/



















































































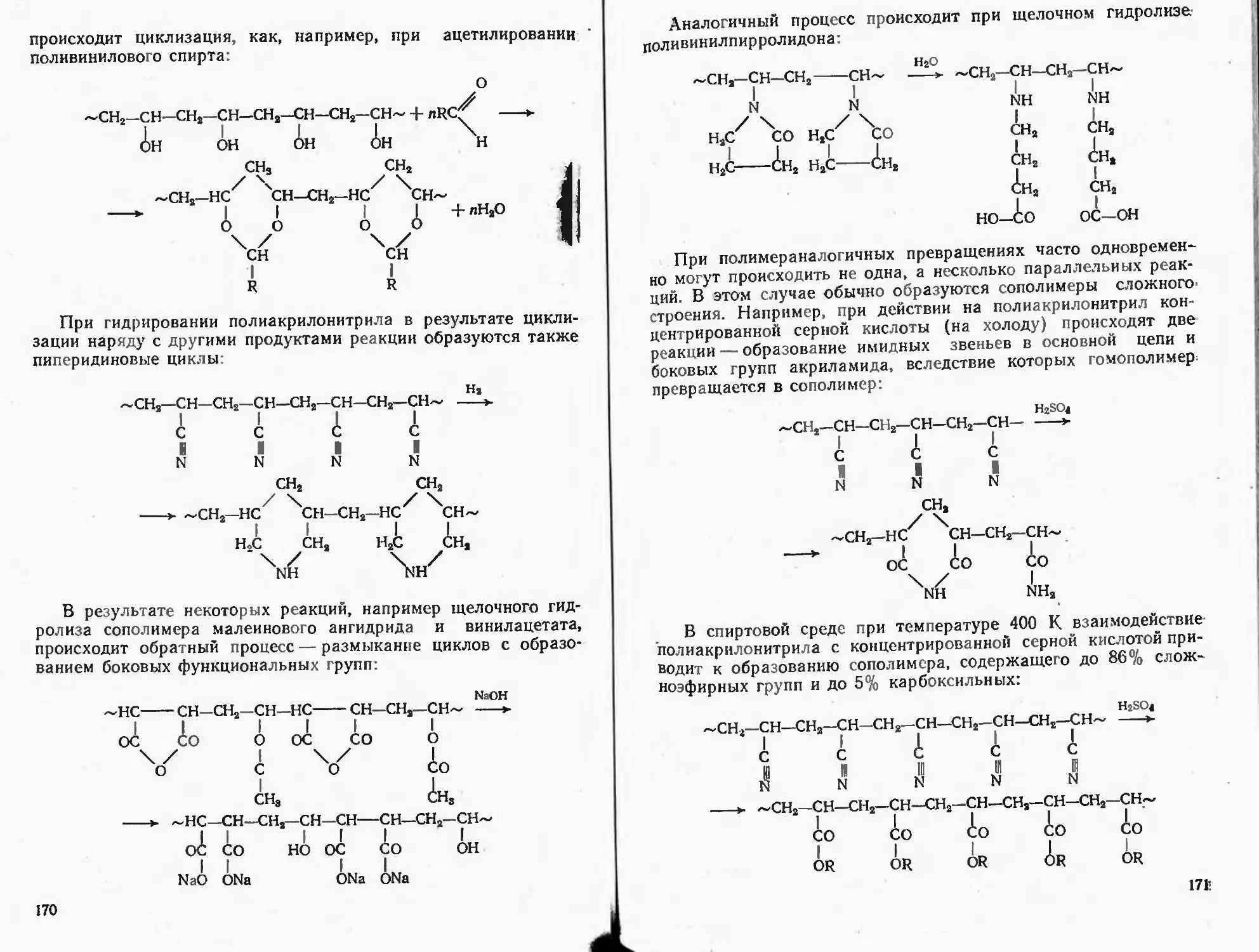
























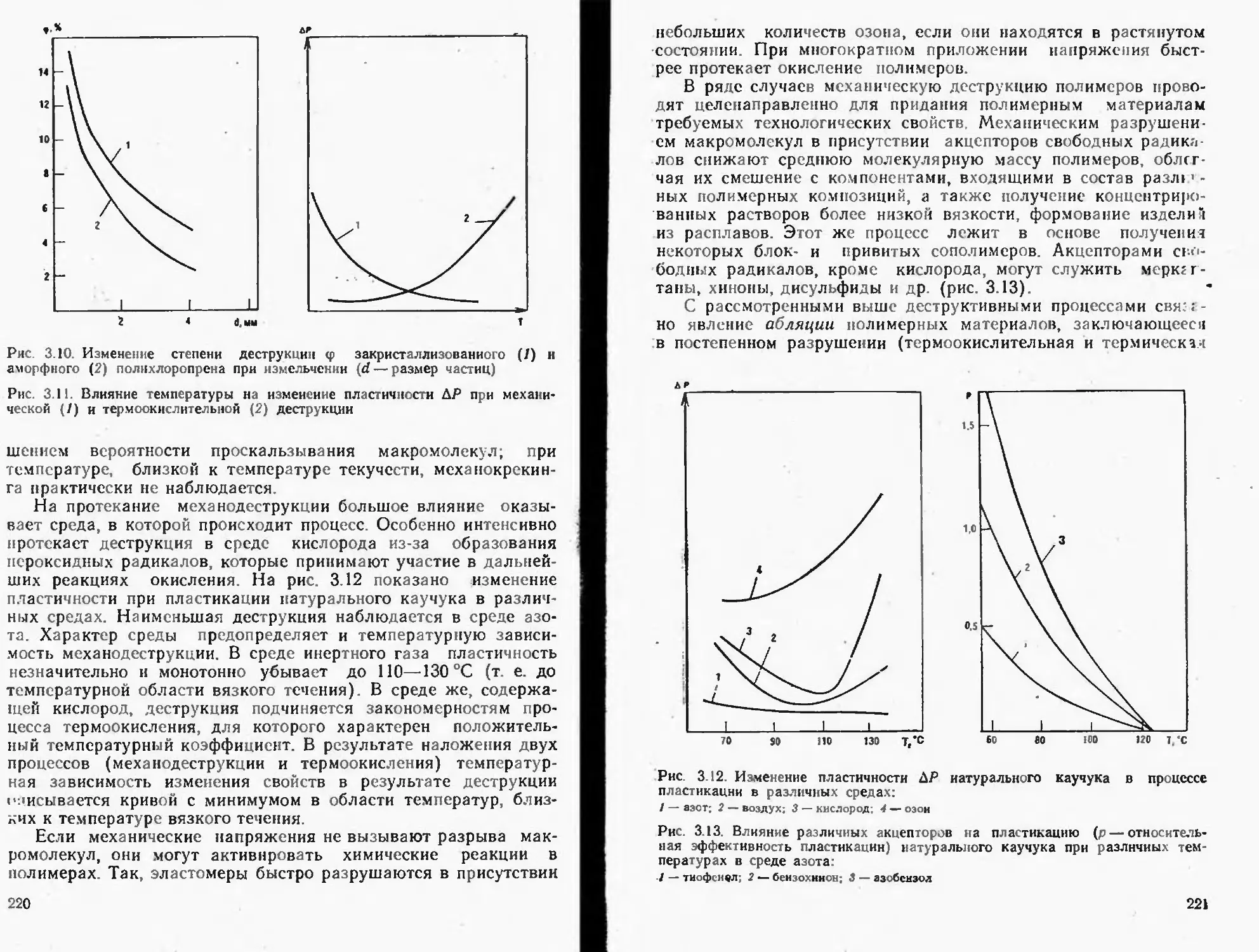






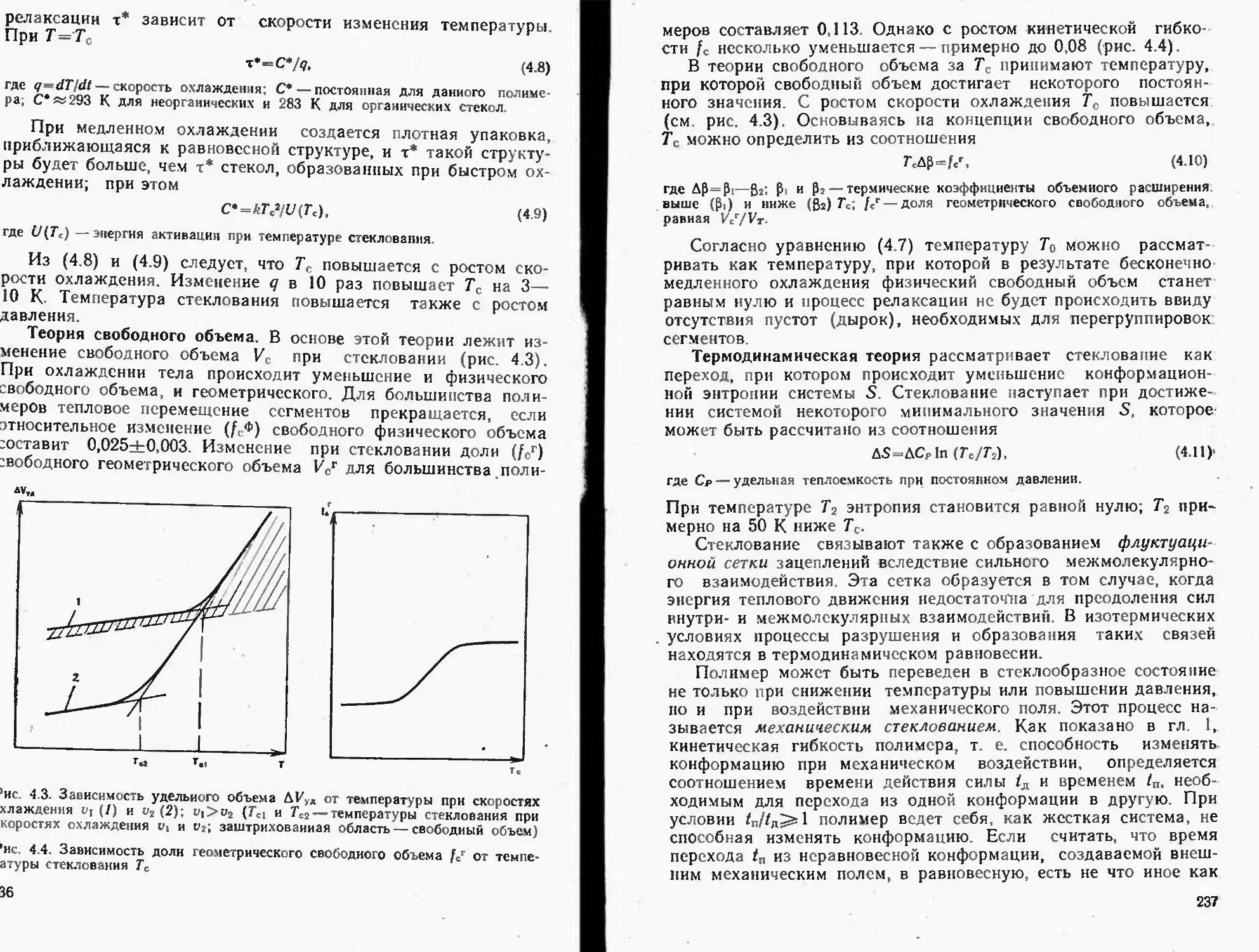





































































































Текст
/
f высшей
школы
И И. Тугое
Г И. Кострыкина
Химия
и физика
полимеров
Допущено Государственным комитетом СССР по на-
родному образованию в качестве учебного пособия для
студентов химико-технологических специальностей вузов
МОСКВА
«ХИМИЯ»
1989
ББК 6П7.55
Т81
УДК 541.6: [54+53] (075.8)
Рецензенты кафедры технологии пластмасс Казанского хнмнко-техно-
логнческого института нм С. М. Кирова и Белорусского ордена Трудового
Красного Знамени технологического института им. С. М. Кирова
Тугов И. И., Кострыкина Г. И.
Т81 Химия и физика полимеров: Учеб, пособие для вузов0 —
М.: Химия, 1989. — 432 с.: ил.
ISBN 5—7245—0243—7
Даны современные представления о строении полимеров, особенно-
стях их свойств (химических, физических н физико химических), мето-
дах исследований! структуры. Рассмотрена связь между строением по-
лимеров и их основными свойствами. Описаны способы получения по-
лимеров. Показана роль физико-химических процессов при переработке,
эксплуатации и разрушении полимеров.
Для студентов химико-технологических специальностей при изуче-
нии курса «Химия и физика полимеров» Может быть использована для
повышения квалификации работников, связанных с получением, перера-
боткой и применением полимеров в народном хозяйстве.
1706000000—075 ,
050(01)—75 75—89
ББК 6П7.55
ISBN 5—7245—0243—7
© Издательство «Химия», 1989 г.
ОГЛАВЛЕНИЕ
Предисловие........................•.............................. 5
Глава 1. Структура полимеров....................................... 6
1.1. Структура макромолекулы....................................... 9
111. Химическое строение..............................* . 9
1.1.2. Молекулярная масса......................................23
1.1.3. Конфигурация макромолекул...............................28
1.1.4. Конформация, размеры н форма макромолекул ... 37
1.2. Надмолекулярная структура ................................. 48
1.2.1. Надмолекулярная структура аморфных полимеров ... 51
1.2.2. Надмолекулярная структура кристаллических полимеров 54
1 2.3 Ориентированное состояние полимеров...................64
1.2.4. Структурная модификация.................................67
1.3. Методы исследования структуры полимеров.......................68
1.3.1. Исследование структуры макромолекулы....................69
1.3.2. Исследование надмолекулярной структуры..................85
1.4. Гибкость полимеров............................................89
1 4 1 Термодинамическая гибкость.......................... 91
1.4.2. Кинетическая гибкость...................................95
Контрольные вопросы..................................• • • 105
Глава 2. Получение полимеров......................................106
21 Полимеризация..................................................107
2.1 1 Радикальная полимеризация...............................111
2.1.2. Сополимеризация........................................120
2.1.3. Ионная полимеризация................................. 121
2.14 Ионно-коордниацнонная полимеризация......................136
2.2. Поликонденсация..............................................144
Контрольные вопросы.....................................••••••«. 154
Глава 3. Химические превращения полимеров.........................155
3.1 Особенности химических реакций полимеров......................155
3.2. Химические превращения, не вызывающие изменения степени по-
лимеризации ..................................................... 163
3.21 Внутримолекулярные превращения........................163
3.2. 2. Полнмераналогнчные превращения........................168
3.3. Реакции, приводящие к изменению молекулярной массы . . . 172
3.3.1. Соединение (сшивание) макромолекул.....................172
3.3.2. Отверждение............................................179
3.4. Реакции, приводящие к уменьшению степени полимеризации н мо-
лекулярной массы..................................................190
3 4.1. Химическая деструкция..................................192
3.4.2. Физическая деструкция..................................198
3.5. Старение и стабилизация полимеров............................222
Контрольные вопросы 225
3
Глава 4. Физические и фазовые состояния и переходы.................226
4.1. Стеклообразное состояние н стеклование....................232
4.1.1. Теории стеклования..................................234
4.1.2. Методы определения температуры стеклования .... 238
4.1.3. Влияние структуры полимера на температуру стеклования 239
4.2. Высокоэластическое состояние..............................241
4.3. Вязкотекучее состояние....................................253
4.3.1. Механизм течения....................................254
4.3.2. Влияние структуры полимера на температуру текучести 257
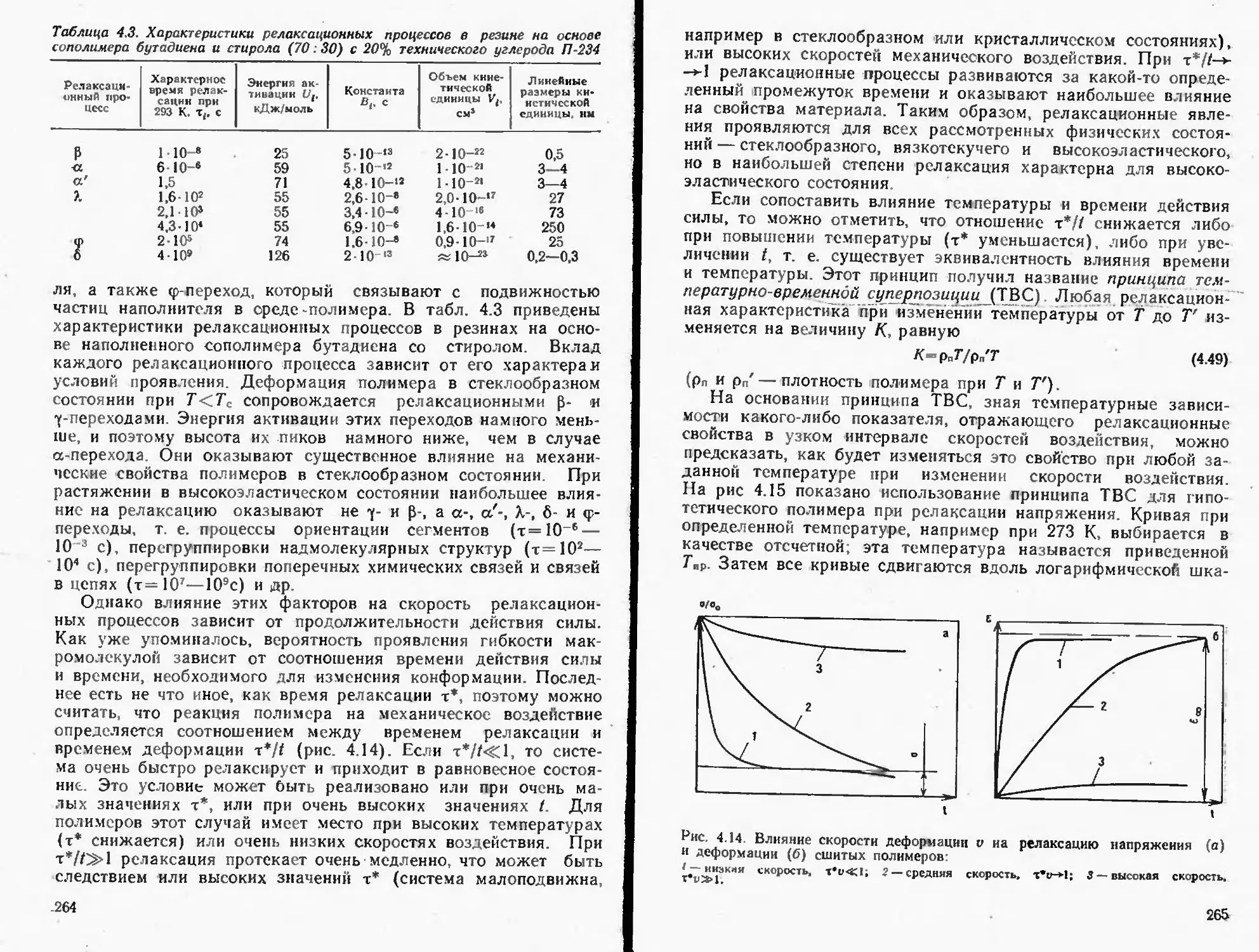
4.4. Релаксационные процессы в полимерах.......................259
4.5 Фазовые переходы...........................................267
4.5.1. Механизм кристаллизации.............................268
4.5.2. Плавление кристаллов................................273
4.5.3. Влияние напряжения на кристаллизацию................274
4.5.4 Влияние структуры полимера на кристаллизацию . . . 276
Контрольные вопросы................................................279
Глава 5. Физические свойства полимеров.............................280
5.1. Механические свойства.........................................280
5.1.1. Деформационные свойства.................................280
5.1.2. Прочностные свойства....................' 316
5.2 Тсплофнзнческие свойства.......................................352
5.2.1. Теплоемкость............................................352
5.2.2. Теплопроводность........................................357
5.2.3. Температуропроводность..................................363
5.2.4. Тепловое расширение ....................................365
5.3. Электрические свойства........................................368
5.3.1. Свойства полимерных диэлектриков........................369
5.3.2. Свойства полимерных полупроводников и электропроводящих
материалов.....................................................383
5.3.3. Свойства полимерных электретов..........................387
Контрольные вопросы......................................... ... 391
Глава 6. Истинные растворы полимеров н коллоидные системы 393
6.1. Истинные растворы............................................394
6.1. 1. Набухание.............................................394
61.2. Термодинамика растворения...............................400
6.1.3 . Термодинамическое сродство полимера и наполнителя . . 402
6.14. Фазовое равновесие в системе полимер — растворитель . 406
6 1.5. Свойства растворов полимеров..........................411
6.2. Коллоидные системы 414
6.2.1 Дисперсии и эмульсин....................................415
6.2.2. Студни.................................................416
6.2.3. Смеси полимеров с пластификаторами.....................418
6.2.4 Смеси полимеров.................................... . 422
6.2.5. Наполненные полимеры...................................425
Контрольные вопросы...............................................428
Список рекомендуемой литературы ................................. 429
ПРЕДИСЛОВИЕ
Можно без преувеличения сказать, что химия, физика, физико*
химия полимеров — одни из наиболее быстро развивающихся
областей науки. Начав существовать как самостоятельный раз-
дел химической науки в 30-х годах нашего столетия, химия и
физика полимеров достигли в настоящее время высокого уров-
ня развития. Крупнейшие отрасли промышленности резиновой,
пластических масс, химических волокон, пленок, лаков и клеев,
электроизоляционных материалов — перерабатывают и приме-
няют полимеры. Развитие практически любой отрасли народно-
го хозяйства сегодня невозможно без применения полимеров.
Широкое использование и высокие темпы роста производст-
ва полимеров обусловлены, в первую очередь, разнообразием
их физических, химических и механических свойств. Для на-
правленного изменения свойств, т. е. для установления связи
состав — структура — свойства, необходимо владеть знаниями о
структуре полимеров и способах се регулирования в процессе
синтеза. Решение этой задачи требует серьезного анализа и
обобщения обширной информации в области химии и физики
полимеров, накопленной за последние годы. Отбирая эту ин-
формацию для учебного пособия, авторы руководствовались
тем, что в какой бы области полимерной науки и технологии
ни работал специалист, он должен владеть знаниями не только
в этой области. Действительно, современный химик-синтетик
должен зиать не только методы синтеза мономеров и полиме-
ров, но и хорошо разбираться в том, как свойства получаемого
им полимера зависят от химической природы исходных ве-
ществ— мономеров. Исследователь, занимающийся физикой и
механикой полимеров, должен иметь четкое представление об
их химическом строении. Наконец технолог, работающий в об-
ласти переработки полимеров, должен знать и химию полиме-
ров, и их физические и эксплуатационные свойства, а также
свойства их растворов.
В предлагаемом учебном пособии изложены современные
представления о структуре полимеров, особенностях их свойств,
способах регулирования структуры. В отличие от других посо-
бий по химии и физике полимеров описаны методы исследова-
ния структуры полимеров, большое внимание уделено их теп-
лофизическим и электрическим свойствам. Рассмотрены спосо-
бы получения полимеров, а также направленной физической и
химической модификации их с целью создания материалов с
требуемыми свойствами. В конце каждой главы даны конт-
рольные вопросы, которые помогут студентам в усвоении прой-
денного материала.
5
ГЛАВА 1
СТРУКТУРА ПОЛИМЕРОВ
Полимеры — это природные и синтетические соединения, моле-
кулы которых, как следует из их названия (поли — много, ме-
ра— часть), состоят из большого числа повторяющихся одина-
ковых или различных по строению атомных группировок, со-
единенных между собой химическими или координационными
связями в длинные линейные или разветвленные цепи.
Группа атомов, с помощью которой можно описать строение
полимера, называется составным звеном. Составное звено, кото-
рое многократно повторяется, называют повторяющимся со-
ставным звеном, а группы на концах цепи — концевыми груп-
пами. Молекула полимера, состоящая из повторяющихся со-
ставных звеньев и концевых групп, называется макромолекулой.
Вещества, из которых образуется полимер, называют моно-
мерами (моно — один). Если при получении полимера мономер
полностью входит в его состав, то составное повторяющееся
звено является мономерным звеном. Если же получение поли-
мера сопровождается выделением низкомолекулярных продук-
тов, например воды, газов, то строение составного (или повто-
ряющегося составного) звена будет отличаться от строения мо-
номера и называть такое звено мономерным нельзя.
Полимеры, полученные из одного мономера, называются го-
мополимерами, а из двух или более — сополимерами.
Число повторяющихся звеньев (я) можно варьировать в ши-
роких пределах — от десятков до десятков тысяч. Как правило,
в одном полимере содержатся макромолекулы различной дли-
ны, т. е. с разным числом составных повторяющихся звеньев.
Переход от низкомолекулярного соединения к полимеру про-
исходит в результате роста числа повторяющихся звеньев При
этом заметно изменяются физические и химические свойства,
но при достижении определенного значения п они перестают
изменяться, несмотря на дальнейшее увеличение числа звеньев.
С этого момента соединение становится полимером. Таким об-
разом, полимер — это соединение, построенное из многократно
повторяющихся одного или более составных звеньев, соединен-
ных между собой химическими или координационными.связями,
число которых достаточно для проявления комплекса свойств,
остающихся практически неизменными при добавлении или
удалении одного или нескольких звеньев.
Промежуточное положение между низкомолекулярными со-
единениями и полимерами занимают вещества, называемые оли-
гомерами (олиго — немного). Они проявляют свойства, харак-
терные как для мономеров, так и для полимеров; при добавле-
6
нии или удалении одного или нескольких повторяющихся звень-
ев наблюдается заметное изменение некоторых их свойств. Чис-
ло повторяющихся звеньев у олигомеров невелико — несколько
единиц или десятков
Названия полимеров образуются из названия мономера с приставкой
поли, а олигомеров — с приставкой олиго. Химическая формула полимера
(олигомера) может быть изображена несколькими способами, например:
полиэтилен
~СН2—СН2~ ---------СН2-СН2----- [-СН2-СН2—
сополимер этилена с пропиленом
СН3
СН3 _т
При одинаковом химическом строении низкомолекулярных
соединений и полимеров последние обладают рядом особенно-
стей:
полимер может существовать только в конденсированном
твердом или жидком состоянии; переход в газообразное со-
стояние невозможен без разрыва молекулы;
растворы полимеров (даже разбавленные) имеют очень вы-
сокую вязкость, значительно превышающую вязкость концент-
рированных растворов низкомолекулярных веществ;
скорость растворения полимеров существенно меньше и рас-
творению, как правило, предшествует набухание; существует
ряд полимеров, которые вообще не растворяются, а только на-
бухают;
при удалении растворителя полимер выделяется не в виде
кристаллов, как низкомолекулярное соединение, а в виде
пленки,
полимеры можно переводить в ориентированное состояние:
например, продавливанием через фильеры можно получить во-
локна;
для некоторых полимеров (эластомеров) характерны боль-
шие обратимые деформации, во много раз превосходящие упру-
гую деформацию низкомолекулярных материалов;
химические реакции полимеров отличаются от аналогичных
реакций низкомолекулярных веществ скоростью и протеканием
большого числа побочных реакций; свойства полимеров резко
изменяются при действии очень небольших количеств реагента.
Специфические свойства полимеров обусловлены особенно-
стями их структуры, знание основных параметров которой не-
обходимо для создания научно обоснованных методов их регу-
лирования.
7
Структурой полимера (как любой сложной системы) назы-
вают устойчивое взаимное расположение в пространстве всех
образующих его элементов, их внутреннее строение и характер
взаимодействия между ними. Каждый структурный элемент в
любом теле подвержен одновременному воздействию многочис-
ленных, непрерывно изменяющихся по величине и направлению
сил (электрических, магнитных, механических и др.), вызыва-
ющих притяжение или отталкивание этих элементов друг от
друга. Находясь в непрерывном (броуновском) движении, каж-
дая структурная единица стремится занять наиболее выгодное,
«удобное» равновесное положение, характеризующееся мини-
мальной энергией и максимальной неупорядоченностью, соот-
ветствующей максимальной энтропии.
В газах структурной единицей являются атомы, поведение
которых подчиняется движениям электронов и управляется за-
конами квантовой механики. В низкомолекулярных жидкостях
и твердых телах структурной единицей становятся молекулы,
движения которых зависят от взаимодействия не только элект-
ронов составляющих их атомов, но и комбинаций атомов и са-
мих молекул друг с другом.
В полимерных телах структурными элементами являются
макромолекулы Движение каждого атома в мономерном звене,
каждого мономерного звена в макромолекуле и каждой макро-
молекулы зависит от совокупности сил, действующих на элект-
ронном, атомном, молекулярном уровнях в каждое данное мгно-
вение.
Так же, как атомы и молекулы, находящиеся в непрерывном
движении, макромолекулы стремятся занять наиболее энерге-
тически выгодное, равновесное положение друг относительно
друга, образуя так называемую надмолекулярную структуру.
При рассмотрении структуры полимеров необходимо учиты-
вать:
строение концевых групп, отличающихся от строения основ-
ного повторяющегося звена; для полимеров высокой молекуляр-
ной массы это несущественно, но для олигомеров достаточно
заметно;
неоднородность по химическому составу, т. е. разнозвенность
полимеров вследствие протекания побочных реакций при их по-
лучении;
неоднородность по числу повторяющихся составных звеньев,
обусловленную статистическим характером протекания реакций
получения полимера;
различное пространственное расположение звеньев в макро-
молекуле;
надмолекулярную структуру.
Рассмотрим структуру полимера на молекулярном (т. е.
структуру макромолекулы) и надмолекулярном уровнях.
8
1.1. СТРУКТУРА МАКРОМОЛЕКУЛЫ
Структура макромолекулы — это сложное понятие, включаю-
щее химическое строение, длину и распределение по длинам и
молекулярным массам, пространственное расположение звень-
ев, форму макромолекулы.
1.1.1. Химическое строение
Характеристикой химического строения макромолекулы являет-
ся химическое строение ее повторяющегося составного звена.
По химическому строению повторяющегося звена полимеры
делятся на органические, неорганические и элементоорганиче-
ские.
Органические полимеры содержат в главной цепи атомы уг-
лерода, а также кислорода, азота и серы. В боковые группы
могут входить водород, галогены, соединенные непосредствен-
но с углеродом, или атомы других элементов, непосредственно
не соединенных с углеродом основной цепи.
Неорганические полимеры состоят из неорганических атомов
и не содержат органических боковых радикалов.
Элементоорганические полимеры — это соединения, макро-
молекулы которых наряду с атомами углерода содержат неор-
ганические фрагменты. По составу главных цепей их делят на
три группы:
соединения с неорганическими цепями, обрамленные боко-
выми органическими группами;
соединения, в главной цепи которых находятся атомы угле-
рода, а боковые группы содержат любые другие атомы, за ис-
ключением азота, серы, кислорода и галогенов, соединенных не-
посредственно с атомом углерода;
соединения с органонеорганическими цепями.
Соединения каждого класса можно разделить на гомоцепные
и гетероцепные. У гомоцепных соединений цепи построены из
атомов одного элемента, у гетероцепных — на разных.
При делении на гомоцепные и гетероцепные полимеры со-
став боковых групп не учитывают. Например, в поливинилаце-
тате группы —СО—О— находятся в боковых ответвлениях
*ч'СН1—сн~
I
о—СО—сн3
а главная цепь макромолекулы полимера образована только из
атомов углерода, и поэтому это соединение относится к гомо-
цепным производным предельных углеводородов, имеющим бо-
ковые сложноэфирные группы. В полиэтилентерефталате труп-
I
лы —СО—О— входят в состав главной цепи, которая состоит из
атомов углерода и кислорода
~ о—со—СО—СН2—сня~
поэтому его относят к кислородсодержащим гетероцепным со-
единениям.
Органические гомоцепные полимеры — это обычно карбоцепные соедине-
ния, главные цепи которых построены из атомов углерода. Они делятся на
алифатические (предельные и непредельные) и ароматические углеводороды,
галогенпроизводиые, спирты, кислоты, эфиры и т. д_:
Алифатические предельные
Полиэтилен
Полипропилен
Поливинилхлорид
Поливинилфторид
Поливиниловый спирт
Поливииилацетат
Полиакролеин
Поливниилметилкетои
Поливнниламии
Полииитроэтилеи
Полиакриловая кислота
Полнметнлакрилат
Полиакриламид
Полиакрилонитрил
—СН2—СН—
I
СН,
—СН2—СНС1 —
—CH»—CHF—
—СН2—СН—
I
ОН
—СН2-СН—
I
о—со—СН,
—сн2-сн—
о=сн
—СНг-СН—
I
О=с—СН,
—СН2—СН—
I
nh2
—сн2-сн—
Аоа
—сна—СН
I
о=с—он
—СНг—СН—
О=С—О-CHs
—СН2—СН—
I
О=С—nh2
—СНг—СН—
I
C=N
10
Алифатические непредельные
Полибутадиен —сн2-сн=сн—сна— СНз
Полиизопрен 1 —СН2—С » СН—СНг— С1
Полихлоропрен 1 —СНг—С=СН—СНг—
Ароматические
Полифенилен
Жирноароматнческие
Полиэтиленфеиилен
-СН
Неорганические гомоцепные полимеры получены только из элементов
III—VI групп Периодической системы, наибольшее практическое значение
имеют полимеры, состоящие из элементов IV и VI групп. С увеличением но-
мера ряда внутри каждой группы возрастает степень делокализации и обоб-
щения электронов, резко снижается энергия о связей между атомами одного
и того же элемента, т. е. способность элементов к образованию прочных
связей
Отсутствие органических обрамляющих боковых групп также оказывает
существенное влияние на свойства неорганических макромолекул. В органи-
ческих полимерах электронные орбитали атомов боковых групп защищают
главную цепь от атаки нуклеофильных или электрофильных агентов, опре-
деляют характер межмолекулярных взаимодействий В неорганических вы-
сокомолекулярных соединениях этот эффект ие проявляется.
Ниже приведены названия и формулы некоторых гомоцепных неоргани-
ческих полимеров:
Карбии
Кумулен
Полисилаи
Полигерман
Полисера
~ S—S—S ~
Число гомоцепных неорганических полимеров довольно ограничено, боль-
шее распространение получили гомоцепные элементоорганические полимеры,
состоящие из неорганических цепей, обрамленных органическими радикалами,
11
или из органических цепей (карбоцепные) с боковыми элементоорганнчески-
мн группами
Полнорганоснланы
Поливинилалкнлснлаиы
Борсодержащие полимеры
R
~ Si^
I
R
~CHz— СН~
I
SiR3
R=CH3
в
I
R
R — алкил или арнл
Органические гетероцепные полимеры делят на классы в зависимости от
природы функциональной группы, повторяющейся в главной цепи; соответ-
ственно различают кислород-, азот-, серусодержащие соединения и др.
Они могут быть алифатическими или ароматическими в зависимости от того»
алифатические или ароматические группировки находятся между функциональ-
ными группами. В табл. 1.1 приведены представители органических гетеро-
цепных полимеров.
Неорганические гетероцепные полимеры построены из атомов элементов
групп III (В, Al), IV (Si, Ge, Те, Pb, Sn), V (Р, As, Sb) и Vl (S, Se, Те)»
а также кислорода и азота; возможны также сочетания элементов III группы
с Р, а V — с В. Ниже приведены некоторые представители этих полимеров:
f
Полиборазол
R — алкил
или арил
Полнкремнневая кислота
Полифосфоннтрил-
хлорнд
I I ?
ООО
- Si—О—Si—О—Si—ОН
i । 1
ОН О О
* г
ci
~P=N~
Cl
Большую группу гетероцепных полимеров образуют элементоорганические
соединения из которых наибольшее практическое значение имеют полимеры,
состоящие из неорганических цепей с органическими боковыми группами
К ним относятся кремнийсодержащие полимеры, главные цепи которых
состоят из чередующихся атомов кремния и кислорода» азота, серы, метал-
лов и т. д.
12
к наиболее распространенным кремнийорганическнм полимерам относятся
следующие:
Полиорганоснлоксаны
Полиорганоснлазаиы
Полналюмооргаиосилоксаиы
Полиборорганоснлоксаны
Полнтнтанорганосилоксаиы
Полимеры, содержащие в основной цепи третий гетероатом—металл»
называются полиметаллоорганосилоксанамн.
К гетероцепным элементоорганическим полимерам с неорганическими
цепями относятся и полиоргаиофосфазены:
R
~P==N~
I
R
Полимеры с органоиеорганнческнми цепями могут быть построены из
атомов углерода, кремния и т. д. К ним относятся.
Поликарбораны
Поликарбосиланы
Поликарбоснлоксаиы
~нс-сн~
Biollio к
13
В полнкарбоснланах н полнкарбоснлоксанах между атомами кремния
могут располагаться алифатические или ароматические звенья:
~Si—(СНа)„—Si
Соединения с органическими цепями и боковыми элементоорганическим»
радикалами имеют главные цепи, построенные из углерода и серы нлн угле-
рода и азота, углерода и кис юрода, а боковые группы — из кремния, фос-
фора, бора, олова, свинца Примером может служить лолиметилен-2 карбора-
миленметилакрилат:
^0—СН2—С^С—СН2—О-СО—R-CO—0^
ВЮНЮ
R- алкил
Рассмотренные выше полимеры и олигомеры состоят в ос-
новном из повторяющихся составных звеньев, в которых атомы
соединены химическими ковалентными связями. Существуют
также полимеры, звенья которых представляют собой внутри-
молекулярные циклы, образованные ионом металла (комплексо-
образователь) и внутрисфернрм заместителем (лиганд) Связь
между ними осуществляется в результате донорно-акцепторного
взаимодействия с образованием координационной связи (побоч-
ная валентность) и ионной связи (главная валентность).
Акцептором электронов являются металлы практи’-ески всех
групп Периодической системы, кроме пятой. Донорами служат
атомы, способные отдавать электроны для образования связи
(О N, S, F, С1). Эти полимеры получили название координа-
ционных внутрикомплексных, хелатных, клешневидных. Они
относятся к гетероцепным полимерам, в зависимости от строе-
ния цепи могут быть органическими и неорганическими.
К органическим относят полимеры, основная цепь которы; содержит
виутрнкомплексиые циклы, состоящие из органических лигандов
Х=О, N, S; R—алкил, арил
Примером может служить полимер нафтазарнна с медью:
16
Неорганические полимеры не имеют в основной цепи атомов углерода,
а обрамляющие группы могут быть неорганическими (I) или органически-
ми (II)
II
Таким образом, на примере рассмотренных типичных пред-
ставителей различных классов полимеров показано, что, несмот-
ря на различную природу атомов в повторяющихся звеньях, их
объединяет общее: связи между атомами и между звеньями яв-
ляются химическими или координационными Они имеют дли-
ну 0,1—0.2 нм и высокую энергию*.
Ниже приведены значения энергии о связей (кДж/моль) в
некоторых гомо- и гетерососдинениях:
Si—Si . 189 С—C 336
Si—С 241 С—В 420
Si—S 256 в—в 257
Si-0 373 c—s 258
Химические связи в макромолекуле по энергии значительно
превышают любые другие связи между структурными едини-
цами:
Тнп связи Энергия свя- Тнп связи Энергия свя-
зи, кДж/моаь зи, кДж/моль
Химическая 590—1050 Водородная До 50
ионная Дисперсионная До 40
ковалентная До 710 Ориентационная До 20
металлическая 110—350 Индукционная До 2
От природы атомов в звеньях зависит не только энергия
связей, но и их полярность. Эти показатели являются очень
важными, поскольку определяют ряд эксплуатационных свойств
полимеров, например стойкость к действию высоких темпера-
тур, агрессивных сред, электрические свойства и др.
* Энергия связи — это энергия, выделяющаяся при образовании данной
связи, или энергия, необходимая для диссоциации данной связи.
2—816
17
В зависимости от полярности связей полимеры делят на не-
полярные и полярные. Количественно степень полярности оце-
нивается дипольным моментом Цо (Кл-м), равным произведе-
нию заряда q на расстояние г между зарядами. Ниже приведе-
ны значения дипольных моментов ряда производных метана
СНз—X, содержащих полярные группы (X):
X Цо-10” Кл-м X Не-10”. Кл-м
СНз 0,00 С1 6,104
сн=сн2 1,167 сосн, 9,640
осн. 4,302 CN 13,142
он 5,637 NHCOCH3 13,743
СООСНз 5,737 SO2CHs 14,810
Дипольный момент макромолекулы равен векторной сумме
дипольных моментов полярных групп, распределенных вдоль
цепи.
К неполярным полимерам относят, например, органические
карбоцепные алифатические полимеры: полиэтилен, полипро-
пилен, полиизопрен, полибутадиен, полиизобутилен. Их диполь-
ный момент равен или близок нулю. Полярные полимеры со-
держат в составе повторяющегося звена группы с полярными
связями (С—ОН, С—СООН, С—NH2, С—CN, С—С1), и их ди-
польный момент отличен от нуля. К полярным полимерам от-
носятся, например, поливиниловый спирт, целлюлоза, крахмал,
содержащие большое количество групп ОН, полинитрилоакрил,
поливинилхлорид и др.
Полярность полимера зависит и от симметричности распо-
ложения полярных групп в повторяющемся составном звене.
При симметричном их расположении электрические поля ком-
пенсируются и цо таких полимеров равен нулю. Сравним три
полимера:
Политетрафторэтилен
Полнвиннлиденхлорнд
Поливинилхлорид
~ CF2—CFz'"'
СН2—СС12~
~CH2—CHC1~
Первый является неполярным, несмотря на наличие сильно
полярных групп С—F, поскольку эти группы расположены сим-
метрично. Полярность второго и третьего полимеров выше, так
как электронная плотность смещена к группе С—С1, причем
ввиду меньшей компенсации электрических полей поливинил-
хлорид является более полярным полимером.
Ниже приведены значения дипольных моментов ц некото-
рых полимеров при температуре 293—298 К (растворитель для
58
всех полимеров, кроме поливинилхлорида толуол, для поливи-
нилхлорида диоксан):
Полнизопрен
Поливиннлнзобути-
ловый эфир
Полиэтнленоксид
Полиметнлметакри-
лат
ц-10”, Кл-м
0,934
3,569
3,636
4,503
Полнхлоропреи
Поливинилхлорид
Поливииилацетат
ц10“. Кл м
4,837
5,3704—5,6039
6,1042
Поскольку каждая структурная единица цепи содержит
электроны и положительно заряженные ядра, она обладает
локальным электрическим полем, которое оказывает влияние
на соседние структурные элементы. В результате этого между
химически несвязанными атомами, принадлежащими одной мак-
ромолекуле или разным, возникает взаимодействие, проявляю-
щееся в притяжении и отталкивании. Назовем это взаимодей-
ствие физическим. На большом расстоянии между несвязанны-
ми атомами действуют силы притяжения, но при достаточном
сближении (исключающем возможность химического взаимо-
действия) проявляются силы отталкивания В результате атомы
располагаются на некотором расстоянии, характеризующемся
минимальной потенциальной энергией. Для многих органиче-
ских соединений эти расстояния составляют 0,3—0,5 нм. Таким
образом, физические связи внутри макромолекул или между ни-
ми, так же как и в низкомолекулярных веществах, имеют элект-
рическую природу Их образование не сопровождается смещени-
ем или переходом электронов и происходит на расстояниях, пре-
вышающих длину химических связей, т. е. для этих связей ха-
рактерно дальнодействие.
В зависимости от строения молекул между ними могут про-
являться дисперсионные, ориентационные и индукционное взаи-
модействие. Дисперсионные связи образуются между молекула-
ми любой структуры и обусловлены возникновением мгновен-
ных диполей в атомах и молекулах при вращении электронов
вокруг ядер.
Для полярных молекул наряду с дисперсионным взаимодей-
ствием характерно диполь-дипольное, или ориентационное,
взаимодействие При взаимодействии полярных молекул (ди-
полей) с неполярными последние могут поляризоваться в поле
диполей. Между постоянным и наведенным диполями возника-
ют силы, называемые индукционными. Энергия дисперсионных
(1/д), ориентационных ((7ОР) и индукционных (С/ии) связей мо-
жет быть оценена из соотношений:
Уд—ЗС/и1С/иаа1ав/(2г6(4/И|+4/иа)); (1.1)
Уор=-3И2,цг2/(З^Гг«); (1.2)
(1-3)
2*
19
где Uni, Uxz — энергии ионизации молекул; ah а2 — поляризуемость, г — рас-
стояние между молекулами; щ» — дипольные моменты; А— постоянная
Больцмана; Т — температура.
Как следует из приведенных уравнений, энергия связей воз-
растает с повышением поляризуемости и дипольного момента,
практически не зависит от температуры (за исключением ори-
ентационного взаимодействия) и резко уменьшается с ростом
расстояния между атомами.
Промежуточное положение между физическими и химиче-
скими связями занимает водородная связь. Она образуется меж-
ду электроотрицательными атомами (обычно F, О, N, реже С1,
S) и атомами водорода (связь обозначается тремя точками)
он- о
нс^ сн
о но^
Длина ее 0,24—0,32 нм, энергия в зависимости от природы
атомов изменяется от 17 до 50 кДж/моль*
С—Н ••• О 17—26
о—н -О 25—50
С—Н ••• N 26—33
F-H • -F 27
При образовании единичных физических связей уровень фи-
зического взаимодействия можно оценить по соотношениям
(1.1) — (1.3). В полимерах вследствие цепного строения макро-
молекул вероятность физических контактов повышается. По-
тенциальная энергия i-й макромолекулы (Л (г) при взаимодей-
ствии с N молекулами, расположенными на расстоянии от
нее, суммируется:
U(r)= £ (k4)
Поэтому суммарная энергия межмолекулярного физическо-
го взаимодействия даже для неполярных молекул может до-
стигать существенных значений, а для полярных — стать соиз-
меримой с энергией химических связей или даже превосхо-
дить ее.
Мерой интенсивности межмолекуляриого физического взаимодействия
является плотность энергии когезии (ПЭК) Она эквивалентна работе уда-
ления взаимодействующих молекул или атомов на бесконечно большое рас-
стояние, что равносильно испарению или возгонке. ПЭК равна потенциаль-
ной энергии единицы объема вещества:
ПЭК=— £/F=— АдЕо/V, (1 5)
где Е— мольная потенциальная энергия вещества; V—мольный объем;
JVa — число Авогадро; EQ— потенциальная энергия одной молекулы.
-20
Иногда для оценки энергии когезии используют показателе называемый
параметром растворимости бр [(Дж/м3)1/2]:
6р = У£ДД (1.6)
Для низкомолекулярных веществ его обычно определяют по величине
теплоты испарения вещества или поверхностного натяжения. Для полимеров
эти методы неприменимы, поскольку полимеры нелетучи и имеют высокую
вязкость. Поэтому ПЭК для полимеров определяют косвенными методами,
в частности по свойствам системы полимер—растворитель. Обычно за бр
полимера принимают 6 жидкости, которая является наилучшнм растворите-
лем этого полимера. (Лучшим считается тот растворитель, в котором степень
набухания максимальна при условии отсутствия теплового эффекта смешения
и изменения объема системы.)
Если в системе проявляются не только дисперсионные силы, но и ориен-
тационные, индукционные, водородные, то вводят понятие о трехмерном
параметре растворимости бт:
*>Т = Уб2д + 6%р + 6%, (1.7)
где бд. бор, б, — параметры растворимости, учитывающие дисперсионное (бд)
ориентационное и индукционное (бор) взаимодействия н образование водород-
ных связей (бн).
В табл 1.2 приведены значения бР для некоторых растворителей Опре-
деляя бп полимера по бр растворителя, необходимо учитывать тип связей,
которые образует полимер с растворителем.
Параметр растворимости полимера определяют и расчетным путем, исхо-
дя из условия аддитивности сил взаимодействия отдельных атомных групп
и радикалов и предполагая, что силы взаимодействия в повторяющемся зве-
не полимера аналогичны силам, действующим в низкомолекулярном соедине-
нии. Значение бг может быть вычислено по формуле Смолла:
6П= (2GJ/V- (рпЩ.)/Л1313> (1.8)
где SGi — сумма констант притяжения отдельных групп; рп — плотность по-
лимера; AfSB—молекулярная масса повторяющегося составного звена (значе-
ния Gt для некоторых групп приведены в табл. 1.3).
Таблица 1.2. Параметры растворимости 6Р [МДж/м3)х/2] некоторых раствори-
телей
Растворитель «д ®ор «г
Ацетон 16,15 10,86 7,24 20,75
Бензол 19,5 1,07 2,13 19,43
Гексан 15,42 0 0 15,42
Дноксан 1981 1,92 7,67 20,72
Изопропилацетат 15,66 3,83 7,88 17,47
Ксилол 18,42 1,07 3,20 18,74
Пропанол 16,51 7,03 18,Н 25,35
Толуол 18,79 1,49 2,13 18 98
Хлорбензол 19 77 4,47 2,13 20.26
Хлороформ 18,42 3,20 5,96 19,68
Циклогексан 17,42 0 0 17,42
Циклогексанон 18.42 8,73 5,33 19,70
Тетрахлорид углерода 18,42 0 0 18,42
Этилацетат 15,85 5,54 9,59 19,26
Этилбензол 18,53 0,64 1,49 18,74
21
Таблица 1.3. Константы притяжения некоторых групп (Gi) при 25°С
Группа (мкДж м*) °»ь Группа Ср (мкДж-м3)°>*
39
57
227
231
1503
1345
388
438
—Вг
—I
—О— (простые
эфиры)
—СО— (кетоны)
—СО—О— (слож-
ные эфиры)
—S— (сульфиды)
CN
SH
307
560
510—550
695
869
143
562
634
460
838
644
40—60
454
583
Этой формулой можно пользоваться для оценки Сп с большой точностью
в основном для неполярных и малополярных полимеров. Особенно большая
погрешность получается в случае образования водородных связей. В табл. 1.4
приведены значения бп некоторых полимеров
Для разнозвенных полимеров прн расчете бп необходимо учитывать со-
держание н структуру аномальных звеньев, а для олигомеров — число н тип
концевых групп.
Итак, в полимерах наряду с химическими связями внутри
повторяющихся составных звеньев и между ними существует
сетка из физических связей. Эти связи подвижны, так как воз-
никают при сближении атомов и разрушаются при их удале-
нии. Поскольку любая система находится в тепловом движении,
Таблица 1.4. Параметры растворимости Сп некоторых полимеров
Полимер вп. (МДж/М») 1/2 Полимер вп. (МДж/м*) 1/*
Политетрафторэти-
лен
Полндиметнлсилок-
•сан
Полиэтилен
Полнизобутнлен
Полннзопрен
Полибутадиен
Полихлоропрен
Полистирол
12,7
14,9-15,5
15,7—17,0
15,9—16,6
16,2—17,0
16,6—17,2
16,8—18,8
17,4—19,0
Полипропилен
Полиметнлметакри-
лат
Полнвниилацетат
Поливинилхлорид
Полиэтилентерефта-
лат
Полиакрнлоннтрил
Полигексаметилен-
адипамид
Целлюлоза
18,8—19,2
18,6—19 4
19,2—19,6
19,2—20,6
21,9
25,6—31,5
27,8
31,9
22
то физическая сетка непрерывно флуктуирует по объему, по-
этому ее называют также и флуктуационной. Таким образом,
химическое строение повторяющегося звена полимера определя-
ет энергию химических связей в звене и между звеньями, тип
и уровень физического взаимодействия (сетки) внутри и между
макромолекулами
1.1.2. Молекулярная масса
Для низкомолекулярных веществ молекулярная масса М явля-
ется константой, характеризующей данное соединение. Для по-
лимеров М определяется как произведение М повторяющегося
составного звена Мзв на число таких звеньев п М—Ммп. Как
уже было сказано выше, полимер состоит из макромолекул, со-
держащих различное число звеньев, а следовательно, имеющих
различные длину и молекулярную массу. Поэтому можно гово-
рить о полидисперсности полимеров. Полидисперсность являет-
ся следствием случайного (статистического) характера реакций
образования полимера, а в некоторых случаях и следствием
разрушения или соединения макромолекул. Поэтому, когда го-
ворят о Л1 полимера, всегда имеется в виду ее усредненное зна-
чение М. Но при одинаковом значении М полимеры могут раз-
личаться полидисперсностью. Этот факт свидетельствует о не-
обходимости рассмотрения в качестве характеристики полиме-
ра кривых распределения по длинам или по молекулярной мас-
се, называемых кривыми молекулярно-числового (МЧР) и мо-
лекулярно-массового распределения (ММР).
Существуют два типа графического изображения молекуляр-
но-массового распределения: интегральная и дифференциаль-
1/NodN*/dM
1 /mp-dmff/’ dM
Рис. 1.1. Кривая интегрального молекулярно-массового распределения
Рнс. 12 Кривые дифференциального молекулярно-числового и молекулярно-
массового распределения
23
(1ЛЗ)
Q —
М каждой фракции, ее мольную и массовую доли. Методы оп-
ределения ММР и МЧР описаны в разд. 13
Кроме рассмотренных усреднений по мольной или массовой
доле молекул, используют другие способы усреднения, опреде-
ляемые методикой измерения молекулярных масс. По зависи-
мости гидродинамических свойств полимеров от молекулярной
массы, например по изменению вязкости, коэффициента диф-
фузии и других свойств, определяют среднегидродинамические
молекулярные массы. К ним относятся средневязкостная
среднедиффузная и др. В общем виде
( Л1«р„ (Л1) dM
— —
( Л1,-1рп (Л1) dM
о
При q—\ получается величина Мп, при q = 2— Mw, а при <7=3
и 4 получают Мг и Ж+ь Могут быть рассчитаны и более высо-
кие степени усреднения; обычно
Средняя молекулярная масса полимеров в зависимости от
требований, предъявляемых к получаемым из них изделиям, из-
меняется в широких пределах —от нескольких десятков тысяч
до миллионов. Варьирование молекулярной массы и ширины
ММР осуществляется изменением условий при получении поли-
мера (см гл. 2) или путем специальной обработки за счет ме-
ханохимического воздействия (см. гл. 3) Ниже приведены зна-
чения среднемассовой молекулярной массы и коэффициента по-
лидисперсности некоторых полимеров:
Полиэтилен
низкого давления
высокого давления
Сополимер этилена с пропиленом
(^50 50)
цис 1 4 Полнбутадиен
цис-' 4 Полннзопрен
Сополимер бутадиена со стиролом
полученный радикальной полиме-
ризацией
полученный ионной полимериза-
цией
A1w10-‘
50-800 1,5-25
30—400 5—25
70—150 2-5
100—350 1,2—2,6
500—1000 3"~*5
400—800
200- 250 2,1-2,5
Олигомеры характеризуются
Олигомеры характеризуются теми же показателями, что и
полимеры: интегральной и дифференциальной функциями чис-
лового и массового распределения, средними значениями число-
вой и массовой молекулярной массы, шириной распределения,
т. е. степенью полидисперсности. Эти показатели, так же как и
для полимеров, зависят от способа получения олигомера. Так,
26
если стереорегулярного олигобутадиена, полученного на ти-
тановом катализаторе, составляет (35ч-38) • 103, то при исполь-
зовании никелевого катализатора она снижается до (1,5ч-3,0)-
-103; MwlMn в первом случае составляет 5—13, а во втором
2—2,5.
Олигомеры с функциональными группами теоретически
должны содержать такие группы на концах молекул, т. е. долж-
ны быть бифункциональными. Но в реальных олигомерах су-
ществуют молекулы с большим числом групп, а также молеку-
лы без функциональных групп. Поскольку концентрация групп
и их распределение определяют ряд важных свойств, то для оли-
гомеров с функциональными группами наряду с молекулярно-
массовыми характеристиками вводят дополнительные.
1. Распределение по типам функциональных групп (РТФ),
которое может быть представлено в виде интегральной или
дифференциальной числовой или массовой функции.
2. Среднечисловая и среднемассовая функциональность:
N N
2 Nifi S W,
fn = -^----; А,—---------- <Ь14>
S Nt 2 MA
1 f—I
(где Ni — число молекул с функциональностью ft) :
3. Степень неоднородности по функциональности, количест-
венной характеристикой которой служит отношение Jw!Jn-
4. Молекулярная масса, приходящаяся на одну функцио-
нальную группу и называемая эквивалентной (Л1Э):
Л1э=Л1ф/Сф • 100
(где Мф—молекулярная масса функциональных групп; Сф— их
концентрация)
Таким образом, полная характеристика олигомеров с функ-
циональными группами должна включать характеристики моле-
кулярной массы и функциональности. Ниже представлены та-
кие характеристики олигобутадиена с различными функциональ-
ными группами:
соон £1 ООН
Ж-Ю-» 2.0—4.0 3,0 2,0
-Мл/ЛТ» 1,5-1,6 1,5 1,6
h 2,0 2 0 1,75
Tw РТФ, % 2,5 2.5 2,0
монофункциональные 10 10 38,2
бифункциональные 75-80 75—80 49,6
трнфункциональные н более 10—15 10-15 12,2
Таким образом, полимеры характеризуются высокой молеку-
лярной массой (т. е. относятся к высокомолекулярным соеди-
нениям) и значительной неоднородностью по длинам и молеку-
лярной массе макромолекул. Неоднородность оказывает зна-
чительное влияние на основные свойства полимеров: низкомо-
лекулярные фракции ухудшают механические, но улучшают
технологические свойства полимеров, а высокомолекулярные
оказывают обратное влияние — обеспечивают высокий уровень
прочности, твердости, но затрудняют переработку.
1.1.3. Конфигурация макромолекул
В общем случае для всех химических веществ понятие конфи-
гурации включает в себя определенное пространственное рас-
положение атомов, составляющих молекулу и не изменяющееся
при тепловом движении. Переход из одной конфигурации в
другую невозможен без разрыва химических связей. Для поли-
мера, состоящего из повторяющихся звеньев, выделяют не-
сколько конфигурационных уровней (подсистем): конфигура-
цию звена, конфигурацию присоединения звеньев '{ближний
конфигурационный порядок), конфигурацию присоединения
больших блоков (дальний конфигурационный порядок), конфи-
гурацию цепи. Ближний порядок — это порядок, который рас-
пространяется только на соседние элементы (звенья), даль-
ний— порядок, который сохраняется на расстояниях, значи-
тельно превышающих размеры элемента.
Конфигурация звена. Для характеристики конфигу-
рации звена используют понятия и определения, принятые в ор-
ганической химии. Так, для полимеров, полученных из углево-
* дородов с сопряженными двойными связями, характерно суще-
ствование звеньев в двух изомерных формах (конфигурациях)
цис-(\) и транс-(П):
HR CHt R
/С==С\ / / /С=С\ /
сн2 сн/7 Н СН2
I II
Стереоизомерным центром таких звеньев является двойная
связь; в цис-форме заместители располагаются по одну сторо-
ну от плоскости двойной связи, в транс-форме— по разные
стороны.
Другим распространенным типом конфигурационной изоме-
рии является I, d-изомерия звеньев строения
Л
R—С*—R',
I
В
28
где R, R' — заместители различного строения, обусловливающие
асимметричность атома углерода С* Этот атом углерода явля-
ется стереоизомерным центром. Например, для полимеров со
звеньями —СН2—CHR—, где R может быть любым радикалом,
возможно существование звена в виде двух изомеров I — лево-
вращающий (I) nd — правовращающий (II):
Конфигурация присоединения звеньев (ближ-
ний порядок) Ближний конфигурационный порядок можно рас-
сматривать в двух аспектах: во-первых, с точки зрения присо-
единения звеньев вдоль цепи полимера (структурная изомерия),
а во-вторых, с позиций пространственного порядка повторяю-
щегося составного звена (пространственная изомерия).
Структурная изомерия. При образовании цепной структуры
звенья могут присоединяться друг к другу несколькими способа-
ми: к концу одного звена (к «хвосту») присоединяется начало
(«голова») другого — присоединение «голова к хвосту»; конец
одного звена присоединяется к концу другого — присоединение
«хвост к хвосту»; начало одного звена соединяется с началом
другого — присоединение «голова к голове». Структурно-регу-
лярным считается полимер, в котором звенья присоединяются
преимущественно по типу «голова к хвосту». Присоединения
«голова к голове» или «хвост к хвосту» являются структурно-
нерегулярными участками цепи Рассмотрим структурную изо-
мерию на некоторых примерах
Если в мономере типа СН2=СНХ группу СН2 считать «хво-
стом» (х), а группу СНХ — «головой» (г), то в полимере будут
содержаться структуры трех перечисленных выше типов:
X Г X Г Г X X Г X г
~СНг—СНХ—СНг—СНХ—СНХ—СНг—СНг—СНХ—СНг—СНХ—
В полимере, полученном из диеновых мономеров, набор
структур будет более многообразным. Мономер типа
CH2=CR—СН=СН2, где R—Н, СН3, С1 и др., может вступать
в реакции или по обеим двойным связям, или по одной. Если в
реакции участвуют обе двойные связи, то полимер может содер-
жать структуры «голова к хвосту», «голова к голове» и «хвост
к хвосту»:
~CH2-CR=CH-CH2-CH2—CR=CH—СН2-СНг-СН=
X X
=CR—СНг—СНг—CR=CH—СНг~
29
Если в реакции присоединения участвует одна двойная связь,
то возможно образование структур типа 1,2-(I) или 3,4-(II).
Эти звенья могут соединяться, друг с другом также по типу «го-
лова к хвосту», «голова к голове» и «хвост к хвосту»:
Г X ГХХ Г Г X
~СН,—CHR—СН,—СН R—CHR—СН,—СН,—CHR—
III I
сн сн сн сн
II II II II
сн, сна сн, сн2
I
—СН,—СН—СН,—СН—сн—сн,—сн,—сн~
III I
CR CR CR CR
II II II
СН, сн2 сн, сн2
II
Для сополимеров сохраняются типы структурной изомерии,
но набор структурных изомеров увеличивается. Так, сочетание
повторяющихся звеньев различного строения может быть слу-
чайным статистическим или упорядоченным в виде диад (два
звена), триад (три звена), тетрад (четыре звена) и т. д. Звенья
внутри диады, триады и т д. могут соединяться так же, как в
приведенных выше полимерах.
Рассмотрим в качестве примера сополимер бутадиена и сти-
рола Для этого сополимера возможно или последовательное
чередование звена бутадиена А и звена стирола В ~А—В—
А—В—А—В'-', или сочетание звеньев в виде диад, тэиад ит. д.,
причем последние могут распределяться регулярно (I) или ста-
тистически (II)
~ВВВ—АА-ВВВ—АА-ВВВ- I
~В—АА—ВВВВ—ААА—ВВ~ II
Пространственная изомерия. Полимер можно считать про-
странственно регулярным (стереорегулярным), еелг последую-
щее звено присоединяется к предыдущему в той же изомерной
форме. Для полимеров, состоящих из повторяющиеся звеньев
типа —СН,—CR=CH—СН2~, характерны стереэизомерные
структуры, состоящие из звеньев, соединенных в положении
цпс-1,4 или троне-1,4:
4 (32
СН, СН, CR=CH
/ \3 2/ \</ \ /
CR=CH СН, СН,
Ч«с-1,4
4 2 4 2
сн2 сн сн2 сн
/ \зУ \1/ \з/^ \ /
CR СН, CR СН,
транс-1,4
30
Для таких полимеров стереоизомерной плоскостью является
плоскость двойных связей.
Как уже отмечалось ранее, для виниловых полимеров, со-
держащих асимметричный атом углерода, характерно сущест-
вование звеньев в виде правых (правовращающих) или левых
(левовращающих) стереоизомеров. При соединении однотипных
стереоизомеров (Z или d) образуются изотактические структу-
ры ближнего порядка, а при последовательном их чередова-
нии— синдиотактические. Стереоизомерной плоскостью для та-
ких полимеров является плоскость основной цепи, состоящей
из асимметричных атомов углерода (рис. 1.4).
а — изотактическая; б — синдиотактическая: в — атактическая
31
Для звеньев типа СНХ—CHY возможны конфигурации ди-
изотактические и дисиндиотактические и, следовательно, ближ-
ний конфигурационный порядок присоединения звеньев с диизо-
и синдиотактической конфигурацией.
Конфигурация присоединения больших бло-
ков (дальний порядок). Для гомополимеров можно го-
ворить о том, что дальний конфигурационный порядок отража-
ет регулярность цепи. Цепь считают структурно- или стереоре-
гулярной, если ближний конфигурационный порядок распрост-
раняется на всю макромолекулу.
Мерой стереорегулярностн полимера является содержание
структур определенной конфигурации. Полимер считается сте-
реорегулярным, если содержание звеньев иной конфигурации не
превышает нескольких процентов и не оказывает влияния на ос-
новные свойства полимера. При соизмеримом содержании цис-
и транс-изомеров полимер будет нестереорегулярным.
Для полимеров с повторяющимся звеном СНг—СНХ харак-
терно разделение на изотактические и синдиотактические поли-
меры, т. е. полимеры, сохраняющие изо- или синдиотактическое
присоединение звеньев на протяжении всей молекулы. Аналогич-
но для полимеров, полученных из мономеров типа СНХ—CHY,
возможно существование диизотактических и дисиндиотактиче-
ских конфигураций дальнего порядка.
Полимеры, в которых изо (диизо)- или синдио(дисиндио) так-
тические группировки соединяются хаотически, являются нере-
гулярными и называются атактическими (см. рис. 1 4), хотя на
каком-то участке для них возможен ближний конфигурацион-
ный порядок.
Как уже указывалось, для сополимеров характерно сущест-
вование ближнего конфигурационного порядка в виде упорядо-
ченного чередования повторяющихся звеньев или их диад, три-
ад, тетрад и других последовательностей. При нерегулярном
чередовании последовательностей получаются статистические
сополимеры (рис. 1.5). Если последовательности имеют доста-
точную длину, т. е. дальний конфигурационный порядок, и ста-
новятся соизмеримыми между собой, то образуются или приви-
тые, или блок-сополимеры.
Привитые сополимеры имеют основную цепь, к которой при-
соединены (привиты) боковые цепи. Основная и боковая цепи
могут быть гомополимерами или сополимерами, могут соеди-
няться непосредственно или через низкомолекулярное соедине-
ние Привитые полимеры не только проявляют свойства со-
ставляющих их макромолекул, но и приобретают новые, не ха-
рактерные для исходных компонентов.
Блок-сополимеры построены из последовательностей звень-
ев, например А и В, имеющих дальний конфигурационный по-
рядок. Блок-сополимеры могут быть нескольких типов А—В,
32
/k _ф_ _ иимоиомкумриое соединена
Рис. 1.5. Схематическое изображение сополимеров
с — статистический; б, е — привитые; г, д, е, ас — блок-сополи-
меры
А—В—А, [—А—В]п Блоки полимеров могут соединяться меж-
ду собой линейно или в виде лучей, а также через низкомоле-
кулярное соединение X (см. рис. 1.5). Каждый из блоков может
быть или гомополимером или сополимером. Как правило, блок-
сополимеры сочетают в себе свойства составляющих их блоков.
Конфигурация цепи определяется соединением по- ,
следовательностей из звеньев или блоков, имеющих дальний
конфигурационный порядок. Такие последовательности могут
соединяться с образованием линейных полимеров Однако число
линейных полимеров невелико. К ним относятся некоторые при-
родные полимеры, например цис-полиизопрен (натуральный
каучук) и целлюлоза Для большинства же полимеров харак-
терно нарушение линейности в результате образования разветв-
ленных структур. Разветвленным полимером называют полимер,
имеющий наряду с основной цепью длиной I боковые цепи дли-
ной /б, связанные с ней химическими связями и состоящие, как
правило, из звеньев того же состава. Разветвленные полимеры
могут иметь короткие ((б<^0 и длинные (/б—►/) ветвления; по-
следние разделяют на регулярные (звездообразные, гребнеоб-
разные) и статистические («древовидные») (см. рис 1.6).
Характеристикой конфигурации цепи может служить раз-
ветвленность, которую оценивают следующими показателями:
функциональностью ветвлений fB — числом ветвей, выходя-
щих из каждого узла разветвления;
плотностью разветвления рв, равной числу разветвленных
звеньев лв, отнесенных к их общему числу л: рв=лв/л;
средним числом ветвей в макромолекуле /в;
фактором разветвленности g.
3—816
33
Рнс. 1.6. Схематичное изображение разветвленных полимеров:
а — с короткими боковыми цепями; б — с длинными боковыми цепями; в — регулярные
звездообразные трех- и четырех лучевые; г — гребнеобразные; д — статистические
Эти величины связаны между собой соотношениями:
1)л.4-1 = (/.-- 1)р^4-1;
ё~2~ (1+пв/7)|/2+4л,/9л при /“31 дЛя монодисперсиого
g-2=(l+nB/6)’/2+4n./3« при f=4j полимера
В ряде случаев плотность разветвления полимеров достига-
ет некоторого критического значения рв, Кр, при котором в си-
стеме возникают сшитые структуры. Сшитыми или сетчатыми
называют полимеры, цепи которых соединены между собой хи-
мическими связями в единую сетку. Сетчатые структуры могут
быть плоскими или пространственными, в сетку могут соеди-
няться две макромолекулы или несколько (вплоть до сшивания
всех макромолекул), сшивающие связи могут быть распределе-
ны регулярно или статистически.
При соединении двух макромолекул могут образоваться
.лестничные полимеры — полимеры со сдвоенной цепью или с
регулярной линейной сеткой (рис 1.7). К ним, как правило, от-
носятся полимеры, макромолекулы которых состоят из конден-
сировапных циклов, соединенных между собой через два или
>более общих атомов, например:
полибутадиен
циклический
N
полиацен
ншгвэяонихнгои
Если циклы соединены через один общий атом, то такие ле-
стничные полимеры называются спирополимерами:
полифенилен
В реальных лестничных полимерах регулярность сетки на-
рушается, и часто конденсированные полициклические фрагмен-
ты чередуются с линейным и разветвленными участками. Такие
полимеры называются полулестничными или блок-лестничными
полимерами.
Если макромолекулы соединяются между собой химическими
связями с образованием двухмерной сетки, то такие полимеры
называют плоскими сетчатыми. Примером таких полимеров мо-
жет служить графит.
Е- узлы полимера А
® - узлы полимера Б
Рис. 1.7. Схематическое изображение сетчатых полимеров:
а — лестничные* б — полулестннчныс; е — плоско-сетчатые; г — пространственно-сетчатые
из одного полимера (/, 2, 3— макромолекулы, темные точки — узлы); д, е— прострел*
сгвеинО’Сетчатыс из полимера А (сплошные линии) и полимера Б (пунктир)
3*
35
Трехмерные сетчатые полимеры,, цепи которых соединены
между собой в трех направлениях, могут быть построены регу-
лярно или характеризоваться статистическим расположением
макромолекул и сшивок. Примером регулярно построенного про-
странственного сетчатого полимера является полимер, состоя-
щий из углеродных атомов, — алмаз.
Среди полимеров наиболее распространены статистические
пространственные сетчатые, которые называют также гелями.
Такие сетки обычно получают или при синтезе полимера, или
специальной обработкой линейных и разветвленных полимеров,
при которой протекают реакции структурирования. В статисти-
ческих сетках распределение узлов сшивания, т. е. точек связы-
вания, приближается к статистическому. В качестве примера та-
ких сеток можно привести сетку из цепей натурального каучу-
ка, сшитую пероксидами. Для характеристики таких сеток наи-
более часто используют следующие параметры:
функциональность узлов /у, определяемую как число цепей,
сходящихся в узле;
молекулярная масса отрезка цепи, заключенного между уз-
лами, Л!с;
число цепей между узлами в единице объема Лгс;
число молей цепей лс, заключенных между узлами;
показатель сшивания — число поперечных связей на одну
макромолекулу;
число узлов в единице объема vc;
степень сшивания — доля сшитых звеньев на одну макро-
молекулу.
Поскольку сшивки распределены в основном статистически,
то все перечисленные показатели являются средними величина-
ми и связаны между собой следующими соотношениями:
Пе-р/Л(с=Я«/ЛГл; vc = GVc7>)/2= (Л'аР/) /2Л1С;
_ _ _ G16)
^св^я/Л4с; Л^зв/Л^с:
где р — плотность полимера; — число Авогадро; Л7П и Л1эв — средняя мо-
лекулярная масса полимера н молекулярная масса мономерного звена п—
степень полимеризации.
Согласно статистической теории бесконечная сетка (гель)
монодисперсных макромолекул образуется в так называемой
точке гелеобразования при условии fc=l и рс«1/п. В случае
сополимеров из повторяющихся звеньев с различной реакцион-
ной способностью в реакциях сшивания рс=(1/л)$> где s —
доля звеньев, способных к сшиванию.
В идеальных сетках все бесконечные цепи должны быть свя-
заны между собой химическими связями и не иметь свободных
концов и несвязанных макромолекул. Однако реальные сетки
отличаются от идеальных наличием растворимой части полиме-
36
i
ра, называемой золь-фракцией и включающей несшитые моле-
кулы и низкомолекулярные фракции несвязанных концов, за-
цеплений, неравномерным распределением поперечных связей.
Эти отклонения, являющиеся дефектами сетки, учитываются по-
казателем, называемым активной долей сетки (Уа):
Ул=1-2(МС/Л1„) (1.17)
Тогда действительное число узлов (эффективное число узлов.
Vs4>) равно
v^-Vc[l—2(ЛГС/Л7„)]. (1.18)
Наибольшая дефектность сетчатой структуры наблюдается в
точке гелеобразования. При этом образуется несовершенная сет-
чатая структура с большим числом свободных концов. Структу-
ра таких сеток, называемых микрогелсм, зависит, в частности,
от типа и способа получения полимера. Так, при получении
статистического сополимера бутадиена и стирола методом
эмульсионной полимеризации образуется рыхлый микрогель,
т. е с невысокой плотностью сшивания, а микрогель полиизо-
прена, полученного полимеризацией в растворе, характеризует-
ся наличием плотного ядра, из которого выходят длинные кон-
цы цепей. По мере увеличения плотности сшивания дефект-
ность сетки снижается и приближается к единице при Л?с—>
—+ЧъМп.
По плотности сшивания сетки можно разделить на редкие и
густые. К редким относятся сетки, имеющие vc до 1029 м~э,
а к густым — сетки с большей плотностью. По структуре разли-
чают сетки, полученные из одного полимера, — однофазные, из
смеси двух полимеров — двухфазные и взаимопроникающие, не
связанные между собой химическими звеньями (см рис. 1.7).
1.1.4. Конформация, размеры и форма макромолекул
Размеры макромолекулы определяются ее длиной I и диамет-
ром d. Если макромолекулу представить в форме вытянутой це-
пи определенной конфигурации, то рассчитать I и d не состав-
ляет труда. Например, для полиизобутилена (ПИБ) диаметр и
длина мономерного звена составляют 0,5 и 0,154 нм соответст-
венно Если число таких звеньев 104, то макромолекула ПИБ
будет иметь длину 0,154-104 нм, а отношение длины к диамет-
ру составит 3100. Однако рассматривать макромолекулу в ви-
де вытянутой цепи в форме плоского зигзага нельзя, поскольку
при этом не учитываются роль взаимодействия (притяжения к
отталкивания) атомов и их групп, в частности боковых, и влия-
ние теплового движения, которое существует при любой темпе-
ратуре, отличной от абсолютного нуля.
37
ад) ад
О СО 120 180 240 300 300 О СО 120 180 240 30) 380 ♦ *
<4 > U < « > Т ц t « Z >
Рис. 1.8. Зависимость потенциальной энергии 1/(ф) молекулы этана (а) и
молекулы дихлорэтана (б) от угла поворота <р метильной группы (ц — цис-
конформацня, т—транс-конформация, г — гош-конформация)
С учетом этих факторов в каждый определенный момент вре-
мени макромолекулы принимают определенные конформации.
Каждая конформация характеризуется только ей одной прису-
щим определенным расположением в пространстве атомов и
групп. Переход из одной конформации в другую осуществляет-
ся за счет вращения, поворота или колебания вокруг одинарных
связей под действием теплового движения или внешних сил и
не сопровождается разрывом валентных химических связей.
Конформационные перестрой ки происходят и в нкзкомолеку-
лярных органических соединениях. В качестве примера рассмот-
рим молекулу этана СН3—СН3. Колебание группы атомов во-
круг связи С—С с частотой a; 1(J10 с-1 сопровождается изменени-
ем потенциальной энергии молекулы, которое описывается сину-
соидальной кривой. Конформации, соответствующие минимуму
энергии, являются устойчивыми, их называют конформерами
или конформационными изомерами. Остальные конформации
представляют собой такие энергетические состояния, которые
молекула должна пройти при переходе из одной устойчивой
конформации в другую. Молекула этана может существовать в
двух положениях: цис и транс:
цис-
транс-
Эти положения энергетически неравноценны: в цис-форме
будут сильнее проявляться силы отталкивания, поэтому транс-
38
форма более энергетически выгодна и ей соответствует минимум
энергии (рис. 1.8). Переход из одной формы в другую, напри-
мер из цис- в транс-, происходит за счет поворота группы СНз
на угол 60°. Потенциальная энергия при этом изменяется от Uu
до Ur. При последующем переходе транс- в цис-форму группа
СН3 должна повернуться еще на 60°, при этом необходимо пре-
одолеть потенциальный барьер вращения Uo за счет накоплен-
ной кинетической энергии (kT). Вращение групп в таких моле-
кулах является практически свободным, т е. незаторможенным
(kT>U0) По мере усложнения структуры вращение групп во-
круг связей становится все более заторможенным, поскольку
изменяется величина потенциального барьера вращения. Так,
в молекуле дихлорэтана СН2С1—СН2С1 свободу вращения и тем
самым существование определенных конформаций ограничива-
ет атом хлора. Наиболее энергетически выгодна транс-форма,
а цис-форма из-за проявления сил отталкивания является не-
устойчивой. Для нивелирования сил отталкивания С1 поворачи-
вается на угол 120°, образуя так называемую «скошенную»
гош-форму (левую 4-120°, правую —120°):
ClffCl
цас-
тронс-
гош левая
хош-правая
Эти гош-формы менее стабильны, чем транс-(иг> UT), но более
устойчивы по сравнению с цис-формой (Ur<Uu). Переход из
одной формы в другую возможен только при условии, что кине-
тическая энергия молекулы kT больше потенциального барьера
вращения £70. Если это не соблюдается, то группы только ко-
леблются относительно положений с минимумом энергии. Для
большинства органических соединений величина потенциально-
го барьера Uo в газовой фазе составляет 4—19 кДж/моль:
СНз—СНз 11,7 СНз—ОН 4,5
СНз—СН2СНз 14,3 СНз—SH 4,45
СНз—СН(СН3)2 16,4 СНз—NHa 8,0
СНз-(СН3)з 18,5 СН3—SH3 7,1
СНз—СН=СН2 8,2 CF3—CFS 18,3
CH3-CF3 15,5 SiHs—S1H3 4,2
ch3-ch2f 13,9 СНз-СН2С1 15,5
Uq зависит от типа заместителя и его полярности: с увеличе-
нием объема заместителей или их полярности (например, при
замене СН3 на С1 или F) значение UQ повышается. Атомы кис-
39
лорода, серы, азота, а также двойные связи снижают эту вели-
чину.
Рассмотрим, каким образом происходит изменение конфор-
маций макромолекулы. Можно условно представить полимер в
виде молекулы этана СНз—СН3, у которой атомы водорода за-
менены на радикалы R и R', являющиеся звеньями макромо-
лекулы. Например, макромолекулу полипропилена можно пред-
ставить как
Н Н
RC—CR', где R=—С~ , R'=—С~
Xi \н3
Звенья могут вращаться вокруг связей только в том случае,
если валентные углы не фиксированы и влияние заместителей
основной цепи отсутствует Звенья такой цепи могут принимать
любые конформации. В действительности такие цепи не суще-
ствуют, их условно называют свободно-сочлененными. В реаль-
ных макромолекулах валентные углы строго фиксированы,
и уже поэтому число конформаций ограничено. Кроме того, ве-
роятность существования тех или иных конформеров, так же
как и для простых химических соединений, будет определяться
соотношением сил притяжения и отталкивания, т. е. внутри- и
межмолекулярным взаимодействием. Это взаимодействие для
полимеров, как мы уже говорили, может быть ближнего (напри-
мер, между соседними атомами и группами) и дальнего поряд-
ка (между группами, расположенными на значительном рас-
стоянии). Существенно большее влияние оказывает взаимодей-
ствие ближнего порядка.
Переход из одной конформации в другую, так же как и для
низкомолекулярных соединений, определяется соотношением по-
тенциального барьера вращения и кинетической энергии моле-
кулы. Существование взаимодействий ближнего и дальнего по-
рядка накладывает настолько существенные ограничения на
вращение звеньев вокруг одинарных связей, что оно становится
заторможенным и вероятны лишь повороты на некоторый угол
<р, величина которого определяется химическим строением и
конфигурацией макромолекулы.
Конформация звена Повторяющиеся звенья боль-
шинства полимеров, как правило, представляют собой смесь по-
воротных изомеров. Так, для виниловых полимеров СНг—СНХ
характерно существование трех поворотных изомеров: транс и
двух гот (левого и правого).
Ближний конформационный порядок проявля-
ется в образовании различных конформаций при присоединении
звеньев друг к другу. Для виниловых и винилиденовых полиме-
40
ров возможно существование соседних звеньев в транс- и гош-
конформациях (а — повторяющееся звено):
г
г
трине
гош-левая
тош-правая
Для диеновых полимеров (полихлоропрена, полнбутадиена)
также были обнаружены такие же транс- и го/^-конформации
относительно групп СН2—СН2.
Ближний конформационный порядок предусматривает и кон-
формационный набор в малых последовательностях, например
диадах, триадах и т. д. Расстояние между группами, одинаково
расположенными в пространстве, называется периодом идентич-
ности. Изменение набора конформеров влечет за собой и изме-
нение периода идентичности. Ниже приведены данные о перио-
де идентичности (тип конформации и величина периода) для
виниловых полимеров (Т— транс, Гл и ГПР — гош левая и пра-
вая; цифры — число повторяющихся конформеров):
Тип конформации
ТГяТГа,
Период, нм
0,25
0,50
0,44
0,36
Тип конформации
(ТГnp)s
(ГГл)з
Т$Г 3Т3Г пр
(ТГЙ)г(ТГпр}2
Период, им
0 62
0,62
0,88
0,85
Дальний конформацио'ннный порядок пред-
усматривает определенное регулярное или нерегулярное сочета-
ние малых последовательностей конформеров на достаточно
большом протяжении цепи. На формирование дальнего конфор-
мационного порядка оказывают существенное влияние распо-
ложение и объем заместителей в цепи. Рассмотрим в качестве
примера виниловые полимеры, простейшим представителем ко-
торых является полиэтилен Минимум свободной для этого по-
лимера энергии достигается в той из трех конформаций (транс-,
еош-левая, гош-правая), которой должно соответствовать мак-
симальное расстояние между достаточно объемными группа-
ми СН2. Это условие выполняется, когда макромолекула на до-
статочно большом протяжении образует зигзагообразную транс-
конформацию (рис. 1.9). В полипропилене атом водорода заме-
щен на группу СНз, имеющую больший объем. Обычно заме-
стители располагаются таким образом, чтобы их электронные
облака не перекрывались и не вызывали тем самым появления
сил отталкивания. Для удовлетворения этому условию макро-
41
Рис.1.9. Схематическое изображение плоской транс-конформации полиэтиле-
новой цепи (а) и различные спиральные конформации в изотактических
полиолефинах (б) с различными заместителями R (светлые кружки):
/ — в тригональных спиралях R ——СИ», — CHj—СН» —С«Н»; II — в гептагональных спи-
ралях R——CHj—CH(Cli»)j, —CHj—CH(CHs)—CHj— СН»; III — в тетрагональных спира-
лях R —СН(СП»)2
молекула изотактического полипропилена вместо плоского зиз-
зага образует спиральную конформацию, в которой на три мо-
номерных звена приходится один виток, а группы СН3 поверну-
ты относительно друг друга на 120°. Для синдиотактического
полипропилена силы отталкивания, создаваемые группами СН3,
практически не проявляются, и поэтому для него сохраняется
конформация плоского зигзага (см. рис. 1.9).
Степень свернутости, или закрученности, спирали оценива-
ется классом спирали Ху, где X— число звеньев, у — число вит-
ков Эти величины определяются природой заместителя и его
расположением Так, если для полипропилена на один виток
приходится три повторяющихся звена, то для политетрафтор-
этилена на каждые восемь повторяющихся звеньев приходится
пять витков.
Конформация макромолекулы (цепи)—это
размеры и конкретные формы, которые макромолекула прини-
мает в результате суммарного влияния теплового движения и
внешних сил. (Следует отметить, что тепловое движение при-
водит к усреднению конформаций, и когда мы говорим о той
или иной конформации, имеется в виду усредненная конформа-
ция.) В зависимости от соотношения этих сил и интенсивности
теплового движения могут реализоваться различные конформа-
ции (рис. 1.10):
42
статистический клубок, т е. более или менее свернутая кон-
формация; такую конформацию обычно принимают макромоле-
кулы полимеров, для которых интенсивность внутреннего тепло-
вого движения превалирует над внешними воздействиями, она
характерна для многих полимеров, например линейных (поли-
этилен, полипропилен, полибутадиен, полиизопрен, тринитроцел-
люлоза и др.), лестничных (полифеииленсилоксан);
конформация спирали; эту конформацию обычно принимают
макромолекулы, у которых дальний конформационный порядок
в виде спирали иммобилизован, например, водородными связя-
ми; спиральная конформация характерна, как правило, для бел-
ков и нуклеиновых кислот;
конформация глобулы, т. е. очень компактной частицы по
форме близкой к сферической; такую конформацию имеют мак-
ромолекулы полимеров с очень сильным внутримолекулярным
взаимодействием, например полимеры, содержащие атомы фто-
ра (политетрафторэтилен),
конформация стержня или струны (обнаружена для неко-
торых алкилполиизоцианатов);
складчатая конформация; характерна для полимеров в кри-
сталлическом состоянии;
конформация коленчатого вала (например, у поли-л-бенз-
амида).
Таким образом, конформация макромолекулы представляет
собой сумму низших конформационных уровней. Например, кон-
формацию макромолекулы полипропилена можно характеризо-
вать следующим образом конформация звена — транс и гош\
ближний конформационный порядок •«— транс и гош\ дальний —
Рнс. 1 10. Конформацни макромолекул*
в — статистический клубок; б —спираль (О — заместители, между которыми образуются
водородные связи); а —глобула; г —струна; д — складчатая; е — коленчатый вал
4а
спираль 3i; конформация макромолекулы — статистический клу-
бок в аморфном состоянии и складчатая в кристаллическом.
Следует отметить, что конформация макромолекулы может
изменяться в зависимости от внешних факторов — температуры,
напряжения и др. При этом затрагиваются все конформацион-
ные уровни: например, при деформации растяжения в макромо-
лекуле полибутадиена изменяется ближний конформационный
порядок (aouz-формы переходят в транс-) и конформация моле-
кулы— статистический клубок переходит в конформацию, при-
ближающуюся к вытянутой струне. Поэтому о конформации
макромолекулы судят обычно в условиях отсутствия возмущаю-
щих факторов. Идеальные условия — это газовая фаза, но по-
скольку макромолекулы полимера не существуют в газообраз-
ном состоянии, то наиболее реальной моделью этого состояния
является разбавленный раствор в так называемом 0-растворите-
ле при О-темпсратуре, когда взаимодействие между полимером
и растворителем отсутствует и цепные макромолекулы имеют
нсвозмущенные размеры.
Для любых конформаций, которые принимает макромолеку-
ла, характеристикой ее длины I является расстояние между кон-
цами г. Теоретический расчет размеров цепи был проведен для
свободносочлененной цени, т. е. цепи, у которой пет жестко фик-
сированных валентных углов и возможно свободное вращение
атомов и групп вокруг одинарных связей (рис. 1.11), Если моле-
кула полностью развернута, то г=1, т. е. длине молекулы, рас-
считанной исходя из длины повторяющегося звена b и числа
этих звеньев п, причем при расчете учитываются длина связей,
значения валентных углов и конфигурация звена. Эту величи-
ну I называют контурной или гидродинамической длиной цепи.
Для предельно свернутой молекулы г—>0, а для любых проме-
жуточных положений 0<г-</.
Рис. 1.11. Схематическое изображение макромолекулы в виде свободно-соч-
лененной цепи (а), цепи, состоящей из независимых сегментов (6) и черве-
образной цепи (в);
г — вектор расстояния между концами цепи; А — сегмент; а — персистентная длина цеп»
44
Рис. 1.12 Кривая распределения рас-
стояний между концами макромолекулы
Поскольку макромолекула
находится в движении, занимая в
каждый момент i-ю конформа-
цию, то величина г является ус-
редненной характеристикой и
рассматривается как вектор, про-
веденный из одного конца цепи
к другому. Среднее значение г по всем конформациям равно
нулю, так как все направления равновероятны. Поэтому рас-
стояние между концами цепи характеризуют или средним
квадратом расстояния г2, или среднеквадратичным расстоянием
<г2>1/2. Кривая распределения абсолютных значений расстоя-
ния между концами макромолекулы имеет вид, представленный
на рис. 1.12, и описывается уравнением
U7 (г) - (4ft»/pi) ехр (- ЬФе в), (1.19)
3 1
2 nt2
где И7(г)—термодинамическая вероятность существования цепи; п— число
повторяющихся звеньев; b — длина такого звена; гсв — расстояние между
концами свободносочлененной цепи.
Анализ этого выражения показывает, что при г—>0 и г—>-со
W(r)—>0, т. е. эти состояния маловероятны. Максимальное
значение №(г)ияКс, т. е наиболее вероятное расстояние между
концами цепи, может быть рассчитано из условий определения
максимума, т. е.
г2св макс = 3/2л Z2j ь = ~। 7“ = V2Л1/Л1ЗВ, (1.20)
1 — cos р
где /с» — длина связи; п=М/М3я (М н Af3B— молекулярные массы полимера
и звена); р— угол, дополнительный к валентному.
Степень свернутости макромолекулы оценивается отношени-
ем 1/{г2)'/2, т. е. также зависит от молекулярной массы поли-
мера.
Расстояние между концами макромолекулы можно также
оценить по персистентной длине макромолекулы. Если макро-
молекулу представить в виде непрерывной червеобразной цепи с
непрерывной кривизной (см. рис. 1.11, в), то а — проекция век-
тора расстояния между концами клубка на направление каса-
тельной к началу клубка — и есть персистентная длина цепи.
Размер молекулы характеризуют также средним радиусом
инерции, или радиусом вращения R2. Для гауссовых клубков
<7?>,/г—1/6<г2св>,/2- (1-21)
45
Реальную цепь можно рассматривать как свободносочленен-
ную, если из длинной цепи выделить условно участки, конфор-
мации которых будут независимы друг от друга и влияние внут-
римолекулярного взаимодействия будет проявляться внутри
этого участка. Такой участок цепи длиной А, положение кото-
рого не зависит от положения соседних участков, называют
термодинамическим сегментом или сегментом Куна. Для реаль-
ных цепей справедливы приведенные выше зависимости, если
вместо длины повторяющегося звена b использовать длину сег-
мента А, а вместо числа звеньев п — число сегментов Z. Тогда
1Г(г)-(4^/)'л)г2ехр (—№2); (1.22)
ьз=А_1_
2 ZA*
(где г2— квадрат среднего расстояния между концами реаль-
ной цепи). В точке максимума (т. е наиболее вероятной) раз-
мер клубка составляет:
„ „ 1 4- cos В 1 4* cos Ф
г2 = 3/2ZA « л/2св—1-----£ -----=T2L
1 — COS р 1— COS ф
(1.23)
(где <р — средний угол, в пределах которого разрешено враще-
ние). При cos ф = 0 (условие свободного вращения) формула
(1 23) переходит в (1.20), а при cos<p=l г2—>-оо, т. е. вероятна
конформация вытянутой цепи. Для всех случаев заторможен-
ного вращения (0<со8ф<0) величина cos ф определяется со-
отношением:
где С/0(ф) — потенциальный барьер вращения; k — константа Больцмана.
Можно выделить два предельных случая:
1) £/о(ф) т. е. вращение практически не заторможено,
макромолекула может принимать бесконечное число конформа-
ций;
2) Uo((p)^>kT, т е. энергии не хватает для перехода из од-
ной конформации в другую, и макромолекула может существо-
вать в форме вытянутых цепей (одна конформация).
Кроме показателя, характеризующего расстояние между кон-
цами макромолекулы, т. е. длины, в понятие размера, как мы
уже говорили, входит и диаметр макромолекулы d — диаметр
цилиндра, описанного вокруг молекулы. Он определяется с уче-
том боковых групп, разветвлений и других конфигурационных
характеристик (рис. 1.13). Так же как и длина, d является
усредненной характеристикой, поскольку форма макромолеку-
ле
1
1
J
Рис. 1.13. Способы определения диаметра макромолекулы d (R — замести-
тели)
лы изменяется вследствие микроброуновского движения. Объ-
ем, занимаемый одной макромолекулой, — это так называемая
координационная сфера, окружающая клубок. Концентрация
полимера зависит от типа конформаций: в статистическом клуб-
ке она невелика и составляет в 0-растворителе «3% Чем силь-
нее взаимодействие внутри клубка, тем больше его плотность и
меньше сфера, т. е. объем, поэтому концентрация полимера
больше при глобулярной конформации по сравнению с конфор-
мацией статистического клубка.
Если макромолекула находится в условиях дальнедействую-
щего взаимодействия (например, при взаимодействии с раство-
рителем), то ее размеры определяются с учетом этих сил. В этом
случае размеры зависят от параметра набухания а:
а=<?2>1/2/<?2в>1/2 (125)
I
(где Ге — расстояние между концами цепи в растворителе).
По мере повышения термодинамического сродства полимера и
растворителя размеры клубка увеличиваются, концентрация по-
лимера в клубке понижается и при близких значениях парамет-
ров растворимости составляет около 1%.
Размеры макромолекулы зависят также от так называемого
исключенного объема Уиск- Это объем, из которого данная по-
лимерная молекула исключает все другие молекулы, что явля-
ется результатом действия сил отталкивания между ними. Меж-
ду Vhck и а существует соотношение:
Птах=АУя« (126)
vhck_,o°
(где х — константа, составляющая от 1 до 6,67; А — постоян-
ная).
Разветвленные молекулы характеризуются меньшими разме-
рами клубка ввиду их большей плотности:
<г2>,'2врВ..=<Я>‘'2ел-.£, (1-27)
где <Я>'л0рЯзв и <?2>,/'еляв — среднеквадратичные расстояния между концами мо-
лекул разветвленного и линейного полимеров; g— фактор разветвленности.
47
Таким образом, конформации и размеры реальных макро-
молекул определяются комбинацией сил ближнего и дальнего
порядков, интенсивностью внутреннего теплового движения, за-
висят от химического строения, молекулярной массы, конфигу-
рации макромолекулы. Вполне естественно предположить, что в
конденсированном состоянии, когда сильно возрастает роль
дальнодействия ввиду высокой кооперативности системы, кон- :
формации макромолекул будут отличны от конформаций изоли-
рованной макромолекулы j
1.2 НАДМОЛЕКУЛЯРНАЯ СТРУКТУРА
В результате действия водородных и межмолекулярных сил
макромолекулы полимеров, так же как и молекулы низкомоле-
кулярных соединений в конденсированном состоянии, вступают
во взаимодействие друг с другом и образуют агрегаты различ-
ной степени сложности и с разным временем жизни Строение
агрегатов зависит от химического состава взаимодействующих
мономерных звеньев макромолекул, числа и размера атомов или
групп, условий (температура, давление, среда и др ) Наиболее
устойчивы структуры, в которых число межмолекулярных и во-
дородных связей максимально В ряде случаев отдельные мак-
ромолекулы объединяются во вторичные образования, вторич-
ные— в образования третьего и четвертого порядка Физиче-
ская структура полимерных тел, обусловленная различными ви-
дами упорядочения во взаимном расположении макромолекул,
называется надмолекулярной структурой.
Взаимное расположение, размеры элементов надмолекуляр-
ной структуры зависят от конфигурации и конформации, хими-
ческого состава мономерных звеньев и макромолекулы в целом,
размеров отдельных атомов и строения их орбиталей, условий,
при которых происходило структурообразование, продолжитель-
ности и скорости структурообразования и многих других факто-
ров
Чем сложнее химическое строение макромолекул, чем раз-
нообразнее условия, в которых синтезировали, перерабатывали
и хранили полимер, тем сложнее и разнообразнее надмолеку-
лярные структуры в нем, менее однородны его свойства По-
скольку состав сырья, а также рецептуры и режимы получения
и переработки полимеров могут колебаться, их надмолекуляр-
ные структуры различаются даже в пределах производственных
партий
Изучение надмолекулярной структуры полимеров крайне ак-
туально, так как она определяет комплекс их физических
свойств, скорость и механизм физико химических и химических
процессов. Вследствие различий в надмолекулярной структуре
48
часто изделия из одного и того же полимера имеют разные
свойства.
Влияние надмолекулярных структур на формирование
свойств можно проиллюстрировать на примере алмаза и гра-
фита— неорганических полимеров, образованных из атомов уг-
лерода. Алмаз представляет собой пространственный алифати-
ческий полимер кристаллического строения. Каждый атом уг-
лерода связан четырьмя ковалентными связями с четырьмя
другими атомами в трех направлениях. Длина связей равна дли-
не обычной простой о-связи С—С, т. е 0,154 нм Строение кри-
сталлической решетки алмаза показано на рис. 1.14, а. Образо-
ванные из атомов углерода, лежащих в одной плоскости, цикло-
гексановые циклы имеют конформацию «кресла» (рис. 1.14,6).
Расстояние между атомами га равно 0,205 нм. Этот полимер от-
личается чрезвычайно высокой прочностью, твердостью, стойко-
стью к агрессивным средам.
а
О - атом углерода
Рнс. 1Л4 Надмолекулярная струк-
тура алмаза (а, б) и графита (в):
а — кристаллическая решетка из атомов
углерода, б — кресловидные конформа-
ции в плоскостях циклогексановых
циклов (заштрихованы два цикла, два
цикла в нижней плоскости не заштри-
хованы); гж — расстояние между ато-
мами С в кристаллической решетке
4—816
49
Графит имеет плоскосетчатое строение, аналогичное строе-
нию конденсированных ароматических соединений. Каждый слой
представляет собой сетку гексагональных циклов, в вершинах
которых расположены атомы углерода. Каждый атом связан с
тремя другими ковалентными связями (расположенными под
углом 2,07 рад друг к другу) длиной 0,142 нм. У всех атомов
углерода четвертые валентные электроны не локализованы.
Они образуют «электронный газ» и могут перемещаться внутри
плоскости, обеспечивая хорошую электрическую проводимость.
Слои расположены друг от друга на расстоянии 0,34 нм и свя-
заны относительно слабыми межмолекулярными силами. Это
обусловливает низкую прочность, способность скольжения сло-
ев один по другому (рис. 1.14, в). Переход электронов из одной
плоскости в другие затруднен, вследствие чего электропроводи-
мость графита анизотропна' проводимость вдоль плоскостей в
1014 раз выше, чем поперек плоскостей.
Наиболее разнообразными являются надмолекулярные струк-
туры органических полимеров По степени упорядоченности эле-
ментов надмолекулярных структур их можно разделить на две
группы: аморфные и кристаллические. Критерием такого раз-
деления может служить дифракция рентгеновских лучей или
электронов, характеризующая степень упорядоченности струк-
тур. В кристаллических структурах существует дальний поря-
док расположения звеньев и цепей, и на рентгенограмме появ-
ляется серия рефлексов (точек, пятен, линий), а на дифракци-
онных кривых — серия максимумов. Аморфные полимеры при
рентгеноструктурном анализе не дают рефлексов, а характери-
зуются аморфным гало (см. разд. 1.3).
В кристаллических полимерах, как правило, существует
трехмерный дальний порядок в расположении атомов, звеньев
и цепей. Аморфные полимеры характеризуются ближним поряд-
ком лишь в расположении звеньев. Дальний порядок в распо-
ложении звеньев и цепей у них отсутствует. Однако такое деле-
ние отнюдь не означает, что аморфные полимеры беспорядочны
и бесструктурны, а кристаллические — имеют бездефектную кри-
сталлическую структуру
Количественно оценить структуру веществ можно с помощью
функции радиального распределения межатомных расстояний
№(га), представляющей собой относительную вероятность на-
хождения соседних атомов на расстоянии га от фиксированно-
го атома. Вид функции V/(ra) для кристаллических и аморфных
веществ показан на рис. 1 15. Вследствие существования даль-
него порядка в кристаллах соседние атомы расположены на оп-
ределенном расстоянии друг от друга, равном периоду кристал-
лической ячейки. Поэтому в случае кристаллических полимеров
функция №(гя) изменяется с периодическими всплесками, т. е.
имеет ряд максимальных значений, соответствующих наиболее
50
Рис. 1.15. Вид функции распределения межатомных расстояний U7(ra) для
кристаллического (а) и аморфного (б) полимеров
вероятным расстояниям между соседними атомами в кристалли-
ческой ячейке. Функция и/(га) аморфных полимеров имеет фор-
му затухающей синусоиды, и после первых максимальных зна-
чений и и Г2 перестает изменяться, что указывает на упорядо-
ченное расположение первого слоя частиц и менее упорядочен-
ное— второго, т. е. свидетельствует о существовании в аморф-
ных полимерах ближнего порядка в расположении сегментов
макромолекул при отсутствии дальнего, характерного для кри-
сталлов
1.2.1. Надмолекулярная структура аморфных полимеров
Наиболее изучена надмолекулярная структура полимеров с вы-
соким уровнем межмолекулярного взаимодействия, таких, как
белки, полисахариды
Как уже указывалось, конформация макромолекул белков
формируется под влиянием боковых полярных заместителей, об-
разуя спираль в а- или p-форме (фибриллярные белки) или гло-
булы (глобулярные белки). Эти структуры (для белков их на-
зывают иногда вторичными, первичной же называют конформа-
цию звеньев ближнего порядка) усложняются скручиванием
двух-трех а-спиралей вместе в двух-трехтяжную спираль, за-
вертыванием шести спиралей вокруг седьмой, свертыванием
спиралей в клубки. Эти структуры называют третичными. Бо-
лее высокий уровень надмолекулярных структур (четвертичная
структура) возникает в результате агрегации нескольких макро-
молекул в общую систему при взаимодействии водородных, со-
левых, дисульфидных и других связей.
В полисахаридах (целлюлоза и крахмал, состоящий из
амилозы и амилопектина) макромолекулы, свернутые в спи-
раль, плотно уложены в пучки. Такая структура вследствие
большого межмолекулярного взаимодействия является доволь-
но устойчивой.
51
Менее определенные формы надмолекулярной организации
наблюдаются у полимеров с невысоким уровнем межмолекуляр-
ного взаимодействия, имеющих макромолекулы в конформации
статистического клубка. Длительное время считали, что в кон-
денсированном состоянии такие полимеры представляют собой
конгломераты хаотически перепутанных клубков, образующих
так называемый «молекулярный войлок». Однако такое пред-
ставление не соответствует свойствам полимеров.
Впервые идея об упорядоченных областях в аморфных поли-
мерах была высказана в 1948 г. Ллфреем На присутствие &
аморфных полимерах областей с упорядоченным расположени-
ем макромолекул указывают следующие факторы.
1. Плотность аморфных полимеров обычно незначительно
отличается от плотности кристаллических полимеров того же
состава (плотность полиэтилена, обладающего идеальной кри-
сталлической структурой, равна 1000, аморфного — 800 кг/м3).
Если бы в аморфном полимере не содержалось упорядоченных
областей, его плотность была бы значительно ниже плотности
кристаллического.
2. Высокая скорость кристаллизации полимеров. При отсут-
ствии областей с упорядоченной структурой продолжительность
кристаллизации аморфных полимеров была бы значительно
выше, чем наблюдается на практике
3. Различными физико-химическими методами (дифракция
рентгеновских лучей и нейтронов под большими и малыми угла-
ми, электронная микроскопия и др.) было показано, что в
аморфных полимерах существуют области с локальной упорядо-
ченностью звеньев, которая сохраняется на расстоянии несколь-
ких нанометров (т. е. ближний порядок), причем между упоря-
доченными и неупорядоченными Областями нет четких границ.
Для объяснения надмолекулярной организации аморфных
полимеров было предложено несколько моделей. В. А. Каргин,
А. И. Китайгородский, Г. Л. Слонимский предложили модель,
согласно которой аморфные полимеры могут состоять либо из
глобул, образованных свернутыми макромолекулами, либо из
развернутых цепей, собранных в пачки. Однако последующие
исследования показали, что «пачечная» теория ошибочна. Она,
в частности, находится в противоречии с основными положе-
ниями кинетической теории высокоэластичности (см. гл. 4), ко-
торая хорошо подтверждается экспериментом. Так, с позиций
этой модели практически невозможно объяснить способность
некоторых полимеров к большим обратимым деформациям.
Одна из возможных моделей строения аморфных полимеров,
предложенная Иехом, показана на рис. 1.16 Домены 1 обра-
зуются из многократно сложенных и параллельно расположен-
ных участков одной или нескольких макромолекул. Степень
упорядоченности доменов (дефектность), их размеры, число в
-252
Рис. 1.16. Модель структуры аморфного полимера:
/ — упорядоченный домен; 2 — междомениое пространство; 3 — «проходиые> макромоле-
кулы
взаимное расположение зависят от условий, при которых про-
исходило охлаждение полимера. Домены могут играть роль за-
родышей кристаллизации, являются переходным типом структу-
ры от аморфного к кристаллическому. Между доменами нахо-
дятся междоменные аморфные области 2, заполненные петлями
и концами цепей, образующими домены, а также макромолеку-
лами низкой степени полимеризации Строение междоменных
областей менее упорядочено, чем доменов, илн вообще не упо-
рядочено. Предполагается, что некоторые макромолекулы вхо-
дят в состав нескольких доменов, т. е. являются «проходными».
«Проходные» макромолекулы <3 соединяют домены и междомен-
ные области в единую трехмерную структуру.
Есть основание считать, что различные домены могут агреги-
роваться и образовывать более сложные «наддоменные» струк-
туры. Такая хорошо упорядоченная структура аморфных поли-
меров наблюдается в блок-сополимерах типа А—Б—А, напри-
мер в блок-сополимерах изопрена со стиролом. В таких сопо-
лимерах блоки одного типа агрегируются в домены Так как
длина блоков в сополимере постоянна, домены образуют квази-
кристаллическую решетку с характерным периодом около 1 мкм,
подобную решетке глобулярных кристаллов.
Модель Перепечко названа автором кластерной моделью.
Под кластером понимают области с более или менее плотной
упорядоченной упаковкой молекул или их частей по сравнению
с основной разупорядоченной и менее плотной частью полиме-
ра Плотность кластера, как правило, должна превышать сред-
нюю плотность полимера, но упорядоченность и плотность кла-
стера ниже, чем кристалла.
53
Морфология (характер расположения) цепей в кластерах
определяется структурой полимера на молекулярном уровне,
т. е. химическим строением его повторяющегося звена, молеку-
лярной массой, степенью разветвленности макромолекул, нали-
чием сетки зацеплений. Если подвижность цепи, молекулярная
масса линейных полимеров или расстояние между узлами для
сетчатых достаточно высоки для образования складчатой кон-
формации, то кластер характеризуется складчатой морфологи-
ей. Если же подвижность цепи ограничена (например, из-за
большого числа полярных групп или сетки зацеплений), то наи-
более вероятной конформацией внутри кластера будет конфор-
мация вытянутой цепи.
Наряду с существованием некристаллизующихся кластеров
предполагают существование так называемых кристаллических.
Если полимер имеет молекулярное строение, обеспечивающее
при определенных условиях трехмерную упорядоченную струк-
туру в кластере, то такие кластеры способны кристаллизовать-
ся и иметь кристаллическую структуру. Если размер кристал-
лических образований меньше так называемых «критических»
размеров зародышей кристаллизации, то кристаллизация на
этих кластерах развиваться не будет и полимер в целом оста-
нется аморфным. При достаточных размерах кристаллического
кластера вероятно образование тех или иных кристаллических
форм надмолекулярных структур.
1.2.2. Надмолекулярная структура кристаллических полимеров
Для кристаллических полимеров характерен большой набор
структурных элементов, характеризующих различные уровни
надмолекулярной организации. Как уже отмечалось, полимер-
ный кристалл, так же как и кристаллы низкомолекулярных
веществ, характеризуется дальним порядком расположения по-
вторяющихся структурных единиц. Таким повторяющимся эле-
ментом кристалла является элементарная ячейка с размерами
а, Ь, с и углами а, £, 7, содержащая определенным образом
сгруппированные атомы. Для большинства известных полиме-
ров эти параметры ячейки определены рентгенографическим ме-
тодом (табл. 1.5).
Расположение макромолекул в кристаллических областях
всегда строго определенно: оси макромолекул параллельны
друг другу, концы их находятся на поверхности кристаллическо-
го образования. Кристаллографическая ось с совпадает с осью
макромолекулы По форме кристаллографические ячейки делят
на несколько типов: кубическая, орторомбическая, гексагональ-
ная и др. В ячейку входит, как правило, не вся молекула, а толь-
ко ее небольшая часть (несколько повторяющихся звеньев), по-
этому элементарная ячейка полимера часто аналогична ячейке
54
Таблица 1.5. Кристаллографические параметры различных полимеров
Полимер Кристалло- графическая система ячейки кри- сталла Параметры элементарной ячейки, нм Число мономерных звеньев а ячейке Конформация цепей
а b с углы
а ₽ V
Полиэтилен Орто 0,74 0,49 0,25 90 90 90 2 Z
монокристалл Три 0,760,56 12 63 71 82 48 Z
Полипропилен изотак- тический Моно 0,66 2,1 0,65 90 99,3 90 12 •^s“3t
Полинзобутилен Орто 0,69 1,20 1,86 90 90 90 16 Ap«=8s
цис-1,4-Полибута- Моно 0,46 0,95 0 86 90 109 90 4 Z
диен цис-1,4 -Полиизопрен Моно 1,25 0,890,81 90 92 90 8 Z
гролс-1,4-Полнхло- ропреи Орто 0,9 0,82 0,48 90 90 90 — —
Полиэтилентерефта- Три 0,46 0,59 1,07 98 118 112 1 z
лат Полиоксиэтилен Моно 0,80 1,31 1,95 90 125 90 28 X,«=78
Целлюлоза Моно 0,83 1,03 0,79 90 84 90 2 z
Гринитрат целлюлозы Орто 1,22 2,5 0,9 90 90 90 4 z
Обозначения: моно — моноклинная, орто — орторомбическая, три — тригональ-
ная. зигзаг, Ху — спираль, цифры — типы спирали.
низкомолекулярных аналогов. Так, у полиэтилена ячейка идеи-
тична ячейке кристаллических «-парафинов. Конформация
звеньев в ячейке соответствует минимуму внутримолекулярной
энергии, определяется силами внутримолекулярного взаимодей-
ствия или водородными связями, т. е. молекулярным строением
полимера, и очень часто совпадает с конформацией изолирован-
ной молекулы. Однако полимерные кристаллы характеризуют-
ся рядом особенностей.
1. Полимеры, как правило, не бывают полностью закристал-
лизованы В кристаллах полимеров наряду с кристаллическими
областями существуют аморфные участки с ближним порядком.
В кристаллической фазе макромолекулы расположены наибо-
лее упорядоченно и плотно, поэтому плотность кристаллической
фазы рКр больше средней плотности полимера и плотности
аморфной фазы рам, т. е. ркр>р>рам. Значения ркр, р и ра.м для
некоторых полимеров (в кг/м3) приведены ниже:
р Ркр
Полиэтилен высокой плотности 940—960 1000 850
низкой плотности 920—930 990 850
Полнэтилентерефталат 1380 1450 1330
Поливинилхлорид 1140 1450 1100
Поли хлортрифтор этилен 2090—2160 2190 2030
Полиформ альдегид 1400 1500 1250
55
Соотношение между содержанием полимера в аморфной и
кристаллической областях оценивается степенью кристаллично-
сти, представляющей собой долю полимера в закристаллизован-
ном состоянии. При комнатной температуре степень кристал-
личности для наиболее распространенных полимеров лежит в
пределах 10—90%:
Полиэтилен Поликапроамид высокой плотности 60—90 Полиэтнлентерефталат низкой плотности 40—60 Поливинилхлорид Полипропилен 40—70 Полнхлоропрен Целлюлоза 30—70 Степень кристалличности Ко (по объему) и Кы можно определить по плотности: 30—60 0—60 0-40 10-30 (по массе)
__ Р Рам .
Рхр —Рам ’ (1.28)
j, Р — Рам Ркр „ Ркр
Ы Ркр — Рам Рам ° Р
Степень кристалличности зависит от регулярности строения
макромолекул и условий кристаллизации. Любой вид нерегу-
лярности (наличие звеньев, находящихся в другой конфигура-
ции, разветвленности, сшивок и др.) снижает степень кристал-
личности (рис. 1.17).
Степень кристалличности ориентированных полимеров, как
правило, повышается. Условия получения полимера, предопре-
деляя регулярность макромолекул, изменяют степень кристал-
личности. Примером может служить полиэтилен, который полу-
чают при высоком и низком давлениях, при низком давлении
образуется менее разветвленный полиэтилен с более высокими
плотностью и степенью кристалличности.
2. Коэффициент молекулярной упаковки, т. е. отношение
-объема, занятого макромолекулами, к общему объему, в поли-
Рис. 1 17 Зависимость степени кристалличности Ко транс-1,4-полихлоро-
прена от числа сшивок Пс и содержания (С) звеньев транс-1,4
56
мерных кристаллах значительно ниже, чем у кристаллов низко-
молекулярных веществ, и находится в пределах 0,65—0,73.
3. Поскольку одна и та же макромолекула многократно вхо-
дит в кристаллографические ячейки, то четкой фазовой грани-
цы между кристаллической и аморфной частями, как правило
не существует.
4. Размер отдельных кристаллических областей с единой
ориентацией кристаллографических осей, так называемых кри-
сталлитов, довольно мал: примерно на два порядка меньше
длины молекулярной цепи
5. Кристаллы характеризуются значительной дефектностью,
т. е. отклонением от правильного трехмерного порядка внутри
кристаллов. Дефектность полимерных кристаллов может быть
обусловлена как дефектами, характерными для низкомолекуляр-
ных соединений (например, включение посторонних атомов, ис-
кажения, вызванные тепловым движением, и др.), как и дефек-
тами, специфичными для полимеров и обусловленными наруше-
нием регулярности макромолекул. Следствием высокой де-
фектности полимерных кристаллов является снижение темпера-
туры плавления по сравнению с теоретически предсказуемой.
6. Для полимерных кристаллов характерна складчатая струк-
тура. В процессе кристаллизации растущий кристалл при дости-
жении определенной длины начинает складываться «сам на се-
бя» и снова входит в кристалл, образуя складку. Моменту об-
разования складки соответствует минимум свободной энергии
системы, т. е. образование складки термодинамически выгодно.
Складчатая структура обнаружена экспериментально для мно-
гих полимеров и олигомеров. При образовании складчатой
структуры дефекты, как правило, «выталкиваются» на поверх-
ность складок, но могут входить и в складчатое образование
(ламель).
7. Один и тот же полимер характеризуется набором кри-
сталлических структур различной морфологии и различной де-
фектности. Причины подобной неоднородности кроются, в пер-
вую очередь, в полидисперсности по молекулярной массе, регу-
лярности, конфигурации макромолекул и т. д Кроме того,
структура полимеров существенно изменяется при переработке
и зависит от ее условий. Следствием этого является существова-
ние интервала температур плавления кристаллов.
Многообразие кристаллических структур и влияние на них
внешних условий можно показать на примере полиэтилена. Он
образует кристаллы различных модификаций с ромбической,
гексагональной, моноклинной и триклинной элементарными
ячейками, различающимися длиной осей и величиной углов
между осями При обычной температуре преобладают макро-
молекулы в форме плоского зигзага, образующие кристаллы с
орторомбической ячейкой. При повышении температуры увели-
57
чивается интенсивность крутильных колебаний макромолекул
вокруг связей С—С и форма ячейки из ромбической переходит
в гексагональную, наиболее удобную для укладки макромоле-
кул. При деформации происходит ориентация цепей и ячейки
кристаллов приобретают моноклинную или триклинную форму.
Наиболее распространенными видами кристаллических
структур являются: кристаллиты, монокристаллы, фибриллы,
сферолиты. Ниже приведены их минимальные (числитель) и
максимальные (знаменатель) размеры (нм):
Макромолекулы 0,2—0.5/102—10*
Кристаллиты 2—10/10—50
Монокристаллы 10/103— 1 О*
Фибриллы в сферолитах 10s—5-10s/10*—10s
Сферолиты — /10*—10»
Кристаллит представляет собой наименьшее кристаллическое
образование с единой ориентацией кристаллографических осей.
Поскольку длина молекулы полимера превышает максимальные
размеры кристаллита, то при его формировании макромолеку-
лы многократно складываются, образуя складчатую конфор-
мацию.
Монокристалл построен, как правило, из элементарных яче-
ек и, так же как и кристаллит, характеризуется единой ориен-
тацией кристаллографических осей. Все монокристаллы отлича-
ются высокой степенью упорядоченности, т е. малой дефектно-
стью По строению монокристаллы можно разделить на три ви-
да: пластинчатые, фибриллярные, глобулярные
Пластинчатые монокристаллы получены для многих полиме-
ров при кристаллизации из разбавленных растворов (концент-
рация полимера 0,01—0,1%). Например, при кристаллизации
линейного полиэтилена из разбавленных растворов в ксилоле
или бензоле при 353—358 К получаются пластинчатые ромбо-
видные монокристаллы (рис. 1.18). Пластинчатые монокристал-
лы состоят из тонких пластинок чаще всего ромбовидной фор-
мы толщиной примерно 10—26 нм и размерами сторон до 1 мкм.
Эти пластины называют ламелями (рис. 1.19, а). Поэтому часто
пластинчатые кристаллы называют ламелярными. Ось с, совпа-
дающая с осью макромолекулы, перпендикулярна плоскости ла-
мели Конформация макромолекулы в ламелях чаще всего бы-
вает складчатой и образуется путем перегибов макромолекул
под углом 3,14 рад (180°). Поэтому в первом приближении мож-
но считать, что кристаллит есть не что иное, как ламель. В за-
висимости от молекулярной массы макромолекула может обра-
зовывать большее или меньшее число складок. Возвращение
цепи в кристалл после выхода из него может происходить по-
разному: цепь возвращается на строго определенном расстоя-
нии от места выхода; цепь возвращается на некотором расстоя-
нии от места выхода; цепь не возвращается в кристалл, обра-
58
Рис. 1.18. Надмолекулярные структуры полимеров:
а — монокристалл (пазиэтплсн); б — кольцевой сферолит; в — радиальный сферолит; г—
сферолнтная лента (изотактический полистирол)
зуя петли, концы. Последние два случая приводят к нарушению
регулярности складывания. Нарушение регулярности складыва-
ния может произойти также и за счет нерегулярных участков
макромолекул, при этом образуются петли внутри складок.
Несмотря на то что монокристаллы являются самыми со-
вершенными кристаллическими структурами, степень кристал-
личности их практически никогда не достигает 100%. Часть мо-
лекул не входит в кристалл, а образует дефектные участки с
низкой степенью упорядоченности, т. е. аморфную часть. Даже
в идеальном складчатом кристалле области складывания не
входят в кристаллит. Их доля может быть оценена отношением
Д/ск//ск (см. рис. 1.19, а, б) и составляет примерно 5—10%. Та-
кой кристалл можно рассматривать как слойку, состоящую из
кристаллических и аморфных областей. По мере роста дефект-
ности толщина кристаллита все больше и больше отличается от
толщины ламели. В общем случае период складывания, или
большой период L, равен полной толщине ламели, т. е. сумме
59”
Рис, 1.19. Модели закристаллизованных полимеров:
а ламель со складчатыми цепями и упорядоченным граничным слоем (a h г_____№
кристаллографической ячейки); 6 - ламель ячейки со склздчатымн цепямк и M3v7Jm-
’□••ениым граничным слоем а-структура <шнш-кебаб> (А -вытянутые мЛ-
,-еферолит (/—центр сфероХ^-^й
Рис. 1 20. Зависимость толщины ламели
полиэтилена L от температуры его кри-
сталлизации
толщин кристаллита и удвоен-
ной толщине дефектных областей
(/Ск+2Д/ск)- Период складыва-
ния зависит от условий кристал-
лизации, в частности от темпера-
туры. Он увеличивается с ростом
температуры и достигает наи-
большей величины при темпера-
туре, близкой к температуре
плавления (рис. 1.20). Поэтому в
равновесных условиях не иск-
лючается возможность образования монокристалла из развер-.
нутых молекул. Такие монокристаллы были получены для
полиметиленоксида при твердофазной катионной полимериза-
ции триоксана. Монокристаллы с выпрямленными цепями — на-
иболее совершенные кристаллические структуры в полимерах,
в них практически отсутствуют дефекты, их плотность прибли-
жается к теоретической плотности бездефектного кристалла.
В определенных условиях могут образовываться кристаллы,
в которых существуют участки с выпрямленными цепями и уча-
стки с цепями складчатой конформации (см. рис. 1.19, в). Эти .
структуры получили название «шиш-кебаб» (шашлык). Они об-
разуются, например, при кристаллизации в условиях переохлаж-
дения предварительно растянутых полимеров (полиэтилена, по-
лиизопрена, полихлоропрсна)
Ламели, являясь первичными элементами монокристаллов,’
могут компоноваться в различных вариантах, образуя разные
морфологические формы: террасоподобныс монокристаллы, по-
лые пирамиды, звездообразные дендриты. Монокристаллы пла-
стинчатого типа могут соединяться друг с другом, образуя
структуры более высокого надмолекулярного уровня: эдриты и
овоиды. Это кристаллические образования, одна из проекций
которых имеет форму многогранника (эдриты) или овала (овои-
ды). Толщина этих структур обычно более 1 мкм, а длина мо-
жет достигать нескольких миллиметров.
Прежде чем рассмотреть строение фибриллярных монокри-
сталлов, остановимся на понятии «фибрилла». Этим термином
обычно называют структуру, представляющую собой агрегат па-
раллельно упакованных цепей. Длина фибриллы, как правило,
намного превышает ее поперечные размеры. Толщина фибрил-
лярных кристаллов обычно составляет 10—20 нм, а длина мо-
жет достигать многих микрон. Фибриллярные кристаллы обра-
зуются из микрофнбрилл, которые, в свою очередь, построены
6Г
Ось
ыихрофябрмлы
Рис. 1.21. Модель строения микрофнбриллы:
а, б — мккрофнбрнлла в монокристалле целлюлозы: в — ммкрофнбрилла в белках; г —
микрофибрилла в сферолите (Д—аморфная часть, К — кристаллическая)
либо из выпрямленных цепей, либо из цепей складчатой кон-
формации. Примером полимера, образующего фибриллярные
монокристаллы, может служить целлюлоза. Отдельное микро-
фибриллы этого полимера представляют собой скрученную в
спираль ленту. Макромолекулы в ленте находятся в сложенном
состоянии, и их ось составляет определенный острый угол с
осью ленты, а ось микрофибриллы параллельна оси макромоле-
кулы (рис. 1.21, а, б) Следует отметить, что микрофибриллы в
монокристаллах практически не содержат аморфных участков,
т. е. по существу бездефектны Для фибриллярных белков ха-
рактерны спирально-кристаллические микрофибриллы (рис.
1.21,е).
В глобулярных кристаллах узлы решетки образованы макро-
молекулами в глобулярной конформации. Такие кристаллы ха-
рактерны для биополимеров, имеющих высокую степень одно-
родности по размерам, что является необходимым условием об-
разования глобулярных кристаллов.
Сферолиты — это поликристаллические структуры, обладаю-
щие симметрией относительно центра (см. рис. 1.19, г), из ко-
торого и начинается рост структуры, как правило, путем соеди-
нения ламелей одинаковой ориентации. При большом числе за-
родышей кристаллизация может ограничиться образованием та-
ких центров, которые называют зернами. При небольшом числе
зародышей и высокой скорости кристаллизации происходит рост
симметричных структур в двух или трех направлениях. Эти
структуры называют лучами. Они могут быть образованы раз-
вернутыми макромолекулами или цепями в складчатой конфор-
мации. Поскольку длина лучей превосходит поперечные разме-
ры, их можно рассматривать как фибриллы. В отличие от мик-
рофибрилл* в монокристаллах в сферолитах они построены из
кристаллических и аморфных участков, которые связаны моле-
кулами, называемыми «проходными цепями» (см. рис. 1.21, г).
62
Дефектность сферолитов существенно больше по сравнению с
монокристаллами и для некоторых полимеров достигает 50—
80%- По особенностям строения сферолиты разделяют на ра-
диальные и кольцевые (см рис. 1 18, б, в). Для кольцевых сферо-
литов характерно чередование аморфных и кристаллических уча-
стков вдоль радиуса сферолита, в радиальных — аморфные уча-
стки (темное поле) образуют характерный «мальтийский крест»
(рис 1.18,6) При высокой степени кристаллизации отдельные
сферолиты соприкасаются своими поверхностями, образуя сфе-
ролитные ленты или заполняя весь объем полимера (рис.
1,18, в, е). Сферолиты невозможно выделить из полимера путем
экстракции растворителем, поскольку они соединены друг с
другом.
Если монокристаллы, как уже отмечалось, образуются при
кристаллизации из разбавленных растворов, то сферолиты мо-
гут получаться при кристаллизации из более концентрирован-
ных растворов и при кристаллизации в блоке.
Рассмотрев особенности кристаллического состояния поли-
меров и морфологию надмолекулярных структур, остановимся
на некоторых моделях, предложенных для кристаллических по-
лимеров. Первой моделью закристаллизованного полимера была
модель «бахромчатой мицеллы», или «бахромчатого кристалли-
та» (см. рис. 1.19,6), предложенная Германом, Генгроссом и
Абитцем. Согласно этой модели кристаллические полимеры со-
стоят из относительно совершенных кристаллитов размером до
нескольких десятков нанометров, которые расположены в
аморфной фазе. Эти кристаллиты связаны с аморфной матри-
цей молекулами, переходящими из одного кристаллита в другой
через аморфные области. Эти молекулы назвали «бахромой».
Такая модель объясняла одну из основных особенностей поли-
мерных кристаллов: низкую степень кристалличности и сосуще-
ствование кристаллических и аморфных областей. Однако на ос-
новании этой модели практически невозможно предусмотреть
существование монокристаллов и сферолитов.
Обращаясь к моделям полимерных моно- и поликристаллов,
можно отметить, что в них входят некоторые элементы модели
«бахромчатой мицеллы», а именно—чередование упорядочен-
ных и разупорядоченных областей Принципиальным отличием
являются представления об упаком<е макромолекул в упоря-
доченных областях и дефектах и их распределении.
В модели «складчатого кристалла», основоположниками ко-
торой считают Тилла, Келлера и Фишера, упорядоченные обла-
сти образованы макромолекулами в складчатой конформации,
а дефекты сосредоточены на торцевых гранях ламелей (см. рис.
1.19, а, б). Это объясняет возможность существования моно-
кристаллов конечных размеров, сосуществование кристалличе-
ских и аморфных, т. е. дефектных, участков, степень кристаллич-
63
ности меньшую, чем 100%, возможность образования в зависи-
мости от условий кристаллизации кристаллов различной степени
упорядоченности и, следовательно, имеющих различные темпе-
ратуры плавления, т. е. все те особенности полимерных кристал-
лов, о которых шла речь выше. Существование монокристаллов
с выпрямленными цепями не противоречит модели складчатого
кристалла. В этом случае длина складки достигает длины мак-
ромолекулы. С помощью этой модели достаточно убедительно
объясняется и строение фибрилл и сферолитов (см. рис. 1.19 и
1.21). Дефекты в сферолитах могут быть сосредоточены как
внутри лучей (микрофибрилл), так и в межлучевом простран-
стве
Если в модели складчатого кристалла можно говорить о ка-
кой-либо локализации дефектов, то модель «паракристалла>
Хоземана предлагает рассматривать любое кристаллическое об-
разование как несовершенный кристалл, т. е. паракристалл с
непрерывным распределением дефектов. Несмотря на определен-
ные преимущества этой модели при расчете степени кристаллич-
ности, ею практически невозможно воспользоваться для поли-
меров, у которых дефектность кристаллов превышает 75%.
Все рассмотренные выше надмолекулярные структуры поли-
меров, начиная с упорядоченных структур ближнего порядка
(домены, кластеры) и кончая совершенными монокристаллами,
в которых реализуется трехмерный дальний порядок, формиру-
ются в основном в условиях доминирующего влияния теплового
движения. При наложении внешних деформирующих напряже-
ний надмолекулярная структура будет изменяться и полимер
будет переходить в особое состояние — ориентированное.
1.2.3. Ориентированное состояние полимеров
Особенность полимеров, которая отмечалась выше, — резкая
анизотропия свойств в продольном и поперечном направле-
нии— характерна, в первую очередь, для полимеров в ориенти-
рованном состоянии.
Ориентированным называют состояние полимеров, при кото-
ром оси макромолекул и надмолекулярных образований преиму-
щественно располагаются вдоль осей ориентации. Ориентирован-
ные полимеры широко распространены в природе волокна хлоп-
ка, льна, шелковые нити, шерсть, сухожилия, мышечная ткань
и др. Синтетические ориентированные полимеры можно полу-
чить в процессе их синтеза, например полимеризацией в твер-
дой фазе, когда мономер существует в форме монокристалла,
полимеризацией жидкого полярного мономера в постоянном
электрическом поле или полимеризацией из газовой фазы на
ориентированной подложке.
В промышленности ориентацию полимеров проводят путем
их одноосного или двухосного растяжения — так называемой
вытяжки. Ориентационная вытяжка заключается в растяжении
при определенных условиях неориентированных полимеров.
Одноосно ориентированные полимеры, чаще всего волокна, по-
лучают растяжением образцов в одном направлении, при этом
увеличивается длина образца, а поперечные размеры уменьша-
ются. Двухосная вытяжка применяется при ориентации пленок.
Она может осуществляться в одну стадию путем одновременно-
го растяжения пленки в двух взаимно перпендикулярных на-
правлениях и в две стадии — путем растяжения пленки вначале
в одном, а затем в перпендикулярном направлении. Под дейст-
вием растягивающих сил все элементы структуры (отдельные
макромолекулы, надмолекулярные образования) ориентируют-
ся в направлении действия этих сил. При этом связи между
макромолекулами нарушаются, макромолекулы изменяют свою
конформацию — распрямляются и сближаются. Распрямление
и сближение макромолекул увеличивает межмолекулярное
взаимодействие, повышает жесткость цепи (рис. 1.22).
Ориентация надмолекулярных структур происходит двумя
путями: поворотом вдоль оси ориентирования или распадом ис-
ходных элементов надмолекулярной структуры, ориентировани-
ем образующихся при распаде элементов и отдельных макро-
молекул и формированием из них новых надмолекулярных об-
разований вдоль оси ориентации.
Особенность ориентированного состояния заключается в том,
что при ориентации полимеров разного химического строения
Рис. 1.22 Схема расположения макромолекул в неориентированном (а)
и ориентированном в направлении действия силы (б) полимерах (/, //, III,
IV — макромолекулы, 1, 2, 3, 4, ... —узлы)
5-816
65
• в
Рис. 1.23. Схема строения фиб-
рилл в аморфном (а) и кри-
сталлическом (б) полимерах
(Л — аморфная часть, Л —
кристаллическая, 1, 2 — микро-
фнбриллы)
образуется однотипная
фибриллярная структура.
Схемы строения фибрилл
в аморфных и кристалли-
ческих полимерах пред-
ставлены на рис. 1.23. В
аморфных полимерах
фибриллы сравнительно
гомогенны, а в кристал-
лических — существует
продольная гетероген-
ность, обусловленная со-
существованием кристаллических и аморфных (проходные мо-
лекулы) областей в микрофибриллах. Микрофибриллы за счет
небольшого числа межфибриллярных цепей объединены в круп-
ные фибриллярные элементы, которые и определяют структуру
ориентированного полимера.
Наиболее распространенной характеристикой степени ориен-
тации является величина cos20, где 0 — угол между осью дан-
ного участка структурного элемента и осью ориентации образ-
ца. В аморфных ориентированных полимерах ориентация ни-
когда не бывает полной и cos20 редко достигает 0,5. Это связа-
но, в первую очередь, со стерическими затруднениями для пере-
группировок и с высокой подвижностью макромолекул.
Кристаллические полимеры имеют две степени молекуляр-
ной ориентации — в кристаллитах она может быть очень высо-
кой, и cos20 достигает 0,95 и даже 1, тогда как в аморфных об-
ластях cos20 не превышает 0,6—0,7. Однако эта величина выше,
чем для полностью аморфного образца. Этот факт является
важным, поскольку объясняет большую прочность ориентиро-
ванных кристаллических структур, при учете того, что разруше-
ние последних происходит по проходным цепям, т. е. по аморф-
ным областям.
На основании рассмотренного выше строения фибрилл объ-
яснима анизотропия свойств вдоль и поперек направления ориен-
тации Наличие большого числа узлов, высокая степень ориен-
тации кристаллической и аморфном части обусловливают высо-
кие значения жесткости и прочности в направлении ориентации.
А небольшое число межфибриллярных проходных молекул при-
водит к слабой связанности микрофибрилл и как следствие это-
го к низкой прочности в поперечном направлении.
66
Возможность образования ориентированной структуры и ее
сохранения после снятия нагрузки определяется температурно-
временными факторами.
Роль температуры неоднозначна: с одной стороны, для из-
менения конформации макромолекулы должны обладать доста-
точным запасом кинетической энергии, т. е. быть достаточно по-
движными, что и достигается повышением температуры; с дру-
гой стороны, тепловое движение разрушает исходную структу-
ру, стремится дезориентировать макромолекулы и вернуть их в
исходные конформации. Однако наряду с этими процессами
идет и процесс образования новой ориентированной структуры
Вероятность сохранения новой структуры после снятия напряже-
ния определяется соотношением прочности этой структуры (оп-
ределяемой силами межмолекулярного взаимодействия, Дефект-
ностью кристаллов и др ) и интенсивностью тепловых флуктуа-
ций.
Рассмотрим два примера. Гибкие полимеры (натуральный
каучук, полибутадиен, полихлоропрен и др.) легко образуют
ориентированную структуру при растяжении, но сохранить ее
могут только под напряжением. После снятия деформирующей
силы внутреннее тепловое движение нарушает достигнутый по-
рядок и возвращает макромолекулы в исходное состояние —
конформацию свернутого клубка, т. е. kT^Uo((p) Для ориен-
тации жесткоцепных полимеров требуется большее напряжение,
но за счет сильного межмолекулярного взаимодействия между
ориентированными макромолекулами ориентированная структу-
ра может сохраниться при условии kT^i Uo(<р).
Вследствие многообразия процессов перестройки структуры
при ориентации этот процесс протекает в течение определенного
времени и, следовательно, требуется определенная скорость де-
формирования. Если скорость ориентации превышает скорости
перестройки и образования новой структуры, то более вероятен
процесс разрушения, чем ориентации. Поэтому для проведения
ориентации требуются оптимальные для каждого полимера тем-
пература и скорость вытяжки.
1.2.4. Структурная модификация
Структурная модификация — это направленное изменение
свойств (физических и механических) за счет преобразования
надмолекулярной структуры под влиянием физических воздейст-
вий при сохранении химического строения макромолекулы. Воз-
можность структурной модификации обусловлена тем, что над-
молекулярная структура полимеров является подвижной систе-
мой: в зависимости от условий одна форма может переходить в
другую Даже для таких малоподвижных систем, как графит,
вероятен переход графита в алмаз в присутствии катализаторов
Б
67
при температуре 1800—2800 К и давлении 4,9—7,8 ГПа Значи-
тельно большей подвижностью обладают надмолекулярные
структуры в органических полимерах. Так, при кристаллизации
полиэтилена из разбавленного раствора в ксилоле образуются
отдельные монокристаллы, кристаллизация из расплава этого же
полимера приводит к образованию поликристаллов — сфероли-
тов, а при кристаллизации в растянутом состоянии образуется
структура типа «шиш-кебаб».
Структурная модификация может осуществляться различ-
ными способами.
1. Воздействием внешнего механического напряжения, приво-
дящего к образованию ориентированного состояния.
2. Изменением температурно временных режимов структуро-
образования. Наиболее часто изделия подвергают термообра-
ботке, в результате которой достигается желаемая надмолеку-
лярная структура. Различают три метода обработки: закалку,
отжиг, нормализацию. Закалку проводят в теплоносителе при
быстром охлаждении (0,8—1,6 К/с). Она снижает степень кри-
сталличности, твердость, повышает эластичность материала От-
жиг проводят в теплоносителе при медленном охлаждении (ме-
нее 0,8 К/с). Он повышает степень кристалличности, прочность,
твердость. Нормализация осуществляется при медленном охлаж-
дении в воздушной среде. Она способствует снижению остаточ-
ных напряжений, широко применяется для аморфных полиме-
ров.
3. Изменением природы растворителя и режимов его уда-
ления.
4. Введением в полимер малых добавок (несколько процен-
тов) веществ, химически с ним не взаимодействующих, опреде-
ляющих морфологию надмолекулярной структуры. К ним отно-
сятся поверхностно-активные вещества, различные неорганиче-
ские или органические соединения, не растворяющиеся в поли-
мере, и др. Введение мелкодисперсных частиц нерастворимых
веществ (например, оксида цинка, технического углерода, ин-
диго и др.) уменьшает размер кристаллических структур за
счет увеличения числа зародышей кристаллизации, повышает
прочностные и деформационные показатели. Например, поли-
пропилен с крупной сферолитной структурой может быть растя-
нут на 100—150%, а с мелкой, образованной в присутствии
1% (масс.) индиго, проявляет способность к растяжению до
500% и более. При этом несколько повышается и прочность.
1.3. МЕТОДЫ ИССЛЕДОВАНИЯ СТРУКТУРЫ ПОЛИМЕРОВ
Термин «Структура полимеров:», как мы уже говорили, включает в себя
структуру самой макромолекулы и надмолекулярных образований, возни-
кающих в результате агрегации макромолекулы Поэтому методы исследова-
68
ния структуры полимеров целесообразно разделить на две группы: методы
исследования структуры макромолекулы и методы исследования надмолеку-
лярной структуры.
л
1.3.1. Исследование структуры макромолекулы
В качестве параметров, определяющих структуру макромолекул обычно ис-
пользуют химическое строение звена, его молекулярную массу, конфигура-
цию и конформацию цепи, причем очень часто одними и теми же методами
оценивают сразу химическое строение звена, природу концевых групп, кон-
фигурацию и конформацию макромолекул.
Химическое строение полимеров определяют обычно химически-
ми и физико-химическими методами, в частности спектральными.
Среди химических методов наиболее распространенным является расщеп-
ление макромолекулы на низкомолекулярные соединения, которые иденти-
фицируют различными способами, принятыми в аналитической химии Этим
методом определено строение природных полимеров, таких, как натуральный
каучук и целлюлоза.
В качестве агента, вызывающего расщепление макромолекулы на низко-
молекулярные продукты, часто используют озон. Так, при взаимодействии
натурального каучука с озоном образуются озониды, которые затем легко
распадаются с образованием стабильных продуктов. Основным продуктом
разложения натурального каучука является левулиновый альдегид и левули-
новая кислота (90%). Их образование указывает на то, что основным пов-
торяющимся звеном являются остатки изопрена, соединенные между собой
последовательно, причем конец одного звена соединяется с началом другого
(присоединение 4,1- или «хвост к голове»):
СНз СНз
I I о-»
~СН2-С = СН-СН2-СНг-С = СН-СН2~ —
1 234 1 234
НзСч о , Нз?х°х' н 0
**СН2- C*'Z^CH -СН2-СН2- С '' СН-СН2~ .н2О2 *
1 2| , |3 4 1 2| I I3 4
о4-о 0-}-°
СНз СНз
-*0 = СН-СН2-СН2-С = 0 -% Н00С-СН2-СН2-С = 0
3 4 12
левулиновый альдегид левулиновая кислота
Кроме этих продуктов при реакции каучука с озоном выделяются незна-
чительные количества уксусной кислоты и ацетальдегида (2%), муравьиной
кислоты и СО2 (1—2%), метилглиоксаля (0с4%) Образование этих продук-
тов дает информацию о природе концевых групп полимера и служит дока-
зательством наличия концевых групп типа
=СН—С1Ь; СН2=С—СН=СН—
СН3
Наличие концевых групп свидетельствует о том, что макромолекула на-
турального каучука имеет линейное строение, а преимущественное образова-
69
мне левулинового альдегида и левулиновой кислоты — о ее высокой регу-
лярности.
Рассмотренный метод позволяет не только определить химическое строе-
ние повторяющегося составного звена и концевых групп, но также оценить
конфигурацию макромолекулы. Например, присутствие в продуктах
распада натурального каучука янтарной кислоты свидетельствует о наличии
структурных изомеров, в которых звенья соединены «хвост к хвосту*:
1 234 4 3 2 f
•^сНг-с=сн-сн^-сНг-сн=с-сНг
^2°?
3 4 4 3 О-
ОСН - сн2 - СНг - нсо---ноос - сн2 - сн2 - соон
Янтарный альдегид Янтарная кислота
Спектральные методы анализа основаны на способности полимера взаи-
модействовать с полем электромагнитного излучения, избирательно поглощая
энергию па определенном его участке. При этом изменяется энергетическое
состояние макромолекулы в результате таких внутримолекулярных процессов,
как переходы электронов, колебания атомных ядер, вращение ядер, электро-
нов, атомных групп, поступательное и вращательное движения молекулы как
целого. Поглощение энергии происходит лишь в том случае, если разность
между двумя энергетическими уровнями ДЕ соответствует энергии кванта:
ДЕ—Лу=Е>—Е2,
где h— постоянная Планка, равная 6,626-10~34 Дж с; v — частота, с-1;
и Е2 — энергии молекулы в конечном (более высоком) и начальном (бо-
лее низком) состояниях.
Для исследования структуры макромолекулы наиболее распространен-
ными являются методы абсорбционной спектроскопии и ядерного магнитного
резонанса.
Абсорбционная спектроскопия охватывает ультрафиолетовую
УФ (0,1— 0,4 мкм), видимую ВИ (0,4—0,8 мкм) и инфракрасную ИК (0,8—
1000 мкм) области излучения.
При пропускании пучка монохроматического излучения через образец
разные группы молекулы поглощают его при различных длинах волн. На
кривой зависимости интенсивности пропускания от длины волны появляется
серия полос с максимумами и минимумами пропускания. Такая кривая назы-
вается спектром пропускания нлн спектром поглощения. Спектр обычно ха-
рактеризуется следующими показателями: положением пика поглощения по
длине волны X или частоте v либо по волновому числу vz в точке макси-
мального поглощения Амане» VMaxc, v'mgkc (Vz — 1/A — v/Vc», ГДе — СКОРОСТЬ
света), интенсивностью полосы поглощения в % пропускания света Т или
в единицах оптической плотности D—lg (То/Т), где TQ—100%-е пропускание,
шириной полосы поглощения АН.
70
Концентрация характеристических групп в молекуле, вызывающих погло-
щение в той или иной области, определяется, как правило, по уравнению
Ламберта — Бугера — Бера
JD=ec/CJJ,
(129)
где в — коэффициент поглощения; С — концентрация групп, вызывающих
поглощение; /Сд— толщина слоя образца
УФ и ВИ спектроскопия При поглощение макромолекулой УФ
или ВИ света определенной энергии валентный электрон переходит со связан-
ной орбитали (л или о) на промежуточную, так называемую разрыхляющую»
характеризующуюся более высокой энергией (лр, ор). Кроме того, возможен
переход несвязанных электронов внешней орбитали (лэ) иа разрыхляю-
щую лр или ср орбиталь
УФ спектроскопия применяется для качественной и количественной оцен-
ки химического строения полимеров, содержащих группы, поглощающие
электромагнитное излучение в УФ и ВИ областях спектра Такие группы
называют хромофорами; типичными хромофорами являются:
?*маке» нм
175; 185
175; 195; 223
160; 185; 280
217
184; 200; 255
Кроме хромофоров полосы поглощения дают функциональные группы,
которые, вступая в сопряжение с хромофором за счет пеподеленной пары
электронов, образуют новый хромофор. Эти группы называют ауксохромами:
^*каке’ им
^*накс’ иМ
—N=O
—NO2
204
300,665
210,271
/S—*0
^C=N-
-C=N
—О—N=O
210
190
>160
230
По спектрам в УФ и ВИ областях можно определить химическое строе-
ние полимера, например наличие двойных (простых или сопряженных) или
тройных углерод-углеродных связей, ароматических колец, кислород-, азот-
или серусодержащих и других группировок, для которых характерны л—
—ор и л»—лр переходы. Оценить же конфигурацию и конформацию макро-
молекулы этим методом практически невозможно.
Следует обратить внимание на довольно значительную ширину полос
поглощения в УФ и ВИ спектре. Это связано с тем, что одновременно с пе-
реходом валентных электронов происходит изменение колебательных и вра-
щательных уровней; кроме того, имеет место наложение полос поглощения,
характерных для различных хромофоров. Это затрудняет интерпретацию
спектров. Химическое строение полимера в большинстве случаев определяют
по атласам УФ и ВИ спектров для полимеров известного строения или по
-спектрам модельных образцов.
Ввиду высокого коэффициента поглощения определяемых групп в УФ
спектрах толщина слоя полимера должна быть невысокой (несколько микро-
метров). Это затрудняет приготовление образцов для съемки. Обычно нс-
71
пользуют растворы полимеров в растворителях, прозрачных в исследуемой
области.
И К спектроскопия охватывает интервал частот 12500—10 см"’,
при этом область частот 650—10 см-1 называют дальной инфракрасной, об-
ласть 4000—12500 см-1— ближней, а область 650—4000 см-1 — просто ин-
фракрасной. Если в УФ и ВИ спектроскопии принято оперировать понятием
«длина волны», то в ИК спектроскопии обычно используют понятие «волно-
вое число». Поглощение в ИК области обусловлено колебаниями атомов,
сопровождающимися изменением межатомных расстояний (валентные колеба-
ния) и углов между связями (деформационные колебания). Эти изменения
обычно не затрагивают электронного состояния молекулы, а сопровождаются
изменением ее дипольного момента, которое зависит от симметрии системы-
Например, если молекула гомоядерная (О^ СЬ и др.), то центры положи-
тельных и отрицательных зарядов при колебаниях совпадают, и дипольный
момент во всех случаях будет равен нулю. Такие колебания не сопровожда-
ются поглощением ИК излучения, и их называют неактивными. При колеба-
ниях в молекулах, состоящих из разных атомов, центры положительных и
отрицательных зарядов не совпадают н в результате этого происходит изме-
нение дипольного момента. Эти колебания называют активными. Они и при-
водят к появлению полос поглощения в спектре. Изменение дипольного мо-
мента молекулы при колебаниях характерно н для молекул, не имеющих
постоянного дипольного момента (например, для тетрахлорида углерода CCW-
Каждая молекула характеризуется колебательной энергией Емл:
£xo«-Av'ae,(i+l/2). (1.30)
где h— постоянная Планка; Vх— волновое число; i—квантовое число, при-
нимающее целые значения: 0: 1; 2 и т. д.
При комнатной температуре большинство молекул находится в состоя-
нии с 1=0; при этом Eko3=iMvzUcb. Основной переход происходит
при поглощении энергии падающего на молекулу излучения, при котором
квантовое число изменяется от 0 до 1. Изменение колебательной энергии при
поглощении излучения определяется соотношением ДЕКОя=Лчг,усв. Отсюда сле-
дует, что положение полос поглощения по шкале волновых чисел зависит от
энергии, необходимой для перехода из состояния с i=0 в состояние с
и зависящей от строения молекулы, ее полярности, массы н других факторов.
Рассмотрим для примера области полос поглощения, обусловленные ва-
лентными колебаниями группы С—X, где Х—Н, О, С! Вероятность измене-
ния дипольного момента при поглощении И К света возрастает при переходе
от II к С1; в этом же направлении уменьшается частота поглощения, обус-
ловленная валентными колебаниями связи С—X. Так, для С—Н обласгь
поглощения расположена при 2970—2890 см*-1, для С—О—сдвигается 1°
1270—1060 см-1. а полосы поглощения групп С—С1 проявляются уже при
800-600 см-1.
Для приближенном оценки влияния массы атомов иа частоту колебаний
можно использовать соотношение, выведенное для двухатомной молекулы
v = ——У/7й, (ья)
2я
где f — силовая постоянная, характеризующая упругие силы, возникающее
при отклонении атомов от положения равновесия; —по-
веденная масса атомов.
Влияние массы наиболее ярко видно на примере группы С—Н при т-
мене водорода на дейтерий. Если у'сн^ЗООО см-1, v'cd составляет 2200 см
Силовая постоянная f возрастает с увеличением кратности связей. Поэтому
валентные колебания связи С—N имеют волновое число «1300 см"1, С=
= N—1640—1690 см-1, a C«N-2250 см'1. .
72
Число полос поглощения в спектре зависит от числа атомов в молекуле.
Для многоатомной молекулы колебательное движение раскладывают на не-
сколько составляющих, каждую из которых можно рассматривать независи-
мо от остальных. Эти компоненты называют нормальными колебаниями
системы. Для молекулы нз Лг атомов характерно 3N степеней свободы, так
как положение каждого атома по отношению к выбранной системе опреде-
ляется тремя координатами. Для характеристики перемещения и вращения
молекулы как единого целого требуется 3+3=6 координат, поэтому для ха-
рактеристики внутреннего движения атомов остается 3/V — 6 степеней свобо-
ды. Число полос поглощения таких молекул также будет составлять 3N —- 6.
На первый взгляд кажется, что спектр полимера должен быть сложным и
содержать большое число полос поглощения. Однако если учесть, что повто-
ряющиеся единицы колеблются в одной фазе, а полимер состоит в основ-
ном из повторяющихся звеньев, то ясно, что спектр полимера будет доволь-
но простым. Если в звене содержится N' атомов, то число колебаний будет
равно 3N' за вычетом 3 колебаний, связанных с поступательным движением
цепи, и одного колебания за счет вращения вокруг осн цепи, т. е.-ЗА" — 4.
Это соотношение справедливо для линейной цепи бесконечной длины. Однако
на практике это наблюдается редко, поскольку кроме основных полос в-
спектре появляются обертоны (кратные значению основной частоты 2v, 3v
и т. д.) и комбинационные тоны (сумма двух колебаний). Это приводит
к увеличению числа полос поглощения. Спектр усложняется также и за счет
колебаний концевых групп.
Однако число полос может и уменьшиться по ряду причин, к которым
относятся: попадание основных частот за пределы области 650—4000 см”1*
наиболее характерной для большинства спектрофотометров; слишком слабые
для наблюдения основные частоты; расположение основных частот близко
друг от друга, что приводит к их слиянию; наличие «вырожденной» поло-
сы от нескольких поглощений одной и той же частоты, но разных знаков
в молекулах с высокой симметрией, возможность проявления некоторых
колебаний из-за отсутствия изменений дипольного момента. В качестве при-
мера рассмотрим спектр линейного полиэтилена. Число колебаний повторяю-
щегося составного звена —СН2—СН2— равно 14, но только пять активны
в инфракрасной области: полосы валентных колебаний групп СН2 при 2851
и 2919 см-1, полосы при 1467 и 1473 см"1, связанные с изменением валент-
ного угла между связями СН в группе СН2, и полосы маятниковых колеба-
ний той же группы (деформационные) в виде дублета прн 720 и 731 см”1.
Таким образом, метод ИК спектроскопии можно с успехом применять
для идентификации химического строения повторяющегося составного звена
полимера. Наиболее достоверная информация может быть получена для вы
сокомолекулярных образцов регулярной структуры с линейной конфигурацией
цепи, когда вклад концевых групп, аномальных звеньев, точек разветвления
и нерегулярности других типов минимален. Однако именно усложнение
спектра за счет влияния концевых групп, составных звеньев другой приро-
ды позволяет определить строение, а в ряде случаев н содержание этих
групп
Качественное определение химического строения полимера (повторяюще-
гося звена» концевых групп, других аномальных звеньев) включает приготов-
ление образцов, определение спектра и его идентификацию.
Образец для съемки может быть получен различными способами рас-
творением в растворителе который не имеет собственных полос поглощения
в исследуемой области; в виде пленки, полученной прессованием или отливом
раствора полимера на подложку; в виде суспензии в иммерсионной жид-
кости; в виде таблеток, прессованных с бромидом калия, и др. Выбор спосо-
ба зависит от природы полимера, которая определяет его растворимость*
способность к пленкообразованию и т. д.
Для определения строения полимера редко проводят полный теоретиче-
ский расчет колебательного спектра, чаще используют метод характеристиче-
73
Таблица 16 Характеристические частоты полимеров (с — сильная полоса,
ос — очень сильная, ср — средней интенсивности, сл — слабая, ш — широкая,
уз — узкая)
Полимер Волновое число, см—1 Интен- сив- ность Шири- на Группа
цис-Полнбутадиен 735—740 с ш —С На—СН 2—
Полибутадиен-1,2 912 С У3 сн2=сн—
ЯЩ>1,4-Полинзопрен 1380 ср У3 —сн,
840 ср ш СНз —с=сн—
транс- 1,4-Полихлоро- 1662 с У3 —СС1=СН
прей Статистический сополи- 1600, 1500 ср У3
мер бутадиена со стиро- лом 912 с У3 сн2=сн—
Статистический сополи- 975 2230 с с У3 У3 —СН=СН-7раяс-1,4 —СН2—СН—
мер бутадиена с нитри- лом акриловой кислоты 912 с У3 An сн2=сн—
975 с У3 —СН=СН-грвнс-1,4
Сложные полиэфиры 1740 с У3 /°— —С=О
Полиизоцианаты 2275 с У3 —N—С—
Полисилоксаны 900-700 с ш О 1 1 —Si—С—
1076—1020 ОС ш 1 1 1 —Si—О— 1
ских частот. Предварительно на модельных образцах известной структуры
определяют ИК спектр, выявляют полосы поглощения, характерные для той
или иной группы атомов, и составляют специальные таблицы, в которых ука-
зывают тип группы, волновое число, характеристику интенсивности (сильная,
средняя, слабая) и ширину полосы. Пользуясь этими таблицами, можно оп-
ределить химическое строение составных звеньев полимера. В табл. 16 при-
ведены некоторые характеристические частоты для ряда полимеров.
При количественном анализе для учета неселективного поглощения, об-
условленного рассеянием света, и учета перекрывания соседних полос погло-
щения используют метод базовой линии, отсекающей неселективное поглоще-
ние. Способы ее построения приведены в монографиях и руководствах по ИК
спектроскопии.
Для проведения сравнительных количественных анализов необходимо
проводить испытания при одинаковой толщине образца. Подобрать такие
образцы довольно трудно, особенно если используется образец в виде плен-
ки, суспензии, таблеток. Поэтому для исключения влияния толщины образца
74
применяют метод внутреннего стандарта. В качестве последнего может быть
использовано вещество, имеющее легко определяемую полосу поглощения.
В ряде случаев в качестве полосы внутреннего эталона может быть исполь-
зована полоса, присущая самому полимеру, которая также легко определима,,
не перекрывается другими полосами и не изменяется в процессе исследова-
ния. В этом случае концентрацию той или иной группы определяют по отно-
шению к оптической плотности полосы внутреннего эталона.
Несомненным достоинством И К спектроскопии является возможность
•определения по ИК спектрам не только химического строения звеньев, ио и
конфигурации и конформации макромолекул полимера на разных уровнях.
От конфигурации звена зависят групповые характеристические частоты.
В качестве примера можно привести некоторые непредельные полимеры:
в полибутадиене звенья в положении транс-1,4- идентифицируют по полосе
967—975 см“’, а для конфигурации qwc-1,4 характерна полоса поглощения
при 730—740 см-1. Блнжиий и дальний конфигурационный порядок в рас-
положении звеньев, а именно структурная и пространственная изомерия так-
же может быть определена по ИК спектрам. Так, для анализа состава со-
полимера этилена с пропиленом используют полосы деформационных коле-
баний (маятниковых) групп СН2 при 720 см“* и группу полос пропилена при
$70 см-1. Положение полосы СН2 в спектре зависит от длины блока
(—СН2—)п. которая определяется соотношением структурных изомеров <го-
лова к голове*, «хвост к хвосту» и «голова к хвосту». Ниже показано, как
зависит положение полосы маятниковых колебаний группы СН2 от длины
блоков сополимера этилена и пропилена
л 1 2 3 4 5
Волновое число, см”1 815 752 735 — 723
Пространственная изомерия (цис-, транс-, изо- и снибно-нзомеры) также
может быть определена по ИК спектрам по стереочувствительиым полосам.
Например, в полипропилене такими полосами являются полосы, связанные
с валентными колебаниями группы СН3 или с деформационными колебаниями
группы СН2. Ниже приведены некоторые стереочувствительные полосы для
снидио- (А), изо- (Б) и атактического (В) полипропилена:
Валентные колебания СНз 2886
Деформационные колеба- * 811
ния СН2
Волновое число, см-1
Б В
2878 —
807 840
Для оценки конфигурации макромолекулы метод ИК спектроскопии
имеет ограниченное применение, поскольку в большинстве случаев структура
точек разветвления мало отличается от структуры групп в основной цепи.
Однако необходимо отметить, что в полиэтилене количественно определяют
степень разветвленности по полосе 1378 см-1 с помощью компенсационного
метода, заключающегося в сравнении разветвленного и линейного поли-
этилена.
Конформацию звеньев определяют с помощью так называемых
конформационных полос, параметры которых зависят только от конформа-
ции звена Типичными примерами конформационных полос являются полосы
при 1450 (траяс-форма) и 1435 см-1 (еош-форма) в спектре полибутадиена.
Полиэтилентерефталат также имеет в спектре полосы, характерные для
транс-конформации группы —О—СН2—СН2—О— при 1473, 1343, 1120, 973*
и 845 см-1, и для гош-кон формации этой группы при 1455, 1370, 1100, 1043
к 898 см-1.
Ближний и дальний конформационный порядок в макромолекуле оцени-
вают по полосам регулярности. Появление этих полос обусловлено следую-
75
щими правилами. В ИК спектре проявляются колебания, при которых сосед-
ние звенья колеблются в одной фазе или при сдвиге фазы на угол закручи-
вания спирали. Наиболее отчетливо конформационные полосы проявляются
в кристаллических полимерах, где обеспечивается дальний порядок (поли-
пропилене и др.). В спектре расплавов (аморфное состояние) большинство
полос пропадает, и на их месте появляются слабые полосы, которые связы-
вают с наличием спиральной конформации ближнего порядка. Например,
в НК спектре полипропилена оптическая плотность полосы при 998 см-1
пропорциональна доле изотактической спирали, содержащей более 12 моно-
мерных звеньев, а полоса при 973 см“‘ характеризует блоки, состоящие из 4
и более звеньев. Конформационные полосы были обнаружены для многих
полимеров: поливинилхлорида (638, 603 см"1), полистирола (1364, 1312, 920»
898 см-’), полиэтилеитерефталата (1387, 1100 см^1) и др.
В последнее время для исследования структуры полимеров применяют
спектроскопию внутреннего отражения Суть метода заклю-
чается в регистрации излучения, полностью отраженного от границы двух
сред, различающихся по оптической плотности. Образец в виде пленки, пла-
стинки приводят в контакт с призмой или пластинкой, изготовленными из
вещества, показатель преломления которого Л| больше, чем у исследуемого
полимера (л2). Падающий свет направляется на покрывающее образец поли-
мера вещество (пластинку) под определенным углом 0 и после попадания
в глубь образца отражается от призмы. При этом образец полимера селек-
тивно поглощает электромагнитные излучения тех или иных частот в зависи-
мости от своей структуры, ослабленный пучок света отражается и измеря-
ется его интенсивность.
При условии 0>0Кр, Окр происходит полное отражение. Путем
многократного прохождения света поглощение можно усилить. Поэтому этот
метод получил название многократного нарушенного полного внутреннего
отражения (МНПВО). Метод МНПВО применяется в УФ, ВИ и ИК обла-
стях. Пластинки для внутреннего отражения изготавливают из материалов
с высоким показателем преломления (германий — для ИК области, титанаты
бария и стронция—для УФ области).
Метод МНПВО используют для исследования полимеров, препарирование
которых сложно. К ним относятся сшитые полимеры, полимеры» наполнен-
ные непрозрачными наполнителями, биологические препараты, полимерные
покрытия на подложках» поглощающих свет, и др.
Несмотря на большую информативность метода ИК спектроскопии, в ря-
де случаев результаты анализа являются неоднозначными по разным причи-
нам, например вследствие перекрывания полос поглощения, характерных для
различных групп, высокой поглощающей способности и др. Поэтому для од-
нозначной характеристики химического строения цепи полимера кроме метода
ПК спектроскопии привлекают метод ультрафиолетовой и видимой спектро-
скопии. а также метод ядерного магнитного резонанса
Ядерный магнитный резонанс (ЯМР). Этот метод основан
на способности образцов, помещенных во внешнее магнитное поле, погло-
-щать электромагнитное излучение в области радиочастот (1—500 МГц). Кри-
вая зависимости интенсивности поглощения радиочастот от напряженности
внешнего магнитного поля дает спектр ЯМР.
Метод ЯМР фиксирует переходы между энергетическими уровнями маг-
нитных ядер во внешнем магнитном поле. Поглощают электромагнитное из-
лучение лишь те ядра» у которых спиновое квантовое число i отлично от ну-
ля. К таким ядрам относятся. *Н, ,$F, 31Р, 13С» ,SN со спином i—Vj, SSC1„
имеющий спин 3/2, и др. Их называют магнитными; они имеют магнитный
момент ц, который определяется соотношением
= (1.32>
где 7 — гиромагнитное отношение, постоянное для данного атомного ядра.
76
Рассмотрим поведение ядра со спином ’А, например *Н. В магнитном
поле напряженностью Но ядра с магнитным моментом р. располагаются на
двух уровнях с энергиями +|лЯ> и —цНо» что соответствует двум ориентаци-
ям* магнитного момента: ориентации, совпадающей с направлением прило-
женного магнитного поля (параллельная ориентация, нижний энергетический
уровень), и ориентации, противоположной направлению приложенного поля
(антнпараллельная ориентация, верхний энергетический уровень). Разность
энергий уровней составляет 2цН0- При воздействии на ядро электромагнитно-
го излучения с частотой v0 протон в параллельной ориентации поглощает
энергию AE=hv0 и может перейти на верхний энергетический уровень с аи-
типараллельной ориентацией (зеемановское расщепление) при условии ра-
венства энергии между уровнями и энергии поглощаемого излучения. Отсю-
да следует, что резонанс будет наблюдаться при условии /iv=2ptf0. В ЯМР
спектрометрах обычно используются постоянное магнитное поле с Но=
= 14 000—71000 Гс, и по этому уравнению можно рассчитать v0 для ядер
различной природы. Например, для ядра при Но—14 000 Гс частота элект-
ромагнитного излучения, при которой имеет место резонанс, составляет
60 МГц, а для ядра ,3С v0=15,l МГц.
Одной из основных характеристик спектра ЯМР является положение
полосы поглощения по оси частот. Радиочастотное поле резонансной часто-
ты vo возбуждает переходы спинов между двумя уровнями с выделением
или поглощением энергии, причем вероятность обоих переходов одинакова.
При выравнивании заселенностей обоих уровней поглощение энергии прекра-
щается и происходит уменьшение резонансного сигнала (т. е. насыщаемость
сигнала ЯМР)- Однако при обычных условиях заселенность обоих уровней
не бывает одинаковой из-за самопроизвольного перехода ядер с верхнего
уровня с большей энергией на нижний уровень с меньшей энергией. Это
явление называется релаксацией В результате заселенность нижнего уровня
выше, и поэтому в целом образец поглощает энергию Поглощенная энергия
определяет вторую характеристику ЯМР спектра — интенсивность сигнала
линии поглощения. Как правило, интенсивность сигнала А пропорциональна
числу ядер N* и квадрату резонансной частоты v0 и уменьшается с ростом .
температуры:
A~Nn(hv0)*/kT. (1.33>
Потеря энергии происходит в результате спин-решет очной релаксации^
когда энергия Е передается на соседние атомы той же молекулы или на
соседние атомы в молекуле растворителя, и вследствие спмн-спиновой релак-
сации, в процессе которой энергия переносится на соседние ядра За время
релаксации тя принимают время, за которое разность заселенностей спино-
вых уровней уменьшится в е раз. Существует зависимость между тя и ши-
риной линии сигнала Av (принцип неопределенности): Avrn—const. Обычно
время спин-спиновой релаксации тЯ2 намного меньше времени спин-решеточ-
ной тЯ|. Поэтому можно приближенно считать Av—1/ta2- Таким образом, треть-
ей характеристикой ЯМР спектра является ширина сигнала.
В твердых телах, в том числе и в полимерах, взаимное распо-
ложение ядер практически не изменяется во времени, обмен энергией проис-
ходит очень быстро и время релаксации очень мало (например, время спин-
спиновой релаксации TR2e10“*—10~5 с). Поэтому согласно принципу неопре-
деленности ширина линии в спектре ЯМР твердых полимеров большая
(104 Гц). Этот метод называют методом ЯМР широких линий. В растворах
полимеров, в которых молекулы совершают интенсивное тепловое движение,,
время релаксации тЯ2 увеличивается и составляет уже несколько секунд, что-
приводит к появлению в спектре очень узких линий поглощения (доли Гц)-
-Этот метод называют ЯМР высокого разрешения
* В общем случае число ориентаций равно И4-1-
77
Для характеристики химического строения полимер ов используют ЯМР
высокого разрешения иа ядрах 1Н, 13С, ,fiF, МС1. Н.аиболее широко изу-
чен метод ЯМР на ядре ‘Н—так называемый протонный магнитный резо-
нанс (ПМР). В последнее время успешно развивается метод ЯМР на 13С
и других ядрах. Интенсивность сигнала ,3С намного м еньше интенсивности
протона, но селективность этого метода выше Для получения спектра ЯМР
полимер растворяют в растворителе, не содержащем ядер, резонанс которых
изучается, чтобы на спектр полимера не накладывался «спектр растворителя.
При интерпретации спектров ЯМР высокого разрешения определяют че-
тыре характеристики: положение полосы поглощения, ее интенсивность, ши-
рину и характеристику расщепления линии (последний показатель характе-
рен только для спектров ЯМР высокого разрешения).
Как уже указывалось, положение линии в спектре ЯМР определяется
типом ядра. Казалось бы, что ядра одного вида должны характеризоваться
одной линией поглощения. Однако в реальных атомах ядра находятся в ок-
ружении электронов, которые в поле постоянного магнита создают вторичное
магнитное поле напряженностью Н', направленное против внешнего поля Но.
Поэтому атомные ядра будут находиться в магнитном поле с напряжен-
ностью меньшей Н иа величину Н'^Н о,
Яэф“Яо-(1-а>), (134)
где Оэ — константа экранирования, зависящая от электронного окружения
ядра чем больше электронов расположено вблизи ядра, тем больше а,.
При определенной частоте резонанс ядра в атоме наступает при больших
полях (на величину ЯО0э)» чем резонанс ядра, полностью лишенного элект-
ронной оболочки. Разность в положении линий поглощения Исследуемого
протона и ядра без электронной оболочки называют химическим сдвигом
(х. с.). На практике для сравнения используют обычно стандартные веще-
ства, относительно которых и измеряют химический сдвиг, поскольку не
существует ядер без электронов. В качестве эталонов в ЯМР спектрах 'Н
и ,3С используют тетраметилсилан (ТМС) или гексаметилдисилаи (ГМДС).
Эталон дает линию высокой интенсивности, расположенную в области
более сильных полей, чем сигналы большинства других протонов. Выражают
химический сдвиг в безразмерных единицах — миллионных долях (м.д.) от
напряженности приложенного магнитного поля или рабочей частоты v<>
спектрометра:
х.с.= (Av/vo) • 10е— (АЯ/Яо) • 10е, (135)
где Av или АЯ — расстояние от интересующей нас линии до линии эталона.
В ЯМР спектроскопии ‘Н и ,3С наиболее распространена шкала б.
В этой шкале линия эталона ТМС принята за нуль, а ясе сдвиги располо-
женные в более слабых полях, имеют положительный знак, Таким образом»
увеличение б соответствует уменьшению константы экранирования
Различия в электронном окружении ‘Н и 13С приводят к тому, что для
протонного ядерного резонанса линии наблюдаются в интервале ~20 м д.,
а в случае резонанса на |3С этот интервал увеличивается до 400 м д. Это
обусловливает ббльшую информативность метода ЯМР иа ядрах |3С.
В табл. 1.7 приведены значения химического сдвига для некоторых функ-
циональных групп. Как видно из таблицы, для одной и той же группы ато-
мов характерна не одна линия, а некоторый интервал химического сдвига-
Это обусловлено влиянием природы заместителей, их расположения в про-
странстве. образованием водородных связей н другими факторами, в той или
иной мере изменяющими константу экранирования. Заместители в линейных
и разветвленных молекулах оказывают разное влияние. Чувствительность хи-
мических сдвигов ,3С к структурным изменениям выше, чем в ЯМР спект-
рах JH, примерно на порядок.
78
Табл^^ 1-7. Химические сдвиги некоторых групп при ЯМР на ядрах 'Н
и
Про/ов'«14>жащне группы С, м, д. Углеродсодержащие группы с, ц. д-
_СИ» 0,8—4,3 Si(CH3)4 0 01^2 1,1—4,7 —С— (алканы) 5—55 I 1 Cf4— 1,4—5,3 ==С— (олефины) ЮО—150: =С1^ ^лефины) 4,0—6,5 =С— 65—90 eQP 2,3—2,9 =С— (ароматические 110—150 соединения) t=Cl^ (ароматические 6,5—8,3 —С=С=С 70—95(С, С) соед1,нен*я) 12 3 12 200—215(0 3 /° \ —СТ альдегиды) 9—10,5 __c==N 30—70 / \ (насыщен- —С—0 иые соедн- 40—85 / нения) —С—S 10—70 —01* (спирты) 2,0—4,5 ^C==N насыщенные соединения 145—165 ненасыщенные соедине- ПО—120 ння 01* (денолы) 4,5—9,0 ^с=О (альдегиды, ке- 175—225 тоны, кислоты, слож- 155—185 ные эфиры) —Of* (вдслоты) 9,0—20 ^С=О (ангидриды) 150—165 (амины) 1,0—2,4 ^Nf* (шиды) 5,0—8,0 C=S (тиоиы) 220—235 —Si1 (сульфиды) 1,2—4,7 —C=N ПО—125
7»
Интенсивность линии в спектре ЯМР характеризует энергию, поглощен-
ною образцом при резонансе, и определяется площадью под кривой лнннн
поглощения Она пропорциональна числу ядер и позволяет оценить их отно-
сительное содержание.
При достаточном разрешении линия поглощения может быть в виде
нескольких компонент, т. е. происходит ее расщепление. Это связано с влия-
нием соседних ядер, которое проявляется через электроны химических связей.
Число пиков (мультнплетность сигнала) зависит от числа ядер соседних
групп и равно п+1, где п — число соседей. Расстояние между линиями каж-
дого дуплета одинаково и называется константой спнн-спинового взаимодей-
ствия J (измеряется в Гц). Соотношение интенсивностей линий при п-1
равно 1 : 1, при п=2 линия расщепляется на триплет при соотношении ин-
тенсивностей 1:2 и т.д. Константа спин-спииового взаимодействия очень
чувствительна к структуре взаимодействующих ядер, зависит от геометрии
связей между ядрами, длины валентных углов и т. д.
Техника ЯМР, так же как и ИК и УФ спектроскопии, включает приго-
товление образца, получение спектра и его интерпретацию. Для того чтобы
получить достаточно узкие сигналы с хорошим разрешением, образец поли-
мера обычно готовят в виде иевязкого раствора невысокой концентрации
л 10%) в растворителях, не содержащих анализируемых ядер. Например,
в ЯМР спектроскопии на ядрах Н паи лучшими растворителями считают
тетрахлорид углерода и сероуглерод. Некоторые полимеры (разветвленные*
сшитые) не могут быть переведены в раствор. Поэтому для их анализа сна-
чала разрушают химическую сетку различными способами (пиролиз, кипяче-
ние с определенными реагентами и т. д.), а затем анализируют продукты
пиролиза. Интерпретацию спектров ЯМР высокого разрешения проводят е
помощью корреляционных таблиц и каталогов спектров ЯМР известных по-
лимеров.
Таким образом, методы ЯМР, ИК и УФ спектроскопии позволяют оценить
химическое строение повторяющихся звеньев н конфигурацию звена, ближ-
ний и дальний конфигурационный порядок в присоединении звеньев, раз-
ветвленность макромолекулы, конформацию звеньев и макромолекулы
в целом. _
Средиечисловую молекулярную массу эксперимен-
тально определяют методами, чувствительными к изменению числа молекул:
мембранной осмометрии, эбулиоскопии, криоскопии и по содержанию конце-
вых групп.
Мембранная осмометрия. В основе этого метода лежит явление осмоса.
Если растворитель отделить полупроницаемой мембраной от раствора поли-
мера в том же растворителе, то химические потенциалы растворителя в обеих
ячейках не будут равны между собой. Это обусловливает перенос раствори-
теля через мембрану в раствор. Разность давлений по обе стороны от мем-
браны в состоянии равновесия называют осмотическим давлением л. Для
разбавленных растворов полимеров в 0-растворителе
л/с=Я77>„, (1.36)
При использовании любых других растворителей среднечисловую моле-
кулярную массу можно рассчитать по данным измерения осмотического дав-
ления растворов полимера различных концентраций, используя выражение
л/с=/?Т(1/Л1п+Л2с+ЛзСг+ (L37)
где л/с — приведенное осмотическое давление; Л2, Л3— внриальные коэффи-
циенты
Поскольку третий член выражения пренебрежимо мал по сравнению
•с другими, то выражение принимает вид
л/с-/?Г/Мя+Л2с. (1.38)
«0
лоивая завис, мости л/е от с представляет собой прямую линию, отсе-
кающую на осн ординат отрезок, равный RT/SIn, с тангенсом угла наклона,
равным А?. Второй вирнальный коэффициент Л2 определяется свойствами
полимера, растворителя н их взаимодействием:
Л2=(Р</Р2"№)(1/2--х), (1.39)
где ps — плотность растворителя; рп — плотность полимера; М* — молекуляр-
ная масса растворителя; х— параметр взаимодействия полимер — раство-
ритель.
Интервал молекулярных масс, определяемых этим методом, составляет
106.
Криоскопия основана на различии между температурами замерзания
раствора н чистого растворителя Изменение температуры замерзания для
разбавленных растворов ДТз—КсЛ, где Кс — криоскопическая константа для
данного растворителя, характеризующая снижение температуры замерзания
(или плавления), вызываемое 1 моль растворенного полимера; л —число
моль Криоскопическая константа равна
(1.40)
где Те и М*— температура замерзания и молекулярная масса растворителя;
рж и Нпз — плотность и теплота плавления растворителя
Среднечисловую молекулярную массу 57« определяют по уравнению
ДТпя/с=Кс(1/ЛГл+Л2с+Лас«+...)» (1.41)
где ДТпл — снижение температуры плавления; с—концентрация раствора;
АГил/с — приведенное снижение температуры плавления.
Поскольку величина Язе2 намного меньше других членов уравнений, ею
можно пренебречь; тогда
ДЛ/с«КД1/Мя+Д£с). (1.42)
В координатах &Т3/с—с эта зависимость представляет собой прямую ли-
нию, отсекающую на оси ординат отрезок, равный Кс!Мп. а тангенс угла на-
клона характеризует А?:
А 2=/?Р/рпДЯп, (1/2 - х) • (1 43)
Криоскопическим методом можно определить до 5-Ю4.
Эбулиоскопия. В основе метода лежит различие между температурами
кипения раствора н чистого растворителя. Изменение температуры кипения
ДГк связано с числом моль п вещества в разбавленном растворе соотноше-
нием ДТк=К«п, где Кк — эбулиоскопическая константа, характеризующая
повышение температуры кипения, вызываемое 1 моль растворенного веще-
ства
KK = RT2&M;/pAH^ (1.44)
где АЯМ—^теплота испарения растворителя.
Расчет А/n проводят по эбулиоскопическим данным, полученным при раз-
личных концентрациях, по соотношению
д7’к/с-Кх(1/.ип+Д2с+Д3с2+...). (1.45)
Мп можно рассчитать графическим методом по зависимости ДТк/с от с
прн с—^0 &Tjc=KK/Jlln. Второй внриальный коэффициент можно опре-
делить по уравнению
A2=RT*t/ptt&HM(l/2 — х)
(1Л6)
Эбулиоскопия применима для определения 100—170 тыс.
6-316
81
Определение ЛГЯ по числу концевых групп. Этот метод используют в ос-
новном для линейных полимеров. Наиболее распространенным способом ко-
личественного определения концевых групп является спектроскопия (ИК
и ЯМР). С помощью ИК спектроскопии определяют природу концевых
групп по характеристическим полосам поглощения и по уравнению Ламбер-
та— Бугера — Бера (129) рассчитывают их концентрацию. Для линейных
молекул среднечисловая молекулярная масса обратно пропорциональна чис-
лу концевых групп в молекуле:
Мп = , (1.47)
.де nt — число молекул с массой ЛЬ; Ц — число звеньев с массой
L/ — число концевых групп i й молекулы; если функциональные группы рас-
положены с обоих концов макромолекулы, то Lt -2, а если полимер содер-
жит только одну определяемую функциональную группу, то L/=l.
Величину Jif„ можно найти по уравнению
(148)
где п — число групп в макромолекуле полимера, которые могут быть опреде-
лены; т — концентрации концевых групп.
При определении Мя по спектрам ЯМР используют так называемый
метод стандарта. К иавеске полимера g неизвестной молекулярной массы
добавляют определенное количество go вещества известной молекулярной
массы Л1о и по данным ЯМР спектров рассчитывают Мп полимера по урав-
нению
AJnQ А/п
£оЛ,о S/Mn
(1.49)
где Ло и Л — интегральные интенсивности стандартного и исследуемого веще-
ства; п0 и п — число ядер, ответственных за линии в спектрах стандартного
и исследуемого веществ.
Среднемассовую молекулярную массу можно определить
экспериментально методами, которые чувствительны к массе частиц, напри-
мер светорассеянием или седиментационным анализом.
Светорассеяние В результате теплового движения макромолекул в лю-
бой среде всегда существуют флуктуации по концентрации и плотности, ко-
торые являются центрами рассеяния света. Рассеяние света молекулами оп-
ределяется уравнением
7//0 = Г-^,
(1.50)
где I н I — интенсивности падающего и прошедшего света; тм — мутность
образца, I — толщина образца.
Рассеивающая способность вещества оценивается коэффициентом рассея-
ния R:
R=Ir*/IQV, (1.51).
где г — расстояние от рассеивающего объекта до наблюдателя; V—рассеи-
вающий объем
Угол, под которым рассеивается свет, называется углом рассеяния 6.
Обычно коэффициент рассеяния обозначается с указанием угла рассеяния
(/?е), например
Если размеры рассеивающих частиц малы по сравнению с длиной волны
света к (менее 1/20), то интенсивность рассеянного света одинакова во
82
всех направлениях. Для этих частиц молекулярная масса может быть най-
дена по уравнениям
Kc/7?e = l/Mw+2A2c;
(1.52)
Нс/х*— 1/Л1ит+2А2с,
где К— константа, равная (2л2л,2) (dn/dc)2/(NAk*)'t ns — показатель преломле-
ния растворителя; anjdc — удельный инкремент показателя преломления; к —
длина волны; Ад—число Лвогадро; Н=16лК/3; с — концентрация рассеи-
вающих частиц; А2 — второй внрнальный коэффициент.
Зависимости Kc/Re н Нс/хы от с выражаются прямыми линиями» из на-
клона которых можно рассчитать второй внрнальный коэффициент, а отре-
зок, отсекаемый этими прямыми на оси ординат, дает значение 1/Afw.
Таким образом, экспериментальное определение молекулярной массы
полимера этим методом сводится к определению показателя преломления
и его изменения с концентрацией dnidc, коэффициента рассеяния Re и мут-
ности тм.
Для макромолекул большего размера (диаметр клубка больше Х/20),
например для виниловых полимеров со степенью полимеризации более 500,
интенсивность рассеянного света зависит от угла, под которым проводится
наблюдение. При оценке рассеяния света от различных участков макромоле-
кулы вводится поправочный фактор рассеяния Ре» который зависит от кон-
формации макромолекулы. Ре определяется отношением интенсивности рас-
сеяния под углом 0 (Re) к интенсивности» экстраполированной к углу 6 —
= 0° (Ро). Для макромолекул любой формы Ре=1 при 0 *0°. С увеличе-
нием 0 значение Р« уменьшается. Для таких макромолекул среднемассовая
молекулярная масса может быть найдена из уравнений
Kef Re » 1 / (AfwPe) + 2Агс;
(1.53)
3A/c/8n₽90= 1/(А/аРэо)+2Л2с.
Для нахождения фактора рассеяния Ре существует два метода обработ-
ки экспериментальных данных: метод асимметрии н метод Зимма. Метод
асимметрии сводится к определению коэффициента асимметрии Z, представ-
ляющего собой отношение интенсивностей рассеяния под углами, симметрич-
ными относительно 90°. Величина Z зависит от концентрации раствора, и для
получения значений Z, не зависящих от с, проводят экстраполяцию величины
1/Z—1 на бесконечное разбавление (с—>0), получая так называемое ха-
рактеристическое значение Z (при с=0), по которому из таблиц находят
значение Ре для соответствующей конформации макромолекулы.
По методу Зимма проводят двойную экстраполяцию: на нулевую кон-
центрацию и на нулевое значение угла Этот метод является более точным
и обычно используется для полимеров с конформацией статистического
клубка.
Таким образом, для определения молекулярной массы методом светорас-
сеяния необходимо определить светорассеяние растворов нескольких кон-
центраций под несколькими углами и инкремент показателя преломления
dnldc. Метод светорассеяния существенно упрощается, если измерение про-
водить в интервале углов 2—10°. Это так называемое малоугловое светорас
сеяние в отличие от рассмотрение™ выше, которое называется широкоугло-
вым По этому методу светорассеяние определяют при одном угле, а значе-
ние рассчитывают по уравнению
Kef Re- 1/Мю+2А2с, (1 54)
которое в координатах Kef Re — с представляет собой прямую линию, отсе-
кающую на оси ординат отрезок, равный 1/Л1», а тангенс угла наклона этой
о* 83
прямой равен Л2. Внриальный коэффициент Л2, характеризующий взаимодей-
ствие полимера и растворителя, равен
’ (ь55>
Его значение может быть найдено и экспериментально по изменению
светорассеяния илн другим методом Если Л2 известен для пары полимер —
растворитель, то рассчитать Mw можно, определив Re при одной концентра-
ции раствора.
Применение малоуглового светорассеяния при использовании в качестве
источника света лазеров делает этот метод удобным для непрерывного конт-
роля молекулярной массы полимера при определении молекулярно-массового-
распределения
Седиментационный анализ. Этот метод (называемый также ультрацент-
рифугированнем) основан на определении состава по массе растворенных
веществ в растворе прн высокой частоте вращения (более 10 000 об/мин)
кюветы с раствором При ультрацентрифугнровании раствора полимера мо-
лекулы движутся в направлении дна кюветы Через определенный промежу-
ток времени происходит разделение раствора на слой чистого растворителя
у мениска н узкий слой полимера вблизи дна кюветы. Граница между эти-
ми слоями нечеткая, поскольку процесс разделения осложняется обратной
диффузией макромолекул, которая начинается сразу же прн возникновении
разности концентраций
Среднемассовую молекулярную массу обычно определяют методом седн-
ментационно диффузного равновесия, т е при достижении равновесия между
седиментацией и диффузией под влиянием слабого центробежного поля. Ве-
личину Aiw можно определить из уравнения
Jr, _______2RT_____Г 3
ю (1 — Уррр) [ с0 (г\— г2,) J ’
где <о — угловая скорость вращения кюветы; Гр— удельный парциальный
объем растворенного вещества; рр — плотность раствора; г — расстояние меж-
ду центром вращения и рассматриваемой точкой раствора; индекс 1 относит-
ся к мениску, индекс 2— к донной части кюветы
Этот метод довольно длительный, например, в слое толщиной 2—3 мм
равновесие устанавливается от нескольких часов до многих суток. Поэтому
чаще определяют методом скоростной седиментации среднеднффузную седи-
ментационную молекулярную массу Мо:
S/D- ЛТ(, + Л4Л^+...) ’ (L57>
где 5 и D — коэффициенты седиментации и диффузии, которые могут быть
определены экспериментально или найдены в справочниках.
Экстраполируя на бесконечное разбавление (с—>-0), определяют эффек-
тивную массу из уравнения Сведберга:
RTS
(1-й^5-- <,'58>
Широкое распространение для оценки средней молекулярной массы по-
лучил вискоз и метрический метод. Молекулярная масса, опреде-
ляемая этим методом, называется средневязкостиой В основе метода ле-
жит зависимость вязкости раствора полимера от его молекулярной массы.
К достоинствам этого метода относятся несложное аппаратурное оформление-
и быстрота определения, недостатком является чувствительность к наличию
разветвлений в полимере, неоднородности макромолекул по химическому
•34
строению и конфигурации, существованию надмолекулярных структур (агло-
мератов) в растворе и др. Наиболее эффективен этот метод Для мотодис-
персных полимеров (см. гл. 6).
Определение молекулярно-числового (МЧР) и молекуляр-
но-массового (ММР) распределения включает фракционирование
полимеров и определение молекулярной массы узкий фракции любым нз рас
смотренных выше методов. Фракционирование — это разделение полндисперс
ного полимера иа различные по молекулярной массе более или менее одно-
родные фракции Фракционирование проводят следующими способами: фрак-
ционным растворением, с помощью хроматографии, седиментации, диффузии
н др. Из этих способов наибольшее распространение получили хроматогра-
фия н седиментация.
Средн хроматографических методов для оценки распределения по мас-
сам чаще всего используют ге л ь-п р о н и к а ю щ у ю хроматографию
Суть этого метода заключается в следующем. Колонку заполняют пористым
гелем с порами разного размера, как правило, большего, чем размер моле-
кул растворителя. Растворитель легко заполняет все поры и промежуточное
пространство между частицами геля. При введении раствора полимера мак-
ромолекулы небольшого размера (т е невысокой молекулярной массы) сво-
бодно диффундируют внутри пор геля и между частицами прн условии, что
диаметр пор (dn) больше диаметра клубка (dM) макромолекулы. При этом
увеличивается длина пробега, и, следовательно, низкомолекулярные фракции
будут удерживаться в колонке дольше, чем фракции полимеров, у которых
Таким образом проводят разделение полидисперсного полимера на
серию узких фракций Как правило, установки для гель-проннкающей хрома-
тографии снабжают анализаторами в сочетании с ЭВМ, *гго позволяет сразу
определить и рассчитать средние молекулярные массы (Afn и узких
фракций и их долю а затем построить кривые МЧР и ММР в интегральной
и дифференциальной форме. В качестве анализирующей ячейки может быть
использована установка светорассеяния или установка для анализа концевых
групп методами УФ илн ИК спектроскопии.
Использование метода скоростной седиментации для опреде-
ления молекулярно-массового распределения основано на различной скоро-
сти седиментации макромолекул различной массы, частицы с большей мас-
сой движутся в направлении дойной части кюветы со скоростью более вы-
сокой, чем макромолекулы с меньшей молекулярной массой. Распределение
скоростей седиментации зависит от градиента концентраций, который уста-
навливается в граничной области, и его изменения во времени. Прн дости-
жении равновесия можно определить молекулярную массу в различных точ-
ках кюветы рассчитать каждой узкой фракции и построить кривую рас-
пределения по молекулярным массам
Размеры макромолекул можно охарактеризовать среднеквад-
ратичным расстоянием между концами цепи <г2>71. Экспериментально невоз-
мущенные размеры цепи можно оценить, определяя характеристическую вяз-
кость [ц] (с.м. гл. 6) разбавленных растворов в 0-растворителе и рассчитать
<г2>71 по уравнению Флори — Фокса:
[п]Л7=:ф<р>з/2 (1.59)
где И —средняя молекулярная масса; Ф — функция, связанная с гидроди-
намическим поведением макромолекул; в растворе для узкой фракции и 0-ус-
ловий Ф = ФО=2,8-1021 моль'1 (Фо — константа Флори).
1.3.2. Исследование надмолекулярной структуры
Способы исследования надмолекулярной структуры включают установление
формы, размеров и относительного расположения различных элементов
надмолекулярных образований различной степени упорядоченности. Их мож-
85
но разделить на две группы визуальные (микроскопические) и интерферен-
ционно-дифракционные.
Визуальные методы основаны иа использовании электромагнитных коле-
баний с длиной волны, намного меньшей размеров изучаемого объекта. Раз-
решающая способность микроскопов d* определяется длиной волны излуче-
ния X, показателем преломления среды между образцом и линзой ппр и уг-
лом приема линзы 6М dM=A/(nnpSinOM). К визуальным методам относятся
световая в электронная микроскопия.
Световая микроскопия позволяет определить форму и размеры
надмолекулярных образований не менее ОД мк.м, поскольку длина волн види-
мого света составляет 0,4—0,8 мкм Метод световой микроскопии в прохо-
дящем и отраженном свете применяют для изучения морфологии поликри-
сталлов, например сферолитов. Чаще всего используют поляризованное излу-
чение, поскольку кристаллизация и ориентация обусловливают эффект двой-
ного лучепреломления В поляризованном свете радиальные сферолиты дают
характерную картину «мальтийского креста», а кольцевые — набор светлых
и темных концентрических линий (см. рис. 1.18).
Для оценки надмолекулярных образований меньшего размера исполь-
зуют электронную микроскопию Просвечивающая электронная
микроскопия (ПЭМ) в принципе аналогична световой, но вместо светового
пучка используют пучок электронов Длина волны пучка электронов л зави-
сит от напряжения U X—(1,5/У)’\ Для ускорения электронов применяют
высокое напряжение (50—100 кВ и даже 1 МВ); электронный пучок фоку-
сируется с помощью электростатических электромагнитных полей. Изображе-
ние формируется с помощью дополнительных электростатических или элек-
тромагнитных лннз и наблюдается на флуоресцентном экране илн фотопла-
стинке. Съемку проводят при глубоком вакууме («Ю~5 мм рт. ст.), пока-
затель преломления лпр в этих условиях равен 1. С повышением напряжения
снижается длина волны пучка электронов н, следовательно, растет разрешаю-
щая способность микроскопа. Наилучшее разрешение у промышленных при-
боров составляет 0,2—0,5 нм, а увеличение 103—105 раз. Возникновение кон-
траста на электронно-микроскопических снимках обусловлено различной рас-
сеивающей способностью ядер разных атомов по отношению к электронному
пучку. Поэтому полимеры, состоящие из легких ядер часто дают неотчетли-
вые снимки Для повышения контрастности их обрабатывают тяжелыми
металлами, такими, как палладий, золото, хром, платина, осмий и др. Образ-
цы для ПЭМ готовят в виде тонких (~0,1 мкм) пленок, тонких срезов или
так называемых «реплик», т. е. отпечатков поверхностей сколов образцов.
Этот метод используют для изучения морфологии кристаллов и аморфных
полимеров. К числу недостатков ПЭМ следует отнести сложность приготов-
ления образцов и возможность ошибок («артефактов») в определении
структуры
Сканирующая электронная микроскопия (СЭМ) позволяет получить изо-
бражение микроскопической поверхностной области образца, причем воз-
можно получение трехмерного изображения. СЭМ дает увеличение 20—
100 000 раз (чаще всего 20 000—50 000). Разрешающая способность СЭМ
несколько меньше ПЭМ и составляет 6—10 нм. Поскольку для СЭМ не тре-
буется специально готовить образцы, этот метод находит все более широкое
применение для изучения морфологии надмолекулярных образований кри-
сталлических и аморфных полимеров.
Интерфереиционно-дифракцнонные методы включают дифракцию рентге-
новских лучей дифракцию электронов, дифракцию нейтронов и рассеяние
света под большими и малыми углами Для изучения надмолекулярной струк-
туры наиболее широко применяют первые два метода.
Рентгеноструктурный анализ основан на взаимодействии ве
щества с рентгеновским излучением, длина волн которого лежит в интервале
от 0,01 до 10 нм. Рентгеновские лучи образуются при бомбардировке метал-
лической мишенн электронами высокой энергии. Для исследования полимеров
-86
Рис. 1 24 Схема отражения лучей
от одномерной структуры (4,
Л', А" — падающие лучи; Д —
дифрагированный луч)
A
применяют рентгеновские лучи с длиной волны 0,154 нм. Степень поглоще-
ния рентгеновских лучей оценивается линейным коэффициентом поглоще-
ния ер, который определяется из соотношения
e₽=(l/01g(/o//), (1.60)
где /о и /—интенсивности падающего и прошедшего пучка рентгеновских
лучей; I — толщина образца.
Массовый коэффициент поглощения равен
Срм^вр/pn» (1 01)
где рп — плотность полимера.
При взаимодействии образца с монохроматическим пучком рентгеновских
лучей возможны два случая: от образца с кристаллической структурой рент-
геновские лучи рассеиваются когерентно без изменения длины волны, т. е.
рассеивание сопровождается дифракцией рентгеновских лучей; от образца
с нерегулярной структурой, т. е. содержащего аморфные и кристаллические
области, рассеяние происходит иекогерентио и сопровождается изменением
длины волны. На этом основано использование рентгеноструктурного анализа
для оценки структурной упорядоченности в расположении макромолекул и их
частей. Прн дифракции рентгеновских лучей появляются дифрагированные
пучкн — результат интерференции вторичного рентгеновского излучения, воз-
никающего прн взаимодействии первичного излучения с электронными обо-
лочками атомов (рис. 1.24). Дифрагированные лучи будут интерферировать
(усиливаться), если выполняется условие Брэгга — Вульфа:
2rnsinGP=nXt (1.62)
где Гп — расстояние между плоскостями; Gp—угол между направлением па-
дающего луча и плоскостью, прн котором интерференция лучей максималь-
ная; п— целые числа (1, 2, 3 и т. д., называемые порядком отражения);
X — длина волны падающего излучения.
Согласно этому уравнению отражение излучения от плоскостей будет
происходить не при любом угле падения, а только в том случае, если некото-
рый набор плоскостей располагается под соответствующим углом к падаю-
щему пучку. В результате отражения от некоторого набора плоскостей на
рентгенограмме появляется пятно (рефлекс). Чтобы увеличить число набо-
ров, образец вращают вокруг вертикальной осн, в результате чего появляется
серия рефлексов. Этн рентгенограммы получили название рентгенограмм
вращения. Они характерны для одноосно ориентированных полимеров с высо-
кой степенью кристалличности.
На рентгенограммах порошкообразных аморфно-кристаллических образ-
цов прн использовании плоской пленки появляется серия концентрических ди
фракционных полос на фоне некогерентиого рассеяния и аморфного гало
Если используется цилиндрический образец, то на рентгенограмме появляется
серия полос на том же фойе — так называемая дебаеграмма (рис. 1.25).
Кроме качественного разделения на кристаллические и аморфные полиме-
ры, рентгеноструктуриый анализ используют для расчета межплоскостных
87
I tt ' ' ч»
Рис. 1.25. Рентгенограммы вращения одноосно-ориентированного полимера
(а) н аморфно-кристаллического на плоской пленке (б) и дебаеграмма (в):
/ — дифракционные кольца; 2 —дифракционные линии; 3 — дифракционные рефлексы^
4 — аморфное гало; 5 — некогсрентнос рассеянно
расстояний в кристаллах и степени кристалличности образцов. Для трехмер-
ной решетки с размерами а, b и с должны выполняться условия:
2а sin Op 1«=лХ; 2b sin 0р 2—тК; 2с sin 0Р 3=Л?<;
(1.63>
cos2 Op <+cos2 Op 2+cos2 Op 3“ 1,
где n, m, k — целые числа; 0Рь Ops» 0p3 — углы между направлением падаю-
щего луча и плоскостью, при которых интерференция лучей максимальна.
Для определения максимума отражения измеряют интенсивность рассея-
ния и строят зависимость этого показателя от угла дифракции 0р (рис. 1.26).
По формулам (1.63) при постоянной длине волны X, но при разных углах
можно рассчитать размеры элементарной кристаллической решетки (малый
период), конформацию спирали (шаг
спирали), т. е. периодичность распре-
деления заместителей вдоль цепи, и так
называемый «большой период», обу-
словленный чередованием областей
дальнего и ближнего порядка, напри-
мер аморфных н кристаллических уча-
стков. Этим трем типам регулярности
структуры соответствуют характерные
углы, прн которых наблюдаются максн-
26,
Рис. 1.26. Рассеяние кристаллической
(/), аморфной (2) фаз и некогерентное
рассеяние (3) аморфно-кристаллических
полимеров
S8
ыумы отражения. Для малых периодов и шага спирали с периодами от 0,1
до 2 нм значения углов составляют от нескольких единиц до десятков гра-
дусов Это так называемая дифракция рентгеновских лучей под большими
углами. При исследовании больших периодов, когда значения гп достигают
десятков и более нанометров, максимумы проявляются в интервале углов от
нескольких минут до 1—2е. В этом случае говорят о дифракции рентгенов-
<ких лучей под .малыми углами (метод МУРР).
Степень кристалличности по рентгенограммам определяется как отноше-
ние суммарного рассеяния кристаллитов к общему рассеянию от аморфных
и кристаллических областей (см. рис. 1.26).
Еще один интерференционно-дифракционный метод — электроногра-
фия — метод исследования структуры, основанный на дифракции электро-
нов. Этот метод, в основном аналогичный рентгеноструктурному анализу,
имеет ряд существенных преимуществ:
1) длина волны электрона («0,006 нм при <7=40 кВ) меньше длины
волны рентгеновского излучения, поэтому максимум интерференции наблю-
дается при очень малых углах, что позволяет исследовать структурные еди-
ницы небольшого размера;
2) интенсивность дифракции выше в 10б—10* раз, что позволяет наблю-
дать картину дифракции прямо на флуоресцентном экране;
3) значительно меньше время экспозиции (несколько минут по срав-
нению с несколькими часами при рентгеноструктурном анализе);
4) для анализа требуется очень небольшое количество материала
(«10“18 г); оптимальная толщина образцов составляет десятки нанометров
(а для рентгеноструктурного анализа — несколько миллиметров).
1.4. ГИБКОСТЬ ПОЛИМЕРОВ
С помощью конформационных характеристик можно описать
важное свойство, характерное только для полимеров — гибкость
цепи.
Гибкость — это способность макромолекулы изменять свою
конформацию в результате внутримолекулярного теплового дви-
жения или вследствие действия внешних сил. Как уже говори-
лось выше, конформации макромолекул изменяются в резуль-
тате заторможенного вращения звеньев вокруг одинарных свя-
зей основной цепи. Энергия активации вращения Uq является
функцией угла поворота ф.
Рассмотрим макромолекулу произвольной формы, скелет ко-
. торой состоит из углеводородной цепи с валентным углом ав,
равным 1,91 рад (109°30'). Под действием внутреннего теплово-
го движения или внешних сил в такой цепи может происходить
вращение звеньев вокруг связей С—С по конусам, изображен-
ным на рис. 1.27. Простейшим представителем полимеров этого
класса является полиэтилен, имеющий плоскую зигзагообраз-
ную транс-конформацию. В разбавленных растворах в 0-усло-
виях вероятен поворот на больший угол ф (по сравнению с твер-
дым состоянием).
В молекулах диеновых полимеров наряду с одинарными име-
ются и двойные связи, поворот вокруг которых невозможен без
разрыва л-связей. Поэтому группу CH=CR можно считать
жесткой, а полимер рассматривать как состоящий из повторя-
8Э
Рис. 1.27. Схема вращения соседних атомов углерода вокруг одинарных уг-
леродных связей в карбоцепных полимерах:
ав — валентный угол; дополнительный к валентному углу; ф — угол внутреннего
вращения; пунктиром н точками Л 2, 3, 4, 5, 6 обозначена траектория вращения свя-
зей в разбавленных растворах, сплошной линией и точками 1, 6, 7 — область возвратно*
поступательного движения (колебаний) в твердом состоянии
ющихся звеньев типа ait а,+2 и т. д.» соединенных простыми свя-
зями, обозначенными а,-ц, а,+з и т. д. Вращение в таких моле-
кулах происходит по конусам, изображенным на рис. 1.28. Бо-
лее сложные макромолекулы имеют набор из большего числа
повторяющихся единиц, но практически все полимеры изменя-
ют конформацию в результате поворота повторяющихся струк-
турных единиц вокруг связей о основной цепи.
Рассмотрим изменение потенциальной энергии U (ф) враще-
ния звена полимера вокруг связи о в зависимости от угла по-
ворота ф при переходе из конформации 1 с энергией Ui в кон-
Рис. 1.28. Схема вращения звеньев в непредельных полимерах типа ~СН»—
—CR—CH СН2~
90
формацию 2 с энергией U2 (рис. 1.29). Как следует из рисунка,
вероятность перехода определяется разностью энергий Ui—U2=
= &U, а также величиной потенциального барьера Uo. В связи с
этим различают термодинамическую и кинетическую гибкость
цепи.
1.4.1. Термодинамическая гибкость
Термодинамическая гибкость характеризует способность цепи
изменять свою конформацию под действием внутреннего тепло-
вого движения и зависит от величины Л(/, т. е. от разности
энергий поворотных изомеров. Чем меньше эта величина, тем
выше вероятность перехода макромолекулы из одной конфор-
мации в другую. Термодинамическая гибкость является равно-
весной характеристикой и определяется в условиях «нёвозму-
щенной» конформации макромолекулы, т. е. в сильно разбав-
ленном растворе в 0-растворителс при 0-температуре. Термоди-
намическая гибкость оценивается несколькими показателями:
параметром жесткости, длиной термодинамического сегмента,
персистентной длиной цепи и параметром гибкости Флори.
Параметр жесткости ож определяется соотношением
Ож=<г2>,/7<^«>,/2. (1-64)
где <г2>'л и <г®свУА— среднеквадратичные расстояния между концами реаль
ной н свободно-сочлененной цепей: <г2св>7’ рассчитывают по формулам (1.4)
(1.5), а <г2>71 определяют экспериментально, например по вязкости разбав-
ленных растворов в 6-растворителе при 6-температуре [см. (1.59)).
Этот параметр используется только для характеристики по-
лимеров, у которых вращение происходит вокруг валентных свя-
зей без их деформации и при условии неизменности надмолеку-
лярной структуры.
Длина термодинамического сегмента макромолекулы А (сег-
мент Куна)—условная величина, представляющая собой такую
последовательность п звеньев,
при которой угловая ориентация
- (п-Н’)-го звена может быть лю-
бой по отношению к i-му звену,
т. е. каждое звено может вести
себя независимо от других звень-
ев (см. рис. 1.11). Для предельно
гибкой цепи А равно длине пов-
торяющегося звена, а для пре-
дельно жесткой А=1, т. е. гид-
родинамической длине макромо-
лекулы.
Рис. 1.29. Зависимость потенциальной
энергии вращения звена Ф(<р) от угла
поворота ф
U(y)
91
Между среднеквадратичным расстоянием между концами
цепи и длиной сегмента Куна существует связь:
<r2>l/2«XZ'/2 (165)
(где Z — число сегментов).
Персистентная длина макромолекулы а также характеризует
гибкость макромолекулы; как правило, а=А/2.
Параметр гибкости Флори fo характеризует содержание гиб-
ких связей в макромолекуле. Переход от макромолекул, неспо-
собных изменять свою конформацию, к макромолекулам, содер-
жащим гибкие связи, т с. связи, вокруг которых возможно вра-
щение, происходит при f0«0,63 (1—1/е—...). Этот параметр мо-
жет служить количественным критерием гибкости, с помощью
которого все полимеры можно разделить на гибкоцепные (Ь>
>0,63) и жесткоцепные (Д><0,63). При />0,63 макромолекулы
ни при каких условиях не могут существовать в конформациях,
для которых характерны вытянутые формы (например, упругая
струна, спираль и др.).
В табл. 1.8 приведены для некоторых полимеров значения
параметров, характеризующих термодинамическую гибкость.
Как видно из таблицы, термодинамическая гибкость определя-
ется химическим строением повторяющегося звена и конформа-
цией макромолекулы, которая, как было показано раньше, так-
же зависит от химического строения На примере полимеров с
одинаковым типом конформации (например, статистического
клубка) можно проследить влияние химической структуры по-
вторяющегося звена Полимеры диенового ряда с повторяю-
щимся звеном —СН2—CR = CH—СН2—(R = H, СН3, Cl) харак-
теризуются большей гибкостью по сравнению с полимерами ви-
нилового ряда —СН2—CHR— (R = H, СН3, Cl, С6Нб, CN и т. д.).
Это обусловлено тем, что разница энергий поворотных изомеров
(транс- и гош-) в диеновых полимерах меньше примерно в 100
раз (At7 для виниловых полимеров составляет «2—3, а для не-
предельных— 0,025 кДж/моль). Такое различие связано с умень-
шением обменных взаимодействий (притяжения — отталкива-
ния) между группами СН2 при введении между ними группы с
двойной связью, имеющей более низкий потенциальный барь-
ер Uq. Аналогичная картина наблюдается и для макромолекул,
содержащих в цепи связи Si—О или С—О.
Следует отметить, что природа заместителей оказывает не-
.. значительное влияние на термодинамическую гибкость. Так,
в силоксановых полимерах замена метильного радикала на фе-
нильный практически нс изменяет гибкости. Та же картина на-
блюдается для непредельных полимеров" замена водорода на
метильную группу или даже на хлор нс приводит к сколь-ни-
будь заметному изменению жесткости макромолекулы В поли-
мерах винилового ряда некоторое увеличение ож и А наблюда-
«2
Таблица 1.8 Показатели, характеризующие термодинамическую гибкость
Полимер Повторяющееся звено • о А нм Число повторяющихся звеньев в сегменте Конформа- ция макро- молекулы
О О
II II
Сложный поли-—(СН?—О—С—СН?—С—О— 1,3— 1 — Статнстиче*
ЭФИР 1.8 ский клубок
Полиамид —NH—(СН2)*—NH—СО— СНз 1,65— 1,85 1,66 6,6 то же
Полнднметнл- силоксан —Si—О— СНз СНз 1,4— 1.6 1,4 4,9 >
Полиметилфе- ннлсилоксан -Si—0- CeHs 1.5 1.4 4,9 >
4«с-1Л-Поли- бутадиен —СН?—СН=сн—сн?— СНз 1,7 1,392-3 »
цис-М-Поли- изопрен 1 —сн2-с—сн—сн?— С1 1.7 1,392—3 »
траме -1,4-Поли- хлоропреи —СН?—С-СН СНз— 1.4 (1.67) 1,392—3 >
Полиэтилен —сн?—сн?— СНз 2,2— 2,4 2,08 8,3 >
Полипропилен 1 —CH?—CH— СНз 2,4 2,17 8,6 >
Полиизобути- лен —СН?-с— 1 СНз 2,2 1,83 7,3 >
Полистирол —сн2—сн— п 2,2- 2,4 2,00 7,9 >
93
Продолжение
Полимер
Повторяющееся звено аж Л, нм Число повторяющихся звеньев в сегменте Конформа- ция макромо- лекулы
Поливинилхло-
рид
Полиакрило-
нитрил
Полиметилме-
такрилат
Полиоктилме-
такрнлат
—СН2—CHCI—
—СН2—СН—
CN
СНз
I
СНз—С—
I
СООСНз
СНз
сн2—с—
соосвн17
2,8 2,9611,7 >
2,6— 3,2 3,17 12,6 >
1,8- 2,2 1,51 6,0 »
2,3 2,0 7,9 »
Этилцеллю-
лоза
R= C0H5
4 20,0 20 >
Полиалкнлнзо-
цианат
Поли-п-бенз-
амид
Биополимеры
R — остаток аминокислот
— 100,0 — Упругая
струна
— 210,0 320 Коленчатый
вал
— 240,0 — Спираль
54
ется лишь при замене водорода на атом С1 и группу CN (поли-
винилхлорид и полиакрилонитрил). Этот факт подчеркивает оп-
ределяющее влияние на термодинамическую гибкость взаимо-
действий ближнего порядка. В полимерах винилового ряда сфе-
ра влияния полярных заместителей затрагивает вращение во-
круг одинарных связей, что усиливает действие сил отталкива-
ния и повышает Д{/. Поэтому гибкость этих полимеров несколь-
ко снижается при введении полярных заместителей.
Наибольшей жесткостью обладают полимеры, содержащие
полярные группы, расположенные на расстояниях, достаточных
для реализации сил взаимодействия, например полиалкилизо-
цианаты. За счет сильных взаимодействий (водородные связи)
в этих полимерах реализуются лишь вытянутые конформации
(типа «упругая струна»), не проявляющие гибкости. Если же
эти группы разделены достаточным числом метиленовых групп,
то их взаимодействие ослабевает (т. е. их действие можно рас-
сматривать уже как дальнодействие), и такие полимеры харак-
теризуются высокой гибкостью (например, алифатические поли-
амиды).
Еще большей жесткостью, чем полиалкилизоцианаты, обла-
дают полимеры ароматического ряда с полярными группами в
основной цепи, например поли-п-бензамид, макромолекулы ко-
торого имеют конформацию «коленчатого вала».
К числу самых жестких полимеров относятся биополимеры,
для которых характерны устойчивые спиральные конформации
макромолекул, иммобилизованные водородными связями высо-
кой энергии
Циклические полиацетали (целлюлоза и ее производные)
относят к полужестким полимерам. В них негативное влияние
взаимодействия полярных групп уравновешивается низким
барьером вращения вокруг связей С—О.
Таким образом, по термодинамической гибкости полимеры
можно разделить на три группы
Гибкие
Полужесткне
Жесткие
А. нм fo
До 10 >0,63
10-35 —
>35 <0,63
1.4.2. Кинетическая гибкость
Кинетическая гибкость отражает скорость перехода макромо-
лекулы в силовом поле из одной конформации с энергией Ui
в другую с энергией U2, при котором необходимо преодолеть
активационный барьер вращения t/0 (см. рис. 1.29).
Подобно тому как термодинамическая гибкость определяет-
ся величиной термодинамического сегмента, для оценки кине-
95
тической гибкости изолированной макромолекулы используется
величина кинетического сегмента, т. е той части макромолеку-
лы, которая отзывается на внешнее воздействие как единое
целое. В отличие от термодинамического сегмента кинетичес-
кий является величиной, изменяющейся в зависимости от тем-
пературы и скорости внешнего воздействия Рассмотрим влияние
этих факторов на кинетическую гибкость изолированной макро-
молекулы.
Активационный барьер вращения зависит от природы ато-
мов, входящих в группировки, окружающие связь, вокруг кото-
рой происходит вращение, и определяющих уровень взаимодей-
ствия На с 39 приведены значения (/0 для низкомолекулярных
веществ в газовой фазе. Несмотря на определенные отличия в
поведении макромолекулы от низкомолекулярных веществ,
можно качественно оценить кинетическую гибкость по величи-
нам Uo низкомолекулярных аналогов Полимеры, состоящие из
звеньев, характеризующихся низкими значениями Uo, проявля-
ют высокую кинетическую гибкость. К ним относятся
карбоцепные непредельные полимеры и полимеры винилово-
го ряда, не содержащие функциональных групп, например
полибутадиен, полиизопрен, полиэтилен, полипропилен, нолиизо-
бутилен и др.;
карбоцепные полимеры и сополимеры с редким расположе-
нием полярных групп, такие, как полихлоропрсн, сополимеры
бутадиена со стиролом или нитрилом акриловой кислоты (при
содержании последних до 30—40%) и др.;
гетероцепные полимеры, полярные группы которых разделе-
ны неполярными, например алифатические полиэфиры;
гетероцепные полимеры, содержащие группы С—О, Si—О,
Si—Si, S—S и др. с низкими значениями Uo.
Увеличение числа заместителей, их объема, полярности,
асимметричность расположения повышают Uq и, следовательно,
снижают кинетическую гибкость. Так, рассматривая ряд вини-
ловых полимеров, по аналогии с низкомолекулярными вещест-
вами можно считать, что замещение водорода на метильную
группу или на полярные атомы хлора или фтора при несиммет-
ричном их расположении повышает Uo и, следовательно, сни-
жает кинетическую гибкость Поэтому в ряду
—СН2—СН2— —СН2—СН —СН2СНС1—
сн3
полиэтилен полипропилен поливинилхлорид
кинетическая гибкость снижается.
Если рядом с одинарной связью находится двойная, то Uo
снижается. Поэтому непредельные полимеры имеют более вы-
сокую кинетическую гибкость по сравнению с полимерами вини*
«6
левого ряда. Так, полибутадиен и полихлоропреп относятся к
гибким полимерам, способным проявлять гибкость при ?ммнат>
ной и более низкой температуре, в отличие от полиэтилена и
поливинилхлорида, кинетическая гибкость которых проявля-
ется только при повышенных температурах. Низкие барьеры
вращения вокруг связей С—О, St—О, С—S обусловливают
очень высокую кинетическую гибкость алифатических полиэфи-
ров, полисилоксанов, полисульфидов. Кинетически жесткими
проявляют себя такие полимеры, как целлюлоза, ароматические
полиамиды и другие полимеры, характеризующиеся высоким
уровнем взаимодействия
Температура не изменяет энергии взаимодействия (за ис-
ключением ориентационного), но влияет на кинетическую энер-
гию молекулы. Если энергия теплового движения оказывается
меньше t/o (U0>kT), то даже термодинамически гибкие полиме-
ры неспособны изменить свою конформацию, т. е. проявить
кинетическую гибкость. Рост температуры, увеличивая кинети-
ческую энергию макромолекулы (kT> £/<>), повышает вероят-
ность преодоления активационного барьера и приводит к увели-
чению кинетической гибкости.
Скорость внешнего воздействия оказывает существенное
влияние на кинетическую гибкость, т. е. на величину кинетиче-
ского сегмента. Для перехода из одной равновесной конформа-
ции в другую необходимо определенное время. Для низкомо-
лекулярных веществ, например этана, это время составляет
10“’° с, т. е. переход конформеров из одного состояния в другое
осуществляется довольно быстро. В полимерах эти переходы
происходят медленнее. Время перехода tn зависит от структуры
макромолекулы чем выше уровень взаимодействия, тем боль-
шее время требуется для изменения конформации.
Рассмотрим влияние скорости деформирования при темпера-
турах, обеспечивающих достаточно высокую кинетическую гиб-
кость (U0<kT). В условиях, когда время действия силы ia
• больше, чем время перехода из одной конформации в другую tn
или ci)/n<l, где со — частота деформации), кинетиче-
ская гибкость довольно высока и кинетический сегмент по ве-
личине приближается к термодинамическому. При очень быст-
рой деформации (а>?п^>1, /Д//П<С1) величина кинетического сег-
мента становится соизмеримой с гидродинамической длиной
макромолекулы, и в этих условиях даже термодинамически
гибкая молекула ведет себя как жесткая. Поэтому кинетиче-
скую гибкость характеризуют внутренней вязкостью В, опреде-
ляемой из соотношения
Г=— B[dl/dt), (1.66)
где В — деформирующая сила; dl/dt — скорость деформирования макромо-
лекулы.
7—816
97
Для оценки кинетической гибкости изолированной макромо-
лекулы определяют вязкоупругие характеристики разбавленных
растворов при низких концентрациях (с) с последующей экстра-
поляцией их к с—>-0. В качестве таких характеристик используют
характеристическую вязкость [ц] (см. гл 6) или характеристи-
ческие значения составляющих динамического модуля [G']
(модуль упругости) и [G"] (модуль потерь) (см. гл 5), опре-
деленные в растворителях с вязкостью при частоте деформи-
рования со:
(П1= Пт[т)уд/с]; [G'] = lim [G'/c]; (1.67>
С—И) с—*0
[G'] = Iim[(G'-<OTl$)/c].
с—0
С повышением температуры показатели вязкоупругих
свойств растворов полимеров в 0-растворителе уменьшаются, а
с увеличением частоты деформирования <о наблюдают их рост
(рис. 1.30). Эти данные подтверждают приведенные выше рас-
суждения об увеличении кинетической гибкости с повышением
температуры и ее снижении с ростом частоты деформации.
Положения о кинетической гибкости, полученные для изоли-
рованной макромолекулы, справедливы и для полимеров в кон-
денсированном состоянии, но имеют ряд особенностей, обуслов-
ленных межмолекулярным взаимодействием, которое вызывает
дополнительное ограничение скорости изменения конформаций.
Мерой кинетической гибкости полимеров в конденсирован-
ном состоянии является величина механического сегмента,.
которую определяют как минимальную длину молекулярной
цепи полимера, начиная с которой проявляется его кинетиче-
-ская гибкость. Его размер (молекулярная масса Л4СГ или сте-
Рис. 1.30. Влияние температуры на характеристическую вязкость [т]] и ча-
стоты деформации со на G' (/) н G" (2) разбавленных растворов полисти-
рола в 0-растворителе
•98
пень полимеризации nfT) приближенно можно оценить по фор-
муле Каргина и Слонимского:
JVer —ехрГ —Гс2 . 1 (1.68) ; Л'сг = л/лСР = М/Л1СГ, (1.69)
La'Tvt'“'c) J
где Net — число механических сегментов в цепи; С и D — константы для
данного полимера, зависящие от режима деформации; Тх и Тс — температу*
ры текучести и стеклования полимера; п и л<т— степени полимеризации
макромолекулы и сегмента; М и Мег— молекулярные массы макромолекулы
и сегмента.
Как следует из формул (1.68) и (1.69), величина механиче-
ского сегмента (Л4СГ или лсг) при постоянной молекулярной мас-
се зависит от температуры стеклования Гс и текучести Гт.
Температура стеклования Тс— это нижняя температурная
граница проявления гибкости; при Т<ТС полимер ни при каких
условиях не способен изменить свою конформацию, даже буду-
чи потенциально гибким (при высокой термодинамической гиб-
кости). Поэтому Тс может служить качественной характеристи-
кой кинетической гибкости полимера в конденсированном со-
стоянии.
Температура текучести Тт — это верхняя температурная
граница изменения конформаций в результате заторможенного
вращения вокруг одинарных связей без изменения центра тя-
жести макромолекулы. При 7’>Тт наблюдается уже перемеще-
ние отдельных сегментов, которое обусловливает перемещение
центра тяжести всей .макромолекулы, т. е ее течение. При
Tc-j-Tt молекулярная масса механического сегмента приближа-
ется к М макромолекулы и полимер ведет себя как кинетически
жесткий. Чем выше Д7’=ГТ—Тс, тем меньше величина механи-
ческого сегмента и, следовательно, выше кинетическая гибкость
полимера в конденсированном состоянии. Поэтому можно счи-
тать, что мерой кинетической гибкости макромолекул в конден-
сированном состоянии является также и величина ДГ (рис. 1.31).
Температуры текучести и стеклования зависят от режима
деформирования, в частности от его скорости. С повышением
скорости (частоты) механического воздействия возрастает как
Тс, так и Тт (рис 1 32), а зависимость ДТ от частоты описыва-
ется соотношением
A 1g (EX/2,3RT2C)&T (1.70)
(где Ет — энергия активации вязкого течения).
Отсюда следует, что с ростом со величина механического
сегмента уменьшается, однако при этом температурная область
проявления кинетической гибкости смещается в сторону более
высоких температур (см. рис. 1.32).
При одинаковых условиях внешнего воздействия кинетиче-
ская гибкость полимеров в конденсированном состоянии не за-
7* 99
Рис. 1.31. Зависимость ДГ—Гс—Гт от соотношения молекулярной массы поли-
мера М и механического сегмента AfCr
Рис. 1.32. Влияние частоты деформации со на температуры стеклования Гс
и текучести Тт
висит от молекулярной массы макромолекулы. Молекулярная
масса не оказывает влияния на величину активационного барье-
ра вращения, так как он определяется только взаимодействием
ближнего порядка. С ростом молекулярной массы повышается
число сегментов. Формально это следует из соотношения (1.68)
с учетом зависимости Тс и Тт от молекулярной массы (рис. 1.33).
Температура стеклования при увеличении молекулярной
массы сначала растет, а затем при определенном значении Л4|ф
dl'JdM-A) Величина Л1ир определяется структурой полимера и
соответствует молекулярной массе механического сегмента. По-
этому иногда механическим сегментом считают такую длину
макромолекулы (молекулярную массу), начиная с которой Тс
практически не зависит от степени полимеризации, т е
dTddM-^Q. Для термодинамически гибких полимеров Мкр со-
ставляет несколько тысяч (для полибутадиена — 1000, поли-
винилхлорида— 12 000, полиизобутилена—1000, полистиро-
ла— 40000). Поэтому для полимеров с Л1~105—106 Тс прак-
тически не зависит от молекулярной массы, т. е. кинетическая
гибкость макромолекул одинаковой природы достаточно высо-
кой молекулярной массы практически одинакова.
Для осуществления конформационных переходов необходи-
мо преодолеть не только потенциальный барьер вращения звень-
ев макромолекулы Uo, но и межмолекулярное взаимодействие,
которое в конденсированном состоянии довольно существенно
и может быть оценено энергией когезии. Поскольку уровень
межмолекулярного взаимодействия определяется не только
химическим строением макромолекулы, но и надмолекулярной
структурой, то и кинетическая гибкость зависит от структуры
полимера на молекулярном и надмолекулярном уровнях.
100
Макромолекулы в аморфном состоянии проявляют большую
кинетическую гибкость, чем в кристаллическом, в котором
вследствие плотной упаковки макромолекул и дальнего поряд-
ка в их расположении реализуется чрезвычайно высокий уро-
вень межмолекулярного взаимодействия. Поэтому макромоле-
кулы гибких полимеров (например, полибутадиен, полихлоро-
прен, полиэтилен и др.) в кристаллическом состоянии ведут
себя как жесткие, неспособные изменять конформацию Гиб-
кость полимеров в ориентированном состоянии также снижа-
ется, поскольку при ориентации происходит сближение цепей и
увеличение плотности упаковки, что повышает вероятность
образования дополнительных узлов между цепями Особенно
это характерно для полимеров с функциональными группами.
В качестве примера можно привести целлюлозу и ее производ-
ные: эти полимеры характеризуются средней термодинамической
гибкостью (см. табл. 1.8), а в ориентированном состоянии не-
способны изменить конформацию ни при каких условиях (Тс
выше температуры разложения).
Рассматривая влияние структуры макромолекулы на моле-
кулярном уровне на кинетическую гибкость, можно еще раз
подчеркнуть, что основные закономерности, выведенные для
изолированных макромолекул, сохраняются и для конденсиро-
ванного состояния
1 Полимеры, содержащие группы, для которых характерны
низкие значения Uo, сохраняют высокую кинетическую гибкость
СбН5
Рис. 1.33. Изменение температур стеклования Тс и текучести Тт с ростом
молекулярной массы (Л4кр — критическая молекулярная масса)
Рис 1.34. Влияние числа стирольных звеньев на температуру стеклования Те
сополимеров бутадиена со стиролом (цифры под кривой — содержание сти-
рольных звеньев, над кривой — температура стеклования)
101
в конденсированном состоянии и характеризуются низкими
значениями Тс:
Полндиметнлсилоксан
Полиэтилен
^«с-1,4-Полнбутадиен
СНз
СНз
~СН;г—СН2~
~ СНг-СН=СН-СН2 ~
7С. К
143
153
168
2. Наличие боковых заместителей, увеличение их числа,
объема и полярности повышает Uo, снижает кинетическую
гибкость и приводит к росту Тс. Влияние числа заместителей
отчетливо видно на примере сополимеров бутадиена со стиро-
лом: с ростом содержания последнего Тс повышается (рис 1.34).
Эффект снижения гибкости можно проиллюстрировать на при-
мере виниловых полимеров:
Полиэтилен
Полипропилен
Полистирол
Поливинилхлорид
Поли акрилонитрил
Поли-2,6-днхлорстирол
~ CHj—CH2~
СНз
~СН—- сн~
~СНг- СН~
I
С6Н5
~СНз—сн~
I
С1
~СНг—СН~
CN
~СН2— СН~
CI С1
тс, К
153
263
373
358
374
440
3. Симметричность в расположении заместителей снижает
и0> повышает кинетическую гибкость и существенно снижает Тс:
Поливинилхлорид
Полнвииилиденхлорид
~СН2—СН~
С1
/С!
*** СН2—с
С1
те. к
358
292
102
Полипропилен
Полннзобутнлен
СНз
I
~СНз- СН~
СНз
~СН2—С~
263
199
I
СН,
Однако для конденсированного состояния наблюдаются и
некоторые особенности, обусловленные межцепным взаимодей-
ствием. Рассмотрим ряд непредельных полимеров полибутади-
ен, полиизопрен, полихлоропрен. Термодинамическая и кинети-
ческая гибкость изолированных макромолекул этих полимеров
практически одинакова, но в конденсированном состоянии ки-
нетическая гибкость снижается при переходе от полибутадиена
к полихлоропрену:
ТС.К
Полнбутадиен
Полиизопреи
Полихлоропрен
~ СНз—СН=сн—сн2 ~
СНз
I
~СНз—С=СН—СН2~
CI
- СНз—С=СН—СНз ~
168
203
233
Полярный атом хлора повышает уровень межмолекулярного
взаимодействия и создает дополнительные препятствия для из-
менения конформаций, что и является причиной повышения Тс-
Все факторы, снижающие межцепное взаимодействие, спо-
собствуют повышению кинетической гибкости. В качестве при-
мера можно рассмотреть ряд полиалкилметакрилатов общей
формулы
СНз
~СН2—
I
ОС—О—R
те. к R тс. К
Поли метил мета- СНз 378 Поли-н-гексилме- С6Нп 268
крилат Полнэтилмегакрн- СзН5 338 такрилат ys.
лат Поли-н-октилме- CsHn 253
Поли-н-пропилме- СзН7 308 такрилат
такрилат Поли-н-бутилме- такрилат с«н, 294 Поли-к-додецнл- метакрнлат CI2H2I 208
Высокая температура стеклования (низкая кинетическая
гибкость) полиметилметакрилата обусловлена сильным межмо-
103
лекулярным взаимодействием полярных заместителей
О-СН3
Длинные боковые алифатические заместители экранируют по-
лярные группы, что снижает эффект их взаимодействия и повы-
шает кинетическую гибкость.
Введение низкомолекулярных веществ, снижающих межмо-
лекулярное взаимодействие, например за счет экранирования
полярных группировок полимера, или увеличивающих расстоя-
ние между макромолекулами, повышает кинетическую гибкость
и снижает Тс. На этом основана пластификация полимеров.
Эффективность ее тем больше, чем ниже гибкость полимерной
цепи, и зависит от Тс самого пластификатора и его объемной
доли:
Гс-ТсфП + Тс,плфпЛ, (1.71)
где Т'с, Тс и Тс.пя — температуры стеклования пластифицированного полиме-
ра, полимера без пластификатора и самого пластификатора; <рп, флл—объ-
емные доли полимера и пластификатора
Особо следует остановиться на кинетической гибкости сет-
чатых полимеров. Во-первых, для таких полимеров можно
говорить только о кинетической гибкости в конденсированном
состоянии, поскольку сетки нерастворимы; во-вторых, формула
(1.29) теряет смысл, поскольку сетчатые полимеры неспособны
к течению. Для таких систем следует говорить не о гибкости
макромолекулы в целом, а о гибкости участка макромолекулы,
заключенного между узлами, с молекулярной массой Мс, Если
намного больше величины механического сегмента, то для
полимера сохраняются выведенные выше зависимости и гиб-
кость его практически не снижается. По мере роста числа
сшивок, т. е. снижения Мс, гибкость снижается, и при Мс, соиз-
меримой с величиной механического сегмента, полимер теряет
способность к изменению конформаций и ведет себя как абсо-
лютно жесткий полимер. Уравнение, характеризующее возра-
стание Тс при сшивании линейных полимеров, имеет вид
(Т'с - Тс)/Тс^КХс/(1 - Хс)~2К/пе., (1.72)
где Т'с и Тс — температуры стеклования сшитого и несшитого полимеров;
Хс — мольная доля сшитых мономерных звеньев иа одну исходную макромо-
лекулу; лсв — среднечисловое содержание одинарных связей в главной цепи
полимера между узлами сетки; К — константа, зависящая от соотношения
сегментальной подвижности и плотности энергии когезии сшитых и несшитых
звеньев, К «1,0—1,2.
104
Для полимеров винилового ряда, в которых на мономерное
звено приходится два атома главной цепи, справедливо соотно-
шение
Хс/(1 — Хс) =2/лс,=Л1эв/Л1с (1.73>
(где Л1зв — молекулярная масса мономерного звена).
Таким образом, структура полимеров достаточно сложная,
и для ее оценки недостаточно знаний химического строения
макромолекул: необходимо определить молекулярную массу,
конфигурацию и конформацию макромолекул, степень их упо-
рядоченности в конденсированном состоянии, т. е. надмолеку-
лярную структуру. Анализ этих параметров подтверждает, что
полимеры представляют собой высокомолекулярные соединения,
имеющие цепное строение, их макромолекулы построены из-
звеньев определенных химического строения, конфигурации и
конформации. В зависимости от строения макромолекула может
принимать ту или иную форму и изменять ее при определенных
условиях, т. е. проявлять гибкость Полимеры крайне неодно-
родны по молекулярной массе, строению звеньев, их конфигу-
рации и конформации, по характеру надмолекулярных структур.
Параметры структуры взаимосвязаны и изменение одного
из них, как правило, влечет за собой изменение остальных.
Умение количественно определять структурные параметры дает
возможность установить связь между структурой и свойствами
полимеров, материалов и изделий из них Это, в свою очередь,
позволяет предсказать комплекс свойств изделий из полимеров
той или иной структуры.
Структура полимера формируется при его синтезе. Знание
механизма этого процесса позволяет целенаправленно управ-
лять формированием структуры и, следовательно, свойствами
полимеров и изделий из них.
Контрольные вопросы
1. Какими параметрами характеризуют структуру макромолекулы?
2. Какие полимеры называют органическими, неорганическими, элементоор-
ганическими, гомо- и гетероцепными?
3. Каким показателем характеризуют энергию когезнн полимеров? Как бу-
дет изменяться параметр растворимости в ряду полимеров -~СН2—СН2~,
~СН2—СН~, ~СН2—СН~, ~CF2—CF~? Для каких полимеров
СН3 CN CF3
можно использовать формулу Смолла для расчета параметра раствори-
мости?
4. Почему для полимеров н олигомеров характерны полиднсперсность и по-
лифункциональность? Какими параметрами характеризуют молекулярную
массу и молекулярно-массовое распределение?
5 Что такое конфигурация повторяющегося составного звена, конфигурация
ближнего н дальнего порядка и конфигурация макромолекулы? Что такое
структурная регулярность и стереорегулярность?
105-
6. Что такое конформация макромолекулы и какие типы конформаций вам
известны? Как можно оценить размеры макромолекул? Почему полиизо-
бутилен не образует стереоизомерных полимеров как, например, полипро-
пилен? Какие стереоизомерные полимеры образует полипропилен? Поче-
му макромолекулы изотактического полипропилена принимают форму
Z-спирали? Почему некоторые полимеры крнсталличиы, а другие
аморфны?
8. Что такое надмолекулярная структура? Зависит лн она от химического
строения макромолекулы3 молекулярной массы, молекулярно-массового
распределения, конфигурации макромолекул и ее конформации? Привести
примеры надмолекулярных структур в аморфных и кристаллических по-
лимерах.
9 Каковы способы модификации надмолекулярных структур?
10. В чем специфические особенности ориентированного состояния по-
лимеров?
11 Что такое термодинамическая и кинетическая гибкость? Почему полиси-
локсан, цис-1,4-полибутадней цис-1,4-полиизопреи, сополимеры 1,4-поли-
бутадиена со стиролом (70:30) и нитрилом акриловой кислоты (70:30)
характеризуются практически одинаковой термодинамической, ио различ-
ной кинетической гибкостью?
12. Какие нз приведенных ниже полимеров будут проявлять гибкость прн
комнатной температуре: полиэтилен, полипропилен, полннзобутилен, по-
лиметилметакрилат, транс-1,4-полнбутадиен. цис-1,4-полибутадиен?
13 При нагревании линейный полимер превращается сначала в разветвлен-
ный, а затем в пространственный Каким образом прн этом будет изме-
няться его гибкость?
14. Прн переработке полимера происходит изменение его химического строе-
ния (например, присоединение амнносодержащего соединения), конфигу-
рации звена (цис-транс-изомеризация) и самой макромолекулы (из линей-
ной она превращается в разветвленную). Какие методы используют для
характеристики этих изменений?
ГЛ АВ А 2
ПОЛУЧЕНИЕ ПОЛИМЕРОВ
В настоящее время наряду с природными материалами все
большее значение приобретают синтетические полимеры Вы-
бор соответствующих исходных продуктов и условий процесса
позволяет проводить направленный синтез высокомолекулярных
соединений и получать их с заранее заданной структурой и не-
обходимым комплексом свойств. При этом можно регулировать
степень полимеризации, полидисперсность, разветвленность, кон-
фигурацию звеньев и порядок их присоединения.
Существуют два основных метода синтеза полимеров — по-
лимеризация и поликонденсация. Кроме того, в последние годы
широко используется возможность изменения свойств полиме-
ров за счет изменения их молекулярного строения в результа-
те химических реакций — так называемых реакций модифи-
кации.
106
2.1. ПОЛИМЕРИЗАЦИЯ
Полимеризацией называют процесс образования макромолекул
путем последовательного присоединения молекул мономеров
М к активному центру М* растущей макромолекулы. При этом
активный центр переходит во вновь присоединившееся звено:
М—М,»+М2 —М—Mi—Мз*
— М—М,—М?-М3Ф
М*+М, —♦- М—М1Ф М—М,*+Мз —»
м—Mj—М2*+Мз —*• М—М,—Mr
М,---М„*+М —М|---М*и+1
В настоящее время в мире полимеризацией получают около
3/< полимеров как гомо-, так и гетероцепных. Процесс протекает
по цепному механизму. Цепными реакциями называются.такие»
в которых образование каких-либо активных частиц (активных
центров) приводит к тому, что каждая из них вызывает цепь
последовательных реакций.
Различают гомо- и сополимеризацию. Гомополимеризация —
это реакция соединения нескольких (п) молекул одного моно-
мера:
лМ —*- [—М—]„
В сополимеризации участвуют молекулы двух (или более}
мономеров и образуется статистический или блок-сополимер:
* [---М----Mi---]л+П>
лМ|+шМ»—
сополимер
> I-M1-]„-[-M2-]m
блок-сополимер
Процесс полимеризации включает следующие основные ста-
дии: образование активных центров, рост цепи, передача цепи,
обрыв цепи. Образование активных центров протекает при взан-
• модействии инициатора или катализатора с мономером. Эта
стадия характеризуется низкой скоростью, требует затраты
энергии. Рост цепи происходит путем присоединения молекул
мономера к активным центрам с передачей активного центра
на присоединившуюся молекулу. Эта стадия обычно идет быст-
ро и сопровождается выделением энергии.
Различают цепь материальную — число составных звеньев
(или степень полимеризации) и кинетическую—»число элемен-
тарных актов присоединения молекул мономера, приходящих-
ся на один свободный радикал, образовавшийся при иницииро-
вании
Обрыв цепи (материальной, но не кинетической) происхо-
дит вследствие дезактивации активных центров, в результате
которой рост данной макромолекулы прекращается. Обрыв цепи
107
осуществляется двумя путями: путем уничтожения активного
центра Мп*, его перехода в неактивное состояние
Мл* — М„
и передачей цепи с одного активного центра на другую молеку-
лу мономера с превращением ее в новый растущий активный
центр*
М„*+М —> Мл+М*
Скорость реакций обрыва обычно лимитируется скоростью
диффузии активных центров в реакционной среде. От соотно-
шения скоростей роста и обрыва цепи зависят степень полиме-
ризации и молекулярная масса образующегося полимера чем
выше скорость роста и ниже скорость обрыва цепи, тем больше
молекулярная масса.
Активными центрами цепной полимеризации могут быть сво-
бодные радикалы (электронейтральные частицы, имеющие один
или два неспаренных электрона), ионы (положительно или от-
рицательно заряженные частицы), ион-радикалы. В соответствии
с характером активных центров различают радикальную и
ионную (анионную, катионную, ионно-координационную) поли-
меризацию.
До середины XX века полимеры получали в основном ради-
кальной полимеризацией. После открытия катализаторов Циг-
лера— Натта началось быстрое внедрение в промышленность
ионных и ионно-координационных процессов, характеризующих-
ся высокими производительностью и стереоселективностью»
возможностью направленного регулирования состава полимера.
Из огромного числа мономеров для производства олигоме-
ров и полимеров используют соединения определенных строе-
ния и состава, не содержащие примесей, способных отравлять
катализаторы или изменять направление основной реакции»
дешевые, доступные, транспортабельные, нетоксичные, пожаро-
и взрывобезопасные, обладающие требуемыми функциональ-
ностью и реакционной способностью.
Для протекания процесса полимеризации молекула мономе-
ра должна содержать кратные связи (ненасыщенные мономеры)»
неустойчивые циклы или реакционноспособные функциональные
группы.
Ненасыщенные мономеры — это соединения с двойными и
тройными связями (/С = С\, —С=С—, /С=О. —CsN
и др)» раскрывающимися при полимеризации (олефины, дие-
новые и ацетиленовые углеводороды, нитрилы и др .), которые
полимеризуются следующим образом:
лА-В —[—А—В—]„
108
К мономерам, полимеризующимся с раскрытием цикла, от-
носятся оксиды олефинов, лактамы, лактоны и др.:
л А—В —> [—А—В—С—]я
Способность мономеров к полимеризации обусловлена тер-
модинамическими и кинетическими факторами. Термодинами-
ческие факторы определяются количеством свободной энергии,,
выделяющейся при полимеризации (вследствие перехода напря-
женных $р2-гибридизованных орбиталей атомов углерода в на-
сыщенные ненапряженные зр2-гибридизованные орбитали) и
энтропией, кинетические — природой активных центров и усло-
виями процесса. Термодинамические и кинетические факторы
не взаимосвязаны: например, этилен имеет наибольшую теплоту
полимеризации, однако до открытия катализаторов Циглера —
Натта он считался инертным мономером; наоборот, изобутилен»
теплота полимеризации которого значительно ниже, чем у эти-
лена, быстро полимеризуется даже при очень низкой темпера-
туре (93 К).
Активность мономера по отношению к свободному радикалу
зависит от природы заместителя при двойной связи. Это харак-
терно как для олефинов, так и для диенов. Влияние заместите-
ля определяется электронными (сопряжение и индукционный
эффект) и стерическими эффектами
Эффект сопряжения вызывает смещение электронного обла-
ка л-связи в сторону заместителя, что приводит к поляризации
двойной связи, к ее ослаблению, а следовательно, к повыше-
нию реакционной способности мономера. Направленное сопря-
жение способствует поляризации по двойной связи, например:
6+ б-
СН2=СН—C«N —> СН2=СН—CN
Положительный индукционный эффект характерен для за-
. местителей — доноров электронов, вызывающих появление избы-
точной электронной плотности на атоме углерода метиленовой
группы
6- б+
СН2=СН—R R—алкил, алкоксигруппа
Отрицательный индукционный эффект наблюдается у заме-
стителей— акцепторов электронов. Избыточная электронная
плотность появляется у того атома углерода, с которым связан
заместитель, например.
б+ 6—
СН2=СН—X X — интрогруппа, галоген, карбонильная группа
Если в молекуле мономера содержится несколько замести-
телей, решающую роль начинают играть стерические факторы:
109
введение заместителей вызывает экранирование двойной связи
и снижение реакционной способности мономера. Имеет значе-
ние размер заместителей, их число и характер их расположе-
ния. Например, виннлантрацен
СН=СН2
баз
менее реакционноспособен, чем стирол.
При симметричном расположении заместителей поляризации
двойной связи не происходит, а экранирующее действие заме-
стителей делает полимеризацию невозможной. При несиммет-
ричном расположении заместителей двойная связь сильно поля-
ризована и мономеры такого типа очень активны. Например,
1,1-дихлорэтилен СНг=СС12 полимеризуется легко, а 1,2-ди-
хлорэтилен С1СН=СН—С1 к ^полимеризации неспособен.
Наличие заместителей в молекулах диена оказывает еще
более сложное влияние на активность мономера (табл 2.1).
Электронодонорные заместители у первого или четвертого уг-
леродного атома снижают скорость полимеризации мономера,
а у второго или третьего — несколько повышают. Электроноак-
цепторные заместители повышают активность мономера, особен-
но если они находятся не у концевых атомов углерода.
Таблица 2.1. Влияние заместителей на относительную скорость полимериза-
ции диенов
Мономер Формула Относитель- ная скорость полимери- зации
Бутадиен-1,3 сн2=сн—сн=сн2 1,0
2-Метилбутадиен-1,3 (изопрен) СН2=С—сн—сн2 1 СН, 1,25
Пентаднн-1,3 (пнперилен) СНз—сн=сн—сн=сна 0,38
2,3- Диметнлбу таднен-113 сн2=с—с=сн2 1 1 СНз СН, 3,75
1-Хлорбутадиеи-1,3 С1—сн=сн—сн=сн2 8,75
2-Хлорбута диен-1,3 (хлоро- прен) сн2=с—сн=сн2 С1 875
2,3-Днхлорбутаднеи-1,3 СН2=С—с=сн2 2500
ii i
по
2.1.1. Радикальная полимеризация
Радикальная полимеризация — один из наиболее распростра-
ненных методов синтеза полимеров При радикальной полимери-
зации активным центром является свободный радикал
Инициирование радикальной полимеризации — это про-
цесс образования свободнорадикальных центров Вследствие
наличия неспаренных электронов на внешних орбиталях они
характеризуются электрофильными свойствами, способны ата-
ковать электронные пары л- и даже о-связи мономера и превра-
щать его в свободный радикал:
С | .
R- + И -----*• R-C-C—
С 1 '
Н
I
R-4-C ----► RH-J- С
/1\ /1\
Энергия, необходимая для образования свободных радика-
лов, снижается в ряду
СН3 СН3
СНз> СН3СНа> /СН > СН9/С
сн/ сн/
первичный вторичный третичный
Устойчивость радикалов (время их жизни) оценивают энер-
гией диссоциации разрываемой связи. Чем меньше энергии тре-
буется для образования свободного радикала, тем легче он
образуется и тем более устойчив Поэтому по устойчивости эти
же свободные радикалы располагаются в обратный ряд:
третичный>вторнчный>первнчный>СН9
Иногда в одной молекуле образуются два свободнорадикаль-
ных центра; такой радикал называют бирадикалом.
Свободные радикалы в процессе полимеризации способны
вступать в различные реакции, указанные в табл. 2.2. При
реакциях 1 и 5 свободные радикалы исчезают, при остальных —
происходит замена одних радикалов другими. В химических
взаимодействиях реакции 2—4 обеспечивают рост цепи и про-
должение процесса, реакции 1 и 5 — его прекра ...ение.
Соотношение констант скоростей диспропорционирования и
рекомбинации 1гЛ и kv для первичных радикалов намного меньше
единицы, т. е. при взаимодействиях радикалов типа СНз,
nt
Таблица 2.2. Реакции свободных радикалов
Реакция Схема реакции Пример 1я|
1. Рекомбинация X-f-Y*—*-Х—¥ CH3-f-CH3—*-СН3—СН3 J 2. Распад на ради- X—Y—Z—►X-f-Y—Z СН3—СН2—СН?—0—► i ^!ия)(ФРЗГМеН" —СНз+СНг-СНг-О ' 1 3. Передача цепи X-f-Y—Z—X-Y-f-Z CF3+CH4—CF3H-f-CH3 ! 4. Присоединение X-f-Y—Z-X—Y—Z CH3-f-CH2=CH2-—СН3— 1 —CHjj—СН2 | 5. Днспропорцио- X-f-Y—Z—W—► СН3+СН3-СН2—►СН4+ | пирование —*-Х—Y4»Z=W 4-СН2=СН2 Я
СН3СН2, (СНз)гСН преобладают процессы рекомбинации Од-
нако для разветвленных радикалов это соотношение превышает
единицу, что указывает на преобладание в данном случае
реакций диспропорционирования. Как рекомбинация, так и дис-
пропорционирование радикалов протекают через стадии образо-
вания различных активированных комплексов.
Свободные радикалы в полимеризационной среде могут
возникать в результате теплового воздействия (термическое
инициирование), под действием света (фотоинициирование),
радиоактивного облучения (радиационное инициирование). Од-
нако эти способы инициирования на практике применяются
редко, поскольку они или не обеспечивают нужной скорости по-
лимеризации, или вызывают побочные процессы. Поэтому в
промышленных условиях применяют метод химического иниции-
рования, при котором используют вещества (инициаторы), легко
распадающиеся с образованием свободных радикалов К ним
относятся пероксиды, гидропероксиды, азо- и диазосоединения,,
окислительно-восстановительные системы.
Основной реакцией генерации свободных радикалов при
использовании пероксидов является гомолитический распад
связи О—О. Скорость распада зависит от строения пероксида и
среды. Типичным представителем пероксидов является пероксид
бензоила, который при 300—330 К разлагается с образованием
двух бензоатных радикалов:
0 0 О
11 : 11 /"А 11
у—с—о—о—С—-----------> 2 С—О-
Распад происходит по связи О—О, обладающей наимень-
шей по сравнению с остальными связями энергией (энергия свя-
зи в кДж/моль равна для О—О— 146, С—С — 345, С—О — 357„
С=О — 741). Образующиеся бензоатные радикалы вновь рас-
112
издаются по наименее прочной связи с образованием фениль-
ного радикала и СО2:
О
Скорость распада пероксида бензоила повышается при на-
гревании, облучении и снижается в присутствии кислорода.
В гидропероксидах свободные радикалы - также образуются
при распаде связи О—О:
R" R"
R'-C-OyO-H -----► R'—С=О+R« + ОН •
R
Типичный представитель этой группы инициаторов — гидрр-
пероксид изопропилбензола — распадается на радикалы следую-
щим образом:
Из азосоединений в качестве инициатора применяют динит-
рил азоизомасляной кислоты (азобисизобутиронитрил), распа-
дающийся по связям С—N на два радикала с выделением
азота:
сн3
сн3
NacC—C-tN==N-t-C—C«N
СН8
2N-C—J- +Na
(Ь8
Из диазосоединений наиболее часто применяют диазоамино-
бензол, распадающийся в две стадии с образованием двух раз-
личных радикалов и азота:
В—816
113
Применение для инициирования радикальной полимеризации
окислительно-восстановительных систем широко распространено
в промышленности. Это связано с существенным снижением
энергии активации распада инициаторов на свободные радика-
лы (50—84 кДж/моль вместо 125—170 кДж/моль при термиче-
ском инициировании), что позволяет проводить полимеризацию
при низких температурах и снижает энергетические затраты.
Рассмотрим некоторые примеры действия окислительно-восста-
новительных систем при инициировании радикальной полиме-
ризации.
В присутствии третичных аминов, имеющих в молекуле не-
поделенную пару электронов на атоме азота, распад пероксида
происходит следующим образом:
R rR
R-N: J- R'—О-О—R' ---> R—N-O—R' R'O“ ------>
LR
R
R—N^ + R'O+R'O"
R
R—N- + R'O"
R-N: + R'O
В качестве восстановителей применяют также соли желе-
за (II), сульфиты, тиосульфаты, оксикислоты. Распад гидропер-
оксида, например, в присутствии солей железа (II) протекает
по следующей схеме
ROOH+Fe3+ —* RO +OH+Fes+
Введение в систему восстановителя (ронгалит OHCFhSC^Na)
позволяет легко переводить ионы железа (III) в ионы железа-
(II), и цикл распада инициатора, таким образом, снова повто-
ряется:
2Fes++2OH+OHCH2SO2Na —> 2Fe2++OHCH2SO3Na+H2O
Процесс инициирования характеризуется двумя после-
довательными реакциями: разложение инициатора In с образо-
ванием свободных радикалов R* и взаимодействие радикала с
мономером М с образованием активного центра свободноради-
кального типа RM<
"ни ин
In ----> 2R-; R-+ М -----------> RM.
114
(где &ип и k'im — константы скоростей соответствующих реак-
ций) В большинстве случаев ^Ш1<А'И1,, поэтому лимитирующей
стадией инициирования является стадия разложения инициато-
ра. Скорость инициирования может быть описана формулой
V нк = ^нв [In] (2.1)
(где [In] —концентрация инициатора).
Рост цепи — основная стадия радикальной полимериза-
ции, ответственная за образование макромолекул На стадии
роста цепи неспаренный электрон переходит от атома углерода
радикала R- к концевому атому углерода последней присоеди-
нившейся молекулы мономера, превращая растущую цепь в
макрорадикал. Каждый акт присоединения ненасыщенных мо-
лекул мономера сопровождается разрывом л-связей мономера
и образованием о-связи с неспаренным электроном свободного
радикала. Второй электрон л-связи при этом остается неспарен-
ным и строение активного центра сохраняется. Происходит
последовательное присоединение молекул мономера к радика-
лам растущей молекулы
RM- + M ---► RMM-
(где kp — константа скорости роста цени).
Если предположить, что активность растущего полимерного
радикала не зависит от длины цепи, то скорость реакции роста
цепи ор описывается уравнением
*р = ММ-1[М], (2.2)
(где [М«] и [М]1—концентрации радикалов и мономера).
Энергия активации реакции роста полимерной цепи состав-
ляет 17—42 кДж/моль. Эта реакция протекает с огромной ско-
ростью: например, за 1 с присоединяется до 10000 молекул
мономера. Скорость реакции роста полимерной цепи зависит от
реакционной способности мономера и активности растущего
полимерного радикала. Наибольшее влияние на скорость роста
цепи оказывает строение свободных радикалов, определяющее
скорость их присоединения к молекулам мономера В табл. 2.3’
Таблица 2.3. Относительная реакционная способность свободных радикалов
в реакциях присоединения к этилену Х-[-СН2=СНа—"XCHz—СНг
Свободный радикал X Энергия активации, кДж/моль Относительная скорость присоеди- нения прн 437 К
сн3сн2сн2сн2 28,01 0,2
СНзСН2 31,80 0,5
СНз 28,42 1,0
СС1з 26,35 1,1
ch2f 17,97 5,6
CFS 10,03 320,0
8* 115
приведены данные о реакционной способности различных ради-
калов в реакциях присоединения к этилену Энергия активации
этой реакции уменьшается, а скорость возрастает с увеличени-
ем электроотрицательности свободных радикалов.
Скорость роста макрорадикалов также зависит от их строе-
ния. Например, константа скорости роста макрорадикалов,
составляет [в м3/(кмоль-с)] в случае полибутадиена—100,
полистирола — 170, полиметилметакрилата — 700, поливинил-
ацетата— 4000, поливинилхлорида— 12 000.
Строение образующегося полимера определяется строением
мономера и условиями его полимеризации. Однако получение
полимеров с регулярным расположением звеньев при радикаль-
ной полимеризации затруднено.
Обрыв цепи — заключительная элементарная стадия про-
цесса полимеризации, на которой происходит уничтожение сво-
бодных радикалов диспропорционированием или рекомбинацией
(см. табл. 2.2) в результате столкновения двух растущих мак-
рорадикалов. Эти реакции протекают в течение всего процесса
полимеризации, но наиболее характерны для начальных этапов,
когда вязкость системы невелика В результате обрыва цепи
образуются макромолекулы различной длины. Обрыв цепи про-
текает со скоростью, значительно превышающей скорость роста
цепи Энергия активации реакции обрыва цепи часто близка к
нулю.
Суммарная энергия активации радикальной полимеризации
равна
£=0,5£ни+ (£₽ - 0,5£ов). (2.3>
где £ни. £р, £<л — энергии активации соответственно реакций инициирования,
роста и обрыва цепи, разность £₽— 0,5£<>в обычно равна 16—29 кДж/моль,
а £ин зависит от способа инициирования.
Скорость обрыва цепи оос выражают уравнением
»об = *об(М.]«, (2.4>
где ЛО6 — константа скорости реакции обрыва; [М-] — концентрация макро-
радикалов.
Обрыв цепи также может происходить в результате взаимо-
действия свободных радикалов с низкомолекулярными соедине-
ниями, так называемыми ингибиторами полимеризации — арома-
тическими аминами, ароматическими нитросоединениями,
хинонами, а также стабильными свободными радикалами, не
взаимодействующими друг с другом, но вступающими в реак-
ции рекомбинации или диспропорционирования с активными
радикалами. Ингибиторы применяют для предотвращения са-
мопроизвольной преждевременной полимеризации при хране-
нии или транспортировании мономеров и олигомеров в период,
между введением инициатора в реакционную среду и началом
реакции. Часто ингибиторы применяют для снижения ско-
116
Рнс 2.1. Влияние замедлителя (2) и
ингибитор* (•$! мэ кинетику полиме-
ризации (/ — полимеризация без ин-
гибитора и замедлителя; Р — количе-
ство образовавшегося полимера)
рости полимеризации. В этом
случае их называют замедли-
телями. Результат действия
ингибиторов и замедлителей
показан на рис. 2.1. Количест-
венно эффективность ингибиторов Э характеризуют соотноше-
нием начальных скоростей полимеризации в отсутствие (и) и в
присутствии (ики) ингибиторов (при одинаковых скоростях ини-
циирования): 3 = v/vKu.
Эффективность ингибиторов зависит как от их строения, так
и от строения мономеров Например, константы скорости инги-
бирования полимеризации стирола [м3/(кмоль-с)] равны: при
применении нитробензола — 30, тринитробензола — 6-Ю3, бен-
зохинона— 5-10*; константа скорости ингибирования нитробен-
золом полимеризации метилакрилата составляет 5, винилацета-
та—2-104.
В процессе полимеризации, как уже говорилось, образуются
макромолекулы разной молекулярной массы. Широкий разброс
значений молекулярной массы обычно приводит к ухудшению
механических свойств полимеров Поэтому при получении по-
лимеров стремятся регулировать их молекулярную массу. Для
этого используют, в частности, реакцию передачи цепи,
которая заключается в том, что вводимое в систему вещество —
регулятор — обрывает растущую цепь, но при этом само стано-
вится свободным радикалом и начинает новую кинетическую
цепь реакции полимеризации. Таким образом, реакция передачи
цепи приводит к продолжению кинетической цепи и прекраще-
нию (ограничению) роста материальной цепи (макромолекулы).
Передача цепи может происходить не только с помощью регу-
ляторов, но и через молекулы растворителя, примеси и т. д.
В качестве регуляторов применяют хлорированные углеводоро-
ды, меркаптаны и др. Особенно широко регуляторы используют-
ся в производстве синтетических каучуков.
Повышение температуры и увеличение количества агента
передачи цепи (например, галогенсодержащих углеводородов)
приводят к резкому возрастанию скорости реакции передачи
цепи, и эта реакция подавляет другие стадии полимеризации,
так что образуются индивидуальные низкомолекулярные веще-
ства, которые можно разделить (реакция теломеризации) Так,
теломеризация этилена в среде тетрахлорида углерода проте-
117
кает с образованием индивидуальных продуктов (тетрахлорпен-
тана, тетрахлоргепта на и др.):
2СН2=СН2+ССЦ —С1—СНг-СНг-СНг—СНг-ССЬ
ЗСН2=СН2+СС14 —С1—СНг-СНг—СН2—СНг-СНг—СНг-СС1з
Появление разветвленных макромолекул также объясняет-
ся передачей цепи на макромолекулу:
R R R R
~СН—СНа—Ан—сн, ~Ан—СН2—Ан—СН3
~СН—СН2—СН—СН2~
I I
R' R
~СН—СН—СН—СН2~
I I
R' R'
Образовавшийся макрорадикал с неспаренным электроном в
середине цепи взаимодействует с мономером, вызывая рост бо-
ковой цепи:
СН2
rAh
~СН—СН—СН—сн2~ + СН=СН, —*- ~сн—Ан—СН—СНа~
III II
R' R' R R' R
Скорость передачи цепи зависит от строения как агента
передачи цепи, так и макрорадикала. Например, константа
скорости передачи цепи на мономер [м3/(кмоль-с)] равна для
пол и метил метакрилата 0,028, полистирола — 0,034, поливинила-
цетата— 4,0. С повышением температуры она возрастает.
Общая скорость реакции полимеризации v определяется
уравнением
v=vm.4-vp4-voe. (25)
В стационарном режиме, когда количества вновь образую-
щихся и исчезающих свободных радикалов равны, т. е. скорость
полимеризации постоянна, имеем:
»1ш = Иоб; (2.6) Аин [In] =*об|М.р; (2.7).
v = vp; (2.8) г = Ар[М.][М]. (2.9)
Определив из уравнения (2.7) значение [М*] и подставив
его в уравнение (2.9), получим:
с/=Ар(^к/Аоб),/2[М] [In]’/2. (2.10>
Поскольку [М] в стационарном периоде можно считать
постоянной величиной, общая скорость радикальной полимери-
зации пропорциональна корню квадратному из концентрации
инициатора
Степень полимеризации п, т. е. средняя молекулярная масса
полимера, определяемая соотношением скоростей роста и обры-
ва цепи, связана с концентрацией инициатора уравнением
п=Л/[1п],/2 (2.11)
(где k — общая константа скорости процесса).
На скорость полимеризации и строение полимеров большое
влияние оказывает температура процесса. С повышением тем-
пературы, как правило, резко возрастают константы скорости
любых химических реакций, причем эффективность повышения
температуры тем выше, чем больше энергия активации реакции
Энергия активации (кДж/моль) различных стадий полимериза-
ции равна: инициирование 84—170, рост цепи 15—40, передача
цепи на мономер 28—32, гибель макрорадикалов 8—20 Вслед-
ствие более высокой энергии активации при инициировании
повышение температуры вызывает значительный рост скорости
инициирования и возрастание концентрации свободных радика-
лов, что приводит к увеличению скорости роста цепи и одно-
временно скорости ее обрыва. Из уравнений (2.2) и (2.4)
видно, что повышение концентрации макрорадикалов вызывает
более сильное увеличение скорости обрыва цени, чем скоро-
сти роста, так как в уравнение для t’oc концентрация макрора-
дикалов входит в квадрате. Вследствие более быстрого увели-
чения скорости обрыва цепи при повышении температуры
снижается средняя степень полимеризации полимера, опреде-
ляемая соотношением vp/vo&.
Изменение температуры при полимеризации влияет также
на механизм обрыва цепей. Поскольку энергия активации дис-
пропорционирования выше, чем рекомбинации, повышение
температуры благоприятствует развитию реакции диспропор-
ционирования, что приводит к снижению степени полимеризации,
т. е. молекулярной массы полимера. Повышение температуры
полимеризации способствует также увеличению вероятности
протекания вторичных реакций разветвления и структурирова-
ния, в результате чего возрастают содержание гель-фракции в
полимере и коэффициент полидисперсности
Температура оказывает влияние на микроструктуру полиме-
ра. Например, в сополимере бутадиена и стирола с понижени-
ем температуры полимеризации наблюдается рост содержания
звеньев в положении транс-1,4; содержание 1,2-звеньев от тем-
пературы полимеризации практически не зависит
373 К 323 К 278 К 253 К
Содержание звеньев» %
quc-l»4 28 14 7 0
транс-1,4 52 62 72 81
1,2 20 23 21 19
119
На структуру полимера в некоторых случаях влияет дав-
ление Например, его повышение при полимеризации этилена
способствует увеличению числа боковых ответвлений, при по-
лимеризации метилметакрилата — росту изотактичности поли-
мера. Изменение давления в пределах нескольких МПа практи-
чески не влияет на скорость полимеризации. Однако примене-
ние высоких давлений значительно ускоряет процесс. Например,
полимеризация метилметакрилата при 370 К протекает при
давлении 10 Па в течение 6 ч, при 300 МПа — в 6 раз быстрее.
2.1.2. Сополимеризация
В последнее время интенсивное развитие получила радикальная
сополимеризация двух, трех или большего числа мономеров.
Получаемые при этом сополимеры состоят из чередующихся
звеньев или блоков звеньев исходных мономеров, обладают
многими ценными свойствами и широко применяются в промыш-
ленности. В качестве примера ниже приведена реакция сопо-
лимеризации бутадиена и стирола, приводящая к получению
бутадиен-стирольного каучука
лСН2=СН-СН=СН2 4- тСН2—СН ----->
свн4
--*. [—СН2—СН—СН—СН2—Г—СН2—СН—1~
CgHj Jrn
Как и гомополимеризация, сополимеризация включает ини-
циирование, рост, обрыв и передачу цепи. Однако все стадии,
значительно осложнены присутствием нескольких мономеров.
Участие в сополимеризации различных мономеров и свобод-
ных радикалов, взаимодействие которых происходит с различ-
ной интенсивностью, влияет на кинетику процесса, состав и.
свойства получаемых сополимеров. Например, при сополимери-
зации двух мономеров М! и М2 образуется не менее четырех
типов свободных радикалов:
• Ац • • Ааа •
1 ► Mg
• Ац • • •
Ma --► ~Mi—1 — > Mj
Поскольку исходные мономеры обладают различной реак-
ционной способностью, константы скоростей роста цепей kp
также различны Относительную активность мономеров и ради-
калов характеризуют константами сополимеризации Г| и г2:
r2=ks2/kn. (2.12)
Константа л представляет собой относительную скорость, с
которой радикал Mi реагирует с мономерами Mi и М2, г2—
120
I
скорость взаимодействия радикала М2 с мономерами М2 и
Mi. Согласно экспериментальным данным произведение кон-
стант сополимеризации всегда равно или меньше 1.
Значения констант сополимеризации позволяют определить
состав сополимера. При Г1Г2= 1 процесс называется идеальной
сополимеризацией. При этом возможно, что г(>1, а г2<1, и
наоборот. Если Г|>1, а г2~0, то это значит, что реакционная
способность Mi значительно выше реакционной способности
М2 и все радикалы будут преимущественно реагировать с
Мь образуя сополимер, обогащенный мономером Mi Обратная
картина наблюдается, если л приближается к 0, а г2>1
При Г1Г2 = 0 процесс называется чередующейся сополимери-
зацией, при которой растущая цепь, состоящая из звеньев
мономера одного типа, присоединяет молекулы другого мо-
номера. При Г|~г2 можно получить регулярно чередующийся
сополимер эквимольного состава. Соотношение 0<Г]Г2< 1 харак-
терно для реальных систем, занимающих промежуточное поло-
жение между идеальными и чередующимися сополимерами
Чем ближе произведение констант к нулю, тем более вероятно
чередование мономеров
При Г1Г2>1 образуется не статистический сополимер, а блок-
сополимер, что не характерно для радикальной сополимериза-
ции. Если Г]>1 и г2>1, то каждый радикал взаимодействует
преимущественно со «своим» мономером, что приводит к обра-
зованию гомополимеров
Зная концентрации мономеров [MJ и [М2] и константы
сополимеризации, можно определить состав сополимера по
уравнению Майо, справедливому при низких степенях полиме-
ризации, когда концентрация всех реагентов постоянна [п —
отношение концентраций звеньев М| и М2 в сополимере):
и _ (MJ г, (MJ + (MJ
(MJ r2 (MJ + (MJ • ' '
2.1.3. Ионная полимеризация
Ионная полимеризация в отличие от радикальной характеризу-
ется гетеролитическим разрывом связей в мономере Разрыв
двойной связи происходит под влиянием катализаторов ионной
полимеризации, образующих ионы Реагируя с молекулой моно-
мера, ионы катализатора переводят ее в состояние иона, и далее
полимеризация идет по механизму цепных реакций.
В зависимости от знака заряда на концевом атоме растущей
цепи различают полимеризацию анионную и катионную. В пер-
вом случае атом углерода растущей цепи имеет отрицательный
заряд (карбанион), во втором — положительный (карбкатион).
Если при ионной полимеризации реакция роста цепи сопровож-
121
(1>
(2>
дается координацией мономера на поверхности катализатора,
то полимеризация называется ионно-координационной. Высокая
координирующая способность катализатора приводит к образо-
ванию линейных макромолекул с высокой степенью регулярно-
сти чередования звеньев, а в ряде случаев и стереорегулярности.
Полученные полимеры характеризуются довольно высокой мо-
лекулярной массой и узким ММР. Классы веществ, способных
к ионной полимеризации, значительно разнообразнее, чем при
радикальной. Кроме виниловых и диеновых мономеров хорошо
полимеризуются соединения с карбоксильными группами, раз-
личные гетероциклы. Схематически процесс инициирования
винилового мономера при анионной (I) и катионной (2) полиме-
ризации может быть представлен следующим образом:
В+А-+СН2=СН —> А—СНг-СН-В*
I I
X X
Н+А-+СН2=СН —СНз—СН+А-
( I
X X
Закономерности ионной 'полимеризации могут быть рас-
смотрены только в общих чертах, так как в каждом конкретном
случае в зависимости от природы мономера, катализатора и
среды процесс имеет свои особенности. Энергия активации ион-
ной полимеризации ниже, чем радикальной, поэтому процесс
идет при низких температурах, часто отрицательных, с очень
высокой скоростью. Ионная полимеризация, как любой цепной
процесс, протекает в три стадии: инициирование, рост цепи,
ограничение роста. Однако в отличие от радикальных процес-
сов функция катализатора не ограничивается только участием
в -реакциях инициирования катализаторы влияют на реакции
роста и обрыва цепи, участвуют в реакциях переноса. Это оп-
ределяет кинетику процесса и структуру получаемого полиме-
ра. При радикальной полимеризации инициатор не оказывает
влияния на структуру полимера.
Активные центры при ионной полимеризации состоят из
растущего иона (R+ или R~) и противоиона (А- или В+). Ста-
бильность и структура ионной пары зависят от их свойств и
сольватирующей способности растворителя; при сильной сольва-
тации ионы могут быть изолированы друг от друга. Различа-
ют три основные формы существования ионов в растворе.
А*В"
контактиая
пара
А+, в- ч=* А++В-
сольватно разде- свободные ионы
ленная ионная
пара
Свободные ионы характеризуются гораздо большей актив-
ностью, чем ионные пары, поэтому реакционная способность
122
активных центров при ионной полимеризации в отличие от
радикальной в значительной степени зависит от свойств реакци-
онной среды.
Ряду систем, полимеризующихся по ионному типу, свойст-
венно отсутствие реакций обрыва и передачи цепи. Образуются
так называемые «живущие» полимеры, макромолекулы которых
после завершения полимеризации (полного исчерпания мономе-
ра) сохраняют активные центры в течение длительного времени
и в связи с этим способны к дальнейшему присоединению
новых порций мономеров, т. е. к продолжению роста цепи.
Катионная полимеризация. Под действием катализаторов
катионного типа полимеризуются циклические соединения, об-
разующие линейные полимеры за счет раскрытия цикла (цик-
лопропан, простые циклические эфиры, циклические формали
и др.) и ненасыщенные соединения, среди которых наибольшей
активностью характеризуются мономеры, имеющие электроно-
донорные заместители у а-углеродного атома при двойной связи
и вызывающие смещение л-электронного облака в сторону
метиленовой группы. Молекула мономера поляризуется и облег-
чается ее взаимодействие с катионом катализатора или расту-
щей полимерной цепью. Этим методом полимеризуются изобу-
тилен, стирол, а-метилстирол, а также мономеры, содержащие
гетероатомы формальдегид, пропиленоксид, эпихлоргидрин,
винилалкиловый эфир и ряд других. Диеновые мономеры по
катионному механизму полимеризуются с гораздо меньшей
скоростью. Так, активность ненасыщенных мономеров снижает-
ся в ряду: винилалкиловые эфиры>изобутилен>стирол>изо-
прен > бутадиен.
Катализаторами катионной полимеризации
служат доноры протонов. Наибольшее применение нашли силь-
ные протонные (H2SO4, НСЮ4, Н3РО4, НС! и др.) и апротонные
(кислоты Льюиса) кислоты общей формулы МеХ„, где Me —
металл, X — галоген (А1С13, SnCI4, T1CI3, BF3 и др ). Протонные
кислоты диссоциируют с образованием протона Н+ по схеме
НС1О4 Н++[СЮ4]-.
При использовании апротонных кислот помимо катализато-
ра в систему вводят небольшие количества сокатализатора (про-
мотора) В качестве сокатализаторов применяют ионогенные
вещества (вода, галогенводородные кислоты, спирты и др).
Образующееся комплексное соединение катализатора и сока-
тализатора имеет свойства сильной кислоты и способно в
определенных условиях отщеплять протоны или ионы карбония,
например:
BF3 H2O H+[BF3OH] я* H++[BFSOH]
123
дается координацией мономе
то полимеризация называетсгЧ
координирующая способност
ванию линейных макромолс Ч'
сти чередования звеньев, а &о *
Полученные полимеры ха Ч Ч
лекулярной массой и узк ЧЧЧ
к ионной лолимеризаци)ЧоЧ7<(>^'
радикальной. Кроме
полимеризуются сое
личные гетероциклы
использованы также
катионов (карбония,
атионной поли-
вшими отрыв
ё с обра-
о ^L-
Закономернее^ <
смотрены тодЧ
случае в зав’ЧЧ
среды проце Ч с-5> Ч
ной поЛимеЧ/ЧЧЧ,
идет при вЧ^ЧЧ^
высокой с
процесс, Ъ/
ограниче ЧЧлЧ
сов фу$?
в -реак Ч
роста3 Ч
РеядеЧЧ
ются
^мерной
/иеров не
1ния поли-
первичных
/йрования: сво-
(К+А~], цвиттер-
координационными
иного ионного харак-
хнициирования — катио-
л пары)
.угреть полимеризацию изо-
Яа стадии иницииро_ва-
ное соединение Н+[ВРзОН]
иономера с образованием ионной
СН8
I
СН3—C+(BF3OH]
i
СНз
к мономеру происходит по месту мак-
плотности в молекуле, например протон
Аон]
о.
хона
энной
к наиболее гидрогенизированному атому (пра-
•икова). Строение образовавшегося активного
»едовательно, и его активность определяются не
риродой катализатора, сокатализатора и их количе-
соотношением, но и свойствами реакционной среды
Ч6*
ра
б
i
гратурой.
ост цепи при катионной полимеризации осуществля-
л путем присоединения молекул мономера к образовавшему-
л катиону В результате гетеролитического разрыва двойной
связи в молекуле мономера каждый акт присоединения сопро-
вождается генерированием карбкатиона на конце цепи. В этом
случае обеспечивается регулярное присоединение звеньев по
типу «голова к хвосту». Поскольку полимеризация проводится
в растворителе с невысокой диэлектрической проницаемостью,
ионная пара в процессе роста цепи сохраняется.
124
Ограничение роста цепи может произойти при
взаимодействии активного центра с противоионом, мономером,
растворителем, полимером. Реакции с противоионом могут
быть двух типов. Во-первых, при уменьшении кинетической под-
вижности макроионов может произойти перестройка ионной
пары с регенерацией каталитического комплекса по схеме
СН, СНа
I _ II
~CHa-C+[BF,OH] —> ~ch2-c + bf8 Н2О
СНз СН,
Во-вторых, сокатализатор может реагировать с растущей цепью
с образованием ковалентной связи Реакция сопровождается
регенерацией катализатора:
СН3 СН3
~СН2—C+[BF3 OH] ~СНа-С—OH + BF,
Обрыв реакционной цепи может происходить в результате
передачи ее иа мономер*
СН3 СН3 СНа СНз
I I И I _
~СНа-C+[BF,OH] -Ь СНа=С -► ~СНа—С -I- СН3—C+[BF,-ОН]
I I II
СН, СН, СН, СНз
В большинстве случаев интенсивность протекания этой реакции
определяет молекулярную массу полимера.
Относительная скорость передачи цепи на мономер растет
с кислотностью катализатора в ряду: Т1С14>$пС14>ЕеС1з>ВРз
и снижается с увеличением полярности среды. Реакции переда-
чи цепи зависят также и от условий процесса, что, по-видимому,
обусловлено ролью противоиона в нем.
Передача цепи на полимер может идти с разрывом
молекул и без него. Например, при катионной полимеризации
циклических ацеталей в результате гидридного переноса про-
исходит разрыв полимерной цепи
-О— СН О СН2~+СНг_^О------»- ~О—СН О
—СН~+СН3—О---- ~О—СН2+О=СН~+СНз—О~
В результате передачи цепи с разрывом от основной поли-
мерной цепи отщепляются небольшие осколки, дающие новые
центры полимеризации. Это приводит к заметному расширению
молекулярно-массового распределения. Данный процесс ис-
пользуется при синтезе разнообразных блок-сополимеров. Пе-
123
В качестве катализатора могут быть использованы также
галогены (Ь, ICI, IBr), соли стабильных катионов (карбония,
оксония), алкил производные металлов и др.
В ряде случаев эффективно инициирование катионной поли-
меризации излучениями высокой энергии, вызывающими отрыв
от молекул мономера или другого реагента электрона ё с обра-
зованием катион-радикала:
AV . + _
R-R' —*• R-R'+e
При взаимодействии катализаторов с мономером образуются
инициирующие ионные частицы, вызывающие рост полимерной
цепи. Большое разнообразие катализаторов и мономеров не
позволяет создать универсальную схему инициирования поли-
меризации. Однако в зависимости от структуры первичных
частиц можно выделить несколько типов инициирования: сво-
бодными катионами К+ или ионными парами [К+А_], цвиттер-
ионами [К+—А-], ион-радикалами К+ и координационными
комплексами, не имеющими ярко выраженного ионного харак-
тера; наиболее распространенный тип инициирования — катио-
нами (свободными или в составе ионной пары)
В качестве примера можно рассмотреть полимеризацию изо-
бутилена в присутствии BF3-H2O. На стадии инициирова-
ния образующееся комплексное соединение H^fBFsOH]
отдает свой протон молекуле мономера с образованием ионной
пары:
СН8 СН3
I I -
CH2=C4-H+(BFsOH1 ----> СНз—С+1ВГ3ОН]
I i
СНз СНз
Присоединение катиона к мономеру происходит по месту мак-
симальной электронной плотности в молекуле, например протон
присоединяется к наиболее гидрогенизированному атому (пра-
вило Марковникова). Строение образовавшегося активного
центра, а следовательно, и его активность определяются не
только природой катализатора, сокатализатора и их количе-
ственным соотношением, но и свойствами реакционной среды
и температурой.
Рост цепи при катионной полимеризации осуществля-
ется путем присоединения молекул мономера к образовавшему-
ся катиону В результате гетеролитического разрыва двойной
связи в молекуле мономера каждый акт присоединения сопро-
вождается генерированием карбкатиона на конце цепи. В этом
случае обеспечивается регулярное присоединение звеньев по
типу «голова к хвосту». Поскольку полимеризация проводится
в растворителе с невысокой диэлектрической проницаемостью,
ионная пара в процессе роста цепи сохраняется
124
Ограничение роста цепи может произойти при
взаимодействии активного центра с противоионом, мономером,
растворителем, полимером. Реакции с противоионом могут
быть двух типов. Во-первых, при уменьшении кинетической под-
вижности макроионов может произойти перестройка ионной
пары с регенерацией каталитического комплекса по схеме
CHS СН2
I . II
~СН2—C+[BFSOH] ► ~CHS—C4-BF. HSO
I I
CHS CH8
Во-вторых, сокатализатор может реагировать с растущей цепью
с образованием ковалентной связи. Реакция сопровождается
регенерацией катализатора
СН3 СН3
~СН2—C+IBFj OH] ---> ~CHS—С—OH4-BF8
I I
CH3 CHj
Обрыв реакционной цепи может происходить в результате
передачи ее на мономер:
СНз СНз СН2 СНз
I I II I
~СН2—C+[BF3OH1 + СН2=С > ~СН,—С -J- СНз—C+IBF8 ОН]
I I II
СН8 СН8 СН8 СНз
В большинстве случаев интенсивность протекания этой реакции
определяет молекулярную массу полимера.
Относительная скорость передачи цепи на мономер растет
с кислотностью катализатора в ряду: TiC14>SnCl4>FeCl3>BF3
и снижается с увеличением полярности среды. Реакции переда-
чи цепи зависят также и от условий процесса, что, по-видимому,
обусловлено ролью противоиона в нем.
Передача цепи на полимер может идти с разрывом
молекул и без него. Например, при катионной полимеризации
циклических ацеталей в результате гидридного переноса про-
исходит разрыв полимерной цепи:
-О-СНг-О—СН2~+СН^О---- ~о—СНг-О—
—CH~+CHs-0---- -О—СН2+О=СН~+СНз— О~
В результате передачи цепи с разрывом от основной поли-
мерной цепи отщепляются небольшие осколки, дающие новые
центры полимеризации. Это приводит к заметному расширению
молекулярно-массового распределения. Данный процесс ис-
пользуется при синтезе разнообразных блок-сополимеров Пе-
125
редача цепи на полимер, не сопровождающаяся разрывом
молекул, приводит к получению разветвленных полимеров
СНг—СН
+ _ ? * QH»
~СН2—CH[BF3OH] + сн2 -------► ~СН2-СН2 + СН2 ---------->-
11 I ।
С4Н9 СН-С4Н, С4Н9 СН-С4Нв
СН2 СНа
I ь
CH-C4He [BF30H]C—С4НР
*
СНа
СН-С4Н,
—* I
СНа
I +
C4He—С—СНа—CH[BF3OH]
? I
С4Нв
Скорость и характер реакций ограничения цепи определя-
ются активностью растущего катиона и стабильностью противо-
иона. При оптимальном сочетании этих величин ограничения
цепи не происходит и образуются «живущие» полимеры.
Кинетика процесса При катионной полимеризации
скорость процесса образования макромолекулы складывается
из скоростей четырех стадий: инициирования иин, роста цепи
ир, обрыва цепи иобр и передачи цепи на мономер ипср. Упро-
щенные уравнения скоростей элементарных стадий имеют
вид
Оии=^нп[Кат] [Сокат] [М], (2.14) иР=Л„[М] [M+J; (2.15)
Ровр«=АобР[М+]1 ©мр«А«мр[М][М+], (2.16)
где [Кэт]. [Сокат], [М], [М+] — концентрации катализатора, сокаталнзатора,
мономера и растущих карбкатионов; k— константы скоростей соответствую
щих реакции
В стационарном периоде при установившейся постоянной
скорости полимеризации
vMW = Vo6P или Агнп [Кат] [Сокат] [М]=АгоСр[М+]. (2.17)
Следовательно, концентрация активных макрорадикалов равна
[М< ]«(^н/Лобр) [Кат] [Сокат] [М] (2.18)
Суммарная скорость процесса иОбщ» т. е. скорость убывания
мономера, выражается уравнением реакции роста цепи:
Ообщ— [МЦМ+]. (2.19)
126
Подставляя в это уравнение значение концентрации расту-
щих карбкатионов, можно выразить скорость катионной поли-
меризации как функцию концентраций катализатора, соката-
лизатора и мономера:
«ов«ц=ЛР(Аип/йоор) [Кат] [Сокат] [М]2=йоощ[Кат] [Сокат] [М]2. (2 20)
Это соотношение не является универсальным, так как не
учитывает влияния температуры и природы среды, которое рас-
пространяется не только на состав активного центра, но и на
характер всех протекающих в системе реакций. Уравнение для
степени полимеризации, отвечающее простейшей схеме, имеет
вид
п=Рр/(оовр+оп«р). (2.21)
Подставляя значения скоростей элементарных стадий, получаем:
1/пяЛпер/ЛрЧ*^©бр/Ар[М]« (2.22)
Из уравнения видно, что средняя степень полимеризации не
зависит от концентрации катализатора. Если скорость реакции
передачи цепи намного выше скорости реакции обрыва (/гПерЗ>
3>&<>бр), то молекулярная масса не зависит от концентрации
мономера; если же Лобр^^пер, то молекулярная масса пропор-
циональна концентрации мономера.
Влияние условий полимеризации. Константа
скорости роста цепи при катионной полимеризации определя-
ется не только природой мономера и температурой, но и зави-
сит от типа инициирующей добавки и полярности среды, т. е.
действие всех этих факторов имеет комплексный характер, и
нельзя их рассматривать изолированно. С понижением темпе-
ратуры скорость процесса уменьшается, но при этом возрастает
диэлектрическая проницаемость среды, в результате чего
уменьшится влияние противоиона на процесс; это может при-
вести к повышению константы скорости роста цепи. Ниже
показано, как изменяется с температурой kp при полимеризации
изобутилена в среде CH2CI2 на катализаторе TiCU-HjO:
Температура, К
243
213
183
Лр. м’Дкмоль-с)
14,3
11,3
12,8
Решающим фактором, оказывающим влияние на ионные про-
цессы, является полярность среды, в которой происходит поли-
меризация Можно выделить два эффекта, обусловленных влия-
нием среды изменение реакционной способности активных
центров и стабилизация образующихся заряженных частиц.
Увеличение полярности среды увеличивает скорость иниции-
127
Таблица 24. влияние диэлектрической проницаемости е растворителя на
скорость полимеризации а-метилстирола и молекулярную массу образующего-
ся полимера
Растворитель е V, моль/мин м
Циклогексан 1,9 1,25 2040
Дихлорэтан 10 3,3 4200
Нитроэтан 28 20,4 4450
Нитробензол 36 150,0 8300
рования и уменьшает скорость обрыва цепи, что приводит к
росту молекулярной массы полимера. В принципе должна воз-
растать и скорость роста цени, однако в этом случае наблю-
дается увеличение среднего расстояния между компонентами
ионной пары и скорость реакции зависит от того, как это по-
влияет на ее реакционную способность. В табл. 2.4 в качестве
примера показано влияние полярности растворителя на поли-
меризацию а-метилстирола.
Сольватирующая способность растворителя также влияет на
кинетику катионной полимеризации. Растворитель, способный
к комплексообразованию, может изменить активность центра
роста. Например, катионная полимеризация стирола в среде
нитротолуола протекает с достаточной скоростью, а в среде
этилового спирта не идет, хотя диэлектрические проницаемости
этих растворителей близки. Подобный эффект связан с образо-
ванием сольватной оболочки вокруг активного центра и его
дезактивацией. Другой пример положительного влияния соль-
ватации на полимеризацию заключается в следующем. Пара-
финовые углеводороды и бензол — растворители, близкие по
диэлектрической проницаемости, однако катионная полимери-
зация в бензоле, как правило, протекает с большей скоростью,
что вызвано сольватационным взаимодействием молекул
растворителя с растущим центром и возникновением сольватно-
разделенных ионных пар, которые более реакционноспособны,
чем контактные. Стабилизация активных центров за счет взаи-
модействия со средой повышает их устойчивость и способствует
росту молекулярной массы полимера.
Заметное влияние на процесс катионной полимеризации ока-
зывают примеси в реакционной среде (в мономере, катализато-
ре, растворителе) При малых концентрациях примеси, как
правило, оказывают сокаталитическое действие и способствуют
увеличению скорости процесса. При больших концентрациях они
участвуют в реакциях ограничения цепи, что дает обратный
эффект
Катионная сополимеризация. Наиболее склонны к катионной
сополимеризации мономеры с электронодонорными заместите-
128
лями. Наблюдается снижение активности мономера в ряду: про-
стые эфиры> изобутилен >стирол, изопрен>этилен При кати-
онной сополимеризации произведение констант сополимеризации
находится в пределах 0,5—5,0 Например, в системе «-ме-
тилстирол— n-хлорстирол Г1Г2=3,78 при 195 К и 5,25 при 273 К;
в системе аценафтилен — винилбутиловый эфир Г!Г2=0,8 при
253 К и 0,4 при 303 К. Таким образом, в отличие от радикаль-
ного процесса, где при повышении температуры Г|Г2->1, в кати-
онных процессах этого нс наблюдается
Важной особенностью катионной сополимеризации является
зависимость констант сополимеризации и строения полимера
от природы катализатора, сокатализатора, полярности среды,
причем зависимости эти более сложные, чем при гомополиме-
ризации Поэтому константы сополимеризации в данном случае
не могут удовлетворительно характеризовать реакционную
способность мономеров.
Существенный вклад в конечный состав и распределение
звеньев в сополимере вносит сольватация активных центров
одним из мономеров. Замечено, что более полярные мономеры
проявляют повышенную активность, что связано с большим их
содержанием в сольватной оболочке активных центров. Важную
роль играет также сокатализатор, формирующий структуру
противоиона. Противоионы различной химической природы не-
одинаково «пропускают» сомономеры к растущему катиону
вследствие стсрических и электростатических препятствий и тем
самым оказывают влияние на структуру основной цепи.
Анионная полимеризация. Этим способом полимеризуется
большое число разнообразных мономеров Мономеры, наиболее
легко полимеризующиеся по анионному механизму, представ-
ляют собой ненасыщенные соединения диенового и винилового
рядов, содержащие электроноакцепторные заместители
(—COOR, —CN, —СбН5 и др.) К анионной полимеризации
способны также карбонилсодержащие соединения, гетероцикли-
ческие мономеры (оксиды, лактоны, лактамы, силоксаны и др.).
Мономеры с электроположительными заместителями полимери-
зуются этим методом значительно труднее. Лишь в определен-
ных условиях по анионному механизму полимеризуется этилен.
Другие олефины, простые и сложные эфиры по анионному ме-
ханизму вообще не полимеризуются Наиболее широко при-
меняемые мономеры по относительной активности в реакциях
анионной полимеризации можно расположить в ряд: акрило-
нитрил > метилметакрилат>стирол>бутадиен>а-метилстирол.
Катализаторами анионной полимеризации могут быть веще-
ства основного электронодонорного характера — щелочные ме-
таллы Li, Na, К, Rb, Cs и производные металлов I и 1,1 групп
Периодической системы амиды, гидриды, органические соеди-
нения (основания, алкилы, алкоголяты и др ).
9—816
129
Анионная полимеризация развивается также по цепному ме-
ханизму, включающему инициирование, рост, обрыв и передачу
цепи.
Инициирование (образование активного центра) прн
ионной полимеризации может происходить по различным меха-
низмам в зависимости от природы катализатора, характера
среды, температуры Существует несколько форм активных цент-
ров, которые находятся в состоянии динамического равновесия:
поляризованная молекула (~Св~—Вб+), ионная пара (~С“,
I I
I
В+), свободные ионы (~С~+В+). Преобладание той или иной
I
формы зависит от строения мономера, природы противоиона,
полярности растворителя, температуры полимеризации Иниции-
рование может происходить за счет присоединения к молекуле
мономера молекулы инициатора, свободного аниона или в ре-
зультате переноса электрона на мономер с инициирующего
анион-радикала или металла Рассмотрим различные механизмы
инициирования в присутствии катализаторов, наиболее широко
используемых на практике.
Инициирование по механизму присоединения свободного
аниона реализуется при полимеризации мономеров в растворах
щелочных металлов или амидов металлов в жидком аммиаке,
выполняющем также роль агента передачи цепи:
СН2=СН + Na+NH2_ -> NHa—СН2—СН Na+
Инициирование путем переноса электрона от инициатора на
мономер происходит при использовании щелочных металлов в
слабополярных средах. Например, при полимеризации натрий-
бутадиенового каучука полимеризация идет в среде жидкого
или газообразного бутадиена. Первоначальным актом иниции-
рования является образование анион-радикала мономера:
CHs=CH-}-Na ---* CHa-CHNa+
СН СН
И II
си2 сна
В результате взаимодействия двух анион-радикалов по сво-
боднорадикальным концам образуется бианион, являющийся
активным центром полимеризации. Подобные реакции реком-
бинации происходят в условиях относительно низкой температу-
ры и в неполярной среде. При изменении условий не исключена
одновременная полимеризация по анионному и радикальному
механизмам. Ниже приводится схема рекомбинации анион-
радикалов:
2NaCH—СН2
I
СН
II
сн2
NaCH—CHj—СН2—СН Na+
СН СН
II II
СН, сн2
При полимеризации виниловых мономеров анион-радикал
образуется в результате присоединения двух молекул мономера:
Na+2CH2=CH —ЙаСНг—СН-СНг-СН
По механизму переноса электрона с инициирующего анион-
радикала на мономер действуют инициирующие системы, обра-
зующиеся при взаимодействии щелочных металлов с полицик-
лическими ароматическими углеводородами, обладающие высо-
ким сродством к электрону. Эта реакция протекает в полярных
средах (эфирные растворители). Так, нафталин восстанавли-
вается металлическим натрием до нафталенида натрия:
Na+
При этом электрон натрия переходит на незанятую молеку-
лярную орбиталь нафталина, характеризующуюся низкой энер-
гией. Электронейтральность обеспечивается связью анион-ради-
кала с катионом натрия. Такие комплексы инициируют анион-
ную полимеризацию, например, стирола. При этом происходит
переход электрона от анион-радикала нафталина на самую низ-
кую вакантную разрыхляющую л-орбиталь винильной группы
стирола с образованием нового анион-радикала и выделением
нафталина:
Na+-bCH2=CH
+ ГСН2—CH1-Na+
Реакция идет с высокой скоростью При комнатной температу-
ре равновесие сдвинуто вправо. Ароматический углеводород вы-
ступает в роли переносчика электронов.
9*
13»
По механизму присоединения к мономеру (по двойной связи)
молекулы инициатора действуют алкилы щелочных металлов
(R—Me). Например, при полимеризации изопрена реакция ини-
циирования может быть представлена следующей схемой:
СН3 СН3
I I _ +
R—Ме+СН2=С—СН=СН2 —ь R-CH2-C=CH—СН2 Me
Рост цепи при анионной полимеризации — это ряд пос-
ледовательных одностадийных актов присоединения мономера
к первичным инициирующим центрам. Для большинства систем
стадию роста можно представить схемой, по которой внедрение
каждой новой молекулы мономера происходит между алкиль-
ным остатком, имеющим отрицательный заряд, и противоионом,
чаще всего ионом металла:
~CH2Me+CH2=CHR —*- -СН?—СНг—CHR Ме+
В случае бианионов присоединение новых молекул мономе-
ра происходит по обоим отрицательно заряженным концам
бииона При реализации такого механизма обеспечивается
строго регулярное присоединение молекул мономера по типу
«голова к хвосту», поскольку поляризованная молекула мономе-
ра перед присоединением ориентируется под влиянием ионной
пары. Однако стереорегулярность при этом не достигается. В то
же время установлено, что в присутствии некоторых металл-
органических катализаторов обеспечивается строго определен-
ное пространственное присоединение мономера Особенно этот
эффект характерен для литийорганических соединений в мало-
полярных средах. Например, при полимеризации изопрена обра-
зуется полимер, содержащий 93—94% цис-звеньев.
Направленное присоединение мономера в этих реакциях
обусловлено образованием циклического комплекса мономера
с ионной парой, в котором мономер имеет /{uc-конфигурацию.
Это связано с тем, что литий имеет наименьший среди щелоч-
ных металлов ионный радиус и самый высокий потенциал
ионизации, что обусловливает наименьшую полярность связи
Li—С. Эта связь сохраняется и в переходном комплексе Обра-
зование шестичленного циклического комплекса происходит по
следующей схеме:
СН3
сн=с
/ \6- 8*
СН2 сн2-------Li
5+дн2 5;дНо
сн—[
СНз
132
Чем выше устойчивость комплекса, тем выше стсреорегуляр-
кость образующегося полимера. При анионной полимеризации
существует несколько механизмов дезактивации активных
центров
1. Перенос гидрид-иона Н~ или другого аниона с конца ра-
стущей цепи на мономер (1) или противоионы (2):
~СН2—СН Me + СН2=СН > ~СН=СН 4-СН3—СН Me (1)
II II
XX XX
~СН2—СН Me > ~СН=СН 4- МеН (2)
X X
Первый тип реакции характерен, например, для полимериза-
ции акрилонитрила, второй — этилена с триэтилалюминием в
качестве катализатора.
2. Отрыв протона растущей цепью от растворителя или мо-
номера. Эта реакция протекает при использовании растворителя,
способного отдавать протон (жидкий аммиак, толуол):
~CIL>—CHMe+NH3 —> ~СН>—CH2+NH2 Me
3. Уменьшение реакционной способности активного центра,
например в результате реакций изомеризации:
СНз
'-'CHj—(L_Me
I
COOCHj
СН3 ОМе
~СН2—С=С,/
хо-сн3
Следует отметить, что во многих случаях процессы анионной
полимеризации протекают без обрыва кинетической цепи с об-
разованием «живущих» полимеров Активность таких полимеров
может сохраняться длительное время, что позволяет использо-
вать их для получения блок-сополимеров.
Полярность среды и температура полимеризации оказывают
влияние на скорость роста цепи и на природу получаемого поли-
мера. Так, при полимеризации стирола при одинаковых темпе-
ратуре и природе противоиона константа скорости роста цепи
возрастает с ростом полярности среды; при снижении температу-
ры скорость полимеризации уменьшается Однако эта корреля-
ция распространяется только на данную конкретную систему и
не может быть перенесена на все процессы, инициируемые ани-
онными катализаторами
133
Ниже показано, как влияют тип растворителя и температура
иа скорость анионной полимеризации стирола (противоион Na+).
Т. К
/Ср, м3/(кмоль-с)
е
Дноксан
298
5
2,2
Тетрагидрофуран
298 275
600 183
7,6 7,6
Большое значение при анионной полимеризации имеет соль-
ватирующая способность растворителя по отношению к ката-
лизатору или к каталитическому комплексу. Прн высокой
полярности растворителя, а следовательно, и высокой сольва-
тирующей способности происходит разделение ионной пары и
образование свободных карбанионов, которые в сотни раз
активнее ионных пар в реакциях роста цени. Однако в этом
случае не проявляется координирующая способность противои-
она, и даже в присутствии лития получаются полимеры диенов,
содержащие до 85% 1,2-звеньев.
Большое влияние на скорость полимеризации оказывает
природа иона щелочного металла: обычно скорость роста цепи
возрастает с увеличением радиуса катиона, что связано с
уменьшением его сольватирующей способности. Так, в ряду
Cs<Rb<K<Na<;Li наименьшая сольватирующая способность
у Cs", наибольшая — у Li+, поэтому скорость полимеризации при
использовании литиевого катализатора наименьшая. Влияние
природы щелочного металла (противоиона) на скорость анион-
ной полимеризации бутадиена в тетрагидрофуране при различ-
ных температурах показано ниже:
Ар, ы’Дкыоль-с) Li+ Na+ К+
283 К 0.85 5,4 70
243 К 0,14 1,8 17
217 К 0,03 0,6 3,1
Природа щелочного металла при анионной полимеризации
оказывает влияние на структуру получаемого полимера (табл.
2 5) Данные таблицы подтверждают, ^то наибольший коорди-
Таблица 2.5. Структура полиизопренов, полученных при анион-
ной полимеризации в присутствии щелочных металлов (раство-
ритель — пентан)
Катализатор Содержанке звеньев. %
цис-1,4 транс-1.4 | 1.2 | 3,4
Li 94 0 0 6
Na 0 43 6 51
К 0 52 8 40
Rb 5 47 8 40
Cs 4 . 51 8 37
134
пирующий эффект достигается в присутствии литиевых катали-
заторов Стереоспецифичность действия катализаторов на осно-
ве лития проявляется также при полимеризации полярных
мономеров в неполярных средах. Например, при действии литий-
органических соединений в углеводородной среде синтезирован
изотактический пол и метил метакрил ат.
Таким образом, при анионной полимеризации открываются
возможности более широкого регулирования структуры полимер-
ной цепи.
Определенная последовательность в присоединении мономер-
ных звеньев, в том числе стереорегулярность, определяется вы-
бором каталитической системы и типа растворителя, если поли-
меризация проводится в растворе. Тип растворителя влияет и
на молекулярную массу полимера. Данные о молекулярной
массе полимера, образующегося при полимеризации изопрена
и бутадиена в различных растворителях (катализатор К), при-
ведены ниже:
м 10~’ Л110-’
Изопрен Бутадиен
бензол 168 бензол 362
толуол 7,4 толуол 41,7
Из этих данных видно, что в среде толуола, который легко
вступает в реакцию переноса цепи, молекулярная масса ниже.
Кинетика анионной полимеризации сложна, так как зависит
от типа катализатора и исследована лишь для простейших си-
стем. Значительные трудности в проведении кинетического
анализа анионной полимеризации обусловлены также отсутст-
вием во многих случаях стадии обрыва цели или протеканием
обрыва под влиянием различных примесей.
Анионная сополимеризация характерна для мономеров с
электроноакцепторными заместителями По степени снижения
активности в анионной сополимеризации наиболее широко
применяемые мономеры можно расположить в ряд: акрилонит-
рил>алкилакрилат>стирол>бутадиен>этилен. Однако ак-
тивность в значительной степени зависит от полярности среды.
Так, в толуоле по снижению активности мономеры располага-
ются в ряд: бутадиен > изопрен >стирол При переходе к поляр-
ному растворителю ряд активности изменяется: стирол>бута-
диен>изопрен. Таким образом, на параметры процесса оказы-
вают влияние характер реакционной среды, тип катализатора,
свойства противоиона, температура.
Как уже говорилось, в ряде случаев в процессе анионной
полимеризации в качестве активного центра образуется анион-
радикал При сополимеризации, например стирола с метилме-
такрилатом в присутствии Li, анион-радикал не рекомбинирует-
ся в биапион, а полимеризация идет с обоих концов На ради-
135
кальном конце образуется статистический сополимер, а на ани-
онном— гомополимер метилметакрилата. Поскольку на процесс
сополимеризации влияет природа не только сополимера,
ио и растворителя, константы сополимеризации не могут быть
использованы в качестве величин, характеризующих реакцион-
ную способность мономера.
2.1.4. Ионно координационная полимеризация
Ионно-координационной полимеризацией называют каталитиче-
ский процесс образования макромолекул, в котором стадии
разрыва связи в мономере предшествует возникновение коор-
динационного комплекса между ним и активным центром.
Характер и структура комплекса зависят от тина катализатора
и строения мономера. Комплексообразование мономер — ката-
лизатор обусловливает возможность синтеза стереорегулярных
полимеров из широкого круга мономеров (сс-олефинов, диенов,
ряда полярных мономеров и др.) Катализаторы, вызывающие
стереорегулирование в процессе присоединения мономерных
звеньев, называют стереоспецифическими. В качестве катализа-
торов наибольшее распространение получили комплексные сое-
динения трех типов соединения Циглера — Натта (открытые
в 1954 г. и названные по имени их открывателей), образующие-
ся при взаимодействии органических производных металлов
I—III групп Периодической системы с солями (обычно хлори-
дами) переходных металлов IV—VIII групп, л-аллильные
комплексы переходных металлов; оксидно-металлические ката-
лизаторы Варьируя состав и способ получения катализаторов,
можно регулировать их каталитическую активность и стереос-
пецифичность действия, т. е. способность «отбирать» при полиме-
ризации мономерные звенья определенной конфигурации и
ориентировать их при подходе к активному центру. Состав этих
катализаторов сложен. Из катализаторов Циглера — Натта в
производстве обычно используют комплексы па основе алюми-
нийалкилов и производных титана и ванадия. Наибольшее
значение эти катализаторы имеют при полимеризации неполяр-
ных олефинов (этилен, пропилен) и диенов (бутадиен, изопрен).
Например, полиэтилен с высокой степенью кристалличности
этим методом может быть получен при низком давлении
Эффективными катализаторами полимеризации а-олефинов,
диеновых и других мономеров служат оксиды переходных ме-
таллов, в частности активированные оксиды хрома и молибде-
на. Активность оксидно-хромовых и оксидно-молибденовых
катализаторов обусловлена частичным восстановлением метал-
лов при предварительной термообработке в промежуточные
валентные состояния: катализаторы, содержащие только Сгб+
или Сг3+ неактивны, содержащие Cr5*—активны. Стереоспеци-
136
фичность оксидно-металлических катализаторов ниже, чем у
комплексных металлоорганических систем, но добавлением
алюмииийалкилов она может быть значительно повышена.
Ионно-координационная полимеризация происходит также
под действием ряда других каталитических систем, включающих
соединения Nt, Со, Ru, Rh, Pd, 1г и др., а также в определенных
условиях и на катализаторах анионной или катионной полиме-
ризации
Большое практическое значение при ионно-координационной
полимеризации имеют координационные комплексы, состоящие
из молекул мономера и катализаторов на основе переходных
металлов (В этом комплексе переходный металл образует цент-
ральный ион, а молекулы мономера являются лигандами.)
В координационной сфере переходного металла молекулы мо-
номера деформируются, вследствие чего удлиняется связь С=С,
изменяются валентные углы у двойной связи и происходит ее
поляризация. Большое значение имеет подбор комплексообра-
зующих катализаторов и мономеров, так как структура и со-
став катализатора и молекул мономера должны соответство-
вать друг другу. Стабильность связи переходный металл —
полимер является решающим фактором активности катализато-
ра В отсутствие мономера эти связи должны иметь определен-
ную устойчивость и время жизни Но при комплексообразова-
нии эта же связь должна дестабилизироваться и «принять»
мономер.
Таким образом, истинными катализаторами в таких системах
являются органические соединения переходных металлов или
в свободном состоянии, или в виде комплексов, или в виде по-
верхностных соединений, встроенных в кристаллическую решет-
ку твердого катализатора.
Процесс присоединения мономера к растущей макромолекуле
при помощи координационных комплексов включает следую-
щие основные стадии: диффузия молекулы мономера к поверх-
ности твердого катализатора, содержащего активный центр;
адсорбция и ориентация мономера на поверхности катализатора
(образование комплекса); соединение мономерного звена, во-
шедшего в комплекс, с активным центром, сопровождающееся
переходом активного центра на вновь присоединившееся звено;
отделение от катализатора полимеризованных звеньев. Рас-
смотрим механизм действия наиболее распространенных
каталитических систем стереоспецифического действия.
Катализаторы Циглера — Натта. Наибольшее промышленное
применение получил комплекс, образующийся при взаимодей-
ствии тетрахлорида титана TiCl4 с триэтилалюминием А1(С2Н5)з.
При таком взаимодействии протекает ряд химических реакций,
в результате которых происходит алкилирование соединения
переходного металла и его восстановление до TiCl3„ Образуется
137
четырехчленный комплекс на поверхности выпавших в осадок
кристаллов трихлорида титана:
С1 С1 С2НБ
'Ч'тГ А!^
CIZ СН, ^СаНв
СН3
Наибольшее влияние на процесс стереорегулирования ока-
зывает строение соединений металлов переменной валентности»
входящих в состав комплекса. Широко применяемый TiCh в
зависимости от технологии его получения может иметь несколь-
ко кристаллических модификаций (а, 0, у, б), из которых наи-
большей стереоспецифичностью характеризуется a-TiCl3. Эта
модификация состоит из трехслойных кристаллических пластин
(вдоль основной кристаллической оси два слоя атомов хлора
чередуются со слоем атомов титана) Рост полимеров происхо-
дит на боковых гранях кристаллов Слоистую структуру имеют
также 7- и 6-формы TiCl3. При полимеризации диенов с приме-
нением а-, у- и 6-TiCls образуются преимущественно транс-1,4-
полимсры; 0-TiCl3 имеет волокнистую структуру, и при полиме-
ризации на нем образуется в основном цис- 1,4-изо.мер
Процессу полимеризации предшествует координация моле-
кулы мономера у атома Ti и внедрение мономера в состав ком-
плекса за счет разрыва связи Ti—Cl При этом мономер высту-
пает в роли донора л-электронов, а переходный металл ката-
лизатора благодаря наличию вакантных d-орбиталей является
акцептором. За счет координации с донором образуется л-ком-
плекс, возникновение которого приводит к ослаблению связи
Ti—Cl в катализаторе, и облегчается внедрение мономера по
этой связи с образованием нового шестичленного комплекса
с последующей его перестройкой в четырехчленный Полимери-
зация производных этилена может быть представлена следую-
щей схемой
'<& C*HS сн5 cu zck —> * Ж< - Cl } сн2 СН4Х-СН2 СНз ХСН=СН2 СНз СК /С2Н5 —> —> C1 сн2 c*Ht X СНз
138
xc2hs
сн—CH2
I I
X CH3
Ck / а г^с2н3
>1^
CLX X2H5
CH2
CH— C2H5
В регенерированном четырехчленном цикле содержится один
из атомов углерода молекулы мономера, соединенный с атома-
ми титана и алюминия, а исходная этильная группа удаляется
из цикла вместе с другим атомом углерода винилового мономе-
ра. Дальнейшее присоединение мономера идет аналогичным
образом и происходит постепенное вытеснение образующейся
полимерной молекулы из структуры комплексного катализато-
ра. Механизм полимеризации предполагает строго определен-
ное пространственное расположение заместителей при атоме
углерода относительно плоскости основной молекулярной цепи
полимера (стереорегулярность)
Наиболее склонны к полимеризации под действием этих ка-
тализаторов мономеры с повышенной электронной плотностью
на двойной связи Кроме того, при оценке активности мономера
следует учитывать роль стерических факторов в реакции внед-
рения мономеров. Так, несмотря на то, что электронная плот-
ность на двойной связи этилена меньше, чем у пропилена, ско-
рость его полимеризации значительно выше, так как отсутству-
ют стерические препятствия при внедрении мономера.
Большая роль в процессе полимеризации диенов отводится
л-аллильным комплексам,. образующимся при взаимодействии
комплексных соединений переходных металлов с диенами.
Предполагается, что атом переходного металла присоединяется
по двойным связям диена, а этильная группа выделяется из
четырехчленного комплекса и присоединяется к одному из кон-
цевых атомов углерода диеновой группы л-Аллильный комплекс
является активным центром каталитической системы, на кото-
ром протекает рост цепи. Рост цепи представляет собой чере-
дование актов образования л-комплексов переходного металла
с диеном, превращения их в л-аллильные комплексы и коорди-
нации следующей молекулы мономера с изомеризацией л-ал-
лилыюго комплекса, т. е. каждая последующая молекула
мономера вытесняет каждое предыдущее мономерное звено по
'Следующей схеме:
образование л-аллильного комплекса
ск хЧН5 /С2н5 уНз
>АК + СН2=С—сн=сн2—>
cr с2н5
13»
сн3—с=сп-сн2— C2HS
Cl сн2 C1 &
\n<Z XzA1(CoH5)2—>ch3-c; >
ск \ / ~ X /
C! CH
с2п5
с2п5
С1
СН2—С2Н5
Л-аллильнни комплекс
восстановление исходной структуры комплекса (вытеснение
мономерного звена)
Cl
CH3-cf4 >тич /Al-
СН ci СгН5
сн2—C2HS
Cl
Rl zc2H5
'C2H5
Cl 19
C=CH—CH2—C0H5
рост цепи (координация следующих молекул мономера и
внедрение их по связи Ti—С)
CI H2C C! Cl
' \ ,. /С2П5 л СНд—С—СП=СНг v <
-------------->СНз—C,
Clz XC2H5
Cl
CH
ai(c2h5)2
С=СН— СНо— С2Н5
СНЯ
СН2 СПз
(!н2-с=сн-сн2)л-с2н5
Подобные каталитические системы обеспечивают формиро-
вание пространственно-регулярных полимеров, причем стерео-
регулярность и конфигурация звеньев зависят от типа мономе-
ра, условий полимеризации и состава комплекса. В качестве
примера ниже приведены данные о структуре полиизопрена
(содержание звеньев, %), полученного в присутствии
катализаторов:
различных
TiCI4+A1R3
VCIj+A1R3
Ti(OR)4+A!R3
цш>1,4
96
транс-1,4
80—99
3.4
94—99
Обрыв цепи при ионно-координационной полимеризации
происходит различными способами:
140
за счет переноса гидрид-иона с конца растущей цепи на про-
тивоион
X ,Х R
X \ /
Меч Мег
* ' СН2 R
I
СН2
к
СН2 = СН
I,
R
R
R
за счет передачи цепи на металлалкил
хХч Хх »Х\ /R
.Ti ;aI(R)2 + R3AI -♦ Тг АГ
Х 'СНг х/ '*'/ 4 R
I t
CH2R
+ R2Al-CH2-CH2-Rr
Взаимодействие с примесями чаще всего приводит к гибели
активного центра.
л-Аллильные комплексы переходных металлов. Весьма эф-
фективными стереоспецифическими катализаторами являются л-
аллильные комплексы ряда переходных металлов (Ni, Сг, Т»,
Rh, Nb, и др.) общей формулы (CwH2n-!)mMe или
(CnH2n-i)mMeX (где X— галоген или другая электроотрица-
тельная группа). Активность и стереоспецифичность этих ком-
плексов при синтезе, например ^ис-М-бутадиепа, резко повы-
шаются в присутствии кислот Льюиса (ZnCl2, AICI3, N1CI2
и др.), а также органических акцепторов электронов. В этих
л-аллильных комплексах связь Me—С стабилизована за счет
взаимодействия вакантных d-орбиталей металла с л-электрона-
ми двойной связи, что обеспечивает устойчивость комплексов.
В качестве примера рассмотрим полимеризацию бутадиена на
л-кротильных комплексах галогенида никеля.
Молекулы катализатора при взаимодействии с бутадиеном
образуют две пространственные изомерные конфигурации в за-
висимости от положения группы СНз относительно плоскости
четырехчленного цикла комплекса.
Пространственная конфигурация полимеризующегося моно-
мера определяется пространственной конфигурацией комплекса:
транс-1,4-полидиены образуются из син-формы, цис- 1,4-полидие-
ны — из анти-формы. Процесс полимеризации бутадиена с
участием л-аллильного комплекса протекаете несколько стадий
Инициирование происходит при взаимодействии стабильного л-
141
аллильного комплекса никеля с мономером, и л-связь катали
затора превращается в о-связь:
СН3—СН=СН—СНг—NiX —
нсх 2Nix
/СИ
СНз
сик-форма
^сн2
Hcf ''NiX
CH3
с«га-форма
CH2
HCf 4>NiX + CHj>=CH-CH=CH2
CH3
CH3-CH=CH~CI Ig-NiX
H2c / \ ch2
нс—CH
оягм-форма
Молекула бутадиена координируется у атома Ni, под дейст-
вием координированной молекулы связь Ni—С ослабляется и
молекула мономера внедряется в комплекс, вытесняя кротиль-
ный радикал При этом восстанавливается четырехчленный л-
аллильный комплекс. В каждом акте присоединения формиру-
ется цепь, в которой звенья находятся в цис-1,4-конфигурации:
,СН2
NiX
Н=СН
Н
С1Ь
СН3
Таким образом, рост цепи происходит аналогично росту цепи
при полимеризации па катализаторах Циглера — Натта путем
последовательной координации и внедрения молекулы мономе-
ра В случае слабых доноров при более восстановленном атоме
никеля координация мономера идет по одной двойной связи*
/да
HCf ^NiX + CHi^CH-CH—СН2—>
CH3-CH==CH-CH2-NiX
/ \
н£с=сн
СНа
син -форма
CU^CHz
142
Рост цепи приводит к образованию транс-1,4-формы:
Ограничение роста цепи возможно за счет передачи цепи на
мономер, что приводит к регенерации активного центра:
^СН2 /Н2
Hcf ')NiX + СН£=СН-СН=СН2—^NiX + ^сн=сн-сн=сн2
ГН . узн
~ СН2 СНз
В ряде случаев рост цепи продолжается до израсходования
мономера, т е образуются «живущие» молекулы.
Структура образующегося полимера и скорость полимери-
зации определяются природой атома металла и лиганда. Введе-
ние электроноакцепторных лигандов приводит к возрастанию*
положительного заряда на атоме металла, что способствует
ускорению процесса роста цепи и приводит к образованию анти-
формы и соответственно цис- 1,4-структуры полимера Такое же
действие оказывают добавки, комплексообразование с кото-
рыми повышает положительный заряд на металле (соли, три-
хлоруксусная кислота и др.). Если лиганды — галогениды, та
их электроноакцепторные свойства выражены в меньшей сте-
пени и, следовательно, меньшее значение имеет положительный
заряд на атоме металла В этом случае образуется сии-форма
л-комплекса и формируется структура транс-1,4
Оксидно-металлические катализаторы эффективны при поли-
меризации сс-олефинов и диеновых углеводородов. Наиболее
распространенным и изученным катализатором такого типа
является оксид хрома, нанесенный на поверхность какого-либо
носителя (силикагель, алюмосиликат, оксид алюминия и др).
В основе каталитического действия оксида хрома лежит спо-
собность хрома изменять свою валентность. Активными цент-
рами при полимеризации на оксидно-хромовых катализаторах
являются ионы Сг5+, находящиеся на поверхности. Полимери-
зация в этом случае идет без индукционного периода, поэтому
используют катализатор, активированный в вакууме, который
содержит Сг6ь. При введении мономера процесс полимеризации
143
сразу не развивается, а во время индукционного периода Сгб4~
восстанавливается мономером до Сг5+. По всей вероятности,
ионы пятивалентного хрома ведут себя, как акцепторы электро-
нов, и при взаимодействии с молекулами мономера образуют
координационные комплексы. Комплексообразование облегчает
присоединение молекул мономера к растущей цепи и способ-
ствует получению стереорегулярных структур. Полиэтилен,
синтезированный на оксчдно-хромовом катализаторе, имеет
более высокие плотность и кристалличность, чем даже полиэти-
лен, полученный на катализаторе Циглера — Натта. При поли-
меризации диенов образуются полимеры, содержащие ~100%
транс-звеньев.
2.2 ПОЛИКОНДЕНСАЦИЯ
Поликонденсацией называют ступенчатый процесс получения
полимеров из би- или полифункциональных соединений, в ко-
тором рост макромолекул происходит путем химического
взаимодействия функциональных групп молекул мономеров друг
с другом и с л-мерами, накапливающимися в ходе реакции, а
также молекул zz-меров между собой. На концах образующихся
макромолекул всегда присутствуют свободные функциональные
группы Каждый акт взаимодействия при поликонденсации
сопровождается исчезновением у реагирующих частиц функцио-
нальных групп Часто (но не всегда) поликонденсация сопро-
вождается выделением низкомолекулярных продуктов реакции.
В табл 2.6 указаны отличительные особенности поликонденса-
ции и полимеризации В общем виде реакция поликонденсации
бифункциональных мономеров описывается уравнением
п(а—А—а)+п(Ь—В—Ь) —>- а—[—А—В—]„—&+(2л— 1)аЬ
где а—А—а, b—В—b — исходные мономеры; а и Ь — функциональные груп-
пы; ab — выделяющееся низкомолекулярное соединение.
Различают гомополиконденсацию и гетерополиконденсацню.
Гомополиконденсацией называют реакции, в которых участвует
минимально возможное для данного случая число типов моно-
меров или только молекулы одного мономера, содержащего два
типа функциональных групп„ Типичным примером гомополикон-
денсации служит синтез полиамидов из аминокислот:
hH2N-R-CO-OH —- Н[—NH-R—СО—1пОН+(л—1)Н2О
Гетерополиконденсацией называют реакции с участием мо-
лекул мономеров, содержащих различные функциональные
группы, способные взаимодействовать друг с другом, например
диаминов с дикарбоновыми кислотами:
лН2Ы—R—NH2+nHO—СО—R'—СО—ОН —*
—* Н[—NH—R—NH—СО—R'—СО-] „ОН + (2л— 1)Н2О
144
Таблица 2.6. Особенности процессов полимеризации и поликонденсации
Параметры процесса Полимеризация Поликонденсация
Характеристика про-
цесса
Механизм процесса
Обратимость реакции
Выделение низкомо-
лекулярных продуктов
Цепной процесс
Последо в ательное при-
соединение молекул мо-
номера к активному
центру растущей поли-
мерной цепи по ионному
или радикальному меха-
низму
Необратима
Не выделяются
Ступенчатый процесс
Замещение (промежуточные
продукты на отдельных ста-
диях могут быть выделены
и охарактеризованы)
Строение составного
звена
Эквивалентно строению
мономера
Исчезновение молекул
мономера
Зависимость средней
молекулярной массы
полимер а от степени
конверсии
В конце процесса
Средняя молекулярная
масса от степени кон-
версии не зависит, и вы-
сокомолекулярный поли-
мер присутствует при
низкой степени конвер-
сии
Возможна обратимость
Выделяются в большинстве
случаев и могут сдвигать
равновесие реакции в сторо-
ну образования исходных
веществ
В большинстве случаев от-
личается от строения исход-
ного мономера вследствие
выделения низкомолекуляр-
ных продуктов
На ранних стадиях реакции
Средняя молекулярная мас-
са зависит от степени кон-
версии, и высокомолекуляр-
ный полимер присутствует
только при высокой степени
конверсии
Реакцию, в которой помимо мономеров, необходимых для ее
протекания, участвуют и другие мономеры, называют сополи-
конденсацией, например:
2nHOCORCO—ОН+л! I2NR'NH2+nH2NR"NH2
4* Н[—HNR'NHCORCOHNR"NHCORCO—]nOH+(4n— 1)H2O
Сополиконденсация может происходить и при взаимодей-
ствии двух мономеров, каждый из которых способен самостоя-
тельно вступать в гомополиконденсацию, например при поли-
конденсации двух амино- или оксикислот.
По пространственному строению получаемых полимеров раз-
личают линейную и трехмерную поликонденсацию. При линей-
ной поликонденсации из бифункциональных мономеров получа-
ют линейные полимеры, при трехмерной — из мономеров с тремя
или большим числом функциональных групп образуются раз-
ветвленные или трехмерные (сетчатые, сшитые) структуры.
В последние годы большое значение приобрела полицикло-
конденсация— двухступенчатый синтез лестничных полимеров
10-816 145
путем внутримолекулярной циклизации продуктов, полученных
на первой стадии
Процессы поликонденсации широко применяются для синте-
за полимеров с рядом специфических свойств термостойких,,
полупроводников, электропроводящих, фотоактивных, биополи-
меров, катализаторов, ионитов и др.
В качестве мономеров при поликонденсации применяются
соединения с двумя или более функциональными группами (ОН,.
OR, NH2, Cl, СООН, COOR, COCI, SiOH, SiOR и др.). Некото-
•>ie из мономеров, имеющие наибольшее практическое значение»
перечислены в табл. 2.7 Бифункциональные мономеры можно
разделить на три основных класса
1) мономеры, содержащие различные функциональные груп-
пы, способные взаимодействовать друг с другом оксикислоты
(НО—R—СООН), аминокарбоновые кислоты (H2N—R—СООН)
и др.;
2) мономеры, содержащие одинаковые функциональные
группы, не способные в условиях реакции взаимодействовать-
друг с другом: диамины (H2N—R—NH2), дикарбоновые кисло-
ты (НООС—R—СООН), их производные и др.;
3) мономеры, содержащие одинаковые функциональные
группы, способные взаимодействовать друг с другом, например
гликоли (НО—R—ОН).
Для поликонденсации в первом случае можно использовать-
один мономер, как, например, при синтезе сложных полиэфиров
из оксикислот:
О О
И II
«НО—(СН2)х—С—ОН —*• Н[—О—(СН2)Х—С—]пОН+(л —1)Н2О
Для поликонденсации во втором случае требуется два моно-j
мера: например дикарбоновая кислота и диамин при получении
линейного полиамида: ,
о О
II II
пНО—С—(СНг)4—С—ОН + nHsN— (CH2)eNHa -->
ГО ОН Н 1
II II I I
---> HOL—С—(СНа)4-С—N-(CH2)e—N—JrtH4- (2л—1)НаО
Наконец, в третьем случае синтез проводят, используя один
мономер с одинаковыми функциональными группами. В качест-
ве примера можно привести синтез простых эфиров из гликоля:
лНО—R—ОН —*• Н[—О—R—]„ОН + (л — 1)Н2О
Основные кинетические закономерности поликонденсации
как ступенчатой реакции взаимодействия функциональных групп
мономеров определяются наличием термодинамического рав-
146
Таблица 2.7. Монтеры для получения некоторых поликонденсационных по-
лимеров и олигомеров
Класс полимеров, полу-
чающихся при поликон-
денсации
Функциональные группы
мономеров
Исходные мономеры
Простые полиэфиры
~R-O-R'~
Сложные полиэфиры
z~-R -С О —
—ОН НО—
—ОМе ОН—
—ОН С1—
—СООН НО—
—COOR НО—
—СОС1 НО—
Полиамиды
~NH—С—R~
II
О
Полиацетали
~О—СН—О~
R
Полиангидриды
~С О С~
- NHj НООС—
—nh2 сюс-
—nh2 ROOC—
-он Н\
он о/0-
Гликоль — гликоль
Ллкоголят — гликоль
Гликоль — дихлоралкан
Днкарбоновая кислота —
гликоль
Диэфнр дикарбоновой
КИСЛОТЫ — ГЛИКОЛЬ
Дихлорангидрнд дикар-
боновой кислоты — гли-
коль
Диамии — днкарбоновая
кислота
Диамин — дихлораигнд-
рид дикарбоновой кис^
лоты
Диамин — диэфир дикар-
боновой кислоты
Гликоль — ди альдегид
—СООН НООС— Дикарбоновые кислоты
—ООСОСН3 Н3СОСОО— Смешанные диаигидрнды
днкарбоновой и уксусной
кислот
-NH2
НООС—
НзСОСО—
Полннмиды
Дндмин — диангндриды
или другие производные
тетракарбоновых кислот
Новесия между начальными и конечными продуктами реакции.
В связи с этим различают поликонденсацию равновесную (об-
ратимую) и неравновесную (необратимую). Принципиальным
отличием обратимых процессов от необратимых является воз-
можность протекания в обратимых процессах обратных реакций,
т. е. взаимодействия полимера с низкомолекулярным продуктом’
приводящего к распаду полимерных цепей. Примерами обрати-
мой поликонденсации служат реакции получения полиэфиров
(*) или полиамидов (2) при нагревании дикарбоновых кислот
с гликолями или диаминами соответственно:
лОН—R—ОН+лОН—СО—R'—СО—ОН —Н[—О—R—О—СО—
—R'—СО—]ПОН+(2л — 1)Н2О (1>
т
nH2N—R—МНг+лОН—СО—R'—СО—ОН —Н[—<NH—R—
—NH—СО-R'—СО—]пОН+(2л— 1)Н2О (2>
В подобных реакциях для сдвига поликонденсационнога
равновесия в сторону образования полимеров исключительна
большое значение имеет вывод из реакционной среды воды или
другого низкомолекулярного продукта реакции.
Необратимыми поликонденсационными процессами являют-
ся реакции образования феноло- и карбамидоальдегидных по-
лимеров, а также полиэфиров и полиамидов.
Процесс образования макромолекул по механизму поли-,
конденсации состоит из следующих основных стадий:
образование реакционных центров как в молекулах моно-
мера, так и в молекулах образующихся олигомеров;
образование макромолекул в результате взаимодействия ре-
акционных центров мономеров, олигомеров, полимеров; про-
цесс имеет статистический характер, т. е. характеризуется не-
зависимостью протекающих в системе реакций; наименьшую-
долю составляют реакции взаимодействия n-меров с мономера-
ми, так как исчезновение последних происходит на сравнитель-
но ранних стадиях; процесс идет за счет взаимодействия ре-
акционных центров различной длины друг с другом; эта ста-
дия определяет все главные характеристики полимера молеку-
лярную массу, ММР, состав и др.;
прекращение образования макромолекул из-за дезактива-
ции концевых функциональных групп, а также снижения ре-
акционной способности функциональных групп в результате
действия физических факторов (высокая вязкость системы, вы-
падение полимера в осадок и т д.)
Направление реакции определяется константой поликонден-
сационного равновесия К, которая представляет собой соотно-
шение констант скоростей прямой и обратной реакций. В слу-
чае реакции, не сопровождающейся выделением низкомолеку-
лярного продукта, т. е. протекающей по схеме
к
А+В С
константа К равна
где (А] и (В] — равновесные концентрации функциональных групп, не вст
пивших в реакцию; [С]— равновесная концентрация связей, образовавших)
при поликонденсации.
148
При реакции с выделением низкомолекулярного продукта Д
к*
А+В чрь С+Д
константа К' равна
[С][Д1
(2.24)
Значение константы поликонденсационного равновесия ха-
рактеризует обратимость процесса. Значения констант равно-
весия обратимых реакций находятся в пределах до 100, необ-
ратимых— значительно выше (^103). Степень завершенности
обратимых реакций лимитируется термодинамическими факто-
рами, необратимых — кинетическими. Скорости этих реакций
и энергии активации также значительно различаются: констан-
та скорости обратимой реакции составляет 10 3—10~5
м3/(кмоль-с), необратимой — до 105 м3/(кмоль-с); энергия ак-
тивации равна 84—167 и 8—42 кДж/моль соответственно
В необратимых процессах поликонденсации распад цепей
подавляется, что может быть обусловлено или низкой ско-
ростью обратных реакций, или тем, что образовавшиеся поли-
меры не взаимодействуют с выделившимися низкомолекуляр-
ными продуктами.
При анализе кинетики поликонденсации используют прин-
цип Флори, предполагая, что активность реагирующих групп
одинакова и не зависит от длины цепи полимера. В случае по-
ликонденсации мономеров, взятых в эквимольных количествах,
кинетическое уравнение изменения концентрации функциональ-
ных групп выглядит следующим образом:
« ГМ = —, (2.25)
1 X
где k — константа скорости реакции; [Ао] — начальная концентрация функ-
циональных групп; х — глубина превращения (доля прореагировавших к даи-
ному моменту функциональных групп).
Это уравнение справедливо для необратимых процессов и
для начальных стадий обратимых в случае, если обратной ре-
акцией можно пренебречь.
Для обратимой гомополиконденсации уравнение зависимо-
сти средней степени поликонденсации п от константы поликон-
денсационного равновесия К' и содержания выделяющегося
низкомолекулярного продукта Д имеет вид
л = УКДДЬ (2.26)
Следовательно, для подавления обратных реакций необходи-
мо непрерывно удалять из реакционной среды низкомолеку-
лярные продукты.
В случаях, когда поликонденсация протекает без выделения
низкомолекулярных продуктов, это уравнение с некоторым уп-
рощением (если не учитывать процессы дезактивации реагиру-
ющих групп) можно представить в виде п—УК, т. е. средняя
степень полимеризации (следовательно, и молекулярная мас-
са) пропорциональна корню квадратному из константы рав-
новесия.
Молекулярно-массовое распределение при поликонденсации
в простейших случаях описывается уравнением
pn=*"-i(l — х), (2.27>
где Рп — доля макромолекул со степенью поликонденсации Я; х—глубина
превращен"?:.
Как мы уже говорили, если в реакции поликонденсации уча-
ствуют мономеры с тремя и большим числом функциональных
групп, то образуются разветвленные или сшитые структуры.
Трехмерная поликонденсация характеризуется точкой гелеобра-
зования (момент образования нерастворимого полимера). Сте-
пень превращения в точке гелеобразования определяется функ-
циональностью мономера. Связь между гелеобразованием и сте-
пенью завершенности реакции х может быть установлена на
основании уравнения Карозерса для эквимольного соотноше-
ния функциональных групп:
*“(2Ле₽)-(2/Я/сР) (2.28>
(fcP — средняя степень функциональности). При л->-оо
Хоо=2//ср* (2 29)
Если /ср=2, то х=1, т. е. реакция идет до полного превра-1
щения с образованием только линейных полимеров, без геле-1
образования; если fcp=3, то степень завершенности в момент|
гелеобразования составляет 2/3; если /ср=4, то реакция до,
гелеобразования протекает всего на 50%. ।
При поликонденсации большое влияние на ход реакции ока-'
зываюг так называемые сопутствующие процессы. Обычно это!
влияние отрицательно, так как подавляется основная реакция»!
ухудшается качество синтезируемого продукта, увеличивают-!
ся расход сырья и себестоимость и т. д. По природе сопутству-1
ющих процессов их можно разделить на химические и физиче-’
ские. К физическим относятся процессы исключения функцио-
нальных групп из реакции вследствие самопроизвольного вы-
падения полимера (или олигомера) в осадок, блокирования
функциональных групп молекулами растворителя, повышения
вязкости системы и т д., к химическим — нежелательные реак-
ции функциональных групп с примесями, растворителями, моно-
функциональными добавками. Например, при взаимодействии
•50
хлорангидридных групп мономера с монофункциональной до-
бавкой
ClOCRCOCl+2HjiNR' —► R'NHCORCONHR'+2HC1
происходит дезактивация функциональных групп. Из химиче-
ских реакций большое значение имеют циклизация, деструк-
ция, структурирование
В зависимости от механизма процессы циклизации разде-
ляют на внутри- и межмолекулярные. Внутримолекулярная
циклизация происходит при взаимодействии функциональных
групп, принадлежащих одной и той же молекуле мономера.
Примером внутримолекулярной циклизации бифункциональных
соединений может служить побочная реакция, протекающая
при синтезе сложных полиэфиров из оксикислот:
основная
реакция
Н— I-O—R-CO-] „ -ОН4- (л — 1) Н20
лНО-R—СО—ОН—
сложный полиэфир
циклизация
---------►
nR--СО+лН2О
О
лактон
Межмолекулярная циклизация происходит при взаимодейст-
вии функциональных групп различных молекул:
CHS—CHS
НО* \>Н СН2—СН,
+ о^ \>-f-2C,HeOH
Н,С2О OC,HS 'ос—co
ОС—co
Наиболее легко циклизуются соединения, образующие 5—
7-членные циклы, хотя возможно образование циклов и из про-
межуточных продуктов поликонденсации, содержащих 20—40
атомов.
Процессы деструкции (разрушения макромолекул) происхо-
дят при взаимодействии высокомолекулярных продуктов поли-
конденсации с низкомолекулярными, т. е. при обратных реак-
циях. Возможен также межцепной обмен, который протекает
или в результате взаимодействия концевых групп одной мак-
ромолекулы с внутренним звеном другой, или при взаимодей-
ствии двух внутренних функциональных групп различного типа.
В этих случаях уменьшение молекулярной массы происходит
не всегда.
151
Соотношение скоростей реакций роста макромолекул и по-
бочных реакций определяет молекулярные характеристики по-
лимера (структуру, молекулярную массу, молекулярно-массо-
вое распределение), содержание примесей и др Это соотноше-
ние зависит главным образом от строения мономеров, степени
их очистки, условий проведения синтеза. Например, внутримо-
лекулярную циклизацию можно предотвратить увеличением
концентрации мономеров и снижением температуры процесса.
Молекулярная масса продуктов поликонденсации зависит от
глубины превращения, соотношения исходных мономеров, кон-
центрации регуляторов молекулярной массы, условий процесса.
Вли—’”' глубины превращения (конверсии) на молекулярную
массу при гомогенном процессе приближенно характеризуется
уравнением
й=1/(1-х), (2.30)
из которого видно, что при х-*-1 молекулярная масса увели-
чивается. Например, при х=0,5 п=2,0; при х=0,995 п=200;
при х=0,999 п= 1000
Таким образом, с увеличением глубины полимеризации мо
лекулярная масса полимера возрастает. В этом отличие по
ликонденсации от реакций цепной полимеризации, в которы:
молекулярная масса не зависит от глубины процесса (рис. 2.2)
Поскольку в гетерогенных системах реакция поликонденсаци!
осложняется процессами диффузии мономера через границ
раздела, высокомолекулярный полимер может образоватьс:
при глубине процесса намного меньше 1 (в расчете на обще
исходное количество реагентов).
Рнс. 2.2. Зависимость средней молекулярной массы Л7ср от степени конвер
сии А прн полимеризации (/) и полнконденсацин (2)
Рис. 2.3 Изменение кривых молекулярно-массового распределения (С* — мас-
совая доля фракции; х — степень полимеризации) в процессе поликонденсация
в зависимости от количества вступивших в реакцию функциональных групп:
/ — 10%. 2—20%: 3 — 50%; 4 — 100%
152
Поскольку в полимере с увеличением продолжительности
процесса возрастает доля макромолекул с большой молекуляр-
ной массой и снижается доля низкомолекулярных олигомеров,
полидисперсность системы непрерывно изменяется (рис. 2 3)
Полимеры с узким ММР образуются на начальных стадиях ре-
акции. С увеличением глубины полимеризации распределение
по молекулярным массам становится значительно шире.
Большую роль в поликонденсации играет соотношение ис-
ходных компонентов. Для достижения высокой молекулярной
массы требуется эквимольное соотношение мономеров. При из-
бытке одного из мономеров процесс поликонденсации протека-
ет до тех пор, пока мономер, присутствующий в меньшем ко-
личестве, не будет израсходован. Тогда избыточный мономер
и все макромолекулы будут содержать на обоих концах оди-
наковые функциональные группы. Поликонденсация прекраща-
ется, не достигнув требуемой глубины. Эквивалентность соот-
ношения исходных веществ в процессе поликонденсации мо-
жет нарушаться вследствие летучести одного из мономеров или
химического изменения функциональных групп и превращения
их в нереакционноспособные в условиях поликонденсации.
В гетерогенных системах может наблюдаться кажущееся на-
рушение правила эквивалентности функциональных групп, од-
нако в зоне реакции эквимольное соотношение компонентов
обязательно должно сохраняться.
Один из надежных способов регулирования молекулярной
массы поликонденсационных полимеров заключается во вве-
дении монофункционального компонента, который взаимодей-
ствует с функциональными группами, дезактивируя их. Моле-
кулярная масса синтезируемых продуктов зависит от содержа-
ния в системе монофункционального соединения: чем больше
его концентрация, тем на более ранних стадиях прекращается
рост макромолекул. Например, при получении полиамидов ре-
акцию прекращают добавлением уксусной кислоты, которая
вступает в реакцию с концевой аминогруппой:
-СО— (СН2) 4—СО—NH— (CI I2)6—NH2+ОН—СО—СНз —+
—Н2О
—- СО- (СН2) 4-CO-N н—(СН2) 6—NH—СО—CHS
Повышение температуры ускоряет поликонденсацию, об-
легчает удаление образующегося низкомолекулярного продук-
та, в результате чего реакция смещается в сторону образова-
ния более высокомолекулярных соединений. Однако после до-
стижения равновесия молекулярная масса выше при более
низкой температуре. Это используется на практике для сокра-
щения продолжительности синтеза полимера: сначала процесс
ведут при более высокой температуре, а после достижения рав-
новесия — при более низкой. Молекулярная масса полимера
153
зависит также от способа проведения поликонденсации (в рас-
плаве, в растворе, на поверхности раздела двух фаз, в твер-
дой фазе). Одним из наиболее производительных, легко авто-
матизируемых и перспективных методов поликонденсации яв-
ляется межфазная поликонденсация. Процесс протекает в ге-
терогенной системе на границе раздела двух несмешивающих-
ся жидкостей, содержащих исходные компоненты и не раство-
ряющих получаемый полимер. Константы скорости межфазной
поликонденсации достаточно высоки—102—Ю6 м3/(кмоль-с),
поэтому процесс можно проводить при относительно низких
температурах, получая полимер с высокой молекулярной
массой.
Контрольные вопросы
1. Какие преимущества имеет нонио-координациоииая полимеризация перед
другими методами полимеризации? Каковы особенности структуры поли-
меров. получаемых этим методом?
2. Сравните методы полимеризации и поликонденсации. В чем основные
преимущества и недостатки метода поликонденсации? Приведите примеры
полимеров, получаемых этим методом
3. Что такое «живые* молекулы? Как они получаются? Какое практическое
применение находят?
4. Мономер СН2=СН—СН = СН2 полимеризуют методом анионной полиме-
ризации н присутствии щелочных металлов Na. К. Li. В чем различия в
структуре образующихся полимеров? В каком случае полимер имеет
более регулярную структуру и почему?
5 Каким способом можно получить стереорегуляриый полимер? Какие ка-
тализаторы используют для этих целей? Приведите схему получения сте-
реорегул яр него полибутадиена.
6 Как называются полимеры, полученные из мономеров
СН2=СН—СН -СН2, СН2= СНС1 и СН2=С (СН3) 2
Какими способами их получают и каковы их свойства?
7. Как можно регулировать структуру полимера (молекулярную массу, мо-
лекулярно-массовое распределение прн радикальной полимеризации и при
ионной?
8. Какой способ синтеза полимеров более перспективен и почему?
9 В чем отличие равновесной поликонденсации от неравновесной? В каком
случае получается полимер с более высокой молекулярной массой и
почему?
10. Какие вы знаете приемы повышения молекулярной массы полимера
прн поликонденсации?
11. В чем различие в структуре мономеров для полимеризации и поликонден-
сации?
ГЛАВЛ 3
ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ ПОЛИМЕРОВ
Химические превращения полимеров позволяют создавать мно-
гочисленные новые классы высокомолекулярных соединений и
в широких пределах изменять их свойства и области приме-
нения (реакции модификации). Некоторые полимеры вообще
нельзя получить непосредственным синтезом из низкомолеку-
лярных соединений вследствие неустойчивости мономеров. Так,
поливиниловый спирт не может быть получен полимеризацией
мономера: его синтезируют омылением готового полимера —
лоливинилацетата. Ацетилированием поливинилового спирта
получают различные поливинилацетали. Только путем взаимо-
действия каучуков с серой и другими полифункцнональными
соединениями (вулканизация) могут быть получены резины.
Наконец, к химическим превращениям относится направленная
деструкция полимеров, часто применяемая для регулирования
их молекулярной массы.
В химии высокомолекулярных соединений различают реак-
ции звеньев полимерной цепи и макромолекулярные реакции
Макромолекулярные реакции всегда приводят к изменению
степени полимеризации, а иногда и строения основной цепи по-
лимера. К этим реакциям относятся реакции деструкции по-
лимеров, сопровождающиеся уменьшением молекулярной мас-
сы, и межмолекулярные реакции, в результате которых обра-
зуются пространственные структуры и возрастает молекуляр-
ная масса полимера.
3.1. ОСОБЕННОСТИ ХИМИЧЕСКИХ РЕАКЦИЙ ПОЛИМЕРОВ
Химические реакции высокомолекулярных соединений в прин-
ципе не отличаются от реакций классической органической хи-
мии, но большие размеры и сложность строения макромолекул
обусловливают специфические особенности этих реакций. Для
полимеров возможны реакции, вообще не имеющие прямых
аналогий с реакциями низкомолекулярных соединений. Эти ре-
акции, например цепная деполимеризация, обусловлены просто
наличием достаточно длинной цепочки однородных звеньев.
При химических превращениях как низкомолекулярных, так
и высокомолекулярных соединений редко достигается 100%-е
превращение. Однако в отличие от реакций низкомолекуляр-
ных соединений, при которых конечные и промежуточные про-
дукты реакции можно отделить от исходных, звенья, по-раз-
ному затронутые данной химической реакцией, входят в состав
одной и той же макромолекулы и при одном и том же их сред-
155-
нем содержании в образце могут располагаться по цепи раз-
личным образом. Характер распределения звеньев по цепи в
пределах одной макромолекулы и композиционная неоднород-
ность полимера в пределах разных макромолекул влияют иа
такие практически важные свойства полимера, как раствори- •
мость, температуры плавления и стеклования, степень кристал- |
личности, плотность, прочность и др.
Направление и механизм любых химических реакций опре-
деляются законами химической термодинамики, условиями про-
цесса. Однако в отличие от свойств низкомолекулярных соеди-
нений химические свойства полимеров зависят не только от
этих факторов, но и от особенностей строения, в частности от
распределения звеньев в цепи и композиционной неоднородно-
сти полимеров, конфигурационных и конформационных эффек-
тов, электростатических взаимодействий, взаимного влияния
функциональных групп на их реакционную способность, физи-
ческого состояния полимера, характера надмолекулярных
структур, сегментальной подвижности макромолекул и т. д.
До недавнего времени считалось, что реакционная способ-
ность функциональной группы не зависит от того, присоедине-
на ли она к длинной цепочке или нет (принцип «равной ре-
акционной способности» Флори). Однако по мере расширения
круга полимерных объектов при изучении химических превра-
щений полимеров накапливалось все больше сведений о том,
что реакционная способность функциональных групп макромо-
лекул отличается от таковой для низкомолекулярных аналогов
и причиной этого является цепная природа полимера. В на-
стоящее время можно сформулировать основные отличия в хи-
мическом поведении макромолекул по сравнению с соответст-
вующими низкомолекулярными аналогами, что сделано
Н. А. Платэ с сотр.
I. Конфигурационные эффекты, включающие: изменение
направленности и степени завершенности реакции благодаря
наличию соседнего звена той же или иной химической или про-
странственной конфигурации и создаваемым из-за этого стери-
ческим затруднениям; изменение кинетики и механизма реак-
ции с низкомолекулярным реагентом из-за различного окруже-
ния данной функциональной группы или звена в начале и в
конце реакции и связанного с этим изменения реакционной
способности функциональных групп с конверсией (так назы-
ваемый «эффект соседа»).
2. Конформационные эффекты, связанные с изменением кон-
(Ьормации макромолекулы в процессе превращения.
3. Изменение локальной концентрации реагирующих групп
вблизи макромолекулы в растворе по сравнению со средней
концентрацией в объеме и связанное с этим изменение скоро-
сти реакции.
1.^6
4 Эффекты, связанные с электростатическим взаимодейст-
вием заряженной макромолекулы с реагирующими частицами;
оно может изменяться с глубиной конверсии, приводя к изме-
нению конформации макромолекулы и скорости реакции
5. Надмолекулярные эффекты, связанные с ассоциацией и
агрегацией реагирующих частиц и приводящие к композицион-
ной неоднородности и изменению химического строения продук-
тов реакции и скорости процесса
Рассмотрим более детально влияние различных факторов на
реакционную способность полимеров.
Конфигурационные эффекты. Любой тип стерео-
изомерии оказывает влияние на реакционную способность при
химических превращениях. Примером влияния стереохимии на
механизм внутримолекулярной реакции может служить цикли-
зация полиакриловой кислоты с образованием полиангидрида.
При обработке тионилхлоридом полиакриловой кислоты син-
диотактического строения ангидридных звеньев не образуется,
тогда как изотактическая полиакриловая кислота легко дегид-
ратируется, давая соответствующий полиангидрид
Реакционная способность функциональной группы может
меняться в зависимости от того, в какую последовательность
звеньев — изо-, гетеро- или синдиотактическую — она входит.
Впервые факт влияния стереоизомерии на кинетику полимерана-
логичных реакций был установлен при исследовании гидролиза
сополимера метакриловой кислоты с л-нитрофенилметакрила-
том; «20% эфирных групп, содержащихся в сополимере, спо-
собны гидролизоваться со скоростью, почти в 10 раз большей,
чем скорость гидролиза остальных эфирных групп. Изотактиче-
ские полиметилметакрилат и полиметилакрилат гидролизуются
быстрее, чем атактические и синдиотактические полимеры. Де-
гидрохлорирование поливинилхлорида и циклизация полиакри-
лонитрила при нагревании, а также гидролиз поливинилацета-
та и поливинилацеталей происходят легче в синдиотактических
полимерах. Цис- и транс-изомеры тоже могут проявлять раз-
личную реакционную способность при химических превращени-
ях. Наличие рядом с реагирующим звеном другого звена той
Же химической природы, но иной пространственной конфигура-
ции также влияет на его реакционную способность.
Расположение функциональных групп по длине макромоле-
кулы влияет на их химические свойства. В частности, стойкость
карбоцеппых полимеров к деструкции зависит от расположения
Функциональных групп: она ниже, если группы расположены
рядом. Например, макромолекулы поливинилового спирта нор-
мального строения
—СНг—СН—СНг—СН—СНг—СН—
I I-’ I
СН сн он
157
не деструктируются под действием иодной кислоты НЮ
и кислорода воздуха, тогда как макромолекулы, содержащие
сс-гликолевые группировки —СН—СН—, в этих условиях дест
I I
он он
руктируются:
ню4
~СН2-СН—СН—СН2----- ~СНг—СН+НС—СН2~
II II II
он он о о
Скорость дегидрохлорирования поливинилхлорида также
зависит от порядка расположения в макромолекуле мономер
ных звеньев —СН2— СНС1—. При обычном расположении
звеньев «голова к хвосту»
~ СНг-СНС!—СН2-СНС1—СН2—СНС1—
термический распад и дегидрохлорирование поливинилхлорида
протекают медленно и по иному механизму, чем при аномаль-
ном строении звеньев ^СН2—СНС1—СНС1—СН2~. В этом
случае дегидрохлорирование сопровождается образованием из
двух соседних звеньев хлористого винила одного звена хлоро
прена
~ СН2—СНС1—СНС1—СН2--- ~ сн2—СН=СС1—сн2 ~
—на
пространственные за
электронные эффекты,
подвижностью электрон
Влияние заместителей, создающих
труднения при химических реакциях,
обусловленные полярностью атомов и
них пар, так называемое ориентирующее влияние заместите
лей в бензольных и нафталиновых циклах — все эти факторы
хорошо известны в классической органической химии низкомо
лскулярных соединений. В полимерах такой фактор, как влия
ние «соседей», приобретает специфические особенности. «Эф
фскт соседа» (под этим термином обычно понимают измененш
реакционной способности данной функциональной группы ил!
звена под влиянием уже прореагировавшей группы, располо
женной по соседству с данной) вызывает изменение скорост!
и механизма реакции в полимерах. В ряде случаев скорост!
реакции при появлении рядом уже прореагировавших груш
может повышаться в 103—104 раза. Прореагировавшие сосед
ние группы могут оказывать и ингибирующее влияние на реак
ционную способность данной группы. Так, щелочной гидроли;
полиметакриламида не проходит до конца из-за блокирован^
амидной группы двумя ионизованными карбоксильными труп
пами. ;
158
Конформационные эффекты Процесс химического
превращения макромолекулы одного типа и строения в моле-
кулы другого типа и строения обязательно связан с изменени-
ем формы макромолекулы в растворе, поскольку меняются ее
химический состав, характер внутри- и межмолекулярного
взаимодействия, потенциальные барьеры вращения и т. д. Если
для осуществления той или иной реакции необходимо сближе-
ние на определенное расстояние функциональных групп мак-
ромолекулы, разделенных десятками звеньев, то произойдет
реакция или нет, будет зависеть от того, реализуется ли необ-
ходимая для этого сближения конформация.
Конформация и степень свернутости макромолекулярного
клубка определяют, с одной стороны, скорость, с которой низ-
комолекулярный реагент достигнет реакционноспособных групп
полимера, а с другой — равновесную концентрацию этого ре-
агента вблизи активных групп. Конформация цепи, обеспечива-
ющая доступность реагента к функциональным группам в на-
чале процесса, может уже не реализоваться на более поздних
стадиях, и реакция замедлится. Возможны и обратные слу-
чаи— ускорение реакции за счет разворачивания цепи
Надмолекулярные эффекты, как правило, тесно
связаны с конформационными, и разделить их чрезвычайно
трудно. Однако их необходимо учитывать при изучении реак-
ций полимеров не только в твердой фазе, но и в растворе.
Примером влияния морфологии полимеров на их химиче-
ские свойства может служить снижение скорости окисления
кристаллизующихся полимеров при их ориентации и кристал-
лизации при растяжении. В качестве примера зависимости ки-
нетики реакции от наличия надмолекулярных образований
можно привести термоокислительную деструкцию полипропи-
лена. Эта реакция идет преимущественно в аморфных обла-
стях. Если же сравнивать кинетику реакций в образцах с раз-
ной кристаллической структурой, то оказывается, что крупно-
сферолитный полипропилен окисляется медленнее, чем мелко-
сферолитный.
При контакте низкомолекулярного реагента с полимером
в реакцию сразу вступают только функциональные группы,
расположенные на поверхности. К функциональным группам,
не расположенным на поверхности полимера, реагент должен
предварительно продиффундировать сквозь слой полимера.
Продолжительность диффузии определяется не только условия-
ми реакции, химическим строением полимера и низкомолеку-
лярного реагента, но и плотностью упаковки макромолекул
полимера. Так как в аморфных областях упаковка макромо-
лекул более рыхлая, чем в кристаллических, продолжитель-
ность контакта и полнота реакции низкомолекулярного реаген-
та с макромолекулами, расположенными в аморфных областях,
>59
выше, чем с макромолекулами в кристаллических областях.
Различная доступность для реагентов функциональных групп
макромолекул или их звеньев, расположенных в аморфных и
кристаллических областях и в надмолекулярных образованиях
с различной плотностью упаковки, является одной из важней-
ших причин неоднородности продуктов реакций в полимерах,
находящихся в стеклообразном или кристаллическом состоянии.
При растворении полимеров или повышении температуры
и переходе полимеров в вязкотекучее состояние возрастает сег-
ментальная подвижность, крупные надмолекулярные образо-
вания, кристаллические области разрушаются, и функциональ-
ные группы становятся более доступными для низкомолекуляр-
ного реагента. Вероятность взаимодействия реагента с любой
макромолекулой значительно возрастает. Поэтому продукты
реакций в растворах или расплавах при температуре текучести
или плавления значительно однороднее, чем полученные при
температуре стеклования. Однако и в этом случае колебания
концентрации растворов или температуры процесса вызывают
существенные нарушения протекания химических реакций, осо-
бенно в высоковязких средах Это объясняется тем, что из-за
высокой вязкости расплавов или концентрированных растворов
замедляется диффузия реагентов к функциональным группам
полимеров, что обусловливает неоднородность продуктов реак-
ций При реакциях растворенных полимеров в процессах взаи-
модействия реагирующих частиц обычно принимают участие
молекулы растворителя и их ассоциаты. Поэтому при опреде-
лении скорости и других параметров химических реакций в
растворах необходимо учитывать молекулярные взаимодейст-
вия исходных частиц, промежуточных комплексов, продуктов
реакции с молекулами окружающей среды. Среда наименее
существенное влияние оказывает на гомолитические реакции
и очень существенное — на гетеролитические. В гомолитических
реакциях, как правило, активными центрами являются свобод-
ные радикалы, в гетеролитических — ионы.
В гомолитических реакциях образование активированных
комплексов не сопровождается значительным перераспределе-
нием электрических зарядов между реагирующими частицами.
Следовательно, при этих реакциях не происходит существенно-
го изменения межмолекуляриых взаимодействий реагирующих
частиц с молекулами растворителя, скорость реакции мало
зависит от природы растворителя.
При гетеролитических реакциях происходит перераспреде-
ление электрических зарядов между реагирующими частицами.
Изменение зарядов ионов вызывает изменение ориентирующе-
го и поляризующего действия электромагнитных полей ионов
или диполей на молекулы растворителя, что приводит к су-
щественной перестройке сольватных оболочек реагирующих ча-
160
стиц в процессе реакции. Поэтому скорость гетеролитических
реакций очень сильно зависит от строения молекул растворите-
ля (иногда она изменяется на 9 десятичных порядков) На-
пример, для растворителей одного гомологического ряда или
смесей, приготовленных из двух определенных растворителей,
установлено, что скорость реакции с увеличением диэлектриче-
ской проницаемости растворителя растет, если взаимодейству-
ют одноименно заряженные ионы, и снижается при реакциях
разноименно заряженных частиц.
На поведение полимеров в различных реакциях и их хими-
ческую стойкость влияют практически всегда имеющиеся в по-
лимере (в результате протекания побочных реакций, сопровож-
дающих любые полиреакции) связи, отличающиеся от связей,
характерных для данного соединения. Наибольшее влияние на
химическую стойкость карбоцепных полимеров оказывают слу-
чайные гетероатомные связи в главных цепях макромолекул,
которые легко разрушаются, что приводит к разрыву макромо-
лекул и значительному снижению молекулярной массы (раз-
рыв 0,01% связей приводит к снижению молекулярной массы
полимера в несколько раз). Существенно снижается химиче-
ская стойкость полимеров и при включении в макромолекуляр-
ные цепи третичных и четвертичных атомов углерода. Приве-
дем несколько примеров.
При нормальном проведении радикальной полимеризации
акрилонитрила процесс идет с образованием макрорадикала:
R—СН2—СН+СН2=СН —> R—СН2—СН—СН2—СН
1 I кг I 1
С =N С N с N С N
Однако отдельные молекулы акрилонитрила могут образовать
бирадикалы:
CHj—CH=C=N
Их участие в полимеризации приводит к образованию гетеро-
атомных кетен-иминных связей в макрорадикалах:
СН2—CH=C=N 4- СН2=СН --► СНа—CH=C=N—СН4—СН
I I
C=N CsbN
В полимерах эти связи при нагревании и воздействии щелочей
легко разрушаются, что вызывает резкое снижение молекуляр-
ной массы полиакрилонитрила. После разрушения кетен-имин-
ных связей в первый период обработки щелочами или нагрева-
ния стойкость полиакрилонитрила к деструкции значительно
повышается.
11-816
161
При полимеризации винилацетата
R—СН2—СН + СН2=СН-> R—СН2—СН—^СН2-СН
II II
О—СО—СН3 О—СО—СН3 о-со-сн3 о—со—СНз
вследствие передачи цепи на боковую группу в главной цепи
макромолекул возникает кислород-углеродная сложноэфирная
•связь, снижающая стойкость полимера к гидролизу:
R'
I
R—СНа—СН + CHS-CO—О—СН >
О-СО-СН, СНа
R*
R'
---► R—СН2—СН2 +СНа—СО-О-СН
I I
о-СО—СНз сн2
I
R'
R' СНа=СН
I • I
НС—О—СО—сн2 + п О—СО—СНз *
СН2
R*
нс—о—со—СН2—Г—СНа—СН—
I I
сн2 L о—со—сн
R'
3Jn
В результате передачи цепи на макрорадикалы при ради-
кальной полимеризации этилена образуется значительное число
боковых ответвлений и третичных атомов углерода:
R' R'
R—СН2—СН2 + HjC^ ---> R—СНа—СН, 4- НС^
Н2С H2i
R' R'
I I
НС-+ лН2С=СН2 --► НС—(—СНа—СН2—СНа—СН2
R’—(!на R—СНа
162
Третичные и четвертичные атомы углерода менее устойчивы
к химическим воздействиям Например, при 570 К вероятность
отщепления водорода от третичного атома углерода в 11 раз
больше, чем от вторичного. Вследствие возникновения третич-
ных атомов в цепях разветвленного полиэтилена при его на-
гревании рвется в два раза больше макромолекул, чем в не-
разветвленном
32 ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ НЕ ВЫЗЫВАЮЩИЕ ИЗМЕНЕНИЯ
СТЕПЕНИ ПОЛИМЕРИЗАЦИИ
К химическим превращениям полимеров, не вызывающим су-
щественного изменения степени их полимеризации, относятся
внутримолекулярные и полимераналогичные превращения.
Внутримолекулярными называют процессы, в результате ко-
торых изменяется строение, а иногда и химический состав мак-
ромолекул, но обычно не сопровождающиеся присоединением
реагентов Они происходят вследствие внутримолекулярных
перегруппировок или взаимодействия атомов, функциональных
групп одной макромолекулы.
Полимераналогичными называют процессы взаимодействия
функциональных групп макромолекул с низкомолекулярными
реагентами, не влияющие на степень полимеризации и строение
основной цепи и приводящие к получению полимераналогов.
3.2.1. Внутримолекулярные превращения
Внутримолекулярные химические превращения происходят под
действием света, излучений высокой энергии, тепла, химических
реагентов (которые ис входят в состав полимера) Внутримо-
лекулярные превращения могут оказывать существенное влия-
ние на механизм реакции, приводить к образованию полимеров
нежелательного строения. Однако в соответствующих условиях
такие превращения позволяют получить наиболее эффектив-
ным способом полимеры нужного строения, синтез которых
другими путями невозможен. Внутримолекулярные превраще-
ния под действием тепловой и лучистой энергии, а также под
действием ряда химических реагентов в ряде случаев являются
побочными реакциями, которые оказывают большое влияние на
строение и свойства полимеров в процессе их получения, пере-
работки и эксплуатации
Внутримолекулярные превращения могут быть нескольких
типов: внутримолекулярные перегруппировки боковых групп;
внутримолекулярные перегруппировки в цепях главных валент-
ностей; изомерные превращения (циклизация, изомеризация,
миграция двойных связей, образование ненасыщенных связей;
сложные превращения).
163
Внутримолекулярные перегруппировки бо-
ковых групп не отражаются на строении цепей главных
валентностей. Они могут иметь место уже при синтезе полиме-
ров. Например, при полимеризации метакриламида при повы-
шении температуры часто происходит циклизация с отщепле-
нием NH3:
СН3 СН3
I I
СН2—С—СН2 С~
I I
ОС со
сн3
СНз
~сн2-с----сн.
—NH3
h2n-co
Поликонденсация, сопровождаемая на определенной стадии
циклизацией полупродукта (преполимера), позволяет получить
наиболее простым способом циклоцепные полимеры с изолиро-
ванными или конденсированными циклами (лестничные поли-
меры), разветвленные или трехмерные. Синтез подобных поли-
меров происходит по схеме
поликонденсация
H2N—HN
NH—NH2
—R-C
С—R'—
О
О
л
О
HN—NH
п
где Х=О, S, NH<
циклизация
О
—R-C
HN—I
—HjO
n
С—R'—
-*п
N---N
Внутримолекулярные перегруппировки боковых групп ха-
рактерны также для полиметакрилатов. При пиролизе (430—
450 К) поли-трет-бутилметакрилата он превращается в поли-
метакриловую кислоту. При этом боковые сложноэфирные
группы разлагаются с выделением изобутилена:
СН3 СН3 СН8
~сн2—сн~ —*- ~сн2—(^н~ + ^=сн2
ОС—о- С(СН8)з ОС—он сн.
164
В качестве примера внутримолекулярных пере-
группировок в цепях главных валентностей
можно привести превращение полиаигидроформальдегиданили-
на в кислой среде в поли-л-бензиламин:
на
-->
~N-CH2-N—СН2 N-CH2~
-СН2—I
Изомерные превращения характерны для полиме-
ров, содержащих ненасыщенные связи в основной цепи или бо-
ковых группах. Элементный состав полимера при этом не из-
меняется. Изомерные превращения зачастую являются побоч-
ной реакцией, сопровождающей процессы переработки эласто-
меров, их химические превращения. Так, изомерные превраще-
ния происходят при серной вулканизации непредельных каучу-
ков и являются одной из причин снижения прочности резин
В то же время ряд продуктов изомеризации находит примене-
ние в технике. Например, циклизованный латекс натурального
каучука используют при производстве латексных изделий. Цик-
локаучуки благодаря их высокой химической стойкости, водо-
и атмосферостойкости применяют для изготовления лакокра-
сочных антикоррозионных и электроизоляционных покрытий.
Циклизованный каучук может быть использован в качестве на-
полнителя в светлых подошвенных резинах с целью повыше-
ния их износостойкости. Частичная цис-транс-изомеризация
цис-полиизопрена и цис-полибутадиена способствует повыше-
нию их морозостойкости, так как замедляются процессы крис-
таллизации. В качестве примера реакции циклизации за счет
ненасыщенных связей в боковых группах можно привести цик-
лизацию полиакрилонитрила при нагревании в отсутствие воз-
духа:
Еще одним примером образования циклов за счет двойных
связей в основной цепи может служить внутримолекулярная
Циклизация каучуков под действием протонных или апротонных
165
кислот, В процессе циклизации в цепи образуются шестичлен-
ные циклы, содержащие двойную связь:
~Н2С СН3 ~Н2С СН3
Х+
Н+ /
НС СН—СН,~ ► Н2С СН—сн2---->
I II I II “Н+
Н2С С-СН3 Н2С С—сн3
Хнг Хн2
~Н2С СНз
ХХ
" > Н2С С—СН2~
I II
Н2С С—СНз
ХХ
/(ас-транс-изомеризация происходит при облучении раство-
ров каучуков УФ светом, 7-излучением и при нагревании в при-
сутствии сенсибилизаторов (веществ, повышающих светочувст-
вительность) — органических бромидов, сульфидов и других
соединений, образующих свободные радикалы. гунс-транс-Изо-
меризация протекает по схеме
присоединение свободного радикала с разрывом двойной
•связи:
2 3
СН=СН
/ \ +R.
~Н2С сн2~
1 4
2 3
• СН—СН—R
~Н2С^ сн2~
I 4
1рс-изомер
радикал quc-изомера
поворот метиленовой группы радикала цас-изомера на
2 3
3,14 рад вокруг одинарной связи С—С с образованием радика-
ла транс-изомера:
2 3
•СН—СН—R :?=*:
радикал
транс-изомера
166
отщепление свободного радикала R* с образованием
трднс-изомера:
4 4
СН2~ СН2—
2 / 2 /
•СН—СН—R CH=CH + R-
/ з / з
~СН2 '"'СНз
I {
траяс-изомср
Миграция двойных связей вдоль полимерной цепи
происходит при взаимодействии ненасыщенных полимеров с ка-
тализаторами ионного типа, например полибутадиена с комп-
лексами солей Со или Ni и алюминийорганических соединений,
применяемыми при ионно-координационной полимеризации:
~СН2СН=СНСН2СН2СН=СНСН2СН2СН=СНСН2----->
—~СН2СН=СНСН2СН2СН=СНСН2СН=С11СН2СН2------*
—» ~СН2СН=СНСН2СН2СН2СН=СНСН=СНСН2СН2~
Образование сопряженных ненасыщенных
связей в цепи главных валентностей происходит под дейст-
вием света, излучений высокой энергии, тепла, в присутствии
кислот, оснований, катализаторов Фриделя — Крафтса и дру-
гих реагентов за счет отщепления низкомолекулярных продук-
тов от некоторых полимеров. Например, при дегидрохлориро-
вании поливинилхлорида алкоголятами щелочных металлов по-
лучают блок-сополимер, состоящий из блоков исходного поли-
винилхлорида и блоков поливинилена:
NaOR
~СН2—СН-СН2-СН—СН2—СН------->
I I I -на
ci ci а
—> ~сн=сн—сн=сн—сн=сн~
поливинилен
~СН2-СН-СНг-СН-СН2-СН~-н>~СН = СН-СН = СН-СН = СН~--‘
I I
ОН ОН ОН т
____________________ - н20>
____________________Д
~СН2-СН-СН2-СН-СН2-СН~->~СН2-НС СН~ +Н20
I I I
он к НгС СНг
сн
I
он
167
Внутримолекулярные превращения могут быть и более
сложными Например, при дегидратации поливинилового спир-
та реакция может одновременно происходить в двух направле-
ниях: образование ненасыщенных связей в цепи главных ва-
лентностей I и циклизация II (см. с. 167).
Две внутримолекулярные реакции протекают также одновре-
менно при термической обработке полиакрилонитрила при 670—
770 К: циклизация (I) и образование двойных связей вследствие
дегидрирования (II):
В результате этих реакций образуются термостойкие полимеры
лестничного типа
-3.2.2. Полимераналогичные превращения
Полимераналогичные превращения приводят к изменению
строения боковых функциональных групп макромолекул, в со-
став которых могут войти атомы или группы атомов из низко-
молекулярных реагентов, но степень полимеризации и строение
основной цепи не изменяются. Большое практическое значение
эти превращения имеют для химической модификации поли-
меров.
В зависимости от механизма реакций при полимераналогич-
ных превращениях могут образовываться новые функциональ-
ные боковые группы, происходить циклизация, раскрытие цик-
лов, различные более сложные превращения. К полимеранало-
гичным превращениям с образованием новых функциональных
групп относится, в частности, получение поливинилового спирта
алкоголизом поливинилацетата в щелочной среде практически
без изменения степени полимеризации. Присутствие воды в ре-
акционной среде тормозит реакцию алкоголиза, усложняет
промывку и стабилизацию поливинилового спирта. Поэтому
рекомендуется проводить процесс в среде абсолютно сухого
•спирта в присутствии 0,2—0,4% раствора едкого натра в каче-
стве катализатора. Процесс алкоголиза сопровождается гидро-
лизом, поскольку на реакцию расходуется лишь 10% едкого
J68
натра от теоретически необходимого. Реакция идет по следую-
щей схеме:
^СН2—СН~ сНзОИ" ^СН2—Сн~ СН3 Na
I NaOH I I I
О —► ОН + О + О
ОС—СНз ОС—сн3 ос—сн3
При действии ацетата серебра на поливинилхлорид хлор
замещается на ацетатную группу, что приводит к получению
поливинилацетата:
~СН2—СН—СНа—СН~ + 2Ag—О—С—СН3 ---►
I I II
С1 С1 О
---> ~СН2-СН-СН2—СН~ 4- 2AgCI
ОС—СНз ci—СН,
При воздействии на поливинилхлорид ароматических соеди-
нений в присутствии катализаторов Фриделя — Крафтса в ре-
зультате полимераналогичных превращений получают заме-
щенные полистиролы. Введение функциональных групп в поли-
этилен можно осуществить путем хлорирования:
С1
Ci2 |
~СН2—СН2—СН2—СН2~ -----> ~СН2—СН—СН2-СН2~
Введение хлора в макромолекулы полиэтилена снижает сте-
пень кристалличности полимера, повышает плотность, изменя-
ет другие свойства. При содержании хлора 25—40% полиэти-
лен приобретает качества, свойственные каучукам.
При одновременном действии газообразного хлора и серни-
стого ангидрида происходит сульфохлорирование полиэтилена,
заключающееся во введении в макромолекулы хлора и хлор-
сульфоновых групп:
SO2C1 Cl
8О« а2 I
~CH2—CH2—CH2—CH2------------ ~CH2—CH—CH2—CH ~
Макромолекула хлорсульфированного полиэтилена содер-
жит одну группу SO2CI на каждые 90 атомов С и один атом
С1 на каждые 7—8 атомов С. Хлорсульфированный полиэтилен
обладает низкой газопроницаемостью, высокой адгезией к раз-
личным субстратам, способностью к вулканизации.
При взаимодействии двух соседних боковых функциональ-
ных групп с одной молекулой низкомолекулярного реагента
169-
происходит циклизация, как, например, при ацетилировании '
поливинилового спирта:
О
—СН2—<СН—CHS—СН—СН,-СН—СН2—СН- 4- nRC^ --►
Ан ОН ОН Ан Н
При гидрировании полиакрилонитрила в результате цикли-
зации наряду с другими продуктами реакции образуются также
пиперидиновые циклы:
—СН2—СН—СН2—СН—СН2—СН—СН2—СН— >
А А А А
N N N N
СН2 СН2
---> ~СН2—НС ^CH-CHj—НС^ ^СН-
I 1 II
н2с сн2 н2с сн,
\lH \hZ
В результате некоторых реакций, например щелочного гид-
ролиза сополимера малеинового ангидрида и винилацетата,
происходит обратный процесс — размыкание циклов с образо-
ванием боковых функциональных групп:
NaOH
-нс—сн-сн2-сн—НС-сн—сн,—сн- ►
ОС со А ОС СО А
\ / I \ / I
О СО СО
СНз Ан3
НС—CH—СН,—CH—CH—CH—CH2—CH—
со
но
ОС
он
NaO ONa
ONa
ONa
о
170
Аналогичный процесс происходит при щелочном гидролизе
поливинилпирролидона:
н2о
—сн,—сн—сн2----СН— —> —сн2—сн—сн2—сн-
А А Ан Ан
HsC^ \о HjC^ \о сн, сн,
н,с---Ан, н2с---Ан, сн8 Ан,
<1на сн2
но—Ао ос—он
При полимераналогичных превращениях часто одновремен-
но могут происходить не одна, а несколько параллельных реак-
ций. В этом случае обычно образуются сополимеры сложного-
строения. Например, при действии на полиакрилонитрил кон-
центрированной серной кислоты (на холоду) происходят две
реакции — образование имидных звеньев в основной цепи и
боковых групп акриламида, вследствие которых гомополимер
превращается в сополимер:
h2so4
-сн,—сн—сн2—сн—сн2—сн------>
A А А
Я I I
N N N
CH,
-СН,—HC CH—СН,—CH-
---► I I I
ОС CO CO
\ / I
NH NH,
В спиртовой среде при температуре 400 К взаимодействие
полиакрилонитрила с концентрированной серной кислотой при-
водит к образованию сополимера, содержащего до 86% слож-
ноэфирных групп и до 5% карбоксильных:
H2SO4
—СН2—СН—СН,—СН—СН,—CH—СН2—СН—СН,—СН----------
A L А А А
И Я III И 01
N N N N N
—*- -сн2—сн—сн,—сн—сн2—сн—сн,—сн-сн,—сн-
I I J. I I
со со со со со
I I I I I
OR OR OR OR OR
1711
3.3. РЕАКЦИИ, ПРИВОДЯЩИЕ К ИЗМЕНЕНИЮ
МОЛЕКУЛЯРНОЙ МАССЫ
Наряду с реакциями, протекающими без изменения молеку-
лярной массы, для полимеров характерны также реакции, при-
водящие к изменению степени полимеризации. Их можно раз-
делить на две группы: реакции, при которых молекулярная
масса растет и при которых наблюдается ее снижение. К пер-
вой группе можно отнести реакции сшивания — соединение
макромолекул поперечными связями (реакции вулканизации
эластомеров, отверждение), получение блок- и привитых сопо-
лимеров
Вторая группа — это реакции деструкции, которые могут
протекать под действием кислорода, а также различных физи-
ческих факторов (тепло, свет, излучение и др.) Особенность
этих реакций — существенное изменение физико-химических
свойств полимера даже при незначительной глубине реакции.
Так, одного акта деструкции на макромолекулу достаточно,
чтобы молекулярная масса полимера уменьшилась приблизи-
тельно вдвое и изменились его механические свойства. Образо-
вания одной меж молекул яр ной связи на макромолекулу доста-
точно, чтобы полимер потерял способность растворяться.
Большинство этих реакций используют в промышленности
для получения технически ценных продуктов. Однако они мо-
гут быть и побочными при химических превращениях поли-
меров.
3.3.1. Соединение (сшивание) макромолекул
Реакции соединения линейных и разветвленных молекул назы-
ваются сшиванием или структурированием и происходят или
при действии на полимеры так называемых сшивающих аген-
тов, или под влиянием тепла, света, радиационного излучения.
Образующиеся сшитые (сетчатые) полимеры теряют способ-
ность к растворению, а также к необратимым пластическим
деформациям. Их физико-механические характеристики, как
правило, повышаются. В частности, по мере увеличения густо-
ты сетки, когда снижается кинетическая подвижность отрезков
сетки между узлами, повышаются твердость, температура раз-
мягчения, термостойкость.
Процессы сшивания широко применяются в промышленно-
сти, например при вулканизации каучуков, отверждении пласт-
масс, высыхании лакокрасочных покрытий, дублении кож.
Вулканизация каучуков — это технологический про-
цесс, при котором каучук превращается в резину в результате
соединения линейных макромолекул поперечными связями в про-
странственную вулканизационную сетку. В результате вулкани-
172
Рис. 3.1. Зависимость прочности
при растяжении оР (/) и относи-
тельного удлинения при разрыве
8р (2) вулканизатов от количе-
ства связанной серы; вертикальны-
ми линиями показано предельное
содержание связанной серы в
мягких резинах (/) и эбонитах (//)
в,. МПа
зании каучук теряет растворимость и термопластичность, приоб-
ретает высокую эластичность, прочность, ряд новых ценных
свойств (рис. 3.1). Продолжительность процесса вулканизации,
необходимая для достижения оптимальных значений наиболее
важных для данного изделия свойств, называется оптимумом
вулканизации. В настоящее время для вулканизации использу-
ют серу, пероксиды, диизоцианаты, оксиды металлов и др. Ее
проводят при нагревании до 410—460 К смесей каучука с необ-
ходимыми ингредиентами и серой.
Основное значение для quc-изопреновых, бутадиеновых и
других каучуков с двойными связями в макромолекулах имеет
вулканизация серой. Сера в обычных условиях состоит из цик-
лов Ss (с энергией' межатомных связей 243—260 кДж/моль),
которые при 413 К распадаются. При взаимодействии с каучу-
ком сера присоединяется к нему по атому, соседнему с двой-
ной связью (а-углеродный атом) или по двойной связи как по>
ионному, так и по свободнорадикальному механизму:
СН3 СН3 снз
~СН—С=СН—СН2-----~СН—С=СН—СН2~ ~снз-с-сн-сн2~^
СН3
---> ~СН—(!=СН-СН,~
I
Sx
~СН2—С—Ан-СНз—
сн3
При содержании связанной серы до 3—5% обычно образу-
ются мягкие, прочные резины, более 25—30%—хрупкий эбо-
173-
нит. Минимальное количество серы, необходимое для вулкани-
зации,— около 0,1%, максимальное количество связываемой
серы — 32%.
Для ускорения процесса вулканизации вводят некоторые
органические вещества, называемые ускорителями вулкани-
зации.
При вулканизации наряду с процессами формирования вул-
канизационной сетки могут одновременно протекать побочные
реакции, среди которых наибольшее значение имеют окисле-
ние и некоторые изомерные превращения, связанные с внутри-
молекулярным присоединением серы. Для подавления побоч-
ных реакций в состав вулканизующей группы вводят так на-
зываемые вторичные ускорители (активаторы) — жирные кис-
лоты и оксиды металлов. Механизм химических реакций при
вулканизации зависит от состава вулканизующей группы, вида
каучука и условий процесса Существенным недостатком сер-
ной вулканизации является низкая термическая и химическая
стойкость образующихся вулканизатов.
Пероксидная вулканизация происходит под действием сво-
бодных радикалов, образующихся при распаде пероксидов при
нагревании их в смеси с каучуком. Наиболее эффективны пер-
оксиды кумила и трет-бутила. Так, пероксид кумила распада-
ется при 411—422 К с образованием свободных радикалов по
следующей схеме:
Образующиеся из пероксидов свободные радикалы вступа-
ют во взаимодействие с наиболее активными а метиленовыми
водородными атомами с образованием макрорадикала:
R + ~сн2—С=СН—СН2~
I
СНЭ
RH+ —СН—С=СН—СН2~
СН3
Макрорадикалы взаимодействуют с новыми макромолекулами
каучука, а также между собой с образованием межмолекуляр-
174
ных связей по следующей схеме:
СН3
. I
~СН,—СН=С—СН~ 4- ~сн—с=сн—сн,-------
<1н3
СНз
I
~СН—С=СН—СН,~
--► ~СН2—СН=С—сн~
СН,
Большое значение пероксиды имеют для вулканизации ди-
метилсилоксановых, этиленпропиленовых и других насыщенных
каучуков, не способных к вулканизации другими соединениями:
< 1 It
0 0 0 0
I . . I I I.
Н3С--Si—СН, 4* СН,—S—СН, 1 * Н,С—Ъ —СН,—СН,—Si—СН,
II । 1
0 0 о о
it it
Пероксидные вулканизаты насыщенных, например кремний-
органических каучуков, можно эксплуатировать при очень низ-
ких и высоких температурах: они характеризуются высокой
химической, радиационной и атмосферостойкостью.
В качестве вулканизующего агента используют также оли-
гомеры, в частности алкилфенолоформальдегидные, например
n-октил- и п-трет-бутилфенолоформальдегидные, содержащие не
менее 3% гидроксиметильных групп СНгОН:
ОН СН,ОН
Эти олигомеры применяют чаще всего при вулканизации
бутилкаучука Они размягчаются при темпера уре приготовле-
ния смесей с каучуком, играя роль мягчителей. Вулканизацию-
олигомерами проводят при 430—450 К. Вулканизация ускоря-
ется хлоридами металлов (SnCl2-2H2O; Р'еС1з-6Н2О, ZnCl2-
•1,5Н2О), хлорсульфированным полиэтиленом в смеси с окси-
175-
дом цинка и другими соединениями, способствующими дегид-
ратации. Основные достоинства резин с алкилфенолоформаль-
дегидными олигомерами — высокие теплостойкость и эластич-
ность.
Вулканизация диизоцианатами применяется для уретановых
каучуков. В качестве вулканизующих агентов используют сле-
дующие:
O=C=N N=C=O
толуилен-2,4 - ди -л изоцианатфеннлмета и
диизоцианат
—N С О
ОСН3
дианизиднндиизоцианат
При вулканизации диизоцианаты взаимодействуют с груп-
пами, содержащими подвижные атомы водорода. Вулканизо-
ванные диизоцианатами уретановые каучуки характеризуются
высокой тем пер атуростой костью.
Диизоцианатами вулканизуют также карбоксилатные кау-
чуки, содержащие карбоксильные группы СООН При взаимо-
действии с карбоксильными группами, принадлежащими раз-
ным макромолекулам, диизоцианаты образуют поперечные свя-
зи типа
R—СО—NH—R—NH—СО—R'
Вулканизация оксидами металлов используется для карбо-
ксилатных и хлоропреновых каучуков, хлорсульфированного
полиэтилена и др. Так, карбоксилатные каучуки вулканизуют-
ся оксидами цинка или магния, для вулканизации хлоропрено-
вых каучуков применяют одновременно и ZnO, и MgO. Реак-
ция протекает на поверхности дисперсных частиц оксида цин-
ка. В процессе вулканизации образуются эфирные мостики в
соответствии со схемой:
миграция двойной связи и хлора ж
~СН2—СС1~ х —~СН2—с~
I II ’
сн=сн2 сн-сн,а
образование ионизированных «подвесок»
~СН2—С~ 4-ZnO --> ~СН2—С~
II
СН-СН2С1 СН—СН2—О—Zn+СГ
176
образование поперечной связи
~СНа—С~ -J- ~СН2—С~ >
II II
СН—СН2—О—Zn+СГ СН—СН2С1
~СН2—С~ ~С—СН2~
---> II II + ZnCI2
сн-сн2—о—сн2-сн
В присутствии хлорида цинка развиваются ионные реакции,
приводящие к образованию поперечных связей С—С, что отри-
цательно сказывается на свойствах вулканизатов
Роль оксида магния сводится к подавлению реакций сшива-
ния, активируемых хлоридом цинка, за счет адсорбции послед-
него на поверхности оксида магния и за счет связывания HCi,
выделяющегося при термическом гидрохлорировании каучука.
Кроме того, оксид магния повышает стойкость смесей к преж-
девременной вулканизации, улучшает качество вулканизатов.
Вулканизующий эффект оксидов возрастает при введении кани-
фоли. По-видимому, абиетиновая кислота, содержащаяся в ней,
образует с металлами соли, облегчающие вулканизацию
При вулканизации хлорсульфированного полиэтилена хлор-
сульфоновые группы SO2CI взаимодействуют с оксидами ме-
таллов, в частности, магния или свинца, в присутствии воды,
а также ускорителей вулканизации, например тиурамов Меха-
низм сшивания можно иллюстрировать следующими схемами
реакций. На первой стадии происходит гидролиз хлорсульфоно-
вых групп в присутствии влаги при нагревании с образованием
групп SO2OH и выделением хлорида водорода:
О
ОН + НС1
Затем группы SO2OH вступают
в реакцию с оксидами:
R-CH2-CH-CH2------СНг-СН-R
SO2 С1
I
О
Нго* РЬ
О
I
S02
R - СН - СН2 - СН2 - СН - R*
I
Cl
12—816
177
Хлорсульфированный полиэтилен вулканизуется также ок-
сидами металлов в присутствии активаторов кислотного ха-
рактера, в частности жирных кислот RCOOH. Процесс вулка-
низации осуществляется за счет хлорсульфоновых групп SO2CL
Механизм реакции можно иллюстрировать следующими схема-
ми. На первой стадии жирная кислота превращается в соль,/
которая затем взаимодействует с каучуком /
2RCOOH4-MgO —► (RCOO)2Mg+H2O /
KaSO2Cl+(RCOO)2Mg —- KaSOjOMgCI+RCOCl / '
Полярные «подвески» каучука адсорбируются на поверхнр-
сти оксида металла с образованием связей, устойчивых при
обычных температурах.
С хлорсульфоновыми группами взаимодействуют с образо-
ванием поперечных связей также многие бифункциональные
соединения, например оловоорганические, олигомеры с гидрок-
симетильными, эпоксидными и другими функциональными
группами.
Радиационно-химическое сшивание осуществляется при дей-
ствии на полимеры ионизирующих излучений: ускоренных
электронов (быстрых электронов), нейтронов, 7- и рентгенов-
ского излучений и др. В промышленности для радиационной
вулканизации используют обычно у-излучения или ускоренные
электроны v-Излучеиие высокой проникающей способности
применяют для вулканизации массивных изделий, быстрые
электроны — для тонкостенных.
Сущность процессов радиационного сшивания макромоле-
кул заключается в их возбуждении при облучении, образова-
нии из возбужденных макромолекул свободных радикалов (ре-
акция 1) и их рекомбинации (реакция 2):
-СН/------- ~СН~4-Н (1)
~сн~—
(2):
Сшивание при радиационном облучении может происходить
также вследствие ион-радикальных реакций и электронных
процессов без участия возбужденных частиц. Одновременно со
сшиванием могут протекать процессы деструкции, циклизации
и др. Повышение сегментальной подвижности, например при
нагревании, ускоряет сшивание, хотя при этом возрастает роль
деструктивных процессов.
Наиболее легко вулканизуются полимеры, не содержащие
боковых групп, т. е. те, в которых отсутствуют стерические пре-
пятствия. Повышение степени кристалличности затрудняет про-
178
цесс сшивания. Сшивание в ряде случаев сопровождается вы-
делением водорода.
Склонность к вулканизации полимеров, содержащих в це-
пи третичные атомы углерода (полипропилен), снижается, а
полимеры с четвертичными углеродными атомами (полиизобу-
тилен, бутилкаучук, полиметилметакрилат) вообще не структу-
рируются при облучении. Наличие в полимере фенильных ядер
снижает эффективность радиационной вулканизации, по-види-
мому, из-за того, что они рассеивают энергию возбуждения при
облучении.
Введение в состав облучаемой композиции полимеров с ви-
нильными группами снижает степень деструкции, а с аромати-
ческими— степень сшивания; это может служить одним из ви-
дов радиационной защиты полимеров.
Скорость радиационной вулканизации зависит также от мо-
лекулярно-массового распределения: наибольшей скоростью ха-
рактеризуются монодисперсные полимеры. Радиационные вул-
канизаты на основе эластомеров в большинстве случаев имеют
пониженные прочностные и деформационные характеристики,
что обусловлено образованием жестких углерод-углеродных
связей Для устранения этих недостатков используют комбини-
рованную вулканизацию под действием серы и ионизирующих
излучений. Высокими физико-механическими и эксплуатацион-
ными показателями характеризуются резины, которые содер-
жат поперечные связи С—С и С—Sx—С, полученные при одно-
временном проведении серной и радиационной вулканизации.
Радиационная вулканизация находит широкое применение
для получения изделий из кремнийорганических каучуков.
Эффективность радиационного сшивания оценивают радиа-
ционно-химическим выходом Gc — числом поперечных связей»
образующихся в полимере при поглощении 100 эВ энергии из-
лучения; для большинства сшивающихся полимеров Gc=l—4.
3.3.2. Отверждение
Отверждением называют необратимое превращение жидких ре-
акционноспособных олигомеров (или их смесей с необходимы-
ми компонентами) в твердые, нерастворимые, неплавкие трех-
мерные полимеры. Отверждение является основной стадией
технологических процессов получения различных реактопла-
стов, герметиков, компаундов, клеев, лакокрасочных покрытий
и др.
^Отверждение происходит в результате химического взаимо-
действия функциональных групп олигомеров между собой или
специально добавляемыми реагентами — отвердителями.
Механизм отверждения зависит от строения олигомеров и от-
12*
179
вердителей, условий процесса и может быть полимеризацион- I
ным, поликондснсационным или смешанным. [
Отверждение включает две стадии. На первой происходит 1
потеря смесью растворимости и текучести вследствие образова- I
иия трехмерной сетки макромолекул. Момент, когда система О
теряет текучесть и переходит из жидкого в студнеобразное со- ’
стояние, называется точкой гелеобразования. Время после до-
бавления отвердителя, в течение которого смеси сохраняют те-
кучесть и способность к переработке, называют жизнеспособ-
ностью Жизнеспособность обычно снижается при повышении
температуры и содержания отвердителя. Кинетика отвержде-
ния и роста вязкости смеси на начальной стадии зависит от
механизма отверждения: при отверждении по механизму поли-
меризации в начальной стадии процесса наблюдается длитель-
ный индукционный период, при котором вязкость существенно
не изменяется. При отверждении по механизму поликонденса-
ции индукционного периода нет и вязкость с момента введения
отвердителя до точки гелеобразования непрерывно возрастает.
На второй стадии отверждения происходит окончательное
структурирование системы. Скорость отверждения после точки
гелеобразования постепенно снижается вследствие исчерпания
функциональных групп и резкого торможения диффузионных
процессов, снижения сегментальной подвижности отверждае-
мого полимера.
Кинетика отверждения существенно зависит от температу-
ры Для полного отверждения необходимо, чтобы температура
была выше температуры стеклования предельно отвержденно-
го полимера. Температура отверждения зависит от экзотермич-
ности реакций. Как правило, высокая экзотермичность реакций
отверждения затрудняет достижение оптимальной температу-
ры, приводит к местным перегревам материала, что обуслов-
ливает его структурную неоднородность, появление в изделиях
внутренних напряжений, а следовательно, ухудшение их каче-
ства.
Большое значение имеет объемная усадка отверждаемого-
изделия и количество выделяющихся низкомолекулярных про-
дуктов реакции. Усадка и внутреннее давление в материале,
создаваемое низкомолекулярными продуктами, вызывают ис-
кажение размеров и формы изделия Усадка возрастает
с уменьшением молекулярной массы олигомеров и увеличени-
ем в них числа функциональных групп, а также при увеличе-
нии количества выделяемых при отверждении низкомолекуляр-
ных продуктов. Так, усадка при отверждении без выделения
низкомолекулярных продуктов составляет 3—6%. а с выделени-
ем—15—25%.
Наряду с собственно отвердителями при отверждении при-
меняют также инициаторы и катализаторы Отвердители всту-
пают в химическое взаимодействие с олигомером и входят в-
трехмерную сетку. В качестве отвердителей используют поли*
функциональные соединения, выбор которых определяется при-
родой и строением функциональных групп отверждаемого оли-
гомера. Например, отвердителями олигомеров с эпоксидными
группами служат первичные и вторичные диамины, низкомоле-
кулярные алифатические полиамиды, ангидриды кислот Ново-
лачные фенолоформальдегидные смолы отверждаются гексаме-
тилентетрамином (уротропином), параформом или эпоксидны-
ми олигомерами. Роль отвердителя могут выполнять некоторые
растворители, например фурфурол для фенолоформальдегид-
ных олигомеров, стирол, метилметакрилат для олигоэфиракри-
латов.
Инициаторы отверждения — это соединения, распадающиеся
с образованием свободных радикалов, служащих инициаторами
процесса отверждения по механизму радикальной полимериза-
ции Часто применяют отверждающую систему, состоящую из
инициатора и ускорителя его распада, а иногда еще и соуско-
рителя (промотора). Например, для отверждения полиэфиров
при 300 К применяют инициатор — пероксид метилэтилкетона
и ускоритель — нафтенат кобальта.
Катализаторы ускоряют взаимодействие олигомеров между
собой или с отвердителями при отверждении по механизму по-
ликонденсации или ионной полимеризации. Они не входят в со-
став трехмерной сетки, но остаются в материале, влияя на его
свойства. Например, отверждение эпоксидных смол или реак-
ции эпоксидных групп с гидроксильными, карбоксильными и
другими функциональными группами катализируются третич-
ными аминами. Активность третичных аминов сильно повыша-
ется в присутствии протонодонорных веществ (спиртов, кислот
и др.) и снижается под влиянием протоноакцепторных (амидов
кислот, альдегидов, кетонов и др.).
По механизму радикальной полимеризации могут отверж-
даться, например, олигоэфиракрилаты. Начальная стадия про-
цесса характеризуется довольно длительным индукционным пе-
риодом, в течение которого вязкость олигомера существенно не
изменяется. Продолжительность этого периода можно регули-
ровать подбором инициатора. В качестве инициатора использу-
ют различные пероксиды Период роста и сшивания макромо-
лекул сопровождается быстрым, практически мгновенным на-
растанием вязкости и потерей текучести. Образуется полимер
пространственно-сетчатой структуры с ценными эксплуатацион-
ными свойствами. Переход от жидкого олигоэфиракрилата к
сетчатому полимеру происходит при степенях превращения
я»0,25—1%. В дальнейшем в связи с заметным уменьшением
подвижности макромолекул наблюдается резкое автоторможе-
ние процесса. Предельная степень превращения, при которой
начинается автоторможение, определяется жесткостью цепей:
181
чем она больше, тем ниже предельная степень превращения.
Схематически отвержденный олигоэфиракрилат можно пред-
ставить следующим образом
X О
О X
о
1
I
с—с—сн»
X о
~СН»-С—с—О—R—О—Г—С—R—С—О—R—О—Ъ
II II
О О Jn
где X—Н, СНз, Hal, CN; R — алкильные или арильные радикалы
Отверждение по механизму поликонденсации широко при-
меняется при производстве изделий из поликонденсационных
полимеров фенолоформальдегидных, эпоксидных, уретановых
и др В зависимости от исходных продуктов и требований к из-
делиям отверждение проводят в кислой, нейтральной или ще-
лочной среде.
При производстве пластмасс на основе фенолоформальде-
гидных смол исходным сырьем являются резолы — олигомеры,
содержащие разветвленные и линейные макромолекулы типа
(т=2—5; m-j-n=4—10) с молекулярной массой 400—1000.
При нагревании резолы отверждаются в результате реакции
поликонденсации, превращаясь в трехмерные полимеры — ре-
зиты.
Отверждение включает три стадии. На первой (стадия А)
олигомер (резол) еще способен плавиться при нагревании, раС"
творяться в щелочах, спирте, ацетоне, на второй (стадия В)
182
образуется полупродукт (резитол), который при нагревании
размягчается, но не плавится, частично растворяется, на треть-
ей (стадия С) — образуется полностью отвержденный полимер
(резит), который не размягчается при нагревании, не растворя-
ется и не набухает в растворителях.
Эпоксидные олигомеры, содержащие в молекуле не менее
двух реакционноспособных эпоксидных или глициди-
ловых —СНг—СН—СН2 групп, а также гидроксильные группы.
могут отверждаться, взаимодействуя с разнообразными соедине-
ниями — мономерами, олигомерами, полимерами. Это позволя-
ет в широких пределах регулировать режим отверждения и
свойства получаемых изделий. Отверждение происходит по ме-
ханизму как поликонденсации, так и полимеризации. В качест-
ве отвердителей при отверждении по реакции поликонденсации
используют первичные и вторичные ди- и полиамины, многоос-
новные кислоты и их ангидриды, фенолоформальдегидные смо-
лы резольного и новолачного типа, многоатомные спирты и фе-
нолы (в количестве до 120% от массы эпоксидного олигомера)
Отверждение может происходить на холоду и при нагревании
(400—460 К), при отверждении не выделяется никаких побоч-
ных продуктов. Например, отверждение алифатическими диами-
нами протекает на холоду по следующей схеме:
—О—СН2—СН—СН;
I I
ОН N-
CHj—СН—CHj—О—С—;
N ОН ”’ СН8
СН
СН—CHj—О—
I
ОН
СН
—О-СН,—CH-CHj
он
N~
R
R
183
Отверждение дикарбоновыми кислотами и их ангидридами
происходит в результате взаимодействия с гидроксильными
группами эпоксидных смол:
~СН2—СН—СН,—ORO
I
ОН
~СН2—СН—СН,—ORO~
СООН
Образующаяся карбоксильная группа СООН, содержащая по-
движный атом водорода, способна взаимодействовать с эпок-
сидной группой, образуя новую гидроксильную группу:
~С1I,—СН—СН,—OR—О~
Эта группа может взаимодействовать со следующей молекулой
ангидрида с образованием пространственного полимера. Меж-
ду эпоксидными и гидроксильными группами протекают и по-
бочные реакции, однако скорость их незначительна.
При отверждении эпоксидных смол дикарбоновыми кисло-
тами протекают реакции карбоксильной группы с эпоксидной.
(1) и гидроксильной (2):
о
ОН
R—СО—О—СН,—СН—СН,~ (1),
R—СО—ОН—
он
I
CHs—сн-сн2
—Н2О
R—СО—О
Н—СН,~ (2)
184
Одновременно происходят взаимодействие эпоксидной и
гидроксильной групп (3), а также гидролиз эпоксидной груп-
пы (4):
—сн2-сн-сн2~ Лн ~сн2—сн—сн2— 1 . RCH,—СН-СН2—О (3) он . ТМ'Ы Н14 ГЧ1 rwj
О RCH2—CH^CHj— НйО
ОН
С наиболее высокой скоростью протекают реакции (1), особен-
но в присутствии щелочных катализаторов.
При отверждении эпоксидных смол по механизму полимери-
зации в качестве отвердителей применяют триэтаноламин и
его производные, комплексы BF3 с аминами (типа BF3-NH2R)
и др. При отверждении эпоксидных смол третичными аминами
происходит полимеризация а-оксидного цикла, протекающая по
ионному механизму:
R3N-f-H2C-СН------> R3N—СН2—СН—
R3N—СН2—СН— 4- Н2С-СН- ----> R3N—СН2—СН— О’ и т. л.
I \ / II
0“ О О—СН2—СН
Процессы отверждения играют важную роль при формиро-
вании лакокрасочных покрытий. Отверждение (высыхание) по-
крытий также может происходить по полимеризационному или
поликонденсационному механизму. По реакции полимеризации
(часто сопровождающейся окислением) происходит отвержде-
ние покрытий на основе растительных масел, алкидных, поли-
эфирных и других смол, по поликонденсационному механизму
отверждаются полиуретановые, фенолоальдегидные и другие
покрытия.
Отверждение масляных покрытий происходит вследствие по-
лимеризации по двойным связям. Процесс высыхания иници-
ируется органическими гидропероксидами, образующимися при
взаимодействии масел с кислородом воздуха. Для ускорения
распада гидропероксидов и высыхания в состав покрытий вво-
дят небольшое количество сиккативов — ускорителей отверж-
дения (линолеаты, резинаты, нафтенаты свинца, марганца, ко-
бальта). Отверждение происходит при комнатной температуре
за 24—30 ч, при нагревании — значительно быстрее
По механизму полимеризации отверждаются покрытия на
основе алкидных смол. Кислород воздуха присоединяется по
185
месту сс-метиловых групп с образованием гидропероксидов, слу-
жащих инициаторами полимеризации.
Для отверждения полиэфирных покрытий применяют раз-
личные мономеры (стирол, метилметакрилат, хлорстирол
и др.), которые сополимеризуются с полиэфирами, или реакци-
онноспособные олигомеры, например олигоэфиракрилаты, при-
меняемые в качестве растворителей полиэфиров.
Отверждение полиэфирных смол протекает по радикально-
му механизму в присутствии пероксидных (пероксид бензоила
и др ) или других инициаторов при 350—420 К В присутствии
ускорителей (третичные амины, кислоты и др.) отверждение
происходит на холоду.
По механизму поликонденсации отверждаются полиурета-
новые покрытия двумя методами. По первому методу покрытия
получают из двух растворов — полиизоцианатов и гидроксил-
содержащих соединений, которые смешивают непосредственно
перед применением В качестве полиизоцианатов применяют
низкомолекулярные продукты превращения диизоцианатов,
главным образом на основе 2,4-толуилендиизоцианатов, в ка-
честве гидроксилсодержащих соединений — простые и сложные
олигоэфиры, эпоксидные, алкидные и другие смолы, содержа-
щие гидроксильные группы. При взаимодействии изоцианатных
групп полиизоцианатов с гидроксильными группами гидроксил-
содержащих соединений с функциональностью больше двух
происходит образование полиуретанов пространственного
строения:
O=C=N—R—N=C=O + СН2ОН-СНОН-СН2ОН >
- С-NH-R NH С-О О=С NH -R-NH—С=О
I I I
О о—сн2—сн-сн2-о о-сн2-сн-сн2-о~
---► NH
I
R
I
NH
о с=о
II I
-С—NH- R—NH—С=О О О=С—NH—R—NH—С=О
О—СН2—СН—сн2—о о-сн2-сн-сн2-о^
186
По второму методу покрытия готовят из одного раствора,
являющегося смесью сложных или простых олигоэфирдиолов
к так называемых «блокированных», или «скрытых», изоциа-
натов. Блокированные изоцианаты образуются при взаимодей-
ствии изоцианатов с нитрофенолами, ацетоуксусным эфиром и
рядом других соединений. Они стабильны при комнатной тем-
пературе, но разлагаются при нагревании. Поэтому подобные
системы отверждаются только при температуре выше 420 К.
Под действием тепла распадаются уретановые связи между
изоцианатами и блокирующими агентами. Блокирующий ком-
понент испаряется, а выделившиеся изоцианатные группы взаи-
модействуют с олигоэфирами, образуя пространственный поли-
мер. В основе отверждения покрытий на основе ароматических
полиимидов лежит реакция циклизации с образованием нерас-
творимого неплавкого полиимида:
НО—ОС СО—NH—R~ СО СО
К химическим превращениям, приводящим к увеличению
молекулярной массы, можно отнести получение блок-сополиме-
ров и привитых сополимеров. Блок-сополимеры могут быть по-
лучены при взаимодействии полимера, содержащего одну или
две активные концевые группы (так называемого макромоле-
кулярного инициатора), с мономером, а также при взаимодей-
ствии двух и более полимеров или макрорадикалов друг с дру-
гом непосредственно или с помощью низкомолекулярного сши-
вающего агента. В первом случае активные концевые группы
в определенных условиях инициируют полимеризацию моно-
мера, образуя второй блок Этот процесс может осуществлять-
ся как по радикальному, так и по ионному механизму. Роль
активных центров могут выполнять псроксидные или гидропер-
оксидные группы, находящиеся на концах макромолекулы. При
определенных условиях полимерные пероксиды разлагаются,
образуя макрорадикалы, инициирующие полимеризацию дру-
гого мономера, добавленного в систему. Таким способом чаще
всего получают блок-сополимеры на основе виниловых моно-
меров.
Однако наибольшее применение для синтеза блок-сополиме-
Ров нашла анионная и ионио-координационная полимеризация.
Наличие «живых» полимеров при анионной полимеризации и
Длительное сохранение активности растущих цепей при ионно-
координационной позволяют проводить этот синтез. В качестве
Примера можно привести получение блок-сополимера изопрена
и стирола «Живой» полимер стирола взаимодействует с изо-
187
лреном по ионному механизму:
СН3
+ - + — I
Na СН2—СН--СН—СН, Na 4 nCHs=C—СН=СН2 ►
сн3
СН3 1
т _ I
► Na СН2—С=СН—СН2-[—СН2—С^СН—СН2—Jrt—
—сн2—сн-
66
Г CHS
СН—СН2—L-CH2—С—СН—СН2—Jm
CHS
.-СН2—i=CH—CH, Na
По второму способу блок-сополимеры получаются в резуль-
тате взаимодействия полимерных или олигомерных блоков по
механизму поликонденсации. Соединение блоков может проис-
ходить или путем непосредственного взаимодействия друг с
другом, или с участием низкомолекулярного реагента:
НО- (R)„—OH+OCN—R'—NCO+HO— (R")m-OH —>
—* Н[—О—(R)n—О—СО—NH—R'—NHCO—(R")m—О—]*Н
Привитая сополимеризация широко используется для моди-
фикации свойств полимеров- она дает возможность сочетать в
одной макромолекуле полимерные последовательности, разно-
образные по свойствам и структуре. Методы синтеза привитых
сополимеров мало отличаются от описанных выше. Процесс
привитой сополимеризации можно осуществить двумя путями:
полимеризацией мономера на активных центрах полимера той
же или другой природы; взаимодействием двух полимеров за
счет их функциональных групп или макрорадикалов. В основе
синтеза привитых сополимеров по первому способу лежит реак-
ция передачи цепи на полимер, т. е. отрыв макрорадикалами
от полимера подвижного атома, например водорода. При этом
в макромолекуле появляются активные центры, на которых
происходит полимеризация добавленного мономера. Эффектив-
ность метода определяется скоростью передачи цепи на поли-
vep, которая зависит от соотношения концентраций мономера
и полимера, подвижности отрываемого атома и реакционной
способности полимерного радикала. В качестве примера мож-
но привести реакцию прививки стирола к полибутадиену. В ре'
188
зультате такой реакции получается ударопрочный полистирол:
~СН2—СН=СН—сн2~ + ~СН2-СН ►
---> -СН—сн=>сн-сн2~+~сн2-сн2
6
~СН-СН=СН—СН,~ -f- лСН4=СН -
—>• -сн—сн=сн—сн2~
I
СН2—сн~
6
Наиболее эффективный способ получения привитого сопо-
лимера — радиационное облучение полимера в среде жидкого
Или газообразного мономера. Если облучение проводится на
воздухе, то кроме свободных макрорадикалов образуются пер-
оксиды и гидропероксиды, являющиеся инициаторами полиме-
ризации добавленного мономера с образованием привитого со-
полимера. Привитые сополимеры могут быть также получены
при полимеризации мономеров в присутствии макромолекул,
•содержащих двойные связи. Реакция передачи цепи на полиме-
ры осуществляется с участием растущих макрорадикалов или
первичных радикалов инициатора.
По второму способу привитые сополимеры получают за
счет взаимодействия функциональных групп (—NH2, —О,
—СООН, —ОН и др.) по методу поликонденсации. В полиме-
ре, к которому производится прививка, функциональные груп-
пы должны располагаться вдоль цепи, а в прививаемом поли-
мере — на концах молекулы. В качестве примера можно приве-
сти реакцию модификации полиакриламида:
-С112—СН2~ + CI (—СН2—СН2—О— )ПН -*-
I НС1
CONH2
~СН2—СН-
* СО—NH(—СН2—СН2—О—)„Н
189
3.4 РЕАКЦИИ, ПРИВОДЯЩИЕ К УМЕНЬШЕНИЮ СТЕПЕНИ |
ПОЛИМЕРИЗАЦИИ И МОЛЕКУЛЯРНОЙ МАССЫ £
Уменьшение степени полимеризации происходит в результате
реакций, протекающих с разрывом связей в основной цепи
полимера и называемых реакциями деструкции. Распад макро-
молекул происходит по различным механизмам, зависящим от
строения полимера и факторов, вызывающих деструкцию. Де-
струкция приводит к значительному изменению свойств поли-
мерных материалов и изделий из них, сокращает сроки их
эксплуатации.
Однако в ряде случаев реакции деструкции используют для
получения из природных полимеров ценных низкомолекулярных
веществ, например глюкозы из целлюлозы и крахмала, для ча-
стичного снижения молекулярной массы полимеров в целях об-
легчения их ттереработки, для определения строения высокомо-
лекулярных соединений.
Наряду с деструкцией, протекающей по закону случая, мо-
жет происходить и деполимеризация — последовательное от-
щепление звеньев мономера, начинающееся с концов макромо-
лекулы. При этом молекулярная масса и другие свойства поли-
меров изменяются значительно медленнее, чем при деструкции
по закону случая (рис. 3.2).
При деструкции полимеров по закону случая в течение вре-
мени т среднестатистическая длина образующихся макромоле-
кул, характеризуемая средней степенью полимеризации пх, за-
висит от степени полимеризации исходного полимера йо и сред-
него числа связей S, распадающихся в каждой макромолекуле
за период времени т
й»=по/(5-{-1).
Скорость реакции деструкции v можно определить по урав-
нению
и = In fl-------—
\ «О
-In 1 - —
\ Пх
где k — константа скорости распада; т — продолжительность распада.
Влияние строения полимера на механизм распада можно
проиллюстрировать на примере виниловых полимеров. Полиме-
ры с третичными атомами углерода склонны к реакциям дест-
рукции по закону случая. При замещении в этих полимерах во-
дорода на группу СНз и другие полимеры приобретают склон-
ность к деполимеризации. При определенных условиях поли-
мер может полностью деполимеризоваться до мономера
Для подавления деполимеризации применяют различные
способы: 1) введение в макромолекулу звеньев, отщепляющих-
ся труднее, чем основные (например, винильных путем сополи-
190
м
рис. 3.2. Кинетика снижения молекуляр-
ной массы полимера в процессе деструк-
ции по закону случая (/) и деполиме-
ризации (2)
меризации формальдегида с ви-
ниловыми мономерами); 2) бло-
кирование концевых групп (на-
пример, ацилирование концевых
гидроксильных групп); 3) введе-
ние стабилизаторов.
Деструкция полимеров может протекать под действием хи-
мических агентов (воды, кислот, спиртов, кислорода, озона и*
т. д.) или под влиянием физических воздействий (тепла, света,
ионизирующих излучений, механической энергии и пр.).
Химическая деструкция наиболее характерна для гетеро-
цепных полимеров и протекает избирательно с разрывом свя-
зи углерод—гетероатом. В ряде случаев конечным продуктом
химической деструкции является мономер. Углерод-углеродная
связь устойчива к действию химических агентов, и поэтому
предельные карбоцепные полимеры обычно мало склонны к хи-
мической деструкции Это возможно только в очень жестких
условиях или при наличии боковых групп, снижающих проч-
ность связей в основной цепи полимера.
Обычно деструкцию вызывают низкомолекулярные вещест-
ва, образующиеся в процессе синтеза данного полимера (при
поликонденсации, сополиконденсации, ионной полимеризации
с раскрытием цикла). Эти реакции оказывают большое влия-
ние на строение макромолекул, их молекулярную массу и мо-
лекулярно-массовое распределение. Степень деструкции про-
порциональна концентрации деструктирующего реагента, а ско-
рость увеличивается с ростом температуры.
Деструкция полимеров под влиянием физических воздейст-
вий не имеет избирательного характера, поскольку энергетиче-
ские характеристики различных связей довольно близки. Она
происходит и в гетероцепных, и в гомоцепных полимерах. Ко-
нечным продуктом физической деструкции являются полиме-
ры с более низкой молекулярной массой. При деструкции под
влиянием физических воздействий протекают свободноради-
кальные реакции, приводящие как к разложению, так и к
структурированию полимера.
В зависимости от характера внешнего воздействия различа-
ет химическую, термическую, радиационную, механическую,
фотодеструкцию. Однако в реальных условиях деструкция
обычно происходит при одновременном воздействии несколь-
факторов
191
Для предотвращения или торможения деструктивных про-
цессов в полимерные материалы вводят свето- и термостабили-
заторы, противоокислители (антиоксиданты), антиозонанты
и т. д.
3.4.1. Химическая деструкция
Гидролиз — расщепление при взаимодействии с водой — наи-
более распространенный вид химической деструкции полиме-
ров. Катализаторами гидролиза являются водородные или
гидроксильные ионы. Гидролиз некоторых высокомолекулярных
соединений ускоряется в присутствии природных катализато-
ров — ферментов.
Склонность к гидролизу определяется природой функцио-
нальных групп и связей в полимере При гидролизе боковых
функциональных групп изменяется химический состав полиме-
ра; при гидролизе связей основной цепи происходит деструкция
и уменьшается молекулярная масса полимера.
Из гетероцепных полимеров особенно легко гидролизуются
полиацетали, сложные полиэфиры и полиамиды. Из полиэфи-
ров легче гидролизуются алифатические эфиры угольной
и щавелевой кислот, труднее — высших дикарбоновых кислот,
особенно ароматических Карбоцепные полимеры, как правило,
гидролизу не подвержены. Кристаллические полимеры гидро-
лизуются медленнее, чем аморфные.
Большое практическое значение имеет гидролиз природных
полиацеталей — полисахаридов. При их полном гидролизе об-
разуются соответствующие моносахариды. Так, при полном
гидролизе крахмала и целлюлозы образуется глюкоза; катали-
заторами в этом процессе служат водородные ионы: I
При гидролизе белков, катализируемом кислотами, щелоча
ми, ферментами, получают а-аминокислоты и другие низкомо
192
.пекулярные продукты, например желатину
н ОН +
н. но-
R—NH—СО—R' -------> R—NHa+ НО-СО-R'
Гидролизом полиамидов в щелочной среде при нагревании
получают лактамы с выходом 75—85%:
Н ОН
NaOH
~NH(CH2)6CO—NH(CH2)sCO~ >
ОС
\сн2)8
I1N
Гидролиз сопровождается выделением газообразных угле-
водородов— метана, этана, этилена и др. Полученные лактамы
вновь используют для синтеза полиамидов. Этот метод имеет
большое значение при регенерации и вторичном использовании
изношенных полиамидных изделий. При гидролизе полиамидов
в кислой среде получают диамины и дикарбоновые кислоты.
Скорость гидролиза определяется скоростью диффузии воды
в полимер и скоростью взаимодействия полимера с ней. Она
чпрямо пропорциональна концентрации воды и ионов водорода
(или гидроксильных) в полимере и содержанию функциональ-
ных групп, подвергающихся гидролизу. Процесс гидролиза
имеет автокаталитический характер, поскольку осколки макро-
молекул, возникшие в результате гидролитического расщепле-
ния. в подавляющем большинстве случаев становятся катали-
заторами последующих актов деструкции.
Ацидолиз происходит под действием безводных кислот.
Так, ацидолиз сложных эфиров при взаимодействии с карбо-
новыми кислотами протекает по схеме
~СН2-О OCR
~СНа-О—COR + R'—СО—ОН -----*- I + |
R'—СО ОН
На расщеплении целлюлозы сверхконцентрированной соля-
ной кислотой основан метод определения выхода глюкозы при
полном гидролизе целлюлозы. Ацидолиз целлюлозы протекает
и при обработке ее уксусным ангидридом в присутствии серной
кислоты при повышенной температуре. Частичный ацидолиз
Целлюлозы происходит и в процессе промышленного получения
ацетата целлюлозы.
Скорость ацидолиза зависит от типа кислоты. Например,
под действием адипиновой кислоты полиакрилаты деструктиру-
и>тся в несколько раз быстрее, чем при воздействии изофтале-
пой. При деструкции более высокомолекулярных полиэфиров
молекулярная масса снижается интенсивнее, чем при деструк-
13—816 193
дни в тех же условиях полимеров с меньшей степенью полиме-
ризации.
Алкоголиз — расщепление полимеров под действием спир-
тов — характерен для полисахаридов, сложных полиэфиров
и др. Катализаторы реакций алкоголиза те же, что и гидроли-
за С наибольшей скоростью протекает алкоголиз поликарбо-
натов. Скорость алкоголиза алифатических сложных эфиров
выше, чем ароматических
При обработке полиэтилентерефталата кипящим этиленгли-
колем образуется дигликолевый эфир терефталевой кислоты
или низкомолекулярный полиэфир с концевыми гликолевыми
группами, которые снова могут принимать участие в реакции
поликонденсации:
Н-ЧЭСН2СН2ОН
---> ~О-(СН2)п-ОН +
-О— (CH2)rt—О—ОС—СвН4—со-
+носн2сн2оос—свн4—со-
Аминолиз и аммонолиз Аминолизу подвержены полиамиды,
анилиноформальдегидные смолы, полиимиды. Процесс проте-
кает, например, при синтезе полимеров в результате действия
диаминов, анилина, аминокислот и других аминосоединений,
служащих мономерами для получения данного полимера:
-NH(CH2)nNH-r СО(СН2),„СО~
-NH(CH2)nNH2 +
H-NH—R—NH2
ОС—(СН2),Л—СО—
+ I
NH—R—NH2
Аммонолиз обычно происходит в полимерах, при синтезе
которых выделяется аммиак, например в полиамидах, синтези-
руемых из амидов аминокислот.
Окислительная деструкция протекает значительно менее
избирательно, чем другие виды химической деструкции. Она
характерна и для гетероцепных, и для карбоцепных полиме-
ров. Отсутствие избирательности в процессе окислительной де-
струкции объясняется тем, что если в первоначальном акте
взаимодействия с окислителем участвуют группы, наиболее
подверженные окислению, то в последующей цепной реакции,
протекающей с участием неспаренного электрона, могут при-
нимать участие другие группы макромолекулы.
194
Окисление полимеров является типичным цепным свобод-
норадикальным процессом, включающим следующие элемен-
тарные стадии:
I) образование свободных макрорадикалов
RH+O2 —> R- + HOO.
2RH + O2 -> 2R-+ lI2Oj
(I)
(2)
2) передача цепи, образование пероксидБых радикалов и
гидропероксидов
R-4-O, --> ROO-
ROO- + RH -> ROOH+R
3) распад гидропероксидов, разветвление цепей
ROOII --> RO- + -OH
ROOH 4- RH --* RO- + R- + ЦО
2ROOH ----* ROO- + RO- + Н2Э
4) обрыв цепи
2R. --► R—R
2ROO- --► ROOR+Oj
R- + ROO- --> ROOR
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
Здесь RH — молекула полимера, содержащая группы с пони-
женной энергией связи С—Н (в карбоцепных полимерах —
сс-метиленовые группы, группы, содержащие водород при тре-
тичном атоме углерода).
Образование свободных радикалов и масрорадикалов на
стадии инициирования может происходить под влиянием со-
держащихся в полимере примесей, остатков инициаторов поли-
меризации, а также при действии света, ме>анических напря-
жений, при нагревании и т. д. Дополнительнее количество сво-
бодных радикалов образуется в результате распада полимер-
ных пероксидов и гидропероксидов по реакциям (5)—(7).
Развитие цепи происходит при взаимодействии пероксидно-
го радикала ROO с полимером (реакция 4) Пероксидный ра-
дикал стабилизируется, отрывая подвижный атом водорода от
молекулы полимера; при этом вновь образуется макрорадикал,
взаимодействующий с кислородом.
Окисление имеет вырожденно разветвленный характер, ко-
торый состоит в том, что образующийся по реакции (4) гидро-
пероксид ROOH нестабилен и распадается с образованием но-
вых свободных радикалов RO-, НО-, R-, ROO , которые так-
же отрывают атомы водорода от молекулы полимера.
13*
195
т
Рис. 3 3. Кинетические кривые погло-
щения кислорода в процессе окисле-
ния несгабнлизированного (/) и ста-
билизированного (2) полимеров
В результате скорость присое-
динения кислорода и окисле-
ния полимера резко возраста-
ет; это явление называется
автокатализом. Кинетические
кривые поглощения кислорода
полимерами имеют S-образ-
ную форму, характерную для всех автокаталитических (само-
ускоряющихся) процессов (рис 3.3V
Обрыв цепи происходит в результате реакций рекомбина-
ции или диспропорционирования. Рекомбинация макрорадика-
лов с концевыми пероксидными группам*» происходит с высокой
скоростью (в отличие от медленной рекомбинации обычных
макрорадикалов в твердом полимере) В аморфных полимерах
выше температуры стеклования и в кристаллических выше тем-
пературы плавления скорость рекомбинации таких макроради-
калов резко возрастает
Следует отметить, что в отличи • от низкомолекулярных уг-
леводородов, при окислении которых все радикалы R- превра-
щаются в пероксидные ROO , в о! исляющемся полимере часть
алкильных макрораднкалов встуш ет в реакции изомеризации,
передачи цепи и др.
Скорость окислительной дестру щии определяется скоростью
диффузии кислорода в полимер и скоростью химического взаи-
модействия полимера с кислородом. Скорость диффузии кисло-
рода наиболее высока, если полимзр находится в растворе или
расплаве. При температурах ннж< температуры стеклования,
а тем более кристаллизации, доступ кислорода в полимер за-
труднен Чем выше степень крист )лличности полимера, тем
меньше скорость диффузии кислорода. Растяжение и ориента-
ция полимера также замедляют дьффузию кислорода. Однако
в этом случае окисление ускоряется за счет механической акти-
вации Низкой скоростью диффузиь кислорода характеризуют-
ся сетчатые полимеры.
Механизм и скорость процесса о сисления зависят также от
строения макромолекул. Так, ненасыщенные высокомолекуляр-
ные соединения окисляются значительно быстрее, чем насы-
щенные, поскольку в них легче образуются пероксиды, благо-
даря чему ускоряется стадия инициирования.
При окислении ненасыщенных полимеров кислород может
присоединяться по двойной связи с последующим разрывом
196
макромолекулы или образованием гидропероксндов. В первом
случае реакция протекает по схеме
-сн,—сн=сн—СН,---->
—СН2—СН==СН—СН,— —► -сн,— сн—сн=сн~
ОО- ООН
—СН,—СН—СН—сн,-
I I
о—о
—СН2—СН—сн—сн,-
I I
о- о-
~СН2—сн + сн—сн,-
Возможно также непосредственное образование гидропер-
оксидов ненасыщенных углеводородов с присоединением кисло-
рода к метиленовой группе, находящейся в сс-положении по от-
ношению к двойной связи:
~СН2—СН-=СН-СН,~ СН,—СН=СН—СН-
ООН
Первичным продуктом окисления полипропилена, содержа-
щего третичный атом углерода, также является гидропероксид:
ООН
о, I
—СИ,—СИ—СН,—СН— > -сн,—с—сн—сн-
(Ь3 <1на <!:н3 сна
Обрыв цепи при окислительной деструкции в результате ре-
комбинации радикалов может приводить к образованию меж-
молекулярных связей. Так, при окислительной деструкции по-
лиэтилена получаются нерастворимые сшитые структуры:
-СН,—СН,—СН—сн,—сн,—
----------------------->
-СН,—СН,—СН—СН,-СН,~
л*СН,^СН,—СН,—СН—СН,-— —СН,—СН,—СН,—СН—сн,—
Следует отметить, что при окислительной деструкции на-
блюдается изменение состава полимера в результате появле-
197
Юз]
Рис. 3 4. Кинетические кривые погло-
щения озона каучуками СКИ-3 (/),
СКД (2) и полихлоропреном (3)
ния карбонильных, карбокси-
льных и других кислородсо-
держащих групп.
Озонирование и озонолиз.
Озонированием называют ре-
акцию взаимодействия озона с
непредельными соединениями, озонолизом — расщепление про-
дуктов озонирования с образованием низкомолекулярных спир-
тов, карбонильных соединений, кислот.
Деструкция ненасыщенных соединений под действием озона
протекает по закону случая в результате атаки озонсм двойной
связи и последующего распада озонидов:
СН3 СН3
I Оз I
~СН2—С=СН—СН2-----> ~СН2—С----сн—сн2~ ---►
о—o-i
Пс продуктам разложения озонидов можно судить о строе-
нии соответствующего ненасыщенного полимера. Кроме того,
процессы- озонирования олефинов широко применяют для син-
теза кислородсодержащих соединений, в частности ненасы-
щенных олигомеров с кислородсодержащими концевыми груп-
пами. •
Кинетические кривые поглощения озона тонкими пленками
полимеров, как и кинетические кривые поглощения кислорода,
имеют S-образную форму (рис. 3.4) На глубоких стадиях озо-
нирования после исчерпания в полимере двойных связей в ре-
акцию с озоном вступают группы СН2 и продукты окисления.
Константы скорости взаимодействия озона с твердыми и рас-
творенными полимерами практически не различаются. Однако
при температуре стеклования происходит изменение реакцион-
ной способности полимеров в твердом состоянии.
3.4.2. Физическая деструкция
Термическая деструкция — один из наиболее распространен-
ных видов деструкции полимеров. Она протекает, как правило,
по цепному свободнорадикальному механизму, хотя распад не-
которых полимеров (поливинилхлорида, полиформальдегида)
198
происходит с участием ионов. В зависимости от строения мак-
ромолекул разрушение связей при нагревании может происхо-
дить как по закону случая, т. е. в любом месте макромолеку-
лы, так и по механизму деполимеризации с образованием мо-
номера. Последний специфичен именно для термической дест-
рукции.
Устойчивость полимеров к нагреванию, скорость термиче-
ского распада и характер образующихся продуктов зависят от
химического строения полимеров. Однако первой стадией про-
цесса всегда является образование макрорадикалов в резуль-
тате разрыва наиболее напряженных и ослабленных связей в
макромолекуле. В зависимости от строения звеньев макроради-
калы или деполимеризуются (одновременно образуется новый,
более короткий радикал)
R—СН,—СН—СН2—СН R—СН2-СН 4-СН, СН
'll I I
XX XX
или вовлекаются в реакцию передачи цепи, т. е. передачи не-
спаренного электрона с образованием нового свободного ра-
дикала и макромолекулы с пониженной молекулярной массой:
R—СН,—СН 4- ~СН—CHi—R' R—СН2—СН2 4- ~С—СН2—R'
XX XX
В последнем случае может происходить также разветвление
макромолекул вплоть до образования сетчатых структур Де-
полимеризация наблюдается тогда, когда передача кинетиче-
ской цепи становится маловероятной, поскольку прочность свя-
зи с замещающими атомами или группами в макрорадикале
выше прочности связей основной цепи. Например, в политетра-
фторэтилене передача цепи на полимер маловероятна вследст-
вие высокой прочности связи С—F, и единственным продуктом
его термического распада является мономер тетрафторэтилен:
~CF2—CF2~ < i nCF2—CF3
Распад до мономера наблюдается и при термодеструкции
полимеров, содержащих в главной цепи четвертичные атомы
углерода, например полиметилметакрилата и поли-а-метилсти-
рола Деполимеризация подобных полимеров объясняется сни-
жением прочности связей С—С в главной цепи и энергии акти-
вации деполимеризации вследствие взаимодействия функцио-
нальных боковых групп.
Полимеры линейного строения, не содержащие четвертич-
ных атомов углерода и больших боковых нетермостойких
групп, как правило, при нагревании не деполимеризуются,
199
а распа хаются по закону случая с образованием сравнительно
высокомолекулярных осколков К таким полимерам относятся,
например, полиэтилен, полипропилен, полиметилакрилат.
Деполимеризация обычно начинается с концевых звеньев
макромолекулы и развивается вдоль цепи. Поэтому, изменяя
структуру концевых звеньев, можно влиять на процесс термо-
деструкцни; например, характер деструкции полиметилметак-
рилата зависит от условий инициирования его полимеризации,
полимер, полученный в присутствии пероксида бензоила, зна-
чительно более устойчив к термическому воздействию, чем по-
лимер, полученный фотоипициированной полимеризацией.
Деполимеризацию можно подавить, вводя в состав макро-
молекул звенья, которые принимают участие в реакции пере-
дачи цепи, например сополимеризуя метилметакрилат с не-
большим количеством акрилонитрила или формальдегид с про-
пиленоксидом. Деполимеризация прекращается, как только
концевым становится звено акрилонитрила в первом сополиме-
ре или пропиленокснда — во втором.
Механизм распада макромолекул при нагревании во многом
определяется теплотой полимеризации. Как правило, теплота
полимеризации а.а-дизамещенных мономеров, приводящих к
Таблица 3.1. Влияние строения полимера и теплоты полимеризации на выход
мономера при нагревании
Под нм ер Формула Теплота по- лимериза- ции. кДж/моль Выход мо- номера. %
Полнмстилметакрнлат сн, 1 ~сн2—с~ 41,8—64,3 90—100
Полиметакрилат о—с—осн, ~СНг—СН~ । 83,6 Почти нет
Поли-а-метнлстнрол О=С—ОСН, /СН, ~СН2—с~ 39,7 >90
Полистирол ^CsHs ~сн2-сн~ 1 71,1 >40
Полиизобутилеи QHS /СН, ~СН2—с~ 65,9 20—50
Полиэтилен Vhs ~CHs— СН2~ 92—104 Почти нет
•200
HCI, ммоль
10 20 30 40 Т. Я
Рис. 3.5. Влияние продолжительности
нагревания поливинилхлорида на от- i;
шепление хлорида водорода (ммоль на
мономерное звено) при 420 К (/).
430 К (2) и 440 К (3)
образованию полимеров с чет- а
вертичными атомами углерода,
относительно мала. Поэтому об-
разующиеся полимеры легко под-
вергаются деполимеризации
(табл. 3.1). <
При нагревании полимеров,
содержащих ацильные боковые
группы (например, поливинил-
ацетата) или атомы галогена
(например, поливинилхлорида) 0
происходит отщепление боковых
группировок от основной цепи с образованием двойных связей
в цепи и выделением кислот, галогенводородов и других соеди-
нений:
~CH2—СН—CH2—СН—СН.>—СН----->
I I ‘ I
С1 С1 С1
---> ~СН=СН—СН2-СН—СН=СН~ + 2НС1
ci
Дегидрохлорирование поливинилхлорида начинается при
нагревании его до 413 К и сильно ускоряется при дальнейшем
повышении температуры (рис. 3.5). Деполимеризации при этом
пе происходит. При полном отщеплении атомов С! образуется
устойчивый полиен с сопряженными двойными связями:
~СН2—СН—СН2—СН—СНз—СН~ ------->
I | | —nHCl
Cl Cl CI
---> -СН СН—СН=СН—сн=сн~
Аналогично протекает термодеструкция поливинилацетата
и поливинилового спирта.
Полимеры, содержащие функциональные группы в основ-
ной цепи, распадаются главным образом вследствие разруше-
ния этих групп с выделением разнообразных продуктов реак-
ции. Например, распад сложных полиэфиров идет по схеме
? т
R—CHj-CHa-r-O—СО—СН2—CH2-R' -->
---> R—СН=СН2 + ОН—СО—СН,—СН2—R'
201
При термической деструкции ароматических полимеров
большое значение имеют реакции присоединения атомарного
водорода и активных свободных радикалов к ароматическим
циклам с последующим распадом продуктов реакции Например,
в результате взаимодействия со свободными радикалами (т. е.
в результате передачи цепи) циклоцепные полимеры, содержа-
щие фениленовые ядра, связанные различными мостичными
группами (SO2, О, NH, S и др ) распадаются следующим обра-
зом:
R
Скорость распада таких полимеров определяется термостой-
костью мостичных групп, связывающих фениленовые ядра, а
иногда и их гидролитической устойчивостью.
При отрыве замещающих групп могут возникать межмолеку-
лярные связи, например:
-СНз—СН~
I
ОН
ОН
~сн2—сн~
~сн2—сн—
г |
—► о
—Н2О |
~сн2—сн~
Примером термической деструкции с образованием цикли-
ческих звеньев с сопряженными двойными связями может слу-
жить деструкция полиакрилонитрила или полиметакрило-
нитрила:
~н,с сн,
СН, 'нс// 'сн-
~сн,-сн'сн- * с с
<J:n cn
При нагревании гетероцепных полимеров протекают очень
сложные процессы, сопровождающиеся уменьшением молеку-
лярной массы и выделением разнообразных продуктов разложе-
ния. Так, при термической деструкции полиамидов при темпе-
ратуре выше 373 К выделяются метан, этан, пропан, бутан,
202
этилен, бутилен, циклопентанон. Деструкция может протекать
по следующей схеме:
—NH—(CH2)e—NH-rCO—СН2—СН2—СН2—СН2—CO^-NH—(СН,)е— NH—
—NH—(CH2)e—NH + CO—CH2—CH2—CH2—CH2—CO + NH—(CH2)e—NH—
Отщепившиеся свободные бирадикалы далее превращаются в
углеводороды или циклизуются, в частности с образованием,
циклопентанона
Н2С—сн2
со-£н,—СН2-СН2-СН2-СО > | |
Н2С СН2
\?о
Стойкость к термической деструкции определяет важнейшее
свойство полимеров — их термостойкость, т е. способность сох-
ранять химическое строение и свойства при высоких температу-
рах. При этом важна не та температура, при которой начина-
ется заметное разложение полимера, а та, при которой полимер
может длительно эксплуатироваться без изменения свойств.
Высокой стойкостью к термодеструкции характеризуются
лестничные полимеры, особенно с сопряженными двойными
связями. Их высокая термостойкость обусловлена тем, что
разрыв макромолекулы требует затраты энергии на разрушение
двух о-связей или двух о- и двух л-связей.
Наиболее высокой термической стойкостью отличаются сет-
чатые полимеры с высокой плотностью сетки, содержащие боль-
шое число ароматических звеньев, в частности сетчатые поли-
метиленфенолы. Гетероцепные и особенно элементоорганические
полимеры с высокой степенью поляризации ковалентных связей
' вдоль основной цепи более устойчивы к термодеструкции, чем>
карбоцепные. Среди гетероцепных полимеров особенно устой-
чивы к термодеструкции полиимиды, полибензимидазолы, поли-
циклогидразины, полиоксифенилен. Следует отметить, что поли-
конденсационные полимеры более термостойки, чем полимери-
зационные.
Термическая деструкция полимерных материалов в процессе
переработки и эксплуатации практически всегда сопровождается
окислением. Разрушение полимера при одновременном воздейст-
вии на него тепла и кислорода, т е. термоокислительная
деструкция, вызывает интенсивное изменение его свойств. На-
пример, полипропилен при нагревании в отсутствие кислорода
только начинает разлагаться при 550—570 К, в присутствии
кислорода он уже при 390—400 К за 30 мин становится непри-
203.
Рис. 3.6. Влияние температуры иа
скорость деструкции при нагревании
полистирола иа воздухе (Л — доля
нер аз ложившегося полимера):
/—470 К: 2 — 520 К: 3 — 570 К; 4 —нагрс-
ванне в азоте при 570 К
годным для использования.
Так же как и при окислении,
присоединение кислорода в
процессе термоокислительной
деструкции происходит по цеп-
ному механизму с участием
гидропероксидов и свободных
радикалов Повышение тем-
пературы приводит к сокращению индукционного периода и
увеличению скорости окисления.
Устойчивость материалов к термоокислительной и другим
видам деструкции характеризуют потерей массы при их нагре-
вании в стандартных условиях (термогравиметрический ана-
лиз— ТГА). Для примера на рис. 3.6 приведены термогравимет-
рические кривые разложения полистирола в атмосфере азота и
кислорода воздуха. Из рисунка видно, что кислород ускоряет
разложение полимера. Более высокая термостойкость полисти-
рола в атмосфере азота по сравнению с кислородом подтверж-
дается также более высоким значением энергии активации
деполимеризации в атмосфере азота она равна 142 кДж/моль,
в кислороде — 41,8 кДж/моль.
Кислород сильно ускоряет также термическую деструкцию
полиамидов, что сопровождается существенным снижением их
прочности. Термодеструкция полиамидов начинается с отрыва
водорода от метиленовой группы, находящейся в а-положении
по отношению к NH-группе, и происходит по схеме
о,
R—СН,—NH—СО—R' > R—СН—NH—СО—R' *
ОО- ООН
---> R—(J:H—NH—СО—R*-> R—СН—NH—СО—R' >
О- ОН
I I
--->. R—СН—NH—СО—R' ► R—СН—NH—СО—R' >
/° 7°
---*• R—С/^ -J- NH,—
\Н XR'
Аналогично протекает термоокислительная деструкция поли*
эфиров. В присутствии кислорода увеличивается скорость де*
204
гидрохлорирования поливинилхлорида, которое может происхо-
дить по схеме
о,
R + ~СН,—СН-СН,—СН----*- RH + ~СН,-С—СН,—СН-----
A A A А
O—O. ООН
I I
---> ~CHa—С—СН,—CH----► ~CH2—C—CH2—CH-------
I I I
Cl Cl Cl Cl
o-
I i
---*- ~CH,-C—СН,—CH~ + OH-->
A: A
---»- -СН,—c=o + СН,—CH--> CH,=CH~ + Cl.
Влияние строения полимеров на их стойкость к термоокисли-
тельной деструкции иллюстрируют данные табл. 3 2, из кото-
рых следует, что наиболее устойчивы к термоокислительной
деструкции политетрафторэтилен и кремнийорганические поли-
меры.
При термической и термоокислителыюй деструкции полиме-
ров выделяется большое количество различных газообразных
продуктов. Например, при деструкции полиэтилена выделяются
бутилен, н-бутан, пропан, этан, пентан и другие продукты, при
деструкции полипропилена — ацетон, метан, этан, этилен и др.
Состав продуктов разложения в значительной степени зависит
от температуры (табл. 3.3)
Фото деструкция. Полимеры в процессе эксплуатации практи-
чески всегда подвергаются действию света. Если длина волны
достаточно мала, то кванты света поглощаются полимером и
вызывают разрыв химических связен в макромолекуле с обра-
зованием свободных радикалов. Наибольшее действие оказы-
вает свет с короткой длиной волны (менее 400 нм).
Фотодеструкция характерна для полимеров, содержащих
группировки, способные поглощать свет с короткой длиной вол-
ны. Это цепной процесс, протекающий по радикальному меха-
низму. Фотодеструкция может идти даже при относительно
низких температурах. Механизм реакции определяется строени-
ем основной цепи полимера и природой боковых хромофорных
групп, способных поглощать солнечный свет
Связи С—С и изолированные двойные С=С не поглощают
свет с длиной волны более 190 нм. Поэтому в карбоцепных по-
лимерах под действием света с такой длиной волны возбужда-
ются и отщепляются боковые группы (чаще водород) и образу-
205
Таблица 32. Стойкость полимеров к термоокислительной деструкции на воз-
духе при 620 К в течение 34 «
Полимер Формула Потеря массы, % ОТ ис- ходного
~ CFj—CFj ~
Политетрафторэтилен
(тефлон)
Полндиметилфенилалю-
мосилоксан
Полидиметилфеинлси-
локеан
СН8 свн5
~ ii—О—Si—О—AI—О~
сн, о-
сн, С8Н5
~Si—О—Si—О
<^н3 о~
он
Фенолоформальдегидиая
смола
Полиакрилонитрил
~сн,
~снг—сн~
I
C = N
2,1
8,8
22,8
68,0
72,0
Полиэтилентерефталат
(лавсан)
Полиамид (капрон)
Политрифторхлорэтилеи
~СО— (СН2)6—NH~
~CFCI—CF2~
О СН-СНг— О—СО—
91,2
94,3
98,9
Таблица 3.3. Состав газов, выделяющихся при деструкции полиэтилена
[% (мол.)]
Температура, К Азот Кислород Оксид углерода Диоксид углерода Метан Этилен Пропилеи Бутены
720 75,5 1,1 3,7 9,3 0,8 1,6 1,3 1,2
810 75,6 1,4 3,6 9,6 0,6 1,5 1,2 1,2
870 77,0 1,4 3,5 9,6 0,5 1,2 1.0 0,7
970 77,5 1.5 2,4 9.7 0,2 0 0,1 0,3
1120 77,5 1,5 0,2 11,3 0 0 0 0
206
ются макрорадикалы, последующие превращения которых могут
привести к фотодеструкции
В результате протекания цепной реакции, инициированной
образовавшимися радикалами, могут изменяться молекулярная
масса, строение и свойства полимеров Например, при действии
ультрафиолетового света на разбавленные растворы изопрено-
вого каучука в атмосфере азота уменьшается молекулярная
масса полимера; в концентрированных растворах после сниже-
ния молекулярной массы наблюдается ее рост, что обусловлено
формированием нерастворимой фракции при соединении макро-
молекул каучука в сетку. Таким образом, при действии на поли-
меры света может происходить не только деструкция, но и
структурирование. При действии ультрафиолетового излуче-
ния на полимеры при повышенной температуре (423 К) скорость
деструкции резко возрастает (фотолиз)
Реакции, протекающие при действии ультрафиолетового све-
та, можно рассмотреть на примере полиизопрена. Как уже было
сказано выше, процесс начинается с отрыва атома водорода с
образованием свободных радикалов:
СН3 CHS
Av I
~СН8—С=СН-СН8-------> ~СН8—С-СН—СН~ 4- н
сн3 сн3
I н I
~сн2—с=сн—сн2-------- ~сн8—с=сн—сн~ + н2
Водород отщепляется от а-метиленовых групп, где энергия
связи С—Н уменьшена за счет сопряжения с двойной связью.
Образующийся свободный радикал аллильного типа может
изомеризоваться, вызывая деструкцию макромолекулы полиизо-
прена:
СН3 СН3
~сн2—с—сн—сн—сн2—с=сн—сн8~------->
сн2 сн3
>1 . I
---*- ~СН8—С—СН-=СН8 4- СНг—С=СН—CHS~
Он может также взаимодействовать с другим макрорадикалом,
что приводит к сшиванию макромолекул:
СН3 СНз
~СН8—сн~ ~сн2—с=сн—сн~
---* I
~СН8—С-СН—СН~ ~СН2 С-СЕ -СН~
<!:н3 <!:н3
В результате одновременно протекающих процессов дест-
рукции и сшивания не только изменяется средняя молекуляр-
207
пая масса полимера, но и расширяется его молекулярно-массо*
вое распределение.
Механизм фотодеструкции полимеров, содержащих боко-
вые группы, поглощающие свет (хромофорные группы), зависит
от строения этих групп Так, фотодсструкция полиметилметакри-
лата протекает по схеме
СН3 СН3 СН3 СН3
I I I ? I
R-CHj—С—СН2—С—R' *- R—СН2—С—СН2-С—R'-I-CO +ОСН3
со со (to
А-сн3 о сн3 А-сн3
сн3 сн3
R—СН2—А=СН2 4- .A—R'
Ао
А—сн3
Даже незначительное изменение строения хромофорных
групп приводит к изменению механизма фотодсструкции. На-
пример, деструкция близкого к пол и мстил мета крила ту по
строению поливинил мстил кетон а происходит не только по сво-
боднорадикальному механизму, но и путем внутримолекуляр-
ной передачи атома водорода:
/IV :
R—СН2—СН—СН2—СН—R' ---► R—СН,—CH—CH2—С—R' --->
I I i I / \
СО со со н со
II II
СН3 СНз СНа СН3
---> R—СНг—СН CH2=C-R'
II I
С-ОН 4- со
I I
сн8 СНз
R—СН8—СН=С—СН3 > R—СН2—СН2—СО—СНЭ
он
При расположении хромофорных групп на небольшом рас-
стоянии друг от друга полимер поглощает меньше света, чем
при больших расстояниях между этими группами.
В ряде случаев фотохимические реакции в полимерах (поли-
олефинах, поливинилхлориде, полистироле) сенсибилизируются
примесями или функциональными группами, возникающими в
макронепях в результате окисления при переработке, хранении
204
или эксплуатации. Наиболее [нарушителем для этих полимеров
свет с длиной волны 300—370 им (300 нм — для полиэтилена,
310 нм — для поливинилхлорида, 318 нм — для полистирола,
370 нм — для полипропилена) Гетероцспные полимеры (напри-
мер, полиуретаны, полиазины, ароматические полиэфиры и по-
лиамиды), как правило, подвергаются интенсивным фотохими-
ческим приращениям, так как хромофорные группы входят в
основную цепь макромолекулы. Целлюлоза при воздействии
света с длиной волны менее 340 нм подвергается фотсдеструк-
ции, при воздействии света с длиной волны больше 340 им —
фотодеструкции, окислительной деструкции, гидролизу. Фотохи-
мические процессы в таких полимерах могут инициироваться
и развиваться непосредственно в основной цепи
Эффективность действия света на полимер оценивают.вели-
чиной квантового выхода — отношения числа квантов, вызываю-
щих деструкцию, к общему числу поглощенных квантов. Кван-
товый выход разрыва макромолекул при облучении пленок из
полимеров обычно значительно ниже квантового выхода при
облучении растворов.
При облучении твердых материалов действие света обычно
ограничивается реакциями на поверхности. Проницаемость све-
та в твердый нолп.мерный материал определяется его оптически-
ми свойствами. Поэтому на скорость фотодсструкцин большое
влияние оказывает цвет, матовость поверхности. Особенно
легко разрушаются при действии света тонкие пленки из поли-
меров
В большинстве случаев фотохимическая деструкция сопро-
вождается гидролизом и окислением под действием влаги и
кислорода воздуха, активированными светом, что обусловлива-
ет весьма сложный характер реакции В присутствии кислорода
резко возрастает скорость деструкции натурального каучука,
происходит его фотоокислительная деструкция, которая вызы-
вает изменение цвета, разрушение повер?;но^ти, ухудшение
механических и других свойств полимеров.
При фогоокнелительпой деструкции образуются пероксид-
ные макрорадикалы, которые распадаются следующих- образом:
оо о
I i 2
R—СН—СН2—Rz ----> R—CH-f-O—CH2—R'
О.
1 II
~CH2—С—СН2~ ----> ~СН2—С—СН2~ + R-
R
В результате подобных реакций при фотоокислительной
деструкции образуются спирты, альдегиды, кетоны, пероксид-
ные и другие соединения.
14—816 209
Наиболее полно механизмы фотоокислительной деструкции
изучены на примере полиолефинов. Установлено, что деструкция
полипропилена начинается с образования гидропероксидов
сн3 сн3 сн3 сн3 сн3 сн3
I I о2 I II
~сн2—сн—сн2—сн—сн2—сн------*• ~си2—СН—сн,—с—сн2—сн~
ООН
которые быстро разлагаются с образованием кетонных групп:
сн3 сн3 сн3
~ СН,—СН—СН,—С—СН,—СН------>
ОтЮН
СН3 СН3
• •
---> ~ СН3—СН—СН2—С—СН2—СН~ + сн3 + ОН
Дальнейшая деструкция окисленного полимера протекает по
механизму фрагментации Норриша (типа I и II) при комнатной
температуре. Общая схема фотодеструкции гидропероксида
полипропилена приведена ниже:
СН? . СН3 реакция
: : I Норриша (I)
СН,—СН—СН,4~СО-гСН,—СН ~ --------►
СН3 СН3
I .1
СН,—СН—СН, + со + сн,—сн
сн3 . сн3 сн3
I И I
~ СН,—СН—CHj—С—СН,—СН-------
I
ООН
гидропероксид полипропилена
СН3 СН3 СН3
| . | = I
---> ~ СНСН—СН, + ОС-CHjfCH ~ + ОН
еакиия
Соррмша (II)
Н3С СНЯ
+ СН2=СН ~
Фотодеструкция полиэтилена начинается с поглощения фо-
тона карбонильной группой С=О (образующейся в полиэтиле-
не вследствие окисления при обработке) с последующей дест-
рукцией по реакциям Норриша.
210
Радиационная деструкция (радиолиз) полимеров протекает
под влиянием излучений высокой энергии (рентгеновские и 7-
лучи, нейтроны, протоны, быстрые электроны, а-частицы и др.).
Энергия этих излучений составляет 9—10 эВ, а энергия химиче-
ских связей в полимерах — 2,5—4,0 эВ Поэтому такие излу-
чения способны вызвать разрыв связей, однако это происходит
не всегда, поскольку часть энергии рассеивается, например в
виде теплоты. Под влиянием ионизирующих излучений в поли-
мерах происходят глубокие структурные и химические измене-
ния. Регулируя интенсивность излучения, можно изменять
свойства полимера в заданном направлении, например перево-
дить их в неплавкое, нерастворимое состояние. Так, облученный
полиэтилен характеризуется очень высокой термостойкостью,
химической стойкостью и другими ценными свойствами.
При действии излучений высокой энергии на полимер проис-
ходит разрыв связей основной цепи, отрыв замещающих групп,
сшивание и др. В отличие от термодеструкции радиолиз не вы-
зывает деполимеризацию полимера и не является цепным
процессом. Радиационная деструкция всегда протекает по за-
кону случая.
Под действием излучений молекулы полимера ионизируются
и возбуждаются. Возбужденная молекула может распадаться
на радикалы, а выделяющийся при радиолизе вторичный элект-
рон— рекомбинировать с образовавшимся ионом полимера
и взаимодействовать с другими молекулами, образуя новые
ионы.
Состав и количество продуктов радиационной деструкции
зависят от химического строения полимеров. Так, при деструк-
ции полиэтилена, полипропилена, полистирола, полибутадиена
основным летучим продуктом деструкции является водород, при
деструкции полимерных кислот и сложных эфиров выделяются
оксид и диоксид углерода, при радиолизе поливинилхлорида и
поливинилиденхлорида — хлорид водорода и хлор.
Как уже было сказано, при радиолизе протекают и деструк-
ция, и сшивание. Это можно видеть на примере полиэтилена:
~ СН2—СНг-----—> - СН2—СНг - 4-е----> ~ СН2—СН2----1
~ СН2—СН ~ + Н- ---► ~ СН=СН ~ 4- Н2
।~ СН2 -f- СН2 ~
~ СН2—CHS ~ Н- ----> ~ СН-СН2 ~ н- н,
~ сн2—сн ~ ~ сн2—сн ~
4- ---*" ।
~ СН2—СН ~ ~ СН2— н ~
В большинстве случаев реакции деструкции и сшивания
при радиационной деструкции протекают одновременно, но в
14* 211
зависимости от химического строения полимера одна из них
может преобладать. Деструкции подвергаются главным образом
полимеры, характеризующиеся низкими теплотами полимери-
зации на основе а,a-за мешенных углеводородов, имеющие в
основной цепи периодически чередующиеся четвертичные угле-
родные атомы (например, полиметилметакрилат, полиизобути-
лен, полн-а-метилстирол и др.), галогенсодержащие полимеры,
целлюлоза. Полимеры с высокой теплотой полимеризации, ма-
лым выходом мономера при нагревании, не имеющие четвер-
тичных атомов углерода в основной цепи, при облучении в ос-
новном структурируются (полиэтилен, полибутадиен, полиизо-
прен, полистирол и др.). Из волокнообразующих полимеров
структурируются полиамиды и полиэфиры. Особенно устойчивы
к радиационным воздействиям полимеры на основе аромати-
ческих углеводородов, так как бензольные кольца способны
поглощать значительную часть энергии излучения. Если бен-
зольные кольца не входят в состав основной цепи полимера,
а являются замещающими группами, то их радиационная стой-
кость проявляется в меньшей степени, и радиолиз вызывает
отрыв атомов водорода, особенно а-водородных атомов, от али-
фатических групп. Деструкция полимеров с ароматическими
заместителями протекает очень медленно. Например, стойкость
полистирола к радиационному воздействию в 80—100 раз выше
стойкости полиэтилена. Поэтому все вещества, которые вводят
в полимеры для защиты от радиационной деструкции (антира-
ды), содержат ароматические кольца.
Количественно соотношение реакций деструкции и сшивания
при облучении оценивается по радиационно-химическому выхо-
ду сшивания (Gc) и деструкции (6Я), т. е. по числу актов раз-
рыва или сшивания при поглощении 100 эВ энергии излучения.
Наибольшим радиационно-химическим выходом деструкции
характеризуются целлюлоза (Сд>10), политетрафторэтилен
(GB«5,5), поли изобутилен (6Д«;5). Наиболее стойкие к ради-
ационной деструкции полимеры имеют радиационно-химический
выход в пределах до 1,5 (например, полистирол — 0 01, полипро-
пилен— 0,8, полиэтилен—1,0—1,5). Число разрывов, а также
число образующихся поперечных связей прямо пропорционально
дозе облучения и не зависит от интенсивности излучения. По-
скольку разрыв макромолекул происходит по закону случая,
молекулярная масса полимера после облучения при одной и
той же дозе не зависит от молекулярно-массового распределения
и определяется только химическим строением полимера. Сред-
нечисловая молекулярная масса при радиолизе уменьшается
по закону
1/Л7„= 1/М®„+1,04 • 10-вгбя,
где Л7„° — молекулярная масса полимера до облучения; г — доза облучения;.
Ga — радиационно-химический выход.
212
Образование поперечных связей в облученных полимерах
особенно интенсивно развивается после перехода полимера из
стеклообразного состояния, в котором он подвергался облуче-
нию, в высокоэластическос. Это объясняется подвижностью
макромолекул в высокоэластическом состоянии, в результате
чего они могут приближаться друг к другу на расстояния, рав-
ные длине химических связей между атомами углерода сосед-
них макромолекул.
Радиационная деструкция происходит более интенсивно при
повышении температуры, а также в присутствии кислорода
воздуха, который в ряде случаев резко ускоряет деструкцию
Например, поливинилиденфторид при облучении в вакууме
структурируется, а при облучении на воздухе деструктируется.
Радиационное окисление связано с присоединением молекул
кислорода к свободным радикалам и образованием пероксид-
ных радикалов Последующие превращения радикалов приво-
дят к образованию устойчивых высокомолекулярных соедине-
ний с кислородсодержащими функциональными группами
(карбонильными, карбоксильными, гидроксильными и др ) или
низкомолекулярных кислородсодержащих продуктов (СО, СО2,
Н2О и др). Процесс радиационного окисления можно иллюст-
рировать следующей схемой:
V-лучи . ♦ Оа
R—СН,—R' ------*• R—CH-R' + H ---> R-CH-R'
io-
о. ООН
I I
R—СН—R'+ R*—СН,—R"' *• R—CH-R' + R’—СН—R'"
I
О-
I
R—СН—R' + ОН
R—CH—R' 4- Н --> R—СН—R'
I I
О- ОН
Скорость и интенсивность радиационно-окислительной дест-
рукции существенно зависят от площади поверхности сопри-
косновения полимера с кислородом, скорости диффузии кисло-
рода и продуктов радиолиза в полимере. После прекращения
облучения полимера в нем остаются различные активные
частицы — свободные радикалы, ионы, которые вызывают проте-
кание (иногда в течение длительного времени) многочисленных
пострадиационных необратимых процессов (окисления, деструк-
ции, сшивания и др ). Например, в полиметилметакрилате при
300 К макрорадикалы сохраняются в течение нескольких ме-
сяцев.
213
Механическая деструкция. В процессе механической перера-
ботки полимеров или их смесей с наполнителем (вальцевание,
измельчение, прессование, каландрование) возникают большие
внутренние напряжения, которые могут привести к разрыву
макромолекул. То же наблюдается и при эксплуатации поли-
мерных материалов под действием механических напряжений.
Разрыв макромолекул приводит к образованию макрорадика-
лов, способных инициировать различные химические реакции
в полимерах, которые называются механохимическими.
Разрыв изолированной макромолекулы при механическом:
воздействии (механокрекинг) происходит вследствие того, что-
приложенное напряжение превышает прочность химической,
связи между атомами основной цепи, величина которой на-
ходится в пределах 4—6 Н/связь В полимере напряжения, воз-
никающие при деформации, распределяются не только на хими-
ческие валентные связи основной цепи, но и на межмолеку-
лярные связи между цепями. Если бы все макромолекулы
были распрямлены и уложены параллельно друг другу, то при
деформации такого пучка они испытывали бы почти одина-
ковое напряжение, были бы равномерно нагружены и для их
разрыва потребовались бы очень большие напряжения, превос-
ходящие средние напряжения, возникающие при переработке
и эксплуатации. Однако в реальных условиях различные мак-
ромолекулы и даже участки одной и той же макромолекулы
расположены в различных направлениях относительно направ-
ления действующей силы. Вследствие этого, а также различной
подвижности сегментов, наличия сил внутреннего трения или.
межмолекулярного взаимодействия, тепловых флуктуаций при
деформации полимерных материалов их отдельные структур-
ные элементы испытывают различное напряжение. В какой-то
точке это напряжение может превысить критическое напряже-
ние, равное прочности химической связи цепи, вследствие
чего эта связь рвется. •
В отличие от деструкции под влиянием теплового или радиа-
ционного воздействия механическая деструкция не затрагивает
замещающих групп или атомов, концевых звеньев
Количественной мерой механодеструкции является степень
деструкции, пропорциональная числу разрывов в макромолеку-
ле. Скорость механодеструкции характеризуют увеличением
числа макромолекул в исходном объеме полимера в единицу
времени Увеличение числа макромолекул х в системе совпа-
дает с количеством связей, разорванных за период времени t:
f 2-_ 2 Y
tn \ nt п0 )’
где N&— число Авогадро; т — молекулярная масса мономерного звена-
no, fit — среднечисловая степень полимеризации до начала деструкции и в.
момент времени t.
214
10< 105
10«
10' JU
Рис 3.7. Влияние продолжительности механической деструкции натурального
каучука на молекулярно-массовое распределение (цифры у кривых — про-
должительность вальцевания в мин, п — относительное содержание фрак-
ций с данной молекулярной массой)
Рис. 3.8. Зависимость молекулярной массы от продолжительности механиче-
ской обработки полимера
В процессе механодеструкции происходит постепенное сни-
жение степени полимеризации. Степень полимеризации, при
которой деструкция резко замедляется или прекращается, на-
зывается пределом деструкции. Предел деструкции для многих
полимеров составляет 100—1000 звеньев. Кроме того, происхо-
дит выравнивание длин макромолекул и, следовательно, суже-
ние молекулярно-массового распределения (рис. 3.7). Мини-
мальная предельная молекулярная масса определяется соотно-
шением энергий химических связей макромолекулы и
межмолекулярного взаимодействия. Кроме того, большое
значение имеет вид механического воздействия, величина
прилагаемой нагрузки, температура и характер среды. Увеличе-
ние степени асимметрии, жесткости и плотности упаковки
макромолекул и концентрации раствора благоприятствуют
механическому крекингу полимеров. И наоборот, повышение
гибкости и подвижности тормозит этот процесс.
Влияние продолжительности механодеструкции на моле-
кулярную массу показано на рис. 3.8 Из рисунка видно, что
существует четкая взаимосвязь между изменением молекуляр-
ной массы и временем механического воздействия Поэтому
характеристиками степени механической деструкции могут
служить также и величины <pi—<рз:
<Pi"(Afo—ЛЬ)/(Л1о — Мм); ф2—ф|(Л!оо/Л1«)»
где Mo, Mt — молекулярные массы в начальный момент и момент t; Мп —
молекулярная масса, соответствующая пределу деструкции.
215
Ниже приведены значения степени деструкции полистирола
при его измельчении в атмосфере азота при низких температу-
рах в зависимости от исходной молекулярной массы при М« =
=700 и Л1,=30 000:
240 000 93 000 34 000
ф! 0,900 0,730 0,148
<Р2 0,210 0,170 0,035
фз 0,234 0,234 0,234
Скорость механической деструкции можно
дующим образом:
_ 2*мдУл *'
уд Л1оо Ле-Л/ + Ли’
вычислить еле-
где ЛМд — константа скорости механодеструкции; Л=Л/0—Af»
Как уже было сказано, механокрекинг приводит к образо-
ванию свободных радикалов с локализацией неспаренного
электрона в зоне разрыва Эти радикалы крайне неустойчивы
и могут существовать только при очень низких температурах.
Число свободных (первичных) радикалов Z равно:
Z = 2m„Nk (—J— — )
* А к Л1<Х. Л1о J
(где /Пд—масса деструктируемого полимера)
Стабилизация свободных радикалов может происходить в ре-
зультате рекомбинации первичных радикалов, миграции не-
спаренных электронов по цепи или между цепями (межцепной
обмен) и т. д. Миграция неспаренных электронов по цепи
приводит, как правило, к образованию разветвленных и сшитых
структур, поскольку вторичные радикалы участвуют в реак-
циях роста цепи В реальных условиях макрорадикалы вступа-
ют в реакции друг с другом и с примесями.
Обрыв цепи при механической деструкции может происхо-
дить при взаимодействии свободных радикалов со специаль-
ными добавками, являющимися акцепторами свободных ради-
калов. Они могут стабилизировать свободный радикал или
сделать его менее активным Подобными свойствами обладает
кислород. При обрыве цепи за счет взаимодействия с кислоро-
дом в системе могут появиться группы, содержащие кислород,
например:
Такие группы были обнаружены в полиэтилене после его од-
ноосного растяжения, в каучуках после их измельчения и в
других случаях.
216
Следствием механокрекинга также является отщепление
мономеров и других низкомолекулярных веществ, например
выделение этана при механодеструкции полиэтилена
- сн,—сн24-сн2—сн3--> ~СН2—СН.+СН4—CHS
СН.—СНа + ~СН2—СН.,—сн.—сн.-----
----------------* ~СН.—СНг—СН—СН2~+СН3—СН3
образование HCN при мсханодсструкции полиакрилонитрила
~CHS-CH-СН.—СН -----------*- ~СН2—СН=СН—СН !- HCN
I > I
C-N C=N C=N
.Механодеструкция полимеров сопровождается эмиссией
электронов, люминесценцией, а в среде, где возможно тормо-
жение электронов, появлением рентгеновского излучения. Эти
процессы, в частности эмиссия электронов, приводят к появле-
нию в макромолекулах довольно стабильных активных центров,
которые существенно повышают активность полимера в после-
дующих реакциях, например окисления, структурирования
В частности, такое явление было обнаружено прн высокоско-
ростном деформировании каучуков и при их измельчении.
Таким образом, следствием мсханодсструкции полимеров
является снижение молекулярной массы, появление новых
концевых групп, образование разветвленных и сшитых структур,
выделение низкомолекулярных веществ, механоактивация.
Среди наиболее важных факторов, влияющих иа механическую
деструкцию, можно выделить физическое состояние полимера
(см. гл. 4) в момент деструкции; структуру макромолекул на
молекулярном и надмолекулярном уровнях; условия проведения
процесса (температура, скорость воздействия, среда, наличие
примесей и др.).
Вероятность мехапокрекинга определяется соотношением
суммы энергий межмолекулярных взаимодействий (^Ем<;) и
энергии химических связей (£»<:) основной цепи. При условии
разрушению будут подвергаться в первую очередь
химические связи, т. е. вероятность мехапокрекинга высока.
Если же ^Ем(:<£Хг, то разрыв макромолекул практически не
должен наблюдаться, поскольку при этих условиях более веро-
ятно скольжение молекул относительно друг друга.
Наибольшее межмолекулярное взаимодействие наблюдает-
ся у полимеров в стеклообразном состоянии, меньшее в высо-
коэластичном, ничтожно малое — в вязкотекучем. Поэтому
механодеструкции в наибольшей степени подвержены полимеры
в стеклообразном состоянии, в меньшей — в высокоэластичном,
в вязкотекучем теоретически мсханодеструкция не должна
217
наблюдаться вообще Однако на практике при переработке
полимеров в вязкотекучем состоянии механокрекинг часто
имеет место. Это обычно обусловлено либо полидисперсностью
полимера, т. е. наличием макромолекул большой длины и,
следовательно, имеющих более высокие температуры течения,
чем температура переработки, либо влиянием больших скоро-
стей деформации, приводящих к переходу полимера в другое
физическое состояние В общем случае справедливо положение:
механокрскинг начинается с момента потери полимером теку-
чести.
Влияние химической природы полимера, определяющей
энергию химических связей в цепи, уровень внутри- и межмо-
лекуляриых взаимодействий, совершенно очевидно: вероятность
механодеструкции снижается с ростом энергии химических
связей в основной цепи. Так, в ряду карбоцепных полимеров
полиэтилен—полипропилен — нолиизобутилен наибольшая сте-
пень деструкции наблюдается у полиизобутилена, что связано
с наличием четвертичного атома углерода, который обусловли-
вает меньшую энергию связи в цепи. Этим же (т. е. наличием
участков с пониженной энергией) обусловлены высокие ско-
рость и степень деструкции полиизопрена, в котором низкой
энергией характеризуются связи между а-метиленовым и груп-
пами
Увеличение плотности энергии когезии приводит, как пра-
вило, к увеличению степени и скорости деструкции. Это убеди-
тельно иллюстрирует изменение твердости при пластикации
(технологический прием целенаправленного снижения молеку-
лярной массы полимера механической деструкцией в присутст-
вии акцепторов свободных радикалов) сополимеров бутадиена
и нитрила акриловой кислоты (НАК) при различном их соот-
ношении:
Содержание НАК. масс. ч.
Параметр растворимости,
(кДж/моль)71
Ts/Го
18 18,1 26 18,9 40 20,2
0.85 0,80 0,70
0,70 0,60 0,23
Примечание. Го н ft — твердость первоначальная и через 5 мин пластикации
— течение твердости, соответствующее пределу деструкции.
Разветвленность макромолекул также влияет на механо-
деструкцию. Так, разветвленные макромолекулы деструкти-
руются легче, чем линейные, поскольку точки ветвления (а это,
как правило, третичный или четвертичный атом углерода)
являются дефектными участками, по которым начинается меха-
нокрекинг. Например, линейные полибутадиены (СКД-1.
СКЛД) не деструктируются при обработке на вальцах в отли-
чие от полибутадиенов разветвленной структуры.
218
Рис. 3.9. Изменение характеристиче-
ской вязкости [П] растворов желати-
на (/) и казеина (2) при механоде-
струкцни
Влияние конформации мак-
ромолекулы на механическую
деструкцию можно проследить
на примере полимеров класса
полипептидов — желатина и
казеина (рис. 3.9). Цепи же-
латина имеют асимметричную
трехспиральную конформа-
цию, а макромолекула казеина
характеризуется глобулярной конформацией. Из рисунка вид-
но, что более жесткий полимер деструктируется интенсивнее.
Одной из причин этого может быть большее число межмолеку-
лярных связей в случае спиральной конформации, что способ-
ствует механодеструкции
Механическая деструкция протекает по-разному в зависимо-
сти от того, находится ли полимер в аморфном, кристалличе-
ском или аморфно-кристаллическом состоянии. В кристалличе-
ских полимерах, почти не содержащих дефектов структуры,
напряжения при деформации равномерно распределяются по
всем связям, и деструкция происходит при более высоких
напряжениях, чем в аморфных. В аморфно-кристаллических
полимерах ввиду высокой дефектности кристаллов при механи-
ческом воздействии первоначальным актом является разруше-
ние кристаллических областей (своеобразное плавление), а
затем разрушение аморфной структуры. Это было обнаружено
при диспергировании кристаллического полиэтилена при низких
температурах. Степень деструкции таких полимеров несколько
выше, чем аморфных. Например, при измельчении закристал-
лизованного полихлоропрена степень деструкции выше, чем
при измельчении в тех же условиях аморфного образца
(рис. 3 10).
Поскольку в закристаллизованных полимерах дефекты со-
средоточены на границе раздела кристалл — аморфная часть,
то механокрекинг прежде всего затрагивает эту область, т. е.
идет по «проходным» цепям в граничных областях. Это также
может служить причиной более высокой степени деструкции
аморфно-кристаллических образцов.
Для большинства полимеров скорость механодеструкции,
характеризуемая повышением пластичности, снижается с ро-
стом температуры (рис. 3.11). Это связано с уменьшением
плотности энергии когезии по мере роста температуры и повы-
219
Рис. 3.10. Изменение степени деструкции ср закристаллизованного (/) н
аморфного (2) полихлоропрена при измельчении (d— размер частиц)
Рис. 3.11. Влияние температуры на изменение пластичности ДР при механи-
ческой (/) и тер.моокиелнтельной (2) деструкции
шепнем вероятности проскальзывания макромолекул; при
температуре, близкой к температуре текучести, механокрекин-
га практически не наблюдается
На протекание механодеструкции большое влияние оказы-
вает среда, в которой происходит процесс. Особенно интенсивно
протекает деструкция в среде кислорода из-за образования
псроксидных радикалов, которые принимают участие в дальней-
ших реакциях окисления. На рис. 3 12 показано изменение
пластичности при пластикации натурального каучука в различ-
ных средах. Наименьшая деструкция наблюдается в среде азо-
та. Характер среды предопределяет и температурную зависи-
мость механодеструкции. В среде инертного газа пластичность
незначительно и монотонно убывает до ПО—130 °C (т е. до
температурной области вязкого течения). В среде же, содержа-
щей кислород, деструкция подчиняется закономерностям про-
цесса термоокисления, для которого характерен положитель-
ный температурный коэффициент. В результате наложения двух
процессов (механодеструкции и термоокисления) температур-
ная зависимость изменения свойств в результате деструкции
списывается кривой с минимумом в области температур, близ-
ких к температуре вязкого течения.
Если механические напряжения не вызывают разрыва мак-
ромолекул, они могут активировать химические реакции в
полимерах Так, эластомеры быстро разрушаются в присутствии
220
небольших количеств озона, если они находятся в растянутом
состоянии При многократном приложении напряжения быст-
рее протекает окисление полимеров.
В ряде случаев механическую деструкцию полимеров прово-
дят целенаправленно для придания полимерным материалам
требуемых технологических свойств. Механическим разрушени-
ем макромолекул в присутствии акцепторов свободных радика-
лов снижают среднюю молекулярную массу полимеров, облег-
чая их смешение с компонентами, входящими в состав разл!1 -
ных полимерных композиций, а также получение концентриро-
ванных растворов более низкой вязкости, формование изделий
из расплавов. Этот же процесс лежит в основе полученич
некоторых блок- и привитых сополимеров. Акцепторами сво-
бодных радикалов, кроме кислорода, могут служить меркгг-
таны, хиноны, дисульфиды и др (рис. 3.13).
С рассмотренными выше деструктивными процессами связ-
но явление абляции полимерных материалов, заключающееся
в постепенном разрушении (термоокислительная и термическая
Рис. 3.12. Изменение пластичности ДР натурального каучука в процессе
пластикации в различных средах:
/ — азот; 2 — воздух; 3 — кислород; 4 — озон
Рис. 3.13. Влияние различных акцепторов на пластикацию (р— относитель-
ная эффективность пластикации) натурального каучука при различных тем-
пературах в среде азота
/ — тиофен йл- 2 — бензохинон; 3 — азе бензол
221
деструкция) и уносе (механохимическая деструкция и эрозия)
поверхностных слоев материала под термическим и абразив-
ным воздействием горячего газового потока, обладающего
агрессивными свойствами и иногда содержащего взвешенные
твердые частицы.
3.5. СТАРЕНИЕ И СТАБИЛИЗАЦИЯ ПОЛИМЕРОВ
Рассмотрение различных реакций полимеров приводит к выво-
ду, что часть из них играет положительную роль и может быть
использована на практике. Так, как мы уже говорили, механи-
ческую деструкцию в присутствии кислорода воздуха или дру-
гих акцепторов свободных радикалов используют для пласти-
кации полимеров с целью облегчения их переработки, для
получения привитых и блок-сополимеров; реакции сшивания
макромолекул приводят к образованию пространственно-сши-
тых структур, отличающихся от линейных значительно более
высокими механическими показателями и повышенной термо-
стойкостью. Однако в большинстве случаев реакции деструкции
приводят к нежелательному уменьшению молекулярной массы,
сопровождающемуся резким снижением механических показа-
телей, появлением текучести при низких температурах и пр.
В процессе хранения и эксплуатации изделий из полимеров
под действием света, тепла, радиоактивных излучений, кисло-
рода может происходить излишне глубокое сшивание макро-
молекул, которое также является причиной ухудшения свойств
полимеров: появляются хрупкость, жесткость, резко снижается
способность к кристаллизации. Это приводит к потере работо-
способности изделий из полимеров. Изменение свойств полиме-
ров под действием различных физических и химических факто-
ров в процессе переработки, хранения и эксплуатации изделий
из полимеров называется старением
В реальных условиях полимерные материалы и изделия из
них обычно подвергаются комбинированному воздействию раз-
личных факторов. Так, только при атмосферном старении, ко-
торому подвержены многие материалы и изделия при хранении
и эксплуатации, на них одновременно воздействуют свет, тепло,
кислород воздуха, перепады температур и т. д Выявить ре-
зультат воздействия каждого фактора практически невозможно,
что сильно усложняет изучение старения и разработк} методов
защиты полимеров от вредных воздействий — методов стабили-
зации полимеров.
Основной способ стабилизации — введение в полимер спе-
циальных добавок (стабилизаторов, ингибиторов), замедляю-
щих старение. Роль стабилизаторов сводится либо к предот-
вращению образования свободных радикалов, либо к взаимо-
222
действию молекул стабилизатора с растущими радикалами и
переводу их в неактивную форму..
Известно много веществ, используемых в качестве стаби-
лизаторов В каждом конкретном случае при выборе стабили-
затора учитывают его эффективность, технологичность примене-
ния, влияние на свойства изделия, токсичность, стоимость и
другие факторы. В зависимости от назначения стабилизаторов
различают антиоксиданты, светостабилизаторы, антирады и др.
Стабилизаторы, применяемые для замедления окислитель-
ной деструкции, называются антиоксидантами. В качестве анти-
оксидантов используют фенолы, ароматические амины, суль-
фиды, меркаптаны и др. Они ингибируют цепной процесс окис-
ления двумя путями либо обрывают цепь окисления, т. е.
взаимодействуют со свободными радикалами на стадии их
образования (антиоксиданты аминного и фенольного типа),
либо предотвращают разложение гидропероксидов по ради-
кальном}7 механизму (сульфиды, тиофосфаты и др.). Антиокси-
данты первой группы имеют в молекуле подвижный атом
водорода, энергия связи которого с углеродом меньше, чем
энергия связи подвижного атома водорода в полимере. Поэто-
му гидропероксидный радикал легче вступает в реакцию с ин-
гибитором, чем с полимером. Образующиеся при этом свобод-
ные радикалы ингибитора малоактивны и не могут вызвать
продолжение цепи радикальных реакций. В процессе окисления
ингибитор расходуется, а часть его присоединяется к полимеру.
Реакции ингибированного окисления можно представить сле-
дующим образом (InH — молекула ингибитора):
ROO- + InH -------> ROOH 4- In-
R- _i_ InH ----> RH + In-
ROO- 4-In--------► ROOIn
R- _|_ in. ----> Rin
In- 4“ In- ----»- In— In
(I)
(2)
(3)
(4)
(5)
Таким образом, действие ингибиторов состоит в обрыве ре-
акционной цепи окисления по реакциям (1) и (2). Образую-
щийся радикал ингибитора малоактивен и не способен оторвать
водород от молекулы полимера. Он дезактивируется сам или
дезактивирует полимерные радикалы по реакциям (3) — (5).
Антиоксиданты второй группы (сульфиды, тиофосфаты, ди-
тиокарбаматы) разлагают гидронероксиды с образованием ста-
бильных молекулярных соединений:
ROOH4-R'—S—R' —* ROH+R'—SO—R
Особенно эффективна стабилизация полимеров смесями ан-
тиоксидантов, называемых синергическими смесями.
Антиоксиданты тормозят окисление только в том случае,
223
когда их концентрация в полимере не превышает определенное
значение, называемое критической, концентрацией.
Перспективным направлением стабилизации полимеров яв-
ляется использование в качестве антиоксидантов стабильных
радикалов, которые малоактивны при обычной температуре и
не могут инициировать деструкцию полимера, а при повыше-
нии температуры взаимодействуют с активными радикалами,
возникающими в процессе окислительной деструкции нолиме-
пов. Так, стабильные радикалы триацетоназотоксида взаимо-
действуют с радикалами, возникающими при окислительной
деструкции полимеров, без образования гндроперокендов:
СН3
СНг—С—СН3
ROO-4-ОС^ /NO-
СНг—С—СН3
сн3
СН3
СНг—С—СН3
ОС^ /NOR
СН2—С—СН3
I
сн3
В качестве стабилизаторов такого типа могут быть исполь-
зованы и полимеры с системой сопряженных связей (полнфе-
нилацетилены, поливинилен и др.), всегда содержащие некото-
рое количество стабильных радикалов.
Применение стабильных радикалов наиболее рационально
для полимеров, которые не содержат подвижных атомов водо-
рода (полиформальдегид, полиамиды), поскольку азотоксидные
радикалы не могут отрывать подвижный атом водорода и ини-
циировать цепной процесс окисления.
Защита от старения под действием озона, в частности резин,
достигается введением так называемых физических (или инерт-
ных) нротивостарителей (парафин, церезин, воски, хлорсуль-
фированный полиэтилен), которые, мигрируя на поверхность
полимерного изделия, покрывают его тонкой пленкой, стойкой
к озону и непроницаемой для нею.
Химически активные антиозонанты также образуют защит-
ную пленку па поверхности резины, однако она возникает в
результате химического взаимодействия антиозонанта с озоном,
которое происходит с большей скоростью, чем взаимодействие
озона с резиной. К таким антиозонантам относятся К’-замешен-
ные ароматические амины, производные фенолов, тиомочевины
и др
Стабилизация полимеров к фотохимической деструкции ос-
нована иа введении в полимер соединений, которые легко пог-
лощают световую энергию в трансформируют ее так, что она
излучается ими квантами меньшей энергии, безопасными для
полимера. Кроме того, выполняя функцию акцептора электрон-
ной энергии возбуждения макромолекулы, вызывающей ее
221
деструкцию, светостабилизаторы превращают эту энергию в
менее опасные для полимера формы, например в тепловую, и
рассеивают ее. В качестве таких светостабилизаторов применя-
ют производные салициловой кислоты (для эфиров целлюлозы,
поливинилхлорида, полиолефинов), бензотриазолы (для поли-
стирола, полиэтилентерефталата, полиолефинов), производные
бензофенона (для полиолефинов, полистирола, поливинилхло-
рида, полиэтиленоксидов, полиэтилентерефталатов).
Светостабилизаторы другого типа, в частности металлоор-
ганические соединения никеля, могут взаимодействовать с об-
разующимися при деструкции свободными радикалами и
тидропероксидами.
Эффективным светостабилизатором для многих полимеров
служит газовый канальный технический углерод (2—5% от
массы полимера) Наибольший эффект дает совместное приме-
нение технического углерода и некоторых антиоксидантов Тех-
нический углерод широко применяется для защиты полиэтиле-
новых изделий черного цвета — труб, мульчирующей пленки
для сельского хозяйства, деталей машин.
Для повышения стойкости полимеров к радиоактивному из-
лучению применяют вещества, способствующие рассеиванию
поглощенной энергии и отнимающие ее от защищаемых полиме-
ров настолько быстро, что последние не успевают разрушиться.
Такие вещества называют антирадами; к ним относятся угле-
водороды с конденсированными бензольными кольцами (наф-
талин, антрацен, фенантрен), амины, фенолы, тиофенолы.
Для защиты полимеров, облучение которых происходит иа
воздухе и сопровождается окислительными процессами, при-
меняют одновременно антирады и антиоксиданты. В случае
каучуков вторичные ароматические амины являю гея одновре-
менно и антиоксидантами, и ингибиторами радиационного
окисления
Контрольные вопросы
1 В чем различие и в чем сходство между полнмераналогнчныуи превра-
щениями и внутримолекулярными реакциями? Приведите примеры реак-
ций каждого типа.
2. Каковы отличительные особенности химических реакций полимеров по
сравнению с реакциями низкомолекулярных соединений?
3. Как влияет структура полимера иа его химическую активность? Какие
группы в полимере характернзуюзея повышенной реакционной способ-
ностью? Какой из полимеров, формулы которых приведены ниже, имеет
большую активность и почему?
СН3 \
—СН2—СН—/т сополимер этилена н пропилена
сн3
~СН2—С=СН—СН2~ чис-полинзопреп
(—СН2—СН2—)rt
15—816
225
4. Какие вы знаете реакции приводящие к увеличению или уменьшению
молекулярной массы?
•5. Полимер имеет строение ААААББББАААА (А и Б — составные звенья).
Как он называется? Каким способом может быть получен?
«6. Что такое деструкция полимеров? Как изменяется молекулярная масса
полимеров в процессе этой реакции? Какие виды деструкции вам из-
вестны?
7. Какая разница между деструкцией и деполимеризацией? Как влияет
структура полимера иа механизм реакции?
8. Как протекает процесс термоокислительной деструкции? Какие соедине-
ния при этом образуются? Какова роль термоокислительиой деструкции
при изготовлении и эксплуатации изделий? Перечислите основные методы
защиты от термоокислительной деструкции
9. Каков механизм действия света на полимеры? Какие полимеры наибо-
лее подвержены действию света и почему? Как защитить полимер от дей-
ствия света?
10. Какие процессы, протекают под влиянием ионизирующих излучений? Как
изменяется структура полимера при действии радиации? Какие полимеры
в наименьшей степени подвержены действию радиации и почему?
11. Какие из перечисленных ниже полимеров (назовите их) в наибольшей
СН3 СН3
I I
~СН2—С~ ~СН2—С~
СН3 СООСН3
сн3
I
~СН2—С =СН—сн2~ ~сн2—сн~
о
степени подвержены радиационной деструкции и структурированию?
12. Что такое гидролиз? Какие полимеры в наибольшей степени подвержены
гидролизу и почему?
13. По какому механизму может происходить отверждение олигомеров?
14. Что из себя представляет процесс вулканизации? Какие полимеры спо-
собны вулканизоваться? Как изменяются свойства полимера в процессе
вулканизации? Назовите основные вулканизующие агенты.
ГЛАВА 4
ФИЗИЧЕСКИЕ И ФАЗОВЫЕ СОСТОЯНИЯ И ПЕРЕХОДЫ
Для правильного выбора условий переработки и эксплуатации
полимерных материалов необходимо знать в каком фазовом и
агрегатном состоянии они находятся.
Низкомолекулярные вещества могут существовать в трех
фазовых состояниях — кристаллическом, жидком и газообраз-
ном, которые отличаются друг от друга порядком во взаимном
расположении молекул.
Кристаллическое фазовое состояние характеризуется нали-
чием кристаллической решетки и трехмерным дальним поряд-
226
ком в расположении атомов или молекул. Жидкое фазовое
состояние часто называют аморфным, поскольку в жидкостях
отсутствует кристаллическая решетка и наблюдается лишь
ближний порядок в расположении атомов и молекул. Плотность
упаковки молекул или атомов аморфных веществ остается вы-
сокой, но несколько ниже, чем у кристаллических тел.
Полимеры существуют в двух фазовых состояниях: кристал-
лическом и аморфном. Для них характерны все признаки фазо-
вых состояний низкомолекулярных соединений, но существуют
и определенные особенности (см. разд. 1.3).
Низкомолекулярные вещества могут находиться в трех аг-
регатных состояниях: газообразном, жидком и твердом, кото-
рые отличаются друг от друга плотностью упаковки, характером
движения атомов и молекул и откликом на механическое
воздействие
Для газообразного состояния характерно поступательное,
вращательное и колебательное движение молекул. Расстоя-
ния между молекулами в газе достаточно велики, т. е. плотность
упаковки мала. Под действием механической нагрузки, напри-
мер при изотермическом сжатии, газ может деформироваться
на сотни процентов, проявляя при этом упругость
Упругость — это способность тела деформироваться под нагрузкой и вос-
станавливать свою первоначальную форму после снятия нагрузки. Деформа-
ция упругого тела подчиняется закону Гука, предусматривающего пропор-
циональность между приложенным напряжением о и относительной дефор-
мацией е
где Е — коэффициент пропорциональности, характеризующий внутреннее со-
противление материала изменению формы и называемый модулем В зави-
симости от вида деформации различают модуль растяжения Е, модуль сдви-
га G, модуль сжатия К и т д Величину, обратную модулю, называют подат-
ливостью J\ ]=\/Е
Упругость газй имеет кинетический характер, т. е. деформа-
ция осуществляется за счет изменения энтропии системы Для
кинетического механизма деформирования газов характерны
выделение тепла, низкий модуль (высокая податливость) и сни-
жение модуля при повышении температуры.
Твердое состояние характеризуется высокой плотностью
упаковки молекул. Поступательное и вращательное движение
молекул практически отсутствует, молекулы или группы атомов
лишь колеблются около центров равновесия Малой подвижно-
стью молекул или атомов и большой плотностью упаковки
объясняется высокое сопротивление твердого тела изменению
формы — высокое значение модуля (т. е. низкая податливость),
снижающееся с повышением температуры деформирования.
Твердые тела деформируются преимущественно упруго, и
величина упругой деформации составляет доли процентов или
несколько процентов. Упругость твердых тел имеет другую при-
227
15*
коэффициент. Это служит убедительным доказательством того,
что под действием приложенного напряжения при температуре
ниже Тс тепловой энергии недостаточно для того, чтобы мак-
ромолекула изменила свою конформацию (Uo>kT). Начиная
с некоторой температуры, kT-^-U^ и появляется вероятность из-
менения конформации за счет вращения сегментов. Полимер
переходит в промежуточную область I', для которой характер-
но существование подвижных («расстеклованных») и неподвиж-
ных («застеклованных») участков макромолекул. Полимер
деформируется в большей степени, чем при Т<ТС, но деформа-
ция его невелика. По мере повышения температуры тепловой
энергии становится достаточно для того, чтобы преодолеть
барьер вращения практически всех структурных единиц (kT>
>Uo), и полимер переходит в область II, которую называют
плато высокоэластичности.
Для него характерны большая величина деформации, кото-
рая остается практически постоянной до температуры текучести.
Полимер в этом состоянии легко изменяет свою конформацию
за счет вращения (заторможенного) сегментов вокруг одинар-
ных связей. При дальнейшем повышении температуры движе-
ние сегментов усиливается и при температуре, близкой к тем-
пературе текучести, наблюдается перемещение молекулярных
клубков относительно друг друга -— начинается течение полиме-
ра (область II') Полимер переходит в область вязкого течения.
Выше Тт полимер находится в вязкотекучем состоянии (об-
ласть III).
Таким образом, характерным отличием полимеров от низко-
молекулярных веществ является наличие трех физических со-
стояний (стеклообразного, высокоэластического и вязкотекуче-
го) и довольно протяженных («10—20°C) переходных обла-
стей. Средние значения температур этих областей принимают
обычно за температуру стеклования Тс и температуру теку-
чести Тт.
Сравнение термомеханических кривых 1 и 2 (см. рис. 4.1, а)
свидетельствует о влиянии гибкости на поведение полимера при
деформации. Чем больше гибкость полимера, тем до более низкой
температуры он сохраняет высокоэластическое состояние. В же-
сткоцепных полимерах характерные температуры переходных
состояний сдвигаются в сторону больших значений. Так, для не-
которых полимеров (например, для целлюлозы) значения тем-
ператур стеклования и текучести находятся выше температуры
•термического разложения
Термомеханическая кривая также очень чувствительна к
изменению молекулярной массы полимера и конфигурации
макромолекул. С уменьшением молекулярной массы плато
высокоэластичности уменьшается, при определенном ее значе-
нии (AfKp) исчезает (см рис. 4.1, б) и поведение материала
230
Рис. 4 2. Термомехаиические кривые
кристаллических полимеров:
1 - 2 - ’Гт>Гпл>5Гс
приближается к поведению
низкомолекулярных веществ.
Это еще раз подчеркивает,
что высокоэластическое сос-
тояние проявляется только у
полимеров.
Сетчатые полимеры ни при
каких условиях не могут пе-
рейти в вязкотекучее состоя-
ние, поскольку сшивки пре-
пятствуют течению. Способность к течению у таких полимеров
проявляется только при разрушении сшивок (химическое тече-
ние) или связей в цепи (деструкция) при температуре выше
температуры деструкции Тд (см. рис. 4.1, в)
Кристаллический полимер при невысоких напряжениях ведет
себя как твердое тело, его деформация мала и зависит от сте-
пени кристалличности, снижаясь по ?иере ее роста Выше
температуры плавления кристаллов Тпя полимер может перейти
или в высокоэластическое (если Т-1>Т11л>Тс), или в вязкоте-
кучее (если Тт<Тпл) (рис 4 2).
Если полимер, способный к кристаллизации, очень быстро
охладить до температуры намного ниже температуры кристал-
лизации, то он перейдет в стеклообразное состояние, а не в
кристаллическое. При Тс полимер переходит в высокоэластиче-
ское состояние, при этом в результате повышения подвижности
звеньев он приобретает способность к кристаллизации. По-
скольку кристаллический полимер характеризуется низкими
значениями деформации, то по мере кристаллизации величина
• деформации будет снижаться до некоторого значения, опреде-
ляемого степенью кристалличности. Далее полимер деформиру-
ется как кристаллический вплоть до температуры плавления
кристаллов, когда деформация резко возрастает, и ои перехо-
дит в вязкотекучее состояние, если Т„я находится в температур-
ной области вязкого течения. Если же ТПл кристаллов заметно
ниже Тг, то на кривой может появиться еще одна область вы-
сокоэластического состояния
В зависимости от фазового или физического состояния при
переработке и эксплуатации все полимерные материалы
можно условно разделить на несколько групп
Пластические массы (пластмассы) — линейные или разветв-
ленные полимеры или олигомеры, которые при переработке
находятся в вязкотекучем или высокоэластическом состоянии,
231
а при эксплуатации — в стеклообразном или кристаллическом,
т е температура стеклования или плавления пластмасс обычно
выше комнатной. Пластмасса называется термопластичной,
если при нагревании она переходит из стеклообразного или
кристаллического состояния в вязкотекучее или высокоэласти-
ческое, т. е. из твердого в жидкое. При охлаждении происходит
обратный переход. Если же при переработке полимер приобре-
тает сетчатое строение (отверждается), то обратный переход
в вязкотекучее состояние невозможен Такие пластмассы
называются термореактивными. К их числу относят и синтети-
ческие смолы, которые получают из олигомеров, отверждаемых
в процессе переработки
Эластомеры — линейные или разветвленные полимеры или
олигомеры, которые перерабатываются в вязкотекучем состоя-
нии, затем сшиваются в трехмерную сетку и эксплуатируются
в высокоэластическом состоянии. Несшитые эластомеры назы-
вают каучуками, а сшитые — чаще всего резинами.
Волокна—так же как и пластические массы, при перера-
ботке находятся в вязкотекучем состоянии, а при эксплуата-
ции— в стеклообразном или кристаллическом Их отличитель-
ной особенностью является высокая степень ориентации мак-
ромолекул и связанная с ней анизотропия свойств.
Для каждой из этих групп характерны свои термомехани-
ческие кривые, по которым можно оценить температурный
интервал их эксплуатации и переработки. Например, для пла-
стических масс Т,. или Тп.-| определяют верхний температурный
предел эксплуатации, при превышении которого изделие теряет
форму, разрушается Для эластомеров важной характеристикой
является Тс, определяющая нижнюю температурную границу
его существования в высокоэластическом состоянии. Любое
изменение структуры, приводящее к повышению Тс, а также
кристаллизация в интервале от Тс до Тт приводят к сужению
области высокоэластичности и ухудшению морозостойкости
Чем ниже Тт полимера, тем легче он перерабатывается и
меньше вероятность его термодеструкции.
4.1. СТЕКЛООБРАЗНОЕ СОСТОЯНИЕ И СТЕКЛОВАНИЕ
Как уже отмечалось выше, стеклообразное состояние (твер-
дое агрегатное состояние) характеризуется высокой плотностью
упаковки макромолекул.
Показателем, отражающим плотность упаковки, является свободный
объем Ус, т. е. объем, не занятый молекулами. Различают физический свобод-
ный объем Ус*, отражающий интенсивность тепловых колебаний молекул,
и геометрический свободный объем Усг, характеризующий неплотность упа-
ковки молекул:
Уо*-ут_уо (4.1)
232
где VT— объем, который занимает полимер, совершающий тепловые колеба-
ния при температуре Т, V0 — объем полимера при О К, рассчитанный на
объемов атомов и групп, входящих в молекулу (в случае полимера — моно-
мерное звено); V0—Л'лЕДУ,- (Л\ — число Авогадро, Дп — объем группы).
Геометрический свободный объем Vcr— это разность между макроскопи-
ческим объемом тела Ут при температуре Т и Уг при той же температуре:
Vrc= уу_ уг_ уг _ (усФ+ yoj. (4 2)-
Vr=Af3B/pn, (4 3)
где Л(„ — молекулярная масса повторяющегося звена; рп — плотность поли-
мера. определяемая экспериментально.
Для оценки плотности упаковки используется также коэффициент упа-
ковки Ку'.
Ку«рг/уг=1_(уег/ут) (4.4)
При расчете объема при температурах Т применяют уравнение, учиты-
вающее его изменение при нагревании или охлаждении:
Vr- Vr,( 1 +р(илн а)ДГ], (4.5)
где Ут и Vr0 — объем при температурах Т и Т’с; ₽ и а — термические коэф-
фициенты объемного и линейного расширения.
По плотности упаковки полимеры в стеклообразном состоя-
нии занимают промежуточное положение между твердыми
кристаллическими и жидкими аморфными, приближаясь к кри-
сталлическим. Так, Ку в кристаллах изменяется в пределах
0,680—0,800, в полимерных стеклах при Тг Ку = 0,667, а в жид-
ких полимерах—может уменьшаться до 0,5. В табл. 4.1 пока-
зано, как структура полимера влияет на параметры упаковки
макромолекул. Из таблицы видно, что свободный объем возра-
стает с ростом объема заместителей (ср. полиэтилен и поли-
стирол и ряд полиметакрилатов)
Для стеклообразного состояния характерны ближний поря-
док в расположении макромолекул и сильно ограниченная сег-
ментальная подвижность.
Вязкость полимера в стеклообразном состоянии превышает
10’2—1013 Па-с, модуль упругости составляет 103—10* МПа,
а деформация — несколько процентов.
Таблица 4.1. Характеристики плотности упаковки полимеров в стеклообраз-
ном состоянии (292 К)
Полимер Vr10* м’/моль Vcr JO», м’/моль УГ10». м*/моль Ку
Полиэтилен 30,4 9,8 20,6 0,676
Полистирол 99,0 33,2 62,8 0,634
Полиметнлметакрилат 85,5 27,0 58,5 0,684
Полиэтилметакрилат 101,4 32,3 69,1 0,680
Полипропилметакрилат 118,7 41,9 76,6 0,646
Полибутнлметакрнлат 134,6 45,0 89,6 0,665
233
По механическому поведению полимерные стекла можно
разделить на хрупкие и нехрупкие. Деформация хрупких стекол
осуществляется по закону Гука, а для нехрупких при опреде-
ленном значении напряжения происходит вытяжка полимера,
сопровождающаяся ориентацией макромолекул.
Для стеклообразного состояния характерна также термоди-
намическая неравновесность, обусловленная очень низкой под-
вижностью структурных единиц, в результате чего в процессе
стеклования образуется неравновесная структура, сохраняюща-
яся в течение длительного времени. Чем выше скорость стек-
лования, тем структура стекла более неравновесна. При после-
дующем охлаждении структура стекла изменяется, стремясь к
равновесной; при этом происходит некоторое повышение плот-
ности полимера и уменьшение его объема.
Стеклообразное состояние полимеров может реализоваться
в результате протекания процессов, которые не относятся к
стеклованию, но приводят к значительному уменьшению скоро-
сти изменения конформаций Это процессы ориентации, сшива-
ния полимеров, находящихся в высокоэластическом состоянии.
Сильно ориентированные или сильно сшитые полимеры ведут
себя, как полимерные стекла, и проявляют все признаки,
характерные для стекол.
4.1.1. Теории стеклования
Стеклование — это переход из жидкого в твердое аморфное
состояние. Для полимеров стеклованием называют переход из
высокоэластического или вязкотекучего состояния в стеклооб-
разное. Если стеклование происходит при понижении темпе-
ратуры или повышении давления, то такой процесс называется
структурным стеклованием. В настоящее время существует
несколько теорий, рассматривающих процесс стеклования с
различных позиций.
Кинетическая релаксационная теория. Если в результате
внешнего воздействия (деформация, изменение температуры
и др.) система выведена из состояния равновесия (неравновес-
ная конформация), то под действием внутренних сил (теплово-
го движения) она изменит конформацию и перейдет в состоя-
ние термодинамического равновесия Процесс перехода системы
из неравновесного состояния в термодинамически равновесное
называется релаксацией. Время релаксации — это время, в те-
чение которого любая физическая величина, являющаяся мерой
отклонения системы от состояния равновесия, уменьшается в
е раз.
При снижении температуры полимера на ДТ равновесное
состояние, соответствующее этой температуре, достигается не
234
сразу, а лишь спустя время т, которое называют временем
структурной релаксации (т*).
Поскольку полимер включает атомы, различные группы, сег-
менты, цепи, надмолекулярные образования различной степе-
ни агрегации, то скорость их перегруппировки различна. При
снижении температуры сначала теряют подвижность элементы
надмолекулярной структуры, затем сегменты, группы, атомы.
Структурное стеклование обусловлено потерей подвижности
сегментов макромолекулы. Этот процесс имеет кооперативный
характер: для того чтобы изменил свое положение один сегмент,
необходимо изменение положения его соседей. Время струк-
турной релаксации сегментов, определяющее скорость измене-
ния конформаций, отнесенное к одной кинетической единице
(сегменту), выражается уравнением
т*-Л exp (U/kT), (4.6)
где А — константа, равная «10-'2 с; U — энергия активации движения сег-
ментов; k — константа Больцмана; Т — температура.
Энергия активации движения свободных сегментов зависит
от температуры:
U==U°° "г~т0~- (4 7)
где 1/<» — постоянная, имеющая смысл энергии активации при Т—»-оо;
То — так называемая характеристическая температура, примерно на 50 К
ниже Тс-
Согласно (4.7) в определенном температурном интервале
происходит резкое увеличение энергии активации Средняя
температура этого интервала является температурой стеклова-
ния Тс.
Как видно из уравнения (4 6) при снижении температуры
время релаксации увеличивается (за счет как снижения Т, так
и роста U) и при достаточно низкой температуре становится
настолько большим, что равновесное состояние полимера, соот-
ветствующее данной температуре, практически не может быть
достигнуто. Полимер переходит в стеклообразное состояние.
Как уже отмечалось, это состояние термодинамически неравно-
весно (так называемое метастабильное состояние), и полимер
может пребывать в этом состоянии достаточно долго. Увеличе-
ние времени структурной релаксации т* может быть достигну-
то и за счет повышения давления вследствие роста энергии ак-
тивации.
Таким образом, релаксационная теория стеклования рас-
сматривает этот процесс как результат замедления релаксаци-
онных процессов с понижением температуры при постоянном
нормальном давлении или с повышением давления при посто-
янной температуре Согласно этой теории время структурной
235
релаксации т* зависит от скорости изменения температуры.
При Т—Тс
t*^C*/q,
(4.8)
где q—dTldt— скорость охлаждения; С* — постоянная для данного полиме-
ра; С* «293 К для неорганических и 283 К для органических стекол.
При медленном охлаждении создается плотная упаковка,
приближающаяся к равновесной структуре, и т* такой структу-
ры будет больше, чем т* стекол, образованных при быстром ох-
лаждении; при этом
С*=АГ?/С/(Ге), (4.9)
где U(Тс) —энергия активации при температуре стеклования.
Из (4.8) и (4.9) следует, что Тс повышается с ростом ско-
рости охлаждения. Изменение q в 10 раз повышает Тс на 3—
10 К Температура стеклования повышается также с ростом
давления.
Теория свободного объема. В основе этой теории лежит из-
менение свободного объема Vc при стекловании (рис. 4 3).
При охлаждении тела происходит уменьшение и физического
:вободного объема, и геометрического. Для большинства поли-
меров тепловое перемещение сегментов прекращается, если
этносительное изменение (Аф) свободного физического объема
доставит 0,025±0,003. Изменение при стекловании доли (/сг)
:вободного геометрического объема Усг для большинства поли-
'ис. 4 3. Зависимость удельного объема AV>a от температуры при скоростях
клаждения (/) и v2 (2); vt>v2 (Tci и Гс2 — температуры стеклования прн
коростях охлаждения и1 и v2; заштрихованная область — свободный объем)
ис. 4.4 Зависимость доли геометрического свободного объема fd от темпе-
атуры стеклования Те
36
меров составляет 0,113 Однако с ростом кинетической гибко-
сти fc несколько уменьшается — примерно до 0 08 (рис. 4.4)
В теории свободного объема за Тс принимают температуру,
при которой свободный объем достигает некоторого постоян-
ного значения. С ростом скорости охлаждения Тс повышается
(см рис. 4.3). Основываясь на концепции свободного объема,
Тс можно определить из соотношения
rcap=fer, (4.10)
где Д₽=р1—Вг; pi и 02— термические коэффициенты объемного расширения;
выше (pi) и ниже (Вз) Тс; fc—доля геометрического свободного объема,
равная Vc/Vt-
Согласно уравнению (4.7) температуру То можно рассмат-
ривать как температуру, при которой в результате бесконечно
медленного охлаждения физический свободный объем станет
равным нулю и процесс релаксации не будет происходить ввиду
отсутствия пустот (дырок), необходимых для перегруппировок
сегментов
Термодинамическая теория рассматривает стеклование как
переход, при котором происходит уменьшение конформацион-
ной энтропии системы S Стеклование наступает при достиже-
нии системой некоторого минимального значения S, которое
может быть рассчитано из соотношения
Д5=АСР1п (Гс/Г2), (411)*
где Ср — удельная теплоемкость при постоянном давлении.
При температуре Т2 энтропия становится равной нулю; Т2 при-
мерно на 50 К ниже Тс.
Стеклование связывают также с образованием флуктуаци-
онной сетки зацеплений вследствие сильного межмолекулярно-
го взаимодействия. Эта сетка образуется в том случае, когда
энергия теплового движения недостаточна для преодоления сил
внутри- и межмолекулярных взаимодействий. В изотермических
. условиях процессы разрушения и образования таких связей
находятся в термодинамическом равновесии.
Полимер может быть переведен в стеклообразное состояние
не только при снижении температуры или повышении давления,
но и при воздействии механического поля. Этот процесс на-
зывается механическим стеклованием. Как показано в гл 1,.
кинетическая гибкость полимера, т е. способность изменять
конформацию при механическом воздействии, определяется
соотношением времени действия силы /д и временем /п. необ-
ходимым для перехода из одной конформации в другую При
условии полимер ведет себя, как жесткая система, не
способная изменять конформацию Если считать, что время
перехода /п из неравновесной конформации, создаваемой внеш-
ним механическим полем, в равновесную, есть не что иное как
237
ремя структурной релаксации т*, то условие механического
геклования будет формулироваться как или
где со — частота действия силы).
Механическое стеклование происходит при воздействии си-
ы или с ультразвуковой частотой при высоких температурах,
ли с инфразвуковой при низких температурах. Температура,
ри которой полимер переходит в стеклообразное состояние в
езультате механического стеклования, называется температу-
эй механического стеклования Тмс. При очень медленных меха-
нческих воздействиях значения Тмс и Тс совпадают, при быст-
х — ГМС>ТС; Л<с зависит от временного режима деформиро-
ания (частоты). Влияние частоты на Тмс натурального каучу-
1 видно из следующих данных:
астота.-с’1 0 0,0167 0,167 1,67 16,7 2 10е 8 10е
К 203 212 217 224 230 259 271
Между температурами Тмс и Тс существует определенная
*язь, например для гибких полимеров
ДГ=ГМС - ГС=А — ВГС, (4 12)
ie А и В— постоянные, определяемые частотой механического воздействия,
1К9 при скорости деформирования 0,013 с“1 Л=10К, £=0,005, а при
>00 Л «35 К. В=0,02.
1.2. Методы определения температуры стеклования
роцесс стеклования полимера сопровождается постепенным изменением его
чзическнх свойств — объема, плотности, диэлектрических и механических
ойств и др Изучая изменение этих свойств от температуры, можно опреде-
ть температуру стеклования полимера. Наибольшее распространение поду-
ли методы исследования удельного объема (дилатометрический метод),
плоем кости, модуля упругости и деформации.
В основе дилатометрического метода лежит снижение удельного объема
>и стекловании (см. рис. 4.3). За Гс принимают точку перегиба на кривой
1ВИСИМОСТН удельного объема от температуры
Метод определения Тс по теплоемкости Ср основан на том факте, что
жидком состоянии Ср существенно выше, чем в твердом. При переходе из
[зкотекучего или высокоэластического состояния в стеклообразное при син-
ении температуры Ср резко уменьшается. Средняя температура области,
которой происходит резкое изменение теплоемкости, обычно принимается
Гс. Характер зависимости Ср от Т и значение Гс определяются скоростью
менення Г с уменьшением скорости охлаждения Гс снижается.
По термомеханическим кривым, представляющим собой зависимость ме-
[нических свойств от температуры, Гс определяют как среднюю температуру
реходной области из стеклообразного состояния в высокоэластическое,
качестве показателей механических свойств используют модуль, деформа-
сю, твердость, податливость, механические потери энергии. Изменение мо-
ля, деформации, твердости и податливости происходит монотонно в пере-
дней области а зависимость тангенса угла механических потерь от тем-
ратуры выражается кривой с максимумом, по которому и определяют тем-
ратуру стеклования.
Перечисленные методы хотя и чувствительны к изменению Гс, ио явля-
йся косвенными, и- Гс, определенная этими методами» значительно зависит
В
от скорости изменения Т. Существуют методы, позволяющие непосредственно
оценить сегментальную подвижность системы. Среди них наибольшее распро-
странение получили метод ядерного магнитного резонанса (ЯМР) широких
линий, диэлектрический метод и метод оадиотермолюмннесценцнн (РТЛ).
В методе ЯМР используется принцип неопределенности (Avre=const, где
&v — ширина линии, тя— время спии-сниновой и спин-решеточной релаксации
ядер) для расчета времени релаксации ядер. При стекловании наблюдается
увеличение времени релаксации и уменьшение ширины линии в некотором
температурном интервале, середину которого принимают за Тс.
Применяется и диэлектрический метод определения Ге, основанный на
изменении диэлектрических потерь tg6 с температурой. При стекловании
полимера на кривой зависимости tg6 от Т появляется максимум, который
и принимают за Те.
Метод радиотермолюминесцеиции заключается в облучении застеклован-
ного образца ионизирующей радиацией (f-лучами). При этом электроны по-
глощаются структурными дефектами полимера и ввиду высокой плотности
стеклообразных полимеров остаются там в течение длительного времени.
С ростом температуры подвижность системы повышается и электроны при-
обретают способность взаимодействовать с ионизированным полимером, вы-
зывая свечение. Точка максимума на кривых зависимости интенсивности све-
. чения от температуры и есть температура стеклования.
4.1.3. Влияние структуры полимера на температуру стеклования
Температура стеклования полимеров определяется, по сущест-
ву, всеми параметрами структуры макромолекулы, которые
влияют на кинетическую гибкость макромолекул и формирова-
ние флуктуационной сетки, а следовательно, на Тс. Все струк-
турные изменения, приводящие к росту гибкости макромолекул,
как правило, способствуют снижению Тс. Чем более развита
флуктуационная сетка в полимере, тем выше его температура
стеклования.
Химическое строение звена оказывает существенное влия-
ние на гибкость цепи и энергию меж молекулярного взаимо-
действия, т. с. на число и тип связей флуктуационной сетки, и,
следовательно, на Тс. Так, на Тс влияет наличие больших по
размеру заместителей, которые затрудняют вращение звеньев,
отчего гибкость цепи практически не проявляется при ком-
натной температуре. Для проявления гибкости такие полимеры
надо нагревать до более высоких температур, т. е. Тс их доста-
точно высока. Число громоздких заместителей также влияет
на Тс: в сополимерах бутадиена и стирола, например, она по-
вышается по мере увеличения числа фенильных ядер в цепи.
Наличие фенильного и метильного заместителей при одном
и том же атоме углерода приводит к еще большим пространст-
венным затруднениям и повышению Тс. Так, поли-а-метилсти-
рол имеет более высокую Тс, чем полистирол.
Неполярные полимеры обладают высокой кинетической гиб-
костью цепи, и, поскольку потенциальный барьер вращения не-
велик, гибкость цепи сохраняется вплоть до очень низких тем-
ператур, а флуктуационная сетка развита слабо, поэтому
239
аполярные полимеры с гибкими цепями характеризуются
1зкими температурами стеклования.
Наличие даже редко расположенных полярных групп увели-
1вает межмолекулярное взаимодействие и повышает Тц. Уве-
рение числа полярных групп повышает Тс в еще большей
епени
Конфигурация макромолекул, т е разветвленность и нали-
ге сшивок, также влияет на Тс. Если молекулярная масса меж-
г точками разветвлений или между узлами сетки (в случае
11ивания) намного превышает М сегмента, то т* и гибкость
ких полимеров близки к т* и гибкости линейных и Тс прак-
чески не изменяется. Например, температуры стеклования
пурального каучука и резины с редкой сеткой на его основе
1инаковы и лежат в области 203—205 К По мере роста числа
ветвлений или сшивок скорость релаксационных переходов
:ижается из-за создаваемых ими стерических затруднений
* повышается) и Тс сдвигается в сторону более высоких тем-
;ратур.
Как было показано в гл 1 (см. рис. 1 33) с увеличением
пекулярной массы полимера Тг повышается сначала очень
!стро, потом медленно и затем достигает постоянного значе-
я. Это происходит, когда молекулярная масса достигает зна-
ния Л/кр, равного молекулярной массе механического сегмен-
, т. е. когда полимер приобретает гибкость. Молекулярная
сса, при которой достигается постоянство Тс, зависит от кине-
ческой гибкости цепи полимера и повышается по мере ее
нжения.
Поскольку в полимеры часто вводят пластификаторы и на-
лнители, важно знать как они влияют на Тс. Пластификато-
уменыпают межмолекулярное взаимодействие и повышают
нетическую гибкость цепи, что приводит к уменьшению т* и
зигу Тс в область меньших значений. Кроме того, нластифи-
горы, как правило, увеличивают свободный объем, что тоже
осит определенный вклад в снижение Тс.
Наполнители изменяют структуру полимера в результате
сорбции сегментов макромолекул поверхностью наполнителя
образования связей полимер — наполнитель. Наполненные
чимеры можно представить себе как двухфазную систему,
:тоящую из твердой фазы, расположенной около частиц на-
знителя, и «мягкой фазы», на которую не распространяется
яяние наполнителя, и т. е по существу она представляет
>ой ненаполненный полимер. Для наполненных полимеров
зактерны две температуры стеклования, соответствующие
жлованию «мягкой фазы» (Тс) и «твердой» (7V); как прави-
ТС'>ТС, так как в «твердой фазе» резко снижена сегмен-
тная подвижность. Разность температур стеклования ДТ=
V—Тс зависит от степени взаимодействия полимер — напол-
нитель, и часто вместо двух Тс наблюдается расширение
интервала стеклования со смещением Тс в сторону более высо-
ких температур.
Повышение Тс при наполнении следует и из концепции сво-
бодного объема: поскольку термический коэффициент расшире-
ния наполненных систем снижается с ростом наполнителя, то,
как следует из (4.10), Тс при наполнении повышается.
4.2. ВЫСОКОЭЛАСТИЧЕСКОЕ СОСТОЯНИЕ
Высокоэластическое состояние, как мы уже говорили, — это
такое физическое состояние, находясь в котором полимер спо- .
собен к очень большим обратимым деформациям, происходя-
щим под влиянием очень небольших нагрузок. Это явление
получило название высокоэластичности, а большие обратимые
деформации называют высокоэластическими деформациями.
Это состояние присуще только полимерам и только в опреде-
ленных условиях. На рис. 4 I, б приведены термомеханические
кривые олигомеров (кривые 1, 2) и полимеров (кривые 3—5)
различной молекулярной массы. Из рисунка видно, что плато
высокоэластичности появляется только для образцов с молеку-
лярной массой выше А/кр.
Температурный интервал, соответствующий высокоэласти-
ческому состоянию, расположен у линейных полимеров между
температурой стеклования Тс и температурой текучести Тт,
а у сетчатых — между температурой стеклования Т(. и темпе-
ратурой термического разложения (см. рис. 4.1,а—в).
Плотность упаковки макромолекул в высокоэластическом
состоянии несколько ниже, чем в стеклообразном, но тоже до-
статочно высока. Например, коэффициент плотности упаковки
Ку для полиизобутилена составляет 0,667 в стеклообразном и
0,628 в высокоэластическом состоянии (при 293 К). С повыше-
нием температуры свободный объем УСФ возрастает:
УСФ= УСФ'[1 Ч-аИТ-Тс)], (4.13)
где Ус** и Ус» — свободный объем при Тс и дайной температуре Т; etc —
коэффициент термического расширения свободного объема.
Сжимаемость полимеров в этом состоянии ниже, чем у
жидкостей, но выше, чем у твердых тел; так, для жидкости (н-
гексана) сжимаемость составляет 1,6-10~9 Па-1, для эластоме-
ров в высокоэластическом состоянии — 5-10~10 Па-1, а для
твердых тел (железо)—7-10“12 Па-1. Для высокоэластического
состояния характерен ближний порядок во взаимном распо-
ложении макромолекул, но существуют надмолекулярные
образования различной степени упорядоченности.
В полимерах, находящихся в высокоэластическом состоя-
нии, наблюдается высокая подвижность всех составляющих ча-
16—816
241
стей макромолекулы: атомов, групп, звеньев, сегментов Макро-
молекулы обычно находятся в конформации статистического
клубка, что характерно для гибких полимеров, относящихся
к классу эластомеров. Под влиянием внешних воздействий
макромолекулы полимеров, находящиеся в высокоэластическом
состоянии, легко изменяют конформацию за счет заторможен-
ного вращения звеньев вокруг одинарных связей.
Высокая подвижность структурных элементов в высокоэла-
стическом состоянии обусловливает легкость их перехода в
, ..зновесное состояние (структурная релаксация). Среднее
время структурной релаксации полимеров в высокоэластиче-;
ском состоянии намного меньше, чем в стеклообразном. Так,
в высокоэластическом состоянии время структурной релакса-
ции сегментов эластомеров при 293 К составляет 10-5—10 6 с
по сравнению с 105—106 с для стеклообразного состояния. Это
и предопределяет термодинамическую неравновесность стекло-
образного и равновесность высокоэластического состояния
Особенности высокоэластичсского состояния полимеров от-
ражаются на их механических свойствах. Для полимеров в
этом состоянии характерны, в частности, высокая податливость,
низкий модуль упругости (Е яг 0,2 МПа) и его увеличение с по-
вышением температуры. Обратимые деформации полимеров в
высокоэластическом состоянии составляют сотни процентов и
деформирование сопровождается экзотермическим эффектом.
Прочность многих полимеров в высокоэластическом состоя-
нии приближается к прочности стеклообразных полимеров.
Например, прочность при растяжении натурального каучука в
стеклообразном состоянии составляет 75—100 МПа, а в высо-
коэластическом достигает 35—40 МПа; прочность полихлоро-
прена составляет 30—40 и 25—30 МПа соответственно в стекло-
образном и высокоэластическом состоянии.
Поскольку наиболее ярко высокоэластическое состояние
проявляется как отклик на механическое воздействие, рассмот-
рим особенности высокоэластической деформации.
Природа высокоэластичности наиболее подробно изучена
на примере эластомеров (каучуков), т. е. полимеров, характе-
ризующихся высокой термодинамической и кинетической гибко-
стью, которые находятся в высокоэластическом состоянии при
комнатной температуре. Рассмотрим с различных позиций де-
формацию (растяжение) макромолекулы каучука.
Для идеального случая (т. е. для так называемого идеаль-
ного каучука) обычно вводят следующие допущения:
вязкотекучая деформация отсутствует из-за сшивания мак-
ромолекул, причем длина цепи между узлами сетки намного
больше длины механического сегмента;
энтропия сетки равна сумме энтропий отдельных цепей;
распределение расстояний между концами цепей сетки В-
242
недсформированном состоянии подчиняется нормальному за-
кону распределения Гаусса;
образец несжимаем, т. е. деформируется при приложении
небольших напряжений без изменения объема;
каждая макромолекула деформируется одинаково по всему
Образцу, т. е. деформация цепей аффинная;
процесс деформирования изотермический.
Молекулярная теория. Равновесному состоянию гибкой мак-
ромолекулы, как уже было сказано, соответствует конформа-
ция статистического клубка. При постоянной температуре спо-
собность к изменению конформации определяется величиной
потенциального барьера Uo. Если энергия внешнего воздействия
превышает величину Uo, то под действием внешних сил макро-
молекула изменяет свою конформацию за счет поворота
звеньев вокруг связей на угол <р, переходя из равновесного
состояния в неравновесное. Поскольку интервал изменения угла
<₽ зависит от структуры полимера и для гибких макромолекул
с низкой энергией активации довольно велик, то при сравни-
тельно небольших напряжениях деформация образца будет
большой. После снятия нагрузки под действием теплового
движения макромолекула, находящаяся в неравновесной кон-
формации, возвращается в равновесную и принимает первона-
чальную форму статистического клубка, т. е. деформация явля-
ется обратимой.
Таким образом, с молекулярной точки зрения высокоэласти-
ческая деформация состоит в изменении конформации (переход
из формы статистического клубка в форму, близкую к струне)
под действием внешних сил и возвращении к исходной конфор-
мации после снятия нагрузки.
Термодинамическая теория. Для деформирования (растяже-
ния) образца длиной I под действием силы f требуется ра-
бота А:
dA=fdl. (4.14)
Согласно первому закону термодинамики внутренняя энер-
гия U системы складывается из теплоты Q, подведенной к си-
стеме, и работы А:
dU=dQ+dA. (4.15)
Для обратимого равновесного процесса в изотермических
условиях dQ = TdS (где S — энтропия системы). Поэтому
dU=dA+TdS. (4.16)
По второму закону термодинамики внутренняя энергия си-
стемы состоит из свободной dFct> и связанной TdS:
dU^dFc»+TdS. (417)
243
16*
Из уравнений (4.14) и (4.17) получаем:
dFcu—dA=fdl; f=(dFit/dl)T. (4.18)-
Следовательно, деформирующая сила — это изменение сво-
бодной энергии при деформировании на единицу длины при
постоянной температуре. Подставляя (4.17) в (4.18), получаем
f~(dU/dl)T — T(dS/dl)T (4.19)
или
Mu+fs. (4.20)
Отсюда следует, что деформирующая сила расходуется на
изменение внутренней энергии fu= (dUIdl)? или энтропии
системы fs=.(dS/dl)T
Деформация твердых тел (например, кристаллов) происхо-
дит с изменением объема в результате деформации валентных
углов и связей. Изменением в относительном расположении
структурных единиц, т. е. изменением энтропии, обычно пре-
небрегают и считают (dS/dl) =0, a f=(dUldl)T.
При высокоэластической деформации объем практически не
изменяется и внутренняя энергия тоже постоянна, поэтому
(dUldl)r=Q. Следовательно, упругая сила в полимере, который
деформируется в высокоэластическом состоянии, обусловлена
изменением энтропии:
f~-T(dS/dl)T. (4-21)
Таким образом, полимер в высокоэластическом состоянии
аналогичен газам: деформация имеет энтропийный характер и
обусловлена изменением порядка в расположении сегментов
макромолекул в полимере.
Статистическая теория. Сущность высокоэластичности мож-
но рассмотреть с позиций статистической теории. Наиболее
вероятным состоянием системы является состояние, характери-
зующееся максимальной энтропией S:
S=*lnW'(f), (4-22)
где W'(r)—термодинамическая вероятность; k— константа Больцмана.
Используя уравнения (4.22) и (1.19) получаем
S=C — 3kr2/2nb2 (4.23)<
(здесь С — постоянная).
Этому состоянию соответствует конформация статистическо-
го клубка при расстоянии между концами макромолекулы г2=
=2{3пЬ2 (п — число повторяющихся звеньев, b — длина одного
звена) При растяжении число возможных конформаций умень-
шается (lV'('r)-+-O] и энтропия системы в растянутом состоянии
становится меньше После снятия нагрузки система переходит
244
Рис. 4.5. Схематическое изо-
бражение трехмерной де-
формации
в устойчивое состояние с максимальной энтропией, т е. снова
сворачивается в клубок.
С помощью уравнений (4.21) и (4.23) можно оценить упру-
гую силу, возникающую при растяжении отдельной цепной
макромолекулы:
f=-T(dS/dr)r=(3Ar/nd«)r. (4.24)
Из уравнения следует, что упругая сила прямо пропорцио-
нальна расстоянию г между концами цепи. При растяжении
(г увеличивается) упругость возрастает, а в нерастянутом со-
стоянии (/=0) цепь сворачивается в клубок и среднее значе-
ние г по всем конформациям будет тоже равно нулю. Другим
важным свойством, следующим из уравнения (4.24), является
прямая пропорциональность между f и Т. Это подчеркивает,
что высокоэластичиость имеет кинетический характер, т. е.
определяется движением звеньев
Перейдем от деформации одной макромолекулы к деформа-
ции образа, имеющего, например, форму куба с длиной сторо-
ны 1 см (1 м и т д.) (рис. 4.5). Растягивающие (нли сжимаю-
щие) силы, приложенные к граням куба, представляют собой
условные напряжения pt, рч и />з, а грани прямоугольного па-
раллелепипеда, который образуется из куба (в результате
деформации) численно равны %i, Хг и А*. Работа, требуемая для
деформации такого образца, равна
з
А = — Т dS = —T(S — S0)= — TdS, (4.25)
So
(So и S — энтропия системы до и после деформирования). Из-
менение энтропии образца объемом 1 м3, состоящего из У
отрезков цепи между узлами сетки, равно:
AS==— >/siVA(A»,+X»8+AW). (4.26)
Тогда
Л = ’/2^Г(Х21+Лг2+Л2з-3) (4.27)
или
А=ЧяС (Л*1+Л2г+Xs»—3),
где G~*NkT.
245
Величина G отражает внутреннее сопротивление деформи-
рованию, т. е. является модулем высокоэластичности при
сдвиге.
Учитывая, что работа деформирования всего образца под
влиянием сил р\, рг, рз складывается из составляющих работы
ъ трех направлениях, имеем:
(М=/7|(/Х| + р2</Х2+рз</Лз. (4 28)
Принимая условие несжимаемости Х1Х2Хз=1, получаем:
=Pi — (Хз/Х|)рз? dA d^2 Р ——(Xj/Xs)/^. (4.29)
Учитывая, что истинное напряжение о=рХ из уравнения
(4.27), получаем:
<Ji — Оз “ G (Х21 — Х2з): ог — 03= G (Х2г — Х2з). (4 30)
Рассмотрим частный случай одноосного растяжения При растяжении об-
разца в Х=1/10 раз ((0 и I — длина до и после деформирования) поперечное
сечение уменьшится в X раз, а для каждого ребра — в | X раз. В этом слу-
чае О1 = а; О2=,Оз=0; Xi=X=///0; Х2=Хз=1/Х.
Тогда из (4.30) получим:
O«G(X2—X-'). (4.31)
Модуль высокоэластичности Е прн растяжении равен
E~3NkT, (4.32)
где W=v/Vn=“Pn/Afc — число отрезков в 1 моль; Уп н рп — мольный объем
и плотность полимера, Л1С — молекулярная масса участка цепи между узла-
ми сетки. Тогда
£=Зр„ЛТ/Л(е. (4.33)
Эта формула подчеркивает кинетический характер высокоэластнческой
деформации (для упругости газов характерно повышение модуля с ростом
температуры, так как увеличивается число столкновений).
Рассмотренный механизм высокоэластичности относится к
идеальному полимеру, который деформируется в идеальных ус-
ловиях. При этом обеспечиваются постоянство объема и темпе-
ратуры при деформировании, отсутствие вязкого течения, аффин-
ный и равновесный характер деформирования Равновесный
характер деформирования следует понимать как вероятность
перехода из неравновесной в равновесную конформацию за
время деформирования; это наблюдается лишь при условии
f->oo, т. е. П-+-0
Таким условиям удовлетворяют полимеры с узким ММР и
гибкими макромолекулами, соединенными редкими сшивками
(при условии, что молекулярная масса отрезка цепи между
сшивками намного больше молекулярной массы механичес-
кого сегмента). Чтобы удовлетворить условию V=const, поли-
мер должен быть некристаллизующимся, так как при кристал-
лизации в результате роста плотности объем уменьшается..
246
Уровень межмолекулярного взаимодействия должен быть так-
же незначителен, а деформирование должно проводиться при
очень низких скоростях.
Деформация реальных полимеров в реальных условиях,
безусловно, отличается от описанной по ряду причин, рассмот-
ренных ниже.
Деформация реальных сеток сопровождается изменением не
только конформационной энтропии, но и внутренней энергии,
т. е. fu^O- Величина fu зависит от температуры и конформации
макромолекулы:
fu—A(&U/kT), (4.34)
где MJ=U\—U2 — разница конформационных энергий при переходе из од-
ной конформации (с энергией U|) в другую конформацию (с энергией Г/2);
А — коэффициент, учитывающий симметрию цепи и соотношение чередующих-
ся валентных углов.
Значения fu для полимеров составляют:
Натуральный каучук 0,22
Полидиметилсилоксан 0,27
Полиэтилен —0,47
Сополимер этилена и —0,42
пропилена
Знак прн fu зависит от соотношения энергий конформеров;
например, при деформации полиэтилена осуществляется пере-
ход из менее компактной еош-формы с большим значением U
в вытянутую транс-форму с меньшей конформационной энерги-
ей, поэтому значение fu<G- При снижении температуры вклад
fu в общую деформацию увеличивается и при ТсТс полимер
деформируется как твердое тело, т. е. деформация имеет пре-
имущественно энергетический характер, и f~fu.
С учетом изменения конформации макромолекул при де-
формировании зависимость между напряжением и деформаци-
ей прн одноосном растяжении имеет следующий вид:
v'feT V }
'о ?>св 1 W ’
(4.35)
где v'— число активных цепей в 1 моль; /Р, Уо — длина и объем иедеформи-
рованиого образца; V — объем деформированного образца; k—константа
Больцмана; <г2> и — средние расстояния между концами цепей сеткн
в недеформированном состоянии и в свободно-сочлененной цепи.
При деформировании реальных полимеров происходит из-
менение их объема Это обусловлено, в частности, изменением
внутренней энергии, поскольку деформация, сопровождающая-
ся изменением внутренней энергии, всегда влечет за собой из-
247
менение (уменьшение) объема. Для оценки этого вклада пред-
ложено уравнение
i
aiz = i/8Kj l(df/dl)rdl, (4.36>
lo
в котором К — модуль объемного сжатия. С ростом степени
деформации относительное изменение объема возрастает.
Еще одно отличие деформации реальных полимеров от де-
формации идеального каучука состоит в том, что процесс де-
формирования не является абсолютно изотермическим. При._
растяжении образца выделяется некоторое количество тепла,
что повышает температуру на 1—2 К; в случае кристаллизую-
щегося полимера тепловыделение увеличивается и повышение
температуры при том же удлинении более значительно — при-
мерно на 10—14 К (табл. 4.2).
Высокоэластическая деформация реальных полимеров прак-
тически всегда сопровождается перемещением макромолекул
относительно друг друга со смещением их центров тяжести —
течением. Это обусловлено тем, что в реальных сетках не все
молекулы сшиты, т. е. существует золь-фракция. Однако это
явление полностью не исчезает, даже если все макромолекулы
соединены в сетку В этом случае течение обусловлено разры-
вом межатомных связей под действием напряжения и тепловых
флуктуаций, т. е. протеканием механохимических реакций.
Следствие этих реакций — появление в системе не связанных
в сетку макромолекул, которые при деформировании проявля-
ют не только высокоэластические, но и вязкотекучие свойства.
Доля вязкотекучей деформации в общей деформации неве-
лика (несколько процентов). Она зависит от числа несвязан-
ных макромолекул (т. е. от степени сшивания, по мере роста
которой она снижается) и от глубины протекания механохими-
ческих процессов Чем ближе температура к Тс, тем больше-
число разрывов.
Таблица 4.2. Тепловые эффекты при деформации полимеров в высокоэласти-
ческом состоянии
Сополимер этилена с пропиленом Натуральный каучук Поллднметнлснлоксан
Л | Q*. Дж/г Л Q. Дж/г К | Q. Дж/г
1,23 —0,0092 1,45 —0,6 3,0 —0,02
1,37 1,78 2,33 —0,0082 0,117 0.52 1,97 3,2** —0,123 —0,820 5,0 —0,06.
• Q—-dU; нагрев: dU>0, охлаждение: dU<V.
•• Образец кристаллизуется.
248
Высокоэластическая деформация в реальных условиях явля-
ется неравновесной, поскольку за реальное время деформиро-
вания макромолекулы не успевают принять равновесную для
данных условий конформацию. Скорость установления равно-
весия оценивается временем релаксации т*, которое тем боль-
ше, чем больше степень сшивания, энергия когезии и т. д.
Таким образом, высокоэластическая деформация имеет релак-
сационный характер.
Рассмотрим высокоэластическую деформацию реальной сет-
ки эластомера с разной длиной цепей (/2>/0 между узлами,
образованными поперечными химическими связями Непрерыв-
ное движение сегментов приводит к свертыванию участков це-
пей между узлами в клубки, размеры которых соответствуют
длине этих участков. Подобное состояние эластомера равно-
весно, и ему соответствует максимальное значение энтропии.
Поперечные связи, соединяющие между собою клубки, статис-
тически распределены по всему объему эластомера во всех на-
правлениях.
При растяжении происходит неравномерное последователь-
ное растягивание цепей между узлами, расположенных в на-
правлении растяжения, с переходом макромолекул из сверну-
того в распрямленное состояние, сопровождающимся снижени-
ем энтропии Из-за дефектов сетки прежде всего распрямляют-
ся и растягиваются наиболее короткие участки Ц, ориентиро-
ванные в направлении растяжения, затем более длинные /2.
В процессе растяжения различные участки макромолекул
испытывают различные по величине и направлению напряже-
ния. Под действием растягивающих сил наибольшие напряже-
ния возникают в наиболее коротких и сильно растянутых,
ориентированных в направлении растяжения цепях Л. После
разрыва этих цепей полимер удлиняется и напряжение переда-
ется на менее растянутые, более длинные цепи /2, которые под
действием растягивающих сил ориентируются в направлении
растяжения и вытягиваются.
Участки сетки большой длины, свернутые в клубки, при этом
испытывают незначительные напряжения Однако после раз-
рыва участков /2 цепи в этих клубках будут ориентированы и
растянуты. Поскольку объем эластомера при растяжении изме-
няется незначительно, увеличение его длины компенсируется
уменьшением поперечного сечения, т. е. растяжение сопровож-
дается поперечным сжатием. Поперечное сжатие вызывает де-
формацию клубков в направлении растяжения. Подобные про-
цессы растяжения цепей, свернутых в клубки, и их разрыва,
ориентации и растяжения новых испей происходят в течение
всего процесса.
В момент, когда последние наиболее длинные участки цепей
будут распрямлены и ориентированы, наступает заключитель-
249
ный этап деформации, предшествующий разрыву эластомера.
При растяжении предельно растянутых участков макромолекул
на этом этапе происходит увеличение валентных углов и рас-
стояний между соседними атомами, что приводит к некоторому
увеличению внутренней энергии системы. При разрыве какой-
либо цепи в процессе растяжения валентные углы между со-
сетними атомами и расстояния между ними мгновенно умень-
шаются, т. е. проявляется упругость, характерная для твердых
тел. После полного разрыва эластомера или прекращения дей-
ствия растягивающей силы растянутые в различной степени
участки сетки оказываются в неравновесном состоянии, так как
обладают ограниченным набором конформаций и более низкой
энтропией, чем в равновесном состоянии. Под действием теп-
лового движения сегментов освобожденные от действия растя-
гивающей силы участки макромолекул между узлами сетки
вновь скручиваются в клубки, что приводит к возрастанию
энтропии и переходу эластомера в первоначальное равновесное
состояние. Скорость образования клубков и достижения полно-
го равновесия определяется строением, температурой и внут-
ренним трением эластомера
При сокращении эластомера после его растяжения образую-
щиеся из свертываемых отрезков цепей клубки стремятся рас-
положиться около поперечных связей, т е. узлов сетки, огра-
ничивающих длину этих отрезков. Вследствие этого после со-
кращения эластомера его структура, пространственное распо-
ложение клубков и поперечных связей почти полностью соот-
ветствуют структуре до растяжения. Если процесс растяжения
эластомера сопровождался разрывом большого числа коротких
участков между узлами сетки, то полного восстановления пер-
воначальной длины не произойдет. В этом случае узлы сетки
и разорванные цепи не смогут возвратиться в первоначальное
положение и длина эластомера увеличится. Прирост длины
эластомера после деформации к его первоначальной длине
(остаточное удлинение) характеризует степень пластичности
эластомера при данных условиях деформации.
Перечисленные выше отклонения от идеального механизма
высокоэластнчности приводят к ограниченному совпадению те-
ории и эксперимента обычно при Х=1.5 (рис. 4.6).
Причиной отклонения от статистической теории деформации
является существование в сшитых полимерах наряду с сеткой,
образованной химическими связями, флуктуационной сетки фи-
зических узлов. Такая сетка есть и в несшитых полимерах, но
при достаточно интенсивном тепловом движении в результате
приложения нагрузки большинство узлов разрушается и сетка
становится нестабильной.
При сшивании макромолекул некоторые физические узлы
становятся «запертыми». Однородная деформация (аффинная)
250
I, МПа
12 3 4 5 >•
Рис. 4 6 Зависимость условного напряже- в
иия f от степени растяжения X для иде-
ального (/) н реального (2) эластомеров
с
всего образца полимера возможна
только в случае жесткого закрепле-
ния всех узлов Если же физичес-
кие узлы свободно флуктуируют «
по полимеру (так называемая
«фантомная» сетка), то система
деформируется неаффинно. Реаль-
ные сетки, в которых наряду со сво- г
бедными существуют и «запертые»
узлы, при малых деформациях де-
формируются аффинно, а при боль-
ших — деформация приближается к о
деформации «фантомной» сетки.
Таким образом, основная причина упругости при деформа-
ции в высокоэластическом состоянии и возникновения напря-
жений в образце заключается в изменении конформации и пе-
реходе из равновесной формы статистического клубка с макси-
мальным значением энтропии в неравновесную с уменьшением
энтропии и обратный переход после прекращения деформации.
Вклад энергетической составляющей в этот процесс невелик,
а для идеальных сеток равен нулю.
Для проявления высокоэластичности, не сопровождаемой
большими пластическими деформациями, необходимы опреде-
ленные условия:
1) высокая гибкость макромолекул и их энергичное тепло-
вое движение, обеспечивающие подвижность сегментов и обра-
зование статистических клубков (Т>ТС);
2) высокая степень полимеризации;
3) соединение макромолекул друг с другом прочными хими-
ческими поперечными связями с образованием пространствен-
ной сетки;
4) минимальное .межмолекулярное взаимодействие между
макромолекулами (низкая энергия когезии);
5) минимальная степень кристаллизации полимера.
Этим условиям удовлетворяют эластомеры, полученные вул-
канизацией высокомолекулярных натурального и синтетичес-
ких каучуков Часто высокоэластичностью обладают не только
сшитые эластомеры, но и линейные высокомолекулярные поли-
меры, например невулканнзованные каучуки В них тоже обра-
зуются пространственные структуры, однако поперечные связи
между линейными макромолекулами каучуков непрочны, имеют
временный характер, являются лабильными, неустойчивыми.
251
''ни образуются при перехлестывании, зацеплениях цепей,
также при переплетениях петель, принадлежащих одной и
:v • же или разным макромолекулам. При деформации прост-
ранственных структур, образованных таким образом, макромо-
лекулы могут перемещаться, скользить относительно друг друга
ч местах переплетений, вследствие чего снижается степень
э. астич ности и увеличивается пластичность полимера.
Часто такие поперечные связи появляются в линейных поли-
мерах, содержащих фрагменты, склонные к сильному межмоле-
кулярному взаимодействию Примером подобных структур яв-
ляются термоэластопласты, которые в условиях эксплуатации
(при 200—350 К) способны к большим обратимым деформаци-
ям, а при повышенных температурах (420—470 К) текут, по-
добна термопластам. Термоэластопласты представляют собой
блок-сополимеры, например, типа АВА, где А — жесткие блоки
термопластов (полистирол и др.), В — гибкие полидиеновые
юки. Молекулярная масса полндиеновых блоков значительно
повышает молекулярную массу жестких блоков термопластов.
При содержании в термоэластопластах более 50% диена поли-
стирольные блоки образуют стеклообразные домены, регу-
лярно расположенные в непрерывной фазе полидиена При
температуре ниже температуры стеклования жесткоцепного
блока (для полистирола 7с = 370К) они играют роль соедини-
тельных узлов, аналогичных химическим поперечным связям.
Но при повышении температуры выше температуры стеклова-
ния жесткоцепного компонента появляется сегментальная под-
вижность, домены размягчаются и перестают выполнять функ
ции соединительных узлов. В результате термоэластопласты
становятся типичными термопластами и при деформации не
проявляют высокоэластичности.
Таким образом, для проявления высокоэластичности линей-
ными полимерами также необходимы высокая гибкость макро-
молекул, высокая степень полимеризации, образование прост-
ранственной структуры. Однако высокоэластичность линейных
полимеров имеет свои специфические особенности, к которым
относятся следующие:
1) быстрая потеря высокоэластичности при нагревании, рас-
творении, перемешивании и действии любых факторов, разру-
шающих малопрочные нестабильные поперечные связи между
линейными макромолекулами;
2) восстановление пространственной структуры, повторное
проявление высокоэластических свойств и снижение пластично-
сти при прекращении действия механических деформаций, на-
гревания и других факторов вызвавших разрушение лабиль-
ных механических или структурных поперечных связей в поли-
мере;
252
4.3. ВЯЗКОТЕКУЧЕЕ СОСТОЯНИЕ
Полимер переходит в вязкотекучее состояние при температурах
выше Тс, и в зависимости от структуры полимера этот переход
может осуществляться из стеклообразного, кристаллического
или высокоэластического состояния; следовательно, 7’т>Тсили
Тт>Тпл. Оно присуще линейным полимерам в интервале 7Т—
Тд, т. е. между температурой текучести Тт и температурой
деструкции Тл Обычно этот интервал бывает узким. Полимеры
в этом состоянии иногда называют расплавами.
Плотность упаковки макромолекул в вязкотекучем состоя-
нии аналогична плотности жидкостей, но ниже плотности упа-
ковки в высокоэластическом состоянии в основном за счет уве-
личения доли свободного физического объема, обусловленного
тепловым движением.
Подвижность кинетически связанных структурных единиц
(сегментов) в вязкотекучем состоянии высокая и превышает их
подвижность в стеклообразном (значительно) и в высокоэлас-
тическом состояниях
Надмолекулярная структура расплавов полимеров характе-
ризуется ближним порядком, свойственным аморфным телам.
Но вследствие более интенсивного теплового движения в вяз-
котекучем состоянии по сравнению с высокоэластическим упо-
рядоченность структуры (доменов, кластеров) ниже.
К числу основных признаков вязкотекучего состояния отно-
сится его реакция на действие напряжения Под влиянием ме-
ханических сил у полимеров в вязкотекучем состоянии разви-
вается деформация течения. Течение — это необратимое пере-
мещение молекул относительно друг друга под влиянием
приложенного извне усилия F, при этом в веществе возникают
силы трения Гт, препятствующие течению, т. е. F=—FT. Внут-
реннее трение полимеров имеет в основном энергетическую
природу, так как связано с преодолением сил взаимодействия
между плотно упакованными макромолекулами Поэтому сетча-
тые полимеры с пространственной структурой, образованной
химическими связями, в вязкотекучее состояние не переходят,
так как эти связи препятствуют свободному перемещению мак-
ромолекул, необходимому для течения. Течение этих систем
возможно лишь при разрушении поперечных связей (химичес-
кое течение)
Таким образом, для вязкотекучего состояния характерны
значительные необратимые деформации, которые резко возрас-
тают при повышении температуры, т. е. deldT^Q.
Модуль упругости полимеров в вязкотекучем состоянии не-
высок. Так, область вязкотекучего состояния определяют как
область, в которой модуль через 10 с после действия нагрузки
составляет 104Л Па. Механическая прочность полимеров в этом
253
Экергш «
Рис. 4.7. Изменение ныеокоэластнческой н вязкотекучей деформации прн низ-
ких скоростях деформирования в областях неустановнвшегося (а) и уста-
новившегося (б) течения
Рис. 4 8 Изменение потенциального барьера течения U под влиянием напря-
жения сдвига от (2) и без напряжения (/)
состоянии также невелика и существенно ниже прочности поли-
меров в стеклообразном и высокоэластическом состояниях
Полимеры в вязкотекучем состоянии представляют собой
вязкоупругие тела, поэтому под действием силы в них разви-
ваются не только необратимые деформации течения, но и обра-
тимые деформации упругой и высокоэластической природы. От-
носительный вклад каждой из них определяется условиями те-
чения (напряжением и скоростью). Простейшим случаем явля-
ется режим установившегося течения — режим, при котором вы-
сокоэластическая деформация достигает постоянного значения
(рис. 4.7)
В условиях установившегося течения (при низких скорос-
тях и большой продолжительности процесса) существует дина-
мическое равновесие между изменением структуры под влияни-
ем напряжения и ее восстановлением под действием теплового
движения, т е. в целом структура равновесна
4.3.1. Механизм течения
Низко- и высокомолекулярные жидкости имеют различную
структуру, соответственно различаются и механизмы их тече-
ния. Движение низкомолекулярных жидкостей происходит пу-
тем мгновенного перескока образующих их атомов или моле-
кул из одного положения в другое. Эти перескоки возможны и
при отсутствии течения В этом случае частота перескоков vo
зависит от высоты потенциального барьера (рис. 4.8), разме-
254
ров молекулярно-кинетических единиц, строения жидкости и
температуры-
v0« exp (— Ет/kT), (4.37)
где £т — энергия активации течения, определяемая высотой потенциального
барьера.
Приложение внешнего напряжения эквивалентно снижению
высоты потенциального барьера на величину А; А — это рабо-
та, которую совершает сила F при перемещении структурной
единицы до вершины потенциального барьера Частота перес-
коков в направлении действия силы в этом случае равна
v+ ехр [— {Ет — А) /kT]; v+ я*vo exp (Л /kT). (4 38
Частота перескоков в обратном направлении равна
V-*» ехр [—(E+A)/kT); v“*“v«exp (—А/kT). (4.39)
Перескок происходит в момент, когда запас тепловой энер-
гии у молекулы больше, чем энергия взаимодействия с сосед-
ними молекулами Это первое условие течения Вторым усло-
вием является наличие флуктуационного (физического) свобод-
ного объема, который определяет наличие так называемой
«дырки», куда может перескочить молекула (рис. 4 9,а). Объ-
ем «дырки» обычно близок к собственному объему молекулы
Таким образом, вероятность перескока Р определяется ве-
роятностью преодоления активационного барьера (Рд) и веро-
ятностью того, что рядом будет находиться «дырка» (Pv): Р=
= PePv. Величину Р£ рассчитывают на основе активационной
теории, и в упрощенном виде уравнение для расчета Р£ может
быть представлено следующим образом:
Ре*1 ехр (— Ev*/RT) (4.40)
(где Ev* — энергия активации при постоянном объеме).
Величину Pv определяют на основе теории свободного объ-
ема:
Pt =exp [—CV*/(Ve*)z], (4.41)
где И*—объем «дырки»: (Vc*)z — средний свободный объем, приходящийся
иа одну частицу: С — коэффициент, введенный для учета возможности пере-
крывания соседних свободных объемов (1 >С>0,5).
Механизм течения полимеров схематически можно предста-
вить так же, но его отличия обусловлены неспособностью длин-
ных макромолекул перемещаться как единое целое (см.
рис. 4 9,6). Все структурные элементы полимеров соединены
химическими или физическими межмолекулярными связями,
поэтому движение любой структурной единицы (боковых групп,
звеньев, сегментов) может осуществляться только кооператив-
но, согласованно с движениями соседних групп, звеньев, сег-
255
X 6 X X
Рис. 4.9. Схема течения низкомолекулярной жидкости (а) и полимера (6);
to, Н» ft — расстояние от условно выбранных осей ординат до центра тяже-
сти молекулы
— "дыр»»’
ментов. Кинетической единицей макромолекул, как известно,
является сегмент. Сегменты находятся в непрерывном тепловом
движении, образуя бесконечное множество конформаций При
этом центр тяжести макромолекулы может оставаться не-
подвижным относительно осей координат или центров тяжес-
ти соседних макромолекул.
Течение полимеров под действием силы осуществляется пу-
тем перескоков сегментов из одного свободного положения в
другое, т. с. в «дырку». Так как все сегменты связаны между
собой, то при перемещении одного из них происходит деформа-
ция всей макромолекулы, т. е. переход ее в неравновесную
конформацию При достаточно свободном объеме вслед за
движением одного сегмента перемещают и другие, что приво-
дит к перемещению всей макромолекулы. Так же как и для
низкомолекулярных жидкостей, частота перемещения сегментов
определяется температурой, высотой потенциального барьера
•256
я величиной приложенного напряжения (от которого зависит
работа Л)
v+= exp [—(£ — A)/kT] ~v0' exp (A/kT), (4.42)
где Vo' — частота перескоков сегмента в отсутствие напряжения, т. е. собст-
венная частота его колебаний.
Наличие свободного объема для реализации течения необ-
ходимо и для полимеров Вязкое течение полимера (т. е. пере-
скоки сегментов) возможно до тех пор, пока физический
(флуктуационный) свободный объем Ксф не уменьшится до
0,025; это значение характерно для стеклообразного состояния.
Рассмотрим теперь течение полимеров при переходе его из
высокоэластического состояния в вязкотекучее с точки зрения
изменения конформации макромолекул. Как уже говорилось,
в состоянии, предшествующем течению, макромолекулы имеют
форму статистических клубков, соединенных между собой сеткой
физических связей (флуктуационной сеткой) Под действием
силы происходит деформация клубков и изменение конформа-
ций макромолекул, которые вытягиваются, ориентируются и
перемещаются в направлении действия силы. Безусловно, при
этом будет разрушаться исходная флуктуационная сетка и об-
разовываться новая, определяемая уже новой конформацией
макромолекул. Таким образом, для осуществления течения по-
лимера необходимо, чтобы молекула имела, во-первых, доста-
точный запас энергии для преодоления активационного барье-
ра перескока и для разрушения флуктуационной сетки и,
во-вторых, достаточный свободный физический (флуктуацион-
ный) объем, обеспечивающий наличие «дырок».
4.3.2. Влияние структуры полимера на температуру текучести
Переход ^полимеров в вязкотекучес состояние происходит в не-
котором интервале температур и начинается с первых переско-
ков сегментов. Теоретически за равновесную температуру сле-
довало бы принимать ту, при которой под действием напряже-
ния смещается центр тяжести макромолекулы, т. е. начинается
течение. При бесконечно низкой скорости деформирования оп
ределяемая экспериментально Тг близка к теоретической. В ре-
альных условиях за Тг принимают среднюю температуру об-
ласти развития необратимых деформаций (см. рис. 4 1).
Наиболее существенное влияние на Гт оказывает молекуляр-
ная масса. С ростом М температура текучести повышается
(см. рис. 1.33 и 4.1.6), но с разной скоростью. При М = Мкр
темп роста замедляется.
Переход в вязкотекучее состояние полимеров с невысокой
молекулярной массой происходит, как правило, из стеклооб-
разного или кристаллического состояния, поэтому Тт = Тс или
17—816
257
Тт=Тпл. На термомеханической кривой таких образцов есть
только два участка, характерные для стеклообразного и вязко-
текучего состояний (см. рис. 4.1,6, кривые 1 и 2) или кристал-
лического и вязкотекучего состояний (см. рис. 4.2, кривая /).
При достижении Л1кр на тер.момехаиических кривых наблюда-
ется «расщепление» Тс и Ту и появляется так называемое пла-
то высокоэластичности. В этом случае переход полимера в вяз-
котекучее состояние происходит уже из высокоэластического.
Влияние молекулярной массы на Тт можно оценить по форму-
ле Каргина и Слонимского:
В(тг — Тс)
1g Af = 1g Мег 4- > (4.43)
где Л1Сг — молекулярная масса сегмента; В н С — константы, зависящие от
структуры полимера н режима деформации.
Таким образом, с увеличением молекулярной массы повы-
шается температура текучести и расширяется интервал сущест-
вования полимера в высокоэластическом состоянии. Например,
при увеличении М полиизобутилена от 1270 до 62500 темпера-
тура текучести повышается от 273 до 573 К, а интервал высо-
коэластнчностн увеличивается с 73 до 270 К Но при этом
уменьшается интервал между температурами текучести и дест-
рукции, что ограничивает возможность переработки полимеров
в вязкотекучем состоянии. Полимеры с широким молекулярно-
массовым распределением характеризуются большей протяжен-
ностью переходного состояния из высокоэластического в вязко-
текучее состояние, поскольку фракции полимеров разной моле-
кулярной массы имеют различную Ту.
На Тг влияет и кинетическая гибкость макромолекул- при
одном и том же значении молекулярной массы Ту тем ниже,
чем выше гибкость макромолекул. Полимеры с гибкими цепя-
ми (эластомеры) начинают течь при сравнительно низких тем-
пературах («350—370 К) и, как правило, перерабатываются
в этом температурном интервале. Температура течения жест-
ких полимеров лежит в интервале 420—500 К и выше. Но
поскольку прн этом существенно
повышается и Тс, то плато высо-
коэластичности у этих полимеров
практически исчезает. В этом
случае говорят о размягчении
полимера и ТС=ТГ.
Рис. 4.10. Влияние скорости деформи-
рования о на температуру текучести Ту
чис-1.4-полнбутадиеиа (/), сополимера
бутадиена с нитрилом акриловой кисло-
ты (2) и чис-1,4-полннзопрена (3)
258
Температура текучести зависит от режима механического
нагружения: напряжения о и скорости его действия. С ростом
о происходит довольно быстрое снижение Тт, а увеличение ско-
рости деформирования о. наоборот, повышает Тт (рис. 4.10).
Это влияние о и v на Тт может быть объяснено на основании
рассмотренных механизмов течения. Повышение о равносильно
снижению энергии активации перемещения сегментов [см. фор-
мулу (4.42)]. С ростом скорости деформирования увеличива-
ется степень ориентации макромолекул и образуется более ус-
тойчивая флуктуационная сетка, для разрушения которой тре-
буется большая тепловая энергия, т. е. более высокая
температура.
4.4. РЕЛАКСАЦИОННЫЕ ПРОЦЕССЫ В ПОЛИМЕРАХ
Выше мы говорили о том, что деформация реальных поли-
меров в любом физическом состоянии имеет неравновесный
характер. Причина этого — наличие сетк.. физических узлов
(флуктуационная сетка), которая не позволяет системе принять
равновесные конформации за время действия силы, т. е. пред-
определяет неравновесный характер деформирования Поэтому
говорят, что деформация имеет релаксационный характер (ре-
лаксация— латинское слово, означающее ослабление, умень-
шение напряжения, отдых). Как уже отмечалось (см. разд. 1 3
и 4.1), процессы релаксации — это процессы перехода системы
из неравновесного состояния к термодинамическому равнове;.
сию под действием внутренних сил, т. е. процессы, в которых
равновесие устанавливается во времени. Различают механиче-
ские, электрические, магнитные и другие релаксационные про-
цессы. Механические релаксационные явления возникают при
нарушении равновесия структурных элементов, электрические—
при нарушении равновесия ориентации электрических диполей,
магнитные— магнитных моментов. Механические релаксацион-
.ные явления могут быть двух видов: релаксация напряжения
и релаксация деформации
При деформации полимера до определенной степени в нем
возникает напряжение, которое зависит от скорости деформи-
рования. Чем выше скорость, тем больше напряжение (при
одинаковой степени деформации) и модуль упругости. Иными
словами, чем быстрее действует на полимер сила, тем больше
сопротивление со стороны полимера, тем жестче полимер в
момент действия силы. Зависимость модуля упругости полиме-
ра от скорости деформации объясняется тем, что при быстрых
деформациях громоздкие макромолекулы и надмолекулярные
структуры не успевают перестроиться в направлении действия
силы и полимер проявляет упругость, свойственную низкомоле-
кулярному твердому телу.
17*
259
Рис. 4.11. Кривые релаксации напряжения о (а) и деформации е (6) сшито-
го (/) и линейного аморфного (2, 2') полимеров при температуре 7\ (/, 2)
н Т2 (2'); T3>Ti (tx — момент удаления нагрузки; е^, е0' — первоначальная;
Свэле'мл — высокоэластнческая, Еостб'ост — остаточная, е«> — равновесная де-
формации)
Однако если 5ыстро растянутый до определенной длины
полимер оставить в таком состоянии на длительное время, то
напряжение, возникшее в нем, постепенно снижается
(рис. 4.11). В растянутом образце происходит релаксационный
процесс перегруппировки структурных элементов, скорость ко-
торого увеличивается с повышением температуры У линейных
полимеров с гибкими цепями релаксация идет быстрее, чем у
полимеров с жесткими цепями, полярных полимеров, разветв-
ленных или сетчатых.
Вследствие изменения конформации макромолекул в рас-
тянутом линейном полимере напряжение быстро снижается,
а в образце сохраняются большие остаточные деформации.
В пространственном полимере поперечные химические связи
между макромолекулами не позволяют им перемещаться, по-
этому релаксация в таких полимерах происходит только до оп-
ределенного напряжения. Чем больше степень сшивания, тем
меньше эффект релаксации.
Процесс нарастания во времени деформации во материала
прн постоянном напряжении называется ползучестью Это яв-
ление наблюдается при растяжении, сжатии и других видах
деформации полимеров. На рис. 4 11,6 показано изменение де-
формации при растяжении сшитого (кривая /) и линейного
(кривая 2) полимеров. Под действием деформирующей силы с
течением времени структурные элементы полимера постепенно
распрямляются, ориентируются в направлении растяжения и
образец медленно растягивается. Наиболее быстро деформация
возрастает в начале процесса. Скорость растяжения значитель-
но увеличивается при повышении температуры и напряжения
в образце и снижается прн наличии у полимера сетки (кри-
вая /). Если через некоторое время (в точке tx) снять растяги-
вающий груз (т. е дать образцам «отдых» без нагрузки), то
•260
растянутые образцы начнут самопроизвольно сокращаться, так
как под действием теплового движения сегментов макромоле-
кулы вновь примут наиболее вероятную и устойчивую форму;
чем больше гибкость макромолекулы и выше температура, тем
быстрее будет происходить сокращение образца. Скорость со-
кращения наиболее высока в начальный момент, а затем зна-
чительно замедляется. Однако как бы долго образцы ни сокра-
щались, они обычно не достигают первоначальных размеров,
т. е. приобретают остаточную деформацию еОст вследствие не-
обратимого течения части макромолекул.
Ползучесть количественно характеризуют показателем по-
датливости J-
]=ы/с, (4.44)
где е<—деформация через промежуток времени t; а —постоянное напря-
жение.
Изменение податливости в процессе ползучести различных
полимеров показано на рис. 4.12. Характерной особенностью
трехмерных полимеров (кривая 4) является существование пре-
дельного равновесного значения податливости. Наличие плато
на кривой 2, а также равновесное оначеиие податливости на
кривой 4 указывают на обратимость деформации, а резкое воз-
растание податливости на кривой /ив конце кривых 2 и 3 —
на текучесть (необратимые деформации).
Релаксационные процессы описываются определенными за-
конами. Например, изменение напряжения при релаксации на-
пряжения подчиняется уравнению Максвелла:
Ф=Со ехр (— //т*), (4.45)
где Gi — напряжение в момент времени /; оо—напряжение в начальный мо-
мент растяжения прн /=0; т* — время релаксации.
По этому уравнению можно рассчитать напряжение ot в об-
разце в любой момент времени /, зная первоначальное напря-
Рис. 4.12. Логарифмическая зависимость
податливости J при ползучести различ-
ных полимеров от времени:
1 — аморфный линейный полимер с низкой М\
2 — разветвленный полимер 3 — аморфный
линейный полимер с высокой М 4 — ппостран
сти-ино сшитый полимер (вулканизованный
каучук); 5—частично кристаллический поли-
мер; 6 — стеклообразный полимер
'«1
261
II
A
Рис. 4.13. Типичный релак-
сационный спектр линейно-
го аморфного полимера
(сплошная линия) при по-
стоянной температуре и его
изменение при сшивании и
наполнении (пунктир):
V—-движение боковых групп*
Р — движение мономерных
звеньев; а — движение сегмен-
тов; к — подвижность надмоле-
кулярных структур: б — по-
движность химических попереч-
ных связей
жение оо и время релаксации т*. При т* = / уравнение (4.45)
принимает вид
ot—Оле-’=ао/е. (4.46)
Из этого уравнения следует, что время релаксации опреде-
ляет продолжительность процесса, в течение которого перво-
начальное напряжение уменьшается в е раз.
В процессах релаксации участвуют различные структурные
элементы полимера, каждый из которых имеет свои времена
релаксации, лежащие в широком интервале — от 10“12 с жо
многих лет. Релаксационные свойства полимеров зависят от
типа и размера релаксирующих в данных условиях элементов
структуры. Поэтому для полной характеристики полимеров не-
обходимо рассматривать сумму всех релаксационных процес-
сов, образующих так называемый релаксационный спектр.
Эти спектры получают или статическими методами (релаксация
напряжения) или динамическими (механические или диэлект-
рические потери). На рис. 4.13 приведен типичный спектр вре-
мен релаксации линейного полимера. Максимумы на релакса-
ционном спектре соответствуют временам релаксации тЛ
каждой релаксирующей единицы при постоянной температуре.
Каждое дискретное время, характеризующее данный переход,
зависит от температуры и снижается при ее повышении вслед-
ствие увеличения подвижности элементов структуры:
т,*=Лехр (4.47)
где Bt — константа, зависящая от размеров структурного элемента; —
энергия активации i-й структурной единицы, равная высоте энергетического
барьера, преодолеваемого при переходе структурного элемента из одного
равновесного состояния в другое.
В общем случае процесс релаксации напряжения при за-
данной деформации описывается уравнением
о(0 = е0Е(/) = е0 У £,ехр( — //т.*), (4-48)
z-i
262
где o(Z) и E(i)—напряжения и модуль через время t прн деформации еР;
г,* — время релаксации i-й кинетической единицы: п — число кинетических
единиц различных типов; Е,-— коэффициент, соответствующий вкладу i-й
кинетической единицы в общий процесс релаксации.
Среднее значение времен релаксации всех структурных еди-
ниц называется средним временем релаксации тср*.
Неоднородность структуры полимера, введение наполните-
лей -и других компонентов, соединение макромолекул друг с
другом, например при образовании пространственной сетки в
процессе вулканизации каучуков, приводит к увеличению числа
структурных элементов, влияющих на релаксационные процес-
сы. В макромолекуле, содержащей разные по структуре и раз-
мерам боковые группы, вместо одного максимума у-процесса
имеется несколько (-р, и т. д.) с низкой энергией активации
(10—20 кДж/моль). Для гомополимера характерен один р-пе-
реход, а для блок-сополимера уже два — р, и В кристалли-
ческих полимерах одна и та же структурная единица обладает
различной подвижностью в аморфной и кристаллической фа-
зах, соответственно наблюдаются два перехода р и р(, харак-
терные для аморфной и кристаллической структуры. Например,
в спектре полиэтилена высокой плотности проявляются р- и Pi-
переходы при 150 и 250 К и частоте 1000 Гц. Очень часто вмес-
то одного а- и 1-максимумов появляется несколько, что обус-
ловлено различной подвижностью сегментов (a-переход) или
надмолекулярных структур (Х-переход). Например, в кристал-
лических полимерах (полиэтилене) существует даже три мак-
симума— а, а.1 и аг. обусловленные сегментальной подвиж-
ностью в аморфной фазе, переходных аморфно-кристалличес-
ких межфазных слоях и аморфных участках лучей сферолитов.
1-Переходы, обусловленные подвижностью надмолекуляр-
ных структур, относятся к медленным релаксационным процес-
сам и связаны с наличием физической сетки. В некоторых по-
лимерах могут существовать физические узлы различной
природы, например в бутадисн-нитрильных эластомерах прояв-
ляется так называемый л-процесс релаксации, обусловленный
диполь-дипольным взаимодействием групп ~CN- -NC~ (при
363 К) (см. рис. 4.13). При образовании водородных связей в
спектре обнаруживается переход, аналогичный л-переходу, но
при более высокой температуре. Переходы, обусловленные ре-
лаксацией химических связей между молекулами (б), также
могут проявляться в виде нескольких максимумов, если суще-
ствуют связи различной энергии. Так, в серных резинах наря-
ду с переходом бс-с имеет место переход б§, обусловленный
подвижностью (т. е. разрывом) химических серных связей.
В наполненных полимерах вместо одного а-перехода появ-
ляется еще один — а', обусловленный подвижностью сегмен-
тов макромолекул, адсорбированных поверхностью наполните-
263
Таблица 4.3. Характеристики релаксационных процессов в резине на основе
сополимера бутадиена и стирола (70:30) с 20% технического углерода П-234
Релаксаци- онный про- цесс Характерное время релак- сации при 293 К. с Энергия ак- тивации кДж/моль Константа Bf. С Объем кине- тической единицы см3 Линейные размеры ки- нетической единицы нм
₽ 1 10-® 25 5-10-’3 2-Ю-2® 0,5
чх 6 ю—6 59 5 10~'® 1 10-®' 3—4
а' 1,5 71 4,8 10-12 1-10-®' 3—4
?. 1,6-10® 55 2,6-10~8 2.0-10-17 27
2,1-10» 55 3.4-10-8 4-10'в 73
4,3-10* 55 6,9-10~е 1,6-10-” 250
ф 2-105 74 1.6-10-8 0,9-10-17 25
6 4-10® 126 2 10-13 10—23 0,2—0,3
ля, а также ф-переход, который связывают с подвижностью
частиц наполнителя в среде-полимера. В табл. 4.3 приведены
характеристики релаксационных процессов в резинах на осно-
ве наполненного сополимера бутадиена со стиролом. Вклад
каждого релаксационного процесса зависит от его характерам
условий проявления. Деформация полимера в стеклообразном
состоянии при Т<ТС сопровождается релаксационными £- и
^-переходами. Энергия активации этих переходов намного мень-
ше, и поэтому высота их пиков намного ниже, чем в случае
a-перехода. Они оказывают существенное влияние на механи-
ческие свойства полимеров в стеклообразном состоянии. При
растяжении в высокоэластическом состоянии наибольшее влия-
ние на релаксацию оказывают не 7- и £-, а а-, а'-, Х-, б- и <р-
переходы, т. е. процессы ориентации сегментов (т=10-6 —
10 3 с), перегруппировки надмолекулярных структур (т=102—
104 с), перегруппировки поперечных химических связей и связей
в цепях (т=107—109с) и др.
Однако влияние этих факторов на скорость релаксацион-
ных процессов зависит от продолжительности действия силы.
Как уже упоминалось, вероятность проявления гибкости мак-
ромолекулой зависит от соотношения времени действия силы
и времени, необходимого для изменения конформации. Послед-
нее есть не что иное, как время релаксации т*, поэтому можно
считать, что реакция полимера на механическое воздействие
определяется соотношением между временем релаксации и
временем деформации т*Д (рис. 4.14). Если т*//<С1, то систе-
ма очень быстро релаксирует и приходит в равновесное состоя-
ние. Это условие может быть реализовано или при очень ма-
лых значениях т*, или при очень высоких значениях I. Для
полимеров этот случай имеет место при высоких температурах
(т* снижается) или очень низких скоростях воздействия. При
т*//^>1 релаксация протекает очень медленно, что может быть
следствием или высоких значений т* (система малоподвижна,
.264
например в стеклообразном или кристаллическом состояниях),
или высоких скоростей механического воздействия. При
-И релаксационные процессы развиваются за какой-то опреде-
ленный промежуток времени и оказывают наибольшее влияние
на свойства материала. Таким образом, релаксационные явле-
ния проявляются для всех рассмотренных физических состоя-
ний — стеклообразного, вязкотекучего и высокоэластического,
но в наибольшей степени релаксация характерна для высоко-
эластического состояния
Если сопоставить влияние температуры и времени действия
силы, то можно отметить, что отношение т*// снижается либо
при повышении температуры (т* уменьшается), либо при уве-
личении /, т. е. существует эквивалентность влияния времени
и температуры. Этот принцип получил название принципа тем-
пературно-временной суперпозиции (ТВС). Любая релаксацион-
ная характеристика при изменений температуры от Т до Т' из-
меняется на величину К, равную
К рпГ/р/Т
(4.49)
(рп и рп' — плотность полимера при Т и Т').
На основании принципа ТВС, зная температурные зависи-
мости какого-либо показателя, отражающего релаксационные
свойства в узком интервале скоростей воздействия, можно
предсказать, как будет изменяться это свойство при любой за-
данной температуре при изменении скорости воздействия.
На рис 4.15 показано использование принципа ТВС для гипо-
тетического полимера при релаксации напряжения. Кривая при
определенной температуре, например при 273 К, выбирается в
качестве отсчетной; эта температура называется приведенной
Тир. Затем все кривые сдвигаются вдоль логарифмической шка-
Рис. 4.14 Влияние скорости деформации v иа релаксацию напряжения (а)
и деформации (б) сшитых полимеров-
J —низкая скорость, т*о«к1; 2 — средняя скорость, 3 — высокая скорость.
T*va>l.
265
t/г.
Рис. 4.15. Обобщенная кривая зависимости релаксационного модуля £=а/е
от времени (а) и зависимость коэффициента ат от температуры (цифры
у кривых — температура. К)
лы времени до их наложения. Кривые при Т>Т„Р сдвигаются
вправо, а кривые при Г<7’пр влево. Обобщенная кривая при
7‘ир охватывает значительно больший период времени, чем экс-
периментальные данные. Расстояние, на которое сдвигались со-
седние кривые вдоль оси абсцисс, называется фактором приве-
дения ат. Температурная зависимость фактора приведения
ат описывается уравнением Вильямса — Ландела— Феоои
(ВЛФ):
1g ат —
G(T-Tnp)
^2 "Т (Т — ТПр)
(4.50)
(Ci и Сг — эмпирические константы).
В качестве температуры приведения Тпр часто выбирают
Тс. В этом случае для большинства аморфных полимеров ат
примерно одинаково и равно
, 17,44(7' — 7'с)
lgfly— 51,6+(Т—Тс) ' (4.51)
Если же за ТПр выбирается другая температура, то константы
Ci и С2 будут иметь другие значения.
266
4.5. ФАЗОВЫЕ ПЕРЕХОДЫ
Фазовыми переходами называются переходы из одного фазо-
вого состояния в другое, т. е. переходы, связанные с измене-
нием взаимного расположения молекул и термодинамических
свойств вещества. Различают фазовые переходы первого и
второго рода. Фазовым переходом первого рода называется пе-
реход, сопровождающийся изменением внутренней энергии,
объема, энтропии и тепловым эффектом. Фазовыми перехода-
ми второго рода называются переходы, при которых изменение
фазы сопровождается непрерывным изменением внутренней
энергии, энтальпии, объема и температуры, а тепло не выделя-
ется и не поглощается Но вторые производные свободной энер-
гии по температуре и давлению претерпевают скачок (отсюда
и название — переход второго рода), следовательно, скачкооб-
разно изменяются теплоемкость вещества, его термический ко-
эффициент объемного расширения и изотермическая сжимае-
мость.
Кристаллизация — это фазовый переход первого рода, кото-
рый характеризуется изменением порядка в расположении мак-
ромолекул и их термодинамических свойств (внутренней энер-
гии, объема, энтропии) и сопровождается экзотермическим эф-
фектом. Кристаллизация полимеров происходит из раствора
или расплава. Способность полимеров к кристаллизации обус-
ловлена особенностями их структуры.
Полимерный кристалл характеризуется трехмерным даль-
ним порядком в расположении звеньев и макромолекул, при-
чем одна из осей кристалла совпадает с осью макромолекулы
Поэтому макромолекула кристаллизующегося полимера долж-
на иметь дальний порядок в расположении звеньев, т. е.
должна быть регулярной.
Упаковка макромолекул в кристалле должна быть макси-
мально плотной. Это достигается, например, в глобулярных мо-
нокристаллах (плотная упаковка шаров), в фибриллярных
белках, в которых спиральные макромолекулы упакованы по
принципу «выпуклость — впадина». Возможна плотная упаков-
ка и длинных макромолекул, имеющих конформацию струны
(полиуретаны, полиамиды). Для многих полимеров плотная
упаковка достигается за счет складывания макромолекул, т. е.
образуются монокристаллы или поликристаллы со складчаты-
ми макромолекулами. Для реализации плотной упаковки в
кристалле такая макромолекула должна быть линейной, по-
скольку разветвления препятствуют плотной упаковке и сни-
жают способность полимера к кристаллизации.
Переход из малоупорядоченного аморфного состояния в кри-
сталлическое со складчатыми цепями требует изменения кон-
формации макромолекул. Поэтому кристаллизующийся поли-
267
мер должен обладать определенной термодинамической и кине-
тической гибкостью Если термодинамическая гибкость
определяется структурой макромолекул, то кинетическую гиб-
кость можно регулировать, изменяя условия кристаллизации
(температуру, давление и т. д). Повышение кинетической гиб-
кости макромолекул в растворах делает возможной кристалли-
зацию полимеров, не способных к образованию кристалличес-
ких структур из расплава.
4.5.1. Механизм кристаллизации
Механизм кристаллизации полимеров аналогичен механизму
кристаллизации низкомолекулярных веществ. Он включает два
этапа: образование зародышей кристаллизации (их называют
также ядрами) и рост кристалла на этих зародышах.
Зародышеобразование. В жидкости (растворе или распла-
ве) существуют тепловые флуктуации, приводящие к образова-
нию или исчезновению структурных ассоциатов (доменов, кри-
сталлических кластеров). Число макромолекул в этих ассоциа-
тах различно. При снижении температуры вследствие уменьше-
ния частоты тепловых колебаний вероятность существования
этих образований повышается, при этом растет и их средний
размер. При температуре, близкой к температуре плавления
кристаллов, ассоциаты довольно стабильны, а при температуре
ниже Т„л становится возможным «их рост.
При образовании зародыша свободная энергия ДГС изменя-
ется в зависимости от общего числа молекул в нем п и дости-
гает максимума, соответствующего критическому размеру заро-
дыша пКр. При Пкр величина ДГС больше нуля и зародыши еще
довольно неустойчивы, но вероятность их роста выше вероят-
ности исчезновения. С увеличением размера зародыша ДГС
переходит в область отрицательных значений, т. е. свободная
энергия системы уменьшается. Таким образом, зародыш стано-
вится устойчивым, если уменьшение свободной энергии при его
образовании больше, чем рост поверхностной энергии при об-
разовании новой границы раздела между кристаллической и
жидкой фазами.
Различают гомогенное и гетерогенное зародышеобразование.
При гомогенном зародышеобразовании возникновение устойчи-
вых зародышей происходит в аморфной фазе самопроизвольно
вследствие агрегации макромолекул. Скорость образования
этих зародышей определяется лишь температурой кристалли-
зации Для ее определения справедливо уравнение, полученное
для неполимерных систем:
N = N оехр I I > (4 * 52)
263
где —число зародышей, образующихся в единицу времени в единице
объема; /?о—константа; AF — свободная энергия активации, т. е. высота
барьера, который должна преодолеть молекула при переходе через границу
раздела зародыш (твердое тело)—жидкость; AFc.xp—изменение свободной
энергии при образовании зародыша критического размера.
Величину ДГС|КР для полимеров рассчитывают с учетом
формы зародыша и конформаций макромолекулы в зародыше
и уравнение (4 52) может быть представлено в виде
ДА Г А£ Г (ТУ,)* ] .. _
N —Л^ехр^ — kT~ ТкрСДТ)* J’ (4.53)
$оа+*
(4-я>
где Г°пл — равновесная температура плавления кристаллов; ДТ — разность
между Т°пл и температурой кристаллизации Ткг; о — средняя энергия обра-
зования границы раздела фаз; ДЯпд — удельная теплота плавления; рк₽ —
плотность закристаллизованного полимера; s и а — коэффициенты, зависящие
от механизма образования и роста зародыша, в частности от его формы
и конформации макромолекул: для гомогенного зародыша цилиндрической
формы при складчатой конформации а=2, s=8n, а для прямоугольного па-
раллелепипеда а=2, s=32.
Как следует из уравнения (4.52), зависимость скорости за-
родышеобразования от температуры описывается кривой с мак-
симумом (рис. 4 16, кривая /). При температурах, близких к
Г°пл, вероятность образования устойчивого зародыша мала
ввиду высокой подвижности макромолекул, а при Ткр, близ-
кой к Тс, внутренней энергии недостаточно, чтобы перевести
макромолекулы в конформацию, необходимую для образования
плотной упаковки. Поэтому скорость зародышеобразования
низка при Ткр, близкой к Тпя и Тс.
Гетерогенное зародышеобразование происходит благода-
ря наличию в жидкой фазе различньиСпр'ймесей или специаль-
но введенных веществ другой природы, чем полимер — так нй'-
. зываемых искусственных структурообразователей. В качестве
зародышей могут выступать упорядоченные области в аморф-
ных полимерах (кристаллические кластеры) или зародыши,
образовавшиеся в других условиях при температурах выше
Тпл. Например, при кристаллизации полидисперсных полиме-
ров в первую очередь будут образовывать зародыши высокомо-
лекулярные фракции и дальнейшее зародышеобразование (мак-
ромолекулами других фракций) будет происходить уже на
этих первичных зародышах, т е. по гетерогенному механизму.
Скорость образования зародышей при этом в значительной
степени определяется скоростью адсорбции макромолекул на
гетерогенных образованиях. Температурная зависимость скоро-
сти гетерогенного зародышеобразования такая же, как и для
гомогенного, и описывается уравнениями, аналогичными урав-
269
N;G;V
Г
Рис. 4Л6. Температурные зависимо-
сти скоростей зародышеобразования
</)» роста кристаллов G (2) и
суммарной скорости кристаллизации
v(3) (Г»,кр — температура макси-
мальной скорости кристаллизации)
нениям (4.53) и (4.54), но с
другими коэффициентами а
и s.
Рост кристаллов. По мере
кристаллизации происходит
присоединение сегментов макромолекул длиной /с к растущей
поверхности первичного зародыша кристалла. Присоединение
каждого последующего структурного элемента изменяет свобод-
ную энергию на постоянную величину ДЕ, которая уменьшает-
ся тем быстрее, чем больше 1С.
Уравнение, описывающее скорость роста кристаллов, ана-
логично уравнению (4.54), но отличается значениями констант
ф" и Go:
г-г Г 4>',(Т°пл)« 1
kT ~ Г(А7’)« ]
(4.55)
... Температурная зависимость скорости роста кристаллов
практически аналогична температурной зависимости зародыше-
образования, но сдвинута по оси абсцисс в сторону меныних
переохлаждений (см рнс. 4 16, кривая 2). Это обусловлено
большим влиянием подвижности макромолекул на рост крис-
таллов по сравнению с влиянием этого фактора на зародыше-
образование.
Рост кристаллов может быть одномерным, двухмерным или
трехмерным, при этом образуются стержни, диски, сферы. Ли-
нейные размеры кристаллов г увеличиваются со временем кри-
сталлизации пропорционально скорости роста кристаллов
G, т. е r=Gi. Рост кристалла прекращается при столкновении
растущих кристаллов и вся масса полимера переходит в новое
фазовое состояние— кристаллическое.
Скорость кристаллизации. Общая скорость кристаллизации
определяется скоростями образования зародышей и роста кри-
сталлов и описывается следующим уравнением:
V —t^l — kT — 7'(Д7')а I (4.56)
(ф и а — константы).
Зависимость скорости кристаллизации от температуры опи-
сывается кривой с максимумом, расположенным при темпера-
270
гуре, которую называют температурой максимальной скорости
кристаллизации Тм.кР. Она определяется структурой полимера,
преимущественно его гибкостью. Ниже приведены значения
Гмкр для некоторых полимеров (в К):
Полисилоксан
цис-1,4 -Поли бут аднен
цис-1,4-Полнизопрен
транс- 1,4-Полихлоро-
прен
203—193
218
248
273—268
При снижении гибкости (в приведенном ряду от полисилок*
сана к полихлоропрену кинетическая гибкость уменьшается) тем-
пература максимальной скорости кристаллизации повышается.
Зависимость скорости кристаллизации от времени удовлет-
ворительно описывается уравнением Авраами — Колмогорова,
полученным для кристаллизации металлов и низкомолекуляр-
ных веществ:
К/= 1 - ехр( -, (4.57)
где Kt — степень кристалличности в момент времени t; kK — константа скоро-
сти кристаллизации; пк — константа, характеризующая механизм зародыше-
образования и роста кристаллов; она может принимать значения от 1 до
4 (табл. 4.4).
Поскольку степень кристалличности полимеров практически
всегда меньше 100%, то вместо Kt используют показатель
KtIKoo, где К<я — степень кристалличности к моменту заверше-
ния первичной кристаллизации.
Как следует из табл. 4.4, параметр пк неоднозначно харак-
теризует механизм роста кристаллов. Так, при пк=3 могут
образовываться или сферы (чаще всего сферолиты) при гете-
рогенном зародышеобразовании, или диски при гомогенном ме-
ханизме образования зародыша. Поэтому для получения од-
нозначных результатов необходимо оценивать морфологию
кристаллов другими методами, например микроскопическими.
Иногда в кинетических расчетах получаются дробные значения
Таблица 4.4. Значения констант пк и kK в уравнении (4.57)
Рост кристаллов Зародышеобразование
гомогенное гетерогенное
«к I «к
Трехмерный (сфера) 4 л/3(1/3Л/) 3 4n/3(VW3) Двухмерный (диск) 3 л/3(РМ) 9 nV2hN3 Одномерный (стержень) 2 л/3(#а) 1 VaN3 Отсутствие роста 1 — — — Примечание: н N3 — число гомогенных и гетерогенных зародышей в едини- це объема V; h — толщина диска а — площадь поперечного сечения стержня.
271
Рис. 4.17. Изотермы кристаллизации (участок ОА — первичная, АВ — вто-
ричная)
«к, что свидетельствует о смешанном механизме кристаллиза-
ции.
Уравнение (4 57) в коорди!атах 1п[—1п(1—Kt/K<x>]—In t
представляет собой прямую линию (рис. 4.17,а), однако на
конечных стадиях кристаллизации наблюдается отклонение
кинетики процесса от линейности, что связано с протеканием
так называемой вторичной кристаллизации, в процессе которой
повышается упорядоченность в кристаллических образованиях
и между ними. Изотерма кристаллизации состоит обычно из
двух участков: S-образной кривой, описываемой уравнением
(4.57), и участка АВ, на котором происходит практически ли-
нейное повышение степени кристалличности во времени
(рис. 4.17,6). Первый из них характеризует первичную крис-
таллизацию, второй — вторичную.
На изотерме первичной кристаллизации .можно выделить
участок, где практически не происходит превращения в крис-
таллическую фазу. Это индукционный период /0, характеризу-
ющий скорость зародышеобразования. Скорость кристаллиза-
ции обычно оценивают полуперюдом кристаллизации Л/2» т. е.
временем, за которое степень первичной кристаллизации дела-
ется равной половине максимальной.
Условия кристаллизации (в частности, степень переохлаж-
дения, а прн кристаллизации вз раствора — и его концентра-
ция) в значительной степени определяют морфологию и де-
фектность кристаллических структур. Чем ближе условия кри-
сталлизации к равновесным, т. е. чем меньше АГ и ниже кон-
центрация раствора, тем более совершенные кристаллы обра-
зуются при кристаллизации. При минимальной А Г образуются
самые совершенные монокристачлы с вытянутыми цепями. Та-
кие монокристаллы полиэтилена получены кристаллизацией
при А 7=1 К в течение несколыих недель С ростом АТ полу-
272
чают более дефектные монокристаллы со складчатой конфор-
мацией цепей, доля дефектов в которых растет с повышением
ДТ. При дальнейшем росте ДТ образуются поликристаллы ти-
па сферолитов сначала менее дефектные—кольцевого типа,
а при больших ДТ — радиальные, причем средний размер сфе-
ролита уменьшается с ростом ДТ из-за увеличения скорости
зародышеобразования. В области температур, где она макси-
мальна, роста сферолитов практически не происходит и кри-
сталлизация заканчивается на стадии неразвившихся центров,
т. е. зерен.
Значительное влияние на морфологию оказывает и кон-
центрация раствора. Как уже указывалось в гл. 1, совершен-
ные монокристаллы можно получить только из очень разбав-
ленных растворов. Повышение концентрации действует анало-
гично росту переохлаждения, т. е увеличивает дефектность
кристаллов и уменьшает их размер.
4.5.2. Плавление кристаллов
Кристаллизация — фазовый переход из жидкого малоупорядо-
ченного состояния в упорядоченное твердое — сопровождается
выделением тепла. Обратный процесс — плавление кристал-
лов — происходит с поглощением тепла и тоже является фазо-
вым переходом первого рода.
За температуру плавления Тил принимают температуру, при
которой происходит плавление кристаллов. Отличительной чер-
той полимерных кристаллов является полиморфность их струк-
тур, которая тем больше, чем выше степень переохлаждения.
Рис. 4.18. Зависимость температуры начала (7) и конца (2) плавления от
температуры кристаллизации Ткр
Рис. 4.19. Влияние напряжения о на полупериод кристаллизации fy, про-
странственно сшитых полимеров:
Ч«с-1,4-полнбутадиен; 2 — греке-1.4-полихлоропрен; 3— ц«е-1,4-полиизопрен; 4 — по-
лнмстялв!ш»ц1ск.токсан; 5—полиуретан (цифры у прямых — Т^р)
18—816
273
Это влечет за собой зависимость Тпп от Ткр и существование
интервала температур плавления (рис. 4.18). Точка пересече-
ния верхней и нижней кривых дает значение Тпл, которую на-
зывают равновесной температурой плавления Т0™, выше кото-
рой существование кристаллов невозможно.
Температура плавления кристаллов зависит от размеров
кристалла, его дефектности, плотности у-паковки и других фак-
торов Так, в случае ламелярного кристалла в первую очередь
плавятся области, образованные складками, а затем плавление
продвигается в глубь кристалла Температура плавления кри-
сталлов может быть определена из соотношения
Тпл = 7Л1Л - Д//пяфкр )• (4,58)
где ог — поверхностное натяжение; &Нпя — удельная теплота плавления; I —
длина складки; рх₽ — плотность кристалла.
Равновесную температуру плавления можно определить из
соотношения T°n> = Д/^пл/Д5пл (где Д5„л — энтропия плавле-
ния) Ниже приведены значения Д5ПЛ и Т°п, некоторых
полимеров:
Т п,- к ДНП_. кДж/моль Дж/ (моль-К)
Полиэтилен 418 4,0 9,8
Полипропилен 445 10,9 24.3
Полистирол 513 8,4 18,3
Бутилкаучук 272 12,7 46,7
Полинзопрен
цис-1,4- 313 4,4 14,1
транс-\А- 347 12,7 36,9
транс-1,4-Полихлоропрен 353 8,8 23,5
Как видно из приведенных данных, низкие Т°Пл характерны
для гибких полимеров, энтропия плавления которых высока.
4.5.3. Влияние напряжения на кристаллизацию
При эксплуатации изделия из полимеров, как правило, нахо-
дятся в напряженном состоянии Рассмотренные выше законо-
мерности кристаллизации и плавления относятся к ненапря-
женным системам, однако напряжение в принципе нс изменяет
механизма кристаллизации. Кристаллизация, так же как и в не-
напряженном состоянии, включает стадии зародышеобразова-
ния и роста кристаллов. Кинетика первичной кристаллизации
описывается уравнением Авраами — Колмогорова, но констан-
та пк с ростом деформации уменьшается в основном за счет
ускорения процесса образования зародышей, о чем свидетель-
ствует уменьшение индукционного периода кристаллизации.
Связь скорости кристаллизации с напряжением описывается
экспоненциальной зависимостью.
*/1“<%ехр (—Во), (4.59)
где /у, и — полупериоды кристаллизации в напряженном и ненапряжен-
ном состоянии; о — напряжение, при котором происходит кристаллизация;
В — константа, зависящая от структуры полимера.
В координатах 1п — о эта зависимость описывается пря-
мой, по наклону которой можно найти В, а отрезок, отсекае-
мый на оси абсцисс, характеризует (рис. 4.19).
Поскольку при кристаллизации под напряжением изменяет-
ся нк, то следует ожидать и изменения морфологии кристаллов.
Действительно, при деформации, особенно при растяжении,
увеличивается доля кристаллов, образованных вытянутыми це-
пями, и соответственно снижается доля кристаллических обра-
зований со складчатой конформацией. Поэтому при кристал-
лизации предварительно растянутых полимеров образуется
структура типа «шиш-кебаб» (см. гл. 1).
Напряжение, приложенное к образцу, повышает и темпера-
туру плавления кристаллов, которые образуются в напряжен-
ном образце. Зависимость Тпл от напряжения описывается эм-
пирическим соотношением:
7’n.-i=T°n«exp (до), (4.60)
где Т^ил — значение Тг.л при о=0; а — константа, отражающая влияние на-
пряжения на Тпл и имеющая порядок 10_* МПа.
Значения а <и Т°„„ можно найти графическим методом, если
уравнение (4.60) представить в координатах 1g Т„п — а;
Т°пл, найденная этим методом, близка к Т°Пл, получаемой
экстраполяцией интервала плавления на нулевую его шири-
ну при 7’пл = 7'|<Р (см. рис. 4 19), и ее можно рассматривать как
равновесную температуру плавления кристаллов.
Повышение температуры плавления кристаллов наблюдает-
ся и при проведении кристаллизации под давлением:
7’пл=Г°пл exp (6р), (4 61)
где Т>„„ — температура плавления при давлении р—0; Ь — константа.
Давление применяют при получении монокристаллов, обра-
зованных выпрямленными макромолекулами. Высокое давле-
ние (500—1000 МПа) повышает Г™ и обеспечивает высокую
скорость кристаллизации при низких ДТ.
Кристаллизация полимеров, в частности эластомеров, мо-
жет развиваться уже в процессе деформирования аморфного
образца, например при его растяжении Если деформирование
проводить при ТКр>7’пл, т. е. из аморфного состояния, то по
мере увеличения степени растяжения Т„л повышается и при
каком-то значении е = еПл Тпя = ТКр По уравнению (4 60) можно
18*
275
найти значение о=аПл, при котором начинается кристаллиза-
ция при растяжении. Величина епл снижается с ростом Т°пл и
а.
Чаще кристаллизация при растяжении начинается при неко-
тором переохлаждении Д7\ и началу ее соответствует деформа-
ция е* = епл4-Де; ДГ и Де зависят от скоростей деформирования
и кристаллизации. Чем выше скорость растяжения, тем
больше ДТ и Де Поэтому существуют такие скорости, при ко-
торых е* достигает значения удлинения при разрыве и возмож-
ность кристаллизации при растяжении не реализуется. Этот
факт объясняет немонотонную зависимость прочности от ско-
рости деформации кристаллизующихся полимеров (см. гл. 5).
Повышение скорости кристаллизации полимера за счет струк-
турных факторов приводит к уменьшению ДТ и Де.
Процесс кристаллизации при растяжении является неизо-
тсрмическим, поскольку по мере деформирования растет ТПл
и, следовательно, степень переохлаждения ДТ. Кроме того,
процесс осложняется дополнительной ориентацией кристаллов,
образовавшихся в начальный момент кристаллизации. Поэтому
кинетика кристаллизации отклоняется от зависимости, выра-
жаемой уравнением (4.57).
Кристаллизация при растяжении имеет большое значение
для полимеров, которые при эксплуатации подвергаются дей-
ствию многократных деформаций растяжения — сжатия, по-
скольку она определяет такие важные свойства, как прочность,
упругость и гистерезис (см. гл. 5).
4.5.4. Влияние структуры полимера на кристаллизацию
Химическое строение полимера, его конфигурация, молекуляр-
ная масса оказывают существенное влияние на процесс крис-
таллизации, в частности на кинетику зародышеобразования и
роста кристаллов. Повышение энергии когезии полимера приво-
дит к уменьшению энергии зародышеобразования и к увеличе-
нию энергии роста кристалла. Определяющее влияние оказыва-
ет второй фактор, в результате чего общая скорость кристал-
лизации снижается. В качестве примера рассмотрим гибкие
полимеры:
Днметилвииялсилоксаи
цис- 1,4-Полибутадиен
цис-1,4 -Полиизопрен
К
193
218
248
G/2’ ыин
0,01—0,1
10—103
102—10*
Влияние заместителей особенно заметно у виниловых поли-
меров. Так, скорость кристаллизации полиэтилена сопоставима
со скоростью охлаждения от Тлп (410—414 К) до комнатной
температуры, и при этих условиях полимер практически пол-
276
ностью закристаллизован, т. е. является твердым телом. По-
лиизобутилен в этих условиях находится в высокоэластическом
состоянии и кристаллизуется крайне медленно даже при низ-
ких температурах (/i/4 при 248—233 К составляет 30 сут).
Увеличение молекулярной массы приводит к росту вязкости
системы, что равносильно росту переохлаждения, поэтом}' ско-
рость зародышеобразования повышается при снижении ско-
рости роста кристаллов. Зависимость скорости кристаллизации
от молекулярной массы имеет экстремальный характер Для
высокорегулярных гибких полимеров увеличение молекулярной
массы обычно ускоряет кристаллизацию, поскольку определяю-
щим является снижение энергии зародышеобразования за счет
большей вязкости высокомолекулярного образца Для менее
гибких полимеров рост молекулярной массы может оказать не-
гативное влияние на общую скорость кристаллизации из-за
снижения скорости роста кристалла вследствие высокой вяз-
кости полимера.
транс-Полимсры кристаллизуются значительно быстрее, чем
цме-полимеры, и имеют более высокие температуры плавления.
Рассмотрим это на примере полиизопренов. Так, цис- 1,4-поли-
изопрен при комнатной температуре кристаллизуется чрезвы-
чайно медленно (полупериод кристаллизации составляет не-
сколько суток) и деформируется как высокоэластический мате-
риал, а транс- 1,4-полиизопрен в этих же условиях представля-
ет собой практически полностью закристаллизованный матери-
ал. траяс-Йзомеры имеют более плотную упаковку кристалла
и более высокие температуры плавления.
Дальний порядок в присоединении звеньев определяет сте-
пень регулярности макромолекул. Следует сразу сказать, что
кристаллизация практически подавляется при содержании ано-
мальных звеньев 15—25%. Структурная и пространственная
нерегулярность отрицательно влияет на скорость кристаллиза-
ции, повышая энергию активации роста кристаллов. С увели-
чением степени нерегулярности снижается скорость зародыше-
образования, так как не достигаются необходимые для образо-
вания зародыша критического размера условия (ввиду малой
подвижности). Поскольку подвижность определяется и кинети-
ческой гибкостью, то отрицательное влияние нерегулярности
будет снижаться то мере роста гибкости полимеров. Так, при
увеличении доли звеньев транс-1,4 в цас-1 4-полибутадиене от
3 до 7% полупериод кристаллизации увеличивается с 0,05 до
б ч Для более гибких полимеров (полисилоксанов) небольшое
количество групп, отличных по строению от основной цепи, ус-
коряет кристаллизацию В области малых содержании (до
0,08) тип группы не влияет на кристаллизацию, а при большем
содержании аномальных звеньев кристаллизация замедляется
тем больше, чем выше объем и полярность группы.
277
Наибольшей скоростью кристаллизации характеризуются
линейные макромолекулы У разветвленных или сшитых поли-
меров кристаллизуются участки макромолекул, расположенные
между узлами сетки или сшивающими связями, поэтому от
числа сшивок зависит гибкость участков макромолекул и, сле-
довательно, скорость кристаллизации. Узлы сетки и сшивки
оказывают двойственное влияние на скорость кристаллизации:
с одно! стороны, они повышают вязкость системы, тем самым
ускоряя зародышеобразование, а с другой—снижают подвиж-
ность '.макромолекул, что приводит к уменьшению скорости
роста кристаллов. Поэтому зависимость общей скорости крис-
таллизации от числа узлов сетки имеет экстремальный харак-
тер. Максимум на кривой соответствует минимальной степени
-сшивания, вызывающей ускорение кристаллизации. Степень
сшивания определяется гибкостью полимера и тем меньше, чем
' гибкость выше. Для гибких полимеров (полисилоксаны) при
низкой плотност»! сшивающих связей наблюдается ускорение
процесса кристаллизации, для .менее гибких (полихлоропрен)
ускорение практически отсутствует. По мере увеличения числа
-сшивок скорость кристаллизации снижается, причем в разной
мере в зависимости от их структуры. Аналогично влияет и раз-
ветвленность макромолекул.
Таким образом, можно отметить общую закономерность:
любой тип нерегулярности (аномальные звенья, сшивки, раз-
ветвления) оказывает одинаковое влияние на скорость крис-
таллизации: при невысоком переохлаждении ускоряя зароды-
шеобразование и замедляя рост кристаллов, а при значитель-
ном — замедляя оба процесса. Вклад каждого из этих
процессов в общую скорость кристаллизации зависит от кине-
тической гибкости макромолекул. Скорость роста кристаллов
более чувствительна к конфигурации цепи, при этом замедляю-
щая роль аномальных звеньев больше, чем сшивок.
Вводимые в полимер пластификаторы и наполнители также
влияют на скорости зародышеобразования и роста кристаллов.
Пластификаторы, повышающие гибкость макромолекул, сни-
жающие число образующихся сшивок и увеличивающие свобод-
ный объем, действуют аналогично снижению степени переох-
лаждения, уменьшая скорость зародышеобразования и повы-
шая скорость роста кристаллов Поэтому зависимость обшей
•скорости от содержания пластификатора имеет экстремальный
характер с максимумом в области невысоких содержаний (не-
сколько процентов).
Наполнители повышают напряжение на границе раздела
полимер — наполнитель, тем самым ускоряя процесс зародыше-
образования и соответственно повышая общую скорость крис-
таллизации, но в ряде случаев (активные наполнители в среде
эластомеров) наполнитель увеличивает степень сшивания по-
278
лимера, вызывая замедление -кристаллизации за счет снижения
скорости роста кристаллов
Контрольные вопросы
1. В каких фазовых и физических состояниях существуют аморфные и кри-
сталлические полимеры? Влияет ли химическое строение, молекулярная
масса и конфигурация макромолекулы на характер термомеханической
кривой?
2. Охарактеризуйте стеклообразное состояние полимеров и процесс стекло-
вания. Зависит лн температура стеклования от скорости деформирования?
3. Температура стеклования полимера находится при 170 К» но он сохраняет
признаки, характерные для твердого состояния, вплоть до 273 К, затем
его деформируемость возрастает и не изменяется с температурой до тем-
пературы разложения (600—800 К). Какой процесс препятствует перехо-
ду полимера в высокоэластическое состояние? Почему этот полимер не
переходит в вязкотекучее состояние? Какова его структура? Какие мето
ды можно использовать для определения температуры стеклования?
4. Перечислите основные признаки высокоэластического состояния. Изменя-
ются ли внутренняя энергия и энтропия полимеров прн их деформирова-
нии в высокоэластическом состоянии? К чему приводит изменение внут-
ренней энергии при деформировании полимера в высокоэластическом со-
стоянии?
5. Охарактеризуйте вязкотекучее состояние полимеров. Каковы различия
в механизмах течения полимеров и низкомолекулярных жидкостей?
6. Зависит ли температура текучести от структуры полимера, напряжения
и скорости деформирования?
7. Полимеры цис-1,4-полиизопрен. сополимер бутадиена с нитрилом акрило-
вой кислоты (60 40). полиэтилен терефталат, полиэтилен при температуре
выше Тс растягивают, затем температуру образца доводят до комнатной
и снимают напряжение. Напишите формулы этих полимеров. Какие из
них кристаллизуются при растяжении? Какова конформация макромоле-
кул до деформирования, при ориентации, кристаллизации и после снятия
напряжения? При растяжении каких полимеров тепловыделение больше?
Какие полимеры и почему сохраняются в ориентированном состоянии
после снятия напряжения?
8. При переработке полимера с Л7ля200 600 в высокоэластическом состоянии
его молекулярная масса уменьшилась до 150 000 и 15 000. Как при этом
изменились температуры стеклования, текучести и интервал высокоэла-
стичности?
’ 9. Что такое релаксация? Перечислите типы релаксационных явлений.
Что такое спектр времен релаксации?
10. Полимер выше Тс характеризуется низкой скоростью ползучести. Как оп-
ределить. является ли причиной этого поперечное сшивание или кристал-
лизация? С помощью каких механических методов испытания можно
установить причину низкой ползучести?
Н. Каково воздействие радиации на релаксацию напряжения сетчатого и
линейного полимеров?
279
ГЛАВА 5
ФИЗИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
При эксплуатации полимерные изделия подвергаются действию
различных внешних полей: механических, тепловых, электриче-
ских, магнитных и т. д. Физические свойства, которые представ
ляют собой отклик на это действие, разделяют на .механичес-
кие, тепловые, электрические и др. Физические свойства по-
лимеров определяются их структурой, физическим и фазовым
состоянием в процессе эксплуатации. Установление связи
между структурой полимеров и их физическими свойствами
позволяет не только определить оптимальные условия их при-
менения, но и проводить направленный синтез новых полимер-
ных материалов с заранее заданными свойствами.
5.1 МЕХАНИЧЕСКИЕ СВОЙСТВА
Механические свойства определяют степень изменения структу-
ры, размеров, формы тела при воздействии на него механичес-
ких сил. В зависимости от величины и продолжительности
действия механических сил полимерные материалы подверга-
ются деформации или разрушению. Соответственно различают
деформационные и прочностные свойства. Деформационные
свойства характеризуют способность полимерных материалов
деформироваться под воздействием механических напряжений,
прочностные — способность сопротивляться разрушению.
Вследствие специфики строения макромолекул и надмолеку-
лярных структур механические свойства полимеров характери-
зуются рядом особенностей и сильно зависят не только от сос-
тава и строения полимера, но и от внешних условий. Работоспо-
собность полимерных материалов во многом определяется ре-
жимом их деформирования, прежде всего характером действия
внешних сил. Различают статические и динамические режимы
нагружения. К статическим относят воздействия при постоян-
ных нагрузках или деформациях, а также при небольших ско-
ростях нагружения, к динамическим — ударные или цикличес-
кие воздействия.
5.1.1. Деформационные свойства
Деформацией тела называется изменение его размеров, объема
и формы под влиянием температуры, внешнего механического
воздействия или внутренних сил. Деформация сопровождается
изменением структуры полимеров и их свойств: чем сильнее
деформация, тем значительнее изменение структуры и свойств
-280
—
Рис. 5.1 Различные виды деформации:
а — простой сдвиг; б — одноосное растяжение; в г — объемное деформирование при сжа-
тии (в) и растяжении (г) (сплошные линии — форма до деформаций; пунктир — после
деформации)
Степень деформации оценивается относительной деформацией е, равной
отношению абсолютной деформации АХ к размеру образца X до деформа-
ции: е=ДХ/Х.
Величина деформирующих сил характеризуется напряжением, т.е.-силой»
приходящейся на единицу площади сечения тела Различают истинное ое
и условное /е напряжения прн деформации:
Oe = P/Se; fe = />/So,
где Р— деформирующая сила; Se, So — площади поперечных сеченнй прн де
формации е и до нагружения.
Эти показатели связаны соотношением
Ce=fe(e+1)
Можно выделить три вида деформации: простой сдвиг, одноосное растя-
жение, всестороннее сжатие (или растяжение) (рнс. 5.1) При простом сдви-
ге деформирование происходит под действием тангенциальных (касательных)
напряжений от, действующих на поверхности образца При этом изменяется
форма образца, а объем остается постоянным. Деформация сдвига 7 опреде-
ляется тангенсом угла а при сдвиге верхней плоскости АВ относительно
нижней 00' в положение А'В\ т. е. тангенсом угла поворота а прямой ОА
(ОЛ — расстояния между плоскостями). Модуль сдвига G Gr/f. Скорость
сдвига i=d^/dt представляет собой величину, характеризующую изменение
деформации во времени
Одноосное растяжение происходит под действием нормальных напряже-
ний он, приложенных перпендикулярно поверхности образца. Прн этом наблю-
даются продольное растяжение еПроД и поперечное сжатие образца еп©пер-
Степень растяжения X равна отношению длин образца до (/о) и после (/)
растяжения: Х=///о.
Относительное удлинение при растяжении еПроД равно
еПрод=(/ — A//Z=X—1
Модуль при одноосном растяжении Е (модуль Юнга) равен
£=Ск/С«род.
Мерой поперечного сжатия при одноосном растяжении является безраз-
мерная величина — коэффициент Пуассона |Л
|Л"8пооер/епрод.
Обычно для эластомеров ц=0,48—0,49, для пластмасс ц=0,2—ОД
Если до деформирования образец имел размеры а, Ь, с, а после — х»
У> г, то степень деформирования составит
Х(-х/а; Х2-0/6;
13—Z/C.
281
Если объем тела не изменяется при деформировании, то abc=xyz
и 7,1X^X3== 1,
При всестороннем сжатии (растяжении) под действием нормальных на-
пряжений происходит изменение объема образца, а форма не изменяется.
В этом случае сжимающим напряжением является давление Р Объемная
деформация еов равна относительному изменению объема ДЕ/Е. Модуль все-
стороннего сжатия К равен:
К=— Р/(ДЕ/Е).
Модули при растяжении Е, сдвиге G и всестороннем сжатии К связаны
соотношением
Е=ЗК(1—2p)-2(l+fi)G
Податливость (величина, обратная модулю) при простом сдвиге равна
/ = l/G, при всестороннем сжатии В — 1/К при растяжении D=\/E.
Деформационные свойства полимеров обычно оценивают по
кривым напряжение — деформация (о—е). На рис. 5.2 приве-
дены диаграммы растяжения для различных полимеров. Нес-
мотря на разный характер кривых, на всех можно выделить,
нача )ьный участок до точки Я, где наблюдается линейная за-
висимость между о и е, т. е. выполняется закон Гука, выве-
денный для твердых тел: о —Ее,, где Е— модуль упругости (мо-
дуль Юнга), чистснно равный о при е = 1, т. е. при изменении
линейного размера в два раза. Напряжение, соответствующее
точке А, называют пределом упругости оУнр, а деформацию —
упругой Еупр
Рис. 5.2. Диаграммы растяжения оргстекла (/), капрона (2), эпоксидного
компаунда (3), полиэтилена высокой плотности 14) и наполненной рези-
ны (5)
Рис. 5.3. Кривые и напряжение—деформация о—е стеклообразных полиме-
ров
/ — полная кривая 2 3 —неполные (а, —предел вынужденной высокоэластнчности)
282
При дальнейшем нагружении при а>аупр (точка А) закон
Гука уже не выполняется. В общую деформацию таких систем,
кроме упругой составляющей еупр входят высокоэластическая
еюл и вязкотекучая еВг:
t = Еупр+ вс »Л + 8вт.
Относительный вклад каждого вида деформации определя-
ется условиями деформирования (температура, скорость), фи-
зическим и фазовым состоянием полимера и его структурой.
На ход кривой о—е сказывает влияние и релаксационный
характер деформации, наиболее ярко проявляющийся в высо-
коэластичсском и вязкотекучем состояниях. Как было показа-
но в гл. 4, релаксационный характер проявляется в отставании
деформации от напряжения при деформировании и наличии
остаточных деформаций после снятия нагрузки. Относительная
остаточная деформация равна еОст= (/—/о)/^о, где I — длина об-
разца после снятия нагрузки.
Иногда по величине еост полимеры делят на пластичные
и эластичные Пластичные сохраняют приданную им деформа-
цию (форму) после удаления деформирующей силы и еОСт==е,
эластичные — восстанавливают размеры и форму до началь-
ных. В этом случае еОСт-»-0. Скорость восстановления опреде-
ляется скоростью релаксации и зависит от структуры, физиче-
ского и фазового состояния полимера.
Деформационные свойства стеклообразных полимеров На
рис. 5.3 приведена характерная кривая (/) о—е стеклообразно
го полимера, а на рис. 5.4 схематически изображены стадии де-
формирования при растяжении. Процесс деформирования мож-
но разделить на три стадии. На стадии 1 до точки А полимер
Деформируется упруго, соблюдается закон Гука (напряжение
пропорционально деформации, форма образца практически не
изменяется). Деформационные свойства оцениваются модулем
упругости Е и упругой деформацией еупр. В точке А (см.
рис. 5.3) начинается стадия //, характеризующаяся сильным
растяжением образцов при незначительном росте напряжения.
Если сопоставить этот участок деформационной кривой с кри-
вой установившегося течения (см. рис. 4.8), то можно провести
аналогию между этими процессами. Поэтому деформацию
стеклообразного полимера на // участке называют «холодным»
течением. В начале этой стадии в точке В напряжение несколь-
ко снижается и образец утончается, возникает так называемая
«шейка» и дальнейшее растяжение образца происходит только
за счет удлинения «шейки» Длина «шейки» растет до тех пор,
пока она не распространится на всю растягиваемую часть об-
разца, причем толщина «шейки» на стадии // остается посто-
янной В точке С весь образец переходит в «шейку» и начи-
нается /// стадия растяжения. Образец в виде «шейки» растя-
283
Л1
М"
м
41
д
о
•с
У
F
г
1
3
н
tfV
Рис. 5.4. Садш, дефориировапи^о характерны релаксационные
стеклообразного полимера:
1 — образец до деформирования 2 — о6ра<,
зек деформируется упруго; 3 — образец вс ’
время образования «шейкн> </ стадия);-
4 5 —рост «шейки» (// стадия); 6 — весь
г-1/Сгх — —
(/// стадия)
7П7Г>
2
гптт
3
1
5
6
II
А
ш
напряжение в дефектном участке релаксирует и распределяет-
я на другие участки. Релаксации напряжения способствуют
*. микропустоты Таким образом, релаксационными явлениями
рисазс%зь₽сш|л_? «точка с 'нфюжно объяснить некоторое снижение а после точки А.
(///стадия) 7~яе*°Рмачия «шейки»! Нельзя не учитывать и изменение активационного барьера
[вращения атомов и их групп вокруг одинарных связей при
наложении напряжения. Прикладываемое напряжение умень-
шает потенциальный барьер, ограничивающий сегментальную
подвижность, и увеличивает вероятность движения в направле-
нии приложенной силы, т е. увеличивается вероятность течения
(«холодной» вытяжки) (см. гл. 4 и рис. 4.8).
Уменьшение потенциального барьера влечет за собой и ус-
корение релаксации Время релаксации т* определяется не
только температурой [см. уравнение (4 47) ] , но и напряжени-
ем:
гивается по всей длине равно-
мерно (вплоть до разрыва i
точке D), при этом напряже-
ния вновь возрастают практи-
чески пропорционально удли-
нению
Таким образом, для стадий
I и III характерна невысокая
обратимая упругая деформация. Деформация, достигнутая на
// стадии, сохраняется после снятия нагрузки, но она не явля-
ется истинно пластичной. Если образец с «шейкой» нагреть или
подвергнуть набуханию (т. е. ускорить релаксацию), то «шей-
ка» исчезнет и образец примет первоначальную форму. Это сви-
детельствует о том, что деформация на II стадии высокоэласти-
ческая. В отличие от обычной высокоэластичности, проявляет
мой при Т>-Гс (см гл. 4), подобная высокоэластичность при
Т<Т., для проявления которой недостаточно энергии тепловогя
движения, а требуется действие больших напряжений, называ т!
ся вынужденной высокоэластичностью, деформации — вынумя
денно-эластическими ев, а напряжение, при котором начина !
проявляться вынужденная высокоэластичность, — пределом вы!
нужденной высокоэластичности ав.
Кривая /, приведенная на рис. 5.3, является полной дефор!
манионной кривой стеклообразного полимера, однако часто об!
разец разрывается на II или / стадиях — в этом случае говоряя
о неполной деформационной кривой (кривые 2, 3).
Механизм образования «шейки», т. е. «холодной» вытяжки!
объясняется с различных точек зрения. На первой стадии уп!
ругое деформирование (по связям и углам) сопровождается
увеличением свободного объема Полимер по своей структуре
неоднороден, в нем существуют слабые (дефектные) и боле
прочные участки При нагружении дефекты структуры являют!
ся концентраторами напряжения, поэтому в полимере происхо!
дят локальные микроразрывы цепей, приводящие к образова-1
нию микротрещин и микропустот размером «2—20 нм, которые
способствуют увеличению свободного объема. I
Рост свободного объема обусловливает возможность изме!
нения конформации под действием напряжения и переход Я
ориентированное состояние, т. е. полимер Тс проявляет спо-Ц
собность к высокоэластическому деформированию, для которо-1
284
U0 — aat
= Biexp—----
(5.1)
(а—константа). При малых напряжениях (аа<§с!7о) урав-
нение (5.1) переходит в (4.47), описывающее релаксацию под
влиянием теплового движения В стеклообразном состоянии
Ua&kT >и величина т* велика Прн больших о (ао->1/0) вре-
мя релаксации снижается и макромолекулы способны изменить
конформацию, т. е способны проявить высокоэластическую де-
формацию.
После перехода всего полимера в ориентированное состоя-
ние (/// участок) в системе значительно возрастает уровень
межмолекулярного взаимодействия, т. е плотность физичес-
кой сетки, за счет сближения цепей (см гл. 1). Внутреннее
сопротивление деформированию у таких систем велико, и они
ведут себя как твердые тела.
Предел вынужденной эластичности ав снижается с ростом
температуры или с уменьшением скорости деформирования,
а величина деформации растет При снижении температуры на-
пряжение Ов, необходимое для реализации вынужденной вы-
сокоэластичности, повышается. При какой-то температуре
Ов достигает значения, при котором происходит хрупкое
разрушение полимера, т. е ов=охр. Эта температура назы-
вается температурой хрупкости Тхр. При этой температуре
кривая а—е становится неполной (см. рис 5 3, кривая 3) и
полимер разрушается хрупко: точки А и D совпадают; Тхр,
так же как и Тс, является неравновесной характеристикой, за-
висит от скорости нагружения и вида деформации (сжатие,
растяжение, сдвиг).
На рис. 5.5 представлены зависимости охр и ав от темпера-
'Уры для трех полимеров, имеющих одинаковую Гс. Полимеры
285
*1
тХр(2) TwPiWi) Mi.2.3) т
Рис. 55 Зависимость хрупкой
прочности Охр и предела вынуж-
денной высокоэластнчностн о, от
температуры (объяснения см. в
тексте)
1 и 2 характеризуются оди-
наковой Охр, но разным тем-
пом изменения ов с темпе-
ратурой [ (dcitd 7) 1 >
>(^,/<77)2] Поэтому
Тхр (I) выше ГхР (2). Поли-
мер 3 имеет такой же темп
изменения ов с температу-
рой, но меньшую Охр, поэтому Т'ХР(3) >ГХР(2). Как видно из
рис. 5.5, ДТв^Гс—ТХр зависит от разрушающего напряжения
при хрупком разрыве оХр и темпа изменения о8 с температурой
(dcJdT) при одинаковой Тс.
Рассмотрим в качестве примера полистирол (ТС=373К) и
пол и метил .метакрил ат (ГС = 383К). Из-за низкого значения охр
и большой скорости роста ов с Т полистирол имеет более высо-
кую Тхр и меньший интервал вынужденной высокоэластичнос-
ти ДТВ, чем полиметилметакрилат (рис. 56). Поэтому полисти-
рол нельзя использовать в изделиях, для которых требуется вы-
сокая стойкость к ударным нагрузкам (ударная вязкость). Для
расширения ДТВ полимеры модифицируют эластомерами, гиб-
кие макромолекулы которых способствуют ускорению релакса-
Рнс. 5.6. Зависимость хрупкой прочности Охр и прелела вынужденной высоко-
эластнчностн о» от температуры для полиметнлмегакрилата (/) и полисти-
рола (2)
Рис. 5.7. Зависимость хрупкой прочности Охр н средела вынужденной высо-
коэластнчностн
(М,<Мг<М>)
о, полимеров различной молекулярной массы от температуры
286
Таблица 5.1 Влияние структуры эластомера на Тхр и Д7"в
Эластомер ахр> МПа Тс, к гхр.к Д7-,-7-е-Тхр, К
цис-1 ,4-Полнбу тадиен 100 173 161 12
цис-1,4 Полнизопрен 100 211 193 18
Сополимер бутадиена со стиролом (70:30) 90 224 138 86
Сополимер бутадиена с НАК (60 : 40) 117 254 163 91
ционных процессов и сдвигают ний, повышая ударную вязкость. Тхр В сторону меньших значе-
Температура хрупкости снижается с уменьшением плотное-
ти упаковки и ростом молекулярной массы При рыхлой упа-
ковке макромолекул увеличивается свободный объем, релакса-
ция ускоряется, что приводит к уменьшению оь при той же
температуре. Это равносильно снижению daBldT Плотность
упаковки определяется наличием заместителей, их объемом и
расположением, например для карбоцепных эластомеров она
убывает в ряду цис-1,4-пол ибутадиен, ци с-1,4-пол и изопрен, со-
полимер бутадиена со стиролом, сополимер бутадиена с нит-
рилом акриловой кислоты (НАК); в этом же направлении уве-
личивается интервал вынужденной высокоэластичности
(табл. 5.1)
Молекулярная масса влияет на Тс, Охр и, следовательно, на
Тхр. Для полимеров, у которых Л4э>Л4сг, Тс практически от мо-
лекулярной массы не зависит, а Тхр снижается ввиду роста охр
(рис. 5.7), что приводит к повышению ДТВ (рис. 5.8).
Таким образом, деформационные свойства полимера в стек-
лообразном состоянии можно охарактеризовать модулем упру-
гости, пределом вынужденной высокоэластичности, его измене-
нием с температурой, величинами упругой и вынужденно-эла-
стической деформации.
Деформационные свойства полимеров в высокоэластичес-
ком состоянии. Как отмечалось в гл. 4, высокоэластическое со-
стояние является равновесным, но для его установления требу-
ется определенное время, т е. деформация в этом состоянии
Рис. 5.8 Зависимость тем-
пературы стеклования Ге
и температуры хрупкости
Тхр от молекулярной массы
(заштрихованная область —
область вынужденной высо-
коэластичности)
287
е
Рис. 5.9. Кривые напряжение — де-
формация о—е сшитого аморфиога
(/—4) и кристаллизующегося (5)
эластомеров:
I Л 4» 5— неравновесное деформирование
при скоростях vj>V2>v<; 3 — равновесное
дефор м Ярова н не
имеет релаксационный харак-
тер. Поэтому в реальных усло-
виях деформация является не-
равновесной. При статических
испытаниях деформационные-
свойства называют упруго-ре-
лаксационными, а при дина-
мических — упруго-гистере-
зисными
На рис 5.9 приведены типичные кривые о—е эластомера,,
полученные в равновесных (кривая 3} и неравновесных (кри-
вая 4) условиях. В сшитых химическими связями полимерах на
сетку этих связей накладывается флуктуационная сетка физи-
ческих связей, характеризующаяся существенно меньшей энер-
гией. В первый момент деформирования (участок I кривой 4)>
внутреннее сопротивление системы определяется общим числом
связей (химических и физических) и напряжение резко воз-
растает. Деформация имеет преимущественно упругий харак-
тер По мере дальнейшего деформирования узлы флуктуацион-
ной сетки распадаются (поскольку напряжение в системе
соизмеримо с энергией физических связей), при этом макромо-
лекулы легко изменяют конформацию, ориентируясь в направ-
лении действия силы, и напряжение изменяется незначительно
(участок //) Но ориентация, в свою очередь, приводит к росту
числа узлов, образующихся между ориентированными макро-
молекулами и, следовательно, к росту напряжения (/// учас-
ток), темп которого зависит от плотности физических сшивок,
образующихся в условиях ориентации Наиболее резко увели-
чивается а с ростом е для полимеров, кристаллизующихся при:
растяжении (см. рис. 5.9, кривая 5).
Степень перестройки структуры за время деформирования:
определяется скоростью релаксации и условиями деформиро-
вания. Чем меньше время релаксации [а оно снижается с рос-
том гибкости и температуры, см. уравнение (4 47)], тем боль-
шая часть макромолекул примет равновесные конформации и-
больше разрушится флуктуационных связей за время деформи-
рования, т. е. напряжение уменьшится. Поэтому кривая о—е в-
равновесных условиях (кривая 3 на рис. 5.9) лежит ниже кри-
вой о—в, полученной в неравновесных условиях (кривая 4)..
Повышение скорости деформирования «отодвигает» систему от
состояния равновесия тем больше, чем выше скорость, и при.
288
(-*0, т. e. при x*/f-*oo (кривые 1, 2), полимер уже неспособен
к высокоэластической деформации.
Отктонение от равновесной кривой на участке II при оди-
наковых внешних условиях определяется структурой полимера.
Повышение полярности и молекулярной массы обусловливает
большие значения а при той же степени деформации вследст-
вие большей плотности физических узлов, и отклонение от рав-
новесных значений возрастает.
Равновесные условия обеспечивают за время деформирова-
ния максимально возможную релаксацию структурных единиц
(звеньев, сегментов, надмолекулярных структур и т. д.). Это,
как правило, достигается при повышении температуры, сни-
жении скорости деформирования, а также при набухании в
растворителе или пластификаторе, которые повышают подвиж-
ность структурных единиц. Но практически полной релаксации
добиться невозможно, так как в спектре релаксации есть очень
большие времена, и поэтому говорят об условно-равновесных
условиях.
Общая деформация системы е<» в этих условиях включает
упругую еУпр и высокоэластическую еВэл составляющие, причем
еупр<ев».кю Для идеальных сеток еВт=0, а для реальных, хотя
8Вт>0, но поскольку евт<Овэл, этой величиной обычно пренеб-
регают; поэтому Воо=евэлоо.
Для расчета модуля упругости используют эмпирические
уравнения Бартенева
a«“G(<>e«>=Go»(X—1) (5.2)
(где о», б». — условно-равновесные значения напряжения и мо-
дуля сдвига) .и Муни:
о« = 2С,(Х» —Л-')4-2С2(Х —Х-а) (5.3)
(где С| и С2 — константы).
В условно-равновесных условиях вклад физических связей
в константу Ct невелик, а константа С2 очень чувствительна к
наличию флуктуационной сетки и характеризует степень откло-
нения от статистической теории высокоэластичности. Как пра-
вило, С2 повышается по мере роста числа физических связей,
т. е. по мере отклонения от равновесных условий. Любой про-
цесс, сопровождающийся их уменьшением, например набуха-
ние, пластификация, приводит к снижению С2 Прн С2=0 урав-
нение (5 3) переходит в (4 31), описывающее деформацию
идеального каучука, ги 2Ci = Goo. Поэтому константа Со харак-
теризует упругие свойства полимера в высокоэластическом со-
стоянии.
Таким образом, деформационные свойства в равновесных
условиях характеризуются величиной высокоэластической де-
формации и условно-равновесным модулем высокоэластичности.
19—816
289
Рис. 5.10. Определение начального и секущего модулей по кривой напряже-
ние — удлинение
Рис. 5 11 Кривые напряжение — деформация а—е сшитого аморфного поли-
мера прн растяжении (/, 2) н сокращении (5, 2) в неравновесных (/, 3)
к равновесных (2) условиях (стрелки указывают направление деформации)
Для неравновесных условий
вулр^ввэл»
О= Оупр+ Ов»л,и>
Оупр<СОвм» (Увэл,м> Овэл.оо-
Поэтому неравновесный модуль упругости в высокоэластичес-
ком состоянии Евэл больше, чем в равновесных условиях Еа>.
В общем случае для неравновесных условий
Ев»з Еа>-}-Ех', бвэл = боо + От,
где Gt, Ех— релаксационные части модулей сдвига и растяжения определяе-
мые степенью релаксации за время деформирования; Gx и Ех зависят от
температуры и скорости деформирования; с ростом температуры и сниже-
нием скорости Ех уменьшается н прн о—>-0 GBM=Ga>.
Упруго-релаксационные свойства в этих условиях характе-
ризуются начальным Ен, мгновенным Ех и секущим Ес модуля-
ми, определяемыми, например, при растяжении. Начальный
модуль Еи — это тангенс угла наклона а прямой о—е (рис.
5.10) на начальном участке (до точки А, см. рис. 5.2)
Ец = Оупр/ Вупр.
Мгновенный модуль характеризует упруго-релаксацион-
ные свойства в любой точке i кривой о—е и равен тангенсу угла
наклона касательной в этой точке, т. е. da/de.
Секущий модуль Ес представляет собой отношение напряжен
ния о,х к удлинению в! и характеризуется тангенсом угла Р
(см. рис. 5.10):
£e=Oej'8b
290
Секущий модуль изменяется по мере деформирования и
только при переходе в область III (см. рис. 5.9) становится
постоянным. Иногда для характеристики упругорелаксацион
ных свойств используют значение напряжения при определен-
ном удлинении, чаще всего при е = 300% (озоо)-
Неравновесный характер деформирования полимеров в вы-
сокоэластическом состоянии определяет явления размягчения
и гистерезиса. Гистерезис — это отставание во времени реак-
ции полимера на изменяющееся внешнее воздействие. Напри-
мер, под влиянием растягивающего напряжения деформация—-
реакция на механическое воздействие — отстает от напряжения.
Рассмотрим явление гистерезиса при растяжении полимера в
статических неравновесных условиях. Растянем образец элас-
томера, макромолекулы которого сшиты редкими химическими
связями, до удлинения е( (точка А на рис. 5 11) и затем со-
кратим его с той же скоростью до а=0. Кривые растяжения
ОА и сокращения АО' не совпадают, образец полностью не со-
кращается, а имеет остаточную деформацию еост=е2.
При растяжении макромолекулы полимера, находящиеся в
конформации свернутых клубков, ориентируются в направле-
нии растяжения. Флуктуационная сетка физических узлов за-
трудняет достижение равновесной конформации, соответствую-
щей напряжению в данный момент, и поэтому реальная дефор-
мация при растяжении е,₽ будет меньше равновесной ей». Та-
ким образом, деформация будет отставать от напряжения,
и кривая растяжения 1 будет лежать выше равновесной кри-
вой 2 (см. рис. 5.11). После снятия нагрузки в точке А обра-
зец начинает сокращаться. Макромолекулы стремятся перейти
из ориентированной неравновесной конформации в конформа-
цию свернутого клубка. Однако достижение равновесной кон-
формации задерживается, так как наличие флуктуационной
сетки тормозит релаксацию структурных единиц. В результате
этого реальная деформация при сокращении е(с будет больше
равновесной е1Оо, а кривая сокращения 3 расположится ниже
равновесной. По мере приближения к равновесным условиям
кривые растяжения и сокращения сближаются, а при условии
практически полного протекания релаксационных процессов за
время деформирования сольются и полимер не будет иметь
остаточной деформации.
Таким образом, при растяжении полимера в неравновесных
условиях наблюдается явление механического гистерезиса, про-
являющегося в отставании деформации от напряжения.
Рассмотрим явление гистерезиса с точки зрения термодина-
мики. Работа А, затрачиваемая на деформирование, возвраща-
ется полностью при упругой деформации и частично превраща-
ется в тепло (Q) при высокоэластической. Поэтому для упру-
гой деформации Араст=АсОкр» а для высокоэластической
19*
291
Драст=ЛСокр+Q. Мерой работы растяжения образца до точки
Л является площадь под кривой о—е (ОЛщ, см рис. 5.11):
ех
Лраст= ode;
ае,= (P/St) (dl,/lQ)=Pdli/V=dA/V, (5.4)
где Р — деформирующая сила; Se — площадь поперечного сечения при де-
формации er, I —исходная длина образца, d/>— приращение длины образца
при деформации; V—объем образца.
Таким образом, Лраст — работа растяжения, приходящаяся
на единицу объема эластомера. При сокращении образца ра-
бота сокращения равна
ei
Лсокр = J ode = (P/V) (dlt - dl2) (5 5)
(dl2— приращение длины образца после сокращения).
Графически Лсокр представляет собой площадь под кривой
О'Ае-2. Как видно из уравнений (5 4), (5.5) и рис. 5 11, при
растяжении затрачивается большая работа, чем при сокраще-
нии Это означает, что в цикле растяжение — сокращение необ-
ратимо теряется часть работы, затраченной на деформирова-
ние, пропорциональная площади под петлей (называемой пет-
лей гистерезиса), ограниченной кривыми ОА и АО' и осью абс-
цисс Эту часть работы называют механическими потерями
(Лмп):
е«
Лмп = J ode.
о
Потери механической энергии происходят при превращении ее
в теплоту, поэтому HMn = Q.
Величина механических потерь зависит от условий дефор-
мирования. Повышение температуры и снижение скорости при-
ближает условия деформирования к равновесным, что приводит
к уменьшению площади петли гистерезиса и, следовательно,
механических потерь. При низких температурах или высоких
скоростях за время деформирования структура изменяется не-
значительно и за время сокращения способна отрелаксировать
практически полностью. Поэтому в данных условиях кривые
растяжения и сокращения совпадают. Таким образом, в усло-
виях, обеспечивающих полную релаксацию, механические поте
ри отсутствуют
Размягчение полимера, или эффект Патрикеева — Маллин-
за, заключается в снижении напряжения при повторных дефор-
мациях (рис. 5 12). Это обусловлено неполным восстановленн-
292
рис. 5.12. Кривые напряжение — дефор-
мация о—е при растяжении (1 2) и со-
кращении Г, 2' прн повторных нагру-
жениях:
/( г — первый цикл; 2, 2' — второй цикл
ем структуры к началу повторно-
го деформирования из-за наличия
флуктуационной сетки. Чем
больше узлов в сетке, чем мед-
леннее идет релаксация в процес-
се деформирования, тем в боль-
шей степени проявляется эффект
размягчения Количественно эф-
фект размягчения оценивают коэффициентом размягчения
(N — порядковый номер повторного цикла растяжения — сокра
щения), равным отношению максимального напряжения при
повторном нагружении к его первоначальному значению при том
*же удлинении Например, коэффициент размягчения при втором
цикле растяжения — сокращения равен ц.О2=О2/щ.
Из рис. 5.12 видно, что площадь петли гистерезиса и оста-
точная деформация за второй цикл растяжения — сжатия мень-
ше, чем механические потери в первом цикле. При последую-
щих циклах механические потери, остаточная деформация и
коэффициент размягчения снижаются и в конечном итоге по-
тери и остаточная деформация достигают предельных значений
и при дальнейшем деформировании практически не изменяются,
а коэффициент размягчения становится близким к единице.
Причина этого в том, что релаксация физических узлов, надмо-
лекулярных образований и других структурных элементов про-
исходит крайне медленно; они разрушаются в первые циклы и
не восстанавливаются за время сокращения. После определен-
ного числа циклов в полимере сохраняется флуктуационная
структура, обладающая способностью практически полностью
релаксировать за время деформирования. Поскольку релакса-
ция ускоряется при нагревании, повышением температуры мож-
но уменьшить вост. Если при деформировании полимера не про-
исходит необратимого изменения его структуры (течения, меха-
нохимической деструкции), то остаточная деформация может
исчезнуть, т. е Вост-^0.
В реальных условиях деформирование всегда сопровожда-
. ется механохимическими реакциями, приводящими к необрати-
мому изменению структуры полимера В этом случае остаточ-
ная деформация складывается из обратимой («задержанной»)
высокоэластической; обусловленной наличием структурных еди-
ниц с большими временами релаксации Девэя, и пластической
293
Депл, связанной с необратимой вязкотекучей деформацией. По
этому
8ост=Д8 »Л+Д8пд.
Особенно велика доля Депл при больших деформациях i
цикле. (>100%), а также для полимеров с редкой химическо(
пространственной сеткой или не имеющих такой сетки. Любо-
внешнее воздействие, ускоряющее релаксацию (нагрев, набу
хание), приводит к снижению только Дев>л. При достаточн
длительном восстановлении Девэл-»-0 и вост определяется толью
пластической составляющей.
Прн эксплуатации полимерные изделия чаще всего испыты
вают невысокие деформации (<100%) и, как правило, рабо
тают в режиме динамического нагружения. Наибольшее расп
ространение получил режим деформирования при сдвиге, npi
котором напряжение и деформация изменяются во времен]
t по синусоидальному закону:
a=0osinw/; (5.6) e=8osincitf, (5.7
где а0> «о — максимальные амплитуды напряжения н деформации; о — круго
вая частота, связанная с периодом колебания v соотношением <o=2n/v.
Как указывалось в гл. 4, полимеры в высокоэластиЧескоп
состоянии являются вязкоупругими жидкостями. При деформи
ровании упругого тела напряжение и деформация изменяются
по синусоиде без отставания синусоид по фазе (рис. 5.13, а),
так как упругое тело мгновенно реагирует на внешнее воздейст-
вие. В этом случае модуль упругости по закону Гука равен
G=o/e=(oosi.n <o/)/(eosin со/) = о0/ео (5.8)
Деформация идеальных жидкостей изменяется с напряжением
по закону Ньютона
o=T](de/d/). (5.9)-
Здесь г] — коэффициент пропорциональности, называемый
вязкостью Для синусоидально изменяющегося напряжения
o = 0osinw/ деформация вязкой жидкости равна
e = mTcos u)/ = ^sin(w/ —л/а)- (5.10)
Таким образом, прн приложении синусоидально изменяюще-
гося напряжения деформирование жидкости также происходит
по синусоидальному закону, но синусоида деформации отстает
от синусоиды напряжения на угол л/2 (см. рис. 5.13,6). Для
вязкоупругих тел также наблюдается отставание деформации
от напряжения, но угол сдвига фаз больше нуля, но меньше
л/2 (см. рис. 5.13, в) Поэтому в общем случае для упругих,
294
Рис. 5.13. Зависимость напряжения о и деформации в прн динамическом
нагружении в режиме синусоидальных колебаний от частоты деформаций для
упругого (а), вязкого (б) и вязкоупругого (в) тел
вязких и вязкоупругих тел уравнения (5.6) .и (5.7) будут иметь
вид
o—Oosin®/; (5.11) e=-ecsin(titf—б), (5.12)
где б — угол сдвига фаз; для упругих тел 6=0°, для вязких 6=л/2, для
вязкоупругих 0<б<л/2.
Отставание деформации от напряжения в этом режиме, так
Же как и в статическом, является следствием заторможенности
протекания релаксационных процессов
Упруго-гистерезисные свойства вязкоупругих полимеров
при деформировании их в синусоидальном режиме оценивают-
ся комплексным динамическим модулем:
G*=o(Z)/e(0 = (oosin ®/)/[еоsin (со/—б)]. (5.13)
295
Испольгзуя прием описания колебательных процессов с по-
мощью комплексных чисел, можно записать:
а
e=e'+ie", (514)
где е' — действительная, a ie" — мнимая часть деформации; вектор е' совпа-
дает по фазе с вектором напряжения, а вектор it," отстает от напряжения на
угол л/2.
Согласию (5.13)
G*=tr/e'+a/ie"- G'+iG", (5.15)-
где G'—действительная, a G" — мнимая часть комплексного модуля; G' —
отражает упругие свойства, поскольку напряжение и деформация совпадают
ио фазе, a G;" — вязкостные, так как деформация отстает от напряжения на
угол л/2 [см . уравнения (5.10)].
При динамическом режиме нагружения, так же как и при
статическом, имеет место механический гистерезис, называе-
мый динамическим. Механические потери работы за цикл де-
формирования составляют
dA = л/ово sin 6. (5.16)-
Поскольку sin6=(7"/G*, а /о = 6*ео, то
dA-n&oG". (5.17)
Если принять максимальную амплитуду ео за единицу, то
Таким образом, мнимая часть модуля характеризует меха-
нические потери работы за цикл, поэтому G" называют моду-
лем потерь.. Поскольку G' является мерой упругости и характе-
ризует количество накопленной при деформировании и отдан-
ной при” разгружении энергии, то G' называют модулем
накопления. Гистерезисные свойства, т. е. механические поте-
ри, при динамическом нагружении характеризуют также углом
б (sin 6 = G''/G*, lgf>=G"/G') и площадью петли гистерезиса,
(рис. 5.14).. Ее строят на основании уравнений (5.11) и (5.12)
при изменении со/ от 0 до 2л.
Модули накопления G' и потерь G" зависят от температуры
и скорости деформирования (рис. 5.15). Гистерезисные (G" и
tg6) потерн минимальны при высокой температуре и медлен-
ной деформации, т. е. в условиях, когда перестройка структу-
ры, успевает произойти за период деформирования, т. с.
т*(о<^1. В этих условиях G невысок и равен равновесному мо-
дулю высокоэластичности. Потери минимальны также при
низкой температуре и быстрой деформации, т. е. в условиях,
когда проявляется упругая деформация Этому состоянию со-
ответствует условие т*<1>^>1 и высокое значение G', характер-
ное для твердых полимерных тел. Гистерезисные потерн мак-
симальны при промежуточных значениях температуры и ско-
296
Рис. 5Л4. Петля динамического ги-
стерезиса прн симметричном синусои-
дальном цикле деформирования G'
н G" — .модули накопления и потерь;
«о» — максимальные амплитуды
деформации и напряжения; 6—угол
сдвига фаз):
/ — со/—О, п—0, е-—Со sin 6; 2— С-
—Go sin б—G"e°. е—0; 3 — л/2, с—а0,
е—во cos б; 4 — о*— л/2+б^с-Оо cos 6-*G'e*
e*e<j 5 — со/*я, о—0, е-& sin б
рости деформирования, когда продолжительность действия
внешней силы сравнима с временем релаксации системы, т. е.
т*<о=1.
Динамический модуль зависит и от амплитуды деформации
со'
C* = G0*e0-ne, (5.18)
где Go* — динамический модуль прн 8о=1; л» — коэффициент, значения ко-
торого находятся в пределах от 0 до 0,6 и определяются плотностью физи-
ческой сетки.
Температурная зависимость деформационных свойств
аморфных полимеров довольно чувствительна к изменению та-
ких структурны^ параметров, как молекулярная масса, число
химических сшивок между молекулами, полярность, к наличию
.добавок (пластификаторов, наполнителей). Повышение молеку-
Рис. 5.15. Зависимость модуля накопления 6' (/). модуля потерь G" (2) и
тангенса угла 6 («?) от температуры (а) и произведения скоростей релаксации
и деформирования т*<о (б)
297
Pi:c. 5.16. Влияние молекулярной массы на температурную зависимость мо-
дуля накопления С' и модуля потерь G" (Мх>Мг>М3)
лярной массы линейных полимеров практически не оказывает
влияния на модули накопления и потерь при Т^.ТС и на рав-
новесный модуль высокоэластичности, но с ростом Мп увеличи-
вается плато высокоэластичности ДТВ, т. е. расширяется интер-
вал работоспособности в высокоэластическом состоянии. Ве-
личина потерь при этом уменьшается (рис 5.16).
Химическое сшивание, так же как и молекулярная масса,
не оказывает влияния на модули G' и G" при ГсТс, но при
Т>ТС заметно повышает модуль накопления G' и снижает гис-
терезисные потери (модуль G") за счет уменьшения доли вы-
сокоэластической деформации. Относительное удлинение при
разрыве с ростом числа сшивок пс снижается (рис. 5.17).
Равновесный модуль сдвига согласно кинетической теории
высокоэластичности связан с молекулярной массой М„ полиме-
ра и молекулярной массой между узлами сетки Мс соотноше-
нием
~гг pRT / 2Мс \
С"'Г’Т7'-тг. ‘519>
г о \ /
где г2о, г2—среднеквадратичные расстояния между концами свободно-сочле-
ненной цепи и между концами цепи сшитого полимера; р — плотность поли-
мера; R — газовая постоянная; Т — температура.
Отношение Мс/Мп учитывает эффект концов цепей, не уча-
ствующих в образовании сетки. Для реальных полимеров с до-
статочно высокими стопенями сшивания этим членом обычно
пренебрегают. Если учесть, что г2/го2«1, то уравнение (5.19)
можно записать в виде
G« .(p/Mc)/?T (5.20)
Полярность полимера определяет уровень внутри- и межмо-
лекулярного взаимодействия, т. е. плотность флуктуационной
сетки. При 7’<ТС модуль накопления G' обычно возрастает с
ростом полярности и плотности упаковки макромолекул. По-
скольку .плотность упаковки, как правило, снижается с ростом
298
Рис. 5.17. Влияние числа узлов лс на температурную зависимость модуля
накопления С', модуля потерь G" и относительного удлинения при раз-
рыве ер:
/ — линейный несшитый полимер; 2, 3 —сшитые полимеры; лс (3)>лс (2)
полярности, то повышение G' за счет увеличения полярности
часто нивелируется вследствие снижения плотности. Для мало-
полярных органических полимеров модули упругости довольно
близки. Наибольшее влияние полярности полимера проявляет-
ся в области температур выше Тс Увеличение полярности, оце-
ниваемой параметром растворимости (т. е. рост плотности
флуктуационной сетки), приводит к повышению G' и гистере-
зисных потерь (рис. 5.18) Поэтому неполярные гибкие поли-
меры характеризуются низкими потерями и теплообразованием
при эксплуатации в неравновесных условиях многократных де-
формаций. В равновесных условиях потери и модуль не за-
висят от полярности полимера, а определяются лишь числом
химических связей.
Наполнители и пластификаторы, вводимые в полимер, вли-
яют на число химических связен, слабых межмолекулярных за
цеплений и на изменение подвижности макромолекул, которая
определяет время их релаксации. Пластификатор сметает пе-
региб на кривой модуль упругости — температура и пик потерь
в сторону низких температур и расширяет область максимума
Т*ис. 5.18 Влияние параметра растворимости бг полимера на температурную
зависимость модуля накопления G' и модуля потерь G"; бп<бТ2<6Гз
299
I}S
Рис. 5.19. Влияние пластификаторов на температурную зависимость модуля
накопления G' и tg 6 сшитых непластифнцнрованного (/) и пластифициро-
ванного (2) полимеров
потерь при стекловании (рис. 5.19). Аналогичный эффект до-
стигается при введении в жесткий полимер гибких звеньев.
Наполнители повышают число химических связей и слабых ад-
сорбционных, причем повышение доли последних более сущест-
венно. Особо заметное влияние оказывают наполнители при
Т>Т<. Они, как правило, сдвигают точку перегиба на кривой
•6'=f(T) в сторону высоких температур, несколько повышают
модуль высокоэластичности в равновесном состоянии и сущест-
венно увеличивают механические потери.
Таким образом, для оценки деформационных свойств поли-
меров в высокоэластическом состоянии необходимо определять
их в равновесных и неравновесных условиях. В равновесных
условиях деформационные свойства характеризуются равновес-
ным модулем высокоэластичности. Релаксационный характер
высокоэластической деформации определяет явления гистерези-
са и размягчения. Упругорелаксационные свойства (при стати-
ческих режимах испытания) оценивают начальным и секущим
модулями, а также величиной механических потерь за цикл,
упругогистсрезисные свойства (при динамических режимах) —
комплексным динамическим модулем, представляющим собой
сумму модуля накопления (упругие свойства) и модуля потерь
(гистерезисные свойства). Гистерезисные свойства оценивают
также величиной динамических потерь, углом сдвига фаз меж-
ду напряжением и деформацией или любой его тригонометриче-
ской функцией.
Деформационные свойства полимеров в вязкотекучем со-
стоянии. Деформационные свойства полимеров при течении
называют реологическими (от греческого слова рео — течь). Рео-
логические свойства характеризуются кривыми течения, пред-
ставляющими собой зависимость вязкости от скорости дефор-
300
манин 7 и напряжения, вызывающего течение. В гл. 4 было
показано, что течение характерно для полимеров, находящих-
ся в вязкотекучем- состоянии. Такие системы называются рос-
плсизами Кроме расплавов в промышленности широко исполь-
зуются и растворы полимеров различной концентрации. Законы
течения как расплавов, так и растворов полимеров отличаются
от соответствующих законов для низкомолекулярных жидкос-
тей.
Полимеры в вязкотекучем состоянии могут испытывать три
вида деформации: сдвиг, одноосное растяжение и объемное
сжатие Наиболее изучено течение полимеров при сдвиге. Пе-
ремещение элементов тела осуществляется под действием тан-
генциальных напряжений от со скоростью ^ = d^ldt (где 7 — де-
формация, t — время) Сдвиг является основным видом дефор-
мирования при переработке полимеров на смесительном обо-
рудовании (смешение с ингредиентами, вальцевание и др ).
Одноосная деформация является определяющей при производ-
стве волокон, а при получении пленок большой вклад вносит и
двухосное растяжение.
При ламинарном течении жидкости в трубе ее слои, распо-
ложенные в центре, движутся быстрее, чем расположенные бли-
же к стенке трубы. Массу движущейся в трубе жидкости мож-
но представить бесконечным множеством тонких параллельных
слоев, соприкасающихся друг с другом, подобно коаксиальным
цилиндрам. Каждый слой движется со своей постоянной ско-
ростью, отличной от скорости соседних слоев. В случае стацио-
нарного течения с постоянным градиентом скорости (эквива=
лентным скорости деформации) тангенциальное напряжение
сдвига От в соответствии с законом Ньютона пропорционально
скорости сдвига 7:
От=Т]Г. п=огт/Т
(где т] — вязкость). Жидкости, подчиняющиеся закону Ньюто-
на, называются ньютоновскими. Вязкость ц ньютоновских жид-
костей не зависит от скорости деформирования, т е. от гради-
ента скорости 7.
Вязкость характеризует внутреннее сопротивление системы
сдвигу, или внутреннее трение. Она изменяется в широких пре-
делах: от 10 2 до 1012 Па с; для полимеров характерны высо-
кие значения вязкости — от 103 до 1012 Па-с.
Вязкость полимеров зависит от 7 и от, причем связь между
1^7 и 1g От является характеристикой конкретной системы Гра-
фическое изображение зависимости 1g 7—1g от называют крн-
4Ь1ми течения.
301
Рис. 5.20. Кривая течения полимеров
нии (6)
(а) и изменение вязкости прн тече-
На рис. 5.20,а 'представлена типичная кривая течения поли-
меров (в установившемся режиме). Она имеет S-образный ха-
рактер, и на ней можно выделить три участка:
/—в области низких скоростей сдвига выполняется закон
Ньютона; вязкость в этой области наибольшая и равна rjo
(рис. 5.20,6), поэтому ее называют областью наибольшей нью-
тоновской вязкости;
// — зависимость от от у нелинейна; это область аномалии
вязкости; для описания зависимости ат от ] в этой области
предложены различные формулы; наибольшее распространение
получило эмпирическое уравнение, которое для простого сдви-
га имеет вид
От“П>фКя> (5.21)
где Цаф — коэффициент вязкости, который называют эффективной вязкостью;
л — индекс течения, характеризующий отклонение зависимости От“/(1) от
закона Ньютона.
///— при больших скоростях сдвига зависимость от=/(*)
имеет опять линейный характер; это область постоянной наи-
меньшей ньютоновской вязкости.
В условиях установившегося течения макромолекулы дефор-
мируются и ориентируются в направлении действия силы. На-
ряду с пластической деформацией, сопровождающейся переме-
щением центра тяжести макромолекулы, определенный вклад в
общую деформацию вносит и высокоэластическая, вызывающая
упругое деформирование макромолекулы. Высокоэластическая
деформация обусловлена гибкостью макромолекул, наличием
302
сетки из физических узлов и имеет релаксационный характер.
Полимеры по своей структуре неоднородны, и каждая струк-
турная единица характеризуется своим временем релаксации
т,*. В целом для полимера характерен спектр> времен релакса-
ции со средним временем релаксации т*.
В области /, т. е. в области низких значений От и у, время
эксперимента <я= 1/f = 'Т“‘ намного больше т;* (т*</д) и мак-
ромолекулы успевают принять равновесную для От конформа-
цию. Между структурой полимера в момент / и структурой в
момент /-+Д/ существует динамическое равновесие. Поэтому
иногда считают, что структура <полимера при течении в облас-
ти / реологической кривой не изменяется.
Поскольку в этих условиях разрушается большая часть уз-
лов флуктуационной сетки, препятствующих Перемещению цент-
ра тяжести макромолекулы, то реализуется большее число пе-
рескоков сегментов и полимер характеризуется высокой вяз-
костью, которая не изменяется до тех пор, пока выполняется
условие т*</д.
С увеличением скорости деформирования время действия
силы /д приближается к времени структурной релаксации. При
условии т*>/д кинетическая единица исключается из течения,
так как за время действия силы не успевает произойти переме-
щение центра тяжести. В первую очередь из процесса течения
исключаются крупные структурные единицы с большими вре-
менами релаксации (например, надмолекулярные образования,
макромолекулы большой длины и т. д.) Поэтому течение осу-
ществляется более подвижными структурными единицами с
малыми временами релаксации. Это влечет за собой снижение
энергии активации перескоков сегментов и, Следовательно, сни-
жение вязкости. Динамическое равновесие сдвигается в
сторону разрушения структуры. При этом Плотность флуктуа-
ционной сетки уменьшается
С этих же позиций объясняют и явление тиксотропии, ха-
рактерное для полимеров. Суть этого явления заключается в
изменении свойств, например снижении вязкс)СТи под действием
механического воздействия, и обратимом их восстановлении при
снижении интенсивности воздействия или при его полном пре-
кращении. Известно, что под действием теплового движения
макромолекула принимает конформацию, равновесную для дан-
ного состояния. При полном прекращении действия силы эти
равновесные конформации приближаются к первоначальным,
образующим флуктуационную сетку с перв--1ачальной плот-
ностью узлов. Поэтому говорят, что восстанавливается исход-
ная структура. Поскольку крупные структурные образования в
полимерах имеют большие времена релаксации, то на их вос-
становление требуется значительное время.
303
В наполненных системах тиксотропные свойства проявляют-
ся сильнее. Это вполне естественно, поскольку структуры напол-
нитель— полимер в большинстве случаев отличаются меньшей
подвижностью, т. е. для них аномалия вязкости проявляется в
большей степени. Ввиду больших т* восстановление структуры
происходит медленнее.
Последовательное исключение из процесса течения различ-
ных по подвижности единиц по мере роста 7 уменьшает долю
макромолекул, способных к перемещению При условии т*>/д
расплав полимера переходит в квазисшитое состояние, для ко-
торого характерна лишь высокоэластическая деформация, т. е.
расплав теряет текучесть. Значения 7 и от, соответствующие
этому переходу, называются критическими (7кр, от,КР) Если по-
лимер содержит структурные единицы с очень малыми време-
нами т*, например низкомолекулярные фракции, которые не
связываются в единую физическую сетку, то течение такого
полимера переходит в область III кривой течения и полимер
течет с низкой наименьшей ньютоновской вязкостью
Приведенная на рис 5.20 кривая течения называется полной;
она обычно характерна для таких систем, как расплавы линей-
ных полимеров невысокой молекулярной массы, разбавленные
растворы полимеров, концентрированные растворы гибкоцепных
полимеров в хороших растворителях и полужсстких — в отно-
сительно плохих. Для расплавов полимеров с достаточно высо-
кой молекулярной массой и широким ММР удается определить
только два участка кривой. При определенных 7 и от полимер-
ный расплав начинает вести себя как квазисшитая система,
т. е. начинает проявлять высокоэластичность, и ламинарное
течение нарушается Важнейшими параметрами, определяющи-
ми поведение таких систем, являются наибольшая ньютоновская
вязкость и эффективная вязкость.
Наибольшая ньютоновская вязкость зависит от температуры
и структуры полимера Поскольку вероятность течения опреде-
ляется наличием свободного объема («дырки») и преодолением
сил межмолекулярного взаимодействия вследствие тепловой
движения и направленного действия силы, вязкость, представ
ляющая собой сопротивление системы перемещению, зависит от
температуры. Температура определяет физический свободный
объем, который тем больше, чем дальше Т отстоит от Тс. Зави-
симость вязкости от величины свободного объема Vc определи
ется соотношением Дулиттла:
lg»fe = a + *—у—
(5-22)
304
где а и b — константы, характеризующие природу жидкости; VM — собствен-
ный объем молекул.
С позиций активационной теории вязкость определяется фор-
мулой Френкеля — Эйринга — Аррениуса (формулой АФЭ):
По-Вехр (Ет//?7), (5.23)
где Е, —энергия активации процесса течения. В —константа
В общем случае вязкость равна:
/ CV* Ет \
По = Лехр (5.24)
\ К* /
где V* — объем «дырки»: Л и С — константы
При низких температурах (Т-*ТС) вязкость полимеров оп-
ределяется свободным объемом и его изменением с температу-
рой и описывается уравнением Вильямса — Ланделла — Ферри
(ВЛФ):
+ (5-25)
ь {1 — 1 с)
При достаточно высоких температурах важное значение при-
обретает скорость активационных процессов и вязкость более
точно подчиняется зависимости АФЭ.
Энергия активации процесса течения характеризует темп
снижения вязкости с температурой и для полимеров практиче-
ски не зависит от молекулярной массы. Это еще раз подчерки-
вает, что течение — это перемещение сегментов, а не всей макро-
молекулы. Поэтому энергию активации обычно рассчитывают
на моль сегментов. Ниже приведены значения энергии актива-
ции вязкого течения £т для некоторых полимеров (кДж/моль):
Полиднметнлсилоксан 15 Полипропилен 46
цис-1,4-Пол нбутадиен 17 Полиизобутилен 54—59
цис-1,4-Полиизопрен 55 Полистирол 118
Полиэтилен: Поливинилхлорид 147
линейный 25—29 Ацетат целлюлозы (в
разветвленный 38-50 растворе) 292
Как видно из приведенных данных и рис. 5.21, на величину
Ех влияют факторы, определяющие гибкость (т. е величину сег-
мента) и взаимодействие макромолекул (наличие аномальных
звеньев, полярных групп, разветвленность и т. д.); особенно она
чувствительна к наличию разветвлений в цепях линейных по-
лимеров.
Поскольку кинетическая гибкость полимера повышается при
введении растворителей (пластификаторов), вполне естественно,
что Ет снижается по мере увеличения доли пластификаторов и
повышения сродства полимера и пластификатора Например,
20—816
305
Рис. 5.21. Зависимость энергии активации вязкого течения Ет от содержания
звеньев цас-1,4 4 в цнс-1,4 полнбутаднене (а), от мольного объема заместите-
лей в полимерах (б) и от параметра жесткости аж (в).
/ — полидиметил силоксан 2 — полибутадиен; 3 — полиэтилен; 4 — полипропилен, 5 — по-
лннзобутилсн; 6 — полпвниилацетат, 7 — полистирол; 8 — полн-а-метнлстнрол
при объемной доле полистирола 0,4 величина £т в этилацетате
(хорошее сродство) составляет «25 кДж/моль, а в четырех-
хлористом углероде (плохое сродство) — повышается др
50 кДж/моль
Сегментальное движение приводит к перемещению макромо-
лекул в целом, поэтому вязкость повышается по мере роста
молекулярной массы (рис. 5.22). Чем прочнее связаны между
Рис. 5.22. Зависимость наибольшей т]0 (1) и наименьшей Имии (2) вязкости
от молекулярной массы (стрелкой указан рост от илн к)
Рис. 5.23. Зависимость Л1кр от мольного объема полимеров:
1 — полиэтилен; 2 — полннзобутнлсн: 3 — полпеннилацетат; 4 — полиметил метакрил ат;
5 — полистирол; 6 — цис-1,4-пол нбу гад иен; 7 цыс-1Л-полннзопрен
306
собой макромолекулы и надмолекулярные структуры (а это
наблюдается при высоких значениях М), тем больше внутреннее
трение в полимере и выше его вязкость. Особенно сильно влия-
ет на вязкость флуктуационная физическая сетка, образованная
в результате межмолекуляриых взаимодействий (зацеплений и
перехлестов) между различными макромолекулами. Поэтому
темп роста с повышением М увеличивается в области высо-
ких М. Точка пересечения двух прямых характеризует критиче-
скую молекулярную массу Мкр, прн которой образуется сетка
в полимере. Значения Мкр (тыс.) для некоторых полимеров
приведены ниже:
Полиэтилен
цис-1,4-Полнбутадисн
1,4-Полинзопрен
Полиизобутилен
3,8-4 ,0
5,5—5,9
10,0
15,2-17
Полнвинилацетат 24,5—22,5
Полнметилметакрилат 27,5
Полистирол 31,2—35
Полнднметнлсилоксан 29
Критическая молекулярная масса увеличивается с ростом
мольного объема полимера и снижается по мере возрастания
гибкости (рис. 5 23). Для линейных полимеров с узким ММР
определена зависимость вязкости от молекулярной массы:
ЛКЛЬф
По(Л1) = {
при
прн
(5.26)
К2м*
где а н £ —константы; а«1, Р«3,4—3,5; Kt и Кг — константы, зависящие
от структуры полимера.
В расчетах чаще всего оперируют среднемассовой молеку-
лявной массой Mw (если в состав полимера не входят фракции
Л1<МКр)- В этом случае i]0 не зависит от ширины ММР При
наличии длинных ветвлений (гребне- и звездообразные полиме-
ры) коэффициент р в уравнении (5.26) может быть выше.
Величина Мкр играет большую роль в поведении полимеров.
Так, только при М>Мкр проявляются большие обратимые (вы-
сокоэластические) деформации, а температура стеклования при-
обретает постоянное значение. Поэтому из рис. 5.22, с одной
стороны, мы получаем информацию о влиянии М на т)о, а с дру-
гой, определяя т]о как функцию от М, можно с высокой точностью
определить Мкр. Таким образом, наибольшая ньютоновская
вязкость т)о является функцией температуры и молекулярной
массы.
В промышленности широко используются полимеры, содер-
жащие наполнители и пластификаторы. Вязкость наполненных
систем т]0.н, как правило, выше, чем ненаполненных, и может
быть рассчитана по уравнению
По.«=По(1 +дФф«+^Ф2фги+- • •). (5.27)
где а, b — константы; Ф— фактор формы (отношение длины частицы или
агрегата наполнителя к его диаметру); фн — объемная доля наполнителя.
20*
307
Это уравнение справедливо при содержании наполнителя до
15% (об.) и отсутствии химического взаимодействия его с поли-
мером
В присутствии пластификаторов вязкость снижается:
g П°>олв S ^плфпл)-р/Сплфпл 1g Ппл> (5.28)
где »]о.пл, 1)пл — вязкость пластифицированного полимера и пластификатора;
Кпл—константа, равная 0,7—0,91 в зависимости от типа полимера и пласти-
фикатора; <р„л — объемная доля пластификатора.
Как было показано выше, в области высоких от и ц нару-
шается пропорциональность между этими параметрами и вяз-
кость зависит от -у. В этой области внутреннее сопротивление
системы характеризуется эффективной вязкостью туЭф, завися-
щей от температуры, молекулярной массы и напряжения сдвига.
Как правило, при увеличении скорости сдвига f эффективная
вязкость снижается тем сильнее, чем больше жесткость и мо-
лекулярная масса полимера. У расплавов низкомолекулярных
полимеров аномалии вязкости не наблюдается, она начинает
проявляться при молекулярной массе более Л1кр. Зависимость
вязкости or М ослабевает с увеличением f и от (см. рис. 5.22).
Снижение температуры повышает аномалию вязкости. Напри-
мер, у расплавов полиэтилена аномалия вязкости почти не про-
является при 560 К, но сильно выражена при 410—470 К
Следует отметить, что аномалия вязкости характерна в боль-
шей мерс для полимеров с широким молекулярно-массовым рас-
пределением. При достаточно высокой температуре (Т»ТС)
полимер с узким ММР ведет себя при течении как ньютонов-
ская жидкость в очень широком интервале у ио: и при 'у’кр(от.кр)
переходит в высокоэластическое состояние, т. е. теряет теку-
честь. В полидисперсном полимере с повышением скорости сдви-
га из процесса течения исключаются сначала фракции с высо-
кой молекулярной массой Mt, затем с меньшей Mt и т. д., т. е.
структурные единицы с временами релаксации Т|*>тг* и т. д.
При этом остаются низкомолекулярные фракции, способные к
течению с меньшей вязкостью В этом и состоит причина ано-
малии вязкости.
Переход таких систем в квазисшитое состояние происходит
при больших 7кР. Таким образом, аномалия вязкости характер-
на для полимеров с широким ММР Но в области Т-*ТС интен-
сивность теплового движения снижается, т* возрастают и ано-
малия вязкости обнаруживается и в случае полимеров с узким
ММР.
308
Таблица 52 Реологические характеристики сополимера бутадиена со стиро-
лом (СКМС-30 АРК), наполненного техническим углеродом (50 масс. ч. на
100 масс. ч. сополимера) при течении через канал диаметром D и длиной L
(LID-20) при Т-393 К
Размер частиц наполнителя, мм п П,ф-10-’ (ПРЯ V-lOOc-1), мПа-c ^т.кр’ *''Г1а
250—300 0,50 1,87 0,240 10,7
50—65 0,52 2,00 0,434 60,3
38—42 0,41 2,05 0,474 58,2
29—32 0,36 V * 2,08 1 • 0,559 86,9
Наполнители оказывают- существенное влияние на величину
эффективной вязкости. С увеличением степени наполнения и с
уменьшением размера частиц наполнителя константа п в урав-
нении (5 21) уменьшается (т]Эф растет) и при высоких значениях
у (^100 с-1) и повышенных температурах т]Эф определяется
только природой полимера. Критические значения 7кр и от.к₽
при этом повышаются (табл. 5.2).
Если наполнитель повышает «связанность» системы, расши-
ряет спектр времен релаксации в сторону больших времен и
вызывает отмеченные выше особенности течения, то пластифи-
каторы оказывают противоположное влияние они уменьшают
степень отклонения течения от ньютоновского и снижают т]Эф;
значение fKp пластифицированных полимеров выше, чем у не-
пластифицированных.
Аномально-вязкие жидкости делят на три основные группы,
псевдопластичные, дилатантные и бингамовские (рис. 5 24).
Псевдопластичные жидкости характеризуются постепенным
уменьшением эффективной вязкости с увеличением скорости
сдвига.
Дилатансией называется увеличение вязкости при сдви-
ге сильно наполненных расплавов полимеров (а также других
систем) вследствие разрушения полимерных прослоек между
частицами наполнителя. Эффективная вязкость дилатантных
Жидкостей увеличивается с возрастанием скорости сдвига.
К бингамовским относятся системы, обладающие в состоянии
покоя определенным пределом текучести ото- Они начинают
деформироваться и течь только тогда, когда напряжение сдвига
От превысит От,о-
При От>от,о течение бингамовских жидкостей подчиняется
зависимости
Сг=От.о+1]б7, (Б.29)
где т]в — постоянная, называемая бингамовской или пластической вязкостью.
309
у
Рис. 5.24 Кривые течения жид-
костей:
1 — ньютоновская; 2 — псеадопластич-
ная; 3 — дилатантная; 4 — бикгамов- <
ская
•
Эффективная вязкость
т]Эф.б бингамовских жидко-
стей снижается при возра-
стании у. К бингамовским
относятся некоторые студни
и наполненные полимеры.
Необратимая пластиче-
ская деформация расплавов
полимеров при их течении
почти всегда сопровождается высокоэластической деформацией,
^поэтому говорят о высокоэластичности расплавов. Особенно ве-
лика доля высокоэластической деформации при течении поли-
меров с жесткими макромолекулами. Эластичность расплавов
связана с изменением не только энтропии, но и внутренней
энергии системы.
Высокоэластичиость расплавов обусловливает ряд специфи-
ческих явлений, имеющих большое значение в технологии пере-
работки расплавов полимеров. К таким явлениям относится,
в частности, эффект Пайссенберга (эффект нормальных напря-
жений), заключающийся в особенностях кругового движения
расплавов, необъяснимых с позиций классической гидродинами-
ки Например, при вращении вала, опущенного в расплав, рас-
плав поднимается по валу вверх. Такое же явление происходит
(рис. 5.25), если в центр вращающегося сосуда с расплавом
опустить неподвижный вал, трубу или диск, способный переме-
щаться по вертикали без вращения При вращении сосуда рас-
плав поднимется по валу, втянется внутрь трубы, соберется под
диском и поднимет его вверх (рис. 5.25,6). Подобные явления
в обычных ньютоновских жидкостях не происходит (рис. 5.25, в).
Вторая особенность расплавов — высокоэластическое восста-
новление. При течении расплавов полимеров в каналах, капил-
лярах, фильтрах макромолекулы ориентируются. При выходе
струи за пределы канала тангенциальные напряжения, вызыва-
ющие эту ориентацию, исчезают и немедленно начинается про-
цесс релаксации. Внешне это проявляется в увеличении диамет-
ра струи (экструдата) по сравнению с диаметром канала, из
которого вытекает экструдат. Это явление и называют высоко-
эластическим восстановлением, Баррус-эффектом, «разбухани-
ем». Процесс протекает во времени, иногда продолжается не-
сколько часов, сопровождается сжатием экструдата по длине —
усадкой. Количественно эффект оценивают коэффициентом вы-
ЗЮ
сокоэластического восстановления et:
e.-(rfc/D)(Pn/p,n),/«
где de — наружный диаметр струи после окончания процесса релаксации;
D — внутренний диаметр канала; рп — плотность полимера при комнатной
температуре; рп'—плотность расплава прн температуре истечения; обычно’
значение ел для расплавов полимеров не превышает 8.
Все технологические приемы, ускоряющие протекание релак-
сационных процессов, снижают усадку. К ним относится повы-
шение температуры, снижение скорости сдвига (рис. 5.26), уве-
личение длины канала при неизмененном расходе, добавление
пластификатора. Усадка зависит от структуры полимера: с рос-
том полидисперсности и разветвленности эластическое восста-
Рис. 5.25. Различные формы проявления эффекта Вайссенберга:
л — состояние покоя; 6 — перемещение расплавов полимеров прн вращении сосуда; в
перемещение низкомолекулярных жидкостей прн вращении сосуда, /—сосуд; 2 — непо-
движный вал; 3 — неподвижная труба; 4 — диск, способный перемещаться по вертикали
311
е.
2,2
13
1,0
102
Рис. 5.26. Влияние скорости сдвига у на коэффициент высокоэластического
восстановления е* полиэтилена низкой плотности при 463 К: отношение L[D
равно 9(/), 82 (2) и 300 (<?)
Рис. 5.27. Зависимости напряжения от от времени течения при различных
скоростях сдвига Cf3>72>7i):
/ — область нестационарного течения; // — область установившегося течения; АЕ и
Л'Я' — области структурной релаксации
новление повышается, поскольку такие полимеры отличаются
от линейных более широким набором времен релаксаций, в том
числе и за счет больших значений т*. Эта же причина является
основной в повышении степени разбухания экструдата напол-
ненных резин при одинаковом содержании полимера в них.
Значение еь учитывают при конструировании профилирующего
инструмента, чтобы предупредить искажение профиля и разме-
ров изделий.
Еще одна специфическая особенность расплавов — неустой-
чивое течение.
Увеличение скорости сдвига при течении
распл
вов полимеров сопровождается не только высокоэластическим
восстановлением. Начиная с определенных значений и скоростей
напряжений сдвига, называемых критическим
(уКр, ат,кр), на по-
верхности экструдата появляются различные дефекты — от «ма-
товости», микрошероховатости до крупных вмятин и даже раз-
рывов струи на отдельные куски Это явление иногда
называют
«срывом струи», эластической турбулентностью, дроблением по-
верхности экструдера. Эффект «срыва струи» усиливается срос-
том молекулярной массы, но при наличии в системе микрогеля,
наполнителя ^кр повышается. При очень большом содержании
пластификатора можно избежать этого явления и добиться те-
чения с наименьшей вязкостью. Для этого применяют следую-
312
щие приемы: увеличение длины канала, т. е. отношения L/D-,
установка конических диффузоров на входе в канал; повышение
температуры экструдата.
Роль высокоэластичности в вязкотекучем состоянии полиме-
ров особенно велика при нестационарном режиме течения.
В первый момент течения преобладает высокоэластическая де-
формация, затем доля высокоэластических деформаций снижа-
ется и возрастает доля пластических.
Переход к установившемуся течению происходит по-разному
при малых и больших скоростях деформации (рис. 5.27) и за-
висит от соотношения времени действия силы /я=7-1 и времени
релаксации т*. При низких скоростях, когда равновесие
устанавливается быстро. При высоких скоростях на кривых за-
висимости От от времени течения появляется максимум (от.макс).
который обусловлен релаксационным поведением полимера. При
условии т*>7~’ деформация отстает от напряжения тем больше,
чем выше 7, флуктуационная сетка нс успевает разрушиться,
макромолекула не успевает принять равновесную конформацию
и в результате этого в системе возникают напряжения, превы-
шающие От при установившемся течении. По мере роста t и де-
формации под действием теплового движения часть связей раз-
рушается и система приходит в равновесное состояние (участки
АЕ и А'Е' на рис. 5 27), т. е. происходит релаксация напряже-
ния. После завершения структурной релаксации полимер пере-
ходит в режим установившегося течения Возрастание от в на-
чальный момент (примерно в 1,5—2 раза) тем больше, чем
больше время релаксации структурных единиц Например, для
жестких полимеров оТмаке намного выше, чем для гибких Это
явление учитывается при переработке полимеров.
Высокоэластичность является причиной механической дест-
рукции макромолекул при течении. При высокоэластической
деформации происходят локальные разрывы связей, что приво-
дит к уменьшению молекулярной массы, увеличению разветв-
ленности и, следовательно, к росту аномалии вязкости.
Механические свойства полимеров в вязкотекучем состоянии
исследуют чаще всего при динамических режимах деформиро-
вания. Деформационные свойства расплавов и растворов (кон-
центрированных и разбавленных) оценивают комплексным ди-
намическим модулем G*, состоящим из модуля накопления
(модуль упругости) G' и модуля потерь G". Комплексный мо-
дуль имеет тот же физический смысл, что и напряжение сдвига
при установившемся течении, и его значение зависит от сопро-
тивления внутреннему трению и сопротивления развитию вы-
"Сокоэластнческой деформации. Значение модуля потерь распла-
313
Рис. 5.28. Кривая напряжение — дефор-
мация о—е кристаллического полиме-
ра (От — предел текучести)
вов выше модуля потерь полиме-
ров в высокоэластическом со-
стоянии Отношение модуля по-
терь к частоте дает значение ди-
намической вязкости т)' :т)'«
= G"/w.
Итак, деформационные (рео-
логические) свойства полимеров в вязкотекучем состоянии при
невысоких напряжениях сдвига характеризуются наибольшей
ньютоновской вязкостью, а при больших—эффективной вяз-
костью. Их значения определяются структурой полимера, тем-
пературой, а эффективная вязкость — и напряжением сдвига.
При течении полимеров наряду с пластической развиваются вы-
сокоэластические деформации Высокоэластичность расплавов
является причиной аномалии вязкости, эффекта Вайссенберга,
высокоэластического восстановления, срыва струи и механохи-
мических реакций прн течении. Наиболее заметно аномалия вяз-
кости проявляется у полимеров с неоднородной структурой,
в частности с широким молекулярно-массовым распределением.
Деформационные свойства кристаллических полимеров. Кри-
сталлические полимеры, как было сказано в гл. 1, состоят из
кристаллических и аморфных участков. Кристаллические участ-
ки деформируются как упругие твердые тела за счет смещения
атомов в решетке, деформации связей и углов. Аморфные про-
слойки в зависимости от условий (температуры и скорости)
могут деформироваться как стеклообразные (при Т^ТС), высо-
коэластические (Тт>Т>7’с) или вязкотекучие (Т>Гт). Кри-
сталлические полимеры отличаются от аморфных повышенными
значениями модуля упругости, пониженной податливостью,
меньшей восстанавливаемостью. Но сочетание жестких кристал-
лических и «податливых» (аморфных) участков делает кристал-
лические полимеры менее хрупкими, чем стеклообразные.
Деформационная кривая кристаллического полимера по внеш-
нему виду напоминает кривую стеклообразного полимера
(рис. 5.28). На ней также можно выделить три участка. На пер-
вой стадии растяжения (линейный участок) развиваются упру-
гие обратимые деформации, увеличивающие свободный объем
в полимере. Модуль упругости (наклон прямой) тем больше,
чем выше степень кристалличности. На этой стадии разрушается
исходная кристаллическая структура На II стадии происходит
перестройка исходной кристаллической структуры и образова-
ние новой в условиях напряженного состояния. Этот процесс
называется рекристаллизацией. Образец в каком-то месте (на
314
микродефектах) утончается и образует «шейку». Иногда «шей-
ка» образуется сразу в нескольких местах (на нескольких мик-
родефектах). Напряжение, при котором начинает образовывать-
ся «шейка», называется пределом текучести от. По мере даль-
нейшего деформирования увеличивается доля материала,
перешедшего в «шейку», и к началу III участка весь материал
находится в «шейке». По внешнему виду «шейка» кристалличе-
ских полимеров напоминает шейку стеклообразных, но механизм
образования их различен. В кристаллических полимерах обра-
зование «шейки» происходит в результате распада исходной
кристаллической структуры и ориентации сегментов аморфных
участков; пластической деформации сегментов в направлении
деформации; ориентации частей кристаллитов в направлении
действия силы.
К моменту полного перехода материала в «шейку» полностью
меняется морфология кристаллов от исходной (чаще всего сфе--
ролитной) в фибриллярную с высокой степенью ориентации в
кристаллических и аморфных участках. Стадия III соответствует
деформации ориентированной структуры — «шейки». Она про-
текает по упругому механизму: па этом участке полимер имеет
высокий модуль и низкую податливость.
Характер кривой определяется исходной степенью кристал-
личности и условиями процесса. Чем ниже степень кристаллич-
ности, тем менее выражена I стадия, и при отсутствии достаточ-
но сформировавшихся кристаллов (возможны только зародыши,
кристаллические кластеры) кривая вырождается в кривую, ха-
рактерную для аморфных полимеров, кристаллизующихся в
процессе растяжения (см. рис. 5.9). Поскольку процесс пере-
стройки структуры имеет временной характер, то деформация
кристаллического полимера зависит от релаксационных свойств
полимера, т. е. от соотношения времени перестройки tn и време-
ни действия силы /д (<д= 1/и) Предел текучести повышается
с ростом скорости v и при снижении температуры. При низкой
Т или высокой скорости (т*/д<&1) от может оказаться выше раз-
рушающего напряжения, и в этом случае кристаллический по-
лимер будет разрушаться хрупко.
Многие полимеры эксплуатируются в кристаллическом со-
стоянии, поэтому устойчивость исходной кристаллической струк-
туры под действием напряжения (т. е. высокое значение от)
является необходимым условием. В процессе эксплуатации в
напряженном состоянии может иметь место рела'кеация напря-
жения или деформация (ползучесть) аморфной части полимера
Степень релаксации в кристаллических полимерах выше, чем у
полимеров в стеклообразном состоянии.
При динамических режимах испытания (синусоидальные не-
затухающие гармонические колебания в отсутствие резонанса)
модуль упругости G' зависит от степени кристалличности, тем-
315
пературы, частоты деформации (рис. 5 29). Эту связь можно
выразить соотношением
lgG-KlgGK+(l — К) 1g Сам. (5.30)
где Ок и Сам — модули упругости кристаллической и аморфной части; К —
степень кристалличности; при К до 50—60% lg G «5,7634-4,77 К.
Механические потери обусловлены в основном наличием
аморфной фазы и проявляются при Тс (a-переход). Высота пик?
потерь снижается с ростом степени кристалличности. Дополни-
тельно к этому пику потерь в аморфной фазе появляется пик
потерь ак при Тс<Т<ТПл, обусловленный процессами, протека-
ющими в кристаллической фазе. Он может быть результатом
деформаций проходных цепей, связывающих ламели, т. е. де-
фектных областей. С ростом длины складок этот переход сдви-
гается ближе к Тпл. Например, в высокоэластическом полиэти-
лене ак-переход проявляется при 373 К, а в разветвленном ПЭ
с меньшей степенью кристалличности — при 333 К. Иногда по-
является пик (у) при Т<ТС, связанный с молекулярным движе-
нием в кристалле.
5.1.2. Прочностные свойства
Прочностью называют свойство материала сопротивляться раз-
рушению под действием механических напряжений. Разруше-
ние — это нарушение сплошности материала, его разрыв, при-
водящий к образованию новых поверхностей. Чтобы разрушить
тело, надо разрушить связи, объединяющие элементы структуры.
Теоретическая прочность твердого тела (от) — это прочность
тела с идеальной структурой (без повреждений и дефектов)
при температуре абсолютного нуля (т. е в отсутствие теплового
Рис. 5.29. Температурные зависимости модуля накопления G' н tg6 кристал-
лических полимеров (степень кристалличности образца 2 выше, чем у об-
разца 1}
I 316
I.
Рис. 5.30. Разрушение однооаю-ориен-
тированного линейного полнме»а
/ — при растяжении вдоль оси ори«тациич
лей полимера // — при растяжениг яерп^ндя.
кулярио оси ориентации цепей полимера-
/// — прн сдвиге вдоль иаправлния поспей
полимера; IV — при сдвиге исрпешикул^ярно
направлению цепей паи им ера
движения) при однородней ••ста-
тической деформации растяжения
и сдвига, обеснечиваюцей рав-
ную нагруженность всех связей и
их одновременный разрыв по
поверхности разрушения Для^ расчета теоретической прочности
обычно используют структурную модель линейного одноосно-
ориентированного поликера. Разрушение таких полимеров мо- •
жет иметь место при растяжении или сдвиге вдоль оси ориента-
ции макромолекул или герпе ндикулярно направлению полимер-
ных целей (рис. 5.30). Гри э.том в / и // случае разрушаются
преимущественно химические.., а во II и III — межмолекулярные
физические связи значительней меньшей энергии. В неориентиро-
ванных полимерах разрешаются как химические, так и слабые
межмолекулярные физшески е связи, т. е. тип разрушения про-
межуточный между I и II >чли III и IV. Теоретическая проч-
ность равна
где N — число атомов или своей, ^приходящихся на единицу площади сече-
ния, Fm — прочность связи (шла Еззанмодействня) двух соседних атомов.
Величина W определится^ степенью ориентации полимерных
молекул: повышение стетени ориентации приводит к росту плот-
ности упаковки макромолекул и, следовательно, к увеличению
числа химических связей на единице площади разрушаемого
тела, а также к росту ч!сла физических узлов. Прочность кова-
лентных связей Fm ощедел яется энергией диссоциации связи
и силовой константой kr, характеризующей валентные коле-
бания атомов
7т=[^д(А,/2Ея)'/2]/2. (5.31)
Прочность межмолекул парных физических связей определя-
ется характером внутрь и межмолекулярных взаимодействий
(см. гл. 1) и намного шже т>1рочности химических связей. Поэто-
му в предельно ориентированном полимере при растяжении
вдоль оси ориентации грочн-юсть будет примерно в 10 раз выше,
чем при растяжении герпе ндикулярно оси ориентации. Ниже
приведены значения теоретической прочности и модуля упруго-
317
сти при растяжении Е некоторых предельно ориентированных
полимеров:
сг_., ГПа £, ГПа
Полнкапрозмид
вдоль оси ориентации 30 188
поперек осн ориентации 2,6 26
Полиэтилен (вдоль оси ориентации) 32—37 285
Алмаз 104—230 1246-2806
Как видно из приведенных данных, между от и модулем
упругости Е наблюдается хорошая корреляция: от = КЕ, где
К—коэффициент корреляции, равный «0,08—0,16.
В полимерах невысокой молекулярной массы разрушение
происходит не столько за счет разрыва химических связей,
сколько за счет скольжения молекул, т. е. преодоления сил
межмолекулярного взаимодействия С ростом молекулярной
массы увеличивается вклад в от химических связей, и при до-
статочно высоких значениях молекулярной массы разрушение
происходит за счет разрыва в основном химических связей и от
уже не зависит от молекулярной массы. В неориентированных
полимерах только часть макромолекул (~‘/з) нагружена, по-
этому от неориентированных полимеров примерно в три раза
ниже От предельно ориентированного полимера. Для них также
характерно снижение от с уменьшением молекулярной массы.
Таким образом, теоретическая прочность определяется энер-
гией разрушаемых связей и степенью ориентации макромолекул.
Прочность реальных полимеров, так называемая техническая
прочность, намного ниже теоретической. Основными причинами
снижения прочности являются тепловое движение атомов и на-
личие слабых дефектных мест В реальных условиях при Т>0К
в любой системе происходит тепловое движение атомов, которое
может являться причиной термофлуктуационного разрыва свя-
зей. Минимальная кинетическая энергия, необходимая для раз-
рыва связи, называется энергией активации разрыва связи С7Р°.
Время ожидания разрыва т* одной связи будет равно
т*=тоехр LFp/kT, (5.32)
где То— период колебания атомов; t/p°— потенциальный барьер (энергия ак-
тивации .разрыва) в отсутствие действия силы; k — константа Больцмана;
Т — температура
При 0 К t/р0 равен энергии диссоциации связи, а с повыше-
нием температуры до температуры деструкции (Тд) £7Р° изме-
няется линейно с ростом температуры:
(7%-У%(0) — qT, (5.33)
где Ц>°(0)—потенциальный барьер при OK; q— константа, равная
«5,4 Дж/(моль-К).
318
Рис. 5 31. Потенциальная энергия U(r} атома полимерной цепи прн разрыве
химической связи:
1 — до разрыва; 2 — при разрыве
Рис. 5.32. Схематическое изображение разрыва изолированной полимерной
цепи н ориентированного полимера с образованием поверхности разрыва АВ
под действием силы F
При наложении механического поля (например, при растя-
жении под действием постоянной силы F) энергия активации
разрыва будет меньше Up° иа величину А, представляющую
собой работу внешних сил (A=KmF) (рис 5.31), где 'Кт— удли-
нение связи при разрыве, т е
yp=l/op—XmF. (534)
Вероятность разрыва связи при этом повышается (т* умень-
шается):
т.-тс ехр [(<7°Р — KmF)/kT], (5.35)
При одновременном разрыве всех связей (т. е. при их рав-
номерном нагружении) вероятность их разрыва т=т*. При меж-
молекулярном расстоянии Хо растягивающее напряжение о~
fnF/'ко2 и при о = const уравнение (5.35) будет иметь вид
т=тоехр [(l/°p— Vao)/AT], (5.36)
где т । — период валентных колебаний атомов в полимерной цепи; для связей
типа С—С То«3-10“'* С; V,—Х02Хт — флуктуационный объем.
Если в полимере имеется лсв связей, то время ожидания
разрыва связи в любом месте цепи т* уменьшается в лСв раз;
тогда
т=т.«(?о/лс») ехр [(1/°р— V&c)/kT}. (537)
Однако одновременный разрыв всех связей является мало-
вероятным, и в реальном полимере обычно связи рвутся после-
319
довательно с образованием поверхности разрыва (рис. 5.32).
Если по пути движения фронта разрыва встречается No связей, I
то x=x»N0 или
T-Totfoexp[(U°p—У.О')/И*], (5.38)
где о'=о(1—l/L), I—длина растущего надреза. L — ширина образца.
Из уравнения (5.38) следует, что с ростом степени ориента-
ции, плотности связей, т. е. с увеличением No, время до разрыва !
увеличивается.
В реальном полимере нагружениость цепей неравномерна,
что определяется коэффициентом перегрузки хо. В идеальном
полимере (при равной нагруженности) хо=1, в реальных —
х0>1 и может достигать больших значений. В связи с этим в
уравнение (5.38) вводят структурный коэффициент /=Рахо>
а величина ю заменяется на постоянную С:
т=Сехр [(U°p — faJ/AT]- (5.39)
Здесь С=т0 для случая одновременного разрыва всех связей в
поперечном сечении и равна 3-10 14 с; при последовательном
разрыве связей С = 8-10-7—3-10-14 с. В реальных полимерах
существуют микротрещины, в вершинах которых возникают
перенапряжения, а также «слабые» места структуры (включения
других атомов, звеньев другого строения и др ) В этом случае
уравнение (5.39) принимает вид
Т=С exp [(U°p —10)/АТ]. (5.40)
где f=f'P= V,x; £ — коэффициент перенапряжения в вершине микро-
трещины; х — коэффициент перенапряжения, учитывающий неравномерность
нагружения и перенапряжение в вершине микротрещины.
Это уравнение Журкова для оценки долговечности твердых тел.
Термофлуктуационная теория рассматривает разрушение]
связей как следствие теплового колебания отдельных атомом
при равномерном распределении силового и температурного
полей. Разрыв связей в реальном полимере рассматривают кам1
результат распространения, взаимодействия и генерирования
фононов — статистически независимых квазичастиц. Под дейст-
вием собственного теплового движения фононы совершают гар-
монические колебания вокруг положения равновесия, и разрыва
не происходит. Разрыв возможен только при больших отклоне-
ниях от положения равновесия, когда тепловые колебания
становятся несимметричными (энгармонизм) Если это проис-
ходит под действием внешних сил, то это силовой энгармонизм,
если при повышении температуры, — температурный энгармо-
низм. В реальных условиях при Т>0 К проявляется как тот,
так и другой энгармонизм.
В системе существуют фононы высокой энергии двух видов:
фононы, вызывающие возникновение положительных и отрнца-
320
тельных (так называемых дилатонов) флуктуаций плотности.
Дилатоны — это области с растянутыми связями и линейными
размерами, близкими к длине свободного пробега фонона. При
растяжении размеры дилатонов увеличиваются и происходит
«подкачка» энергии из окружающей среды. Это приводит к ло-
кальному повышению температуры и последующему удлинению
связей вплоть до разрыва Таким образом, процесс разрыва
связей можно представить как двухстадийный: на первой стадии
флуктуации приводят к возникновению дилатона, живущего
короткое время (несколько периодов атомных колебаний), на
второй — происходит разрыв связи в дилатоне, причем энергии
активации разрыва меньше, чем при одностадийном разрыве
Ангармонизм (силовой и температурный) влияет на Up0 и
постоянные у и С в уравнении (5.40):
U%=GmKM0/2g-, 7=xVa = xK??o/2g, (5.41)
где ат — теоретическая прочность; К — силовой коэффициент; — межмоле-
кулярное расстояние; g — коэффициент ангармоничности
Из уравнения (5.41) следует, что чем сильнее выражен ан-
гармонизм, тем меньше энергия активации разрыва и структур-
ный коэффициент *у; чем меньше силовой коэффициент К, тем
меньше флуктуационный объем Va, а следовательно, меньше
структурный коэффициент у. Константа С уменьшается с ростом
коэффициента теплового энгармонизма q:
А=С exp (—q/k), (5.42)
где q— коэффициент, учитывающий температурный ангармонизм и пропор-
циональный термическому коэффициенту объемного расширения, k — констан-
та Больцмана.
Таким образом, для реальных полимеров, в которых возмо-
жен разрыв связей на бездефектных участках, в вершинах мик-
ротрещин, т е. в областях перенапряжения (разрыв определя-
ется не средним напряжением в системе о, а локальным напря-
жением в вершине трещин о* = хо), а также в слабых местах
структуры, при наличии силового и температурного энгармониз-
ма вероятность разрушения описывается уравнением Бартенева:
т=Д ехр [(£/%- га)АЛ- (5.43)
Из уравнения видно, что с повышением температуры, когда
kT^nUp (Up=Uv°—y°)> т е. уже первое колебание атомов
может привести к разрушению. Аналогичная картина наблюда-
ется и с ростом о При очень высоких напряжениях разрушение
может происходить мгновенно. При одновременном повышении
Тио вероятность разрыва увеличивается (т снижается). По-
этому обычно различают кратковременную и длительную проч-
ность. Данные (истинное напряжение) о кратковременной (про-
должительность нагружения 1 мин) и длительной (продолжи-
21—816
321
тельность нагружения 12 мес) прочности некоторых полимерных
материалов приведены ниже (при 293 К):
Кратковременная
прочность, МПа
Длительная проч-
ность, МПа
Эластомеры:
мягкие
жесткие
Резины наполненные
Пластмассы
Химические волокна
3-10
30—50
100—200
100—200
500—1000
0,3—2
3—10
20—30
20—40
100—300
Как видно, длительная прочность значительно ниже кратко-
временной. Кратковременную прочность определяют при нагруз-
ке, приближающейся к разрушающей, а длительную — при на-
грузках, намного меньших разрушающих. Длительную прочность
также называют долговечностью или статической усталостью,
которую оценивают временем до разрушения т по уравнению
(5 43). Разрушающее напряжение, т. е. кратковременную проч-
ность, также можно рассчитать по уравнению (5.36) и (5.43).
Для идеального полимера, в котором цепи одинаково напряжены
и рвутся одновременно (х=1, 7 = Уа)
- (I / V») [t/°P - 2.3*7 1g (т/то)]. (5.44)
Величина оп называется предельно достижимой прочностью:
при Т=0К On = i/p°/Va, а при ир°=Ел (Ел— энергия диссоциа*.
ций связей) получаем теоретическую прочность о„,. В реальных
полимерах
0 - (1 /7) [£Л„ - 2,3*7 1g (т/Л) ]. (5 45)
Как следует из этого уравнения (при учете того, что 7=УаХ,
а х>1), в реальных полимерах значения о ниже как оп, так
И Ст'
Полиэтилен
Полипропилен
.Поливинилхлорид
Поликапроамнд
Полиформальдегид
от (0 К). ГПа а„ (293 К). ГПа а (293 К). ГПа
26—27 4,0-6,5 0,02—0,04
11—12,5 2,0—3,5 0,08—0,20
14—17 2,7—4,0 0,05—0,10
23—27 4,0—7,0 0,06—0,16
21,5 22,5 3,5—5,5 0,07—0,072
Снижение прочности по сравнению с теоретической и пре-
дельно достижимой происходит именно вследствие неравномер-
ной нагрузки цепей, существования коротких и длинных цепей
и их различной ориентации, неоднородности структуры на моле-
кулярном и надмолекулярном уровнях, приводящей к существо-
ванию слабых и прочных связей, наличию микротрещин и ан-
гармоничного действия температуры и напряжения. Из этого
следует, что одним из путей получения высокопрочных полиме-
-322
ров является создание ориентированной бездефектной однород-
ной структуры.
Для расчета долговечности и кратковременной прочности по
уравнениям (5.43) и (5.45) необходимо знать значения Up0, 7
и А Для их определения уравнение (5 43) представляют в виде
ПО V
1g х = lg А +• —-----<3. (5.46)
К 2,3*7 2,3*7 ' ’
В координатах lgx=a это уравнение изображается прямой
линией при Т—const, а при разных Т — серией прямых
(рис. 5.33). Из наклона прямых определяют коэффициент у по
формуле; 7 = 2,3fcTtga (где tga — тангенс угла наклона пря-
мой). Экстраполяция прямой на ось о (а=0) дает значение
1g т°= 1g Л + Uf>0/2,3kT, из которого можно найти Up°. Экстра-
поляция прямых долговечности в координатах 1g т—1/Т на ось
1g т позволяет рассчитать значение константы А в уравнении
(5.46)
Уравнение (5.46) показывает, какое большое влияние на
прочность оказывает равномерность распределения напряжений,
дефектность, наличие микротрещин Прн одинаковых значениях
Up0 прочность тем больше, чем ниже 7, что достигается при
равномерном распределении нагрузки по всем разрываемым
связям. При наличии одновременно напряженных и ненапря-
женных связей коэффициент 7 возрастает и тело легко разру-
шается при небольших значениях о. Для идеальных твердых
тел коэффициент 7 должен быть одинаковым независимо от ма-
териала тела и равен объему атома («10~23 см3). Реальное
значение 7 для полимерных материалов значительно выше.
Ориентация полимеров вызывает заметное снижение этой вели-
Рнс. 5.33. Долговечность полимеров прн различных температурах (а; Т|<
<7г<Т3<А<75) и различных напряжениях (б; ®|>04>0з)
21*
323
чины: так, * для неориентированного капрона равен 128-10-23,
для высокоориентированного — 20-10 см3.
Таким образом, разрушение реальных полимеров происходит
в результате тепловых флуктуаций, а роль внешних нагрузок
сводится к ускорению термофлуктуационного разрыва связей.
Повышение степени ориентации, плотности упаковки, снижение
коэффициента перенапряжения, увеличение прочности связей —
все это способствует повышению прочности, а рост дефектности,
в том числе и микротрещин, снижает прочность. Чтобы выяснить
причины снижения прочности при наличии в теле микротрещин,
рассмотрим термодинамику разрушения и некоторые теорети-
ческие положения, объясняющие эти причины.
Разрушению реальных полимеров, как уже было сказано,
предшествует деформирование образца Величина и характер
деформации зависят от физического или фазового состояния по-
лимера. При деформировании подведенная механическая энер-
гия накапливается в образце в виде упругой энергии №о- При
образовании в образце микротрещины энергия изменяется, и ее
изменение dW равно
W — Го=— л02/оЛ/4£, (5.47)
где W— упругая энергия образца после образования микротрещины; о—на-
пряжение; h — толщина образца; /© — длина микротрещины; Е— модуль уп-
ругости (Юнга).
Величина dW отрицательна, так как появление трещины при-
водит к убыли упругой энергии. Запасенная энергия dW идет
на образование новой поверхности при разделении образца на
части, т е. на увеличение свободной поверхностной энергии dFce,
и частично превращается в тепло dQ, которое рассеивается в
пространстве. За малый промежуток времени dt величина
—dW=dFCB+dQ Суммарная энергия образца №=— ' dW, где
т — время до разрушения образца
При разрушении потери dQ складываются из деформацион-
ных потерь dQi (потери, сопровождающие высокоэластическую
и вязкотекучую деформации), динамических потерь dQ2 (вы-
званных переходом части упругой энергии в кинетическую энер-
гию раздвижения стенок растущей трещины или в кинетическую
энергию разлетающихся осколков) и потерь dQ3, связанных с
рассеянием энергии при разрыве связей Вклад dQ в разрушение
полимеров определяется механизмом разрушения, физическим
и фазовым состоянием полимера В идеально упругом теле dQ =
= 0, в реальных телах dQ^=C. Особенно велика роль члена dQ
при разрушении полимеров в высокоэластическом и вязкотеку-
чем состояниях.
Одной из причин снижения реальной прочности по сравнению
с предельно достижимой или теоретической является наличие
324
Рис 534. Концентрация напряжений иа различных типах микротрещин:
/ — краевая острая; 2 — краевая эллипсовидная; 3— эллипсовидная в объеме
Рис. 5.35. Диаграмма прочностных состояний в координатах напряжение с
— длина дефекта /0:
1 — безопасное напряжение (То: 2 —критерий разрушения Гриффита; 3 — критическое на-
пряжение <ткр; /--область безопасных напряжений; // — область термофлуктуациоиио-
го роста трещин; /// — область атермического роста трещины
микротрещин. Их роль в разрушении полимеров наиболее полно
рассмотрена в теории Гриффита, в основе которой лежат сле-
дующие положения:
1) материал представляется как идеально упругий, содержа-
щий микротрещины различной формы, сосредоточенные на по-
верхности и в объеме (рис. 5 34);
2) на краях микротрещин возникает локальное перенапря-
жение а*, которое намного превосходит среднее напряжение,
приходящееся на сечение образца; при о*—ат происходит ката-
строфическое разрастание трещин со скоростью v*, близкой к
скорости звука, и образец разрушается, т. е разделяется на
части, — образуется магистральная трещина; приложенное в
этот момент напряжение называется критическим акр или мак-
симальной реальной прочностью; при о*<от, т. е. когда о<оКр,
трещина не растет и образец не разрушается;
3) вся работа за время dt внешних сил (—dW) расходуется
на увеличение свободной поверхностной энергии dFCB, т. е.
—dW=dFC9-, при этом роль теплового движения не учитывается,
т. е. процесс разрушения считается атермическим;
325
4) критерий разрушения Гриффита определяется соотно-
шением:
Oc = /((aE//0)V2 (5.48)
где К — коэффициент зависящий от вида микротрещины ее расположения
и толщины образца; для тонкой пластинки с внутренней эллипсовидной мик-
ротрещиной длиной /о /(=1,13; при краевой трещине /(=0,8; круглой с диа-
метром /0 /(= 1,8; а — удельная (т. е. приходящаяся на единицу площади)
свободная поверхностная энергия; /0 — длина (для узкой) или диаметр (для
круглой) трещины.
Экспериментальные значения а выше рассчитанных по урав-
нению (5 48), поскольку, во-первых, процесс разрушения не яв-
ляется атермическим, во-вторых, в теории принимается dQ=0,
т. е не учитываются потери энергии. Потери dQi и dQz зависят
от скорости роста трещины и при о*->0 dQt и dQ2-'-0, но потери
-dQ3 не исчезают даже в том случае, если С ростом ско-
рости деформирования величина dQ существенно возрастает.
Из этого следует, что катастрофическое разрушение (с о*, при-
ближающейся к скорости звука) будет происходить не при
о = ос (критерий разрушения), а при о>оо- При напряжениях,
равных ос, разрушение не имеет катастрофического характера
и скорость распространения трещины меньше скорости звука
Поэтому для реальных полимеров критерий разрушения ор
равен
= (5.49)
где а* — обобщенная поверхностная энергия, включающая свободную энер-
гию и механические потери dQ, отнесенные к единице площади свободной
поверхности.
На основании термодинамического подхода и термофлуктуа-
ционной теории (кинетический подход) можно построить диа-
грамму прочностных состояний в координатах о—/о, которая
имеет большое практическое значение, так как определяет гра-
ницы безопасных напряжений и размеров дефектов (рис. 5.35).
Для ее построения необходимо определить зависимости от дли-
ны трещины /© следующих параметров:
1) критерия разрушения Гриффита ос по уравнению (5.48);
2) критического напряжения оКр, рассчитываемого по фор-
муле
а«Р=^°р/Г.х, (5.50)
где Up0 — энергия активации разрушения; V* — флуктуационный объем; х—
коэффициент перенапряжения, х»-хоР; Р— коэффициент коицентрацин на-
пряжения в вершинах микротрещин, зависящий от длины трещины; напри-
мер, для короткой краевой трещины (когда длина ее меньше ширины образ-
ца) р» l-f-0.79tW^*. где (Ко — межмолекуляриое расстояние) для од-
иоосиоориентнрованного и Х*=ЗХ0 для неориентированного образцов.
326
3) безопасного напряжения о0, при котором вероятности
разрыва и восстановления связей равны
- Vsa0 = l/p0' + Vao0, (5.51)
где Up0 и С/р0'— потенциальные барьеры при о«=0 прн разрыве и восстанов-
лении связи; а —локальные растягивающие напряжения, действующее на
связи в вершине трещины: o0=(t/p°—UP° )/(2pV«)—для неориентированного
полимера; o0= (f/p°—Up0 )/(2xo₽Va)—для ориентированного полимера.
Если известна свободная поверхностная энергия а, то Оо=
=а/рхт — удлинение химической связи при разрыве)
Таким образом, о0— это напряжение, при котором не прояв-
ляется ускоряющее влияние о на разрушение.
Из рис. 5.35 видно, что кривые оо=/(/о) и оо=/(М располо-
жены очень близко друг к другу; это свидетельствует о том, что
критерий Гриффита отражает уровень безопасных напряжений,
а не разрушающих. Ниже кривой 1 оо=/(М на рис. 5 35 распо-
лагается область безопасных напряжений и безопасных трещин
и т-*0 Между кривыми 2 и 3 находится область термофлуктуа-
ционного разрушения, где начальная трещина растет со скоро-
стью тем большей, чем выше о Выше кривой 3 располагается
область атермического разрушения, где трещина растет со ско-
ростью, близкой к скорости звука. Из диаграммы следует, что
сильно дефектные материалы даже при низких о могут разру-
шаться атермически, а малодефектные — выдерживать значи-
тельные напряжения Зная эксплуатационную нагрузку материа
ла или изделия, можно определить безопасный размер трещин
и, наоборот, при известном размере микротрещин определить
безопасные напряжения
Тот факт, что в нагруженных полимерных материалах за-
долго до их разрушения наблюдается распад межатомных свя-
зей с образованием свободных радикалов, неоднократно был
доказан методами электронного парамагнитного резонанса
(ЭПР), инфракрасной спектроскопии (ИКС) и масс-спектромет-
рии летучих продуктов распада. Образующиеся свободные ра-
дикалы вступают в химические реакции с соседними связями,
вследствие чего в области наиболее перенапряженных участков
протекают цепные реакции, происходит распад новых химиче-
ских связей. Экспериментально показано, что на каждый сво-
бодный радикал, образовавшийся при распаде связей, прихо-
дится до 1000 разорванных концов макромолекул. В момент
распада наиболее перенапряженных связей нагрузка, которую
они несли, перераспределяется на другие участки полимера,
перенапряженными становятся новые связи, а после их разры-
ва — следующие, и т. д.
Чем больше напряжение и продолжительность деформации,
тем больше возникает в полимере зон разрушения, называемых
субмикротрещинами, которые имеют форму «чечевичек» разме-
327
ром 0,1—0,01 мкм; их плоскость ориентирована перпендикулярно
оси растяжения. Концентрация субмикротрещип в предразрыв-
ном состоянии достигает огромного значения. Ю16—1017 тре-
щин в 1 см3. Они располагаются вплотную друг к другу и, сли-
ваясь, образуют одну или несколько трещин большого размера,
по которым н происходит разрушение полимера.
Таким образом, разрушение полимеров под нагрузкой про-
исходит в несколько стадий:
1) растяжение межатомных связей под влиянием механиче-
ской нагрузки при о^Оо,
2) разрыв возбужденных связей под действием термических
флуктуаций (создаваемых тепловым движением) с образовани-
ем свободных радикалов;
3) цепные реакции в зоне разорвавшихся связей, иницииро-
ванные свободными радикалами, приводящие к образованию
летучих продуктов, разрушенных микрообластей (дефектных
зон), возникновение субмикротрещин;
4) накопление субмикроскопических трещин, их слияние в
магистральную трещину и разрушение полимера.
Соотношение длительности этих стадий определяется физи-
ческим и фазовым состояниями полимера при разрушении. На
рис. 5.36 приведена диаграмма прочностных состояний аморф-
ного полимера, в различных физических состояниях
Разрушение стеклообразных полимеров. Стеклообразные по-
лимеры при температуре, близкой к 0 К, находятся в хрупком
состоянии и разрушаются по атермическому механизму. Пове-
дение таких материалов описывается теорией Гриффита. Крат-
ковременная прочность зависит от модуля упругости и характе-
Рис. 5.36. Температурная зависимость разрывного напряжения а₽ (а) н кри-
вые а—е (б) при различных механизмах разрушения:
I— атермический, //— хрупкий; /// — вынужденный высокоэластичсский; IV — высоко-]
эластический; V — пластический VI — вяэкотскучее состояние 7*xp, Тц, Гт—j
температуры атермического разрушения хрупкости, стеклования пластичности, вязкого*
течения)
328
ра дефекта [см. уравнение 5.48)] Долговечность т определяется
напряжением а и равна
т- (а/Схр) arcsin (ор/сг). (5.52)
Как было показано в разд. 5.1.1, при Т^Тхр для стеклооб-
разных полимеров характерно явление вынужденной высоко-
эластичности. Но при некоторых условиях (например, при Т<
<Тхр) высокоэластическая деформация в полимерах не разви-
вается. Такие полимеры называют хрупкими стеклами в отличие
от нехрупких, для которых характерно явление вынужденной
высокоэластичности. Хрупкие стекла (Т<7’хр) разрушаются по
хрупкому механизму, особенности которого перечислены ниже.
1. Разрушению предшествуют лишь упругие деформации и
в зоне разрушения нет следов пластических деформаций.
2 Разрушение происходит в две стадии; первая связана с
медленным ростом начальной микротрещины (о*а; 10~4—
10~5 м/с), приводящей к образованию «зеркальной», гладкой
зоны разрушения, вторая — с прорастанием первичной и образо-
ванием вторичных микротрещин, распространяющихся в объем
(при этом образуется шероховатая поверхность разрушения —•
«шероховатая» зона) со скоростью, близкой к скорости звука.
На первой стадии действует термофлуктуационный механизм,
на второй — атермический Соотношение этих стадий зависит
от растягивающего напряжения, температуры и длины первона-
чальной трещины. Чем больше о, тем короче первая стадия и
меньше «зеркальная» зона. При а=оКр «зеркальная» зона исче-
зает и разрушение принимает сразу критический характер, т. е.
идет по атермическому механизму Поскольку большая часть
времени тратится на первую стадию, то прочность тем выше,
чем больше доля «зеркальной» зоны.
3. Трещина растет в основном (особенно при атермическом
разрушении) нормально растягивающей силе.
Кратковременную прочность охр оценивают по величине раз-
рушающего напряжения:
Охр=Ор^ (1/М) [^Г- 2.3ЛТ 1g (т/Л)], (5.53)
где Va — флуктуационный объем; (5 — коэффициент концентрации напряже-
ний в вершине трещины: U°v — энергия активации разрыва при Т—►О;
k — константа Больцмана; т — время до разрушения; А — постоянная.
Долговечность определяется согласно термофлуктуационной
теории по уравнению
т-=Лехр[(У% —(5.54)
(То< Окр»
где ао, GKp — безопасные и критическое напряжения.
Разрушение нехрупких стекол характеризуется следующими
особенностями.
329
Рис. 5.37. Микротрещнны «серебра» при
разрушении полимера в стеклообразном
состоянии (показано начало разруше-
ния)
1. Разрушению предшествуют
значительные деформации — вы-
нужденные высокоэластические
(см. рис. 5.3) под действием ло-
кальных напряжений и вызываю-
щие сегментальную подвижность
в микрообластях, прилегающих
к вершине микротрещины.
2 Разрушение происходит
также в две стадии. Первая —
образование микротрещин, назы-
ваемых трещинами «серебра»,
и их накопление. Микротрещины
«серебра» — особый вид дефектов, которые возникают при де-
формировании стеклообразных нехрупких тел по механизму вы-
нужденной высокоэластичности. В микротрещине происходит
расслаивание полимера на микротяжи и вся микротрещина ока-
зывается заполненной чередующимися областями ориентирован-
ного полимера и микропустотами (рис. 5.37). Наличие этих
трещин практически не влияет на прочность и модуль, так как
трещина не растет, поскольку створки ее скреплены тяжами,
которые принимают на себя нагрузку и снижают перенапряже-
ние в вершинах трещины. Микрорасслаивание материала свя-
зано с a-процессом релаксации, протекающим под действием
напряжения. В результате релаксации происходит снижение ко-
эффициента концентрации напряжений.
Вторая стадия представляет собой разрушение — прораста-
ние магистральной трещины, или разрушение по трещинам «се-
ребра» с предварительным растяжением тяжей в трещине при
напряжении о, равном предельно достижимому оп, или разру-
шение по неориентированному материалу. При этом механизм
разрушения является термофлуктуационным. Следовательно,
полное разрушение включает в себя два механизма: релакса-
ционный и термофлуктуационный.
3. В зоне разрыва имеются следы остаточной деформации,
но они исчезают со временем, что свидетельствует о ее высоко-
эластической релаксационной природе.
Кратковременная прочность нехрупких стекол характеризу-
ется двумя показателями: пределом вынужденной эластичности
Овэл и разрушающим напряжением оР. Долговечность описыва-
ется соотношением, аналогичным уравнению (5.43) при о<ов»л.
Структурный коэффициент в уравнениях (5 43) и (5.45)
330
для нехрупких стекол ниже, чем для хрупких, так как снижа-
ется коэффициент перенапряжения х.
Таким образом, чем больше образуется микротрещин «сереб-
ра», т. е чем выше вклад вынужденной высокоэластичности, тем
труднее разрушить образец.
Условия проявления вынужденной высокоэластичности опре-
деляются соотношением времени структурной релаксации т* и
времени продвижения микротрещины за одну флуктуацию Тф.
Если Тф<т*, то вынужденная высокоэластичность не развивает-
ся; если же тф>т‘, то в вершине микротрещины вначале будет
развиваться высокоэластическая деформация, а затем происхо-
дить разрыв полимерных цепей. Температура хрупкости в этом
случае — температура, при которой выполняется условие Тф = т*.
Для повышения прочности хрупких стекол в них вводят эла-
стомеры, расширяющие релаксационный спектр полимера.
Разрушение полимеров в высокоэластическом состоянии. По-
лимер находится в высокоэластическом состоянии при Т>ТС и
в этих условиях высокоэластическая деформация начинает раз-
виваться практически сразу с начала деформирования, поэтому
разрушению предшествуют значительные высокоэластические
деформации, имеющие релаксационный характер. Механизм,
разрушения полимеров в высокоэластическом состоянии назы-
вают «вязколокальным». Он реализуется при Тп>Т>Тс, где
Тп<.Т1 (Тп — температура, при которой появляются локальные
области вязкого течения).
Для полимеров в высокоэластическом состоянии также ха-
рактерны две стадии разрушения (рис. 5.38). Первая (медлен-
ная) начинается с образования очага разрушения, в месте ко-
Рис. 5.38. Стадии разрушения полимера в высокоэластическом состоянии:
в—до разрушения </о —дефект); б — образование микротяжей в зоне дефекта; е—раз-
рыв; If — «шероховатая» зона; / — «гладкая» зона; АВ — путь трещины
торого начинает расти «надрыв». Его можно рассматривать как
аналог трещины в нехрупких стеклах. Очаги разрушения воз-
никают в наиболее дефектных слабых местах: на микродефек-
тах (поры, трещины и т д.) и структурных дефектах (неодно-
родные атомы в главной цепи, неупорядоченные участки на
границе раздела аморфная часть — кристалл в аморфно-кри-
сталлических телах, в наиболее рыхлоупакованных областях в
аморфных полимерах, на границе раздела полимер — наполни-
тель при их низкой адгезии друг к другу и т д.). Первый «над-
рыв» возникает в месте наиболее опасного дефекта, затем по
мере деформирования в зоне «надрыва» образуются микротяжи,
состоящие из ориентированного в направлении действия силы
материала При образовании микротяжей происходит преодоле-
ние межмолекулярных сил, и поэтому напряжения на первой
стадии невелики. По мере углубления зоны надрыва тяжи раз-
рываются в слабых местах, распределенных по случайному за-
кону. Поэтому на первой стадии образуется «шероховатая» зона
разрушения. Когда практически весь полимер перейдет в пре-
дельно ориентированное состояние и напряжение будет превы-
шать суммарную прочность межмолекулярных связей, произой-
дет нагружение химических связей и их быстрое разрушение.
Трещина растет перпендикулярно направлению силы с очень
высокой скоростью, и поэтому вторая стадия характеризуется
гладкой поверхностью («гладкая» зона) и высокими значениями
энергии разрушения.
Основной вклад в долговечность вносит первая медленная
стадия. Долговечность определяется по уравнению
T=Cia”fc, (5.55)
где Ct=Cexp(Up°/^T); Ь — структурно-чувствительный показатель, не завися-
щий от температуры; b несколько растет с увеличением степени сшивания:
для несшитых полимеров Ь«2,7, а для сшитых t>=3,4—3,2.
Это уравнение справедливо при средних значениях о; при
очень больших о полимер ведет себя как твердый стеклообраз-
ный, и зависимость т от о подчиняется экспоненциальному за-
кону (5.43), при очень малых о^оо т-*оо. Кратковременная
прочность в режиме т=const равна
о₽- (С/т) 'f» exp (l/P/(W)]- (5-56)
Как уже отмечалось, вклад потерь, обусловленных релакса-
ционными процессами, в разрушение наиболее заметен для
эластомеров, которые деформируются по высокоэластическому
механизму. Повышение доли dQ снижает вероятность разруше-
ния (т е. образования новой поверхности). Поэтому чем больше
потери, т. е. чем шире набор релаксирующих единиц в момент
разрушения, тем выше кратковременная и длительная проч-
332
ность. Релаксация приводит к снижению перенапряжений, дис-
сипации напряжения по образцу и, следовательно, к снижению
среднего напряжения. Основную роль в разрушении играют
a-переход, связанный с подвижностью сегментов в области /,
близкой к Тс, и Х-пере.ходы, обусловленные изменениями надмо-
лекулярной структуры.
Расширение спектра релаксации, например, за счет введения
наполнителей (а'-переход) или присутствия полярных групп,
(л-переход), приводит к увеличению прочности. Так, незапол-
ненный аморфный цис-1,4-полибутадиен имеет кратковременную
прочность при растяжении 1—1,5 МПа, а для ненаполненного
сополимера бутадиена с нитрилом акриловой кислоты (60:40)
прочность повышается до 8 МПа. Наполнение цис- 1,4-полибу-
тадиена техническим углеродом приводит к еще большему росту
прочности (до 20 МПа).
Поскольку при деформации для релаксационных характерис-
тик справедлив закон температурно-временной аналогии, то ц.
при разрушении прочность определяется скоростью деформиро-
вания и температурой и для прочностных характеристик ха-
рактерен принцип температурно-временной суперпозиции при
условиях неизменности структуры (отсутствие кристаллизации)
и механизма разрушения. Зависимость разрушающего напряже-
ния оР от температуры и скорости деформирования подчиняется
уравнению
ap-40e,/mexp (UP/mkT), (5-57)
где Ло= (/пС1£п»л),/т; С| — константа в уравнении (5.56); Епя—.модуль
высокоэластичности; е — скорость деформации; ;п=1+5 (Ь — константа в
уравнении (5.56)].
Таким образом, разрушение полимеров в высокоэластическом
состоянии определяется в основном релаксационными процесса-
ми, а вклад термофлуктуационпого разрыва невелик в отличие
от хрупкого разрушения, где термофлуктуационный разрыв хи-
мических связей является основным.
Повышение температуры приводит к снижению прочности
полимеров, повышение скорости деформирования, наоборот, по-
вышает прочность. Для оценки области безопасных напряжений
и удлинений для эластомеров при различных Т и е обычно
строят кривую, называемую «.огибающей разрывов» (рис. 5.39).
В области 1 полимер сохраняет способность сопротивляться раз-
рушению, а в области II— разрушается.
Разрушение полимеров выше температуры пластичности.
Разрушение при Т^Тп сопровождается появлением участков
вязкого течения, т. е образованием «шейки» в наиболее слабом
месте. При этом существует вероятность образования «микро-
шеек» сразу в нескольких местах. Несмотря на некоторое их
333
t
чае происходит по «шейке» при
Рис. 5 39 «Огибающая разрывов»
(Д, Б, С, D, Е, F — точки разрыва;
стрелка — повышение скорости или
снижение температуры)
/ — область безопасного деформирования;
// — область разрушения
сходство с «шейкой», возни-
кающей при вынужденно-эла-
стической деформации, струк-
тура их различается. Пласти-
ческая «шейка» характеризу-
ется неупорядоченной струк-
турой. Разрушение в этом слу-
невысоких напряжениях В зоне
разрыва четко проявляются следы необратимой пластической
деформации. Прочность характеризуется пределом текучести от,
который, как правило, совпадает с разрушающим напряжени-
ем оР. Иногда ор называют когезионной прочностью. Механизм
разрушения при Т^Ти имеет релаксационный характер и зави-
сит от температуры и скорости деформирования. С увеличением
скорости деформирования уменьшается доля пластического раз-
рыва и вероятность образования пластических «шеек», и при
1, МПа
о. МПа
достаточно высоких значениях скорости разрушения полимер
разрушается как высокоэластическое тело (рис. 5.40). При Т>
>ТГ начинается вязкое течение полимера
Разрушение кристаллических полимеров. Разрушению кри-
сталлических полимеров или полимеров, кристаллизующихся в
процессе растяжения, предшествуют значительные деформации.
Полимер характеризуется
очень высокой ориентацией
цепей и ведет себя как твер-
дое упругое тело. Поэтому
механизм разрушения кри-
сталлических полимеров
приближается к термофлук-
туационному и при высокой
степени ориентации поли-
меры могут иметь предель-
Рис. 5.40. Кривые напряжение де-
формация /—е линейного цис-1,4-
полиизолрена(/, 2,3) и нерегу-
лярного полибутадиеиа (Г, 2', 3')
при различных скоростях растя-
жения при 295 К
/. /'—0.16 м/с; 2, 2' — 4.5 м/с: 3. 3'
— 30 м/с; S— начало образования пла-
стической шейки; 4— момент разрыва
334
Рис. 5 41. Зависимость прочности оР от
скорости растяжения и кристаллизующего-
ся полимера
но достижимую прочность. При раз-
рушении эластомеров, кристалли-
зующихся при растяжении, опреде-
ленный вклад вносит и релаксаци-
онный механизм деформирования,
характерный для высокоэластиче-
ского разрушения Повышение проч-
ности кристаллизующихся полиме-
ров связано как с увеличением чис-
ла цепей, проходящих через зону
разрыва (т. е. повышение степени
ориентации), так и с вкладом гистерезисных потерь за счет ре-
лаксационного характера деформирования.
О влиянии релаксационных явлений на прочность кристалли-
зующихся эластомеров свидетельствует немонотонная зависи-
мость прочности от скорости растяжения (рис. 5.41). На участ-
ке АБ происходит кристаллизация полимера (образование фиб-
риллярной структуры), при этом повышается степень ориента-
ции молекул и в кристаллической части, и в аморфной. Трещины
или надрывы зарождаются в аморфной области или на границе
кристалл — аморфная часть, и прочность определяется прочно-
стью аморфных участков Поскольку при кристаллизации повы-
шается степень их ориентации, а следовательно, и прочность,
то можно считать, что кристаллизация приводит к упрочнению.
В процессе деформирования на участке БВ макромолекулы не
успевают принять необходимую для кристаллизации конформа-
цию и кристаллизация замедляется, а на участке ВГ полимер
не кристаллизуется и прочность определяется степенью ориен-
тации макромолекул.
Таким образом, кристаллические полимеры разрушаются по
механизму, характерному для твердых ориентированных тел с
малодефектной структурой Разрушение аморфно-кристалличе-
ских полимеров происходит по аморфной части по механизму,
определяемому условиями процесса (температурой и скоростью).
Разрушение полимеров при динамических нагрузках. Разру-
шение полимеров под действием, циклических деформаций про-
исходит в результате динамической усталости или утомления.
Динамическая усталость — это снижение прочности под влияни-
ем многократных периодических нагрузок
Испытание полимеров проводят в трех режимах: при посто-
янной амплитуде деформации eo=const; при постоянной ампли-
туде напряжения oo=const; при постоянной работе цикла
= const. Динамическая усталость характеризуется временем до
зза
разрушения тц или числом циклов до разрушения Afu; Wu=tu®
(где со — частота деформации) Напряжение, при котором про-
исходит разрушение после действия заданного числа циклов,
называется усталостной прочностью сц. Кривые зависимости
Си—J^u называют кривыми усталости Между ow и JVtt сущест-
вуют эмпирические соотношения
для сшитых эластомеров О№ОР^_,/₽; (5.58)
для пластмасс N^ — Се °N •, (5.59)
для волокон и пленок 0№Ор — Л lg N,, (5.60)
где р С, D, А — эмпирические константы, характеризующие скорость сниже-
ния прочности прн циклических деформациях.
Разрушение полимеров при многократном циклическом на-
гружении определяется теми же факторами, что и при статиче-
ских режимах; между временем разрушения т и напряжением
a=const при статическом режиме временем до разрушения тц
и максимальным напряжением за цикл о0 при циклическом су-
ществуют следующие соотношения
тот=С\; T«Oom“Cj, (5.61)
где m — константа, одинаковая для обоих режимов; Сг и Сг— постоянные,
С\
Отсюда следует, что при динамическом нагружении, так же
как и прн статическом, определяющим является напряжение и
разрушение обусловлено термофлуктуационным распадом свя-
зей, наличием дефектов и релаксационными процессами.
Статический режим является более «мягким» по сравнению
с динамическим несмотря на то, что в первом случае полимер
все время находится в напряженном состоянии, а во втором —
периодически разгружается Причин, обусловливающих более
низкую динамическую выносливость, несколько. Во-первых, это
релаксационные (гистерезисные) потери. При статическом ре-
жиме процесс релаксации напряжения или деформации за время
испытания протекает практически полностью до постоянных
значений ом или 8<». При периодических нагрузках за время де-
формации I/ю (о> — частота) перенапряжения в полимере не
успевают отрелаксировать и накапливаются с каждым циклом.
С ростом частоты деформации гистерезисные потери возрастают
и достигают максимума при условии т<о==1. Эти потери, как уже
говорилось, оказывают одновременно и положительное влияние
на долговечность, и отрицательное вследствие выделения тепла.
При низкой теплопроводности полимеров и плохом отводе тепла
саморазогрев может быть значительным. В зависимости ст ре-
жима нагружения гистерезисные потерн, а следовательно, и сте-
пень саморазогрева будут изменяться с ростом модуля упруго-
сти полимера Е по-разному: с увеличением Е в режиме е0=
336
Рис. 5 42. Зависимость усталост-
ной прочности <J.v от числа цик-
лов N при динамическом нагру-
жении в отсутствие (/) саморз-
зогрева и при саморазогреве (2)
(объяснение см. в тексте)
=const потери возрастают,
в режиме о0=const —
уменьшаются, а при Д1Г=
= const не изменяются
Влияние саморазогрева на
динамическую усталость
особенно значительно в об-
ласти средних значений
W и о (рис. 5 42). В области I образец выдерживает небольшое
число циклов до разрушения, саморазогрев мал и Л^ц=/(ор),
т. е. определяется кратковременной прочностью. В области III
напряжение приближается к безопасному, релаксация протека-
ет практически полностью и саморазогрев незначителен; Л'ц=
=о)т (т — время до разрушения). Область // характеризуется
наибольшими гистерезисными потерями. Если учесть негатив-
ную роль саморазогрева, обусловленного гистерезисными поте-
рями, то становится ясным, что в режиме eo=const лучшей вы-
носливостью будут обладать мягкие низкомодульные полимеры,
а в режиме o0=const— жесткие, высокомодульные.
Вторая причина снижения динамической выносливости по
сравнению со статической — локальные разогревы в микрооб-
ластях у вершин микротрещин при их реете. Например, при
неизменной средней температуре образца локальное повышение
температуры при разрушении твердых органических полимеров
составляет 120—200 К при скорости роста трещины 0,9—1,5 м/с.
С повышением скорости роста трещины до 200—600 м/с локаль-
ные перегревы возрастают до 500 К у органических полимеров
и до 3000 К у неорганических. В результате значительных ло-
кальных разогревов в микрообластях размером около 1 мкм
прочность полимера в этой области снижается, что при нарас-
тающем числе таких областей приводит к уменьшению динами-
ческой выносливости. Еще одной причиной снижения динамиче-
ской выносливости являются механохимические реакции, со-
провождающие деформирование и разрушение и приводящие к
появлению в системе свободных радикалов, взаимодействующих
с полимером, кислородом и т. д. Напряжение оказывает акти-
вирующее влияние на эти реакции. Ускорение химических реак-
ций в полимере под действием механического напряжения обус-
ловлено увеличением внутренней энергии системы, смещением
уровней энергии внутри- и межмолекулярных взаимодействий.
22—816
337
Например, при растяжении полимерных пленок внутренняя
энергия увеличивается на несколько кДж/моль, при воздействии
ударных волн — на десятки кДж/моль Вследствие неоднород-
ности структуры полимера избыточная энергия неравномерно
распределяется по его объему. В аморфных участках и в обла-
стях, где находятся наиболее напряженные макромолекулы,
плотность избыточной энергии может быть на один-два порядка
выше среднего уровня и делается равной энергии химических
связей.
Механические напряжения могут ускорять химические про-
цессы по многим причинам. Так, уменьшение энергии химиче-
ских связей в напряженном материале вызывает уменьшение
энергии активации химических реакций Кроме того, прн цик-
лических нагрузках, когда время релаксации полимера больше
продолжительности действия внешней силы, избыточная упру-
гая энергия превращается в энергию колебательного возбужде-
ния химических связей, что увеличивает их реакционную спо-
собность. Вследствие гистерезисных потерь в местах выделения
избыточной’ энергии развиваются высокие давление и темпера-
тура, что также ускоряет химические процессы. Роль гистере-
зисных потерь иллюстрирует следующее уравнение:
N* = (7?Г/Д£Z) In (Ор/Оо), (5 62)
I
где \U—изменение потенциального барьера разрушения за цикл;
(i\Q — гистерезисные потери; Q — коэффициент, характеризующий долю гисте-
резисных потерь, расходуемых на изменение потенциального барьера разру-
шения.
Наконец, При разрывах макромолекул, как мы уже сказали,
образуются свободные радикалы, ионы и другие активные цент-
ры химических реакций и протекают многочисленные электрон-
ные процессы, сопровождающие деформацию полимера Резуль-
татом протекания механохимических процессов является необ-
ратимое изменение структуры полимера
Еще одной причиной снижения динамической выносливости
является наложение микропапряжений, возникающих в данном
цикле, на остаточные микронапряжения, возникающие в преды-
дущих циклах и связанные со структурными перестройками в
микрообъемах. Отсюда следует определяющая роль числа цик-
лов при определении динамической усталости.
Таким образом, снижение динамической выносливости про-
исходит вследствие релаксационных потерь, приводящих к са-
моразогреву, локальных разогревов: повышения микронапряже-
ний в микрообъемах, протекания механохимических реакций и
необратимого изменения структуры. Вклад того или иного фак-
тора определяется механизмом разрушения, зависящим от ре-
жима нагружения, физического состояния полимера и др.
338
Динамическая усталость является сложной характеристикой
и определяется кратковременной прочностью, релаксационными
гистерезисными потерями, стойкостью к окислению, зависит от
.амплитуды и частоты деформации:
N4 = (<Тр/а0)₽о; = (ер/е0)₽е, (5.63),
‘Ор и Ср — напряжение и удлинение при разрыве; о0 и Со — амплитуды на-
пряжения и деформации; и ре— коэффициенты динамической выноелнво-
•сти, характеризующие снижение прочности с увеличением числа циклов; ко-
эффициенты и ре можно рассчитать, если уравнения (5.58) и (5.63) пред-
ставить в логарифмических координатах 1g — 1g У и 1g М,—- 1g о0; зави-
симости О№=/(Л’) и Wu=f(ao) в этих координатах выражаются прямыми
линиями, по тангенсу угла наклона которых к оси абсцисс можно рассчи-
тать величину р (рнс. 5.43).
К числу факторов, от которых зависит коэффициент р, отно-
сятся стойкость полимеров к реакциям окисления и гистерезис-
ные потери. Чем больше р, тем выше сопротивление полимера
действию многократных деформаций.
Даже более прочные в статических условиях резины могут
иметь меньшую усталостную выносливость при невысоких зна-
чениях р (см. рис. 5 43). Высокой динамической выносливостью
характеризуются высокопрочные полимеры, стойкие к окисле-
нию, с невысокими гистерезисными потерями.
Из уравнения (5.63) и рис. 543 следует, что снижение ам-
плитуды деформации е0 или напряжения оо повышает динами-
ческую выносливость Но скорость снижения с ростом в0 или
•оо непостоянна и изменяется в области больших и малых зна-
чений амплитуд. Как видно из рис. 5.44, существует значение е0
(или оо), при котором Л'ц практически не зависит от амплитуды.
•Это безопасные амплитуды е0* (о0*), и при ео<8о* или о0<оо*
полимер может эксплуатироваться в течение длительного вре-
Рис. 5.43. Зависимость усталостной прочности on от числа циклов N (а)
и динамической усталости NK от амплитуды напряжений о0 двух резин
(оР>, aw — кратковременная прочность)
22*
339
Рис. 5.44. Зависимость числа циклов до
разрушения NK от амплитуды деформации
е« в режиме е0»= const
мени Существуют и такие значения
амплитуд, выше которых образец
разрушается очень быстро. Это кри-
тические значения напряжений ао*р
и деформаций 8окр; при е>еокр или
о>аокр динамическая усталость оп-
ределяется в основном уровнем
кратковременной прочности. В об-
ласти амплитуд 8о*<ео<8окр динамическая усталость зависит
от прочностных, гистерезисных свойств и от стойкости к окис-
лению.
Влияние температуры на динамическую выносливость неод-
нозначно и зависит от режима деформирования и среды, в кото-
рой эксплуатируется полимер. В инертной среде, где скорость
механохимических реакций невысока, член ДЛ7 в уравнении
(5.62) невелик, и повышение температуры приводит к росту ди-
намической усталости Эго обусловлено ускорением релаксаци-
онных процессов, что приводит к снижению напряжения в системе
и сдвигу в сторону более мягких режимов. В среде кислоро-
да, озона влияние температуры зависит от режима деформиро-
вания. При 8о>8о* наблюдается наибольший саморазогрев,
увеличивается А (7 и усталостная выносливость снижается. При
ео<8о* саморазогрев незначителен и усталостная выносливость
определяется температурой окружающей среды. Повышение
температуры в интервале, в котором вероятность термодеструк-
ции мала, способствует выравниванию локальных перенапряже-
ний и приводит к росту динамической выносливости.
Влияние частоты деформации определяется изотермичностью
процесса. Повышение частоты в неизотермических условиях
(например, при многократных деформациях массивных изделий,
изготовленных из материала с плохой теплопроводностью) при-
водит к снижению числа циклов до разрушения ввиду высокого
теплообразования в системе и интенсивного протекания реакций
окисления. Если же деформирование полимера происходит в
изотермических условиях (например, тонкостенных изделий с
хорошей теплопроводностью) при наличии агрессивного реаген-
та (например, озона), то скорость усталостного разрушения Мц
определяется зависимостью
Л=ф(//)+й>3/<о. (5.64)
где ф(//)—член, характеризующий скорость разрастания дефекта под влия-
нием механических факторов; со3 — концентрация озона; ю — частота нагру-
жения.
340
При ео<ео* $(Н)<&Со3/(>) и усталостная выносливость опре
деляется концентрацией агрессивного агента, а при Co3 = const
динамическая усталость повышается с ростом <о; при жестких
режимах нагружения выносливость определяется механическим
фактором ф(Я), т. е. упругогистерезиснымн и прочностными
свойствами. При высоких частотах нагружения гистерезисные
потери минимальны и практически не изменяются, поэтому и ди-
намическая долговечность в интервале скоростей 50—500 циклов
в минуту также не изменяется.
Рассмотрим особенности динамической выносливости стекло-
образных и высокоэластических материалов В стеклообразном
состоянии полимеры характеризуются высоким модулем упру-
гости и очень малыми гистерезисными потерями. Для этих ус-
ловий уравнение (5.62) будет иметь вид
Nn-RTln (ар/ао). (5.65)
Отсюда следует, что при незначительных локальных перегре-
вах повышение температуры положительно сказывается на ди-
намической выносливости. Существенное влияние оказывает и
режим испытания: так, при Oo=const высокомодульные полиме-
ры (8=о/£) характеризуются большей долговечностью. Поэтому
пластмассы, которые в условиях эксплуатации находятся в
стеклообразном состоянии, имеют большую динамическую вы-
носливость в режиме ao = const. При многократных деформациях
в режиме eo=const динамическая выносливость высокомодуль-
ных материалов снижается.
У полимеров, находящихся в высокоэластическом состоянии,
гистерезисные потери намного выше по сравнению со стеклооб-
разными. Поэтому все рецептурные и технологические факторы,
приводящие к снижению потерь (замена каучука на более гиб-
кий, повышение гибкости за счет введения небольшого количе-
ства пластификатора и др.), способствуют повышению динами-
ческой выносливости. Мягкие резины с невысоким модулем ха-
рактеризуются большей выносливостью при работе в режиме
Eo = const, а жесткие — в режиме oo = const Наполнители, напри-
мер технический углерод, оказывают сложное влияние на дина-
мическую усталость: при е0^е0* определяющим фактором
является способность наполнителя ускорять или ингибировать
окисление, а при eo~^eoKp влияние наполнителя на Nu зависит
от его влияния на уровень гистерезисных потерь — чем в боль-
шей степени наполнитель увеличивает потери, тем больше сни-
жаются усталостная прочность и динамическая выносливость.
Экспериментальное определение прочности полимеров в ста-
тических и динамических условиях показывает, что существует
значительный разброс показателей. Из теории Гриффита сле-
дует, что прочность зависит от вида дефекта, его размера, фор-
341
u
XU)
Рис. 5.45. Распределение микро-
трещин р(/0) по размерам /0
мы и места расположения.
Поэтому разброс показате-
лей является следствием
многообразия дефектов и
слабых мест в полимере.
Статистическая теория проч-
ности объясняет этот факт
следующим образом:
1) в каждом теле есть дефекты разного вида, различной
степени опасности, которые распределяются по объему стати-
стически (рис. 5.45);
2) разрушение начинается с наиболее опасного дефекта;
3) с увеличением объема образца увеличивается число де-
фектов и вероятность наличия опасного дефекта;
4) поскольку распределение тепловых флуктуаций по объ-
ему имеет случайный характер, с уменьшением объема образца
вероятность термофлуктуацнонного развития трещины сннжа-
ется и прочность растет.
Таким образом, из статистической теории следует, что раз-
брос показателей прочности является закономерным Кривые
распределения по прочности представлены на рис. 5.46. Центр
тяжести площади, ограниченной кривой и осью абсцисс (точки
А, В, С), характеризует среднюю прочность, а До', До", До'" —
разброс показателей. С увеличением толщины образца увеличи-
ваются среднее значение прочности и разброс показателей. За-
висимость прочности от объема образца называют «масштабным
фактором» Он характеризует не только кратковременную проч-
ность, но и долговечность в статических и динамических усло-
виях. В табл 5.3 приведены данные о прочности капронового
волокна в зависимости от длины наиболее опасной трещины 1о
Приведенные дан-
ные еще раз подчерки-
вают существенную
роль дефектов в сни-
жении реальной проч-
Рис. 5.46 Кризые распреде-
ления р(ор) по ор для об-
разцов сшитых эластомеров
(СКС-30) различной тол-
щины:
/ — 2.2 мм; 2—1.2 мм; 3 —
3.4 мм
342
Таблица 53. Зависимость прочности капронового волокна от длины трещины
/о-IO4. мм ар. МПа % Pi-On/Op
29 142 6 2761
2,8 422 20 g макротрещины
1.8 512 31 7,61
1.3 585 36 6,7 ) микротрещины
0,9 690 39 5,7 J
*0,6 820 43 4,81
О.З 1050 57 3,7 } субмикротрещины
0.2 1230 77 3,2 J
.Примечание, — коэффициент концентрации напряжений.
кости по сравнению с предельно достижимой оп. Особенно зна-
чительно снижаются прочность и относительная деформация
при разрыве ер при наличии грубых дефектов — макротрещин.
Прочность полимеров, в которых имеются субмикротрещины,
ближе к предельной.
Итак, разрушение полимеров под действием нагрузки проис-
ходит в результате проскальзывания макромолекул относитель-
но друг друга и разрыва химических связей (назовем это меха-
ническим фактором) и сопровождается необратимым изме-
нением структуры вследствие интенсивного протекания
механохимических реакций (химический фактор). Прочность
повышается с ростом степени ориентации макромолекул в на-
правлении действия силы и снижается с увеличением дефектно-
сти материала.
Влияние структуры полимера и условий испытаний на проч-
ность. При эксплуатации полимерных изделий их разрушение
происходит в самых разнообразных условиях- при растяжении,
сжатии, изгибе, срезе, в результате проколов, надрезов, истира-
ния и т. д. Поэтому прочностные свойства характеризуют обыч-
но несколькими показателями, определяемыми при разных
условиях деформирования Поскольку прочность зависит от
скорости и температуры испытания, прочностные показатели
определяют при постоянных скорости деформирования и темпе-
ратуре. Кратковременную прочность оценивают по разрушаю-
щему напряжению при растяжении, сжатии, изгибе, срезе в
обычных условиях при невысоких скоростях деформирования
(~0001—0,5 м/мин). Для некоторых полимеров определяют
сопротивление разрушению при ударных воздействиях нагрузки
со скоростью 2—4 м/с. Этот показатель называется ударной
вязкостью (или ударной прочностью). Он представляет собой
отношение работы разрушения Лраэр к площади поперечного
сечения образца <S0.
343
Разрушение полимеров при истирании характеризуют исти-
раемостью а, представляющей собой отношение потерянной за
определенное время истирания массы Д/n (или объема ДУ)
образца к работе, затраченной на истирание А, или показателем,
обратным истираемости — сопротивлением истиранию 3-
Прочность образцов, содержащих дефекты в виде надрезов,
проколов, принято оценивать сопротивлением раздиру (отноше-
ние разрушающей нагрузки к толщине образца) или удельной
энергией раздира, представляющей собой работу образования
единицы площади поверхности образца.
Длительную прочность, долговечность, усталость в статиче-
ских условиях определяют как правило, временем до разруше-
ния т, а в динамических условиях — усталостной прочностью оу
(т. е. кратковременной прочностью образца при растяжении,
сжатии и т. д. после действия на него N циклов напряжения)
или числом циклов до разрушения образца.
Кратковременная прочность определяется преимущественно
механическим фактором, поскольку за время действия силы не-
обратимые изменения структуры полимера вследствие протека-
ния механохимических реакций минимальны. На длительную
прочность существенное влияние оказывает и химический
фактор.
Влияние структуры на кратковременную прочность опреде-
ляется физическим и фазовым состоянием полимера. При Т<
<ГХр полимеры разрушаются по хрупкому механизму и проч-
ность оценивается разрушающим напряжением оХр. Хрупкое
разрушение сопровождается малыми деформациями, при этом
не происходит изменения исходной надмолекулярной структуры
и дополнительной ориентации макромолекулы. Поэтому опреде-
ляющими факторами являются прочность химических связей в
полимере и его дефектность. При одинаковой степени дефектно-
сти Охр повышается с ростом энергии химических связей в основ-
ной цепи полимера. Так, для карбоцепных полимеров (полиизо-
прена, полнхлоропрена, сополимеров бутадиена со стиролом или
нитрилом акриловой кислоты) оХр находится на одном уровне
и составляет примерно 100 МПа. Введение в углеводородную
цепь атомов меньшей энергии приводит к снижению оХр. У по-
лисульфидов, содержащих в цепи серные связи, характеризую-
щиеся меньшей, чем углеродные, энергией, охр снижается до
50—20 МПа.
Особо заметное влияние на охр оказывает дефектность мате-
риала [коэффициент х в уравнении (5.50) может достигать
значений до 1000], поскольку в этом состоянии релаксационные
процессы замедлены и диссипация перенапряжений затруднена.
Дефекты структуры, особенно микротрещины, действуют как
эффективные концентраторы напряжения: напряжение в верши-
344
не трещины резко возрастает и разрушение наступает при мень-
ших значениях прикладываемого напряжения.
Возможность образования микротрещин в полимерах связа-
на с наличием в них значительного свободного объема (см. гл 4).
Микротрещины возникают, как правило, на границах надмоле-
кулярных образований и в дефектных участках самих структур.
Поэтому чем меньше размеры надмолекулярных структур в
аморфных и кристаллических полимерах, чем выше плотность
упаковки макромолекул в надмолекулярных структурах и самих
структур, тем в меньшей мере снижается прочность по сравне-
нию с предельно достигаемой. Кристаллические полимеры ха-
i рактеризуются большой плотностью упаковки по сравнению с
‘ аморфными, и для них охр, как правило, выше и существенно
зависит от степени кристалличности и морфологии кристаллов
Ниже приведены значения охр некоторых полимеров в аморфном
(А) и кристаллическом (К) состояниях-
Структура сгр, МПа
Полиакрилат К, глобулярная 64
К, фибриллярная 74
Полихлоропрен А 3!
К, сферолитная, К —15%
£>*<1 34
D=l—3 25
£)=40-60 18
К. сферолитная, Ко=21% 48
К» фибриллярная 217
Полиэтилеитерефталат А 130
К 124
• D — диаметр сферолита, мкм
Из приведенных данных следует, что охр увеличивается с
ростом степени кристалличности за счет повышения пло'Тности
упаковки и упорядоченности системы. Однако наличие границы,
раздела кристалл — аморфная часть создает на границе разде-
ла перенапряжения, которые тем больше, чем больше размер
кристаллов, поэтому охр снижается с увеличением размеров
кристаллов. В некоторых случаях перенапряжения могут быть
определяющим фактором и охр кристаллических полимеров
остается на уровне прочности аморфных.
Повышение прочности (ав 10 раз) при переходе от сферо-
литной к фибриллярной структуре связано со значительной
ориентацией макромолекул в фибриллярных кристаллах Еще
в большей мере проявляется роль ориентации при разрушении
стеклообразных полимеров (нехрупкие стекла) в области тем-
ператур 7'хр<7'^7'с, где прочностные свойства определяются
способностью материала образовывать «шейку». В этом случае
345
показателем прочности является разрушающее напряжение Ор.
Все факторы, способствующие развитию ориентационных про-
цессов (повышение молекулярной массы, конфигурационной ре-
гулярности), приводят к росту прочности. Негативная роль пе-
ренапряжений на дефектах структуры в этих условиях снижа-
ется, поскольку становится возможной релаксация напряжения,
в вершине микротрещины и выравнивание напряжения в си-
стеме.
Прочностные свойства полимеров в высокоэластическом со-
стоянии (при Ти>Т>Тс) определяются дефектностью структу-
ры, релаксационными и ориентационными эффектами (определя-
ющая роль при разрушении в процессе растяжения). Дефектные
участки создают перенапряженные зоны, но ввиду релаксацион-
ного механизма разрушения полимеров уровень опасности де-
фекта зависит от степени релаксации напряжения. Чем выше
скорость релаксации, тем значительнее диссипация напряжения,
тем ниже перенапряжение в вершине микротрещин и, следова-
тельно, выше прочность.
При деформировании низкомолекулярных линейных полиме-
ров разрушение происходит не в результате разрыва макромо-
лекул, а вследствие их скольжения относительно друг друга.
С увеличением степени полимеризации растут длина макромо-
лекул, число межмолекулярных связей, вероятность образования
механических зацеплений. При определенных, достаточно высо-
ких значениях степени полимеризации лкр, вследствие ориента-
ции и плотности упаковки силы межмолекулярного взаимодей-
ствия по всей длине макромолекулы делаются равными прочно-
сти химических связей в цепи. Начиная с этого момента (Мкр)
прочность ориентированного полимера при увеличении степени
полимеризации не возрастает. Расширение молекулярно-массо-
вого распределения за счет наличия низкомолекулярных фрак-
ций всегда приводит к снижению прочности. Увеличение раз-
ветвленности макромолекул повышает число дефектных мест
(точек разветвления), снижает степень ориентации макромоле-
кул, увеличивает время релаксации, т. е. уменьшает диссипацию
напряжений. Все это также способствует снижению прочности
Химические узлы между линейными макромолекулами пре-
пятствуют их скольжению под действием механических нагрузок
и, следовательно, способствуют повышению прочности. Чем
больше таких узлов, тем выше напряжение и меньше удлинение
при разрыве. В наибольшей степени влияние сетки химических
связей на прочность проявляется при разрушении в высокоэла-
стическом состоянии, т. е. для эластомеров. Зависимость проч-
ности от степени сшивания в этом случае описывается кривой
с максимумом при оптимальном числе узлов псопт. Величина
псопт определяется гибкостью полимера и молекулярной .массой
механического сегмента Л1СГ. Если молекулярная масса между
-346
узлами сетки Мс намного превышает Мсг, то сетка не препятст-
вует ориентации макромолекул при деформации, снижает веро-
ятность скольжения макромолекул при нагружении и, следова-
тельно, упрочняет материал. При большой плотности сетки,
когда Мс-*-Мсг, резко снижается способность макромолекул к
ориентации, растет дефектность структуры (вследствие развития
реакций окисления, изомеризации, взаимодействия с различны-
ми добавками и др.) и прочность снижается. С ростом гибкости
цепи Mvr повышается, и поэтому лсопт сдвигается в сторону боль-
ших значений. При МС=МСГ полимер теряет способность к ори-
ентации, переходит в псевдостеклообразное состояние и разру-
шается по хрупкому механизму
Характер поперечных связей также оказывает определенное
влияние на прочностные свойства- более гибкие связи обеспечи-
вают большую прочность при более высоких значениях лсопт.
Повышение доли активной сетки приближает материал к иде-
альному, т. е. приводит к росту прочности. На разрушающее
напряжение оказывает влияние уровень межмолекулярного вза-
имодействия (плотность флуктуационной сетки), определяемый
присутствием полярных групп, их расположением, полярностью.
При одинаковом числе поперечных химических связей прочность
и удлинение при разрыве сополимеров бутадиена с нитрилом
акриловой кислоты (НАК) повышаются с ростом содержания
НАК в сополимере (в ряду СКН-18, СКН-26, СКН-40). Поло-
жительная роль связей, обладающих низкой энергией, в разру-
шении полимеров обусловлена релаксационными процессами,
которые наиболее сильно проявляются у полярных полимеров.
Влияние межмолекулярных связей особенно велико при растя-
жении в направлении, перпендикулярном ориентации макромо-
лекул, или при сдвиге в направлении ориентации.
Когезионная прочность линейных полимеров при Тп<Г<7'т
определяется плотностью флуктуационной сетки и способностью
макромолекул ориентироваться при деформации. Увеличение
плотности физических узлов в результате роста полярности по-
лимера, его молекулярной массы, разветвленности п других
факторов повышает предел пластичности оп, но снижает способ-
ность к ориентационному упрочнению. Поэтому когезионную
прочность некрнсталлизующихся полимеров обычно повышают
модификацией полимера, приводящей к росту плотности флук-
туационной сетки, а у кристаллизующихся — за счет усиления
способности к ориентации и кристаллизации. Так, синтетический
цис-1,4-полиизопрен (СКИ-3) имеет значительно меньшую ко-
гезионную прочность по сравнению с натуральным каучуком.
Модификация СКИ 3 полярными агентами (нитрозосоединения-
ми, акрилатами и др.), введение микрогеля повышают плотность
флуктуационной сетки и, следовательно, когезионную прочность
до уровня прочности натурального каучука.
347
Прочность кристаллических или аморфных полимеров, кри-
сталлизующихся при деформации, определяется способностью
полимера к перестройке исходной надмолекулярной структуры
при деформировании.
Повышение молекулярной массы, возникновение редких хи-
мических поперечных связей препятствуют скольжению макро-
молекул и тем самым способствуют ориентации, кристаллизации
и упрочнению. Если исходная кристаллическая структура поли-
мера такова, что при деформации возможно ее разукрупнение
и скольжение образующихся блоков в направлении деформиро-
вания без их разрыва, то прочность и разрывное удлинение до-
вольно высоки. Так, при уменьшении размера сферолитов в изо-
тактическом полипропилене с 300—500 до 10—20 мкм происхо-
дит повышение прочности при разрыве от 6 до 30 МПа,
а относительное удлинение растет от 5—7 до 600%.
Свойства полимерных материалов можно регулировать, из-
меняя их состав. Наибольшее влияние на механические свойства
оказывают пластификаторы, наполнители, армирующие мате-
риалы. Введение пластификаторов способствует снижению тем-
пературы стеклования полимера (что расширяет температурную
область эксплуатации полимерных материалов), но снижает
модуль упругости и прочность, увеличивает долю пластических
деформаций и текучесть в вязкотекучем состоянии. Влияние
наполнителей на прочность полимеров неоднозначно. С одной
стороны, введение твердых частиц в полимерную матрицу соз-
дает на границе раздела полимер — наполнитель дополнитель-
ные перенапряжения (дефектные зоны), которые снижают проч-
ность. Уровень дефектности определяется прочностью связи
полимер — наполнитель. С другой стороны, наполнитель изме-
няет структуру: в наполненных материалах увеличивается доля
слабых адсорбционных связей и повышается ориентация макро-
молекул в направлении действия нагрузки, что способствует
росту прочности. В стеклообразном состоянии наполнители сни-
жают прочность, в высокоэластическом — проявляется их упроч-
няющая роль; в последнем случае зависимость прочности от
содержания наполнителя описывается немонотонной кривой с
максимумом при оптимальной концентрации <рОпт, которая оп-
ределяется структурой полимера (в основном гибкостью)
и физико-химическими свойствами наполнителя (размером час-
тиц, свойствами их поверхности). Чем ниже гибкость поли-
мера и больше активность наполнителя (например, меньше раз-
мер частиц), тем меньше фОПт- Снижение прочности при кон-
центрациях наполнителя, превышающих оптимальную, обу-
словлено уменьшением ориентирующего влияния наполнителя.
Зто объясняет тот факт, что кристаллизующиеся полимеры или
сильно сшитые резины (эбониты) не упрочняются при наполне-
нии
34S
В кристаллизующихся полимерах наполнитель сосредоточи-
вается преимущественно в аморфной части, где его оптимальная
концентрация оказывается превышенной. Это затрудняет ори-
ентацию макромолекул в аморфной части, и прочность напол-
ненных кристаллизующихся каучуков не только не увеличива-
ется, но даже несколько снижается по сравнению с прочностью
ненаполненных. В эбонитах подвижность макромолекул настоль-
ко снижена из-за высокой плотности химических сшивок и внут-
римолекулярного присоединения вулканизующего агента, что их
можно рассматривать как стеклообразные полимеры, в которых
эффект упрочняющего действия наполнителей отсутствует.
Среди наполнителей особую группу образую! армирующие
материалы К ним относятся стеклянные, асбестовые, борные,
углеродные волокна, монокристаллы оксида алюминия, карбида
кремния и др Отличительной особенностью полимерных компо-
зиций, содержащих волокна, является анизотропия свойств.
Поэтому для характеристики деформационных и прочностных
свойств используют несколько показателей Если волокна ори-
ентированы преимущественно в одном направлении, то опреде-
ляют продольный модуль Юнга EL (растягивающее напряжение
а направлено вдоль оси ориентации волокон), трансверсальный
модуль Юнга Ет (о направлено перпендикулярно оси ориентации
волокон); при сдвиге также определяют GL и GT.
Для композиций с длинными ориентированными в одном на-
правлении волокнами (£/£>>100) определяют продольную проч-
ность при растяжении op,t, трансверсальную ор.т и сдвиговую от-
Прочность композиции зависит от угла расположения волокон
0, при 0^5° разрушение определяется величиной вр.с, при 0 в
интервале 5—45° — величиной от, а при 0^45° характер раз-
рушения определяется величиной ар,т:
Op.t оР.пфп-|-ор.»ф»; (5.66)
ат*«0,5от.п; Gp >e = Op.»/sin 0. (5 67)
(Здесь индекс «п» относится к полимеру, «в» — к волокну).
Таким образом, продольная прочность повышается при вве-
дении армирующего наполнителя, а трансверсальная — зависит
от угла укладки волокон.
Для однонаправленных волокнистых композиций характерно
значительное повышение модуля в направлении действия силы
(j^b 10 раз), но в других направлениях модули могут быть
низкими. Для повышения жесткости композиции в нескольких
направлениях волокна либо распределяют хаотически, либо
укладывают под разными углами (слоистые структуры).
Армирующие волокна в полимере выполняют двоякую роль:
с одной стороны, они повышают степень ориентации системы,
в том числе и полимера, а с другой — точки контакта волокна
с полимером являются концентраторами напряжения. Поэтому
349
прочность композиций зависит от длины волокна и прочности
связи на границе раздела полимер — волокно, т е. адгезионной
прочности.
Существует критическая длина волокна LKf>, ниже которой
упрочняющий эффект не проявляется. Для достижения макси-
мальной прочности отношение L/D должно быть не менее 100.
Адгезионная прочность оказывает наиболее существенное
влияние на трансверсальную прочность, а на продольную —
только в случае очень коротких волокон. Обычно ор.т ниже проч-
ности полимерной матрицы, ио за счет повышения адгезии мож-
но добиться роста ор,т до уровня прочности полимера или даже
превысить ее.
Влияние структуры и состава полимера на длительную проч-
ность (долговечность, усталостную выносливость) осложняется
действием химического фактора, в частности реакций окисления.
Как известно, скорость окисления значительно повышается с
ростом температуры и напряженности макромолекул:
к—ооехр—(Uo— tc}/RT, (5.68)
где Vo—постоянная; Uo—энергия активации; о — напряжение; f — коэффи-
циент концентрации напряжений.
В статических условиях повышения температуры при испы-
таниях практически не наблюдается и скорость реакций опре-
деляется воздействием механических напряжений. В режиме
динамических нагружений существенный вклад в ускорение
окисления вносит и повышение температуры [см уравнение
(5.68)], особенно при эксплуатации толстостенных изделий в,
условиях нестационарного режима (явление саморазогрева).
Механическое напряжение оказывает сложное влияние на
скорость реакций окисления полимера. На стадии инициирова-
ния в индукционном периоде напряжение приводит к снижению
энергии активации, и тем в большей степени, чем выше о, при-
чем в первую очередь активируется распад наиболее слабых
связей.
Ускорение реакций окисления может быть вызвано появле--
нием в напряженном полимере свободных радикалов, а также
повышением реакционной способности связи, сопряженной с
деформированными участками основной полимерной цепи.
Деформирование приводит, как правило, к обеднению кон-
формационного набора и уменьшению подвижности макромоле-
кул. В результате этого затрудняется передача реакционноак-
тивного центра на другую цепь, и реакция протекает с более
высокой энергией активации, т. е. ингибируется на стадии авто-
катализа. Таким образом, напряжение может и ускорить, и за-
медлить реакции окисления.
Влияние структурных параметре® на механическую прочность
л термодеструкцию часто неоднозначно. Например, линейный
350
гибкий полиизопрен характеризуется высокими прочностными
показателями и небольшими гистерезисными потерями, но очень
высокой скоростью окисления. Для снижения скорости окисле-
ния в состав композиций, работающих в условиях статических
или динамических нагрузок, вводят специальные добавки (про-
тивоутомители), замедляющие окисление в напряженных образ-
цах Однозначно можно сказать, что полимерные материалы с
хорошими прочностными свойствами, характеризующиеся уме-
ренными механическими потерями и высокой стойкостью к окис-
лению, будут обладать и высокой длительной прочностью
Полимерные материалы эксплуатируются в самых разнооб-
разных условиях. При выборе материала и способа изготовления
из него изделия необходимо учитывать условия эксплуатации,
свойства полимера и их изменения в процессе эксплуатации.
Например, линейным полимерам (полиэтилен, поливинилхлорид
и др.) при действии механических напряжений, особенно при
повышенных температурах, свойственна ползучесть, т. е накоп-
ление остаточных деформаций, что приводит к деформации из-
делия (например, к утонению стенок и увеличению диаметра
труб, вздутию поливинилхлоридного линолеума и пр ). Поэтому
изделия, подвергающиеся многократным деформациям, целесо-
образно изготавливать из сетчатых или армированных полиме-
ров Для предупреждения преждевременного деформирования
изделий необходимо, чтобы они работали при напряжениях, не
превышающих предела текучести в условиях эксплуатации.
Наибольшее влияние на свойства полимеров оказывает тем-
пература, величина и частота нагружения. Оптимальные темпе-
ратуры эксплуатации линейных полимеров должны быть не ниже
температуры хрупкости и не выше температуры механического
стеклования (для аморфных полимеров) или температуры плав-
ления (для кристаллических). Нижний предел температурного
интервала эксплуатации сетчатых эластомеров обычно не дол-
жен быть ниже температуры механического стеклования или
температуры хрупкости; верхний — температуры начала терми-
ческого разложения. Способность полимерных материалов со-
хранять эксплуатационные свойства при низких температурах
называют морозостойкостью, при высоких — теплостойкостью.
Одним из показателей морозостойкости является температура
хрупкости Тхр. Степень сохранения необходимых свойств при
низкой температуре характеризуют также коэффициентом мо-
розостойкости Км, представляющим собой отношение какого-ли-
бо показателя при низкой температуре к этому же показателю
при комнатной. Поскольку потеря эластических свойств у элас-
томеров связана с их стеклованием или кристаллизацией в ус-
ловиях эксплуатации, для получения морозостойких изделий
используют некристаллизующиеся полимеры с низкой темпера-
турой стеклования.
351
Для твердых полимеров (стеклообразных и кристаллических)
теплостойкость характеризуют температурой, при которой в ус-
ловиях действия постоянной нагрузки деформация образца не
превышает заданную величину. Для эластомеров количествен-
ной характеристикой теплостойкости служит коэффициент теп-
лостойкости Кт — отношение какого-либо показателя (например,
прочности при растяжении, относительного удлинения при раз-
рыве и др.) при повышенной температуре к его значению при
комнатной.
Наиболее распространенные полимерные материалы на ос-
нове карбоцепных полимеров обычно могут работать при тем-
пературах не выше 370—420 К; при более высоких температурах
резко снижаются их прочность, твердость, жесткость, возрастает
ползучесть. При температурах до 570—700 К могут эксплуати-
роваться некоторые гетероцепные полимеры, например кремний-
органические. Для работы при более высоких температурах
пригодны только специальные термостойкие полимеры.
5.2 ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА
Тепловые явления сопровождают фазовые переходы, деформи-
рование и разрушение полимеров. Основным источником тепло-
вых эффектов являются разрывы макромолекул, пластические
деформации, заторможенность конформационных превращений
под влиянием нагрузки и др. Теплофизические свойства полиме-
ров, которые следует рассматривать как реакцию на темпера-
турное поле, возникающее в полимере, специфичны из-за осо-
бенностей строения макромолекул: большая длина, гибкость,
локальная анизотропия силового поля, обусловленная резким
различием сил, действующих внутри макромолекулы (химиче-
ские связи) и между молекулами (физические связи), и др.
К теплофизическим свойствам обычно относят теплоемкость,
тепло- и температуропроводность, изменение размеров при из-
менении температуры.
5.2.1. Теплоемкость
Теплоемкость — это количество тепла, необходимое для нагре-
вания тела на 1 К. Различают удельную и мольную теплоемко-
сти. Удельная теплоемкость — количество тепла, необходимое
для нагревания на 1 К единицы массы (Дж/(кг-К)], мольная —
количество тепла, необходимое для нагревания на 1 К одного
моля вещества [Дж/(моль*К)]. Различают также теплоемкость
при постоянном давлении (СР) и постоянном объеме (Су):
a*VT
СР = (dH/dT)y, Cv = (dU/dT)P; Ср = Су + —-------,
Асж
где Н — энтальпия; U — внутренняя энергия; V — объем; а — термический
352
коэффициент объемного расширения; Ксж — коэффициент изотермического
сжатия.
Наконец, различают истинную и среднюю теплоемкость.
Истинной мольной теплоемкостью называют отношение беско-
нечно малого количества тепла dQ, подводимого к 1 молю ве-
щества, к бесконечно малому приращению температуры dT,
которое при этом наблюдается, т. е. C=dQ/dT Средней мольной
теплоемкостью С в интервале температур от Т, до Т2 называют
отношение количества тепла Q, подведенного к 1 молю вещест-
ва, к разности температур, т. е. C — Q/(T%—7\). В термодина-
мических расчетах обычно пользуются истинной теплоемкостью.
Теплоемкость характеризует способность поглощать энергию,
которая выделяется в систему вследствие молекулярного дви-
жения, т. е. определяется колебательным спектром. Учитывая
цепной характер макромолекул, теплоемкость твердых полиме-
ров (стеклообразных и кристаллических) представляют адди-
тивной функцией двух составляющих, обусловленных решеточ-
ными скелетными колебаниями основной цепи и характеристи-
ческими колебаниями отдельных боковых атомов и групп в
повторяющемся звене. Решеточные колебания являются низко-
частотными, акустическими, вносят основной вклад в теплоем-
кость твердых тел и зависят главным образом от массы повто-
ряющегося звена Характеристические колебания боковых ра-
дикалов проявляются в области более высоких частот
(в частности, оптические — в ИК области) и, следовательно,
более высоких температур. Они зависят от соотношения масс
атомов основной цепи и бокового заместителя. Если полимер
неоднороден по конфигурации или конформации, то появляется
третья составляющая теплоемкости — конформационная, обус-
ловленная различными энергетическими состояниями изомеров.
Для теоретического определения теплоемкости рассчитывают
колебательный спектр полимера в широком диапазоне частот.
Макромолекула при этом моделируется в виде тонкой струны
или гибкого стержня, к которому жестко прикреплены боковые
Труппы Такие расчеты были проведены для полиэтилена при
частотах 0—2500 см-1. Было установлено, что низкотемператур-
ная теплоемкость определяется лишь акустической частью ко-
лебательного спектра («до 500 см-1). а вклад оптических
колебаний (500—2500 см-1) составляет не более 2%. На колеба-
тельный спектр оказывает влияние не только внутрицепное
взаимодействие, но и межцепное. В области частот до 100 см-1
вид спектра зависит от межмолекулярного взаимодействия,
а в области 100—500 см-1 определяется только химическим
строением цепи (внутрицепное взаимодействие). Таким образом,
в определенной области температур («до ЮК, а иногда до
50—70 К) низкотемпературная теплоемкость кристаллических
тел зависит от внутри- и межмолекулярного взаимодействия,
53—816
353
и теплоемкость изменяется пропорционально кубу температуры
по закону Дебая (рис. 5 47, область /):
Су«Р (5.69)
В интервале 50—150 К межцепным взаимодействием прене-
брегают и теплоемкость изменяется с температурой в соответ- I
ствии с соотношением
Су«Г+Г'/2, (5.70)
в котором первый член характеризует продольные колебания
(деформация связей), а второй — вклад поперечных колебаний,
обусловленных деформацией углов. Для модели полимера в ви- j
де струны (одномерная структура) определяющим является
первый член, т. е. а для двухмерных структур (модель I
гибкого стержня) теплоемкость пропорциональна корню квад- 1
ратному из Т.
Влияние фазовой структуры полимера на теплоемкость про-
является лишь в низкотемпературной области. Для аморфных I
полимеров теплоемкость в области 1 выше, а зависимость С= |
=f(T) отклоняется от закона Дебая и определяется соотно- I
шением ,
Cv=C17'+C2T3+C3E(0f/7’), (5.71)
где Ci, Cs, С3. 6е — константы; E(Qe/T)— функция теплоемкости Эйнштейна.
Таким образом, в неупорядоченных аморфных полимерах
теплоемкость повышается за счет так называемой избыточной
теплоемкости (ДС), которая обусловлена меньшей плотностью
аморфного полимера и ближним порядком в расположении
структурных единиц, т е конформационным вкладом в тепло-
емкость.
Для аморфно-кристаллических полимеров также характерно
отклонение от закона Дебая и
появление избыточной теплоем-
кости; их теплоемкость моно-
тонно повышается при сниже-
нии степени кристалличности.
По мере повышения темпе-
ратуры различие в теплоемко-
сти кристаллических и аморф-
ных полимеров нивелируется
и при Т>50 К вплоть до Гс
теплоемкости становятся оди-
Рис. 5.47. Зависимость теплоемкости
от температуры кристаллического (7)
и аморфного (2) полиэтилена
354
каковыми (область II на рис. 5 47). В этой области температур
теплоемкость не зависит от межмолекулярного взаимодействия,
а определяется только внутримолекулярным, обусловленным
подвижностью скелета цепи и боковых радикалов, и линейно
изменяется с температурой. Линейный характер зависимости
теплоемкости от температуры позволяет оценить С при любой
температуре по значениям, которые определены при какой-то
одной температуре (например, комнатной) с учетом темпера-
турного коэффициента, примерно равного 3-10-3. Теплоемкость
некоторых полимеров при 298 К приведена ниже:
Полиэтилен
Полипропилен
атактический
изотактический
Ср. Ср.
Дж,(моль К) ДжДмолнЮ
49,6 Политетрафторэтилен 96,6
Полистирол 128,2
68,3 Полиметилметакрилат 138,6
90,7 Полиэтилентерефталат 218,4
Как видно из приведенных данных, теплоемкость возрастает
по мере увеличения числа и объема боковых групп.
При температуре структурного стеклования Тс происходит
переход аморфных полимеров из стеклообразного в высокоэла-
стическое состояние, сопровождающийся скачкообразным уве
личением теплоемкости СР (см рис. 5 47). Изменение теплоем-
кости является суммой трех вкладов
ДС=ДО+ДСЯ+ДО*
Здесь ДСК отражает конформационную теплоемкость, возника-
ющую вследствие перехода замороженных ниже Тс конформа-
ций в конформации с более высокой энергией (например, пере-
ход из транс- в гош-конформацию); ДСд связан с разморажи-
ванием свободного объема и возникновением выше Тс новых
дырок; ДСКОЛ отражает изменение частоты и амплитуды колеба-
ний структурных единиц, т. е. суммарные изменения в колеба-
тельном спектре вследствие изменения термического коэффици-
ента расширения объема при Тс Значения этих величин для
некоторых полимеров приведены ниже
АСК ДСд дск°л
Полиизобутилен 31—85 40 13
Полиметилметакрилат 13—75 40—61 33
Поливинилхлорид 18—78 52—71 25
Наиболее существенный вклад в скачок Ср дают первые два
члена (примерно 60—90% от общего увеличения теплоемкости).
Таким образом, изменение теплоемкости в области стеклования
обусловлено размораживанием сегментальной подвижности при
Тс (a-переход), которое сопровождается конформационными
23*
355
переходами, размораживанием свободного объема и увеличени-
ем числа дырок.
В аморфно-кристаллических полимерах также имеет место
скачок теплоемкости, но по мере роста степени кристалличности
он становится все менее заметным и при высоких степенях кри-
сталличности исчезает. Согласно двухфазовой модели аморфно-
кристаллических тел степень кристалличности Л связана со
скачком теплоемкости соотношением
Л=1—ДСр/ДСр ,8М, (5-72)
где ДСр и АСр ам — скачок теплоемкости при Тс в аморфно-кристаллическом
и аморфном полимерах.
При высоких степенях кристалличности (/(>50%) снижение
скачка теплоемкости со степенью кристалличности будет боль-
ше, чем следует из соотношения (5.72). Это связано с заметным
увеличением степени ориентации аморфной фазы при кристал-
лизации и, следовательно, обеднением конформационного набо-
ра структурных единиц в этой фазе и уменьшением члена ДСК.
При высокой степени ориентации конформационный вклад во-
обще исчезает.
Выше Л аморфный полимер может находиться в высоко-
эластическом или вязкотекучем состоянии В этой области тем-
ператур для аморфных полимеров, так же как и для кристал-
лических, существует линейная зависимость теплоемкости от
температуры с температурным коэффициентом dCP/dT, в сред-
нем равным 1,2-10~3. Ниже приведены значения мольной тепло-
емкости Ср некоторых полимеров при 293 К [Дж/(моль К)]:
Ч«с-1,4-Полибутадиен (СКД) 96,6
Ч«с-1,4-Полиизопреи (НК) 129
Сополимеры бутадиена:
со стиролом СКС-30 (70 : 30) * 110
с нитрилом акриловой кислоты
СКН-18 (82: 18) 95,8
СКН-26 (74 : 26) 111
СКН-40 (60,40) 102,4
Сополимер изобутилена с изопреном (бутилкаучук, 95:5) 110
транс-1,4 -Полихлоропрен 193
Теплоемкость полимеров при Т>ТС определяется химиче-
ским строением звена и повышается по мере роста отношения
массы боковых заместителей к массе основной цепи (скелета)
полимера.
В наполненных системах теплоемкость полимеров меняется
по закону аддитивности.
Ср=С₽,а+<р(Ср,в — Ср.п), (5.73)
где Ср и и Ср1П — теплоемкости наполнителя и полимера, ф — объемная доля
наполнителя.
-356
С г—------------------------ -"IX
Тс Тс Г
Рис. 5.48. Температурная зависимость теплоемкости
Рис. 5.49. Температурная зависимость теплопроводности кристаллических (/).
и аморфных (2) полимеров
Выше Тс характер температурной зависимости теплоемкости
может осложниться вследствие фазовых переходов первого ро-
да— кристаллизации и плавления (рис. 5.48). Кристаллизация
сопровождается экстремальным уменьшением теплоемкости с
максимумом при температуре максимальной скорости кристал-
лизации, а плавление — экстремальным ростом теплоемкости с
максимумом при температуре плавления. После плавления кри-
сталлов зависимость теплоемкости от температуры снова при-
обретает линейный характер и прн высокой температуре тепло-
емкость всех тел составляет ?»25 Дж/(моль-К) (закон Дюлон-
га — Пти).
5.2.2. Теплопроводность
Теплопроводностью называют процесс переноса тепла от более
нагретых частей тела к менее нагретым, приводящий к вырав-
ниванию температур. Возникновение в теле градиента темпера-
туры приводит к появлению теплового потока, который будет
существовать до тех пор, пока вследствие переноса энергии
градиент не окажется равным нулю. Теплопроводность харак-
теризуют коэффициентом теплопроводности л, равным количе-
ству тепла Q, протекающего в единицу времени через единицу
площади поверхности, перпендикулярной к направлению потока
тепла при перепаде температуры в 1 К на единицу длины в этом
направлений, т. е. Х=—dQ/dT. Размерность коэффициента
теплопроводности Вт/(м-К). Теплопроводность зависит от тем-
пературы, физического и фазового состояния и структуры
полимера.
357
В отличие от теплопроводности металлов, в которых перенос
тепла осуществляется электронами,теплопроводность полимеров,
относящихся к диэлектрикам, определяется решеточными коле-
баниями сетки полимера. Для описания теплопроводности поли-
меров в твердом агрегатном состоянии (кристаллическом и стек-
лообразном) используют основные положения фононной теории,
разработанной для твердых тел. (Фонон — это квазичастнца,
представляющая собой квант упругих колебаний среды.) Со-
гласно этой теории теплопроводность л определяется взаимодей-
ствием (перебросом) фононов и зависит от теплоемкости С,
средней скорости распространения фононов г>ср и средней длины
их пробега /ср:
УзСу^ср/ср. (5 74)
Температурная зависимость теплопроводности идеальных
кристаллических полимеров описывается кривой /, представ-
ленной на рис. 5.49. При низких температурах (до 30 К) тепло-
проводность определяется преимущественно переносом фононов
на границах кристалла и зависит от теплоемкости (?ср и уср
практически постоянны, так как в процессах переброса участ-
вует очень малое число фононов). Поскольку в этой области
Cv^T3, то и теплопроводность Z~7’3 (область /). При высоких
температурах увеличиваются число фононов и взаимодействие
между ними, длина пробега снижается и теплопроводность экс-
поненциально уменьшается с ростом Т При нормальных и вы-
соких температурах_(выше 100—200 К) возбуждено очень много
фононов, величина Гс₽ невелика и практически не зависит от Т,
эффективность взаимодействия фононов высока, сопротивление
переносу пропорционально температуре, а теплопроводность
Л«1/Г (область //).
Итак, в идеальных кристаллах перенос тепла осуществляется
за счет переброса фононов внутри и на границе кристаллов.
В реальных кристаллах теплопроводность ниже вследствие рас-
сеяния части фононов на дефектах кристалла. Таким образом,
можно считать, что теплопроводность реальных полимеров яв-
ляется релаксационным процессом.
Аморфные полимеры ниже температуры стеклования нахо-
дятся в твердом стеклообразном состоянии. Для описания тем-
пературной зависимости теплопроводности стекол также исполь-
зуются положения фононной теории. Теплопроводность стекол
растет с Т немонотонно (см. рис. 5.49) и в области низких тем-
ператур существенно ниже теплопроводности кристаллических
полимеров. Это обусловлено большим рассеянием фононов из-за
отсутствия дальнего порядка в аморфных полимерах, т. е. явле-
нием релаксации Кроме того, отсутствие дальнего порядка при-
водит к неоднородности распространения фононов, т. е. к появ-
лению определенных флуктуаций, что также повышает рассеи-
358
Рис. 5.50. Модель решетки аморфных
полимеров (двойные линии — внутри-
молекулярные ковалентные связи,
одинарные — межмолекулярные физи-
ческие связи)
вание фононов. При высоких
температурах (выше 100—
200 К) длина свободного про-
бега фононов находится на
уровне ближнего порядка в
стеклах (несколько десятых
нанометра) и сопоставима с
1ср в кристаллических полиме-
рах. Поэтому при таких темпе-
ратурах значения теплопроводности
лического тел близки.
стеклообразного и кристал-
При температуре стеклования наблюдается излом зависимо-
сти A=f(7’). Ниже Тс теплопроводность имеет небольшой поло-
жительный температурный коэффициент d>^dT~>Qt а после Тс —
отрицательный d}./dT<0. Это объясняют изменением механизма
переноса тепла в аморфных полимерах в области температур
выше Гс. Предполагают, что в высокоэластнческом состоянии
перенос энергии осуществляется не за счет распространения
упругих волн (переброса фононов), а в результате передачи
энергии путем внутри- и межмолекулярного взаимодействия,
т. е. по механизму, характерному для жидкостей. В этом случае
теплопроводность
X=2ftv«d-2,
(5.75)
где k — константа Больцмана, ож — скорость звука в жидкости; d — среднее
расстояние между молекулами, равное (M/L рп)'й; М— молекулярная масса;
L — постоянная.
Полимер при этом рассматривают как квазирешетку (квази-
сетку), образованную ковалентными внутримолекулярными и
физическими межмолскулярными связями (модель Айсрмана,
рис. 5.50), а длину пробега фонона Zcp заменяют средним рас-
стоянием между молекулами d, не зависящим от Т. Внутримо-
лекулярная теплопроводность а0 намного выше (примерно на
порядок) межмолекулярной При статистическом расположе-
нии валентных связей теплопроводность всей сетки пропорцио-
нальна усредненному значению коэффициента теплопроводности
межмолекулярных физических связей
(5.76)
где k'> I — коэффициент, не зависящий от температуры и учитывающий
вклад А*.
359
Расчет Хм можно провести на основе уравнения (5 74), в ко-
тором макроскопические параметры заменены на молекулярные
характеристики:
Cv-3kl<Pb- 'vcp = tfV^m; 7cp = d; Хм = . (5.77)
где f» — константа упругости межмолекулярной связи; т — масса повторяю-
щегося звена; b — длина химической связи; d — расстояние между молекула-
ми; k — константа Больцмана, К — постоянная.
С ростом температуры уменьшается вероятность передачи
энергии через межмолекулярные связи, что и является причиной
снижения теплопроводности и отрицательного температурного
коэффициента йк/АТ.
Существенное различие между внутри- и межмолекулярной
теплопроводностью обусловливает зависимость X от молекуляр-
ной массы. Согласно модели Айермана
1/Х=1/Х«+Л/Л1, (5.78)
где X а» — теплопроводность полимера с бесконечно большой молекулярной
массой М, А — константа.
Большее совпадение с экспериментальными результатами
дает уравнение
1/X=l/X«+B/Af/S (579)
(В — постоянная).
Причина насыщения теплопроводности при увеличении мо-
лекулярной массы связана с тем, что при достаточно большой
длине цепей доля межмолекулярных связей растет и возрастает
вклад передачи энергии через межмолекулярные связи. В случае
достаточно длинных макромолекул суммарная плотность физи-
ческих межмолекулярных связей определяется конформацией
макромолекул, а при одинаковой конформации — мало зависит
от М. Например, молекулярная масса, при которой наступает
насыщение X, составляет «105 для полиэтилена при 413 К и
полистирола при 373 К.
Увеличение разветвленности макромолекулы, появление
длинных боковых заместителей создает дополнительное сопро-
тивление передаче энергии и приводит к снижению теплопро-
водности и увеличению отрицательного коэффициента dX/dT.
Теплопроводность некоторых полимеров при 293 К приведена
ниже:
Z, Вт/(м К) Л. Вт/(м К)
Полиэтилен низкой сти (К)* высокой плотно- 0,380 плотно- 0,470 изотактический (К) 0,230 цис-1,4-Полиизопрен (А) линейный 0,130 сшитый вулканизат 0,167
сти (К) Полипропилен атактический (А)* ОД75 вулканизат, иапол- 0,289 иеиный техническим углеродом П-321
* К — кристаллический, А — аморфный.
360
Модель Айермана объясняет также некоторое повышение теп
лопроводности при химическом сшивании макромолекул, т. е.
при изменении линейной конфигурации на сетчатую.
Для аморфно-кристаллических полимеров теплопроводность
зависит от степени кристалличности Если допустить, что кри-
сталлиты распределены в аморфной матрице статистически и
имеют форму, близкую к сферической, то для определения теп-
лопроводности может быть использована формула Максвелла:
= К f (5.80)
X -р 2Хам + 2?.ам
где Хам и Хкр— теплопроводность полностью аморфного и кристаллического
полимеров; К — степень кристалличности.
Теплопроводность полимеров с низкой степенью кристаллич-
ности (К<0,4) определяется главным образом теплопроводно-
стью аморфной фазы, т. е величиной Хам, и для них характерно
возрастание теплопроводности с температурой вплоть до появ-
ления пологого максимума. При высоких степенях кристаллич-
ности (К^0,7) температурная зависимость теплопроводности
определяется кристаллической частью полимера и снижается с
ростом температуры (см. рис. 5.49).
Для ориентированных кристаллических и аморфных полиме-
ров характерно явление анизотропии теплопроводности, прояв-
ляющееся в том, что теплопроводность в направлении ориента-
ции (Хв) выше, чем в направлении, перпендикулярном ориента-
ции (Х±). Физической причиной появления анизотропии
теплопроводности аморфных полимеров является переход из
конформации статистического клубка в конформацию вытянутой
струны (фибриллярную структуру), что приводит к увеличению
доли ковалентных связей, расположенных вдоль осн ориентации,
и повышению проводимости энергии за счет межмолекулярных
связей, так как ориентация приводит к росту их числа вдоль
направления действия силы:
Хг’+гХх-^ЗХ-'. (5.81)
Коэффициент анизотропии Хц/Хх в аморфных полимерах не-
высок и обычно не превышает 2. В кристаллических полимерах
коэффициент анизотропии существенно выше и может достигать
50 и более. Это связано с тем, что при ориентировании кристал-
лических полимеров происходит ориентация кристаллитов и
макромолекул в аморфной части полимера (проходных цепей
и надмолекулярных структур — кластеров), а также образова-
ние дополнительного числа межмолекулярных связей (так на-
зываемых кристаллитных мостиков) между кристаллитами со-
седних микрофибрилл. Это и приводит к существенному росту
Хц и коэффициента анизотропии.
361
Наполнители изменяют теплопроводность полимеров Степень
влияния наполнителей на Z зависит от размера и формы частиц
наполнителя, их распределения в матрице полимера и взаимо-
действия наполнителя с полимером. Существуют уравнения,
позволяющие рассчитать теплопроводность наполненных поли-
меров. Прн регулярном распределении частиц наполнителя в
среде полимера и невысоком их содержании справедливо урав-
нение Гамильтона — Гроссера:
(I ф)/И -1- ^о(^н — >-о)
(5.82)
где Ло — коэффициент теплопроводности ненаполнеииой системы; <р — объем-
ная доля наполнителя; л — коэффициент, характеризующий форму частиц
(для шарообразных частиц л=3, для частиц другой формы л>3).
В области средних содержаний наполнителя X рассчитывают
по формуле Миснара:
X = ?.о 11 + 7—„1/гЛ л---rJ • (5 • 83)
I 1 —Х>)1
Прн повышенной концентрации наполнителя справедливо
уравнение Дульнева:
2vc( 1 — с)
ХД0 = & -L v( 1 - с)« +-4 (5 84)
VC -|- 1 — с
где v=X1(/Xo; с — переменная, зависящая от концентрации наполнителя а;
2с5—Зс24-1«=<р.
При использовании наполнителей, способных к физическому
и химическому взаимодействию с полимером, наблюдается от-
клонение экспериментальных данных от расчетных. В этом слу-
чае применяют эмпирические соотношения. Так, для эластоме-
ров, наполненных техническим углеродом, справедливо урав-
нение
Х=Хо+Кх/п, '(5.85)
где т — массовая доля технического углерода; — коэффициент, зависящий
в основном от физико-химических свойств технического углерода-
Марка технического угле- К-354 ft-234 П-324 П-514 П-803
рода
А'л-105, Вт/(.м К масс. ч) 151 203 227 235 216
Фазовые переходы (кристаллизация и плавление) изменяют
теплопроводность полимера. Кристаллизация приводит к росту
теплопроводности тем большему, чем выше степень кристаллич-
ности. Плавление кристаллов сопровождается сильным умень-
шением теплопроводности до постоянного значения, характерно-
го для аморфных полимеров
36 2
5.2.3. Температуропроводность
Температуропроводность характеризует скорость изменения
температуры в материале под действием теплового потока в не-
стационарных температурных условиях. Температуропровод-
ность определяется коэффициентом а (м2/с):
G» X/ (рСр),
(5.86)
где X — коэффициент теплопроводности; р — плотность материала; Ср —
удельная теплоемкость при постоянном давлении.
Температурная зависимость коэффициента температуропро-
водности аморфных н кристаллических полимеров приведена на
рис. 5 51. В области стеклообразного состояния температуропро-
водность аморфного полимера снижается с ростом температуры
до Тс; при Тс наблюдается скачкообразное возрастание а, а при
Т>ТС (в области высокоэластического или вязкотекучего со-
стояния) величина а практически не изменяется. Скачок тем-
пературопроводности в области стеклования является следстви-
ем резкого возрастания теплоемкости при стекловании (см.
рис. 5.48) и слабого изменения теплопроводности в этой об-
ласти.
Температуропроводность кристаллических полимеров заметно
уменьшается с ростом температуры, затем проходит через ми-
нимум в области плавления и после завершения плавления
снова увеличивается до значений, примерно равных значениям а
до плавления. После плавления температурный коэффициент
da/dT близок к нулю. Такой характер зависимости a=f(T)
обусловлен изменением теплоемкости, теплопроводности и плот-
ности полимера при плавлении. На кривых температурной за-
висимости температуропроводности наблюдается экстремум,
обусловленный плавлением, и скачок прн температуре стекло-
вания. Скачок температуропроводности проявляется только при
низких степенях кристалличности. С увеличением степени кри-
сталличности температуропроводность возрастает при одно-
временном увеличении температурного коэффициента dajdT
Поскольку теплоемкость и
теплопроводность зависят от
структуры полимера, то и тем-
пературопроводность также
зависит от молекулярной мас-
сы, конфигурации, химическо-
Рис. 551. Температурная зави-
симость температуропроводности
аморфных (/) и кристаллических (2)
полимеров
363
го строения звена полимера, наличия наполнителя. Значения а
для некоторых полимеров приведены ниже:
а-!0*. м*/с
Полиэтилен низкой плот- 14
ности Поливинилхлорид 12.1
Полнметилметакрнлат с ЛЫ0< 100 Н.9
2,3 10,5
0,66 8,9
0,02 6
Полидиметилсилоксан 10,8
Полистирол 9,9
цис-1,4-Полиизопреи 8,9—9,0
цис-1,4-Полибут ад иен п.з
Сополимеры
бутадиена со стнро- 11,5
лом (СКС-30), 70 : 30
бутадиена с нитри-
лом акриловой кис-
лоты
СКН-18 (82:18) 10.8
СКН-26 (74 : 26) 9,0
СКН-50 (60 : 40) 10,2
изобутилена с изопре- 7.26
иом (бутилкаучук)
транс-1,4-Пол их лоро- 9.3
прей
а-101, м3/с
В области малых и средних молекулярных масс температуро-
проводность аморфных полимеров увеличивается пропорцио-
нально корню квадратному из М,а затем ее рост с увеличением
М замедляется, и при достаточно высоких значениях коэффи-
циент а практически не изменяется. Такая зависимость обус-
ловлена преимущественным влиянием молекулярной массы на
теплопроводность.
Повышение разветвленности за счет увеличения размеров
боковых заместителей приводит к снижению температуропровод-
ности аморфных полимеров Например, коэффициент а снижа-
ется в ряду виниловых полимеров полиэтилен — поливинилхло-
рид— полистирол или в ряду диеновых полибутадиен — поли-
изопрен — полихлоропрен. Это обусловлено снижением
теплопроводности с увеличением массы звена цепи за счет бо-
ковых групп.
Переход от линейной конфигурации к сетчатой в результате
химического сшивания макромолекул приводит к некоторому
росту температуропроводности.
Введение наполнителей, которые, как было сказано выше,
влияют на теплоемкость и теплопроводность, изменяет и коэф-
фициент температуропроводности. Существует эмпирическая
зависимость а наполненных полимеров от содержания и типа
наполнителя (технического углерода):
a=a0+Kom, (5 87)
где ао — температуропроводность иенаполиениого сшитого полимера; m — со-
держание технического углерода, масс. ч. иа 100 масс. ч. полимера; Ка— эм-
пирический коэффициент, зависящий от типа наполнителя; для технического
углерода марки К-324 Ла—8,2 Ю-10 м2/(с масс. ч.); для других .марок техни-
ческого углерода Ко имеет значения 13-10_,°—13.5-10~10 м2/(с-масс. ч.).
364
5.2.4. Тепловое расширение
В равновесном состоянии твердые тела занимают объем, соот-
ветствующий минимуму свободной энергии При повышении тем-
пературы увеличивается амплитуда колебаний атомов, их сред-
нее смешение от положения равновесия. Вследствие этого твер-
дое тело будет изменять свои размеры до тех пор, пока его
объем не станет таким, что ему будет соответствовать минимум
потенциальной энергии. Количественной характеристикой тепло-
вого расширения полимеров служат термические коэффициенты
объемного (а) и линейного (0) расширения, определяемые при
постоянном давлении. Термический коэффициент объемного
расширения равен
a=(l/V)(aV/aT)p. (5.88)
Термический коэффициент линейного расширения равен
(5.89)
Для изотропных тел а=Зр>0.
С теплоемкостью термический коэффициент объемного рас-
ширения связан уравнением
a-t(CvK7/V), (5.90)
где 7 — константа; Cv — удельная теплоемкость при постоянном объеме;
хг — изотермическая сжимаемость; Хт=1/Кг (Кт — модуль объемного
сжатия).
Из уравнения (5.90) следует, что термический коэффициент
объемного расширения прямо пропорционален теплоемкости.
Так же как и теплоемкость, аир зависят от температуры,
физического и фазового состояния и структурных характеристик
полимера. Температурная зависимость термических коэффици-
ентов а и р по характеру аналогична температурной зависимо-
сти теплоемкости Для аморфных полимеров в области низких
температур значение а невелико и при Т-»-0 а-*0. До Тс терми-
ческие коэффициенты расширения аир примерно равны между
собой и несколько повышаются с ростом температуры. При Тс
наблюдается резкое увеличение а и р (скачок термического ко-
эффициента) в узком температурном интервале (2—5 К). Ниже
приведены коэффициенты линейного расширения некоторых по-
лимеров выше и ниже Тс:
цис-1,4-Полнбутадиен
цис-1,4-Полиизопрен
гране-1,4-11олихлоропреи
Полнметнлвинилснлоксан
т. к ниже Гс выше Т_ V
171—161 0,5 2,4
205—201 0,7 2,3
233—231 0,6 1,7
148—143 0,9 4,2—3,5
365
Т. К МО*, ниже Т_ V- 1/К выше Гс
Сополимеры бутадиена
со стиролом
СКМС-10 (90: 10) 199—195 0,9 2,5
СКС-30 (70:30) 222—217 0,8 2,3
СКС-50 (50:50) 243 0,8 2,3
с нитрилом акриловой кислоты
СКН-18 (82:18) 226—223 0,8 2,4
СКН-26 (74.26) 233—231 0,7 2.3
СКН-40 (60:40) 253—248 0,7 2,2
Между коэффициентами а в высокоэластическом (а8)
и стеклообразном (ас) состояниях и Тс существует определенная
связь:
(а. - ас) Тс=Ус, (5.91)
где Ус—доля свободного объема, примерно равная 0,113.
В высокоэластическом состоянии (при Т>ТС) значения ко- ,
эффициентов аир отличаются друг от друга, определяются I
химическим строением звена полимера и практически не зави-
сят от температуры. Однако в последнее время установлено, что I
прн некоторых Х-переходах (см. гл 4), обусловленных пере- I
группировкой надмолекулярных структур, происходит измене I
нпс коэффициента линейного расширения примерно в 1,5— I
2 раза. I
Тепловое расширение полимерных кристаллов анизотропно jl
и характеризуется суммарными линейными коэффициентами 0 lj|
в трех направлениях а, Ь, с: Л
a=Pa+Pt>4-Pe, (5.92) I
что обусловлено цепным строением макромолекул. Для многих II
полимеров установлено существование отрицательного термине- Ц
ского коэффициента линейного расширения £ вдоль оси макро- И
молекул, совпадающей с направлением ориентации. л
Причина наличия отрицательных коэффициентов расшире- И
ния вдоль оси макромолекулы — вращение звеньев вокруг свя I
зей С—С, которое усиливается с ростом температуры и приво- 1
дит к сокращению макромолекулы. Так, для проявления сокра-
щения полиэтилена угол вращения должен быть не менее 13°.
В изотропных (блочных) образцах аморфно-кристаллических ’
полимеров влияние отрицательных коэффициентов на суммар- 1
ный коэффициент, по существу, не проявляется ввиду их мень- i
шей величины и статистического распределения кристаллов ;
в аморфной среде. Кроме того, высокий термический коэффици- |
ент в аморфной фазе вносит весомый вклад в суммарный тер- |
366
мический коэффициент. В сильноориентированных полимерах,
где преобладает фибриллярная структура с высокой ориентаци-
ей кристаллических и аморфных участков, вклад отрицатель-
ного |3 будет уже существенным.
Как правило, при нагревании выше Тс ориентированные по-
лимеры сокращаются, а при Т<.ТС расширяются, поскольку
аморфные области находятся в стеклообразном состоянии, для
которых (J положителен. С ростом ориентации степень анизотро-
пии повышается:
а=3₽=Р|-|-₽х.
(5.93)
>де Рл н — термические коэффициенты линейного расширения вдоль на-
правления ориентации и перпендикулярно ему.
При введении в полимеры наполнителей, таких, как техниче-
ский углерод, мел, коллоидная кремниевая кислота и др., при
их объемной доле <р до 0,3 коэффициент а равен
a=oto — ф(а<> — <хн).
(5.94)
где а<| и а„ — термические коэффициенты линейного расширения полимера
и наполнителя.
При <р>0,3 (а для других наполнителей при ф>0,1). экспе-
риментальные значения а становятся меньше рассчитанных по
уравнению (5.94), что связано с возникновением термоупругих
напряжений на границе полимер — наполнитель. Для некоторых
наполненных систем (например, с волокнистым наполнителем
асбестом) наблюдается аномальное изменение а при Л.
При переходе из стеклообразного в высокоэластическое со-
стояние а нс только нс уменьшается, но даже увеличивается.
Это связано с отслаиванием полимера от наполнителя под дей-
ствием термоупругих напряжений на границе раздела.
Таким образом, изменение а или р с температурой можно
использовать не только для оценки физических переходов, но
и для определения структуры наполненных систем.
Итак, теплофизические свойства полимеров зависят от тем-
пературы, фазового и физического состояния полимеров и опре-
деляются структурными параметрами полимера и составом ком-
позиций. Знание этих характеристик наряду с механическими
свойствами необходимо для выбора полимера и для изготовле-
ния изделий, работающих в контакте с материалами, имеющими
другие теплофизические характеристики, для правильного про-
гнозирования температурных полей при вулканизации изделий
и т. д. Высокую чувствительность теплофизических свойств
к структурным изменениям широко используют для определения
структуры полимеров.
367
Механические свойства наиболее распространенных полимер-
ных материалов приведены ниже:
ар, МПа Е, МПа ®отн
Резина из натурального каучука 1.3*
ненаполнениая 30 850
наполненная Полиэтилен 34 7* 650
низкой плотности 12—16 150—250 100-600
высокой плотности 22—33 550—800 200—900
Фснолоформ альдегидная смола от- вержденная 40—50 2000—2500 2-3
Полистирол 30—70 2800—3500 1.5
Полиметнлметакрилат 70 2700 4
Капрон неориентированный 55—70 800-1000 100-150
Капроновое волокно Полиэтилентерефталат 740—880 3500 15—20
пленка 170 3500 100
волокно Эпоксидные пластики, армированные волокном 800—1000 5000—16000 10-20
стеклянным 1080 3500** 0
борным — 250000** 0
* Секущий модуль прн растяжении на 300%.
*• Продольный модуль и прочность.
5.3 . ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
Поведение полимеров в электрическом поле характеризуется его
электрическими свойствами. Способность полимера пропускать
электрический ток при приложении электрического напряжения
называется электрической проводимостью
Количественно электрическая проводимость т» оценивается удельной диффе-
ренциальной электрической проводимостью, равной dj/dEa, где dj — плотность
тока, текущею через образец, Еп— напряженность постоянного электрическо-
го ноля. В случае выполнения закона Ома 7»=j/£n- Величина, обратная
электрической'проводимости, нпзыпа<'тся удельным дифференциальным элек-
трическим сопротивлением Различают удельное объемное pv н по-
верхностное ps сопротивления н соответственно удельную объемную Tfv
и поверхностную 7$ электрическую проводимость. Объемная электрическая
проводимость выражается в С.м/м (I См«=1 Ом"'), а удельная поверхностная
проводимость — в См; соответственно удельное объемное сопротивление
выражается в Ом-м, а удельное поверхностное—в Ом.
Перенос электрических зарядов в полимерах осуществляется
ионами, отдельными заряженными макромолекулами или их
ассоциатами (так называемыми моль ионами) и электронами.
Соответственно различают ионную, моль-ионную и электронную
проводимость. Суммарная электрическая проводимость опреде-
ляется числом п,, зарядом qi и подвижностью х< всех видов но-
сителей, т. е.
358
По величине электропроводимости все тела, в том числе и
полимеры, разделяют на три класса: проводники^ полупровод-
ники и изоляторы (диэлектрики)
Vp. См/м
Проводники >10"3
Полупроводники 10-3—10“*
Диэлектрики <10“ *
Большинство полимеров являются диэлектриками, т. е. ха-
рактеризуются большим объемным сопротивлением и ничтожно
малой электрической проводимостью Развитие ряда отраслей
промышленности вызвало необходимость создания полимерных
изделий, обладающих высокой проводимостью и выполняющих
роль проводников или полупроводников электрического тока.
Этого удается достигнуть изменением структуры или состава
полимерной композиции. В последнее время нашли широкое при-
менение в народном хозяйстве новые материалы — диэлектрики,
способные длительно сохранять заряд на поверхности после
электризации, так называемые электреты.
5.3.1. Свойства полимерных диэлектриков
Электрические свойства диэлектриков характеризуются удель-
ной электрической проводимостью, диэлектрической проницае-
мостью, диэлектрическими потерями и электрической проч-
ностью.
Электрическая проводимость. Удельная объемная электриче-
ская проводимость (или обратная ей величина — удельное объ-
емное сопротивление) определяется наличием свободных заря-
дов и их подвижностью. Для диэлектриков характерна главным
образом ионная проводимость.
Присутствие ионов в полимерах обусловлено электролитической дисаг
циан ней ионогенных участков макромолекул, а также инжекцией (холодной
эмиссией) ионов в полимер из электродов. Источником ионов, химически не
связанных с макромолекулой, являются низкомолекулярные примеси. Ионная
проводимость характерна для полимеров, не содержащих сопряженных двой
ных связей, а также для растворов полимерных диэлектриков.
В полях высокой напряженности (более 10 кВ) возможен
вклад и электронной проводимости вследствие инжекции элек-
тронов вблизи электродов
Электронная проводимость характерна для диэлектриков, содержащих
сопряженные двойные связи.
Электронная проводимость в полимерных материалах осуществляется
также электронами проводимости электропроводящих ингредиентов, специ-
ально вводимых в полимерный материал (например, порошки никеля, сереб-
ра и других металлов). Кроме того, под воздействием ионизирующих излу-
чений, тепла, сильного электрического поля может происходить ионизация
24—316
369
даже макромолекул с насыщенными связями (полиэтилен и др.), сопровож-
даемая образованием свободных электронов. Подвижность электронов в по-
лимеоах иа 2—6 порядков превышает подвижность ионов.
Электронная проводимость полимерных материалов возрастает при повы-
шении температуры, внешнего давления, интенсивности радиационного облу-
чения. Она обусловлена переходом электронов из валентной зоны в зону
проводимости. Для такого перехода необходима энергия, определяемая ши-
риной запрещенной зоны перехода. Вакансии в валентной зоне называются
дырками и рассматриваются как положительные частицы.
Число свободных ионов и электронов в диэлектриках чрез-
вычайно мало, поэтому их электропроводность незначительна
и обусловлена преимущественно подвижностью макромолекул
или их частей, определяющих, в свою очередь, подвижность ио-
нов. Электрическая проводимость диэлектриков зависит от на-
пряженности электрического поля, температуры, давления, фи-
зического и фазового состояния полимера, его структуры и со-
става.
С ростом напряженности поля электрическая проводимость
повышается за счет увеличения числа инжектируемых носителей
зарядов (ионов и электронов) в диэлектрик и образования ин-
жектированного объемного заряда. Повышение температуры уве-
личивает электрическую проводимость согласно экспоненциаль-
ному закону:
у = Ae~u»fRT , (5.95)
где А — коэффициент, практически не зависящий от Т; £7»— энергия актива*
цин переноса заряда; R — универсальная газовая постоянная; в случае элек-
тронной проводимости U* характеризует ширину запрещенной зоны или глу-
бину залегания дефектов структуры, которые могут быть ловушками для
электронов.
В координатах 1g 7—1/Т зависимость 7=/(1/Г) описывается
прямой линией, наклон которой к оси ординат характеризует
энергию активации переноса заряда. При температуре стекло-
вания происходит резкое изменение температурного коэффици-
ента dyldT. Электропроводимость начинает расти намного быст-
рее с повышением температуры. Это связано с увеличением по-
движности звеньев и сегментов макромолекул и снижением
энергии активации переноса зарядов при Т>ТС. С ростом внеш-
него давления значение ионной проводимости полимеров снижа-
ется, а электронной — растет.
При кристаллизации неполярных полимеров происходит сни-
жение проводимости тем большее, чем выше степень кристал-
личности. Это еще раз подчеркивает, что проводимость в ди-
электриках имеет преимущественно ионный характер, так как
электронная проводимость увеличивается при кристаллизации,
а ионная — снижается.
Повышение полярности полимеров приводит к росту вклада
в общую проводимость электронной составляющей и увеличению
370
суммарной проводимости. Так, для виниловых полимеров с не-
полярными заместителями ширина запрещенной зоны велика,
и их электронная проводимость ничтожна по сравнению с ион-
ной У полимеров, содержащих боковые группы, образующие
цепь сопряженных связей (полиимиды, полибензоксазолы и др.),
электронная проводимость заметно выше. Значения электропро-
водимости некоторых полимеров при 293 К и относительной
влажности воздуха 40—60% приведены ниже:
yv, См/м Vs. См
Поливинилхлорид 10~12— ю-‘« 10-12—ю-*3
Полаэтилентерефталат 1 о-13—ю-14 10-‘4—10-15
Полиэтилен ю-1*—10-15 10-16—ю-'Г
Полистирол 10--14— ю-‘6 Ю-'«—10->?
Поли гетрафторэтилен 10-!$—10-’6 Ю-'б—1U-17
Электрическая проводимость существенно зависит от соста-
ва полимерной композиции, например от наличия наполнителей
и пластификаторов Наполнение полимеров электропроводящи-
ми наполнителями, такими, как графит, технический углерод,
металлические порошки и др., повышает электрическую прово-
димость диэлектриков. Электропроводимость наполненных ди-
электриков зависит ст содержания наполнителя, размера его
частиц и физико-химических свойств его поверхности, распреде-
ления наполнителя в полимере. Пластификаторы уменьшают ве-
роятность контакта наполнитель — наполнитель и тем самым
снижают электрическую проводимость наполненных полимеров
Диэлектрическая проницаемость и диэлектрические потери.
В отсутствие внешнего электрического поля заряженные части-
цы распределены статистически и результирующий электриче-
ский момент равен нулю. Под действием внешнего электрическо-
го поля равновесие нарушается, в результате чего возникает
электрический момент, т. е. наступает поляризация диэлектрика.
Количественно поляризацию Р характеризуют вектором, равным
электрическому моменту J единицы объема полимера:
(5-96)
z-o
где Qi — заряд диполя; г,- — радиус-вектор, определяющий положение i-ro за-
ряда; N — число диполей.
Величина Р представляет собой сумму двух составляющих:
деформационной Рдеф и ориентационной Рор поляризации;
Рдеф — поляризация, обусловленная деформацией электронных
оболочек, смещением атомных ядер или ионов; Рор — поляриза-
ция, обусловленная ориентацией слабосвязанных ионов внутри
диэлектрика или постоянных диполей, характеризующихся по-
стоянным дипольным моментом.
24*
371
Роль диполей в полимерах обычно выполняют полярные заместители,
•связанные ковалентными связями с основной цепью макромолекулы или
с боковыми группами. Дипольный момент любого тела р0 равен произведе-
нию величины заряда q на расстояние между зарядами /. Дннольный момент
макромолекулы определяют как векторную сумму составляющих векторов —
дипольных моментов звеньев макромолекул. Химическая структура, конфигу-
рация и конформация макромолекул оказывают влияние на величину ди-
польного момента.
В неполярных полимерах основным видом поляризации яв-
ляется ориентационная. Диполи, которые возникают вследствие
деформационной поляризации, называются наведенными. В этом
случае Рдеф=а£‘п, где а—коэффициент пропорциональности,
называемый поляризуемостью.
Основным видом поляризации полярных полимеров является
ориентационная, но определенный вклад вносит и деформацион-
ная составляющая. Поэтому общая поляризация полярных по-
лимеров больше, чем у неполярных. В результате возникнове-
ния поляризации происходит увеличение напряженности суммар-
ного поля, действующего на атомы или молекулы, на величину,
пропорциональную поляризации. Поэтому емкость конденсато-
ра (С), между обкладками которого помещен диэлектрик,выше
емкости того же конденсатора, у которого между обкладками
находится вакуум (Со). Отношение С/Со называется диэлектри-
ческой проницаемостью е. Диэлектрическую проницаемость
также можно определить как величину, показывающую, во сколь-
ко раз сила взаимодействия двух зарядов меньше в среде ди-
электрика по сравнению с вакуумом.
Вектор поляризации Р связан с напряженностью поля £п со-
отношением:
Р=еоХя£п. (5.97)
где ео — электрическая постоянная, равная 8,854 Ф/м; Хл — безразмерный
коэффициент, называемый диэлектрической восприимчивостью.
Величины % и е/ связаны между собой соотношением Хд+1 =
= е'. Для неполярных полимеров величины Хл и е' зависят от
поляризуемости а и числа наведенных диполей пЛ-
лда
Хд = й---: при Пд“ <О ~ "да;
Пд— (Лд«/3)
(в'— 1)/(е'-*-2)=ляа/3.
Деформационная поляризация устанавливается достаточно
'быстро (10-‘4—10~’2 с), ориентация же диполей в электриче-
ском поле происходит не мгновенно, а в течение определенного
времени вследствие цепного строения макромолекул и сильного
внутри- и межмолекулярного взаимодействия Это явление по-
лучило название диэлектрической релаксации, а время, в тече-
ние которого поляризация диэлектрика уменьшается в е раз,
называется временем диэлектрической релаксации тд. В жидко-
372
стях ориентация диполей происходит достаточно быстро и тд~
~ 10-10—10_|2с. В твердых телах, где соседние молекулы огра-
ничивают подвижность диполей и процессы ориентации проте-
кают медленно, скорость релаксационных процессов невелика
и время релаксации достигает больших значений. Если при
мгновенной ориентации диполей суммирующая напряженность
составляет D=En + P, то при запаздывании ориентации суммар-
ная напряженность в момент t будет меньше D за счет необра-
тимого рассеивания электрической энергии и превращения ее
в тепло. Эти потери называют диэлектрическими.
Для прохождения переменного электрического тока не тре-
буется переноса активных центров, а достаточно небольших ко-
лебаний зарядов вблизи некоторого положения равновесия При
этом возникают так называемые токи смещения. Для описания
поведения диэлектриков в таком переменном электрическом поле
при отсутствии резонанса диэлектрическая проницаемость ха-
рактеризуется показателем, который называется комплексной
или обобщенной диэлектрической проницаемостью в*:
е*-в'-Не", (598)
где е'— действительная часть проницаемости, характеризующая диэлектриче-
скую проницаемость диэлектрика; се" — минмая часть в*, характеризующая
диэлектрические потерн.
Диэлектрические потери численно равны количеству тепла,
выделяющегося в единице объема диэлектрика при прохожде-
нии электрического тока через него. В идеальном диэлектрике
при отсутствии потерь энергии вектор тока опережает вектор
приложенного напряжения на 90°. В диэлектрике, где часть
энергии рассеивается, угол между векторами меньше 90° на ве-
личину 6. Этот угол называется углом диэлектрических потерь.
Тангенс его равен отношению е,,/8/, или отношению активной
и реактивной составляющих электрического тока. Причиной рас-
сеивания энергии является заторможенность ориентационной по-
ляризации, поскольку при деформационной поляризации наве-
денные диполи возникают настолько быстро, что их смещение
проявляется при любых частотах поля со Поэтому деформаци-
онная поляризация не зависит от со. Для неполярных полимеров
диэлектрические проницаемость и потери также не зависят от
частоты электрического поля:
е'=«2о: ъ"~1Е2п/&л, (5.99)
где по—показатель преломления полимера: f — удельная электропроводность
диэлектриков, определяемая по остаточному току в постоянном электрическом
поле; поскольку значения к малы, то и потери в неполярном полимере неве-
лики.
В случае полярных полимеров значения е' и е" зависят от
температуры и частоты со электрического поля. Между е', г"
373
:и круговой частотой со существует определенная зависимость;
е'=е«. + (вст — ете)/[1 + (со-Гд)2]; (5.10и)
(е„ —ете)огд/[1 + ((отд)!], (5.101)
где Сет — статическая диэлектрическая проницаемость, т. е. е' прн <о—>-о;
е» — высокочастотная диэлектрическая проницаемость, т. е. е' прн ©—>-оо>
тд — время релаксации.
Пои <отл<С1 (когда тд или со малы) е/-»-ест, а в"->0; при
<отд>>1 а е"-»-0. Физический смысл этого сводится
к следующему. При низких частотах поляризация проявляется
полностью и е' достигает значения еСт", величина потерь мала,
е"-*0. При очень высоких частотах (больших (Омаке) диполи нс
успевают ориентироваться (их можно рассматривать как непо-
движные) и ориентационная поляризация отсутствует. Поэто-
му при со-*-оо величина е"-*-0, а значение е7 стремится к етс.
При отд=1 на кривой e,=f(co) есть точка перегиба, а кривая
зависимости e77=f(co) проходит через максимум.
Влияние температуры на диэлектрическую проницаемость
проявляется через влияние се на тА. При высоких Т подвиж-
ность диполя велика. тд мало и е77-»-0, а е,->8сТ. При низких
температурах тд настолько велико, что даже при небольшой час-
тоте диполи не успевают ориентироваться и е7-*-е«., а е,7->0.
В области средних температур, когда период колебаний сопоста-
вим со временем релаксации, т. с. при (отд-*-0, на кривой е7=
—j(T) имеется перегиб, а диэлектрические потери максимальны.
В области максимума е7 и е" составляют
£/маке= (во+8<х>)/2; 8//какс= (во-* ®м)/2.
(5.102)
Соотношения (5.100) — (5.102) справедливы при условии, что
все диполи одинаковы, нс взаимодействуют друг с другом и, сле-
довательно, характеризуются одним временем релаксации.
Но реальным полимерам присуще распределение времени ре-
лаксации, которое оценивают релаксационным спектром. По-
этому комплексная диэлектрическая проницаемость равна
[1 + ('«тд)’-«]Р ‘
(5.103)
при а-;
Здесь а — коэффициент, учитывающий спектр времен релакса-
ции, а р — асимметрию функции распределения времен релакса-
ции; при а=0 полимер имеет одно время релаксации,
= 1 — бесконечно широкий спектр времен релаксации;
С учетом СС ЗНЗЧСНИЯ Е макс И Е макс рЗВНЫ
<5'1(1,1
\ х / \ X J
374
Для полимеров характерны два механизма диэлектрической
релаксации: дипольно-сегментальный (ДС, или a-процесс), обу-
словленный ориентационными поворотами сегментов и прояв-
ляющийся при Т=ТС, и дипольно-групповой (ДГ, или 0-про-
цесс), связанный с ориентацией полярных боковых групп и про-
являющийся при T<zTc. Энергия активации ДГ процесса не пре-
вышает 5—15 кДж/моль, а энергия активации ДС поляризации
на порядок выше (130—590 кДж/моль). Если полимер содер-
жит несколько полярных групп, способных ориентироваться не-
зависимо друг от друга, то на температурных зависимостях е,
ъ" или tg6 появляется несколько перегибов, или максимумов
дипольно-групповых потерь -у, б и т д.
Диэлектрическая проницаемость и диэлектрические потери
определяются структурой полимера: химическим строением его
звена, конфигурацией и конформацией макромолекулы и над-
молекулярной структурой. Влияние химического строения со-
ставного повторяющегося звена проявляется через влияние его
на полярность полимера. Значение е' в неполярных и малопо-
лярных полимерах пропорционально квадрату показателя пре-
ломления при длинных волнах видимого света, не зависит от
частоты поля и слабо зависит от температуры. Ниже приведены
значения г' и Пр для некоторых полимеров:
Полиэтилен
плотностью 0,918 кг/мэ
плотностью 0,959 кг/м’
Чис-1,4-Полиизопреи (НК)
транс- 1,4-Полиизопрен (гуттаперча)
Политетрафторэтилен
е' по
2,273 2,278
2,357 2,361
2,36 2,27
2,40 2.383
2,009 1,966
Статическая диэлектрическая проницаемость этих полимеров
составляет 2,8—4,0.
Для полярных полимеров значения е' намного выше и опре-
деляются величиной дипольного момента, который, в свою оче-
редь, зависит от полярности групп, их числа и расположения,
температуры и частоты электрического поля. Ниже приведены
значения е' для некоторых полярных полимеров при частоте
103 Гц и 398 К:
Поли метил метакрилат
Поли этилентерефта лат
Поливинилхлорид
2,84
3,12
3,0—3,3
Триацетат целлюлозы
Полиуретан
Трицнанэтнлцеллюлоза
3,2
6,7—7,5
13
Как уже отмечалось выше, диэлектрические потери в непо-
лярных и малополярных полимерах незначительны и определя-
ется электрической проводимостью диэлектрика. Ниже приве-
375
дены значения tg6Ma«c некоторых полимеров:
Частота. Гц 18 ®ы.кс
Неполярные полимеры: полиэтилен 10’—10» 2-10-4
полистирол I О’—10е
полибутадиен 103 9 10-4
полиизолрен 103 (10-5-30)-10-
Полярные полимеры: поликарбонат 10’ 0,001
10» 0,01
поливинилхлорид 10’ 0,02
эпоксидные смолы отвержденные 103 0,02
10е 0,2
В полярных полимерах потери намного больше и влияние по-
лярности проявляется через влияние ее на времена ДС и ДГ
релаксации. Чем выше полярность полимера, тем меньше по-
движность звеньев, больше тд и выше температура, при которой
наблюдаются максимумы ДС и ДГ релаксации. Определяющее
влияние на тя оказывают число и природа полярных групп, раз-
меры заместителей, изомерия бокового радикала и другие фак-
торы. увеличивающие меж- и внутримолекулярное взаимодей-
ствие. Время релаксации возрастает, а следовательно, растут ди
электрические потерн при увеличении содержания полярных
групп в макромолекуле, их полярности, повышении объема за-
местителей и при образовании внутримолекулярных связей у бо-
ковых заместителей При этом максимум tg6 сдвигается в сто-
рону более высоких температур. Конфигурация цепи, в частно-
сти стереорегулярность, также оказывает влияние на диэлект-
рические потери. Например, переход от синдио- к изотактиче-
скому полиметилметакрилату сопровождается сдвигом tgSMaKc
ДС и ДГ потерь в сторону более низких температур и умень-
шением доли ДГ потерь. У атактического полимера tg6HaKc
ДГ почти вдвое превышает tg6MaK<^C потерь Суммарная вели-
чина потерь у этого полимера намного выше по сравнению с ре-
гулярными полимерами.
Переход от линейной конфигурации макромолекулы к раз-
ветвленной или пространственно-сетчатой изменяет е' и е" по-
разному в зависимости от физического состояния полимера.
В высокоэластическом состоянии е' и е" уменьшаются с ростом
степени разветвления или сшивания. При этом максимум потерь
сдвигается в сторону более высоких температур и уменьшается.
В стеклообразном состоянии точки разветвления и сшивки
снижают плотность упаковки и уменьшают межмолекулярное
взаимодействие, тем самым способствуют повышению доли мак-
ромолекул, способных релаксировать, приводя к росту е' и е”.
Существенное влияние на диэлектрическую проницаемость
и диэлектрические потерн оказывает уровень надмолекулярной
376
организации. Кристаллизация полимера приводит к упорядоче-
нию системы, возрастанию межмолекулярного взаимодействия
и уменьшению доли полимера, способного к релаксации.
Предположим, что процесс диэлектрической релаксации
в аморфно кристаллическом полимере представляет собой сум-
му релаксационных процессов в полностью аморфной и пол-
ностью кристаллической фазах и в кристаллической фазе по-
движность макромолекул, ее сегментов и групп настолько мала,
что релаксация полностью подавлена. В этом случае е' и е"
уменьшаются с ростом степени кристалличности. Так как ско-
рость снижения е" с ростом степени кристалличности больше
скорости снижения е', то tg6 также уменьшается. Однако для
ряда кристаллических полимеров подвижность частей макромо-
лекулы в кристалле сохраняется и протекают процессы релак-
сации. Если их интенсивность выше интенсивности релаксаци-
онных процессов в аморфной фазе, то е' и е" могут увеличи-
ваться с ростом степени кристалличности.
Введение пластификаторов, как правило, ухудшает диэлек-
трические свойства полимеров Изменение е.', е" и tg6MaKc
с температурой и их значения зависят от полярности пластифи-
катора и его термодинамической совместимости с полимером.
При хорошей совместимости пластификатора с полимерохм поло-
жение tg 6макс смещается в сторону меньших температур с рос-
том содержания пластификатора Абсолютные значения потерь
зависят от полярности самого пластификатора. В случае непо-
лярных пластификаторов, диэлектрическая проницаемость и по-
тери которых малы, значения е', е." и tg6MaKc пластифицирован-
ного полимера снижаются. Введение же полярных пластифика-
торов, имеющих высокие г' и е", приводит к росту диэлектриче-
ской проницаемости и потерь пластифицированной композиции.
При ограниченной совместимости полимера с пластификатором
снижение tg6M»Kc происходит только до определенного предела.
Обнаруженная четкая корреляция между диэлектрическими
свойствами (проводимостью, диэлектрической проницаемостью
и диэлектрическими потерями) и структурой полимера, его фи-
зическим и фазовым состоянием позволяет использовать эти
свойства, их частотную или температурную зависимость для
оценки структуры полимеров и ее изменения.
Электрическая прочность. При повышении напряжения, при-
ложенного к диэлектрику, наступает момент, когда при опреде-
ленном значении напряжения Unfl диэлектрик теряет свои элек-
троизоляционные свойства, в нем образуется проводящий элек-
трический ток канал, т. е. наступает разрушение диэлектрика —
его пробой, При пробое выделяется большое количество энергии,
материал в области пробоя разогревается настолько, что рас-
плавляется или загорается. Электрическая прочность ЕПр харак-
теризует сопротивление диэлектрика разрушению его в электри-
377
ческом поле. В зависимости от вида напряжения, прикладывае-
мого к диэлектрику, различают электрическую прочность при
постоянном и переменном напряжении, в однородном и неодно-
родном полях, .а в зависимости от продолжительности действия
напряжения — кратковременную и длительную Наиболее изу-
чена электрическая прочность в однородных полях, поэтому она
и будет рассмотрена ниже.
Значения Епр, определенные при переменном напряжении,
меньше Еир при постоянном напряжении. Кратковременная
электрическая прочность — это напряженность электрического
поля при пробое в условиях постепенного повышения напряже-
ния с заданной скоростью (~1—2 кВ/с). Длительная электриче-
ская прочность — это напряженность электрического поля при
пробое при заданном времени выдержки под напряжением или
время жизни (тж) диэлектрика при заданных значениях напря-
женности электрического поля В однородном электрическом
поле электрическая прочность равна отношению пробивного на-
пряжения Unp к толщине диэлектрика h: Enp—Unp/h.
Кратковременная электрическая прочность, так же как и ме-
ханическая, зависит от наличия дефектов в структуре материала
и степени его разогрева, т. е. от термофлуктуационных явлений,
а также от формы и размеров образцов (масштабный фактор).
Ниже приведены значения кратковременной электрической проч-
ности (МВ/м) некоторых полимеров при 298 К в переменном
поле частотой 50 Гц:
Полиэтилен
Полипропилен
Поливинилхлорид
Полистирол
Полиэтилентерефталат (степень кристал-
личности К=60%)
Поликарбонат
аморфный
кристаллический (Д'=26%)
Полисилоксаны
Сополимер бутадиена с 18% нитрила акри-
ловой кислоты (сшитый)
28—40 (2)*
150±30 (0,02)
45—55 (1)
20- 25 (2)
150—180 (0,02)
350—450 (0,02)
21—23 (2)
150—200 (0,02)
70—1000 (0,05)
10—15 (2)
15—40 (2)
* В скобках указана толщина образца в мм.
Механизмы разрушения полимеров в электрическом поле,
т. е. механизмы пробоя, различны для разных полимеров. Разли-
чают электронный пробой, тепловой и пробой вследствие газо-
вых разрядов.
Электронный пробой характерен для малодефектных мате-
риалов, которые не разогреваются при испытании. Как уже
отмечалось выше, при приложении высокого напряжения в ди-
378
электрик из электродов инжектируются электроны, ускоряю-
щиеся в электрическом поле. Полимер в стеклообразном или
кристаллическом состоянии можно считать малодефектным ма-
териалом, так как дефекты его «заморожены». В этих условиях
инжектируемые электроны захватываются кристаллической ре-
шеткой и остаются в ней. Электронная проводимость (взаимо-
действия электрон — электрон) мала, в основном преобладает
взаимодействие электронов с полимером (взаимодействие элек-
трон— фонон). С повышением температуры это взаимодействие
усиливается и электрическая прочность таких материалов воз-
растает с температурой. Однако редко удается наблюдать повы-
шение Епр с ростом температуры: чаще всего Fnp мало изменя-
ется с температурой до Тс или ГПЛ- При температурах выше Тс
или Тпл возрастают подвижность макромолекул и вероятность
«выбивания» электронов из мелких ловушек, размораживаются
дефекты. Все это приводит к резкому увеличению числа элек-
тронов в зоне проводимости, преобладанию взаимодействия
электрон — электрон, повышению электропроводимости и, следо-
вательно, к снижению £Пр с ростом температуры
В результате притяжения разноименно заряженных электро-
дов диэлектрик сжимается, что повышает вероятность пробоя,
и образец может разрушиться раньше, чем произойдет электрон-
ный пробой. Такой механизм пробоя называют электромехани-
ческим.
Величина Епр в этом случае зависит от диэлектрических
свойств и жесткости материала и увеличивается с ростом мо-
дуля упругости Е и при снижении диэлектрической проницае-
мости е
£np~O,6(E/eeo)’/s. (5.105)
Тепловой пробой характерен для материалов, сильно разо-
гревающихся при приложении электрического поля. Вследствие
нагрева растет проводимость и образец разогревается до тех
пор, пока в каком-нибудь (наиболее дефектном) месте не про-
изойдет пробой. Степень разогрева зависит также от диэлек-
трических потерь и эффективности отвода тепла и наиболее
значительна при переменном напряжении. Поскольку диэлек-
трические потери максимальны при переменном напряжении
в области довольно высоких температур и частот, то вероят-
ность теплового пробоя наибольшая в этих условиях.
Диэлектрические потери зависят от структуры полимера,
а эффективность теплоотвода определяется теплопроводностью
полимера и толщиной образца. Чем больше диэлектрические
потери, хуже теплопроводность и больше толщина образца, тем
при более низких значениях напряженности электрического поля
произойдет пробой, т. е. тем ниже электрическая прочность.
Повышение температуры наибольшее внутри образца и зависит
379
от продолжительности действия напряжения и его величины.
При невысоких напряжениях температура внутри образца по-
вышается незначительно в первый момент приложения напря-
жения и практически не зависит от продолжительности его дей-
ствия. При напряжениях выше некоторых критических значе-
ний скорость роста температуры увеличивается и dTjdt-^oo.
Это приводит к резкому снижению электрической прочности
и пробою. Если температура приближается к температуре хи-
мической деструкции, то пробой сопровождается разложением
полимера.
Электрический пробой вследствие газовых разрядов проис-
ходит в диэлектриках, имеющих микродсфекты в виде полостей,
наполненных газом, например воздухом (внутренний пробой),
или в случае неплотного прилегания электродов к поверхности
образцов (внешний пробой). Электрическая прочность газов
ниже электрической прочности твердых диэлектриков, поэтому
при приложении высокого напряжения в первую очередь проис-
ходит электрический разряд в зазоре электрод — диэлектрик
и в полостях внутри диэлектрика. Напряжение пробоя в газах
определяется размером зазора и плотностью газа при постоян-
ной плотности ипр снижается с увеличением зазора и размеров
микродефектов. При разряде развивается высокая температура
и выделяется озон, что является причиной деструкции диэлек-
трика и приводит к снижению пробивного напряжения. Особен-
но опасны внутренние газовые пробои, приводящие к возникно-
вению разветвленных эрозионных каналов от полости к электро-
ду. Влияние разрядов на прочность диэлектрика наиболее
существенно при переменном напряжении.
С ростом температуры электрическая прочность полимеров
в переменном электрическом поле при любом механизме про-
боя снижается: сначала (до Тс или Tnj}) незначительно, а в об-
ласти Тс или Тпя происходит ее резкое уменьшение. В постоян-
ном электрическом поле в области Тс иногда наблюдается мак-
симум ЕПр, обусловленный дипольной поляризацией, ослабляю-
щей напряженность приложенного поля.
Длительная электрическая прочность в значительной мере
определяется интенсивностью так называемого электрического
старения, которое происходит под влиянием разрядов, и связан-
ного с ними повышения температуры, а также озона и заключа-
ется в необратимом изменении структуры полимера (химической
деструкции). Старение приводит к росту электрической прово-
димости (за счет увеличения числа носителей тока), и пробой
наступает при меньших значениях напряженности электрическо-
го поля Такой пробой называется электрохимическим.
Время жизни диэлектрика тж в постоянном поле зависит от
температуры:
тж=тоехр (ДТР'/ЛГ), (5.106)
380
где Го — постоянная; ДW — коэффициент, для различных полимеров состав-
ляющий 0,5—2,0 эВ; k — константа Больцмана.
Время жизни диэлектрика в переменном поле обратно про-
порционально частоте приложенного поля.
Статическая электризация. Для диэлектриков характерно яв-
ление статической электризации, т. е. способность накапливать,
и сохранять электрические заряды в процессе получения, пере-
работки и эксплуатации. Поверхностная плотность зарядов а»
на образце полимера в момент t зависит от скорости накопле-
ния (генерации) статических зарядов и скорости их спада
аэ(0 = dt - f dt (5.107)
j dt j dt
Максимальная плотность зарядов аэ.ыакс, возникающих на
поверхности, определяется пробивной прочностью воздуха (или
любой другой среды). При нормальных условиях (давлений
0,101 МПа и влажности 65%) Оэ.макс Для разноименно заряжен-
ной пластины толщиной h равна (Кл/м2)
<*э, макс = 100е0 (200 + Узё/Л)2. (5.108),
Потенциал на поверхности С/Макс равен: С/макс = Оэмакс/еое-
Причина генерации и возникновения статических зарядов
кроется в процессах, происходящих на границе раздела двух
материалов. При контакте двух металлических тел, находящих-
ся на расстоянии й', происходит перетекание электронов из ме-
талла с более высоким уровнем Ферми (Ф1) в металл с более
низким уровнем (Ф2). После разрыва контакта плотность за-
ряда оз равна
о»= (ео/Л') (Ф| — Ф2). (5.109)
При контакте металла с полупроводником также происходит
перераспределение зарядов, но этот процесс затрагивает толь-
ко приповерхностные области полупроводников. Поверхностная
плотность зарядов составляет
[2ш (Фы - Фиг) ],/2. (5.110)
где ги—концентрация носителей в полупроводнике с диэлектрической про-
ницаемостью е; Фы и Фпп — уровни Ферми в металле и полупроводника.
При контакте металла с диэлектриком плотность зарядов на
диэлектрике зависит от уровня Ферми металла и составляет
Сз=ё£»,Д(Фм-Фд), (5.111)
где е — заряд электрона; Ds—плотность поверхностных состояний, равная
«(0,7+1,35) 10-'6 эВ_,.м~2 для различных полимеров, Фл—уровень Ферми
в диэлектрике.
381
При установлении и разрыве контакта между двумя полиме-
рами без трения на них возникает плотность зарядов, равная
е(Ф, _ фг)
1/DS14- 1/0S3 *
(5.112)
Ряд полимеров, расположенных последовательно таким об-
разом, что каждый последующий заряжается отрицательно по
отношению к предыдущему при контакте с ним, называется три-
боэлектрическим рядом. При разрыве контакта между заря-
женными телами возникает разность потенциалов (7; если U
окажется выше максимально возможной, то может произойти
пробой.
Все материалы можно разделить на электрофобные и элект-
рофильные. Электрофобные материалы не электризуются без
трения, а электрофильные электризуются всегда Трение приво-
дит к локальному повышению температуры, улучшению электри-
ческой проводимости и превращению изолятора в проводник.
Электрические заряды образуются при трении полимеров меж-
ду собой или с другими материалами, при деформировании по-
лимеров, в результате пьезоэффекта (возникновение электриче-
ских зарядов на поверхности диэлектрика и электрическая по-
ляризация внутри его при воздействии механических нагрузок).
Величина электрического заряда зависит от многих факторов:
уровней Ферми в контактирующих материалах, скорости трения,
диэлектрической проницаемости и электрического сопротивле-
ния материала. Электризуемость определяется скоростью спада
заряда do^cueAaldt. Заряд на полимерном образце опадает по
экспоненциальной зависимости:
с,-а0 ехр (— //?,)>
(5.113)
где Go — постоянная; t — время; т> — скорость спада заряда (полупериод
утечки), равный
т,= [еое($/Л) +СГ] [(S/Л) pv+ (S/Л) ps], (5.114)
где S и ft — площадь н толщина образца с диэлектрической проницае-
мостью е; объемным pv и поверхностным рз сопротивлениями; Сг — гермети-
ческая емкость относительно земли.
Если проводимость определяется объемным сопротивлением,
то т, = £оерг и Оэ=Ооехр(—Z/eoepv).
Значения pv= I012 О.м см и р$=1012 Ом являются граничны-
ми, выше которых материал электризуется, а ниже — не элек-
тризуется.
В ряде случаев для предотвращения электризации полиме-
ров применяют специальные вещества—антистатики, повышаю-
щие электрическую проводимость и облегчающие утечку заря-
дов с поверхности.
382
5.3.2. Свойства полимерных полупроводников
и электропроводящих материалов
Полупроводниками называют полимеры, имеющие электриче-
скую проводимость 10~®—10~3 См/м. К ним относятся полимеры
с сопряженными двойными связями, полимерные комплексы
с переносом заряда (КПЗ), некоторые биополимеры, а также
диэлектрики, наполненные токопроводящими наполнителями.
Полупроводники имеют признаки, характерные как для диэлек-
триков, так и для проводников электрического тока Например,
в переменных полях полупроводники характеризуются больши-
ми диэлектрическими потерями (так же, как и диэлектрики),
а некоторые полупроводники имеют проводимость, характерную
для проводников. Механизм электропроводимости полупроводни-
ков может быть зонным, туннельным и механизмом перескоков.
Зонный механизм заключается в следующем. Электроны
в кристалле обладают строго определенными значениями энер-
гии. Наивысший энергетический уровень — зона, в которой на-
ходятся электроны, называется валентной. Наряду с разрешен-
.ными зонами существуют так называемые запрещенные, соот-
ветствующие тем значениям энергии, которыми электрон не мо-
жет обладать. Для перехода с одного энергетического уровня на
другой электрон должен преодолеть запрещенную зону. В ме-
таллах валентная зона не полностью заполнена электронами
Наличие в этой зоне свободных уровней позволяет электрону
переходить легко на них с заполненных уровней той же зоны,
что и обусловливает хорошую электропроводимость металлов.
В диэлектриках и полупроводниках валентная зона заполне-
на полностью и для протекания тока необходимо, чтобы элек-
троны попали в зону, свободную от них (зону проводимости).
Но при переходе электрон должен преодолеть энергетический
барьер Д(7Э (запрещенную зону). Полупроводник отличается от
диэлектрика меньшей шириной запрещенной зоны, т. е. меньшей
высотой энергетического барьера, которая составляет 2—3 эВ
Для преодоления этого барьера электрон должен получить из-
вне дополнительную энергию в виде тепла (термическая элект-
ропроводимость), кванта света (фотопроводимость) или под-
вергнуться действию высокого напряжения (пробой диэлектри-
ка ). При низких температурах и в отсутствие фотопроводимости
полупроводники ведут себя как диэлектрики. По мере нагрева-
ния или облучения все большее число электронов приобретает
энергию, достаточную для перескока через активационный барь-
ер из валентной зоны в зону проводимости (эл гтронная прово-
димость). Покидая заполненную валентную зону, электроны
оставляют в ней свободные энергетические уровни, называемые
«дырками». Появление «дырок» создает возможность последова-
тельного перехода на них электронов с более низких энергети-
383
ческнх уровней, что эквивалентно перемещению «дырок» в об-
ратном направлении. Возникает так называемая «дырочная»
проводимость.
При наличии примесей, энергетические уровни которых рас-
положены в запрещенной зоне, носители тока могут возникать
в результате переходов между уровнями примеси и разрешен-
ной зоной. Если примесь отдает электрон в зону проводимости,
то возникает электронная проводимость (полупроводник
н-типа), если же примеси акцептируют электрон из валентной
зоны, то носителями тока являются «дырки» (полупроводник
p-типа). Отличие полупроводников от электропроводящих ма-
териалов (в частности, металлов) заключается в различном влия-
нии на проводимость температуры: в металлах электропроводи-
мость от температуры практически не зависит, поскольку кон-
центрация носителей тока, равная числу атомов в единице
объема, также не зависит от температуры. Подвижность же носи-
телей при этом обычно несколько уменьшается. В полупровод-
никах с ростом температуры резко увеличивается число носите-
лей тока, которое тем больше, чем меньше ширина запрещен-
ной зоны. Изменение электрической проводимости с температу-
рой описывается соотношением
х=и«ехр (— bEJkT), (5.115)
где Хо — постоянная, не зависящая от температуры, не зависящая от кон-
центрации и подвижности носителей тока; ДЕ» — энергия активации прово-
димости, равная для полупроводников без примеси половине ширины запре-
щенной зоны, а для примесных проводников — половине расстояния между
уровнем примеси и краем зоны проводимости (л-тип) или валентной зоны
(p-тип), k— константа Больцмана.
Величина Д£э для полупроводников, не содержащих приме-
сей, намного выше, чем у полупроводников с примесями. В пере-
менном поле энергия активации снижается с ростом частоты.
Зонный механизм проводимости характерен для полупро-
водников с низким сопротивлением и высокой подвижностью
носителей тока. Туннельный механизм электропроводимости
обусловлен вероятностью безактивационного перехода (проса-
чивания) электронов через энергетический барьер. Этот меха-
низм характерен для полупроводников с очень высокой подвиж-
ностью носителей тока.
Механизм перескоков предусматривает проводимость элек-
трического тока путем перескоков носителей с одного уровня
на другой и проявляется в системах с высоким сопротивлением
и низкой подвижностью носителей тока [~10"2—10-3 см2/(В-с)].
Для этого механизма характерно также повышение электриче-
ской проводимости с ростом температуры, но связано это не
с увеличением концентрации носителей тока, а с повышением их
подвижности [в некоторых случаях в сотни и тысячи раз до
10—100 м2/(В-с)].
384
Механизм электрической проводимости полупроводников
определяется их структурой (химическим строением макромо-
лекулы и надмолекулярной структурой, определяющей уровень
взаимодействия между молекулами). Так, для полупроводни-
ков с сопряженными связями вдоль макромолекулы наиболее
характерен механизм перескоков, согласно которому ток пере-
носится путем активационных перескоков из одной полисопря-
женной области в другую над диэлектрическим барьером, созда-
ваемым неупорядоченными (не имеющими сопряженных связей)
участками. Переход электрона внутри полисопряженной обла-
сти осуществляется практически безактивационно. С ростом
температуры повышается подвижность носителей и электриче-
ская проводимость увеличивается.
Энергия активации Д£э снижается с ростом числа кратких
связей в молекуле, но при определенном их содержании («50)
Д£9 практически перестает зависеть от их числа и становится
настолько низкой, что уже при комнатной температуре молекула
способна проявить электрическую проводимость. Однако, если
проводимость этих соединений достаточно велика вдоль макро-
молекулы, то в перпендикулярном направлении она существен-
но ниже из-за высокого межмолекулярного энергетического
барьера
Проводимость полупроводников с сопряженными связями
можно изменять, изменяя их надмолекулярную структуру Если
образование надмолекулярной структуры связано с повышением
компланарности расположения двойных связей, то Д£э снижа-
ется и проводимость растет; при нарушении компланарности
проводимость, как правило, уменьшается в связи с увеличением
энергетического барьера перескока между молекулами. Это объ-
ясняет увеличение проводимости с ростом степени ориентации
и при повышении давления.
Полимерные полупроводники, относящиеся к полимерным
комплексам с переносом заряда, характеризуются высокой про-
водимостью как вдоль макромолекулы, так и между молекула-
ми. Перенос тока в них осуществляется преимущественно по
зонному механизму с невысокой шириной запрещенной зоны
(«0.1—0,3 эВ). К полимерным комплексам относятся системы,
включающие мономерные звенья, играющие роль доноров
электронов, и соединения, выполняющие роль акцепторов Об-
разование донорно-акцепторных комплексов сопровождается
частичным или полным переносом электрона с орбитали донора
на орбиталь акцептора. Электропроводимость этих соединений
зависит от степени взаимодействия компонентов. Увеличение
донорно-акцепторного взаимодействия приводит к уменьшению
расстояния между компонентами и повышению электрической
проводимости.
На основе полимеров с сопряженными связями можно со-
•25—816 385
>дать электропроводящие полимеры. Они должны иметь упоря-
юченную структуру, состоящую из длинных сопряженных
участков с соответствующими заместителями, способными со-
эдавать и поддерживать в цепи сопряжения особое возбужден-
ное состояние. Такие полимеры способны проявлять высокую
электропроводимость даже при обычных комнатных температу-
рах (явление сверхпроводимости)
В качестве полупроводников могут быть использованы ди-
электрики, наполненные токопроводящими наполнителями: ме-
таллическими порошками, графитом, техническим углеродом.
В качестве металлических наполнителей используют серебро,
никель и другие металлы, не подвергающиеся окислению и не
вызывающие химического разрушения полимеров Механизм
электропроводимости наполненных систем (полупроводников
и диэлектриков) более близок к туннельному, хотя не исключа-
ется возможность эмиссии электронов от частицы к частице.
Туннельное сопротивление определяется толщиной прослойки
полимера, которая зависит от содержания и размера частиц,
их распределения и других факторов С уменьшением толщины
прослойки сопротивление снижается. Его значение зависит так-
же от диэлектрической проницаемости полимера, разделяющего
частицы: при уменьшении проницаемости оно снижается В об-
ласти слабых полей сопротивление практически не зависит от
напряжения, а при высоких значениях напряжения сопротивле-
ние уменьшается
Электропроводимость полимеров, содержащих электропрово-
дящие наполнители, зависит от количества и характера распо-
ложения частиц наполнителя в матрице полимера, а также от
контактного сопротивления между частицами. Если на контакт-
ных поверхностях отсутствует пленка диэлектрика, то величина
контактного сопротивления /?к равна отношению удельного со-
противления проводника pv к радиусу «контактного пятна» а:
RK=pv/a. Если же между контактирующими частицами сущест-
вует пленка диэлектрика, имеющая удельное сопротивление
рк.лл и толщину hnn, то равно (Гпл — площадь соприкосно-
вения)
Rk = pv/2a+ рУ.плЙлл/^пл- ( Но)
Величину pv,n.iAnjI можно заменить на опл (Ом-м2)—сопро-
тивление, приходящееся на единицу площади пленки. Если по-
верхность соприкосновения представляет собой окружность с ра-
диусом а, то Rk равно
/?к=ру/2а+Спл/яя2- (5-117)
С увеличением толщины пленки вклад второго члена (Опл/ла2)
в общее контактное сопротивление будет расти и при толщинах,
превышающих 10 нм, /?к = /?пл=0пл/ла2.
386
При механическом диспергировании наполнителей чаще все-
го наблюдается статистическое распределение их в полимере.
При этом зависимость удельного сопротивления материала от
содержания наполнителя <рн описывается сложной кривой,
имеющей три участка: первый характеризуется постоянным зна-
чением сопротивления, которое определяется свойствами поли-
мерной среды, на втором происходит заметное снижение сопро-
тивления с ростом количества наполнителя, третий характери-
зуется очень слабой зависимостью pv от <рн. Первый перегиб со-
ответствует концентрации наполнителя, при которой начинает
образовываться его непрерывная цепная структура, второй —
моменту, когда формирование этой структуры завершено. Зави-
симость р^=/(срн) на втором участке может быть выражена со-
отношением
pv*“ l,6(/?Kd,./<p;>) (5.118}
(dM — диаметр частиц наполнителя).
На формирование сетки электропроводящего наполнителя
оказывает влияние и взаимодействие полимера с наполнителем.
Проводящая цепная структура наполнителя образуется лишь
в том случае, когда энергия взаимодействия частиц наполните-
ля с полимером превышает энергию взаимодействия полимер —
полимер, но при условии, что на поверхности наполнителя есть
участки, по которым осуществляется контакт, и энергия взаи-
модействия наполнитель — наполнитель выше энергии взаимо-
действия наполнитель— полимер.
Таким образом, изменяя содержание наполнителя, характер
его распределения в полимере, уровень взаимодействия поли-
мер— наполнитель, контактное сопротивление между частицами,
можно в широких пределах варьировать электропроводимость
наполненных композиций, превращая диэлектрик в полупровод-
ник или в электропроводящий материал.
‘S-
5.3.3. Свойства полимерных электретов
Практически всем диэлектрикам, в том числе и полимерным,
присуще электретное состояние, т. е. такое, при котором на по-
верхности диэлектрика возникают поверхностные заряды под
влиянием внешних факторов, таких как электрическое поле,
облучение электронами, ионами- и др. Свойства полимерных
электретов характеризуются эффективной плотностью зарядов
Оэф и временем жизни электрета тж. Значения оЭф электретов
составляют Ю-9—10~7 Кл/см2, а тж— 3—10 лет и более.
Физические свойства электретов зависят от свойств диэлект-
рика, способа и режима его изготовления (температуры, напря-
женности поля, времени поляризации). По способу получения
электреты разделяют на две группы. К первой относятся элек-
25*
387
треты, заряды которых обусловлены поляризацией диполей
(термо-, хемо- и механоэлектреты), ко второй — электреты с ин*
жектированными зарядами (короно- и электроэлектреты, техно-
логические, радиационные). Электреты первой группы обладают
равными и разноименными по отношению к электроду заряда-
ми на противоположных сторонах (гетероэлектреты), а электре-
ты второй группы — равными и одноименными зарядами (гомо-
электреты).
Термоэлектреты, например, получают следующим образом.
Образец полимера нагревают до температуры поляризации Тпсл,
при которой реализуется высокая подвижность диполей и ионов
(7'пол>Л), прикладывают постоянное электрическое поле напря-
женностью Епол и выдерживают в этом поле в течение опреде-
ленного времени /пол- Затем образец охлаждают до температу-
ры хранения Тх, при которой подвижность диполей и ионов не-
значительна или подавлена вообще (обычно это температура
близкая к Тс или ниже ее). Механизм образования электретов
можно представить следующим образом. Под влиянием поля
постоянного электрического тока начинается процесс поляриза-
ции. Диполи и ионы ориентируются и смещаются в соответствии
со знаком потенциала электродов. Это приводит к тому, что на
поверхностях образца возникают заряды, противоположные по.
знаку потенциалу электрода. Если образец охладить до Т*
и снять напряжение, то образуется гетероэлектрет. Если же
дальше повышать напряженность поля, может произойти пробой
прослойки воздуха между поверхностью образца и электродом
и ионизация молекул воздуха, сопровождающаяся выделением
электрона и образованием положительно заряженного иона.
Кроме того, электроны могут быть инжектированы с катода.
Эти электроны (инжектированные и полученные в результате
ионизации воздуха) вступают в последующие реакции (В — мо-
лекула воздуха):
В —*• В++ё; ё+В — ->• В-;
ё+В —*• В++2ё и т. д.
Ионы и электроны захватываются молекулами полимера
и вступают с ними в реакции (П — молекула полимера):
В-+П —В+П-; В-+П —> В+П+ё;
В-+П —> ВП-; В-+П —* ВП+ё;
П+ё —*• П~ и т. д.
Но ионы и электроны могут и не взаимодействовать с поли-
мером, а попадать в ловушку и задерживаться там длительное
время. Ловушками могут служить различные дефекты структу-
ры полимера (концевые группы, границы раздела между кри-
сталлической и аморфной фазами и др.) и ориентированные ди--
ну (несколько молекулярных слоев от поверхности), электроны
проникают глубже, но в целом гомозаряд сосредоточен вблизи
поверхности образца. После охлаждения и снятия напряжения
получаются гомоэлектреты
В термоэлектретах фиксирование ориентированных диполей
и смешанных ионов происходит в результате снижения их по-
движности при охлаждении. Если подвижность снизить за счет
химического сшивания, то получают хемоэлектреты.
В результате механического воздействия на полимеры полу-
чаются механоэлектреты Например, при сжатии полярных по-
лимеров наряду с ориентацией макромолекул происходит их по-
ляризация в направлении, перпендикулярном плоскости ориен-
тации.
Охлаждение в сжатом состоянии как бы замораживает ди-
поли, и после снятия деформации получается заряженный
электрет. Электреты, получаемые при разрушении адгезионной
связи между металлической подложкой и полимером (в том чис-
ле и неполярным), получили название технологических. В этих
электретах образование зарядов обусловлено инжекцией носите-
лей зарядов из металлической подложки. Электреты, получае-
мые при воздействии поля высокой напряженности без термиче-
ской обработки, называют электроэлектретами, заряженные
в коронном разряде, когда одним из электродов является иони-
зированный воздух (плазма), — короноэлектретами, а под дей-
ствием пучка заряженных электронов — радиационными. В тех-
нологических, электро- и короноэлектретау заряд обусловлен
инжекцией носителей заряда.
Эффективная поверхностная плотность заряда оЭф зависит
ют соотношения поляризованных и инжектированных зарядов,
т е. от количества связанных агетеро и свободных Огояо зарядов:
Озф — Огон о “ Огетсро ИЛИ Озф = О*р —~Р, (5.119)
где ол’=Ос»яэ+<7с»об — компоненты гомозаряда, обусловленные захватом но-
сителей ловушками (Освяз) и смешением носителей во внутреннем поле элек-
трета (сак*); Р—Р1+Р3 (Pi — остаточная поляризация, Р3 — поляризация,
обусловленная внутренним полем электрета).
Инжектированные заряды при хранении перемещаются
в объеме электрета вследствие диффузии. Скорость их переме-
щения зависит от того, в какие ловушки попали заряды: если
в глубокие, то вероятность перемещения мала, а уменьшение
Оэф во времени происходит за счет собственных носителей за-
рядов электрета.
Скорость изменения гомозаряда составляет
E»/pv, (5.120)
где pv — удельное объемное электрическое сопротивление, £> — напряжен-
ность поля электрета; для незакорочеиного электрета £»“(А“=о»ф/е0е; для
электрета толщиной Л, с электродом иа расстоянии I от его поверхности
Е» - о»ф/ [ео (е+Лэ//) ].
Максимальная поверхностная плотность зарядов электретов
зависит от пробивного напряжения окружающей среды, которое
определяется давлением При нормальном давлении зависи-
мость максимальной плотности заряда омакс от толщины образ-
ца h описывается уравнением (5.108). Снижение давления и по-
вышение влажности окружающей среды приводит к снижению
эффективности зарядов.
Заряды на поверхности электретов распределяются с различ-
ной плотностью: оЭф растет от центра к краям образца. Резко
неоднородное распределение зарядов после получения электре-
тов приводит к их спаду из-за компенсации зарядов разного
знака.
Время жизни электрета тж зависит от скорости изменения
а»Ф во времени dc^!dt\
da^/dt^-d&Jdt+dP/dt. (5.121).
В простейшем случае уменьшение ар* и Р происходит по экспо-
ненциальному закону:
Р(0 = Рмаке (1 - е~**9); (5.122).
о9р(/) = бэр, макс О — 8 ) » (5-123)
где и Ом«кс — максимальные значения остаточной поляризации и сво-
бодного заряда для данного диэлектрика прн £„<» и Г„ол; т» — время ди-
электрической релаксации (для простоты условно принимается, что полимер
имеет одно время релаксации); время установления Рм*кс зависит от струк-
туры полимера на молекулярном и надмолекулярном уровнях.
С повышением температуры скорость поляризации повыша-
ется, причем не монотонно, а ступенчато. Сначала разморажи-
ваются полярные группы (механизм ДГ-релаксацин), затем сег-
менты (ДС-релаксация). Формирование остаточной поляриза-
ции связано с ориентацией диполей, которая резко возрастает
после размораживания сегментов, т е. при 7'пол>7’с, и замора-
живанием этой ориентации при ТХ<ТС:
Р макс = Сэ[е(7’кол ) —е(Гх)]Епол, (5.124)
где е(ТГОл) и е(Тх) —диэлектрическая проницаемость электрета при темпе-
ратурах поляризации и хранения; ЕПОл—напряженность поля поляризации.
Поскольку Тпол выше, а Тх ниже Тс (т. е. температуры, при
которой размораживается сегментальная подвижность), то мож-
но принять е(Гпол) равной еСт, а е(Гх) равной е<». Тогда
Рмакс = 8о[(8с» ““ е«.)Епол] (5.125)
Величина Де = ест—зависит от температуры и структуры
диэлектрика.
Де=К,пцгЭф/7; (5.126)
390
где /С»— постоянная: п — число молекул в единице объема; рцф— эффектив-
ный дипольный момент равный gofg (Рс— статический дипольный момент,
g— коэффициент корреляции тем больший, чем ниже подвижность диполя,
т. е. меньше угол вращения).
При низких £п, когда смакс весьма мало, зависимость оЭф(0
определяется изменением Р(1) и снижается с ростом t. При
высоких Еп основной вклад в о9ф вносит оэрмаКс и зависимость
0эф(О определяется изменением о’р.максЮ. т е. отрицательный
заряд снижается.
В процессе хранения происходит не только спад оэр,Макс
и Рмакс, но и образование оР и Р в поле электрета, значения ко-
торых зависят от диэлектрических свойств электрета.
Влияние температуры на стабильность электретов обуслов-
лено ее влиянием на скорость изменения гетеро- и гомозарядов.
Как правило, с повышением температуры хранения скорость
спада зарядов увеличивается и тж уменьшается. Проникающая
радиация вызывает накопление носителей зарядов в результате
захвата заряженных частиц извне и образования заряженных
частиц в период облучения. В результате этого в диэлектрике
растет электропроводимость, снижается поверхностная плот-
ность зарядов и тж уменьшается.
Механические свойства электретов отличаются от механиче-
ских свойств диэлектриков той же структуры. При разрушении
стеклообразных полимеров существенно возрастают предел вы-
нужденной эластичности и напряжение при разрыве. Релакса-
ция напряжения в механоэлектретах (например, в поликарбона-
тах) зависит от степени деформации: при малых деформациях
релаксация протекает быстрее, а при больших — медленнее.
Причина заключается в предварительной ориентации макромо-
лекул (и диполей) и увеличении размеров упорядоченных обла-
стей под влиянием электрического поля. Предварительно ори-
ентированные механоэлектреты быстрее релаксируют, но при
высоких степенях деформирования между упорядоченными
структурами образуется довольно прочная флуктуационная
сетка, которая затрудняет протекание релаксационных процес-
сов и снижает их скорость.
«
Контрольные вопросы
I. Какими показателями характеризуются деформационные (упругие и ре-
лаксационные) свойства полимеров в стеклообразном, высокоэластиче
ском, вязкотекучем и кристаллическом состояниях. Перечислите особен-
ности деформирования полимеров в различных фазовых и физических
состояниях.
2. Что такое гистерезис? Перечислите причины гистерезисных потерь при
деформировании полимеров.
3. Оказывают ли влияние температура, скорость деформирования и струк-
тура полимера на зиачеиня модулей к гистерезисных потерь?
391
I I. Как можно доказать, используя механические испытания, имеет ли дело
экспериментатор с кристаллическим или сетчатым полимером?
5 Будут ли различаться модули упругости (модули Юнга) вдоль и поперек
направления ориентации одноосно-ориентированного полиамидного волок-
на? Если будут, то как?
6 Будут ли отличаться кривые зависимости модуля Юнга от температуры
для линейного полимера высокой молекулярной массы от аналогичных
кривых для того же полимера, сшитого химическими связями?
7 Полимер имеет три инка механических потерь. Как определить, какой из
них связан с наличием кристаллической фазы?
Ч. Один из полимеров имеет два пика механических потерь при Т<ТС
<й-переход) и при Тс (a-переход), а другой — одни, только прн Гс. Ка-
кой из этих полимеров будет иметь меньшую температуру хрупкости?
9. Что характеризует кривую течения полимера. В чем причина аномалии
вязкости? Каким уравнением описывается течение полимеров в области
аномалии вязкости?
10. Влияет ли структура полимера на наибольшую ньютоновскую и эффектив
иую вязкость?
il. Чго такое высокоэластичность расплавов полимеров?
12. Как изменяются реологические свойства полимера прн введении в него
до 20% (об.) наполнителя, который образует флуктуационную сетку и
снижает подвижность макромолекул?
13. Каковы причины уменьшения усадки линейных полимеров при введении
в них небольшого количества микрогеля?
14 Что такое теоретическая кратковременная и длительная механическая
прочность? Влияет лн температура испытания, уровень напряженности и
стойкость полимера к окислению на длительную и кратковременную
прочность? Снизится ли долговечное 1ь полимера при введении в него
пластификатора?
15. Изменится ли механизм разрушения полимеров при повышении темпера-
туры от 0 К до температуры текучести полимера? Каковы основные
особенности разрушения полимеров в различных физических и фазовых
состояниях?
16. Каковы причины разброса показателей прочности полимеров при испы-
таниях и способы снижения разброса?
17. Два сшитых полимера (натуральный каучук и цас-1,4-полибутадиен)
разрушаются при скоростях деформирования от 0,004 до I03 м/с и
температурах от Тс до Л+200 К. Как будут изменяться кривые напря-
жение— деформация в зависимости от температуры и скорости? Какой
вид будут нметь огибающие разрывов?
18. Каковы особенности разрушения полимеров прн динамическом нагру-
жении?
19. Почему царапины на поверхности образцов снижают их выносливость
при многократном деформировании?
20 В процессе усталостных испытаний полимеров модуль упругости умень
шаегся, а модуль потерь возрастает. Обусловлено ли это постепенными
изменениями структуры образца или свидетельствует о его близком раз-
рушении?
21 Как будет изменяться усталостная выносливость полимеров с редкой хи-
мической сеткой (натуральный каучук цнс-1,4-полибутадиен. его сополи-
меры с 18, 26 и 40% нитрила акриловой кислоты) при нспытаиин в ре-
жиме синусоидальных гармонических колебаний при постоянной амплиту-
де деформации? Как изменится усталостная выносливость полимеров прн
расширении молекулярно-массового распределения и увеличении числа по-
перечных связен: I) в режиме постоянных амплитуд деформации;
2) в режиме постоянной амплитуды напряжения?
22 Изменится лн теплоемкость полибутадиена, его сополимера с нитрилом
392
акриловой кислоты (60 40) и полиакрилонитрила при повышении тем-
пературы от 233 до 473 К? Если изменится, то почему?
23 Изменятся ли теплопроводность и температуропроводность полимера при
наполнении его 50% (масс.) технического углерода?
24 Какие из приведенных ниже полимеров
CI
~СН2—СН2~, ~СН2-СН~СН-СН2~, ~СН2—С==СН—СНо~.
СН2=(=С==С-С=)П=СН2
относятся к классу диэлектриков? Какой из них имеет большие диэлек-
трические потерн7
25 Как изменится электропроводимость композиции, состоящей из низкомоле-
кулярного диенового полимера и технического углерода, если в резуль-
тате химического взаимодействия между компонентами образуются сопря-
женные двойные связи?
ГЛЛВА 6
ИСТИННЫЕ РАСТВОРЫ ПОЛИМЕРОВ
И КОЛЛОИДНЫЕ СИСТЕМЫ
%
Процессы взаимодействия полимеров с низкомолекулярными
жидкостями имеют большое значение при синтезе полимеров,
их переработке и эксплуатации в различных жидких средах.
При взаимодействии полимера с низкомолекулярными жидко-
стями в зависимости от степени диспергирования его могут об-
разовываться истинные растворы и коллоидные системы. Ниже
приведены основные признаки истинного раствора и коллоидной
системы
Истинные растворы
1 . Наличие сродства между компо-
нентами
2 Самопрозвольное образование
3 . Молекулярная или ионная дис-
персность
4 Термодинамическая устойчивость
5 Увеличение степени дисперсности
во времени
6 Агрегативная устойчивость
7 . Одиофазность
8 . Отсутствие поверхности раздела
9 Обратимость
Коллоидные системы
1 . Отсутствие сродства между ком-
понентами
2 . Принудительное образование
3 Коллоидная дисперсность
4 Термодинамическая неустойчивость
5 Уменьшение степени дисперсности
во времени
6 - Агрегативная неустойчивость
7 . Двухфазность
8 Наличие поверхности раздела
9 Необратимость
Если между компонентами системы есть сродство, то при
контакте друг с другом без затраты внешней энергии они начи-
нают самопроизвольно диспергироваться друг в друге, что при-
водит к постепенному увеличению степени дисперсности до мо-
лекулярной или ионной» Самопроизвольное диспергирование,
393
in растворение, как всякий самопроизвольный процесс, проно-
сящий при постоянных давлении и температуре, сопровожда-
йся уменьшением свободной энергии и ростом энтропии.. При
го.м образуется однофазная система, в которой отсутствует
оверхность раздела.
В истинном растворе (если ои не бесконечно разбавлен)
результате взаимодействия молекул растворенного вещества
руг с другом образуются ассоциаты, обратимо разрушающиеся
од влиянием теплового движения. Это обусловливает возмож-
ость обратимых изменений свойств раствора при изменении
нешних условий. Так, истинный раствор можно нагреть, охла-
дить, разбавить, сконцентрировать, но при заданных темпера-
уре и давлении концентрация раствора, его свойства и струк-
ура будут одними и теми же независимо от способа приготов-
1еиия. Равновесие, не зависящее от пути его достижения, назы-
зается истинным (отсюда и название растворов).
Свойства и структура коллоидных систем, как правило, за-
аисят от способа их приготовления В коллоидных системах
всегда идут процессы агрегирования, они являются агрегативно
неустойчивыми, в результате чего распадаются на две фазы.
Из любого полимера в зависимости от его сродства к той
'или иной жидкости можно получить истинный раствор или кол-
лоидную систему. Например, натуральный каучук самопроиз-
вольно растворяется в алифатических углеводородах, полисти-
рол— в бензоле, при этом образуются истинные растворы.
Но эти же полимеры не могут самопроизвольно растворяться
в воде или метаноле—в этих жидкостях они образуют колло-
идные системы.
*
6 1 ИСТИННЫЕ РАСТВОРЫ
Истинные растворы полимеров имеют специфические особенно-
сти, отличающие их от растворов низкомолекулярных веществ
и обусловленные огромной разницей в размерах молекул поли-
мера и растворителя. К этим особенностям относятся явление
набухания, высокая вязкость даже разбавленных растворов,
ряд отклонений от классических законов и уравнений термоди-
намики.
6.1.1. Набухание
•
При соприкосновении полимера с низкомолекулярной жид-
костью ее молекулы начинают быстро проникать в фазу поли-
мера, а макромолекулы за это время не успевают перейти
в фазу растворителя: прежде чем раствориться, полимер на-
бухает.
394
Набухание — это процесс поглощения или сорбции низко-
молекулярных жидкостей (или их паров) полимером. При на-
бухании молекулы низкомолекулярной жидкости проникают
между элементами надмолекулярной структуры полимера, вы-
зывая межструктурное набухание, или внутрь структур, раздви-
гая макромолекулы, — внутриструктурное набухание. Следова-
тельно, набухание—это сорбция (поглощение) низкомолеку-
лярного вещества полимером, сопровождающаяся увеличением
его массы, объема и изменением структуры Между молекула-
ми полимера и диффундирующего в него растворителя происхо-
дит взаимодействие, которое называется сольватацией. Проник-
новение растворителя в полимер быстрее всего происходит в об-
ластях с наиболее рыхлой упаковкой макромолекул по меха-
низму капиллярного всасывания Одновременно с относительно
быстрым заполнением пор, пустот, каналов идет более медлен-
ная диффузия растворителя в надмолекулярные образования.
Скорость проникновения растворителя от поверхности в глубь
полимера зависит от степени термодинамического сродства
растворителя и полимера, уровня межмолекулярного взаимо-
действия в полимере, температуры и других условий процесса.
На начальной стадии набухания распределение растворителя
в объеме полимера неоднородно: поверхностные слои, непосред-
ственно контактирующие с растворителем, содержат наибольшее
его количество, в средних слоях растворителя нет. Естественно,
что на этой стадии набухания образец полимера сильно дефор-
мируется, в нем возникают большие внутренние напряжения,
вызывающие разрыв наиболее растянутых участков макромо-
лекул. Однако при достижении растворителем центральных об-
ластей набухающего образца его концентрация в полимере по-
степенно выравнивается
Процесс набухания можно представить как одностороннее
смешение, а набухший полимер — как две сосуществующие
фазы, разделенные поверхностью раздела и находящиеся в рав-
новесии: раствор низкомолекулярного растворителя в полимере
и чистый растворитель или разбавленный раствор полимера
в низкомолекулярном растворителе. Различают ограниченное
и неограниченное набухание (рис. 6 I)
Неограниченное набухание—это набухание, самопроизволь-f, 3
но переходящее в растворение. Оно аналогично неограниченно-
му смешению жидкостей, например воды и этилового спирта.
Неограниченное набухание характерно для линейных аморфных
полимеров с невысокой степенью полимеризации, сольватиро-
ванные макромолекулы которых легко и быстро могут перехо-
дить в раствор Степень набухания, после которой начинается
растворение, должна быть достаточной для полной сольватации
макромолекул и их отделения от остальной массы набухающе-
го полимера, т. е. растворения Таким образом, вокруг набу-
395
ающего образца полимера образуется слой раствора полимера.
• результате диффузии макромолекулы равномерно распреде-
яются по всему объему растворителя и в конце растворения
бразуют однофазную гомогенную систему. Кинетика неограни-
енного набухания некоторых полимеров показана на рис 6.2.
1о точки а для всех полимеров наблюдается постепенно замед-
ляющееся увеличение степени набухания (вследствие более ин-
енсивного набухания в начале процесса) В точке а скорость
>астворения становится равной скорости набухания и некоторое
>ремя степень набухания не изменяется. В точке в скорость
тастворения начинает превышать скорость набухания, и масса
образцов уменьшается. Между точками айв образцы имеют
максимальную степень набухания QMaKC в течение времени Дт.
Лз сопоставления кривых набухания можно сделать вывод, что
зем ниже молекулярная масса, меньше разветвленность макро-
молекул и межмолекулярное взаимодействие и выше термоди-
намическое сродство между полимером и растворителем — тем
меньше Омаке И Дт.
При очень высокой молекулярной массе или сильном меж-
молекулярном взаимодействии некоторые полимеры (например,
белки) растворяются крайне медленно (кривая 3) и могут со-
хранять максимальную степень набухания в течение длитель-
ного времени, т. е. характеризуются ограниченным набуханием.
Для растворения таких набухших полимеров требуется переме-
шивание, повышение температуры; при этом межмолекулярное
Рис. 6 1. Кривые неограниченного (/) и ограниченного (2) набухания полиме-
ров (Q — степень набухания, т — продолжительность)
Рис. 6.2. Кинетика неограниченного набухания линейных полимеров при
290 К:
1— иеразветвленные нлн с невысокой молекулярной массой; 2— с высокой молекуляр-
ной массой или разветвленные; 3 — с высокой молекулярной массой н сильным межмо-
лекулярным взаимодействием
3%
Рис 6.3. Кинетика ограниченного
набухания сетчатых полимеров:
/ — редкосетчатые; 2 — густосетчатые;
3 — сетчатые, содержащие экстрагируе-
мые компоненты
взаимодействие ослабляет-
ся и повышается подвиж-
ность макромолекул, что
ускоряет их растворение.
Ограниченное набуха-
ние — процесс взаимодей-
Q
ствия полимеров с низкомолекулярными жидкостями, не сопро-
вождающийся растворением. Это наблюдается при невысоком
термодинамическом сродстве полимера и растворителя, а так-
же характерно для полимеров, макромолекулы которых соеди-
нены прочными поперечными связями в пространственную сет
ку. Редкие поперечные связи между макромолекулами на пер-
вой стадии набухания полимера не затрудняют диффузию
в него молекул растворителя. Поэтому в первый период набу-
хание происходит с максимальной скоростью. Однако сольвата-
ция растворителя звеньями макромолекул, расположенными
между узлами сетки, снижает их подвижность, приводит к уве-
личению расстояний между ними, к растяжению и распрямле-
нию макромолекул, уменьшению энтропии системы, появлению
сильных механических напряжений и разрыву некоторых пере-
напряженных участков; скорость набухания при этом умень-
шается.
При определенном давлении набухания (см. с. 399) процесс
прекращазтея. В этот момент система приходит в равновесие,
при котором увеличение энтропии вследствие перемешивания
молекул растворителя и сегментов макромолекул равно умень-
шению энтропии в результате растяжения сегментов.
С увеличением числа поперечных связей, т. е. густоты про-
странственной сетки, степень и скорость набухания снижаются.
Кинетика ограниченного набухания сетчатых полимеров пока-
зана на рис. 6.3. Своеобразный вид кривой 3 объясняется экст-
ракцией из набухающего сетчатого полимера растворимых низ-
комолекулярных компонентов смеси и их растворением.
На участке а—b скорости набухания и растворения одинаковы,
на участке Ь — с преобладает растворение, на участке с — d
растворение заканчивается. Степень набухания соответству-
ет максимуму- набухания смеси сетчатый полимер + раствори-
мый компонент, Qi — максимуму набухания сетчатого полимера,
Q,— Qi—количеству растворившегося компонента. В результа-
те вымывания из полимера растворимых примесей при набуха-
нии наблюдается не увеличение, а уменьшение массы образца.
397
L
Процесс набухания характеризуют несколькими показателя-
ми: степенью набухания, скоростью, кинетикой набухания,
контракцией, давлением набухания и др.
Степень набухания характеризует увеличение массы (QM)
или объема (Qo) полимера в результате набухания его в опре-
деленных условиях (форма и размеры образца, продолжитель-
ность, температура и др.). Расчет ведут по формулам
q _ . 100%; Qo = ГнТ~~ • 100% •
т0 Уо
где т„, т0 — масса стандартного образца полимера после и до набухания;
VK. Vo — объем стандартного образца полимера после и до набухания.
Скорость набухания зависит от скорости диффузии раство-
рителя в полимер. Она может быть оценена по увеличению мас-
сы (им) или степени набухания (oq) образца полимера за за-
данный отрезок времени:
_ nL _ дТг . _ Q ~ Qi _
1м~ т2— ?! — Дтг ’ Vq~ т2— q — д-t/ ’
где нц, т2 — масса образца в момент времени Т| и т2; Ql и Q?—степень на-
бухания в момент времени Ti и т2.
Скорость набухания может также характеризоваться углом
наклона а кривой зависимости степени набухания от времени
к оси абсцисс: чем больше а, тем выше скорость (см. рис. 6.3).
Как видно из рис. 6.3, начиная о определенного времени, сте-
пень набухания становится постоянной. Степень набухания, ко-
торой соответствует появление горизонтального участка на кри-
вой, называется максимальной или равновесной степенью на-
бухания. Скорость набухания полимера в парах значительно
меньше, чем в жидкости, но максимальная степень набухания
нс изменяется.
Анализ изменения степени набухания в различных условиях
позволяет судить о структуре полимера и его эксплуатационных
свойствах. Форма кинетических кривых набухания зависит от
структуры полимера, степени термодинамического сродства его
к растворителю. При изменении внешних условий ограниченное
набухание может перейти в неограниченное, и наоборот.
При набухании полярных полимеров в полярных растворите-
лях увеличение объема полимера обычно сопровождается
уменьшением объема всей системы (суммы объемов полимера
и растворителя до набухания), т. е. Ун<Ко4-Кр, где Кр — объем
растворителя, вызвавшего увеличение Vo до Ук Такое уменьше-
ние объема системы называется контракцией (от латинского
contractio — сжатие, сжимание). Это явление обусловлено ори-
ентацией молекул растворителя и их более плотным располо-
жением на поверхности макромолекул при сольватации, чем
в растворе, а также заполнением растворителем микропор внут-
398
ри аморфных Областей П ЛИ-------a. l\UHipdnunn ~~v спи л iv i v* &м л
определена по эмпирическому уравнению
V=am/(^+rn),
где а и 0 — константы; т — масса растворителя, поглощенного прн набуха-
нии 1 г полимера.
Контракция прямо пропорциональна интегральной теплоте
набухания <7ННТ> т. е VfqHur=const.
При набухании полимерных материалов в условиях сохране-
ния постоянного объема полимера внутри образца развивается
очень высокое давление — давление набухания. Оно зависит от
энергии взаимодействия полимера с растворителем, концентра-
ции полимера в набухшей системе, условий процесса и может
быть определено по уравнению
Pp=PaCk
или в логарифмической форме:
In Рр—in p0+k in с,
где Рр — давление, развиваемое растворителем внутри полимера; р0 — кон-
станта. определяемая химическим строением полимера и растворителя, тем-
пературой процесса; с — содержание сухого полимера в набухающем об-
разце, k — константа.
Обычно чем больше значение константы р0, тем выше равно-
весная степень набухания при ограниченном набухании. Набу-
хание полимерных изделий приводит не только к увеличению их
объема и размеров, искажению формы, но и к резкому сниже-
нию прочности. Изменение свойств полимера при набухании
в значительной степени зависит от природы полимера и раство-
рителя, с которым он соприкасается. Так, действию паров воды
и водных растворов кислот, солей и других веществ наиболее
подвержены полимеры с полярными функциональными группа-
ми, например целлюлоза, белки и др. Равновесное содержание
влаги в полимере (в % к его массе при данной влажности воз-
духа) минимально у полиолефинов (полиэтилен — 0,1%), более
значительно у аминопластов и полиамидов (капрон — до 4%),
очень высокое у белков (10% и более). Влажность существенно
влияет на свойства полимеров, особенно при высокой темпера-
туре, в частности снижает прочность, диэлектрические показа-
тели, прозрачность.
Вода в полимерах способствует протеканию гидролиза и дру-
гих химических реакций, облегчает подвижность структурных
элементов, что может привести к их разрушению, часто способ-
ствует разрушению полимеров под действием плесени. С водой
в полимер попадают различные растворенные в ней примеси.
Поэтому при изготовлении изделий из полимеров тщательно
.контролируют и регулируют влажность полимеров и всех дру-
399
Рис. 6 4. Изменение степени набухания Q
н прочности а вулканизатов в процессе на-
юо бухания в органическом растворителе
75
гих компонентов. В процессе экс-
5С плуатации изделий из полимеров
вода может вымывать из них раз-
г5 личные компоненты. Например, при
.соприкосновении с водой подиви
нил хлор и дней пленки из нее вымы-
ваются стабилизаторы и пласти-
фикаторы.
На рис. 6.4 показано, как изменяется прочность вулканиза
тов в процессе набухания в органическом растворителе. Из ри-
сунка видно, что прочность (как и другие физико-механические
показатели) быстро снижается в первый период набухания за-
долго до достижения максимальной степени поглощения. Следу-
ет отметить, что степень набухания полимеров в органических
растворителях зависит и от молекулярной массы растворителя-
чем она ниже, тем больше степень набухания.
Процесс набухания сопровождается разрушением межмоле-
кулярных и водородных связей, разрывом наиболее напряжен
пых макромолекул, что приводит к возникновению свободных
радикалов, которые могут инициировать реакции деструкции
Интенсивная окислительная деструкция полимера наблюдается
в том случае, если сам растворитель легко окисляется. При этом
прочность полимера снижается весьма существенно. Интенсив-
ность деструкции возрастает при повышении температуры
В ряде случаев, когда сам полимер не набухает в раствори-
теле, изделие из него под действием этого растворителя может
разрушаться из-за вымывания пластификатора, стабилизаторов
или каких-либо других компонентов Действие растворителей
усиливается при воздействии тепла, механических деформаций,
различных факторов, интенсифицирующих процессы экстраги-
рования компонентов из изделий, их разрушение.
6.1.2. Термодинамика растворения
Растворением, как мы уже сказали, называют самопроизволь-
ный процесс образования термодинамически устойчивой гомо-
генной (однофазной) системы. При самопроизвольном растворе
нии полимеров происходят следующие процессы: диффузия мо-
лекул растворителя в матрицу полимера, сольватация молекул
растворителя на активных центрах макромолекул; распад над-
молекулярных образований вследствие сольватации и ослабле-
ния межмолекулярного взаимодействия; отделение предельно
400
сольватированных макромолекул от поверхности наоухшего но-
лимера; диффузия сольватированных макромолекул из полиме-
ра в раствор.
Обязательное условие растворения — уменьшение свободной
энергии системы при смешении компонентов. Особенностью рас-
творения полимеров является очень большая роль энтропии,
так как введение растворителя в полимер повышает вероятность
изменения конформации молекул. Изменение энтропии при рас-
творении полимеров в растворителе определяется уравнением
Флори — Хаггинса:
Д$=— 7?(Л| Intpj+nzlncpj),
(6.1)
где /? — универсальная газовая постоянная; «1 и л2—число молей компонен-
тов ф! и фз — их объемные доли.
Поскольку растворы полимеров являются термодинамически
устойчивыми и обратимыми системами, при растворении поли-
мера происходит уменьшение изобарно-изотермического потен-
циала AG или (при постоянных температуре и давлении) умень-
шение свободной энергии, что возможно при условии, если
в уравнении —ГД5СМ величина ДГсм<0. Это реали-
зуется в нескольких случаях
I) ДЯсмСО и Д5см>0—растворение экзотермическое, со-
провождается возрастанием энтропии: при этом энергия взаи-
модействия между разнородными молекулами больше, чем меж-
ду однородными;
2) Д//с«<0 и ASvmCO при условии |Д//с>.|>ITAS™] — рас-
творение также экзотермическое, но сопровождается уменьше-
нием энтропии вследствие иммобилизации растворителя в обра-
зующихся сольватных оболочках вокруг звеньев макромолекул;
это наблюдается, например, при растворении полярных полиме-
ров в полярных растворителях:
3) Д//см<0 и Д£см>0 при условии |Д//См|<|ТД5€М| —
растворение эндотермическое, сопровождается возрастанием эн-
тропии; наблюдается в неполярных растворителях;
4) Д//сМ = 0 и Д£см>0 — растворение атермическое, сопро-
вождается возрастанием энтропии, условие A/VCM—О обычно
означает, что энергия взаимодействия и средняя плотность упа-
ковки молекул при растворении полимера не изменяются.
Теплота смешения на единицу объема материала при дан-
ной концентрации связана с плотностью энергии когезии сле-
дующим уравнением:
ДЯ/( 1/ф,<ра) - [ (ДЕ1/У1)7* - (AEj/Va) J \
(6.2)
где Д// — полная теплота смешения; V — общий объем смеси; <₽| н фа —
объемные доли компонентов; AEt н ДЕ2 — изменение энергии когезии в про-
цессе смешения; и У2— объемы компонентов.
26—816
401
Отношение ДЕ/V называется плотностью энергии когезии
(или плотностью энергии молекулярного притяжения), а вели-
чина (ДЕ/V)1/2 — параметром растворимости и обозначается
буквой 6. Таким образом,
AW=V,y2(6l — 6?)2. (6.3)
Если 61—6г=0 (т. е. 6j=62), то ДН=0, и при растворении ос-
новную роль играет энтропийный фактор.
6.1.3. Термодинамическое сродство полимера и растворителя
Способность к набуханию и растворению полимеров в тех или
иных растворителях зависит от строения их молекул. Эмпири-
ческое правило «подобное растворяется в подобном» подтверж-
дается тем, что обычно неполярные полимеры легко растворя-
ются в неполярных растворителях и не растворяются в поляр-
ных; наоборот, полярные полимеры не растворяются в неполяр-
ных растворителях, но растворяются в полярных.
Набухание и растворимость полимера в том или ином рас-
творителе зависят от взаимодействий функциональных групп
или атомов, в результате которых возникают донорно-акцеп-
торные и другие связи, приводящие к образованию устойчивых
комплексов макромолекул полимера с молекулами растворите-
ля. Например, ароматические полимеры вследствие подвижности
л-электронов бензольного ядра образуют с молекулами арома-
тических или хлорсодержащих растворителей л-комплексы.
Процесс растворения (набухания) идет только в том случае,
если компоненты могут взаимно смешиваться или взаимно рас-
творяться, т. е. зависит от того имеется ли между ними термо-
динамическое сродство. В зависимости от степени термодина-
мического сродства растворителей к полимерам их подразде-
ляют на термодинамически совместимые с полимерами
и несовместимые. Количественную оценку термодинамического
сродства компонентов друг к другу проводят по степени сниже-
ния в результате взаимодействия их химических потенциалов р.
Химический потенциал равен приращению свободной энергии
раствора данной концентрации при добавлении к нему бесконеч-
но малого количества i-го компонента при постоянных давлении
Р и температуре Т и постоянном числе молей всех остальных
компонентов пр.
Vi=(dG/dnt)P'TП} (6.4)
Химический потенциал характеризует парциальную мольную
свободную энергию данного компонента Gp, для чистого вещест-
ва \ii-Gi. Химический потенциал компонента в растворе р/
должен быть меньше его химического потенциала до растворе-
402
ния р,0 и, следовательно, разность этих величин должна быть
отрицательной (Дц,<0).
Процесс идет в направлении смешения компонентов только
в том случае, если между ними имеется сродство, поэтому кри-
терии сродства и направленности одни и те же (AG<0 и Ац,<
<0): чем больше абсолютные значения этих величин, тем боль-
ше термодинамическое сродство между компонентами, тем пол-
нее идет процесс их взаимодействия.
Химические потенциалы определяют, измеряя давление пара
растворителя над раствором, осмотическое давление, и другими
методами.
Определение Др, по давлению пара растворителя При оди-
наковой температуре парциальное давление паров жидких ком-
понентов над раствором р, всегда меньше давления насыщенно-
го пара р,° над индивидуальным компонентом. Отношение р,7р»°
называется относительным давлением пара. Оно связано с хи-
мическим потенциалом уравнением
Др,-=ЯГ1п (р./р,°). (6.5)
Поскольку р,7р,°<1, то Др;<0, и растворение происходит са-
мопроизвольно.
Существует понятие идеального раствора. Это раствор, для
которого во всей области концентраций химический потенциал
каждого компонента определяется только его мольной долей х-,
в растворе:
Дщ—РГ In Xi, (6.6)
т е. для идеальных растворов выполняется соотношение
Pi/pF^xi, (6.7)
которое называется законом Рауля. Идеальные растворы обыч-
но образуются при смешении веществ, сходных по химическому
строению и размерам молекул. В этом случае энергии взаимо-
действия однородных и разнородных молекул и их объемы близ-
ки, поэтому при смешении не происходит изменения внутренней
энергии, энтальпии и объема, т. е. свойства идеального раствора
складываются аддитивно из свойств компонентов.
Большинство реальных (неидеальных) растворов не подчи-
няется закону Рауля Для них также по мере увеличения со-
держания растворенного вещества парциальное давление пара
растворителя снижается, однако при этом наблюдаются поло-
жительные (pi/pt°>Xi) или отрицательные (р,7р«0О<) отклоне-
ния от закона Рауля (рис. 6.5).
Отклонения от закона Рауля объясняются энергетическим
взаимодействием молекул компонентов раствора и изменением
энтропии системы при растворении. Так, если молекулы какого-
либо компонента могут располагаться среди молекул др)того
26»
403
Рис. 6.5 Зависимость относительного давления пара р;/л° растворителя от
копиейграцпн полимера в растворе хг.
/ — идеальный раствор, подчиняющийся закону Рауля; 2 — отрицательное отклонение
от закона Рауля при «хорошем» термодинамическом сродстве растворителя к полимеру;
3 — положительное отклонение при «плохом» термодинамическом сродстве растворителя
к полимеру
Рис. 6.6. Зависимость осмотического давления л от концентрации с раствора
низкомолекулярного вещества (/) и полимера (2)
компонента большим числом способов, чем среди себе подоб-
ных, то энтропия реального раствора будет выше энтропии
идеального раствора, что приведет к отрицательному отклоне-
нию от закона Рауля Подобный эффект наблюдается прн рас-
творении полимеров с гибкими макромолекулами. В противо-
положном случае энтропия раствора становится меньше энтро-
пии идеального раствора. Это способствует переходу молекул
растворителя в парообразную фазу, т. е. росту plt что дает по-
ложительное отклонение от закона Рауля.
Полимеры не переходят в парообразное состояние, поэтому
давление над растворами полимеров является давлением пара
растворителя и наблюдается резкое отрицательное отклонение
от закона Рауля Чем выше термодинамическое сродство рас-
творителя к полимеру, тем меньше при данной концентрации
раствора значение р, и больше абсолютное значение Др,-.
Определение Др, по осмотическому давлению. Если раство-
ритель отделить от раствора полупроницаемой мембраной, то он
будет проникать через нее до тех пор, пока в растворе не воз-
никнет гидростатическое давление, компенсирующее давление
растворителя на мембрану. Это давление называется осмотиче-
ским. Осмотическое давление л связано с химическим потен-
циалом растворителя Др, соотношением
ДГ(.= - яй/(1 bPrv)’
\ £ /
(6.8)
404
где Vi° — парциальный объем растворителя при внешнем атмосферном дав-
лении иа раствор; fir — коэффициент изотермической сжимаемости раство-
рителя в растворе.
Из уравнения следует, что осмотическое давление — это по-
казатель термодинамического сродства между компонентами,
отнесенный к единице объема растворителя: чем больше осмоти-
ческое давление, тем больше абсолютное значение Др,, т. е. тем
выше термодинамическое сродство растворителя к полимеру.
В разбавленных растворах низкомолекулярных веществ ос-
мотическое давление л подчиняется закону Вант-Гоффа:
n=cRT, (6.9)
где с — концентрация растворенного вещества; R— универсальная газовая
постоянная; Т — температура
В растворах полимеров каждый сегмент макромолекулы пе-
ремещается независимо от других, ведет себя как кинетически
самостоятельная единица. Поэтому осмотическое давление рас-
творов полимеров не подчиняется закону Вант-Гоффа и зави-
сит от гибкости макромолекул: чем выше гибкость, тем короче
сегменты и больше их содержится в макромолекуле, тем выше
осмотическое давление и сильнее отклонение от закона Вант-
Гоффа. Кроме того, осмотическое давление растворов полимеров
возрастает не пропорционально их концентрации, как в случае
низкомолекулярных веществ, а значительно быстрее (рис. 6.6).
Поэтому для описания зависимости осмотического давления от
концентрации полимеров пользуются не уравнением Вант-Гоф-
фа, а уравнением Ван-дер-Ваальса в виде вириального разложе-
ния, т е. по степеням концентрации с:
л«/?Г(А|С+Л2с2+Д3с3+. .) (6.10)
или л/с=/?Г(Л1+Л2С-Мзсг+...)» (6Л1)
где с — концентрация полимера в растворе; Л|, А3, As— первый, второй, тре-
тий вириальиые коэффициенты, зависящие от формы и размеров молекул
растворенного вещества и действующих между ними сил; значения коэффи-
циентов можно вычислить по экспериментальной зависимости я/с«/(с).
Первый внриальный коэффициент Ai непосредственно связан
с молекулярной массой М полимера соотношением Л|=1/М.
Третий и последующий члены степенного ряда незначительно
сказываются на результатах, поэтому уравнение (6.9) можно
записать в виде
n/c=RT/M+A3c. (6.12)
Для растворов одного и того же полимера в различных рас-
творителях зависимость л/с==/(с) выражается серией веерооб-
разных прямых (рис. 6.7), наклон которых определяется значе-
нием второго вириального коэффициента А?. Этот коэффициент
является мерой термодинамического сродства растворителя к по-
405
Рис. 6.7. Зависимость осмотического
давления от концентрации растворов по-
лимеров, отличающихся значением вто-
рого вириальиого коэффициента Z2:
(идеальный растоор): 2 —Л±<0
(«плохое» термодинамическое сродство); 3, 4,
5—Лт>0 («хорошее» термодинамическое срод-
ство)
лимеру. Чем выше положитель-
ное значение Д2, тем лучше рас-
творитель, т. е. для семейства
линий 3, 4, 5 термическое срод-
ство растворителя возрастает
в ряду 3<4<5. Таким образом, если Д2>0,— растворитель
«хороший», Д2<0—растворитель «плохой». В случае идеаль-
ного раствора Д2 = 0 и уравнение (6.10) переходит в уравнение
Вант-Гоффа (6 7)
6.1.4. Фазовое равновесие в системе полимер — растворитель
Устойчивость системы (фазовое равновесие), как правило, опре-
деляется степенью термодинамического сродства компонентов,
зависит от их химического состава и строения, внешних условий,
в частности от температуры. Основным законом равновесия
многофазной многокомпонентной системы является правило фаз
Гиббса, которое устанавливает взаимосвязь между числом
фаз Ф, числом компонентов в системе К и числом ее степеней
свободы С:
С=К—Ф+2.
Число степеней свободы показывает, сколько термодинамиче-
ских переменных, определяющих состояние системы (давление,
температура и др.), можно изменить произвольно, не вызывая
изменения числа фаз в системе, т. е не нарушая ее равновесия.
В системах, в которых компоненты находятся только в жид-
ком и твердом состоянии, изменение давления незначительно
сказывается на свойствах, поэтому давление можно считать по-
стоянным, и уравнение правила фаз принимает вид
С-Л'—Ф+1.
Согласно этому уравнению двухкомпонентная однофазная
-система имеет две степени свободы (состояние системы опреде-
ляется температурой и концентрацией одного из компонентов).
При наличии двух фаз (Ф = 2) двухкомпонентная система имеет
одну степень свободы. Это значит, что изменение температуры
вызывает изменение концентрации обеих фаз. При некоторой
406
температуре эти фазы могут слиться с образованием однофаз-
ного гомогенного раствора. Наоборот, однофазный гомогенный
раствор при определенной температуре может расслоиться или
разделиться на две фазы Температура, при которой происходит
расслоение, называется температурой фазового расслоения или
фазового разделения (ГфР). Раствор каждой концентрации име-
ет свою ТфР, зависимость которой от состава раствора выража-
ется кривой взаимного смешения, или пограничной кривой, от-
деляющей область однофазных растворов от двухфазных.
В зависимости от состава однофазных двухкомпонентных
жидких растворов при их охлаждении возможны два случая
разделения на составляющие компоненты: жидкостное и кри-
сталлическое. При жидкостном расслаивании одна жидкая фаза
расслаивается на две жидкие, при кристаллическом — из рас-
твора выделяется компонент в виде кристаллической фазы.
На рис 6.8 представлены типичные диаграммы фазового равно-
весия, отвечающие этим двум механизмам расслаивания. Тем-
пература Т\ (рис. 6.8, а, б) представляет собой температуру
фазового расслаивания раствора, имеющего состав, характеризуе-
мый абсциссами точек Д и В, т. е при этой температуре сосу-
ществуют две фазы состава А и В. Температура Т2 — это тем-
пература фазового расслаивания раствора состава, определяе-
мого положением точек А' и В'. Кривые, соединяющие точки А
и А', В и В', являются кривыми взаимного смешения, или по-
граничными кривыми. В условиях, соответствующих области
между кривыми АА' и осью ординат, а также между кривы-
ми В1¥ и осью ординат, раствор однофазен. В области, распо-
ложенной между кривыми АА' и ВВ', раствор расслаивается на
две фазы. Горизонтальные линии, пересекающие кривые вза-
имного смешения и соединяющие сосуществующие в равновесии
фазы, называются нодами. При повышении (рис. 6.8, а) или
Состав
Состав
Рис. 6.8. Диаграммы состояния жидкость — жидкость (а, б) и жидкость —
кристалл (в)
407
пи »меняй (рис. о.б,о) температуры длина иод уменьшается.
При какой-то температуре Т3 ноды вырождаются в точку, назы-
ваемую критической, и обе кривые (А А' и В В') сливаются
в одну АА'ВВ', называемую бинодалью. Если эта точка лежит
в вершине кривой взаимного смешения (рис. 6.8,а), она назы-
вается верхней критической температурой смешения (ВКТС)
или растворения (ВКТР), если в нижней части кривой
(рис. 6.8,6)—нижней критической температурой смешение
(НКТС) или растворения (IIKTP)
Для растворов двух низкомолекулярных компонентов бино
дали имеют форму, близкую к параболе, ее ветви никогда н-.
пересекают ось ординат и идут параллельно ей. В условиях
соответствующих областям вне бинодалей (т. е. слева, сверху
справа от бинодали), раствор сохраняет гомогенность; внутрен
нее пространство бинодали соответствует условиям, при которые
раствор расслаивается
Бинодаль совпадает с линиями взаимного смешения толью,
для двухкомпонентных растворов. Присутствие третьего компо
нента искажает форму бинодали, приводит к смещению ВКТР
Поскольку полимеры полндисперсны и в их растворах при
сутствуют макромолекулы различной степени дисперсности (что
делает их многокомпонентными), ВКТР и НКТР полимерных
растворов всегда сдвинуты в область малых концентраций, т. е
критическая концентрация растворов полимеров очень мала. Это
объясняется также огромной разницей в размерах макромоле-
кул полимера и молекул растворителя
При кристаллическом разделении системы на две фазы
(рис. 6.8, в) критические температуры растворения (смешения)
отсутствуют Температурам плавления чистых компонентов со-
ответствуют точки Т] и Т2- Растворимость компонентов харак-
теризуется линиями ликвидуса, например линия Т}Е характери-j
зует равновесие раствора (или расплава) с кристаллами ком-
понента /, т. е. растворимость компонента 1 в системе придан-
ной температуре. Растворимость компонента 2 характеризуется
линией Т^Е. Линии ликвидуса отличаются от бинодалей тем,
что они пересекают оси ординат при 100 %-м содержании компо-
нента.
Диаграмма фазового состояния, приведенная на рис. 6.8, в,
может быть построена не только для раствора, но и для распла-
ва Точка Е пересечения линий ликвидуса Т\Е и ТгЕ соответ-
ствует температуре и составу расплава, который одновременно
находится в равновесии с кристаллами компонентов 1 и 2. Она
называется эвтектической точкой. Расплав, соответствующий эв-
тектической точке, называется эвтектическим расплавом, темпе-
ратура эвтектической точки ТЕ— эвтектической температурой.
Соответствующая эвтектической температуре прямая АВ назы-
вается эвтектической прямой или линией солидуса.
408
При температуре выше линий ликвидуса, т. е. в области над
кривой Т\ЕТг, расплав однофазен. При температуре ниже эвтек-
тической происходит кристаллизация обоих компонентов Смесь
кристаллов этих компонентов, выпадающая при эвтектической
температуре, называется твердой эвтектикой и состоит из двух
твердых фаз. При температуре в интервалах между линиями
ликвидуса и линией солидуса расплав содержит кристаллы
соответствующих компонентов в области, ограниченной точка-
ми 1\ЕА, — кристаллы компонента /, в области, ограниченной
точками ТгЕВ, — кристаллы компонента 2.
В зависимости от строения полимера и растворителя разбав-
ленные растворы расслаиваются как при охлаждении, так и при
нагревании. При охлаждении, в частности, расслаиваются
с ВКТР растворы диацетата целлюлозы в хлороформе и тетра-
хлорэтане, полистирола в циклогексане и декалине, полиизо-
бутилена в бензоле. Многие растворы полимеров расслаивают-
ся при нагревании, т. е. обладают НКТР, которая может быть
двух типов Первый тип НКТР характерен для растворов, между
компонентами которых образуются водородные связи, например
нитрата целлюлозы в этиловом спирте. НКТР этих растворов
ниже температуры кипения растворителя. Второй тип НКТР
характерен для растворов, компоненты которых характеризуют-
ся большой разницей в термических коэффициентах расширения.
Вследствие этого при нагревании ассоциируют как молекулы
растворителя, так и макромолекулы полимера, что приводит
к расслаиванию системы. НКТР этих растворов выше темпера-
туры кипения растворителя. К ним относятся растворы полисти-
рола в циклогексане, бензоле и этилбензоле, ацетата целлюло-
зы в ацетоне и др. Некоторые растворы полимеров одновремен-
но обладают и ВКТР, и НКТР, например раствор полистирола
расслаивается и при охлаждении, и при нагревании:
ВКТР. к НКТР. к
Полистирол — циклогексан зоз 453
Полистирол — цнклопемтан 279 423
Полистирол — этилбензол 267 568
Полвизобутилен — бензол 296 433
Поливннилацетат — метанол 270 479
Поливиниловый спирт — вода — 504
Для таких систем могут быть два случая, схематически пред-
ставленные на рис. 6.9, а, б. В первом случае диаграмма состоя-
ния представляет собой замкнутый контур, и ВКТР по шкале
температур выше НКТР; во втором случае наблюдается НКТР
второго типа, которая значительно выше ВКТР Возможен тре-
тий случай (рис. 6 9,в), когда критической температуры вообще
не наблюдается. Во всех случаях область вне бинодалей отве-
чает неограниченному смешению, т. е. однофазной системе.
409
Рис. 6.9. Возможные случаи жидкостного расслаивания системы полимер —
растворитель
ВКТР и НКТР зависят от молекулярной массы полимера
(рис. 6 10). С увеличением молекулярной массы ВКТР возрас-
тает, а НКТР снижается, т. е. область гомогенного состояния
растворов сокращается; критическая концентрация и раствори-
мость полимеров с ростом молекулярной массы снижаются.
Устойчивость трехкомпонентных систем, например состоя-
щих из двух растворителей и полимера, зависит от их состава.
Так, нитрат целлюлозы (содержащий 10—12% азота) ограни-
ченно набухает в этиловом спирте и этиловом эфире, но в смеси
этих растворителей неограниченно растворяется, образуя гомо-
генный раствор. В некоторых случаях растворяющая способ-
ность растворителя улучшается при добавлении к нему незна-
чительного количества нерастворителя. При большом содержа-
нии нсрастворителя полимер выделяется из раствора.
С
Тип критической температуры
растворения (ВКТР или НКТР)
предопределяется характером из-
менения термодинамического срод-
ства растворителя к полимеру с из-
менением температуры. Расслаива-
ние системы во всех случаях про-
исходит вследствие ухудшения тер-
модинамического сродства между
компонентами, что характеризует-
ся снижением положительного зна-
чения второго вириального коэф-
фициента Л2 и отрицательного зна-
чения потенциала Д(7. Расслаива-
ние с ВКТР происходит при охлаж-
Рис. 6.10 Зависимость ВКТР и НКТР от
молекулярной массы полимера (Af«>Af2>
410
дении системы и вообще при эндотермических процессах и воз-
растании энтропии, расслаивание с НКТР — при нагревании,
при экзотермических процессах и уменьшении энтропии. Раство-
ры ограниченно растворимых полимеров могут расслаиваться на
две фазы как с ВКТР, так и с НКТР. При расслаивании в обе-
их фазах устанавливается определенная концентрация полиме-
ра, зависящая от температуры. Концентрация полимера в та-
кой системе остается при данной температуре постоянной и не
зависит от того, охлаждали или нагревали раствор. Таким об-
разом, растворы ограниченно растворимых полимеров подчиня-
ются правилу фаз и являются обратимыми.
6.1.5. Свойства растворов полимеров
Ряд свойств растворов полимеров в значительной степени за-
висит от их концентрации. В связи с этим различают разбав-
ленные н концентрированные растворы (реологические свойства
растворов см. гл. 5).
Разбавленными называют растворы, в которых макромолеку-
лы находятся друг от друга на расстояниях, превышающих их
собственные геометрические размеры, т. е. не взаимодействуют
между собой. Концентрация с разбавленных растворов обратно
пропорциональна молекулярной массе полимера, характеризуе-
мой характеристической вязкостью (см. с. 413):
Концентрированными называют растворы, в которых с>
>1/[Ч] и макромолекулы растворенного полимера взаимодей-
ствуют друг с другом. Это приводит к резкому возрастанию вяз-
кости. Часто к концентрированным относят растворы с относи-
тельной вязкостью (представляющей собой отношение вязкости
раствора к вязкости растворителя) более 100 Концентрация та-
ких растворов находится в пределах от долей процента для
длинных жестких цепей до 1% Для гибких полимеров низкой
молекулярной массы.
Результатом взаимодействия макромолекул в таких раство-
рах является образование лабильных ассоциатов, состав кото-
рых непрерывно изменяется. Средний период жизни ассоциатов
высокомолекулярных соединений значительно больше, чем пери-
од жизни ассоциатов низкомолекулярных жидкостей, так как
отрыв и присоединение сегментов макромолекул происходят го-
раздо медленнее, чем в случае молекул низкомолекулярных ве-
ществ. Размеры ассоциатов и продолжительность их жизни за-
висят от температуры, концентрации раствора, строения поли-
мера и растворителя. При повышении температуры увеличива-
ется сегментальная подвижность макромолекул, что способству-
ет распаду ассоциатов; повышение концентрации, снижение
температуры раствора приводят к увеличению размеров и про-
должительности существования ассоциатов
411
С
Рис. 6.11. Зависимость вязкости от кон-
центрации раствора натурального кау-
чука
В определенных условиях
(например, при высокой молеку-
лярной массе или концентрации,
низкой температуре, в отсутст-
вие перемешивания) возможно
взаимодействие между различ-
ными ассоциатами или соедине-
ние их между собой макромоле-
кулами («проходными»), входя-
щими одновременно в состав
разных ассоциатов. В этом слу-
чае в концентрированном растворе образуется пространствен-
ная сетка из взаимосвязанных ассоциатов. Например, концен-
трированные растворы ацетатов целлюлозы представляют со-
бой трехмерную сетку, в ячейках которой находится раствори-
тель Тип растворителя определяет структуру и густоту сетки.
Появление пространственной сетки в концентрированных рас-
творах приводит к аномалии их вязкости и другим специфиче-
ским свойствам, превращению жидких растворов в твердое со-
стояние— студни, или гели (см. с 417). Таким образом, ассо-
циаты можно рассматривать как зародыши новой фазы.
Важнейшей отличительной особенностью растворов полиме-
ров является их аномально высокая вязкость и быстрый ее рост
с увеличением концентрации (рнс. 6.11). Коэффициент вязко-
сти т] растворов может быть определен в капиллярном виско-
зиметре и рассчитан по уравнению Пуазейля:
лДР/?1
811/ Х‘
(6.13)
где R и L— радиус и длина капилляра; ДР — разность давлений на концах
капилляра; V — объем шарика с раствором; т — время истечения раствора.
Обычно на практике пользуются не абсолютной вязкостью,
а так называемой удельной, которую рассчитывают по формуле
Пуд =
Не—Но
По
(6-14>
где ijc — вязкость раствора определенной концентрации; Г) — вязкость рас-
творителя.
Отношение удельной вязкости к концентрации называют
приведенной вязкостью г]уд/с, т. е. вязкостью, отнесенной к еди-
нице концентрации. Определяя удельную вязкость растворов
412
различной концентрации, можно построить графическую зави-
симость 1]Уд/с от концентрации и экстраполяцией прямой к оси
ординат найти значение характеристической вязкости [т|], т. е.
вязкости раствора при бесконечном разбавлении, когда макро-
молекулы можно рассматривать как обособленные, изолирован-
ные друг от друга (рис 6 12):
[ц]= lira (||уд/с). (6.15)
с—О
Между характеристической вязкостью и молекулярной мас-^
сой существует зависимость, выражаемая уравнением Марка —
Хаувинка — Куна:
Р]]=Ж (6.16>
|де К — постоянная, зависящая от температуры и природы полимера и рас-
творителя; а — показатель, характеризующий конформацию макромолекул
в растворе.
Вязкость растворов при повышении температуры снижается, 2
причем особенно интенсивно у более концентрированных рас-
творов (рис. 6.13). Вязкость растворов полимеров зависит от
состава раствора, присутствия посторонних веществ, характера
взаимодействия растворителя с полимером. Чем лучше полимер
растворяется в жидкости, тем больше его уровень сольватации.
Вследствие этого снижаются межмолекулярное взаимодействие
.между макромолекулами, затрудняется их свертывание в ком-
Рис. 612 Зависимость приведенной вязкости т|уд/с разбавленного раствора
полимера от концентрации с
Рис. 6.13. Влияние температуры на вязкость концентрированных растворов
натурального каучука
1 — 10%-й. 2 - 20%-й; 3 - 30% й
413
пактные плотные клубки, что приводит к повышению вязкости
растворов В «плохих» растворителях, слабо взаимодействую-
щих с полимером, сольватация выражена слабо. В этих услови-
ях макромолекулы без затруднений образуют плотные клубки
небольших размеров, что обусловливает снижение вязкости рас-
творов. Поэтому добавление к раствору полимера нераствори-
теля приводит к снижению его вязкости; это широко использу-
ется иа практике.
Растворы полимеров способны рассеивать свет, что обуслов-
лено непрерывным изменением концентрации в микрообъемах
системы вследствие образования и распада ассоциатов. Изме-
нения концентрации приводят к отклонению показателя прелом-
ления от его среднего значения. Пр величине светорассеяния
разбавленных растворов полимеров, макромолекулы которых
свернуты в клубки диаметром меньше 0,1 длины волны рассеян-
ного света, можно определить молекулярную массу полимера
(см. гл. 1).
Растворы полимеров способны также избирательно погло-
щать световые лучи По ультрафиолетовым и инфракрасным
спектрам поглощения судят о присутствии в полимерах сопря-
женных двойных связей, определенных атомных групп, что по-
могает установить строение макромолекул.
6.2. КОЛЛОИДНЫЕ СИСТЕМЫ
Наряду с растворами полимеров широкое применение находят
и различные полимерные гетерогенные коллоидные системы, ха-
рактеризующиеся коллоидной степенью дисперсности Это озна-
чает, что частицы в таких системах представляют собой не от-
дельные макромолекулы, как в растворах, а их агрегаты Эти
агрегаты нерастворимы в жидкой среде, называемой дисперси-
онной средой, и образуют в ней отдельную дисперсную фазу.
Состав и свойства коллоидных систем существенно отличаются
от состава и свойств истинных растворов.
Специфической особенностью дисперсных систем является
их агрегативная неустойчивость, т. е. способность к разруше-
нию, разделению на отдельные фазы — дисперсионную среду
и дисперсную фазу. Разрушение коллоидных систем легко про-
исходит при введении в них электролитов, а также при изме-
нении температуры и других факторов. При разрушении систе-
мы отдельные частицы дисперсной фазы соединяются друге дру-
гом, что приводит к снижению степени их дисперсности, отде-
лению дисперсной фазы от дисперсионной среды Процесс раз-
рушения коллоидной системы с выделением из дисперсионной
среды дисперсной фазы называют коагуляцией, а выделившую-
ся дисперсную фазу — коагулятом (коагулюмом).
414
Неустойчивость коллоидных систем объясняется большой,
всегда положительной свободной поверхностной энергией, со-
средоточенной на межфазной поверхности раздела. Поверхност-
ная энергия Gs представляет собой произведение поверхностно-
го натяжения о на площадь поверхности раздела фаз S. В со-
ответствии с законами термодинамики такие системы неравно-
весны и стремятся перейти в состояние, соответствующее мини-
мальной свободной энергии, т. е. разделиться на отдельные фазы
с минимальной поверхностью раздела.
В реальных условиях устойчивость коллоидных систем игра-
ет громадную роль. Она зависит от сроков и условий их транс-
портирования, хранения, переработки. Изменения структуры
коллоидных систем, приводящие к их разрушению, в различных
условиях различны и зависят от соотношения и природы сил,
действующих между диспергированными частицами. Это могут
быть силы сцепления и силы отталкивания. Силы сцепления
обычно проявляются при наличии межмолекулярного взаимо-
действия. Они сильно возрастают при сближении частиц, вы-
зывая их слияние, коагуляцию. Поэтому устойчивость коллоид-
ных систем резко снижается при увеличении концентрации. От-
талкивание частиц друг от друга происходит по нескольким
причинам. Большое значение имеет электростатическое отталки-
вание частиц, имеющих одинаковый электрический заряд. Сбли-
жению частиц препятствует также образование на поверхности
раздела сольватных оболочек, состоящих из молекул дисперси-
онной среды, поверхностно-активных веществ, играющих роль
эмульгаторов, стабилизаторов, часто специально вводимых
в коллоидные системы, и т п.- Подбором рецептуры, способов
приготовления, хранения и переработки коллоидных полимерных
систем добиваются значительного повышения их устойчивости.
Коллоидные системы образуются в результате перехода од=
нофазного раствора в двухфазную систему при снижении тем-
пературы, введении нерастворителя или синтезе полимера в сре-
де нерастворителя, а также за счет диспергирования полимера
в низкомолекулярном компоненте или низкомолекулярного ком-
понента в полимере. Во всех этих случаях образуются гетеро-
генные системы с высокоразвитой поверхностью раздела фаз.
6.2.1. Дисперсии и эмульсии
По строению и степени дисперсности гетерогенные коллоидные
полимерные системы подразделяют на диспер'*уи и эмульсии.
Дисперсии и эмульсии — устойчивые коллоидные системы с раз-
мерами частиц 0,1 мкм—1 нм; дисперсная фаза в дисперси-
ях— твердая, в эмульсиях — жидкая. В состав этих систем вхо-
дят три компонента — дисперсная фаза, дисперсионная сре-
415.
да, эмульгатор. Молекулы эмульгатора имеют полярные и нспо;
лирные участки, взаимодействующие с разными фазами.
Агрегативная устойчивость эмульсий обусловлена многими
факторами. В определенных условиях они могут самопроизволь-
но образовываться в двухкомнонентной гетерогенной системе
(без эмульгатора) Например, гетерогенная система вода — фе-
нол самопроизвольно переходит в термодинамически устойчивую
эмульсию, если межфазное натяжение настолько мало, что оно
полностью компенсируется энтропийным фактором. Таким же
свойством обладают коллоидные системы поверхностно-актив-
ных веществ (ПАВ) и растворы полимеров. Добавление поверх-
ностно-активных веществ вызывает сильное снижение поверхно-
стного натяжения в системе, что способствует образованию тер-
модинамически устойчивых (самопроизвольно образующихся)
в обычных условиях эмульсий. Такие эмульсии характеризуют-
ся очень высокой дисперсностью.
Однако большинство эмульсий — это микрогетерогенные
термодинамически неустойчивые системы, разрушающиеся при
хранении, изменении температуры и т. д. Для стабилизации та-
ких эмульсин применяют эмульгаторы. Они не только повыша-
ют агрегативную устойчивость дисперсий и эмульсий, но и изме-
няют их электрические свойства, уменьшают работу образова-
ния новых поверхностей, т. е. облегчают диспергирование. В слу-
чае сильного снижения межфазного натяжения дисперсная фаза
может самопроизвольно диспергироваться с образованием мик-
рочастиц размером 60—100 нм даже под действием теплового
движения. Скорость и степень дробления дисперсной фазы оп-
ределяются площадью поверхности раздела между фазами,
свойствами эмульгаторов. Например, иногда неионогенные
эмульгаторы при эмульсионной полимеризации обеспечивают
более высокую степень диспергирования мономера, чем ионоген-
ные эмульгаторы.
В отличие от растворов полимеров коллоидные системы об-
ладают низкой вязкостью даже при высокой концентрации, лег-
ко разрушаются при замораживании или действии электроли-
тов, обладают незначительным осмотическим давлением.
6.2.2. Студни
Студнями (или гелями) называют двухкомпонентные системы
полимер — растворитель, представляющие собой пространствен-
ные сетчатые структуры, образованные из сольватированных
макромолекул и их агрегатов, в которых распределены моле-
кулы растворителя. Такие сетки при достаточно высокой кон-
центрации могут образовываться и в растворе. Переход в об-
416
расть ограниченной совместимости приводит к застудневанию
раствора и образованию гетерогенной двухфазной системы
с коллоидными размерами частиц одной из фаз. Основное отли-
чие студней от концентрированных растворов заключается
р том, что в растворах сетки непрерывно разрушаются и обра-
зуются под влиянием теплового движения, т. е. имеют флуктуа-
ционный характер, а в студнях сетки в данных условиях устой-
чивы и не разрушаются под действием теплового движения.
Однако в некоторых студнях при повышении температуры про-
исходит разрушение поперечных связей и студень превращается
в жидкость. Этот процесс называется плавлением студня
Существуют два типа студней. Студни первого типа — это
системы, в которых пространственная сетка образована хими-
ческими связями (набухшие резины, некоторые ионообменные
смолы и др ), студни второго типа — системы, в которых про-
странственная сетка образована связями различной природы,
в частности растворы белков, производных целлюлозы и др. При
нагревании студни первого типа не плавятся, второго — плавят-
ся Поэтому студни первого типа называют термонеобратимыми,
второго — термообратимыми.
Студни первого типа образуются при самопроизвольном на-
бухании пространственно-сшитых полимеров, при трехмерной
полимеризации или поликонденсации в растворе, в процессе хи-
мических реакций сшивания в присутствии растворителей. Мо-
мент, когда растворы теряют текучесть и превращаю!ся в сту-
день, называется точкой гелеобразования (желатинирования,
желирования, гель-точкой).
Свойства студней первого типа зависят от строения поли-
мера и растворителя, концентрации поперечных связей, степени
набухания. Прочность, долговечность, другие физические свой-
ства набухающих систем (например, вулканизатов) достигают
минимальных значений задолго до достижения предела набуха-
ния (см. рис. 6.4). Студни первого типа — устойчивые гомоген-
ные системы, они не имеют критических температур растворе-
ния, их строение не зависит от температуры вплоть до гермо-
распада.
При охлаждении студней иногда происходит выделение ча-
сти растворителя вследствие снижения равновесной степени на-
бухания. Процесс отделения растворителя от студня называется
синерезисом.
Строение и устойчивость студней второго типа зависят от
температуры; они могут иметь как ВКТР, так и НКТР При
изменении температуры (или во времени) происходит разделе-
ние фаз (синерезис). В конечном итоге такие неравновесные си-
стемы должны расслоиться на две фазы — набухший полимер
и растворитель или очень разбавленный раствор полимера. Та-
ким образом, студни второго типа являются системами с неза-
27—816
417
вершенным расслаиванием Некоторые характеристики студней’ обоих типов приведены ниже: /
I н
Характер системы Гомогенная Гетерогенная
Природа обратимой деформации Энтропийная Энергетическая
Скорость релаксации Высокая Низкая
Термическая обратимость Необратимы Обратимы
Обратимость синерезиса Обратим Необратим
Причины синерезиса Изменение условий Самопроизволь-
равновесия ное разделение фаз
6.2.3. Смеси полимеров с пластификаторами
Пластификаторы — это твердые или жидкие вещества, введение
которых в полимеры повышает их пластичность, снижает вяз-
кость до требуемого уровня, обеспечивающего снижение темпе-
ратуры переработки, облегчение и улучшение диспергирования
в полимерных композициях сыпучих (особенно ком .ующихся)
компонентов, высококачественное формование при заданных
технологических параметрах, минимальную усадку изделий при
хранении и др.
Чаще всего пластификатор вводят в полимер в жидком со-
стоянии. При проникновении жидкого пластификатора в фазу
полимера может происходить молекулярное или коллоидное его
диспергирование. Если пластификатор имеет сродство к поли-
меру, то происходит молекулярное диспергирование, т. е. само-
произвольно образуется истинный раствор пластификатора в по-
лимере— полимер набухает в пластификаторе. Если пластифи-
катор не имеет сродства к полимеру, он самопроизвольно в него
не проникает, т. е. набухания не происходит. В этом случае
пластифицированный полимер является коллоидной системой,
в которой дисперсионной средой служит полимер, а дисперсной
фазой — пластификатор. Образующаяся эмульсия является тер-
модинамически и агрегативно неустойчивой, и поэтому может
расслаиваться. Вследствие высокой вязкости системы расслаи-
вание происходит медленно, иногда при хранении или эксплуа-
тации изделия Это проявляется, например, в выделении капель
пластификатора на поверхности изделия.
При образовании истинного раствора пластификатора в по-
лимере говорят об их совместимости. Поскольку пластификатор
вводят в количестве 30% от массы полимера, то нет необходи-
мости в неограниченном смешении пластификатора с полиме-
ром: они могут смешиваться и ограниченно, т. е. полимер может
только набухать в пластификаторе, но не растворяться.
Механизм пластификации зависит от характера надмолеку-
лярной структуры полимера, химического строения полимера
и пластификатора, их термодинамической совместимости, уело-
418
вий процесса. В зависимости от термодинамической совместимо-
сти полимера и пластификатора различают внутриструктурную
и межструктурную пластификацию.
Внутриструктурная пластификация происходит при высоком
термодинамическом сродстве пластификатора к полимеру, силь-
ном межмолекулярном взаимодействии молекул пластификато-
ра с макромолекулами полимера, набухании и растворении по-
лимера в пластификаторе, когда молекулы пластификатора про-
никают внутрь любых надмолекулярных структур, постепенно
их разрушая.
Ьсли пластификатор является «плохим» растворителем поли-
мера и смешивается с ним в очень небольших количествах, то
его молекулы проникают только в .межструктурные пространст-
ва, и пластификация называется межструктурной. При меж-
структурной пластификации истинно растворяется (или совме-
щается) с полимером ничтожное количество пластификатора,
молекулы которого адсорбируются на межструктурной поверх-
ности раздела, образуя тончайшие мономолскулярные слои так
называемой граничной смазки, облегчающей подвижность над-
молекулярных структур
Количественной оценкой действия пластификаторов является
снижение температуры стеклования пластифицированного поли-
мера по сравнению с непластифицированным (ДТС). Особенно
это характерно для полимеров с жесткими цепями, где ДТС мо-
жет достигать 100—160 К. Значительно менее эффективно плас-
тификатор действует на полимеры с гибкими молекулами: тем-
пература стеклования полярных каучуков может быть снижена
на 30—40 К, а неполярных — всего на 10—20 К. Чем больше
ДТС, тем эффективнее пластификатор.
При межструктурной пластификации наблюдается значи-
тельное снижение температуры стеклования при введении не-
больших количеств пластификатора. Для внутриструктурнсй
пластификации характерно непрерывное снижение Тс с увели-
‘ чением содержания пластификатора. Это означает, что в при-
сутствии пластификаторов повышается морозостойкость и мате-
риал сохраняет высокоэластическис свойства при более низких
температурах, чем непластифицированный полимер.
Введение пластификатора вызывает также снижение темпе-
ратуры текучести, поскольку вязкость полимера при введении
в него менее вязкого компонента, как правило, снижается, и,
следовательно, система способна течь при более низкой темпе-
ратуре.
Изменение Тс и Тт зависит от концентрации пластификатора.
При небольшом его содержании Тс снижается более резко, чем
Тт, и разность Т,—Тс возрастает; при дальнейшем увеличении
содержания пластификатора более резко снижается Тт, поэтому
27*
419
разность Гт—Гс уменьшается, т е. сокращается область высоко-
эластического состояния.
Изменение прочности и других механических свойств плас-
тифицированных полимеров зависит от механизма пластифика-
ции. При внутриструктуриой пластификации по мере увеличения
содержания пластификатора прочность и модуль упругости по-
лимера уменьшаются, а эластичность возрастает. При межструк-
турной пластификации в области малых содержаний пластифи-
катора наблюдается некоторое повышение прочности и модуля,
которые при дальнейшем добавлении пластификатора уменьша-
ются. Повышение прочности при введении пластификаторов
объясняется увеличением подвижности надмолекулярных струк-
тур, которые при растяжении ориентируются, что всегда способ-
ствует упрочнению полимера.
При введении в жесткоцепные полярные полимеры, находя-
щиеся в стеклообразном состоянии (например, в поликарбона-
ты), небольших количеств некоторых хорошо совмещающихся
с ними веществ иногда наблюдается возрастание модуля упру-
гости при снижении удлинения при разрыве и ударной вязко-
сти Такое явление называется ант и пластификацией. Наиболее
эффективные антипластификаторы — совместимые с полимера-
ми вещества, содержащие хлор, азот, кислород и другие поляр-
ные атомы и имеющие высокую температуру стеклования
Введение пластификаторов вызывает снижение долговечно-
сти полимерных материалов, при этом энергия активации раз-
рыва не изменяется, а увеличивается структурно-чувствитель-
ный коэффициент у. Это связано с проникновением молекул
пластификатора между макромолекулами или надмолекуляр-
ными структурами, что сопровождается изменением только меж-
молекулярного взаимодействия
Пластификаторы влияют и на диэлектрические свойства по-
лимеров Как правило, введение пластификаторов ухудшает
диэлектрические характеристики. Изменение диэлектрической
проницаемости и максимума тангенса угла диэлектрических
потерь tg 6 зависит от полярности пластификатора и его термо-
динамической совместимости с полимером. Если пластифика-
тор истинно растворим в полимере, то tg6MaKc смещается в об-
ласть более низких температур. При этом абсолютные значения
tg 6 и диэлектрической проницаемости е' зависят от полярности
пластификатора, т. е. от его собственной диэлектрической про-
ницаемости. При введении неполярных пластификаторов, ди-
электрическая проницаемость которых мала, е,' и tg6MaKc плас-
тифицированного полимера уменьшаются, а введение полярных
пластификаторов может привести к возрастанию этих пока-
зателей.
Помимо специфических требований к пластификаторам, оп-
ределяемым назначением изделия и условиями его эксплуата-
420
цин, все пластификаторы должны удовлетворять ряду общих
требований: хорошо совмещаться с полимером; иметь высокую
температуру кипения (в случае жидких пластификаторов), что-
бы не испаряться в процессе переработки или эксплуатации из
„телия. и невысокую температуру размягчения (для твердых),
чтобы легко расплавляться или размягчаться в процессе пере-
работки; быть нетоксичными; не вызывать коррозии оборудова-
ния и не вступать в химические реакции с компонентами поли-
мерной композиции при хранении и эксплуатации и т. д.
К числу важнейших пластификаторов для пластмасс отно-
сятся: эфиры ароматических кислот, например фталаты; эфиры
алифатических кислот, например адипинаты, себацинаты, стеа-
раты; эфиры гликолей и монокарбоновых кислот, например эфи-
ры триэтиленгликоля и алифатических монокарбоновых кислот
Сб—С$; эфиры фосфорной кислоты, например трикрезилфосфаты
(эти пластификаторы придают изделиям негорючесть); полиэфи-
ры дикарбоновых кислот и полиолов; эпоксидированные расти-
тельные масла, например соевое, и др.
В качестве пластификаторов могут применяться также по-
лимеризационноспособные олигомеры, способные вступать в по-
лиреакции Их применение особенно эффективно, так как при
смешении с полимером они повышают его пластичность, мяг-
кость, улучшают другие технологические свойства; при этом по-
является возможность уменьшить содержание других пласти-
фикаторов, снижающих прочность и другие показатели полимера
Д1ли вообще их нс применять). К таким полимеризационно-
способным олигомерам относятся некоторые фснолоформальде-
гидные смолы, совмещающиеся с резиновыми смесями, олиго-
эфиракрилаты — с поливинилхлоридом и др. В процессе струк-
турирования полимср-олигомерной системы в зависимости от
условий процесса олигомеры одновременно могут вступать
в разнообразные реакции, например сополимеризации с полиме-
ром (играя роль сшивающего агента), гомополимеризации с об-
разованием микровключений гомополимера (играющего роль
наполнителя) или образовывать пространственную сетку, рас-
пределенную в пространственной сетке полимера.
Пластифицирующая способность олигомеров зависит от их
строения и молекулярной массы. Скорость реакций олигомеров
в присутствии полимерной матрицы обычно снижается. Изменяя
состав и концентрацию олигомеров в полимерном материале,
достигают существенного изменения их свойств. Например, вве-
дение в олигомеры ароматических ядер приводит к значитель-
ному повышению теплостойкости материалов, увеличение длины
и гибкости цепи олигомеров — к снижению температуры стекло-
вания и модуля упругости, повышению адгезии, твердости,
•сопротивления динамическим нагрузкам, бензостойкости.
Свойства пластифицированного материала зависят от спосо-
421
ба введения пластификатора в полимер, определяемого методом
производства, конструкцией оборудования, видом сырья. Наибо-
лее часто применяются следующие способы пластификации по-
лимерных смесей:
растворение полимера в пластификаторе (производство по-
лимерных пленок, искусственной кожи, лакокрасочных покрытий
и др );
сорбция пластификатора полимером из эмульсий или рас-
творов пластификатора (производство производных целлюлозы,
поливинилхлорида и др.);
добавление пластификаторов к мономерам перед их полиме-
ризацией или поликонденсацией (производство фенолоформаль-
дегидных, карбамидоформальдегидных полимеров, полиэфиров
и др.);
введение пластификатора в эмульсию полимера;
смешение пластификатора с полимером.
6.2.4. Смеси полимеров
/Многие изделия изготавливают с применением не одного, а не-
скольких полимеров, их смеси. Свойства таких изделий зависят
от состава смеси, свойств ее компонентов, режимов смешения
и последующих технологических процессов, от совместимости
полимеров Совместимостью называют способность различных
полимеров образовывать в определенных условиях однородные
смеси с хорошими механическими свойствами Известны одно-
фазные и двухфазные смеси полимеров, существенно различаю-
щиеся структурой и свойствами
Однофазные смеси представляют собой твердые растворы
взаиморастворимых полимеров, обладающих хорошим термоди-
намическим сродством друг к другу в условиях смешения, спо-
собностью к взаимному самопроизвольному растворению с об-
разованием гомогенных систем. Взаиморастворимые полимеры,
образующие однофазные гомогенные твердые растворы, назы-
ваются термодинамически совместимыми В однофазных смесях
двух полимеров наблюдается появление одной температуры
стеклования вместо двух, присущих каждому исходному поли-
меру. Оценить совместимость полимеров можно по параметру
§ = (6i—бг)2; полимеры совместимы при §<0,0697 Дж/см3.
Совместимость полимеров может быть оценена по поведению
растворов смесей. Разбавленные растворы смесей полимеров,
как правило, не расслаиваются. Следовательно, в разбавлен-
ных растворах все полимеры образуют однофазные смеси. При
больших концентрациях подобные растворы расслаиваются, при-
чем концентрация раствора, при которой происходит расслаи-
вание, определяется природой растворителя и полимера.
422
Образование однофазных систем из двух полимеров сопро-
вождается уменьшением или свободной энергии, или изобарно-
изотермического потенциала
Дбем = ДНсм — ГД5см. (6.17)
Поскольку у полимеров число молекул в единице объема
мало, изменение энтропийного члена Д$см незначительно
и уменьшение изобарно-изотермического потенциала определя-
ется только величиной Д#См-
Согласно теории Скотта термодинамический потенциал сме-
шения для полимеров с высокой молекулярной массой опреде-
ляется по формуле
ДбсМ=/?7'ф1ф2х12, (6.18)
где <fi и <рз — объемные доли полимеров в смеси; Хи— параметр взаимо-
действия полимеров.
Из этого уравнения следует, что чем больше xi2. тем полиме-
ры более совместимы. Параметр взаимодействия в критической
точке (в вершине бинодали, см. рис. 6.8) можно рассчитать по
формуле
Х12.«₽=1/2(1/х1’а+1/х2'/’) (6.19)
(xt и Х2 — степени полимеризации полимеров). Из уравнения
видно, что чем больше молекулярная масса, тем ниже х>2.кр
и тем больше вероятность расслаивания полимеров. Повышение
температуры несколько увеличивает растворимость полимеров
друг в друге. Однако неограниченная взаимная растворимость
полимеров наблюдается крайне редко. В определенных услови-
ях она имеет место при совмещении поливинилхлорида и бута-
диен-нитрильного каучука СКН-40, поливинилацетата и нитра-
та целлюлозы.
Большинство полимеров растворяются друг в друге в коли-
честве долей процентов, а критическая температура смешения
очень высока. Поэтому при температуре эксплуатации и пере-
работки мы имеем дело с двухфазной коллоидной системой
Однако олигомеры с молекулярной массой, близкой к молеку-
лярной массе статистического сегмента макромолекулы, как
правило, характеризуются неограниченной взаимной раствори-
мостью Поэтому многие полимеры, не способные к полному
взаиморастворению с образованием гомогенного термодинами-
чески устойчивого раствора, при смешении проявляют способ-
ность к сегментальной растворимости на поверхности их кон-
такта. Сегментальная растворимость заключается в образова-
нии промежуточного переходного слоя между различными поли-
мерами вследствие взаимной диффузии наиболее подвижных
участков их макромолекул (например, концевых сегментов, бо-
ковых ответвлений и др.). Сегментальная растворимость харак-
423
терна для большинства несовместимых полимеров. Толщина
переходного слоя может составлять десятки нм и зависит от раз-
ности энергий когезии: чем последняя меньше, тем толщина слоя
больше. /Межфазный слой, в котором проявляется сегменталь-
ная растворимость, представляет собой «раствор» сегментов
одного полимера в другом Образование переходного слоя при-
водит к уменьшению энергии когезии смеси и повышению ее
устойчивости.
Наряду со слоем сегментальной растворимости полимеры
могут образовывать протяженные граничные слои с измененны-
ми конформацией макромолекул и надмолекулярными структу-
рами, которые могут включать часть сегментов более удален-
ных макромолекул другой фазы.
Кристаллические полимеры совместимы, если они изоморф-
ны и способны кристаллизоваться в одной кристаллической ре-
шетке; при этом их структурные параметры не должны разли-
чаться более чем на 0,02—0,03 нм.
Вследствие ничтожной взаиморастворимости большинство
полимеров при совмещении образуют гетерофазные системы.
Число фаз в этих системах определяется числом полимерных
компонентов. Наиболее часто используют двух- или трехфазные
смеси. К ним относятся смеси цис- и трпнс-полинзопренов, поли-
метилакрилата и полиэтилакрилата, линейного и разветвленно-
го полиэтиленов и др.
В зависимости от соотношения полимеров в смеси и режимов
ее приготовления каждый полимер может быть как дисперсной
фазой, так и дисперсионной средой. Как и во всякой коллоид-
ной системе, при изменении соотношения компонентов происхо-
дит обращение фаз. При добавлении небольшого количества
одного полимера к другому образуется дисперсия его в матрице
второго полимера. При увеличении концентрации дисперсной
фазы до 30—70% она образует матрицу (дисперсионную среду),
а исходный компонент становится непрерывной фазой. Обраще-
ние фаз в смесях полимеров возможно при изменении не только
соотношения компонентов, но и условий перемешивания. При-
рода непрерывной фазы определяет комплекс физико-механиче-
ских и химических свойств смесей
Свойства двух- и многофазных полимерных смесей опреде-
ляются не только их составом, но и способом изготовления, ре-
жимом переработки, физическим состоянием полимеров в мо-
мент смешения. Наивысшая степень диспергирования одних по-
лимеров в других достигается при их смешении в виде латексов,
дисперсий с последующей коагуляцией или при смешении рас-
плавов одинаковой вязкости. В случае аморфных полимеров
степень дисперсности тем больше, чем ближе химическое строе-
ние и ниже молекулярная масса смешиваемых полимеров.
Вследствие гигантских размеров макромолекул и надмолеку-
424
лярных структур и высокой вязкости подобные системы крайне
медленно расслаиваются, их структура и свойства практически
нс изменяются в процессе эксплуатации изделий. Стабильность
многофазных полимерных систем еще более возрастает в случае
сегментальной растворимости и образования переходных слоев
на поверхности раздела фаз.
Двух- и многофазные полимерные смеси характеризуются
высокими показателями механических свойств Например, дол-
говечность многих смесей при многократной деформации в де-
сятки раз выше долговечности исходных полимеров, модуль
упругости резин резко возрастает при введении в них 20—30%
пластмасс; введение 5—10% каучука в пластмассы значительно
улучшает их механические свойства (ударопрочный полисти-
рол). Основную роль в улучшении механических свойств подоб-
ных смесей играет межфазный переходной слой, препятствую-
щий концентрации напряжений в какой-либо одной точке систе-
мы, т. е. способствующий распределению напряжения по всему
объему.
6.2.5. Наполненные полимеры
Для того чтобы придать полимерным материалам необходимые
технологические и эксплуатационные свойства, в них часто вво-
дят наполнители. Они равномерно распределяются в объема
полимерной матрицы и образуется система, в которой полимер
является дисперсионной средой, а частицы наполнителя — дис-
персной фазой.
Благодаря высокоразвитой поверхности дисперсной фазы
в ней протекают все специфические физико-химические процес
сы, характерные для коллоидных систем. Особенно большое
значение наполнение имеет при получении резин на основе не-
кристаллизующихся каучуков, характеризующихся низким
• уровнем межмолекулярного взаимодействия, а также компози-
ций на основе термореактивных полимеров, при отверждении
которых наблюдается значительная усадка.
Свойства наполненных полимерных композиций во многом
определяются степенью дисперсности наполнителя, взаимодей-
ствием на поверхности раздела фаз и т. д. Как правило, чем
выше степень дисперсности, сильнее межмолекулярное взаимо-
действие на поверхности контакта, тем эффективнее воздействие
наполнителя на свойства полимера. В качестве наполнителей
в зависимости от назначения готовых изделий применяют газо-
образные, жидкие, твердые порошкообразные и волокнистые ве-
щества.
Газообразные наполнители используют в производстве лег-
ких пористых изделий с изолированными или сообщающимися
425
порами: пенопластов, пористых ионообменных смол, резиновых
губок и т. п. Жидкие наполнители применяют при получении
твердых пористых материалов из жидких эмульсий.
Наиболее часто применяют твердые порошкообразные и во-
локнистые наполнители, обеспечивающие повышение прочности,
модуля, температур плавления, необходимую электропровод-
ность, снижение степени набухания и растворимости. Кроме
того, наполнители снижают расход полимеров и себестоимость
изделий. Применение волокнистых наполнителей позволяет из-
менять свойства изделий в заданном направлении. Такие изде-
лия называются армированными, t / ( .с.
Наибольшее значение в промышленности имеют органиче-
ские волокнистые и порошкообразные наполнители (древесная
мука, целлюлоза, натуральные и синтетические волокна), угле-
родные (графит, кокс, технический углерод), металлы и их ок-
сиды, силикаты и т. д.
По эффективности воздействия на свойства полимера, в част-
ности на его прочность, наполнители условно подразделяют на
активные (упрочняющие, усиливающие) и неактивные (инерт-
ные) Например, активными для резин являются некоторые
виды технического углерода, инертными — мел, каолин. Напол-
нитель тем активнее, чем больше энергия адгезии полимера
к наполнителю превышает энергию когезии полимера. Этот вы-
вод основан на том, что при условии нарушения адгезионного
контакта (т. е. при разделении фаз) исчезает поверхность раз-
дела между полимером и наполнителем с образованием равных
по площади поверхностей обеих фаз. Математически это можно
следующим рбр^вом:
представить
(6.20)
где Е< и Е» — свободные поверхностные энергии полимера и наполнителя;
Eis — свободная межфазная энергия.
Если (по аналогии с жидкостями) принять, что Ек— свобод-
ная энергия когезии полимера равна 2ЕЬ то
E.-EK=E2-Et-E,2. (6.21)
Упрочнение будет наблюдаться в том случае, если (F12+
+ £(—£г)<0. Поскольку £2 большинства наполнителей выше,
чем £1, введение наполнителей приводит, как правило, к упроч-
нению полимера. Поэтому наиболее эффективно применение на-
полнителей в полимерах с гибкими макромолекулами, т. е.
с низким уровнем межмолекулярного взаимодействия.
На поверхности активного наполнителя (например, техниче-
ского углерода) имеется большое число активных центров:
ребра и углы кристаллитов, свободные (неспаренные) электро-
426
-
ны, частично связанные с активными центрами, карбоксильные
и лактонные группы. Различная природа активных центров обу-
словливает широкий спектр взаимодействий между наполните-
лем и полимером- от чисто физических (ван-дер-ваальсовых) до
водородной и химических связей. Последние образуются в ре-
зультате реакции активных центров на поверхности наполните-
ля и свободных макрорадикалов, возникающих при механиче-
ской деструкции полимера в процессе переработки.
Обоснованной, общепризнанной теории усиления полимеров
наполнителями, позволяющей математически описать изменение
свойств любого полимера с изменением степени наполнения лю
бым наполнителем, пока не разработано. К одной из основных
причин усиливающего действия наполнителя относится ориенти-
рующее влияние активных центров на поверхности его частиц
на макромолекулы, приводящее к ограничению их подвижностч
и образованию в силовом поле вокруг частиц топких адсорбци-
онных слоев из упорядоченных надмолекулярных структур
Вследствие этого наполнение полимеров сопровождается умень-
шением числа возможных конформаций макромолекул в этих
слоях, возрастанием средних времен релаксации, расширением
релаксационных спектров, повышением температур стеклования,
плотности упаковки молекул, изменением условий кристаллиза-
ции (для кристаллизующихся полимеров)
Введение наполнителей приводит к изменению типа и разме-
ров кристаллитов. При малых концентрациях наполнитель яв-
ляется искусственным зародышем кристаллизации (см. гл 5),
при больших — степень кристалличности уменьшается.
Усилению полимеров способствует также возникновение тон-
кой прослойки полимера между частицами наполнителя. Обра-
зование такой прослойки способствует возникновению мелкокри-
сталлической структуры и уменьшает вероятность образования
дефектов структуры, являющихся очагами разрушения.
Изменения свойств полимеров при их адсорбции на поверх-
ности наполнителя столь значительны, что наполненный поли-
мер можно рассматривать как трехкомпонентную систему: на-
полненный полимер, адсорбированный полимер, образующий
граничный слой с измененными свойствами иа поверхности на
полнителя, и наполнитель
В наполненной системе образовавшиеся связи полимер—на-
полнитель при деформации разрушаются и вновь восстанавли-
ваются в новом положении, в результате чего происходит вы-
равнивание местных перенапряжений, т. е. усиление полимера.
Дополнительный вклад в упрочнение вносит повышенный меха-
нический гистерезис, обусловленный снижением подвижности
макромолекул у поверхности наполнителя и разрушением свя-
зей полимер — наполнитель и наполнитель — наполнитель.
Вследствие повышенного гистерезиса степень релаксации иапря-
427
жения наполненных полимеров больше, чем ненаполненных,
в области больших деформаций в вершине разрастающейся
трещины.
К упрочнению приводит и взаимодействие частиц наполните-
ля между собой с образованием агрегатов, а в ряде случаев
и непрерывных сеток в полимерной матрице.
Рассмотренные выше факторы относятся главным образом
к усилению эластомеров, т. е полимеров с гибкими макромоле-
кулами. Упрочнение жесткоцеиных полимеров обусловлено в ос-
новном не ориентирующим действием наполнителя, а измене-
нием условий перенапряжения на краях растущих трещин. Рост
развивающейся макротрещины, встречающей на пути наполни-
тель, ограничивается, и дальнейшее ее развитие требует повы-
шения напряжения.
Свойства композиций при армировании определяются свой-
ствами полимеров и армирующих материалов в изделии и глав-
ным образом адгезией полимерной матрицы к их поверхности.
Для изготовления прочных изделий необходимо создать требуе-
мые ориентацию и степень натяжения всех армирующих эле-
ментов, что обеспечит их равномерное напряжение при работе;
выбрать оптимальную форму и размеры армирующих элемен-
тов, позволяющих обеспечить максимальную удельную поверх-
ность контакта со связующим и предотвратить их разрушение
в процессе переработки; выбрать связующее, химическая термо-
стойкость, адгезия, модуль, эластичность и другие физико-ме-
ханические показатели которого в условиях эксплуатации изде-
лия должны иметь оптимальные значения.
Большие перспективы имеет метод полимеризанионного на-
полнения, при котором наполненный полимер получают в про-
цессе синтеза из мономеров на поверхности наполнителя. При
этом имеющиеся на поверхности частиц наполнителя функцио-
нальные группы превращаются в активные центры полимериза-
ции и каждая частица наполнителя покрывается слоем поли-
мера нужной толщины. Способ полимеризационного наполнения
позволяет получить сыпучие гомогенные композиции заданного
состава, легко перераоатываемые в изделия.
Контрольные вопросы
1 В чем сходство и различия растворов низко- и высокомолекулярных
соединений?
2. Что такое «хороший» н «плохой» растворитель? Какой показатель харак-
теризует качество растворителя?
3 Что такое термодинамическое сродство компонентов раствора? Какие
свойства раствора оно характеризует? Как оценивается термодинамиче-
ское сродство?
4. Какие растворы полимеров считаются концентрированными, какие раз-
бавленными?
428
5. Каковы особенности свойств разбавленных растворов полимеров^ чем
они обусловлены?
6 Какие виды взаимодействия существуют в растворах полимеров и как
они сказываются на свойствах растворов?
7 Что такое характеристическая вязкость? Как се определяют?
8. В чем различие между истинными и коллоидными растворами? Приведи-
те примеры коллоидных систем.
9. Расскажите о структуре смесей полимеров. Какова особенность межфаз-
ной границы в этих системах?
10. Какова роль пластификаторов и полимеров? Каким показателем оцени-
вают эффект пластификации?
II. В чем особенности структуры наполненных полимеров? Как изменяются
свойства полимеров при введении наполнителей? Назовите основные фак
торы усиления
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Андрианов К. А., Хананашвили Л. М. Технология элемеитооргаинческнх
мономеров и полимеров. М Химия, 1983 416 с.
Аскадский А. Я. Деформация полимеров. М.: Химия, 1973 448 с.
Аскадский Я. Я.. Матвеев Ю. И Химическое строение и физические свой-
ства полимеров. М Химия, 1983. 248 с.
Барамбойм Н. К Мехакохимия высокомолекулярных соединений. М
Химия, 1978. 384 с.
Бартенев Г. М Структура и релаксационные свойства эластомеров. М.:
Химия. 1979. 288 с
Бартенев Г. М. Прочность и механизм разрушения полимеров. М Хи-
мия. 1984. 279 с
Бартенев Г. М., Зеленев Ю В. Курс физики полимеров. М.: Химия. 1976,
288 с.
Бартенев Г. М., Зеленев Ю. В Физика и .механика полимеров М.: Выс-
шая школа. 1983. 391 с.
Берлин Ал. Ял., Вольфсон С Я.» Ениколопян Н. С. Кинетика полимерн-
зационных процессов М.: Химия, 1978. 320 с.
Бухана М. Ф. Кристаллизация каучуков и резин. М.: Химия, 1973. 240 с.
Бухина М Ф. Техническая физика эластомеров. М.: Химия. 1984, 224 с.
Бюллер Г. К. Тепло- и термостойкие полимеры: Пер. с нем ./Под ред.
я. С Выгодского. М.‘ Химия. 1984 1056 с.
Ван Кревелен Д. В Свойства и химическое строение полимеров: Пер.
с англ./Под ред. А. Я. Малкина. М.: Химия, 1976. 414 с.
Василенок Ю И Предупреждение статической электризации полимеров
Л.: Химия. 1981. 208 с.
Виноградов Г. В Малкин Я. Я. Реология полимеров. М.; Химия. 1977.
438 с.
Воюцкий С. С. Курс коллоидной химии М.: Химия, 1975. 505 с.
Годовский Ю. К. Теплофнзическне свойства полимеров. М.: Химия. 1971).
216 с.
Гддовский Ю. К. Теплофизика полимеров. М.: Химия, 1982. 280 с. _
Гуль В. Е Структура и прочность полимеров. М.: Химия, 1978. 350 с.
Гуль В. Е., Кулезнев В Н. Структура и механические свойства полиме-
ров. М.: Высшая школа. 1979. 351 с.
Гуль В Е.. Шенфилъ Л. 3 Электропроводящие полимерные композиции
М.: Химия, 1985 240 с.
Гуняев Г. М Строение и свойства полимерных волокнистых компози-
ционных материалов. М.: Химия, 1981. 210 с.
429
----л. л. процессы структурирования эластомеров. М.: Химия, 1978
287 с.
Джейд Ф X. Полимерные монокристаллы: Пер. с англ./Под ред.
С Я Френкеля М.: Химия, 1968. 552 с.
Елисеева В. И. Полимерные дисперсии. М.: Химия. 1980. 296 с.
Ерусаммский Б Л., Любецкий С. Г. Процессы ионной полимеризации.
Л Химия, 1974. 256 с.
Кестельман В. М Физические методы модификация полимерных материа-
лов. М.: Химия, 1980. 224 с.
Козлов П В.. Панков С. П Физико-химические основы пластификации
полимеров. М.: Химия» 1982. 234 с.
Кулезнев В. И Смеси полимеров. М.: Химия, 1980. 304 с.
Кулезнев В Н., Шершнев В А. Физика н химия полимеров. М.: Высшая
школа, 1988. 313 с.
1980^260™* Ю' Межфазные явления в полимерах. Киев: Наукова думка.
Лосев И. П., Тростянская Е Б. Химия синтетических полимеров. 3-е из-
дание. М.: Химия, 1971. 616 с.
Лущейкин Г. Л. Полимерные электреты М.: Химия, 1984 184 с.
Лущейкин Г. Л. Методы исследования электрических свойств полимеров.
М.: Химия, 1988. 160 с.
Минскер К. С., Федосеева Г. Т. Деструкция и стабилизация поливинил-
хлорида. М.: Химия, 1979. 272 с.
Марихин В Л., Мясникова Л. П. Надмолекулярная структура полимеров.
Л.: Химия, 1977 238 с.
Морган П. У. Поликонденсационные процессы синтеза полимеров Пер.
с англ. Я С Выгодского и Б. Р. Лившица. Л.: Химия, 1970. 448 с.
Нильсен Л Механические свойства полимеров и полимерных компози-
ций Пер. с англ. П. Г Бабаевского. М Химия, 19 312 с.
Оудиан Дж. Основы химии полимеров. Пер. с англ./Под ред. В В. Кор-
шака М. Мир, 1974 614 с.
Перепечко И. И. Акустические методы исследования полимеров. М : Хи-
мия. 1973. 296 с.
Перепечко И. И Введение в физику полимеров. М.: Химия, 1978. 312 с.
Перепечко И И. Свойства полимеров при низких температурах. М.: Хи-
мия, 1977. 272 с.
Платэ Н. Л., Литманович Л Д., Ноа О. В Макромолекулярные реакции.
М.: Химия, 1977 286 с.
Практикум по высокомолекулярным соединениям/Под. ред. В А. Каба-
кова. М.: Химия. 1985 224 с.
Привалко В. П. Молекулярное строение и свойства полимеров. М.: Хи-
мия, 1986. 238 с.
Рабск Я. Экспериментальные методы в химии полимеров. Пер. с англ.
Под ред. В. В. Кормака. М.: Мир. 1983. Т. 1. 382 с.; т. 2. 479 с.
Реакции в полимерных снстемах/Под ред. С С. Иванчева. Лл Химия,
1987. 304 с.
Регель В Л., Слуцкер Л. И.. Томашевский Э. Е. Кинетическая природа
прочности твердых тел. М.: Наука, 1974. 560 с.
Саеада X Термодинамика полимеризации: Пер с англ. М.: Химия, 1979.
312 с.
Соколов Л Б Основы синтеза полимеров методом поликонденсации.
М.: Химия, 1979. 264 с.
Стрепихеев А Л Деревицкая В А Основы химки высокомолекулярных
соединений. Изд. 3-е. М.: Химия, 1976. 137 с.
Тагер Л Л. Физикохимия полимеров. Изд. 3-е. М.: Химия. 1978. 544 с,
Тюдзе Р Каван Т. Физическая химия полимеров: Пер. с ял. М. Химия,
1977. 296 с.
Уорд И. Механические свойства твердых полимеров: Пер. с англ./Под ред.
А. Я Малкина. М.: Химия, 1975.
430
Хэм Д. Сополимеризация Пер. с аигл./Под ред. В. А. Кабанова. М.:
.Химия, 1971. 616 с.
Чирков Н. М., Матковский П. Е., Дьячковский Ф. С Полимеризация иа
комплексных металлоорганических катализаторах. М.: Химия, 1976, 416 с.
Шляпников Ю А., Дирюшин С Г., Марьин А. П Аитиоксислительная
стабилизация полимеров. М.: Химия, 1986. 256 с.
Шур А. М. Высокомолекулярные соединения. М Высшая школа, 1981.
656 с.
Электрические свойства полнмеров/Под ред. Б. И. Сажина. Л.: Химия,
1986. 226 с.
Энтелис С Г.» Евреинов В В., Кузаее А. И Реакционноспособные олиго-
меры. М.: Химия, 1985. 304 с
Учебное издание
Тугое Иван Иванович
Кострыкина Галана Ивановна
ХИМИЯ и ФИЗИКА
ПОЛИМЕРОВ
Редакторы Г. М. Медникова, Л. А. Чеканова
Художественный редактор К. К. Федоров
Технические редакторы Б М. Молодцов, С. Ю. Титова
Корректоры Т С. Васина, М В. Черниховская
ИБ № 2312
Сдано в наб. 23.01.89. Поди в печать 09.08.89. Формат бумаги
60X88Vi«. Бумага офс. А* 2 Печать офсетная Гарнитура
литературная Уса. печ. л 26.46. Усл. кр.-отт. 26.46. Уч.-нзд.
л. 29.55. Тираж 9600 экз. Заказ As 816. Цена 1 р 30 к
Ордена «Знак Почета» издательство «Химия»
107076. Москва, Стромынка. 21
Московская типография № П Союзполиграфпрома при Госкомиздате СССР, Москва
113105, Нагатинская ул.» д. I