/
















































































































































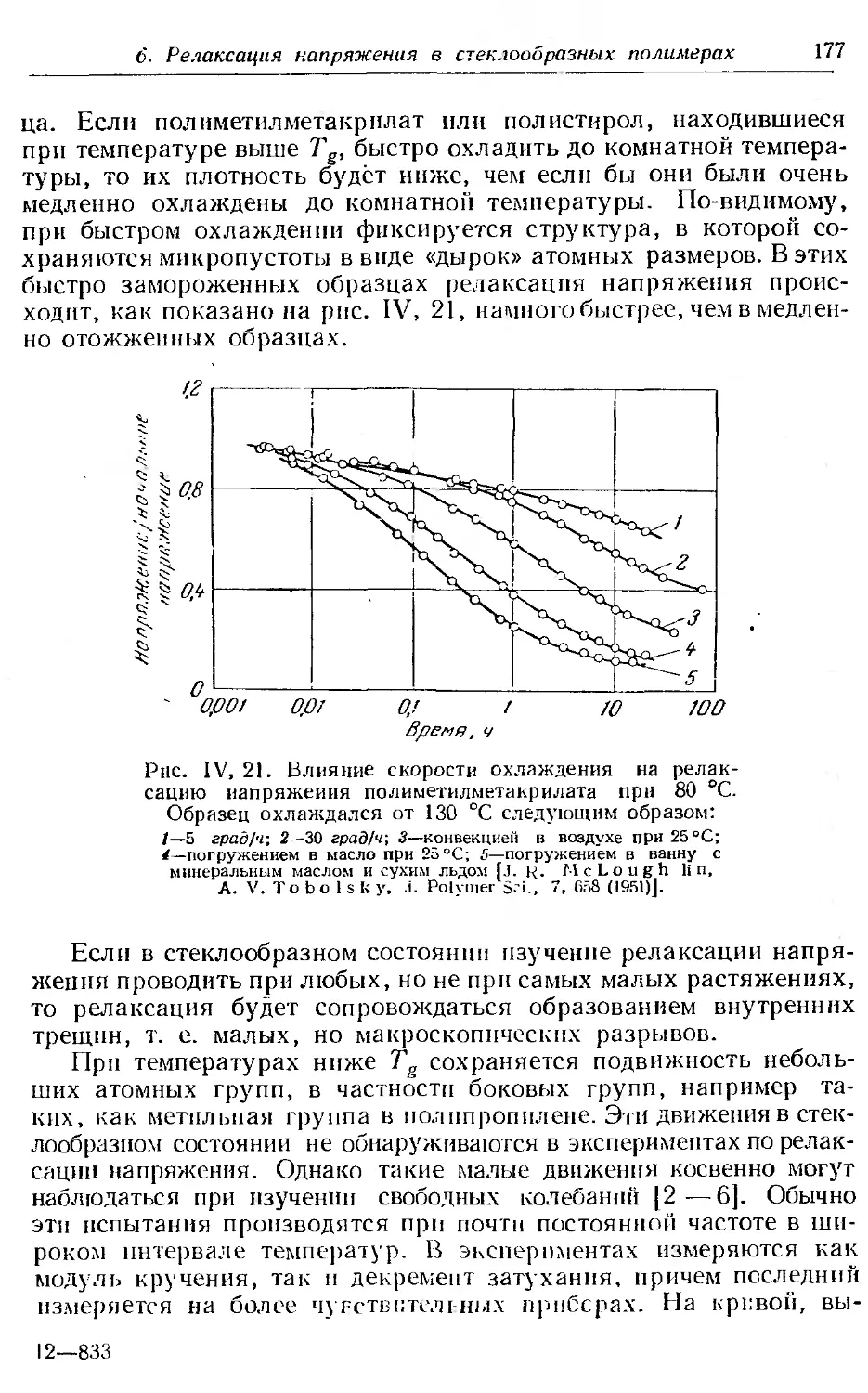









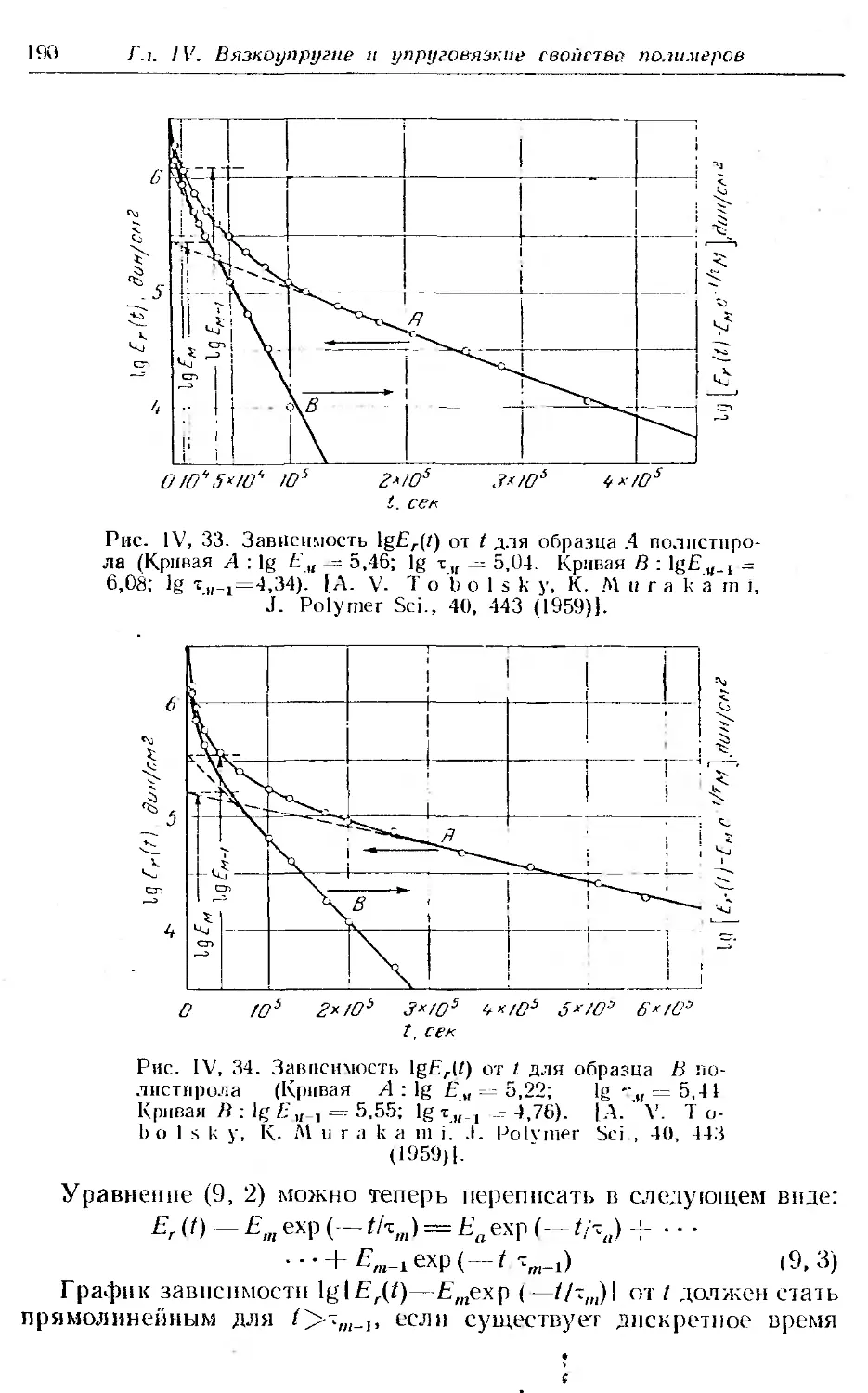

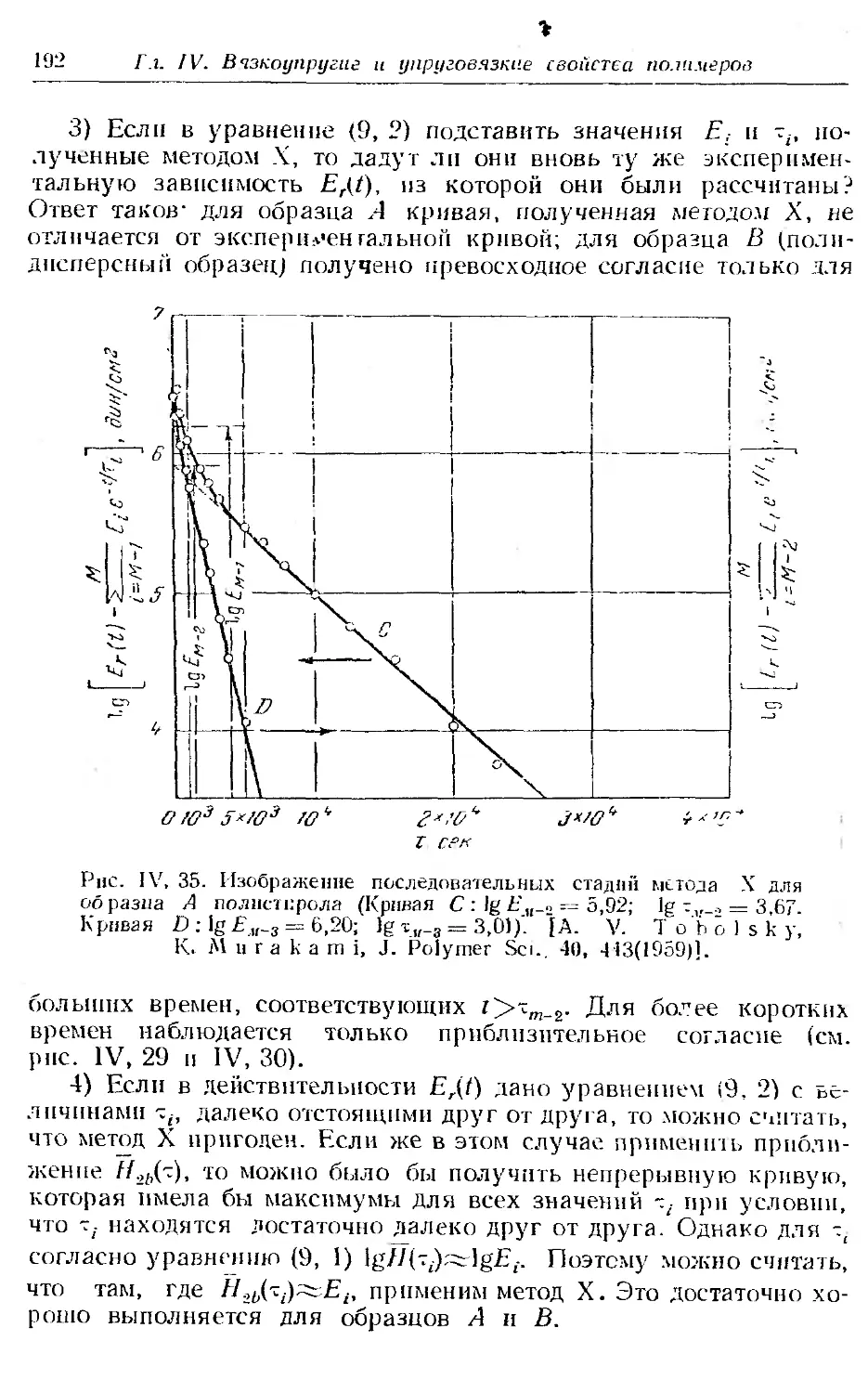








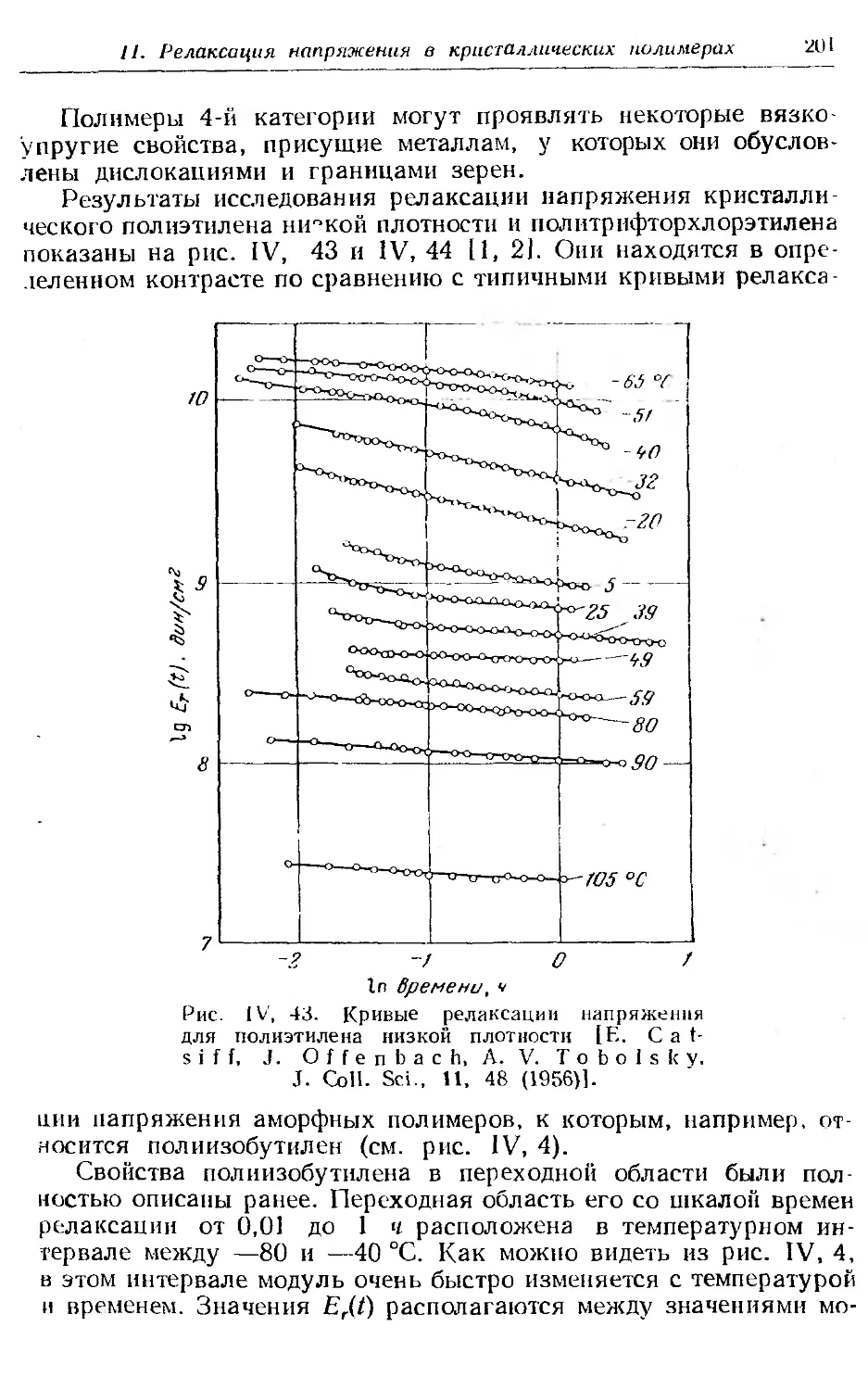
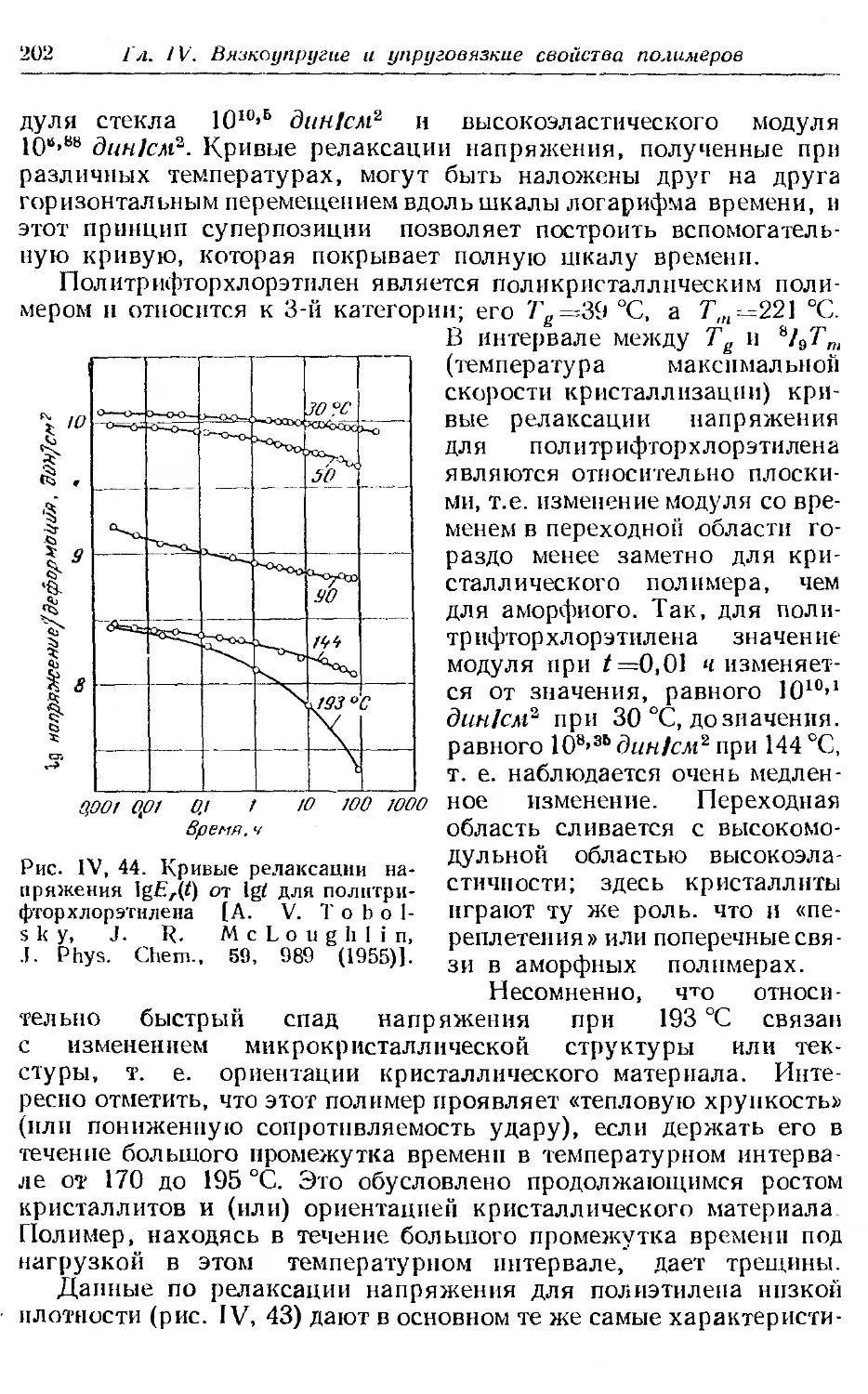





















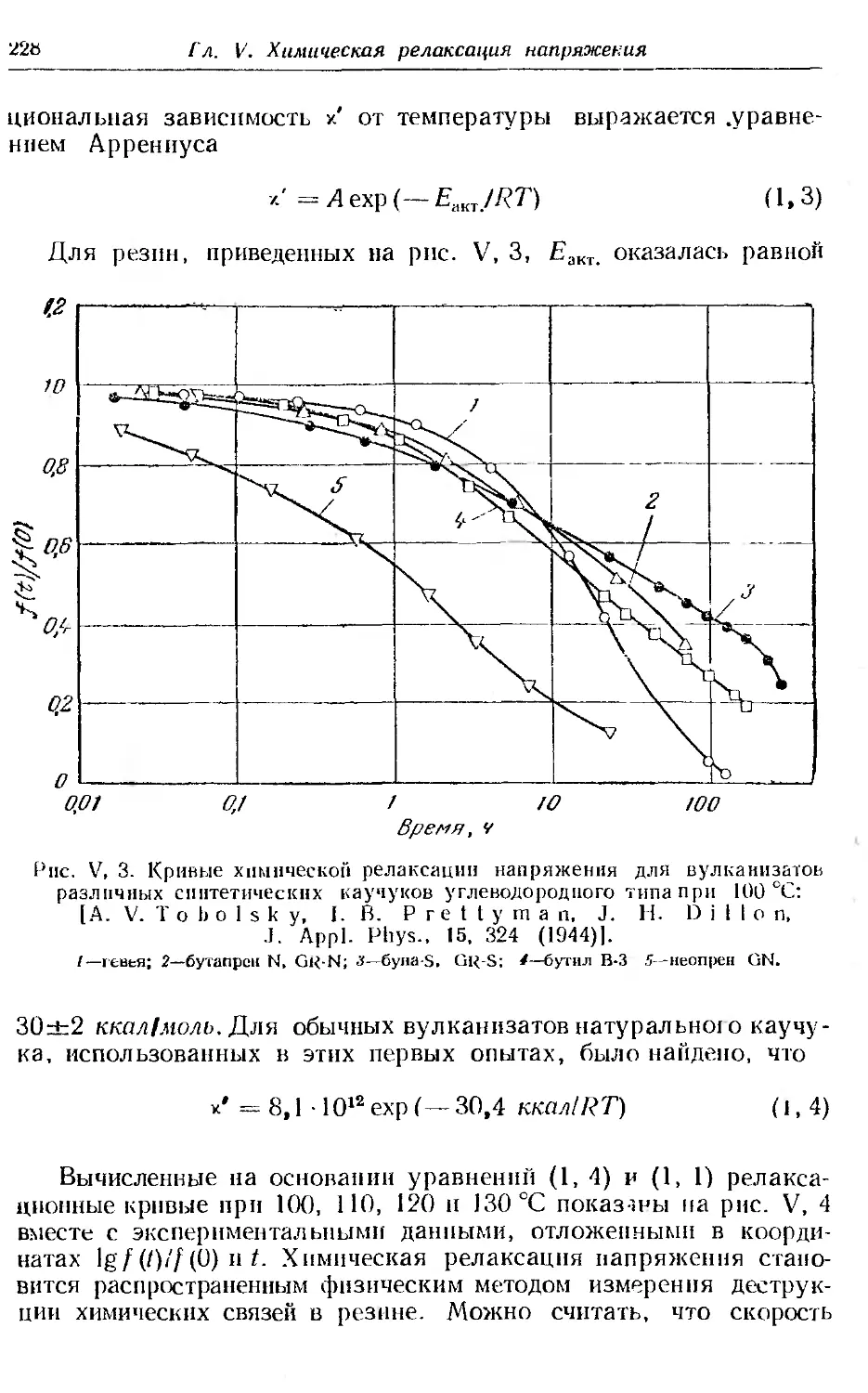



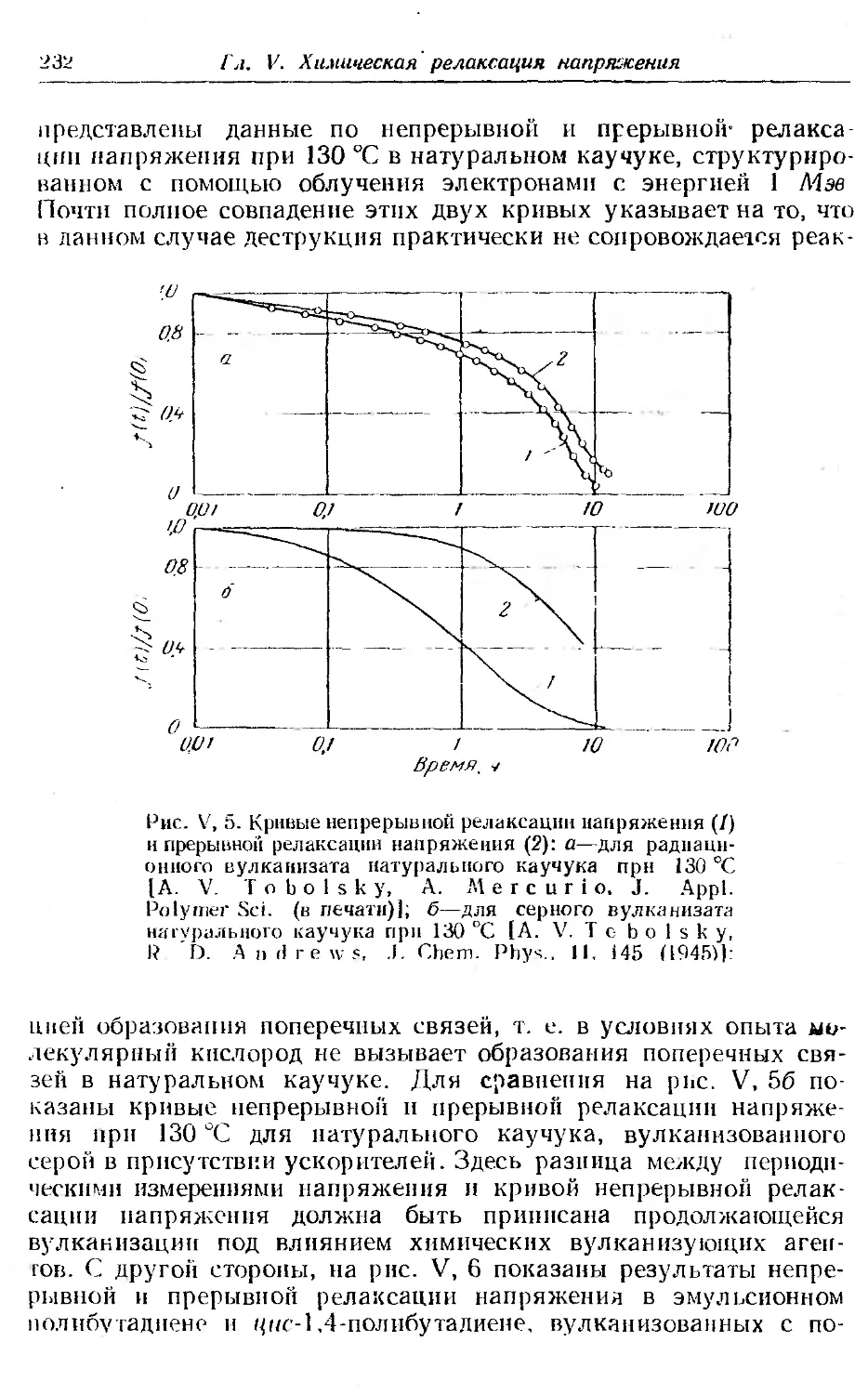
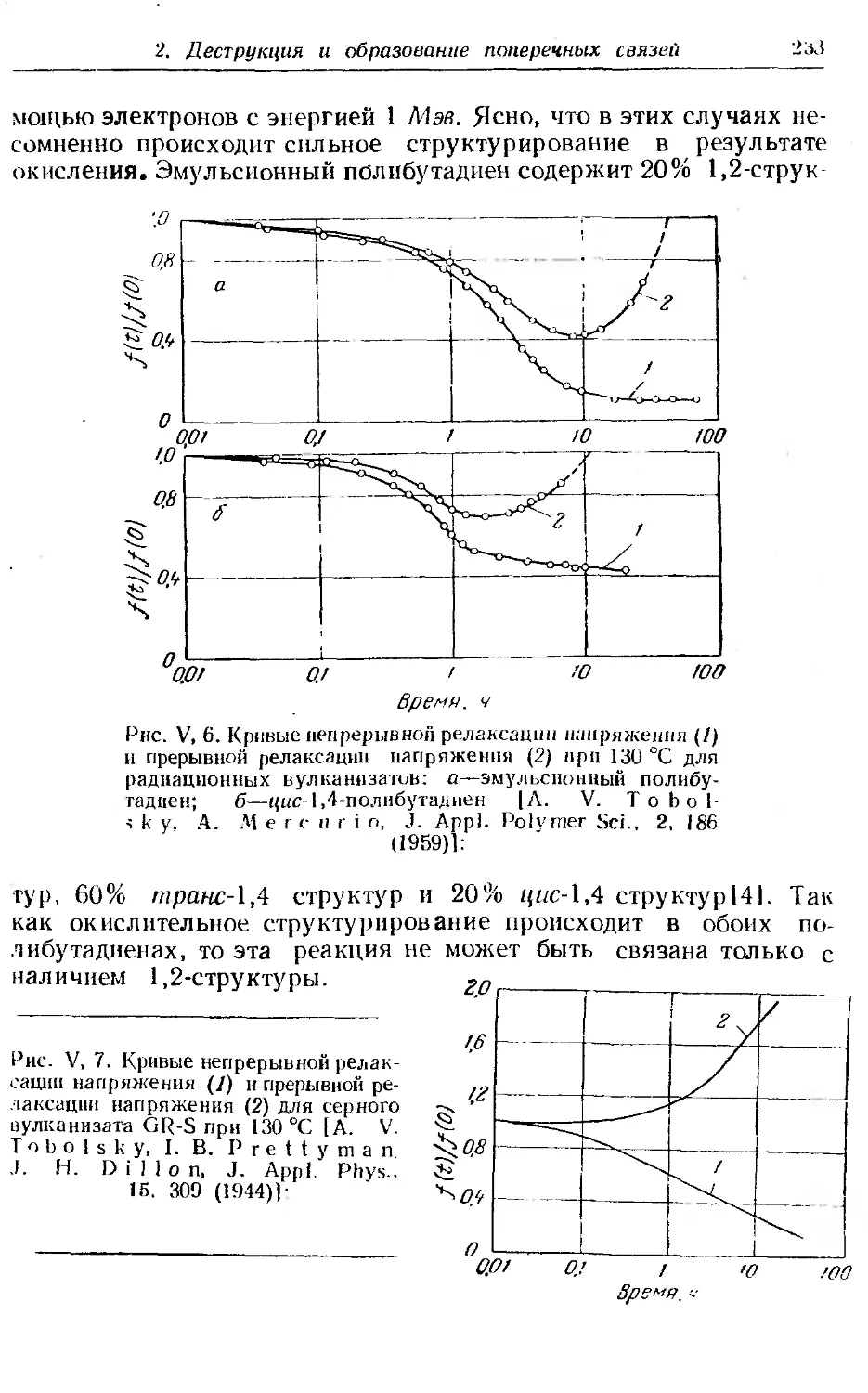








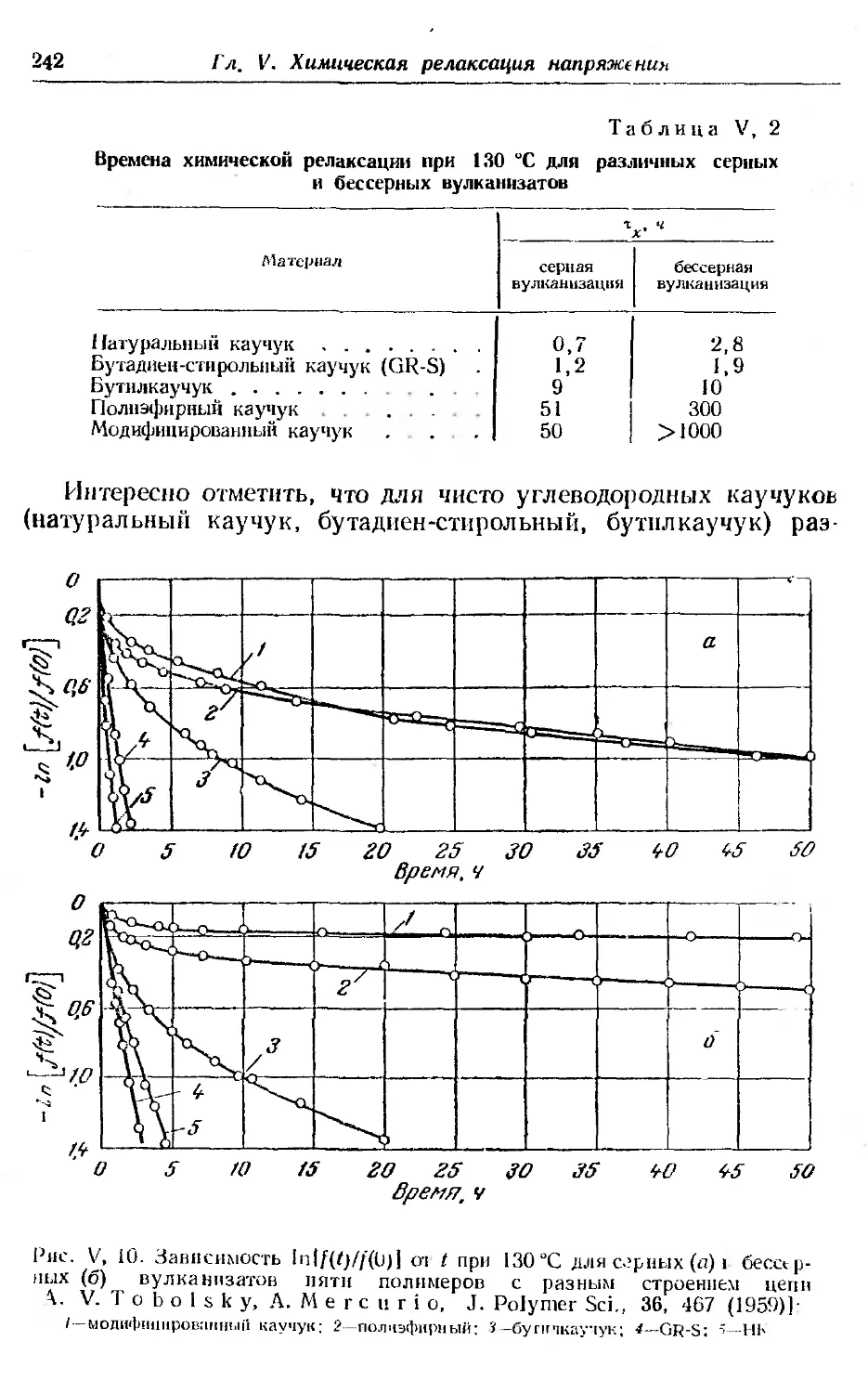








































































Текст
А. ТОБОЛЬСКИЙ
СВОЙСТВА И СТРУКТУРА
ПОЛИМЕРОВ
ПЕРЕВОД (. АНГЛИЙСКОГО
под редакцией Г. Л. Слонимског.
и Г. М. Бартенева
ИЗДАТЕЛЬСТВО «ХИМИЯ»
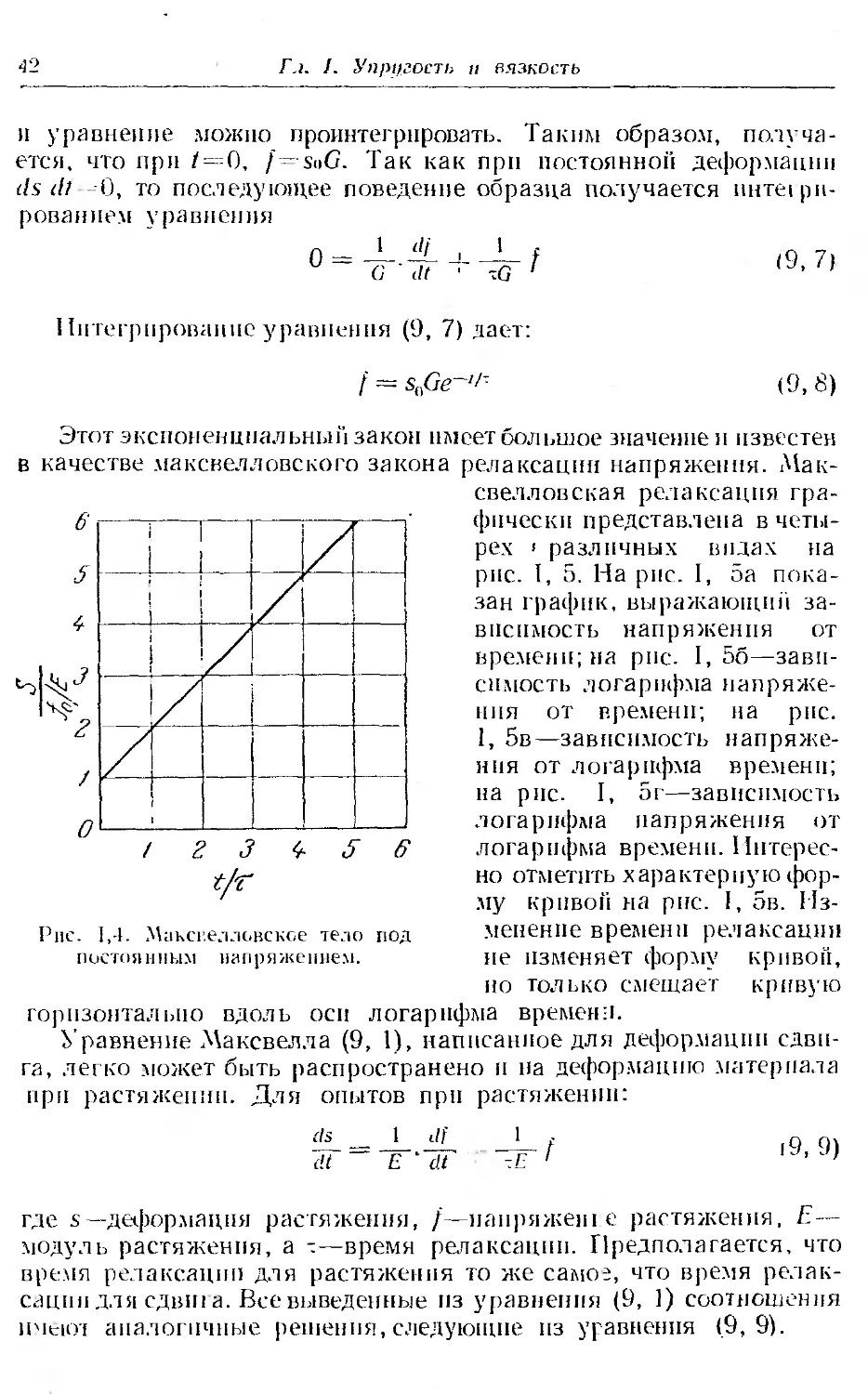
Москва 1964
УДК 541.6
Автор книги — представитель широко известной в США
научной школы, разрабатывающей проблемы физики и хи-
мии полимеров. В книге, написанной как учебное пособие,
па высоком научном уровне, но без излишне сложного ма-
тематического аппарата, рассмотрены вопросы упругости,
высокоэластнчиости и текучести полимерных материалов,
имеющие большое значение для процессов переработки по-
лимеров, а также для расчета изделий из-них.
Кинга представляет большой интерес для широкого круга
специалистов, работающих в области физнко-химин полиме-
ров и переработки полимерных материалов; опа может слу-
жить учебным пособием для студентов соответствующих спе-
циальностей технических вузов.
PROPERTIES AND STRUCTURE
OF POLYMERS
BY ARTHUR V. TOBOLSK.Y
Professor
Department of Chemistry
Princeton University
NewYork — London
John Wiley & Sons, Inc.
СОДЕРЖАНИЕ
Предисловие редакторов .... 7
Предисловие автора................................................. 8
Глава I. Упругость и вязкость
I. Сжатие изотропных веществ. Уравнение состояния.
Л1одуль объемного сжатия..................................... 6
2. Статистическая термодинамика объемного сжатия 12
3. Уравнение состояния для молекулярных кристаллов . . 17
4. Упругие свойства изотропных тел при малых деформациях 21
5. Упругие константы и колебания решетки.................... 24
6. Термодинамическая трактовка простого растяжения 28
7. Упругость идеальных каучуковых сеток 31
8. Вязкость жидкостей....................................... 35
9. Упруговязкость: модель Максвелла 40
f’j.T а в а II. Основы физики полимеров
1. Конформации линейной полимерной цепи 45
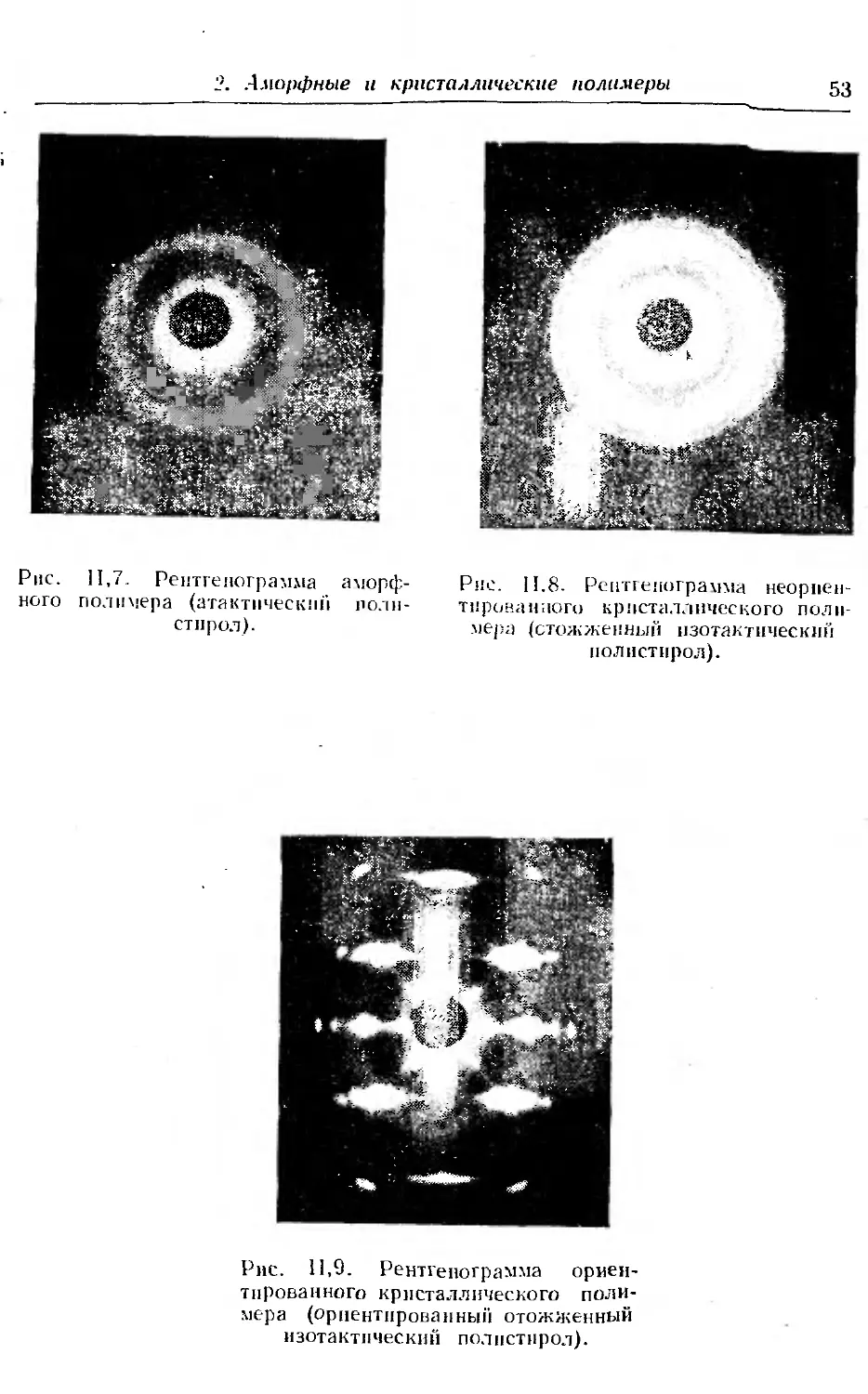
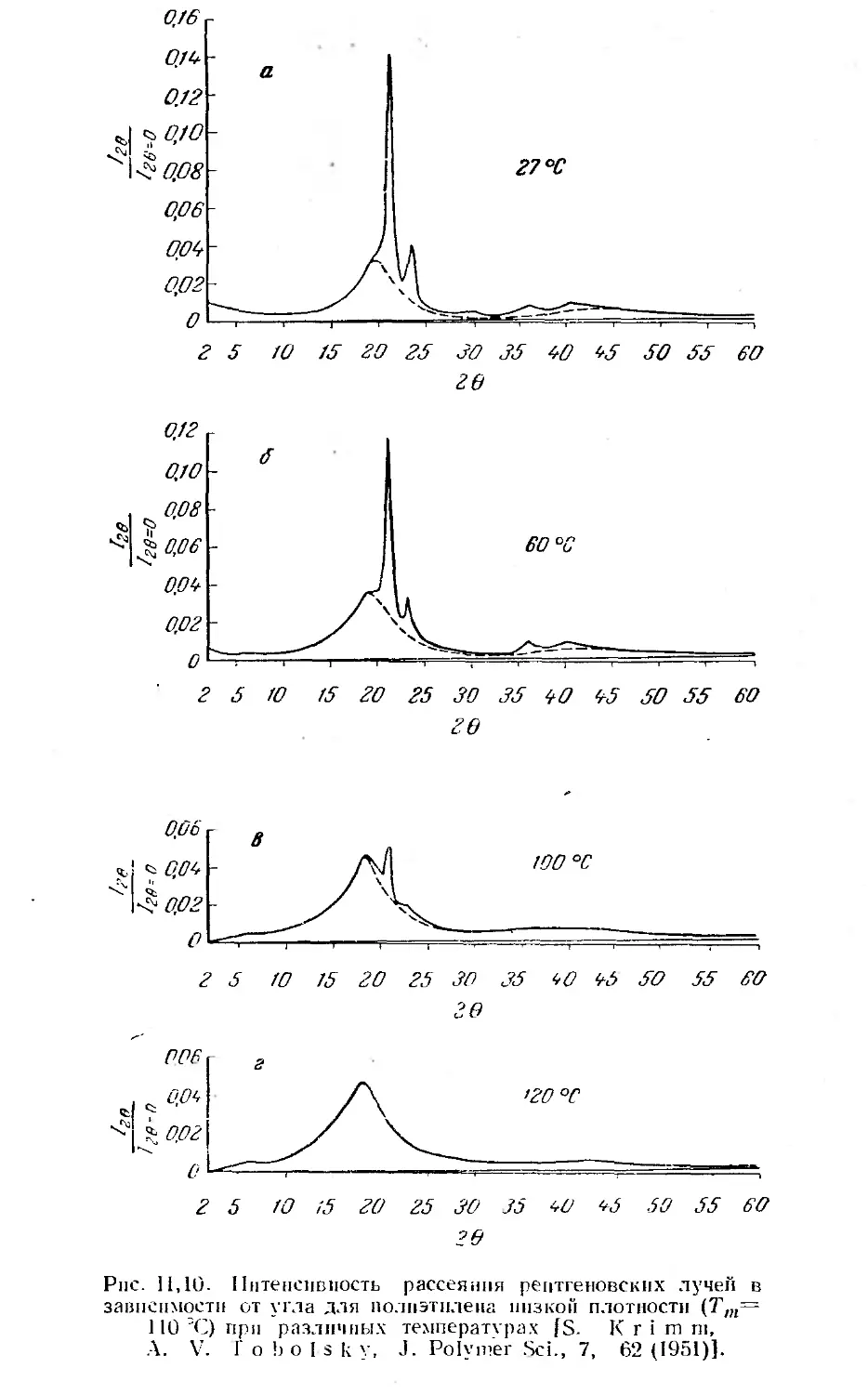
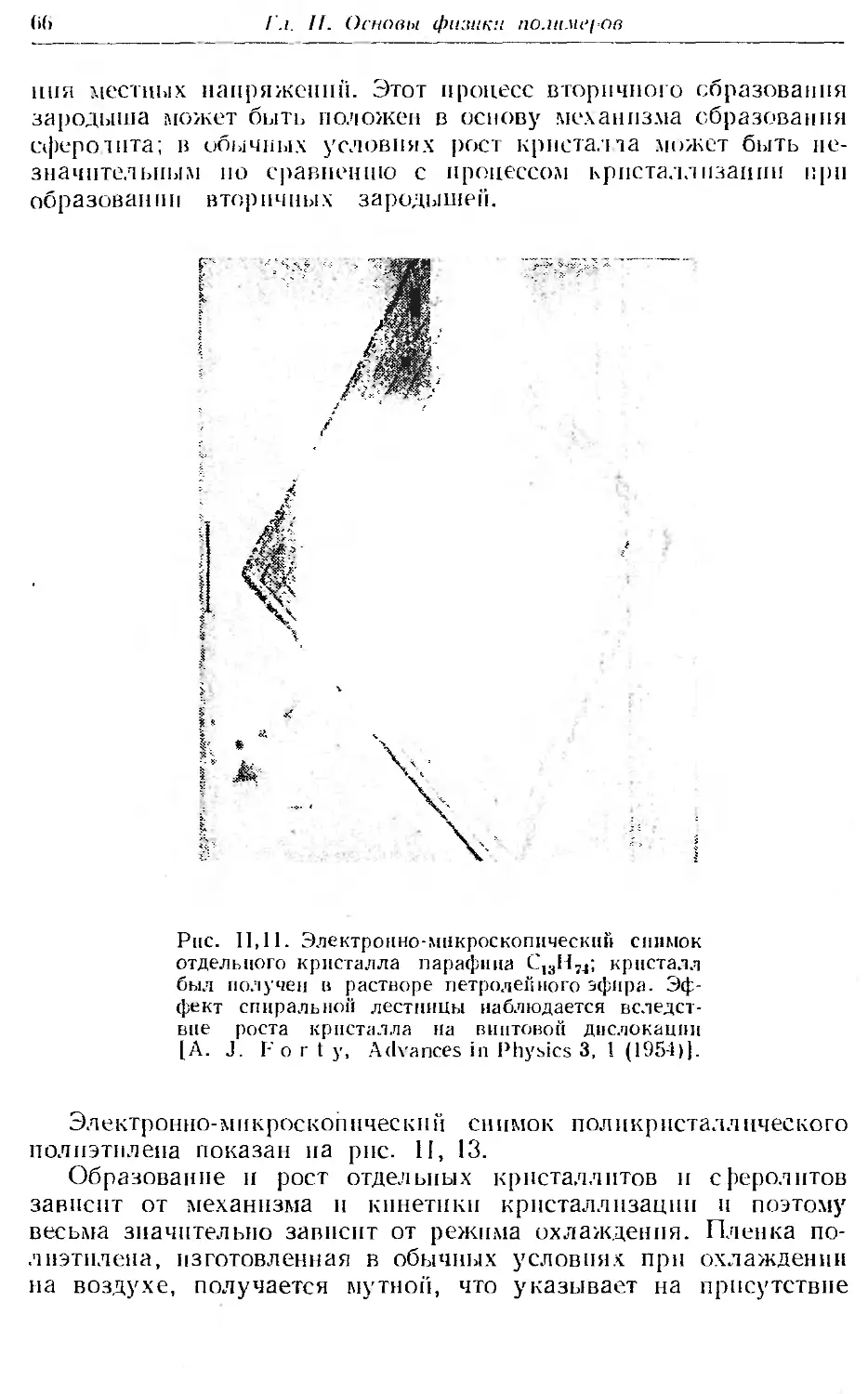
2. Аморфные и кристаллические полимеры , 50
3. Термодинамика кристаллизации............................. 60
4. Кинетика кристаллизации.................................. 64
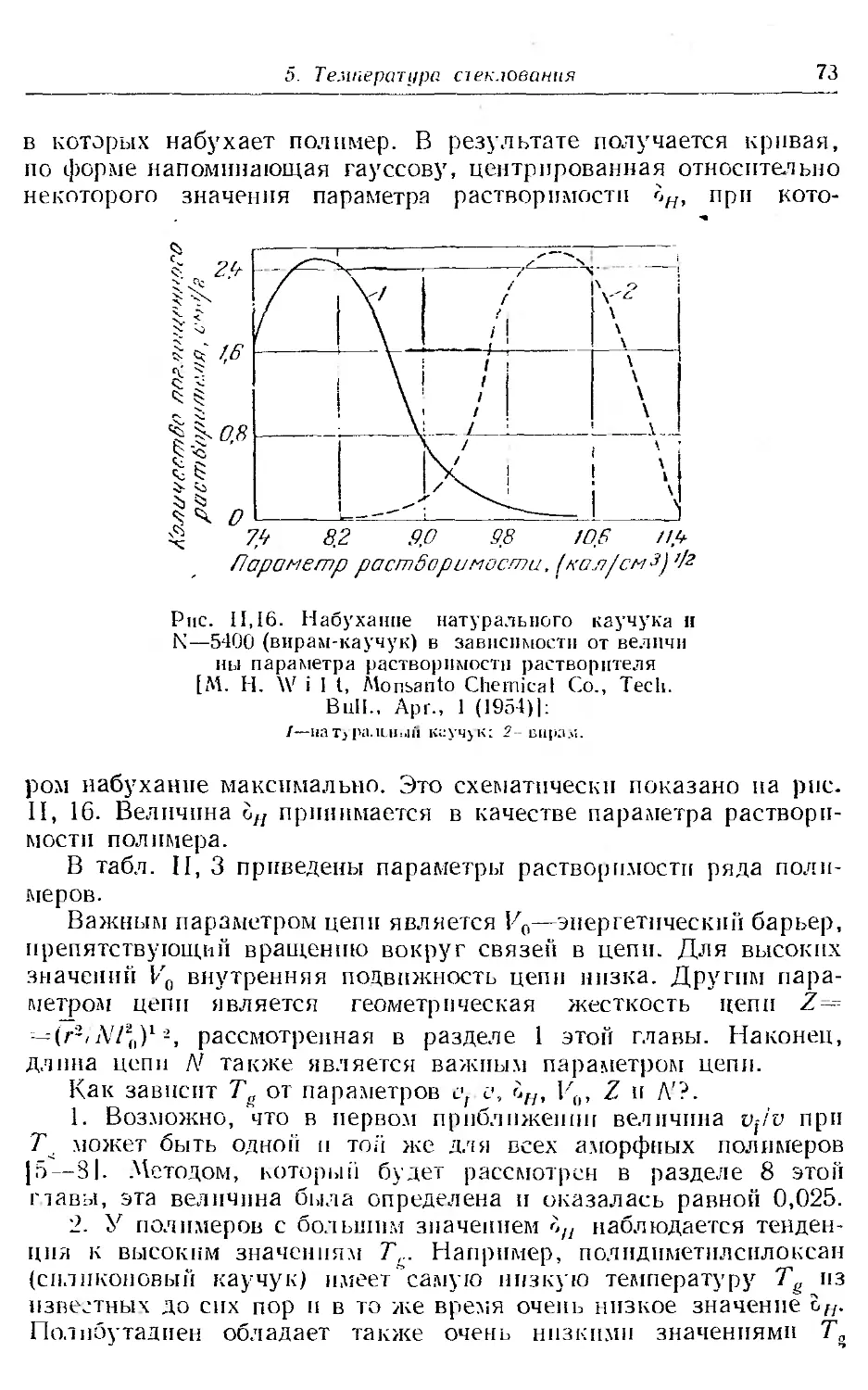
5. Температура стеклования ................................. 68
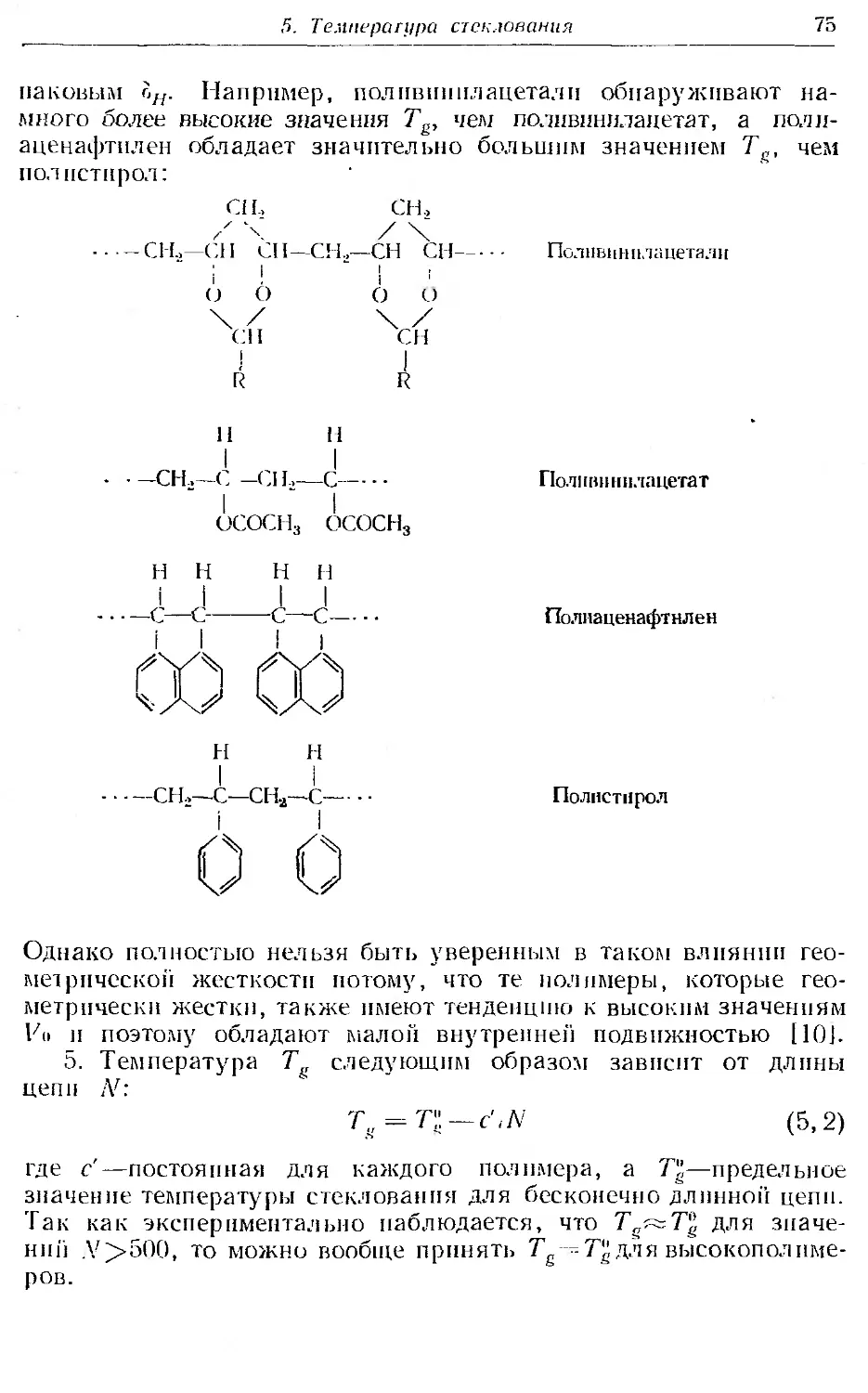
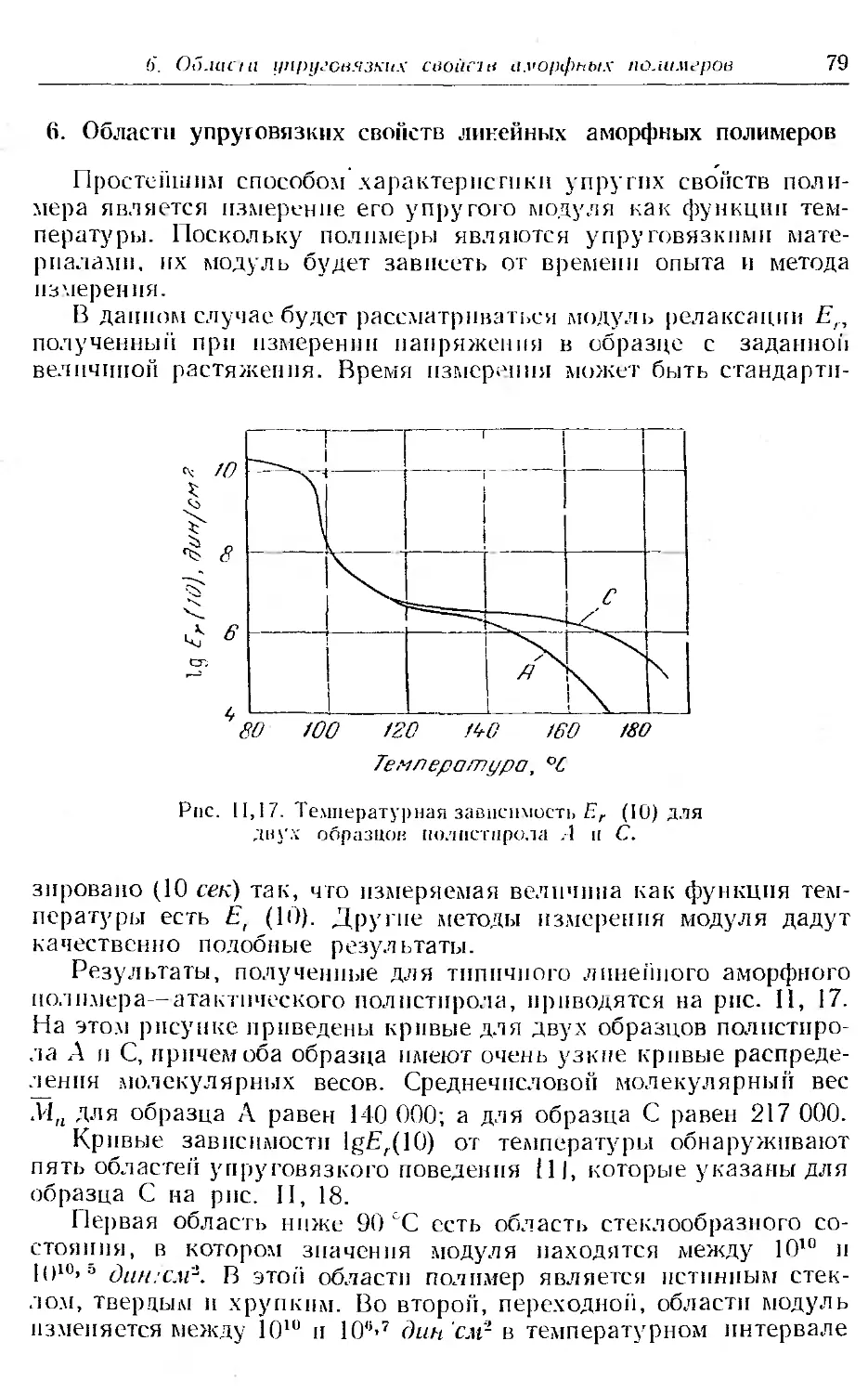
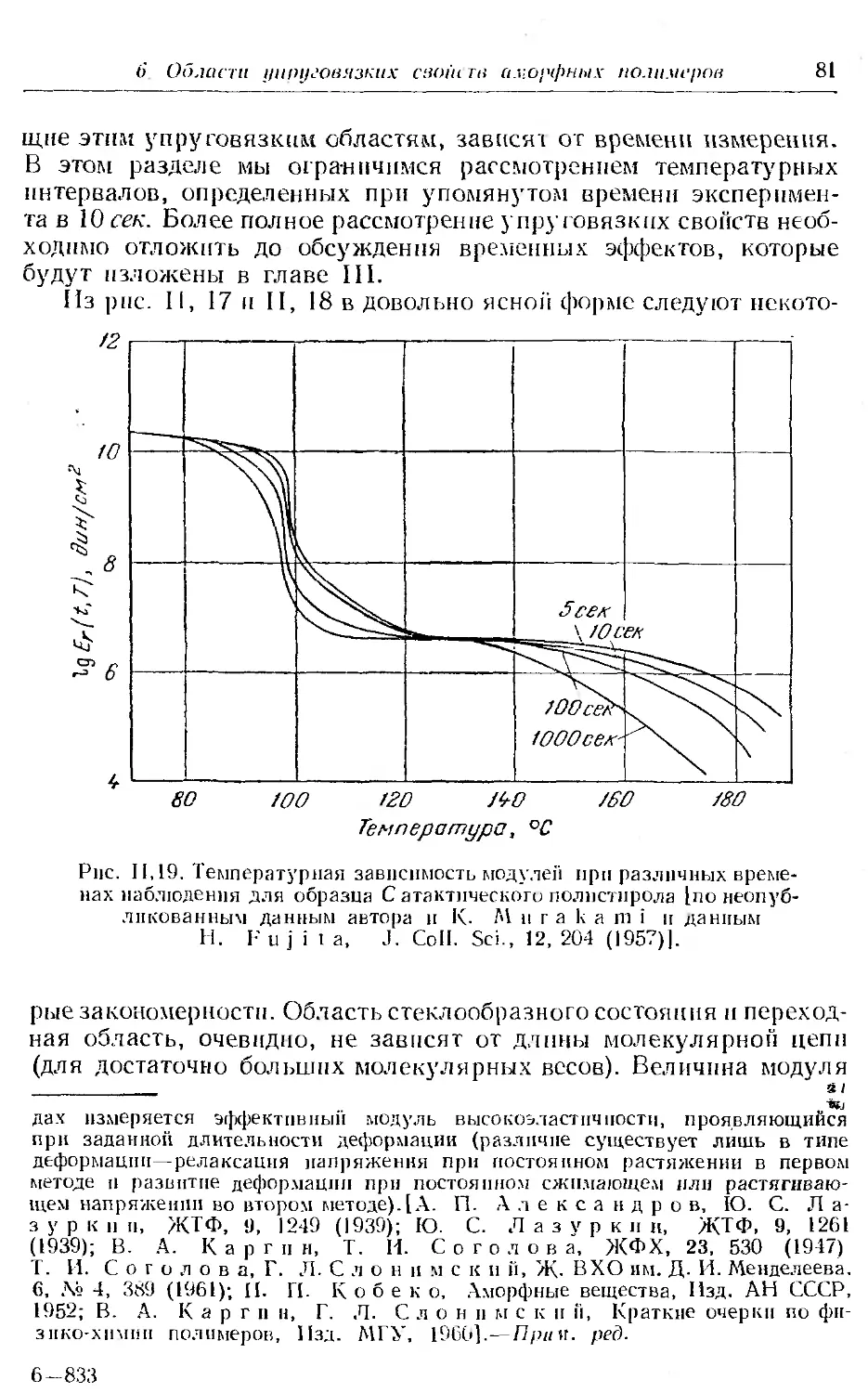
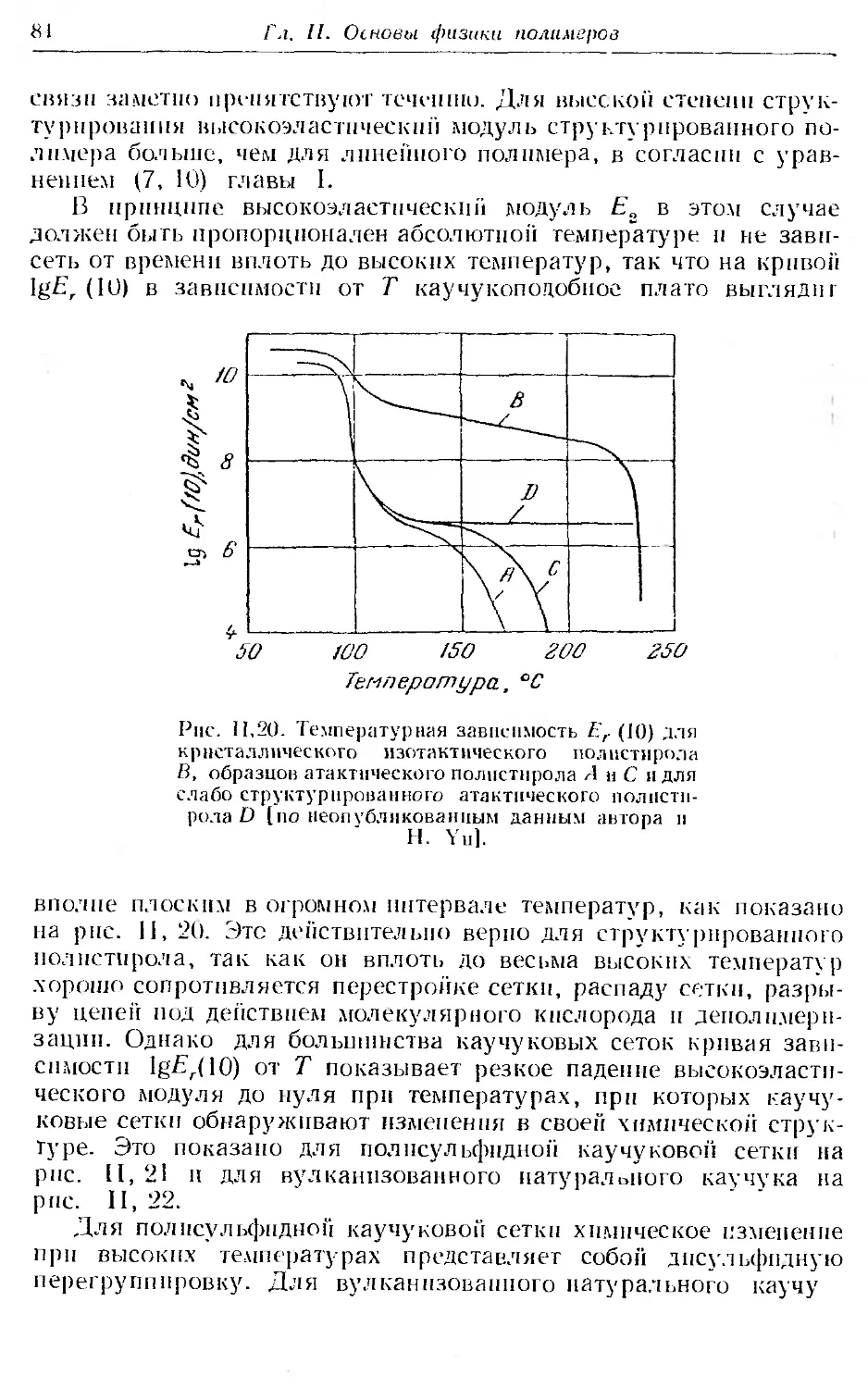
6. Области упруговязких свойств линейных аморфных полимеров 79
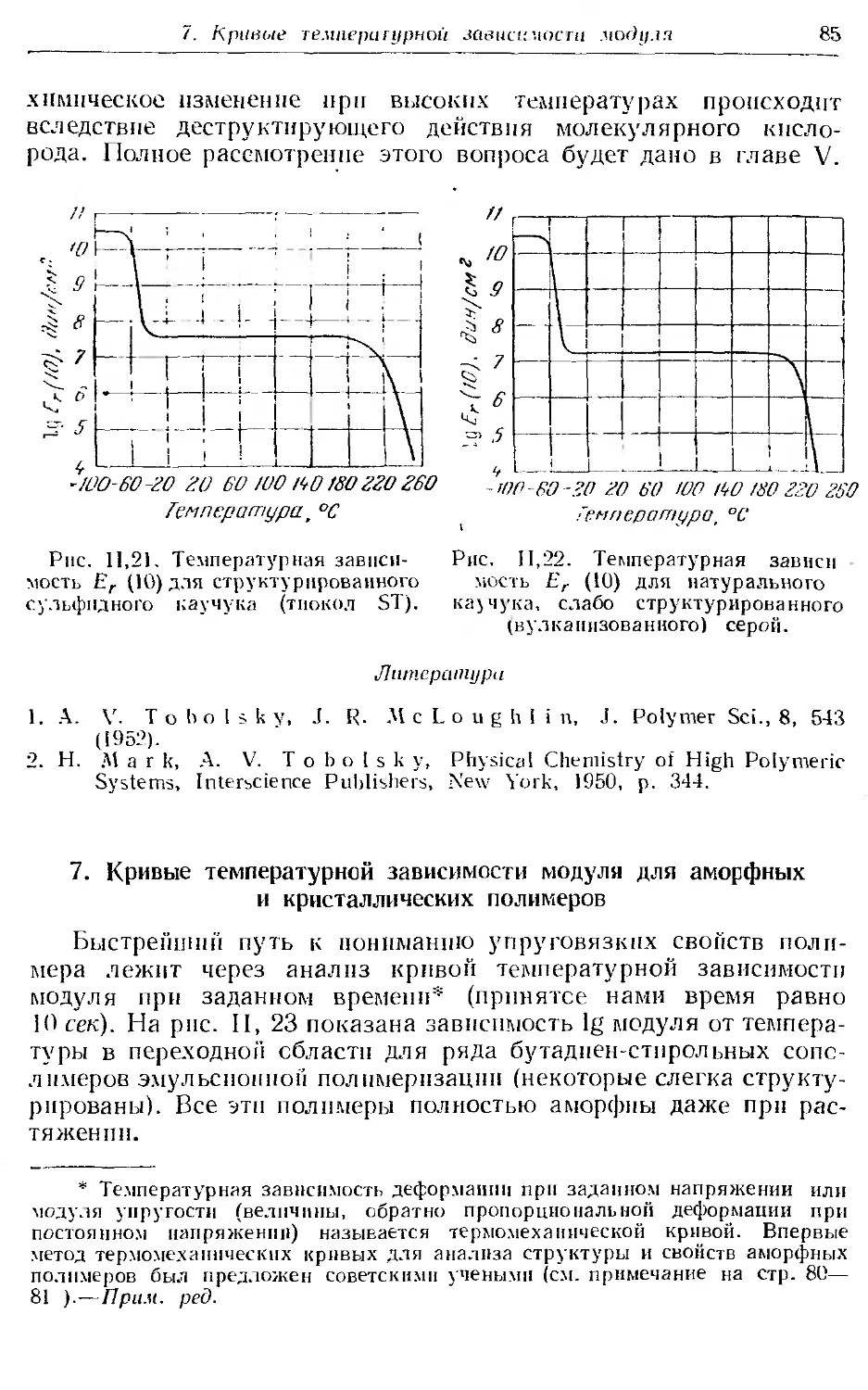
7. Кривые температурной зависимости модуля для аморфных и кри-
сталлических полимеров...................... . б5
8. Вязкое течение линейных аморфных полимеров .... 90
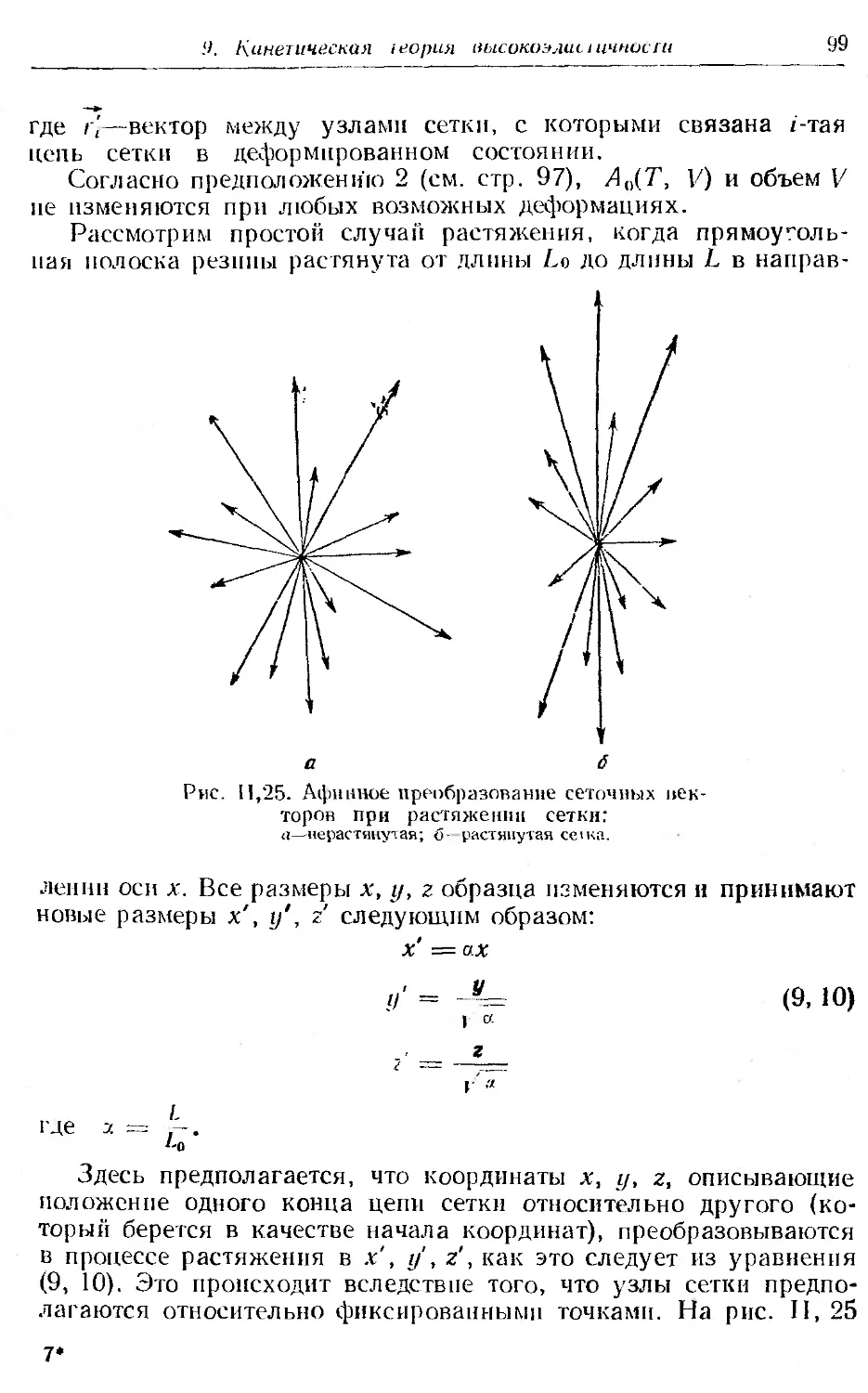
9. Кинетическая теория высокоэ частичности . . .......... 96
Глав a Ill. Математическая теория линейной вязкоупругости
и упруговязкости полимеров
1. Определение и измерение вязкоупругих характеристик 106
2. Линейная вязкоупругость и принцип Больцмана 111
3. Модели .Максвелла—Вихерта 115
4. Обобщенная модель Фойгта............................... 121
5. Динамический модуль и обобщенная модель Максвелла . . . 124
6. Расчет распределения времен релаксации из данных по релакса-
ции напряжения...................... . 125
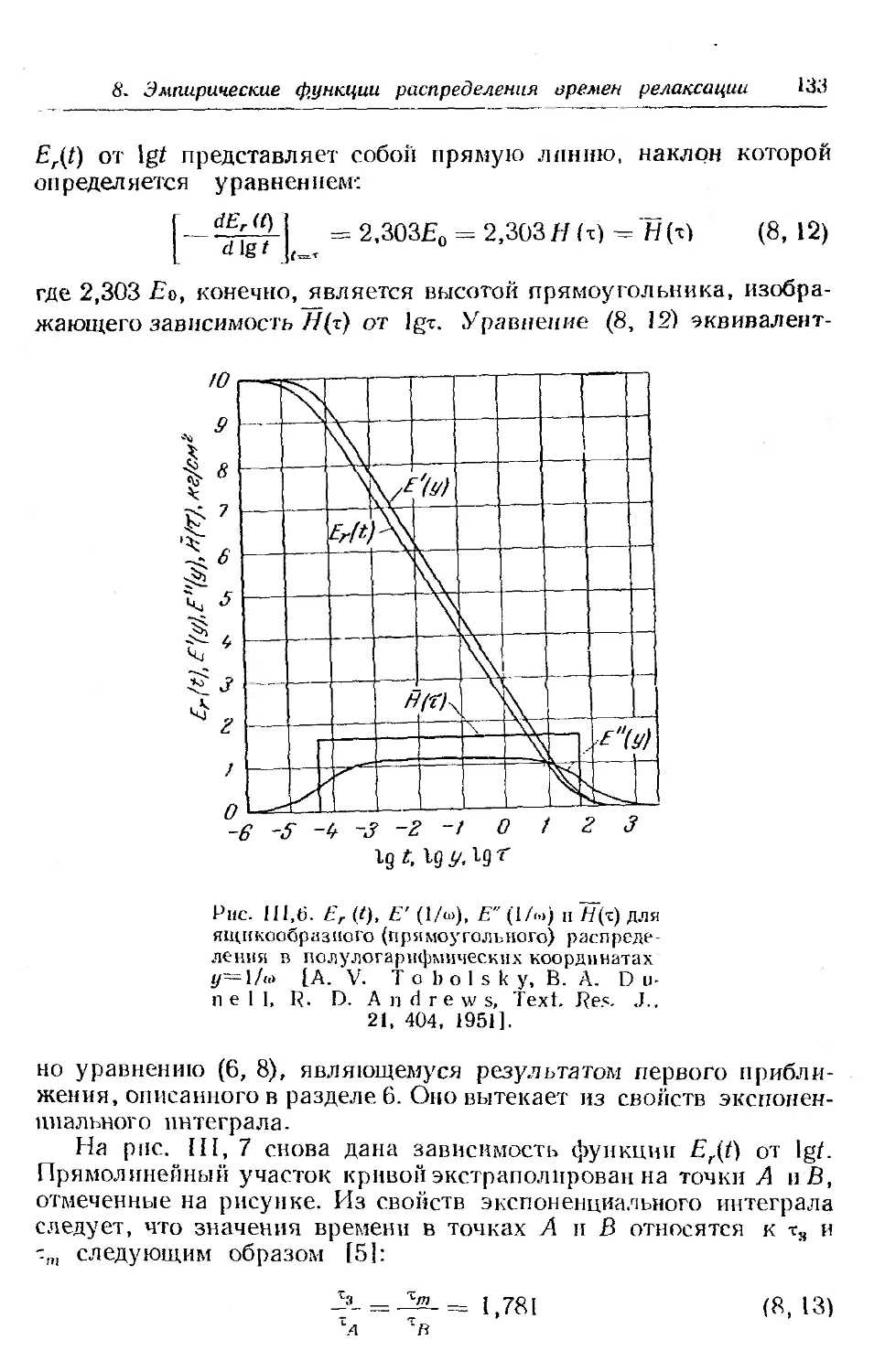
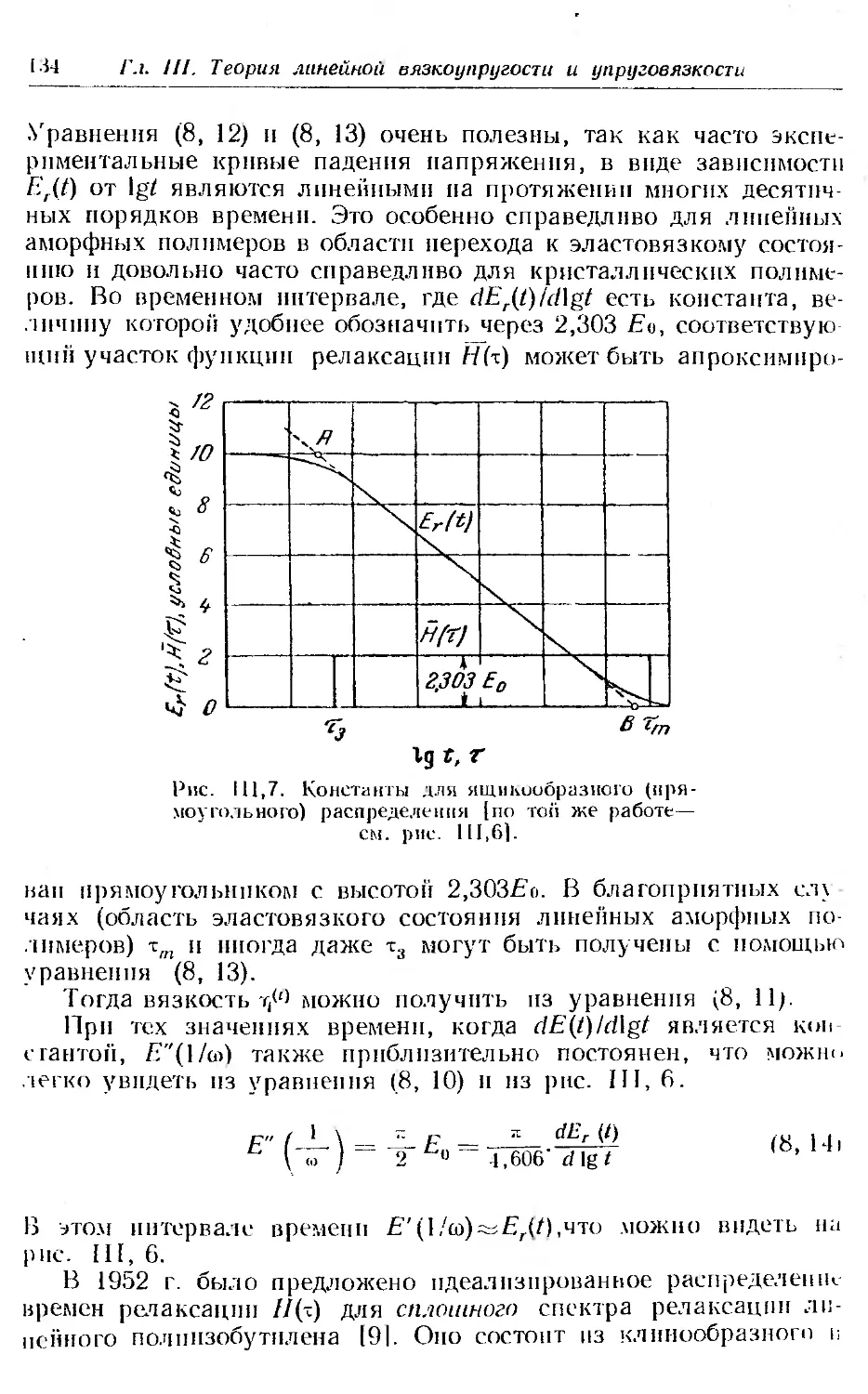
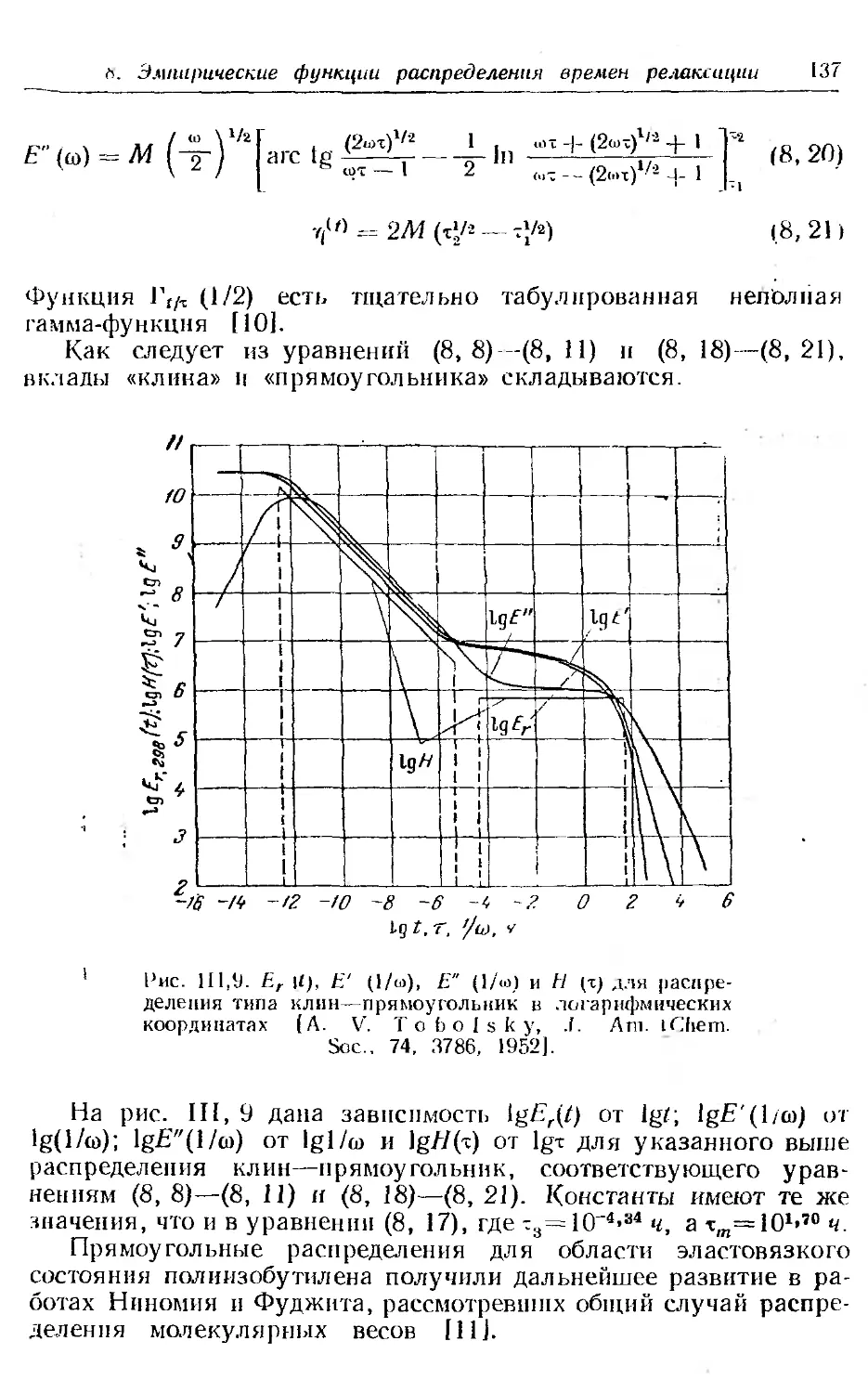
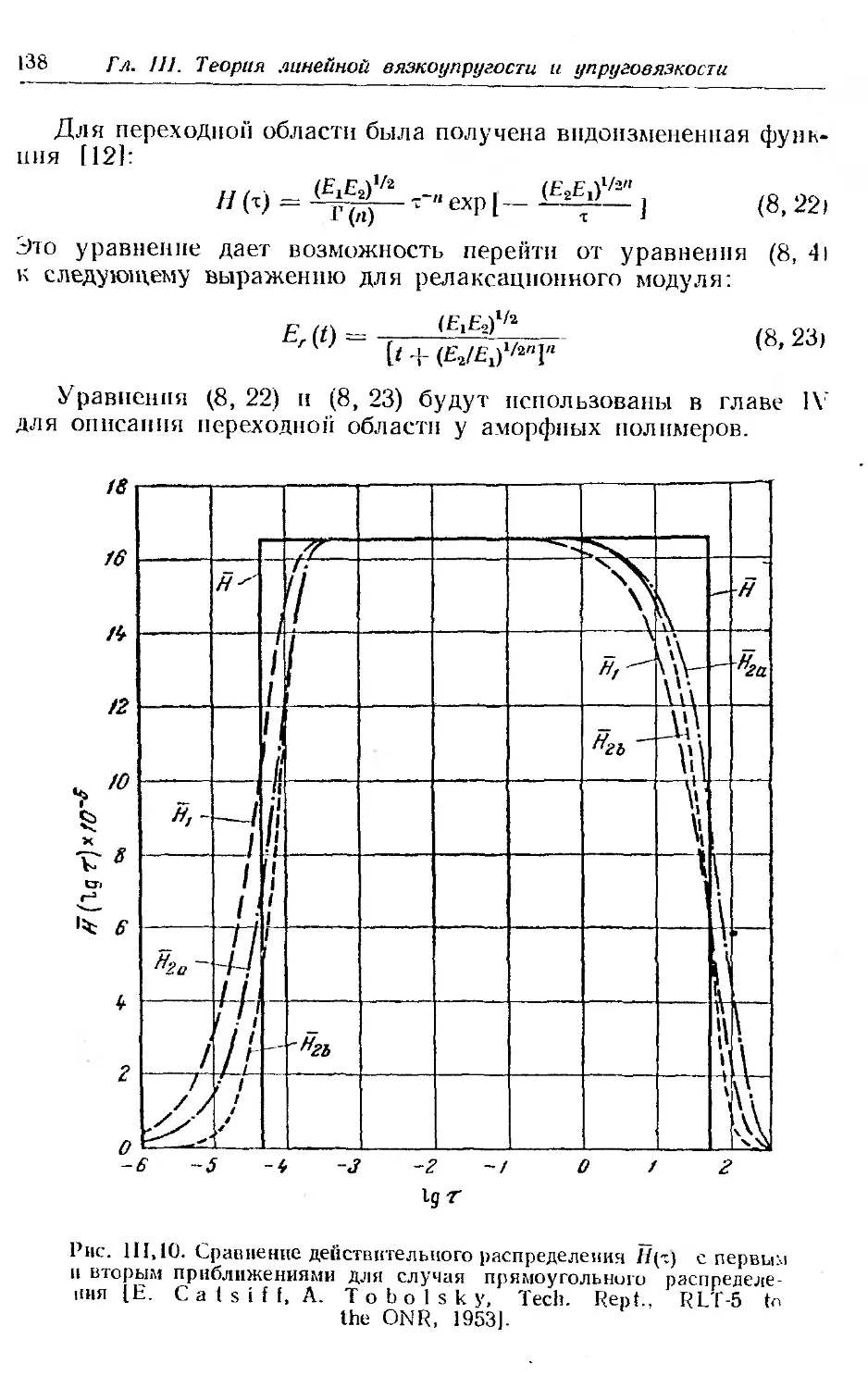
7. Преобразование £-,.(/) в другие функции................ 128
8. Некоторые эмпирические функции распределения времен ре-
лаксации . .......... . ......... . . 131
9. Упруговязкпе свойства максвелловского тела...........,. 141
4
Содержание
Глава IV. Вязкоупругие и упруговязкие свойства полимеров
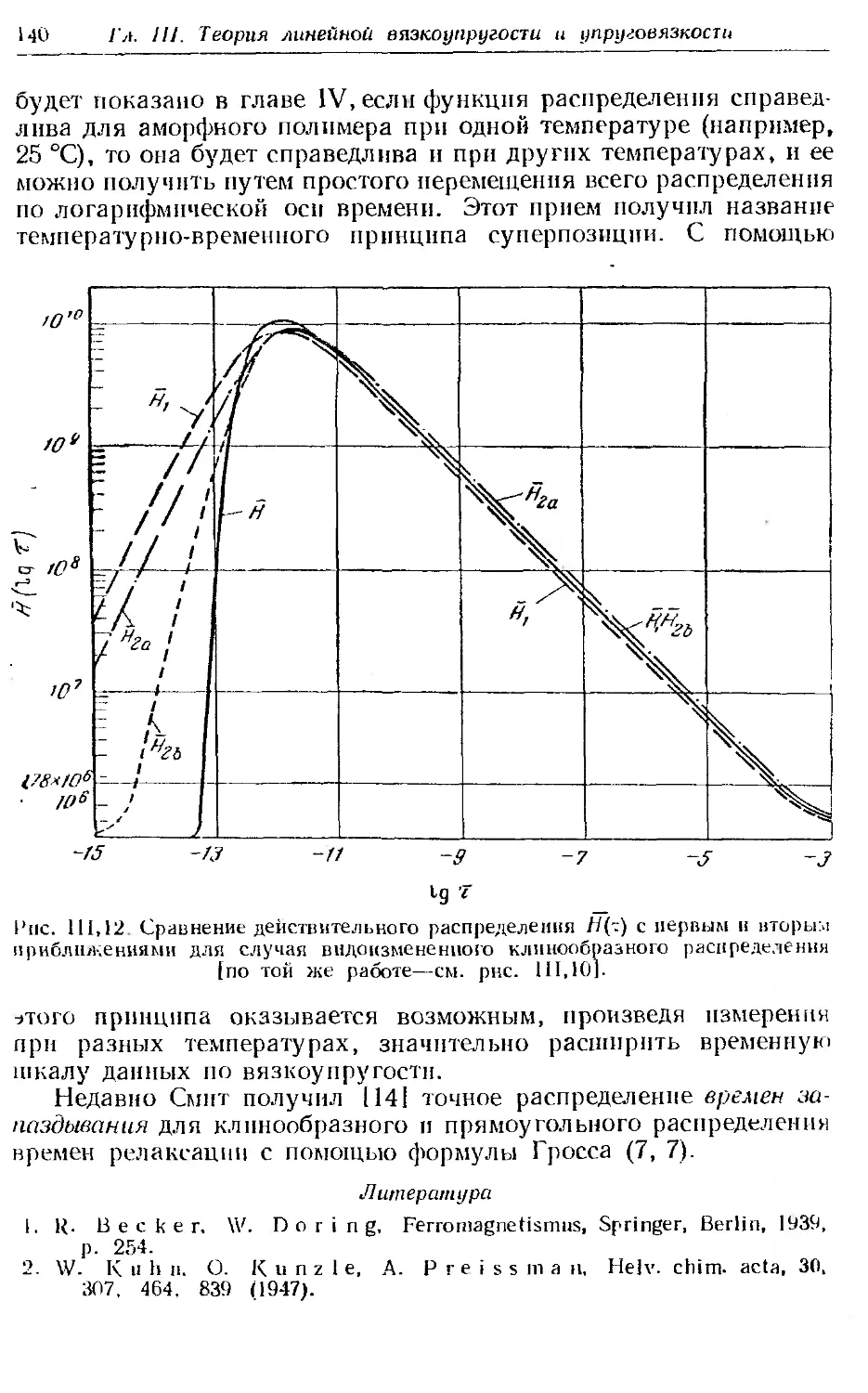
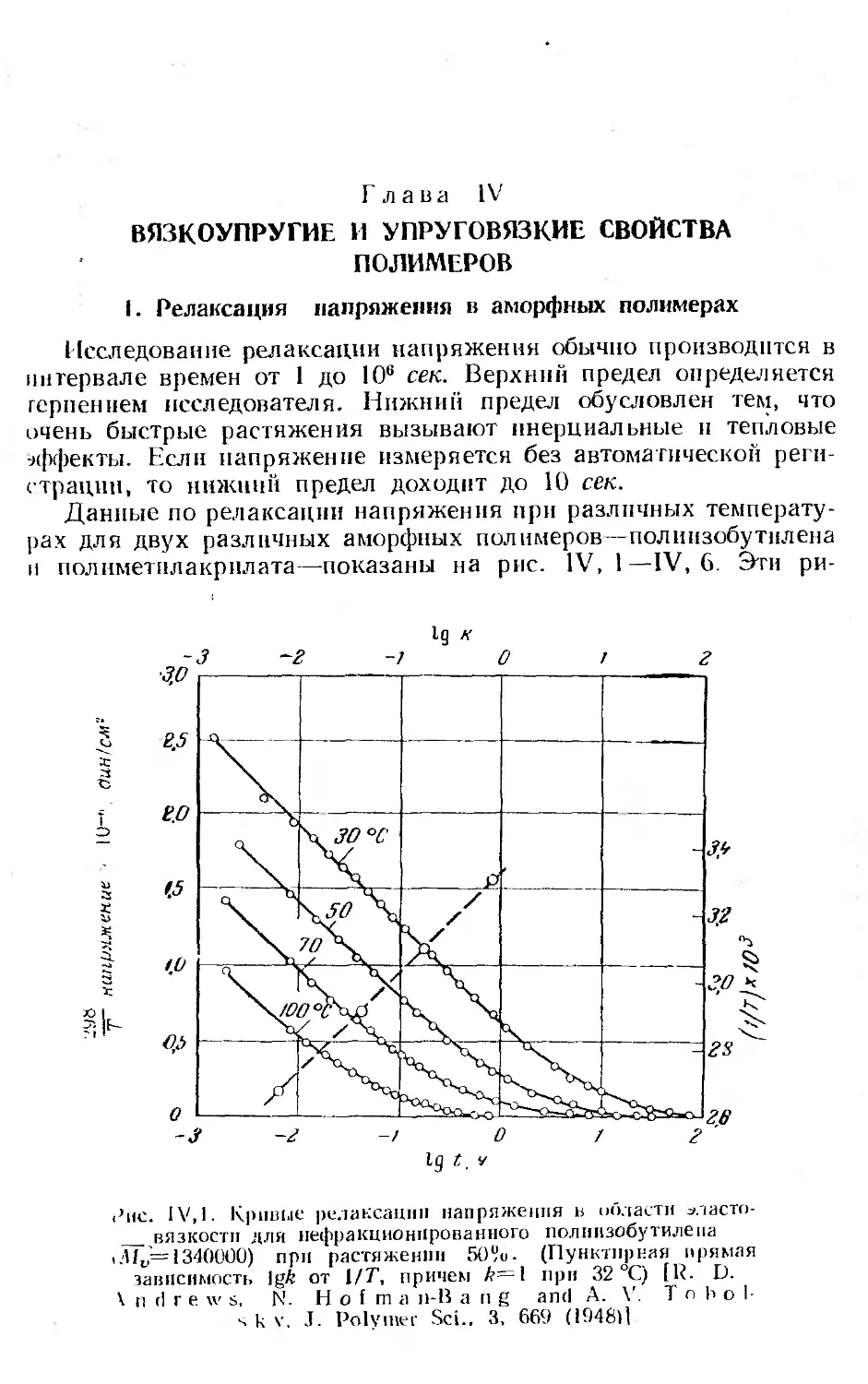
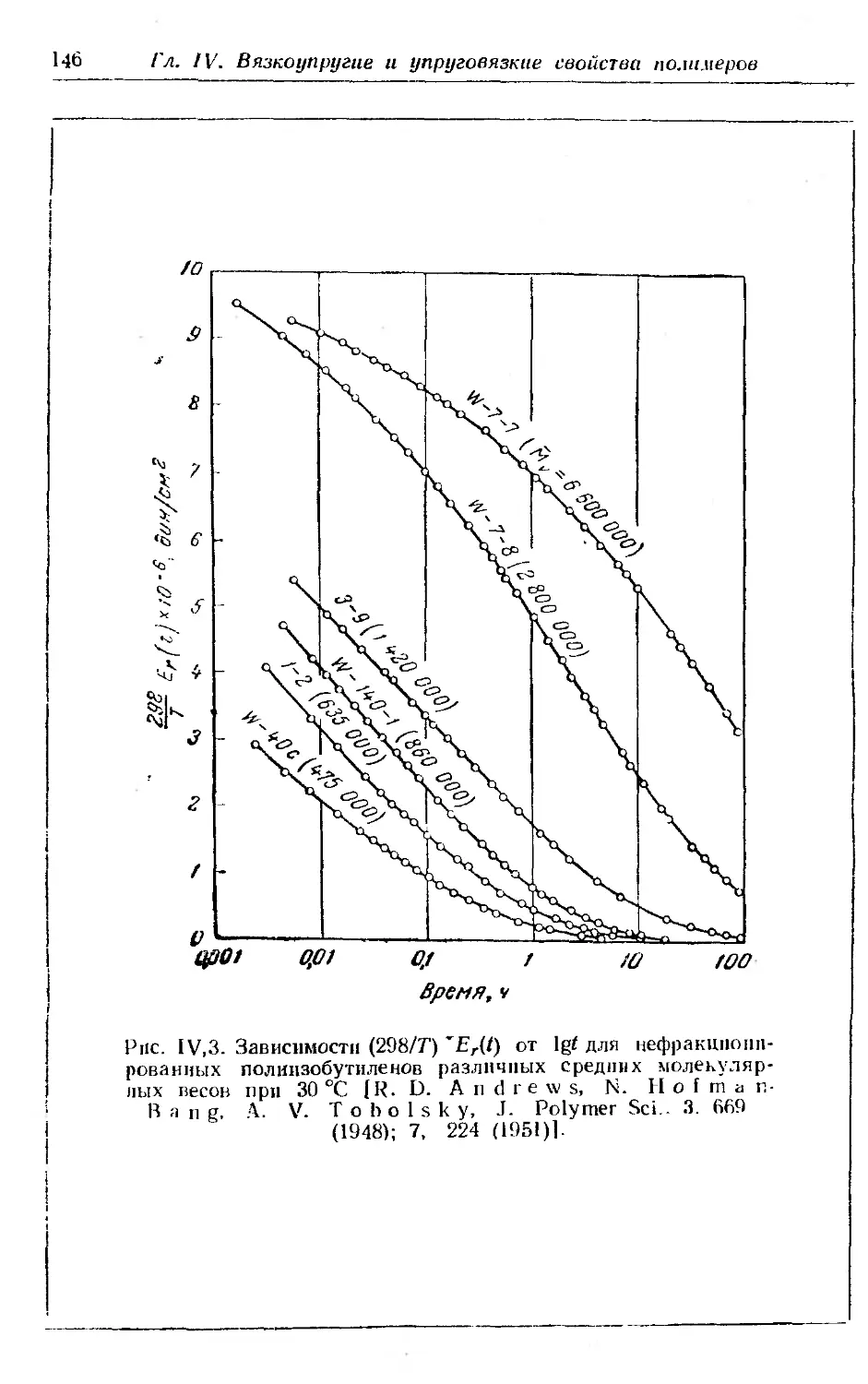
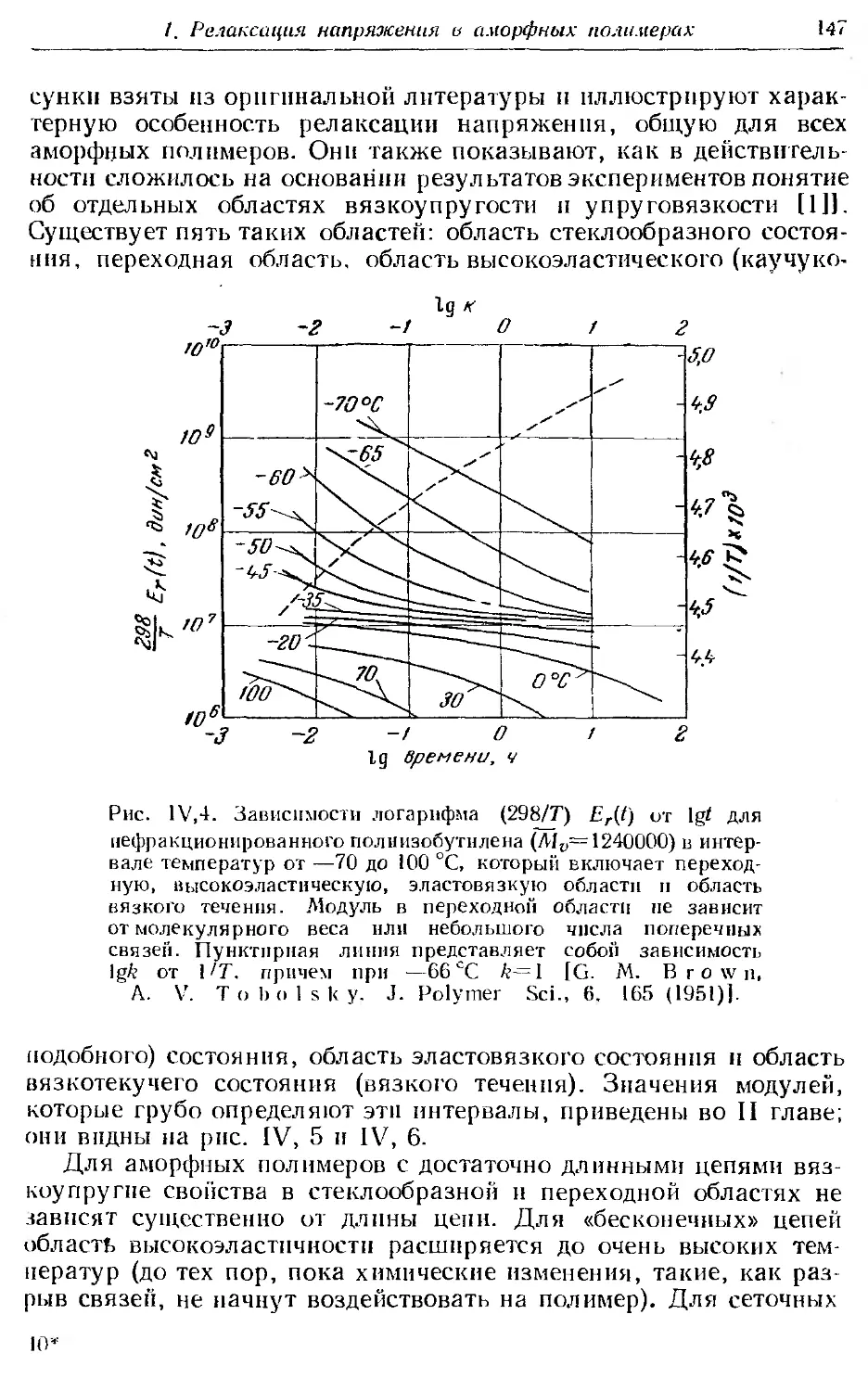
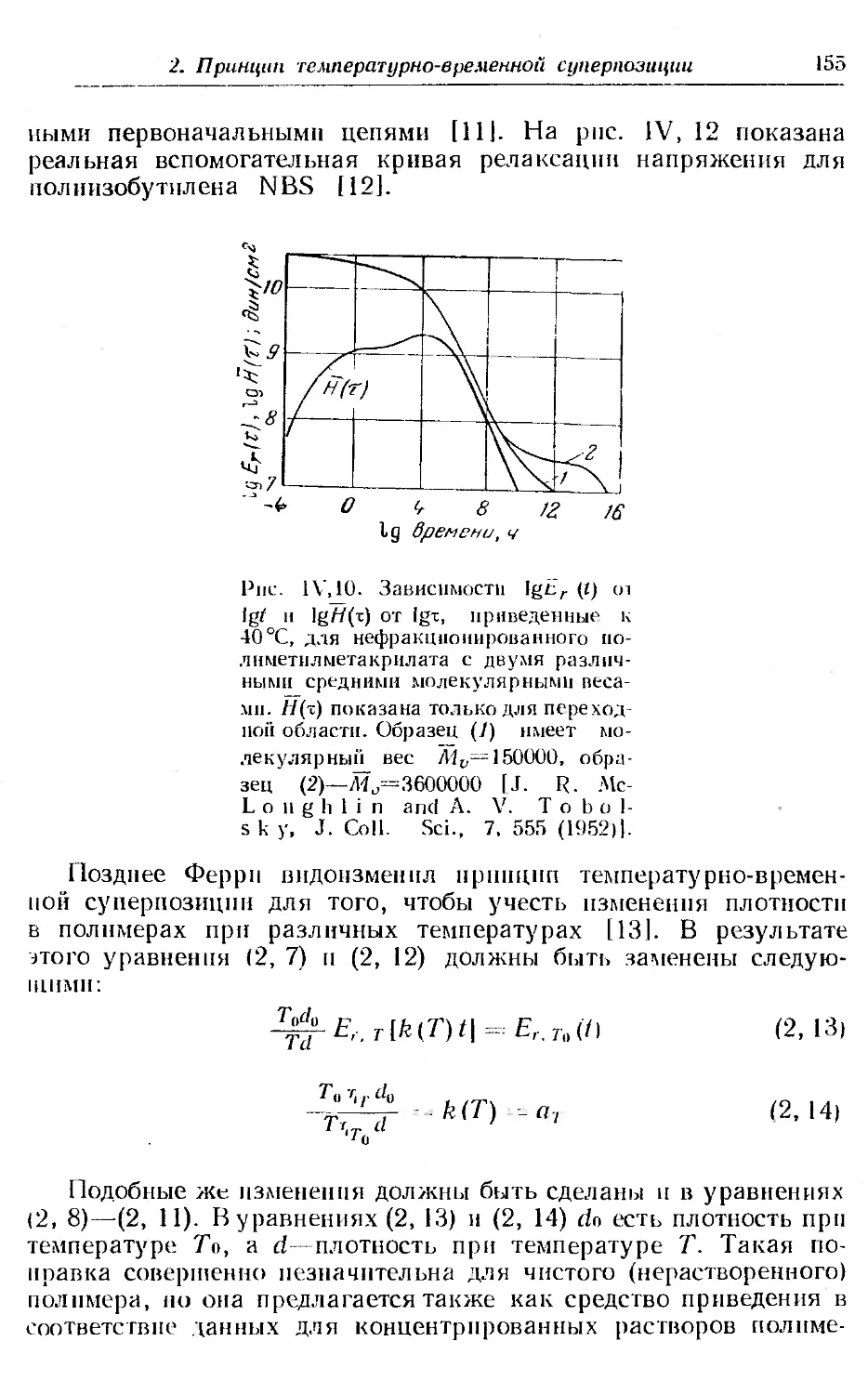
1. Релаксация напряжения в аморфных полимерах.............. 144
2. Принцип температурно-временной суперпозиции н вспомога-
тельная кривая.............................. . . . . . 149
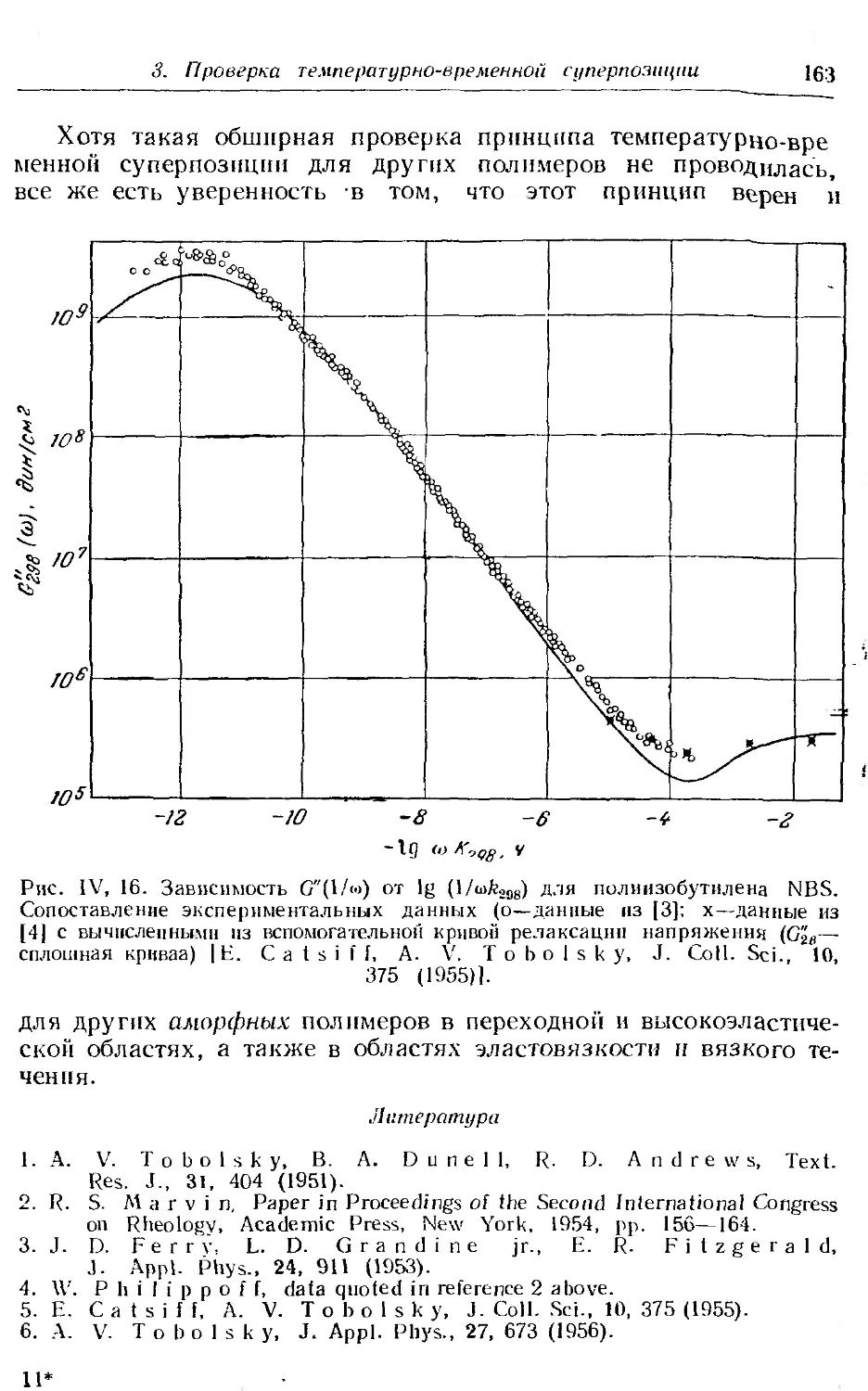
3. Проверка принципа температурно-временной суперпозиции 158
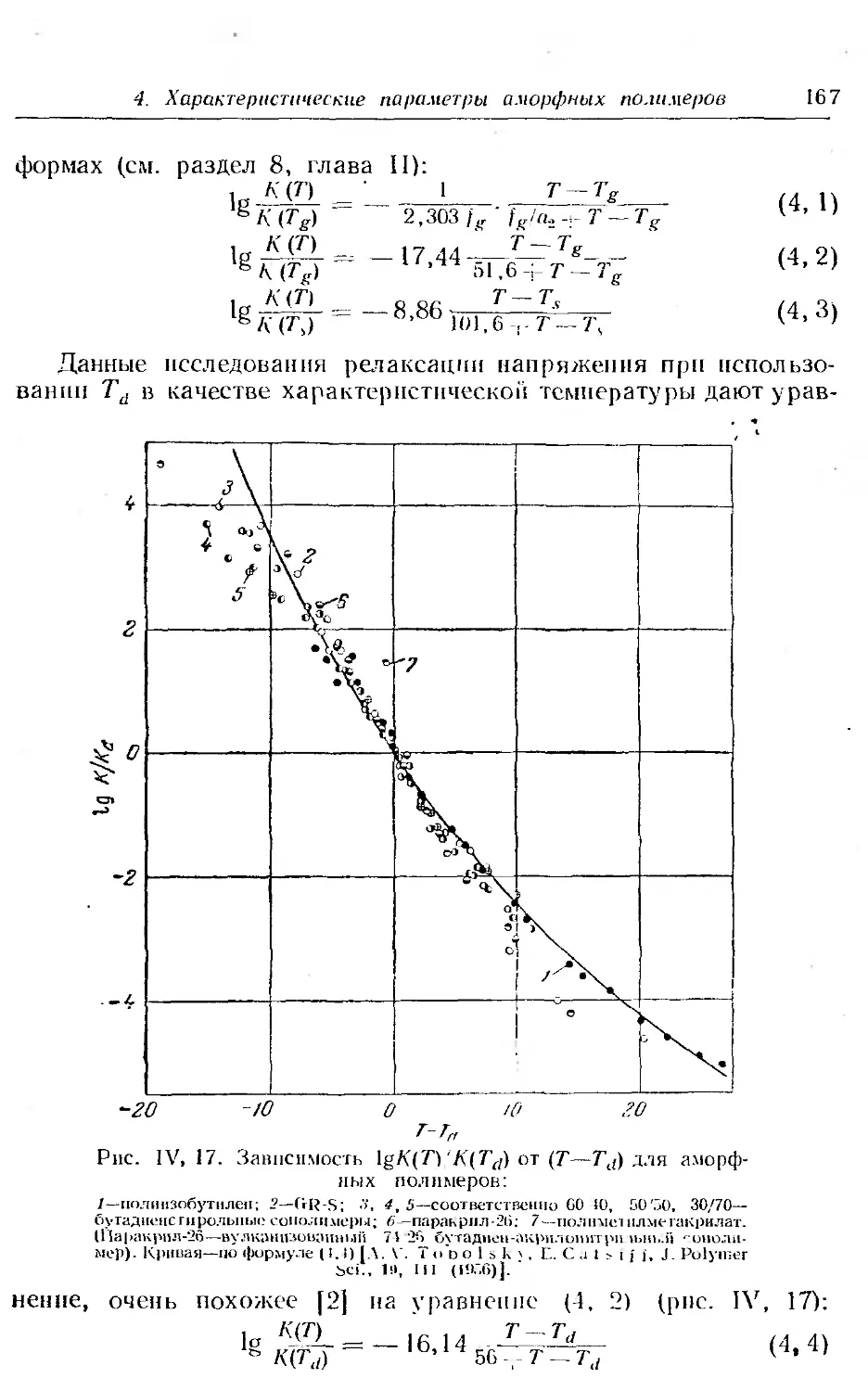
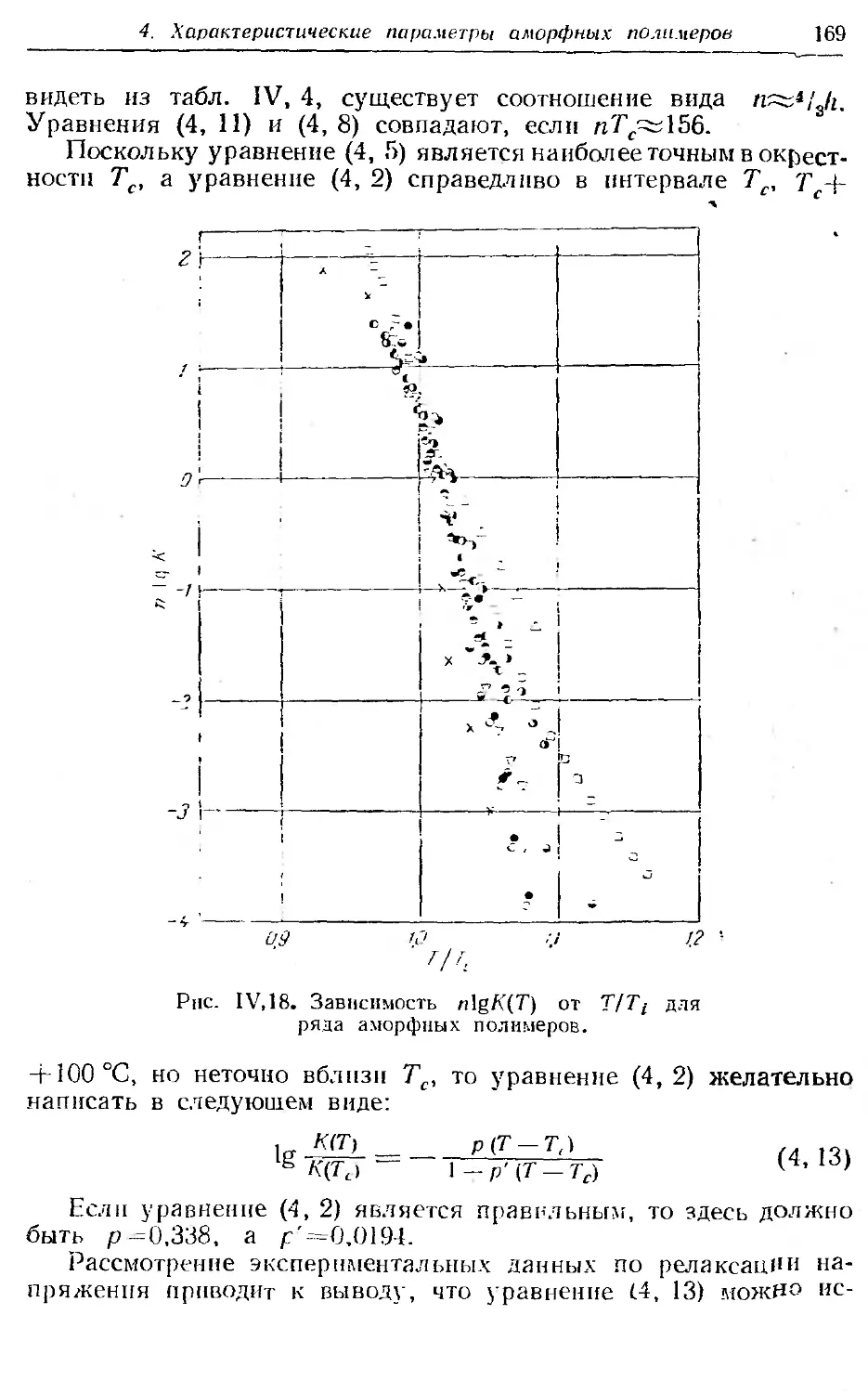
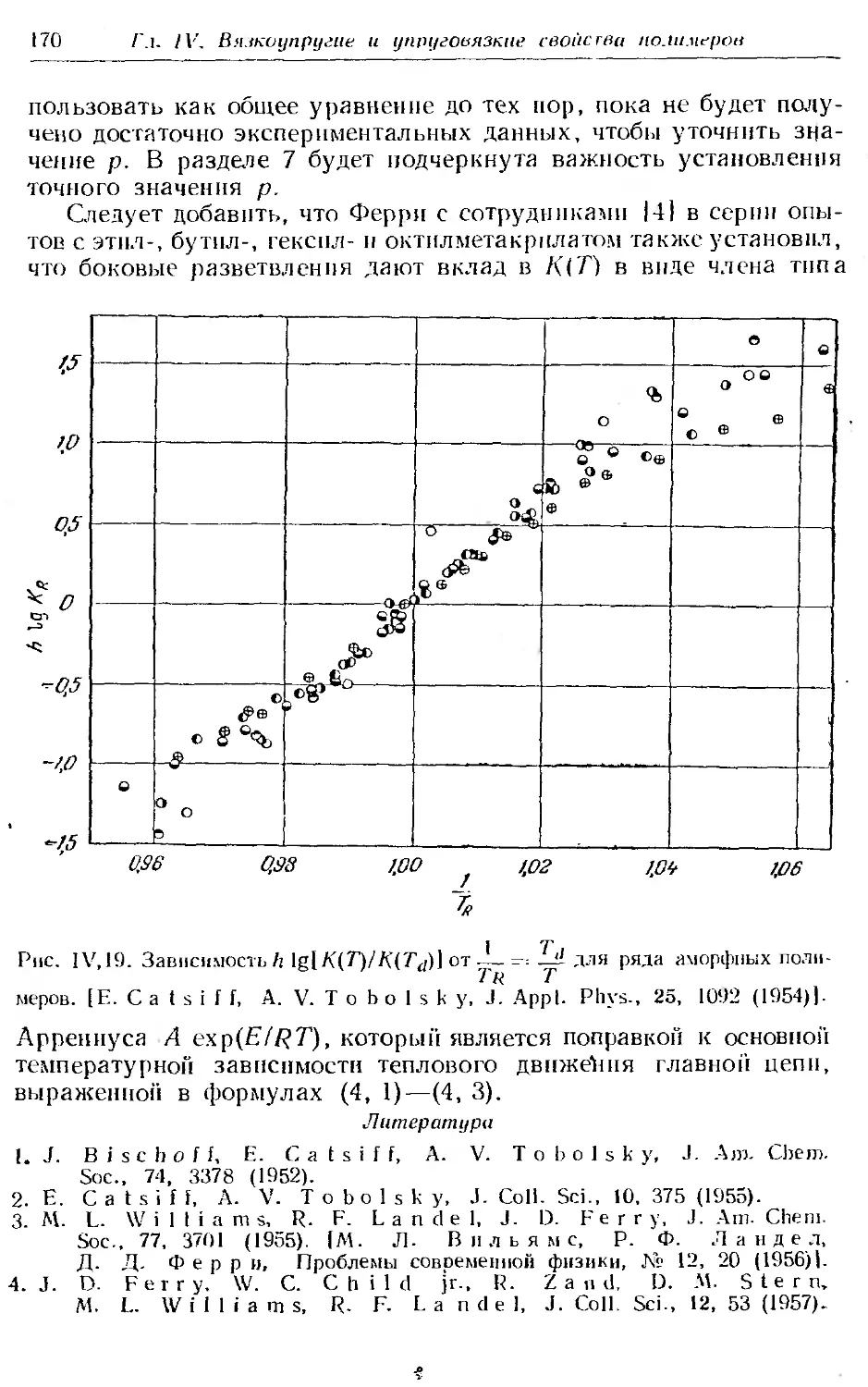
4. Характеристические параметры аморфных полимеров . . 164
5. Молекулярная теория вязкоупругости и упруговязкости по-
лимеров ................................................... 171
6. Релаксация напряжения в полимерах, находящихся в стекло-
образном состоянии....................................... 176
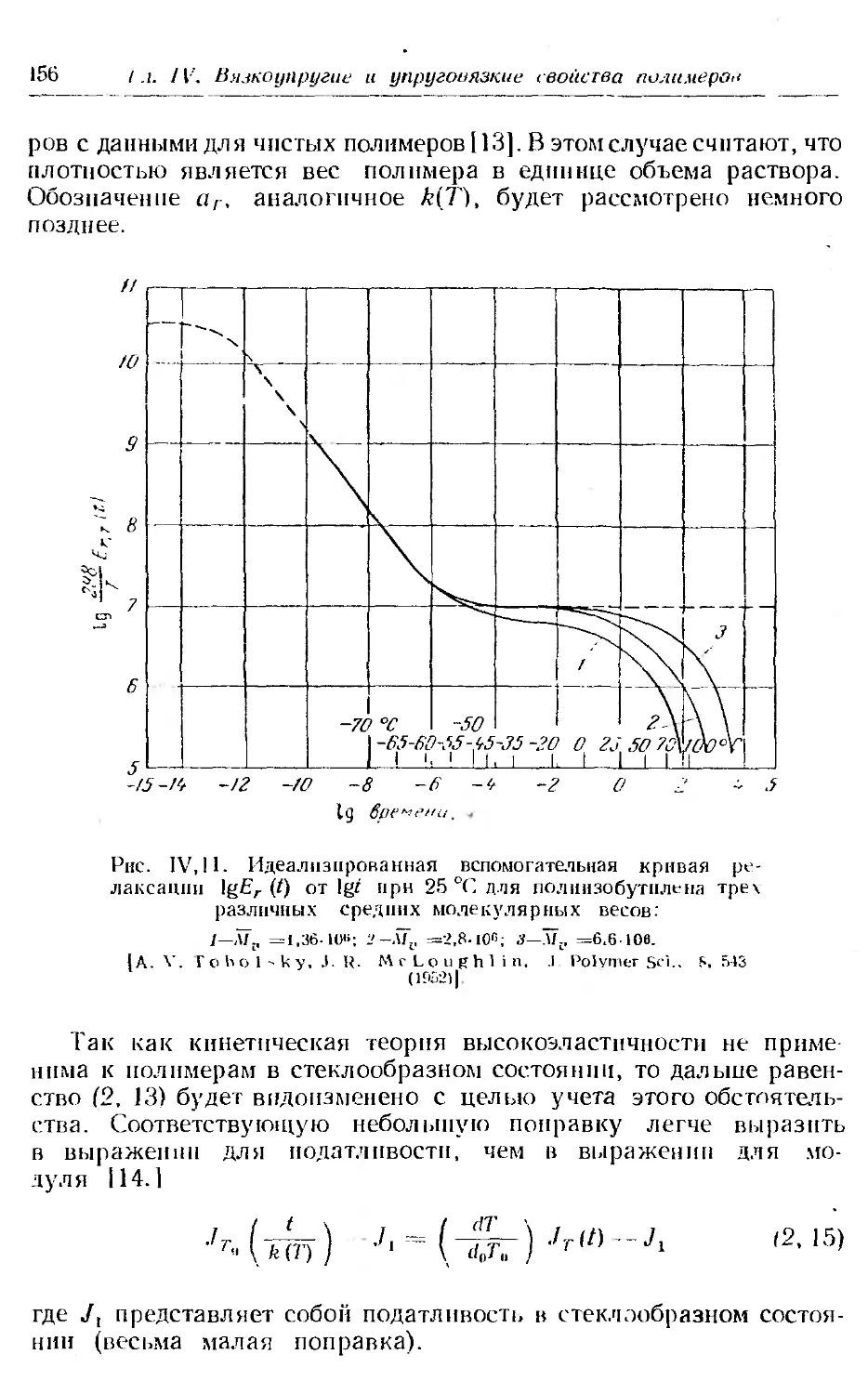
7. Переходная область н зависимость модуля от температуры . 179
8. Переходная область и функция ошибок..................... 181
9. Область эластонязкостн и область вязкого течения; дискрет-
ный спектр времен релаксации ......................... ... 185
10. Теплообразование в каучуках . 196
11. Релаксация напряжения в кристаллических полимерах 199
12. Релаксация напряжения в перестраивающихся сетках 207
13. Вязкоупругость н упруговязкость перестраивающихся сеток 213
14. Ползучесть при постоянной нагрузке в случае перестраиваю-
щихся сеток.................................... . . 216
15. Изучение двойного лучепреломления ...................... 218
Глава V. Химическая релаксация напряжения
1. Химическая релаксация напряжения в резинах из углеводо-
родных каучуков............................................ 225
2. Деструкция и образование поперечных связей ............. 230
3. Определение скорости деструкции ио релаксации напряжения 234
4. Распад поперечных связей п распад цепей сетки ..... 237
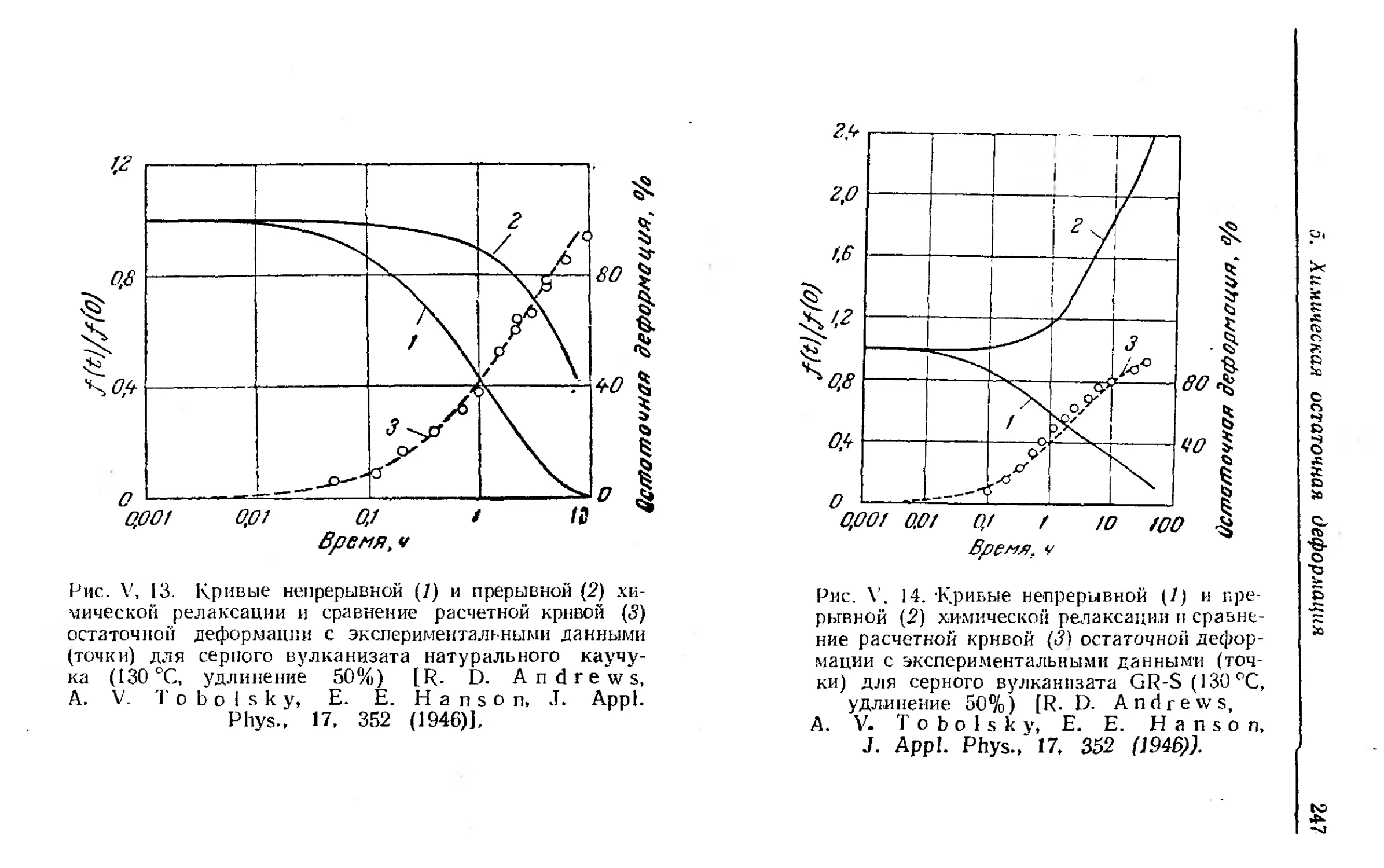
5. Химическая остаточнап деформация........................ 244
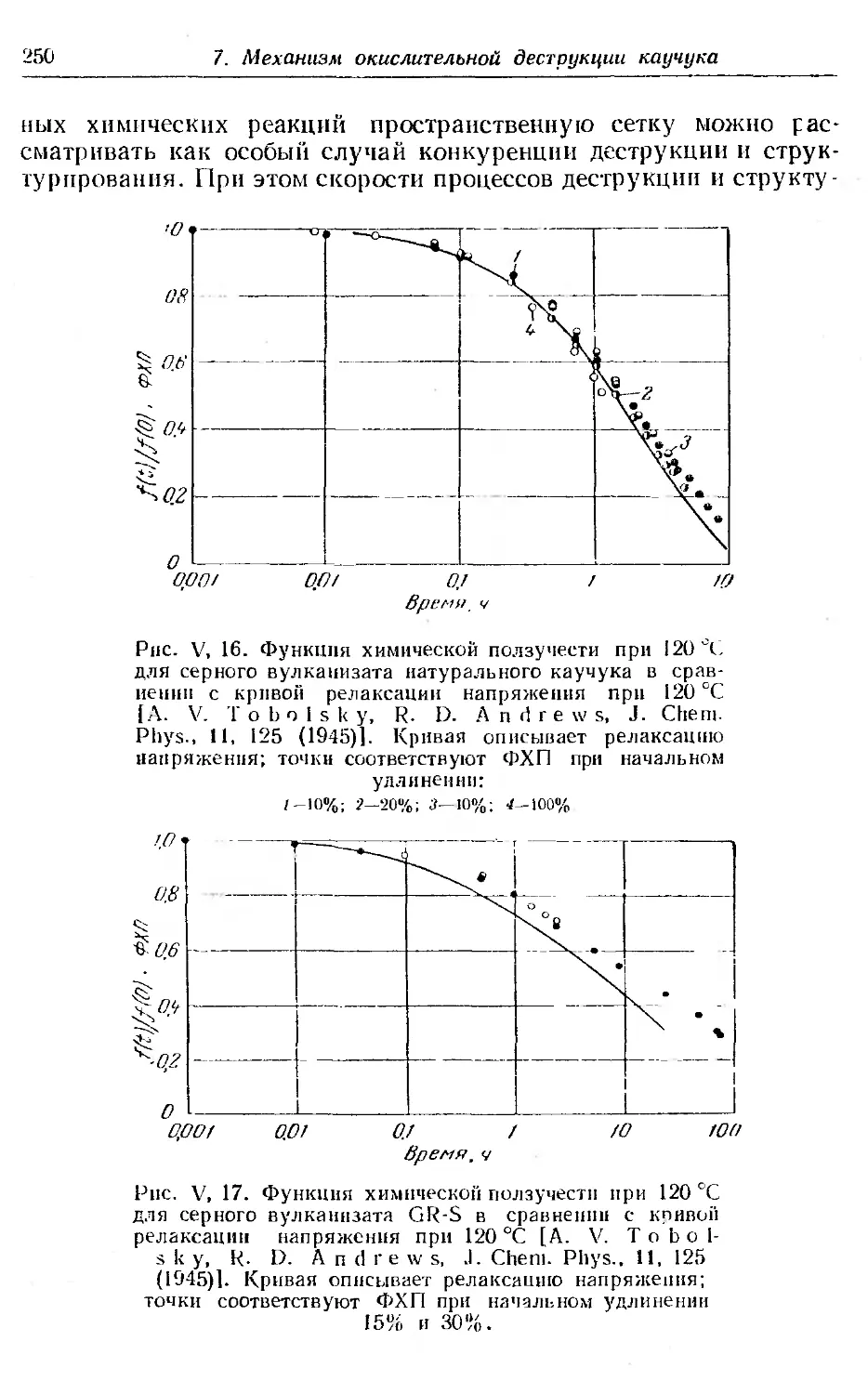
6. Химическая ползучесть при постоянной нагрузке . . 248
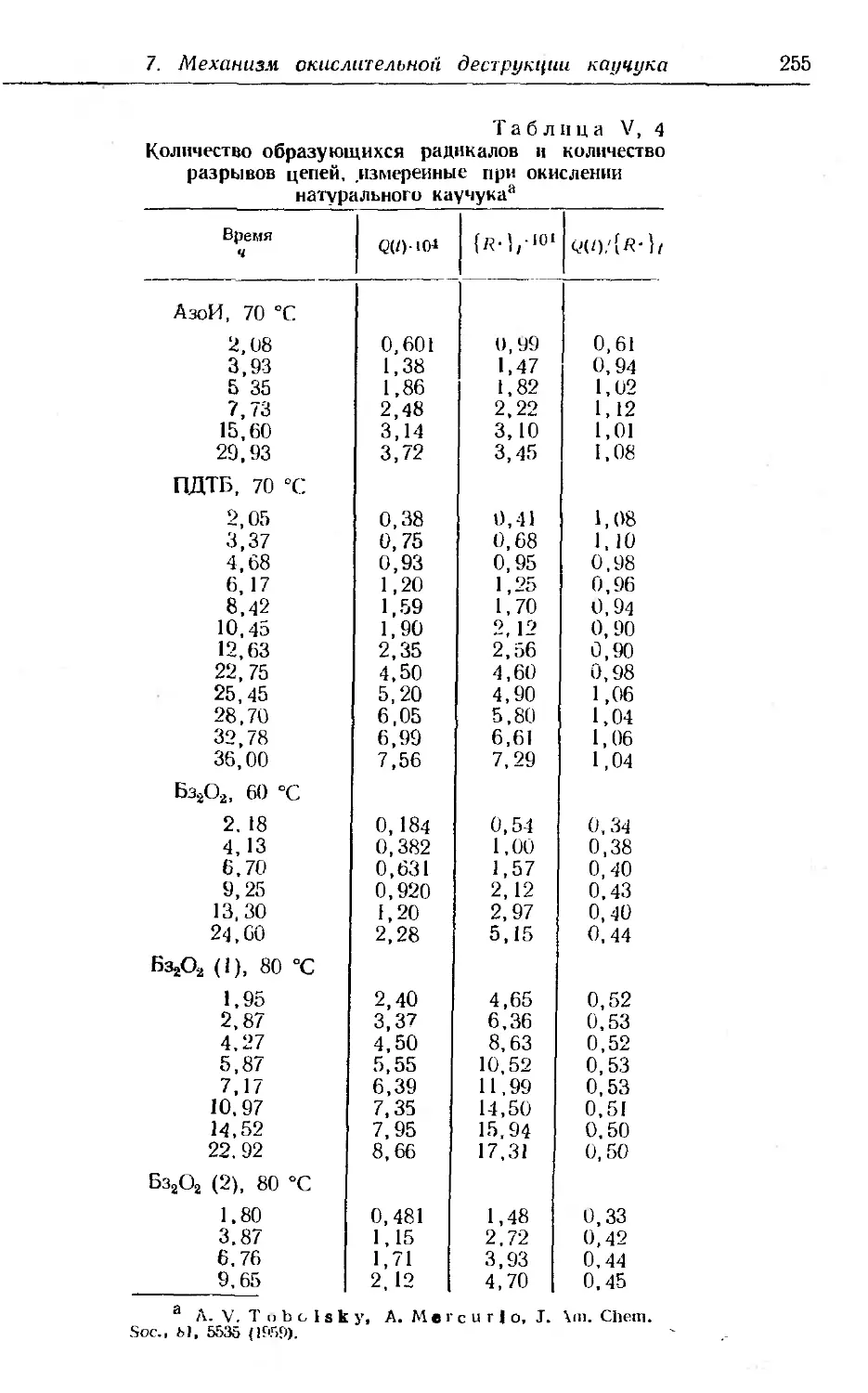
7. Механизм окислительной деструкции натурального каучуки 251
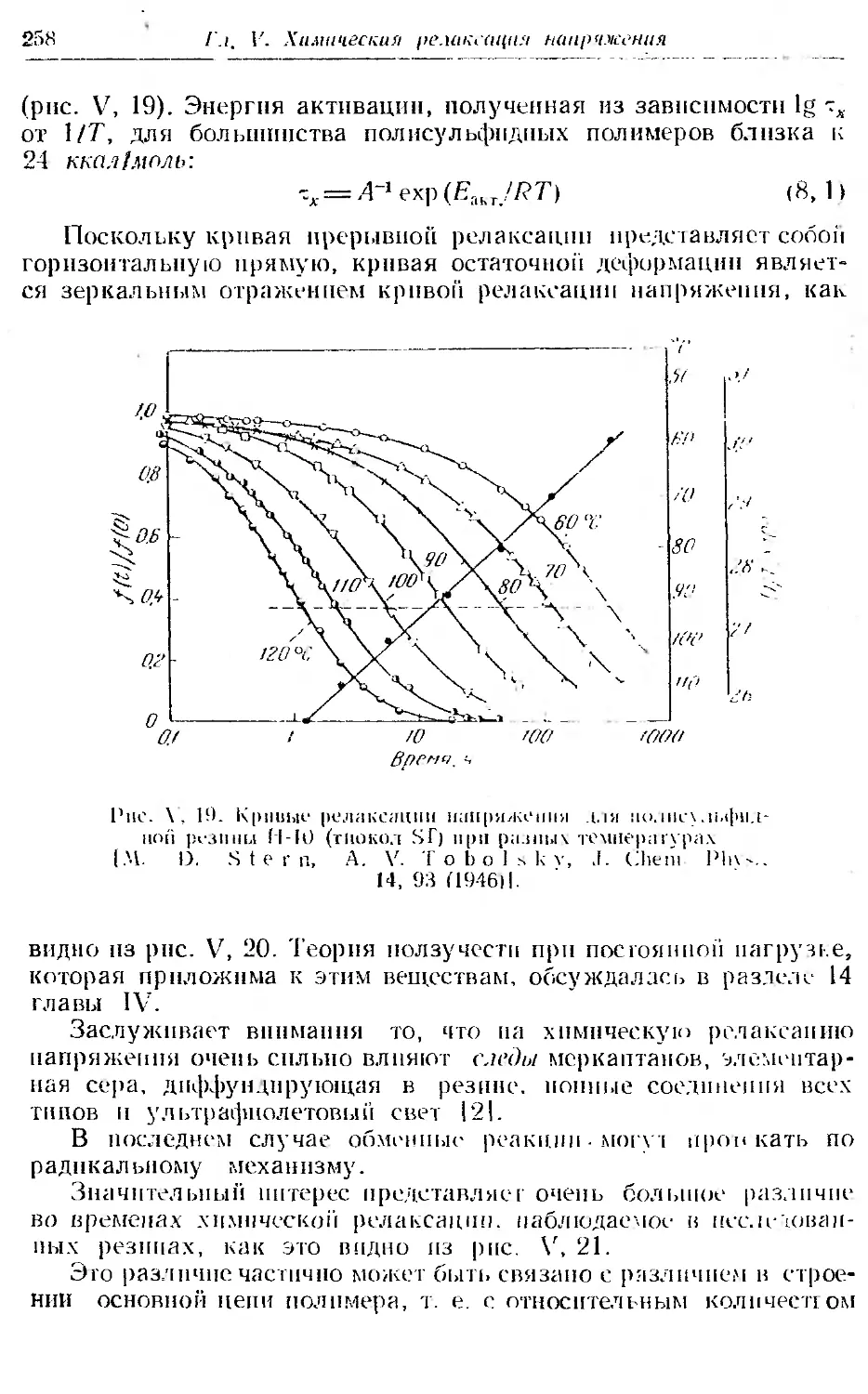
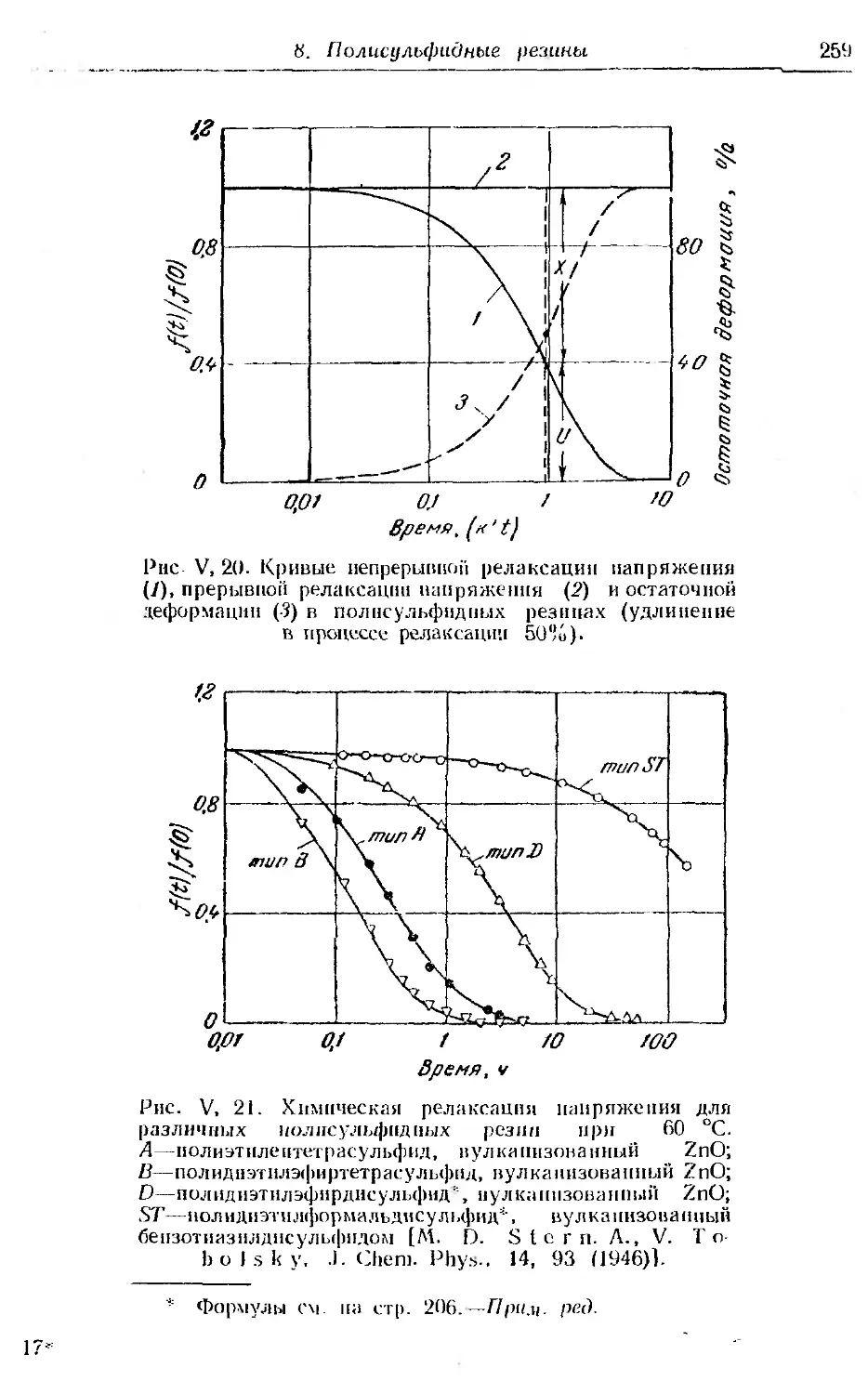
8. Полнсульфидные резины ... . 257
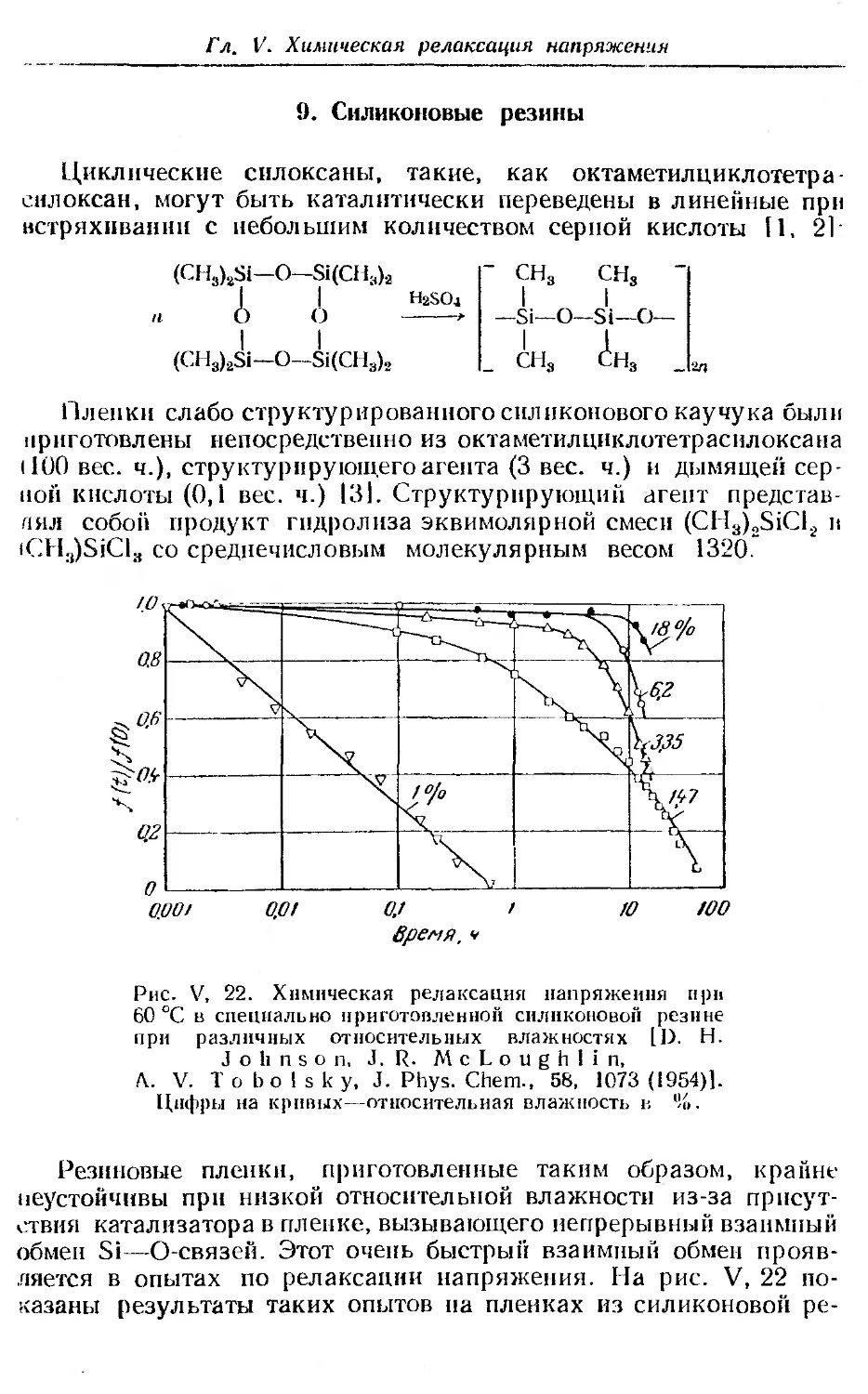
9. Силиконовые резины ... ...................... 262
10. Полиуретановые резины........................ ........ 264
Глава VI. Полимеризационное равновесие
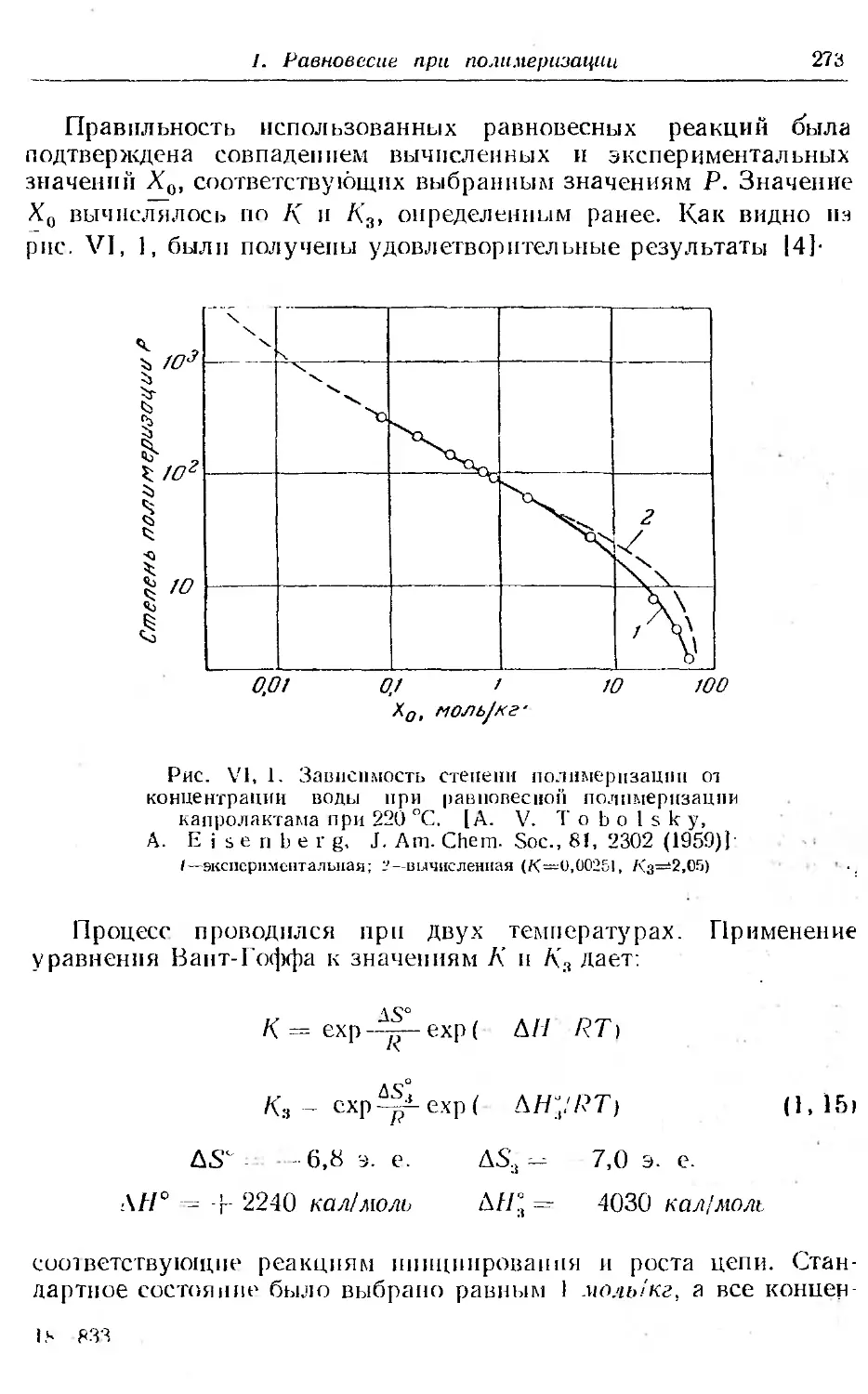
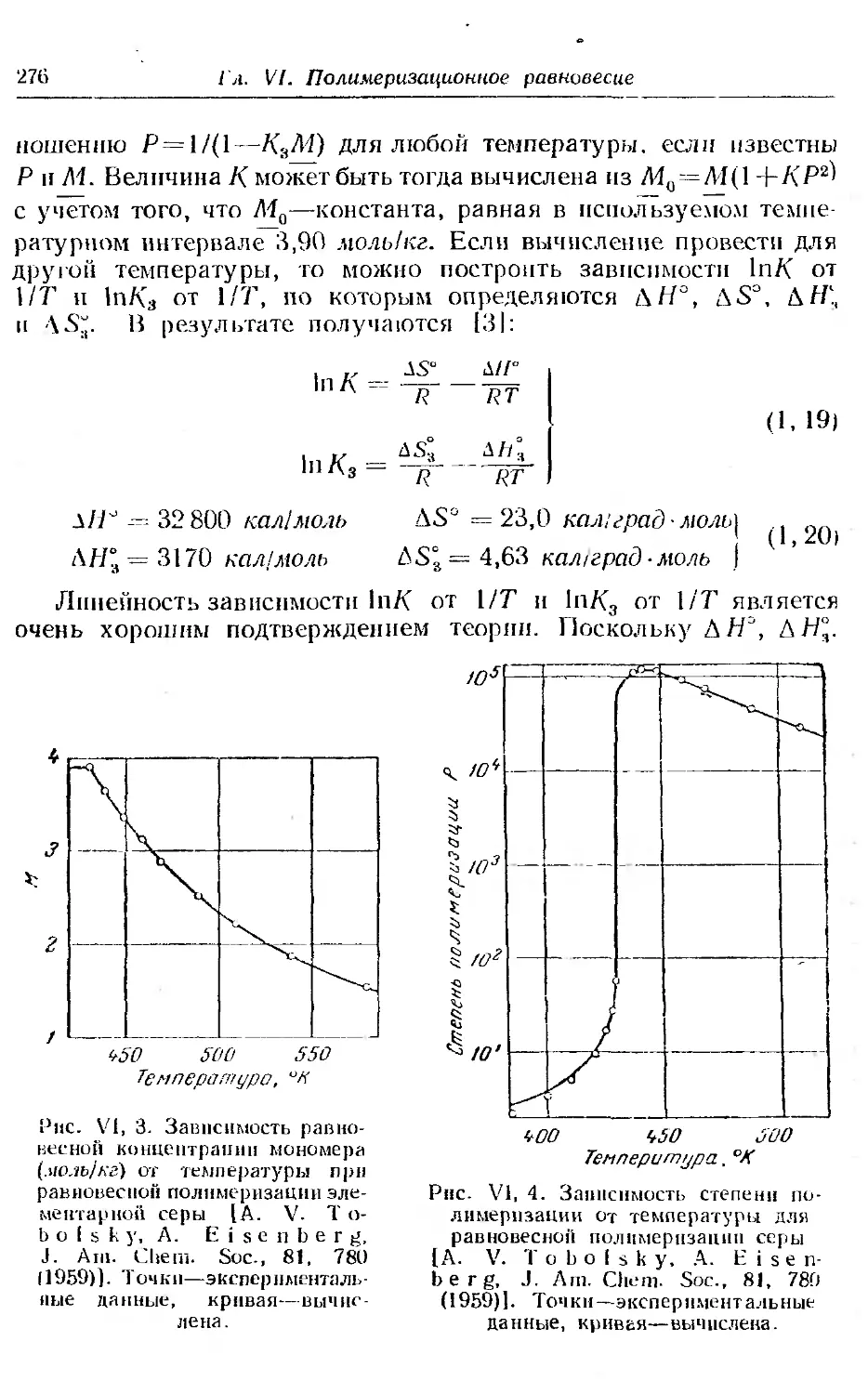
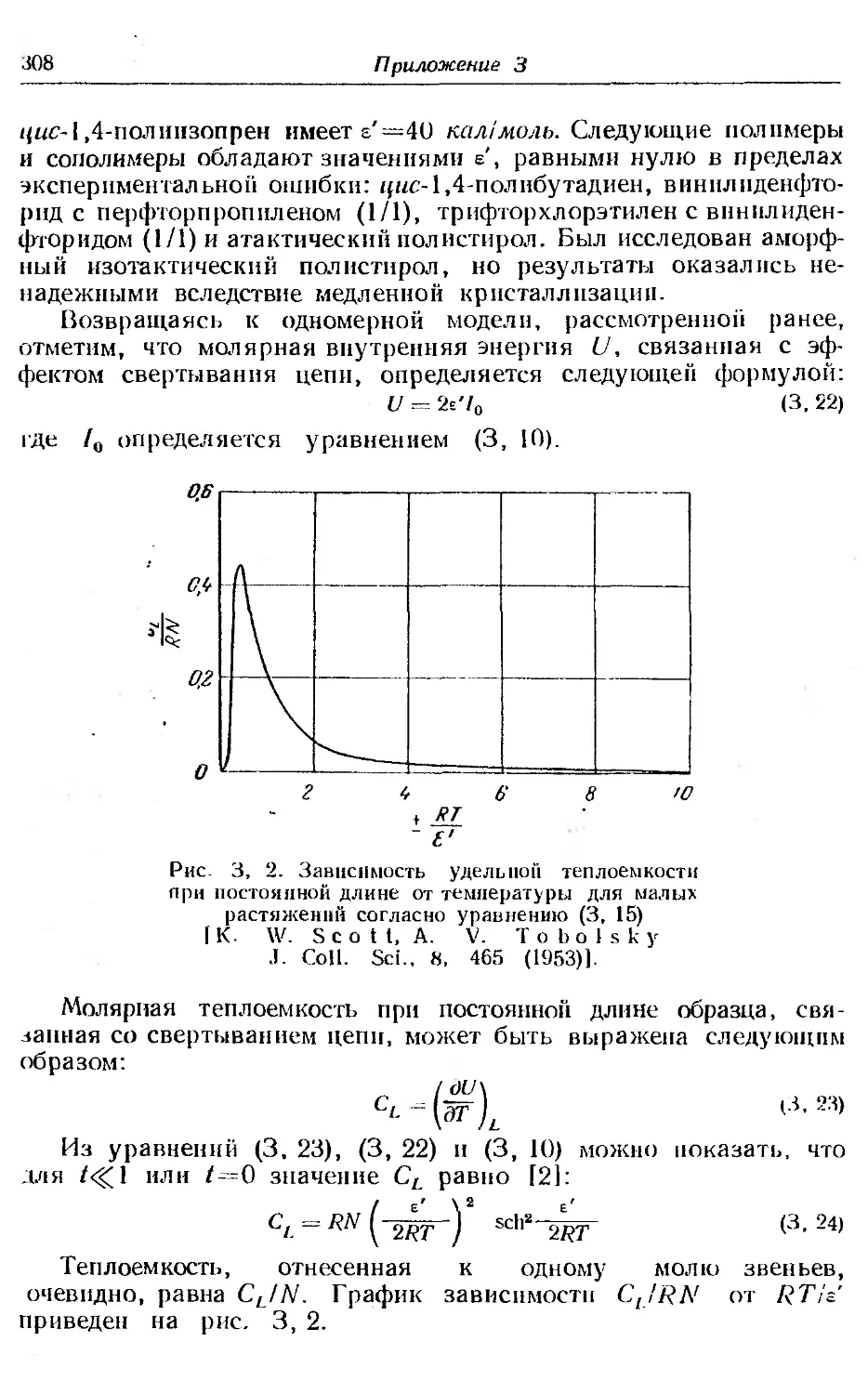
1. Равновесие при полимеризации.............. ... 268
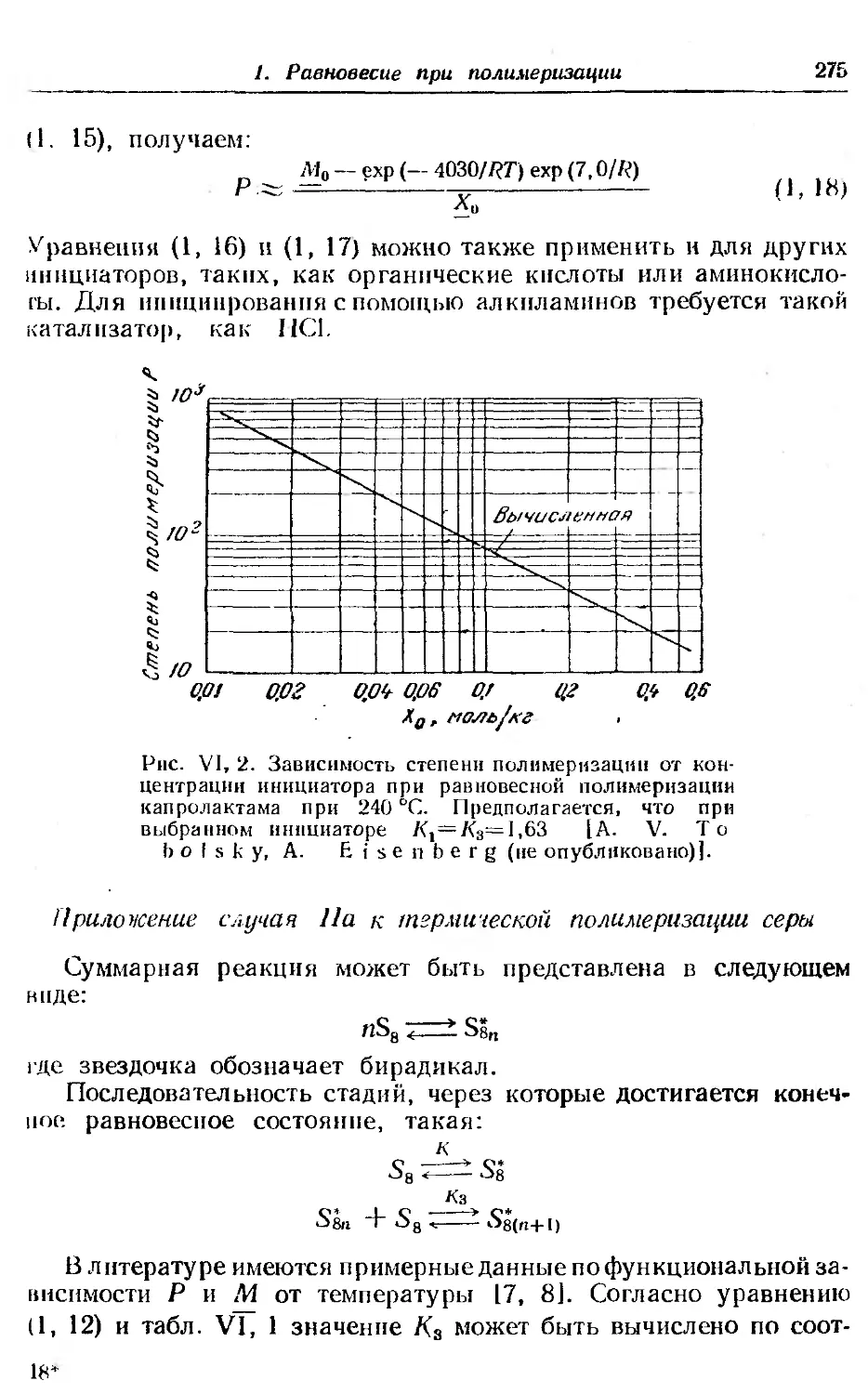
2. Равновесие при сополимеризации ..... . 281
Приложения:
А. Статистическая сумма состояний........................... 284
Б. Конфигурационная энтропия газа........................... 286
В. Уравнение состояния молекулярных кристаллов.............. 287
Содержание
5
Г. Еаивероятнейший квадрат длины цепи углеводородного типа 289
Д. Гауссово распределение расстояний между концами цепей . . 294
Е. Распределение длин молекул в линейных полимерах . . 295
Ж. Лфинное преобразование сферы ........................... 301
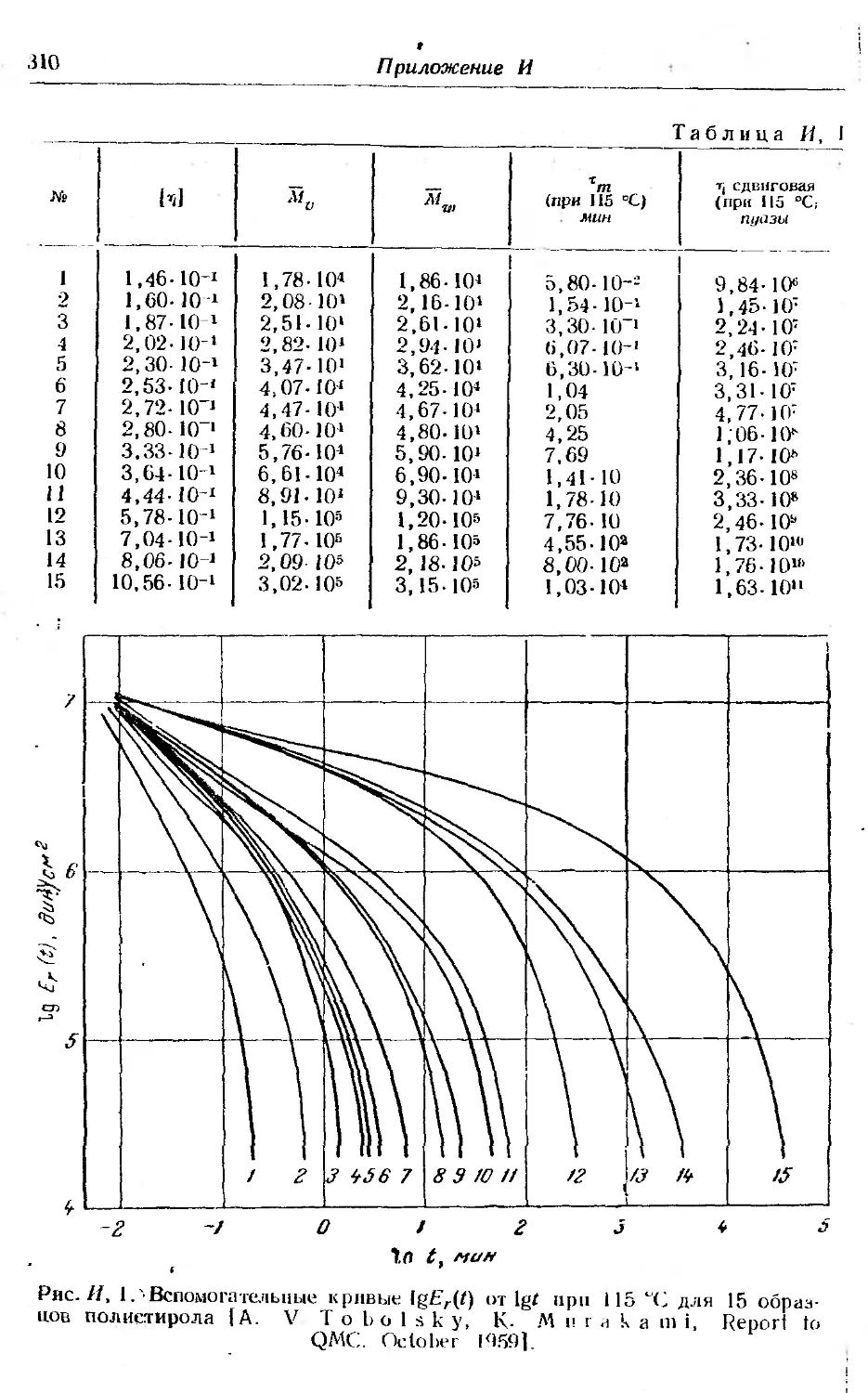
3. Уравнение состояния полимерных цепей и сеток, учитываю-
щее влияние энергии и энтропии ... ......... 302
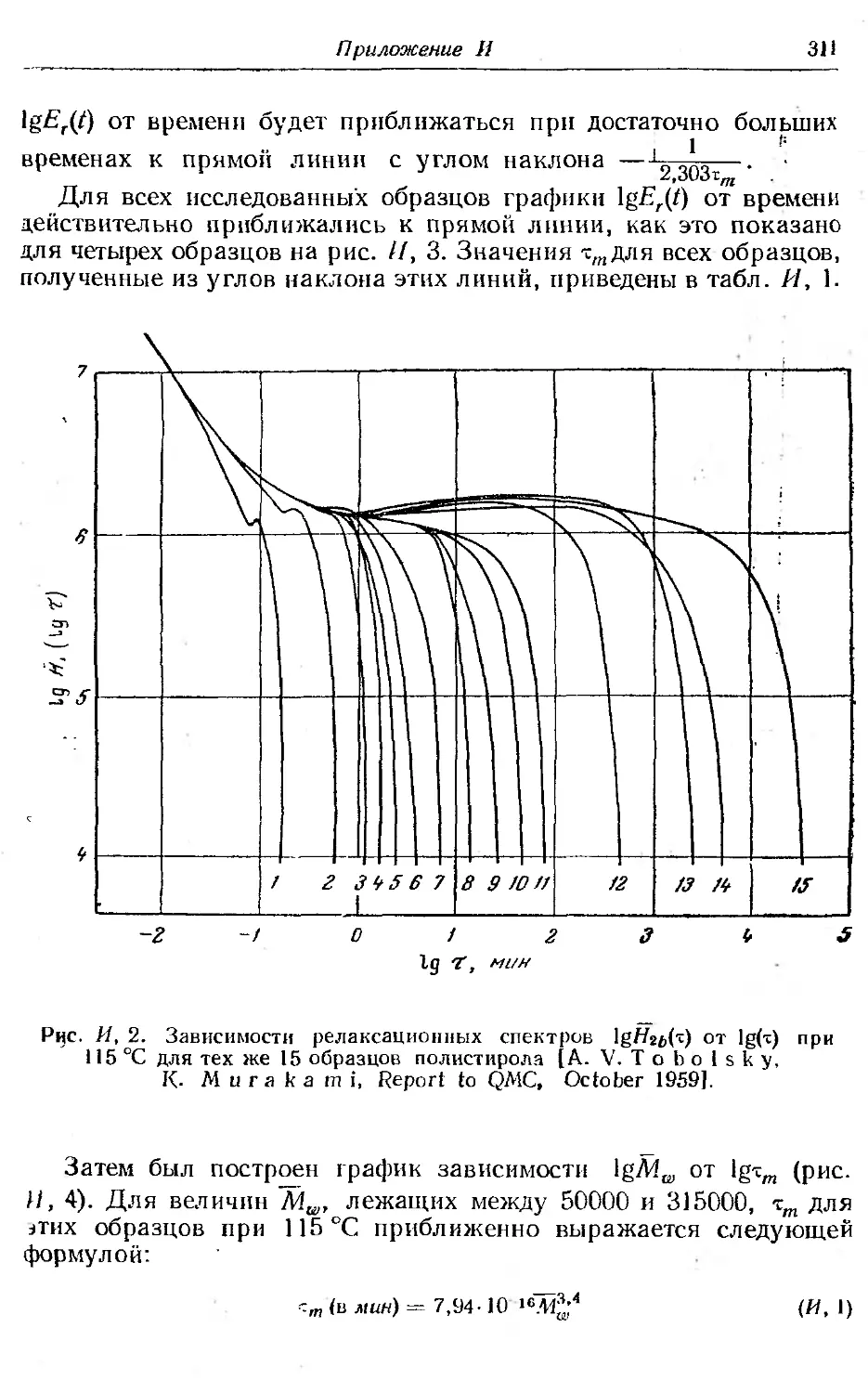
И. Максимальное время релаксации........................ . . 309
К. Значения Tj и Tg для некоторых полимеров . . . . . 314
Предметный указатель............................................ 316
*
ПРЕДИСЛОВИЕ РЕДАКТОРОВ
В последние годы в связи с быстрым развитием производства
полимеров и необычайным расширением их применения интерес
к механическим свойствам полимерных материалов резко возрос.
К сожалению, в мировой литературе пока еще имеется очень не-
бел ыное число книг, квалифицированно излагающих эти слож-
ные и важные вопросы. Одной из таких книг является киша
Тобольского, в которой в живой форме систематически изложены
многие сравнительно недавно полученные экспериментальные и
теоретические результаты исследований упругости, высокоэластпч
ности и текучести аморфных н кристаллических полимеров.
Эта область науки, давно развивающаяся в Советском Союзе
(П. П. Кобеко, А. П. Александров, Я. И. Френкель, В. А. Кар-
гин и их сотрудники и др.), была фактически вторично открыта (с
опозданием на несколько лет) и активно разрабатывается запад-
ными учеными, среди которых А. Тобольский занимает одно из
ведущих мест.
Следует заметить, что автор книги среди цитируемых ориги-
нальных исследований заметно выделяет собственные работы и
почти полностью игнорирует аналогичные более ранние работы
советских ученых. В связи с этим редакторами сделаны приме
чанпя с указанием этих публикаций.
Нельзя также обойти молчанием имеющиеся в книге много
численные мелкие ошибки и опечатки, которые, по возможности,
были при переводе исправлены.
Необходимо также заметить, что А. Тобольский пользуекя тер-
мином «viscoelasticity» для обозначения релаксационных явлений
как в текучих, так и в иетекучпх телах. Между тем в советской
научной литературе различаются упруговязкпе и вязкоупругие
тела. Первые—это вязкие, текучие тела, обладающие упругостью
(высокоэластические жидкие полимеры и их растворы), вторые—
это упругие тела, лишенные текучести, но обладающие внутрен-
ним трением (пространственно-структурированные полимеры, студ-
Предисловие редакторов 7
ни). В связи с этим при переводе, в соответствии с содержанием,
использовались оба термйна—«упруговязкость» и «вязкоупру-
гость».
Издание книги А. Тобольского иа русском языке, несмотря на
указанные недостатки, будет весьма полезным для широкого кру-
га научных работников, инженеров, аспирантов и студентов, ин-
тересующихся основными физическими закономерностями меха-
нического поведения полимеров и современными способами рас-
чета механических свойств релаксирующих полимерных тел.
Главы I и II переведены Г. М. Бартеневым, главы III и IV, а
также Приложения—10. В. Зеленевым, главы V и VI—10. С. Зуе-
вым.
Г. Л. Слонимский. Г. М. Бартенев
ПРЕДИСЛОВИЕ АВТОРА
Эта книга излагает связь основных закономерностей механи-
ческого поведения полимеров со строением и движением молекул.
Представлялось целесообразным развить основное содержание
проблемы путем перехода от знакомых понятии к наиболее но-
вым представлениям и при этом обойти утомительные п сложные
математические выкладки, не жертвуя строгостью и глубиной
изложения. Однако некоторые параграфы неизбежно оказались
математически довольно трудными.
В книге умеренного размера могли быть рассмотрены только
избранные темы. Их выбор, естественно, определялся моими
собственными научными исследованиями. Множество оригиналь-
ных экспериментальных данных, приведенных в тексте, показывает,
как па их основе развивались или возникали теоретические пред-
ставления.
Эта книга посвящается моим ученикам и сотрудникам, сов-
местно с которыми была создана новая область знания.
Последние десять лет мои исследования в области вязкоун
ругостп 11 упруговязкостп частично поддерживались Научно-
исследовательским Управлением военно-морских сил США.
Артур В. Тобольский
Принстон
май ! 960 г.
Глава 1
УПРУГОСТЬ и вязкость
1. Сжатие изотропных веществ. Уравнение состояния.
Модуль объемного сжатия
Упругое поведение вещества при сжатии (объемном) является
одним из наиболее известных явлений для изучающего физику,
будь он химик, физик или инженер. В самом деле, современная
атомно-молекулярная теория вещества возникла в равной сте-
пени на основе трактовки Дальтоном закона кратных отношений
и трактовки Авогадро газовых законов Бойля й Шарля. В совре-
менной терминологии принцип Авогадро выражается следующим
образом:
NkT
р = —
(1, 1)
где р—давление, которое оказывают А' молекул идеального газа
при температуре 7' на стенки резервуара, имеющего объем V;
k—универсальная постоянная Больцмана, равная
1,3810"1в эрг/град. Для одного моля газа величина N в урав-
нении (1, 1) равна числу Авогадро Дл = 6,023-1023.
С другой стороны, уравнение состояния идеального газа
можно записать:
п/?Т
Р у
(1,2)
где п—число молей газа п Д kNA есть универсальная газовая
постоянная, которая численно равна 8,31 107 эрг/град-моль.
Вообще для любого вещества, твердого, жидкого или газо-
образного, можно записать соотношение между давлением, объе-
мом и температурой, которое описывает упругие свойства при
объемном сжатии. Такое соотношение известно как уравнение
состояния. Для любых стабильных веществ объем при постоян-
ной температуре уменьшается при увеличении давления. В этой
главе будут рассмотрены уравнения состояния различных си-
стем из большого числа N молекул (или п молен) в газообразном,
жидком нлп твердом состояниях. Во многих случаях уравнение
состояния относят к одному молю вещества, вследствие чего
число N заменяют на N А.
9
10
Гл. I. Упругость и вязкость
Хороню известно, что металлы или ионные кристаллы текут
или разрушаются при деформации растяжения или сдвига, со-
ставляющей малую долю их первоначальной длины, в то время как
жидкости и газы вообще не могут быть подвергнуты какому-либо
упругому обратимому растяжению пли сдвигу*. Напротив,
объемное сжатие твердых тел, жидкостей п газов возможно до
весьма больших давлений термодинамически обратимым путем.
Временными эффектами при объемном сжатии можно пренебречь
во многих условиях эксперимента.
Простота понятия объемного сжатия, вместе с полным отсут-
ствием временных эффектов при статическом объемном сжатии**,
позволяют выбрать его в качестве исходной точки при рассмот-
рении упругих свойств вещества.
Прежде всего необходимо определить первую упругую кон-
станту, а именно, изотермический модуль объемного сжатия:
<‘-3'
.Модуль объемного сжатия обратно пропорционален термоди-
намической сжимаемости. В системе единиц CGS (см-гсек) его
размерность выражается в динах на квадратный сантиметр и
является размерностью единицы давления или же энергии, при-
ходящейся на единицу объема. Модуль объемного сжатия есть
мера упругого сопротивления вещества сжатию, поэтому трудно
сжимаемые вещества имеют высокий модуль обьемного сжатия.
Модуль объемного сжатия для любых значений р, V, Т мо-
жет быть получен из уравнения состояния. Например, модуль
объемного сжатия для идеального газа может быть получен не-
посредственно из уравнений (1, 1) и (1,3):
В = Р
(1,4)
” Известно, что жидкости обладают упругостью на сдвиг при относитель-
но быстрых деформациях. При достаточно быстрых нагружениях они ведут
себя как обычные упругие гела. Для этого лишь необходимо, чтобы время
приложения тангенциальной силы было мало по сравнению с временем молеку-
лярных перегруппировок (или временем релаксации), зависящим от структуры
жидкости, температуры и давления. [М. К о р н ф е л ь д, Упругость и проч-
ность жидкостей, Гостехтеоретиздат, 19511.—Прим, ред.
** Эго утверждение в какой-то мере справедливо для твердых тел при от-
носительно низких температурах. Для жидкостей и стекол (вблизи ТУ), и осо-
бенно для аморфных полимеров, необходимо учитывать релаксационные про-
цессы и изменение структуры в ближнем порядке с изменением давления.
[П. 11. К о б е к о, Аморфные вещества. Изд. АН СССР, гл. 7 (1952):
Н. И. Шишкин, ЖТФ, 26, 1461, (1956)1.—Прим. ред.
I. Сжатие изотропных веществ. Уравнение состояния
11
Для идеального газа при давлении, равном 1 атлг.
В = 1 ат'м = 1,013 - 10ъ дин!см2
Идеальный газ при давлении 1 атм является легко сжимаемым
веществом и поэтому имеет низкое значение В. Большинство
жидкостей при атмосферном давлении имеют модуль объемного
сжатия примерно в 104 раз больший, а именно, 1010 дин/см2. Наи-
менее сжимаемые твердые тела, такие, как алмаз, родий, вольфрам
и иридий, имеют величину В при атмосферном давлении значи-
тельно большую, чем 1012 дин!см2. С другой стороны, модуль объем-
ного сжатия при атмосферном давлении большинства наиболее
сжимаемых твердых тел, таких, как твердый неон, азот, аргон и
цезий, имеют модуль только немногим больше 1010 диш'см2.
Хотя модуль объемного сжатия идеального газа изменяется
пропорционально давлению, модули объемного сжатия жидкостей
и твердых тел являются очень слабыми функциями давления.
В действительности нет большой разницы в значениях В при дав-
1енип, равном нулю, и при давлении в 100 атм для большинства
жидких и твердых тел. Температурная зависимость В для жид-
ких, и особенно для твердых тел, в общем также очень слаба. На-
пример, модуль объемного сжатия кристалла MaCI равен
3,0-1011 дин/слг при 0сК и 2,4-1011 динкм- при комнатной тем-
пературе. Соответствующие величины для КС1 равны
2,1-1011 дин/см’ при 0 °К и 1,8-Н)11 дцн/см2 при комнатной тем-
пературе.
Для сравнения в табл. 1, 1 приводятся величины В при атмо-
сферном давлении и при комнатной температуре (пли при дру-
гих близких температурах) некоторых жидких и твердых тел.
Твердые тела для удобства разделены на молекулярные, метал-
лические, ионные и атомные кристаллы.
Несмотря на то, что мы главным образом рассматриваем вели-
чину В при атмосферном давлении, интересно также принять
во внимание влияние очень высоких давлений.
Весьма любопытным свойством сжатия жидкостей при высо-
ких давлениях является их одинаковое поведение. Большинство
органических жидкостей и вода сжимаются от 20 до 30% по срав-
нению с их первоначальным объемом, если давление увеличивается
от 1 до 20 000 атм, и от 36 до 39% при увеличении давления до
30 000 атм [1]. В то же время при атмосферном давлении значе-
ния модулей объемного сжатия твердых тел различаются на
3 порядка величины, а при давлении в 100 000 атм область зна-
чений В всех твердых тел лежит в весьма узких пределах со сред-
ним значением, равным 1012 дин!см- II].
12
Гл. I. Упругость и вязкость
Таблица 1, 1
Модуль объемного сжатия некоторых распространенных веществ
Вещество дин/CAfi Вещество B- l(T12 дин/см-
Жидкости Металл ы (продолжение)
Ацетон (0 °C) Бензол (35 °C) Хлороформ (20 °C) Эфир (34,8 °C) Этиловый спирт (20 °C) Глицерин (14,9 °C) Ртуть (0 °C) Вода (0 °C) Вода (20 °C) 0.0124 0.0101 0,0086 0,0049 0,0081 0.0461 0,259 0,0193 0,0204 Mg Al Fe Си Ag Pb Ni Ti W 0,33 0,66 1,6 1,5 0,97 0,42 2,3 1,2 3,6
Молекулярные к р Неон Аргон Азот Атомные крист С (алмаз) Be Ge Bi Мета л л ы Li Na К и с т а л л ы 0,010 0,016 0,019 галлы 5,5 1,2 0,70 0,33 0,11 0,062 0,027 LiF LiCl LiBr LiJ NaF NaCl NaBr NaJ KC1 KBr KJ NaCl(0 Ион н °K) ыe co п и 0,65 0,29 0,23 0, 17 0 46 0,24 0,20 0, 14 0, 18 0, 15 0,12 0,30
Литература
1. S.
D. Hamann, Physico-chemical Effects of Pressure, Ch. 3, Academic
Press, New York, 1957, pp. 36—60.
2. Статистическая термодинамика объемного сжатия
Первый и второй законы термодинамики применительно 'к
изотропным веществам, подвергнутым чистому объемному сжатию,
можно представить в виде следующих двух уравнений:
dU = oQ — оГ (2,1)
dU = TdS — pdV Г (2,2)
где U—внутренняя энергия вещества, 6Q—теплота, полученная
системой, 61F—работа, произведенная системой против внешних
сил, S—энтропия вещества и р—давление.
2. Статистическая термодинамика сжатия
Уравнение (2, 2) может быть преобразовано следующим обра-
зом:
/ dS \ idU\ __ ( дА \
{W)T \dv h I dV )т
(2, 3)
В уравнении (2, 3) A=U—TS есть свободная энергия Гельм-
гольца.
Наиболее строгий теоретический метод, применяемый для
расчета уравнения состояния любого вещества, основан на пред-
положении, что энергетические уровни вещества известны. Он
дает возможность рассчитать А как функцию объема и темпера-
туры, исходя из статистической суммы состояний Q (см. Прило-
жение А). Отсюда может быть получено уравнение состояния:
д In Q •
" uV )т
(2,4)
Внимательное изучение уравнения (2, 3) дает возможность
сделать дальнейшие выводы. Ясно, что давление р вещества
состоит из двух слагающих: члена 7'(c)S/c)V)r, который можно
назвать энтропийной составляющей или кинетическим давлением,
и члена—(dU/dV)r, который можно назвать энергетической со-
ставляющей или внутренним давлением. В определенных случаях
S п U могут быть рассчитаны как функции объема и температуры
из очень простых моделей и весьма простыми методами, как это
будет показано впоследствии, что является другим способом
нахождения уравнений состояния из теоретических соображений.
Для любого данного вещества желательно иметь чисто экспе-
риментальный способ оценки кинетического и внутреннего дав-
ления по данным р, V, Т. Это достигается благодаря использо-
ванию хорошо известного термодинамического соотношения:
,-JS_
( dV
(2,5)
Отсюда кинетическое давление T(dpldT)v может быть получе-
ногпросто измерением темпера!урной зависимости давления при
постоянном объеме:
Кинетическое давление = Т
др \
ffrlV
(2,6)
Вычитая кинетическое давление из общего давления, полу-
чаем внутреннее давление:
Внутреннее тавление— р
\ 37 /у
(2,7)
11
Гл. I. Упругость и вязкость
Другой очень удобной термодинамической формулой для вы-
ражения кинетического давления является;
<2.»'
где и “Коэффициент объемного расширения (1/E)(dlz!^Т) и В-
модуль объемного сжатия.
Эго дает возможность другим способом определи гь кинетическое
давление из опытных данных.
Экспериментальное изучение кинетической составляющей
T(dp/dT)v для многих веществ показывает, что имеется два край-
них случая:
1. Если давление при постоянном объеме пропорционально
абсолютной температуре, т. е. р ~сТ, то при подстановке в урав-
нение (2, 6) оказывается, что кинетическое давление также рав-
но еТ, тогда как по уравнению (2, 7) внутреннее давление равно
нулю.
2. С другой стороны, если давление при постоянном объеме
не зависит от температуры, то из уравнения (2, 6) явствует, что
кинетическое давление T(dS/dV)T равно пулю.
Если внутреннее давление — (0UldV)r равно нулю, то вполне
очевидно, что U является функцией только температуры. Отсюда
<2’9)
(если применимо первое условие и внутреннее давление равно
нулю).
Для идеального газа при постоянном объеме давление пропор-
ционально абсолютной температуре согласно закону Шарля. По
первому условию внутреннее давление равно нулю. Отсюда по-
лучается уравнение (2, 9).
Чтобы применять уравнение (2, 9), необходимо иметь выраже-
ние для энтропии как функции объема. Для системы А молекул
с заданной энергией U и объемом И энтропия S определяется с
помощью известного принципа Больцмана следующим образом:
S = feln *2 (И, Г) (2,10)
где 2—число квантовых состояний системы, реализующихся
при заданных значениях U, V п А'. В классической статистической
механике 2 пропорционально объему фазового пространства си-
стемы. В простейшей физической трактовке 2 представляет со-
бой число микросостояннй, которое соответствует данному термо-
динамическому мак росостоя н ню.
2. Статистическая термодинамика сжатия
15
Для идеального газа
U=zf(UWN (2,11)
где f(U)—функция только внутренней энергии.
Пропорциональность между 2 и V'N интуитивно очевидна,
как показано в Приложении Б, что вполне достаточно для обо-
снования сказанного.
Комбинируя уравнения (2,9), (2, 10) и (2, 11), можно легко
получить уравнение состояния p = NkTIV.
Изложенная выше трактовка предполагает, что молекулы
имеют точечные размеры и что нет сил, действующих между ними.
Давление на стенки сосуда является истинным кинетическим
давлением, возникающим только вследствие теплового движения
молекул и их столкновений со стенками сосуда. 1
Для молекулярной модели, способной объяснить /сйойства
реальных газов, в особенности газов при высоких^ давлениях,
необходим учет сил притяжения и отталкивания между молеку-
лами. Эти силы приводят к отклонениям свойств реальных; га-
зов от свойств идеального, в особенности при высоких|давленЦях.
Для реальных газов уравнение состояния для одного, моля газа
при низких и средних давлениях может быть выражено ‘так;на-
зываемым вириальным рядом: 1 '•
+ .. (2;12)
Величины (2, т3, и т. д. называются вторым, третьим, чет-
вертым и т. д. вирпальными коэффициентами газа. Они могут быть
найдены как функции температуры с помощью измерений р, V
и Т.
Из общих законов статистической механики [1] может быть
записано простое соотношение между вторым вириальным коэф-
фициентом у2 и энергией взаимодействия н(г) между двумя изо-
лированными молекулами, находящимися на расстоянии г:
Т2(Л = 2кМ
(1
ехр [—п(г)/йТ|) r2dr
(2.13)
Для химически насыщенных молекул, таких, как аргон, азот
двуокись углерода и метан, энергия взаимодействия и(г) между
двумя изолированными молекулами может быть выражена как
функция межмолекулярного расстояния г. Наиболее удобным
приближением является формула Леннарда-Джонса [2]:
i/(r) =
(2, 14)
in
Гл. 1. Упругость и вязкость
Первый член с показателем степени 12 описывает потенциал
сил отталкивания между молекулами, а второй с показателем
степени 6 выражает потенциал сил притяжения. Величина г*
представляет собой минимум потенциальной энергии при расстоя
нпп между молекулами, равном г* (рис. 1, 1). Величины г* иг* сле-
дует рассматривать как основные
характеристики молекул.
Если уравнение (2, 14) подста-
вить в уравнение (2,13), то получим
определенное соотношение между
у2,г* и г* [2]. Это соотношение бы-
ло детально табулировано для ис-
пользования в численных расчетах
131. Это значит, что из экспери-
ментально установленных значений
второго вириальното коэффициента
газов можно получить фундаменталь-
ные величины е* н г*. В табл. 1, 2
приводятся значения этих констант
для некоторых молекул 14].
Величины г* и г* можно исполь-
зовать также для того, чтобы полу
чить уравнение состояния для газов
I'.ic. 1.1. Потенциал Ченкарда при очень высоких давлениях [5].
Джонса. Это уравнение состояния выражено
в приведенных переменных р/р*.
I7E* и 77Т*, и в этих приведенных переменных оно имеет одни
н ют же вид для всех химически насыщенных веществ1 *.
Величины р*. V* п 71* определяются следующим обратом:
р* = 2г*/г*3
Е* = /V/'*3, |/2
7’* • г*/Р
(2, 1 го
Два вещества, имеющие одинаковые величины pip'"", Е/Е' и
l;lf, находятся в соответственных состояниях. К сожалению,
уравнение состояния газов при очень высоких давлениях
сложно в математическом отношении и далее усложняется кван-
товыми эффектами, важными для легких молекул, таких, как
11.,, IX и Не, при низких температурах. Для молекул, где квапто-
1 Т. е. для веществ, молекулы которых химически не взаимодействую!
и не разлагаются под действием температуры, давления и других факторов
в исследуемых интервалах. — Прим. ред.
Уравнение состояния для молекулярных кристаллов
17
Таблица 1, 2
Характеристические величины . г’ для некоторых химически насыщенных
молекул
Газ г*-1015, С г\ А г», -К \ , СМ^/АЮЛЬ р*, атм
Не [4] 1,41 2,87 10,22 10,06 84,5
Не 4,91 3,09 36,3 13,51 220
Аг 16,54 3,82 120,3 24,0 415
Кг 23,6 4,04 171 28,3 503
Хе 30.5 4,60 221 41,5 440
Н. 5, 11 3,29 37,0 15, 12 203
D, 5,11 3,29 37,0 15, 12 203
N., 13,2 4,17 96,6 31,3 254
о2 16,3 3,89 118 24,9 390
NO 18,1 3,56 131 19.3 562
со2 28,3 4,57 205 40,6 416
сн4 20,5 4,29 148,5 33,6 364
вые эффекты несущественны, теория предсказывает следующие
шачения для критических констант рс, Vc и Тс:
ТС1Т* = 1,33
= 2,0
ре1р* = 0,47
(2, 16)
Эти величины хорошо согласуются с экспериментальными
данными [4|.
Интересно заметить, что для жидкостей при атмосферном дав-
лении как кинетическое, так п внутреннее давление очень вы-
соки и почти уравновешивают друг друга.
Литература
1. .1. Е. М ajer, М. Ci. М а у е г, Statistical Mechaiiics, ch. 12, John Wiley a-
Sons, New York, 14-10.
2. J. E. 1. e n n a r d-J о n e s, Proc. Roy. Soc. London, AI06, 463 (1924).
3. .1. O. rl i rsc li le 1 de r, F. Curtiss. R. В. В i r d, Molecu-
lar Theory of Gases and Liquids, John W iley a. Sons, New York, 1954.
4. ,S. D. Hama n n, Physico-chemical Etlects of Pressure, ch. Ill, Academic
Press, New York, 1957, pp. 42—48.
5 J E. Lennar d-J ones, A F. D e v о n s h i T e. Proc. Roy. Soc.
(London), M63, 53 (1937).
3. Уравнение состояния для молекулярных кристаллов
Очень важные с точки зрения понимания свойств твердых тел
величины е* и г* могут быть также использованы для нахожде-
ния уравнения состояния и модуля объемного сжатия молеку-
2—833
18
Гл. 1. Упругость и вязкость
лярных кристаллов. Легче всего производить вычисления при
о ° к.
Формула для А(Т, V) молекулярного кристалла следующая:
А(Т, V) = UL(V) + UZ(V) + USV, Г) - TS(V, Т) (3, 1)
В уравнении (3, 1) величина UL(V) -энергия решетки, в которой
каждая молекула находится в ее равновесном положении, 6^(к)—ко-
лебательная энергия при абсолютном нуле, C/V, Т)—тепловая ко-
лебательная энергия и S(V,T)—энтропия, зависящая от колеба-
ний (и в некоторых случаях добавочная, конформационная эн-
тропия). При 0°К Ut(V, Т) и —TS(V, Т) равны нулю.
Для очень легких молекул, таких, как водород, гелии и воз-
можно неон, квантовые эффекты, относящиеся к члену UZ(V).
весьма существенны. Для других кристаллов членом UZ(V) можно
пренебречь, а давление п модуль объемного сжатия при О °К
могут быть выражены в хорошем приближении как:
р = — dUL (V)/dV при 0°К (3,2,
B=Vd2t/L(V)/dr при 0°К (3,3,
Используя формулу Леннарда-Джонса {уравнение (2, 14)].
можно получить формулу для молярной энергии кристалличе-
ской решетки IJL(V) молекулярных кристаллов (аргон, азот,
двуокись углерода, метан и т. д.). Рассмотрим случай гранецент-
рнрованной кубической решетки. При О °К NA молекул занимают
точные положения—узлы кристаллической решетки. Межмоле-
кулярные расстояния г,-,- между любыми двумя узлами решетки
/ и j легко выражаются через расстояние между ближайшими
соседями г. Соотношение между объемом И и расстоянием между
ближайшими соседями г есть:
Энергия UL(r) или UL(V) может быть записана как функция
парных взаимодействий и(г(|):
UL(V) = 4"N^'n(r'i) (З.Гм
Штрих при сумме указывает на суммирование по всем узлам ре-
шетки, исключая j-м. Уравнение (3, 5) отражает тот факт, что
сначала подсчитывается общая энергия взаимодействия t-той
молекулы со всеми ее соседями /. Для того чтобы получить пол-
;л Уравнение состояния для молекулярных кристаллов
19
ную энергию, необходимо умножить этот результат на NA и
затем разделить на 2, так как все парные взаимодействия учте-
ны дважды.
В гранеценгрированной решетке имеется 12 ближайших сосе-
дей на расстоянии г, 6 следующих ближайших соседей на расстоя-
нии | 2 г и т. д. Сумму, указанную в уравнении (3, 5), можно
численно рассчитать, учитывая последовательно влияния бли-
жайших соседей, следующих ближайших соседей и т. д. При вы-
числении этим способом сумма быстро сходится.
Результат вычислений следующий |1 2|:
UL(V) =- NAz* f 8,62 17,24 | (3, 6)
В этом уравнении Ии является функцией величины г* (по-
тенциал Леннарда-Джонса):
Ио = 0,65/V./*3 = 0,91 GV* (3, 7)
Из уравнений (3, 6) и р -—dUJdV легко можно вывести урав-
нение состояния для молекулярного кристалла при 0°К:
р = 37,64р*|(-^)5 р|’| (3,8)
—
ш = 1 2 ...
Из уравнения (3, 8) видно, что величина 10, введенная в урав-
нениях (3, 6) и (3, 7), фактически есть молярный объем при
1 --0 °К и р - О.
Модуль объемного сжатия при 0 °К может быть получен как
функция давления из уравнений (3,8) и (1,3):
В(р) = 1,07-102 1~7р
(3,9)
При обычном давлении второй член в уравнении (3, 9) пре-
небрежимо мал по сравнению с первым, так что при 0 °К и дав-
лении ниже 100 атм модуль объемного сжатия Во дается как:
Во= 1,07-102-~ (3,10)
Молярная энергия испарения £i,cn. при 0 °К и р^=0 равна
- UL (Ио). Она может быть сразу получена из уравнения (3, 6)
подстановкой И -=И«:
£Scn, = 8,62Л^* (3,11)
20
Гл. 1. Упругость и вязкость
Новое выражение для модуля объемного сжатия Во поэтом}
может быть дано таким, чтобы в него входили экспериментально
измеримые Е°кп и Со. Как можно видеть из уравнении (3, 7),
(3, 10) и (3, 11)’
_ 8,О4ЕцСП
(3, 12-
При более высоких температурах, как это следует из экспери-
ментальных данных, модуль объемного сжатия приблнзшельнс
равен Ви. К несколько лучшему приближению приводит формула:
В(Т) =
8,04£ПСп.
V
(3, 13}
где Eiicn и V молярная энергия испарения и молярный объем
при данной температуре.
В табл. I, 3 представлены величины молярного объема, энер-
гии испарения, модуля объемного сжатия, величин з* и г* для
некоторых молекулярных кристаллов.
Таблица 1. 3
Сравнение рассчитанных и измеренных характеристик молекулярных
кристаллических решеток
Вещество Ne Аг Ki Хе М2 0'2 NO СОо сн4
I015 г*, эрг г*. А I'o рассч.3 измер.6 £11СП, зре/люль-Ю"10. рассч.3 6|1сп , арг/люль- Ю10, измер.6 Ва, дин/см~ КГ10, рассч.1' В, дин/смг-10“10, измер.6 0, °К, рассч.-1 А. 'К, измер.6 4.91 3,09 11,4 2,55 1,82 1,70 1,0 60 63 16.54 3,82 21.8 16,76 8,58 6,47 3,16 1,6 63 85 23,6 4,04 25,8 32,19 12,2 9,7 3,83 30,5 4,60 38,0 42,94 15,8 13,0 3,35 13,2 4, 17 28,3 13,65 6,86 5,6 1,94 1,9 61 68 <6,3 3,89 23,0 25,20 8,48 6,8 2,99 69 <8,1 3,56 17,6 9,41 4,29 82 119 28,3 4,57 37,3 38, 16 14.7 25,2 3,17 65 20,5 4,29 30’,7 37,76 10.7 81,9 2,7г 98
а> Из уравнения (3, 7).
Все экспернменгальные величины взяты из литературных исго*шпков при самых низких
температурах, которые там указывались, а не экстраполированы к О °К; I" — ыолярньп
объем в точке кипения.
Из уравнения (3, II).
г> Из уравнения (3, 12).
Из уравнения (5, 17).
4. Упругие свойства изотропных тел при малых деформациях
21
Литература
1 . S. D. Haman n, Physico-chemical Eflects of Pressure, ch. Ill, Academic
Press, New York, 1957, pp. 42—48.
2 J. E. L e n n a r d-J о n e s, A. E. Ingham. Proc. Roy. Soc. (London),
A107, 636 (1925).
4. Упругие свойства изотропных тел
при малых деформациях
Имеется гри весьма элементарных вида упругой деформации
изотропных тел: всестороннее (объемное) сжатие, простое растя-
жение и простой сдвиг. Всестороннее сжатие обсуждалось в пер-
вых двух параграфах. Теперь мы введем понятия о растяжении н
сдвиге.
.Пример простого растяжения приведен на рис. I, 2. Прямо-
угольная призма из изотропного упругого материала с размерами
х, у, г подвергается действию двух уравно-
вешенных растягивающих сил X, как показано
на рисунке. Под воздействием этих
растягивающих сил образец изменяет свои
размеры до x-]dx, y\dy и z+dz. Деформации
sx, Sy и s, в направлениях х, у, z определяются
следующим образом (рассматриваются очень
малые деформации):
Рис. 1,2. Простое
растяжение.
Образец, конечно, будет иметь положи-
тельную деформацию только в направлении оси х.
Для изотропного тела в поперечных направ-
лениях наблюдается некоторое сокращение размеров:
sv = sz = —os*
(4,2)
где о--коэффициент Пуассона, являющийся важной характери-
стикой упругих тел. Величина о изменяется от нуля (в случае
отсутствия поперечного сокращения) до 0,5 (если объем не из-
меняется при растяжении). Величина о для большинства каучу-
коподобных материалов близка к 0,5. Для большинства кристал-
2'2
Гл. I. Упругость и вязкость
тических материалов ц стекол значения коэффициента Пуассона
располагаются между 1li и Ча.
Напряжение растяжения в эксперименте простого растяжения
(см. рис. I, 2) равно:
f = XlA (4,3)
где A—yz—площадь поперечного сечения образца, а X — растя-
гивающая сила.
I /я упругих веществ при растяжении напряжение и деформа-
ция связаны между собой
та коном Гука:
/ =• Esx (4,4;
где /? называется модулем
Юнга.
Пример простою сдвига
показан на рис. I, 3. Прямо-
угольная призма, размерами
.г, у, z, прочно прикреплена
к неподвижному основанию
гранью ху. На противоположной стороне в направле-
нии .V действует тангенциальная сила, которая заставляет тело де-
формироваться. Деформация сдвига s определяется как
^dx!z. Для малых деформаций tgT~y, отсюда
ь tg р р (4, 5)
Напряжение сдвига / определяется как сила X, деленная на
площадь .4 -=ху.
f = X/A (4,6)
Доя упругих веществ деформация и напряжение при сдвиге
связаны метках' собой талоном Гука:
I = 0s (4, 7)
где О’—модуль сдвига.
Закон Гука, выраженный уравнениями (4,4 ) л (4, 7), действи-
телен только при оче.нь малых деформациях. Многие стекла и
кристаллы могут быть подвержены растягивающим пли сдвиго-
вым деформациям только на величину 0,001 или меньше без
разрушения, пластического течения или проявления других не-
обратимых неупругих эффектов. Для этих тел изучение упругих
свойств при сдвиге и растяжении должно быть ограничено малыми
цеформацпями.
Различные упругие константы, приведенные в этом разделе,
находятся в следующем соотношении [1]:
Е --= ЗВ(1 — 2Д = 2(1 ф з)0 (4,8)
4. Упругие свойства изотропных тел при малых деформациях
23
Другими словами, только две из четырех упругих констант
Е, В, G и а в действительности независимы. Поскольку мы уже
рассмотрели зависимость модуля объемного сжатия В от фун-
даментальных молекулярных параметров, по крайней мере для
молекулярных кристаллов, то интересно проследить, как изме-
няются Е/В и G/В с изменением коэффициента Пуассона □.
В табл. I, 4 приведены эти отношения для различных возможных
величин а.
Таблица I, 4
Отношения Е/В, О/В и Е/G при различных значениях коэффициента
Пуассона а
5 Е/В G/B E/G Примечание
0,00 3 1,50 2
0,10 2,4 1,09 2,2
0,20 0,25 1,8 1,5 0,75 0,60 2,4 , 2,5 1 Область кристаллнче-
0,30 1,2 0,463 2,6 | ских материалов и
0,333 1,0 0,375 2,666 I стекол
0 40 0,6 0,214 2,8
0,4996 0,0024 0,008 2,9992 1
0,4997 0,0018 0,0006 2,9994
0,4998 0,0012 0,0004 2,9996 1 Область каучукоподоб-
0,4999 0,0006 0,0002 2,9998 | ных материалов
0,49995 0,0003 0,0001 2,9999
0,50 0,000 0,000 3,00 1
Общее определение напряжения и деформации в трех измере-
ниях представляет интерес, но это по существу геометрическая
проблема, которая не будет здесь рассматриваться, поскольку она
хорошо изложена во многих других учебниках 11]. Для наиболее
общего анизотропного тела закон Гука выражается шестью ли-
нейными уравнениями, связывающими каждую из шести компо-
нентов напряжения с шестью компонентами деформации. В самом
общем случае имеется 21 независимая упругая константа.
Число независимых упругих констант снижается в кристал-
лах, имеющих элементы симметрии. Тем не менее даже кубиче-
ский монокристалл имеет три независимых упругих константы.
С точки зрения простоты математической обработки, к счастью,
большинство макроскопических образцов кристаллических ве-
ществ находится в поликристаллической форме, т. е. состоит из
тысяч мельчайших беспорядочно ориентированных относительно
друг друга кристалликов. Такое поликрпсталлическое вещество
можно считать изотропным телом, не вдаваясь в рассмотрение
того, какова кристаллическая симметрия его индивидуальных
'-!! I .i. i. Упругоаь и вязкость
кристаллов. Неорганические н органические стекла и каучуки
вообще являются изотропными телами (в нерастянутом состоянии).
Все эти вещества можно рассматривать, как имеющие только две
независимые упругие константы. Соотношения между В, Е, 6
и с представлены уравнением (4, 8).
Для такого вещества, как резина, которая является обратимо
деформируемой в широком интервале величии длины L, можно
экспериментально найти уравнение состояния относительно рас-
тягивающей силы X, длины L я температуры Т (при атмосфер-
ном давлении). При растягивающей силе, равной нулю, вещество
приобретает свою первоначальную длину Ln. Это уравнение со-
стояния обычно называется законом деформации резины и яв-
ляется вообще нелинейным, так как закон 1ука в этом случае
неприменим за исключением очень малых величин (L—La)lLn.
В случаях больших обратимых растяжений деформа! ия более
cipoio должна определяться в дифференциальной форме как dL/L,
а не в форме (L -Lo)/Lo.
Литература
I. С Kittel, Introduction to Solid State Physics, 2nded., ch. 4, John Wiley
a. Sons, New York, 1956. [Ч. Ккттел ь. Введение в физику твердого
тела, Гостехтеоретиздат, 1957].
5. Упругие константы и колебания решетки
Представим себе, что кристаллическая решетка состоит из
NА узлов с массой т, занятых атомами, нонами пли молекулами.
Молярный объем связан с расстоянием до ближайших соседей
следующим образом:
V = cNArs (5,1)
где с- -1 /1/2 для гранецентрированной кубической решетки,
=1 для простой кубической решетки.
При 0°К энергия решетки С/.(И) может быть представлена ря-
дом Тейлора для значения V - VT, являющегося равновесным объе-
мом при давлении, равном нулю:
(5, 2)
Второй член исчезает, так как р^=—dU/d\' при О °К, а по
условию р=0 при l/=Vo. Следующий член может быть упрощен
>. Упругие константы и колебания решетки
25
поскольку Во~- Vo(d2U/dV2) при 0 ° К. Отсюда, пренебрегая чле-
нами высшего порядка,, имеем:
UL(V) = UL{V0) 4 4- V-(Д,/)2 <5’ 3»
Из уравнения (5, 1) получаем дифференцированием:
ЛЕ = 3(V4cr2(Ar) ЗЛглсг2(Аг) (5, 4)
где го—значение г, если объем равен Ео. Подставляя (5, 4) в
(5, 3), получаем:
чдг
UdV) = ^(V'o) -I (5. 5>
Эйнштейн предположил, что реальная кристаллическая ре
шетка из NA точечных масс может быть заменена соответствую-
щим набором NA трехмерных гармонических осцилляторов или
эквивалентно набором 3/¥л независимых линейных осцилляторов
[1J, так как каждая точечная масса имеет три степени свободы,
поскольку она колеблется в трех измерениях.
Если силовая константа каждого из этих гармонических ос-
цилляторов с массой m есть К, то энергия совокупности осциллято-
ров равна’
U = Uo + 3Na ~ К (А7)2 (5, 6)
где Uo—энергия осцилляторов в равновесных положениях и
А г2—среднее квадратичное смещение осцилляторов из положения
равновесия. Если взять набор 3NA осцилляторов как эквивалент
всей кристаллической решетки, то можно соответствующие члены
уравнений (5,6) и (5,5) приравнять. Отсюда получим весьма
простое соотношение
Во = (5. 7)
0 Зсгп v ’
Частота колебаний v£ осцилляторов связана с силовой кон-
стантой /( и массой m соотношением
1 / К \ V2
2х ( ш )
(5, 8)
Сравнивая уравнения (5, 8) и (5, 7), получаем:
_ 1 (3сгоВо 'Л'2
'Е~~ 271
(5,9)
F
26 Г.i. /. Упругость и вязкость
Подставляя уравнение (5, 1) в уравнение (5, 9), получим в ре-
зул ьтате:
gVa t,*/e
V = 2,33 • 10’ -Чг— (5, 10)
/И1/з
для простой кубической решетки
D,/2 >/б
-,£= 2,1-10’ (5,11)
м
для гранецентрщюванной кубической решетки
где v—удельный объем (объем одного грамма), а М—молекуляр-
ный вес молекул, атомов или ионов, находящихся в узлах ре-
шетки.
Эйнштейн далее предположил, что разрешенные значения
энергии, приписываемые каждому осциллятору, подчиняются
закону Планка s„=n/jvt-, где п—принимает значения п = 1,
2, 3- • и т. д., a h—постоянная Планка, равная 6,62-10-2’ эрг сек.
Затем он нашел среднее значение всей энергии ЗА'Л осцилляторов,
которую принял в качестве тепловой колебательной энергии
кристалла:
U,(V, Т} = 3N , V е exP(-£n/feT-L =---(5, 12)
' 1 i-J " ~ exp h<JkT\ — 1 ’
}>хр(-,Ж)
п= 1
Пз уравнения (5, 12) сразу может быть получена теплоемкость
кристалла (в приближении Эйнштейна) путем дифференцирова-
ния по Т\
р __ ГдО',(1/ Т) I _ ^Ak(^E/kTyexp(h-lE/kT)
v'~ | дТ |v [exp (Ir^/kT) — 1 p ’
Уравнение (5,13) для теплоемкости кристалла было первым
применением квантовой теории к свойствам вещества. При под-
ходящем выборе уравнение (5, 13) обеспечивает допустимое,
но не строгое описание экспериментальной кривой Cv от-
носительно Т, полученной для одноатомных твердых тел.
С точки зрения механических свойств представляет большой
интерес то обстоятельство, что лучшее значение эйнштейновской
частоты >Е, полученной пз экспериментальной кривой теплоем-
кости и уравнения (5, 13), приводит к значению В, найденному
по уравнению (5, 10) или (5, 11). Это иллюстрируется данными
табл. 1, 5. Теория Эйнштейна для твердых тел дает приближен-
ное соотношение между тепловыми свойствами и механическими
э. Упругие константы и колебания решетки
27
свойствами, которое не зависит от закона сил между атомами в
узлах решетки кристалла. Другие упругие константы Е и G
могут быть получены из В и коэффициента Пуассона с.
Таблица 1,5
Сравнение экспериментальных и теоретических
(по Эйнштейну) значений модуля объемного сжатия В
Вещество
в“ч иг12
СЛ<2
^эксп.
^расч.а
Na 0,062 0,61
Al 0,66 0,64
Си G 1,5 5 5 0,81 0,33
Au 1,5 1,1
Pb 0,42 1,2
Bi 0,33 0,74
\V 3,6 0,59
Ag 0,97 0,89
NaCl 0,30 0,57
KC1 0,21 0,66
Л — рассчитано из уравнения (5,1!); у —рассчитано из
Е
уравнения (5,15).
Как хорошо известно, Дебай улучшил эйнштейновскую тео-
рию теплоемкости, введя распределение З.¥л частот, характери-
зуемое максимальной частотой vm (приложение А). Это приводит
к соотношениям между В и и между Cv и несколько более
точным, но также, и более сложным, чем уравнения Эйнштей-
на (2|.
Величины которые, если их ввести в уравнение Дебая
для C.v, дают лучшее совпадение с данными по теплоемкости,
подробно табулированы для многих веществ. Чаще всего таблич-
ные данные выражают с помощью дебаевской температуры 6D,
определяемой следующим образом:
6 о = h'imlk
(5, 14)
Эйнштейновское значение частоты которое дает нанлучшее
совпадение с данными по теплоемкости, находится с широко табули-
рованной дебаевской температурой 0Л) твердого тела в следующем
п р пбл ижен н ом соотн ош ен и и:
4h 'л'
2
4
(5, 15)
28
Гл. /. Упругость и вязкость
Из соотношения между модулем объемного сжатия п эйнштей-
новской частотой [уравнение (5, 11)1 и из уравнений для модуля
объемного сжатия и молярного объема молекулярных решеток
[уравнения (3, 7) и (3, 10)J автор получил новое соотношение:
(5, 16)
где ш -масса молекулы. Это приводит к интересному соотноше-
нию между тепловыми свойствами и молекулярными константа-
ми е’ и г* для молекулярных решеток. Законность этого урав-
нения проще всего подтвердить, если заменить на 0д, исполь-
зуя уравнение (5, 15):
1)^1,17-10*711/
(5, 17)
Экспериментальные значения 0Д были получены для некото-
рых молекулярных решеток путем измерений удельной теплоем-
кости. Эти экспериментальные величины сопоставляются в табл.
I, 3 с предсказанными величинами, рассчитанными из е*, г*
и молекулярного веса М по уравнению (5, 17).
Соотношения, приведенные в этом разделе и в разделе 3, почти
достаточны, чтобы получить уравнение состояния, пригодное для
молекулярных кристаллов не только при 011\, по также при
всех других температурах. Такое уравнение выведено в
Приложении В.
Литература
1. А. Е 1 п s t е i п, Ann. с!. Phvs., 22, 180 (1907); ibid., 34, 170 (1911).
2. J. С. Slater, Introduction to Chemical Physics, ch. 14, McGraw-Hill
Book Co., New York, 1939, p. 238.
6. Термодинамическая трактовка простого растяжения
Если применить первый и второй законы термодинамики к
деформации растяжения, то получим следующее уравнение:
dU = TdS+ XdL — pdV
(6,1)
где А'—растягивающая сила, a L—длина образца. Это уравнение
может быть переписано в следующем виде:
(6, 2)
6. Термодинамическая трактовка простого растяжения
29
Из уравнения (6, 2) следует, что равновесная растягивающая
сила, которая действует на упругое тело, имеющее длину L,
состоит из двух частей: а) кинетической, обусловленной изме-
нением энтропии —r{dSldL)T,Vi и б) энергетической, обуслов-
ленной изменением внутренней энергии (dU/dL)r v.
Величина растягивающей силы кинетического происхождения
может быть рассчитана из следующего термодинамического урав-
нения:
Хотя уравнение (6, 3) является точным, однако коэффициент
\dXldT)v L трудно получить из простого эксперимента. Конечно,
сохранять постоянную длину L при изменении температуры легко,
но если температура изменяется при обычных атмосферных усло-
виях, то объем также изменяется. Для того чтобы скомпенсиро-
вать это, нужно приложить изменяющееся гидростатическое дав-
ление, что встречает серьезные экспериментальные затруднения.
Были проведены различные, по существу эквивалентные, теоре-
тические исследования для решения этой сложной задачи
11, 2, 3|. В случае сильно растягивающихся веществ, которые
имеют физические свойства, качественно описываемые как ка-
учукоподобные, простейшим выражением этих теоретических
рассмотрений является |3] следующее приближенное (но доволь-
но точное) уравнение:
где а—относительное растяжение (кратность растяжения).
Величина а определяется следующим образом:
« = Г (6,5)
Г-0
где Lo—длина при напряжении, равном нулю, при давлении р
(обычно в 1 аиш) и температуре Т. Длина Lo есть функция тем-
пературы вследствие теплового расширения.
В случае каучу когюдобных материалов для определения
(дХ/дТ)р „ существует два подходящих метода: 1) длина L может
устанавливаться при любой температуре такой, чтобы LILo в
действительности являлась константой и 2) измерение силы как
функции температуры может производиться при постоянной
длине. Во втором случае для отнесения результата к постоянному
удлинению необходимо вносить рассчетную поправку, вытека-
ющую из значения коэффициента теплового расширения и кри-
вой напряжение—деформация для каучукоподобных материа-
лов 11, 4, 5J.
30
Гл. I. Упругость и вязкость
При больших удлинениях, когда а примерно равна двум или
больше, эта поправка становится относительно малой.
Уравнение (6, 2) теперь может быть преобразовано следую-
щим образом:
(6, 6)
Если при постоянном растяжении а напряжение пропорцио-
нально абсолютной температуре, т. е. Л' —аТ (в температурном
интервале высокой эластичности), то тогда по уравнению (6, 6)
слагающая растягивающей силы (dUldL)T, v равна нулю, п
напряжение возникает только от кинетической силы. Это было
точно установлено для вулканизованного натурального каучука
и для многих вулканизованных синтетических каучуков и при-
нимается в качестве определения идеальной резины.
Для идеального газа, подвергнутого обратимому изотермиче-
скому сжатию, давление возрастает вследствие уменьшения энтро-
пии при уменьшении объема. Совершенно аналогично в случае
идеального каучука, подвергнутого обратимому изотермическому
растяжению, сила, сопротивляющаяся растяжению, возникает
вследствие уменьшения энтропии при увеличении растяжения:
для идеального газа
(6, 8)
для идеальной резины.
Тот факт, что сила, поддерживаемая в резине при постоянном
высоком растяжении, пропорциональна абсолютной температуре
и что длина образца каучука при постоянной силе сокращается
с увеличением температуры, хорошо известен более столетия.
Термодинамический смысл этого результата, как это видно из
уравнения (6, 8), также был давно известен. Однако только после
того, как в 20-х годах текущего столетия было установлено макро-
молекулярное цепное строение каучука, Мейер, Сузих н Валко
[61 раскрыли смысл уравнения (6, 8) методом молекулярной ста-
тистики, который был быстро применен и развит многими другими.
История развития этих исследований, а также само изложение
молекулярной статистики упругости каучука даны в трудах
Флори [71 и Трелоара [8].
В следующем разделе будет рассмотрено уравнение состоя-
ния идеальной резины, а в главе II (раздел 9)—приведена совре-
менная трактовка его на основе молекулярной статистики.
7. Упругость идеальных каучуковых сеток
31
Если растягивающая сила X при постоянном удлинении не
зависит от температуры, то из уравнения (6, 4) следует, что ки-
нетическое напряжение равно нулю. Напряжение в этом случае
возникает только вследствие увеличения внутренней энергии
при растяжении. Это встречается в кристаллах и стеклах при
абсолютном нуле. Из уравнения (6,2) ясно (и a fortiori из третьего
закона термодинамики*), что если Т стремится к нулю, то
tdU\
X=(dL) (6,9)
для кристаллов и стекол при О °К.
Полученный результат совершенно аналогичен объемному
сжатию кристаллов или стекол при О °К, которое рассматривалось
для молекулярных кристаллов в разделе 3. Вклад слагающей
(д1ЛдЬ)ту становится значительным при. рассмотрении упругих
свойств полимеров в их стеклообразном состоянии, которое всегда
достигается при относительно низких температурах.
Литература
1. D. R. Elliott, S. A. Lippma пп, J. Appl. Phys., 16, 50 (1945).
2. G. Gee, Trans. Farad. Soc., 42, 585 (1946).
3. P. J. Flor y, Principles of Polymer Chemistry, Cornell University Press,
Ithaca, New York, 1953, pp. 489—491.
4. К- H. Meyer, C. Ferry, Helv. Chim. acta, 18, 570 (1935).
5. R. L. Anthony, R. II. Caston, E. Guth, J. Phys. Chem., 46,
826 (1942).
о. К- H. Meyer, G. von Susie h, K. Vai k o, Koll. Z., 59, 208
(1932).
7. P. J. Flor y, Principles of Polymer Chemistry, ch. 12, Cornell University
Press, Ithaca New York, 1953.
8. L. R. G. T re 1 о a r, The Physics of Rubber Elasticity, Clarendon Press,
Oxford, England, 1949; 2nd ed., 1959. [Л. T релоар, Физика упру-
гости каучука, Издатинлит, 1953].
7. Упругость идеальных каучуковых сеток
Если модуль Юнга кристаллических твердых тел и стекол ле-
жит в области от 1010 до 1013 дин/см2, то модуль Юнга натураль-
ного каучука и большого количества синтетических каучуков
находится примерно в области от 10® до 108 дин/см2. Более того,
если предел упругости многих кристаллических и стеклообраз-
ных твердых тел существенно меньше 1%, то натуральный и син-
* Имеется в виду, что для чистых веществ энтропия при абсолютном нуле
равна нулю, поэтому из уравнения (6, 2) следует уравнение (6, 9).—Прим. ред.
32
Гл. I. Упругость и вязкость
тетнческие каучуки часто могут быть обратимо растянуты на
несколько сотен процентов. Мы обычно будем употреблять слово
«каучук» для характеристики состояния вещества, а не как назва-
ние соответствующего химического вещества, полученного из
каучукового дерева*.
Весьма важным ключом для объяснения механизма растяжения
каучукоподобпых материалов являлся тот факт, что напряжение
при постоянном растяжении а пропорционально абсолютной темпе-
ратуре в температурной области высокой! эластичности. Из рас-
смотрения, приведенного в разделе 6, сразу становится очевидным,
что напряжение в растянутом идеальном каучуке возникает вслед-
ствие присутствия энтропийного члена в уравнении (6, 2).
Все многочисленные синтетические каучуки, так же как и
натуральный каучук, имеют общую молекулярную морфологию.
Они образуются из длинных гибких ценных молекул, иногда
сшитых поперечными связями, что создает трехмерную сетку.
В тниичпом «сшитом» каучуке (резине) с хорошими свойствами
приходится примерно одна поперечная связь на несколько сотен
атомов цепи. Линейные полимеры с достаточно большим молеку-
лярным весом также проявляют каучукоподобные свойства в
соответствующем температурном интервале. В этих случаях
«запутанности» цепей действуют как подвижные поперечные свя-
зи. Поэтому линейный полимер будет течь лишь при существен-
но высоких температурах. Например, невулканпзованный на-
туральный каучук является линейным полимером, который, хотя
каучукоподобен при комнатной температуре, однако имеет огра-
ниченную применимость, так как течет при высоких температурах.
Для того, чтобы изготовить необходимое изделие, натуральный
каучук смешивают с вулканизующими агентами, такими, как сера
или перекиси, формуют или прессуют в желаемой форме прг:
высоких температурах и в конце концов структурируют (вулка-
низуют) при высоких температурах. Последний процесс состоит
в химическом поперечном сшивании макромолекул каучука
для создания сетки. В этом разделе рассматриваются главным об-
разом химически сшитые сетки с учетом также и подвижных попереч
ных связей, которые могут быть образованы вследствие перепу-
танности цепных молекул, наличия мельчайших кристаллитов,
а также возможно исключительно сильными межмолекуляриыми
вза 11 моде йств и я м и.
В нерастянутом состоянии цепь сетки (часть молекулярной
цепи между смежными поперечными связями) беспорядочно свер-
нута и фактически быстро изменяется от одной конформации к
* Автор употребляет слово «каучук» вместо слова «эластомер», обозначая
им тело, находящееся в высокоэластическом состоянии.—Прим. /:ед.
7. Упругость идеальных каучуковых сеток
33
другой при сохранении среднего расстояния между поперечными
связями. Если резина растянута, то среднее расстояние между
поперечными связями увеличивается и возможное число кон-
формаций гибкой цепи между ее фиксированными концами (по-
перечными связями) уменьшается от 2о до О. Изменение энтро-
пии между растянутым состоянием и нерастянутым состоянием
связано с числом конформаций в растянутом и нерастянутом
состояниях по формуле:
S— S0 = fcln<J/90 (7,1)
Вычисление числа конформаций Со и Q многочисленными
методами, полностью представленными в ссылках 1 и 2, приводит
к следующему уравнению:
1 Г/ I \2 I
= + 2-^-3] (7.2)
где Лъ—число цепей сетки в образце, начальная (исходная) дли-
на и площадь поперечного сечения которого есть Lo н Ло, а длина
и площадь поперечного сечения его в растянутом состоянии есть
L и А. Экспериментально доказано, что объем изменяется весьма
слабо даже при двукратном растяжении, поэтому L0A0=LA.
Уравнение (7, 2) будет выведено элементарным методом в
главе II (раздел 9).
Растягивающая сила X получается пз уравнения (7, 2) и урав-
нения
пз которого легко получается
NDkT
Lo
(7,3)
(7, 4)
Кривая истинное напряжение—растяжение для идеальной ре-
зины получается делением обеих частей уравнения (7, 4) на А —
площадь поперечного сечения растянутого образца длиной L:
X
А
= tiRT
у
L ]
(7, 5)
где п—число молей цепей сетки в единице объема (в кубических
сантиметрах) резины, равное Л;о/(Л<>£<>)NA.
Очень часто уравнение состояния идеальной резины выражают
через величину «напряжения» F, которое определяется как
Л'/Ло, т. е. как сила, отнесенная к единице площади поперечного
сечения нерастянутого образца. Из уравнения (7, 4) получается:
у (У
Е = — nRT
(7, 6)
3—833
3 4
Гл. I. Упругоеi it и низкое i и
Часто вводится новая величина Л1. (среднечпсловой молеку-
лярный вес цепи сегкп), которая определяется соотношением:
nMt = d (7, 7)
где d—плотность резины (в граммах в кубическом сантиметре).
Уравнения (7, 5) и (7, 6) могут быть выражены гораздо лучше
через вечичпну Мс, чем через п:
Изотермический модуль Юнга для любого участка кривой на-
пряжение—деформация может быть определен следующим обра-
зом:
E — L(djidL)T (7,10)
Из уравнений (7, 10) и (7, 5) получаем:
Е — пЕ.т\2(~\ +4] (7,11)
[ \ Lo / |
Для весьма малых деформаций модуль Юнга можно получить
из уравнения (7, 11) подстановкой L^Lo:
dRT
Е (при малых деформациях) = 3/i/?Г = 3 — (7, 12)
При 298' К значение модуля Юнга Е в динах на квадратный
сантиметр равно:
£ = 7,43 1010 -4- =7,43- 10|0/г (7,13)
Если плотность резины равна единице, то модуль равен
7,43-10®, если Л/с=10 000 или /г=10“4. Величины Л1С между
5000 и 10 000 являются весьма типичными для многих «сшитых»
натуральных и синтетических каучуков.
Допустим, что сетка образована из исходных бесконечно длин-
ных линейных молекул при добавлении с молей четырехфункцпо-
налыюго сшивающего агента на каждый кубический сантиметр
резины. От каждой поперечной связи отходят четыре цепи сетки,
но каждая цепь сетки принадлежит двум поперечным связям.
Отсюда имеем 2с молей цепей сетки в каждом кубическом санти-
метре пространственной сетки:
2c = n = dlMc (7,14)
8. Вязкость жидкостей
35
Если линейные молекулярные цепи до поперечного сшивания
имеют конечный среднечнсловой молекулярный вес Мп, то сет-
ка, образованная с молями сшивающего агента в одном кубиче-
ском сантиметре резины, будет содержать некоторое количество
концевых цепей сетки, которые не участвуют в деформации и
находятся в ненапряженном состоянии.
Те же самые теоретические соображения, которые приводят
к уравнению (7, 5) для деформации растяжения, приводят к
следующему уравнению для деформации сдвига:
(7,15)
где [,—напряжение сдвига, у—деформация сдвига и п—число
цепей сетки в 1 си3 резины.
Это уравнение теоретически и экспериментально пригодно
для очень широкой области деформаций сдвига. Модуль сдвига
для сетки идеальной резины равен:
G = nRT = dRT!Mc (7,16)
Литература
1. Р. J. Flory, Principles of Polymer Chemistry, ch. XI, Cornell University
Press, Ithaca, New York, 1953.
2- L. R. G. Treloar, The Physics of Rubber Elasticity, Clarendon Press, Oxford,
England, 1949; 2nd ед., 1959. [Л. Трелоар, Физика упругости каучука,
Издатиилит, 1953.]
8. Вязкость жидкостей
Жидкость может быть определена как конденсированная фаза,
которая при обычных условиях эксперимента реагирует на на-
пряжение сдвига в процессе течения в значительно большей сте-
пени, нежели в условиях заданной деформации сдвига*. При су-
щественно низких напряжениях сдвига как жидкости, так и газы
подчиняются закону Ньютона:
f = r^s.dt (8,1)
где f—напряжение сдвига, dsidt—скорость сдвига, a -q—коэф-
фициент вязкости. Если f—выражено в динах на квадратный сан-
тиметр, время t в секундах, то коэффициент вязкости, найденный
* Имеется в виду, что при заданной деформации напряжение в жидкости
быстро релаксирует и .механическое воздействие, а следовательно и вязкое
течение, исчезают, тогда как при заданном напряжении сдвига вязкое течение
и накопление остаточной деформации будут наблюдаться все время. Для твер-
дых упругих тел безразлично, задавать ли постоянное напряжение или постоян-
ную деформацию, так как оба условия нагружения приведут к одному и тому
же результату—упругой деформации.—Прим. ред.
36
Гл. /. Упругость и вязкость
из уравнения (8, 1), выражается в пуазах. Вязкость имеет раз-
мерность (масса/длпна - время].
Вискозиметрами называются приборы, которые использу-
ются для измерения вязкости. Они могут быть различных кон-
струкций в зависимости от области вязкости, которая должна
быть исследована. Следующие методы являются распространенны-
ми: течение через капилляр; падающий (или поднимающийся)
шарик, пузырек пли капля масла; вращающиеся концентричес-
кие цилиндры; пластометр с плоско-параллельными пластинами.
Интервал вязкостей жидкости весьма велик. При комнатной
температуре вязкости воды, касторового масла и смолы прибли-
зительно равны 10-2, 101 и 1010 пуаз соответственно. Для сравне-
ния приведем вязкость воздуха при 23 СС, которая равна 1,84- 10~4
пуаз.
Вязкость газов при давлениях менее 100 а/пм удовлетворитель-
но рассчитывается из основных положений кинетической теории.
Если иод воздействием внешнего напряжения сдвига одни пло-
ский слой молекул в газе движется быстрее, чем прилегающий
слой молекул, то каждый пз них будет стремиться тянуть другой.
При этом скорость относительного движения их б^дет уменьшать-
ся, если напряжение сдвига удалить. В газе вязкая сила возни-
кает следующим образом. Если молекулы быстро движущегося
плоского слоя переходят в медленно движущийся слой, то они
передают избыток количества движения молекулам этого слоя
путем соударений; если же молекулы медленно движущегося слоя
переходят в быстро движущийся слой, то они уменьшают коли-
чество движения молекул этого слоя также путем соударений.
Таким образом, состояние относительного движения стремится
исчезнуть. Формула, вытекающая из прямого применения кине-
тической теории газов, имеет вид:
1 I 2 \3/- (mkT)1'-
3 \ п I (2г)2
(8, 2)
где m—масса молекулы, а 2г—диаметр молекулы. Следует отме-
тить, что вязкость газа не зависит от давления и возрастает с тем-
пературой.
В жидкости прилегающие слон находятся в сцеплении вследствие
очень сильных межмолекулярных сил. Если эти два слоя движут-
ся относительно друг друга, то сила трепня будет зависеть от
сил притяжения между слоями.
Молекулярные теории вязкости жидкостей были развиты Бор-
ном и Грином [1, 21, Кирквудом с сотрудниками [3] и др. Эти тео-
рии весьма сложны и только окончательные результаты могут быть
здесь представлены. Они применимы к жидкостям, образованным
химически насыщенными и сферическими молекулами. Важной
8. Вязкость жидкостей
3 7
характеристикой молекул является энергия взаимодействия и (г)
между парой изолированных молекул:
п(г) = щ(г) — иа(г) (8,3)
В уравнении (8, 3) член иг(г) представляет взаимодействие оттал-
кивания между молекулами, а иа(г)—взаимодействие притяжения
между молекулами. Для химически насыщенных молекул потен-
циал Леннарда-Джонса является хорошим приближением, даю-
щим:
= (8,4)
пЙ(г)^2зЧ^у (8,5)
Молекулярная теория вязкости жидкости приводит к простым
результатам только после многочисленных упрощении. Один из
таких результатов следующий 11, 2, 61:
4 = 0,48 41- ехр (_ ц{Г1)1кТ) (8,6)
Lm
где vm—объем молекулы, m—масса молекулы, а гг—некоторое
частное значение г, которое оказывается несколько большим,
чем г*.
В этом месте следует вернуться к выражению для частоты ко-
лебаний осцилляторов эйнштейновского твердого тела, рассмо-
тренному в разделе 5:
= 2,37-4 VИ (8, 7)
' f m
Путем комбинации четырех последних уравнений приближенно
получается:
где 'iE—эйнштейновская частота соответствующего твердого тела.
Уравнение (8, 8) очень сходно с формулой, которая была пред-
ложена Андраде {4, 51 для вязкости жидких металлов в их точках
плавления и оказалась очень хорошо совпадающей с эксперимен-
тальными результатами:
.Молекулярная теория вязкости жидкостей в форме уравнения
(8, 6) и в более точном выражении, данном Кирквудом, была
38
Гл. 1. Упругость и вязкость
применена к жидкому аргону п жидкому метану, причем было най-
дено хорошее согласие с экспериментальными данными в преде-
лах множителя, равного двум 161.
Сделав определенные упрощающие предположения, можно
расширить эту молекулярную теорию с тем, чтобы найти значение
вязкости для более сложных .молекул 161. Однако при этом требу-
ется много приближений.
Весьма полезна обработка данных по вязкости жидкостей при
помощи более эмпирических приближений, которые позволяют
учесть многие важные факты.
Бачинский [71 в результате исчерпывающего эксперименталь-
ного изучения вязкости жидкостей пришел к заключению, что
вязкость при постоянном удельном объеме практически не зависит
от давления и температуры. Он предложил уравнение:
'c’(t£—Л’о) — cvf (8, Ю)
где с—константа данной жидкости, и—удельный объем жидкости
(объем, приходящийся на один грамм), а уо—удельный объем
жидкости, экстраполированный к абсолютному нулю без изме-
нения фазового состояния. Некоторые численные методы такой
экстраполяции рассмотрены Дулитлом [8].
Что действительно желательно, так это определить объем одно-
го грамма «плотно-упакованных» молекул, хотя способ упаковки
их точно не определен. Величина у^=у—уо называется удельным
«свободным объемом». Установлено, что эта величина имеет
большое значение для расчетов вязкостей.
Более точное эмпирическое уравнение, в котором использо-
вано понятие свободного объема, было предложено Дулитлом [81:
1п In Л 4- Bv0;vf
(8, 11)
где Я и В зависят от природы жидкости (В близко к единице),
а Уо и vf были рассмотрены выше.
Следовало бы подчеркнуть, что температура не входит в урав-
нение (8, И) в явном виде, но неявно проявляется через темпера-
турную зависимость v-t (а отсюда и у). Следовало бы также отме-
тить, что эмпирическое уравнение (8, 11) пригодно в области ма-
лых удельных объемов (и малых свободных объемов), где урав-
нение (8, 6), полученное на основе молекулярной теории, вероят-
но несправедливо. Уравнение (8, 11) поэтому очень полезно для
расчетов вязкости переохлажденных жидкостей или вязкости
жидкостей под большими давлениями.
Другой путь рассмотрения свойств жидкостей заключается в
предположении, что молекулы в жидкости образуют весьма не-
совершенную квазирешетку, которая имеет много вакантных
8. Вязкость жидкостей
39
узлов решетки (или «дырок»)*. Размеры «дырки» не обязательно
равны размерам молекулы. В этой квазирешетке молекулы могут
очень просто передвигаться пз одного положения в другое, пре-
одолевая энергетический барьер. Легкость этого диффузионного
движения находится в обратной зависимости к вязкости. Эта
«активационно-диффузионная модель» приводит к следующей
формуле Эйрпнга для вязкости жидкостей (9]:
4 = -фД ехр JRT) (8, 12)
где V—молярный объем жидкости, а ДАвязк есть свободная энергия
активации моля молекул, перемещающихся из одного равно-
весного состояния в другое.
Почти для ста простых жидкостей при атмосферном давлении,
включая ассоциированные жидкости, на опыте было найдено, что
АЛяз=Л,сп./2,45, где Е1|СП.—молярная теплота испарения жид-
кости. Поэтому при атмосферном давлении [91
Д' h
4 = ехр (Е11СП /2,45/?Т) (8. 13)
Были сделаны попытки объединить активационную теорию и
теорию свободного объема с тем, чтобы получить уравнение вяз-
кости, имеющее более универсальное значение. Они приводят к
уравнениям в форме [61:
4 = f (yf) ехр (E'^JRT) (8,14)
где f[vf) некоторая функция свободного объема, а Евязк.—пред-
ставляет собой энергию активации диффузии при постоянном
объеме (или при постоянном свободном объеме).
Литература
1. М. Born, Н. S. Green, Proc. Roy. Soc. (London), A190, 455 (1947).
2. H. S. Green, The Molecular Theory of Fluids, Interscience Publishers,
New York, 1952.
3. J. G. Kirk w о о d, F. P. В u f f, M. S. G r e e n, J. Chem. Phys.
17, 988 (1949).
* Эти представления, развитые в 20-х годах текущего столетия Я. И. Френ-
келем и II. Дебаем, а позднее Г. Эйрингом, были в свое время весьма прогрес-
сивными (Я. II. Френкель. Кинетическая теория жидкостей. Изд. АН
СССР, 1945, гя. IV-.
В настоящее время теория жидкости опять подчеркивает сходство жидкого
п газообразного состоянии, так как и газовая фаза обладает некоторой ближней
упорядоченностью (кроме случая бесконечного разрежения). Современная
теория, развитая Н. Н. Боголюбовым, М. Борном, X. Грином, Дж. Киркву-
дом н др., описывает физические свойства и структуру жидкости с помощью
функции распределения положений групп частиц.—Прим. ред.
40
Г.1. /. Упругость и вязкость
4. Е. N. da С. А и d г a de. Phil. Mag., 17, 497, 698 (1934).
5. Е. N. da C. Andra d e, Proc. Roy. Soc. (London), A215, 36 (1952).
6. A. Bondi, ch., 9 in Rheology: Theory and Applications, F. R. Eirich fed)
Academic Press, New York, 1956. [А. Бойд л, гл. 9 в кн. «Реология:
теория и применение», под ред. Ф. Эйриха, Издатинлит, 1962].
7. A. J. В a t с h i п s к i, Z. physik. Chem., 84, 643 (1913).
8. А. К. Doolitil е, J. Appl. Phys., 22, 1471 (1951); ibid., 23, 236 (1952).
9. 11. E у r i n g, 1<- La idler, S. Glasstone, The Theory of Rate
Processes, ch. IX, McGraw Hill Book Co., New York, 1941. [C. T.iec-
c т о it, К. Л e и д л e p, Г. Э и p и н г, Теория абсолютных скоро-
стей реакции, гл. 9, Издатинлит, 1948].
9. Упруговязкость: модель Максвелла
Джэмс Клерк Максвелл был одним из первых, кто рассмотрел
количественно упруговязкость вещества. Он заметил, что такие
вещества, как смола пли деготь, не могут рассматриваться ни
как идеальные упругие твердые тела, ни как вязкие жидкости,
но, по-видимому, совмещают свойства тех и других. В результате
он предложил уравнение, носящее теперь его имя и которое, как
он полагал, будет достаточным для объяснения поведения этого
типа тел [1]. При весьма быстро приложенных напряжениях смо-
ла ведет себя, как упругое тело. Она испытывает деформацию,
пропорциональную напряжению, которая полностью удаляется,
если напряжение снимается очень быстро. Если напряжение
приложить медленно или же оно действует в течение длитель-
ного времени, то смола ведет себя, как настоящая вязкая жидкость:
она будет давать непрерывное нарастание деформации с течением
времени, а скорость деформации будет пропорциональна прило-
женному напряжению, как следует из уравнения Ньютона.
Максвелл пытался объединить закон Гука и закон Ньютона
в единое уравнение. Он предложил:
—— f (9 I)
dt G dt'-.G ' ’ '
где s—деформация сдвига, /—напряжение сдвига, G—модуль
сдвига, а т—величина, называемая временем релаксации.
Для понимания уравнения Максвелла (9, 1) наилучшим пу-
тем является применение его к некоторым простым эксперимен-
там. Предположим сначала, что в момент времени /=и внезапно
приложено постоянное напряжение сдвига /о к максвелловскому
телу [веществу, которое подчиняется уравнению (9, 1)]. В крат-
ковременном интервале, в течение которого / возрастает от нуля
до /о, df 'dt чрезвычайно велика и первый член правой части урав-
нения (9, 1) гораздо больше, чем второй член; отсюда
_L (О 94
dt — С dt 1 '
9. Упругость: модель Максвелла
41
(для времен, гораздо меньших, чем время релаксации).
Интегрирование уравнения (9, 2) дает s=/o/G в конце малого
интервала времени, в течение которого развивается действие
полного напряжения /о. Последнее может быть принято в качестве
начального условия для t=0. Интегрируя теперь уравнение
(9, 1) при и вводя это начальное условие, получаем:
*-4 + 4' <9'3>
Другими словами, при воздействии постоянного напряжения
fu максвелловское тело обнаруживает вначале мгновенную упру-
гую деформацию, а в последующем вязкое течение, для которого
деформация линейно возрастает со временем. Для больших ин-
тервалов времени упругая составляющая деформации ста-
новится относительно малой, и поэтому
s = -“- (9,4)
Закон Ньютона, связывающий напряжение сдвига и деформа-
цию сдвига в вязкой жидкости, выражается уравнением:
s = A/A. (9,5)
Сравнивая уравнение (9, 4) с уравнением (9, 5), находим, что
вязкость максвелловского тела равна:
v( = tG (9,6)
Вероятно, все ньютоновские жидкости, даже такие, как вода
и бензол, которые весьма текучи, обладают как упругими, так и
вязкими свойствами. Однако наибольшее время релаксации бен-
зола или воды так мало (вероятно меньше, чем 10~12 сек), что
если приложить постоянное напряжение (либо сдвиг, либо растя-
жение), то вязкая составляющая уравнения (9, 3) перекрывает
упругую составляющую по величине в неизмеримо короткое вре-
мя (по крайней мере для этого вида эксперимента).
График зависимости деформации от времени для максвеллов-
ского тела, подвергнутого действию постоянного напряжения,
показан на рис. 1, 4.
Другой интересный опыт заключается в том, что максвеллов-
ское тело подвергается действию постоянной деформации зо в
момент времени t=0. Предполагается, что деформация может
быть приложена мгновенно, т. е. образец может быть растянут до
величины so за время, весьма малое сравнительно с временем ре-
лаксации т. При этих условиях вязкая составляющая незначи-
тельна по сравнению с упругой составляющей уравнения (9, 1)
42
Г.1. I. Упругость п вязкость
и уравнение можно проинтегрировать. Таким образом, получа-
ется, что при /=0, f=snG. Так как при постоянной деформации
ds di 0, то последующее поведение образца получается пнте|рп-
рованием уравнения
Интегрирование уравнения (9, 7) дает:
/ = snGe~'/--
(9, 8)
Этот экспоненциальный закон имеет большое значением известен
в качестве максвелловского закона релаксации напряжения. Мак-
t/T
Рис. 1,1. Максг.елльвсксе тело под
постоянным напряжением.
свелловская релаксация гра-
фически представлена в четы-
рех । различных видах на
рис. 1, 5. На рис. I, 5а пока-
зан график, выражающий за-
висимость напряжения от
времени; на рис. I, 56—зави-
симость логарифма напряже-
ния от времени; на рис.
1, 5в—зависимость напряже-
ния от логарифма времени;
на рис. I, 5г—зависимость
логарифма напряжения от
логарифма времени. Интерес-
но отметить характерную фор-
му кривой на рис. I, 5в. Из-
менение времени релаксации
не изменяет форму кривой,
ио только смещает кривую
горизонтально вдоль осп логарифма времени.
Уравнение Максвелла (9, 1), написанное для деформации сдви-
га, легко может быть распространено и на деформацию материала
при растяжении. Для опытов при растяжении:
rfs 1 Щ 1 •
dt Е ‘ dt -Е '
|9, 9)
где х -деформация растяжения, /—напряжен! е растяжения, Е—
модуль растяжения, а -—время релаксации. Предполагается, что
время релаксации для растяжения то же самое, что время релак-
сации для сдвп! а. Все выведенные из уравнения (9, 1) соотношения
имеют аналогичные решения, следующие из уравнения (9,9).
9. Упругость: модель Максвелла
43
При постоянном напряжении растяжения деформация растя-
жения выражается следующим образом:
Нактон графика зависимости s от времени равен fo/~E. Одна-
ко так как мы имеем дело с напряжением растяжения, а по с на-
Ркс. 1,5. Максвелловская релаксация напряжения при постоянном растяже-
нии.
пряжением сдвига, то величина -Е является вязкостью при
растяжении, а не вязкостью при сдвиге. Соотношение между
7( и коэффициентом Пуассона z следующее:
т^=^т.Е\
= ~ =2(1 4-о) (9, И)
г G
Для несжимаемого вещества з —1 2 и т/'' —
44
Г.1. I. Упругость и пязиость
Механическая модель, которая описывает максвелловское
тело, состоит из пружины и вязкого амортизатора, соединенных
последовательно, как показано на рнс. I, 6. Здесь напряжение
в обоих элементах модели то же самое, а деформа-
ция всей модели есть сумма деформации пружи-
ны и амортизатора. Предполагается, что пружина
подчиняется закону Гука Esx, а амортизатор—
закону Ньютона ~~.E(ils.y,'dn. Ввиду 1010, что
полная*деформация s- s, следует
ds _ 1 dj
~dt~—~lT' di
(9,12)
что идентично уравнению (9, 9).
Литература
Pne. 1,6.
.Модель .Макс-
велла.
1. J. С. М а х \v е 1 1, Phil. Trans. | Roy. । Soc. (London),
1о7, 52 <1867); Scientific Papers, 2, 26; Canibridge
University Press. 1890.
Глава II
ОСНОВЫ ФИЗИКИ ПОЛИМЕРОВ
1. Конформации линейной полимерной цепи [1]*
Структурной основой полимерных молекул является гибкая
линейная цепь. Крайне идеализированным представлением такой
структуры является цепь, образованная из N звеньев длиной /0,
каждое из которых связано с предыдущим звеном так, чтобы обе-
спечивалось полное свободное вращение. Это позволяет каждому
звену принимать любые направления относительно предыдущего. Та-
кая находящаяся в тепловом движении молекулярная цепь прини-
мает очень большое число конформаций**. Звенья предполага-
ются бесконечно тонкими, так что стерпческими взаимодействия-
ми между различными частями цепи можно пренебречь.
Если предположить, что все конформации цепи равновероят-
ны, то можно найти среднее квадратичное расстояние между
концами цепи. Пусть последовательность звеньев в цепи при дан-
ной конформации будет обозначена векторами /х,
имеющими длины, равные /о. Вектор расстояния г между началом
и концом цепи для отдельной конформации равен (рис. II, 1 и
11,2):
7=Л+72 + --- -1-7, (1,1)
Квадрат расстояния между концами цепи выражается, как
N
(1,2)
О
(1,3;
* См. также монографию М. В. Волькенште п н а «Конфигура-
ционная статистика полимерных цепей», Изд. АН СССР, 1959.
** Конформация цепи—одна из пространственных форм, которую может при-
нимать вследствие внутренних вращений гибкая подвижная цепная макромоле-
кула. В литературе иногда встречается другой, неправильный с точки зрения
хтпка, термин—конфигурация цепи.—Прим. ред.
46
Г.1. II. Основы физики полимеров
Первый член в правой части уравнения (1,3) один и тот же для
всех конформаций цепи. Типичный член суммы в .этом уравнении
равен /Jcosci,-, где а;/ представляет собой угол между вектором
/, и вектором I. Так как
предполагается абсолютно сво-
бодное вращение, то угол -
равноценен углу при сум-
мировании по всем конформа-
циям. Поскольку для всех углов
Рис. 11,1. Свободна сочлененная цепь.
z
а:
Рис. 11,2. Одна из конформаций по in-
мер noii цепи, у которой один конец
принят за начало координат.
соча,,.—cos(rто второй член в правой части уравнения (1,3)
исчезает. Среднее квадратичное расстояние г- от одного конца
цепи до другого для свободно вращающихся звеньев есть [2, 3]:
г2=Л702 (1,-1)
Свободно сочлененная полимерная цепь
Если соседние звенья в цепи представляют собой химические
связи с фиксированным углом 0 относительно друг Apyia (где
О--—о), но при этом они свободно вращаются по конусу с со-
хранением этого угла, то величина г- есть 131:
Звенья цепи свободно вращаются при сохранении вален того угла ч
В частном случае углеводородной цепи, как, например, в
полнметплспе, валентный угол 0 есть тетраэдрический угол, а
именно угол 109,5. Уравнение (1,5) принимает вид:
?=2/V/j; (1,6)
Звенья цепи свободно вращаются вокруг тетраэдрической связи
1. Конформации линейной полимерной цепи
47
Для реальной цепи углеводорода вращение вокруг тетраэдри-
ческого угла несвободно, но вместо этого имеется Три преимущест-
венных положения на конусе вращения (рис. 11,3—П, 5). Так,
у'-тая связь может быть или в плоскости связен j—1 и у—2 (транс),
пли она может находиться в двух других положениях, отклонен-
•слп мы примем, что разностьэнер-
ных от плоскости на 120° (гош). 1
гни между готн-конформаци-
ей п транс- конформацией
равна е на моль, то может
быть выведено точное выра-
жение г2 [4, 5] (см. Прило-
жение Г):
- (4+ (’>7)
Если трянс-позицня со-
ответствует наименьшей энер-
гии, то цепь будет стремить-
ся принять вытянутые плос-
кие зигзагообразные конфор-
мации, а г2 будет стремить-
ся принять большие значе-
ния.
Если гош—положениям
соответствует более низкая
энергия, то г- будет мень-
ше, чем по уравнению (1, 6).
Если г=0, то /2 имеет зна-
чение, соответствующее у рав-
нению (1, 6).
В дополнение к различию
Рис. 11,3. Пространственное представ-
ление углерод-углеродной цепи с ор-
динарными связями между атомами
(по Флорн [1]).
энергий трех потенциальных ми-
нимумов, соответствующих трем типам колебаний около связи
С—С, имеется также (средний) энергетический барьер высотой
То, разделяющий потенциальные минимумы. Для заторможен-
ного вращения вокруг центральной связи С—С в бутане То рав-
но 3,3 ккал/моль. Высота этого энергетического барьера есть мера
обратной величины скорости, с которой цепь может переходить
от одной конформации к другой (скорость —ехр(—Ео/7?Г)|.
Некоторые диаграммы энергетических барьеров для заторможен-
ного вращения приводятся на рпс. II, 5.
Вследствие конечности поперечных размеров звеньев в реальной
полимерной цепи наблюдается важный стерический эффект, так
называемый эффект «исключенного объема».
48
Гл. II. Основы физики полимеров
Этот эффект не ограничивается только соседними элементами
в цепи, ио включает также и связи, которые удалены по цепи друг
от друга. Этот эффект приводит к увеличению г2, а также значи-
Рпс. 11,4. Расположение, соответствующее минимуму
энергии для этана (а) и для участка цепи пол и четплена
(б), если смотреть вдоль связи С—С, для которой рас-
сматривается торможение при вращении вокруг нее.
Штриховые участки изображают атомы или заместители,
связанные с нижним атомом углерода связи С—С (по
Флори [11).
Рис. 11.5. Зависимость потенциальной
энергии 1'(Ф) от угла вращения Ф
вокруг связи С—С [W. J. Tavlor,
J. Chem. Phys.. 16, 257 (1948)].
тельно усложняет его вычис-
ление. Предполагается, что
в случае «исключенного объ-
ема» [61:
= (1,8)
где ( изменяется между 1,0
и 2,0.
Действительная величина
г2 может быть получена из
экспериментальных измере-
ний, таких, как угловое
рассеяние света в разбав-
ленных полимерных раство-
рах, пли при одновремен-
ном измерении напряжения
п двойного лучепреломления
в твердом полимере в его вы-
сокоэластическом состоянии.
1. Конформации линейной полимерной цепи
49
Жесткость Z реальной полимерной цепи может быть опреде-
лена как квадратный корень из действительной величины г2,
деленной на величину г2 для свободно сочлененной цепи с тем же
количеством звеньев и той же длиной звена /о:
МтУ’ (1.9)
Если принять один конец цепи за начало отсчета, то можно
ввести функцию, которая выражает вероятность того, что другой
конец цепи находится в области пространства dx dy dz |7—91
(см. Приложение Д п рис. II, 2):
W (д-, у, z) dxdydz -= (b/n1/-)3 ехр [ — Ь'2- (х'2 -р у2 -р z2)] dxdy dz (1, 10)
где величина b2 весьма просто связана с действительным средне-
квадратичным расстоянием
&2 = -|г (МП
Вероятность того, что цепь имеет длину от конца до конца в
интервале между г и r-rdr, независимо от направления, равна:
W (г) dr = (b^)3 [ ехр (—Ь'2г2)] 4№ dr (1,12)
Наиболее вероятная длина от конца до конца гвер легко полу-
чается нахождением максимального значения Пу(г):
1 / 2Р \‘ 2
Цепь реального полимера со сложным влиянием угла между
связями, заторможенности вращения, стерических препятствий
и т. д. может быть для расчета заменена эквивалентной свобод-
но сочлененной цепью с таким же расстоянием между концами цепи
17]. Реальная цепь из N звеньев каждое длиной /о, имеющая сред-
неквадратичную длину между концами г2, заменяется свободно
сочлененной цепью пз Ns «статистических сегментов» длиной 15,
так что:
d2=Nsl2 (1,14)
При этом
Даст. (1,15)
где грасг является максимальной длиной растянутой реальной
цепи. Уравнения (1, 14) и (1, 15) определяют Ns и /s.
4—833
Гл. II. Основы физики полимеров
Размеры полимерной цепи могут характеризоваться не только
количеством связей вдоль основной цепи, но также и числом хи-
мически идентичных повторяющихся единиц (степенью полимери-
зации), а также молекулярным весом. Определение этих величин
усложняется тем, что масса полимерного материала содержит мно-
го молекул, которые, однако, редко бывают одного и того же раз-
мера. Вопрос о распределении по размерам (распределение по
молекулярным весам), а также смысл и значение среднечпслового
молекулярного веса Мп и средневесового молекулярного веса
ЛЕ, рассматриваются в Приложении Е.
Литература
1. Р. .1. I’ 1 о г у, Principles о( Polymer Chemistry, ch. X. Cornell University
Press, Ithaca, New York, 1953.
2. F. T. Wail, J. Chem. Phys., tl, 67 (1943).
3. II. E у r i n g, Phys. Rev., 39, 746 (1932).
4. \V. J. T а у 1 о r, J. Chem. Phys., 16, 257 (1948); M. В. В о л ь к е н-
111 т е ii и, ЖФХ, 26, 1072 (1952).
5. А. V. Г о bo 1 s k v, J. Chem. Phys., 31, 387 (1959).
6. Р. Л. Flory, .1. Chem. Phvs., 17, 303 (1949).
7. W. К u h n, Koll.-Z., 76, 258 (1936); 87, 3 (1939).
8. 11. M. .1 a rn e s, E. Gut h, J. Chem. Phys., 11, 470 (1943).
9. W. Kuhn, F. Gru n, Koll.-Z„ 101, 248 (1942).
2. Аморфные и кристаллические полимеры
Полимеры могут быть очень строго разделены на две группы:
полностью аморфные и кристаллические. Некоторые полимеры
полностью аморфны при любых условиях; кристаллические по-
лимеры, конечно, могут быть аморфными при определенных усло-
виях (выше точки плавления или если полимер быстро охлажден
из расплавленного состояния).
Эти две группы полимеров легко отличить друг ог друга при
помощи рентгенограмм пли путем измерений счетчиком Гейгера
интенсивности рассеянных рентгеновских лучен прг различных
углах рассеяния. Аморфные полимеры показывают такого же
тина диффузное гало па рентгеновских снимках, кап и простые
жидкости. Локальная структура аморфного полимера пли про-
стой жидкости не обладает высокой степенью пространственного
порядка, характерной для кристаллов. 13 действительности, од-
нако, в аморфной структуре па расстоянии приблизительно до
15 А от любой данной точки имеется однотипная пространствен-
ная упорядоченность как в аморфных полимерах, так и в простых
жидкостях и органических стеклах. При рассмотрении больших
2. Аморфные и кристаллические полимеры
61
областей пространства структура аморфного полимера может
быть сравнена со сваренными тонкими макаронами, находящимися
в чашке. «Макароноподобные» молекулы, конечно, находятся в
непрерывном хаотическом движении, амплитуда п скорость
которого зависят от температуры.
Рентгенограммы кристаллических полимеров показывают се-
рию резких дифракционных колец, наложенных на некоторое
диффузное рассеяние. Диффузное рассеяние возникает в аморф-
ных областях полимеров; острые же кольца на рентгеновском
снимке возникают от областей с кристаллическим порядком
(кристаллиты), вкрапленных в аморфную матрицу. Отдель-
ные кристаллиты малы; типичные размеры их могут быть
100 <200 '200 А-
Отдельные кристаллиты обычно группируются вместе в боль-
шие агрегаты, называемые сферолитами, которые часто дости-
гают размеров, легко видимых невооруженным глазом, если их
наблюдать между скрещенными поляроидами (рис. II, 6).
Необходимо иметь в виду, что в кристаллических высокопо-
лпмерах одна и та же молекулярная цепь может проходить через
кристаллит, входя затем в разупорядоченную аморфную область
между кристаллитами, снова входить в другой кристаллит и т. д.*.
Линейные размеры отдельного кристаллита малы по сравнению
с линейными размерами высокомолекулярной цепи. В тех слу-
чаях, когда это неверно (т. е. в кристаллических полимерах с
малой длиной цепи), полимер обладает физическими свойствами
воска.
Типичные рентгенограммы аморфных и кристаллических поли-
меров показаны на рис. II, 7—II, 9.
Кристаллические полимеры характеризуются определенным
расположением атомов в элементарной ячейке, долей крпсталли-
* Излагаемые А. Тобольским широко распространенные представления
о двухфазной структуре кристаллических полимеров, о прохождении цепных
макромолекул через несколько кристаллических п аморфных областей п др.
подверглись серьезной критике со стороны советских ученых. Новые экспери-
ментально проверенные представления о пачечной структуре полимеров и
большом разнообразии надмолекулярных структур, существенно влияющих на
механические и другие физические свойства кристаллических полимеров,
получили в Советском Союзе широкое распространение в результате исследо-
ваний, проводимых В. А. Каргиным и его сотрудниками (см. например,
В. А. К а р г и и, А. II. Кита й городе к и й, Г. Л. Слои и м -
с к и й, Коллоид, ж., 19, 131 (1957); В. А. К а р г и и, Г. Л. Слон и м-
с к и и, Усп. хим., 24, 785 (1955); В. А. Карг и и, Г. Л. Слон и м-
с к и ii. Краткие очерки по фпзнко-химпп полимеров, очерк VII, Изд. МГЬ-
1960; В. А. К а р г и н, Современные проблемы пауки о полимерах, ИЗД-
МГУ. 1962; А. А. С т р е п и х е е в, В. А. Д е р е в и ц к а я. Основы
химии высокомолекулярных соединений, Госхимнздат. 1961, стр. 199—200].
Прим. ред.
52
Гл. II. Основы физики полимеров
ческою материала, размером, формой, ориентацией п агрегацией
кристаллитов, а также температурой плавления Тт, выше ко-
торой исчезают все кристаллические области.
Рис. 11,6. Сферолитная структура кристаллического полимера, види-
мая через скрещенные поляроиды [F. Р. Price, Growth and
Perfection of Crystals, New York, 1958, стр. 466).
Аморфные и кристаллические полимеры
53
Рис. 11,7. Рентгенограмма аморф-
ного полимера (атактический поли-
стирол).
Рис. 11.8. Рентгенограмма неориен-
тированного кристаллического поли-
мера (отожженный изотактический
полистирол).
Рис. 11,9. Рентгенограмма ориен-
тированного кристаллического поли-
мера (ориентированный отожженный
изотактический полистирол).
54
Гл. II. Основы физики полимеров
Температура плавления часто определяется путем наблюде-
ния тонкой пленки полимера на нагревательном столике поляри-
зационного микроскопа. Если температура нагревательного сто-
лика превышает Тт, то свет не проходит через пленку, помещен-
ную между скрещенными поляроидами.
Температура Тт может быть определена также снятием рентге-
нограмм при различных температурах и измерением темпера-
туры, при которой полностью исчезают кристаллические дифрак-
ционные кольца (рис. II, 10).
Температура плавления твердых тел с низким молекулярным ве-
сом (лед, камфора и т. д.) точно определяется при заданном дав-
лении, например при атмосферном давлении. Действительно,
точка плавления льда при атмосферном давлении использует-
ся для определения фиксированной точки 0° на стоградусной тем-
пературной шкале. Явление плавления для этих простых ве-
ществ является фазовым переходом первого рода. При темпера-
туре плавления наблюдается скачкообразное изменение объема
и скачкообразное изменение энтальпии, эквивалентное теплоте
плавления.
Скачкообразное изменение объема также наблюдается при
температуре плавления Тт кристаллических высокополнмеров
при условии, если кристаллический полимер должным образом
отожжен при температурах несколько ниже температуры плав-
ления, и при условии, если скорость нагревания в процессе изме-
рения температурной зависимости удельного объема чрезвычай-
но мала. При этих условиях резкое изменение объема вблизи
температуры плавления наблюдается удивительно отчетливо,
причем около 80% кристаллического материала плавится в ин-
тервале от 3 до 4° [11. Имеется точно определенная температура
Т,п, при которой резко исчезают последние следы кристаллич-
ности. Оказывается, что плавление полимеров лучше всего опи-
сывается как диффузный переход первого рода. На рис. II, 15
показаны некоторые vT кривые вблизи Тт.
Весовая доля аморфного материала в кристаллическом поли-
мере может быть получена из рентгеновского исстедованпя, поз-
воляющего получить зависимость интенсивности рассеяния от угла.
Для этого измеряется интегральная интенсивность рассеяния
в аморфном гало (интенсивность рассеяния в кристаллических
пиках во внимание не принимается). Тот же самый полимер затем
подвергается облучению рентгеновскими лучами выше температуры
плавления Тт. При этом должна быть уверенность в том, что он
подвергается действию того же общего количества рентгеновского
излучения, что и в первом случае. Интегральная интенсивность
рассеяния измеряется опять в этих новых условиях; никакого
кристаллического рассеяния выше температуры Т,п, конечно, нет.
?е
Рис. 11,10. Интенсивность рассеяния рентгеновских лучей в
зависимости от угла для полиэтилена низкой плотности (7,п=
110 'Q при различных температурах fS. Krim ni,
Л. V. Tobolsk у, J. Polymer Sei., 7, 62 (1951)].
56
Гл. IL Основы физики полимеров
Доля аморфного материала, находящаяся в полимере в условиях
первого опыта, считается равной отношению интегральных ин-
тенсивностей, измеренных в этих двух опытах.
Удельный объем и коэффициент теплового расширения аморф-
ных областей может быть найден путем измерений выше темпера-
туры плавления. Удельный объем и коэффициент теплового рас-
ширения кристаллических областей могут быть определены из
параметров решетки, полученных из дифракционных колец. Из-
меренный при любой температуре удельный объем и(Т) находится
в следующем соотношении с удельным объемом кристаллических
областей <(Г) и удельным объемом аморфных областей иА(Т):
v(T) = (i-}.)vr(T)+Zu4(T) (2,1)
где /.—объемная доля аморфного материала в полимере при тем-
пературе Т.
Из приведенного выше уравнения следует:
I = L> 9)
vA(T)-vc(T) 1
Поэтому измерения удельного объема приводят к весьма
простому методу определения объемной доли кристаллического
материала при данных условиях.
Теперь важно рассмотреть структурные особенности, влияю-
щие на свойства аморфного или кристаллического полимера, и
в последнем случае найти факторы, определяющие кристалличе-
скую долю материала.
Способность полимерной цепи упаковываться в правильную
кристаллическую решетку прежде всего определяется регуляр-
ностью полимерной цепи. Например, полистирол может быть при-
готовлен в двух формах: атактической и изотактической. В атакти-
ческой форме расположение фенильных групп в «верхнем» и
«нижнем» положениях вдоль цепи беспорядочно. В изотактиче-
ской структуре фенильные группы вдоль цепи находятся либо все
в «верхнем», либо в все «нижнем» положениях:
С6Н5 С6Н5 СвН5
I I I
----СН2—СП-С1С-СН—СН.,—СН—СН,—СН—сн,—сн---------
I I
Сви6 С6Н5
атактический полистирол
----СН2—СН—сн.>—СИ—СН2—СИ—СН2—СН—сн,—СИ---------
I I I I I
СвН5 С6н5 свн5 сон5 С6Н5
изотактический полистирол
Атактический полистирол является совершенно аморфным;
никому не удалось когда-либо привести его хстя бы в частично
2. Аморфные и кристаллические полимеры
57
кристаллическое состояние. С другой стороны, изотактический
полистирол обычно получается в частично кристаллическом со-
стоянии (возможно около'50% кристалличности). Его точка плав-
ления около 250 СС. В случае быстрого охлаждения или выпадения
пз определенных растворов или при определенных полимериза-
ционных процедурах он может быть получен полностью в аморф-
ном состоянии при комнатной температуре. Однако это—мета-
стабпльиая форма, которая может быть переведена в кристал-
лическое состояние путем соответствующего режима отжига.
Были также изготовлены синдиотактические полимеры. Они
имеют правильное чередование боковых групп «в верхнем» и
«нижнем» положениях.
Другой пример влияния регулярности на способность к кри-
сталлизации следует из большой склонности симметричных ви-
нилиденовых полимеров к кристаллизации. Двумя замечатель-
ными примерами этого являются полпвинилиденцпаннд и поли-
винилнденхлорид. Оба эти полимера более кристаллпчны, чем
соответствующие атактический полнвинплцианид (полиакрило-
нитрил) и атактический поливинилхлорид:
----CH2C(CN)2—CH2C(CN)2—CH2C(CN)2----
полившшлпденцианид
----CH2CH(CN)—СН2СН(С\’)—CH2CH(CN)-----
паливив1|Лциаш1д
----CH2CCI2—CH2CCI2—CH2CCI2----
полившшлидеихлорид
----CH2CHCI—CH2CHC1—CH2CHCI------
поливинилхлорид
Важность регулярности строения цепи для возникновения
кристаллизации отчетливо видна пз отрицательного влияния со-
полимеризации на кристаллизацию. Например, поливинилиден-
фторид и политрнфторхлорэтилен оба являются высоко кристал-
лическими полимерами:
----ch2cf2—ch2cf2—ch2cf2-----
поли винилиденфтор ид]
----cf2cfci—cf2cfci—cf2cfci----
пол итрн фторх л орэти лен
Два мономера'CH2—CF2 и CF2=CFC1 также могут быть сов-
местно полимеризованы во всей области составов. При этом обра-
зуются нерегулярные сополимеры типа:
----CU2CF2—CFCICFo—CFCICF2—. •
Вблизи состава 1 : 1 сополимеры совершенно аморфны ш
каучукоподобны при комнатной температуре). Для сополимеров,
составы которых имеют большой перевес в группах —CFC1CF2—,
58
I .i. II. Основы физики по.шмеров
наблюдается кристалличность. Однако такие сополимеры имеют
более низкую кристалличность и более низкую точку плавле-
ния, чем чистые гомополимеры. То же самое можно сказать п
о сополимерах, в составах которых имеют большой перевес груп-
пы - - СН.СЕ,—.
Введение примеси в чистый гомополпмер с очень большим (в
принципе бесконечным) молекулярным весом понижает точку
плавления полимера. Этой примесью может быть одни из сомо-
номеров, низкомолекулярный растворитель пли концы цени
полимера.
Формула, выражающая понижение точки плавления сополи-
мера, в котором повторяются структурные единицы А и В, имеет
вит:
где Tin—точка плавления чистого гомополимера А; Тт—точка
плавления сополимера, который содержит молярную долю ХА
мономерных единиц А из числа всех А и В единиц (где Хл близ-
ка к единице), R—газовая постоянная н АН,,—теплота плавления,
отнесенная к одной структурной единице А.
При введении нерегулярности, такой!, как сополимер изую-
щаяся единица, понижается не только точка плавления Т,п, но
также уменьшается и равновесная объемная доля кристаллической
части материала. Это доказывается сравнением процентного содер-
жания кристаллической части в полиэтилене, полученном новым
процессом при низком давлении с гетерогенным ионным катализато-
ром, с процентным содержанием кристаллической части в полиэти-
лене, синтезированном старым свободнорадикальным процессом
полимеризации при высоком давлении (2000 am). Первое вещество
является истинно линейным полпметнленом с темп. пл. 137 “С
и степенью кристалличности около 90%. Второе содержит в сред-
нем две бутильных и одну этильную боковые группы на каждые
100 атомов углерода в цепи, имеет темп. пл. 110 °C и является кри-
сталлическим на 50%. Плотность линейного полиэтилена равна
0,97 г смя; плотность разветвленного полиэтилена равна около 0,92.
Если процентное содержание сополимерной примеси сущест-
венно велико, то сополимер становится совершенно аморфным,
хотя каждая составляющая сама по себе может образовать кри-
сталлический гомополпмер. Требуется только восемь этильных
плп бутильных групп на каждые 100 углеродных атомов цепи,
чтобы полностью разрушить кристалличность полиэтилена.
Необходимо добавить, что не все сополимерные единицы В
приводят к снижению точки плавления полимера, образованного
единицами А. Если В является изоморфным с А и может входить
2. Аморфные и кристаллические полимеры
59
в цепь без нарушения кристаллической решетки чистых А це-
пей, н аналогично, если единицы А входят в В цени без нарушения
кристаллической решетки, образованной цепями из чистых еди-
ниц В, то сополимеры А и В будут иметь температуры плавления,
линейно изменяющиеся между температурами плавления чистых
А и В. Это же наблюдается п в случае твердых растворов, обра-
зованных из низкомолекулярных кристаллических веществ.
Другим фактором, который оказывает влияние на склонность
к кристаллизации, является объемность боковых групп. Линей-
ный полиэтилен высококрнсталлпчен; атактический поливинило-
вый спирт также высококрнсталлпчен; поливинилхлорид, полу-
ченный радикальной полимеризацией, и полиакрилонитрил яв-
ляются менее кристаллическими и содержат очень несовершен-
ные кристаллиты. Кроме того, есть основания полагать, что
эти вещества являются не полностью атактическими, а на корот-
ких отрезках имеют некоторую небольшую изотактическую или
синдиотактическую регулярность [2]. Атактический полипропи-
лен, атактический полибутен-1 и атактический полистирол яв-
ляются совершенно аморфными, так как громоздкость боковых
групп не позволяет сформироваться кристаллической решетке,
даже, если боковые группы расположены в одном и том же
направлении.
Геометрическая конформация отдельных полимерных молекул
в элементарной ячейке полимерного кристаллита очень сильно
зависит от деталей молекулярной геометрии и энергии. Для по-
лиэтилена молекулярной конформацией внутри элементарной
ячейки кристалла является плоская зигзагообразная конформа-
ция (по крайней мере для коротких отрезков цепи). Это происхо-
дит частично благодаря большой положительной величине е
для «-парафинов (около 600—800 кал/моль). Для изотактиче-
ского полипропилена отталкивание метильных групп в плоской
зигзагообразной конформации очень велико. Предполагается, что
молекулы скорее имеют гош-, чем //фонс-конформацпп, и поэтому
имеют винтовую конформацию внутри элементарной ячейки кри-
сталлитов. Возможны многие виды винтовой структуры. Систе-
матизация [31 этих структур с точки зрения молекулярной геомет-
рии является весьма увлекательным и сложным предметом.
В случае молекул протеина особенно интересна очень сложная
винтовая структура, называемая а-сппралью 141.
Литература
1. L. М a n d е 1 к е г n, Chem. Rev., 56, 903 (1956); Усп. хим. 27, 193 (1958).
2. В. D. Cole ni а п, J. Polymer Sci., 31, 155 (1958).
3. G. Na t la, P. Corra di n o, J. Polymer Sci., 38, 29 (1959).
4. L. Pauling, R. B. Core v, H. R. В r a n s о u, Proc. Nate. Acad.
Sci. U. S., 37, 205 (1951).'
СО
Гл. II. Основы физика полимеров
3. Термодинамика кристаллизации
Статистическая термодинамика кристаллизации кристалличе-
ских полимеров широко развита главным образом П. Дж. Флори
в его обстоятельных работах 11]. В этом разделе будут представ-
лены важнейшие формулы без их вывода, но значение их будет
рассмотрено. Многие из этих формул легко воспринимаются ио
аналогии.
Точка плавления низкомолекулярного твердого тела Д по-
нижается при введении примеси, растворимой в расплаве, со-
гласно формуле:
у-----(3,1)
' «I ' ш f
ме —температура плавления чистого твердого тела А, Т„ —
температура плавления в присутствии примеси. AHf—теплота
плавления, Л?—газовая постоянная и аЛ—активность А в рас-
плаве в присутствии растворенной примеси. При существенно
малых концентрациях примеси активность аА примеси равна
молярной доле А, находящейся в расплаве.
Точка плавления кристаллического полимера соответственно
понижается такими «примесями», как низкомолекулярные рас-
творители, концы цепей и беспорядочно расположенные вдоль
цепи сополимера мономерные единицы. Соответствующие форму-
лы, аналогичные уравнению (3, 1), имеют следующий вид 11):
J_____________1_ =
Т,п т;,, А//„ • V, 1 1 RTr,! /
I I _ R 2
(3,2)
(3.3)
(3.4)
‘5 В приведенных выше уравнениях АНи—теплота плавления
моля повторяющихся структурных единиц полимерной цепи.
В уравнении (3, 2) \7, u V',—молярные объемы структурной еди-
ницы полимера и растворителя,Ф,—объемная доля растворителя
в полимерном растворе и В'—постоянная, которая характеризует
энергию взаимодействия в системе полимер—растворитель. В урав-
нении (3, 3) Р„—среднечпсловая степень полимеризации полиме-
ра. т. е. число повторяющихся структурных единиц в цепи. В урав-
нении (3, 4) Л'4—молярная доля единиц Я в сополимере, содержа-
щем малое количество единиц В, беспорядочно сополимеризован-
пых с единицами А.
3. Термодинамика кристаллизации
61
Все уравнения от (3, 1) до (3, 4) включительно сводятся к
одному общему уравнению при существенно малых концентра-
циях примесей:
Т----(3.5)
гдечХв—молярная доля «примеси», т. е. растворителя, концов
цепей или 1единиц сомономера.
Величины АНи для некоторых полимеров были получены глав-
ным образом по уравнению (3, 2). Из А//„ и величины ТП1 можно
получить AS,,, воспользовавшись соотношением:
<3’6*
где AS,,—энтропия плавления моля повторяющихся структур-
ных единиц в полимерной цепи.
Таблица II, 1
Термодинамические величины, характеризующие плавление полимеров"
Полимер т /п сС повторяющихся единиц j на связь на единицу л5« на связь
Полиметилен ..... 137 785 785 1,90 1,90
Полиэтиленоксид ... 66 1980 660 5.85 1,95
Нату|>альньш каучук . . 28 1050 350 3,46 1,15
Г уттаперча 74 3040 1013 8,75 2,92
Полихлоропрен 80 2000 667 5,7 1,9
Политрифторхлорэтилеи . 210° 1200 600 2,50 1,25
Трибутират целлюлозы 207 3000 1500 6,2 3, 1
Полипропилен 176
Политетрафторэтилен . . Полигексаметиленадип- 327
амид 260
Полиэт илентерефталат 267
‘Р L. М а п <1 е I k е г n, Chem. Rev., S.i, 903 (1956); Усп. хим., 27, 193 (I9..8).
°) Последние данные 220—223 JC; J. D Н о । f m a n n. J. J. Weeks, J. Res. Nat. Bur.
Stand., W, 465 (I'.Cb).
В табл. II, 1 приводятся для некоторых полимеров Тт, АНи,
ASU, а также АНи п AS,,, отнесенные к одной связи [2]. Величи-
на, рассчитанная па одну связь, получается делением АНи и
соответственно Д Su на число связей в главной цепи повторяющейся
структурной единицы. В частности, интересно заметить, что
величина AS,,, рассчитанная на одну связь, является весьма по-
стоянной для многих полимеров и равной но порядку величины
У? кал/град моль.
62 Гл. II. Основы физики полимеров
Величина AH„ может быть получена калориметрическим изме-
рением теплоты плавления кристаллических полимеров. Она долж-
на быть вообще меньше АН.,, рассчитанной из уравнений (3, 2)—
(3, 5), так как полимеры содержат как аморфные, так и кристал-
лические области. Для кристаллических на 100*’о полимеров
AН„ и АН,, должны быть равны. В других случаях АНй/АНц^
= 1-/. есть объемная доля кристаллического материала.
Формула (3, 6) может быть переписана в следующем виде:
т =.- А//» <[1,1 связь) 7,
m XS„ (на свяж) ’ ’ '
Величина AS„ (на связь) поддается некоторым молекулярным
интерпретациям. Прежде всего ASU (па связь) =ASp (на связь) , ASC
(на связь), где AS., возникает в результате изменения объема при
плавления, a ASC вследствие изменения конформационной энтро-
пии при плавлении. Было подсчитано, что для натурального кау-
чука ASj, (на связь) равно 0,60 кал!град-моль, поскольку ASC
(на связь) равно примерно 0,55 кал,град-моль [21. Вполне правдо-
подобно, что величина ASU (на связь) является одной и той же
для большинства полимеров.
В кристаллах парафина молекулярные цепи «заморожены» в
виде плоской зигзагообразной конформации. При плавлении
каждая связь может принимать три выгодных положения по от-
ношению к предыдущей связи, а не одно, лежащее в плоскости
двух соседних связей. Поэтому конформационная энтропия, рас-
считанная на моль связей, равна ТДпЗ, что несколько больше, чем
величина ASt (иа связь) = ),30 кал/град-моль, полученная для
полиметплена. Последняя величина получена из Д5„ (на связь) =
= 1,90 (см. табл. II, 1) и ASv (на связь) -0,60.
Возможно, что не все три положения единичной связи явля-
ются полностью достигаемыми даже в расплавленном состоянии.
Поэтому конформационная энтропия, отнесенная к одному молю
связей, фактически существенно меньше, чем АЧпЗ.
Другим толкованием Д5С (на связь) является представление
о том, что она представляет собой энтропию смешения с «дыр-
ками», возникающими в полимерной структуре в расплавлен-
ном состоянии (2|.
Естественно ожидать, что Д//„ (на связь) будет находиться
в определенной корреляции с величиной молекулярной когезии.
Эта последняя может быть получена из параметра растворимости,
рассмотренного в разделе 5 этой главы. Весьма удивительно,
однако, что экспериментальные результаты не подтвердили этого.
Например, было установлено, что полиэфиры имеют значение
АНи выше, чем соответствующие полиамиды [3], хотя параметры
3. Термодинамики кристаллизации
63
растворимости полиэфиров меньше параметров растворимости
полиамидов.
Так как полиамиды плавятся при температурах более высо-
ких, чем соответствующие полиэфиры, они должны иметь также
значительно меньшую энтропию плавления. Действительно, было
установлено, что высокие температуры плавления связаны с низ-
кими энтропиями плавления гораздо в большей степени, чем с
высокими теплотамп плавления. Кристаллическая структура
полиамидов, по-видимому, допускает больший конформационный
беспорядок, чем структура полиэфиров, и это может объяснить
низкие энтропии плавления, обнаруженные в полиамидах |3|.
Для кристаллизующихся сеточных полимеров точка плавле-
ния зависит от относительного растяжения a^L/Ln следующим
образом 111:
(3,8)
(3, 9)
где Т3,н—температура плавления нерастянутой сетки, а Т,п—тем-
пература плавления растянутой сетки; ,VS—число статистических
сегментов в цепи находящейся между поперечными связями. Фор-
мула (3, 8) неверна при я=1, но будет справедливой при высоких
степенях растяжения. Объемная доля аморфного материала в
растянутой кристаллизующейся сетке выражается следующими
формулами [1, 2|:
(3, 10)
(3, Н)
Формула (3, 10) справедлива только при относительно высо-
ких растяжениях.
Ориентированные кристаллические полимерные волокна будут
сокращаться п терять свою ориентацию при температуре плавле-
ния Т,п кристаллитов. Внешнее напряжение, приложенное к
ориентированному полимеру, будет весьма значительно увеличи-
вать температуру плавления (пли температуру сокращения). Это
может быть объяснено очень просто путем прямого применения
6-1
Гл. II. Основы физики полимеров
уравнения КлапеГфона—Клаузиуса 14, 5]:
где Агравн.—растягивающая сила, необходимая для того, чтобы
поддерживать равновесие между ориентированным (кристалличе-
ским) п неориентированным (аморфным) состоянием волокон при
температуре Т и давлении р. Величины AS и AL есть изменения
энтропии и длины, связанные с фазовым переходом при плавле-
нии кристаллического волокна.
Длина ориентированных кристаллических полимеров, частич-
но сшитых поперечными связями в трехмерную сетку, сокращает-
ся как только они достигают температуры, лежащей выше точки
плавления. Это сокращение длины частично обратимо 16], что
указывает на присутствие начальных ориентированных зароды-
шей в расплаве.
Фазовый переход первого рода кристалл—кристалл был опи-
сан [7], по крайней мере, для одного синтетического полимера,
а именно для транс-1,4-полпбутадпена
Литература
1. Р. J. Flor у, Principles of Polymer Chemistry, Cornell University Press»
Ithaca, New York, 1953, pp. 565— 576.
2. I.. Mande I kern, Chem. Rev. 56, 903 (1956); Усп. хим., 27, 193 (1958).
3. P. J. Flory, H. D. В e d о n, E. H. Keefer, J. Polymer Sci.,
28, 151 (1958).
4. G. Gee, Quart. Rev. Chem. Soc., 1, 265 (1947).
5. J. F. M. Oth, P. J. Flory, J. Am. Chem. Soc., 80, 1297 (1958).
6. I.. Ma n de 1 ke r n. D. E. Roberts, A. F. D i о r i c, A. S. P о s-
ne r, J. Am. Chem. Soc., 16, 4148 (1959).
7. G. Na tta, P. С о r r a d i n o, J. Polymer Sci., 38, 29 (1959).
4. Кинетика кристаллизации
Скорость роста кристаллического материала в полимерах яв-
ляется вопросом большой важности. Для определенных полиме-
ров, таких, как натуральный каучук или изотактический поли-
стирол, возможно, минуя область кристаллизации, быстрое охлаж-
дение от расплавленного до стеклообразного состояния без како-
го-либо процесса кристаллизации. Для других полимеров, таких,
как полиметилен, еще не доказана возможность быстрого охлажде-
ния полимера с переходом его в полностью аморфное состояние
ниже точки плавления Тт. Тем не менее степень кристалличности,
размеры кристаллитов и агрегатов кристаллитов (сферолитов),
даже в случае полиметилена во многом будут зависеть от
тепловой истории и режима отжига. Поскольку механические
4. Кинетика крисшл.ииации
65
свойства весьма сильно зависят от этих факторов*, было бы хоро-
шо дать краткое обсуждение этих явлений.
Скорость образования кристаллического материала пропор-
циональна скорости образования зародышей, а также скорости
их последующего роста. Скорость образования зародышей, ко-
торому благоприятствует переохлаждение, имеет максимальное
значение при температуре значительно ниже Тт. Линейная ско-
рость роста кристаллов, сильно зависящая от диффузии, имеет
максимальную величину только немного ниже Тт. Скорость роста
кристаллического материала в целом, будучи произведением этих
скоростей, также имеет определенную температуру, при которой
наблюдается ее максимальное значение. В качестве приближен-
ного правила найдено, что эта температура равна % Тт Ц].
Температурная область, в которой наблюдается заметная ско-
рость роста кристаллического материала, неширока. Так, натураль-
ный каучук имеет максимальную скорость кристаллизации при-
мерно при —25 °C. Скорости кристаллизации становятся весьма
малыми при —40 °C и —10 СС.
Полпкристаллпческая текстура кристаллических полимеров
зависит от кинетики кристаллизации. При кристаллизации из раз-
бавленных растворов в тех случаях, когда скорость образования
зародышей весьма мала по сравнению с их ростом, может быть
выращен единичный кристалл макроскопических размеров.
Кристаллы парафина и полпметилена растут, по-впдпмому, по
механизму винтовой дислокации. В результате получается спи-
ралеподобная ступенчатая структура пластинчатого кристалла
(рис. II, 11 и II, 12).
При нормальных условиях кристаллическая текстура полиме-
ров устанавливается в процессе охлаждения горячего расплава.
При этих условиях формируются очень тонкие (субмикроскопи-
ческие) кристаллиты. Каждый отдельный кристаллит является
частью плотно упакованного образования, называемого сфероли-
том, который часто может быть макроскопических размеров (см.
рис. II, 6).
Образование единичного зародыша в полимере, охлажденном
до температуры ниже Тт, благоприятствует образованию другого
зародыша в его непосредственной окрестности в результате созда-
* В последние годы обнаружено существование многих форм надмолеку-
лярных образований: пачек макромолекул, пачек, сложенных в ленты и пла-
стины, монокристаллов, сферолитов, лентообразных и пластинчатых образова-
ний из сферолитов н др. Показана также существенная зависимость механиче-
ских свойств кристаллических полимеров от характеристик надмолекулярной
структуры, очень чувствительной к тепловой и механической предыстории
[В. А. К а р’г и и, Т. И. С о г о л о в а, Г. Ш. Та л и п о в, ДАН СССР,.
142, 627, 844 (1962)).—Прим. ред.
5—833
(ill
Гл. II. Основы физики nO.Ul.Ul'1-Ов
ния местных напряжении. Этот процесс вторичного образования
зародыша .может быть положен в основу механизма образования
сферолита; в обычных условиях рост кристалла может быть не-
значительным по сравнению с процессом кристаллизации при
образовании вторичных зародышей.
Рве. 11,11. Электронно-микроскопический снимок
отдельного кристалла парафина С)3П74; кристалл
был получен в растворе петролейиого эфира. Эф-
фект спиральной лестницы наблюдается вследст-
вие роста кристалла на винтовой дислокации
[A. J. I-' о г t у, Advances in Physics 3, I (1954)].
Электронно-микроскопическии снимок поликристаллического
полиэтилена показан на рис. II, 13.
Образование п рост отдельных кристаллитов н сферолитов
зависит от механизма п кинетики кристаллизации и поэтому
весьма значительно зависит от режима охлаждения. Пленка по-
лиэтилена, изготовленная в обычных условиях при охлаждении
на воздухе, получается мутной, что указывает на присутствие
4. Кинетика кристаллизации
67
больших сферолитов. С другой стороны, эта же самая пленка мо-
жет быть получена почти прозрачной, если ее быстро охладить
в холодной воде. В последнем случае процесс образования заро-
Рис. 11,12. Электронно-микроскопический снимок от-
дельного кристалла полиэтилена [A. Keller, Growth
and Perfection of Crystals, New York, 1958, p. 499].
дышей, вероятно, является более быстрым, что приводит к пре-
дельно большому числу малых сферолитов. Физические свойства
этой пленки также, как п оптические свойства ее, зависят от ми-
крокристаллической текстуры.
Вопросы механизма п кинетики роста кристаллического мате-
риала в полимерах находятся в настоящее время в стадии быстрого
5*
68
Гл. II. Основы физики полимеров
развития. Результаты, полученные до 1956 г., отражены в работе
[11, а более поздние исследования (1958) представлены в рабо-
те 12).
Рис. 11,13. Электронно-микроскопический снимок
тонкой пленки полиэтилена, демонстрирующий тонкую
структуру сферолитов.
Литература
1. L. Mandel kern, Chein. Rev., 56,903 (1956). Усп. хим., 27, 193
(1958).
2. R. Н. Do re щ u s, B. W. Roberts, D. Turnbull, (eds.).
Growth and Perfections of Crystals, John Wiley a. Sons, New York, 1958.
5. Температура стеклования T
Вернемся к аналогии между структурой аморфного линейного
полимера п структурой находящихся в чашке сваренных топких
макарон.. Чтобы сделать аналогию более полной, нужно вообра-
зить, что при достаточно высоких температурах макароны нахо-
дятся в непрерывном хаотическом движении, что делает эту мо-
дель похожей на спутанный клубок земляных червей, хотя эта
аналогия п неприятна.
5. Температура стек.юаания
69
Беспорядочное сегментальное движение линейных полимерных
молекул обусловлено тепловой энергией п существованием свобод-
ного объема в полимерах. При достаточно низких температурах
свободный объем становится очень малым. Тепловая энергия kT
Рис. 11,14. Температурная зависи-
мость удельного объема вблизи Tg.
стеклообразным.
оказывается при этом также ма-
лой по сравнению с высотой
потенциальных энергетических
барьеров, препятствующих
вращательным п поступатель-
ным перемещениям полимерных
сегментов. В результате этого
вращательные и диффузионные
движения полимерных молекул
«замораживаются» и полимер-
ные сегменты и атомные груп-
пы могут совершать только ко-
лебательные движения, как в
идеальных твердых телах. Это
состояние полимера называется
Имеется узкий температурный интервал или, возможно, опре-
деленная температура Т„, ниже которой полимер находится в
стеклообразном состоянии*. Самым распространенным методом
определения Т,, является измерение температурной зависимости
удельного объема полимера и(Т). Измерения дают возрастающую
кривую, как схематически показано на рис. II, 14. На этих кри-
вых наблюдается определенный перелом (в идеальном случае—
скачок производной dv'dT). Температура, соответствующая этому
перелому, определяется как Tg. Насколько резким является этот
перелом и как он зависит от скорости нагревания или охлажде-
ния с экспериментальной и теоретической точек зрения,—вопросы,
которые рассматриваться здесь не будут**. Достаточно сказать, что
* Как было показано впервые советскими учеными, переход в стеклооб-
разное состояние имеет кинетическую природу и поэтому, в отличие от равно-
весных фазовых превращений I п II рода, происходит в некотором температур-
ном интервале. Температура стеклования Tf!, часто обозначаемая в советской
литературе Тс, является лишь условной характеристикой положения этого
интервала на температурной шкале [см. П. П. К о б е к о, Аморфные веще-
ства, Изд. АН СССР, 1952]. — Прим. ред.
Зависимость температуры Tg от режима нагревания и охлаждения весь-
ма существенна для понимания природы стеклования как релаксационного
процесса и для практических расчетов в технологии отжига и закалки. Зако-
номерности этого явления приводятся в книге ]1] ц статьях |2, 3], а теория
стеклования—в работе [4]. [1. Г. М. Бартенев, Механические свойства
и тепловая обработка стекла, Госсгройпздзт, гл. 1, I960. 2. Г. М. Барте-
н е в. И- А. Л у к ь я н о в, ЖФХ, 29, 1486 (1955). 3. Г. М. Барте-
нев, Ю. А. Г о р б а т к п и а, Высокомол. соединения, I, 769 (1959).
4. М. В. Волькенште и н, О. Б Питы и. ЖТФ, 26, 2204 (1956)].—
Прим. ред.
70
Гл. II. Основы физики полимеров
метод, описанный выше, дает разумные величины Т„, обычно
определяемые с точностью до нескольких градусов. Для сравне-
ния на рис. II, 15 приводятся температурные зависимости удель-
ного объема для некоторых кристаллических полимеров.
Температура стеклования является, пожалуй, наиважнейшей
характеристикой аморфных полимеров (1, 2]. Как будет видно из
' Температура, °C
Рис. 11,15. Температурная зависимость удельного
объема вблизи температуры плавления Тт
[L. М а п <1 е 1 к е г n, Chem. Revs., 56, 911
(1956); Усп. хим.. 27, 200 (1958)].
дальнейшего, аморфные полимеры подчиняются закону соответ-
ственных состояний, в которых Т!Т„ или Т—Та являются основ-
ной приведенной переменной. Так как Т„ является важной ха-
рактеристикой полимеров, то, конечно, весьма интересно знать,
как Tg зависит от структурных п энергетических свойств поли-
меров*.
* Здесь п в дальнейшем автор рассматривает только структурное стекло-
вание полимера при охлаждении и соответственно температуру структурного
стеклования 7*Т|’. Этот процесс стеклования характерен как для простых
жидкостей, так п для расплавов полимеров; происходит в отсутствие внешних
механических воздействий и заключается в «замораживании» структуры блпж-
5. Температура стеклования
71
Беспорядочно перепутанная совокупность полимерных моле-
кул может быть определена простыми структурными параметра-
ми. Прежде всего пз естественных соображении возникает понятие
об удельном «свободном объеме». Свободный объем vjt приходя-
щийся на 1 г полимера, определяется как v—vs, где v—реальный
удельный объем, a t's—удельный объем совершенно плотно упако-
ванных полимерных молекул. Это понятие уже рассматривалось
в связи с вязкостью жидкостей в гл. I (раздел 8).
Чтобы описать энергетические взаимодействия между сегмен-
тами полимерных молекул, заимствуем одно понятие из теории
жидких растворов [31. Величина, известная под названием плот-
ность когезионной энергии (ПКЭ), является важной характери-
стикой свойств жидкостей. Величина ПКЭ определяется как мо-
лярная энергия испарения, деленная на молярный объем; обе
эти величины легко измеряются. ПКЭ является мерой когезии
единицы объема жидкости.
Параметр растворимости о равен квадратному корню из плот-
ности когезионной энергии:
й = (ПКЭ)1'* = [Д'К'---у/2 (5,1)
Величины параметра растворимости ряда простых жидкостей
приведены в табл. II, 2.
Параметры растворимости для полимеров не могут быть полу-
чены прямо из уравнения (5, 1). Для их определения приготавли-
вается слабо сшитый полимер, идентичные образцы которого
помещаются в ряд жидкостей с известными о [4]. Полимер набу-
хает во всех этих жидкостях, но не растворяется из-за наличия
поперечных связей. Величина набухания может быть графически
изображена в зависимости от параметра растворимости жидкостей,
него порядка при температуре стеклования. При механических воздействиях
выше этой температуры аморфный полимер в отличие от простых жидкостей
обнаруживает новое свойство—высокую эластичность. С понижением темпе-
ратуры высокая эластичность исчезает. Температура потери высокой эластич-
ности 7'“ех’ также часто называется температурой стеклования полимера, но
она не совпадаете 7'|тр‘. Оба процесса стеклования имеют одну и ту же молеку-
лярно-кинетическую природу, однако «механическое» стеклование имеет свою
специфику. Например, 'f^lx зависит от частоты периодических деформации и
тем больше отличается от 7^1р , чем больше частота механических воздействий.
Во всех случаях -7'‘;,р'. [ Более подробные сведения о двух процессах стек-
лования можно найти в статьях: Г. М. Барте и е в, ДАН СССР, ПО, 805
(1956); Со. «Стеклообразное состояние» Изд. АН СССР, 1960, стр. 147—153.]—
Ирам. ред.
Г.i. П. Основы физики полимеров
Т а блиц а И, 2
Параметры растворимости некоторых жидкостей1
/1\НДКОС1Ь Параметр рас- творимости S Жидкость Пар.1.-..с>р рас- твори ,:истп 3
Линейные диметнлсилокса- 5,90—4,97 Диметилсульфид 9.4
иы (от 2 до 11 атомов Si) Бутиллактап 9,4
Алифатические фторнро- 5, .5—6,2 Пенгахлорэтан 9,4
ва иные у гл свод о роды
Ароматические фторнро- 7,5-8,2 Me гплацетат 9,6
ванные углеводороды Метиленхлорид 9,7
Неопентан 6,3 Эт илендихлорпд 9.8
н-Гексан 7,3 Циклогексанон 9,9
Гексен-1 7,4 2-Этокснэтанол (целло- 9,9
Диэтнловый эфир 7,4 ЗОЛЬВ)
Диизобутилеи 7,7 Диоксин 9.9
Трикрезплфосфат 8,2 Этиллактат 10.<>
Этилампн 10.0
Циклогексан 8,2
Ацетон 10,0
Скипидар 8,1
Уксусная кислота 10,1
Дипентек 8,5
Бутилацетат 8,5 Метилформиат 10, 1
Четыреххлористый углерод 8,6 Анилин 10,3
Диоктилсебацинат 8,6 Метнлцеллозольв 10,8
Окись этилена 11,1
Целлозолькацетат 8, 7
Ксилол 8,8 Циклогексанол 11,4
Толуол 8,9 Бутанол И.4
Бромистый этил 8,95 Ацетонитрил 11,7
Этилаиетаг 9,1 Диметилформамид 12,1
Диацетоновый спирт 9,2 Нитрометан 12,4
Метилцеллозольвацетат 9,2 Дн.метилфосфит 12,5
Бензол 9,2 Этанол 12,7
Метанол 14,5
Этнлмеркантан 9.2о
Метилэтилкетон 9,3 Аммиак 16,3
Хлорбензол 9,3 Вода 24,2
Хлороформ 9,3
J. Н. 11 i 1 d cb га и d, R. I.. Scot!, Solubility « f Ncneledr lytes, Rlicinhuld Publishing»
Corp.. New Yuk. 1950. II. Burel I, IrHerthem. Rev., 14. 31 (1935). M. II. Will, Use of
G. hesive Energy Densitx, Monsanto Chemical Co. Tech-, Bull, I5'\Il 1955.
5. Температура стеклования
73
в которых набухает полимер. В результате получается кривая,
по форме напоминающая гауссову, центрированная относительно
некоторого значения параметра растворимости ?,н, при кото-
Рпс. 11,16. Набухание натурального каучука и
N—5400 (вирам-каучук) в зависимости от величи
ни параметра растворимости растворителя
[М. Н. W i I t, Alonsanto Chemical Co., Tech.
Bull., Apr., 1 (1954)|:
/—натуральный каучук: 2- вирам.
ром набухание максимально. Это схематически показано на рис.
II, 16. Величина 6Н принимается в качестве параметра раствори-
мости полимера.
В табл. II, 3 приведены параметры растворимости ряда поли-
меров.
Важным параметром цепи является Ио—энергетический барьер,
препятствующий вращению вокруг связей в цепи. Для высоких
значений Ко внутренняя подвижность цепи низка. Другим пара-
метром цепи является геометрическая жесткость цепи Z =
~(г2, Д'Д,)1 -, рассмотренная в разделе 1 этой главы. Наконец,
длина цепи N также является важным параметром цепи.
Как зависит от параметров с, с, аи, lz0, Z и Л'?.
1. Возможно, что в первом приближении величина vfiv при
Т< может быть одной и той же для всех аморфных полимеров
|5—8|. Методом, который будет рассмотрен в разделе 8 этой
гтавы, эта величина была определена и оказалась равной 0,025.
2. У полимеров с большим значением наблюдается тенден-
ция к высоким значениям ТНапример, полидиметилснлоксан
(силиконовый каучук) имеет самую низкую температуру Tg из
известных до сих пор и в то же время очень низкое значение Ън.
Полнбутадиен обладает также очень низкими значениями
74
/ л. II. Основы физики полимеров
Т а 6 д л ц а II, .4
Параметры растворимости для некоторых полимеров-1
i Ю.шм.р Параметр рас 1 L,ipi,
Тефлон (поли гетрафторэгн.п н) 6. 2
Силиконовый катчтк I поднайме ш.к'и.ыкс.тн) . Г . >
11о.1итоГ>\ шлеи. о\ iii.iK.o ч\к . , Л
Полиэтилен 1 . м
Натуральный каучук 8, 1
Полноттадпен К. 1
GR-S (оугаднеп-стпиол 75,25) 8, 1
Полистрол 8.56
Пеопреи (полпхлонопреп) 6
Буна-Х (6\ ia;iin и акрилони! рил ‘'5 25) Н. 9
I lo.iii'ieiH.nieiaKpiLiai 9,08
I колнвннилаиешт ... 9.4
Поливинилхлорид . . 9,55
Этилцеллюлозл . . . Ь),3
Полиметакрнлонпгрот Ю.7
11ол) । э 1 плентерс-фтала г . 1п. 1
Диане га I целлюлозы 1U.9
Эпоксидная смола 111.4
Пилпппиилпдепхлорид . . 12.2
Нанлои-6.6 ... 13.6
Полиакрилонитрил ... 15 |
’ И. В ti г е 1 I. Iiller.hem. Rev., 11. 3 (!97>); М Н. Wilt, Use .,f Chaise E .ogy
IicibUx, М нвлпю Chesni_.il С’>. Teh. Bull., 1~.Vll 1955; (г M Bri stow, W. r. Wai - n.
Тип-, I’.riti. Soc., 51, November (193b).
и 'Jfl. С другой стороны, полиакрилонитрил, полнметакрилоннтрил
ii полиакриловая кислота имеют большие значения н высокие
значения Т„.
3. Полимеры с низким значением Уо (большая гибкость цепей)
имеют низкие значения Т„. В этой связи интересно сравнить зна-
чения 7'с силиконового каучука (—120 °C), полиэтилена (около
—85 °C) п политетрафторэтилена (>20 ~С). Все эти вещества
имеют низкое значение оП, причем самое низкое значение он имеет
политетрафторэтилен. Однако вращение вокруг связи Si—О—
почти свободно; барьер, препятствующий вращению вокруг свя-
зей —СН.,—СИ.,—, равен 3,3 ккал/моль, а барьер, препятствующий
вращению вокруг связи —CF.,—CF2—, превышает 4,7 ккал ноль
1101. Аморфная сера с очень большим значением имеет 7\ око-
ло -20 С вследствие относительно свободного вращения вокруг
связи -S—S —.
4. Полимерные цепи, которые геометрически жестки (высокое
значение Z), возможно, имеют более высокие значения Т , чем бо-
лее гибкие полимеры аналогичного химического строения, н с одп-
5. Температура стеклования
75
иаковыл г,И. Например, поливинилацетали обнаруживают на-
много более высокие значения Tg, чем полнвнниланетат, а поли-
аценафтилен обладает значительно большим значением Т„, чем
полистирол:
СП, СН,
---СИ,—CI I CII—СН,—СН ^СН-----
i ! ' I «
(J О О С)
\:н
! )
R R
Поливинилацетали
Поливпнилацетат
Полиаценафтнлен
Полистирол
Однако полностью нельзя быть уверенным в таком влиянии гео-
метрической жесткости потому, что те полимеры, которые гео-
метрически жестки, также имеют тенденцию к высоким значениям
Ин и поэтому обладают малой внутренней подвижностью [10].
5. Температура Т следующим образом зависит от длины
цепи N:
T^T'i — c'.N (5,2)
где с'—постоянная для каждого полимера, а Т"—предельное
значение температуры стеклования для бесконечно длинной цепи.
Так как экспериментально наблюдается, что T^Tg для значе-
ний .V>500, то можно вообще принять Tg ^Т^для высокополпме-
ров.
7
Г.1 11. ОснПиы физики полимера i
Значение Г, для аморфных сополимеров можно прпблнзпгель
ио предсказать, если температуры стеклования 7’31) и 732)
обоих гомонолнмеров известны. Одна из эмпирических формул,
которая была найдена достаточно хороню приложимой во многих
случа ях, следующая [111:
1 __ Ц'| , ^'2
Tg 7\(1) ' 7Д, (2)
(О, 31
где ыд п ьд,—весовые доли каждой составл-нощей сополимера.
Эта формула легко может быть распространена на трехзвен-
ные сополимеры и т. д. Предполагается, что мономерные единицы
каждой составляющей распределены вдоль цепи более или менее
беспоряточно. Уравнение (5, 3) является лишь приблизительным
для некоторых систем, а более общие формулы оылп предложены
в работе |12|.
Формула (5, 3) очень полезна для вычисления температуры
стеклования Т.г кристаллических полимеров. Часто па кривых г—Т
кристаллических полимеров виден неясный излом, соответствую-
щий ТВ этом случае, получая серию аморфных сополимеров,
температуры стеклования которых могут быть легко измерены,
можно рассчитать величины Т„( 1) и 7'л,(2).
Хорошим примером являются гомополпмеры и сополимеры
трнфторхлорэтплена и виннлнденфторида. Оба эти гомополимера
высококристаллпчиы, но в то же время можно легко приготовить
ряд соответствующих аморфных сополимеров, величины Т ко-
торых легко измерить. Строя график 1/7', от ид для этих сополи-
меров, можно получить несколько экспериментальных точек в об-
ласти 0,4<ид<0,6. Экстраполяцией прямой линии, полученной
в этой области, к значениям ид=0 пид = 1, получаются величины
Г (1) и 7'О(2). Таким образом, оказывается, что Т„ для полптрн-
фторхлорэтилена равна примерно 40 °C, a Tg для поливинилиден-
фторида примерно равна —39 °C 1131. Подобными методами ве-
личина Tg для полиперфторпропплена была найдена равной 11 С
1101. Используя известные величины Т„ для полисульфпдных
каучуков, таких, как —CH.,CH,,SSSS—, вместе с Tg полиэтилена,
можно определить величину Т„^ — 27 "С для линейных цепей
серы, согласующуюся со значением, найденным для аморфной
серы.
Некоторые кристаллические полимеры быстрым охлаждением
могут быть переведены из расплава в аморфное состояние. В этих
случаях величина Тр аморфного полимера легко может быть изме-
рена. Величина Т„ изотактического кристаллического полипро-
пилена может быть получена при изучении величины Tg аморф-
ного атактического полипропилена.
.5. Темпераiypu ciеклования
Интересно заметить, что для кристаллических полимеров ока-
зывается существует простое численное отношение между Т и
Тт U4]:
V--0.5 (5,4)
-* tn
для симметричных полимеров, таких, как полиэтилен или поливинилиденхлорид
2k. —о,67
1 т
для несимметричных полимеров, таких, как полптрпфторхлорэтнлен
или изотактический полипропилен
(5, 5)
Возможно, было бы более надежно предложить в качестве об-
щего правила:
0,5Д„ < 7\. < 0,677;,
(5, 6)
В табл. II, 4 приводятся температуры стеклования отдельных
полимеров. Величина аг есть коэффициент объемного теплового
Таблица II, 4
Температуры стекловании некоторых полимеров11
Полимеры г„, -с огют «о--104
Силиконовый каучук (иолидиметилсило-
ксан) — 123 12 2,7
Полибутадпен (эмульсионный, 50 СС) . —85 7,8 2
Полиизобутилен0 —70 —- —
Натуральный каучук Т‘4 6,16 2,07
Полибутилакрнлат —56 6,0 2,6
Поливинилиденфторид —39 — •—-
Полиэти лак рилат 22 6,1 2,8
Полиметилакрилат 9 5,6 2,7
По.типерфторпропплен И — —-
Поли-н-бутплметакрплат . 22 6,3 3,7
Поливииилацетат 29 5,98 2.07
11оли-н-пропилметакрнлат 35 5,80 3,51
Полптрпфторхлорэтнлен 45 — —-
Полиэти.тметакрнлат .... 65 5,40 2,75
Поливинилхлорид 82 5,2 2,1
Полистирол 100 5,5 2,5
Полиметил.метакрилат ... 105 5.0 1,95
Полиакриловая кислота 106
По.тп.метакрилопитрнл0 120
3) L. А. XV О о d. J. Polymer Sci.. 2Ь. 319 (19.‘.8).
"I P. J. El ory. Principles of Polymer Chemistry, Ccrnvll University Press, Jiliaca, New
York., 1953, pp. 52 -53
I'.t. II. Основы- физики но.шмерив
расширения в каучукоподобпом состоянии, а щ—коэффициент
объемного теплового расширения в стеклообразном состоянии.
Непредвиденные затруднения, возникающие при определении
Т\ высококрпсталлпческп.х полимеров при помощи кривых
объем—температура пли по механическим свойствам, видны на
примере политетрафторэтилена. При определении Те подобными
методами было установлено, что величина равна —112 С 115].
В противоположность этому было установлено, что истинная тем-
пература Т., должна быть выше комнатной температуры, как пред-
сказывается уравнением (5, 6) и следует из незначительной внут-
ренней подвижности полимера |9|.
Здесь мы имеем дело с очень важным практическим вопросом.
Если Г политетрафторэтилена практически равна —112 С’С, то
аморфные сополимеры тетрафторэтилепа и подходящих других
фторированных мономеров должны обладать каучукоподобпымн
свойствами при очень низких температурах. Является сомни-
тельным, что это соответствует истине. Для того чтобы получить
хорошие низкотемпературные свойства веществ из класса фтор-
содержащих эластомеров, необходимо иметь такую гибкость це-
пи главных валентностей, какая присуща макромолекулам поли-
меров, синтезированных пзСН., -CF„; CH.,=CH(CF3); СН„ =CF(CF3)
п подобных мономеров. Они могут быть сополпмеризованы с под-
ходящими мономерами с целью получения аморфных сополиме-
ров.
Литература
1. R 1' В о у е г, R. S. S р е и с е г. Advances in Colloid Science, vol.
II. ch. 1, Interscience Publishers, New York, 1916.
2. N. Bekkedahl, J. Res. Nat. Bur. Stand., 13, III (1934).
3. J. II. Hildebrand, R. L. Scott, Solubility of Nonelectrolytes,
Rlnenhold Publishing Corp., New Yok, 1950.
4. (i. (i e e, Thermodynamics of Rubber Solutions and (Seis, chapter in «Advan-
ces in Colloid Science», vol. 11, Interscierice Publishers, New York, 1946.
5. J. D. I-errj, R. F. L a n d e 1, Koll.-Z., 148, 1 П956).
6. 31. 1.. \V i 1 1 i a in s, R. F. L a n <1 e 1. J. D. Ferri. Xin. Chem
Soc., 77, 3701 (1955).
7. II. F ii| i la, A. 1\ i s h i in о I o, J. Coll. Sci. (in press).
6. T. (i. Fo x, P. .1. F 1 о r v. J. Appl. Phvs., 21, 581 1195'9; -1 Am. Chem.
Soc., 70. 2384 (1948); J. Phys Chem., 55.’ 2'21 (1951); J. Polcm. Sci.. 14.
315 (1954).
9. A. V. T о b о 1 s k y. J. Polymer Sci., 35, 555 (1959).
ID. Ci. J. .1 a n z. Estimation oi Thermodynamic Propertieso; Organic Compounds
Academic Press, New York, 1958, p. 161.
11. T. (1. Fox, Bull. Am. Phys. Soc., I. 3. 123 (1956).
12 L. A. Woo d, .1. Polymer Sci., 28, 319 (1958).
13. L. 31 a nilel ke r u, G. M. Marti si, F. A. Quin n. jr., J. Res.
Nat. Bur. Stand., 58, 137 (1957).
14. R. Ci. Bea in a n, J. Polymer Sci., 9, 470 (1952).
15. M. В a с c a r e d d a, E. В u t t a, J. Polymer Sci.. 31. 189 (1958).
6. Oo.tuciii упруговязких свойств аморфных полимеров
79
6. Области упруговязких свойств линейных аморфных полимеров
Простейшим способом характеристики упругих свойств поли-
мера является измерение его упругого модуля как функции тем-
пературы. Поскольку полимеры являются упруговязкпми мате-
риалами, их модуль будет зависеть от времени опыта и метода
измерения.
В данном случае будет рассматриваться модуль релаксации £,,
полученный при измерении напряжения в образце с заданной
величиной растяжения. Время измерения может быть стандартп-
Рпс. 11,17. Температурная зависимость (10) для
двух образцов полистирола .1 и С.
знровано (10 сек) так, что измеряемая величина как функция тем-
пературы есть Et (10). Другие методы измерения модуля дадут
качественно подобные результаты.
Результаты, полученные для типичного линейного аморфного
полимера—атактического полистирола, приводятся на рис. II, 17.
На этом рисунке приведены кривые для двух образцов полистиро-
ла А и С, причем оба образца имеют очень узкие кривые распреде-
ления молекулярных весов. Среднечисловоп молекулярный вес
Л1,г для образца А равен 140 000; а для образца С равен 217 000.
Кривые зависимости lg£r(10) от температуры обнаруживают
пять областей упруговязкого поведения ill, которые указаны для
образца С на рис. II, 18.
Первая область ниже 90 СС есть область стеклообразного со-
стояния, в котором значения модуля находятся между 10го и
дин.см2. В этой области полимер является истинным стек-
лом, твердым и хрупким. Во второй, переходной, области модуль
изменяется между 1010 и 10'*’7 дин см2 в температурном интервале
‘81)
/ л. II. Основы физики полимеров
Темпероглура. °C
Рис. 11,18. Пять областей уп-
руговязкого поведения (для об-
разца С полистирола).
от 90 до 120 гС. В этой области быстро изменяющегося с темпера-
турой модуля физические свойства полимера могут рассматри-
ваться как кожеподобные. В третьей! области модуль, практически
не изменяясь с температурой, находится в пределах от 10“>т до
10й’J дишсм". Температурный интервал, охватывающим эту об-
ласть, зависит от длины N молекулярных цепей полимера. Для
образца С этот интервал находится между 120 и 150 "С. Эта об-
ласть называется каучу коподобпым
плато или областью высокоэластиче-
ского состояния, и свойства полиме-
ра в этом интервале истинно высо-
коэластнческпе. В четвертой обла-
сти модуль изменяется от Ю”’1 до
105’5 дин'см2. Здесь полимер вы-
сокоэластичеи, но имеет также
заметную составляющую течения.
Эта область, которая может быть
названа эластовязкоп, для образца
С простирается от 150 до 177 'С. На-
конец, в пятой области, с модулем
меньше 103’5 динкм2, полимер ис-
пытывает очень малые высокоэла-
стпческпе деформации и обнаружи-
вает явно выраженное состояние
вязкого течения.
Температурные интервалы этих областей упруговязких свойств
зависят от произвольно выбранного упомянутого выше вре-
мени опыта 10 сек. Это видно на рис. II, 19, где данные для образ-
ца С полистирола изображены при временах измерения 5, 10,
100 п 1000 сек.
Итак, области упруговязкпх свойств лучше всего определяют-
ся по величине модуля*; температурные интервалы, соответствую-
* Методы изучения деформируемости полимеров в широком интервале
температур были впервые разработаны в СССР. А. 11. Александровым и
Ю. С. Лазуркипым был предложен частотно-температурный метод, а В. А. Кар-
гиным и Т. II. Соголовой—термомеханический метод. Авторами термомеуанн-
ческого метода впервые были выполнены систематические исследования пере-
ходов одного п того же полимера (пол и изобутилена) из стеклообразного со
стояния в высокоэластическое и из высокоэластического в вязкотекучее. При
это л было установлено, что при повышении температуры линейные аморфные
полимеры последовательно проходят через три физических состояния — стекло-
образное, высокоэластпческое п вязкотекучее.
Три температурных интервала существования этих состояний, совместно
с двумя областями переходов между ними, и являются пятью областями упру-
ювязкого поведения полимеров, о которых пишет А. Тобольский.
Метод измерения температурной зависимости модуля релаксации по суще-
ству вполне эквивалентен термомеханическому методу, так как в обоих мето-
б Области цируговизких ceoiu те аморфных полимеров
81
щпе этим упруговязким областям, зависят от времени измерения.
В этом разделе мы ограничимся рассмотрением температурных
интервалов, определенных при упомянутом времени эксперимен-
та в 10 сек. Более полное рассмотрение упруговязких свойств необ-
ходимо отложить до обсуждения временных эффектов, которые
будут изложены в главе III.
Из рис. II, 17 и II, 18 в довольно ясной форме следуют некото-
Рпс. 11,19. Температурная зависимость модулей при различных време-
нах наблюдения для образца С атактического полистирола 1по неопуб-
ликованным данным автора и К. М и г a k a m i и данным
Н. Е и j i t a, J. Coll. Sci., 12, 204 (1957)].
рые закономерности. Область стеклообразного состояния и переход-
ная область, очевидно, не зависят от длины молекулярной цепи
(для достаточно больших молекулярных весов). Величина модуля
___________ " а
KJ
дах измеряется эффективный модуль высокоэластпчностц, проявляющийся
при заданной длительности деформации (различие существует лишь в типе
деформации—релаксация напряжения при постоянном растяжении в первом
методе и развитие деформации при постоянном сжимающем пли растягиваю-
щем напряжении во втором методе).[А. П. Александров, IO. С. Л а-
з у р к и и, ЖТФ, 9, 1249 (1939); Ю. С. Л а з у р к и и, ЖТФ, 9, 1261
(1939); В. А. Карги н, Т. 11. С о г о л о в а, ЖФХ, 23, 530 (1947)
Т. И. С о г о л о в а, Г. Л. С л о н и м с к и п, Ж- ВХО им. Д. И. Менделеева.
6, № 4 , 389 (1961); II. Г1. К о б е к о, Аморфные вещества, Изд. АН СССР,
1952; В. А. Карг н н, Г. Л. Слон н м с к и й, Краткие очерки по фи-
зпко-хпмип полимеров, Изд. МГУ, i960].—Прим. ред.
6-833
82
Г.t. II. Отоны физики no.iuMepoi’
в области каучукопэдобнэго плато также не зависит от длины
цепи. .Между тем области эластовязкого п вязкого течения суще-
ственно зависят от длины цепи полимера.
Полезно определить некоторые параметры, которые могут быть
получены непосредственно из кривых модуль-температура для
аморфных полимеров. Можно без труда определить Ех— предель-
ный модуль для стеклообразного состояния п Е„—модуль для
области каучукоподобиого плато Также можно определить тем-
пературу перегиба кривой 1\, при которой lgE;(10) —(IgE,
lgE„). Для полистирола величины Е,. Е„ и 7\ соответственно
равны !()’">35 дин.см2, !<)“>Ii5 дин см2 и 98 С.
Необходимо также определить температуру текучести Т,,
при которой максимальное время релаксации еоо гвет с пп ст
10 сек. Обсуждение понятия «максимальное время релаксации»
будет дано в главе IV.
В стеклообразном состоянии сегменты полимерной цепи замо-
рожены в определенных положениях—узлах раз^ порядоченной
квазпрешеткп. Опп колеблются около этих фиксированных поло-
жений точно так же, как молекулы в молекулярных решетках.
Тем не менее они едва ли подвергаются какому-либо тн||х])узпон-
ному перемещению пз одного места решетки в другое. Диффу-
зионное движение, характерное для жидкостей, наблюдается
только выше Е„.
Следует ожидать, что модуль объемного сжатия полимера в
стеклообразном состоянии может быть рассчитан тем же путем,
как п для молекулярной решетки, а именно (см. раздел 3
главы I):
В=8,04-^_ (6.1)
Вместо величины Е,,.,, Г подставим плотность когезионной
энергии или квадрат параметра растворпмосди, как указано в
разделе 5 этой главы. 11з табл. II, 3 следует что ПКЭ для по-
листирола равна 83 кал см3 -3,-17- 10й эрг см3. На основе уравне-
ния (6,1) можно предсказать, что В должно быть равно
2,78 К)1" дин.'см2. Это находится приблизительно в согласии
с экспериментальным значением, равным 3,79- Id1" дан см2.
В стеклообразном состоянии коэффициент Пуассона вероятно
лежит между 0,25 п 0,33. Поэтому модуль Юнга Е, в этой области
имеет тот же порядок величины, как и В. Отсюда следт ет ожидать,
что
Е, 8,0-1 8,04'? (6, 2)
6. Области упруговязких своиав аморфных полимеров
83
Экспериментальное значение Ej равно 2,24 l()lu диц:см-, что
опять-таки находится в приближенном согласии с уравнением
(6, 2).
В переходной области сегменты полимерной цепи испытывают
.\111Кр(>брпун1'<вское диффузионное движение. Время, необходимое
для диффузии сегмента из одного «узла решетки» в другой, есть
величина порядка 10 сек (время нашего эксперимента). В переход-
ной области модуль изменяется очень быстро со временем, также
как и с температурой.
Температура перегиба Tf, конечно, зависит от выбранного
памп времени 10 сек. При этом условии Т- для аморфных полиме-
ров оказывается в пределах нескольких градусов совпадающей с
Т„, определенной из п.Г-крнвых.
В каучукоподобной области мпкроброуновское диффузионное
движение полимерных сегментов происходит очень быстро. Тем
не менее перемещение молекул в целом, включающее совместное
перемещение многих сегментов цепи, заторможено, в частности,
из-за перепутанности и зацеплений между цепями, которые дей-
ствуют как временные поперечные связи. Используя результаты
кинетической теории высокоэластпчностп, представленные урав-
нением (7, 10) главы I, можно рассчитать величину Мс, т. е. мо-
лекулярный вес цепи между двумя зацеплениями, исходя из ве-
личины Е„. Например, для полппзобутплена—первого полимера,
у которого было обнаружено каучукоподобное плато,—автор на-
шел, что молекулярный вес отрезка цепи между зацеплениями
[2| равен 8000. Для полистирола Мс между зацеплениями равен
20000.
В области эластовязкого течения перемещение молекул как
целое становится существенным. Большие конформационные изме-
нения всей молекулы, включая скольжение зацеплений, наблю-
даются за время порядка 10 сек.
Наконец, в области вязкого течения самые медленные изме-
нения конформаций молекул происходят за время, меньшее чем
10 сек. Высокоэластпческое восстановление ничтожно мало, если
напряжения п деформации действуют в течение времени, больше-
го 10 сек.
Области эластовязкого и вязкого течения могут полностью
исчезнуть, если вместо временных молекулярных зацеплений
создать постоянные узлы сетки—химические поперечные связи.
На рис. 11, 20 приведена кривая модуль—температура для поли-
стирола, структурированного в процессе сополимеризации с
0,25 мол. % дпвннплбепзола (образец D).
Для очень малого количества химических поперечных связей
величина высокоэластического модуля структурированного по-
лимера та же самая, что и для линейною полимера, но поперечные
81
Гл. П. Основы физики полимеров
связи заметно препятствуют течению. Для высокой степени струк-
турирования высокоэластический модуль структурированного по-
лимера больше, чем для линейного полимера, в согласии с урав-
нением (7, 10) главы I.
В принципе высокоэластнческий модуль Е„ в этом случае
должен быть пропорционален абсолютной температуре п не зави-
сеть от времени вплоть до высоких температур, так что на кривой
lg£r (10) в зависимости от Т каучукопоцобное плато выглядит
Рис. 11,20. Температурная зависимость Ег (10) для
кристаллического изотактического полистирола
В, образцов атактического полистирола Л и С и для
слабо структурированного атактического полисти-
рола D [по неопубликованным данным автора п
Н. Yu],
вполне плоским в огромном интервале температур, как показано
на рис. 11, 20. Это действительно верно для структурированного
полистирола, так как он вплоть до весьма высоких температур
.хорошо сопротивляется перестройке сетки, распад}' сетки, разры-
ву цепей под действием молекулярного кислорода и деполимери-
зации. Однако для большинства каучуковых сеток кривая зави-
симости lg£r( 10) от Т показывает резкое падение высокоэластп-
ческого модуля до нуля при температурах, при которых каучу-
ковые сетки обнаруживают изменения в своей химической струк-
туре. Это показано для полпсульфпдноп каучуковой сетки на
рис. 11,21 и для вулканизованного натурального каучука на
рис. 11,22.
Для полпсульфпдноп каучуковой сетки химическое изменение
при высоких температурах представляет собой дисульфидную
перегруппировку. Для вулканизованного натурального каучу
7. Кривые темперцп/р.чой acieuci: мости .модуля
85
химическое изменение при высоких температурах происходит
вследствие деструктирующего действия молекулярного кисло-
рода. Полное рассмотрение этого вопроса будет дано в главе V.
Рис. 11,21. Температурная зависи-
мость Ег (10) для структурированного
сульфидного каучука (тиокол ST).
Рис. 11,22. Температурная завнеп
мость Ег (10) для натурального
каучука, слабо структурированного
(вулканизованного) серой.
Литература
1. А. V. Т о b о 1 s k v, J. R. М с L о u g h I i n, J. Polymer Sci., 8, 543
(1952).
2. H. M a г к, A. V. T о b о 1 s к у, Physical Chemistry of High Polymeric
Systems, Interscience Publishers, New York, 1950, p. 344.
7. Кривые температурной зависимости модуля для аморфных
и кристаллических полимеров
Быстрейший путь к пониманию упруговязкпх свойств поли-
мера лежит через анализ кривой температурной зависимости
модуля при заданном времени* (принятое нами время равно
Ю сек). На рис. II, 23 показана зависимость 1g модуля от темпера-
туры в переходной области для ряда бутадиен-стирольных сопо-
лимеров эмульсионной полимеризации (некоторые слегка структу-
рированы). Все эти полимеры полностью аморфны даже при рас-
тяжении.
* Температурная зависимость деформации при заданном напряжении или
модуля упругости (величины, обратно пропорциональной деформации при
постоянном напряжении) называется термомеха пической кривой. Впервые
метод термомехапических кривых для анализа структуры и свойств аморфных
полимеров был предложен советскими учеными (см. примечание на стр. 80—
81 ).—Прим. ред.
86
Г.1. II. Основы физики полимеров
Геблера mg о а, °C
Рис. 11,23. Температурная зависимость
Г.г (10) для бутадиеи-стпрольпых сополиме-
ров. Цифры па кривых—соотношение бута-
диена п стирола в вес. ‘.’о.
Интересно заметить, что эти кривые приблизительно одинако-
вы. В этом .можно убедиться, если их горизонтально перемещать
вдоль осп температур так, чтобы величины Т, (пли 7\) совпали.
Для аморфных полимеров имеются различия в величине наклона
s графика зависимости IgE,. (10) oi температуры при Tt (эти ве-
личины в основном находятся в интервале от s 0,125 до s -0,333).
Однако в общем для всех полимеров переходная область прости-
рается приблизительно в пределах ЗО С. По существ}' тот же
самый тип кривой 1g модуль —температура получен почти для всех
аморфных полимеров.
Это приводит естес-
твенно к весьма важ-
ному качественному пред-
ставлению о механических
свойствах линейных аморф-
ных полимеров. Автор
высказал пр ед п ол ожен не,
что два линейных аморф-
ных полимера с одинако-
вым распределением це-
пей по длинам (пли с оди-
наковым распределением
величин г-) механически
почти эквивалентны при
соответственных темпера-
турах 11, 2|. Эти же со-
ображения применимы и к
аморфным структурированным полимерам с одинаковой степенью
поперечного сшивания и одинаковой концентрацией концов
цепей.
«Эквивалентное» поведение относится не только к унруговяз-
кпм свойствам, но также и к таким свойствам, как прочность на
разрыв, удтипенпе при разрыве, ударная прочность п т. д. 12|.
Это применимо только к полимерам, которые полностью аморфны
даже при растяжении.
Здесь мы встретились с проблемой определения понятия со-
ответственных температур. Это может быть сделано введением
приведенной температуры Т!ТС или Т—Тс, где Тс—характери-
стическая температура полимера. Мы уже ввели две температуры,
7„ и Ткоторые являются весьма подходящими величинами для
выбора характеристической температуры. Если принцип соот-
ветственных упруговязкпх состояний рассматривать как качест-
венный принцип, то или Т„ пли Г, могут быть пригодны одинако-
во дтя этой дети, так как характеристическая температура, взя-
тая тем или иным образом, будет отличаться только на несколько
7. Кривые температурной мв:итт чотти моду |я
87
градусов. Для получения более точных результатов должны быть
рассмотрены другие пути выбора характеристической температу-
ры, как это и будет сделано в главе IV. Однако для точного коли-
чественного рассмотрения применимости этого принципа необхо-
димо иметь в виду такие факторы, как жесткость цепи, подвиж-
ность боковых групп и т. д, которые вызывают существенные от-
клонения 01 принципа соответствен пых температур.
Основным содержанием этого закона соответственных темпе-
ратур является то, что два аморфных полимера могут считаться
механически «эквивалентными» для практических целей, если они
имеют одинаковые Tg и одинаковые длины молекулярных цепей.
Бутадиен-стирольные сополимеры применяются почти во всей
области практического использования аморфных полимеров. На
одном конце шкалы находится вулканизованный полибутадпеи с
Tg=—85 "С, используемый в качестве морозостойкой резины, в
частности, в Арктике п Антарктике. Вулканизованный 75/25 со-
полимер с 75 вес. ч. бутадиена и 25 вес. ч. стирола в настоящее вре-
мя является в США наиболее широко используемой резиной для различ-
ных целей. Она имеет величину Tg=—57 С. Сополимер 30/70 с 7^=
18 С применяется в эмульсиях (с добавлением пигмента) в ка-
честве красок на водной основе или (без пигмента) в качестве
грунтовки для различных поверхностей. При комнатной
температуре он образует тягучую гибкую пленку. Сополи-
мер 10/90 с 7^68 СС представляет собой твердую стеклообразную
пластмассу, используемую как формуемый термопластичный ма-
териал для получения листов п многочисленных формуемых изде-
лий. Хотя это и твердый полимер, однако он обладает некоторой сте-
пенью внутренней гибкости, на что указывает умеренная величина
прочности на удар. Атактический полистирол, с Т^--100 °C, так-
же используется как формуемый термопластичный материал. Полу-
чается твердый чисто стеклообразный пластик с отличными элект-
рическими свойствами, но весьма нестойкий к ударам. В добавле-
ние к этому отметим, что различные смеси этих сополимеров про-
изводятся н продаются, как будет сказано об этом далее.
Следует указать, что величина Т„ для бутадиен-стирольных
сополимеров подчиняется эмпирическому уравнению (7, 1) луч-
ше, чем обычно применяемому эмпирическому уравнению (5, 3):
Г„ = а1Г„,| (7,1)
где А'1 и х2—молярные доли стирола и бутадиена в сополимере,
7\л 11 Т'чл—значения Т„ для полистирола и полпбутадиена.
С точки зрения принципа механической эквивалентности поли-
меров при соответственных температурах, не удивительно, что
два других главных каучука, используемых в шинной промыт-
88
Гл.
II.
Основы
физики полимеров
ленностп—натуральный каучук п бутилкаучук имеют 7’„, весьма
близкую к сополимеру 75,25. Практически применяемые латекс-
ные краски па базе сополимеров этплакрплата п метилметакрила-
та, с одной стороны, и винилацетата и дпбутплфу.марата, с другой
стороны, имеют величину Тц, близкую к сополимеру 30 70 бутади-
ена и стирола, а именно 18 С. Полпметилметакрнлат с 7'Ё—105~С
очень часто применяется как и полистирол с Т.~ 100'С. Чтобы
получить продукт, который механически подобен 10 90 бута-
диен-стирольному сополимеру, метилметакрилат сополимер изует-
ся с подходящим количеством этплакрплата плп бутплметакрп-
лата с тем, чтобы получить сополимер с Tg^68 С.
По своему смыслу принцип соответственных упруговязкпх
состояний аморфных полимеров ограничивает механические при-
менения этих материалов. Во многих случаях желательно иметь
материал с высоким модулем, а также с весьма высокой ударной
прочностью. Аморфные полимеры проявляют эти свойства в тем-
пературном интервале между 7^ п Г„~г20 °C. Выше 7'£д-20 мо-
дуль становится слишком низким; ниже Т£ полимер хрупок п
имеет низкую ударную прочность.
Для того чтобы расширить температурную область высоко-
модульных п высокоударопрочных свойств, часто прибегают к
смешиванию аморфных полимеров 131. Надлежащее смешение мо-
жет создать полимерный материал, кривая температурной зависи-
мости модуля которого обнаруживает наличие двух переходных
областей. Если два полимера полностью совместимы, то смесь
имеет свойства обычного аморфного полимера с единственной
переходной областью п с промежуточной температурой стекло-
вания и подчиняется закону соответственных упруговязкпх со-
стояний. Если полимеры, подвергаемые смешению, полностью
несовместимы, то они не смачивают друг друга и ио существу не
могут смешаться даже в виде грубой гомогенной среды. Наплуч-
шпй эффект достигается, если полимеры находятся па границе сов-
местимости-несовместимости, так что система образует две
тонко раздробленные непрерывные фазы, причем одна фаза кау-
чукоподобная (при комнатной температуре), другая—твердая.
Смешанные друг с другом полимеры широко используются для
изготовления подметок для обуви. В этом случае применяется
смесь на основе двух частей 75/25 бутадиен-стирольного сополи-
мера н одной части 10/90 бутадиен-стирольного сополимера. Дру-
гая широко используемая смесь состоит из одной части 75 25
сополимера и трех частей полистирола. Опа оказывается почти
той же самой твердости, что п полистирол, по обладает хорошим
сопротивлением удару в результате присутствия каучукоподобной
фазы. Такие же физические характеристики могут также дости-
гаться в блок- и привитых сополимерах. Блоксополимеры имеют
7. Кривые температурной зависимости модули
89
в цени большие последовательности А единиц н большие после-
довательности В единиц, расположенных друг за другом. Приви-
тые сополимеры содержат длинные цепи из В единиц в виде боко-
вых ответвлений от главной цепи, состоящей из А единиц.
Кривые температурной зависимости модуля для смесей поли-
меров приведены на рис. 11,24. Полимерами, образующими смеси,
в этом случае являлись полистирол и 30 '70 бутадиен-стирольный
сополимер.
Очень широкая температурная область, в которой сосущест-
вуют высокие значения модуля и прочности па удар, встречается
Рис. 11,24. Температурная зависимость
Ег (10) для смесей полистирола и 30/70 бута-
диен-стирольного сополимера. Цифры на
кривых—содержание полистирола в смеси
в вес. %.
в кристаллических полимерах в очень широкой области темпера-
тур, лежащих между Tg и Тт. Для линейного полиэтилена Тт
равна 137 °C, a Tg приблизительно равна —85 °C (эта последняя
цифра несколько дискуссионна)*. В этой относительно большой
температурной области линейный полиэтилен имеет высокую твер-
дость (высокий модуль) п хорошую ударную прочность. Для по-
лиэтилена с низкой плотностью (три разветвления на 100 угле-
родных атомов) соответствующая температурная область лежит
от —85 до ПО °C, но при комнатной температуре и вблизи нее
разветвленный полиэтилен менее тверд, чем линейный полиэтилен,
* См. Приложение К.
9.)
Гл. II. Основы физики по.ш.иероя
так как он менее кристаллпчен. Между 1g модуля н степенью кри-
сталличности полиэтилена имеется эмпирически найденная ли-
нейная связь в определенной области кристалличности.
Изотактический полистирол, полученный в кристаллическом
состоянии, имеет Г —100 'С п Т,„ около 250 °C. В этой температур-
ной области кристаллический изотактический полистирол обла-
дает желаемым сочетанием высокой твердости и хорошей сопро-
тивляемостью удару. Кривая £Д10) в зависимости от Т для кри-
сталлического изотактического полистирола приведена на
рис. 11, 20 для сравнения с атактическим линейным и атактиче-
ским структурированным полистиролом (оба, разумеется, аморфны).
Интересно отметить поведение £,(!()) в интервале между Т.,
п 7',,, для кристаллического полимера. Детальная форма кривой
зависит от тепловой истории образца, в частности от того, каким
способом он охлажден из расплавленного состояния. При пре-
дельно больших скоростях охлаждения пз расплава изотактический
полистирол может быть получен в полностью аморфном состоя-
нии. В этом случае он имеет ту же самую кривую температурной
зависимости модуля и ту же самую структурную рентгенограмму,
как н атактический полистирол.
Два кристаллических полимера механически «эквивалентны»
для практических целей, если они имеют те же самые величины
Г„ и Тт, одинаковые длины цепи, равные степени кристалличности
п одинаковую кристаллическую текстуру. Такое совпадение, ка-
залось бы, должно быть очень редким. Между тем, если изучать
имеющиеся кристаллические полимеры, которые используются
с одинаковым успехом в тех же механических условиях, будь
то пленка, волокно или литье, то, к удивлению, можно заметить,
что часто эта «эквивалентность» структурных свойств приближен-
но наблюдается.
Литература
1. А.
2. А.
3. R.
V. Tobolsk у,
V. Tobolsk у,
А. М. Border s,
В и с h d а 11 I, L.
Е. С a t s i f f, J. Am. Chem. Soc., 76, 4204 (1952).
Riibb. Chem. Technol., 30, 437—438 (1957); s. a.
R. D. J и v e, Ind. Eng. Chem., 38, n(>6 (1946).
Nielsen, J. Polymer Sci., 15. 1 (1955).
8. Вязкое течение линейных аморфных полимеров [ 11
В ряду линейных полимер гомологов с возрастанием молеку-
лярного веса обнаруживается огромное увеличение вязкости.
Это наиболее ярко проявляется у полппзобутплепа, который мо-
жет быть получен в обширной области молекулярных весов. При
комнатной температуре дппзобутплен представляет собой весьма
подвижную жидкость, вязкость которой имеет порядок величины
8. Вязкое течение линейных аморфных полимеров
91
ИГ2 пуаз. Если продвигаться вверх до молекулярных весов по-
рядка 103, 105, 107, то при этом встречаются соответственно вяз-
кие жидкости, клейкие каучуки и, наконец, каучукоподобные ма-
териалы с вязкостью, превышающей 1013 пуаз, которые ведут себя
при комнатной температуре практически таким образом, как
если бы они были вулканизованы.
Было найдено, что вязкость линейных полимер гомологов мо-
жет быть выражена в следующих эквивалентных формах:
1й7(=Ф*(Т)-|-Е(Л')>Е>* (8,1)
(8,2)
где Ф*(Т)—функция только температуры, F(N’)—функция толь-
ко длины цени п £)*—константа (причина того, почему D* не
включена в Ф*(Т), будет вскоре выяснена).
Тот факт, что вязкость может быть выражена как произведе-
ние члена, зависящего от температуры, и члена, зависящего от
длины цепи, приводит к выводу, что течение полимерной моле-
кулы происходит путем активационного диффузионного перескока
полимерных сегментов. Этот сегментальный перескок является
общим для всех членов полпмергомологпческого ряда и объясняет
наличие общего температурного фактора для цепей всех размеров.
Фактор, учитывающий зависимость от длины цепи, показывает,
что для того, чтобы имело место течение, необходима координи-
рованная последовательность сегментальных перескоков. Эта ко-
ординация становится все более трудной по мере увеличения дли-
ны цепи. Если длина цепи настолько велика, что происходит
перепутывание цепей, то эта координация становится действи-
тельно очень трудной, так как теперь в нее включается и совмест-
ное движение сегментов различных цепей.
График 1g т, от IgA' показывает, что для существенно больших
длин цепей (;V>500) наблюдается прямая линия с наклоном, рав-
ным 3,4. Для малых величин /V наклон примерно равен 1. Длина
цепи, при которой кривая вязкости приближается к наклону,
равному 3,4, по [2] есть длина цепи, при которой образование
сетки вследствие перепутанности цепей становится заметным.
Следовательно, для достаточно больших величин N:
F (N) = 3,4 lg N (Л/>500) (8,3)
Для того чтобы удобнее использовать закон соответственных
упруговязкпх состояний, рассмотренный в предыдущем парагра-
фе, желательно выразить температурную зависимость ц через
Т—Тс или Т, 7\, где Тс—некоторая характеристическая темпера-
тура для каждого полимера. Если выразить зависимость в одной
92
Гл. II. Основы (разики полимеров
из этих форм, то уравнение (8, 1) примет вид (для полимеров с
достаточно большим Д'):
lgT1==<]>(T — Тс) л. 3,4 IgA' + D* (8,4)
Весьма подходящее выражение для Ф(Т—Тс) было предло-
жено Впллпамсом, Лапделом и Ферри 131 с использованием Т„
в качестве характеристической температуры полимера. Авторы
использовали для этой цели уравнение Дулитла 1(8, 11), глава I):
In = In А -- В ~ (8, 5)
Так как полимеры вообще являются сильно переохлажден-
ными жидкостями (вспомним, что 0.5Т„;<Те<0,67 Т,п для кри-
сталлических полимеров), то величина по может быть заменена
п—удельным объемом полимера. Отсюда уравнение (8, 5) мо-
жет быть выражено следующим образом при температурах Т
11 Те:
1П7ДТ) = 1пД + В-^П- (8,6)
•7 U)
In 7 (Г) = lnA + B^L (8,7)
’ЩВычптая уравнение (8, 7) из уравнения (8, 6) и полагая В
равным единице, что по порядку величины наблюдается иа опыте,
получим:
1g = _______Li]
g ДТД 2.303 ( f
f _ U/(T) . c __ Vf (Tg) (
' a(T) ’ 's v(Tg) j
(8, 8)
В уравнении (8, 8) величина f есть доля свободного объема
при температуре Т. а величина f„ есть доля свободного объема
при Т„.
Зависимость свободного объема от температуры связана с
различием коэффициентов теплового расширения а, п а„ выше
и ниже температуры стеклования. Отсюда:
f = f + (ar - о J (Т - Т) = + п, (T - Tg)
(8,9)
8. Вязкое течение линейных аморфных полимеров
93
то полу-
(8, 10)
полпме-
следую-
(8, И)
Если уравнение (8, 9) подставить в уравнение (8, 8),
чнм.
1Р <> (Т) ____1 Т--Т,
ё д(7„) 2,303/g ’ T - Tg
Опытные данные по многочисленным органическим
рам п некоторым неорганическим стеклам приводят к
тему приближенному эмпирическому уравнению 121:
и (Г) = -17.44(7'7а,)
Tt(Tg) 51.6-j-Г — 7'^
Приравнивая соответствующие коноапты уравнении (8, 10) и
(8, 11), получим:
7^ = 0,025; а2 = аг — п^ = 4,8-10 4 грат-1 (8,12)
Здесь отчетливо видно, что температура стеклования различ-
ных полимеров соответствует состоянию с постоянной долей
свободного объема; величина а2 находится в согласии с экспе-
риментальными данными по многочисленным полимерам.
Лучшая эмпирическая формула, выражающая изменение с
температурой, получена в результате замены в уравнении (8, 4)
величины Тс на новую характеристическую температуру Ts. Ве-
личина Ts для полипзобутилена произвольно выбрана равной
243 СК. Величины Ts для других полимеров были затем выбраны
произвольно, но так, чтобы Ф(Т—Ts) в уравнении (8, 4) оказа-
лась универсальной функцией для всех полимеров. Найдено, что
хорошим приближением для Ф(Т—7\) является 121:
Ф(Т — Ts)
_ — 8.86 (Т — Ts)
101,6-р Г — 7\
(8, 13)
Значения величии Ts для некоторых полимеров приведены в
табл. 11,5. Величина Ts примерно на 50 °C выше температуры Tg
полимера. Вероятно, Ts—это температура, при которой данный
полимер имеет тот же самый относительный свободный объем, как
и полпизобутилен при 243 °К.
Эмпирическая формула для вязкости полимеров с весьма
большими цепями имеет вид*:
lg -fl (Т) = 3,4 lg N
8.86 (Г -7\)
101.6 -Г —Г,
(8, 14)
* Следует заметить, что еще в 1948—1949 гг. советскими авторами была
предложена эмпирически найденная для полипзобутилена формула, связываю-
щая молекулярный вег линейиэто аморфного полимера с разностью темпера-
94
Гл. If. Основы физики полимеров
Т а 6 лица II, 5
Величины 7\ для некоторых полимеров-1
Полимер г., '-К А 7,„
Полннзосл силен . . 243 2( >2
I Кт.тиметн.такрнлат 378 324
11олив1111П.'1ацетат . . 349 301
11олистирол 408 373
Полпметпл.метакрплат 433 378
Поливинилацеталь .... 380 —
Бу гадиен-стпрол:
75 '25 268 216
60 40 283 235
50'50 296 252
30’70 328 291
‘ ' T.ti. Г х, S- <1 rate h. S. L о s h a e k, ih. Г’ in Rhe >Iogy: The ry md Appli.-.itions,
vol I. Г. R.Eiri.li, (ed.). A.aduinc Pre^s, New York, 1936 [ Г. Фокс, С. Гратч, С. До-
шек. гл. 12 в an. «Реология: теория и применением, под ред. Ф. ЭПрнха. 1 Ьдатннлпт. 1462].
Для того чтобы получить хорошее совпадение с эксперимен-
тальными данными, необходимо допустить, чтобы «константа» D
слабо зависела от температуры, особенно для Т>Т„\ 100".
Пока мы не учитывали влияние полпднсперсностн, т. е. форму-
лы предполагались пригодными для полимеров, состоящих из
молекул с одинаковой длиной цепи N. В общем случае полимер
состоит из молекул с различными длинами иепей. Если весовая
доля молекул с длиной цепи / равна тс,-, а число их равно Nit
то средневесовая длина цепи определяется как
Л'и, = У кд,- = ----- (8, 15)
“* ' i Ni
тур текучести 11 стеклования {В. А. Каргин, Т. И. С о г о л о в а, ЖФХ,
536 (1949); В. А. К а р г и н, Г. Л. С л о и и м с к и й, ДАН, 62. 242 (1948)
ЖФХ. 23, 569 (1949)1:
В (Tf - - ГН
где 11с, В н С—постоянные, характерные для данного полнмергомологического
ряда. В тех же работах эта формула была получена и с ьо.мощыо расчета меха-
нической модели полимерного тела.
Тесная связь этого соотношения с полученными позднее более общими
формулами (8, 11) и (8, 14) этого раздела, а также с формулой (4, 2) в главе IV
очевидна.—Прим. ред.
о. Вязкое течение линейных аморфных полимеров 95
Другая обычно применяемая средняя длина цепи есть средне-
числовая N„, определимая как
V ЛУ,
^,, = 1'*,.= ^— (8,16)
1 -V,
где
есть молярная доля /-мера.
На опыте было найдено, что формулы этого раздела, предло-
женные для моноднсперсного полимера, применимы и для полн-
дпсперсного полимера, если N заменить па ЛП,. Отсюда формула
(8, 14) может быть записана для полиднсперсного полимера сле-
дующим образом:
1g т) = 3,4 ]g<,- + D (8,17)
Эмпирические формулы, выражающие температурные зависи-
мости вязкости многочисленных полимеров в более или менее
ясной форме, приведены в монографии 111.
Величина D в уравнении (8, 17) значительно различается при
переходе от одного полимера к другому. Это, пожалуй, нанлуч-
шпм образом иллюстрируется при сравнении вязкости расплава
полиэтилена и расплава полипропилена. Хотя величина Tg по-
лиэтилена значительно ниже, чем полипропилена, п, следователь-
но, величина Ts, без сомнения, также ниже, вязкость полиэтиле-
на выше, чем полипропилена при той же самой температуре и
при той же самой длине цепи. Это происходит, вероятно, потому,
что полиэтиленовая цепь является более жесткой, чем полипропи-
леновая цепь.
По предположению автора для вязкости полимеров с длин-
ными цепями может быть применимо следующее уравнение:
+ в (^8>
1 С) v I , U I 1 о
В уравнении (8, 18) г- есть средний квадрат длины полимерной
цепи (определенный методом светорассеяния пли измерением харак-
теристической вязкости в подходящем растворителе), а /„—длина
связи. Величина 3 может изменяться между 3,0 и 3,4 в зависимости
от того, произведены ли измерения г'- в хорошем плп плохом рас-
УЬ
Гл. If. Основы физики полимерии
творптеле. Предполагается, что величина В в уравнении (8, 18)
возможно является более постоянной величиной для различных
полимеров, нежели величина D в уравнении (8 17).
Уравнение (8, 18) может быть, очевидно, выражено через Ts
так же, как и через Tg.
Литература
1. Т. G. F ox, S. Grate h, S. l.o shae k, ch. 12 in Rheology: Theory
ami Applications, F. R. Eirich (ed.), Academic Press, New York, 1-956.
[Г. Фо к с, С. Г р а т ч, С. Л о ш е к, гл. 12 в ни. «Реология: тео-
рия н применение», под ред. Ф. Эйрпха, Издатпнлпт, 1962].
2. F. В пес h е, J. Cheni. Phys., 20, 1959 (1952).
3. At. L. \V i 1 1 i a in s, R. F. L a n d e 1, J. D. F e r r y, J. Am. Chem,
See., 77, 3701 (1955).
9. Кинетическая теория высокоэластичности
Соображения о напряжении в растянутом образце резины как
о величине, возникающей в результате теплового движения макро-
молекул, были рассмотрены в главе I, разделы 7 и 8. Расчет та-
кой кинетической силы на основе молекулярной модели сетки
резины может рассматриваться как одно из важнейших достиже-
ний науки о полимерах. Было сделано много ценных теоретиче-
ских разработок этой проблемы, каждая пз которых вносила но-
вое понимание. Среди крупных исследователей, внесших вклад
в эту проблему, были Кун, Гут, Марк, Джемс, Уолл, Трелоар и
Флори*. История этих исследований и различные теоретические
выводы даны в [ 1J—131
В этом разделе представлены некоторые расчеты кинетической
силы 14, 51, математически вполне элементарные, по внимание в
которых концентрируется на неполностью решенной проблеме
высокоэластичности.
Для углеродной цепочки из N звеньев, у которой /-тая связь
может быть в трех положениях относительно j— 1-связп и /—2-
связи (одна транс- и две гош-конфнгурашш), полное число кон-
формаций равно:
<|> = 34--’ (9,1)
В приложении Д 1уравнение (Д, 9)1 выведено следующее выра-
жение для нормированного распределения конформаций поли-
* А также советские исследователи Я- II- Френкель, С. Е. Бреслер,
М. В. Волькепштепп п др.—Прим. ред.
9. Кинетическая теория высокоэластичности
97
мерной цепи в свободном пространстве (г—вектор между конца-
ми цепи):
со (г) = ~^=) ех9 ~ Ь2'2}
(9, 2)
3
Для идеализированной углеродной цепочки, имеющей все
три положения относительно связи С- С с одинаковой энергией,
гг=2Л7о‘, отсюда /г З/4Л7о.
Число конформаций углеродной цепочки в свободном прост-
ранстве, характеризующихся вектором между концами цепи г.
равно:
<0 (Г) 3Л'"2 (
exp (— Ь2г2)
(9, 3)
В уравнении (9, 3) предполагается, что все величины г воз-
можны. Более общее выражение имеет вид:
с,- (г) —с ехр (
(9. 4)
Уравнение (9, 4) не обязательно ограничивается углеродной
цепочкой, оно может быть применено к любому типу полимерной
цепи со своими значениями b и с. В уравнении (9, 4) можно также
считать, что константа с имеет свойства нормировочной постоян-
ной, даже если допустить, что г принимает отдельные дискретные
значения (последнее верно, например, если представить углерод-
ную цепочку как некоторую физически эквивалентную цепочку,
полученную путем произвольного обхода решетки алмаза, что
рассмотрено в Приложении Г).
Рассмотрим пространственную каучуковую сетку, состоящую
из Nc цепей сетки, в которой: 1) узлы сетки относительно фик-
сированы, но имеется сравнительно свободное движение отрез-
ков молекулярных цепей, соединенных между собой узлами сет-
ки; 2) внутренняя энергия и объем сетки не изменяются при дефор-
мации (нет сжимаемости); 3) напряжения, возникающие при дан-
ной деформации, являются результатом одного только изменения
конформационной энтропии.
Далее предположим, что сетка в недеформированном состоя-
нии задается Nc сеточными векторами г,, где ri—вектор между
узлами сетки, с которыми связана z-тая цепь сетки.
7—833
98
Гл. II. Основы физики полимеров
Предположим, что число конформаций t-той цепи сетки
определяется выражением:
ы (/-,) = с, exp (—bi rj) (9, 5)
где ct зависит от bt, ио ие зависит от г(. Величина Ь? (равна
З/4/V/o для идеализированной углеродной цепочки) должна
зависеть от длины цепи между узлами сетки.
Полное число В конформаций в иедеформировапном состоянии
равно:
-
- = II w (гр (9, 6)
i = 1
Согласно принципу Больцмана конформационная энтропия
сетки равна-
-Д Nc
Sc = k In 9! = k V In ct — k V b'i ц (9, 7)
i=l t=i
Свободная энергия Гельмгольца .-4 сетки в недеформнрованном
состоянии равна:
-vc Nc
A(T,V)=Ati[T,V) -kT V Inc,+ kT У bfZ (9,8)
i=l i=l
где АЬ(Т, V) есть свободная энергия сетки, исключая конформа-
ционную свободную энергию. Она включает внутреннюю энергию,
колебательную энергию и энтропию и т. д.
Для того чтобы получить действительные значения г, в ие-
деформированном состоянии, необходимо найти минимум .4(7, Г)
относительно всех вариаций г;, которые не приводят к разрыву
любого из узлов сетки. Ясно, что знание только одной величины
l>i недостаточно для определения этих расстояний и что детальное
знание Ао(Т, V) также необходимо. Между тем ниже станет оче-
видным, что для того, чтобы получить уравнение состояния,
необходимо знать только Nc и среднее значение bjr'j в недеформн-
рованном состоянии, если оно изотропно.
В деформированном состоянии свободная энергия Гельмгольца
.4' равна:
Nc Nc
А' (Т, Г) = Л„ (7, V) - kT У In ci + kT £ (7i)2 (9,9)
i ~ 1 i = I
9. Кинетическая теория иысокоэлистичпосги
99
где /'—вектор между узлами сетки, с которыми связана /-тая
цепь сетки в деформированном состоянии.
Согласно предположению 2 (см. стр. 97), А0(Т, У) и объем V
не изменяются при любых возможных деформациях.
Рассмотрим простой случаи растяжения, когда прямоуголь-
ная полоска резины растянута от длины Lo до длины L в направ-
Рис. 11,25. Афинное преобразование сеточных век-
торов при растяжении сетки:
а—нерастянутая; <5- растянутая ceiua.
леннн оси х. Все размеры х, у, г образца изменяются и принимают
новые размеры х', у1, z следующим образом:
х' = ах
у' = (9, 10)
। “
г
Z = ---
г'«
/.
где а = Г.
‘-о
Здесь предполагается, что координаты х, у, г, описывающие
положение одного конца цепи сетки относительно другого (ко-
торый берется в качестве начала координат), преобразовываются
в процессе растяжения в к', у , z', как это следует из уравнения
(9, 10). Это происходит вследствие того, что узлы сетки предпо-
лагаются относительно фиксированными точками. На рис. II, 25
7*
100
Гл. II. Основы физики полимеров
показано, как сферически симметричный набор сеточных векторов
в нерастянутом состоянии преобразуется соотношениями (9, 10)
в набор векторов, обладающий эллиптической симметрией.
Сеточные векторы г{ в недеформированном состоянии распре-
делены сферически симметричным способом вследствие того, что
образец является изотропным в нерастянутом состоянии. Если
сеточные векторы rt в недеформированном состоянии отнести к
общему началу, то векторы с одинаковой величиной г и тон нее
самой величиной Ь, образуют сферическую поверхность. Такая
сферическая поверхность радиуса г при применении преобразо-
ваний (9, 10) переходит в эллипсоид с осями аг, г!\ а, г!\ а.
В этом случае легко показать, что среднее значение (г')2 по всей
поверхности эллипсоида равно (см. Приложение ЖУ-
(Г)2= + (9,11)
(Отсюда
Nc ___
V bl == £ Г1 a4 '2/ i = ( у2 + 1} (9,12)
£-1 <=1
где Ь"г2 есть среднее значение по всем цепям сетки. Подставляя
уравнение (9, 12) в уравнение (9,9). найдем:
Д’ (Т, В) = А0(Г, V) -kT V inc. 4- kTNc-^-[o2 +-|-| (9, 13)
i -=I
Растягивающая сила X находится из уравнения
Х==(ттЧ (9, 14)
\ <iL L„ \ дз ! у, ।
Сила, отнесенная к единице площади начального поперечного
сечения Ло, равна:
Е-Х/Д, (9,15)
Комбинируя последние три уравнения, получим:
F - NtkT | у.
19, 16)
где N1=NCIV—число цепей сетки в единице объема.
9. Кинетическая теория высокоэластичности
101
Более детальное рассмотрение величин 2(62г2)/3 дано в форму-
ле, приведенной ниже:
2 3 ~ 3 “ D‘ z Nr
(9, 17)
суммируется
ПО ШеМ
величинам
суммируется
по вг ем кон-
формациям / для
дашюго
Если цепи сетки в ненапряженном состоянии сетки имеют то
же самое конформационное распределение, что и аналогичный
набор молекулярных цепей в свободном пространстве, то тогда
сопоставление уравнении (9, 17) и (9, 2) показывает, что
2(/>-z’’)/3 равно единице. В этом случае (приближенный случай сво-
бодных цепей) уравнение (9, 16) сводится к уравнению (7, 6)
главы I. Это то уравнение, которое действительно получено в
большинстве работ по кинетической теории высокоэластичности
11, 2, 31.
Для реальной сетки распределение расстояний rt не обязатель-
но подчиняется уравнению (9, 2).
Как упомянуто ранее, действительные растояния rt в недефор-
мированном состоянии сетки можно получить только нахожде-
нием минимума А(Т, Е) в уравнении (9, 8) относительно всех ва-
риаций г,-, которые не приводят к разрыву узлов сетки, так что
знания 6,- недостаточно для определения г,. Если это утверждение
верно, то величина 2(Л2г'-)/3 не обязательно равна единице при
всех условиях, а широко распространенное 11—31 в настоящее
время уравнение (7, 6) главы I справедливо только в особых
условиях. Эта критическая точка зрения разделялась автором
уже давно 14, 51. К этим же выводам пришли Джемс и Гут путем
совершенно иных теоретических соображений [6].
Величина 2(7?2г2)/3 фактически будет зависеть от детальных
условий процесса поперечного сшивания, т. е. зависит от того,
находится ли сетка в деформированном пли отрелаксированном
состоянии во время образования поперечных связей; вводятся
ли поперечные связи в присутствии растворителя, который может
или же не может затем испариться; находился ли полимер в ча-
стично кристаллическом состоянии в процессе сшивания.
Для аморфной сетки, сшитой в условиях полностью отрелак-
сированной системы, 2(№/2)/3 может быть величиной, близкой к
единице, если плотность поперечного сшивания низка При очень
больших плотностях поперечного сшивания необычные стериче-
ские условия изменяют ситуацию, так как из каждого узла исходят
102
Гл II. Основы физика полимеров
четыре объемных полимерных цепи. В данном случае может
быть, что ближайшая поперечная связь должна быть также смеж-
ной поперечной связью сетки, т. е. взаимное проникновение эле-
ментов сетки становится невозможным. Тогда 2(/гг2)/3 не может
быть равно единице.
Чтобы оценить величину 2(/>2г2)/3 сеточного полимера при по-
мощи других приближений, нежели приближение для случая
свободной цени, требуются некоторые дальнейшие предположения.
Допустимо, что можно заменить все b'j на Ь~, где Ь2 (равное 3/4 Л7~
для идеализированной углеводородной цепочки) соответствует
среднему числу N звеньев цени, находящихся между уз-
лами сетки. Величина /V, как известно, легко рассчитывается пз
плотности поперечных связей. В этом случае уравнение (9, 17i
упрощается следующим образом (этот простой результат был ис-
пользован в 1944 г.) 151:
3 3 >- (в свободном пространстве) ’
Рассмотрим теперь весьма важную проблему связи между
концентрацией поперечных связей в сетке п концентрацией цепей
сетки. Предположим, что сетка образована из исходных бесконеч-
но длинных линейных молекул путем введения с молен четырех-
фуикциональных поперечных связей в каждый 1 с.н3 каучука
Из каждой поперечной связи исходят четыре цепи сетки, по каж-
дая цепь сеткн принадлежит двум поперечным связям. Поэтом)
имеется 2с молей цепей сетки в каждом 1 см3 попречно-сшнтоп сет-
ки. Напомним определение Л4Г как среднечпслового молекуляр-
ного веса цепей сетки. Отсюда d!Mc равно п—числу молей цепей
сетки в кубическом сантиметре каучука:
« =-лг-= Тиу = 2с (9,19)
Если линейные молекулярные цепи имеют конечный средне-
числовой молекулярный вес /И,г до сшивания, то сетка, образован-
ная с молями поперечных связей в кубическом сантиметре кау-
чука, будет содержать некоторое количество концевых цепей,
г. е. цепей, которые идут от поперечной связи к концу молекулы,
а не к другой поперечной связи. Такая сетка является несовер-
шенной сеткой, и не все цепи сетки поэтому будут вносить свой
вклад в напряжение.
С целью решения этой проблемы Флори [71 предположил,
чго вначале используется некоторое количество поперечных сши-
9. Кинетическая теория высокоэласшчности
103
вок, связывающих все линейные молекулы конечных размеров
в одну «бесконечную макромолекулу». Так как имеется d/Mn
молей исходных молекул в 1 см3 линейного полимера, то в 1 см3
должно быть (ЦМп молей поперечных связей (более точно
d/Mn—1/Л/л), чтобы образовать одну «бесконечную макромолеку-
лу». Такая бесконечная молекула еще не является сеткой, спо-
собной поддерживать напряжение. Только те поперечные связи,
которые возникают после этого, образуют истинную сетку. Сле-
довательно, число молей этих эффективных поперечных связей
в 1 см3 равно:
ce = c — d/M., (9,20)
По тем же причинам, на основе которых было выведено урав-
нение (9, 19), в сетке на две эффективные цепи приходится одна
эффективная поперечная связь:
пе — 2с — 2d/ М,
(9,21)
Исходя из определения Мс*:
J
Мс
= 2с
(9, 22)
получим после подстановки пе вместо п в уравнение (7, 6) гла-
вы 1:
F-_.±RTJ\ 2 ме \1 £
Мс I, /[ Ц ~ । L ) |
(9, 23)
Из уравнения (9, 23) следует, что когда Мп =2Mc=dfc, истин-
ной сеточной структуры не образуется и равновесный модуль сет-
ки равен нулю. Тем не менее поправочный множитель 1—2Л/С/И„
хотя и широко применяется 111, является, по мнению автора,
теоретически неверный, если Лф,ЛИ£.^>1.
Представим себе сетку (или плотность поперечного сшивания,
характеризуемую Л1с), которая образована из бесконечно длин-
ных молекул. Далее представим, что вводится небольшое число
* Следует иметь в виду, что под М6. по-прежнему понимается молекулярный
вес пепей бездефектной сетки, образованной исходным полимером с бесконечно
большим молекулярным несом, причем d—плотность этого полимера. Молеку-
лярный вес /Иг эффективной цепи н реальной сетке, образованной исходным
полимером с конечным молекулярным весом \1п. в общем случае не равен /И,.
Например, при Л1Я=2МГ молекулярный вес пени реальной сетки М’г— х>, а
для достаточно густой сетки ,\1п' Л1е приближенно можно полагать, что Л1'схМе.
Это допущение использовано А, Тобольским при выводе формулы (9, 24).—
Прим. ред.
10-1
Гл. И. Основы физика полимеров
случайно распределенных разрезов (q молен на 1 см3). Каждый
разрез приводит к образованию двух новых концов цепи. Эта сет-
ка должна быть поставлена в соответствие с сеткой (той же самой
плотности поперечного сшивания Л/J, образованной случайно
распределенными поперечными связями в полимере из молекул
с конечным среднечисловым молекулярным весом Л1„.
Соответствие будет количественным, если q=--dli\ln, так как
в этом случае обе сетки имеют то же самое число концов цепей.
Между тем для малого числа разрывов каждый разрез умень-
шает число эффективных цепей на одну. Поэтому число молей
эффективных цепей в 1 см3 равно dlMc—q=dlMc—d!Mn. Уравне-
ние состояния для этого приближения будет иметь вид:
р - dRT (1____\ I _ (Lu \ “ 1 /п 24»
Л1е \ ’ X ) | Lo \1.) I 1 ’
приближение, верное для Мп/Мс'^- 1
Поправочный фактор 1—2 Л'1с/Л1„ является, вероятно, точным
для Л4П/Л1^2,О, в то время как поправочный фактор 1—MjMn
является точным, вероятно, для Л4П/Л1СХ> 1 Оба фактора дают пра-
вильное выражение для Л1п/Л1с->со. Точная теория, в которую
должны будут войти оба эти выражения (как предельные формулы),
еще не развита.
Справедливость уравнения (9, 23) может быть проверена экспе-
риментально. Величина Мп исходного несшитого полимера может
быть определена измерением осмотического давления в разбав-
ленном растворе. Число молей с сшивающего агента может кон-
тролироваться, если возможно найти сшивающий агент, который
реагирует количественно с линейным полимером. Зная Л1П и Мс,
где Alc=d/2c, можно определить напряжение при любых величи-
нах L!L0 по уравнению (9, 23).
В тех редких случаях, когда проверялась правильность мно-
жителя 1—2MJMn, было получено очень хорошее согласие между
теорией и экспериментом [81, хотя возможные отклонения при
больших значениях Мп1Мс практически не были проверены.
Определение абсолютных значений F из и Л1П и L/Lo дает
разные результаты для различных резин. Экспериментальные
значения F постоянно стремятся превысить теоретические значе-
ния, даваемые уравнением (9, 23), со средним отклонением около
50%. Зацепления цепей, находящиеся между постоянными попе-
речными химическими связями, могут рассматриваться как воз-
можное объяснение этого отклонения 171. Возможно, величина
2(й2г2)/3, рассмотренная ранее в этом разделе, позволит найти иное
объяснение этого отклонения.
Успех кинетической теории высокоэластичности, без сомне-
ния, замечателен. Уравнение (9, 23) может применяться с боль-
у. Кинетическая теория высокоэласт ичносги
105
шпм успехом для расчета (приближенного) числа молей попереч-
ных связей в 1 сма каучука при помощи простой процедуры изме-
рения величины F, необходимой для поддержания в образце ре-
зины (с начальной длиной Lo) состояния растяжения L. Так как
большинство каучуковых сеток образуется в результате химиче-
ского процесса сшивания, который количественно не контроли-
руется, то эти уравнения дают очень важный метод характеристи-
ки каучуковых сеток*.
Если поперечные связи образуются посредством ионизирую-
щей радиации, такой метод определения с (или Л4С) является един-
ственным.
Кинетическая теория высокоэластичности, рассмотренная в
этом разделе, основана на предположении, что все конформации
полимерной цепи имеют равные энергии. Это предположение спра-
ведливо для многих типов полимерных цепей. Тем не менее опре-
деленные полимерные цепи имеют заметную тенденцию к преиму-
щественно вытянутым конформациям, в то время как другие обна-
руживают тенденцию к преимущественно свернутым конформа-
циям. В этих случаях нельзя уже допускать, что внутренняя энер-
гия изолированной цели не зависит от расстояния между ее кон-
цами или что внутренняя энергия каучуковой сетки не изменяется
при растяжении. Эти очень важные вопросы рассматриваются
для некоторых полимеров в Приложении 3.
Литература
1. Т. A I f ге у, Mechanical Behavior of High Polymers, ch. C, Interscienct
Publishers, New York, 1948. [T. А л ф p e й, Механические свойства
высокополимеров, Издатинлит, 1952).
2, L. R. G. Т г е 1 о а г, The Physics of Rubber Elasticity, Clarendon Press,
Oxford, England, 1949. [Л. T p e л о a p. Физика упругости каучука.
Издатинлит, 1953].
3. Р. J. Flory, Principles of Polymer Chemistry, ch. XI, Cornell Uni-
versity Press, Ithaca, New York, 1953.
4. M. Green, A. V. Tobolsk y, J. Chem. Phys., 14, 80 (1946).
5. A. V. Tobo I sk y, Ph. D. thesis, Princeton University, 1944.
6. H. M. James, E. Guth, J. Chem. Phys., 11, 455 (1943); ibid., 21
1039 (1953); ibid., 15, 669 (1947).
7. P. J. Flory, Chem. Rev., 35, 51 (1944).
8. P. J. Flory, Ind. Eng. Chem., 38, 417 (1946).
* Кроме этого, предложен и другой метод расчета числа цепей сетки в
единице объема по равновесному модулю (метод равновесного модуля н его
применение описаны в указанных ниже работах). Этот метод основан на уравне-
нии £сс—N-/3, где —число цепей сетки в единице объема сеточного полиме-
ра. |Г. МАБ а рте не в, ЖТФ, 22, 1154 (1952), Сб. «Вулканизация резин»,
Госхимнздат, 1954, стр. 196; А. С. Новиков, Г. М. Бартенев,
Ф. А. Гали л-О г л ы, ДАН СССР, 94 , 253 (1954); Г. М. Бартенев.
Т. Н. X а з а н о в п ч, Высокомол. соединения, 2, 20 (I960)]. — Прим. ред.
Глава Ш
математическая теория ЛИНЕЙНОЙ ВЯЗКОУПРУГОСТИ
И УПРУГОВЯЗКОСТИ ПОЛИМЕРОВ
1. Определение и измерение вязкоупругих характеристик
Если идеально упругое тело растянуть до определенной задан-
ной длины L и затем поддерживать ее неизменной, то внешняя
растягивающая сила X, необходимая для того, чтобы сохранить
это растяжение постоянным, также будет неизменной. Деформация
растяжения s есть (L—Lo)/Lo, где Lo—длина в ненапряженном со-
стояния. Модуль Юнга не зависит от времени и дается выражением
вида:
Е= — (1,1)
где /—напряжение, равное XIА (Л—площадь поперечного се-
чения).
В этой главе в основном рассматриваются малые деформации
твердых тел*. В некоторых редких случаях, когда зависимость
между деформацией и напряжением линейна при больших дефор-
мациях (например, идеальная резина при сдвиге), это ограниче-
ние, вероятно, отпадает. Поведение полимеров при больших де-
формациях будет кратко рассмотрено в разделе 2.
Для любого реального упругого тела, растянутого до опреде-
ленной длины L и оставленного при этой неизменной длине,
уравновешивающая сила растяжения не будет неизменной, а
будет убывающей функцией времени X(t) 1в некоторых редких
случаях X(t) может возрастать]. Деформация s— по-прежне-
му отношение (L—Lo)/Lo; напряжение в момент времени t есть
/(/) =Х(/)/А. Новая величина—релаксационный модуль Е,((\—
определяется следующим уравнением:
Er(0 = /W* (1,2)
Опыт, описанный выше, называется релаксацией напряжения
при постоянной деформации 111. Этот метод исследования вяз-
* Поэтому автор не различает условноеп истинное напряжения растяже-
ния. — Прим. ред.
1. Определение и измерения еязкорпругих характерна ин
107
коупругих свойств полимеров используется в данной книге чаще
всего.
С другой стороны, если вязкоупругое тело подвергается дей-
ствию постоянного растягивающего напряжения то в общем
случае его длина есть возрастающая функция времени L(/).
Деформация в любой момент времени определяется как s(/) =
= lL(t)—Lol/Lo, где Lo—длина в нерастянутом состоянии. Модуль
ползучести, или крипа, Е((1) можно определить следующим образом:
ЕЛО = ,(',) (1.3)
Измерение s(t) при постоянном напряжении / называется иссле-
дованием процесса ползучести, или крипа 121. Иа практике боль-
шинство таких опытов проводят при постоянной растягивающей
нагрузке X. Так как поперечное сечение изменяется в течение
времени растяжения, это является лишь опытом при прибли-
женном постоянстве напряжения.
Опыты но релаксации напряжения или опыты по ползучести
в принципе легче выполнить в случае деформации сдвига, чем
растяжения. Зависимость от времени модулей сдвига Gr(t) и
Gc(t) можно было бы выразить совершенно аналогично уравне-
ниям (1, 2) и (1, 3).
Очень часто ползучесть при растяжении выражают с помощью
упругой податливости )(/). При постоянном напряжении / за-
висимость деформации от времени дается выражением вида:
s (/) = '/(/)/ (1,4)
Для материалов, подчиняющихся закону Гука, податливость
при растяжении является обратной величиной модуля Юнга.
Податливость при сдвиге обозначают ./(/).
Третий вид методов исследования вязкоупругих свойств—изу-
чение вынужденных или свободных колебаний. Предположим на
мгновение, что вязкоупругое тело можно представить как клас-
сический гармонический осциллятор с затуханием (более подроб-
ный анализ будет дан позднее). Допустим, что образец вязко-
упругого тела, имеющий форму прямоугольного параллелипипеда
площадью .4, высотой й, жестко скрепленный с внешней массой
М, подвержен колебаниям под действием внешней силы сдвига
f'o costi)/, амплитуда которой равна /’п, а угловая частота есть
(рис. III, 1).
Уравнение движения имеет вид:
MiPx , A dx , А „ г , 1 г \
dp + V dT + ~h 6 х cos ®г (1 ’ о)
108
Гл. III. Теория линейной вязкоупругости и упруговязкости
где х, рассматриваемая как функция времени x(t\, есть поперечное
смещение образца в направлении действия приложенной силы
сдвига Focos со/.
Уравнение (1, 5) можно записать иначе, введя деформацию
s(/) =х(/)/й, напряжение /Ocosa>/ =(F0M)cos со/ и приведенную мас-
су rn =(A'UA)h:
m ' Y‘ ~dt + G s = COS 0,Z
(1,6)
Величины TJwi„ и бД1|11. называются соответственно динамической
вязкостью и динамическим модулем сдвига вязкоупругого тела
Рис. 111,1. Схема опыта вынужденных коле-
баний (при сдвиге).
131. Эги величины зависят от угловой частоты колебаний со, од-
нако при фиксированном значении со они могут считаться в урав-
нениях (1,5) и (1,6) постоянными.
Уравнение (1, 6) можно также рассматривать как уравнение
колебаний единичного объема вязкоупругого материала, связан-
ного с массой т. Предполагая, что s(/) имеет вид s0cos(ci>/—ср),
где f часто называют углом сдвига фаз, находим решение урав-
нения (1, 6):
_ ____________/о cos (<п/ — у)____________
1 (бдии. — «‘"2)2 -И м21,2Д1ш.11/2
(1.7)
<071днн.
^Дин. — /Лбз2
Если возбуждающая сила меняется по закону /ocosco/, а внеш-
няя масса варьируется от опыта к опыту, то из формулы (1,7)
можно видеть, что максимальная амплитуда (в резонансе) по-
лучается при некотором значении массы nipe1., определяемом
из выражения
^Тдин. — РТрез (1.31
I. Определение и измерения вязкоупругих характеристик
109
Поскольку mpe.,. является величиной, которую можно определить
экспериментально, уравнение (1, 7) дает метод измерения динами-
ческого модуля СД1|Н.. Для любого вязкоупругого тела величина
бднн., определенная описанным выше методом, будет зависеть от
частоты о) действующей силы. При резонансе величина амплитуды
so, определенная из выражения для s(/), будет равна:
Равенство (1, 9) позволяет определять этим методом значение ве-
личины т|Д11Н_. Она также может быть определена по форме графика
зависимости s0 от массы m пз уравнения (1, 7) или прямым экспе-
риментальным измерением угла сдвига фаз (р.
Энергия 117, рассеиваемая в виде тепла в единице объема за
цикл в процессе колебаний силы /Ocos wt, определяется интегри-
рованием по времени /Ocos wtds(f) от нуля до 2к/«>:
IE . /)д|1Н 8в
”Ы1ГДИН. Л) _________
(<7дш1. — пи»2)- -|- 1"2т|ДИН.
(1.10)
1де s0—амплитуда деформации, a f0—амплитуда напряжения.
Единичный объем вязкоупругого материала, связанный с внеш-
ней массой tn, можно подвергнуть свободным колебаниям, сооб-
щив начальную деформацию сдвига so. Уравнение движения имеет
вид:
d2s ds . -
dt2 1 т1иш. ~7д Ь О,1Нц s б
Решением этого уравнения является выражение вида.
s = s0 [ехр (—члии./2т)/] cos(0/
где
io 1°Д н. / т|дин. \а 1/2.— /Сдпн. \1/а
[ in ( 2m ) " \ m )
(1.11)
(1.12)
(1. 13)
Если затухание невелико, то <в =(Од,н./т)1/2. Отношение амплитуд
последовательных колебаний в этом случае равно:
Отношение амплитуд == ехр (-.
\ ^дии.
(Е 14)
Отношение упругих энергий последовательных затухающих ко-
лебаний равно квадрату отношения амплитуд.
Поэтому динамические модули п динамическую вязкость можно
рассчитывать из частоты свободных колебаний, отношения после-
довательных амплитуд и массы. Обычно на практике свободные
110 Г л. HI. Теория линейной вязкоупругости и упругое язкости
колебания осуществляются на простых крутильных маятниковых
системах.
Лучше всего гармонические колебания описываются с помоптыо
комплексной периодической функции:
eiu,t = cos»)/ ф- isinы/
Допустим, что внешняя периодическая сила равна действительной
части комплексной величины foe
Уравнение (1,6) преобразуется при ^том к виду:
S’ + + Ga^.s = foe‘“: (1, 15)
Если решение в виде периодической функции s—slle'“'' (где s0—
комплексное число) подставить в равенство (I, 15), то получим:
- mco3s Е I, s + G,,,,, s = /ое""' (1,16)
- (»2/ns„ + йодs., i G1И11. s0 = f„ (l,17i
При этом решением, имеющим физический смысл, будет действи-
тельная часть выражения soe“"'.
Поведение вязкоупругого тела при колебаниях часто описы-
вается при помощи комплексного динамического модуля G' ф/G",
где G' соответствует компоненте «в фазе», a G" компоненте
«не в фазе» 14]. Уравнение движения для единичного кубика ма-
териала имеет вид:
Aoе (1,181
Первый член в правой части уравнения (1, 18) соответствие! внеш-
ней силе, вызывающей колебания, второй член в правой части
представляет' собой противодействующее вязкоупругое напряже-
ние. Связь между f и деформацией (здесь у, и -ком-
плексные числа) имеет вид:
f^ = (0' +Ki’’}s (1,19)
Если уравнение 11, 19) подставить в уравнение (1, 18), то по-
лучим:
- т w2 s ф iG's !- G's = (1,20)
Сравнивая уравнение (1, 20) с уравнением (1, 16), для случая
деформации сдвига получаем:
G'= GU1„. (1,21)
G" = сод,„„. (1,22)
2. Линейная вязкоупругость и принцип Больцмана
111
Совершенно аналогично можно описать колебания для случая
растяжения и всестороннего сжатия используя соответствующие
комплексные модули Е'-\ЛЕ" и B'-]iB".
Величины G' и 6”/> являющиеся функциями частоты колебания
со, широко используются в литературе, посвященной описанию
вязкоупругих свойств полимеров. Чаще G' и G" выражаются как
функции 1/ю, так как 1/ю имеет размерность времени.
Можно ожидать, и это подтверждается практикой, что модули
£r((), Ec(t) и довольно близки по величине, т. е способ
измерения вязкоупругости не имеет большого значения, если
время измерения одинаково 15]. То же самое остается справедли-
вым и для Gr(/), Gc(t) и G'(t). В следующих разделах будут даны
точные соотношения между этими величинами п указаны пределы
пригодности упомянутого выше приближения. Измерения релак-
сации и ползучести для изучения вязкоупругости очень удобно
производить в пределах от 1 до 10е сек, измерения в динамическом
режиме удобны для области временной шкалы от 1 до 10~в сек.
Литература
1. А. V. Т о b о 1 s к у, J. Appl. Phys., 27, 673 (1956).
2. Н. Lea de гша п, Elastic and Creep Properties of Filamentous Materials,
The Textile Foundation, Washington, D. C., 1943.
3. S. D. G e h m a n, J. Appl. Phys., 13, 402 (1942); M. Mooney,
R. H. G e г к e, Rubb. Chem. Technol., 14, 35 (1941); J. H. Dillon,
S. Gehman, India Rubb. World, 115, 61 (1946).
4. J. D. Fe r r y. Die Physik der Hochpolymeren, Vol. IV, Ch. VI, H. A. Stuart
(ed.), Springer, Berlin, 1956.
5. T. A 1 f г e у, P. M. Dot y, J. Appl. Phys., 16. 700 (1945).
2. Линейная вязкоупругость и принцип Больцмана
Пусть идеально упругий материал, подчиняющийся закону
Гука, был подвержен серии последовательных деформаций растя-
жения s0, Sp s2, •••, sn_lt sn в моменты времени t0=Q, tlt
(„_ъ tn- Напряжение в конце этого процесса нагружения будет
равно:
f = Est. -i Е (st - s0) 4- Е (s2 — st) +
4-£(s3 s2)4-----4-Esn (2,1)
Если материал вязкоупругий, то напряжение, создаваемое
каждой из этих последовательных деформаций, зависит от вре-
112
Г.i. III. Теория линейной вязкоупругости и упруговязкости
мени через модуль релаксации Er(t). По Больцману 111, состоя-
ние линейной вязкоупругости описывается следующим уравнением
Г (Г) = Ег (0 [sj + Er (t -1L) [ (S1 - s0) I +
4- Er(t -ts) [(s2 - S1)J + Er (t -13) [(s3 - s2)] +
J-------------|_Д(/ _ /„)[($„— S„_!)J
(2, 2)
Для непрерывного процесса деформации этот принцип Больц-
мана выражается в виде интегрального уравнения*:
t
f(t) = Er(t -0)s(0)i/b
i)
(2, 3)
где 0—текущее время истории процесса деформации, s(0) есть
ds(O)/dO, a t—заданное время (постоянное при интегрирова-
нии). Предполагается, что «текущее время» 0 отсчитывается от
нуля (момента начальной деформации).
Уравнения, совершенно аналогичные уравнениям (2, 2) и
(2, 3), можно написать и для линейного вязкоупругого тела,
подверженного последовательным напряжениям /0, Д, /2, • • -,
/п-i. fn в моменты времени to=0, tlt Е, tn_lt tn
Для этих уравнений мы используем временную зависимость
упругой податливости, определенную в уравнении (1.4):
s(0-HWoH Y(t -МКЛ /0)Н—
+ V(/ -иКЛ /„-JJ
(2, 4)
Для непрерывного нагружения определение линейной вязкоупру-
гости можно выразить при помощи другого интегрального урав-
нения:
[
s(0 = Р (/ O)f(O)dO (2,5)
о
* Уравнение (2, 2) можно записать в виде суммы
п
\т is,-
f (0 = L -д77 ДС-
i =0
которая при непрерывном процессе деформации (обозначая tj — Ь, Ss, = ds,
= d6) переходит в интегральную форму (2, 3).—Прим. ред.
2. Линейная вязкоупругости и принцип Больцмана
113
где 0—«текущее время» истории процесса нагружения, Д0) есть
df(6)lcie, a t рассматривается как константа при интегрировании.
Одной характерной особенностью уравнений (2, 3) и (2, 5),
определяющих линейную вязкоупругость, является то обстоя-
тельство, что и кривые релаксации напряжения и кривые
ползучести s(/)/f не зависят от величины постоянной деформации
или величины постоянного напряжения, при которых они полу-
чены. Это свойство присуще также £,.(/). и Y(t), введенным
в разделе 1.
Как показал Вольтерра 12, 31, уравнения (2, 5) и (2, 3) яв-
ляются однозначными следствиями друг друга.
Если тело обладает линейной вязкоупругостью, определяемой
уравнениями (2, 3) и (2, 5), то его состояние можно описать бес-
конечным набором линейных дифференциальных уравнений
13—71. Имеется неограниченное количество способов нахождения
этих уравнений, но все они эквивалентны, что является важным
обстоятельством, подчеркнутым Алфреем и Доти 151. Два из этих
способов описаны в следующих двух разделах.
История развития теорий линейной вязкоупругости до 1943 г.,
начиная с интегральных уравнений Больцмана и дифференциаль-
ных уравнений Вихерта и др., была остроумно и творчески рассмо-
трена Лидерманом [3]. За последние 100 лет многими выдающими-
ся физиками были получены очень интересные эксперименталь-
ные и математические факты. Неожиданно возникающие матема-
тические проблемы способствовали бурному развитию прикладной
математики 121.
Дальнейшие попытки (с 1943 г. по 1948 г.) развить молекуляр-
ные и феноменологические теории линейных вязкоупругих свойств
полимеров достаточно сжато и ясно описаны Алфреем в его
монографии [41*.
Обобщенное математическое рассмотрение линейной вязко-
упругости было сделано Сипсом 16] и затем более полно развито
1 'россом [7].
Можно ожидать, что линейная вязкоупругость наблюдается
у тел, основная структура которых не меняется за время экспе-
римента.
* С этой оценкой Тобольского трудно согласиться, так как книга Алфрея
написана без должного критического рассмотрения использованного литера-
турного материала и содержит поэтому много внутренних противоречий и
поверхностных суждений. Кроме того, в ней полностью игнорируются работы
советских ученых, еще в 1939—1941 гг. установивших огромное значение ин-
тегральных уравнений Больцмана и функционалов Вольтерры для описания
механических свойств вязкоупругих и упруговязких тел. [Г. Л. С л о н и м-
с к и й, ЖТФ, 9, 1791 (1939); В. А. Каргин, Г. Л. Слонимский,
КТФ, 11, 341 (1941); А. П. Вронский, Прикл. мат. и мех., 5, № I,
(1941).—Прим. ред.
3—833
114 Гл. 1П. Теория линейной вязкоупругости и упруговязкости
Больцман и его ученики показали, что принцип суперпозиции
в виде уравнения (2, 4) справедлив для стекла. Лидерман провей
несколько тщательных опытов, которые показали, что уравнению
(2, 4) подчиняются вискоза, шелк, найлон-66, шерсть ангорской
козы. Все они—ориентированные волокна. Он обнаружил также
заметные отклонения от принципа Больцмана во многих экспери-
ментах, которые были правильно объяснены тем, что ряд веществ
при нагружении обнаруживает дальнейшую ориентацию и кри-
сталлизацию, вследствие чего в конце опыта их структура сильно
отличается от первоначальной. Принцип Больцмана применим
к этим веществам, только при особых условиях (при очень малых
нагрузках и при температурах, при которых кристаллизация и
ориентация практически не развиваются).
С другой стороны, было обнаружено, что принцип Больцмана,
выраженный уравнением (2, 2), остается справедливым для поли-
изобутилена—аморфного полимера 181и’для GR-S 19]. Несомнен-
но, что принцип Больцмана справедлив и для всех других аморф-
ных полимеров.
Наиболее полное описание «состояния обобщенной линейной
вязкоупругости» можно дать па основе опытов по релаксации на-
пряжения при различных относительных растяжениях a=L/Lo.
Материал обнаруживает «состояние обобщенной линейной вязко
упругости», если при постоянном относительном растяжении
выполняется соотношение:
= (2,6,
где Ф;а) стремится к единице для очень малых значений а—1 =
/La —s. В пределе для малых деформаций уравнение
(2, 6) идентично уравнению (1, 2). Для линейного полимера в
вязкоэластнческоп области Ф(а) дается выражением вида
(2, /)
в соответствии с уравнением состояния идеальных каучукоподоб-
иых полимеров.
Из уравнения (2, 6) вытекает, что влияние величины растяже-
ния, при котором наблюдается релаксация напряжения, состоит
в простом умножении кривой спада напряжения на постоянный
коэффициент. При этом изменения формы кривой релаксации не
происходит. Влияние характера соотношения между напряжением
и деформацией в «нулевой» период времени отделяется от процес-
сов релаксации, протекающих во времени.
3. Модели Максвелла—Вихерта
115
Тобольским и Эндрюсом было найдено, что уравнение (2, 6)
справедливо для вулканизата GR-S [9] в нижней части переход-
ной области при значениях а вплоть до 2 и для линейного поли-
изобутилена в области эластовязкого состояния при а<2. Несом-
ненно, что данное уравнение остается справедливым в этих обла-
стях и для других аморфных полимеров.
Этот результат имеет очень большое практическое значение и
повышает роль методов исследования процессов релаксации напря-
жения.
Уравнение вида (2,6) не является справедливым для ползу-
чести при растяжении, так как форма кривой ползучести зависит
о г величины растягивающей нагрузки, вплоть до нагрузок, про-
изводящих очень малые начальные деформации растяжения.
Нели использовать такие малые нагрузки, то заметное и сложное
влияние будет оказывать и вес образца полимера (в области
эластовязкого состояния).
Литература
1. L Boltzmann, Pugg. Ann. Phys., 7, 624 (1876).
2. V. V о 1 t e г г a, Theory of Functionals, Dover, 1959.
1 H. L e a <1 e r in a n, Elastic and Creep Properties of Filamentous Materials
and Other High Polymers, The Textile Foundation, Washington,
D. C„ 1943.
4. T. A 1 f r e y, Mechanical Behavior of High Polymers, Interscience Publi-
shers, New York, 1948 [T. А л ф p e it, .Механические свойства высоко-
полнмерон, Издатннлит, 1952].
5. I. A lire у, Р. М- Dot у, J. Appl. Phys., 16, 700 (1945).
6. R. Si p s, J. Polymer Sci., 5, 69 (1950).
7. В Gross, Mathematical Structure of the Theories of Viscoelasticity, Her-
mann, Paris, 1953.
8. R. D. Andrew s, N. Hof m a n-B a n g, A. V. Tobolsk y,
J. Polymer Sci., 3, 669 (1948); also extensive unpublished experiments
of A. V. T о b о 1 s k у and R. D. Andre w s.
О. A. V. T о b о 1 s k y, R. D. A n d r e w s, J. Chem. Phys., 13, 3 (1945).
3. Модели Максвелла—Вихерта
Дифференциальные уравнения линейной вязкоупругости, осо-
бенно удобные при описании опытов по релаксации напряжения,
были получены Вихертом 111 как обобщение уравнения Максвел-
ла (раздел 8 главы I). Механические модели, соответствующие
уравнениям Вихерта, показаны на рис. Ш, 2, и III, 3. Модель,
изображенная на рис. III, 2, состоит из очень большого (или бес-
конечного) числа элементов Максвелла, соединенных параллель-
но. Модель на рис. III, 3 состоит из очень большого (или беско-
нечного) числа элементов Максвелла, соединенных параллельно,
плюс пружина без поршня.
8*
116 Гл. III. Теория линейной вязкоупругости и упруговязкости
Математические уравнения, соответствующие этим моделям,
имеют вид:
ds dt ds dt 1 df. E, ' dt t 1 j = £/'d/ + A _! f (3, 1)
ds dF _ 1 dfm j — Em' dt •'
i A + A + ' ' + fm
ds 1 dfi , 1 f dt z1E1 '*
dT 1
ds 1 dh , 1 f
dF~ dt ' -1Ег /2
ds 1 dfm । r ‘ dt ‘ т Г 'л/ 4nLjm
dF^
ds_ _ 1 ‘V.
dt ' dt
(3,2)
/ = Л + А+ /2+ • + /„ (3,3)
В уравнениях (3, 1) и (3, 2) Д. является парциальным напря-
жением, действующим на t-ты» элемент, £(- и т|. являются соот-
ветственно модулем и временем релаксации / того элемента, f есть
общее напряжение, a s—деформация. Отдельная пружина с Е.
на рис. 111,3 может рассматриваться как элемент с бесконечным
временем релаксации.
Рис. III,2. Модель Вихерта для уnpyi«вязкого тела.
3. Модели Максвелла—Вихерта
117
Практически, когда механические модели, изображенные
на рис. Ill, 2 и III, 3, или-уравнения (3, 1) и (3, 2), используются
для описания свойств материала, нужно помнить, что един-
ственными величинами, доступными наблюдению, являются де-
формация s и общее напряжение f. Парциальные напряжения fh
а также величины Et и макроскопически не наблюдаемы, но
их можно вычислять из экспериментальных данных.
ЛАодель, изображенная на рис. 111,2, и система уравнений
(3, 1) пригодны для несшитых, или линейных полимеров, так как
при постоянной деформации эта модель показывает уменьшение
Рис. 111,3. Модель Вихерта для вязкоупругого тела.
напряжения вплоть до нуля (упруговязкое поведение). Из урав-
нения (3, 1) при постоянной деформации sfl (начальные условия:
при t =0) следует:
гп
f (t) — So 5] Ei exp ( -6%) (3, 4)
Z-l
m
Er(t) — У £,exp( -f/t,.) (3,5)
Если в течение периода времени, гораздо более длинного, чем
наибольшее время релаксации напряжение fo поддерживается
постоянным, то все величины (1 /E^fd^/dt) в уравнениях (3, I)
становятся исчезающе малыми*. Поэтому имеет место выражение
вида:
m
/о = I//= $ (3.6)
* Вследствие того, что процесс деформации становится стационарным и
все /f=const.—Прим. ред.
Не
Гл. III. Теория .шнеиной вязкоупругости и упруговязкосси
Следовательно, если модель Вихерта (рис. 111,2) долгое время
находится при постоянном напряжении, то опа ведет себя как
ньютоновская жидкость, обладающая постоянной скоростью де-
формации Константа Дс(Е,- есть т(('>—вязкость при
растяжении, связанная с вязкостью при сдвиге д и коэффициентом
Пуассона а следующим образом:
= =20 -f- (3,7)
Для линейных полимеров в каучукоподобном состоянии а—1/2,
ПОЭТОМУ 7((,>=3V|.
Если величины п £, можно получить на основе анализа кри-
вой релаксации напряжения, то вязкость д’й можно получить
из уравнения (3, 7). Это один из характерных примеров, когда
одни вид эксперимента по упруговязкости может быть использо-
ван для предсказания результатов второго; в этом случае релак-
сация напряжения используется для предсказания состояния те-
чения при постоянном напряжении, действующем в течение дли-
тельного промежутка времени. Многие другие примеры будут
приведены в разделе 7.
Модель, изображенная на рис. 111, 3, и уравнение (3, 2) при-
годны для полимеров с поперечными сшивками, так как при по-
стоянной деформации эта модель дает напряжение, спадающее
до определенной величины, не равной пулю (вязкоупругое пове-
дение).
Из уравнения (3, 2) при постоянной деформации su следует
IH
I (/) = s0£\ J- s0 Е,ехр (—//г, ) (3, 8)
i --1
Е, (I) — Е. I- 1Е( exp ((3,9)
Если постоянное напряжение /о действует в течение промежутка
времени, очень большого по сравнению с самым большим временем
релаксации, то в конце концов будет достигнута постоянная де-
формация*:
s, = (3,10)
Гела такого типа обладают замедленной упругостью (вязко-
упругостью), но не истинным вязким течением.
Уравнения (3, 1) и (3, 2) можно обобщить (или аппроксимиро-
вать) при помощи непрерывного набора дифференциальных урав-
нений. Для всех значений т, лежащих между нулем и бесконеч-
’ В отечественной научной литературе принято называть эту деформацию
<< рав новее нон ».—Прим. ред.
3. Модели Максвелла—Вихерта
119
ностыо, мы определяем непрерывные функции Е(Д и так, что
для вязкоупругого тела получим следующие уравнения:
dt F(t)’ dt +
f.=Ens; = v \ fit,-:) dz
где Eco и, следовательно, /Д могут быть равны нулю.
Уравнения (3, 5) и (3, 9) принимают вид:
Е, (/) -Е„ = ( Е(т)ехр(—tl~.)d.':
(3, 11)
(3, 12)
о
где Есо может принимать нулевое значение (для модели III, 2).
Функция £'(т) называется распределением времен релаксации.
Хотя в уравнении (3, 12) интегрирование производится по всем
временам релаксации от нуля до бесконечности, Е(т) может исче-
зать для значений т ниже некоторой минимальной величины
и для значений выше некоторой максимальной величины тт>.
Заметим, что величинами, имеющими смысл напряжения, явля-
ются а не [(/,-), и что E(t)d-: также имеют размерность
напряжения или модуля упругости.
В принципе, если ЕД/)—ЕЛ известно для всех значений /,
то уравнение (3, 12) может быть решено относительно Е(Д. Этот
тип уравнения непосредственно связан с известным преобразова-
нием Лапласа.
Для вязкоупругого поведения при сдвиге можно написать
уравнения, совершенно аналогичные уравнениям (3, 1—3, 11).
Времена релаксации одинаковы и для сдвига и для растяжения.
Модули Е/ или Е(т) должны быть заменены на G,- или <7(т), где
G,-=Е,/2(1 фо), а С(Д=Е(Д/2(1 (-о).
Для модели, изображенной на рис. Ill, 2, в случае непрерыв-
ного распределения времен релаксации уравнение (3, 7) прини-
мает вид:
zE(z}dz (3. 13)
б
Для распределения времен релаксации при сдвиге имеем
/3,14),
О
1211
Гл. 111. Теория линейной вязкоупругости и уируговязкости
В принципе, если функция Е(~) известна, то ее можно исполь-
зовать для предсказания результатов любого другого эксперимен-
та по изучению механических свойств: например, такого, когда
деформация Sj прилагается в промежутке времени от /=0 до
деформация s2—в интервале от Е до t2 и т. д. Из £(т) можно
рассчитать комплексный динамический модуль путем интегриро-
вания, а модуль ползучести—с помощью совершенно точных, но
довольно сложных математических соотношений, что будет пока-
зано впоследствии.
Можно легко показать тождественность 121 формулировки
Вихерта (3,11) и формулировки Больцмана [уравнение (2, 3)].
Для данного значения т уравнение (3, 11) есть линейное уравнение
первого порядка, и оно легко интегрируется. Если для простоты
мы примем при t=Q, то получим:
/(Г, т) = Е(т) j ехр
6
s(6) db
(3, 15?
Для f(l) имеем следующее выражение (приняв для простоты
Е. =0):
/(0 =- ( =
- j| j £(x)exp( dxjs(O) db
о о
Теперь введем Er(t—6) с помощью равенства
Er(l b) = f £(-) exp ( (E
0
тогда
U)s(b)db
b
(3,16)
(3, 17)
(3, 18)
а это уравнение идентично уравнению (2, 3).
В 1939 году 131 В. Кун использовал модель Bnxepia для опи-
сания механического поведения полимеров, затем она была
4. Обобщенная модель Фойгта
121
развита Р. Сима 141 и многими другими исследователями, что
предвещает дальнейшее широкое развитие модельных представ-
лении.
Литература
1. Е. Wiech ert, Wied. Лпп. Physik, 50, 335, 546 (1893).
2. R. Sips, J. Polymer Sci., 69 (1950).
3. W. Kuhn, Z. physik. Chem., B42, 1 (1939); K. Bennewitz,
H. R 6 t g e r, Phys. Z., 40, 416 (1939); W. Holzmuller,
E. Jenckel, Z. physik. Chem-, A186, 359 (1940).
4. R. Si mha, J. Appl. Phys., 13, 201 (1942).
4. Обобщенная модель Фойгта
Дифференциальными уравнениями линейного вязкоупругого
состояния, особенно пригодными для описания опытов по пол-
зучести, являются обобщенные уравнения Фойгта—Кельвина [11.
Соответствующие математические модели (двух вариантов)
изображены на рис. 111,4 и 111,5.
Рис. 111,5. Модель
Фойгта—Кельвина для
упруговязкого тела.
Рис. III,4. Модель
Фойгта—Кельвина для
вязкоспругого тела.
122
Гл. III. Теория линейной вязкоупругости и унруговязкости
Математические уравнения, соответствующие модели, изобра-
женной на рис. Ш,4, имеют вид:
(У)3' г ' 1ц Щ Сц 4 4 4 T1^1S1 t2£2s.
f EfUsrn "t ~ t £* s’ '’ar^irPtn
S — Sq 4“ S'j + s2 + ‘ ' i
(4, 1)
где sz d'i^dt.
Уравнения, соответствующие модели, изображенной на рис.
111, 5. имеют вид:
/ = Еч\
f ~ ^isi F
/' = £,s, -г -2£2s2
I =-
S 51 H- 52 -j- • • ; Slf[ 4" S,
(4, 2)
В этих уравнениях есть модуль (или упругая константа)
/-того элемента, а есть время запаздывания /-того элемента
является вязкостью при растяжении для /-того поршня).
Очевидно, общая деформация является суммой парциальных де-
формации всех элементов. Величина появляется вместо rlt
гак как мы имеем дело с растяжением, а не со сдвигом. Для вяз-
коупругого поведения в случае сдвига можно написать уравнения,
аналогичные уравнениям (4, 1) и (4, 2), имея в виду, что Д'» нужно
заменить па т/, а па G;. Времена запаздывания для сдвига п
растяжения предполагаются равными.
При постоянном напряжении кривая ползучести для уравне-
ний (4, 1) имеет вид:
m
s=- 4^ • У 4” 11 - ехр( —/Л,-)] (4.3)
Первый член в правой части уравнения (4, 3) есть мгновенная
упругость, второй член представляет собой запаздывающую
4. Обобщенная модель Фойгта Г23;
упругость. При постоянном напряжении /о кривая ползучести
для уравнении (4, 2) имеет вид:
m
»- Г-+ ф), (4.4)
/=1
Третий члер в правой части описывает установившееся течение
материала при постоянном растягивающем напряжении.
Уравнение (4, 3) описывает вязкоупругий материал, который
под действием постоянного напряжения в конце концов дости-
гает предельной упругости; уравнение (4, 4) описывает упруго-
вязкие вещества, которые при постоянном напряжении обнару-
живают вначале мгновенную упругость и в конце концов состоя-
ние течения. Уравнение (4, 3) пригодно для сшитых аморфных
полимеров, а уравнение (4, 4) для линейных аморфных полиме-
ров.
Модуль мгновенной упругости Е., из обобщенных моделей
Фойгта III, 4 п III, 5 связан с парциальными модулями обобщен-
ных моделей Максвелла 111,2 и 111,3 следующим образом:
m
Е9 = Е.|У.Е; (4,5)
«=1
где Е,. может принимать нулевое значение (как в модели III, 2).
Уравнения (4, 1—4, 4) проще записать при помощи упругой
податливости, а не упругого модуля. Например, уравнение (4, 4)
можно записать в виде:
m
V/ohU- ехр( -(.МФ (4,6)
1
С помощью функции У'(т) можно также описать непрерывное рас-
пределение времен запаздывания:
©о
/оУ + A) f V(T)|1 -ехр< —(4,7)
о 1
где У'(т) может быть равно нулю, если т меньше или больше
тт. Обобщенная модель Фойгта была использована Александро-
вым и Лазуркииым еще в 1939 г. для объяснения релаксационных
свойств полимеров 12].
Литература
1. W. Voigt, Abhandl. Akad. Wiss. Gottingen, 36 (1899).
2. А. П. Александре в, Ю. С. Лазу р к и и, .ЖТФ, 9, 1249 (1939)-
124
Гл. III. Теория линейной вязкоупругости и унруговязкости
5. Динамический модуль
и обобщенная модель Максвелла
Предположим, что кубик упруговязкого метериала единичных
размеров связан с внешней массой т, а вся система подвержена
колебаниям под действием силы сдвига, меняющейся по закону
foe‘wt. В дальнейшем мы будем считать, что упруговязкип мате-
риал подчиняется обобщенной модели Максвелла (см. рис. Ill, 2).
Уравнение движения имеет вид 1см. уравнения (1,5), (1, 6) и
(1, 1К)1:
/п ’ f™- (5> !)
где /Э1<—упруговязкое напряжение в материале, a s—деформация
растяжения. Упруговязкое напряжение определяется уравнениями
<i = А- + J>— G, ',G, >
-t-^ 01 и (5, 2)
^rn frn1 “t” f п/ ~п£*т
f = h*. = /i -1- /2 4 !“ f,n
Предположим, что под действием внешнего напряжения, из-
мепяющегося по закону устанавливаются парциальные
напряжения, изменяющиеся по закону: , а также
Лл. =/эл.ое'ш/ и 5=5ое'ш/, где /,,0, /эл.о и so—комплексные числа.
Из уравнения (5, 2) находим:
t soiu>zfit
>i0 = “1 .|-^7
^.o = ^.o + s0^ (5,3)
Выражение для существующего в материале упруговязкого
напряжения можно записать при помощи комплексного динами-
ческого модуля:
/эл.,о = s0 [G' (<>) + iG" («>)]
(5,4)
Приравняв мнимые и действительные части уравнений (5,4)
и (5, 3), получим следующие выражения, впервые выведенные
5. Динамический модуль
125
Тобольским и Эйрингом [И с использованием обозначений Сдии.
И ^дин.: G'=X1^-= (5,5) хт G" у - = V ГГ-^ - dx (5> 6)
11одобный вывод можно произвести и для непрерывного спектра
времен релаксации, заменив при этом суммы, фигурирующие в
уравнениях (5, 5) и (5, 6), интегралами, указанным способом.
Те же выкладки можно проделать и для переменных растяги-
вающих напряжений. Единственное изменение, которое нужно
произвести в этих уравнениях, состоит в замене модуля G( на
Если в уравнение (n. 1) подставить решения s=Soe‘*“', /эл. =
=1эл.,пе‘ы* и /эл.,о из уравнения (5, 3), то получим уравнение сле-
дующего вида:
- m Ао + /So2 + s0S = f0 (5, 7)
Используя уравнения (5, 5) п (5, 6), имеем:
— тш-з0 + iG"s0 + G's0 = f0 (5, 8)
Если уравнение (5, 8) сравнить с уравнением (1, 17), то снова
подтверждается, что G" =йп|ДИИ , a G' — СД1|Н..
Литература
1. А. V. Tobolsk у, Н. Е у г i n g, J. Chem. Phys., 11, 125 (1943).
6. Расчет распределения времен релаксации
из данных по релаксации напряжения
Соотношение между модулем /:,.(/), определенным из опытов
до релаксации напряжения, и распределением времен релаксации
Е(т) было дано в разделе 3:
(0 = j £(у)ехр(—(6,1)
о
где £(') может принимать нулевые значения при т ниже и вы-
ше
126
Гл. HI. Геория линейной вязкоупругости и упру, овязкости
С математической точки зрения соотношение, выраженное
уравнением (6, 1), тесно связано с преобразованием Лапласа.
Если Еr(t) известно для всех значений t от нуля до бесконечности,
то Е(-) в принципе можно рассчитать; многие аналитические функ-
ции E(f) известны и составлены таблицы преобразованных функ-
ций Ег(~). Примеры будут даны позже.
С другой стороны, £,.(/) получается экспериментально в вид»,
графика; ио даже если графические данные выражены в виде ана-
литической функции, соответствующие лапласовы преобразования
для них могут быть неизвестны. Поэтому весьма желательно иметь
хотя бы приближенный метод получения Е{~) из Er(t).
Более того, для использования преобразования Лапласа нужно
знать Er(f) по всей шкале времен от t —0 до /=оо. В главе IV мы
покажем, как это можно выяснить путем построения «вспомога-
тельных кривых», полученных из данных по релаксации напряже-
ния при различных температурах. Однако довольно часто
£,(/) известна "олько в ограниченном интервале времени. В этом
случае невозможно решить уравнение (6, 1), но можно получить
соответствующее графическое приближение.
В первом приближении функцию ехр(—th) в уравнении
(6, 1) можно заменить ступенчатой функцией, равной 1 для ~>i
и нулю, если Следовательно
Е (t) Е(-.)<Е: (6.2)
't
Дифференцируя уравнение (6, 2), имеем;
-[^гЦ (6,3>
Мы можем определить новую функцию, которая называется пер-
вым приближением распределения времен релаксации, следующим
образом:
с . , Г dE (I) I .
(6.41
Формула (6, 3) была впервые предложена Алфреем [11. Второе
и следующие более высокие приближения были получены Ферри
и Вильямсом 121, Эндрюсом 131, Шварцлем и Ставермапом 141:
их результаты будут рассмотрены ниже.
Распределение времен релаксации можно выразить с помощью
другой функции /У(-), определяемой следующим образом* *:
Н (?) = т£ (?) (6,5)
* Размерность этой функции распределения вновь та же, что и размер-
ность модуля (дан,'саг).— Прим. ред.
6. Расчет распределения времен релаксации
127
Свойства И часто выражают с помощью независимого переменного
1m вместо т, с заменой /7(1т)=//(т), означающей, что эти две
различные функции равны для данного значения т. Функция
//(1m) с/(1п ) есть парциальный модуль тех элементов модели Мак-
свелла, которые имеют время релаксации, лежащее между 1m
и 1m4 d(lm). Очевидно, из уравнения (6, 5) следует:
//(In-)rf(ln-) = E(z)dr
(6, 6)
Первое приближение для И(~), обозначаемое через следует
из уравнения
», с*—м
Гак как Er(t) или часто откладываются в зависимости от
1g/. то полезны следующие определения:
1
2,303
\dEr (f) I _ р . [dig Е,(0 I
| dlgt |z=x ~ dipt J,=z
(6,8)
Второе приближение согласно методу Эндрюса [3], имеет вид:
1
2,303
I dEr (I)
I dlgT
4- 0,109
I d'2Er (QI
L'lg/2 |,=T
(6,9)
Второе приближение, соответствующее методу Ферри и Вильямса
(2], имеет вид:
№-'<»
где (6,11) dig- V '
а Г—хорошо известная гамма-функция (для целых значений гп,
0(14-«О =«*!)•
Следует заметить, что в наших статьях до 1 января 1959 г.
использовано обозначение для //(т), несколько отличное от
введенного здесь. Ранее применяемое //(-) будет теперь заменено
на /У(т), причем //(*) будет использоваться па всех рисунках этой
книги, кроме рис. Ill, 9, где отложено //(-).
Определяется Н(~) следующим образом:
Я(1ёт)Лё- = Е(тНт
(6, 12)
128
Гл. III. Теория линейной вязкоупругости и упруговязкости
Следовательно, соотношение между Н(-), Н(~.) и Е(т) имеет bill:
/7 (?) = 2,303/7 (?) = 2,303-Е (f)
(6, 13.
Снова мы употребляем систему обозначений и /;(*) с уче-
том тождества в том смысле, что эти две различные
функции равны для данных значений -. Таким же образом ис-
пользуется система обозначений Е'(«) и Е'(1/ы), взаимозаменяе-
мых в том смысле, что эти две различные функции равны для
данных значений «».
Введение двух функций //(?) и //(?) желательно для того, что-
бы предупредить недоразумение, которое может возникнуть при
сравнении с работами других авторов.
Метод получения дискретного спектра времен релаксации
из экспериментальных данных по релаксации напряжения был
развит Тобольским н Мураками. Он описан в главе 4, раздел 9.
Литература
1 Г. А 1 f г е у, Mechanical Behavior of High Polymers, Appendix П, Intercsience
Publishers, New York, 1948, p. 533 (T. А л ф p e й, Механические свои
ства высокополимеров, Издатинлит, 1952; Приложение II, стр. 556]
2. ,1. D. Ferry, М. L. Williams, J. Coll. Sci., 7, 347 (1952):
M. L. Williams, J. D. F e r r y, J. Polymer Sci., 11, 169 (19531.
3. R. D. Andrews, Ind. Eng. Chem., 44, 707 (1952).
4. F. S c h w a r z I, A. J. S t a v e r m a n, Physica, 18, 791 (1952); ^ppl.
Sci. Research, A4. 127 (1953).
7. Преобразование Er (/)
в другие функции
Предположим, что опыты по релаксации напряжения дают
вспомогательную кривую Er(t) по всей шкале времени. Как ис-
пользовать эту кривую для получения других важных характе-
ристик вязкоупругих и упруговязких свойств?
Вспомним соотношения между Е'(ы), Е"(ы) и Е(?):
//9 == | H(T)dT (7, И
Е'М>)= р/(.)тГ^_4Лпт (7,2.
Е" к») = С /7 (?) d Inc (7. 3.
7. Преобразование Er(t) в другие функции
129
В этих уравнениях можно заменить И на Н1г Н2а пли Н2Ь,
рассмотрев приближения, описанные в предыдущем разделе,
и выполнить интегрирование графически или с помощью числен-
ных расчетов.
Однако оказывается возможным сделать несколько более про-
стые приближения. Они необходимы в тех случаях, когда Er(t)
и Д(т) известны только в ограниченном интервале времени. На-
пример, для Е"(ы), принимая во внимание, что ыт/(1-)-<и2т2)
есть функция с максимумом при т=1/ю, имеет место следующее
приближение:
Е" (со) [Н (Д]х=1/.о j* d In т — -Д [/У (х)],_1/ш (7, 4)
Вместо // в уравнении (7, 4) можно снова подставить /Д, H2J
или Н2Ь. !
Для вязкости т/0, определяемой уравнением (7, 1), наибольшее
значение имеет тот факт, что величины Н, Ht, Е.,а или H2b должны
быть очень точно определены в области тга. Однако при т<тП1/10
влияние Д(т) на т/0 невелико, и им часто можно пренебречь, если
оно точно не известно.
В грубом приближении Er(t), Ec(t) и Е'(1/ы) равны друг другу.
Для данных значении t (или 1/ы) величина Ec(t) несколько мень-
ше, чем Er(t), a Er(t) несколько меньше, чем Е'(1/«>). Эти прибли-
жения справедливы в том случае, если Er(t) асимптотически стре-
мится к ненулевому значению Ег,~, при У, стремящемся к бесконеч-
ности.
Если Er(t) стремится к нулю, эти приближения справедливы
при /<тя>, где т,п—максимальное время релаксации.
Динамический модуль Е'(1/ы) можно совершенно точно под-
считать из следующего уравнения [1]:
Е' (1 /со) — [Е,.(/)], =1/ш =[т (щ) Н2[/ (т)|х=1/„ (7, 5 )
Y (т) = cosec
В уравнении (7, 5) т есть величина, определяемая уравнением
(6, 11). Значения функции у(т) даны в табл. Ill, 1.
Аналогично имеем:
Е"( 1 /Ш) = -Д sec /У2Ь(г) (7, 6)
Связь между модулями ползучести и релаксации более сложная.
Распределение времен запаздывания Е(~), совершенно аналогичное
9-833
130
Гл. ///. Теория линейной вязкоупругости и упруговязкости
Функция y(wt)
Таблица III, Г*
0 1 2 3 4 5 О 7 8 У
—m 0,0 0,2 51 53 56 58 61 64 67 70 74 77
1 80 84 88 91 95 99 *03 *07 *12 *16
2 0,3 20 25 30 35 40 46 52 58 64 70
3 77 83 90 98 *05 *13 *20 *29 *38 *47
'1 0,4 57 67 77 87 98 *10 *22 *35 *48 *61
5 0, 575 590 606 662 640 658 676 696 718 740
6 763 788 814 841 872 902 935 970 *008 *048
7 1, 090
+<п 0,0 0,2 51 48 46 44 42 40 38 35 33 31
1 29 28 26 24 22 21 20 18 17 15
2 14 13 12 И 10 08 07 06 05 05
3 04 03 02 01 01 00 *99 *99 *98 *98
4 0, 1 97 97 97 97 96 96 96 96 95 95
5 95 95 95 95 95 96 96 96 96 96
6 97 97 97 98 98 99 99 *00 *01 *01
7 0,2 02 03 04 04 05 06 07 08 09 10
8 12 13 14 16 17 18 20 21 23 25
9 27 28 30 32 35 37 39 41 44 46
1.0 48 51 53 56 59 62 65 68 71 74
1 78 81 85 88 92 96 *01 *05 *10 *14
2 0,3 18 23 28 34 39 45 51 57 63 69
3 76 83 90 £8 *05 *13 *22 *31 *40 *49
4 0,4 59 69 79 90 *02 *13 *25 *38 *51 *65
5 0, 580 595 612 628 646 665 685 706 727 750
6 774 798 825 855 885 917 950 986 *023 *064
7 1, 108
а Е. Cats iff, А. То bo I sky, J. Coil. S3, 11,375 (195э). Звездочки означают,
по числа, у которых они стоят, относятся к следующему ряду.
//(-), может быть получено пз опытов по ползучести. Соотноше-
ние между Л(т) и Н(~), выведенное Гроссом [2], имеет вид:
/7(т)
(7,7)
[//(р)/(т —n)]dr
Если исходные экспериментальные данные получены пз опы-
тов по ползучести пли из испытаний в динамическом режиме, то
для них можно использовать подобные же способы приближения.
Целесообразно ввести и другие виды функций распределения
[такие, как Z.(t)J в зависимости от исходных экспериментальных
данных по вязкоупругости. Удобная сводка литературы дана в
обзоре [3].
8. Эмпирические функции распределения времен релаксации
131
Литература
1. Е. Cats iff, А. V. Т о Ъ о 1 s к у, J. Coll. Sci., 10. 375 (1955).
2. В. Gross, Mathematical Structure of the Theories of Viscoelasticity, Her-
mann, Paris (1953); R. S i m h a, J. Appl. Phys., 13, 201 (1942).
TH. L e a d e r m a n, in Rheology: Theory and Applications, vol. Il,
F. R. Eirich (ed.), Academic Press, New York. 1956. See ch. I, especially
Table III, p. 49
8. Некоторые эмпирические функции распределения
времен релаксации
Для того чтобы более глубоко разобраться в методах исследо-
вания вязкоупругих свойств полимеров, желательно рассмотреть
несколько спектров времен релаксации Н(т), оказавшихся по-
лезными при изучении вязкоупругих свойств полимеров.
При обсуждении гистерезисных свойств ферромагнитных ма-
териалов Беккер и Доринг 11] ввели спектр времен релаксации,
который мы будем называть «ящикоподобным» (прямоугольным)
спектром.
Кун, Кунцле и Прайсман ввели тот же самый спектр для опи-
сания вязкоупругих свойств полимеров с помощью модели Ви-
херта ]2]. Используя этот спектр, они показали следующее:
если кривая ползучести описывается эмпирическим уравнением
Ee(t) = (8,1)
го комплексный динамический модуль в этом же временном ин-
тервале имеет вид:
Е"
г.Ь^
2а2
(8,2)
Математические методы, необходимые для получения этого
соотношения, очень сложны, и так как приложение модели Ви-
херта к экспериментальным данным по ползучести затруднено,
то было необходимо сделать несколько приближений. Тем не ме-
нее это соотношение оказалось первым численным соотношением
для данных по истинной ползучести п по испытаниям в динами-
ческом режиме.
А. Тобольский и сотрудники использовали ящикоподобный
спектр [3] для получения численных соотношений между данными
по релаксации напряжения и данными по испытаниям в динами-
ческом режиме |4, 5], между данными по релаксации напряжения
п данными по вязкости при течении [6], а также для расчета в
первом приближении полного спектра времен релаксации аморф-
9-:
132
Гл. Ш. Теория линейной вязкоупругое г и и упруговязкости
кого полимера (последнее для полиметилметакрилата) [7]. По-
скольку модель Вихерта по своей природе легко применима для
данных по релаксации, расчеты оказались очень ясными.
Ящикоподобный спектр определяется уравнениями вида:
// (т) = Е(); т3 < - < т,„ ।
//(Q0. г<тя; т>т„,|
(8.3)
Величины Er(t), Е'(со), Е"(ы) и т/б можно получить из уравнений:
Ег (() - j И (т) ехр (—tit) d In т
Е' Н =
Е"(ш)= [ H(t) -гс/1пг
I I -f- ш-т4
= ( Н (т) dt
(8, 4)
(8.5)
(8,6)
|8.7)
Подставляя в эти уравнения /7(т) из (8, 3), находим:
ЕД/) - Еа [El ( --- -) -Ei(—ЛУ| (8,8)
Гн..)’ (8’9'
Е" (w) = Ео (arc tg сот,,, — arc tg ют3) (8, 10)
д(/) = Ео (тт — т3) Еотт (8,11)
В уравнении (8, 8) функция Ei есть хорошо известный и оца-
тельио табулированный экспоненциальный интеграл |8].
На рис. III, 6 приведены зависимости Er(t) от 1g/; Е' (1/ш) от
lg (l/u>); Е" (1/ю) от 1g (1/ы) и Н(т) от IgT для функций (8, 3) и
(8, 8)—(8, 10). В этом случае Ео принят равным 7,2-10Б дин!см2\
т3 взято равным 10-4’34 </; т,„ — равным 1О‘>70 ч. Л1одуль на
«плато» равен Ев 1п(т,п/та).
Если lgT3 и lgT,„ отличаются больше, чем па единицу, как это
имеет место в данном случае, то центральная часть зависимости
8. Эмпирические функции распределения времен релаксации 133
Er(t) от 1g/ представляет собой прямую линию, наклон которой
определяется уравнением:
^it~\ = 2,303Ео = 2,303/7 П) = 77(0 (8,12)
где 2,303 Ео, конечно, является высотой прямоугольника, изобра-
жающего зависимость//(т) от 1g?. .Уравнение (8, 12) эквивалент-
Рис. 111,6. Ег (/), Е' (1/“). £" (!/'>) И НО) для
ящикообразиого (прямоугольного) распреде-
ления в полулогарифмических координатах
у— 1/ы [А. V. Т о 1> о 1 s к у, В. A. D и-
п е 1 1, R. D. Andrews, Text. J?es. J..
21, 404, 1951].
но уравнению (6, 8), являющемуся результатом первого прибли-
жения, описанного в разделе. 6. Оно вытекает из свойств экспонен-
циального интеграла.
На рис. 111,7 снова дана зависимость функции Er(t) от 1g/.
Прямолинейный участок кривой экстраполирован на точки А и В,
отмеченные на рисунке. Из свойств экспоненциального интеграла
следует, что значения времени в точках А и В относятся к тя и
следующим образом [51:
4а- = -^-= 1,781
а -Ц
(8, 13)
134
/’.i. III. Теория линейной вязкоупругости и упруговязкости
Уравнения (8, 12) и (8, 13) очень полезны, так как часто экспе-
риментальные кривые падения напряжения, в виде зависимости
Er(t) от 1g/ являются линейными па протяжении многих десятич-
ных порядков времени. Это особенно справедливо для линейных
аморфных полимеров в области перехода к эластовязкому состоя-
нию и довольно часто справедливо для кристаллических полиме-
ров. Во временном интервале, где dEr(t)id\gt есть константа, ве-
личину которой удобнее обозначить через 2,303 £о, соответствую
шин участок функции релаксации НЕ} может быть апроксимиро-
Рнс. 111,7. Константы для ящикиобразиого (пря-
моугольного) распределения [по топ же работе—
см. рис. 111,6].
вап прямоугольником с высотой 2,ЗОЗ£о. В благоприятных сл\
чаях (область эластовязкого состояния линейных аморфных по-
лимеров) т„г и иногда даже т3 могут быть получены с помощью
уравнения (8, 13).
Тогда вязкость т/() можно получить из уравнения (8, И).
При тех значениях времени, когда dE(t)/d\gt является кон
сгантоп, Е"(1/ы) также приблизительно постоянен, что можно
.легко увидеть пз уравнения (8, 10) и из рис. III, 6.
1 \ - р __ - dEr (/)
м ) 2 ° ~ 4,606’ dig t
(8, 14i
В этом интервале времени Е' (1 ,!а>)^Ег(() ,что можно видеть на
рис. III, 6.
В 1952 г. было предложено идеализированное распределение
времен релаксации //(-) для сплошного спектра релаксации ли-
нейного полипзобутилена [9]. Ойо состоит из клинообразного и
8. Эмпирические функции распределения времен релаксации
135
прямоугольного «ящиков», если на графике отложена зависимость
lg/7(x) от Igx. Клин является частью спектра времени со стороны
релаксации быстрых процессов (связанной с переходной областью)
и не зависит от молекулярного веса. «Прямоугольник» передает
участок, соответствующий большим временам релаксации, свя-
занным с областью эластовязкого состояния; сплошной «прямо-
угольник» перемещается по логарифмической шкале времени при
изменении молекулярного веса полимера. Для полимеров с боль-
шим молекулярным весом клин и прямоугольник отделяются друг
от друга. Для полимеров с низким молекулярным весом клин и
прямоугольник сливаются вместе. При 25° клинообразная и
прямоугольная функции распределения времен релаксации поли-
изобутплеиа, имеющего случайное распределение молекулярных
весов (см. Приложение £), приобретают вид:
Клин:
11W = ~Т7Г; Ti < т < т2 (8’ 15)
Прямоугольник:
Я(т) = £0; (8,16)
Для всех других значений т функция Н(х) равна нулю.
Если релаксационный модуль выразить в динах на квадратный
сантиметр, а время в часах, то константы в указанных выше урав-
нениях будут иметь следующие значения:
/И = 8,9-103; £0 = 7,2-105; = 10-12>5; т3=10-М;
т3 = 9,65 1О"28 Л13;30; хт= 1,06 IO"20 <’30 (8,17)
В уравнениях (8, 17) есть средневесовой молекулярный вес.
На рис. III, 8 показано идеализированное распределение Я(т)
типа клина и прямоугольника при 25 °C на примере полиизобу-
гилеиа с «случайным распределением», для которого т3=10-4’34 ч,
а хт— 101,7° ч. Для сравнения на рисунке дан график действитель-
ного распределения H2b(x), полученного для нефракционнрован-
ного образца полпнзобутилена (полппзобутплен NBS), который
выпускает Бюро стандартов США. Средневесовой молекулярный
вес такого образца равен Mw= 1,6- 10е. Если это значение Mw
подставить в уравнение (8, 17), то получим значения т3 и хт, очень
близкие, но не полностью совпадающие с теми значениями, кото-
рые приведены на рис. Ill, 8, как «наиболее приемлемые». Идеали-
зированное распределение типа клина и прямоугольника дейст-
вительно дает очень хорошее приближение к реальному распреде-
лению.
Показанный пунктиром прямоугольник на рис. III, 8 есть
идеализированная функция для области эластовязкого состояния
130 Гл. 111. Теория линейной вязкоупругости и упруговязкости
члюнодисперсниго» полиизобутилена с тем же значением -г,,,. Нан-
депо, что для «монодисперсных» полимеров Ео = 1,06 • 10е, а т,„
п высокоэластический модуль на «плато» имеют ту же величину,
что и у образцов с «случайным распределением», но с тем же мо-
лекулярным весом Мш |61. Из последнего соображения можно
заключить, что площади пунктирного и сплошного прямоугольни-
ков одинаковы.
Рис. III,8. Идеализированное распределение ти-
па клин—прямоугольник //(х) для полипзобутпле-
на NBS (Л1Ш= 1,60-10е) при 25 °C. Прямоуголь-
ник, изображенный пунктиром, представляет со-
бой идеализированное распределение в области
эластовязкого состояния для случая весьма узкой
фракции полимера с тем же Mw. Для сравнения
показано распределение Н2в (т), полученное из
Er (I) [А. V. Tobolsk у, J. Am. Chem. Soc.,
74. 3786 (1952)]; J. Арр). Phys., 27, 673, 1956].
Уравнение (8, 15) для «клина» позволяет получить простые
зависимости для /?,(/), £'(ы), Е"(ьл) и
(8, 18)
(8, 19)
л. Эмпирические функции распределения времен релаксации
137
„„ /л лл I " \1/2 (2<ог)1/2 1 .
Е (со) = М arc lg ------------------r In
от -|- (2<о-)1/д -j- 1 “Г"2
<оТ --- (2<от)*^2 j- 1
(8, 20)
== 2М (тУ2 — 7}/2)
(8,21)
Функция Г(/х (1/2) есть тщательно табулированная неполная
гамма-функция [10].
Как следует из уравнений (8, 8) —(8, 11) и (8, 18)—(8, 21),
вклады «клина» и «прямоугольника» складываются.
Рис. 111,9. Er |l), Е' (1/ы), Е" (1/<о) и И (т) для распре-
деления типа клин—прямоугольник в логарифмических
координатах (А. V. Tobolsk у, .1. Am. iCIiem.
Soc., 74, 3786, 1952].
На рис. Ш, 9 дана зависимость Ig/^/) от IgZ; Ig/ET' (1/со> от
lg(l/co); lg.E"(l/со) от Igl/со и 1g//(т) от IgT для указанного выше
распределения клин—прямоугольник, соответствующего урав-
нениям (8, 8)—(8, 11) и (8, 18)—(8, 21). Константы имеют те же
значения, что и в уравнении (8, 17), где т3=10~4'34 ч, а х,„== 101»70 ч.
Прямоугольные распределения для области эластовязкого
состояния полипзобутилена получили дальнейшее развитие в ра-
ботах Ниномия и Фуджита, рассмотревших общий случай распре-
деления молекулярных весов [11].
138
Гл. HI. Теория линейной вязкоупругости и упруговязкости
Для переходной области была получена видоизмененная функ-
ция [12]:
«(Ч = -^<.ехр1-<£4>'±) (8>22>
Это уравнение дает возможность перейти от уравнения (8, 4)
к следующему выражению для релаксационного модуля:
Er (t) ~ (8,23)
Уравнения (8, 22) и (8, 23) будут использованы в главе IV
для описания переходной области у аморфных полимеров.
tgr
Рис. 111,10. Сравнение действительного распределения Н(~.) с первым
и вторым приближениями для случая прямоугольного распределе-
ния [Е. С а 1 s i f f, А. Tobolsky, Tech. Rept.. RLT-5 to
the ONR, 1953].
6. Эмпирические функции распределения времен релаксации 139
Одна очень интересная возможность, вытекающая из точного
знания и Er(t), предусматриваемого этими идеализирован-
ными распределениями («прямоугольник», «клин» и «видоизменен-
ный клин»), состоит в том, что можно испытать действенность
приближенных методов, используемых для вывода Нг (с), //2а(т)
и HSb(x) из £,(/) с помощью уравнений (6, 8), (6, 9) и (6, 10). С по-
мощью известных.релаксационных функций Er(t) для прямоуголь-
ника, клина и видоизмененного клина были рассчитаны значения
и Я2(,(т) и результаты сравнены с известными дейст-
вительными величинами Н(~) [131. Это показано для прямоуголь-
ника на рис. III, 10, для клина на рис. Ill, 11, а для видоизменен-
ного клина на рис. III, 12. Приближения хороши в центре распре-
делений, по недостаточны в некоторых краевых участках. Как
Рис. 111,11. Сравнение действительного распределения Я(т) с первым и вторым
приближениями для случая клинообразного распределения [по тон же работе-
см. рис. 111,10].
140
Гл. III. Теория линейной вязкоупругости и упруговязкости
будет показано в главе IV, если функция распределения справед-
лива для аморфного полимера при одной температуре (например,
25 °C), то она будет справедлива и прп других температурах, и ее
можно получить путем простого перемещения всего распределения
по логарифмической осп времени. Этот прием получил название
lg T
Рис. 111,12 Сравнение действительного распределения //(.) с первым и вторым
приближениями для случая видоизмененного клинообразного распределения
[по той же работе—см. рнс. 111,10].
угого принципа оказывается возможным, произведя измерения
при разных температурах, значительно расширить временную
шкалу данных по вязкоупругости.
Недавно Смит получил [141 точное распределение времен за-
паздывания для клинообразного и прямоугольного распределения
времен релаксации с помощью формулы Гросса (7, 7).
Литература
1. R. Becker, W. Boring, Ferroniagnetismus, Springer, Berlin, 1939,
p. 254.
2. W. К u h u. О. К u n z 1 e, A. P re i ss ma ii, Helv. chim. acta, 30,
307 , 464 , 839 (1947).
9. .Упруговязкие свойства .максвелловского тела
141
3. R. D. Andrews, N. Hof in a n-B a n g, A. V. Tobolsk y,
J. Polymer Sci., 3, 669 (1948).
4. B. A. D u n e 1 I, A. V. T о b о 1 s к y, J. Chem. Phys., 17, 1001 (1949).
5. A. V. Tobolsk y, R, D. Andre w s, B. A. D u n e 1 1, Text.
Res. J., 21, 404 (1951).
6. R. D. Andrews, A. V. Tobolsky, J. Polymer, Sci., 7, 221 (1951).
7. J. R. McLoughlin A. V. Tobolsky, J Coll. Sci.. 7. 555 (1952).
8. Tables of Sine, Cosine and Exponential integrals, 2 vols., WPA Tables, New
York, 1940.
9. A. V. Tobolsk y, J. Am. Chem. Soc., 74 , 3786 (1952).
10. Karl P.e a r s о n, Tables of the Incomplete Г Function, His Majesty’s
Stationery Office, London, 1922.
U.K. NinOmiya, H. Fujita, .1. Coll. Sci , 12, 204 (1957); J. Poly-
mer Sci., 24, 233 (1957).
12. A. V. T о b о 1 s к у, J. R. Me Lo uglil i n, J. Polymer Sci., 8, 543
(1952).
13. E. C a t s i f f, A. V. 1 о b о I s к у, Tech. Rept. RLT-5 to the Office of
Naval Research, Contract Ne 6—onr-27021, Project № 6 onr-27021, 1953.
14. T. 1.. Smit h, Trans. Soc. Rheol., 2, 131 (1958).
9. Упруговязкие свойства максвелловского тела
Поскольку приближенные методы, описанные в предыдущем
параграфе, наиболее точны в случае мало меняющейся функции
распределения Н(т), представляется интересным рассмотреть
свойства максвелловского тела, для которого Н(т) является раз-
рывной функцией. Для максвелловского тела имеем:
Н (т) — -.Е = tpE; при т т;, ।
Н (т) = 0; при т Тр I
О, 1)
Различные функции упруговязкости имеют вид.
Er (I) = Е ехр ( (9, 2)
Г
= (9,3)
р
= <9’4)
£"(У) = 4^4- (9,5)
У i р
В уравнениях (9,4) и (9,5) у~ 1/и. Зависимости lg£r(/l.
lgEc(/), IgE'(y) и JgE"(r/) от ]gt или Jgt/ даны на рис. III, 13.
Из уравнения (6, 7) первое приближение Я(т) дается выраже-
нием вида:
|¥nr?L= —ЕехрИДр) (9,6)
42
Гл. 111. Теория линейной вязкоупругости и упруговязкости
Рис. 111,13. Er(t), Ec(t), Е’(у) и £"(1/)для
максвелловского тела (у—\/ы).
Рис. 111,14. Сравнение Ер, Нх (т) н //.2„ для
ма ксвелловс кого тела.
9. Упруговязкие свойства максвелловского тела
143
Интересно сравнить //L (т) с точным значением Я(т), даваемым
уравнением (9, 1). Также стоит заметить, что при т=тр имеет
место:
Ях(тр) = 0,368£
Нг (т„) 2,303 HL (тР) = 0,847£
ig 77, (*₽) - 1е"772ь - 1g е W12
(9,7)
(9,8)
(9,9)
Применимость первого приближения а также второго
приближения Н2Ь(т) к Er(f) для максвелловского тела и сравнение
с Е показано на рис. III, 14.
Глава IV
ВЯЗКОУПРУГИЕ И УПРУГОВЯЗКИЕ СВОЙСТВА
ПОЛИМЕРОВ
I. Релаксация напряжения в аморфных полимерах
Исследование релаксации напряжения обычно производится в
интервале времен от 1 до 10е сек. Верхний предел определяется
герпением исследователя. Нижний предел обусловлен тем, что
очень быстрые растяжения вызывают инерциальные и тепловые
эффекты. Если напряжение измеряется без автоматической реги-
страции, то нижний предел доходит до 10 сек.
Данные по релаксации напряжения при различных температу-
рах для двух различных аморфных полимеров—полипзобутплена
и полнметплакрилата—показаны на рис. IV, 1 —IV, 6. Эти ри-
?не. IV, I. Кривое релаксации напряжения в области эласто-
вязкостп для нефракционированного полиизобутилепа
,31^=1340000) при растяжении 50%. (Пунктирная прямая
зависимость lg£ от 1/Г, причем k= 1 при 32 °C) [R. D.
\ п <1 г е w s, N. Holm а n-B a n g and A. V. Tobol
s k v, J. Polymer Sci., 3, 669 (194811
I. Релаксация напряжения в аморфных полимерах
145
время, v
Рис. IV,2. Зависимость (298/Z1) Er (I) от IgZ для не-
фракциоиированиого полипзобутилена (Мv=6600000) в области
эластовязкости. Здесь ясно видна область высокоэлас-
тичности, а также участок переходной области с низкими зна-
чениями модулей ]). Andrews, А. V. Tobol sky.
Л. Polymer Sei.. 7. 224 (1951)1
0—833
146
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
Время, ч
Рис. IV,3. Зависимости (298/Г) rEr(t) от lg< для нефракцнопп-
рованиых полиизобутиленов различных средних молекуляр-
ных весов при 30 °C IR. D. A n d г е w s, N. Hof ma n-
' R a n g. A. V. Tobo Isky, J- Polymer Sci.. 3. 669
(1948); 7, 224 (1951)].
I. Релаксация напряжения и аморфных полимерах
147
сункп взяты из оригинальной литературы и иллюстрируют харак-
терную особенность релаксации напряжения, общую для всех
аморфных полимеров. Они также показывают, как в действитель-
ности сложилось на основании результатов экспериментов понятие
об отдельных областях вязкоупругости и упруговязкости [1]].
Существует пять таких областей: область стеклообразного состоя-
ния, переходная область, область высокоэластического (каучуко-
Рнс. IV,4. Зависимости логарифма (298/Т) Er(t) от 1g/ для
иефракционированного полиизобутилена (Л1v—1240000) в интер-
вале температур от —70 до 100 °C, который включает переход-
ную, высокоэластнческую, эластовязкую области п область
вязкого течения. Модуль в переходной области не зависит
от молекулярного веса пли небольшого числа поперечных
связей. Пунктирная линия представляет собой зависимость
lg/с от VT. причем при —66 °C k=l [G. М. В г о w п,
А. V. Т о b о 1 s 1< у. J. Polymer Sci., 6. 165 (1951)].
подобного) состояния, область эластовязкого состояния п область
вязкотекучего состояния (вязкого течения). Значения модулей,
которые грубо определяют эти интервалы, приведены во II главе;
они видны на рис. IV, 5 и IV, 6.
Для аморфных полимеров с достаточно длинными цепями вяз-
коупругие свойства в стеклообразной и переходной областях не
зависят существенно от длины цепи. Для «бесконечных» цепей
область высокоэластпчностп расширяется до очень высоких тем-
ператур (до тех пор, пока химические изменения, такие, как раз-
рыв связей, не начнут воздействовать на полимер). Для сеточных
10”
148
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
полимеров достигается истинное равновесное высокоэластнческое
состояние, в котором значение модуля зависит от числа попереч-
ных связей. Здесь также область высокоэластичности простирает-
ся до очень высоких температур, пока химические изменения не
начнут влиять на пространственную сетку. Области эластовязкости
и вязкого течения в отсутствие химических изменений в сеточных
полимерах совершенно не проявляются.
Рис. IV.5. Зависимости lg£r (7) от 1g/
для нефракционированного полпизо-
бутилена NBS (41г,= 1350000) в интер-
вале температур от —83 до 25 °C
(Е. С a t s i f f, A. V. T о b о 1 s k у,
J. Coll. Sci., 10, 375 (1955)1.
Рис. IV,6. Зависимости (z) от
lg( для нефракционированного поли-
метилметакрилата (Л1р= 3600000) в ин-
тервале температур от 40 до 135 °C
[J. R. М с L о u g h 1 i n,
Л. V. Tobolsk у, J. Coll. Sci.,
7. 555 (1952)].
Для линейных полимеров с конечными длинами цепей как об-
ласть эластовязкости, так и область вязкого течения заметно зави-
сят от длины цепи (и распределения длин цепей), как это видно
из рис. IV, 3, п что будет подробнее рассмотрено в разделе 7.
Литература
1. А. V. ToboCky, .1. R. McLoughlin, J. Polymer Sci., 8, 543
(1952).
J. Принцип 1емпературно-врел1еннО11 суперпозиции
149
Рис. IV,6а. Релаксометр.
2. Принцип температурно-временной суперпозиции
и вспомогательная кривая
Как уже отмечалось, практическим интервалом шкалы времен
для измерений релаксации напряжения является промежуток
времени от 10 до I06 или 10е сек. Определенно известно, что при
заданной температуре весь набор вязкоупругих и упруговязких
150
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
свойств не перекрывается этим интервалом из четырех пли пяти
десятичных порядков времени. Очевидно, что вопросом крайней
важности является нахождение удовлетворительного метода
экстраполяции.
Впервые такой метод был предложен Лпдерманом [1, 2],
который нашел, что экспериментальные данные но ползучести,
полученные при различных температурах 131, могут быть совме-
щены горизонтальным переносом вдоль осп логарифма времени’’.
Это равноценно утверждению, что действие температуры на вязко-
упругие и упруговязкпе свойства заключается в умножении (пли
делении) временной шкалы на определенный для каждой темпера-
туры коэффициент. Например:
Ec,T\k(T)t\--= Е1Л, (!)
(2, I)
о го
= Ес_ 1 (t)
(2,2)
t
В уравнениях (2, 1) и (2, 2) Ес, r(t) есть модуль ползучести
при температуре Т\ Ec,To(t) есть модуль ползучести при произ-
вольно выбранной характеристической температуре То. Вели-
чина k прямо получается из экспериментальных кривых ползуче-
сти (в зависимости от логарифма времени) путем измерения вели-
чины смещения их вдоль шкалы времени до совмещения с кривой,
соответствующей температуре То. Величина k, называемая коэф-
фициентом времени, является функцией только температуры и
выбирается равной единице при характеристической температуре
То, причем k является функцией, убывающей по мере увеличения
температуры. Согласно уравнению (2, 2) функция E~(tlk) не за-
висит от температуры, если опа изображена графически в зависи-
мости от t/k пли lg(t/k).
Если применимы законы линейной вязкоупругости или упру-
говязкостп, то равенства, аналогичные (2, 1) и (2, 2), действитель-
ны для любых релаксационных измерений. Например, аналогич-
’ Следует заметить, что основные идеи принципа температурно-времен-
ной суперпозиции были впервые в совершенно отчетливой форме высказаны в
работах А. П. Александрова и IO. С. Лазуркина (ЖТФ, 9, 1249, 1261 (1939)).
давших также простейшие количественные соотношения. Эти идеи были ши-
роко использованы во многих более поздних работах советских ученых. Не-
сомненной заслугой Лидермана, Тобольского и других зарубежных ученых
является детальная разработка метода расчета перехода от одной темпера-
туры к другой и широкое экспериментальное исследование температурно-вре-
менных зависимостей для многих полимеров.—Прим. ред.
2. Принцип температурно-временной суперпозиции
151
ними формуле (2, 2) являются уравнения:
£ т [ — r'т° [ k (Т) j — Е, т (t) (2,3)
F' Г * 1 Lt° [ыА (Т) j =я (4j (2,4)
Ет» [ МТУ |; = Ег(-> (2. 5)
У _ X = k(T) (2, 6)
Принцип температурно-временной суперпозиции впервые был
применен к экспериментальным данным в точных числовых расче-
тах Тобольского и его сотрудников (Эндрюс и др.) [4—81. Они
также ввели коэффициент времени k(T), дали его таблицы и
видоизменили принцип суперпозиции с тем, чтобы учесть пропор-
циональность модуля абсолютной температуре следующим образом:
^Er,T(kt) = Ег,Го(/) (2,7)
Ег,т0 г t [А (Г) II
Ес, То 1 / 1 А(Т) ] _ т« т
Ет [- [а 1 1 >А (Г) ] II
Ет 1 _ A(T)j II
Er.-rit)
Е'т
Ет I7)
= k(T)
Т^о
(2.8)
(2, 9)
(2, Ю)
(2, 11)
(2, 12)
Можно построить вспомогательную кривую, охватывающую
гораздо более широкий интервал логарифма времени, перемещая
кривые (Го/Т)ЕГ'Т(Г) [или \g(To/T)EriT(t)] вдоль оси 1g/ до со-
вмещения их с ЕГ'Т (/) или [lg£'r,r (/)[. То же самое, конечно,
можно проделать п для (TolT)Ec(f),°(TolT)E' (1/со) и т. д.
Впервые наиболее полная вспомогательная кривая была полу-
чена для вулканизата GR-S (в 1945 г.) [41.
Первоначальный график представлен на рис. IV, 7, где ис-
пользована размерность дин/см2- вместо фунтов на квадратный
дюйм и обозначение (298/7)£'с(7) вместо приведенного модуля пол-
зучести для того, чтобы получилось соответствие с обозначениями
этой книги.
152
Гл. IV. Вязкоупругие и упруговязкиг свойства полимеров
Этот график показывает зависимость lg (298/7')£'t(T) и
—Igft(T) от lg/. Для того чтобы получить вспомогательную кривую
для любой температуры, необходимо перемещать шкалу 1g/ вдоль
Рис. IV,7. Вспомогательная кривая,
выражающая зависимость IgE^ (/) от
1g/ для GR-S при температуре
— 40°С. [А. V. Tobolsky, R D.
Andrews, J. Chem. Phys.,
13,3 (1945)].
зучести, полученные в
горизонтальной осп до тех
пор, пока значение lg/ = ()
не совпадет с заданной
температурой на шкале
lgZ> ('/’). В первоначальной
публикации были приведе-
ны данные, показывающие,
что экспериментальные
значения/г для ползучести
и релаксации напряжения
приблизительно равны. В
1952 г. были получены бо-
лее полные данные по ре-
лаксации напряжения на
подобном же образце
вулканизата GR-S 191.
Вспомогательная кривая,
дающая зависимость Ег
(f) от 1g/, представлена
на рис. IV, 8. В табл.
IV, 1 сопоставляются
значения k для релак-
сацип напряжения и пол-
зпаченпями, полученными на
1945 г., со
Рис. IV,в. Вспомогательная кривая, выражающая
зависимость lg£,. (/) от lg(//A-) для GP-S U
R. Bischolf, Е. С a t s i f f. A. V. To-
bolsk). .1. Am. Chem. Soc.. 74. 3378 ,1952)]
2. Принцип температурно-временной суперпозиции
153
основе более новых данных по релаксации. Учитывая историю
этих исследований, согласие следует считать превосходным.
Таблица IV,!
Значения k, полученные из данных по ползучести
и релаксации для GR-S
Релаксация3 (1952 г.) Релаксация0 (1945 г.) п б Ползучесть (1945 г.)
ig но Ig k (Г) 1g * (О
—64,0 —60,0 7,7 6, 37
-61,0 6,9
-58,8 6,3
-57,6 5,7
-55,0 4,68 4,60 4,60,
-52,5 3,84
— 50,0 -49,5 -47,5 -45,0 2,72 2,18 2,48 2,55 0,70
-44,0 0,80
—40,0 0,00 0,00 0,00
-32 8 -0.6
30,0 — 1.05
-21,0 —2,0
-20,0 — 1.37
-15,0 - 1,75
- 8,5 -3,15
а) J. В i sc h о f f, E. Cats iff, A. V. Tobolsky,
J. Am. Chem. Sec., 74, 3378 (1952).
°) A. V. Tobolsky, R. D. Andrews, J. Chem.
Phys., 13 , 3 (1945).
В 1948. г. были получены первые вспомогательные кривые для
эластовязкой области и области вязкого течения для линейного
иол и изобутилена (рис. IV, 9); было показано, что кривые вязкости
и упругости перемещаются с температурой одинаково [5, 6, 10].
Было точно установлено существование квазистатического высо-
коэласгнческого модуля [5, 6, 10]. Для линейного аморфного
154
Гл. /I. Вязкоупругие и упруговязкие свойства полимеров
полимера первая наиболее полная вспомогательная кривая полу-
чена для полнметилметакрилата (рис. IV, 10).
Для аморфных полимеров форма кривых релаксации напря-
жения не зависит от растяжения образца, как показано в разделе 2
главы III. По этой причине принцип температурно-временной су-
перпозиции можно непосредственно применять к данным по ре-
лаксации напряжения, полученным при различных температурах
lg t. Т, ч
Рис. IV,9. Вспомогательная кривая, выражающая зависи-
мость lg£r(Z) от 1g/ для высокоэластической области
полипзобутилена для образна со средпевесовым моле-
кулярным весом 6600000. Светлые кружки обозначают
экспериментальные данные, полученные при 30 СС;
сплошные кружки показывают экстраполированные
значения, полученные при других температурах и
приведенные к 30 "С с помощью принципа температур-
но-временной суперпозиции. Сплошные кривые соот-
ветствуют идеализированному прямоугольному распре-
делению. Пунктирные кривые представляют собой фак-
тическую вспомогательную кривую и соответствующую
ей кривую для //(т) [R. D. Andrews, А. V-
Tobol'skv, J. Polymer Sci., 7, 221 (1951); 3,
669 (1948)].
и высоком заданном растяжении. Многие пз прежних вспомога-
тельных кривых, построенных по релаксации напряжения, были
получены для напряжения при растяжении на 50%, а не для мо-
дуля. Те кривые, которые воспроизведены в этой книге, преобразо-
ваны в соответствующие зависимости релаксационного модуля
(использованием уравнения кинетической теории для выражения
связи между деформацией и напряжением) для того, чтобы сохра-
нить однотипной систему обозначений.
На рис. IV, 11 приведены идеализированные вспомогатель-
ные кривые (соответствующие «клинообразно-прямоугольному»
распределению) для полипзобутилена с тремя различными дли-
нами цепей и для слабо сшитого полипзобутилена с очень длин-
2. Принцип температурно-временной суперпозиции
15э
ними первоначальными цепями [11]. На рис. IV, 12 показана
реальная вспомогательная кривая релаксации напряжения для
полиизобутнлена NBS [12].
Рис. IV,10. Зависимости lg£r (I) от
IgC н lg//(x) от igr, приведенные к
40°C, для нефракционированного по-
лнметилметакрилата с двумя различ-
ными средними молекулярными веса-
ми. 7/(т) показана только для переход-
ной области. Образец (/) имеет мо-
лекулярный вес Mv= 150000, обра-
зец (2)—/17^=3600000 [J. R. .Mc-
Loughlin and А. V. Т о b о I-
s к у, J. Coll. Sci., 7.555 (1952)1.
Позднее Ферри видоизменил принцип температурно-времен-
ной суперпозиции для того, чтобы учесть изменения плотности
в полимерах при различных температурах [131. В результате
этого уравнения (2, 7) и (2, 12) должны быть заменены следую-
щими:
Е,. T[k(T)t\~ Ег Тл(Е (2,13)
7 о т, г d0
~Т\Г1Г
'о
k(T) - a-i
(2, 14)
Подобные же изменения должны быть сделаны н в уравнениях
(2, 8)—(2, 11). В уравнениях (2, 13) и (2, 14) do есть плотность при
температуре То, a d—плотность при температуре Т. Такая по-
правка совершенно незначительна для чистого (нерастворенного)
полимера, по она предлагается также как средство приведения в
соответствие данных для концентрированных растворов полиме-
156 t л. IV. Вязкоупругие и упругомзкие свойства полимеров
ров с данными для чистых полимеров [13]. В этом случае считают, что
плотностью является вес полимера в единице объема раствора.
Обозначение аг, аналогичное А’(7 ), будет рассмотрено немного
позднее.
Рнс. IV,11. Идеализированная вспомогательная кривая ре-
лаксации \f>Er (/) от Igr при 25 °C для полннзобутплена трех
различных средних молекулярных весов:
1—М., =1,36-10«: =2,8.Ю»; 3—ЛГ. =6.6-10».
IА. V. Г о b о 1 - к у, .1. R. McLoughlin. .1 Polymer Sci.. S, 543
(1K2)|
Так как кинетическая теория высокоэластичности не приме
ннма к полимерам в стеклообразном состоянии, то дальше равен-
ство (2. 13) будет видоизменено с целью учета этого обстоятель-
ства. Соответствующую небольшую поправку легче выразить
в выражении для податливости, чем в выражении для мо-
дуля 114.1
где Jx представляет собой податливость в стеклообразном состоя-
нии (весьма малая поправка).
2. Принцип температурно-временной суперпозиции
157
Ферри II его сотрудники использовали экспериментально из-
меренные вязкости как средство получения из равенства (2, 14)
коэффициента времени
k(T). Этот способ может
быть применен только для
несеточных полимеров,
которые имеют измеримую
вязкость. Они использо-
вали также 19] обозначе-
ние ат вместо обозначе-
ния k{T), которое раньше
было введено для вспомо-
гательных кривых релак-
сации напряжения [51.
Предположили 18], что
k будет применяться для
обозначения коэффициен-
тов времени полученных
из опытов по релаксации
пли из других вязкоупру-
Рис. IV, 12. Действительная вспомо-
гательная кривая lgEr(l) от 1g/ для
полипзобутилена NBS при 25 °C
[ Е. С a t s i 1if, Л. V. To bo Isky,
J. Polymer Sci., 19, 111 (1956)].
гих данных, в то время
как ат будет представлять собой характеристики, определяемые
из данных по вязкому течению, согласно уравнению (2, 14).
Литература
1. Н. Leaderman, Text. Res. J-, И, 171 (1941).
2. Н. Leaderman, Elastic and Creep of Filamentous Materials, The Textile
Foundation. Washington, D. C., 1943, pp. 16, 30, 76, 100.
3. Г1. Г1. К о б e к о. E. В. К У в ш и н с к и й, Г. И. Г у р е в и ч,
Изв. АН СССР, Сер. физ., № 3, 1937. стр. 329.
4. А. V. Tobol sky, R. D. Andrews, J. Chem. Phys., 13, 3 (1945).
5. R. D. Andrews. N. H о f m a n-B a n g, A. V. T о b о 1 s k y,
J. Polymer Sci., 3, 669 (1948); G. M. В ro w n. Ph. D. thesis, Princeton
University, 1948; K. W. Scott, Ph. D. thesis, Princeton University,
1950.
6. R. D. Andre w s. A. V. To bo I s k r. J Polymer Sci., 7, 221
(1951).
7. J. R. McLoughlin. A. V. T о b о I s k y, J. Coll. Sci., 7, 555 (1952).
8. A. V. Tobol sky- .1. R- McLoughlin, J. Polymer Sci., 8, 543
(1952).
9. J. Bischoff, E. C a t s i f I, A. V. To bolsky, J. Am. Chem.
Soc., 74, 3378 (1952).
10. IL Mark, A. V. Tobolsky, Physical Chemistry of High Polymeric
Systems, Interscience Publishers, New York, 1950, pp. 333—348.
II. R. S. Marvin, paper in Proceeding of the Second International Congress
on Rheology, Academic Press, New York, 1954, pp. 156—164.
12. A. V. T о b о 1 s k у, E. C a t s i f f, J- Polymer Sci., 19, 111 (1956).
13. J. 13. Ferry, J. Am. Chem. Soc., 72, 3746 (1950).
II. .I. D. Ferry, E. R. Fitzgerald. J. Coll. Sci., 8, 224 (1953).
158
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
3. Проверка принципа температурно-временной суперпозиции
Принцип температурно-временной суперпозиции, который
гак важен для экспериментального изучения упруговязкостп и
вязкоупругости, в настоящее время все еще является чисто эмпи-
рическим. Для объяснения справедливости этого принципа не
было еще выдвинуто достаточно убедительной теоретической кон-
цепции.
Следовательно, этот принцип очень важно проверить экспе-
риментально, чтобы увидеть, насколько его можно считать вер-
ным.
Первая экспериментальная проверка этого принципа 111 за-
ключалась в том, что была построена вспомогательная кривая,
выражающая зависимость — Е(/) от 1g/ при данной характеристи-
ческой температуре (30 °C). Она была получена на основе данных
для полпизобутилепа при нескольких значениях температуры в
интервале времени от 10-2 до 10 ч. Эта вспомогательная кривая
включала в себя область эластовязкости и область вязкого тече-
ния. Она была сопоставлена с динамическими данными зависи-
мости G'(l/w) от lg(l/«)), полученными при 30 °C в интервале вре-
мени 1/ы от 10 7’5 до 10'8 ч. Теория линейной упруговязкостп пред-
сказывает, что-^-E(0~G' (1/®)для широкого распределения вре-
мен релаксации, встречающегося в области эластовязкого со-
стояния. Это и было обнаружено, как показано на рис. IV, 13.
Оказывается, что для данного значения /=-1/а> величина G'(l/<o)
немного отличается от -^-Е(/), однако, учитывая эксперименталь-
ные трудности, следует считать, что в области высокоэластпч-
иости принцип температурно-временной суперпозиции соблю-
дается достаточно хороню.
Стандартные образцы полипзобутплена, полученные из Нацио-
нального Бюро Стандартов, будут обозначаться здесь как поли-
изобутилен NBS. Были сопоставлены результаты измерений вяз-
коупругости, включающие в себя релаксацию напряжения, вы-
нужденные колебания, свободные колебания и ползучесть, полу-
ченные несколькими объединенными лабораториями; при этом
оказалось, что по указанным данным можно построить непрерыв-
ную вспомогательную кривую [2|.
Ферри, Грендпн и Фитцджеральд 131 в опытах по вынужден-
ным колебаниям, Филлипов 141 в опытах по свободным колеба-
ниям, Катсифф и Тобольский [51 в опытах по исследованию ре-
лаксации напряжения провели широкую проверку применимости
принципа температурно-временной суперпозиции для полиизобу-
"члена в переходной н высокоэластпческой областях. Был применен
1
Рис. IV, 13. СравнениеЕг(/) с G'(l/<.>) для иолипзобу-
гилена в эластовязкой области при 30 °C. Точки—данные
по динамическому сдвигу, кривая—составная кривая ре-
лаксации (-д- [А. V. Т о b о 1 s к у, В. A. D и-
n е 1 1, R. D. A n d г е w s, Text. Res. J., 21, 404 (1951)].
Рис. IV. 14. Кривые Er(i) (точки внизу) и ЗО'(1/“) (точки
вверху) для полиизобутнлена NBS при —44 °C. Сплош-
ная кривая представляет собой вспомогательную
кривую релаксации напряжения. [А. V. Tobolsk}',
.1. Appl. Phys., 27, 673 (1956)].
-л#**
160 /л. /V. Вязкоупругие и упруговязкие свойства полимеров
обычный способ построения вспомогательной кривой Er(t) по
данным релаксации напряжения, полученным при различных тем-
пературах в интервале времени от 10"2 до 101 ч [51. Эта кривая
при —44 °C вместе с действительными экспериментальными ре-
лаксационными данными при —44 °C приведена на рис. IV, 14.
Приведены также значения динамического модуля 3G'(1 /со),
полученные при —44 °C в интервале времени 10_8>2<1/<о 10"7 ч.
Рис. IV, 15. Зависимость <?'(l/w) от lg(l/oj/?2e8) для полиизобутнлена
NBS. Сопоставление экспериментальных данных (о—данные из [3]; х—дан-
ные из (4]) с вычисленными из вспомогательной кривой релаксации напряжения
(G^b—сплошная кривая). [Е. Catsiff, А. V. Tobolsk у, J. Coll. Sci.,
10 , 375 (1955)].
Подтверждением принципа температурно-временной суперпози-
ции является тот факт, что значения 30'(1/<о) очень близки к зна-
чениям Er(t) [6].
Подобная, но более детальная проверка была проделана Кат-
сиффом и Тобольским [5], которые использовали вспомогательные
кривые ЕД/) при 298 °Кдля того, чтобы предсказать G'(a>) и С"(ы),
использовав метод второго приближения, описанный в III главе.
Эти расчетные данные сопоставлены с экспериментальными дан-
ными на рис. IV, 15 и IV, 16, а также в табл. IV, 2. В табл. IV, 3
даны значения k и К (см. раздел 4).
3. Проверка температурно-временной суперпозиции
161
Таблица IV, 2
Механические характеристики (в. дин'см2) полиизобутилена NBS, полученные
из опытов по релаксации напряжения*
lg t 1g - — Ip IU часы Ig£r-2S8(O6 1g Н 0s -) 1g ш' (•>) lg 36" (.»)
Расч. Эксп.в Расч. Эксп.«
— 14.4 10,48,
2 10,48 8.77
0 10,46, 8,96 10.50
— 13,8 10,45, 9,12 10,49,
6 10,44. 9,27 10,49
4 10,43 9.41, 10,48
9 10,41 9,55 10,47 9,49
0 10,39 9.66- 10,45, 9,57,
—12,8 10,37 9,77 10,43 10,45 9.64, 9,83
6 10,34 9,84. 10,41 10,42 9,70- 9,88,
4 10,30 9,91 10,38 10.38 9,75“ 9,93
О 10,25, 9,96- 10.34, 10,32, 9,78 9,96,
0 10,20, 10.01 10,30 10,27 9,81 9,99
— 11.8 10,15 10.03, 10,24, 10,20 9,82 10,00
6 10,07, 10,05, 10, 17, 10, 12 9,82 9,99,
4 9,98 10,04- 10,09 10,03 9.81 9,96,
О 9.88, 9.99, 10,00 9,91, 9,78 9,91
0 9, 77-, 9.93, 9,90 9,79 9,73- 9,82
—10,8 9,65, 9,87 9,78 9.67, 9,67, 9,73,
6 9,52, 9,76 9,66 9,55 9,62 9,64
4 9,39, 9,64 9,52, 9,43 9,53, 9,53
9 9,26 9,50 9,39 9,33, 9,45 9,42,
0 9,12, 9,35 9 2-Ч 9,22 9.35 9,32
—9.8 8,99 9,21- 9.Н, 9,11 9,24 9,22
6 8,86, 9.09 8,99 8.99, 9,12 9.Ю,
4 8, 73, 8.95 8,86 8,88 9,00 9,00
О 8.60-, 8,83 8,72, 8,76, 8,88 8,88
о 8,47 8,70 8,59, 8,62 8,76 8,77,
—8,8 8,33 8,57, 8,46, 8,48, 8,63 8,66,
6 8, 19, 8,43, 8,33 8,34 8,50 8,53
4 8.05, 8,27, 8,19, 8,21 8.37, 8,41
О 7,92, 8, 11., 8,04 8,06, 8,25 8,27,
0 7,80, 7,96 7.92 7,95 8, 12 8,14
—7.8 7,69, 7,83 7,80, 7,83 7,99 8,00
6 7, 58-, 7,71 7,69 7,72 7,85 7,86
4 7,48- 7,58, 7,57 7,62 7,71, 7,73
2 7,38- 7,46 7.-17, 7,52 7,58, 7,58,
0 7,29- 7,33 7,38, 7,42 7,45“ 7,46,
—6,8 7,21 7,20 7,29, 7,34 7,31, 7,33,
6 7,14 7,05, 7,21, 7,26 7. 18 7,21
4 7,08, 6,88 7,13, 7.20 7.05 7,08
О 7,04 6,72 7,07, 7, 14, 6,91 6,96,
0 7,00, 6,60 7,03 7,09, 6,77 6,85
—5,8 6,98 6,48 7,00 7,05 6,64 6,72
6 6,96 6,35, 6,97, 7,02 6,51 6,60
11—833
162
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
Продолжение
1g' 1g - 1g ... часы iecr.2UnV)v IB 11 (1g -) 1g 3G' (-I 1g 3G" (<")
Pac’i. Эксп.” Расч. Экс и.в
-1 6,94 6,26 6,95, 6,99 6,37 6,48
2 6,92., 6,16- 6,94 6,97 6,25 6,36,
0 6,91, 6,07 6,93 6,95- 6,12, 6,25
—1.8 6,90, 5,97 6,91. 6,94, 6,01 6,15
6 6,90 5,875 6,91 6,93, 5,90 6,06
-1 6,89, 5,78 6,90, 6,92, 5.81, 5,98
2 6,89 5,68 6,90 6,91, 5,73, 5,91
l) 6.88, 5,58, 6,89- 6,91 n,66 5,86
— 3,8 6,88 5,49 6,89, 6,90, 5.62 5,83-
6 6,87, 5,41., 6,89 6.89, 5,63 5,83
4 6,87 5,45- 6,88. 6,88- 5,66 5,83,
2 6,85, 5,75, 6,88 6,88 5.72, 5,85
0 6,84 5,96 6.86, 6,87 5.80, 5.86,
—2,8 6,82, 6,07 6,85 6,86 5,86, 5,88,
6 6,81 6, 14 6,83- 6.8-1 5,91 5,90
•1 6,79, 6,18 6,81- 6,82- 5,95 5,92-
9 6,78 6,20 6,79, 6,81 5,98 5,9o
I) 6,75-, 6,20, 6,77. 6.79 6,00 5,97,
1 ,8 6,73, 6,21 6,75, 6,77 6,01 6,00
6 6,71 6,215 6,73. 6,75 6,02 6,02,
4 6,68, 6,216 6.71., 6,73 6,03 6,05
9 6,65-, 6,21 з 6.69 6,71 6,03, 6,07
0 6,62- 6,22 6,66 6,68 6,04 6,09
—0,8 6,59 6,22 6,63 6,65, 6.04, 6,11
6 6,55 6,22 6,59- 6,62 6,04, 6,12,
4 6.50, 6,22 6.56 6.58, 6,04 6,135
9 6,455 6,22 6,52 6,53- 6,03, 6, 14
0 6,39, 6 22 6,47, 6,48 6,03 6,145
0,2 6,33 6,22 6,41 6,41, 6,02 6,14,
4 6,26 6,22 6,34, 6,00,
6 6,17, 6,22 6,27 5,99
8 6,08 6,19 6,19 5,96
1,0 5,97 6, 14 6,09, .5,92,
2 5,85 6,08 5,98 5.87,
4 5,70 6,00, 5,85 5,81
6 5,54 5,89, 5,71 5,73,
8 5,34 5,74 5,54 5,64
2.0 5, 18 5,!57, 5,35 5,53,
2 4,9 5,38 5.13 5,41
4 4,5 5,18 4,86 5,28
6 4.0 4,98 4,5 5,12,
8 ОС — oo X) 4,96
3,0 4,78,
_А. V. Т о b о 1 s к у. Е. С a I s i [ [, J. Pulymel Sci., IP, Ш ()9,t>).
о) Е. Cats iff, А. V. Т о b о 1 s к у, J. Coll. Sci.. 10, 37,> (19л>).
Ь1 J. Ferry, L. 1J. lir г a n d i n е jr., Е. R. F i t z g е г а 1 <1. J. Appl. 1’lrys., 24, 911
3. Проверка температурно-временной суперпозиции
163
Хотя такая обширная проверка принципа температурно-вре
менной суперпозиции для других полимеров не проводцЛасЬ1
все же есть уверенность в том, что этот принцип верен и
Рис. IV, 16. Зависимость G''(l/<o) от 1g (1/ш/г.2О8) для полиизобутнлена NBS.
Сопоставление экспериментальных данных (о—данные из [3]: х—данные нз
[4] с вычисленными из вспомогательной кривой релаксации напряжения (G'ze—
сплошная крнваа) |Е. С a t s i f 1, A. V. Tobolsky, J. Coll. Sci., 10,
375 (1955)].
для других аморфных полимеров в переходной и высокоэластиче-
ской областях, а также в областях эластовязкости и вязкого те-
чения.
Литература
1. А. V. Tobolsky, В. A. D unell, R. D. Andrews, Text.
Res. J., 31, 404 (1951).
2. R. S. M a r v i n. Paper in Proceedings of the Second International Congress
on Rheology, Academic Press, New York, 1954, pp. 156—164.
3. J. D. Ferry. L. D. G r a n d i n e jr., E. R. Fitzgerald,
J. Appl. Phys., 24, 911 (1953).
4. IV. Philippoff, data quoted in reference 2 above.
5. E. C a t s i f f, A. V. Tobolsky, J. Coll. Sci., 10, 375 (1955).
6. A. V. Tobolsky, J. Appl. Phys., 27, 673 (1956).
11
61 Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
Таблица 1V,3
Значения 1g k2ss и 1g К для полиизобутилена NBSa
т, ас ig ^2se 1g К Т. °C 1g 1g к
—82,6 12,05 2,73 —34,7 3,69 — 5,63
—81,7 11,89 2,57 —33,4 3,44 - 5,88
—80.8 11,53 2,21 —29,8 3,22 — 6,10
—79,8 1 1,95 2,63 —28,5 3,06 — 6,26
—79.3 11,51 2,19 —25,0 2,84 — 6,48
—77,3 1(1,89 1,57 —23,0 2,68 — 6,64
—76,7 10,70 1,38 —20,2 2,34 — 6,98
—76,5 10,47 1,15 — 19,9 2,45 — 6,87
—74, 1 9,70 0,38 — 14,8 2,08 - 7,24
—71,7 9,13 —0,19 — 9,9 1,78 - 7,54
—70,6 8,85 —0,47 — 5,0 1,51 — 7,81
—69,15 8,46 —0,86 — 0,1 1,22 — 8,10
—66,5 7,93 — 1,39 4,9 0,98 — 8,34
—65,4 7,62 — 1,70 9.8 0.72 — 8,60
—62,0 7,06 —2,26 14,8 0,51 — 8,81
—60,9 6,79 —2,53 19,9 0,27 — 9,05
—58,8 6,55 —2,77 2э,0 0,00 — 9,32
—56,2 6,04 —3,28 30,2 —0,21 — 9,53
—54,0 5,77 —3,55 34,9 —0,42 — 9,74
—51,4 5,47 —3,85 40,0 —0,62 — 9,94
—49,6 5,31 —4,01 50,0 —0,99 — 10,31
—44,6 4,75 —4,57 59,8 —1,31 — 10,63
—44,3 4,72 —4,60 80,0 — 1,90 — 11,22
—40,4 4,23 —5,09 99,9 —2,29 — 11,61
—40,1 4,20 —5,12
А. \ . Т о b о 1 s к у, Е. С a ts i и, J. Polymer Sci., IS, HI (1S.6).
4. Характеристические параметры аморфных полимеров
Как показано па примере данных, приведенных на рпс.
IV, 5—IV, 9, при помощи вспомогательных кривых можно полу-
чить некоторые параметры, характеризующие вязкоупругие свой-
ства аморфных полимеров. При достаточно малых временах мо-
дуль достигает значения, характерного для стеклообразного со-
стояния. Это предельное значение обозначим Ег. Для полимеров с
достаточно длинными цепями можно определить квазистатпче-
ский высокоэлаетпческий модуль для сеточных полимеров су-
ществует равновесный модуль высокоэластичности, зависящий от
4. Характеристические параметры аморфных полимеров
165
числа поперечных связен в единице объема, который мы, однако,
также обозначим через Е„.
Для любой температуры можно определить характеристиче-
ское время релаксации К\Т) как время, необходимое для того,
чтобы значение lg£r(/) упало до значения, равного (lg£x+lg£2)/2.
Функция К(Т) прямо пропорциональна k(T), но в то время как
абсолютное значение k(T) зависит от значения, выбранного для
характеристической температуры То, функция К(Т) определяет-
ся из опытов по релаксации напряжения для каждого полимера
однозначно.
Другим определяемым параметром является отрицательный
наклон вспомогательной кривой lg£r(/) от 1g/ в переходной об-
ласти, измеряемый в точке перегиба. /Мы назовем эту величину п.
Экспериментальные данные показывают, что и колеблется для
аморфных полимеров в пределах от 0,5 до 1,1.
. Наиболее важным параметром каждого аморфного полимера
является характеристическая температура, которую можно выб-
рать многими различными способами. Температура стеклования
Tg является очень подходящей характеристикой. Однако Tg оп-
ределяется во многих случаях по температурной зависимости
объема, а не пз вязкоупругих данных и кроме того зависит от
скорости охлаждения пли нагревания образца.
Другим возможным значением для характеристической тем-
пературы могла бы быть Ti—температура, соответствующая пере-
гибу, при которой К(Т) равно 10 сек (в главе II об этом уже было
сказано). Она имеет то преимущество, что просто и однозначно
определяется вязкоупругими измерениями, но зависит от произ-
вольно выбранного значения времени 10 сек.
Определение, которое, кажется, дает единственное и не произ-
вольно выбранное значение [1], состоит в следующем: строится
график зависимости dln!\(T)/d(l/T) от температуры; в общем
случае этот график имеет ясно определяемый максимум, который
обозначим через Td. Величина r/ln/C(T)/J(lIT) равна tXHaKxlR,
где есть кажущаяся теплота активации для характеристи-
ческого времени релаксации. Эта кажущаяся теплота активации
не является постоянной; она проходит через максимум при Td.
Обозначим характеристическое время релаксации К(Т) при Td
через Kd пли K\Td).
В разделе 8 главы II дано другое определение характеристи-
ческой температуры. Значение Ts=243 гК было выбрано произ-
вольно для полипзобутилена. Для других полимеров 1\ выбирает-
ся так, чтобы K(T}/I\(TS) [или a(£),a(Tj] были единой функцией
or Т—Ts для всех аморфных полимеров.
Характеристические температуры Т„. Ti и Та для данного по-
лимера оказываются очень близкими друг к другу (±5С). Харак-
166
<5
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
теристическая температура Ть приблизительно на 50 °C больше,
чем Те.
При построении приведенного уравнения состояния вязко-
упругости все характеристики Tg, Tit Td и Т\ могут быть исполь-
зованы в равной степени.
Таблица IV,4
Параметры, характеризующие релаксацию напряжения аморфных полимеров11
Полимер г°к d 1g El 1g Ei n h lgxd
Полпметилметак рилат . . . 384 10,35 7,35 0,52- 0,31 — 1,5
Паракрил 26 (буi ./акр) . . 241 10,10 7,40 0,63 0,416 -1,54
GR S (75/25 бут./стир.) . . 220 10,24 7,44 0,71 0,4э — 1,50
(60/40 бут./стир.) 237,1 10,27 8,03 0,505 0,40 — 1,94
(50/50 бут./стир.) 250,8 10,212 7,558 0,54s 0,364 —1,12
(30/70 бут./стир.) 285,1 9,955 7,255 0,55 0,36 — 1,13
11олнизобутилен 197,0 10,48 6,88 0,745 0,367 + 1,04
Натуральный каучук (невулк.) 205,3 10,42 7,20 1,07 0,495 —1,90
Натуральный каучук (вулк.) 206,3 10,39 7,26 1,10 0.555 — 1,095
А. V. Т о Ъ о 1 s к у, Е. Са t si (f, J. Polymer Sri., 19, 111 (1956).
В табл. IV,4 даны значения характеристических параметров
нескольких полимеров. Значение Е± модуля полимера, находя-
щегося в стеклообразном состоянии, оказывается более или менее
постоянным для большинства полимеров; его величина была при-
ведена в разделе 6 главы 11. Значение F, зависит от числа попереч-
ных связей в единице объема сеточного полимера, а в линейных
полимерах—от концентрации «переплетений»*, упрощенно опи-
сывающих сложное явление. По-видимому, величина п близка
к 0,5 для ряда жестких полимеров (таких, как полиметилмета-
крилат) и имеет наибольшее значение, близкое к единице, для
резины из натурального каучука, имеющего очень гибкие
цепи. Приведен в таблице также и другой характеристический
параметр Л, тесно связанный с н. Его смысл будет выяснен в
разделе 6.
Крайпе важна зависимость /\(7') от температуры.
Поскольку К(Т)!К{Т^ и имеют одни п те же тем-
пературные зависимости, то можно ожидать, что уравнение
Вильямса—Ландела—Ферри 131, будет применимо в различных
* Лучше говорить о вторичных узлах сетки, которые могут возни-
кать как в местах переплетений макромолекул, так и в результате образования
локальных полярных поперечных связей.—Прим. ргд.
4. Характеристические параметры аморфных полимеров
167
формах (см. раздел 8, глава II):
&K(TS) 2,303 )g/a. Т — Tg V ’
^,2)
<4’3>
Данные исследования релаксации напряжения при использо-
вании Td в качестве характеристической температуры дают урав-
Рис. IV, 17. Зависимость lgK(T) 'K(Td) от (Т—Tj) для аморф-
ных полимеров:
/—полиизобутилен; 2—GR-S; «>, 4, 5—соответственно 60 10, 50'30, 30/70—
бутадиене гн рольные сополимеры; 6—па рак рил-26; 7—полнмс! илмегакрилат.
(Паракрнл-26—вулканизованный 71 26 бутадиен-акрилом нт ри 1ыи..н гоноли-
мер). Кривая—по формуле (1J) [А. \ . Tobolsk'. £. С a t > i f f, J. Polymer
ЬС!.. 19, III (1936)].
нение, очень похожее [2] на уравнение (4, 2) (рис. IV, 17):
1а ЛЖ = _|6 14 (4 4)
& KIT.!) ’ 5й--Т-Т.1 ( '
168
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
В пределах нескольких градусов вблизи Tg, Td и Tt можно
ожидать, что (согласно разложению в ряд Тейлора)
18тга=“'’(7'-Д> = -«(^-1) <4’5»
где в качестве Тс можно взять Т Td пли Tt без заметного изме-
нения значения константы р, так как эти три температуры очень
близки друг к другу.
Если уравнения (4, 1), (4,2) и (4, 4) разложить в ряд в области
температуры Tg пли Та, то можно получить уравнения точно та-
кой же формы, как и (4, 5), со следующими значениями р:
из (4,1) р — а,.2,303/; (4,6)
пз (4,2) р = 0,338 (4,7)
пз(4,4) р-- 0,288 (4,8)
На рис. IV, 18 приведены зависимости nlg[l\(T)iK(Tc)] от
Т1ТС, полученные для ряда полимеров из опытов по релаксации
напряжения.
Видно, что для 0,95< Г/7'с< 1,05
л18^В‘==-45(^--1)==-С’(Г”^) (4,9>
Для экспериментальных данных, приведенных на рис. IV, 18,
Тс взята равной 7\ (и поэтому = сек), хотя Td или Tg
можно использовать примерно с тем же результатом. Несколько
раньше [1] было получено уравнение, похожее на выражение
(4, 9), в котором в качестве характеристических параметров были
использованы h и Td [1] (рис. IV, 19):
ftlg-^1 =36(^4- 1^-Ц(Т-Т.) (4,10)
где Тс берется в данном случае равной Td.
Сравнив уравнения (4, 9) и (4, 10) с уравнением (4, 5), получим
дтя р Два более простых выражения:
₽ = (4.11)
Р - (4. 12)
Уравнения (4, 11) и (4, 12), полученные пз одних п тех же дан-
ных, не являются одинаковыми, поэтому между h и п, как можно
4. Характеристические параметры аморфных полимеров
169
видеть из табл. IV, 4, существует соотношение вида
Уравнения (4, 11) и (4, 8) совпадают, если пТс^А5&.
Поскольку уравнение (4, 5) является наиболее точным в окрест-
ности Тс, а уравнение (4, 2) справедливо в интервале Тс, т -\-
+ 100 °C, но неточно вблизи Тс, то уравнение (4,2) желательно
написать в следующем виде:
1А-_________Р(Т-Т,}
g — 1-Р'(У-+)
Если уравнение (4, 2) является правильным, то здесь должно
быть р -=0,338, а р'==0,0194.
Рассмотрение экспериментальных данных по релаксации на-
пряжения приводит к выводу, что уравнение (.4, 13) можН° !1С-
170
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
пользовать как общее уравнение до тех пор, пока не будет полу-
чено достаточно экспериментальных данных, чтобы уточнить зна-
чение р. В разделе 7 будет подчеркнута важность установления
точного значения р.
Следует добавить, что Ферри с сотрудниками |41 в серии опы-
тов с этил-, бутил-, гексил- и октилметакрнлатом также установил,
что боковые разветвления дают вклад в К(Т) в виде члена типа
Рис. IV,19. Зависимость/г 1g] А'(7')/Л'(7',;)] от— — _4 для ряда аморфных поли-
Tr Т
меров. [Е. С a t s i f f, A. V. To bo 1 s k у, J. Appt. Phys., 25, 1092 (1954)].
Аррениуса A exp(EfRT), который является поправкой к основной
температурной зависимости теплового движения главной цепи,
выраженной в формулах (4, 1)—(4, 3).
Литература
1. J. Bischoff, Е. С a t s i f f, A. V. Tobolsky, J. Am. Chem.
Soc., 74, 3378 (1952).
2. E. C a t s i f f, A. V. Tobolsky, J. Coll. Sci., 10, 375 (1955).
3. M. L. William s, R. F. Lande 1, J. 1). F e r r v, J. Am. Chem.
Soc., 77, 3701 (1955). (M. Л. Вильямс, P. Ф. Ландел,
Д. Д. Ферр и. Проблемы совоеменной физики, № 12, 20 (1956)].
4. J. D. Ferry. W. С. Child jr., R. Zand, D. M. Stern,
M. L. Williams, R. F. L a n d e 1, J. Coll. Sci., 12, 53 (1957),
5. Теория вязкоупругости и упруговязкостп
171
5. Молекулярная теория вязкоупругости
и упруговязкостп полимеров
В молекулярную теорию вязкоупругости и упруговязкостп
полимеров внесли весьма существенный вклад теоретические иссле-
дования Рауса [ЦпБпкки [2|*. Эта работа была далее продол-
жена Цпммом |3|, Серфом
14] и др. 15—81. *
Эти авторы рассматрива-
ли гидродинамическую про-
блему полимерной молекулы
в разбавленном растворе, на
которую действует внешняя
периодическая (динамиче-
ская) сила. Модель полимер-
ной молекулы представля-
лась как ожерелье ИЗ сфе- р|1С .Модель, примененная Раусом,
рпческих бусин, соединен-
ных упругими пружинами. Каждая бусина и соответ-
ствующая пружина представляют собой гауссов сегмент
полимера. Пружина имеет упругую константу kT (при сдвиге),
определяемую на основе кинетической теории упругости резины
при применении ее к сегменту. Бусина, двигающаяся через рас-
твор, подчиняется закону Стокса и моделирует сопротивление,
препятствующее движению сегмента. Это классическая задача
нагруженной струны 121.
Результаты Рауса можно сопоставить с зависимостями, полу-
чающимися из модели Максвелла—Вихерта. Рассмотрим единич-
ный объем (1 с.п3) раствора, содержащий Л\ молекул, каждая из
которых содержит z одинаковых свободно вращающихся сегментов.
Каждый сегмент состоит из достаточного числа звеньев, так что
распределение по расстояниям между концами является гауссов-
ским. На рис. IV, 20 приведена модель Максвелла—Вихерта (при
-сдвиге), описывающая вязкоупругий вклад полимерных молекул
во все свойства системы. На рисунке изображено z элементов Макс-
велла, соединенных параллельно. Упругая постоянная каждого
элемента Максвелла равна \\kT. Времена релаксации т1( т2, --
* Значительно раньше Рауса и Бпкки советскими учеными была предло-
жена н рассмотрена аналогичная модель полимера, основанная на современ-
ных представлениях о строении и свойствах макромолекул, которая позволила
описать основные упруговязкие свойства [В. А. Каргин, Г. J1. Слон и м-
с к и й, ДАН СССР, 62, 239 (1948); ЖФХ, 23, 563 (1949)]. Эта модель была
существенно усовершенствована в 1961 г. [Г. Л. С л о н и м с к п й, ДАН
СССР, 140, 343 (1961)].—Прим. ред.
172
Г.1. IV. Вязкоупругие и упруговязкие свойства полимеров
г даются формулой вида:
г-2,а
- = l-1 г ______ -та
Р 6~-2kT р2 р2
р= 1, 2, 3,...,г
(5, 1)
Следует заметить, что здесь т,, -ч......тг имеют обратный
порядок, где т, максимальное, а тг минимальное время релакса-
ции:
- =
'nSi 6r.2feT
т = Т = —tL- е. "max
г min 6ЙГ Z2
(5, 2)
(5, 3)
Величина /, появившаяся в уравнениях (5, 1)—(5, 3), является
коэффициентом трения. Коэффициент трения, умноженный на
относительную скорость между растворителем и полимерным сег-
ментом, дает силу, действующую на отдельный сегмент. Для ша-
рика радиуса го закон Стокса определяет коэффициент трения
следующим выражением:
f — f5,4)
Величина з2 в формулах (5, 1)—(5, 3) есть средняя квадратичная
длина недеформированных свободновращающихся сегментов.
Общая вязкость системы -ц является суммой вязкости раство-
рителя 7(s и вязкостей каждого из поршней (см. рис. IV, 20):
(5, 5)
2
0=1
Поскольку величина z очень велика
fN^z2
^-^ = “36-
Следовательно
(5, 6)
(5, 7)
(5, 8)
Формула (5, 8) для добавочной вязкости, возникшей в резуль-
тате наличия в растворителе молекул полимера, тождественна с
формулой, полученной для этой же характеристики с помощью
5. Теория вязкоупругости и упруговязкости
173
«свободно протекаемой» модели Хаггинса [9], Дебая [10] и др.
Нз уравнения (5, 8) вытекает, что вязкость, обусловленная
полимерными молекулами, находящимися в 1 см3, пропорциональ-
на квадрату длины цепи. Характеристическая вязкость разбав-
ленного раствора полимера оказывается пропорциональной длине
цепи, что находится в согласии с моделью свободносочлененных
сегментов. Исходя из этого, можно установить, что изменения,
введенные Циммом 13] в теорию Рауса, весьма существенны, так
как он рассматривает молекулы как не вполне «свободнопротекаемые»,
при этом предполагается, что линии тока растворителя значи-
тельно видоизменяются внутри полимерного клубка. Другие
характеристики упруговязкпх свойств, обусловленных наличием
макромолекул, могут быть получены из модели Рауса, изображен-
ной на рис. IV, 20, и уравнения (5, 1) посредством методов, опи-
санных в разделе 5 главы III. Например:
= (5,9)
1 » ’° -р
р==|
г
(5,10)
у = 1
Если предположить, что коэффициент Пуассона равен Ч2, то
2
Ег (/) = 3NJT У ехр (~^/тшах) (5,11)
p=i
При /=0 максимальное значение модуля равно:
£(1Л) = ЗгА^Т (5,12)
Ясно, что в уравнении (5, 12) есть число свободно вращаю-
щихся сегментов, приходящихся на 1 см3.
В уравнении (5, 11) для значений t больших, чем Tmin, но мень-
ших, чем ттах) сумму можно заменить интегралом:
2 С?
— ехр (—tp2,7max) J exp (— tp2/ттах) dp
б
„lo-1 2
'* "max 'in in .
‘min '• vmax
(5,13)
174 Г.i. IV. Вязкоупругие и упруговязкие свойства no.iuv.epou
Подставляя уравнение (5, 13) в уравнение (5, И), а также ис-
пользуя определение тиИп из уравнения (5, 3), получим:
Er(t) -izN^T [ Л^?) (5,1-1)
где
A'<K1 СО = (v) чп п = g7- /5,15)
Величину К<К|(Т) можно назвать характеристическим време-
нем релаксации, определяемым из теории Рауса. Оно зависит от
температуры, так как, кроме множителя \,Т, в уравнении (5, 15)
коэффициент трения также зависит от темпера гуры. Это .хорошо
видно из уравнения (5, 4), где / пропорционален вязкости i...
которая явно зависит от температуры.
Теорию упруговязкостп Рауса для разбавленных растворов
полимеров можно применить также к нераствореиным аморфным
полимерам. .Можно считать, что сегменты полимера погружены в
жидкость, состоящую из других полимерных сегментов. Кроме
изменения общей концентрации сегментов z.\\, необходимо от-
метить только то, что вязкость растворителя существенно входит
в математические выражения лишь через уравнение типа (5, 4)
для коэффициента трения. Поэтому в отсутствие растворителя
коэффициент трения / пли, что даже еще более удобно, T,nill пли
Л(К,(Й) могут быть взяты в качестве фундаментального свойства
системы, определяемого экспериментально.
Как видно, для зависимости lg/2’,(7) от 1g/ теория предсказывает
наклон, равный —Ч, (т. е. характеристический параметр п равен
1Л). Здесь получается тот же самый результат, который был
прежде эмпирически предположен в случае «клинообразного»
спектра времен релаксации для переходной области (раздел 7
главы Ill). Таким образом, было очень соблазнительным считать,
что теория Рауса—Бикки применима и в переходной области к
участку с относительно низким модулем. Возможно, что здесь
можно применить молекулярную модель. Однако ни верхняя
часть переходной области (высокие модули), ни высокоэластпче-
ская область, ни эластовязкая область, ни область вязкого тече-
ния линейных полимеров высокого молекулярного веса не могут
быть объяснены удовлетворительно теорией Рауса, если в нее не
внести изменения.
Первое видоизменение ее может быть сделано для случая по-
перечно-сшитых трехмерных сеток |2, 5, 6|. При этом считается,
что цепей сетки в 1 см3 играют ту же самую роль, что и
молекул в 1 см3. Результаты, следующие из рис. IV, 20 и урав-
5. Теория вязкоупругости и упруговязкости
175
нения (5, 1), полностью применимы, если максимальное время
релаксации заменить бесконечно большим. Конечно, считать, что
изменяется только максимальное время, а все другие времена
релаксации не меняются—это значит допускать определенное
упрощение. Однако экспериментально проверено, что короткие
времена релаксации, соответствующие переходной области, не
меняются при наличии поперечных связен, как это было показано
на рис. IV, 11 при сопоставлении данных для полипзобутилена и
для резины на основе бутилкаучука II11.
В разделе 8 главы III было показано, что у линейных аморф-
ных полимеров с высоким молекулярным весом все большие вре-
мена релаксации зависят от молекулярного веса в степени 3,3
(^3,4); это был участок релаксационного спектра, приблизитель-
но описываемый прямоугольным распределением. Высокоэластп-
ческая область описывалась с помощью величины молеку-
лярного веса отрезков цепей, находящихся между вторичными
узлами («переплетениями») |12|.
Для модификации теории Рауса с целью ее применения к вы-
сокоэластической и эластовязкой областям и к области вязкого
течения для линейных полимеров с высоким молекулярным
весом, Бпкки [13] предложил считать, что коэффициент
трения в уравнении (5, 1) должен быть пропорциональным мо-
лекулярному весу в степени 3,4 для значений р больших, чем
Рпереплет,- гДе Рперсплет.—ЧИСЛО СегМСНТОВ, СООТВвТСТВуЮЩИХ ВЫСОКО-
эластическому модулю£,. Автор считает [13, 14|, однако, что та-
кое рассмотрение не лучше, чем рассмотрение распределения типа
клин—прямоугольник, приведенное в разделе 8 главы III. Обе
трактовки являются, по существу, математическим обоснованием
экспериментальных данных, а не выводятся из априорных сообра-
жений. Нужна намного более убедительная теория влияния пере-
путанности молекул на их диффузию для того, чтобы описать
высокоэластпческую область, эластовязкую область и область
вязкого течения.
(Можно считать, что релаксационный спектр в высокоэластичес-
кой области и эластовязкой области зависит от разрушения и
восстановления необычайно сильных вторичных валентностей и
(или) «переплетений». Грин и Тобольский [151 использовали это
допущение в качестве математической модели упруговязкости. Оно
оказалось очень удачным для объяснения упруговязких свойств
полимеров с перестраивающимися химическими сетками, где
имеется лишь одно время релаксации. Это будет описано в разде-
лах 12—14. Однако все же не был достигнут убедительный
априорный вывод распределения времен релаксации, соответ-
ствующих скольжению и восстановлению молекулярных пе-
реплетений.)
176
S
Гл. IV. Вязкоупругие и упруговязкие свойстве полимеров
Теория Рауса—Викки недостаточна и для описания высоко
модульной части переходной области. Если экспериментальное
значение модуля Ег (модуль стеклообразного состояния) взять
равным 310i0 дин/см2 и приравнять модулю в уравнении
(5, 12), то рассчитанное общее число свободно вращающихся сег-
ментов zA\ в 1 си3 окажется 2,4-1028. Это невероятно высокое
число, соответствующее молекулярному весу свободно вращаю-
щихся сегментов, равному 2,5 для полимера с плотностью, рав-
ной единице.
Измерения двойного лучепреломления, описанные в разделе 15
этой главы, указывают на изменение молекулярной природы на-
пряжения при приближении к несомненно, изменение заклю-
чается в переходе от упругости, связанной с энтропией, к упру-
гости, связанной с внутренней энергией. Никакая молекулярная
теория, основанная только на конформационной энтропии, не бу-
дет адэкватно описывать весь спектр вязкоупругости в области
вблизи стеклообразного состояния.
Литература
1. Р. Е. Rouse, J. Chem. Phys., 21, 1272 (1953).
2. F. В u e c h e, J. Chem. Phys., 22, 603 (1954).
3. В. H. Z i in m, J. Chem. Phys., 24, 269 (1956); Б. Ц и м м, Co. «Физика
полимеров», Издатинлит, 1960, стр. 379.
4. R. Cerf, J. Polymer Sci., 23, 125 (1957). Р. С е р ф, сб. «Физика полиме-
ров», Издатинлит, 1960, стр. 416.
5. О. N a k a d a, J. Phvs. Soc. Japan, 10, 804 (1955).
6. A. Miyake, J. Polymer Sci., 22, 560 (1956).
7. Y. H. P ao, J. Chem. Phys., 25, 1294 (1956).
8. M. Copic, J. Chim. phys., 53, 440 (1956).
9. M. L. Huggins, J. Phys. Chem., 42, 911 (1938); 43, 439 (1939).
10. P. Debye, J. Chem. Phys., 14, 636 (1946).
11. G. M. Brown, A. V. Tobols к у, J. Polymer Sci., 6, 165 (1951).
12. H. M ark, A. V. Tobolsk y, Physical Chemistry of High Polymeric
Systems, Interscience Publishers, New York, 1950, p. 344.
13. F. В и e c h e, J. Appl. Phys., 26, 738 (1955); Ф. Б п к к и, сб. «Физика
полимеров», Издатинлит, 1960, стр. 15; J. Chem. Phys., 25, 599 (1956).
14. J. D. Ferry, R. F. Lande 1, M. L. Williams, J. Appl. Phvs.,
26, 359 (1955).
15. M. S. Green, A. V. Tobolsk y, J. Chem. Phys., 14, 80 (1946);
M. Y a m a m о t o, J. Phys. Soc. Japan, 11, 413 (1956).
6. Релаксация напряжения в полимерах,
находящихся в стеклообразном состоянии
Изучение релаксации напряжения в аморфных полимерах ниже
7\ дает некоторое представление о природе стеклообразного состоя-
ния. Например, обнаружено, что ниже Т„ кривые релаксации на-
пряжения сильно зависят от способа отжига полимерного образ-
б. Релаксация напряжения в стеклообразных полимерах
177
ца. Если пол иметилметакр плат пли полистирол, находившиеся
при температуре выше Tg, быстро охладить до комнатной темпера-
туры, то их плотность будёт ниже, чем если бы они были очень
медленно охлаждены до комнатной температуры. По-видимому,
при быстром охлаждении фиксируется структура, в которой со-
храняются микропустоты в виде «дырок» атомных размеров. В этих
быстро замороженных образцах релаксация напряжения проис-
ходит, как показано на рпс. IV, 21, намного быстрее, чем в медлен-
но отожженных образцах.
Рис. IV, 21. Влияние скорости охлаждения на релак-
сацию напряжения полиметилметакрилата при 80 °C.
Образец охлаждался от 130 °C следующим образом:
I—5 wpab}4't 2—30 град/ч\ 3—конвекцией в воздухе при 25 °C;
/—погружением в масло при 25 °C; 5—погружением в ванну с
минеральным маслом и сухим льдом [J. R. McLough И п,
А. V. То bo 1 s к у, J. Polymer Sci., 7, 658 (1951)J.
Если в стеклообразном состоянии изучение релаксации напря-
жения проводить при любых, но не при самых малых растяжениях,
то релаксация будет сопровождаться образованием внутренних
трещин, т. е. малых, но макроскопических разрывов.
При температурах ниже Tg сохраняется подвижность неболь-
ших атомных групп, в частности боковых групп, например та-
ких, как метильная группа в полипропилене. Эти движения в стек-
лообразном состоянии не обнаруживаются в экспериментах по релак-
сации напряжения. Однако такие малые движения косвенно могут
наблюдаться при изучении свободных колебаний [2—6]. Обычно
эти испытания производятся при почти постоянной частоте в ши-
роком интервале температур. В экспериментах измеряются как
модуль кручения, так и декремент затухания, причем последний
измеряется на более чукствктел!пых приборах. На кривой, вы-
12—833
178
f'.i /Г. Вязкоупругие и упруговязкие свойства по.шмеров
ражающей зависимость декремента затухания от температуры,
всегда имеется максимум, который расположен по соседству с тем-
пературой стеклования. Однако при температурах ниже темпера-
туры стеклования имеются другие меньшие максимумы затухания.
Полагают, что они возникают благодаря вибрациям и крутильным
движениям небольших групп атомов. Новейшим методом обнаруже-
ния движений небольших атомных групп полимера в твердом со-
стоянии (включая стеклообразное, кристаллическое и высоко-
эластическое состояния) является ядерный магшггпый резонанс
|7—191. Детальное описание техники этого метода (и других ме-
тодов, использующих электромагнитную теорию) выходит за
рамки этой книги. Отметим лишь, что сущность метода заключается
в нахождении ширины резонансной линии протона как функции
температуры. (Помимо этого иногда измеряется ширина резонанс-
ных линий и ядер других атомов, например таких, какфгор.) Умень-
шение ширины резонансной липни указывает на повышение ин-
тенсивности молекулярного движения. Обычно предполагают,
что это движение состоит из кручения и вращения отрезков цепей.
Частоты молекулярного движения, необходимые для того, чтобы
вызвать сужение резонансной кривой, имеют порядок 104 сек *.
Температурная область этого сужения соответствует молекуляр-
ному движению приблизительно этой частоты.
Ядерный магнитный резонанс используют также для расчета
степени кристалличности полимеров. Это определение основано
па предположении о том, что имеется температура, при ко-
торой среднее время корреляции для молекулярного движения
в области кристаллизации много больше, чем 10 4 сек, тогда
как соответствующее время корреляции в аморфной области
много меньше, чем 10~4 сек. Если эти условия имеют место,
то поглощение при ядерном магнитном резонансе (поглощение
графически дается в зависимости от напряженности магнитного
поля) проявляется в виде накладывающихся узких и широких
резонансных линий. Степень кристалличности может быть опре-
делена измерением площадей, ограниченных каждой кривой.
Для изучения молекулярных движений в полимерах исполь-
зуют также частотные зависимости диэлектрической проницае-
мости п диэлектрических потерь. 11а частотных зависимостях
диэлектрических характеристик имеются области дисперсии, со-
ответствующие частотам молекулярного движения. Однако в
электрических измерениях проявляются только те движения, ко
торые вызваны изменением дипольного момента. Для одних и
тех же полимеров были проделаны интересные сопоставления ре-
зультатов диэлектрических и вязкоупругих измерений. В част-
ности, показано, что температурно-временные факторы 1А’(Т)
или одинаковы для обоих методов измерений 1201.
7. Переходная оолаыь и модуль
179
Литература
1. J. R. McLoughlin, А\ V. Т о b о 1 s к х, J. Polymer Sci., 7, 658
(1951).
2. К. W о If, К. S с h in i е d е г, Ricerca sci. Suppl., 25, 732 (1955).
3. J. H i e j b о e r, KolL Z., 148, 36 (1956); Chem. Weekbl., 52, 481 (1956).
4. J. A. S a u e r, D. E. К line. J. Poh mer Sci., 18, 491 (1955).
5. D. E. Eli n e. .1. A. Sane г, A E W о о <1 w a r cl, .1 Polvmer
Sci.. 22. 455 (1956).
6. E. But t a, J. Polymer Sci.. 25, 239 (195/), IN. G. McCrnm, J. Poly
mer Sci., 34, 355 (1959).
7. E. R. A ndrew, Nuclear Magnetic Rc-u nance. Cambridge University
Press. Cambridge, England, 1955.
8. R. New in ,i n, .1 Chem. Phys., 18, 14)3 (1950)
9. L. V. Il о Iroi d, B. A. ,'d i о v, e E. G u t h, Phvs. Rev., 79,
1026 (1950).
10 I . \r 11 о 1 г о у d. R. S. G о d r i n g t о и, В. Л. Mrowca,
.1 . G ii I h, .1 Appl. Pins.. 22. 696 (1451).
II II. S. (in towsky, E. 11. Meyer, .1. Chem. Phys., 21, 2122 (1953).
12. V. R. II о и no 1 d, F. M с C a f f r e \, В A. M г о w c a, J. Appl.
Ph\s.. 25. 1219 (1954).
13. W. P. Slichtc r, .1. Appl. Phy< . 25, 1219 (1954).
14 .1. A. S. Smit h. Disc. Faraday Sue. 19, 207 (1955).
15. I. .1. Loire. L. O. Brown, R E. Norberg, Bull. Am. Phvs.
Soc.. 30. 16 (1955).
16. .1. G. P о w 1 e s. Proc. Phys. Soc. 1 ondon. 69, 281 (1956).
17. \V. P. S I i c h t e r. ,1. Polvmer Sci.. 21, 173 (1957); ibid., 26, 171 (1957).
18. C. W. Wilson, G. E. Раке. J. Polvmer Sci , 10, 503 (1953).
19. W. P. Slichter. .1. Pop mer Sci., 25, 230 (1957).
20. .). D. F e r r y, S. S t r e 1 I a, J. Coil. Sci., 13, 459 (1958).
7. Переходная область и зависимость модуля
от температуры
Простая эмпирическая функция, приблизительно описываю-
щая модуль релаксации, наблюдающийся в переходной области,
может быть выражена уравнением, напоминающим уравнение
«клина» 111:
Ег, т (П- Е„ = —
- |/ Л
(/./?' 2
(///•>)' И"
(7, 1)
Все параметры, использующиеся в уравнении (7, 1), а именно;
Ер Е.,; К(Т) п п, являются характеристическими параметрами,
уже рассмотренными в разделе 4. Распределением времен релак-
сации, которое соответствует Е,,7(/)—Е2 в уравнении (7, 1),
является;
180 Г.1. /V. Вязкоупругие и упруговязкие свойства полимеров
Логарифмируя обе части уравнения (7, 1), получим:
1g [£,. т (t) - £s] = 4(lg Е1+lg £2) - n lg[4 + (-|)1/2"] <7’ 3>
В самой переходной области Е2<С.Ег,т поэтому урав-
нение (7, 3) примет следующий приближенный вид:
1g £,, НО = 4 Ой £1 + >g е2) ~ п lg t + »’g К (Т)
Е, < £г, т (0 £i
(7, 4)
Наибольший интерес уравнение (7, 4) представляет вблизи Тс
(где Т'. есть ТЁ, Tt или Td). В этой связи интересно переписать
уравнение (4,5) в следующей форме-
lg£(T) = lgA’(£f)-p(T-Tf) (7,5)
Если (7, 5) подставить в уравнение (7, 4), то в результате по-
лучим:
1g £,-. т (t) = 4 (1gEi + lgE.J -n\gt +
+ n lg E (Tc) — пр (T — Tc)
(7, 6)
Уравнение (7, 6) можно использовать для объяснения зависи-
мости модуля от температуры вблизи Тс. Особенно удобно выбрать
Тс равной 7'р где Tt—температура, при которой Е(Т{) равняется
10 сек.
Если в уравнении (7, 6) сохранять t постоянным и равным
10 сек, то можно предсказать зависимость модуля от темпера-
туры:
lg£,_ 7 (10 сек) — 4(*-£‘ - lg £J-«Р (£ -£,) (7,7)
Уравнение (7, 7) предсказывает вблизи 1\ линейную зависи-
мость логарифма модуля от температуры. Показано, что это спра-
ведливо для случая аморфных полимеров. При этом можно считать
отрицательный наклон s участка зависимости lg£r.; ПО) от Т
вблизи Tt еще одним характеристическим параметром.
Согласно уравнению (7, 7)
s — np (7,8)
В разделе 4 были приведены различные эмпирические уравне-
ния для р 1(4, 6)—(4, 8), (4, 11) и(4, 12)1.
Пеиехооная область и функция ошибок 181
Если уравнение (4, 11) справедливо, а его мы нашли в резуль-
тате наших собственных исследовании, то:
s = 4] (7,9)
В настоящее время целесообразно считать, что уравнение
(7, S) более предпочтительно по сравнению с уравнением (7, 9)
вследствие недостаточно точных сведений относительно значения р.
Фактически было бы желательно оценивать р через эксперимен-
тальные значения s и и, используя для это1 о уравнение (7, 8).
Интересно заметить, что вязкость при деформации растяжения,
полученная из распределения (7,2), равна:
д(') = ЛЧП£11^'^71)-(£1^)’/2П (7,10)
Таким образом, вязкость, полученная только из одного рас-
пределения времен релаксации типа видоизмененного клина,
уже имеет аномально большие значения. Фактически т(<;>(7',)>
1013 пуаз, и эго, конечно, при полном исключении го-
раздо более важного вклада, оказываемого временами релакса-
ции, входящими в прямоугольное распределение. Об этом следует
напомнить, потому что (близкую по своему значению к Т()
часто определяют как температуру, при которой вязкость равна
1013 пл.
Литература
1. А V. Т о b г I s к , .1 R. М с I. о и е h 1 i n, J. P'>)vnier Sci., 8, 543
(11)52).
8. Переходная область и функция ошибок
Другой) эмпирической функцией, которая пригодна для опи-
сания вязкоупругих данных в переходной области, является |1!:
igE,. 7i/> = -4 (igE, -г IgEJ -^(IgE,- IgfJerf {'z Ig-^) (8, 1)
В уравнении (8,1) обозначение erf соответствует табулиро-
ванной функции ошибок Гаусса. Величина /г, появившаяся в
уравнении (8, 1), уже приводилась в качестве одного из возмож-
ных характеристических параметров для Er(t). Эксперименталь-
ные данные Е//) от Z, нанесенные- на специальную бумагу с ве-
роятностной сеткой, должны ложиться на прямую линию, если
справедливо уравнение (8, 1). Это показано на рис. IV, 22 и
IV, 23. По наклону прямой быстро определяется h. Как упо-
миналось раньше, йт^3/4 п.
182
Iл. /Г. Вязкоупругие и упруговязкие свойстве полимеров
В разделе 4 |см. уравнение (4,10)1 было установлено, что для
значений Т, близких к Td, можно записать:
(8, 2)
Рис. 1\. 22. Зависимость Ег(/) от 1g/ для полипзобутилена XBS в переход-
ной области (график па специальной верой! пост пой сетке) |Е. С a 1-
к i 1 1, А. V. Tobolsk)’, J. Appl Phys., 25, П>92 (1954)1.
Для серии бутадиен-стирольных сополимеров было найдено,
что
hTd^ 100
(8, 3)
1g К (Td) = — 1,45 (в часах)
Если скомбинировать уравнения (8, 1), (8,2) и (8, 3), го полу-
им универсальное уравнение для Ег(/) как функции от T/Td
Переходная область и функция ошибок
183
(или Т —Tj) п Td, которое действительно в переходной области для
всех аморфных бутадиен-стирольных сополимеров, синтезиро-
ванных в условиях радикальной полимеризации:
1
Г (0 "у- lg Е,/:., . j, ]
-----~1----=------= -erfHydg? l,45)-36f-<-id (8,4)
“2“ ' ‘‘ '
Рис. IV,23. Зависимость Er(t/K) от 1g t К для 30 -70 бу-
тадпеп-стпролыюго вулканизата (график па специаль-
ной вероятностной сетке) |Е. С a t > i f f,
A. V. Tobolsk y, J. Appl. Phys., 25, 1092 (1954).
Уравнение (8, 4) можно изобразить на вероятностной бумаге,
как показано на рис. IV, 24. Г1а этом графике приведены все дан-
ные по релаксации напряжения для четырех различных бутадиен-
стпрольных сополимеров; они действительно показывают, что
уравнение (8, 4) удовлетворительно описывает весьма различные
данные. Принимая t =10секв уравнении (8, 4), можно получить зави-
симость модуля от температуры, которая будет являться функцией
от Т и Td. На рис. IV, 25 и IV, 26 показаны такие зависимости
для ряда значений Td.
184
Гл. /Г. Ьязкоупругиё и упруговяз/гие свойства полимеров
Рис. IV, 24. Приведенное уравнение вязкоупругих свойств бутадиен-
стирольных сополимеров в переходной области. Приведены данные для
следующих сополимеров: 75/25; 60/40; 50/50; 30/70. Включены также дан-
ные для бутадиен-акрилонитрилового сополимера 74/26. Все сополимеры
слегка сшиты поперечными связями [А. V. Tobols к у, Е. Cal-
s i f f, J. Arn. Chem. Soc., 76. 4204 (1954)].
Температура, °C
Рис. IV, 25. Зависимость 10-секундпого модуля бутадиен-сти-
рольных сополимеров от температуры согласно приведенному
уравнению вязкоупругих свойств [А. V. Т о 1> о i s к у,
Е. С a t s i I f, J. Am. Chem. Soc., 76, 4204 (1954)].
9. Области эластовязкости и вязкого течения 165
Рис. IV, 26. Зависимость 10-секундного модуля бутадиен-сти-
рольных сополимеров от температуры согласно приведенному
уравнению вязкоупругих свойств [Л. V. Т оЪ о 1 s к у,
Е. С a t s i f f, J. Ain. Chem. Soc., 7(5, 4204 (1954)].
Литература
I. J. Bischoff, E. C a t s i f f, A. V. Tobolsk y, J. Am. Chem.
Soc. 74 , 3378 (1952).
2. A. V. Tobolsky, E. C a t s i f f, J. Ain. Chem. Soc., 76, 4204 (1954).
9. Область эластовязкости и область вязкого течения;
дискретный спектр времен релаксации
В главе III уже было приближенно описано распределение
времен релаксации Н(~) в области эластовязкости, имеющее вид
прямоугольного «ящика». Для времен измерения, превышающих
максимальное время релаксации, поведение аморфного полимера
может рассматриваться как течение жидкости*.
Было также показано, что ширина «ящика» зависит от того,
насколько узким оказывается распределение по молекулярным
весам.
Для того чтобы получить более детальные сведения, относя-
щиеся к областям эластовязкости и вязкого течения, два образца
атактического полистирола, обозначенные как Д и В, подверга-
лись широкому исследованию (1]. Образец А приготавливался
таким образом, чтобы он мог рассматриваться как моноднсперс-
нып полимер. Средневесовой молекулярный вес его был равен
АО, = 164000. Отношение Al^/A'l,, оказалось равным 1,17. Образец
В был приготовлен таким образом, чтобы распределение по моле-
кулярным весам давалось выражение?»! =A1W-,(1--р)2, где
* Однако п в этом случае течение сопровождается высокоэластическими
деформациями,—Прим. ред.
ч
186 Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
Х(М) является молярной долей полимерных цепей с N звеньями,
а р есть параметрическая константа. Для этого распределения от-
ношение А1г,,/Л1„ равно l,F>. Для образца В значение ,\1^, равня-
лось 171000. Приближенно можно считать образец .4 монодпсперс-
иым, а образец В полиднсперсным, нос достаточно узким распре-
делением молекулярных весов. На рис. IV, 27 и IV, 28 приведены
данные но релаксации напряжения для образцов А и В. На рис.
IV, 29 и IV. 30 приведены вспомогательные кривые релаксации
напряжения для эластовязкон области полистирола (образцы
.4 п В).
До спх пор все математические анализы результатов иссле-
дования вязкоупругих и упруговязкпх свойств проводились в
зависимости от предполагаемого непрерывного распределения вре-
мен релаксации //(").
В главе III рассматривались методы первого и второго прибли-
жений для получения 11(~) из экспериментальных данных. Если к
данным для образцов А п В применить метод второго приближения
1где Hib—2,303 то получим результаты, приведен-
ные на рис. IV, 31 п IV, 32.
Методы приближения для Н(~) хорошо отвечают требованиям в
тех случаях, когда Н(~) медленно изменяется в зависимости от т.
Там, где Н(~) изменяется быстро, //)(-) н Н21, (т) уже не являются
достаточно удовлетворительными приближениями. Это было ра-
нее показано при применении этих приближении к Er(t}, получен-
ному из прямоугольной функции распределения //(т) (см. рис.
III, 10 п IV, 36). Тот же самый результат был получен еще более
убедительно, когда первое и второе приближения были приме-
нены к простому элементу Максвелла (рис. Ill, 14).
Было также показано |см. уравнение (9,9) главы НИ, что
1g /М\,) = 1g = lg£P-0,072 (9, 1)
Если распределение времен релаксации в области эластовязкости
выражается дискретным распределением, то имеем:
£г (/) = £„ ехр (—/ та) 4-
, £mexp( -/т„,) (9,2)
График зависимости lg£,(Z) от t должен стремиться к прямой
линии для />т„„ если на самом деле существует максимальное
время релаксации. Наклон липни равен 1/2,303т,„; отрезок, от-
секаемый прямой на осн ординат, равен lg£,„.
Если lg£,i/) изобразить графически в зависимости от I для об-
разцов полистирола .4 и В, то действительно получается прямая
линия. Эго показано на риг. IV, 33 и IV, 34. Таким образом,
можно вычислить значения т,„ и Ет для этих полимеров.
fi'l
8
Рис. IV, 27. Зависимость IgEr(/) от ]g/ в эласто-
вязкоп области для образца А полистирола.
Рис. IV, 28. Зависимость lgEr(Z) от 1g/ в эласто-
вязкой области для образна В полистирола.
Рис. IV, 29. Вспомогательная кривая £,.(/) в эластовяз-
кой области для образца Л полистирола при 115 °C.
Показаны также вклады отдельных времен релаксации,
вычисленные методом X, сумма которых точно описы-
вает экспериментальную вспомогательную кривую, изоб-
раженную сплошной линией [Л. V. Т о 1> о I s й у,
К. М п г а 1< a mi, J. Polviiier Sci.. 40, 443 (195'1).
l.q Г сгк
Рис. IV, 30. Вспомогательная кривая релаксации в
эластовязкой облает и для образца В полистирола при
115 °C. Показаны также вклады отдельных времен ре-
лаксации, полученные методом X. Суммарная вспомо-
гательная кривая, полученная методом X, показана
пунктирной линией. Экспериментальная вспомогательная
кривая показана сплошной линией | А. V. Т о b о 1 s к у,
К. М н га к а ш i. .1. Polymer Sei., 40, 443 (1959)j.
Гл. IV Вязкоупругие и упруговязкие свойств-! полимеров
9. Области эластовязкости и вязкого течения
189
Рис. IV, 31. Зависимость от Igc (при 115 °C) для
образца А полистирола (изображена сплошной линией).
Приведены также значения Е, и т,, полученные мето-
дом X [А. V. Tobolsk у, К. Murakami,
J. Polymer Sci., 40, 443 (1959)].
Рис. IV, 32. Зависимость Нц,(-) от 1ут (при 115 °C) для
образца В полистирола (изображена сплошной линией).
Также показаны значения Е- и т(, полученные мето-
дом X [А. V. Tobcls к у. К М и г а к a m i,
J. Polymer Sci.. 4o, 443 (1959)].
190
Г.1. IV. Вязкоупругие и упруговязкие свойство полимеров
Рис. IV, 33. Зависимость lg£r(Z) от t для образца .4 полистиро-
ла (Кривая А : 1g £„ _= 5,46; 1g т„ = 5,04. Кривая В : lg£„_i -
6,08; 1g т„ р -1,34). [А. V. Tobolsk у, К. М ига ка rn 1,
J. Polymer Sci., 40, 443 (1959)).
Рис. IV, 34. Зависимость lg£r(/) от t для образца В по-
листирола (Кривая А : 1g £„ = 5,22; 1g -„ = 5,41
Кривая В : lg £„_, = 5,55; lgT„_1 -4,745). |А. V. To-
bolsk у, К- М и гака ni i. .1. Polvmer Sci , 40, 443
(1959)).
Уравнение (9, 2) можно теперь переписать в следующем виде:
<0 ~ £„, ехР (— = £„ exp (— //та) 4- • • •
----H£m-iexp(—/ (9,3)
График зависимости 1gIEr(t)—E„,exp ( —I от l должен стать
прямолинейным для если существует дискретное время
9. Области эластовязкости и вязкого течения
191
релаксации :„н1, существенно отличающееся по своему значению
от ~щ н После определения т,.,_1 и £,„„! (по наклону пря-
мой и месту ее пересечения с осью ординат) можно повторить
эту процедуру с тем, чтобы найти т„,_2, Е„,_., н т. д. Это назы-
вается методом X. В табл. IV, 5 даны значения £; и получен-
Таблица IV7, 5
Дискретный спектр времен релаксации для образцов А и В полистирола3
Л\ОНОД11С1И[»СШ.1Н llO,llk rilpu I .1 1кьШД1К‘1!С|ХНЫЙ ПО HtCTlipO.1 li
i * ью, If.' lg
III 5,46 5,04 r, OO 5.44
III - 1 6,08 4.34 4,76
Hl 2 5,92 3,6/ 5.95 4,09
in - - 3 6,20 3.01 6,19 3,54
hi - 4 6,31 2,20 6,39 2,91
in — 5 6,50 1.14 6.68 2.33
III — 6 6,95 0,40 7.03 1.51
ill — 7 7,03 —0,30
J A. \ . T i boh k у, К. M i rakaini. J.P,I •mer Sc... 40. 443 (i9"9).
ные для образцов А п В этим методом. Па рис. IV, 35 для образца
А показаны следующие одна задругой стадии расчета методом X.
Применимость этого метода для получения максимального
времени релаксации ~1П и соответствующего ему Е,п по большей
части определяется точностью экспериментальных измерений на-
пряжения при малых значениях его. Существует некоторое сом-
нение в том, будет ли монодпсперсный полимер иметь хорошо оп-
ределяемое максимальное время релаксации.
Еще труднее установить справедливость метода X для дру-
гих значений £, и
Достоверность результатов метода X проверяется методами
прикладной математики; программа этой проверки приведена ниже
в 6 пунктах.
1) Достигают ли кривые lgl£r(7) — £,„ехр(— —
E,ll_laxp(—(/-„..А—... I от/практически прямолинейных участков в
широком интервале времен? Является ли различие между lg£,.(/)
н асимптотой lg£m—Z/2,303~„, малой величиной при /-=“,„ и наб-
людается ли подобная картина и для других т,.? Представляет-
ся, что все это имеет место, к -к видно пз рис. IV, 35.
2) Получаются ли времена релаксации существенно отличны-
ми друг от друга? Ответ, содержащийся в забл. IV, 5, заклю-iaei-
c'i в том, что интервалы между временами релаксации допускают
пх разделение.
192
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
3) Если в уравнение (9, 2) подставить значения Е, и по-
лученные методом X, то дадут ли они вновь ту же эксперимен-
тальную зависимость Er(Z), из которой они были рассчитаны?
Ответ таков- для образца J кривая, полученная методом X, не
отличается от экспериментальной кривой; для образца В (.поли-
дисперсный образец) получено превосходное согласие только для
Рис. IV, 35. Изображение последовательных стадий метода X для
образна А полистирола (Кривая С : 1g Ь'„_2 5,92; ig т,,_; = 3,67.
Кривая £): Ig Е„_3 = 6,20; Ig т„_3 — 3,0))'. )А. V. Tobolsky,
К. М u г a k a m 1, J. Polymer Sci.. 40, 413(1959)1.
больших времен, соответствующих z>xm_2. Для более коротких
времен наблюдается только приблизительное согласие (см.
рис. IV, 29 и IV, 30).
4) Если в действительности Ez(Z) дано уравнением (9, 2) с ве-
личинами далеко отстоящими друг от друга, то можно считать,
что метод X пригоден. Если же в этом случае применить прибли-
жение то можно было бы получить непрерывную кривую,
которая имела бы максимумы для всех значений при условии,
что т;. находятся достаточно далеко друг от друга. Однако для ~1
согласно уравнению (9, 1) lgE(T()^]gE,-. Поэтому можно считать,
что там, где //^(-cj^E,-, применим метод X. Это достаточно хо-
рошо выполняется для образцов А и В.
9. Области эластовязкости и вязкого течения
19:1
5) Если метод X был применен к функции Er(t), которая по-
лучена из непрерывного распределения времен релаксации, то по-
кажут ли изложенные выше критерии 1—4, что дискретный спектр,
найденный с помощью метода X, является искусственным? Для
проверки этого метод X был применен к Er(t), полученному из
прямоугольного распределения; в результате получился дискрет-
ный спектр, показанный на рис. IV, 36 (соответствующую табли-
цу см. в [ 1J). Все четыре критерия, изложенные выше, показали, что
метод X менее пригоден, чем тогда, когда его применяли к экспе-
рт:. IV, 36. Значения Et и определенные методом
К из ЕМ = Ей Ei ( —--------] —El (--------11, получен-
\ тз I >, T.il /J
ной из прямоугольного распределения. Сплошная кри-
вая показывает величину вычисленную из того
же Er(t). Параметрами прямоугольного распределения
являются:
£‘о=1,86-10е дин/сл®, t3=9I,2 сек. т/7|=1,1-105 сек. |Л. V. Tobol
sky, К. М и га kain i, J. Polymer ScL. 40. 443 (1959)].
риментальным кривым Er(t} для образцов А и В. «Прямые линии»
критерия 1 оказались сильно разбросанными; полученные значе-
ния времен релаксации располагались очень тесно, особенно вбли-
зи краевых значений т3 и ~т; в противоречии с критерием 4 ока-
залось, что когда значения т, и полученные ме-
тодом X, были подставлены в уравнение (9, 2), то точная кривая
E/t), соответствующая прямоугольному распределению, СыЛа
воспроизведена не совсем удовлетворительно. На рис. IV. 37
приведено это сравнение. Все критерии показали, что значения
13—833
194
/ .1. 1l-'. Вязкоупругие и упруговязкие свойства полимеров
~1 и Et, полученные методом X, являются искусственными,
хотя их математическое выражение качественно согласуется с
функцией Я(т), даваемой прямоугольным распределением,
6) Дает ли метод X, применяемый различными исследовате-
лями, одни и те же значения и Е-? При независимых расчетах
очень хорошее совпадение было получено для значений ~т,
Е1п и Е,^ образцов А и В, приведенных в табл. IV, 5.
Метод X можно применить к известному дискретному распре-
lg Г, сел
Рис. IV, 37. Сравнение Er(t), полученного из пря-
моугольного распределения (кривая 1), с результа-
тами по методу X (кривая 2) [А. V. Tobol-
5 к у. К- М и г а к a m i, J. Polymer Sci., 40. 443
(1959).
уже известные величины. Это было проделано с тремя последними
членами распределения Рауса [уравнения (5, 1) и (5, 11)1:
Е, (!) = Ет-э ехР (— М',п-2) +
Д ехР (— ^\и-т) + Е,и ехр (— t/~m)
Р ______ Р — р - т ™ • т —
^т-2 ‘•'т-1 т-2 32 ’ т-1 О'2
(9, 4)
Для определенности Ет было взято равным 2,88 • 10Б дин[см\
атт равным 1,1 106г'7С. Как показано на рис. IV, 38 и в !11, метод
X, примененный к уравнению (9, 4), дает очень хорошие резуль-
таты. Когда в уравнение (9, 4) были подставлены т(. и Elt получен-
ные методом X, то оно точно воспроизвело, как показано па рис.
IV, 39, данную кривую Er(t). На рис. IV, 40 приведены Il SA и
9. Области эластовязкости и вязкого течения
195
t, сел-
Рис. IV, 38. Применение метода X к уравнении
Рауса (9,4):
A-lgEr (/); B-lg[Er(/) — Ем exp (-//глП]; С-1g [Ел (/)-
—Ед/—I ехР ( </-л/—1)—Ем ехр (—//тл/)]
[ А. V. Tobolsky, К- Murakami, I PolYiner So.
40, 443 (1959)].
Рнс. IV, 39. Сравнение Er(f), полученного из
функции релаксации Рауса, с результатом по
методу X [А. V. Tobolsky, К. Мн г а-
k a m i, J. Polymer Sci., 40. 443 (1959))
3*
190
I л. IV. Вязкоупругие и упруговязкие свойства. полимеров
Wsi(t), полученные из уравнения (9, 4), для сравнения с Ej и
полученными методом X.
Указанная математическая обработка экспериментальных дан-
ных приводит, таким образом, к выводу, что метод X, применен-
ный к монодисперсиому полимеру, с большой вероятностью дает
Рис. IV, 40. Величины ffapiz), Et и т,- для
функции Рауса )А. V. Tobolsk у, К. М и г а-
k a m i, J. Polymer Sci., 40, 443 (1959)).
значения дискретных времен релаксации тш и тгп_1 и возможно
позволяет выделить еще больше времен релаксации, имеющих
разумные значения. Для полидисперсного образца В метол X
приводит к значениям тт и тт_и которые являются, вероятно,
достоверными, тогда как расчет других времен релаксации, по-
видимому. дает искусственные значения.
Литература
1 А V 1 о b о I s к у, К. Murakami, J. Polymer Sci., 40, 443 (1959).
10. Теплообразование в каучуках
Исторически очень важная и интересная проблема вязкоупру-
гости была выдвинута на передний план второй мировой войной*.
* В Советском Союзе эта проблема возникла значительно раньше в связи
с отсутствием собственного натурального каучука.
Именно поэтому работы советских ученых в области исследования меха-
нических свойств полимеров на много лет опередили работы зарубежных уче-
ных, незнакомство которых с советской научной литературой привело к ряду
повторных открытий.—Прим. ред.
10. Геплооиразование в каучуках
197
Было обнаружено, что резина из GR-S (бутадиенстирол 75/25)
выделяет при испытании на изгиб больше тепла, чем резина из на-
турального каучука. По этой причине она была признана не
совсем удовлетворительной для шин грузовиков больших мощ-
ностей в самолетов.Этот недостаток все еще полностью не преодолен.
В уравнении (1, 10) главы 111 было показано, что количество
теплоты, образующееся за цикл в единице объема при вынужден-
ных колебаниях по закону soe‘“', равно:
£r(t). оин/с^г
IR - ™м)Д11Н sg - r.G"si
(10,1)
Рис. IV, 41. Er(t) для вулканизатов
каучука GR-S (/) и натурального
каучука (2) при 25 °C [Е. С a t s i f f,
A. V. Tobolsky, Tech. Rept.
RLT-21 to the ONR, Dec.. 18(1956)]
Рис. IV. 42. Зависимости £"(1/ы) для
вулканизатов каучука GR-S (/) и нату-
рального каучука (2) при 25 °C, полу-
ченные расчетом из кривых Ег(1)
[Е. С a t s i f f, A. V. Tobolsky,
Tech. Rept. RLT-21 to the ONR, Dec..
18 (1956)].
Тепловые потери h в единице объема за единицу времени
равны:
(10,2)
Поэтому мы предполагаем, что для шин одной и той же формы и
конструкции, работающих в одинаковых условиях, но сделанных
из разных сортов резины, образующаяся теплота будет пропор-
циональна G" (или Е"), которые отражают свойства резины.
Были получены данные по релаксации напряжения для ву^1'
канизатов натурального каучука и GR-S, далее были на идей111
значения k и построена вспомогательная кривая, из которой опре‘
19b
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
Таблица IV, 6
Температурно-временные коэффициенты смещения для
каучука GR-S и натурального каучука, приведенные
к 25 °C
GR-S Натуральный каучук
Г, °C 1g А'298 Г, °C 1g &298
—64,0 13,20 —75,3 13,675
—61,0 12,60 —74,9 13,31
-58,8 П,895 —72,6 13,045
-57,6 11,40 —69,9 12,32
-55,0 10,38 —66,6 11,236
—52,5 9,55,, —64,6 Ю,375
— — —61,1 9,575
-49,5 8,44-, —59,6 9,20
—47,7 7,99 —55,3 8,355
—44,0 6,52 —50,9 7,51
—40,0 5,95
—32,8 4,05
Таблица IV .
Релаксационные зависимости, приведенные к 25 °C
1g времени ч Ig ЕГ (0 ig И (г) Ig Е' (!/<) 1g Е" (!/'«)
GR-S НК (гевея) GR-S НК (гевея) CR S НК (гевея) GR-S НК (гевея1
— 16,5 10,40
— 16,0 10,38 8,83 10,39
—15,5 10,35 9,32 10,37
— 15,0 10,22 10,30 9,67 10,33 9,53
— 14,5 10,21 10,19 8,89 9,91 10,23 10,26 9,70
— 14,0 10,17 10,01 9,32 10,04 10,20 10,12 9,78
— 13,5 10,10 9,66 9,62 10,05 10,15 9,85 9,45 9,73
— 13,0 9,96 9,10 9,82 9,50 10,04 9,38 9,45 9,52
— 12,5 9,77 8,54 9,83 8,85 9,87 8,78 9,57 9,15
— 12,0 9,50 8,10 9,68 8,33 9,61 8,27 9,48 8,77
— 11,5 9,16 7,81 9,42 7,89 9,30 7,90 9,32 8,36
— 11,0 8,78 7,61 9,05 7,55 8,93 7,68 9,06 7,94
- 10,5 8,41 7,45 8,59 7,28 8,55 7,50 8,75 7,55
—10,0 8,13 7,34 8, 18 6,93 8,23 7,38 8,41 7,31
- 9,5 7,92 7,28 7,85 6,53 7,99 7,30 8,07
— 9,0 7,78 7,26 7,58 7,83 7,27 7,73
- 8,5 7,68 7,25 7,34 7,72 7,25 7,40
— 8,0 7,60 7, 12 7,63 7,14
— 7,5 7,55 6,92 7,57 6,92
— 7,0 7,51 6,79 7,53 6,73
— 6,5 7,47 6,71 7,49
— 6,0 7,43 6,62 7,45
- 5.5 7,39 7,41
11. Релаксация напряжения в кристаллических полимерах
199
делены значения Е'(®) 11 приведенные к 25 °C 111. Резуль-
таты графически показаны на рис. IV, 41 и IV, 42 и в табл. IV, 6
и IV, 7. Вспомогательная кривая релаксации напряжения гораз-
до круче для натурального каучука, чем для GR-S, и имеет мак-
симальный отрицательный наклон, равный 1,1 (соответственно
0,74 для второго каучука). Динамические потери Е" дают более
острый максимум для резины из НК, чем для резины из GR-S.
Пик для резины из НК соответствует также мепыним значениям
времени, ибо резина из НК имеет более низкое значение Tg (см.
табл. II, 4).
Шина, имеющая окружность около 3 м, двигающаяся со ско-
ростью 90 км/ч, делает 30 000 полных оборотов в час. Следо-
вательно, т,^200 ООО рад/ч, a Igco =5,3. Эта частота лежит справа
от области частот, в которой проведены измерения, но представ-
ляется возможным плавно экстраполировать кривые, что приво-
дит к значениям модуля потерь для резины из GR-S по крайней
мере в 10 раз большим, чем для резины из Н1\.
Больший модуль потерь резины из CR-S при частотах, имею-
щих место в шинах, обусловлен наличием более высокой темпера-
туры стеклования и широкого релаксационного спектра. Возмож-
но также, что релаксация мест зацеплений 121 и «болтающихся»
цепей сетки оказывает влияние на Е" в интервале частот, харак-
терных для шин. Установлено, что частицы наполнителя (а в
шины для усиления всегда добавляют значительное количество
сажи) взаимодействуют и скользят как относительно друг друга,
так и относительно цепных молекул каучука, выделяя при этом
теплоту.
Литература
I. Е. G a t s i f f, A. V. Г о b о 1 s k у, Tech. Rept. RLT-21 to the Office
of Naval Research, Contract Nonr—1858(07), Project NB 356—377, Dec.
18, 1956.
2, I.. J. Z a p a s, S. 1. Shuler,? W DeWif 1. .1 Polymer, Sci.,
18, 245_(1955).
11. Релаксация напряжения в кристаллических полимерах
Вязкоупругие свойства кристаллических полимеров гораздо
более сложны, чем аморфных. Во-первых, к ним не всегда приме-
нимы принцип суперпозиции Больцмана и общие соображения о
линейной вязкоупругости. Особенно это верно, если параметры
кристаллического полимера изменяются во время проведения
эксперимента, например вследствие роста новых кристаллов по-
лимера.
20U
1 л. 1К Вязкоупругие и упруговязкие свойства полимеров
Кристаллические полимеры необходимо подразделять по край-
ней мере на четыре категории с тем, чтобы сделать более четким
обсуждение их вязкоупругих свойств.
1. Слабо закристаллизованные полимеры: кристалличность
от 5 до 10% с очень небольшими кристаллическими областями.
Примерами таких полимеров являются некоторые эластические
полиамиды. Последние получаются частичным замещением водо-
рода в амидных группах на метильные группы, а также при на-
рушении регулярности расположения амидных групп, возникаю-
щем при поликонденсации смесей адипиновой и себациновой кис-
лот с гексаметпленднамином.
2. Полимеры с умеренной степенью кристалличности: около
50%. Полиэтилен низкой плотности является очень важным при-
мером этого класса полимеров.
3. Полимеры высокой кристалличности: около 90%. Хорошим
примером их являются новые линейные полиэтилены.
4. Полимеры очень высокой степени кристалличности: >90%
Возможно, что к этой категории можно отнести некоторые новые
изотактические полимеры, такие, как изотактический поли-(3-ме-
тнл)-бутен-1.
Полимеры первой категории ведут себя подобно аморфным
полимерам, слегка сшитым поперечными связями, ибо кристал-
лизация проявляет себя так же, как и поперечные связи. «Попе-
речные связи» являются довольно устойчивыми во времени (исклю-
чая область максимальной скорости кристаллизации), но не устой-
чивы по отношению к температуре. При приближении к точке
плавления кристалличность начинает изменяться и полностью исче-
зает при температуре Тт. Выше этой температуры полимеры (в за-
висимости от их молекулярного веса) переходят в жидкое пли
высокоэластнческое состояние.
У полимеров 1-й категории переходная область очень растянх
та по температурной шкале.
Полимеры 2-й и 3-й категорий похожи на полимеры 1-й кате-
гории за исключением того, что они имеют более высокий модуль
во всей области между Т„ и Т,п. Полимеры 1-й категории являются
«каучукоподобиыми» в большей части этой области; 2-й катего-
рии— «кожеподобными», а 3-й категории—«твердыми». В этом
интервале температур все полимеры имеют высокую проч-
ность на удар, хотя к 3-й категории могут относиться н
хрупкие полимеры, в том случае, когда кристаллиты пли сферо-
литы становятся очень большими и ориентированными под дей-
ствием напряжения в температурном интервале роста кристал-
лов. Примером этого явления может быть образование трещин
под действием напряжений в пластмассовых трубках, сделан-
ных нз полиэтилена.
11. Релаксация напряжения в кристаллических полимерах
201
Полимеры 4-й категории могут проявлять некоторые вязко-
упругие свойства, присущие металлам, у которых они обуслов-
лены дислокациями и границами зерен.
Результаты исследования релаксации напряжения кристалли-
ческого полиэтилена низкой плотности и политрнфторхлорэтилена
показаны на рис. IV, 43 и IV, 44 [1, 2]. Они находятся в опре-
деленном контрасте по сравнению с типичными кривыми релакса
Рис. IV, 43. Кривые релаксации напряжения
для полиэтилена низкой плотности [Е. Cat-
s i f f, J. Offenbach, A. V. Tobolsk v.
J. Coll. Sci., 11, 48 (1956)].
яиц напряжения аморфных полимеров, к которым, например, от-
носится пол и изобутилен (см. рис. IV, 4).
Свойства полиизобутилена в переходной области были пол-
ностью описаны ранее. Переходная область его со шкалой времен
релаксации от 0,01 до 1 ч расположена в температурном ин-
тервале между —80 и —40 °C. Как можно видеть из рис. IV, 4,
в этом интервале модуль очень быстро изменяется с температурой
и временем. Значения Er(t) располагаются между значениями мо-
202
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
? 9
100 1000
(1001 0,01 OJ
Время, ч
IV, 44. Кривые релаксации на-
Рис.
пряжения lg£z(0 от igl для политрл-
фторхлорэтилена
s к у, J. R.
.1. Phys. Chem.,
дуля стекла 1010>Б дин!см? и высокоэластического модуля
10“,88 дин]см2. Кривые релаксации напряжения, полученные при
различных температурах, могут быть наложены друг на друга
горизонтальным перемещением вдоль шкалы логарифма времени, и
этот принцип суперпозиции позволяет построить вспомогатель-
ную кривую, которая покрывает полную шкалу времени.
Политрифторхлорэтнлен является поликрпсталлическим поли-
мером и относится к 3-й категории; его Tg=39 °C, а Т,п— 221 °C.
В интервале между Tg и ЧйТт
(температура максимальной
скорости кристаллизации) кри-
вые релаксации напряжения
для политрифторхлорэтилена
являются относительно плоски-
ми, т.е. изменение модуля со вре-
менем в переходной области го-
раздо менее заметно для кри-
сталлического полимера, чем
для аморфного. Так, для поли-
трифторхлорэтилена значение
модуля при t =0,01 « изменяет-
ся от значения, равного 1010’1
дин1см2 при 30 °C, до значения,
равного 108>ЗБ дин!см? при 144 °C,
т. е. наблюдается очень медлен-
ное изменение. Переходная
область сливается с высокомо-
дульной областью высокоэла-
стичности; здесь кристаллиты
играют ту же роль, что и «пе-
реплетения» или поперечные свя-
зи в аморфных полимерах.
Несомненно,
спад напряжения при
структуры
[А.
М с
59,
V. Т о b о 1-
Loughlin,
989 (1955)].
что относи-
193 °C связан
микрокристаллической структуры или тек-
ориентации кристаллического материала. Инте-
тельно быстрый
с изменением 1
стуры, т. е.
ресно отметить, что этот полимер проявляет «тепловую хрупкость»
(или пониженную сопротивляемость удару), если держать его в
течение большого промежутка времени в температурном интерва-
ле от 170 до 195 °C. Это обусловлено продолжающимся ростом
кристаллитов и (или) ориентацией кристаллического материала
Полимер, находясь в течение большого промежутка времени под
нагрузкой в этом температурном интервале, дает трещины.
Данные по релаксации напряжения для полиэтилена низкой
плотности (рис. IV, 43) дают в основном те же самые характеристик
11. Релаксация напряжения в кристаллических полимерах
20 3
ки. Относительно быстрое уменьшение 0,01-часового модуля,
когда полимер приближается к точке плавления (110 °C), объясняет-
ся быстро изменяющейся в этом температурном интервале сте*
пенью кристалличности. Действительно, для полиэтилена были
предложены эмпирические соотношения между модулем и сте-
пенью кристалличности.
Так как с изменением температуры наблюдаются изменения
микрокристаллической структуры и механизма упругости, то
в связи с этим, по мнению автора, простой принцип температурно-
временной суперпозиции, действительный для аморфных полиме-
ров, не будет справедлив без жестких ограничений для кри-
сталлических полимеров.«Это не только горизонтальное перемеще-
ние вдоль оси логарифма времени, связанное с изменяющимися
скоростями молекулярных движений при изменении температу-
ры, ио, что значительно более важно, п вертикальное смещение
вдоль оси Er(t) (особенно около точки плавления), обусловленное
изменяющейся структурой и другими факторами» [1].
Некоторые авторы предполагали, что принцип температурно
временной суперпозиции практически применим к кривым релак-
сации напряжения для кристаллических полимеров при очень
небольших растяжениях, при температурах значительно ниже
точки плавления и для не очень продолжительных времен [3, 41.
Полностью удостовериться в этом можно только при широком
сравнении экспериментальных данных по релаксации напряжения
и изменению динамического модуля, как было сделано для полиизо-
бутнлена (см. раздел 3). Распределения времен релаксации для
поликристаллических полимеров, полученные при помощи неиз-
мененного принципа температурно-временной суперпозиции, ока-
зываются особенно широкими (рис. IV, 45 и IV, 46).
Совсем недавно было предложено [51 количественное уравне-
ние, описывающее вертикальное перемещение вдоль оси Er(f)
для кристаллических полимеров. Для того чтобы получить вспо-
могательную кривую для выбранной стандартной температуры
Го, кривые Er(t) при других температурах должны сначала быть
приведены к температуре То посредством следующей формулы:
Ig Er (t, привел.) = Ig Ег (0 + 1g -у- —- 1g + 1g (Н/D
В уравнении (11, 1) разность (1— Ха) есть степень кристаллич-
ности полимера при температуре То, а (1—X)—степень кристал-
личности при температуре Т.
Чтобы получить вспомогательную кривую, сначала производит-
ся перемещение вдоль оси lg£r(/), затем данные, полученные ука-
»анным способом, подвергаются горизонтальному перемещению
204
Г л. 1U. Вязкоупругие и упруговязкие свойства полимеров
1g Времени, ч
Рис. IV, 45. Вспомогательные кривые lg/:r(/) от
1g/ при 25 °C:
/—кристаллический полипропилен; 2—линейный полиэтилен
высокой плотности; 3—разветвленный полиэтилен низкой
плотности; 4—аморфный полипропилен и 5—полиизобутилен
[J. A. Fan с 11 er, Trans. Soc. Rheol.. 3, 81 (1959)].
I g времени, ч
Рис. IV, 46. Зависимости от )g/ при
25 °C:
/—кристаллический полипропилен; 2—полиэтилен выси
кой плотности; 3—полиэтилен низкой плотности; 4—аморф
иый полипропилен и 5—полиизобутилен NBS.
11. Релаксация напряжения в кристаллических полимерах
205
вдоль оси 1g/ обычным способом. Эта процедура успешно приме-
нялась для полиэтилена высокой плотности 151.
Результаты исследования релаксации напряжения иевулка
низованного натурального каучука в температурном интервале
от —50 до 30 °C, приведенные на рис. IV, 47 и IV, 48, показывают
трудности, связанные с исчерпывающей интерпретацией данных по
релаксации напряжения кристаллических полимеров. Натураль-
Рис. IV, 47. Кривые релаксации напряжения для невулканизованного нату-
рального каучука при растяжении 50% в температурном интервале кристал-
лизации [А. V. Tobolsk у, G. М. В г о w n, J. Polymer Sci..
17, 547 (1958); G. М. Bro \v n, Ph. D. thesis, Princeton University. 19481.
ный каучук имеет максимальную скорость кристаллизации при
—20 °C. Когда длина образца исследуемого материала поддер-
живалась постоянной в температурном интервале от —40 до 0 °C,
напряжение в образце падало до нуля вследствие увеличения
ориентации кристаллического материала [6,71. Более того, образ-
цы со временем увеличиваются в длине, превышая длину перво-
начального растяжения (явление, известное под названием
«спонтанного удлинения»). Это происходит так, как будто сварен-
ные макароны, лежащие в чашке, подвергнутые легкому растя
206
1.1. IV. Вязкоупругие и упруговязкие свойства полимеров
женою, внезапно «вспомнили», что они были ранее упакованы в
длинной узкой коробке.
Релаксацию напряжения, происходящую вследствие присут-
ствия ориентированного кристаллического материала, можно
обнаружить, как описано в разделе 15, изучением двойного луче-
преломления.
Для того чтобы кристаллический полимер полностью прояв
лял высокую прочность и способность сопротивляться ударным на-
грузкам. необходимо, чтобы в среднем полимерные цепи Пересе-
Рис. IV, 48. Кривые релаксации напряжения при —20 ’С
невулканизоваииого натурального каучука при
различных растяжениях [А. V. Tobolsky,
G. М. В г о w п, J. Polymer Sci., 17, 547 (1958)].
кали хотя бы два кристаллита. В противном случае кристалличе-
ский полимер будет иметь физические свойства воска. 5то значит,
что для получения оптимальных свойств кристаллических поли-
меров требуется, чтобы длина молекулярных цепей не снижалась
ниже некоторого минимального размера. Желательно также, чтобы
кристаллиты были небольшими, так как при этом одна полимерная
цепь может пересекать большее число кристаллитов*. Кристалли-
* Излагаемые здесь представления о связи свойств со структурой кри
сталлнческнх полимеров в настоящее время устарели. Интенсивно изучаемые
в последние годы надмолекулярные структуры в полимерах оказались гораздо
более разнообразными, чем можно было ожидать, и гораздо более значитель-
но влияющими иа механические свойства, чем предполагали вначале.—Пои.»
сед
12. Релаксация напряжения в сетках
207
ты небольших размеров могут быть получены правильным прове-
дением отжига или, что еще лучше, нарушением регулярности
цепей путем введения в них небольшого количества сомономера,
например, введение в политрнфторхлорэтилен менее чем 5% ви-
нилиденфторида заметно повышает его сопротивление ударным
нагрузкам и особенно понижает его хрупкость, если такой поли-
мер выдерживался при температуре 180 °C.
Замечательно то, хотя это еще не объяснено, что многие кри-
сталлические полимеры даже ниже температуры стеклования об-
ладают хорошим сопротивлением ударным нагрузкам. Примерами
таких полимеров являются политрифторхлорэтилен, поливинил-
хлорид, найлон-6, найлон-66, политетрафторэтилен (все они с
значением Tg выше комнатной температуры). В связи с этим важ-
но напомнить, что для правильного определения Тё кристалличе-
ских полимеров необходимо, чтобы эти полимеры были получены
в аморфном состоянии переохлаждением или сополимеризацией
Различные утверждения в литературе о том, что Tg этих полиме-
ров лежат ниже комнатной температуры, ошибочны.
Дальнейшее обсуждение вопросов, относящихся к кристалли-
ческим полиэтилену и полипропилену, приводится в Приложе-
нии К.
Литература
1 .4 V. Т о Ь о 1 s к у, .1. R. McLoughlin, J. Phys. Chem., 59, 989
(1955).
2 . Е. С a t s i f f, J. Offenbach, A. V. T о b о 1 s к y, J. Coll. Sci .
11, 48 (1956).
3 . Г. Y о s h i t о m i, K- Naga ma tsu, K. Kos iy a ma, J. Polymer
Sci., 27, 335 (1958).
4 K. Nagamatsu, T. Yoshitomi, T. Takemoto, J. Coll
Sci., 13, 257 (1958).
5 K. Nagamatsu, T. Takemura, T Yoshitomi, T. Ta
к e m о t o, J. Polymer Sci., 33, 515 (1958); T. Takemura, J. Po
lymer Sci., 38, 471 (1959).
6 . G. M. Brown, A. V. Tobo Isky, J. Polymer Sci., 17, 547 (1955):
G. M. Brown, Ph. D. thesis, Princeton University, 1948
7.4 N. Gent, Trans. Farad. Soc., 50, 521 (1954).
12. Релаксация напряжения в перестраивающихся сетках
Классом веществ, которые по своим свойствам приближаются
к идеальным максвелловским телам, являются полисульфидные
резины [1, 21. Они представляют собой сетки, состоящие из по-
лисульфндных цепей, структура которых имеет следующий вид-
---CH2CH2SSCH2CH2SS---
---CH„CH2OCH2CH.,SSCH2CH2OCH2CH2SS—
- CH.,CH2OCH2OCH,CH2SSCH2CH2OCH2OCH2CH2SS—
208
.i. / И. Вязкоупругие и упруговязкие свойства полимеров
Хотя в основных полимерных цепях дисульфидные звенья
преобладают, но имеются также моносульфидные звенья —S—
трпсульфидные звенья —SSS и тетрасульфпдные звенья
-SSSS—. Узлы сетки обычно состоят из трех ковалентных связей
Концами обрамляющих сетку цепей являются SH-связи. Следует
добавить, что, кроме этих главных деталей сеточной структуры,
существует небольшое количество связей типа — S~- 7РЬ+ • "S-
внутри цепи или —S“- • Na+ на концах цепей, обрамляющих сетку
При действии быстрых напряжении и деформаций эти резины
подчиняются кинетической теории высокоэластичности, рассмот-
ренной в главе II. Однако н сетках происходят химические пере-
стройки (вследствие реакций обмена), которые становятся замет-
ными, если резина поддерживается при заданной постоянной
деформации в течение длительного времени Ill. Встречаются
химические перегруппировки следующего рода:
R—S—S—R катализатор R—S S—R
-------" I К I
R'—S—S—R’ R'—S S-R’
R—S—Н катализатор R—S Н
--------- I + I
R'-S— S—R' R'—S S—R'
R-S-Na R-S Na
+ — I 4- I
R'—S-S-R' R'-S S-R'
R—S—Pb—S-R R -S—Pb S—R
— 14 1
R'—S—S—R' R'-S S-R'
(12. 1)
(12. 2)
(12, 3)
(12. 4)
где R и R' представляют собой полимерные цепи.
В эти перегруппировки связей вовлекаются также трпсуль
фидные и тетрасульфпдные звенья. Катализаторы в формулах
(12,1) или (12, 2) являются ионными веществами (кислотами,
основаниями или солями), остающимися после процессов поли-
меризации и вулканизации.
Если бы каучук всегда поддерживали в недеформированном
и ненапряженном состоянии, то было бы невозможно никакими
простыми физическими средствами определить, происходят ли
-гп обменные перегруппировки.
Однако если каучук растянуть до определенной длины и
поддерживать его при этой деформации, то эти перегруппировки
приведут сеточную структуру к структуре, находящейся в равно-
весии с новым состоянием растяжения, и поэтому будет пропсхо
дить релаксация напряжения, которая постепенно полностью за
вершится и напряжение станет равным нулю. Ситуация аналогич-
на явлению, имеющему место при течении жидкости (но в другой
12. Релаксация напряжения в сеткал
209
шкале времени). Не сразу очевидно, что молекулы воды, налитой
в мензурку, непрерывно меняют свои места. По это становится
совершенно очевидным, когда воду переливают в сосуд другой
формы. Так же как и вода, перелитая из одного сосуда в другой,
ио своим свойствам остается тождественной (кроме изменения
формы), так'и растянутая полисульфидная резина, которая имеет
необходимые условия для того, чтобы напряжение в ней упало до
нуля, тождественна по свойствам с первоначальным образцом н
отличается от него только формой. Скорость перегруппировки свя-
teu не зависит от деформации.
Для простоты мы будем считать, что перегруппировки связей,
происходящие в данном образце полнсульфидной резины, пол-
ностью соответствуют случаю, представленному уравнением
(12,3) Для растянутого образца резины элементарным процес-
сом спада напряжения является следующий:
(12.5)
Сразу после растяжения образца от длины Lu до длины L
напряжение (отнесенное к действительной площади поперечного
сечения) выразится:
= Nt(0)kT (-±-|
(12,6)
। де .'V, (0)—число эффективных цепей сетки в 1 сма резины (не вклю-
чая концевые цепи) при времени /=0, и Т - температура 1см.
уравнение (7, 5) гл. I].
Если резина поддерживается при постоянной длине L в течение
времени /, то только A\(Z) из Д\(0) цепей сетки не подвергнутся
обменной перегруппировке вида (12, 5).
Предположим, что цепь, участвующая в процессе перестрой-
ки связен, релаксирует полностью и не будет больше давать
вклад в напряжение образца, растянутого до заданной длины L.
Только iVj (/) цепей сетки, которые еще не участвовали в перегруп-
н- ьзз
210
Гл. IV. Вязкоупругие и упруговязкие свойства полимеров
ппровке связей, будут давать вклад в напряжение. Поэтому
можно записать:
Ht) = NAt)kTy^y _А| (12,7}
Следует ожидать, что обменная реакция подчиняется следую-
щему закону:
- (12,8)
где пг—среднее число звеньев в каждой цепи, способных к акту
перегруппировки (или общее число дисульфидных, трисульфид-
ных и тетрасульфидных звеньев), т2—число S—Na концов в 1 см®
резины, а —константа скорости реакции. Начальным условием
является Лгг(/) =AfI(0) при /=0.
Интегрируя уравнение (12, 8), получим:
А\ (/) = Д\ (0) ехр (—х() = (0) ехр (-- tl-) (12, 9)
где х=л1/?г2х'; т=1/х.
Подставив уравнение (12, 9) в (12, 7) и разделив результат
на (12,6), окончательно получим-
= ехр (— -/-О = ехр (— t[r) (12,10)
Уравнение (12, 10) есть тот же самый закон релаксации напря-
жения при постоянном растяжении, какой был получен из урав-
нений Максвелла для упруговязкого сдвига и растяжения:
ds__ 1
dt ~ G ' dt + 4G
(12, 11)
ds 1 _ df , 1
dt ~~ E dt ' zE '
(12, 12)
Действительно, ранее было показано, что уравнения
(12, 11) и (12, 12) теоретически обоснованы для полпсулофидных
резин при малых деформациях при условии, что G равно N^OjkT,
а Е равно 3N1(0)kT. Экспериментально было обнаружено, что ре-
лаксация напряжения при постоянном растяжении очень близко
следует уравнению (12, 10). На рис. IV, 49 приведены данные по
релаксации напряжения при постоянном растяжении для типич-
ного полисульфидного каучука и сопоставлены с максвелловским
законом релаксации напряжения (12, 10). Найдено, что относи-
тельное падение напряжения совершенно не зависит от
степени растяжения образца вплоть до наибольших изученных
растяжений (150% растяжения). Это подтверждает наше предпо-
12. Релаксация напряжения в сетках
411
ложение о том, что скорость обмена связей не зависит от деформа-
ции. В главе V будет широко рассмотрен процесс химической Ре-
лаксации напряжения полисульфидных резин, причем большое
внимание будет уделено химической стороне проблемы.
Теоретические соображения, представленные, здесь и в следую-
щем разделе, можно применять и к каучукам, не относящимся
к полнсульфидным. в которых также протекают обменные реак-
ции. Например, в силиконовых резинах, основная цепь которых
имеет следующую структуру
СН3 СН, СН, СН., СН,
V «’ о «/ о
I I I I I
----Si—О—Si—О—Si—О—Si—О—Si—О-------
Рис. IV, 49. Кривая релаксации напряжения полисуль-
фндного каучука Н-11 при различных растяжениях и
график функции ехр (— t/x) при 60 ‘С: •—10% растяже-
ния, С—30% растяжения, □—50% растяжения;
------------ехр (—t/x) при т=3 ч[М. D. Stern,
A. V. Tobol sky, J. Chem. Phys., 14, 93 (1946)).
также могут протекать обменные реакции типа
R—О—Si—R катализатор
R'—Si—О—R'
R-O
I
R'—Si
Si—R
-F I
О—R'
(12. 13)
где катализаторами являются кислота, основание или соль, ос-
тающиеся после полимеризации или вулканизации [3, 4].
Указанные перегруппировки могут происходить не только в
Цепях, но и в поперечных связях, причем некоторые химические
14
212 1л. IV. Вязкоупругие и упруговязкие свойства полимеров
поперечные связи, такие, как солевые типа
О О
II _ + - II
—С—О...М+т..О—С-
очень подвижны, и их можно считать способными к обменным
реакциям [5, 61. (Солевые связи могут находиться и внутри се-
точных цепей.) Конечно, в этих случаях перегруппировку связен
можно считать двухстадийным процессом: разрушение и затем об
разоваиие поперечных связей, причем разрушение и восстановле-
ние связей протекают с одинаковыми скоростями.
Если сеточный полимер поддерживается в растянутом состоя
пии, то разрушение поперечных связей вызывает разгрузку цепей
сетки, соединенных этими поперечными связями. Вследствие вое
становления поперечных связей образуется то же число цепей сет
ки, но они находятся в недеформированном состоянии, если даже
сетка резины в целом еще находится под напряжением. Такой
тип перегруппировки поперечных связей приводит к тому же
уравнению упруговязкостп, как и перегруппировка типа цепь-
цепь 1схема (12, 13)1 или типа конец—цепь [схема (12, 5)]. Это
будет показано в следующем разделе. Более то"о, между полимер-
ными цепями могут образовываться временные «физические» по-
перечные связи, которые разрушаются и восстанавливаются, но
могут играть ту же самую роль, что и подвижные химические по-
перечные связи. Возможно, что такое положение может возник-
нуть в полимере, содержащем небольшое количествофункциональ-
ных групп, способных к сильным локальным взаимодействиям
с близлежащими цепями (например, водородные или солевые
связи) [61. В действительности, наряду с ковалентными —S—S—
поперечными связями 171 с такими связями можно встретиться
в набухших шерстяных волокнах, а также в полимерах, содержа-
щих карбоксильные группы [6]
Однако межмолекулярные физические взаимодействия в поли-
мерах с высоким молекулярным весом лучше всего представлять
как сложные «переплетения». Эти «переплетения» действуют как
временные поперечные связи, которые разрушаются и восстанав-
ливаются, но в этом случае упруговязкое поведение является бо-
лее сложным, чем то, которое можно описать посредством одного
времени релаксации. По-видимому, здесь требуется найти теоретиче-
ский подход к определению фактического распределения времен
релаксации.
Литература
1. М. D. Stein, А. V. Tobolsk у, J. СЬеш. Phys., 14, 93 (1946).
2. М. М о с h u 1 s k v, А. V. Tobolsk у, Ind. Eng. Chem., 40, 2155
(1948).
3. D. H. Johnson, J. R. McLoughlin. A. V. Tobolsk y. .1
Phys. Chem , 58, 1073 (1954).
13. Вязкоупругость и упруговязкость сеток
213
4 R С- О s t h о 1 1, А. М. В и е с ii е, W. 1 Grubb, J. Am. Chem.
Soc., 76, 4659 (1954).
5. M. S. Greer, A. V. T- о b о 1 s к у, J. Chem. Phys., 14, 80 (1946).
6. W. Cooper, J Polymer Sci., 28, 628 (1958).
7. S. M. Katz, A. V. T о b о 1 s к y, Text. Res. 20, 87 (1950).
13. Вязкоупругость и упруговязкость
перестраивающихся сеток
Рассмотрим сетку, в которой происходят обменные перегруп-
пировки типа цепь—цепь, конец—цепь или поперечных свя-
зей (включая двустадийный процесс разрушения и восстановле-
ния связей) [1]. Вначале мы будем рассматривать только один
тип связи, участвующей в этих перегруппировках. Поэтому при
постоянном растяжении или сдвиге закон релаксации напряжения
запишется в виде 1см. уравнение (12. 10)1
^| = exp(-z/) (13,1)
Объяснение процесса релаксации напряжения при постоянном
растяжении или постоянной деформации сдвига, который наблю-
дается, несмотря на то, что разрушение и восстановление связей
происходит с одинаковой скоростью, заключается в том, что раз-
рушение связей приводит к разгрузке и сокращению напряжен-
ных цепей, а восстановление связей происходит таким образом,
что не приводит к увеличению напряжения при постоянном рас-
тяжении. Вполне можно предположить, что образование связей
приводит к появлению отрелакснрованных цепей сетки независимо
от того, какие размеры имеет образец и какие напряжения на него
действуют.
Представим, что произойдет, если образец подвергался пред-
варительным деформациям. Можно разделить цепи полимерной
сетки на классы, содержащие в 1 см3 N' цепей, образовавшихся за
время /',в течение которою образец резины поддерживался при
длине £(/') или при деформации сдвига ?(/') Согласно кинетиче-
ской теории упругости каучука и предположению о том, что цепи,
образованные при длине £(/') или деформации сдвш a 7(C), находят-
ся в отрелаксированном состоянии, соотношение между текущим
истинным напряжением /(/) и текущей длиной /.(/) или сдвигом
7</) имеет вид:
f(t) =kT\N' T77TilexPi z,/ ,13’2)
' f (t) = kT У (t)- т(Г')|ер[ /(Z .’ )i (13,3)
214
I л. 1\'. Вязкоупругие и упруговязкие свойства полимеров
Величины Л" удовлетворяют следующим уравнениям:
dN'
dt'
ZN’ = Nl
= *(A\ -N')
(13,4)
(13, 5)
где iMi есть число цепей сетки всех классов TV' в 1 си3. Согласно
нашим предположениям величина Nx является постоянной. В урав-
нении (13, 5) dN' есть число цепей сетки, образующихся в интер-
вале времени /'-►/' , когда длина образца равна £(/') или сдвш
образца равен ?(/'). Число цепей—dN'pa3p, класса N', которые раз-
рушаются в 1 см3 за интервал времени t-^td-dt, дается уравнением
-rfWpa3P.
dt
(13, б)
= z.A'
Полное число цепей всех классов, разрушающихся в 1 слг за
интервал времени dt, равно —dN\tVa3p. и определяется урав-
нением
^1, р«зр.
dt
*N
(13, 7)
Если мы предположим, что образец первоначальной длины £(|
внезапно растягивается до длины £(0) при t=0 и с этого времени
его длина L(t') претерпевает с течением времени f непрерывное
изменение, то уравнение (13,2) перепишется так:
/«= ехр(-«р + ([£»
О
-у-О-1 ехр [— z (t — Г)] v.dt'
Ь (t) J
(13,8)
Уравнение (13, 8) является интегральным уравнением, которое
можно преобразовать в дифференциальное уравнение второго по-
рядка и решить при определенных начальных условиях [II. В раз-
деле 14 дается решение для ползучести при постоянной нагрузке.
Если образец при начальном сдвиге, равном нулю, подвергает-
ся непрерывной деформации сдвига ‘((f), то уравнение (13. 3)
перепишется в виде:
t
f (0 = J [у (0 — -((f)exp[— f.(t — f')) 7. di (13, 9)
о
13. Вязкоупругость и упруговязкость сеток
215
Интегрирование по частям дает*:
f (0 = NJT f М- ехр I - X (2 - f)] dt’
'о
(13, 10)
Уравнение (13,10) можно преобразовать в следующее диф-
ференциальное уравнение:
__ 1 .(If I I
dt “ G dt ' tG
(13, 11)
где G=N}kTii т=1/л. Уравнение (13, 11) является уравнением
Максвелла, рассмотренным в разделе 9 главы I.
Предположим, что мы имеем дело с сеткой, в которой происхо-
дят обменные перегруппировки поперечных связен, но эти попе-
речные связи различных типов j и характеризуются различными
константами скоростей реакций х-. Пусть М;- цепей сетки в 1 см9
соответствуют поперечным связям типа /, причем соблюдается
следующее условие: Уравнение (13, 10) при этом примет
вид:
t
fit} == O\df (13,12)
'о
где
n
Gr(t —f) = X NjkTexpl-Mt -f)] (13,13)
Уравнение (13, 12) можно преобразовать в систему дифференциаль-
ных уравнений следующего вида:
ЛМ _ (1/б/)^у(0 , f (')
dt dt r T,G;
i = 1, 2, 3,... n.
где
Gj^NjkT',
h(t) j^{expH x,-(f t'}\\dt'
(13, 14)
(13, 15)
* К сожалению, математические преобразования, излагаемые в разделе
13, содержат много неясностей, исправление которых невозможно без перера-
ботки значительной части текста этого раздела [например, уравнение (13, 10)
не следует из уравнения (13, 9); уравнение (13, 8) противоречит уравнению
(14, 3)].—Прим, ред
216
Гл. ! I7. Вязкоупругие а упруговязкие свойства полимеров
Уравнение (13,12) является одной из форм записи принципа
суперпозиции Больцмана для линейной упруговязкости, тогда
как уравнение (13, 14) является записью формулировки Вихерта
для линейной упруговязкости; оба эти выражения уже рассматри-
вались в главе 111. В главе III [см. уравнение(2, 3)] формулиров-
ка Больцмана дана в виде фундаментального постулата. Форму-
лировка Вихерта (раздел 3 главы III) выводится из формулировки
Больпмана (и наоборот). Интересно отметить, что обе эти форму-
лировки можно получить из физической модели, использованной
в этом разделе.
Тот же самый вывод применим и к растяжению, если мы огра-
ничиваемся очень небольшими деформациями растяжения. Мо-
дули сдвига G- при этом заменяются модулями растяжения
E^SNjkT.
Литература
1. М. S. G г е е n, А. V. Т о b о I s к у, J. Chem. Phys.. 14, 80 (1946).
14. Ползучесть при постоянней нагрузке в случае
перестраивающихся сеток
Если к полоске резины с первоначальным поперечным сечением
.4П приложена постоянная нагрузка X, то при £=0 возникает бы
строе упругое растяжение от первоначальной длины £0 до дли-
ны /.(0). Это выражается уравнением состояния вида:
F = ^- = ^,674-^
I La
I
(14, 1)
Если резина обладает упруговязкими или вязкоупругими свой-
ствами, то она будет со временем продолжать растягиваться. Это
явление названо ползучестью. Величина F=XIA0 во время этого
процесса остается постоянной, но напряжение /(7), рассчитанное
на истинную площадь поперечного сечения, будет зависеть от
времени /:
(14,2)
где L(t)—длина образца в момент времени t.
В разделе 13 показано, что соотношение между f(t) и £(() для
перестраивающихся вследствие обменных реакций сеток имеет
14. Ползучесть при постоянной нагрузке
217
ВИД:
/(') .. ехр(~ vt) /[£W Г „ _A_I + f z II AW-Г
\\kT ~exP< £0 | £(/)| || £(f)J
6
--4^.1 exp(- ,(t
\4 }
(14, 3)
Чтобы получить выражение для ползучести в перестраиваю-
щейся сетке при постоянной нагрузке, можно просто подставить
уравнение (14, 2) в уравнение (14, 3). Результирующее уравнение
может быть преобразовано в дифференциальное уравнение [1J.
Для простоты сделаем следующие изменения в системе обозначе-
ний:
У г > i/o I ’
Ч, М
(14,4)
Дифференциальное уравнение примет вид:
где
Уо- 1 РЛ
(14, 5)
(14, 6)
а д' и у" соответственно являются первой и второй производ-
ными по х.
Решение этого уравнения при помощи преобразования Лап-
ласа имеет вид [11:
ехр (х() = Р
Г 2L(t)
I RLa
„ 12/.(£) L | I I p I L„ Lv 11
P£„ ' ‘ Iе-1 1 I 6(0) £(/) |j
p____ Уо 2уо -г 3
— ~ " Чуо
О i (2-Уо R) (1/ii ~ 2Уо ~г 3)
fyo + R (2уи R) Ryn
(14,7)
На рис. IV, 50 для нескольких значений ло=£(О)/£о даны гра-
фики этой функции в форме логарифмической зависимости
ln£(/)/L0 от х/. Особенно следует отметить, что скорость ползу-
чести очень существенно зависит от нагрузки (измеренной при
218
Гл. IV. Вязкоупругие и упруговязкие свойства полимеро>
начальном растяжении), даже если и предполагается, что молеку-
лярная релаксация не зависит от напряжения. Следует помнить,
что эти кривые ползучести применимы только к перестраивающим-
Piic. IV, 50. Зависимость In[L(t)/L0] от >7 для различ-
ных значений ло=£(0)/£о согласно уравнению (14, 7)
(Р. .1. В I a t z, А. V. Т о b о 1 s к у. ,1. Chem.
Phys., 14, 113 (1946)].
Литература
1. Р. J. В 1 a t z, А.
V. Tobolsk у, J. Chem. Phys.. 14, 113 (1946).
15. Изучение двойного лучепреломления
При растяжении резины сетка становится оптически анизо-
тропной и вызывает эффект двойного лучепреломления. Теория
этого явления была разработана Куном 11] и Трелоаром 12].
Было получено следующее выражение для двойного лучепрелом-
ления А образца, растянутого от длины до L (a=LlL0Y.
/\ = Са + 2)а
А = гх — гу
(15,1)
В уравнении (15, 1) гх— показатель преломления в направ-
лении растяжения, гу—показатель преломления в направлении,
15. Изучение двойного лучепреломления
219
перпендикулярном растяжению, и г—средний показатель прелом-
ления, т. е. показатель преломления в нерастянутом состоянии,
являющийся легко измеримой величиной; есть число цепей
сетки в единице объема, а 6ц и Ь. —это главные поляризуемости
«статистического сегмента» вдоль и перпендикулярно контуру по-
лимерной цепи. «Статистический сегмент» определяют как гипо-
тетическую единицу полимерной цепи, которая обладает жест-
костью, но может свободно ориентироваться в любом направлении
по отношению к другим статистическим сегментам. Вычисления,
произведенные при выводе уравнения (15, 1), основаны на тех же
самых физических и геометрических соображениях, посредством
которых в разделе 9 главы II подсчитывается уменьшение энтро-
пии сетки растянутого каучука
Уравнение для напряжения (па истинное поперечное сечение)
для образца пз идеальной резины имеет вид:
(15,2)
Разделив уравнение (15, 2) на (15, 1), получим
f — _________________________45kr_____ .. г о
ГД 2гс (г2-j-2)2 (Ьц — Ь , )
Величина flTh является константой, характеризующей по-
лимер и зависящей только от структуры полимерной цепи. Она
дает возможность получить величину (&ц—b, ) для данного поли-
мера.
Трелоар показал для натурального каучука, как можно срав-
нить (6ц—bi_), полученное из уравнения (15, 3), с рассчитанным
значением (/?ц—Ь±)о для повторяющейся единицы цепи 13]. По-
следняя величина подсчитывается из главных поляризуемостей
всех связей. Вычисляемое значение (£>ц—£>, )0 зависит от предполо-
жений, принятых относительно конфигурации мономерного звена.
Трелоар предположил, что в натуральном каучуке остаток изо-
прена принимает плоскую жесткую конфигурацию. Противополож-
ное допущение состоит в предположении, что имеется свободное
вращение вокруг всех ординарных связей мономерного звена.
Действительная конфигурация занимает промежуточное по-
ложение между двумя положениями, указанными выше [4].
Если предположить, что конфигурация плоская, то для изопрено-
вого звена в натуральном каучуке (£>ц—£>±)0 будет равно
30,8 10-26 сл13; если же предположить свободное вращение вокруг
ординарных связей, то (£>ц—£>±)0 будет равно 17,8- 10~2В см3.
В общем случае найдено, что действительное значение (£>ц—6Х),
полученное из уравнения (15, 3), больше, чем вычисленное] зна-
220
[л. /I. Вязкоупругие а упруговязкие свойства полимеров
чение (£>ц—Ь±)о для повторяющегося мономерною звена. Это яв-
ляется слелствием жесткости полимерной цепи и результатом
таких факторов, как асимметричное заторможенное вращение и
стерические препятствия, которые обсуждались в разделе 1 гла-
вы 11. Например, Трелоар 13] находит, что (£ц—Ь.',1(Ьц—Ь±)а
равно 1,52. По его мнению, это указывает на то, что олии стати-
стический сегмент модели цепи оптически эквивалентен 1,52
изопренового звена. Конечно, значение величины
?=-.=(/j|l—bj.)/(b||~ Z)±)o будет отчасти зависеть от метода вычисле-
ния (6ц—Ь±)о.
В табл. IV, 8 приводятся несколько значений (&ц—Ь.>, по-
лученных рля натурального каучука (гевея), и несколько рассчи-
танных значений z. Для изопренового сегмента значение (Ь\\—Ь±)а
было взято равным 24 • Ю'^см3, которое является средним из соот-
ветствующих значений для плоской конфигурации и случая сво-
бодного вращения.
Уравнения (15, 1)—(15, 3) были использованы Тобольским
и Стейном 15] для определения молекулярной природы напряже-
ния в растянутом полимере, а также молекулярного механизма
релаксации напряжения (рис. IV, 51)
11олиизобутпленовые образцы поддерживались при постоян-
ном удлинении и постоянной температуре в интервале от 50 до
—40 °C; при этом были проведены игмерения как напряжения,
так п двойного лучепреломления. Было найдено, что двойное
лучепреломление уменьшается точно таким же образом, как и
напряжение*, и фактически величина [IT& являлась постоянной,
не зависящей от времени, температуры и удлинения 16].
Таблица IV, Я
Оптическая анизотропия статистического сегмента для натурального каучука
f/TS’iin дин/см” Т, °C \)’1025 г
1.1а 30 73 3,0
2,0° 20 40- 1,7
1,7“ 25 46,7 2,0
1,4Г 50 50 2. 1
а AV. Kuhn, Н, Ст 1 и н, Kol.-Z., 101,248 (1942); W- Kuhn, J. Ри Kiner Sci., I,
380 ('916).
6 J. J. Herman s, Kcl.-Z., 103. 210 (1943).
u L. R. G. T r e I о a r. The Physics of Rubber Elasticity, Oxford Press, 1949, p. 148.
[Л. Tp елоар, Физика упругости каучука. Издатинлит, 1953]
1 R. S. S t е I п, А. V. Tobolsky, Тех. Res. J., 18 , 201, 302 (1948).
* Подобие оптической и механической релаксации известно очень давно.—
Прим. ред.
la. Изучение двойного лучепреломления
221
в вязкое течение или диффузия
1g времени
1. о времени
Г исчезновение местных искривлении
молекул, кристаллов
Область искривлений
1g времени
1g времени
Рис. IV, 51. Типы элементарных релаксационных процессов, наблюдае-
мые в полимерах. Кривые, сопровождающие схемы, иллюстрируют харак-
тер изменения напряжения и двойного лучепреломления, ожидаемый для
каждого из этих элементарных процессов IK. S. Stein, А. V. Т о-
b о I s к у. Text. Res. J., 18, 201 (1948)].
222
1л. JV. Вязкоупругие и упруговязкие свойства полимеров
Для полиизобутилена температурный интервал от 50 и до - -40 °C
охватывает область эластовязкости, высокоэластическую область
и часть переходной области с низким значением модуля. При
растяжении образца в этих областях напряжение в деформиро-
ванном образце возникает за счет уменьшения конформационной
энтропии при растяжении. Релаксация напряжения при постоян-
ной деформации возникает в результате диффузии полимерных це-
пей, восстанавливающей вновь изотропное макросостояние, обла-
дающее наибольшей термодинамической вероятностью. Уравнения
(15,1)—(15, 3) действительны в этих областях вследствие того,
что напряжение и двойное лучепреломление рассчитываются,
исходя пз одних и тех же геометрических соображений. Для
того чтобы константа //ТА была независящей от температуры в
этом интервале, как это наблюдалось для иол и изобутилена, не-
обходимо, чтобы величина и оптические свойства «статистиче-
ского сегмента» не изменялись с изменением температуры.
Величина fiTА также оставалась постоянной, как п предпо-
лагалось, в процессе релаксации напряжения в полпсульфидных
резинах (перестраивающиеся сетки) и в вулканизованном нату-
ральном каучуке (см. главу V) [4, 51.
В случае лактопренового каучука, который является по
существу сшитым полиэтил акр платом, наблюдали, что выше
—17,5 °C отношение напряжения к двойному лучепреломлению
остается постоянной величиной во время наблюдения релаксации
напряжения при заданной температуре (Tg для полиэтилакрплата
равна —22 °C). Однако двойное лучепреломление этого каучука
при заданном значении a=L/L0 является функцией температуры
в интервале от —20 до 30°С, и согласно уравнению (15, 1) это мо-
жет означать только то, что изменяются оптические свойства
«статистического сегмента». Величина f]T& также меняется
с температурой, значительно и почти скачкообразно изменяясь
вблизи Tg. В области Tg двойное лучепреломление меняет знак,
а поэтому flTi\ ведет себя так, как это показано на рнс. IV, 52.
По-видимому, не удивительно, что в той части переходной об-
ласти, для которой характерны высокие значения модулей, на-
блюдается постепенное изменение f IT А с температурой. Оптиче-
ские свойства «статистического сегмента» могут изменяться по
двум причинам: 1) «статистический сегмент» может изменять раз-
мер, когда температура понижается; так как жесткость цепи воз-
растает, то требуется допустить включение большего числа атомов
в статистический сегмент, чтобы можно было его рассматривать
как свободно вращающийся; 2) даже если «статистический сег-
мент» сохраняет свои размеры с температурой, то его оптические
свойства могут изменяться благодаря изменениям вращения бо-
ковых групп.
15. Изучение двойного лучепреломления
223
Когда достигается температура стеклования и полимер Пере-
ходит в стеклообразное состояние, то оказывается невозможным
производить расчет напряжения по уравнению (15, 2) и расчет
двойного лучепреломления по уравнению (15, 1). В стеклообраз-
ном состоянии напряжение возникает далеко не исключительно
за счет вклада энтропии, также и оптическую анизотропию уже
нельзя считать полностью зависящей от беспорядочно ориенти-
рующихся «статистических сегментов». Поэтому не удивительно,
чтОдЧасто f IT А подвергается резкому изменению уже около Tg.
Температура, аС
Рис. IV, 52. Изменение отношения приращения двойно-
го лучепреломления к приращению напряжения с тем-
пературой для вулканизата лактопрена. Образец был
растянут на 30% от первоначальной длины при 30 °C и за-
тем при охлаждении поддерживался при этой же дли-
не [ R. S. Stein, S. К г i m m, А. V. Tobol-
s k у, Text. Res. J., 19, 8 (1949)].
Обычно двойное лучепреломление полимера в стеклообразном со-
стоянии принято описывать с помощью оптического коэффициен-
та упругости А//, а не с помощью величины flTh.
Оптический коэффициент упругости (коэффициент фотоупру-
гости) в стеклообразном состоянии сильно зависит от местных
конфигурационных особенностей главной цепи [7]. Это представ-
лено в табл. IV, 9. Замещение групп в главной цепи создает за-
метные изменения в оптическом коэффициенте упругости, тогда
как подобное замещение, произведенное вне главной цепи, дает
лишь небольшой эффект.
В кристаллических полимерах в процессе релаксации на-
пряжения при постоянном удлинении двойное лучепреломление
может возрастать (в абсолютном смысле), так как релаксация
224
I л. IV. Вязкоупругие и упруговязкие ееойсгеа полимеров
Таблица IV, 9
Оптический коэффициент упругости полимеров в стеклообразном
состоянии3
Полимер
Л//- 1013 CM-Zdlb.
Атактический полистирол . . + 10
Атактический ноли- л-тлретл-бутилсгирол . . + Н
Атактический поли- а-метилстнрол 9
Изотактический полистирол (аморфное состояние до-
стигалось быстрым охлаждением) + 12
К. 1). Andrew s, private communication.
напряжения можетбыть результатом ориентации кристаллических
областей и ориентированного роста зародышей и кристаллитов. Эти
соображения были применены для изучения указанных эффектов
в желатиновых гелях. Так как эти гели сохраняют двойное луче-
преломление и после полной релаксации напряжения при задан-
ной постоянной деформации, то был сделан вывод, что в подоб-
ных «каучуковых» гелях падение напряжения сопровождается
ориентацией и ростом кристаллических областей 18].
Стейн с помощью сложного расчета дал количественную ин-
терпретацию двойного лучепреломления в растянутых кристал-
лических полимерах, причем им было рассмотрено влияние на
двойное лучепреломление как кристаллических, так и аморфных
областей 19].
Литература
1. W. К п h п, Н. G г и п, Koll.-Z., 101, 248 (1942); W. К п h п, J. Po-
lymer Sci., I, 380 (1946).
2. I.. I?. G. T r e 1 о a r, Trans. Farad. Soc., 43, 277, 289 (1947).
3. I.. R. G. Treloar, The Physics of Rubber Elasticity, Clarendon Press,
Oxford, England, 1949, pp. 147—150. (T p e л о a p, Физика упругости
каучука, Издатиилш, 1953].
4. R. S. Stein, A. V. To bo 1s k y, J. Polymer Sci., 11, 285 (1953).
5. R S. S t e i n, A. V. Tobolsk y. Text. Res. J,, 18, 201 (1948); ibid..
18, 302 (1948); R. S. Stein, S. К r i m m, A. V. T о b о 1 s k у
Text. Res. J., 19, 8 (1949).
6 R. S. S t e i n, F. H. Holmes, A. V. T nbolsky, .1. Polymer
Sci., 14, 443 (1954). S. a. R. S. S t e i ii. Ph. D. thesis, Princeton Univer-
sity, 1948.
7. R. D. Andrews, Private communication.
8. A. V. Tobolsk y, J. Phys. Chem., 59, 575 (1955).
9. R S. Stein. .1 Polymer Sci., 27, 567 (1958).
Г лава V
ХИМИЧЕСКАЯ РЕЛАКСАЦИЯ НАПРЯЖЕНИЯ
1. Химическая релаксация напряжения
в резинах из углеводородных каучуков
Явление химической релаксации напряжения было обнаружено
при исследовании вулканизованных каучуков углеводородного
типа [1] (натуральный каучук, неопрен, бутил-каучук, сополимеры
бутадиена со стиролом и бутадиена с акрилонитрилом).
Было найдено, что в температурном интервале от 100 до
150 °C у вулканизатов этих каучуков, находящихся при постоян-
ной деформации, наблюдается очень быстрое уменьшение напря-
жения вплоть до нуля. Так как в пространственно-структуриро-
ванном каучуке, находящемся в высокоэластическом состоянии,
физическая релаксация напряжения может иметь место только
в небольшой степени и, конечно, не до нулевого напряжения, это
явление было связано с химическим разрывом пространственной
сетки резины. Такой разрыв можно было приписать специфическо-
му влиянию молекулярного кислорода, так как в условиях очень
низких давлений кислорода (< 10_* атм) скорость релаксации на-
пряжения заметно уменьшалась. Однако при умеренно низких
давлениях кислорода скорость химической релаксации была такой
же, как и в атмосферных условиях. Этот результат аналогичен
давно известному факту, что в жидкой фазе скорость взаимодей-
ствия углеводородов с кислородом не зависит от давления кислоро-
да вплоть до очень малых давлений. Существенно отметить, что
образцы резин, использованные в этих опытах, были достаточно
гонкими для того, чтобы обеспечить стационарные условия для
диффузии кислорода; в противном случае скорость химической
релаксации напряжения определялась бы скоростью диффузии.
Одновременно с тем, как была обнаружена химическая релак-
сация напряжения, было показано, что ползучесть резин при вы-
соких температурах также вызывается присутствием молекуляр-
ного кислорода 12]. В ранних исследованиях химической релак-
сации напряжения был установлен и ряд других важных фактов
[II. Так, было найдено, что кривые относительного уменьшения
напряжения где /(/)—напряжение ко времени /, а /(0)—
15—833
226
Г л. Iх. Химическая релаксация напряжения
начальное напряжение, не зависят от величины удлинения, при
котором испытывается резина, по меньшей мере вплоть до 200%
удлинения (относительная длина, или кратность растяжения
х=3). Некоторые данные, подтверждающие это для резины из
натурального каучука, приведены на рис. V, 1.
Было найдено, что па кривые относительного уменьшения на-
пряжения /(/)//(0) практически не влияет присутствие или отсут-
ствие сажи пли других наполнителей (рис. V, 2).
Рис. V, 1. Влияние удлинения на химическую релак-
сацию напряжения серных вулканизатов натурального
каучука при 100 °C:
/—20, Б0 II 100%; 2—400%; S—700% |А. V. Tobolsky, |. В
Prettyman, J. Н. D i I I о n, J. Appl. Phys., 15, 3'24 (1941)]
На рис. V, 2 также видно, как заметно уменьшается скорость
релаксации напряжения при тщательном удалении кислорода.
Полное его исключение, однако, невозможно, так же как невоз-
можно простым эвакуированием удалить перекисные группы,
содержащиеся в цепи углеводорода.
Для резины из натурального каучука кривые химической ре-
лаксации подчиняются закону Максвелла, т. е. имеют вид:
Д- = ехр(-х7)
[(I. 1)
Характерным для спада напряжения по Максвеллу является то,
что если построить график /(/)//(0) от 1g/, то спад напряжения про-
исходит за промежуток времени, несколько превышающий 2
1. Химическая релаксация напряжения в резинах
2'2.1
порядка, как показано на рис. 1, 5. Это очень точно соблюдается
для резины из натурального каучука (см. рис. V, 1). Для резины
из других углеводородных каучуков (дивинилстирольного, нео.
преиа, бутилкаучука, дивинилнитрилакрилового и др.) было
найдено, что уменьшение напряжения вследствие химической
релаксации, хотя и не следует точно закону Максвелла, однако
Рис. V, 2. Кривые химической релаксации напряжения
(120 °C) для серных вулканизатов натурального каучука
без наполнителя (1 и 5) и с 33 1/3 вес. % усиливающей
углеродной сажи (2 и 4). Кривые 1 и 2—в среде тща-
тельно очищенного азота; кривые 3 и 4 па воздухе.
[А. V. Т о b о I s к у, I. В. Р г е t t у m а п, J. Н
D i I 1 о п, J. Appl. Phys., 15, 324(1944); W. С. Sc h п еД
d e г, Ph. D. thesis, Princeton University (1944)]:
/—гевея ненапллненная 120 °C); 2—гевея наполненная (TVq,
120° С); 3—гевея пенаполиенная (воздух, 120 °C); 4—гевея наполнен-
ная (воздух, 120 °C).
также завершается за промежуток времени в 2—3 порядка, как
показано па рис. V, 3. Для всех этих резин можно описать влия-
ние температуры на химическую релаксацию следующим обра-
зом:
МО-
НО)
= Ф(-/.7)
(1.2)
где х' зависит только от Т. Это значит, что если Д/)/Д0) для дан-
ной резины отложить относительно 1g/, то релаксационные кри-
вые для различных температур могут быть получены перемещением
точек вдоль осп логарифмов времени. Было найдено, что функ-
15”
22й
Гл. к. Химическая релаксация напряжения
циональная зависимость х' от температуры выражается .уравне-
нием Аррениуса
х' =Лехр(~ЕаКТ//?П
(1,3)
Для резин, приведенных на рис. V, 3, £акт. оказалась равной
Рис. V, 3. Кривые химической релаксации напряжения для вулканизатов
различных синтетических каучуков углеводородного типа при 100 °C:
[А. V. Т о Ь о 1 s к у, I. В. Prettyman, J. Н. Dillon,
J. Appl. Phys., 15, 324 (1944)].
/—гевея; 2—бутапрен N, Gl<-N; T- Cyiia S. /—бутил В-3 5— неопрен GN.
30±2 ккал/моль. Для обычных вулканизатов натурально! о каучу-
ка, использованных в этих первых опытах, было найдено, что
к' = 8,1 • 1012 ехр (— 30,4 ккал/RT)
(1,4)
Вычисленные на основании уравнений (1,4) и (1, 1) релакса-
ционные кривые при 100, ПО, 120 и 130 °C показаны на рис. V, 4
вместе с экспериментальными данными, отложенными в коорди-
натах Ig/(t)/f (0) и t. Химическая релаксация напряжения стано-
вится распространенным физическим методом измерения деструк-
ции химических связей в резине. Можно считать, что скорость
Химическая релаксация напряжения в резинах
229
этой реакции деструкции совершенно не зависит от сил, действую-
щих на пространственную сетку резины, или от величины дефор-
мации сетки (вплоть до очень больших удлинений).
Рис. V, 4. Влияние температуры на хими-
ческую релаксацию напряжения вулканиза-
та натурального каучука. Прямые вычис-
лены но уравнениям (I, 1) и (1, 4), точ-
ки—экспериментальные данные (А. V.
Tobolsky, I. В. Р г е t I у in а п,
J. Н. D i 1 I о п, J. Appl. Phys., 15, 324
(1944); R. D. Andre w s, R. В. M e s-
r о b i a n, A. V. Tobolsky, India
Rubber World, 112, May, 1945].
Литература
1. A. V. Tobolsky, I. В. P r e t t v m a n, J. H Dillon, J. Appl
1 Phys., 15, 380 (1944).
2 M. Mooney, W. E. Wo his te nho I me, D. S. Villars. J
Appl. Phys., 15. 324 (1944).
230
Гл. I'. Химическая релаксация напряжения
2. Деструкция и образование поперечных связей
Наблюдалось, что во время опытов по химической релакса-
ции напряжения в области температур 100 150 °C некоторые
вулканизаты, например из бутилкаучука, постепенно размягча-
ются, другие (вулканизаты пз дивннплстирольного каучука) по-
степенно затвердевают, а резины пз натурального каучука снача-
ла размягчаются, а затем затвердевают. Был сделан вывод, что
при таких высоких температурах одновременно протекают два
процесса—деструкция и структурирование и что скорости как
деструкции, так и структурирования совершенно не зависят от
напряжения или удлинения образца 11]. Если резина находится
при постоянном удлинении, ясно, что реакция деструкции вызы
вает спад напряжения. Предполагалось, что в первом приближе-
нии реакция структурирования не влияет на напряжение в об-
разце, находящемся при постоянной деформации, до тех пор, пока
структурирование не разовьется так сильно, что приведет к
уменьшению объема (усадке). Другими словами, мы считаем, что
поперечные связи должны возникать таким образом, что образую-
щаяся сетка оказывается в ненапряженном состоянии независи-
мо от того, был образец растянут или пет. С другой стороны, из-
менение длины образца после реакции является наглядным до-
казательством того, что имело место образование поперечных свя-
зей. На основе этих соображений был разработан метод раздель-
ного определения эффектов деструкции и структурирования
11, 2] по измерениям непрерывной релаксации напряжения и из-
мерениям периодического напряжения. Техника непрерывного из-
мерения релаксации напряжения является обычным методом, при
котором образец выдерживается при заданном удлинении, напри-
мер при 50%, и напряжение измеряется как функция времени.
В соответствии с нашими предположениями этот метод дает воз-
можность определят!, только результат деструкции, по крайней
мере в первом приближении. Образцы, предназначенные для из-
мерений периодического прерывистого напряжения, выдерживаются
в ненапряженном состоянии при заданной постоянной температу-
ре. Через значительные промежутки времени резина быстро растя-
гивается до постоянного удлинения (например, до 50%), измеряет-
ся равновесное напряжение и образец немедленно возвращается к
исходной длине. Иными словами, производится периодическое изме-
рение модуля упругости пространственной сетки в условиях, когда
время измерения и действия напряжения очень мало по сравнению
с промежутками времени между измерениями. Поэтому можно
считать, что почти все акты деструкции и структурирования со-
вершаются в условиях ненапряженного состояния. Измерения
прерывистых напряжений отражают суммарный результат дест-
2. Деструкция и образование поперечных связей
231
рукции и структурирования. Иногда для краткости измерения не-
прерывной релаксации напряжения и измерения прерывистого
напряжения обозначают общим термином—измерения «непрерыв-
ной и прерывной релаксации напряжений:'.
Предположим, что начальная структура сетки характеризует-
ся наличием с(0) молей эффективных четырехфункциональных по-
перечных связей (узлов) в 1 си3. Количество молей добавочных по-
перечных связей в 1 си3, образовавшихся позже ко времени t, обо-
значим с(/), а количество молей, распавшихся в 1 см3, через q(t).
Если образец в нерастянутом состоянии имеет длину Lo, при измере-
нии напряжения длину L, а прерывистое напряжение в момент
времени t равно ft (/), то
Л(0 = 12с(0) 4- 2с(0—
_Ма А|
бо ) ' l j
(2, п
С другой стороны, напряжение f{f), смеренное в опытах по не-
прерывной релаксации, в которых образец в продолжение всего
опыта растянут до длины L, равно:
f (0 3-= [2с (0) — q (/)] RT [( ~) ’ - |
(2, 2)
В обоих случаях начальное напряжение /(0) равно:
f (0) = 2с (0) RT
(2, 3)
Если имеет место только деструкция, кривые непрерывной и
прерывной релаксации напряжения должны совпадать. Во всех
первоначальных экспериментах было найдено, что напряже-
ние в каждый момент времени при прерывном измерении
больше, чем в опытах по непрерывной релаксации. Эта раз-
ница является результатом наложения реакции структури-
рования. Структурирование может быть вызвано двумя причи-
нами: во-первых, продолжающимся сшиванием за счет действия
в.^И'.каннзующих агентов, полностью не израсходованных в про-
цессе вулканизации; во-1'Л‘, ''"чх, структурирование является
частью тех же самых окислительных цепных реакций, которые
вызывают деструкцию. Недавно было показано, как наложение
этих двух процессов при структурировании может быть разде-
лено |3]. Можно получить пространственную сетку в натуральном
каучуке, полибутадиене и дру1 их полимерах действием на них иони-
зирующих излучений. Большое преимущество такого метода заклю-
чается в том, что в полимер не вводится никаких загрязнений, т. е.
веществ, которые могут действовать как структурирующие
агенты после окончания процесса вулканизации. На рис. V, 5а
232
/л. V. Химическая релаксация напряжения
представлены данные по непрерывной и прерывной- релакса-
ции напряжения при 130 °C в натуральном каучуке, структуриро-
ванном с помощью облучения электронами с энергией 1 Мэв
Почти полное совпадение этих двух кривых указывает на то, что
в данном случае деструкция практически не сопровождается реак-
Рис. V, 5. Кривые непрерывной релаксации напряжения (/)
и прерывной релаксации напряжения (2): а—для радиаци-
онного вулканизата натурального каучука при 130 °C
[А. V. Tobolsky, A. Mercurio, J. Appl.
Polymer Sci. (в печати)]; б—для серного вулканизата
натурального каучука при 130 °C [А. V. Т с b о 1 s к у,
R D. Andrews, .1. Chem. Phys., 11. 145 (1945)]:
пней образования поперечных связей, т. е. в условиях опыта мо-
лекулярный кислород не вызывает образования поперечных свя-
зен в натуральном каучуке. Для сравнения на рис. V, 56 по-
казаны кривые непрерывной и прерывной релаксации напряже-
ния при 130 °C для натурального каучука, вулканизованного
серой в присутствии ускорителен. Здесь разница между периоди-
ческими измерениями напряжения и кривой непрерывной релак-
сации напряжения должна быть приписана продолжающейся
вулканизации под влиянием химических вулканизующих аген-
тов. С другой стороны, на рис. V, 6 показаны результаты непре-
рывной и прерывной релаксации напряжения в эмульсионном
полнбутаднене и цнс-1,4-полибутадиене, вулканизованных с по-
2. Деструкция и образование поперечных связей
233
мощью электронов с энергией 1 Мэв. Ясно, что в этих случаях не-
сомненно происходит сильное структурирование в результате
окисления. Эмульсионный полибутадиен содержит 20% 1,2-струк
Рис. V, 6. Кривые непрерывной релаксации напряжения (/)
и прерывной релаксации напряжения (2) при 130 °C для
радиационных вулканизатов: о—эмульсионный полнбу-
гадиен; б—цис-1,4-полибутаднен [А. V. Tobol-
i к у, А. М е г с и г i о, J. Арр]. Polvmer Sci., 2, 186
(1959)1:
гур, 60% транс-1,4 структур и 20% цнс-1,4 структур 14]. Так
как окислительное структурирование происходит в обоих
либутадиенах, то эта реакция не
может быть
наличием 1,2-структуры.
ПО-
связана только с
Рис. V, 7. Кривые непрерывной релак-
сации напряжения (/) и прерывной ре-
лаксации напряжения (2) для серного
вулканизата GR-S при 130 °C [А. V.
Т о b о 1 s к у, I. В. Р г е t t у m а п.
.1. Н. D i 1 1 о п, J. Appi. Phvs..
15 . 309 (1944)]-
234
Гл. V. Химическая релаксация напряжения
На рис. V, 7 приведен пример непрерывно возрастающей
(с самого начала) кривой, полученной при прерывном измерении
напряжения при 130 °C для GR-S, вулканизованного серой
Литература
1. А. V. Т о b о 1 s к у, 1. В. Pretty m а и. J. Н. Ditto в. J. Appl.
Phys., 15, 309 (1944).
2. А. V. Tobolsky, R. D. Andrew s, J. Chem. Phys., 11, 125 (1945).
3. A. V. Tobolsky, A Mercurio, J. Appt.^Polymer Sci. (в печати).
4. J. L. В i n d e r, Anal. Chem., 26, 1877 (1954).
3. Определение скорости деструкции
по релаксации напряжения
При наличии в резине пространственной сетки с п(0) молями
отрезков цепей между узлами в 1 см3 при температуре Т соотно-
шение между напряжением (сила на единицу поверхности истин-
ного поперечного сечения) и относительным растяжением а =
=LIL0 выражается следующим образом:
f = П (0) А7’|«2 — -L
(3,1)
Величина п(0) может быть определена из измерений /, а и Т при
соответствующих условиях.
Предположим, что пространственная сетка резины находится
при заданном относительном растяжении а. Пусть ко времени t
происходит разрыв q(t) молей в 1 см3 и остается n(q) молей отрез-
ков цепей первоначальной сетки в 1 СЛ13, которые поддерживают
напряжение в образце. Мы будем идентифицировать n(rj) [можно
также ввести обозначение /г(()1 с такой сеткой, которая совершенно
не подвергалась разрыву за время t. Далее, n(q) может быть из-
мерено непосредственно при непрерывной релаксации из следую-
щего соотношения:
/(/) _ n(q) _ n(t) „
f(0) п(0) — п(0)
1де /t0)—напряжение в момент времени, равный нулю, и zi(0)
концентрация цепей сетки при (=0, т. е. перед деструкцией.
Теперь необходимо установить соотношение между n(q) и q так.
чтобы q можно было получить из экспериментально определяемых
значений [(t)jt(O). Рассмотрим два случая.
3. Определение скорости деструкции по релаксации напряжения 235
Случай А. Беспорядочный разрыв связей
пространственной сетки
Прежде всего следует предположить, что разрыв цепей
происходит беспорядочно по всей пространственной сетке и что
имеется большое количество уязвимых мест для разрыва вдоль
каждой цепи сетки. Это значит, что цепь в сетке, однажды разор-
ванная, может быть с равной вероятностью разорвана снова. Как
уже указывалось, n(q)—количество цепей в 1 см3 неразорванных
ко времени t. В момент t вероятность того, что новый разрыв про-
изойдет среди n(q) цепей равна n(q)ln(QY Поэтому соотношение
между n(q) и q будет иметь вид [1]
(1ч (0 = » К) /о о.
dq • п(0) ' ’ ’
После интегрирования с учетом начального условия n(q) =и(0)
при <7=0 получается:
Подставляя уравнение (3, 2), получаем:
q (t) = -- п (0) In (3, 5)
Следовательно, q(t) может быть определено прямо из данных по
релаксации напряжения п величины равновесного напряжения
при t =0 [для вычисления п(0)1.
Для ряда идентичных резин, отличающихся только густотой
пространственной сетки, можно ожидать, что q(t) будет одинако-
вым. Однако если уравнение (3, 5) правильно, то должно
быть в этом случае различным. Это является наиболее правиль-
ным критерием того, имеет ли место в каждом данном случае бес-
порядочный разрыв сетки или разрыв происходит по попереч-
ным связям. Если скорость разрыва постоянна, т. е. если q—q'ot,
тогда, используя уравнение (3, б), получаем:
f Wf (0) = ехр (— qlfin (0)) (3, 6)
что, конечно, выражает собой спад напряжения по Максвеллу.
Кинетика реакции деструкции и, следовательно, форма функ-
циональной зависимости <;(/) будут всецело зависеть от химизма
реакции разрыва.
В некоторых случаях может наблюдаться постоянная скорость
разрыва, однако кинетика деструкции может быть значительно
более сложной, особенно если деструкция является пепион реак-
236
Гл. к. Химическая релаксация напряжения
цией. Поэтому спад напряжения по Максвеллу не является ни
необходимым, ни достаточным признаком беспорядочной деструк-
ции звеньев цепей пространственной сетки.
Случай Б. Распад по местам поперечных связей
Предположим, что распад происходит только по месту по-
перечных связей или по прилегающим к ним участкам. Для удоб-
ства будем считать, что распад поперечной связи или соседнего
с ней участка приводит к отделению одной цепи и что каждая цепь
имеет только одно уязвимое для разрыва место
Соотношение между n(q) и q следующее:
n(<7) = n(0) — q
или при подстановке уравнения (3, 2) получается:
q(t) = n(O) [1-|g-]
_Ж_ . ч(Б
НО)-"1 «(0)
(3, 7)
(3, 8)
(3, 9)
В этом случае мы ожидаем, что общее число разрывов в I слР.
происходящее ко времени I, т. е. q(t), будет пропорционально
п(0) для ряда идентичных резин, отличающихся только количе-
ством поперечных связей. Это следует из того факта, что, как
предполагается, места разрыва в данном случае ограничены толь-
ко поперечными связями. Поэтому можно считать, что q(t) должно
быть пропорционально и(0) для ряда таких резни, но что
/(/)//(0) будет независимым от п(0), как можно видеть из урав-
нения (3, 9) 121:
Простейшие кинетические соотношения, которые можно ожи-
дать в случае Б, имеют следующий вид:
-5 й-=«"(«>
н (q) = п (0) ехр ( - k't)
-|g-==exp(— k't)
(3, 10)
(3, П)
(3, 12)
Поэтому разрыв в местах поперечных связей может вести к спад\
напряжения по Л^аксвеллу.
Однако реакция деструкции не должна во всех случаях под-
чиняться уравнению (3, 10), так как кинетика разрыва связей не
4. Распад поперечных связей а цепей сетки
237
обязательно должна протекать по уравнению реакций первого
порядка. Разрыв по узлам пространственной сетки не является
ин необходимым, ни достаточным условием для выполнения закона
Максвелла. Таким образом, по форме кривых релаксации напря-
жения нельзя заключить, происходит ли случайный разрыв про-
странственной сетки на участках, заключенных между попереч-
ными связями, или разрываются сами поперечные связи
Литература
1 А. V. Tobolsky, D. J. Metz, R. В. М е s г о b i a n, J Am Chem
Soc., 72, 1942 (1950).
2. A. V. Tobolsky, J. Appl. Phys., 27. 673 (1956).
4. Распад поперечных связей и распад цепей сетки
В ранних исследованиях химической релаксации напряжения
было найдено, что вулканизаты натурального каучука с серой и
с динитрозобензолом имеют очень близкие скорости деструкции
С другой стороны, ряд резин с разной структурой цепи, но вулка-
низованных одинаковым образом, имеют совершенно различные
релаксационные кривые. Из этого можно было заключить, что
деструкции подвергаются цепи, а не поперечные связи [1].
Это заключение в дальнейшем было подвергнуто сомнению на
основании анализа формы релаксационных кривых. Утвержда-
лось, что в вулканизатах натурального каучука, полученных с
помощью серы или перекисей, деструкция происходит по месту
поперечных связей 121. Для устранения сомнений в правильности
ранее высказанных соображений был проведен ряд дополнитель-
ных исследований.
А. Окислительная релаксация напряжения радиационного
вулканизата натурального каучука [<?]
Натуральный каучук в виде тонких пластинок, полученных из
латекса, вулканизовался при помощи электронов с энергией
I Мэв от источника Ван де Граафа. Такие образцы очень удобны
для исследования химической релаксации, так как они содержат
минимум веществ, которые могут играть роль как инициаторов, так
и ингибиторов окисления. Применение «химической» вулкани-
зации с этой точки зрения значительно менее удобно. Были при-
готовлены образцы с разной плотностью поперечных связей, т. е
имеющие различные значения и(0). Величина л(0) определялась
238
Гл. Iх. Химическая релаксация напряжения
измерением равновесного напряжения при 100% удлинения с
использованием уравнения состояния резины:
Л>1
L j
/(0) =» (0)/?Т
-Г
La )
(4, 1)
Было найдено, что на каждые 100 эв поглощенной энергии обра-
зуется 1,9 поперечных связей.
Рис. V, 8. Кривые релаксации напряжения при 130 °C
для радиационного вулканизата натурального каучу-
ка с различной густотой пространственной сетки
[А. V. Т о b о I s й у, A. Mercurio, Н. Yu,
J. Appl. Polymer Sci. (готовится к печати)].
Время облучения пучком электронов с энергией
1 Мэе прн 1 мка:
1—2 мин; 2—5 мин; 3—10 мин
Результаты последующих опытов по релаксации напряжения
при 130 °C приведены на рис. V, 8. Эти же данные в виде зависи-
мости q(t) от времени показаны на рис. V, 9 \q(t) получено из
уравнения (3, 5)].
Результаты явно воспроизводят случай беспорядочной деструк-
ции цепей пространственной сетки, рассмотренной в раз-
деле 3 (случай А), а не разрыв поперечных связей. Ясно видно, что
q(t) не зависит от п(0), а кривые от t зависят от н(0), как
это предсказывалось для случая беспорядочного разрыва цепей
4. Распад поперечных связей и цепей сетки 231-
простраиственной сетки [1]. Такой же результат был получен для
натурального каучука, вулканизованного с помощью азодикарб-
оксилата [51.
sky, A. Mercurio, Н. Yu (в печати)].
Время облучения электронами 1 Alss при 1 мка-
1-2 мин; 2—5 мин; 3—10 мин
Б. Скорость деструкции невулканизованногс
натурального каучука [3]
Деструкция невулканнзованного натурального каучука при
130 °C на воздухе измерялась по изменению среднечислового мо-
лекулярного веса со временем. Применялись образцы в виде тон-
ких пленок; молекулярный вес как функция времени определял
ся на отрезаемых от них маленьких кусочках по характеристи-
ческой вязкости. Количество Q(/) молей разрывов на 1 слг3 каучука
определялось по уравнению
Q(0 = d[=J—
_____।
(0)
(4, 2i
В уравнении (4, 2) d— плотность каучука, Mn(t)—среднечисловои
молекулярный вес ко времени t и Л4п(0)—начальный среднечисло
вой молекулярный вес.
240 Г л. V. Химическая релаксация напряжения
Сравнение q(f), полученного из релаксации напряжения в ра
диационном вулканизате натурального каучука при 130 СС,
с Q(t), полученным из данных по деструкции невулканизованиого
натурального каучука прн той же температуре, показано на рис
V, 9. Из рисунка видно, что имеется хорошее количественное со
гласие между этими данными. Это должно означать, что деструк-
ция, измеренная с помощью релаксации .напряжения, действи-
тельно является результатом случайного разрыва полиизопре-
новых цепей, а це только поперечных связей (по крайней мере в
случае пространственной сетки радиационного вулканизата).
В. Спад ‘.напряжения в серных и бессерных вулканизатах пяти
каучуков различного строения [6]
Пять углеводородных каучуков, сильно отличающихся построе-
нию молекул, вулканизовались с помощью элементарной серы и се-
русодержащих ускорителей, а также без серы (излучением высо-
кой энергии, перекисью дикумила и н-хинондиоксимомс перекисью
свинца). Во всех невулканпзованных каучуках имелось по край-
ней мере один или два молярных процента двойных С-С-связей,
так что места, в которых могли образовываться поперечные свя-
зи, до некоторой степени подобны. Выбранные два класса вул-
канизующих агентов несомненно образуют совершенно различные
типы поперечных связей: можно предполагать, что при серной
вулканизации образуются тиоэфирные и другие содержащие серу
поперечные связи; излучение высокой энергии и перекись дику-
мнла, очевидно, вызывают образование углерод-углеродных по-
перечных связей. Строение молекул использованных каучуков
(натуральный каучук, бутадиен-стирольный (GR-S), бутил-
каучук, полиэфирный каучук и модифицированный каучук] по-
казано в табл. V, 1.
Вулканизаты были приготовлены без антиоксидантов и перед
изучением релаксации напряжения подвергались экстракции
растворителем. Концентрация поперечных связей как в серных,
так и в бессерных вулканизатах одних и тех же каучуков была
одинаковой, о чем свидетельствовали близкие значения, получен-
ные для п(0). Кривые релаксации напряжения откладывались в
виде зависимости lg/(/)//(0) от t, которая для спада напряжения
по Максвеллу должна представляться в виде прямой линии. Ре-
лаксационные кривые при 130 °C в этой форме, представлены на
рис. V, 10.
В табл. V, 2 приведены времена химической релаксации для
10 исследованных образцов. Время химической релаксации —
это время, которое требуется для достижения значения /(/)//(0) =
= 1/е =0,368 в процессе химической релаксации.
16—833
Строение некоторых каучуков
Таблица V, I
Материал Структура
Натураль- ный каучук бутадиен- стироль- ный кау- чук (GK-S) Бутил- каучук 11оли- эфирный каучук Модифи- цирован - ный каучук сн3 сн3 сн3 сн3 —СН.—к=^сн—СН.—сн2—С=СН—СН2—СН2—С=СН—СН.—СН.—i=CH—СН.- цис- J, 4-полиизопрев —CH- CH.-CH. СН= СН—СН.—СН.—СН=СН—СН.-сн.—СН=СН—СН.—СН--СН. 1 ' 1 С„Н,, С„Н, нерегулярный сополимер 75% бутадиена и 25% стирола СН3 CHS CHS СН3 СН., Ill II -СН2—С—СН2—С—СН2—С—СН.—С=СН—СН.—сн.,—с—сн,—с- 1 1 1'1 1 1 снэ сн. сн., сн3 сн3 сн„ сополимер полиизобутнлена и 2°изопрена О О о о II II II II -О—(СН2)Х—О—С—(СН.)Х—С—О—(СН2)Л—о- С-(СН2)Х-СН=СН—(СН2)Х— С—О—(СН.)Х —о— алифатический сложный полиэфир, содержащий несколько процентов ненасыщенных звеньев -СН2—СН—СН2—СН,—СН2—СН—СН.—СН.—СН»—СН=СН—СН.- 1 1 SCHs SCH3 полибутадиен, в котором 97% двойных связей насыщены метилмеркаптаном
* Вулканизованные каучуки, использованные в этой работе [6], перед испытанием тщательно экстрагировались для удаления анти-
оксиданта. вопреки утверждению Дана и др. [J. R. Dunn. J. Scanlan. W. F. Watson, Trans. Farad. Soc.. 55, 667 (1959)].
i. Распад поперечных связей и цепей сетки
№
242
Гл. V. Химическая релаксация напряжения
Таблица V, 2
Времена химической релаксации при 130 "С для различных серных
н бессерных вулканизатов
Материал Хх' 4
серная вулканизация бессерная вулканизация
11атуралы1ын каучук 0,7 2,8
Бутадиен-стирольный каучук (GR-S) 1,2 1,9
Бутилкаучук . 9 10
Полиэфирный каучук 51 300
Модифицированный каучук 50 >1000
Интересно отметить, что для чисто углеводородных каучуков
(натуральный каучук, бутадиен-стирольный, бутилкаучук) раз-
Время, v
Рис. V, 10. Зависимость Inlf(t)/f(O)] от t при 130 °C для серных (a) i бессер-
ных (б) вулканизатов пяти полимеров с разным строением цепи
V V. То bol sky, A. Merc urio, J. Polymer Sci., 36, 467 (1959)]-
модифицированный каучук: 2—полиэфирный: '<-бупгчкаучук; 4—GR-S: '—НК
4. Распад поперечных связей и цепей сетки
243
личие во временах релаксации между серными и бессерными
вулканизатами невелико, максимум в 4 раза.
Был испытан широкий набор таких вулканизатов, и оказалось,
что найденное различие во временах релаксации является наи-
большим. Добавление небольших количеств антиоксиданта к нату-
ральному пли бутадиен-стирольному каучуку действует так, что
разность в тл. между серными и бессерными вулканизатами почти
исчезает [7]. Интересно отметить, что время химической релакса-
ции вулканизата бутилкаучука практически не зависело от спр-
соба вулканизации. С другой стороны, как для серных, так и
для бессерных вулканизатов каучуков различного строения имеет-
ся очень заметная разница во временах химической релаксации.
Для серных вулканизатов время химической релаксации изменяет-
ся от 0,7 до 50 ч, для бессерных вулканизатов —от 2,8 до более
чем 1000 ч. В случае серных вулканизатов оказывается, что наи-
большее время химической релаксации (50 ч) наблюдается у по-
лиэфирного каучука и у модифицированного каучука.
Ясно, что у натурального, дивннилстиролыюго и у бутилкау-
чука деструкция должна протекать вдоль углеводородных це-
пей. В противном случае наблюдалось бы намного большее
различие между серными и бессерными вулканизатами.
Для полиэфирного и модифицированного каучуков, молекулы
которых деструктору ются с большим трудом, местами разрыва
могут быть поперечные связи, по крайней мере в таких вулканиза-
тах, как серные.
Исследование химической релаксации напряжения и скорости
поглощения кислорода у широкого набора резин показало, что
электропоакцепторные группы в полимерной цепи как будто со-
общают стойкость к окислительной деструкции, в то время
как электронодонорные группы делают полимер более чувстви-
тельным к окислению [8, 91. Исследования химической релакса-
ции напряжения могут быть использованы для оценки эффектив-
ности химических антиоксидантов [101.
Литература
1 . А. V. Tobolsky, I. В. Prettyman. J Н. Dillon,
J. Appl. Phys., 15, 324 (1944).
2 J. P. Berry, W. F. Watson, .I. Polymer Sci., 18, 201 (1955);
J. Polymer Sci., 25, 493, 494, 497 (1957).
3 A. V. Tobolsky, A. Mercurio, H. Y u (manuscript in prepa-
ration).
4 A. V. Tobolsky, D. .1. Metz. R. В. M e s г о b i a n, J. Am
Chem. Soc., 72, 1942 (1950).
5 D. J. Metz, R. B. Mesrobian, J. Polymer Sci., 11, 83 (1953).
6 A. V. Tobolsk v. A. M e r c u r i o, .1. Polymer Sci.. 36. 467
(1959).
16*
244
Гл. У. Химическая релаксация напряжения
7. A. Mercurio, Ph D. thesis, Princeton University, 1959.
8. R. B. Mesro bia n, A. V. Tobolskv, J. Polymer Sci., 2, 463
(1947).
9. A. V. Tobolsky, Disc. Farad. Soc., 2, 384 (1948).
10. R. B. Mesrobian, A. V. Tobolskv. Ind Eng Chem . 41. 1496
(1949).
5. Химическая остаточная деформация
В образцах резни, находящихся при постоянном рас-
тяжении в температурных условиях, при которых проявляется
их хемореологпческое поведение, после снятия напряжения наблю-
дается остаточная (необратимая) деформация. Остаточная дефор-
мация возникает по двум причинам: во-первых, исходная про-
странственная структура постепенно разрушается при деструк-
ции таким образом, что упругие силы, стремящиеся возвра-
тить образец к исходной длине, постепенно уменьшаются; во-
вторых, с помощью возникающих поперечных связей образуется
новая структурная сетка, фиксирующая деформированное состоя-
ние образца и поэтому сопротивляющаяся всякому изменению его
новой формы. Когда образец освобождается от растягивающего
напряжения, он принимает длину, при которой эти две силы
уравновешиваются. Эти две противоположно направленные силы
могут быть связаны с величинами U и X, получаемыми из кри-
вых непрерывной и прерывной релаксации, как показано на
рис. V, 11 и поясняется ниже.
Обозначим длину нерастяпутого и растянутого образца ре-
зины через Lo и Lt соответственно и остаточную длину после снятия
напряжения через Ls. После того как образец резины освобожден
и принял новую длину Cs, можно написать для двух состояний
цепей в пространственной сетке резины уравнения:
(5,1,
г, -F-I (5,2|
В этих уравнениях /и—напряжение (на единицу истинного
поперечного сечения), обусловленное пепямп, находящимися в
равновесном ненапряженном состоянии при длине образца to, и
по—число неразорванных цепей пространственной сетки в 1 см3.
находящейся в равновесии в нерастянутом состоянии при длине Lv
Соответственно [х и пх относятся к сетке, цепи которой находятся
в равновесии (не напряжены) при длинеLx. Далее, условием равнове-
5. Химическая остаточная деформация
24!
сия в образце с остаточной длиной Ls является равенство сок-
ращающей силы, развиваемой цепями типа «О», и растягивающей
силы, развиваемой цепями тина «х»:
/о = fx (5> 3)
Из уравнений (5, 1) и (5, 3) после упрощения получаем-
(5, 41
Остаточная деформация (которую мы будем обозначать ОД) обыч-
но выражается как процент деформации, сохраняемой образцом,
т. е.:
ОД = ф—100 = ' 100 (5, 5)
Lv Lq Lx/ Lq — I
Из уравнений (5, 4) и (5, 5) получаем:
О
' I'3
i
ОД = С3
(и0/лд) С2 4- i
(5.6)
где Сп Cs и Сэ просто выра-
Осповной идеей теории оста- lg г
точной деформации является л ,,,,,,
тп что t, hi -1НХ ГПА // п Рис- V’ 1L U 11 А’ опРелеляемые по теории
in, ни ‘'«л, 1дс xj н химической остаточной деформации
X получены из данных по [R. D. Andrews, А. V. Tobols-
непрерывной И прерывной к у. Е. Е. Hanson, J. Appl. Phys.,
релаксации напряжения к , 17, 352 0946)0
1 1 /—непрерывная; 2—прерывная релаксация,
моменту времени t, как пока-
зано на рис. V, 11. Это соотношение следует из сущности прерыв-
ной и непрерывной релаксации напряжения и используется в тео-
рии «двух сеток». Таким образом, уравнение для остаточной дефор-
мации принимает следующий вид:
ОД— С3| ((4//Л)с2+ 1 ” Ч
(5, 8)
1
246
Гл. Г. Химическая релаксация напряжения
Графики зависимости остаточной деформации от lg(t//X) для ряда
различных по величине удлинений показаны на рис. V, 12.
Необходимо сделать несколько замечаний относительно пред-
положения о том, что образование поперечных связей в растяну-
том образце происходит между цепями в ненапряженном состоя-
нии. Нельзя математически удовлетворительно описать отдельный
процесс поперечного связывания; эффект скорее следует рассмат-
ривать как статистический, а образование новых связей, ненапря
жснных при той степени удлинения, при которой они возникают,
считать только средним результатом.
Рис. V, 12. Зависимость химической остаточной деформации от U/Х для
различных удлинений [R. D. Andrews, А. V. Tobolsky,
Е. Е. Hanson, J. Appl. Phys., 17, 352 (1946)].
Экспериментальные данные, подтверждающие теорию остаточ-
ной деформации, показаны на рис. V, 13—V, 15. Было получено
очень хорошее соответствие между теорией и экспериментом
Подробности эксперимента полностью описаны в литературе [1].
Для достижения истинного значения остаточной деформации,
после того как напряжение снято с образца, необходимо выждать
должное время. В тех резинах, где разрыв молекул вызывается
диффузией молекулярного кислорода, должны использоваться
достаточно тонкие пленки, чтобы реакция протекала равномерно
в массе всего материала, а не ограничивалась поверхностным
слоем. Следует подчеркнуть, что данная теория применима к
Рис. V, 13. Кривые непрерывной (7) и прерывной (2) хи-
мической релаксации и сравнение расчетной кривой (3)
остаточной деформации с экспериментальными данными
(точки) для серного вулканизата натурального каучу-
ка (130 °C, удлинение 50%) [R. D. Andrews,
А. V. Tobolsky, Е. Е. Hanson, J. Appl.
Phys., 17, 352 (1946)].
Рис. V. 14. Кривые непрерывной (1) и пре-
рывной (2) химической релаксации и сравне-
ние расчетной кривой (3) остаточной дефор-
мации с экспериментальными данными (точ-
ки) для серного вулканизата GR-S(130cC,
удлинение 50%) [R- D- Andrews,
А. V. Tobolsky, Е. Е. Hanson,
J. Appl. Phys., 17. 352 (1946)).
248
Гл. V. Химическая релаксация напряжения
образцам, изучаемым при температурах достаточно высоких для
того, чтобы упруговязкое поведение в основном определялось хе-
мореологическпм процессом. Многие испытания по определению
остаточной деформации, которые используются в промышленной
практике и часто проводятся при 70 °C, не удовлетворяют этому
требованию. С другой стороны, многие из этих экспериментов,
проводимые при 100 °C или выше, должны отражать влияние реак-
Рис. V, 15. Кривые непрерывной (7) н прерывной (2)
химической релаксации и сравнение расчетной кривой (3)
с экспериментальными данными (точки) для серного вул-
канизата бутилкаучука (130 °C, деформация 50%)
1R. D. А п d г е w s, А. V. Т о b о 1 s к у, Е. Е. Н а п-
s о и, J. Appl. Phys., 17, 352 (1946)].
цин поперечного связывания вследствие продолжающегося дей-
ствия вулканизующих агентов. Образцы, применяемые в промыш-
ленной практике, однако, обычно слишком толсты, чтобы в них
происходила равномерная реакция деструкции.
Литература
1 R. D. Andrews, А. V. Tobolsky, Е. Е Н a n s о n. J. Лрр1-
Phys., 17, 352 (1946).
6. Химическая ползучесть при постоянной нагрузке [1]
Если образец вулканизованной резины подвергнуть действию
постоянной нагрузки в температурной области хемореологпческо-
го поведения, то вначале будет иметь место эластическая деформа-
ция, а затем образец будет продолжать удлиняться вследствие
6. Химическая ползучесть при постоянной нагрузке
249
разрушения пространственной сетки. Это явление можно назвать
химической ползучестью.
Простейшим для рассмотрения случаем является такой, когда
происходят только разрывы пространственной сетки. Удобно
начать анализ с уравнения эластичности резины, выраженного
в форме:
F=.n(O)RT[-^- (6,1)
где F напряжение (рассчитанное на начальное поперечное сече-
ние), которое в условиях опыта остается все время постоянным;
п(0)—количество молей цепей пространственной сетки
в 1 с.«8 в начальный момент времени; Lo—длина нерастянутого
образца, £.(0)—длина образца сразу после приложения напряжения
F. Для момента времени t уравнение может быть написано в сле-
дующем виде:
F = п (t) RT
I М)
(6.2)
где L(t)—длина образца в момент t и /1(7)—число цепей в
I сл18, не разорванных ко времени /. Из уравнений (6, 1) и (6, 2)
получаем;
п (/) _ L (0)//.„ - [Lo/L (О)]2
«(0) ~ L — [Д/Z. (I)]2
(6, 3)
Уравнение (6, 3) выведено в предположении, что имеет место
только разрыв цепей без их поперечного связывания, т. е. для
крайне редкого случая. Назовем правую часть уравнения (6, 3),
являющуюся экспериментально измеряемой функцией, функцией
химической ползучести (ФХП):
ФХП — -£ (6, 4)
В отсутствие процесса поперечного связывания цепей сравнение
уравнений (6, 3) и (3, 2) показывает, что функция химической
ползучести и функция химической релаксации напряжения
/(/)//(0) должны быть идентичными. Этого можно ожидать даже
и при наличии процесса поперечного связывания, если преобла-
дает реакция деструкции (как это показано на рис. V, 16). Если
преобладает структурирование, то ФХП уменьшается намного
медленнее, чем функция химической релаксации напряжения
(как показано на рис. V, 17). Изменяющуюся вследствие обмен-
250
7. Механизм окислительной деструкции каучука
пых химических реакций пространственную сетку можно рас-
сматривать как особый случай конкуренции деструкции и струк-
турирования. При этом скорости процессов деструкции и структу-
рно. V, 16. Функция химической ползучести при 120%.
для серного вулканизата натурального каучука в срав-
нении с кривой релаксации напряжения при 120 °C
[А. V. Tobolsk у, R. D. A nitre w s, J. Chem.
Phys., 11, 125 (1945)]. Кривая описывает релаксацию
напряжения; точки соответствуют ФХП при начальном
удлинении:
Z—10%; 2—20%; 3—10%; Z—100%
время. v
Рис. V, 17. Функция химической ползучести при 120 °C
для серного вулканизата GR-S в сравнении с кривой
релаксации напряжения при 120 °C [А. V. Tobol-
s к у, R. D. Andre w s, .1. Chem. Phys.. 11, 125
(1945)]. Кривая описывает релаксацию напряжения;
точки соответствуют ФХП при начальном удлинении
15% и 30%.
7. Механизм окислительной деструкции каучука
251
рирования должны быть точно равны. Мы уже рассматривали
ползучесть таких пространственных сеток, в которых происходят
обменные реакции при постоянной нагрузке, в разделе 14 гла-
вы IV*.
Литература
1. Л V Т о b о 1 s к у, R D. Andrews, J. Chem Phys., 13, 125 (1945).
7. Механизм окислительной деструкции натурального каучука
Окисление натурального каучука молекулярным кислородом
является радикальной цепной реакцией 111, протекающей по
механизму, подобному тому, который предполагается для объяс-
нения окисления большинства углеводородов 12]. В случае, когда
имеется внешний инициатор (источник свободных радикалов), при
* Следует заметить, что развитые Тобольским с сотрудниками пред-
> сзвлеиия о химической релаксации напряжения, химической ползучести
и т. и. явлениях ограничены рассмотрением изменений строения напряжен-
ных пространственно-структурированных полимеров, возникающих вследствие
развития окислительных, обменных и других химических реакций. При этом
Тобольский особо подчеркивает независимость этих реакций, ведущих к
разрыву и образованию цепей в сетке, от величин деформации или напря-
жения, т. е. от механических воздействий (см., например, раздел 13 главы
IV и раздел 1 главы V), и рассматривает только влияние независимо про-
текающих химических реакций на состояние напряженного полимера.
Однако хорошо также известно обнаруженное советскими учеными
явление химического течения [В. А. Каргин, Т. II. Со голова, ДАН
СССР, 108, 662 (1956); «Проблемы физической химии», вып. 1, Госхнмиздат
1958, стр. 18], при высоких механических напряжениях развивающееся
вследствие чисто механических разрывов напряженных цепей макромоле-
кулярной сетки и сопровождающееся сложной системой цепных реакций
деструкции и структурирования полимера (инициированных свободными
макрорадикалами, возникшими при таких механических разрывах химиче-
ских связей). у.^1
Хорошо известно также влияние механических напряжений на скорость
окисления резин и другие химические реакции в полимерах [(А. С. Кузь-
минский, М. Г. М а й з е л ь с, Н. Н. Лежнев. ЛАН СССР, 71, 2. 319
(1950); А. С. Кузьминский, Л. И. Л ю б ч а н с к а я, Сб. «Старение и
утомление каучуков и резин и повышение их стойкости», Госхнмиздат, 1955.
стр. 89; В. А. Каргин, Г. Л. Слонимский, Л. И. Голубей ков а
ЖФХ, 30, 2436 (1956)].
Подобного рода механо-химические явления, имеющие особенно боль-
шое значение при тяжелых механических воздействиях, например в про-
цессах переработки полимеров в изделия, встречаются, естественно, наряду
и в тесном взаимовлиянии с явлениями химической релаксации и ползуче-
сти (рассмотренными в IV н V главах), роль которых становится опреде-
ляющей лишь при достаточно малых напряжениях.—Пппм. ред.
252
Г л. к’. Химическая релаксация напряжения
среднем или высоком давлении кислорода, может быть предложен
следующий механизм:
(а) Инициатор > 2R'» )
(a') R'« + RII > R. -р R'H J
(б) R. 4- О2 --> RO2.
(в) Г<О2. 4- RH----» ROOH + Re
(г) RO2. + RO2.-----> Продукты 1 (J6
рекомбинации J
Инициирование
j Развитие цып:
В этих уравнениях RH представляет собой натуральный кау-
чук (—СН2—СН =C(CHS)CH2—)„. Радикалы, возникающие при
инициировании, взаимодействуют с каучуком по реак-
ции (а') с образованием R* либо вследствие отрыва водорода от
а-метпленовов группы, либо при действии на двойную связь. При
развитии цепи с участием радикалов RO.,« могут иметь место сле-
дующие внутримолекулярные побочные реакции 131:
ОО.
-СН2—С(СН3)=СН—СН— СН»—СГСН3)=СН-СН2 - --->
I----О—О------1
I I 02
--СН2—С(СН3)=СН— СН—СН2—С(СН3)— сн—сн2-—
о-—I
---СН2—С(СН3)=СН—СН—СН2—С(СН3)—сн-сн2---
io.
Весьма замечательным следствием окисления является разрыв
молекул полппзопрепа, на что мы обращали внимание в преды-
дущих разделах. Эта реакция деструкции совсем недавно была
объяснена существованием определенной стадии в кинетике про-
цесса, разбиравшейся выше. Последнее было четко показано при
исследовании кинетики деструкции натурального каучука в виде
разбавленного раствора в бензоле, куда непрерывно подавался
молекулярный кислород под давлением в 1 атм. Исследование
проводилось в температурном интервале 60—80 °C, причем ини-
циирование окислительных цепей осуществлялось с помощью
хорошо изученных катализаторов—перекиси бензоила (Бз2О2),
перекиси дптретпчного бутила (ПДТБ) и дипитрила азо-бис-пзомас-
ляной кислоты ( Азо! 1). Скорости образования радикалов в присут-
ствии этих катализаторов очень хороню известны из других экспе-
риментов. Окисление натурального каучука в отсутствие внешних
инициаторов (и сопровождающая его деструкция) сильно иниции-
руется гидроперекисями, образующимися в молекулах каучука. Это
7, Механизм окислительной деструкции каучука
253
(7, 1)
Q (О = d
ясно выраженная автокаталитическая реакция, и поэтому окис-
ление в отсутствие внешних инициаторов называется автоокцелени-
ем. В условиях рассматриваемых здесь кинетических исследова-
ний автоокисление и автоок целительна я деструкция пренебрежи-
мо малы по сравнению с окислением и сопутствующей деструкцией,
вызываемыми внешними инициаторами. Деструкция измерялась по
величине среднечислового молекулярного веса каучука как функ-
ции времени. Общее количество молен разрывов па 1 см3 каучука,
которое произошло ко времени t, обозначим через Q(/).
Его можно получить из уравнения (7, 1):
1 1
Мп (7) Мп (0)
где Mn(t)—среднечисловой молекулярный вес ко времени I,
/ИДО)—начальный среднечисловоп молекулярный вес и d—плот-
ность.
Скорость Ri (в молях па 1 см3 в 1 сек), с которой применяемый
инициатор дает радикалы, выражается уравнением (7, 2):
Я, = 2/?а|Иниц.] | (7
rff! 1ннц.],Л = &а[11нпц.| |
где ka—константа скорости самопроизвольного гомолитического
разложения индивидуального инициатора, протекающего по реак-
ции первого порядка.
Предполагается, что индуцированное разложение применяе-
мых инициаторов происходит в этих условиях слабо.
Интегрирование уравнения (7, 2) дает:
{/?*Jz = 2[IIhhu.]0[1 — ехр(-/?Д)| (7,3)
где —общее количество молей радикалов в 1 см3, которое
образуется ко времени t. Значение ka хорошо известно для этих
инициаторов при используемых температурах; следовательно,
можно вычислить {/?•},.
В табл. V, 3 приведены концентрации инициатора, концент-
рации каучука в бензоле и температуры, при которых проводились
исследования деструкции.
Таблица V. 3
Экспериментальные условия при исследовании деструкции
Инициатор [Ии>|ц.]о* Ю4 MOAblcM'd каучука RH конц. СА!3/ 4 Температура °C сек 1
Азо И 1,76 1,16 70 3,80-10~6
ПДТБ 19,5 1,39 70 1,44- И)8
Бз2О2 1,01 1,02 60 3,34- Ю 6
Бз.,О.,(1 ) 0,89 0,944 80 4,3- IO"5
Бз2О2(2) 3,02 1,00 80 4.3-1°“6
254
Гл. I'. Химическая релаксация напряжения
В табл. V, 4 приведены данные, полученные для {/?•)< из
уравнения (7, 3) и для Q(t) из уравнения (7, 1) при реальных
экспериментальных условиях. Видно, что для всех трех инициа-
торов порядок величины Q(t)l\R»}t близок к единице. Отсюда
ясно, что каждая окислительная цепь должна соответствовать
одному акту деструкции, a может служить мерой
эффективности инициатора. Вследствие большого различия ис-
пользованных скоростей инициирования разрыв молекул может
иметь место при развитии окислительной цепной реакции, только
если при всех наших условиях инициирования длина кинетиче-
ской цепи будет равна единице. Но это должно было бы означать,
что один акт деструкции приходится на одну поглощенную моле-
кулу кислорода при всех условиях инициирования. Однако было
показано, что при этих температурах при чисто термическом ини-
циировании на один акт разрыва приходится около 30 молекул
поглощенного кислорода, что исключает возможность деструкции
на стадии развития цепи.
Радикалы /?'•, создаваемые инициатором, либо могут реаги-
ровать с углеводородом каучука RH, давая начало окислительной
цепи, либо они могут присоединять кислород с образованием
R'O2*. Прямое взаимодействие R'« с RH не может вызвать де-
струкцию, так как было показано, что в отсутствие кислорода
деструкции почти не происходит. Подобным же образом очень
трудно предположить, чтоR'O,», реагируя с RH в начале окисли-
тельной цепи, будет вызывать разрыв RH. При наличии этого
должна была бы также наблюдаться деструкция па стадии раз-
вития цепи RO2*-4-RH-»ROOH |-R«, так как трудно себе пред-
ставить, что R'O2« для всех использованных инициаторов сущест-
венно отличается от ROa«.
Из этого можно заключить, что деструкция происходит на ста-
дии обрыва цепи, когда столкновение двух перекисных радикалов
приводит к двум актам деструкции.
Исследование деструкции проводилось также на радиацион-
ных вулканизатах натурального каучука, в которые вводилось
известное количество перекиси бензоила [51. Количество молей
разрывов в I см3 каучука q(t) определялось с помощью релаксации
напряжения с использованием уравнения (3, 5). Эти значения
сравнивались с вычисленными значениями {/?•}, по известной
величине для перекиси бензоила при 80 °C. Такое сравнение
показано на рис. V, 18, где экспериментальные точки для q(t)
лежат довольно близко к кривой для значений 0,7 {R»}t. Здесь
опять количество разрывов, деленное на общее количество ради-
калов, образуемых инициатором, дает величину порядка едини-
цы. Величина 0,7, вероятно, соответствует эффективности ини-
циатора в этих условиях.
7. Механизм окислительной деструкции каучука
255
Таблица V, 4
Количество образующихся радикалов и количество
разрывов цепей, измеренные при окислении
натурального каучука3__________
Время ч <5(0-ют {« Ъ ‘°* Q(O.'(R-b
АзоИ, 70 °C
2,08 0,601 0,99 0,61
3,93 1,38 1,47 0.94
5 35 1,86 1,82 1,02
7,73 2,48 2,22 1,12
15,60 3,14 3,10 1,01
29,93 3,72 3,45 1,08
ПДТБ, 70 °C
2,05 0,38 0,41 1,08
3,37 0,75 0,68 1,10
4,68 0,93 0,95 0,98
6, 17 1,20 1,25 0,96
8,42 1,59 1,70 0,94
10,45 1,90 2,12 0, 90
12,63 2,35 2,56 0,90
22,75 4.50 4,60 0,98
25,45 5,20 4,90 1,06
28,70 6,05 5,80 1,04
32,78 6,99 6,61 1,06
36,00 7,56 7,29 1,04
Бз2О2, 60 °C
2. 18 0, 184 0,54 0,34
4,13 0,382 1,00 0,38
6.70 0,631 1,57 0,40
9,25 0,920 2, 12 0,43
13,30 1,20 2,97 0,40
24,60 2,28 5,15 0,44
Бз2О2 (1), 80 °C
1,95 2,40 4,65 0,52
2,87 3,37 6,36 0,53
4.27 4,50 8.63 0,52
5,87 5,55 10,52 0,53
7,17 6,39 11,99 0,53
10,97 7,35 14,50 0,51
14,52 7,95 15.94 0.50
22. 92 8,66 17,31 0,50
Бз2О2 (2), 80 °C
1.80 0,481 1,48 0,33
3.87 1.15 2,72 0,42
6.76 1,71 3,93 0,44
9,65 2 12 4,70 0,45
а Л. V. Tobolsk у, Л. М е г с u г I о, J. \m. Chem.
Soc., Ы, 5535 (1959).
256
Гл. к’. Химическая релаксация напряжения
В отсутствие более эффективного источника радикалов таким
источником для окисления, а следовательно, н для деструкции
служит гидроперекись каучука ROOH. Этот источник иницииро-
вания особенно важен при высоких температурах и в действи-
тельности является причиной инициирования окисления при
исследованиях релаксации напряжения, результаты которых
приведены на рис. V, 8 и V, 9. Кинетика автоокислительных
Рис. V, 18. Зависимость q(t) от t и 0,7 (/?•), от t
при окислительной деструкции (инициатор Бз2; 80°С)
радиационного вулканизата натурального каучу-
ка; q (/) измерялось с помощью релаксации на-
пряжения [A. V. Tobolsk у, A. Mercu-
rio, J. Am. Chem. Soc., 81, 5,539 (1959)]:
/-0,7(R.)f;
реакций имеет некоторые очень интересные характерные черты,
в частности существование стационарной концентрации гидропе-
рекиси [6].
Литература
1. Reviewed in N. G г a s s i е, «The Chemistry ol High Polymer Degradation
Processes», Ch. 4, Interscience Publishers, New York, 1956 [H. Грасс»,
Химия процессов деструкции полимеров, Издатинлит, 1959].
2. J. Bolland, G. Gee, Trans. Farad. Soc., 42, 236 (1946).
3. J. Bolland, H. Hughe s, J. Chem. Soc., 1949, 492.
4. A. V. Tobolsk v, A. M e r c u r i o, J. Am. Chem. Soc., 81, 5535
(1959).
5. A. V. To b о 1 s k y, A. Mercurio, J. Am. Chem. Soc., 81, 5539 (1959).
6 A V. 1 о b о 1 s k y, D. Metz, R. В. M e s г о b i a n, J. Am. Chem.
Soc., 72, 1942 (1950).
8. По.шсу.1ьфис/ные ренины
257
8. Полисульфидные резины
Основные причины, вызывающие химическую релаксацию на-
пряжения в полнсульфидных резинах, уже обсуждались в разделе
12 главы IV. Релаксация напряжения в этих резинах происходит
вследствие реакций взаимного обмена ди-, три- или тетрасульфпд-
ных связей друг с другом пли с меркаптаинымн и меркаптидными
связями. Наличие обменных реакций между связями типа ди-
сульфид -дисульфид или дисульфид—меркаптан было одновре-
менно обнаружено как при исследовании релаксации напряжения
II, 2], так и по образованию сополимеров 131 из смеси латексов
полнсульфидных каучуков. Эти наблюдения были сначала сде-
ланы на небольших молекулах пропилдисульфида и децилмеркап-
тана |4|:
СЛ I,C1 LCILSSC1LCILCI I,, ' СН.,(С1 1..ДС1 LSI I-
---L('| LSSCIL(Cn2)sCH3 I CH,Cl LCI LSI 1
Было найдено, что такой обмен происходит в течение нескольких
часов при 130 °C, если ингредиенты очень чистые, но в присутствии
ионных катализаторов он завершается быстро даже при
комнатной температуре. Аналогично при полимеризации цикли-
ческих дисульфидов в линейные полимеры было найдено, что по-
лимеризация протекает медленно даже в присутствии следов воды
пли меркаптана. Однако в присутствии ионных реагентов (основа-
ний, таких, как натрийбутилмеркаитид, или кислот Льюиса,
таких, как FeCl:i со следами воды) полимеризация происходит
быстро 151:
|С1 LCI L( И J LC1 LSS[V -
После этих работ было получено очень много данных отно-
сительно обмена связей дисульфид—дисульфид, которые недавно
рассмотрены в обзоре [61.
Периодические измерения напряжения в полнсульфидных рези-
нах не показывают изменения свойств со временем, т. е. разрыв свя-
зей и их восстановление не сказываются на физических свойствах
недеформированной резины. Уменьшение напряжения при по-
стоянной деформации весьма близко следует закону Максвелла и
не зависит от степени удлинения, а также от того, присутствует
ли или отсутствует кислород. Времена химической релаксач1111, ~х.
при различных температурах подчиняются закону Аррениуса
17—833
25S /д. К Химическая релаксация напряжения
(рис. V, 19). Энергия активации, полученная из зависимости 1g тА.
от 1/Г, для большинства полисульфпдпых полимеров близка к
24 ккал1моль:
zx==A~'exp(EakT,/RT) (8, 1)
Поскольку кривая прерывной релаксации представляет собой
горизонтальную прямую, кривая остаточной деформации являет-
ся зеркальным отражением кривой релаксации напряжения, как
Рис. 19. Кривые релаксации напряжения для полису .п,фнл-
иоп резины Н-10 (тиокол ST) при разных темпера гурах
(М. l5. Stern, А. V. Tobolxkv, J. Chem Ph\'..
14, 93 (1946)1.
видно пз рис. V, 20. Теория ползучести при постоянной пагрузье,
которая приложима к этим веществам, обсуждалась в разделе 14
главы IV.
Заслуживает внимания то, что на химическую релаксацию
напряжения очень сильно влияют следы меркаптанов, элементар-
ная сера, диффундирующая в резине, ионные соединения всех
типов и ультрафиолетовый свет 121.
В последнем случае обменные реакции • могу i upon кать по
радикальному механизму.
Значительный интерес представляет очень большое различие
во временах химической релаксации, наблюдаемое в псс.ц-тован-
пых резинах, как это видно пз рис. V, 21.
Это различие частично может быть связано с различием в строе-
нии основной цепи полимера, т. е. с относительным количестгом
8. Полисульфидные резины
259
Остаточная деформация, °/а
Рис- V, 20. Кривые непрерывной релаксации напряжения
(/), прерывной релаксации напряжения (2) и остаточной
деформации (?) в полисульфндных резинах (удлинение
в процессе релаксации 50%).
Рис. V, 21. Химическая релаксация напряжения для
различных полпсульфпдпых резни при 60 °C.
А—полиэтилеитетрасульфид, вулканизованный ZnO;
В—полидиэтилэфнртетрасульфпд, вулканизованный ZnO;
D—полиднэтнлэфнрдисульфид', вулканизованный ZnO;
ST—нолидиэтилформальднсулвфид4, вулканизованный
бепзотиазнлднсульфпдом [М. Г). Stern. Л., V. Го
bolsky. J. Chem. Phy.-,.. 14, 93 (1946)).
Формулы см. на стр. 206. —Прим. ре<).
/60
Гл. V. Химическая релаксация напряжения
дисульфидных, трпсульфпдных пли тетрасульфпдных связей. Но
в основном такая разница во временах химической релаксации
объясняется случайными следами катализаторов ионного типа,
вводимых при вулканизации. Поэтому полпсульфпдная резина,
совершенно свободная от загрязнений ионного характера, а так-
же, насколько это возможно, свободная от меркаптанных и осо-
бенно меркаптидных групп, в принципе должна быть почти не
способной к химической релаксации напряжения.
Для проверки такого предположения был приготовлен низко-
молекулярный полпсульфпдпый полимер следующего строения.
17, 81:
I1S—• -SS- -SS- SS-----------SS--—SS-----— SH
I
SS----SS------SH
Между дисульфидными связями в цепи молекулы помещались
группировки днэтплформаля —СН2С112С)СН2ОСН2СН2—.
Среднечпсловая степень полимеризации была 11, а содержание
трифункцнональиых узлов (точек разветвления) —2 мол. %.
Вещество представляет собой жидкость с вязкостью 560 пуаз
при 30 °C/
Этот жидкий полимер вулканизовался с образованием трех-
мерной пространственной сетки с помощью реакции, при которой
связывалась концевые группы —SH:
। ---SPbS---- О П2О
(А) --SH PbOg HS-----—
1-^ ... -S—S—• 4 -Н2О
---s----+PbS + H..O
(Б) -SH + PbO2 + HS--—
1_». ... -s-s-----Р Н.,О
(В) SH |-MnO2+HS-------->---S S—• 4 МпО 4-Н.,О
(Г) —SH I V3HON=/^ .--NOH ! HS-----•>
--->----S-S----+ */3H2N— NH« + %Н2О
(Д) • -SH -I- О=С—N—R—N—С=О -р IIS- • •->
---> ---S—СО—NH—R—NH—СО—S--------
Результаты определения времен химической релаксации 181
при 80 °C в пространственных сетках, образовавшихся при вул-
канизации жидких полисульфидов методами А— Д, показаны в
табл. V, 5. Ясно виден очень большой разброс в значениях тл.
Вулканизат А, который содержит сильно ионизированные попе-
речные связи из меркаптида свинца, показывает наименьшее
8. Полисульфидные резины
261
Таблица V, 5
Время химической релаксации + при 80 °C для полнсульфидных резин,
вулканизованных различными химическими агентами* 1 2 3 4 5 6 7 8
Вулканизующий агент
Количество вулканизующего
агента
вес. ч. на 100 вес. ч. полимера
А. Перекись свинца . . 7,3 0,68
Б. Перекись свинца (5 суток нагрева-
ния при 80 °C) 7,3 15
В. Двуокись марганца + морфолин . . 18,9+2,0 32
Г. п-Хннонднокснм ... 2,5 15
Ц. 2,4-Толуиленднизоцианат N-ме-
тил-2-иироллндон 7.0+0,5 200
а А. V. Tobolsky, Р. С. Со loci и у, J. Appl. Polymer Sci., 2, 39 (1959)
время релаксация. Вулканизаты Б, В и Г имеют промежуточные
значения хх. Вулканизат Л. в котором не содержатся ионизирую-
щиеся вещества, имеет наибольшее значение tv. Интересно, что
энергия активации химической релаксации у вулканизата Д
равна 36,6 ккал в отличие от обычно наблюдаемых значений
24 ккал. Химическая релаксация напряжения в этом случае воз-
можно происходит вследствие разрыва нзотиоуретановых связей
— S—CONII—R—NHCO -S-
а не дисульфидных связей.
Литература
1. Al. D. S t е г п, А. V. Tobols к у, Chem. Phys., 14, 93 (1946).
2. AS. Mochul s к у, A. V. Tobolsk у, Ind. Eng. Chem., 40, 2155
(1948).
3. E. AS. Fe ttes, F. O. Davis, private communication to the author in
1946; E. Al. F e t t e s, J. S. Jorcza k, Ind. Eng. Chem., 42, 2217
(1950).
4. G. Gori ii, A. V. Tobols к у, G. D о u g h e r t у, J. Am. Chem.
Soc., 71, 3155 (1949); G. Gori n, Ph. D. thesis, Princeton University,
1949.
5. A. V. Tobolsk y, F. Leona r d, G. P. R oeser, J. Polymer Sci.,
3, 604 (1948).
6. E. At. F e t t e s, F. O. Davis E. Bertozzi, J- Polymer Sci.,
19, 17 (1956).
7. J. S. Jorczak, E. Al. F e t t e s, Ind. Eng. Chem., 43, 324 (1951), .
8. P: C. Colodnv, A. V. Tobolskv. .1. Appl. Polymer Sci., 2,39
(1959). '
Гл. V. Химическая релаксация напряжения
9. Силиконовые резины
Циклические силоксаны, такие, как октаметилциклотетра -
силоксан, могут быть каталитически переведены в линейные при
встряхивании с небольшим количеством серной кислоты [1, 21
(CH3)2Si-O-Si(CH3)2
I I H2so4
и О О --------------------->
(CH3)2Si—О—Si(CH3)2
- СН3 СН, •
—Si—О—Si—о—
_ сн3 (^Н3 .
Пленки слабо структурированного силиконового каучука были
приготовлены непосредственно из октаметилциклотетрасилоксана
(100 вес. ч.), структурирующего агента (3 вес. ч.) и дымящей сер-
ной кислоты (0,1 вес. ч.) 131. Структурирующий агент представ-
лял собой продукт гидролиза эквимолярной смеси (CH3)2SiCl2 и
iCH3)SiCl3 со среднечисловым молекулярным весом 1320.
Рис. V, 22. Химическая релаксация напряжения при
60 °C в специально приготовленной силиконовой резине
при различных относительных влажностях [D. Н.
Johnson, J. R. McLoughlin,
Л. V. Т о b о 1 s к у, J. Phys. Chem., 58, 1073 (1954)].
Цифры на кривых—относительная влажность в %.
Резиновые пленки, приготовленные таким образом, крайне
неустойчивы при низкой относительной влажности из-за присут-
ствия катализатора в пленке, вызывающего непрерывный взаимный
обмен Si—О-связей. Этот очень быстрый взаимный обмен прояв-
ляется в опытах по релаксации напряжения. На рис. V, 22 по-
казаны результаты таких опытов па пленках из силиконовой ре-
9. Силиконовые резины
263
шны при 60 °C и при разных относительных влажностях. Ясно,
что пары воды в атмосфере над этими пленками стабилизируют
катализатор и тем самым замедляют обмен связей. Влияние паров
воды, однако, полностью обратимо, пленки, находящиеся при
высоких относительных влажностях и поэтому стабилизованные,
становятся снова лабильными при уменьшении относительной
влажности. Длительная стабилизация этих пленок может быть
достигнута при их пропитывании пиридином.
Промышленные силиконовые резины получаются при структу-
рировании линейного полидиметилсилоксаиа с помощью перекиси
бензоила. Для увеличения модуля и прочности резин применяются
различные минеральные наполнители. Для этой цепи использу-
ются высокодисперсная двуокись кремния, двуокись титана,
окись железа и т. п.
Было обнаружено, что времена химической релаксации спе-
циально приготовленных силиконовых резин сильно изменяются
в зависимости от наполнителя и условий изготовления резины
14]. Было сделано заключение, что в этих резинах химическая
релаксация напряжения происходит вследствие катализируемого
взаимного обмена Si—О-связей, причем катализаторами явля-
ются некоторые неизвестные ионизированные соединения [4].
Было показано, что наиболее важным катализатором таких
реакций обмена в промышленных резинах является бензойная
кислота 151.
Перекись бензоила, используемая в качестве вулканизующего
агента, действует на метильные группы двух соседних силокса-
новых цепей с образованием поперечной связи следующим обра-
зом:
О О
I—сн3+ СН3— I + СвН6С—ОО-СС6Нв--> |—СН2СН2—| + 2СвН3СООН
Бензойная кислота, выделяющаяся в качестве побочного про-
дукта, действует как катализатор в реакции обмена Si—О-свя-
зей и является причиной химической релаксации напряжения
при высоких температурах, а также образования циклических
летучих силоксанов.
С помощью радиационной вулканизации линейных силокса-
новых цепей можно получить резину, не содержащую кислых или
основных побочных соединений. Было показано, что такие ра-
диационные вулканизаты значительно более устойчивы к хими-
ческой релаксации напряжения, чем резины, полученные при
помощи перекиси бензоила [5].
26-j
Гл. V. Химическая релаксация напряжения
Литература
1. \V Pain о d е, D. 1'. W i 1 с о с к, .1. Ain. Chem. Soc., 68, 358 (1946)
2 А. V Tobolsky. I- Leonard, G. Р Roeser, J. Polymer Sci.
3, 604 (1948).
3 D. И. Johnson, J. R. McLoughlin, Л. V. Tobolsky
J. Phys. Chem., 58, 1073 (1954); J. R. McLoughh n, Ph. D. thesis
Princeton University, 1951.
1 A V. Tobolsk y, G. M. Bro w u, reported in Ph. D. thesis b\
G. M. Brown, Princeton University, 1948.
5 R C. Ostholl, A. M. В и e c h e. W. 1. Grubb. J. Am Chem
Soc.. 76. 4659 (1954).
10. Полиуретановые резины
Полиуретановые каучуки являются важным новым классом
эластомеров, получаемым пз низкомолекулярных сложных поли-
эфиров и дпизоцнанатов или пз низкомолекулярных простых
полиэфиров и дипзоцпапатов [1]. Кроме простых и сложиоэфпр-
ных связей, пространственные
с од ер жа т сл еду ющ не связи:
о
— XII—с—о---
о
- МН —С—-NH • •
О о
!i И
NH—С—N -C-NH-
сетки резин пз этих каучуков
уретановая спять
связь типа дпзамепепиоп
мочевины
биуретовая связь
Исследования химической релаксации напряжения прово-
дились па образцах промышленных полиуретановых резни, из-
готовленных на основе простого полиэфира 1 и па основе, сложного
полиэфира 2, а также для сложного полиэфира без уретановых
связей 3 [2]. Полиэфирный полимер 3 представлял собой высоко-
молекулярное соединение, приготовленное пз этиленгликоля,
пропнленглпколя, адипиновой кислоты и нескольких молярных
процентов малеиновой кислоты. Последняя вводилась для созда-
ния участков, способных к образованию поперечных связей, а
в качестве вулканизующего агента использовалась перекись бен-
зоила.
Времена химической релаксации при 120 °C для резни 1, 2 и 3
соответственно были 2,1; 3,0 и свыше 500 ч. Ясно, что слабые
связи в резинах 1 и 2, от которых зависит спад напряжения,
характерны для резни 1 и 2 и отсутствуют в резине 3. Такими
10. Полиуретановые резины
265
связями могут быть только уретановая, биуретовая или связь
типа дпзамещенной мочевины, изображенные выше.
Для того чтобы установить, которая из этих трех видов связей
является слабой, ответственной за спад напряжения при 120 °C,
был приготовлен [3J ряд резин 4, 5 и 6.
Типичный сложный полиэфир, использованный для приготов-
ления полиуретанового каучука, был синтезирован из адипино-
вой кислоты и смеси этилен- и пропилеи гликоля, взятых в моляр-
ном отношении 4:1. Применялся небольшой избыток гликолей отно-
сительно кислоты, что привело к получению полиэфира со средне-
числовым молекулярным весом 1580, имевшего концевые гидрок-
сильные группы. Этот полиэфир обозначим «Р». Для дальнейшей
полимеризации использовался типичный диизоцианат, а именно
2,4-толуилендппзоцнанат
СН3
I
N=C=O
Ниже приведены основные реакции, происходящие при поли-
меризации с участием диизсцнаната:
(А) N=C=O + HO--------->----NH—СО-О------
уретановая связь
(Б) --N=C=O-|-HOH + O=C=N----------->
---> • • —NH—СО—NH— • • + СО2 связь типа дпзамещенной
мочевины
(В) N=-C=O+------NH—СО—NH---------,
---'» —NH—СО—N—СО—NH—•• биуретовая связь
При взаимодействии полиэфира Р с избытком 2,4-толуилендиизо-
цпаната в качестве окончательного продукта был получен поли-
эфир с концевыми изоцианатными группами, который мы обозна-
чили как «форполимер» А. Молекулы А имеют уретановые связи,
каждая пз которых расположена рядом с остатком изоцианатной
группы. Как полиэфир Р, так и форполимер А представляют собой
вязкие жидкости. Они вулканизовались до образования резин,
обозначенных как 4, 5, 6, следующим образом:
Р4 метплтрпфенилтрипзоциаиат (4)
А 1,2,6-гексаптрпол (5)
А 4- вода (6)
Резина 4 содержит, очевидно, только сложные эфирные и
уретановые связи. То же относится и к резине 5. Резина 6, одна-
26Ь
Гл. V. Химическая релаксация напряжения
ко, содержит уретановые, биуретовые связи и связи типа диза-
мещениой мочевины. Общее число уретановых связей в массе ре
зины 6 равно сумме числа связей типа дизамещенной мочевинь,
н числа биуретовых связей.
Исследования химической релаксации напряжения проводи
лнсь па резинах 4, 5 и 6 при 120 °C. Результаты этих исследова-
ний сравнительно с данными для образцов 1, 2 и 3 приведены па
рис V, 23.
Rper-w. ч
Рис. V, 23. Химическая релаксация напряжения
при 120 °C различных полиуретановых резин и по-
лиэфирного каучука, вулканизованного перекисью
бензоила. Цифры на кривых—типы резин
1—6 (см. в разделе 10 и табл. V, 6) [J. A Of-
fenbach, А. V. Tobolsk у, J. Coll.
Sci., 29, 39 (1956); Р. С. С о 1 о d п у, Л. V. To-
bolsk у, J. Am. Chem. Soc., 79, 4320 (1957)1
Ясно, что наиболее слабыми связями в полиуретановых рези-
нах должны быть связи типа дизамещенной мочевины плп би-
уретовые. Возрастание времени химической релаксации три 120 <
в 10 раз может быть достигнуто при замене этих связей
Анализ кривых химической релаксации напряжения двух
промышленных резин 1 и 2 (адипрен В и вулколлан А) сравни-
тельно со специально приготовленными резинами 4, 5 п 6 ясно
показывает, что промышленные резины, использованные в этих
исследованиях, также имеют связи типа дизамещенной мочешпп.
и биуретовые.
В табл. V, 6 приведены времена химической релаксации
при 120 °C, резин 1—6.
10. Полиуретановые резины
267
Таблица V, Ь
Времена химической релаксации -д. при 120 СС полиуретановой и полиэфирной
резины
i ’гчнна Гии СВЯЗИ 4
f Эфирная, уретановая, типа дизамещенной мочевины, биуретовая 2, 1
/ Сложноэфирная, уретановая, тина дизамещенной мочевины, биуретовая 3,0
•5 Сложноэфирная 500
4 Сложноэфирная, уретановая 14,0
5 То же 9,7
6 Сложноэфирная, уретановая, типа дизамещенной мочевины, биуретовая 1.6
Литература
I E. Muller, О. Bayer et al., Rubb. Chem. Technol., 26, 493 (1953).
2. J. A. Offenbach, A. V. Tobolsky, J. Coll. Sci., 11, 39 (1956).
i. P. C. Colodny, A. V. T о b о 1 s k v, .1. Am. Chem. Soc., 79, 4320
(1957).
Глава VI
ПОЛИМЕРИЗАЦИОННОЕ РАВНОВЕСИЕ
1. Равновесие при полимеризации
В предыдущей главе рассмотрена химическая релаксация
напряжения как способ измерения химических изменений, кото-
рые происходят в полимерной пространственной сетке при повы-
шенных температурах.
Некоторые из этих изменений, такие, как разрыв и образо-
вание поперечных связей под действием кислорода, являются
необратимыми. С другой стороны, реакции взаимного обмена,
подобные происходящим в пространственной сетке полисульфид-
ной или силиконовой резины, являются случаем динамического
равновесия.
Интересно рассмотреть равновесие между мономером и поли-
мерными молекулами всех размеров с точки зрения чистой термо-
динамики 11—5]. Следует различать три основных случая рав-
новесия, которые мы обозначим как случаи I, II и III.
Обозначим мономер через М, инициатор полимеризации
(там, где он присутствует) через ХУ, их концентрации (обычно в
моль/кг) через М, ХУ и т. д.
Случай 1
Ха
ХУ + м хм\
хь
XMY -ф /И ХМ
С л у ч а и
ХЯ.-.У -ГА! ХМП)
II
Ки
Al , ...ci М*
X/i
/VI* + М М'2
/И*_( .1 м м;
1. Равновесие при полимеризации
269
(звездочка обозначает активированное состояние—бирадикал или
цвиттерион).
С л у чай III
/VI + М 7=^ м2
м2 + М 7=2 М,
Ч, !- /VI т== Mn+i
Примерами первого случая является полимеризация е-капро-
лактама, инициированная водой пли амином.
Пример 1а
СО---------NH
а | | + Н2О Поликапроамид
СН2—(СН2)3—СН2
При м е р 16
СО---------NH
п | I + RNI1-2 < ? Поликапроамнд
СН2-(СНг)з—СН2
Примером второго случая является полимеризация цикличе-
ской серы в полимерный бирадикал.
Пример Па
nSB «[SSSSSSSS],,»
Примерами третьего случая являются равновесная полимери-
зация виниловых мономеров без инициаторов и равновес-
ная полимеризация е-капролактама в циклы большего размера.
Пример Ша
nCH2=CHZ —± CH2-=CZ-[CH„ СН71, .2-СН,-СНг2
Пример 1116
СО--------Nil
п I | “77 Цикл ич п зпепьен
СПг—(СН2)3—сн2
С экспериментальной точки зрения наиболее важными величи-
нами, характеризующими равновесие, являются:
1) равновесная концентрация мономера /И;
2) равновесная среднечисловая степень полимеризации Р,
3) равновесная концентрация инициатора XY в тех случаях,
когда присутствует инициатор.
270
Г.г. VI. Полимеризационное равновесие
Для того чтобы получить удобное соотношение между М, /<
XY и константами равновесия Ка, Кь, Кс, ...» Кп, , мы вводим
два физически обоснованных приближения:
а) Ка = К
Kb = K(.^K(t = --==Kn = -- = K9
б) Ха = Кь = Хс = • •• = К„ — •• = К3
Подходит ли данное равновесие под приближение (а) или (б),
можно иногда определить интуитивно. Пример случая 1а—по
димеризация е-капролактама с помощью воды—явно относится
к приближению (а) и поэтому обозначается нами, как случай 1а.
Пример случая 16—полимеризация е-капролактама с помощью
амина, очевидно, относится к'приближению (б) и поэтому обозна-
чается нами, как случай 16.
Полимеризация серы совершенно очевидно относится к прибли-
жению (а) и поэтому обозначается, как случай Па. Полимериза-
ция е-капролактама в циклы большего размера, вероятно, отно-
сится к приближению (б) и поэтому обозначается, как случай
П16. Полимеризация винильных соединений в отсутствие инициа-
тора временно обозначена, как случай Ша 151.
Математическая задача, которая должна быть решена,—это
установить соотношение между Р, М, с одной стороны, и К, К3
и Мо—с другой, гдеЛД—начальная концентрация мономера. Для
случая 1 мы имеем соотношение между Р, М, XY и К, К3, Мо, Х{,
(короче,—для ХК0), где Хо—начальная концентрация инициатора
Подробная математическая интерпретация случая II 1а при-
ведена ниже:
ЛД - КМ1 (1.Н
Al i .И Д1, К =
м.. Кз М < • 4Д Л;, = л/3
<Иа-Л1
л-1., АЗ А'з М*
-р /VI — /VI, /МЭЛ1
А1а = К3ЛДЛ1 = КК3М3 (1.21
ЛД =Х3М3М -КК1М* (1.31
Кз
Л1„-1 Al М+) Л1„ + ,
- ККз'*Ми для п >2 (1,4)
1. Равновесие при полимеризации
271
Пусть Л/—общая равновесная концентрация полимерных мо-
лекул:
,М = £ ККпЛ~2Мп
= КМ2 + ЛЖаЛ43 -ь КК^Р_ -j- • (1.5)
= W2 [1 + KaM -h /®и2 I K'iM2 | )
K№
Пусть W—общая равновесная концентрация мономерных сег-
ментов, входящих в полимер;
№=2 пКК'Г2ЛР
п=2
= КМ2 [2 + ЗК3М + 4ХЖ 11
КЛГ2 (к ~_L)
(1 — Л^Л!)2 \Лз М /
Очевидно, что P — W/N (где Р—среднечисловая степень полиме-
ризации): —
«7 2 — К3М I , „
Р = ~ 1 - /<а/И ~ "Г “/QVI ’ 1'
Легко заметить, что
Л'Л12(2 — К3М) км2
Мо = Л1+^^^+-=!4-^--^Л14- (1Л>
где Л40—начальная концентрация мономера.
Объединив уравнения (1,7) и (1,8) и положив (что
допустимо для Р>1), получим: ~~
/ МаК1 к \>/2 ,,
р — ( —“ 3___Ils 1 (1,9а ।
\ К К
Приведенные выше результаты в равной степени приложимы и к
случаю II16 с тем упрощением, что ^=^3, так как все константы
11'2.
Ля. V.i. Полимеризационное равновесие
в приближении (б) идентичны. Уравнение (1, 9а)‘при этом преоб-
разуется следующим образом:
Р-(М0/<3)1/2=« (-^-)*/2 (1,96)
Необходимые соотношения для остальных случаев рассмотре-
ны в других работах 11—4].
Результаты суммированы в табл. VI, 1 [51.
Таблица VI, 1'
у'равней не | Случай р л>о Хо р
(1,10) 1а 1/(1- КаМ) Л1(1-|-/<ХР2) Х(1+/<МР) (Мй-М)/(Хй—X)
(1.11) 16 1/(1—/С3Л4) Л1(Ь|/(3ХР2) Х(1+Х,МР) ~(Л1и-Л1)/Х0
(1.12) Па 1/(1—7\3Л1) Л1(1+№) ^[(МоКз-О/Х]1'2
(1.13) П1а 1/(1-К3М) Л1(1+/<Л-1Р2) ^|(Л10/<2 Рз)/Л'1,/а
(1.14) Шб 1/(1—Р3Л4) Л4(1+К,МР2) ^(Л1„/Л4)>/2
* Выражение для Р, приведенное в последней колонке, не является независимым, а может
быть выведено из остальных соотношений. В последних четырех случаях выражения для 1'
действительны только для значений Р;у 1.
Приложение случая 1а к равновесной полимеризации
t-капролактама, инициированной водой
Суммарная реакция может быть представлена в следующем
виде:
СО----------NH
п I | + Н2О Поликапроамид
сн3—(сн2)8——сн2
Последовательность равновесий, использованная для описа-
ния этого процесса, такова:
СО----------NH к
I | +Н2О z=± HOOC(CH2)5NH.
СН2-(СН2)3—СН2
HOOC(CH2)3NH|—CO(CH2)6NH]„_1—CO(CH2)3NH2->
СО-----------Nil К3
+ | | =z±HOOC(CH2)5NHf—CO(CH2)6NH]„—CO(CH2)6NH2
C.H2-(CI-I2)S—CH2
n = 0, 1, 2, 3, 4...
При данной температуре и выбранных значениях Л1о и X
ветчины Р и М были определены по литературным источникам
16]. Используя уравнение (1, 10) табл. VI, 1, можно легко получить
Ка пз Р^=1/(1—/(3Л1). X получается пз Р = (Л40—A4)/(Xfl—А).
Наконец, А было определено из Xn-X(lКМР).
I. Равновесие при полимеризации
273
Правильность использованных равновесных реакций (тыла
подтверждена совпадением вычисленных и экспериментальных
значений Хо, соответствующих выбранным значениям Р. Значение
Хо вычислялось по Д' и А'3, определенным ранее. Как видно из
рис. VI, 1, были получены удовлетворительные результаты 141-
Рис. VI, 1. Зависимость степени полимеризации от
концентрации воды при равновесной полимеризации
капролактама при 220 "С. [А. V. Tobolsk у,
A. Eisenberg, J. Am. Chem. Soc., 81, 2302 (1959)1
/—экспериментальная; 2—вычисленная (/<—0,00251, Кз=*2,05)
Процесс проводился при двух температурах. Применение
уравнения Вант-Гоффа к значениям Л’ и Кя дает:
К == ехр ехр ( A// RT>
К3 - схр-^—ехр( \irjRT) (1,15)
AS" 6,8 э. е. AS3^ 7,0 э. е.
Х1Г ~= г 2240 кал/моль ХН°Л = 4030 кал'моле
соответствующие реакциям инициирования и роста цепи. Стан-
дартное состояние было выбрано равным 1 моль/кг, а все конпен
18 833
Гл. VI. Полимеризационное равновесие
грации, приведенные в этом разделе, также выражены в моль/ке.
Так как Р —1/(1—/\3/И), то для больших значений Р
<1,16)
— Лз
Зледовагельно, в области больших степеней полимеризации
не зависит от концентрации инициатора.
П рименение случая 16 к равновесной полимеризации
а-капролактама, инициированной алкиламином
Суммарная реакция может быть представлена в следующем
виде:
СО----------NH
и | | + RNH2 t > Поликап роампд
СП2-(СН2)8—сн2
Последовательность реакций, которые используются для
описания этого процесса, такая:
А'з
RNIL +КЛ г—± R (КЛ) NH2
Кз
R (КЛ)„МН2+КЛ R (КЛ)/1+1ЫН2
п = 1, 2, 3, 4...
где 1<Л обозначает капролактам, а (КЛ) обозначает —NHCO(CH2)5
В этом случае мы полагаем весьма точно К -К3, так как в
равновесии участвуют те же функциональные группы. Для это-
го случая в литературе отсутствуют удовлетворительные данные
ио равновесию. Мы предполагаем, что это равновесие может быть
описано в рамках случая 1а простой заменой К на К3. При этом
первое важное следствие получается нз уравнения (1, 16), а имен-
но, что Из этого ясно следует, что равновесная концент-
рация мономера является функцией одной температуры и не зави-
сит от природы инициатора. Поэтому можно ожидать, одно и то же
шаченпе М независимо от того, что применяется в качестве ини-
циатора: вода или амин.
Второе важное следствие вытекает из уравнения, приведен
ного ниже, которое может быть выведено 15] из уравнения (1, 11):
Мо-Л1 1//<а
(1. 17)
Уравнение (1, 17) при 240 °C графически представлено на рис.
VI, 2. Выражая К3 как функцию температуры из уравнения
1. Равновесие при полимеризации
275
(1, 15), получаем:
Л40 — ехр (— 4030//?'/') ехр (7,0//?)
(1, IB)
Уравнения (1, 16) и (1, 17) можно также применить и для других
инициаторов, таких, как органические кислоты или аминокисло-
ты. Для инициирования с помощью алкпламинов требуется такой
катализатор, как НО.
Рис. VI, 2. Зависимость степени полимеризации от кон-
центрации инициатора при равновесной полимеризации
капролактама при 240 °C. Предполагается, что при
выбранном инициаторе /<,= /<3=1,63 [А. V. Т о
b о I s к у, A. Eisenberg (не опубликовано)].
Приложение случая На к термической полимеризации серы
Суммарная
виде:
реакция может быть представлена в следующем
nS8 Ss„
где звездочка обозначает бирадикал.
Последовательность стадий, через которые достигается конеч-
ное равновесное состояние, такая:
к
Ss^Sl
Кз
Ssn + S8,—I Setn+o
В литературе имеются примерные данные по функциональной за-
висимости Р и М от температуры 17, 8J. Согласно уравнению
(1, 12) и табл. VI, 1 значение Ка может быть вычислено по соот-
18
276
Гл. VI. Полимеризационное равновесие
ношению Р=1/(1—КаМ) для любой температуры, если известны
Р и Л1. Величина Л может быть тогда вычислена из - Л1( 1 +/\Р2^
с учетом того, что Мо—константа, равная в используемом темпе
ратурном интервале 3,90 моль/кг. Если вычисление провести для
другой температуры, то можно построить зависимости 1п/\ от
\IT II 1п/\3 от ИТ, по которым определяются l\H°, ;\Sa, &Н\
и Л-Sj. В результате получаются [3|:
In Г
1пГ3
Д/Г = 32 800 кал! моль
i\H°A = 3170 кал!моль
Д5° Ы[°
~R 1ТГ
as° дл°
RT
(1, 19)
AS0 =23,0 кал;град моль\
AS° = 4,63 кал / г рад -моль |
(1,20)
Линейность зависимости 1п/\ от l/Т и 1п/\3 от l/Т является
очень хорошим подтверждением теории. Поскольку АН’, ДЯ°.
Рис. VI, 3. Зависимость равно-
весной концентрации мономера
(.ио.гь/’лг) от температуры при
равновесной полимеризации эле-
ментарной серы [А. V. Т о-
b о I s к у, A. Eisenberg,
J. Ain. Chem. Soc., 81, 780
11959)). Точки—эксперименталь-
ные данные, кривая—вычис-
лена .
Рнс. VI, 4. Зависимость степени по-
лимеризации от температуры для
равновесной полимеризации серы
[А. V. Tobolsky, A. Eisen-
berg, J. Am. Chem. Soc., 81, 780
(1959)1. Точки—экспериментальные
данные, кривая—вычислена.
1. Равновесие при полимеризации
277
AS° и AS° совершенно не зависят от температуры, точное выра-
жение для К и /у, можно в принципе получить из измерений Р и М
только при двух температурах, которые должны лежать выше
«переходной» температуры серы.
В этом случае по уравнению (1, 12), (см. табл. VI, 1), взятому
вместе с уравнениями (1, 19) и (1, 20), можно предсказать значе-
ния М и Р при всех температурах, как показано на рис. VI, 3 и
VI, 4. При сравнении этих вычисленных кривых с эксперимен-
тальными данными, приведенными в литературе [7, 8J, полу-
чено очень хорошее согласие. Интересно отметить, что соотно-
шение между Р, К и /(3 в явном виде может быть получено при
исключении М из двух - зависимостей, приведенных в уравне-
ниях (1. 12):
<| 2|>
которое для значений Р>1 имеет вид:
(/ИрЛГз- 1) |'/2
I
(1,22)
Приложение случая Ша к равновесной полимеризации
виниловых мономеров
Будем считать, что это равновесие достигается через следую-
щие стадии:
к
СН2=С1 IX + СН2=СНХ 7-* Димер
Кз
СН2=СХ(СН2СНХ)„—СН,СН2Х СН2=СНХ 7—»
СН2=СХ (С112СНХ)„ +1СН2СН2Х
п = 0, 1, 2, 3...
Следует иметь в виду, что форма написания равновесия ничего
не говорит о механизме процесса. Равновесие зависит только от
исходного и конечного состояний. Априори можно сказать, что
К должно быть равно /\;1, и поэтому к данному равновесию в рав-
ной степени применим случай 1116. С какой степенью точности
проводится при этом расчет, можно определить только на основе
э ксп ер и мен та л ьн ы х данных.
При изучении равновесной полимеризации виниловых мономе-
ров, особенно метилметакрилата, Дайитон и Ливии, а также
Байуотер [9, 101 молчаливо допускали возможность использова-
ния только одной константы и вывели другими методами соотно-
278
Гл. VI. Полимеризационное равновесие
шенне, которое в наших обозначениях выглядит так (см. уравне-
ние (1, 16)]:
Байуотер также проводил измерения М и, следовательно, Ks при
разных температурах (10].
Из табл. VI, 1 ясно, что это соотношение действительно либо
для случая 1116 с одной константой, либо для случая II 1а с двумя
константами.
Если рассматривать этот случаи как одноконстантный, то сте-
пень полимеризации должна быть связана с равновесной концен-
трацией мономера Л1 следующим образом (см. табл. VI, 1):
/ Мо\'/2
Р = / |
I м I
По экспериментальным данным Байуотера должно было бы н
этом случае получиться максимальное значение Р, равное 5 при
130 °C, однако в его работе не приведено никаких значений Р.
Мы повторили опыты Байуотера и действительно получили
данные, очень близкие к его результатам для «равновесной»,
концентрации мономера, с некоторыми отклонениями, указанными
ниже. Однако мы определили, что Р в этой системе равно от 500
до 650 в полном противоречии с одноконстантной теорией [5].
Такой результат, однако, не является несовместимым с двухкон-
стантной теорией, по которой
Г(«-^з)Т«
К ~ L К J
Фактически из измерений Р может быть вычислена величина К,
которая равна около 3- 10~Б (ср./(3^-3). Расхождение в экспери-
ментальных результатах, о котором говорилось выше, заключается
в том, что мы в опытах по Байуотеру обнаружили зависимость
«равновесной» концентрации мономера от интенсивности света.
Поэтому результаты Байуотера, может быть, следует рассматри-
вать скорее для фотостацнонарного состояния, чем для равновес-
ного 15].
Приложение случая Шб к полимеризации ^-капролактама
в большие циклы
Образование систем больших циклов в процессе полимериза-
ции е-капролактама—хорошо известное явление. Мы считаем,
что это равновесие подходит под случай II16, так как по существу
1. Равновесие при полимеризации
279
только один тип реакций приводит к образованию больших цик-
лов; как мы предполагаем, это реакции взаимного обмена связей:
I i кз i i
2NH(CH2)5CO -—* NH(CH2)8CONH(CH2)6CO
1 I j i Кз I I
[NH(CH2)8CO|V—I + NH(CH2)5CO ? I-[NH(CH2)6COJx+1- I
x = 2, 3, 4, 5, 6...
В идеальном случае это равновесие можно было бы изучитьс пол-
ностью сухим е-капролактамом при подходящей температуре, что
исключило бы влияние реакции взаимного превращения цикл
цепь.
В нормальных условиях (т. е. при инициировании водой) од-
новременно имеют место два равновесия, т. е. равновесие цикл—
цепь и равновесие цикл—большой цикл. Так как большие цик-
лы присутствуют в относительно малых концентрациях, ими мож-
но пренебречь при изучении систем цикл—цепь, как это и делается
(см. случай 1а). Поскольку для этих упрощенных систем М из-
вестно, его можно рассматривать как Мо для равновесия в си-
стеме цикл—большой цикл.
Некоторые результаты последних исследований
Было показано, что в тетрагидрофуране а-метнлстирол нахо-
дится в равновесии с его полимером при использовании в каче-
стве инициатора нафтила натрия [ 11J. В нашей лаборатории
было получено очень хорошее согласие с этими результатами,
кроме того, были проведены исследования для широких
интервалов начальной концентрации мономера и инициатора (12).
Измерение величин М и Р при равновесии дает очень хорошее
согласие со случаем 1а при упрощающем предположении, что К
очень велико.
Иногда две молекулы нафтила натрия реагируют коли-
чественно с мономером на стадии инициирования. При
этом Р^2(Л4О—Л4)/Хо. Если в качестве инициатора ис-
пользуется натрийдифенилацетилен, случай 1а приложим без
указанного упрощения 1121. Было найдено, что М в этом случае
такое же, как и при использовании нафтила натрия. Однако
для данного Хо значение Р было больше, чем с таким же коли-
чеством нафтила натрия, так как Р~2(М0—М)/(Х0—X) п
величиной X в данном случае пренебречь нельзя. .Значение X
может быть вычислено из Р, а К—из соотношения
Х0 = Х(1 + 2КХМ2Р)
280-
Гл. 17. Полимерииационное равновесие
Случай 11а может быть применим к полимеризации селена в по-
лимерные бирадикалы [131. Довольно ограниченные эксперимен-
тальные данные можно очень хорошо объяснить с помощью точно
такого же математического приближения, какое принималось
для серы. Для селена мы нашли:
Л//' = 25000 нал-моль Л8° = 23,0 кал!град моль
&Н°3= 2270 кал!моль Д8° = 5,47 кал/град моль
Табличные данные для некоторых теплот п энтропий полимери-
зации имеются в литературе [141. Теоретические предпосылки,
на которых они основаны, не учитывают равновесия при иницииро-
вании. В терминах рассматриваемой здесь теории эти табличные
значения могут обычно довольно точно рассматриваться, как
\НЯ и Д83.
Использование весовых долей пли объемных долей вместо
активностей в законе действующих масс, конечно, является при-
ближением, которое приемлемо только в случае постоянства вы-
числяемых констант равновесия. Это также не совсем законно с
точки зрения общей теории растворов полимеров [15, 161.
Литература
1. А. V. Tobols к у, J. Polymer Sci , 25. 220 (1957).
2. А. V. Tobolsky, .1. Polymer Sci., 31, 126 (1958).
3. .A. V. Tobolsky, .A. Eisenberg, J. .Am. Chem. Soc 81, 780
(1959).
4. A. V. Tobolsky, .A. Eisenberg, .1. xAm. Chem. Soc., 81, 2302
(1959).
5 A. V. T о b о 1 s к y, xA. Eisenber g, .1. xAm. Chem. Soc.. 82, 289
(I960).
6. F. W i 1 о t h, Z. Phys. Chern., 4, 66 (1955); P. H. H erm a n s,
A. .1. S t a v e r m a и et al., Recueil, 74, 1376 (1956).
7. G. Gee, Trans. Farad. Soc., 48, 515 (1952).
8 F. F a i r b г о t h e r, G. G e e, G. T. Merrill,.!. Polj mer Sci. 16,
459 (1955).
9. F. S. D a i n t о n, K. .1. I v i n, .Nature, 162, 705 (1948).
10. S. By wale r, Trans. Farad. Soc., 51, 1267 (195x5).
11. D. T. W о r s f о 1 d, S. В у w a 1 e r, .1. Polymer Sci.. 26, 299 (1957);
IE W. McCormick, ibid., 25, 488 (1957).
12. A. V. Tobolsk y, A. R e m b a u m, A. Eisenberg (manu-
script in preparation).
13. A. E i s e n b e r g, A. V. Tobols к у (manuscript in preparation).
14. F. S. D a i n t о n, K. J. I v i n, Quart. Rev., 12, 67 (1958)
15. A. V. 1 о b о 1 s к у, J. Chem. Phys., 12, 402 (I944i.
16 P .1. Flor y, E Chem. Phys ., 12, 425 (1944).
2 Равновесие при сополимеризации
281
2. Равновесие при сополимеризации [1]
Исследования релаксации напряжения показали существова-
ние реакций обмена связей в структурированных полимерах.
Реакции обмена, имеющие место как в цепях сополимеров, так
и в пространственных структурах, состоящих из сополимерных
цепей, будут, несомненно, приводить к равновесию, т. е. к равно-
весному распределению сочетаний различных по структуре участ-
ков вдоль цепей. Интересно рассмотреть этот сравнительно мало-
обсуждавшпйся вопросе совершенно общей точки зрения, вклю-
чая экспериментальные условия, при которых может быть до
стигнуто равновесие при сополимеризации.
При обычном методе получения виниловых сополимеров со-
став и структура конечного продукта определяются относительной
скоростью реакции двух мономеров с двумя типами активных
(в виде радикалов) растущих концов цепей.
Различные мономеры сильно отличаются по реакционной спо-
собности, так что в общем этим методом крайне трудно получить
сополимеры желаемого состава и структуры. С другой стороны,
известно, что при достаточно высоких температурах некоторые
виниловые полимеры способны к реакциям деполимеризации, про-
текающим по цепному радикальному механизму, обратному
механизму полимеризации. Некоторые полимеры, например ме-
тилметакрилат, способны почти полностью превращаться в мо-
номер при нагревании их в вакууме. Можно было предположить,
что в полимерах такого типа равновесие полимеризация—депо-
лимеризация может быть связано с молекулярновесовым распре-
делением при очень высоких температурах. По некоторым при-
знакам можно ожидать, что если сополимер обрабатывается та-
ким образом, процессы деполимеризации и полимеризации будут
определять как молекулярновесовое распределение, так и распре-
деление по расположению отдельных компонентов вдоль цепи
молекул сополимера. Несомненно, что во многих случаях при
этих температурах могут и будут происходить побочные реакции,
такие, как передача цепи, ведущие к структурированию. Однако
эти вторичные реакции не принимаются во внимание. Следует
ожидать, что при использовании таких мономеров, как метилмет-
акрилат, эти побочные реакции могут быть доведены до миниму-
ма, и можно предполагать, что равновесие полимеризация—де-
полимеризация будет устанавливаться при достаточно высоких
температурах и будет определять как структуру и состав сопо-
лимера, так и кривую распределения по молекулярным весам.
Получить равновесные сополимеры возможно нагреванием при,
достаточно высоких температурах: Г) смеси двух мономеров,
2) механической смеси двух полимеров, 3) смеси одного мономера
282
Гл. VI. Полимеризационное равновеси:-
с другим полимером или 4) сополимера из двух мономеров. Сле-
дует указать, что для достижения равновесия со значительной
скоростью приходится во многих случаях так высоко повышать
температуру, что при этом образуется материал с довольно низ-
ким молекулярным весом.
Необходимо также иметь в виду, что при этом возникаю!
шачнтельные трудности, связанные с взаимной растворимостью
и диффузней одного материала в другой.
Применение сильных ионных катализаторов полимеризации,
которые нашли использование в последние годы, открывает но-
вые возможности. Так как эти вещества могут быть в одинаково/!
степени сильными катализаторами полимеризации и деполимери-
зации, возможно, что при их применении равновесие при сополи-
меризации сможет быть достигнуто в смеси двух мономеров даже
при сравнительно низких температурах.
Высказанные соображения в равной степени относятся нс
юлько к сополимеризации виниловых соединений, но и к сопо-
лимеризации циклических соединений, конденсационной сопо
лпмеризации и т. п.
В первую очередь математически рассмотрим случай, когда мо-
лекулы сополимера имеют бесконечную длину, н определим рас-
пределение звеньев по цепи. Это было сделано двумя независимы-
ми методами, приведшими к одинаковому конечному результа-
ту (11:
АЪв
~ к (20И ехр (\ЕАВ/RT)
\ *АВ /
(2,1)
1де А/л—число мономерных единиц типа A, Nв—число мономер-
ных единиц типа В, МАВ—число АВ соседних пар вдоль
цепи, Л'—константа равновесия, которая выражена через коле-
бательные статистические суммы состояний fAA, fBB и fAB и
энергию обмена ДЕЛВ. Функции fAA и fBB могут включать фак-
тор симметрии 2. Значение Д ЕАВ определяется как 2ЕАВ—ЕАА —Евв,
|де Еаа—энергия моля связей А—А, Евв—энергия моля свя-
зей В—В, а Елв—энергия моля связей А—В. Энергия ЕАА
примерно равна теплоте полимеризации моля мономера А, то
же соответственно относится к Евв и ЕАВ. Распределение одно-
Z. Равновесие при сополимеризации
28.5
родных участков цепей по длинам дается формулой:
п* =^ЛрхАЛ\\ Рад)2
пх -- МвРвв\\ — Рвв\
(2, 2i
где /И—число имеющихся в сополимерной цепи последователь-
ностей мономеров Д„ соединенных в участок длиной х, пх—чис-
ло последовательностей мономеров В, соединенных в участок
длиной х, а рЛЛ и рвв определяются уравнениями:
Проблема равновесия при сополимеризации математически иден-
тична задаче Изинга 121 (в феромагнетизме) и, конечно, приво-
дит к тем же выводам.
Литература
i. 1. А 1 I г е у, I. V. lobolsky, J. Polymer Sci.. 38, ‘269 (1959).
2. Е. Ising, Z Phys., 31, 253 (1925).
ПРИЛОЖЕНИЕ А
СТАТИСТИЧЕСКАЯ СУММА СОСТОЯНИЙ
Статистическая сумма состояний системы, состоящей из N
молекул, определяется следующим образом:
Q = Есхр(- EJkT) (А, 1)
i
где Ez-—энергия i-того состояния системы.
Можно показать, что для системы из N молекул одноатомного
газа, занимающей объем И при температуре Т, имеет место сле-
дующее выражение I1J:
Qra3. = 2>
где m—масса молекулы, g0—степень вырождения электрона в
основном состоянии.
Уравнение (А, 2) справедливо, если среднее число молекул,
находящихся в каждом молекулярном квантовом состоянии, мало.
Это выполняется для всех газов (кроме электронного газа) при
гемиературах выше 5 СК.
Чтобы получить затем уравнение состояния, используют сле-
дующие основные формулы:
Л = —kTluQ (А.З)
из которых легко выводится закон идеального газа:
pV = NkT
Для кристалла Эйнштейна, состоящего из NA атомов, можно
показать [2], что
QKP. = ехр [— UL (V);kT] ехр (— ЗА/Л/гч2/гТ) х
X [ 1 - ехр (— hv/kT)]~3NA (А, 5)
Приложение А
285
где UL (V)—энергия решетки молярная для состояния, когда каж-
дый атом находится в равновесном положении, NA—число Аво-
гадро, '/—эйнштейновская частота, являющаяся функцией
объема [3].
Используя уравнения (А, 3)—(А, 5), получим:
А = UL (V) Ал/г/ + 3NAkT In [ 1 — ехр ( h'//kT)\ (А, 6)
— -2 NAh
Р - -
) ,-3NAh (^ )r (ехр h'dkT - 1)-‘ (А, 7)
Два последних члена в правой части уравнения (А, 6) представ-
ляют собой Ае—вклад колебании решетки в свободную энергию
в приближении Эйнштейна. Из равенства (А, 6) следует:
A = Ul(V) + Ae(T, V) (А, 8)
По дебаевской теории кристалла предполагается, что вместо
ЗЛ'Л одинаковых частот имеется распределение частот (V(v)dv,
определяемое следующим уравнением:
0<v<vOT
\1}1 /
‘ N (v) cl'i = 3N л
о I
Все ранее выведенные уравнения для кристалла Эйнштейна
можно применять к кристаллу Дебая, используя следующее
преобразование:
3№л/ (v)---> | N (A f (A d'< (А, 10)
о
где ДО—любая функция частоты.
Дебаевская температура 0D определяется формулой
0/> = (А, 11)
Литература
1. К. G. Denblg li, The Principles of Chemical Equilibrium, Ch. 12, Cam-
bridge University Press, New York, 1955.
2. E. A. G u g ge n h e i m, Boltzmann’s Distribution Law, Ch. 8, Inlerscience
Publishers, New York, 1955.
3. J. E. Maye r, M. G. Mayer, Statistical Mechanics, Ch. 11, John Wiley
a. Sons, New York, 1940; R. Fowler, E. A. G u g g e n h e i m,
Statistical Thermodynamics, Ch. IV, Cambridge University Press, Cambrid-
ge. England, reprinted 1956.
286
Приложение Б
ПРИЛОЖЕНИЕ Б
КОНФИГУРАЦИОННАЯ ЭНТРОПИЯ ГАЗА
Рассмотрим два сосуда с объемами и V, окруженных тепло-
непроницаемой оболочкой и соединенных трубкой. Поместим в
эту систему N молекул идеального газа и выясним, какова ве-
роятность нахождения N молекул в объеме (состояние 1),
н сравним с вероятностью нахождения N молекул в объеме
=У2 (состояние 2). Вероятность присутствия любой одной
молекулы в объеме Pj определяется выражением: Вероят-
ность Р, определяющая присутствие всех N невзаимодействую-
щих молекул в объеме У1, определяется выражением
(Б, 1)
Вероятность Р можно также рассматривать как отношение
числа микросостояний (конфигураций) молекул в состоянии 1
к числу мпкросостоянпй (конфигураций) В2 молекул в состоянии
2. Так как все конфигурации (или микросостояния или комплексии)
равновероятны, то
N
р „ —L
-
(Б, 2)
Из принципа Больцмана следует, что разность энтропий
газа в этих двух состояниях равна:
о у
S, — S2 = k in •— = Nk In -ту- (Б, 3)
--2 l2
Отсюда легко можно получить уравнение состояния идеально-
ю газа.
Точное выражение для В как функции U и V для «классиче-
ского» одноатомного газа имеет вид [11:
==go
V \N / , mU \3N/"
N ) 31 AV )
(Б.4)
где tn—масса молекулы, go—степень вырождения основного со-
стояния, U—внутренняя энергия, являющаяся функцией толь-
ко Т
Литература
1 J. Е. Mayer, М. G. Mayer, Statistical Mechanics, John Wiley a.
Sons, New York, 1940, p. 116.
Приложение В
287
ПРИЛОЖЕНИЕ В
УРАВНЕНИЕ СОСТОЯНИЯ МОЛЕКУЛЯРНЫХ КРИСТАЛЛОВ
Выражение для свободной энергии кристалла можно записать
следующим образом 1см. уравнения (А, 8) и (А, 10)1:
Д = {7ДУ) + Д„(Т,У) (В.1)
где Ad(T, У) представляет собой свободную энергию колебаний
кристаллической решетки в дебаевском приближении.
В дебаевском приближении из уравнений (А, 6), (А, 8) и
(А, 10) следует, что AD(T, У) равна температуре Т, умноженной
на функцию от 0/Т, где 0—дебаевская температура, определяе-
мая как h»„Jk, а зависимость AD от объема может быть
через зависимость 0 от объема:
Давление определяется формулой
dli,(V) / dAD \ / ае \
р = dV \Sdjr, v\ dv /т
Используя уравнение (В, 2), можно получить 111:
(MD\ d(AJT) id(ADIT) }
\ aw /r.v [ a(i/T) Je. v
Из известной термодинамической формулы следует,
,,
д(1/Т) |у г’
учтена
(В, 2)
(В, 3)
(В, 4)
что
(В, 5)
Таким образом, используя уравнения (В, 3)—(В, 5), получим:
dUL (V) yUD
dV ’’ V
(В, 6)
где UD—внутренняя энергия, обусловленная колебаниями ре-
шетки в дебаевском приближении, у—константа Грюнайзеиа,
определяемая формулой
/ a in а \
т= a in u /j.
(В, 7)
Предположим, что константа Грюнайзена практически не за-
висит от температуры. Кроме того, из соотношения между дебаев-
288
Приложение В
ской температурой н модулем всестороннего сжатия [глава I,
уравнения (5, 11) и (5, 14)1 следует, что
о —
(В, 8)
откуда, используя уравнения (В, 8) и (В. 7), легко видеть [21
что
2 1 . / д"Р \ I , дР '
Y = ~ 3 2 1 \dVi')r[ ( dV j-.
(В, 9)
Далее предположим, что константу Грюпайзена можно опре-
делить с достаточной точностью при всех температурах, вычис-
ляя (д2р1д\г2)т и (dpldV)r при Т =0 °К. Для этого нужно исполь-
зовать уравнение (3, 8) главы I. Оказывается, что отношение
\'(diPldV2)l(dPldV} = —9. Следовательно, константа Грюпайзена
для молекулярного кристалла равна:
у = 3,83
(В, 1и>
Подставив эту величину и уравнение (3, 8) главы 1 в уравне-
ние (В, G), получим новое уравнение:
Р = 37,64/,* | (-£ ) ’ - ( 3 у 3,83 |
(В, 11)
Функция Дебая U D—элементарная функция, которая табу-
лирована для всех значений О/Т. Здесь 0 рассматривается как
константа (см. раздел 5 главы 1). Для молекулярных кристаллов
температура Дебая дается уравнением (5, 17) главы I.
Такое же рассмотрение может быть сделано в приближении
Эйнштейна, при котором в уравнении (В, 11) UD заменяется на
UЕ. Все математические соотношения, приведенные в этом при-
ложении, могут быть применимы как в приближении Дебая, так
и в приближении Эйнштейна.
Зависимость объема от температуры при нулевом давлении
можно получить из уравнения (В, 11), если положить р=0.
Дифференцируя уравнение (В, 6) по Т при постоянном Г получим:
/ Др \ _ I / Др \ Ц/JVX I уСу
\гг)у~ Ц др ~
следовательн о
(В, 12)
(В, 13,1
где а—коэффициент объемного расширения [\IV)(dV 1дТ)р,
В—модуль всестороннего сжатия и С\,—удельная теплоемкость,
Приложение Г
289
Так как у и В почти не зависят от температуры, то а изменяется
с температурой таким же образом, как и Cv.
Если у=3,83 и В =Во=1,07-102 t*/r*3 [из уравнения (3, 10)
главы I], то получим:
(-jjr \ = 3,58-10~2 — Cv/ (В, 14)
' ' р е
Интегрируя уравнение (В, 14) при постоянном (нулевом)
давлении, получим:
Т(П - V'o(l |-3,58-10~a-= <в- 15)
Уравнение (В, 15) для молекулярного кристалла полностью
определяется величинами е* и г*, так как [из уравнения (3,7)
главы II Vo равно 0,65 NAr*3, a UD—универсальная функция 770,
причем 0 определяется из уравнения (5, 17) главы 1. Таким обра-
зом, мы можем записать:
/ Т \ UL) (Т/Щ
r Hr = 1 +5 ’55,10~2
(В, 16)
При больших величинах Т/0 величина UD равна 3NAkT. Пре-
дельная величина коэффициента расширения при высоких темпе-
ратурах (большие 770) приблизительно равна:
а = 0,167 (В, 17)
Литература
1. Ch. Ki t 1 е I, Introduction to Solid State Physics, 2nd ed., John Wiley
a. Sons, New York, 1956, pp. 153, 154. [4. К п т т e л ь, Введение в фи-
зику твердою тела, 1957].
2. J. С. Slate г, Introduction to Chemical Physics, McGraw-Hill Book Co.,
New York, 1939, pp. 238 and 239.
ПРИЛОЖЕНИЕ Г
НАИВЕРОЯТНЕЙШИЙ КВАДРАТ ДЛИНЫ ЦЕПИ УГЛЕВОДОРОДНОГО
ТИПА ]1]
Вычислим нанвероятнейпшй квадрат расстояния между кон-
цами случайным образом изогнутой цепи углеводородного типа,
состоящей пз X звеньев (связей), которые определяются векто-
рами (Х<!>, ЕЮ , Z1'!), i—0, 1, 2, 3, ..., N—1. Каждый вектор
представлен одной из восьми комбинации (±1; ±1; ±1), и по-
следовательные звенья отличаются друг от друга только одним
знаком [21. Для упрощения вычислений средняя длина компонент
19—833
290
Приложение Г
векторов принята за единицу; в конце вычислений мы учтем, что
действительные средние длины компонент равны IJ31!2. где /0—дли-
на звена.
Вероятность того, что у одной н той же координаты происхо-
дит два последовательных изменения знака, так что (Х(‘+2<,
уи+2), /(/+2)) идентична с (Х(‘>, У(‘>, Z(/>), равна а; повторное
измененне знака имеет место у одной из двух других координат
вектора с вероятностью (1—а)/2. Эти три случая соответствуют
транс- и двум аош-конформациям. Нанвероятнейшая длина цени
углеводородного тина, состоящей из N звеньев, была найдена уже
давно Энрннгом 131 при условии, что энергии, а следовательно,
вероятности всех трех конформаций одни и те лее. Это соответст-
вует особому случаю, когда о =1/3. Мы попытаемся дать общее
решение 111.
Можно выбрать такую координатную систему, что
Г(0), /к»)) =(1,1,1) ц (ХИ), yd), Z‘»)=(— 1. 1, 1). Следующее
звено имеет такие возможные координаты: (1, 1, 1), (—1, —1, 1»
и (1, —1, —1) с вероятностью а, (1—сг)/2, (1—а)/2.
Вычислим наивероятнейший квадрат длины, который обозна-
чим через EN:
Е {(Х(0> 4---р x(N-H)- ? у(Л' -Ц)2
N—2 Л-1
. ( j . .. । z(,v~-|))2| = з,у-i-2 Y V i; i-z*'*.
!=•-<> j I -1-1
Л'—2 N— 1—i
3iV r2Y Y (xl”>x(fc) I- У1<)) Y{f:}(Г. I)
i=-u k=a
Пусть
Lk = E p Z(°)Z<A>} (Г 2|
113 полученных результатов следует, что
Г - = 3 Li = 1 L,— 1 Д- (Г, 3)
В дальнейшем сЛ,=ЗД' | мы может упростить выражение N--2 N— 1— i N— 1 Л —1—i 2 Y У l-k ЗА I- 2 У У /ч. Z—о д>=; i=o ч=о N—1 = —ЗЛЧ-2 \(Л- k)Ll{ для Е (Г. ||
fc-0
Пусть (Л'<°), I'W, Z'°>)=(1, 1, 1) и
Тогда
Li = E (Uf)
Приложение Г
291
Переменная U(V> может иметь четыре значения ±3, ±1. Рас-
смотрим последовательность пар (U0U^, (UXU^, (U2U3),
(IJ.... Un). Каждый член этой последовательности
есть состояние с возможными значениями (3, 1), (1, 3), (1, - 1),
( 1, 1), ( 1, —3) и ( -3, — 1). Последовательность пар есть цепь
Маркова 141 со следующей матрицей Р вероятностей перехода:
3 1 1 3 1—1 11 1 3 3 — 1
3 1 0 а 1 а О 0 0
1 3 1 0 0 0 0 0
р 1 1 0 0 0 (1 1 «)/2 (1 «)/2 0 (Г, 5)
1 1 (1 (1 - rt)/2 (1 + «)/2 О 0 (1
- 1 3 0 0 0 0 0 1
— 3 1 0 0 0 1 а а 0
.Матрица вероятностей перехода, соответствующая переходу
О-*/ в последовательности, обозначена через Р‘. Если первый ряд
Pir> имеет элементы /4'?> ’ РЙ> то вероятность того, что
б/('"’=3,1,—1,—3 равна’ соответственно р\‘>, p’/l+P1/’- РтН'Р’а-
р(/,! и, следовательно
P (Pi'l I-Pi3 /’и’ р',^
.Метод выражения элементов pi'z! матрицы РИ) через извест
пые элементы р(7с матрицы Р имеется для общего случая цепи
.Маркова в книге Феллера [41.
Эти решения выражены через собственные значения п соб-
ственные векторы матрицы Р. Матрица Р [уравнение (Г, 5)1
имеет три собственных вектора симметричной формы (х, у, г,
2, у, д'), которые не дают вклада в Л;, и три собственных вектора
антисимметричной формы (х, у, г, —г, --у, —х). Это значитель-
но упрощает вычисления. Собственные значения, соответствую-
щие антисимметричным собственным векторам, равны —1, Q'
и Q", где Q', Q" корни уравнения:
(Г, 7)
Величины полученные методом, рассмотренным выше
(см. 141), выражены через Q’ и Q". Если их подставить в урав-
292
Приложение Г
нение (Г, 6), то получится следующее выражение:
11 I — За .]
С = 13 (Q")'+l — 3 (Q'),+1 ~ (<2")'-(<2')' =
DQ - (3Qi + 1 ~ Q‘ ) ” ’ 8)
Из этого уравнения ясно видно определение оператора D.
Напомним, что выражение для E.v coj Л'—1 Е (N—k)Lk. Заметим, что А-0 М-1 V ь 'V Q'v+1 Q 7j(A - k)Q - j Q + (1 др ([_( A---0 Следовательно 2N 1 3Q 1 — За 1 (ержало член ?)2 (Г. 9) i- (Г, 10) (Г, 11»
+ ~DQ ll ~~~DQ ~ Чтобы вычислить OCHOF 1 DQ ~ 3QV+ (1 - Q ( 3Q2 2 1 - 3o 2 • 1 3o 2 ' 1 — Q 0,v+^ (1 - Q)2 Q ‘ (1 -Q)2 им, что
| (I - Q)2 2 шоп член, замет 1 1
-1 Q D Q D - + { Q" 1 Q’ 1 a DQ
- 1 — Q Откуда n ( 3Q 1 - 30 1 '• 1 - Q L 1 1 3«1 II. 1 - J — a DQ& -3a) . „ 13) а |Г-
- 4 Q 2 ’ 1 - Q / Таким образом ЗЛ' 1 —« V’ " 2 , 5 }-3«)УУ (2 -p 6< 1 — a 1
Точную наивероятненшую длину можно получить путем
дальнейшего преобразования уравнения (Г, 10):
(2-;-&,)(У 9«2+30о —7 2 £ .,+2 7(1 — За) .
К 1—о ~ 2(1—о)2 - a)*'DQ 'iQ ~ 2 'г
. 5(1 - 3u)2QA (1 - 3o)3Qv-' :
+-------4-----—----------Я------I ,Г- lt>>
Приложение Г
293
Вспомним, что длина каждой компоненты, которую для упро-
щения приняли за единицу, в действительности равна /0/31/2-
Выражение для на и вероятнейшего квадрата расстояния между
концами цепи которое очень точно соответствует действитель-
ности для больших значений N, получается из уравнения (Г, 14k
(2 + 6О)М2
3(1 о) ’
Если энергия (или более точно, свободная энергия) транс-
конформацин принята за нуль и энергия каждой из двух гош-кон-
формацнп принята за е (на моль), то величина а определяется
формулой
1
" 1 + 2 ехр (— s/RT) <г- 17)
Следовательно
-12 4 1
г1 "3" 'S' T ехР (e/R7’> Nl» Г.
Если г равно нулю, то а равно‘/3 и уравнение (Г, 16) сводится
к уравнению Эйринга 13]:
2Л72 г. о (Г, 19)
Если г//^Т очень велико и положительно, т. е. гош-конформации
почти исключаются, то а приблизительно равно 1—2ехр(—s!RT)
и мы получаем следующее асимптотическое выражение для г2:
- I 4 I , г
'2 = ~з~ exp (./RT) Nl* > 1 (Г. 20)
Если sjRT велико и отрицательно, т. е. /прпнс-конформация поч-
ти исключена, топ приближается к нулю и мы получим следующее
выражение для г~:
-2 г
г- з Л/(5 gj, ,1 (Г, 21)
Учет исключенного объема при таком вычислении не произ-
водится.
Уравнение (Г, 18) может быть выведено из простых рассужде-
ний, развитых Тейлором 151 и Волькешлтейиом 16]. Метод, рас-
смотренный в этом Приложении, дает, однако, полное и точное
решение задачи, которое также справедливо и для коротких пе-
ней—см. уравнение (Г, 15) II].
Волькенштенн дал очень интересные формулы для г2 в случае
изотактических п синдиотактических цепей 171, а также для
коротких цепей 161.
294
Приложение Д
. Литература
1. A'-'V Го bo 1 sky, J. Chem. Phys., 31, 387 (1959).
2. A. 1 V. Tobols к у, R. E. Powell, H. E у r i n g, Ch. 5 in «The
CJipmistry of Large Molecules», R. E. Burk and O. Grummitt (ed.), Inter-
science Publishers, New York, 1943. [А. Тобольски й. P. П a y-
ilu'ji', Г. Э й p и н г, Химия больших молекул, Сборник № 2. Издат-
инлит, 1948, стр. 2061.
3. Н. Е у г i n g, Phys. Rev., 39, 746 (1932).
4 \V. Feller, Au Introduction to Probability Theory ind its Applications,
v. 1, 2nd ed., John Wiley a. Sons, New \ork, 1957, pp. 338 319, 380-
; 385.
5. W.v J. Taylor, J. Chem. Phys., 16, 257 (1948).
6. M. В, В о л ь к e и ш т e й u, ДАН СССР, 78, 879 (1951); ЖФХ. 26, 1072
‘(1952).
7. M. В. В о л ь к е н ш т е й п, J. Polymer Sci., 29, 441 (1948).
1
ПРИЛОЖЕНИЕД
. 1ДУССОВО РАСПРЕДЕЛЕНИЕ РАССТОЯНИЙ МЕЖДУ КОНЦАМИ ЦЕПЕЙ
Рассмотрим,свободносочлененную цепь из N сегментов. Дли-
на каждого из них равна /0. Какова вероятность 1Р(х) того, что
вектор, соединяющий концы, имеет компоненту х вдоль произ-
вольного направления, выбранного в качестве оси х? Данная за-
дача является одномерной. Рассмотрим случайное блуждание,
состоящее из N шагов в одном измерении. Каждый шаг имеет
величину ±1(1/31н> причем знаки «плюс» и «минус» равновероят-
ны. Вероятность того, что будет сделано N+ положительных ша-
гов и АД отрицательных равна:
<л"
при условии, 4TQ
Д+ | AL Л (Д.2)
Для .удобства введем величину т:
(Д, 3)
Расстояние х, достигаемое в конце блуждания, равно:
' : \ (A+~W)/0 mi
\ ‘Я, 4)
Уравнение (Д, 1), выраженное через т, запишем следующим
обрдзом:•
(l/2)w AI
Г (А, т) = [(yv + ш)/2|![(А — м)/2|! (Д’ 5)
Приложенис Е
295
Используя в уравнении (Д, 5) приближениеСтирлингд.ддя боль-
ших величин /V!=^(2rr)1/2 AW-Vn/gX п учитывая условие. mtN<^\,
получим:
/ 2 \ '/л
IV' (TV. т) ~ I —тт ) exp (— nr/2N) (Д, 6V
Установим соответствие между W(m, N) и 1Р(х), причем по-
следняя функция означает вероятность того, что расстояние,
достигаемое после N шагов, равно х. А1ы предполагаем, что
П7(х)—непрерывная функция, чтобы использовать ее для реше-
ния молекулярной задачи, поставленной вначале этого Прило-
жения. По определению имеем:
Г (х) =
W (т, А)
Ах
(Д, 7)
Заметим, однако, что наименьшая величина Am равна 2 и
величина Ах, соответствующая Ат =2, равна Ах=2/о/31/2. Под-
ставив эту величину Ах в уравнение (Д ,7) и заменив т==3,/2х//0
в уравнении (Д, 6), получим следующее выражение [1, 21:
/ 3 \1/з/ 1 \
W(x)dx - \7jW)exv(~ 3*V2W/o)rfA (Д. 8)
Любая реальная цепь с заторможенным вращением, со связя-
ми, ограниченными валентными углами, и т. д. может быть при-
ближенно заменена эквивалентной цепью с Ne свободно вращаю-
щимися сегментами, каждый из которых имеет длину 1е (см. раз-
дел 1 главы И). Уравнение (Д, 8) можно также обобщить для
трех измерений х, у, z\
/ ь V
IV (х, у, z) dxdy dz - ( Ji/5) exp]—ba(x2 i - tp |- z2)] dx dy dz (Д, 9)
3 3
b~ 2^ •’
Литература
I E. Gut li, II. M ark, Monatsh., 65, 93 (19341. 1
2 W. К ii h n, KrH|.-Z., 68, 2 (1934).
ПРИЛОЖЕНИЕ E
РАСПРЕДЕЛЕНИЕ ДЛИН МОЛЕКУЛ В ЛИНЕЙНЫХ ПОЛИМЕРАХ
Размеры линейной молекулы полимера можно выразить через
число связей в цени, через степень полимеризации или через
молекулярный вес полимерных молекул.
296
Приложение Е
В природе многие белковые молекулы, например инсулин,
имеют точно определенный молекулярный вес. Систему полимер-
ных молекул одного п того же молекулярного веса называют мо-
нодисперсной; систему полимерных молекул с множеством различ-
ных молекулярных весов называют полидисперсной.
Все синтетические полимеры полндисперсны, так как в действи-
тельности всегда образуется непрерывное распределение молеку-
лярных весов. Иногда это распределение молекулярных весов
очень узко, и синтетический полимер приближается к монодиспер-
сиому.
Для определенности выразим распределение размеров через
степень полимеризации, хотя можно также использовать число
связей в цепи или молекулярный вес. Двумя основными функ-
циями являются Х(л)—молярная доля полимера со степенью по-
лимеризации х и Wz(x)—весовая доля полимера со степенью по-
лимеризации х. Обе эти функции нормированы к единице.
Часто, встречающиеся распределения молекулярных весов
определяются так называемыми случайными распределениями:
X (х) = px~l (1 — р) (Е, 1)
IV' (х) pxl (1 --- р)~ IЕ, 2)
В уравнениях (Е, 1) и (Е, 2) величина р является параметром,
который определяет распределение и величина которого лежит
между нулем и единицей. Это распределение по закону случая
встречается при деструкции полимеров с большим молекулярным
весом 111, при случайной бифункциональной поликонденсации
12] и при случайных реакциях обмена |2|.
В процессах полимеризации винпльпых соединений, где не
присоединение, а передача цепи и диспропорционирование ре-
гулируют молекулярный вес, распределение по закону случая
указывает на то, что полимер получен в очень узком интервале
превращений [3]. Величина р может изменяться в зависимости
от глубины реакции, и поэтому распределение, образовавшееся
в полимере при достижении некоторой глубины реакции, полу-
чается в результате интегрирования уравнений (Е, 1) и (Е, 2) по
соответствующим значениям р.
В реакциях полимеризации винпльпых соединений, в которых
молекулярный вес регулируется сочетанием растущих радика-
лов, полимер, полученный в очень узком интервале превраще-
ний, характеризуется следующими комбинационными распреде-
лениями 14, 51:
Х(л)-лр'1(1 ~р)2 (£. 3)
(1 — р)3
Г(х) = х*р‘ I -ТфДГ" ^.4)
Приложение Е
'297
В указанных выше уравнениях полимер RA1X_1R определяется
как х-мер (R—инициирующий радикал и М—полимеризующийся
мономер). Здесь р также Может изменяться с глубиной реакции, и
поэтому окончательное распределение может быть гораздо шире.
Распределение Пуассона получают 16] при образовании поли-
мера присоединением мономера к инициирующей молекуле в отсут-
ствие реакций обрыва. Оно имеет вид:
е ~zx 1
х W = (х— 1)7
(£. 5)
(Е,6)
z г12
№ 17=7)1
В указанных выше уравнениях z—число мономерных звеньев,
прореагировавших с молекулой инициатора; при определении х
остаток инициатора принимают за единицу.
Распределение Пуассона получено для случая полимеризации
циклических соединений, например окиси этилена, полиме-
ризация которой инициируется этиленгликолем [71. Оно имеет
место также и для ванильной полимеризации, если отсут-
ствует реакция обрыва цепи, т. е. для синтеза так называе-
мого «живого полимера», в котором используются анионные ини-
циаторы [8]. Некоторые выводы этих распределений приведены в
работе [91.
Двумя важнейшими средними величинами, характеризую-
щими распределение макромолекул по размерам, являются
средиечпсловая степень полимеризации Рп и средневесовая
степень полимеризации Pw, которые определяются следующим
образом:
Р„ . .vxX(x)-.-4^- Щ,7>
Р№ =- ХхГ (х) -уух~(-ху- 'Е. 8)
Подобные определения используются для среднечислового
молекулярного веса Мп, средневесового молекулярного веса Mw,
среднего числового значения числа звеньев в цепи Nn и средпеве-
сового числа звеньев цепи Nw.
Для ряда монодисперсных полимергомологов используется
следующее эмпирическое соотношение, устанавливающее связь
29Н
Приложение Е
между характеристической вязкостью раствора и размером мо-
лекул х:
[Д = Кх‘‘ (F, 9)
где а лежит в интервале от 0,5 до 1,0.
Если это соотношение применить к полидисперсному полиме-
ру. то получим [101 средневязкостиую степень полимеризации Р.:
Аналогичное определение дается н для средневязкостного мо-
лекулярного веса Mv. Очевидно, если п=1, то Fv=Pa. В общем
а приблизительно равно 0,7, и поэтому Pv лежит между Рп и Р..
причем ближе к Ра.
Определения Ра и Ри, данные в уравнениях (Е, 7) и (Е, 8).
можно применить к различным ранее упомянутым распределениям.
Для случайного распределения:
г'п' 1 — р 1 + р Pw -=г~^ - 1 — р Р,„ -= 1 г- Р Р г п Для распределения, определяемого уравнениями (I-.. Н) (Е. 12) (Е. 13) (Е, 3) и
(Е, 4). получим: /3 L+a ' « “ 1 — р р.. 1 , 1 - р- Д, 1 + 1/> б Р2 рп (• 4 РУ- (Е, 14) (Е. 15) (Е. 1ь>
Для распределения Пуассона: Рл г -4- 1 (Е, 17)
_ 4 Рп • U + 42 (Е. lb, (Е, 19)
Приложение Е
299
Величина PwtPn, которую часто называют показателем гете-
рогенности, определяет ширину распределения. Для монодисперс-
ного распределения P^fPn равно единице; чем больше PwlPn,
гем шире распределение. Распределения, рассмотренные здесь,
не очень широки. Максимальное значение Р^/Р„ для случайного
распределения равно 2,0 (когда р=1); для комбинационного рас-
пределения максимальное значение Р^/Р,, равно 1,5 (когда р=1).
Распределение Пуассона является значительно более узким рас-
пределением: когда z=99, Pw/Pn =1,0099, а для больших величин
z, PW‘Pn еще меньше. Полимеры с таким распределением иногда
называют «монодисперсными».
Очень широкие распределения можно получить в случае ви-
нильной полимеризации в присутствии агента переноса цепи для
полимера в целом, а не для полимера с узкой областью превраще-
ния.
Ниже мы выясним, что процесс переноса цепи на модифика-
тор S (с постоянной переноса с) является единственной важной
стадией, контролирующей размеры молекул при полимеризации
винилового мономера М [11]. Начальные концентрации равны So
и /У1о, концентрации во время полимеризации—S и М, доля пре-
вращения у равна (7И0—
Величина Р'п для полимера, полученного немедленно, опреде-
ляется следующим образом:
1 /л1\
р'‘ — ( т) <Е-20)
Гак как быстро полученный полимер имеет случайное распре-
деление, то Р'а, приближенно равно 2P,'t:
Концентрация .S определяется 1121 формулой:
(Е, 21)
(Е, 22)
Полное значение Рп и Рш дастся следующими уравнениями:
Л10
Мо
(Мо М) Ри, -.=. j PudM (Е, 24)
М
Зои
Приложение Е
Подставляя значения Р’п и Р'а, в уравнения (Е, 23) и (Е, 24л
н исключая S с помощью уравнения (Е, 22). получим 111) сле-
дующие выражения:
(M0/S0)y Мо-М
- 1 _ (1 _ у)с • Л10 <| -а»
- (2M0/cS0)|l-(l -i/ГЧ
=-------7(2^7)--------- <fc- 2Ь»
Показатель гетерогенности для полимера равен (для с - 2)\
Pw 2 1 — (1 — (1 —
Рп - с (2- г)’ .г (
Такой частный вид распределения специально применим к по-
лимерам винилового и диенового типа с низким молекулярным
весом, полученным полимеризацией в присутствии заметных ве-
личин активного модификатора. Эти вещества имеют особое зна-
чение как способные к литью жидкости, которые можно превра-
тить в каучуки (131.
Если модификатор имеет константу переноса цепи, равную
единице, то показатель гетерогенности будет иметь величину.
равную двум, независимо от доли превращения у. Если эта кон-
станта заметно отличается от единицы, то полимер будет иметь
показатель гетерогенности, равный двум ври нулевой доле пре-
вращения и далее увеличивающийся по мере повышения доли
превращения до единицы, что указывает на возрастающую шири-
ну распределения.
Лр чич примером случая широкого распределения является
n<Ai >< .Л<_р. IHaTj ЧйЮШИИСл iipi. * м’ЛоГ сг.пОь Ьх.г.ЛЛ:.-
пых мономеров в условиях истощения катализатора. Этот случай
рассматривается в теории полимеризации, заканчивающейся обра-
зованием малоактивных радикалов 1141.
Литература
1. \\ . К п h и. Вег., 63, 1503 (1430).
2. Р. .1. Flory, J. Am. Chem. Soc., 58, 1877 (1936); 64. 2205 (1942)
3. G. V. Sc h u I z, Z. Phys. Chem.. B30, 379 (1935).
4. G. V. Schulz. Z. Phys. Chem., B43, 25 (1939).
5. B. Bay sal, A. V. Tobolsky, .1. Polymer Sci., 9, 171 (1952).
6. fl. Dostal, H. M a r k, Z. Phys. Chem., B29, 299 (1935).
7. P. J. Flory, J. Am. Chem. Soc., 62, 1561 (1940).
8. R. Waack, A. Reinbaum, J. D. С. о о in b e s, Al S/warc.
.1. Am. Chem. Soc., 79, 2026 (1957).
9. P .1. Flory, Principles of Polymer Chemistry, ch 8, Cornell University
Press, Ithaca, New York, 1953.
К). P. .1. Flory, J. Am. Chem. Soc.. 65, 372 (1943)
I 1 - A \ I о b о 1 S k у. R. C. belles. D. 11 J о I; ii *. <i i; pri ilcel< >n
Univ. Plastics i.ab. Tech. Repl . 17A (1950)
Приложение Ж 301
12. W. V. Smith, J. Am. Chem. Soc., 68, 2059 (1946).
13. V Conwell, G. P. Roeser, A. V. Tobolsk}. J. Polymer
Sci. 4, 309 (1949).
14. A. V.’ Tobolsk}', .1. Am. Chem. Soc., 80, 5927 (1958)
ПРИЛОЖЕНИЕ Ж
АФИННОЕ ПРЕОБРАЗОВАНИЕ СФЕРЫ
Рассмотрим сферическую поверхность, определяемую уравне-
нием
л-а ; С/2-1 г~ (Ж, I)
Далее, пусть эта сфера подвергается афинному преобразова-
нию:
х' = ах у' --- а~х^гу г' = оС^г (Ж, 2)
Сфера становится поверхностью эллипсоида:
х'2/а2 ау'- -р az'3 = г2 (Ж, 3)
Найдем среднюю величину г'2 =д:'2 -рз'2. Для этого пре-
образуем х, у, z и х', у', z' в сферические координаты:
X = гй cos В
у = гй sin В sin Ф z r() sin 0 cos Ф ' г' - arG cos В у' = а ''-г0 sin В sin >1 z' =. a l/2r1) sin b cos < r'~ -= .C2+ z/'2 + z'~ a 2rjj COS2 в -L a Чтобы найти среднюю величину г' считая их равновероятными: г. г'2 = (a2/-2 COS2 В р a 1 О = ~Г2 j | < 1 1- (а2 - 0 1 = 4" с; ра 1 : (а2 —а (Ж. 4) ’ (Ж, 5) sin2 В 2, учтем все ориентации, 0; sin2 В)2л sin В dH = a J) cos2 в] d cos В =. •)x2|dx (Ж, 6)
302
Приложение 3
или окончательно:
(Ж. 7)
ПРИЛОЖЕНИЕ 3
УРАВНЕНИЕ СОСТОЯНИЯ ПОЛИМЕРНЫХ ЦЕПЕЙ И СЕТОК,
УЧИТЫВАЮЩЕЕ ВЛИЯНИЕ ЭНЕРГИИ И ЭНТРОПИИ
Рассмотрим одномерную цепь, сегменты которой имеют оди-
наковую длину х0 и ориентируются либо в положительном, либо
в отрицательном направлении. Когда последовательные сегменты
ориентированы в противоположных направлениях, то будем счи-
тать, что возникает взаимодействие, которое измеряется энергией
взаимодействия s' (на моль). Нулевая энергия приписывается
конформации, состоящей из последовательности сегментов, имею-
щих одну и ту же ориентацию, т. е. растянутой форме. Если Е
отрицательно, то наблюдается взаимодействие и, следовательно,
преобладает свернутая форма цепи. Если е' положительно, то
свернутые конформации не имеют предпочтения и преобладает
вытянутая форма цепи. Статистическое рассмотрение такой цепи
было сделано одновременно Скоттом и Тобольским [1, 2], Гут-
том и Джемсом 13L
В какой степени физическая реальность может быть связана
с этой одномерной моделью? Было указано [2], что эта модель мо-
жет находиться в соответствии с цепью углеводородного типа,
где транс- и гош-конформации имеют неравные энергии, причем
энергия транс-конформации принята за нуль, а энергия двух
гош-конформаций—за е. Этот тип цепи был рассмотрен в Прило-
жении Г, где было показано, что среднеквадратичное расстояние
г2 равно:
-12 4 1
/2 = ~ ехр (г//??)\Nlt (3.1)
Для среднеквадратичного значения х2 проекции углеводе»
родной цепи на ось х, имеем:
г 2 4 I l
& = I -3- exp (HRT) А ~~ (3.2)
Для одномерной цепи, которая рассматривается в этом разде-
ле как аналог углеводородной цепи, можно легко показать при
помощи методов, рассмотренных в Приложении Г. что
№ = [ехрб77?7’)1 Nxn (3. 3)
Приложение 3
303
Следовательно, две модели аналогичны, если
2 4
-5- Ь -3-exp P/RT) = exp H'/RT)
(3, 4)
(3, 5)
ll
Одномерную модель можно также использовать Дди таких
молекул, как белки, у которых наблюдается внутримолекулярная
свернутость, обусловленная водородными связями между соседни-
ми функциональными группами цепи.
Перейдем теперь к выводу уравнения состояния Цепи из N
сегментов, в которой Р сегментов имеют положительное и Q
сегментов— отрицательное направление. Для удобства математи-
ческого рассмотрения предположим, что два конечных сегмента
имеют противоположные направления и что Р>(}. Длина цепи
равна:
L — (Р - Q) — Rx'jj
Чтобы удержать цепь при растяжении L (т. е. при фиксиро-
ванной величине Р и Q), требуется приложить растягивающую
силу X. Величину X можно найти из уравнения:
kT / dlnZ \ kT (д In Z/dR)T
\ dL / т — л0
(3,7)
где Z—статистическая сумма состояний цепи.
Различные макросостояния, соответствующие фиксированным
величинам Р и Q, можно определять при помощи параметра /.
Этот параметр определяется как число последовательностей поло-
жительных сегментов в данной конформации цепи. Так как каж-
дая последовательность положительных сегментов следует за
последовательностью отрицательных сегментов, то 1 есть также
число последовательностей отрицательных сегментов.
Например, в приведенном ниже случае величины /, Р и Q
равны 5, 18 и 12 соответственно:
1- 4- + 4-4-4 1 + -I---------1---Р 4' 4“ 4- 4~ л-4“ 4--------
Полное число взаимодействии (изменений от положительных
к отрицательным последовательностям и изменений от отрица-
тельных к положительным последовательностями очевиДно> Рав-
304
Приложение 3
но (2/—1). В приведенном выше примере число взаимодействий
равно 9.
Энергия (на моль) любой данной конформации цепи равна
s'(27—1). Предположим, что Р, Q и 1 очень велики и что в по-
следующих уравнениях можно пренебречь единицей по сравнению
с Р, Q и I.
Число комплексий IVZ(7) макросостояпия, соответствующего
фиксированной величине I, определяется формулой
__у — _____V/ — ч-_
-- (/ - 1)! (Р1)! (Q - /)! -
ЛУ+ R\ /N — R\ / [N~P R . [N — R ,\ , ,
= (—) ! (—2~~J । / “'J ! (“Г- <3-8*
Из уравнения (3, 8) следует, что W(I) есть число способов
разбивки Р сегментов на / групп, каждая из которых содержит
по крайней мере один сегмент, умноженное на число способов
разбивки Q сегментов на I групп, причем каждая группа содер-
жит по крайней мере один сегмент.
Статистическая сумма состояний цепи Z определяется сум-
мой членов, соответствующих всем возможным величинам I:
N/2
Z = X W (/) ехр (— 2е7/ЯТ) (3. 9)
/=1
Только максимальный член этой суммы, соответствующий ве-
личине 1=1й, вносит заметный вклад в эту сумму. Величина /0
находится дифференцированием по I общего члена суммы (при
постоянном R) и приравниванием нулю полученного результата.
Для вычисления факториалов в уравнении (3, 8) используется
теорема Стирлинга. Величина /0 определяется пз соотношения
Отсюда Z определяется выражением
Z ~\V (/0) ехр (- 2PIJRT) (3,11)
Из уравнений (3, 7), (3, 8), (3, 10) и (3, 11) можно получить
уравнение состояния [I, 2, 3], справедливое для малых величин t:
kT ~ ц___ехР ( /RT) ~ t ехр (— .’/RT) (3, 12)
N ~ Nx0
Приложение 3
305
Для большинства значений е'/RT уравнение (3, 12) можно
приближенно записать следующим образом:
X = kT ехр (— t'!RT) (—
\ xo
kTL
x-
(3, 13)
На рис 3, 1 показаны зависимости напряжения от температуры
при постоянном /(/=0,1) для е'=0,е'<0 и е'>0, построенные как
функции величины ±NЛХхо1г от ±RTls .
Рис. 3, 1. Зависимость силы от температуры для
/=0,1 и е'=0, е'>0, г'<0 согласно уравнению
, (3, 12)* [К. W. Scott, А. V. Tobolsky.
J. Coll. Sci., 8, 465 (1953)].
Очевидно, что температурная зависимость напряжения в сет-
ке каучука при постоянном относительном растяжении может быть
такой же, как зависимость силы от температуры для цепи при пос-
тоянном t. Имея это в виду, были сделаны измерения напряже-
ния в зависимости от температуры для сшитых бутилкаучука
[4] и лактопренкаучука [5]. Результаты измерений исследо-
вались с помощью уравнений (3, 12) и рис. 3, 1, чтобы вы-
яснить, имеются ли отклонения от кинетической теории высо-
коэластичности [6]. Было найдено, что в этих каучуках напряже-
ние при постоянном удлинении прямо пропорционально абсолют-
ной температуре в температурной области каучукоподобного
состояния. Это находится в согласии с прежней кинетиче-
ской теорией высокоэластичности, для которой г' =0 (см. рис. 3, 1)
и показывает отсутствие разности энергий между изогнутыми
и вытянутыми конформациями.
* При е'==0 по осп ординат отложена величина, пропорциональная NAXxh.
а по оси абсцисс величина, пропорциональная ЦТ.—Прим. ред.
20--833
306-
Приложение 3
Конфигурационная свободная энергия Ае цепи может быть
получена из выражения для X, данного в уравнении (3, 13).
Г / 2Ь2/.2 \
Ас = I X dL = kT ( —3—)
1 27/2 \
. ,^ ... ( =?= -3- = М^ехр(е7/?Г) (3.14)
Это можно легко обобщить для трехмерной цепи |7|:
I •V-a-PA '31S’
где г2—квадрат действительного расстояния между концами
цепи и г'о—среднее квадратичное расстояние в свободном состоя-
нии.
из Уравнение (3, 15) можно применить к трехмерной сетке, если
рассматривать г2 как квадрат расстояния между узлами сетки.
Математическое рассмотрение, данное в разделе 9 главы II, пред-
ставленное уравнениями (9, 8—9/ 18), сохраняется без изменения
и приводит к следующему выражению:
• F = nRT
ri
7щ~ ехр
(3, 16)
где г2—среднеквадратичное расстояние между узлами сетки.
Из Уравнения (3, 16) получим:
7 /Э1п (Д/П\ 17
; Г \ <9(1/77 )а~ R 1,)
Недавно Флори, Хойве и Киферри решили трехмерную задачу
для упругости сетки каучука, приняв во внимание энергетиче-
ские эффекты, обусловленные валентными углами. При этом они
использовали математический метод для расчета конформации
цепи [81, развитый М. В. Волькенштейиом и О. Б. Птицыным 19].
Результат имеет следующий вид:
(3, 1«)
В уравнении (3, 18) величина г‘- представляет собой средне-
квадратичное значение действительного расстояния между узла-
ми сетки в ее изотропном состоянии, а величина rl—среднеквад-
Приложение 3
307
ратичное значение расстояний между концами цепей .сетки, на-
ходящихся в свободном состоянии при температуре Т; ;Необхо-
димо отметить идентичность уравнений (3, 18) и (3, 16). с-урав-
нениями (9, 16) и (9, 18) главы II. •>..
Так как г? практически не зависит от температуры, то из>-урав-
нения (3, 18) следует, что ••
/д In (//Т) \ d In r'f, .q 19)
\ д'Г )a- ~ dT ’
Кроме того, для углеводородных цепей:
^ = y/s|4+4 еХР<Е-^Г) | vu,;
Используя уравнения (3, 19) и (3, 20), получим:
д In (F/Т) е Г 1 V1 ! £
-• a(T/n~ = “r'[1 +“2“ехр<~ £/^r)j « -r (3.21)
Результат, выраженный уравнением (3, 21), хорошо согла-
суется с уравнением (3, 17).
Флори, Хойве и Кпфферри использовали уравнения (3, 19) и
(3, 20) для получения значений s для сшитого силиконового
каучука и сшитого полиэтилена в температурном интервале высо-
коэластического состояния. Они обнаружили, что силиконовый кау-
чук подчиняется кинетической теории высокоэластичности, т. е.
е=0. С другой стороны, для сшитого полиэтилена они нашли,
что е—540 кал!моль.
Эта относительно большая величина е для сшитого полиэтиле-
на находится в хорошем согласии с оценкой разности энергий для
транс- и гош-конформацпи н-бутана, полученной из спектров
комбинационного рассеяния. Однако это находится в резком не-
соответствии с прежними результатами, полученными для бу-
тилового и лактопренового каучуков [4—6].
Недавно мы проверили свои прежние результаты 110J для бу-
тилового и лактопренового каучуков и нашли, что они экспери-
ментально правильны, т. е. е'^0. Многие другие значения е'
были получены [10] для слабо сшитых полимеров по температур-
ным кривым напряжения при использовании уравнения (3, 17).
Было найдено, что этиленпропиленовый сополимер (молярное
отношение 2/1) имеет значение е', равное 380 кал!моль. Полиэтил-
акрплат, полибутилакрилат и полиоктилакрилат дают значения
е','^0. Полибутилметилакрилат и полноктилметакрилат имеют ве-
личины е' =—500 кал/моль, что указывает на преобладание свер-
нутых конформаций. Сополимеры бутилметакрилата й эТилакрилата
(молярные отношения 3/1, 1/1 и 1/3) дают г'^0. Синтетический
20*
308
Приложение 3
цис-l,4-полиизопрен имеете'=40 к,ал!моль. Следующие полимеры
и сополимеры обладают значениями е', равными нулю в пределах
экспериментальной ошибки: цис-\,4-полибутадиен, винил иденфто-
рид с перфторпропиленом (1/1), трифторхлорэтилен с вннилиден-
фторндом (1/1) и атактический полистирол. Был исследован аморф-
ный изотактический полистирол, но результаты оказались не-
надежными вследствие медленной кристаллизации.
Возвращаясь к одномерной модели, рассмотренной ранее,
отметим, что молярная внутренняя энергия U, связанная с эф-
фектом свертывания цепи, определяется следующей формулой:
U = 2е70 (3, 22)
где /0 определяется уравнением (3, 10).
Рис. 3, 2. Зависимость удельной теплоемкости
при постоянной длине от температуры для малых
растяжений согласно уравнению (3, 15)
( К. W. S с о t t, А. V. Tobolsk у
.1. Coll. Sci., 8, 465 (1953)].
Молярная теплоемкость при постоянной длине образца, свя-
занная со свертыванием цепи, может быть выражена следующим
образом:
/ ои\
С1- " \&г) 13 • 23)
Из уравнений (3, 23), (3, 22) и (3, 10) можно показать, что
для /<^1 или /=0 значение CL равно [2]:
/ е' \2 е'
Су = RN 1 ) sch2 CART (3.
Теплоемкость, отнесенная к одному молю звеньев,
очевидно, равна CJN. График зависимости CJRN от RT/z'
приведен на рис. 3, 2.
Приложение И
309
Аномалия удельной теплоемкости, показанная на рис. 3, 2,
может наблюдаться для трехмерной сетки, состоящей из цепей,
у которых транс- и гош-конформации имеют неодинаковые энер-
гии. Интересно выяснить, наблюдается ли этот эффект в аморф-
ных этиленпропиленовых сополимерах.
Литература
1. К. W. Scott, А. V. Tobolsky, Am. Phys. Soc. Meeting, New York,
January 1948.
2. K. W. Scott, A. V. Tobolsky, J. Coll. Sci., 8, 465 (1953);
K. W. Scott, Ph. D. thesis, Princeton University, 1950.
3. E. Gut li, H. M. James, Am. Phys. Soc. Meeting, New York. January
1948.
4. G. M. Brown, Ph. D. thesis, Princeton University, 1948.
5. R. D. Andrews, Ph. D. thesis, Princeton University, 1948.
6. K. W. Scott, Ph. D. thesis, Princeton University, 1950.
7. H. James, E. Guth, J. Chem. Phys., 11, 455 (1943).
8. P. J. F 1 о г у, C. A. J. H о e v e, A. C i f e r r i, J. Polymer Sci., 34,
337 (1959).
9. M. В. Волькенште й и, О. Б. Птиц ы н, ЖТФ, 25, 649 (1955).
10. А. V. Tobolsky. D. С а г 1 s о n, N. I n il i с t о г, manuscript in
preparation.
ПРИЛОЖЕНИЕ И
МАКСИМАЛЬНОЕ ВРЕМЯ РЕЛАКСАЦИИ |11
Было приготовлено 15 образцов полистирола блочной полиме-
ризацией при 60 °C с различными концентрациями динитрила азо-
бис-изомасляной кислоты как инициатора. Полимеризация произво-
дилась при низких конверсиях (меньше чем 1096). Такие полимеры
обладают узким молекулярно-весовым распределением (см. урав-
нения (Е,3) и (Е, 4)] с Mw/Mn= 1,5. Средневязкостные молекуляр-
ные веса были получены измерением характеристической вязкости
в бензоле при 25°C 12], а затем были вычислены средневесовые мо-
лекулярные веса [3]. Эти величины представлены в табл. И, 1.
Изучение релаксации напряжения проводилось в температур-
ном интервале эластовязкости. Были вычислены вспомогательные
кривые Ег(/), приведенные к 115 °C. Они показаны на рис. И, 1.
Предположив непрерывное распределение времен релаксации,
было вычислено второе приближение H2b(y) при 115 °C; резуль-
таты приведены на рис. И, 2.
В разделе 9 главы IV были рассмотрены общие представления
о дискретном максимальном времени релаксации. Если такое
максимальное время релаксации существует, то зависимость
310
Приложение И
Таблица И, I
Л» I’ll Л1с гт (при И5 °C) мин т4 сдвиговая (при 115 °C; пуазы
1 1,46-10*1 1,78-104 1,86-10» 5,80-10~- 9,84- 10®
2 1,60-10 1 2,08 10» 2, 16-10» 1,54-10» 1,45- 10'
3 1,87-10 1 2,51-10» 2,61-10» 3,30- КГ' 2,24- 10-
4 2,02-10 » 2,82-10* 2,94-10» 6,07-10-' 2,46- 10»
5 2,30- 10~l 3,47- 10' 3,62-10' 6,30-10» 3, 16- !()•
6 2,53- it)1 4,07-Ю4 4,25-10» 1,04 3,31-10»
7 2,72- 10~i 4,47-10» 4,67-10» 2,05 4,77-10»
8 2,80-10 1 4,60-10» 4,80-10» 4,25 1,06-ИУ
9 3,33-101 5,76-10» 5,90- 10' 7,69 1,17-I05
10 3,64-10-1 6,61-10» 6,90-10» 1,41-10 2,36-Ю8
11 4,44-10» 8,91- 10» 9,30-10» 1,78-10 3,33-10*
12 5,78-Ю-1 1,15-Ю5 1,20-Ю5 7,76- 10 2,46- Ю"
13 7,04-Ю-1 1,77- Ю5 1,86- Ю5 4,55-10» 1,73- Ю»5
14 8,06-10 1 2,09 105 2, 18-105 8,00-10» 1,76-10“
15 10,56-10-' 3,02-Ю5 3,15-105 1,03-10» 1,63-10"
Ряс.//, 1 Вспомогательные кривые lg£r(/) от Igt при 115 СС для 15 образ-
цов полистирола [А. V Tobolsk у, К. М u г a k a m i, Report io
QMC. October 1959]
Приложение И
311
lg£fr(Z) от времени будет приближаться при достаточно больших
временах к прямой линии с углом наклона —3р3х .
Для всех исследованных образцов графики lg£r(/) от времени
действительно приближались к прямой линии, как это показано
для четырех образцов на рис. //, 3. Значения т,„для всех образцов,
полученные из углов наклона этих линий, приведены в табл. И, 1.
ig -z", Ml/И
Рис. И, 2. Зависимости релаксационных спектров от lg£) при
115 °C для тех же 15 образцов полистирола [А. V. Т о Ь о 1 s к у,
К- Murakami, Report to QMC, October 1959].
Затем был построен график зависимости IgAJ^, от lgTm (рис.
И, 4). Для величин Mw, лежащих между 50000 и 315000, тт для
jthx образцов при 115 °C приближенно выражается следующей
формулой:
(в мин) = 7,94-10 16Л1’Л
(И, 1)
312
Приложение И
Рис. И, 3. Зависимости lgEr(0 от t для четырех образцов полистирола (12,
13 14 и 15) (А. V. Tobolsk)’, К- Murakami, Report to QMC.
October 1959].
Рис. И, 4. Зависимость— максимального времени
релаксации lg-г,,, от lgMw при 115сС для 15 об-
разцов полистирола ]А. V. Т о b о! s к у,
К. М и г а к a m i. Report to QMC, October
1959]
Приложение И
313
Вязкость полимера при сдвиге вычисляется из Н2Ь
с помощью следующего уравнения:
4 “ 3 2 303
о
Интеграл вычислялся графически. Результаты этого расчета
даны в последней графе табл. И, 1.
Величины тт как функции молекулярного веса были вычисле-
ны также для полиизобутнлена и поливпнилацетата на основе
опубликованных данных по релаксации напряжения [4, 5].
Было найдено следующее соотношение для Nw, Т„ и Т для
этих полимеров и полистирола [6]:
, 17,44(7’ — Tg)
Ig[сек]-Ig /1 si 64'7_____Т 4-3,4 1g
(И, 3)
Второй член в правой части представляет собой знакомое урав-
нение Вильямса, Ландела и Ферри. Величина Na означает сред-
невесовое число звеньев в цепи. Величины lg/4 для этих трех по-
лимеров мало отличаются друг от друга. Если в уравне-
нии (/7,3) тт выражено в секундах, то lg/4==—0,52 для поли-
изобутнлена, —0,70 для полистирола и 0,86 для поливинилаце-
тата.
Если этот результат верен и для других полимеров, то уравне-
ние (И, 3) дает простой способ нахождения приближенного сред-
невесового молекулярного веса полимера путем определения
и измерения Tg при условии, что значение IgTl известно для дан-
ного полцмера.
Между тт, ri), Т и Tg получено следующее соотношение:
17,44 (Т~ Tg) о , / \
Ig [сел1 — Ig S 5| g_j_y + 3,4 Ig у (77, 4)
Здесь го—среднеквадратичное расстояние между концами цепи
полимера в 0-растворе (раствор, в котором полимер находится
почти в состоянии осаждения) и —длина связи С—С, равная
1,54 А.
В этом случае IgB один и mom же для всех трех полимеров
и равен —2,15. Если уравнение (И, 4) верно и для других полимеров,
то, следовательно, этот метод применим для расчета г$ непосред-
ственно из данных по релаксации напряжения.
314
Приложение К
Литература
1. А. V. Tobolsky, К- Murakami, Report to QMC, Contract DA-19- !
129-QM-1306. October 1959.
2. В. H. Z i m m, J. Chem. Phys., 18, 830 (1950).
3. В. В a у s a 1, A. V. Tobolsky, J. Polymer Sci , 9, 171 (1952).
4. R. D. Andre w s, A. V. Tobolsky, J. Polymer Sci., 7, 221 (1951)
5. K. Nino rn i у a, J. Coll. Sci., 14, 49 (1959).
6. A. V. Tobols к у, К. M nrakami. manuscript submitted to J Coll
Sci.
ПРИЛОЖЕНИЕ A
ЗНАЧЕНИЯ Tj И Тк ДЛЯ НЕКОТОРЫХ ПОЛИМЕРОВ
Были определены величины для различных аморфных по-
лимеров и проводилось их сравнение с величинами Tg. Для удоб-
ства температура Т4- была выбрана как температура, при которой
10-секундный модуль достигает величины 109 дин/см2 [для боль-
шинства полимеров 10s дин!см~]. Величины 7\ пред-
ставлены в табл. К, 1 наряду с величинами Tg, в большинстве
случаев взятыми из литературы. Интересно отметить, что отличие
величин Tg от Д составляет в общем несколько градусов. Значе-
ние Т1 для аморфного найлона (тройной сополимер) весьма инте-
ресно.
Таблица К, 1
Пилим ер Гг, °C г,.. °C
Натуральный каучук ...............
Полиизобутилен ............................. .
Сополимер этилена и пропилена (2/1).............
GR-S ...........................................
Аморфный атактический полипропилен
Полибутплметакрилат ............................
Аморфный найлон (тропной сополимер найлоиа-6,
найлона-66, найлона MXD-6) .....................
Полистирол (атактический) ...........
Полистирол (аморфный изотактический) ...
Полпметплметакрнлат ............. . .
—72
—70
- 59а
—57
—20
09
100
105
—68
—66
—59
—53
— 18
25
46
95
97
109
Л. V. Т <» Ь о 1 s к у, D, С а г I s <» п. A. Eisenberg (не опубликовано).
Величины Tg, равные —20 и —125 °C, были получены из изме
рений [1] зависимости удельного объема от температуры для
аморфного полипропилена и кристаллического полиэтилена со-
ответственно. Из измерений v, Т мы получили для аморфного эти-
лен-пропиленового сополимера (молярное отношение 2/1). что
Приложение К
315
Tg равна —59 °C. Используя это значение, а также величину
Tg=—20 °C для аморфного полипропилена [1J и применяя урав-
нение (5, 3) главы II, получим, что для аморфного полиэтилена
Tg=—81 °C. Это расходится с величиной —125 °C, взятой из ли-
тературных данных [1], но мы считаем, что величина —81 °C
болеедостоверна. Изгиб кривой v, Т при —125 °C, найденный для
кристаллического полиэтилена, может возникнуть не в результате
перехода аморфных областей в стеклообразное состояние, а вслед-
ствие какого-нибудь другого перехода.
Литература
1. М. L. Dannis. J. Appl. Polymer Sci., 1, 121 (1959).
ПРЕДМЕТНЫЙ У КА 3.4 ТЕ Л Ь
Авогадро пр.пиши 9
Александрова—Л азуркина частотно-
температурный метод 80
Аморфные полимеры 50 сл.
определение 63
Андраде уравнение 37
Аррениуса уравнение 228, 258
Афпппое преобразование
сеточных векторов 99
сферы 301, 302
Бачинского уравнение 38
Викки, молекулярная теория вязко-
упругости и у.пруговязкостн
171176
Блоксополимеры 88
Больцмана
константа 9
принцип 14, 33, 98, 286
принцип суперпозиции 111—114,
215, 216
Вант-Гоффа уравнение 273
Вихерта модели 115—120
Вильямса—Ландела—Ферри уравне-
ние 166, 313
Внутреннее вращение 46—48, 69, 75
Внутреннее давление 13
Времени факторы 149—163
Время запаздывания 121 —123, 140
Время релаксации 40—44, 115—120,
125—128, 131 — 140, 309—313
дискретный спектр 185—195
максимальное 82. 132—140, 171 —
174, 185—195, 309—313
минимальное 132—140, 171 —174
распределение 113—115, 125—127',
131 — 140, 152, 153, 161, 162,
171 — 175, 179, 180, 185—199,
202, 203, 309—313
характеристическое 165—170, 174
химической 240, 242, 257—261
Вспомогательные кривые 149—157
Вынужденные колебания 107—НО
Высокоэластпчность полимеров 144,
145, 164—171, 175
кинетическая теория 96—105
Вязкое течение 80, 83, 309—313
аморфных полимеров 91—96
Вязкость 9 сл., 118. 119, 172. 181, 298
газов 36
динамическая 108. 109
жидкостей 35 сл.
полимеров 91—93, 309—313
при растяжении 43, 118, 119, 121
характеристическая 173
Вязкоупругость 144—148
линейная 111—115
молекулярная теория 171 — 176
определение и измерение 106—111
пепесграпвакицнхся сеток 213-
216
Гамма-функция неполная 137
Гауссово распределение 294
Гейгера счетчик 50, 55
Гельмгольца свободная энергия 13,
18, 98
Гросса уравнение 130
Грюнайзена константа 287. 288
Гука закон 22, 44
Гуттаперча 61
Двойное лучепреломление 176. 218-
224
Дебаевская температура 27, 285— 287
Дебая уравнения 27
Деполимеризация 281
Деструкция 230—233
поперечных связей 225—243
скорость 234—236, 239
Деформация химическая остаточная
244 сл.
Динамический модуль 106—111, 124.
125, 158—163, 199-207
Дискретный спектр времен релакса
цин 128, 185 сл.
Диэлектрические характеристики по-
лимеров 178
Длина полимерной цепи см. Цепи
размеры
Дулитла уравнение 38. 92
Предметный указатель
317
Жесткость полимерной цепи 49, 73,
95, 218—224
Законы
Аррениуса 228, 258
Гука 22, 44
деформации резины 24
идеального газа 284
Ньютона 35, 41, 44
Стокса 172
Планка 26
термодинамики 12, 28
Заторможенное вращение 47
«Зацепления» в сетке («переплете-
ния») 83, 91, 175, 176, 212
Идеальная резина 30
Инициаторы 251—257
Ионизирующее излучение, действие
на полимер 105, 231, 232, 238,
239, 254
Каучуки
вспомогательные кривые 152
кривые модуль—температура 84,
85
параметры растворимости 74
релаксация напряжения 166, 183,
197, 205, 206
строение 241
температура перегиб;! кривой 314
— плавления 61
-- стеклования 74—77, 314
термодинамические величины 61
химический релаксация напряже-
ния 219—236, 240
К аргина— Соголовой термо механиче-
ский метод 80
Квантовые эффекты 18
Кинетики кристаллизации 64 сл.
Кинетическая теория высокоэластпч-
носпи 96
Кинетическое давление 13
Клапейрона—Клаузиуса уравнение 64
Колебания решетки 24 сл.
Константы
Больцмана 9
газовая 9
Грюнанзена 287, 288
для «ящикоподобного» распреде-
ления 134
критическая
молекулярные 16, 28
нормировочная 97
Планка 26
Константы
равновесия 268—281
силовая 25
универсальная газовая 9
упругие 22, 24 сл., 122
Конфигурация цепи 45
Конформации цепи 33, 45 ел., 96
Коэффициенты
вириальные 15
времени 150, 151, 157
объемного расширения 14, 77, 288
Пуассона 21, 23, 43, 82, 118, 173
смещения 198
трения 172
фотоуп ругости 223
Кратность растяжения (относитель-
ное растяжение) 29
Кривые
вспомогательные 149—157
деформация—время 41
модуль—температура 79, 81,
83—85, 179—185
напряжение—растяжение 33
остаточной деформации 247
ползучести 122, 123, 131, 218
релаксации напряжения 310
химической релаксации напряже-
ния 227, 238, 247, 258, 259
Крип см. Ползучесть
Кристалл единичный полимера 64—68
Кристаллизация полимеров
и объемность боковых групп 59
и регулярность цепи 56
кинетика 64 сл.
термодинамика 60 сл.
Кристаллические полимеры 50 сл.
температура плавления 60—62
— стеклования 76
Кристалличность полимеров, опреде-
чение 56
Леннарда-Джонса
потенциал 16, 37
уравнение 15
Лидерман, принцип температурно-
временной суперпозиции 150
Максвелла
модели 115—120, 124
уравнение 40, 42, 210, 215, 226,
235, 236
Максвелловское тело 40, 42
Маркова цепь 291 сл.
Матрица вероятностей перехода 291
Межмолекм.тярные силы 15, 17—20,
36
318
Предметный указатель
Метод X 191 сл.
«Механическое» стеклование 71
Модели
Вихерта 115—120
Извнга 283, 301—304
Каргина—Слонимского 171
А1аксвелла 40, 115—120, 124, 125
Максвелла—Вихерта 171
Рауса 171
свободиосочленеиных сегментов
173
Слонимского 171
Фойгта—Кельвина 121 —123
Модификаторы цивильной полимери-
зации 299, 300
Модуль
высокоэластичности 81, 164
динамический 108 —110, 124. 125,
129, 131, 160
комплексный 110, 111, 124, 125
мгновенной упругости 123
обьемного сжатия в стеклообраз-
ном состоянии 82, 83
— — для молекулярных кри-
сталлов 18—20, 27, 288
----жидких и твердых тел 11,
12
----идеального газа 11
---- определение 9
ползучести 107, 150
релаксации 79, 106, 112, 125, 138
— в переходной области 179
— преобразование 128—131
сдвига, определение 21, 22
— для резин 35
стеклообразного состояния 164,
171 — 176
Молекулярно-весовое распределение
см. Распределение по молеку-
лярным весам
Молекулярные кристаллы 17—21,
287—290
Молекулярный вес
средпевесовой 50, 94, 95, 297
средневискозиметрический 298,
309, 310
срелпечисловон 34, 102—105, 297
цепей сетки 34, 102—104
Молярная энергия
испарения 19, 20
кристаллической решетки 18—20
Молярный объем 20, 24
«Моподисперсные* ^"--имеры 136,
185—196, 297—299
Набухание полимеров 73
Надмолекулярные структуры 51. 65.
206"
Найлон-6,6 74, 207, 314, 315
Напряжение, определение 21, 22
Нормировочная постоянная 97
Ньютона закон вязкости 35. 41, 44
Окисление 219—248
Окислительная деструкция 251—256
Окислительная релаксация напряже-
ния 237
Оптическая анизотропия 220 сл.
Оптический коэффициент упругости
223, 224
Ориентированные полимеры 53
Отжиг кристаллических полимеров 64
Относительное растяжение (крат-
ность растяжения) 29
Параметры растворимости 71
полимеров 74
Парафина кристалл 66
Перекиси 251 сл.
Переноса цепи, агент 299, 300
«Переплетения» в сетке («зацепле-
ния») 83, 91, 175, 176, 212
Перестраивающиеся сетки 213—216.
257—264
Переходная область 83, 86, 125—131,
147, 179—185
и температурная зависимость мо-
дуля 179
и функция ошибок 181
Планка закон 26
Плотность когезионной энергии 7'1.
82
Податливость 107. 156
Показатель
гетерогенности 300
преломления 218
Ползучесть 216, 225
при растяжении 107
химическая 248
Полиакрилаты 75
кривые релаксации 148
температура стеклования 77, 222
— характеристическая 94
Полиакрилонитрил 74
Полиамиды 63
Полпаценафтилен 75
Полибутадяен
параметр растворимости 74
температура стеклования 77, 87
Поливинилацеталь 94
Предметный указатель
319
Поливинилацетат
параметр растворимости 74
температура стеклования 77
— характеристическая 94
Поливинилиденфторид
регулярность цепи 57
температура стеклования 76, 77
Поллвипилиденхлорид
параметры растворимости 74
регулярность цепи 57
Поливинилиденциаиид 57
Поливиниловый спирт 59
Поливинилхлорид
параметр растворимости 74
регулярность цепи 57
температура стеклования 77
Поливинилцпанид 57
Полигексаметиленадипамид
температура плавления 61
термодинамические величины 61
Полидисперсность 95
Полипзобутилен 74
вспомогательные кривые 159, 163,
204
кривые релаксации 144—146, 155,
157
механические характеристики
161, 162
релаксация напряжения 166, 182
температура перегиба кривой 314
— стеклования 77, 314
— характеристическая 94
Полиизопрен 251—256
Поликапролактам 269, 272
Полиметакрилат 75
кривые релаксации 155
параметр растворимости 74
релаксация напряжения 166, 177
температура перегиба кривой 314
- - стеклования 77, 88. 314
— характеристическая 94
Полиметакрилонитрил
параметр растворимости 74
температура стеклования 77
Полиметилен
температура плавления 61
термодинамические величины 61
Полиперфторпропилен 76, 77
Полипропилен
винтовая конформация 59
вспомогательные кривые 204
температура перегиба кривой
314
— плавления 61
— стеклования 314
термодинамические величины 61
Полистирол 75
времена релаксации 191
вспомогательные кривые 188
изомерные формы 56
кривые модуль—температура 79
оптические коэффициенты упру-
гости 224
параметр растворимости 74
плотность когезионной энергии 82
релаксация напряжения 187,
310—312
рентгенограммы 53
температура перегиба кривой
314
— плавления 57
— стеклования 77, 87, 88, 90,
317
характеристическая 94
11олцсу.1ьфндные резины 257—
262
Политетрафторэтилен 74
параметр растворимости 74
температура плавления 61
— стеклования 78
термодинамические величины 61
Политрифторхлорэтилеи
регулярность цепи 57
релаксация напряжения 201
температура плавления 61
— стеклования 76
термодинамические величины 61
Полиуретановые резины 264—266
Полихлоропрен
параметр растворимости 74
температура плавления 61
термодинамические величины 61
Полиэтилен
вспомогательные кривые 204
зигзагообразная конформация 59
параметр растворимости 74
плотность 58
релаксация напряжения 201
рентгенограммы 55
температура стеклования 74, 89
эчектронно-микроскопические .ис-
следования 66—68
Полиэтнпеноксид 61
Полиэтилен терефталат 61, 74
Полиэфиры 63, 264—267
Поляризуемость сегмента 218—224
Постоянная см. Константа
Потенциал Леннарда-Джонса 16, 37
Привитые полимеры 89
Принцип
Больцмана 14, 33, 98, 286
320
Предметный указатель
11ршщип
соответственных температур 86,
87
суперпозиции Больцмана 111,
112, 114, 215, 216
температурно-временной суппер-
позиции 149—154, 203
— — проверка 158—163
Принцип Авогадро 9
Пуассона коэффициент 21, 23, 43,
118, 173
Равновесие полимернзацнонное
268 сл.
Равновесная сополимеризация 281 —
283
Распределение
времен запаздывания 129, 140
— релаксации 119, 125—128,
131—140, 179—185
-----и трехмерные сетки 174—
176
-----«случайное» 135, 295—300
— — «ящикоподобное» (прямо-
угольное) 131 —140
длин молекул 295
комбинационное 296—300
конформаций нормированное 96
по молекулярным весам 295—301
Пуассона 297, 298
типа клина и прямоугольника
135—140
Растяжение
определение 21, 22
термодинамика 28
Рауса модели 171
молекулярная теория вязкоупру-
гости и упруговязкости 171—
176
Релаксационный модуль см. Модуль
репаксацп и
Релаксация напряжения 81, 106, 198,
210, 213, 221
в аморфных полимерах 144
в перестраивающихся сетках
207—212
кристаллических полимеров 199—
207
максвелловская 42, 43
«прерывная и непрерывная»
230—234
при постоянной деформации 106
стеклообразных полимеров 176—
178
Релаксометр 149
Рентгенограммы полимеров 51—55
Свободное вращение 46
Свободно сочлененная цепь 46, 49,294
Свободные колебания 107—110
Свободный объем 38, 39, 71, 73, 91,
92
Сдвиг, определение 21, 22
Сегмент 219
статический 49, 63, 218—224, 294.
295
Сегментальный перескок 91
Сжатие 9
изотропных веществ 9
термодинамика статическая 12
Сжимаемость 10
Силиконовые резины 262, 263
Скорость образования зародышей
65—67
Смеси полимеров 88, 89
Соответственные состояния 70, 86—
90
Соответственные температуры 86—90
Сополимеризация 57, 60, 61, 281—284
Сополимеры
вязкоупругие свойства в переход-
ной области 184
кривые модуль—температура 85
кристаллизация 57
температура перегиба кривой 314
— плавления 58
— стеклования 75, 87, 88, 314
— характеристическая 94
температурная зависимость мо-
дуля 89
Спектр времен релаксации 134, 311
«Спонтанное удлинение» 205
Статистическая сумма состоянии 13,
284—286. 302 сл.
Стеклообразное состояние полимеров
69, 79
н релаксация 176—178
определение 69
Степень полимеризаты см. Цепи
размеры
Стокса закон 172
Структурирование 230—233
Структурное стеклование 70
Сферолиты 52, 64—68
«Сшитые» полимеры 32 84, 85, 102—
105, 225—256
«Тепловая хрупкость» 202
Тепловые потерн в каучуке 197
Теплоемкость
кристаллов 26—28, 288
цепей 308. 309
Предметный указатель
321
Теплообразование в каучуках 196—
199
Теплота полимеризации 273—280
Температуры
дебаевская 27, 285—286
«механического» стеклования 71
перегиба кривой 82, 83, 179—185,
314, 315
плавления 52—64
— и режим нагревания 69
— кристаллических полимеров
60—62
— методы определения 54
— сеточных полимеров 63
соответственные 86—90
стеклования 68 сл., 314, 315, 317
- - и длина цепи 75
- и жесткость кепи 74
— и параметр растворимости 73
— и «свободный объем» 73
— и удельный объем 69
— и энергетический барьер 74
— определение 69
структурного стеклования 70
характеристические 86, 91—94,
. 150. 164—171
Эйнштейна 26—28, 37, 287—289
Температурная зависимость модуля
85, 179—181, 184, 185
Термодинамика
кристаллизации 60 сл.
полимеризации 208 сл.
растяжения 28
Термомеханпческая кривая 85
Термомехапический метод исследо-
вания полимеров 80
Сечение вязкое см. Вязкое течение
Тобольского уравнение для опреде-
ления вязкости 95
Тобольского—Эйринга уравнения 125
Трещины 177, 202
Угол сдвига фаз 108
Ударная прочность 88
Удельный объем 56
Упругая податливость 107, 123
Упругие константы 21—24
Упругие свойства изотропных тел 21
Упруговязкость 40. 106 сл., 144—148
максвелловского тела 141
молекулярная теория 171—176
основные области 79—85, 144—
148 :
перестраивающихся сеток ,213—
216
Упругость 9 сл.
жидкостей (при сдвиге) 10
запаздывающая 122
идеальных каучуковых сеток
31 сл.
мгновенная 122
Уравнение
Андраде 37
Аррениуса 228, 258
Бачинского 38
Больцмана 14, 33, 98, 111, 112,
286
Вант-Гоффа 273
Вильямса—Лаидела—Ферри 166,
313
вязкости 39
Гросса 130
движения 107, 109, ПО, 124
двойного лучепреломления 218
Дебая 27
деформации сдвига 35
Дулитла 38, 92
Каргина—Соголовой—Слоним-
ского 94
Клапейрона-Клаузиуса 64
колебаний единичного объема
108
Леннарда-Джонса 15
линейной вязкоупругости 112
Максвелла 40, 42, 210, 215, 226,
235, 236
податливости 156
ползучести 217
состояния 13, 104, 216
— идеального газа 9
— изолированных цепей 302—
305
— каучукоподобчых веществ
29—35, 96—105, 302
— кристалла Дебая 285
-Эйнштейна 284
— молекулярных кристаллов 17
— реальных газов 15
Тобольского для определения
вязкости 95
Тобольского—Эйринга 125
Эйринга 39
«ящиколодобного» (прямоуголь-
ного) спектра 132
Фактор времени 149—163
Физика ' полимеров 45 сл.
«Физические» поперечные связи 212
Фойгта модель 121—123
Фойгта—Кельвина модели 121
21 833
322
Предметный указатель
Функция
Дебая 288
ошибок 181—185
химической ползучести 249, 250
Характеристическая вязкость 173
Характеристическая температура 86,
91—95
Характеристические параметры поли-
меров 164—170
Характеристическое время релаксации
164 170, 174
Химическая остаточная деформация
244 сл.
Химическая ползучесть 248—250
Химическая релаксация
каучуков 229, 225, 226, 232, 233
напряжения 225 сл., 240, 259, 262,
264, 267
Химическое течение 247, 251
«Хрупкость теплоная» 202
Целлюлоза и ее производные 61, 74
Цепи
жесткость 49, 7'3
концевые 102
Маркова 291
размеры 32, 33, 45—50, 75, 80,
95—102, 131 — 137, 144—147,
185—196, 289—300, 302—319
сетки, определение 32
— — числа молей 103—105
Частота
колебаний 25, 37
Частота Эйнштейна 26—28, 37, 287—
289
Частотная зависимость диэлектриче-
ской проницаемости 178
Частотно температурный метод иссле-
дования полимеров 80
Число
Авогадро 9, 285
комплексам мнкросостоянпя 304
цепей в сетке 214
Эйнштейна температура 26—28, 37.
287—289
Эйнштейновская частота см. Частот;
колебаний
Эйринг, иаивероятнейшая длина пени
290
Эйрпнга уравнение 39
Эластовязкое состояние 80
течение 83
Эластовязкость 79—90, 144- 147.
185—196, 309—313
Энергетический барьер 47. 73
Энергия
диссипации 109
когезионная 71, 82
кристаллической решетки 18 -20
осцилляторов 25
свободная Гельмгольца 13, 18, 98
Энтропия 33
газа 14, 15, 286, 287
каучукоподобных веществ 29 -35
96—105, 302—309
полимеризации 273—280
расплава 60—64
Эпоксидные смолы 74
Эффективные поперечные связи 103
Эффекты
временные 81
«исключенногэ объема» 47
квантовые 18
Юнга модуль 22, 31. 34. 106
Ядерпын магнитный резонанс 178
Державна наууо»*
еивлютека
Ih Kgpojiemka.Xapk/r
г>‘ vuf- gv
А. Тобольский
ОПЕЧАТКИ
Стр. Строка Напечатано Должно быть
14 34 39 49 В В В уравнении (2,9) уравнении ' (7,8) 14 сверху уравнении (1.9) (р- dRT “ \МС д8 \ Т 3V )г = Г/ LA2 А] A L«! L j 7исп./2,45 (А у/2 Р / as \ — Т (— А — ~ 1 \ dV ]т~ dRT Г/ L \2 Lo 1 Мс Л L J ] — £цсп./2,45 / 0/2 \ Nt2 /
127 160 4 сверху 7 сверху Н (In т) d (In) 10-8.2 < 1/ш 10-’ ч 10 //(InT)d(ln т) -8.8 < 1/ч> < 10“7 Ч
271 В уравнении (1.6) КМ2 / j \ КАР j — А3Л4)2 ’ Кз ' М
(1 — К3Л4)2 1Лз ~ М j ~ (1
277 В уравнении (1,22). Р = Л'з Р=~К
Заказ 833.