979. Т. 2,
574 с.
49. Козлов П. В., Кабанов В. А., Фролова А. А.—ДАН QCCP, 1959, т. 125,
№ 1, с. 118—121.
50. Muller F. Н., Engelter Ad. — Koll.-Ztschr., 1957, Bd 150, N 2, S. 156—157.
51. Андрианова Г. П., Каргин В. А., Кечекьян А. С.—Высокомол. соед., 1970,
сер. А, т. 12, № 8, с. 2424—2434; J. Polymer Sci., 1971, pt А-2, vol. 9, N 10,
p. 1919—1933.
52. Баренблатт Г. И. — Изв. АН СССР. Мех. тв. тела, 1970, № 5, с. 121 —
131.
53. Баренблатт Г. И. — Прикл. мат. и мех., 1964, т. 28, № 6, с. 1048—1060.
54. Matkowsky В. J., Sivashinsky G. I. — Quart. Appl. Math., 1979, vol. 37, N 1,
p. 23—34.
55. Pakula T., Fischer E. W. — In: 26th International Symposium on Macromo-
lecules, IUPAC, Mainz, 1979. Preprints. Vol. 3, p. 1405—1408.
56. Андрианова Г. П., Попов Ю. В., Артамонова С. Д., Арутюнов Б. А. — Вы-
сокомол. соед., 1977, сер. А, т. 19, № 6, с. 1230—1236.
57. Физические процессы пластической деформации при низких температурах/
/Отв. ред. В. И. Старцев и В. 3. Бенгус. Киев, Наукова думка, 1974. 384 с.
58. Клявин О. В. Докт. дис. Л., ФТИ им. А. Ф. Иоффе, 1975.
59. Малыгин Г. А. — Физика металлов и металловед., 1975, т. 40, № 1, с. 21 —
28.
50. Петухов Б. В. — ФТТ, 1977, т. 19, № 2, с. 397—401.
61. Estrin К, Kubin L. Р. — Scripta Metallurgica, 1980, vol. 14, N 12, p. 1359—
1364.
62. Иванченко Л. Г., Солдатов В. П. — Физика металлов и металловед., 1981,
т. 52, № 1, с. 183—188.
Глава 6
ТЕПЛОФИЗИКА РАЗРУШЕНИЯ ПОЛИМЕРОВ
Разрушение полимеров неизбежно сопровождается тепловыми
явлениями. Источниками тепловых эффектов, сопровождающих
разрушение, могут являться как деформационные процессы, так
и разрывы макромолекул. Чисто упругое (хрупкое) разрушение
твердых тел является исключением, а не правилом [1]. Обычно
образование первичных источников разрушения трещин сопро-
вождается локальными пластическими деформациями. Значи-
тельная доля работы, связанная с этими локальными пластиче-
скими деформациями, превращается в тепло, и следствием это-
го превращения являются локальные разогревы при прораста-
нии трещин в твердых материалах. В отличие от низкомолеку-
лярных твердых тел, для которых пластическая деформация в
зоне роста трещин является основным механизмом локальных
температурных изменений при разрушении, в полимерах воз-
можен еще и другой механизм рассеяния энергии, обусловлен-
ный цепным строением макромолекул. Разрыв упругонапряжен-
ной макромолекулы или достаточно длинного напряженного
241
участка макромолекулы по какой-либо одной из напряженных
связей должен неизбежно приводить к рассеянию энергии, за-
пасенной во всех остальных нагруженных связях. Этот процесс
также может являться источником локальных температурных
изменений.
Разрушение, обычно имеет локализованный характер, что не
всегда позволяет на основе макроскопических тепловых из-
мерений воссоздать картину тепловых процессов, протекающих
в местах локализации разрушения. Именно этим, по-видимому,
объясняется тот факт, что экспериментальные исследования
тепловых явлений, сопровождающих разрушение твердых тел, в
том числе и полимерных, стали проводиться лишь в последние
годы после развития высокочувствительных бесконтактных ме-
тодов регистрации тепловых процессов.
КИНЕТИЧЕСКИЙ ПОДХОД К ПРОБЛЕМЕ
ПРОЧНОСТИ ТВЕРДЫХ ТЕЛ. ЭНЕРГЕТИЧЕСКИЕ ЭФФЕКТЫ,
СВЯЗАННЫЕ С РАЗРЫВОМ МАКРОМОЛЕКУЛ
К настоящему времени сложились два основных подхода к
проблеме прочности твердых тел. Согласно одному из них —
статическому элементарные процессы разрушения протекают в
статически неподвижной матрице твердого тела под действием
внешних сил, сконцентрированных на различных дефектах
структуры. Такими дефектами — концентраторами напряже-
ния — могут являться микротрещины, скопления дислокаций,
инородные включения, границы зерен и т. п. При таком подхо-
де тепловое движение атомов не принимается во внимание, и
акты разрушения представляются как простое механическое
преодоление взаимодействия ближайших соседей. Проблема теп-
ловых явлений при таком подходе фактически не возникает.
В основе другого подхода — кинетического, в наиболее от-
четливом виде сформулированного Журковым [2], лежит пред-
ставление о разрушении как о термофлуктуационном процессе,
в котором решающая роль принадлежит тепловому движению
атомов твердого тела. В соответствии с кинетической концеп-
цией разрушение твердых тел под нагрузкой рассматривается
как процесс последовательного разрыва напряженных межатом-
ных Связей тепловыми флуктуациями [2, 3]. При таком подходе
долговечность нагруженного тела как фундаментальная харак-
теристика механической прочности отражает усредненную ско-
рость протекания разрушения, связанного с накоплением по-
вреждений в твердом теле. Систематические экспериментальные
исследования долговечности твердых тел различной природы,
в том числе и полимерных, привели к установлению основной
закономерности, связывающей напряжение, абсолютную темпе-
ратуру и долговечность т. В случае одноосного растяжения,
который только и будет рассматриваться далее, эта связь пред-
242
(6.1}
(6-2)
ставляется формулой Журкова {9J:
Uo — Ужа
т = т0 ехр —-
«Б'
где и0 — энергетическая константа материала, совпадающая по значению с
энергией распада межатомных связей; То—константа, равная периоду тепло»
вых колебаний атомов («10-13 с); у®— структурно-чувствительный коэффи-
циент.
Формула (6.1) позволяет представить реальную прочность,,
т. е. напряжение, разрывающее образец за время т, в виде
1 / т X къТ х
а. = — КД — khT In — = амакс —----- In —
Уж \ хо / Уж хо
Формула (6.2) наглядно показывает роль теплового движе-
ния в прочности твердого тела: при заданной продолжительно-
сти действия нагрузки прочность должна линейно снижаться
с ростом температуры. Экспериментальная проверка темпера-
турно-временной закономерности прочности, выражаемая фор-
мулами (6.1) и (6.2), проведенная на металлах, неорганических
стеклах, твердых полимерах, горных породах и др., подтвердила
справедливость этой закономерности и позволила, учитывая
значение и физический смысл параметров Uq и то, представить
механизм разрушения твердых тел как термофлуктуационный
процесс распада межатомных связей и возникновения первич-
ных разрывов сплошности — субмикро- и микротрещин [3, 4].
Наличие разрывов межатомных связей при разрушении
полимеров доказано в многочисленных работах прямыми физи-
ческими методами (ЭПР, ИК, масс-спектрометрия) [2, 3, 5].
Тем не менее количественная интерпретация экспериментальных
данных по разрушению твердых тел с позиции кинетической
концепции прочности обычно осложняется тем, что процесс
разрушения самым тесным образом связан с деформационными
процессами. Многолетняя дискуссия о соотношении этих про-
цессов дтри разрущении привела к двум противоположным точ-
кам зрения. Согласно точке зрения, изложенной выше, разрыв
межатомных связей и возникновение в результате этого первич-
ных очагов разрушения — субмикроскопических трещин — ос-
новное звено в многоэтапном процессе разрушения [3]. Другая^
точка зрения основана на представлении, что разрушение под-
готавливается пластической деформацией и является ее следст-
вием (6]. Эта неоднозначность порождена тем, что деформиро-
вание также обычно является термокинетическим процессом,
описываемым близкими по виду к уравнению (6.1) соотноше-
ниями, причем параметр Uq может быть сопоставлен с энергией
различных атомных перестроек,! сопутствующих пластической
деформации. Таким образом, согласно второй точке зрения
температурно-временная зависимость прочности твердых тел
обусловлена несовершенством их структуры и долговечность!
определяется временем перестройки этой структуры и парал-|
243-
Рис. 6.1. Схема растяжения межатомной
связи в ангармонической цепочке атомов.
дельного накопления повреждений. Ограничившись по очевид-
ным соображениям этим кратким изложением существа кине-
тических подходов к проблеме прочности твердых тел, рас-
смотрим теплофизические аспекты этой проблемы.
Вернемся к формулам (6.1) и (6.2) и обратим внимание на
тот факт, что, хотя эти формулы формально отражают роль
теплового движения в прочности, в них в явном виде никак не
•фигурируют фундаментальные теплофизические характеристики
теплового движения — теплоемкость (гармонические колеба-
ния) и тепловое расширение (ангармонические колебания).
Константы UQ и т0 в уравнениях имеют достаточно ясный физи-
ческий смысл. Что касается третьего параметра в уравнении
{6.1) —уж, то его действительный физический смысл долгое
время оставался неясным и он рассматривался как элементар-
ный флуктуационный объем, в котором под действием концент-
рации напряжений происходит разрыв связей.
В последнее время Журков [7, 8] высказал и обосновал но-
вую физическую идею относительно физического смысла коэф-
фициента уж, что позволило также уточнить смысл и значение
параметра Uo. Эта идея дала возможность ввести в уравнение
(6.1() такие тепловые характеристики, как теплоемкость, тепло-
вое расширение и параметр Грюнайзена. Рассмотрим сущность
этих новых идей и вытекающих из них представлений о молеку-
лярных явлениях, лежащих в основе разрушения твердых тел.
Физический смысл параметра уж удалось! выяснить, рас-
смотрев механизм термофлуктуационного разрыва напряженной
межатомной связи в одномерной модели, учитывающей энгар-
монизм межатомных колебаний [7]. Подобная модель уже ис-
пользовалась нами для анализа тепловых свойств (см. с. 106
и ПО). Воспользуемся также выражением для упругой силы,
учитывающим энгармонизм колебаний атомов в линейной це-
почке [уравнение (3.63)]:
f = k&a — g(bay> (6.3)
График этой зависимости (рис. 6.1) показывает, что мак-
симальная сила fth, достигаемая при удлинении Да*, характе-
ризует теоретическую прочность. При происходит атер-
мический разрыв связи, при fk<fth разрыв может произойти
только по термофлуктуационному механизму. В работе (7]
244
предполагается, что именно энгармонизм порождает дополни- fe
тельную флуктуирующую силу fg. Исходя из силового критерия У
разрыва связи
fk + fg=ftk (6.4)
флуктуационное удлинение связи Да, приводящее к разрыву,
определяется соотношением Да = Ла*—Да*.
Анализ вероятности флуктуаций силы приводит к зависимо-
сти от температуры и времени флуктуирующей силы fg
fg=(g!k)ksT 1п(т/т0) (6.5)
и внешней силы fk:
fk = ftk~(g/k)kETln(T/t0) (6.6)
Переходя к напряжению f*/a2o = xno (где о — среднее напря-
жение в теле, а хп — коэффициент перенапряженности связи),
получаем:
о = омакс—-4“ In ('/’,) (6.7)
Сравнивая (6.2) и (6.7), видим, что они совпадают при
Уж=(*а;/£)хп (6.8)
Таким образом, полученное описанным способом уравнение
долговечности твердого тела под нагрузкой указывает на то,
что термофлуктуационный разрыв связи обусловлен ангармо-
ничностью тепловых колебаний и определяется отношением
g/k. В гармоническом приближении (g=0) температурно-вре-
менная зависимость прочности должна отсутствовать. Восполь-
зовавшись соотношением Грюнайзена (3.11), постоянные g и
k, а следовательно, у® можно выразить через коэффициент
линейного теплового расширения 0, теплоемкость Cv и модуль
Юнга [8].
Параметр Грюнайзена
ga0
?=-—= —— (6.9)
к Су
откуда
?ж=77хп (6.10)
Таким образом, в уравнении долговечности стали фигуриро-
вать фундаментальные параметры теплового движения твердого
тела. Согласно (6.10) коэффициент уж в эмпирическом уравне-
ниц лрочности (6.1) является мерой локальной перегрузки хп,
а коэффициентом пропорциональности между и хп служит
отношение CvIfiE, включающее эти параметры. При таком пред-
245
ставлении уж удается уточнить и смысл параметра Uq [8]. Под-
становка (6.10) в (6.2) дает:
№ k^E т
а, =----—----------In — (6.11)
СуХп СуИп
Первый член в правой части этого уравнения представляет
Омаке [уравнение (6.7)], что позволяет представить его в виде
хпОмакс = о';/1 = £'е# (где е* —относительное удлинение, при кото-
ром межатомная связь теряет устойчивость). Это дает возмож-
ность выразить параметр Uo в виде
UQ = it(Cv/V) (6-12)
Анализ экспериментальных значений энергетического пара-
метра Uq для ряда металлов и ионных кристаллов подтвердил
существование такой зависимости [8]. Для полимеров аналогич-
ный анализ пока, по-видимому, не проводился, хотя в работе
[9] отмечается, что предварительные данные как будто под-
тверждают выполнимость этого соотношения и для полимеров.
Выражение (6.11) можно представить в другой форме [8],
учитывая, что Cv~3/jb (в классической области температур):
О. = _Ш£ (6.,з>
хп \ з т,, / Хп \ Зе, т0 /
Из этих уравнений очевидно, что реальная разрывная проч-
ность представляется суммой двух компонент: атермической
оо = £'е*/хп и зависящей от температуры и времени оу.т =
= (р£Т/Зхп)1п(-г/то). Атермическая компонента o0=otfc при хп =
= 1. Приведенные в [8] оценки приводят к ath ~ 0,2 Е, что не
противоречит общепринятым представлениям.
Теперь уместно обратить внимание на одно важное обстоя-
тельство. При рассмотрении термоупругих явлений в твердых
телах, проведенном в гл. 4, в частности, для случая одноосного
растяжения, который мы сейчас рассматриваем, было исполь-
зовано простейшее уравнение состояния, прямо учитывающее
ангармонический характер колебаний составляющих его частиц,
поскольку в него входит кроме обычного для теории упругости
параметра — модуля Юнга — коэффициент теплового рас-
ширения. Но в таком случае, если отвлечься от временных эф-
фектов, не рассматриваемых в теории термоуиругости, тепло-
вые и температурные эффекты, полученные из анализа молеку-
лярной модели (линейная цепочка атомов) и из анализа фено-
менологического уравнения состояния, должны быть формально
тождественны. Тепловые эффекты возникают при деформирова-
нии твердых тел в результате изменения теплового движения
в деформированном твердом теле, а в термофлуктуационной
теории рассматривается роль теплового движения в прочности
твердого тела. И в том, и в другом случае речь идет об изме-
нении колебательного состояния атомов при упругом нагруже-
нии твердого тела. Это ясно видно из рис. 6.1.
246
При анализе термоупругости твердых тел в гл. 4 мы инте-
ресовались фактически, какими явлениями сопровождается из-
менение равновесного расстояния между частицами на величи-
ну Да* под действием силы fk, предполагая, что это изменение
мало по сравнению с Да*. Но очевидно, что аналогичные явле-
ния будут наблюдаться, если изменения равновесного расстоя-
ния осуществляются тепловой флуктуацией. В конце концов, <
безразлично, что деформирует межатомную связь — внешняя[
сила или тепловая флуктуация. С этой точки зрения оба этих
подхода фактически используются для рассмотрения двух эта-
пов одного и того же процесса. Объединяющим началом, несом-
ненно, является ангармонизм межатомных сил в твердых те-1
лах. В гл, 4 показано, что тепловые эффекты при упругом де-[
формировании твердых тел появляются лишь при условии, что
коэффициент теплового расширения отличен от нуля. Именно
существование теплового расширения определяет1 переход от
теории упругости к теории термоупругости. Точно так же един-
ственным и достаточным условием исчезновения температурно-
временных эффектов в прочности, т. е. перехода от термофлук-
туационного подхода к статической теории, является отсутствие
энгармонизма колебаний и, следовательно, отсутствие теплово-
го расширения. Этот вывод прямо следует из уравнений (6.13).
Коэффициентом пропорциональности между тепловым эф-
фектом и деформацией при одноосном растяжении является
произведение $ТЕ [уравнение (4.55)]. Замечательно, что именно
эта величина входит во второй температурно-временной член
выражения для прочности. Таким образом, в гармоническом
приближении в твердых телах должны отсутствовать не только
тепловые явления при деформировании, но и температурно-вре-
менные эффекты в прочности.
В гл. 3, 4 (с. 124) показано, что произведение рЕ определя-
ет изменение энтропии при одноосной деформации и кинетиче-
ское давление. Уже отмечалось, что кинетическое давление
в твердых телах велико (сотни МПа) и может приводить
к огромным локальным внутренним напряжениям, значительно
превышающим среднее напряжение от внешней нагрузки. Жур-
ков [8] приводит следующую оценку для алюминия с проч-
ностью о* = 0,1 ГПа: при комнатной температуре и длительно-
сти испытания т= 1 с величина Тг,т= (рЕ773)1п(т/то) =2-1СНХ
ХЮ2-3-102-30/32 = 6 ГПа. Это значение соизмеримо с теорети-
ческой прочностью.
Развитые представления о важной роли энгармонизма коле-
баний в прочности твердых тел позволяют по-новому взглянуть
на некоторые проблемы прочности [8]. /Прежде всего оказыва-
ется, что прочность материала может понижаться не только,
из-за наличия концентраторов механических напряжений хп,I
которыми обычно считаются многочисленные дефекты кристал-
лической структуры (скопления дислокаций, микротрещины и
т. п.), но и из-за термофлуктуационной составляющей. Особен-:
247
не существенным оказывается тот факт, что понижение проч-
ности на одно и то же значение может быть достигнуто за счет
значительно меньшего изменения термофлуктуационной состав-
ляющей. Другими словами, термофлуктуационный фактор, точ-
нее, более высокий коэффициент линейного теплового расшире-
t ния вблизи дефектов оказывается значительно более эффектив-
) ным концентратором напряжений, чем хп. В связи с этим ста-
' новится очевидной важная роль поверхности в разрушении
Vi твердых тел, поскольку, как показывают оценки и эксперимен-
i\ тальные результаты некоторых прямых физических методов
| исследования поверхностей (8], коэффициент теплового расши-
рения на поверхности может быть в несколько раз больше, чем
: в объеме. Локальные коэффициенты теплового расширения
I вблизи дефектов также должны играть важную роль в прочно-
i сти твердых тел. Вблизи полос скольжения, в вершинах микро-
трещин, вблизи скоплений дислокаций, границ зерен или бло-
ков вследствие наличия больших градиентов напряжения коэф-
фициент теплового расширения может заметно отличаться от
коэффициента теплового расширения в бездефектных областях.
Эти изменения могут быть существенными.
Теоретический анализ влияния деформации на коэффициент
теплового расширения показывает [10], что при наличии дефор-
мации коэффициент теплового расширения может быть пред-
ставлен в виде
р = ро(1 4-Ле) (6.14
где Ро — коэффициент теплового расширения в отсутствие деформации (е=0);
А= (хгЕу)/3; (хг — изотермическая сжимаемость).
Коэффициент А, вычисленный для ряда металлов [10], бли-
, зок к 2. Это означает, что-тепловое расширение напряженного
f твердого тела может быть заметно больше теплового расшире-
ния при отсутствии деформирующего, напряжения.
\ Эта новая физическая идея относительно роли теплового
' расширения в прочности твердых тел уже стимулировала по-
становку экспериментальных [11] и теоретических [9, 12, 13]
исследований проблемы теплового расширения. В этих теорети-
ческих исследованиях развиты представления о фононном Меха-
низме разрушения, впервые выдвинутые в работе [14]. На мо-
дели одномерной нагруженной цепочки колеблющихся атомов
удалось показать, что разрыв такой цепочки происходит в два
этапа. На первом этапе система находится в ожидании достаточ-
но мощной локальной отрицательной флуктуации плотности.
При наличии же такой флуктуации начинается быстрый подвод
энергии в эту область из остальной части цепочки с помощью
потока несущих тепло фононов. За счет такого притока энергии
возникает разрушение в области флуктуации плотности. Долго-
вечность образца в такой модели практически определяется
первым этапом.
24в
Новые идеи рождают новые проблемы. В связи с идеей о
решающей роли энгармонизма колебаний в механизме прочно-
сти возникает один принципиальный вопрос, особенно важный
для полимеров. При выводе уравнений (6.11) и (6.13) предпо-
лагается, что коэффициент линейного теплового расширения
Р является положительной величиной. Для большинства твер-
дых тел любой природы он действительно положителен. Однако
Р может принимать и отрицательные значения, что, как пока-
зано в гл. 3 и 4, особенно характерно для полимеров. Фор-
мально отрицательное значение р означает, что термофлуктуа-
ционный фактор действует в направлении, противоположном
направлению действия внешней силы. Упругое растяжение твер-
дого тела с отрицательным р сопровождается выделением теп-
ла (см. с. 124). Физически трудно представить зависимость
прочности от знака коэффициента теплового расширения, в
частности возрастание прочности по сравнению с Ее». Ответ,
по-видимому, нужно искать в том, что отрицательные коэффи-
циенты р для твердых тел обычно наблюдаются при достаточ-
но низких температурах, т. е. в квантовой области, а не в клас-
сической, для которой получены выражения (6.11) и (6.13).
Для полимеров учет квантовых эффектов при рассмотрении
разрывов нагруженных цепей оказывается особенно необходим,
на что впервые обращено внимание в работах [Ф5, 16]. Необхо-
димость этого учета вызвана следующими обстоятельствами.
Во всех рассмотренных выше теоретических расчетах [3, 7, 9,
12] предполагается, что при достаточно большом натяжении
цепи существуют лишь продольные флуктуации и именно они
приводят к разрыву цепи. Однако в результате анализа темпе-
ратурной зависимости теплоемкости (гл. 1) и теплового расши-
рения (гл. 3) было установлено, что в температурной области
<300 К продольные колебания для большинства полимерных
цепей практически не возбуждены из-за высокой дебаевской
температуры («1500 К), что, в свою очередь, связано с высо-
кими значениями продольного модуля упругости макромолекул.
Тем не менее разрывы макромолекул являются реальностью, и
встает вопрос о причине этих разрывов*. В |[16] в связи с этим
подчеркивается, что при малом по сравнению с предельным
натяжением цепи учет квантовых эффектов совершенно необхо-
дим. При относительно больших натяжениях может наблюдать-
ся снижение характеристической температуры под действием
больших натяжений, в результате температура, при которой
проводится испытание, может оказаться близкой к дебаевской.
Очевидно, что напряжения в макромолекулах, которые могут
приводить к такому эффекту, должны быть близкими к их тео-
ретической прочности. Экспериментальные основания для по-
явления такого эффекта существуют, так как регистрируемые
* Следует обратить внимание на очень низкое значение параметра Грю-
чайзена для продольной колебательной моды в ПЭ (см. табл. 3.1).
17—440
249
методом ИК перенапряжения достигают очень высоких значе-
ний [3].
Вместе с тем возможно, что решающее значение в разрывах
макромолекул принадлежит вообще не продольным, а попереч-
ным (крутильным и деформационным) колебаниям, возможная
роль которых в долговечности напряженных полимерных цепей
пока теоретически не исследовалась. Однако и в этом случае,
как отмечено в [17], учет квантовых эффектов в динамике на-
пряженных макромолекул необходим вследствие достаточно вы-
соких значений дебаевских температур деформационных коле-
бательных мод.
Энергетика разрыва напряженных макромолекул. Упруго-
напряженная макромолекула или ее значительный участок
являются чрезвычайно мощным потенциальным источником
упругой энергии [18]. Действительно, нагружение внешней си-
лой макромолекулы сопровождается накоплением упругой энер-
гии в каждой из ее скелетных химических связей. Накопленная
энергия тем больше, чем ближе растяжения и напряжения на
валентной связи к пределу молекулярной прочности. Пока все
связи напряженной макромолекулы целы, она находится в рав-
новесии и способна совершать внешнюю работу. Однако, если
хотя бы одна из связей растянутой цепочки вдруг разорвется,
равновесие мгновенно окажется нарушенным, и запасенная в
«половинках» разорванной макромолекулы упругая энергия
должна будет превратиться в теплоту из-за невозможности со-
вершения внешней работы. Работа внешних сил, затраченная
на диссоциацию одной разрывающейся связи и образование
двух макрорадикалов, при достаточно большой длине макромо-
лекулы будет составлять лишь малую долю общей запасенной
упругой энергии. Диссипация этой энергии будет локализована
в очень малом объеме и должна иметь взрывообразный харак-
тер. Эти качественные соображения об энергетических измене-
ниях при разрыве упругонапряженной макромолекулы могут
быть проиллюстрированы количественными оценками на одно-
мерной модели [3, 19, 20].
Рассмотрим участок (сегмент) макромолекулы из п связей
в предположении, что взаимодействие соседних атомов доста-
точно хорошо описывается потенциалом Морзе. Пусть этот
участок нагружен одинаковой силой f, а за пределами этого
участка сила равна нулю. Упругая энергия, сосредоточенная в
каждой из химических связей, выражается соотношением
,6J5)
где D — энергия диссоциации связи; ftit = bDI2Ai3Q0 ГПа; b—коэффициент
уравнения Морзе. ч
Упругая энергия, запасенная в цепи из п связей, равна Wv =
= nwy.
При отсутствии растягивающей силы термическая диссоциа-
ция сегмента по одной из связей требует затраты энергии D,
250
Jll '
Puc. 6.2. Энергетические эффекты при
разрыве цепочки атомов при разном рас-
тягивающем усилии [3];
/ — деформированное состояние; // — область
•разрушения; /// — энергетическое состояние
после разрушения.
которая может быть почерпнута
из внутренней энергии окружаю-
щего пространства в виде тепло-
ты. Таким образом, при отсутст-
вии растягивающего напряжения
разрыв сегмента является эндо-
термическим процессом (мы не рассматриваем для простоты
вторичные химические реакции), и внутренняя энергия системы
увеличивается на величину D. Хотя разрыв напряженного сег-
мента будет происходить легче, чем ненапряженного, тем не ме-
нее и в этом случае для разрыва необходимо наличие внешне-
го источника энергии при всех значениях Это означает,
что исходное и разорванное состояния сегмента разделены
энергетическим барьером (рис. 6.2). Разрыв сегмента по одной
связи в любом случае переводит его в энергетическое состоя-
ние, уровень которого определяется величиной D. Поэтому теп-
ловой эффект разрыва напряженного сегмента
Q = D-Wy=D-
(6.16)
Характер тепловых эффектов при разрыве сегмента в зави-
симости от его длины и соотношения действующего напряжения
и предела молекулярной прочности показан на рис. 6.3. При
/=0 разрыв связи в сегменте обусловлен ее термической дис-
социацией, тепловой эффект положителен (Q>0) и равен D.
По мере увеличения внешнего напряжения тепловой эффект
разрыва меняет знак: распад сильнонапряженного сегмента
сопровождается экзотермическим эффектом (Q<0). При таких
напряжениях, когда <2<0, распад сегмента энергетически выго-
ден, поскольку в результате распада его энергия снижается.
Предельные значения экзоэффектов при распаде напряженного
сегмента достигаются при f = fth. Для достаточно длинных сег-
ментов распад даже не очень сильнонапряженного сегмента яв-
ляется экзотермическим процессом.
Из уравнения (6.16) и рис. 6.3 видно, что распад сегмента
может протекать и без теплового эффекта. Этому случаю соот-
ветствует условие Wy = D. Оно формально отвечает энергетиче-
скому критерию Гриффитса в приложении к элементарному
акту разрыва. Для макроскопического процесса разрушения
этот критерий будет рассмотрен дальше, здесь же можно отме-
тить, что для элементарного акта разрыва связи критерий
Гриффитса выполняется при напряжениях значительно мень-
ших, чем предел молекулярной прочности [19].
17* ----------------------------
Рис. 6.3. Силовая зависимость теплового
эффекта при разрыве цепочек атомов с
различным числом звеньев [3, 19, 20]:
1 — 10 звеньев; 2 — 100 звеньев.
Тепловой эффект разрыва
сегмента может быть соотнесен
с кинетическими характеристи-
ками разрушения. Для этого не-
обходимо знать зависимость
энергии активации разрыва на-
пряженного сегмента от дейст-
вующего напряжения. Расчеты,
в других исследованиях [3] для
выполненные в [19], а также
цепи с закрепленными концами, показывают, что энергия акти-
вации распада Uo является нелинейной функцией f. Для того
чтобы представить порядок величин тепловых эффектов при
разрушении напряженных цепей, воспользуемся оценками, при-
веденными в [19, 20]. При комнатной температуре и при ти-
пичных временах нагружения т= 100 с эффективный разрыв
цепей будет протекать тогда, когда энергия активации распада
связей понизится до 75 кДж/моль. Для сегмента длиной 100
С—С-связей, характеризующихся энергией диссоциации
330 кДж/моль, такое понижение энергетического барьера будет
достигаться при напряжениях f=Ofifth (см. рис. 6.3). В этих
условиях тепловой эффект, сопровождающий разрыв сегмента,
примерно в 3 раза превысит энергию диссоциации связи и до-
стигнет 103 кДж/моль. При быстром нагружении или при низ-
ких температурах, когда Uo—>0 и f—»fth, тепловой эффект
разрыва такого сегмента будет превышать энергию диссоциа-
ции связи примерно в 30 раз. Аналогичные оценки приведены
в монографии [5].
Таким образом, разрыв достаточно напряженной макромо-
лекулы должен быть подобен микровзрыву, последствия кото-
рого нетрудно представить |[3, 5]. Во-первых, он может резке»
ускорить распад соседних с разрывающимся сегментом цепейг
и инициировать вторичные свободнорадикальные процессы в-
Напряженных цепях. Во-вторых, локализация таких огромных
энергий в субмикроскопических объемах, определяемых разме-
рами сегментов, должна сопровождаться значительными ло-
кальными повышениями температуры, следствием которых мо-
жет быть как возрастание молекулярной подвижности, так и не-
посредственная термодеструкция соседних макромолекул.
Тепловой эффект при разрыве макромолекул является не
единственным энергетическим следствием разрыва. В резуль-
тате разрыва должна возрасти и внутренняя энергия системы
[21]. Это изменение внутренней энергии может быть «сосредо-
точено» как в промежуточных активных (свободные радика-
лы), так и в конечных пассивных (стабильные группы) про-
дуктах разрушения. Образование каждого радикала увеличи-
ОКО
ЁйёТ энергию системы на £>/2. Если в системе Окажется ста8и-
лизированными Мрад радикалов, то общее возрастание внутрен-
ней энергии, обусловленное их присутствием, составит МрадХ
Х(Р/2). Возникающие при разрывах макромолекул свободные
радикалы вызывают химические процессы цепного характера.
В результате их протекания полимер претерпевает необрати-
мые химические изменения, которые состоят в основном в по-
явлении химически стабильных концевых групп типа СН3 и
СН = СН2. Образование насыщенных метильных групп, связан-
ное с перемещением атомов водорода, мало меняет энергетиче-
ское состояние системы (не более чем на ip кДж/моль), в то
время как образование двойной связи увеличивает энергию си-
стемы примерно на 80—100 кДж/моль 1[3, 5]. Если число таких
новых групп с двойными связями, образующихся в полимере-
при разрушении, составляет NT$, то повышение энергии систе-
мы составит (£>с=с—Дс-с)А^гр.
Каков порядок энергетических изменений в результате хи-
мических превращений в разрушенной системе? Типичные зна-
чения концентрации свободных радикалов в ориентированных
полимерах перед разрывом могут достигать 1017—1018 1/см3, а
число вновь образовавшихся двойных связей —1019 1/см3 (2, 3].
Итак, энергия, связанная с образованием радикалов, имеет
порядок нескольких десятых долей Дж/см3, а «принадлежащая»
образовавшимся двойным связям равна 1—1,5 Дж/см3. Таким
образом, механохимические процессы, протекающие при разру-
шении цепных молекул, действительно могут заметно изменить
внутреннюю энергию полимера.
В каких условиях рассмотренный процесс диссипации упру-
гой энергии при разрыве напряженного сегмента может дейст-
вительно иметь место в полимерах? Из всего предыдущего рас-
смотрения, особенно из анализа термоупругости ориентирован-
ных полимеров (см. с. 179 и далее), очевидно, что наиболее
вероятно появление разрывов достаточно длинных напряжен-
ных сегментов в сильноориентированных кристаллических по-
лимерах. В таких полимерах существуют «внутренние зажимы»—
кристаллиты с закрепленными проходными участками макромо-
лекул, благодаря жесткости которых можно достаточно сильно
нагрузить эти проходные макромолекулы. Длина напряженно-
го участка цепи (число повторяющихся связей), который после
разрыва по одной из связей может сокращаться и приводить
к диссипации упругой энергии, определяется межмолекулярным
взаимодействием, структурой полимера и его жесткостью. Меж-
молекулярные силы в полимерах значительно слабее, чем внут-
римолекулярные, поэтому длина напряженного участка не мо-
жет быть малой, т. е. величина п должна быть значительной,
что как раз и является условием экзотермического разрушения.
Микромеханика деформирования и разрыва цепей, закреп-
ленных в кристаллитах, впервые теоретически была рассмотре-
на Чавычеловым [22, 23], дальнейшая детализация предложен-
253
ной им теории проведена Каушем [5, гл. 5]. Теория построена для
модели микрофибриллы ХБ (рис. 4.27). В ней рассматривается:
1) до каких напряжений можно нагрузить проходящую через
аморфную область полностью вытянутую цепь, прежде чем
участки макромолекулы, находящиеся в двух соседних кристал-
литах-зажимах, начнут вытягиваться из кристаллитов; 2) на
какое расстояние в периодическом потенциальном поле меж-
молекулярного взаимодействия в кристаллитах распространя-
ется напряжение в натянутой макромолекуле; 3) как эти напря-
жения зависят от межмолекулярного взаимодействия в кристал-
литах.
Не вдаваясь в детали, приведем лишь необходимые для по-
следующих оценок результаты расчетов. При типичных разме-
рах кристаллитов и аморфных прослоек в ориентированном ПЭ
и ПКА расчеты приводят к следующим величинам [5, 22, 23].
Предельное напряжение, которое бездефектный кристаллит мо-
жет передать на вытянутую цепь в аморфной прослойке, состав-
ляет для ПЭ 7,5 ГПа, а для ПКА —22,4 ГПа. Более высокие
значения статической прочности кристаллитов ПКА обусловле-
ны существованием сильных водородных связей. Этим объясня-
ется также тот факт, что напряжения в кристаллитах ПКА
«затухают» на расстоянии «3,5 нм, в то время как в ПЭ — на
расстоянии «5 нм. Предельное напряжение для ПЭ — пример-
но 30% молекулярной прочности, а для ПКА оно близко к мо-
лекулярной прочности. Типичная длина цепи, проходящей че-
рез два ближайших кристаллита и аморфную область между
ними, в микрофибриллах составляет «200 С—С-связей. Длина
участка цепи в аморфной области составляет несколько десят-
ков связей. Если считать, что напряжения в цепях снижаются
до нулевых значений примерно к центрам кристаллитов, то дли-
на перенапряженного участка должна составлять «100 связей.
В соответствии с приведенными выше расчетами (см. рис. 6.3)
разрыв такого сегмента будет экзотермическим процессом. Это
особенно относится к ПКА с более жесткими кристаллитами.
Таким образом, приведенные расчеты и оценки дают осно-
вание ожидать, что при разрушении ориентированных кристал-
лических полимеров должны протекать очень мощные локаль-
ные экзопроцессы, обусловленные разрывом напряженных про-
ходных цепей. Выше рассматривались лишь внутрифибрилляр-
ные проходные цепи, однако все эти соображения можно отне-
сти и к межфибриллярным напряженным аморфным участкам
цепи. Для аморфных полимеров и неориентированных кристал-
лических оценить длину перенапряженного участка труднее.
Есть основание считать, что и в этих случаях она примерно со-
ставляет 100 С—С-связей [19].
Теория Чавычелова заставляет обратить внимание еще на
одну важную деталь. При больших напряжениях и больших от-
носительных деформациях аморфных прослоек основным меха-
низмом упругости цепей кристаллических ориентированных по-
254
лимеров является вытягивание участков цепи из кристаллитов.
Это вытягивание будет сопровождаться локальным экзоэффек-
том. Разгрузка образца также будет сопровождаться экзоэф-
фектом, если при этом участок вытянутой цепи опять будет
втягиваться в кристаллит. В гл. 4 такой же механизм упругости
ориентированных кристаллических полимеров рассматривался
на надмолекулярном уровне, когда при значительно меньших
напряжениях предполагалось обратимое проскальзывание мик-
рофибрилл друг относительно друга.
Согласно кинетической концепции прочности разрыв меж-
атомной связи является первым актом в многостадийном про-
цессе разрушения. В ориентированных кристаллических поли-
мерах разрыв макромолекулы в аморфной области сопровож-
дается протеканием целой гаммы механохимических процессов
в нагруженных цепях, завершающихся разрывом большого чис-
ла соседних макромолекул [3]. Этому в значительной мере спо-
собствует тепловыделение при разрывах макромолекул. Таким
образом, разрыв одной молекулы в аморфной прослойке может
приводить к ускоренному разрушению всей аморфной прослой-
ки и появлению первого очага разрушения — субмикротрещины.
Возникновение субмикротрещин, их объединение в микротре-
щины и образование макротрещин являются последовательными
этапами, приводящими к окончательному разрыву образца. За-
кономерности локализации процесса разрушения в виде трещин
на различных уровнях структуры тесно связаны с энергетиче-
скими процессами, протекающими при образовании и росте
трещин, к рассмотрению которых мы и переходим.
ТЕПЛОВЫЕ ЯВЛЕНИЯ ПРИ ОБРАЗОВАНИИ
И РОСТЕ ТРЕЩИН В ТВЕРДЫХ ТЕЛАХ
Теоретическая прочность идеального твердого тела очень вели-
ка и по общепринятым оценкам составляет примерно 0,1 Е.
Реальная прочность достаточно массивных образцов обычно на
два-три порядка меньше, и это расхождение приписывают на-
личию различного рода дефектов, среди которых центральное
место отводится трещинам. Для объяснения расхождения зна-
чений теоретической и реальной прочности твердых тел, разру-
шающихся хрупко (стекло), Гриффитс впервые постулировал
существование в них трещин и предположил, что разрушение
происходит путем распространения этих трещин под действием
приложенных к нему напряжений [1, 24]. Согласно Гриффитсу,
энергетика процесса распространения трещины состоит в пере-
ходе работы упругого деформирования тела в поверхностную
энергию трещин. Таким образом, свободная энергия тела скла-
дывается из объемной упругой энергии ТГОб и поверхностной
энергии Гпов. Если упругое тело растянуто напряжением о, дей-
ствующим нормально к расположенной в теле эллиптической
255
трещине шириной 2/ (/ — значительно меньше размеров тела),
то свободная энергия
(fl
Е = -Wo6 + Wn0B= --^12 + 4Г/ (6.17)
E
где Г—'поверхностная энергия.
Критерий неустойчивого равновесия и начала роста трещины
получается из условия
dpjdl = — (2aU/E) + «40 (6.18)
Величина о0, при которой трещина равновесна (dF/dl=0),
представляет собой критическое напряжение
а0= (2£Г//)1/2 (6.19)
'При о>'Оо dFldl<Q, и существующая трещина данной ширины
начинает самопроизвольно неограниченно разрастаться. Для
плоского деформированного состояния путем более точного рас-
чета Гриффитс получил точное выражение для критического
напряжения |[24, 25]: j
г 2£Т 11/2
ао = Н771----JT <6-20)
L ш(1 — р.2) j
где ц— коэффициент Пуассона.
Формула (6.20) получена на основе анализа баланса энергии
(первый закон термодинамики) и фактически не включает в
себя никаких представлений об идеальной прочности твердого
тела и о молекулярных процессах при образовании свободных
поверхностей трещины и в ее вершинах (энергетический подход).
Она применима к идеально хрупким телам, при разрушении
которых единственным энергетическим процессом является пере-
ход упругой энергии тела в энергию образующихся поверхно-
стей. Таким образом, в теории Гриффитса рассматривается рост
идеально упругих трещин и проблемы диссипативных потерь
при их росте не возникает*.
Из формулы (6.20) следует, что для данного твердого тела
а0/л/ = = const (6-21)
V 1 --- fjt,2
С уменьшением длины трещины разрушающее напряжение
повышается. Предельное значение I определяется размером
межатомных расстояний. Уравнение (6.21) определяет условие
разрастания неравновесной трещины, которая по достижении
критического значения освобождающейся энергии (4 Г для
трещины с двумя вершинами) подвергается лавинообразному
разрушению.
* См., одиако, анализ условий квазистатического роста трещин в хруп-
ких телах методами термодинамики необратимых процессов [25, 26].
256
Хрупкое разрушение, ближе всего приближающееся к по-
стулированному в теории Гриффитса, наблюдается в неорганиче-
ских стеклах, в которых поверхностная энергия разрушения
может быть близкой к поверхностной энергии, определенной не-
зависимым путем. Многочисленные исследования разрушения
материалов других классов (металлы, керамика, твердые поли-
меры) _показывают [1, 5, 24, 27], что для них зависимость
о0~1/У/ качественно наблюдается, однако значения поверхно-
стей энергии Г, получаемые на основе экспериментальных дан-
ных, сильно (на несколько порядков) отличаются от вычислен-
ных теоретически. Последующие модификации этой теории,
предложенные Ирвином и Орованом, привели к выводу, что
получаемая в экспериментах величина Г характеризует не
только чисто хрупкое образование новой поверхности, но и дру-
гие процессы, среди которых особенно важна пластическая де-
формация. Некоторые пластичные при обычных механических
испытаниях материалы, в частности металлы, при образовании
трещин ведут себя подобно хрупким материалам. Они разру-
шаются таким образом, что пластическая деформация сосредо-
точивается в тонком слое у поверхности трещины. Найденная
по уравнению Гриффитса величина Г оказывается на два-три'
порядка выше той теоретической величины, которую можно по-
лучить из оценок, сделанных с учетом лишь сил сцепления.
В связи с этим возникло представление, что вместо Г в урав-
нении (6.20) следует просто использовать некоторое эффектив-
ное значение ГЭф, включающее в себя работу пластической де-
формации. С учетом этого формулу (6.20) можно представить
в виде
а0 =
(Гп 4- Гпл) £ГЭф
и/(1 —[л2) nl( 1 — [л2)
(6.22)
В отличие от Гп величина ГПл не поддается простым теоре-
тическим оценкам и до сих пор теоретически не определена.
Таким образом, представления, развитые для идеально хрупких
тел, перенесены и на тела, разрушающиеся квазихрупко, т. е.
хрупкопластически. При этом константа Г, входящая в форму-
лы, описывающие трещины в идеально хрупких телах, рассмат-
ривается как обобщенная величина, учитывающая эффект
пластических деформаций. Так как работа пластической дефор-
мации при росте трещины намного больше поверхностной энер-
гии Гпл>Гп, последней вообще можно пренебречь, так что
ГЭф в действительности контролируется работой пластической
деформации.
Характер разрушения твердого тела (хрупкое или хрупко-
пластическое) зависит от характера напряженного состояния
в вершине трещины 1(24, 25]. Напряжения вблизи вершины тре-
щины определяются межатомными силами. Эти силы опреде-
ляют также соотношение прочности при отрыве и сдвиге, от
которого зависит, будет ли твердое тело раскалываться хруп-
267
ко или пластически деформироваться в вершине трещины. Ин-
тегральный энергетический подход Гриффитса к изменению
упругой энергии твердого тела при внесении в него трещины
может быть использован и в локальной форме для анализа
энергетического состояния в вершине трещины, особенно важ-
ной для механики разрушения |[24, 25]. Такой анализ был про-
веден Ирвином, который ввел представление о коэффициенте
интенсивности напряжений — величине, полностью характери-
зующей локальное деформирование и разрушение в вершине
трещины (силовой подход). Для плоской деформации коэффи-
циент интенсивности напряжений
*с = ]/ ^ГэФ2; (6-23)
у л(1 — р-2)
Тот факт, что в этом выражении не фигурируют размер тре-
щины и макроскопическое напряжение о, подчеркивает опреде-
ляющую роль вершины трещины и критерия К с, при силовом
подходе, /(с определяет условие роста трещины: после достиже-
ния значения Кс, определяемого уравнением (6.23), трещина
начинает быстро распространяться. В отличие от (6.20) условие
(6.23) имеет локальный характер.
В отличие от идеально хрупкого разрушения квазихрупкое
должно сопровождаться заметными тепловыми явлениями, свя-
занными с локальными пластическими деформациями при росте
трещин. При быстром росте трещины генерируемое при пласти-
ческой деформации тепло может приводить к сильным локаль-
ным перегревам. Соответствующие теоретические оценки при-
ведены ниже.
Рассмотренные представления о хрупком и квазихрупком
разрушении применяют и для анализа разрушения полимеров
[5, 27, 28]. Как и для других материалов, значение поверхност-
ной энергии разрушения твердых полимеров намного (два —
четыре порядка) превышает значения, полученные из теорети-
ческих оценок. Полагают, что и для полимеров решающим
вкладом в величину эффективной поверхностной энергии раз-
рушения является вклад пластической деформации в поверхно-
стных слоях микротрещин. Наличие таких слоев обнаруживает-
ся при морфологических исследованиях поверхностей разруше-
ния. Однако кроме пластической деформации вклад в
поверхностную энергию разрушения полимеров могут вносить
разрывы напряженных макромолекул в вершинах трещин и
механические потери, обусловленные вязкоупругими характери-
стиками полимеров 1[28]. Теория роста трещин в вязкоупругих
материалах развита в работах |[29, 30].
Таким образом, эффективная поверхностная энергия разру-
шения полимеров может быть представлена суммой нескольких
вкладов. Кауш [5] полагает, что наиболее важными вкладами
в поверхностную энергию разрушения полимеров являются ра-
258
бота пластической деформации, удельная поверхностная энер-
гия, энергия упругого втягивания макромолекул в матрицу,
энергия химических реакций, вызванных разрывом цепи. Тем
не менее нужно еще раз подчеркнуть, что наиболее важными
вкладами в поверхностную энергию разрушения является рабо-
та пластической деформации и вклад от разрыва полимерных
цепей. Решить вопрос о соотношении этих двух важнейших
вкладов в поверхностную энергию разрушения можно лишь
экспериментальным путем.
Перейдем теперь к оценкам экзотермических эффектов, ге- .
нерируемых при росте трещин. Диссипация энергии при росте [
микротрещины должна сопровождаться локальными экзоэффек-
тами, величина которых будет зависеть прежде всего от доли
работы пластической деформации, превратившейся в теплоту,
размера зоны пластической деформации и скорости роста тре-
щины. Известен ряд попыток теоретического расчета локаль-
ного подъема температуры в вершине растущей трещины твер-.
дого полимера {31—35]. Однако полученные в этих расчетах
значения локального подъема температуры в сильной мере за-
висят от деталей используемой при расчете модели.
Обычно в расчетах предполагается, что поверхностная энер-
гия разрушения, рассчитываемая из молекулярных параметров
полимера, полностью превращается в теплоту. При решении
задачи о теплопроводности решающим для оценки скачка тем-
пературы в вершине трещины является выбор размера зоны
пластической деформации. Роль скорости распространения тре-
щины в генерировании теплоты в ее вершине можно уяснить из
обсуждавшихся в предыдущей главе локальных экзоэффектов,
вызванных пластической деформацией при ориентации. Типич-
ные значения скоростей распространения трещин в твердых
полимерах, обычно используемые в теоретических оценках,
составляют сотни метров в секунду. При таких скоростях про-
цесс диссипации можно считать адиабатическим, и даже гру-
бая экстраполяция любой из зависимостей, приведенных на
рис. 5.15, приводит к выводу, что скачок температуры в таких
условиях должен составлять многие сотни градусов. Это совпа-
дает с теоретическими оценками, полученными в работах [31—
35]. Так, Леви и Райс [32] для ПММА при скорости роста тре-
щины 250 м/с получили значение 1200 К. Близкое значение полу-
чено в работе [34]: при скорости роста трещины 200 м/с разогрев
составляет 900 К- Однако для этого же полимера Камбур и
Баркер [31] для скорости 200 м/с и выше приводят значение
локального разогрева 210 К, а Уильямс 1[33] оценивает макси-
мальную величину подъема температуры в вершине трещины в
ПММА в 11|5 К. Таким образом, расхождения в теоретических
оценках локального разогрева в вершине трещины достигают
порядка величины.
Следуя работе '[35], рассмотрим основные принципы, на ко-
торых строятся такие теоретические оценки. Предположим, что
259
6
Рис. 6.4. Схема расчета теплового поля в вершине растущей трещины [35]:
а — зона, в которой генерируется теплота в вершине трещины; б — зависимость нормиро-
ванной температурной функции 6 от параметра скорости роста трещины ф при различ-
ном расположении источника тепла.
генерирование теплоты осуществляется в зоне вершины расту-
щей трещины, аппроксимируемой прямоугольником шириной
26 и длиной d (рис. 6.4,а), причем эта зона движется со ско-
ростью с. Физические свойства материала, в частности тепло-
проводность и температуропроводность, предполагаются пос-
тоянными. Считается также, что соблюдается классический
закон теплопроводности (закон Фурье). В приведенной на
рис. 6.4,а системе координат, движущейся вместе с источником
тепла, распределение температуры будет стационарным. Оно
определяется выражением
О / х у d \
7’(х,у) = Г()+-^- в — • -f- Ф (а) (6.24)
28Срр \ о о о /
Ф = с8/2а (б)
где а — температуропроводность; р — плотность; 0 — температурная функция,
нормированная относительно размеров теплового источника и параметра ско-
рости ф.
Решение этого уравнения в терминах 0 в зависимости от
параметра скорости ф может быть представлено в виде обоб-
щенной диаграммы (рис. 6.4,6), учитывающей положение теп-
лового источника в зоне генерирования теплоты. Из этой диаг-
раммы следует существование трех различных областей. При
ф<0,1 существует область изотермических состояний. Внутри
зоны с тепловым источником значение 0<С1 и не зависит от
положения источника. Изотермы в этой области являются
окружностями, температурные градиенты в зоне малы. При
0,1<ф<10 существует переходная область. В этой области с
повышением ф значения 0 сначала во всех участках зоны воз-
растают, однако в центре зоны (точка 2) и в хвосте зоны (точ-
ка /) этот рост существенно больший, чем на фронтальной по-
верхности (точка 3). Примерно при значении ф = 0,3 темпера-
тура на фронтальной поверхности достигает максимума и затем
начинает снижаться, в то время как в точках 2 и 1 она продол-
260
жает расти. Наконец, при ф>10 существует адиабатическая
область, в которой на фронтальной поверхности 0 равна нулю,
а в центре зоны составляет 0,5 значения, достигаемого в хво-
сте зоны. Дальнейшее повышение параметра скорости не ме-
няет распределения температур в зоне. Таким образом, макси-
мальное значение 0 во всех областях наблюдается в хвосте
зоны.
Теплота, генерируемая в вершине растущей трещины, явля-
ется источником повышения температуры материала вблизи
трещины. Максимальный подъем температуры на расстоянии
г/3>2б при условии, что скорость роста трещины достаточно вы-
сока, может быть рассчитан из уравнения
Т’макс - То = (2ле)-1/2( 1/рСр) (Q/y) (6.25)
(е — основание натуральных логарифмов), вытекающего из за-
дачи о теплопроводности с мгновенным линейным тепловым ис-
точником [36], а время достижения максимума температуры
т = ;/2/2а (6.26)
Эти уравнения можно использовать для определения величи-
ны Q материалов, разрушающихся квазнхрупко (металлы, орга-
нические стекла). Для таких материалов поверхностная энер-
гия разрушения имеет порядок 103—1;05 Дж/м2. Для неорганиче-
ских стекол, разрушение которых ближе всего соответствует
представлениям о хрупком разрушении, она находится на уров-
не 10 Дж/м2.
Как отмечалось выше, при оценке температурных скачков
вблизи вершины растущей трещины обычно предполагают, что
величина Q в уравнении (6.25) и поверхностная энергия разру-
шения равны как для хрупкого, так и для квазихрупкого раз-
рушения. Хотя основной вклад в величину ГЭф вносит работа
пластической деформации, она как у низкомолекулярных твер-
дых тел, так и у твердых полимеров лишь частично превраща-
ется в теплоту (см. гл. 5). Коэффициент трансформации нахо-
дится в пределах 0,6—0,9. Поэтому более корректно в таких
•оценках использовать Q—(0,6—0,9) Г, что было учтено в оцен-
ках, приводимых в [35].
При очень больших скоростях распространения трещин па-
раметр ф оказывается большим, и условия в вершине распро-
страняющейся трещины можно считать адиабатическими (см.
рис. 6.4,6). Для этих условий уравнение (6.24) принимает вид
7’макс-7’о = (2/(28Срр) (6.27)
Единственным параметром в этом уравнении является шири-
на зоны 26. Диаграмма, определяющая максимальное повыше-
ние температуры в вершине трещины, растущей с очень боль-
шой скоростью, когда выполняется условие ф^>1 (рис. 6.5),
показывает, что при соответствующих размерах зоны локаль-
ный разогрев в вершине трещины может действительно состав-
261
2StM
Рис. 6.5. Зависимость максимальной темпе-
ратуры вблизи вершины растущей трещины
от размера зоны, в которой генерируется
теплота, при [35]:
1 — сталь; 2 — ПММА; 3 — неорганическое стекло.
лять многие сотни градусов. К этому следует добавить, что в
этих расчетах не принимается во внимание второй мощный
тепловой источник — теплота, генерируемая разрывающимися
упругонагруженными цепями. Учет этого фактора может еще
более повысить локальный разогрев при быстром прорастании
трещины.
ТЕПЛОВЫЕ ЯВЛЕНИЯ ПРИ РАЗРУШЕНИИ ТВЕРДЫХ ПОЛИМЕРОВ
Согласно кинетической концепции прочности твердых тел в
них задолго до того, как магистральная трещина начинает раз-
делять образец на отдельные части, накапливаются разрывы
отдельных межатомных связей, приводящие к возникновению
разрывов сплошности субмикроскопического размера [3, 4]. Это
означает, что уже на стадии образования таких субмикротре-
щин должны существовать диссипативные потери и протекать
локальные тепловые явления. Размеры этих первичных разры-
вов сплошности находятся на уровне нескольких десятков нм,
что чрезвычайно усложняет регистрацию прямым методом теп-
лового эффекта или изменения температуры в объемах указан-
ного масштаба. На нынешнем этапе экспериментальных иссле-
дований информация об этих явлениях может быть получена
лишь путем регистрации интегральных эффектов от процессов,
протекающих в огромном числе субмикротрещин. Для этого мо-
жет быть использована высокочувствительная калориметрия
[38]. Перейти от интегральных характеристик тепловых явлений
к микроскопическим можно лишь при наличии дополнительной
информации о числе и размерах микротрещин, числе разорван-
ных макромолекул, надмолекулярной структуре объекта. Таким
образом, в этом случае необходим комплексный подход.
Слияние и укрупнение первичных разрывов сплошности при-
водит в конце концов к появлению и развитию дефектов мак-
роскопических размеров — магистральных трещин. Размеры
этих дефектов уже таковы, что для регистрации протекающих
при их развитии тепловых явлений могут быть использованы
локальные методы регистрации температур. В ранних исследо-
ваниях для этой цели применяли миниатюрные термопары или
термонндикаторы. В последние годы стали регистрировать крат-
ковременное избыточное равновесное ИК-излучение нагретой
262
зоны. Пространственное разрешение этого метода может состав-
лять 10 мкм, что позволяет использовать его для надежной
регистрации локальных тепловых процессов в вершине магист-
ральной трещины. Отсутствие контакта между объектом иссле-
дования и регистратором температурных изменений и малая
инерционность регистратора (временное разрешение—несколько
мкс) делает этот метод чрезвычайно эффективным для реги-
страции максимальных локальных разогревов при больших
(околозвуковых) скоростях роста трещин. При медленном раз-
витии разрушения чувствительность локальных методов резко
снижается.
Оба метода — интегральный и локальный — уже применя-
лись для изучения энергетики разрушения твердых полимеров.
Ниже будут рассмотрены полученные результаты и проведено
их сравнение.
Макроскопические тепловые явления
при разрушении полимеров
Разрыв одноосно-растянутого полимера. В гл. 4 детально рас-
смотрена термодинамика одноосного растяжения конденсиро-
ванных тел с двумя типами упругости — высокоэластической и
характерной для твердых тел. Теперь эти результаты можно ис-
пользовать для описания тепловых явлений, которые будут
возникать при их разрушении. Простые рассуждения приводят
к выводу, что в идеальном случае разрыв одноосно-растянутого
эластомера или твердого полимера эквивалентен рассмотрен-
ным в гл. 4 случаям их мгновенного сокращения без соверше-
ния внешней работы. Убедиться в этом можно на простом при-
мере.
Предположим, что находившийся в равновесных условиях
однородно-растянутый стержень твердого полимера или эласто-
мера мгновенно разорвался на две части. Оставляя в стороне
причины и возможность разрыва в таких условиях, оценим
энергетические изменения в полимере в результате разрыва.
Очевидно, что единственным ощутимым эффектом такого раз-
рыва должно быть рассеяние энергии, запасенной в образце при
растяжении, поскольку энергетическими и энтропийными эф-
фектами, обусловленными собственно разрывами макромолекул
и образованием новой поверхности, можно пренебречь. Действи-
тельно, если принять, что поперечное сечение стержня 1 см2, то
число разорванных макромолекул в предельном случае, когда
все цепи расположены перпендикулярно поверхности разрыва,
будет составлять 1014—1015 г/см2. Предположим, что конечным
результатом разрыва является превращение всех разорванных
ординарных С—С-связей в концевые ненасыщенные двойные
связи. Как уже отмечалось, энергия таких связей примерно на
80 кДж/моль больше, чем ординарных связей, и потому повы-
шение внутренней энергии, вызванное образованием К)15 нена-
263
сыщенных двойных связей, будет иметь порядок 10-7 Дж. Тако-
го же порядка будет и энергия образования новой поверхности.
Таким образом, этими эффектами в макроскопическом масшта-
бе действительно можно пренебречь и рассмотреть энергетику
разрушения в этих двух идеальных случаях, воспользовавшись
полученными в гл. 4 результатами. Эти же соображения, есте-
ственно, справедливы и для случая, когда разрыв полимера
происходит непосредственно в процессе растяжения.
Разрыв эластомерного стержня. Теплота Q*, со-
провождающая разрыв одноосно-растянутого эластомерного
стержня, определяется уравнением (4.92), которое теперь удоб-
нее рассматривать в виде
Q* = — Д1/ (6.28)
где Д(7— изменение внутренней энергии растянутого стержня.
Из этого уравнения очевидны следующие выводы. Разрыв
идеального энтропийноупругого стержня (AU=0) не будет со-
провождаться никакими тепловыми явлениями, так как, хотя
вся работа, затраченная на растяжение, будет превращаться
в теплоту, она будет полностью скомпенсирована теплотой, по-
глощаемой при сокращении разорванных половинок образца.
Разрыв неидеального эластомера будет сопровождаться либо
нагреванием разорванных частей (At/>0), либо охлаждением
(iAf7<0). Оценить температурные изменения при разрыве мож-
но из простого отношения
ДТ = ьи/Ср = — Q*/Ср (6.29)
предполагающего адиабатический характер тепловых процес-
сов при разрыве.
Приведенные ниже результаты калориметрического иссле-
дования [37] тепловых эффектов, сопровождающих разрыв рас-
тянутых эластомеров, подтверждают сделанный выше основной
вывод, что знак теплового эффекта определяется знаком энерге-
тических изменений растянутого эластомера:
скэп НК пдмс пив
А. Q’, Дж/г X, Q*. Дж/г к Q*. Дж/г X. Q", Дж/г
1,23 —0,0092 1,45 —0,06 3,0 —0,02 3,0 «0
1,37 0,0082 1,97 —0,123 5,0 —0,06 5,0 «0
1,78 0,117 3,2 —0,820 — — — —-
2,33 0,52 1— — — — — —
Большинство этих данных получено путем мгновенного раз-
резания тонкой проволоки, с помощью которой образец растя-
гивался и в таком состоянии удерживался в ячейке микрокало-
риметра. Аналогичные эффекты наблюдались в тех случаях,
когда растянутый эластомер сам разрывался на части. Полу-
264
ченные таким методом значения Q* могут быть использованы
для расчета энергетической составляющей в эластомерах [см.
уравнение (4.92) и экспериментальные данные, приведенные в
[37]. При небольших растяжениях тепловые и температурные
эффекты, обусловленные разрывом эластомеров, малы. При
больших растяжениях значение Q* (или AU) может достигать
нескольких Дж/г и соответственно нагрев или охлаждение мо-
гут составлять 1—2 К.
Разрыв твердого стержня. Аналогичный анализ
можно провести и для твердого растянутого полимера. Ранее
было показано (см. с. 195), что мгновенная разгрузка твердого
стержня с ₽>0 будет сопровождаться рассеянием энергии (на-
греванием). Для стержней с р<0 мгновенное прекращение дей-
ствия растягивающего напряжения может приводить и к охлаж-
дению (е<еИн). Однако в реальных условиях разрыв обычно
происходит при е>еин, когда и для стержней с 0<О должно
наблюдаться нагревание. Возрастание внутренней энергии в
полимерных стержнях при деформациях, близких к разрывным,
может достигать нескольких десятков Дж/г. Макроскопическое
повышение температуры образца при разрыве в этом случае
может составлять 1—10 К. Такие эффекты легко обнаружить
как калориметрическими измерениями, так и любым методом
регистрации температурных изменений (38].
Усталостное разрушение твердых полимеров. Вязкоупругий
характер деформирования полимеров и их низкая теплопровод-
ность являются причиной теплового разрушения при цикличе-
ских деформациях полимеров. Гистерезисные потери при цикли-
ческом деформировании могут оказаться сильным тепловым
источником, что при недостаточном теплоотводе может приве-
сти к непрерывному саморазогреву полимера и тепловому раз-
рушению типа теплового взрыва [39].
Задача о саморазогреве твердых полимеров при циклическом
деформировании, рассматривавшаяся в ряде работ, ссылки на
которые можно найти в монографии [4], сводится к составле-
нию уравнения теплового баланса:
Cpf(dT/dt) — fe©no0e0sinS — as(T — To) (6.30)
где <в — частота нагружения; ао и е<>—амплитуда напряжения и деформа-—
ции; 6 — угол сдвига фаз между о0 и е0; То—температура окружающей сре-
ды;' а — коэффициент теплоотдачи; s — поверхность образца; k — коэффи-
циент, характеризующий долю гистерезисных потерь, трансформировавшихся
в теплоту.
Гистерезисные потери и, следовательно, мощность теплового
источника разогрева зависят от температуры через зависимость
характеристик полимера от температуры (б, упругий модуль
и модуль потерь). Согласно уравнению (6.30) могут существо-
вать два режима гистерезисного саморазогрева полимера при
циклическом деформировании. Первый — стационарный — ха-
рактеризуется такими условиями, при которых разогрей
18—440 265
Рис. 6.6. Кинетика разогрева при
циклическом деформировании
ПОМ [39]:
I — стационарный режим (механическое
разрушение), а0=32 МПа; 2 — нестацио-
нарный режим (тепловое разрушение),
(То=37,5 МПа; (0=50 Гц; Го=15°С.
А7’=7’—Т через определенное время после начала деформиро-
вания достигает постоянного значения, что соответствует
Т = const. Физически это означает, что генерируемая при цикли-
ческом деформировании теплота успевает целиком отводиться
в окружающее пространство, хотя полимер при этом находится
при Т>Т0. Второй — нестационарный — режим соответствует
нестационарному решению уравнения (6.30), который отвечает
тепловому разрушению полимера. В этом случае А7’=/(т), т. е.
T=T0+f(r).
Зависимости саморазогрева от времени (числа циклов), от-
вечающие этим двум режимам (рис. 6.6), характеризуют два
типа усталостного разрушения. В стационарном режиме долго-
вечность полимера определяется воздействием нагрузки, а в
нестационарном — выносливость образца определяется продол-
жительностью подъема температуры до критического значения.
Это критическое значение саморазогрева тесно связано с тем-
пературой стеклования или с температурой начала снижения
кристалличности полимера.
Теоретический анализ гистерезисного разогрева в жестких
полимерных материалах, учитывающий их локализацию в вер-
шинах растущих трещин, проведен в работе [40].
Микроскопические тепловые явления
при медленном разрушении полимеров
К настоящему времени предпринята единственная попытка вы-
явить роль разрывов макромолекул в энергетическом балансе
при медленном разрушении полимера методом высокочувстви-
тельной калориметрии и использовать эти результаты для де-
тализации процесса разрушения [21]. Идея экспериментов осно-
вана на определении полного энергетического баланса в не-
скольких последовательных циклах растяжение — сокращение.
Метод последовательных нагружений широко используется при
изучении термокинетической природы прочности твердых тел.
Он основан на вытекающих из кинетической концепции прочно-
сти представлениях, что при нагружении твердого тела до за-
данного напряжения разрывы межатомных связей наблюдаются
главным образом при первом нагружении. Это подтверждается
как феноменологическими исследованиями с перерывами в
266
действии нагрузки, так и (особенно) прямыми физическими ме-
тодами регистрации разрывов макромолекул (ЭПР) и появле-
ния субмикротрещин (малоугловая рентгеновская дифракция)
{3]. Все последующие нагружения до первоначального напря-
жения дополнительным разрушением практически не сопровож-
даются. Таким образом, прием повторных нагружений с энерге-
тической точки зрения означает, что при первом нагружении в
энергетическом балансе должна присутствовать составляющая,
обусловленная разрывом макромолекул, в то время как при
повторных нагружениях энергетический баланс должен опреде-
ляться только деформационными процессами. При этом, однако,
необходимо иметь в виду, что такая структура энергетического
баланса должна быть характерна не только для чисто упругого
растяжения и разрыва макромолекул. Если наряду с разрывами
макромолекул при первом нагружении будет наблюдаться пла-
стическая деформация, то она также может привести к появле-
нию в энергетическом балансе составляющей, качественно не
отличающейся от составляющей, обусловленной разрывами
макромолекул. Поэтому такой подход требует тщательного
контроля обратимости деформации и в случае появления пла-
стической деформации — раздельного определения вкладов в
энергетический баланс разрыва макромолекул и пластической
деформации. Имея в виду эти соображения, рассмотрим ре-
зультаты калориметрического исследования медленного разру-
шения полимеров.
Из приведенного в начале этой главы материала очевидно,
что надеяться обнаружить тепловые явления, обусловленные
Рис. 6.7. Энергетические эффекты в циклах нагрузка — разгрузка для ориенти-
рованного ПКА [21]:
а — механический гистерезис 6W за цикл (ТРр— работа растяжения; IFC — работа сокра-
щения); б — тепло, рассеянное за цикл (/ — первый цикл, 2 — второй цикл); образцы
получены ориентационной вытяжкой в 5,5 раза при 210 °C; диаметр цилиндрического об-
разца 0,27 мм, Прочность при растяжении около 0,8 ГПа, разрывное удлинение 16—17%;,
скорость деформирования 0,116 мм/с.
18’
267
Рис. 6.8. Энергетические эффекты, свя-
занные с разрушением ПКА [21].
30
<20
'AW /к
V/ АО
ли
6 8
б-10'* НПа
ю
б
4
разрывом макромолекул, можно
; в первую очередь в сильноориен-
тированных кристаллических по-
S лимерах с достаточно высокой
3 прочностью. Энергетические Ха-
S. рактеристики для двух последо-
вательных циклов растяжение —
сокращение высокопрочных во-
локон ПКА (рис. 6.7) совпадают
о лишь при напряжениях
<7^400 МПа. Дальнейшее растя-
жение существенно меняет кар-
тину: 8W и Q при первом нагру-
жении становятся систематиче-
ски больше, чем при втором.
В области напряжений, близких к разрушающим (о = 700—
750 МПа), 6W и Q при первом нагружении уже в несколько
раз болыце, чем при повторном. При этом важно отметить, что
различия между энергетическими параметрами возникают лишь
на стадии растяжения, поскольку стадии сокращения во всех
циклах являются энергетически эквивалентными.
Энергетические различия между первым и последующими
нагружениями при о>400 МПа, представленные на рис. 6.8,
связаны с необратимыми явлениями, протекающими при первом
нагружении. Примечательно, что во всей области рассеянная
теплота меньше механической работы, так что наблюдается
повышение внутренней энергии. Зависимость энергетических
параметров (ДИ7, AQ, At/) от напряжения является экспонен-
циальной (для примера на графике приведена лишь соответст-
вующая зависимость lgAQ—о). В связи с этим" необходимо от-
метить, что исследование зависимости концентрации радикалов
от напряжения в таких же высокопрочных образцах ПКА по-
казывает, что их появление удается зарегистрировать лишь при
первом нагружении при сг>400 МПа, причем зависимость кон-
центрации радикалов от напряжения также является экспонен-
циальной. Таким образом, существующая качественная корре-
ляция зависимости энергетических параметров и концентрации
радикалов от напряжения, а также изменение внутренней энер-
гии, которое можно представить как результат химических из-
менений при разрывах макромолекул, позволяют отнести рас-
сматриваемые необратимые изменения на счет разрывов макро-
молекул.
Для окончательного заключения необходимо рассмотреть
роль пластической деформации. Тщательный анализ размеров
образцов показывает, что после достаточно длительного отдыха
268
размер образцов, нагруженных до сг<400 МПа, полностью об-
ратим. При нагрузках, превышающих эти напряжения, в образ-
цах появляется остаточная деформация, которая при
<т=750 МПа достигает 1,5% (общая деформация при этом со-
ставляет «15%). Закономерность нарастания необратимой де-
формации с увеличением напряжения качественно подобна за-
висимостям энергетических характеристик (см. рис. 6.8).
Таким образом, энергетические параметры необратимых
изменений включают как составляющую, связанную с разрывом
макромолекул, так и составляющую, обусловленную пластиче-
ской деформацией. Вклад последней оценить достаточно легко,
предположив, что вся необратимая деформация развивается
при максимальном для заданного нагружения напряжении. Тог-
да работа пластической деформации определяется произведе-
нием этого максимального напряжения на величину необратимой
деформации. Для предразрывных напряжений работа пласти-
ческой деформации равна №Пласт=13 Дж/г, что составляет
«45% общей необратимой работы. Принимая во внимание
данные по ориентационной вытяжке ПКА (см. табл. 5.1), будем
считать, что 80% работы пластической деформации рассеива-
ется в виде теплоты, а оставшиеся 20% расходуются на повы-
шение внутренней энергии при пластической деформации. Та-
ким образом, энергетические параметры, обусловленные собст-
венно разрывом макромолекул в предразрывном состоянии, для
которого ниже будут приведены соответствующие оценки, округ-
ленно следующие: А1ГРазр=16 Дж/г; AQPa3p=14,5 Дж/г;
АЦ>азр=1,5 Дж/г*.
Используем теперь эти значения энергетических параметров
для детализации процесса разрушения высокопрочного ПКА.
Прежде всего оценим число разрывов макромолекул. Считая,
что Af/разр целиком обусловлено образованием концевых групп
с двойной связью и полагая (см. с. 253), что на каждый разрыв
расходуется 1,7 -10“19 Дж (100 кДж/моль), то число разрывов
в предразрывном состоянии составит Аразр = АДразр/1,7-10“19 =
= 1019г-‘.
Зависимость Аразр от напряжения симбатна АДразр, и к пред-
разрывному состоянию число разрывов достигает 1019 г-1, что
удовлетворительно согласуется с числом разрывов, определен-
ных в аналогичных образцах вискозиметрическим методом [21].
Представляет интерес также оценить длину перенапряжен-
ного фрагмента макромолекул, разрыв которого по одной из
«вязей приводит к диссипации запасенной в этом фрагменте
упругой энергии. Зная работу АИ7разр, затраченную на упругое
растяжение этих фрагментов макромолекул, и число разрывов,
* В работе (21] вкладом пластической деформации в изменение внутрен-
ней энергии пренебрегали. Однако, как видим, в действительности ее вклад со-
ставляет 60% и лишь 40% приходится на разрывы макромолекул. В связи с
этим приводимые ниже оценки несколько отличаются от сделанных ранее, од-
нако общие выводы не изменяются.
26»
можно оценить среднее значение механической энергии, сосре-
доточенной в каждом фрагменте:
Д^разр^-Д^/Хразр
Проведя такую оценку А№"разр для различных напряжений
(рис. 6.9), можно отметить интересное обстоятельство: коли-
чество механической энергии, приходящейся на один разрываю-
щийся фрагмент, практически не зависит от напряжения. Важ-
ным следствием этого факта является следующее: разрывы
молекул в напряженном полимере происходят при более или
менее определенном локальном напряжении на фрагментах не-
зависимо от внешней нагрузки. По достижении этого уровня
начинаются разрывы макромолекул, и дальнейшее увеличение
макроскопического напряжения на образце лишь увеличивает
число разрывающихся фрагментов.
Из рис. 6.9 видно, что АИ7разр = (14—16)-10~19 Дж. Это»
означает, что запасенная в напряженном участке макромоле-
кулы энергия превышает и энергию диссоциации С—С-связи
(«4-КН9 Дж), и особенно энергию перехода к стабильным
концевым группам (1,7-1019 Дж). Следовательно, разрыв на-
пряженной макромолекулы, как и предсказывает приведенный
выше иллюстративный расчет (см. с. 253), есть экзотермический
процесс. Это видно и из того факта, что отношения AQpa3p/
/At/разр, а также AU7pa3p/At/Pa3p составляют «10. ИК-спектро-
скопические исследования показывают [3], что локальные на-
пряжения на таких сегментах достигают 15—20 ГПа. Эти зна-
чения соответствуют f/fth~0,5. Тогда в соответствии с форму-
лой (6.15) упругая энергия деформированных связей
ХЮ~20 Дж. Отсюда n = AIT'paap/IT« 100. В соответствии с этим
значением и оценочная длина перенапряженного фрагмента
составляет «13—15 нм. Это значение хорошо согласуется со
структурными данными: для обсуждаемых образцов ПКА боль-
шой период составляет 12 нм [21].
Проведенные оценки демонстрируют вполне разумное согла-
сие результатов энергетических исследований медленного раз-
рушения высокопрочного кристаллического полимера с данны-
ми прямых физических методов и подтверждают существование
в энергетическом балансе разрушения составляющей, обуслов-
ленной разрывами макромолекул. В связи с этим здесь уместно
обсудить энергетические последствия
С, МПа
разрывов макромолекул. В соответ-
ствии с последней предпосылкой при
нагружении полимера в нем натяги-
ваются, а затем по достижении опре-
Рис. 6.9. Механическая энергия, сосредоточен-
ная в одном разрывающемся фрагменте моле-
кулы ПКА [21].
270
деленных перенапряжений под действием тепловых флуктуа-
ций разрываются фрагменты макромолекул длиной порядка
десятка нм, причем большая часть (примерно 90%) запасен-
ной в «половинках» разорвавшегося фрагмента упругой энер-
гии выделяется в виде тепла. Последнее, пожалуй, и представ-
ляет наибольший интерес с позиции локализации разрушения.
Поскольку разрушение ориентированных кристаллических по-
лимеров локализовано в «слабых» аморфных прослойках, наи-
больший интерес представляет оценка локального подъема тем-
пературы в таких прослойках после разрыва макромолекулы.
Акты разрыва напряженных макромолекул инициируют хи-
мические свободнорадикальные процессы [3]. Поэтому вслед за
распадом первой молекулы передача свободных радикалов мо-
жет привести к быстрому распаду десятков соседних напряжен-
ных макромолекул и, следовательно, к «групповому» тепловы-
делению. Такой процесс уже квалифицировался выше как свое-
образный тепловой микровзрыв. Это тепловое явление, несом-
ненно, должно способствовать образованию зародышевых
разрывов сплошности—субмикротрещин, обнаруживаемых экспе-
риментально [3, 4]. В аналогичных образцах ПКА число субмик-
ротрещин Л/тр среднего диаметра d=15 нм в предразрывном со-
стоянии достигает 5-1016 L/см3 [4]. Согласно этим структурным
исследованиям число образовавшихся субмикротрещин хорошо
коррелирует с числом радикалов. Каждый разрыв цепи ини-
циирует образование отдельной субмикротрещины, локализован-
ной вблизи места первичного разрыва фрагмента макромолеку-
лы, причем образование субмикротрещин имеет «взрывоподоб-
ный» характер. Это позволяет оценить локальный подъем тем-
пературы в месте локализации разрыва фрагмента макромоле-
кулы. Разрыв фрагмента должен привести к «мгновенному»
сбросу упругой энергии, и для оценки кратковременного локаль-
ного разогрева можно предположить «микроадиабатический»
характер процесса. Тогда
ЬТ = — —« 750 К (6.31)
MpVтррСр
Мы видим, что локальное повышение температуры вследствие
разрыва макромолекул может достигать многих сотен градусов.
Мы уже занимались оценками локального повышения темпе-
ратуры, вызванного эффектами пластической деформации (вы-
нужденной высокоэластичности) при ориентационной вытяжке
твердых полимеров, и пришли к выводу, что при малых скоро-
стях ориентации температурные эффекты малы. В приведен-
ных выше оценках они огромны. Это различие обусловлено раз-
личной скоростью диссипации механической энергии. При пла-
стической деформации подъем температуры определяется ско-
ростью деформирования (см. с. 227). Разрыв макромолекулы
независимо от скорости деформирования должен сопровождать-
ся диссипацией энергии с околозвуковой скоростью, что оправ-
271
дывает предполагаемый микроадиабатический характер тепло-
вых явлений при разрыве. Проблема состоит в идентификации
объема локализации сброса энергии. В рассмотренном выше
примере этот объем удается идентифицировать на основании
• рентгеноструктурных данных. Несомненно, схема зарождения
субмикротрещины, рассмотренная выше, является идеализиро-
ванной, однако можно считать, что сделанные на основе этой
схемы оценки локальных разогревов вследствие разрывов мак-
ромолекул дают правильный порядок величины.
Рассмотренные результаты подтверждают предположение О'
существенном вкладе необратимых разрывов макромолекул в
/поверхностную энергию разрушения и показывают, что в поли-
'мерах этот вклад может не только быть сравнимым с вкладом
пластической деформации, но и превышать последний. Иденти-
/фицировать локализацию пластической деформации на стадии
/ зарождения субмикротрещин, по-видимому, значительно слож-
нее, чем места разрывов макромолекул. Можно полагать, что
пластическая деформация будет также локализована вблизи
перенапряженных участков макромолекул. При медленном раз-
рушении диссипация энергии пластической деформации не бу-
дет, по-видимому, влиять на величину АТ, хотя она и вносит
основной вклад в изменение внутренней энергии. При больших
скоростях разрушения локальные разогревы вследствие пласти-
ческой деформации могут суммироваться с локальными разо-
1гревами в результате разрывов макромолекул, что еще больше
[увеличивает оценочное значение АТ и приближает его к верхне-
му пределу теоретических оценок. Таким образом, совместное
эассмотрение энергетических параметров медленного разруше-
ния ориентированного кристаллического полимера и данных
Структурно-физических методов не противоречит представлению
о возможности протекания локальных микровзрывов при разру-
шении. Посмотрим теперь, в какой мере это заключение под-
тверждается результатами методов локального изучения теп-
ловых явлений при разрушении полимеров.
Локальные экзопроцессы при росте трещин
в твердых полимерах
Для многих полимеров характерно существование трех областей
развития трещин: медленной, переходной и быстрой [5]. Этим
областям соответствуют три области температурных изменений
(см. рис. 6.4,6). Наибольшие локальные разогревы при росте
.трещин должны, естественно, наблюдаться в адиабатической
области. Скорость роста трещин в этой области можно оценить
из выражения температурной функции 0 [уравнение (6.24)].
Для типичных значений коэффициента температуропроводности
полимеров а=10-3 см2/с и размера зоны 6=10 мкм для ф>10,
соответствующей адиабатическому развитию трещин, получаем
нижиее значение скорости адиабатического развития трещины в
272
Таблица 6.1. Локальное повышение температуры
при разрушении твердых полимеров
Полимер Скорость роста трещины, м/с Метод ре- гистрации температуры Локальное повышение температу- ры ДТ, к Размер зоны б, мкм Литера- турные ссылки
Полиметил метакрил ат 200—650 ик 500 0,5 (42J
140 Термопара 230 0,8 [43J
Полистирол Полиэтилентерефта- лат 380—550 ик = 400 0,5 [42]
неориентирован- 40 ик ^250 20’ [44]
ный 0,9—1,5 ик 180± 15 —— [45, 46]
ориентированный (Л=4) 0,9-1,5 ик 170± 15 — [45, 46]
неориентирован- ный облученный 0,9—1,5 ик 120±20 — [45, 46]
неориентирован- ный (сканиро- вание через тяж) 0,9—1,5 ик До 215 [45, 46]
Неорганическое стек- ло 170 Регистра- ция светово- го излуче- ния на не- скольких длинах волн «3000 «0,002 [35]
• Действительный размер зоны
заметно завышено, и, следовательно,
точно неизвестен, приведенное значение,
ДГ>250 К сильно занижено.
вероятно,
твердых полимерах — с=200 м/с. Таким образом, максимальных
локальных разогревов следует ожидать при околозвуковых
скоростях распространения трещин.
Характерные результаты экспериментального определения
локального повышения температуры при росте трещин в твер-
дых полимерах суммированы в табл. 6.1. Они вполне согласу-
ются с теоретическими оценками локальных разогревов и с при-
веденными выше оценками, основанными на калориметрической
регистрации интегральных тепловых эффектов при медленном
разрушении полимеров,, и доказывают тот факт, что при боль-
ших скоростях распространения трещин в твердых полимерах
температуры вблизи вершины трещины на многие сотни граду-
сов превышают температуру полимера. Эти локальные разогре-
вы настолько велики, что превосходят не только температуру
перехода из твердого в размягченное состояние, но и значи-
тельно превышают температуру термического разложения. Не-
смотря на кратковременный характер локальных разогревов
(порядка сотен мкс), такие тепловые микровзрывы должны
приводить и к характерным морфологическим изменениям по-
верхности разрушения, и к появлению продуктов деструкции
полимера. Морфологические и масс-спектрометрические иссле-
273
дования процесса разрушения ПММА и ПС вполне согласуются
с этим выводом [3, 42].
Локальное повышение температуры АТ позволяет оценить
количество теплоты Q, генерируемой при росте трещины. Эта
величина равна или пропорциональна поверхностной энергии
разрушения Г. Соответствующие оценки приводят к значению
Q= 103—104 Дж/м2 для стеклообразных полимеров типа ПММА
и ПС (43, 44, 47]. Эти величины того же порядка, что и полу-
чающиеся на основании анализа зависимости напряжения от
размера трещины [27], что является независимым подтвержде-
нием высоких значений поверхностной энергии разрушения по-
лимеров.
Величина поверхностной энергии разрушения Q, полученная
локальными тепловыми методами, может быть рассмотрена
также с позиции соотношения вкладов работы пластической
деформации и разрыва фрагментов напряженных макромоле-
кул [41, 44]. Наблюдаемые тепловые эффекты вследствие дис-
сипации упругой энергии после разрыва напряженных фрагмен-
тов макромолекул удается непротиворечиво объяснить, только'
допустив объемный характер разрушения связей в окрестности
растущей трещины. Это вполне согласуется с данными о повы-
шенной концентрации субмикротрещин в вершине магистраль-
ной трещины и о повышенноой концентрации разорванных свя-
зей в области, прилегающей к поверхности разрушения [3, 4J.
Оценка минимальной ширины области разрушения, которая
пропорциональна длине фрагмента и соотношению интеграль-
ного теплового эффекта Q и упругой энергии, запасенной во
фрагментах, приводит к значению 1—3 мкм. Объемный харак-
тер разрушения вблизи поверхностей трещины способствует
увеличению размера этой минимальной зоны разрушения.
В этих оценках учитывали данные приведенных выше кало-
риметрических результатов и исходили из одинакового вклада
работы пластической деформации и разрыва макромолекул в
поверхностную энергию разрушения. Экспериментально опреде-
лить соотношение этих вкладов удается, изучая локальные теп-
ловые явления при разрушении, протекающем со скоростью, да-
лекой от скорости звука. Такое исследование, выполненное в
последнее время на ПЭТФ (45, 46], позволило кроме максималь-
ного значения температуры в зоне разрушения (см. табл. 6.1|)
определить размеры и температурный профиль нагретой зоны
и получить на его основании соотношение между двумя основ-
ными вкладами в поверхностную энергию разрушения.
В адиабатической области распространения трещин поли-
I мер разрушается достаточно «хрупко». Зоны пластической де-
формации в этом случае достаточно тонкие. Если скорость
роста трещин намного меньше скорости звука в полимере, так
что разрушение происходит в промежуточной температурно-
скоростной области (см. рис. 6.4,6), сопротивление трещинооб-
разованию может возрасти [5, 35] вследствие увеличения раз-
274
Рис. 6.10. Температурный профиль горячей зоны &Т,К
при разрушении ПЭТФ (D — расстояние от края
трещины) [45].
мера зоны пластической деформации и 160 ' I
характер разрушения может стать более \
пластическим. Скорости распростране- \
ния трещин, при которых можно ожи-1 по - \
дать максимального сопротивления тре- \
щинообразованию, как показывают । I
оценки [35], для стеклообразных поли- । \
меров (ПММА) лежат в области -А м/с. ! 80 ~ \.
Температурный профиль зоны разруше- \
ния ПЭТФ исследовался как раз на этих \
скоростях. _ \
Характерный температурный про- \
филь (рис. 6.10) зоны разрушения пока- \
зывает, что наибольший разогрев наб- 0 ____ \
людается на краю магистральной тре- о j л-10'2тм
щины, а затем следуют резкий спад и
участок пониженных примерно в 2 раза по сравнению с мак-
симальной температурой. Такой температурный профиль согла-
суется с представлением о существенном вкладе диссипации
упругой энергии при разрывах фрагментов макромолекул в ве-
личину разогрева. Об этом свидетельствует также ряд полу-
ченных в работах [45, 46] косвенных данных, основанных на
сравнении температурного профиля зоны разрушения неориен-
тированного и ориентированного образцов и на сравнении ло-
кальных разогревов при разрушении облученных и необлучен-
ных образцов.
Количественный анализ температурных профилей зоны раз-
рушения показал, что соотношение эффектов пластической де-
формации и разрыва макромолекул составляет 0,45 и 0,55 со-
ответственно, что совпадает с калориметрическими оценками.
Величина локального разогрева вследствие пластической дефор-
мации хорошо коррелирует с температурными эффектами раз-
вития ориентационной вытяжки в ПЭТФ (см. рис. 5.15).
Нехрупкий характер разрушения полимера при скоростях,
далеких от скорости звука, позволяет рассмотреть роль локаль-
ных разогревов в долговечности образцов. Такой анализ пока-
зывает, что основной процесс разрушения развивается не при
температурах проведения эксперимента, а при существенно бо-
лее высоких. Это согласуется с представлением о возможности
разрушения по механизму теплового взрыва.
ЛИТЕРАТУРА
1. Разрушение/Под ред. Г. Либовица. Пер. с англ. Под ред. А. Ю. Ишлинско-
го. М„ Мир, 1973. Т. 1, 616 с.
2. Журков С. И. — Вести. АН СССР, 1968, № 3, с. 46—52.
275
3. Регель В. Р., Слуцкер А. И., Томашевский Э. Е. Кинетическая природа проч-
ности твердых тел. М., Наука, 1974. 560 с.
4. Тамуж В. П., Куксенко В. С. Микромеханика разрушения полимерных ма-
териалов. Рига, Зинатие, 1978. 294 с.
5. Кауш Г. Разрушение полимеров. Пер. с англ./Под ред. С. Б. Ратнера. М.,
Мир, 1981. 440 с.
6. Индебом В. Л., Орлов А. Н. — Физика металлов и металловед., 1977, т. 43,
№ 3, с. 469—492.
7. Журков С. Н., Петров В. А.—ДАН СССР, 1978, т. 239, № 6, с. 1316.
8. Журков С. Н. — ФТТ, 1980, т. 22, № 11, с. 3344—3349.
9. Кусов А. А., Веттегрень В. И,-—Там же, с. 3350—3357.
10. Гарбер Р. И., Гиндин И. А. — ФТТ, 1961, т. 3, № 1, с. 176—177.
И. Банщиков А. Г., Корсуков В. Е. — ФТТ, 1980, т. 22, № 8, с. 2368—2373.
12. Кусов А. А. — ФТТ, 1979, т. 21, № 10, т. 3095—3099.
13. Веттегрень В. И., Кусов А. А., Михайлин А. Я. — ФТТ, 1981, т. 23, № 5.
14. Бартенев Г. М., Разумовская И. В. — Физико-хим. мех. материалов, 1969,
т. 5, № 11, с. 60—68.
15. Салганик Р. Л. — ДАН СССР, 1969, т. 185, № 1, с. 97—100.
16. Салганик Р. Л. — ФТТ, 1970, т. 12, № 5, с. 1336—1343.
17. Дадобаев Г., Слуцкер А. И.—-ФТТ, 1981, т. 23, № 7, с. 1936—1942; Высо-
комол. соед., 1982, сер. А, т. 24, № 8, с. 1616—1622.
18. Патрикеев Г. А. — ДАН СССР, 1958, т. 120, № 3, с. 339—342.
19. Томашевский Э. Е. — ФТТ, 1970, т. 12, № 11, с. 3202—3207.
20. Tomashevskii Е. Е., Egorov Е. A., Savostin A. Ya. — Intern. J. Fract., 1975,
vol. 11, N 5, p. 817—827.
21. Годовский Ю. К. и др. — ФТТ, 1971, т. 13, № 8, с. 2289—2295.
22. Чавычелов А. Д. — Высокомол. соед., 1966, т. 8, № 1, с. 49—55.
23. Чавычелов А. Д.—Мех. полимеров, 1966, № 5, с. 664—670; 1967, № 1.
24. Келли А. Высокопрочные материалы. Пер. с англ. М., Мир, 1976. 262 с.
25. Качанов Л. М. Основы механики разрушения. М., Наука, 1974. 311 с.
26. Rice J. R.—J. Meeh. Phys. Solids, 1978, vol. 26, N 2, p. 61—78.
27. Берри Дж. П. — В кн.: Разрушение/Под ред. Г. Либовица. Пер. с англ. Под
ред. Ю. Н. Работиова. М., Мир, 1976. Т. 7, ч. 2, с. 7—65.
28. Бартенев Г. М.—Мех. полимеров, 1966, № 5, с. 700—721.
29. Williams М. L. — Intern. J. Fract. Meeh., 1965, vol. 1, N 4, p. 292—310.
30. Schapery R. A.— Intern. J. Fract., 1975, vol. 11, N 1, p. 141—159.
31. Kambour R. P., Barker R. E. — J. Polymer Sci., 1966, pt A-2, vol. 4, N 3.
32. Levy N., Rice J. R. — In: Physics of Strength and Plasticity/Ed. by A. S. Ar-
gon. Cambridge, MIT, Press, -1969, p. 277.
33. Williams J. D. — Intern. J. Fract. Meeh.. 1972, vol. 8, N 4, p. 393—411.
34. Weichert R., Schdnert K'—J. Meeh. Phys. Solids, 1974, vol. 22, N 1, p. 127.
36. Weichert R., Schdnert K. — J. Meeh. Phys. Solids, 1S78, vol. 26, N 3, p. 151.
36. Карслоу Г. С., Егер Д. Теплопроводность твердых тел. Пер. с аигл./Под
ред. А. А. Померанцева. М., Наука, 1964. 487 с.
37. Godovsky Yu. К — Polymer, 1981, vol. 22, N ,1, p. 75—86.
38. Годовский Ю. К. Теплофизические методы исследования полимеров. М.,
Химия, 1976. 216 с.
39. Ратнер С. Б., Коробов В. И, — Мех. полимеров, 1965, № 3, с. 93—100; Рат-
нер С. Б., Коробов В. И., Агамалян С. Г. — Физико-хим. мех. материалов,
1969, т. 5, № 1, с. 88—93.
40. Баренблатт Г. Я. и др. — Ииж. ж. Мех. тверд, тела, 1966, № 6, с. 47—58.
41. Tomashevskii Е. Е. е. а. — Intern. J. Fract., 1975, vol. 11, N 5, p. 803—815.
42. Fuller K. N. e. a.—Proc. Roy. Soc., 1975, Ser. A, vol. 341, p. 537—557.
43. Doll W. — Koll-Ztschr. u. Ztschr. Polymere, 1972, Bd 250, N 11/12, S. 1066—
1073; приведенное значение 7=230 К цитируется по [5], с. 382.
44. Егоров Е. А. и др. — ФТТ, 1975, т. 17, № 1, с. 111—117.
45. Егоров Е. А., Жиженков В. В., Безладнов С. Н. и др. — Высокомал. саед,,
1980, сер. А, т. 22, № 3, с. 582—588.
46. Egorov Е. А. е. а. —Acta Polym., 1980, vol. 31, N 9, p. 541—545.
47. Doll W. — Intern. J. Fract., 1976, vol. 12, N 4, p. 595—605.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аморфная текстура 190 сл., 232 сл., 237 сл.
Ангармоиизм 40, 50, 88 сл., 107 сл., 123,
244 сл.
Анизотропия
теплопроводности 61
аморфных полимеров 79, 80
кристаллических полимеров 80—84
ориентированных полимеров 79—84
потенциалов взаимодействия 89, 90
теплового расширения 103, 104
термоэластнчности ориентированных поли-
меров 194 сл.
Блок-сополимеры 156—163
Большой период 75 сл., 182 сл.
Бутадиеи-стирольные термоэластопласты
158, 159, 161
Внутренние напряжения 213 сл., 221, 222
Виутреиияя энергия 85, 86 , 88, 120—124, 128,
130—136, 144, 167—169
Виутрифибрилляриые аморфные области
179 сл.
Всестороннее расширение (сжатие) 118 сл.
Вынужденная эластичность 157 сл.
Высокомодульиые полимеры 192, 193
Гауссовы сетки 127 сл.
Гетерофазные эластомеры 155 сл.
Гибкость цепей 15
Гистерезис механический 157, 158
Гистерезисный разогрев 265, 266
Давление
внутреннее 87, 91, 122 сл., 129
кинетическое (термическое) 87, 91,
122 сл., 129
Деформация 203 сл.
высокоэластическая 126 сл.
вынужденная высокоэластическая 157 сл.,
205 сл.
изотермическая 119, 120
микрофибрилл 180 сл.
молекулярных сеток 126-сл.
ориентированных кристаллических поли-
меров 167 сл.
обратимая 167, 168
пластическая 203 сл., 241 сл., 268, 269
скачкообразная 233 сл.
упругая 117 сл.
Дисперсионная кривая 11, 12
Диссипация энергии
при ориентации 203 сл., 223—225
— разрушении 250 сл., 267 сл.
Длина
корреляции 59, 65 сл.
свободного пробега 56 сл.
Долговечность 242 сл.
Домены 156 сл.‘
Закон
Гука 118,' 124
Дебая 19 сл.
дисперсии
воли изгиба 22, 23
изотропной среды 18
крутильных колебаний 11, 22
поперечных колебаний 10, II, 23, 25
продольных колебаний 10, 23
Дюлоига — Пти 11, 19 сл.
логарифмический скоростей 206 сл.
теплового расширения 118, 124
Фурье 260
Изоэиергетические цепи 174
Инверсия термоупругая
внутренней энергии
твердых полимеров 121—123, 125, 167,.
168
эластомеров 132—134, 147, 148
теплоты эластомеров 132—134, 147, 148,
174
Кластеры 83 сл.
Колебания (моды)
акустические 11, 19, 25; 96
амплитуда ПО, 123
деформационные 16, 99, 106 сл.
крутильные 11, 22, 39, 90, 106 сл., 250
оптические 11, 12, 96, 98
поперечные 10, 11 сл., 59, 68 сл., 106
продольные 10 сл., 59, 68 сл., 250
характеристические 8, 38
Константы упругости
валентных связей 10
— углов 10
межмолекуляриого взаимодействия 13
Континуум 18 сл., 65 сл.
Концентраторы напряжения 242, 248
Коэффициент
ангармоничности 110, 244
анизотропии 80 сл., 194 сл.
интенсивности напряжений 258
квазиупругой силы ПО, 244
перенапряжениости 245 сл.
Пуассона 18, 122, 129, 169, 181 сл., 256 СЛ-
структурио-чувствительиый 243 сл.
теплового расширения 87, 117, 118, 101 сл.,
131—133, 165 сл., 195 сл., 246—249
•теплопроводности 61, 62
Кристаллы с вытянутыми цепями (КВЦ)
189 сл.
Кручение 125, 126, 135, 136
Межмолекулярное взаимодействие 12, 13,
89, 90, 171, 178
Межфибрилляриые аморфные области
181 сл.
Мембранный эффект 103
Микровзрыв 252 сл., 271 сл.
Микрофазовое разделение 156 сл., 163 сл.
Мнкрофибриллы 82, 179 сл., 196 сл., 217,
232
Модель
агрегатная 83
Айермана 61
Боиарта'и др. 172
Вада и др. 90
Гесса — Стэттоне — Херла (ГСХ) 179 сл.
Дебая 18 сл.
двумерных пачек 92
двухфазная кристаллических полимеров
51, 71, 112, 175, 195
дырочная 41, 94 сл.
избыточной теплоемкости стекла 30 сл.
Клеменса 58, 75
Кобаяси 231
Лифшица 23 сл., 103 сл.
локализованных резонансных колебаний
32, 66 сл.
Максвелла 71 сл., 81
Моргаиа — Смита 65
одномерной макромолекулы 10, 244, 280
Петерлииа — Преверзека 183 сл.
поворотио-изомериая 45, 53, 144 сл.
полужесткового «ожерелья» 109
расчета температурных эффектов прн вы-
тяжке 223 сл.
277
Модель
статистических кристаллических мости-
ков 82 сл., 192 сл., 193
Стокмейера — Хечта 14 сл., 90, 106
субцепей Рауза 63
Такаянаги 183
Тарасова 20 сл.
Тивери и др. 36
^унельиого перехода 32 сл., 66
Хоземана — Бонарта (ХБ) 181 сл.
Чоя и др. 106 сл.
Модуль
объемный 87, 93, 97, 118
сдвига 24, 125 сл.
спектроскопический макромолекулы 64
упругости (Юнга) 18, 24, 83, 102, 110,
124 сл., 169 сл., 189 сл., 245 сл.
Монокристалл
надмолекулярный 160 сл.
полиоксиметилена (ПОМ) 111 сл.
Натуральный каучук (НК)
изменение объема 148. 149
тепловое расширение 105
теплоемкость 50
термоупругая инверсия теплоты 147, 148
энергетическая составляющая 141, 144—
146, 165, 264
Обратимые деформации 167—170
Ориентационная вытяжка 205 сл.
и внутренние напряжения 215
источники энергетических изменений 213—
216
и конформационные изменения 214, 215
образование шейки 216
и степень кристалличности 2J4, 218, 219
структурные процессы 217, 218
Ориентационная кристаллизация 236 сл.
Отжиг 187 сл., 194
Параметр Грюнайзена 47, 85 сл., 245 сл.
решеточный 96, 97, 99
термодинамический 96, 97, 99
Переориентация 209, 210, 222
Поверхностная энергия разрушения 258.
259, 261
Податливость 235
Полиамиды 79, 105, 210, 227
Полибутилентерефталат (ПБТФ)
плавление 221, 222
скрытая энергия ориентации 209, 219, 231,
232
термоупругость 169, 191
Поливинилацетат (ПВА) 45
Поливинилиденфторид (ПВДФ) 80, 178
Поливинилидеихлорид (ПВДХ) 37, 110
Поливиниловый спирт (ПВС) 37, ПО, 111
Поливинилхлорид (ПВХ)
локальный разогрев 225, 227, 228
скрытая энергия ориентации 209, 213
теплоемкость 45, 50
теплота растворения 213
теплопроводность 80
энергетическая составляющая 161
Полидиметилсилоксан (ПДМС)
теплоемкость 45
теплопроводность 62
термоупругая инверсия теплоты 148
энергетическая составляющая 141, 144, 145.
154, 161, 162, 264
’Полиизобутилен (ПИБ) 45, 145, 161, 264
Полиизопрен 50
Поликапроамид (ПКА)
локальный разогрев 227—229, 232, 233, 237,
238
тепловое расширение 105, ПО, 111
термоупругость 168—170, 183, 189, 197, 198
скрытая энергия ориентации 208, 209, 219
Поликапроамид
энергетические эффекты при разрушении
266-271 *
Поликарбонат (ПК)
локальный разогрев 227, 238
теплоемкость 30, 45
теплопроводность 69
скрытая энергия ориентации 208, 209
Полиметнлметакрилат (ПММА)
локальный разогрев 259, 269, 273—275
параметр Грюнайзена 96, 97, 100
тепловое расширение 194
теплоемкость 30, 37, 45
теплопроводность 60, 62, 68—70, 80, 81
Поли-а-метилстирол 45
Полиморфизм при деформации 191
Полиоксиметилен (ПОМ).
разогрев при циклических деформациях
266
тепловое расширение 111, 112, 176 178
179
теплоемкость 52
теплопроводность 70, 72, 73, 79, 80
Полипропилен (ПП)
высокомодульиый 189, 190, 192
локальный разогрев 227—231, 237, 238
параметр Грюнайзена 100
тепловое расширение 177, 178, 182 184
186
теплоемкость 45
теплопроводность 70, 73, 79, 80
термоупругость 168—170
Полистирол (ПС)
локальный разогрев прн росте трещин
273, 274
параметр Грюнайзена 97, 100
скрытая энергия ориентации 222
тепловое расширение 194
теплоемкость 30, 37, 45
теплопроводность 63, 64, 69^ 80
термоупругость 166
Политетраметиленоксид 146
Политетрафторэтилен (ПТФЭ)
параметр Грюнайзена 100
теплоемкость 35, 37, 39, 52
Полифеииленоксид (ПФО) 45
Полихлоропрен 105, 144. 146, 148
Полиуретановые эластомеры 163, 164
Полихлортрифторэтилеи (ПХТФЭ) 70
Полиэтилен (ПЭ)
высокомодульиый 82, 83, 192
дисперсионные кривые изолированных це-
пей И, 12
кристаллы с вытянутыми цепями (КВЦ)
189, 193
локальный разогрев 221
параметр Грюнайзена 96, 98—101
плавление 220, 221
плотность спектрального распределения
11, 12
скрытая энергия ориентации 206—209, 219
стеклование 51, 52
тепловое расширение 105—111, 176—179,
198
теплоемкость 27, 28, 35—38, 45, 52, 53
теплопроводность 71, 73, 75—83
термодинамические функции деформации
транс-кристалла 193
термоупругая инверсия внутренней энер-
гии 168, 169
термоупругость 166—170, 182, 184—188, 197
уравнение состояния 91
энергетическая составляющая 145, 154,
155
Полиэтилеиоксид (ПЭО) 62, 70, 100, 146
Полиэтилеитерефталат (ПЭТФ)
локальный разогрев при росте трещин
273-275
----в шейке 225—227, 229, 231
скрытая энергия ориентации 209, 210
тепловое расширение 105
теплоемкость 31, 33, 34
:278
Полиэтилентерефталат (ПЭТФ)
теплопроводность 69—71, 73, 74, 78—81
термоупругость 166, 168, 169, 171—173,
190, 191, 198 •
Полнэтилакрилат (ПЭМА) 50, 51
Приближение
обобщенное 15
скелетное 8, 14 сл.
Проскальзывание
микрофибрилл 183 сл.
цепей 254 сл.
Проходные цепи 71, 175, 181
Прочность 242 сл.
Работа
деформации 120, 121, 123, 126, 128, 147
ориентации 206 сл., 216
Разогрев
при медленном разрушении 266 сл.
— ориентации 223 сл.
— разрыве стержней 264, 265
— росте трещины 259 сл., 272 сл.
— скачкообразной деформации 233 сл.
— циклической деформации 265 сл.
тепловые эффекты 262 сл.
Разрушение
усталостное 265, 266
хрупкое 241 сл., 255 сл.
хрупкопластическое 256 сл.
Растяжение
гауссовых сеток 127, 128
одноосное 124, 125
Расширение тепловое
монокристаллов ПОМ 111 сл.
ориентированных полимеров 173 сл.
растянутых эластомеров 174 сл.
цепных и слоистых структур 103 сл.
Рекристаллизация 229 сл.
Релаксация внутренних напряжений 215,
222
Свободная энергия деформированного со-
стояния 123
Свободный объем 41 сл., 132 сл.
Сдвиг 125, 126
Сетка(и)
аффинная 151
с бимодальным распределением 154
гетерогенные иегауссовы 167
с короткими цепями 154, 155
фантомная 151
Сжатие 118 сл.
всестороннее 118 сл.
гауссовых сеток 127, 128
одноосное 124, 125
Сжимаемость 43 сл., 139
анизотропия 150
Скрытая энергия
деформации 204, 205, 215, 216
ориентации 206, 216, 218, 221, 222
Смеси эластомеров 156 сл., 164 сл.
Сополимер этилена с пропиленом (СКЭП)
144, 145, 147, 148, 152, 161, 165, 264
Стеклование 40 сл., 51 сл., 60 сл., 166 сл.,
211 сл., 225, 231
Степень
анизотропии 194, 195
вытяжки 175—177, 193
кристалличности 209, 214, 221, 222
при ориентационной вытяжке 214, 218,
219
и теплоемкость 26 сл., 51—56
непрерывности кристаллической структу-
ры 192, 193
ориентации 194—196
Структурное рассеяние 58
Структурный фактор 75 сл.
Теория
Айермаиа 60 сл.
Баренблатта 236
Волькенштейиа — Птицына 53 сл., 144 сл.
Гиббса — Ди Марцио 42
Гриффитса 255 сл.
Дебая 18 сл.
Ди Марцио — Даула 46 сл.
кинетической прочности 242 сл.
Клеменса 58, 65, 75
Литлла 78
Лифшица 23 сл., ЮЗ сл.
локализованных резонансных колебаний*
32, 66 сл.
локальных разогревов 259 сл., 266
Моргана — Смита 65, 75 сл.
Оскотского — Смирнова 66
Роэ — Тонелли 43 сл.
статистической высокоэластичности
127 сл.
Стокмейера — Хечта 15
Такено — Года 37
Флори 151
Хансена и Хоу 62, 80
Чавычелова 253 сл.
Шварца 154
Тепловое сокращение
высокомодульных кристаллических поли-
меров 194
кристаллических макромолекул 104 сл.
ориентированных кристаллических поли-
меров 169 сл.
растянутых эластомеров 173 сл.
цепных и слоистых структур 103 сл.
Теплоемкость 8 сл., 87, 88, 91, 97, 98, 117~
118, 219, 220
взаимодействующих цепей и слоев 22^
24
дырочная 41 сл.
избыточная 29—37
изолированной цепи 9 сл.
и колебательные спектры 8 сл.
конформационная 42 сл., 53, 54
межмолекулярная 91, 97 сл.
невзаимодействующих цепей н слоев 1L.
23
низкотемпературная 11, 15, 23, 24, 26 сл.
в области стеклования 26, 27
растянутых цепей 53
скелетная 8, 23, 36, 39
при стекловании аморфных полнмеро»;
40 сл.
----кристаллических полимеров 51—54
и степень кристалличности 26 сл., 51—
56
и структура полимеров 26 сл.
температурная зависимость 27 сл.
цепных и слоистых структур 20 сл.
частично-кристаллических полимеров 28,.
29, 33, 35
Теплопроводность 56 сл.
аморфных полимеров 56, 57, 60 сл.
внутримолекулярная 61, 62
кристаллических полимеров 56—59, 70 сл,
межмолекулярная 60—62
и модуль упругости 83
— молекулярная масса 62. 63
низкотемпературная 65 сл., 74 сл.
и плотность 73
сверхориентированных полимеров 82 сл.
и степень кристалличности 73 сл.
температурная зависимость 56, 57; 60 сл..
фононный механизм 56 сл.
Теплота
деформации 120—122, 124, 130, 131—134,.
147, 166, 167
ориентации 207 сл., 214, 215
плавления 186, 220 сл.
разрушения 264 сл.
растворения 211 сл.
Термоупругость 166 сл.
аморфной текстуры 190 сл.
279*
Термоупругость
аморфных областей кристаллических по-
лимеров 168 сл.
высокомодульиых волокон 192 сл.
транс-кристалла ПЭ 193
ориентированных полимеров 183 сл.
— пленок 191
стержней 117, 124, 128 сл.
твердых эластичных волокон 189
Термоэластнчность
гауссовых сеток 127 сл.
гетерофазных эластомеров 155 сл.
негауссовых сеток 136 сл.
ориентированных полимеров 194 сл.
Термоэластопласты 155—157, 163
Тиксотропия 159
Трещины
макроскопические 262
микроскопические 247 сл., 255 сл.
субмнкроскопические 243 сл.
Уравнение
Бонди 64
Вада 91
Валаииса — Ландела 138
Гриффитса 256
Грюнайзена 87
Гута — Смолвуда 160
Дебая 19, 56, 88
Журкова 243 сл.
Клайперона — Клаузиуса 230
Клеменса 59
Крнгбаума — Конеко 137 •
Литлла 78
Лнфшица 23, 24
Максвелла 71
Ми—- Грюнайзена 87
Мунн — Ривлина 138
Ноуза 44
Пэстайна 91
Симха 95
Смита 137
состояния молекулярной сетки 127
— твердых тел 119, 124, 125
Тарасова 20 сл.
Тобольского — Шена 134
Френкеля 110
Чогла и др. 136
Эйнштейна 9
Усадка 188, 189, 222
Усталостное разрушение 265, 266
Фибриллизация 217
Фибриллярная структура ориентированных
полимеров 180 сл.
Фронт-фактор 127 сл., 134 сл., 151
Функция
Дебая 19 сл.
ориентации 80 сл., 195 сл.
распределения расстояний между конца-
ми цепей 127, 136 сл., 152 сл.
— частот 9 сл.
Эйнштейна 9
Холодная вытяжка 205, 206, 228
Целлюлоза 105
Шейка 157, 203 сл., 216
автоколебательный режим 233 сл.
равномерное распространение 225 сл.
Эластомеры
гетерофазные 155 сл.
деформация 126 сл.
наполненные 156 сл.
Энергетическая составляющая 128 сл.
коротких цепей 154, 155
наполненных эластомеров 161 сл.
симметричных цепей 144
смесей эластомеров 164
термоэластопластов 162 сл.
цепей с чередующимися валентными уг-
лами 145 сл.
Энергия '
деформации 203—205
конформационная 214, 215
поверхностная 256 сл.
разрушения 259 сл., 273 сл.
решетки
фибрнллизации 216
Энтальпия 219—221
при деформировании 125, 130
Энтропийная составляющая 128 сл.
Энтропия
при деформации 123, 128, 130—132, 135,
136, 144, 147, 148, 204
колебательная 123, 129, 204
конфигурационная 204
конформационная 128 сл., 214, 215
Эффективная константа упругости 107
Юлий Кириллович Годовский ИБ № 1070
ТЕПЛОФИЗИКА
ПОЛИМЕРОВ
Редактор
Г. М. Медникова
Художник
Е. В. Бекетов
Художественный редактор
Н. В. Носов
Технический редактор
В. М. Скитина
Корректор
П. Б. Иваницкая
Сдано в наб. 26.07.82. Подп. в печ. 20.10.82.
Т-17555. Формат бумаги 60X90‘/ie. Бумага тип. № 2.
Гари, литературная. Печать высокая.
Усл. печ. л. 17,5. Ус. кр.-отт. 17,5. Уч.-нзд. л. 20,20.
Тираж 4000 экз. Заказ № 440. Цена 3 р. 30 к.
Изд. № 1956.
Ордена «Знак Почета» издательство «Химия».
107076, Москва, Стромынка, 13.
Московская типография № 11 Союзполиграфпрома
при Государственном комитете СССР по делам
издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., д. 1.