/


























































































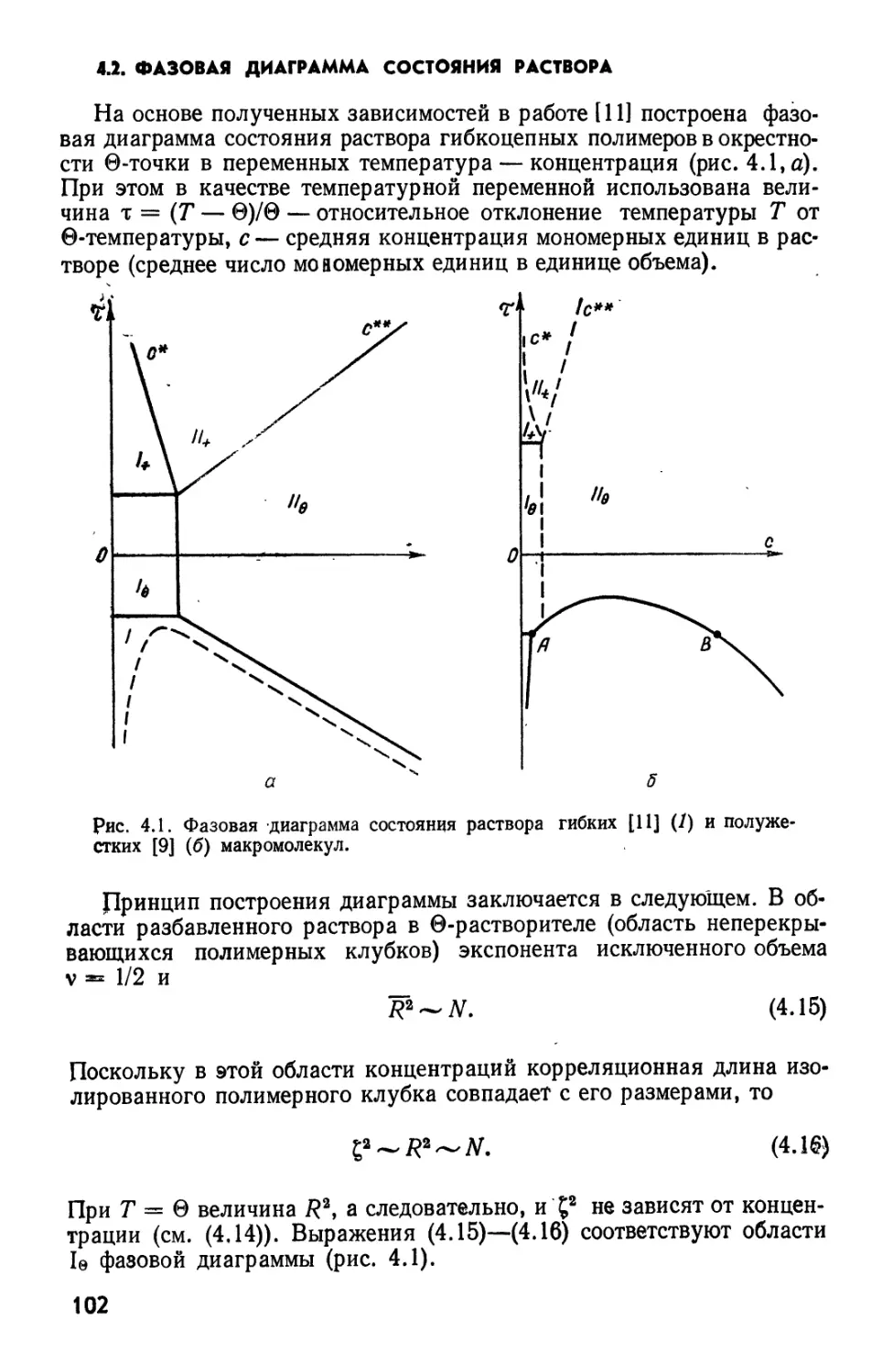










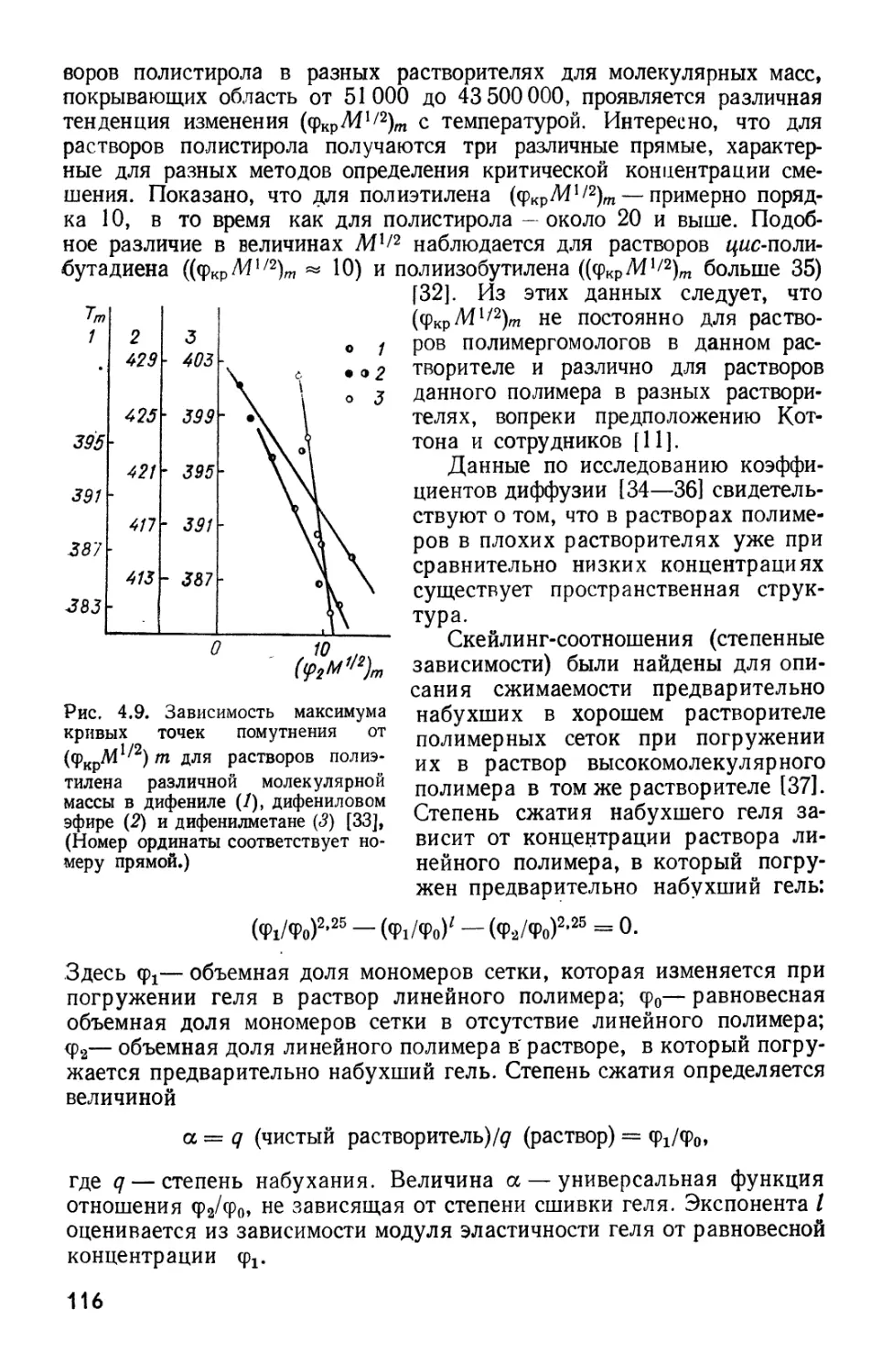



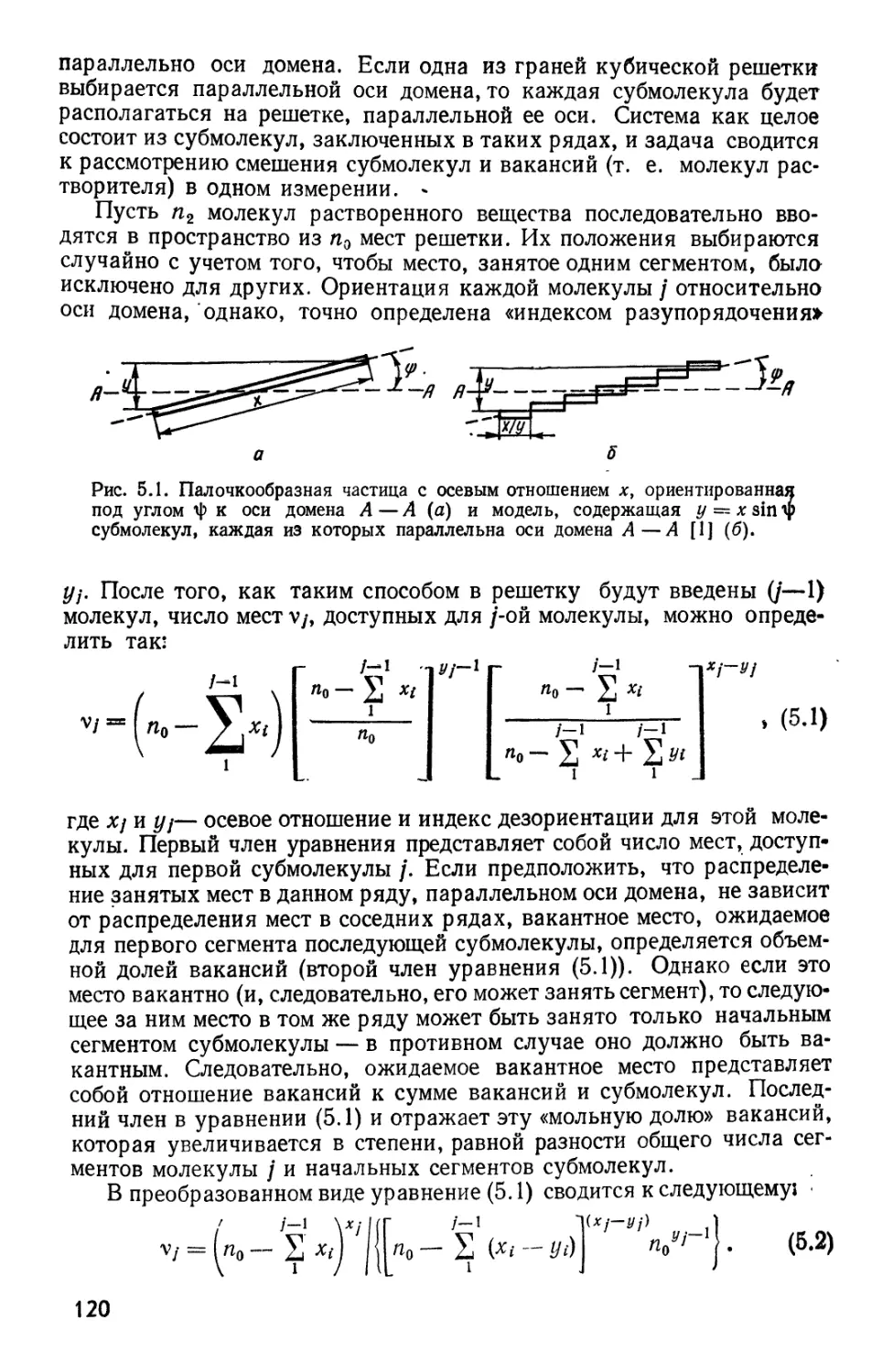






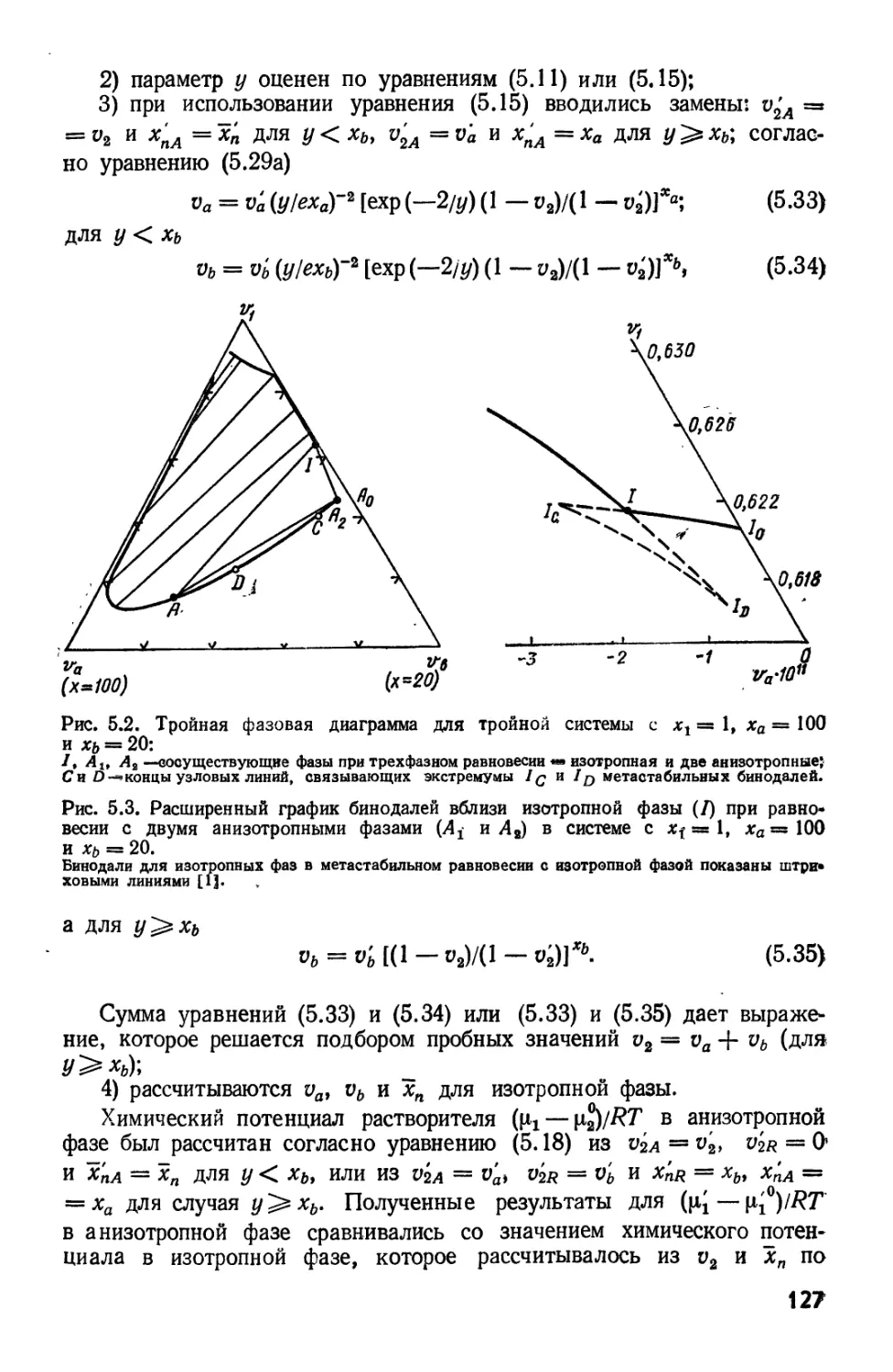



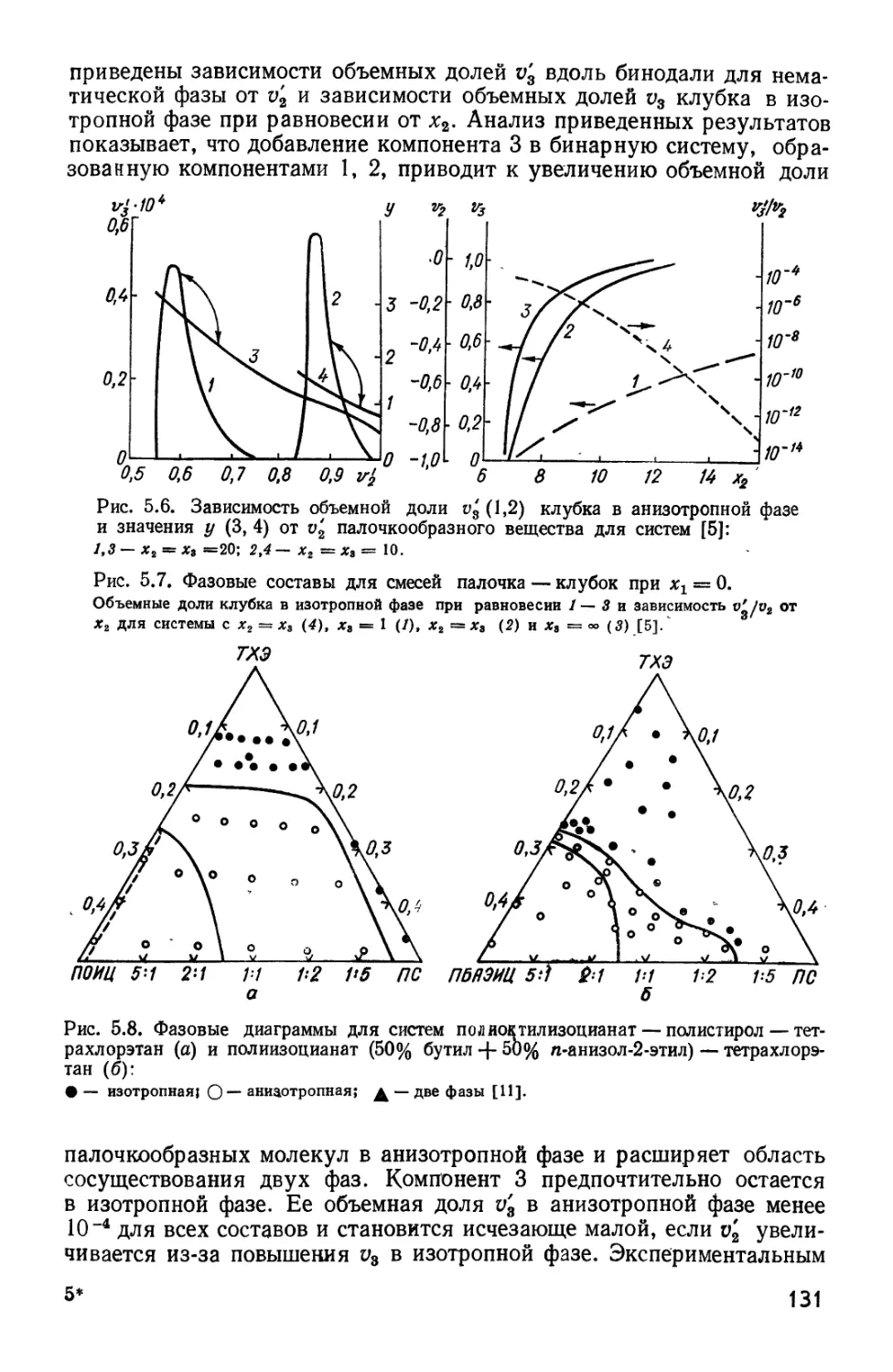






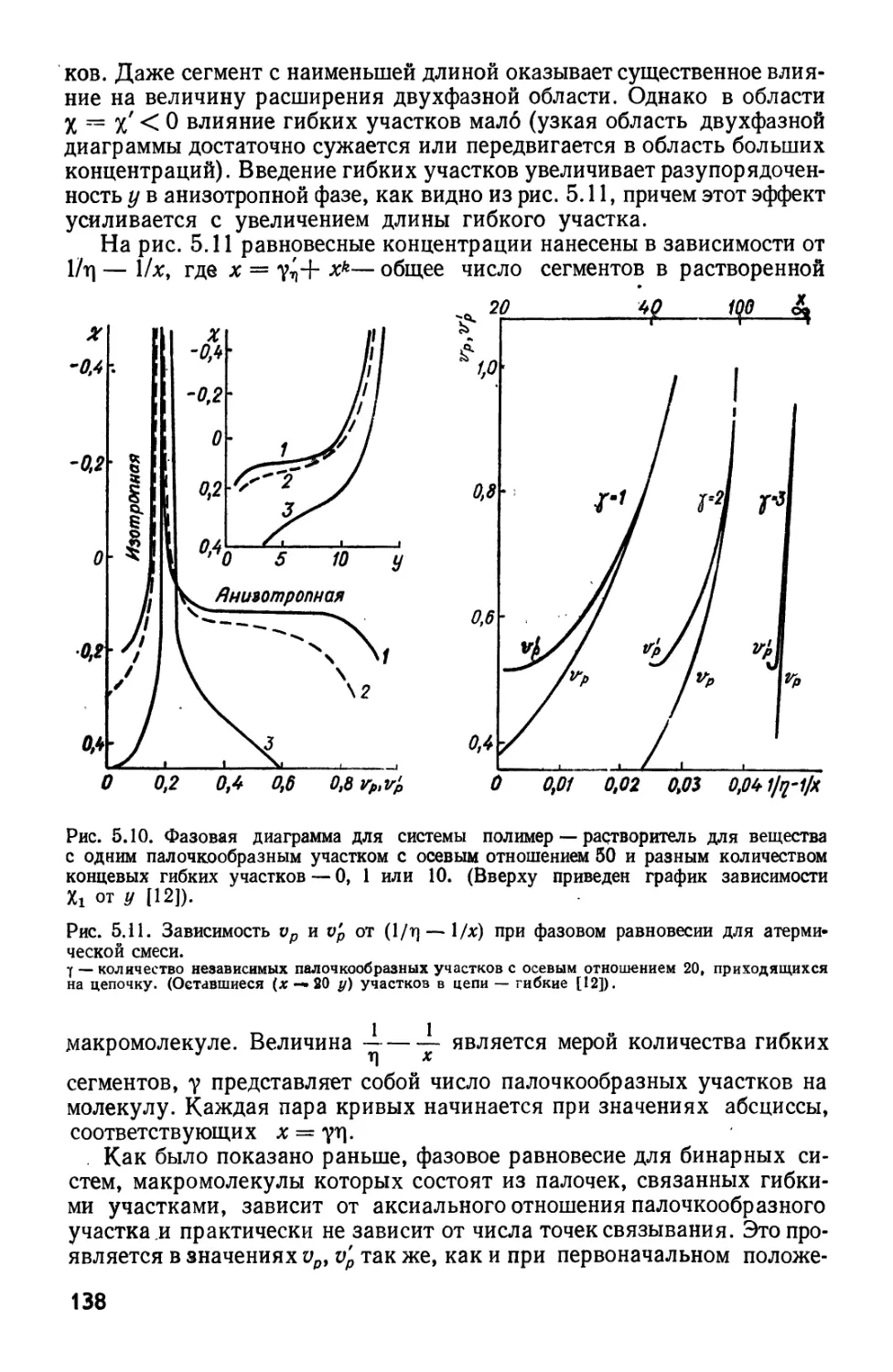
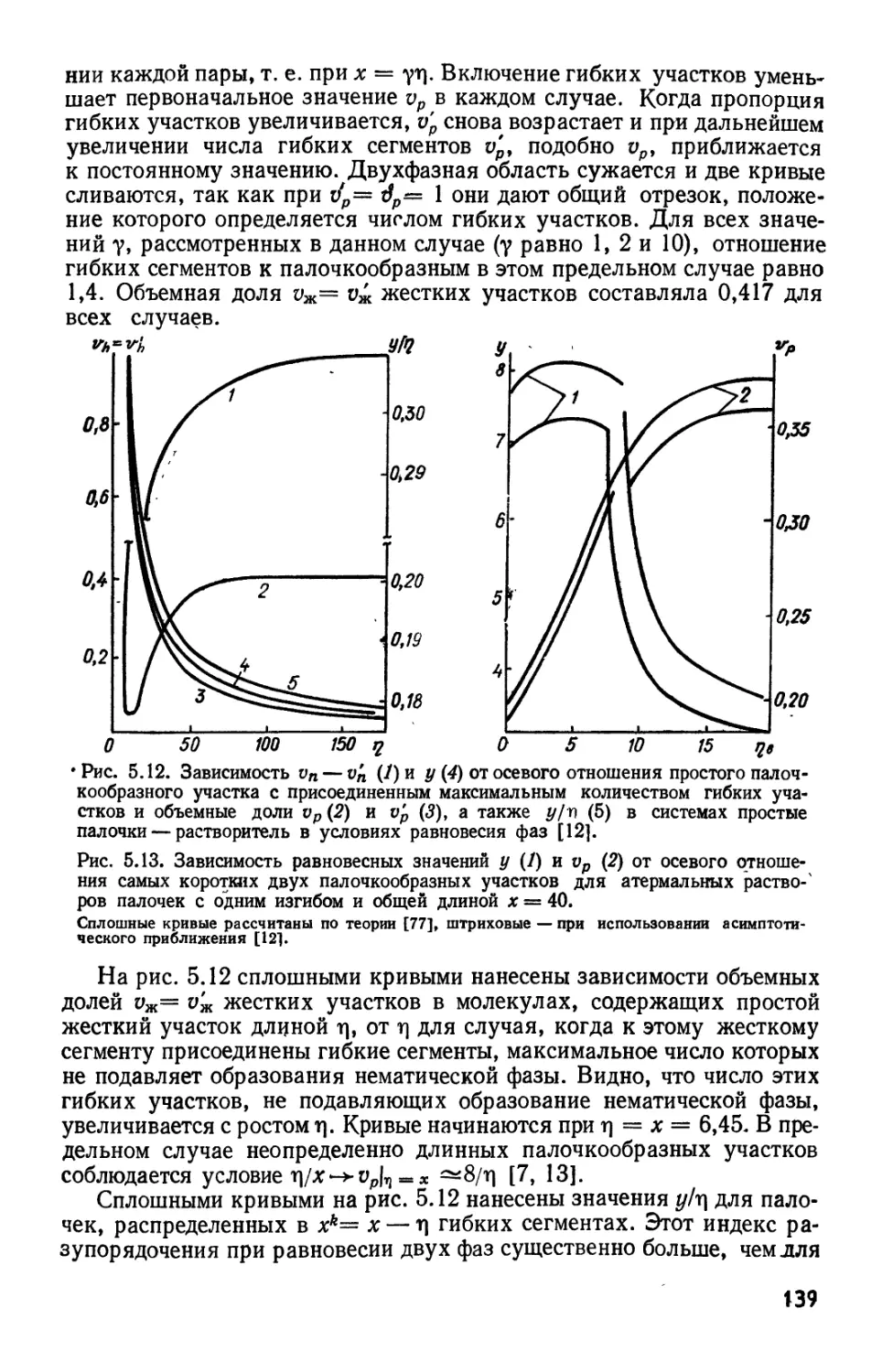








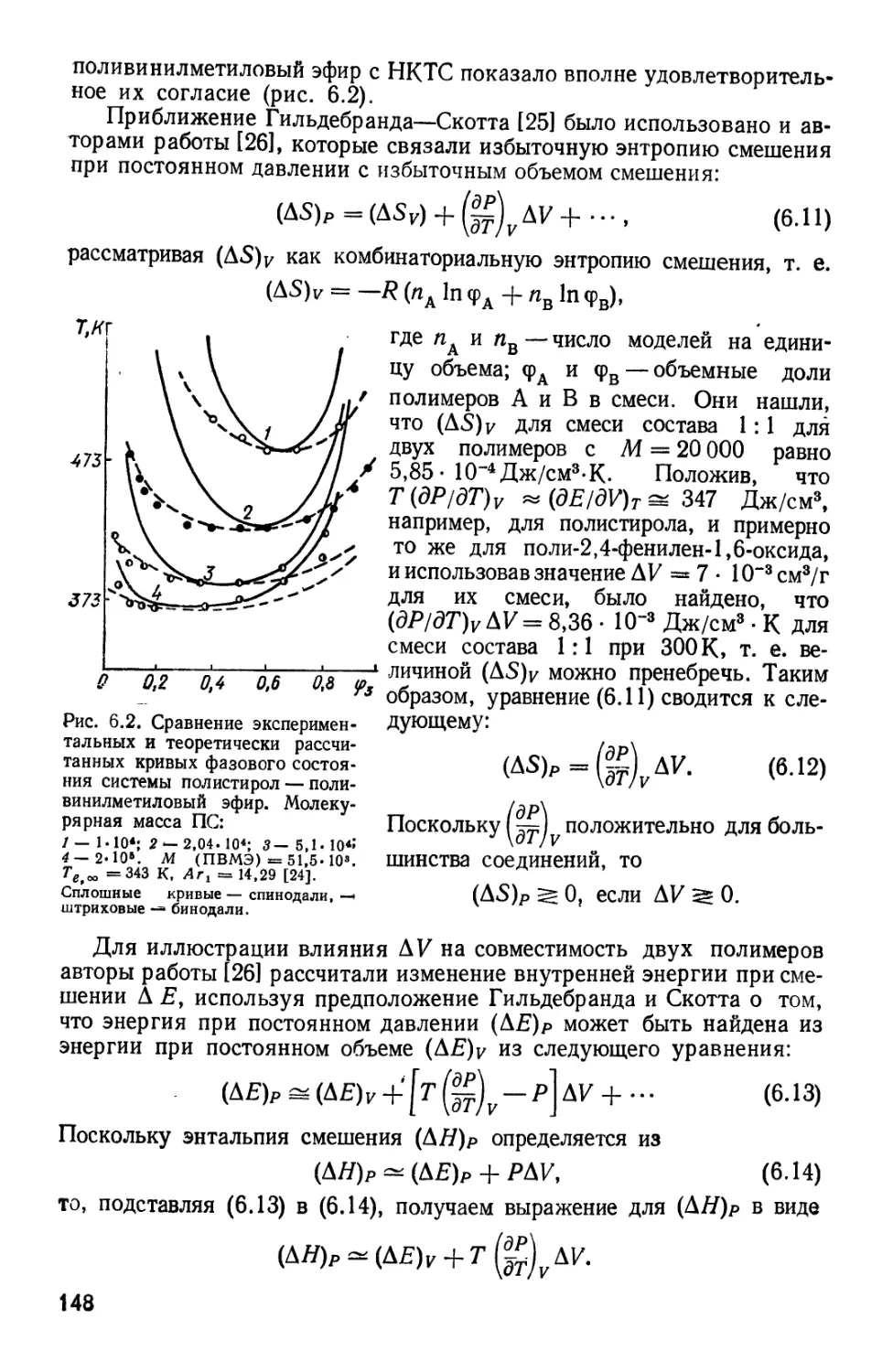

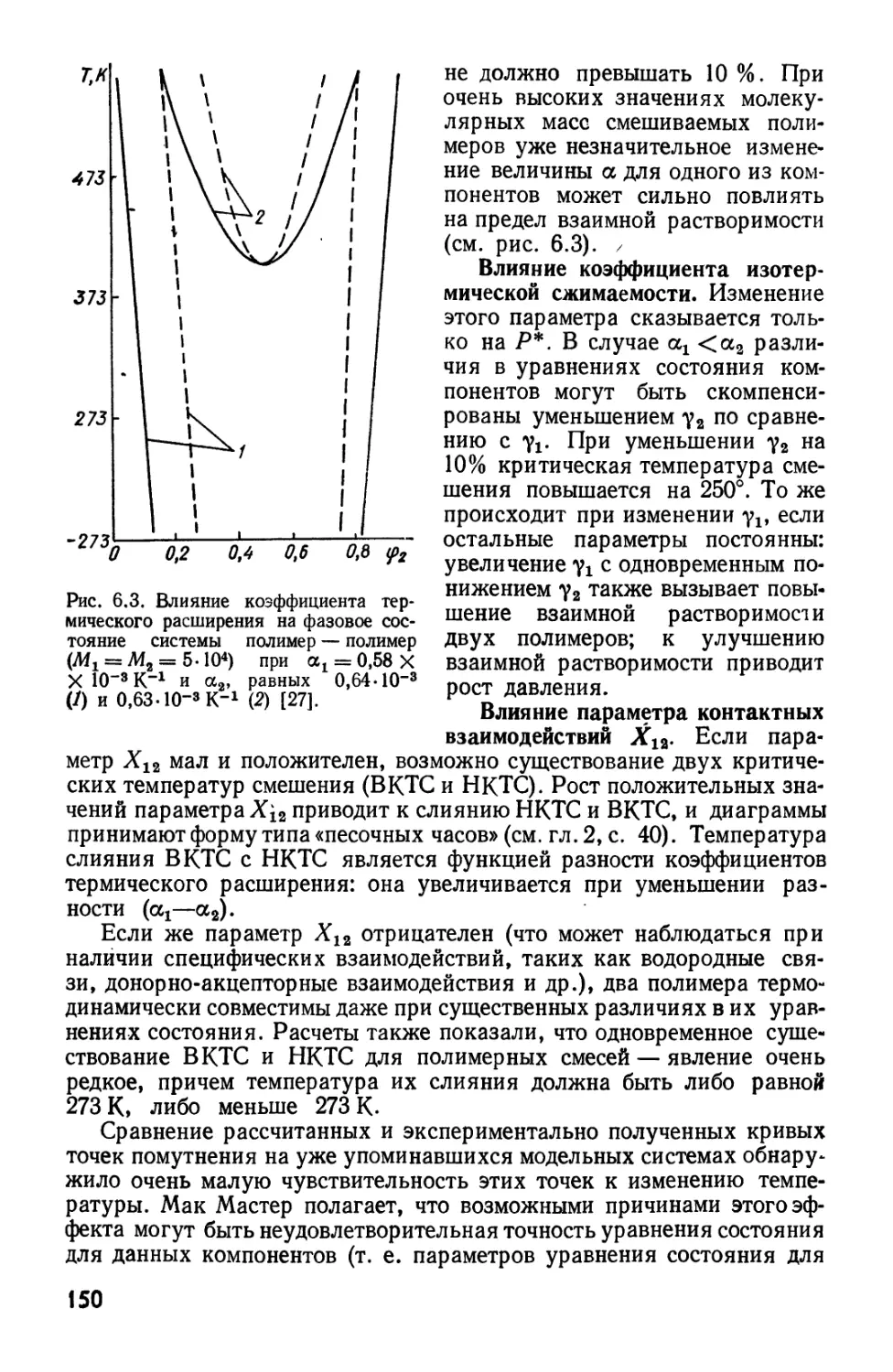

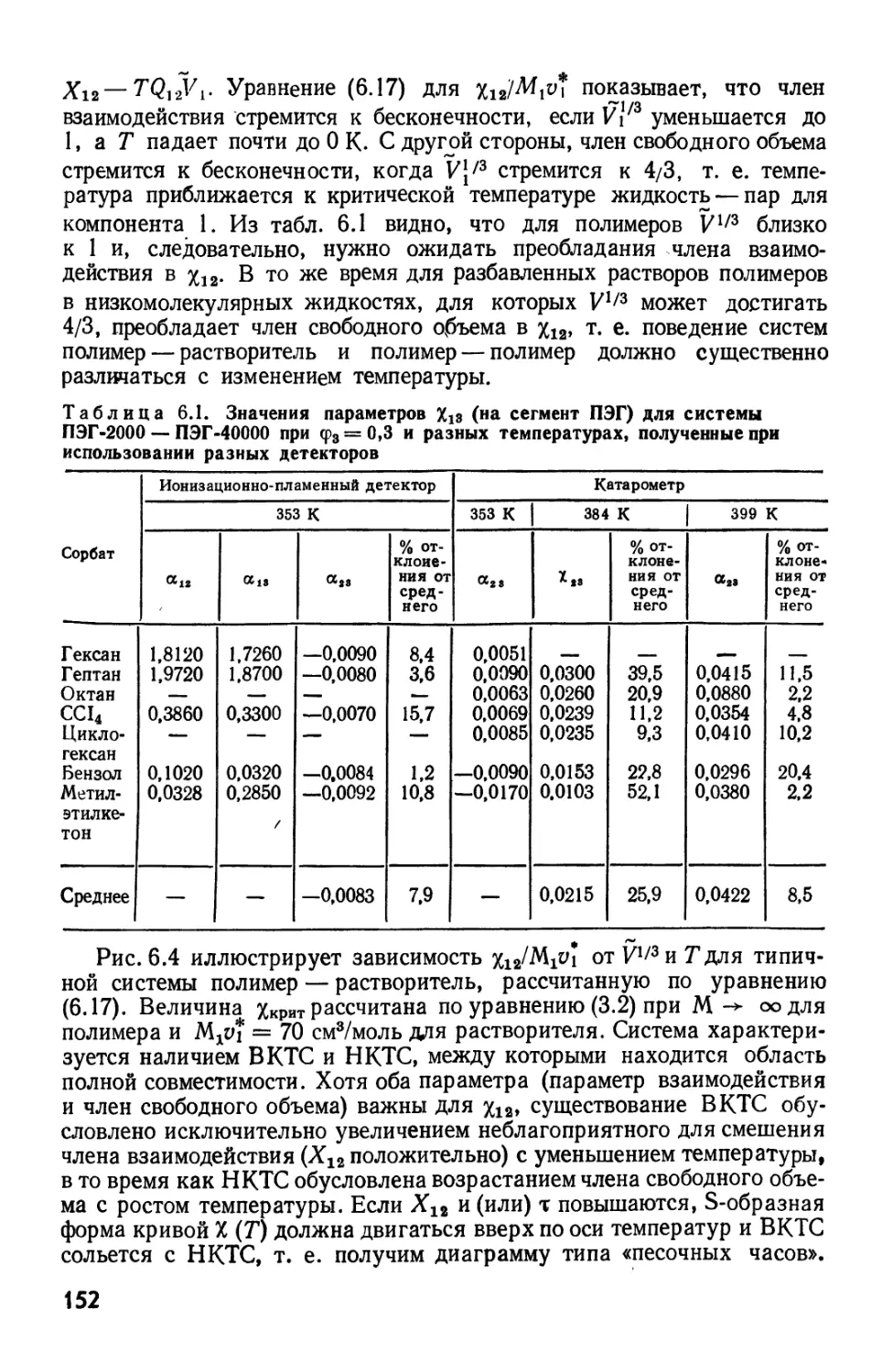
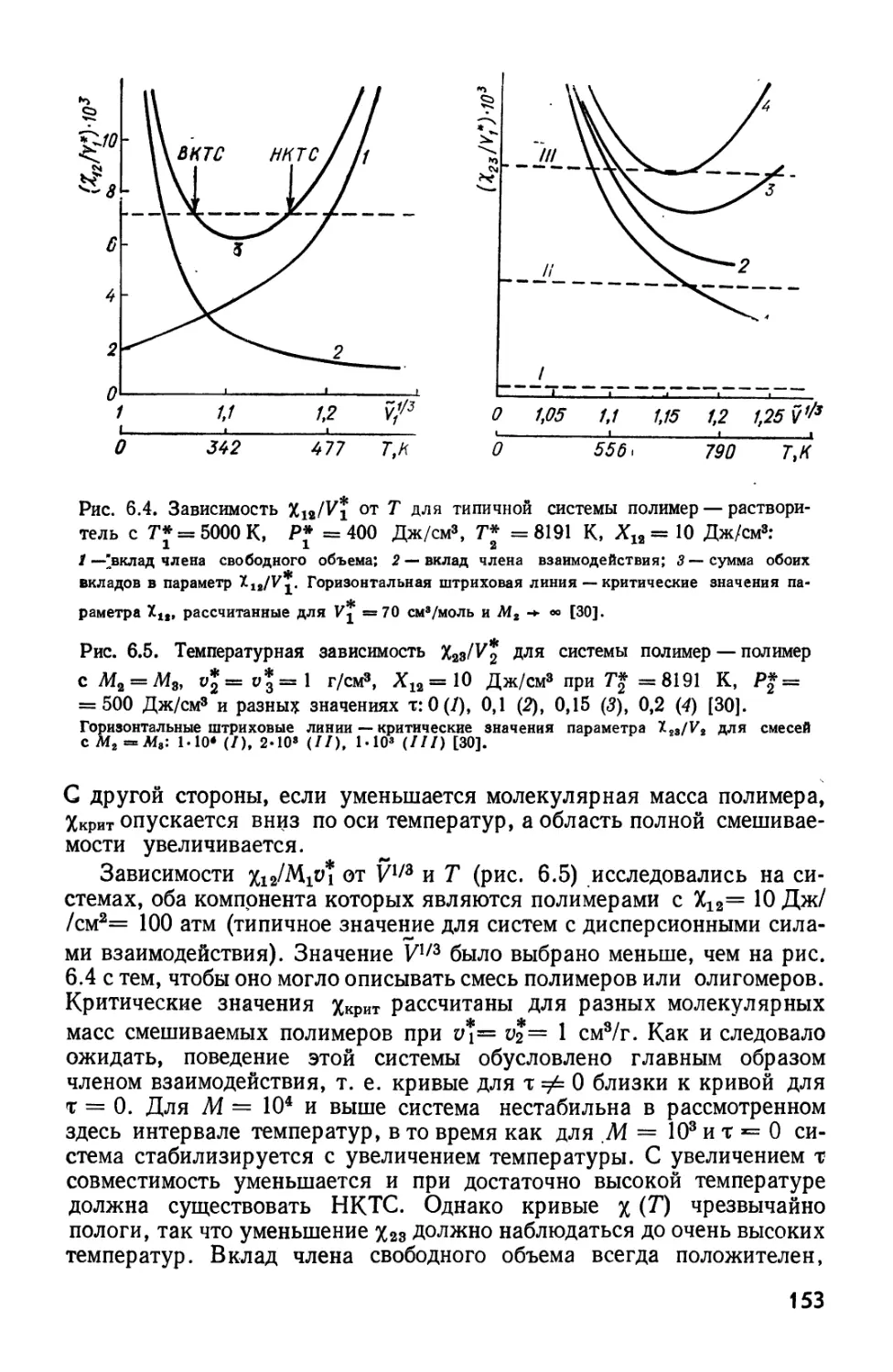



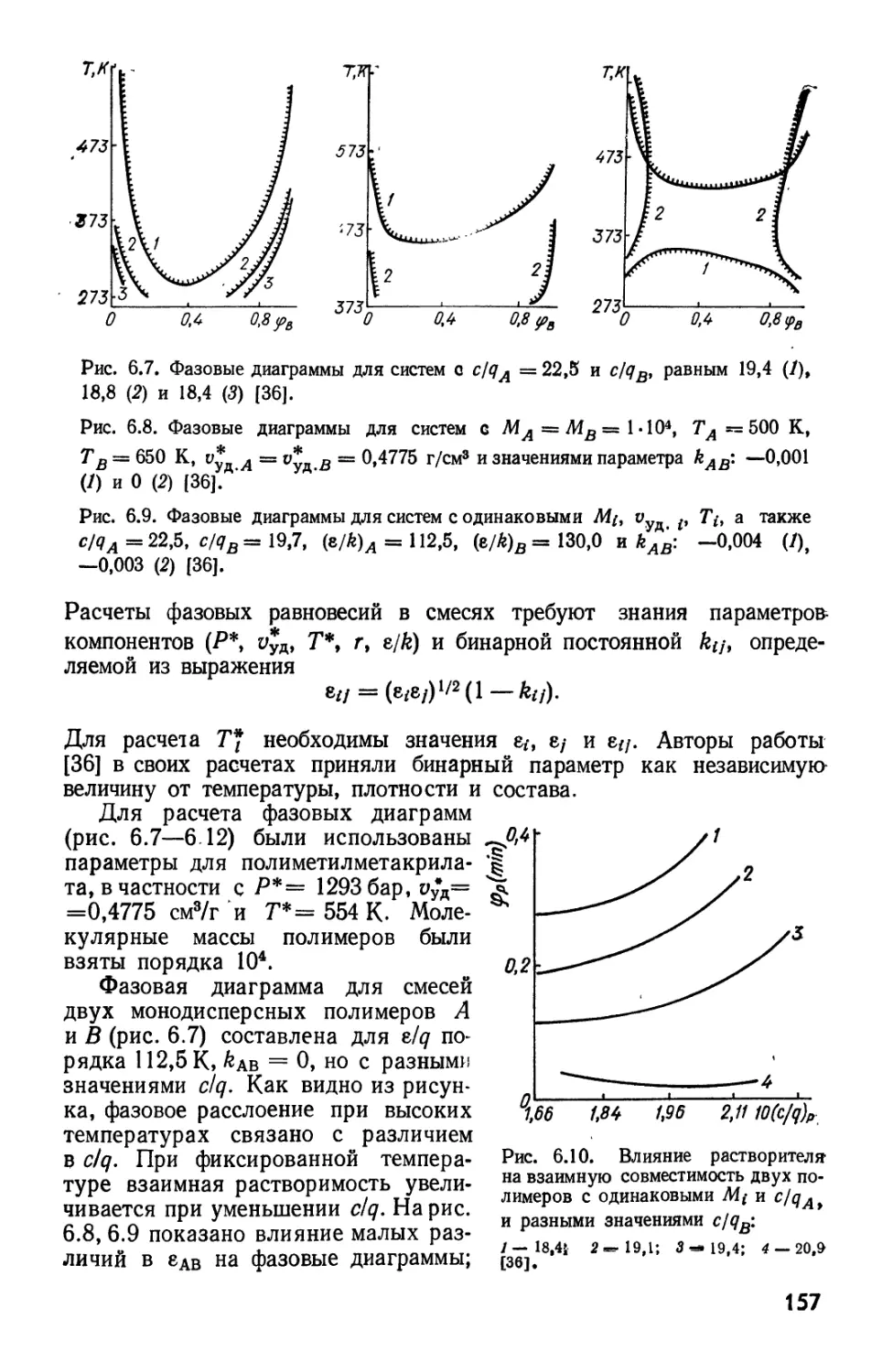
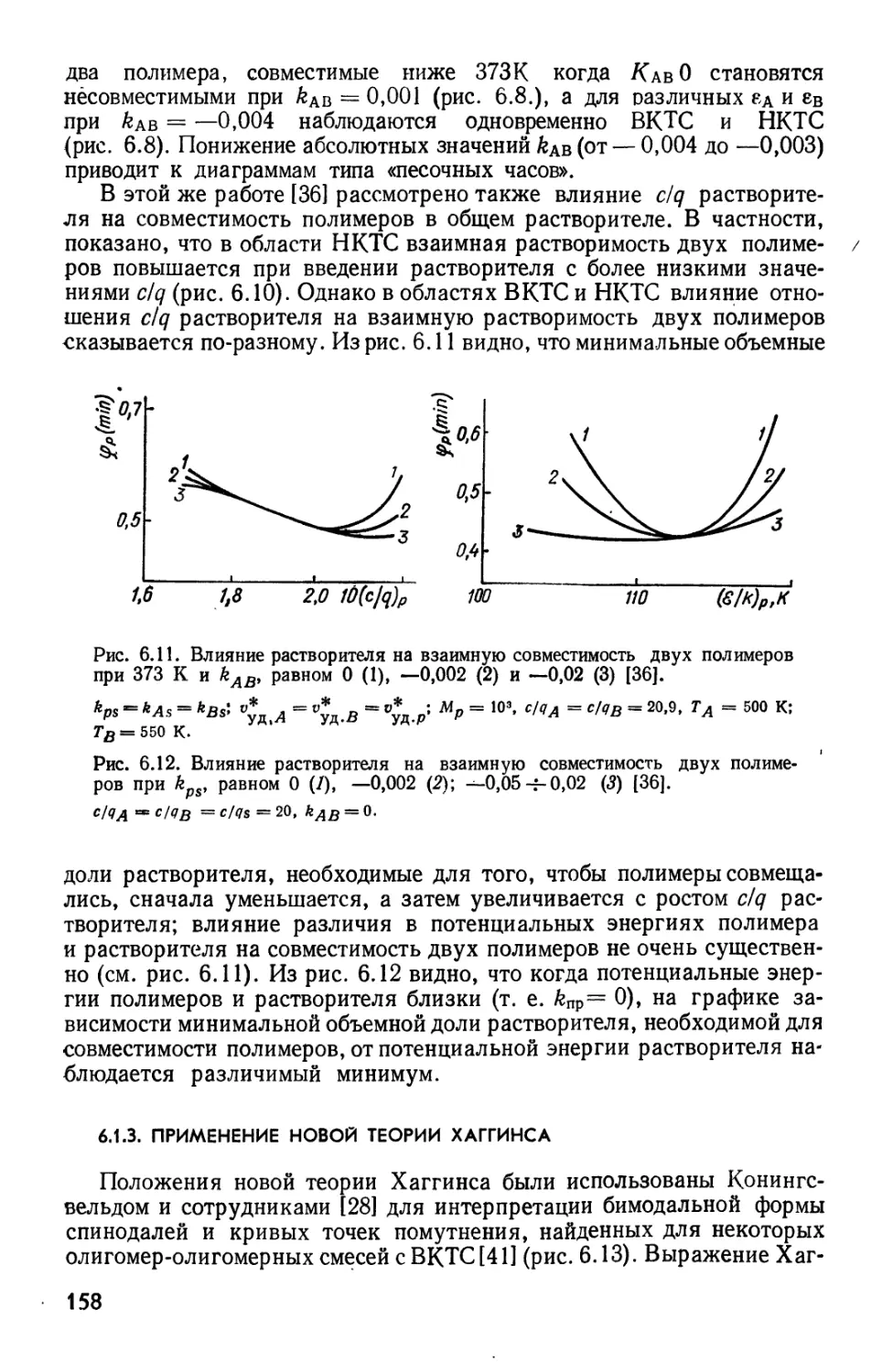









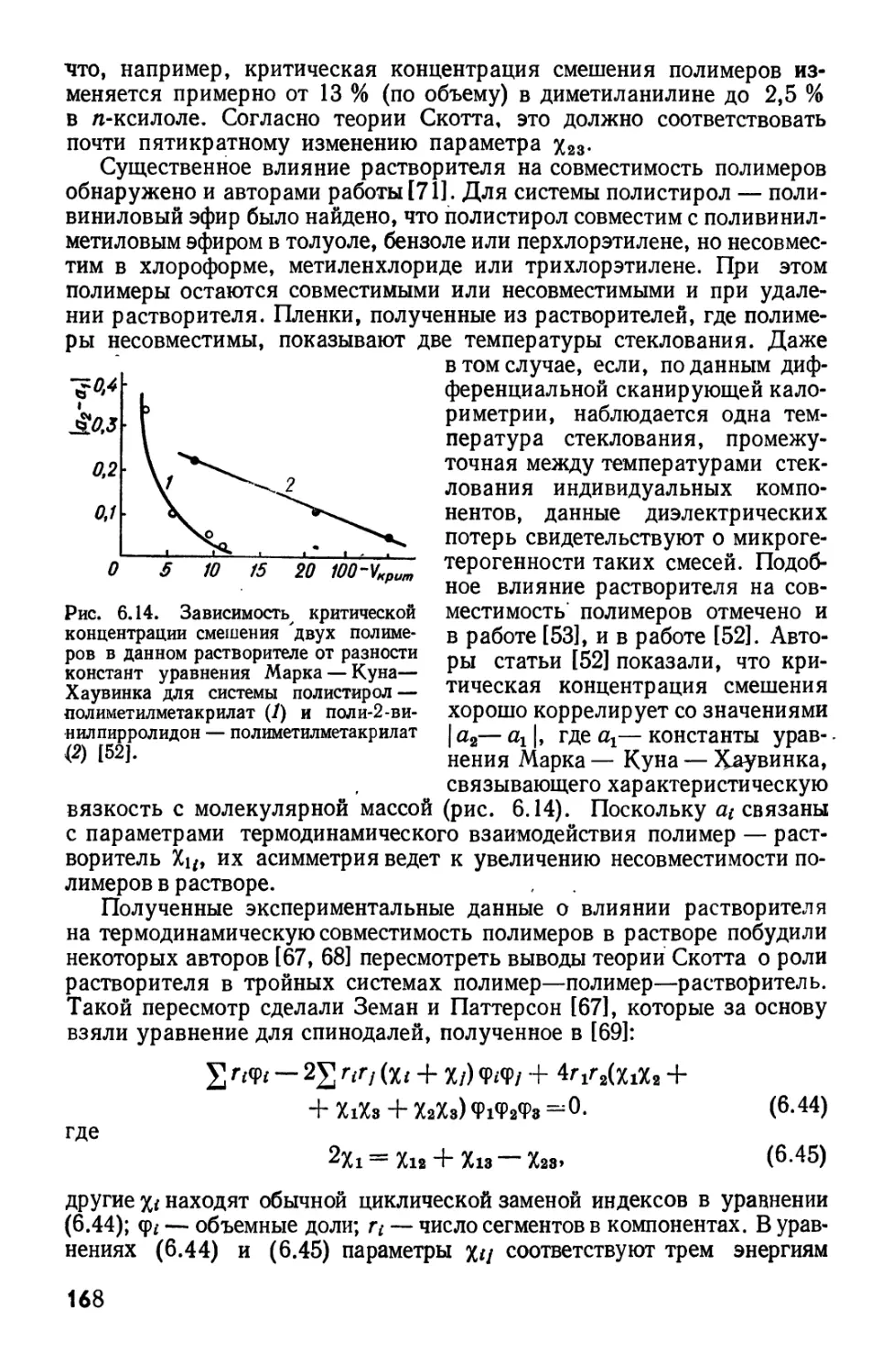



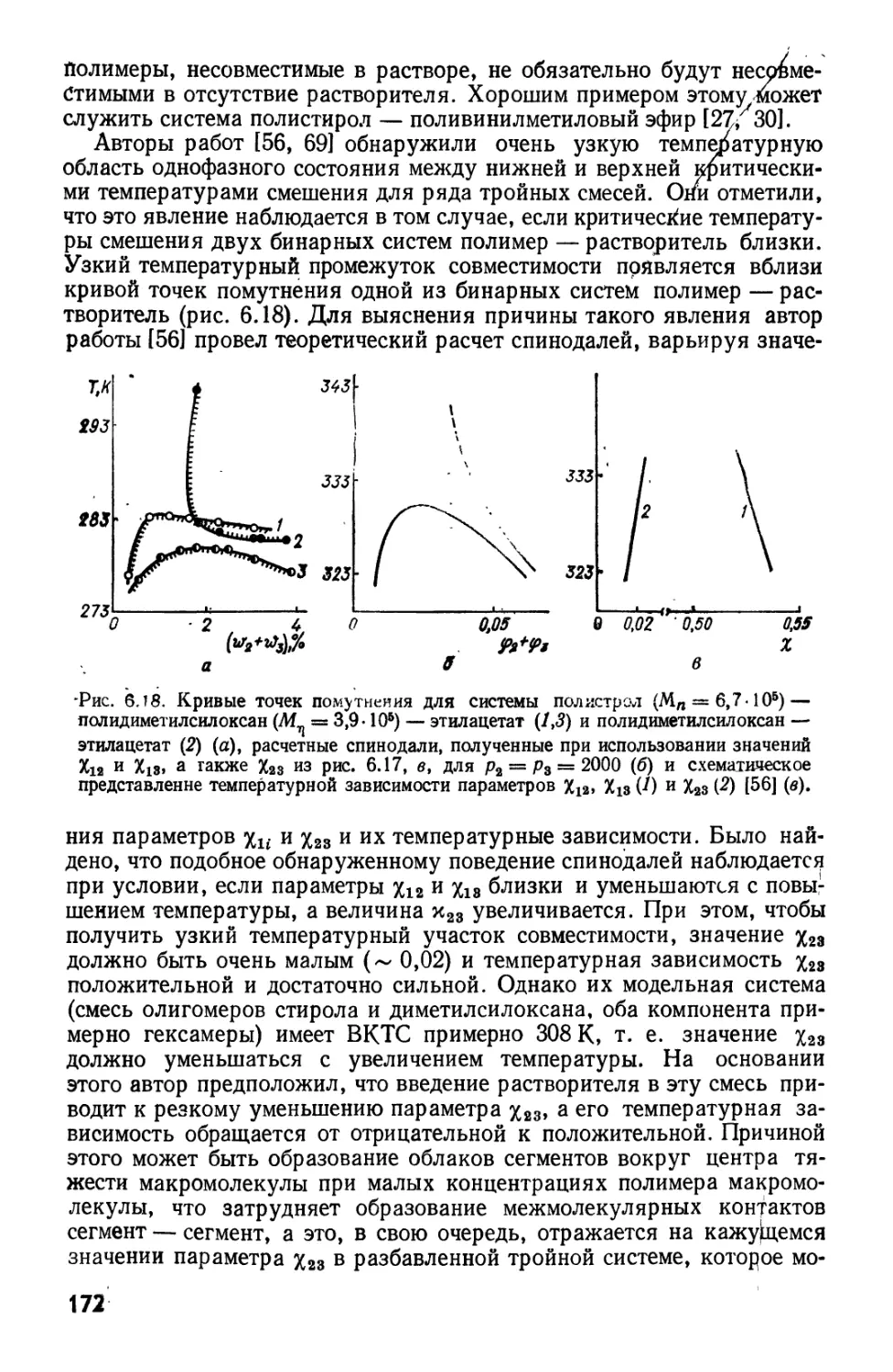

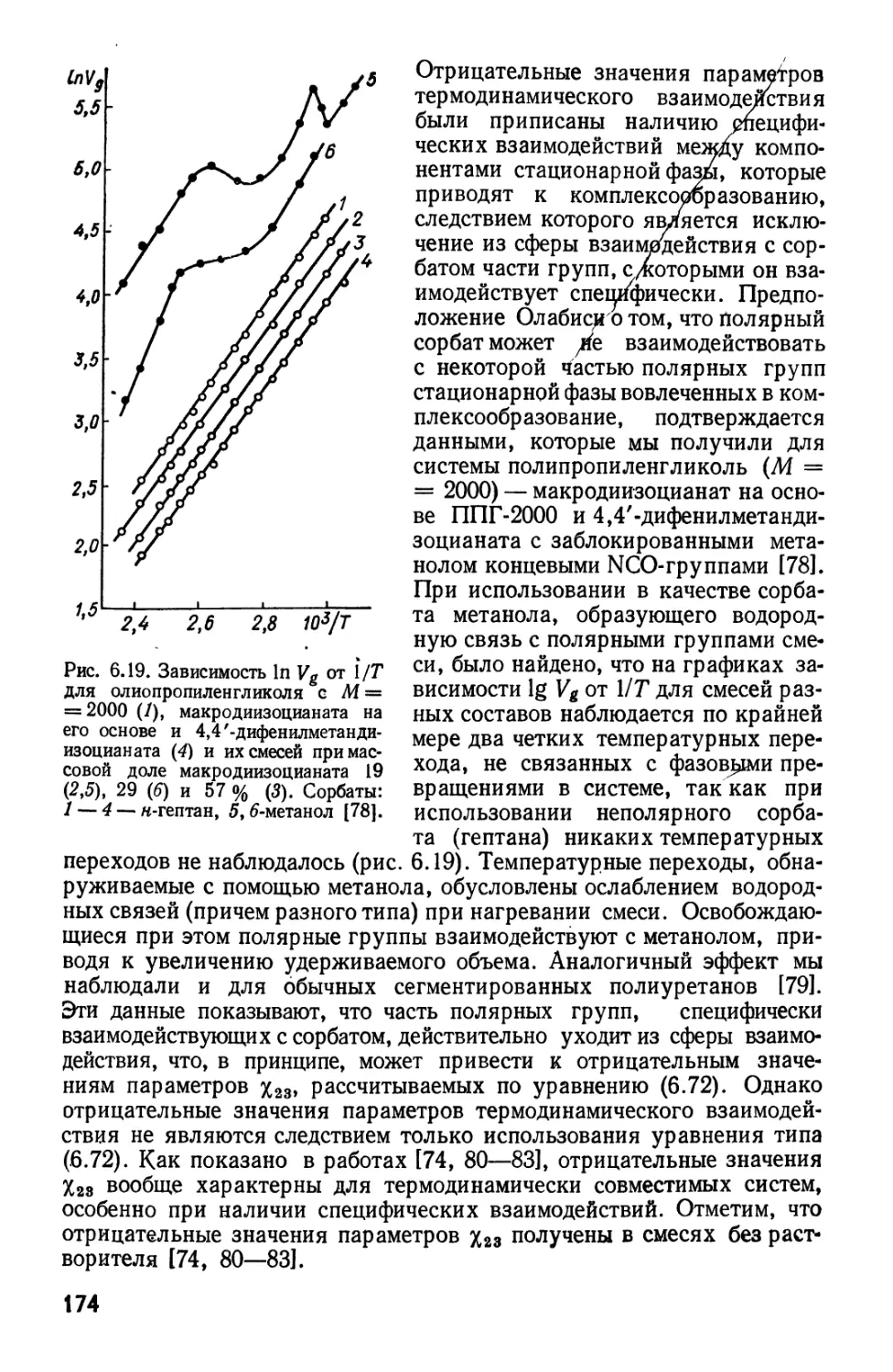













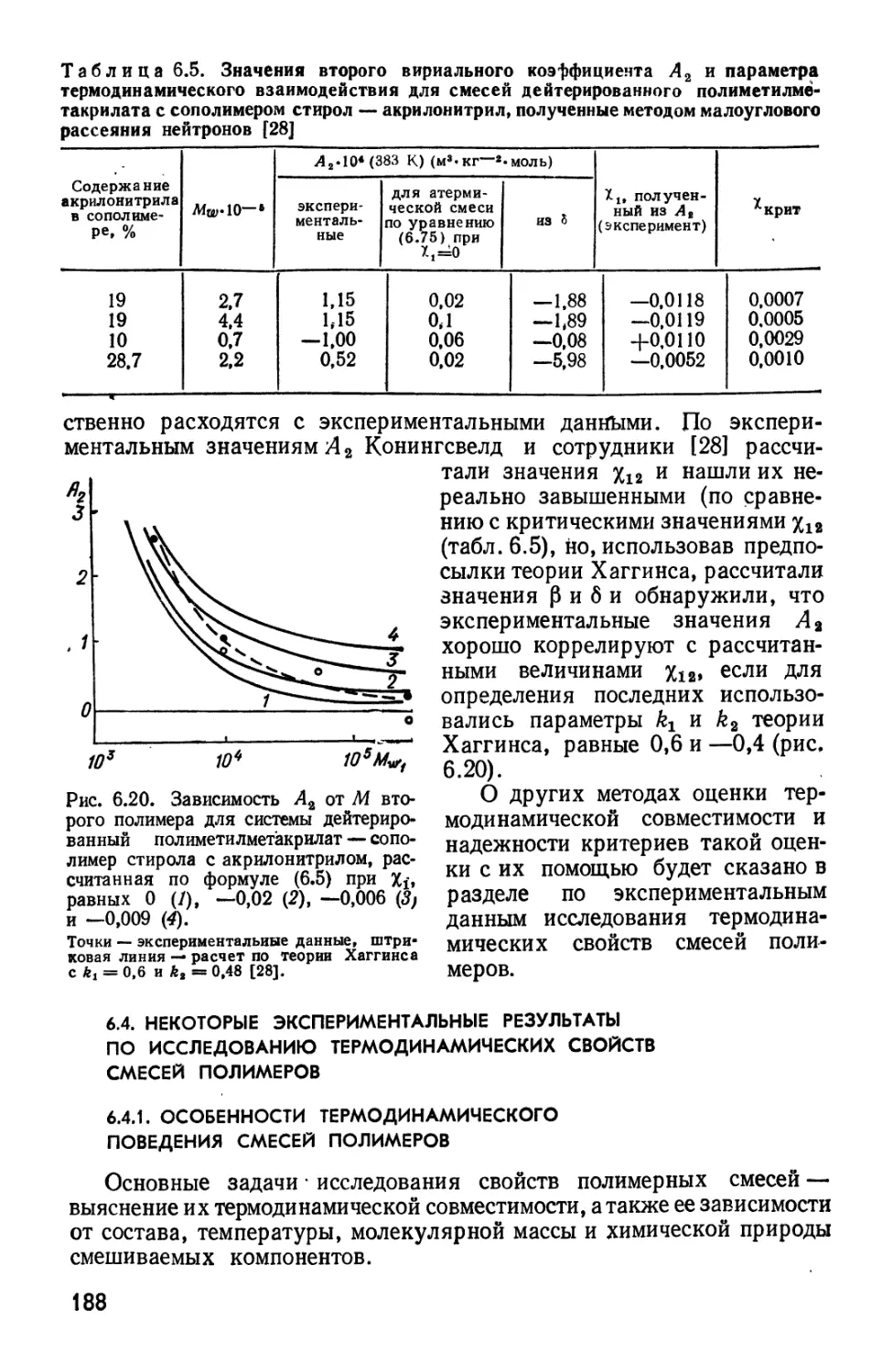















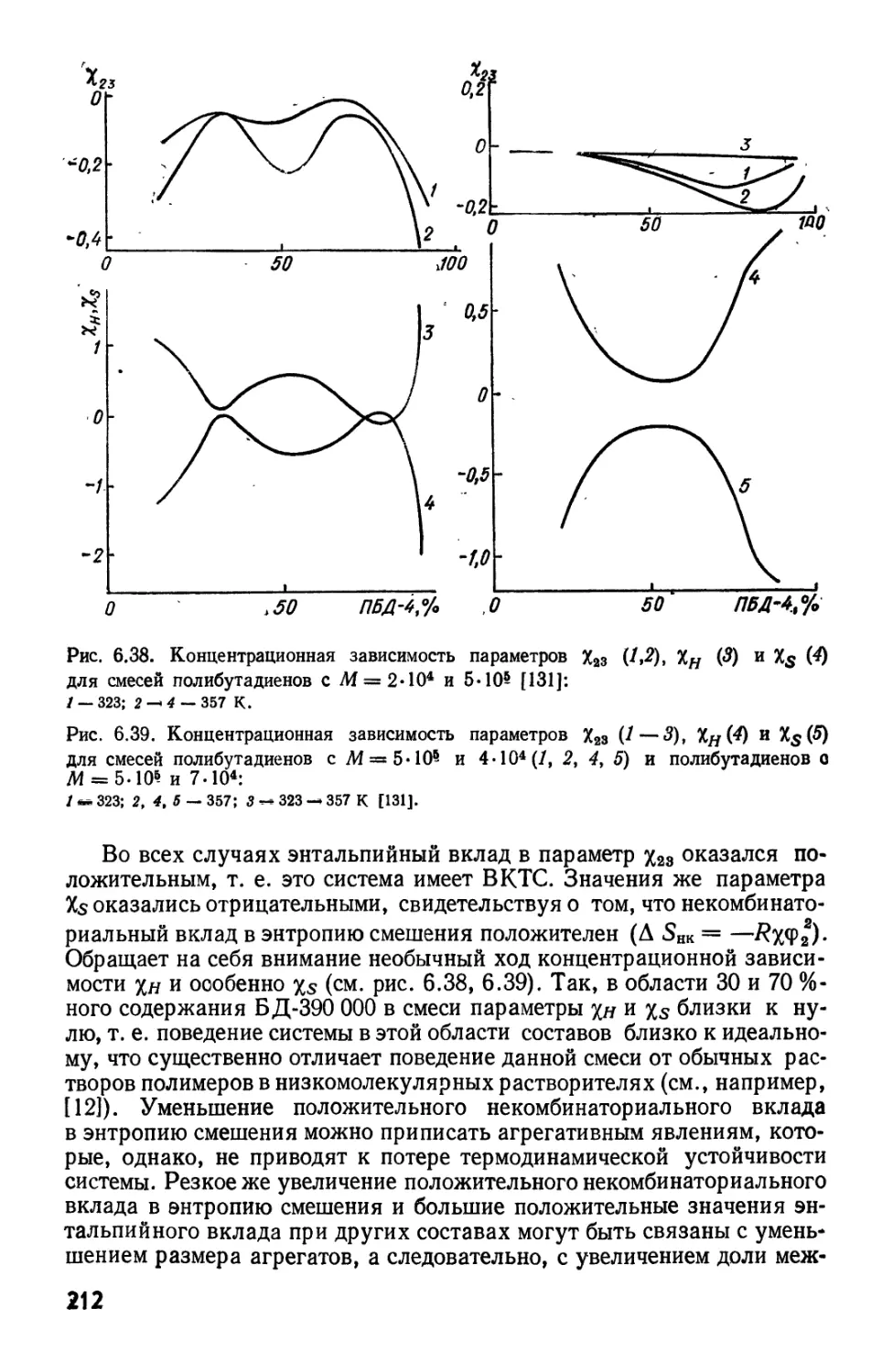


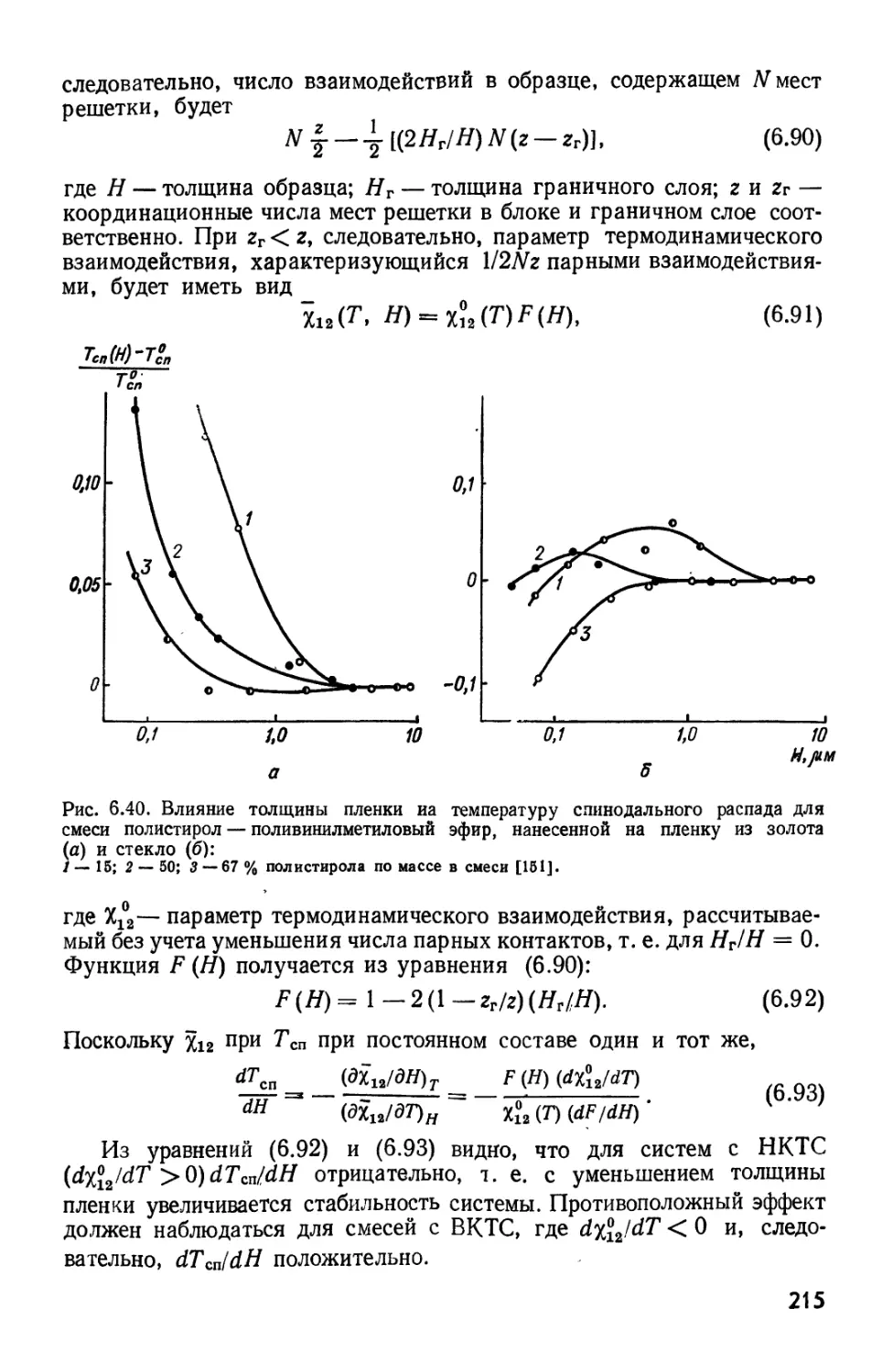









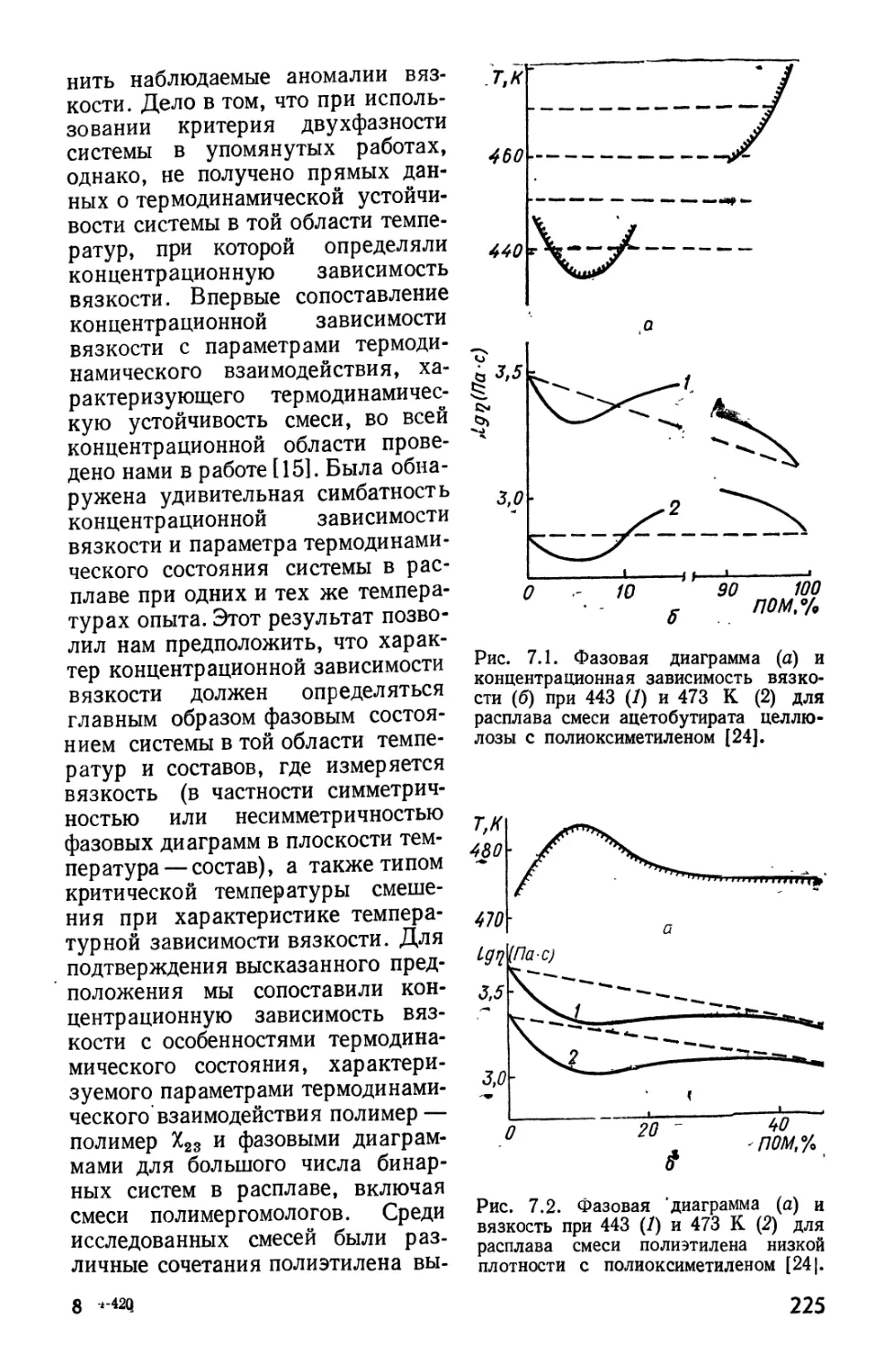
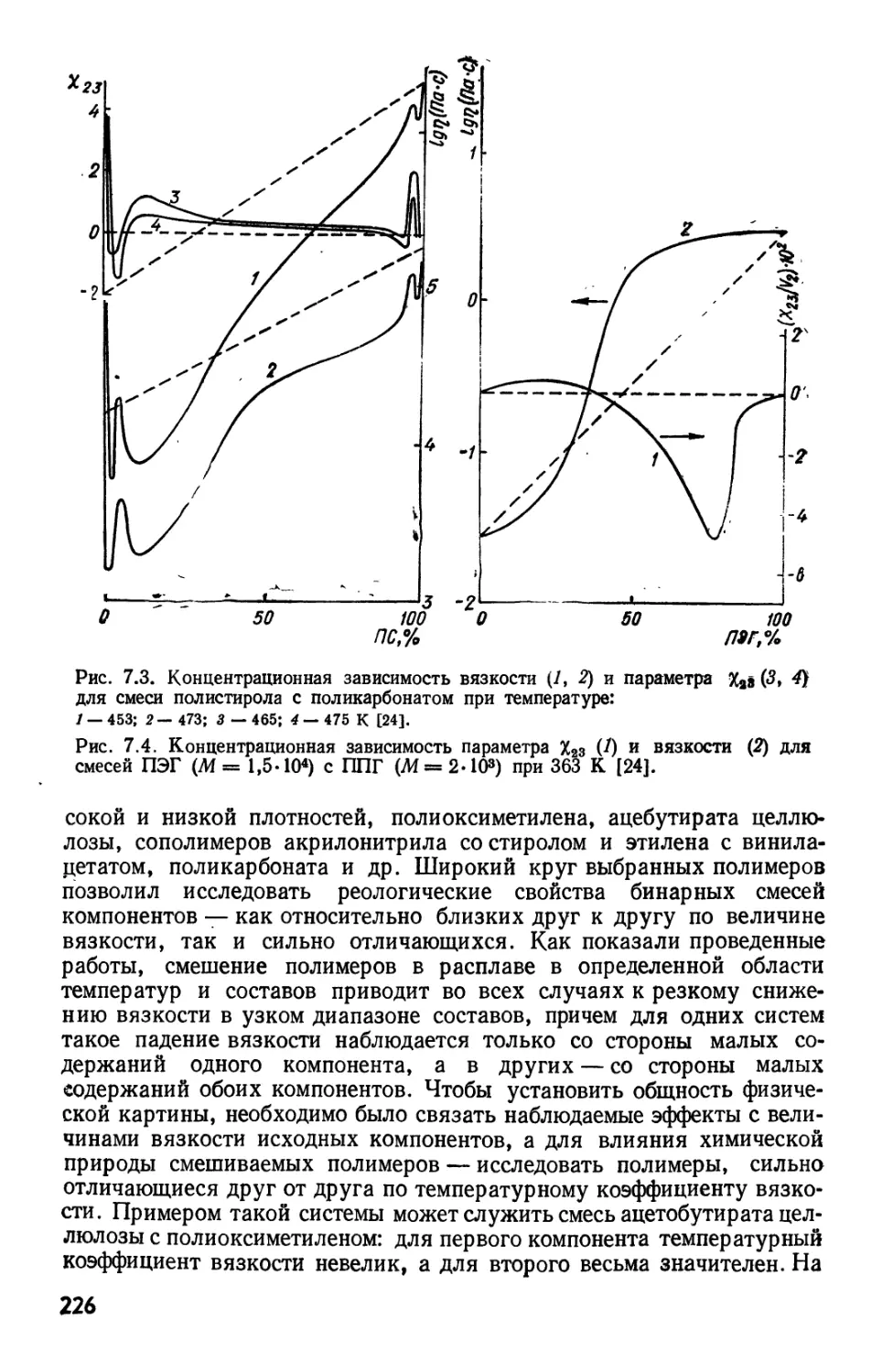











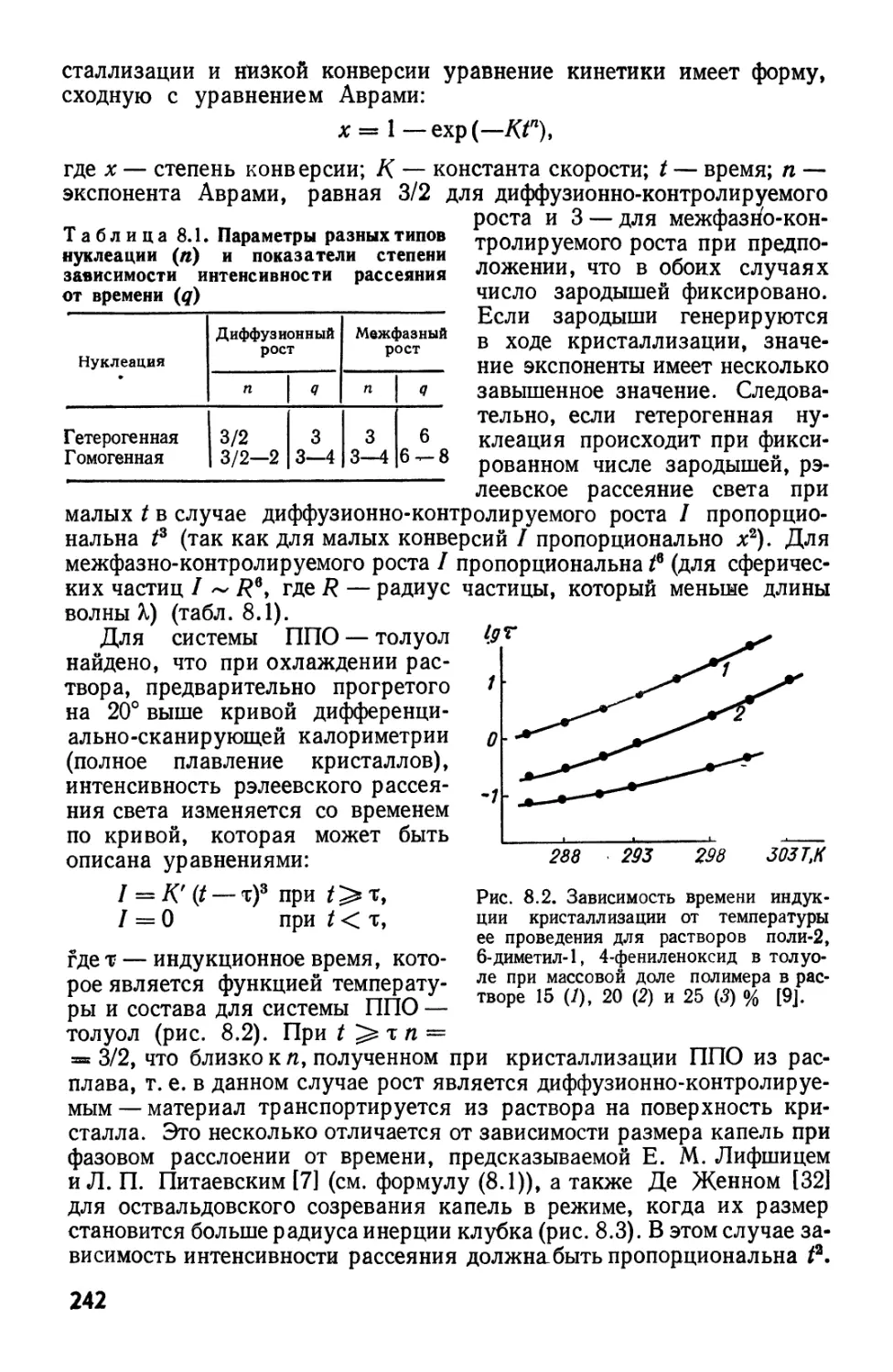










































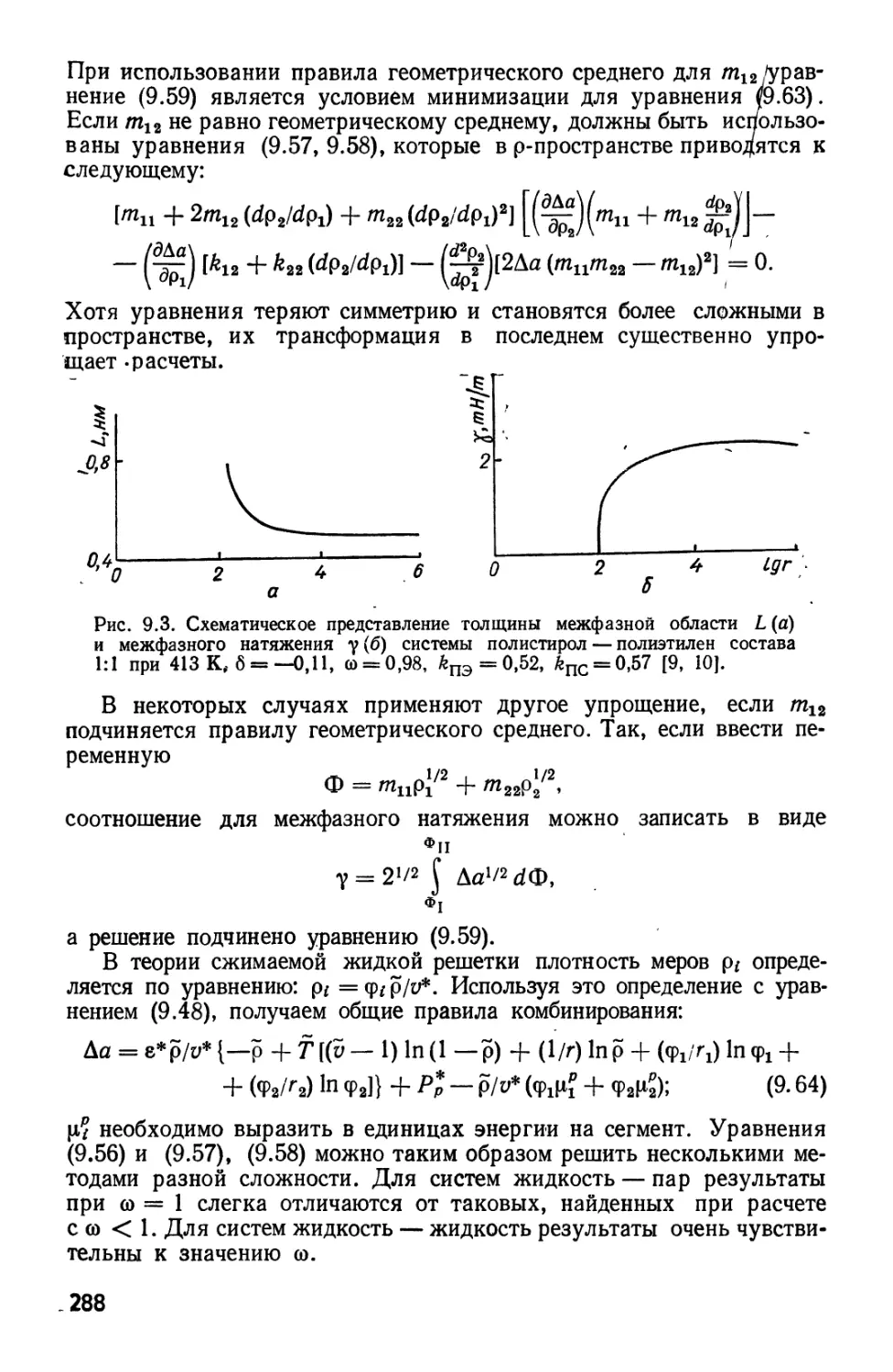




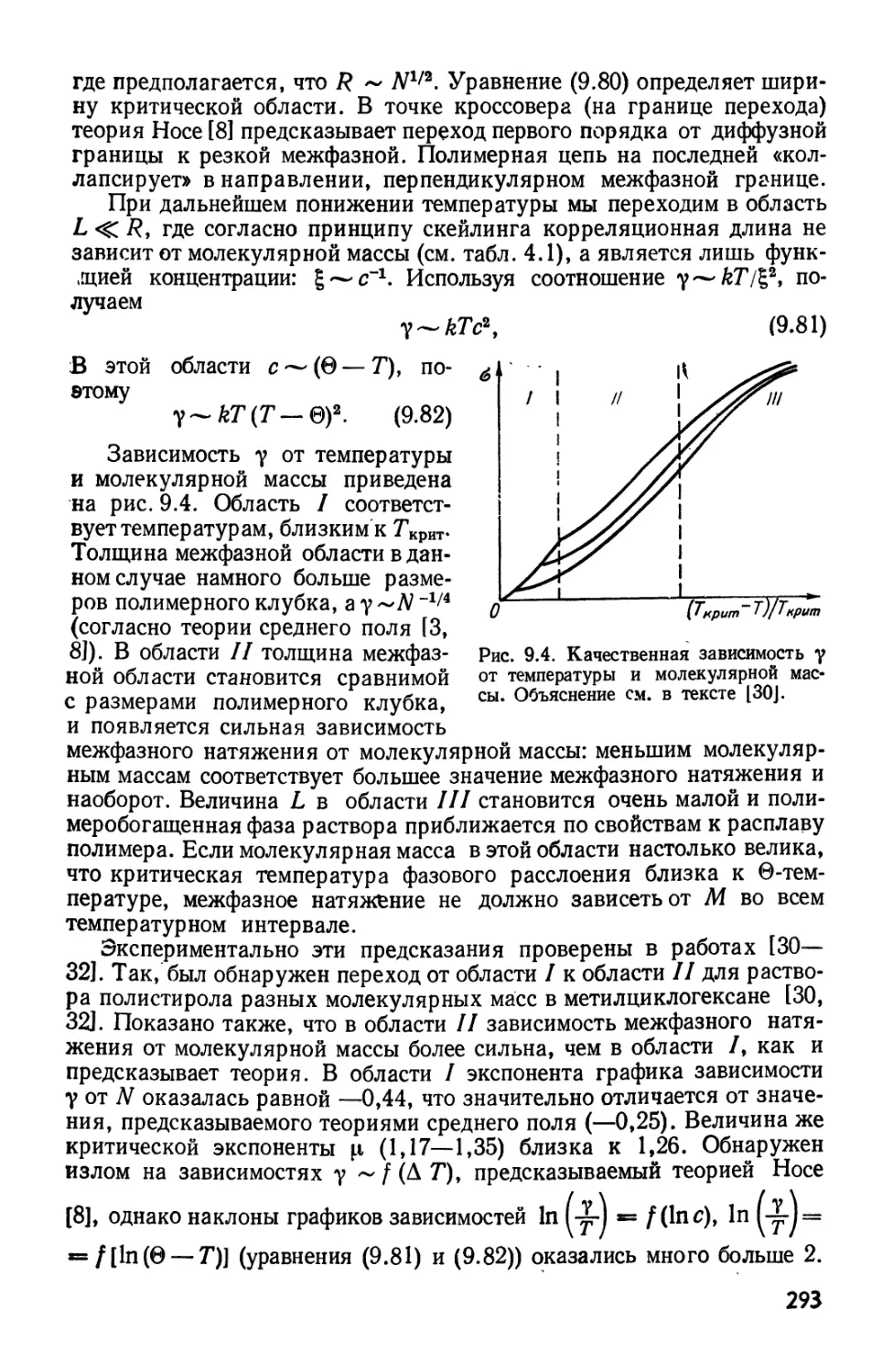






Текст
АКАДЕМИЯ НАУК
УКРАИНСКОЙ ССР
ИНСТИТУТ ХИМИИ
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
А-Е.НЕСТЕРОВ
Ю.С.ЛИПАТОВ
динамика
РАСТВОРОВ
И СМЕСЕЙ
ПОЛИМЕРОВ
КИЕВ НАУКОВА ДУМКА 1984
УДК 541.64
Термодинамика растворов и смесей полимеров /А. Е. Нестеров, Ю. С.
Липатов.—- Киев : Наук, думка, 1984.— 300 с.
В монографии исследовано современное состояние термодинамики
растворов и смесей полимеров. Изложены основные положения классических и новых
статистических теорий растворов полимеров в низкомолекулярных жидкостях
и их применение к описанию термодинамических свойств смесей полимеров.
Рассмотрены современные представления о механизмах фазового
расслоения в растворах и смесях полимеров и теории межфазной границы в
расслаивающихся растворах и смесях полимеров. Приведены многочисленные
экспериментальные данные по исследованию термодинамических свойств растворов и смесей
полимеров.
Для специалистов в области физической химии и физики полимеров.
Ил. 114. Табл. 9. Библиогр. в конце глав.
Ответственный редактор Л. М. Сергеева
Рецензенты А. Ф* Скрышевский, В. П. Привалю
Редакция химической литературы
1807000000^614
Н М221<04)-84 213"84
E)Издательство «Наукова думка», 1984
ПРЕДИСЛОВИЕ
Физическая химия полимеров отделилась от химии
высокомолекулярных соединений первоначально как физико-химия растворов
полимеров. Именно благодаря изучению растворов полимеров были установлены
основные особенности поведения макромолекул в растворах, позволившие
впоследствии перейти к описанию свойств изолированных полимерных цепей и
построить общую теорию структуры полимеров, существенно определяемой конфор-
мационными состояниями цепей.
Интерес к растворам был вызван тем, что в определенный период получение
полимерных материалов осуществлялось преимущественно через стадию
использования растворов (искусственные волокна, покрытия, клеи и пр.). Изучение
свойств растворов стимулировало развитие работ по термодинамике растворов
полимеров, получивших особенно большое развитие после того, как в 1936 г.
В. А. Каргин, С. П. Папков и 3. А. Роговин установили применимость к
растворам полимеров правила фаз и показали, что полимеры способны образовывать
истинные термодинамически устойчивые растворы. Современному читателю
может теперь показаться непонятным, как можно было полагать, что растворы
полимеров не подчиняются общим термодинамическим закономерностям. Но не
надо забывать, что учение о растворах полимеров возникло на базе представлений
о лиофильных коллоидах.
Благодаря отмеченным особенностям развития исследований в области
растворов полимеров соответственным образом рождалась и монографическая
литература. На русском языке первой специализированной монографией,
посвященной растворам полимеров, была книга А. А. Тагер «Растворы
высокомолекулярных соединений» (М.: Госхимиздат, 1951), хотя основные положения теории
растворов полимеров были даны еще в учебнике С. М. Липатова «Физико-химия
коллоидов» (М.: Госхимиздат, 1948). В то же время за рубежом вышла книга Майе-
ра «Синтетические и природные высокополимеры» A942), а в 1953 г. появился сразу
ставший классическим (к сожалению, не переведенный на русский язык) труд
лауреата Нобелевской премии П. Флори «Принципы полимерной химии» (New
York, Cornell univ. press., Ithaca, 1953), содержащий обширное изложение
термодинамики полимеров. Несмотря на выход в свет еще нескольких крупных
работ зарубежных авторов (Tompa H. Polymer solutions.— London: Butterworths,
1956; Huggins M. L. Physical chemistry of nigh polymers.— N. Y., John Wiley
and Sons, 1958; Prigogine I. The molecular theory of solutions.—N. Y.: Interscien-
ce, 1959), литература по растворам полимеров, особенно по термодинамике
растворов полимеров, на русском языке продолжает оставаться довольно скудной.
Небольшое пособие С. С. Воюцкого «Растворы высокомолекулярных соединений»
(М.: ГНТИ химической литературы, 1960), краткое изложение начальных основ
термодинамики растворов полимеров в учебнике А. А. Тагер «Физикохимия
растворов полимеров» C-е изд., перераб.— М.: Химия, 1978), монография С. Р. Ра-
фикова, В. П. Будтова и Ю. Б. Монакова «Введение в физикохимию растворов
полимеров» (М.: Наука, 1978) — это все, чем мы сегодня располагаем.
Между тем за последние годы исследования в области термодинамики
полимеров приобрели интенсивный размах и в нашей стране, и за рубежом. Совершился
отчетливый переход от описания термодинамических свойств макромолекул
в растворах к описанию фазовых равновесий. Этому во многом способствовали
промышленная переориентация и отказ от переработки полимеров в виде
растворов с переходом на переработку расплавов и смесей полимеров в расплаве.
Возникла новая область термодинамики полимеров — термодинамика
совместимости полимеров в расплавах и твердом состоянии, были выполнены крупные
теоретические работы, частично обобщенные в недавно переведенной на русский
язык книге: «Полимерные смеси», т. 1, под ред. Д. Пола и С. Ньюмена
(М.:Мир).
Получили интенсивное развитие исследования в области механизма фазового
разделения не только в простых бинарных смесях линейных полимеров, но и в
таких сложных системах, как взаимопроникающие полимерные сетки, в
химически формирующихся полимерных системах на разной стадии их
развития.Наконец, развитие де Женном концепций скейлинга внесло новую свежую струю в
термодинамику многокомпонентных полимерных систем.
Все это побудило авторов данной монографии предпринять попытку
систематически изложить термодинамику растворов и смесей полимеров с детальным
анализом существующих в настоящее время теорий.
Мы хотим предупредить, что настоящая книга никак не может
рассматриваться как введение в область термодинамики растворов и смесей полимеров,
а предполагает знание читателем не только химической термодинамики вообще»
но также основ физической химии и термодинамики полимеров. Эта книга
представляет собой обширный обзор современного состояния вопроса, существующих
теорий (иногда противоречивых, иногда имеющих некоторые нерешенные
вопросы). Ее достоинство, на наш взгляд,— весьма подробный охват работ,
выполненных в области термодинамики полимеров, особенно за последнее время. Ее
основной недостаток — подробное изложение работ, которые лет через пять — семь
окажутся устаревшими (редко какая работа сохраняет полную значимость в
науке через столь долгий срок — даже Флори пересмотрел многие свои концепции).
Сказанное выше определило принцип написания книги, рассчитанной на
специалистов в области высокомолекулярных соединений, физической химии
и физики полимеров. Она должна помочь читателю ориентироваться в бурно
развивающейся области, так как, основываясь только на многочисленных
оригинальных публикациях, войти в эту область и охватить ее — мы знаем это по
собственному опыту — очень трудно.
При работе над монографией авторы пользовались исключительно
оригинальными работами, не прибегая к помощи специализированных монографий
или обзоров, что позволит найти в книге также обширную библиографию. Вместе
с тем мы избегали приводить в книге большое количество табличных или цифровых
данных, учитывая, что такой материал можно найти в «Справочнике по
физической химии полимеров» (А. Е. Нестеров «Свойства растворов и смесей полимеров»,
г. 1, под ред. Ю. С. Липатова. К.: Наук, думка, 1984).
Разумеется, в книге нашли отражение и результаты собственных
исследований авторов и их сотрудников в области термодинамики растворов, и особенно
смесей полимеров.
Мы понимали, что написание такой книги сопряжено с большими
трудностями и невозможностью избежать тех или иных недостатков и не надеялись,что нам
удалось их избежать, но старались, чтобы их число было не очень велико, и
позволило после прочтения книги заинтересоваться этой очень важной областью
современной физической химии полимеров.
ГЛАВА 1
ОСНОВНЫЕ ПОНЯТИЯ
ТЕРМОДИНАМИКИ РАСТВОРОВ
1.1. ПАРЦИАЛЬНЫЕ ВЕЛИЧИНЫ
В открытых термодинамических системах, т. е. таких, которые
обмениваются массами с окружающей средой, внутренняя энергия
и свободная энергия смещения Гиббса (изобарно-изотермический
потенциал) изменяются с изменением масс компонентов:
t/ = /(S, V, nlf п2, ...),
G = / (Л 7\ п19 п2, ...).
Полные дифференциалы этих величин (dU и dG) выражаются
суммой частных дифференциалов:
dU = TdS — PdV + ii1dn1 + \i2dn2 + ••• = TdS — PdV+ ?
dG = VdP — SdT + ii1dn1 + \i2dn2+ ... =VdP-SdT+ V
где |i; — химические потенциалы компонентов.
Величины Ut = №) , 5, = Ш , Vf = (Л и ^ =
\дгц}р9т% ( \дТ/р,т' 1 \дщ/р,т ri
-g^-J при постоянных Р и Т называются парциальными.
Основное уравнение для парциальных величин таково:
? * = 0 (Л 7 = const).
Поскольку на практике абсолютные значения V\ S, G, а также i/j»
A;, St- не определяются, имеют дело с их разностью при данных
условиях и некотором стандартном состоянии при тех же температурах
и давлениях. Тогда
1.2. ОСНОВНЫЕ ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ
РАСТВОРА. ТЕРМОДИНАМИЧЕСКОЕ СРОДСТВО
МЕЖДУ КОМПОНЕНТАМИ
Основными термодинамическими функциями раствора являются
энтальпия образования раствора Д Н и энтропия A S. Изобарно-
изотермический потенциал (свободная энергия смещения Гиббса) —
функция этих двух величин: Д G = АЯ- Т A S.
При постоянных давлении и температуре растворение —
самопроизвольный процесс, идущий в направлении уменьшения свободной
энергии смешения Гиббса. Следовательно, раствор должен иметь
меньшую свободную энергию, чем сумма свободных энергий компонент:
GP-pa < S nfi°i- Разность AG =* Gp.Pa — X nfi\ называется избыточной
свободной энергией смешения и при самопроизвольном растворении
должна быть отрицательной: AG < 0. Соответственно химический
потенциал компонента в растворе \i{ должен быть меньше его
химического потенциала до растворения |i, < (aJ, т. е. Д^ < 0. Значения
AG<0, A\i{< 0 являются критериями сродства между
компонентами, величина которого определяется абсолютной величиной разностей
AG или A[i?.
1.3. ИДЕАЛЬНЫЕ И НЕИДЕАЛЬНЫЕ РАСТВОРЫ
Идеальный раствор можно представить как смесь компонентов,
молекулы которой одинаково взаимодействуют между собой и свойства
которой складываются аддитивно из свойств компонентов. Для таких
растворов выполняются законы Генри и Рауля:
р2 = Кх, A.1)
Pi = ^(l-*)> A.2)
где рг и /?2— парциальные давления компонент: х — мольная доля
компонента в смеси. Поскольку
Aii^RTlnpt/pl A.3)
то
A.4)
т. е. химический потенциал каждого компонента определяется только
его мольной долей в растворе.
В большинстве случаев растворы не подчиняются уравнениям
A.1)—A.4) и называются неидеальными. Отклонения от
идеальности (—¦?- = #,] могут быть положительными (—->хЛ и отрица-
\*! / \Р\ 1
(р \
-~- < хЛ. Для оценки степени отклонения от идеальности
вводится понятие об избыточных величинах — избытках свойств над
идеальной величиной
А^Г6 = А^ — д^?д = RT In У г
bsf* = 5, - sr = -if in Tl - rt
где yt — коэффициент активности компонента i в реальном растворе
(yi = ai/xi), если активность а{ выразить через RT \na{ = \i? — \10^ '
Тогда
ДСиз6 = Yixi ^^Г6 = КГ 1С xi 1П
^j #/Д«з* = к T^Xj in 7/ —
1.4. РЕГУЛЯРНЫЕ РАСТВОРЫ. УРАВНЕНИЕ
ГИЛЬДЕБРАНДА — СКЕТЧАРДА
Раствор, при образовании которого А Н Ф О, Гильдебранд назвал
регулярным. При этом принимается, что распределение молекул
смешиваемых компонентов совершение беспорядочное, т. е. энтропия
смешения равна идеальной энтропии смешения, а смешение
компонентов происходит без изменения объема. Энергия смешения чистых
жидких компонентов А Есы равна разности между межмолекулярной
энергией моля раствора Яр.ра и суммой межмолекулярных энергий
чистых жидких компонент Е}.
00
/ 2 С 2 Л
Д?см = 2nN0VqIqJ ( \ e12p12>* ar —
мо
ОО 00
If 1 Г \
2~ \ 8llPlir2^ Г \ ?22Р22Г2^Ч > (*'5)
* X t/ » 2 t) I
О О
где V — объем моля раствора; еш е22 и е12— взаимные энергии двух
молекул для разных пар молекул; рп, р22 и р12 — вероятности того,
что случайно выбранная в элементе объема молекула окажется
молекулой 1-го, 2-го и т. д. рода; г — расстояние от центральной,
произвольно выбранной молекулы; ф3 и ф2— мольно-объемные доли
компонентов, постоянные в массе раствора: фх= Vx(l—xyiV^l — х) +
+ V2x]\ ф2= У2х1[Уг{\ — х) + V2x\. Используя выражение для e{f
к
в виде —jf и правило геометрического среднего k12 = {knkl2)^2r
уравнение A.5) получаем в виде
Д?см = Уф1ф2 [(E\IVJ12 (E°2/V.2)l/2]\ A.6)
Это и есть уравнение Гильдебранда — Скетчарда для регулярных
растворов. Отношение E?/Vt называется плотностью энергии когезии
компонентов, а величина б = (Я,/7J1/2 была названа Гильдебрандом
коэффициентом растворимости или «параметром растворимости»
компонента. Разность величин, заключенных в квадратные скобки в
уравнении A.6), определяет отклонение раствора от идеального поведения.
Одним из недостатков теории регулярных растворов является
допущение о независимости расположения молекул в растворе от
теплового эффекта, т. е. допущение идеальной энтропии смешения при
ДЯ^О. Следовательно, эта теория пригодна лишь для растворов
компонентов, молекулы которых малополярны, имеют молекулярное
поле шаровой симметрии и смешиваются с незначительным изменением
объезда.
Учет неравномерности распределения молекул компонентов лег
в основу теории строго регулярных растворов. Теория
таких растворов основана на решеточной модели, в которой приняты
следующие допущения:
1) структура считается квазикристаллической (ближний порядок),
частицы жидких компонентов и растворов имеют одно и то же
координационное число г;
2) взаимодействие учитывается только между ближайшими
молекулами;
3) потенциальная энергия разбивается на конфигурационную
(энергия размещения всех молекул по узлам решетки) и
акустическую (энергию колебаний молекул около центра равновесия).
Колебательная энергия молекулы не зависит от того, находится она в
чистой жидкости или в растворе;
4) каждая молекула занимает одно место в решетке (следовательно,
молекулы должны иметь одинаковый объем и форму); потенциальная
энергия обмена молекул различных жидкостей местами берется в виде
При обмене местами рвутся г связей 1—1 с энергией Еп и г связей
2—2 с энергией ?яа, а образуется 2z связей с энергией ?12. Если
энергия взаимообмена равна нулю, то система является атермической,
если не равна нулю — неатермической.
Нулевое приближение теории строго регулярных растворов
приводит к выражениям
Д# = #0 A - *Цда,12 _ Г
AG = Nox(l —x)Aw12.
1.5. ФАЗОВОЕ РАВНОВЕСИЕ В РАСТВОРАХ.
СПИНОДАЛИ И БИНОДАЛИ
Невозмущенная равновесная система состоит только из одной фазы.
Возмущение системы приводит к появлению в ней небольшого
количества новой фазы. Интенсивные свойства этой новой фазы (парциальный
мольный объем, состав и др.) могут отличаться от свойств равновесной
системы либо на бесконечно малую, либо на конечную величину. При
этом могут возникать следующие варианты1: 1) исходная фаза устой-
1 Пригожий #., Дефэй Р. Химическая термодинамика.—Новосибирск?
Наука, 1966.—509 с.
8
чива по отношению ко всем другим, независимо от того, отличаются
они от нее по своим свойствам на бесконечно малую или на конечную
величину; 2) исходная фаза устойчива по отношению ко всем
бесконечно мало отличающимся от нее фазам, но в системе имеется по
крайней мере одна фаза, по отношению к которой исходная фаза
неустойчива; 3) исходная фаза неустойчива по отношению к фазам, бесконечно
мало отличающимся от нее.
В первом случае фазу называют устойчивой (стабильной), во
втором — метастабильной, в третьем — неустойчивой (нестабильной).
Если фаза неустойчива по отношению к фазам, бесконечно мало
отличающимся от нее, она исчезнет и возникнет одна или несколько
близких к ней новых фаз. Этот процесс будет продолжаться до тех
пор, пока не образуется фаза, устойчивая по отношению к
примыкающим к ней фазам.
При наличии метастабильных фаз система неопределенно долго
остается в равновесии, и новая фаза в ней не появляется. Если же в
систему внести зародыши новой, более устойчивой фазы, происходит
переход в стабильное равновесное состояние.
В случае растворов устойчивость системы зависит от диффузии,
приводящей к появлению неоднородности по составу в первоначально
однородной двойной системе. Согласно [1], для всех систем,
устойчивых по отношению к диффузии, d2g/dx2i>0 (g — мольная свободная
энергия Гиббса). Это условие эквивалентно условию: д\х1/дх1 > 0.
Поскольку
д^/дх2 = — х^8/дх1 A.7)
то
д\хь/дх2<0 A.8)
и, следовательно,
дрл/дхг<0. A.9)
Эти условия равновесия по отношению к диффузии
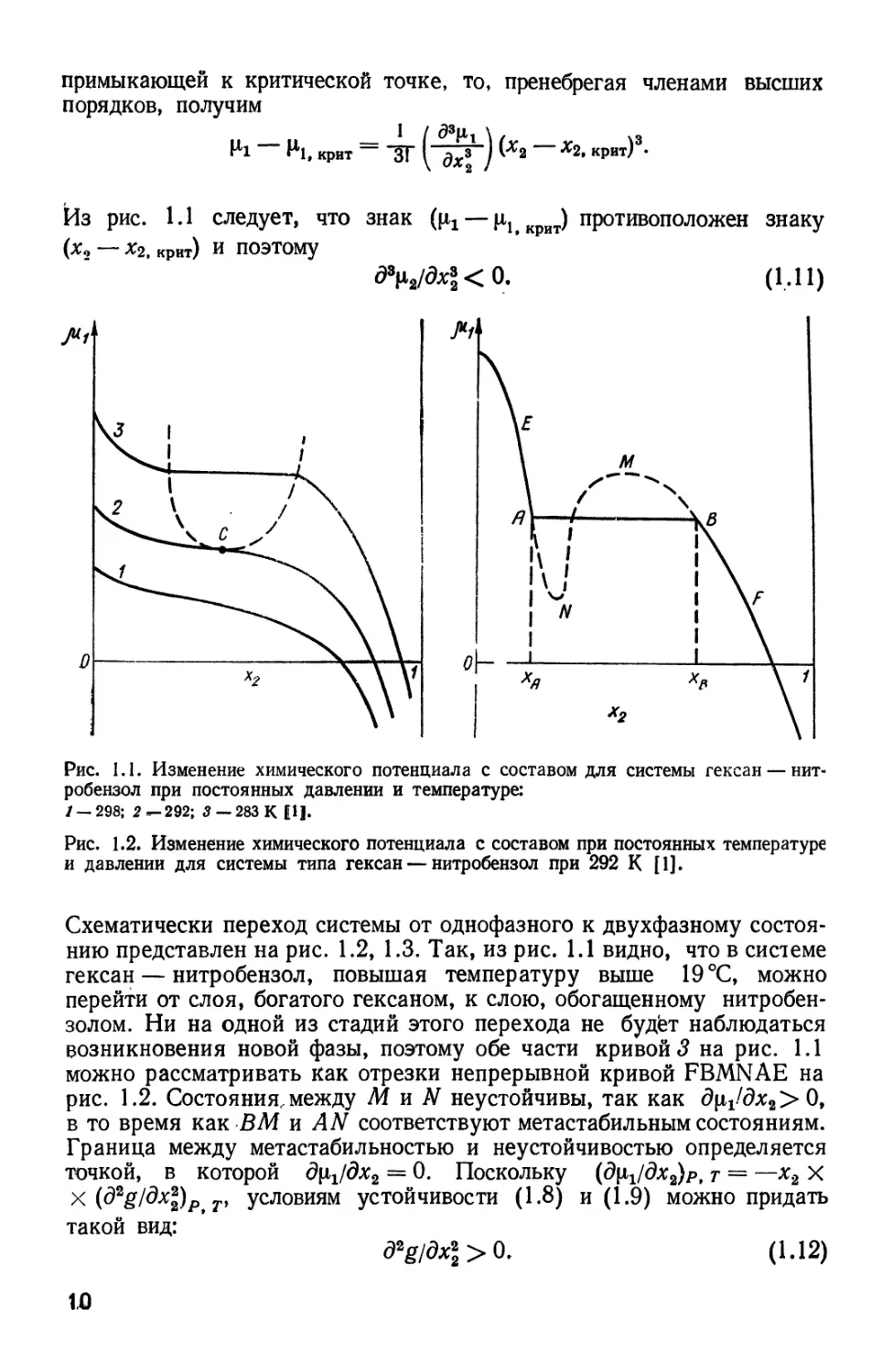
проиллюстрированы в [1] на примере системы гексан — нитробензол. Если для
нескольких температур изобразить химический потенциал гексана как
функцию мольной доли нитробензола при постоянном давлении,
получится семейство кривых (рис. 1.1). Выше 19 °С (кривая 1) имеется
только одна фаза, и условия A.8), A.9) всегда выполняются. Ниже 19°С
кривая (например, кривая 3) состоит из трех участков: первый
соответствует слою, богатому нитробензолом, второй — слою, богатому
гексаном, третий — горизонтальная прямая, соединяющая эти
участки, характеризует одновременное наличие двух фаз. Кривая при 19 °С
образует границу между этими двумя типами кривых. Горизонтальный
отрезок на ней выродился в одну точку перегиба С, характеризуемого
условиями
^O. A.10)
Критическое состояние устойчиво, так как (д*\12/дх1) < 0.
Действительно, если химический потенциал \ix разложить в ряд в области,
9
примыкающей к критической точке, то, пренебрегая членами высших
порядков, получим
**1 - 1*1. крит = Ж (ifcjr) (*» - **- )8
Из рис. 1.1 следует, что знак (\хг— |*ltKpHT) противоположен знаку
(*о — Х2, крит) И ПОЭТОМУ
O. A,11)
Рис. 1.1. Изменение химического потенциала с составом для системы гексан —
нитробензол при постоянных давлении и температуре:
/ — 298; 2^292; 3 — 283 К 1Ц.
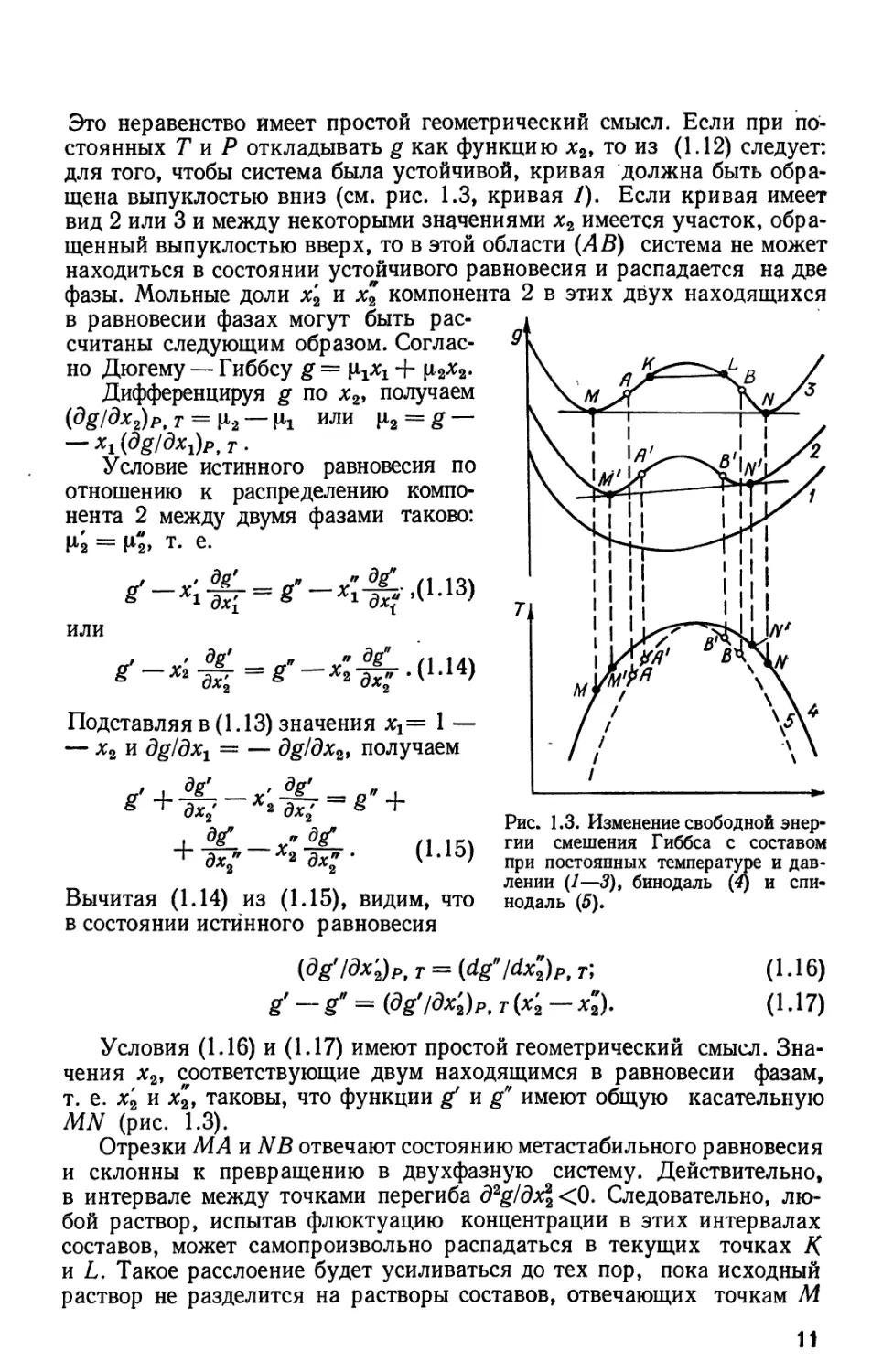
Рис. 1.2. Изменение химического потенциала с составом при постоянных температуре
и давлении для системы типа гексан — нитробензол при 292 К [1].
Схематически переход системы от однофазного к двухфазному
состоянию представлен на рис. 1.2, 1.3. Так, из рис. 1.1 видно, что в системе
гексан — нитробензол, повышая температуру выше 19 °С, можно
перейти от слоя, богатого гексаном, к слою, обогащенному
нитробензолом. Ни на одной из стадий этого перехода не будет наблюдаться
возникновения новой фазы, поэтому обе части кривой 3 на рис. 1.1
можно рассматривать как отрезки непрерывной кривой FBMNAE на
рис. 1.2. Состояния,между М к N неустойчивы, так как д\х1/дх2>09
в то время как ВМ и AN соответствуют метастабильным состояниям.
Граница между метастабильностью и неустойчивостью определяется
точкой, в которой d^Jdx2 = 0. Поскольку (dii1/dx2)pf т = —х2 X
X (d2g/dxl)pr условиям устойчивости A.8) и A.9) можно придать
такой вид:
>0. A.12)
10
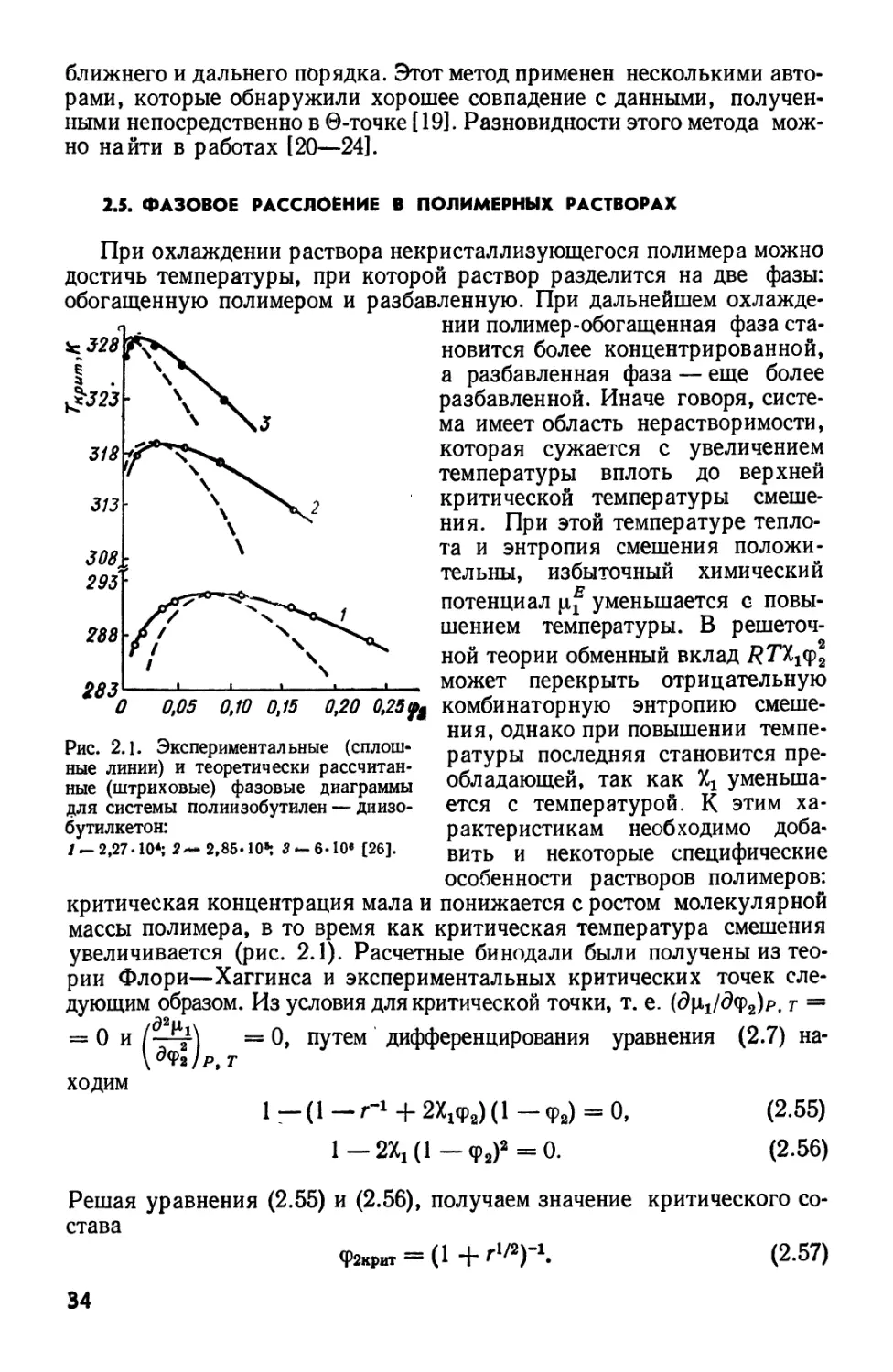
Это неравенство имеет простой геометрический смысл. Если при
постоянных Т и Р откладывать g как функцию х2у то из A.12) следует:
для того, чтобы система была устойчивой, кривая должна быть
обращена выпуклостью вниз (см. рис. 1.3, кривая 1). Если кривая имеет
вид 2 или 3 и между некоторыми значениями х2 имеется участок,
обращенный выпуклостью вверх, то в этой области (АВ) система не может
находиться в состоянии устойчивого равновесия и распадается на две
фазы. Мольные доли х'2 и х2 компонента 2 в этих двух находящихся
в равновесии фазах могут быть
рассчитаны следующим образом.
Согласно Дюгему — Гиббсу g = H^i + Щ*2-
Дифференцируя g по х2, получаем
{dgldx2)Pt т = М-2 — hi или ^2 = ? —
— *i {dgldx^p, т .
Условие истинного равновесия по
отношению к распределению
компонента 2 между двумя фазами таково:
\i'2 = \i2, т. е.
или
.A.14)
Подставляя в A.13) значения хх= 1 —
— *2 и dgldx-L = — dgldx%, получаем
d*2'
дх;
A.15)
Вычитая A.14) из A.15), видим, что
в состоянии истинного равновесия
Рис. 1.3. Изменение свободной
энергии смешения Гиббса с составом
при постоянных температуре и
давлении (/—3), бинодаль D) и спи-
нодаль E).
(dg'/dx'2)Pt т = {dgn/dx)P,
A.16)
A.17)
Условия A.16) и A.17) имеют простой геометрический смысл.
Значения х2, соответствующие двум находящимся в равновесии фазам,
т. е. х2 и х%% таковы, что функции g' и g" имеют общую касательную
MN (рис. 1.3).
Отрезки МА и NB отвечают состоянию метастабильного равновесия
и склонны к превращению в двухфазную систему. Действительно,
в интервале между точками перегиба d2gldx\<0. Следовательно,
любой раствор, испытав флюктуацию концентрации в этих интервалах
составов, может самопроизвольно распадаться в текущих точках К
и L. Такое расслоение будет усиливаться до тех пор, пока исходный
раствор не разделится на растворы составов, отвечающих точкам М
11
и N. Геометрическим местом точек М и ЛГ, N и N' и т. д. для кривых
g = f (x2), построенных при разных температурах, является кривая 4,
названная бинодалью, разграничивающая области устойчивых
и метастабильных состояний системы. В каждой ее точке d2g/dx22> О,
a \i\= fJtg. Геометрическим местом точек Л и Л', В и В' и т. д. является
кривая 5, названная спинодалью, разделяющая области
метастабильных и неустойчивых состояний. В каждой ее точке d2g/dxl = 0.
В связи с условием A.7) выражениям A.10) и A.11),выполняющимся
в критической точке, можно придать вид
0. A.18)
ГЛАВА 2
КЛАССИЧЕСКИЕ ТЕОРИИ
РАСТВОРОВ ПОЛИМЕРОВ
С точки зрения общей теории растворов
особенно интересными являются растворы полимеров. Молекулы
полимера в растворе представляют собой длинную цепочку из одинаковых
сегментов, которые во многих случаях по химической природе и
взаимодействию близки к молекулам низкомолекулярного растворителя.
Естественно поэтому, что в случае одинаковой химической природы
сегментов полимера и молекул растворителя энергия взаимодействия между
молекулой полимера и растворителя в таком растворе будет близка
к таковой в растворе низкомолекулярных компонент. Следовательно,
теплота смешения здесь будет близка к нулю. Растворы же с такими
теплотами смешения относятся к классу атермических, т. е.
растворам, термодинамические свойства которых определяются изменением
энтропии при смешении, в отличие от регулярных растворов с A SH3E,
равной нулю.
Для атермических растворов, образованных компонентами с
молекулами, сильно различающимися по размерам (а такими растворами
и являются растворы полимеров в низкомолекулярном растворителе),
энтропия смешения не равна идеальному значению. Во многих
случаях в таких растворах величина A SHs6 очень велика, что
соответствует большим отклонениям от идеальности.
Поскольку атермические растворы — одни из простейших,
теоретические расчеты сначала были выполнены для атермического
смешения полимера с низкомолекулярной жидкостью. Впервые такие
расчеты выполнили независимо друг от друга Флори и Хаггинс.
Первая теория Флори—Хаггинса сыграла большую роль в
понимании процессов растворения высокомолекулярных соединений и стала
базой, на основе которой были развиты другие статистические теории
растворов полимеров.
2.1. ТЕОРИЯ ФЛОРИ — ХАГГИНСА
Основы статистической термодинамики растворов полимеров были
заложены в начале 40-х годов независимо друг от друга Флори [1—3]
и Хаггинсом [3], которыми была получена формула для энтропии
смешения, известная как комбинаторная энтропия Флори—Хаггинса и
играющая в термодинамике растворов полимеров такую же роль, как
уравнение Ван дер Ваальса для реальных газов.
13
2.1.1. ЭНТРОПИЯ СМЕШЕНИЯ
Для вычисления энтропии Флори и Хаггинс использовали
решеточную модель жидкости (раствора), которую для растворов
полимеров видоизменили так, чтобы молекула полимера занимала ряд
примыкающих друг к другу ячеек решетки, тогда как молекула
растворителя — только одну. При этом в ячейке помещали один из отрезков
(сегментов) макромолекулы, величину которого выбирали одинаковой
с размером молекулы растворителя. Далее рассчитывали число конфор-
маций, доступных каждой цепи, путем последовательного введения
в решетку сегментов цепи полимера. Так, если в решетке уже имеется
N цепей молекул полимера или эквивалентное число молекул
растворителя, то для остальных элементов остаются свободными (п0—rN)
ячеек. Следовательно, первый сегмент (N + 1)-ой цепи может быть
расположен (п0— rN) способами. Второй сегмент при закрепленном
первом расположится тогда [г (п0 — rN)/no\> третий [(z — 1) (п0 —
— rN)/n0] способами и т. д. Общее число возможных конформаций,
которые может принять цепь из г сегментов, определяются так:
v _ 1 (п0 - rN) z (п0 - rN) (z - ly-» К - rNY~*
или — после преобразований —
Далее рассчитывается термодинамическая вероятность состояния
раствора W, выражаемая как произведение числа способов размещения
всех N цепей и всех молекул растворителя:
N
W — П v B.2)
Здесь N\— число перестановок между цепями полимера или между
молекулами растворителя — устраняется, так как они не дают новых
способов размещения. Подставляя уравнение B.2) в B.1), применяя
формулу Стерлинга и делая ряд преобразований, получаем
Логарифмирование уравнения B.3) приводит к значению энтропии
раствора — Sp_pa. Аналогичный расчет проводится для получения
числа конформаций в исходном состоянии,т. е. для чисто аморфного
полимера. В общем случае энтропия аморфного вещества
рассчитывается по уравнению B.3) в предположении,что на решетке расположены
только цепи полимера (п = 0), либо только молекулы растворителя
(N = 0). В этом случае
AS = k(r— 1) N [In B—1) — l] — k In 2.
14
Тогда энтропия смешения выражается уравнением
где Nrl(n + Nr) и п/(п + rN) приравниваются объемным долям
полимера и растворителя соответственно, т. е. <р2 и <рх. Для одного
моля мест решетки
&SM = — R (ф! In фх + <ряг* In ф2). B.4)
При этом предполагается, что молекулы располагаются в решетке
раствора совершенно хаотично, смешение происходит без изменения
объема, а величина г определяется как отношение молярных объемов
полимера и растворителя г = У2/Уг. Расчет этих авторов был сделан
для атермического раствора.
2.1.2. ТЕПЛОТА И СВОБОДНАЯ ЭНЕРГИЯ СМЕШЕНИЯ.
ХИМИЧЕСКИЙ ПОТЕНЦИАЛ И ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ РАСТВОРА
Для неатермических смесей в решеточной модели теплота смешения
рассчитывается как обменный вклад, который просто добавляется
к комбинаториальной энтропии смешения. Наличие теплоты смешения
обусловлено различием в суммарной энергии межмолекулярных
взаимодействий в чистых компонентах и в растворе. Поскольку силы
взаимодействия в жидкостях между незаряженными молекулами быстро
убывают с расстоянием, то при рассмотрении парных взаимодействий
в обычных жидкостях можно ограничиться учетом лишь
непосредственных соседей данной молекулы. Пусть wn, иу22 и w12— энергии
взаимодействия соответствующих пар молекул. При образовании одного
контакта типа 1—2 (полимер — растворитель) изменение энергии
записывается так:
A ( + a>) w
Следовательно, теплота смешения
?де р12— число контактов типа 1—2. Поскольку каждая молекула
растворителя имеет гф2 соседних молекул полимера, а всего таких
молекул Nl9 то
Флори ввел безразмерный параметр
. B.5)
характеризующий избыточную энергию взаимодействия в
растворе, приходящуюся на одну молекулу растворителя (kT%1 представляет
собой разность энергий молекулы растворителя, погруженной в поли-
15
мер и в чистый растворитель). Тогда теплоту смешения Nt молекул
растворителя с N2r сегментами можно определить из уравнения
&НМ = zAw12N^2 = kTx^^.
Для одного моля мест решетки изменение свободной энергии при
смешении будет равно
AGM/RT = фх In ф! + W1п Фг + ЭЬФхФ*- B-6)
Дифференцирование уравнения B.6) приводит к следующей величине
химического потенциала для растворителя:
Ih = Й + RT fin A - Фз) + A - г-*) Фз + Xi922]. B.7)
Поскольку активность растворителя равна exp[(fij—
/=з
при разложении логарифма в уравнении B.7) в ряд по ф2. Легко по-
к азать, что — ф2/г асимптотически приближается к нулю при
бесконечном разбавлении (Хг-+ 1). Таким образом, при бесконечном
разбавлении растворы подчиняются закону Рауля, а избыточный
химический потенциал fif зависит только от второй и высших степеней
концентрации. С этой точки зрения полезно определить другой избыточный
потенциал: jif = ЯТ[\паг + Ф2/г], без рассмотрения частной формы
lnav Он отличается от p,f членами ф2,//-, получающимися при
разложении 1пХ1#
Поскольку осмотическое давление я = —(|хх — \x°)/Vv то произведя
замену в B.7): ф2 = с2д, щ/гУ1 = cvlrV1 = с2/М (где v—удельный
парциальный объем полимера, а М — его молекулярная масса),
получим
[±D) D)] B.8)
Неидеальность раствора отражена в этом уравнении наличием членов,
следующих в квадратных скобках за 1/М.
2.2. РАЗБАВЛЕННЫЕ РАСТВОРЫ ПОЛИМЕРОВ
Комбинаторный расчет, приводящий к уравнению B.4), очевидно»
приемлем лишь для концентрированных растворов полимеров,
поскольку только в этом случае распределение сегментов в растворе
приближается к однородному на микроскопическом уровне. Это
справедливо, когда участки цепей различных макромолекул в растворе
переплетены.
16
Для очень разбавленных растворов полимеров решеточная модель
должна быть неадекватной, так как в таких растворах набухшие мак-
ромолекулярные клубки находятся на расстояниях, превышающих
их размеры, вследствие чего существуют области, резко
различающиеся по концентрации полимерных сегментов: одни области раствора
содержат чистый растворитель, другие — набухшие макромолекуляр-
ные клубки.
Эта неадекватность решеточной модели привела Флори в конце
40-х годов к формулировке теории разбавленных растворов, которая
учитывает неоднородную структуру раствора. В обоих трактовках
макромолекулярный клубок характеризуется сферически симметричным
распределением сегментов относительно его центра тяжести. В первой
модели предполагается однородная плотность сегментов в объеме
клубка [4], а во второй — более реалистичное гауссовское распределение
сегментов около центра масс [5]. Клубки предполагаются
взаимопроницаемыми, а энергия взаимодействия между двумя такими клубками-
рассчитывается как функция расстояния между их центрами масс.
Для этого используются решеточные расчеты в малых областях, в
которых распределение сегментов считается однородным из-за
взаимоперекрывания клубков. В пределах каждой микрообласти энергия
взаимодействия сегментов пропорциональна квадрату плотности
сегментов. Результатом этих расчетов является функция распределения,
которая, как и в статистических теориях реальных газов, приводит
ко второму вириальному коэффициенту в уравнении состояния, в
данном случае — к уравнению для осмотического давления. Рассмотрим
вывод этого уравнения для разбавленных растворов.
Представим молекулу в виде шара. Тогда два таких шара не могут
подойти друг к другу на расстояние г <d (d— диаметр шара).
Таким образом, для шара получаем исключенный объем, равный и =
= -Q- d3, где d = 2г. Число способов размещения в растворе
составляет: для первой молекулы — ^F; для второй — ^ V — и, для третьей —
^V — 2и, для 1-й молекулы — <=^V — ш. Число способов размещения
для всех молекул определяется так:
Q = const + П (V—lu)
П
при учете лишь парных контактов между молекулами (N2l
перестановок N2 молекул исключено; в данном случае оно фиксировано и
включено в константу).
Энергию смешения тогда можно вычислить по уравнению
AG = —kT\nQ = —kT Ц In (К— iu)+ const =*
= -kT [N2 In V + ^ In (l - -?)] + const
(A H здесь не фигурирует, так как она учтена исключенным объемом).
17
Поскольку мы рассматриваем разбавленный раствор, то исключенный
объем мал, т. е. лишь небольшая часть раствора исключена для
других молекул. Поэтому In A — у ) можно разложить в ряд
-± ~2+...]
+Const
с точностью до первого приближения. Тогда осмотическое давление
определяется выражением
Учитывая, что N2/V = cNa/M, получаем
Г 2 Л/
n==RT[ivt + "Ж •^+-
где
Аг = 1/М, А2 = NAu/2MK B.9)
Такое разложение осмотического давления в ряд называется ви-
риальным, а коэффициенты — вириальными коэффициентами. Это
•общее соотношение, справедливое и для низкомолекулярных веществ,
<и для полимеров. Если взаимодействия между молекулами
отсутствуют (идеальный раствор), нет и исключенного объема, т. е. и = 0. В
общем же случае Л2=0 и является мерой межмолекулярного
взаимодействия.
Для вывода выражения, описывающего свободную энергию
взаимодействия всех сегментов двух макромолекул, которые попадают в
малый элемент объема раствора 8v> находятся на расстояниях тк и rt от
^центров инерции макромолекул k и /, а средняя плотность
сегментов в bv принимается одинаковой, используется то же рассмотрение,
что и при выводе комбинаторной энтропии смешения в случае
однородного раствора. Отличие заключается в том, что в элементе объема
8v находятся не целые молекулы, а лишь ограниченные участки их
цепей. Поэтому при размещении сегментов в ячейки решетки
необходимо учитывать то, что для любого сегмента одна из ячеек первой
координационной сферы уже занята предшествующим сегментом. Если
в бп0 ячеек объема 8v нужно разместить 8х сегментов, то средняя
вероятность нахождения свободной ячейки для (/ + 1)-го сегмента
равна (бп0—/)/5п0 и число размещения всех сегментов —
или
_ б/ipl Гг-П5*
- Fпо-бх)\[ бп0 \ •
Последовательность операций, которая была использована при
переходе от формулы B.2) к B.4), приводит к соотношению
18
здесь бпг и ф!*— число молекул и объемная доля растворителя в
элементе объема 8v.
Свободная энергия смешения сегментов с растворителем в объеме
8v получается добавлением к — T8(kSM) члена kTx^n^^ Это
приводит к
M = кТ[Ьпг 1пA -ф2)
где объемная доля полимера ф2 относится лишь к объему 8v.
Дифференцируя B.10) по бп1э получаем изменение химического-
потенциала растворителя:
Д|*1 = RT fin A - ф2) + ф2 + Xlq>|]. .
При малых ф2, разлагая In A — ф2) в ряд по ф2, будем иметь
д^ = _/?7[A/2 -Х1)Ф22 +Ф|/ 3+ ...]. B.11)
Сохраняя лишь квадратичный член уравнения B.11) и полагая, что
1
Xi — у = «1 — ^i»
получаем
где kx и ярх— соответственно энтальпийный и энтропийный параметры:
Д#х = RTktfl ASX = RfyfPl
Введя «идеальную температуру»
запишем
Д^ = _*74h A - 0/7) ф^. B.13>
Как видно из этого уравнения, при Т = 0 имеем kt = ipl9 — -j + Xi =
= 0, т. е. Д[л = 0, следовательно, при 0-температуре поведение
раствора становится идеальным. При 0-температуре при сближении
макромолекул каждая из них накладывает ограничения на размещение
другой молекулы, т. е. это энтропийно невыгодно, но зато выгодно
энергетически (энергия контакта полимер — полимер больше энергии
контакта полимер — растворитель). При 0-температуре энтропийный
фактор компенсируется энергетическим, т. е. происходит компенсация
сил притяжения и отталкивания: клубок принимает так называемые
невозмущенные размеры.
2.3. ВЫЧИСЛЕНИЕ ИСКЛЮЧЕННОГО ОБЪЕМА
Для вычисления эффективного исключенного
объема определяется изменение свободной энергии, когда
молекула / полимера сближается с молекулой k на расстояние а. Возьмем
два элемента объема (bvt для молекулы / и 8vk для молекулы k)
19
•и подсчитаем энергию смешения при слиянии их в один объем
при сближении молекул до расстояния а. Если pz и pfe — плотности
сегментов в элементах объемов, vc — объем сегмента, то
После сближения молекул до расстояния а объемная доля полимер-
«ых сегментов обеих молекул в элементе объема определяется так:
Ф2*/ = (Рл + Р/) fc
Числа молекул растворителя в этих элементах объема вычисляются
из уравнений:
бс;A — p^j
опи = ; bnlk =
где vx— объем молекулы растворителя.
Используя уравнение B.10), подсчитаем свободную энергию до
и после смешения сегментов соответственно:
FG), + {Ю)к = kT^-[(l- Pivc) InA - Plvc) + A -pkvc) x
x in (l — рЛис) + XiP/fe (l — p/»c) + Xip*0c (l — p*»c)]; BЛ4)
= ЛГ -y- {[1 - (p^c + PkVc)] 1П [1 - (ftOc + p*l>c)] +
+ Xi [P/^c + P^c] [1 — (9iVc + pkvc)]}. B.15)
Поскольку концентрация сегментов pvc достаточно мала,
логарифмы в B.14) и B.15) можно разложить в ряд,ограничиваясь членами
второго порядка. Вычитая B.14) из B.15), находим изменение свободной
энергии в объемах при сближении молекул до расстояния а:
2 2
= 2kT(l/2- хО plPk -^ 80 = Ш (^ - kx) 9l9k -^ 8v =
Суммируя по всем элементам объема, получаем всю энергию
/t»! J P/Pfc во. B.16)
2
Примем, что плотность сегментов убывает от центра тяжести
молекулы по закону Гаусса. Тогда число сегментов (молекул) в некоем слое
6S для ненабухшей молекулы
а для набухшей молекулы
20
ЪЛеК4п& 6s,
У я/
где Р = ро/а; ро = C/2Я*I/2; (#*I/2 — радиус инерции макромолекулы;
а — коэффициент набухания, т. е. а = (R2/Rl)l/2. Тогда плотность
сегментов в набухшей макромолекуле можно выразить так:
B.17)
или
р (s) = N (-?=А*е-Р*\ B.18)
где
Используя B.17) и интегрируя уравнение B.16) по всему объему,
занимаемому молекулами / и ky получим
Здесь р2 = —=r- , Nvc = -jj—, vx = ^. Учитывая это, получаем
B.19)
т
- е/П
Как видно из уравнений B.19) и B.20), знак Д Ga зависит от
знака X, т. е. от соотношения между энтальпийным и энтропийным
параметрами, г|)х и k± (уравнение B.12)). При уменьшении расстояния a
монотонно возрастает AGO, достигая при a = 0 значения XkT.
Изменение AGa определяет вероятность (отнесенную к единице объема
раствора) /в того, что молекула / находится на расстоянии а от молекулы
k (при этом предполагается, что /«,= 1 и что другие молекулы вблизи
/ отсутствуют):
Произведение fa(bna2da) представляет собой, таким образом, объем,
доступный для молекулы / вблизи k, а A—fa)(ina2da) — объем,
исключенный для нее. Весь исключенный объем получается путем
суммирования последнего произведения по всем элементам объема:
00 00
и = j A — e~"°alkT) 4яа2 da = J A — е-*'""*) 4па2 da. B.21)
о о
21
Подставляя в B.21) значение Л Gfl, находим полный исключенный
объем:
оо
« = iS-(*2K/2J(i-<
Интеграл в уравнении B.22) не вычисляется. Флори и Орофино дали
приближенное значение этого интеграла:
JS(?) B.23)
где
*-тDГ^Г"#г*.П-*П. B.24)
При ©-температуре X = 0 и w = 0. Таким образом,
«тета»-температур а является температурой, при которой молекулы как бы
перестают взаимодействовать. Это значит, что вблизи 0-температуры
молекулы не мешают друг другу, а вдали от нее (Т >> 0) представляют собой
твердые сферы с объемом, равным истинному объему . молекулы, и
непроницаемые для других молекул.
Подставляя уравнение B.23) в B.9), получаем, что
Из уравнения B.25) видно, что при Т = 0 Л2= 0, так как и = 0,
т. е. раствор ведет себя как идеальный. Но при 0-температуре, как
уже было сказано раньше, происходит компенсация энтропийных
и энергетических взаимодействий, т. е. макромолекула принимает
состояние невозмущенного клубка. Следовательно, коэффициент
набухания макромолекулы а равен единице. Но в то же время в этих
условиях происходит и компенсация энергетических и энтропийных
взаимодействий при контактах разных молекул, поскольку
взаимодействия между сегментами, принадлежащими одной цепи, одинаковы
с таковыми между сегментами, принадлежащими разным
макромолекулам. Таким образом, компенсация сил притяжения и отталкивания
при внутримолекулярных взаимодействиях, обусловливающих
коэффициент набухания а, и компенсация межмолекулярных
взаимодействий, определяющих Л2, происходят одновременно. Следовательно,
А 2 и коэффициент набухания взаимосвязаны. Для нахождения этой
взаимосвязи надо исключить 0/71 и связать Л2 с а. Преобразуем
выражение для X:
(?2)з/2 = a3 (R20)W = а3 (Л02K/2/63/2. B.26)
Подставляя B.26) в B.24), получаем
^z?L3 = 2(a2-l) B.27)
22
и после подстановки последнего выражения в B.25) имеем
Л Щ— In [ 1 + — (а2 — 1)J. B.28)
Все величины, входящие в уравнение B.28), можно измерить на
опыте. Из уравнения B.28) видно, что коэффициент набухания а
растет с ростом Л2. Это означает, что ос, так же как и Л2, убывает с
повышением в. Поскольку в = 2zA w12/k, то в возрастает с А ш12, т. е.
увеличивается проигрыш энергии при смешении полимера с
растворителем (ухудшается растворение полимера в данном растворителе).
Если это хороший растворитель, то Aw12 может быть либо
отрицательным (растворение энергетически выгодно), либо положительным
(растворение энергетически невыгодно), но малым. Тогда в/Т = 2zA w12/
(/kT и при zA ay12<?C kT 0 <с Т. Следовательно, 0 является очень
низкой (она может быть даже отрицательной, что лишено физического
смысла). Поэтому в хорошем растворителе всегда Л2> 0 и а > 1, т. е.
силы отталкивания преобладают над силами притяжения. Молекула
полимера как бы растягивается, иммобилизируя в себя растворитель.
Ухудшая растворитель, можем прийти к ситуации, когда Т = 0
и А 2=0, а <х= 1. Дальнейшее ухудшение качества растворителя
может привести к Т < 0, а следовательно, к Л 2 < 0 и а<1: молекулы
не разбухают, а сжимаются, выталкивая из себя растворитель. Однако
в плохом растворителе молекула не сжимается до бесконечности, она
просто не растворяется (выпадает в осадок). Поэтому ©-точка является
одновременно и критической температурой выпадения полимера в
осадок при М -* оо.
2.4. КОЭФФИЦИЕНТ НАБУХАНИЯ МАКРОМОЛЕКУЛЫ.
ТЕОРИЯ ЗИММА И ШТОКМАЙЕРА
Набухание отдельной макромолекулы в растворе переводит ее
в менее вероятную конформацию. Такой, более развернутой конфор-
мации соответствует упругая сила. Следовательно, набухание
макромолекулы прекращается, когда осмотическая сила уравновешивается
упругой силой.
Как и при вычислении исключенного объема, будем
рассматривать макромолекулу как облако сегментов, распределенных
относительно центра тяжести по гауссовскому закону. Это значит, что можно
разбить весь объем, занимаемый макромолекулой, на концентрические
слои с постоянной плотностью, которая убывает по мере удаления от
центра тяжести:
dN . ч /3 \з/2 - —
ш($)Ш е Rdv-
Число сегментов до набухания в /-том слое определяется выражением
6W2/ = N (?Ц 3 е~^ 4ns}Ss/, B.29)
23
где ро_ константа, выбираемая таким образом, чтобы R*
соответствовал своему истинному значению
f]-^U± B.30)
Это уравнение является условием для нахождения |30.
Пусть цепочка набухает равномерно, т. е. если она набухла в а
раз, то а2 = Я2//?*. ТогДа все сегменты, находящиеся до набухания
в сферическом слое от (s/, s/ + 6s;), перешли в слой (as/, as,- + a8s/).
Следовательно, в новом слое оказались те же 8N2j сегментов.
Поскольку в результате набухания раствор разбавляется, вокруг цепочки
увеличивается число молекул растворителя и объем слоя записывается
так: 8V = 4na2S/6s/. Если <р2/— объемная доля полимера, а A — ф2/)—
объемная доля растворителя, то число молекул растворителя в слое
— <p2/)
>
Полная энергия смешения AG полимерных сегментов одной
макромолекулы с растворителем состоит из суммы &GM/ всех элементов
объема макромолекулы и члена AGynp, связанного с изменением ее
конформации
ДО - Е AGm/ + Дбупр.
Условие равновесия имеет вид
ИЛИ
да )т. Р '
ЗДО_ уЗАОдц , аА°упр_л
5« - Zj -^Г~ + да ~ U-
Энтропия сегментов до набухания равна S0(s/)> а после набухания —
S (asf). Число сегментов в /-том слое выражается уравнением B.29).
В этом случае мы имеем
Дбупр = — Т 2 #2/ Is (as,) — s0 (sg)]..
Поскольку до набухания
So (s/) = k In tf2/ (sy) = С - fePo s • + ft In (s)8sf),
и после набухания
S (as7) = ft In tf2/ (as/) = С — feP2a2s2- + ft In tffis,) + ft In a*,
то выражение для AGynp можно записать так:
A j3 j e-Wi $1$ (а? - 1) - In a3] s? 6S/ -
= kT [3/2 (a2 - 1) - In a3]. B.31)
24
Найдем полное изменение свободной энергии
да — 4J дFпи) ' да + да
Число сегментов 6Л/2/ остается постоянным, а меняется
(увеличивается) число молекул растворителя при набухании.
Поскольку AGMj относится к малому объему, среднюю плотность
сегментов внутри которого можно считать всюду одинаковой, то
dAGMj/d(bn\j) в этом объеме совпадает с локальным избыточным
химическим потенциалом:
- ^ - fi? = kT f In A - Ф2/) + «ft, + Х1Ф1/].
Объемная доля полимера очень мала, поэтому
Обозначая энтропийный член г/2 через i|)x и учитывая, что в = 2x1^,
имеем
Используя B.29), определяем объемную долю ф2/:
-Эо1 B.34)
Производная d(bn\j)/da выражается уравнением (полагая фг/С 1)
5(fi«i/) д [4Jia3s26S/A — ф2/)
da 5a [ flj
Подставляя B.31), B.33) и B.34) в B.32), получаем
Учитывая, что
h "A A
окончательно будем иметь
*j? = -бСмкТ^ f 1 — J) !^ + 3kT (a -), B.35)
oa \ i / a \ a /
где
27 V /MW2
25
Видно, что свободная энергия смешения состоит из двух членов»
Первый из них отрицателен при Т >0 и увеличивается
пропорционально V My т. е. в этом случае смешение более выгодно. Если в ~
— A w12> 0, весь правый член будет больше или меньше нуля в
зависимости от соотношения энтропийной и энергетической
составляющих. Второй член связан с упругостью, поэтому он всегда положителен
(энтропийно выгодно, энергетически невыгодно). При равновесии
(dAG/da = 0) уравнение B.35) превращается в
а6 — а3 - 2СМ^1 A — в/Г) У~М. B.36)
Это окончательный результат теории Флори. Из уравнения B.36)
следует несколько важных выводов. Так, коэффициент набухания а
медленно растет с М и Т и зависит от двух термодинамических
параметров: энтропийного ifi и энергетического в. В в-точке а =1 и А2='
= Лв; следовательно, размеры цепи определяются только ее гиб"
костью, а не взаимодействием с растворителем. Это означает, что
размеры должны быть одинаковыми в любом 0-растворителе, т. е.
гибкость цепи не зависит от растворителя. Однако, как было показано в
работах [6—8], это утверждение неверно.
Далее размеры макромолекул растут с улучшением качества
растворителя, так как с А 2 растет а. Этот вывод подтверждается
экспериментом [9, 10]. С ухудшением качества растворителя ниже в-темпера-
туры макромолекула сжимается (а < 1). При Т > в значение а растет
с повышением М, т. е. коэффициент набухания изменяется
пропорционально М. В результате этого размеры макромолекул растут более
быстро с М, чем в идеальном растворителе. Если а очень велико,то,
пренебрегая а3, получили бы а6 — УИ0»50, а для малых а, а — М0»1,
т. е. (Л2I/2 = в(ЛоI/2~ А*0*6. Поэтому, согласно Флори, (Л2I'2 ~
1-И
~Af 2 , где 0^8 <;0,2. Этот вывод также согласуется с
экспериментом [11—13].
Теория предсказывает связь Л2 с а. Поэтому чрг из А2 и а должна
была бы определяться одинаково. Однако это не подтверждается
экспериментально. Следовательно, теория дает неправильную зависимость
Л2 от а. Это видно из следующего примера. Разложим А2 вблизи
Т = 0 (уравнение B.25)) в ряд по X:
В 1958 г. Штокмайер [14], приняв, что вблизи в-температуры
величина /42=^-1|IA—в/Г) универсальна и постоянна, а также
попользовав данные Кригбаума и Флори для растворов полиизобутилена
в бензоле [15J, получил значение ifo, равное 0,3. С другой стороны,
26
разложив уравнение B.36) в ряд по а2 вблизи 0-температуры, будем
иметь выражение
а2 = 1 + 2СмУрг A — в/Г) VM B.37)
На основе B.37) для той же системы полиизобутилен —
бензол oh оказался равным 0,15. Верно только первое значение, не
основанное на теории Флори, т. е. теория Флори дает ошибку в 2 раза.
Следовательно, надо брать См= См/2. Строгая статистическая теория
(Штокмайера и Зимма) приводит к формуле, близкой к B.36), но с
коэффициентом, в два раза меньшим. Основная причина количественного
несовпадения теории Флори с опытом заключается в том, что она
относится не к единой цепи полимера, а к нескольким разрозненным
сегментам.
С 1940 г. начались попытки улучшить теорию Флори. В 1953 г.
такая теория была развита для частного случая, она рассматривала
взаимодействия дальнего порядка как небольшую поправку. Это
теория Зимма и Штокмайера [16, 17]. В этой теории рассматривается
модель «жемчужного ожерелья» для полимерной цепи: бусинки,
соединенные бестелесными нитями. При этом бусинки взаимодействуют
между собой, а нити — нет. Для такой модели можно найти функцию
распределения между концами цепи, / (h). Пусть имеется N сегментов,
их совокупность {N}, a f{N) — полная функция распределения для
координат всех сегментов цепи. Чтобы найти функцию
распределения, необходимо взять полную функцию и проинтегрировать ее по
всем координатам, кроме h:
ff^ B.38)
dh
Полная функция распределения для координат всех сегментов
цепи имеет такой вид:
v{n)
f{N} = f»{N}e *т , B.39)
где /° {N} — функция распределения при отсутствии взаимодействия;
V {N} — полная функция взаимодействия всех сегментов в смеси
(энергия взаимодействия всех сегментов):
%UG{j) B.40)
(U (га) — энергия взаимодействия пары сегментов, быстро убывающая
функция расстояния между сегментами).
Тогда подставив B.40) в B.39), получим
27
Поскольку Гц очень малы, удобно ввести функцию
кт
которая отлична от нуля при малых Гц и равна нулю при больших Гц.
Возвращаясь к формуле B.38), имеем
dh
i< i
dh - - к,
Ограничиваясь вторым членом, получаем
S dhdru
Интегрирование по всем координатам, кроме Л и г*/, приводит
к выражению
/(Л) = /°(А) - fS /°(^Й
Следует иметь в виду, что ф(г,-/) резко изменяется с изменением Гц,
f° (h, гц) — функция плавная. Поэтому /° (й, гц) можно вынести за
знак интеграла, вычислив ее в точке, где Гц = 0. Тогда
|drw
(^0— исключенный объем). Такое вынесение и0 за знак интеграла
означает, что любая энергия взаимодействия двух пар сегментов одинакова.
Таким образом
где /°(Л)—функция распределения при отсутствии взаимодействия,
а /°(Л, О if) — сумма вероятностей соударения пары сегментов для
фиксированной 1и Нормированная функция имеет вид
28
fo(h)-vn
Oif)-f°(h)x
Знаменатель в B.41) является результатом интегрирования v0. С
помощью нормировочной функции можно вычислить распределение-
между концами цепи:
j
= hi - у0 S. [(Ло)о,/ - h20] Г (On) + • •., B.42)
где f°(O{j) — гауссовская функция распределения. Накладываем
условие г{/ = 0. Тогда /° (Л, Oij) = /° (О//) /о/у (Л) — вероятность
одновременного наступления двух событий, a fo{f(h) —то же при условии*
Гц = 0. Подставив эти величины в явном виде в B.42), получим
N
|3/2 ~А J —~kU* ^ ^о + "з" \2h) Лз V° • B.43>
1
Гауссовская функция распределения имеет вид
[о 13/2 «^ ЗГ;/ г о
л t 6 е 2(/-о^2 = i
2n(j — i)A2\ Bя(/—/)Л2
Ъ2 -
при гц = 0, Л2 = -^ и hi = Л^Л2. При соприкосновении двух
сегментов цепь упорядочивается (hl)oif = [N — (j — i)] A2.
Уравнение B.43) можно переписать в виде
13/2
J
Л2 =hl(l+±z +."), B.44)
где
Видно, что поправочный член пропорционален |/W, т. е. У
В этом наблюдается согласие с теорией Флори: с увеличением М уси~
ливаются взаимодействия дальнего порядка.
2»
Рассмотрим физический смысл v0. Пусть бусинки представляют со-
<юй твердые шарики с диаметром d. Тогда
Таким образом, эффективный исключенный объем реального шарика
равен объему шара, построенного на его диаметре. Для реальной
молекулы разобьем v0 на два интеграла:
К—Щгул во р — Ц(Г)-\
\—ект \r*dr + 4nUl-e kT \r2dr,
где первый интеграл соответствует силам отталкивания( U > 0),
а второй — силам притяжения (U < 0). Следовательно, эффективный
исключенный объем состоит из положительной и отрицательной
составляющих. Обе части по-разному зависят от температуры: для первой
части U велико ( U >?Т), поэтому она не изменяется с температурой,
а для второй части U ~ кт и влияние температуры в данном случае
существенно. Поэтому
Vo = а — Ь/Т.
В теории Флори был введен членг|IA —в/Г), связанный с
энтропийным и энергетическим членами, т. е. в теории Флори уже
учитывались отталкивание и притяжение. Очевидно, что и в этом случае
можно найти такую температуру, при которой силы притяжения и
отталкивания будут таковы, что vo= 0. Это 0-температура.
Выразим v0 через термодинамические параметры. Как было
сказано выше, А 2= NA и /2М2(и — эффективный исключенный объем
макромолекулы). Вблизи в-температуры «в= 0, а вдали от нее и
пропорционально геометрическому объему. Каждый сегмент молекулы 1
исключает N сегментов для всей молекулы 2, а вся молекула 1
исключает а = N2v0 сегментов и
А? = NAN2v0!2M2.
При удалении от ©-температуры
B.46)
где
= 1 — 2,8652 -\ (по Зимму), B.47)
F(z) = 1 — 1,152 Н (по Флори)
и
Л2 = ^1 ^ A _ 6/7) F (X). B.48)
30
При Г -*• О F (г) -» 1 и F (X) -* 1. Оба выражения для Л2 связаны
лишь с термодинамическими параметрами взаимодействия и не
зависят от структуры цепи. Поэтому можно приравнятьB.46) и B.48):
? ^ ^ ^ = ^2% A-6/7),
B.49)
так как Mv/NA = Nvc.
Для простой решеточной модели % = 1/2, г>0 = yt и у0 = »сA —
— в/Г). Подставим B.49) в B.45) и B.44)
= 1 + 2См Ь A — в/Г) Ум, B.50)
Из сравнения B.37) и B.50) видно, что характер зависимостей
тот же, но численный множитель в B.37) в два раза больше, т. е.
в B.50) С/ц= См/2. Следовательно, точная теория дает вблизи в-тем-
пературы то же выражение, что и теория Флори, но с другими
коэффициентами, что согласуется с экспериментом [14]. Таким образом,
влияние взаимодействий дальнего порядка на размеры макромолекул
в действительности в два раза слабее.чем предсказывает теория
Флори. Область применимости уравнений B.44) и B.50) невелика.
Поскольку z~VN, а У~М — очень велико (~100), то и 2 тоже
большое число и пользоваться уравнениями в широком интервале
температур нельзя. Это возможно лишь в том случае, когда v0 очень
мало, т. е. вблизи в-точки. Следовательно, точная теория применима
лишь в очень узких пределах, а теория Флори дает приближенные
значения.
Следующую попытку создать еще одну приближенную теорию
осуществил Фиксман [18].
Рассмотрим формулу
Эта формула представляет собой разложение в ряд по vOi
При этом
(f) 4C/2я)з/^
А
Напишем выражение для (dh2/dv0) в любой точке, где УОФ 0.
Принимаем, что если размеры молекул за счет взаимодействий дальнего
порядка увеличиваются в а раз, то
JJ2 _. а2/Д. ^5! = -i- C/2дK/2
а
d№ T? rfot2 dec*
Отсюда
'Таким образом получаем дифференциальное уравнение с
разделенными переменными относительно а. Интегрирование его приводит к
выражению:
2f = ± C/2я)з/2 &- + const (о» = 2г + С).
При z = 0 а = 1 и, следовательно, С = 1. Тогда
а3=1+2г. B.51)
Это уравнение является разумным приближением уравнения B.44),
При малом z из B.44) получаем а3 = 1 +1> z- Причем при малых г
формула B.51) точна, а при больших z — приближенная. Сравнивая
формулу Флори с B.51), имеем а6 — а3^ J-z. Следовательно, обе
формулы дают существенно различную зависимость а от М. Согласно
Флори, при а» 1
а*~г~УЖ а — М0'1; (/?I/2~М0'60, B.52>
-а по формуле B.51) при а> 1
Т. е. по статистической теории зависимость а от М сильнее, чем по
теории Флори. Кроме того, из первой следует, что при a = 1
а при
«где Л — характеристика гибкости цепи.
Из этих результатов можно сделать вывод, что в хороших
растворителях размеры макромолекулы зависят не от гибкости цепи, а
только от М2 и эффективного исключенного объема v0, т. е. размеры
макромолекулы вдали от 0-точки вообще не зависят от гибкости цепи и объем
макромолекулы определяется лишь исключенным эффективным объемом.
Этот вывод имеет очень важное значение для гидродинамики растворов
полимеров.
32
Так, коэффициент поступательного трения шарика по Стоксу
записывается уравнением: / == 6tcy\0R . Макромолекула также ведет себя
как непротекаемое твердое тело [68]: / = Рт]о(А2I/2 (где Р = 5,11).
Величина / может быть измерена экспериментально или по диффузии:
или по седиментации:
S - Л* A —l>po)/NAf ~ М^1Р+~*'*>.
На основании уравнения B.51) можно сделать определенные
заключения о поведении макромолекулы вблизи в-точки и в хорошем
растворителе.
С другой стороны, для суспензии шариков по Энштейну
2,5NVM,
где N — число молекул; Vm— объем молекулы. Уравнение для
характеристической вязкости тогда будет таким:
[г\] — 2,5р (v — удельный объем молекулы), т. е. в этом случае
молекула ведет себя так, как будто растворитель обтекает ее только снаружи.
Иначе говоря, не только в поступательном, но и во вращательном
трении молекула ведет себя как твердое тело:
[г]] = Ф(/?K/2/ЛГ, B.53)
где Ф = 2,8 • 1028. Уравнение B.53) известно как формула Флори.
Таким образом и теория Флори, и статистическая теория дают вполне
определенное предсказание о зависимости гидродинамических свойств
макромолекул от молекулярной массы. Выражение B.61) и формула
Флори позволяют разделить влияние взаимодействий ближнего и
дальнего порядка на размеры макромолекулы. Так, согласно формуле
B.51)
или при подстановке h2
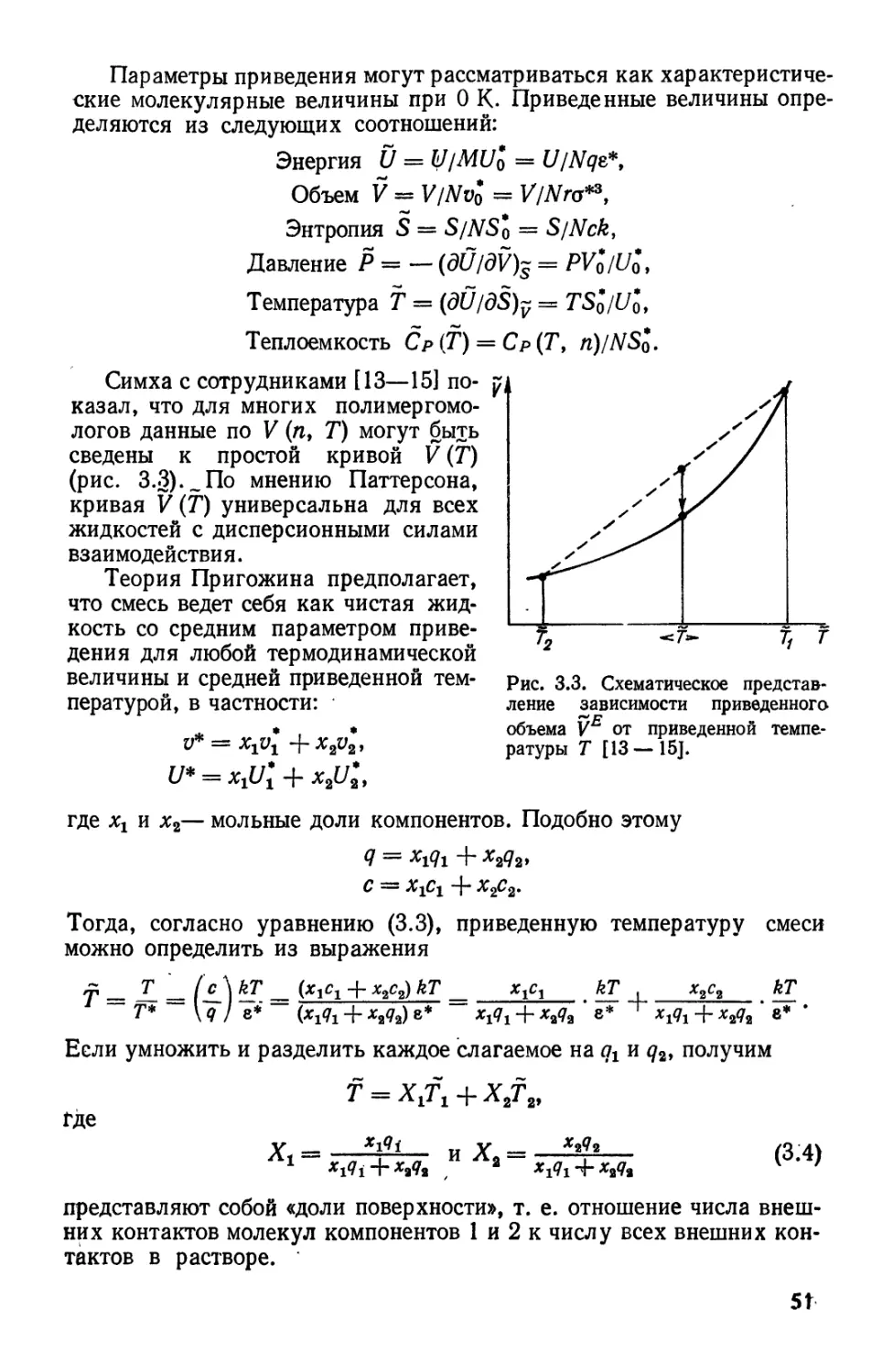
/q \3/2
(/,2K/2 = лгз/2лз + 2 ^у N2v0. B.54)
Первый член зависит от гибкости цепи, а второй — от
эффективного исключенного объема сегмента. Уравнение B.64) можно
переписать в виде
Г Л8 + 2
Если теперь для нескольких фракций измерить молекулярную
массу и построить зависимость (h/Nf/2, то можно разделить взаимодействия
4-420
33
ближнего и дальнего порядка. Этот метод применен несколькими
авторами, которые обнаружили хорошее совпадение с данными,
полученными непосредственно в в-точке [19]. Разновидности этого метода
можно найти в работах [20—24].
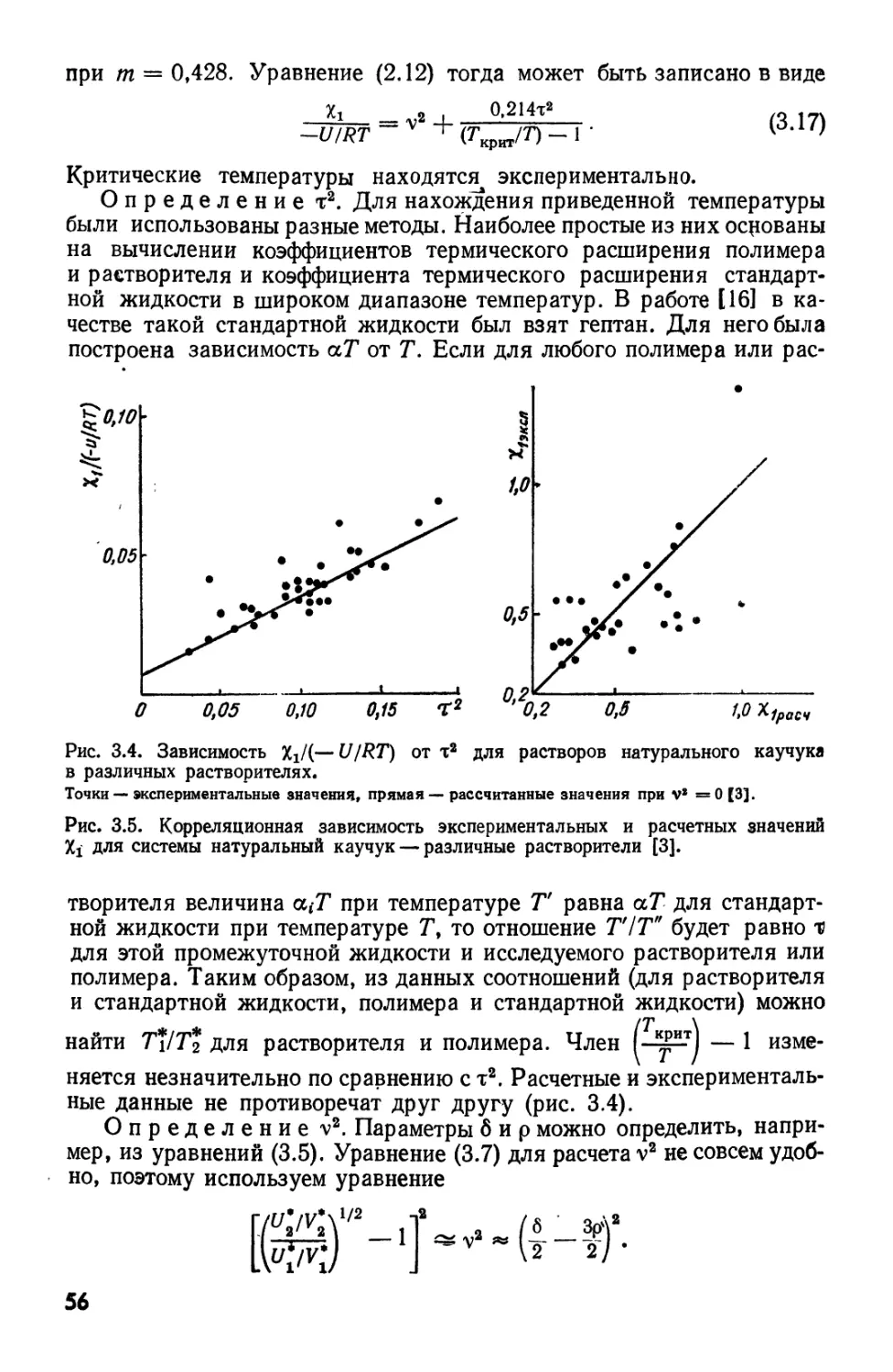
2.5. ФАЗОВОЕ РАССЛОЕНИЕ В ПОЛИМЕРНЫХ РАСТВОРАХ
При охлаждении раствора некристаллизующегося полимера можно
достичь температуры, при которой раствор разделится на две фазы:
обогащенную полимером и разбавленную. При дальнейшем
охлаждении полимер-обогащенная фаза
становится более концентрированной,
а разбавленная фаза — еще более
разбавленной. Иначе говоря,
система имеет область нерастворимости,
которая сужается с увеличением
температуры вплоть до верхней
критической температуры
смешения. При этой температуре
теплота и энтропия смешения
положительны, избыточный химический
потенциал |xf уменьшается с
повышением температуры. В
решеточной теории обменный вклад RTX^l
может перекрыть отрицательную
0,20 0,25<рг комбинаторную энтропию
смешения, однако при повышении
температуры последняя становится
преобладающей, так как 1Х
уменьшается с температурой. К этим
характеристикам необходимо
добавить и некоторые специфические
особенности растворов полимеров:
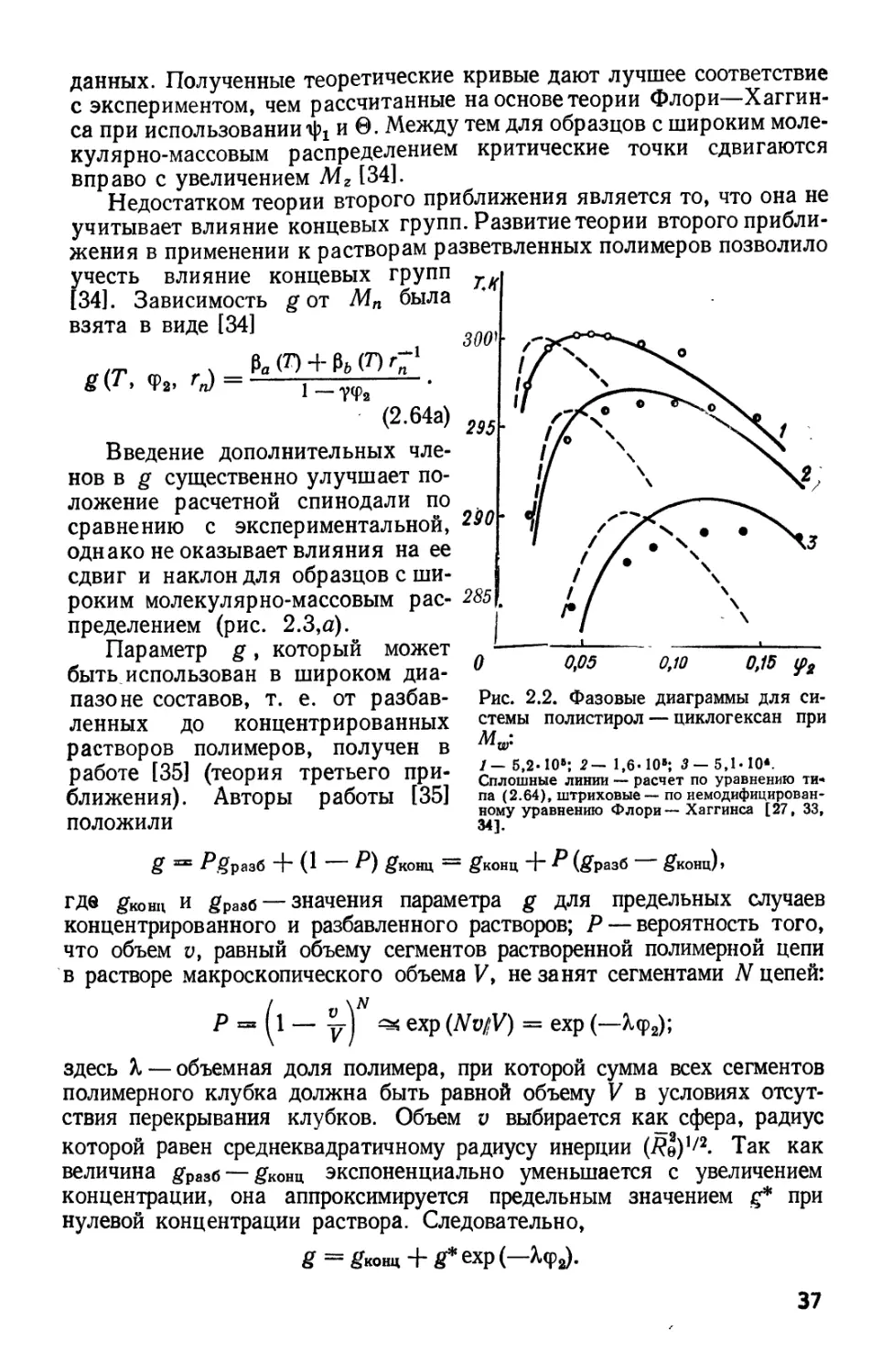
критическая концентрация мала и понижается с ростом молекулярной
массы полимера, в то время как критическая температура смешения
увеличивается (рис. 2.1). Расчетные бинодали были получены из
теории Флори—Хаггинса и экспериментальных критических точек
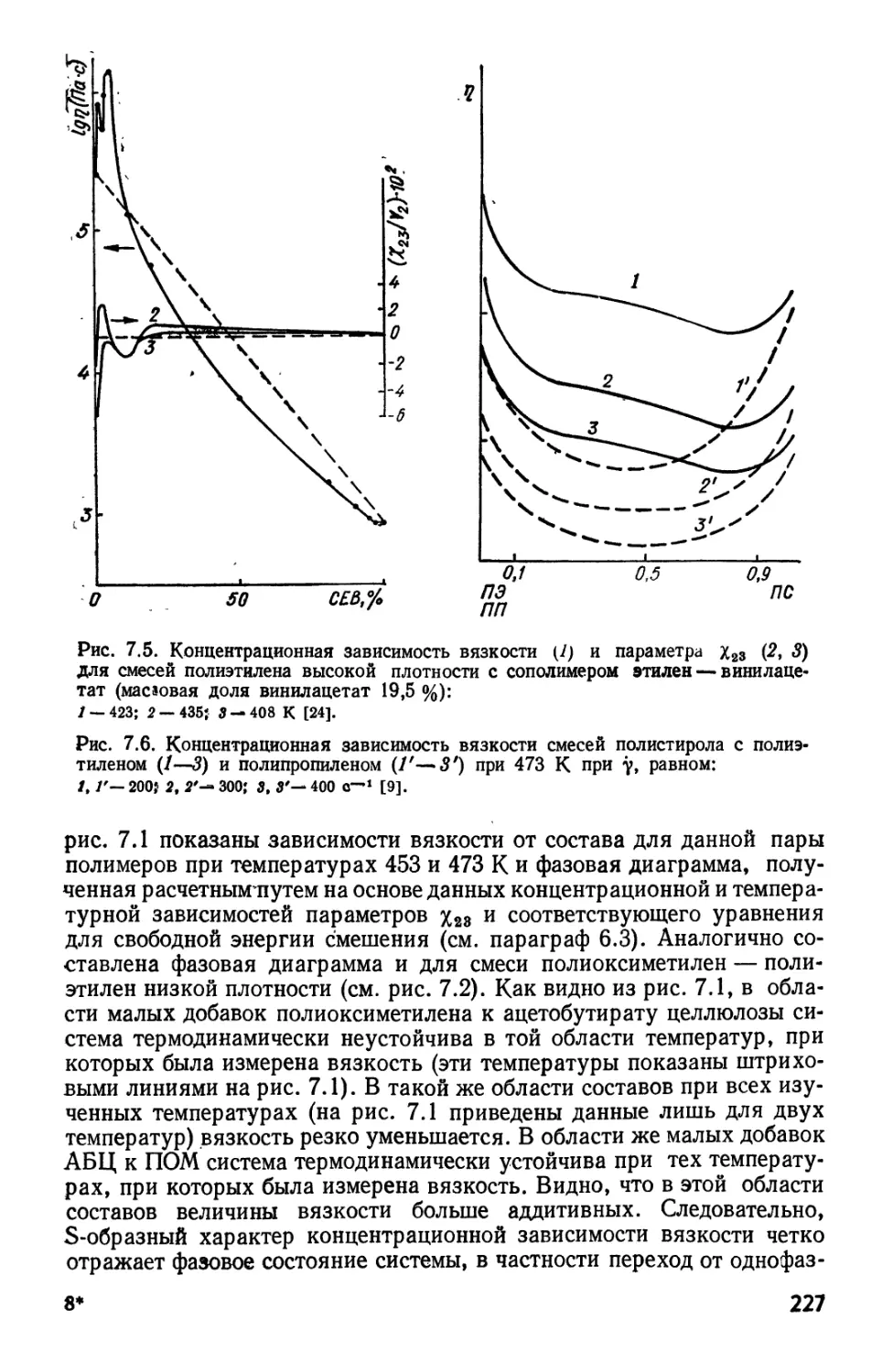
следующим образом. Из условия для критической точки, т. е. (д\11/дц>2)р9
^328
Лз23
318
313
308.
293
288
283
О 0,05 0,10 0,15
Рис. 2.1. Экспериментальные
(сплошные линии) и теоретически
рассчитанные (штриховые) фазовые диаграммы
для системы полиизобутилен — диизо-
бутилкетон:
/.-2,27.10*; 2~* 2,85.10»; З^б-Ю* [26].
= 0 и/
ходим
—^А = 0, путем дифференцирования уравнения B.7) на-
дч>1)
р9т
B.55)
B.56)
Решая уравнения B.55) и B.56), получаем значение критического
состава
A+/-1/2)-1. B.57)
34
Уравнение, связывающее критическую температуру с G-темпера-
турой, энтропийным параметром взаимодействия г|)х и длиной цепи,
выраженной как число сегментов г, Флори и Фокс получили в виде
]| B.58)
— 1 1 1
График зависимости Ткрит от —j— + — должен быть линейным.
По углу наклона и длине отрезка этой зависимости получают ви^,
подставляют в уравнение B.7) и рассчитывают бинодали, т. е.
температуры и составы, где разные фазы находятся в равновесии (см.
уравнение B.7)). Процедура довольно проста, но длительна, так как
равновесные составы не могут быть четко выражены как функция
температуры [2].
Как видно из рис. 2.1, сдвиг критических точек в область низких
концентраций при увеличении молекулярной массы согласуется с
уравнением B.57), а зависимость Ткрит от молекулярной массы в основном
количественно отвечает уравнению B.58). Расчетные бинодали
качественно согласуются с экспериментальными, но область растворимости
много уже, а Фг, крит — ниже, чем полученные экспериментально (см.
также [25]). Уравнение B.58) предсказывает, что 0-температура
является предельным значением ТКрИТ для бесконечно длинной цепи,
и экспериментальные данные подтверждают тот факт, что при этой
температуре второй вириальный коэффициент А2 равен нулю [2, 27,
28]. В работе [26] показано, что предельное поведение уравнения
B.58) может быть получено и без использования смешанной формы
для свободной энергии смешения, а лишь при предположении, что
вириальное разложение вблизи критической точки верно.
Несмотря на хорошее согласие между в и предельной критической
температурой смешения, значение г|?1э полученное из данных
критической точки, много больше, чем определенное по температурной
зависимости второго вириального коэффициента (см. уравнение B.36)).
Причина этого различия неясна, но, возможно, это связано с тем, что
в вириальном разложении не учитывается третий вириальный
коэффициент. Штокмайер показал [14], что наклон зависимости 1/ГКрИт от
1/г1/2 включает тройные молекулярные кластерные (гроздевые) вклады,
которые не исчезают при Л2= 0.
Штриховыми линиями на рисг2.1 показаны спинодали, т. е.
пределы метастабильности в однофазной области, определяющие
положение точек, для которых (ф-1/дф2)я, г = 0 (или d2AG/dcpf =0).
Значения щ (или AG) могут быть рассчитаны по уравнениям типа B.7)
или B.6). Из рис. 2.1 видно, что максимумы расчетных спинодалей
находятся при меньших концентрациях, чем полученные
экспериментально, т. е. расчетные спинодали не совсем хорошо совпадают с
экспериментальными.
Дальнейшие усилия усовершенствовать теоретические расчеты
фазовых диаграмм на основе решеточной модели были направлены на
коррекцию параметра %г. Для этого параметр %х обычно заменяется
2* 35
общим концентрационно зависимым параметром свободной энергии
g (Tf ф2), включающим энтальпийную и энтропийную составляющие:
AG/RT = ф1 Inф1 + ф2гх In ф2 + g(Г, ф2)ф1ф2. B.59)
Параметр Xi B уравнении B.7) связан с g соотношением
*i (ф2> Т) = g - A - ф2) (dg/dy2)T. B.60)
Концентрационная зависимость параметра g аппроксимируется
полиномом по ф2 (или w2). Так, Конингсвелд и сотрудники [29]
использовали следующее выражение для g:
(/ = 0, 1, 2, ... , я), B.61)
в котором каждый коэффициент g/ может быть функцией
температуры в форме
Si = ёп + gnlT + gjsT + gh In T.
Если в системе нет полярных или водородных связей, в уравнении B.61)
п = 0 и g не зависит от w2. Если в этих случаях температурная
зависимость также незначительна, расчетные значения свободной энергии
смешения находятся в хорошем согласии с экспериментом (см.,
например, [29]). Для систем же, в которых имеются полярные межсегментные
взаимодействия, параметр g должен зависеть от w2 и Т [30, 31]. Для
систем с полярными взаимодействиями между компонентами можно
использовать уравнение B.61) с п = 1:
B.62)
где g0 и gx могут зависеть от температуры. Поскольку полярные
взаимодействия сильно зависят от температуры, температурная зависимость
параметров может быть взята в виде
B.63)
где gj\ принимает отрицательные и положительные значения.
Используя уравнения B.62) и B.63),-значения Д G могут быть рассчитаны при
разных температурах по уравнению B.59). Двойное
дифференцирование кривых Д G = / (ф2) позволяет определить бинодали или спинода-
ли (см. параграф 3 гл. 6). В работе [32] этим способом было получено
шесть типов бинодалей.
В других работах [27, 33] Конингсвелд и сотрудники
использовали уравнение для g с четырьмя параметрами (так называемое второе
приближение теории):
( |I, B,64)
которое в общих чертах оказалось пригодным для описания
данных по критической температуре и составу для системы
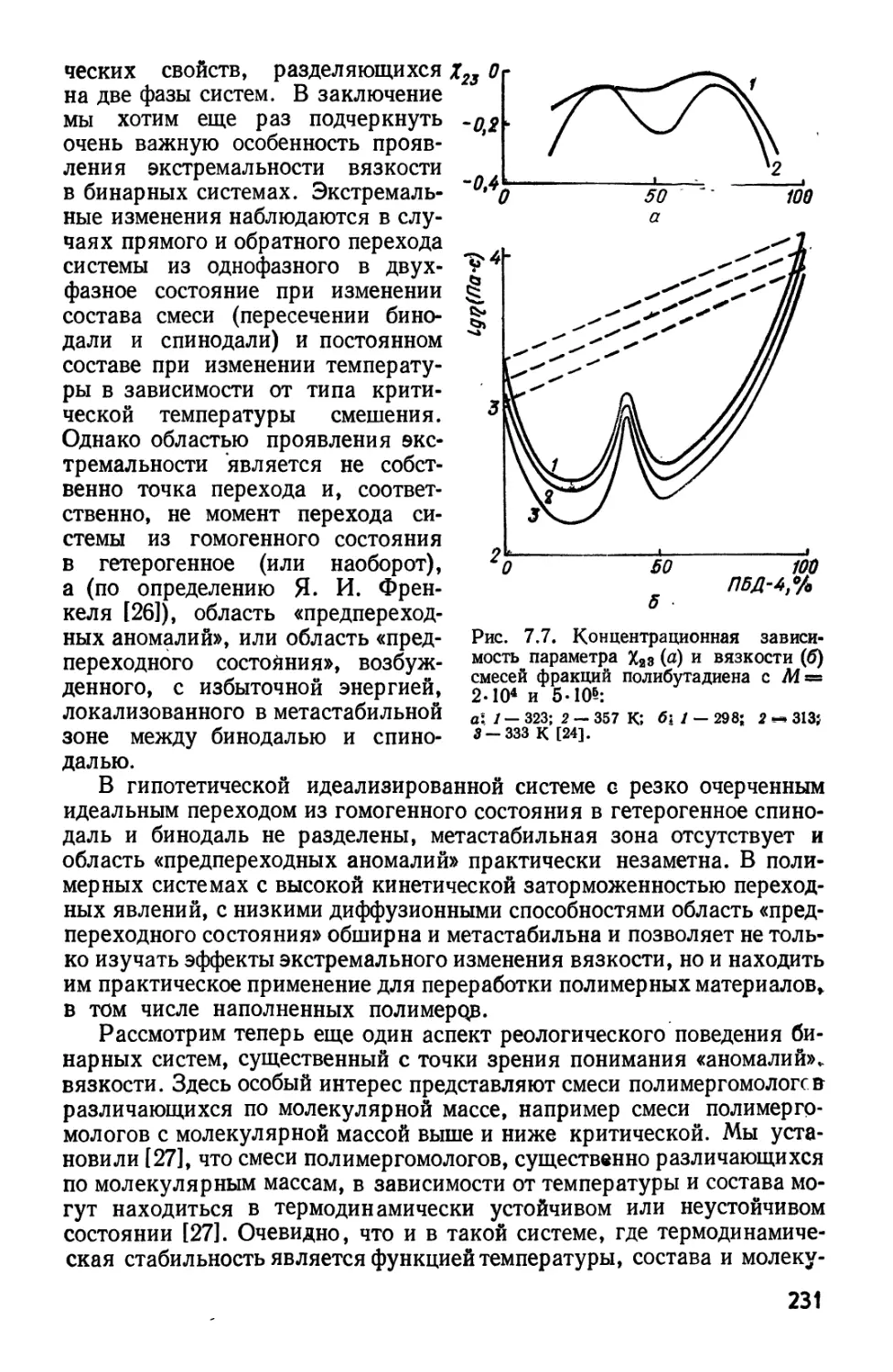
полистирол — циклогексан [27, 33] (рис. 2.2). Постоянные в B.64) являются
просто связующими параметрами для описания экспериментальных
36
данных. Полученные теоретические кривые дают лучшее соответствие
с экспериментом, чем рассчитанные на основе теории Флори—Хаггин-
са при использовании ifo и G. Между тем для образцов с широким моле-
кулярно-массовым распределением критические точки сдвигаются
вправо с увеличением Мг [34].
Недостатком теории второго приближения является то, что она не
учитывает влияние концевых групп. Развитие теории второго
приближения в применении к растворам разветвленных полимеров позволило
учесть влияние концевых групп
[34]. Зависимость g от Мп была
взята в виде [34]
s(r,*!,,J_t<?>j±M^.
2 B.64а)
Введение дополнительных
членов в g существенно улучшает
положение расчетной спинодали по
сравнению с экспериментальной,
однако не оказывает влияния на ее
сдвиг и наклон для образцов с
широким молекулярно-массовым рас- 285
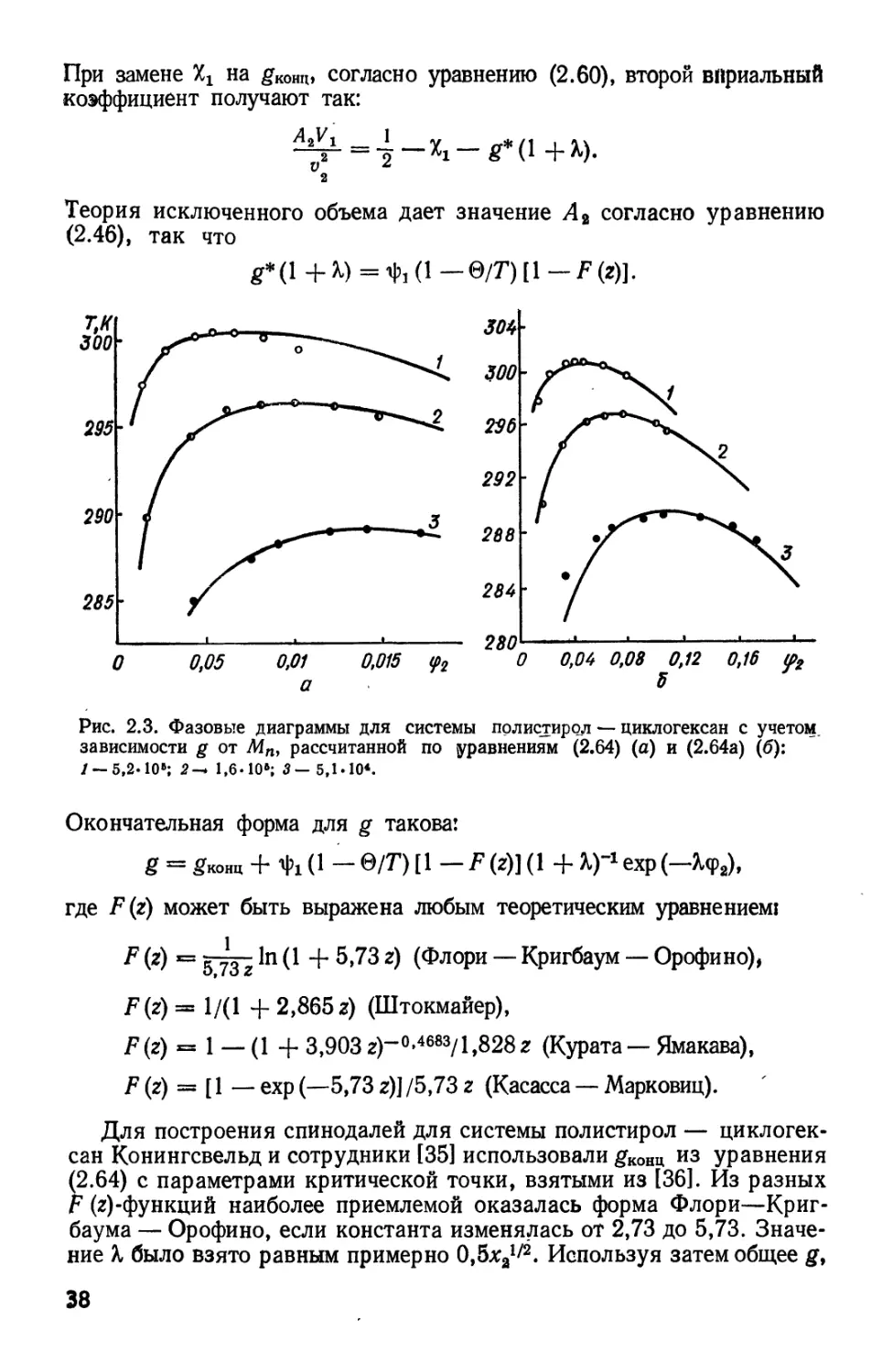
пределением (рис. 2.3,я).
Параметр g, который может
быть использован в широком
диапазоне составов, т. е. от
разбавленных до концентрированных
растворов полимеров, получен в
работе [35] (теория третьего
приближения). Авторы работы [35]
положили
Рис. 2.2. Фазовые диаграммы для
системы полистирол — циклогексан при
W
/— 5,2.10»; 2 — 1,6.10е; 3—5,1-10*.
Сплошные линии — расчет по уравнению
типа B.64), штриховые— по немодифицирован-
ному уравнению Флори— Хаггинса [27, 33,
g ¦» Л§разб + A — Р) ?конц = ?конц + Р (#разб — ?конц),
где #конц и gpa36 — значения параметра g для предельных случаев
концентрированного и разбавленного растворов; Р — вероятность того,
что объем v, равный объему сегментов растворенной полимерной цепи
в растворе макроскопического объема У, не занят сегментами N цепей:
; exp (NvfiV) = ехр (—Яф2);
здесь % — объемная доля полимера, при которой сумма всех сегментов
полимерного клубка должна быть равной объему V в условиях
отсутствия перекрывания клубков. Объем v выбирается как сфера, радиус
которой равен среднеквадратичному радиусу инерции (Rl)]/2. Так как
величина g-pa36 — gK0HUi экспоненциально уменьшается с увеличением
концентрации, она аппроксимируется предельным значением g* при
нулевой концентрации раствора. Следовательно,
g = ?конц + g* exp (-
37
При замене tt на gKOmb согласно уравнению B.60), второй вприальный
коэффициент получают так:
Теория исключенного объема дает значение А2 согласно уравнению
B.46), так что
300
295
290
285
304
500
292
0t05
0,01
а
0,015
280
г*
Рис. 2.3. Фазовые диаграммы для системы полистирол — циклогексан с учетом,
зависимости g от Мп, рассчитанной по уравнениям B.64) (а) и B.64а) (б):
/ — 5,2.10е; 2-> 1,6-10»; 3 — 5.Ы0*.
Окончательная форма для g такова:
g = Яконц + i|>i A — в/Г) [1 — F (z)] A + Я,) ехр (—Яср2),
где F(z) может быть выражена любым теоретическим уравнением!
F (г) = ттрг- In A + 5,73 z) (Флори — Кригбаум — Орофино),
F(z) = 1/A + 2,8652) (Штокмайер),
F(z) « 1 — A + 3,903z)~0'4683/1,828z (Курата — Ямакава),
F (z) = [ 1 — ехр (—5,73 z)] /5,73 г (Касасса — Марковиц).
Для построения спинодалей для системы полистирол —
циклогексан Конингсвельд и сотрудники [35] использовали gK0IW из уравнения
B.64) с параметрами критической точки, взятыми из [36]. Из разных
Р (г)-функций наиболее приемлемой оказалась форма Флори—Криг-
баума — Орофино, если константа изменялась от 2,73 до 5,73.
Значение % было взято равным примерно QfixJP. Используя затем общее gf
38
спинодаль получили путем двойного дифференцирования уравнения
B.59). Полученные таким образом кривые (рис. 2.3,6) лучше
согласуются с экспериментальными спинодалями независимо от того, какая
форма использована для F (г)-функции.
В рамках этих приближений была сделана попытка описать
критические данные для смесей полистирола и полиэтилена в Балканах и
а-замещенных алканах [37]. Поскольку в данном случае имеется
большое число параметров, эффектами концевых групп пренебрегают,
оставляют только шесть различных параметров взаимодействия,
характеризующих различные возможные типы контактов. Функция
g (Ту Ф2) идентична той, которая дается уравнением B.64), а параметр
р имеет сложную зависимость от шести упомянутых параметров.
Рассматривая смесь как бинарную, авторы работы [37], используя
критические условия, нашли
При введении некоторых приближений в это уравнение и в уравнение
для р, выражение для критического состояния можно записать в более
простой форме:
Y?*$yst B.65)
Р = (Z - 2) [A - 2/71) gbp + 'Г' (gelP + geiP - getb - gegb) +
2 + 2getb - getet)h B.66)
где через ghk = {ghks' + ghkH/T) обозначены параметры для h — k
контактов. Имеются срединные группы (Ь) и два типа концевых групп
(ег и е2) в алкановой молекуле и полимерные сегменты р в полистироле.
Приближение включает условия: уг<?1; z — 1^г — 2, е ^ 1/гх; 2/гг <^
<^z — 2. Если две концевые группы в молекуле растворителя идентичны
(ех = е2 = ё), число параметров уменьшается до трех. Это должно быть
применено к критическим данным для системы полистирол — и-алкан,
причем уравнения B.65) и B.66) в этом случае сводятся к следующему:
Y/(z - 2) ?8 a Yrj2 = A - 2rf) gbp + 2r~l (gep - geb) + ^2geb.
Рассчитанные этим способом кривые приведены на рис. 2.4
штриховыми линиями. Интересно, что значения gebs и gebH, полученные
таким образом, предполагают наличие нижней критической температуры
смешения для системы полиэтилен — w-алканы. Это хорошо
согласуется с экспериментом [38].
Из других работ, в которых была использована решеточная модель
-раствора, следует отметить работы Кеннеди и других [39, 40].
Теория Флори—Хаггинса в свое время сыграла очень важную роль
в понимании процессов растворения высокомолекулярных соединений
и внесла существенный вклад в общую теорию растворов, однако она
не лишена некоторых недостатков.
39
Т,К
493
473
483
4Z3
413
393
373
353
%K
A 53
413
373
333
10 15 n О
0,1 0,2 0Kip%
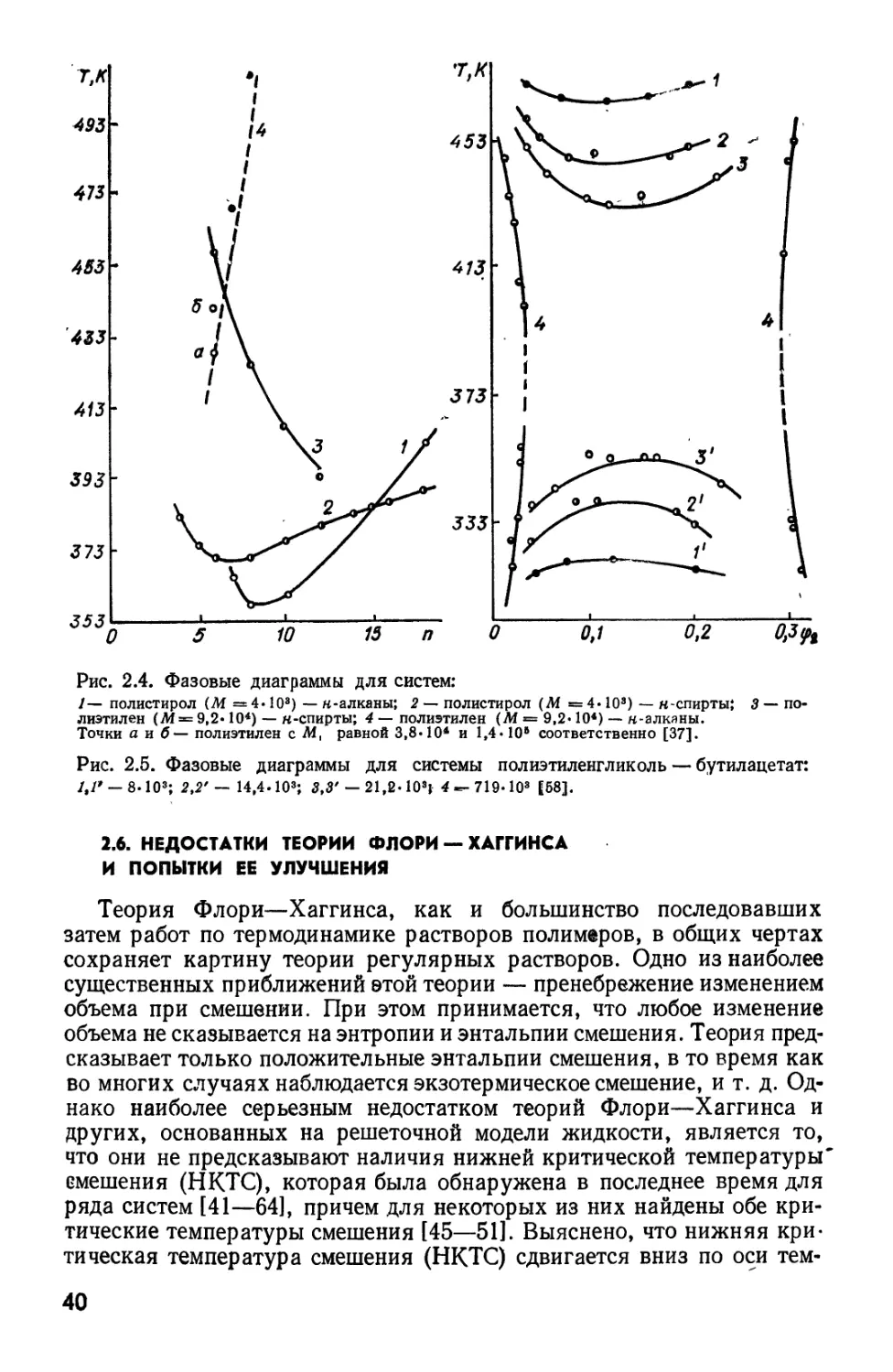
Рис. 2.4. Фазовые диаграммы для систем:
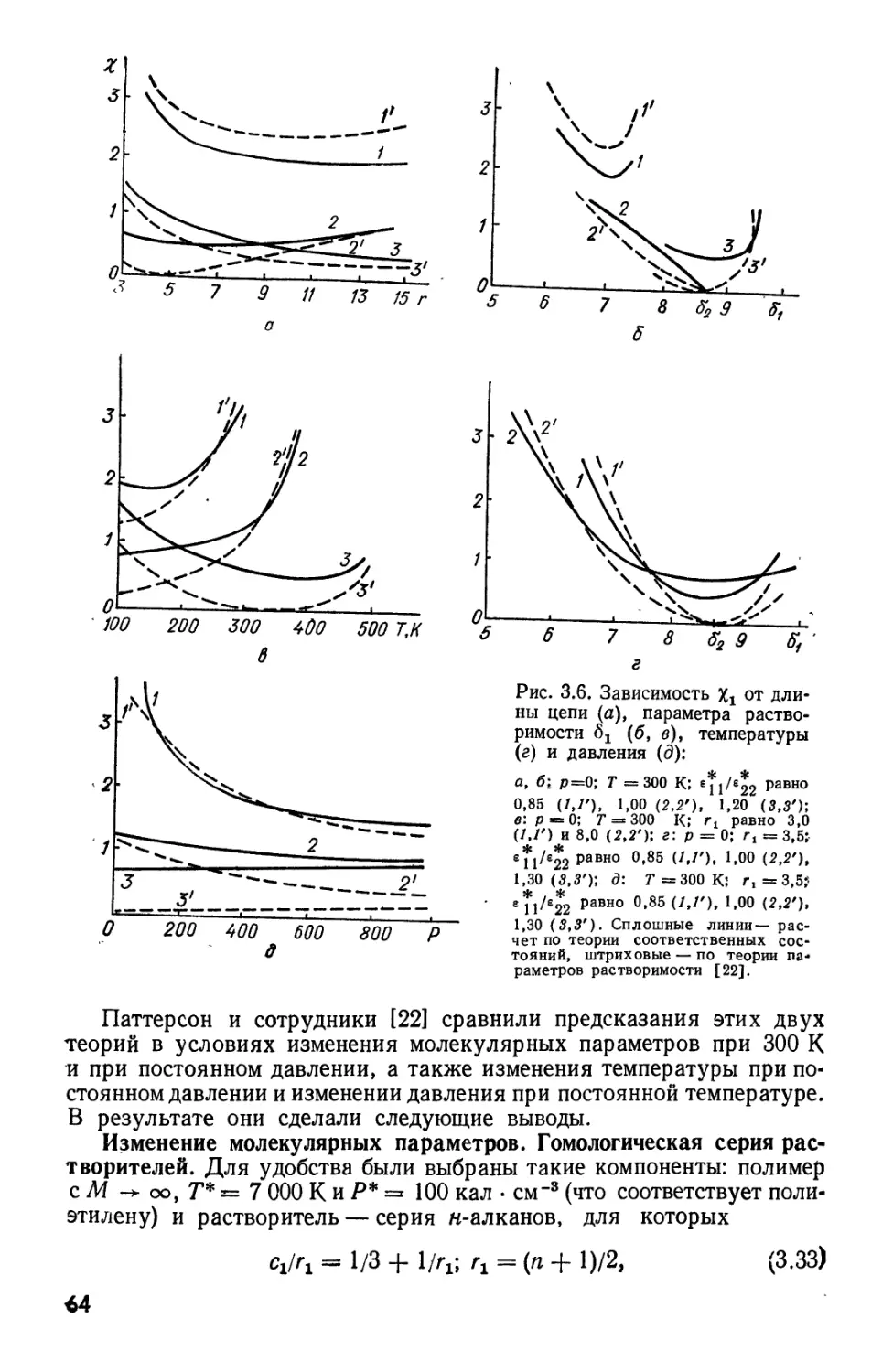
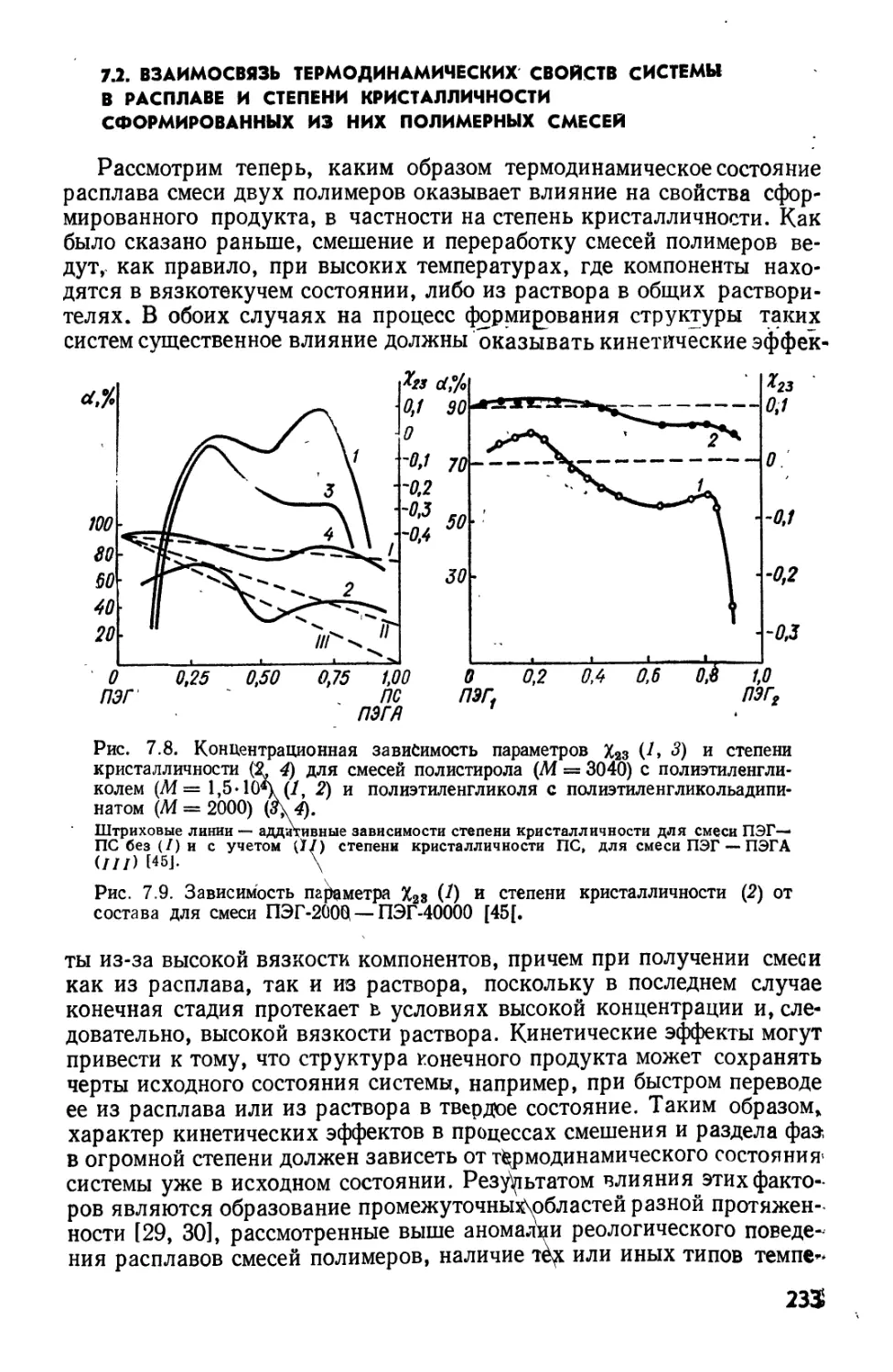
/— полистирол (М = 4.103) — «-алканы; 2 — полистирол (М = 4.103) — к-спирты* 3 —
полиэтилен (М = 9,2-10*) — «-спирты; 4 — полиэтилен (М *= 9,2* 10*) — и-алканы.
Точки а и б— полиэтилен с М, равной 3,8-10* и 1,4*10* соответственно [37].
Рис. 2.6. Фазовые диаграммы для системы полиэтиленгликоль — бутилацетат:
1,1* — 8- Ю3; 2,2' — 14,4.103; 3,3' — 21,2-108* 4 — 719- Ю3 158].
2.6. НЕДОСТАТКИ ТЕОРИИ ФЛОРИ — ХАГГИНСА
И ПОПЫТКИ ЕЕ УЛУЧШЕНИЯ
Теория Флори—Хаггинса, как и большинство последовавших
затем работ по термодинамике растворов полимеров, в общих чертах
сохраняет картину теории регулярных растворов. Одно из наиболее
существенных приближений этой теории — пренебрежение изменением
объема при смешении. При этом принимается, что любое изменение
объема не сказывается на энтропии и энтальпии смешения. Теория
предсказывает только положительные энтальпии смешения, в то время как
во многих случаях наблюдается экзотермическое смешение, и т. д.
Однако наиболее серьезным недостатком теорий Флори—Хаггинса и
других, основанных на решеточной модели жидкости, является то,
что они не предсказывают наличия нижней критической температуры*
смешения (НКТС), которая была обнаружена в последнее время для
ряда систем [41—64], причем для некоторых из них найдены обе
критические температуры смешения [45—51]. Выяснено, что нижняя
критическая температура смешения (НКТС) сдвигается вниз по оси тем-
40
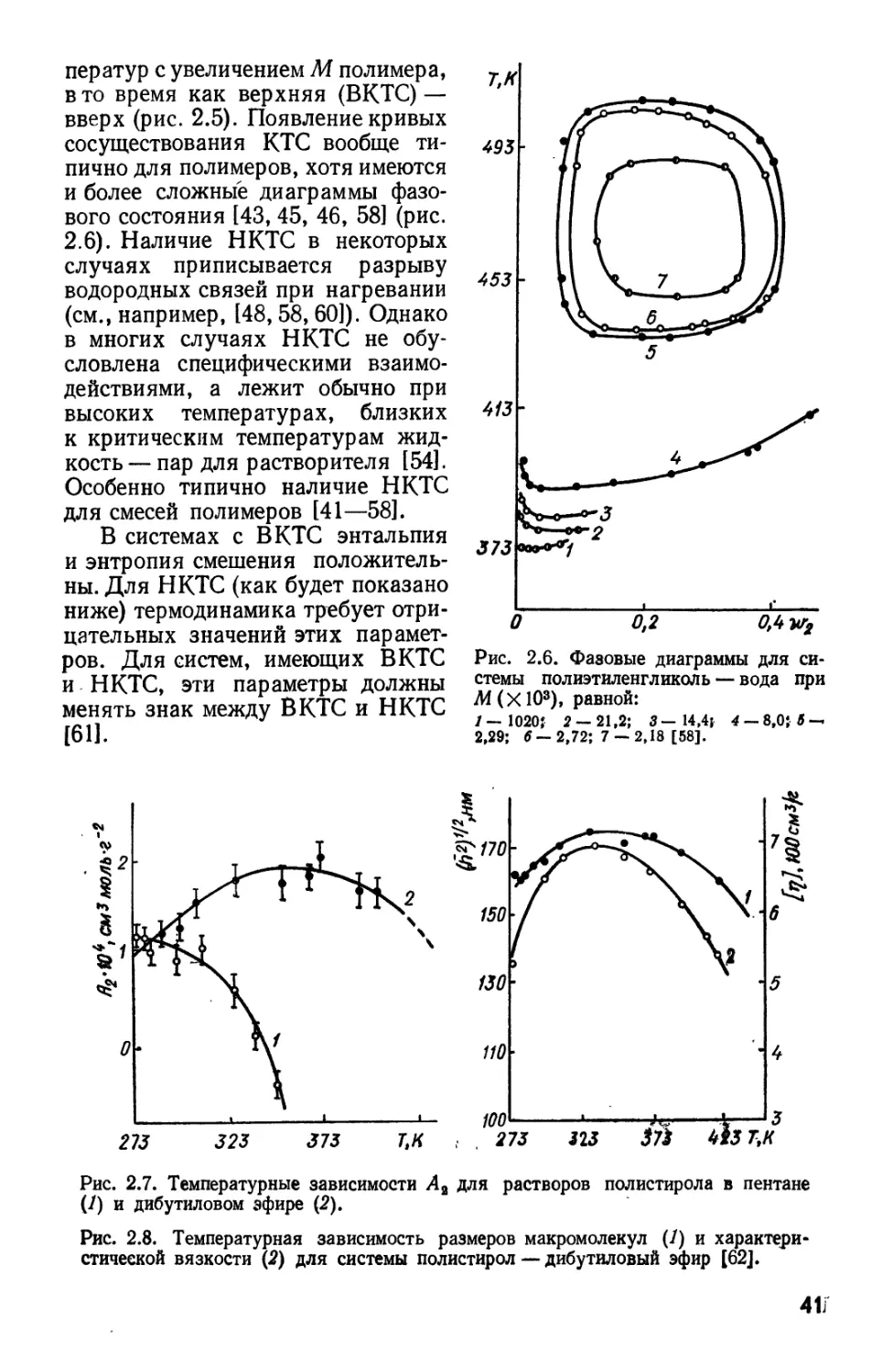
ператур с увеличением М полимера,
в то время как верхняя (ВКТС) —
вверх (рис. 2.5). Появление кривых
сосуществования КТС вообще
типично для полимеров, хотя имеются
и более сложные диаграммы
фазового состояния [43, 45, 46, 58] (рис.
2.6). Наличие НКТС в некоторых
случаях приписывается разрыву
водородных связей при нагревании
(см., например, [48,58, 60]). Однако
в многих случаях НКТС не
обусловлена специфическими
взаимодействиями, а лежит обычно при
высоких температурах, близких
к критическим температурам
жидкость— пар для растворителя [54].
Особенно типично наличие НКТС
для смесей полимеров [41—58].
В системах с ВКТС энтальпия
и энтропия смешения
положительны. Для НКТС (как будет показано
ниже) термодинамика требует
отрицательных значений этих
параметров. Для систем, имеющих ВКТС
и НКТС, эти параметры должны
менять знак между ВКТС и НКТС
[61].
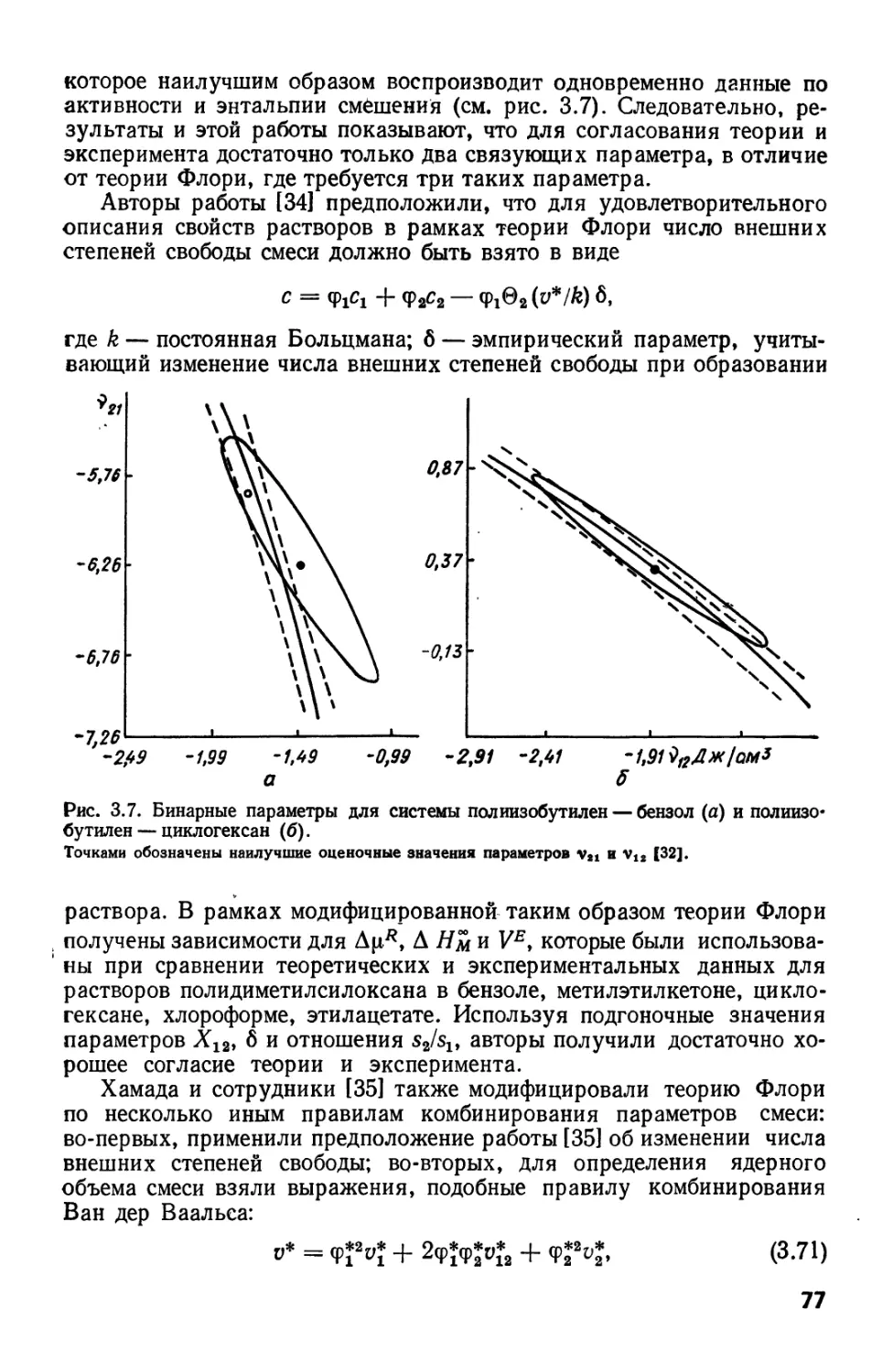
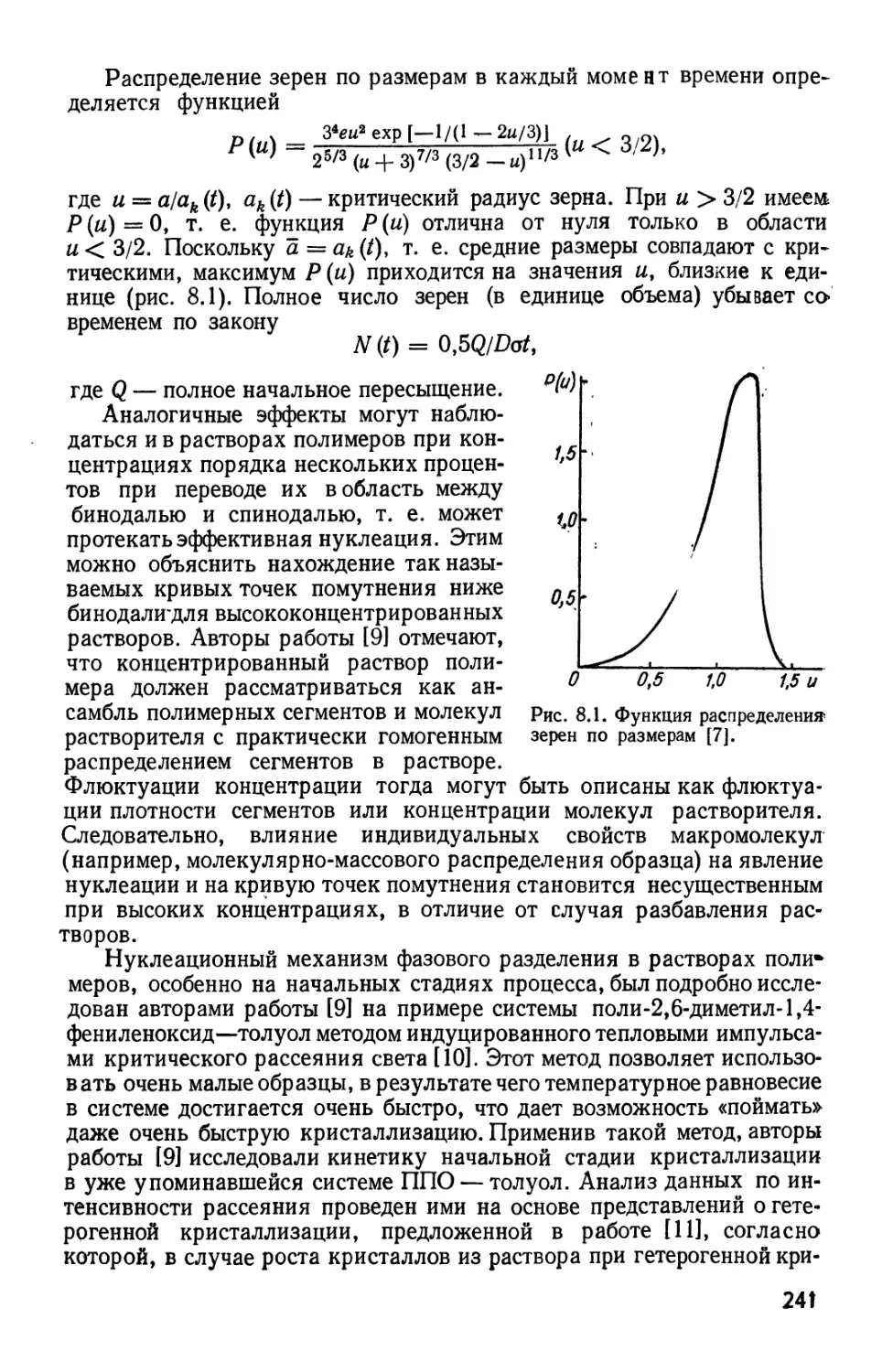
7, К
493
453
О 0,2
Рис. 2.6. Фазовые диаграммы для
системы полиэтиленгликоль — вода при
М(хЩ, равной:
7—1020* 2 — 21,2; 3—14,4* 4 — 8,0; б —
2,29; 6—2,72; 7 — 2,18 [58].
1
0
I
4j 2
ч
I
§/7*
150
130
110
213
323
373
Т.К
100
273 ПЗ
Лз к
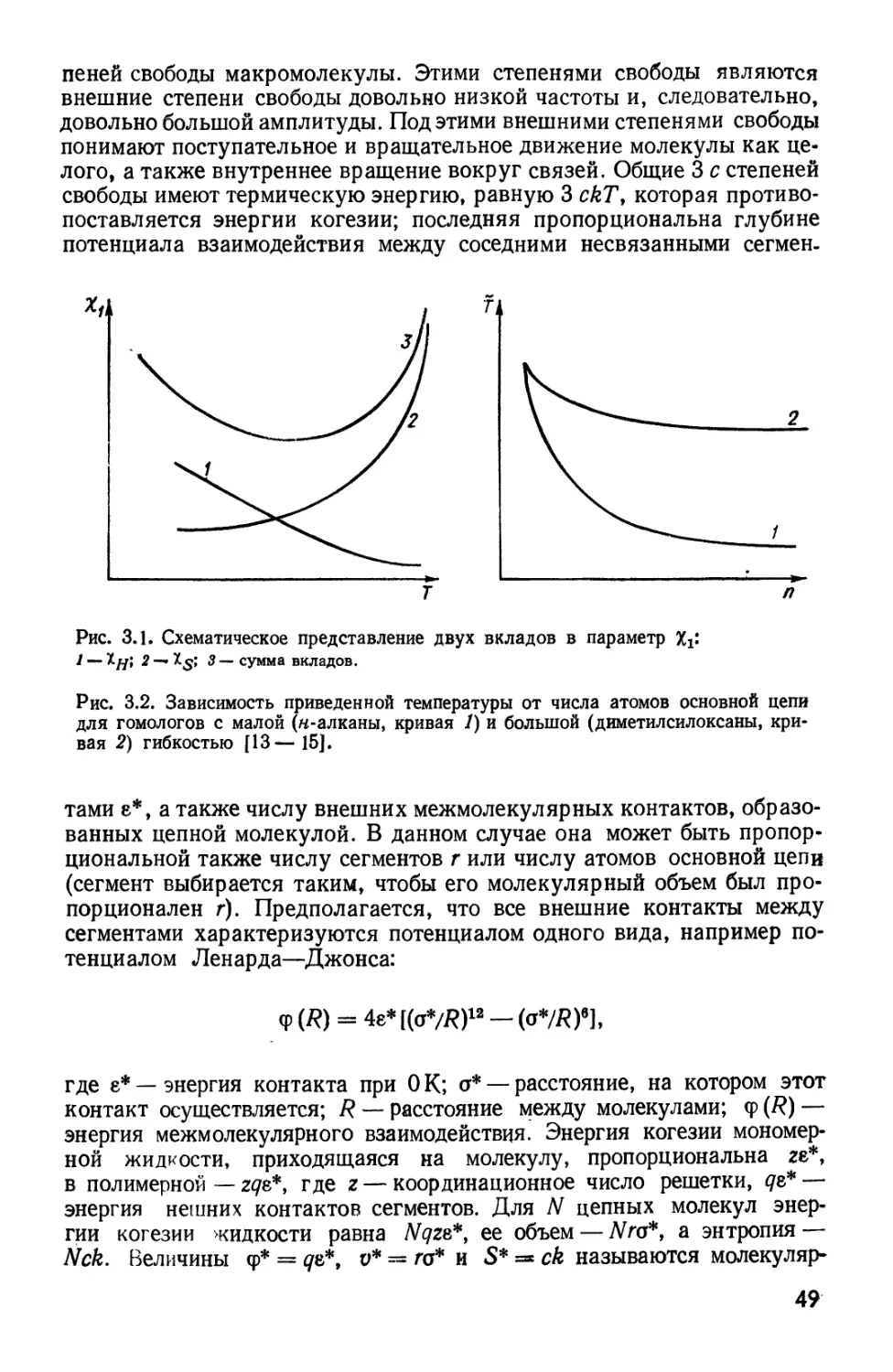
Рис. 2.7. Температурные зависимости А2 для растворов полистирола в пентане
A) и дибутиловом афире B).
Рис. 2.8. Температурная зависимость размеров макромолекул A) и
характеристической вязкости B) для системы полистирол — дибутиловый эфир [62].
41
Если система имеет две КТС, второй вириальный коэффициент А%
должен быть равен нулю и при приближении к НКТС (которую
обычно обозначают как. 0Н, наподобие вв), т. е. ожидается определенная
симметрия свойств между В КТС и НКТС (рис. 2.7, 2.8) [62].
Два различных соотношения типа уравнения B.67)
- в/Г); А, = A'tF (г) B.67)
должны быть корректны вблизи каждой из КТС, но не совсем
корректны в некоторой температурной области между В КТС и НКТС.
Следовательно, переменная z не может быть дана таким простым уравнением,
как уравнение B.45). Для получения корректной функции г (Т) Эйхин-
гер [61] ввел температурно-зависимый параметр грх. Согласно
определению, Rtytyl является парциальной молярной энтропией разбавления.
Интегрирование ведет к избыточному химическому потенциалу:
т
-Яф22 J ¦, (Т) от - - rt (Ь-) А;Ф;, B.68)
имеющему Л2, полученное из уравнения B.67) при у$>г = const. Эйхингер
ввел параметр ?, =**Т (di|ydT)p, приняв в качестве первого
приближения ? постоянным со средним значением f в диапазоне от 0В до @н*
В случае температурно-зависимого г^ имеем
т
% (Л = 4>ieB + J {ЦТ) dT = г|?1вв -1 In (в/П B.69)
где г|Iвв — значение г^ вблизи ВКТС. Тогда, подставив i|>i(T) в
уравнение B.68) и проведя интегрирование в пределах от 0В до 6Н>
получим выражение
t = 1 + A - l-J1 In (вв/вн), B.70)
по которому значение ? может быть рассчитано, если известны 0В, вн
и фх. Подстановка значения f, выраженного уравнением B.70), в
уравнение B.69) приводит к следующему:
в (Ц)\( У) Ц B.71)
Поскольку
то подставив А'2 из уравнения B.71) в уравнение B.72), получим
корректную температурную зависимость параметра 2. Используя уравнения
B.70), B.71) и B.69), экспериментальные значения 6В, 0Н и г^ для
42
какой-либо системы, по уравнениям B.70), B.71) и B.69) можно
рассчитать теоретическую зависимость г = /A/Т) для другой системы
в предположении, что уравнения B.46), B.47) и B.72)
предусматривают одинаковую функциональную зависимость ЛИАРДА2K/2 от г для
всех систем полимер — растворитель. В работе [64] было
показано, что универсальность такой зависимости действительно
наблюдается в системах полимер — растворитель.
Успехами в более полном описании термодинамических свойств
растворов полимеров с НКТС мы обязаны новым статистическим
теориям растворов полимеров, в основу которых положен принцип
соответственных состояний [64].
1. Flory P. J. Thermodynamics of polymer solutions.—J. Chem. Phys., 1941,
9, N 3, p. 660—664; 1942, 10, N 1, p. 51—53.
2. Flory P. J. Principles of polymer chemistry.— New York: Cornell univ. press..
1953.—594 p.
3. Huggins M. L. Physical chemistry of polymers.— New York: Interscience,
1958.— 175 p.
4. Flory P. /. Thermodynamics of dilute solutions of high polymers.— J. Chem.
Phys., 1945, 13, N 2, p. 453—457.
5. Flory P. J. Statistical mechanics of dilute polymer solutions.—Ibid., 1949,
17, N 7, p. 1347—1348.
6. Dondos Л., Benoit Я. Unperturbed dimensions of polymers in binary solvent
mixtures.— Eur. Polym. J., 1968, 4, N 5, p. 561—570.
7. Bianchi U.9 Magnasso V., Rossi C. Dimensions of high polymers in solution.—
Chim. e ind., 1958, 40, N 3, p. 263—266.
8. Schulz G., Kirste R. Uber die Temperature- und die MolekulargewichtsalhSn-
gigkeit des 2. Osmotischen Virialkoeffizienten (A2) von Polymethylmethacry-
latlosungen.— Z. phys. Chem., 1961, 27, N 5/6, S. 301—325.
9. Outer P., Can C, Zimm В. Я. Light-scattering investigation of the structure
of polystyrene.— J. Chem. Phys., 1950, 18, N 2, p. 830—835.
10. Schulz G.t Graubner H. Thermodynamic properties and molecular dimensions
in macromolecular solutions.— Z. Elektrochem., 1959, 63, N 2, p. 301—308.
11. Эскин В. ?., Волков Т. И. Светорассеяние и вязкость растворов поли-2,5-
дихлорстирола в диоксане.— Высокомолекуляр. соединения. Сер. А, 1963,
5, N 4, с. 614—619.
12. Цветков В. Я., Фаттахов К. 3., Каллистов О. В. Исследование растворов
линейных полимеров методом рассеяния света. 2. Молекулярные веса и
размеры молекул полиметилметакрилата в ацетоне.— Жури. теор. и эксперим.
физики, 1954, 26, № 3, с. 351—361.
13. Справочник по химии полимеров/Ю. С. Липатов, А. Е. Нестеров, Т. М.
Гриценко, Р. А. Веселовский.— К. : Наук, думка, 1971.— 536 с.
14. Stockrnayer W. Я. Problems of the statistical thermodynamics of dilute
polymer solutions.— Makromol. Chem., 1960, 35, S. 54—74.
15. Krigbaum W. P., Flory P. J. Statistical mechanics of dilute polymer solutions.
4. Variation of the osmotic second coefficient with molecular weight.—J. Amer.
Chem. Soc, 1953, 75, N 8, p. 1775—1778.
16. Zimm В. Я., Stdckmayer U7. Я., FixmanM. Excluded volume in polymer
chains.— J. Chem. Phys., 1953, 21, N 10, p. 1716—1723.
17. ZimmB. Я., Stockmayer W. H. Dimensions of chain molecules containing
branches and rings.— Ibid., 1949, 17, N 3, p. 1301—1314.
18. Fixman M. Excluded volume in polymer chains.— Ibid., 1955, 23, N 9,
p. 1656—1659.
19. Нестеров Л. ?. Справочник по физической химии полимеров. Т. 1. Свойства
растворов и смесей полимеров / Под ред. Ю. С. Липатова : В 3-х т. Киев :
Наук, думка, 1984. 376 с.
20. Dondos Л., Benoit Я. A new representation of viscosity data as a function of
molecular weight.— Polymer, 1977, 18, N 11, p. 1161—1163.
43
21. Flory P. J., Fox T. G. Treatment of intrinsic viscosities.—J. Amer. Chem.
Soc, 1951, 73, N 5, p. 1904—1908.
22. Cowie M. G. Estimation of unperturbed polymer dimensions from viscosity
measurements in non-ideal solvents.— Polymer, 1966, 7, N 10, p. 487—495.
23. Stockmayer W. #., Fixtnan M. On the estimation of unperturbed dimensions
from intrinsic viscosities.—J. Polym. Sci. C, 1963, N 1, p. 137—141.
24. Berry G. C. Thermodynamic and conformational properties of polystyrene.
2. Intrinsic viscosity studies on dilute solutions of linear polystyrene.— J.
Chem. Phys., 1967, 46, N 4, p. 1338—1352.
25. Нестеров А. Е. Критическая опалесценция растворов полимеров : Автореф.
дис. ... канд. физ.-мат. наук.— Л., 1966.— 16 с.
26. Shultz A. R., Flory P. J. Phase equilibria in polymer-solvent systems.— J.
Amer. Chem. Soc, 1952, 74, N 19, p. 4760—4767.
27. Koningsveld R., Kleintjens L. A. Liquid-liquid phase separation in multicompo-
nent polymer systems. 10. Concentration dependence of the pair—interaction
p.arameter in the system cyclohexane— polystyrene.— Macromolecules, 1971,
4, N 5, p. 637—641.
28. Orofino Т. Л., Mickey J. W. Dilute solution properties of linear polystyrene
in 0-solvent media.—J. Chem. Phys., 1963, 38. N 10, p. 2512—2520.
29. Koningsveld R., Staverman A. /. Determination of critical points in multi-
component polymer solutions.— J. Polym. Sci. C, 1967, 16, p. 1775—1786.
30. Scholte Th. G. Determination of thermodynamic parameters of polymer-solvent
systems by light scattering.— Eur. Polym. J., 1970, 6, N 8, p. 1063—1074.
31. Rehage G., Mdller D., Ernst 0. Entmischungserscheinungen in Losungen von
molekularuneinheitlichen Hochpolymeren.— Makromol. Chem., 1965, 88,
5. 232—255.
32. Van Emmerik P. 7\, Smolders C. A. Phase separation of polymer solutions.
The calculation of the cloud point curves with a concentration and
temperature— dependent free energy correlation parameter.— Eur. Polym. J., 1973,
9, N 2, p. 157—167.
33. Kennedy J. W., Gordon M., Koningsveld R. Generalization of the Flory-Huggins
treatment of polymer solutions.— J. Polym. Sci. C, 1972, N 39, p. 43—69.
34. Gordon M., Chermin H. A. G., Koningsveld R. Liquid—liquid phase separation
in multicomponent polymer solutions. 7. Relations for the splnodal and
critical loci.—Macromolecules, 1969, 2, N 2, p. 207—210.
35. Liquid—liquid phase separation in multicomponent polymer systems. 11.
Dilute and concentrated polymer solutions in equilibrium / R. Koningsveld, W. H.
Stockmayer, J. W. Kennedy, L. A. Kleintjens.— Ibid., 1974, 7, N 1, p. 73—
79.
36. Koningsveld /?., Kleintjens L. Л., Shultz A. R. Liquid — liquid phase
separation in multicomponent polymer solutions. 9. Concentration dependent pair
interaction parameter from critical miscibility data on the system
polystyrene—cyclohexane.— J. Polym. Sci.: Polym. Phys. Ed., 1970, 8, N 8, p. 1261 —
1273-
37. Koningsveld R.f Kleintjens L. A. Thermodynamics of polymer mixtures.— J.
Polym. Sci. C, 1977, N 61, p. 221—249.
38. Orwoll R. A., Flory P. J. Equation-of-state parameters for normal alkanes.
Correlation with chain length.— J. Amer. Chem. Soc, 1967, 89, N 26, p. 6814—
6822.
39. Kennedy J. W. Thermodynamic limitations on interaction functions and the
theta temperatures of polymer solutions.— J. Polym. Sci. C, 1972, N 39, p. 71 —
81.
40. The heats of dilution of polystyrene solutions / Y. Baba, K. Fujimoto, A. Ka-
gemoto, R. Fujischiro.—Makromol. Chem., 1977, 178, N 5, p. 1439—1444.
41.. Freeman /., Rowlinson J. S. Lower critical points in polymer solutions.—
Polymer, 1960, 1, N 1, p. 20—26.
:42. Upper and lower critical solution temperatures in polystyrene solutions / S. Sae-
ki, N. Kuwahara, S. Konno, M..Kaneko.—Macromolecules, 1973, 6, N 2, p.
246—250.
Ш. Allen G., Baker С. Н. Lower critical phenomena in polymer — solvent system.—
Polymer, 1965, 6, N 4, p. 181—183.
44
44. Liddetl А. #., Swinton F. L. Thermodynamic properties of some polymer
solutions at elevated temperatures.— Discuss. Faraday Soc, 1970,* N 49,
p. 115—120.
45. Cowie J. M. G., Maconnachie Л., Ranson R. J. Phase equilibria in cellulose
acetate—acetone solutions. The effect of the degree of substitution and
molecular weight on upper and lower critical solution temperatures.— Macromolecu-
les, 1971, 4, N 1, p. 57—61.
46. Stow K. S.t Delmas G., PattersonD. Cloud—point curves in polymer solutions
with adjacent upper and lower critical solution temperatures.—Ibid., 1972,
5, N 1, p. 29—34.
47. A study of the thermodynamic properties and phase equilibria of solutions of
polyisobutene in л-pentane / C. H. Baker, W. B. Brown, G. Gee et al.—
Polymer, 1962, 3, N 2, p. 215—230.
48. Upper and lower critical solution temperatures in polystyrene solutions / S. Sae-
ki, N. Kuwahara, S. Konno, M. Kaneko.— Macro molecules, 1973, 6, N 4,
p. 589—593.
49. Upper and lower critical solution temperatures in polystyrene solutions/
N. Kuwahara, S. Saeki, T. Chiba, M. Kaneko.— Polymer, 1974, 15, N 12, p.
777^782.
50. Upper and lower critical solution temperatures in polystyrene solutions. 3.
Temperature dependence of the %i parameter / S. Saeki, S. Konno, N. Kuwahara
et al.-— Macromolecules, 1974, 7, N 4, p. 521—527.
51. Upper and lower critical solution temperatures in polystyrene solutions. 4. Role
of configurational heat capacity/S. Konno, S. Saeki, N. Kuwahara et al.— Ibid.,
1975, 8, N 6, p. 799—804.
52. Cowie J. W. G., McEwen I. J. Polymer—cosolvent systems. 4. Upper and lower
critical solution temperatures in the system methylcyclohexane — diethyl
ether —polystyrene.—Ibid., 1974, 7, N 3, p. 291—296.
53. Patterson D.t Delmas G., Somsinsky T. A comparison of lower critical solution
temperatures of the some polymer solutions.— Polymer, 1967, 8, N 10, p. 503 —
517.
54. Delmas G., Patterson ?>., Somsinsky T. Thermodynamics of polyisobutylene-
/i-alkane systems.—J. Polym. Sci. C, 1962, N 57, p. 79—98.
55. Delmas G., Sant-Romain P. de Upper and lower critical solution temperatures
in polybutadiene — alkane systems. Effect of the surface-to-volume ratio of
the polymer.— Eur. Polym. J., 1974, 10, N 12, p. 1133—1140.
56. Нижние критические температуры растворения полистирола в бензоле и
этилбензоле/В. М. Андреева, А. А. Аникеева, С. А. Вшивков, А. А. Тагер.—
Высокомолекуляр. соединения. Б., 1970, 12, № 11, с. 789—793.
57. Вшивков С. А,, Тагер А. А., Гайфуллина Н. Б. Исследование фазового
равновесия растворов полимеров вблизи точек помутнения и измерения объема
сосуществующих фаз и их состава.— Там же, 1976, 18, № 1, с. 25—28.
58. Upper and lower critical temperatures in polyethylene glycol) solutions /
S. Saeki, N. Kuwahara, M. Nakata, M. Kaneko.—Polymer, 1976, 17, N8, p.
685—690.
59. Bailey F. ?., Callard R. W. Some properties of poly (ethyl ene oxide) in aqueous
solution.— J. Appl. Polym. Sci., 1959, 1, N 1, p. 56—62.
60. Влияние температуры на процесс формирования полиуретановой сетки из
олигомеров / Т. Э. Липатова, А. Е. Нестеров, В. К. Иващенко, Ю. С.
Липатов.— Высокомолекуляр. соединения А, 1970, № 5, с. 1039—1043.
61. Eichinger В. Е. Heat capacity of dilute polymer solutions.—J. Chem. Phys,,
1970, 53, N 2, p. 561—566.
62. Delmas G., Patterson D. The temperature dependence of polymer chain
dimensions and second virial coefficients.— Polymer, 1966, 7, N 10, p. 523—525.
63. Casassa E. Thermodynamics of polymer solutions.— J. Polym. Sci. C, 1976,
N 54, p. 53—83.
64. Prigogine /., Trappeniers N.t Mathot V. Statistical thermodynamics of r-mers
and r-mer solutions.— Discuss Faraday Soc, 1953, N 15, p. ?3—107.
66. Цветков В. #., Эскин В. ?., Френкель С. #. Структура макромолекул в
растворах.— М. : Н-аука, 1964.— 719 с.
ГЛАВА 3
НОВЫЕ СТАТИСТИЧЕСКИЕ
ТЕОРИИ РАСТВОРОВ ПОЛИМЕРОВ
В выражении для свободной энергии
смешения AG = А Н — ТА S предполагалось, что только А Я зависит от
природы молекул или характера межмол'екулярных взаимодействий,
характеризуемых эмпирическим параметром %х. Однако при
экспериментальном определении Хг из A G и А Н оказалось, что его значения
не одинаковы. Поэтому величина А w12, которой выражается параметр
5Ci (см. уравнение B.5)), была вновь интерпретирована как обменная
энергия с энтальпийным и энтропийным вкладами:
Аш12 = Aw12 (H) - TAw12 (S); C.1)
(H) zAw12(S)
Поскольку при образовании контактов 1—2 взамен контактов 1—1
и 2—2 при смешении энтропия увеличивается, то Awl2(S)
положительна и, следовательно, %s является малой отрицательной поправкой
к параметру %х. В новых статистических теориях эта поправка
непосредственно вытекает из учета изменения объемов при смешении.
Ранее внимание исследователей было сконцентрировано на
различии в энергиях когезии между сегментами полимера и молекулами
растворителя разной химической природы. Новым фактором, который
был введен с целью определения различия между молекулами
полимера и растворителя, стала разность между свободными объемами
полимера и растворителя, в частности, тот факт, что растворитель обычно
сильнее расширяется при нагревании, чем полимер. Это различие в
свободных объемах обусловлено разными размерами и длинами цепей
полимера и растворителя. Раствор полимера в химически идентичном
растворителе должен иметь одинаковые значения wllt w22 и w12, но
различные свободные объемы, что приводит к новым
термодинамическим следствиям.
Учет свободного объема в теории растворов полимеров был введен
в 50-х годах Пригожиным с сотрудниками [1], которые предложили
картину смешения, отличную от той, которая следовала из теории Флори—
Хаггинса. Теория Пригожина была дальше развита в работах Паттер-
сона и сотрудников [2—4] и стала основной базой теории Флори для
жидких смесей [5, 6].
46
3.1. ТЕОРИЯ ПРИГОЖИЙ А — ПАТТЕРСОНА
В работе [7] Паттерсон рассмотрел основные идеи учета свободного
объема и дал интуитивное введение в теорию Пригожина. Рассмотрим
эти идеи.
Известно, что теория регулярных растворов оперирует растворами
сферических молекул. Растворы полимеров отличаются от последних
тем, что полимер состоит из сегментов, связанных в цепочку. Число
способов размещения полимерных молекул в решетке раствора
отлично от такового для раствора сферических молекул и, следовательно,
не приводит к идеальной энтропии смешения. Теплота смешения в
строго регулярных растворах постоянна, так что она пропорциональна
обменной энергии: это энергия образования контакта между.
молекулой растворителя и сегментом полимера, т. е. Д w12. Параметр %i
выражается уравнением B.5), a A w12 состоит из двух вкладов: Дш12(#)
и Д w12(S) (в терминологии Флори %и и %s обозначаются как kt и -у—
—Хт). Однако типичные значения параметров Xi показывают, что %s
теперь положительна и по величине много больше, чем %н. Поэтому
значения %i обычно находятся в пределах 0,35—0,5, независимо от
того, каким бы малым ни было значение %н. Новый положительный
знак %s соответствует отрицательному значению Д wl2(S) (см.
уравнение C.1)) и не объясняет большого увеличения беспорядка при
образовании контактов полимер — растворитель. Ясно, что
взаимодействие полимер — растворитель качественно изменяется из-за того, что
сегменты связаны в цепь. Традиционные теории этого не объясняют.
Новые трактовки эффекта исключенного объема тоже не решают этого
затруднения. Взаимодействие полимер — растворитель
характеризуется исключенным объемом v09 связанным с энергией взаимодействия
сегментов U (г). Поскольку U (г) не может быть оценена точно, v0
по старым теориям связывается с Хг [8]:
где Vx — молярный объем растворителя, Na — число Авогадро.
Большие трудности при использовании традиционных теорий, как
уже было сказано раньше, возникают при объяснении наблюдаемого
фазового расслоения раствора при повышении температуры.
Температурная зависимость параметра Xi не является монотонно убывающей
функцией %i(T) в виде %1=а+ЫТ (в теории Флори—Хаггинса
-^—Xi= ^i(l — в/Т)). Новая критическая температура смешения
(НКТС) была названа нижней критической температурой.
Термодинамическими условиями наличия НКТС являются АН < 0 и A S < 0,
т. е. действительно система при смешении упорядочивается. Таким
образом, комбинаторная Д SK может быть перекрыта большим
отрицательным членом Д w12(S) (или положительным %s). Это условие
реализуется в очень многих смесях молекул близких размеров, например
47
в водных растворах аминов, где упорядочение молекулы воды вокруг
молекулы растворенного вещества приводит к отрицательному
значению A w12(S). Однако этот эффект характерен многим растворам
полимеров. Причина НКТС, как и положительных значений %s> как будет
показано ниже, заключается в различии свободных объемов компонент,
в частности плотной полимерной жидкости и более рыхлого
растворителя. Растворитель в плотной полимерной среде не может
расшириться так, как в чистом виде при данной температуре, поэтому
свободный объем раствора меньше аддитивного, т. е. общее изменение объема
при смешении отрицательно. С этим связан экзотермический эффект
смешения и отрицательный вклад в Д S.
Каждая функция смешения представляет собой сумму двух
вкладов, один из которых связан с различием в контактной энергии (т. е.
wn и до22), а второй обусловлен разными свободными объемами
компонент. Знак %! или A G будет зависеть от относительного вклада %и
и %s (рис. 3.1). Первый вклад (т. е. связанный с разностью в
контактных энергиях) может быть оценен из теории Флори—Хаггинса. Он
положителен и уменьшается с температурой. С повышением
температуры, однако, увеличивается разница в свободных объемах полимера
и растворителя, а следовательно, растет и новый вклад в %г. В
результате %i будет повышаться, и при критическом значении хькрит будет
наблюдаться НКТС.
Рассмотрим характеристики НКТС. Согласно теории Флори—
Хаггинса, критическое значение %1,Крит определяется из выражения
где г — отношение молярных объемов полимера и растворителя. Как
видно из уравнения C.2), %1,крит увеличивается с уменьшением
молекулярной массы; т. е., как и в случае систем с ВКТС, имеется
возможность фракционирования полимеров путем повышения
температуры в сторону НКТС [10].
Поскольку 5Ci растет при приближении к НКТС, размеры
макромолекул и вторые вириальные коэффициенты уменьшаются [11]. На рис.
2.8 приведены данные для раствора полиизобутилена в w-пентане при
изменении температуры от 273 до 349 К. В данном случае изменение
%i в основном обусловлено лишь различием в свободных объемах
компонент. Поскольку молекулярная масса полиизобутилена очень
велика (М « 9 • 106), НКТС должна соответствовать %х= 1/2 и в-тем-
пературе. Если учесть некоторую разность в контактной энергии
взаимодействия между полиизобутиленом и растворителем, взяв в
качестве последнего бутиловый эфир, будут проявляться оба вклада в %и
а общая зависимость Xi от Т изобразится кривой 3 на рис. 3.1.
Размеры макромолекул и второй вириальный коэффициент проходят
через максимум, как показано на рис. 2.8.
Учет вклада различия в свободных объемах компонент в параметр
%г дает теория Пригожина [1]. Расширение жидкости, состоящей из
цепных молекул, отражает термическую энергию только некоторых сте-
48
пеней свободы макромолекулы. Этими степенями свободы являются
внешние степени свободы довольно низкой частоты и, следовательно,
довольно большой амплитуды. Под этими внешними степенями свободы
понимают поступательное и вращательное движение молекулы как
целого, а также внутреннее вращение вокруг связей. Общие 3 с степеней
свободы имеют термическую энергию, равную 3 ckT, которая
противопоставляется энергии когезии; последняя пропорциональна глубине
потенциала взаимодействия между соседними несвязанными сегмен-
Рис. 3.1. Схематическое представление двух вкладов в параметр %х:
1 — Xfj; 2 -*• Х$; 3 — сумма вкладов.
Рис. 3.2. Зависимость приведенной температуры от числа атомов основной цепи
для гомологов с малой (я-алканы, кривая /) и большой (диметил сил океаны,
кривая 2) гибкостью [13—15].
тами е*, а также числу внешних межмолекулярных контактов,
образованных цепной молекулой. В данном случае она может быть
пропорциональной также числу сегментов г или числу атомов основной цепи
(сегмент выбирается таким, чтобы его молекулярный объем был
пропорционален г). Предполагается, что все внешние контакты между
сегментами характеризуются потенциалом одного вида, например
потенциалом Ленарда—Джонса:
где е* — энергия контакта при О К; ff* — расстояние, на котором этот
контакт осуществляется; R — расстояние между молекулами; ф (R) —
энергия межмолекулярного взаимодействия. Энергия когезии
мономерной жидкости, приходящаяся на молекулу, пропорциональна ге*,
в полимерной — z^e*, где г — координационное число решетки, qe* —
энергия нешних контактов сегментов. Для N цепных молекул
энергии когезии жидкости равна Nqze*, ее объем — Nro*, а энтропия —
*
р q
Nek. Величины ф* = </е*, у* = ш* и S*
ck называются молекуляр-
49
ными единицами, причем q ^ г. Свободный объем жидкости зависит
от отношения двух указанных энергий, названного приведенной
температурой:
~ т
m * термическая
1 когезионная
/ С \ kT Т
В этом выражении с/г представляет собой однутреть числа внешних
степеней свободы на сегмент. Для серии химически идентичных
гомологов с/г уменьшается с увеличением г или п. Например, мономер имеет
три внешних степени свободы, так что с/г = 1. Только две внешние
степени свободы добавляются для димера, поскольку одна из степеней
свободы связана с высокочастотными колебаниями сегментов и не
учитывается. Следовательно, для димера с/г = 5/6. Для длинных молекул
параметр с/г зависит от протяженности участка цепи, для которого
крутильные колебания не зависят от других участков, т. е. от гибкости
цепи (сегмента Куна). Отношение с/г изменяется линейно с 1/г, т. е.
с/г = А + b/r, где А — мера гибкости цепи (сегмент Куна). Для
сферических молекул (мономеров) f зависит только от параметра энергии
когезии е*, так как с/г «= 1. Для жидкости из цепных молекул 71 и
свободный объем зависят еще от того, связаны ли сегменты в цепочку
(например, от длины и гибкости цепи через параметр с/г, названный
«структурным параметром»).
Схематическая зависимость Т от п для двух серий гомологов (рис.
3.2) показывает, что в первом случае вращение вокруг связей
относительно ограничено (в алканах), во втором — цепь очень гибкая (ди-
метилсилоксаны). Видно, что различие в свободных объемах при
любой температуре больше между, например, полиэтиленом и пентаном,
чем между полидиметилсилоксаном и его гомологом с п = 5 (гексаме-
тилдисилоксан). Это отражается и на НКТС. Для системы
полиэтилен — пентан НКТС равна 363 К и составляет 0,77 от критической
температуры пентана [9], а система полидиметилсилоксан — гекса-
метилдисилоксан имеет НКТС, равную 484 К, что составляет 0,95 от
критической температуры гексаметилдисилоксана [12].
Согласно теории Пригожина, температура входит в функцию
распределения частиц в конфигурационном пространстве (см. уравнение
B.39)), но в приведенной форме. При постоянном приведенном
давлении (обычно нулевым для жидкости при давлении насыщения паров)
молярные или удельные термодинамические величины связаны с
приведенными величинами параметрами приведения, зависящими только
от длины цепи
V (л, Т) - Vl (n) V (f) - rv*V (f)9
U (л, Т) = Uо (п) 0 (?) - гг*0 (Г),
G(n, T)~G(f)U*(n)=*re*G(f).
Величина U представляет собой конфигурационную или внешнюю
молекулярную энергию жидкости; она отрицательна.
50
Параметры приведения могут рассматриваться как
характеристические молекулярные величины при О К. Приведенные величины
определяются из следующих соотношений:
Энергия U = U/MUo = U/Nqs*9
Объем V = V/Nvq. = VlNro**,
Энтропия S = S/NSl = S/Nck,
Давление Р = — (dU/dVh = PVllUl,
Температура 7 = {dU/dS)~ = TSo/U*o,
Теплоемкость CP(f) = CP(Tt n)/NSo.
Симха с сотрудниками [13—15] по- у{
казал, что для многих полимергомо-
логов данные по V (п, Т) могут быть
сведены к простой кривой V (Т)
(рис. 3.3). _ По мнению Паттерсона,
кривая V (Т) универсальна для всех
жидкостей с дисперсионными силами
взаимодействия.
Теория Пригожина предполагает,
что смесь ведет себя как чистая
жидкость со средним параметром
приведения для любой термодинамической
величины и средней приведенной тем- рис. 3.3. Схематическое представ-
пературой, в частности: ление зависимости приведенного
m * * объема Vе от приведенной темпе-
v ~xxvx+x2v2, ратуры Т [13— 15J.
где xt и #а— мольные доли компонентов. Подобно этому
С = Х]
Тогда, согласно уравнению C.3), приведенную температуру смеси
можно определить из выражения
f = — = (l\ ~ =
T* \q) e*
kT
хлсл
о* I
Т* \д I 8* (xtfi + *2?а)8* *i?i + *A 8* ' *Л + ^Л 8*
Если умножить и разделить каждое слагаемое на ог и q2, получим
где
C.4)
представляют собой «доли поверхности», т. е. отношение числа
внешних контактов молекул компонентов 1 и 2 к числу всех внешних
контактов в растворе.
5Г
Рассмотрим два компонента [4]: 1 — растворитель, а 2 — полимер,
которые взяты из двух разных гомологических рядов и
характеризуются, естественно, разными параметрами, но подчиняющимися
принципу соответственных состояний.
Три параметра (б, р и К) описывают различие между полимером
и растворителем:
б = Ег/г2 — 1,
Р = а2/а1—1, C.5)
Первые два параметра не зависят от молекулярной массы
полимера; третий должен быть положительным и увеличиваться
асимптотически с увеличением разности в длинах цепей полимера и
растворителя. Для полимера с бесконечной молекулярной массой этот параметр
значительно больше, чем аир для большинства систем. Если
определить «поверхностную долю» так, как дано уравнением C.4), то для
-смеси
о* = хг8г -f- x2S2,
[/* = XJJ[ + x2U; - ггХгХ^ (x1q1 + x2q2).
Нелинейный член в этом уравнении приводит к уменьшению коге-
зии в смеси, обусловленной обменной энергией образования контакта
1—2 (б-эффект) и смешением сегментов разного размера (р-эффект).
Коэффициент v2 может быть представлен как сумма этих двух эффектов.
Если для простоты предположить правило геометрического среднего
ДЛЯ 812, ТО
v2^62/4 + 9p2. C.6)
Точное значение коэффициента перед р2 неопределенно. Тогда
1_4-(Я + б)Х2^тХ2, C.7)
где
* - 1- (Tl/Tl) = (К + б)/A + б) ~ X + б, C.8)
если 6 = р =* 0, уравнение C.7) является точным.
Некомбинаторная часть свободной энергии смешения Гиббса
тогда имеет вид
AG = U*G(f, P)-x1U{G(fv PJ-xJJlQif^ P2), C.9)
так как р = 0.
Полагая Р = 0, из уравнения C.9) можно получить
AGi = fcWirf - U[ [-О (f)] v2*! + U{ [F (f) - F(f)x + (8.1Q)
+ (T1-f)dF/fff] .
52
x = kxRT<A = U[ [-U (f) + TCp] v*x2 + U\ [U (f) -
- U G\) + G\ - f) dU/df]. C.11)
Здесь предполагается, что X2-^q>2, поскольку q -»- г.
Величины Д Gx и Д Hj состоят из двух частей, которые вносят вклад
в %i и kv Причем, Д Gx зависит от параметров энергии и размера
(б и р). Это аналогично г Aw12 или %г и kx в теории Флори—Хаггинса.
Теперь вклад в %г изменяется аналогично величине— U(T),
которая может быть определена как теплота испарения усредненного
сегмента раствора. С увеличением концентрации раствора
приведенная температура Т уменьшается, а величина —U увеличивается в
соответствии с уплотнением раствора. Таким образом, параметр Xi
повышается с ростом концентрации полимера в растворе.То же
происходит и с параметром Ах.
Второй вклад в уравнениях C.10) и C.11) зависит только от
«структурного параметра» Я в т и характеризует смесь гомологов
разной длины цепи, где б = р = 0, т. е. т = Я.
Разлагая уравнения C.10) и C.11) в ряд по^ и используя
уравнение C.8), получаем выражения для %{ и kt:
C.12)
C.13)
- 1/2 = К ~ Хх = (Cp/R) v> - (Ср + ™р/П) т>, C.14)
Td(CpIT)dT з
^ 9
C.15)
Каждый параметр содержит два вклада: первый обусловлен
различием в энергиях когезии и размерах между сегментами полимера
и растворителя, второй — только величиной К, т. е. «структурным
фактором».
3.2. РОЛЬ РАЗЛИЧНЫХ ВКЛАДОВ В ПАРАМЕТРЫ СМЕШЕНИЯ
Паттерсон [4] рассмотрел роль разных вкладов в параметры
взаимодействия.
Влияние энергии когезии и размера на Xi и *i- Значения — U,
—U + ТСр и Ср положительны для любого растворителя.
Следовательно, вклады энергии когезии и размера в %i и kx положительны, что
и следовало ожидать (это вытекает и из модели жесткой решетки,
53
принятой в теории Флори—Хаггинса). Величины —U и т. д.
пропорциональны длине цепи для серии гомологов растворителя; таким
образом, их вклады в термодинамические параметры подобны таковым
в модели жесткой решетки.
Структурный вклад в Xi- Пусть б = р = 0. Следовательно,
параметр %i будет зависеть лишь от структурного вклада, оцениваемого
параметром Я.
Симха с сотрудниками [13—15] показали, что данные по V (Т) для
разных полимеров и олигомеров могут быть представлены в
приведенной форме, и получили величины Т\1Т% для разных систем
полимер — растворитель. Поэтому % может быть найден из уравнения C.6).
Например, для системы полиэтилен—гептан % равен 0,40, полидиметил-
силоксан — димер диметилсилоксана — 0,25, полистирол —
толуол — 0,35. Величина Ср для органических растворителей вблизи
комнатной температуры составляет примерно 40—67 Дж/К • моль. При
% = 0,35 и Ср= 58,5 Дж/К • моль структурный вклад в %г будет
равным 0,42, т. е. даже без учета б и р получается очень большое значение
5Са- Можно сделать вывод, что только различие в длинах цепей
полимера и растворителя приводит к большим значениям %v
Из уравнения C.12) видно, что лучшим растворителем для
полимера является не олигомер, а компонент с р == 0 и отрицательным
значением б, т. е. с большей молекулярной энергией когезии, чем у
полимера. Параметр %г проходит через минимум, когда
а минимальное значение Xi выражается так:
_? -цср/т
Xl"" 4Д * СР + (-U/2T) •
Используя те же значения X и Ср и полагая, что —U = 35,5 кДж/
/моль при Т = 300 К, получим %г= 0,22.
При приближении к критической точке жидкость — пар для
растворителя Ср-+ оо и dCpldT ->¦ оо. Таким образом, структурный вклад
в %i увеличивается беспредельно, поэтому при повышении температуры
(НКТС) должно наблюдаться фазовое расслоение. Структурный вклад
в kx из уравнения C.13) отрицателен и его величина растет с
повышением температуры. Теплота смешения, таким образом, становится
отрицательной при достаточно высоких температурах, близких
к НКТС. Однако во многих случаях для летучих растворителей вместе
с малыми значениями б и р температура 298 К очень близка к
критической температуре жидкость — пар, поэтому kx не всегда отрицательно.
Другим следствием структурного вклада при НКТС является то,
что общая энтропия %, т. е. сумма комбинаторной A/2) и
некомбинаторной (ifi— 1/2) энтропии смешения отрицательна (см. уравнение
C.14)). Даже при 298 К для высокомолекулярного полиизобутилена
в пентане отрицательная общая энтропия была получена. Для
большинства же систем при обычных температурах г^, однако, положи-
54
тельна, но ^г—172 отрицательна. Первый член уравнения C.14)
положителен, он характерен для смесей мономерных жидкостей, где
некомбинаторная энтропия смешения действительно положительна.
Для растворов полимеров она содержит отрицательный структурный
вклад, который приводит к %—1/2 <0.
Структурный вклад при низких температурах. Изменение Ср при
обычных температурах, далеких от критических температур
жидкость — пар, различно для простых и сложных жидкостей (т. е.
высших гомологов). Для первых Ср монотонно растет до бесконечности
при критической точке жидкость — пар, так что не существует
качественного различия между высокой и низкой температурами. Для
вторых cPCp/dT2 при всех температурах положительно, но Ср сначала
уменьшается при низких температурах, а затем начинает
увеличиваться до бесконечности; минимальные значения Ср находятся при
температуре выше 298 К.
Эта неопределенность в изменении Ср не позволяет даже
качественно предсказать изменение параметров в низкотемпературной области.
Например, для я-алканов наблюдали именно такой характер
изменения СР [16].
Таким образом, согласно уравнению C.13), kt (т. е. Д Н)
положителен при низких температурах и отрицателен при высоких
температурах, где dCpldT > 0. Возможность положительного структурного
вклада в %г является неожиданной. Если Ср увеличивается с
уменьшением температуры, приводя к повышению Xi» существует возможность
получить фазовое расслоение системы при низких температурах даже
при б = 0 и р = 0. Этой температурой должна быть ВКТС или в-точ-
ка. Возможно, что именно таким поведением и объясняются свойства
системы низкомолекулярный полиизобутилен — пентан [17],
полистирол— этилбензол [18], а также смеси полимергомологов
полистирола [19].
Параметры Х2 и #2. Влияние %2 на фазовое расслоение растворов
полимеров хорошо известно: кривая сосуществования фаз около
в-точки соответствует более высокой концентрации, чем по теории Фло-
ри [20]. Член энергии когезии и размера макромолекулы в уравнении
C.15) положителен, так что значение %2 должно быть выше для плохих
и в-растворителей. Структурный вклад, вероятно, тоже положителен,
в частности при низких температурах, где dCpldT может быть
отрицательно. Звдк kt предсказать еще труднее, поскольку при d2Cp/dT2>0
первая производная может быть и отрицательной и положительной.
Однако для смеоей я-алканов k2> 0 и действительно в этих системах
теплота смешения может менять знак от отрицательного к
положительному, когда концентрация алкана с более длинной цепью
увеличивается [16].
Количественные расчеты параметра %г.
Значения — U определяются из теплот испарения. Для определения
Ср можно воспользоваться эмпирическими уравнениями
—(/-const A—Т/Ткрт)>"9
T) C.16)
55
при т = 0,428. Уравнение B.12) тогда может быть записано в виде
—U/RT (^кри/П —] ' W-A'J
Критические температуры находятся^ экспериментально.
Определение т2. Для нахождения приведенной температуры
были использованы разные методы. Наиболее простые из них основаны
на вычислении коэффициентов термического расширения полимера
и растворителя и коэффициента термического расширения
стандартной жидкости в широком диапазоне температур. В работе [16] в
качестве такой стандартной жидкости был взят гептан. Для него была
построена зависимость аТ от 7\ Если для любого полимера или рас-
:0,W
0,05
1,0
0,5
0,05 0,10 0,15 Г2 '0,2
0,5
!,0Х
1расч
Рис. 3.4. Зависимость Xi/(— U/RT) от т2 для растворов натурального каучука
в различных растворителях.
Точки — экспериментальные значения» прямая— рассчитанные значения при v2 =0 [3].
Рис. 3.5. Корреляционная зависимость экспериментальных и расчетных значений
%i для системы натуральный каучук — различные растворители [3].
творителя величина а{Г при температуре 7" равна <хТ для
стандартной жидкости при температуре 7\ то отношение TIT' будет равно t
для этой промежуточной жидкости и исследуемого растворителя или
полимера. Таким образом, из данных соотношений (для растворителя
и стандартной жидкости, полимера и стандартной жидкости) можно
найти Т\1Т% для растворителя и полимера. Член (-^-т) — 1
изменяется незначительно по сравнению с т2. Расчетные и
экспериментальные данные не противоречат друг другу (рис. 3.4).
Определение v2. Параметры 6 и р можно определить,
например, из уравнений C.5). Уравнение C.7) для расчета v2 не совсем
удобно, поэтому используем уравнение
Ku'/v*\l/2 i2
w J
V2
6 SpV*
56
В этом уравнении использовано приближение теории параметров
растворимости. Действительно, U*/V* выражает плотность энергии
когезии жидкости при О К. Изменение плотности энергии когезии
жидкости, обусловленной ее термическим расширением, в этом уравнении не
учитывается. Значение U*/V* можно найти при использовании любой
величины, имеющей размерность отношения энергии к объему,
например, плотности энергии когезии, внутреннего давления или
сжимаемости, если такие данные имеются в наличии для стандартной
жидкости в широком диапазоне температур. В работе [16] такие данные были
получены для гептана. Согласно принципу соответственных
состояний, плотность энергии когезии, разделенная на U*/V*9 зависит
только от температуры приведения, или а7\ Таким образом, отношение
плотности когезии стандартной жидкости к плотности энергии когезии
растворителя равно отношению их значений -f—?, если асТс = a±Tv
Такое же отношение можно найти для полимера и стандартной жид-
кости, т. е. —*—f для системы полимер — растворитель находится так
же, каких2. Изданных по v2 с помощью уравнения C.17) находится %г.
На рис. 3.5 приведены экспериментальные и рассчитанные с
помощью уравнения C.17) значения %i- Как видно из рисунка,
наблюдается определенная корреляция теоретических и экспериментальных
значений, но полученные данные показывают, что рассчитанные
значения v2 ошибочны, по-видимому, из-за необоснованности правила
геометрического среднего. Особенно это характерно для галогенсодержа-
щих растворителей. Для получения —U и СР может быть
использовано несколько моделей, которые позволяют вычислить а, плотность
энергии когезии или сжимаемость — как функцию приведенной
температуры. Поэтому использование стандартной жидкости в этом
случае необязательно.
Пригожий и сотрудники [1] использовали две модели —
усредненного потенциала Ленарда—Джонса и гармонического потенциала [21,
22]. При температурах ниже 0,75 7крит величина Ср для обеих моделей
прямо пропорциональна Т. Это приближение использовано для
описания теплот смешения [23] и дало хорошее предсказание наличия НКТС.
Флори и сотрудники [5, 6] использовали потенциал Гишфельдера—
Эйрингас — U& VV; в такой модели Ср = 1/D/3У/3 — 1). Все
модели показывают увеличение СР до бесконечности при приближении
к критической точке и, следовательно, предсказывают НКТС. Однако
при низких температурах все модели предсказывают dCpldT > 0,
поэтому найденное значение kx может оказаться некорректным.
3.3. ВЛИЯНИЕ ДЛИНЫ ЦЕПИ РАСТВОРИТЕЛЯ
НА ПАРАМЕТРЫ ТЕРМОДИНАМИЧЕСКОГО ВЗАИМОДЕЙСТВИЯ
В работе [23] Паттерсон и сотрудники использовали
термодинамические функции, полученные Бальменсом и Нар-Колиным [21] для
раствора мономеров А с Гв-мерами, имеющими Зс = (гв + 3) степеней
57
свободы, при предположении, что размеры сегментов А и В равны, т. е.
ЛАА = ГВВ
Для общего случая смеси гА-и гв-меров число внешних степеней
свободы равно соответственно ЗсА и Зсв, кроме того, они использовали
простой усредненный потенциал вместо гармонического потенциала
ячеечной модели. Было найдено, что
где
. ДЯ' = zAw12 (qANA + qBNB) ХАХВ; qAz - rA (z - r) + 2.
Доля поверхностей
a Aw12 выражается уравнением типа C.18):
A^i2 = (еп + 822) — ад C.18)
Д w12— подобно «обменной энергии», но оценивается при минимуме
потенциальной энергии, т. е. при абсолютном нуле. Если
использовать правило геометрического среднего, т. е.
«Ав - (еАА^вI/2>
то Aw12 — положительная величина.
Используя в = 8зВ/8дА—1. получаем
Величина АН11 вводится для учета расширения раствора и чистых
компонент при данной температуре:
ДЯ" (у СА у СВ\ Л
2Ш (qANA + gsNB) ~ [Л А qk + Л* ^j l ~
-ХаС-*Та-ХъС^Тв. C.19)
4 А ЧВ
Здесь Т, Tt — приведенные температуры для смеси и чистых
жидкостей!
2г ^А^А +
г. - кТ
Ta-Ra
^ kT
58
Пусть еАА==8вв==8* и 1Г = 0- Тогда уравнение C.19) упрощается:
kT /сА св\2
"~2i^fe-^j ЛаАв. C.20)
Рассмотрим случай, когда гв > гд и гА Ф 1 и примем, что
компоненты имеют одинаковую гибкость, которая выражается уравнением
limc/r= 1/3. Тогда
ск<Кх ~ W = { 1 - 2 [3 (г - 2)ГЧ дАК
Если Z-* оо, то
где/ — малая численная постоянная. Если А — мономер, а В —
полимер, то из условия, что г = 1, с = 1 и с/г = 1, следует
AH/(rANA + rBNB) Фафв/^ = AH/rBNB<pA/N, C.21)
ДЯДчисло полей базового полимера X ц>А) = А — ВA/гАJу где А и
Б —константы: A = zAw12N= гг*АА&ЫА/8; В= lO,5(k2lz&*)/NA; NA—
число Авогадро.
Энтальпийный параметр Флори тогда равен
С другой стороны, когда растворителем является мономер (гд= 1),
правая часть уравнения C.21) должна быть заменена на Л — 4/9 ВТ2.
Из этих формул видно, что Д Я зависит от температуры и
становится отрицательной при высоких температурах.
Согласно [21], некомбинаториальная энтропия смешения есть
ГДЯнекомб = —2ДЯП. Следовательно, из уравнения C.20) при гв > гА
следует
* ЛСнекомб . = А+В(Т/гЛ\
Параметр %t тогда равен
или
R%t = А (гА/Т) + В (Т/гА). C.22)
Снова, когда гд= 1, член В умножается на 4/9.
В уравнении C.22) зависимость %х от Т и длины цепи молекулы
растворителя отличается от той, которая выражается уравнениями C.23)
и C.24):
C.23)
C.24)
59
хотя А может быть достаточно большим. Если Л = 0, уравнение C.22)
показывает минимум для %х при изменении гА в случае постоянной Т
при г2 = (В/А) Т2 или с изменением Т при постоянном гд и Т2=(А/
Ниже и выше То качество растворителя становится хуже и при
критических температурах Xi должно приближаться к некоторому
критическому значению. Приняв за критическое значение Хкрит
величину 1/2 для бесконечно большого гв, имеем
ЯХкрит = А (/уторит) + В (TKvm/rA) = R/2.
Решая это уравнение, находим, что Ткрт/гА не зависит от гА при
= Я/2 ±
Если А =0, т. е. компоненты отличаются только длиной цепи, ВКТС
равна 0, а НКТС определяется так:
Ткрит/гА = R/2B.
Если 4 А В < A/2#J, существуют обе КТС, если 4АВ > A/2#J,
то полимер нерастворим в данном растворителе ни при каких
концентрациях.
Отметим, что критическое состояние очень чувствительно к г.
Поэтому выражение для Хкрит должно быть записано в общем виде:
1 крит ' А \?А/ \* ~~ "/ \ "V " \^ ~* Я
Обычно Гл выбирается равным (л + 1)/2, где /г — число углеродных
атомов в цепи. Величины А и В могут быть получены из теплот
смешения, из Xi> определенному по набуханию, из НКТС и др.
В рамках более детализированных моделей уравнение типа C.22)
несколько видоизменяется. Так, при использовании модели
усредненного потенциала
а для г-> оо
/?Xi = А' (I) гв + В' (Т) A + | б) + CrB + D.
Первые два члена соответствуют уравнению C.22), но С и D — члены,
появление которых связано с ненулевым значением р. В рамках этих
постоянных можно объяснить формально, что НКТС не строго
пропорциональна гд.
60
3.4. СРАВНЕНИЕ ПРЕДСКАЗАНИЙ ТЕОРИИ, ОСНОВАННЫХ
НА ПРИНЦИПЕ СООТВЕТСТВЕННЫХ СОСТОЯНИЙ,
И ТЕОРИИ ПАРАМЕТРОВ РАСТВОРИМОСТИ
В работе [22] Паттерсон и сотрудники сравнили предсказания
теории, в основу которой положен принцип соответственных состояний,
с приближением теории параметров растворимости.
В последней величина %i выражается разностью параметров
растворимости смешиваемых компонент
X, - %¦ № -<W + Р = (if [l -s?]' + P. C25)
[^f, C.26,
где A Ev и V — молярная энергия испарения и молярный объем
компонента; р — эмпирический параметр, который отсутствовал в
первоначальной теории для смесей квазисферических молекул, но
необходим при рассмотрении растворов полимеров. Член в скобках в
уравнении C.25) отражает обменную энергию при образовании контакта 1—2
взамен 1—1 и 2—2, т. е. A w = -пЧеи+ 612) — 8i2- Использование
правила геометрического среднего приводит к выражению Aw^ y^li2*—
— е1^2J. Таким образом, Xi связан с различием в энергиях контактов
8U и е22 в чистых жидкостях. На первый взгляд, может показаться,
что теория параметров растворимости игнорирует эффекты свободного
объема. Однако в первых работах Гильдебранда—Скатчарда был
использован параметр растворимости для получения изменения
свободной энергии смешения (A G) при постоянном общем объеме системы,
т. е. некоторое гипотетическое давление нужно приложить к
раствору, чтобы А Усм оставалось равным нулю. Энтропия смешения при
этих условиях предполагается комбинаториальной, так что %i>
представляющий некомбинаториальную часть свободной энергии смешения,
должен быть полностью энергетическим по своему происхождению и
равен первому члену уравнения C.25). Было показано, что AG и AS
можно модифицировать, но таким путем, чтобы изменение объема при
смешении не входило в выражение для свободной энергии смешения,
Таким образом, %г снова выражается уравнением C.25), но первый
член уже нельзя рассматривать как чисто энергетический
(эмпирическая поправка ft была введена как поправка к комбинаториальной'
энтропии в теории Флори и несмотря на то, что она комбинаториальна
по происхождению, введена в %19 чтобы сохранить выражение для
химического потенциала в форме, Данной Флори).
'Как уже было сказано, эффект изменения объема в термолинами-
ческие параметры вводится иным путем. Как отметили Флори и
сотрудники [51, изменение объемов компонент при смешении может привести
и к А V = 0, но эти изменения дадут вклад в другие
термодинамические функции смешения. Свободная энергия смешения будет теперь
61
содержать ощутимый вклад из-за изменения объемов, что в конце
концов приведет к НКТС. Это указывает на неадекватность теории
параметров растворимости, однако успешное ее применение (в частности,
уравнения C.25)) в термодинамике растворов полимеров требует
пояснения.
Как следует из уравнения C.26), б должен зависеть от свободного
объема жидкости через AEV и V. Скрытый эффект этого изменения
объема приводит к качественному сходству в предсказании поведения
раствора по уравнению C.25) с тем, что следует из теории, в основу
которой положен принцип соответственных состояний. Например, для
серии гомологов с увеличивающейся молекулярной массой должны расти
значения б, потому что уменьшается свободный объем гомологов.
Высокомолекулярные смеси с гомологами, как растворителями,
должны, таким образом, иметь отличное от нуля значение (бг—б2J,
обусловленное разностью свободных объемов компонент. Далее,
теория дает качественно корректную зависимость параметра %г от
температуры и давления через
!д In 6\ (д ]
где а и рг— коэффициенты термического расширения и
изотермической сжимаемости. Оба эти параметра намного выше для растворителей,
чем для полимеров. Значение б уменьшается с увеличением
температуры более резко для растворителя, чем для полимера, так что при
высоких температурах (Ьг—б2J должно приближаться к таким значениям,
которые характерны при фазовом расслоении. Подобно этому, с
повышением давления %г уменьшается, когда бх<б2, и понижается, когда
6j> б2, что хорошо согласуется с экспериментальными данными. Для
количественных сравнений необходимо выразить параметр Xi в обеих
теориях через одинаковые параметры.
Теория соответственных состояний приводит к уравнению для
%г в виде C.12). В приведенной форме оно записывается так:
tJ-I^ + t1*- <3-27>
Как следует из определения 71*, параметр т в этом уравнении зависит
от отношения контактных энергий компонент, а также — через
структурный фактор с/г — от относительной гибкости и длины цепи.
Параметр v2 является мерой относительной слабости контакта 1—2 по
отношению к средним контактам 1—1 и 2—2. Обычно v2 находится из
экспериментальных данных при предположении истинности правила
геометрического среднего: v2= [1 — (в22/&11I^]2. Отношение е22/еп может
быть получено из Р%1Р% при а2= аг. Для простоты можно положить,
что v^ll — iPt/P*I/2]2.
Выражение, подобное уравнению C.12), было получено для
зависимости 1г от давления [3]:
^%[%|] C28)
62
где
Флори и сотрудники [5, 6] получили выражение для %г при нулевом
давлении в виде
C.29)
В модели Флори была взята вандерваальсовская жидкость с —0 =
= У~\ которая приводит к уравнению состояния (вывод см. ниже):
Ц- = 1/A _ у^1/з) _ l/Vft C.30)
Тем не менее уравнения C.29) и C.12) очень похожи. Уравнение
C.30) вместе с — U=*V~% показывает, что в модели Флори
а уравнение C.29) может быть взято в виде
(р* с у \
~ т а -—) и приняв
s2/sx = 1 и модель Флори для чистого растворителя, можно получить
где
cj1 =* (l — 1 V-lA — 2 A — V~~l/3)/(PV2 + 1).
Положив Я = 0, получим уравнение для нулевого давления,
эквивалентное уравнению C.27) с термодинамическими свойствами
растворителя, взятыми из модели Флори.
Возвращаясь к теории параметров растворимости, из уравнений
C.26) и условия — U = F-1 имеем
Вводя в уравнение C.25) параметр Xi как функцию температуры и
давления, получаем простое выражение
63
500 TtK
Рис. 3.6. Зависимость %г от
длины цепи (а), параметра
растворимости дг (б, в), температуры
(г) и давления (д):
a, 6i р=0; Т =300 К; 6*i/e*2 Равно
0,85 (/,/'), 1,00 {2,2'), 1,20 C,3');
e:p«=0; T = 300 К; rt равно 3,0
(/,/') и 8,0 B,2'); г: р = 0; rt =3,5;
?П/е22 равно °'85 (/';/)' 1'00 B>2')>
1,30C,3'); д: Т =300 К; гг=3,5?
etl/?22 Равно 0.85 (/,/'), 1,00 B,2'),
1,30 {3,3'). Сплошные линии—
расчет по теории соответственных
состояний, штриховые — по теории
параметров растворимости [22].
Паттерсон и сотрудники [22] сравнили предсказания этих двух
теорий в условиях изменения молекулярных параметров при 300 К
и при постоянном давлении, а также изменения температуры при
постоянном давлении и изменении давления при постоянной температуре.
В результате они сделали следующие выводы.
Изменение молекулярных параметров. Гомологическая серия
растворителей. Для удобства были выбраны такие компоненты: полимер
с М -* со, 71* = 7 000 К и Р* = 100 кал • см (что соответствует
полиэтилену) и растворитель — серия «-алканов, для которых
C.33)
где п — число углеродных атомов. Цепи полимера и растворителя,
следовательно, имеют одинаковый параметр гибкости (а = 1/3).
Зависимость %i от'длины цепи растворителя приведена на рис. 3.6,а для
трех значений ej^/ejj. Это соотношение и уравнение C.23) позволяют
рассчитать Т*, Р* и Т%. С помощью уравнения состояния Флори
C.30) находят Уг и У2. Следовательно, параметр %г можно рассчитать
по уравнению C.32) из теории соответственных состояний с Р = 0.
На рис. 3.6,6 нанесены значения %х в зависимости от параметров
растворимости растворителя. Когда г*г/г*%> 1 (например, для растворов
полидиметилсилоксана в серии я-алканов), обе теории предсказывают
минимальное значение для %х при Ьг= 62 в теории параметров
растворимости и при значении 8Х, очень близком кб2 в теории соответственных
состояний. Отличием является лишь абсолютное значение параметра
Xi, который на 0,3 больше, чем по теории соответственных
состояний. Полученный результат предполагает, что эмпирическая
постоянная Р [25] должна интерпретироваться как привносимая теорией
параметров растворимости в %г по аналогии с той, что следует из теории
соответственных состояний.
Когда еп/е|а= 1 (например, для полиэтилена и полиизобутилена
в я-алканах с длинной цепью, т. е. когда растворитель идентичен
полимеру), обе теории предсказывают нулевое значение %г, если р = 0.
В области же обычных длин цепей растворителей теория
соответственных состояний, однако, предсказывает значение %х на 0,3 выше, чем
теория параметров растворимости, что интерпретируется как
оправдание введения параметра р. Для е*х/е*2< 1 снова появляется минимум
на'кривой зависимости Хх от параметра растворимости, но при меньшем
значении 61э чем для полимера. Таким образом, не всегда корректно
приписывать полимеру значение б2, равное бх растворителя, в котором
наблюдается максимальное набухание полимера. Это видно, например,
из результатов по набуханию каучуков, таких как натуральный
каучук, неопрен и бутадиенстирольный каучук в я-алканах [26].
Параметры растворимости каучуков составляют 9—10, но максимальная
степень набухания обычно наблюдается в гептане, для которого
бх = 7,4.
Растворители с разными энергиями когезии, но постоянной
длиной цепи. Поскольку когезионные силы в растворителях
увеличиваются, вырастают и их параметры растворимости. Для обеих теорий %х
достигает в этом случае минимального значения при близком значении
параметра растворимости растворителя. Между тем минимум не
должен наблюдаться в обеих теориях при отношении &\11г%2= 1 (условие,
соблюдающееся для полимера и растворителя, принадлежащих к
одной гомологической серии*, например, полиэтилен и гептан). Но
параметр растворимости отражает и свободный объем жидкости, а,
следовательно, и длину молекулы. Таким образом, бх= 62 будет в том
случае, если Е*г несколько больше е*2. Большая величина энергии когезии
растворителя, обусловленная тем, что его свободный объем становится
близким к свободному объему полимера, понижает значение Xi при
4-420
65
уменьшении этой разности, но при увеличении г*г растет и Xi- Коррект-1
ный баланс разности свободных объемов для минимального значения1
%i находится при 8г= 62 в теории параметров растворимости. Такой же
баланс между разностью контактных энергий и свободными объемами
получается и по теории соответственных состояний (рис. 3.6,в). Так,
когда е*х/е22 = 1, параметр v2 исчезает, но член свободного объема
в Хг остается большим. Увеличение отношения e*i/e*2 приводит к не-т
нулевому значению v2, но 1г продолжает уменьшаться из-за
понижения члена свободного объема. Интерпретируя Р как разность между Хг.
определяемыми по двум теориям, отметим, что Р отрицательно для
малого отношения 811/622. Однако в этой области полимер нерастворим
в растворителе и, следовательно, выбор Р ~ 0,34 для обычных систем
полимер — растворитель не совсем корректен.
Гибкость полимера. Обе теории показывают, что параметр Xi
зависит от гибкости компонент. Если изменять cjr2 полимера при
постоянном значении г*х/-е*2 и длине цепи растворителя, то при умень^
шении(с2/т2)соответственно понижаются гибкость и свободный объем
полимера, а б2 будет увеличиваться, оказывая соответствующее
влияние на Хи рассчитанный из теории параметров растворимости. В
теории соответственных состояний член свободного объема изменяется,
таким же образом, и обе теории дают сходные результаты по
изменению Xi с изменением гибкости полимера.
Температурная зависимость параметра Xi« Одним из важных
достижений теории соответственных состояний является предсказание увет
личения параметра Xi с повышением температуры. На рис. 3.6,г при-,
ведена зависимость Xi от температуры при фиксированной длине цепи
растворителя и гибкости полимера, но стремя различными
значениями е*/е?.
Для е*/е|2=1член контактной энергии в теории соответственных
состояний исчезает, но возрастает член свободного объема, а
следовательно, увеличивается и параметр Xi .Такое же поведение
предсказывает и теория растворимости. Для е*/е*2= 1 справедливы 61<6Ж
и с повышением температуры бх уменьшается быстрее, чем ба.
Следовательно, (б2—б2J растет с температурой, вызывая увеличение %г.
Таким образом обе теории могут предсказать фазовое расслоение при
НКТС.
Для е*/е*2>1 (например, для полиизобутилена и бензола) 61>б2
при нормальных температурах. Быстрое уменьшение 8г с ростом
температуры приближает его значение к б2, и теория параметров
растворимости предсказывает снижение параметра Xi До значений меньших*
чем при НКТС. Увеличение Xi с температурой в обычной
температурной области обе теории предсказывают при ё*/е*2 < 1, т. е. при бх< б2.
Примером может быть система полистирол— циклогексан, для которой
Xi уменьшается с повышением температуры вблизи 377 К (в —
температура для этой системы), что также предсказывается обеими
теориями, но при нереально низких температурах. Эта ошибка в теории
соответственных состояний (и, возможно, в теории параметров растворимо-1
66
ста), по-видимому, обусловлена большой переоценкой члена
свободного объема (см., например, [27]).
Зависимость %г от давления. Из теории соответственных состояний
следует зависимость %i от давления благодаря воздействию последнего
на свободный объем компонент. Рис. 3.6, д показывает, что зависимость
%i от давления предсказывается так же, как и в теории параметров
растворимости, где 6Х относительно сильно сжимаемого растворителя
увеличивается с давлением быстрее, чем для полимера._ Уравнение
состояния Флори C.30) дает возможность рассчитать Уг и К2, которые
могут быть использованы в уравнении C.32) теории параметров
растворимости и в уравнении C.31) теории соответственных состояний.
Если е*/е*2 <: 1, то 81<8^ и, следовательно, d^JdP <0 в теории
параметров растворимости и в теории соответственных состояний. Если
е*/е*2 заметно больше единицы, но бх> б2, растворитель становится
менее сжимаемым, чем полимер. Тогда в результате повышения
давления может возрасти член (8Х— б2), приводя к д%1/дР> 0.
Влияние параметра (s2/si). В приведенных выше расчетах параметр
) принимался равным единице в уравнении C.31). Если
использовать значения (sjs^ меньше единицы, что может наблюдаться в си-
ртемах полимер — растворитель [6], то форма кривых на рис. 3.6 не
изменится, но уменьшится абсолютное значение параметров %i> что
приведет к уменьшению параметра р. Однако делать вывод о подобии
результатов, полученных на основе обеих теорий, пока
преждевременно.
Таким образом, видно, что feopия параметров растворимости
также может быть полезна при трактовке эффектов свободного объема.
Авторы работы [22] считают, что широкое использование
приближения параметров растворимости для предсказания термодинамических
свойств растворов полимеров вполне оправдано.
Рассмотренная в этом разделе теория Пригожина — Паттерсона
широко используется в последние годы для интерпретации данных по
растворам полимеров наряду с новой теорией Флори для жидких
смесей.
3.5. НОВАЯ ТЕОРИЯ ФЛОРИ
Флори и сотрудники [5, 6] использовали ту же ячеечную функцию
распределения, что и Пригожий, но в отличие от последнего для
конфигурационной энергии применили приближение Гирщфельдера—s
Эйринга вандерваальсовской жидкости, т. е. Е = —1/V.
В некоторых работах показано, что это приближение лучше
описывает экспериментальные зависимости конфигурационной энергии
от объема, чем приближение, полученное при использовании
потенциала Леннарда—Джонса.'
Рассмотрев одномерную систему из частиц длиной
/^распределенных в конфигурационном пространстве длиной L и приняв, что
частицы взаимно не перекрываются, для общего конфигурационного про-
3* 67
странства, занимаемого системой, Флори использовал функцию рас- .
пределени я Тонкса:
Q = A — Nl*)"/Nl & [(I — /*) e]N, C.34)
где /*— длина жесткого ядра частицы; I = LIN — пространство,
занимаемое частицей. Общая энтропия вводится фактором eN.
Для распространения этих результатов на трехмерное пространство
вводится параметр (yv)l/3 = / и G^*I/3 = /* (у = V/N, V — общий объем
системы; у — численный фактор; и* — ядерный объем частицы). Для
обобщения этого результата N молекулы разделяются на г сегментов.
Тогда v* идентифицируется как характеристический или ядерный объем
сегмента, т. е. v = V/r, где V — объем молекулы.
Следуя Пригожину, Флори вводит параметр с, так что 3 с
представляет собой число степеней свободы на сегмент. Функция распреде-,
ления тогда имеет вид
Q = \ye*v* (vU3 — l)*]Nrcy C.35)
(v = v/v*). Средняя межмолекулярная энергия, входящая в
конфигурационный интеграл для такой системы C.36)
Z=Qexp(-E0/kT), C.36)
выражается так:
E0 = —Nrsr\/2v, C.37)
где s — число контактных мест на сегмент (пропорционально площади
поверхности сегмента); N — число молекул;— \\/v —
межмолекулярная энергия на контакт. Вводя приведенную температуру в виде
Т = 777* = 2v*ckT/si), C-38)
получаем функцию распределения
Z = const (vl<* — IK"" exp (Nrc/vf)f C.39)
из которой следует уравнение состояния
PVlf = vl/3 /(vl/3 — 1) — l/vf, C.40)
где
Я= Р/Р* = 2Pv*Vsr\. C.41)
Параметры приведения (или характеристические параметры) Р*>
v* и 7* связаны между собой уравнением
P*v*=ckT*. C.42)
При Р = 0 уравнение состояния приводится к упрощенной форме:
f = (Ь1/3 — 1)/1?4/3 > C.43)
а затем к виду
у 1/з _ 1 = №\ I (I + аТ), C.44):
68
где
а = (д In v/ЯТ)
При известном а этих уравнений достаточно, чтобы найти ~v и Г.
Температура приведения Т* получается из уравнения C.38), a v* —
из v и v. После дифференцирования уравнения C.40) с
соответствующей подстановкой членов уравнений C.41) и C.42) получаем
р* = T7V,
где у = (дР/дТ)у- Величина Р*, оцененная по изотермическому
коэффициенту сжимаемости путем использования этого уравнения,
полностью характеризует чистую жидкость. Остальные параметры (с и sr\j
можно определить с помощью уравнений C.38), C.41) или C.42).
Для бинарной смеси функция распределения может быть записана
в виде
Z = Zkom6 Q exp {—E0/kT)> C.45)
где ZKOM6 — комбинаториальный фактор для смеси двух компонент,
a Q выражаются уравнением C.34). Пренебрегая различием между
свободными энергиями Гельмгольца и Гиббса для конденсированной
фазы, согласно уравнению C.45) свободную энергию смешения Гиббса
можно представить выражением
где АСкомб = — Т^Д^комб; GR —так называемая остаточная свободная
энергия, следующая из функции распределения Qexp(—E0/kT).
Сегменты обеих молекул смеси обычно выбирают равными ядерным
объемам. Этот выбор неограничен, поскольку Q ехр (—EJkT) не
зависит от формы и расположения молекул. Функция распределения для
смеси тогда будет иметь вид
Z = const • Zkom6(vl'z— l)*FN~cexp(—EQjkT),
Эти соотношения позволяют интерпретировать ср2 и <рх как сегментные
доли, а не объемные. Тогда
ф2 = 1 — фх = r2N2/rN
и уравнение C.46) можно переписать в виде
с =
Определения, приведенные выше, позволяют рассчитать <рг иным
путем. Молярные характеристические объемы компонент i равны
^* = rtv* и> таким образом, r2/r1 = Kf/V*. Здесь V* определяется
из 5t. = VVV7, гДе Vt = rtv — молекулярный или молярный объем.
Можно тогда сегментные доли выразить следующим образом:
ИЛИ
ф2 = Ш2У*Д. 2/(аУ2<>УД. 2 + ^1^уд. l),
где w{ — масса компонента г, v*A.t — характеристический удельный
объем. Энергия Ео смеси берется в такой же форме, как и для
чистого компонента:
—Ео = (Апг\п + Л22т]22 + Л12т]12)М C.47)
где А^ — число контактов, каждый из которых характеризуется
энергией —1\ti/v. Из определения следует (см. уравнение C.47)), что
2Л22 + А12 = s2r2N2\
для однородной смеси
А12 = Vi^A
Межмолекулярная энергия описывается выражением ь
—Eo/rN = [вхЛц + в2л22 — вхвгДг!] (s/2v), C.48)
где
Дл = %i + Л22 — 2^12, C.49)
s = (s1r1N1 + sfaN2)/TN ^фл + Ф252,
а доли мест вх и в2 определяются уравнением
G2 = 1 — Gx = s2r2N2/(s1r1N1 + s2r2N2) =
+ (s2/Si) Ф2] •
По аналогии с энергией для чистого компонента, для смеси ее
значение может в быть представлено в виде
—E0/rN - P*v*/v = ckT*/v. C.50)
В результате сравнения C.50) с уравнениями C.48) и C.41) имеем
выражение
/>*^Ф1/1 + Ф2Я*-Ф1в2Х12, C.51)
где
Х12 = SiA^/rv*2.
Поскольку уравнение C.42) применимо и к смесям, с его помощью
ияожно получить
??4^.. C.52,
Функций распределения для смеси имеет ту же форму, что и для
удостой жидкости. Следовательно, уравнение состояния идентично
выражению C.40). Однако приведенные переменные Р = Р/Р* иГ-
70
To TJI*nm* ШеСИ зависят от состава ^гласно уравнениям C,51) щ
C.52). Для малых давлении достаточно заменить энтальпию смешений
энергией: "№
ДЯ = ?Nv* [фхР^х + <Р2Р$/Ъъ — P*/v], C.53)
или, из определения Р* по уравнению C.51),
АЯ = FNv* [ф1Я: EГ1 - S) + Ф2Р* (?~1/2 -
— у-1) + Ф^Х^гГ1]. C.54)
Выражение для интегральной теплоты смешения
где В выбирается постоянной, не зависящей от концентрации, не
соответствует уравнениям C.53) и C.54). Постоянная В обычно
определяется из теплоты смешения блочного полимера с большим избытком рас-
творителя. Применение полученного таким образом значения В для
расчета энтальпии по уравнению C.55) при высоких концентрациях не
оправдывается. Параметр В, определенный по уравнению C.55},.
как видно из C.53) и C.54), должен зависеть от концентрации.
Разложение уравнения C.54) в ряд по ф2 для случая предельного
разбавления приводит к выражению
- (-?) Hi -' -щТ(' ~
Это уравнение облегчает расчет Х12 из энтальпии смешения полимера
с избытком растворителя. Значение В, полученное таким образом, не
может быть использовано в уравнении C.55) для расчета А Нм при
высоких концентрациях. При aj>a2 вклад «уравнения состояния» (в
квадратных скобках уравнения C.56)) отрицателен. Энтальпия
смешения при большом избытке растворителя будет отрицательной, если)
Х12 недостаточно большое, чтобы превысить член в квадратных скобках.
Выражение для избыточного объема смешения получается из
стого соотношения: аддитивный объем
отличается от объема смеси Ъ добавкой
vE = v — 50, C.58)
которая связана с состапом через Г* и уравнение C.57). Определение
изменения объема при смешении (AV) и объема до смешения (Vo)
приводит к выражению vE/vb = &V/V0.
Представленная теория предсказывает также существенный вклад
в энтропию смешения, вызванный различием в параметрах уравнения
71
состояния компонент. Из функции распределения и уравнения C.53)
следует
S* = -3(^*1^/7?) In [(у!/3 - 1)/E1/3- 1)] -
- 3(N2PtV*2/Tt) In [C2/3 - 1 )/(и -1I. C.59)
Эта «остаточная» энтропия добавляется к комбинаториальным членам,
выраженным уравнениями C.60) или C.61):
ASK0«6 = —k [Nt In фх + N2 In фJ, C.60)
ASK0M6 = -ftMhKPi + tf.linpd + ft (#!¦!•) ln[l -2ф2A -y)jz] +
C.61)
Парциальные молярные величины получаются из уравнений C.54)
и C.59)
C.62)
TSl = -РУ* {375! In [ф/3 — 1)/E1/3 - 1)] -
- (у) (^2 - Л/*5} + «Г (F*X12/5) ej. C.63)
где а — коэффициент термического расширения смеси, связанный с
v уравнением C.44). Комбинация уравнений C.62) и C.63) приводит к
(i*i - А)* = р\К (ЗЛ in №'3 - 1)/E>/з _ 1)] +
+ (йг1 - г-1)} + (vtx12/5) ej. C.64)
Разложение этой величины в ряд приводит к следующему:
(l*i - ^)* =» (^^I/3i) ИЧ772 + Ku| Ф22 + О (ф1), C.65)
где
Л = A - Tt/Tt) (Pt/P*t) - (s2/Sl) Xlt/Pt
Подобным же путем получаем
^=(/>*УГ/51)[Г12A+а1Т)-
- B/3) {AaxTf A + аТ)] Ф22 + О (Ф32), C.66)
f ^ D/3) ахГ х
Для использования этой модели необходимо, чтобы комбинатори-
альная энтропия вытекала из смешения сегментов. Предыдущие рыра-
жения, полученные с помощью метода решеточных моделей или дру-
72
гих методов, могут быть использованы при замене объемных долей сег^
ментными долями. При этой замене химический потенциал
растворителя в полимерном растворе можно выразить суммой комбинаториаль-
ной и «остаточных» частей, т. е.
= \паг = In A - ф2) + A — 1/г)ф2 +
где ах — активность растворителя; <р2 — сегментная доля. Параметр 1г
будет тогда идентифицирован как приведенный остаточный химический
потенциал:
Если его представить в виде ряда, то
Xi = Xio + ХФ
Величина, соответствующая приведенной парциальной молярной
остаточной энтальпии, записывается уравнением
kx = 77f lRTy\ = Я? /RTvl = k1Q + k&2 + • • •
Приведенная молярная парциальная энтропия
ДМф! = -Пп 0 ~ Ф2) -+- Ф2]/ф22 + S?IR<&.
При высоких разбавлениях это выражение сводится к следующему:
где il?!—энтропийный параметр взаимодействия для разбавленных
растворов полимеров. Таким образом, я^-* 1/2 представляет собой
приведенную парциальную молярную остаточную энтропию при
0
3.6. МОДИФИКАЦИЯ НОВОЙ ТЕОРИИ ФЛОРИ
Приложение описанной теории к интерпретации
экспериментальных данных по растворам полимеров было сделано Флори и
сотрудниками [6], а также другими авторами. Сравнение предсказаний теории
с экспериментом в основных чертах подтвердило основные ее
положения, например, наличие НКТС, концентрационной зависимости
параметров Xi и др. Однако во многих случаях наблюдались существенные
расхождения между прогнозами этой теории и экспериментальными
данными, что потребовало введения третьего связующего (помимо
Х12 и s^Si) параметра Q12. Причина такого расхождения теории и
эксперимента состоит в том, что теория Флори разработана при
предположении однородного смешения, т. е. допущения, что полимерный
сегмент (или молекула растворителя) выбирает ближайшего соседа
случайно, без предпочтения одного другому. Упрощенный учет
неоднородности смешения впервые сделан Прусницем и Ренунсио [28], которые
73
предложили изменить уравнение C.47) путем введения поправки на
локальный состав при расчете избыточных функций смешения. В
результате в эти выражения вошли два параметра, связанных с
локальными долями мест компонентов. Полученные ими выражения были
применены к описанию термодинамических характеристик ряда систем
полимер — растворитель^ и показали хорошее согласие с
экспериментальными результатами.
Более строгий учет неоднородности смешения сделан в работах
132, 33], поэтому рассмотрим их более подробно. Так, в работе [32]
было показано, что учет неоднородности смешения, в отличие от
результатов Прусница и Ренунсио, несколько видоизменяет форму
уравнения состояния, предложенного Флори.
Функция распределения в этой работе была взята в виде
Z = ZK
Om6
где ZKOm6 — комбинаториальный фактор; w(i) — средняя
межмолекулярная энергия ячейки типа i.
Здесь w(i) — энергия, освобождающаяся при переходе сегмента
молекулы i в гипотетическую жидкость i, где он получает окружение
.из stQH соседей из компонент i и из s,e# соседей из компонент /:
621г|21),
где \-fv-потенциальная энергия на контакт мест/ и/; ©и и в*/ —
средние доли локальных поверхностей:
+ в/т,/); в,/ = тдв/Де, + в/т/,), C.67)
в« + в/, = 1 (i, /=1, 2).
Уравнение C,67) связывает долю средних локальных поверхностей
с общими долями поверхностей
и параметрами
%lt = exp [I s. (т)«. - rfa)/(RTv)} = exp {»% [vR Л),
где ф, — сегментные доли, а \ц определяется из выражения
-н = \^{^ЯЩ (*, / = 1. 2).
74
Приведенное уравнение состояния, получающееся. в результате-
этих соотношений, представляется в виде
PV -,
ТЛ fv
Это уравнение отличается от соответствующего уравнения состояния
Флори наличием третьего члена справа, где
А = ^{фхвцвп (-^-) + Ф2©22©
12
Р* = Ф1^* + Ф2^* + Ч&иУп + Ф2©^12; C.69)
с = -тЗ?- = ФЛ + ф2с2. C.70)
Характеристические давления для чистых компонент определяются
так:
Р* * о п° /п*2* Р* -L
Из уравнений C.69) и C.70) следует
ЦТ* = (ч>Л/т;
где
71* = | wljRciV*; T*2=±s2y\°22/Rc2v*.
Уравнение C.68) отличается от уравнения состояния, данного в
работе [8], знаком третьего члена справа. Различие в знаке обусловлено
иным определением доли средних локальных поверхностей.
Рассмотрим, например:
для 0х= 02 и v2i> 0 имеем 021> 012, т. е. контакты 2—1,
окружающие сегменты молекулы 1, более многочисленны, когда интенсивность
взаимодействий 2—1 выше, чем взаимодействий 1—1.
Для этой модели авторами работы [32] получены и соответствующие
термодинамические функции:
энтальпия смешения
4
АНМ = Nrv* UP* (« - 4-) + Ф2Р* (J- - 1) -
— у (Ф1в2^21 + Ф2012^12)];
остаточная свободная энергия и энтропия смешения
AGR = Wrv* [ъДТг In ^5| + ф2/? 7* In ^ZiJ + д нм-
75
остаточный химический потенциал
приведенная парциальная энтальпия смешения
приведенная парциальная молярная
энтропия смешения
^ + fn^
vl/3-1 v [ Tv \[ Т
коэффициент объемного расширения
интегральная теплота смешения
при бесконечном разбавлении
На основе развитой модели авторы работы [32] проанализировали
свойства четырех систем полимер — растворитель, таких как полиизо-
бутилен — бензол, полиизобутилен — w-пентан, полиизобутилен —
циклогексан и полистирол — ацетон. Было показано, для определения
аг и В невозможно найти единственное сочетание параметров v21 и v12:
их можно аппроксимировать поверхностью, характеризуемой
множеством положений бинарных параметров (рис. 3.7). Эти параметры
воспроизводят в пределах экспериментальных ошибок данные по
активности, энтальпии смешения и волюметрические данные при низких
давлениях. Обычно выбирается такое сочетание бинарных параметров,
которое наилучшим образом воспроизводит одновременно данные по
активности и энтальпии смешения (см. рис. 3.7). Следовательно,
результаты и этой работы показывают, что для согласования теории и
эксперимента достаточно только два связующих параметра, в отличие
от теории Флори, где требуется три таких параметра.
Авторы работы [34] предположили, что для удовлетворительного
описания свойств растворов в рамках теории Флори число внешних
степеней свободы смеси должно быть взято в виде
с = ФА + щс2 — <ргв2 (v*/k) 6,
где k — постоянная Больцмана; б — эмпирический параметр,
учитывающий изменение числа внешних степеней свободы при образовании
0,87
0,37
-0,13
-5,76
-6,26
-6,76
-7 ОЙ
\ \
\>
Г
VV
\\
Vs
\
V
\\
\}
\
\
\
\
, \
\ \
л\\
\ \
-2,49 -1,99 '1,49
а
-0,99 -2,91 -2,41
Рис. 3.7. Бинарные параметры для системы полиизобутилен — бензол (а) и полиизо-
бутилен — циклогексан (б).
Точками обозначены наилучшие оценочные значения параметров v2i и v12 [32].
раствора. В рамках модифицированной таким образом теории Флори
, получены зависимости для Д|д^, А #м и Vе\ которые были использова-
1 ны при сравнении теоретических и экспериментальных данных для
растворов полидиметилсилоксана в бензоле, метилэтилкетоне, цикло-
гексане, хлороформе, этилацетате. Используя подгоночные значения
параметров Х12, б и отношения s2/5i» авторы получили достаточно
хорошее согласие теории и эксперимента.
Хамада и сотрудники [35] также модифицировали теорию Флори
по несколько иным правилам комбинирования параметров смеси:
во-первых, применили предположение работы [35] об изменении числа
внешних степеней свободы; во-вторых, для определения ядерного
объема смеси взяли выражения, подобные правилу комбинирования
Ван дер Ваальеа:
Ф*Х, C.71)
77
где
а ? равна единице для жестких сфер.
По аналогии с C.41) и C.71) определяли
Р
/>* = ф^р* (v*/v*f + ф^Р»*J + 2ф?02/>?2 (v*2/v*)\
Для числа внешних степеней свободы использовали выражение
с = фл + ф*с2 — Ф*в«А2, C.72)
где с12— параметр, характеризующий отклонение числа внешних
степеней свободы от аддитивности. Из уравнений C.50) и C.72) получали
выражение для Т*
1/7* = [Ф*Р? (vt/v^/Tt + ЩР1 (v*2/v*)/T*2 - <p*el2kc12/v*}/P*.
Тогда для избыточного химического потенциала уравнение C.64)
приобретает вид
(Hi - V®* = ЗРХ [ 1 /2 In (mjm) - j In (v*/v•)] -
— 3/2<p*/>*FT (mx — mj/m + 3rlCliRTSl lnK +
+ ZPytfi In \(ЪТ - 1 )f(vl/3 - 1)] + 3rlCl2i?022 In C>/8 - 1) +
+ PtKllfa - (T^f)/©! + (V*/v) {P* (o*/w*)((p*/9t) [2 (о >*) 62 -
- f 2/f ] + 2/>*2 (t)*2/t;*)a 62 - P* (в2/ф?)} + 2ф2Р*У* (j-
X [Ф>! - (Ф* — Ф2) Vu — Ф?»Й/о* + Qzhc^RT l&T),
где
m = ф!/^! + ф2т2;
/п^ — масса сегмента; h — постоянная Планка. Энтальпия смешения
тогда равна
АНМ s AE0 ^ ?N [—C/2) (pt
+ ViPivt/Vt + 4>%Ptvt/V
а при бесконечном разбавлении
= (Р*1,*д. а/5,) (у>2)A + ЩТ) I(s2/st) + 2(u*2/ut)- 2] +
д. 2 [ОхГ (ГГ/Г5/5х + 1/5J- 2 (Я* »;д. а/5,) (v$/v*2) (v^/vtf x
X (s2/Sl)A + ат) - (c12RTVyA, 2/v*2) (s2/Sl) [3/2 + a^/ivjfj],
где у*д 2 — удельный характеристический объем компонента 2; ах
коэффициент термического расширения компонента 1.
78
Отношение избыточного объема Vе к сумме объемов чистых
компонентов V0 таково:
На примере растворов полидиметилсилоксана с различными
растворителями авторы работы [35] показали, что использование двух
параметров (Р*2 и с12) позволяет иметь хорошее согласие теоретически
рассчитанных и экспериментально полученных концентрационных
зависимостей %1У А Я00 и VеIV0.
В работе [36] было рассмотрено влияние полярных эффектов на
термодинамические функции новых статистических теорий, а в
работе [39] сделана попытка описать термодинамику смешения полимеров
на основе концепции свободного объема, но без использования
решеточной модели. С этой целью автор работы [39] положил, следуя Гиль-
'дебранду, что процесс смешения эквивалентен необратимому
расширению центра масс молекул от свободного объема, характерного для
чистых компонент, к свободному объему раствора. Сравнение этой
теории с теорией Флори показало, что для атермической смеси
наблюдается качественное согласие данных теории и опыта, в частности,
обе теории приводят к близким значениям праметра %s.
Смеси полимер гомологов разной длины рассмотрены в работе [37].
3.7. ХАРАКТЕРИСТИЧЕСКИЕ ЗНАЧЕНИЯ
ПАРАМЕТРА ОБМЕННЫХ ВЗАИМОДЕЙСТВИЙ
ДЛЯ НЕКОТОРЫХ ХИМИЧЕСКИХ ГРУПП
В работе [38] сделана попытка получить простое выражение для
параметра контактных взаимодействий Х12 в членах
характеристических обменных параметров Хц между химически разными сегментами
(/, / ...), входящими в молекулы компонентов 1 и 2. При этом каждому
сегменту или химической группировке приписывается определенное
значение доли (ai9 а/, ...)мест (или поверхностей) в общей поверхности
молекулы. Выражение для Х12 представляется в виде
> ^12 ~ 1л (ал 1 — а(, 2) (а/, 1 — а/, г) Хц.
Для оценки доли мест (а,-, ...) используется приближение Бонди
для оценки площади поверхности групп [29]. Так, для групп —СН3—,
—СН2—, —СОО—, — <Г у —, —О— площади поверхностей
равны соответственно 3,51, 2,24, 3,65, 8,85 и 1,25 нм2. Для простоты
молекуле, подобной молекуле сложного алифатического эфира,
приписывается два типа поверхностей: «алифатическая» (ал), включающая
метальные и метиленовые группы, и «сложноэфирная» (слэ).
Например, для 1,2-этандиолацетата аслэ^О^Э и аал = 0,61, для толуола
«аром = 0,72 и аал=0,28, для 1,4-диоксана аал = 0,79 и аэф = 0,22
и т. д. '
Используя калориметрические данные, авторы работы [38]
оценили значения Х12 для многих смесей сложных алифатических эфиров
79
в толуоле, а также для многих смесей простых алифатических эфиров
в бензоле. Было показано, что график зависимости Х12= f (аслэ),
полученный экспериментально, хорошо аппроксимируется уравнением
Л12 — ХаЛщ слэ • О&слэ — 0,72 (Аал# аром + Хал. слэ — Харом, слэ) <Xcji3 +
+ 0,5184Хал.аром,
если Х^. сд, = 700, Харом. слэ = 350 и Хал, аром = 40 Дж/см3 для сме-
сей сложных алифатических эфиров с толуолом.
Для смесей простых алифатических эфиров с бензолом
экспериментальные данные описываются уравнением
А12~ О^эф^ал. эф —аэф(^ал. аром ~Ь ^ал. эф—<^аром. эф) ~Ь ^
ал. аром»
если Харом. зф = 940, Хсл. эф = 100 и Хал. эф = 1400 Дж/см3.
Предполагается [38], что полученные результаты можно перенести
на растворы полимеров, в частности растворы сложных алифатических
полиэфиров и простых алифатических полиэфиров. Так, из данных
калориметрии для раствора полиизобутилена в бензоле Х12= 42,
а расчетное значение Х12 равно 40 Дж/см3, для полистирола в толуоле
и этилбензоле Х12 равны 0,6 и 4,0, а расчетные значения — 0 и 1 Дж/
/см3 соответственно, т. е. согласие расчета и эксперимента вполне
удовлетворительное.
3.8. НОВАЯ ТЕОРИЯ ХАГГИНСА
В последние годы Хаггинс [40—44] предпринял попытку
использовать идеи, положенные в основу описания термодинамики растворов
полимеров, на растворы вообще, т. е. включая растворы и полимеров,
и низкомолекулярных веществ. Цель, поставленная Хаггинсом,
заключалась в попытке нахождения параметров, необходимых для
описания концентрационной зависимости термодинамических свойств
растворов полимеров, из экспериментальных данных, полученных для
растворов низкомолекулярных соединений. Для этого он
использовал модель, в которой каждая молекула имеет поверхность,
находящуюся во взаимном контакте с поверхностями окружающих
молекул. Если молекулы состоят из химически разных сегментов, то
каждому сегменту приписываются определенные поверхность и энергия
контакта. Например, молекулу нормального алкана можно
рассматривать как состоящую из двух метальных сегментов и (п — 2) метилено-
вых сегментов. Сегменты могут контактировать как с себе подобными,
так и с сегментами другого типа. Это предполагает, что для каждого
типа контакта сегмент — сегмент средняя энергия контакта на
единицу площади контакта постоянна при данной температуре, несмотря на
изменение типа и площади других контактов. Относительные площади
контактов должны быть такими, чтобы свободная энергия была
минимальной. Поэтому считают, что относительные поверхности контактов
связаны одной или несколькими константами равновесия, которые
определенным образом зависят от величин энергии взаимодействия на
единицу поверхности контакта.
80
С помощью этих приближений Хаггинс получил уравнения,
связывающие энергию когезии в данной системе с числом сегментов разного-
типа, средней поверхностью контакта сегмент — сегмент каждого
типа и с одной или несколькими константами равновесия, если
имеется два или больше сортов сегментов. Параметры, определенные таким*
образом, в принципе, должны быть применены ко многим сегментам,
содержащим тот же тип сегментов или сегмент-сегментных контактов.
Следуя Хаггинсу, рассмотрим вывод этих основных уравнений.
3.8.1. ВЫВОД ОСНОВНЫХ ТЕРМОДИНАМИЧЕСКИХ ФУНКЦИЙ
Пусть типы сегментов обозначаются буквами а, р и т. д., а типы
контактов сегмент — сегмент — буквами аа, рр, ар, ... и площади
контактирующих поверхностей через аа, ар, ... (в одном моле
вещества или смеси), среднюю площадь контактирующей поверхности'
на 1 моль простых сегментов типа а (или Р) — через сг? (или а°)
и т. д., среднюю площадь контактов на 1 моль разных типов
сегментов — через сгоа, о"зэ» сгар, ... , а средние энергии на единицу площади
контакта — через еаа, еРр, .... В общем случае имеются в виду
энергии притяжения, т. е. отрицательные величины. Единица
соответствующей площади не специфицируется, так как получаемые величины
являются любым произведением в форме ае или отношением типа о$/оа.
Если система состоит из сегментов одного типа, т. е. имеются
только одинаковые контакты, общая межмолекулярная энергия
взаимодействия (отрицательная энергия когезии) на 1 моль записывается
выражением
Е = баа8аа = лаа&аа/2, C.73>
где па — число сегментов а-типа на одну молекулу.
Если в системе имеется два типа сегментов, т. е. три типа контактов,
то
а3еар, C.74)-
= 2ааа + (Таз, C-75)
C.76)
Константа равновесия, связывающая площади контактов трех
типов, определяется уравнением
Коэффициент 4 включен в знаменатель для того, чтобы сделать К
равным единице при совершенно хаотичном образовании контактов, т е.
при отсутствии предпочтительности в образовании любого из двух
возможных контактов а (или Р) сегментов в гипотетической эквимо-
лярной смеси. Константа равновесия может быть связана с энергиями
контактов уравнением вида
C.78)
8t
где
Де = 2га3 — ваа — Ч$- СЗ-79)
В случае, если в системе имеется больше, чем два типа сегментов,
необходимо ввести больше констант равновесия. Величины /С, Ау k
и Д е в уравнениях C.77)—C.79) должны быть тогда записаны как
/Сар, ит.д.Дв уравнении C.78) не равна постоянной Больцмана, так
как зависит от выбора единицы площади. Коэффициент А связан со
стерическими или другими ограничениями. Обычно уравнение C.78)
яе используется и поэтому нет необходимости определять ни Л, ни k.
Из уравнений C.75), C.76) и C.77) можно получить
аа3 = -2 (а« + а3) [1 - A + уI'*]/К', C.80)
СТаа = <Та/2 ~\ ^7
где
1), C.83)
Подставляя эти уравнения в C.74), получаем молярную энергию
взаимодействия в следующем виде:
Е =
Для простых жидкостей, например, СС14, С14 и т. д., па — \.
Величина молярной молекулярной энергии ааеаа может быть оценена
из стандартной теплоты испарения: АЩ = AE°V + PAV°9 где AE°V =
= — ?мол. взаим + А?внутр + Д^внешн (Д^внутр — раЗНОСТЬ Между КОЛе-
бательными и вращательными энергиями атомов или групп атомов
в молекуле в газообразном и жидком состояниях и, вероятно, равна
нулю; Д?"внешн — разность трансляционных и вращательных энергий
целых молекул в газовой фазе и колебательными и вращательными
энергиями целой молекулы в жидком состоянии; предполагается, что
она равна — 3RT/2, однако неточность в Е не влечет неточности
в трактовке теплоты смешения и других свойств); AV0 — разность
молярных объемов газа и жидкости; Д?мол. взаим — межмолекулярная
энергия жидкости (отрицательная энергия когезии), обозначенная
в уравнении C.75) как Е.
Таким образом,
Избыточная энтальпия на 1 моль или молярная теплота смешения
равна изменению молярной энергии при смешении и является разно-
82
стью истинной энтальпии (или энергии) 1 моля раствора и суммы
молярных энтальпий (или энергий) компонент, каждая из которых
умножена на ее мольную долю в растворе:
НЕ =Н — хлН1 — х2Н2 = Е — х1Е1 — х2Е2.
Из уравнения C.73) с ла = 1 для чистых компонентов и уравнения!
C.84) с аа и ар, равными хгаа и х2о$, получаем
или
где
г =
Если К точно равна 1, то К' = 0, и уравнение C.85) сводится к
следующему:
_? о°аАе\ г(х2-х§ ]
Н = -Г- LT+^Л)J •
Теплота смешения зависит, таким образом, от концентрации %
и трех параметров: К (или /('), г и произведения о&Д е, которые могут
быть определены из точных измерений НЕ в зависимости от состава
раствора.
Уравнение C.85) можно записать в сокращенном виде, заменив
Аго^/2 на ед, а г выразив через доли контактирующих поверхностей
г, следующими соотношениями:
C.87)
Тогда
^тЕ /о QQ\
ti = s&XiZ$gk. (o.ooy
нии C.79) получается выражени<
х/Е /о оп\
V = v^Zfrgk, (о.оУ)'
При замене е на у в уравнении C.79) получается выражение для
избыточного объема смешения
где
В принципе, значения параметров г и К применимы как к Я?, так и к
Vе.
81
В соответствии с термодинамическими свойствами растворов
полимеров использование уравнения Флори—Хаггинса позволяет найти
параметр термодинамического взаимодействия Хх:
АС?! ДЯ, — TASt
-г = —Ч—- =
для чего Xi целесообразно представить в виде
Тогда после дифференцирования и замены Л^ и N2 на лгх и х2 получим
ф 2
С учетом этих преобразований будем иметь
%н = "ЛГ^" W + V3«V»)f C'90)
для /С = 1
Параметр %s представляет собой некомбинаториальный вклад
в энтропию смешения, включающий и ориентационную энтропию,
которая определяется концентрационной зависимостью ориентации
молекул или сегментов относительно окружающих их молекул, и
комбинаторную энтропийную поправку на неоднородное смешение. Эти два
вклада были рассчитаны на основе тех же приближений, что и при полу-
яении уравнения для энтальпии смешения.
3.8.2. ОРИЕНТАЦИОННАЯ ЭНТРОПИЯ
Многие молекулы не жестки; один или больше участков (например,
сегментов или межатомных связей) может изменять ориентацию
относительно другого участка. Существует поэтому некоторая случайная
ориентация и для раствора, состоящего из таких молекул; этим и
обусловлен вклад в энтропию смешения. Рассмотрим смесь жестких
молекул растворителя (тип 1) с гибкими цепными молекулами (тип 2),
каждая из которых состоит из. п жестких сегментов, соединенных (п—1)
гибкими связями. Пусть v° и v°(l—ks) — значения средней
случайной ориентации сегментов в каждой цепи полимерной молекулы по
отношению к ориентации предыдущего сегмента в цепи, когда эта
молекула находится в бесконечно разбавленном растворе в данном
растворителе и в чистом полимере соответственно. При промежуточной
концентрации среднюю ориентацию можно выразить как v°(l—ksP),
где Р — вероятность того, что единица площади контактирующей
поверхности сегмента находится в контакте преимущественно с
поверхностью полимерного сегмента, а не с поверхностью молекулы раство-
84
рителя. Энтропия сегментной ориентации тогда определяется
уравнением
Зориент = k 1П [V° A - ksP)<»-l>«*]. C.91)
Если молекулу растворителя рассматривать как простой жесткий
сегмент типа а, а каждый из п сегментов полимерной цепи — как
жесткий сегмент типа Р, то вероятность Р является отношением средних
поверхностей РР и оф контактов между сегментными поверхностями
Эти площади поверхностей контактов связаны с контактирующими
площадями аир типов уравнениями C.78) — C.84). Для К = 1,
/С'= 0 (полная однородность смешения)
- ^L, C.92)
и при К = gk = 1 имеем Р = zp. Подставляя C.92) и C.91) и заменяя
(п — 1) на /г, получаем значение избыточной энтропии смешения на
1 моль
= Sop — SopMlS=0 = RnN2 In [l + y-^- zagk\. C.93)
Дифференцируя C.93) no Nt для получения А 52 и деля результат на
—&Ф2, получаем значение ориентационной энтропийной части
параметра:
где
Для /С = ^ = 1
* US ориент =
для предельно разбавленного раствора
а для системы с одним типом сегментов
%S ориент = —ksTal^v
3.8.3. КОМБИНАТОРНАЯ ЭНТРОПИЙНАЯ ПОПРАВКА
НА НЕОДНОРОДНОСТЬ СМЕШЕНИЯ
Свободная энергия взаимодействия молекулы (или сегментов)
и ее окружения в растворе зависит от типа соседних молекул, поэтому
смешение неоднородно. Величина отклонения комбинаторной эн-
85
тропий раствора от рассчитанной в предположении совершенно
однородного смешения должна быть функцией константы равновесия /О
Пусть имеем лишь два типа сегментов —аир. Общие площади
контактирующих поверхностей обозначим через оа и стр. Рассмотрим
образование контактов трех типов единицами площадей а- и Р-поверх-
ностей. Поправка к комбинаторной энтропии смешения тогда может
быть записана в виде
где Pa-*a— вероятность того, что единица площади контакта a
с любой единицей поверхности контакта Р образует контакт aa;
Ра+$ = 1 — Pa-*a и т. д.; Qaa и а33 — общее количество контактов aa и РР
и т. д. Символами со штрихами в знаменателе обозначены параметры,
рассчитанные при предположении К = 1. Фактор 1/2 введен для
устранения двойного учета каждого контакта. Площади контактов и
вероятности связаны соотношениями
0а*а-»а/^> * a-»-a == -Afaa/Ga»
а площади поверхностей с константами равновесия — уравнениями:
где га, гр и g1^ — определены уравнениями C.86) и C.87). Поскольку
при gk = 1
Qua = аага/2, а33
то, подставляя их в уравнение C.95), получаем
Sin = 4 ((Ta A — ZaSk) Ь A — гз^) + aa A — г«^) In A - z«gk) +
— v*Za In 2a — а3гэ In z3 — ааг3 In (гагз)]. C.96)
Если имеется п\а сегментов а-типа и п^ р-типа на одну молекулу типа 1,
а также яга сегментов a-типа и П23 Р-типа на одну молекулу типа 2,
то
a
Для 1 моля раствора
86
Следовательно, k в уравнении C 96) можно заменить газовой
постоянной R и произвести замены:
— га In 2а — гз In (гагз)] + (п\$хх + п2$х2) х
X га [A — га?*) + Zagk In
Тогда
[A — Zfgk) In A — 23gfe) —
Для полимерных систем, состоящих только из р-сегментов и молекул
растворителя, состоящих только из а-сегментов
у 1
Поправки, данные уравнениями C.94) и C.97), в случае растворов
полимеров добавляют к величине комбинаториальной энтропии
смешения, выражаемой обычно формулой Флори—Хаггинса.
Параметры го и К определяют по измерениям избыточных
энтальпий или избыточных объемов смешения. Для этого используют
отношения НЕ или Vе при двух концентрациях [43]
Щ» (или V^ П + <"' " 1) ** 1! -A + «'*№
[1 + (пга - 1) х2] [1 - A + К'гаг$1'*\
Из двух таких отношений (Уф'/Ф и Vfrw, Щ'/<? и Ну»/Я)>)
рассчитывают значения го и К'. Используя средние значения этих
параметров (га и К) вместе с оценочными значениями Vq> и Щ>, вычисляют
третий член (ед или va) для каждой из этих двух величин:
(или ,д) =
_.
2 [1 + (пга - 1) х2] [1 - A + /CVPI/2J
Уравнения C.88) и C.89) с полученными параметрами
характеризуют в этом случае зависимость НЕ и Vе от концентрации. Подставляя
га, К1 и ед в уравнение C.97), находим концентрационную зависимость
параметра %#. Вычитая Хн из Хх для одной и той же концентрации,
МОЖНО ПОЛуЧИТЬ %S- ЕСЛИ ПОЯВЛЯеТСЯ боЛЬШОЙ ВКЛаД В XS ОТ Х^ориент
при этой концентрации, уравнение C.94) используется для расчета k's,
с которым можно рассчитать концентрационную зависимость Х5, %н
и %1 и сравнить ее с экспериментальными данными.
Параметры га, /С, ks и а°а определяют из данных, полученных на
других системах, содержащих тот же тип сегментов и
сегмент-сегментных контактов. Следовательно, есть надежды получить из параметров
87
на простых системах, состоящих из низкомолекулярных компонентов,
параметры для растворов полимеров. А это существенно упрощает
эксперимент, особенно в случае смесей полимеров. Применение этой
теории к некоторым системам показало хорошее совпадение ее с
экспериментом.
3.9. ТЕОРИЯ САНШЕЗА И ЛАКОМБА
Саншез и Лакомб предложили новые уравнения состояния для
жидких систем и растворов полимеров [45]. Их теория, которую можно
охарактеризовать как теорию решеточной жидкости, требует, как и
теория Флори, оценки трех параметров уравнения состояния для
чистых компонент. На основе предложенной теории они получили
выражения для химических потенциалов, теплот и объемов смешения,
критических температур смешения, энтальпийных и энтропийных вкладов
в химические потенциалы.
Уравнение состояния. Для конфигураций, доступных системе из
N молекул с конфигурационной энергией Е и объемом 1/, Саншез и
Лакомб получили выражение
где со — число конфигураций, доступных сегменту в
плотноупакованном состоянии; /0= N0/(N0+ rN) — доля свободных мест; / = rN/
/(No+ rN) — доля занятых мест в решетке. Конфигурационная
статистическая сумма выражается в виде
Z (Л Т) = ? 2 Q (Е, V, N) ехр [- (Е + PV)/kT]. C.98)
V Е
Поскольку Е и V являются только функциями числа свободных мест
в решетке, двойное суммирование в C.98) заменяется одинарным
суммированием по No:
оо
Z(P,T)= 2 Qexp[—(E + PV)/kT].
/Vo=0
Свободную энергию смешения тогда можно записать так:
G = Е +PV — kT In Q.
Как и в теории Флори, объем плотноупакованного сегмента выбирается
равным у*, объем плотноупакованной молекулы — равным ги*.
Параметры у* и гу* связаны с плотностью р*, удельным объемом и*д в
плотноупакованном состоянии и молекулярной массой М
соотношением
гу* = М/р* = Му*д. C.99)
Общий объем определяется выражением
V - (Л/, чг rN)v*,
88
а приведенные величины — соотношениями
v = иуд/^уд; р = 1/5 = р/р*;
v = V/V* = у/у*; V* = rNti*. C.100)
Долю мест / можно найти через относительную плотность, так как из
уравнений C.99) и C.100) следует, что
_ масса __ NM ___ rNp* __ - #
Р "~" объем ~~ (#0 + гЛ/) »• ~~ (Nn+rN) "~ 'Р '
следовательно, f=p/p*=p. Полное число парных взаимодействий
равно (Afo+ rN)zl2. Обозначая энергию взаимодействия между
сегментами через е, среднюю межмолекулярную энергию взаимодействия
можно представить в виде
или
E/rN = — ре*,
где /2— вероятность того, что два места окажутся заняты сегментами;
е*= гг/2—полная энергия взаимодействия на сегмент.
Используя эти соотношения, свободную энергию смешения можно
выразить в относительных (приведенных) величинах:
G/(rNs*)=G = — р + РЪ + f [(д— 1) 1пA — р) + A/г) 1п(р/со)];
f = Т/Т*\ Г* = e*/k; Р = Р/Р*; Р* = е*/у*.
Дифференцирование G по v и приравнивание результата нулю
приводит к уравнению состояния решеточной жидкости:
r =o. C.101)
Из уравнения состояния видно, что реальную жидкость полностью
можно охарактеризовать тремя параметрами: 71*, Р* и р*. Для
полимерной жидкости г -* со, и уравнение состояния C.101) сводится к
Параметры уравнения состояния получают из PVT данных, в
частности из экспериментальных значений плотности, коэффициента
термического расширения и сжимаемости:
При нулевом давлении
р* = ЪЧу = ь*Таф.
89
Смеси жидкостей. Распространение теории на смеси жидкостей
связано с использованием следующих правил смешения (правил
комбинирования).
1. Если молекула i занимает г\ мест в чистом состоянии и имеет
плотноупакованный молекулярный объем тр ь, то она должна занимать rt
мест в смеси:
Это правило гарантирует аддитивность плотноупакованных объемов:
V* = Wrf + r°2N2v* = (r.N, + r2N2) v*.
2. Общее число парных взаимодействий в плотноу пакованной
смеси равно сумме парных взаимодействий в плотноупакованном чистом
состоянии, т. е.
B/2) № + r°N2) = B/2) (r.N, + r2N2) = (г/2) rN;
здесь z — координационное число решетки, а
г == хгг° + х%г\ == ххгг + х2г2,
хг = N2/N =1 — х2 (мольная доля),
N = NX + N2.
3. Характеристические давления в плотноупакованной смеси
Р* = ФЛ* + ЩР1 - Ф1Ф2АЯ*, C.102)
ДР* в р* + р* - 2Р*2, C.103)
ф1 = riNxlrN = 1 — Фа. C.104)
Первые два правила смешения приводят к следующему
соотношению для среднего плотноупакованного объема сегмента:"
где
Концентрации фх и ф2 выражаются так:
Из определения ф1э данного уравнением C.104), и первого правила
смешения легко показать, что эта величина является объемной долей
плотноупакованного компонента i, т. е.
__ «i/p*
1 шг/о* +щ/о*'
где тх и /п2 — массовые доли. Объем смешения
1/4>:(#0+ rlV)v*=: Vfv.
90
Приведенный объем v может быть выражен через удельный объем
смеси руд или плотность р:
где
v *д = mi/p* + m2/p* = 1 /р*.
Энергия взаимодействия между молекулой растворителя и сегментом
полимера
е* =
е* = W = (ф1Р* + ф2Р* - ф1ф2ДЯ*) («pjtf + Ф^*). C.106)
Другие приведенные величины находятся из соотношений
f=T/T*; Г* = 6*/*; Р = Р/Р*;
здесь е* выражена уравнением C.106), а Р*— уравнением C.102). Из
уравнения C.106) и C.105) 71* можно выразить в виде
Г/Г* = у = [Фх/Гх + гфг/Т^Дфх + тф2) - ф1ф2Х
при X = AP*v*/kT.
Свободная энергия и химические потенциалы. Для бинарной
системы конфигурационная свободная энергия Гиббса
G = r№*G\
где
G = -р + Рд + fv [A - р) In A - р) + 1- In p] +
Дифференцирование G по плотности или объему и приращцвание
результата нулю приводит к уравнению состояния смеси, подобному
уравнению состояния для чистой жидкости (уравнение C.101)), но с Г*,
Р* и /-, определенными для смеси (для последней — = ^ + —). Хими-
ческии потенциал (д,х выражается в виде
-( dG \
h = kT [In Фа + A - rjrj ф2 +
где
Выражение для \i2 л^гко получается заменой индекса* 1'на индекс 2.
Химические потенциалы характеризуются следующими свойствами.
91
1. Они трансформируются при фх= 1 или ср2= 1 в соответствующие
молярные величины, характерные для чистых компонентов, например
2. При низких температурах или высоких давлениях приведенные
плотности стремятся к своим максимальным значениям, близким
к единице. В этом предельном случае (при р и Р]-*» 1) получаются
химические потенциалы Флори—Хаггинса:
A4 - \&!кТ-+ 1пф1 + A -/уг2)ф2 + г\Х^\.
3. Имеется лишь один параметр — ДЯ* или Х1$ который
характеризует бинарную смесь, все остальные известны из свойств чистых
компонентов. Энергию взаимодействия принято выражать в членах
безразмерного параметра ?, который является мерой отклонения
Р*2 от среднего геометрического:
при этом величина ДР* будет определяться соотношением
АР* = />* + р* _ 2? (Р^*I/2
Функции смешения. Избыточный объем смешения
А Усм/Уо = 5/(ФЛ + Фг^2) — 1 -
Энтальпия смешения Д#ш при низком давлении (пренебрегается член
PAV)
ДЯСМ//?Г = г {рф,ф2Х + и* [«fcPjfa — р) + Ф2Р2 (р2 - P)\/RT).
Энтропия смешения
= -г \Ь\п ф1 + Sf in ф2 + Ъ [A - р) In A - р) + f In р]} +
A - р.) In A - р2) + -JJF- In pt| .C.107)
-JJF- In pt|
Первые два члена в уравнении C.107) представляют собой
комбинаторную энтропию смешения Флори—Хаггинса.
Стабильность фазы и спинодаль. Отрицательная свободная энер* ия
смешения не является достаточным условием совместимости. Для
бинарной смеси необходимыми и достаточными условиями
смешиваемости во всем концентрационном диапазоне являются такие: d\i1/dx1 > 0;
dii2/dx2> 0. Для бинарной смеси решеточных жидкостей при
постоянной температуре и постоянном давлении
92
где г|> — безразмерная функция, выражаемая так:
d(\/r) b
dtp +v
(ф1 +
"y(v-l)(<h +
a P — изотермическая сжимаемость смеси — определяется таким образом:
77>*Р = v[l/(v— 1) + 1/г — 2/5ГГ1 > 0.
Система устойчива при условии
где [A/г1Ф1) + A/г2ф2)] — вклад
комбинаторной энтропии; рХ—
энергетический вклад; 1 /2pty2TP*$ —
неблагоприятный для смешения
энтропийный ьклад, вытекающий из
уравнения состояния. На рис. 3.8
схематически представлена
зависимость этих трех вкладов от
температуры.
Для бесконечно разбавленного
раствора
Д//~ =
Рис. 3.8. Схематическая зависимость
4
X рг|?2ГЯ*а E), также суммы рХ +
от температуры [45J.
C.108)
где Ми— молекулярная масса повторяющегося звена; Мг—
молекулярная масса растворителя. При выводе уравнения C.108)
использовано соотношение
Значения параметров Х{ для бесконечно разбавленного (ф2 -> 0) и
центрированного (Фз -+¦ 1) растворов выражаются уравнениями
К
1)
+ (Pi - р2)/^1 + E, -1) х
X In A - рг) - E» - 1) In A - pj + 4- In
550
500
А50\
400\
о 1
о 4
об
•6
Рассмотренная здесь теория была опробована для нескольких систем
полимер — растворитель и было найдено хорошее согласие
теоретически рассчитанных параметров смешения с экспериментальными.
3.10. НЕКОТОРЫЕ ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ
ИССЛЕДОВАНИЯ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
РАСТВОРОВ ПОЛИМЕРОВ
К настоящему времени в области исследования термодинамических
свойств растворов полимеров накоплен огромный экспериментальный
материал: работы по определению теплот растворения и разбавления,
вторых вириальных коэффициентов,
параметров термодинамического
взаимодействия, в-температур,
критических температур смешения и др.
Многие из такого рода данных
приведены в Справочнике по физической
химии полимеров вместе с
соответствующей библиографией [24]. Поэтому
в данном параграфе остановимся,
главным образом, на результатах тех
работ, в которых в наибольшей
степени обнаружены общие
закономерности термодинамического поведения
систем полимер — растворитель, а
также проведено сопоставление
экспериментальных данных с
предсказаниями существующих
термодинамических теорий растворов полимеров.
Сюда относятся работы Кувахары и
сотрудников [50], Ж- Дельма с
соавторами [30, 31, 46], Д. Паттерсона
с группой ученых [7, 12, 17, 23], статьи
по обращенной газовой хроматографии
[47, 48] и др. [49, 51].
Одними из существенных
результатов многих работ являются
обнаружение нижней критической
температуры смешения [10, 12, 17, 30, 31, 46]
и отрицательных значений энтропии и
энтальпии смешения в области НКТС
[10]. Это, как уже было сказано
раньше, предсказали современные
статистические теории растворов
полимеров [1—7], причем существование НКТС связывается с различием
в коэффициентах термического расширения полимера и растворителя.
Обобщив многие данные по НКТС, Ж. Дельма и сотрудникам [31]
удалось найти хорошую корреляцию между НКТС и относительной
плотностью смешиваемых компонентов (рис. 3.9).
350\
0,6
0,7
0,8 dp/cln
Рис. 3.9. Зависимость нижней
критической температуры смешения
различных полимеров в различных
растворителях от отношения
удельных объемов полимера и
растворителя:
1 — полипропилен; 2 — полиизобутил ен;
3 — полибутбн-1; 4 — поли-4-метил пен-
теин-1; 5 — полипентен-1; 6 —
полиэтилен [31].
94
Во многих работах обнаружены одновременно НКТС и ВКТС
[17, 50], причем с ухудшением термодинамического качества
растворителя, обусловленного большими различиями в свободных объемах
компонентов, ВКТС и НКТС сближаются и образуют диаграмму типа
«песочных часов» [10, 50]. Для таких систем наблюдаются отклонения'
от графика Шульца—Флори [50]. Повышение давления приводит
к уменьшению различия в свободных объемах компонентов, в
результате чего растворимость полимера в плохих растворителях
увеличивается [10]. Характерным для такого рода систем является тот факт„
что ВКТС и НКТС являются зеркальным отображением друг друга,
за исключением систем с широким молекулярно-массовым
распределением [10]. Во многих случаях обнаружена слабая зависимость
критической концентрации от молекулярной массы (см. рис. 2.6).
Интересные результаты получены в работах [41, 46], в которых
сравнены растворимости полибутадиена в линейном и разветвленных
алканах. Установлено, что качество растворителя увеличивается
с ростом длины цепи линейного алкана. Это улучшение
термодинамического качества растворителя связано с уменьшением разности
свободных объемов полимера и растворителя. Замена линейного алкана
на разветвленный сильно ухудшает растворимость полибутадиена.
Авторы этих работ предлагают объяснить такой эффект либо
уменьшением плотности энергии когезии алкана из-за его разветвленносхи
(т. е. увеличением разности между энергиями когезии полимера и
растворителя), либо сходством структурной упорядоченности линейных
алканов в жидком состоянии и полимеров. Различие в качестве
растворителей можно объяснить также присутствием ориентационного
порядка в полимере. Сравнение полученных результатов этой работы
и других данных с предсказаниями теорий Пригожина—Паттерсона
и Флори показало, что лучшее согласие теории и эксперимента для
толстых цепей (ПС, ПДМС и др.) получается при использовании
малых значений параметров s = ^/sj @,5—0,8), а для тонких полимерных
цепей (ПЭ, ПБД и др.) — при s — 1.
Авторы работы [47] с помощью методов обращенной газовой
хроматографии оценили параметры термодинамического взаимодействия
полиизобутилена с М от 400 до 2,5 • 104 с гептаном, гексаном и
октаном в области температур 308—368 К. Обнаружено, что параметр Хг
растет с М, стремясь к пределу при М = 2,5 • 103. При этом, как и в
статьях [41, 46], показано, что растворимость полимера увеличивается
с ростом молярного объема растворителя, т. е. длины цепи.
Полученные экспериментальные зависимости параметра %i 0T температуры,
молекулярной массы и природы растворителя сравнили с
предсказаниями теорий Пригожина — Паттерсона и Флори. Оказалось, что
теория Пригожина—Паттерсона лучше описывает экспериментальные
результаты при высших температурах, тогда как теория Флори в
некоторых случаях приводит к более реальным результатам.
Тем же методом в работе [48] получены параметры
термодинамического взаимодействия для растворов полидиметилсилоксана и полиди-^
этилсилоксана во многих растворителях в области температур от 333
до 453 К. Анализ полученных данных в рамках теории Флори и При-
95
гожина—Паттерсона привели к следующим выводам: параметр %1$
рассчитанный по теории Флори, уменьшается с температурой быстрее,
чем полученный экспериментально, и имеет отрицательные значения
в низкотемпературной области. Интерпретация данных по теории
Пригожина—Паттерсона приводит к более разумным результатам,
•согласующимся с экспериментальными данными [48].
1. Prigogine /. The molecular theory of solutions.— New York: lnterscience,
1959.— 479 p.
2. Patterson D., Delmas G. Corresponding states theory and liquid models.—
Discuss. Faraday Soc, 1970, N 49, p. 98—105.
3. Patterson D, Role of free volume in polymer solution thermodynamics.—
Pure and Appl. Chem., 1972, 31, N 1/2 p. 133—149.
4. Patterson D. Role of free volume changes in polymer solutions
thermodynamics.— J. Polym. Sci. C, 1968, N 16, p. 3379—3389.
5. a) Flory P. /., Orwoll R. A., Vrij A. Statistical thermodynamics of chain
molecular liquids. 1. An equation of state for normal paraffin hydrocarbons.—
J. Amer. Chem. Soc, 1964, 86, № 17, p. 3507—3515;
b) Flory P. J. Statistical thermodynamics of liquid mixtures.— Ibid., 1965,
87, N 9, p. 1833—1838;
c) Abe A.t Flory P. J. The thermodynamic properties of mixtures of small,
nonpolar molecules. —Ibid., 1965, 87, N 9, p. 1838—1846.
6. Eichinger B. ?., Flory P. J. The thermodynamics of polymer solutions.—
Trans. Faraday Soc, 1968, 64, N 8, p. 2035—2072.
7. Patterson D. Free volume and polymer solubility. A qualitative view.— Mac-
romolecules, 1969, 2, N 6, p. 673—678.
8. Stockmayer W. H. Chain dimensions near the Flory temperature.— J. Polym.
Sci., 1955, 15, N 80, p. 595—598.
9. Ehrlich P., Kurpen J. J. Phase equilibrium of polymer—solvent systems at
high pressures near their critical loci: polyethylene with /г-alkanes.— J.
Polym. Sci. A, 1963, 1, N 10, p. 3217—3229.
10. Allen G., Baker С. Н. Lower critical phenomena in polymer—solvent systems.—
Polymer, 1965, 6, № 4, p. 181—193.
11. Delmas G., Patterson D. The temperature dependence of polymer chain
dimensions and second virial coefficients.— Ibid., 1966, 7, N 10, p. 513—525.
\% Patterson D., Delmas G., Somsinsky T. A comparison of lower critical solution
temperatures of the some polymer solutions.— Ibid., 1967, 8, N 10, p. 503—517.
13. Simha R., Havlik A. On the equation of state of oligomer and polymer
liquids.— J. Amer. Chem. Soc, 1964, 86, N 2, p. 197—204.
14. Nanda V. S., Simha R. Equation of state related properties of polymer and
oligomer liquids.—J. Phys. Chem., 1964, 68, N 18, p. 3158—3163.
15. Nanda V. S., Simha R., Somsinsky T. Principle of corresponding states and
equation of state oligomer liquids and glasses.— J. Polym. Sci. C, 1966,
N 12, p. 277—295.
16. Bhattacharyya S. N., Patterson D., Somsinsky T. Tne principle of
corresponding states and excess functions of N-alkane mixtures.— Physica, 1964, 30,
N 7, p. 1276—1292.
17. A study of the thermodynamic properties and phase equilibria of solutions of
polyisobutene in л-pentane/C. H. Baker, W. Brown, G. Gee et al.— Polymer,
1962, 3, N 2, p. 215—230.
18. Исследование методом светорассеяния положения спинодали и
термодинамических параметров взаимодействия компонентов
умеренно-концентрированных растворов полистирола/В. М. Андреева, А. А. Тагер, И. С.
Фоминых, О. Л. Замараева.— Высокомолекуляр. соединения. Сер. А, 1976, 18,
№ 2, с. 286—290.
19. Липатов Ю. С, Нестеров А. Е., Игнатова Т. Д. Термодинамические свойства
смесей полимергомологов.— Докл. АН СССР, 1976, 229, № 6, с 1382—1386.
20. Нестеров А. Е. Критическая опалесценция растворов полимеров: Автореф.
дис ... канд. физ.-мат. наук.— Л., 1966.— 16 с.
96
81. Bellemans A., Naar-Colin С Application of the cell modeb|to^the statistical
thermodynamics of r-mers solutions. 2.— J. Polym. Sci. C, 1955, 15, N 79,
p. 121—132.
22. Biros Л, Zeman L., Patterson D. Prediction of the parameter by the
solubility parameter and corresponding states theory.—Macro molecules, 1971, 4,
N 1, p. 30—35.
23. Delmas G., Patterson D., Somsinsky T. Thermodynamics of polyisobutylene-n-al-
kane systems.— J. Polym. Sci. C, 1962, N 57, p. 79—98.
24. Нестеров А. Е. Справочник по физической химии полимеров. Т. 1.
Свойства растворов и смесей полимеров/Под ред. Ю. С. Липатова.— Киев: Наук.
думка, 1984.—476 с.
25. Blanks R. F.JPrausnitz J. M, Thermodynamics of polymer solubility in
polar and nonpolar systems.— Ind. Chem., 1964, 3, N 1, p. 1—8.
26. Bristow G. M.t Watson W. F. Cohesive energy densities of polymers. 1.
Cohesive enerqy densities of rubber by swelling measurements.— Trans.
Faraday Soc, 1958, 54, N 5, p. 1731—1741.
27. Robard Л., Patterson D. Thermodynamics of polymer compatibility.— Mac-
romolecules, 1977, 10, N 6, p. 1021—1025.
28. Renuncio J. A. /?., Prausnitz J. M. An approximation for non-randomness
polymer-solution thermodynamics.— Idid., 1976, 9, N 6, p. 898—903.
29. Bondi A. Physical properties of molecular crystals, liquid and glasses.— New
York: Wiley, 1968.
30. Delmas G., Sant-Romain P. de. Upper and lower critical solution
temperatures in polybutadiene — alkane systems. Effect of the surface to — volume
ratio of the polymer.— Eur. Polym. J., 1974, 10, N 12, p. 1133—1140.
81. Charlet G., Ducasse R., Delmas G. Thermodynamics properties of polyolefin
solutions at high temperature. 2. Lower critical solubility temperatures for
polybutene-1, polypentene-1, and poly D-methyl pentene-1) in hydrocarbon
solvents and determination of the polymer —solvent interaction parameter for
PBI and one ethylene — propylenecopolуmer.— Polymer, 1981, 22, N 9, p.
1190—1198.
82. Brandani V. Effect of nonrandomness in polymer — solution
thermodynamics.— Macromolecules, 1979, 12, N 5, p. 883—889.
33. Brandani V. Effect of nonrandomness mixing on Flory's equation of state,—
Ibid., 1978, 11, N 6, p. 1293—1299.
34. Theoretical consideration of the number of external degrees of freedom and
its application to polydimethylsiloxane solutions/K. Fujisawa, T. Shiomi,
F. Hamada, A. Nakajima.— Polym. Bull., 1980, 3, N 5, p. 261—266.
86. Statistical thermodynamics of polymer solutions based on free volume theo-
ry/F. Hamada, T. Shiomi, K. Fujisawa, A. Nakajima.—Macromolecules,
1980, 13, N 3, p. 729—734.
86. Sugamiya /C. Polar effect in free volume theory for polymer solution
thermodynamics.— Makromol. Chem, 1977, 178, N 2, S. 565—581.
87. Orwoll R. A, Flory P. J. Equation-of-state parameters for normal alkanes correla*
tion with chain length.—J. Amer. Chem. Soc, 1967, 89, N 26, p. 6814—6822.
88. Crescenzi V., Manzini G. Exchange-enthalpy parameters for binary mixtures
of low molecular weight and simple chain-like compounds.— J. Polym. Sci.
C, 1976, N 54, p. 315—325.
89. Dayantis J. Entropy of mixing of polymer and solvent as derived from the
free —volume concept.—Ibid., 1972, N 39, p. 35—41.
40. Huggins M. L. The thermodynamic properties of liquid, including
solutions. 1. Inter molecular energies in monotonic liquids and their mixtures.—J.
phys. Chem., 1970, 74, N 2, p. 371—379.
41. Huggins M. L. The thermodynamic properties of liquid, including solutions.
2. Polymer solutions considered as ditonic systems.— Polymer, 1971, 12, N 6,
p. 389—400.
42. Huggins M. L. The thermodynamic properties of liquid, including solutions.
4. The enthalpy of mixing.— J. phys. Chem., 1971, 75, N 9, p. 1255—1260.
43. Huggins M. L. The thermodynamic properties of liquid, including solutions.
5. Poly (propylene oxide) in carbon tetrachloride.—Macromolecules, 1971,
4, N 3, p. 274—279.
4 4'420 97
44. Huggins M. L. The thermodynamic properties of liquid, including solutions;
14. Solutions of normal alkanes: model for oligomer solutions.— Brit. Polym.
J., 1977, 9, N 3, p. 189—195.
45. Sanchez 7., Lacombe R. H. Statistical thermodynamics of polymer solutions.—
Macromolecules, 1978, 11, N 6, p. 1145—1156.
46. Charlet 0., Delmas G. Thermodynamic properties of polyolefin solutions at
1 high temperature. 1. Lower critical solubility temperature of polyethylene,
polypropylene and ethylene—propylene copolymers in hydrocardone solvens.—
Polymer, 1981, 22, N 9, p. 1181—1189.
47. Marcille P., Audebert #., Quivoron С Etude des interactions pol у mere — sol-
vant par chromatographie in phase gazeuse. Applications aux couples poly*
isobutene-/i-alkane.— C. r. Acad. Sei. C, 1973, 277, N 1, p. 9—12.
48. Galin M, Gas-chromatographic investigation of the thermodynamic
interactions of poly(dimethylsiloxane) or poly(diethylsiloxane) with some solvents
between 60 and 180°C— Macromolecules, 1977, 10, N 6, p. 1239—1244.
49. Upper and lower critical solution temperature for star branched polystyrene in
cyclohexane/J.M. G. Cowie, A. Horta, I. J. McEwen, K. Prochazka.— Polym.
Bull., 1979, 1, N 5, p. 319—335.
50. Upper and lower critical solution temperature in polystyrene solutions/N. Kuwa-
hara, S. Saeki, T. Chiba, M. Kaneko.— Polymer, 1974, 15, N 12, p. 777-782.
51. Hugtin M., Idris /.f Sokro M. B. Miseibility of oligomeric pol у dimethyl si-
loxane with long chain unbranched alkanes.— Makromol. Chem. Rapid Com-
muns, 1981, 2, N 1, p. 17—24.
ГЛАВА 4
ПРИНЦИП СКЕЙЛИНГА
И КОНЦЕПЦИЯ БЛОБОВ В ОПИСАНИИ
СВОЙСТВ РАСТВОРОВ ПОЛИМЕРОВ
В последние годы для описания свойств
растворов полимеров успешно используется так называемый принцип
скейлинга, сущность которого заключается в следующем.
Постулируется степенная зависимость какого-либо свойства системы от
соответствующего параметра. Например, в случае растворов полимеров
это может быть зависимость радиуса инерции макромолекулы от
концентрации, температуры, степени полимеризации и др. Поскольку
размер макромолекулы в разных концентрационных диапазонах
имеет разную степенную зависимость от молекулярной массы, весь
концентрационный диапазон разбивается на участки, где эти зависи*
мости известны. Например, в случае разбавленного раствора завися*
мость радиуса инерции макромолекулы от М выражается степенным
законом с показателями степени, равным 1/а. В области же
полуразбавленного или концентрированного раствора, где начинают проявляться
эффекты исключенного объема (см. теорию Флори—Хаггинса), эта
зависимость характеризуется показателем степени, равным 3/5. В
области перехода от разбавленного к полуразбавленному или
концентрированному раствору эти зависимости «сшиваются» и находится!
общая степенная зависимость радиуса инерции от концентрации во
всей концентрационной области.
4.1. ПРОЦЕДУРА СКЕЙЛИНГА
Упрощенно процедуру скейлинга можно представить следующим
образом. Рассмотрим раствор полимера в хорошем растворителе, кон*
центрация которого выше критической концентрации перекрывания
клубков [1]:
C*~W> D-1)
где М — молекулярная масса полимера; R — радиус инерции
макромолекулы.
В трехразмерном пространстве (d = 3, как следует из теории
Флори—Хаггинса), где показатель степени в зависимости от радиуса
инерции макромолекулы от молекулярной массы (так называемая
4» 99
экспонента исключенного объема) равен v (см. уравнение B.36)), из
уравнения D.1) следует, что
с*~Л^-^ D.2)
(N — число повторяющихся единиц в цепи). В хорошем растворителе,
согласно теории Флори—Хаггинса [2], v = 3/5, а в в-растворителе,
где исключенный объем равен нулю, v = 1/2.
При концентрации раствора с > с* среда, которая образована
перепутанными макромолекулами, может быть разделена на сферы или
блобы с радиусом, равным длине корреляции плотности ?, входящей
в функцию корреляции плотности. Эта корреляция впервые получена
Де Женном, обнаружившим аналогию между статистикой
изолированной вдпи и статистическими закономерностями поведения решетки
спинов в ферромагнетике вблизи температуры фазового перехода
[3].
Для раствора полимера функция корреляции плотности имеет
вид [4]
[с @) с (г)) — с2 = Ас @) Ас (г) = const с2 j
где г — расстояние между точками, где рассматривается
концентрация; b — размер мономерной единицы (длина связи). По аналогии
с длиной корреляции спинов в ферромагнетике вблизи критической
точки (т. е. зависимости длины корреляции от (Т—Ткр)/Ткр, где Гкр—
критическая температура, при которой происходит спонтанное
намагничивание ферромагнетика), длина корреляции плотности
предполагается зависящей от концентрации [5].
Зависимость ? от с можно определить следующим образом. Если
полимерная цепь находится в полуразбавленном растворе, ее средний
радиус инерции может быть представлен в виде
Ж* ? = а1\ D.3)
где #б— среднее число повторяющихся единиц в блобе.
Следовательно, в уравнении D.3) блоб аналогичен сегменту Куна в модели свобод-
носочлененной цепи [6]. Отличие заключается в том, что в данном
случае число повторяющихся единиц в блобе предполагается зависящим
от концентрации раствора (как уже упоминалось, это предположение
взято из аналогии между статистикой полимерных цепей и
корреляционной длиной в ферромагнетике [3]). Таким образом, а = N/Nt
блобов можно описать гауссовской статистикой подобно тому, как это
делается для Af сегментов в модели свободносочлененной цепи [6].
С другой стороны, принимается, что участки цепей внутри блоба
должны подчиняться статистике с исключенным объемом:
t?~Nlvb2 D.4)
и
R2~NN2J-lb2. D.5)
100
Как уже было сказано, предполагается, что число повторяющихся
единиц в блобе является функцией концентрации:
N6~cx. D.6)
Поэтому, подставляя уравнение D.6) в уравнение D.5), получаем
#*~№:<2v-i)*. DJ)
При концентрации ?*, используя уравнение D.2), имеем
<2v-i)*+i, D.8)
При этой концентрации статистические характеристики цепи,
подчиняющиеся уравнению D.8), должны подчиняться и обычному
соотношению для цепи с исключенным объемом:
W — N2*, D.9)
т. е. концентрация с* выбирается как точка «сшивания» двух границ
концентраций (переход от статистики изолированной цепи к
статистике полимерной цепи с исключенным объемом). Приравнивая
экспоненты в уравнениях D.8) и D.9), находим, что
* = A— vd). D.10)
где х будет экспонентой в зависимости, например, ?2 или R2 от
концентрации во всей концентрационной области. Тогда из уравнения
D.10) при учете выражений D.4) и D.5) следует, что
?2^,c2v/(\-vd)t D.11)
Поскольку в хороших растворителях v = 3/5, для трехразмерного
пространства (d = 3) из уравнений D.10) и D.11) получаем
D.12)
D.13)
Тогда из уравнения D.7) получаем, что
и соответственно
При Т = 0 v =
= 1/2 и
X
?2
л: =
г;2-
= -5/4
с-3/2.
= -2,
— С 2.
рр м 14v
\N при Г = в. DЛ4>
Подобным образом экспоненциальные зависимости были получены
и для других характеристик растворов полимеров: осмотических
давлений и сжимаемости константы седиментации, коэффициента
кооперативной диффузии Dc и др. [5, 7, 8]. Вид этих зависимостей будет
рассмотрен при обсуждении (с учетом жесткости цепи) результатов
сопоставления теории скейлинга с экспериментом (табл. 4.1).
10t
4.2. ФАЗОВАЯ ДИАГРАММА СОСТОЯНИЯ РАСТВОРА
На основе полученных зависимостей в работе [11] построена
фазовая диаграмма состояния раствора гибкоцепных полимеров в
окрестности 6-точки в переменных температура — концентрация (рис. 4.1, а).
При этом в качестве температурной переменной использована
величина т = (Т — 8)/в — относительное отклонение температуры Т от
0-температуры, с — средняя концентрация мономерных единиц в
растворе (среднее число моаомерных единиц в единице объема).
\
1
\
\
Н+ ,
/
а,
У
1С* ;
1
1с**
Рис. 4.1. Фазовая диаграмма состояния раствора гибких [11] (/) и
полужестких [9] (б) макромолекул.
Принцип построения диаграммы заключается в следующем. В
области разбавленного раствора в 0-растворителе (область
неперекрывающихся полимерных клубков) экспонента исключенного объема
v - 1/2 и
]?~N. D.15)
Поскольку в этой области концентраций корреляционная длина
изолированного полимерного клубка совпадает с его размерами, то
tf~R*~N. D.1§)
При Т = 6 величина #2, а следовательно, и'?а не зависят от
концентрации (см. D,14)). Выражения D.15)—D.16) соответствуют области
Ь фазовой диаграммы (рис. 4.1).
102
При Т >® полимерные клубки подчиняются статистике с
исключенным объемом (см. уравнение Флори B.28)). Экспонента последнего
v = 3/5. Следовательно, с учетом уравнений B.28) и D.9)
D.17)
и соответственно
Эти условия реализуются в области 1+ фазовой диаграммы на рис.
4.1. Граница перехода от области 1@ к области 1+ находится
приравниванием уравнений D.15) и D.17). Из этого условия следует
В области полуразбавленного раствора (с > с*) при Т = 0 (см.
уравнение D.14))
R*~N. D.18)
Однако размеры блобов подчиняются уравнению D.13). На диаграмме
состояния эти условия соответствуют области Не, расширение которой
с концентрацией обусловлено уменьшением размеров блобов в
соответствии с уравнением D.13). Граница области перехода от Ь к IT»,
находимая из уравнений D.18) и D.13), соответствует
Для Т > 0 в области полуразбавленного раствора степенные
зависимости находятся путем использования принципа
термодинамического подобия, заключающегося в данном случае в том, что величины
R2 или ?2 зависят лишь от относительной удаленности данной точки от
точки «сшивания» (в нашем случае от точки перекрывания клубков
с*):
{N ^ оо) D.19)
и
?2 ~ Л^Ч2/5 (с/с*)*, D.20)
где х.— экспонента в зависимости R2 и С2 от с, а с* с учетом эффектов
исключенного объема (уравнения D.2) и B.28)) выражается так:
С*~#-4/5т-3/5 D.21)
Подставляя в уравнения D.20) и D.21) соответствующие экспоненты
из уравнений D.14) и D.12), находим
R*~N%l'*(r-W9 D.22)
?»~Т-1/2 0-3/2 D.23)
Этим зависимостям соответствует область П+ фазовой диаграммы.
Граница перехода из области 1+ в Н+ находится путем приравнивания
уравнений D.17) и D.22):
103
Граница перехода от П+ к IIe находится из сравнения уравнений
D.18) и D.22) или уравнений D.13) и D.23):
?** Т.
Приведенные зависимости изложены здесь в основном так, как они
рассмотрены в работе [9]. В ней же получены степенные зависимости
различных характеристик с учетом параметра жесткости р на
основании результатов статьи [12], где было показано, что параметр 2б,
характеризующий объемные эффекты в блобе, является функцией
параметра жесткости /?, определяемого отношением персистентной длины
цепи к ее поперечнику, т. е. 2б~ тЛ^б1/2^~3/2. В области разбавленного
раствора г$ переходит в обычный параметр z [2]. Полученные
соответствующие выражения с учетом жесткости цепи приведены в табл. 4.1,
Таблица 4.1. Характеристика цепей и блобов в различных областях фазовой
диаграммы [3,10]
Q6-
ласть
i«
iT
ит
»е
I
Si
Np
(NHp)Vb
Nii^pc)-11*
Np
—
С2
Np
(NHpJ/5
(%c*p)-V2
—
n/T
c/N
c/N
(с3ртK/4
c3p3
—
N
N
(т3р3сбГ1/4
p-3<r-2
—
Границы
т*~ (Np~3)-l/2
с*-(Л^Ч3р3)-1/5
т* ~ pzc
C** r-> T/7~3
с* ~ (Npa)~l/2
а на рис. 4,1, б представлена соответствующая диаграмма состояния
раствора полужестких макромолекул. Анализ соотношений,
представленных в табл. 4.1, проведенный в работе [10], показал, что
средняя плотность звеньев в «блобе» не зависит от жесткости и объемных
эффектов и определяется только концентрацией раствора:
Рб ~ ?Г ~ с-
В то же время, если выделить участок цепи, состоящий из п звеньев^
плотность каждого звена этого участка будет тем меньше, чем жестче
цепь и больше т, которое характеризует объемные эффекты:
т. е. убывание п вызывает увеличение средней плотности ре.
Поскольку рб(я) — r~z(n)> то, выделяя вблизи контура цепи объемы
разного размера, получаем большую плотность, чем меньше г.
В работе [9] получена и кривая расслоения раствора, которая
показывает, что критическая температура фазового расслоения выше
температуры перехода клубок — глобула, а критическая концентрация
104
выше концентрации перекрывания клубков. На кривой расслоения
раствора при температуре перехода клубок — глобула
предсказывается излом. Получены формулы зависимости температурных интервалов,
выраженные в виде приведенной температуры т = | АГ |/0, от N и р,
в частности, температурный интервал между критической и 0-точкой
(т~ЛМ/2), точкой перехода клубок — глобула и 0-точкой (т ~ ръ^ х
ЛЛ~1/2), областью перехода I® — 1т и 0-точкой (т — p3/2#-i/2) И др.
Отметим, что в противоположность случаю Т > 0, полученные
автором работы [9] соотношения для Т < 0 не были проверены. В
частности, пока никем не наблюдался излом со стороны как низких, так
и высоких концентраций на кривой расслоения в области температуры
перехода клубок—глобула. Возможно, что отсутствие такого излома
на многочисленных кривых фазового расслоения, полученных в
разных работах, связано с полидисперсностью образцов. Во всяком
случае, для доказательства полученных А. Р. Хохловым [9] результатов
необходимы дальнейшие исследования. Предсказываемый же в
некоторых теоретических работах «коллапс» макромолекул вблизи
критической температуры расслоения недавно был подтвержден в работе
[13] для растворов полимеров и в [14] для термодинамически
несовместимых полимеров в области малых добавок одного из полимеров.
4.3. ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА СООТНОШЕНИЙ
СКЕЙЛИНГА И КОНЦЕПЦИИ БЛОБОВ
4.3.1. КОЭФФИЦИЕНТ СЕДИМЕНТАЦИИ В ПОЛУРАЗБАВЛЕННЫХ
РАСТВОРАХ ПОЛИМЕРОВ В ХОРОШИХ РАСТВОРИТЕЛЯХ
Если предположить, что в полуразбавленных растворах (с^с*)
вместо индивидуальной макромолекулы седиментирующей единицей
является блоб (М = с?3) и если этот «блоб» непроницаем для
растворителя, то, согласно закону Стокса, коэффициент трения
пропорционален ? (так как S ~ Mlf):
(см. уравнение D.12)). Это выражение означает, что 5 в области с^ с*
должен быть независимым от М растворенного вещества. Для 6-рас-
творителя (см. уравнение D.13))
В работе [7] авторы попытались установить, во-первых, независимость
5 от М в области полуразбавленного раствора, во-вторых, наличие
степенной связи между S и с (S ~ с^~а)у а если последняя
подтвердится, сравнить предсказанное числовое значение экспоненты с
полученным экспериментально.
Для образцов полистирола с Mw, равным 3,2 • 10б, 6,2 • 10в и
9,6- 105 в бромбензоле и полиметилметакрйлата с Mw= 1,3- 106
в бензоле авторы работы [7] действительно обнаружили, что в области
с > с* коэффициент седиментации не зависит от М, а зависимость его
105
от концентрации подчиняется степенному закону S ~ с*а. Однако
наблюдаемая экспонента а @,82) много больше, чем предсказывается
теорией (~0,5).
4.3.2. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
В разбавленных растворах осмотическое давление может быть
представлено в виде
1^=1+А2Мс+.., D.24)
где второй вириальный коэффициент Л2 связан с R и М соотношением
А2 = 4nWNA~zh (г) Щ?? ; 5 = г/а3. D.25)
3 хороших растворителях величина zh (z) почти постоянна (равна
0,21) и не зависит от молекулярной массы. Поскольку концентрация
раствора, при которой происходит перекрывание макромолекул, равна
с* = М/D/Зя) (R2)V2NA, D.26)
то из уравнений D.24) — D.26) получаем [15]
^ = 1,0 +1,12 (с/с% D.27)
где с/с*— приведенная концентрация, определяющая степень
перекрывания макромолекул.
Введем определение кажущегося второго вириального
коэффициента:
S = (л/с — RT/Mn)/cRT. D.28)
JJ вириальном разложении кажущийся второй вириальный
коэффициент дается уравнениями
S = А2 + Аъс + ... ,
S = A2
где А3 —третий вириальный коэффициент; g — численная постоянная,
равная g= As/AlM. Ограничиваясь разложением до третьего
вириального коэффициента и полагая g= 1/4 [15], получаем
= 1 +0,28 (с/с*).
Авторы работы [15] исследовали разные формы представления
осмотического давления, в частности, следующие из теорий Фиксмана [16,
|7], Ямакавы [18], Конингсвелда и сотрудников [19] и соотношений
скейлинга, полученные в [5, 8] в виде
^г = F (cAl»^)t D.29)
где сх — молярная концентрация полимера, т. е. сг=с/М\ v —
экспонента исключенного объема, определяющая зависимость 1?2от М
106
((R2y/2 — Mv). Вводя в выражение D.29) критическую концентрацию
перекрывания макромолекул с*, имеем
ЗХ/W С г / 7~19\
Согласно Де Клуазо [5, 8] в умеренно-концентрированных растворах
где К— постоянная. Таким образом
^ = ^(c/c*)i/<3v-i>. D.30)
Кажущийся второй вириальный коэффициент при большом М может
быть выражен так: х
S(*yu*l)M D.31)
Комбинируя D.31) с D.26) и D.25) и полагая, что "lift (г) —величина
постоянная, получаем
S/A2 = К' (с/с*)-* Cv-2)/Cv-d .
где К" — константа.
Как видно из уравнения D.30), в теории Де Клуазо nM/(cRT)
или S/A2 являются функциями только с /с* и не зависят от М. В
теории Фиксмана [16, 17]
S = 2яЛ0а3 (B')-*/*M-*NA [1 — A + в)~б/2], D.32)
ас—1 =0,1777 А [1 —15,87 J IF)d6\>
5 (А,0)
где / (б) — функция б, которая в свою очередь является функцией А
и у:
^
В' =9,61/6^
(р — плотность полимера; у'— безразмерная величина концентрации).
Комбинируя D.32) с D.28), D.26) и D.25), можно получить
где б —функция с/с* и 5. Видно, что ^ или S/A2 являются
функциями не только с/с*, как в теории_Де Клуазо, но и параметра
исключенного объема z. Если с, z и (#еI/2 известны, можно рассчитать
5 или я.
107
Теория Ямакавы [181, с учетом выражений D.28), D.26) и D.25),
приводит к уравнениям:
я? с, zc)(c/c*)
S/A2 = H(c, 2c)/AB),
здесь Н (с, zc) является функцией с/с* и гс. Если даны с, М, а, г
и Rl, можно рассчитать S или п. Согласно теории Конингсвелда
и сотрудников [19]
S = (vVVJ A /2 - X) + (W) с/3 + (vVVJ с/4+ ...
с
X = Xi
где
() но и параметров термодинамического взаимодействия Xi» X2»
а также Л2. Теоретические значения 5 или я можно рассчитать, если
известны с, Л4, Xi и х2-
В работе [15] приведено сравнение предсказаний концентрационной
зависимости -^ и S/A2 согласно рассмотренным теориям и
эксперименВидно, что ^« или S/i4a являются функциями не только с/с*
тальным зависимостям, полученным для растворов поли-а-метилсти-
рола разной молекулярной массы в толуоле при 298 К. Обнаружено,
что экспериментальные значения ^— и S/A2 хорошо описываются
теоретической кривой, рассчитанной по уравнениям D.30) и D.27).
При с > с* зависимость 2— пропорциональна (с/с*I»325, что хорошо
Сг\1
согласуется с предсказанием теории скейлинга. В то же время теории
Фиксмана, Ямакавы, а также Конингсвелда и сотрудников
предсказывают более резкое увеличение S с концентрацией для более
высокомолекулярных образцов. Значения S для них выше, чем для йизко-
молекулярных. Существенное различие в предсказаниях старых
теорий и теории скейлинга наблюдается как раз в тех областях
концентраций, которые рассматриваются в этих теориях. В теории скейлинга
экспериментальные данные в области с > с* сравнимы с выводами
теории и этим предполагается, что теория вириального разложения в этом
случае достаточна для описания данных в области с < с*. В старых
теориях форма вириального разложения осмотического давления
модифицирована для описания данных в этой области. Если концентрация
становится выше с*, теоретическая зависимость S от с ослабевает или
подчиняется теории Флори—Хаггинса. Область «полуразбавленных
растворов» теории екейлинга не описывается старыми теориями.
108
4.3.3. ИЗМЕНЕНИЯ РАДИУСА ИНЕРЦИИ
С ТЕМПЕРАТУРОЙ И МОЛЕКУЛЯРНОЙ МАССОЙ
По простейшей теории «блобов», переход от гауссовой статистики
до статистики с исключенным объемом для парной корреляционной
функции происходит при N = N6. Среднеквадратичное расстояние
между сегментами i и / тогда записывается в таком виде [20]:
~2if=\i-j\b\ |/-/|<ЛГб, D.33)
Введение этих выражений в уравнение для радиусов инерции
макромолекулы Rg (определяемый в статических условиях) и RD
(гидродинамический радиус инерции, определяемый из коэффициента диффузии
А.)
= 2^2 2j / f
i I
а также замена суммирования интегрированием, приводит к
выражениям
г — p)dp—I Л^б ( ат-| X
о
N 2v
§(fy (N-p)dp]
0
l
о
где интеграл от Ыб до Л^ выражается как разность интегралов от 0 до
N и от 0 до Мб- Аналитический результат получается в виде
Г 2у-' /ТГ1 2у-1 /IV2 1
/j_\2v]
2)\a:J J'
/ТГ
[2Bv+l)\x) 3Bv + 2)U
D.34)
Фарну и сотрудники [22] получили выражение для коэффициента
линейного расширения ав = Re (x)/Rg (в), где х— Ne/N, a
109
В уравнении D.34) х изменяется от N'1 до 1, когда изменяется
качество растворителя от «хорошего» к «плохому». В хорошем
растворителе при х < N'1
R b
До « 0f377WV°'e (v = 3/5)
и
aG = 0,923^°^.
Изменение aG с лГ1 представлено на рис. 4.2 для значения v =*= 3/5.
Асимптотическое изменение aG при больших N/Ne из уравнения D.34)
имеет вид
aG-> [3/Bv + 1) (v + 1)]*/2(N/N6)*-»'*, „
0,923 (ЛГ/ЛГбH'1 (v = 3/5). К 0)
Авторы работы [21] сравнили уравнение D.35) с уравнением Флори
ы% — oCq = yz, где параметр исключенного объема г выражается в
форме
а у в модифицированной теории Флори равно 1,276 [23]. Поскольку
N6~(bs/vJ9 можно написать:
(С — константа пропорциональности). Подбирая асимптотическое
поведение уравнения D.35) так, чтобы a,G~>(yzI/B, можно оценить С как
С = <у2/0>923 [24], которое становится равным С =3,63 для 7= 1,276,
и больших z (рис. 4.2). Видно, что теория Флори предсказывает более
плавное приближение к асимптотическому поведению а<?, чем теория
блобов. В теории блобов область степенной зависимости достигается
при N^5Ne. Поскольку теория Флори воспроизводит точный
результат теории возмущений первого порядка в области малых г,
модифицированная ее форма должна приводить к более точным значениям
для малых N(N/Nc< 1).
Для гидродинамического радиуса макромолекулы соответствующее
уравнение было приведено в работах [9, 24]:
Ш'п [2 (' -т) + г^<*-' - ч-гЬ с*-1 -«))•
В в-растворителе для
а для хорошего при х < 1
Rh = [F^/2/12] A _ V) B - v) bN\
П /у\
Коэффициент линейного расширения ан (х) = DH /?к\ тогда получается
«\ иг V&)
в виде
ан (х) = -щ h C — л:) + 3 (*7-"^' ~ ^~7^)Г * D'36^
110
Зависимости ан (х) от NiNq, рассчитаны по уравнению D.36) с v
3/5 (рис. 4.3). Асимптотическое поведение ан (х) при больших N
таково:
Видно, что данные не попадают в область асимптотического поведения.
На кривой зависимости а<? от т = (I — &)/Т для полистирола в
бензоле и циклогексане видны три температурные области, в-область
распространяется до температуры перехода
Ю'2 W'f 1 10 W2 1OZ N/N5 W'2 W'f 1 10 W2 1OSN/N&
Рис. 4.2. Сравнение изменения aG с N/N6, рассчитанных по уравнению D.35)
G) и уравнению Флори B) [24].
Рис. 4.3. Зависимость ан от N/N6, рассчитанная по уравнению D.36) [24].
где а — постоянная, входящая в связующий параметр при
количественном сравнении теории и эксперимента для <xq при замене N/N
на
Л — молекулярная масса мономерного звена; п — число мономеров
в статистическом отрезке цепи, которую можно получить расчетным
путем [21]. Для постоянной а в случае полистирола было взято
значение около 0,2. Верхняя температура перехода означает предел
перехода к хорошему растворителю, в котором наблюдается максимум
набухания цепи для данного растворителя
, D.37)
когда а < 1. Согласно уравнению D.37) для бензола Тт^ 130°С при
@ = _50 °С и а = 0,2, т. е. этот грубый результат свидетельствует
о том, что полистирол в бензоле не находится в условиях хорошего
растворителя, где ад становится независимым от температуры. Для
больших N авторы [21] нашли при Т > Тм
111
при Тт<Т<Тм,
aG = [3/(v + 1) Bv
ипри7<Гт
Рассмотренные результаты свидетельствуют о том, что в
переходной области N ~ N6 теория блобов неадекватна. Последнее
обусловлено скачкообразным переходом от гауссовской статистики к
статистике с исключенным объемом при моделировании г?, (см. уравнения
D.33)). Кроме того, как видно из рис. 4.4 в области в-температуры
размеры макромолекул практически не должны изменяться (т. е.
Rg и Rh должны оставаться постоянными в широком диапазоне т).
2,0
1,5
1,0
Лду/dJN
V
0,9
-2,5 -2 -1,5 -/ -0,5 LgT '303
313
323
353ТЯ
Рис. 4.4. Зависимость aG от т = G| — 9)/Г для полистирола (Mw = 44,4* 10е)
в бензоле и циклогексане [24].
Рис. 4.5. Зависимость ан от Т для растворов полистирола различной молекуляр*
ной массы в циклогексане при Mw: *
1^ 5,35-10«; 2 — 3,16.10*; 5— 1,25- 10е; 4— 6,3- 10е; 5*- 1,75.10е; 6^3,55-10* [27].
Между тем имеющиеся в настоящее время экспериментальные
результаты противоречивы. Так, на основе данных малоуглового рассеяния
тепловых нейтронов авторы статьи [25] сделали вывод, что размеры
макромолекул остаются почти неизменными в конечном температурном.
интервале вблизи в-температуры. В работах [26, 27] показано, что
гидродинамический радиус полистирола с Mw= 179 000 в пгранс-Д?-
калине линейно увеличивается с температурой в области от 20 до 40 °С,
а для образца с Mw— 12 • 106 эта зависимость становится плавнее при
более высоких температурах. В статье [27] приведены данные по
измерению гидродинамического радиуса полистирольных фракций с Mw
от 1,8 • 10б до 5,05 • 106 в циклогексане в области температур от точки
осаждения до 333 К с переходом через 6-температуру. Обнаружено,
что гидродинамический радиус клубка плавно увеличивается с
температурой. При этом скорость роста является возрастающей функцией
молекулярной массы и убывающей функцией температуры (рис. 4.5).
Повышение Rg с температурой в окрестности в-температуры
отмечалось и в работе [28] для поли-Р-нафтилметакрилата с Mw= 2,6 • 10е
112
в тетралине. Эти результаты согласуются с многими теориями
набухания полимерного клубка. Теория же блобов в данном случае
оказывается непригодной для описания наблюдаемых эффектов увеличения-
размеров макромолекул в окрестностях 0-температуры. Дело в том^
что модель блобов подразумевает, что при любой температуре имеется
характеристическая длина полимерной цепи (блоб), которая
описывается гауссовской статистикой. Эта длина бесконечна в 0-точке и
уменьшается при удалении от нее как функция т = | Т — в | /в.
Следствием этой модели является существование конечной 6-области для
цепей конечной длины [29] (область 10 фазовой диаграммы на рис.
4.1). Экспериментальные результаты, как уже говорилось, не
подтверждают существование такой области,
4.3.4. ОСМОТИЧЕСКАЯ СЖИМАЕМОСТЬ
Осмотическая сжимаемость (дп/дс)р т может быть рассчитана по
уравнению
Ш
1Ш _ Ш
\дс)р, т /г, (в)
где Rc (9) — избыточная рэлеевская постоянная, получаемая из
абсолютной избыточной интенсивности рассеяния при экстраполяции к ну-
/ 4я2я2\
левому углу рассеяния; Н — константа рассеяния Н = \ ); п0 —
\ na\)
показатель преломления растворителя; дп/дс — инкремент показателя
преломления раствора; с — концентрация; Яо — длина волны
падающего света.
При бесконечном разбавлении (дп/дс)р т = RT/MW. Однако при
высших концентрациях вириальное разложение
(дп/дс) р т = RT/MW + 2A2RTc
не оправдывается, как показано на рис. 4.6.
Следствием диаграммы состояния, полученной в [11], является то,
что в области разбавленного раствора (дп/дс)р т — с+°, в области
полуразбавленного (дп/дс)р т~ с+5/4 и в концентрированном растворе
(дп/дс)Pf т~с+2\ т. е. согласно теории скейлинга должен
наблюдаться прерывистый характер концентрационной зависимости (дп/дс)р т.
Авторы работы [30] на примере раствора полистирола в транс-
декалине показали, что прерывистая форма зависимости (dn/dc)Pj от
с для раствора конечной молекулярной массы в в-растворителе (на
10° выше в-температуры) не проявляется. Однако в области
разбавленного раствора (дп/дс)р Т — с0, а при высших концентрациях
можно выделить область, в которой (дп/дс)р т — с+5»4; эта область
переходит в конечном итоге к зависимости вида (дп/дс)р> г~ с2, т. е.
наблюдается качественная картина скачкообразной зависимости
113
ijSnJdc) p т от с теории скейлинга, которая справедлива для полимеров
бесконечной молекулярной массы. Выразив концентрацию
перекрывания макромолекул в виде
{ps— плотность растворителя; Rq— радиус инерции
макромолекулы), можно отметить, что экспонента, близкая к 5/4, действительно
0,10
-3,5
0,01
0,1
4,0 3,5 3,0 2,51дс,г/см'
Рис. 4.6. Зависимость осмотической сжимаемости от концентрации для растворов
полистирола с М= 1,79» 10б в mpawc-декалине при 303 К [30].
Рис .4.7. Зависимость осмотической сжимаемости от концентрации для
растворов полистирола с М = ЫО7 в четыреххлористом углероде и транс-декалине
при 291 К A) и в транс-декалине при 313 К B) [32].
появляется вблизи с* в очень узкой области с. Концентрация
перекрывания клубков с*, согласно определению, соответствует переходу к
области пол у разбавленного раствора. Это позволяет допустить, что
область полу разбавленного раствора
приходится на массовую долю,
равную ГО %. Авторы считают, что
основной причиной расхождений
в данном случае является
конечность молекулярной массы
полимера, поскольку степенные
зависимости скейлинга установлены для
предельного случая бесконечно
больших молекулярных масс по-
г200
100
О 0,02
/"у
о// /'"улимеров.
,/о (по массе) в работе [5] приведена зависи-
Рис. 4.8. Зависимость длины корреля- мость c{dn/dc)Pf т от с для очень
длинных цепей (М = 7-106). Как
видно из рис. 4.6, наклон прямой
зависимости с (дп/дс)р т от с
рации от концентрации для растворов
полистирола е М = ЫО? в четырех-
хлористом углероде при 291 К и
транс-декалине при 313 К [32].
вен 0,25 ± 0,04, т. е. (дп1дс)р> Т —
~я5/4 в согласии с теорией скейлинга. Для очень длинных цепей
найдена зависимость (дп/дс)р т от с и в статье [31]; в в-растворите-
ле, таком как т/?а«с-декалин, авторы также не обнаружили
скачкообразного перехода на графике зависимости (дп[дс)р^ т = f(c).
114
Несколько неожиданные результаты были получены Чу и
сотрудниками [32] при исследовании концентрационной зависимости
осмотической сжимаемости и кажущегося радиуса инерции (корреляционной-
длины L) для растворов полистирола с М ~ 1 • 107 в СС14. Так, в
промежуточной области концентраций (между разбавленным и
полуразбавленным растворами), где клубки в растворе начинают
перекрываться, наблюдается аномальный ход концентрационной зависимости
осмотической сжимаемости (рис. 4.7, 4.8).
В области разбавленного раствора (с/с* <с 1) кажущийся радиус
инерции, определенный из данных рассеяния света по соотношению
(в числителе — начальный наклон, в знаменателе — начальный
отрезок) графика зависимости Hell от sin20/2, уменьшается с
концентрацией в соответствии с предсказаниями теорий, в том числе и теории
скейлинга. Однако в области с ~ 0,01 % на кривых зависимости
кажущегося радиуса инерции (корреляционной длины L, которая в области
с/с*< 1 может быть идентифицирована как jR*, а в области
полуразбавленного раствора как величина ?, и определяется по уравнению
где q = -j- sin 9/2) наблюдается минимум, не предсказываемый
теоретически. В области концентраций от 1 • 10~4 до 5,6 • 10~4 г/см3
поведение клубков соответствует свойствам индивидуальных (хотя и не
изолированных) частиц, т. е. клубки, хотя и проникают друг в друга,
но не образуют заметного количества зацеплений. Наличие же
минимума на зависимостях L и (дп/дс)р т от концентрации при том
составе раствора, где зацепления еще отсутствуют, по мнению авторов,
связано с взаимным проникновением полимерных клубков и взаимным
экранированием гидродинамических взаимодействий.
4.3.5. ДРУГИЕ ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ ПРИНЦИПА СКЕЙЛИНГА
ДЛЯ ОПИСАНИЯ СВОЙСТВ РАСТВОРОВ ПОЛИМЕРОВ
Коттон и сотрудники [11] в рамках теории скейлинга
предположили, что, если нанести кривые точек помутнения в координатах
(Tm — в)М1я* 10~3 = (фкрМV% (Tm — температура осаждения, фкр—
критическая объемная доля полимера в растворе), получится кривая,
независимая от молекулярной массы. Следовательно, кривые,
полученные для разных гомологов того же полимера, будут накладываться
друг на друга. В максимуме кривых точек помутнения, таким
образом, должно выполняться условие (фкрЛ11/2)т = const.
Используя литературные данные, Агарони [33] показал, что
величина (фкрМ1/2)^ не постоянна, а меняется с температурой (рис. 4.9).
Так, для растворов полиэтилена значения (фКрМ1/2)т уменьшаются с
увеличением М для разных систем полимер — растворитель. Для раст-
115
1
395
391
.387
383
2
429
425
421
417
413
3
- 403
• 399
- 395
¦ 391
- 387
о /
¦\ \ ° J
Vv °\
\ \
\ к
М Я
.к
воров полистирола в разных растворителях для молекулярных масс,
покрывающих область от 51000 до 43 500 000, проявляется различная
тенденция изменения (фкрЛ41/2)т с температурой. Интересно, что для
растворов полистирола получаются три различные прямые,
характерные для разных методов определения критической концентрации
смешения. Показано, что для полиэтилена (фкрМ1/2)т — примерно
порядка 10, в то время как для полистирола — около 20 и выше.
Подобное различие в величинах М1/2 наблюдается для растворов цис-полк-
бутадиена ((фкрМ1/2)т « 10) и полиизобутилена ((фкрМ1/2)т больше 35)
[32]. Из этих данных следует, что
(фкР/И1/2)т не постоянно для
растворов полимергомологов в данном
растворителе и различно для растворов
данного полимера в разных
растворителях, вопреки предположению Кот-
тона и сотрудников [11].
Данные по исследованию
коэффициентов диффузии [34—36]
свидетельствуют о том, что в растворах
полимеров в плохих растворителях уже при
сравнительно низких концентрациях
существует пространственная
структура.
Скейлинг-соотношения (степенные
зависимости) были найдены для
описания сжимаемости предварительно
набухших в хорошем растворителе
полимерных сеток при погружении
их в раствор высокомолекулярного
полимера в том же растворителе [37].
Степень сжатия набухшего геля
зависит от концентрации раствора
линейного полимера, в который
погружен предварительно набухший гель:
(Фх/ФоJ'25 - (Ф1/Фо)/ - (Ф2/ФоJ'25 = 0.
Здесь фх— объемная доля мономеров сетки, которая изменяется при
погружении геля в раствор линейного полимера; ф0— равновесная
объемная доля мономеров сетки в отсутствие линейного полимера;
Ф2— объемная доля линейного полимера в' растворе, в который
погружается предварительно набухший гель. Степень сжатия определяется
величиной
а = q (чистый растворитель)/^ (раствор) = Фх/Фо,
где q — степень набухания. Величина а — универсальная функция
отношения ф2/фо> не зависящая от степени сшивки геля. Экспонента /
оценивается из зависимости модуля эластичности геля от равновесной
концентрации ср1в
Рис. 4.9. Зависимость максимума
кривых точек помутнения от
(ф М1^2) т для растворов полиэ-
массы в дифениле (), ф
эфире B) и дифенилметане C) [33],
(Номер ординаты соответствует
номеру прямой.)
116
На примере сеток стирол — дивинилбензол, полученных анионной
полимеризацией, авторы работы [37] нашли хорошее согласие
предсказаний теории с экспериментом.
1. Debye P. The intrinsic viscosity of polymer solution.— J. Chem. Phys., 1946,
14, N 10, p. 636—639.
2. Flory P. J. Principles of polymer chemistry.— New York: Cornell univ. press,
1953.—594 p.
3. Де Жен П. Идеи скейлинга в физике полимеров.— М.: Мир, 1982.—368 с.
4. Brochard F., de Gennes P. G. Dinamical scaling for polymer in theta solvents.—
Macromolecules, 1977, 10, N 5, p. 1157—1161.
5. Solutions of flexible polymers. Neutron experiments and interpretations/M.
Daoud, J. P. Cotton, B. Farnoux et al.— Ibid., 1975, 8, N 6, p. 804—818.
6. Цветков В. #., Эскин В. Е., Френкель С. #. Структура макромолекул в
растворах .—М.; Л.: Наука, 1964.— 719 с.
7. Pouyet G.y Dayantis. Velocity sedimentation in tne semidilute concentration
range of polymer dissolved in good solvents.— Macromolecules, 1979, 12, N 3,
p. 293—296.
8. Des Cloizeaux J. On tne absence of Flory terms in the energy and in the entropy
of a polymer chain.— J. Phys. (France), 1976, 37, N 5,p. 431—434.
9. Хохлов А. Р. Диаграмма состояний изотропного раствора полугибкоцеп-
ных макромолекул вблизи 0-точки.— Высокомолекуляр. соединения А,
1978, 20, № 12, с. 2754—2761.
10. Бирштейн Т. М., Скворцов А. М., Сарибан А. А. Структура растворов
полимеров. Скейлинг и моделирование на ЭВМ.— В кн.: Докл. на 11 Всесоюз.
совещ. «Математические методы для исследования полимеров», Пущино,
1981, 23—25 июня.—30 с.
11. Dependence determination of the temperature — concentration diagram of
flexible polymer solutions by neutron scattering/J. P. Cotton, M. Nierlich,
F. Boue et al.— J. chem. Phys., 1976, 65, N 3, p. 1101—1108.
12. Бирштейн Т. М., Скворцов А. М., Сарибан Л. Л. Эффективный
исключенный объем в макромолекулах различной жесткости.— Высокомолекуляр.
г соединения Б, 1975, 17, № 8^ с. 607—611.
13. Stepanek P., Konak С, Sedlacek В. Coil-globular transition single polystyrene
chain in diocthyl phtalate.— Macromolecules, 1982, 15, N 4, p. 1214—1216.
14. Experimenteller Nachweis des molekulardispersen Charakters der Mischung
von zwei Polymeren und Bestimmung des chemischen Potentials in diesen Mi-
schungen/W. A. Kruse, R. G. Kirste, J. Haas et al.— Makromol. Chem., 1976,
177, N 4, S. 1145—1160.
15. Thermodynamic properties of moderately concentrated solutions of linear po-
lymers/I. Noda, N. Kato, T. Kitano, H. Nagasawa.— Macromolecules, 1981,
14, N 3, p. 668—676.
16. Fixman M. Osmotic pressure of moderately concentrated polymer solutions.—
J. Chem. Phys., 1960, 33, N 2, p. 370—381.
17. Fixman M. Osmotic pressure and dimensions in moderately concentrated
polymer solutions.—J. Polym. Sci., 1960, 47, N 149, p. 91—98.
18. Yamakawa H. Theory of moderately concentrated polymer solutions.—
J. Chem. Phys., 1965, 43, N 4, p. 1334—1344.
19. Liquid—liquid phase separation in multicomponent polymer systems.
11. Dilute and concentrated polymer solutions in equilibrium/R. Konings-
veld, W. H. Stockmayer, J. W. Kennedy, L. A. Kleitjiens.—Macromolecules,
1974, 7, N 1, p. 73—79.
20. Franqois J., Schwartz T.t Weill G. Crossover from the 0 to excluded volume
single chain statistics: new experimental evidences and modified blob
model.— Ibid., 1980, 13, N 3, p. 564—570.
21. Akcasu A. Z., Han С. С Molecular weight and temperature dependence of
polymer dimensions in solutions.— Ibid., 1979, 12, N 2, p. 276—280.
22. Cross-over in polymer solutions/B. Farnoux, F. Boue, J. P. Cotton et al.—
J. Phys. (France), 1978, 39, N 1, p. 77—86.
117
23. Yamakawa И. Modern theory of polymer solutions,— New York: Harper,
1971.-319 c. F
24. Benmona M., Akcasu A. Z. Temperature effects on the dynamic structure
factor in dilute polymer solutions.— Macromolecules, 1978, 11, N 6, p.
1187-1192. ' V
25. Nierlich M., Cotton J. P., Farnoux B. Odservation of the collapse of a polymer
chain in poor solvent by small angle neutron scattering.—J. Chem. Phys., 1978,
69, N4, p. 1379—1383.
26. Nose 7\, Chu B. Static and dynamic properties of polystyrene in trans-deca-
line. 1. NBS 705 standard near в conditions. 3. Polymer dimensions of dilu«
te solution in the transition region.— Macromolecules, 1979, 12, N 4, p. 590—
599; 12, N 6, p. 1122—1129.
27. Pritchard M. J.t Caroline D. Dinamic radius of polystyrene around the в-te-
mperature.— Ibid., 1981, 14, N 2, p. 424—426.
28. Нестеров Л. Е. Критическая опалесценция растворов полимеров: Автореф.
дис... канд. физ.-мат. наук.—Л., 1966.—16 с.
29. Daoud M.f Jannink G. Temperature-concentration diagram of polymer
solutions.—J. Phys. (France), 1976, 37, N 7/8, p. 973—979.
30. Chu В., Nose T. Static and dynamic properties of polystyrene in trans-deca-
line.—Macromolecules, 1979, 12, N 2, p. 347—348.
31. Chu В., Nose T. Static and dynamic properties of polystyrene in trans—deca-
line 4. Osmotic compressibility, characteristic lengts, and internal and pseudogel
motions in the semidilute regime,— Ibid., 1980, 13, N 1, p. 122—132.
32. Static and dynamical properties of polystyrene in carbone tetrachloride.
2. Osmotic pressure and radius of giration in the dilute, intermediate, and
semidilute regions/B. Chu, K. Kubota, M.-J. He ,Y.-H. Lin.—Ibid., 1981, 14,
N 2, p. 392—395.
33. Aharoni S. M. On the molecular weight independence of cloud point curves.—
Ibid., 1977, 10, N 6, p. 1408—1410.
34. Chu В., Nose T. Static and dynamic properties of polystyrene in toms-deca*
line. 2. Correlation fuction profile a analysis by the histogram method,—
Ibid., 1979, 12, N 4, p. 599—606.
35. Chu В., Nose T. Static and dynamic properties of polystyrene in frafts-deca-
lin. 1. NBS 705 standard near в conditions.— Ibid., p. 590—599.
36. Nose Т., Chu B. Hydrodynamic and gel models of a concentrated solution of
low molecular weight polystyrene in foms-decaline.— J. Chem. Phys., 1979,
70, N 11, p. 5332—5334.
37. Bastide J.f Candau S., Leibner L. Osmotic deswelling of gels by polymer
solutions.—Macromolecules, 1981, 14, N 3, p. 319—326.
38. Shultz A. #., Flory P. J. Phase equilibria in polymer-solvent systems.—
J. Amer. Chem. Soc, 1952, 74, N 19, p. 4760—4767.
ГЛАВА 5
СТАТИСТИЧЕСКАЯ
ТЕРМОДИНАМИЧЕСКАЯ ТЕОРИЯ
РАСТВОРОВ ЖЕСТКИХ
И ПОЛУЖЕСТКИХ ЧАСТИЦ
В предыдущих главах мы рассмотрели
теории, разработанные для растворов гибкоцепных макромолекул. В
последние годы Флори и сотрудники [1—7] развили статистическую
термодинамическую теорию растворов жестких и полужестких частиц,
в основу которой положена решеточная модель. Изложим основные
положения этой теории для бинарных и тройных растворов жестких
палочкообразных частиц, смесей жестких и гибких частиц в общем
растворителе и бинарных растворов полужестких макромолекул.
5.1. СТАТИСТИЧЕСКАЯ ТЕОРИЯ СМЕСЕЙ ПАЛОЧКООБРАЗНЫХ
ЧАСТИЦ [1]
5.1.1. ВЫВОД ФУНКЦИИ РАСПРЕДЕЛЕНИЯ
' Для нахождения функции распределения, которая служит основой
для вычисления термодинамических функций (например, химических
потенциалов), рассмотрим систему, состоящую из смеси
палочкообразных частиц с осевым отношением х и растворителя. Принимаем, что
палочкообразные частицы имеют одинаковый диаметр, но разную
длину. Для упрощения расчетов диаметр молекулы растворителя
выбираем равным толщине палочкообразной частицы, т. е. Хр-рителя = 1.
Сегменты растворенного вещества считаем равными по объему с
молекулой растворителя, т. е. х является и осевым отношением, и числом
сегментов растворенного вещества.
Рассматривается домен типа жидкокристаллического или фаза,
в которой растворенные частицы ориентируются предпочтительно вдоль
цилиндрической оси домена. Этим способом ориентируются лишь
частицы с определенным предельным значением х% частицы с меньшим
осевым отношением х распределяются хаотически, в пределах
некоторого пространственного угла.
Пусть молекула растворенного вещества / с осевым отношением х\
ориентирована под углом *ф/ к соответствующей оси домена, как
показано на рис. 5.1, а. Для проведения комбинаторного анализа с
использованием решеточной модели разделим молекулу на yj~ x/sini|?/
субмолекул, как дано на рис. 5.1,6. Каждая субмолекула располагается
11?
параллельно оси домена. Если одна из граней кубической решетки
выбирается параллельной оси домена, то каждая субмолекула будет
располагаться на решетке, параллельной ее оси. Система как целое
состоит из субмолекул, заключенных в таких рядах, и задача сводится
к рассмотрению смешения субмолекул и вакансий (т. е. молекул
растворителя) в одном измерении. -
Пусть п2 молекул растворенного вещества последовательно
вводятся в пространство из п0 мест решетки. Их положения выбираются
случайно с учетом того, чтобы место, занятое одним сегментом, была
исключено для других. Ориентация каждой молекулы / относительна
оси домена, однако, точно определена «индексом разупорядочения»
Рис. 5.1. Палочкообразная частица с осевым отношением *, ориентированная
под углом i|) к оси домена А — А (а) и модель, содержащая у = х sin ip
субмолекул, каждая из которых параллельна оси домена А—А [1] (б).
yj. После того, как таким способом в решетку будут введены (/—1)
молекул, число мест V/, доступных для /-ой молекулы, можно
определить так:
/-1
«о —
п0
«о —
т
• EЛ)
где xj и у!— осевое отношение и индекс дезориентации для этой
молекулы. Первый член уравнения представляет собой число мест,
доступных для первой субмолекулы /. Если предположить, что
распределение занятых мест в данном ряду, параллельном оси домена, не зависит
от распределения мест в соседних рядах, вакантное место, ожидаемое
для первого сегмента последующей субмолекулы, определяется
объемной долей вакансий (второй член уравнения E.1)). Однако если это
место вакантно (и, следовательно, его может занять сегмент), то
следующее за ним место в том же ряду может быть занято только начальным
сегментом субмолекулы — в противном случае оно должно быть
вакантным. Следовательно, ожидаемое вакантное место представляет
собой отношение вакансий к сумме вакансий и субмолекул.
Последний член в уравнении E.1) и отражает эту «мольную долю» вакансий,
которая увеличивается в степени, равной разности общего числа
сегментов молекулы / и начальных сегментов субмолекул.
В преобразованном виде уравнение E.1) сводится к следующему* •
" = Лп —
F.2)
120
Выражая это уравнение через факториалы, получаем
v/ =
(п0 — ]TJ xt) \ [п0 — JJ (*i — yt
1 1
(п0 — 2J *г) ! [л0 — 2 fa — ^)]
E.3)
Комбинаторная часть конфигурационной функции распределения
смеси выражается через [2]
п2 п
1 » *> У >
где пх% у — число молекул с осевым отношением х и индексом
дезориентации у\ (дх>у — отношение пространственного угла, занятого
компонентами этих молекул (субмолекулами), к пространственному углу
для у = 1 (совершенное упорядочение всех молекул вдоль доменной
оси). Подстановка уравнения E.3) в уравнение E.4) приводит к
х, у
Заменяя п0 на пг + 2 хпх> где пг и пх — число молекул растворите-
х
ля и растворенного вещества соответственно, получаем
пх\ \\ <опхх/у
E.5)
(f/д;— среднее значение у для л:-меров).
Последний член в уравнении E.5) учитывает дисперсию различных
ориентации ух для разных компонент. Как показано в работе [7],
для у < х/2
пх\ П со**'//я*, у\ « у?*-
Использование этого приближения приводит к следующему:
где произведения и суммы охватывают все значения х.
E.6)
121
Введя формулу Стирлинга для факториалов и пренебрегая незна*
чительными ошибками, получим
— 1П ZM = Пг In Vt + ^Пх In Ux — (nt + %ухПх) In X
где vt = —-^— и vx = —-^ объемные доли растворителя
и #-мера соответственно. Уравнение E.7) можно переписать в виде
— In ZM — Я! In v1+^ixn In vx — (пг + %хпх) [1 — v2 A —
. — ynFxn)\\n[\-v2(\-ynrxn)]-Y,nx\\n(xyl)-(yx-\)]y E.8)
где v2 = 2 0* — объемная доля всех растворенных компонент, ?„ =
^тг", а Уп=\ ухпх у пх. Если у*=1 для всех а; (совершен-
2j л* ^-j / ^-j
ное упорядочение всех молекул вдоль оси домена), уравнение E.7) и
подобное ему E.8) определяют идеальное смешение. В случае
полностью хаотических ориентации растворенных молекул ух = х для
всех х. Тогда уравнения E.7) и E.8) сводятся к выражению
—InZM = nt Innt + 2XInVx — Yi^x[In*—(* — 1)] — SnxIn*2, E.9)
которое подобно полученному ранее для растворов гибкоцепных
полимеров с учетом энтропии дезориентации [8], если последний член
уравнения E.9) заменить обычным решеточным членом Ъпх\п (z — 1), где
г—координационное число решетки.
При выводе Zm не учитывались некомбинаторные вклады, но при
необходимости их можно учесть добавлением обычного члена в %,
который формально представляет собой обменные взаимодействия, но
может содержать и другие вклады [9, 10].
5.1,2. РАВНОВЕСНОЕ ЗНАЧЕНИЕ у
Дифференцируя уравнение E.7) или E.8) по ух и приравнивая
результат нулю, получаем
ехр (—2/ух) = 1 - v2 A - уп/Хп). E.10)
Это уравнение имеет два реальных решения для у при условии, что
концентрация v2 и среднее осепое отношение хп достаточно велики.
Согласно уравнению E.10) значение ух, при котором достигается
максимум Zm, т. е. равновесное значение у, не зависит от х.
Следовательно, индекс при ух может быть опущен и уравнение E.10)
заменено на
ехр (—2/у) = 1 — v2 A - ynlxn), E.11)
где параметр разупорядочения у характеризует средние ориентации
всех компонентов с х > у. Для компонентов с х < у значение Zm
монотонно увеличивается с ух, вплоть до физического предела,
122
т. е. для таких компонент ух= х. В уравнении E.11) уп является
средним для всех растворенных компонентов. Поэтому в общем случае
необходимо различать две категории растворенных частиц в смесях,
для которых уравнение E.11) дает решение для у и которые
соответственно являются анизотропными при равновесии. Первая категория
содержит «упорядоченные» компоненты с х > у, ориентация которых
скоррелирована с доменной осью (обозначенной через АА на рис.
5.1). Вторая категория состоит из «случайных» компонентов с х < у,
если такие присутствуют. Они неориентированны, их направления по
отношению к оси домена совершенно случайны. Первые и последние
обозначаются соответственно индексами А и R.
Объемные доли и средние числа осевых отношений обозначим
следующим образом:
» V2A = 2 vx;
х>у
E12)
E.13)
xnR = 2 xnx/ 2 пх\ хпА = 2 хпх/ Ц пх.
х<у х<у х>у х>у
Тогда
х>у
ИЛИ
Гуп/Хп) v2 = v2R + {y/xnA) v2A. E.14)
Подстановка уравнений E.12) и E.14) в выражение E.11) приводит
к соотношению
ехр (-2/у) = l-v2A(l- y/xnA). E.15)
Таким образом, параметр разупорядочения у является только
функцией концентрации и среднего осевого отношения взаимно
ориентированных компонентов (это соотношение может быть выведено путем
дифференцирования уравнения E.16)).
Равенство ух = у для компонентов с х > у /не свидетельствует об
одинаковой ориентации для всех компонентов. Наоборот, средняя
угловая дезориентация \рх для х-меров с х>у равна <р* = sin" (у/х)
и уменьшается с увеличением х.
Одинаковые значения ух = у для всех компонентов с х > у очень
помогают анализировать полидисперсные системы, и этот факт
позволяет вместо я^в качестве меры разупорядоченности использовать у,
так как он характеризует анизотропную фазу в целом.
Модель, с помощью которой была выведена функция
распределения, требует, чтобы у были положительными. В частности, у < 1
может быть приемлемым, поскольку у = 1 характеризует совершенное
упорядочение палочкообразных частиц.
123
Надо отметить, что функция распределения, выражаемая
уравнениями E.7) и E.8), и все результаты, выведенные из этих уравнений,
являются непрерывными функциями у. Никакой сингулярности не
должно наблюдаться в области 0 < у < х. В этом смысле трактовка у
как непрерывной переменной вполне законна. Законность же у < 1,
выражаемых уравнениями E.10) и E.15), при некоторых
обстоятельствах более проблематична. Значения у в этой области не могут быть
получены в рамках решеточной модели.
В следующих статьях [3—61 параметру у приписываются любые
значения у > 0, требуемые уравнениями E.10) или E.15). Между тем
и для условий, определяющих у< 1, имеются результаты
альтернативных расчетов, в которых у= 1 выбирается в качестве нижнего
предела.
5.1.3. ХИМИЧЕСКИЕ ПОТЕНЦИАЛЫ
В соответствии с результатами предыдущей части пересчет
уравнения E.8) для комбинаторной функции распределения приводит к
следующему:
—In ZM =n1lnv1 + Yi nx In (vx/x) — (nx + ? xnx) x
X П ~V2A 0 -У/*плI 1П П - V2 0 )\ +
V2A 0
+ Ц (x—l)nx- 2 nx\nx2-(\ny*-y + 1) ? nx. E.16)
x<y x<y x>y
Химический потенциал растворителя в анизотропной фазе дается
выражением
(Hi - t*°i)/RT = - (д In ZM/dnt)T, v, {nx]eq =
= — (д\г\1м1дпх)т, v, {nx}, у —
— (д In ZM/dy)T> vt {пх], п, {ду/дпг)т, v, {nx}eq,
где {пх) означает набор числа молей для всех растворенных
компонентов, а индекс eq — равновесное разупорядочение. Поскольку
(д In 2м/ду)т, v, {пх}, я, = 0 для смеси с равновесной степенью разупо-
рядочения у, то
Дифференцируя уравнение E.16) и подставляя E.15) и E.14),
получаем
0*1 - WRT = In A - t»2) - In [1 - у2Л A - y/xnA)] +
После подстановки E.15) в уравнение E.17) имеем
(IH- Vl)/RT - In A - v%) + 2/у + v2R A - l/xnR) + v2A (у - l)/xnA.
E.18)
124
Выражения для химических потенциалов растворенных веществ
выводятся тем же способом для х < у:
(I** — P°x)/RT = In (vx/x) + 2х/у — In x2 +
+ xv2R A - l/xnR) + xv2A (y - l)/xnA, E.19)
и для х > у:
(Ц* — vlVRT = In (vx/x) + 2 — In f +
+ xv2R(l - WnR)+xv2A (y- \)lxnA. E.20)
Химические потенциалы для идеальной смеси с концентрациями,
выраженными в объемных долях, даются следующими уравнениями:
(Vi-\h)/RT = 1пA - v2) - ln[l - Ml - l/3f»)J E.21)
(Vx - vh/RT = In (vx/x) - In [ 1 - v2 A - l/xn)]. E.22)
Подстановка у = 1, у2я= 0 и у2л= у2 в уравнение E.17) дает
уравнение E.21). Те же подстановки в уравнение E.19) (здесь не включены),
полученные прямо путем дифференцирования E.16), без подстановки
E.11), приводят к выражению E.22). Химические потенциалы,
которые получаются из уравнения E.9) для изотропной фазы, таковыг
= In A - eg + A - l/xn) v2, E.23)
(ft* - V?x)IRT = In (vx/x) + x(l — l/Xn) v2 - In a:2. E.24)
5.1.4. РАВНОВЕСИЕ МЕЖДУ ИЗОТРОПНОЕ И АНИЗОТРОПНОЙ ФАЗАМИ
При равновесии между изотропной и анизотропной фазами для
всех х
^=[4, E.25)
I** = ni, E.26)
где штрихом обозначена анизотропная фаза. Уравнения E.25) и E.26)
используются для расчета отношения объемных долей компонентов
в анизотропной и изотропной фазах.
Подставляя уравнения E.23) и E.24) в E.18) и E.19)
соответственно для \iv \ixy \i} и |х^, для сличая х <: у получаем уравнение
ln[(l-i>i)/(l -о,)] = A - lfxn)vt-2/y — B E.27)
и
In (vxlvx) = х A — 1 fxn) v2 — 2x/y — Bx, E.28)
где
В = v2R> A — 1/JCni?') + v2a' {У
Когда х>у, использование уравнения для \ix (уравнение E.20))
приводит к
In (px;vx) = x(\ — 1/*„) ^2 — 2 [1 + In (x/y)\ —Вх. E.29)
125
Результатом подстановки уравнений E.27) в E.28) будет выражение
v'x/vx == [A — v2)/(l — v2)]x = {v[/vx)x для x < у,
или
fli/0* = exp (—?*) для * < */,
где
g - -1п[A —»;)/A -у2)] = 5 + 2/у— A - 1/**)»,. E.29, а)
Для х>у путем подстановки уравнения E.27) в E.29) получаем
vJx/vx = (у/exJ [exp B/t/) A — i>2')/(l — v2)]*,
или
»>* = (t//e*J ехр (г)*), E.30)
где
?, Г)=A — 1/Xn)v2 — В.
Значения ? и т), оцененные этим способом, положительны.
Для 1>ад,<»я и (у —1)/*^
Л — v2
<и уравнение E.30) сводится к
v'x/vx ^ (у/^л:J exp(v2x) (x > у).
Если анизотропная фаза идеальна (совершенное упорядочение,
у < 1), то, подставляя значения химических потенциалов \i[ и \\>х из
уравнений E.21) и E.22) в уравнение E.25) и E.26) (условия
равновесия), имеем
In [A - i>a')/(l - v2)] = A - 1/Зс„) у2 + In [1 - wi A -
и
v'x/Vx = а: [ 1 — v2 A — 1 /U)] ехр (т1*л:),
Полученные соотношения были использованы для описания
свойств тройных систем, содержащих жесткие и палочкообразные
молекулы с осевыми отношениями ха и хь и растворитель. Рассмотрена
атермическая система. Для выбранных значений ха, хь(ха >хъ) и
объемной доли у' растворенного вещества в анизотропной фазе
y'=vt/vi E.31)
были рассчитаны концентрации компонентов в двух фазах. При этом
среднее число х'п в анизотропной фазе определяется этими тремя
параметрами согласно соотношению
1/*А«A-7')/*в + т7*б. E.32)
Соответствующие численные решения были получены следующим
образом:
1) выбиралось пробное значение v2 и vi, sl v'a = v2—Vb и Хп
были оценены из у' и v'2 по уравнениям E.31) и E.32);
126
2) параметр у оценен по уравнениям E.11) или E.15);
3) при использовании уравнения EЛ5) вводились замены: v^
v2 и хпА = хп для у < хЬу v'2A = v'a и *^ = *а для
52
но уравнению E.29а)
va - va {y/exa)-2 [exp (—2/у) A — v2)l( 1 — Ог)
для у < хь
![ехр(-2/г/)A-у2)/A_У2')
соглас-
E.33)
E.34)
1**20)
Q
Va-1Qn
Рис. 5.2. Тройная фазовая диаграмма для тройной системы с х< = 1, х„ = 100
и ^6 = 20:
/, i44, А2 —сосуществующие фазы при трехфазном равновесии «¦ изотропная и две анизотропные?
Си D-^концы узловых линий, связывающих экстремумы Iq и /d метастабильных бинодалей.
Рис. 5.3. Расширенный график бинодалей вблизи изотропной фазы (/) при
равновесии с двумя анизотропными фазами (Л^ и Л2) в системе с *f = L лга = 100
и^ = 20.
Бинодали для изотропных фаз в метастабильном равновесии с изотропной фазой показаны штпи*
ховыми линиями [П-
а для
E.35)
Сумма уравнений E.33) и E.34) или E.33) и E.35) дает
выражение, которое решается подбором пробных значений v2 = va + vb (для;
У>)
)
4) рассчитываются vaf vb и хп для изотропной фазы.
Химический потенциал растворителя (\it — i4)/RT в анизотропной
фазе был рассчитан согласно уравнению E.18) из v^a—v'z, V2R == О
И Х'ПА = Хп ДЛЯ у < ХЫ ИЛИ ИЗ v'M = ^, ^ = Vb И ^ = ^6f ХпА =
= л:а для случая у^хь. Полученные результаты для (\х'г —- [i[0)lRT
в анизотропной фазе сравнивались со значением химического
потенциала в изотропной фазе, которое рассчитывалось из v2 и 1сп по
127
тхэ
ЛОИп 54
0,4
1'ЗПбЯЭИЦ
Рис. 5.4. Тройная фазовая диаграмма
для системы полиоктилизоцианаг — по-
Лиизоцианат E0<% бутил + 50% л-ани-
зол-2-этил) — тетрахлорэтан [11].
уравнению E.23). Выбиралось
любое пробное значение у2' и стадии
1) — 4) повторялись до тех пор
пока не удовлетворялось условие
|гх = \х[. Расчеты были выполнены
для смесей компонент с ха = 40
и хь = 20, ха = 100 и хь = 20,
ха == 100 и хь = 10 соответственно.
На рис. 5.2 и 5.3 приведена одна
из рассчитанных тройных диаграмм
для тройной системы. Было
показано, что область сосуществования
обеих фаз (изотропной и
анизотропной) расширяется при добавлении
второго компонента. Компонент
с осевым отношением ха = 100
практически уходит из изотропной
фазы при добавлении компонента
с меньшим осевым отношением
{хь = 10 или 20) в систему, содержащую несколько процентов
компонента а.
Экспериментальные результаты, в некоторой степени
согласующиеся с теоретическими предсказаниями, получены Агарони [11] на
примере тройной смеси полиоктилизоцианата ([ц] =2,4 дл/г в хлороформе)
•с поли E0 % бутил + 50 % /г-анизол-2-этил) изоцианатом ([г]] =
«= 0,93 дл/r в хлороформе) в тетрахлорэтане (рис. 5.4).
5.2. СИСТЕМА КЛУБКООБРАЗНАЯ ЦЕПОЧКА —
ПАЛОЧКООБРАЗНАЯ ЧАСТИЦА — РАСТВОРИТЕЛЬ
В работе [5] Флори рассмотрел случай тройной смеси, содержащей
растворитель и два компонента, один из которых — палочкообразные
частицы, а второй — клубкообразные цепочки. Осевое отношение
палочкообразной частицы равно х2, а контурная длина клубкообразной
частицы — хВУ т. е. молекулярные объемы компонентов смеси
находятся в соотношении 1 : х2: х3.
Комбинаторный анализ для тройной смеси, рассмотренный ранее
B, 3], приводит к функции распределения
X
(По - х2п2 - xsn3)\ п3\ я
E.36)
где
128
a Zs— внешняя конфигурационная функция распределения для
клубка. Первый член в скобках уравнения E.36) выражает ожидаемое
число конфигураций для палочкообразных частиц в пустой решетке. Он
соответствует уравнению E.6). Второй член учитывает конфигурации,
достижимые клубками, последовательно добавляемые в решетку.
Введение приближения Стерлинга для факториалов приводит к
—lnZM = n1lnv1 +n2lnv2 + n3lnv3 = no[l— v2(l —
— y)/x2] In[1 — v2A — y)/xz] + n2(y—l)—n2ln(xjf) +
+ «в (*з - 1) - «8 In (xsZs), E.37)
где vlt v2 и v3— объемные доли соответствующих компонентов.
Приравнивая д\п2м/ду нулю, получаем
ехр (-2/у) = l-vs(l-y/X2). E.38)
Химические потенциалы, найденные из уравнения E.37), равны
(Hi — Hj)/OT = InVl + v2(y- l)/x,+ v3(l — \/xz) — In[1 — v2 A —
~У)/х2], (V2-\1§/RT =\n(v2/x2) +v2(y-l)+ v3x.2(\ - l/x,) —
- у In [ 1 - v2 A - y/xj] —2\ny, (us - iiyRT = In (vs/x3) +
+ v2 (x3/x2) (y—l) + v3 (x3 — 1) — xa In fl — v2 A — y/x2)] — In Z3,
где под у подразумевается равновесное значение, данное уравнением
E.38).
Подстановка уравнения E.38) в каждое из этих уравнений
приводит к выражениям
(f-4 - \tylRT = In Vl + v2 (у — \)lx2 + v3 A — 1 /x8) + 2/yt
(ii2 - ytylRT = In (v2/x2) + v2 (y - 1) + v3x2 A - l/x3) +2A- In y),
In (V3/x3) + v2 (x3/v2) (tf—l) + v3 (x3 — l)+2x3/y—In Z3.
Для изотропной фазы химические потенциалы равны
(^- vyRT = In Vl + vtA - l/x2) + va(l- Vx3),
(Ц2 — V$/RT = In (p2lx2) + v2 (x2 — 1) + v3x2 A — 1 /x3) — In x\,
(fi3 - [ip/RT = In (va/x3) + v2x3 A — l/x2) + v3 (x3 — 1) - In Z3,
При равновесии между изотропной и анизотропной фазами
In (v[/v x) = А — В — 2/у, E.39)
In {v'2/v 2) = (A — B)x2 — 2ln (х2е/у), E.40)
In (v'3/v3). = (A — B)x3 — 2x3ly, E.41)
где
А = v2 A - l/x2) + v3 A - 1 /*8), E.42)
B = v'2(y-l)/x2 + v3(l-l /x3) E.43)
(уравнения E.39), E.40) и E.41) получаются путем приравнивания
химических потенциалов для анизотропной фазы к химическим по-
129
генциалам для изотропной фазы). Комбинация уравнений E.40) и
E.41) приводит к
In (v'2/v2) — (х2/х3) In (v3/v3) = 2х2/у — 2 In (x2e/y).
Для выбранных х2, х3 и v2 эти уравнения могут быть решены
относительно у, v3, v2 и v3 следующим образом. Сначала получаем у
из уравнения E.38) с v2, замененным на v2. Выбор пробных значений
для v2 и v3 позволяет оценить А согласно уравнению E.42).
Подставляя у, v2 и v'3 в уравнение E.43), получаем В. Уравнение E.40)
можно тогда использовать для получения значения v2, которым
заменяется v2. Значение v2, в которое включается выбранное и3, может
Рис. 5.5. Фазовые диаграммы для системы с х9 -.
= оо (б):
20, хь =
/ — растворитель; 2 — палочка! 3 -*• клубок [51. (Бинодали показаны сплошными
толстыми линиями.)
быть тогда определено методом последовательных приближений.
Подстановка v[ = I — v2 — v3 в уравнение E.39) дает значение vl9
которое сравнимо с vx = 1 — v2 — v3. Тогда выбирают новое значение v3
и процедуру повторяют до тех пор, пока получится
удовлетворительное решение.
Для концентрированной бинарной смеси компонентов 2 и 3, т. е.
для v±= О по уравнению E.39), требуется, чтобы v[ также было равным
нулю. Уравнения E.40) и E.41) в этом случае приводятся к
выражениям
In {v'2/v2) = v2 (x2 — y)+ (v2 — v2) A — х2/х3) — 2 In (x2e/y), E.44)
In (v'3/v3) = In A — v2)l(\ - v2) = (x3/x2) [v2 (x2 -y) +
E.45)
соответственно. Для выбранных х2 и х3 уравнения E.44) и E.45)
в связи с уравнением E.38) можно решить одновременно для v2 и v2
и, следовательно, для v3 = 1 —v2 и v'b = 1 —v2.
На рис. 5.5 и 5.6 приведены фазовые диаграммы для тройных
систем. Бинодали для изотропной и анизотропной фаз обозначены
сплошными линиями, а узловые точки— штриховыми. На рис. 5.6 и 5.7
130
приведены зависимости объемных долей v's вдоль бинодали для нема-
тической фазы от v'2 и зависимости объемных долей vz клубка в
изотропной фазе при равновесии от х2. Анализ приведенных результатов
показывает, что добавление компонента 3 в бинарную систему,
образованную компонентами 1, 2, приводит к увеличению объемной доли
тг2
0,5 0,6
-3 -0,2
-0,4
-0,6
-0,8
О -1,0
8
10
12 14 х2 '
Рис. 5.6. Зависимость объемной доли v$ A,2) клубка в анизотропной фазе
и значения у C, 4) от v'2 палочкообразного вещества для систем [б]:
1,3— х2 = хг =20; 2,4— х2 = ха = 10.
Рис. 5.7. Фазовые составы для смесей палочка — клубок при хг = 0.
Объемные доли клубка в изотропной фазе при равновесии / — 3 и зависимость v^/v2 от
х2 для системы с х2 = хъ D)f #3 = 1 (/), х2 = xz B) и хг = ~ C) [5]/
ТХЭ
о.*,
0,1
тхэ
0,4
ПОИЦ 5-1 2-1 1-1 Ь2 V5 ПС ПШИЦ 5Й
а
/••/ Ь2 Ь5 ПС
6
Рис. 5.8. Фазовые диаграммы для систем по л иоктилизоцианат — полистирол — тет-
рахлорэтан (а) и полиизоцианат E0% бутил + 50% л-анизол-2-этил) — тетрахлорэ-
тан (б):
• — изотропная} О~ аниаотропная; д — две фазы [И].
палочкообразных молекул в анизотропной фазе и расширяет область
сосуществования двух фаз. Компонент 3 предпочтительно остается
в изотропной фазе. Ее объемная доля v's в анизотропной фазе менее
10 для всех составов и становится исчезающе малой, если v'2
увеличивается из-за повышения v3 в изотропной фазе. Экспериментальным
131
подтверждением этих выводов могут служить данные уже
упоминавшейся работы Агарони [11] для смесей полиоктилизоцианата с
полистиролом (М = 400) в тетрахлорэтане (рис. 5.8,а) и поли E0 %
бутил +50 % п-анизол-2-этил)изоцианата с полистиролом в
тетрахлорэтане (рис. 5.8,6).
5.3. СТАТИСТИЧЕСКАЯ ТЕРМОДИНАМИКА СМЕСЕЙ ПОЛУЖЕСТКИХ
МАКРОМОЛЕКУЛ С ПАЛОЧКООБРАЗНЫМИ УЧАСТКАМИ,
ЗАФИКСИРОВАННЫМИ В ОПРЕДЕЛЕННЫХ МЕСТАХ ЦЕПИ
В работе [12] Матесон и Флори развили статистическую
термодинамическую теорию для растворов полужестких цепей, состоящих из
гибких и жестких участков. Примером таких макромолекул могут
быть*, например, сополипептиды, которые содержат длинные, строго
Нематическая ось Нематическая ось
а 6
Рис. 5.9. Схематическое представление полужесткой цепи с тремя
палочкообразными и одним гибким участками (а) и представление ее на решетке (б).
(Субучастки выстроены в прямую линию.) [12].
спиралевидные участки, разделенные блоками, не образующими
спиралей (глицин или пролин). Схематически такая макромолекула
представлена на рис. 5,9,а. Она содержит три участка жесткой
палочкообразной формы, которые, в принципе, могут быть разделены гибким
участком переменной длины. На основе решеточной модели для
растворов таких макромолекул получены выражения для химических
потенциалов и условия равновесия между нематической и изотропной
фазами.
Конфигурационная функция распределения. Для использования
решеточной модели сегменты определяются как часть цепи, имеющей
длину, равную ее диаметру. Длина растворенной макромолекулы
определяется числом х сегментов. Для упрощения молекула растворителя
предполагается квазисферической и равной по размеру сегменту.
Следовательно, каждая ячейка решетки может быть занята либо
молекулой растворителя, либо сегментом растворенного вещества. Далее
используется схема представления жестких сегментов, так, как это
показано на рис. 5.9,6, т. е. субучастки располагаются параллельно
основной оси нематической фазы, которая выбирается параллельной
основной оси решетки.
Функция распределения Zm предполагается состоящей из суммы
нескольких частей:
ехр (—%v8 J] хпх), E.46)
X
132
где ZK€>M6 — комбинаториальный или «стерический» фактор; Zopiiem—
учитывает ориентацию палочкообразных участков, а третьим фактором
вводится обменная свободная энергия взаимодействия между
растворителем и растворенным веществом; vs — объемная доля растворителя;
пх — число молекул растворенного вещества, состоящих из х
сегментов; X — общеизвестный параметр взаимодействия.
Комбинаториальный фактор записывается в виде [2—6]
пР к
где v/ — число способов размещения /-ой молекулы растворенного
вещества на решетке; пр — общее число молекул растворенного
вещества; ZK — внешняя конфигурационная функция распределения гибких
сегментов по отношению к Zn = 1 для палочкообразных сегментов;
х) и х1} — число гибких и жестких сегментов соответственно. Общее
число сегментов в молекуле определяется так:
Вид уравнения E.47) зависит от вывода выражения для v/.
Используя процедуру, описанную раньше [2—6J, для v/ имеем
1 "- J
i — > . X,
/1
«о 2 &1-
где п0 — общее число мест решетки, т. е.
п0
xt = ns+ %xnx
. E.48)
E.49)
ns — субучастки молекул растворителя; у(—число субучастков,
необходимое для того, чтобы расположить палочкообразный участок
в направлении основной оси решетки, как показано на рис. 5.1,6; т. е.,
величина у{ служит мерой дезориентации молекулы i. Первый член
уравнения E.48) выражает число вакантных мест решетки для первого
сегмента /-ой цепи. Второй член (в квадратных скобках) определяет
ожидаемое вакантное место для гибкого сегмента или для первого
сегмента жесткого субучастка. Последний член уравнения представляет
собой условную ожидаемую вакансию, необходимую для достижения
ожидаемой вакансии при условии, что предыдущее место вакантно.
Оно выражается отношением числа вакансий к сумме вакансий и
субучастков. Заменяя члены уравнения E.48) отношениями факториалов
и используя уравнения E.46), получаем
у хк
F.50)
133
Суммирование может быть выполнено путем использования
отношений
где Эп — доля жестких сегментов в молекулах растворенного
вещества;^— среднечисленная длина цепи; у—число жестких участков
т) сегментов на длину молекулы размера х; 2л*Т*.ч —общее число
X
участков длины tj; уп—средняя дезориентация для таких участков.
Величина у зависит только от длины г\ участка, а не от размера всей
молекулы, которой он принадлежит [13]; г)— представляет собой
среднюю длину палочкообразного участка, усредненную по всем
молекулам, а 1}—среднюю дезориентацию всех участков. Подставляя
уравнения E.49) и E.51) в E.50), имеем
комб - (nJbi^Mrfi-^i ¦ E'52)
Если допустить, что распределение ориентации жестких участков
зависит только от их длины т), можно положить, что
у
ZopHeHx = ПП к,„?лл*.ч |2лЛ*.ч, Л* **'П'У , E.53)
где Vjp.rj.y — ожидаемое число палочкообразных участков длины ц
и дезориентация у в молекуле размера х\ со^ ^ — априорная
вероятность этой дезориентации; щ, у отражает область жесткого участка,
связанного с у для оценки значения г). Авторы работы [12J
выражение для ZopHeHT использовали в виде
В предельном случае малых средних дезориентаций уравнение E.53)
сводится к уравнению E.54) со значением С, равным [13]
С-21п(я^/8) = 0,131. E.55)
Для высоких степеней разупорядочения, однако, предпочтительнее
использовать С = 0 [7].
Свободная энергия смешения и равновесное разупорядочение.
Подставляя уравнения E.52) и E.54) в E.46) и используя приближение
134
Стирлинга для факториалов для приведенной свободной энергии
смешения получаем
ЫЗ/RT = — In ZM = ns In vs + 2 nx In ipjx) + (nx + xnv) x
4- ivsvp] —US n/tx, ч (In t/* + C). E.56>,
Здесь vn = 0nt>p и ук = A —0п)^р — объемные доли жестких и гибких
сегментов соответственно; vp = vK + vn — объемная доля растворенного
вещества;
E.57)
хУх. ч (Л — Уч)/(п. + *лр) E.68)
Дифференцируя уравнение E.56) по уп и приравнивая результат к нулю,
имеем
ехр (-2/уп) = 1 - vn A - у/Ц) = 1 — Q, E.59)
что соответствует условию равновесного разупорядочения,
найденному ранее [1—7, 13] для систем из несвязанных палочек. Равновесное
значение у^ полученное таким образом, не зависит от т|.
Следовательно, можно опустить индексы и положить уп= у при условии т] > у.
Для участков с г\ < у свободная энергия уменьшается монотонно от
уп к пределу уч= т], поэтому решений уравнения E.59) не существует.
Для таких участков параметр разупорядочения достигает верхнего
предела при у = т). Поэтому эти участки неориентированы. Используя
процедуру, предложенную ранее [2, 3], неориентированные участки
можно выделить в отдельную категорию, приписывая им индекс а
? ,ч
х ц<у х i\>y х т)
= (vru/vn) т| + (У/Ца) (van/vn) л, E.60)
где Vn и Уп — объемные доли жестких участков, которые
ориентируются хаотично (т] <: у) и упорядоченно (г) > у)\ ца — средняя длина
ориентированных участков, т. е.
1а =2 2 ЩхУх.'п/Л 2 ЯхТх,ч. E.6i>
Af f\>y X t\>y
Подставляя E.60) в E.57) и E.58), имеем
Q = i>n(l-?/?), E.62)
) [2 2л,(Л — У)Т*.чЬ E.63)
Подставляя уравнение E.62), E.63) в E.59) и заменяя уп на у, по*
лучаем
ехр (-2/у) =1-^A- |//ла) = 1 - Q> E.64)
V35
т. е. у определяется объемной долей и средней длиной вытянутых
участков. Остается разделить последний член уравнения E.56). Для
Я < у используются такие замены: ^= (/ и С = 0, для ц > у имеем
#7]= У у а С остается таким же, как и в уравнении E.55).
В результате свободную энергию можно вычислить в виде, удобном
для дифференцирования путем подстановки уравнения E.63) для E.56)
ц путем выражения всех величин в членах числа
компонентов.Результатом этого является уравнение
AQ/RT » ns lnn8 + 5Х In л, — (п8 + %пх) In (пв +%xnx) +
+ ?(*-i)"*-["s + ?*"*-? S (y-r\)nxyXtn]inx
[*ш+Яхпх— 2 S
— ? 2 **w*,4 —2 м*—2чт*.чIп ^к—2 2 л*
- In у2 - С) 2 2 nxyXt ч + Хя8 2 *ях/(л. + 2 хпж). E.65)
Химические потенциалы и условия фазового равновесия. Химический
потенциал растворителя получают обычным способом, в частности,
дифференцированием уравнения E.65) и приравниванием результата
к нулю для получения равновесного значения, т. е. нахождения
значения дАвм/ду = 0. Это приводит к
(|if- \i°s)/RT = In vs + A - l/x) vp - In A - Q) - Q + %vl E.66)
Подстановка уравнения E.64) дает следующее:
0if - \i°s)/RT - In A - vp) + A - 1/S) у, + 2/y - [1 - exp (-2/t/)] +
+ xoj. E.67)
Аналогично для химического потенциала растворенного вещества
дифференцированием уравнения E.65) по пх получаем выражение
In (vx/x) + (l-l/x)xvp-[x-
- 2 (Ъ-У) Ix. ч] ln(l - Q) -Q*- (^- 2 ЛТ*.Ч)In ZK-
— 2 Ъ, ч In Л2 - (In / + C) 2 Y*. ч + nv\> E.68)
которое при подстановке уравнения E.64) сводится к
(Ik - ^)/^Г = In (vx/x) + A - l/x) wp + 2x/y - 2 2 (Л/У ™ 1) Y*. ч-
— *[1—«p(—2/y)] —(*—24Y*.4)lnZK— 2 Y^.^nn2 —
- (In */2 + Q 2 Y*. -n+x%(l- vpf. E.69)
136
Для анизотропной смеси уп*= ц для всех tj и, следовательно, Q = о
и уравнения E.66) и E.68) заменяются на
(ft - vfy/RT = In A - wp) + A - Цх) vp + %v2p E.70)
и
(ft — ft)/^' = ln (»*/*) + 0 — VX) wp — (x — ]? чу,. „) In ZK -
- 2 ТлчМ' + яхО-»„)"•
Используя условие сосуществования фаз \i's = \iai из уравнений E.66)
и E.67) получим
In [A - v'p)/(l - vp)} « A - 1/S) i>, - A - MY) v'p +
+ 1—2/У — exp {—2/y) + %v*p — %'v'p2, E.71)
а из уравнений E.68) и E.69) для \хх = \ix
ln (*>,) = [A - 1 lx) vp - A - 1 /х) v'p + 1 - 2/y -
- exp {-2/y) + % A - и,J - %' A - t;;J] ^ +
S - 1] - 2 ln (ц/у) + C}9 E.72)
где индексом (') обозначается неупорядоченная фаза.
Уравнения E.71), F.72) и E.64) являются условиями
сосуществования изотропной и анизотропной фаз.
Все растворенные компоненты данного размера х предполагаются
идентичными [1—7]. Это ограничение по отношению к свободной
энергии и химическим потенциалам можно устранить интерпретацией yXt ^
как ожидаемым числом участков длины ц в #-мере.
Авторы работы [12] на основе развитой теории рассчитали фазовые
диаграммы для растворов полужестких макромолекул с целью
продемонстрировать влияние длины гибких участков, их положения в цепи,
значения параметра взаимодействия и других на положение кривых
сосуществования изотропной и анизотропной фаз на фазовых
диаграммах.
На рис. 5.10 представлена фазовая диаграмма, рассчитанная с
использованием асимптотического приближения (т. е. со значением С,
взятым из уравнения E.55)) для трех систем растворенное вещество —
растворитель, где растворенное вещество представляет собой
макромолекулу, в которой жесткий участок находится между гибкими
участками и xs=l. В каждой молекуле аксиальное отношение жесткого
участка т] равно 50. Этот жесткий блок расположен между гибкими
участками с 0,1 и 10 сегментами соответственно, т. е. х = г) + хк « 50, 51
и 60 для трех пар кривых, представляющих бинодали на рис. 5.10.
Параметры взаимодействия %, выбранные одинаковыми в обеих фазах
(изотропной и анизотропной), нанесены в зависимости от объемной доли
vp и v'p растворенного вещества в двух фазах. Как видно из рис. 5.10,
наблюдаемое расширение двухфазной области в том случае, когда
качество растворителя ухудшается (% становится положительным),
становится более узким при введении в жесткую палочку гибких участ-
137
ков. Даже сегмент с наименьшей длиной оказывает существенное
влияние на величину расширения двухфазной области. Однако в области
X = %' < 0 влияние гибких участков мало (узкая область двухфазной
диаграммы достаточно сужается или передвигается в область больших
концентраций). Введение гибких участков увеличивает разупорядочен-
ность у в анизотропной фазе, как видно из рис. 5.11, причем этот эффект
усиливается с увеличением длины гибкого участка.
На рис. 5.11 равновесные концентрации нанесены в зависимости от
1/г) — Их, где х = Уч+ хь— общее число сегментов в растворенной
. 20 40 100 ?>
0,8
0,6
0,4
0,4 Ofi OS Yp>v'p
О 0,0/ 0,02 0,03 O,Obi/q-i/x
Рис. 5.10. Фазовая диаграмма для системы полимер — растворитель для вещества
с одним палочкообразным участком с осевым отношением 50 и разным количеством
концевых гибких участков — 0, 1 или 10. (Вверху приведен график зависимости
%г от у [12]).
Рис. 5.11. Зависимость vp и v'p от A/г) — \/х) при фазовом равновесии для атерми-
ческой смеси.
7 — количество независимых палочкообразных участков с осевым отношением 20, приходящихся
на цепочку. (Оставшиеся (х -~ 20 у) участков в цепи — гибкие [12]).
макромолекуле. Величина является мерой количества гибких
Т] X
сегментов, у представляет собой число палочкообразных участков на
молекулу. Каждая пара кривых начинается при значениях абсциссы,
соответствующих х = уг\.
Как было показано раньше, фазовое равновесие для бинарных
систем, макромолекулы которых состоят из палочек, связанных
гибкими участками, зависит от аксиального отношения палочкообразного
участка и практически не зависит от числа точек связывания. Это
проявляется в значениях vp> v'p так же, как и при первоначальном положе-
138
нии каждой пары, т. е. при х = уП- Включение гибких участков
уменьшает первоначальное значение vp в каждом случае. Когда пропорция
гибких участков увеличивается, vp снова возрастает и при дальнейшем
увеличении числа гибких сегментов vpf подобно vp, приближается
к постоянному значению. Двухфазная область сужается и две кривые
сливаются, так как при v'p= др*= 1 они дают общий отрезок,
положение которого определяется числом гибких участков. Для всех
значений у, рассмотренных в данном случае (у равно 1, 2 и 10), отношение
гибких сегментов к палочкообразным в этом предельном случае равно
1,4. Объемная доля иж= v^ жестких участков составляла 0,417 для
всех случаев.
yfq у
•- 0,20
0 50 Ю0 150 % 0 5 10 15 qe
•Рис. 5.12. Зависимость vn — v'n (/)и у D) от осевого отношения простого
палочкообразного участка с присоединенным максимальным количеством гибких
участков и объемные доли vp B) и vp C), а также у/ п E) в системах простые
палочки — растворитель в условиях равновесия фаз [12].
Рис. 5.13. Зависимость равновесных значений у A) и vp B) от осевого
отношения самых коротких двух палочкообразных участков для атермальных раство-'
ров палочек с одним изгибом и общей длиной х = 40.
Сплошные кривые рассчитаны по теории [77], штриховые — при использовании
асимптотического приближения [12].
На рис. 5.12 сплошными кривыми нанесены зависимости объемных
долей vx= v'm жестких участков в молекулах, содержащих простой
жесткий участок длиной т|, от ц для случая, когда к этому жесткому
сегменту присоединены гибкие сегменты, максимальное число которых
не подавляет образования нематической фазы. Видно, что число этих
гибких участков, не подавляющих образование нематической фазы,
увеличивается с ростом т). Кривые начинаются при т) = х = 6,45. В
предельном случае неопределенно длинных палочкообразных участков
соблюдается условие ц/x^-Vp^^x ^8/г) [7, 13].
Сплошными кривыми на рис. 5.12 нанесены значения yl\\ для
палочек, распределенных в xk= х — ц гибких сегментах. Этот индекс ра-
зупорядочения при равновесии двух фаз существенно больше, чемлля
139
Изотропная
простых палочек в чистом растворителе (xk= 0), значения отношения
у/ц = у/х для которых показаны на рисунке кривой 5. Далее авторы-
рассматривают палочки с одной точкой излома, содержащие 40
сегментов. Предполагается, что % = %'== 0. На рис. 5.13 приведены
зависимости vp и у от положения точки излома палочки, характеризуемой
ци, т. е. длиной самого короткого участка. Индекс разупорядочения у
тоже приведен на этом рисунке. Сплошными линиями нанесены
кривые, полученные путем приближения, использованного в [7, 13] (т. е
С = 0), а штриховыми — найденные путем использования
асимптотического приближения [13].
Из рассмотрения сплошных кривых видно, что при изломах вблизи
центра молекулы, т. е. для г]и> 10, ни vp ни у не зависят от положения
излома. При движении этого излома
к концу макромолекулы параметр
разупорядочения, однако, быстро
растет до тех пор, пока не достигнет
насыщения при г)и= у = 7, 3. Для
меньших значений г)и индекс
разупорядочения у превышает длину самой
короткой палочки, которая полностью
разу пор ядочивается [2]. При
увеличении длины палочки индекс
разупорядочения уменьшается, снижается
и крутизна зависимости vp от т)и.
Расчеты с использованием
асимптотического приближения (штриховые
кривые) качественно подобны
полученным с использованием
приближения из работ [1—6, 13].
Следовательно, поскольку это приближение
ограничивается малыми степенями
разупорядочения [13], оно дает менее'
удовлетворительные результаты, чем приближение работ [7] вблизи
насыщения, где один из компонентов полностью разупорядочивается.
На рис. 5.14 представлены расчеты для тройной системы,
содержащей компоненты с л:а= 40 и г)а= 39, и компонент в с хь = ць = 20,
а также растворитель s. Введение только одного гибкого сегмента
оказывает существенное влияние на ход фазовой диаграммы.
Фазовая диаграмма, приведенная на рис. 5.15, получена для
тройной системы, в которой компонент большего размера состоит из
палочкообразных участков, охватывающих лишь половину длины
молекулы, а остальные участки гибкие, т. е. ч\а = 20, а ха = 40. Двухфазная,
область для бинарной системы s—fc, содержащей растворитель и
палочкообразный компонент в с хь = ць =¦ 20, расширяется при добавлении
компонента й, как это видно из передвижения узловых точек, две из
которых показаны штриховыми линиями в этой области диаграммы.
Концентрацию растворителя в этих сосуществующих фазах уменьшают
путем введения компонента а, который исключается из анизотропной
фазы (va< 10~4) таким же образом, как и для смесей клубков с палочко-
2)
Рис. 5.14. Фазовая диаграмма для
артемальной смеси с ха = 40, % =
sat 39, хь = г\ь =* 20. Толстые сплош*
ные линии — бинодали, тонкие
сплошные линии — узловые [12].
140
образными макромолекулами [51. Дальнейшее увеличение содержания
компонента а приводит к уменьшению объемной доли ve в изотропной
фазе, которая достигает предельного значения C • 10 ~4) в точке /.
Эта часть бинодали для изотропной фазы расположена на оси а—г
на рис. 5.15,6 в большем масштабе. Она прерывается в точке / из-за
возникновения анизотропной фазы Аг. Точки / и Ах находятся
действительно очень близко к составам, характеризующим
сосуществование фаз для бинарных систем s—а, а двухфазная область для этой
системы мала (рис. 5.15,6). Левая часть прямой 1Аг характеризует
вторую двухфазную область. Узкая область, ограниченная ломаной
линией 1АгА2 на рис. 5.15,6, является трехфазной. Бинодаль, опреде-
Изотропная,
йбухфазная,
0,83,
Анизотропная
0,01
Рис. 5.15. Фазовая диаграмма системы палочка (% = 20 сегментов, соединенных
20 гибкими сегментами), — простая палочкообразная компонента (хь = Щ = 20) —
растворитель (а) и расширенный участок трехфазной области [12] (б).
Для /1 с>а = 0,8263, 1^ = 3,1-10—*; для Ati va = 0,8370, vb = 1,3-10—»; для Аг\ va = 0,978
ляемая линиями Аг и Л2, лежит очень близко к предельной узловой
линии АгА2у поэтому она не показана на рис. 5.15,а. Таким образом,
влияние соединения обоих (гибкого и жесткого) участков в одной
молекуле сильно сказывается на фазовой диаграмме, как это можно видеть
при сравнении рис. 5.15,а с рис. 5.14.
Расчеты, сделанные на основе уравнений E.67) и E.69), приводят
к следующим выводам.
1. Гибкие участки, введенные в жесткую макромолекулу,
оказывают существенное влияние на свойства обеих фаз, существующих
в равновесии. В результате введения этих гибких участков в
макромолекулу увеличивается разу пор ядочение анизотропной фазы и сужается
двухфазная область. Для изученных аксиальных отношений палочек
влияние гибких участков больше при % > 0, чем при % < 0.
2. Введение гибких участков подавляет процесс выделения жестких
частиц в анизотропную фазу, и, как следствие, способствует
расширению двухфазной области. Если длиш более короткого жесткого
141
участка в модели палочки с одной точкой изгиба существенно
отличается от равновесного значения у согласно уравнению E.64), то
протяженность области разупорядочения нечувствительна к г)и.
1. Flory P. У., Abe A. Statistical thermodynamic of mixtures of rodlike
particles. 1. Theory for polydisperse systems.—Macromolecules, 1978, 11, N6,
p. 1119—1122.
2. Flory P. /., Abe A. Statistical thermodynamics of mixtures of rodlike
particles. 2. Ternary systems.—Ibid., p. 1122—1126.
3. Flory P., J.t Abe A. Statistical thermodynamics of mixtures of rodlike
particles. 3. The most probable distribution.— Ibid., p. 1126—1133.
4. Flory P. J. Abe A. Statistical thermodynamics of mixtures of rodlike
particles. 4. ThePoisson distribution.— Ibid., p. 1134—1138.
5. Flory P. J., Abe A. Statistical thermodynamics of mixtures of rodlike
particles. 5. Mixtures with random coils.— Ibid., p. 1138—1141.
6. Ftory P. /., Abe A. Statistical thermodynamics of mixtures of rodlike
particles. 6. Rods connected dy flexible joints.— Ibid., p. 1141 — 1144.
7. Flory P. J. Statistical thermodynamics of semi-flexible chain molecules.—
Proc. Roy. Soc. London, A, 1956, 234, N 1196, p. 60—73.
8. Flory P. J. Principles of Polymer Chemistry.— New York: Cornell univ. press.,
1953.— 594 p.
9. Flory P. J. Statistical thermodynamics of liquid mixtures.— J. Amer. Ghem.
Soc, 1965, 87, N 5, p. 1833—1838.
10. Flory P. /. Thermodynamics of polymer solutions.— Discuss. Faraday Soc,
1970, N 49, p. 7—29.
11. Aharoni S. M. Rigid backbone polymers. 6. Preliminary ternary phase
diagrams of poly (isocyanates). — Macromolecules, 1979, 12, N 3, p. 537—538.
12. Matheson R. R., Flory P. /. Statistical thermodynamics of mixtures of
semirigid macromolecules. Chains with rodlike sequences at fixed locations.— Ibid.,
1981, 14, N4, p. 954—960.
13. Flory P. /. Rotica G. Theory of systems of rodlike particles. 1. Athermal
systems.— Mol. Cryst. and Liquid Cryst., 1979, 54, N 3/4, p.. 289-310.
ГЛАВА 6
ТЕРМОДИНАМИКА СМЕШЕНИЯ
ПОЛИМЕРОВ
6.1. БИНАРНЫЕ СМЕСИ
С самого начала развития термодинамики процессов смешения
полимеров исследователи пытались найти пределы взаимной
растворимости компонентов и связать их с химической природой и
молекулярной массой полимеров.
Поскольку в большинстве случаев смешение и переработку
полимеров проводят при высоких температурах, компоненты смеси находятся
в вязкотекучем состоянии и по структуре должны быть близки к
высоковязким жидкостям. Естественно поэтому, что их смешение
необходимо рассматривать как смешение жидкостей.
Согласно законам термодинамики, образование термодинамически
устойчивой системы должно сопровождаться уменьшением изобарно-
изотермического потенциала (свободной энергии смешения Гиббса):
AG=kH -TAS. F.1)
Необходимое, но недостаточное условие стабильности системы
AG<0
выполняется в том случае, если Д # < О, а Т A S >0, или если
Д#>0, но |ГА5| >|Д#|.
Если имеется возможность определить A G для всех возможных
составов смеси, можно рассчитать значение температуры и состава,
при которых смесь будет стабильной, т. е. однофазной. Однако
экспериментальное определение A G для смесей полимеров связано с большими
трудностями, так как некоторые методы, используемые для
нахождения этого параметра в системах полимер — растворитель, в данном
случае оказываются непригодными. Авторы [1] предположили, что при
смешении полимеров больших молекулярных масс ответственной за
смешение является энтальпия, поскольку вклад энтропии смешения
в A G чрезвычайно мал. В этом случае считали, что при Д Н < 0
полимеры совместимы, а при Д#>0 — несовместимы. Как правило,
для растворов полимеров в низкомолекулярных жидкостях условие
А Н < 0 отвечает совместимости компонентов. Однако для смесей
полимеров Слонимский и Струминский показали [2, 3], что это
наблюдалось не во всех случаях,* следовательно, существенная роль должна
принадлежать энтропийному вкладу в A G. Расчеты, проведенные Фло-
143
ри и сотрудниками [4], подтвердили
уменьшение энтропии смешения, т. е.
Д 5 < 0. Впервые вклад энтропии
смешения во взаимную растворимость
полимеров экспериментально оценили
А. А. Тагер и сотрудники [5—8],
которые нашли, что энтропия смешения двух
полимеров не равна нулю, а может
достигать больших значений и иметь
разные знаки (рис. 6.1). Они обнаружили,
что для совместимых полимеров Д 5 < 0.
На этом основании А. А. Тагер в
качестве одного из критериев совместимости
двух полимеров предложила условие
A S < 0, но это на самом деле
некорректно, так как условие A S < 0 не
способствует образованию
термодинамически устойчивой системы.
В большинстве случаев для оценки
термодинамической совместимости
использовались (и используются)
приближения теории регулярных растворов.
В последние годы для описания термодинамических свойств
полимеров начали успешно применять новые статистические теории
растворов полимеров, развитые Пригожиным [10], Паттерсоном [11],
Флори [12] и др.
Рис. b.l. Концентрационные
зависимости свободной энергии
смешения Гиббса A), энтальпии
B) и энтропии C) для системы
ацетат целлюлозы —
нитроцеллюлоза [64].
6.1.1. ПРИБЛИЖЕНИЯ ТЕОРИИ РЕГУЛЯРНЫХ РАСТВОРОВ
В ТЕРМОДИНАМИКЕ СМЕШЕНИЯ ПОЛИМЕРОВ
Теоретическое рассмотрение процесса смешения полимеров сделано
впервые Скоттом [13] и Томпа [14], которые использовали уравнение
Флори—Хаггинса для свободной энергии смешения Гиббса:
F.1а)
где V — общий объем смеси; VQ — объем сегмента, выраженный
равным объему повторяющейся единицы цепи, причем одинаковым для
обоих полимеров; гА и гв — число сегментов полимеров А и В; тг =
= Vt/Vc (V{ — молярный объем компонента /); фд и фв — объемные
доли компонентов А и В в смеси; %дв — параметр термодинамического
взаимодействия, относящийся к энтальпии взаимодействия между
сегментами, каждый из которых имеет объем Vc.
Смесь оказывается в критической точке, когда
144
а критические значения концентрации и параметра взаимодействия
равны соответственно
(фА)крит = 1/[1 + {Гъ1Гк)Щ, F.2>
(ЗСав)крнт = ~ [Ы-1/2 + (гв)'2!2. F.3>
Как видно из последнего уравнения, (хлв)крит при больших
молекулярных массах смешиваемых полимеров должно быть очень малым
(около нуля) и при М ->¦ оо полимеры будут несовместимы при любой,
даже очень малой положительной теплоте смешения. При этом
уравнение спинодали имеет вид
d*AG л / ч *
-щ- = 0 и (ХАВ)с„ - 7[
где индекс «сп» относится к спинодали. Из предположения, что
химические потенциалы полимеров одинаковы в двух сосуществующих
фазах, можно получить следующие основные уравнения бинодали:
**а - *Ч> = 1п Фа + 0 - га/гв) Фв + хавгаФв>
1*в - *Ч = 1п Фв + (! - V^a) Фа + Хав^вФа' F.6)
где |i{ — \i{0—химические потенциалы t-ro компонента смеси.
Для использования уравнений F.4) и F.6) параметр оСав
рассчитывают из соотношения
Хав = ^ (вА - 6вJ, F.7)
где бд и бв — параметры растворимости компонентов. Как видно из
уравнения F.7), расчетное значение параметра %дв всегда положительно.
Поскольку же %авкрит близко нулю, а вклад комбинаторной энтропии
(первые два члена уравнения F.1а)) пренебрежимо мал, приближение
Скотта (см. уравнения F.1 а) и F.7)) свидетельствует о том, что найти
термодинамически совместимую пару — безнадежное дело. Лишь для
случая очень близких значений бд и бв расчетное значение %ав может
быть меньшим хавк иГ На этом основании в работах [15, 16] был
предложен способ предсказания совместимости двух полимеров в
твердом состоянии. Так, если хав < Хавкрит> то два полимера совместимы
при том же процентном соотношении, которое выбирается для
расчета параметров взаимодействия. В работе [16] приведены результаты
расчета значений АХдв, для которых величина АН меньше, чем TAS.
В частности, авторы [16] предположили, что если различие в
плотностях энергии когезии двух смешиваемых полимеров меньше 67 Дж/см3,
полимеры будут совместимы независимо от знака теплоты смешения.
Для предсказания совместимости полимеров в твердом состоянии
Шнейер [17] использовал другое приближение, основанное на соотно-
145
шении, предложенном Джи [18] для теплоты смешения Ясм каучуков
при набухании,
где Vo и б, — молярный объем и параметр растворимости жидкости
и каучука; A—ф0) — объемная доля набухшего каучука.
Для смесей полимеров Шнейер "получил уравнение
Д//см = [wiMfo FХ - 82)* [-JUL. м2р2 - A - w
где Mt и pt-— молекулярная масса мономерной единицы и плотность
полимера. Для проверки этого соотношения он использовал данные
работы [20], с помощью которых показал, что в случае совместимых
полимеров значения А Ясм находятся в диапазоне от 10~3 до 10~2 Дж.
Для расчета А #см он применил результаты, опубликованные в
статье [ 19], а в качестве М — молекулярную массу повторяющегося звена
полимерной цепи. Несмотря на удовлетворительное качественное
согласие экспериментальных данных по совместимости с результатами
теоретических предсказаний, все же найдены исключения: для
некоторых систем значения А Нш были меньше 10 ~2 Дж, но полимеры
в твердом состоянии являлись термодинамически несовместимыми [20].
Для предсказания совместимости полимеров Шоу [21] использовал
двухразмерный параметр растворимости. Предположение о
необходимости использования многоразмерных параметров растворимости
для описания фазового поведения смесей полимеров, обладающих
большим дипольным моментом, было высказано еще в работе [22].
Приняв, что полимеры образуют регулярные растворы, а
основным взаимодействующим элементом служит повторяющееся звено цепи,
условие равенства химического потенциала компонента 1 в фазах А
и В Шоу получил в виде
+ A ™ VJVj фА1 + Vx ^^ФА, =
« lnq>Bi + A - WVt)<PBi + Vi^W^^
В этом уравнении ф — мольная доля; V — молярный объем; 6 —
параметр растворимости. Аналогичное уравнение можно записать и для
компонента 2. Первых два члена F.8) определяют энтропию смешения
регулярного раствора, а третий член связан с взаимодействиями.
Выражение F.8) практически не отличается от аналогичного уравнения
Флори—Скотта для бинодали (см. уравнение F.6)) и также
предсказывает увеличение растворимости с ростом температуры. Если
предположить, что энергия взаимодействия обусловлена только
дисперсионными и диполь-дипольными взаимодействиями, то энтальпию
взаимодействия можно записать так: (8^ — 82di) + (8pt — б?2), где d относится
к дисперсионным, а р к полярным взаимодействиям. Тогда для
высокой растворимости полимеров необходимо хорошее соотношение между
полярными и дисперсионными компонентами энергии когезии обоих
146
полимеров. Возможность этого метода продемонстрирована для систем
полимер — растворитель в работе [22].
Для расчета Ьр используется схема [22], по которой
б» "- 2\i*/3kT + 3/а*/4
где |л и a — дипольный момент и поляризуемость; / — первый
ионизационный потенциал молекулы, обычно выбираемый равным 10 эВ.
Параметр б рассчитывается по данным работ [23].
Проверка этого приближения показала, что с увеличением
молекулярной массы растворимость уменьшается при фиксированном
значении параметров растворимости в соответствии с уравнением F.8).
Однако температурные исследования дали противоречивые
результаты. Проверку совместимости проводили на основании данных
исследования термомеханических свойств пленок, полученных из общего
растворителя. Использовав приближение Скотта (уравнение типа
F.1а)) и выразив значения параметра взаимодействия в виде
F.9)
Ниши [24] получил уравнение для расчета спинодали в виде
L
1_\ Г, Ш (-J _ , -L-W
где Ткрнт ~ = — B/AR соответствует критической температуре смешения
полимеров с бесконечной молекулярной массой. Гкрит и Гкрит» при
критических условиях связаны друг с другом соотношением
) /2]*}. F.10)
КрИТ °о/ '
Комбинируя уравнения F.5), F.6) и F.9), получаем выражение
Ф2' получается из решения уравнения
2 [(?) - A /г2)] (Ф; - Ф;> + [B - ф; - ф^/rj in A - Ф;)/A -
ф; = о.
Критические значения Гкрит и Фгкрит могут быть определены из
фазовой диаграммы, если ММР каждого полимера не очень широко. Тогда
уравнения F.2) и F.10) превращаются в выражение
-f^— = (тг—\ П - A/2Лтх) A/A - ф2крит)]2,
1 крит \ ' сп в* /
где Гкритвв и 2Art получают из графика зависимости 1/ГкрИт= 1/A —
— ф2критJ-
Сравнение рассчитанных епинодалей, бинодалей и критических
точек с экспериментально полученными для системы полистирол —
147
поливини л метиловый эфир с НКТС показало вполне
удовлетворительное их согласие (рис. 6.2).
Приближение Гильдебранда—Скотта [25] было использовано и
авторами работы [26], которые связали избыточную энтропию смешения
при постоянном давлении с избыточным объемом смешения:
ДУ+..., F.11)
рассматривая (AS)y как комбинаториальную энтропию смешения, т. е.
(AS)V = —R (nA In фА + пв In фв),
473
где пА и пв
— число моделей на
единицу объема; фА и фв — объемные доли
полимеров А и В в смеси. Они нашли,
что (AS) у для смеси состава 1:1 для
двух полимеров с М = 20 000 равно
5,85- 10~4Дж/см3-К. Положив, что
T(dP/dT)v ~(dE/dV)T^ 347 Дж/см3,
например, для полистирола, и примерно
то же для поли-2,4-фенилен-1,6-оксида,
и использовав значение AV = 7 • 10 см3/г
для их смеси, было найдено, что
(dP/dT)v AV= 8,36 • Ю-3 Дж/см3 • К для
смеси состава 1:1 при 300 К, т. е. ве-
о о? 0 4 об 08 личиной (AS)V можно пренебречь. Таким
' ' ' ' У*3 образом, уравнение F.11) сводится к сле-
Рис. 6.2. Сравнение эксперимен- Дующему:
тальных и теоретически рассчи- {дР\
тайных кривых фазового состоя- (AS)p = I ~~?) AV. F.12)
ния системы полистирол — поли- \o*/V
винилметиловый эфир. Молеку-
рярная масса ПС:
/ — 1. io*j 2 — 2,04.10*; 3-5,1.10*!
4—2» Ю». М (ПВМЭ)j==51,5.103.
373
дР
() 5,
Те$оо =343 К, Art = 14,29 [24].
Сплошные кривые — спинодали, —*
штриховые — бинодали.
Поскольку^! положительно для
большинства соединений, то
(AS)P a=E 0, если AV 5* 0.
Для иллюстрации влияния AV на совместимость двух полимеров
авторы работы [26] рассчитали изменение внутренней энергии при
смешении А Еу используя предположение Гильдебранда и Скотта о том,
что энергия при постоянном давлении (АЕ)р может быть найдена из
энергии при постоянном объеме (АЕ)у из следующего уравнения:
(AE)p~(AE)v + [t(^)v-p]aV + ... F.13)
Поскольку энтальпия смешения (АН)р определяется из
(АН)р ~ (АЕ)Р + РАV, F.14)
то, подставляя F.13) в F.14), получаем выражение для (АН)Р в виде
148
Подставляя же уравнение F.12) и F.14) в F.1) имеем
» {АЕ)р < 0.
В большинстве случаев для термодинамически совместимых систем
наблюдается А V < 0, поэтому при смешении должно уменьшаться
число внешних степеней свободы, а следовательно, и уменьшаться
подвижность цепей по сравнению со средней подвижностью
индивидуальных компонентов. Эти предположения были подтверждены авторами
работы [26] на примере термодинамически совместимой системы
полистирол — поли-2,4-фенилен-1,6-оксид.
6.1.2. ПРИМЕНЕНИЕ НОВЫХ СТАТИСТИЧЕСКИХ ТЕОРИЙ
ДЛЯ ОПИСАНИЯ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
СМЕСЕЙ ПОЛИМЕРОВ
Положения новой теории Флори [12] для описания
термодинамического поведения смесей полимеров первым использовал Мак Мастер
[27]. На основе модифицированного им уравнения состояния Флори
получил выражения для химических потенциалов, спинодалей и
критических точек для бинарных смесей моно- и полимолекулярных
полимеров. В последнем случае были применены результаты Конинг-
свельда и сотрудников [28]. Уравнения эти слишком громоздки,
поэтому здесь мы их не приводим.
С помощью полученных соотношений Мак Мастер рассчитал бино-
дали и спинодали для модельных систем: полистирол — поливинилме-
тиловый эфир, сополимер стирол — акрилонитрил (массовая доля
акрилонитрила 0,28) — поликапролактон, и сравнил с
экспериментально полученными кривыми точек помутнения для этих систем. На
указанных модельных системах с помощью полученных уравнений
Мак Мастер исследовал качественно влияние различных параметров
уравнения состояния на взаимную растворимость полимеров.
Основные выводы его расчетов могут быть сведены к следующему.
Влияние молекулярных масс смешиваемых полимеров. С
увеличением молекулярной массы каждого компонента взаимная
растворимость двух полимеров ухудшается. При этом, когда отношение длин
цепей компонентов гг/г2 уменьшается, критический состав на фазовой
диаграмме постепенно сдвигается в сторону уменьшения концентрации
полимера с более высоким значением молекулярной массы. Это,
собственно, следует и из теории Флори—Хаггинса для растворов
полимеров.
Влияние различия в коэффициентах термического расширения
компонент. Изменение а приводит к изменению всех параметров
уравнения состояния: при Т = 300 К и увеличении а на 6 % Г*
уменьшается на 4,6 %, Я* увеличивается на 2 %, а v* понижается на 1 %.
Чтобы два полимера с молекулярными массами порядка 200 000
образовали термодинамически совместимую систему, разность в
коэффициентах термического расширения этих полимеров не должна
превышать 4 %. Для смесей двух полимеров с молекулярными массами
порядка 50 000 это различие в коэффициентах термического расширения
149
473
373-
273 -
-273
о,д tp2
не должно превышать 10 %. При
очень высоких значениях
молекулярных масс смешиваемых
полимеров уже незначительное
изменение величины а для одного из
компонентов может сильно повлиять
на предел взаимной растворимости
(см. рис. 6.3). /
Влияние коэффициента
изотермической сжимаемости. Изменение
этого параметра сказывается
только на Р*. В случае ах <а2
различия в уравнениях состояния
компонентов могут быть
скомпенсированы уменьшением у2 по
сравнению с yv При уменьшении у2 на
10% критическая температура
смешения повышается на 250°. То же
происходит при изменении Yi, если
остальные параметры постоянны:
увеличение уг с одновременным
понижением 7г также вызывает
повышение взаимной растворимости
двух полимеров; к улучшению
взаимной растворимости приводит
рост давления.
Влияние параметра контактных
взаимодействий Х12. Если
параметр Х12 мал и положителен, возможно существование двух
критических температур смешения (ВКТС и НКТС). Рост положительных
значений параметра Х{2 приводит к слиянию НКТС и ВКТС, и диаграммы
принимают форму типа «песочных часов» (см. гл. 2, с. 40). Температура
слияния ВКТС с НКТС является функцией разности коэффициентов
термического расширения: она увеличивается при уменьшении
разности (аг—а2).
Если же параметр Х12 отрицателен (что может наблюдаться при
наличии специфических взаимодействий, таких как водородные
связи, донорно-акцепторные взаимодействия и др.), два полимера
термодинамически совместимы даже при существенных различиях в их
уравнениях состояния. Расчеты также показали, что одновременное
существование ВКТС и НКТС для полимерных смесей — явление очень
редкое, причем температура их слияния должна быть либо равной
273 К, либо меньше 273 К.
Сравнение рассчитанных и экспериментально полученных кривых
точек помутнения на уже упоминавшихся модельных системах
обнаружило очень малую чувствительность этих точек к изменению
температуры. Мак Мастер полагает, что возможными причинами этого
эффекта могут быть неудовлетворительная точность уравнения состояния
для данных компонентов (т. е. параметров уравнения состояния для
Рис. 6.3. Влияние коэффициента
термического расширения на фазовое
состояние системы полимер — полимер
(М1 = М2 = 5.Ю4) при 04 = 0,58 X
ХЮ^К и а2, равных 0,64-Ю
A) и 0,63-Ю-3 К B) [27].
150
компонентов модельной системы), полидисперсность компонентов и
наличие специфических взаимодействий. Одним из главных результатов
этой работы Мак Мастер считает экспериментальное подтверждение
наличия НКТС в смесях двух полимеров.
Полученные Мак Мастером уравнения для спинодали
использовал Олабиси [29] при расчете фазового поведения смеси поликапролак-
тон — поливинилхлорид. Было показано, что если варьировать
только величину Х12, то для больших отрицательных значений Х12 можно
получить бимодальность фазовой диаграммы. Если же изменяются
оба значения Х12 и s2/slt наблюдается асимметрия бинодали, а если
только отношение sa/si при постоянном значении Х12— практически
зеркальное отражение первоначально рассчитанной спинодали. Эти
результаты указывают на исключительно важную роль параметров Х12
и s2/sx в описании фазового поведения смесей полимеров (напомним,
что в большинстве случаев величину s2/s1 принимают равной 1).
Уравнения Мак Мастера очень сложны для практического
использования. Более того, как показали Паттерсон и Робард [30], Мак
Мастер слишком переоценивает вклад члена свободного объема в параметр
%12. Поэтому Паттерсон и Робард [30] использовали первоначальную
посылку теории Пригожина — Флори в более простом приближении,
т. е. в том виде, как это сделано при описании фазовых диаграмм для
систем полимер — растворитель при обычных [31, 32] и высоких [33,
34] давлениях. Главное внимание они уделили взаимодействиям,
благоприятствующим смешению, стабилизирующим смесь при низких
температурах и ослабевающим при повышении температуры. Они
предположили, что первопричиной появления НКТС не является
чрезвычайно неблагоприятный вклад члена свободного объема, как
в обычных системах полимер — растворитель. Суть их приближения
заключается в следующем.
Согласно теории Пригожина — Флори [10—12], выражение для
параметра термодинамического взаимодействия %12 (нормированного
к молярному объему компонента 1) выражается уравнением C.12).
Поскольку в модели Флори используется вандерваальсовская
жидкость, для которой
-Ut = P*V*VT\ F.15)
/D/3 - V/3)> F.16)
то, используя соотношение типа (З.Г2) и пренебрегая
концентрационной зависимостью %12, из уравнений C.12), F.15) и F.16) получаем
х12 _ ?1Г JVL (хЛ + Я" J F 17)
Mtf - RT* Lv1/3—I {P*J+2 D/3 - У}/3) J • Л °
Здесь для выполнения условия симметрии, т. е. %и = х//, параметр %12
отнесен к единице объема. Если учесть параметр Q12, введенный
Флори для коррекции энтропии, то к уравнению C.12) необходимо добавить
член, равный — Q12/#, или заменить Х12 эквивалентной величиной
151
Х12 — T'QiJ'i- Уравнение F.17) для %12/Mxv* показывает, что член
взаимодействия стремится к бесконечности, если V\/z уменьшается до
1, а Т падает почти до О К. С другой стороны, член свободного объема
стремится к бесконечности, когда V\/3 стремится к 4/3, т. е.
температура приближается к критической температуре жидкость — пар для
компонента 1. Из табл. 6.1 видно, что для полимеров Vl/Z близко
к 1 и, следовательно, нужно ожидать преобладания члена
взаимодействия в %12. В то же время для разбавленных растворов полимеров
в низкомолекулярных жидкостях, для которых V1/3 может достигать
4/3, преобладает член свободного объема в %12, т. е. поведение систем
полимер — растворитель и полимер — полимер должно существенно
различаться с изменением температуры.
Таблица 6.1. Значения параметров %13 (на сегмент ПЭГ) для системы
ПЭГ-2000 —- ПЭГ-40000 при ф8 — 0,3 и разных температурах, полученные при
использовании разных детекторов
Сорбат
Гексан
Гептан
Октан
СС14
Цикло-
гексан
Бензол
Метил-
этилке-
тон
Среднее
Ионизационно-пламенный детектор
а13
1,8120
1,9720
0,3860
—
0,1020
0,0328
—
35С
а18
1,7260
1.8700
0,3300
—
0.0320
0,2850
—
—0,0090
—0,0080
—0,0070
—
-0.0084
—0,0092
—0,0083
%
клонения от
среднего
8,4
3,6
.
15,7
—
1.2
10,8
7,9
Катарометр
353 К
«2 8
0,0051
0,0090
0.0063
0.0069
0,0085
—0.0090
—0.0170
—
384
_
0,0300
0,0260
0,0239
0.0235
0.0153
0.0103
0,0215
К
%
клонения от
среднего
_
39,5
20,9
11.2
9.3
22,8
52,1
25,9
399
_
0,0415
0,0880
0,0354
0,0410
0.0296
0,0380
0,0422
К
%
клонения от
среднего
Н.5
2,2
4,8
10,2
20,4
2,2
8,5
Рис. 6.4 иллюстрирует зависимость %12/№iu* от V1/3 и Г для
типичной системы полимер — растворитель, рассчитанную по уравнению
F.17). Величина %кРит рассчитана по уравнению C.2) при М -* оодля
полимера и JAxv* = 70 см3/моль для растворителя. Система
характеризуется наличием ВКТС и НКТС, между которыми находится область
полной совместимости. Хотя оба параметра (параметр взаимодействия
и член свободного объема) важны для %12, существование ВКТС
обусловлено исключительно увеличением неблагоприятного для смешения
члена взаимодействия (Х12 положительно) с уменьшением температуры,
в то время как НКТС обусловлена возрастанием члена свободного
объема с ростом температуры. Если Х12 и (или) т повышаются, S-образная
форма кривой X (Т) должна двигаться вверх по оси температур и ВКТС
сольется с НКТС, т. е. получим диаграмму типа «песочных часов».
152
1,2
v/3
0 1fl5 f,1 ftf5 1,2 1,25 Vf/J
342
477 T,K
556.
790
Рис. 6.4. Зависимость X12/V* от Т для типичной системы полимер —
растворитель с Г* = 5000 К, Р* =400 Дж/см3, Г* =8191 К, Х12= 10 Дж/см3:
112
t —"вклад члена свободного объема; 2 — вклад члена взаимодействия; 3 — сумма обоих
вкладов в параметр X18/F*. Горизонтальная штриховая линия — критические значения
параметра XUf рассчитанные для V* =70 см8/моль и М2 ¦*¦ « [30].
Рис. 6.5. Температурная зависимость %28/У* для системы полимер — полимер
t;*=t;*=l г/см3,
3
Дж/см3 при Г* = 8191 К, Р* =
5 02 [3
2
= 500 Дж/см3 и разные значениях т: 0 (/), 0,1 B), 0,15 E), 0,2 D) [30].
Горизонтальные штриховые линии — критические значения параметра X28/V2 для смесей
с M2=MS: МО* (/), 2-Ю8 (//), МО3 (///) [30].
С другой стороны, если уменьшается молекулярная масса полимера,
Хкрит опускается вниз по оси температур, а область полной
смешиваемости увеличивается.
Зависимости х^/^М* от Vl/S и Т (рис. 6.5) исследовались на
системах, оба компонента которых являются полимерами с Х12= 10 Дж/
/см2= 100 атм (типичное значение для систем с дисперсионными
силами взаимодействия). Значение Vm было выбрано меньше, чем на рис.
6.4 с тем, чтобы оно могло описывать смесь полимеров или олигомеров.
Критические значения Хкрит рассчитаны для разных молекулярных
масс смешиваемых полимеров при af= у*= 1 см3/г. Как и следовало
ожидать, поведение этой системы обусловлено главным образом
членом взаимодействия, т. е. кривые для т Ф 0 близки к кривой для
т = 0. Для М = 10* и выше система нестабильна в рассмотренном
здесь интервале температур, в то время как для М == 103 и т = 0
система стабилизируется с увеличением температуры. С увеличением т
совместимость уменьшается и при достаточно высокой температуре
должна существовать НКТС. Однако кривые % (Т) чрезвычайно
пологи, так что уменьшение %2з должно наблюдаться до очень высоких
температур. Вклад члена свободного объема всегда положителен,
153
поэтому совместимость имеет^ место при отрицательных значениях
члена взаимодействия, т. е.^для Х12<0. Далее, на рис. 6.7 приведена
зависимость х^/М^* от V1/3 и Т лля полимерной пары совместимых
полимеров высокой молекулярной массы, где Хкрит=О и Х12<0.
Таким образом, условиям совместимости отвечает отрицательное
значение Xi2> а несовместимости — положительное. Поскольку ХХ2< О,
член взаимодействия, благоприятный для смешения, можно
сбалансировать членом свободного объема, неблагоприятным для смешения.
Приравнивая в уравнении F.17) оба члена, получаем
" 2D/3 — V\IS~~
В случае т ^ 0,15 и Р*^ 400 Дж/см3 достаточно очень малого
отрицательного значения параметра Х12(~ —2 Дж/см3) для того, чтобы
полимеры стали совместимыми (более строго, отрицательным должно быть
выражение! Х12— IQV2V\/S). S-образная форма кривой % (Т) на рис. 6.7
типична для полимерных пар. При низкой температуре система
совместима, однако при повышении температуры величина члена
взаимодействия, благоприятствующего смешению, уменьшается быстро до тех
пор, пока его значение не перекроется неблагоприятным для смешения
членом свободного объема, и при %12= 0 должна наблюдаться НКТС.
В противоположность НКТС для систем полимер — растворитель,
в данном случае она обусловлена главным образом членом
взаимодействия, уменьшающимся с повышением температуры.
Дифференцируя уравнение F.17), находим отношение
температурных зависимостей обоих вкладов в d%/dT при НКТС:
~^f/ ""Зг2"* 4 (v{/3 _ 1) ~3'
Поскольку V\/z для полимеров близко единице, член
взаимодействия изменяется больше, чем член свободного объема. Роль
последнего, таким образом, меньше, чем следует из работы Мак Мастера [271,
где на него делается главный упор, полагая в большинстве случаев
Х12= 0.
Если Q12<0, то взаимодействие характеризуется эффективным,
значением Х12, т. е. Х12— TQltV\&. С увеличением температуры
значение этого параметра изменяется от отрицательного к
положительному, приводя к более быстрому, чем при Q12= 0, температурному
изменению параметра X. В этом случае роль члена свободного объема
должна быть даже меньшей, чем при Q12= 0. Может оказаться, что
НКТС будет даже ниже, чем ВКТС, что действительно наблюдалось
для некоторых систем полимер — растворитель [27].
Рассмотрим теперь значения т и ХХ2> при которых типичные
полимерные системы должны быть совместимыми или несовместимыми при
изменении молекулярной массы. В качестве примера возьмем
полистирол с Р*= 500 Дж/см3, Г*= 1891 К, У}'3= 1,067 при 408 К, v*= v%=
= 1 см3/г. Значения т для разных М при постоянном Х12 были получе*
154
ны приравниванием уравнения F.17)
критическому значению %12,
рассчитанному по уравнению C.2) (рис. 6.6).
Значения т и М справа и выше любой
кривой соответствуют фазовому расслоению,
а значения ниже и слева —
несовместимости. Кривые являются критическими
линиями и соответствуют системам при
критических точках. Когда Х12<0,
системы совместимы при М -> оо, причем
достаточно лишь очень малого
отрицательного значения параметра Х12 для
совместимости полимеров при высоких
М для всех систем, имеющих т <=ц 0,15.
Точки вдоль кривых для Х12= 0
соответствуют НКТС, а совместимость
возможна только при низких молекулярных
массах и низких значениях т. Если Х12
увеличивается, то максимальная
молекулярная масса, при которой
компоненты совместимы, быстро уменьшается.
Для Х12> 0 кривые соответствуют ВКТС,
за исключением очень высоких значений
т, где они превращаются в НКТС. На.
каждой кривой с постоянным Х12
существует значение т, при котором ВКТС
и НКТС сливаются.
Дифференцируя %12, находим
значения t/(X12/P*) и V1/3 для получения
НКТС, когда Х]2>0:
F.18)
0,4
0,2
0,1
6 LgM
Рис. 6.6. Фазовое состояние
системы полимер — полимер при
Т = 408 К и разных значениях
параметров т и Х12, равных
0 (/), —1,0 B), —2,0 C), —4,0
D), 4,0 E), 10 F) Дж/см3 [30].
Штриховая линия разделяет
системы с ВКТС (слева) и с НКТС (
справа); точки на ней — слияние ВКТС
с НКТС. Ниже и слева сплошных
кривых — области совместимых
систем.
Положив Й/в= 1,067 иР] = 500 Дж/см3, по уравнению F.18)
находим т = 0,77 для Х12= 10 Дж/см3 или т = 0,31 для Х12= 4 Дж/см8.
Эти значения являются очень большими для систем полимер —
полимер, так что НКТС не должна наблюдаться. Наибольшее достижимое
значение для смеси полимеров может быть ~ 1,15, соответствуя Т =
==703 К для полистирола. Если т = 0,15 — наибольшее значение,
ожидаемое для систем полимер — полимер, уравнение F.18)
показывает, что Х12 должен быть меньше, чем 5 Дж/см3, что является низким
для дисперсионных сил взаимодействия. Эти расчеты
свидетельствуют о том, что системы с обеими КТС (ВКТС и НКТС) практически вряд
ли могут быть обнаружены, в то время как для смесей полимер —
растворитель такое встречается довольно часто [106].
Роль «структурного фактора» clq (q = г), названного
«подвижностью контакта», изучена и в работе [36]. Поскольку Т*= (Blk)(c/q),
где e/k — потенциальная энергия взаимодействия, выраженная в еди-
155
ницах абсолютной температуры; с = 1/3 числа внешних степеней
свободы; q — величина, пропорциональная площади молекулярной
поверхности, то, согласно уравнению C.12), совместимость
необязательно связывать с еп= е22, если clq для двух полимеров различны.
Анализ влияния clq на совместимость двух полимеров основан на теории
жестких возмущенных цепей [37, 38], в которой функция
распределения взята в виде
Q(N, V, D-j
где N — число молекул при температуре Т и объеме V\ Л — длина
волны де Бройля; Vf— свободный объем; Ф — потенциальное поле;
(йг, v — вклады ротационных и вибрационных степеней свободы
(с—число внешних степеней свободы в интерпретации Пригожина).
В применении к смесям функция распределения должна подчиняться
следующим граничным условиям: 1) второй вириальный коэффициент
должен быть квадратичной функцией мольных долей; 2) при высоких
плотностях энтропия смешения должна сводиться к комбинаторной
энтропии Флори—Хаггинса; 3) избыточные величины должны
определяться по отношению к идеальному газу при тех же объеме,
температуре и составу смеси; избыточный химический потенциал растворителя
в концентрированном растворе не должен быть равным нулю при
увеличении длины цепи растворителя до бесконечности (т.е. при
переходе к смеси полимеров); 4) для учета кластеризации молекул, когда
смесь содержит молекулы (или сегменты) с существенно
различающимися потенциальными энергиями, свободная энергия Гиббса,
получаемая из функции распределения, должна содержать результаты
теории возмущений Хендерсона [39] для смесей сферических молекул.
При этих условиях свободная энергия Гиббса G разлагается в ряд
по температуре:
где член G@) учитывает силы отталкивания и рассчитывается по
уравнению [40]
G@) __ N~cC%p — 4) тр
ЩТ ~ A — ТрJ
(т= 0,7405; р = v*/v — приведенная плотность). Другие члены
уравнения F.19) отражают вклады сил притяжения. Они записываются
в форме
G/=//(p, Tu с), / = 1, 2, 3, 4.
Химический потенциал компонента / рассчитывается стандартным
образом по уравнению
1 \dNi)NiU*l),TtV '
156
473
Z73
273
0,8 (рв
Рис. 6.7. Фазовые диаграммы для систем с c/qA = 22,5 и c/qB, равным 19,4 (/),
18,8 B) и 18,4 C) [36].
Рис. 6.8. Фазовые диаграммы для систем с
МВ=ЫО4, 7^=500 К,
Тв = 650 К, V* А = v* в = 0,4775 г/см3 и значениями параметра kAB: —0,001
(/) и 0 B) C6]. '
Рис. 6.9. Фазовые диаграммы для систем с одинаковыми
иуд
k
7*/, а также
—0,003 B) [36].
уд с
= 112fi, (e/k)B = 130,0 и kAB: -0,004
Расчеты фазовых равновесий в смесях требуют знания параметров-
компонентов (Я*, 0*д, 71*, г, s/k) и бинарной постоянной ?*/,
определяемой из выражения
0,2
Для расчета Т* необходимы значения е,-, е/ и ец. Авторы работы
[36] в своих расчетах приняли бинарный параметр как независимую
величину от температуры, плотности и состава.
Для расчета фазовых диаграмм
(рис. 6.7—6.12) были использованы
параметры для полиметилметакрила-
та, в частности с Р*= 1293 бар, РуД=
=0,4775 см3/г и 7*= 554 К.
Молекулярные массы полимеров были
взяты порядка 104.
Фазовая диаграмма для смесей
двух монодисперсных полимеров А
и В (рис. 6.7) составлена для г/q
порядка 112,5 К, &ав = 0, но с разными
значениями c/q. Как видно из
рисунка, фазовое расслоение при высоких
температурах связано с различием
в c/q. При фиксированной
температуре взаимная растворимость
увеличивается при уменьшении c/q. На рис.
6.8, 6.9 показано влияние малых
различий в 8АВ на фазовые диаграммы;
1t96 2,1110(c/q)p
Рис. 6.10. Влияние растворителя
на взаимную совместимость двух
полимеров с одинаковыми М( и c/qA>
и разными значениями c/qB:
2 «19,1; 3-М9,4; 4 — 20,9
/ - 18.4S
[36].
157
два полимера, совместимые ниже 373К когда /Cab 0 становятся
несовместимыми при ?Ав = 0,001 (рис. 6.8.), а для различных ед и ев
при &ав = —0,004 наблюдаются одновременно ВКТС и НКТС
(рис. 6.8). Понижение абсолютных значений &Ав (от — 0,004 до —0,003)
приводит к диаграммам типа «песочных часов».
В этой же работе [36] рассмотрено также влияние clq
растворителя на совместимость полимеров в общем растворителе. В частности,
показано, что в области НКТС взаимная растворимость двух
полимеров повышается при введении растворителя с более низкими
значениями clq (рис. 6.10). Однако в областях ВКТС и НКТС влияние
отношения clq растворителя на взаимную растворимость двух полимеров
сказывается по-разному. Из рис. 6.11 видно, что минимальные объемные
0,5
1,6
.5
4*5
0,5
Ofi
1,8 2,0 iO(cjq)p 100
110
Рис. 6.11. Влияние растворителя на взаимную совместимость двух полимеров
при 373 К и kAB, равном 0 A), —0,002 B) и —0,02 C) [36].
7? = 550К. УА| УА# УДР
Рис. 6.12. Влияние растворителя на взаимную совместимость двух
полимеров при kps, равном 0 (/), —0,002 B); —0,05-?-0,02 C) [36].
= c/qs = 20, k л в = 0-
доли растворителя, необходимые для того, чтобы полимеры
совмещались, сначала уменьшается, а затем увеличивается с ростом clq
растворителя; влияние различия в потенциальных энергиях полимера
и растворителя на совместимость двух полимеров не очень
существенно (см. рис. 6.11). Из рис. 6.12 видно, что когда потенциальные
энергии полимеров и растворителя близки (т. е. knp= 0), на графике
зависимости минимальной объемной доли растворителя, необходимой для
совместимости полимеров, от потенциальной энергии растворителя
наблюдается различимый минимум.
6.1.3. ПРИМЕНЕНИЕ НОВОЙ ТЕОРИИ ХАГГИНСА
Положения новой теории Хаггинса были использованы Конингс-
вельдом и сотрудниками [28] для интерпретации бимодальной формы
спинодалей и кривых точек помутнения, найденных для некоторых
олигомер-олигомерных смесей с ВКТС [41] (рис. 6.13). Выражение Хаг-
158
в(т,
гинса для параметра
термодинамического взаимодействия g G\ ср)
Конингсвельд [28] применил в виде
РП-О-р)ф.]-
л In A +kxzj —
k2zx), F.20)
где р = (а2/(Тх) {rVt i/rVt п) (ап —
контактная поверхность сегментов в
полимере); /v, п = Vn/nuVL {Vn —
мольный объем полимера; пи — число
сегментов в макромолекуле, Vu —
мольный объем участков решетки);
*2 = 1 — гл = РФ2/[1 — A — Р) Ф2]>
Р sg (г — 2)Дш12//?Т, Дш12 изменение
энергии при образовании одного
контакта между двумя сегментами,
принадлежащими разным
полимерам. Последние два члена в
уравнении F.20) отражают влияние
окружения полимерного сегмента
на его гибкость. При надлежащем
выборе параметров kx и k2 удается
получить бимодальную форму спино-
дали или параметров Р (см. рис. 6.13).
Впрочем, бимодальная форма
фазовой диаграммы может быть получена
и при использовании параметра
взаимодействия gG\ ф) в виде его
разложения в ряд по температурам
и концентрациям:
Ф) =
39 ЗУ
373
353
333
Рис. 6.13. Кривые фазового
расслоения для системы полиизобутилен
(М = 370) — полистирол (М = 2200):
/ — экспериментальная; 2 — рассчитанная
по уравнению типа F.33) с kt = 0,6 и k2 =
= — 0,4; 3 — рассчитанная для g = 0,354 +
-j- 321/7 « 0,158ф2 + 0,784 ф^ [41].
При этом gi должно быть < 0, a g2 > 0 и | g2 \ > | gL \ (см. рис. 6.13).
Интересную интерпретацию представления Хаггинса о влиянии
окружении молекул одного полимера на гибкость цепей другого дали
Конингсвелд и сотрудники в работе [42]. Основываясь на теории Сил-
берберга [43] и Хаггинса, они получили соотношение между
некоторыми параметрами, характеризующими энтропию смешения. Суть этой
интерпретации заключается в следующем.
Согласно теории Силберберга [43], общее число конфигураций
системы, представляющей собой смесь полимолекулярных гомополиме-
ров 1 и 2, записывается в виде
й1+2 — "
159
где Nn и N2j— числа молекул фракций i и / в полимере 1 и в
полимере 2. Относительные длины цепей (ги и r\j) определяются обычным
•способом: молекула растворителя служит мерой отсчета.
«Параметрический объем»У* был введен Силбербергом для коррекции переоценки
числа конфигураций, входящих в член, содержащий факториал.
Поскольку первый сегмент полимерной цепи размещен в общем объеме К,
то объем, доступный для следующих (г — 1) сегментов, ограничен
параметрическим объемом предыдущего сегмента. Если предположить
V* независимым от объема смеси, получается выражение для энтропии
смешения, идентичное уравнению Флори—Хаггинса. Авторы работы
[42] учли концентрационную зависимость V* и получили выражение
для свободной энергии смешения Гиббса A G на 1 моль сегментов
обычным путем — добавляя к энтропийному члену ванлааровскую теплоту
-смешения g(f>i4>2:
&G/RT = ? фнгЦ11пфц + ? Ф2/Г2/11пф2/ + gWP2 —
— 2 <Pu(l — ru1) In ти- ? ф2/A — гГ/1) 1пт2/, F.21)
i !
где ф — объемная доля полимера (?ф*/=ф*), а в %ы входит член
VI Ш/Vti.
Уравнение Хаггинса для свободной энергии смешения Гиббса
с учетом двух ориентационных вкладов в энтропию смешения имеет
вид
&G/RT = ? фигГ/ In фи + ? ф2/г7/ In ф2/ + ?фгф2 —
) —Фя1пA +k2chQ~i)t F.22)
где Q = [1 — A — р)ф2]; р = (tfa/aJ/^/fi); <fc и vk — поверхность
контакта и молекулярный объем сегмента полимера k. Имеется, таким
образом, возможность учета различия между взаимодействующими
поверхностями (объемами) сегментов 1 и 2. Два параметра, ks\ и kS2,
определяемые соотношением kst = kc/(l + ki), Хаггинс интерпретировал
как эмпирические. Как уже отмечалось, Конингсвелд 128], показал,
что уравнение типа F.22) может описывать бимодальную форму спи-
;нодали, причем для получения бимодальной формы спинодали
необходимо приписать отрицательное и положительное значения параметрам
kx и k2. Сравнивая уравнения F.21) и F.22) и пренебрегая г",
а также полагая т независимым от длины цепи, получают вырагкени!
для тх и т2
*i = V* Ы/Vi A) ее 1 +klP4>2Q-\ F.23)
% t . F.24)
Силберберг [43] предположил, что параметрический объем может
быть взят равным или соизмеримым с объемом полимерного клубка.
Используя это предположение, надо ожидать уменьшения размера
•клубка полимера 1 при уменьшении его концентрации в смеси. В
предельно разбавленном растворе полимера 1 в полимере 2 (ф2= 0, ф1= 1)
160
имеем т2= 1 + kl9 тогда как блочный полимер будет
характеризоваться значением тх= 1 (ф!= 1, фа= 0). Наоборот, для полимера 2,
разбавленного в полимере 1, т2= 1 + k2 (ф2= 0, фх= 1), а при фа= 1
и фх= 0 т2= 1 (для блочного полимера 2). В отношении блочного
полимера уравнения F.23) и F.24) дают разумные результаты.
Действительно, данные нейтронографии [44] показали, что клубок имеет
невозмущенные размеры в аморфной области (т = 1).
Отрицательные же значения kx предполагают, что тх< 1 при бесконечном
разбавлении полимера 1 в полимере 2, т. е. уменьшение размеров клубков
по сравнению с невозмущенными размерами в блоке. Имеющиеся
в настоящее время данные показывают, что такое уменьшение размеров
клубков действительно наблюдается. Так, в работе [45] было отмечено,
что размеры клубков поли-а-метилстирола, растворенного в полиме-
тилметакрилате, очень малы, т. е. происходит «коллапс»
макромолекулы поли-а-метилстирола.
6.1.4. ТЕОРИЯ САНШЕЗА
Для характеристики стабильности систем, состоящих из двух
высокомолекулярных соединений, Саншез развил теорию, в основу
которой положил модель сжимаемой решетки [47, 48]. Эта теория
способна описать изменение объема при смешении и предсказать наличие
НКТС [46—48]. В теории Саншеза предполагается, что сегменты
полимера представляют собой жесткие ядра, а из шести энергетических
параметров, с которыми оперирует эта теория, четыре могут быть
определены из PVT-свойств для индивидуальных компонентов. На основе
этой теории Саншез получил выражения для химических потенциалов,
условий стабильности фаз и спинодалей, а также для межфазного
натяжения (см. также [48]).
Химические потенциалы. Для получения химических потенциалов
рассматривается смесь из Ыггг, N2r2> ... и Nmrm сегментов. Общее их
число в смеси записывается уравнением
где N( — число молекул компонента i; ti — число сегментов в
молекуле компонента i.
Свободная энергия смешения Гиббса для смеси из N компонентов
G = ГЫ (#комб + ^некомб) == rNg,
где ?комб и ?Некомб — комбинаторная и некомбинаторная свободные
энергии на сегмент смеси:
а _ре* + pvv* + kTV[(I -?) In A - р) +1-Inp],
4420 161
где
<Pi = rtNt/rN, I/r = 2 q>,/r,,
е*=ЕЕф^Ф/аК)/1/*, F.25)
у* = ?2ф*ф/*?/> F.26)
а р и V — приведенные плотность и объем смеси соответственно (У =
= 1/р). Параметры е*/ и ац являются энергетическими параметрами,
которые характеризуют взаимодействие сегмент — сегмент, причем все
сегменты имеют плотноупакованный объем V*= о?/, где а*/ —ближайшее
расстояние сближения между t-м и /-м сегментами. Сила притяжения
между сегментами пропорциональна е*/.
Из условия равновесия
(dg/dV)p,T* = (д?некомб/<5У)/>,7> = О
получается уравнение состояния смеси
р2 + р + f [in (i _ р) + (i _ 1/г)й = о,
где f и Р — приведенные температура и давление:
Т = Г/Г*; Т* =
Химический потенциал компонента ^г- выражается в форме
здесь q/ означает, что все ф^, за исключением одного, должны быть
постоянными. Отметим, что е*, 1/г и V* — гомогенные функции
нулевого, первого и второго порядков соответственно. Следовательно,
2 Ф/ (д?некомб/<5ф/)ф', р« 2РУ* + (?77r) In p. F.28)
Подстановка F.28) в F.29) ведет к
|х, = *Г[1п ф, + A - rtlr)\ + г/ {- р [е* + (de*/dq>V] +
+ PV [(ЗУ*/аФ,)ф: + I/*] + Wf fA - р) In A - р) +1 In p]}, F.29)
где е* и V* даются уравнениями F.25) и F.26). Они могут быть
выражены в разной функциональной форме. Для бинарной смеси
8* + (де*/дф1)ф2 = еп - Х12<р22 F.30)
и с меньшими ошибками имеем
(<^*/<Эф2)Ф, -.V* = Ft + (I/? + Vt) fitf, F.31)
162
где
Х12 = (P*iPZy>*Vt(Vt/Vi)* [а + Ь126
а = (t/vI/2 4- (v/tI/2 — (v1/2 + v-l
&12 = (Ф1 B + V<px) (т1/2 - ?) — <p\ (| — T-V2)] (vl/2
t v =
Подставляя уравнения» F.30) и F.31) в F.29), получаем значение
химического потенциала для одного компонента в бинарной смеси:
\iJkT = In <рг + A — а\/г2) ф2 + гхх12ф2,
где
F.32)
Химический потенциал второго компонента получается простым
обменом индексов 1 и 2 (отметим, что Х12ф Х21). При атмосферном
давлении два члена уравнения F.32), связанных с давлением, можно
опустить.
Как видно из приведенных выше уравнений, для расчета
химических потенциалов требуется лишь два параметра взаимодействия:
I и б (или 8^2 и а1а); I выражается уравнением F.31а), а физический
смысл б довольно прост — это характеристика отклонения объема
реальной системы от идеальной (простая аддитивная схема для объема):
<0 У*< V*,
=0 V* = V*C F.33)
где
При температуре 0 К знак объема смешения должен определяться
знаком б. Следовательно, уравнение F.33) предполагает, что 6i2 =
= Ф\г+ б*2)/2.
Стабильность фазы и спинодаль. При постоянных Т и Р
необходимым и достаточным условием совместимости бинарной системы во всей
области составов является зависимость g от состава, которая
описывается вогнутой кривой:
d*g/d<p! > 0.
6* 163
Интерпретируя dg/dtpi как функцию cpj и V (при Г = const иР
const) и используя соотношение [47]
= —VTP*$ [д2 (g/kTyd^dh
можно показать, что
d*{g/kT) .
td*(g/kT)l
где Р — изотермическая сжимаемость смеси, которую можно найти
из выражения
-р) + 1-2р/т]
]~\
Следовательно, кривая зависимости g от состава является вогнутой,
если во всем концентрационном диапазоне выполняется неравенство
1/2 [тк+тк] ~ ~р Iх+i **~тр**} >0> F-34)
где
P
1 _
»-,Ф. [ «Эф
Используя уравнение F.30), имеем
Саншез [48] рассмотрел также возможность наличия бимодальной
формы спинодали. Рассуждения его сводятся к следующему.
Поскольку для олигомер-олигомерных смесей комбинаториальный
вклад в неравенство F.35) относительно мал, спинодаль должна быть
очень чувствительна к концентрационной зависимости X и W2.
Наличие бимодальной спинодали указывает, что при концентрации срт,
где наблюдается минимум на кривой зависимости Т—ср, смесь
термодинамически более устойчива при фт, чем при несколько больших
или меньших концентрациях. Эта добавочная стабильность должна
наблюдаться в том случае, если X отрицательна вблизи фт, но
положительна при других составах. Для этого нужно, чтобы величина d (\IT)li
/d принимала три раза нулевые значения во всем концентрационном
164
интервале @; 1). Можно показать, что нулевые значения d(\IT)ld^t
определяются квадратичным уравнением
Tl/2 _ т-1/2 + (ф1
+ (Vl/2 + V-V2) [(VTI^ + (гт)-1/2ф2]б}, F.36)
где Д = (vtI/2 + (vt)^1/2 — (v1/2 + v-1/2) A + 6).
Таким образом, три нулевых значения X во всей
концентрационной области невозможны. Однако при низких давлениях* и больших
гх и г2, W2 пропорциональна [d2(l/f)/dq>l]. Когда й^/ТУФрх
принимает нулевое значение при фт, исчезает неблагоприятный член
сжимаемости.
Необходимыми, но недостаточными условиями наличия бимодальной
спинодали являются следующие:
0 F.37)
и
d*(\/T)/d<pl>0. F.38)
Эти условия удовлетворяются даже в случае v = 1 и 8 = 0; тогда
уравнение F.36) сводится к
Фт = (Г-1'2 - Э/(Т^ + Х-*/2 - 26),
а уравнение F.35) — к
В общем случае даваемое теорией необходимое условие наличия
бимодальной спинодали при низких температурах (ВКТС) является
очень приближенным и требующим, чтобы гг и г2 были примерно
одинаковыми и выполнялись условия F.37) и F.38).
Если d2(l/T)/d<f)l<0 в концентрационной области @; 1) и
выполняется условие. F.38), можно ожидать бимодальную спинодаль для
олигомер-олигомерных или полимер-полимерных смесей при высоких
температурах (НКТС). Последнее обстоятельство более вероятно,
чем в случае ВКТС, так как НКТС обусловлена преимущественно
членом сжимаемости. Экспериментальной проверки выводов Саншеза
по данному поводу пока нет.
Критерий совместимости полимер-полимерной смеси. Как уже
упоминалось выше, для смеси высокомолекулярных соединений комбина-
ториальный вклад в спинодаль очень мал, так как член сжимаемости
всегда приводит к понижению стабильности фазы. Совместимость
двух полимеров может наблюдаться в том случае, если d2(l/f)/dq>i< 0
во всем концентрационном диапазоне @; 1). Это условие эквивалентно
требованию отрицательной теплоты смешения при низких
температурах. Теплота смешения выражается следующим образом.
= rN [— ре* + рхфД + РлФав22].
165
При низких температурах (полагая Рх = р2 = р =» 1)
ДЯСМ (Г = 0) = rtf [—е* + ФхвЙ + Ф2е*2]; F.39)
4^см всегда отрицательна, если d2e*/d<p? < 0 во всем
концентрационном диапазоне @; 1). Уравнение F.39) может быть записано в виде
Д#см (Г = 0)/rN = ф1ф2 (VfVt/V*) {ДР* +
.+ 6 [ДР* + Pt (vq>x - ф2) - Р% (ф1 - ф^-1)]], F.40)
где
ДР* = (Р*Я*) а = Р? + Р2* — I (vV2 + v-i/«) (Р?Р*У'К
Теплота смешения будет отрицательной при ДР* < 0 или если
8 > Бш»
Низкое значение ?мин должно способствовать смешению,
поскольку облегчает получение неравенства F.41). Исследование неравенства
F.41) показывает, что значения ?мин при т > 1 и v > 1 будут ниже,
чем при т > 1 и v < 1. Член, пропорциональный б в F.40), меньше по
величине, чем Д Р*, будет изменять знак в концентрационном
интервале @; 1). В концентрационном интервале, где член 6 вносит
положительный вклад, I должна иметь большее значение, чем |мин, чтобы
получить отрицательную теплоту смешения. В общем случае полная
смешиваемость высокомолекулярных соединений возможна при Д#см < 0.
Свойства чистых компонентов, которые способствуют смешению,т. е.
приводят к отрицательной теплоте смешения, Даются неравенством
F.41). Однако, если система совместима при низких температурах*
можно ожидать, что она будет расслаиваться при высоких
температурах из-за увеличения вклада члена сжимаемости, неблагоприятствую-
щего смешению.
Таким образом в теории Саншеза была найдена связь параметров
смеси с молекулярными параметрами чистых компонентов как и в
новой теории Хаггинса. Для бинарной смеси необходимым и
достаточным условием фазовой стабильности (совместимости) является
неравенство F.34), а наличия бимодальной спинодали — неравенства F.37)
и F.38). Предполагается, что бимодальные фазовые диаграммы более
вероятны для смесей с НКТС. Важным обстоятельством является то,
что для наблюдения бимодальных фазовых диаграмм необходимы
высокие молекулярные массы смешиваемых компонентов (отметим, что
такие бимодальные фазовые диаграммы наблюдались и для смесей
низкомолекулярных компонентов [49]). Полная совместимость двух
полимеров требует отрицательной теплоты смешения, для чего должно
выполняться неравенство F.41) для чистых компонентов.
6.2. ТРОЙНЫЕ СИСТЕМЫ ПОЛИМЕР — ПОЛИМЕР — РАСТВОРИТЕЛЬ
Большинство работ по определению термодинамической
совместимости полимеров были выполнены в присутствии растворителя [2, 3,
5, 50—66]. Теоретически такие системы исследовали Скотт [13] и Том-
па [14], которые рассмотрели так называемый симметричный случай,
166
т. е. когда взаимодействие каждого полимера с данным растворителем
одинаково (Ххл= Xis)> а гл= гв= г.
Уравнение для свободной энергии смешения для тройной смеси
имеет вид [13]
А/, RTV[ фА qw
Д° = Т~~ Фр Фр Ч- — In фА + — In Фв + ХавФаФв + ХарФаФр +
Р [ 'А ГВ
+ ХврфвФр],
где Vp— молярный объем растворителя. Соответствующее уравнение
для бинодали записывается так:
d*AG\/d*AG\ __( d2AG
Для симметричного случая Скотт получил такое уравнение бинодали:
In в; + фхав A - Фр) (ввJ = In вв + ФХав A -ФР) (вк)\ F.42)
где
в - Фа - в' = Фв
А Фа + Фв ' В Фд+Фв"
Вид бинодали, получаемой по уравнению F.42), не зависит от
Xib и %1л, т. е. фазовое расслоение в таких системах связывается
лишь с неблагоприятными взаимодействиями между сегментами
полимеров (хав > 0), а роль растворителя сводится лишь к разбавлению
смеси и, таким образом, к уменьшению эффективного числа
неблагоприятных контактов между сегментами полимеров разного типа и к
уменьшению параметра взаимодействия фав A —фр).
При фр-* 1 эффективный параметр взаимодействия стремится
к нулю и вся система становится однофазной. Соответствующее
критическое значение для параметра термодинамического взаимодействия
в тройной системе можно определить по уравнению
(Хав)крйт = 1 [ Й
Из уравнения F.43) следует, что критическое значение параметра
достигается для данной пары полимеров независимо от природы
растворителя при фр, одинаковом для разных растворителей. Отсюда
следовал вывод о том, что если два полимера несовместимы в данном
растворителе, они не будут совместимы и во всех других.
Этот вывод и кажущееся его подтверждение данными Добри и
Бойер-Кавеноки [50] почти на два десятилетия затормозили поиск
совместимых полимерных пар. Между тем в некоторых работах было
установлено, что влияние растворителя на совместимость двух
полимеров не сводится лишь к простому разбавлению системы. Так, уже в
работе Керна [51], где были приведены данные по измерению
критических концентраций смешения полистирола с полиметилметакрилатом
в большом числе растворителей разной природы, было обнаружено,
167
0,2
ft/
"что, например, критическая концентрация смешения полимеров
изменяется примерно от 13 % (по объему) в диметиланилине до 2,5 %
в n-ксилоле. Согласно теории Скотта, это должно соответствовать
почти пятикратному изменению параметра Хгз-
Существенное влияние растворителя на совместимость полимеров
обнаружено и авторами работы [71]. Для системы полистирол —
поливиниловый эфир было найдено, что полистирол совместим с поливинил-
метиловым эфиром в толуоле, бензоле или перхлорэтилене, но
несовместим в хлороформе, метиленхлориде или трихлорэтилене. При этом
полимеры остаются совместимыми или несовместимыми и при
удалении растворителя. Пленки, полученные из растворителей, где
полимеры несовместимы, показывают две температуры стеклования. Даже
в том случае, если, поданным
дифференциальной сканирующей
калориметрии, наблюдается одна
температура стеклования,
промежуточная между температурами
стеклования индивидуальных
компонентов, данные диэлектрических
потерь свидетельствуют о
микрогетерогенности таких смесей.
Подобное влияние растворителя на
совместимость полимеров отмечено и
в работе [53], и в работе [52].
Авторы статьи [52] показали, что
критическая концентрация смешения
хорошо коррелирует со значениями
I а2— #11» где ах— константы урав- *
нения Марка — Куна — }?аувинка,
связывающего характеристическую
вязкость с молекулярной массой (рис. 6.14). Поскольку щ связаны
с параметрами термодинамического взаимодействия полимер —
растворитель Хц, их асимметрия ведет к увеличению несовместимости
полимеров в растворе.
Полученные экспериментальные данные о влиянии растворителя
на термодинамическую совместимость полимеров в растворе побудили
некоторых авторов [67, 68] пересмотреть выводы теории Скотта о роли
растворителя в тройных системах полимер—полимер—растворитель.
Такой пересмотр сделали Земан и Паттерсон [67], которые за основу
взяли уравнение для спинодалей, полученное в [69]:
О 5 10 15 20 100-Чкрит
Рис. 6.14. Зависимость^ критической
концентрации смешения двух
полимеров в данном растворителе от разности
констант уравнения Марка — Куна—
Хаувинка для системы полистирол —
полиметилметакрилат (/) и поли-2-ви-
нилпирролидон — полиметилметакрилат
B) [52].
где
+ Шз + Х2Хз)ф1Ф2<Рз=:0-
9v —— V | л/ ___ v
^Al А12 1 Л13 Л23»
F.44)
другие %t находят обычной циклической заменой индексов в уравнении
F.44); ф/ — объемные доли; г* — число сегментов в компонентах. В
уравнениях F.44) и F.45) параметры х*/ соответствуют трем энергиям
168
образования контактов между сегментами в противоположность
параметрам Флори, в которых контакты относятся к целой молекуле
первого полимера, т.е %ij (Флори) = гф} (Томпа) (см. уравнение F.69)).
На основе уравнения F.44) Земан и Паттерсон построили спинодали
для разных вариантов изменения параметров %и и %23 и
молекулярных масс смешиваемых полимеров. Из анализа полученных
результатов следуют достаточно интересные выводы.
1. Для симметричного случая, т. е. при %12= Xie и постоянном %23,
спинодали не зависят от xi/. Почти идентичные спинодали получаются
при малой разности в хи» показывая лишь большую область
несовместимости (рис. 6.15). Интересно, что улучшение качества растворителя
Рис. 6.15. Спинодали для тройных систем с Х2з = ^» Xi2 = 0,50, Х13 = 0,30,
Pl = 1, р2 = pg, равных 220 (/), 250 B), 500 C), 1000 {4).
При р2 =Рз == 200 наблюдается полное смешение [67].
Рис. 6.16. Спинодали для тройных систем' при р1==1, р2 = /?3 = 1000, %12 =
= 0,40 и Х13, равном:
/ ^- о,4О; 2 — 0,45; 2'— 0,35; 3 — 0,50; 3'— 0,30 (Хя8 = 0,0025); 4 — 0,50; 4' — 0,30 (Х23= 0).
дЛя полимера 3 приводит к тому же эффекту, что и его ухудшение
(кривые 2 и 2'). Введение большей разности в %и вызывает еще большее
расширение области несовместимости (кривые 3 и »?'). При %2з=* 0 и %12=
= %13 спинодали исчезают, т. е. полимеры совместимы как в растворе,
так и без растворителя. Если же ввести большую разность в %н>
область несовместимости полимеров сдвигается к меньшим
концентрациям (кривые 4 и 4'), т. е. область несовместимости остается и при
%2з— 0 и даже при Хгз< 0» но спинодали имеют вид замкнутых петель,
которые не пересекают концов треугольной фазовой диаграммы, т. е,
бинарные смеси полностью совместимы.
В этой модельной системе (и в реальной тоже [70]) ограничение
совместимости в области малых концентраций связано с неравенством
взаимодействий полимер — растворитель, в то время как в области
высоких концентраций несовместимость связана с неблагоприятными,
взаимодействиями полимер — полимер.
Ш
2-. Влияние неравенства взаимодействий полимер — растворитель,
так же как и неблагоприятные взаимодействия полимер—полимер,
характерны для высокомолекулярных образцов. Уменьшение
молекулярных масс смешиваемых полимеров приводит к тому, что даже
при существенном различии в %ц (например, при Х23= 0,.рис. 6.16),
замкнутые петли, определяющие область несовместимости, быстро
сужаются и при существенном уменьшении молекулярных масс
полимеров вовсе исчезают. Увеличение молекулярной массы одного из
смешиваемых полимеров расширяет область несовместимости, сдвигая
ее главным образом в область высоких концентраций полимера с
низкой молекулярной массой. ,.
3. В случае неполной совместимости в бинарных системах
полимер — растворитель при одновременном изменении %12 и %13 с
определенным инкрементом в случае %23= О критические точки при высоких
концентрациях остаются на дне фазовой диаграммы, а при низких
концентрациях сдвигаются в область чистых смесей 1—2 или 1—3
(концу фазовой диаграммы) на спинодали, приводя к появлению
критической температуры с %икрт--=~{1 + (/У2I/2}2и фикрИТ - 1A +
+ {fi^i)l/2} (см. рис. 6.16, кривая 5). Дальнейшее повышение Xi2
ведет к тому, что критическая точка для этой бинарной смеси
находится вне фазовой диаграммы. Еще большее увеличение %12 и %13
приводит к появлению критической точки, характерной для бинарной
смеси 1—3 (см. рис. 6.16, кривая 3), которая с дальнейшим ростом %и
выйдет вне рамок фазовой диаграммы.
4. Фазовые диаграммы замкнутого типа, как следует из этого
рассмотрения, должны быть, в принципе, очень редким явлением, так как
для их появления необходим ряд специальных условий, в частности,
различие в %и и чтобы %28 было близко к нулю или отрицательно. По
мнению авторов настоящей работы, такая ситуация может наблюдаться
при образовании тройной смеси полимер—полимер — растворитель
для полимеров, имеющих подобную химическую природу (где можно
предположить %28^ 0), но отличающихся гибкостью цепей, которая
определяет различие в свободных объемах компонентов или во вкладах
члена уравнения состояния в %12 и %13. В принципе, такими системами
могут быть смеси полимергомологов, существенно различающихся по
молекулярным массам и, следовательно, по коэффициентам
термического расширения [72]. Для таких систем можно ожидать, что х12ф
Ф x13. И действительно, как мы обнаружили [73], параметр
термодинамического взаимодействия для системы олигоэтиленгликоль (М =»
= 2000) — бензол при 353 К равен 0,102, а для системы
олигоэтиленгликоль (М = 40 000)— бензол при той же температуре — 0,0320.
При этом параметр термодинамического взаимодействия между
компонентами бинарной смеси этих олигогликолей отрицателен, так что,
в принципе, для тройной смеси этого типа можно получить фазовую
диаграмму типа замкнутых петель.
В настоящее время обнаружено уже достаточно много систем
с Хз8<° [61, 74, 75]. Следовательно, для этих систем такж^ могут
быть характерны диаграммы типа замкнутых петель. И действи-
17©
сна
тел^но, фазовые диаграммы этого типа были получены
экспериментально [30, 70] (рис. 6.17). Особенно интересные данные,
свидетельствующие об определяющей роли неравенства параметров %и во
взаимной растворимости двух полимеров в общем растворителе,
приведены в работе [30], авторы которой методом обращенной газовой
хроматографии определили параметры %и для полистирола и поливинилме-
тилового эфира в широком диапазоне температур и нашли, что если,
величина Д % = 1%12— %ie | уменьшается с повышением
температуры, область несовместимости сужается. Поскольку система имеет
НКТС [27], параметр х2з с ростом температуры должен
увеличиваться, и если бы совместимость компонентов в растворе определялась
параметром Хгз» как следует из
теории Скотта, то в данном случае
должен был бы наблюдаться
совершенно противоположный
эффект, а именно — расширение
области взаимной несовместимости
полистирола с поливинилметиловым
эфиром. Эти теоретические
результаты полностью подтвердились
экспериментально (см. рис. 6.17).
Следовательно, этот результат
является наиболее убедительным
аргументом в доказательстве
определяющей роли неравенства %и в
несовместимости двух полимеров
в общем растворителе.
Необходимо отметить еще одно
важное обстоятельство. При
формировании бинарной смеси двух
полимеров из общего растворителя,
в котором фазовая диаграмма имеет вид замкнутых петель, при
испарении растворителя система проходит область концентраций вне
области несовместимости, т. е. полученная из таких растворов бинарная
смесь двух полимеров должна быть гомогенной. Однако из-за резкого
повышения вязкости системы при концентрировании раствора, когда
растворитель удаляется из смеси, бинарная система может оказаться
неравновесной и характеризоваться микрогетерогенной структурой,
как это было показано в работе [71].
К заключению о существенной роли неравенства %i2—%13 во
взаимной растворимости полимеров в общем растворителе пришли также
Хсу и Прусниц [68] на основе расчета бинодалей при различных
вариациях параметров %и, Хгз и молекулярных масс смешиваемых
полимеров. Кроме того, они получили несколько кривых, определяющих,
пределы совместимости в зависимости от параметров %23 и | Xi2— OCis I»
Таким образом, рассмотренные данные достаточно четко
показывают, Что роль растворителя в тройных системах полимер—полимер —
растворитель более существенна, чем это следовало из первоначальной
теории Скотта. Более того, эти данные подтверждают вывод о том, что.
ПС
0,5
ПВМЭ
Рис. 6.17. Фазовая диаграмма для
системы полистирол (М =* 2100) — поли-
винилметиловый эфир (М =11 000) —
хлороформ при температурах:
/ — 307; 2— 317; 3 — 323 [70].
Полимеры, несовместимые в растворе, не обязательно будут
несовместимыми в отсутствие растворителя. Хорошим примером этому может
служить система полистирол — поливинилметиловый эфир [27* 30].
Авторы работ [56, 69] обнаружили очень узкую температурную
область однофазного состояния между нижней и верхней
критическими температурами смешения для ряда тройных смесей. От отметили,
что это явление наблюдается в том случае, если критические
температуры смешения двух бинарных систем полимер — растворитель близки.
Узкий температурный промежуток совместимости появляется вблизи
кривой точек помутнения одной из бинарных систем полимер —
растворитель (рис. 6.18). Для выяснения причины такого явления автор
работы [56] провел теоретический расчет спинодалей, варьируя значе-
293
№
273
343
333
333
- 2
а
0,05
Q 0,02 ' 0,50
в
0,55
X
-Рис. 6.18. Кривые точек помутнения для системы полистрол (М„ = 6,7«106) —
полидиметилсилоксан (М^ = 3,9- 10б) — этилацетат A,3) и полидиметилсилокеан —
этилацетат B) (а), расчетные спинодали, полученные при использовании значений
Xi2 и %i3» а также %2з из рис. 6.17, в, для р2 = р3 = 2000 (б) и схематическое
представление температурной зависимости параметров Х12» Х13 СО и Х2з B) [56] (в).
ния параметров %и и %2з и их температурные зависимости. Было
найдено, что подобное обнаруженному поведение спинодалей наблюдается
при условии, если параметры %i2 и %1з близки и уменьшаются с
повышением температуры, а величина х23 увеличивается. При этом, чтобы
получить узкий температурный участок совместимости, значение %гз
должно быть очень малым (~ 0,02) и температурная зависимость %гз
положительной и достаточно сильной. Однако их модельная система
(смесь олигомеров стирола и диметилсилоксана, оба компонента
примерно гексамеры) имеет ВКТС примерно 308 К, т. е. значение Хгз
должно уменьшаться с увеличением температуры. На основании
этого автор предположил, что введение растворителя в эту смесь
приводит к резкому уменьшению параметра %гз> а его температурная
зависимость обращается от отрицательной к положительной. Причиной
этого может быть образование облаков сегментов вокруг центра
тяжести макромолекулы при малых концентрациях полимера
макромолекулы, что затрудняет образование межмолекулярных контактов
сегмент — сегмент, а это, в свою очередь, отражается на кажущемся
значении параметра %2з в разбавленной тройной системе, которое мо-
172
жет быть даже меньше, чем истинное его значение в бинарной системе
полимер — полимер. С увеличением общей концентрации полимеров
в смеси эффекты конформации цепей могут играть все большую роль
в изменении параметра Хгз- Оказава [56] рассмотрел результаты Ко-
нингсвелдаи сотрудников [69], которые обнаружили узкую
температурную область совместимости лишь для одной из исследованных
систем изотактиЧеский полипропилен — полиэтилендифениловый эфир
(второй была система атактический полипропилен — полиэтилен —
дифениловыйэфир). Оказава предположил [56], что это связано с тем,
что радиус инерцик изотактического полипропилена на 10 % больше,
чем у атактического,к а 0-температура в дифениловом эфире для
изотактического полипропилена ца 8° ниже, чем для атактического.
Поэтому вероятность образования межсегментных контактов в атактиче-
ском полипропилене меньше, чем в изотактическом.
Помимо упомянутых явлений, в тройных системах полимер —
полимер — растворитель Агарони [59] обнаружил бимодальность
замкнутых кривых точек помутнения. Для трех изученных им систем
бимодальность кривых точек помутнения он наблюдал в области составов
1:1. Поскольку эти бинарные смеси полимер — полимер можно
считать совместимыми [59], Агарони сделал предположение, что
молекулярное притяжение между полимерами больше, чем между каждым
полимером и растворителем, причем наибольшая роль этого эффекта
проявляется при составах 1:1. Надо все же отметить, что причина
появления бимодальности кривых точек помутнения в тройных
системах до конца не выяснена, так как эти эффекты наблюдались и в
случае термодинамически несовместимых полимеров, в частности, для
системы полистирол — полибутадиен — тетралин [57].
Из других работ по исследованию тройных полимерных систем
можно упомянуть работу Квея и сотрудников [76], которые показали,
что, если два полимера взаимно несовместимы, но каждый в
отдельности совместим с третьим полимером, то введение в смесь этих двух
несовместимых полимеров третьего полимерного компонента приводит
к образованию совместимой тройной системы. Следовательно, здесь
наблюдается аналогия с введением низкомолекулярного растворителя
в смесь двух термодинамически несовместимых полимеров.
Рассмотренные в этом разделе данные относятся непосредственно
к оценке критерия корректности данных по параметрам
термодинамического взаимодействия, определяемым методами на основе
использования растворителя (методами Аллена, Тагер, обращенной газовой
хроматографии). Рассмотрим имеющиеся в настоящее время
экспериментальные данные.
Уже в первой работе по использованию обращенной газовой
хроматографии для оценки параметров термодинамического
взаимодействия между компонентами в смесях низкомолекулярных компонентов
[77] было обнаружено заметное влияние природы сорбата на величину
Х2з- Олабиси [29] обнаружил, что при использовании неполярных сор-
батов значения параметров термодинамического взаимодействия между
компонентами смеси поли-е-капролактон — поливинилхлорид
положительны, а при использовании полярных сорбатов — отрицательны.
173
5,5
5,0
4,5
4,0
15
3,0
2,5
2,0
1,5
2,4 2,6 2,8 105/Т
Отрицательные значения параметров
термодинамического взаимодействия
были приписаны наличию
специфических взаимодействий меж^у
компонентами стационарной фазы, которые
приводят к комплексоооразованию,
следствием которого является
исключение из сферы взаимодействия с
сорбатом части групп, сДоторыми он
взаимодействует специфически.
Предположение Олабисн о том, что йолярный
сорбат может fie взаимодействовать
с некоторой частью полярных групп
стационарной фазы вовлеченных в ком-
плексообразование, подтверждается
данными, которые мы получили для
системы полипропиленгликоль (М =
= 2000) — макродиизоцианат на
основе ППГ-2000 и 4,4'-дифенилметанди-
зоцианата с заблокированными
метанолом концевыми NCO-группами [78].
При использовании в качестве сорба-
та метанола, образующего
водородную связь с полярными группами
смеси, было найдено, что на графиках
зависимости lg Vg от VT для смесей
разных составов наблюдается по крайней
мере два четких температурных
перехода, не связанных с фазошдми
превращениями в системе, так как при
использовании неполярного сорба-
та (гептана) никаких температурных
переходов не наблюдалось (рис. 6.19). Температурные переходы,
обнаруживаемые с помощью метанола, обусловлены ослаблением
водородных связей (причем разного типа) при нагревании смеси.
Освобождающиеся при этом полярные группы взаимодействуют с метанолом,
приводя к увеличению удерживаемого объема. Аналогичный эффект мы
наблюдали и для обычных сегментированных полиуретанов [79].
Эти данные показывают, что часть полярных групп, специфически
взаимодействующих с сорбатом, действительно уходит из сферы
взаимодействия, что, в принципе, может привести к отрицательным
значениям параметров %23, рассчитываемых по уравнению F.72). Однако
отрицательные значения параметров термодинамического
взаимодействия не являются следствием только использования уравнения типа
(.6.72). Как показано в работах [74, 80—83], отрицательные значения
%23 вообще характерны для термодинамически совместимых систем,
особенно при наличии специфических взаимодействий. Отметим, что
отрицательные значения параметров %гз получены в смесях без
растворителя [74, 80—83].
Рис. 6.19. Зависимость In Vg от \/Т
для олиопропиленгликоля с Л1 =
= 2000 (/), макродиизоцианата на
его основе и 4,4'-дифенилметанди-
изоцианата D) и их смесей примас-
совой доле макродиизоцианата 19
B,5), 29 F) и 57 о/о E). Сорбаты:
/ — 4 — я-гептан, 5,6-метанол [78].
174
Зависимость параметров %23 от природы сорбата обнаружена и в
работах^ 29, 75, 84, 85]. Так, в статье [85] для системы поливинилхлорид—
диоктрЦфталат были получены очень большие отрицательные
значения параметров %23, что указывает на высокую степень совместимости
компонентов вплоть до объемной доли диоктилфталата около 0,25. При
увеличений концентрации диоктилфталата этот параметр становится
менее отрицательным, а затем и положительным при объемной доле
диоктилфталата примерно 0,55. Переход от отрицательных значений
%2з к положительным авторы [85] объясняют тем, что при больших
концентрациях дцоктилфталата большинство его является
«мобильным», т. е. находите^ в контакте с другими молекулами
диоктилфталата, и меньше находился в контакте с поливинилхлоридом. Когда
проба входит в стационарную фазу, контакты диоктилфталат — диоктил-
фталат разрушаются легче, чем контакты диоктилфталат —
поливинилхлорид или поливинилхлорид — поливинилхлорид, если используется
такой сорбат, как я-ялкан. Следовательно, совместимость пробы с ди-
октилфталатом больше, чем с поливинилхлоридом, или со смесью
диоктилфталат — поливинилхлорид, из-за селективного взаимодействия
ее со свободными молекулами диоктилфталата. Поэтому параметр
%123, найденный по уравнению F.72), ниже, чем таковой для случая,
если бы проба свободно взаимодействовала со всеми компонентами
смеси. Уравнение же F.72) дает значение х23 путем вычитания %123 из
суммы (ср2%12 + Фз%1з)- Значит, понижение %123 может привести
к более положительным значениям %23. Естественно, что сорбаты
разной природы будут селективно разрушать контакты разного типа, и это
может стать причиной наблюдаемой зависимости параметра %23 от
природы сорбата, используемого для его определения. Зависимость
параметра %23 от природы сорбата, наблюдавшегося и авторами
работы [70], объясняется предпочтительным взаимодействием сорбата с тем
полимером, для которого %и меньше. Этот эффект был назван ими
«А X-эффектом».
Можно ожидать поэтому, что если %и близки (что может быть в
случае смеси полимергомологов), то независимо от используемого сорбата
определяемые значения параметров %23 будут одни и те же; т. е., если
использовать в качестве стационарной фазы смесь полимергомологов,
то, в принципе, можно в определенной степени судить о погрешности
определения параметра %23 методом обращенной газовой
хроматографии. С этой целью мы провели специальные эксперименты на смесях
полимергомологов этиленгликоля. В условиях отсутствия влияния
природы подложки, толщины покрытия, скорости газа-носителя и
симметричности газохроматографических пиков на величину удельного
удерживаемого объема найдено, что точность экстраполяционных
значений удельных удерживаемых объемов зависит от чувствительности
используемого детектора. Полученные в табл. 6.1 данные относятся
к системе с близкими параметрами %12 и %13. Но и для других
систем, для которых %12 и %13 существенно различны, газохроматографи-
ческий метод дает корректные значения параметров %13, как это видно
из сравнения данных, приведенных в табл. 6.2 и 6.3 (параметры %13э
данные в табл. 6.2, получены методом обращенной газовой хромато-
175
Таблица 6.2. Значения параметров термодинамического взаимоде йствия/для
смесей ПЭГМ5000 — ППГ-1050 при температуре 350 К [96] /
ППГ 1 ПЭГ, %
(по массе)
82,2:17.8
69.5:30.5
53.0:47,0
40,7: 59,3
31,0:69,0
эксперимент
0,121
0.121
0,120
0,118
0,114
расчет
0,071
0,073
0,070
0,075
0,075
*„ (Я)
эксперимент
0,411
0,488
0,642
0,798
0,978
расчет
0,075
0,077
0,082
0,093
0,095
экспери- /
мент J
—0Ш
-Д367
/0,522
/-0,680
/ —0,864
расчет
—0,004
—0,004
—0,012
—0,018
• —0,020
Таблица 6.3. Значения
параметров термодинамического
взаимодействия для смесей ПЭГ —ППГ,
рассчитанные из фазовых диаграмм [97]
П*р имечание. Параметры взаимодействия рассчитаны на сегмент ПЭГ.
графии, а в табл. 6.3 — по составу сосуществующих фаз [97]). Кроме
того, в работе [154] для аналогичной системы (ПЭГ-1000 и ПЭГ-4000)
были получены значения х2зтого же порядка величины и того же
знака, что и в нашем случае для смеси
ПЭГ—ПЭГ.
А. А. Тагер и В. С. Блинов [98]
пришли к выводу, что для
определения Д G (а следовательно, и %23)
необходимо использовать хорошие
растворители. Согласно их
данным, свободная энергия смешения,
определяемая по
термодинамическим циклам, не зависит от природы
используемого растворителя. Одна-
Примеч ание. Параметры взаимо- vn R nafvvrp FQQ1 чиячрнист пяпямрт-
действия рассчитаны на сегмент ПЭГ. КО В Р^ООТе IOTJ ЗНачеНИЯ ПарамеТ-
ров Х23 для привитых сополимеров,
оцененные по тем же термодинамическим циклам, оказались
зависимыми от природы растворителя, используемого для определения Хгз-
Система
ПЭП-600 +ППГ-1000
ПЭГ-600 + ППГ-1500
ПЭГ-600 + ППГ-2025
0,13
0,16
0,17
6.3. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ОЦЕНКИ ТЕРМОДИНАМИЧЕСКОЙ
СОВМЕСТИМОСТИ СМЕСЕЙ ПОЛИМЕРОВ
Наиболее достоверную с количественной точки зрения картину
процесса смешения полимеров могут дать лишь чисто термодинамические
методы, которые позволяют определить соответствующие
термодинамические параметры, характеризующие взаимную растворимость
компонентов (свободную энергию смешения Гиббса, энтальпию и
энтропию смешения, параметр термодинамического взаимодействия).
Самое же наглядное представление о фазовом состоянии смеси полимеров
дают фазовые диаграммы. Поэтому здесь мы рассмотрим подробно
лишь методы получения фазовых диаграмм и определения
термодинамических функций.
176
46.3.1. ПОЛУЧЕНИЕ ФАЗОВЫХ ДИАГРАММ
Бинодали для бинарных и тройных систем можно получить любым
оптическим методом, если исходные смеси прозрачны в видимой
области cnektpa длин волн, путем построения так называемых кривых
точек помутнения, которые рассматриваются в данном случае как
бинодали. Дл* построения спинодалей обычно используют метод
светорассеяния, лЦбо получение концентрационных и температурных
зависимостей свободной энергии смешения Гиббса и на их основе
определяют условие спинодали, т. е. д2А G/dq>22= 0. Из этих же данных,
в принципе, находятся и бинодали.
Рассеяние света\(метод Шолте [86]). Согласно теории Смолухов-
ского — Энштейна —• Зернике интенсивность рассеянного света
многокомпонентной системой определяется выражением
W — — в..
Zj La dct dc} ч
kT -'
L— д Vt
где в детерминант \b\ входят элементы (d2AG/dcKdci).
В точке спинодали \Ь\ = 0, поэтому при приближении к спинодали
величина [7?^]е=о стремится к бесконечности, а следовательно 1/[/??]е=о—
к нулю. Совершая серию измерений при постоянной концентрации
и разных температурах, а также строя график зависимости 1/[#?]е=о от
температуры, можно найти точку на спинодали для любой
концентрации путем экстраполяции 1/[/?|]в=о к нулю. Если параметры
термодинамического взаимодействия (W, g или х) линейно зависят от 1/7\
то зависимость f—) р—1| (/?§) _ от 1/Т должна быть линейной1.
Этот метод можно использовать для определения спинодали
квазибинарной системы, например полимерного раствора или смеси двух
полимеров (раствор полимера или смесь двух полимеров
рассматривается как квазибинарный, поскольку индивидуальный полимер
представляет собой смесь полимергомологов с тем или иным молекулярно-
массовым распределением). Возможно также определить спинодаль
и квазитройной системы, например, раствора полидиспёрсного
полимера в смеси двух растворителей; раствора двух полимеров в общем
растворителе или смеси трех полимеров. Преимуществом этого метода
является то, что он не основан на какой-либо модели раствора.
Концентрационная и температурная зависимость А G. Если
определены значения свободной энергии смешения Гиббса в широком
диапазоне температур и составов, то точки на спинодали могут быть
получены путем двойного графического дифференцирования кривых
Д G = / (ф) при разных температурах и экстраполяции
зависимостей д2Д О/дфа при данной концентрации к д%А Gldy\=* 0.
1 Кувахара и сотрудники [87] нашли, что зависимость [Rq] e==0 = f{T)
подчиняется уравнению /?^=G1—TcnI>26f где ^„—температура на спинодали.
177
Схематически эту процедуру можно представить следующим
образом. Для кривых зависимости A G от ф2, полученных при
разных/температурах, путем графического дифференцирования находятуюрвые
производные (дД G/dy2)p, т и наносят на график зависимости д АО/
/<?Ф2 от ф2 для разных температур. Затем определяют вторые .производ-
ные (d2AG/d(pl)p) т таким же методом по кривым зависимости <ЗД G/
/дф2 от ф2 и наносят на график зависимости д2ДС/дф2 от я^ также для
разных температур. При постоянных значениях ф, гароят графики
зависимости д2Д Gldq>\ от \1Т и полученные прямые ^экстраполируют
к значениям д2Д Gldy\-+ 0. Температуры, при которых д2Д Gldy\ = 0,
соответствуют точкам спинодали для определенных концентраций.
По характеру температурной зависимости <?2Д G/дф2, от \1Т можно
судить о типе критической температуры смешения. Положительный
наклон графика зависимости д2Д Gld^\ от 1/Г характеризует систему
с нижней, а отрицательный наклон — систему с верхней критической
температурой смешения (см. условия устойчивости A.12)).
Из графиков зависимостей Д G от ф2 легко получить и бинодали
(см. рис. 1.3).
Экспериментально определить Д G в широком диапазоне температур
и составов очень трудно, но можно для этих условий определить
параметр термодинамического взаимодействия (например, методом
обращенной газовой хроматографии) и рассчитать значения A G по
соответствующим уравнениям, связывающим A G с %2з (например,
уравнение Флори—Скотта [13]).
Определение АО, А Я и A S. Для определения этих параметров
А. А. Тагер и сотрудники [8] предложили использовать
термодинамический цикл Гесса. Сущность этого метода заключается в следующем:
1 г полимера I + растворитель = раствор I — AGi ,
1 г полимера II + растворитель = раствор II — AGn,
1 г полимера I + 1 г полимера II = раствор III — AG*,
Раствор III + растворитель = раствор I + раствор II —AGiv.
Отсюда
_ AG* = — AGi — AGn + AGm. F.47)
Для расчетов по приведенным циклам необходимо
экспериментально определить изотермы сорбции общего растворителя для
каждого полимера и смеси, по которым рассчитывается величина
химического потенциала растворителя A \ix. По закону Гиббса—Дюгема
вычисляется химический потенциал полимера Д \х2 или смеси:
—J 5
и по ним — значения AGb AGn и AGm. Рассчитанные значения
подставляют в уравнение F.47) и находят AGX Если смешивать тх г
полимера I с т2 г полимера II, то
— AGX = {т± + т2) AGm — (пгг Ad
178
Поделив все на (тг + /я2), получают среднюю свободную энергию
смещения Agx на 1 г смеси полимеров:
Agx = AGui — (w2AGi + w2AGu)-
По аналогичным циклам находят энтальпию смешения
= ДЯ, + Д#„ — ДЯщ.
Из обычного соотношения AS = (АН — AG)fT определяют энтропию
смешения.
6.3.2. ОЦЕНКА ПАРАМЕТРОВ ТЕРМОДИНАМИЧЕСКОГО
ВЗАИМОДЕЙСТВИЯ
Депрессия температуры плавления. Квей и Фриш [81],
рассматривая систему, состоящую из аморфного (компонент 1) и
кристаллического (компонент 2) полимеров, для определения разности свободной
энергии на сегмент между кристаллом и переохлажденной жидкостью
предложили уравнение
AGG\ Б) = [F(T, I) - F(T, БоМ + IF G1, So) - Fa G\ Фх = 0)] +
где ? — число сегментов между двумя ламеллярными поверхностями
кристалла; Fa— характеристика аморфного состояния; фх— объемная
доля компонента 1 в жидкости. Индексы 0 и 2 относятся к компоненту
2, закристаллизованному в отсутствие компонента 1. Три члена в
квадратных скобках в правой части этого уравнения характеризуют
соответствующие изменения свободной энергии, связанные с
морфологическими эффектами, плавлением и смешением соответственно. Каждый
из этих членов записывается следующим образом:
F(T91) - F(T, Бо)
ЦТ, Бо) - ^ (Г, ф1 = 0) - - АНт (Г°пл - Тпл)/Т1, F.48)
F* (Т, ф1 = 0) - Fa (Г, ф1) = ^[ In ф2 + Фх - g Фх + Хп In Ф?],
где рх и р2— степени полимеризации компонентов 1 и 2
соответственно; 2ве— поверхностная свободная энергия на группу сегментов в ла-
мелле; А Япл— энтальпия плавления на моль звеньев
кристаллизующегося полимера; параметр термодинамического взаимодействия %21
связан с длинами цепей соотношением
X2'i = W/>2 = W/>i. F-49)
Полагая A F G\ I) = 0 в точке плавления Тпл, комбинируя
уравнения F.48), F.49) и разлагая In в ряд по ф2, получают
АН( Т° JJ - («1 -^-)ч>? +
Г6.50)
179
т
' пл
Если [Boe/Q — Bae/l)°\ = 0, a p2 очень велико, то уравнение
F.50) сводится к форме, предложенной Флори [88]:
1
1
а если /?! очень большое, получаем уравнение, предложрйное Ниши
и Вангом [89]:
1 -М
——ТО
пл пл у
Считая член [Boe/Q — {2ee/Q°] функцией ср* исчезающей при
Фх — 0, а его величину пропорциональной фх с коэффициентом про-
яорциоральности С, зависимость g от фх можно записать в виде
и
Bа,/Е)
для
1-
Принимая далее, что температурная зависимость параметра
взаимодействия имеет обычный вид: % = а + Ь/7\ и что а «С Ь/71 вблизи
точки плавления Гпл, конечное уравнение записывают в форме
д#пл (Т1 - Тпл)/Ф! RTI - bl - ф1 Is. e с//? - 6ф1. F.51)
Зависимость левой части этого уравнения от фх тогда описывается
•прямой линией с наклоном —Ь и отрезком С/7? (табл. 6.4).
Таблица 6.4. Параметры термодинамического взаимодействия и
морфологические эффекты для некоторых смесей полимеров [81]
Система
Изотактический поли-2, 6-диметил-1,
4-фениленоксид (Мп = МО4; Mw—
= 4,35• Ю4; Мг =* 9,72• 104) + атак-
тический полистирол с
молекулярной массой
800
2200
10 000
37 000
78000
Атактический поли-2, 6-диметил-1,
4-фениленоксид + изотактический
полистирол
Температурная
область, К
433—510
455—502
473—511
496—512
333—374
473
ь
41,5
16,0
3,0
1.0
X
0,095—0,081
0,036—0,032
0,0063—0,0059
0,0020—0,0019
0
0
лкрит
0,196
0,042
0,010
0,006
С
196,7
—
Метод сосуществующих фаз (метод Аллена [90]). Параметр
термодинамического взаимодействия определяется из состава сосуществую-
180
щихфаз в расслоившихся тройных растворах полимер — полимер —
растворитель по уравнению
In (ф2/Ф'2) + (l/V3) In
где ф2, фз» фг и фз— объемные доли полимеров в фазах; %12 и %13—
параметры термодинамического взаимодействия растворителя с
полимером 2 и полимером 3; Vl9 V2 и V3— молярные объемы растворителя
и полимеров. Объемную долю одного из полимеров в какой-либо фазе
определяют методом/ детектирующим этот полимер и его
концентрацию, а объемную долю второго полимера находят по разности между
общим содержанием полимеров в смеси и количеством детектируемого
полимера.
Если для данной тройной смеси получена спинодаль, параметр %2з
можно рассчитать с домощыь уравнения спинодали (например, типа
предложенного Конингсвелдом и сотрудниками [155]):
(х,з - л, - х») - - ?Ь+{(^
где индекс «сп» относится к составу на спинодали.
Рассеяние света (метод Шолте [86]). Используется уравнение типа
F.46). Уравнение Флори—Хаггинса для свободной энергии смешения
берется в виде
Ш = Ё Щ Wi 1п щ + 2 Щ uiln и*
где Mi и Mf — молекулярные массы первого и второго полимеров смеси;
Wt и щ — массовые доли этих полимеров б смеси; р — плотность смеси;
AV — элемент объема; i|) = w(l — w)g; g — параметр взаимодействия,
связанный с параметром %w соотношением
Соответствующее уравнение для рассеяния света имеет вид
l l (dn\2\J_, 1 , ^il
- ю
Параметры взаимодействия тогда находят интегрированием члена
/d2 в определенных пределах концентраций.
Для двух полидисперсных полимеров в одном растворителе
уравнение Флори—Хаггинса берут в виде
[A —ш —а) 1п A—ш—«)+ 2ж;1п
здесь wi—массовая доля t-ro компонента первого полимера; w**
и V до/ — общая массовая доля первого полимера в смеси; и\ —мас-
181
совая доля другого полимера, т. е. его /-го компонента, и = ^ щ -*-
общая массовая доля полимера 2 в смеси. Член взаимодействия # (w, и)
является функцией двух концентраций — w и и.
Соответствующие дифференцирования уравнения F.52) дают
следующие выражения:
d*AG/dw* = RTpAV \j^m{ +KU], F.63)
d*AG/dwtdw, = RTAVpKn i^j, F.54)
д*АО/ди) = RTpAV [ф: + Км], F.55)
d2AG/dUidu, =ВТрАУК22{1ф)), F.56)
d*AGldwidu, = RTpAVKla, F.57)
где
к _ ' Г 1 .. д*$]
д" ~Я;[l-w-u + dS5J»
А- - 1 Г
Д22 - м0 L
_ш —ы
если предположить, что
Подставляя уравнение F.53)—F.57) в детерминант уравнения F.46)
и в выражение для Bih получаем
\b\=(RTpAV)p+«x
здесь /7 и 9 — числа компонентов в первом и втором полимерах
соответственно, и
S *f Щ в„ = (ю-рдп**- [(?)' ? «л»,
Уравнение F.46) тогда преобразуется в следующее:
х A + #ru«ML") - 2 (g) (g ^шМ'ЧАЙ»^! + Kn
X A
182
Уравнение F.58) прямо связано с производными d2ty/dw2, dty/du?
и d^/dwdu. В принципе, возможно теперь по измерению [/?ec)Je=o при
разных w и и определить после интегрирования г|? как функцию w и и-
В работе [91] предложен более простой метод оценки параметров
термодинамического взаимодействия между полимерами по рассеянию
света тройной смесью полимер — полимер — растворитель.
Приведенная интенсивность рассеяния многокомпонентной системой берется
в виде [92]
$ MiCi - 2 2 2 (?), ($, М<М,В<м + . • •, F.59)
где Вц — вторые вириальные коэффициенты; Н — константа рассеяния;
\ш) —инкремент показателя преломления компонента / смеси,
причем суммирование проводится по всем молекулам. Для тройной смеси
с монодисперсными полимерами 1 и 2 в растворителе уравнение F.59)
вместе с выражением для избыточного второго вириального
коэффициента [93]
приводится к форме
X [(?) № + Ш ^B ] + 2 [?) Ш ММДВ
F.60)
где
с=*с2-\- с3; Х{ = Ci/c (i = 2 или 3).
Тогда при условии, что
(ёJ^Ма = -(ёK^М8> F.61)
имеем
f 7J + • • •, F.62)
У = (х2М2)/(х3М3). F.63)
Видно, что второй член в уравнении F.62) содержит только Д В2В
и независимо определяемый параметр у. Таким образом, измерения
рассеяния света при условиях, удовлетворяющих уравнению F.61),
должны дать величину Д В2з-
В рамках теории Флори—Хаггинса параметр Д 523
пропорционален параметру термодинамического взаимодействия %2з между
неподобными сегментами, если удельные объемы v2 и v3 полимеров
идентичны. Поскольку это не является общим критерием, уравнения,
приведенные выше, должны быть модифицированы, чтобы определить %28
183
согласно описываемому методу. Второй вириальный коэффициент
связан с Xi2 и %1з уравнением [93]
Bti = A — Х12 - Х13 + %i№fl>№vv (xti = 0) ' F.64)
где vt — удельный объем растворителя.
Тогда при выполнении условия, данного уравнением F.64),
выражения F.60) и F.64) приводятся к следующим:
(dn/dc)\Hc/R0 = [х2М2 A + y')]-1 - Bvh23c)/[vL(l + Y'JJ + • • •,
F.65)
V' = (x2M2vl)/(x8M3vl), F.66)
(dn/dcJx2M2v2 = — (dn/dc)Bx3M3v3. F.67)
В'принципе, эти уравнения пригодны для смесей монодисперсных
полимеров. Однако, приняв Вц независимым от молекулярной массы,
можно показать, что и для полидисперсных полимеров уравнения типа
F.60), F.61)—F.63) и F.65)—F.67) законны при замене М2 и М3
на M2w и Мът соответственно. Следовательно, если измерить
рассеяние света в этих условиях, то кажущийся второй вириальный
коэффициент, получающийся из графика Зимма [94], будет пропорционален
~АВ23 или —%2з- Для этого предварительно нужно подобрать
растворитель, в котором (dn/dcJ примерно равен —{dnldcK. Например, для
систем полистирол — полиметилметакрилат и полистирол —
полиизопрен (или полибутадиен) такими растворителями являются бромбен-
зол, хлорбензол или дихлорбензол [94].
Метод Рйонг-Жун Ро [95]. Рйонг-Жун Ро показал, что для
определения параметров термодинамического взаимодействия между двумя
полимерами эффективно использовать полимер-сополимерные смеси,
один из компонентов которых — тот же гомополимер. При этом опре-
деляется не параметр %ав, а параметр Аав = у—, согласно уравнению
AG = RT [(ТОф! In ф1 + A/К2) ф2 In ф2] + Лф1Фа.
Из фазовых диаграмм находят величину Л12 для смесей сополимеров
или смесей полимер — сополимер, которая связана с параметром
взаимодействия полимер — полимер Л^я соотношением
Л12 = Лдв {f а, — ?а2J = ААВ (?в — /в2J,
где fA i и [в. — объемные доли сомономеров А и В соответственно
в Ш компоненте. Тогда
f
для смесей сополимеров или
ААв = A12/f%t
для смесей полимер — сополимер.
Обращенная газовая хроматография [77, 96]. В основу этого
метода положено уравнение Флори—Хаггинса для химического
потенциала тройной смеси полимер — полимер — растворитель [88], приме-
184
ненное Томпа [14] и Скоттом [13], и которое в условиях бесконечного
разбавления (q>i>0) при введении активности может быть записано
в виде
= [ 1 - (гг/г%)] Фа + [ 1 — (/угаМ Ф8 +
F.68)
где ъ — количество сегментов в компонентах, которое в случае
полимолекулярных полимеров выбирается как среднечисловая величина
гг = (Vi)n/v ((Vi)n и v—молярные объемы компонента i и сегмента,
выбранного одинаковым для компонентов/ и/смеси);
х*/—традиционный параметр термодинамического взаимодействия Флори—Хаггинса,
определяемый соотношением
(Х//)флоРи = rtzAwij/kT. F.69)
При таком определении %ц пропорционален числу сегментов
компонента / и, следовательно, (х*/)флоРи ф (х</)томпа. С другой стороны,
Томпа использовал величину %,/, отнесенную к единичному сегменту
компонента i или /, которые выбираются одинаковыми по объему,
следовательно, %и симметричен (безразлично, какой сегмент считать
первым):
() Томпа = ZkWij/kT = (Х*/)флорн/Г*.
Величина (х//)томпа неудобна тем, что существенно зависит от
произвола в выборе значения объема сегмента v. Поэтому взаимодействие
обычно относят к единице объема компонента:
Используя выражение для lnl^-1 метода обращенной газовой
хроматографии [77, 96]
IV
'«(I)'
(см)
и комбинируя F.68) с F.69), Д. Паттерсон и сотрудники [77]
получили уравнение
Хш
8 - ?, (Вц - V,); F.70)
здесь Vg(cu) — удерживаемый объем сорбата смесью двух полимеров;
ф2, ф3 и У2> vs— объемные доли и удельные объемы полимеров; w2
и w3— их весовые доли в смеси; Р1— давление насыщенного пара
сорбата при температуре измерения; Vx и Vt— молярные объемы сорбата
и полимеров; Вп— второй вириальный коэффициент сорбата. Значе-
185
ния %12 и %1S рассчитываются из данных хроматографии в раздельных
экспериментах по уравнению
Комбинируя F.71) с F.70), легко получить уравнение, удобное для
практического использования:
ln V* (см) +2 Ф* In % - In 2 wfit]9 F.72)
где Vgi— удерживаемые объемы сорбата индивидуальными
компонентами смеси. Как видно из последнего уравнения, для оценки
параметра %2з*достаточно определить удельные удерживаемые объемы сорбата
смесью и отдельными компонентами смеси.
На основании формулировки теории Флори—Пригожина некомби-
наториальную свободную энергию Гиббса на грамм тройной смеси
можно соотнести с размерностью приведенной величины:
где U*— параметр приведения на 1 г для величин, имеющих
размерность энергии:
U* = w^lfi + w2U* + w3Ut -wx®<pXX12 - ^03t;iX13 — w2®8v*2X23-
F.73)
(v* — параметр приведения объема; Si — молекулярная поверхность,
деленная на объем).
Величину U* можно записать также в членах параметров
приведенного давления и объема;
Hi =PV
Параметр Хц является параметром контактного^ взаимодействия,
а приведенная температура тройной смеси связана g ft чистых
компонентов соотношением
Приведенная температура смеси обозначается через То.
Дифференцируя уравнение F.73), Паттерсон и сотрудники [77] получили
уравнение для расчета параметра контактных взаимодействий в виде
*- - Ш ®>+fir) @з - fe)H [
+ [I/ (То) - U (Т,) + ЪТг {S(f0 - 5 (f,)} J [^Щ F.74)
В модели Флори получаем параметры U = — F-1; S = ln^1/3— 1);
Т = ~4/з— , которые поставляются в уравнение F.74). Величины Х12
186
и Х18 для индивидуальных компонентов рассчитываются по уравнению
типа
Хи - {хн-
6.3.3. ИСПОЛЬЗОВАНИЕ МАЛОУГЛОВОГО РАССЕЯНИЯ
РЕНТГЕНОВСКИХ ЛУЧЕЙ И ТЕПЛОВЫХ НЕЙТРОНОВ
ДЛЯ ОЦЕНКИ СОВМЕСТИМОСТИ ПОЛИМЕРОВ
В последние годы для оценки термодинамических свойств смесей
полимеров начали успешно использоваться методы малоуглового
рассеяния рентгеновских лучей и тепловых (медленных) нейтронов
[101—103]. В первом случае в цепь одного из смешиваемых полимеров
необходимо ввести тяжелые атомы (например, хлора или брома), а во
втором случае один из смешиваемых полимеров следует дейтерировать.
В качестве критерия термодинамической совместимости полимеров
в данном случае служат определяемые этим методом вторые вириаль-
ные коэффициенты Л2, а также рассчитываемые на их основе
параметры термодинамического взаимодействия %12. Кроме того, по характеру
температурной зависимости Л2, а также определяемых этими
методами размеров макромолекул можно, в принципе, судить о наличии той
или иной критической температуры смешения. Значения Л2 и размеры
макромолекул оцениваются этими методами так же, как и методом
рассеяния света, т. е. путем построения графика двойной экстраполяции
Зимма [94]. Преимуществом этих методов по сравнению с методом
светорассеяния для оценки Л 2, размеров макромолекул и молекулярной
массы является то, что они пригодны для исследования смесей (и
растворов) олигомеров. Это связано с тем, что в данном случае длина
волны соответствующего излучения (рентгеновских лучей ~ 0,15 нм,
тепловых нейтронов ~ 1—1,5нм) на несколько порядков меньше
длины волны видимого излучения, поэтому выше их разрешающая
способность.
Обработку данных по результатам экспериментальной оценки
второго вириального коэффициента А2 обычно проводят в рамках
уравнения типа
A2 = vlVTl A/2-rlXl2), F.75)
где %i2~ So—gi> ^2—удельный объем полимера 2; Vt—молярный
объем первого полимера, пропорциональный гг (степени
полимеризации р). Из уравнения F.75) видно, что вторые вириальные
коэффициенты в полимерных смесях должны быть очень малыми из-за необычно
больших значений Vx и зависеть от молекулярной массы матрицы
(растворителя), т. е. от rv Обсуждая полученные результаты в
приближении, что %i2 не зависит от ф, авторы [28] сравнили экспериментальные
значения А2 с рассчитанными на основании уравнения F.75), полагая
%12= 0 или выразив теплоту смешения через параметр
растворимости 6. Найдено, что в обоих случаях рассчитанные значения Л2 суще-
187
Таблица 6.5. Значения второго вириального коэффициента Л2 и параметра
термодинамического взаимодействия для смесей деитерированного полиметилмё-
такрилата с сополимером стирол — акрилонитрил, полученные методом малоуглового
рассеяния нейтронов [28]
Содержание
акрилонитрила
в
сополимере, %
19
19
10
28.7
Л1Ш.1О—*
2.7
4.4
0.7
2,2
Л2.10*C83 К) (м«.кг—2.моль)
ментальные
1,15
1,15
—1,00
0.52
для атерми-
ческой смеси
по уравнению
F.75) при
0.02
0,1
0.06
0.02
из Ь
—1.88
—1,89
-0.08
-5.98
Xlt
полученный из Л е
(эксперимент)
—0.0118
—0.0119
+0,0110
—0,0052
*крит
0.0007
0,0005
0.0029
0.0010
ственно расходятся с экспериментальными данными. По
экспериментальным значениям А2 Конингсвелд и сотрудники [28]
рассчитали значения %12 и нашли их
нереально завышенными (по
сравнению с критическими значениями %12
(табл. 6.5), но, использовав
предпосылки теории Хаггинса, рассчитали
значения р и б и обнаружили, что
экспериментальные значения А2
хорошо коррелируют с
рассчитанными величинами %12, если для
определения последних
использовались параметры кх и k2 теории
Хаггинса, равные 0,6 и —0,4 (рис,
6.20).
О других методах оценки
термодинамической совместимости и
надежности критериев такой оцен-
Рис. 6.20. Зависимость Л2 от М
второго полимера для системы дейтериро-
ванный полиметилметакрилат —
сополимер стирола с акрилонитрилом,
рассчитанная по формуле F.5) при
0 G) 002 (J?) 000
считанная по формуле «м,, при х„ ™ с их помощью будет сказано в
равных о (/), —0,02 B), —0,006 C, разделе
р ()
и —0,009 D).
Точки — экспериментальные данные,
штриховая линия — расчет по теории Хаггинса
с kx = 0.6 и ?2=0,48 [28].
по экспериментальным
данным исследования
термодинамических свойств смесей
полимеров.
6.4. НЕКОТОРЫЕ ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ
ПО ИССЛЕДОВАНИЮ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
СМЕСЕЙ ПОЛИМЕРОВ
6.4.1. ОСОБЕННОСТИ ТЕРМОДИНАМИЧЕСКОГО
ПОВЕДЕНИЯ СМЕСЕЙ ПОЛИМЕРОВ
Основные задачи • исследования свойств полимерных смесей —
выяснение их термодинамической совместимости, а также ее зависимости
от состава, температуры, молекулярной массы и химической природы
смешиваемых компонентов.
188
Первой работой в этом направлении следует считать работу Добри
и Бойер-Кавеноки [50], которые оценили совместимость 98 полимерных
пар. В качестве критерия термодинамической совместимости двух
полимеров эти авторы рассматривали прозрачность растворов смеси двух
полимеров состава 1 : 1 в общем растворителе и пленок, полученных
из таких растворов. Из 98 изученных ими полимерных пар лишь около
10 % оказались, по их данным, термодинамически совместимыми.
Прозрачность пленок смеси двух полимеров, полученных либо из
расплава, либо из раствора в общем растворителе, в качестве критерия
совместимости использовали и авторы других работ [50—54]. В
некоторых случаях были получены и тройные фазовые диаграммы [54, 56—
59, 104]. В основном же о термодинамической совместимости полимеров
вудили по данным динамических механических измерений,
дифференциальной сканирующей калометрии, термомеханики, деструкции,
вязкости растворов смесей полимеров или вязкости их расплавов,
изменения плотности и др. Результаты большинства из этих работ, а
также соответствующая библиография приведены в обзорах [15, 105]
и справочнике [106].
Характеризуя результаты этих работ в целом, отметим следующее.
Как показано в параграфе 6.2 этой главы, несовместимость двух
полимеров, обнаруживаемая в данном растворителе, еще не означает, что
два смешиваемых полимера будут несовместимы без растворителя
и наоборот [67, 70, 71]. Пленки прозрачны, если показатели
преломления смешиваемых полимеров одинаковы или близки, несмотря на то,,
что полимеры несовместимы; т. е. результаты этих работ можно
рассматривать лишь как качественные. Чтобы более надежно судить о
термодинамической совместимости полимеров по результатам
исследования тройных растворов полимер — полимер — растворитель,
желательно иметь полную фазовую диаграмму. Особенно ненадежны
данные исследования диэлектрической релаксации, динамических
механических свойств и в некоторых случаях калориметрии (ДСК), поскольку
они зависят от условий приготовления образцов [107]. Из-за высокой
вязкости расплавов смесей полимеров или растворов смесей полимеров
в общем растворителе на конечных стадиях испарения последнего ва
многих случаях может сформироваться структура, характерная для
исходного расплава или раствора смесей полимеров в общем
растворителе. В частности, если два полимера совместимы в расплаве или
в общем растворителе, быстрое охлаждение расплава или быстрое
удаление растворителя приведет к тому, что сформированный образец,
будет проявлять свойства, типичные для совместимой системы, если
в расплаве или растворе два полимера были совместимы [96] из-за
«вынужденной» совместимости компонентов, если при комнатной
температуре эти полимеры несовместимы. При фиксированной структуре
переохлажденного расплава или раствора все зависит от кинетики
фазового расслоения в процессе охлаждения расплава или испарения
растворителя. В другом случае, если два полимера совместимы без
наличия растворителя, но в каком-либо из растворителей они
несовместимы, то из-за кинетических эффектов пленка может иметь
неравновесную структуру, отвечающую условиям несовместимости. Очень
189
наглядный пример этому —данные работы [71], детальное
обсуждение которой приведено в параграфе 6.2. При малых концентрациях
одного из полимеров при фазовом расслоении по ну^леационному
механизму (см. главу 8) формируется высокодисперсная структура [108],
которая может и не обнаружить себя в виде второго релаксационного
перехода при динамических механических измерениях, либо второго
излома на кривых ДСК, и др. Следовательно, к данным такого рода
необходимо подходить достаточно осторожно.
Много работ посвящено оценке тепловых эффектов процесса
смешения полимеров [2, 3, 65]. Впервые такие оценки для большого числа
полимерных смесей проведены группами во главе с Г. Л. Слонимским
{2, 3],С. М. Липатовым [9], С. С. Воюцким[109] и др. [110, 111]. В
частности, Г. Л. Слонимский и сотрудники [2, 3] обнаружили, что не во
всех случаях условие А Н < 0 отвечало наличию термодинамической
совместимости компонентов. С. М. Липатов [9] высказал
предположение, что в процессах совместимости полимеров важная роль должна
принадлежать энтропии. Флори и сотрудники [4] расчетным путем
показали, что при небольших экзотермических тепловых эффектах
вклад некомбинаторной энтропии смешения (в их случаях она
оказалась отрицательной) может привести к A G > 0, что обусловливает
несовместимость двух полимеров. Экспериментально энтропия
смешения двух полимеров впервые была определена А. А. Тагер и
сотрудниками [6—8]. Оказалось, что для термодинамически совместимых систем,
с которыми они работали, энтропия смешения была отрицательной, что
послужило А. А. Тагер основанием предложить в качестве
необходимого условия совместимости двух полимеров отрицательное значение
энтропии смешения [6]. Между тем в работе [112] на примере системы
полистирол — полибутилметакрилат отмечалось, что с уменьшением
молекулярной массы одного из полимеров параметр %23 становился
отрицательным, т. е. наблюдалась совместимость этих полимеров.
Когда молекулярные массы велики, параметр Х23 мал, улучшение
совместимости их при уменьшении молекулярной массы одного из
полимеров свидетельствует о существенном вкладе энтропии, которая
должна быть в общем положительной. С. М. Липатов [9] связывал этот
энтропийный вклад с различием в плотностях упаковки исходных
компонентов и смеси.
Вывод о том, что для совместимых полимерных пар энтропия
смешения должна быть отрицательной, связан с наблюдаемыми для таких
систем отрицательными значениями избыточного объема смешения
А Vе [115—118], который связан с энтропией смешения (формула
Гильдебранда и Скотта [25]).
- ..., F.76)
где Д Sy может рассматриваться как комбинаторная энтропия
смешения, значениями которой в случае смесей высокомолекулярных
образцов можно пренебречь [90]. Тогда уравнение F.76) записывается в виде
190
Поскольку {dPIdTY для большинства веществ положительно, то
из F.77) следует:
A Sp^9 если А Vе ^ О,
т. е. для систем, где А Vе < 0, можно ожидать, что и ASP будет
отрицательной.
Следует отметить, что сопоставление значений AVE с
термодинамической совместимостью смесей полимеров было сделано в основном для
систем с НКТС, где теория действительно предсказывает АУ?<0 и
ASp<0 [27]; в случае же, если для смешиваемых систем А Vе> 0,
ASP тоже будет больше нуля.
Теплоты смешения полимеров были определены и в работах [ПО,
111]. Наибольшее же количество работ посвящено определению
параметров термодинамического взаимодействия между компонентами
полимерных смесей, т. е. %23,. Многие из экспериментальных данных
приведены в табл. 6.6; обращает на себя внимание тот факт, что значения
параметров %23, полученные при использовании общего растворителя,
очень малы. Для объяснения этого факта Аллен, Джи и Нихольсон
[90] предположили, что в тройных растворах полимер — полимер —
растворитель два полимера реально полностью не смешиваются, т. е.
число контактов 2—3 намного меньше, чем должно быть при свободном
смешении. Стремление избежать перекрывания клубков должно
приводить к их отталкиванию и тенденции к сегрегации двух полимерных
компонентов. Ямакава и Фуджии [119] полагают, что взаимодействие
полимерных сегментов в разбавленном растворе отличается от
такового в случае малых молекул и приписывают это меньшему, чем у
несвязанных в цепочку малых молекул, числу степеней свободы. Во всяком
случае, для полимерных пар параметр Х23 (на сегмент) очень мал даже
при критических условиях и уменьшается с увеличением
молекулярной массы смешиваемых полимеров (см. табл. 6.6). Эти факты
согласуются с выводами Флори [88] о том, что полимеры с высокими
молекулярными массами совместимы в основном при отрицательном %23,
т. е. когда взаимодействия типа 2—3 преобладают над
взаимодействиями 2—2 и 3—3, что может наблюдаться при наличии в системе
специфических взаимодействий.
В работе [55] обнаружено, что %23 уменьшается с М2 при М3= const,
%23 сильно зависит от М в области малых М и слабо зависит от него
в области больших М (см. табл. 6.7); это было объяснено энтропийным
вкладом в %2з- Сильная концентрационная зависимость Х23 отмечена
в работе [61, 120, 125] (см. табл. 6.8).
Для некоторых систем параметры Х23 были определены по депрессии
температуры плавления (см. табл. 6.6, рис. 6.21); они оказались
отрицательными, что, впрочем, и неудивительно, так как все системы, для
которых %23 находили этим способом,— это системы с НКТС и со
специфическими взаимодействиями между компонентами. Значения
параметров %23 вычисляли по уравнению F.51), которое в общем случае не
предусматривает концентрационной зависимости параметра %23 при
достаточно больших молекулярных массах смешиваемых полимеров
(второй член в правой части мал) и отличается от аналогичного уравнения
191
Таблица 6.6. Значения параметров термодинамического i
разных систем
Система
Полистирол A,94) — полиизобутилен
A,5) —толуол
Полистирол E,26) — полиизобутилен
F.7) — толуол
Полистирол B4,0) — полиизобутилен
B4,4) — толуол
Полистирол — полибутадиен
Полистирол — полиметилметакрилат
Полистирол A0,0) — полиизобутилен
A5,0)—ССЦ
Полистирол — поливинилметиловый
эфир
Атактический полистирол B,07) — изо-
тактический полистирол E,9)
Изотактический полистирол — поли-2,
6-диметилфениленоксид
Полистирол — полибутилметакрилат
(высокие М)
Поливинилхлорид — полибутилакрилат
Поливинилхлорид—полипропилакрилат
Поливинилхлорид — полиэтилметаклат
Поливинилхлорид — полипропилмет-
акрилат
Поливинилхлорид—полибутилметакрилат
Поливинилхлорид — полипентилметакри-
лат
Полиметилметакрилат — поливинил-
иденфторид
Полиметилметакрилат (изотактиче-
ский) — поливинилиденфторид
Полиметилметакрилат (атактический)—
поливинилиденфторид
Полиметилметакрилат (синдиотактиче-
ский) — поливинилиденфторид
Полиэтилметакрилат — поливинилиден-
Лтооил
Полиэтиленоксид @,006) —
полипропилен оксид @,01)
Полиэтиленоксид @,006) — полипропи-
леноксид @,015)
Полиэтиленоксид @,006) — полипропи-
леноксид @,0202)
Полиэтиленгликоль @,15) — полипро-
пиленгликоль @,0105)
Х28 (на сегмент)
0,0289
0,0314—0.0405
0,0163
0,0181—0,0224
0,008
0,007—0,0106
0,03
0,01
0,11
От—0,75 до —0,2
(в зависимости от
состава)
От —0,04 до 0.4
(в зависимости от
состава)
0
0,1
0,98
0,38
—3,03
—1.52
—0,96
—0.02
—0.295
—0.13
—0,10
—0,06
—0,34
0,13
0,16
0,17
0,12
1заимодействия для
Метод оценки
По бинодали [121]
По спинодали [121]
По бинодали [121]
По спинодали [121]
По бинодали [121]
По спинодали [121]
Цит. по [122]
Цит. по [122]
[90]
По сорбции паров
бензола [61J
По депрессии Т^
[123]
То же [124]
Обращенная газовая
хроматография [ 112]
То же [125]
»
ъ
»
По депрессии Гпл
[89]
То же [126]
»
» [80]
Из фазовых
равновесий [97]
То же
»
Обращенная газовая
хроматография [96]
Примечание. Цифры в скобках — молекулярные массы полимеров (ЛЬ 10е).
192
Таблица 6.7. Зависимость
параметров термодинамического взаимодействия
от молекулярной массы смешиваемых
полимеров [55]
Таблица 6.8. Зависимость
параметра Х2з °т состава для смесей
полистирола с поливинилметиловым
эфиром при 303 К [61]
10~*
полистирола
М*\0—* поли-
диметилсилок-
сана
Содержание ПВМЭ,
% (по массе)
2.7
1,35
0.6
Бензол
0,011
0,0185
0,045
0.057
0,011
0,0185
0,045
0,011
0,0485
0,078
1.71
Этилац етат
0,22
0,16
0,15
0,115
0,25
0,205
0,185
0,255
0,185
0,155
0,145
35,06
45,30
55,0
65,0
—0.75
—0.69
—0,60
—0,60
0.6
0,011
0,045
0,057
1.71
0,44
0,315
0,255
0,195
Флори для систем полимер —
низкомолекулярный растворитель [88]
знаком в правой части. Из этого
следует, что в случае
положительных значений параметра %23 для
расплавов смесей полимеров
должно наблюдаться повышение их
температуры плавления по сравнению
с таковой компонента с более
высокой 7"пЛ. В настоящее время
данных такого рода пока нет.
Имеющиеся же результаты показывают
либо понижение температуры
плавления смеси [89, 123—126], либо ее постоянство [124]. В нескольких
работах [112, 120,123,127] отмечена существенная концентрационная
зависимость параметров термодинамического взаимодействия,
которая не предусматривается уравнением F.51). Исключением
является работа [123], где определенные по депрессии температуры
плавления параметры %23 для смесей атактического и изотактического
полистиролов с Mw = 2,07 • 105 и Mw= 5,9 - 10* соответственно
изменяются от —0,04 до 0,4 в диапазоне содержания атактического
полистирола примерно от 0,225 до 0,5 % (по объему).
Некоторые значения параметров %23 были определены методом
обращеннэй газовой хроматографии (см. [126] и табл. 6.6). Обнаружены
сложные концентрационные (рис. 6.22—6.24) и температурные [112,
120] зависимости параметров термодинамического взаимодействия,
особенно для смесей олигомеров, к рассмотрению которых мы перейдем
в следующем параграфе.
Относительно достоверности данных о термодинамической
совместимости полимеров, получаемых косвенными методами (оценки
прозрачности пленок, наличия одной или нескольких Гст и др.) было уже
сказано раньше. В случае же использования для оценки
термодинамической совместимости параметров %2з или A G необходимо отметить
следующее. Как правило, их значения (то же, кстати, относится и к
оценке А Н) вычисляют при одной какой-либо температуре и
определенном составе смеси. Если при данном составе %2з<0 или AG <0,
4-420
193
о
-0,1
-0,2
•0,3
-0,4
'0,5
-0,д
л-р-лл,лэс,лэд
\ллл
лкл\
Несодместимь!
- **
Совместимы
\лмлпл
1 1
0,2
О
-0,2
-0,4
5 6 7
CHJCOO
20 40 60 Ыпс
Рис. 6.21. Зависимость параметров Х2з от числа алифатических
углеводородов на эфирную связь в структуре полиэфира для смесей полавинилхлорида
с поли-Р-пропиолактоном (я-р-ПЛ), полиэтилвинилсукцианатом (ПЭС),
полиэтилена дипинатом (ПЭА), полипивалолактоном (ППЛ), поли-а-метил-а-я-про-
пил-Р-пропиолактоном (ПМППЛ), полибутиленадипинатом (ПБА), поли-е-кап-
ролактоном (ПКЛ) и полигексаметиленсебацинатом (ПГС) [49].
Рис. 6.22. Концентрационная зависимость параметра Х2з Для системы
полистирол (Мп = 1,709 • 103) — полибутилметакрилат (^«=3,75 • 104, Mw=3,2.105)
при 413 К [112].
0/2
0,4
0,6
ЛИГ
330
350 370
5
390 Т,К
Рис. 6.23. Концентрационные (а; и температурные (б) зависимости параметра
9С2з Для системы полипропиленгликоль (М = 1050)—полиэтиленгликольадипинат
(Ж = 2000):
а: 7 — 335; 2 — 330,8; 3 — 328,2 К; б: 7— 88,0; 2 — 78,0; 3 — 65,0% ПЭГА в смеси [120].
то система термодинамически совмес- %гз
тима, и наоборот. Но при данной 02
температуре совместимость
компонентов является функцией состава (см.,
например, фазовую диаграмму на
рис. 6.2), поэтому система, совмести-
мая при одном составе, может быть
несовместимой при другом, и наобо-
рот. Следовательно, для суждения
о термодинамической совместимости
двух полимеров желательно иметь
полную концентрационную зависи-
мость Хгз или Д G- Но тут возникает
вопрос, а что же определяет на самом
деле значение %2з или A G? Согласно
законам термодинамики условие
AG < 0 характеризует процесс
образования термодинамически
устойчивой системы, критерием устойчивости
которой является d2A Gldq>\ = 0. Под
совместимостью же, как правило,
понимают молекулярное диспергирование полимера в среде другого
полимера. Но поскольку условие A G < 0 — критерий протекания
любого самопроизвольного процесса, приводящего к образованию
термодинамически устойчивой системы, оно может характеризовать
и процесс, например, самопроизвольного эмульгирования одного
полимера в среде другого полимера, приводящего к образованию
термодинамически устойчивых коллоидных систем.
Рассмотрим, каким же образом процесс самопроизвольного
эмульгирования влияет на термодинамические функции. Для этого примем
A28], что термодинамические параметры системы изменяются не
только в результате диспергирования (например, при растворении
полимера в низкомолекулярном растворителе или среде другого полимера),
но и из-за того, что при образовании новой межфазной поверхности при
диспергировании часть вещества дисперсионной среды и
дисперсионной фазы переходит в межфазные слои. Произведем учет изменения
энтропии системы при упомянутом переходе, используя известную
в термодинамике поверхностных явлений зависимость межфазного
натяжения у от температуры и состава смеси
о,1
-0,1
-0,2
-0,3
-0,4
320 340 360 380 400 420 Т,К
Рис, 6.24. Температурные и
концентрационные зависимости %2з для
системы полипроленгликоль (М =
= 1050) — полиэтиленгликоль (М =
= 1,5-10*) при 12 A), 31 B), 41 C)
и 69 D) % ППГ в смеси [120].
ady = (s12^- —.
F.78)
В этом уравнении а — молярная поверхность (поверхность,
приходящаяся на 1 моль вещества в межфазном слое), S12 и у12— изменение
энтропии и объема при переходе 1 моля вещества из фазы 1 в фазу 2
195
соответственно; SlT и y1T— то же при переходе из фазы 1 в межфазный
слой; Nt— мольная доля 1-го компонента. Верхние индексы
обозначают A) — фазу 1, (у) — межфазный слой, G — мольный
термодинамический потенциал Гиббса.
В нашем случае будем считать фазу 1 дисперсионной средой, 2 —
дисперсной фазой. Для бинарной смеси уравнение F.78) переходит
в изобарических условиях в выражение
^«- FJ9)
Для определенности пронумеруем компоненты так, чтобы первый
образовывал дисперсионную среду, а второй — дисперсионную фазу.
Для несовместимых бинарных полимерных смесей, где N\= 1, в фазе
2 — ее поверхностном слое — первый компонент практически
отсутствует (т. е. N\= N^ = 0) и оба компонента нелетучи, уравнение
F.79) принимает наиболее простой вид
где аг и S\ — молярная поверхность и мольная энтропия межфазного
слоя со стороны фазы 1. Аналогичное уравнение получается и для
фазы 2, поэтому
AS(t) = dy/dT,
где
AS(t) = [(S< о + 5B>) - (S^ + SBT))]/(^ + a2) F.80)
есть изменение энтропии двухфазной полимерной системы при
образовании единицы поверхности межфазного слоя (поверхностная
плотность изменения энтропии). Как видно, для определения энтропии
необходимы экспериментальные измерения зависимости межфазного
натяжения от температуры.
Если воспользоваться уравнением, описывающим изменение
свободной энергии AG системы при диспергировании
AG = -TAS* + Y ?Ait F.81)
то, учитывая изменение энтропии за счет образования межфазных
слоев, получим
k+(y )S
F.82)
В уравнениях F.81) и F.82) ASk — изменение комбинаторной
энтропии при диспергировании, А{ — площадь поверхности i-й
частицы в дисперсной фазе.
Уравнение F.82) существенно меняет наши представления о
процессах, приводящих к образованию устойчивых дисперсных систем.
196
Действительно, поскольку в имеющихся до сих пор случаях величина
dy/dT для полимерных пар отрицательна (см., например, [106]), то
даже без учета вклада комбинаторной энтропии в соответствии с
уравнениями F.80) и F.82) значение A G будет уменьшаться с самого
начала диспергирования, если выполняется условие
TAS(t)>y. F.83)
Как видно из [106], для всех исследованных полимерных пар dy/dT
находится в пределах от—0,002 • 10Дж/м2 К в области температур
от 400 до 500 К. Вычисленные значения ТА SW по этим данным равны
@,8—10) • 10 -3 м2, что близко к величинам межфазного натяжения
с учетом разброса экспериментальных
данных. Из этих рассуждений следует,
что процесс самопроизвольного
эмульгирования (диспергирования) — не
такое уж редкое явление при
формировании бинарных полимерных смесей. Из
условия F.83) вытекает, что
диспергирование облегчается с ростом
температуры, так как левая часть неравенства
при этом растет, а правая (межфазное
натяжение) падает.
Остановимся теперь на причинах,
препятствующих самопроизвольному
диспергированию до уровня отдельной
молекулы. Для этого рассмотрим
простейшую модель сферической частицы
дисперсной фазы (рис. 6.25,я).
Поскольку толщина межфазного слоя т
определяется характеристиками межфазного
взаимодействия, то в первом
приближении она не зависит от радиуса частицы г.
Поэтому при диспергировании доля меж-
\
\
\
\
\
\
\
Ты
\
1-
\\
*^» ^_
г
а
—.
=^\
1I
&>'
0,1 0,2 0,3 О А
5 I
Рис. 6.25. Модель сферической
частицы радиуса г с граничным
слоем со стороны дисперсионной
среды (та) и частицы (т«) (а),
а также изменение допустимой
степени диспергирования с
увеличением содержания дисперсной
фазы (б).
фазных слоев в общем объеме системы растет, т. е. увеличивается
отношение т/г. .Отсюда понятно, что при определенной степени
дисперсности и полимер дисперсионной среды, и полимер дисперсной фазы
переходят целиком в межфазный слой, вследствие чего уменьшается,
а затем и исчезает энтропийный выигрыш перехода полимера в
межфазный слой. Для полимера дисперсной фазы условие перехода его
в межфазный слой в соответствии с рис. 6,25, а определяется
соотношением х //¦ =¦ 1
Для полимера дисперсионной среды аналогичное условие можно
получить с применением модели в виде кубической решетки, в узлах
которой расположены частицы дисперсной фазы одинакового радиуса,
Поскольку для данной модели число частиц дисперсной фазы (число
узлов решетки) в единице объема определяется выражением
F.84)
197
то объем полимера дисперсной среды, находящегося в межфазном слое,
можно оценить из уравнения.
Ф2 [у я {г + тх) - j я/-3] /1 яг3. F.85)
Переход полимера дисперсной среды в межфазные слои
прекращается при перекрывании (касании) этих слоев для соседних частиц,
откуда с учетом плотнейшей упаковки получаем
Из соотношений F.85) и F.86) видно, что если для дисперсной фазы не
имеет значения, какова объемная доля компонента, то для
дисперсионной среды ее содержание непосредственно определяет допустимую
степень диспергирования дисперсной фазы.
На рис. 6.25,6 показано, как изменяется допустимая степень
диспергирования с увеличением содержания дисперсной фазы. Условие
F.84) позволяет оценить минимальную концентрацию дисперсной
фазы, выше которой будет происходить резкий рост размеров частиц.
Это вывод объясняет причину образования дисперсных систем чаще
всего в области малых содержаний одного из компонентов смеси для
случая, если устойчивость дисперсной системы определяется только
энтропийными эффектами, сопровождающими процесс
диспергирования. Что же касается самих значений толщин межфазного слоя, то
только адсорбционный слой полимера при адсорбции из
концентрированных растворов имеет толщину порядка 0,1 мкм, следовательно,
межфазный слой еще толще и минимальный размер частиц дисперсной
фазы для бинарных полимерных систем должен существенно
превосходить размеры отдельной молекулы.
Другая причина, вызывающая изменение услрвий при
диспергировании,— изменение кривизны граничной поверхности. Для оценки
влияния кривизны воспользуемся видоизмененным уравнением F.79),
учитывающим смещение двухфазного равновесия при наличии
искривленной поверхности [129]
df = ~ + f^
где
/(')=;*ViT)-t>i). F.89)
В этих уравнениях vx и v™ — молярные объемы вещества внутри f-ой
фазы и в межфазном слое соответственно (рассматриваются
несовместимые полимерные компоненты). Как видно из уравнений F.87)—
F.89), ожидаемый эффект зависит от знака разности v™— vx и
производной dr/dT. На основании вывода сделанного при обсуждении
соотношения F.83), можно утверждать, что знак dr/dT отрицателен. Что
198
же ка сается знака разности v^ —vu то здесь можно сослаться на
литературные данные о том, что для жидкостей рост энтропии в
поверхностном слое сопровождается падением плотности в этом слое
[ 129]. Если и для границы раздела полимер — полимер это так, то при
существенном уменьшении г величина ASW будет падать как из-за
понижения ее поверхностной плотности, так и из-за уменьшения (по
абсолютной величине) производной dyldT. В свою очередь,
уменьшение энтропийного выигрыша вызовет появление минимума на графике
зависимости A G = f (г), т. е. диспергирование прекратится.
Полученные выводы вполне согласуются с экспериментальными
результатами исследования термодинамических параметров смесей
полимеров и олигомеров, в
частности, с результатами
температурных зависимостей параметра Хгз
[120, 130, 131], по которым для
многих систем мы рассчитали энталь-
пийную (хя) и энтропийную (is)
составляющие этого параметра. Как
видно из рис. 7.10 и 7.11, при
уменьшении содержания одного из
компонентов наблюдается резкий
рост — %s и %Ну что приводит, в
конечном итоге, к %2з < 0» а
следовательно, и к AG < 0. Резкое
увеличение некомбинаторной энтропии
смешения (ASHK = — RT%$yl),
следовательно, обусловлено
протеканием процесса самопроизвольного
эмульгирования, в результате которого
растет величина доли межфазной
области.
340
330
320
0,2 0,4 0,6 0,8
Рис. 6.26. Зависимость температуры
плавления A) и параметров
термодинамического взаимодействия от
концентрации для системы ПЭГ-2000 —
ПЭГ-40000, полученных по депрессии
температуры плавления B) и методом
обращенной газовой хроматографии C)
[120].
Таким образом, для достоверности суждения о том, действительно
ли при %23 < 0 или AG < 0 происходит молекулярное диспергирование
одного из компонентов смеси, необходимы дополнительные данные,
например о размерах макромолекул. Для смесей полимеров такие
данные могут быть получены методом малоуглового рассеяния
рентгеновских лучей или тепловых нейтронов [44]. Впрочем, как уже
отмечалось, эти методы могут быть использованы и для определения второго
вириального коэффициента, по которому можно оценить значения %2з-
Например, в работе [101] с помощью методов малоуглового рассеяния
тепловых нейтронов было показано, что для смесей полийодстирола
с полистиролом А2< 0, а радиус инерции полийодстирола в среде
полистирола растет с его концентрацией, т. е. рост частиц
полийодстирола с концентрацией при Л2<0 свидетельствует о существовании
кластеров (агрегатов) полийодстирола в среде полистирола.
Многие авторы оценивали значения параметров %23 по депрессии
температуры плавления [123, 124, 126]. Несмотря на предсказуемость
термодинамической совместимости компонентов (см., например, рис.
199
6.21), этот метод все же не дает полной картины термодинамического
поведения системы во всем интервале концентраций. Это связано с тем,
что по уравнению F.51) нельзя судить о концентрационной
зависимости %23 (рис. 6.26).
Таким образом, для оценки термодинамической совместимости
двух полимеров по различным методикам (кроме построения фазовых
диаграмм) необходимо, во-первых, исследовать весь диапазон
концентраций (в случае, если речь идет о %2з или АО)» во-вторых, получить
разными методами совпадающие характеристики, определяющие
наличие совместимости. Особенно важны бинодали и спинодали, так как,
помимо фазового состояния системы, эти характеристики
представляют собой ценный материал для исследования механизмов фазового
расслоения.
В большинстве случаев фазовые диаграммы были получены для
тройных смесей полимер — полимер — растворитель. Многие из
таких диаграмм приведены в Справочнике по физической химии
полимеров [96], поэтому здесь не рассматриваются. Отметим лишь, что
обнаружены системы с ВКТС [57, 58, 104], НКТС [56], а также получены
диаграммы замкнутого типа [59, 67]. Во многих случаях полученные
диаграммы согласуются с предсказаниями теории Скотта [13], Земана
и Паттерсона [67], рассмотренными в параграфе 6.2 этой главы.
Экспериментальных фазовых диаграмм для систем полимер —
полимер в настоящее время очень мало. Это связано с большими
экспериментальными трудностями их получения: высокими температурами
перехода полимеров в вязкотекучее состояние, высокой вязкостью
расплавов, необходимостью работы с оптически прозрачными
образцами и др. В основном обнаруженные совместимые полимерные смеси
характеризуются наличием НКТС [27, 105]. Это даже послужило
основанием Мак Мастеру сделать вывод о том, что все совместимые
системы должны иметь НКТС [27]. Однако в последние годы были
найдены и системы с В КТС [28, 34, 132], причем даже с двумя КТС [133],
а также диаграммы типа «песочных часов» [133], т. е. практически все
типы фазовых диаграмм, характерных для обычных систем полимер —
растворитель.
Зависимость фазового состояния от состава и молекулярной массы
смешиваемых полимеров, как следует из этих работ, хорошо
описывается существующими термодинамическими теориями растворов
и смесей полимеров. Однако обнаружены и некоторые особенности
поведения таких систем, не укладывающихся в рамки существующих
теорий, причем как старой теории Флори—Хаггинса, так и новых
статистических теорий: бимодальность фазовых диаграмм [28] и
смещение фазовой диаграммы в область концентраций, обогащенную
полимером с большей молекулярной массой [134].
Интересные результаты описаны в статье [134]. Ее авторы заметили,
что для смеси поливинилиденфторид — полиэтилметакрилат (М
соответственно равна 100 000 и 4 000) критическая концентрация смешения
находится в области составов фаз, обогащенных поливийилиденфтори-
дом, несмотря на то, что его молекулярная масса намного больше
молекулярной массы полиэтилметакрилата (рис. 6.27, а). Т. е. наблюдается
200
картина, прямо противоположная предсказаниям старой теории Фло-
ри—Хаггинса и новых статистических теорий растворов полимеров,
а также экспериментальным результатам по исследованию фазового
состояния растворов полимеров в низкомолекулярных жидкостях (см.
параграф 2.5). Авторы работы [134] показали, что такое поведение
системы можно описать в рамках модифицированной теории Флори,
которая состоит в следующем. В уравнении состояния системы число
внешних степеней свободы принимается не аддитивным (вспомним, что
600
475
450
375
550
450
0,2
0,6
350
О 0.2 0,4 6,6 0,8 1J0
ь/2,П9МЙ
в
Рис. 6.27. Экспериментальная (а) и теоретические (б) фазовые диаграммы
для системы поливинилденфторид (М =* 1 • 10$) — полиэтилметакрилат (М =
=*4«103), рассчитанные при Х12 равных —-0,5 (/), —1,0 B), —1,5 C), —2,0
D), —2,5 E), а также /?1/р2 = 0,66 и ci2 = 0,02 [134].
Сплошные линии — для SJSt = 0,55, штриховые •— для SJSt = 1,0.
это приближение использовал Хамада для описания свойств растворов
полимеров [135]). Число степеней свободы на сегмент смеси берется
в виде
о
Тогда можно применить оригинальное уравнение состояния Флори,
введя обозначения
f « т/т* = (jL
Используя уравнение для спинодали и параметров Х12, предложенных:
Мак Мастером, авторы [134] рассчитали спинодали путем варьирова-
201
ния параметра Х12 при условии с12> 0. Выбор с12 > 0 обусловлен тем,
что смесь поливинилиденфторид — полиэтилметакрилат
характеризуется специфическими взаимодействиями, приводящими к уменьшению
объема при смешении, т. е. уменьшению числа внешних степеней
свободы, по сравнению с аддитивными значениями. На рис. 6.27, б
приведены рассчитанные этими авторами спинодали для двух значений:
Vsiи Сц=* 0,02. Из этого рисунка видно, что при отрицательных
значениях Х12У s1/s2= 1 и с12= 0, получаются фазовые диаграммы с
минимумом в области составов фаз обогащенных полиэтилметакрилатом,
причем эти диаграммы расположены значительно выше
экспериментальных. Следовательно, для согласования фазовой диаграммы с
экспериментальной необходимо использовать положительные значения с^..
Т,К
393
323
313
20 40 60 60 100
а
0 20 40 60
6
Рис. 6.28. Фазовые диаграммы для системы полиметилметакрилат (Mw =
= 1,7.104, Мл = 5,6-104) — хлорированный полиэтилен со степенью
хлорирования 47,8 % (А[ш = 1,2.105, Мя = 1,8-10*) (а) и 27,4 % (Mw =
= 1,08.10?, Mrt= 1,52.10*) (б) [133]. -
Лучшее совпадение расчетной и экспериментальной фазовых диаграмм
получается при /yV2*» 0,66, фъ= 0,55 и с12= 0,02, т. е. отклонение
степеней свободы от аддитивности примерно такое же, какое нашел Ха-
мада [135] для систем полидиметилсилоксан — растворитель. В данном
случае самый лучший результат получается при Х1%=* — 6,27 Дж/см3.
Отметим, что подобная картина (смещение фазовой диаграммы
в область составов, обогащенных полимером с большей молекулярной
массой) наблюдали еще Аллен и сотрудники [136] для смесей с верхней
критической температурой смешения. Согласно данным работы [135],
для систем, изученных Алленом и сотрудниками, с12>0, поскольку
для исследованных в их работе смесей (полиизобутилен с Л4 = 250 и
полидиметилсилоксан с М = 850) Гпив/гПдмс< 1, т. е. «неправильная»
фазовая диаграмма может быть связана с неаддитивностью числа
внешних степеней свободы.
В работе [133] впервые описано наличие обеих критических точек
смешения, а также получена диаграмма типа «песочных часов». Так,
у р
для смеси полиметилметакрилата
202
а,
11,7 . Ю4, Мл=*5,6. 104)
и хлорированного полиэтилена со степенью хлорирования 47,8 %
(Mw= 1,2 . 105, Мп = 1,8 • 104) наблюдались две КТС, а для смеси
полиметилметакрилата с хлорированным полиэтиленом со степенью
хлорирования 27,4% (Mw= 1,08- 105, М„ = 1,52 .
104)—диаграмма типа «песочных часов» (рис. 6.28).
6.4. 2. Особенности концентрационной и температурной
зависимостей параметров термодинамического
взаимодействия в смесях некоторых олигомеров
В последние годы существенный прогресс в технологии синтеза
полимерных материалов был достигнут благодаря использованию в
качестве реакционных систем олигомеров (олигоэфиров, олигоэфиракри-
латов и др.). Основная особенность большинства этих олигомеров —
наличие сильных межмолекулярных взаимодействий, приводящих
330
0,8 Щ
ПЭГ
Рис. 6.29. Температурные (а) и концентрационные (б) зависимости параметров Хгз
для системы полиэтиленгликольадипинат (М = 2000) —- полиэтиленгликоль (М =
= 1,5-104) [120];
а:/ —10; 2—20; 3 — 30; 4 — 50; 5—70; 5—80% ПЭГА; б:/ — 377,6; 2 — 384,8; 3 — 392,4;
4 — 400 К-
к явлениям ассоциации или агрегации компонентов в исходной
реакционной системе. Последнее оказывает существенное влияние как на
механизм реакции, так и на структуру конечного продукта [137—140].
Этим оправдывается интерес, связанный с изучением особенностей
термодинамического поведения такого рода систем в широком интервал
ле составов и температур. Знание этих особенностей в некоторых
случаях позволяет направленно регулировать уровень межмолекулярных;
взаимодействий в исходной реакционной системе, а следовательно,,
изменять механизм реакции и, в конечном итоге, структуру и свойства
получаемых из олигомеров полимерных материалов [78, 140],
203)
Некоторые представители таких систем, в частности смеси простых
и сложных олигоэфиров, используемых для получения линейных
и сетчатых полиуретанов, и явились объектом наших исследований.
Для смесей, таких как смеси полиэтиленгликоля (М = 1,5 - 104)
с полипропиленгликолем (М = 1,05 • 108) (ПЭГ — ППГ) и полиэтилен-
гликольадипинатом {М = 2000) (ПЭГ — ПЭГА), а также смеси ПЭГА
с ППГ, методом обращенной газовойхроматографии были определены
параметры термодинамического взаимодействия Хгз в широком
диапазоне температур и составов (см. рис. 6.23, 6.24, 6.29). Как видно из
рис. 6.23, для системы ППГ — ПЭГА кривая зависимости %23 от Т
проходит через минимум в области температур 343—363 К и достигает
максимума при 385—-390 К. Наличие минимума на графике завистимости
Хаз от Т делает поведение этой системы сходным с таковым для
бинарных смесей с верхней и нижней критическими температурами смешения
(ВКТС и НКТС). Так, в области температур 328—343 К (см. рис.
6.23) %н= — T(d%/dT)p> 0, что характерно для систем с верхней,
критической температурой смешения, а в области 370—390 К (рис.
6.23): хя= — Т (d%/dT)p< 0, что типично для систем с нижней
критической температурой смешения. В то же время максимум на кривой
зависимости хаз от Т при 385—390 К (рис. 6.23) свидетельствует о том,
что в данном случае повышение Хгз в области 370—390 К не является
признаком наличия КТС в этой системе.
В настоящее время известно, что специфические взаимодействия
в системе полимер — растворитель способствуют совместимости
компонентов. Ослабление же этих взаимодействий, например водородных
связей при повышении температуры, приводит к расслоению системы
из-за разрыва водородных связей [141]. Следовательно, появление
НКТС в такого рода системах — результат ослабления
специфических взаимодействий. То же происходит и в системе ППГ—ПЭГА,
характеризующейся специфическими взаимодействиями: понижение
термодинамической устойчивости системы с повышением температуры (от
350 до 385 К) также может быть обусловлено ослаблением водородных
связей. Однако это не приводит к расслоению системы на две фазы,
о чем свидетельствует уменьшение %2В с дальнейшим повышением
температуры (рис. 6.23). В одной из работ [120] мы показали, что при
отсутствии фазового расслоения в системе и фазового перехода
существенное ослабление водородных связей между компонентами неподвижной
фазы приводит к резкому возрастанию удерживаемого объема сорбата,
способного к специфическому взаимодействию полярных групп
неподвижной фазы, благодаря образованию дополнительного числа
водородных связей с освобождающимися группами неподвижной фазы
(см., например, рис. 6.19). В результате резко увеличивается
относительный удерживаемый объем полярного и неполярного сорбатов
Об^отн = lgVn/Vim). Для смеси ППГ — ПЭГА также была получена
температурная зависимость относительного удерживаемого объема
у
lg^oTH= lg метанола, т. в. части сорбата, способного образовывать
гентана
водородные связи g полярными группами неподвижной фазы, и части
204
2,0
0,2
-0,2
1,2
сорбата, неспособного к специфическим взаимодействиям с такими
группами. В области максимума %2Ъ (рис. 6.30) четко наблюдается
максимум на кривой зависимости lg КОтн = f(T), причем особенно
четко он проявляется для предварительно термообработанной смеси.
Следовательно, появление максимума на кривых %23 = f(T) может быть
приписано ослаблению водородных связей в этой области температур.
На графиках зависимостей %23 от Т в области средних составов
и для смесей ПЭГ—ППГ получены максимумы (рис. 6.24), которые, по-
видимому, обусловлены той же причиной. Изменение максимумов
с составом смеси можно объяснить изменением доли связей,
образованных индивидуальными и
разнородными ' компонентами смеси. Минимумы
на кривых зависимости %2з от Т
наблюдаются и для смесей ПЭГ— ПЭГА
(см. рис. 6.29). Однако в данном
случае отсутствует максимум на кривых
9С23— / (Т) из-за возможной
деструкции ПЭГ при дальнейшем повышении
температуры. Поэтому здесь нельзя
судить о том, обладает ли данная
система НКТС, или ее поведение
должно быть сходным с поведением
системы ППГ—ПЭГА.
Для системы ППГ—ПЭГ мы
рассчитали энтальпийные и энтропийные
значения параметра %2в в области
температур 340—370 К, а также
параметры %2Ъ{Н) и ХгзE), для чего
предварительно оценили значения параметра
контактных взаимодействий Х23.
Результаты эксперимента и расчетов
приведены в табл. 6.2, из которой
видно, что значения %2з(Щ
положительны и растут с увеличением
содержания ПЭГ в смеси ППГ—ПЭГ, а значения %23(S) отрицательны.
Отметим, что положительные значения энтальпий смешения были получены
для аналогичных систем и в работе [97].
Обращает на себя внимание тот факт, что расчетные и
экспериментальные значения %н и %$ существенно различны. Это может быть
связано со специфическими взаимодействиями между компонентами в
такого рода системах, наличие которых не учитывается в теории. Тем не
менее теория Флори правильно предсказывает знак этих параметров.
Обнаруженные аномалии температурной зависимости параметров
Хзз Для некоторых систем такого рода свидетельствуют о том, что не
во всех случаях по характеру изменения %23 с температурой можно
однозначно судить о наличии той или иной КТС, особенно НКТС, если
измерения проводятся в узком интервале температур. Аномалии
температурной и концентрационной зависимости параметров
термодинамического взаимодействия в таких системах могут быть связаны с ассо-
348
378 408 Т,К
Рис. 6.30. Температурные
зависимости In Уотн A,2) и Х23 C,4) для си-
стемы ППГ-1050 — ПЭГА-2000 при
массовой доле ППГ 30 % в смеси:
7,5 — исходные образцы; 2,4 .«
предварительно прогретые [120].
205
циативными явлениями, обусловленными специфическими
взаимодействиями между компонентами, а также с существенным расхождением
экспериментальных данных с предсказаниями существующих теорий
взаимной растворимости полимеров.
6.4.3. СМЕСИ ПОЛИМЕРГОМОЛОГОВ
Новые статистические теории растворов полимеров предсказывают,
что одной из существенных причин несовместимости компонентов
бинарной лимерной смеси является различие в параметрах уравнения
состояния компонентов, в частности, различие в свободных объемах.
Даже при одинаковых значениях контактных энергий, различие в
свободных объемах может привести к фазовому расслоению системы. Для
выяснения роли этих двух вкладов в термодинамические свойства
бинарных смесей полимеров необходимо иметь системы, где можно был®
бы исключить один из них. С этой точки зрения особый интерес
представляют смеси полимер гомологов (т. е. практически любой
синтетический полимер), которые до сих пор традиционно считали
термодинамически устойчивыми системами, исходя из подобия их химической
природы и параметров растворимости. Между тем для смесей полимер-
гомологов с существенно различающимися молекулярными массами
различие в коэффициентах термического расширения компонентов
может быть довольно велико (например, для ПС с М = 2000 и М =
=30000), коэффициенты термического расширения равны
соответственно 0,821 • 10 и 0,515-10"8[72]. С другой стороны, для таких систем, как
полиэтиленгликоли, различие в коэффициентах термического
расширения, например, для фракций с молекулярными массами 2 • 103и4 • 104,
мало, но из-за разного удельного содержания ОН-групп существенно
различаются энергии взаимодействия [106]. Следовательно, на примере
этих двух типов систем можно оценить роль двух вкладов в
термодинамические свойства смесей; поэтому они и были выбраны в качестве
объектов исследования.
Мы изучили концентрационные и температурные зависимости
параметров %2з для этих систем, используя метод обращенной газовой
хроматографии [96]. Полученные данные для систем полиэтиленгликоль
(ПЭГ)-2000-полиэтиленгликоль (ПЭГ)-40 000 приведены на рис. 6.31.
Из рисунка видно, что термодинамическая устойчивость системы
является функцией температуры и состава. Наблюдается даже
бимодальный характер концентрационной зависимости параметров
термодинамического взаимодействия. Параметр термодинамического
взаимодействия увеличивается с повышением, температуры, что может быть
обусловлено существенным ослаблением водородных связей.
Для неполярной системы ПС-2000 — ПС-30 000 мы получили очень
большие положительные значения параметров %23 (рис. 6.32),
значительно превосходящие критические значения Хгзкрит- Для этой системы
характерно уменьшение параметра %2з с повышением температуры.
В принципе, для системы, где компоненты имеют существенное различие
в коэффициентах термического расширения и малое в параметрах
растворимости, новые статистические теории растворов полимеров
206
предсказывают наличие НКТС, а следовательно, и увеличение %23
с температурой. Как было уже показано выше, примеров такого рода
очень много, но в основном это системы полимер — растворитель,
где вклад члена свободного объема во взаимную растворимость
компонентов очень велик. Для смесей же полимеров, как показали Паттер-
сон и сотрудники [301, эффект изменения свободного объема не такой уж
большой. Мы тоже воспользовались представлениями новых
термодинамических теорий о роли двух вкладов в параметр %23, и рассчитали
значения последнего по уравнениям типа F.15)—F.17). Из значений
коэффициентов термического расширения для компонентов смеси,
рассчитанных по данным работы [72], были получены Vt и Г, по
уравнению C.44). Из 7\вычислили Т\ иГ3,а следовательно, и т для
смесей. Выбрав Р\ « 500 Дж/см3, типичное для ПС, а также используя
0,05
-0,05
О
ПЭГ,
0,8 tp3
ПЭГ,
Рис. 6.31. Концентрационные зависимости параметра Х23 для системы
ПЭГ-2000--ПЭГ-40000 при температурах:
/—345; 2 — 370; 5 — 409 К [73].
Рис. 6.32. Концентрационные зависимости параметров Х2з Для смеси ПС-2000—
ПС-30000 при температурах:
/ — 415; 2 — 454 К [120]. . -
рассчитанные значения т по уравнению F.17), мы нашли величину
параметров %^IV\ в зависимости от температуры для системы ПС-
2000— ПС-30 000, а также для смесей разных функций ПС с ПС-30 000
и друг с другом. При этом, учитывая, что для фракций ПС параметры
растворимости должны быть близки, величину параметра контактных
взаимодействий Х23 мы взяли на порядок ниже A Дж/см3), чем обычно
наблюдаемое значение для дисперсионных сил взаимодействия. Как
видно из рис. 6.33, даже при таком малом значении параметра Х23
величина 1чЪ1У1 Для системы ПС-2000 — ПС-30 000 много больше
критического значения, рассчитанного согласно соотношению
(%2з)К
V*
(Для выполнения условия симметричности, т. е. %ц == 1ц, параметр
%23 отнесен к единице объема.) Величина параметра %23/У% становится
меньше критической лишь для системы ПС-10000-ПС-ЗОООО при очень
207
'0,7
0,5
0,3
0,1
0,8
0,6
0,2
1.0
1,1
12
Рис. 6.33. Температурные зависимости параметров Х2з/^*» рассчитанных по
уравнению F.17) (сплошные линии) при Х12 = 1 Дж/см3, и критические значения
параметров (штриховые) для смесей фракций полистирола с М = ЪА№ и полистирола
сЛ1ит соответственно равных:
/, V — 2000 и 0,16; 2, 2* — 3000 и 0,13? 3, 3' — 5000 и 0,068; 4, 4'-* 7000 и 0,055; б, 5'-* 10 000
н 0,034 [73].
Рис. 6.34. Температурные зависимости параметров Хгз/У* (сплошные линии) и
критические значения параметров Х2з/У* (штриховые) при Х12 = 1 Дж/см3 для смесей
фракций полистирола с молекулярной массой:
/, /' _ 2000— 5000; 2, 2'— 3000 — 5000; 3, 3'— 3000 *- 7000; 4, 4' «- 3000 «— 10 000.
высоких температурах. При этом с уменьшением молекулярной массы
фракции, используемой в качестве второго компонента смеси,
температура, где х2з < (Х2з)кРит, существенно увеличивается (табл. 6.9). Для
системы же ПС-3000 — ПС-30 000 и ПС-5000 — ПС-30 00Q значения
параметров X23/V* вообще не достигают критических, поэтому смеси
термодинамически несовместимы. Как видно из рис. 6.33, во всех
случаях с понижением температуры параметр х2з/^* растет, что
согласуется с экспериментально наблюдаемой картиной для системы
ПС-2000 — ПС-30 000 (см. рис. 6.32).
Интересно рассмотреть изменение параметра Хгз^г Для смесей
фракций с М=30 000 с ростом молекулярной массы фракции. Видно
(рис. 6.34), что при достижении молекулярной массы второго
компонента величины 10 000 при низких температурах х2з^2 > (Х2з)крит/^ .
Если же в качестве опорного компонента взять ПС-30 000, то в
зависимости от молекулярной массы второго компонента вклад только
208
Члена Свободного объема В %2з ИЗО- Таблица 6.9. Соотно ше ние моле-
браЗИТСЯ кривыми 1—5 на рис. "Улярной массы полимера и темпера-
6.35. Если • даже энергетический *УР??ЯХР* разных значен иях v*
член Х28 равен нулю (что в принци- * ' • '
пе возможно ожидать для смесей
полимергомологов), то и в таком
случае член свободного объема
может обусловить несовместимость
компонентов (кривая /).
Отметим, что при небольших
значениях т практически
невозможно достичь реальных нижних
критических температур смешения
(см. рис. 6.35, кривую 5). Для
систем же со специфическими взаимодействиями, в частности для
системы ПЭГ—ПЭГ, энергетический член с повышением
температуры становится более положительным (обычно для таких систем о»
отрицателен), что вместе с ростом члена свободного объема
приводит к увеличению параметра %2В с повышением температуры (он ста-
м
2000
3000
5000
7000
10000
1,05
6
45
64
73
80
1,10
191
225
286
301
313
1.15
309
390
429
448
464
1,20
390
473
516
538
555
0,4'
0,2
ч
0,5
0,1
-G3
1,0
1,2 Vjfi
1,1
1,2
679 444 657 606 Т,К
Рис. 6.35. Температурная зависимость члена свободного объема параметра
Хгз/У* ^и = 0) (сплошные линии) и критические значения параметра Хзз/У*
(штриховые) для смесей фракций полистирола с М = 3«104 и полистиролом.
с молекулярной массой:
7. /' — 2000; 2, Г— 3000; 3, 3' — 5000: 4, 4>*~ 7000* 5, 5'*. 10 000 [73].
Рис. 6.36. Температурная зависимость суммарного параметра Хав/У* W» члена
свободного объема (II) и члена взаимодействия (III) параметра Х2з/^* Дл*
системы ПЭГ—ПЭГ при Х1а = — 0,87 Дж/см3 и т = 0,1.
Штриховые линии — критические значения параметра t^/V^ для смесей ПЭГ с
молекулярными массами: 2000-^40 000 (/), 3000-40 000 {2), 5000 - 40 000 C) и во ^40 000
D) 173].
209
новится менее отрицательным) (рис. 6.36). В данном случае величина
т принята постоянной, так как разность свободных объемов между
ПЭГ-2000 и ПЭГ-40 000 незначительна, но в принципе этот параметр
должен увеличиваться при уменьшении молекулярной мйссы одного из
компонентов смеси при постоянном значении молекулярной массы
второго. Это согласуется с полученными нами данными для системы
ПЭГ-2000—ПЭГ-40 000, для которой все же характерно небольшое
различие в коэффициентах термического расширения компонентов [106].
Таким образом, полученные данные свидетельствуют о том, что,
с одной стороны, при больших различиях в коэффициентах
термического расширения компонентов смеси полимергомологов даже при
небольшой разнице в параметрах контактных взаимодействий (или
параметрах растворимости) смесь может оказаться термодинамически
несовместимой, если нет специфических взаимодействий: с другой
стороны, для полярных систем основной причиной термодинамической
несовместимости является существенное ослабление специфических
взаимодействий, даже если различие в коэффициентах термического
расширения компонентов невелико. Вполне возможно, что
несовместимость фракций в полимолекулярном образце и может быть причиной
структурной гетерогенности, если образец достаточно полидисперсен
или имеет бимодальную форму молекулярно-массового распределения.
Одна из характерных обнаруженных нами особенностей системы
ПЭГ-2000 — ПЭГ-40 000 — бимодальный характер концентрационной
зависимости параметра %23 (точек помутнения или спинодалей); для
объяснения этого существуют разные предположения. (например,
[28]), однако до сих пор физический смысл наблюдаемого явления все
еще остается неясным.
Как уже упоминалось в параграфе 6.44, в работе [48] были
сформулированы необходимые условия наличия бимодальной спинодали (см.
уравнения F.37) и F.38)), в частности, трех нулевых значений d (\IT)I
/dcp! во всем концентрационном диапазоне @; 1). В работе [48] и в
теории Флори [12] величина Т одна и та же, поэтому для ее определения
можно использовать выражение C.43), C.51), C.52) в виде
Pj г2
Заменив объемные доли q>, сегментными ср*, величину d(l/T)/d<f)*
получаем в виде
d (l/f) Pf-Pt-
или ~ d (ср? 09Х1Й)
d(l/T)/dtf АВ ™ Г
где А и В —константы. Поскольку для смесей полимергомологов
доля мест в2 равна q>*, то вид концентрационной зависимости
dO/TJ/dq)]. будет определяться формой концентрационной зависимости
величины
210
Для системы ПЭГ — ПЭГ мы получили концентрационную
зависимость параметра Х12 и затем по экспериментальным значениям
построили зависимость d (q>*q>JXa)/d<pJ от q>* (рис. 6.37). Производная
v \ ~ *— действительно будет принимать три нулевых
значения во всей концентрационной области @; 1), следовательно,
соблюдается необходимое условие наличия бимодали, предложенное в работе
[48]. Причина аномальной концентрационной зависимости параметра
дХ12/д<рг (или d(l/f)/d(Pi) пока неизвестна; в некотором приближении
объяснением могут быть данные,
полученные нами для смесей поли- Х12
мергомологов бутадиена. Мы
определили параметры
термодинамического взаимодействия для смесей
фракций бутадиена (БД) с молеку- -2
лярными массами 3,9 • 105 (БД-
390 000), 7-10* (БД-70 000), 4х
X Ю4 (БД-40000) и 2-104 (БД- ~*
20000). Как видно из рис. 6.38, -5
6.39 в случае смесей БД-390 000 ^
и БД-70 000, БД-40 000 и БД-20 000 °
в измеренном диапазоне
температур значения параметров термоди- -8
намического взаимодействия ком-
0,2 0,4 0,6 0,8 pf
-2
Рис. 6.37. Концентрационная
зависимость параметра Х12 (/) и его
производной d tof ф*Я12)/дф* B) для
системы ПЭГ-2000 — ПЭГ-40 000 [73].
понентов смеси %23 отрицательны,
что свидетельствует о
термодинамической устойчивости систем. Однако
концентрационная зависимость %23
изменяется при изменении
молекулярной массы второго компонента. Так, для смеси фракций БД-20000 и
БД-390 000 наблюдается бимодальный характер концентрационной
зависимости параметра %23 (рис. 6.38). По мере увеличения молекулярной
массы второго компонента бимодальность концентрационной зависимости
параметра %23 исчезает (рис. 6.39), после достижения значения 70000
величина %23 с концентрацией практически не меняется. Для всех
изученных смесей значения параметров %23 уменьшаются при
повышении температуры (становятся более отрицательными), кроме смеси
БД-390 000 — БД-70 000, для которой в измеренном диапазоне
температур параметры %23 практически не изменяются. По температурным
зависимостям %2В для смесей фракций БД-390 000 — БД-20 000 и
БД-390 000 — БД-40 000 мы определили энтальпийную часть
параметра %23 (%н = — Т (¦gf-8))» a также рассчитали значение
энтропийного вклада в %23 по известному соотношению
где %s— некомбинаториальный вклад в энтропию смешения.
211
-0,4
,50
ПБД-4,% О
50
ПбД~49°/о
Рис, 6.38. Концентрационная зависимость параметров Х2з (Д2), %н C) и %s
для смесей полибута диенов с Л1 = 2 • 104 и 5-105 [131]:
/ — 323; 2^4 — 357 К.
Рис. 6.39. Концентрационная зависимость параметров Х2з (^ — 3), Х//D) и
для смесей полибутадиенов с Af = 5»105 и 4-104(/, 2, 4, 5) и полибутадиенов о
М = 5.105 и 7.Ю4:
/ ** 323; 2, 4, б — 357; 3 -* 323 — 357 К [131].
Во всех случаях энтальпийный вклад в параметр %гз оказался
положительным, т. е. это система имеет ВКТС. Значения же параметра
%s оказались отрицательными, свидетельствуя о том, что некомбинато-
риальный вклад в энтропию смешения положителен (A SHK = —#%ф22)-
Обращает на себя внимание необычный ход концентрационной
зависимости %н и особенно %s (см. рис. 6.38, 6.39). Так, в области 30 и 70 %-
ного содержания БД-390 000 в смеси параметры %н и %5 близки к
нулю, т. е. поведение системы в этой области составов близко к
идеальному, что существенно отличает поведение данной смеси от обычных
растворов полимеров в низкомолекулярных растворителях (см., например,
[12]). Уменьшение положительного некомбинаториального вклада
в энтропию смешения можно приписать агрегативным явлениям,
которые, однако, не приводят к потере термодинамической устойчивости
системы. Резкое же увеличение положительного некомбинаториального
вклада в энтропию смешения и большие положительные значения эн-
тальпийного вклада при других составах могут быть связаны с
уменьшением размера агрегатов, а следовательно, с увеличением доли меж-
212
фазной области, следствием чего и является рост некомбинаториаль-
ной энтропии смешения [128]. По крайней мере, для более строгой
трактовки этих данных необходимы сведения о размерах макромолекул
в смеси или об уровне гетерогенности системы, определить которые
для смесей полимергомологов пока не представляется возможным из-за
одинаковых значений показателя преломления компонентов1.
Изменение же уровня гетерогенности системы в зависимости от состава и
может быть причиной аномальной зависимости параметров
термодинамического взаимодействия %23-
Таким образом, рассмотренные данные показывают сложное
термодинамическое поведение даже таких простых смесей, как полимер-
гомологи. В настоящее время из-за ограниченного количества
экспериментальных данных еще нельзя говорить об общности такого
поведения смесей полимергомологов. Однако уже имеющиеся данные
показывают, что такие смеси могут быть хорошими моделями для выяснения
роли разных факторов в термодинамике процесса смешения полимеров.
Кроме того, исследование систем подобного типа важно и в том смысле,
что практически все синтетические полимеры являются смесями
полимергомологов с тем или иным молекулярно-массовым распределением,
включая и бимодальное. Структура же и физико-химические
характеристики таких полимеров существенно зависят от термодинамической
совместимости отдельных фракций [130].
В заключение следует еще упомянуть работу Сузуки [143], в
которой было показано, что для смесей двух полимергомологов с сильно
различающимися молекулярными массами в общем растворителе
значения вторых вириальных коэффициентов, характеризующих
взаимодействие между компонентами полимергомологов, т. е. Л2з>
отрицательны. Эти данные противоречат предсказаниям теорий второго
вириального коэффициента Л123» по которому рассчитывается Л28-
Согласно таким теориям [144, 145], кривая зависимости второго
вириального коэффициента Л12з> описывающего взаимодействие
растворителя со смесью двух полимеров, от состава должна иметь максимум,
а А 2з должен быть положительным. Зависимость Л123 от состава
проходит через минимум, а Л28 в некоторых случаях оказывается
отрицательным. Учитывая представления Лондона о силах притяжения между
частицами, Сузуки [143] пришел к выводу, что в такого рода системах,
в которых компоненты двух полимергомологов существенно
различаются по молекулярным массам (размерам макромолекул), силы
притяжения Лондона способствуют агрегации нескольких молекул малого
размера вблизи поверхности макромолекулы большого размера и смесь
становится гетерогенной.
Следует отметить, что идея о возникновении молекулярных
агрегатов различных фракций была выдвинута С. М. Липатовым еще в 30-х
годах [146].
1 В принципе, такие измерения могут быть выполнены методом
малоуглового рассеяния рентгеновских лучей или медленных (тепловых) нейтронов [44],
если один из компонентов дейтерирован. Однако дейтерирование одного из
компонентов может оказать существенное влияние на термодинамику процесса
смешения [142].
213
6.4.4. ВЛИЯНИЕ ТОЛЩИНЫ ПЛЕНКИ И ПРИРОДЫ ПОДЛОЖКИ
НА ТЕМПЕРАТУРУ ФАЗОВОГО РАССЛОЕНИЯ
В СМЕСЯХ ПОЛИМЕРОВ
Во многих работах (например, [147—149]) было показано, что на
границе раздела полимера с твердой подложкой возникает межфазный,
или граничный слой большой протяженности со свойствами,
существенно отличающимися от свойств полимера в массе. Результатом
проведенных нами исследований зависимости свойств полимера от толщины
пленки, нанесенной на твердую подложку, и природы подложки был
следующий вывод: с уменьшением толщины пленки существенно
изменяются температура стеклования (растет или уменьшается в
зависимости от природы полимера и подложки), степень кристалличности,
параметры термодинамического взаимодействия полимер —
растворитель, и др. [148, 149]. Такие изменения свойств были приписаны
изменениям подвижности макромолекул на твердой подложке, а
следовательно, и набора их конформаций и т. п.
При исследовании адсорбции и структуры граничных слоев,
полученных при адсорбции из смесей полимеров на высокоэнергетической
подложке, мы выяснили [150], что в случае смесей термодинамически
несовместимых полимеров происходит избирательная адсорбция
компонента, имеющего более высокое поверхностное натяжение. При этом
компонент, преимущественно сорбирующийся на подложке, «тянет»
за собой молекулы другого компонента, что приводит к
неравномерному распределению полимерных компонентов различной природы в
граничном слое по его толщине; т. е. в данном случае должна существенно
изменяться концентрация компонентов в граничном слое по сравнению
с исходной концентрацией смеси. Поскольку же температура фазового
расслоения является функцией концентрации компонентов, фазовое
расслоение в граничном слое должно происходить при температуре,
отличной от температуры фазового расслоения исходной смеси. И это
действительно было обнаружено авторами работы [151], которые
попытались оценить влияние толщины пленки, нанесенной на твердую
подложку, и природы подложки на температуру фазового расслоения
для смесей двух полимеров (полистирола и поливинилметилового
эфира). Они действительно установили различие температур фазового
перехода для тонких пленок, нанесенных на твердую подложку, и
смеси полимеров в массе (рис. 6.40). При этом в зависимости от природы
подложки этот эффект оказался разным (на подложках из золота и
стекла). Считая, что подложка из золота не оказывает какого-либо
селективного воздействия на оба компонента, эти авторы попытались
дать простейшую термодинамическую трактовку наблюдаемому
изменению температуры фазового расслоения пленок на подложке —
объяснить это изменение уже упоминавшимся влиянием подложки на
изменение набора конформаций макромолекул. Рассуждения авторов
сводятся к следующему.
Согласно решеточной модели для бинарных смесей
координационное число мест решетки однородно по всей решетке, но в тонких
пленках в поверхностном слое оно должно быть меньшим, чем в массе, и,
214
следовательно, число взаимодействий в образце, содержащем Ммест
решетки, будет
f —j [BHr/H)N(z - *г)],
F.90)
где Н — толщина образца; Нг — толщина граничного слоя; ги 2г —
координационные числа мест решетки в блоке и граничном слое
соответственно. При zr < z, следовательно, параметр термодинамического
взаимодействия, характеризующийся l/2Nz парными
взаимодействиями, будет иметь вид _
* F.91)
0,1
-0,1
0,1
1,0
10
H,jum
Рис. 6.40. Влияние толщины пленки на температуру спинодального распада для
смеси полистирол — поливинилметиловый эфир, нанесенной на пленку из золота
(а) и стекло (б):
i — 15; 2 — 50; 3 — 67 % полистирола по массе в смеси [151].
где хД— параметр термодинамического взаимодействия,
рассчитываемый без учета уменьшения числа парных контактов, т. е. для НТ1Н = 0.
Функция F (Н) получается из уравнения F.90):
= 1-2A-гР/г)(Яг/Я). F.92)
Поскольку Xi2 ПРИ
dr
СП
ж
при постоянном составе один и тот же,
(д%12/дН)т F(H)
F.93)
(д%12/дТ)н Xi2 (T) (dF/dH)
Из уравнений F.92) и F.93) видно, что для систем с НКТС
(d%l2/dT >Q)dTCn/dH отрицательно, т. е. с уменьшением толщины
пленки увеличивается стабильность системы. Противоположный эффект
должен наблюдаться для смесей с ВКТС, где d%{JdT < 0 и,
следовательно, dTcn/dH положительно.
215
Для предсказания функциональной зависимости температуры
фазового расслоения (Гсп) от толщины пленки параметр
термодинамического взаимодействия при Тси берется в виде
Х12 (Гс„) =4 (! + ?!), F.94)
где ф^— объемная доля компонента /; V{ — его молярный объем.
Подставляя уравнения F.90) и F.92) в F.94), получаем
Для систем с НКТС (например, системы полистирол — поливи-
нилметиловый эфир, исследованной авторами настоящей работы)
параметр термодинамического взаимодействия может быть взят в виде
X° X°X/7\ Xе. Xi>0. F.96)
Подставляя уравнение F.96) в уравнение F.95), один раз для Н = 0,
а другой раз для конечных Н9 имеем
Т°сп Тсп (Я) ~ 2Х12 [щ + V2 Фа; [F (Н)
где То, — температура на спинодали при #г/# = 0. Если F(H) не
очень отличается от 2, то 1/Тсп должно быть линейной функцией 1/Я:
Это уравнение при соответствующем выборе значений Н позволяет
предсказать толщину пленки, при которой Гсп будет отличаться от
Тсп для смеси в блоке.
Как видно из рис. 6.40,я, Теп действительно увеличивается при
уменьшении толщины пленки, нанесенной на подложку из золота.
Используя литературные данные по Х12 для этой системы, авторы
работы [151] нашли, что эффект повышения ГСп должен наблюдаться для
цленок, начиная с толщин 0,4—0,8 нм, что хорошо согласуется с
экспериментальными данными.
Для пленок на стекле те же авторы отмечали различную
зависимость Гсп от толщины пленки для разных составов смеси (рис.6.40,6).
Причину такой зависимости они объясняют дестабилизирующим
влиянием электростатического взаимодействия компонентов с подложкой
и селективной адсорбцией полистирола на стекле. В общем эти данные
не противоречат высказанным выше предположениям, что при
селективной адсорбции одного из компонентов состав граничного слоя будет
отличен от состава исходной смеси и это должно привести к изменению
температуры фазового расслоения. Прямым ответом на вопрос, каким
образом должна изменяться Гсп при селективной адсорбции одного из
компонентов на твердой подложке, могут быть результаты
исследования изменения Тсп пленки, нанесенной на твердую подложку, от
состава граничного слоя и типа фазовой диаграммы для данной системы.
216
Решение этого вопроса связано с проблемой создания
композиционных материалов на основе связующих «второго поколения», т. е.
использования в этом качестве смесей полимеров, иначе говоря —
о связи условий фазового разделения с условиями взаимодействия
бинарных полимерных систем с наполнителем. В этом плане пока
имеется лишь ограниченное число работ [152]. Но уже сейчас можно
утверждать, что введение наполнителя может дать два результата. Во-
первых, если один из компонентов бинарной смеси преимущественно
взаимодействует с поверхностью наполнителя (Д G2i< A G23, где 1 —
наполнитель, 2 и 3 — компоненты полимерной смеси), наполнитель
способствует возрастанию скорости фазового разделения. Во-вторых,
если энергии взаимодействия компонентов с поверхностью одинаковы,
то вследствие адсорбционных эффектов и общего понижения
молекулярной подвижности вблизи границы раздела [153] процессы фазового
разделения будут тормозиться. Ответить на этот вопрос можно лишь
на основе экспериментальных результатов исследования влияния
наполнителя на процессы фазового расслоения бинарных полимерных
смесей.
I. Gee G, Some thermodynamic properties of high polymers and their molecular
interpretation.— Rev. Chem. Soc, 1947, 1, N 1, p. 265—298.
2. Слонимский Г. Л., Струминский Г. В. О взаимной растворимости
полимеров. 4. Влияние плотности упаковки молекул на их взаимную растворимость—
Журн. физ. химии, 1956, 30,, № 10, с. 2144—2148.
3. Стру минский Г. В., Слонимский Г. Л. О взаимной растворимости
полимеров. 3. Теплоты смешения полимеров. Там же, № 9, с. 1941—1947.
4. Flory P. /,, Eichinger В. E.t Orwoll R. A. Thermodynamics of mixing
polyethylene and polyisobutylene. —Macromolecules, 1968, 1, N 3, p. 287—288.
5. Термодинамика смешения полимеров / А. А. Тагер, Т. И. Шолохович,
И. М. Шарова и др.— Высокомолекуляр. соединения, А, 1975, 17, № 12,
с. 2766—2773.
6. Тагер Л. Л. Термодинамика смешения полимеров и термодинамическая
устойчивость полимерных композиций. Там же, 1977, 19, с. 1659—1669.
7. Тагер А. Л. Шолохович Т. И. О методе оценки совместимости полимеров в
растворах и фазовом равновесии систем полимгр—полимер./Там же, 1976,
18, № 5, с. 1175—1181.
8. Тагер А. А. Шолохович Т. #., Цилипоткина М. В. Оценка
термодинамической устойчивости системы полимер — полимер./Там же, 1972, 14, № 6,
с. 1423—1424. *
9. Липатов С. М. Липатова Г. В. Фазовое расслаивание в системах полимер —
полимер — растворитель.— Коллоид, журн., 1959, 21, № 5, с. 517—521.
10. Prigogine I. The molecnlar theory of solutions.— New York: lnterscience,
1959.—470 p.
II. Patterson D. Role of free volume changes in polymer solutions
thermodynamics.— Polym. Sci. C, 1968, N 16, p. 3379—3389.
12. Eichinger B. ?"., Flory P. J. Thermodynamics of polymer solutions.— Trans.
Faraday Soc, 1968, 64, N 8, p 2035—2072.
13. Scott R. L. The thermodynamics of high—polymer solutions. 4. Phase equi-
bria in ternary system polymer — liquid — liquid.— J. Chem. Phys.,
1949, 17, N 2, p. 268—279.
14. Tompa H. Polymer solutions.— London : Butterworths, 1956.—325 p.
15. Krauze S. Polymer compatibility.— J. Macromol. Sci. C, 1972, 7,N 2, p. 261 —
314.
16. Pazonyi 71., Dimitrov M. Preparation and properties of polymer mixtures.—
Rubber Chem. and Technol., 1967, 40, N 4, p. 1119 —1125.
17. Schneier B. Polymer compatibility.—J. Appl. Polym. Sci., 1973, 17, N 10,
p. 3275-3185.
217
18. Gee G. The interaction between rubber and liquids. 11. The thermodynamic
. basis of the swelling and solution of rubber.— Trans. Faraday Soc, 1942, 38,
N 2, p. 276—284.
19. Small P. A. Some factors affecting the solubility of polymers.—J. Appl.
Chem., 1953, 3, N 2, p. 71-88.
20. Bohn L. Incompatibility and phase formation in solid polymer mixtures and
graft and block copolymers.— Rubber Chem. and Technol., 1968, 41, N 2,
p. 495—513.
21. Shaw M. T. Studies of polymer — polymer parameter approach.— J , Appl.
Polym. Sci., 1974, 18, N 2, p. 449-472.
22. Hansen С. М. The three — dimensional solubility parameter — key to
component affinities. 2. Solyants, plastisizers, polymers and resins. 2. Dies, emul-
sifiers, mutual solubility and compatibility, and pigments.—J. Paint.
Technol., 1967, 39, N 505, p. 104—117; N 511, p. 505—510.
23. Bunn C. W. The melting poiting of chain polymers.— J. Polym. Sci.,
1955, 16, N 82, p. 323—343.
24. Nishi T. Experimental aspects of compatible polymer mixtures.— J. Macro-
mol. Sci. B, 1980, 17, N 3, p. 517—542.
25. Hildebrand J. Я., Scott R. L. Regular solutions.— New York: Prentice — Hall,
1962.— 327 p.
26. Yee A. F., Maxwell M. A. Mechanical properites of polymer mixtures: effect
of compatibility.— J. Macromol. Sci. B, 1980, 17, N 3, p. 513—564.
27. McMaster P. L. Aspects of polymer — polymer thermodynamics. — Mac-
romolecules, 1973, 6, N 5, p. 760—763.
28. Koningsveld Я., Kleintjiens L. A. Thermodynamics of polymer mixtures.—
J. Polym. Sci. C, 1977, N 61, p. 221-249.
29. Olabisi O. Polymer compatibility, by gas — liquid chromatography.— Macro-
molecules, 1975, 8, N 3, p. 316—322.
30. Robard A., Patterson D. Thermodynamics of polymer compatibility.—Ibid.,
1977, 10, N 7, p. 1021—1025.
31. Siow K- 5., Delmas G., Patterson D. Cloud — point curves in polymer
solutions with adjacent upper and lower critical solution temperatures.— Ibid.,
1972, 5, N 1, p. 29—34.
32. Upper apd lower critical solution temperatures in polystyrene solutions/
S. Saeki, N. Kuwahara, S. Konno, M. Kaneko.— Ibid., 1973,6, N 2, p. 246—250.
33. Pressure effects in polymer solution phase equilibria. 1. The lower critical
solution temperature of polyisobutylene and polydimethylsiloxene in lower al-
kanes/L.. Zeman, J. Biros, G. Delmas, D. Patterson.—J. phys. Chem., 1972,
76, N 8, p. 1206—1213.
34. Wolf В. А.у Blautn G. Dependence of oligomer — oligomer incompatibility
of chain length and pressure. 1. OHgo-isobutene oligopropylene glycol
and oligo-styrene/oligo-ethylene glycol.— J. Polym. Sci., C, 1977, N 61,
p. 251—270.
35. Upper and lower critical solution temperatures in polystyrene solutions S. Saeki,
N. Kuwahara, S. Konno, M. Kaneko,—Macromolecules, 1973,6, N 4, p. 589—593.
36. Liu D., Prausnitz «/. M. Moleculat thermodynamics of polymer
compatibility: effect of contact agility.— Ibid., 1979, 12, N 3, p. 354—459.
37. Beret 5., Prausnitz Л М. A generalized van der Waals equation for
polymers and other fluids.— Ibid., 1975, 8, N 6, p. 878—882.
38. Donohue M. D., Prausnitz J. M. Perturbed hard chain theory for fluid
mixtures: thermodynamic properties for mixtures in natural gas and petroleum
thechnology.— AlChE Journal, 1978, 24, N 5, p. 849—860.
39. Henderson D. Perturbation theory for a mixture of hard spheres and square —
well molecules.— J. Chem. Pnys., 1974, 61, N 3, p. 926—931.
40. Carnahan N. F.9 Starling /С. Е. Equation of state for nonattracting rigid
spheres.—Ibid., 1969, 51, N 2, p. 635—636.
41. Koningsveld R., Kleintjiens L. A., Liquid-liquid phase separation m multi-
compenent polymer systems. 15. Thermodynamic of polymer compatibility.—
Brit. Polym. J., 1977, 9, N 3, p. 212—215.
42. Koningsveld R., Stepto R. F. T. On polymer mixture thermodynamics.— Mac-
romolecules, 1977, 10, N 5, p. 1166—1167.
218
43. Silberberg Л. Polymer solutions. General discussions.— Discuss. Faraday Soc,
1970, N 49, p. 162.
44. Allen G.9 Maconnachie A. The contribution of neutron scattering to polymer
science.— Brit. Polym. J., 1977, 9, N 3, p. 184—188.
45. Experimenteller Nachweis des molekulardispersen Charakters der Mischung
von zwei Polymeren und Bestimmung des chemischen Potentials in diesen Mis-
chungen/—Makromol. Chem., 1976, 177, N 4, S. 1145—1160.
46. Sanchez I. C, Lacomse R. H. An elementary molecular theory of classical
fluids. Pure fluids.—J. phys. Chem. 1976, 80, N 21, p. 2352—2562.
47. Sanchez I. C, Lacombe R. H. Statistical thermodynamics of polymer
solutions.—Macromolecules, 1978, 11, N 6, p. 1145—1156.
48. Sanchez I. C, Lacombe R. H. Statistical thermodynamics of bulk and
surface of polymer mixtures.— J. Macromol. Sci. B, 1980, 17, N 3, p. 565—589.
49. Ziska J. /., Barlow /. W., Paul D. R. Miscibility of PVC — polyester blends.—
—Polymer, 1981, 22, N 7, p. 918—924.
50. Dobry A., Boyer-Kawenoki F. Phase separation in polymer solution.—
J. Polym. Sci., 1947, 2, N 1, p. 90—100.
51. Kern R. J. Component effects in phase separation of polymer — polymer —
solvent systems.—Ibid., 1956, 21, N 98, p. 19—25.
52. Huglin Ch., Dondos A, Systemes ternaires: polymere A — polymere B-
solvant. Influence de la nature du solvant sur Pincompatibilite des poly-
meres.—Makromol. Chem., 1968, 126, S. 206—216.
53. BristowG. M. Phase separation in rubber — poly (methyl methacrylate) —
solvent systems.—J. AppL Polym. Sci., 1959, 2, N 4, p. 120—122.
54. Berek D., Lath D., Durdovic V. Phase relations in the ternary system atactic
polystyrene — atactic polypropylene — etolene.— J. Polym. Sci. C, 1967,
N 16, p. 659-667.|
55. Strazielle C. Etude du parametre d'interaction polymere — polymere pour
le systeme ternaire solvant — polydimethyl — siloxane — polystyrene.
Influence de la concentration et de la masse moleculaire.— Eur. Polym. J.,
1979, 15, N 1, p. 55—59.
56. Okazawa T. On the narrow miscibility gap in polymer l-polymer 2-solvent
ternary systems.—Macromolecules, 1975, 8, N 3, p. 371—373.
57. Weligan D. C., Burns С. М. The effect of molecular weight and
temperature on phase separation in the ternary system polystyrene/polybuta-
diene/ tetraline.—J. Appl. Polym. Sci., 1974, 18, N 2, p. 521—529.
58. Clampitt В. Н. The 1974 SPE polyolefin division Award Adress: phase
behavior of polybutene — 1 plus polystyrene in carbon tetrachloride,— Polym.
Eng. Sci., 1974, 19, N 12, p. 827—830.
59. Aharoni S. M. Bimodal closed cloud point curves of ternary systems.—
Macromolecules, 1978, 11, N 1, p. 277—279.
60. Djadoun S., Goldbeg R. N., Morawetz H. Ternary systems containing an
acidic copolymer, a basic copolymer, and a solvent. 1. Phase equilibriums.— Ibid.,
1977, 10, N 5, p. 1015—1020.
61. Kwei T. /G, Nishi Г., Roberts R. F. A study compatible polymer mixtures.—
Ibid., 1974, 7, N 5, p. 667—674.
62. Влияние молекулярного веса на взаимную растворимость полимеров/
В. Н. Кулезнев, Л. С. Крохина, Ю. Г. Оганесов, А. М. Злацен.— Коллоид,
журн., 1971, 33, № 1, с. 98—105.
63. Кулезнев В. Н. Крохина Л. С. Структурообразование в растворах смесей
полимеров.— Высокомолекуляр. соединения А, 1973, 15, JNJb 4, с. 906—916.
64. Tager А. Л., Scholokhovich Т. /., BessonovYu. S. Thermodynamics of mixing of
polymers.— Eur. Polym. J., 1975, 11, N 4, p. 321—326.
65. Тазер А. А., Бессонов Ю. С. Теплоты смешения расслаивающихся
полимерных бинарных композиций.—Докл. АН СССР, 1972, 205, № 5, с. 1146—
1148.
66. Тагер А. Л., Бессонов Ю. С, Руденко И. В. Энтальпия и энтропия смешения
некоторых ароматических полимеров с эпоксидной смолой.—
Высокомолекулярные соединения, 1980, 22, № 6, с. 438—439.
67. Zeman L., Patterson D. Effect of the solvent on poLymer incompatibility
in ternary systems.—Macromolecules, 1972, 5, N 4, p. 513—516.
219
68. Hsu С. С, Prausnitz J. M. Thermodynamics of polymer compatibility in
ternary systems.— Ibid., 1974, 7, N 3, p. 320—324.
69. Koningsveld R., Chermin H. Л., Gordon M. Liquid — liquid phase
separation in multicomponent polymer solutions. 8. Stability limits and consolute
states in quasiternary mixtures.— Proc. Roy. Soc. London, A, 1970, 319,
N 1538,p. 331—349.
70. Robard Л., Patterson D., Delmas G. The «A%-effect» and polystyrene — poly-
(vinyl methyl ether) compatibility in solution.— Macro molecules, 1977, 10,
N 3, p. 706—708.
71. Bank M., Leffingwell /., Thies C. The influence of solvent upon the
compatibility of polystyrene and poly(vinyl methyl ether).—Ibid., 1971, 4, N 1,
p. 43—46.
72. Effects of chain ends and molecular weight on specific volumes of ani-
onic polystyrene melts/ A. Rudin, R. A. Wagner, К. К. Chee et al.—
Polymer, 1977, 18, N 2, p. 124—129.
73. Липатов Ю. С, Нестеров А. Е., Игнатова Т. Д. О некоторых особенностях
концентрационной и температурной зависимости параметров
термодинамического взаимодействия между компонентами в смесях олигомеров и полимер-
гомологов.— Высокомолекуляр. соединения, А, 1980, 22, № 10, с. 2284—
2291.
74. Roerdink ?., Challa G. LCST behaviour in blends of poly(vinylidene fluoride)
and isotactic poly(ethyl methacrylate).— Polymer, 1980, 21, N 10, p. 1161 —
1166.
75,-Walsh D. J.t McKeown J. G, Compatibility of polyacrylates and polyme-
thacrylates with poly(vinyl chloride). 2. Measurement of interaction
parameters,—Ibid., N 11, p. 1335—1339.
76. Ternary polymer mixtures/T. K. Kwei, H. L. Frisch, W. Radigan, S. Vogel.—
Macromolecules, 1977, 10, N 1, p. 157—161.
77. Thermodynamic interactions in polymer systems by gas — liquid chromato-
graphy. 4. Interactions between components in a mixed stationary phase /
D. D. Deshpande, D. Patterson, H. P. Schreiber, С S. Su.— Ibid., 1974, 7,
N 4, p. 530—536.
78. Formation of polyurethane networks on polypropyleneglycol and 4,4'-dipheny-
Imethane diisocyanate / A. E. Nesterov, T. E. Lipatova, K. Dusek et al.—
Angew. makromol. Chem., 1976, 52, S. 39—52.
79. Липатов Ю. С, Нестеров А. Е., Игнатова Т. Д. Исследование
множественных переходов в полиуретанах методом газовой хроматографии.— Укр.
хим. журн., 1975, 41, № 9, с. 939—945.
80. Kwei Т. К., Patterson G. D., Wang Г. Т. Compatibility of mixtures poly-
(vinylidene fluoride) and poly(ethyl methacrylate).—Macromolecules, 1976,
9, N 5, p. 780—785.
81. Kwei T. K-* Frisch H. L. Interaction parameter in polymer mixtures.— Ibid.,
1978, 11, N 6, p. 1267—1271.
82. Runt J, P. Polymer blends containing isotactic polystyrene: thermal
behavior of model systems.— Ibid., 1981, 14, N 2, p. 420—433.
83. Липатов /О. С, Нестеров А. Е, Термодинамика взаимодействий в смесях
полимеров.— Композиц. полимер, материалы, 1971, вып. 1, с. 5—20.
84. Su С. S., Patterson D. Determination by gas — liquid chromatography of
the polystyrene-poly(vinyl methyl ether) interaction.— Macromolecules, 1977,
10, N 3, p. 708—710.
85. Su С S., Patterson D.y Schreiber H. P. Thermodynamic interaction and
properties of polyvinylchloride — plasticizer systems.—J. Appl. Polym. Sci.,
1976, 20, N 4, p. 1025—1034.
86. Scholte Th. G. Determination of thermodynamic parameters of polymer — so-
- lvent systems by light scattering.— Eur. Polym. J., 1970, 6, N 8,
p. 1063—1074.
87. Upper and lower critical solution temperatures in polyethylene solutions/
N. Kuwahara, S. Saeki, T. Chiba, M. Kaneko.— Polymer, 1973, 15, N 2,
p. 777—781.
88. Flory P. J. Principles of polymer chemistry.— New York: Cornell univ. press.
1973.—594 p.
220
89. Nishi 7\, Wang Т. Т. Melting point depression and kinetic effects of cooling
crystallization in poly(vinylidene fluoride) — poly(methyl methacrylate)
mixtures.— Macro molecules, 1975, 8, N 6, p. 909—915.
90. Allen G., Gee G., Nicholson J. P. The miscibility of polymers. 1. Phase
equilibrium in systems containing two polymer and mutual solvent.— Polymer,
1960, 1, N 1 p. 56—62.
91. Tanaka 7\, Inagaki M. Light scattering from polymer — polymer —
solvent ternary systems. A simple and reliable method estimating the
interaction between unlike polymers.— Macromolecules, 1979, 12, N 6, p. 1299.
92. Stockmayer W. H. Light scattering in multicomponent systems.—J. Chem.
Phys., 1950, 18, N 1, p. 58—61.
93. Stockmayer W. #., Stanley H. E. Light — scattering measurement of
interactions between unlike polymers.— Ibid., p. 153—154.
94. Эскин Т, Е. Рассеяние света растворами полимеров.— М.: Наука, 1973.—
350 с.
95. Roe R.-J., Zin W.-C. Determination of the polymer — polymer interaction
parameter the polystyrene — polybutadiene pair,— Macromolecules, 1980,
13, N 6, p. 1221—1228.
96. Нестеров A. E.t Липатов Ю. С. Обращенная газовая хроматография в
термодинамике полимеров.— Киев.: Наук, думка, 1976.— 128 с.
97. Friday Л., Cooper D. R.t Booth С. Mixing of ethylene oxide and propylene
oxide olygomers. 2. Phase separation.— Polymer, 1977, 18, N 2, p. 171—174.
98. Блинов В. С, Тагер А. А. О влиянии природы сорбата на величину энергии
смешения Гиббса полимеров, рассчитанную из данных статической сорбции,—
Высокомолекуляр. соединения, А, 1981, 23, № 9, с. 2122—2128.
99. Параметры термодинамического взаимодействия диацетата целлюлозы с
некоторыми полимерами/Б. А. Голендер, 3. Г. Сагдиева, Ш. Фузаилов,
С. А. Ташмухамедов. Там же, Б, 1976, 18, № 9, с. 688—691.
100. Wignall G. D., Child H. #., Li — Aravena F. Small — angle neutron
scattering studies of a compatible polymer blends atactic polystyrene — poly
B,6 — dimethylphenyleneoxide).—Polymer, 1980, 21, N 2, p. 131 — 134.
101. Russell T. P., Stein R. S. Scattering studies of amorphous polymer blends.—
Arrier. Chem. Soc. Polym. Prepr., 1979, 20, N 1, p. 24—27.
102. Calorimetric and neutron scattering studies of mixture polystyrene with poly
B,6 dimethyl-1, 4-phenylene oxide) and its brominated derivatives/R. P.
Kambour, R. С Bopp, A. Maconnachie, W. J. MacKnight.— Polymer, 1980,
21, N 2, p. 133—134.
103. Structure and thermodynamics of polymer blends from neutron scattering
experiments/J. Jelenic, R. G. Kirste, B. J. Schmitt, S. Schmitt-Steker.—
Makromol. Chem., 1979, 18. N 8, S. 2057—2059.
104. Кулезнев В. Я. Смеси полимеров.— Химия, 1980.— 303 с.
105. Paul D. R., Barlow J. W. Polymer blends (or alloys).— J. Macromol. Sci. C,
1980, 18. N 1, p. 109—168.
106. Нестеров Л. Е. Справочник по физической химии полимеров. Т. 1.
Свойства растворов и смесей полимеров / Под ред. Ю. С. Липатова.— Киев:
Наук, думка, 1984.—528 с.
107. Elmqvist С, Svanson S. E. An NMR investigation of compatibility in blends
polyvinylchloride and ethylene — vinylacetate copolymer.— Eur. Polym. J.,
1975, 11. N 11, p. 789—793.
108. McMaster L. P. Aspects of liquid — liquid phase transition phenomena in
multicomponent polymeric systems.— Amer. Chem. Soc. Prepr., 1974, 15.
N 1, p. 254-259.
109. Воюцкий С. С. Растворы высокомолекулярных соединений.— М. : Гос-
химиздат, I960.— 132 с. \
ПО. Novakov P., Konstantinov Ch.t Mitanov P. A study of the compatibility of
synthetic rubber with phenolic resins.—J. Appl. Polym. Sci., 1972, 16.
N 7, p. 1827—1832.
111. Akijama 5., Miasa /0 The heat of mixing of both poly(vinyl nitrate) with
poly(vinyl pivalate) and poly(vinyl nitrate) with polyethylene - со - vinyl
acetate) mixtures.— Polym. J., 1979, II, N 2, p, 157—158.
221
112. Di — Paola Baranyi G., Degre P. Thermodynamic characterization of
polystyrene - poly(n - butyl methacrylate) blends.— Macromolecules, 1981, 14,
N 5, p. 1456 — 1460.
113. Кулезнев В. #., Игошева К- М. Исследование плотности смесей полимеров.—
Высокомолекуляр. соединения, 1962, 4, № 12, с. 1858—1862.
114. Eried J. R.. Karasz F. E.. MacKnight W. /.Compatibility of poly B,6 -
dimethyl - 14 - phenylene oxide) PPO / polystyrene-co-chlorostyrene) blends.
1. Differential scanning calorymetry and density studies.—
Macromolecules, 1978, 11. N 1, p. 150—158.
115. Rancy B. G. Two — component polymer systems: physical properties as related
to compatibility and interaction.— J. Polym. Sci. C, 1975, N 51, p. 89—104.
116. Hickman J. J.. Ikeda R. M. Studies of polymer blends. 1. Compatibility of
poly(vinyl chloride) and poly(ethylene — со — vinyl acetate — со — sulfur
dioxide).—J. Polym. Sci. : Polym. Phys. Ed., 1973, 11, N 9, p. 1713—1721.
117. Исследование смеси полиэтилена с воском / А Г. Сирота, М. Д. Пукшан-
ский, Е. Л. Виноградова и др.— Высокомолекуляр. соединения, Б, 1975,
.17. № 9, с. 695—698.
118. Savard S.. Levesque D., Prud'homme R. E. Partial miscibility of cellulose—
polуacrylonitrile blends.— J. Appl. Polym. Sci., 1979, 23. N 7, p. 1943—1949.
119. Yamakawa H., Fujii M. Binary claster integrals in tne theory of dilute
polymer solutions.—J. Chem. Phys., 1973, 58. N 4, p. 1523—1528.
120. Lipatov Yu. S.t Nesterov A. ?., Ignatova T. D. Some peculiarities of
thermodynamic behavior of olygomeric mixtures with specific interaction between
components.— Eur. Polym. J., 1979, 15, N 8, p. 775—780.
121. Van den Esker M. W. /., Vrij A. Incompatibility of polymer solutions. 1. Bino-
dals and spinodals in the system polystyrene + polyisobutylene +
toluene.—J. Polym. Sci.: Polym. Phys. Ed., 1976, 14. N 11, p. 1943—1952.
122. Helfand E.. Tagami Y. Theory of interface between immiscible polymers.—
J. Polym. Sci. B, 1971, 9. N 10, p. 741—746.
123. Thermal transitions in cold — crystallized blends of isotactic and atactic
polystyrene / E. Martuscelli, G. Demma, E. Drioli et al.— Polymer, 1979,
20. N 5, 571—576.
124. Bergmans #., Overbergh N. Crystallization and melting of the system
atactic polystyrene + polyB,4 - dimethyl-1,4-phenylene oxide).— J. Polym.
Sci. B, 1977, 15. N 10, p. 1757—1767.
125. Walsh D. J., Mckeown J. G. Compatibility of polyacrylates and polymethy-
1 methacrylates with poly(vinylchloride). 2. Measurement of interaction
parameter.— Polymer., 1980, 21. N 11, p. 1335—1339.
126. Roerdink E., Challa G. Influence of tacticity of poly(methyl methacryl ate)
on compatibility with poly(vinyl chloride).—Ibid., 1978, 19, N 2, p. 173—177.
127. Липатов Ю. С, Нестеров А, Е.. Игнатова Т. Д. Термодинамические
параметры взаимодействия между компонентами в смесях олигомеров.— Докл.
АН СССР, 1975, 224, № 3, с. 634—637.
128. Липатов Ю. С, Файнерман А. Е. О возможности образования
термодинамически устойчивых дисперсных полимерных систем.— Там же, 1983,
268, № 4, с. 1270—1275.
129. Русанов А. И. Фазовые разновесия и поверхностные явления.— Л. :
Химия, 1967.— 388 с.
130. Термодинамическая совместимость компонентов в расплаве и степень
кристалличности формирующихся из них полимерных смесей / Ю. С. Липатов,
А. Е. Нестеров, Т. Д. Игнатова, А. А. Лашук.—Высокомолекул. соединения,
А, 1980, 22, № 12, с. 2665—2671.
131. Comparison of thermodynamic and rheological properties of binary polymer
mixtures / Yu. S. Lipatov, A. E. Nesterov, V.F. Shumski et al.— Eur. Polym.
J., 1982, 18, N 11, p. 981—986.
132. Чалых А. ?., Сапожникова И.Н. Исследование зон взаимодиффузии в
системе поливинил хлорид — полиметилметакрилат.— Композ. полимер,
материалы, 1980, вып. 7, с. 46—49.
133. Walsh D. /., Laingle S., Zhiknan C. A pair of high molecular weight
compatible polymers showing upper critical solution temperature behaviour.—
Polymer, 1981, 22. N 8, p. 1005—1007.
222
134. Equation of state theory applied to mixtures of isotactic poly(ethyl metha-
crylate) and poly(vinylidene fluoride) / G. Ten Brinke, A. Eshuis, E. Ro-
erdink, G. Challa.—Macromolecules, 1981, 14, N 3, p. 867—870.
135. Statistical thermodynamics of polymer solutions based on free volume
theory / F. Hamada, T. Shiomi, K. Fujisawa, A. Nakajima A.—Ibid., 1980,
13. N 3, p. 129—134.
136. Allen G., Gee G., Nicholson J. P. The miscibility of polymers. 2. Miscibi-
lity and heat of mixing of liquid polyisobutenes and silicones.— Polymer,
1961, 2, N 1, p. 8-17.
137. Взаимодействие бинарных смесей олигоэфиргликолей с диизоцинатами /
А. С. Зубко, А. Е. Нестеров, Ш. Г. Венгеровская, Л. С. Шейнина.— Высоко-
молекуляр. соединения, А, 1975, 17, № 5, с. 1054—1057.
138. Ягфаров М. ДО., Мухутдинов А. А., Игонина Н. С. Исследование кристал-
лизуемости уретановых эластомеров на основе полиэтилен - и полидиэти-
ленадипината.— Синтез и физико - химия полимеров, 1973, вып. 12,
с. 94-97.
139. Гудзера С. С, Спирин Ю. Л., Магдинец В. В. Исследование сополимеров
олигоуретанакрилатов и олигоэфиракрилатов.— Там же, с. 101 —103.
140. Липатова Т. Э. Каталитическая полимеризация олигомеров и
формирование полимерных сеток.— Киев : Наук, думка, 1974.— 208 с.
141. Bailey F. E.t Callard R. W. Some properties of polyethylene oxide) in
aqueous solution.—J. Appl. Polym. Sci., 1959, 1. N 1, p. 56—62.
142. Koningsveld R., Kleintjiens L. A. Thermodynamics of polymer mixtures.—
J. Polym. Sci. C, 1977, N 61, p. 221—249.
143. Suzuki H. On the second virial coefficient for solutions of polystyrene
mixtures.— Brit. Polym. J., 1977, 9, N 3, p. 222—227.
144. Krigbaum W. R.t Flory P. J. Statistical mechanics of dilute polymer
solutions.—J. Chem. Phys., 1950, 18, N 5, p. 1086—1090.
145. Kirkwood J. G., Goldberg R. J. Light scattering arising from component
fluctuations in multicomponent systems.— Ibid., N 1, p. 54—57.
146. Липатов С. M. Высокомолекулярные соединения (лиофильные
коллоиды).— Ташкент: Изд - во АН БССР, 1943.— 160 с.
147. Липатов Ю. С. Физическая химия наполненных полимеров.— Киев :
Наук, думка, 1977.— 304 с.
148. Lipatov Y и. S*f N ester ov Л. Е. The influence of thickness of polymeric
stationary phase on its properties determined by gas chromatography.— Macro-
molecules, 1976, 8, N 6, p. 889—895.
149. Адсорбция полимеров из концентрированных растворов и температура
стеклования адсорбционных слоев / Ю. С. Липатов, А. Е. Нестеров, Л. М.
Сергеева и др.— Укр. хим. журн., 1975, 41, № 10, с. 1075—1080.
150. Семенович Г. М., Липатов Ю. С, Сергеева Л. М. Исследование структуры
граничных слоев в системе каучук — эпоксидная смола— хлористый
аммоний.— Высокомоле кул яр. соединения, А, 1978, 20, № 10, с. 2375—2380.
151. Reich S., Cohen Y. Phase separation of polymer blends in thin films.— J.
Polym. Sci.: Polym. Phys. Ed., 1981, 19, N 8, p. 1255—1267.
152. Липатов Ю. С, Файнерман A. E.t Шифрин В. В, О влиянии минерального
наполнителя на поверхностное натяжение бинарной смеси полимеров.—
Докл. АН СССР, 1977, 234, № 3, с. 596—599.
153. Фабуляк Ф. Г. Исследование молекулярной подвижности полимерных
цепей в поверхностных слоях в зависимости от природы поверхности :
Автореф. дис. ... д-ра хим. наук.— К., 1976.— 44 с.
154. Prud'homme R, E. Fractionation in mixtures of polyethylene oxide fractions
during crystallization.—J. Polym. Sci. : Polym. Phys. Ed., 1982, 20, N 2,.
p. 307—317.
155. Koningsveld /?., Staverman A. J, Liquid— liquid phase separation in multi
component polymer solutions.—J. Polym. Sci. A, 1968, 2, N 6, p. 349—366-
ГЛАВА 7
ТЕРМОДИНАМИКА
ВЗАИМОДЕЙСТВИЙ КОМПОНЕНТОВ
И СВОЙСТВА БИНАРНЫХ
ПОЛИМЕРНЫХ СМЕСЕЙ
Имеющиеся в настоящее время данные
свидетельствуют о тесной связи между термодинамическим состоянием
системы в расплаве и ее вязкостью, кинетикой и механизмом фазового
расслоения со структурой и свойствами конечного продукта и др.
В данном разделе мы продемонстрируем примеры такой
взаимосвязи для обычных полимеров и для взаимопроникающих полимерных
сеток.
7.1. ТЕРМОДИНАМИКА И ВЯЗКОСТЬ РАСПЛАВОВ СМЕСЕЙ ПОЛИМЕРОВ
В некоторых работах по исследованию концентрационной и
температурной зависимости вязкости расплавов смесей полимеров
обнаружено аномальное ее падение в узкой области концентраций,
преимущественно при малом содержании одного из компонентов [1—7]. В
нескольких случаях обнаружен S-образный характер кривых вязкость —
состав [7, 8], резкое падение вязкости с обеих сторон
концентрационного диапазона [9—13] и другие отклонения от поведения, типичного
для смеси двух низкомолекулярных жидкостей. Наличие таких
аномалий объясняется в основном возникновением в узкой области
составов двухфазной структуры смеси [2, 3, 13, 14]. Предложены разные
механизмы для объяснения наблюдаемых аномалий, сущность которых
сводится к представлениям о том, что наблюдаемый эффект
сопровождает прямой и обратный переход системы из однофазного состояния в
двухфазное в области температур, где компоненты термодинамически
несовместимы [8, 13, 15]. В принципе, резкое падение вязкости должно
было бы наблюдаться для смесей полимеров с обеих сторон
концентрационного диапазона, по крайней мере, для смеси полимеров с близкими
молекулярными массами, фазовая диаграмма для которой, вообще
говоря, должна быть близка к симметричной [16]. В то же время для
некоторых систем резкое падение вязкости наблюдается лишь с одной
стороны концентрационного диапазона (рис. 7.1—7.5), с другой
стороны в некоторых случаях, как указывалось выше, обнаружено
значительное падение вязкости на обоих концах концентрационного
диапазона (рис. 7.6). Все эти факты свидетельствуют о том, что лишь
переходом системы в двухфазное состояние вряд ли однозначно можно объяс-
224
нить наблюдаемые аномалии
вязкости. Дело в том, что при
использовании критерия двухфазности
системы в упомянутых работах,
однако, не получено прямых
данных о термодинамической
устойчивости системы в той области
температур, при которой определяли
концентрационную зависимость
вязкости. Впервые сопоставление
концентрационной зависимости
вязкости с параметрами
термодинамического взаимодействия,
характеризующего
термодинамическую устойчивость смеси, во всей
концентрационной области
проведено нами в работе [15]. Была
обнаружена удивительная симбатность
концентрационной зависимости
вязкости и параметра
термодинамического состояния системы в
расплаве при одних и тех же
температурах опыта. Этот результат
позволил нам предположить, что
характер концентрационной зависимости
вязкости должен определяться
главным образом фазовым
состоянием системы в той области
температур и составов, где измеряется
вязкость (в частности
симметричностью или несимметричностью
фазовых диаграмм в плоскости
температура— состав), а также типом
критической температуры
смешения при характеристике
температурной зависимости вязкости. Для
подтверждения высказанного
предположения мы сопоставили
концентрационную зависимость
вязкости с особенностями
термодинамического состояния,
характеризуемого параметрами
термодинамического взаимодействия полимер —
полимер %23 и фазовыми
диаграммами для большого числа
бинарных систем в расплаве, включая
смеси полимер гомологов. Среди
исследованных смесей были
различные сочетания полиэтилена вы-
8 -*-42Q
460
440
Рис. 7.1. Фазовая диаграмма (а) и
концентрационная зависимость
вязкости (б) при 443 (/) и 473 К B) для
расплава смеси ацетобутирата
целлюлозы с полиоксиметиленом [24].
480
470
Цч
3,5
3,0
(Па-с)
20
40
P0Mt%
Рис. 7^2. Фазовая 'диаграмма (а) и
вязкость при 443 (/) и 473 К B) для
расплава смеси полиэтилена низкой
плотности с полиоксиметиленом [241.
225
лс,%
50
WO
Л9Г,%
Рис. 7.3. Концентрационная зависимость вязкости (/, 2) и параметра Хай («?> 4}
для смеси полистирола с поликарбонатом при температуре:
/ — 453; 2— 473; 3 — 465; 4 — 475 К [24].
Рис. 7.4. Концентрационная зависимость параметра %2з (-0 и вязкости B) для
смесей ПЭГ (М = 1,6-10*) с ППГ (М= 2-103) при 363 К [24].
сокой и низкой плотностей, полиоксиметилена, ацебутирата
целлюлозы, сополимеров акрилонитрила со стиролом и этилена с винила-
детатом, поликарбоната и др. Широкий круг выбранных полимеров
позволил исследовать реологические свойства бинарных смесей
компонентов —- как относительно близких друг к другу по величине
вязкости, так и сильно отличающихся. Как показали проведенные
работы, смешение полимеров в расплаве в определенной области
температур и составов приводит во всех случаях к резкому
снижению вязкости в узком диапазоне составов, причем для одних систем
такое падение вязкости наблюдается только со стороны малых
содержаний одного компонента, а в других — со стороны малых
содержаний обоих компонентов. Чтобы установить общность
физической картины, необходимо было связать наблюдаемые эффекты с
величинами вязкости исходных компонентов, а для влияния химической
природы смешиваемых полимеров — исследовать полимеры, сильно
отличающиеся друг от друга по температурному коэффициенту
вязкости. Примером такой системы может служить смесь ацетобутирата
целлюлозы с полиоксиметиленом: для первого компонента температурный
коэффициент вязкости невелик, а для второго весьма значителен. На
226
50
С?В,%
ПС
Рис. 7.5. Концентрационная зависимость вязкости {1) и параметра х23 B, 3)
для смесей полиэтилена высокой плотности с сополимером этилен — винилаце-
тат (массовая доля винил ацетат 19,5 %):
7 — 423; 2 — 435; 3 — 408 К [24].
Рис. 7.6. Концентрационная зависимость вязкости смесей полистирола с
полиэтиленом (/—5) и полипропиленом (/'—«?') при 473 К при -у, равном:
/, Г— 200? 2, 2'^ 300; 3, 3'-* 400 в—1 [9].
рис. 7.1 показаны зависимости вязкости от состава для данной пары
полимеров при температурах 453 и 473 К и фазовая диаграмма,
полученная расчетнымиутем на основе данных концентрационной и
температурной зависимостей параметров %2з и соответствующего уравнения
для свободной энергии смешения (см. параграф 6.3). Аналогично
составлена фазовая диаграмма и для смеси полиоксиметилен —
полиэтилен низкой плотности (см. рис. 7.2). Как видно из рис. 7.1, в
области малых добавок полиоксиметилена к ацетобутирату целлюлозы
система термодинамически неустойчива в той области температур, при
которых была измерена вязкость (эти температуры показаны
штриховыми линиями на рис. 7.1). В такой же области составов при всех
изученных температурах (на рис. 7.1 приведены данные лишь для двух
температур) вязкость резко уменьшается. В области же малых добавок
АБЦ к ПОМ система термодинамически устойчива при тех
температурах, при которых была измерена вязкость. Видно, что в этой области
составов величины вязкости больше аддитивных. Следовательно,
S-образный характер концентрационной зависимости вязкости четко
отражает фазовое состояние системы, в частности переход от однофаз-
8*
227
ного состояния в двухфазное; т. е. в данном случае такая зависимость,
вязкости является следствием несимметричности фазовой диаграммы
что является, скорее, правилом, чем исключением, будучи следствием
существенной разности в молекулярных массах смешиваемых
компонентов, ММР образцов и др. [16, 17] (рис. 6.2). Такие несимметричные
фазовые диаграммы были получены для многих систем полимер —
полимер, в частности с НКТС [18]. Естественно ожидать, что в случае
симметричных фазовых диаграмм (в такие фазовые диаграммы тоже
были получены, причем как для систем с ВКТС, так и с НКТС [19, 20])
будет наблюдаться и симметричность концентрационной зависимости
вязкости.
Для системы ПЭНП — ПОМ в области малых. добавок второго
к первому в температурной области измерения вязкости (на рис. 7.2
приведена концентрационная зависимость вязкости для двух
температур) система термодинамически неустойчива (в области выше 20 % -ного
содержания ПОМ определить надежно точки спинодали не
представляется возможным из-за очень слабой температурной зависимости
параметра %23, а следовательно, и A G). В этой области малых содержаний
ПОМ наблюдается и падение вязкости (см. рис. 7.2).
Более сложный характер концентрационной зависимости вязкости
отмечается для системы полистирол — поликарбонат. Из рис. 7.3
(кривые 1 и 2) видно, что введение в расплав ПС до 1 % ПК вызывает
значительное уменьшение вязкости системы. Увеличение содержания
ПК в смеси до 5 % приводит к интенсивному росту вязкости, хотя ее
экспериментальные значения и ниже аддитивных величин. При
дальнейшем росте концентрации ПК вязкость смеси снова понижается и
при 10 %-ном содержании ПК на кривой вязкость — состав
появляется второй минимум. В области от 10 до 50 % ПК вязкость смеси
возрастает, однако при дальнейшем росте концентрации ПК ее рост
замедляется. При содержании 90—95 % ПК снова вязкость резко
возрастает, а при 97—98 %-ном содержании ПК резко снижается. При
содержании 0,5 % ПС в ПК на диаграмме вязкость — состав
отмечается третий минимум. Характерно, что во всем диапазоне составов
вязкость смеси ПС — ПК ниже аддитивных значений. Несмотря на такую
сложную концентрационную зависимость вязкости для этой системы,
она очень хорошо коррелирует с концентрационной зависимостью
параметра термодинамического взаимодействия между компонентами
смеси в данной области температур (см. рис. 7.3, кривые 3 и 4). Так,
в области малых добавок ПК в ПС меньшим значениям параметра х23
соответствует повышение вязкости системы и наоборот, т. е. в этом
случае уменьшение термодинамической совместимости системы приводит
к понижению вязкости, а увеличение термодинамической
устойчивости — к ее повышению. Таким образом, наблюдаются закономерности,
обнаруженные для первых двух рассмотренных систем.
Аналогичная корреляция концентрационной зависимости вязкости
с параметрами термодинамического взаимодействия характерна и для
смесей полиэтиленгликоль — полипропиленгликоль (см. рис. 7.4),
а также для системы сополимер этилен — винилацетат — полиэтилен
высокой плотности (см. рис. 7.5). Интересной особенностью системы
228
СЭВ—ПЭВП является уменьшение ее термодинамической устойчиво*
сти с повышением температуры во всем концентрационном диапазоне,
кроме 10 %-ного содержания СЭВ (см. рис. 7.5). Эта система неполярна,
следовательно, никаких специфических взаимодействий в ней не
должно быть, тем не менее ее поведение типично для систем с НКТС.
Возможной причиной такого поведения может быть увеличение вклада
члена «свободного объема» в параметр Х23 с повышением температуры
[21]. Дело в том, что для СЭВ в области температур 413—493 К
удельный объем практически не меняется (т. е. коэффициент объемного
расширения близок к нулю [22]), а для ПЭ он в данной области температур
растет. Увеличение различия в свободных объемах компонентов и
может стать причиной наличия НКТС в системе [21]. Особенности же
температурной и концентрационной зависимости вязкости ддя этой
системы четко коррелируют с концентрационной зависимостью параметров
Хаз ПРИ разных температурах (см. рис. 7.5).
Данные по концентрационной зависимости вязкости при разных
температурах показывают, что для многих систем эффекты падения
вязкости не связаны с величинами вязкостей исходных систем, а также
мало зависят от температуры в определенной температурной области.
В частности, при исследовании концентрационных зависимостей
вязкостей для некоторых систем, термодинамическая совместимость
которых была оценена косвенным образом, обнаружено, что для одних
систем вязкость понижается очень сильно, а для других очень слабо
или вовсе не изменяется [6, И, 12, 15, 23]. Причиной таких различий
может являться тип критической температуры смешения (верхняя или
нижняя) [24]. В случае систем с НКТС, если смешение компонентов
происходит в расплаве в области температур, отвечающих фазовому
разделению, вязкость при температурах, лежащих ниже спинодали,
может и не проявлять никаких аномалий. Это характерно для систем
с кристаллизующимися полимерами, имеющими НКТС выше
температуры плавления образца. Наличие или отсутствие резкого падения
вязкости может быть связано с механизмом фазового разделения [24]:
если оно протекает по нуклеационному механизму (см. гл. 8), следует
ожидать резкого падения вязкости в области неустойчивости системы,
а если по спинодальному,— образование взаимосвязанных структур,
типичных для спинодального механизма распада системы, не приводит
к резким изменениям вязкости. Здесь опять-таки все зависит от формы
фазовой диаграммы и температурной области измерения вязкости, т. е.
от того, в какой области фазовой диаграммы находится система в
условиях измерения — между бинодалью и спинодалью, или под спино-
далью. Поэтому даже для одной и той же пары полимеров эффект
изменения вязкости может сильно зависеть от температурной области.
Лишь в случае большой крутизны бинодалей и спинодалей нуклеацион-
ный механизм может реализоваться в широком температурном
интервале. При меньшей крутизне (что наиболее часто встречается —см.,
например, рис. 6.2) понижение температуры для системы с ВКТС и
повышение ее в случае системы с НКТС может привести к изменению
механизма фазового разделения и, соответственно, эффекты изменения
вязкости в переходной области от однофазного состояния к двухфаз-
229
чюму будут выражены по-разному. С одной стороны, получающиеся
аномалии вязкости можно объяснить видом фазовой диаграммы и
типом КТС. С другой стороны, по типу концентрационной зависимости
вязкости, по-видимому, можно в определенной мере судить о форме
фазовой диаграммы, а по результатам измерений температурной
зависимости вязкости при данной концентрации — о типе КТС.
Дальнейшие исследования в этом плане, на наш взгляд, должны быть
достаточно интересными.
Какие же факторы влияют на понижение вязкости? По нашему
мнению, такими факторами являются неоднородность распределения
концентраций компонентов и возникновение при расслоении
дополнительного свободного объема, локализующегося в межфазной области.
Экспериментальные данные по вязкости исходных полимеров для
разных температур и различных полимерных пар мы сравнили с
расчетными значениями, полученными по уравнению Фогеля—Таммана [12]
ур$ Ау В и То—эмпирические константы, причем То представляет
йобой некую критическую температуру, при которой свободный объем
Исчезает. Из полученных данных можно вычислить долю свободного
^бъема при температуре стеклования и при температуре
эксперимента, исходя из соотношения
а также энергию активации течения по уравнению
Обнаружено, что во всех смесях доля свободного объема возрастает
по сравнению с ее значениями для исходных компонентов, причем
прослеживается тенденция к последующему уменьшению этой величины
с повышением содержания одного компонента в другом. Известно, что
вязкость — величина, существенно определяемая свободным объемом,
поэтому увеличение последнего является важным фактором,
приводящим к понижению вязкости. Очевидно, что вследствие микрофазового
разделения в метастабильной области свободный объем локализуется
на межфазной границе, вернее, в межфазной области. Именно в
области метастабильного состояния системы в соответствии с
представлениями о механизме нуклеации возникают зоны, обедненные одним из
компонентов, иными словами, фактически переходные области между
двумя фазами. Эти области вследствие механизма разделения (в данном
случае и нуклеационного, и спинодального) характеризуются меньшей
плотностью, т. е. повышенным свободным объемом. Таким образом,
молекулярная причина понижения вязкости заключается в механизме
формирования новой фазы.
Термодинамическая теория процесса нуклеации разработана Гибб-
сом, а спинодального распада — Канном и Хиллардом [25], однако эти
процессы не рассматривались с точки зрения механических и реологи-
230
-0,2
ческих свойств, разделяющихся х25 О
на две фазы систем. В заключение
мы хотим еще раз подчеркнуть
очень важную особенность
проявления экстремальности вязкости
в бинарных системах.
Экстремальные изменения наблюдаются в
случаях прямого и обратного перехода
системы из однофазного в
двухфазное состояние при изменении
состава смеси (пересечении бино-
дали и спинодали) и постоянном
составе при изменении
температуры в зависимости от типа
критической температуры смешения.
Однако областью проявления
экстремальности является не
собственно точка перехода и,
соответственно, не момент перехода
системы из гомогенного состояния
в гетерогенное (или наоборот),
а (по определению Я. И.
Френкеля [26]), область «предпереход-
ных аномалий», или область «пред-
переходного состояния»,
возбужденного, с избыточной энергией,
локализованного в метастабильной
зоне между бинодалью и спино-
далью.
В гипотетической идеализированной системе с резко очерченным
идеальным переходом из гомогенного состояния в гетерогенное спино-
даль и бинодаль не разделены, метастабильная зона отсутствует и
область «предпереходных аномалий» практически незаметна. В
полимерных системах с высокой кинетической заторможенностью
переходных явлений, с низкими диффузионными способностями область «пред-
переходного состояния» обширна и метастабильна и позволяет не
только изучать эффекты экстремального изменения вязкости, но и находить
им практическое применение для переработки полимерных материалов,
в том числе наполненных полимеров.
Рассмотрим теперь еще один аспект реологического поведения
бинарных систем, существенный с точки зрения понимания «аномалий»,
вязкости. Здесь особый интерес представляют смеси полимергомологсв
различающихся по молекулярной массе, например смеси полимергр-
мологов с молекулярной массой выше и ниже критической. Мы
установили [27], что смеси полимергомологов, существенно различающихся
по молекулярным массам, в зависимости от температуры и состава
могут находиться в термодинамически устойчивом или неустойчивом
состоянии [27]. Очевидно, что и в такой системе, где
термодинамическая стабильность является функцией температуры, состава и молеку-
231
100
ПБД-4,%
Рис. 7.7. Концентрационная
зависимость параметра Х23 (а) и вязкости (б)
смесей фракций полибутадиена с Л1 =
2-10* и 5.10*:
а: 1 — 323; 2 — 357 К; б: 1 — 298; 2^313$
3 — 333 К [24].
лярной массы, в принципе, возможно проявление «аномалий» вязкости.
Мы изучили связь термодинамического состояния систем с их
вязкостью, используя смеси различных узких фракций полибутадиена (ПБ)
с крайними представителями гомологического ряда, в частности, с
молекулярной массой 2 . 104 (ПБ-1) и 5 . 10б (ПБ-4) (рис. 7.7).
Исследуемые смеси характеризуются отрицательными значениями
параметра Ъз» указывающими на их термодинамическую стабильность.
Концентрационная зависимость параметра %23, однако, оказывается
зависящей от молекулярных масс компонентов и их соотношения. Так, для
смеси ПБ-1—ПБ-4 эта зависимость бимодальна, что указывает на
различную термодинамическую устойчивость системы разного состава.
При увеличении молекулярной массы одного из компонентов смеси
аномальный характер концентрационной зависимости параметра %23ис-
чезает и при М = 7 • 104 значения %23 практически не изменяются
с концентрацией, оставаясь отрицательными во всем
концентрационном диапазоне (см. рис. 6.38). Энтальпийный вклад в параметр %23
для системы ПБ-1—ПБ-4 везде положителен, а энтропийный
—отрицателен, хотя зависимости этих характеристик от состава также
бимодальны. Только при составе 30 и 70% эти параметры близки к
нулю, т. е. поведения системы близки к идеальному. Сопоставление
таких данных с вязкостью (см. рис. 7.7) показывает, что последняя
везде ниже аддитивной и характеризуется определенной аномалией,
проявляя два минимума, совпадающие по концентрации с максимумами
концентрационной зависимости параметра %23. Таким образом,
наблюдаемые здесь закономерности в целом аналогичны обнаруженным
нами для смесей полимеров разной химической природы. Одна из
возможных причин такой аномалии заключается в том, что в силу
ограниченной термодинамической совместимости фракций разной
молекулярной массы в системе возникают молекулярные агрегаты различных
фракций (идея С. М. Липатова 30-х годов [28]), и смешение двух
фракций поэтому не происходит на истинно молекулярном уровне. Из
общеизвестных данных по наибольшей ньютоновской и наименьшей
неньютоновской вязкостям видно, что разделение полимерного объема
на области с преимущественным содержанием высоко- и
низкомолекулярных фракций дает объяснение существования этих mini и maxi.
В области невысоких скоростей сдвига деформируются и текут и те, и
другие. В области же высоких скоростей сдвига высокомолекулярная
часть не релаксирует и ведет себя как единое целое, а перемещение
и движение осуществляются только за счет более подвижных
молекулярных зон. Отсюда — наименьшая вязкость.
Данные, полученные для смесей полигомергомологов, не только
подтверждают общую закономерность изменения вязкости в
зависимости от термодинамического состояния системы (для смеси
высокомолекулярных фракций нет аномалий термодинамического поведения — см.
(рис. 6.38) — и нет аномалий вязкости [8]), но и показывают, что
эффекты изменения реологических характеристик, реализуемые путем
введения в один полимер небольших количеств другого, могут быть
достигнуты определенным смешением фракций разной молекулярной
массы, что имеет существенное практическое значение.
232
7.2. ВЗАИМОСВЯЗЬ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ СИСТЕМЫ
В РАСПЛАВЕ И СТЕПЕНИ КРИСТАЛЛИЧНОСТИ
СФОРМИРОВАННЫХ ИЗ НИХ ПОЛИМЕРНЫХ СМЕСЕЙ
Рассмотрим теперь, каким образом термодинамическое состояние
расплава смеси двух полимеров оказывает влияние на свойства
сформированного продукта, в частности на степень кристалличности. Как
было сказано раньше, смешение и переработку смесей полимеров
ведут, как правило, при высоких температурах, где компоненты
находятся в вязкотекучем состоянии, либо из раствора в общих
растворителях. В обоих случаях на процесс формирования структуры таких
систем существенное влияние должны оказывать кинетические эффек-
0J5 WO
ПС
лэгд
Рис. 7.8. Концентрационная зависимость параметров Х2з (Л $) и степени
кристалличности (&, 4) для смесей полистирола (М = 3040) с полиэтиленгли-
колем (М — 1,5-10*\ (/, 2) и полиэтиленгликоля с полиэтиленгликольадипи-
натом (М = 2000) (к 4).
Штриховые линии — аддитивные зависимости степени кристалличности для смеси ПЭГ-
ПС без (/) и с учетом (II) степени кристалличности ПС» для смеси ПЭГ — ПЭГ А
[45J. \
Рис. 7.9. Зависимость параметра Х2з V) и степени кристалличности B) от
тава для смеси ПЭГ-20001 — ПЭГ-40000 "'""
состава
[45[.
ты из-за высокой вязкости компонентов, причем при получении смеси
как из расплава, так и из раствора, поскольку в последнем случае
конечная стадия протекает ь условиях высокой концентрации и,
следовательно, высокой вязкости раствора. Кинетические эффекты могут
привести к тому, что структура конечного продукта может сохранять
черты исходного состояния системы, например, при быстром переводе
ее из расплава или из раствора в твердое состояние. Таким образом*
характер кинетических эффектов в процессах смешения и раздела фаз*,
в огромной степени должен зависеть от термодинамического состояния*
системы уже в исходном состоянии. Результатом влияния этих
факторов являются образование промежуточны^чобластей разной
протяженности [29, 30], рассмотренные выше аномалии реологического
поведения расплавов смесей полимеров, наличие те^ или иных типов темпе-
233
OJS
uo
ПЭГ
О 0,25
Парафин
Рис. 7.10. Зависимость степени
кристалличности (/) и параметров
Хаз^Л, %нC) я%8D) от состава
смеси парафин (М = 270) — поли-
этиленгликоль (М = 1,5* 10*) [45].
ратурных переходов [29, 31],
морфологических и иных особенностей
сформированного материала [23, 32].
Рассмотрим корреляции между
термодинамическим состоянием
расплава смеси двух полимеров и
степенью кристалличности
сформированного из такого расплава
композиционного материала. Смеси
формировали путем охлаждения расплава,
полученного при температуре, при
которой измерялись значения пара^
метра %28> Д° комнатной температуры
(рис. 7.8—7.11). Анализ этих данных
позволил прийти к следующему
заключению. Если в расплаве смесь
термодинамически неустойчива, степень
кристалличности системы больше
аддитивных значений. При
повышении термодинамической устойчивости
системы в расплаве степень кристалличности смеси при ее охлаждении
уменьшается. Это особенно четко прослеживается на таких системах,
как ПЭГ—ПС, ПЭГ—ПЭГА, ПЭГ—ПЭГ, ПЭГ — парафин (см. рис.
7.8—7.10), т. е. эти данные свидетельствуют о том, что при
кристаллизации из расплава, где компоненты смеси термодинамически
совместимы, при равных скоростях охлаждения фазовое расслоение
заторможено, чем и обусловлено понижение степени кристалличности
системы» В противном случае (когда смесь термодинамически неустойчива),
разделение на фазы идет более полно, поэтому степень/кристалличности
повышается. Неясным остается лишь факт превышения степенью
кристалличности аддитивных значений. В работе 03] было высказано
предположение о том, что в двухфазных смесях, Индивидуальные
компоненты которых существенно полидисперсн^, возможна миграция
низкомолекулярных фракций полимера, являющегося дисперсионной
средой, в межфазную область, чдо, в принципе, должно приводить к
повышению степени кристалличности среды.
, Кажущимся противоречием с выводом о большей степени
кристалличности термодинамически несовместимых смесей могут быть данные
рис. 6.23 для системы ППГ—ПЭГА, из которого видно, что для этой
системы, сформированной из расплава, где %2з>0> степень
кристалличности меньше аддитивных значений. На самом деле это система со
специфическим взаимодействием (водородные связи), резкое
ослабление которых происходит при повышении температуры [34], приводя
к %28>0- При низких температурах %23< 0, следовательно, система
становится совместимой при/Охлаждении.
Причиной уменьшения степени кристалличности смесей,
полученных из расплавов в условиях термодинамической совместимости
компонентов, может быть резкое замедление кинетики фазового
расслоения при охлаждении системы, Согласно уравнению (8.43), размер до-
134
менов при разделении фаз будет
тем меньшим, чем ниже
температура (т. е. чем больше разность
между температурой,
соответствующей спинодали, и температурой, до
которой система охлаждена)..
Следовательно, быстрое охлаждение
системы может привести к
ситуации, когда домены вообще не
могут образоваться, и мы получим
структуру переохлажденного
расплава [29] из-за большой вязкости
системы. Мак Мастер, например,
показал, что даже при температуре,
близкой к температуре фазового
расслоения, прогрев пленок из сме-
?и сополимера стирол — акрило-
нитрил с ПММА в течение 2 ч
приводит к увеличению доменов лишь
в 2 раза. Следовательно, в
результате быстрого охлаждения системы
можно получить метастабильное
состояние системы, которое может
сохраняться очень длительное
время. В ©том случае такие системы
могут рассматриваться как
«квазиравновесные» [35]; примером может
быть система ПС — поливинилметиловый эфир [36]. Система,
сформированная в условиях термодинамической совместимости
компонентов, в определенных интервалах температур расслаивается на две
фазы в течение недель и даже месяцев. Эти закономерности, в принципе,
характерны и для системы ПЭНП—ПОМ (рис. 7.11), охлажденной от
температуры 453 К, которой соответствует зависимость %2з от состава
(см. кривую рис. 7.11). Видно, что в области 10—90 % -ного
содержания ПОМ %23 > 0; значит, система термодинамически несовместима
при данной температуре. В этой же области степень
кристалличности смеси практически близка к аддитивной (кривая /). В области
малых содержаний ПЭНПд23< 0, что характерно для
термодинамически совместимых систем; в этой же области составов степень
кристалличности смеси меньше аддитивной, т. е. небольшие добавки ПЭНП к ЛОМ
приводят к резкому торможению процесса кристаллизации ПОМ
(вероятно, и ПЭНП) из расплава. Эти небольшие добавки ПЭНП,
по-видимому, могут рассматриваться как стабилизаторы дисперсной фазы,
возникающей при кристаллизации ПОМ из расплава. При малых
содержаниях ПОМ и ПЭНП наблюдается значительное повышение сте*
пени кристалличности системы по сравнению с аддитивными
значениями (см. рис. 7.11). Такое существенное увеличение степени
кристалличности связано, вероятно, с тем, что к^йисталлизация ПЭНП
протекает в присутствии уже сформировавшихся твердых частиц ПОМ (раз*-
Рис. 7.11. Зависимость степени
кристалличности (/) и параметров
Хаз B — 5), %нF) и %sG) от состава
для смеси полиэтилен низкой
плотности— полиоксиметилен при
температурах: 476 B), 465 E), 453 D) и 445 К
E) [45].
235
личие в температурах кристаллизации ПОМ и ПЭНП составляет
примерно 60°), которые могут рассматриваться как твердый наполнитель.
Поскольку из-за высокой вязкости системы частицы ПОМ не могут
образовать сплошной второй фазы, их можно рассматривать как
центры кристаллизации (зародыши) по аналогии с обычными минеральными
наполнителями [37]. Понижение степени кристалличности наблюдали
и авторы работы [38].
Таким образом, рассмотренные результаты свидетельствуют об
аналогии изменения термодинамического состояния систем в
расплавах их реологических свойств и свойств композиций в твердом
состоянии. Как правило, области минимума вязкости (т. е. условия
несовместимости полимеров) соответствует область максимума механических
свойств [39] (в области малых добавок одного из компонентов). Это,
очевидно, связано с тем, что структура, возникающая в метастабиль-
ном состоянии, замораживается при переходе через температуру
плавления или стеклования, в результате чего в твердом состоянии
появляется своеобразная структура наполненного полимера с тем
отличием, что варьируя соотношение составов (и температур), можно
регулировать размеры микрофазовых частиц или микрофазовых областей
переменного состава, их распределение в объеме и характер
взаимодействия друг с другом.
Следовательно, термодинамическое состояние системы в расплаве
не только влияет на реологические свойства (что позволяет широко
использовать эти эффекты при переработке пластмасс), но и улучшает
физико-механические свойства получаемых композиционных
материалов.
В заключение остановимся на следующем.
Ниши [40], рассматривая смеси двух кристаллизующихся
полимеров, предположил, что они должны иметь точку эвтектики. Так, если
два кристаллизующихся полимера совместимы при высокой
температуре в расплаве, то при охлаждении смеси они будут кристаллизоваться.
Температура плавления каждого из кристаллизующихся полимеров
может быть тогда выражена в виде
1/Тпл> 1—^ (-^1/ДВД)Х12Фа2,
'пл, 1
1/Тпл, i—J- =-(RV2/AH2V1)%n<?l,
Упл,2
где V{ и АН{ — молярный объем мономерного звена i-ro компонента
и энтальпия плавления моля сегментов этого полимера соответственно.
При некотором составе температуры плавления обоих полимеров
должны совпадать (температура эвтектики). Если (АЯХ/ДЯ2) = (F2/^iJ» то
объемную долю одного из полимеров в эвтектической точке можно
записать так:
236
В некоторых работах точки эвтектики были обнаружены для систем
полимер — растворитель [41—44], однако для смесей полимеров
такого рода данные пока отсутствуют.
1. Влияние термической предыстории на вязкостные свойства бинарнкх
полимерных систем / Г. В. Виноградов, А. Я. Малкин, В. Н. Кулезнев,
В. Ф. Ларионов.— Коллоид, журн., 1966, 28, № 6, с. 809—813.
2. Кулезнев В. Н. Особенности структуры и свойств смесей полимеров.— В кн.:
Композиционные полимерные материалы.— Киев: Наук, думка, 1975,
с. 93—109.
3. Свойства смесей кристаллизующихся полимеров в вязко-текучем
состоянии / В. Ф. Шумский,. А. С. Дорожкин, Ю. С. Липатов, И. П. Гетманчук.—
В кн.: Смеси и сплавы полимеров. Киев. : Наук, думка, 1978, с. 79—86.
4. Липатов Ю. С, Шумский В. Ф., Гетманчук И, П. Реологические свойства
смесей аморфных кристаллизующихся полимеров на примере смесей
полиамида с сополимером акрилонитрила со стиролом.— Высокомолекуляр.
соединения А, 1979, 21, № 9, с. 2093—2098.
5. Липатом Ю. С. Межфазные явления в полимерах.— Киев : Наук, думка,
1980.— 260 с.
6. Липатов Ю. С, Шумский В. Ф., Гетманчук И. #.. Реологические свойства
смеси полиэтилена с полистиролом.— Высокомолекуляр. соединения А, 1981,
23 ,№ 1, с. 44—49.
7. Липатов Ю. С, Шумский В. Ф.,г Гетманчук И. П. Вязкость смесей полипро-
пиленгликоля и полиэтиленгликоля различного молекулярного веса.—
Докл. АН СССР, 1976, 229, № 6, с. 1382—1385.
8. Кулезнев В. Н. Смеси полимеров.— М. : Химия, 1980.— 303 с.
9. Нап С. ZX, Yu. Т. С. Rheological properties of molten polymers. 2. Two —
phase systems.—J. Appl. Polym. Sci., 1971, 15, p. 1163—1180.
10. Dobrescu V., Cobzaru V. Some rheological properties of polycarbonate blends.—
J. Polym. Sci. C, 1978, N 64, p. 27—42.
П. Шумский В, Ф., Лебедев Е. Б., Липатов Ю. С. Вязкоупругие свойства смесей
кристаллизующихся полимеров.— Высокомолекуляр. соединения А, 1979,
21, № 5, с. 992—999.
12. Viscoelastic properties of polystyrene — polycarbonate blends in melt / / Yu.
S. Lipatov, V. F. Shumsky, A. N. Gorbatenko et al.— J. Appl. Polym. Sci.,/
1981, 26, N 2, p. 499—508.
13. Термодинамические и реологические свойства расплавов смесей полимеров /
Ю. С. Липатов, А. Е. Нестеров, Т. Д. Игнатова и др.— Высокомолекуляр.
соединения А, 1982, 24, № 3, с. 549—554.
14. Shih С. /С. Rheological properties of incompatible blends of two elastomers.—
Polym. Eng. $ci., 1976, 16, N 11, p. 742—746.
15. Вязкость и термодинамика смесей расплавов полимеров / Ю. С. Липатов,
В. Ф. Шумский, Е. В. Лебедев, А. Е. Нестеров.— Докл. АН СССР, 1979,
244, № 1, с. 148—151.
16. Flory Р. J. Principles of Polymer Chemistry.— New York : Cornell univ.
press., 1953.— 594 p.
17. Koningsveld R.f Kleintjens L. A. Thermodynamics of polymer mixtures.— J.
Polym. Sci. C, 1977, N 61, p. 221—249.
18. Me Master P. L. Aspects of polymer — polymer thermodynamics.— Mac-
romolecules, 1973, 6, N 5, p. 760—773.
19. Чалых А. Е., Сапожникова И. Н. Исследование зон взаимодиффузии в системе
поливинилхлорид — полиметилметакрилат.—Композиц, полимер, материалы,
вып. 7, с. 46—49.
20. Friday A., Cooper D. R., Booth С. Mixing in ethylene oxide and propylene
oxide oligomers. 2. Phase separation.— Polymer, 1977, 18, N 2, p. 171—174.
21. Robard Л., Patterson D. Thermodynamics of polymer compatibility.— Mac-
romolecules, 1977, 10, N 7, p. 1021—1027,
22. Теплофизические и реологические характеристики полимеров / Под ред.
Ю. С. Липатова.— Киев : Наук, думка, 1977.— 222 с.
237
23. Реологические свойства смесей кристаллизующихся полимеров / В. Ф. Шум-
ский, А. С. Дорожкин, Ю. С. Липатов и др.— Коллоид, жури., 1976, 38,
№ 5, с. 949—954. /
24. Comparison of thermodynamic and rheological properties of binary polymer
mixtures / Yu. S. Lipatov, A. E. Nesterov, V. F. Smimsky et al.— Eur.
Polym. J., 1982, 18. p. 981—986. /
25. Cahn /. Spinodal decomposition.— Trans. Met. Soc. ДШЕ, 1968, 242, N 2,
166—180. /
26. Френкель Я- #• Кинетическая теория жидкостей*— Л. : Наука, 1975.—
592 с.
27. Lipatov Yu. S., Nesterov A. E.t Ignatova T. D. Some peculiarities of
thermodynamic behaviour of oligomeric mixtures with specific interaction between
component.— Eur. Polym. J., 1979, 15, N 8, p. 775—780.
28. Липатов С. М. Высокомолекулярные соединения (лиофильные коллоиды).—
Ташкент : Изд-во АН БССР, 1943.— 160 с.
29. Липатов Ю. С., Нестеров Л. ?., Игнатов Т. Д. О возможности
сегментальной растворимости в смесях термодинамически несовместимых
полимеров. — Доп. АН УРСР. Сер. Б, 1975, № 7, с.629—631.
30. Воюцкий С. См Каменская А. #., Фодиман Н. М. Прямые доказательства
само и взаимодиффузии при образовании адгезионной связи между
полимерами.— Механика полимеров, 1966, № 3, с. 446—452.
31. Elmqvist С, Swanson S. R. An NMR investigation of compatibility in blends
polyvinylchloride and ethylene—vinylacetate copolymer.— Eur. Polymer.
J., 1975, 11, N 11, p. 789—793.
32. К термодинамике взаимопроникающих полимерных сеток / Ю. С. Липатов,
А. Е. Нестеров, Л. М., Сергеева и др.—Докл. АН СССР, 1975, 220, №3,
с. 637—640.
33. Robeson L. M., Furtek А. В. Miscible blends of poly(butylene terephtalate)
and polyhydroxyether bisphenol A.—J. Appl. Polym. Sci., 1979, 23, N 3,
p. 645—659.
34. Игнатова 7\ Д. Термодинамические свойства смесей полимеров и олигсь
меров : Автореф. дис. ...канд. хим. наук.— К., 1978.— 16 с.
35. Пригожий #., Дефэй Р. Химическая термодинамика.—Новосибирск; Наука,
1961,— 509 с.
36. Kwei Т. /О, Nishi 7\, Roberts R. F. A study of compatible polymer mixtures.—
Macromolecules, 1974, 7, N 5, p. 667—674.
37. Липатов Ю. С. Физическая химия наполненных полимеров.— М.: Химия,
1977.— 304 с.
38. Siegmann A. Crystalline — crystalline polymer blends: some structure — pro-»
perty relationships.— J. Appl. Polym. Sci., 1979, 24, N 12, 2333—2347.
39. Лтатов Ю. С, Лебедев в. В., Шумський В. Ф. Аномал11 реолопчних i
ф!зико-мехашчних властивостей бшарних сумшей mwiiMepiB.— BicH.
АН УРСР, 1981, № 12, с. 22—31.
40. Nishi Т. Experimental aspects of compatible polymer mixtures.— J. Macromoh
Sci. B, 1980, 17, N 3, p. 517—542.
41. Папков С. Я. Типы диаграмм состояния полимерных систем с
кристаллизующимся растворителем.— Высокомолекуляр. соединения А, 1978, 20, № 11,
с. 2517—2520.
42. Hager S. L., MacRuly Т., В. Investigation of phase behaviour and water
binding in poly(alkylene oxide) solutions.—J. Appl. Polym. Sci., 1980, 25, N 8,
1559—1571.
43. Pennings A. J., Smith P. Eutectic crystallization in polymer solutions. — Brit.
Polym. J., 1975, 7, N 6, p. 460—465.
44. Изучение твердых растворов низкомолекулярных веществ в полипропилене
/ В. М. Мясникова, В. Д. Самарская, Е. К. Оболонкова, Е. Ф. Титова.—
Высокомолекуляр. соединения А, 1980, 22, № 9, с. 1937—1943.
45. Термодинамическая совместимость компонентов в расплаве и степень
кристалличности формирующихся из них полимерных смесей / Ю. С. Липатов,
А. Е. Нестеров, Т. Д. Игнатова, А. А. Лашук.— Там же, № 12, с. 2665—
2671.
ГЛАВА 8
МЕХАНИЗМ ФАЗОВОГО РАЗДЕЛЕНИЯ
В МНОГОКОМПОНЕНТНЫХ
ПОЛИМЕРНЫХ СИСТЕМАХ
Подавляющее большинство работ по
термодинамике растворов и смесей полимеров было посвящено изучению
равновесных термодинамических свойств, в частности процессов
растворения и совместимости, фазового состояния, кинетики
кристаллизации и морфологии полимерных кристаллов, растущих из растворов,
и т. д. В последние годы все больший интерес вызывает исследование
самого механизма фазового расслоения растворов полимеров, и главным
образом полимерных композиционных материалов различного типа.
Этот повышенный интерес к исследованию такого рода процессов
обусловлен, по нашему мнению, тем, что в создании композиционных
материалов намечается четкая тенденция к использованию так называемых
связующих «второго поколения». Такими связующими могут быть
сплавы и смеси полимеров, либо системы типа взаимопроникающих,
полувзаимопроникающих сеток, сегрегированных сеток [1—3], которые
должны рассматриваться как гибридные связующие. Особенностью
же связующих «второго поколения» является то, что по существу они
сами представляют собой композиционные материалы. Имеющиеся в
настоящее время данные показывают, что определяющим фактором струк-
турообразования в таких системах и формирования всего комплекса
физико-механических свойств материалов на их основе может
являться тип фазового расслоения таких систем.
Общий подход к описанию фазового разделения в негомогенных
системах был развит в работах Ван дер Ваальса, Харта, Кана и Хиллар-
да [4, 13—19]. С. П. Папков [5], наблюдая конечные стадии фазового
разделения, причину образования «геля» или жидких слоев в аморфных
полимерах приписал разной ширине области совместимости на
диаграмме состав — температура и вязкотекучим свойствам
концентрированной полимерной фазы. Образование «геля», по его мнению,
должно наблюдаться в том случае, если температура текучести полимера
достаточно высока и добавление растворителя не очень сильно ее
понижает. Это сводится к тому, что гель образуется в результате
разделения и роста микрообластей разбавленной полимерной фазы, оставляя
концентрированную фазу как постепенно ожесточающую матрицу
вплоть до достижения конечной равновесной концентрации; т. е. это
типичный механизм нуклеации и роста.
239
Кани Хиллард[ 13—19] описали второй тип фазового расслоения —
спинодальный распад гомогенного раствора. Этот тип распада может
приводить к типичным сетчатым структурам в изотропной среде,
например в неорганических стеклах [15, 16]. Как показали дальнейшие
исследования многих авторов, такой механизм фазового разделения
может наблюдаться в растворах и смесях полимеров.
Рассмотрим оба типа фазового расслоения, а также
экспериментальные результаты исследования механизмов фазового разделения в
многокомпонентных полимерных системах.
8.1. НУКЛЕАЦИЯ И РОСТ
Переход от однофазной системы к двухфазной связан с усилением
флюктуации состава и развитием на их основе микрообластей новой
фазы. Это происходит в том случае, если система переводится в мета-
стабильную или нестабильную области фазовой диаграммы (см.главу 1,
параграф 1.5). Несмотря на то, что в метастабильной области
диффузионные потоки компонентов направлены в сторону уменьшения
флюктуации, выделение новой фазы начинается именно здесь.
Энергетически невыгодный в данном случае эффект образования межфазной
поверхности раздела приводит к тому, что система оказывается
нестабильной только к флюктуациям, размеры которых больше
критической величины, а сами флюктуации близки по составу к новой фазе.
Такие флюктуации называются критическими зародышами. Здесь
процесс сходен с возникновением, ростом и коалесценцией
микрообластей выделяющейся новой фазы при испарении, конденсации или
кристаллизации.
Эти процессы были описаны в работах Я. И. Френкеля [6], а также
Е. М. Лифшица и Л. П. Питаевского [7]. Мы рассмотрим лишь
основные закономерности роста зародышей и их распределения на поздней
стадии, когда флюктуационное возникновение новых зародышей
практически исключено, например, при выпадении растворенного вещества
из пересыщенного раствора, где критерием метастабильности являлась
степень пересыщенности раствора [7]. Итак, когда возникновение
новых зародышей исключено, критические размеры зародышей
увеличиваются за счет процесса «поедания» мелких зерен крупными — растут
более крупные зерна за счет растворения мелких. В этих условиях
средний размер зерна возрастает со временем по асимптотическому
закону:
\1/3
(8-1)
где D — коэффициент диффузии растворенного вещества; а = 2ои/ X
Хсооо/Т; а — коэффициент поверхностного натяжения на межфазной
границе; v'— молекулярный объем растворяемого вещества; со~ —
концентрация насыщенного раствора над плоской поверхностью
растворяемого вещества t — время.
240
Распределение зерен по размерам в каждый момент времени
определяется функцией
3W[l/(l2/3)) 2
" < 3/2)>
где u = alak(t), ak(t) — критический радиус зерна. При и > 3/2 имеем
р (ц) = 0, т. е. функция Р (и) отлична от нуля только в области
и< 3/2. Поскольку а = а*(/), т. е. средние размеры совпадают с
критическими, максимум Р(и) приходится на значения м, близкие к
единице (рис. 8.1). Полное число зерен (в единице объема) убывает со
временем по закону
1,6
0,5
0,5 1,0
1,5 и
Рис. 8.1. Функция распределения
зерен по размерам [7].
где Q — полное начальное пересыщение.
Аналогичные эффекты могут
наблюдаться и в растворах полимеров при
концентрациях порядка нескольких
процентов при переводе их в область между
бинодалью и спинодалью, т. е. может
протекать эффективная нуклеация. Этим
можно объяснить нахождение так
называемых кривых точек помутнения ниже
бинодали-для высококонцентрированных
растворов. Авторы работы [9] отмечают,
что концентрированный раствор
полимера должен рассматриваться как
ансамбль полимерных сегментов и молекул
растворителя с практически гомогенным
распределением сегментов в растворе.
Флюктуации концентрации тогда могут быть описаны как
флюктуации плотности сегментов или концентрации молекул растворителя.
Следовательно, влияние индивидуальных свойств макромолекул
(например, молекул яр но-массового распределения образца) на явление
нуклеации и на кривую точек помутнения становится несущественным
при высоких концентрациях, в отличие от случая разбавления
растворов.
Нуклеационный механизм фазового разделения в растворах поли*
меров, особенно на начальных стадиях процесса, был подробно
исследован авторами работы [9] на примере системы поли-2,6-диметил-1,4-
фениленоксид—толуол методом индуцированного тепловыми
импульсами критического рассеяния света [10]. Этот метод позволяет
использовать очень малые образцы, в результате чего температурное равновесие
в системе достигается очень быстро, что дает возможность «поймать»
даже очень быструю кристаллизацию. Применив такой метод, авторы
работы [9] исследовали кинетику начальной стадии кристаллизации
в уже упоминавшейся системе ППО — толуол. Анализ данных по
интенсивности рассеяния проведен ими на основе представлений о
гетерогенной кристаллизации, предложенной в работе [11], согласно
которой, в случае роста кристаллов из раствора при гетерогенной кри-
24Г
сталлизации и низкой конверсии уравнение кинетики имеет форму,
сходную с уравнением Аврами:
*=*1—ехр (—/»").
где х — степень конверсии; К — константа скорости; t — время; п —
экспонента Аврами, равная 3/2 для диффузионно-контролируемого
роста и 3 — для межфазно-кон-
^еации («) иРпок1зат?лиНЫстТепе™ тролируемого роста при првДПО-
зависимости интенсивности рассеяния ложении, что В обоих случаях
от времени (q) число зародышей фиксировано.
Если зародыши генерируются
в ходе кристаллизации,
значение экспоненты имеет несколько
завышенное значение.
Следовательно, если гетерогенная ну-
клеация происходит при
фиксированном числе зародышей, рэ-
леевское рассеяние света при
малых t в случае диффузионно-контролируемого роста /
пропорциональна f (так как для малых конверсии / пропорционально л;2). Для
межфазно-контролируемого роста / пропорциональна t* (для
сферических частиц / ~ R9, где R — радиус частицы, который меньше длины
волны X) (табл. 8.1).
Для системы ППО — толуол
найдено, что при охлаждении
раствора, предварительно прогретого
на 20° выше кривой
дифференциально-сканирующей калориметрии
(полное плавление кристаллов),
интенсивность рэлеевского
рассеяния света изменяется со временем
по кривой, которая может быть
описана уравнениями:
Нуклеация
Гетерогенная
Гомогенная
Диффузионный
рост
п
3/2
3/2—2
Q
3
3—4
Межфазный
рост
п
3
3—4
я
6
6^-8
288 293
3031 М
/ = 0
при
при t < т,
Рис. 8.2. Зависимость времени
индукции кристаллизации от температуры
ее проведения для растворов поли-2,
6-диметил-1, 4-фениленоксид в
толуоле при массовой доле полимера в
растворе 15 (/), 20 B) и 25 C) % [9].
где х — индукционное время,
которое является функцией
температуры и состава для системы ППО —
толуол (рис. 8.2). При /^тп =
= 3/2, что близко к я, полученном при кристаллизации ППО из
расплава, т. е. в данном случае рост является диффузионно-контролируе-
мым — материал транспортируется из раствора на поверхность
кристалла. Это несколько отличается от зависимости размера капель при
фазовом расслоении от времени, предсказываемой Е. М. Лифшицем
и Л. П. Питаевским [7] (см. формулу (8.1)), а также Де Женном [32]
для оствальдовского созревания капель в режиме, когда их размер
становится больше радиуса инерции клубка (рис. 8.3). В этом случае
зависимость интенсивности рассеяния должна быть пропорциональна t2.
242
Авторы [9] исследовали процесс кристаллизации ППО из
растворов в толуоле, варьируя разные температурные режимы
предварительного прогрева уже начинающегося кристаллизоваться раствора
Так, если после предварительного фазового расслоения раствор
некоторое время прогреть при температуре, соответствующей концу
кривой дифференциально-термического анализа, индукционный период
исчезнет. Этот тип кристаллизации может быть назван «засеянной
кристаллизацией». При не очень длительном прогреве смеси (т е когда
Igt
12 3 4 t.MUH
"три оствальдовском созревании от
Рис. 8 4 Зависимости интенсивности рассеяния света системой поли-2, 6-ди-
метил-1, 4-фениленоксид — толуол от времени кристаллизации. Объяснения
см. в тексте 1У|«
кристаллы полностью еще не расплавились) после ее охлаждения
сначала наблюдается линейное увеличение интенсивности рассеяния (см.
рис. 8.3). После прохождения точки, до которой первоначально была
проведена кристаллизация (точка Б на кривой АБВГ, рис. 8.3),
снова получаем нормальную кривую кристаллизации (точка ?' на кривой
t рИС'А4''Т* е* Все кРивые ПРИ сдвиге по оси / могут быть совмещены.
dTOT эффект объясняется неполным плавлением кристаллитов или
неполной диффузией*материала от поверхности старого кристалла в
раствор. Повторная кристаллизация этого раствора, вероятно,
происходит в границах старого кристалла. Отсутствие рассеяния света при всех
случаях прогрева и наличие индукционного времени при полностью
расплавленной системе могут быть обусловлены тем, что
кристаллизация ППО протекает по двум механизмам. По-видимому, перед
кристаллизацией происходит конформационное превращение молекул.
В «засеянной кристаллизации» (здесь прогрев также приводит к
исчезновению рассеянного света) кристаллы могут исчезать, но материал все
еще обогащен соответствующими конформациями цепей, так что
кристаллизация начинается без индукционного периода.
Эти результаты были получены в том случае, когда растворенное
вещество при фазовом разделении выкристаллизовалось. Интересно
провести подобное исследование фазового расслоения по типу
жидкость — жидкость, в растворах полимеров и в смесях двух полимеров.
243
Особенно интересным представляется изучение кинетики фазового
расслоения при формировании сетчатых структур. Результаты такого
рода исследований пока единичны [12].
Перейдем к рассмотрению другого типа фазового расслоения в
многокомпонентных полимерных системах — спинодального распада
однофазной системы.
8.2. СПИНОДАЛЬНЫЙ МЕХАНИЗМ ФАЗОВОГО РАССЛОЕНИЯ
В РАСТВОРАХ И СМЕСЯХ ПОЛИМЕРОВ
8.2.1. ТЕОРИЯ СПИНОДАЛЬНОГО РАСПАДА
Теория спинодального распада гомогенной смеси на начальных
стадиях "процесса была развита в работах Кана и Хилларда [13—18].
Согласно этой теории на начальных стадиях фазового расслоения
возникающие флюктуации концентрации рассматриваются как набор
синусоидальных волн с фиксированной длиной Я, которая и является
размером образующихся структур при спинодальном распаде. Следуя
Кану [14—16], рассмотрим основные положения этой теории.
Пусть имеется раствор, в котором существуют малые градиенты
состава. Для такого раствора свободная энергия может быть взята
в виде
j (8.2)
где g (ф) — свободная энергия смешения единицы объема гомогенного
раствора состава <р; К (VcpJ— добавочная свободная энергия,
обусловленная градиентом состава. Разлагая g (ср) в ряд по малым флюктуа-
циям концентрации
и принимая, что
разность свободной энергии между гомогенным и негомогенным
растворами можно получить в виде
af=Яг Ш(ф - ф°)г+к (уф2)]dV- (8-3)
Константа К предполагается равной нулю. Видно, что раствор
устойчив ко всем чрезвычайно малым флюктуациям, если ^>0. С
другой стороны, если d2g/dy2 < 0, то раствор нестабилен к таким
флюктуациям, для которых первый член уравнения (8.3) будет
преобладающим. Далее обычным путем проводится Фурье-анализ локальных
флюктуации, который с физической точки зрения представляет собой
описание теплового движения в жидкости как наложение сверхзвукси
244
вых волн различных направлений и длин волн. Для определения
вклада флюктуации в величину AF суммируются вклады каждой
Фурье-компоненты. Например, вклад в AF от одной такой
компоненты Acosfix представляется величиной
] <8-3а>
Видно, что присутствие Фурье-компоненты с достаточно большой
длиной волны (или достаточно малым волновым числом Р) приводит к
уменьшению свободной энергии, если раствор находится в метаста-
бильной области. Максимальное волновое число ркрит выражается
в виде
, Кинетику начальной стадии фазового расслоения можно получить
тогда простым решением уравнения диффузии
JB = -JA=MV(ixA-iiB)f (8.4)
где Jb— диффузионный поток; М — подвижность. Последняя должна
быть положительной, если диффузия вещества при наличии градиента
химического потенциала приводит к уменьшению свободной энергии.
Величина \хА—\1в получается из частной производной F в уравнении
(8.2):
+ члены высшего порядка. (8.5)
Подставляя (8.5) в (8.4), получаем уравнение диффузии в виде
v<p-2MKVV (8.6)
Поскольку коэффициент при V2<p может быть отождествлен с
коэффициентом диффузии Z), последний на спинодали меняет знак с
положительного на отрицательный (для спинодали (^4) = 0). Решение
уравнения (8.6) находим в виде
где так называемый «фактор уширения» #(Р) равен
R (Р) - -М @) р - 2MW. (8.7)
Поскольку уравнение однородно и линейно с ф, сумма решений
и является возможным решением
;)], (8.8)
ще р и rt — векторы волнового числа и единицы объема L
245
Кан [16] показал, что в процессе спинодального распада
флюктуации концентрации с волновыми числами Р ниже критического
волнового числа ркрит увеличиваются по амплитуде с временем, тогда
как флюктуации концентрации с Р < ркрит уменьшаются по
амплитуде и исчезают. Амплитуды А и В оцениваются при t = О, т. е.
в момент перехода системы через спинодаль, которая находится из
условия ^-f = 0. Значения А и В не изменяются со временем или
температурой. Фактор уширения /?(Р) принимает максимальное
значение при
рт = РкР„Ж2 = 1[-(^)/^]1/2. (8.9)
Поскольку зависимость амплитуды звуковой волны от времени t
экспоненциальна (экспонента равна R (р)), можно игнорировать все
Фурье-компоненты, кроме имеющих волновое число рт, при котором
R (р) достигает максимума. Это приводит к такому преобразованию:
Ф-Фо= S
все 0
где Rm может быть дано несколькими эквивалентными выражениями:
Следовательно, на начальных стадиях спинодального распада
должно наблюдаться экспоненциальное возрастание интенсивности
синусоидальных флюктуации состава, длина волны которых
определяется соотношением (8.9). Между оптимальной и критической длинами
волны спинодального распада существует соотношение [16]
Для более поздних стадий спинодального распада, т. е. при
больших значениях ср—ф0, выражение (8.2) оказывается грубым
приближением, так как оно предсказывает непрерывное возрастание величины
Ф—ф0 со временем. На самом же деле, разность концентраций не
может превысить предельного значения, соответствующего выделению
микрообластей чистых компонентов. В работе [19] было показано, что
для поздних стадий спинодального распада диффузионное уравнение
принимает вид
-gp = DxV2(p — ?>2У4ф + D[ (\7фJ + нелинейное выражение более
высоких порядков, (8.10)
где D, = -?[-|?]; D2 = 2MK/NV; D[ = ^ (Nv - число ато-
мов в единице объема).
246
В тех случаях, когда объем системы зависит от ее состава,
последовательность бесконечно малых флюктуации состава в твердом теле
должна приводить к появлению упругих деформаций [18]. Кан
показал, что в результате этого появляется дополнительный вклад в
свободную энергию системы, описываемый выражением т^— (ф — Ф0J>
[17, 18],
где Е — модуль Юнга; v — коэффициент Пуассона; г\ — линейное
расширение, соответствующее единичному изменению состава. С
учетом этого соотношения вклад в Д F Фурье-компоненты с длиной волны
2д/рт может быть представлен выражением
1
VA2'
Следовательно, для системы с упругими деформациями, которые
являются результатом локального изменения состава, предел устойчивости
определяется условием
-Г = 0, (8.11)
где Y = 2rfE/(l— v). Таким образом, упругие напряжения
стабилизируют систему. Соотношение (8.11) определяет положение так
называемой когерентной спинодали в отличие от химической, определяемой
только химической природой компонентов и выражаемой условием
Теория Кана в настоящее время продолжает оставаться основной
для изучения кинетики фазового разделения по спинодальному
механизму в стареющих сплавах металлов [20], неорганических стеклах
{21] и т. д. Соответствующие эксперименты основаны на применении
метода малоуглового рассеяния рентгеновских лучей, а также
угловой зависимости рассеяния света.
Применение теории Кана—Хилларда для интерпретации
экспериментальных данных, полученных этими методами, дает возможность
получить такие характеристики, как оптимальная длина волны спи-
нодального распада, вторая производная свободной энергии по
составу, величина rfE/(l —v) при наличии упругих напряжений и
значение R (Р) в уравнении (8.2). Проводимые с этой целью расчеты
основаны на анализе временной зависимости интенсивности рассеяния
рентгеновских лучей или рассеяния света. Как было показано в [22],
такая зависимость определяется из соотношения
/(р, 0 = /(Р, 0)ехр[ЗД(Р)а (8-12)
где Л(Р)- —(/>/ 0) @ + 2п«27/A - v) + 2/ф*) Ра (Р - коэффи-
диент взаимодиффузии компонентов). Из теории следует, что в
качестве основных признаков спинодального распада можно выделить
следующие! прямолинейность зависимостей R (р)/р2 от р8и In /(P,
247
t) от /, наличие интерференциального максимума на угловой
зависимости интенсивности рассеяния. Проявление этих эффектов дает
возможность сделать вывод о наличии или отсутствии спинодального
распада, а анализ соответствующих зависимостей — определить
некоторые параметры спинодального распада (см. ниже).
При установлении наиболее характерных особенностей
спинодального распада большую роль сыграли работы по математическому
моделированию фазового разделения в области неустойчивых состояний
фазовой диаграммы. Несмотря на сравнительно небольшое число
подобных исследований, их результаты оказались весьма
показательными, поскольку дали возможность проследить во времени процесс
возникновения, обогащения, а в отдельных случаях и коалесценции
микрообластей гетерогенной структуры [23, 24]. В частности, Шендел-
ман и б'Туль показали, что при определенном составе системы имеется
температура, соответствующая максимальной скорости образования
взаимосвязанных микрообластей фазового расслоения. В этом случае
формирование модулированных структур при спинодальном распаде
соответствует краткому периоду развития взаимодействующих ядер.
В течение более поздних стадий распада важную роль начинает играть
гидродинамически инициированная коалесценция. Эти авторы
показали, что существенной особенностью результатов, полученных при
численном решении диффузионного уравнения типа (8.10), следует
считать возможность модулирования процесса разделения фаз и в мета-
стабильной, и в нестабильной областях фазовой диаграммы, а также
возможность получения сведений о коалесценции микрообластей на
более поздних стадиях распада.
В последнее время появились работы по теоретическому
исследованию, цель которых — более общее описание явления спинодального
распада, чем на основе теории Кана—Хилларда. Необходимость более
общего подхода к анализу явлений спинодального распада прежде
всего связана с тем, что основное уравнение в данной теории (уравнение
типа [8.2]) неприменимо в общем случае, поскольку оно из соображений
симметрии не принимает в расчет вклады, пропорциональные у<р.
В качестве одной из альтернативных формулировок теории для
описания фазового разделения в области неустойчивых состояний фазовой
диаграммы была предложена теория [25], основанная на наборе
феноменологических коэффициентов, определяемых из условия корреляции
с экспериментальными данными. Другой подход, развитый в последнее
время, основан на обобщении теории возмущений для жидкого
состояния на случай неоднородных жидкостей [26]. Численное решение
соответствующего диффузионного уравнения уже для одномерного случая
показывает заметные отклонения от теории Кана—Хилларда вне
линейного режима.
Один из наиболее строгих вариантов современной теории фазового
разделения, включающий описание процессов нуклеации и
спинодального распада, развит в работах [27, 28]. Он состоит в рассмотрении
этих процессов как релаксационных явлений, подчиняющихся
марковскому уравнению. Для термодинамической системы, подлежащей
такому релаксационному процессу (например, жидкость в определенном
248
состоянии), скорость изменения локальной плотности р (rj)
определяется уравнением эволюции
M, (8.13)
где К — константа, связанная с температурой и характеристическим
временем переходов; Q — большой термодинамический потенциал;
X — расстояние, проходимое молекулой жидкости за
характеристическое время.
Уравнение (8.13) описывает скорость наиболее вероятной
эволюции флюктуации плотности жидкости. В приближении малой вариации
р (г, t) по сравнению с А, справедливо уравнение, впервые полученное
Каном:
S&L <8-14'
Из анализа соотношений (8.13) и (8.14), видно, чтоЙ может быть
идентифицирована с функцией Ляпунова. При этом особый интерес
представляет изучение количества и свойств соответствующих
стационарных решений. Направление наиболее вероятной эволюции системы
определяется условием
В частности, если мы имеем два стационарных состояния — рг(г)
8 Рз(г)> то на пути от первого ко второму для флюктуации Ар (г)
справедливо выражение
При stom стационарное состояние определяется условием
Однородные состояния соответствуют тривиальному решению р (г) =
= р уравнения (8.15). В точке, где такое решение становится
неустойчивым, происходит ветвление, или бифуркация.
Соотношение (8.15) дает условие неустойчивости для каждого
однородного решения в спинодальном районе, т. е. от каждой точки
ниже спинодали ответвляется бесконечное семейство неоднородных
стационарных состояний. Неустойчивости однородных решений
определяются в терминах либо механической (большой отклик на внешнее
воздействие), либо термодинамической аналогии (отсутствие
однозначно определенного минимума Q[p (r)]). Было установлено, что
периодические стационарные состояния ветвятся с определенной величиной
первоначальной длины волны %с. Несколько спонтанных флюктуации
может вывести систему из ставшего неустойчивым однородного
состояния через указанные периодические состояния в метастабильное.
В дальнейшем благодаря нуклеации возможен^переход из метастабиль-
ного в равновесное однородное состояние. Свойство п [р (г)] как функ-
249
ции Ляпунова (8.13) определяет направление в последовательности
стационарных состояний, проходимых вдоль наиболее вероятного пути
после перевода системы в спинодальную область фазовой диаграммы.
Таким образом, явление спинодального распада ассоциируется с
большим числом путей, по которым проходит релаксация системы к
равновесию. Во всех случаях после закалки система развивает
периодическую неоднородность с волновым вектором, соответствующим
первоначальному состоянию. С течением времени эта неоднородность
возрастает по амплитуде и длине волны до тех пор, пока не начнется
нуклеация после достижения метастабильного состояния. В
цитируемой работе показано, что в этом состоянии флюктуации в
различных областях пространства не являются зависимыми друг от друга,
они приводят к развитию нуклеационного процесса, не описываемого
в рамках локального приближения Кана—Хилларда.
8.2.2. ПРИМЕНЕНИЕ ТЕОРИИ СПИНОДАЛЬНОГО РАСПАДА
К ПОЛИМЕРНЫМ СИСТЕМАМ
Для описания кинетики спинодального распада в растворах
полимеров Ван Аартсен использовал модель концентрированного
раствора, т. е. совокупность сегментов, однородно распределенных в среде
растворителя [8]. Это позволило ему применить уравнение Флори—
Хаггинса для свободной энергии смешения (уравнение типа [2.6]).
Далее Ван Аартсен использовал выражение для свободной энергии
Гельмгольца для двухкомпонентной системы, предложенное Дебаем
[30]:
J
где <р—ф0— разность между локальной концентрацией q> и средней
концентрацией <р0 раствора; AV — элемент объема; vx и v2—
молекулярные объемы молекул растворителя и полимера; Q и Н — параметры
взаимодействия. Вклад в свободную энергию A F, вносимый флюктуа-
циями состава, таков:
[±{$} ±] (8.17)
здесь g — свободная энергия смешения гомогенного раствора
(первый член в уравнении (8.16)).
Сравнение соотношений для А/7, полученные Дебаем и Каном
(8.3), показывает, что они идентичны, если К = A/2)#.
Из уравнений (8.17) и (8.3) видно, что AF отрицательна, если
первый член в скобках отрицательный и больше по абсолютной величине,
чем второй. Согласно теории Кана, флюктуации концентрации с Р <
< Ркрит должны спонтанно увеличиваться со временем, причем ско-
250
рость увеличения зависит от р. Наиболее часто реализуются
флюктуации с волновым числом рт (см. уравнение (8.9)). Чтобы оценить pmf
необходимо найти д^/ду2. Используя уравнение Флори — Хаггинса
(8.10), имеем
&-L ' RT [vk + VAf=^> ~
Полагая %т = Л + (fivJRT) и имея в виду, что при некоторой
температуре Гсп (температуре на спинодали) выражение (8.18) должно стать
равным нулю, получаем
^(?) (8Л9,
Если известно значение /С, можно рассчитать рт из уравнений (8.9)
и (8.19). Для расчета К было использовано представление Дебая о
флюктуациях концентрации как о периодических функциях типа
Я(Р) =- Т1в(Р) sin фх).
При измерении интенсивности рассеянного света, ее величина
должна быть пропорциональна у$ (Р), где Э = -~^- • s == —- sin 0/2,
ft = Ф — Фо-
Величина Т|о(Р) может быть найдена в виде
<» 1 СО
?12о (Р) = J !Й ехр (- AF/VkT) dt]0 / J exp [- AE/VkT] d^ =
^Т- <8-20>
Подставляя (8Л9) в (8.20), имеем
Сравнивая (8.21) с соответствующими уравнениями Дебая [30],
получаем, что
К/В = /V6, (8.22)
I — радиус действия межмолекулярных сил Дебая. Комбинируя
(8.9), (8.19) и (8.22), находим
[JX|jI/2. (8.23)
Длина волны наиболее часто реализуемой флюктуации такова:
Хт - 2яР^ = 2я/ [311 - JL | ]"/2. (8.24)
Необходимо отметить, что величина Хт зависит не просто от /, а являет-
ся функцией термодинамического качества растворителя,
выражающейся величиной /.
251
С использованием уравнения Кана для флюктуации концентрации
негомогенного раствора (8.8), в работе [31] получено выражение для
интенсивности рассеянного света такой системой в зависимости от
времени:
^ (8.25)
где
h0— длина волны падающего света; (да/дц2) — флюктуация
поляризуемости раствора, вызванная флюктуациями концентрации. Величина
/С2 представляет собой интенсивность рассеяния стеклянными
стенками ячейки, в которой находится образец.
Угловая зависимость рассеянного света может быть записана
в виде
где Р = -у- sin 0/2; X — длина волны света в среде.
Из уравнения (8.25) видно, что зависимость In {dlvjdt) от t должна
быть линейной, а кривая иметь наклон, равный 4/?2(Р). Кроме того,
как следует из уравнения (8.7), зависимость R (Р)/р2 от Р2 при спино-
дальном распаде также должна быть линейной. Определив из наклона
зависимости -^ = / (Р2) величину МК = М (-gjr) = А по
управлению (8.23) можно рассчитать Ркрит и %т — по (8.24).
Теория спинодального распада смеси двух высокомолекулярных
компонентов была дана в работах Де Женна [32] и Пинкуса [33]. Для
описания динамики флюктуации в таких системах Де Женн
использовал концепцию формы движения молекул, названную «рептацией»,
или переползанием в трубе, образованной лабальными узлами
зацеплений [34—37]. Свободная энергия системы была взята в виде
расширения соотношения уравнения Флори—Хаггинса на неоднородные
системы [34]:
-У = ¦?- In Ф„ + -g- in ф* ^
Временные изменения локального состава системы связаны с
потоком компонента А уравнением непрерывности
?g! + div7A = 0. (8.26)
Фурье-компонента потока для волнового вектора ^л(Р) определяется
на основании соответствующей Фурье-компоненты градиента
химического потенциала данного компонента y\i ф) из уравнения
^(P) = -[A(P)/ftTHV|i(P)), (8.27)
где Л(Р) — коэффициент Онзагера.
252
Находя V(x и оставляя в выражении для него только линейные
члены по бф(г), имеем
i. (8.28)
На основе соотношений (8.26) — (8.28) получена так называемая
центральная релаксационная формула
ы
Релаксационный спектр, находимый по уравнению (8.29),
определяет первоначальную нестабильную моду в бинарной системе и
скорость ее роста. В предположении единственного времени релаксации
для моды волнового вектора р в расплаве двух полимеров коэффициент
Онзагера, полученный Де Женном, имеет вид
(8.30)
(Dx—коэффициент микроскопической диффузии). Из этого
соотношения следует, что коэффициент Онзагера пропорционален квадрату
волнового вектора, т. е. резко возрастает с уменьшением длины волны.
При переводе системы в двухфазную область, где N <С 5СХ<С 1,
— _L = const D^XfiW B — р2/2), (8.31)
тз
где / = ja%71/2 — толщина межфазного слоя теории Хелфанда и
Тагами [38, 39].
Из уравнения (8.31) видно, что при р/ < 21/2 флюктуации
усиливаются. Поскольку (l/R0J= 1/foA^cl (?0—радиус инерции клубка),
/ много меньше размеров клубка. Выражение (8.31) отличается от
уравнения Кана для спинодального распада сплавов тем, что в него входит
фактор р4 вместо р2 у Кана. Устойчивый рост флюктуации наблюдается
при
р = ф*аХГ1/2, (8.32)
т. е. оптимальная длина волны много меньше радиуса инерции
полимерного клубка.
С учетом (8.32) оптимальная скорость роста флюктуации
определяется соотношением
где %г— время микроскопической диффузии элемента цепи.
На последних стадиях роста (оствальдовское созревание) средний
размер капель увеличивается согласно уравнению
R2dR/dt =2A(p)a
253
где or — межфазная энергия, равная
а Л(р) определяются выражениями
Л @) = ВД^флфв (Р#о < 1);
Л (р) s ф^фвЛ^А (paJ (Р#о > О
при ф =» фкрит. Для РЯО > 1
^ Фкрит)
чг~const т
В этовд режиме /? (/) увеличивается пропорционально *1/5. Когда /? (t)
становится большим, чем Ro (т. е. Л(Р) =Л@)), то
ОН 2ND оа*
Кл -щ- = ^ фкрит A — фкрит)
и размеры растут пропорционально tl/s (см. рис. 8.3).
Пинкус [33], основываясь на центральной релаксационной формуле
Де Женна, исправил выводы последнего относительно величины
оптимальной длины волны спинодального распада. Пинкус учел, что для
волнового вектора невзаимодействующих цепей в расплаве имеется
не одна, а по крайней мере две независимые моды релаксации [40].
В результате для коэффициента Онзагера при р#0 > 1 он получил
Л (Р) = 27V! (р/Г [1 - ехр (- Р2#2/6)] Флфв, (8.33)
где (лх— подвижность мономерной единицы.
Таким образом, коэффициент Онзагера оказывается
пропорциональным обратной величине квадрата волнового вектора. Комбинируя (8.28)
с (8.33), для скорости релаксации к равновесному состоянию получаем
следующее выражение:
J- = \ (TvlJL*) [1 - ехр (- р2#2/6)] [N-i + 2Х1флФв + (Ра)а/36], (8.34 )
где L — контурная длина цепи.
При тр < 0 наблюдается экспоненциальный рост флюктуации
состава. При резком переходе от Xi<0 к Хг> Хсп первоначальная
неустойчивая мода дается экстремумом (8.34), т. е.
при быстром переводе системы ниже спинодали. В нестабильной
области фазовой диаграммы вблизи спинодали имеем
ft"*"* / D /f\ I /*5\ /V /V 1 \**1 /2» /V V \ /*V ^d^ 1
Для оптимальной скорости роста флюктуации в первом случае
получаем
'I' * /Л/ /Л/ \ Л
254
во втором случае —
7" « rg* (Xi/Xcn - 1); (%, - ЭССП)/ХСП < 1;
здесь Грепт — время, необходимое для выхода молекулярной цепи из
трубы зацеплений.
Таким образом, Пинкус показал, что неустойчивая мода имеет
длину волны, сравнимую с радиусом молекулярного клубка и
изменяется пропорционально N1/2. Соответствующая данной моде скорость
роста флюктуации состава пропорциональна константе рептационной
диффузии в расплаве, изменяясь как#~2, и имеет концентрационную
зависимость, отражающую форму спинодали.
Анализ явлений спинодального распада в смесях гомополимеров,
проведенный Пинкусом, показывает, что лишь для очень глубокого
охлаждения однофазной смеси полимеров возможна реализация
микрофазового разделения на шкале порядка Ne.
8.3. НЕКОТОРЫЕ ЭКСПЕРИМЕНТАЛЬНЫЕ РЕЗУЛЬТАТЫ
ИССЛЕДОВАНИЯ СПИНОДАЛЬНОГО РАСПАДА
В РАСТВОРАХ И СМЕСЯХ ПОЛИМЕРОВ
Теория Кана—Хилларда [13—18] и других авторов [22—28]
описывает главным образом начальную стадию процесса спинодального
распада. Если зафиксировать картину спинодальногр распада на
более поздних стадиях, которая может сохраниться из-за сильно
увеличивающейся вязкости (например, высококонцентрированного
раствора, а тем более расплава полимеров), можно наблюдать однообразную
структуру с расстояниями между полимеробогащенными областями,
равными кт. Морфологическая структура должна быть более или
менее уплотненной, состоящей из взаимосвязанных областей
концентрированной фазы (здесь мы рассматриваем растворы полимеров),
погруженной в разбавленную полимерную фазу (или чистый растворитель).
Кан предположил, что для поддержания взаимосвязанной структуры
необходимо, чтобы в этой структуре объемная доля второй фазы
составляла 15 %. Это значит, что для полимерных растворов такая
взаимосвязанная структура может наблюдаться при массовых объемных
долях полимера, равных нескольким процентам (например, при
концентрации полимера в растворе больше 4,5 % взаимосвязанная структура
должна содержать более 10 % полимерной фазы). Если полимеробога-
щенная фаза достаточно подвижна при всех концентрациях,
первоначально сформировавшиеся области могут образовываться в
агрегаты и в конце произойдет распад на две жидкие фазы.
Экспериментально механизм спинодального распада был описан
во многих работах [41—50]. Одной из первых работ в этом направлении
была работа Смолдерса [41]. Для системы поли-2, 6-диметил-1,4-фе-
ниленоксид (ППО, М = 2,1 • 10б) — капролактам (КЛ) при 15 %-ном
содержании ППО методом светорассеяния ими было обнаружено, что
при некоторых условиях наблюдается спинодальный механизм
фазового расслоения; значения %т уменьшаются при увеличении разности
255
Тсп — Т в соответствии с предсказаниями уравнения (8.23). При Тсп —
400 К и / = 20 нм по уравнению (8.23) были рассчитаны значения
Хт для разных температур и найдено, что в диапазоне 399—360 К Ят
изменяется от 0,23 до 1,43 \х\ это находится в хорошем согласии с
данными электронной микроскопии (~ 0,5 — 2 \х) [42].
Результаты исследования этой системы показали, что расслоение
начинается по спинодальному механизму, приводя в течение
нескольких минут к образованию взаимосвязанной структуры, не имеющей
равновесного состава. На последующей, более поздней стадии
процесса, концентрация в разных местах образца продолжает изменяться,
и фаза обогащается одним из компонентов. Этот процесс
сопровождается изменением объема, и при относительно высокой температуре (для
¦обеспечения подвижности макромолекул в фазе) из полимеробогащен-
ной.фазы в матрице капролактама образуются сферы.
Авторы работы [43] методом электронной микроскопии изучили
механизм фазового разделения в системе ППО — толуол. Было
обнаружено, что если раствор охладить до температуры ниже бинодали,
но выше спинодали, появляются зародыши новой фазы, т. е. расслоение
осуществляется по механизму нуклеации. Если же раствор быстро
перевести" в область неустойчивого состояния (ниже температуры на
спинодали), образуются взаимосвязанная структура и некоторое
количество зародышей в результате конечного времени прохождения мета-
стабильной области при охлаждении. Чем выше скорость прохождения
метастабильной области, тем меньшее число зародышей успевает
образоваться. Следовательно, структура системы на начальных стадиях
фазового расслоения зависит от скорости прохождения
метастабильной области. Было обнаружено, что при охлаждении системы до
температуры, соответствующей области метастабильного состояния, для
образования зародышей требуется определенное время индукции,
которое увеличивается с повышением температуры; при температуре,
равной Тсп> оно практически равно нулю.
Приняв /=20 нм, Тсп==311 К, авторы [43] рассчитали размер
спинодальных структур при 258 К для 30 %-ного раствора ППО в
толуоле по уравнению (8.23). Значения Хщ оказались равными примерно
0,16 мкм, что согласуется с данными электронной микроскопии.
Скорость роста зародышей при нуклеационном механизме фазового
разделения для этой системы drldt оказалась равной ~ 0,15 мкм/мин. Спи-
нодальный механизм фазового расслоения наблюдался и в растворах
сополимер этилен-винилацетат D0 % винилацетата) — капролактам
при температурах намного ниже Тсп [42].
На примере системы полистирол — декалин А. А. Тагер и
сотрудники [44] показали, что критерии спинодального распада соблюдаются
лишь для раствора критической ^концентрации. При с > скрит эти
критерии соблюдаются только на первых стадиях процесса фазового
расслоения. Обнаружено, что время жизни епинодальных структур
тем больше, чем ниже температура (т. е. Тсп —Т). Методом
светорассеяния определены размеры спинодальных структур, а также
коэффициенты взаимодиффузии, которые, как и предсказывалось
теоретически, оказались отрицательными.
256
Спинодальный механизм фазового расслоения наблюдали и для
полимерных гелей. Так* Пайнс и Принс [45] дифракционный
максимум интенсивности рассеянного света гидрогелей агарозы приписали
образованию регулярно расположенных областей фазового
разделения. Детально фазовое разделение при переходе золь — г§ль
исследовали Феке и Принс [46].
Уэллингоф и сотрудники [47] изучили механизм фазового рас*
слоения в способных к гелеобразованию растворах атактического
и изотактического полистиролов, полифениленоксида, и их смесей
в разных растворителях. Они показали» что образование геля
обусловлено не кристаллизацией, а тем, что при переводе системы в область
неустойчивого состояния получается тонкая дисперсия обогащенных
полимеров микрообластей, температура стеклования которых
достаточна для фиксации концов различных полимерных цепей в одной за-
стеклованной области. В качестве доказательства спинодального
механизма фазового расслоения по типу жидкость — жидкость они
использовали величину микрообластей и скорость их формирования. Когда
микрообласти расстекловываются, связанные отрезки цепей
освобождаются и гель плавится.
Для полимер-полимерных смесей сшшодальные структуры впервые
были обнаружены Мак Мастером [48]. Он нашел, что такие структуры
для системы сополимер стирола и акрилонитрила B8 % акрилонитри-
ла) — полиметилметакрилат, имеющей НКТС, наблюдаются при
переходе от однофазной области в двухфазную при критической
концентрации. Размер доменов, образовавшихся на начальных стадиях
процесса расслоения, равен 50—100 нм (электронная микроскопия).
Распад взаимосвязанной структуры происходит в том случае, если
домены, образовавшиеся на начальных стадиях процесса, увеличиваются
в размерах на несколько порядков. Полностью распавшаяся структура
состоит из капель размером около 500 мкм. Скорость роста частиц
постоянная вплоть до разрушения взаимосвязанной структуры, после
чего она увеличивается по экспоненте.
При концентрациях, далеких от критической, фазовое расслоение
происходит по механизму нуклеации и роста. Было замечено, что после
двухчасовой выдержки системы вблизи Тсп размер капель
увеличивается всего вдвое. Следовательно, при фазовом расслоении по
механизму нуклеации и роста существенной коалесценции не происходит
из-за большой вязкости системы, поэтому образуется очень
высокодисперсная структура.
Мак Мастер оценил и коэффициенты диффузии, для чего исполь-,
зовал соотношение R (km) = I — Q-^j2n3/^ следующее из теории
Кана — Хилларда. Он определил, что при 453 К D = 2,6 • 10~18, а при
483 К D = —3,3 • I07 м2/с
1 Таким образом, Мак Мастер получил йаиболее характерные
признаки ранних стадий спинодального * распада в системе полиме𠦻—
полимер. При этом оказалось, что масштабный уровень фазового
разделения больше, чем для систем на основе низкомолекулярных
соединений.
i/4 9 4-420 257;
Ниши, Ванг й Кйей [49, 50], используя метод ЯМР и оптическую
микроскопию, исследовали механизм фазового расслоения в системе
полистирол — поливинилметиловый эфир, имеющей НКТС На
основе морфологических данных авторы показали, что в этой системе
возможны два типа фазового разделения. Поскольку это система с НКТС»
то при повышении температуры микрофазовая структура является
более тонкой. При этом на последней стадии разделения фаз
микрообласти резко увеличиваются, сохраняя взаимосвязанность, а затем либо
разбиваются на малые, либо слипаются в макрофазы. В случае, если
фазовое разделение происходит медленно, размеры микрообластей
невелики по сравнению с периодами взаимосвязанной структуры.
Данные ЯМР подтвердили непрерывное изменение состава
микрообластей со временем, что согласуется с предположением о спинодальном
механизме фазового расслоения.
Другим доказательством наличия спинодального распада в этой
системе является значение R (PJ в уравнении Кана—Хилларда,
определяющего кинетику изменения состава микрообластей фазового
расслоения по соотношению.
ф - Фо = А (pm) exp {R (PJ t) cos pm x, (8.35)
где R (Pm) = -i Pm#. Здесь Ф представляет собой концентрацию поли-
винилметилового эфира в фазе, обогащенной полистиролом. Из
соотношения (8.35) следует, что общее уменьшение содержания поливинил-
метилового эфира в данной фазе Q в процессе фазового разделения
можно вычислить так:
[|
Следовательно, In Q = 3 In \j^ A (Pm)J + 3R (PJ tf т. е. тангенс угла
наклона прямой, описывающей зависимость In Q от t9 равен 37? (PJ.
Величину lnQ авторы оценили из данных ЯМР и обнаружили, что
R (Р) положителен, т. е. в системе, содержащей 50 % поливинилмети-
лового эфира, при температуре 403 К флюктуации усиливаются со
временем, свидетельствуя о спинодальном механизме фазового расслоения.
Если оценить рт из морфологических данных, то для коэффициента
диффузии получается значение D = — 27? (pm)/Pm = — 2,8 • 107 м2/с
при #(PJ = 3,82 • 10-* с и рт = 5,24. 10е м.
Величина Х19 найденная на основе теории Ван Аартсена, равна
—0,42 при 373 К, а Тт для 50 %-ного состава равна 391 К, / = 58НМ>
Как и у Мак Мастера, в данном случае периодичность
модулированной структуры, приведенная в работах [49, 501, оказалась намного
выше» чем для низкомолекулярных компонентов.
Из других работ, авторы которых исследовали влияние различных
факторов на механизм фазового расслоения, следует упомянуть статьи
Когена и Райха [51], а также Прайса, оценившего влияние стеклова-
25а
*ния и присутствия низкомолекулярных примесей на процесс фазового
-разделения полимерных смесей в системе полифениленоксид —
сополимер /г-хлорстирола с о-хлорстиролом [52J и др. [-53].
Количественные данные по кинетике фазового разделения в смесях
полимеров были получены в работах [71—72]. Так, Каваи с
сотрудниками [71], исследовавшие начальную и конечную стадии спинодального
распада, показали, что начальная стадия спинодального распада
хорошо описывается линейной теорией Кана — деЖена [32].
Аналогичные результаты получили и авторы работы [72]. В то же время для
смесей олигомеров начальная стадия процесса фазового разделения не
подчиняется линейной теории Кана: зависимость интенсивности
рассеянного света от времени отклоняется от экспоненциальной,
предсказываемой этой теорией. Различие в поведении смесей олигомеров и
полимеров авторы работы [71] объясняют существенной разностью
коэффициентов самодиффузии, которые для смесей олигомеров на
несколько порядков больше, чем для смесей высокомолекулярных
компонентов. Сделано предположение [71], что линейный режим спинодального
распада в смесях олигомеров может наблюдаться лишь при очень
малых отклонениях температуры эксперимента от температуры спино-
- дального распада, или на более ранних стадиях процесса.
8.4. ОСОБЕННОСТИ ФАЗОВОГО РАЗДЕЛЕНИЯ
В БЛОЧНЫХ СИСТЕМАХ
В общем случае блок-сополимеры можно рассматривать как
многокомпонентные полимерные системы. Как и в случае смесей
полимеров, в блок-сополимерах при определенных условиях наблюдается
фазовое расслоение, т. е. выделение микрообластей равновесного
состава, размеры и форма которых зависят от «архитектуры»
молекулярных цепей и объемной доли каждого из блоков [54—64]. Многие из
экспериментальных данных, свидетельствующих о наличии такого
расслоения блоков, можно найти в справочнике по физической химии
полимеров [70].
Вместе с тем для такого рода систем следует ожидать и некоторые
отличия от поведения обычных смесей полимеров. Очевидно, что в тех
случаях, если релаксация блок-сополимера из пространственно
однородного состояния происходит достаточно медленно, образование
структуры может осуществляться по механизму спинодального
распада. Этот вопрос впервые с достаточной строгостью был рассмотрен
И. Я. Ерухимовичем [62, 63], который установил, что если условие
неустойчивости в смесях полимеров рассматривать с точки зрения
неустойчивости к разделению на бесконечно однородные фазы (с
волновым числом р, равным нулю), то для блок-сополимеров это условие
ориентируется на устойчивость относительно разделения на
микрофазы с характерным периодом пространственной неоднородности,
определяемым волновым числом р = $0Ф 0. В области устойчивости
пространственно однородного состояния любая малая неоднородность
экспоненциально затухает пропорционально характеристическому
времени релаксации. С приближением к спинодали значения времени ре-
i/2 g 4-420 259
лаксации возрастают и на спинодали становятся бесконечно большими.
В области неустойчивых состояний амплитуда неоднородностей,
которым соответствуют отрицательные собственные значения матрицы
Gjf1 (обратная матрица матрицы корреляционных функций в р-
пространстве), экспоненциально увеличивается, приводя к разрушению
пространственно однородного состояния. При этом должны расти
периодические неоднородности с волновыми числами, лежащими в
определенном интервале Pi<p <Р2» а флюктуации с р < рх и Р > р2
по-прежнему рассасываются. С течением времени указанный интервал
волновых чисел сужается вплоть до формирования гетерогенностей
структуры с характерным периодом, и экспоненциально повышается
амплитуда неоднородности. Для «сильно перемешанных» гетерополиме-
ров величина периода гетерогенной структуры равна примерно
радиусу корреляции в системе разорванных связей (звеньев). Увеличение
средней длины химически однородных участков приводит к
возрастанию равновесных значений периодичности.
Интересная аналогия в поведении блок-сополимеров и смесей
полимеров в области неустойчивых состояний обнаружена Рйонг-Жун Ро
и сотрудниками [64]. Для системы типа стирол—бутадиен и стирол—
бутадиен—стирол они наблюдали переход однофазного раствора
блоков в состояние микрофазового разделения. При этом более низкой
температуре соответствовала большая степень сегрегации блоков, что
проявлялось в постепенном увеличении интенсивности малоуглового
рассеяния рентгеновских лучей с понижением температуры. В общем
случае такой эффект характерен и для спинодального распада [22].
Кривые малоуглового рассеяния рентгеновских лучей для этих систем
оказались весьма похожими: наряду с увеличением максимумов
наблюдалось понижение интенсивности рассеяния «хвостовой части»
кривых рассеяния и наличие общей точки пересечения всех кривых.
Учитывая это обстоятельство, авторы [22] для определения
интенсивности малоуглового рассеяния света в случае блочных систем,
находящихся в состоянии равновесия при различных температурах,
предложили следующее уравнение:
/ (р, зу = / (р, 0)ехр (а (Р)эу,
где а (Р) соответствует фактору усиления, используемому для
описания процесса спинодального распада. Для температурной зависимости
параметра термодинамического взаимодействия предполагается
выражение типа %х= А + (BVJRT). Множитель а (Р) имеет максимум при
Р = рт и равен нулю при р = ркрит. Отношение ркРит/р близко к
1,5, т. е. близко к ркрит/р = У'2 в случае спинодального распада.
Температурная зависимость In / (р, Хх) для ди- и триблочных
сополимеров прямолинейна, т. е. наблюдается совершенно однозначная
аналогия с изменением интенсивности рассеяния рентгеновских лучей
при спинодальном распаде.
Аналогию в поведении кривых малоуглового рассеяния,
обнаруженную Роу для сополимеров с длинными блоками, мы наблюдали и
для блок-сополимеров, представляющих собой сравнительно короткие
260
центральные блоки (М = 4500) и концевые жесткие блоки с М ~ 300.
Эти данные свидетельствуют о том, что множитель (у|- — 2/Ссф2),
характеризующий размеры микрообластей фазового разделения в
случае смесей полимеров, по-видимому, применим и для описания фазового
разделения блоков в блок-сополимерах.
8.5. НЕКОТОРЫЕ ОСОБЕННОСТИ ФАЗОВОЙ СТРУКТУРЫ СИСТЕМ
СО СТАБИЛЬНЫМИ УЗЛАМИ ЗАЦЕПЛЕНИЙ
Наличие в системе «стабильных» узлов зацеплений препятствует
полному разделению молекулярных фрагментов различной
химической природы и удерживает их в состоянии «вынужденного» смешения,
что приводит к дополнительному вкладу в свободную энергию системы.
В этом случае условием неустойчивости является выражение типа
(8.11), в котором положительная величина У отражает вклад в разность
свободных энергий (см. уравнение (8.3а)) упругой энергии,
появляющийся в результате спинодального разделения системы. Уравнение
(8.11), таким образом, отражает реальные условия фазового
разделения и положение спинодали при этом условии (например, для систем
с ВКТС) соответствует более низким температурам, чем положение
так называемой химической спинодали, которая определяется
условием d2g7d<p2<0. В случае таких полимерных систем, как полиблочные
сетчатые полимеры или взаимопроникающие сетки, область
неустойчивых состояний под реальной спинодалью лежит намного ниже
химической спинодали и может быть вообще недоступна. Вполне очевидно,
что вне химической спинодали существуют однофазные системы с
равновесным распределением флюктуации, а ниже реальной спинодали —
двухфазная структура с определенной степенью дисперсности. В
области же между химической и реальной спинодалями также имеются
микрообласти негомогенного распределения компонентов. Однако их
нельзя рассматривать в качестве равновесных флюктуации состава,
поскольку термодинамические силы, стремящиеся к усилению
флюктуации, сдвигают систему в сторону двухфазного состояния. Несмотря на
незавершенность процессов разделения фаз, область состояний между
химической и реальной спинодалями относительно стабильна. Но
в концепции «однофазная — двухфазная система» таким
промежуточным состояниям аналога в бинарных системах не имеется. Поэтому
эти системы необходимо выделить из совокупности одно- и
двухфазных систем. При рассмотрении состояния системы между химической
и реальной спинодалями выражение «флюктуация концентрации» не
имеет, по-видимому, такого смысла, как в однофазных системах.
Микрофазными областями их тоже нельзя назвать, так как эти области
характерны только для двухфазных систем. Поскольку области
негомогенности в системах, промежуточных между одно- и двухфазными,
способны переходить в микрофазовые области, их можно рассматривать
в качестве виртуальных микрофазовых частиц и называть фейзонами
(phaseon). В таком случае системы, находящиеся в области состояний
*/i 9* 261
между химической и-реальной спинодалями, являютея системами фей—
зонного типа. Вопрос теперь заключается лишь в том, каким образом
реально получить такое состояние системы, т. е. войти в область между
химической и реальной спинодалями. Дело в том,, что при
формировании любой реальной полиблочной системы сетчатого типа реакция
протекает за конечное время (от нескольких минут до нескольких часов).
Поэтому в ходе формирования таких систем мы неизбежно пересекаем
бинодаль, где механизм формирования микронеоднородностей иной,
чем в области под спинодалью. Естественно, что в этих условиях
появляются структуры смешанного типа с. нерегулярным расположением
их в системе, что и наблюдается в эксперименте [69, 70]. По-видимому,
единственной реальной возможностью получить структуры феизонного
типа йвляется выбор таких условий, при которых система переводится
В'ббласть между химической и реальной спинодалями при критическом
составе. Другой путь — резкое замораживание системы на разных
этапах протекания реакции с анализом возникающих структур.
8.6. РАССМОТРЕНИЕ СПИНОДАЛЬНОГО РАСПАДА В РАМКАХ
ФОРМАЛИЗМА НЕЛИНЕЙНОЙ ТЕРМОДИНАМИКИ
НЕОБРАТИМЫХ ПРОЦЕССОВ
Как видно из рассмотренных в предыдущих параграфах
экспериментальных данных, возникающие в результате спинодального
распада в полимер-полимерных системах структуры остаются затем в ме-
тастабильном состоянии, т. е., как правило, процесс спинодального
рдспада не доходит до стадии полного разделения системы на две
сосуществующие фазы. Отсюда следует, что наиболее адекватное описа-.
ние возникающих структур, очевидно, может быть дано с позиций не-;
линейной,термодинамики необратимых процессов и концепции дисси--
пативных структур. Как было показано в работах Пригожина [65, 66],
вдали от равновесия возникают новые, так называемые «диссипатив-
ные структуры», устойчивые благодаря взаимодействию с внешней
средой. Линденмейер [67, 68] обнаружил аналогию между чередованием
низкоэнергетических и высокоэнергетических областей, характерных
для диссипативных структур, и возникновением модулированных
структур при епинодальном распаде. Выразив термодинамические
силы и потоки, через градиенты концентраций, он показал, что для
приращения производства энтропии вблизи стационарного состояния
системы справедливо соотношение
АР J
V 0
где 2j Ra$ V(paVq)|3 + ••• = $] JaXa\ Ja — кинетические потоки; Ха —
соответствующие термодинамические силы; 1/2 62Р — избыточное
производство энГропии 165]. Для бинарной системы последний член уравнения
(8.36) аналогичен /((УфJ в уравнении (8,3), а 0,5(d2g/dq>2)(ф — Ф0J
262
= J [ 18*Я + J] Яа^ФЛ + • • Л dV, (8.36)
V а0
соответствует 0,562Р в разложении приращения производства энтропии>
Таким образом, спинодаль можно рассматривать как условное
равенство нулю избыточного производства энтропии. Сопоставление
указанных соотношений позволяет выразить длину волны модулированной
структуры при спинодальном распаде так:
Хт = [ — 8я (Ф - ФоJ ? #а& + . • • ]/? bJabXa.
а,3 а
Следовательно, формирование диффузных микрообластей
промежуточного состава в результате фазового разделения между исходной
однофазной и конечной двухфазной системами может рассматриваться
как результат нарушения устойчивости процесса разделения фаз ниже
спинодали. Термодинамические силы при этом стремятся к минимуму,
а кинетические потоки могут возрастать в одной части системы и
убывать в другой, приводя к возникновению диссипативных структур.
1. Рентгенографические методы изучения полимерных систем / Ю. С. Лиггатов,
В. В. Шилов, Ю. П. Гомза, Н. Е. Кругляк.— Киев : Наук, думка, 1982,—
295 с.
2. Липатов Ю. С, Сергеева Л. М. Взаимопроникающие полимерные сетки,—;
Киев : Наук, думка, 1979.— 160 с.
3. Липатова Т. Э. Каталитическая полимеризация олигомеров и формирование
полимерных сеток.— Киев : Наук, думка, 1974.— 208 с.
4. Hart E. W. Thermodynamics of inhomogeneous systems.— Phys. Rev., 1959,
113. N 2, p. 412—416.
5. Папков С, П. Студнеобразное состояние, полимеров.— М. : Химия, 1974.—
-' 255 с. *
6. Френкель Я- И. Кинетическая теория жидкостей.— Л. : Наука, 1975.,—
592 с.
7. Лифшиц Е. М., Питаевский Л. Я. Теоретическая физика. Т. 10.
Физическая кинетика.— М. : Наука, 1979.— 528 с.
8. Van Aartsen J. J. Theoretical observations on spinodal decomposition of polymer
solutions.— Eur. Polym. J., 1970, 6, N 7, p. 919—934.
9. Koenhen D. M., Smolders С. Л., Gordon M. J. Phase — separation phenomena
in solutions of poly B,6-dimethyl — 1,4 - phenylene oxide). 3. Pulse induced!
critical scattering of solutions in toluene.— J. Polym. Sci. C, 1977, N §1,
p. 93—100. .
10. Kennedy J W., Gordon M., Alvarez G. A. Polydyspersyjnpsc, a Jndukcwan^
impulsami rozproczenie krytyczne.— Polymery, 1975, N 10, p. 463—471. , ,
11. Turnbull D. Solid-state physics.— New York : Heah. ;press.,;' 1956.— 623 ;pv
12. О гетерогенной структуре полимерных сеток из олигоизопрендигидразидов
и эпоксидного олигомера / В. В. Шилов, Ю. С. Липатов, Б л А. Богданович
и др:._ Докл. АН УССР. Сер. Б, 1981, № 7, с. 50—54. ;
13. Cahn J. W.y Hillard J. E. Free energy of a nonuniform system.;!. lnterfaci^I
free energy.— J.-Chem. Phys., 1958, 28, N 2, p. 258—267., . .
14. Cahn J: w. Free energy of a nonuniform system. 2* Thermodynamio basis.-
Ibid., 1959, 30, N 5, p. 1121—1124.
15. Cahn J. W.t Hillard J. E. Free energy of a nonuniform system. 3. Nucleation
in a two — component incompressible fluid.—Ibid., 31, N 3, p. ,688—699
16. Cahn J. W. Phase separation by spinodal decomposition in isotropic system.—
Ibid., 1965, 42, N 1, p. 93—99. ' , ,
17. Cahn J. W. Spinodal decomposition.—Trans. Met. Soc. tAIMrE, 4968, 242,
n 2, p. лев—iao. ¦ .
18. Cahn J. W. On spinodal decomposition. —Acta Met., 1961, 9, N 4, p. 795—801.
19. Cahn J. W. The later stages of spinodal decomposition arrd the beginnings of
particle coarsening.— ffiifi:, 1966, 14, N 12, р.Ч685-М692. и • '
20: Чуистдв К- В. Модулированные структуры в стареющих сплавах.—*. Киев :
Наук, думка, 1975.— 231 ,с. '
263
21. Явления ликвации в стеклах / Н. С. Андреев, О. В. Лазурин, Е. А. Порай-
Кошиц и др.— Л. : Наука, 1974.—273 с.
22. Rundman К. В., Hilliard J, E. Early stages of spinodal decompositions in
Al — Zn alloys.— Acta Met., 1967, 15, N 10, p. 1035—1038.
23. Swanger L. A., Gupta P. /C., Cooper Л. R. Computer simulation of one —
dimensional spinodal decomposition.— Ibid., 1970, 18, N 1, p. 9—14.
24. Shendelman L. #., O'Toole J. T. Nucleating and coarsening in binary conden-
ced phases.— J. Colloid and Interface Sci., 1968, 27, N 2, p. 145—160.
25. Tiller W. A.t Pound G. M.t Hirth J. P. A critique on the mathematical theory
of spinodal decomposition.— Acta Met., 1970, 18, N 2, p. 225—232.
26. Langlois W. ?., Abraham F. F. On the dynamics of spinodal decomposition:
a numeral solution of a generalized diffusion equation.—Chem. Phys. Lett.,
1977, 52, N 1, 129—132.
27. Metiu //., Kitahara /C., Ross J. A derivation and comparison of two equations
(Landau — Ginsburg and Cahn) for kinetics of phase transitions.— J. Chem.
Phys., 1976, 65, N 1, p. 393—397.
28. Varea C, Rodledo Л. Nucleation, spinodal decomposition, and interface
motion in the van der Waals fluid.—Ibid., 1981, 75, N 10, p. 5080—5089.
29. Lindenmeyer P. H. The inhomogeneous structure of glass.— Amer. Chem. Soc.
Polym. prepr., 1980, 21, N 2, p. 3—4.
30. Debye P. Angular dissimetry of the critical opalescence in liquid mixtures.—
J. Chem. Phys., 1959, 31, N 3, p. 680—687.
31. Van Aartsen J. /., Smolders C. A. Light scattering of polymer solutions
during liquid — liquid phase separation.— Eur. Polym. J., 1970, 6, N 5,
p. 1105—1113.
32. De Gennes P,G. Dinamics fluctuations and spinodal decomposition in polymer
blends.— J. Chem. Phys., 1980, 72, N 9, p. 4756—4763.
33. Pincus P. Dinamics of fluctuations and spinodal decomposition in polymer
blends. 2.— Ibid., 1981, 75, N 4, p. 1996—2000.
34. De Gennes P. G. Qualitative features of polymer demixtion.— J. Phys. Lett.
(France), 1977, 38, N 21, p. 441—443.
35. Daoud M.. De Gennes P. G. Some remarks on the dinamics of polymer melts.—
J. Polym. Sci.: Polym. Phys. Ed., 1979, 17, N 11, p. 1971 — 1981.
36. Klein J. The onset of entagled behaviour in semidilute and concentrated
polymer solutions.— Macromolecules, 1978, 11, N 5, p. 852—858.
37. De Gennes P. G. Scaling concepts in polymer physics.— New York : Cornell
univ. press., 1979.— 324 p.
38. Helfand ?., Tagami Y'. Theory of the interphase between immissible
polymers — J. Polym. Sci.: Polym. Lett., 1971, 9, N 3, p. 741—746.
39. Helfand E.t Sapse A. M. Theory of concentrated polymer solution — solvent
interface.— J. Polym. Sci. C, 1976, N 54, p. 289-297.
40. De Gennes P. G. Coherent scattering by on reptating chain.— J. Phys. (France),
1981, 42, N 5, p. 735—740.
41. Van Emmerik P. 7\, Smolders С A. Phase separation in polymer solutions.
I. Liquid — liquid phase separation of PPO poly B,4-riimethyl-l,4-pheny-
lene oxide) in binary mixtures with toluene ternary mixtures with toluene
and ethyl alcohol.— J. Polym. Sci. C, N 38, p. 73—80.
42. Smolders С. Д., van Aartsen J, /., Steenbergen A. Liquid—liquid phase
separation in concentrated solutions of noncrystallizable polymers by spinodal
decomposition.— Kolloid-Z.+Z. Polym., 1971, 243, N 1, p. 14—20.
43. Van Emmerik P. Т., Smolders С. Л., Geymeyer W. Liquid—liquid phase
separation in concentrated polymer solutions studied by electron microscopy.—
Eur. Polym. J., 1973, 9, N 4, p. 309—316.
44. Исследование спинодального механизма фазового разделения растворов
полистирола в декалине / В. М. Андреева, А. А. Тагер, И. С. Тюкова, Л. Ф.
Голенкова.— Высокомолекуляр. соединения А, 1977, 19, № 11, с. 2604—
2611.
45. Pines ?., Prins W. Structure — property relations of thermoreversible mac-
romolecular hydrogels.— Macromolecules, 1973, 6, N 6, p. 888—896.
46. F-eke G. 71., Prins W. Spinodal phase, separation in a macromolecular sol —
gel transition.— Ibid., 1974, 7, N 4, p. 527—530.
264
47. Wellinghoff S., Shaw /., Beer E. Polymeric materials from the gel state. The
development of fringed micelle structure in a glass.— Ibid., 1979, 12, N 5,
p. 932.
48. McMaster J. P. Aspects of liquid —liquid phase transition phenomena in
multicomponent polymeric systems,— Amer. Chem. Soc. Polym. Prepr.,
1974, 15, N 1, p. 254—259.
49. Nishi 7\, Wang Т. 7\, Kwei Т. K. Thermally induced phase separation
behavior of compatible polymer mixtures.— Ma его molecules, 1975, 8, N 1,
p. 227-231.
50. Nishi T. Experimental aspects of compatible polymer mixtures.— J. Macro-
mol. Sci. B, 1980, 17, N 3, p. 517—542.
51. Reich S.9 Cohen J. Phase separation of polymer blends in thin films.— J.
Polym. Sci.: Polym. Phys. Ed., 1981, 19, N 8, p. 1225—1267.
52. Price F. P. The search for spinodal decomposition in binary systems of high
molecular weight polymers.—J. Polym. Sci.: Polym. Symp., 1978, N 63,
p. 13—31.
53. Липатов Ю. С, Шилов В, В. О фазовом разделении полимерных систем,—
Композиц. полимер, материалы, 1981 вып, 11, с. 55—69.
54. Meier D. J. Theory of block copolymers. 1. Domain formation in А—В block
copolymers.— J. Polym. Sci. C, 1969, N 26, p. 81—98.
55. LearyD. F.. Williams M. C. Statistical thermodynamics of ABA block
copolymers. 3. Microstructural transition and model verification,— J. Polym.
Sci. : Polym. Phys. Ed., 1974, 12, N 2, p. 265—287.
56. Krause S. Microphase separation in block copolymers. Zeroth
approximation including surface free energies. — Macromolecules, 1970, 3, N 1,
p. 84—87.
57. Helfand E.t Wasserman Z. R. Block copolymer theory. 4. Narrow interface
approximation.— Ibid., 1976, 9, N 3, p. 879—888.
58. Leibler L. Theory of microphase separation in block copolymers.— Ibid.,
1980, 13, N 6, p. 1602—1617.
59. Keller A.t Pedemonte E.f Willmouth F. M. Macrolattice from segregated
amorphous phases of a three block copolymer.— Kolloid. Z -\- Z. Polym., 1970,
238, N 1/2, S. 385—389.
60. Lewis R, L.,Price С The morphology of (styrene)* (butadiene)^ (styrene)*
block copolymers.— Polymer, 1971, 12, N 4, p. 258—270.
61. Hadziioannou G., Mathis Л., Scolios Л. Obtention de «monocristaux» de copo-
1 у meres trisequences styrene (isoprene) styrene par cisauillement plan.—
Colloid and Polym. Sci., 1979, 257, N 2, p. 136—139.
62. Ерухимович Я. Я- Флуктуация и образование доменной структуры в гете-
рополимерах.— Высокомолекуляр. соединения А, 1982, 24, № 9, с. 1942—
1949.
63. Ерухимович Я. Я. Влияние химического строения двухкомпонентных
расплавов гетерополимеров на образование в них доменной структуры.— Там
же, с. 1950—1957.
64 Roe /?.-/. Fishkis M., Chang J. С. Small —angle X-ray diffraction study of
thermal transition in styrene — butadiene block copolymers.—
Macromolecules, 1981, 14, N 4, p. 1091—1103.
65. Гленсдорф Я.. Пригожий Я. Термодинамическая теория структуры,
устойчивости и флюктуации.— М. : Мир, 1973.— 280 с.
66. Николис Г.. Пригожий Я. Самоорганизация в неравновесных системах.—
М. : Мир, 1979.— 280 с.
67. Lindenmeyer P. Я. Polymer morphology as dissipative structure.— Polym. J.,
1979, 11, N 8, p. 677—679.
68. Lindenmeyer P. H. Polymer solidification as dissipative process.— J. Polym.
Sci. : Polym. Phys. Ed., 1979, 17, N 10, p. 1965—1970.
69. Липатов Ю. С, Шилов В. Т, Коллоидная структура гетерофазных
полимерных систем.— В кн. : Успехи коллоидной химии. Киев : Наук, думка,
1983, с. 180—193.
70. Нестеров А. Е. Справочник по физической химии полимеров. Т. 1.
Свойства растворов и смесей полимеров / Под ред. Ю. С. Липатова.— Киев :
Наук, думка, 1984.— 376 с.
265
71. Hashimoto ^.., Kumari J.t Kawai H. Time—resolved light scattering studies
on kinetics of phase separation and phase dissolution of polymer blends : I.
Kinetics of phase separation of a binary mixture of polystyrene and poly(vinyl
methyl ether).— Macromolecules, 1983, 16, N 4, p. 641—648.
72. Snyder H: L:, Meakin P., Reich S. Dynamics of phase separation in polymer
blends.— Ibid., N 5, p. 957—962.
.73. Nojima S.. Tsutsumi /(., Nose T. Phase separation process in polymer systems.
' I.'Light scattering studies on a polystyrene and poly(methylphenylsiloxane).—
Polym. J., 1982, 14, N 3, p. 225—232.
ГЛАВА 9
МЕЖФАЗНАЯ ГРАНИЦА
В РАССЛАИВАЮЩИХСЯ РАСТВОРАХ
И СМЕСЯХ ПОЛИМЕРОВ
Образование любой гетерогенной системы
всегда сопровождается возникновением межфазного или граничного слоя,
наличие которого во многом определяет физико-химические и
механические свойства этой системы. В настоящее время имеется достаточно
большое количество работ, посвященных изучению взаимодействий
(межфазного поверхностного натяжения) и свойств межфазного слоя
(концентрационного профиля и толщины межфазного слоя). Обзор
работ о механизме формирования межфазного слоя и его влияния на
свойства многокомпонентных полимерных систем можно найти в
монографии [1]. Здесь же мы остановимся главным образом на результатах
работ, посвященных исследованию межфазного натяжения и
структуры переходного слоя в расслаивающихся растворах и смесях
полимеров, а также рассмотрим работы, в которых приведено сравнение
выводов соответствующих теорий межфазного натяжения с
экспериментальными данными.
Теории межфазного натяжения и структуры межфазного слоя
в растворах и смесях полимеров были развиты в работах авторов [2—
10]. Так, Фрей [2, 3], основываясь на теории Кана и Хилларда [4] о
межфазной границе в смесях низкомолекулярных веществ, получил
уравнения для межфазного натяжения между расслаивающимися
растворами и нашел качественное согласие с экспериментом. Авторы
работы [5] развили теорию межфазной границы для несовместимых
симметричных смесей, используя теорию среднего поля, авторы [61
применили ее к асимметричным системам. Они предсказали, что
толщина межфазного слоя в системе полимер — полимер равна примерно
1 нм и зависит от параметра термодинамического взаимодействия Фло-
ри—Хаггинса X. Рйонг-Жун Ро [7], основываясь на своей старой
теории неоднородных систем, развитой на основе модифицированной
теории Флори—Хаггинса, разработал теорию межфазной границы между
смесями полимеров и растворами полимеров, в которой межфазное
натяжение и толщина межфазного слоя выражается в виде зависимости от
(X—Хкрит) и длины цепи, где Хкрит — параметр % при критической
температуре смешения. Носе [81» также основываясь на теории Кана и
Хилларда, развил теорию межфазного натяжения и толщины
межфазного слоя в расслаивающихся полимерных системах, учитывая
изменение размеров полимерного клубка на межфазной границе. В основу
267
теории межфазной границы Саншез [9, 101 положил градиентную
теорию 111] и модель сжимаемой жидкой решетки [9, 12, 13]. Приближения
скейлинга для описания свойств межфазной границы использованы
в работах [15—17]. Основные вопросы, решаемые в этих теориях, —
установление зависимостей межфазного натяжения от температуры и
молекулярной массы полимера. Рассмотрим некоторые из этих теорий.
9.1. ТЕОРИЯ ФРЕЯ И РОБЕСОНА
Фрей и Робесон рассмотрели концентрационный профиль
межфазной области и межфазную свободную энергию (межфазное натяжение)
для растворов расслаивающихся полимеров вблизи критической
температуры смешения, где деформация полимерных клубков мала [3].
Для характеристики гомогенных фаз они использовали уравнение
Флори—Хаггинса для комбинаторной энтропии смешения 5fe на
единицу объема в виде
—Sk/k = П2 In (V2n2) + Пх 111 (Ufa),
где tt\ и м2—количества молекул на единицу объема растворителя
и полимера соответственно: vx и v2— объемы, занимаемые
молекулами растворителя и полимера. Они предположили, что свободная
энергия смешения зависит также от локальной структуры раствора
и является* таким образом, функцией локальной плотности
сегментов:
a/kT= п2\п (v2n2) + %ln (vxnx) + g (nC9T); (9.1)
здесь пс— локальное число сегментов.
Плотность сегментов полимера и растворителя они связали
соотношением
vcnc + v1n1 = I, (9.2)
предполагая аддитивность объемов (ис—объем сегмента).
В гомогенной системе уравнение (9.2) эквивалентно выражению
v1n1= mvcn2+ v1n1= 1,
где m = v2/v1. Для негомогенной системы псф гп2. Предполагается,
что среднее пространственное распределение сегментов около центра
масс клубка сферически симметрично и определяется зависимостью
% (г) (г — радиальное расстояние от центра клубка), тогда локальная
плотность сегментов в элементе объема dx2 при х2 следует из линейной
суперпозиции вкладов полимерных молекул с центрами при бтх около
бт2:
* W - J Е (l>i - г2\) п2 (хх) dTv (9.3)
где интервал берется по объему всей системы. Когда изменение от пп
() расстояния сравнимо с размером клубка.
(9.4)
268
Интегрирование по угловым координатам уравнения (9.3) приводит
к тому, что линейный член уравнения (9.4) исчезает, а оставшиеся члены
выражаются в виде
1 /Я2- \
?2 + • • • , (9.5)
где радиус инерции клубка R определяется из выражений
во оо
#2 = пг1 J г21 (r) 4nr2dr, т = J I (г) ЫгЧг.
6 о
3 межфазной области п2(х) имеет S-образный характер (рис. 9.1).
Этим подразумевается, что при некотором положении ха d2n2/dx2= О
и пс(х) = тп2(х). В более разбавленной области пс> тп2, а в более
концентрированной п <т2п2.
Общее число сегментов в межфазном
слое в m раз больше числа цент- 1 ! //
ров макромолекулы:
и ш
\ ncdx = m j n2dx\
i i
это следует из частного
интегрирования d2n2/dx2 и из условия dnj
Ых = 0 в гомогенных фазах.
Для расчета поверхностной
энергии и концентрационного
профиля негомогенный межфазный
слой разделяют на слои толщиной
dx, перпендикулярные к
координате х при (vI/3<t.dx < R.
Свободную энергию смешения Гиббса
получают тогда путем интегрирования а из уравнения (9.1) по всей
межфазной границе:
Рис. 9.1. Схематическое представление
изменения плотности полимерных
сегментов в расслаивающемся растворе
полимера:
/ — разбавленный раствор; //— межфазная
область; /// — концентрированный
раствор [3j.
in
AkT
(9.6)
где А — поверхность межфазной границы.
Дальше предполагается, что d2n2/dx2 достаточно мало, поэтому
второй член в уравнении (9.5) также мал. Подставляя уравнение (9.5)
в (9.1), получаем
где
о'/ *Г= - ~ m {^.j [ In A - v2n2) - 5^]„в - mn,.
10 4-420 2«9
a°/kT = n2 In v2n2 + v~l A — v2n2) x
X ln(l — iyia) +g{(l -tyz2),T};
Подстановка a'lkT в уравнение (9.6) показывает, что этот член
после частного интегрирования может быть заменен (при интегрировании)
выражением
kf Т^м5F/ \v\ A — ^2) v2dn\ « — w«i- (9-7)
Тогда уравнение (9.6) преобразуется в следующее:
G/AkT :
ш
) kf + kf)
dx.
(9.8)
Член a*/kT можно рассмотреть как «градиентный» вклад в свободную
энергию смешения Гиббса в трактовках Ван дер Ваальса или Кана.
Теперь из G необходимо вычесть
свободную энергию смешения Гиббса для
гомогенной системы Go:
(9.9)
р2(Ш)
Рис. 9.2. Изменение избыточной
свободной энергии смешения при
переходе от разбавленного
(область /) к концентрированному
(область III) раствору при
фазовом расслоении (Ла — изменение
свободной энергии в межфазной
области) [3].
где Nt и Af2— количество молекул
компонентов 1 и 2 во всей системе: щ(/?) =
= MD = Hi (HI) и |*,(р) = MD =
= fx2 (III) — химические потенциалы
смешения в двух равновесных фазах.
Для уравнения (9.9) можно написать
ш
III
n2dx.
(9.10)
Значения \itip) и \i2(p) можно получить дифференцированием
уравнения (9.1). Из уравнений (9.10) и (9.8) следует, что
<9Л1>
где
(p) ^ Vi [l— v2n2(p)\
v* [пш -n2(p)) + (m-l) [n2 - n2 (p) ]. (9.12)
Пределы интегрирования могут быть выбраны как —оо и +оо,
поскольку подынтегральное выражение не исчезает только в межфазной
области. Можно использовать либо л2A), либо п2(П\) для определения
пъ(р) (рис- 9-2). Теперь G—Go еще зависит от п2(х), т. е. от выбора
профиля. Видно, что функция G—Go принимает минимальное значение,
270
тогда G—Go равно о А, где а — межфазное натяжение или межфазная
свободная энергия.
Вариационные расчеты приводят к уравнению Эйлера, как
необходимому условию для экстремума:
Аа(*2) _ R* • (апЛ21 ! , m?g] = а*
~~?г " у2 ^j |»i A — v2n2) ^ V2 dn2j w фЛд)
Добавочным условием истинного минимума является критерий Ле-
жандра: вторая производная подынтегрального выражения по dnjdx
должна быть не отрицательной между пределами интегрирования.
В данном случае это условие выражается так:
(Да (я,) а* (я,, dn2fdx)
d(dn2/dxJ У kT ^ kT
= — T |мь
что верно только в случае
Это условие удовлетворяется в области малых отклонений (обычно
~ 1 % или меньше) от критической температуры, поэтому
применимость данной теории строго ограничена.
Подставляя (9.13) в (9.11), имеем
+~ +1."
а = 2 J Aa(n2)dx=2 J a*dx. (9.15)
— оо — оо
Используя уравнение (9.13) для замены переменной х на /г2,
окончательно получаем
^^^ 1 ?^g)]1/2^2 (9.16)
«id)
Видно, что это уравнение решается только в случае, если
удовлетворяется условие (9.14).
Уравнения (9.13), (9.15) и (9.16) были использованы для расчета
а и п2(х) при двух разных выражениях для g:
(9.17)
где Q — постоянная (в гомогенной фазе <рс = vcnc == v2nz =Ф2) и
Ф2) (9Л8)
с g, не зависящей от"ф2. Выражение, данное уравнением (9.18), было
использовано в работах [18, 19].
Полагая формулировку а в уравнении (9.1) справедливой вблизи
критической точки, его можно разложить в ряд Тейлора. Этим подразу-
Ю* 271
мевается, что мы находимся в рамках «классических» теорий
критического состояния [4]:
а = акрит + ех (Т — Ткрит) + е2 (Т — 7крит) (фз — ФкритJ +
+ е3(Ф2 — Фкр«тL+ .... (9.19)
где гг = (За/аГ)криТ; Ч = у (д3я/<ЭГдф*)крит; е3 = ^ (д4я/дф24)криТ;
индекс «крит» характеризует значения в критической точке. Уравнение
(9.19) симметрично относительно (ф — ф2 крит), так что бинодаль и
спинодаль получаются соответственно из условий да/дц>2 = 0 й
д2а/ду1 = 0. Записав
^ = ф2
имеем для бинодали
а для спинодали
Выражение для Аа (см. уравнение (9.12) и рис. 9.2) теперь имеет вид
а
«крит = 4 (kTKpmR*V2/V2 крит) (d?
Из уравнений (9.7), (9.13) и (9.16) следует, что
|ф2б — ф2крит| = y (У2> ш ~ V2' ^ =
или а = Л2G1крит — T)Vt; Л2 = Be2/e3)vs a для профиля межфазной
области —
«2 — у (^2, i — п2, ш) = у Ап2 tg g" [2 (д; — xo)/L]
в Г ^крит^2 1 в Г
138Зи2Ф2крит(Аф2J] L
V * >
где L — толщина межфазной области.
Согласно Дебаю [14],
'•И. = ! + m4v> (Й^/^крит) = A + т-1/'J = ФГ
272
Используя уравнение (9.17), можно записать
6=
у
18 /2 V 1ф/к8рит
A _ Г/Гкрит) Vt •
Эти уравнения предсказывают слабую зависимость а от /я, т. е. от
молекулярной массы. В частности, а~ т1/^(А(р2K—m~'/*[l—77
Ткрит]*\ a L — const(Дф2Г1~т1/4A—Г/ГкритГ1А для m»l,
принимая, что R^zm1/*. Значение экспоненты, равное 1/4, было найдено
Носе [81. Он же установил корреляцию между толщиной межфазного
слоя и персистентной длиной корреляций флюктуации. Возможность
такой корреляции вытекает из того факта, что свободная энергия
флюктуации концентрации концептуально сходна с межфазной
свободной энергией.
Согласно теории [20], угловая зависимость интенсивности
рассеяния света выражается уравнением
где G — угол рассеяния; X — длина волны света в вакууме; Lk— пер-
систентная длина корреляций флюктуации, определяемая из
соотношения
Вблизи (но выше) ТКрит и при Ф2 = ф2крит из уравнения (9.19)
дим
k Т /?2
г К 1 КрИТП
*~ е2У2ф2(Г-Гкрит)-
Сравнение с уравнением (9.20) позволяет увидеть сходство
физической природы Lk и L. Введением радиуса действия межмолекуляр-
(т
7р 1) , для случая ре-
1 крит '
гулярных растворов было найдено [20], что /2= 2/?2ф2Крит- Из этого
следует, что Z, и таким образом Lk, пропорционально т1/*.
Зависимость I 01 m такого рода была экспериментально найдена при
исследовании явлений критической опалесценции в растворах полимеров
[20].
273
*.2. ТЕОРИЯ КАММЕРА
В основу теории межфазной границы Каммера [21—23] положены
условия Гиббса о равновесии между двумя фазами при существовании
между ними граничного слоя; следовательно, химические потенциалы
фаз и слоя равны: \is{= ц,*= \x2r Принимается, что состав межфазного
слоя является функцией температуры и, соответственно, межфазное
натяжение зависит от состава слоя и температуры. Для межфазного
слоя вводятся молярные значения поверхностей разделов составляющих
компонентов Als и A2s. Получено следующее соотношение,
характеризующее зависимость состава слоя от температуры и давления,
пригодное для любой двухкомпонентной системы:
п— 1
I" / ] \%i — X{ ) -p \Xi — X{ ) I 1 — ~z— / I dikdXk == Of
i, n=\
где индексами 1, 2 и s отмечены фазы и межфазный слой, а *\ k и п —
компоненты системы; х — молярная доля компонента.
Молярная свободная энергия и энтропия выражаются равенствами:
здесь V12 = V2 — V1 + (дУ/дхгУ; V — молярный объем. Величины Sls
и Vis определяются из соотношений типа
где Y=S, V и /=s, 2.
Каммер полагает, что два полимера находятся в жидком состоянии
и несовместимы, а в межфазном слое происходит лишь
незначительное взаимопроникновение макромолекул. Поверхность межфазного
(граничного, в определении Каммера) слоя а связана с его толщиной
соотношением a~Vs/d. Принимая сначала, что количество молей
компонентов в граничном слое постоянно при его любом составе и
что с составом изменяются только молярный объем V и толщина
слоя d, автор получает, что A\s = V\d; ^2S= V\ld.
Концепция компонента в слое может быть выражена так:
где у — межфазное натяжение.
274
Расчет, проведенный для пары полимеров (полиамид —
полиэтилен), дал значение х2= 0,92, которое можно рассматривать как
среднее для всего слоя, откуда следует, что плотность компонента в слое
изменяется сначала очень медленно, а далее быстро падает до нуля.
При получении расчетных уравнений для толщины межфазнога
слоя автор рассматривает граничный слой как идеальный раствор
полимеров и вводя степени полимеризации компонентов гх и г9
получает
где V*20— молярный объем поверхностного слоя чистого второго
компонента; фь = 1 — [r^l/faxl + r2xs2)].
Подстановка числовых значений дает для системы ПА—ПЭ толщину
граничного слоя порядка 7,0 нм, согласующуюся с расчетными
данными Хелфанда и Тагами. Теория Каммера позволяет вычислить
температурную зависимость параметров слоя.
В дальнейшем [21, 22] Каммер использовал для расчетов теорию
Флори—Хаггинса, которая для определения химического потенциала
учитывает степени полимеризации и параметр термодинамического
взаимодействия, т. е. полимерную специфику и термодинамику
растворов полимеров. В этом случае уравнение для толщины слоя имеет
вид
5 5, (9.21)
где V — геометрическое среднее молярных объемов двух
компонентов. Использование этого уравнения при гх= г2= г требует
специальных расчетов параметра X. В этих условиях для разных пар (ПЭВД—
ПС, ПЭ—ПС, ПЭ—ПА, ПС—ПММА) толщина межфазного слоя лежит
в пределах 7—10 нм.
Каммер [24, 25] развил также общую теорию негомогенной
межфазной области, учтя градиенты плотностей компонентов и свободную
энергию (как и в рассмотренных выше теориях Фрея, Ноузе, Саншеза
и др.). Межфазное натяжение в рамках теории Флори—Хаггинса
с учетом размеров макромолекул, характеризующихся
среднеквадратичным расстоянием между концами цепи (А2I/2, дается здесь как
где X вычисляется из параметров растворимости; Ь — эффективная
длина мономерного звена, определяемая из соотношения
Ь^тУЦН/М1/*) (9.23)
(М — молекулярная масса полимера; т — мономерного звена).
275
В уравнение (9.22) входит величина z, являющаяся неопределенной,
так как степень полимеризации в межфазном слое неизвестна. Эту
величину автор определяет из выражения %z = я2/3- Т*/Т9 где Г*—
параметр приведения (характеристическая температура), находимый
путем сравнения с экспериментальными данными по межфазному
натяжению. В этом случае
где Р = с2Д2, с и к — константы, характеризующие взаимодействие
компонентов. В упрощенном виде имеем
Как и раньше, находят толщину межфазного слоя, выражаемую
теперь как
2 b (Г*/ТI/2
3/3
I ^
Расчеты d по этому уравнению дают хорошее согласие с данными,
получаемыми по уравнению (9.21) [22, 23].
Существенным результатом теории Каммера является
термодинамическое обоснование того, что толщина слоя пропорциональна
эффективной длине мономерного звена (см. уравнение [9.23]) и обратно
пропорциональна X, т. е. взаимопроникновение реализуется на очень
небольшом расстоянии, значительно меньшем по сравнению с размерами
макромолекул.
Каммер получил также уравнения для толщины слоя в случае
ограниченной совместимости компонентов [24].
9.3. ТЕОРИЯ ХЕЛФАНДА
В ряде работ Хелфанда [5, 6, 26—29] развита теория межфазной
границы, основанная на статистическом рассмотрении проблемы в
рамках решеточной мадели. В общем виде структура межфазного слоя
определяется следующими факторами: энергией контактов между
звеньями полимера и выигрышем конформационной энтропии. Поэтому
задача сводится к статистическому вычислению энтропии для данного
профиля концентраций компонентов с последующей минимизацией
свободной энергии.
Мономерные единицы макромолекул А и В (их количество
соответственно Na и Nb) располагаются на решетке из п ячеек таким
276
образом, чтобы заполнить все места на решетке. Эта решетка
представляет собой tiL слоев, параллельных границе раздела с ns ячейками
в каждом слое так, что п = п8пь- Каждое звено взаимодействует с z
соседями, доля таг которых находится в смежных слоях, а доля
A—2т) — в том же слое. Принимается, что степень полимеризации N
обоих компонентов одинакова; п полимерных единиц располагав
ются на решетке в определенной последовательности: сначала z
звеньев молекулы А, затем от (z + 1)до 2z звеньев ? и т. д., чередующим*
ся образом.
Вследствие существования потенциала отталкивания между едини*
цами А и В в соседней ячейке система разделяется на фазы А и В
и межфазную область между компонентами. Математически это озна-г
чает, что существует L слоев, в которых доля занятых мест ф^ компо*
нента К (К обозначает А или В) равна единице для многих слоев, а за*
тем, после перехода через малое количество слоев, обращается в нуль.
Полная занятость решетки требует соблюдения условия ф^л+ ф^д533 Ь
Эти величины и должны быть выбраны таким образом, чтобы
минимизировать свободную энергию системы.
Центральной частью теории является вычисление вклада конфор-
мационной эйтропии в негомогенность системы SHr в рамках
классической теории Флори — Хаггинса. Конформационная энтропия связана
с потерей конформационной свободы в результате того, что молекулы
вблизи границы раздела должны возвратиться в свою собственную
фазу. Свободная энергия, связанная с негомогенностью системы РИГ,
является суммой —TSur и энергии смешения, под которой
понимается смешение слоев, т. е. FHr = Епг— TSHr. Величина ?Нг может
быть описана как свободная энергия контактов и их обмена.
Минимизация энергии дает функцию распределения плотности
в межфазной области и величину межфазного натяжения у,
определяемую из условия: уа= FHr, где а — поперечное сечение ячейки решетки.
Применение методов вычислительной техники позволило для
различных значений -параметров взаимодействия Флори—Хаггинса %
рассчитать объемную долю единиц А в первом слое фм со стороны
поверхности В из условия, что минимальная свободная энергия
соответствует симметрии, т. е. 4>L в — *P\-.L в-
Существенно, что в основном переход из одной фазы в другую
происходит в слоях 1, 0, 1 и 2 для % > 0,05, а преимущественно — в
слое 0 и 1.
Далее з теории рассматривается иной масштаб негомогенности [6],
когда она распространяется на большие расстояния, чем расстояния
между слоями в решетке. Для постепенного, перехода от А к В
получается следующее упрощенное выражение для профиля негомогенности
с учетом того, что величина L принимается как непрерывная
переменная:
<PLtA = {l+ exp [2(X/mI/2J [IT17*]},
а межфазное натяжение уа = kT (Х/лI/2.
277
Согласно Хелфанду, величина 2 (%//пI/2 является
характеристической толщиной межфазного слоя. Хотя теория не была сопоставлена
с экспериментом из-за неопределенности величины т, в качестве
основного результата она дает весьма узкую межфазную область.
Численные решения уравнений, полученных Хелфандом для ряда
рассмотренных случаев, даны в работе [28]. В частности, на основе
решеточной модели дается решение для полимера, находящегося в
контакте с непроницаемой местной границей, соответствующей условию
<pLP= I, L^ 1, для L до 4. В последнем случае также происходит
потеря энтропии от слоя к слою.
В последующем Хелфанд [26, 28, 29] развил теорию границы
раздела между концентрированным раствором полимера и растворителем
и для двух растворов, в которой (также на основе статистического
рассмотрения поведения макромолекул на решетке с учетом теории Фло-
ри—Хаггинса) были определены величина межфазной области и
профиль плотностей.
В работе [5] теория межфазной границы развита на основе
приближения самосогласованного поля. Профили плотностей двух полимеров
в межфазной области определяются полями, которые модифицируют
конфигурационную статистику цепей в этой области. В свою очередь,
конфигурационные вероятности могут быть использованы для расчета
профилей плотностей. Проблема сводится, таким образом, к
определению самосогласованных полей и профилей плотностей. Для этого
вводится функция, описывающая статистику полимерных молекул на
межфазной границе и представляющая собой отношение функции
распределения участков макромолекул средней длины Ь, находящихся
в поле сегмента U (г), к функции их распределения в нулевом поле:
. q(r0, t) = j drt ... drMP(rQ, rlt ... , rM) x
м
X exp (— J] -r^jr) IJ drx ... drMP (r0, ..; , rM). (9.24)
Принимается, что степень полимеризации макромолекулы 2, участки
цепи имеют один конец в точке г0. а другой — в точке г, а также
rt последовательных положений (t — масштабный параметр,
заключенный между 0 и 1). В уравнении (9.24) M=-zt, r{ — положение
конца /-го сегмента; Р выражается гауссовской статистикой:
Функция q (r, t) выражает отношение плотности сегментов в точке г
межфазной области к плотности сегментов в чистой фазе. При малом
278
b v U функция q (r, i) удовлетворяет модифицированное уравнение
диффузии
dj^J) J e |
Для вывода уравнения среднего поля на межфазной границе
рассматривается следующий случай смеси полимеров с симметричными
свойствами: 1) степень полимеризации z, которая полагается стремящейся
к бесконечности; 2) эффективная длина мономерной единицы,равная Ь,
выбирается таким образом, чтобы среднеквадратичное расстояние
между концами цепи равнялось zb2; 3) плотность чистого полимера равна
р0 (число мономерных единиц в единице объема); 4) сжимаемость к
составляет ~ 5 • 10~*10 м2/Н. Если свойства А и В не идентичны,
используется правило геометрического среднего.
Рассмотрим участок молекулы А. Этот участок находится в
эффективном поле, создаваемом средним молекулярным окружением.
Относительно чистой Л-фазы это поле состоит из следующих частей:
первая — kTXpB (r)/p0 — обусловлена неблагоприятными контактами
сегментов А с сегментами В\ вторая — [(l/p^x) — (kT/zp0)] [pA (r) +
+ Рд (г) — Pol — вытекает из тенденции системы проталкивать
полимер в области, где общая плотность (рл + рв) меньше, чем р0, и
выталкивать из областей, где плотности больше, чем р0.
Тогда уравнение для q(r, t) можно записать в виде
6 V ЧА \ Г> 1>
dt 6
L Рв W , t \Ра (г) , Рв (г) ]\
S = A/РоАТ4^) — A/2).
Аналогичное уравнение может быть записано и для qB(r, t):
где
S = A/РоАТ4^) — A/2). (9.26)
Начальные условия таковы:
QA(r> 0)=<7вС. О)^1- (9.28)
Уравнение (9.25) (или (9.26)) получено введением вместо U(r) эф*
фективного потенциала \ь*А (г) — \кед (р0, 0):
279
полученного расчетом работы добавления сегмента А в межфазную
область в точке г с использованием лишь первых двух членов уравнения
(9.29).
Функция q (г, t) равна отношению плотности концов полимерных
молекул типа А с длиной zt в точке г к плотности сегментов в чистой
фазе. Каково же отношение плотностей для zZ-сегмента по цепи Л, если
цепи имеют длину z? Этот сегмент может рассматриваться как начало
двух участков: один длиной г/, а другой — z A — t). Таким образом,
относительная плотность равна
4а(г> *)'Ча(г> !—О- (9.30)
Интегрируя по t9 находим
^ =§dtqk(r, \-t)qk(t, t) (k~A или В). (&.31)
о
Это уравнение вместе с (9.25)—(9.28) и граничными условиями:
ЯА (оо, t) = qB(— oo, 1) = 1 и qA (— оо, /) = ?в(оо, t) = 0
могут быть использованы для расчета профиля плотности в
межфазной области.
Авторы работы [5] дали приближенное решение, приняв, что
система несжимаема и X/g мало (%/?-*0), а сегменты имеют
одинаковое распределение плотности по молекуле. Математически это
сводится к выражению qk(r, t)-+qk(r). Из уравнения (9.31) тогда
следует, что pk (г) = poq* (r),
| Ь2 (d*/dx*) qk(x) -\q\, (x) qk (x) - g \q\, (x) + q\ (x) - 1] qk (x) = 0
(k = Л, В, в то время как №=* В, А). Тогда
РЛ W/Po = ?2/(i + i2); PBW/Po = 1/A + g2);
В качестве толщины межфазного слоя может быть взята величина
(9.32)
C.3 нм для типичных значений Ь — 0,7 нм, % = 0,03).
Для межфазного натяжения получено уравнение в виде
(9.33)
После расчета из данных параметров растворимости значения X для
некоторых полимерных пар по уравнению % = (8л— Ьв)УрокТ с
помощью уравнения (9.33) были оценены значения межфазного
натяжения и сравнены с экспериментальными данными. Как видно из табл.
9.1, расчетные и экспериментальные данные хорошо коррелируют
друг с другом. Толщина межфазного слоя сопоставима с размерами
мономерного звенад. е. согласно представлениям, развитым Хелфан-
дом, на межфазной границе возможна только ограниченная диффузия
280
Таблица 9.1. Сравнение рассчитанных и измеренных значений межфазного
натяжения [5]
Система
Полистирол — полиметилметакрилат
Полиметилметакрилат — полибутил метакрилат
Полибутилметакрилат — поливинилацетат
Т. мН/м
расчет
1.0
2.0
1,9
эксперимент
1.5
1.8
1.9
Ъ, нм
0.65
0.61
0.63
X
0.01
0.07
0.05
96
114
107
сегментов цепей в очень малой области, что исключает, естественно,
образование переходной области, представляющей собой раствор
сегментов молекул одного типа в сегментах поверхности другого полимера.
9.4. ТЕОРИЯ НОСЕ
Эта теория [8] также основана на теории Кана и Хилларта, но не
учитывает изменение размеров молекулярного клубка на межфазной
границе. Как и у Фрея и Робесона, взаимодействия между
полимерами (или полимером и растворителем) приписываются
взаимодействиям между сегментами. Полагается, что молекулы могут
взаимопроникать друг в друга. В неоднородной системе локальная плотность
сегментов и локальная плотность макромолекул связаны друг с
другом градиентом концентрации. Для учета изменения размеров
макромолекулы вблизи границы раздела вводится локальный коэффициент
объемного расширения, являющийся лишь функцией направления,
перпендикулярного к межфазной границе. Рассчитывается вклад
в свободную энергию, связанный с изменением конформации цепи,
и повторяется процедура расчетов градиента плотности свободной
энергии, подобная уже рассмотренной. Опуская эти расчеты, проведем лишь
окончательный результат. Так, значение межфазного натяжения
выражается в виде
уо 2{1+(/
(9.34)
где т — число сегментов в макромолекуле; v — объем сегмента;
Ь2 = (а\ 'ж*)Ща — размер полимерного клубка, X — длина межсегмент*
ных взаимодействий [4]), Т = Т/Ткрит.
Для симметричной полимерной системы, т. е. mt = m2=m
и bt = b« = b имеем
Р=^Ц. = 2A+ mb*I/2m-i (I - ff2 с
K1 критА
L/k = 2 A + b2m)V2 A - f)-1/a.
(L/K — толщина межфазной области).
'> (9.35)
(9.36)
281
Для несимметричной
yv о ,, о .
ът \ ~ v ~гm
K1 критЛ
LA =2{1 •
системы
^ 26/п~"
-fmfe2(l-
(система полимер
-1I/2 A -j- Ш1/2)—1/:
i/4(i т-K/2
-f- ml/2)}1/2 A —'
— растворитель)
2A ГK/2^
f pl/2 _
Как видно из уравнения (9.34) и (9.36), критические экспоненты
для межфазного натяжения и толщины межфазного слоя равны 3/2
и — 1/2 соответственно.
В случае симметричной полимерной системы (полимер — полимер),
согласно уравнению (9.35), величина у/[(Ткрт)/A—ГK/21
уменьшается с увеличением молекулярной массы в степени — V2> в то время
как y/A—ТK/2 растет с молекулярной массой в степени 1/2, т. е.
пропорциональна невозмущенному размеру полимерного клубка.
Для ^растворов полимеров обе величины, у/ {Ткрт /A — ГK/2} и
у/A — ТK/2, уменьшаются с повышением молекулярной массы в
степени — 1/4 в области высоких молекулярных масс (см. уравнение
(9.37)). Толщина межфазного слоя L увеличивается с ростом
молекулярной массы для систем полимер — полимер и полимер —
растворитель, но с разной степенью. Так, в симметричной системе L
пропорциональна невозмущенным размерам клубка, т. е. изменяется с m в
степени 1/2, в то время как для полимерного раствора L пропорциональна
m в степени 1/4. Следовательно, теория Носе приводит к тем же
зависимостям у и L от температуры и молекулярной массы, что и теория
Фрея и Робесона [3], где не учитывается изменение размеров
макромолекул в межфазной области. Экспериментально теория Носе была
проверена в работах [30—32], в которых отмечено удовлетворительное
совпадение выводов теории с экспериментальными результатами.
9.5. ТЕОРИЯ САНШЕЗА
В основу теории Саншеза [9, 10] положены градиентная теория (как
и в других рассмотренных выше теориях межфазной границы) и
модель сжимаемой жидкой решетки. Энтропия негомогенной системы
также предполагается функцией только локальной плотности, а для
оценки влияния градиентов плотности на потенциальную энергию
использовано приближение среднего поля. В частности,
потенциальная энергия на единицу объема жидкости из m компонентов на
расстоянии R в предположении парной аддитивности выражается в виде
M#); (9-38)
i i
etj (R) = 9i (R) 1 P/ iR + s) gti (s) U и (s) ds, (9.39)
282
где &i(R) — плотность компонента i при R; p/ (R + s) — плотность
компонента / при R + s; s = \s\ — межодолекулярное расстояние;
Utj(sy\—межмолекулярный потенциал между молекулами i и /;
Stj (s) г" Функция распределения. Предполагается, что Uq —
сферически симметричен, т. е. зависит только от s. В приближении
среднего, щля пренебрегаются корреляции близкодействия и условия
0
где сц -+ параметр, характеризующий «ядерную» часть парного
потенциала {<№, — ядерный объем, на который распространяются силы
отталкивания между компонентами i и /).
Если вдали от межфазной границы жидкая смесь гомогенна,
значение ец не зависит от R и пропорционально средней плотности
компонентов i и /. При наличии градиента концентрации p.(R + s)
можно разложить в ряд Тейлора около s = 0:
Р, (R + s)= p, (R) + (VP/ • s)s=o + |
где, например,
Подставляя уравнение (9.40) в (9.39) и интегрируя, получаем
оо
en (R) = —Pt (R) P/ (R) tn'o' + p, (Я) У -?¦, (9.41)
* I * Л^ш ТЫ
где /n^—положительная постоянная (мохмент нулевого порядка
оо
= — 4я J s*U{j(s)ds, (9.42)
Поскольку [/// предполагается сферически симметричным, он является
функцией Sx, Sy и 5Z. Поэтому подынтегральные выражения в
интегралах уравнений (9.42) являются либо четными, либо нечетными
функциями sx, sy и sz. Тогда промежуточно
On четное,
=^0 п нечетное.
283
Для п = 2 интервалы, вовлекающие смешанные производные,,
исчезают и получаем
/2/ = — if-vV/, (9.43)
где т*1 положительная постоянная (момент второго порядка (///):
оо
тЧ = - -J- j sWij (s) ds. (9.44)
Предположив энтропию m-компонентной жидкости функцией только
локальной плотности, не зависящей от градиентов плотности,
локальную плотность свободной энергии a (R) можно выразить путем
комбинирования уравнений (9.38), (9.41) и (9.43):
i /
где а0 (R) — локальная плотность свободной энергии при отсутствии
градиентов плотности
t f
a S = S/V—плотность энтропии. Общая свободная энергия
Для системы с плоской межфазной границей с поверхностью So
ч объемом LS0{L->~оо) имеем
G=S0 $ a(x)dx. (9.46)
-L/2
Пренебрегая членами четвертбго и высших порядков в уравнении (9.45),
интегрируя по частям и используя граничные условия (dpjdx = О»
когда л:->+ оо), уравнение (9.46) приводят к виду
L/2
-L/2
i i
Межфазное натяжение у определяется тогда через
YS(G Gp)/S0= J [An+ -^22 «У (?)(?)]&; (9-47)
—~ г /
Да = а0 — Gp/V = а« — Др, (9.48)
284
где G\— равновесная свободная энергия гипотетической гомогенно*
системы при отсутствии межфазной границы. Она может быть
выражена в членах равновесных химических потенциалов \i и внешнего
давления \РР:
Поэтому Д а представляет собой избыточную свободную энергию ив
единицу объема жидкости состава plf p2, ..., рт при отсутствии
градиентов плотности. При наличии вкладов градиентов плотности
добавочный вклад в локальную избыточную свободную энергию
представляется двойной суммой в уравнении (9.47). Поскольку dpjdx и Д а ->• О*
при L -*» оо, пределы интегрирования можно расширить до
бесконечности в уравнении (9.47). Минимизация интеграла в последнем
приводит к т дифференциальных уравнений (уравнения Эйлера—Лагранжа):
Эти т уравнений могут быть получены также прямо из уравнения
(9.6) с использованием условия равновесия, которое требует, чтобы
химические потенциалы каждого компонента были постоянными
в пределах межфазной области. Умножая уравнение (9.49) на dpjdx
и суммируя по I, получаем
Интегрируя по х и используя граничные условия (Да и
при х -> оо), находим, что при равновесии
-т IE-?(?
Подставляя уравнение (9.51) в (9.47), находим равновесное натяжение
{dpi/dx)(dpI/dx)dx = 2 J Aadx.
Представленную теорию можно применить для расчета межфазноп*
натяжения и концентрационного профиля для многокомпонентных
смесей путем применения любой теории среднего поля. Поузер и Сан-
шез [10] использовали теорию сжимаемой жидкой решетки, где pi
заменяется на плотность сегмента компонента i:
a Uij(s) интерпретируется как потенциал взаимодействия сегмент —
285
сегмент. При обратной степени зависимости части этого потенциала,
связанной с силами притяжения, имеем
{g(o,;/5r itZ (9-52)
Таким образом,
mf/ = 4w*0/oJ//(« - 3) ->- 2г
m*l = 4ns<<a> ./(л - 5) -* 2 [?=
где g дается суммой уравнений (9.53) и (9.54), а \х% — уравнением
гКо„б = ^?г71пф<; (9-53)
= - Ре* + Я5о» + йГо [A - р) In A - р) +1 In р]; (9.54)
(9.55)
Для бинарной системы уравнение для межфазного натяжения можно
записать в виде
Г f /1 \ Г ]\
Y= I ]A^+ hr/ miiPi2 + 2т12рхр2 +m22p'2 [ dx, (9.56)
подчиняющемся условиям
дАа м а Л /л r^v
л— ~~~ ^nPi — ^iaP2 == ^» ^У.о/J
CPj
U/ли it *> r\ tr\ rro\
Н— —^i2Pi —^гРг == ^> ^y.OoJ
где введены следующие оценки:
Далее используется обратный степенной закон для части потенциала,
связанного с силами притяжения (уравнение (9.52)). В общем,
экспонента зависит от l—j взаимодействия, так что из уравнения (9.44).
получаем
mit^totfifimtf, (9.58а)
где
в?/ = 2яво"/(/1,/ —3)
286
= Цпц — 3)/(/i/y — 5)]/6.
Определяя а и как v*> величину т12 можно оценить, используя
правила комбинирования в виде
I5/3 ~
^12»
»0(в + I)]'
где /л12 иржет трактоваться как связующий параметр. Полезные
упрощения в уравнениях Эйлера достигаются при использовании для т12
закона геометрического среднего: т12= (тит22I/2. Тогда уравнения
(9.50), (9151) приводят к алгебраической форме
Для сравнения теории с экспериментом авторы работы [10]
использовали безразмерную величину
o) = m12/(m11m22)I/2> (9.60>
характеризующую отклонение т12 от геометрического среднего.
Значение со = 1 представляет собой высший предел достижимых значений
т12. Это видно из рассмотрения уравнений (9.50), (9.51) для бинарной
смеси:
Ля = 4 [т12р[2 + 2m12p;p2 + m22p22]. (9.61>
Ясно, что если у положительно, Д а также положительно, так что
должна быть положительной и квадратичная форма правой стороны
уравнения (9.61). Поскольку производные р1 и р2 по х могут быть как
положительными, так и отрицательными, т12< {тг1т22I12 или © < I
является необходимым условием, если квадратичная форма
положительна.
Численный и аналитический анализы упрощаются трансформацией
уравнений из пространства х в пространство р. Такая трансформация
изменяет пределы интегрирования в уравнении (9.56) от
бесконечности к конечным пределам. Записав уравнение (9.61) в виде
о> _ . Г 2Ло I1'2
Pl ~ ~ 1Щг + 2«и (Ф2/Ф1) + ^22 (*2/dpiJJ
и произведя замену переменных в уравнении (9.56), получим
р»
7 = 2'/2 J [ntu + 2m12 (dp2/dPl) + m22 dpzldpxf] ^ До1/2 dPl, (9.63)
р!
где I и II относятся к двум равновесным фазам. Уравнение (9.62)
может быть формально проинтегрировано для получения профиля
межфазного натяжения:
Pi (*)
12 (dp2/dPl) + /n22 (d
287
При использовании правила геометрического среднего для
т12/уравнение (9.59) является условием минимизации для уравнения (9.63).
Если т12 не равно геометрическому среднему, должны быть исг/ользо-
ваны уравнения (9.57, 9.58), которые в р-пространстве приводятся к
следующему:
12
и + 2m12 (dp2/dPl) + m22(dp2/dPlJj [(^?)(mn + m1
i2 + ft» (dp2/dPl)) - (^2)[2Да (тит22 - т12J] = 0.
Хотя уравнения теряют симметрию и становятся более сложными в
пространстве, их трансформация в последнем существенно
упрощает -расчеты.
А*
0А,
4 6 0 2 4
а 5
Рис. 9.3. Схематическое представление толщины межфазной области L(a)
и межфазного натяжения у (б) системы полистирол — полиэтилен состава
1:1 при 413 К, 6 = — 0,П, @ = 0,98, kU3 = 0,52, ?пс = 0,57 [9, 10].
В некоторых случаях применяют другое упрощение, если т12
подчиняется правилу геометрического среднего. Так, если ввести
переменную
Ф = тир{/2 + т22р2/2,
соотношение для межфазного натяжения можно записать в виде
7 = 2!/2 ? Да1/2 dO,
а решение подчинено уравнению (9.59).
В теории сжимаемой жидкой решетки плотность меров р,
определяется по уравнению: р, =ф,р/у*. Используя это определение с
уравнением (9.48), получаем общие правила комбинирования:
Аа = е*рД;* {—р + f [E - 1) In (I - р) + A/г) 1пр + (qyrj In Vl +
/г2) In Ф2]} + Р*р - р/У* (фХ + фя|х2); (9.64)
я! 21ечобходимо выразить в единицах энергии на сегмент. Уравнения
(9.56) и (9.57), (9.58) можно таким образом решить несколькими
методами разной сложности. Для систем жидкость — пар результаты
при со = 1 слегка отличаются от таковых, найденных при расчете
с со < 1. Для систем жидкость — жидкость результаты очень
чувствительны к значению о.
288
При исследовании применимости теории к системам^ полимер —
полимер авторы положили а равным среднему значению, а со = g.
На рис\ 9.3 приведена молекулярно-массовая зависимость
поверхностного натяжения и толщины межфазного слоя, полученная согласно
теории. Кривые рассчитаны для системы полиэтилен — полистирол
с I = со = 0,98 и гг= г2= г. Толщина определяется так:
L = (dx/dpj^w, (9.65)
где pj— плотность сегмента компонента 1, приведенная к
равновесному значению р2 в фазе, в которой компонент 1 преобладает. Поскольку г
уменьшается, смешение становится более благоприятным вплоть до
некоторого критического значения г (в данном случае ~ 100), когда
система становится полностью совместимой. Следовательно, толщина
межфазной границы должна стремиться к бесконечности, а пОзерхност-
ное натяжение — к нулю с уменьшением г. Если значение ?
уменьшается, критическое значение г и асимптотические значения толщины
межфазной границы понижаются, а межфазное натяжение
увеличивается. Как видно из рис. 9.3, межфазное натяжение между двумя
полимерами приближается к асимптотическому пределу, когда
молекулярные массы обоих полимеров очень велики. Для двух несовместимых
полимеров растворимость полимера А в полимере В пропорциональна
Mj\ а растворимость В в А пропорциональна М~?. Поэтому, когда
МА и Мв очень велики, две фазы близки по составу к чистым
полимерам А и В.
Эти результаты были использованы Поузером и Саншезом [10]
для получения приближенной формулы для межфазного натяжения
и толщины межфазной области двух бесконечно высокомолекулярных
полимеров. Предполагается, что плотность межфазной границы
изменяется линейно с составом:
dp/dpx = const = 1 + dp2/dpi- (9.66)
Постоянная в уравнении (9.66) может быть определена
интегрированием, а градиенты — из выражений
(р? — pi)/pS; (9.67)
Ф2/Ф1 = -P2/PJ, (9-68)
где р? и р2 — плотности сегментов чистых компонентов 1 и 2:
(9.69)
Следовательно, уравнения (9.57), (9.58) заменяются уравнением (9.69).
Подставляя уравнение (9.67) в (9.63), получаем
1
V = 2 V* [Kfp?J - 2m12p?p» + rfpSJ] f Да»/» Oft. (9.70)
о
289
Для простоты было положено, что тц = 1/2 и а = 0. Тогда, рсполь-
зуя уравнение (9.58а), находим
у = B/яоI/2 (Y*Y*)i/4 (^о5I/« J да»/» dplt (9.71)
где т0—безразмерная величина:
то= х1'2 v-Ve p2 _ 2о)р!р2 + т-1/2 v1/6 р*5;
со определяется уравнением (9.60), а у*— характеристическое
поверхностное натяжение чистых компонентов
Из уравнения (9.64) определяют Да:
Да = р/а* [а (р, f) г* - ф1а (р, 7Х) е*г - ф2а (р2, f 2) е*2],
где
а (р, Г) = -р + f A - р) In A - р)/р.
Значения рх и р2 вычисляют из уравнения состояния при их
соответствующих приведенных температурах 7\ и Т2 с г-> оо и Р = 0.
Величина Да будет достигать максимального значения вблизи р =
== (Pi + Рг)/2- Поэтому интеграл в уравнении (9.70), (9.71) может
быть принят приблизительно равным (Aai/2I/2/2, где
(9.72)
Поскольку р = рх + р2 = р/о*, а фх = рх/р, уравнение (9.72) при 6 = 0
приводится к следующему;
Afli/2 = Pi/2 {a (pi/2, f i/з) Р* - [а (Рх, Г0 в^Т + а (р2, Т2) в2я1]},
где
Pl/2 = (Pi + Р2)/2,
= 02XP* + в^Р* + 6 (V*/2 + v-1/2) вхв2 (Pt
Окончательно уравнение для межфазного натяжения двух полимеров
с бесконечно большими молекулярными массами принимает вид
y = jBfh0Aai/2y*iy*2y*9 (9.73)
где 5i/2 — безразмерная свободная энергия, определяемая через
выражение
Из уравнений (9.61) и (9.65) толщина межфазного слоя равна
L = (йо/2Да1/2)(^|I/6. (9.74)
290
Межфкзное натяжение и толщина межфазного слоя для системы
полимер V— полимер могут быть рассчитаны из уравнений (9.73) и (9.74).
если известны значения ? и со.
Авторы работы [10] провели такие расчеты, положив со = 0, а
значения g выбирали таким образом, чтобы воспроизводились
экспериментальные данные наилучшим образом. Они обнаружили, что
расчетные значения межфазного натяжения очень чувствительны к выбору
параметра взаимодействия ?: несмотря на то, что g равно примерно
0,99 для многих полимерных пар, оно никоим образом не является
универсальной величиной для всех использованных полимерных пар.
Рассмотрев случай несжимаемой решетки, авторы [10] оценили
вклады эффектов сжимаемости. Для такой решетки р2 = р2 = р1/2 = 1,
a(pi = 1, Т) = —1, Дя1/2 = А^*> где
ДР* __ р* _|_ р* t /vl/2 _|_ v— l/2Wp*p*\l/2^
Величина ДР* = 0, если I = g0, где
?0 = [(t/vI/2 + (t/v)~1/2]/(v1/2 -f- v~1/2), (9.75)
При значении ?, выраженном уравнением (9.75), для несжимаемой
решетки теплота смешения равна нулю. Для этих значений ? авторы
нашли, что такое приближение, названное ими «приближением чистой
сжимаемости», хорошо описывает только системы с низким
межфазным натяжением (меньше 6 мН/м), т. е. полимерные пары, где
энергетическими вкладами можно пренебречь (табл. 9.2). Для сравнения
в табл. 9.2 приведены также данные, рассчитанные на основе теории
Хелфанда [23], в которой эффекты сжимаемости игнорируются, так же
как и в теории Носе.
Таблица 9.2.
Межфазное
некоторых полимерных пар
Система
ПС-ПММА
ПБМА—ПММА
ПБМА—ПВА
ПС—ПВА
ПБМА—ПДМС
ПИБ —ПДМС
ПЭНП - ПДМС
ПС—ПДМС
ПВА—ПДМС
-
0,9951
1,0027
0.9970
0,9751
1,0150
1,0007
0;9999
0,9947
1.0224
натяжение
и толщина межфазного
7, мН/м
эксперимент
17
Г,9
2,9
3,7
3,8
4,2
5.1
6.1
7,4
расчет при
?=?о [21]
1.0
2.0
1,2
4,0
4,6
4,6
5,4
6,8
3.8
расчет
[23]
0,3
1.5
3.0
1.9
3,2
—-.
7.2
L,
расчет при
[21]
10,0
4,8
8.0
2.3
1.6
1.5
1.3
1.1
2,0
слоя для
им
расчет
[23]
16,0
2,9
1.6
3,1
1.3
—
—
—
0,8
291
9.6. ПРИНЦИП СКЕЙЛИНГА В ОПИСАНИИ МЕЖФАЗНОЙ ГРАНИЦЫ
Как было рассмотрено выше, теории межфазной границы п^едска-
зывают зависимость межфазного натяжения от молекулярной массы
и температуры вблизи критической температуры смешения. Для
оценки критических экспонент таких зависимостей соотношения скейлинга
были использованы в работе [30]. Так, зависимость у от (Гкрит — Т)
вблизи Гкрит можно записать в виде
Т)» (9.76)
(|х — некая критическая экспонента). Согласно принципу скейлинга
К (9.77)
где k — постоянная Больцмана; d — размерность пространства; g —
корреляционная длина, которая была введена де Женном из аналогии
поведения решетки спинов в ферромагнетиках и изолированной
цепи [33]
T)~\ (9.78)
где v — другая критическая экспонента, описывающая изменение
корреляционной длины вблизи критической точки.
Из уравнений (9.76) — (9.78) получаем
Для трехразмерного пространства при v = 0,630 [34] ji = 1,260.
Градиентные теории межфазной границы [3, 8] для систем
полимер — растворитель вблизи Гкрит приводят к следующей зависимости
у от молекулярной массы и температуры:
при которой \i = 1,5. Толщина межфазного слоя определяется из
выражения
LNWT T)-W, (9.79)
т. е. v == 1/2, если L идентифицировать как ?.
Многие теории предсказывают, что ? ~ N1/4; тогда, полагая, что
| изменяется с N:
для межфазного натяжения получаем выражение в виде
При понижении температуры толщина межфазного слоя
уменьшается и при некоторой температуре Т становится сравнимой с размером
полимерного клубка. При этом ожидается явление кроссовера
(возникновение границы перехода на фазовой диаграмме, ем. рис. 4.1).
Идентифицируя L как \ и применяя уравнение (9.79), получаем
^крит-Г-Л^-1/2, (9.80)
292
где предполагается, что R ~ N1^. Уравнение (9.80) определяет
ширину критической области. В точке кроссовера (на границе перехода)
теория Носе [8] предсказывает переход первого порядка от диффузной
границы к резкой межфазной. Полимерная цепь на последней «кол-
лапсирует» в направлении, перпендикулярном межфазной границе.
При дальнейшем понижении температуры мы переходим в область
L <c R, где согласно принципу скейлинга корреляционная длина не
зависит от молекулярной массы (см. табл. 4.1), а является лишь
функй 1 И kTl2
цией концентрации:
лучаем
с
( ) фу
Используя соотношение y~kT/l29
по(9.81)
В этой
этому
о
(I
крит
1крит
Рис. 9.4. Качественная зависимость у
от температуры и молекулярной
массы. Объяснение см. в тексте [30j.
области с — (в — 71), по-
V— kT(T — вJ. (9.82)
Зависимость у от температуры
и молекулярной массы приведена
на рис. 9.4. Область /
соответствует температурам, бЛИЗКИМ К Гкрит-
Толщина межфазной области в
данном случае намного больше
размеров полимерного клубка, а у ~N ~1/4
(согласно теории среднего поля [3,
8]). В области // толщина
межфазной области становится сравнимой
с размерами полимерного клубка,
и появляется сильная зависимость
межфазного натяжения от молекулярной массы: меньшим
молекулярным массам соответствует большее значение межфазного натяжения и
наоборот. Величина L в области /// становится очень малой и поли-
меробогащенная фаза раствора приближается по свойствам к расплаву
полимера. Если молекулярная масса в этой области настолько велика,
что критическая температура фазового расслоения близка к 0-тем-
пературе, межфазное натяжение не должно зависеть от М во всем
температурном интервале.
Экспериментально эти предсказания проверены в работах [30—
32]. Так, был обнаружен переход от области / к области // для
раствора полистирола разных молекулярных масс в метилциклогексане [30,
32]. Показано также, что в области // зависимость межфазного
натяжения от молекулярной массы более сильна, чем в области /, как и
предсказывает теория. В области / экспонента графика зависимости
у от N оказалась равной —0,44, что значительно отличается от
значения, предсказываемого теориями среднего поля (—0,25). Величина же
критической экспоненты р A,17—1,35) близка к 1,26. Обнаружен
излом на зависимостях у ~ / (А 71), предсказываемый теорией Носе
[8], однако наклоны графиков зависимостей In (-~-1 = /(Inс), In (-|г) =
= /[1п(в — Т)] (уравнения (9.81) и (9.82)) оказались много больше 2.
293
Авторы работы [23] это расхождение теоретических и
экспериментальных данных связывают с недостаточно высокими молекулярными
массами (вспомним, что соотношения скейлинга, т. е. степенные
зависимости свойств, справедливы в случае очень больших N). Возможно»
что и температуры, при которых проводили эксперимент, не были
слишком далеки от ГкрИт.
Согласно теории Носе [8], значение у0 в уравнении (9.76) записы*
вается в виде
То = B/3) фУр2ит kTKvm (]&/M)lV(M/Ny/*(l + N-W + N-iyv/v, (9.83)
где (A|I/2— среднеквадратичное расстояние между концами цепи:
М — молекулярная масса полимера; N — число сегментов в молекуле;
фкрит — критическая концентрация, v — объем сегмента. Используя
значения M/N = 146, (hl/MI'2 = 6,6- 10~2 нм, v = 136 см3/моль,
экспериментальные значения Гкрит и фкрИт для полистирола в мегад-
циклогексане, по уравнению (9.83) авторы работы [32] рассчитали
значение 7о- Оно оказалось равным 3,63 мН/м, что близко к
полученному экспериментально C,23 мН/м), т. е. найдено хорошее согласие
теории и эксперимента.
1. Липатов Ю. С. Межфазные явления в полимерах.— Киев : Наук, думка,
1980.— 260 с.
2. Vrij Л. Equation for the interfacial tension between demixed polymer
solutions. — J. Polym. Sci. A-2, 1968, 6, N 11, p. 1919—1932.
3. Vrij Л., Robeson G. /. Inhomogeneous polymer solutions: theory of the
interfacial free energy and the composition profile near the consolute point.— J.
Polym. Sci. : Polym. Phys. Ed., 1977, 15, N 1, p. 109—125.
4. Cahn W.9Hillard J. E. Free energy of a nonuniform system. 1. Interfacial
free energy.— J. Chem. Phys., 1958, 28, N 2, p. 258—267.
5. Helfand ?., Tagami Y. Theory of the interface between immiscible polymers.—
Ibid., 1972, 56, N 7, p. 3592—3601.
6. Helfand E.t Sapse A. M. Theory of concentrated polymer solution —solvent
interface.— J. Polym. Sci. C, 1976, N 54, p. 289—297.
7. Roe /?.-/. Theory of the interface between polymer or polymer solutions.— J.
Chem. Phys., 1975, 62, N 2, p. 490—499.
8. Nose 7\ Theory of liquid—liquid interface of polymer solutions.— Polym.
J., 1976, 8, N 1, p. 96—113.
9. Sanchez /. С Lacombe R. H. Statistical thermodynamics of bulk and surface
properties of polymer mixtures.— J. Macromol. Sci. B, 1980, 17, N 3,
p. 565—589.
10. Poser C. /., Sanchez I. G. Interfacial theory of low and high molecular weight
liquid mixtures.— Macromolecules, 1981, 14, N 2, p. 361—370.
11. Bongiorno V.9 Scriven У. ?., Davis H. T. Molecular theory of fluid interfaces.—
J. Colloid and interface Sci., 1976, 57, N 3, p. 462—475.
12. Sanchez I.С Lacombe R. H. An elementary molecular theory of classical
fluids. Pure fluids.—J. phys. Chem., 1976, 80, N 21, p. 2352—2362.
13. Sanchez L. С Lacombe ft. H. Statistical thermodynamics of polymer
solutions.—Macromolecules, 1978, 11, N 6, p. 1145—1156.
14. Debye P. Angular dissimetry of the critical opalescence in liquid mixtures.—
J. Chem. Phys., 1959, 31, N 3, p. 680—687.
15. Widom B. Surface tension and molecular correlations near the critical point.—
Ibid., 1965, 43, N 11, p. 3892—3897.
294
1 6. Fisk S., Widom B. Structure and free energy of the interface between fluid
phases in equilibrium near the critical point.— Ibid., 1969, 50, N 8, p. 3219—
3227.
17. Rice О. К- Fluctuations, density gradients, and interfaces near the critical
point of one—component fluids.— J. phys. Chem., 1979, 83, N 14, p. 1859—
1863.
18. Koningsveld R.9 Kleintjens L. Л., Shultz A. R. Liquid—liquid phase
separation in multicomponent polymer solutions. 9. Concentration—dependent pair
interaction parameter from critical miscibility data on the system
polystyrene—cyclohexane.—J. Polym. Sci. A-2, 1970, 8, N 8, p. 1261 — 1278.
19. Scholte Th. G. Thermodynamic parameters of polymer—solvent systems from
light—scattering measurements below the theta temperature.— Ibid., 1971,
9, N 9, p. 1553—1578.
20. Vrij А.у van den Esker M. W. J'. Critical opalescence of polymer solutions.— J.
Chem. Soc. Faraday Trans. Part 2, 1972, 68, N 3, p. 513—525.
21. Kammer H. W. Thermodynamik der Grenzschicht zwischen flussigen Polyme-
ren.— Wiss. Z. Techn. Univ. Dresden, 1975, 24, N 1, S. 35—39.
22. Berger W., Olbricht K., Kammer H. W. Grenzflachenerscheinungen in Poly-
merschmelzen. 1. Oberflachen — und Grenzflachenspannungen von Polymer-
schmelzen.—Faserforsch. und Textiltechn., 1976, 27, № 1, S.9—14.
23. Grenzflachenerscheinungen in Polymerschmelzen. 2. Einfluss von Additiven
auf die Oberflachenspannung von Polymerschmelzen / H. W. Kammer, O. Sa-
mek, K. Wietusch, W. Berger.— Ibid., S. 15—17.
24. Kammer H. W. Grenzflachenerscheinungen in Polymerschmelzen. 4. Theorie
der Grenzschicht zwischen Polymeren.— Ibid., 1978, 29, N 7, S. 459—466.
25. Kammer #. W. Grenzflachenerscheinungen in Polymerschmelzen. 3. Eine
quasithermodynamische Theorie der Oberflachenregion von geschmelzen Poly-
meren.— Ibid., 1977, 28, N 1, S.27—36.
26. Helfand E. Tagami Y. Theory of the interface between immiscible polymers.—
J. Polym. Sci.: Polym. Lett., 1971, 9, N 3, p. 741—746.
27. Helfand E. Theory of inhomogeneous polymers. Lattice model for solution
interfaces.— Macromolecules, 1976, 9, N 6, p. 879—888.
28. Weber 7\, Helfand E. Theory of inhomogeneous polymers. Solutions for the
interfaces of the lattice model.— Ibid., N 2, p. 311—316.
29. Helfand E. Theory on inhomogeneous polymers: lattice model for polymer—
polymer interfaces.— J. Chem. Phys., 1975, 63, N 5, p. 2192—2198.
30. Interfacial tension of demixed polymer solutions near the critical
temperature; polystyrene + methylcyclohexane/K. Shinozaki, T. V. Tan, У. Saito, T.
Nose.— Polymer, 1982, 23, N 5, p. 728—734.
31. Shinozaki /C., Abe M., Nose T. Interfacial tension of demixed polymer
solutions over wide range of reduced temperature.— Ibid., p. 722—727.
32. Nose Т., Tan T. V. Interfacial tension of demixed polystyrene—methylcyc-
lohexane solution.— J. Polym. Sci. : Polym. Lett., 1976, 14, N 12, p. 705—
712.
33. De Gennes P. G. Scaling concepts in polymer physics.— New York : Cornell,
univ. press., 1979.—324 p.
34. Le Guillou J. С Zinn—-Justin J. Critical exponents for the л-vector model
in theree dimensions from field theory.— Phys. Rev. Lett., 1977, 39, N 2,
p. 95-98.
ОГЛАВЛЕНИЕ
Предисловие 3
Глава L Основные понятия термодинамики растворов 5
1.1. Парциальные величины 5
1.2. Основные термодинамические характеристики раствора.
Термодинамическое сродство между компонентами 5
1.3. Идеальные и неидеальные растворы 6
1.4. Регулярные растворы. Уравнение Гильдебранда—Скетчарда .... 7
1.5. Фазовое равновесие в растворах. Спинодали и бинодали 8
Глава 2. Классические теории растворов полимеров 13
2.1. Теория Флори—Хаггинса 13
2.1.1. Энтропия смешения 14
2.1.2. Теплота и свободная энергия смешения. Химический
потенциал и осмотическое давление раствора 15
2.2. Разбавленные растворы полимеров 16
2.3. Вычисление исключенного объема 19
2.4. Коэффициент набухания макромолекулы. Теория Зимма и Штокмай-
ера 23
2.5. Фазовое расслоение в полимерных растворах 34
2.6. Недостатки теории Флори—Хаггинса и попытки ее улучшения ... 40
Глава 3. Новые статистические теориии растворов полимеров 46
3.1. Теория Пригожина — Паттерсона 47
3.2. Роль различных вкладов в параметры смешения . . . 53
3.3. Влияние длины цепи растворителя на параметры термодинамического
взаимодействия . : 57,
3.4. Сравнение предсказаний теорий, основанных на принципе
соответственных состояний, и теории параметров растворимости 61
3.5. Новая теория Флори 67
3.6. Модификация новой теории Флори 73
3.7. Характеристические значения параметра обменных взаимодействий для
некоторых химических групп 79
3.8. Новая теория Хаггинса 80
3.8.1. Вывод основных термодинамических функций 81
3.8.2. Ориентационная энтропия 84
3.8.3. Комбинаторная энтропийная поправка на неоднородность
смешения 85
3.9. Теория Саншеза и Лакомба 88
3.10. Некоторые экспериментальные результаты исследования
термодинамических свойств растворов полимеров 94
Глава 4* Принцип скейлинга и концепция блобов в описании свойств
растворов полимеров 99"
4.1. Процедура скейлинга . 99
4.2. Фазовая диаграмма состояния раствора 102
4.3. Экспериментальная проверка соотношений скейлинга и концепции
блобов 105
4.3.1. Коэффициент седиментации в полуразбавленных растворах
полимеров в хороших растворителях 105
4.3.2. Осмотическое давление . . 106
4.3.3. Изменение радиуса инерции с температурой и молекулярной
массой 109
4.3.4. Осмотическая сжимаемость ИЗ
4.3.5. Другие примеры использования принципа скейлинга для описания
\Щ свойств растворов полимеров 115
Глава 5. Статистическая термодинамическая теория растворов жестких и
полужестких частиц 119
5.1. Статистическая теория смесей палочкообразных частиц [1] . . . .119
5.1.1. Вывод функции распределения 119>
5.1.2. Равновесное значение у 122
5.1.3. Химические потенциалы .j 124
5.1.4. Равновесие между изотропной и анизотропной фазами .... 125*
5.2. Система клубкообразная цепочка — палочкообразная частица —
растворитель 12?
5.3. Статистическая термодинамика смесей полужестких макромолекул с
палочкообразными участками, зафиксированными в определенных местах
цепи 132
Глава 6. Термодинамика смешения полимеров 143
6.1. Бинарные смеси 143
6.1.1. Приближения теории регулярных растворов в термодинамике
смешения полимеров 144
6.1.2. Применение новых статистических теорий для описания
термодинамических свойств смесей полимеров 149*
6.1.3. Применение новой теории Хаггинса 158»
6.1.4. Теория Саншеза - 161
6.2. Тройные системы полимер — полимер — растворитель 166
6.3. Экспериментальные методы оценки термодинамической совместимости
смесей полимеров 176
6.3.1. Получение фазовых диаграмм 177
6.3.2. Оценка параметров термодинамического взаимодействия . .179
6.3.3. Использование малоуглового рассеяния рентгеновских лучей
и тепловых нейтронов для оценки совместимости полимеров . . 187
6.4. Некоторые экспериментальные результаты по исследованию
термодинамических свойств смесей полимеров 188
6.4.1. Особенности термодинамического поведения смесей полимеров 188
6.4.2. Особенности концентрационной и температурной зависимостей
параметров термодинамического взаимодействия в смесях
некоторых олигомеров 203
6.4.3. Смеси полимергомологов 206
6.4.4. Влияние толщины пленки и природы подложки на температуру
фазового расслоения в смесях полимеров 214
Глава 7. Термодинамика взаимодействий компонентов и свойства бинарных
полимерных смесей 224
7.1. Термодинамика и вязкость расплавов смесей полимеров 224
7.2. Взаимосвязь термодинамических свойств системы в расплаве и степени
кристалличности сформированных из них полимерных смесей . . . 233'
Глава 8. Механизм фазового разделения в многокомпонентных полимерных
системах 239
8.1. Нуклеация и рост 240
8.2. Спинодальный механизм фазового расслоения в растворах и смесях
полимеров 244
8.2.1. Теория спинодального распада 244
8.2.2. Применение теории спинодального распада к полимерным
системам 250
5.3. Некоторые экспериментальные результаты исследования
спинодального распада в растворах и смесях полимеров 255
8.4. Особенности фазового разделения в блочных системах 259
8.5. Некоторые особенности фазовой структуры систем со стабильными
узлами зацеплений ... 261
$.6. Рассмотрение спинодального распада в рамках формализма нелинейной
термодинамики необратимых процессов . . . .... . 262
•
Глава 9. Межфазная граница в расслаивающихся растворах и смесях
полимеров 267
-9.1. Теория Фрея и Робесона 268
9.2. Теория Каммера 274
*9.3. Теория Хелфанда 276
¦9.4. Теория Носе 281
9.5. Теория Саншеза 282
9.6. Принцип скейлинга в описании межфазной границы 292
АНАТОЛИЙ ЕВТИХИЕВИЧ НЕСТЕРОВ
ЮРИЙ СЕРГЕЕВИЧ ЛИПАТОВ
ТЕРМОДИНАМИКА РАСТВОРОВ
И СМЕСЕЙ ПОЛИМЕРОВ
Утверждено к печати ученым советом
Института химии высокомолекулярных
соединений АН УССР
Редактор Э. П. Конева
Оформление художника В. Г. Самсонова
Художественный редактор В. П. К у з ь
Технический редактор И. А. Р а т н е р
Корректоры Л. М. Тищенко, Л. Н, Регета
Информ. бланк № 6467
Сдано в набор 02.04.84. Подп. з печ. 12.09.84. БФ 01995.
Формат 60x90/16. Ьум. тип. № 1. Лит. гарн. Вые.
печ. Усл. печ. л. 18,75. Усл. кр.-отт. 19,31. Уч.-изд. л.
20,31. Тираж 1350 экз. Заказ 4-420. Цена 3 р. 40 к.
Издательство «Наукова думка», 252601 Киев 4, ул. Репина, 3.
Отпечатано с матриц книжной фабрики им. М. В. Фрунзе
на книжной фабрике «Коммунист», 310012, Харьков-12,
Энгельса, И.