/






































































































Текст
ю. к. годовский
ТЕПЛОФИЗИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЛИМЕРОВ
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ» 1976
УДК 678.01:536.21
Годовский Ю. К.
Тепло физические методы исследования полимеров. М.,
«Химия», 1976.
В книге рассматриваются теплофизические методы (кало-
риметрия и дилатометрия, стационарные и нестационарные
методы определения теплопроводности и температуропроводно-
сти) исследования полимеров и излагаются основные теорети-
ческие и экспериментальные результаты исследований теплофи-
зических процессов в полимерах при разных температурах, а
также тепловых эффектов, сопровождающих механические де-
формации.
Книга рассчитана на научных и инженерно-технических ра-
ботников, а также аспирантов и студентов, специализирующих-
ся в области физики, физико-химии и технологии полимеров.
216 с., 10 табл., 88 рис., список литературы 495 ссылок.
31410-050
Г 050(01 )-76
© Издательство «Химия», 1976 г.
СОДЕРЖАНИЕ
Предисловие ............................................. 5
РАЗДЕЛ I. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ....................... g
Глава 1. Калориметрия и дилатометрия полимеров ... 8
Адиабатическая калориметрия.............................. 8
Дифференциальный термический анализ......................10
Динамическая калориметрия................................12
Микрокалориметрия....................................... 17
Деформационная калориметрия..............................19
Дилатометрия ............................................27
Линейная дилатометрия ............................. 27
Объемная дилатометрия...............................28
Глава 2. Методы исследования теплопроводности и темпе-
ратуропроводности полимеров .......................31
Стационарные методы.................................... 31
Нестационарные методы....................................34
Методы определения анизотропии теплопроводности и темпера-
туропроводности .......................................39
Л и т е р а т у^а........................................40
РАЗДЕЛ II. ТЕПЛОВОЕ ДВИЖЕНИЕ В ПОЛИМЕРАХ .... 45
Глава 3. Теплоемкость полимеров..........................45
Теплоемкость твердых полимеров...........................45
Теоретический анализ теплоемкости...................45
Экспериментальные результаты........................56
Теплоемкость полимерных расплавов........................63
Глава 4. Перенос энергии в полимерах (теплопроводность
и температуропроводность полимеров) ... 65
Температурная зависимость теплопроводности ............. 65
Аморфные полимеры...................................69
3
Кристаллические полимеры........................
Изменение теплопроводности в области фазовых пере-
ходов .............................................
Теплопроводность и молекулярные параметры...............
Молекулярная масса.................................
Разветвленность ...................................
Структура цепи ....................................
Анизотропия теплопроводности ......................
Влияние давления на теплопроводность....................
Температурная зависимость температуропроводности .
Температуропроводность и молекулярные параметры
Глава 5. Термические особенности переходов и релаксацион-
ных процессов в полимерах...............................
Плавление и кристаллизация .............................
Плавление..........................................
Кристаллизация ....................................
Стеклование ............................................
Превращения в стеклообразном состоянии и промежуточные
превращения.............................................
Литература .............................................
РАЗДЕЛ III. ТЕПЛОФИЗИЧЕСКИЕ ПРОЦЕССЫ ПРИ ДЕФОРМА-
ЦИИ ПОЛИМЕРОВ.........................................
Глава 6. Обратимые деформации.........................
Тепловое расширение полимеров.........................
Термодинамика обратимых деформаций....................
Термоэластичность твердых полимеров...................
Термоэластичность каучуков............................
Глава 7. Необратимые деформации.......................
Ориентационная вытяжка полимеров......................
Разрушение полимеров..................................
Силовое размягчение наполненных резин.................
Литература ...........................................
ПРЕДИСЛОВИЕ
Среди физических методов исследования полимеров
важное место принадлежит теплофизическим методам,
позволяющим изучать особенности теплового движения
в . полимерах, термические характеристики переходов и
релаксационных процессов, тепловые процессы, проте-
кающие при приложении механических нагрузок к поли-
мерам, и другие свойства и процессы. Калориметриче-
ские и дилатометрические методы, сравнительно давно
применяемые для изучения полимеров, в последние годы
особенно интенсивно развиваются и внедряются в иссле-
довательскую практику. Особенно это относится к кало-
риметрии. Были разработаны принципиально новые при-
боры для калориметрических измерений и значительно
усовершенствованы уже применявшиеся методы и при-
боры. Основными достоинствами новых приборов явля-
ется возможность с их помощью изучать на образцах
малых размеров термодинамику и кинетику быстрых и
медленных процессов, протекающих как в динамических,
так и в статических условиях, получая при этом доста-
точно точные результаты. Современные теплофизические
методы позволяют выполнять широкую программу ис-
следований важнейших физических и химических про-
цессов в полимерах.
Первой задачей предлагаемой книги является крат-
кое изложение физических основ методов, описание при-
боров для исследования теплофизических процессов в
полимерах и обсуждение возможных направлений их
дальнейшего развития. Этому посвящен первый раздел
книги.
В развитии теплофизических исследований полиме-
ров можно выделить два этапа. На первом этапе основ-
5
ное внимание уделялось исследованию важнейших фено-
менологических закономерностей теплофизических про-
цессов и изучению влияния отдельных молекулярных
параметров на теплофизические характеристики и про-
цессы. С этих позиций исследовались теплоемкость, теп-
ловое расширение, теплопроводность, разрабатывались
теплофизические методы оценки кристалличности полиме-
ров, анализировались кинетические и термодинамиче-
ские особенности переходов и превращений и т. д. *
Следующий этап в развитии теплофизических иссле-
дований был связан с интенсивным развитием в конце
50-х — начале 60-х годов структурных исследований
полимеров, когда стало очевидно, что природа твердого
состояния полимеров может быть понята лишь при ус-
ловии детального исследования их структурной гетеро-
генности и динамики структурных превращений. Имен-
но развитие молекулярных и надмолекулярных струк-
турных представлений стимулировало постановку новых
теплофизических исследований и поиски новых экспе-
риментальных методов, которые позволили бы проводить
количественную оценку тонких структурных превраще-
ний в полимерах.
К этому времени относятся появление и развитие
динамической (сканирующей) калориметрии, позволяю-
щей исследовать метастабильную структуру полимеров
и термокинетику превращений в неравновесной струк-
туре; применение микрокалориметрии для изучения дли-
тельных тепловых процессов, столь характерных для по-
лимеров, разработка нестационарных (комплексных)
методов исследования теплофизических характеристик и
разработка «гибридных» теплофизических методов, та-
ких, как деформационная калориметрия.
Таким образом, в современной теплофизике полиме-
ров начали наряду с феноменологическими широко ис-
пользовать и структурные представления. Поэтому вто-
рой задачей книги является краткое систематическое из-
ложение основных экспериментальных и теоретических
результатов исследований теплофизических свойств по-
лимеров и процессов, протекающих в них, на основе со-
временных структурных представлений. Этому посвяще-
ны второй и третий разделы книги.
Исследования теплового движения в полимерах теп-
лофизическими методами (теплоемкость, теплопровод-
6
ность, тепловое расширение), описанные во втором раз-
деле, достаточно многочисленны и получают все большее
распространение в связи с развитием эксперименталь-
ной техники. Напротив, механотепловые исследования,
изложению которых посвящен третий раздел, проводятся
пока лишь в нескольких специализированных лаборато-
риях. Это связано с значительными экспериментальными
трудностями прецизионного измерения очень малых
тепловых эффектов. Однако, несмотря на малые значе-
ния этих эффектов, в них заложена глубокая информа-
ция о молекулярных изменениях, происходящих при де-
формации. Использование механотепловых измерений
для изучения полимеров представляется особенно важ-
ным и перспективным, если учесть микрогетерогенность
полимерных тел, обусловленную наличием в них обла-
стей с различными типами упругости, и большой диапа-
зон морфологических изменений при деформации, неиз-
бежно сопровождающихся энергетическими превраще-
ниями. В связи с этим еще одной задачей книги явля-
ется- привлечение внимания к подобным теплофизиче-
ским исследованиям.
Основу книги составляют исследования, проводив-
шиеся автором в 1962—1975 гг. в лаборатории физики
полимеров ордена Ленина Института элементоорганиче-
ских соединений АН СССР. Постановка и проведение
этих исследований стали возможны благодаря поддерж-
ке проф. Г. Л. Слонимского. Приятный долг автора —
выразить ему искреннюю признательность.
Автор приносит благодарность И. И. Дубовик и
А. В. Волынской за помощь в подготовке рукописи к пе-
чати.
РАЗДЕЛ 1
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
Глава 1
КАЛОРИМЕТРИЯ И ДИЛАТОМЕТРИЯ ПОЛИМЕРОВ
Адиабатическая калориметрия
Прецизионные измерения теплоемкости обычно про-
водят в адиабатическом калориметре. Образцу сооб-
щается определенная порция тепла и регистрируется
соответствующее изменение температуры [1, т. 2, с. 292;
2; 3]. Основная трудность состоит в адиабатизации са-
мого калориметра. При условии, что все подведенное
тепло расходуется на нагрев образца массой т, удель-
ная теплоемкость может быть определена на основании
соотношения
AQ
СР~~ /иАТ (1‘
где Ср — теплоемкость, средняя для данного температурного интер-
вала, AQ—подведенное к образцу тепло; АТ — повышение темпе-
ратуры образца.
В зависимости от рабочей температуры адиабатиче-
ские калориметры могут быть условно разделены на две
категории; низкотемпературные (Т < 300 К) и высоко-
температурные (250—600 К). Измерение теплоемкости
полимеров при помощи калориметров обоих типов свя-
зано с рядом экспериментальных трудностей, обуслов-
ленных главным образом низкой теплопроводностью
полимеров, их низкой плотностью, вследствие чего отно-
шение теплового значения полимерного образца к теп-
ловому значению собственно калориметра мало, и нали-
чием дрейфа температуры, вызванного протеканием за-
медленных релаксационных процессов в полимерах. При
использовании высокотемпературных калориметров боль-
шое значение имеет способность полимеров к окислению
8
и разложению, а также возможность прилипания их к
металлу после плавления.
Конструктивные особенности отдельных адиабатиче-
ских калориметров изложены в специальных руковод-
ствах [1, 2], а устройство адиабатических калориметров,
разработанных специально для исследования полимеров,
описано в оригинальных статьях и обзорах [3—8].
Обычно при измерениях теплоемкости образец нагре-
вают ступеньками по 1—20 °C, причем тепло подводится
таким образом, чтобы скорость нагрева была невелика
(<С1°С/мин). После каждого нагрева следует длитель-
ная выдержка для достижения теплового равновесия.
Точность определения теплоемкости достигает 0,1%. Для
измерений используются обычно навески полимеров в
20—80 г.
Характерное для полимеров наличие метастабильных
состояний и протекание в них замедленных релаксаци-
онных процессов часто заставляет отказываться от из-
мерений с длительными перерывами между отдельными
стадиями нагрева образцов, поскольку в эти периоды
в них происходят существенные необратимые изменения
и, таким образом, каждое новое измерение проводится
фактически на образце, отличном от исходного. Это об-
стоятельство особенно существенно при измерениях в
области переходов и структурных превращений. Поэто-
му во многих случаях лишь измерения в условиях доста-
точно быстрого непрерывного нагрева позволяют избе-
жать необратимых изменений в образцах.
Оригинальный малоинерционный адиабатический ка-
лориметр, пригодный для измерений теплоемкости
полимеров в условиях непрерывно изменяющейся тем-
пературы, был разработан Журковым и Левиным [9].
Тот же принцип с некоторыми изменениями впоследст-
вии был использован Волькенштейном и Шароновым
[10]. Малая инерционность, необходимая для измере-
ний при непрерывно меняющейся температуре, достига-
лась специальным расположением образца, нагревате-
ля и термометра сопротивления. В отличие от применяе-
мых обычно блочных образцов полимер наносился из
раствора тонким слоем на три изолированные проволо-
ки, одна из которых служила нагревателем, а две дру-
гие — для измерения температуры и создания адиаба-
тических условий. В результате многократного нанесе-
9
ния раствора на проволоках осаждался слой полимера
толщиной 0,15—0,20 мм. Тепловое равновесие в приго-
товленном таким способом образце достигалось за доли
секунды при скорости нагревания 0,5 °С/мин. Эта мето-
дика позволяла проводить измерения со скоростями на-
грева до 2—3°С/мин. Навески полимера были снижены
по сравнению с применяемыми при использовании обыч-
ных адиабатических калориметров примерно до 8 г.
Разработаны и другие адиабатические калориметры
с непрерывным нагревом, в которых скорости нагрева
доведены до 6°С/мин, а навески уменьшены до 1г
[11, 12]. Уменьшение навесок и увеличение скоростей,
а также отступление в значительной мере от адиабати-
ческих условий привело к снижению точности измере-
ний до 0,5—5%. Однако часто этой точности бывает до-
статочно для количественного сравнения результатов.
Дифференциальный термический анализ
В последние годы дифференциальный термический
анализ [ДТА] получил широкое распространение в фи-
зико-химических исследованиях полимеров [13—17].
Принцип этого метода заключается в измерении темпе-
ратурной зависимости разности температур исследуемо-
го образца и термически инертного сравнительного ве-
щества при непрерывном нагреве или охлаждении [18,
19]. Метод ДТА применяется для регистрации термиче-
ских переходов, при которых изменяется либо энтальпия
(фазовые переходы), либо теплоемкость вещества (стек-
лование). Типичный прибор для ДТА (рис. 1.1) состоит
из блока с ячейками для образца и инертного вещества,
электропечи, программатора температуры и регистрато-
ра температуры и разности температур.
Процессы, сопровождающиеся тепловыми эффектами,
приводят к возникновению разности температур между
образцом и инертным веществом, что фиксируется на
термограммах в виде пиков. Если превращения связаны
лишь с изменением теплоемкости (стеклование), то на
термограммах могут появиться характерные резкие от-
клонения (скачки) разности температур. Навески поли-
мера, необходимые для исследования методом ДТА,
обычно составляют от нескольких десятков до несколь-
ких сотен миллиграммов, а скорости нагрева чаще все-
10
Рис. 1.1. Схема прибора для
ДТА:
1,2 — регистраторы температуры
и разности температур; 3 — ком-
бинированная термопара; 4 —
блок; 5 — печь; 6 — сравнитель-
ная ячейка; 7— рабочая ячейка;
8, 9 — регулирующая термопара
с регулятором.
го — от 1 до 10°С/мин. В последнее время навески стре-
мятся уменьшать до миллиграммов, а скорости нагрева
увеличивать до 50 °С/мин и более. Методом ДТА прово-
дят исследования в широком интервале температур: ниж-
ним пределом обычно является температура жидкого
азота, верхний же практически не ограничен. Приборы
для ДТА выпускаются серийно [18, 20, 21].
Основной количественной информацией в ДТА явля-
ются температурные характеристики превращений.
Обычная точность опреде-
ления температуры ±0,5°C.
Кроме того, по термограм-
мам могут быть оценены
тепловые эффекты превра-
щений. Эти оценки основа-
ны на анализе площадей
пиков в предположении,
что теплота процесса про-
порциональна площади пи-
ка. Для определения коэф-
фициента пропорционально-
сти необходимо
предварительные
вечные
практика
ДТА для
определения тепловых эф-
фектов показывает, что этот
коэффициент от опыта к
опыту удается воспроизве-
сти лишь с небольшой точ-
провести
калибро-
Однако
опыты.
использования
количественного
ностью, поскольку он зависит от большого числа часто
трудно контролируемых факторов (например, размер,
форма и плотность образца, расположение термопары
в образце, скорость нагрева и др.) [18, 19, 21]. Хотя
проведено большое число работ по исследованию влия-
ния различных факторов на количественные характери-
стики, определяемые методом ДТА [18, 19, 21, 22], ре-
альная точность определения тепловых эффектов невы-
сока и в лучших случаях может достигать 10—15%.
В связи с этим следует отметить, что в некоторых ра-
ботах по применению ДТА количественные возможности
этого метода явно переоцениваются [13].
11
Динамическая калориметрия
Попытки повысить точность определения тепловых
эффектов методом ДТА и приблизить ее к точности ка-
лориметрических методов, с одной стороны, и стремле-
ние уменьшить затраты времени на измерения в преци-
зионной калориметрии, с другой стороны, привели к раз-
витию динамической калориметрии. Динамические кало-
риметры, применяемые в настоящее время для исследо-
вания полимеров, можно разделить на две групцы.
Принцип работы калориметров первой группы основан
на регистрации тепловых потоков, поступающих в об-
разец в процессе непрерывного нагрева. В отличие от
обычного ДТА при использовании калориметров эта ре-
гистрация осуществляется вне образца. Ко второй груп-
пе относятся приборы, работа которых основана на авто-
матической компенсации возникающей разности темпе-
ратур между измерительной и сравнительной ячейками
непосредственно в ячейках калориметра таким образом,
чтобы в течение всего опыта температура рабочей и
сравнительной ячеек поддерживалась постоянной.
Для повышения точности определения тепловых эф-
фектов методом ДТА и использования его для количест-
венного определения теплоемкости необходимо выпол-
нять следующие требования: дифференциальная термо-
пара, регистрирующая разность температур между ис-
следуемым и сравнительным веществами, должна быть
расположена вне этих веществ и строго зафиксирована
по отношению к держателям образца и эталона и по от-
ношению к источнику тепла; должна быть обеспечена
стабильность и воспроизводимость температурной про-
граммы во всех опытах. Исходя из этих требований, на
основе обычных приборов для ДТА было разработано
большое число приборов, отличающихся конструкцией
ячеек и способами фиксации в них термопар [16, 18, 19,
23—27]. Приборы такого типа в отличие от приборов
для ДТА стали называть дифференциальными динамиче-
скими калориметрами.
На рис. 1.2 показана измерительная калориметриче-
ская ячейка, используемая в дифференциальном термо-
анализаторе «Du Pont 900», который широко применяет-
ся для исследования полимеров. Этот прибор позволяет
определять теплоты переходов в температурном интер-
12
вале от —120 до 4-600 °C на образцах массой от 1 до
200 мг при скоростях нагрева от 1 до 30°С/мин. Точ-
ность определения тепловых эффектов составляет ±5%.
Ягфаровым [28, 29] предложен термографический
метод определения теплоемкости вещества (метод теп-
лового места), на основе которого был разработан при-
бор для исследования полимеров [28, 30]. Схема изме-
рительного блока такого прибора приведена на рис. 1.3.
Тепловой поток в рабочую и сравнительную ячейки по-
дается в основном через стержни (тепловые мосты).
Рис. 1.2. Схема калориметрической
ячейки термоанализ атор a «Du
Pont 900»:
/ — рабочая ячейка; 2— регистратор
разности температур; 3 — регистратор
температуры; 4 — печь.
Рис. 1.8. Схема блока для из-
мерения теплоемкости [30]:
1, 2 — закрытые сосуды, в один из
которых помещается исследуемое
вещество; 3, 4 — стержни («тепло-
вые мосты»); 5 — блок-нагреватель;
6 — крышка; 7 — торцовые теплоизо-
ляторы; 8, 9 — термопары.
Ячейки одновременно являются и спаями дифференци-
альной термопары. В процессе квазистационарного на-
грева регистрируются разность температур между изме-
рительной и сравнительной ячейками и разность тем-
ператур между сравнительной ячейкой и блоком. Этих
измерений достаточно для определения теплоемкости и
тепловых эффектов. Прибор позволяет проводить изме-
13
рения на образцах массой 10—20 мг в температурном
интервале от —196 до +200 °C. Воспроизводимость из-
мерений составляет ±0,3%.
Среди методов динамической калориметрии, основан-
ных на регистрации (а также регулировании) теплового
потока, поступающего в образец в процессе нагрева,
наиболее полно обоснован теоретически и разработан
практически метод диатермической оболочки [31—38].
Он сочетает в себе достоинства и калориметрии, и ДТА.
Измерение (интегрирование) теплового потока в этом
методе производится путем регистрации температурного
перепада во многих точках оболочки малой теплопро-
водности, окружающей исследуемый объект. Регистра-
ция этого перепада осуществляется дифференциальной
термобатареей, равномерно покрывающей поверхность
оболочки таким образом, чтобы «холодные» спаи нахо-
дились на одной ее поверхности, а «горячие» — на дру-
гой. Обычно в такой батарее имеются десятки или сотни
дифференциальных термоспаев. По своим калориметри-
ческим возможностям этот метод регистрации теплового
потока идентичен методу Тиана—Кальве [39], принцип
которого будет рассмотрен далее. Последний, однако,
теоретически обоснован лишь для условий постоянной
температуры, в то время как в обоснование метода диа-
термической оболочки в работах Барского [34, 37] при-
водится теория измерения теплоемкости и тепловых эф-
фектов для существенно переменных температурных
условий.
Измерения по методу диатермической оболочки про-
водятся в условиях квазистационарного режима. Тепло-
емкость определяется по уравнению
АТ
тср = К — h (I- 2)
где т — масса образца; К — коэффициент теплопередаЧ|И через обо-
лочку; АТ— перепад температуры на оболочке; v—скорость нагре-
ва; h — константа (термический балласт).
Тепловой эффект определяется из соотношения
Q = k\bTdt = kS (1.3)
h
где Л —Константа; время; S —площадь пика, ограниченного
кривой температурного перепада. . . .
14
Константы k и h могут быть определены калибровоч-
ными опытами. Они не зависят от свойств исследуемого
объекта, а определяются лишь свойствами и размерами
оболочки.
В дифференциальном варианте метода применяются
две оболочки, одна из которых окружает исследуемое
вещество, а другая — инертное. В этом случае сравни-
ваются тепловые потоки, поступающие в процессе на-
К рееастатору разности
температур
Ри'с. 1.4. Схема калориметриче-
ской установки, принцип дей-
ствия которой основал на ме-
тоде диатермической Оболочки
[401 •
1 — калориметрический блок; 2 —
термопара, регистрирующая темпе-
ратуру образца; 3 — диатермическая
оболочка; 4 — образец; 5 — термоба-
тареи.
Рис. 1.5. Схема дифференци-
ального сканирующего кало-
риметра DSC-1B [48]:
1 — блок; 2 — рабочая ячейка; 3 —
сравнительная ячейка; 4 — регистра-
тор разности температур; 5 — нагре-
ватели.
грева к образцу и инертному веществу. Расчетные урав-
нения при этом не изменяются, причем АТ представляет
собой разность температурных перепадов на оболочках.
На основе метода диатермической оболочки были
разработаны автоматические калориметрические уста-
новки [рис. 1.4] для исследования теплоемкости и теп-
ловых эффектов в полимерах [40—43]. В калоримет-
рическом блоке располагались две или четыре цилинд-
рические микрокалориметрические ячейки, включающие
керамические оболочки малой теплопроводности с рас-
положенными на их поверхностях термобатареями, со-
15
держащими обычно 100—150 дифференциальных термо-
спаев. Обычные размеры ячеек 0,5—3 см3. Температур-
ный интервал работы от —180 до +300 °C. Типичные
скорости нагрева 1—5°С/мин. Точность определения
теплоемкости и тепловых эффектов на образцах массой
0,5—1,0 г составляет ±(24-3)%.
Работа динамических калориметров второй группы,
в которых компенсация происходит непосредственно в
ячейках калориметра, основана на принципе, впервые
реализованном Клербро с сотр. [44] для измерения ла-
тентной энергии деформации металлов. Впоследствии
этот же принцип был использован при создании ряда
динамических калориметров [45—48], из которых наи-
большее распространение получил прибор типа DSC-1B,
выпускаемый с 1963 г. серийно американской фирмой
«Perkin ELmer» и названный дифференциальным скани-
рующим калориметром [48—51]. Непривычный для ка-
лориметрии термин «сканирующий» был использован
с целью подчеркнуть способность этого прибора давать
автоматическую развертку тепловой мощности, необхо-
димой для компенсации температурных изменений при
различных скоростях нагрева. В широком смысле слова
сканирующим является прибор, позволяющий регистри-
ровать изменение экстенсивной величины при непрерыв-
ном изменении интенсивной. В этом смысле все динами-
ческие калориметры и приборы для ДТА являются ска-
нирующими приборами, так как позволяют непрерывно
записывать соответствующую разность температур в за-
висимости от температуры.
Принцип действия динамических калориметров с
компенсацией поясняется схемой 1.5. Калориметриче-
ский блок с двумя микроячейками нагревается с посто-
янной скоростью. Два автономных микронагревателя,
расположенные в каждой из ячеек, автоматически вы-
равнивают разность температур, возникающую в про-
цессе нагрева между измерительной и сравнительной
ячейками. Тепловая мощность, необходимая для этого,
автоматически регистрируется. Энтальпия процесса оп-
ределяется по площади под графиком зависимости теп-
ловой мощности от времени (температуры).
Таким образом, фундаментальным отличием диффе-
ренциального калориметра с компенсацией от обычных
приборов для ДТА является регистрация непосредствен-
16
но тепловой мощности процесса. При этом соблюдается
полное внешнее сходство термограмм. Этот калориметр
дает возможность определять и температурную зависи-
мость теплоемкости путем сравнения результатов изме-
рения на образце с известной массой с соответствую-
щими результатами измерений для стандартного веще-
ства (например, А12О3). Калориметр позволяет прово-
дить измерения в температурном интервале от —100 до
+ 500 °C при скоростях нагрева (охлаждения) 0,62—
80°С/мин. Масса образца 1—50 мг. Точность измере-
ния тепловых эффектов 1—2%.
В последнее время фирма «Perkin Elmer» разрабо-
тала улучшенную модель калориметра DSC-2 с расши-
ренным температурным интервалом (от —175 до
+ 725 °C) [20]. В нашей стране разработан калориметр,
основанный на аналогичном принципе компенсации, и
с близкими к DSC-2 параметрами [52].
Важным достоинством этих калориметров является
возможность проведения измерений в изотермическом
режиме. Малая термическая инерция позволяет исполь-
зовать эти калориметры для исследования кинетики фи-
зических и химических процессов на основе анализа ки-
нетики выделения или поглощения тепла [51].
Микрокалориметрия
Для изучения процессов, сопровождающихся малы-
ми тепловыми эффектами или характеризующихся ма-
лой тепловой мощностью, применяются микрокалори-
метры [1, 39, 53]. Для исследования полимеров в по-
следние годы стали широко использоваться микрокало-
риметры типа Кальве [39]. Эти калориметры, работа
которых основана на методе регистрации тепловых по-
токов дифференциальными термобатареями (метод Тиа-
на—Кальве), обладают высокой чувствительностью (по
температуре — до 10~6°С, по тепловому потоку — до
10~7 Дж/с и выше), что позволяет изучать процессы
длительностью в несколько часов или даже десятков ча-
сов при общем тепловом эффекте около 1 Дж.
Типичный калориметр, работа которого основана на
этом методе (рис. 1.6), состоит из массивного блока,
в двух симметричных цилиндрических полостях которо-
го расположены микрокалориметрические ячейки также
2—264 . r 17
- • «4
цилиндрической формы, отделенные от поверхности по-
лости воздушными зазорами. Вся боковая поверхность
ячеек покрыта спаями термопар. Спаи противоположно-
го знака находятся на поверхности полости блока. В за-
висимости от назначения калориметра батареи могут
содержать от сотен до тысячи термопар. Термобатареи
ячеек соединены по дифференциальному принципу: в
одной из ячеек проводится изучение теплового явления,
Рис. 1.6. Микрокалориметр Кальве [39]:
1 — калориметрический блок; 2—калориметрические ячейки;
3 — нагреватель. Отдельно показана калориметрическая ячейка
с термобатареями.
а другая ячейка является «свидетелем». При протека-
нии в рабочей ячейке теплового процесса между внут-
ренними поверхностями ячеек возникает разность темпе-
ратур, которая автоматически регистрируется, и на ее
основании могут быть определены как интегральный
тепловой эффект, так и тепловая мощность, т. е. кине-
тика теплового процесса. Эти калориметры являются
теплопроводящими, поскольку практически все тепло
процесса из рабочей ячейки отводится в металлический
блок и рассеивается в термостате. Кроме измерительной
термобатареи в ячейках имеются специальные термоба-
18
тареи для компенсации возникающих разностей темпе-
ратур при помощи эффектов Пельтье и Джоуля [39],
что позволяет проводить изучение процессов практиче-
ски в изотермических условиях. Индивидуальное изго-
товление этих калориметров сложно. В настоящее время
они производятся серийно для работы в температурном
интервале от —196 до +800°C [2, 39, 54].
Различные варианты микрокалориметров, работа
которых основана на этом принципе, были успешно при-
менены для исследования кинетики и термодинамики
кристаллизации полимеров [41, 43, 55], для изучения
полимеризационных процессов [42, 57], для исследова-
ния растворов полимеров [58] и тепловых процессов при
деформации полимеров [59, 60].
Деформационная калориметрия
Для регистрации тепловых процессов, сопровождаю-
щих механические деформации твердых тел, используют
два типа измерений. Первый связан с регистрацией тем-
пературных изменений в процессе деформации, а второй
состоит в прямом калориметрировании возникающих при
этом тепловых эффектов. Простые оценки показывают,
что при разумном выборе размеров образца упругая де-
формация таких твердых тел, как металлы или полиме-
ры, сопровождается изменением температуры порядка
10"1—10“2°С, и это обычно соответствует тепловым эф-
фектам порядка 10~3 Дж. Очевидно, что регистрация
таких изменений температуры является задачей более
простой, чем измерение столь малых количеств тепла.
Температурные измерения могут быть использованы
для расчета тепловых эффектов, если считать процесс
адиабатическим. Однако экспериментальное выполнение
этого требования часто связано со значительными труд-
ностями (имеет место отвод тепла по проводам, расход
тепла на нагрев термопары и др.). Задача еще более
осложняется при локальном развитии деформации, на-
пример при образовании «шейки» в металлах или поли-
мерах, поскольку в этом случае изменение температуры
также носит ярко выраженный локальный характер. По-
этому для точной количественной оценки тепловых эф-
фектов при деформации целесообразно проводить их не-
посредственное калориметрическое определение.
2* 19
Малые значения тепловых эффектов, возникающих
при деформации твердых тел, требуют применения вы-
сокочувствительных калориметрических методов. Низ-
кая теплопроводность полимеров, а также влияние мас-
штабного фактора на характер деформации делают не-
обходимым применение образцов малых размеров.
Простейший способ регистрации температурных из-
менений при деформации твердых тел состоит в исполь-
зовании термопар. Впервые этот способ был применен
Джоулем [61] при исследовании изменений температу-
ры в каучуках и металлических проволоках и с тех пор
неоднократно применялся для этих целей [62—64]. При-
менение термопар позволяет надежно зафиксировать
изменения температуры в сотые доли градуса.
При измерении температурных изменений в обла-
сти образования «шейки» при ориентационной вытяжке
твердых полимеров большое значение имеет способ
крепления термопар к образцам. Мюллер с сотр. [65,
66] провел большое число температурных измерений в
области образования «шейки» с использованием раз-
личных способов крепления термопар й пришел к выво-
ду, что такого рода измерения следует рассматривать
лишь как качественные.
Поиски более корректного метода измерения повы-
шения температуры в зоне «шейки» привели к приме-
нению для этих целей фосфоров или термокрасок в ка-
честве индикаторов распределения температуры по об-
разцу в зоне «шейки» [67, 68]. Окраска фосфора и цвет
термокраски, нанесенных на поверхность образца, зави-
сят от температуры, что позволяет по спектру опреде-
лить распределение температур. Однако количественные
расчеты и в этом случае оказываются затруднительными
главным образом из-за микроразмеров зоны «шейки»,
в которой происходит собственно ориентационная вы-
тяжка, поскольку размер зерен фосфора или термокрас-
ки примерно на порядок превышает размер «микро-
шеек», вследствие чего этот метод не может давать
истинную величину температурных изменений в зоне
«шейки» [66, 69, 70].
Оба описанных способа измерения температуры при
деформации основаны на непосредственном контакте
датчиков температуры с образцом. Новые большие воз-
можности в этой области открываются с применением
20
бесконтактного метода измерения температуры, основан-
ного на регистрации теплового (инфракрасного) излу-
чения. Этот метод характеризуется достаточно высокой
интегральной чувствительностью и чрезвычайно малой
инерционностью, что позволяет использовать его не
только для оценки интегральных температурных измене-
ний при деформации, но и для регистрации кинетики
температурных изменений при быстрых процессах де-
формации.
Рис. 1.7. Блок-схема установки для регистрации тепло-
вого излучения [71]:
1 — приспособление для растяжения; 2 — регистратор инфракрас-
ного излучения (болометр); 3 — электронная схема.
Установка, работа которой основана на этом методе,
разработана недавно Бутягиным с сотр. [71]. Блок-схе-
ма ее приведена на рис. 1.7.Установка состоит из приспо-
собления для одноосного растяжения, приемника ИК-из-
лучения (болометра) и электронной схемы. В установке
использован болометр БМК-3, чувствительный в интер-
вале длин волн от 0,3 до 25 мкм, с двумя термочувстви-
тельными сопротивлениями: приемным и компенсацион-
ным. Чувствительность установки с различными боло-
метрами составляет (14-3) • 10-3 °С/мм шкалы.
Деформационная калориметрия полимеров получила
развитие в последние 15 лет. В основу ее легли работы
Мюллера [72—74], впервые сконструировавшего для
этой цели соответствующее калориметрическое устрой-
ство и привлекшего внимание к такого рода измерениям
на полимерах. Работа разработанного Мюллером мил-
ликалориметра основана на принципе газового термо-
21
метра (рис. 1.8). В одном из двух металлических ци-
линдров, соединенных между собой дифференциальным
капиллярным манометром, расположен образец полиме'
ра, а в другом — сравнительный нагреватель. Цилиндры
герметичны и заполнены воздухом или другим газом
(например, водородом). При механических воздействиях
на образец, задаваемых автоматической системой де-
формации, изменяется температура образца и окружаю-
Рис. 1.8. Схема газового дефор-
м аци о н ног о м и л л ик ал о р и м етр а
Мюллера [72]:
1,2 — регистраторы усилий и элек-
трической мощности; 3 — нагружаю-
щее устройство; 4 — тензометр; 5,
6 — следящая система; 7 — диффе-
ренциальный манометр; 8 — обра-
зец; 9 — нагреватель.
Рис. 1.9. Схема газового де-
формационного 'ка лор и м е гр а
[77]:
1 — нагружающее устройство; 2 —-
образец; 3 — регистратор разности
температур; 4 —- камера; 5 — нагре-
ватель; 6 — регистратор напряжения
и деформации; 7 — регистратор га-
зового потока; 8 — термостат; 9 —
регулятор давления.
щего его газа. Последнее влечет за собой изменение дав-
ления газа, что регистрируется дифференциальным ма-
нометром. Специальная следящая система стремится вы-
равнять давления в цилиндрах путем подачи тока в на-
греватель сравнительного цилиндра. По величине этого
тока рассчитывается тепловой эффект. Для регистрации
эндотермических эффектов калориметр предварительно
нагревают с постоянной скоростью. При поглощении
тепла образцом исходное состояние изменяется, и тепло-
вой эффект, необходимый для сохранения равномерного
нагрева, регистрируется. Чувствительность этого милли-
22
калориметра составляет примерно 4-10-8 кДж/с при ка-
либровках постоянной тепловой мощностью, а мини-
мальный тепловой импульс, который удается зафиксиро-
вать, равен 2-10~6 кДж. Точность определения тепловых
эффектов, равных (4-4-8) • 10~5 кДж, составляет ±5%.
Для меньших тепловых эффектов она достигает ±10%.
Описанный калориметр был использован для одно-
временной регистрации зависимости между напряже-
нием и деформацией при одноосном растяжении и теп-
ловых эффектов, возникающих при растяжении. Если
напряжение в образце создается достаточно быстро, на-
пример в случае упругой (обратимой) деформации
твердых полимеров, то удается определить лишь инте-
гральный тепловой эффект процесса, поскольку сущест-
вует определенное запаздывание между установлением
давления и возникновением теплового эффекта. При
изучении процессов развития ориентационной вытяжки
с постоянной скоростью можно определить не только
интегральный тепловой эффект, но и скорость выделения
тепла.
Важнейшей проблемой устойчивой работы газового
калориметра является термостатирование цилиндров.
Для достижения указанной точности измерений необхо-
димо термостатирование с точностью 10-4°С. В связи
с этим область работы калориметра не превышает
±20 °C по отношению к комнатной температуре.
В первоначальную конструкцию вносились соответ-
ствующие конструктивные изменения, тем не менее су-
ществуют методические источники ошибок измерений,
детально рассмотренные в работах Мюллера [67, 74].
Принцип калориметрирования, разработанный Мюлле-
ром, был использован рядом авторов при конструирова-
нии соответствующих устройств [75, 76].
Другое калориметрическое устройство, в котором ра-
бочей средой также является газ, можно отнести к ка-
тегории проточных газовых калориметров [77]. Образец
(рис. 1.9), закрепленный в зажимах в цилиндрической
камере, омывается потоком сухого термостатированного
газа, поступающего с постоянной скоростью. Электриче-
ский нагреватель, расположенный вблизи образца, не-
сколько повышает температуру газа, создавая опреде-
ленную разность температур входящего и выходящего
газовых потоков. Тепловой поток, возникающий при де-
23
формации образца, изменяет эту предварительно уста-
новившуюся разность температур. Для поддержания
этой разности температур в нагреватель подается элек-
трический ток, мощность которого автоматически конт-
ролируется и регистрируется; мощность пропорциональ-
на тепловому эффекту деформирования образца.
Чувствительность и качество регистрации существен-
но зависят от скорости газа и его природы. Увеличение
скорости газового потока и наличие турбулентности уве-
личивают время быстродействия прибора, однако одно-
временно повышается уровень шумов, что снижает чув-
ствительность. Это требует оптимизации скорости газа.
При расходе газа 4500 см3/мин уровень шумов при ком-
натной температуре составляет 0,6 мВт, что позволяет
регистрировать тепловые потоки мощностью около
20 мВт при разности температур газа на входе и выходе
0,35 °C. Константа времени прибора зависит от размеров
образца (главным образом толщины) и параметров га-
зового потока и колеблется от 10 до 55 с. Воспроизводи-
мость интегральных значений тепловых эффектов ±6%.
Описанные деформационные газовые калориметры
довольно сложны в работе и имеют недостаточно высо-
кую чувствительность. Работа деформационного кало-
риметра более совершенной конструкции [59] основана
на принципе Тиана — Кальве. Калориметр состоит из
двух основных блоков: собственно микрокалориметра и
блока растяжения. Блок-схема установки приведена на
рис. 1.10.
Датчиками температурных изменений являются тер-
мобатареи, содержащие по 810 дифференциальных тер-
моспаев медь — константан. Эти термобатареи смонти-
рованы в калориметрических ячейках диаметром 10 мм
и высотой 125 мм. Регистрация температурных измене-
ний осуществляется электронным потенциометром после
предварительного усиления сигнала на фотокомпенса-
ционном усилителе Ф116/1. Регистрация растягивающих
усилий производится тензометром с тензометрическим
усилителем, сигнал с которого автоматически регистри-
руется электронным потенциометром. Микрокалориметр
помещен в специальный термостат; использованные ма-
териалы позволяют работать до температур 80—90 °C.
Электрическими калибровками было установлено, что
устойчивая чувствительность при температуре 20 °C со-
24
ставляет около 4-10~7 Вт. Точность определения тепло-
вых эффектов равна ±(2—3) %. Константа времени
пустой микрокалориметрической ячейки составляет при-
мерно 30—35 с. При введении полимерных образцов она
возрастает. Ее значение для каждого образца может
быть определено калибровкой.
Многие измерения при изучении теплового поведения
полимеров при деформации могут быть выполнены в
баллистическом режиме (например, растяжение упруго-
Рис. L10. Блок-схема деформационного микрокалори-
метра [69]:
1 — регистратор усилий и деформаций; 2 — регистратор разности
температур; 3 — нагружающее устройство; 4 — тензометр; 5, 6 —
термостатирующее устройство; 7 — калориметрический блок;
8 — образец.
го материала). Изучение баллистических свойств мик-
рокалориметра [39, 59] показало, что если время тепло-
вого процесса /^0,2тЭф (тЭф — эффективная константа
времени), то AT/Q= const с точностью примерно до 1%
и не зависит от времени (ДТ — максимум пика балли-
стической кривой, Q — количество тепла).
Таким образом, тепловые эффекты процессов дли-
тельностью до 10—15 с (тЭф обычно составляет около
45—60 с) могут быть определены по величине максиму-
ма пика после предварительного определения отношения
&T/Q. Калориметр позволяет регистрировать тепловые
25
эффекты процессов длительностью менее 1 с. При этом
минимальный тепловой импульс, который удалось за-
фиксировать, составлял <С4-10~7 кДж. Изучение балли-
стических характеристик микрокалориметра показало
также, что теплота, определенная по площади под бал-
листической кривой с использованием константы по от-
клонению, точно соответствует тепловому эффекту, по-
лученному на основании анализа высоты пика.
В связи с использованием баллистических режимов
измерений важно оценить характеристическое время до-
стижения равномерного распределения температуры по
толщине образца. Это время может быть примерно оце-
нено по отношению §2/а, где а — температуропровод-
ность (для твердых полимеров и каучуков она равна
примерно ГО7 м2/с) и 6 — толщина образца. Для 6 =
= 0,01 см это время составляет около 0,1с, а для
6 = 0,03 см оно равно примерно 1 с.
Использование баллистического метода позволяет
анализировать в первом приближении случаи, когда
протекают два последовательных тепловых процесса,
один из которых баллистический (например, быстрое
растяжение упругого материала до постоянных дефор-
маций и последующий релаксационный процесс). В этом
случае интегральный тепловой эффект может быть опре-
делен по площади под кривой теплового процесса, а теп-
ловой эффект упругого растяжения — по максимуму
пика. Эксперименты показывают, что запаздывание мак-
симума пика после отключений баллистической тепло-
вой мощности составляет примерно 5 с. Поэтому влия-
нием теплоты релаксации напряжения на тепловой эф-
фект упругого растяжения в первом приближении мож-
но пренебречь.
Описанный принцип деформационного калориметра
был использован в установках для исследования тепло-
вых эффектов при деформации массивных полимерных
образцов [60] и волокон [78]. В последнем случае вме-
сто термобатареи термопар использованы термометры
сопротивления, равномерно навитые на наружных по-
верхностях рабочего и сравнительного цилиндров.
Следует признать, что более перспективными для
развития деформационной калориметрии полимеров
являются не газовые калориметры, а калориметры, ра-
бота которых в основана на методе Тиана — Кальве.
26
В отличие от газовых калориметров теория калоримет-
ров типа Тиана — Кальве,хорошо разработана [39] .и
продолжает развиваться [79], и уже сейчас существуют
надежные методы восстановления истинных термокине-
тических кривых быстрых процессов на основании запи-
санных кривых.
Дилатометрия
Среди тепловых методов исследования полимеров
распространены методы исследования теплового расши-
рения. Совокупность методов регистрации изменения
размеров и объема тел под влиянием температуры или
в результате протекающих в них физических или хими-
ческих процессов объединяется термином «дилатомет-
рия». Измерение теплового расширения полимеров ис-
пользуется для обнаружения и идентификации темпера-
турных переходов, для изучения динамики таких процес-
сов в полимерах, как плавление, кристаллизация, стекло-
вание, полимеризация, а также для установления урав-
нений состояния. Чрезвычайно важным является и тех-
ническое приложение таких измерений, поскольку поли-
меры обладают большими коэффициентами теплового
расширения по сравнению с другими твердыми телами.
Линейная дилатометрия
Изменения линейных размеров твердых тел регист-
рируются в линейных дилатометрах. Насчитывается
большое число разнообразных конструкций этих дилато-
метров [80]. Для измерений используются образцы в
виде цилиндров, нитей, пленок. Ряд дилатометров для
исследования полимерных цилиндрических образцов не-
больших размеров в широком температурном интервале
разработан Бартеневым с сотр. [6, 81—83]. Принци-
пиальная схема одного из них приведена на рис. 1.11.
Цилиндрический образец, помещенный в трубку, прижат
к ее дну толкателем, соединенным с индикатором пере-
мещений, по показаниям которого определяется измене-
ние размеров образца. Специальная автоматическая си-
стема позволяет охлаждать и нагревать образец с по-
стоянной скоростью. Для создания и обеспечения в про-
цессе работы контакта образца с трубкой дилатометра
27
и штоком используются пружины с противовесами. Ма-
лые усилия на образец (50—150 Н) обеспечивают до-
статочно высокую точность измерений (2—5%). Скоро-
сти изменения температуры в таком дилатометре малы,
что связано с необходимостью избегать больших гра-
диентов температур по образцу.
Измерения линейного теплового расширения в широ-
ком интервале скоростей изменения температуры про-
водят на тонких полимерных пленках и нитях. Для та-
кого типа образцов Сидоровичем и Кувшинским [84]
была разработана высокочувствительная установка,
позволяющая нагревать и охлаждать образцы с по-
стоянной скоростью в пределах от 0,5 °С/ч до 2 °С/мин,
Рис. 1.11. Линейный дилатометр [81—83]:
/ — регулирующая термопара; 2— рабочая камера; 3 — охлаждающее устройст-
во; 4 — образец; 5 — трубка; 6 — толкатель; 7 — детали подвески толкателя;
8—электрический контакт; 9 — индикатор; 10 — измерительная термопара.
а также поддерживать изотермические условия с точно-
стью до +0,2 °C в течение нескольких суток. Измерения
проводятся в вакууме на свободно висящих образцах,
и наряду с дилатометрическими измерениями может
быть проведено и взвешивание образцов. Типичные раз-
меры пленок: длина до 100 мм, ширина 2—3 мм. Регист-
рация изменения длины образца осуществляется двумя
катетометрами. Иная конструкция дилатометра для пле-
нок и волокон разработана Кайминем [85].
Объемная дилатометрия
Линейная дилатометрия исключает возможность ис-
следования жидких или легко деформируемых тел. Для
этих целей используют объемные дилатометры. Они ши-
роко применяются и для исследования твердых полиме-
28
ров. Простейший объемный стеклянный дилатометр со-
стоит из колбы, в которую помещают исследуемый
объект, и калиброванного капилляра, соединенного с
колбой. Эта система заполняется под вакуумом дилато-
метрической жидкостью. В качестве последней чаще все-
го используется ртуть (для исследований в температур-
ном интервале от —38 и примерно до +300 °C) или
жидкости (до —100°C и ниже). Диаметр капилляра
(обычно равный 0,5—2,0 мм) и общий объем дилатомет-
ра выбирают в соответствии с требуемой точностью из-
мерений. Объем образца рассчитывается по известным
значениям общего объема и объема заполняющей жид-
кости (с соответствующими поправками). Во время из-
мерений дилатометр находится в термостатирующем
устройстве, которое позволяет поддерживать с необходи-
мой точностью температуру или изменять ее по задан-
ной программе. Скорости нагрева и охлаждения в дила-
тометрах незначительны (^1°С/мин), что обусловлено
возможностью возникновения довольно больших темпе-
ратурных градиентов в образцах при высоких скоростях
нагрева.
Детально конструкции стеклянных дилатометров для
исследования полимеров и методика работы с ними опи-
саны Беккедалем [86] и Коваксом [87]. Точность опре-
деления объема зависит от точности регистрации высо-
ты мениска дилатометрической жидкости и температу-
ры. При регистрации высоты мениска с точностью до
0,2 мм и температуры с точностью до 0,01 °C точность
измерения удельного объема может достигать 1%!. В по-
следние годы дилатометрические измерения стремятся
полностью автоматизировать, для этого применяют раз-
личные электрические и электромеханические системы
контроля изменения объема [88, 89].
Объемная дилатометрия с большим успехом исполь-
зуется не только для определения теплового расширения
твердых тел и жидкостей, но и для исследования фазо-
вых превращений в полимерах. Физической основой
применения дилатометра в этих целях является проис-
ходящее при фазовых превращениях изменение объема,
обусловленное изменением расстояния между атомами и
молекулами. Дилатометрия часто является наиболее про-
стым, доступным и точным методом исследования кинети-
ки фазовых превращений, в частности кристаллизации
29
полимеров. Кинетика процесса может быть оценена на ос-
новании .соотношения высот мениска в начальный, конеч-
ный и промежуточный моменты времени. Такие относи-
тельные измерения особенно просты, поскольку не тре-
буют абсолютной калибровки дилатометра.
Ряд факторов может явиться источником ошибок в
этих простых измерениях. Важнейшими из них являют-
ся произвольность выбора начала и конца процесса и
неизотермичность процесса за счет теплоты кристалли-
зации [90, с. 56; 91]. Юберрайтер и Стейнер исследо-
вали влияние размеров образца, неизотермичности про-
цесса, неопределенности в выборе точки начала процес-
са и других факторов на дилатометрические кинетиче-
ские параметры и показали, как можно свести к мини-
муму влияние этих факторов [92]. Некоторые из ука-
занных источников ошибок можно исключить, используя
микродилатометры с образцами массой 0,1—0,5 г. Та-
кие дилатометры находят все более широкое применение
[93—95].
Дилатометры специальных конструкций позволяют
исследовать кинетику кристаллизации растянутых эла-
стомеров [95], а также изменение объема, сопровож-
дающее деформацию [96, 97].
Экспериментальные возможности дилатометрии мо-
гут быть значительно расширены при использовании ее
наряду с другими методами в единой установке. Такая
комплексная установка для исследования кинетики кри-
сталлизации полимеров объединяет в себе микрокалори-
метр и микродилатометр [98]. Она позволяет проводить
одновременно измерения кинетики выделения тепла при
кристаллизации и изменения объема на одном и том же
образце в одинаковых температурных условиях. Это в
значительной мере помогает идентифицировать резуль-
таты.
Еще один метод регистрации изменения плотности
при кристаллизации состоит в гидростатическом взве-
шивании образца в жидкости, находящейся при темпе-
ратуре кристаллизации [99]. Дополнительным источни-
ком ошибок при пользовании этИхМ методом являются
конвекционные потоки в жидкости.
В последние годы дилатометрия стала применяться
для изучения объемных эффектов в растворах макромо-
лекул [100].
30
Глава 2
МЕТОДЫ ИССЛЕДОВАНИЯ ТЕПЛОПРОВОДНОСТИ
И ТЕМПЕРАТУРОПРОВОДНОСТИ ПОЛИМЕРОВ
Полимеры являются плохими проводниками тепла,
т. е. имеют низкую тепло- и температуропроводность.
Экспериментальные методы определения теплопроводно-
сти полимеров могут быть разделены на две группы
[101]. К первой группе относятся методы, основанные на
закономерностях стационарного, а ко второй :— неста-
ционарного теплового потока. Температуропроводность
непосредственно может быть определена лишь в неста-
ционарных тепловых режимах. Хотя тепло- и температу-
ропроводность связаны простым соотношением, методы
их измерения принципиально различаются. Для опреде-
ления теплопроводности необходимо получить абсолют-
ное или сравнительное значение теплового потока, в то
время как для определения температуропроводности до-
статочно одних лишь температурных измерений.
Теоретической основой экспериментальных методов
является уравнение теплопроводности, связывающее
временные и пространственные изменения температуры
под действием теплового потока. В общем случае при
отсутствии внутренних источников тепла это уравнение
имеет вид [102—104]
дТ А / д2Т д*Т д2Т \
СрР dt =Цдж2 + ду2 + dz2 ] (1.4)
где сР — удельная теплоемкость; р—плотность; Т — температура;
t—-время; к — коэффициент теплопроводности.
Стационарные методы
В приборах, работа которых основана на стационар-
ных методах [105—122], распределение температуры в
образце не зависит от времени. Решение уравнения (1.4)
для тел простой геометрической формы, в которых тем-
пературное поле одномерно, с учетом закона Фурье при-
водит к уравнению [102, 103]
Q
к ~ пр _пр Кф (I. 5)
где Q—количество тепла, проходящего в единицу времени от изо-
31
термической поверхности с температурой 1\ к изотермической по-
верхности с температурой Т2\ Лф—коэффициент формы образца.
Экспериментально Л определяется для известной геомет-
рии образца на основании измерения теплового потока Q
и разности температур 1\—Т2. Уравнение (1.5) предпо-
лагает независимость % от температуры, и это застав-
ляет ограничивать величины градиентов температур в
образце несколькими градусами.
Основные трудности при использовании приборов, ра-
бота которых основана на стационарных методах, свя-
заны с созданием равномерного одномерного теплового
потока: должны быть устранены утечки тепла и обеспе-
чен идеальный контакт образца с другими элементами
прибора. Главным недостатком стационарных методов
является длительность установления необходимого теп-
лового режима при каждой заданной температуре. Это
практически исключает возможность применения ста-
ционарных методов для исследования полимеров при по-
вышенных температурах, когда длительная подготовка к
измерению может сопровождаться окислительными и
деструкционными процессами. Однако стационарные ме-
тоды являются наиболее точными и позволяют надежно
определять коэффициент теплопроводности как при от-
сутствии переходов, так и в области переходов и струк-
турных превращений.
Приборы, работа которых основана на закономерно-
стях стационарного теплового потока, различаются гео-
метрией образцов, способами учета и компенсации уте-
чек тепла, характером нагрева, размещением нагревате-
лей и др. Наряду с абсолютными методами [105—119],
позволяющими на основании измеряемых величин опре-
делять значение коэффициента теплопроводности, ис-
пользуют также относительные методы [120—122], в ко-
торых для определения теплопроводности применяют
эталонный материал с известными тепловыми характе-
ристиками.
Для измерения коэффициента теплопроводности -при
низких температурах обычно используется [115, 119]
следующая схема (рис. 1.12). При установившемся теп-
ловом режиме определяется разность температур АТ
между двумя фиксированными точками цилиндрического
образца, соединенного по торцам с источником и стоком
тепла. Длина образца выбирается в пределах 5—10 см
32
при диаметре 1—2 см. Тепловые потоки выбираются
таким образом, чтобы АТ составляла 2—8% от темпе-
ратуры образца. Точность определения коэффициента
теплопроводности зависит от температуры и составляет
4—10%. ... -
Большое число измерений коэффициента теплопро-
водности полимеров в температурном интервале от —180
до +100°C выполнено на приборе [108, ПО], схема ко-
торого приведена на рис. 1.13. Два образца в виде пла-
Рис. 1.12. Стационар-
ный метод измерения
теплопроводности при
низких температурах
[119]:
1 -- цилиндрический об-
разец; 2 — источник теп-
ла; 3 — теплоотводящая
пластина; 4 — регистра-
тор разности температур.
Рис. 1.13. Стационарный метод измерения
теплопроводности [110]:
1 — образцы; 2 —- медные блоки; 3 — нагреватель;
4 —• изоляционная фольга.
стин толщиной 5—10 мм, расположенные симметрично
относительно нагревателя, зажаты между медными пла-
стинами. Эта измерительная ячейка помещена в термо-
стат. Перепад температур на образцах определяется
термопарами, зачеканенными в медные пластины и на
поверхности нагревателя. Абсолютная точность опреде-
ления коэффициента теплопроводности составляет ±2%.
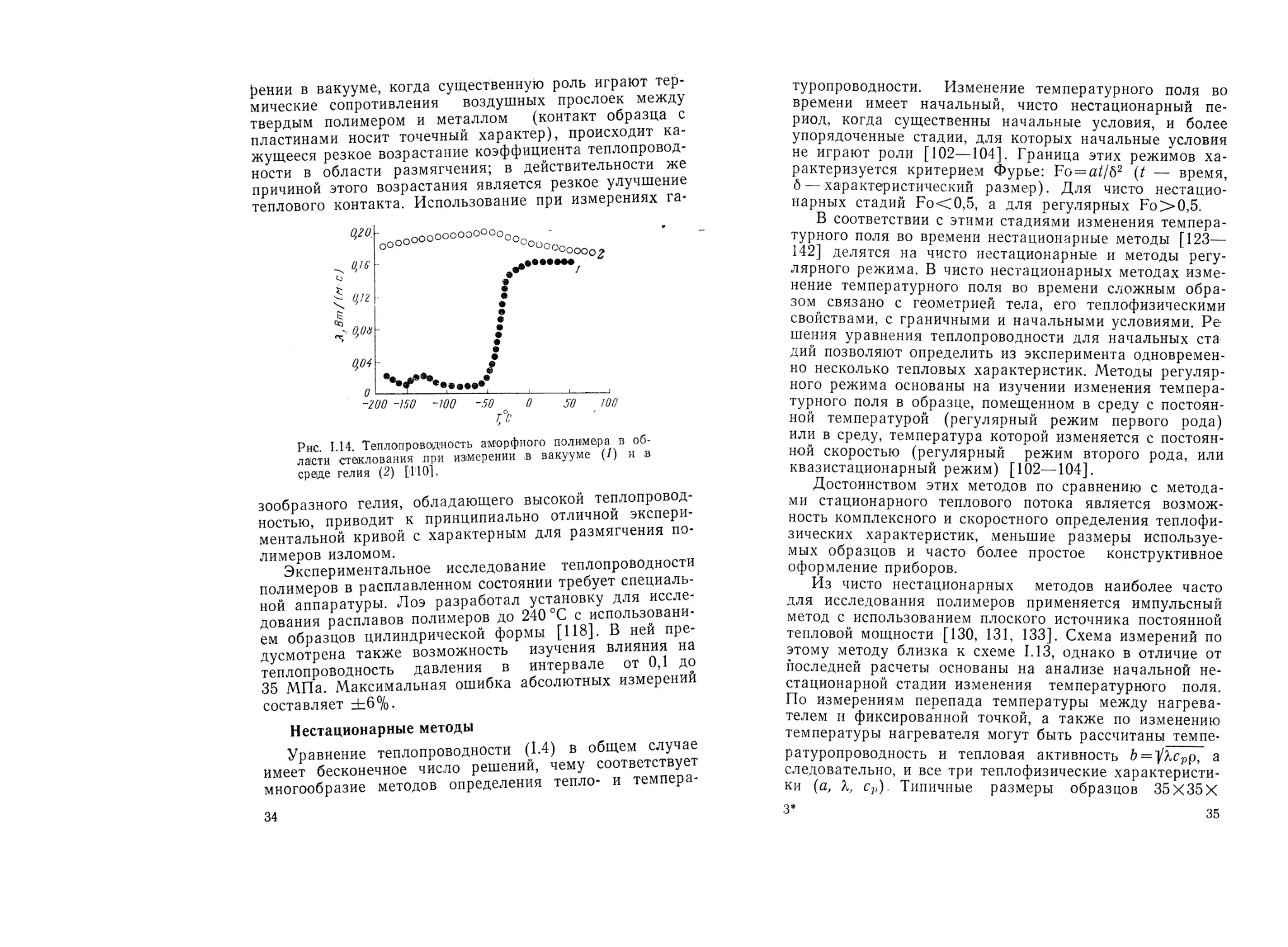
Насколько важен хороший тепловой контакт образ-
цов с нагревателем и пластинами, особенно для полиме-
ров в твердом состоянии, видно из рис. 1.14. При изме-
3—264 33
рении в вакууме, когда существенную роль играют тер-
мические сопротивления воздушных прослоек между
твердым полимером и металлом (контакт образца с
пластинами носит точечный характер), происходит ка-
жущееся резкое возрастание коэффициента теплопровод-
ности в области размягчения; в действительности же
причиной этого возрастания является резкое улучшение
теплового контакта. Использование при измерениях га-
0оооооооооооо0о -
°0о°оо000о2
7
~200 -150 -100 -50 0 50 100
Со
Рис. 1.14. Теплопроводность аморфного полимера в об-
ласти стеклования при измерении в вакууме (/) ив
среде гелия (2) [110].
зообразного гелия, обладающего высокой теплопровод-
ностью, приводит к принципиально отличной экспери-
ментальной кривой с характерным для размягчения по-
лимеров изломом.
Экспериментальное исследование теплопроводности
полимеров в расплавленном состоянии требует специаль-
ной аппаратуры. Лоэ разработал установку для иссле-
дования расплавов полимеров до 240 °C с использовани-
ем образцов цилиндрической формы [118]. В ней пре-
дусмотрена также возможность изучения влияния на
теплопроводность давления в интервале от 0,1 до
35 МПа. Максимальная ошибка абсолютных измерений
составляет ±6%.
Нестационарные методы
Уравнение теплопроводности (1.4) в общем случае
имеет бесконечное число решений, чему соответствует
многообразие методов определения тепло- и темпера-
34
туропроводности. Изменение температурного поля во
времени имеет начальный, чисто нестационарный пе-
риод, когда существенны начальные условия, и более
упорядоченные стадии, для которых начальные условия
не играют роли [102—104]. Граница этих режимов ха-
рактеризуется критерием Фурье: Fo = ^//52 (t — время,
д—характеристический размер). Для чисто нестацио-
нарных стадий Fo<0,5, а для регулярных Fo>0,5.
В соответствии с этими стадиями изменения темпера-
турного поля во времени нестационарные методы [123—
142] делятся на чисто нестационарные и методы регу-
лярного режима. В чисто нестационарных методах изме-
нение температурного поля во времени сложным обра-
зом связано с геометрией тела, его теплофизическими
свойствами, с граничными и начальными условиями. Ре
шения уравнения теплопроводности для начальных ста
дий позволяют определить из эксперимента одновремен-
но несколько тепловых характеристик. Методы регуляр-
ного режима основаны на изучении изменения темпера-
турного поля в образце, помещенном в среду с постоян-
ной температурой (регулярный режим первого рода)
или в среду, температура которой изменяется с постоян-
ной скоростью (регулярный режим второго рода, или
квазистационарный режим) [102—104].
Достоинством этих методов по сравнению с метода-
ми стационарного теплового потока является возмож-
ность комплексного и скоростного определения теплофи-
зических характеристик, меньшие размеры используе-
мых образцов и часто более простое конструктивное
оформление приборов.
Из чисто нестационарных методов наиболее часто
для исследования полимеров применяется импульсный
метод с использованием плоского источника постоянной
тепловой мощности [130, 131, 133]. Схема измерений по
этому методу близка к схеме 1.13, однако в отличие от
последней расчеты основаны на анализе начальной не-
стационарной стадии изменения температурного поля.
По измерениям перепада температуры между нагрева-
телем и фиксированной точкой, а также по изменению
температуры нагревателя могут быть рассчитаны темпе-
ратуропроводность и тепловая активность Ь = ^Ксрр, а
следовательно, и все три теплофизические характеристи-
ки (а, К, ср). Типичные размеры образцов 35X35X
3*
35
Х60 мм. Продолжительность опыта 5—7 мин. Точность
определения теплофизических характеристик по этому
методу составляет: для а — 2%, для % — 2,5%, для
сР — 3% [33], однако реальная точность, по-видимому,
значительно ниже.
В приборах, работа которых основана на методе ре-
гулярного режима первого рода, проводится изучение
закономерностей изменения распределения температуры
в образце, внесенном в среду с постоянной температу-
рой Тс, отличной от температуры образца Т [104]. Ре-
шение основного уравнения теплопроводности при соот-
ветствующих граничных условиях представляется в виде
ряда [ЮЗ]
Т Тс — А/тЦп ехр ( (1.6)
7t«»0
где Ап — постоянные, зависящие от формы тела и определяемые
из начальных условий; Un — функции координат; шп — положи-
тельные постоянные числа, зависящие от формы и размеров тела
и его теплофизических характеристик, причем тп-\ < тп < тп+\.
Этот ряд быстро сходится, и начиная с некоторого
момента распределение температур в теле описывается
выражением
Т — Тс ~ AU exp ( — mt) (I. 7)
На этой стадии величина т, называемая темпом охлаж-
дения, сохраняет постоянное значение для любой точки
тела. В общем случае темп охлаждения представляется
в виде
т — f (a, Bi, 5) (I. 8)
где Bi—аб/л— критерий Био (X — коэффициент теплоотдачи); б —
хар актер истический размер.
Наиболее простое решение уравнение (1.8) имеет в слу-
чае интенсивного теплообмена на поверхности образца
со средой, для которой Bi—>оо. В этом случае
а = т#ф (1.9)
где —коэффициент формы.
При конечных значениях Bi используют один из ва-
риантов метода, основанный на том, что отношение
36
(Ti—7С)/(Т2—Тс) постоянно для любых двух точек те-
ла, где Т2>ТЬ a
(Л — 71с)/(71а - Гс) = В = const (1.10)
Для образцов простой геометрической формы извест-
ны аналитические значения В [104]:
В = cos 8 (пластина)
В = Jo (8) (цилиндр) (1.11)
В = 1 /8 sin 8 (шар)
где /о — функция Бесселя первого рода нулевого порядка; & = ^mla.
Нахождение коэффициента температуропроводно-
сти а заключается в экспериментальном определении
величины В по отношению (1.11), расчете величины 8 и
расчете темпа охлаждения m по измерениям температу-
ры в некоторой точке тела для двух моментов времени
t\ и /2 по уравнению, вытекающему из уравнения (1.7):
In [Т (^) - Тс] - In [Т (/2) - Тс]
------------------------- (1.12)
После этого коэффициент температуропроводности полу-
чается из соотношения
а = т&№ (1.13)
Установки для определения температуропроводности
полимеров, работа которых основана на методе регуляр-
ного режима первого рода, описаны в работах [127, 128,
139, 140]. Для нахождения коэффициента теплопровод-
ности по методу регулярного режима необходимо про-
вести дополнительные измерения коэффициента тепло-
отдачи. Точность определения коэффициента теплопро-
водности составляет 4—8%.
Среди нестационарных методов определения коэффи-
циента теплопроводности наиболее распространены ква-
зистационарные методы, позволяющие определять тем-
пературную зависимость теплофизических характеристик
в процессе нагрева с постоянной скоростью. В этом слу-
чае решение одномерного уравнения теплопроводности
(1.4) для тел простейшей формы при соответствующих
граничных условиях начиная с определенного момента
имеет вид [103]
V
Т(х, 0 - То = и/- 2^-(1 + 2/Bi) - (I. 14)
37
где Го — начальная температура тела; v — постоянная скорость на-
грева; ^ф — коэффициент формы, равный соответственно 1,2,3 для
неограниченных пластины, цилиндра и шара; 2R — толщина пласти-
ны, диаметр цилиндра или шара (начало координат — в центре
тела).
Стадия нагревания, описываемая этим уравнением,
характеризуется постоянной скоростью изменения тем-
пературы всех точек тела. Перепад температуры между
двумя произвольными точками тела Х\ и х2
в этом случае обратно пропорционален температуропро'-
водности:
(1.15)
Таким образом, для определения температуропроводно-
сти необходимо измерить разность температур ДТ меж-
ду двумя точками образца при постоянной скорости на-
грева. Такой метод измерения температуропроводности
полимеров использован, например, в работе [132].
Для определения коэффициента теплопроводности по
методу квазистационарного режима необходимо кроме
перепада температуры по толщине образца определить
тепловой поток, пронизывающий образец в процессе на-
грева. Это может быть сделано путем измерения скоро-
сти нагрева эталонного стержня с известной теплоемко-
стью [129, 136, 137]. Приборы, работа которых основана
на этом принципе, применяются для определения тепло-
физических характеристик полимеров в температурном
интервале 300—650 К. Точность определения X равна
5%.
Обладая значительными преимуществами перед ста-
ционарными методами, нестационарные методы имеют
одно существенное ограничение. В большинстве случаев
теория этих методов предполагает слабую зависимость
теплофизических характеристик от температуры. Поэто-
му их применение для изучения теплофизических харак-
теристик в области резкого изменения с температурой
(фазовые переходы и структурные превращения) тре-
бует специального обоснования. Использование же обыч-
ных расчетных соотношений может приводить к ошибоч-
ным результатам.
Методы определения анизотропии
теплопроводности и температуропроводности
Ориентированные полимеры обладают ярко выра-
женной анизотропией теплопроводности и температуро-
проводности. Экспериментальное исследование анизо-
тропии теплопроводности в ориентированных полимерах
дает важную информацию о молекулярном механизме
переноса тепла в таких системах. Для измерений на мас-
сивных образцах применяются стационарные методы оп-
ределения теплопроводности [116—121, 143—145].
Ориентированные образцы, находящиеся ниже темпера-
туры стеклования или плавления, рассекаются на от-
дельные прямоугольные стержни, из них составляются
пластины, большие грани которых либо перпендикуляр-
ны, либо параллельны направлению растяжения. При
исследовании эластомеров часть ориентированного об-
разца зажимается в металлической рамке, предохраняю-
щей его от усадки, и в таком виде образец используется
для измерений.
Анизотропия теплопроводности и температуропровод-
ности может быть оценена простым и изящным методом
де Сенармонта, основанным на анализе фигур плавле-
ния тонкого покрытия, наносимого на пленку, при под-
ведении к пленке точечного источника тепла. Форма
оплавленной фигуры позволяет оценить анизотропию
теплопроводности и температуропроводности. Если ани-
зотропия теплофизических характеристик отсутствует, то
фигура плавления представляет собой круг. При нали-
чии анизотропии фигура плавления обычно является
эллипсом. Решение соответствующей плоской задачи
теплопроводности [131, 146] показывает, что
Кх -ах
ау Ь*
(I.W)
где Ьх и Ьу—соответственно большая и малая полуоси эллипса.
Такой метод оценки анизотропии теплопроводности
ориентированных полимеров был впервые применен Кар-
i иным и Слонимским с сотр. [147—149], которые ис-
пользовали в качестве покрытия парафин. В настоящее
время применяются специальные термоиндикаторы
|146].
39
ЛИТЕРАТУРА
1. Скуратов С. М., Колесов В. П., Воробьев А. Ф. Тер-
мохимия. М., изд-во МГУ, 1964, ч. I, 302 с.; ч. II, 434 с.
2. W i 1 h о i t R. C., J. Chem. Education, 1967, v. 44, № 7, p. A571;
№ 8, p. A629; № 9, p. A685; № 10, p. A853.
3. Уэструм Э., Мак-Каллаф Дж. В кн.: Физика и химия
твердого состояния органических соединений. Пер. с англ. Под
ред. Ю. А. Пентина. Т. I. М., «Мир», 1967, с. 9—160.
4. Попов М. М., Колесов В. П., ЖОХ, 1956, т. 26, с. 2385—
2390.
5. С о ч а в а И. В., Трапезникова О. Н., Вестник ЛГУ, 1958,
т. 13, № 16, с. 65—72.
6. Бартенев Г. М., Горбаткина Ю. А., Лукьянов И. А.,
Пласт, массы, 1963, № 1, с. 56—64.
7. Дол М., «Химия и технология полимеров», 1962, № 1, с. 3—49.
8. Karasz F. Е., O’Reilly J. М., J. Rev. Sci. Instr., 1966, v. 37,
p. 255.
9. Журков С. H., Левин Б. Я. В кн.: Юбилейный сборник
к 70-летию акад. А. Ф. Иоффе. М. — Л., изд-во АН СССР, 1950,
с. 260—267.
10. В о л ь к е н ш т е й н М. В., Шаронов Ю. А., Высокомол.
соед., 1961, т. 3, № 11, с. 1739—1745.
11. Hell we ge К. Н., Knap ре W., Wetzel W., Koll.-Z, 1962,
Bd. 180, S. 126.
12. T a u tz H. e. a., «Plaste u. Kautschuk», 1963, Bd. 10, S. 648.
13. К и Б. В кн.: Новейшие методы исследования полимеров. Пер. с
англ. Под ред. В. А. Каргина и Н. А. Платэ. М., «Мир», 1966,
с. 286—340.
14. Тейтельбаум Б. Я-, Аношина Н. П., «Успехи химии»,
1967, т. 36, № 1, с. 142—166.
15. Годовский Ю. К. В кн.: Энциклопедия полимеров. Т. I. М.,
«Сов. энциклопедия», 1972, с. 729—733.
16. Techniques and Methods of Polymer Evaluation. V. 1. Ed. by
F. E. Slade and L. T. Jenkins. N. Y., Marcel Dekker, 1966.
253 p.
17. W u n d e r 1 i c h B. In: Differential Thermal Analysis. Ed. by
A. Weissberger, B. W. Rossiter. V. 1, pt. V, ch. 8, Phys. Methods
of Chemistry. New York, Wiley — Intersciense, 1971.
18. Берг Л. Г. Введение в термографию. М., «Наука», 1969.
395 с.
19. Пилоян Г. О. Введение в теорию термического анализа. М.,
«Наука», 1964. 232 с.
20. Wendlandt W. W., J. Chem. Education, 1972, v. 49, № 11,
p. A623.
21. Differential Thermal Analysis. Ed. by R. C. Mackenzie. New
-York, Academic Press, 1970, v. 1, 775 p.; 1972, v. 2, 607 p.
22. Differential Thermal Analysis. Basel, Birkhauser Verlag, v. 1,
631 p.; v. 2, 800 p.; v. 3, 711 p.
23. Boer sm a S. L., J. Am. Ceram. Soc., 1955, v. 38, p. 281.
24. Schwiete H. E., Ziegler G., Ber. deut. keram. Ges. e. V.,
iqcq Rd 35 4 1Q4
25. Arndt R.’A.*, Fujita F. E., J. Rev. Sci. Instr., 1963, v. 34,
p. 868. _ . _ ____________________
40
26. Ozawa T., Bull. Chem. Soc. Japan, 1966, v. 39, p. 2071.
27. Muller F. H., Martin H., Koll.-Z., 1960, Bd. 172, S. 97.
28. Ягфаров M. III., Ж. неорг., хим., 1961, т. 6, № 11,. с. 2440—
2443.
29. Я г ф а р о в М. Ш., Изв. Казанского филиала АН СССР. Сер.
хим. наук, 1961, т. 6, с. 239—243.
30. Ягфаров М. Ш., ДАН СССР, 1968, т. 179, № 3, с. 581—
584.
31. Smith С. S., Trans. AIME, 1940, v. 137, р. 236.
32. Шурыгина Е. П. Канд, дис., М., 1941.
33. К а п у с т и н с к и й А. Ф., Барский Ю. П., Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1950, т. 20, с. 317—319.
34. Барский Ю. П., «Труды НИИСтройкерамики», 1953, вып. 8,
с. 143—166; Капустинский А. Ф., Барский Ю. П. В кн.:
Труды I совещания по термографии. М. — Л., изд-во АН СССР,
1955, с. 82—84.
35. Барский Ю. П., Фридман Н. Г., Ивницкая Р. Б. Там
же, с. 87—90.
36. Барский Ю. П., «Труды НИИСтройкерамики», 1960, вып. 16,
с. 149—162.
37. Б а р с к и й Ю. П., «Труды НИИСтройкерамики», 1961, вып. 18,
с. 126—142.
38. Барский Ю. П., «Труды НИИСтройкерамики», 1962, вып. 20,
с. 99—117.
39. К а л ь в е Э., П р а т А. Микрокалориметрия. Пер. с фр. Под
ред. Л. А. Николаева и К. П. Мищенко. М., Издатинлит, 1963.
477 с.
40. Г о д о в с к и й Ю. К-, Барский Ю. П., Пласт, массы, 1965,
№ 7, с. 57—59.
41. Годовский Ю. К. Канд, дис., М., НИФХИ им. Л. Я. Карпо-
ва, 1965.
42. Б а р к а л о в И. М., Гольданский В. И., Рапопорт В. Б.,
ДАН СССР, 1965, т. 161, № 6, с. 1368—1372.
43. Годовский Ю. К., Левин В. Ю., Слонимский Г. Л.,
Жданов А. А., Андрианов К- А., Высокомол. соед., 1969,
А, т. 11, № 11, с. 2444—2449.
44. Clarebrough L. М. е. a., Proc. Roy. Soc., 1952, A. v. 215,
р. 507.
45. Eyraud Ch., «Technica» (Lyon), 1954, v. 22, № 777, p. 2.
46. Noddack W., Jakobi R., Z. anorg. allg. Chem., 1956,
Bd. 284, S. 208.
47. Thomasson С. V., Cunnigham D. A., J. Sci. Instr., 1964,
v. 41, p. 308.
48. W a t s о n E. S. e. a., Anal. Chem., 1964, v. 36, p. 1233.
49. O’Neill M. J., Anal. Chem., 1964, v. 36, p. 1238.
50. Analytical Calorimetry. Ed. by R. S. Porter, J. F. Johnson. New
York, Plenum Press, 1968, v. 1, 332 p.; 1970, v. 2, 460 p.; 1975, v. 3,
811 p.
51. Techniques and Methods of Polymer Evaluation. V. 2. Ed. by
F. E. Slade and L. T. Jankins. New York, Marcel Dekker, 1970. 218 p.
52. Вестник АН СССР, 1974, № 7, c. 141.
53. S w i e t о s 1 a w a k i W. Microcalorimetry. New York, Reinhold,
1946. 199 p.
54. Вестник АН СССР, 1972, № 11, c. 29; 1973, № 8, c. 131.
41
55. Г о д о в с к и й Ю. К., Барский Ю. П., Высокомол. соед.,
1966, т. 8, № 3, с. 395—403.
56. Г у с а к о в с к а я И. Г. Автореферат канд. дис., М., ИХФ АН
СССР, 1968.
57. Андрианов К. А., Слонимский Г. Л., Годов-
ский Ю. К., Жданов А. А., Зав ин Б. Г., Свисту-
нов В. С., Высокомол. соед., 1973, Б, т. 15, № 11, с. 837—841.
58. Бессонов Ю. С. Канд. дис. Свердловск, 1974.
59. Г о д о в с к и й Ю. К., Слонимский Г. Л., А л е к с е е в В. Ф.,
Высокомол. соед., 1969, А, т. 11, № 5, с. 1181—1186.
60. М о л ч а н о в Ю. М., Молчанова Г. А., «Механика полиме-
ров», 1970, № 5, с. 579—585.
61. Joule J. Р., Trans. Roy. Soc., London, 1859, A, v. 149, p. 91.
62. V i 11 a r i E., Pogg. Ann., 1871, v. 144, p. 274.
63. W i 11 i a m s J., Ind. Eng. Chem., 1929, v. 21, p. 872.
64. D a r t S. L., Anthony R. L., Guth E., Ind. Eng. Chem.,
1942, v. 34, p. 1340.
65. Binder G., Muller F. H., Koll.-Z., 1961, Bd. 177, S. 129.
66. Jackel K., Koll.-Z., 1954, Bd. 137, S. 130.
67. M u 11 e r F. H. In: Rheology. V. 5. Ed. by F. R. Eirich, N. Y.,
London, Academic Press, 1969, ch. 8.
68. Brauer P., Muller F. H., Koll.-Z., 1954, Bd. 135, S. 65.
69. Muller F. H., «Kunststoffe», 1954, Bd. 44, S. 569.
70. M u 11 e r F. H., «Kautschuk u. Gummi», 1956, Bd. 9, S. 197.
71. Бутягин П. Ю., Гаранин В. В., Кузнецов А. Р., Вы-
сокомол. соед., 1974, А, т. 16, № 2, с. 333—340.
72. Muller F. Н., Coll. Czech. Chem. Comm., 1957, v. 22, p. 66.
73. Engel ter Ad., Muller F. H., Koll.-Z., 1958, Bd. 157,
S. 89.
74. Engelter Ad., Muller F. H., Rheol. Acta, 1958, v. 1,
p. 39.
75. Foster H. O., Benner R. E., Fourth Int. Cong, on Rheol.
Pt. 2. New York, J. Wiley a. Sons, 1963, p. 121.
76. Morbitzer L., Hentze G., В on art R., Koll.-Z. u.
Z. Polymere, 1967, Bd. 216—217, S. 137.
77. Duvdevani I. J., Biesenberger J. A., Gogos C. G.,
Polymer Eng. Sci., 1969, v. 9, p. 250.
78. С e p г e e в Ю. А., Ф а й н б e p г Э. 3., Михайлов Н. В., Вы-
сокомол. соед., 1972, А, т. 14, № 1, с. 250—254.
79. Гальперин Л. Н., Колесов Ю. Р., Г о н т к о в с к а я В. Т.,
Озерковская Н. И. В кн.: Пятая конференция по кало-
риметрии (расширенные тезисы докладов). М., изд-во МГУ, 1971,
с. 409—413.
80. А м а т у н и А. Н. Методы и приборы для определения темпера-
турных коэффициентов линейного расширения материалов. М.,
изд-во стандартов, 1972, 138 с.
81. Бартенев Г. М., Лукьянов И. А., ЖФХ, 1955, т. 29, № 8,
с. 1486—1498.
82. Б а р т е н е в Г. М., Горбаткина Ю. А., Высокомол. соед.,
1959, т. 1, № 5, с. 769—775.
83. Бартенев Г. М., Гарцман В. И., Зав. лаб., 1962, т. 28,
№ 2, с. 245—247.
84. Сидорович А. В., Ку вши некий Е. В., Зав. лаб., 1959,
т. 25, № 9, с. 1124—1126.
4?
85. Кай минь И. Ф., Пласт, массы, 1966, № 9, с. 62—63.
86. Bekkedahl N., J. Res. NBS, 1949, v. 42, p. 145.
87. К о v a c s A. J., Ric. Sci. Suppl., 1955, v. 25, p. 668.
88. Равич Г. Б., Егоров Б. К., Изв. АН СССР, ОХН, 1963,
№ 3, с. 481—486.
89. Автоматический дилатометр для исследования температурных
переходов в полимерах. Информ, письмо № 19 ИХВС АН УССР,
Киев, «Наукова думка», 1973.
90. Ш а р п л е з А. Кристаллизация полимеров. Пер. с англ. Под
ред. Н. Ф. Бакеева. М., «Мир», 1968. 200 с.
91. Ца хм ан Г., «Химия и технология полимеров», 1966, № 5,
с. 3—77.
92. U е b е г г е i t е г К., Steiner К., Makromol. Chem., 1966,
Bd. 91, S. 175.
93. Banks W. e. a., «Polymer», 19Q3, v. 4, p. 61.
94. T u n g L. H., J. Polymer Sci., 1967, pt. A-2, v. 5, p. 391.
95. Г о д о в с к и й Ю. К., Слонимский Г. Л., Г а р б а р Н. М.,
Высокомол. соед., 1973, А, т. 15, № 4, с. 813—897.
96. Марей А. И. и др., «Механика полимеров», 1970, № 5, с. 780—
784.
97. Holt W. L., McPherson, J. Res. NBS, 1936, v. 17, p. 657.
98. Г о д о в с к и й Ю. К., Г а р б а р Н. М., Слонимский Г. Л.,
Высокомол. соед., 1972, А, т. 14, № 8, с. 1833—1837.
99. Kolb Н. J., Izard Е. F., J. Appl. Phys., 1949, v. 20, р. 564.
100. Замяткин А. А. Дилатометрия растворов белков. М., «Нау-
ка», 1973, 101 с.
101. Годовский Ю. К- В кн.: Успехи химии и физики полимеров.
М., «Химия», 1970, с. 173—205.
102. Ч у д н о в с к и й А. Ф. Теплофизические характеристики дисперс-
ных материалов. М., Физматгиз, 1962. 456 с.
103. Лыков А. В. Теория теплопроводности. М., «Высшая школа»,
1967. 599 с.
104. К о н д р а т ь е в Г. М. Тепловые измерения. М., Машгиз, 1957.
244 с.
105. Schallamach A., Proc. Phys. Soc., 1941, v. 53, p. 214.
106. Holzmiiller W., Miinx M., Koll.-Z., 1958, Bd. 159, S. 25.
107. 3 а н e м о н e ц H. А., Фогель В. О., «Каучук и резина», 1959,
№ 2, с. 21—24.
108. Knappe Е., Z. angew. Phys., 1960, Bd. 12, S. 508.
109. Holzmiiller W., Lorez J., «Plaste u. Kautschuk», 1961,
Bd. 8, S. 351.
110. Eiermann K-, Werner K-, Z. angew. Phys., 1962, Bd. 14,
S. 484.
111. Kline D. E., J. Polymer Sci., 1961, v. 50, p. 441.
112. Sheldon R. P., L a n e K-, «Polymer», 1965, v. 6, p. 77.
113. S h о u 1 b e r g R. H., S het ter J. A., J. Appl. Polymer Sci.,
1962, v. 6, № 23, p. 532.
114. Physik der Kunststoffe. Berlin, Akademie-Verlag, 1961. 652 S.
115. Anderson A. C., Reese W., Wheathley J. C., Rev. Sci.
Instr., 1963, v. 34, p. 1386.
116. Pasquino A. D., Pils warth M. N., J. Polymer Sci., 1964,
B, v. 2, p. 253.
117. Anderson D. R., Chem. Rev., 1966, v. 66, p. 677.
118. Lohe P., Koll.-Z. u. Z. Polymere, 1965, Bd. 203, S. 115.
43
119. Koi ouch R. J., Brown R. G., j. Appl. Phys., 1968, v. 39,
p. 3999.
120. Erk S., Keller A., Poltz H., Phys. Z., 1937, Bd. 38,
S. 394.
121. Tautz H., Exper. Techn. Phys., 1959, Bd. 7, S. 1.
122. Tautz H., Koll.-Z. u. Z. Polymere, 1961, Bd. 174, S. 128.
123. Kris ch er O., Esdorn H., «VDI-Forschungsheft», 1954,
Bd. 450, S. 28.
124. Gast T., Hellwege К.-H., Kohlhepp E., Koll.-Z. u. Z.
Polymere, 1957, Bd. 152, S. 24.
125. Hellwege К.-H., Knappe W., Semjonov V., Z. angew.
Phys., 1959, Bd. 11, S. 285.
126. Ч e p к а с о в а Л. H., Вестник электропром., 1957, № 6, с. *54—
57.
127. 3 а м о л у e в В. К-, Пласт, массы, 1960, № 8, с. 46—48.
128. Г е н г р и н о в и ч Б. И., Фогель В. О., «Каучук и резина»,
1957, № 9, с. 27—30.
129. Eiermann К., Hellwege К--Н., Knappe W., Koll.-Z.
u. Z. Polymere, 1961, Bd. 174, S. 134.
130. Вертинская А. Б., H о в и ч е н о к Л. Н., Инж.-физ. ж., 1960,
т. 3, № 9, с. 65—68.
131. Новиченок Л. Н., Шульман 3. П. Теплофизические свой-
ства полимеров. Минск, «Наука и техника», 1971. 116 с.
132. Shoulberg R.-H., J. Appl. Polymer Sci., 1963, v. 7, p. 1597.
133. S t e e r e R. C., J. Appl. Phys., 1966, v. 37, p. 3338.
134. Фогель В. О., Алексеев П. Г., Инж.-физ. ж., 1962, т. 5,
Д[Ь 1 с 35____39
135. Kri’scher О., VDI-Z., 1958, Bd. 100, № 23, S. 1085.
136. Платунов Е. С., Изв. вузов. Приборостроение, 1961, № 4,
с. 90—95.
137. К у р е п и н В. В., Пл а тунов Е. С., Изв. вузов. Приборо-
строение, 1961, № 4, с. 119—123.
138. Л ев кович Л. В., Пл ату нов Е. С., Изв. вузов. Приборо-
строение, 1962, № 4, с. 85—89.
139. Кириченко Ю. А., Олейник Б. Н., Чадович Т. 3.,
Инж.-физ. ж., 1964, т. 7, с. 70—74.
140. Hattori М., Koll.-Z. u. Z. Polymere, 1965, Bd. 202, S. 11.
141. U eb er г ei ter K-, Ortmann H. J., Z. Naturforsch., 1950,
Bd. 5a, S. 101.
142. Tautz H., J. Polymer Sci., 1968, pt. C, № 16 (7), p. 3723.
143. Eiermann K., «Kunststoffe», 1961, Bd. 51, S. 512.
144. Hennig J., Koll.-Z. u. Z. Polymere, 1964, Bd. 196, S. 136.
145. Hennig J., Knappe W., J. Polymer Sci., 1964, pt. C, № 6,
p. 167.
146. Hellmuth W., Kilian H.-G., Muller F. H., Koll.-Z. u.
Z. Polymere, 1967, Bd. 218, S. 10.
147. Каргин В. А., Слонимский Г. Л., Липатов Ю. С.,
ДАН СССР, 1955, т. 104, № 1, с. 96—98.
148. С л о н и м с к и й Г. Л., Дика рев а Т. А., Высокомол. соед.,
1965, т. 7, № 7, с. 1276—1278.
149. Слонимский Г. Л., Дикарева Т. А., Павлов В. И.,
Высокомол. соед., 1966, т. 8, № 10, с. 1717—1721.
РАЗДЕЛ 11
ТЕПЛОВОЕ ДВИЖЕНИЕ В ПОЛИМЕРАХ
Глава 3
ТЕПЛОЕМКОСТЬ ПОЛИМЕРОВ
Теплоемкость твердых полимеров
Теоретический анализ теплоемкости
Согласно квантовой теории теплоемкость твердого
тела в гармоническом приближении равна [1—3]
где k— постоянная Больцмана; h—постоянная Планка; v — чисто-
та; Т — абсолютная температура; p(v)—функция распределения ча-
v
max
стот, причем Jp(v)dv = 3N (где N — число Авогадро, vmax — мак-
о
симальная частота).
Простейшее предположение о том, что все атомы
обладают одинаковой частотой (модель Эйнштейна)
приводит к следующему выражению (в символической
форме): j
/ в \
= —j (ц .2)
штейна; 0 = hv^/R—эйнштейновская характеристическая температу-
ра (ve — эйнштейновская частота).
Реальные твердые тела характеризуются большим
набором частот — спектром частот, и в соответствии с
уравнением (II. 1) для теоретического расчета теплоем-
кости необходимо знать колебательный спектр твердого
45
тела. Для теоретического определения спектра частот
используются два подхода [1—3] — континуальный
подход Дебая и подход, основанный на теоретическом
анализе спектра частот колебаний по методу Борна—
Кармана. Оба эти подхода используются и для расчета
теплоемкости полимеров.
В дебаевском приближении твердое тело рассматри-
вается как гомогенный упругоизотропный континуум.
Внутренняя энергия такого континуума представляется
как сумма энергий стоячих волн. Такой континуум обла-
дает бесконечным числом собственных колебаний, но,
поскольку реальное твердое тело построено из конечного
числа частиц, спектр ограничивается максимальной ча-
стотой Vmax, причем последняя находится из условия
нормировки [см. уравнение (II. 1) ]. Рассмотрев скорости
распространения продольных и поперечных волн, Дебай
получил функцию распределения частот в виде
9N
p(v) = -^-v2, гдеОООр (П. 3)
Таким образом, функцией распределения частот в моде-
ли Дебая является парабола, ограниченная сверху мак-
симальной дебаевской частотой vp. Подстановка урав-
нения (II.3) в уравнение (II.1) дает выражение для
теплоемкости:
ция Дебая; 0р = hv^jk — температура Дебая.
Из уравнения (II.4) при Т^О,10р следует известный
закон кубов Дебая:
4л4 / Т \3
cv = 3R р. ( g j ( 11. 6)
46
а при T 6р — классический закон Дюлонга — Пти:
Сп = 37? (II. 7)
В промежуточном интервале температур изменение теп-
лоемкости описывается уравнением (П.4). Закон кубов
Дебая, естественно, может быть справедливым лишь для
области температур, где возбуждены колебания, длины
волн которых значительно больше расстояний между
соседними структурными элементами твердого тела, т. е.
при очень низких температурах.
Для многих твердых тел теория Дебая хорошо опи-
сывает изменение теплоемкости с температурой, однако
известно много примеров отклонения от этой теории
[2, 4]. Среди них особую группу составляют анизотроп-
ные твердые тела, имеющие слоистую или цепную струк-
туру. Для этих структур характерно резкое различие сил
взаимодействия внутри цепей и слоев и между ними. По-
лимеры являются типичными представителями такого
рода твердых тел.
Основой для развития квантовой теории теплоем-
кости анизотропных структур послужили работы Тара-
сова [5—9]. Использовав континуумный подход Дебая,
он теоретически рассмотрел законы распределения ча-
стот собственных колебаний для цепных и слоистых
структур и показал, что эти законы различаются: для
линейного континуума частоты равномерно распреде-
лены по всему диапазону от 0 до vmax, а для двухмерно-
го — линейно возрастают от 0 до vmax. Обобщенная
функция распределения частот для m-мерного конти-
нуума, по Тарасову, имеет следующий вид:
р (v) dv == 3mNv~™vm'~1dv (11. 8)
При рассмотрении невзаимодействующих слоев и цепей
на основе этих распределений частот было получено
обобщенное уравнение теплоемкости:
(П. 9)
из которого следуют все три возможных частных случая:
а) для трехмерного континуума Дебая; б) для слоев,
47
или двухмерных континуумов; в) для цепей, или одно-
мерных континуумов.
Так же, как и трехмерная функция Дебая £>3, двух-
мерная D2 и одномерная функции были табулирова-
ны [8]. При Т—>0 уравнение (II.9) переходит в пре-
дельные законы:
4л4
сз = g-
/ Т \2
Сд = 43,277^ I “q )
3
(II. 10а)
*-
(II. 106)
(II. Юв)
Таким образом, теплоемкость при низких температурах
твердых тел, составленных из невзаимодействующих
слоев, изменяется по Г2-закону, а твердых тел, состоя-
щих из невзаимодействующих цепей, — по Р-закону.
Для перехода к взаимодействующим цепям и слоям
был использован формальный прием суммирования де-
баевского распределения частот для трехмерного конти-
нуума с распределениями для двух- и одномерного кон-
тинуума. Характер функций распределения частот для
всех рассмотренных выше случаев представлен на
рис. II. 1.
Для интересующего нас случая взаимодействующих
цепей уравнение теплоемкости имеет вид
(П.И)
Анализ температурной зависимости теплоемкости, про-
веденный на основе этого уравнения, позволяет выде-
лить несколько характерных областей.
1. Область очень низких температур, когда возбуж-
дены лишь низкочастотные колебания. В этом случае
анизотропность тела не проявляется, спектр соответст-
вует дебаевскому распределению частот, что математи-
чески выражается в переходе уравнения (П.П) при
в выражение
03 4 _ ( Т V
С1,3(Т->О) ~3 ех 5 п \ 03 /
(II. 12)
Согласно Тарасову, кубическая зависимость теплоемко-
сти от температуры должна наблюдаться до Т«0,1 03.
2. Переходная область температур 0,1 03^Т^О,50$,
в которой появляются и высокочастотные колебания.
В этой области теплоемкость изменяется в соответствии
с уравнением (11.11).
3. Область температур, в которой возбуждены все
низкочастотные колебания и с повышением температу-
ры возбуждаются высокочастотные колебания, харак-
Ри'с. ПЛ. Плотность 'опектраиь’норо распределения для
различных моделей твердых тел {3, 8]:
/ — модель Эйнштейна; 2 — трехмерная модель Дебая-, 3 — мо-
дель Тарасова (невзаимодействующие слои); 4—модель Тарасо-
ва (невзаимодействующие цепи); 5 — модель Тарасова (взаимо-
действующие слои); 6 — модель Тарасова (взаимодействующие
цепи).
терные для одномерного континуума. В этой области
взаимодействием цепей можно пренебречь; здесь тепло-
емкость изменяется линейно в зависимости от Т. Эта об-
ласть ограничивается температурой 7^1/7 0ь
Таким образом, согласно модели Тарасова, теплоем-
кость твердого тела, образованного цепными макромо-
лекулами, при очень низких температурах изменяется в
соответствии с дебаевской ^-зависимостью; в области
низких температур существует область линейного изме-
нения теплоемкости с температурой, и, наконец, тепло-
емкость достигает постоянного классического значения.
Физическая сторона теории теплоемкости Тарасова
неоднократно вызывала возражения [10-11]. Физиче-
4—264
49
ская необоснованность ее основных предпосылок была
особенно отчетливо показана в работах Лифшица [12—
14], посвященных теплофизическим свойствам цепных
и слоистых структур при низких температурах. Рассмот-
рим существо этих работ и сравним их с работами Тара-
сова.
Изучив теплоемкость тонких пластин (пленок) и
стержней (игл) при низких температурах, Лифшиц [13]
показал, что определяющую роль в теплоемкости таких
структур играют волны изгиба с необычным законом
дисперсии cd~F. Наличие таких волн изгиба приводит
к своеобразным температурным зависимостям теплоем-
кости при достаточно низких температурах: для тонких
стержней С оказывается пропорциональной Т1/2, а для
тонких пластин — Т1. Этот важный результат был учтен
при анализе теплофизических свойств сильноанизотроп-
ных твердых тел, имеющих цепные и слоистые структу-
ры. Для случая невзаимодействующих цепей с учетом
волн изгиба было получено следующее выражение для
теплоемкости:
/ т \ 1/2 г / т \ 1/2 "
Cv — Nkyt ( I 1 0“ j
(II. 13)
где Yi и T2 — безразмерные параметры, ’Определяемые поперечной
жесткостью цепей.
При Т<0
(II. 14)
Этот результат существенно отличается от уравнения
(П.10в), полученного Тарасовым для случая невзаимо-
действующих цепей при очень низких температурах без
учета волн изгиба.
Переход к термодинамике реальных цепных структур
проведен Лифшицем путем учета взаимодействия между
цепями на модели сильноанизотропного кристалла гек-
сагональной системы. Расчеты показали, что при очень
низких температурах учет взаимодействия между цепями
приводит к появлению кубической зависимости тепло-
емкости от температуры cv~ (Г/0)3, а в области низких
температур всегда можно выделить температурную об-
ласть, в которой взаимодействием между цепями можно
50
пренебречь и в которой теплоемкость должна зависеть
от температуры по уравнению (11.13).
Таким образом, как расчеты Тарасова, так и Лифши-
ца для взаимодействующих цепей предсказывают куби-
ческую зависимость теплоемкости от температуры в об-
ласти очень низких температур и постоянное классиче-
ское значение при высоких температурах, так же как и
теория Дебая. Расхождение между теориями имеет ме-
сто в области температур, где межцепным взаимодейст-
вием можно пренебречь. Учет лишь колебаний с обычным
законом дисперсии приводит к линейной зависимости
теплоемкости от температуры, а дополнительный учет
волн изгиба существенным образом изменяет темпера-
турную зависимость теплоемкости — она становится про-
порциональной квадратному корню из температуры. Сле-
дует подчеркнуть, что волны изгиба являются более
мягкими по сравнению с обычными колебаниями сжатия
и растяжения и при низких температурах вносят основ- .
ной вклад в теплоемкость цепных структур. Необходимо
также отметить, что в теории Тарасова учет взаимодей-
ствия проведен путем довольно формального комбини-
рования трехмерного континуума с одномерным, в то
время как в теории Лифшица это сделано строго.
Заканчивая рассмотрение теорий теплоемкости цеп-
ных структур, основанных на континуальном подходе,
следует особо подчеркнуть, что в них рассматривается
лишь скелетная теплоемкость макромолекул, а любые
боковые радикалы автоматически предполагаются жест-
ко связанными со скелетом и не обладающими независи-
мыми от него движениями.
Недавно Баур [15] еще раз детально проанализиро-
вал основные предпосылки теории теплоемкости цеп-
ных-структур Тарасова и пришел к выводу, что «значи-
тельный вклад в теплоемкость цепей вносит их гибкость,
не учитываемая в теории Тарасова» и что «в прибли-
жении сплошной среды полимерная цепь в большей сте-
пени представляет собой тонкий стержень, чем одномер-
ную струну». Как видим, эти выводы целиком совпадают
с заключением Лифшица.
Рассмотренные теории теплоемкости твердых полиме-
ров основаны на приближении сплошной среды. Другой
подход состоит в прямом расчете функций распределе-
ния частот в уравнении (II. 1) по методу Борна—Карма-
4*
51
на. Первая попытка теоретического расчета теплоемко-
сти полимеров на основе анализа их колебательных спек-
тров была предпринята в работах [16, 17]. При расче-
тах была использована модельная тетрагональная ре-
шетка с жесткими повторяющимися структурными эле-
ментами в узлах, характеризующаяся различными кон-
стантами взаимодействия ближайших соседей. При этом
была сделана попытка учесть (путем выбора соответст-
вующих соотношений между константами, характеризую-
щими взаимодействие) две важнейшие особенности
твердых тел, образованных полимерными молекулами:
сильную анизотропию и гибкость цепей.
Анализ валентной и деформационных частотных ветвей
колебательных спектров позволил выделить три темпера-
турные области характерного изменения теплоемкости:
область кубической зависимости теплоемкости при очень
низких температурах, переходную область, в которой
^теплоемкость меняется в соответствии с соотношением
cv = aiT3 + а2Г5/2, и область, в которой cv = а2Т + а4Т1/2.
Первый член в последнем выражении является вкладом
валентной колебательной ветви, а второй — двух де-
формационных. Это выражение совпадает с уравнением
Лифшица. В результате анализа был сделан вывод, что
для твердых тел, образованных цепными молекулами,
закон кубов Дебая должен соблюдаться в более узком
температурном интервале по сравнению с другими твер-
дыми телами.
Проведенные впоследствии расчеты колебательных
спектров полимерных молекул, детально проанализиро-
ванные недавно Вундерлихом и Бауром [3], показали
значительное отличие этих спектров от полученных на
основе рассмотрения моделей цепных структур. При
этом среди других вопросов был подвергнут анализу и
вопрос о соотношении скелетной теплоемкости и тепло-
емкости, связанной с подвижностью боковых групп. При
низких температурах подвижность боковых групп вы-
ражается в колебательных движениях около положений,
соответствующих наиболее устойчивому изомеру. Эта
подвижность эквивалентна заторможенному вращению.
Теплоемкость, соответствующая такому движению, мо-
жет быть рассчитана на основе уравнения
CV кол ~ RE (II* 15)
52
Часто полное «размораживание» колебаний боковык
групп происходит при очень низких температурах, так
что даже при низких температурах вклад в теплоем-
кость колебательных движений боковых групп является
классическим, т. е. cv=R.
По мере повышения температуры все больше возра-
стает возможность поворотной изомеризации, соответ-
ствующая в пределе свободному вращению боковых
групп. Предельное значение теплоемкости свободно
вращающихся боковых групп составляет cvcp=R/2, т. е.
половину от величины, характерной для колебаний. Тем-
пературная область перехода от колебаний к свободно-
му вращению определяется прежде всего высотой по-
тенциального барьера и моментом инерции радикала.
Для такого заторможенного ротатора появление макси-
мумов на температурной зависимости теплоемкости воз-
можно лишь при условии не слишком высоких барьеров
и узкого температурного интервала, в котором этот пе-
реход будет происходить [18].
Готлиб и Сочава [19] провели расчет колебательных
спектров двух простейших полимеров — полиэтилена и
политетрафторэтилена — с использованием значений си-
ловых постоянных, полученных из инфракрасных коле-
бательных спектров. Расчет позволил выделить колеба-
тельный спектр скелета и соответствующую теплоем-
кость из всего частотного спектра и теплоемкости поли-
мера. При расчетах теплоемкости для полиэтилена кро-
ме крутильных и деформационных колебаний скелета
были учтены лишь внешние деформационные (маятни-
ковые) колебания СН2-групп, поскольку вклад осталь-
ных ветвей колебательного спектра в теплоемкость поли-
этилена в рассматриваемом интервале температур ни-
чтожен. При расчете теплоемкости политетрафторэтиле-
на кроме скелетных колебаний оказалось необходимым
учесть большое число колебаний, обусловленных движе-
ниями тяжелых боковых групп. Вычисления привели к
выводу, что для полиэтилена вклад скелетных колеба-
ний является определяющим, поскольку доля маятнико-
вых колебаний СН2-групп в общей теплоемкости стано-
вится заметной (3—5%) лишь выше 150 К. Что касает-
ся политетрафторэтилена, то для него вклад скелетных
колебаний в суммарную теплоемкость при низких темпе-
ратурах составляет в среднем 40%. Готлиб и Сочава
53
полагают, что примерно такого же соотношения следует
ожидать для большинства полимеров с тяжелыми боко-
выми группами.
Таким образом, исходя из химической структуры по-
лимеров, в их колебательном спектре можно выделить
две характерные группы колебаний. Первая группа —
скелетные колебания. При низких температурах эти ко-
лебания представлены двумя колебательными ветвя-
ми — поперечных (деформационная ветвь) и крутиль-
ных колебаний, поскольку ветвь продольных (валент-
ных) колебаний высокочастотна и при низких темпера-
турах не возбуждена [3, 19]. Скелетные колебания
являются акустическими колебаниями и вносят основной
вклад в низкотемпературную теплоемкость полимеров,
особенно полимеров с легкими боковыми радикалами.
В этом случае отдельные звенья цепи могут рассматри-
ваться как точечные массы, и для оценки теплоемкости,
обусловленной акустическим спектром полимера, могут
быть использованы приведенные выше теории теплоем-
кости.
Другую группу колебаний составляют колебания
отдельных групп атомов, вклад которых в теплоемкость
зависит от соотношения масс атомов основной цепи и
боковой группы и от температуры. Их вклад в тепло-
емкость может быть рассчитан по уравнению (II.2) для
одной степени свободы из данных инфракрасной спек-
троскопии или на основании изучения низкомолекуляр-
ных соединений. Колебания боковых радикалов являют-
ся оптическими колебаниями и характеризуются значи-
тельно большими частотами, чем скелетные колебания.
В связи с этим их вклад в теплоемкость становится
ощутимым, начиная с умеренно низких температур [3].
Наличие в полимерах изомеров с различными энергия-
ми, отличающимися на 400—8000 Дж/моль [20], делает
вероятным появление еще одной составляющей теплоем-
кости, обусловленной двумя возможными энергетически-
ми состояниями. Если соотношение между изомерами,
разделенными энергетическим барьером ДЕ, изменяется
при повышении температуры, то связанная с этим пере-
ходом теплоемкость может быть определена из простого
статистического соотношения [3, 21]:
cv = R (ДЕ/RT)2 ехр
ДЕ
RT
(И.16)
54
Температурная зависимость cv, рассчитанная по форму-
ле (11.16) и представленная на рис. II.2, имеет максимум
при соотношении AE/RT~2,4, величина которого дости-
гает примерно 4 Дж/моль. Температура, при которой на-
блюдается этот эффект, зависит от ДЕ и с возрастанием
ДЕ от 400 до 4000 Дж/моль повышается с 20 до 200 К.
Уравнение (11.16) получено в предположении, что
распределение молекул в двух состояниях подчиняется
закону Больцмана. В кристаллических полимерах обыч-
но «выкристаллизовыва-
Рис. II.2. Теплоемкость системы с
двумя энергетическими уровнями,
различающимися на величину ДЕ,
как функция относительной тем-
пературы [3],
ется» и сохраняется до
плавления лишь один из
изомеров, а в стеклооб-
разном состоянии, хотя и
«заморожены» различные
изомеры, переход между
ними также невозможен.
Поэтому для основной
цепи этот вклад в тепло-
емкость в твердом состо-
янии обычно не реализу-
ется. Тем не менее мож-
но предположить, что для
боковых групп этот вклад
может иметь место в
твердых полимерах [22].
Исходя из теоретических соображений, следует ожи-
дать зависимости теплоемкости от температуры и при
обычных температурах. Для полимеров этот вопрос рас-
смотрен Бауром [3]. Из проведенного им анализа выте-
кает, что, кроме отмеченных выше вкладов в теплоем-
кость твердых полимеров при низких температурах, при
обычных и повышенных температурах следует прежде
всего ожидать вклада, связанного с увеличением энгар-
монизма колебаний, который для полимеров может быть
особенно существенным из-за больших значений
коэффициентов теплового расширения. Кроме того, опре-
деленный вклад в теплоемкость должны вносить различ-
ные характерные для твердых полимеров дефекты, кото-
рые могут существенно изменить колебательный спектр
и привести к появлению так называемых локальных ко-
лебаний. Количественный анализ теплоемкости полиме-
ров с этих позиций еще не проводился.
55
Теоретические расчеты позволяют получить теплоем-
кость при постоянном объеме cv. Однако в эксперимен-
тальных исследованиях обычно измеряется ср. Связь
между этими двумя теплоемкостями через другие тер-
модинамические величины выражается соотношением
ср — cv = t&V/k (II. 17)
где а — коэффициент объемного расширения; х— коэффициент изо-
термического сжатия.
Часто для расчетов используют формулу Нернста -Лин-
демана, вытекающую из уравнения (11.17):
cp — cv — Ac*T (11.18)
где Л—А01Тпл (здесь Ло — универсальная константа, равная
0,0849 Дж/моль).
Следует отметить, что для твердых тел различие меж-
ду ср и cv проявляется лишь в области температур выше
100 К и становится ощутимым при средних и повышен-
ных температурах.
Экспериментальные результаты
Очень низкие температуры (<30 К). Хотя исследо-
вания в этой области температур немногочисленны, техм
не менее они позволяют составить представление о ха-
рактере изменения теплоемкости твердых полимеров с
температурой в зависимости от структуры полимера и от
некоторых других параметров. Наиболее детально в
этой области температур исследован полиэтилен, и ре-
зультаты, полученные различными авторами, суммиро-
ваны и проанализированы Ризом и Такером [23, 24] и
Вундерлихом [3]. В этой области температур наблю-
дается линейная зависимость теплоемкости от степени
кристалличности (рис. П.З). Эта зависимость, очень
резкая при температурах ниже примерно 15 К, умень-
шается при повышении температуры, исчезая примерно
при 50 К. Экстраполяция на 100%-ную кристалличность
и в область полностью аморфного полиэтилена показы-
вает, что закон кубов Дебая выполняется лишь для пол-
ностью кристаллического полиэтилена при температурах
ниже 9 К. Для аморфного полиэтилена и трех исследо-
ванных образцов этот закон не выполняется; для них
56
наблюдается появление максимума при 5 К, который ха-
рактерен для аморфной части полиэтилена (он линейно
возрастает с понижением кристалличности)* Поскольку
такие же максимумы были обнаружены для других
аморфных полимеров (полистирол, полиметилметакри-
лат), а также для ряда простых аморфных веществ [24],
был сделан вывод о том, что такое поведение характер-
но для аморфных тел. Предполагается, что наиболее ве-
Ри'с. П.З. Зависимость низкотемпературной теплоемкости поли*
этилена Г231:
а — от степени кристалличности при различных температурах (1—5 К;
2 — 1,5 К; 3—10 К; 4 — 15 К; 5— 20 К; 6 — 25 К); б — от температуры
для различных степеней кристалличности (1 — 0%: 2 — 46%; 3 — 75%;
4 — 84%; 5-100%).
роятной причиной этих отклонений является наличие
неакустических вкладов в теплоемкость аморфных тел
при гелиевых температурах. Хотя формально изменения
теплоемкости удается описать введением дополнитель-
ных эйнштейновских слагаемых, физическая причина
появления этих слагаемых остается неясной.
Анализ обобщенных данных о теплоемкости кристал-
лического полиэтилена на основе модели Дебая при тем-
пературе ниже 10 К привел к значению температуры
Дебая 0р=260 К [15].
57
Низкие температуры (50—250 К). Детальное иссле-
дование температурной зависимости теплоемкости ли-
нейных полимеров в области низких температур, про-
веденное Сочавой и Трапезниковой [25—30], показало,
что соответствующие кривые для всех исследованных
ими полимеров единообразны (рис. II.4). Близкие по
смыслу результаты получены и для других полимеров
[3]. Для всех исследованных полимеров в области не
очень низких температур существует значительный ин;
тервал, в котором изменение теплоемкости близко к ли-
Рис. II.4. Зависимость теплоемкости полимеров от тем-
пературы [28, 29]:
/ — полиметилметакрилат; 2 — полистирол; 3 — поливинилиден-
хлорид; 4 — политетрафторэтилен; 5 — поливиниловый спирт;
6 — полиэтилен.
Хотя экспёриментальные закономерности изменения
теплоемкости с температурой близки к линейным в об-
ласти низких температур, это не может служить под-
тверждением теории и расчетов Тарасова, поскольку в
этой области температур экспериментальные значения
теплоемкости у большинства полимеров превышают
«классические» значения, вычисленные при условии, что
теплоемкость определяется лишь поперечными колеба-
ниями скелета [29]. Это различие может быть обуслов-
лено лишь теплоемкостью, связанной с колебаниями бо-
ковых групп. Как отмечалось выше, это означает, что
экспериментально определяемые значения теплоемкости
в рассматриваемом интервале температур представляют
собой суммарную теплоемкость, обусловленную как ко-
58
лебательным спектром скелета, так и колебаниями бо-
ковых групп. Лишь в случае полиэтилена, в согласии с
теоретическими расчетами [3, 19], экспериментальные
значения теплоемкости при этих температурах практиче-
ски целиком определяются колебаниями скелета, что
оправдывает сравнение экспериментальных данных с
теоретическими расчетами, относящимися к колебатель-
ному спектру скелета. Такое сравнение показывает [29],
что температурная зависимость теплоемкости ближе к
уравнению Лифшица (т = 0,79), чем к уравнению Та-
расова.
Таким образом, линейная зависимость эксперимен-
тальной теплоемкости от температуры в области низ-
ких температур является результатом наложения нели-
нейно изменяющихся с температурой вкладов от низко-
частотных составляющих колебательного спектра скеле-
та и боковых групп. В связи с этим естественно, что в
большинстве случаев она не может служить критерием
правильности модельных расчетов, относящихся лишь
к скелету полимерной цепи. Для проведения такого срав-
нения из общей теплоемкости необходимо выделить
теплоемкость, обусловленную колебательным спектром
скелета полимерной цепи.
Такое разделение было выполнено для ряда полиме-
ров [3], и к оставшейся части теплоемкости, связанной
лишь с акустическим спектром скелета, было применено
уравнение Тарасова [уравнение (11.11)]. Для некоторых
полимеров, особенно для полиэтилена, было достигнуто
хорошее совпадение расчетных и экспериментальных
значений теплоемкости [24, 31]. Ниже приведены харак-
теристические температуры 01 и 03, входящие в уравне-
ние Тарасова [32]:
01, К 03, К
Полиэтилен 540 147
Полистирол 230 41
Полипропилен 480 1
Полибутадиен . 580 1
Полиизопрен 580 ——
Политетрафторэтилен ..... 270 46
Поливинилиденхлорид ..... 260
Поливинилхлорид 350 175
Несмотря на достаточно хорошее совпадение теоре-
тических и экспериментальных значений теплоемкости,
59
нельзя, вероятно, сделать никаких заключений относи*
тельно физической реальности модели Тарасова, так как
теплоемкость нечувствительна к тонким изменениям ко-
лебательного спектра, особенно в области высоких час*
тот. В связи с этим даже самое грубое приближение для
спектра частот может привести к удовлетворительному
совпадению расчетных и экспериментальных данных.
Еще раз детально проанализированная недавно экспе-
риментальная температурная зависимость теплоемкости
кристаллического полиэтилена [33] лучше выражается
уравнением Лифшица, что совпадает с заключением Со*
чавы и Трапезниковой.
Теперь кратко рассмотрим возможное влияние боко-
вых радикалов на температурную зависимость теплоем-
кости и на ее значение. Выше уже отмечалось, что пере-
ход от заторможенного вращения радикалов к свобод-
ному может сопровождаться появлением максимумов на
температурной зависимости теплоемкости. В действи-
тельности же такие максимумы обычно не появляются
(рис. II.4), что обусловлено, по-видимому, недостаточной
кооперативностью такого перехода. В связи с этим тем-
пературная зависимость теплоемкости оказывается мало
чувствительной к вращению радикалов. Тем не менее
очень важная информация о подвижности радикалов и
об их вкладе в общую теплоемкость может быть полу-
чена на основании анализа экспериментальной теплоем-
кости и результатов теоретических расчетов.
Плодотворность такого подхода для полимеров была
продемонстрирована в работах Сочавы и Трапезниковой
[26, 29], в особенности на примере изучения подвиж-
ности радикалов в акриловых полимерах [30]. С целью
выяснения вклада в низкотемпературную теплоемкость
подвижности боковых радикалов были исследованы
три акриловых полимера: полиметилметакрилат
(СН2ССН3СООСН3), полиметакрилат (СН2СНСООСНз)
и полиметакриловая кислота (СН2ССНзСООН). Первый
полимер отличается от второго наличием метильной
группы в основной цепи, а от третьего — наличием ме-
тильной группы в эфирном радикале. Изменение тепло-
емкости при переходе от полиметакрилата и полимет-
акриловой кислоты к полиметилметакрилату может быть
обусловлено увеличением доли скелетной теплоемкости
за счет увеличения массы мономерного звена, низкоча*
60
стотными колебаниями самой метильной группы и, на-
конец, вращением метильной группы вокруг оси симмет-
рии третьего порядка. Сравнение молярных теплоемко-
стей полиметилметакрилата и полиметакрилата показы-
вает, что замена атома водорода метильной группой
очень мало влияет на теплоемкость в области низких
температур. Это позволило заключить, что метильная
группа, соединенная со скелетом цепи в полиметилмет-
акрилате, не вращается и что ее наличие тормозит воз-
можное внутреннее вращение как эфирного радикала,
так и входящей в его состав метильной группы. Напро-
тив, сравнение теплоемкости полиметилметакрилата с
теплоемкостью полиметакрилата и полиметакриловой
кислоты позволило сделать вывод, что метильная груп-
па эфирного радикала в полиметилметакрилате облада-
ет свободным вращением вокруг оси симметрии и вокруг
направления связи СО.
Таким образом, сравнительный анализ теплоемкостей
трех акриловых полимеров при низких температурах по-
казал различие в подвижности метильных групп в поли-
метилметакрилате.
Исходя из теоретических расчетов теплоемкости
твердых полимеров, при низких температурах должна
существовать область температур, в которой межмоле-
кулярным взаимодействием можно пренебречь и где
теплоемкость определяется лишь внутримолекулярными
параметрами. После анализа экспериментальных дан-
ных для многих полимеров стало очевидно, что выше
примерно 60 К различие в кристалличности, тактично-
сти, плотности и других параметрах, характеризующих
межмолекулярное взаимодействие, оказывает слабое
влияние на теплоемкость [3]. Эти параметры опять на-
чинают играть существенную роль выше температуры
стеклования из-за различного теплового движения в
аморфных и кристаллических областях (рис. II.5). Та-
ким образом, при низких температурах имеется значи-
тельный температурный интервал, в котором различия
в теплоемкостях твердых полимеров обусловлены массой
повторяющихся звеньев макромолекулы и вкладами бо-
ковых групп. В связи с этим изменения теплоемкости,
обусловленные изменением акустического спектра при
переходе от одного карбоцепного полимера к другому,
можно объяснить изменением массы повторяющегося
61
звена, поскольку частоты обратно пропорциональны
корню квадратному из массы.
Сочава и Трапезникова проанализировали ' с этой
точки зрения изменения скелетной теплоемкости ряда
полимеров (политетрафторэтилен, полистирол, полиме-
тилметакрилат) по отношению к простейшему полиме-
ру — полиэтилену и пришли к заключению, что эти из-
менения действительно обусловлены главным образом
изменением массы повторяющегося звена [26]. Это* за-
ключение послужило в дальнейшем основой аддитивной
схемы расчета теплоемко-
сти твердых полимеров при
:
L
Рис. II.5. Теплоемкость атакти-
ческого (/) и изотактического
(2) полипропилена со степенью
кристалличности около 0,6.
Пунктиром показана теплоем-
кость кристаллического поли-
этилена [3].
низких температурах, пред-
ложенной Вундерлихом и
Джонсом [32].
Средние и повышенные
температуры. Большинство
исследований теплоемкости
твердых полимеров выпол-
нено в этом температурном
интервале. Анализ теплоем-
кости в этой температурной
области осложняется нали-
чием переходов и релакса-
ционных процессов, кото-
рые могут проявляться на
температурной зависимости теплоемкости как в виде хо-
рошо выраженных аномалий (пики, скачки, горбы), так
и в замедленных дрейфах теплоемкости, обусловленных
медленными тепловыми процессами. Характер анома-
лий теплоемкости в полимерах будет рассмотрен от-
дельно.
Для очень многих полимеров, как аморфных, так и
кристаллических, в значительных интервалах температур
наблюдается линейная зависимость теплоемкости от тем-
пературы [34] со средним температурным коэффициен-
том dcPldT = 3-10~3 [35]. Это позволяет оценивать теп-
лоемкость твердого полимера при любой температуре
на основании одного известного значения. Обычно быва-
ют известны теплоемкости при комнатной температуре,
которые могут быть определены как экспериментально,
так и на основании аддитивной схемы вкладов отдель-
ных групп атомов. Такие таблицы вкладов, основанные
62
на данных для низкомолекулярных соединений, состав*
лены ван Кревеленом [35]. Для многих кристаллических
полимеров аморфные области в этом температурном ин-
тервале находятся выше температуры стеклования. Теп-
лоемкости стеклообразных и каучукоподобных полиме-
ров значительно различаются, и это служит основой для
расчета фс — степени кристалличности на основании
данных о теплоемкости кристаллических полимеров.
Расчеты основаны на предположении об аддитивности
вкладов в теплоемкость аморфных и кристаллических
областей (двухфазная модель) и проводятся по уравне-
нию [36, 37]
фс = ,.Ср’ а ~Ср.._ (П. 19)
СР, а к
где Ср — измеренная теплоемкость, а индексы «а» и «к» относятся
соответственно к аморфной и кристаллической фазам.
Это уравнение справедливо при температурах, при кото-
рых степень кристалличности постоянна.
Теплоемкость полимерных расплавов
Согласно кинетической теории жидкости плавление
приводит лишь к количественным изменениям в подвиж-
ности структурных элементов, а качественная картина
теплового движения в жидкости остается сходной с кар-
тиной теплового движения в кристаллическом состоянии
[38, с. 108]. Это подтверждается незначительным уве-
личением объема и теплоемкости при плавлении твердых
тел, в том числе и полимеров [35, 36]. В связи с этим
колебательный спектр полимера практически не меняет-
ся при переходе в расплавленное состояние, и, если
спектр известен, теплоемкость может быть определена
на основании уравнения (1.1). В большинстве случаев
полный колебательный спектр полимеров в расплавлен-
ном состоянии неизвестен, тем не менее даже грубые
расчеты теплоемкости на основе упрощенного колеба-
тельного спектра, в котором все высокочастотные коле-
бания усреднены, приводят к вполне приемлемым ре-
зультатам [3, 39]. Таким образом, изменение подвижно-
сти в результате плавления может привести лишь к
63
Очень Незначительному изменению Феплоемкости распла-
ва по сравнению с твердым состоянием.
Более важным является рассмотренный в предыду-
щем параграфе вклад в теплоемкость, обусловленный
переходом макромолекул в результате плавления из вы-
тянутых конформаций в свернутые. Величина этого
вклада может быть оценена на основании уравнения
(11.16).
Вероятно, наиболее существенный вклад в теплоем-
кость расплава связан с возникновением дырок. Дыроч-
ные теории жидкого состояния приводят к следующему
уравнению для теплоемкости, связанной с возникнове-
нием дырок в системе [38, 40]:
(&N \ f 8 \
exp ( (П.20)
где 8Д —энергия образования дырки; ЛГД » Af0
равновесное число дырок при температуре Т и атмосферном дав-
лении (NQ, Vo — соответственно число и объем молекул при выбран-
ных соответствующим образом повторяющихся единиц в жидкости;
Уд объем дырки); п = Vq/V.
Это уравнение было успешно применено для описания
скачка теплоемкости при стекловании аморфных поли-
меров [32, 41], которое будет рассмотрено далее. Не-
давно оно было использовано для анализа температур-
ного изменения теплоемкости полимеров в расплавлен-
ном состоянии [42]. Исследования теплоемкости поли-
мерных расплавов довольно ограниченны, что связано
с экспериментальными трудностями измерений на поли-
мерах при повышенных температурах, хотя применение
динамической калориметрии позволило заметно поднять
верхнюю границу измерений. Экстраполяционная тепло-
емкость аморфного полиэтилена (рис. II.6) проанализи-
рована Вундерлихом [39]. Линейное возрастание теп-
лоемкости с температурой выше температуры стеклова-
ния является результатом нелинейного изменения от-
дельных вкладов. По этим данным можно сделать за-
ключение о соотношении различных вкладов в экспери-
ментальную теплоемкость. Возможно, что эти соотноше-
ния типичны вообще для полимерных расплавов.
Из результатов исследования многих полимеров сле-
дует, что температурный коэффициент линейного возра-
стания теплоемкости расплавов изменяется в значитель-
но больших пределах, чем у твердых полимеров. Ван
Кревелен [35] на основании
анализа данных для 17 поли-
меров приводит значение
дс)дТ—1,2-10~3 1/К со сред-
ним отклонением ±30%.
Данные для некоторых поли-
мерных расплавов проанали-
зированы Бонди [43] на осно-
ве развиваемого им полуэмпи-
п------1---------L—
и200 300 400
рического подхода.
Систематические измере-
ния теплоемкости расплавов
четырех полимеров до темпе-
ратур начала разложения [44]
позволили сравнить результа-
ты теоретического анализа с
т,к
Рис. II.6. Теплоемкость
аморфного полиэтилена и
ее отдельные составляю-
экспериментальными данны-
ми. Теоретический расчет теп-
лоемкости был проведен пу-
тем суммирования вкладов от
колебательных спектров и от
щие, обусловленные - вкла-
дами [3]:
I — скелетных колебаний;
II— оптических колебаний;
III — разностью Ср—-cv\ IV-?
гош-транс-изомерией; V —
возникновением дырок.
изменения теплоемкости, свя-
занного с появлением дырок. Даже такой сравнительно
простой теоретический расчет приводит к весьма удов-
летворительному совпадению с экспериментальными ре-
зультатами.
Глава 4
ПЕРЕНОС ЭНЕРГИИ В ПОЛИМЕРАХ
(ТЕПЛОПРОВОДНОСТЬ И ТЕМПЕРАТУРОПРОВОДНОСТЬ)
Температурная зависимость
теплопроводности
. Существующие теории температурной . зависимости
теплопроводности полимеров делятся на две категории.
В одной’ исходят из рассмотрения коллективных коле-
5—264
65
баний структурных элементов цепи и анализируют про-
цессы рассеяния упругих волн, ограничивающие перенос
энергии. Такой подход характерен для теорий теплопро-
водности твердых диэлектриков [45, 46].
В другой группе теорий анализируют перенос энергии
в предположении некоррелированной передачи ее между
соседними атомами или молекулами через химические и
физические связи. Такой подход характерен для теорий
теплопроводности жидкостей [45, 46].
Представления о передаче энергии путем распрост-
ранения упругих волн были введены Дебаем [47], ко-
торый получил формулу для определения коэффициен-
та теплопроводности X кристаллической решетки диэлек-
трика:
Л = (П.21)
где cv — объемная теплоемкость; и — скорость распространения
волн; I — длина свободного пробега упругих (волн в твердом теле
(расстояние, на которое распространяется упругая волна, прежде
чем ее значение уменьшится в е раз по отношению к начальному
значению).
Дебай установил, что тепловое сопротивление твер-
дого тела (IFP = 1/Z) обусловлено ангармонизмом коле-
баний атомов. Пайерлс [48] ввел квантовые (фононные)
представления в теорию Дебая и показал, что тепловое
сопротивление решетки возникает в результате особого
воздействия между фононами (процессы переброса).
Основная проблема при использовании формулы
(11.21) состоит в определении длины свободного пробега
упругих волн (фононов). Значение I определяется глав-
ным образом двумя видами взаимодействия: фонон-фо-
нонного и фононов с дефектами. Для диэлектриков фо-
нон-фононное взаимодействие определяет теплопровод-
ность при высоких температурах (выше температуры
Дебая) и приводит к теоретической зависимости тепло-
проводности от температуры (Х~1/Т). При Т<<0р ве-
роятность рассеяния фононов за счет процессов пере-
броса экспоненциально понижается, что приводит к бы-
строму возрастанию теплопроводности (Х~ее/Г). Одна-
ко экспериментальные исследования показали, что при
низких температурах у кристаллических диэлектриков
наблюдается резко выраженный максимум теплопровод-
66
ности. Появление этого максимума связано с рассеянием
фононов на границах кристалла [49, 50]. Этот механизм
является доминирующим при температурах ниже темпе-
ратуры максимума, причем длина свободного пробега
для всех фононов становится постоянной и определяется
геометрическими размерами кристалла. Поэтому тепло-
проводность в этой области температур, как и теплоем-
кость, пропорциональна Т3. Типичный вид температур-
ной зависимости теплопроводности чистого кристалла
представлен на рис. II.7.
т
Рис. II.7. Температурная зависимость теплопроводности
кристалла:
/-Х-ГЗ; 2 —3 — К-1/Т.
Последующие теоретические и экспериментальные ис-
следования показали, что теплопроводность в области
максимума очень чувствительна к наличию различных
дефектов в кристаллической решетке (дислокаций, при-
месей, точечных дефектов и др.), которые являются ис-
точником дополнительного рассеяния фононов [51].
Теория низкотемпературной теплопроводности позво-
ляет количественно оценивать величину X. Этого нельзя
сказать о физической теории теплопроводности в обла-
сти средних и высоких температур, для которых теория
надежно предсказывает температурную зависимость теп-
лопроводности (Х~1/Т); числовые же значения X обыч-
но находятся на основе полуэмпирических зависимостей
Описанные представления развивались для кристал-
лических решеток, незначительно искаженных примеся-
«5* 67
ми и дефектами. Затем они были распространены и на
некристаллические твердые тела, прежде всего стекла
[52]. Теплоемкости твердых тел в кристаллическом и
аморфном состоянии почти не различаются, что объяс-
няется наличием ближнего порядка в стеклах и сходст-
вом сил, действующих между атомами в кристаллах и
аморфных телах. Однако теплопроводность кристалли-
ческих и аморфных тел очень сильно различается по
абсолютной величине и температурной зависимости [45,
51]. Отсутствие трансляционной симметрии в аморфных
телах исключает рассеяние за счет процессов переброса.
Расчеты на основе уравнения Дебая показывают, что
при известных %, cv и и значения I по порядку величи-
ны оказываются сравнимыми с размерами структурных
элементов в стеклах и не зависят от температуры [51,
52]. Поэтому теплопроводность стекол должна с пони-
жением температуры следовать за изменением cv. Это
хорошо выполняется для многих стекол до температур
20—50 К. При более низких температурах эта закономер-
ность резко изменяется. Для многих стекол при 10—
20 К значение % практически перестает зависеть от Т, а
при дальнейшем понижении температуры уменьшается,
но не так резко, как при к~Т3. Расчеты по формуле
(11.21) для этой области температур показывают, что
длина свободного пробега возрастает с понижением
температуры.
Клеменс теоретически рассмотрел процесс передачи
тепла в стеклах, введя представления о рассеянии фо-
нонов на структурных элементах стекол («структурное
рассеяние») [52]. Эта теория удовлетворительно объяс-
няла уменьшение X до 20—50 К, однако возникли
серьезные трудности при объяснении характера изме-
нения теплопроводности при более низких температу-
рах. Для области температур, где const, было по-
стулировано, что поперечные колебания рассеиваются в
значительно большей степени, чем продольные. Это при-
вело к следующему выражению для теплопроводности:
Д, = cvutIt (II. 22)
где Zl — вклад в теплопроводность продольных волн; ит, 1т — соот-
ветственно (скорость распространения и длина пробега поперечных
волн.
68
В теориях теплопроводности жидкостей исходят из
рассмотрения передачи тепла через атомы или молеку-
лы, связанные со своими соседями силами Ван-дер-
Ваальса. Бриджмен [53] провел анализ упрощенной мо-
дели передачи энергии между молекулами жидкости и
получил формулу для теплопроводности Xw:
= 2ku>Kd~2 (II. 23)
где k — констаита Больцмана; —’скорость звука ® жидкости;
d — среднее расстояние между молекулами, равное (Л4/Ар)х/з
(М — молекулярная масса, L — число Лошмидта).
Эта формула представляет собой преобразованную
формулу Дебая, в которой длина свободного пробега
предполагается пропорциональной межмолекулярному
расстоянию, а атомная теплоемкость считается постоян-
ной. Соотношения такого вида получены и другими ав-
торами [45, 54, 55].
Изложенные общие представления лежат в основе
существующих теорий температурной зависимости теп-
лопроводности полимеров. Подход, основанный на рас-
смотрении коллективных колебаний структурных единиц,
оправдан для низких температур, когда длина свобод-
ного пробега упругих волн велика по сравнению со
средними расстояниями между ними. С повышением
температуры длина свободного пробега уменьшается и
становится одного порядка со средними расстояниями
между атомами и молекулами. В этом случае правоме-
рен и «жидкостный» подход.
Аморфные полимеры
Очень низкие (водородные и гелиевые) температуры.
К настоящему времени проведено крайне мало измере-
ний при этих температурах, и основные результаты по-
лучены для полиметилметакрилата [24, 49, 56] и поли-
стирола [24, 56]. Результаты для полиметилметакрила-
та суммированы на рис. II.8. Риз [24, 56] провел теоре-
тический анализ этих экспериментальных данных. Полу-
ченные им расчетные зависимости также представлены
на рис. II.8. Анализ данных ниже 50 °C основан на мо-
дифицированной теории Клеменса, Поскольку экспери-
ментальные результаты плохо согласовывались с суще-
ствующей теорией [уравнение (11.22) ], поперечная со-
69
ставляющая теплопроводности была представлена в ви-
де суммы трехмерной (Хз) и одномерной (Xi) составляю-
щих. Это разбиение основано на соображениях Тара-
сова.
Рассчитанные значения теплопроводности отличают-
ся от экспериментальных не более чем на 10%. Важно
также то, что они хорошо передают общую температур-
ную зависимость теплопроводности. При высоких темпе-
ратурах доминирующей является составляющая М, в.то'
время как при низких температурах — две другие со-
ставляющие. Расчеты показывают, что при высоких тем-
пературах теплопроводность вдоль полимерной цепи
Рис. II.8. Температурная зависимость теплопроводности
полиметил’метакрилата [24]. (Пояснения в тексте.
превышает межмолекулярную теплопроводность на по-
рядок. Это важное заключение использовано в модель-
ных представлениях механизма переноса тепла в аморф-
ных полимерах. Однако ограниченность эксперименталь-
ных данных по низкотемпературной теплопроводности
аморфных полимеров не позволяет пока судить о том,
в какой мере проведенные расчеты являются общими
для полимерных стекол.
Низкие и средние температуры (от —180 до
+ 150 °C). Характер изменения теплопроводности в этой
температурной области исследован достаточно подроб-
но [57—59]. Типичная температурная зависимость теп-
лопроводности в этой области приведена на рис. II.8.
Для изменения теплопроводности аморфных полимеров
70
характерно наличие излома при температуре стеклова-
ния. Ниже температуры стеклования X имеет небольшой
положительный температурный коэффициент, а выше —
отрицательный. Такое изменение теплопроводности на-
блюдалось для многих полимеров [57—65]. Эксперимен-
тальные данные в этой температурной области могут
быть представлены [35] в обобщенном виде (рис. II.9).
При этом нормировка теплопроводности проводится по
максимальному значению X, а температуры — по темпе-
ратуре стеклования. В качестве температурного пара-
метра может быть использовано и более сложное выра-
жение [66].
Рис. II.9. Обобщенная зависимость теплоп1ро1водно1сти
аморфных полимеров [35]:
□ — полидиметилсилоксановый каучук; — полиизобутилен; О—
натуральный каучук; Д — полипропилен; V—политрифторхлор-
этилен; X — полиэтилентерефталат; ▼ — поливинилхлорид;
ф — полиметилметакрилат; + — поликарбонат; А — поливинил-
карбазол.
Следует, однако, отметить, что в некоторых работах
по теплопроводности аморфных полимеров вместо плав-
ного излома отмечалось резкое ступенчатое изменение
теплопроводности в области размягчения [67]. Прове-
денное недавно специальное исследование показало, что
резкие провалы теплопроводности в области размягче-
ния появляются лишь при применении нестационарных
методов и являются, по-видимому, результатом методи-
ческих особенностей нестационарных методов измерения
в областях переходов [68].
Для объяснения экспериментальной зависимости теп-
лопроводности аморфных полимеров от температуры
был проведен теоретический анализ структурной модели,
в основу которой были положены следующие предполо-
71
жёния {58]. Аморфный полимер представляет собой сет-
ку, образованную двумя типами термических сопротив-
лений. Ковалентные связи между атомами в цепи харак
теризуются низким термическим сопротивлением, а
ван-дер-ваальсовы связи между цепями — большим тер-
мическим сопротивлением. Из-за статистического направ-
ления валентных связей предполагается, что коэффи-
циент теплопроводности всей сетки X пропорционален
усредненному значению коэффициента теплопроводно-
сти ван-дер-ваальсовых связей W
X = kpKw (11.24)
где kP не зависит от температуры.
Расчет Kw проведен на основе уравнения Дебая, в
котором макроскопические параметры заменены на мо-
лекулярные характеристики:
- cv = 3£/(cd2/i); и = о Y Мт
где fw—константа жесткости ван-дер-ваальсовой связи; т — масса
элементарного (структурного элемента; «=/.
Для расчета fw использован 12/6-потенциал взаимо-
действия U {fw=d2u/d(d2). Подстановка определенного
таким образом значения fw в уравнение (П.24) приво-
дит к выражению, связывающему теплопроводность со
средним расстоянием между атомами. Это позволяет
выразить относительный температурный коэффициент
теплопроводности у • через объемный коэффи-
циент теплового расширения а и соответственно измене-
ние Да в
a/-L
\ X ’dT)
области размягчения — через изменение
. Таким образом, именно различие в коэф-
фициентах теплового расширения аморфных полимеров
до и после размягчения является физической причиной
изменения температурного коэффициента теплопровод-
ности.
Поскольку изменением валентных связей с изменени-
ем температуры можно пренебречь по сравнению с из-
менением ван-дер-ваальсовых связей, окончательное
уравнение имеет вид
[(11.25)
72
Это уравнение достаточно хорошо описывает взаимо-
связь между изменением Да в области размягчения .и
экспериментально обнаруженным пологим максимумом
на температурной зависимости теплопроводности [58].
Кристаллические полимеры
Очень низкие (водородные и гелиевые) температуры.
Для кристаллических полимеров, как и для аморфных,
до сих пор проведено очень мало измерений теплопро-
водности при этих температурах. Характерные данные
для полиэтиленов различной плотности приведены на
рис. 11.10. Анализ результатов по определению низко-
температурной теплопроводности кристаллических поли-
меров показывает, что температурная зависимость X
в этой области хорошо передается следующим выраже-
нием [24, 69, 70, 80]:
Л &Т / А \ Г xle* dx
“ )] , ( А \(tiu \ (IL26)
где Л =Л/2л (здесь h— постоянная Планка); Д/а—эксперименталь-
но определяемый параметр с размерностью см-1; x—hvl\(kT}\
Л2— средняя длина свободного пробега; и — продольная скорость
звука.
Существует предположение [69], что отклонение от
линейного изменения теплопроводности с температурой
обусловлено наличием дополнительных источников рас-
сеяния, причем процесс рассеяния характеризуется
средней длиной свободного пробега Лг, которая не за-
висит от температуры. Риз и Такер [69] предположили,
что этими внутренними источниками рассеяния в кри-
сталлических полимерах являются сферолиты. Констан-
та Аг для исследованных полимеров, определенная экс-
периментально из данных по теплопроводности, в темпе-
ратурном интервале 1—4,5 К имеет порядок 10-4 см. Это
соответствует размеру сферолитов в исследованных об-
разцах. Проведенное впоследствии исследование на по-
лиэтиленах и найлоне 6,6 не привело к такому соответ-
ствию [70]. Определенное экспериментально в темпера-
турном интервале 1,2—4 К значение Лг было значитель-
но меньше размеров сферолитов и хорошо совпадало
73
с размером ламелярных кристаллов. Таким образом, со-
вокупность имеющихся экспериментальных результатов
по температурной зависимости теплопроводности кри-
сталлических полимеров в области очень низких темпе-
ратур указывает на важную роль надмолекулярной
структуры полимера в процессах рассеяния фононов.
Рис. 11.10. Температурная
з а.в ис и м о с т ь теп лоп ров о дм о -
сти полиэтилена различной
плотности [24]:
/ — 0,98 Мг/м3; 2 — 0,96; 3 —
0,92 Мг/м3.
Низкие и средние температуры (от —180 до+150 °C).
По характеру температурной зависимости теплопровод-
ности в этой области температур кристаллические поли-
меры можно разделить в основном на две группы [59,
71—74]. К первой группе относятся полимеры, тепло-
проводность которых с повышением температуры падает
(полиэтилен, полиоксиметилен, полиоксиэтилен, най-
лон 6). Для полимеров второй группы характерно повы-
шение теплопроводности с повышением температуры
(полиэтилентерефталат, изотактический полипропилен,
политетрафторэтилен, полихлортрифторэтилен). Для
обеих групп характерно увеличение теплопроводности с
ростом степени кристалличности. По абсолютным зна-
чениям теплопроводность полимеров первой группы вы-
ше, чем полимеров второй группы.
Первая группа. Особенности изменения тепло-
проводности полимеров этой группы изучены на примере
полиэтилена. Экспериментальные результаты для образ-
цов различной 1ПЛОТНОСТИ приведены на рис. 11.11.
Для объяснения найденных закономерностей измене-
ния теплопроводности с изменением температуры и сте-
пени кристалличности была использована двухфазная
74
модель строения кристаллических полимеров [59]. При
расчете теплопроводности смеси кристаллических и
аморфных областей использовалась формула Максвелла
для смеси, в которой шарообразные частицы равномер-
но расположены в окружающей массе. Этой формулой
удовлетворительно описываются данные по теплопровод-
ности смесей и в случае частиц несферической формы.
Формула имеет вид
2Ха -р Хд Ф (Хк — Ха)
i iil 27)
где Ха и Хк—теплопроводность соответственно аморфной и кри-
сталлической фаз; ф— степень кристалличности.
Для расчетов использовалась также и более простая
формула для теплопроводности смеси [75], аналогичная
формулам для других характеристик (объем, теплоем-
кость) двухфазной полимерной системы [35]:
Х = фХк+(1—ф)аа (11.28)
На основе этих формул по известным значениям теп-
лопроводности для данной
Р ис. 11.11. Темп ер ату р н ая
зависим ость теплопроводно -
сти полиэтилена [71] раз-
личной плотности (при
20 °C):
1 — 0,982 Мг/м3; 2 — 0,962; 3 —
0,961; 4 — 0,951; 5 — 0,923; 5 —
0,918 Мг/м3 (пунктир — быстро
охлажденные образцы, сплош-
ные линии — отожженные об-
разцы); / — теплопроводность
кристаллической решетки; II —
теплопроводность аморфного
полиэтилена.
степени кристалличности
можно определить Ха и и сравнить их с соот-
ветствующими зависимостями для аморфных и кристал-
лических веществ. Такие расчеты были выполнены на
основании формулы (11.27) для той области температур,
где степень кристалличности остается неизменной.
75
Для согласования экспериментальных данных с рас-
четными потребовалось введение предположения о на-
личии в высококристаллических образцах областей с ну-
левой теплопроводностью (пузырьков воздуха). Это
предположение имеет экспериментальные основания
[76]. Соответствующие расчетные зависимости представ-
лены на рис. 11.11. Изменение Лк с температурой хорошо
описывается гиперболическим законом (Х~1/Г), харак-
терным для низкомолекулярных кристаллов. Параметры
уравнения 1/А,к=а+6Т для некоторых полимеров при-
ведены ниже [59]:
а, м-К/Вт 6-103, м/Вт
Полиэтилен............—0,026 4,6
Полиоксиметилен .... 0,225 4,47
Найлон 6, найлон 6,6 .. . 1,8 1,77
Изменение теплопроводности высококристаллических
образцов полиэтилена с температурой и полученная рас-
четным путем гиперболическая зависимость лк от темпе-
ратуры свидетельствуют о том, что механизм передачи
тепла в полимерных кристаллах не отличается принци-
пиально от механизма передачи тепла в низкомолеку-
лярных кристаллических диэлектриках. Разница заклю-
чается лишь в том, что для полимерных кристаллов
можно ожидать существенной анизотропии теплопровод-
ности. Средняя длина пробега фононов вдоль макромо-
лекулы достигает длины складки в кристалле. В то же
время в направлении, перпендикулярном оси макромоле-
кул, теплопроводность существенно меньше и зависит
от термического сопротивления физических связей. Сред-
няя длина свободного пробега фононов определяется
размерами кристаллитов в этом направлении.
Изменение теплопроводности аморфной фазы Ла по-
лиэтилена с температурой в общих чертах характерно
для аморфных полимеров. Примерно вблизи —20 °C на-
блюдается плавный излом; изменение температурного
коэффициента теплопроводности при этом составляет
1,9 % /°C, что близко к среднему значению для аморфных
полимеров. Анализ теплопроводности на основе двух-
фазной модели был проведен и для полиамидов [59].
Несмотря на то что описанный метод позволяет предска-
зывать зависимость теплопроводности от температуры,
он вызывает возражения. Прежде всего, необходимо от-
метить, что расчеты приобретают смысл лишь в предпо-
76
ложении, что кристаллические участки равномерно рас-
положены в аморфной матрице. Обратное предположе-
ние — аморфные участки равномерно размещены в кри-
сталлической матрице — приводит к лишенному физи-
ческого смысла результату: при низких температурах
для теплопроводности аморфной фазы получаются отри-
цательные значения. Для такого высококристаллическо-
го полимера, каким является полиэтилен, предположе-
ние о размещении кристаллической фазы в аморфной,
имеет мало смысла. Очевидно, что описанный подход к
анализу экспериментальных данных по теплопроводно-
сти кристаллических полимеров является чисто формаль-
ным приемом. Это же относится и к расчетам темпера-
турного хода степени кристалличности по уравнению
(11.27) на основании измерений теплопроводности [77].
Вторая группа. Температурный ход теплопро-
водности полимеров этой группы напоминает изменение
теплопроводности аморфных полимеров. При анализе
характера закономерностей теплопроводности полиме-
ров этой группы также используется двухфазная модель.
Расчетные зависимости Ак для полимеров этой группы
не подчиняются зависимости а изменяются по-
добно теплопроводности аморфных полимеров ниже тем-
пературы размягчения. На основании этого сделано
предположение, что длина свободного пробега фононов
в таких полимерах сопоставима с размерами расстоя-
ний между отдельными атомными группами, и потому
к ним можно применить модельный анализ, проведенный
для аморфных полимеров. При этом более высокая теп-
лопроводность кристаллической фазы по сравнению с
аморфной объясняется различием в их плотностях: в бо-
лее плотной кристаллической фазе силовые постоянные
связей больше и, следовательно, меньше тепловое сопро-
тивление. На этой основе теплопроводность связывается
с плотностью. Расчеты показывают, что
*а ~ Т Ра
Аа Ра
где ф—5,8 [как и в уравнении (11.25)].
(II. 29а)
(II. 296)
77
Изменение теплопроводности
в области фазовых переходов
Систематические исследования характера изменения
теплопроводности в области переходов в настоящее вре-
мя отсутствуют, а приводимые в литературе данные ча-
сто противоречивы. Наиболее существенные изменения
теплопроводности происходят в области плавления. Для
Рис. 11.12. Температурная зависи-
мость теплопроводности полиэти-
лена различной плотности в обла-
сти плавления [59]:
1 — 0,976 Мг/м3; 2 — 0,945; 3 — 0,921
Мг/м3.
полимеров как первой,
так и второй группы от-
мечается сильное умень-
шение теплопроводности
при плавлении [58, 71,
75]. Примером могут
служить данные для раз-
личных полиэтиленов
(рис. 11.12). Как и в слу-
чае стеклования аморф-
ных полимеров, нестацио-
нарные методы приводят
иногда к резким экстре-
мумам теплопроводности
в области плавления
[78], что, скорее всего,
связано с ошибками из-
мерений [68]. Стеклова-
ние аморфной фазы кри-
сталлических полимеров практически не отражается на
температурной зависимости теплопроводности.
Изменение теплопроводности в области переходов в
твердом состоянии исследовалось главным образом для
политетрафторэтилена. Озава и Канари [79] на прак-
тике проверили результаты различных авторов и срав-
нили их со своими данными. В области первого фазово-
го перехода при 20 °C наблюдается резкое уменьшение
теплопроводности, в то время как изменения при втором
фазовом переходе при 30 °C находятся в пределах экс-
периментальных ошибок. Оценки изменения теплопро-
водности в области первого перехода на основании из-
вестного скачка плотности при этом переходе с исполь-
зованием уравнения (11.296) привели к согласующимся
с экспериментом результатам [71].
78
Теплопроводность и молекулярные параметры
Молекулярная масса
Низкое термическое сопротивление валентных связей
по сравнению с физическими связями должно иметь
своим следствием возрастание теплопроводности с уве-
личением молекулярной массы, что и отмечалось в ран-
них работах для полимергомологов стирола [81, 82].
Тем не менее единый взгляд на зависимость теплопро-
водности от молекулярной массы в настоящее время
еще не определился. Айерман [58] на основе развитых
им модельных представлений о теплопроводности аморф-
ных полимеров получил следующее соотношение для
такой зависимости:
(II. 30)
где Хоо—теплопроводность полимера бесконечно большой молеку-
лярной массы; А — константа; Р — степень полимеризации.
Это соотношение получено в предположении, что при
расчленении макромолекулы по валентным связям кон-
формации ее участков остаются неизменными, а в ме-
стах разрыва валентные связи заменены на физические.
Проверка этого соотношения на полидиметилсилоксанах
и полиэтиленгликолях показала, что оно не выполняется
и что при больших молекулярных массах теплопровод-
ность оказывается мало чувствительной к степени поли-
меризации [83]. В связи с этим были предприняты по-
пытки теоретического обоснования этой эксперименталь-
ной зависимости.
Лоэ [83] рассмотрел упрощенную модель располо-
жения клубкообразных макромолекул в объеме жидкого
полимера, аппроксимируя объем, занимаемый макромо-
лекулой, кубиком со стороной й = Р1/з. Учитывая допол-
нительное термическое сопротивление между соседними
кубиками и считая, что внутри кубика теплопроводность
Хоо определяется лишь степенью полимеризации макромо-
лекулы, Лоэ получил следующее соотношение, отличное
от уравнения (11.30):
1 1 В
т = —+ W (IL31)
79
Таким образом, рассмотренная модель приводит к _ли-
нейной зависимости термического сопротивления от Р"1/з
во всей области молекулярных масс и не предсказывает
замедления роста теплопроводности при больших моле-
кулярных массах. Это, по-видимому, связано с тем, что
рассмотренная модель вряд ли реалистична при значи-
тельных молекулярных массах. Тем не менее анализ
экспериментальных данных в терминах соотношения
(11.31) для полидиметилсилоксанов и полиэтиленглико-
лей, казалось бы, подт-
верждает эту зависи-
мость [83]. Однако было
отмечено [84], что прове-
м иг
nw
Рис. ПЛЗ. Зависимость теплопро-
водности от молекулярной массы
Г751:
1 — полистирол; 2 — полиэтилен.
денный Лоэ анализ не
может служить доказа-
тельством справедливо-
сти соотношения (11.31),
так как использованные
данные относятся лишь
к области низких моле-
кулярных масс и не
включают результатов
для высоких.
Хансен и Хоу [75] теоретически рассмотрели зависи-
мость теплопроводности полимеров от молекулярной
массы и от ориентации макромолекул. Их рассмотрение
основано на «жидкостной» модели теплопроводности и
учитывает различную степень взаимодействия соседних
звеньев, связанных между собой химическими и физиче-
скими связями. Эта теория приводит к заключению, что
в области низких молекулярных масс теплопроводность
должна линейно зависеть от Р1/2, а затем будет наблю-
даться значительное замедление этого роста. Имеющие-
ся экспериментальные результаты лучше всего отвечают
этой зависимости (рис. 11.13).
Различия в температурном изменении теплопровод-
ности разных полимеров обусловлены различием в хи-
мической структуре макромолекул. Так, для полистиро-
ла отклонения от линейного хода проявляются более
резко, чем для полиэтилена. Это объясняется относи-
тельно большим влиянием бензольного кольца на пере-
дачу тепла между звеньями соседних макромолекул.
80
Разветвленность
Систематическое исследование влияния разветвлен-
ности цепи полимера на теплопроводность проведено на
примере полиолефинов [85]. Для выявления этого влия-
ния в чистом виде была исследована теплопроводность
расплавов. Из рис. 11.14 видно, что теплопроводность
линейного неразветвленного полиэтилена монотонно
уменьшается при увеличении числа метильных групп
на 1000 атомов углерода главной цепи. Попытка теоре-
тического обоснования этой закономерности основана
0Р----—I----L------1----1—
' 0 250 500 750 1000
Число групп СНз
на 1000 атомоО С
Рис. 1Ы4. Влияние разветвленно-
сти на теплопроводность распла-
вов полиолефинов [85]:
1 — полиэтилен высокой плотности;
2 — полиэтилен низкой плотности; 3 —
сополимер этилена с пропиленом; 4 —
полипропилен; 5 — полиизобутилен; / —
давление 0,1 МПа; // — 30 МПа.
Пропилен Зтилон
Метилмотпприлит Стирол
Рис. 11.15. Зависимость теп-
лопроводности сополимеров
от состава Г59]:
/ — сополимер этилена с пропи-
леном; 2 — сополимер метил-
метакрилата со стиролом.
на модельных представлениях о передаче тепла в
аморфных полимерах. Так как элементарная теплопро-
водность ван-дер-ваальсовых связей равна
то увеличение массы элемента цепи за счет боковых
групп должно приводить к понижению теплопроводно-
сти. В общих чертах такое поведение характерно и для
акриловых полимеров [59, 86].
Структура цепи
Статистическое введение в цепь различных атомов и
атомных группировок приводит к большому разнообра-
6—264
81
зию физических связей и соответственно элементарных
термических сопротивлений. Характер изменения тепло-
проводности с изменением состава может быть рассмот-
рен на примере сополимеров метилметакрилат — стирол
и полиэтилен — полипропилен (рис. 11.15) [59, 86].
В первом случае наблюдается линейное изменение теп-
лопроводности в зависимости от содержания компонен-
тов. Однако для других аморфных сополимеров (напри-
мер, метилметакрилат — акрилонитрил) такая линей-
ность отсутствует. Поэтому линейное соотношение для
сополимера метилметакрилат — стирол может быть
следствием того, что их молекулярные массы практиче-
ски равны, а температуры стеклования соответствующих
гомополимеров близки. Для сополимеров, компоненты ко-
торых способны к кристаллизации, изменение теплопро-
водности при изменении содержания компонентов имеет
иной характер. Уменьшение теплопроводности сополи-
мера по сравнению с гомополимерами обусловлено
уменьшением степени кристалличности. При сравнимых
соотношениях компонентов сополимеры этилена с про-
пиленом являются аморфными.
Анизотропия теплопроводности
В ориентированных аморфных и кристаллических
полимерах, а также в растянутых каучуках появляется
анизотропия теплопроводности [87—92]. При этом сте-
пень анизотропии сильно зависит от степени ориентации.
Теплопроводность в направлении растяжения оказы-
вается во всех случаях выше, чем теплопроводность в
изотропном состоянии, а также в направлении, перпен-
дикулярном ориентации.
Для ориентированных аморфных полимеров пробле-
ма анизотропии была рассмотрена теоретически. Одно-
осное растяжение приводит к увеличению числа сегмен-
тов макромолекул, ориентированных преимущественно
в направлении растяжения. Поэтому повышенная тепло-
проводность вдоль направления растяжения у ориентиро-
ванных полимеров объясняется преимущественно пере-
носом тепла вдоль ориентированных макромолекул.
Айерман [94] рассмотрел изменение соотношения теп-
лопроводностей вдоль (% ц) и перпендикулярно (Z_l)
82
оси растяжения и получил уравнение
1 2____з
Л (1 + %о
где Хо — теплопроводность изотропного материала.
(11.32)
Это соотношение может быть использовано при лю-
бых степенях растяжения. Хансен и Хоу [75] теоретиче-
ски рассмотрели эту же проблему и получили выраже-
ние, отличное от соотношения (11.32):
(II. 33)
Результаты экспериментальной проверки этих соотно-
шений для некоторых полимеров приведены в табл. II. 1.
Таблица II.1. Теплопроводность ориентированных полимеров [68]
Полимер Степень растяжения, % Теплопроводность при 25 °C, Вт/(м-К) Ар X 1 / к || \ 1/2 \ /
Л и х0 рас- чета . х0 экспер.
Полистирол 400 0,173 f 0,154 0,160 0,162 1,06 1,03
Полиметилметакрилат 157 0,238 0,181 0,200 0,197 1,08 1,10
275 0,280 0,168 0,195 0,197 1,17 1,19
Поливинилхлорид . . 85 0,228 0,149 0,168 0,168 1,13 1,17
165 0,279 0,140 0,167 0,168 1,20 1,29
Экспериментальные и расчетные данные достаточно хо-
рошо совпадают для обоих соотношений. Формула
(11.32) приводит, по-видимому, к несколько лучшему
соответствию, чем формула (11.33).
Еще больших эффектов, связанных с анизотропией
теплопроводности, следует ожидать для ориентирован-
ных кристаллических полимеров, в которых наряду с
ориентацией участков макромолекул в аморфных обла-
стях происходит ориентация кристаллитов. Немногочис-
ленные качественные [87—89] и количественные резуль-
таты [91] свидетельствуют о большой анизотропии теп-
лопроводности ориентированных кристаллических поли-
меров.
6* 83
Влияние давления на теплопроводность
Теплопроводность полимеров, так же как и низко-
молекулярных жидкостей [53], увеличивается под дей-
ствием давления [78, 83, 95]. Для расплавов некоторых
полимеров (полиолефины, полиметилметакрилат, поли-
стирол, полиамид 6, пентон) в области давлений до
30 МПа относительное увеличение теплопроводности со-
ставляет 1,6-10-4 [83].
Так как в изотропных полимерах теплопроводность
определяется преимущественно термическим сопротив-
лением физических связей, увеличение теплопроводности
под давлением обусловлено, очевидно, изменением упру-
гости физических связей (увеличение fw).
Температурная зависимость
температуропроводности
Температурная зависимость температуропроводности
некоторых аморфных полимеров приведена на рис. 11.16.
В области стеклообразного состояния температуропро-
Рис. 'IL16. Температурная зависимость температуропро-
водности аморфных полимеров:
1 — полиэтилентерефталат ,[99]; 2 — поликарбонат [99]; 3 — по-
ливинилхлорид [98]; 4 —полистирол [98]; 5 — полистирол [97]
6 — полиметилметакрилат [104].
водность уменьшается с понижением температуры, в об-
ласти стеклования наблюдается ее резкое падение, а в
области вязкотекучего состояния она остается почти по-
84
стоянной в широком интервале температур [96—100].
Падение температуропроводности в области стеклования
может служить характеристикой стеклования. Оно явля-
ется следствием резкого возрастания теплоемкости при
стекловании, поскольку теплопроводность в этой области
изменяется очень незначительно.
Рис. 11.17. Температурная зависимость температуропро-
водности кристаллических полимеров:
1 — политетрафторэтилен [105]; 2 — полиэтилен низкой плотности
[102]; 3 — полиэтилен высокой плотности [102]; 4 — полипропи-
лен [108].
Характерные экспериментальные результаты по тем-
пературной зависимости температуропроводности кри-
сталлических полимеров приведены на рис. 11.17 [98,
101—103]. Повышение температуры сначала приводит
к заметному падению температуропроводности, затем
в области плавления она проходит через минимум, а по-
сле завершения плавления достигает примерно тех же
значений, что и перед плавлением, обнаруживая при
этом очень слабую зависимость от температуры. Такой
вид аномального поведения температуропроводности в
температурном интервале плавления можно предсказать
на основании сопоставления поведения теплоемкости,
теплопроводности и плотности.
Температурная зависимость температуропроводности
политетрафторэтилена имеет экстремумы в области пе-
реходов при 20 и 30 °C [96, 99, 104]. На примере поли-
трифторхлорэтилена было изучено влияние степени кри-
85
сталличности на температурную зависимость температу-
ропроводности в температурном интервале от 20 до 90 °C.
С повышением степени кристалличности температуро-
проводность возрастает, при этом одновременно изме-
няется и скорость уменьшения температуропроводности
с возрастанием температуры: для образцов с более вы-
сокой степенью кристалличности она больше. Изменение
степени кристалличности отражается на температуро-
проводности в значительно меньшей степени, чем'на
теплопроводности.
Температуропроводность
и молекулярные параметры
Систематические исследования температуропроводно-
сти, охватывающие широкий круг полимеров различно-
го химического строения, в настоящее время отсутст-
вуют. Тем не менее влияние отдельных молекулярных
факторов на температуропроводность уже изучено на
примере некоторых полимеров, и результаты исследова-
ний позволяют составить определенное представление
об этом влиянии.
Хотя в отдельных работах отмечалось, что влияние
молекулярной массы на температуропроводность очень
незначительно [81, 82], проведенное недавно системати-
ческое исследование [100] на 11 фракциях в полймер-
гомологическом ряду полиметилметакрилатов свиде-
тельствует о том, что такое влияние все-таки имеет ме-
сто (рис. 11.18). Кроме заметного влияния молекуляр-
ной массы на температуропроводность эти результаты
демонстрируют появление перехода в области жидкого
состояния полиметилметакрилатов. Этот переход для
всех фракций наблюдается примерно через 60 °C после
перехода в высокоэластическое состояние. В результате
анализа возможных причин появления этого перехода
был сделан вывод, что он связан со стеклованием син-
диотактических последовательностей в цепи полиметил-
метакрилата [100, 105]. Так же как и в случае тепло-
проводности, в области малых и средних молекулярных
масс температуропроводность линейно зависит от корня
квадратного из молекулярной массы, а затем рост тем-
пературопроводности резко замедляется. Такая же тен-
денция отмечалась и для температуропроводности рас-
86
творов полистирола в гексахлордифениле [106]. Это за-
ключение хорошо согласуется с теоретическими пред-
ставлениями, развитыми недавно для объяснения темпе-
ратурной зависимости температуропроводности аморф-
ных полимеров [107].
Хаттори [98] исследовал температуропроводность
трех образцов полиэтилена с молекулярными массами,
равными соответственно 21-103, 80-103 и 120-103. При
температурах от 20 до 100 °C наблюдается линейное
уменьшение температуропроводности для всех трех об-
разцов. При комнатных температурах температуропро-
Рис. 11.18. Влияние молекулярной массы и температуры
на температуропроводность полиметилмета1кр1илата
[100]. Молекулярная масса (в скобках — начальные
значения температуропроводности) :
/ — 106 (0,900); 2 — 23 300 (0,945); 3— 11 700 (0,980); 4 — 8700
(0,967); 5 — 6600 (0,978); 5— 5400 (0,994); 7 — 3600 (0,980)- 8 — 1600
(0,950); 9 — 1370 (0,940); /5 — 800 (1,055); // — 200 (1,165).
водность полиэтилена самой высокой молекулярной мас-
сы значительно превосходит температуропроводность
двух других образцов, причем температурная зависи-
мость для этого образца более резкая. При 80 °C зна-
чения температуропроводности всех трех образцов близ-
ки между собой. Однако полученные зависимости могут
быть скорее связаны с тем, что образец высокой молеку-
лярной массы имел, по-видимому, большую степень кри-
87
сталличности по сравнению с другими образцами, а не
с различием в молекулярных массах.
Изучение влияния на температуропроводность размеров
боковых заместителей полимеров винилового ряда пока-
зывает, что температуропроводность в расплавленном
состоянии тем меньше, чем больше боковые группы [97].
На примере поливинилхлорида исследовалось влия-
ние пластификатора на температуропроводность [98, 99].
Наличие пластификатора заметно снижает температуро-
проводность чистого полимера в стеклообразном состоя-
нии и практически не изменяет ее в области высокоэла-
стического состояния. Повышение степени сшивания по-
листирола дивинилбензолом повышает температуропро-
водность системы как в стеклообразном, так и в высо-
коэластическом состоянии.
Было исследовано влияние гидростатического дав-
ления на температуропроводность [78, 107] и показано,
что повышение давления приводит к возрастанию тем-
пературопроводности во всем исследованном темпера-
турном интервале.
Глава 5
ТЕРМИЧЕСКИЕ ОСОБЕННОСТИ ПЕРЕХОДОВ
И РЕЛАКСАЦИОННЫХ ПРОЦЕССОВ
В ПОЛИМЕРАХ
Согласно термодинамической классификации фазо-
вых переходов (превращений) порядок (род) перехода
определяется условием прерывности соответствующих
производных термодинамического потенциала по темпе-
ратуре и давлению при непрерывном изменении самого
термодинамического потенциала [109, с. 47]. При этом
производные более высокого порядка обращаются в бес-
конечность. Обычно ограничиваются рассмотрением пе-
реходов первого и второго рода, часто встречающихся
в природе. Фазовые переходы первого рода протекают
при определенной температуре в условиях равновесного
сосуществования обеих фаз. Они характеризуются раз-
рывами на температурных зависимостях энтальпии,
энтропии и объема (рис. 11.19). Основными термодина-
88
мическими уравнениями для фазовых переходов перво-
го рода являются уравнения
Q = T (S2 —Sr) = TSS (11.34)
dP S2-St AS
dT ~ ~ AV (н.гю;
где Q, AS и AV — изменение соответственно теплоты, энтропии и
объема при переходе; Р и Т— соответственно температура и дав-
ление перехода [109, с. 48; 110].
Типичными фазовыми переходами первого рода явля-
ются процессы плавления и испарения.
Рис. II. 19. Изменение термодинамического потенциала и его произ-
водных при фазовых переходах первого рода (а), второго рода (б)
и стекловании (в).
Для фазовых переходов второго рода характерны не-
прерывные кривые энтальпии, энтропии и объема и раз-
рывы на температурных зависимостях вторых производ-
ных: теплоемкости, теплового расширения и сжимаемо-
сти. К, фазовым переходам второго рода относятся пере-
ходы порядок — беспорядок в некоторых сплавах, изме-
нения в ферромагнитных и антиферромагнитных веще-
89
ствах в точке Кюри и переход в жидком гелии при тем-
пературе 2,2 К [ИО]. Все эти переходы протекают в
пределах одной фазы. Основным соотношением между
термодинамическими характеристиками для переходов
второго рода является уравнение Эренфеста:
TV (Да)2 = ДсрДх
(11.36)
где а— коэффициент объемного теплового расширения; х — изотер-
мическая сжимаемость [НО].
Характер аномалии теплоемкости вблизи этих пере-
ходов имеет Z-форму, определяющую постепенное раз-
витие и внезапное окончание перехода, в связи с чем
эти переходы часто называют Х-точками.
Казалось бы, что на основании формы температурной
аномалии теплоемкости или коэффициента теплового
расширения переходы легко классифицировать. Однако
положение осложняется тем, что имеется много аномаль-
ных изменений теплоемкости, близких по виду к ^-точ-
кам, в которых теплоемкость постепенно возрастает до
довольно острого максимума, но не претерпевает разры-
ва ни в одной точке [111]. Такие переходы классифици-
руют как размытые фазовые переходы первого рода,
в которых степень кооперативности меньше, чем в гипо-
тетических фазовых переходах первого рода. В связи
с этим классификация переходов на основании формы
аномального изменения теплоемкости не является одно-
значной. Это положение вполне подтверждается эмпи-
рической классификацией переходов, встречающихся в
органических соединениях [112].
Основой термодинамической классификации перехо-
дов является понятие о фазе. Одновременно с фазовым
переходом может происходить и изменение агрегатного
состояния вещества (плавление, испарение). Эта связь,
очевидно, не является обязательной. Известные перехо-
ды второго рода и многие переходы первого рода проис-
ходят в пределах одного и того же агрегатного состоя-
ния — твердого (исключением является переход в жид-
ком гелии). Однако возможна и обратная ситуация —
изменение агрегатного состояния без фазового перехода.
Известным примером является превращение жидкости
в стеклообразное состояние (переход из жидкого в твер-
дое агрегатное состояние в пределах одной и той же
фазы — жидкой). Характер изменения термодинамиче-
90
ских характеристик при таком превращении приведен
на рис. II. 19,в. Природа этого превращения будет рас-
смотрена далее.
Необходимо обратить внимание на то, что термин
«переход» часто употребляется как для характеристики
фазовых переходов, так и для характеристики превра-
щений типа стеклования. Описанные выше общие пред-
ставления используются и для описания переходов в по-
лимерах, хотя уже давно отмечалось, что классическое
понятие фазы в применении к полимерам в значительной
мере утрачивает определенность, характерную для низ-
комолекулярных веществ [113, 114]. Тем не менее этот
вопрос до сих пор остается неразрешенным, и исследо-
ватели вынуждены использовать существующие пред-
ставления. Кроме того, в последнее время в работах по
полимерам термин «переход» стали совершенно необос-
нованно применять для обозначения любой аномалии на
температурной зависимости практически любого изучае-
мого свойства, несмотря даже на то, что природа анома-
лий часто остается совершенно неизвестной и что мно-
гие из этих аномалий физических свойств полимеров
имеют очевидный релаксационный характер [115].
В связи с этим необходимо ясно понимать условность
термина «переход» в применении к полимерам.
Согласно классификации Бойера [116], температур-
ные переходы и релаксационные явления в полимерах
могут быть сгруппированы следующим образом: 1) плав-
ление (ТПл); 2) стеклование (7"с); 3) превращения в
стеклообразном состоянии (7,<ТС); 4) промежуточные
превращения (Тс<Г<Тпл). Следуя этой классифика-
ции, рассмотрим термические особенности переходов и
релаксационных процессов в полимерах, обращая осо-
бое внимание на кинетический аспект превращений.
Плавление и кристаллизация
Плавление
Хотя и высказывались различные точки зрения на
природу плавления и кристаллизации полимеров [114,
117—123], в настоящее время принято считать, что,
несмотря на частичную кристаллизацию, плавление
(кристаллизация) полимеров представляет собой фазо-
91
вый переход первого рода, сопровождающийся поглоще-
нием (выделением) теплоты и изменением удельного
объема, т. е. является процессом той же природы, что
и плавление (кристаллизация) низкомолекулярных ве-
ществ [124]. Однако плавление и кристаллизация поли-
меров имеют ряд характерных особенностей, отличаю-
щих их от соответствующих процессов в низкомолеку-
лярных соединениях.
При медленном нагревании кристаллических полиме-
ров задолго до температуры плавления наблюдается по-
степенное уменьшение степени кристалличности с ростом
Рис. 11.20. Температурная зависимость теплоемкости (а)
и энтальпии (б) изотактического полипропилена в об-
ласти плавления [126]. Пунктиром отмечен гипотетиче-
ский ход кривой для случая плавления в точке.
температуры, причем этот эффект обратим. Это явление
называют частичным плавлением [125]. Верхняя грани-
ца частичного плавления обычно принимается за темпе-
ратуру плавления.
Частичное плавление в полимерах приводит к харак-
терному изменению теплоемкости в области плавления.
На температурной зависимости теплоемкости это отра-
жается следующим образом. Значения удельной тепло-
емкости начинают возрастать (сначала медленно, а за-
тем быстрее), и на кривой появляется пик с конечным
максимумом (рис. 11.20). Такое изменение теплоемко-
сти в области плавления полимеров внешне очень похо-
же как на Х-аномалии теплоемкости, так и на изменения
теплоемкости в диффузных переходах первого рода, что
92
и служило причиной дискуссий о природе этого процес-
са в полимерах.
Температурный интервал частичного плавления за-
висит от химической структуры и условий кристаллиза-
ции. Для нефракционированных полимеров частичное
плавление может протекать в температурном интервале
в несколько десятков (вплоть до сотни) градусов. Повы-
шение температуры и продолжительности кристаллиза-
ции может заметно сузить интервал частичного плавле-
ния. Особенно эффективным здесь оказывается отжиг
полимеров.
Исследования плавления узких фракций полимеров
показали, что кривые плавления их в значительно
меньшей степени отличаются от кривых плавления низ-
комолекулярных соединений и что плавление узких
фракций является очень резким [24, 127, 128].
Предложено несколько объяснений частичного плав-
ления полимеров [125]. Часто существование интервала
плавления объясняется распределением кристаллитов по
размерам, обусловленным кинетическими факторами.
Физической причиной такой зависимости является влия-
ние размеров на температуру плавления кристаллитов.
Анализ этой зависимости для ламелярных кристаллитов,
высота которых h мала, а энергиями боковых граней
которых можно пренебречь, приводит к уравнению
[129—131], аналогичному формуле Томсона для шаро-
образных кристаллов:
/ 2ае \
7’пл = Г0л 1—(11.37)
где ?пл — температура плавления 'кристаллов бесконечных размеров;
ве—(Поверхностная энергия; ДЯПл—теплота плавления.
Таким образом, меньшие по размеру кристаллиты
плавятся при более низкой температуре по сравнению
с более крупными, что и приводит к широкому темпе-
ратурному интервалу плавления полимера.
В последнее время обсуждается возможность частич-
ного плавления ламелярных кристаллитов, образован-
ных складчатыми макромолекулами, основанная на
предположении о том, что с повышением температуры
увеличивается толщина дефектного граничного слоя> об-
93
разованного складками макромолекул [132, 133]. При
этом предполагается, что между поверхностной дефект-
ной областью и кристаллитом может существовать мета-
стабильное термодинамическое равновесие. Расчеты
числа конформаций закрепленной кристаллитами цепи
на основании решеточной модели показали, что свобод-
ная энергия кристаллической системы минимальна тог-
да, когда незакристаллизовавшиеся участки цепей в де-
фектных областях имеют некоторую среднюю «равно- ~
весную» длину. При наличии складчатых цепей эта рав-
новесная длина зависит как от температуры (вследствие
изменения конфигурационной энтропии), так и от рас-
стояния между закрепленными концами незакристалли-
зовавшихся участков цепи. Представления о поверхност-
ном плавлении включают также предположение о том,
что степень кристалличности зависит не только от тем-
пературы, но и от размера кристаллитов. Это дает воз-
можность понять влияние термической предыстории на
частичное плавление. Предположение о том, что с повы-
шением температуры увеличивается толщина дефектно-
го слоя, подтверждается некоторыми результатами ди-
латометрических и рентгенографических исследований
[133]. Ряд других предположений относительно причин
частичного плавления (наличие примесей, широкое мо-
лекулярно-массовое распределение и др.) имеет менее
общий характер.
Изотермическая кристаллизация полимерных распла-
вов при нормальном давлении приводит к образованию
набора неравновесных (метастабильных) кристаллитов,
средний размер которых вдоль оси макромолекул зави-
сит от температуры кристаллизации, возрастая с ее уве-
личением. При повышении температуры расплав, обра-
зовавшийся в результате плавления мелких и несовер-
шенных кристаллитов, может начать кристаллизоваться
с образованием кристаллитов с более высокой темпера-
турой плавления. Такой процесс называется новой кри-
сталлизацией или рекристаллизацией [125].
Еще одна возможность совершенствования кристал-
лической структуры в температурном интервале между
температурами кристаллизации и плавления обуслов-
лена тем, что в исходных неравновесных кристаллитах
возможно протекание процессов реорганизации и без их
плавления (в твердой фазе), в результате может увели-
94
чиваться толщина кристаллитов и соответственно тем-
пература плавления [3, с. 153]. Хорошо известный факт
повышения экспериментальной температуры плавления
при медленном нагревании полимера тесно связан с про-
цессами рекристаллизации и реорганизации.
Таким образом, в области плавления в кристалличе-
ских полимерах возможно протекание по меньшей мере
трех процессов: плавления, рекристаллизации и реорга-
низации. Каждый из этих процессов сопровождается со-
ответствующим изменением энтальпии и объема, и это
должно отражаться на форме кривых плавления. Прояв-
ление процессов реорганизации и рекристаллизации на
фоне частичного плавления зависит от соотношения ско-
ростей этих процессов и скорости изменения температу-
ры при измерении. При использовании адиабатической
равновесной калориметрии и проведении очень медлен-
ных измерений в дилатометрических исследованиях раз-
делить эти процессы невозможно, и регистрируемые кри-
вые изменения теплоемкости и удельного объема явля-
ются отражением единого процесса частичного плавле-
ния. Принципиальная возможность разделения этих про-
цессов появилась лишь с развитием динамической кало-
риметрии и ДТА полимеров.
Было обнаружено, что для многих полимеров кривая
плавления не является монотонной (как в адиабатиче-
ской калориметрии), а характеризуется наличием муль-
типлетных пиков плавления [134—140]. Чаще всего —
это дублеты или триплеты. Примером появления дуб-
летных пиков плавления является калориметрическая
кривая плавления полиуретана (рис. 11.21). Форма
мультиплетной кривой плавления зависит от условий
кристаллизации и скорости изменения температуры при
измерениях. Как уже отмечалось выше, условия кристал-
лизации определяют начальное распределение кристал-
литов по размерам и степени совершенства. Реоргани-
зация и рекристаллизация в процессе нагрева могут
появиться на кривой плавления в виде полностью или
частично разрешенных пиков лишь при условии, если
скорости этих процессов сравнимы со скоростями нагре-
ва. В двух предельных случаях кривая плавления будет
отражать плавление кристаллитов исходного распреде-
ления или распределения; вновь возникающего в про-
цессе медленного нагрева.
95
2
Рис. 11.21. Теплоемкость быстро охлажденных линейных поли-
уретанов на основе гексаметилендиизоцианата и диэтиленгли-
коля (/) и триэтиленгликоля 1(2) [11'35, 136].
а
4 Т°С
6
Рис. 11.22. Кривые плавления полихлоропренового каучука
(температура кристаллизации: а —2 °C, б — 20°C), получен-
ные при разных скоростях нагревания [141]:
/ —0,1°С/мин; 2 — 0,5; 3 — 3; 4—10; б- 20; 6 — 5 °С/мин.
Детальное исследование кривых плавления быстро
и медленно кристаллизующихся полимеров с использо-
ванием метода динамической калориметрии проведено
Сочавой с сотр. [141 —143]. Влияние условий кристалли-
зации и скорости повышения температуры при измере-
ниях на кривые плавления отражено на рис. 11.22. Ха-
рактерной особенностью кривых плавления медленно
кристаллизующегося полихлоропренового каучука явля-
ется дублетность пика плавления. Эта особенность со-
храняется при всех вариациях скорости нагрева, темпе-
ратуры и длительности кристаллизации, температуры и
длительности отжига; она имеет кинетическое происхож-
дение и обусловлена одновременно протекающими про-
цессами реорганизации и рекристаллизации. Дублетная
форма кривой плавления характерна и для быстро кри-
сталлизующихся полимеров (полиуретан на основе гек-
саметилендиизоцианата и диэтиленгликоля, гуттаперча),
подвергнутых «холодной» кристаллизации. В этом слу-
чае дублетность кривой плавления обусловлена лишь
одним механизмом совершенствования — реорганиза-
цией в твердом состоянии. Однако для быстро кристал-
лизующихся полимеров, закристаллизованных из рас-
плава при умеренных переохлаждениях, структура кри-
вых плавления при всех вариациях скорости нагрева и
практически при всех вариациях температуры кристал-
лизации является триплетной. Последнее, по мнению
Сочавы, обусловлено различием в кинетике реорганиза-
ции и рекристаллизации из-за очень высоких скоростей
реорганизации в твердом состоянии.
Таким образом, в большинстве случаев мультиплет-
ность кривых плавления имеет кинетическое происхож-
дение и в конечном счете обусловлена наличием широ-
кого набора кристаллитов по размерам и их метаста-
бильностью. Однако положение может осложняться тем,
что в некоторых полимерах наблюдаются фазовые пере-
ходы в твердом состоянии, которые также могут при-
водить к мультиплетной кривой плавления.
Переходы типа кристалл — кристалл кроме хорошо
исследованных методом теплоемкости в тефлоне {34, 36]
и полиамидах [36] наблюдаются в полибутене-1, у кото-
рого обнаружены три кристаллографические модифика-
ции, причем температуры переходов в твердом состоя-
нии близки к температуре плавления самой высокотем-
7—264
97
пературной модификации [115, с. 322], в гуттаперче
'143, 144], в некоторых полиэфирах и полиуретанах
145]. Поскольку переход одной формы в другую сопро-
вождается тепловым эффектом, на температурной зави-
симости теплоемкости появляется пик, как и в случае
плавления.
Температурные области существования каждой из
фаз могут быть велики, и тогда на кривых ср(Т) на-
блюдаются два пика, характеризующие плавление каж-
дой из кристаллических модификаций. Чаще встречают-
ся случаи, когда низкотемпературная модификация не
успевает полностью перейти в высокотемпературную,
так как последняя сразу же начинает плавиться. В та-
ких случаях на кривых теплоемкости могут наблюдаться
неразд ел яющиеся пики, переходящие один в другой
(гуттаперча, полибутен-1).
Соотношение между отдельными пиками на кривой
плавления, полученной динамическим калориметриче-
ским методом или методом ДТА, также может зависеть
от скорости нагрева из-за кинетических условий перехо-
дов кристалл—кристалл, и потому окончательное заклю-
чение о природе мультиплетности кривой плавления мо-
жет быть сделано лишь после исследования с использо-
ванием метода равновесной калориметрии (в этом слу-
чае также должна наблюдаться мультиплетность) и
рентгеноструктурного исследования возможности сущест-
вования различных кристаллографических модифика-
ций.
Одним из важнейших вопросов, касающихся плавле-
ния полимеров, является вопрос о температуре плавле-
ния. Описание температуры плавления термодинамиче-
ским соотношением (11.34) обычно осложнено зависимо-
стью температуры и интервала плавления полимеров от
условий кристаллизации и от условий, в которых прово-
дился эксперимент по определению температуры плав-
ления. Поэтому в полимерах различают эксперимен-
тальную Тпл и равновесную Пл температуры плавле-
ния [146]. Для пластинчатых кристаллитов эти темпе-
ратуры связаны между собой уравнением (11.37).
Для многих практических случаев достаточными ока-
зываются экспериментальные значения Тпл, однако ча-
сто необходимо знать и Тпл. На основании уравнения
(11.37) значение ТцЛ может быть определено по зависи-
98
мости Гпл от толщины кристаллита. В то время как тол-
щина кристаллитов может быть рассчитана по данным
рентгеноструктурного анализа, определение температу-
ры плавления исходных кристаллитов различной толщи-
ны представляет большие трудности по причине их
реорганизации и рекристаллизации в процессе измере-
ния. К тому же зависимость температуры плавления от
скорости нагрева не является однотипной для кристалли-
тов различной морфологии [3]. Для корректного опре-
деления 7ПЛ в этом случае необходимо применение очень
высоких скоростей нагревания при измерении, которые
позволили бы предотвратить протекание этих процессов.
^кр
Рис. 11.23. Определение равновесной температуры плав-
ления на основании зависимости температуры плавле-
ния от температуры кристаллизации.
Поэтому уравнение (11.37) обычно используют для
экстраполяционного определения Тпл на основании за-
висимости экспериментальной температуры плавления
от температуры кристаллизации. Его детальный анализ
с этих позиций проведен Хоффманом и Уиксом [130,
148]. Они исходили из кинетической теории кристалли-
зации, согласно которой наименьшая толщина ламеляр-
ного кристаллита h определяется размером критического
зародыша:
- дяпл(Тпол-Т)
(11.38)
где Т — температура кристаллизации.
7*
99
Тогда на основании уравнений (11.37) и (11.38)
+ Л (П.39)
Размер конечного кристаллита может превышать раз-
мер критического зародыша в у раз, где у может изме-
няться в пределах примерно от 1 до 5. Поэтому наибо-
лее часто используемым уравнением является
(11.40)
а равновесная температура плавления Тпл определяет-
ся как точка пересечения экспериментальной зависи-
мости 7’пл = /(7’) и прямой ТПЛ = Т (рис. 11.23). Этот
метод широко используется в последние годы для опре-
деления равновесных температур плавления кристал-
лических полимеров.
На рис. 11.24 показана зависимость температуры
плавления от молекулярной массы полимера. Данные
такого рода могут быть использованы для экстраполя-
ционного определения равновесной температуры плав-
ления полимера с бесконечно большой молекулярной
массой [124]. Однако начиная с определенной молеку-
Рис. 11.24. Зависи-
мость температуры
плавления полиэти-
леноксида от мо-
лекулярной массы
р127, 128] по данным
калориметрии (•),
дилатометрии ( О ) и
оптической микроско-
пии (Д).
лярной массы температура плавле-
ния начинает зависеть и от разме-
ра кристаллитов вдоль оси макро-
молекул.
Для решения проблемы влияния
молекулярной массы и размеров
кристаллов на температуру плавле-
ния большое значение имеют про-
веденные Вундерлихом [147, 149]
и сел е д ов а н ия ф изических свойств
кристаллов, образованных вытяну-
тыми макромолекулами. Полимеры
с относительно низкими молекуляр-
ными массами могут быть закри-
сталлизованы с полностью вытя-
нутыми макромолекулами при
нормальном давлении вблизи тем-
пературы плавления. Для высоко-
молекулярных полимеров соответствующие результаты
могут быть достигнуты при кристаллизации под высо-
кими давлениями.
100
Кристаллы, образованные вытянутыми цепями мак-
ромолекул, ближе всего соответствуют термодинамиче-
ски равновесным полимерным кристаллам. Плавление
таких кристаллов в равновесных условиях происходит
очень резко, а экспериментальные температуры плавле-
ния близки к равновесным. Ниже приведены темпера-
туры плавления полимерных кристаллов с вытянутыми
макромолекулами [149]:
Полимер
Т СС
7 пл’ с
Полиэтилен...................... 141,4
Политетрафторэтилен ........... 327,0
Полиоксиметилен................. 182,5
Поликапрола'ктам...............228
Селен.......................... 220,5
Все исследованные к настоящему времени кристал-
лы с вытянутыми цепями склонны к заметному пере-
греву, если измеряется температура плавления в динами-
ческих условиях. Этот эффект обусловлен замедленной
кинетикой плавления таких кристаллов.
Экспериментальная теплота плавления полимеров
может быть использована для анализа степени кри-
сталличности [36, 37]. Этот анализ основан на соотно-
шении, вытекающем из двухфазной модели кристалли-
ческого полимера:
= дя;л/дяпл (п.41)
где фн—степень кристалличности; Д#пл—теплота плавления пол-
ностью кристаллического полимера; ДТ/^л —калориметрическая теп-
ло'та плавления.
Для независимого определения степени кристаллич-
ности полимеров необходимо знать теплоту плавления
полностью закристаллизованного полимера. Такие дан-
ные для полимеров, как правило, отсутствуют. Лишь
для линейного полиэтилена эта величина определена
путем экстраполяции значений теплот плавления низ-
комолекулярных н-парафинов на цепь бесконечной
длины; она составляет примерно 293 кДж/кг [147].
Поэтому обычно для расчета на основании этого урав-
нения используют другой путь. Степень кристаллично-
сти находят при определенной температуре другими
методами (по данным измерения плотности, рентгенов-
ским методом и др.) и, используя эти значения, рас-
считывают ДЯпл-
101
Этот способ оценки кристалличности по теплотам
плавления основан на предположении, что степень
кристалличности является однозначной характеристи-
кой полимера и не зависит от метода ее определения.
Однако экспериментальные данные свидетельствуют
о том, что при больших степенях кристалличности
такая зависимость, по-видимому, существует. Так, для
наиболее полно изученного всеми методами линейного
полиэтилена тщательный анализ значений степени кри-
сталличности, полученных на основании измерений
теплоемкости и плотности высококристаллического
образца, показывает, что примерно на 15% выше,
чем фр [148]. Значения степени кристалличности, опре-
деленные методами рентгенографии и инфракрасной
спектроскопии [149], оказываются примерно на 10%
ниже значений, полученных по данным теплот плавле-
ния [148]. Для эластомеров с низкой степенью кри-
сталличности (<30%) эти же методы приводят к хо-
рошо согласующимся результатам [124]. Как метод
относительной оценки измерение теплот плавления яв-
ляется, по-видимому, самым чувствительным методом
обнаружения кристалличности.
Кристаллизация
Кристаллизация полимеров, как и кристаллизация
низкомолекулярных веществ, включает образование
зародышей кристаллизации и их рост [124, 125, 151,
152]. Тепловые методы не позволяют разделить эти
процессы и регистрируют суммарную (валовую) ско-
рость кристаллизации. Изотермы кристаллизации
в этом случае могут быть проанализированы на осно-
ве уравнений формальной кинетики кристаллизации.
Проблема заполнения объема различными структур-
ными элементами (шары, диски, стержни) в связи
с формальной кинетикой кристаллизации рассматрива-
лась многими учеными [153—157]. Обобщенное кине-
тическое уравнение изотермической кристаллизации,
часто называемое уравнением Аврами, имеет вид
’Ф . - 1 — e~~Ki * (11. 42)
гд е ф — доля в ещ ест® а, п од в ер г ш е г ос я ф а >з о® ом у п р ев р а щ ен и кф к с
времени Кп— константа, содержащая параметры зародыш^обра-
102
зования и роста; п—константа, зависящая от природы процессов
зародышеобразования и роста.
Значения констант Кп и п приведены в табл. II.2.
Таблица 11.2. Значения констант Кп и гг*
Тип растущего зародыша Гомогенное зародыше- образование Гетерогенное зародыше- образование
п «п п
Шар 4 л з G3/ 3 4л „ -x-ffW О
Диск (пластина) . 3 -^-G2Ih О 2 riPhN
Цилиндр .... 2 5 г—< со 1 GfN
«• ' Г* 1
* Обозначения: G — линейная скорость роста зародышей; I — число за-
родышей, образующихся в единице объема в единицу времени; 4N — число гетеро-
генных зародышей в единице объема; h — толщина диска (пластины); f— поперечное
сечение цилиндра.
С учетом того, что полимеры обычно не подвергают-
ся полному превращению, уравнение такого вида долж-
но описывать и процесс изотермической кристаллиза-
ции полимеров [124, 125].
Основным источником экспериментальных данных
о кинетике кристаллизации полимеров в блоке явля-
ются тепловые методы — дилатометрия и калоримет-
рия. Типичная дилатометрическая изотерма кристалли-
зации, схематически показанная на рис. 11.25, включает
основной S-образный участок и пологую часть, растя-
нутую на значительный интервал времени. S-образный
участок изотермы характеризует первичную кристалли-
зацию, соответствующую превращению исходного рас-
плава в кристаллический материал с определенной
степенью кристалличности. На этой стадии процесса
кристалличность должна развиваться в соответствии
с уравнением (11.42). Пологая часть изотермы соответ-
ствует вторичной кристаллизации, в процессе которой
происходит медленное совершенствование образовав-
шейся аморфно-кристаллической структуры.
юз
Анализ изотерм кристаллизации на основе уравне-
ния (11.42) часто проводят с целью определения формы
растущих кристаллических образований. Поскольку
параметр п неоднозначно характеризуется формой ра-
кристаллизации
Первичная стадия
кристаллизации
3J
- — Начальная высота
мениска
t~o
Вторичная ста'-
дия кристаллизации
Время t
Рис. 11.25. Типичная ди-
латометрическая изотер-
ма кристаллизации поли-
меров.
стущих частиц, отдельно приходится решать вопрос
о природе зародышеобразования. Экспериментальные
исследования кинетики кристаллизации свидетельст-
вуют о том, что для полимеров в блоке характерно
гетерогенное зарождение кристаллической фазы [148,
151, 160].
Применение уравнения Аврами для анализа изо-
полимеров показывает, что для
некоторых полимеров оно хо-
рошо описывает кинетику кри-
сталлизации (рис. 11.26), в то
время как для других полиме-
ров наблюдаются значитель-
ные отклонения. Наиболее ха-
рактерными признаками от-
клонений являются дробные
значения показателя п, не
имеющие ясного физического
смысла, и появление показа-
теля п, непрерывно изменяю-
щегося по мере развития
пр оцесс а кр иста л л и з ации
(рис. 11.27). Возможен также
>ie стадии кристаллизации описы-
ваются уравнением (11.42) с целыми значениями пока-
зателя п, а затем наблюдаются заметные отклонения
(рис. П.28). Имеются попытки объяснения отклонений
от уравнения Аврами с использованием представлений
об одновременном действии гомогенного и гетерогенно-
го зародышеобразования, о непостоянстве плотности
растущих частиц, о непрерывном изменении формы ча-
стиц в процессе роста, о влиянии молекулярно-массово-
го распределения на форму изотерм кристаллизации
[161—165].
В результате многочисленных дилатометрических
исследований установлено, что в тех случаях, когда
кинетика кристаллизации подчиняется уравнению
(11.42), значения показателя п часто составляют 3 или
4 [124, 125]. Сравнение этих значений с теоретически-
случай, когда ;
104
Рис. 11.26. Изотермы кристаллизации:
а — полипропилена [158] (/ — 117 °C; 2—120; 3 — 122,5; 4—125; 5—127,5;
6 — 130; 7 — 132,5; 8—135; 9 — 137,5 °C); б — полиуретана на основе гексамети-
лендиизоцианата и триэтиленгликоля [159] (/ — 69 °C; 2— 72; 3 — 75; 4 — 78;
5 — 81; 6' —85 °C).
ми приводит к выводу, что они соответствуют образо-
ванию трехмерных (шаровых) структур — сферолитов.
В связи с этим было сделано заключение, что в поли-
мерах показатель п указывает на геометрию сферолитов,
а не отдельных кристаллитов. Однако по мере разви-
тия исследований по кинетике кристаллизации стало
очевидным, что значение показателя п может зависеть
от многих факторов (формы и размера образца, гете-
рогенного влияния стенок, температуры расплава
и кристаллизации, концентрации гетерогенных зароды-
шей и их распределения по размерам и др.), изменяясь
в широких пределах, часто даже в пределах от 1 до 4,
т. е. принимая все возможные теоретические значения.
Это ставит под сомнение возможность использования
уравнения Аврами для идентификации структур на
основе формального анализа изотерм кристаллизации
полимеров [160].
Положение осложняется зависимостью значения по-
казателя Аврами от метода исследования, установлен-
ной при детальном сравнительном анализе калоримет-
рических и дилатометрических изотерм кристаллизации
некоторых полимеров [127, 128, 144, 160]. Калоримет-
рические изотермы для некоторых полимеров полностью
описываются уравнением (11.42) со значением показа-
теля п = 2, в то время как дилатометрические изотермы
для тех же образцов подчиняются уравнению (11.42)
при п = 3. Влияние перечисленных выше факторов на
величину п было исключено, поскольку такое же рас-
хождение зафиксировано также в процессе одновре-
менной регистрации изменения объема и выделяюще-
гося тепла. Важно отметить, что кристаллизация была
сферолитной и развивалась н<а гетерогенных заро-
дышах.
Таким образом, калориметрические изотермы свиде-
тельствуют о развитии в процессе кристаллизации
плоских структурных элементов, а дилатометриче-
ские— об одновременном развитии сферолитов. По-
видимому, это расхождение имеет физический смысл,
являясь отражением морфологических особенностей
кристаллических полимеров: кинетика выделения тепла
в процессе кристаллизации указывает на образование
ламелярных кристаллитов, а кинетика изменения объе-
ма, включающая и упаковку кристаллитов в сферо-
107
литы, свидетельствует об образовании сфероли-
тов.
Суммируя результаты многих исследований, можно
заключить, что определение формы растущих структур-
ных элементов на основе формального анализа изотерм
валовой скорости первичной кристаллизации с исполь-
зованием уравнения Аврами не является однозначным.
Важнейшее свойство кристаллизационных процес-
сов— сильная зависимость от температуры, являющая-
ся следствием зависимости от температуры скорости ~
зародышеобразования и роста зародышей [124, 148].
Поскольку рост зародышей также контролируется вто-
ричным зародышеобразованием, температурные зависи-
мости J и G однотипны. Выше уже отмечалось, что для
полимеров в блоке характерно гетерогенное зарожде-
ние кристаллических структур, и потому скорость роста
контролируется произведением NGn (см. с. 126). Выра-
жение для скорости роста имеет вид [148, 151]
(П.43)
где AF — энергия активации процесса переноса на поверхность раз-
дела кристалл—расплав; ДФ*—работа образования зародышей
критических размеров.
Конкретный вид этого уравнения зависит от формы
вторичных зародышей. Теоретический анализ различ-
ных моделей приводит к следующему обобщенному
уравнению роста [148, 166]:
G = Goexp ( — — \ exp
Т (АТ)™
(II. 44)
где С—константа, включающая энергетические характеристики
(теплоту плавления, поверхностные энергии); m — константа [т=4
для модели с когерентной укладкой поверхностных зародышей со
сложенными макромолекулами, т = 2 для модели с некогерентной
укладкой вторичных зародышей (трехмерные зародыши)]; ДГ =
= ТЙл-Т.
Выражение для скорости роста при т = 2 совпадает
с уравнением для скорости первичного зародышеобра-
зования /. В результате критического рассмотрения
различных приближений для транспортного и нуклеа-
ционного членов выяснено [167], что конкретное выра-
жение для скорости роста в модели с когерентным по-
108
верхностным зародышеобразованием, хорошо согласую-
щейся с современной морфологической концепцией,
является достаточно удовлетворительным:
G — Go exp ( --
А/?ВЛФ \-Avn f _ пл \
RT Г \ МНилТЛТ /
(11.45)
где Д^влф— энергия активации процесса переноса; bQ — толщина
поверхностного зародыша, определяемая параметрами кристалличе-
ской решетки; о и ое — свободные поверхностные энергии поверх-
ностей, параллельных и перпендикулярных осям макромолекул.
Для выражения энергии активации процесса пере-
носа использовано значение энергии активации вязкого
течения из соотношения Вильямса—Лэндела—Ферри.
Температурные зависимости скорости образования
и роста зародышей, а следовательно, и скорости вало-
вой кристаллизации, имеют колоколообразный вид.
В области температуры плавления скорость кристалли-
зации сначала сильно возрастает с понижением темпе-
ратуры, а затем достигает максимума. Это является
отражением решающей роли нуклеационных процессов
в данной температурной области. Напротив, ниже мак-
симума скорость кристаллизации уменьшается с пони-
жением температуры, что обусловлено влиянием транс-
портного члена. Максимальная скорость кристаллиза-
ции для гибкоцепных полимеров достигается между
температурами плавления и стеклования при 7тах =
= (0,824-0,83) Тцл независимо от структуры полимерной
цепи и температуры плавления [168, 169]. Эксперимен-
тально определить значение максимальной скорости
кристаллизации обычно удается лишь для медленно
кристаллизующихся полимеров, главным образом кау-
чуков. В большинстве случаев экспериментальные ис-
следования ограничены областью вблизи температур
плавления, а для полимеров, которые закалкой можно
перевести в аморфное стеклообразное состояние, такие
исследования проводятся и вблизи температуры стекло-
вания (холодная кристаллизация) [158—160, 170].
Соотношение (11.45), описывающее скорость роста
ламелярных сферолитов, позволяет выразить константу
скорости валовой кристаллизации Кп в виде [144, 160]
_JL_ ]сг к I А^ВЛФ „ д _ kgTim /тт 4g\
n 1g п+ 2,ЗТ?Т п 2,ЗТДТ ‘
109
где 773—; An — параметр, включающий число гетерогенных
kan пл
центров кристаллизации и максимальную скорость роста зародышей
Go.
М.. 10z
т-дт
Рис. 11.29. Зависимость суммы нормированных констант
скоростей валовой кристаллизации и транспортного чле-
на от температурного параметра Т^Л/(ТАТ):
1 — полипропилен (Тпл==185 °C); 2 — полипропилен (Т'пл =176 °C);
3 — полипропиленоксид; 4 — полиуретан на основе гексаметилен-
диизоцианата и диэтиленгликоля; 5 — полиуретан на основе ге-
ксаметилендиизоцианата и триэтиленгликоля; 6 — полидиметил-
силоксан; О — калориметрические данные; ф, □ и А — дилато-
метрические данные [160, 171, 172].
Вообще говоря, концентрация гетерогенных зароды-
шей кристаллизации также зависит от температуры
кристаллизации. Однако если температуры предвари-
тельного плавления являются одинаковыми для всех
опытов, что обычно имеет место на практике, то вблизи
температуры плавления этой зависимостью можно пре-
небречь по сравнению с очень сильной зависимостью
скорости роста зародышей. Если исследования прово-
110
дятся в широком температурном интервале, зависи-
мость числа гетерогенных центров от температуры ста-
новится также сильной, и этим пренебречь уже нельзя.
Основной целью анализа валовой скорости кристал-
лизации в соответствии с уравнениями (11.45) и (11.46)
Рис. 11.30. Зависимость суммы нормированных констант
скоростей валовой кристаллизации и транспортного
члена от температурного параметра (Т^Л)2/[Т(АТ)2].
Обозначения те же, что на рис. 11.29.
является определение вида вторичного зародышеобра-
зования по величине показателя m и поверхностных
энергий кристаллитов от и ае. В качестве константы
скорости часто используется величина, обратная време-
ни полукристаллизации /0>5. На рис. 11.29 приведены
калориметрические и дилатометрические данные для
ряда полимеров. Для всех полимеров наблюдается ли-
нейная зависимость обобщенного выражения скорости
от соответствующего температурного параметра, что
отвечает значению m = l. Зависимость той же величи-
ны от \Т~2 в большинстве случаев нелинейна
111
(рис. 11.30). Этот анализ позволяет заключить, что для
приведенных полимеров рост зародышей осуществля-
ется путем поверхностного зародышеобразования. Сде-
лать такой определенный вывод относительно природы
вторичных зародышей удается далеко не всегда [173],
поскольку часто зависимости оказываются линейными
в обоих случаях, как, например, для полиуретана на
основе гексаметилендиизоцианата и диэтиленгликоля.
На основании величин kg, определенных „ из
рис. 11.29, может быть рассчитано произведение ож
поскольку значения Ьо и Д/7Пл известны из независимых
исследований. Важным параметром при таком анализе
является выбранное значение Пл [160, 173]. О влиянии
равновесной температуры плавления на величину Kg
свидетельствуют данные для полипропилена. Основные
параметры плавления и кристаллизации некоторых
полимеров приведены в табл. II.3. Для расчета значе-
ний в используется эмпирическое соотношение [148]
а = рМЯпл (И-47)
где Р — константа, обычно принимаемая для полимеров равной 0,1.
В последней колонке табл. П.З приведены значения
работы складывания макромолекул, рассчитываемые по
соотношению [148]
q = 2Ace (11.48)
пде А—площадь поперечного сечения макромолекулы, определяемая
на Основании кристаллографических данных.
Для большинства исследованных в настоящее вре-
мя полимеров величина kg слабо зависит от химиче-
ской структуры полимера и обычно находится в интер-
вале от 200 до 300 [160, 167—173].
Важным выводом из анализа скорости кристаллиза-
ции полимеров является заключение о наличии в поли-
мерах поверхностей раздела, характеризующихся не-
обычно высокими для органических соединений значе-
ниями поверхностных энергий. Значения поверхностных
энергий вдоль оси макромолекул а не отличаются от
соответствующих значений для низкомолекулярных со-
единений, поскольку цепное строение макромолекул не
оказывает влияния на эти значения. Оно проявляется
лишь в областях, перпендикулярных осям макромоле-
112
Таблица П.З. Основные параметры плавления и кристаллизации полимеров
Л
Я» кДж/moj 25,1 17,6 23,8 19,6 20,5 9,2 9,2 о *>> 00 25,0 1 1 5,0 10,0 13,0 с Я S- Ч’ и. Я <и
Ч Я F- <Т> Я О. f- Я
ае103, Дж/м2 58,5 41,7 56,5 46,0 о оо ’Я4 20,4 20,4 62,9 47,0 «к о см 22,7 11,0
<м со 10,0 00 00 •>> ». 00 00 см е. о о 05 00 05 00 см to о 00 •ч 05 05 00 см СО Н-М t—Ч >> с
10 ч
«о 2 ,s 585 00 ь- СО 05 СО ’Я4 515 480 182 СМ 00 327 656 197 202 68 к ч о я к Ч
о ч Я
Ьо 1 262 UO Tf т—1 0} см см 205 215 1 95 95 205 268 55 । 106 73 0) ч я н я я я
еь
5,95 6,56 6,56 5,95 5,95 6,08 6,08 4,97 6,78 4,65 6,08 10,00 1 >> Е
к Е5
- (N о< 35,5 i 00 00 U0 Ю СО СО 35,5 35,5 37,0 37,0 24,5 46,0 21,7 37,0 о о о о я я ч U я 05 ч
ДЯ„„ Дж/см3 167 Tf Tf СО СО 00 00 ^—Ч 167 146 146 100 209 209 146 63 я ?Ч S я О. F-
Я
X о Ьч 295 О о со со см см 272 260 220 220 201 213 198 223 150 Я F- Я я я я я о я Я я я я <ь» ч
X ч О С Ьч 480 о> оо Tf U0 Tf Tf 413 393 361 361 357 353 343 328 238
я
ч к Ьч 456 СО СО 396 383 353 349 346 347 339 320 230 ф S я о я
Полимер Полиуретан (ПУ-1)* . . Полипропилен .... Полиуретан (ПУ-П)* Полиуретан (ПУ-Ш)* 1 юлидекаметиленсебаци- нат Полиэтиленсебацинат . . Полипропиленоксид . . Гуттаперча 11олиэталеноксид (ПЭО-20) Полиэтиленадипинат . . Полидиметилсилокса н * Полиуретаны на основе ля (ПУ-Ш).
8—264
кул, приводя к значениям поверхностных энергий
в 5—10 раз. более высоким. Сейчас .уже почти не оста-
ется сомнений в том, что эти высокие поверхностные
энергии связаны со складыванием макромолекул на
больших поверхностях кристаллитов, в результате чего
на этих гранях образуются разупорядоченные слои,
в которых, собственно, и запасена эта энергия.
Рис. 11.31. Зависимость
скорости роста сфероли-
тов G (а), калориметри-
ческой (б) и дилатомет-
рической (в) скоростей
кристаллизации полиэти-
леноксида от молекуляр-
ной массы при значении
ДГ, равном:
/ — 8 °C; 2—10; 3 — 15; 4 —
20 °C; различные точки —
данные разных авторов [127,
128].
Этот вывод вытекает из многочисленных структур-
но-термодинамических [148, 174] и кинетических иссле-
дований [148, 160, 163, 167]. Особенно важными в этом
направлении являются исследования структурных и ки-
нетических изменений, происходящих в гомологическом
ряду полимера при переходе от обычных низкомолеку-
лярных соединений к полимерам. Комплексное исследо-
вание такого рода, проведенное недавно на узких фрак-
циях полиэтиленгликоля [127, 128], обнаружило нали-
114
чие резкого уменьшения скорости кристаллизации,
приходящегося на область молекулярных масс около
4-Ю3 (рис. 11.31). Фракции с молекулярной массой
меньше 4-Ю3 при указанных переохлаждениях кристал-
лизуются с выпрямленными цепями, а больше 4-103 —
со складыванием цепей. Не останавливаясь на молеку-
лярных причинах замедления кристаллизации, отметим,
что при этом в зависимости от длины цепи может обра-
зоваться либо одна складка, либо несколько. Подсчи-
тать число складок удается на
основании анализа величин
больших периодов в закристал-
лизованных полимерах и пу-
тем сравнения их с длиной
макромолекул в вытянутом со-
стоянии. Эти данные демонст-
рируют прямую структурную
чувствительность термокине-
Рис. 11.32. Зависимость
произведения аае от мо-
лекулярной массы для
фракций полиэтиленокси-
да [128]:
О — усредненные калоримет-
рические и дилатометриче-
ские данные; * — на осно-
тических методов.
Анализ результатов термо-
кинетических исследований в
рамках описанной выше схе-
мы приводит к следующему
заключению (рис. 11.32). Низ-
комолекулярные фракции,
с полно-
молекула-
вании данных по скорости
роста сферолитов.
кристаллизующиеся
стью вытянутыми
ми, характеризуются обычны-
ми для низкомолекулярных органических соединений
поверхностными энергиями. Если предположить, что все
грани кристаллитов, образованных низкомолекулярны-
ми фракциями, обладают одной и той же поверхностной
энергией, то экспериментальные результаты приводят
к значениям поверхностной энергии 0,005—0,007 Дж/м2,
что близко к значениям для низкомолекулярных пара-
финов [148]. Появление способности макромолекул к
складыванию при кристаллизации приводит к резкому
возрастанию поверхностной энергии о и соответственно
произведения аоге, которое по мере дальнейшего повы-
шения молекулярной массы практически не зависит от
нее, поскольку при этом увеличивается лишь число пе-
регибов макромолекулы при кристаллизации. Таким об-
разом, указанные результаты достаточно наглядно де-
8*
115
монстрируют связь высоких поверхностных энергий со
складыванием макромолекул при кристаллизации.
Способность различных макромолекул к складыва-
нию может быть оценена путем сравнения величин ра-
боты складывания q, которая связана с kg соотноше-
нием [130, 148]
kg^2$q/k (11.49)
Выше мы отмечали, что kg слабо зависит от химической
структуры полимера. То же самое можно сказать
и о работе складывания. Но при этом работа складыва-
ния должна коррелировать с 72л [130]. Такая тенден-
ция действительно существует (см. табл. II.3).
Для широкой области температур, включающей
и область вблизи температуры стеклования, экспери-
ментальные калориметрические данные по валовой ско-
рости кристаллизации некоторых полимеров не удается
удовлетворительно описать теоретическими уравнения-
ми на основе обычно применяемых приближений для
транспортного члена [160]. Эти данные расходятся
с данными по скорости роста сферолитов [167], что
является, очевидно, в значительной мере результатом
резкого увеличения числа зародышей кристаллизации
вблизи Тс.
Рассмотренные выше результаты относились к пер-
вичной кристаллизации, т. е. к той части изотерм
(см. рис. 11.25), которые представляются S-образными
кривыми и которые соответствуют образованию кри-
сталлической решетки в полимере и обычно его сферо-
литизации. Вторичная кристаллизация полимеров, опи-
сываемая пологой частью изотерм, исследована значи-
тельно меньше [125, 151, 177]. На самом деле оба
процесса протекают, вероятно, одновременно, однако
скорость первичной кристаллизации обычно значитель-
но превосходит скорость вторичной, а увеличение сте-
пени кристалличности при вторичной кристаллизации
мало по сравнению с первичной [178—180]. У некото-
рых полимеров (полипропилен, некоторые линейные
полиуретаны, полиэтилентерефталат и др.) вторичная
кристаллизация практически отсутствует. Причины
такого разделения неясны.
Увеличение степени кристалличности в процессе
вторичной кристаллизации может быть описано урав-
116
нением (11.40) при значениях п, меньших или близких
к единице [151, 180, 181]. При вторичной кристаллиза-
ции обычно происходит либо совершенствование кри-
сталлитов, либо медленная кристаллизация трудно
кристаллизующихся компонентов, находящихся в меж-
сферолитных областях. В последнее время все больше
склоняются к тому, что действительный механизм вто-
ричной кристаллизации включает оба процесса [177].
Стеклование
Как уже отмечалось выше, стеклование представ-
ляет собой переход из жидкого (высокоэластического)
состояния в твердое стеклообразное и наблюдается как
в низкомолекулярных соединениях, так и в полимерах.
С точки зрения структуры оба состояния являются
аморфными и характеризуются наличием лишь ближ-
него порядка в расположении звеньев полимерной цепи
в пространстве [11, 182]. Различие между этими двумя
состояниями заключается в том, что им отвечают раз-
ные виды молекулярного теплового движения. В твер-
дом стеклообразном состоянии атомы низкомолекуляр-
ного соединения или полимерных цепей закреплены
в точках нерегулярной пространственной решетки
и колеблются относительно своего положения равно-
весия.
Высокоэластическое состояние как аналог жидкого
состояния характеризуется наличием определенных ви-
дов молекулярного трансляционного движения. В отли-
чие от низкомолекулярных жидкостей, где в результате
теплового движения происходит перемещение целых
молекул, в полимерах, находящихся в высокоэластиче-
ском состоянии, кинетическими единицами движения
являются статистические сегменты макромолекул.
В результате трансляционного группового движения
сегментов ближний порядок в элементарном объеме
непрерывно изменяется вследствие непрерывного разру-
шения и создания возможных структур. Поэтому для
жидкого и соответственно высокоэластического состоя-
ния можно говорить о среднем ближнем порядке по
времени или по всему объему. Каждой температуре
(при постоянном давлении) отвечает определенное рав-
новесное пространственное расположение частиц, т. е.
117
определенная структура с соответствующим ближним
порядком. Поэтому состояние жидкости обычно харак-
теризуется не только температурой и давлением, но
и параметром Z(T, Р), определяющим структуру жид-
кости [11, 182, 183].
На перестройку структуры жидкости расходуется
определенная энергия, что выражается в более высо-
ком значении теплоемкости жидкости, чем стекла. Ско-
рость перестройки структуры определяется уровнем
развития молекулярной структуры, т. е. соотношением
Рис. 11.33. Температурная зависимость теплоемкости полиуретана на
основе гексаметилендиизоцианата и диэтиленгликоля в области стек-
лования образцов, закристаллизованных на разную глубину. Тепло-
та предварительной кристаллизации:
1 — 0; 2 — 31,9; 3 — 39,9; 4 — 60,3; 5 — 68,5 кДж/кг. Стрелками отмечены тем-
пературы стеклования [135, 136].
энергии теплового движения частиц и энергии их взаи-
модействия. При достаточно высоких температурах
перестройка структуры осуществляется практически
мгновенно. Однако по мере понижения температуры
и повышения вязкости системы скорость перестройки
структуры постепенно уменьшается и ниже некоторой
температуры становится столь незначительной, что
в жидкости «замораживается» структура Z(T*, Р),
соответствующая этой температуре Г*. Это «замора-
живание» структуры означает отвердевание, или стек-
лование. При дальнейшем охлаждении структура стек-
118
ла не изменяется, и его тепловые параметры, так же
как и тепловые параметры кристаллических тел, опре-
деляются характером ангармонических колебаний ато-
мов и групп атомов.
Таким образом, согласно современным представле-
ниям процесс стеклования является постепенным пере-
ходом от равновесной структуры жидкости к метаста-
бильной (квазиравновесной) вследствие «заморажива-
ния» трансляционной подвижности молекул или их ча-
стей [И, 182—184].
Проблеме стеклования посвящен ряд исчерпываю-
щих обзоров и монографий [80, 116, 182—187]. Ниже
мы рассмотрим характер изменения тепловых парамет-
ров полимеров (с и а) в области стеклования и пока-
жем, в какой степени наиболее распространенные тео-
рии позволяют описать наблюдаемые эксперименталь-
ные зависимости.
Многочисленные исследования температурной зави-
симости теплоемкости и термического коэффициента
расширения низкомолекулярных и высокомолекулярных
стекол показывают, что в области стеклования наблю-
дается скачкообразное изменение этих величин
(рис. 11.33, 11.34). Скачкообразному изменению ср и а
соответствует появление излома (перегиба) на темпе-
ратурной зависимости функций состояния, в частности
энтальпии Н и объема V. Стеклование всегда происхо-
дит в некотором температурном интервале (примерно
3—20 К). На кривых температурной зависимости V, Н,
а и ср можно выделить
температуры начала и
конца затвердевания, от-
вечающие отклонению
указанных величин от
соответствующих зави-
симостей, и характеризо-
вать ими температурную
область стеклования. Од-
нако обычно область
стеклования характеризу-
ется условной величи-
ной — температурой стек-
лования Тс, соответству-
ющей точке пересечения
Рис. 11.3'4. Температурная зависи-
мость термического коэффициента
объемного расширения полицикло-
гекс1илметак|рилата в области стек-
лования 1'226].
119
продолжений линейных участков температурных зави-
симостей V и Н или точке перегиба на температурных
зависимостях ср и а. К настоящему времени накоплен
большой экспериментальный материал по значениям
скачков Аср и Аа в области стеклования и установле-
ны важные корреляции между основными тепловыми
характеристиками [116, 188, 189]. Результаты для не-
которых полимеров приведены ниже [183]:
7-с, К Да-10—4, К-1 кДж/(кг-К)
Полистирол 375 3,3 0,376
Поливинилхлорид .... 355 4,8 0,628
Поливинилацетат .... 305 4,5 0,503
Полиизобутилен .... 200 4,2 0,398
Полиметилметакрилат . . 385 3,0 0,293
Натуральный каучук . . 200 4,1 0,462
Значение скачка теплоемкости Аср зависит от хими-
ческой структуры полимера. Однако в расчете на наи-
меньшую единицу, способную к самостоятельному пере-
мещению (бусинку), инкремент теплоемкости при стек-
ловании оказывается практически постоянным и состав-
ляет 11,3±2,1 Дж/(моль-К) [41]. Использовав дыроч-
ную теорию стеклования, Вундерлих показал, что рост
удельной теплоемкости при температуре стеклования
составляет 12,4 Дж/(моль-К). Таким образом, среднее
значение увеличения теплоемкости при Тс может рас-
сматриваться как характеристика стеклования аморф-
ных веществ.
Два других эмпирических соотношения связывают
величину скачков теплоемкости и теплового расшире-
ния у аморфных полимеров с температурой стеклова-
ния [116, 189]:
АсрТс ~ 115 кДж/кг (II. 50а)
ДаТс = 0,113 (11.506)
Согласно дырочным теориям жидкости [38, 40],
структура жидкости, соответствующая каждой темпера-
туре, характеризуется определенным числом дырок
и их размерами. Изменение объема жидкости при
изменении температуры может быть представлено как
следствие увеличения общего объема дырок и ангармо-
ничности колебаний атомов. Естественно, что общий
объем дырок является параметром, свидетельствующим
120
определенным образом о соотношении энергий межмо-
лекулярного взаимодействия и теплового движения.
Именно это положение является физической основой
концепции свободного объема [187]. В соответствии
с этой концепцией трансляционное движение в жидко-
сти замораживается при уменьшении свободного объе-
ма до определенной величины. Согласно представле-
ниям Фокса и Флори [190], температуре стеклования
у всех полимеров соответствует одно и то же значение
свободного объема. Определенным подтверждением
этого представления является эмпирическая зависи-
мость Симхи и Бойера между Тс и скачком коэффици-
ента теплового (расширения Ла (11.506) и постоянство
значения константы с% в уравнении Вильямса—Лэнде-
ла — Ферри [187], которой придается смысл парциаль-
ного свободного объема:
lg ат------2,3/с
(П.51)
Его значение при Тс составляет 0,025. Разница в значе-
ниях величины свободного объема связана с различиями
в определении понятия свободного объема.
Исследования последних лет показывают [191—
193], однако, что существуют значительные отклонения
от значений свободного объема Симхи и Бойера
и Вильямса — Лэндела — Ферри. Такое положение пред-
ставляется вполне естественным, так как ряд дополни-
тельных факторов кроме свободного объема несомнен-
но должен оказывать влияние на трансляционную под-
вижность кинетических единиц в жидкости. Одним из
таких факторов может быть соотношение поверхностей
контакта макромолекул и пустот [191].
Важнейшей особенностью поведения тепловых пара-
метров в области стеклования, подтверждающей релак-
сационную природу этого превращения, является зави-
симость температуры стеклования от скорости нагрева
и охлаждения и наличие гистерезисных процессов. Наи-
более показательными являются результаты калоримет-
рического исследования размягчения и отжига полимер-
ных стекол [194—197]. В отличие от обычной аномалии
теплоемкости для нормально охлажденного стекла тем-
121
пературная зависимость динамической теплоемкости
при размягчении отожженных стекол характеризуется
наличием пиков (рис. 11.35). Величина, положение
и острота этих пиков существенно зависят от характера
предварительной обработки и скорости нагревания при
измерениях. Максимум на кривой ср(Т) практически
отсутствует только в том случае, если полимер не под-
вергался предварительному отжигу. При этом его появ-
ление не может быть вызвано повышением скорости
нагревания при измерениях. Увеличение же продолжи-
тельности отжига приводит к очень острому пику
Рис. 11.35. Температурная зависимость теплоемкости по-
лив1И'нилацетата при нагревании со скоростью 0,5 °С/мип
после отжига при температуре 21 °C [196]; продолжи-
тельность отжига:
1 — 1 сут; 2 — 2—8 сут; 3 — 40 сут. Цифры у кривых указывают
максимальное значение теплоемкости.
с большим значением теплоемкости в максимуме. Ана-
логичные по смыслу зависимости наблюдаются и при
изучении закономерностей изменения объема в области
стеклования [183, 198, 199]. Таким образом, отжиг
приводит к тому, что в аморфном полимере удается
наблюдать превращение, имитирующее фазовый пере-
ход первого рода. Следовательно, к трактовке анома-
лий динамической теплоемкости и динамических коэф-
фициентов расширения следует подходить осторожно.
Описанное поведение аморфных стекол в области
размягчения связано с кооперативным характером дви-
122
жения частиц в конденсированном состоянии, т. е.
с зависимостью кинетических свойств любой данной
частицы от состояния ее соседей. Появление максиму-
мов на кривых (Г) и а (Г) после длительного отжига
обусловлено тем, что состояние стекла при этом при-
ближается к равновесному. При последующем нагрева-
нии та часть суммарной теплоемкости, которая обуслов-
лена перестройкой структуры при размягчении, не
успевает достичь равновесного значения, и на кривых
появляются максимумы, которые, таким образом, явля-
ются отражением кинетики процесса размягчения. При
этом необходимо отметить, что равновесие в стеклооб-
разном состоянии достигается лишь при температурах,
близких к Тс (обычно на 10—20 К ниже Тс). При даль-
нейшем понижении температуры продолжительность
перехода к равновесному состоянию резко возрастает
и стремится к бесконечности.
Многочисленные экспериментальные и теоретиче-
ские исследования релаксации объема [183, 198—201],
среди которых выделяются обширные исследования
Ковакса [183, 202], рассматривавшего релаксацию
объема при отжиге как объемное течение в условиях
гидростатического давления, а также исследования
релаксации энтальпии [196, 197, 203] показали, что
скорость релаксации тепловых параметров не является
экспоненциальной во времени, а зависит от величины
и знака отклонения объема и энтальпии от их равно-
весных значений. В некоторых работах была обнаруже-
на зависимость скорости релаксации объема и от пути
достижения начального состояния, т. е. от термической
предыстории [199].
Уже в первой модельной кинетической теории стек-
лования, развитой Волькенштейном и Птицыным [204],
было предсказано появление гистерезисных эффектов
при нагревании и охлаждении (даже если они прово-
дятся при одинаковых скоростях) и качественно объяс-
нено появление максимумов на температурной зависи-
мости теплоемкости ср в области стеклования и их
зависимость от скорости нагревания, несмотря на то
что в основе этой теории лежит представление об
аморфном теле как об ансамбле невзаимодействующих
частиц (или дырок в соответствии с дырочной теорией
жидкости [38, 40]), которые могут находиться лишь
123
в двух энергетических состояниях, разделенных энерге-
тическим барьером. Эта упрощенная трактовка кинети-
ческих процессов, протекающих при стекловании, не
учитывает, таким образом, кооперативного характера
движения частиц в конденсированной фазе. Дальней-
ший учет этой кооперативности, проведенный Птицы-
ным и Готлибом [205, 206], показал, что наблюдаемый
экспериментально неэкспоненциальный характер кри-
вых стабилизации стекол и их асимметрия являются
следствием кооперативности. К числу важных теорети-
ческих выводов этих работ следует отнести вывод о том,
что законы релаксации объема и энтальпии при стаби-
лизации стекол могут различаться. Качественно карти-
ны релаксации объема и энтальпии похожи [199],
однако тщательные количественные исследования кине-
тики стеклования с этих позиций, по-видимому, не про-
водились.
Эти экспериментальные и теоретические исследова-
ния составляют основу кинетического подхода к про-
цессу стеклования полимеров. Однако наряду с кине-
тическим (релаксационным) подходом к стеклованию
полимеров существует совершенно иная трактовка это-
го превращения, а именно термодинамическая, в кото-
рой стеклование рассматривается как фазовый переход
второго рода [116]. Такое заключение было впервые
сделано [207] на основании формального анализа изме-
нения теплоемкости в области стеклования, поскольку
наблюдаемое скачкообразное изменение ср (и а) при
стекловании формально соответствует закономерностям
изменения этих величин при фазовых переходах второ-
го рода (см. рис. 11.19, в). Однако вся совокупность
экспериментальных данных по стеклованию позволяет
с очевидностью отвергнуть эту трактовку, поскольку
даже чисто формальное рассмотрение аномалии тепло-
емкости говорит об ошибочности этого заключения (при
стекловании наблюдаются обратные по знаку скачки
Дер и Да; при фазовом переходе второго рода фазы по
обе стороны перехода находятся в термодинамическом
равновесии, в то время как при стекловании такое рав-
новение отсутствует) [11].
Положение осложнилось после того, как Гиббс и Ди
Марцио [208, 209] теоретически показали существова-
ние фазового перехода второго рода для гипотетическо-
124
го ансамбля полимерных цепей с заторможенным внут-
ренним вращением звеньев и межмолекулярным взаи-
модействием, характеризуемым некоторой энергией.
Этот переход должен происходить при температуре Т2,
при которой возможное число способов размещения
полимерных цепей в трехмерной решетке*, т. е. число
конфигураций системы, становится равным единице
и соответственно конфигурационная энтропия становит-
ся равной нулю (рис. 11.36). Температура этого гипоте-
тического перехода Г2 определяется главным образом
Рис. 11.36. Схематичеюкое изображение изменений теп-
лоемкости 1(а) и энтропии (б) в области стеклования.
величиной энергетического барьера — внутреннего вра-
щения в цепях, т. е. характером увеличения жесткости
цепи при понижении температуры.
Признавая кинетическую природу наблюдаемого в
реальных условиях превращения высокоэластического
полимера в стеклообразный, Гиббс и Ди Марцио за-
ключают, что оно является кинетическим проявлением
термодинамического перехода второго рода. При беско-
нечно медленном охлаждении этот переход может
быть достигнут, а следовательно, может быть достиг-
* Анализ конфигурационных изменений проводился на основе
квазирешеточной модели [210].
125
нуто равновесное стеклообразное состояние, энтропия
которого будет практически равна энтропии кристалла.
Вопрос состоит в том, может ли такое состояние быть
достигнуто в реальных условиях. Развивая этот подход,
Адам и Гиббс [211] теоретически оценили соотношение
между наблюдаемой температурой стеклования Тс
и гипотетической температурой перехода второго рода
Г2; они установили, что Гс/Г2=1,30, а Тс—Т2 = 55°С.
Как уже отмечалось выше, в реальных полимерах л
при температурах значительно выше теоретической
температуры перехода Т2 вязкость системы и соответ-
ственно времена релаксации структуры становятся
столь значительными, что происходит замораживание
трансляционных перемещений кинетических единиц,
и полимер переходит в стеклообразное метастабиль-
ное состояние. В силу указанных выше причин, по
крайней мере, при практически осуществимых скоро-
стях охлаждения достичь равновесного стеклообразно-
го состояния при Т2 нельзя, и потому нельзя ответить
на вопрос, возможно ли действительно такое состояние.
Более того, существует мнение, основанное на анализе
поведения реальных полимерных систем, что в обыч-
ных условиях такой гипотетический переход вообще не
происходит [184, 212]. С другой стороны, экстраполя-
ционные оценки энтропии при Т2 и положения этой
температуры перехода на температурной шкале по дан-
ным калориметрических измерений теплоемкости со-
гласуются с теоретическими предсказаниями [213, 214].
Таким образом, реально наблюдаемое стеклование
полимеров является, несомненно, кинетическим пре-
вращением, а не термодинамическим. Вопрос же прин-
ципиальной возможности достижения равновесного
перехода второго рода при Т2 остается открытым.
Важным звеном в экспериментально-теоретическом
анализе стеклования является вопрос о соотношении
между основными термодинамическими параметрами*
причем этот вопрос также имеет как термодинамиче-
ский, так и кинетический аспект. При переходе второга
рода термодинамические параметры связаны уравне-i
нием (11.36). Однако различными методами было пока?
зано, что такое же соотношение должно соблюдаться^
и при отсутствии термодинамического равновесия, если
структуру системы при температуре перехода характ§|
126
ризовать параметром упорядоченности [121, 184, 215].
При этом, если существует более одного такого пара-
метра упорядоченности, считается, что равенство
(II.36) превращается ib неравенство [215]:
ДсрДх > TV (Да)2
(11.52)
Однако совсем недавно Ди Марцио [216] теоретически
показал, что равенство должно иметь место при любом
числе параметров упорядоченности и, таким образом,
классификация превращения лишь на основании фор-
мального анализа соотношения между термодинамиче-
скими параметрами невозможна. К тому же точность
расчетов по этим уравнениям невелика главным обра-
зом из-за низкой точности определения Дх. Поэтому
в литературе встречаются как хорошо согласующиеся
данные, так и данные, различающиеся в два раза и бо-
лее [183, 186].
Изменения тепловых параметров, связанные с пере-
ходом из стеклообразного состояния в высокоэластиче-
ское, наблюдаются и у кристаллических полимеров.
В этом случае они, естественно, относятся лишь к
аморфной части. Данные о влиянии степени кристал-
личности на Тс довольно противоречивы и свидетель-
ствуют о том, что Тс может увеличиваться, оставаться
постоянной и уменьшаться при возрастании степени
кристалличности [116, 185]. Характерные результаты
приведены на рис. 11.33. По мере увеличения глубины
закристаллизованное™, характеризуемой в данном слу-
чае степенью завершенности холодной кристаллизации,
температура стеклования несколько возрастает, а ска-
чок теплоемкости при этом уменьшается.
Связь скачка теплоемкости (или теплового расши-
рения) со степенью кристалличности может быть ис-
пользована для оценки степени кристалличности при
обычных для двухфазной модели предположениях на
основании соотношения [218—220]
t == 1 - Ьср1ЬсР;м
(II. 53)
где — экспериментальный сказок теплоемкости для полимера со
степенью кристалличности Ф; АсРам—скачок теплоемкости в аморф-
ном полимере.
Для малых и умеренных степеней кристалличности
такая пропорциональность действительно наблюдается,
127
Рис. II.37. Зависимость темпера-
туры стеклования от молекуляр-
ной массы для индивидуальных
соединений и фракций в гомоло-
гическом ряду полиэтиленоксида
[1127].
однако она, по-видимому, исчезает при больших степе-
нях кристалличности [221, 222]. Кроме того, для поли-
меров высокой степени кристалличности (полиэтилен,
полипропилен) скачок теплоемкости в области предпо-
лагаемого стеклования часто практически отсутствует
(см. рис. III.28). Еще большие сложности возникают
при попытке применить соотношение (11.53) к оценке
кристалличности ориентированных полимеров, в кото-
рых даже при умеренной
кристалличности сказки
теплоемкости в области
предполагаемого стекло-
вания могут отсутство-
вать (см. рис. III.29).
Температура стекло-
вания зависит от молеку-
лярной массы (рис. 11.37),
подобно температуре
плавления, и эта зависи-
мость является следстви-
ем влияния концевых
групп: с уменьшением
числа концевых групп
(увеличение молекуляр-
ной массы) температура
стеклования повышается.
Наиболее простая анали-
тическая зависимость температуры стеклования от моле-
кулярной массы имеет следующий вид [116, 185, 223]:
ТС = ТСОО-~К/М (11.54)
20pW
где К=~ (здесь 0 — добавочный свободный объем, связанный с
Да
концевыми группами; р— плотность; N — число Авогадро); М —
молекулярная масса.
При необходимости эта зависимость используется
для оценки ТСоо по данным низкомолекулярных гомоло-
гов. Большое число концевых групп, способных к спе-
цифическим взаимодействиям, может приводить к замет-
ным отклонениям от этой простой зависимости [185].
Из контролируемых параметров наибольшее влия-
ние на температуру стеклования полимеров оказывает
128
добавление низкомолекулярных соединений — пласти-
фикаторов, следствием которого является либо экра-
нирование полярных радикалов, либо увеличение веро-
ятности образования слабых или лабильных узлов
[И, 224]. В обоих случаях наблюдается снижение Тс.
Тепловые методы оказываются весьма информативными
Рис. 11.38. Температурная зависи-
мость теплоемкости поликарбона-
та, пластифицированного диметил-
фталатом (а) [содержание пла-
стификатора: Х-—0, О—25, Д—!35,
А — 45 % 1 (м асе.) ] из а1висимость
температуры стеклования от со-
держания пластификатора (б).
при количественном исследовании процесса пластифи-
кации полимеров. Рассмотрим характерные данные по
пластификации поликарбоната (рис. 11.38).
Повышение содержания пластификатора приводит
не только к снижению температуры стеклования, но
и к повышению подвижности, результатом которой яв-
ляется кристаллизация полимера. В отсутствие пласти-
фикатора поликарбонат даже в самых благоприятных
температурных условиях кристаллизуется в течение
9—264
129
многих суток. Начиная с содержания примерно 30%
пластификатор перестает полностью совмещаться с по-
лимером и выделяется в виде микрофазы, которая
плавится в области температур —15 °C. Можно легко
определить предельные концентрации пластификатора,
при которых он совмещается с полимером. Близкие по
смыслу результаты получаются и для других полиме-
ров [225].
Превращения в стеклообразном состоянии
и промежуточные превращения
Плавление и стеклование полимеров являются
в высшей степени кооперативными процессами и легко
регистрируются всеми физическими методами, хотя
количественного соответствия при этом часто не наблю-
дается. Кроме этих переходов в полимерах в темпера-
турном интервале 4—600 К наблюдается большое число
аномалий в физических свойствах, часто очень незначи-
тельных в сравнении с плавлением и стеклованием. Не-
смотря на то что молекулярные причины таких анома-
лий в большинстве случаев остаются неизвестными, эти
аномалии обычно также считают проявлением опреде-
ленных превращений в полимерах и называют перехо-
дами или температурными переходами [115, 116].
Основная сложность при анализе этих переходов со-
стоит в том, что их проявление и разрешение сильно
зависят от метода исследования. Наиболее часто они
регистрируются методами механических и диэлектриче-
ских потерь в виде дискретных пиков на зависимости
тангенса угла потерь от температуры. Полагают, что
их появление в стеклообразных полимерах связано
с развитием молекулярных движений и прежде всего
с размораживанием движений боковых групп и неболь-
ших последовательностей в основной цепи. В кристал-
лических полимерах дополнительными механизмами,
приводящими к появлению мультиплетных пиков по-
терь, могут являться движения дефектов в кристаллах,
движения в молекулярных складках или движения
концевых групп на поверхности кристалла, а также
переход из одной решетки в другую.
Пики на спектрах механических потерь обычно луч-
ше разрешаются при низких частотах. Это дает основа-
130
ние полагать, что вторичные переходы будут особенно
хорошо проявляться в изменении тепловых свойств теп-
лового расширения, теплоемкости и теплопроводности.
Однако это предположение во многих случаях не под-
тверждается.
Большинство ранних измерений теплоемкости и теп-
лового расширения многих полимеров не обнаруживало
никаких заметных изменений, кроме связанных со
стеклованием и плавлением. Однако в процессе развития
исследований механических релаксационных процессов
2/0
2/0
1/0
1/0
0/0
0/0
0/0
О ^'рооооо ооос
о'—1—L—L-
"180-160-^0-120-100-00-00-^0-20 0 20 40 60 80 100 120
_CL
Оооооооооо^
) оо ООООООООО/
noooo^ qQ:
M Сноо4
2oQO°00°^ h
pOOO°
l^noooooooooooo1
о
о
-200
У/0
-2/0
±f/0
~ 2,00
±f/0
> 1/0
- 1/0
- 0/0
-0/0
- 0
о
о
Л
Рис. 11.39. Температурная зависимость термического коэффи-
циента линейного расширения:
А — поли-п-бутилметакрилат; Б — поли-п-пропилметакрилат; В — поли-
этилметакрилат; Г — полиметилметакрилат. Стрелками указаны темпе-
ратуры переходов [228].
были выполнены специальные исследования тепловых
свойств с целью обнаружения вторичных переходов
тепловыми методами [227—230]. Тщательное измере-
ние теплового расширения совместно с динамическими
механическими измерениями на нескольких полиалкил-
метакрилатах (рис. 11.39) привело к заключению о на-
личии вторичных превращений в этих стеклообразных
полимерах. В отдельных случаях наблюдалась опреде-
ленная корреляция между тепловым расширением
и низкочастотными механическими потерями, однако
полного соответствия между этими методами получить
не удалось. Величина и положение на температурной
шкале небольших скачков в тепловом расширении при
низких температурах зависит от размера боковых
9* 131
групп. Постоянство свободного объема (0,113) в обла-
сти стеклования при наличии переходов в стеклообраз-
ном состоянии не наблюдается. Таким образом, в этих
полимерах свободный объем не является постоянной
величиной ниже температуры стеклования.
Эти данные указывают на необходимость тщатель-
ных измерений в области предполагаемых вторичных
переходов, поскольку изменения в них незначительны
Рис. 11.40. Температурная зависимость тангенса угла
механических потерь (О) и скорости звука *) при
резонансной частоте 115 Гц (а) и температурная зави-
симость линейного теплового расширения (б) полифе-
Н1илизобутил1силсесквиоксана |[J231]; р равен:
7 — 14.10-5 К-1. 77—16.-10-5; III -17,5-10~5; ZV-14,0-10-5 К"1.
и могут быть приняты за разброс экспериментальных
данных. Возможно, что во многих ранних работах имен-
но это имело место.
Развитие подвижности в стеклообразных полимерах
может происходить в широком температурном интервале
и представлять собой набор последовательных перехо-
дов. Это должно быть особенно характерно для жест-
коцепных полимеров. На рис. 11.40 приведены резуль-
таты исследования механических потерь, скорости зву-
ка и теплового расширения для жесткоцепного лестнич-
ного полимера цолифенилизобутилсилсесквиоксана
132
в широком температурном интервале [231]. Множест-
венный характер температурного перехода, соответст-
вующего последовательному появлению значительной
гибкости цепи, является отражением дискретного ха-
рактера развития внутримолекулярных движений в ле-
стничных полимерах. Вероятно, эта дискретность вооб-
ще характерна для жесткоцепных полимеров. В частно-
сти, ярко выраженная множественность переходов на
температурной зависимости теплового расширения
наблюдается у триацетата целлюлозы [232]. Результа-
ты исследования теплового расширения отражают, по-
видимому, общее правило: постепенное увеличение
межмолекулярных расстояний с повышением темпера-
туры приводит к возможности развития новых типов
молекулярных движений.
Особого внимания заслуживает сравнение тепло-
емкости со спектром механических потерь. Выше уже
отмечалось, что хотя в полимерах ниже температуры
стеклования возможно несколько механизмов размора-
живания молекулярных движений, однако, в отличие
от спектра механических потерь, на равновесной тепло-
емкости они не проявляются в виде пиков, горбов или
скачков, а приводят к монотонному возрастанию тепло-
емкости. На основании этого иногда делается вывод,
что теплоемкость нечувствительна к возникновению
подвижности в полимерах при низких температурах.
Этот вопрос уже рассматривался при анализе поведе-
ния теплоемкости при низких температурах. Здесь
лишь необходимо отметить, что такое простое сравне-
ние теплоемкости со спектром механических потерь,
по-видимому, затруднительно.
Это заключение, сделанное недавно Бауром [3],
основано на следующих рассуждениях. Температурная
зависимость теплоемкости тела неизменной структуры
является отражением его термостатических свойств.
Она определяется появлением дополнительных степеней
свободы и интенсивностью теплового движения. С точ-
ки зрения фононных представлений это соответствует
появлению дополнительных фононов определенной
энергии. Взаимодействие же фононов не должно давать
существенного вклада в теплоемкость. Напротив, имен-
но это взаимодействие служит причиной появления
механических потерь, которые являются сугубо дина-
133
мическим свойством системы, поскольку появляются
при выведении системы из одного равновесного состоя-
ния и переходе ее в другое равновесное состояние.
С этой точки зрения прямое сравнение температурной
зависимости равновесной теплоемкости и спектра меха-
нических потерь в большинстве случаев невозможно.
Справедливость этих аргументов, по-видимому, в
значительной степени ослабевает при попытке сопоста-
вить динамическую (неравновесную) теплоемкость со
спектром механических потерь. Из-за неравновесности
динамических измерений теплоемкости в областях раз-
мораживания подвижности элементов молекулярной
структуры теперь уже возможно проявление динамики
размораживания. Экспериментальным доказательством
этой возможности является переход при 50 °C в стекло-
образном полистироле, который удается зафиксировать
чувствительным методом ДТА и который не проявляет-
ся при равновесных измерениях, хотя и предполагается,
что он связан с дополнительным размораживанием
подвижности фенильных групп [226]. Метод динамиче-
ских потерь также фиксирует наличие заметного макси-
мума потерь в этой температурной области.
Еще одним экспериментальным подтверждением су-
щественной роли динамических измерений теплоемко-
сти для выявления вторичных переходов служит анома-
лия, обнаруженная у расплава атактического полисти-
рола при использовании комплекса динамических
методов исследования, в том числе и метода ДТА
[115, с. 305]. Предполагается, что этот переход связан
с повышенной подвижностью полимерной цепи при
высоких температурах и способностью макромолекул
проявлять себя как единое целое из-за значительного
свободного объема.
Кажущаяся нечувствительность равновесной тепло-
емкости к переходам в стеклообразном состоянии по-
служила стимулом для более детального анализа ре-
зультатов измерения температурного хода теплоемкости
стеклообразных полимеров [233]. В местах предпола-
гаемых переходов в стеклообразном полистироле был
проведен анализ наклонов температурной зависимости
теплоемкости (рис. 11.41). Хотя скачки на ср отсутст-
вуют, однако наклоны на отдельных участках несколько
различаются. Формально такое поведение теплоемкости
134
должно быть характерно для переходов третьего рода,
что и послужило основанием для Бойера отождествить
переходы в стеклообразном полистироле с переходами
третьего рода. Так же как и для случая отождествле-
ния стеклования с переходами второго рода, это заклю-
чение имеет мало смысла.
Суммируя результаты применения тепловых методов
для исследования переходов в аморфных полимерах
ниже температуры стеклования, следует отметить, что
в настоящее время для многих полимеров существуют
значительные расхождения между данными, получен-
ными тепловыми и другими физическими методами.
100 150 200 250 300 350
Рис. 11.41. Температурная зависимость теплоемкости сР
и ее температурной производной для полистирола
[233].
Для многих кристаллических полимеров в спектрах
механических потерь между температурами стеклова-
ния и плавления наблюдаются максимумы потерь. Для
полимеров, способных находиться в нескольких кри-
сталлических состояниях, характеризующихся различ-
ными кристаллическими решетками, максимумы потерь
связаны с фазовыми переходами от одной решетки
к другой. Эти переходы хорошо проявляются в изме-
нении тепловых свойств. Если такие переходы в твер-
дом состоянии отсутствуют, максимумы потерь обычно
объясняют упомянутыми выше причинами. Появление
аномалий на температурной зависимости теплоемкости
у некоторых полимеров в этой области (атактический
полипропилен) объясняют плавлением стереорегуляр-
135
ных последовательностей, присутствующих в атактиче-
ских цепях. Многие из этих промежуточных переходов
сильно зависят от термических условий приготовления
образцов. Сравнительное изучение одних и тех же об-
разцов тепловыми и механическими методами до сих
пор, по-видимому, не проводилось. Возможно, что
такое исследование позволило бы установить, имеются
ли основания для отождествления этих аномалий с
упомянутыми выше механизмами.
ЛИТЕРАТУРА
1. Борн М., Хуан Кунь. Динамическая теория кристалличе-
ских решеток. Пер. с англ. Под ред. И. М. Лифшица. М., Из-
датинлит, 1958. 488 с.
2. Лейбфрид Г. Микроскопическая теория механических и теп-
ловых свойств кристаллов. Пер. с англ. Под ред. Б. Я. Мойжеса.
М., Физматгиз, 1963. 312 с.
3. Вундерлих Б., Баур Г. Теплоемкость линейных полимеров.
Пер. с англ, и нем. Ю. К. Годовского. М., «Мир», 1972.
238 с.
4. Blackman М. In: Handbuck der Physik. Bd. VII, T. I. Red.
S. Flugge. Berlin, Springer, 1955.
5. T a p а с о в В. В., ДАН СССР, 1945, т. 46, № 1, с. 22—25.
6. Т а р а с о в В. В., ДАН СССР, 1947, т. 58, № 4, с. 577—580.
7. Тарасов В. В., ЖФХ, 1950, т. 24, № 1, с. 111—128; 1952,
т. 26, № 5, с. 759—763.
8. Тарасов В. В. Новые вопросы физики стекла. М., Госстрой-
издат, 1959. 270 с.
9. Т а р а с о в В. В., Юницкий Г. А., ЖФХ, 1965, т. 39, № 11,
с. 2077—2080.
10. Степанов П. Е., ЖФХ, 1952, т. 26, № 11, с. 1642—1658.
11. Ко бек о П. П. Аморфные вещества. М. — Л., изд-во АН СССР,
1952. 431 с.
12. Лифшиц И. М., ЖФХ, 1953, т. 27, № 2, с. 294—295.
13. Лифшиц И. М, ЖЭТФ, 1952, т. 22, № 4, с. 471—474.
14. Лифшиц И. М., там же, с. 475—486.
15. Baur Н, Koll.-Z. u. Z. Polymere, 1970, Bd. 241, S. 1057.
16. Stockmayer W. H., Hecht С. E., J. Chem. Phys., 1953,
v. 21, p. 1954.
17. G e n e n s k у S. M., Newell G. F., J. Chem. Phys., 1957, v. 26,
p. 486.
18. Герцберг Г. Колебательные и вращательные спектры много-
атомных молекул. Пер. с англ. Под ред. М. А. Ельяшевича. М.,
Издатинлит, 1949. 648 с.
19. Готлиб Ю. Я., Сочава И. В., ДАН СССР, 1962, т. 147, № 3,
с. 580—583.
20. Бирштейн Т. М., Птицын О. Б. Конформации макромоле-
кул. М., «Наука», 1964. 391 с.
21. Киттель Ч. Введение в физику твердого тела. Пер. с англ.
А. А. Гусева. М., Физматгиз, 1963. 696 с.
136
22. О’Р е й л и Д ж. М., К а р а ц Ф. Е. В кн.: Переходы и релакса-
ционные явления в полимерах. Пер. с англ. Под ред. А. Я. Мал-
кина. М., «Мир», 1968. 384 с.
23. Tucker J. Е., Reese W., J. Chem. Phys., 1967, v. 46, p. 1388.
24. Reese W., J. Macromol. Sci., 1969, A, v. 3, p. 1257.
25. Сочава И. В., Трапезникова О. Н., ДАН СССР, 1957,
т. 113, № 4, с. 784—786.
26. С о ч а в а И. В., Трапезникова О. Н., Вестник ЛГУ, 1958,
т. 13. № 16, с. 65—72.
27. Сочава И. В., ДАН СССР, 1960, т. 130, № 2, с. 126—128.
28. Сочава И. В., Вестник ЛГУ, 1961, т. 16, № 10, с. 70—72.
29. Сочава И. В., Вестник ЛГУ, 1964, т. 19, № 10, с. 56—64.
30. С о ч а в а И. В., Трапезникова О. Н., Вестник ЛГУ, 1965,
т. 20, № 22, с. 71—74.
31. Wunder li ch В., J. Chem. Phys., 1962, v. 37, p. 1207.
32. W u n d e r 1 i c h B., Jones L. D., J. Macromol. Sci., 1969, B,
v. 3 (1), p. 67.
33. Baur H., Koll.-Z. u. Z. Polymere, 1971, Bd. 244, S. 293.
34. Г о д о в с к и й Ю. К. В кн.: Успехи химии и физики полимеров.
М., «Химия», 1970, с. 173—205.
35. К г е v е 1 е n D. W. van. Properties of Polymers. Amsterdam —
London — New York, Elsevier Publ. Comp., 1972. 427 p.
36. Дол M., «Химия и технология полимеров», 1962, № 1, с. 3—49.
37. Дол М., «Химия и технология полимеров», 1967, № 1, с. 62—70.
38. Френкель Я. И., Собр. избр. трудов. Т. III. М. — Л., изд-во
АН СССР IQ^Q Д^Я г
39. Wunderlich’ В., J.’chem. Phys., 1962, v. 37, р. 2449.
40. Н i г a i N., Eyring Н., J. Polymer Sci., 1959, v. 37, p. 51.
41. W u n d e r 1 i c h B., J. Phys. Chem., 1960, v. 64, p. 1052.
42. Bares V., Wunderlich B., J. Polymer Sci., 1973, pt. A-2,
v. 11, p. 1301.
43. Bondi A., Ind. Eng. Chem. Fundementals, 1966, v. 5, p. 442.
44. Bares V., Wunderlich B., J. Polymer Sci., 1973, pt. A-2,
v. 11, p. 861.
45. M и с н a p А. Теплопроводность твердых тел, жидкостей, газов
и их композиций. Пер. с франц. Под ред. Э. Шпильрайна. М.,
«Мир», 1968. 464 с.
46. Thermal conductivity. Ed. by R. P. Tye. London — New York,
Academic Press, 1969, v. 1, 422 p.; v. 2, 353 p.
47. D e b у e P. Vortrage fiber kinetische Theorie der Materie und
Electrizitat. Teubner, 1914.
48. П а й e p л с P. Квантовая теория твердых тел. Пер. с англ. Под
ред. А. А. Абрикосова. М., Издатинлит, 1956. 259 с.
49. В г eman R., Proc. Roy. Soc., 1951, v. 208, p. 90.
50. 3 а й м а н Д. Электроны и фононы. Пер. с англ. Под ред.
В. Л. Бонч-Бруевича. М., Издатинлит, 1962. 488 с.
51. Оскотский В. С., Смирнов И. А. Дефекты в кристаллах
и теплопроводность. Л., «Наука», 1972. 159 с.
52. К 1 е m е n s Р. G. In: Non-Crystalline Solids. Ed. by J. A. Prins.
Amsterdam, North — Holland Publ. Comp., 1965.
53. В r i d g m a n P. W. The Physics of High Pressure. London,
G. Bell, 1952. 445 p.
54. Пре дв о дител ев А. С., ЖФХ, 1948, т. 22, № 3, с. 339—
348.
137
55. Herrocks J. К., McLaughlin E., Trans. Faraday Soc.,
1960, v. 56, p. 206.
56. Reese W., J. Appl. Phys., 1966, v. 37, p. 864.
57. Eiermann K., «Kunststoffe», 1961, Bd. 51, S. 512.
58. Eiermann K., Koll.-Z. u. Z. Polymere, 1964, Bd. 198, S. 5.
59. К n a p p e W., «Fortschritte der Hochpolymeren-Forschung»
(Adv. Polymer Sci.), 1971, Bd. 7, S. 477.
60. Schallamach A., Proc. Phys. Soc., 1941, v. 53, p. 214.
61. Shoulberg R. H., Shelter J. A., J. Appl. Polymer Sci.,
1962, v. 6, p. 532.
62. Hо 1 zmu 11 er W., Miinx M., Koll.-Z. u. Z. Polymere, 1958.
Bd. 159, S. 25.
63. Holzmuller W., Lorenz J., «Plaste u. Kautschuk», 1961,
Bd. 8, S. 351.
64. Eiermann K., Koll.-Z. u. Z. Polymere, 1962, Bd. 180, S. 163.
65. Eiermann K., Hellwege К.-H., J. Polymer Sci., 1962,
v. 57, p. 99.
66. F r i s c h H. L., Rogers С. E., J. Polymer Sci., 1966, pt. C,
№ 12, p. 297.
67. Мамедалиев Г. Г., Абдинов Д. Ш., Алиев Г. М.,
ДАН СССР, 1970, т. 190, № 6, с. 1393—1395.
68. Г а в р и л о в А. Н., Володин В. П., К у в ш и н с к и й Е. В.,
Высокомол. соед., 1972, Б, т. 14, с. 687—690.
69. Reese W., Tucker J. Е., J. Chem. Phys., 1965, v. 43, p. 105.
70. К oil ouch R. J., Brown R. G., J. Appl. Phys., 1968, v. 39,
p 3999
71. Eiermann K., Koll.-Z. u. Z. Polymere, 1965, Bd. 201, S. 3.
72. Черкасова Л. П., ЖФХ, 1959, т. 33, № 9, с. 1928—1932.
73. Hattori M„ Koll.-Z. u. Z. Polymere, 1962, Bd. 185, S. 27.
74. Wil ski H., «Kunststoffe», 1963, Bd. 53, S. 363.
75. H a n s e n D., Но С. C., J. Polymer Sci., 1965, A, v. 3, p. 659.
76. Matsuoka S., J. Appl. Phys., 1961, v. 32, p. 2334.
77. Eiermann K-, Koll.-Z. u. Z. Polymere, 1964, Bd. 198, S. 96.
78. Арутюнов Б. А., Биль В. С., «Механика полимеров», 1966,
№ 5, с. 793—796.
79. О z a w а Т., Kanari К., J. Polymer Sci., 1967, В, v. 5, р. 767.
80. В о n d i A. Physical Properties of Molecular Crystalls, Liquids
and Glasses. N. Y. — L., J. Wiley and Sons, 1968. 502 p.
81. U eb err ei ter K., Nens S., Koll.-Z., 1951, Bd. 123, S. 92.
82. U e b e r r e i t e г К., О 11 о - L a u p e n m u h 1 e n E., Z. Natur-
forsch., 1953, Bd. 8a, S. 664.
83. Lohe P., Koll.-Z. u. Z. Polymere, 1965, Bd. 204, S. 7.
84. Hansen D., Wash о В. D., Koll.-Z. u. Z. Polymere, 1966,
Bd. 210, S. 111.
85. Lohe P., Koll.-Z. u. Z. Polymere, 1965, Bd. 205, S. 1.
86. К n a p p e W., Lohe P., W u t s c h i n g R., Angew. makromol.
Chem., 1969, Bd. 7, S. 181.
87. Каргин В. А., Слонимский Г. Л., Липатов Ю. С.,
ДАН СССР, 1955, т. 104, № 1, с. 96—97.
88. С л о н и м с к и й Г. Л., Дик а рев а Т. А., Высокомол. соед.,
1965, т. 7, № 7, с. 1276—1280.
89. Слонимский Г. Л., Дика рев а Т. А., Павлов В. И.,
Высокомол. соед., 1966, т. 8, № 10, с. 1717—1721.
90. Hennig J., Koll.-Z. u. Z. Polymere, 1964, Bd. 196, S. 136.
138
91. Hennig J., Knappe W., J. Polymer Sci., 1964, pt. C, № 6,
p. 167.
92. H e 11 m u t h W., Kilian H.-G., Muller F. H, Koll.-Z. u.
Z. Polymere, 1967, Bd. 218, S. 10.
93. Hell we ge К.-H., Hennig J., Knappe W., KolL-Z. u.
Z. Polymere, 1963, Bd. 188, S. 121.
94. Eeirmann K., Koll.-Z. u. Z. Polymere, 1964, Bd. 199, S. 125.
95. Fox J. S., Imber M, J. Appl. Polymer Sci., 1968, v. 12,
p. 571.
96. Кириченко Ю. А., Олейник Б. H., Чадович T. 3.,
Инж.-физ. ж., 1964, т. 7, № 1, с. 70—74.
97. Shoulberg R.-H, J. Appl. Polymer Sci., 1963, v. 7, p. 1597.
98. Hattori M, Koll.-Z. u. Z. Polymere, 1965, Bd. 202, S. 11.
99. St ее re R. C., J. Appl. Polymer Sci., 1966, v. 10, p. 1673.
100. Ueberreiter K-, Naghizadeh J., Koll.-Z. u. Z. Polyme-
rP 1079 Rrj 9^0 Q Q97
101. St ее re R. C, J. Appl. Phys., 1966, v. 37, p. 3338.
102. Tautz H, J. Polymer Sci., 1968, pt. C, № 16 (7), p. 3723.
103. Friel i ng s dor f K., Chem.-Ing.-Techn, 1960, Bd. 32, S. 291.
104. Егоров Г. M. Канд. дис. M, ВНИИФТРИ, 1973.
105. Ueberreiter К., Koll.-Z. u. Z. Polymere, 1967, Bd. 216—217,
S. 217.
106. U e b e r r e i t e r K., Purucker S., Koll.-Z.-u. Z. Polymere,
1950, Bd. 144, S. 120.
107. Naghizadeh J., Ueberreiter K-, Koll.-Z. u. Z. Polyme-
re, 1972, Bd. 250, S. 932.
108. A p у т io н о в Б. А., «Механика полимеров», 1965, № 6, с. 122—
124.
109. Керзон Хуанг. Статистическая механика. Пер. с англ. Под
ред. Ю. А. Церковникова. М, «Мир», 1966. 520 с.
НО. Эпштейн П. Курс термодинамики. Пер. с англ. М.—Л., Гос-
техиздат, 1948. 419 с.
111. Mayer J. Е., Streeter S. F., J. Chem. Phys., 1939, v. 7,
p. 1019.
112. У э стр ум Э, Мак-Каллаф Дж. В кн.: Физика и химия
твердого состояния органических соединений. Пер. с англ. Под
ред. Ю. А. Пентина. М, «Мир», 1967, с. 9—160.
113. Каргин В. А., Слонимский Г. Л., «Успехи химии», 1955,
т. 24, № 7, с. 785—800.
114. Мюнстер А. В кн.: Проблемы современной физики. М, Из-
датинлит, 1956, с. 96—109.
115. Переходы и релаксационные явления в полимерах. Под ред.
Р. Ф. Бойера. Пер. с англ. Под ред. А. Я. Малкина. М, «Мир»,
1968. 384 с.
116. Boyer R. F, Rubb. Chem. Technol., 1963, v. 36, p. 1303.
117. Enkel E, R inkens H., Z, Electrochem., 1956, Bd. 60,
S. 96.
118. С e м e н ч e н к о В. К., Коллоид, ж., 1962, т. 24, № 3, с. 323—
331.
119. Tobol sky А. V., J. Chem. Phys, 1962, v. 37, p. 1139.
120. Tobol sky A. V, J. Polymer Sci, 1965, pt. C, № 9, p. 157.
121. Флори П. В кн.: Проблемы современной физики. М, Издат-
инлит, 1956, с. 88—96.
139
122. Бартенев Г. М., Ремизова А. А., ЖФХ, 1957, т. 31, № 11,
с. 2534—2546.
123. Бартенев Г. М., ЖФХ, 1960, т. 34, № 3, с. 618—628.
124. Манделькерн Л. Кристаллизация полимеров. Пер. с англ.
Под ред. С. Я. Френкеля. М., «Химия», 1966. 336 с.
125. Ц а хм ан Г., «Химия и технология полимеров», 1966, № 5,
с. 3—77.
126. Слонимский Г. Л., Г о д о в с к и й Ю. К., Высокомол. соед.,
1965, т. 7, № 4, с. 621—625.
127. Годовский Ю. К., Слонимский Г. Л., Гарбар Н. М.,
Высокомол. соед., 1972, А, т. 14, № 4, с. 813—827.
128. Godovsky Yu. К., Slonimsky G. L., Garbar N. M.,
J. Polymer Sci., 1972, pt. C, № 38, p. 1.
129. Mandelkern L., J. Polymer Sci., 1960, v. 47, p. 494.
130. Hoffman J. D., Weeks J. J., J. Chem. Phys., 1962, v. 37,
p. 1723.
131. Wunderlich B., «Polymer», 1964, v. 5, p. 611.
132. Fischer E. W., Koll.-Z. u. Z. Polymere, 1969, Bd. 231, S. 458.
133. Фишер E. В. В кн.: Физическая химия полимеров за рубежом.
Пер. с англ. Под ред. 3. А. Роговина и А. Я. Малкина. 1970,
с. 9—50.
134. Годовский Ю. К., Слонимский Г. Л., Высокомол. соед.,
1967, А, т. И, № 4, с. 845—850.
135. Годовский Ю. К., Липатов Ю. С., Высокомол. соед.,
1968, А, т. 10, № 1, с. 32—39.
136. Годовский Ю. К-, Липатов Ю. С., Высокомол. соед.,
1968, Б, т. 10, № 5, с. 323—326.
137. Тейтельбаум Б. Я., Аношина Н. П., Высокомол. соед.,
1965, т. 7, № 12, с. 2112—2116.
138. Lovering Е. G., Wooden D. С., J. Polymer Sci., 1969,
pt. А-2, v. 7, р. 1639.
139. Pelzbauer Z., Manley R. S. T. J., J. Polymer Sci., 1970,
pt. A-2, v. 8, p. 649.
140. Keller A., M a r t u s c e 11 i E., Makromol. Chem., 1972, Bd. 151,
S. 189.
141. С о ч а в а И. В., Церетели Г. И., Смирнова О. И., ФТТ,
1972, т. 14, № 2, с. 553—561.
142. Сочава И. В., ФТТ, 1973, т. 15, № 5, с. 1559—1565.
143. Сочава И. В., Смирнова О. И., ФТТ, 1973, т. 15, № 10,
с. 3003—3005.
144. Г одовский Ю. К. Докт. дис. М., НИФХИ им. Л. Я. Карпова,
1972.
145. Т е й т е л ь б а у м Б. Я., Аношина Н. П., «Успехи химии»,
1967, т. 36, № 1, с. 142—166.
146. Г о д о в с к и й Ю. К- В кн.: Энциклопедия полимеров. М., «Сов.
энциклопедия», 1974, т. 2, с. 607—609.
147. W u n d er 1 i ch В. Macromolecular Physics. V. 1. N. Y. — L.,
Academic Press, 1973. 549 p.
148. Hoffman J. D., SPE Trans., 1964, October, p. 353.
149. W u n d e r 1 i c h B., Koll.-Z. u. Z. Polymere, 1969, Bd. 231,
S. 605.
150. Hen d us H., Schnell G., «Kunststoffe», 1961, Bd. 51, S. 69.
151. Шарп лез А. Кристаллизация полимеров. M., «Мир», 1968.
200 с.
140
15t2 . Годовский Ю. К. В кн.: Энциклопедия полимеров. М., «Сов.
энциклопедия», 1972, т. 1, с. 1178—1186.
153. Колмогоров А. Н., Изв. АН СССР. Сер. математ., 1937
№ 3, с. 355—358.
154. Avrami М., J. Chem. Phys., 1939, v. 7, р. 1103; 1940, v. 8,
р. 212; 1941, v. 9, р. 174.
155. Сирота Н. Н., ДАН СССР, 1942, т. 36, № 6, с. 192—196.
156. Ерофеев Б. В., ДАН СССР, 1946, т. 52, № 1, с. 515—518.
157. Evans R. U., Trans. Faraday Soc., 1945, v. 41, p. 365.
158. Годовский Ю. K-, Слонимский Г. Л., Высокомол. соед.,
1966, т. 8, № 3, с. 395—402, 403—411; № 4, с. 718—721; 1967,
А, т. 9, № 4, с. 863—869.
159. Го д ов ски й Ю. К., Липатов Ю. С., Высокомол. соед.,
1968, А, т. 10, № 4, с. 741—749.
160. Godovsky Yu. К., S 1 о n i m s к у G. L., J. Polymer Sci.,
1974, pt. A-2, v. 12, p. 1053.
161. Banks W., Sharpies A., Hay J. N., J. Polymer Sci., 1964,
pt A, v. 2, p. 4059.
162. Price F. P., J. Polymer Sci., 1965, pt. A, v. 3, p. 3079.
163. Hillier J. H., J. Polymer Sci., 1965, pt. A, v. 3, p. 3067.
164. Danusso F., Tieghi G., Felderer V., Europ. Poly-
mer J., 1970, v. 6, p. 1521.
165. Gordon M., Hillier J. H., Trans. Faraday Soc., 1964, v. 60,
p. 743.
166. Hoffman J. D. e. a., Koll.-Z. u. Z. Polymere, 1969, Bd. 231,
S. 564.
167. Suzuki T., Kovacs A. J., Polymer J., 1970, v. 1, p. 82.
168. Г о д о в с к и й Ю. K-, Высокомол. соед., 1969, А, т. 11, № 10,
£ 2129____2135
169. Gandica A., Magill J. Н., «Polymer», 1972, v. 13, р. 595.
170. Г о д о в с к и й Ю. К., Левин В. Ю., Слонимский Г. Л.,
Жданов А. А., Андрианов К. А., Высокомол. соед., 1969,
А, т. 11, № 11, с. 2444—2451.
171. F а 1 k a i В., Makromol. Chem., 1960, Bd. 41, S. 86.
172. Marker L. e. a., J. Polymer Sci., 1959, v. 38, p. 33.
173. Kim H., Mandelkern L., J. Polymer Sci., 1968, pt. A-2,
v. 6, p. 695.
174. Fischer E. W., Koll.-Z. u. Z. Polymere, 1967, Bd. 218, S. 97.
175. Gonthier A., Vidotto G., Kovacs A. J., IUPAC Sym-
posium. Leiden^ 1970; Abstracts, 1970, v. 2, p. 797.
176. Kovacs A. J., Gonthier A., Koll.-Z. u. Z. Polymere, 1972,
Bd. 250, S. 530.
177. Keith H. D., Koll.-Z. u. Z. Polymere, 1969, Bd. 231, S. 421.
178. Андрианов К. А., Жданов А. А., Годовский Ю. К.,
Левин В. Ю., Цванкин Д. Я., Москаленко В. А., Вы-
сокомол. соед., 1970, Б, т. 12, № 4, с. 722—723.
179. Andrianov К. A., Slonimsky G. L., Zhdanov А. А.,
Levin V. Y u., Godovsky Y u. К<, Moskalenko V. A.,
J. Polymer Sci., 1972, pt. A-l, v. 10, p. 1.
180. M ock а л енк о В. А., Цванкин Д. Я-, Галил-Ог-
лы Ф. А., Высокомол. соед., 1970, № 3, А, т. 12, с. 548—552.
181. Петерлин А., «Химия и технология полимеров», 1966, № 10,
с. 49—73.
182. Kauzmann W., Chem. Rev., 1948, v. 43, p. 219.
141
183. Kovacs A. J., «Fortschritte der Hochpolymeren-Forschung»,
1963, Bd. 3, S. 394.
184. The Physics of Glassy Polymers. Ed. by R. N. Haward. London,
Applied Sci. Publ., 1973. 620 p.
185. Shen M., Eisenberg A., Rubb. Chem. Technol., 1970, v. 43,
p. 95, 156.
186. G о 1 d s t e i n M. In: Modern Aspects of the Vitreous State.
V. 3. Ed. by J. D. MacKenzie. London, Butterworths, 1964,
ch. 4.
187. Ферри Дж. Вязкоупругие свойства полимеров. Пер. с англ.
Под ред. В. Е. Гуля. М., Издатинлит, 1963. 535 с.
188. W г a s i d 1 о W., «Fortschritte der Hochpolymeren-Forschung»,
1974, Bd. 13, S. 3.
189. Boyer R. F., J. Macromol. Sci., 1973, v. B7 (3), p. 487.
190. Fox T. G., Flory P. J., J. Appl. Phys., 1950, v. 21, p. 581.
191. К a nig G., Koll.-Z. u. Z. Polymere, 1969, Bd. 233, S. 54.
192. M i 11 e r A. A., J. Polymer Sci., 1964, pt. A, v. 2, p. 1095.
193. Sharma S. C., Man del kern L., Stehling F. C., J. Po-
lymer Sci., 1972, pt. B, v. 10, p. 345.
194. Журков С. H., Левин Б. Я. В кн.: Юбилейный сборник к
70-летию акад. А. Ф. Иоффе. М. — Л., изд-во АН СССР, 1950,
с. 260—267.
195. В ольке и штейн М. В., Шаронов Ю. А., Высокомол.
соед., 1961, т. 3, № 11, с. 1739—1745.
196. Ш а р о н о в Ю. А., В о л ь к е н ш т е й н М. В., Высокомол.
соед., 1962, т. 4, № 6, с. 917—921.
197. Petrie S. Е. В., J. Polymer Sci., 1972, pt. A-2, v. 10, p. 1255.
198. Сидорович А. В., Кувшинский E. В., ФТТ, 1964, т. 6,
Ко 3, с. 888—895.
199. Сидорович А. В., Кувшинский Е. В. В кн.: Релакса-
ционные явления в полимерах. Л., «Химия», 1972, с. 63—76.
200. А1 f г е у Т., G о 1 d f i п g е г G., Mark Н., J. Appl. Phys., 1943,
v. 14, p. 700.
201. Spencer S., J. Coll. Sci., 1949, v. 4, p. 229.
202. Kovacs A. J., J. Polymer Sci., 1958, v. 30, p. 131.
203. Шаронов Ю. А., Волькенштейн M. В., ФТТ, 1963, т. 5,
№ 2, с. 590—598.
204. Волькенштейн М. В., Птицын О. Б., ЖТФ, 1956, т. 26,
№ 10, с. 2204—2222.
205. Птицын О. Б., ДАН СССР, 1955, т. 103, № 6, с. 1045—1048.
206. Готлиб Ю. Я., Птицын О. Б., ФТТ, 1961, т. 3, № 11,
с. 3383—3388.
207. Boyer R. F., Spencer R. S., Adv. Coll. Sci., 1946, v. 2,
p. 1.
208. Gibbs J. H., DiMarzio E. A., J. Chem. Phys., 1958, v. 28,
p. 373.
209. D i M а г z i о E. A., Gibbs J. H., J. Polymer Sci., 1963, pt. A,
v. 1, p. 1417.
210. F1 о г у P. J. Principles of Polymer Chemistry. Ithaca, New York,
Cornell Univ. Press, 1953. 672 p.
211. Adam G., Gibbs J. H., J. Chem. Phys., 1965, v. 43, p. 139.
212. Breuer H., Rehage G., Koll.-Z. u. Z. Polymere, 1967, Bd. 216,
S. 159.
142
213. Passaglia E., Kevorkian K., J. Appl. Phys., 1963, v. 34,
p. 90.
214. Bestul A. B., Chang S. S., J. Chem. Phys., 1964, v. 40,
p. 3731.
215. De vies R., Jones G., Proc. Roy. Soc., 1953, pt. A, v. 217,
p. 26.
216. DiMarz io E. A., J. Appl. Phys., 1974, v. 45, p. 4143.
217. Zachman H. G., Koll.-Z. u. Z. Polymere, 1969, Bd. 231, S. 504.
218. К a r a z s F. E., Bair H. E., J. Phys. Chem., 1965, v. 69, p. 2607.
219. Ягфаров M. Ш., Высокомол. соед., 1969, А, т. 11, № 6,
с. 1195—1201.
220. Привалко В. П., Липатов Ю. С., Керча Ю. Ю., Высо-
комол. соед., 1969, А, т. 11, с. 237—246.
221. O’Reilly J. М., Karazs F. Е., Polymer Prepr., 1964, v. 5,
р. 351.
222. Hobbs S. Y., Mankin G. J., J. Polymer Sci., 1971, pt. A-2,
v. 9, p. 1907.
223. U e b e r r e i t e r K., Kanig G., Z. Naturforsch., 1951, Bd. 6a,
S. 551.
224. Журков С. H., ДАН СССР, 1945, т. 47, № 7, с. 493—496;
т. 49, № 3, с. 201—204.
225. М а р т ы н е н к о Л. Я., Рабинович И. Б., Овчинни-
ков Ю. В., Маслова В. А., Высокомол. соед., 1970, А, т. 12,
№ 4, с. 841—848.
226. Wunderlich В., Bodily D. М., J. Appl. Phys., 1964, v. 35,
р. 103.
227. Неу dem an Р., Gui eking H. G., Koll.-Z., 1963, Bd. 193,
S. 16.
228. Holden R., S i m h a R., J. Appl. Phys., 1968, v. 39, p. 1890.
229. Witmann J. C., Kovacs A. J., J. Polymer Sci., 1969, pt. C,
№ 16, p. 4443.
230. O’R e i 11 у J. M., Karazs F. E., Bair H. E., J. Polymer Sci.,
1963, pt. C, № 6, p. 109.
231. Квач ев Ю. П., Папков В. С. и др., ДАН СССР, 1974,
т. 215, № 6, с. 1373—1375.
232. Козлов П. В. и др., Высокомол. соед., 1967, А, т. 9, № 9,
с. 2047—2061.
233. Boyer R. F. In: Thermal Analysis. Proceedings of the Third In-
tern. Conference in Thermal Analysis. Basel und Stutgart,
Birkhauser Verlag, 1972, v. 3, p. 3—18.
РАЗДЕЛ III
ТЕПЛОФИЗИЧЕСКИЕ ПРОЦЕССЫ
ПРИ ДЕФОРМАЦИЯХ ПОЛИМЕРОВ
Глава 6
ОБРАТИМЫЕ ДЕФОРМАЦИИ
Тепловое расширение полимеров
Тепловое расширение является простейшим случаем
изменения размеров и формы тела под действием одной
лишь температуры (тепловая деформация). Представ-
ление о гармонических колебаниях связанных между
собой частиц, используемое при теоретическом анализе
теплоемкости твердых тел, оказывается недостаточным
для объяснения теплового расширения, поскольку
в гармоническом приближении твердое тело вообще
не обладает тепловым расширением. Анализ показы-
вает, что тепловое расширение появляется лишь в си-
стеме взаимодействующих частиц, характеризуемой
асимметричной кривой потенциальной энергии взаимо-
действия при учете ангармонических членов в разло-
жении потенциальной энергии по смещениям частиц
из положения равновесия [1, с. 237; 2, с. 179; 3, с. 404;
4, с. 130]. Тепловое расширение твердых тел связано
с другими термодинамическими характеристиками соот-
ношением Грюнайзена:
где ₽—термический коэффициент линейного расширения; у г—па-
раметр Грюнайзена; Хг — изотермическая сжимаемость; V — объем.
Параметр Грюнайзена, который в простейшем слу-
чае предполагается не зависящим от температуры, для
твердых тел, для которых единственным вкладом в теп-
144
лоемкость является вклад от колебаний решетки, опре-
деляется соотношением
д In Op
Тг = “ din У- 2)
Если пренебречь изменением ut/V с температурой, ко-
торое обычно невелико, то температурная зависимость
теплового расширения пропорциональна температурной
зависимости теплоемкости. Поэтому при приближении
к абсолютному нулю тепловое расширение стремится
к нулю. Для многих твердых тел параметр Грюнайзена,
представляющий отношение усредненного свойства
твердого тела, определяемого ангармоничностью коле-
баний частиц, к свойству, связанному с гармоническими
колебаниями, практически не зависит от температуры,
по крайней мере в температурном интервале от 6р до
O,20d, и по величине близок к 2.
Тепловое расширение кристаллов (за исключением
кубических) анизотропно. Оно описывается при помо-
щи трех главных коэффициентов расширения. Терми-
ческий коэффициент объемного расширения равен сум-
ме главных коэффициентов расширения. Для большин-
ства кристаллов главные термические коэффициенты
расширения положительны, что приводит к появлению
эллипсоида расширения. Однако у ряда монокристал-
лов (кальций, теллур, цинк, селен) некоторые коэффи-
циенты отрицательны, что приводит к сложным поверх-
ностям теплового расширения. Наиболее точным спосо-
бом измерения анизотропии теплового расширения кри-
сталлов является рентгеновский метод измерения пара-
метров решетки. Тензор теплового расширения анизо-
тропных структур характеризует анизотропию сил,
действующих в кристалле. Поликристаллические тела
обычно являются макроизотропными по отношению
к тепловому расширению, хотя они и построены из
заведомо анизотропных микроблоков. Для большинства
поликристаллических веществ термический коэффи-
циент расширения положителен. Интересен в этом
отношении иодид серебра, кристаллы которого в интер-
вале температур от —10 до +70 °C обладают отрица-
тельными коэффициентами по всем главным осям, так
что и термические коэффициенты объемного и линей-
ного расширения поликристаллического иодида серебра
10—264
145
оказываются отрицательными [5, с. 93]. Таким обра-
зом, тепловое расширение твердых тел является отра-
жением сил, действующих между частицами, и осо-
бенностей динамики соединенных между собой ча-
стиц.
Цепное строение макромолекул, являющееся причи-
ной высокой локальной анизотропии полимеров, может
явиться источником и общей анизотропии всего поли-
мерного тела (ориентационная способность полимеров)?
В связи с этим (во многих случаях) следует ожидать
и высокой анизотропии теплового расширения поли-
меров.
Еще в работе Хенгстенберга и Марка [6] впервые
было установлено, что кристаллические решетки цел-
люлозы и натурального каучука обладают отрицатель-
ными термическими коэффициентами расширения вдоль
оси макромолекул. Изучение температурной зависимо-
сти параметров кристаллических решеток полимеров
позволило впоследствии обнаружить наличие отрица-
тельных термических коэффициентов расширения вдоль
оси макромолекул (ось ориентированного образца)
для ряда других полимеров [7—12]. В то же время два
других коэффициента оказываются положительными
и изменяются в широких пределах (табл. III. 1). Невы-
сокие значения Ра для полиамидов обусловлены нали-
чием водородных связей. Этого можно было ожидать,
исходя из данных для молекулярных кристаллов низко-
молекулярных соединений [13, с. 335]. В ранних иссле-
дованиях по изучению температурных изменений пара-
метров решетки полиэтилена трудно было выяснить,
является коэффициент расширения вдоль оси макромо-
лекулы нулевым или отрицательным. В недавних рабо-
тах было однозначно установлено, что, как и для дру-
гих полимеров, для полиэтилена наблюдается сокраще-
ние параметра решетки вдоль оси макромолекул [11,
12]. По данным Кобаяси и Келлера, для полиэтилена
в температурном интервале 20—120 СС коэффициент
рс = — 1,8-10“5 К”1. Таким образом, можно полагать, что
отрицательный термический коэффициент расширения
вдоль оси макромолекул у кристаллических решеток по-
лимеров — обычное явление. Недавно отрицательный ко-
эффициент вдоль оси макромолекулы впервые обнару-
жен и исследован дилатометрическим методом на поли-
146
Таблица I II.1. Термические коэффициенты линейного расширения
кристаллических решеток полимеров (средние значения
для температурного интервала от —196 до +20 °C) [8]
Полимер Тип решетки Степень растяжения образца Термический коэффициент л. шейного расширения р-105, К-1
Целлюлоза . . . Моноклинная (ось волокна — «Ь») — О О* м II II II ND [ 05 00 4^ о Сп
Полиэтиле нтерефта лат Триклинная (ось волокна — «Ь») 5,99 Ра = 17,1 Pb = —2,2 5с = 12,7
Найлон 6 . . . . Моноклинная (ось волокна — «Ь») То же 2,99 (Р-форма) 5,48 (а-форма) -со о ю сг II II II 1 ’ ND 4^ 4^ СП
Найлон 6,6 ... Триклинная (ось волокна — «Ь») 5,99 (а-форма) си сс О В5 II II ND ND ND’ О
Полиэтилен высокой плотности . . . Орторомбическая (ось волок- на— «с») 6,0 Ра = 14,7 Рь = 6,0
Полихлоропрен . То же 6,0 Ра = 13,1 Рс = -4,1
Натуральный каучук Моноклинная (ось волокна — «с») 5,0 Г. ст SS 1 1 ND — 1 О ND 00’ - СП СП
диацетиленовых монокристаллах, полученных полиме-
ризацией в твердой фазе [14].
Предположения относительно физической причины
появления отрицательных коэффициентов расширения
вдоль оси макромолекул сводятся в основном к посту-
лированию возрастающего с температурой вращения
вокруг С—С-связей, приводящего к сокращению
[11, 14]. В связи с этим необходимо отметить, что
Лифшиц [15] предсказал появление отрицательного
коэффициента вдоль цепи в цепных структурах при
низких температурах в результате возбуждения воли
изгиба с отличным от обычного законом дисперсии, что
одновременно должно приводить к появлению анизо-
тропной температурной зависимости тензора термиче-
ских коэффициентов расширения. Хотя этот вывод
относится лишь к низким температурам, можно пола-
гать, что тенденция к уменьшению длины с повышением
температуры сохранится в цепях, находящихся в поле
10*
147
сил кристаллической решетки, и при высоких темпера-
турах. Такое поведение было экспериментально обнару-
жено у цепочных кристаллов теллура, образованных
спиральными макромолекулами теллура [16]. Таким
образом, ценность теоретического предсказания Лиф-
шица состоит в его общности, не требующей рассмотре-
ния молекулярных особенностей цепной структуры,
предполагающей лишь высокую анизотропию сил взаи-
модействия между цепями и внутри цепей.
Возвращаясь к предположению о возрастающем*
с температурой вращении вокруг С—С-связей, можно
отметить, что величина постулированного первоначаль-
но угла вращения, равная 3° [11], при последующем
более детальном анализе [17] оказалась существенно
большей; этот угол должен быть не менее 13°, чтобы
именно вращением можно было объяснить эксперимен-
тально найденное сокращение молекулы полиэтилена.
Теперь обратимся к рассмотрению последствий, ко-
торые могут иметь для кристаллических полимеров
отрицательные коэффициенты вдоль оси макромолекул
в кристаллических решетках. Для изотропных (блоч-
ных) кристаллических полимеров их влияние не может
сказаться на общем макроскопическом термическом
коэффициенте расширения по крайней мере по двум
причинам. Во-первых, отрицательные коэффициенты
значительно меньше по абсолютной величине положи-
тельных коэффициентов в других направлениях, что
приводит к положительному коэффициенту объемного
расширения. Учитывая статистическое распределение
кристаллитов и соответственно осей макромолекул, сле-
дует полагать, что и коэффициенты расширения по
любому из направлений в макрообразцах окажутся
положительными.
Вторая причина заключается в наличии аморфных
областей в кристаллических полимерах. Выше темпера-
туры стеклования тепловое расширение кристалличе-
ских полимеров существенно зависит от степени кри-
сталличности. Наличие даже не очень существенной
доли аморфных областей приводит к весомому их
вкладу в общий коэффициент расширения ввиду значи-
тельно большего теплового расширения каучукоподоб-
ных полимеров по сравнению с твердыми. Оставляя
в стороне возможное влияние дефектов решетки на теп-
148
Ловое расширение, можно заключить, что в макрообраз-
цах кристаллических полимеров отрицательные терми-
ческие коэффициенты расширения вдоль осей макро-
молекул не должны проявляться, что и наблюдается
экспериментально.
Положение может коренным образом измениться
при переходе к ориентированным полимерам — плен-
кам и волокнам. Согласно современным представле-
ниям, структура ориентированных
кристаллических полимеров представ-
ляется 'системой микрофибрилл с по-
следовательным чередованием вдоль
них кристаллических и аморфных об-
ластей (рис. III.1). При этом макро-
молекулы в кристаллитах расположе-
ны параллельно оси ориентации. По-
этому при нагревании размеры кри-
сталлитов вдоль оси ориентации будут
уменьшаться, что должно приводить
к отрицательному вкладу в общий
термический коэффициент расшире-
ния ориентированного полимера.
В отличие от изотропного состоя-
ния роль аморфных областей в теп-
ловом расширении ориентированных
полимеров значительно сложнее.
В зависимости от степени вытяжки
участки макромолекул в аморфных
областях также оказываются в той
или иной мере вытянутыми и при тем-
Рис. III.1. Схема-
тическое изобра-
жение структуры
о р и е нт и р о в а нн ого
кристаллического
полимера:
1 — кристаллит; 2 —
аморфная область
(стрелками показана
ось ориентации).
пературах выше температуры стеклования должны со-
кращаться при нагревании. При этом вклад аморфных
областей в общий отрицательный коэффициент расши-
рения несильно ориентированного полимера будет опре-
деляющим по величине. Здесь возможны весьма различ-
ные соотношения, и можно думать, что способность к
сокращению ориентированных кристаллических полиме-
ров выше температуры стеклования будет изменяться
в широких пределах в зависимости от температуры и
условий ориентации. Ниже температуры стеклования
сокращение должно смениться расширением из-за опре-
деляющей роли аморфных областей, находящихся, Од-
нако, теперь в стеклообразном состоянии.
149
Ориентированные аморфные полимеры по вполне
очевидной причине будут сокращаться при нагревании
выше температуры стеклования, в то время как ниже
температуры стеклования они должны расширяться
вдоль оси ориентации. Однако и в случае аморфных
ориентированных полимеров возможно, по-видимому,
появление отрицательных коэффициентов расширения
в стеклообразном состоянии по двум причинам. Преж-
де всего, стеклование не означает потерю полной под-
вижности, и потому между температурой стеклования*
и температурой хрупкости в стеклообразных полиме-
рах сохраняется определенная подвижность. Она может
явиться причиной сокращения при повышении темпера-
туры. А кроме того, на наш взгляд, в очень сильно
ориентированных стеклообразных полимерах возможно
появление отрицательных коэффициентов расширения
за счет наличия волн изгиба (по Лифшицу) даже без
образования кристаллической решетки.
Как для кристаллических, так и для аморфных
полимеров с повышением степени ориентации должна
увеличиваться анизотропия теплового расширения. Тер-
мический коэффициент объемного расширения одно-
осно-ориентированного полимера равен, очевидно,
а = р и +2pj_. Если ориентация не сопровождается кри-
сталлизацией, то термический коэффициент объемного
расширения не зависит от степени ориентации, что при-
водит к простому соотношению
р(1 +2₽± = зр = а (Ш.З)
Возрастание отрицательного коэффициента расширения
вдоль оси ориентации должно сопровождаться пропор-
циональным увеличением коэффициента расширения
перпендикулярно оси ориентации.
Макроскопические отрицательные термические ко-
эффициенты расширения неоднократно наблюдались
у кристаллических полимеров [18—22] (полиэтилен,
полипропилен, полиамиды, политрифторхлорэтилен,
поливиниловый спирт, триацетат целлюлозы). В каче-
стве характерных рассмотрим результаты [22], полу-
ченные на триацетатцеллюлозной пленке (рис. III.2).
Ниже температуры стеклования (Тс = 162оС) тепловое
расширение как вдоль оси ориентации, так и поперек
150
положительно с коэффициентами расширения рц==
= 1,5-10"*5К”1 и Рх = 15-КУ-5К""1. После размягчения
пленки, вырезанные в направлении ориентации, сокра-
щаются, а в направлении, перпендикулярном ему, —
сильно расширяются. Этот эффект практически пол-
ностью обратим при переходе к охлаждению. Степень
* б
Рис. III.2. Изменение геометрических размеров при нагревании за-
кристаллизованных триацетатцеллюлозных пленок [22]:
а: 1 — изотропная при нагревании и охлаждении; 2, 2' — одноосно-ориентиро-
ванная по направлению ориентации; 3, 3' — перпендикулярно оси ориентации;
<5:0 — изотропная, 1, 2, 3, 4 и Г, 2', 3', 4' — одноосно-ориентированная по на-
правлению и перпендикулярно оси ориентации, соответственно растянутая на
15 (1, 1'), 30 (2, 2'), 40 (3, 3') и 50 (4, 4') %.
вытяжки существенно влияет как на характер измене-
ния тепловой усадки вдоль оси ориентации, так и на
характер теплового расширения по направлению, пер-
пендикулярному ориентации.
151
Интересные результаты получены на высокоориен-
тированных (100% вытяжки) аморфных триацетат-
целлюлозных пленках. Хотя при температурах ниже
100 °C трудно решить, сокращается ли образец при
нагревании, выше этой температуры наблюдается
отчетливое сокращение, которое резко усиливается пос-
ле размягчения. Таким образом, эффект обратимого
термического сокращения характерен, по-видимому, как
Рис. II 1.3. Зависимость относи-
тельного коэффициента линейного
расширения от относительной де-
формации [24] вдоль (1—4) и по-
перек —4') направления ориен-
тации:
1, 1' — поликарбонат; 2, 2' — поливинил-
хлорид; 3, 3' — полиметилметакрилат;
4, 4' — полистирол.
для ориентированных
кристаллических, так и
предельно ориентиро-
ванных аморфных поли-
меров.
Большая часть иссле-
дований такого рода ог-
раничивалась положи-
тельными температурами.
В связи с этим представ-
ляют интерес результа-
ты, полученные на раз-
ветвленном полиэтилене
(около 3,4 разветвления
на 100 атомов углерода,
плотность равна пример-
но 0,92 Мг/м3) в интер-
вале температур от ком-
натной до жидкого азота
[23]. Достаточно ориен-
тированный образец со-
кращается во всем этом
температурном интервале.
Этот крайне интересный
экспериментальный факт
свидетельствует о том, что стеклование не оказывает
заметного влияния на характер и величину сокраще-
ния, несмотря на примерно равное соотношение кри-
сталлической и аморфной фаз в этом образце. Это на-
водит на мысль о важной роли сильно ориентированных
проходных макромолекул в механизме сокращения по-
лимеров. Оценка среднего для всего температурного ин-
тервала коэффициента сокращения приводит к величи-
не, равной примерно —2-10~5 К”1, что практически сов-
падает с данными для высоких температур.
152
Изучение анизотропии теплового расширения ориен-
тированных полимеров проводилось на аморфных поли-
мерах [24]. Отрицательные коэффициенты при этом
не обнаружены, что можно отнести за счет недоста-
точно высокой ориентации. Приведенные на рис. Ш.З
и в табл. Ш.2 результаты по влиянию степени вытяж-
ки на относительное изменение коэффициентов расши-
рения ряда стеклообразных полимеров в температурном
интервале от —80 до +45 °C свидетельствуют о доста-
точно точном совпадении измеренных коэффициентов
с рассчитанными по формуле (Ш.З). Эти результаты
находятся в очень хорошем соответствии с данными по
анизотропии теплопроводности, полученными в этой же
работе и рассмотренными в предыдущем разделе.
Таблица III.2. Термические коэффициенты расширения
аморфных полимеров [24]
Полимер Степень ориента- ции, % Термический коэффициент линейного расширения х 105, К—1
₽| 3 (расчет) 3 (экспери- мент)
Полистирол 400 7,35 7,98 7,77 7,74
Полиметилметакрилат 157 5,59 8,58 7,59 7,59
275 4,07 9,30 7,56 7,59
Поливинилхлорид . . . 85 4,45 7,68 6,61 6,63
165 2,44 8,63 6,57 6,63
Поликарбонат .... 67 2,31 8,25 6,27 6,25
Термодинамика обратимых деформаций
Приложение напряжения к упругому материалу со-
провождается обратимым изменением его температуры
и соответствующими тепловыми эффектами. Изложе-
ние термодинамики упругих явлений в твердых телах
целесообразно начать с рассмотрения термоупругости
стержней. Уравнением состояния стержня является
соотношение вида f=f(/, Т). Это уравнение, объединяю-
щее зависимость длины стержня I от температуры Т
(тепловое расширение) и от растягивающей силы f
(закон Гука) в области упругих (обратимых) деформа-
ций в достаточно широком интервале температур, дале-
153
ком от температуры плавления, имеет вид [25, с. 52]
^-£Л[/О(1 + 0Л —1
(III. 4)
где Е — модуль Юнга; А—площадь поперечного сечения стержня;
/о—длина стержня в отсутствие растягивающей силы при исходной
температуре; р—термический коэффициент линейного расширения
стержня.
При анализе термодинамики стержней величины Е
и р считают постоянными и пренебрегают величинами,
второго порядка малости по рГ и изменением попереч-
ного сечения при растяжении*. В пределах такой точ-
ности уравнение состояния стержня проще применять
в виде
f = ЕЛ
(1 — ₽Т) — 1
(III. 5)
Используя соотношения между термодинамическими
коэффициентами и уравнение состояния стержня, вы-
ведем основные соотношения для термомеханического
эффекта.
Изменение температуры при адиабатическом нагру-
жении выражается соотношением
дТ \ __ Т / д1 \
df Л ~ cf \дт If
(III.6)
где Cf — теплоемкость при постоянной силе.
Обозначив термический коэффициент линейного
расширения р/ нагруженного силой f образца через
t dl \
1 нт соотношение (III.6) можно привести к виду
dTs = — Tfyldf/cf
или для конечных изменений (поскольку
температуры малы) к виду
ATS= -TMf/cf « -TfW/cf
изменения
(III.8)
Уравнение (III.8) было впервые получено Томсоном
(Кельвином) [26], создавшим основы теории термо-
упругости. Впоследствии вопросами термоупругости за-
* Учет этого изменения дает эффекты (второго порядка малости
по относительной деформации.
154
нимались известные ученые, и соотношение (III.8) не-
однократно выводилось из самых различных предполо-
жений [27, с. 74; 28—30; 31, с. 15].
Из уравнения (III.8) следует, что знак изменения
температуры определяется знаком термического коэф-
фициента расширения Р/. Тела с положительным тер-
мическим коэффициентом расширения должны охлаж-
даться при растяжении и нагреваться при сжатии.
Еще в классических опытах Джоуля [32, 33] по
определению механического эквивалента теплоты было
экспериментально установлено, что мгновенное прило-
жение (или снятие) нагрузки к твердому телу сопро-
вождается обратимым изменением температуры на
небольшую величину. При этом было установлено, что
металлические стержни и проволоки охлаждаются при
нагружении и нагреваются при снятии напряжений.
Сжатие металлических стержней приводило к противо-
положным по знаку изменениям температуры.
Экспериментальная проверка линейного соотноше-
ния между изменением температуры и приложенной си-
лой проводилась неоднократно [5, с. 461] и приводила
к полному соответствию с этим соотношением в тех
случаях, когда измерение всех величин, входящих
в уравнение, проводилось на одном и том же образце.
Среди прочих были исследованы и стержни из иодида
серебра, обладающего отрицательным макроскопиче-
ским термическим коэффициентом расширения. В пол-
ном согласии с уравнением (III.8) они нагревались при
растяжении и охлаждались при сжатии.
Уравнение (III.8) позволяет оценить теплоту дефор-
мации. Если произведение дТс/ рассматривать как ко-
личество тепла, поглощаемого или выделяемого телом
при наступлении его равновесия с окружающей сре-
дой, то
dQ = —cfdT = fyTldf (III. 9)
или в первом приближении*:
(III. 10)
Знак теплоты деформации также определяется зна-
ком Р/.
* Использование в уравнении (ШЛО) вместо /0 величины 1—10Х
Х(1 + в) приводит к появлению небольшого поправочного члена,
квадратично зависящего от f.
155
Теплота, поглощаемая телом, когда его нагружают
при постоянной температуре, определяется формулой
Томсона [2]:
(Ш. 11)
Здесь W представляет собой работу, совершаемую при
переходе от начального ненагруженного состояния к ко-
нечному в изотермических условиях. Для стержней ра'*
бота растяжения определяется соотношением
f2
W= 2ЕА3
(III. 12)
Учитывая уравнение состояния стержней, можно полу-
чить следующее выражение для теплоты, поглощаемой
при изотермическом растяжении:
Q = Tp/f (III. 13)
Это выражение совпадает с уравнением (III.10) при
условии Р/~р, что имеет место в реальных условиях.
Уравнение (III.11) применимо для любого вида нагру-
жения. Оно, в частности, позволяет оценить тепловые
эффекты, связанные с чисто сдвиговыми деформациями.
Надаи показал, что тепловой эффект чисто сдвиговых
деформаций по порядку величины значительно ниже теп-
ловых эффектов, связанных с изменением длины при
растяжении [31, с. 64]. Поэтому указанными эффекта-
ми можно пренебречь. При этом он использовал урав-
нение состояния сдвига в форме
Т = т[1 + 0 (Т — To)]/Go
где у — деформация сдвига; т — напряжение сдвига; Go — изотерми-
ческий модуль сдвига при температуре Т=Т0.
Основные термодинамические функции стержня
определяются следующим образом. Исходя из стан-
дартного выражения для энтропии
(III. 14)
и используя соотношения Максвелла
/ dS \ / df \
\ /п \ дт /п /
\ 'р>т \ 'pt I
156
и уравнение состояния, находим:
df
дТ
= — pEAZ/Z0
р, I
Отсюда
dS = cidT/T + ^EAldl/l^
где ci — теплоемкость при постоянной длине.
(III. 15)
(III. 16)
Из уравнения (III. 16) для адиабатических условий
имеем:
TfiEAldl
(III. 17)
Поскольку различие между теплоемкостями Cf и с/,
определяемое соотношением
cf — ci = P2TEAZ0
(III. 18)
является величиной второго порядка малости по рТ
и им в нашем приближении можно пренебречь и так
как
д1
ЕА
Zo
то формулы (III.7) и (III.17) совпадают. Для изотер-
мических условий
dQ = TdS = T^EAldl/lb = T$ldf (III. 19)
что для конечных значений совпадает с формулой
(III.13), полученной из уравнения (Ш.П). Выражение
для внутренней энергии имеет вид*
( / \
dU = dQ + dW = TdS + fdl = TfiEA -j— f } dl =.
\ *o /
I — Zo
= EA —dl
lo
(III. 20)
Рассмотрим изменение внутренней энергии стержня
по отношению к некоторому произвольному состоянию,
* ;В'Н1утренн1Я1я энергия практически тождественна энтальпии
Н ъ И, поскольку обычно при деформации твердых тел вкладом
р A v можно пренебречь, если р — атмосферное давление.
157
характеризующемуся параметрами I* и Т*. Из выраже-
ния (III.20) находим?
(III. 21)
Согласно этой формуле разность U—U* уменьшается
от нулевого значения при I = Z* до минимума при 8mm,
определяемого из условия d(U—U*)/dl=0, затем начи-
нает возрастать и снова обращаться в нуль при условии
(Z — /о)2 — G* — /о)2 = О
Случай Z = Z* соответствует отсутствию деформации,
а из второго условия, учитывая, что Z = Z*(l+e) и
Z* = Z0(l+₽r*), можно получить следующее соотно-
шение:
26Т*
£инв = - -1 jZ r't*~ ~ (П1. 22)
Таким образом, разность U—U* обращается в нуль
в начале координат и при значении еИнв, выражаемом
Рис. II 1.4. Изменение внутренней энергии при упругой
деформации твердых тел, обладающих положительным
(/) и отрицательным (2) термическим коэффициентом
линейного расширения в направлении деформации.
уравнением (III.22). При этом знак еИнв зависит от зна-
ка р. Отрицательные значения 0 приводят к положи-
тельным значениям еИнв, и наоборот. Характер измене-
ния внутренней энергии деформации показан на
рис. Ш.4. Растяжение упругих тел с отрицательным
158
термическим коэффициентом расширения и сжатие
упругих тел, обладающих положительным термическим
коэффициентом расширения, характеризуется инвер-
сионным изменением внутренней энергии. Значение де-
формации, при которой наблюдается минимум внут-
ренней энергии, определяется выражением
рт*
8 = 8mjn == — 1 । ~ — РТ*
(III. 23)
а величина £/тщ— условием
t/min = ~ ЕА10 фТ)»/2
(III. 24)
Отмеченная особенность изменения внутренней энергии
с деформацией является, конечно, результатом различ-
ного изменения отдельных слагаемых — работы и теп-
лоты — с деформацией. Работа является параболиче-
ской функцией деформации:
Г = E&I2
а тепло линейно зависит от деформации:
Q = Т$Ег
(III. 25а)
(III. 256)
Характер изменения отдельных составляющих внутрен-
ней энергии с деформацией показан на рис. III.5.
Из соотношений (III.25) следует, что для упругих
тел зависимость отношения теплоты Q к работе w от
деформации является гиперболой:
Q 2рт
- е
(III. 26)
Это отношение полезно для анализа изменения внут-
ренней энергии с деформацией, поскольку одним из ва-
риантов записи выражения внутренней энергии являет-
ся следующий:
U = w(\±-^r\ (III. 27)
Знак плюс здесь относится к растяжению тел с поло-
жительным термическим коэффициентом расширения.
Из этого уравнения следует, что относительное измене-
ние внутренней энергии с деформацией U/W определя-
ется относительным изменением теплоты.
159
При переходе от одноосного растяжения упругих
тел к трехмерным (объемным) деформациям следует
в соответствующих выражениях заменить длину стерж-
ня / на объем V, силу на напряжение о, термический
коэффициент линейного расширения [3 на термический
коэффициент объемного расширения а = 3р, упругий мо-
дуль Е на модуль объемного сжатия (растяжения) К
Рис. II 1.5. Зависимость работы (/, /') и теплоты (2,2',
3,3') упругой деформации и отношения теплоты к ра-
боте (4,4', 5,5') от относительной деформации (2 — за-
висимость Q от е для тел с положительным коэффици-
ентом линейного расширения, 3— с отрицательным); 1—
5 — растяжение; 1'—5' — сжатие.
и относительную деформацию на относительное измене-
ние объема е. Таким образом, уравнение состояния
и основные уравнения термоупругости для случая
всестороннего
[31, с. 23]
растяжения или сжатия имеют вид
(1 — ссТ) — 1
Q = TaVo
— 2аТ/е
(III. 28)
(III. 29)
(III. 30)
(III.31)
а = Я
— TaV^/c^
Закономерности изменения термодинамических функ-
ций при всесторонней деформации такие же, как и при
одноосной.
160
Рассмотренные выше случаи обратимых деформа-
ций предполагали независимость термического коэффи-
циента расширения и модуля упругости от деформации
и температуры. Эти условия хорошо выполняются для
большинства твердых тел и жидкостей в достаточно
широких интервалах температур. Однако имеется одна
группа тел, способных к обратимым деформациям,
а именно каучукоподобные тела, для которых эти усло-
вия не выполняются. Замечательной особенностью тер-
момеханического поведения каучуков является зависи-
мость термического коэффициента линейного расшире-
ния от степени растяжения (или растягивающей силы).
Зависимость эта такова, что положительный термиче-
ский коэффициент линейного расширения нерастянуто-
го каучука при одноосном растяжении очень быстро
падает с увеличением степени растяжения и затем по
мере растяжения становится сильно отрицательным.
Джеймс и Гут [34] показали теоретически, что
1 X3 —1 а
Р и------Т ‘ X3 + 2 + V 4- 2
1 V —1 а А3 4-1
2Т * X3 4- 2 + 2 ’ X3 4- 2
(III. 32а)
(III. 326)
где — степень растяжения.
По мере увеличения растяжения член, содержащий
а, быстро уменьшается по сравнению с первым членом,
и при значительных растяжениях им можно пренебречь.
При Х=1 имеет место равенство р» = |3_i_ = 1/За,
как и должно быть для изотропного тела. Таким обра-
зом, растянутый каучук обладает ярко выраженной
анизотропией теплового расширения с коэффициентами
расширения порядка 1/Т, т. е. сравнимыми с коэффи-
циентами расширения газов. Кроме того, в отличие от
обычных твердых тел, у которых модуль упругости не-
сколько уменьшается с повышением температуры, что
является следствием теплового расширения, у каучу-
ков модуль упругости оказывается пропорциональным
температуре. Указанные особенности приводят к каче-
ственному изменению термомеханических эффектов
в каучуках.
Для теоретического анализа этих эффектов необхо-
димо иметь уравнение состояния, отражающее упомя-
11—264
161
йутые выше особенности деформационного поведения
каучука. Современная статистическая теория эластич-
ности каучука приводит к следующему уравнению
[34—36; 37, с. 420; 38]:
f = cT^l-~^ = сТ1й(^— -j^-) (III.33)
kN f г*\
где с = —ft- -=— ; I и V — соответственно длина и объем растя-
*о \ h2 )
нутого образца; /0 и Vo — соответственно длина и объем нерастя-
нутого образца; k — постоянная (Больцмана; N— число цепей в об-
разце, деформирующихся при растяжении образца; г2—квадрат
Среднего расстояния между концами цепи в изотропном образце;
А2 — квадрат среднего расстояния между концами свободной цепи
в идеальном растворителе.
Для цепей со свободным (вращением константа с не за-
висит, а для цепей с заторможенным вращением зави-
сит от температуры. Предполагается также абсолютная
несжимаемость каучуков, т. е. V=Vq.
Если пренебречь тепловым расширением, то термо-
динамические функции каучука имеют наиболее про-
стой вид. Этот случай соответствует деформации
в условиях постоянного объема. Используя соответст-
вующие термодинамические соотношения, определим
термодинамические функции каучука для этого случая.
Изменение энтропии при изотермическом растяжении
определяется уравнением Максвелла [25, с. 41]:
(Ш.34)
На основании этого уравнения имеем:
(AS)y> т = — [т 4- с j ~Ь Z/o /о (П1. 35)
Таким образом, изменение энтропии при растяжении
в этом случае описывается монотонно убывающей
функцией, обращающейся в нуль при / = /0. Работа де-
формации равна
Г
fdl = сТ
(III. 36)
Так же как и выражение (III.35), уравнение (III.36)
162
обращается в нуль при / = /0. Отношение тепла к рабо-
те равно
Q \ / TAS \ Т дс
W )„ т~~\ V - ~ 1 “ с ' дТ =
_। гр д In с __ j । р д In h2
~дТ + дТ
а изменение внутренней энергии:
(U)v т = W (1 + = WT —
v ,v,‘ \ W } дТ
или в относительной форме:
/ U \ _ т д In h2
\Ж Л, т ~ дТ
(III. 37)
(III. 38)
(III .39)
Уравнения (III.35) — (III.39) выражают основную
идею «газовой» статистической теории высокоэластич-
ности. Для цепей со свободным вращением работа рав-
на уменьшению энтропии при растяжении, и потому
изменение внутренней энергии равно нулю, а для цепей
с заторможенным вращением определенная доля за-
траченной на растяжение работы расходуется на изме-
нение внутримолекулярной энергии, связанной с пере-
ходом одних конформаций в другие. Статистическая
теория высокоэластичности приводит к выводу, что знак
производной d\nh~2/dT определяется видом потенциала
внутреннего вращения, т. е. молекулярной структурой
цепи, и может быть как положительным, так и отрица-
тельным [37, с. 408; 39, с. 258]. Отрицательные значе-
ния температурного коэффициента среднего расстояния
между концами цепи характерны для конформаций,
обладающих минимальной энергией в вытянутом со-
стоянии, и наоборот. Таким образом, растяжение поли-
меров в высокоэластическом состоянии может сопро-
вождаться как увеличением, так и уменьшением внут-
ренней энергии.
Формулы (III.35) — (III.39) получены в предположе-
нии отсутствия теплового расширения каучука. Это,
конечно, является лишь грубым приближением, по-
скольку реальные каучуки в недеформированном со-
стоянии при нагревании расширяются, причем термиче-
ский коэффициент расширения их близок к тер-
11*
163
мическим коэффициентам расширения жидкостей.
Определим термодинамические функции каучука на
основе уравнения состояния с учетом теплового рас-
ширения.
Работа деформации по-прежнему выражается урав-
нением (III.36), а изменение энтропии составит:
/ dS \ _ / df \ / дс \
\ д1 ) ~ \ ПЭТ ) , \с + Т дТ 11 +
\ /р,Т V /p,z х !
( дс dV \ 1
+ ( с + Т ftp -\~Тс j (III. 40)
или в интегральном виде:
(AS)p>7 — —
(III. 41)
В отличие от уравнения (III.35) в этом случае энтро-
пия сначала возрастает (начиная с нуля при Z = Zo) до
максимального значения, определяемого условием
dS/dl = 0 (изометрическая инверсия):
(III. 42)
Для цепей со свободным вращением
/из = Г(1+аЛ'/«^ (1+“Г7’
(III. 43)
1 dV
где а = -г?— -tff- — термический коэффициент объемного расшире -
НИЯ.
После достижения максимального значения при /Из эн-
тропия начинает уменьшаться, обращаясь в нуль (адиа-
164
Рис. III. 17. Зависимость теплоты релаксации напряжения в ориен-
тированных кристаллических полимерах от относительной деформа-
ции [47, 48]:
1 — полиэтилен высокой плотности; 2 — полипропилен; 3 — капрон; 4 — поли-
этилен низкой плотности.
релаксация связана с поглощением тепла. При этом
относительная глубина падения напряжения в этих
полимерах заметно больше, чем в капроне и полиэти-
лене низкой плотности. Этот результат можно понять,
Рис. III.18. Зависим ость теплоты сокращения ориентированных кри-
сталлических полимеров от относительной деформации:
а — капрон (/) и полипропилен (2); б — полиэтилен высокой плотности, рас-
тянутый в 20 раз (/), и полиэтилен высокой плотности, растянутый в 10 раз,
после релаксации в течение 6 мин (2) и в течение 1 ч (3).
181
лишь (предположив, что релаксация напряжения в таких
системах связана не столько с установлением равновес-
ной конформации цепи в аморфной прослойке, сколько
с разрушением структуры аморфных областей микрофиб-
рилл. Что касается различия в поведении полиэтилена
высокой плотности и полипропилена по сравнению с по-
ведением капрона и полиэтилена низкой плотности, то его
можно, очевидно, объяснить различием в степени кри-
сталличности и в тонкой структуре микрофибрилл.
Рис. III. 19. Термограммы растяжения (1,2,3) и сокращения (1',2Г,
3') ориентированных полимеров. Пунктиром показаны термограм-
мы сокращения, которые должны были бы наблюдаться без эффекта
тиксотропии [48]:
Мы видим, что деформация ориентированных кри-
сталлических полимеров может сопровождаться сущест-
венными тиксотропными эффектами. Эти эффекты осо-
бенно ярко проявляются в тепловых процессах при со-
кращении образца до первоначальной длины
(рис. III. 18). Разгрузка капрона во всем интервале
растяжений сопровождается поглощением тепла, и весь
цикл растяжение — выдержка — сокращение не сопро-
вождается изменением внутренней энергии. Для поли-
этилена высокой плотности и полипропилена поглоще-
ние тепла при сокращении наблюдается лишь после
малых растяжений, а затем знак теплоты сокращения
изменяется. Весьма показательным здесь являются
термограммы тепловых эффектов, однозначно свиде-
тельствующие о двух последовательных тепловых про-
цессах при сокращении (рис. III. 19). Первый — эндо-
термический — является обычным процессом поглощения
182
тепла при сокращении упругого тела, обладающего
отрицательным термическим коэффициентом расшире-
ния вдоль оси растяжения, а второй — экзотермический—
проявляется в области исчезновения напряжения в об-
разце от внешнего воздействия за счет рассеяния упру-
гой энергии, связанной с тиксотропным восстановле-
нием структуры.
У сильно ориентированных образцов переход от по-
глощения к выделению тепла происходит резко и не ос-
тавляет сомнений в изменении механизма деформации
в этой области растяжений (см. рис. III. 18,6).
Вполне естественно, что тиксотропное разрушение
и восстановление структуры существенно зависят от
продолжительности нахождения полимера под нагруз-
кой. Хотя зависимость экзотермического эффекта тик-
сотропного восстановления структуры от деформации
после часовой релаксации напряжения и оказывается
качественно похожей на соответствующую зависимость
после шестиминутной релаксации, количественное раз-
личие между ними очень велико.
Характерным у ориентированных полиэтилена вы-
сокой плотности и полипропилена оказывается и энер-
гетический баланс деформационного цикла: при увели-
чении деформации внутренняя энергия резко повыша-
ется. Наиболее вероятными причинами этого являются,
по-видимому, упругое последействие и тиксотропное
разрушение структуры. Определенная доля затрачен-
ной на деформацию энергии оказывается запасенной
в медленных релаксационных процессах, как обычных,
столь характерных для полимеров, так и в процессах
разрушения и восстановления структур.
Тепловые эффекты этих медленных процессов нахо-
дятся за пределами чувствительности установки и не
могут быть зафиксированы экспериментально.
Термоэластичность каучуков
Первые количественные исследования в области
термоупругости каучука были проведены Джоулем
[32, 33], который использовал для этой цели измере-
ния температурных изменений при быстром растяже-
нии. Его результаты для натурального каучука
183
(рис. III.20), подтвержденные затем в других работах
[34, 56, 57], показывают, что начальное охлаждение
каучука сменяется нагреванием. Точка изометрической
инверсии находится при 8 — 7%, а адиабатической —
при 8«14%. Изменения энтропии при растяжени^и на
основании такого рода температурных изменений могут
быть рассчитаны по уравнению (III.9).
Хотя эти экспериментальные результаты достаточно
хорошо совпадали с теоретическими, температурные из-
мерения не нашли широкого применения для точных-
количественных оценок. Им была отведена роль на-
глядных качественных иллюстраций энтропийной при-
роды упругости каучука, а для точных оценок был из-
бран другой путь — определение температурной зависи-
мости равновесных напряжений, основанный на том,
что растягивающая сила f является суммой двух со-
ставляющих — энергетической fu и энтропийной fs
[36, 40]:
/ dU \ / df \
f = fu + h = {~dT] -т ЬН (ш.53)
\ ! T,V \ ' V, I
Наклон температурной зависимости равновесной силы
в каждой точке дает изменение энтропии при растяже-
нии, а экстраполяция этого наклона на Т=0 К позво-
ляет определить изменение внутренней энергии. Этот
простой экспериментальный метод определения отдель-
ных составляющих связан с необходимостью экстрапо-
ляции экспериментальных данных на больший темпе-
ратурный интервал, так как обычно температурную
зависимость равновесной силы надежно удается опреде-
лить лишь в интервале температур, равном приблизи-
тельно ДТ=100°С, а измерения, как правило, проводят-
ся в температурном интервале 350± 100 К. Тем не
менее уже в течение 40 лет этот подход является основ-
ным экспериментальным методом исследования термо-
упругости каучука, и при помощи его были сделаны
выводы о природе отдельных составляющих уравнения
(III.53).
В ранних работах был сделан вывод об идеальной
энтропийной упругости многих каучуков, поскольку
обычно оказывалось, что составляющая fu~0. Однако
дальнейшие теоретические и экспериментальные иссле-
дования термоупругости каучука привели к выводу, что
184
Рис. II 1.20. Изменение тем-
пературы при растяжении
каучука [401 (различные
точки — измерения разных
авторов).
Этот экспериментальный
упругость большинства каучукоподобных полимеров
не является идеально энтропийной, а связана с возник-
новением энергетической составляющей исключительно
внутримолекулярного происхождения, значение которой
определяется химической структурой макромолекул
[37, с. 364; 38; 39; 257]. Экспериментальные и теоре-
тические результаты по исследованию зависимостей
напряжение — температура в каучуках изложены
в большом числе монографий и обзоров [36, 37—40,
58, 59], и мы не будем на
них останавливаться более
подробно, а сосредоточим
внимание на результатах
калориметрического иссле-
дования термоупругости
каучука и сравним их с экс-
периментальными данными
зависимостей напряжение
(сила) —температура.
Наряду с упомянутыми
выше экспериментальными
возможностями определе-
ния изменения энтропии
при деформации каучука
существует еще один путь—
прямое калориметрирова-
ние тепловых эффектов, со- '
провождающих деформацию,
подход был использован Диком и Мюллером [60], и
уже в этом первом калориметрическом исследовании
было показано, что натуральный каучук не является
каучуком, обладающим идеальной энтропийной упру-
гостью, так как, по данным калориметрии, доля энерге-
тической составляющей общей силы при растяжении на
150—250% при комнатной температуре составляет при-
мерно 35%.
Начиная с 1969 г. в лаборатории физики полимеров
ИНЭОС АН СССР автором проводились калориметри-
ческие исследования термоэластических свойств каучу-
ков. Некоторые результаты этих исследований сумми-
рованы в этом разделе.
Типичные зависимости работы, теплоты и внутрен-
ней энергии от растяжения в области умеренных растя-
13—264
185
пичные размеры образцов 25X3,5X1 мм.
’ ер, содержащий 50% акрилонитрила.
жений приведены на рис. III.21. Исследования прово-
дили как методом непосредственного растяжения до
необходимой деформации, так и методом ступенчатого
растяжения с «шагом» примерно 10—30%. Скорости
растяжения составляли около 10—100%/мин. Для
определения изменения внутренней энергии было ис-
пользовано также сокращение образца без совершения
внешней работы (разрезание тяги). Зафиксированный
при этом тепловой эффект по знаку и величине равен
изменению внутренней энергии при растяжении.
Рис. III.21. Зависимость работы
внутренней энергии (5) от степени
(/), теплоты (2) и
растяжения для кау-
чуков:
О - СКИ; 1 - СКН.
Характер изменения работы, теплоты и внутренней
энергии при растяжении вполне соответствует получен-
ным выше формулам. В табл. III.4 приведены значения
энтропийной и энергетической составляющих механиче-
ской работы при растяжении для ряда высокоэластиче-
ских полимеров. Лишь для бутадиен-стирольного кау-
чука изменение внутренней энергии близко к нулю в
указанном интервале растяжений, для всех остальных
полимеров наряду с изменением энтропии наблюдается
изменение внутренней энергии. Этот вывод полностью
согласуется с данными новейших термомеханических
исследований [38, 58] и с теоретическим анализом
13*
187
[37, 39]. Для большинства исследованных высокоэла-
стических полимеров наблюдается хорошее количест-
венное соответствие результатов калориметрических
исследований с термомеханическими данными. Что ка-
сается натурального каучука, ^с-1,4-полиизопрена, то
наши калориметрические значения U/W несколько пре-
вышают соответствующие усредненные результаты
большинства термомеханических данных, однако они
очень хорошо согласуются с независимыми калориметр
рическими результатами [60, 61] и термомеханически-
ми данными Шена [59, 62], полученными в эксперимен-
тах, отличающихся особой тщательностью проведения
и обработки. Значения д1пй2/дТ, вычисленные по фор-
муле (III.39), находятся, естественно, в таком же соот-
ветствии с данными, полученными термомеханическим
методом.
Знак энергетической составляющей силы U/W нахо-
дит свое объяснение в конформационных особенностях
полимеров [37, с._409; 39, с. 259]. Положительные зна-
чения U/W и d\nh2/dT характеризуют растяжение мак-
ромолекул, для которых вытянутые конформации энер-
гетически менее выгодны. Из исследованных нами
полимеров лишь сополимер этилена с пропиленом ха-
рактеризуется большим отрицательным значением
U/W, что, очевидно, является следствием того, что как
транс-конформации в полиэтилене, так и спиральная
конформация в полипропилене имеют более низкую
энергию. С другой стороны, хотя такое же энергетиче-
ское соотношение между конформациями имеет место
и в макромолекулах полидиметилсилоксана, энергетиче-
ская сила в них положительна, что объясняется особен-
ностями конформационных изменений в макромолеку-
лах с чередующимися значениями валентных углов
[39, с. 272; 63, с. 191].
Мы видим, что калориметрические результаты опре-
деления энергетической составляющей упругой силы
согласуются с данными термомеханических и других
измерений.
Изложенные выше результаты относятся к умерен-
ным степеням растяжения. Переход к большим растя-
жениям приводит к существенному изменению соотно-
шений между величинами Q, W и U. Характер измене-
ния зависимостей этих величин от степени растяжения
188
для ^цс-1,4-полиизопрена, включая большое растяже-
ние, показан на рис. III.22*. Резкое увеличение выде-
ления тепла и соответственно уменьшение внутренней
энергии в области удлинений Л —4,5 у ^цс-1,4-полиизо-
прена при растяжении, безусловно, связано с кристал-
лизацией. Развитие кристаллизации вызывает появле-
ние значительных гистерезисных эффектов, менее вы-
раженных при умеренных
лых скоростях растяже-
ния. Примечательно, что
гистерезисный ход вели-
чин W и Q приводит к
возрастанию внутренней
энергии, что, по-видимо-
му, связано с резким за-
медлением релаксацион-
ных процессов в образце
с только что расплавив-
шимися кристаллитами.
Очевидно, что длина об-
разца при Х=1 должна
значительно превышать
начальную длину (перед
растяжением), что наб-
людается визуально. По-
сле полной разгрузки об-
разца происходит мед-
степенях растяжения и ма-
Рис. III.22. Зависим ость рабо-
ты (/), теплоты ;(2) и внутрен-
ней энергии (3) от степени
растяжения для г<«с-!1,4-поли-
изопрена.
ленное выделение тепла,
связанное с релаксацией
размеров (длины) образ-
ца. Эти результаты сви-
детельствуют о больших возможностях калориметрии в
исследовании динамических процессов развития и ис-
чезновения деформации.
Приведенные выше уравнения предсказывают неза-
висимость энергетической составляющей внутримолеку-
лярного происхождения от степени растяжения высоко-
эластического полимера. Это предсказание должно
быть особенно справедливым при малых растяжениях,
поскольку в этой области деформаций с большим при-
* Опыты в этом случае проводились главным образом методом
дифференциального растяжения с «шагом» около 25—30%.
189
ближением должно выполняться гауссово распределе-
ние. В исследованиях последних лет, в которых прово-
дилось экспериментальное определение энергетической
составляющей напряжения в каучуках, наблюдалось
систематическое возрастание энергетической составляю-
щей при уменьшении степени растяжения для ряда
полимеров (натуральный каучук, полидиметилсилоксан,
сополимер этилена с пропиленом, полиалкилметакри-
латы) [38, 64, 65]. Делались попытки связать зависим
Рис. II 1.23. Энергетическая составляющая механической
работы для каучуков при степени растяжения X < 2:
О, ф — цис-1,4-полиизопрен; Л, ▲ — сополимер этилена с про-
пиленом; □ , И — полидиметилсилоксановый каучук; О, Д, □ —
(UIW)p т', Ф А, — [см. уравнение (III.50)]; гори-
зонтальные линии соответствуют значениям (U/W) v ? при боль-
ших к (см. табл. III.4).
мость энергетической составляющей от деформации
с молекулярной и надмолекулярной структурой эласто-
меров [38, 65], однако специальные исследования на-
турального каучука показали, что эта зависимость при
малых деформациях является, по-видимому, отраже-
нием экспериментальной ситуации, при которой для
корректного определения значения fu/f требуется очень
высокая точность нахождения или поддержания задан-
ной деформации [62]. Исследование термоэластичности
натурального каучука в условиях постоянного объема,
190
когда исчезает необходимость введения зависящей от
деформации поправки на изменение объема, также
привело к выводу о независимости энергетической со-
ставляющей возвращающей силы fu/f от степени растя-
жения, по крайней мере, в области степеней растяже-
ния к— 1,14-2,1 [66].
Результаты калориметрического исследования тер-
моэластичности некоторых эластомеров в области рас-
тяжений Л<2 (рис. III.23) позволяют отметить следую-
щее. Большинство значений энергетической составляю-
щей (U/W)yiT при растяжениях А, < 2 заключено
в интервалах, характерных для больших растяжений.
Заметный разброс в области малых деформаций обу-
словлен недостаточной точностью определения дефор-
мации, поскольку поправочный член очень чувствите-
лен в этой области к величине деформации. Отдельные
выпады значений наблюдаются также в области адиа-
батической инверсии, где теплота деформации опреде-
ляется как разность двух сравнимых по величине зна-
чений. Таким образом, калориметрическое исследование
термоэластичности эластомеров при малых деформа-
циях приводит к выводу о практической независимости
значений внутримолекулярной энергетической состав-
ляющей от степени растяжения.
Глава 7
НЕОБРАТИМЫЕ ДЕФОРМАЦИИ
В отличие от обратимых деформаций необратимые
деформации всегда связаны с рассеянием в виде тепло-
ты части затраченной на деформацию механической
работы. Естественно, что применение первого начала
термодинамики к таким процессам позволяет составить
энергетический баланс, т. е. определить изменение
внутренней энергии или энтальпии в результате дефор-
мации, поскольку уравнение
U(H) = W + Q (III. 54)
применимо ко всем процессам.
При этом, очевидно, возможны три случая: 1) Q<W,
2) Q>W и 3) Q = W. Простейшими примерами третьего
191
случая являются течение жидкости и идеальная пласти-
ческая деформация, при которых вся работа против сил
межмолекулярного трения полностью рассеивается
в виде теплоты. Очевидно, что никаких дополнительных
сведений о молекулярных механизмах этих процессов
при изучении их калориметрическим методом получить
не удается.
Два первых случая в деформационных процессах
позволяют получить дополнительную по сравнению*
с чисто механическими измерениями информацию об
изменении состояния тела в результате деформации.
Так, изучение энергетического баланса пластической
деформации металлов, приводящей к поглощению ча-
сти механической работы и упрочнению, позволяет
охарактеризовать механизмы этого упрочнения
[67—69]. Однако проблема здесь в конечном счете
состоит в том, что теплота является суммой обратимой
и необратимой частей [70]:
Q = Qi + Q2 (HI. 55)
Чтобы полностью термодинамически охарактеризовать
такой процесс, их необходимо разделить. Для этого
кроме экспериментального (калориметрического) опре-
деления энергетического баланса необходимо отдельно
изучение энергетического состояния материала до
и после деформации. Это может быть сделано, напри-
мер, путем измерения температурной зависимости теп-
лоемкости или путем определения теплот растворения
исходных и деформированных образцов. Ниже будут
рассмотрены наиболее характерные случаи энергетиче-
ского баланса необратимых деформаций полимеров.
Ориентационная вытяжка полимеров
Если растяжение неориентированных полимеров не
ограничивать обратимыми деформациями, то характер
энергетических эффектов существенно изменяется [48].
Характерное замедление роста напряжения, наблюдае-
мое для кристаллических полимеров при 8 = 54-10%, со-
провождается отклонением теплового эффекта от ли-
нейной зависимости. Типичные результаты показаны
на рис. III.24. Поскольку зависимость теплоты сокраще-
ния от деформации оказывается с достаточной степенью
192
приближения линейной, то отклонения от нее при
растяжении свидетельствуют о последовательном нало-
жении на эндотермический эффект упругого растяже-
ния дополнительного экзотермического эффекта, свя-
занного с появлением пластических деформаций и под-
готовкой к началу ориентационной вытяжки. По мере
растяжения этот процесс проявляется все заметнее
и обычно приводит к появлению «шейки», т. е. к ориен-
W
Р*ис. II 1.24. Зависимость тепло-
ты (1) и сокращения (2) от
относительной деформации для
полиэтилена высокой плотно-
сти [48].
t
Рис. II 1.25. Изменение тепло-
вой мощности .во времени (а)
и соответствующая кривая рас-
тяжения (б) при ориентацион-
ной вытяжке.
тационной вытяжке. Это сопровождается изменением
знака интегрального теплового эффекта за счет того,
что экзотермический эффект пластической деформации
становится доминирующим. Рис. III.25 характеризует
тепловые изменения при переходе в область ориента-
ционной вытяжки. По мере ее развития рассеиваемая
тепловая мощность стабилизируется в соответствии
с постоянной скоростью ориентации, что позволяет
сравнивать на этой стадии тепловую и механическую
мощности. Полный энергетический баланс перехода из
неориентированного в ориентированное состояние мо-
жет быть получен интегрированием.
Была изучена зависимость работы ориентации
и теплоты, рассеянной в процессе ориентации, от ско-
рости растяжения при комнатной температуре для ряда
кристаллических полимеров [48] (рис. III.26). И рабо-
та, и теплота линейно возрастают с логарифмом скоро-
193
сти растяжения. Для механической работы это является
прямым следствием логарифмического закона скоро-
стей, согласно которому предел текучести (для метал-
лов), вынужденной эластичности и рекристаллизации
(для полимеров) линейно зависят от логарифма скоро-
сти растяжения [71, с. 31; 72; 73]. Однако отношение
теплоты к работе от скорости растяжения практически
не зависит* и в большинстве случаев меньше единицы
(табл. III.5).
Рис. III.*26. Зависимость работы ориентации (а) и теп-
лоты, рассеянной при ориентации (б), от скорости рас-
тяжения (в скобках указана относительная деформа-
ция) [48]:
1 — полипропилен (500%); 2 — полиэтилен высокой плотности
(400%); 3 — капрон (250%); 4 — полиэтилен низкой плотности
(300%).
Это свидетельствует о возрастании энтальпии в резуль-
тате растяжения. При этом поскольку
W = a + &lgVp (III, 56а)
то
Н =F(1 — Q/IF) = r-const = a' 4-У lgVp (III. 566)
т. e. H также возрастает линейно с логарифмом скоро-
сти. Сравнение нормированных значений ДЯюо% пока-
зывает, что наибольшей способностью поглощать энер-
* Для капрона Мюллер наблюдал необъяснимою стремительное
уменьшение отношения тепловой мощности к механической от 0,62
до 0,25 в области скоростей растяжения 1 мм/с [70].
194
гию при ориентации обладает капрон, а наименьшей —*
полипропилен. На примере полиэтилена видно также,
что повышение степени кристалличности увеличивает
примерно пропорционально способность поглощения
механической энергии при ориентации и ее зависимость
от скорости растяжения.
Для стеклообразного полиэтилентерефталата на-
блюдается заметное превышение рассеянной теплоты “
над затраченной работой. Такое соотношение при ука-
занной скорости имеет место как для пленок, так и для
волокон и свидетельствует о значительном понижении
энтальпии в результате ориентации.
Рис. II 1.27. Зависимость нормированных на 100% относитель-
ной дефю|рмации значений рассеянной теплоты (а) и механи-
ческой работы, затраченной при переориентации (б), от угла
переориентации [48]:
1 — полипропилен; 2 — капрон; 3 — полиэтилентерефталат.
Изучение энергетических характеристик переориен-
тации ориентированных образцов показало, что работа
и теплота существенно зависят от угла переориентации,
а их отношение практически не зависит от него
(рис. III.27). При этом в отличие от первичной ориен-
тации при переориентации Q/W= — 1 ±0,05 для углов
90 и 60° и Q/W = — 1 ±0,08 для углов 45 и 30°. Такое
соотношение означает, что процесс переориентации под
большими углами практически не сопровождается по-
глощением энергии. Для углов 45 и 30° такой вывод
может быть сделан лишь с большой осторожностью
ввиду значительного разброса экспериментальных
данных.
196
Таким образом, при ориентационной вытяжке поли-
меров реализуются все три отмеченные в начале этого
раздела соотношения диссипированной теплоты и меха-
нической работы. Для определения обратимой части
теплоты был использован метод динамической тепло-
емкости. Измерения, проведенные на образцах, хранив-
шихся после ориентации в жидком азоте для исключе-
ния возможных релаксационных процессов, показали
(рис. III.28), что поведение ориентированных образцов
заметно отличается от неориентированных лишь в обла-
сти плавления. От —100 °C и до начала плавления их
теплоемкости практически равны (точность определе-
ния ср составляла ±2,5%). Параметры плавления при-
ведены в табл. III.6.
Таблица III.6. Термодинамические параметры плавления
неориентированных и ориентированных полимеров
Полимер Неориентиро- ванный Ориентиро- ванный Mt "я схч О й о т е « Д О' АН в результате вы- тяжки*, кДж/кг кДж/кг
К 4 в ь» И* К О 4 с Д и 0.4 О И О
Полиэтилен высо- кой плотности . Полиэтилен низкой 405 177,5 411 190,0 -12,5 15,9 —28,4
плотности . . . 385 118,3 385 133,0 -14,7 6,3 ——20,9
Полипропилен . . 438 85,7 440 72,4 13,3 10,1 2,9
* Эти значения относятся к — 0,1165 мм/с, при которой проводилась ориен-
тация образцов для опытов по определению температурной зависимости теплоем-
кости.
Можно полагать, что в кристаллических полимерах
энергия поглощается просто за счет механического
плавления отдельных кристаллитов. Это должно приво-
дить к уменьшению степени кристалличности ориенти-
рованных полимеров и уменьшению теплоты плавления
по сравнению с неориентированными. Данные, приве-
денные в табл. 6, показывают, что лишь для полипро-
пилена результаты соответствуют этому предположе-
197
нию. Для полиэтиленов теплота плавления ориентиро-
ванных образцов оказывается заметно больше теплоты
плавления изотропных образцов. Кроме того, у них
наблюдается значительное повышение температуры
плавления после ориентации. Повышение теплоты и
Рис. III.28. Темпер.атур1ная зависимость теплоемкости
изотропных (пунктир) и ориентированных (сплошная
линия) полимеров [48]:
а — полиэтилен низкой плотности; б — полиэтилен высокой плот-
ностп; в — полипропилен.
температуры плавления ориентированного полиэтилена
было установлено в ряде работ [74—76]. Исходя из
понижения сорбционной способности ориентированного
полиэтилена и уменьшения коэффициента диффузии
в нем, Петерлин [77] высказал предположение, что
198
энтальпия и энтропия аморфных областей ориентиро-
ванного полиэтилена вследствие значительной ориента-
ции в них участков макромолекул должна быть замет-
но ниже, чем для изотропного полимера и, таким обра-
зом, эти области должны вносить существенный вклад
в теплоту плавления ориентированных полимеров. Дан-
ные по теплотам и температурам плавления ориентиро-
ванного полиэтилена, полученные нами, в этом смысле
хорошо согласуются с результатами других работ.
Таким образом, для полиэтилена существует какой-то
механизм поглощения энергии при ориентации, не свя-
занный с понижением степени кристалличности.
В связи с этим здесь уместно отметить, что много-
численные энергетические исследования пластической
деформации низкомолекулярных веществ, и прежде
всего металлов, показывают, что металлы тоже обла-
дают способностью поглощать энергию при деформации
[67—69]. Латентная энергия сильно зависит от усло-
вий деформации, степени деформации и температуры,
чистоты металла, скорости и вида деформации и дру-
гих факторов, и значение ее лежит в пределах от не-
скольких процентов до 20—25% от затраченной на
деформирование механической работы, т. е. в тех же
пределах, что и у твердых полимеров. Латентная энер-
гия деформации металлов локализуется в скоплениях
дислокаций и связана с вакансиями и дислоцированны-
ми атомами. В процессе последующего нагрева дефор-
мированного металла происходит исчезновение вакан-
сий, распад скоплений дислокаций на отдельные дисло-
кации и уменьшение их числа. Каждая из этих стадий
приводит к выделению соответствующей части скрытой
энергии деформации в этом температурном интервале,
что может быть зафиксировано, например, сканирую-
щим калориметром [78]. В отличие от металлов, у ис-
следованных полимеров такого выделения латентной
энергии не наблюдается (см. рис. III.28). Еще одно
важное различие между ними состоит в том, что кри-
сталлические полимеры обладают способностью погло-
щать энергию при значительно более высоких относи-
тельных (гомологических) температурах Т1Тил
(табл. III.7). Зависимость поглощенной энергии от
гомологической температуры у металлов имеет гипер-
болический характер. Способность поглощать энергию
199
при деформации у них полностью исчезает при значе-
ниях Т/Тпл=0,55-?-0,6, что объясняется высокими скоро-
стями процессов возврата и рекристаллизации [67].
Таблица Ш.7. Латентная энергия деформации металлов
и полимеров
Материал Температура плавления ^пл’ Латентная энергия деформации при е == 25%, Дж/моль Гомологи- ческая температура, ^пл
Сталь 3 1803 100,0 0,186
Медь 1353 39,8 0,217
Алюминий 932 5,5 0,315
Свинец 600 2,5 0,488
Олово . 504 0 0,582
Капрон . 498 209,0 0,587
Полипропилен 438 26,8 0,668
Полиэтилен высокой плот-
ноети 405 33,0 0,725
Полиэтилен низкой плот-
НО'СТИ 385 17,5 0,760
Исследования молекулярных механизмов ориентаци-
онной вытяжки кристаллических полимеров, установ-
ленных путем применения прямых структурных мето-
дов, убедительно свидетельствуют о том, что ориента-
ционная вытяжка кристаллических полимеров сопро-
вождается глубокими структурными перестроениями
на всех уровнях надмолекулярной структуры [79]. Так,
для наиболее полно исследованного с этой точки зрения
полимера — полиэтилена установлено, что задолго до
начала образования шейки в области малых значений
обратимых деформаций пластинчатые кристаллы, из
которых построены сферолиты, начинают постепенно
поворачиваться, двойниковаться и рекристаллизовать-
ся, переходя из одной модификации в другую. Даль-
нейшее растяжение приводит к полному распаду
исходной сферолитной структуры на отдельные блоки
со сложенными цепями. Эти блоки оказываются соеди-
ненными между собой общими участками макромоле-
кул и при последующей ориентации образуют харак-
терную фибриллярную структуру. Таким образом, хотя
на уровне кристаллитной организации в полимерах
200
могут протекать все элементарные процессы, характер-
ные для пластической деформации низкомолекулярных
тел, макроскопический механизм больших деформаций
низкомолекулярных кристаллических тел и полимеров
оказывается различным, и дислокационный механизм
поглощения энергии не может, по-видимому, играть
существенной роли для кристаллических полимеров.
В то время как для металлов основным механизмом
пластических деформаций, приводящим к поглощению
энергии, является скольжение по кристаллографиче-
ским плоскостям и перемещение дислокаций, в полиме-
рах этим механизмом может быть обусловлено лишь
несколько десятков процентов начальной необратимой
деформации. Дальнейшая деформация полимеров обес-
печивается распадом ламелярных кристаллитов на от-
дельные блоки. При этом отдельные участки макромо-
лекул могут терять свою складчатую конформацию.
Эти участки связывают блоки в аксиальном направле-
нии в микрофибриллы. Они оказываются сильно вытя-
нутыми, что может быть достигнуто за счет конформа-
ционного гош-трбшс-перехода. Такой переход должен
сопровождаться выделением энергии. Проходные макро-
молекулы определяют упругие свойства ориентирован-
ных полимеров и обеспечивают геометрическую обрати-
мость деформации полимеров при нагреве этих полиме-
ров до области температур, близкой к температуре
плавления.
Хотя эта общая схема больших деформаций кри-
сталлических полимеров считается в настоящее время
наиболее реальной, отдельные стадии и детали ориен-
тационной вытяжки остаются мало изученными. Это
не позволяет установить и общий механизм поглощения
энергии при ориентации. Можно предполагать, что по-
глощение энергии за счет возникновения искажений
и неравновесных дефектов (подобно металлам) имеет
место в стеклообразных полимерах, в которых эти де-
фекты оказываются замороженными из-за низкой под-
вижности. В области размягчения латентная энергия
должна выделяться и приводить к аномалиям, напри-
мер, в теплоемкости. Детальные исследования с этих
позиций ориентированных стеклообразных полимеров
не проводились, хотя аномалии в теплоемкости дейст-
вительно наблюдались [80]. Попытка выяснить судьбу
14—264
201
поглощенной энергии при ориентации поликарбоната
путем определения теплот растворения не увенчалась
успехом [81].
Переориентация полимеров происходит практически
без поглощения энергии. Принимая во внимание фиб-
риллярное строение ориентированных полимеров, мож-
но полагать, что полное превращение механической
работы в теплоту является следствием в значительной
степени реологического характера переориентации без.
Рис. II 1.29. Температурная зависимость теплоемкости
изотропного ( X ) и ориентированного (О) полиэтилен-
терефталата [48].
существенного разрушения фибриллярной структуры.
Такая возможность может возникать благодаря нали-
чию межкристаллитных и межфибриллярных аморфных
областей, обусловливающих высокую подвижность кри-
сталлитов. Рентгеноструктурные исследования элемен-
тарных структурных процессов при переориентации
ориентированного полиэтилена показали, что исходные
стопки ламелярных кристаллитов за счет поворотов
и скольжений с незначительными разрушениями пере-
ходят во вновь образующуюся фибриллярную структу-
ру переориентированного полимера [82, 83].
202
Отдельного рассмотрения заслуживает поведение
стеклообразного полиэтилентерефталата при ориента-
ционной вытяжке, поскольку это единственный из
имеющихся случаев, когда ориентация сопровождается
существенным понижением энтальпии. Последнее долж-
но означать наличие процесса упорядочения при ориен-
тации. Ориентированный при комнатной температуре
(на 60 °C ниже Гс) полиэтилентерефталат обладает
характерной аморфной текстурой, обусловленной нали-
чием транс-конформации макромолекул, сдвинутых от-
носительно друг друга вдоль оси текстуры вполне за-
кономерно, как и в кристаллическом полиэтилентере-
фталате. Однако азимутальные повороты звеньев макро-
молекулы и межцепные расстояния в экваториальной
плоскости оказываются сильно нарушенными [84].
Калориметрические характеристики таких текстури-
рованных образцов (рис. Ш.29) приведены ниже:
Неориентированный Ориентирован
(аморфный) ный образец
образец
Температура стеклования
Тс, К..................
Температура плавления
Тпл, К.................
Теплота «холодной» кри-
сталлизации, кДж/кг
Теплота (рекристаллиза-
ции, кДж/кг . . . .
Теплота плавления,
кДж/кг.................
354 —
531 528
28,5 2,5
6,3 —
39,3 23,5
Если отвлечься от предыстории ориентированного
образца, которая нам известна, и рассматривать лишь
температурную зависимость его теплоемкости и приве-
денные количественные данные, то очевидный вывод из
этих результатов будет следующим: этот образец яв-
ляется кристаллическим, так как стеклование у него
отсутствует. Однако рентгеноструктурные данные одно-
значно указывают на то, что после ориентации этот
образец является текстурированным, но аморфным.
Это можно понять, предположив, что основная часть
теплоты кристаллизации выделилась при ориентации,
т. е. при образовании текстуры. Эта текстура является
своего рода кристаллической «заготовкой» [85]. Для
ее окончательного перехода в кристаллическую струк-
туру необходимы лишь повороты отдельных повторяю-
14*
203
Щихся звеньев и встройка их в кристаллическую решет-
ку. Эта стадия должна быть менее энергоемкой, чем
стадия образования параллельно упакованных, но разу-
порядоченных в экваториальной плоскости участков
цепей.
Действительно, теплота плавления ориентированного
полиэтилентерефталата составляет 23,5 кДж/кг. Раз-
ность между теплотой плавления и понижением энталь-
пии при вытяжке равна 8,8 кДж/кг и представляет со-**
бой тепловой эффект установления порядка в ориенти-
рованной структуре при нагревании. Около 2,5 кДж/кг
из этого теплового эффекта относится к кристаллиза-
ции в области 50—100 °C. Остальная часть связана,
очевидно, с медленной кристаллизацией в области тем-
ператур от 100 до 200 °C, о наличии которой свидетель-
ствует более низкое значение теплоемкости по сравне-
нию с теплоемкостью изотропного закристаллизован-
ного образца. Более низкие температура и теплота
плавления ориентированных образцов свидетельствуют
о большей дефектности их кристаллической структуры.
Этот пример является хорошей иллюстрацией плодо-
творности комплексного изучения физических процес-
сов в полимерах теплофизическими и структурными
методами.
В заключение рассмотрим вопрос о повышении тем-
пературы в зоне образования шейки. В большинстве
случаев ориентационная вытяжка развивается локаль-
но — путем возникновения и последовательного распро-
странения «шейки» в неориентированном материале,
и эта локализация деформации может приводить к су-
щественным разогревшим материала за счет превраще-
ния работы пластической деформации в теплоту. Пред-
положение об адиабатическом повышении температуры
выше температуры размягчения в области «шейки»
было одним из первых объяснений процесса ориента-
ционной вытяжки стеклообразных полимеров [86—89]
и повлекло за собой экспериментальную проверку рас-
пределения температуры вдоль образца, подвергаемого
ориентационной вытяжке. Независимые измерения
с применением термопар [72, 88, 90] и фосфоров [70]
привели к заключению, что повышение температуры
в зоне образования «шейки» существенно зависит от
скорости ориентации; при обычных лабораторных ско-
204
ростях растяжения (1—0,001 мм/с) оно не превышает’
20—25 °C и составляет несколько градусов или даже
десятые доли градуса (рис. Ш.ЗО), т. е. на один-два
порядка меньше того, которое необходимо для размяг-
чения полимера.
Калориметрические данные по энергетическому ба-
лансу ориентационной вытяжки также использовались
для оценки повышения температуры при образовании
«шейки» [91]. При этом Мюллер исходил из схемы,
согласно которой все тепло, возникающее в зоне пла-
стической деформации
«шейки», идет на повы-
шение температуры близ-
лежащей изотропной ча-
сти образца и лишь по-
сле этого отводится в
окружающее пространст-
во. В соответствии с та-
кой схемой повышение
температуры ДТ в иде-
альном случае выража-
ется уравнением
11,1 57)
при стационарном развитии
Рис. Ш.ЗО. Повышение темпера-
туры в зоне образования «шейки»
для полиэтилена [90] (О), рези-
ны СКН-40 [72] ( X ) и капрона
[88] (©).
где е — относительное удлинение; Q — тепловая мощность; р — плот-
ность; Ср—удельная теплоемкость; А—поперечное сечение; VP —
скорость растяжения.
Вычисленное повышение температуры для поливи-
нилхлорида при скорости растяжения 0,34 мм/с соста-
вило 59 °C, для полиамида — 30 и 45 °C при скорости
растяжения 1,1 и 2,2 мм/с. Однако при таком расчете
необходимо учитывать зависимость теплоемкости от
температуры, особенно в области размягчения, нали-
чие охлаждения изотропной части образца и, что самое
главное, отвод теплоты путем излучения и теплообмена
между различными частями образца. На это обстоя-
тельство, как на решающий фактор неадиабатичности
процесса, в свое время указывал Лазуркин [72]. По-
этому реальные значения ДГ по сравнению с рассчи-
танными таким способом значительно ниже. Оценить
205
йх довольно сложно ввиду неопределенности значений
коэффициента теплопередачи. Грубые расчеты градиен-
та температур по образцу в предположении полного
превращения механической работы в теплоту для ско-
рости деформации Ур = 2,5-10~3 мм/с дают значения
6°С/мм для поливинилхлорида и 0,75 °С/мм для поли-
этилена [92]. Это величины того же порядка, что и экс-
периментальные.
На основании всей совокупности эксперименталь-
ных данных по повышению температуры в области
«шейки» гипотеза последовательного размягчения, по
крайней мере для указанной области скоростей дефор-
мации, может быть отвергнута [72, 92, 93]. Тем не
менее проблема определения истинного разогрева
в области «шейки» не может считаться решенной, так
как локальные разогревы при пластической деформа-
ции в микрообъемах могут быть значительно выше экс-
периментально определяемых усредненных макровели-
чин. Они, по-видимому, могут быть зафиксированы
с применением метода регистрации инфракрасного из-
лучения. Измерения с такими целями на полимерах до
сих пор не проводились.
Разрушение полимеров
Разрушение твердых тел неизбежно связано с дис-
сипативными процессами, обусловленными принципи-
альной необратимостью процесса разрыва перенапря-
женных межатомных связей тепловыми флуктуациями
[94—96]. В отличие от низкомолекулярных тел, для
которых при упругом разрушении предполагается лишь
один механизм рассеяния энергии — создание новых
поверхностей [97, 98], в полимерах возможен еще
один механизм рассеяния энергии при чисто упругом
деформировании и разрушении, обусловленный цепным
строением макромолекул [99.] Разрыв упругонапря-
женной макромолекулы или ее отрезка (с достаточно
большим содержанием валентных связей) по одной ка-
кой-либо связи должен неизбежно приводить к рассея-
нию огромной энергии, запасенной в нагруженных
валентных связях, из-за отсутствия внешних сил на
«половинках» разорванной макромолекулы и невоз-
можности поэтому совершать механическую работу.
206
Оценки и расчеты энергетики распада механически
напряженной цепочки атомов (макромолекулы) приво-
дят к выводу, что ее разрыв должен быть подобен микро-
взрыву [99, 100].
Чисто упругое разрушение твердых тел является
скорее исключением, чем правилом [97, 98]. Обычно
же в твердых телах образование новой поверхности
(трещин) сопровождается локальными пластическими
деформациями [96—98]. Это, естественно, является
дополнительным механизмом диссипации механической
энергии. Все это свидетельствует о том, что изучение
структуры энергетического баланса подвергаемых раз-
рушению твердых тел может дать определенную инфор-
мацию о соотношении отдельных диссипативных меха-
низмов. Хотя изучение разрушения твердых полимеров
с позиций решающей роли гистерезисных потерь при
усталостных деформациях с использованием простейше-
го приема — регистрации температурных изменений
образцов термопарами — проводится сравнительно дав-
но [101, 102], первая попытка прецизионного калори-
метрического определения энергетического баланса при
разрушении полимеров была предпринята лишь в по-
следнее время [48, 103]. Она имела своей целью преж-
де всего выявить роль необратимых разрывов макро-
молекул в энергетике разрушения.
Наиболее детальное изучение процесса разрушения
с позиций кинетической концепции разрушения прове-
дено на одноосно-ориентированных кристаллических
полимерах, обладающих одномерным строением на
молекулярном (длинные цепные молекулы) и надмоле-
кулярном (фибриллоподобные образования) уровнях
[95]. Для калориметрического исследования был вы-
бран хорошо охарактеризованный прямыми физически-
ми методами объект — предельно ориентированные
моноволокна капрона с разрушающим напряжением
при растяжении около 800 МПа в расчете на исходное
сечение и относительным удлинением при разрыве
16—17% (при комнатной температуре). Для выявления
энергетических эффектов, связанных с разрывами
макромолекул, был использован прием повторных на-
гружений, вытекающий из кинетической концепции раз-
рушения и экспериментального изучения разрушения
полимеров [94, 95].
20?
Основные результаты этого исследования сводятся
к следующему. С возрастанием напряжения механиче-
ские потери и теплота, рассеянная за цикл растяже-
ние — сокращение, возрастают как при первом, так
и при последующих нагружениях (рис. III.31). При
<7^400 МПа эти характеристики для первого и второго
нагружений практически совпадают. Дальнейшее повы-
шение напряжения существенно меняет картину:
б, НИ а б, НИ а
б
Рис. II 1.31. Зависимость (механических потерь (а) и
рассеянной теплоты (б) от максимального напряжения
в цикле растяжение — сокращение [103]:
/ — первый цикл; 2 — второй цикл.
и Qi для первого нагружения становятся систематиче-
ски больше, чем 6IF2 и Q2 для второго нагружения.
В области напряжений 700—750 МПа (близких к раз-
рушающим) эти параметры расходятся в несколько
раз. При этом важно подчеркнуть, что различия между
энергетическими параметрами возникают лишь на
стадии растяжения, поскольку стадии сокращения при
всех нагружениях оказываются практически эквива-
лентными.
Различия в значениях и Qi обусловлены необ-
ратимыми процессами. Это подтверждается видом зави-
симости доли механической работы растяжения, рассе-
янной в виде теплоты (рис. III.32). Наблюдающаяся
разница в структуре энергетического баланса — резкое
нарастание тепловых потерь при первом нагружении
и постоянство их при втором — свидетельствует о су-
208
щественном различии Молекулярных явлении, происхо-
дящих в полимере при первом и втором нагружениях.
Значительное нарастание относительного количества
тепла, выделяющегося в растягиваемом первый раз
полимере, теперь уже трудно согласовать с картиной
упругого растяжения участков макромолекул, проходя-
щих через аморфные области из одного кристаллита
в другой, хотя, как было показано в предыдущем раз-
деле, в области обратимых деформаций этот процесс
является доминирующим. Уровень гистерезисных по-
терь Q/IVp = 0,27 ~ 0,3 ха-
рактерен для обратимых
деформаций капрона в об-
ласти низких напряжений
при первом нагружении и
для всех напряжений при
втором и последующих на-
гружениях и обусловлен ре-
лаксационными процесса-
ми [48]. Эти результаты
свидетельствуют о том, что
в энергетическом отноше-
нии первый цикл растяже-
ние — сокращение неэкви-
валентен последующим.
В соответствии с пред-
ставлениями кинетической
Рис. III.32. Зависимость до-
ли механической работы,
рассеянной в виде тепла,
от максимального напряже-
ния в цикле растяжение —
сокращение [1031:
1 — первый цикл; 2 — второй
цикл.
теории прочности можно
полагать, что отмеченные различия в энергетике перво-
го нагружения по сравнению с последующими обуслов-
лены необратимыми разрывами макромолекул. В этом
предположении различие в энергетическом балансе
между первым циклом (i~ 1) и последующими может
быть отнесено на счет разрывающихся макромолекул
(рис. Ш.ЗЗ):
\Ui = - AQZ = 6IFZ - 61FZ+1 - Qz + Qz+1 (III.58)
Необходимо выяснить также вклад в энергетику
разрушения полимеров необратимых (пластических) де-
формаций, сопровождающих разрушение при комнат-
ной температуре даже предельно ориентированных
образцов. Измерения показали, что незначительные
обратимые удлинения (от 1 до 1,5%) появляются лишь
20g
Рис. 111.33. Энергетические
эффекты, обусловленные
разрыйами макромолекул
[103].
при о>6 МПа, а при более низких напряжениях они
практически отсутствуют. Если предположить, что вся
необратимая деформация возникает при наибольшем
для данного цикла напряжении, то работа пластиче-
ской деформации даже для таких жестких условий
не должна превышать 30—40% от приведенных значе-
ний. Это позволяет сделать следующее заключение:
хотя работа пластической деформации составляет опре- .
деленную долю общей ра-
боты разрушения, она не
является определяющей.
Сложнее оценить вклад
пластической деформации
в величину АСУ. Если пола-
гать, что механизм погло-
щения энергии при пласти-
ческой деформации в обла-
сти разрушения такой же,
как и при ориентационной
вытяжке капрона, то Д£7~
— 0,21^плас , что примерно
соответствует соотношению
между Д[7 и для про-
цесса разрушения. В таком
случае пластическая дефор-
мация в предразрывных об-
ластях должна, по-видимо-
му, приводить к определен-
ным структурным измене-
ниям. Такие изменения дей-
ствительно имеют место, и
это проявляется в том, что при вторичном нагружении
до той же деформации (а не напряжения) зависимость
о—8 пересекает первичную кривую а—е в области на-
чала появления пластических деформаций в первом
цикле. Этот экспериментальный факт можно понять,
лишь предположив, что пластическая деформация при-
водит к определенному упрочнению [104]. Таким обра-
зом, вопрос о вкладе пластической деформации в вели-
чину ДС7 при разрушении остается открытым. Очевидно,
что при определенных условиях пластическая деформа-
ция может начать играть определяющую роль в энерге-
тическом балансе разрушения. Энергетические эффекты,
10
связанные с разрушением «мягких» капроновых моно-
волокон, свидетельствуют о том, что работа пластиче-
ской деформации составляет значительно более полови-
ны работы разрушения [48].
Энергетические эффекты разрушения предельно
ориентированных моноволокон капрона были использо-
ваны для ряда оценок, связанных с разрывом макро-
молекул. Наибольший интерес представляют оценки
локального повышения температуры вследствие рас-
сеяния большей части работы разрушения в виде
тепла. Поскольку разрушение оказывается локализо-
ванным главным образом в неупорядоченных областях
полимера [95], выделение такого большого количества
энергии должно приводить к огромным локальным
перегревам независимо от механизма диссипации. Гру-
бые оценки на основе полученных интегральных тепло-
вых эффектов показывают, что локальные температуры
могут достигать значений, на сотни градусов превы-
шающих среднюю температуру полимера. Последствия
таких перегревов очевидны: эти своеобразные микро-
взрывы могут просто выжигать места, в которых они
локализованы, приводя к образованию зародышевых
разрывов сплошности — субмикротрещин, обнаруживае-
мых экспериментально [95]. Следует отметить, что
использование метода инфракрасного излучения позво-
лило экспериментально зафиксировать локальное по-
вышение температуры вблизи растущей трещины
в полиметилметакрилате и полистироле, которое со-
ставило 450 °C [105]. Это вполне совпадает с нашими
оценками.
Силовое размягчение наполненных резин
Эффект силового размягчения резин состоит в том,
что для повторного растяжения наполненной резины до
одного и того же удлинения требуется значительно
меньшее усилие по сравнению с первым растяжением,
следствием чего является появление гистерезисной пет-
ли в координатах напряжение — деформация [106,
107]. Этот эффект необратим. Существуют две доста-
точно различные точки зрения на механизм этого эф-
фекта. Одна из них связывает его с необратимым раз-
рывом связей полимер — наполнитель, и ее сторонники
211
считают, что именно на это расходуется механическая
энергия, соответствующая гистерезисной петле [108—
110]. С другой точки зрения, причина размягчения,
заключается в скольжении физических узлов полимер-
ная молекула — наполнитель по поверхности частичек:
наполнителя, приводящем к уменьшению числа корот-
ких цепей вследствие перераспределения длин цепей
в сетке [111, 112]. В соответствии с этим механизмом
гистерезис связан с преодолением сил трения полимер-**
ных цепей о поверхность частичек наполнителя и вся
связанная с гистерезисом работа рассеивается в виде
тепла. Мы видим, что здесь необходимо решить прежде
всего принципиальный вопрос, который может быть
в общем виде сформулирован следующим образом:
происходят ли при первичном растяжении необратимые
изменения структуры, сопровождающиеся увеличением
внутренней энергии, или образующаяся новая структу-
ра энергетически эквивалентна старой. Это требует
определения полного энергетического баланса про-
цесса.
Недавно такое исследование было впервые прове-
дено на наполненных полидиметилсилоксановых резинах
[113]. Его результаты показали, что при действии ме-
ханической нагрузки на наполненные резины происхо-
дит перестройка ее исходной пространственной струк-
туры в новую, энергетически эквивалентную, но отли-
чающуюся от исходной структуры механическим
поведением. Это дает основание считать более вероят-
ным механизм размягчения, связанный с перегруппи-
ровкой цепей на поверхности наполнителя - вследствие
их скольжения. При этом, однако, было отмечено, что
в действительности механизм размягчения должен быть
значительно более сложным, чем простое перераспреде-
ление длин цепей в полимерной сетке вследствие их
скольжения по поверхности наполнителя, как это обыч-
но предполагается [111, 112]. В дополнение к этому
эффекту возможно, например, перемещение самих ча-
стиц в полимерной матрице, что должно приводить
к соответствующему изменению пространственной
структуры наполненной резины и появлению у нее
анизотропии механических свойств. Появление такой
анизотропии у размягченных полидиметилсилоксано-
вых резин было обнаружено экспериментально.
212
ЛИТЕРАТУРА
1. Лейбфрид Г.' Микроскопическая теория механических и теп-
ловых свойств кристаллов. Пер. с нем. Под ред. Б. Я. Мойжеса.
М. — Л., Физматгиз, 1963. 312 с.
2. Киттель Ч. Введение в физику твердого тела. Пер. с англ.
А. А. Гусева. М., Физматгиз, 1962. 626 с.
3. Жданов Г. С. Физика твердого тела. М., изд-во МГУ, 1962.
4. Н а й Дж. Физические свойства кристаллов. Пер. с англ.
Л. А. Шувалова. М., «Мир», 1967. 385 с.
5. Хв о льс он О. Д. Курс физики. Изд. V. Т. 3. Берлин, ГИ, 1923.
6. Henstenberg J., Mark Н., Z. Kristallogr., 1928, Bd. 69,
S. 271.
7. С о 1 e E. A., Holms D. R., J. Polymer Sci., 1960, v. 46, p. 245.
8. W a k e 1 i n J. H., Sutherland A., Beck L. R., J. Polymer
Sci., 1960, v. 42, p. 278.
9. Muller A., Proc. Roy. Soc., 1930, A, v. 127, p. 417.
10. Swan P. R., J. Polymer Sci., 1962, v. 56, p. 403.
11. Kobayashi Y., Keller A., «Polymer», 1970, v. 11. p. 114.
12. De vis G. T., Eby R. K., Colson J. P., J. Appl. Phys., 1970,
v. 41, p. 4316.
13. Китайгородский А. И. Молекулярные кристаллы. M.,
«Наука», 1971. 424 с.
14. В a u g h m a n R. Н., T u г i Е. A., J. Polymer Sci., 1973. pt. A-2,
v. 11, p. 2453.
15. Лифшиц И. M., ЖЭТФ, 1952, т. 22, № 4, с. 475—486.
16. Новикова С. И., ФТТ, 1968, т. 10, № 11, с. 3439—3441.
17. В a u g h m a n R. Н., J. Chem. Phys., 1973, v. 58, p. 2976.
18. Фельдман P. И., Коллоид, ж., 1959, т. 21, № 2, с. 238.
19. Колобов Е. И., ЖФХ, 1960, т. 34, № 4, с. 716—725.
20. Meredith R., Bay Sing Hsu, J. Polymer Sci., 1962,
v. 61, p. 271.
21. Малинский Ю. M., Гузеев В. В., Зубов Ю. А., Кар-
гин В. А., Высокомол. соед., 1964, т. 6, № 6, с. 1116—1119.
22. К о з л о в П. В., Кай минь И. Ф., Каргин В. А., ДАН
СССР, 1966, т. 167, № 6, с. 1321 — 1324.
23. К i m В. Н., Batist R., J. Polymer Sci., 1973, pt. В, v. 11,
p. 121.
24. Hell we gw К.-H., Hannig J., Knappe W., Koll.-Z. u.
Polymere, 1963, Bd. 188, S. 121.
25. P у м e p Ю. Б., Рывкин M. Ш. Термодинамика, статистиче-
ская физика и кинетика. М., «Наука», 1972. 400 с.
26. Thomson W. Mathematical and Physical Papers. V. 1 (art. 48).
London, Cambridge University Press, 1882. 529 p.
27. Умов H. А. Избранные труды. M.—Л., ГИТТЛ, 1950, 553 с.
28. Шиллер Н. Н., ЖРФХО, 1879, т. 11, № 6, с. 55—77.
29. Planck М. Physikalische Abhandlungen und Vortrage. Bd. 1.
Braunschweig, Friedrich Vieweg und Sohn. 1958.
30. Voigt W. Lehrbuch der Kristallphysik. Leipzig — Berlin, Teubner,
1928. 978 S.
31. H а д а и А. Пластичность и разрушение твердых тел. Пер. с англ.
Под ред. Г. С. Шапиро. Т. 2. М., «Мир», 1969. 863 с.
32, Joule J. Р,, Proc. Roy. Soc., 1857, v. 8, p. 564.
213
33. Joule J. P., Phil. Mag., 1858, ser. 4, v. 15, p. 538.
34. James H. M., Gut E. J. Chem. Phys., 1943, v. 11, p. 455.
35. J a m e s H. M., Gut E., J. Polymer Sci., 1949, v. 4, p. 153.
36. Flory P. J. In: Principles of Polymer Chemistry. Ithaca, New
York, Cornell University Press, 1953, p. 134—491.
37. Волькенштейн M. В. Конфигурационная статистика поли-
мерных цепей. М. — Л., изд-во АН СССР, 1959. 466 с.
38. Krigbaum W. R., Roe R.-J., Riibb. Chem. Technol., 1965,
v. 38, p. 1039.
39. Б и p ш т e й н T. M., Птицын О. Б. Конформации макромо-
лекул. М., «Наука», 1964. 391 с.
40. Тр е л о а р Л. Физика упругости каучука. Пер. с англ. Под ред.
Е. В. Кувшинского. iVL, Издатинлит, 1953. 240 с.
41. Gee G., Trans. Faraday Soc., 1946, v. 42, p. 585.
42. Flory P. J., Trans. Faraday Soc., 1961, v. 57, p. 829.
43. Physik der Kunststoffe, Herausgegeb. W. Holzmiiller und K. Alten-
burg. Berlin, Akademie-Verlag, 1961. 652 S.
44. Липатов Ю. С., Нестеров A. E., Гриценко T. M.,
Веселовский P. А. Справочник по химии полимеров. Киев,
«Наукова думка», 1971. 535 с.
45. Журков С. Н., Слуцкер А. И., Я стреби некий А. А.,
ДАН СССР, 1963, т. 153, № 2, с. 303—306.
46. Слуцкер А. И. Докт. дис. Л., ФТИ им. А. Ф. Иоффе, 1968.
47. Годов ск ий Ю. К., Слонимский Г. Л., Папков В. С.,
Ди кар ев а Т. А., «Механика полимеров», 1970, № 5, с. 785.
48. Годове к ий Ю. К. Докт. дис. М., НИФХИ им. Л. Я. Карпо-
ва, 1972.
49. G о d о v s к у Y и. К. е. а. ШРАС Intern. Sympos. on Macromole-
cules, Leiden, 1970, v. 2, p. 669.
50. Peterlin A., J. Polymer Sci., 1966, pt. C, № 15, p. 427.
51. Treloar L. The Physics of Rubber Elastisity. 2nd ed. Oxford
University Press, 1958. 342 p.
52. Papkov V. S., Godovsky Yu. K., Slonimsky G. L.
8 IUPAC Microsympos. on Macromolecules, Prague, 1971, Abstr.
E4.
53. С а д ы к о в а Ф. X., Хим. волокна, 1971, № 9, с. 45—50.
54. Morbitzer L., Hentze G., В on art R., Koll.-Z. u. Z. Po-
lymere, 1967, Bd. 216—217, S. 137.
55. Bondi A. Physical Properties of Molecular Crystalls, Liquids and
Glasses. N. Y., J. Wiley a. Sons, 1968. 502 p.
56. V i 11 a r i E., Pogg. Ann., 1871, v. 143, p. 88.
57. Dart S. L., Anthony R. L., Guth E., Ind. Eng. Chem., 1942,
v. 34, p. 1340.
58. Mark J. E., Rubb. Chem. Technol., 1973, v. 46, p. 593.
59. A k 1 о n i s J. J., M а с К n i g h t W. J., Shen M. Introduction
to Polymer Viscoelasticity. New York, Wiley-Interscience, 1972.
60. Dick W., Muller F. H., Koll.-Z., 1960, Bd. 172, S. 1.
61. Goritz D., Muller F. H., Koll.-Z., 1973, Bd. 251, S. 679.
62. Shen M., Blatz P. J., J. Appl. Phys., 1968, v. 39, p. 4937.
63. Ф л о p и П. Статистическая механика цепных молекул. Пер. с
англ. Под ред. М. В. Волькенштейна. М., «Мир», 1971. 440 с.
64. Roe R.-J., Krigbaum W. R., J. Polymer Sci., 1962, v. 61,
p. 167.
65. X а л и к о в Д. X. Канд. дис. М., МГУ, 1970,
214
66. A 11 e n G. e. a., Trans. Faraday Soc., 1971, v. 67, p. 1278.
67. Большанина А., Панин В. E. В кн.: Исследования по фи-
зике твердого тела. М., изд-во АН СССР, 1957, с. 193—233.
68. Клер бр о М., Хар грив с М. Е., Лоретто М. X. В кн.:
Возврат и рекристаллизация металлов. М., «Металлургия», 1966,
с. 69—85.
69. Б о д я ч к о М. Н., А с т а н ч и к С. А., Я р ошев и ч Г. Б. Тер-
мокинетика и рекристаллизация. Минск, «Наука и техника»,
1968. 251 с.
70. М u 11 е г F. Н. In: Rheology. V. 5. Ed. by F. R. Eirich. N. Y. —
London, Academic Press, 1969, ch. 8.
71. Падай А. Пластичность и разрушение твердых тел. Т. 1. Пер.
с англ. Под ред. Г. С. Шапиро. М., Издатинлит, 1954. 647 с.
72. Л а з у р к и н Ю. С. Докт. дис. М., ИФП АН СССР, 1954.
73. Kumar A., J. Appl. Polymer Sci., Appl. Polymer Symposia, 1969,
№ 12, p. 67.
74. Peter lin A., Me in el G., J. Appl. Phys., 1965, v. 36, p. 3028.
75. Fischer E. W., Hinrichsen G., Koll.-Z. u. Z. Polymere,
1966, Bd. 213, S. 28.
76. 111 e r s К.-H., Angew. makromol. Chem., 1970, Bd. 12, S. 89.
77. P e t e r 1 i n A., J. Polymer Sci., 1967, pt. C, № 18, p. 123.
78. Clearebrough L. M. e. a., Proc. Roy. Soc., 1952, A, v. 215.
79. Peter lin A., J. Mater. Sci., 1971, v. 6, № 6, p. 490.
80. Muller F. H, Koll.-Z., 1959, Bd. 165, S. 96.
81. Muller F. H., Osterr. Chemiker-Z., 1962, Bd. 63, S. 158.
82. Г e н и н Я. В., Герасимов В. И., Ц в а н к и н Д. Я., Высо-
комол. соед., 1973, А, т. 15, № 8, с. 1798—1808.
83. Генин Я. В., Герасимов В. И., Гинзбург Б. М., Сул-
танов Н., Ц в а н к и н Д. Я., Высокомол. соед., 1973, А, т. 15,
№ 1, с. 85—90.
84. Казарян Л. Г., Ц в а н к и н Д. Я-, Высокомол. соед., 1967, А,
т. 9, с. 377—384.
85. Френкель С. Я. Дополнения I и II в книге Ф. X. Джейла:
Полимерные монокристаллы. Л., «Химия», 1968. 551 с.
86. Miiller F. Н., Jackel К., Makromol. Chem., 1953, Bd. 9,
S 97
87. Muller F. H., Jackel K., Koll.-Z., 1952, Bd. 129, S. 145.
88. Jackel K-, Koll.-Z., 1954, Bd. 137, S. 130.
89. M a r s c h a 11 J., Thompson A. B., Proc. Roy. Soc., 1954, A,
v. 221, p. 541, 1147.
90. Nakamura M., Skinner S. M., J. Polymer Sci., 1955, v. 18.
91. E n g e 11 e r Ad., Muller F. H., Koll.-Z., 1958, Bd. 157, S. 89.
92. V i n c e n t P. J., «Polymer», 1960, v. 1, p. 7.
93. Robertson R. E., J. Appl. Polymer Sci., 1963, v. 7, p. 443.
94. Журков С. H., Вестник АН СССР, 1968, № 3, c. 46—52.
95. P ere ль В. P., Слуцкер А. И., Томашевский Э. Е.
Кинетическая природа прочности твердых тел. М., «Наука», 1974,
560 с.
96. Бартенев Г. М., Разумовская И. В., Физ. хим. меха-
ника полимеров, 1969, т. 5, с. 60—72.
97. The Physics of Glassy Polymers. Ed. by R. N. Haward. London,
Applied Science Publishers, 1973. 620 p.
215
98. Разрушение твердых полимеров. Пер. с англ. Под ред. В. Е. Гу-
ля. М., «Химия», 1971. 528 с.
99. Патрикеев Г. А., ДАН СССР, 1958, т. 120, № 2, с. 339.
100. Томашевский Э. Е., ФТТ, 1970, т. 12, № 11, с. 3202.
101. Ратнер С. Б. Докт. дис., М., ИХФ, АН СССР, 1971.
102. Лексовский А. М., Ре гель В. Р., «Механика полимеров»,
1968, № 6, с. 1008—1036.
103. Г о д о в с к и й Ю. К., П а п к о в В. С., Слуцкер А. И., То-
машевский Э. Е., Слонимский Г. Л., ФТТ, 1971, т. 13,
№ 8, с. 2289—2294.
104. Bonart R., Schultze-Gebhardt F., Angew. makromol.
Chem., 1972, Bd. 22, S. 41.
105. Fox P. G., Fuller K. N., «Nature-Physical Science», 1971,
v. 234, № 44, p. 13.
106. Mullins L. J., J. Rubb. Res., 1947, v. 16, p. 275.
107. Mullins L. J., J. Phys. Coll. Chem., 1950, v. 54, p. 239.
108. Blanchard A. F., Parkinson D., Ind. Eng. Chem., 1952,
v. 44, p. 799.
109. Blanchard A. F., J. Polymer Sci., 1954, v. 14, p. 355.
110. Bueche F., J. Appl. Polymer Sci., 1960, v. 4, p. 107.
111. Dannenberg E. M., Trans. IRI, 1966, v. 42, p. T26.
112. Rigbi Z., Rev. gen. caoutch. plast., 1968, v. 45, p. 625.
113. Па пк OB В. С., Годовский Ю. К., Булкин А. Ф., Жда-
нов А. А., Слонимский Г. Л., Андрианов К. А., «Ме-
ханика полимеров», 1975, № 3, с. 387—392.
ЮЛИЙ КИРИЛЛОВИЧ годовский
ТЕПЛОФИЗИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЛИМЕРОВ
Редактор Л. И. Галицкая
Технический редактор Г. И. Косачева
Художник А. К. Малкин
Корректоры Л. П. Волкова, Г. М. Гольбиндер
Т 09821. Сдано в наб. 2/Ш 1976 г. Подп. к печ. 3/V 1976 г.
Формат бумаги 84Х1087з2. Бумага тип. № 2. Усл. печ.
л. 11,34. Уч.-изд. л. 11,06. Тираж 4300 экз. Зак. 264.
Изд. № 222. Цена 1 р. 25 к.
Издательство «Химия». 107076, Москва, Стромынка, 13
Московская типография № 11 Союзполиграфпрома при
Государственном комитете Совета Министров СССР по
делам издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., 1.