/













































































































































































































































































Автор: Аскадский А.А. Кондращенко В.И.
Теги: химия ядерная, атомная и молекулярная физика химия высокомолекулярных соединений полимеры физика
ISBN: 5-89176-077-0
Год: 1999




Текст
РОССИЙСКАЯ АКАДЕМИЯ НАУК
Институт элементоорганических
соединений им. А.Н. Несмеянова
А.А. Аскадский, В.И. Кондращенко
КОМПЬЮТЕРНОЕ
МАТЕРИАЛОВЕДЕНИЕ
ПОЛИМЕРОВ
т.1. Атомно-молекулярный уровень
МОСКВА
Научный мир
1999
УДК 541.64:539.199
А-90
ББК 24.7
ISBN 5-89176-077-0
Аскадский А.А., Кондратенко В.И.
Компьютерное материаловедение полимеров, т.1. Атомно-молекулярный
уровень. - М.: Научный мир, 1999. - с. 544
В монографии изложен подход для количественного анализа влияния химического
строения линейных и сетчатых полимеров на их свойства. Подход основан на представлении
повторяющегося звена полимера в виде набора ангармоничных осцилляторов, которые
описывают термическое движение атомов в поле внутри- и межмолекулярных сил, включая
слабые дисперсионные силы, диполь-дипольные взаимодействия, водородные и химические
связи. Описываются ЭВМ-программы, основанные на данном подходе, которые позволяют
производить расчеты более 50 фундаментальных физических и химических констант ли-
нейных и сетчатых полимеров, а также низкомолекулярных органических жидкостей. Про-
граммы позволяют решать прямую задачу, те. проводить количественную оценку
физических свойств полимеров на основе их химического строения, и обратную задачу,
т е. проводить компьютерный синтез полимеров с заданными физическими свойствами.
Для химиков, физико-химиков, научных сотрудников, аспирантов, студентов.
Илл. 123. Библ. 224. Табл. 62.
Р
Публикуется при финансовой поддержке
Российского фонда фундаментальных исследований
(грант № 98-03-46001)
A. A. Askadskii, V.I. Kondraschenko
Computer-Based Materiology of Polymers. Volume 1. Atomic and Molecular
Line of Approach. - Moscow, Scientific World, 1999. - p. 544
Developed in this monograph is a line of approach to quantitative structure -property
relationships appropriated fer Id th ihear po kiners an dp ok mer networks The line of approach
is based on polymer repeating unit represented as a set of anharmonic oscillators which describe
thermal motion of atoms in the field of intra-and intermolecular forces, including weak dispersion
forces, dipole-dipole interactions, hydrogen and valence bonds. Also presented in this paper are
computer programs based on the given approach. These programs enable one to compute more
than 50 fundamental physical and chemical constants of linear and network polymers as well as
low molecular liquids. The programs make it possible to solve a direct problem, i.e. quantitative
evaluation of physical properties of polymers based on their chemical strucure, and a reverse
problem, i.e. a computer synthesis of polymers with prescribed physical properties.
The monograph is intended for chemists, physicists-cum-chemists. researchers, post-graduate
students and students.
111. 123. Bibl. 224. Tabl. 62.
© А. А. Аскадский, В.И. Кондратенко 1999
© Научный мир, 1999
ISBN 5-89176-077-0
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ ...................................... 9
ВВЕДЕНИЕ .......................................... 12
Глава I. КРАТКИЕ СВЕДЕНИЯ О ТИПАХ ПОЛИМЕРОВ И ИХ
ХИМИЧЕСКОМ СТРОЕНИИ............................... 19
Глава II. УПАКОВКА МАКРОМОЛЕКУЛ И ПЛОТНОСТЬ
ПОЛИМЕРОВ ....................................... 29
II. 1. Метод инкрементов и основные
физические представления .............. 29
II.2. Связь между свободным объемом полимеров,
коэффициентом молекулярной упаковки и
пористой структурой ................... 54
Глава III. ТЕМПЕРАТУРНЫЙ КОЭФФИЦИЕНТ ОБЪЕМНОГО
РАСШИРЕНИЯ ............................... 74
Глава IV. ТЕМПЕРАТУРА СТЕКЛОВАНИЯ ПОЛИМЕРОВ ..... 85
IV.1. Термомеханический и другие методы определения
температуры стеклования пэлимеров ..... 85
IV.2. Механизм стеклования ...............112
IV.3. Расчет температуры стеклования
линейных полимеров ........................127
IV.4. Расчет температуры стеклования
сетчатых полимеров ........................153
Глава V. ТЕМПЕРАТУРА ТЕКУЧЕСТИ АМОРФНЫХ
ПОЛИМЕРОВ .......................................202
Глава VI.ТЕМПЕРАТУРА ПЛАВЛЕНИЯ ПОЛИМЕРОВ ........206
4
Оглавление
Глава VII. ТЕМПЕРАТУРА НАЧАЛА ИНТЕНСИВНОЙ
ТЕРМИЧЕСКОЙ ДЕСТРУКЦИИ ПОЛИМЕРОВ ...........................216
Глава VIII.ОПТИЧЕСКИЕ И ОПТИКО-МЕХАНИЧЕСКИЕ
СВОЙСТВА ПОЛИМЕРОВ ..............................230
VIII . 1. Показатель преломления ...................230
VI II.2. Коэффициент оптической чувствительности
по напряжению ............................236
Глава IX. ДИЭЛЕКТРИЧЕСКАЯ ПРОНИЦАЕМОСТЬ
ПОЛИМЕРОВ И ОРГАНИЧЕСКИХ
РАСТВОРИТЕЛЕЙ ......................................257
Глава X. РАВНОВЕСНЫЙ МОДУЛЬ ВЫСОКО-
ЭЛАСТИЧНОСТИ СЕТЧАТЫХ ПОЛИМЕРОВ ....................270
Х .1. Расчетный способ оценки равновесного модуля .270
Х. 2. Разномодульные и градиентные полимеры ........281
Глава XI. ОПИСАНИЕ РЕЛАКСАЦИОННЫХ ПРОЦЕССОВ В
ПОЛИМЕРАХ ..........................................293
XI. 1. Релаксация напряжения .......................293
XI.2 . Процессы сорбции и набухания ................320
Глава XII. РАСТВОРИМОСТЬ ПОЛИМЕРОВ .........................327
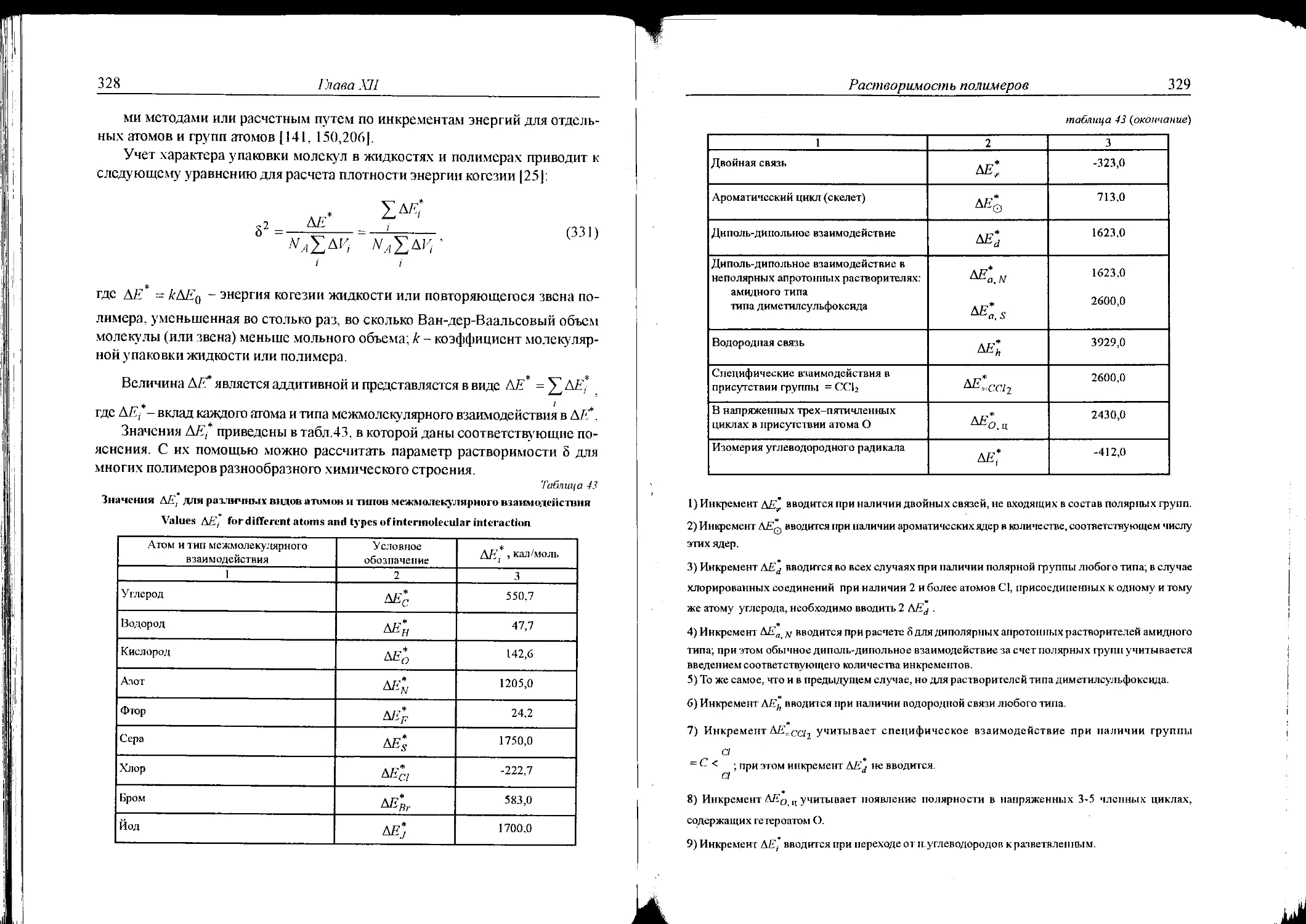
XII . 1. Плотность энергии когезии органических жидкостей
и полимеров. Параметр растворимости Гильдебранда ...327
XI 1.2. Критерий растворимости .....................333
XII .3. Влияние молекулярной массы и ориентации
макромолекул на растворимость ............346
Глава XIII. ПОВЕРХНОСТНЫЕ СВОЙСТВА ОРГАНИЧЕСКИХ
ЖИДКОСТЕЙ И ПОЛИМЕРОВ ...........................352
XIII . 1. Поверхностное натяжение
органических жидкостей ...................353
XIII. 2. Поверхностное натяжение полимеров .........362
Глава XIV. СОВМЕСТИМОСТЬ ПОЛИМЕРОВ .........................374
Глава XV. ВЛИЯНИЕ КОНЦЕВЫХ ГРУПП НА СВОЙСТВА
ПОЛИМЕРОВ .......................................383
Глава XVI. ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ .....392
XVI. 1. Теплоемкость ...............................392
XVI.2. Температуропроводность и теплопроводность ...394
Оглавление
5
Глава XVII. МОЛЕКУЛЯРНЫЙ ДИЗАЙН И КОМПЬЮТЕРНЫЙ
СИНТЕЗ ПОЛИМЕРОВ С ЗАДАННЫМИ
СВОЙСТВАМИ .......................................397
ПРИЛОЖЕНИЯ
Приложение 1.Пример решения прямой задачи оценки свойств
полимеров по их химическому строению ..............425
Приложение 2.Пример решения обратной задачи
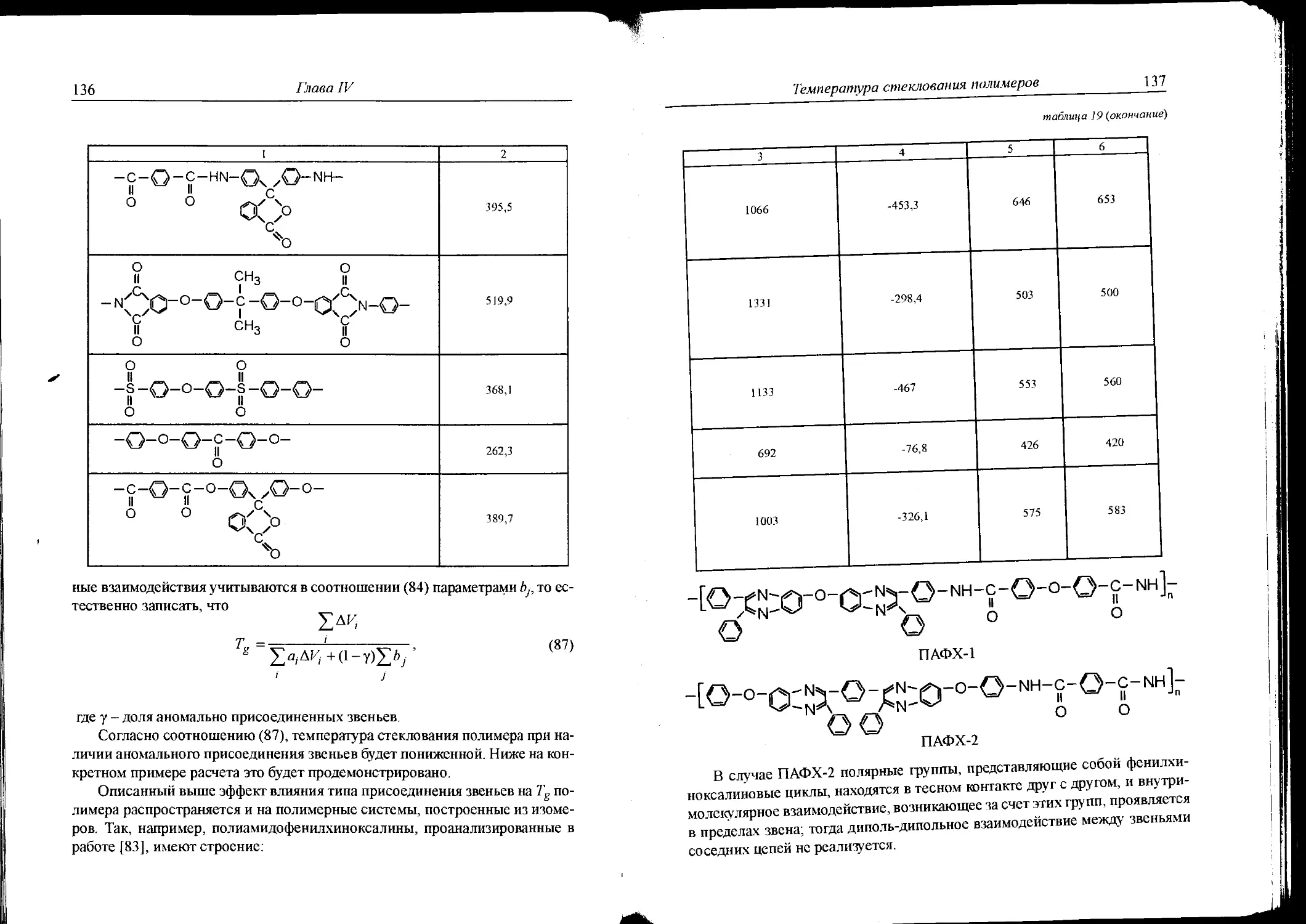
синтеза полимеров .................................440
Приложение З.Пример решения смешанной задачи -
анализ химического строения
фенолформальдегидной смолы ............450
Приложение 4. Применение подхода к многокомпонентным
сополимерам .......................................467
Приложение 5. Влияние сильного межмолекулярного взаимодействия,
возникающего между двумя разнородными
полимерами, на их совместимость ..................472
SUMMARY ..........................................496
ЛИТЕРАТУРА .......................................517
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ .............................534
CONTENTS
PREFACE .............................................................. 9
INTRODUCTION ........................................................ 12
CHAPTER I. BRIEF INFORMATION ON TYPES OF POLYMERS
AND THEIR CHEMICAL STRUCTURE .......................... 19
CHAPTER II. PACKING OF MACROMOLECULES AND POLYMERS
DENSITY ............................................... 29
II. 1. Increments method and basic physical assumptions . 29
11.2. Relation between empty volume of polymers, their
coefficient of molecular packing and porous structure . 54
CHAPTER III. THERMAL COEFFICIENT OF
VOLUMETRIC EXPANSION ................................................ 74
CHAPTER IV. GLASS TRANSITION TEMPERATURE
OF POLYMERS ........................................... 85
IV. 1.Methods of evaluation
of the glass transition temperature .................... 85
IV.2.Mechanism of glass transition .........112
1V.3.Calculation of the glass transition temperature
of linear polymers ....................................127
IV.4.Calculation of the glass transition temperature
of polymer networks ...................................153
CHAPTER V. TEMPERATURE OF TRANSITION INTO
THE VISCOUS FLOW STATE FOR AMORPHOUS
POLYMERS ...............................................202
CHAPTER VI. MELTING POINT OF POLYMERS ..................206
Contents
7
CHAPTER VII.TEMPERATURE OF ONSET OF INTENSE TERMAL
DEGRADATION OF POLYMERS ...................................216
CHAPTER VIII. OPTICAL AND OPTICO-MECHANICAL
PROPERTIES OF POLYMERS ....................................230
VIII. 1. Refractive index ........................230
V1II.2. Sterss-optical coefficient ...............236
CHAPTER IX. DIELECTRIC CONSTANT OF POLYMERS AND
ORGANIC SOLVENTS .........................................257
CHAPTER X. EQUILIBRIUM RUBBERY MODULUS
FOR POLYMERS NETWORKS ....................................270
X. 1. Calculations of the equilibrium modulus.....270
X.2. Heteromodular and gradient-modulus polymers .281
CHAPTER XI. DESCRIPTION OF RELAXATION PROCESSES
IN POLYMERS ..............................................293
XI. 1 .Stress relaxation .........................293
XI.2.Sorption and swelling processes .............320
CHAPTER XII. SOLUBILITY OF POLYMERS ..........................327
XII. 1 .Density of cohesive energy of organic liquids and
polymers. Solubility parameter of Hyldebrand ....327
XII.2.Solubility criterion .......................333
X11.3.Influence of molecular mass and degree
of macromolecules orientation on the solubility .346
CHAPTER XIII. SURFACE PROPERTIES OF ORGANIC LIQUIDS
AND POLYMERS ..............................................352
XIII. 1. Surface tension of organic liquids ......353
XIII.2. Surface tension of polymers ..............362
CHAPTER XIV. MISCIBILITY OF POLYMERS .......................374
CHAPTER XV. INFLUENCE OF THE END GROUPS
ON THE PROPERTIES OF POLYMERS ............................383
CHAPTER XVI. THERMAL-PHYSICAL PROPERTIES
OF POLYMERS ...............................................392
XVI. 1.Heat capacity .............................392
XVI.2.Thermal conductivity and heat conductivity .394
CHAPTER XVII. MOLECULER DESIGN AND COMPUTER
SYNTHESIS OF POLYMERS WITH
PREDETERMINED PROPERTIES .................................397
8 Contents
APPENDICES
Appendix 1. Examples of decision of direct problem
of polymers synthesis .......................................425
Appendix 2. Examples of decision of reverse problem
of polymers synthesis ......................................440
Appendix 3. Application of the approach
to the multi-component copolymers ..........................450
Appendix 4. Influence of strong intermolecular interaction between
two heterogeneous polymers on their compatibility ...........467
Appendix 5. Influence of the strong intermolecular interaction occuring
between two dissimilar polymers on their miscibility.496
SUMMARY ...................................................496
REFERENCES ................................................517
INDEX .....................................................534
ПРЕДИСЛОВИЕ
В журнале “Химия и жизнь” N°_2 за 1981г. была напечатана моя статья,
которой редактор дал следующее название: “Атом плюс атом плюс тысяча
атомов”. В этой статье речь шла о возможности расчета ряда физических
свойств полимеров на основе химического строения повторяющегося звена
(тогда была возможность рассчитывать свойства лишь линейных полимеров).
В заключении этой статьи, которое называлось “Немного фантазии”, было
написано: “Итак, многие свойства полимера можно предсказать, не зная ничего
кроме структурной формулы соответствующего мономера. Это немало: уже
сегодня такие расчеты позволяют избавить химиков от тяжелого труда по
синтезу бесперспективных мономеров. Раньше, при чисто эмпирическом
подборе материалов, таких мономеров приходилось делать немало. Но все-
таки расчеты пока приходится выполнять вручную. А вот когда их удастся
перевести на машинный язык, традиционные при любой химической дискуссии
мел и доску сможет заменить электронный “карандаш”. Нарисует им химик
на экране дисплея формулу предполагаемого мономера - а ЭВМ сразу ответит,
есть ли смысл браться за синтез. Другая обратная задача представляется еще
более увлекательной. Ведь если ЭВМ сможет определять свойства по фор-
муле, то, видимо, машину удастся научить, и, наоборот, выдавать формулу
подходящего мономера (или несколько формул на выбор) по сообщенному ей
любому, даже самому противоречивому набору свойств. И тем самым заменить
химика в самойголово домной части его работы, где сегодня успех определяет-
ся только опытом, интуицией и удачей.” Это была фантазия, и было трудно
предположить в то время, что когда-либо в обозримом будущем эти идеи могут
быть реализованы. Однако события стали развиваться очень быстро,особенно
после появления персональных компьютеров большой мощности. Прежде чем
рассказать об этапах этой большой работы, следует кратко остановиться на
10
Предисловие
методах количественной оценки физических свойств полимеров, которая
осуществляется на основе их химического строения. В настоящее время сущест-
вует три основных подхода к такой оценке. Один из этих подходов, развитый
Ван Кревеленом [214], основан на идее так называемых “групповых вкладов”,
согласно которой записываются простейшие эмпирические выражения адди-
тивного типа, причем данная группа, находясь в разных полимерных звеньях,
вносит один и тот же вклад в рассчитываемую характеристику (например, в
температуру стеклования, плавления и т.д.). Это - чисто эмпирический подход,
как отмечает его автор, который позволяет с хорошей точностью рассчитывать
физические свойства многих линейных полимеров.
Другой подход, развиваемый длительное время автором данного преди-
словия совместно с Ю.И. Матвеевым [28, 128], является полуэмпирическим.
Согласно этому под ходу, уравнения для расчета физических свойств получены
на основании представлений физики твердого тела, а калибровка метода
осуществляется с помощью физических характеристик полимерных
стандартов, свойства которых хорошо изучены. В результате параметры
уравнений имеют определенный физический смысл (эн ергия дисперсионного
взаимодействия, энергия сильного межмолекулярного взаимодействия,
включая водородные связи, Ван-дер-Ваальсовый объем и т.д.). Использование
такого подхода позволяет с достаточной точностью оценивать многие
физические характеристики полимеров (сейчас их уже около 60), и при этом
количество полимеров самого разнообразного строения не ограничено.
Третий подход, развиваемый Дж. Бицерано [133], появился совсем не-
давно, он основан на так называемых индексах связанности, что на прак-
тике свелось к поиску различных корреляций физических свойств со множест-
вом правил, как находить коэффициенты корреляционных зависимостей.
В данной монографии мы будем рассматривать принципы подхода,
развитого А. А. Аскадским и Ю.И. Матвеевым, причем существенное внимание
будет уделено именно компьютерной реализации данного метода расчета
физических свойств полимеров. Первая ЭВМ-программа была написана
сотрудниками лаборатории квантовой химии Института элементоорганичес-
ких соединений им. А.Н. Несмеянова РАН Е.Г. Гальперн, И.В. Станкевичем и
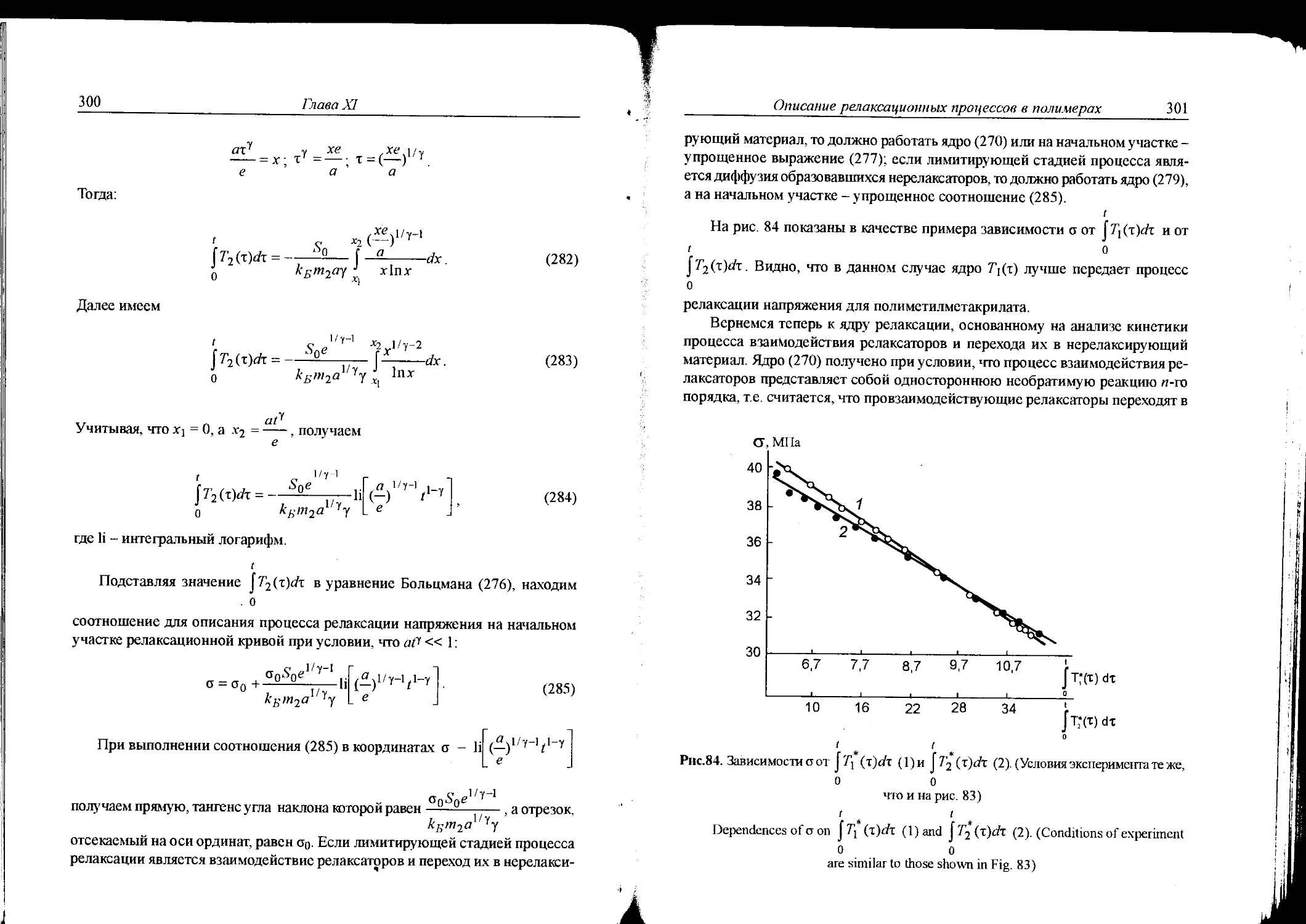
А.Л. Чистяковым. В первоначальном варианте этой программы компьютерный
“синтез” полимеров осуществлялся из так называемых крупных заготовок,
которые представляли собой остатки мономеров, вовлекаемых в реакцию
синтеза. Во втором варианте компьютерный “синтез” проводился из мель-
чайших заготовок, из которых конструировалось повторяющееся звено
полимера. Это существенно расширило возможности ЭВМ-программы как
для решения прямой задачи (расчет свойств полимера по его химическому
строению), так и обратной задачи (компьютерный “синтез” полимеров с заранее
Предисловие
11
заданными свойствами, интервалы которых введены в компьютер), поскольку
количество “синтезируемых” полимеров резко возросло. Затем А.Ф. Клинских
была написана принципиально новая программа, в которой химическое
строение повторяющегося звена полимера “конструируется” из атомов. При
этом пользователю лишь нужно изобразить химическое строение полимера
на экране дисплея, как это делается химиком на бумаге, и после этого компью-
тер выдает все физические свойства полимеров, предусмотренные в программе
(всего около 60 свойств). Эта программа предусматривает также расчет ряда
свойств низкомолекулярных органических соединений, а также, что очень ва-
жно, свойств полимерных сеток. Предусмотрено и решение обратной задачи.
Особо следует отметить возможность расчета свойств сополимеров и их смесей,
предсказания растворимости и совместимости полимеров .построения зависи-
мостей свойств от температуры, молекулярной массы, степени кристал-
личности, микротактичности (особенно важны зависимости температуры
стеклования и текучести от молекулярной массы).
Разумеется не все проблемы решены Предстоит повысить точность расчета
и различных предсказаний поведения полимеров при растворении и смешении
друг с другом, разработать расчетные схемы для оценки новых свойств
полимеров и осуществить их компьютерную реализацию, и т.д.
Совершенно очевидно, что данная монография не лишена недостатков.
Авторы с благодарностью примут все замечания, появившееся после ознаком-
ления с книгой.
ВВЕДЕНИЕ
Как было отмечено выше, подход для оценки физических свойств поли-
меров, рассматриваемый в данной монографии, является полу эмпирическим.
В случае оценки термических характеристик полимеров, таких как темпера-
тура стеклования, температура плавления, предполагается, что повторяющее-
ся звено построено из набора ангармонических осцилляторов, представляю-
щих собой пары атомов, связанных межмолекулярными физическими связя-
ми. Критическая температура такого набора ангармонических осцилляторов
и определяет упомянутые выше две температуры переходов. К этим характе-
ристикам тесно примыкает коэффициент термического расширения. В случае
такой характеристики, как температура начала интенсивной термической дес-
трукции, звено полимера рассматривается в виде набора ангармонических
осцилляторов, связанных химическими связями. Критическая температура та-
кого набора осцилдяторов характеризует температуру начала интенсивной тер-
мической деструкции при заданной скорости нагрева (естественно, что при
другой скорости нагревания температура начала интенсивной деструкции изме-
нится, те. кинетические эффекты здесь играют существенную роль). На пер-
вый взгляд может показаться странным, что процесс термической деструкции
здесь рассматривается не как кинетический, что общепринято, а как своеоб-
разный фазовый переход, при котором, однако, из продуктов термического
распада нельзя снова получить исходное вещество простым охлаждением.
Уравнения для расчета других физических характеристик основаны на
физических подходах, которые детально изложены ниже, и мы не будем здесь
на них останавливаться.
Общим для всех этих уравнений является суммирование ряда атомных
констант, характеризующих вклады в энергию межмолекулярного взаимодей-
ствия, энергию химических связей, Ван-дер-Ваальсовый объем и т.д. Строго
говоря, данный подход не может быть назван ад дитивным в обычном понима-
Введение
13
нии этого слова, поскольку рассчитываемые свойства не являются аддитив-
ными по отношению к атомам и группам, из которых построено повторяюще-
еся звено полимера.
Аддитивность применяется здесь только к таким характеристикам, кото-
рые действительно являются аддитивными (Ван-дер-Ваальсовый объем, мо-
лекулярная масса, энергия межмолекулярного взаимодействия и т.д ). Опи-
сываемый подход позволяет рассчитывать свойства неограниченного числа
полимеров, и с помощью разработанных и описанных в монографии ЭВМ-
программ проводить компьютерный синтез полимеров с заданными свойства-
ми, что не позволяют другие существующие сейчас программы.
Как было отмечено выше, подход, изложенный в монографии, является
полу эмпирическим, причем калибровка метода основывается на так называе-
мых полимерных стандартах, свойства которых детально изучены и хорошо
известны. Сущность калибровки рассмотрим на примере уравнения для рас-
чета температуры стеклования линейного полимера Tg:
z^
Tg xalAV'+YbJ
' J
где a - атомные константы; Ь - константы, связанные с энергией сильного
межмолекулярного взаимодействия (диполь-дипольного, водородных связей),
возникающего между полимерными цепями за счет имеющихся в них поляр-
ных групп; - Ван-дер-Ваальсовый объем повторяющегося звена по-
лимера, который складывается из Ван-дер-Ваальсовых объемов атомов, вхо-
дящих в это звено.
Преобразуем данное уравнение к виду
Ya^vi + Ybj=J~ZAf<
< j 1 g i
На основе такого уравнения составляется избыточная система линейных
уравнений, которая выглядит следующим образом:
+ °2Д1/1,2 + -.. + а„АИ1я +аА +₽i^2 +---+7ibk =——(£ДИ,)[ ;
12,1 /
а1Д^2,1 + Й2Д^2,2 + ..- + а„ДИ2„ +а2^1 + 02^2 +--- + ?2^ = ~-(ZA^')2 >
4,1 i
а1^т,1 +а2^т,2 + -+ап^т,п +aml,l +(М’2 +---+1 mhk = ~-----------(ZA^)« •
12,1 /
14
Введение
Далее составляется матрица коэффициентов при неизвестных этой избы-
точной системы уравнений
АГц ДИ1>2 ... ДИ1;И cq р! ... У1
ДК2;1 A^2,2 Д^2,л а2 ₽2 ••• У 2
А^т,! ^^т,2 ••• т,п Pm ••• Ут
и столбцовая матрица свободных членов этих уравнений
В =
yg,i /
^(^',)2
7g.2 /
—i— (У ДК)_
rr~f * ' т
1 g,m i
Далее строится транспонированная матрица А , которая перемножается
на исходную - А А , а также на столбцовую матрицу - А В, в результате чего
получается каноническая система уравнений. Эта каноническая система ре-
шается, например, методом Гаусса. Вся процедура калибровки осуществляет-
ся с помощью стандартных ЭВМ-программ.
Не рассматривая особенности такого регрессионного анализа, отметим
лишь, что полимеры, которые выбираются для калибровки метода, должны
обладать экспериментальными значениями анализируемых физических ха -
ракгеристик как можно в более широких пределах, а химическое строение
полимерных стандартов должно существенно различаться. Обычно решается
избыточная система, составленная из 30-40 уравнений, что соответствует
30К0 полимерам. Далее по полученным коэффициентам рассчитываются свой-
ства всех других полимеров. При этом определяются также такие характери-
стики, как энергия слабого дисперсионного взаимодействия, энергия сильных
диполь-дипольных взаимодействий и водородных связей, их относительная
доля и многие другие физические параметры системы.
Перейдем к краткому описанию содержания отдельных глав монографии .
В первой главе приведены сведения о современной классификации поли-
меров и их химическом строении. Исключительный1 интерес,вызванный осо-
бенностями химического строения и областью применения, представляют
интерполимеры, дендритные и лестничные полимеры.
Введение
15
Во второй главе обсуждается подход к компьютерному материаловеде-
нию полимеров на атомно-молекулярном уровне, основанный на методе инк-
рементов. Рассчитаны инкременты различных атомов и их основных групп.
Приведены основные физические представления о структуре какромэ лекул
полимеров и определяющих ее параметрах. Дана методика расчета такой важ-
ной характеристики структуры полимера, как коэффициент молекулярной упа-
ковки. Установлена связь между свободным объемом полимера, коэффици-
ентом молекулярной упаковки и параметрами его пористой структуры. Для
экспериментального определения характеристик микропористой структуры
полимеров использован метод аннигиляции позитронов, с использованием
которого выявлены структурные изменения в полимерах при их релаксации.
В третьей главе с учетом слабых дисперсионных и сильных (диполь-ди-
польных и водородных связей) взаимодействий получены формулы для рас-
чета термического коэффициента объемного расширения в зависимости от
химического строения полимера. При этом вид атомов полимерной цепи и
тип межмолекулярного взаимодействия оценивается ограниченным числом
соответствующих инкрементов, численные значения которых определены.
В четвертой главе подробно освещен термомеханический метод опреде-
ления температуры стеклования и текучести полимеров, проанализированы
особенности интерпретации термомеханических кривых для аморфных и кри -
сталлических полимеров, приведен расчетный метод определения по хими-
ческому строению полимера величины механического сегмента. Рассмотре-
ны две основные концепции механизма процессов застекловывания полиме-
ров - релаксационная и межмолекулярная. Рассматривается более
универсальный, чем широко распространенный групповой подход расчета
свойств полимера по их химическому строению, “атомистический” подход, с
использованием которого получены аналитические выражения для расчета по
химическому строению температуры стеклования линейных и сетчатых поли-
меров. Выполнен анализ влияния типов разветвлений линейных полимеров, а
для сетчатых полимеров - числа звеньев между узлами сшивки, типа и стро-
ения этих узлов, наличия и вида дефектов сетки на температуру стеклования
полимеров.
В пятой главе дается методика расчета температуры текучести аморфных
полимеров и температурного интервала высокоэластичности полимеров по их
химическому строению, а также выясняются условия появления состояния
высокоэластичности полимера в зависимости от его молекулярной массы, что
важно при переработке полимеров.
В шестой главе освещены два подхода к расчетному определению темпе-
ратуры плавления полимеров по химическому' строению повторяющегося зве-
на первый подход основан на экспериментально установленном факте близо-
16
Введение
сти долей пустого объема при плавлении кристаллического полимера и при
переходе из стеклообразного в высокоэластическое состояние аморфного по-
лимера того же строения, а второй - на рассмотрении повторяющегося звена
полимера как набора ангармонических осцилляторов.
В седьмой главе рассмотрена важнейшая характеристика термостойкости
полимеров - температура начала их интенсивной термической деструкции,
получена формула для расчета такой температуры исходя из химического стро-
ения полимера, выявлены условия опережения термодеструкции полимера его
застекловыванию или плавлению, отмечена необходимость учета образую-
щихся продуктов термодеструкции, которая начинается с распада концевых
групп макромолекул полимера.
В восьмой главе на основании формулы Лоренц-Лорентца получены
уравнения для расчета показателя преломления полимеров и сополимеров
по их химическому строению. Для определения коэффициента оптической
чувствительности по напряжению предложены эмпирический и полуэмпири-
ческий подходы, в которых оценивается вклад каждого атома и типа межмо-
лекулярного взаимодействия соответствующим инкрементом. С использова-
нием полученных зависимостей величины коэффициента оптической чувстви-
тельности по напряжению от химического строения повторяющегося звена
полимера оценен вклад различных атомов и полярных групп на величину та-
кого коэффициента, и предложен полимер с уникальными для метода динами-
ческой фотоупругости свойствами.
В девятой главе приведена схема расчета диэлектрической проницаемос-
ти полимеров и органических жидкостей по их химическому строению, что
важно как для синтеза полимеров с требуемой величиной диэлектрической
проницаемости, так и для прогноза растворимости полимера в органических
жидкостях. Учет не только вклада различных полярных групп в величину ко-
эффициента диэлектрической проницаемости полимеров и жидкостей, но и
различного вклада полярной группы в данном классе жидкостей позволил
получить ранее не достигавшееся согласование экспериментальных и расчет-
ных значений диэлектрической проницаемости для широкого спектра органи-
ческих полимеров и жидкостей.
В десятой главе на основе представлен ия сетчатого полимера в виде упру-
гой и поворотно-изомерной подсистем и с учетом его строения в виде линей-
ных фрагментов и узлов получены формулы для расчета равновесного модуля
высокоэластичности и молекулярной массы межузлового фрагмента полиме-
ра. Последующий анализ полученных зависимостей позволил сформулиро-
вать условия получения полимеров с необычными свойствами - разномодуль-
ных и градиентных, имеющих широкий диапазон изменения равновесного
модуля высокоэластичности при низкой температуре стеклования. Наличие
Введение
17
таких необычных свойств находит экспериментальное подтверждение для
синтезированных сетчатых полиизоциануратов.
В одиннадцатой главе получены аналитические выражения ядер релакса-
ции, необходимые для определения релаксации напряжений и деформаций
ползучести полимеров. При этом рост энтропии релаксирующей системы пред-
ставлен переходом релаксаторов - кинетических единиц полимера различной
природы - в нерелаксаторы вследствие их взаимодействия или диффузии, при-
чем механизм взаимодействия релаксаторов при релаксации напряжения ока-
зался превалирующим. Разработанный аппарат для описания релаксацион-
ных явлений в полимерах применен к описанию процессов сорбции и набуха-
ния. Причем, в отличие от релаксации напряжений, при сорбции преоблада-
ющим является механизм диффузии релаксаторов.
Двенадцатая глава посвящена проблеме повышения предсказания раство-
римости полимеров в органических жидкостях. Показано, что предсказатель-
ная способность критерия растворимости рассчитываемого по химическому
строению полимера и растворителя, резко повышается с учетом типа надмо-
лекулярной структуры полимера и степени его полимеризации.
В тринадцатой главе дана методика расчета важнейшего свойства органи-
ческих жидкостей и полимеров - поверхностного натяжения, исходя из хими-
ческого строения вещества. Развиваемый подход, в отличие от аддитивной
схемы суммирования парохоров,характеризующих вклад отдельных атомов в
поверхностное натяжение, позволяет оценить вклад отдельных полярных групп
и специфического молекулярного взаимодействия в величину поверхностного
натяжения и связать эту величину с параметром растворимости и плотностью
энергии когезии веществ.
В четырнадцатой главе с привлечением идеи “растворимости” одного гомо-
полимера в другом предложен критерий для оценки совместимости полиме-
ров по данным о химическом строении отдельных компонентов. Анализ при-
менения критерия для совместимых, частично-совместимых или несовмести-
мых полимеров показывает высокую его предсказательную способность.
В пятнадцатой главе на примере расчета Ван-дер-Ваальсового объема,
молярной рефракции, теплоемкости и других свойств для ряда полимеров
показана роль химического строения концевых групп макромолекул и важ-
ность их учета при изучении закономерностей изменений свойств полимеров
от их молекулярной массы.
В шестнадцатой главе приведена методика расчета молярной теплоемкос-
ти по химическом^' строению полимеров. В основу методики положено пред-
положение о том, что вклад каждого атома в теплоемкость пропорционален
его Ван-дер-Ваапьсовому объему. Отмечается, что теплоемкость, температу-
ропроводность и теплопроводность полимеров зависит не только от их хими-
18
Введение
ческого строения, но и физического, а также фазового состояний полимерно-
го тела.
В семнадцатой главе описаны методологические приемы решения пря-
мой задачи определения на ЭВМ физических характеристик полимеров и низ-
комолекулярных жидкостей по их химическому строению и обратной задачи -
компьютерному синтезу полимеров с заданным комплексом свойств. Решение
этих задач выполнено методами фрагментов и отдельных атомов. Разработа-
ны соответствующие программы, позволяющие рассчитать свыше 50 хим и-
ческих свойств линейных и сетчатых полимеров и сополимеров, а также ряд
важнейших свойств низкомолекулярных жидкостей. Обсуждается методика
построения диаграмм совместимости свойств полимеров использование ко-
торых может существенно упростить решение прямой и, особенно, обратной
задач компьютерного материаловедения.
В Приложениях продемонстрированы возможности описанного в моно-
графии подхода к определению свойств ряда природных полимеров (пример
решения прямой задачи синтеза полимеров) по их химическому строению
(Приложение 1); поиску химических структур полиэфиркетонов (пример ре-
шения обратной задачи синтеза полимеров), свойства которых должны ле-
жать в заданном интервале (Приложение 2); решению смешанной задачи
синтеза полимеров на примере анализа химического строения фенолоформаль-
дегидной смолы, когда последовательно решается прямая задача - оценка
свойств идеальных структур такой смолы по их химическим формулам, и об-
ратная задача - поиск такого сочетания структур, при котором полученная
химическая формула фенолоформальдегидной смолы обеспечивает экспери-
ментально наблюдаемые значения ее свойств (Приложение 3); анализу струк-
туры и свойств сополимеров, состоящих от трех до пяти сомономеров (При-
ложение 4), а также дается анализ влияния сильного межмолекулярного взаи-
модействия, возникающего между двумя разнородными полимерами, на их
совместимость (Приложение 5).
Глава I
КРАТКИЕ СВЕДЕНИЯ О ТИПАХ ПОЛИМЕРОВ
И ИХ ХИМИЧЕСКОМ СТРОЕНИИ
Огромное число полимеров можно подразделить на три основных класса,
лежащих в основе принятой сейчас классификации. К первому классу отно-
сится обширная группа карбоцепных полимеров, макромолекулы которых
имеют скелет, построенный из атомов углерода. Типичными представителя-
ми полимеров этого класса можно назвать полиэтилен, полипропилен, поли-
изобутилен, полиметилметакрилат, поливиниловый спирт и множество дру-
гих. Фрагмент макромолекулы первого из них имеет следующее строение:
[-сн2-сн2-]й
Ко второму классу относится не менее обширная группа гетероцепных
полимеров, макромолекулы которых в основной цепи помимо атомов углеро-
да содержат гетероатомы (например, кислород, азот, серу и др.). К полимерам
этого класса относятся многочисленные простые и сложные полиэфиры, по-
лиамиды, полиуретаны, природные белки и т.д., а также большая группа эле-
ментоорганических полимеров. Химическое строение некоторых представи-
телей этого класса полимеров выглядит так:
[-сн2-сн2-о-]„
[-(СНЖ-О-С-О-с-О-]Й
о о
[- NH-(CH2)6-NH-C-(CH2)4- с-]„
о о
СН3
[-Si-O-]„
СНз
полиэтиленоксид
(простой полиэфир);
полиэтилентерефталат
(сложный полиэфир);
полиамид;
полидиметилсилоксан
(элементоорганический
полимер);
20
Глава I
Cl
i полифосфонитрилхлорид
[- N = P~ ]„ (неорганический полимер).
Cl
Третий класс полимеров - высокомолекулярные соединения с сопряжен-
ной системой связей. К ним относятся различные полиацетилены, полифени-
лены, полиоксадиазолы и многие другие соединения. Примерами таких поли-
меров могут служить:
[-СН = СН-]„ полиацетилен;
полифенилен;
N-N
р.11 11 _
[—\=/_С С ]„ полиоксадиазол.
О
К этому же классу относится интересная группа хелатных полимеров, в
состав которых входят различные элементы, способные к образованию коор-
динационных связей (они обычно обозначаются стрелками). Элементарное
звено таких полимеров часто имеет сложное строение, как например:
Л 'А
р
CH3-q' YVCH3
Среди многочисленных полимерных материалов наибольшее практичес-
кое применение пока находят материалы на основе представителей первого
класса полимеров - карбоцепных высокомолекулярных соединений. Из кар-
боцепных полимеров можно получить ценнейшие материалы - синтетичес-
кие каучуки, пластмассы, волокна, пленки и т.д., и исторически именно эти
полимеры нашли первое практическое применение (получение фенолофор-
мальдегидных смол, синтетического каучука, органического стекла и др.).
Многие из карбоцепных полимеров стали впоследствии классическими объек-
тами для исследования и создания теории механического поведения полимер-
ных тел (например, полиизобутилен, полиметилметакрилат, полипропилен,
фенолоформальдегидная смола и т.д.).
Краткие сведения о типах полимеров и их химическом строении 21
Впоследствии большое распространение получили материалы на основе
гетероцепных полимеров - полиамидные и полиэфирные волокна, пленки,
лаки, покрытия и другие материалы и изделия. Это дало толчок к исследова-
нию свойств и формированию представлений, в частности, об анизотропных
телах, обладающих совершенно различными свойствами в разных направле-
ниях. Особое место в ряду этих полимеров заняли высокомолекулярные эле-
ментоорганические соединения.
И, наконец, представители третьего класса - полимеры с сопряженной
системой связей - нашли применение для изготовления электропроводящих
материалов.
До сих пор, рассматривая в общих чертах химическое строение полиме-
ров разных классов, мы по существу говорили о структурной формуле повто-
ряющегося звена макромолекулы. Однако наличие множества таких звеньев
в макромолекуле сразу же усложняет картину. Начнем хотя бы с того, что каж-
дое звено в процессе элементарного акта роста макромолекулы может присо-
единяться к соседнему звену по-разному; в этом случае говорят о присоедине-
нии “голова к голове”, “хвост к хвосту” или “голова к хвосту”. Различные
варианты присоединения звена к растущей макромолекуле возможны для не-
симметричных мономеров типа СН2=СН- У которых имеются заместители R
R
у одного из атомов углерода. Здесь возможны варианты “голова к голове”
---сн2-сн-сн-сн2-сн2-сн-сн-сн2—
R R R R
и “голова к хвосту”,
---сн2-сн-сн2-сн-сн2-сн----
R R R
Возможно чередование типов присоединения, т.е. в пределах одной-мак-
ромолекулы звенья могут присоединяться друг к другу различным образом.
Наличие большого количества звеньев в полимерной цепи и возможность всего
лишь нескольких вариантов их присоединения создает огромное количество
изомеров уже по отношению ко всей макромолекуле. Иными словами, поли-
мер может содержать (и действительно содержит)макромолекулы не строго
одинакового химического строения, но смеси большого количества макромо-
лекул, что, конечно, сразу же отличает его от низкомолекулярных веществ,
построенных из совершенно одинаковых молекул.
Не будем говорить о том, что в ряду замещенных предельных углеводо-
родов с возрастанием числа углеродных атомов (т.е. с ростом молекулы) ко-
22
Глава I
личество возможных изомеров быстро увеличивается, и даже при неболь-
шом (по сравнению с полимерами) их числе достигает огромной величины.
Легко представить, что когда количество звеньев достигает многих десятков
или сотен тысяч, число возможных изомеров будет выражаться астрономи-
ческими числами [80].
Вернемся вновь к монозамещенным непредельным углеводородам. При
построении полимерной цепи в процессе полимеризации заместители R мо-
гут располагаться относительно плоскости одинарных связей различным об-
разом. В одном из возможных случаев эти заместители располагаются беспо-
рядочно по обе стороны плоскости; такие полимеры называют нерегулярны-
ми или атактическими:
В других случаях синтез можно провести так, чтобы заместители распо-
лагались либо по одну сторону плоскости главных связей
MRMRMRMRMRMRriRMR
либо по обе стороны, но с правильным чередованием направления заместите-
лей:
Полимеры, построенные из звеньев с регулярно чередующимся направле-
нием заместителей, получили название стереорегулярных. Если заместители
расположены до одну сторону плоскости главных связей, стереорегулярные
полимеры называются изотактическими, если по обе стороны - синдиотакти-
ческими.
Несколько сложнее обстоит дело с полимерами, синтезированными из
дизамещенных мономеров. Уже в самом мономере заместители могут распо-
лагаться по одну сторону плоскости первичных связей (цис-изомер), или по
обе стороны (транс-изомер):
Краткие сведения о типах полимеров и их химическом строении 23
Н Н
Н R'
R Н
R R’
Синтез макромолекул из цис-изомеров приводит к получению эритро-ди-
изотактических полимеров
а из транс-изомеров - трео-диизотактических полимеров
Н pH н н pH ?.Н ?.Н у
L—I | I | I | I Т I Г-Д. Y | .Y | Y /
R hRhRhR hRhR h^hR н
Разумеется, возможны и другие, более сложные модификации, которые
немедленно влекут за собой изменение свойств полимерных материалов.
Материалы, созданные из стереорегулярных полимеров, часто легко кри-
сталлизуются, что дает возможность регулировать их физическую структуру
и свойства.
Здесь мы впервые столкнулись с модификацией свойств полимерных ма-
териалов, к которой приводят практически всякие изменения в химическом
строении макромолекул и физической структуре полимерного тела. Часто фи-
зическая модификация обусловлена изменением химического строения, а иног-
да полностью им определяется.
Один из главных приемов модификации - синтез сополимеров, когда в
реакции участвует не один мономер, а несколько, и поэтому макромолекула
оказывается построенной из разнородных звеньев. Эти звенья могут непре-
рывно чередоваться:
-Л - S-Д- В-А- В-А- В-А-В- чередующийся сополимер,
но чаще всего они располагаются беспорядочно:
-А - А- В - А~ В - В~ А - А~ А ~В~ статистический сополимер.
Звенья могут соединяться также в отдельные блоки, которые затем стыкуют-
ся:
-А -А —А -А —А -В -В -В-В—В — блок -сополимер .
Совершенно очевидно, что в каждом блоке может содержаться различное
число звеньев. Это сразу же сказывается на свойствах будущего полимерного
24
Глава I
кого тела. Сейчас процесс сополимеризации становится регулируемым. За-
бегая несколько вперед, заметим, что механические смеси полимеров и сопо-
лимеры одинакового молярного состава могут часто обладать весьма различ-
ными свойствами, но иногда - практически одинаковыми.
Рассмотренные схемы присоединения звеньев в процессе роста макро-
молекулы отражают лишь случай сополимеризации двух типов мономеров.
И хотя даже в этих простейших случаях реализуется множество сочетаний,
их количество неизмеримо возрастает при переходе к трем мономерам (или
типам звеньев) или к большему их количеству.
Все изображенные нами до сих пор цепи полимеров представляют собой
линейные образования. Однако легко осуществить синтез разветвленных мак-
ромолекулярных цепей. Для этого не обязательно даже вводить в состав цепи
многофункциональные соединения. Разветвление происходит и при полиме-
ризации ненасыщенных углеводородов, вообще не содержащих никаких фун-
кциональных групп. Если не принимать специальных мер, продукты полиме-
ризации этилена, пропилена, изобутилена и других подобных соединений все-
гда будут содержать некоторое количество цепочек, ответвляющихся от
главной цепи. Что касается продуктов поликонденсации (см.выше о поли-
эфирах и полиамидах), то введение трехфункционального соединения в ос-
новную цепь всегда приводит к получению разветвленных полимеров:
..._д_д_д_д-_д_д_д_д_д_д_д_...
I
А
I
А
I
А
I
А
I
Разумеется, полимерное тело на основе разветвленных макромолекул бу-
дет отличаться по структуре и свойствам от тела, построенного из линейных
макромолекул. Однако спешить с заключением о характере физического струк-
турирования разветвленных полимеров не следует. На первый взгляд кажет-
ся, что присутствие больших ответвлений будет препятствовать более плот-
ной упаковке цепей, а также помешают процессу кристаллизации или вообще
упорядочению макромолекул. В одних случаях именно так и происходит. В
других случаях наблюдается прямо противоположная картина. Все зависит от
химического строения главной цепи и ее ответвлений,которое определяет объем
звеньев, силы взаимодействия между ними и между соседними цепями и т.д.
В последние время существенное внимание уделяется структуре и свой-
ствам так называемых дендритных полимеров, макромолекула которых схе-
Краткие сведения о типах полимеров и их химическом строении 25
Рис.1. Схематическое изображение дендритного полимера
Schematic representation of dendric polymer
матически изображена на рис.1 [98, 212]. Ниже мы более подробно остано-
вимся на влиянии типов разветвлений на свойства получаемых полимеров.
Ответвления могут быть построены различно. Они могут содержать те
же звенья, что и в главной цепи. Однако большое распространение получают
“привитые” сополимеры; они образуются прививкой заранее полученных цепо-
чек определенного строения к главной цепи совершенно другого строения:
---А- А-А-А'- А-А-А-А- А- А-А----------
I
В
I
В
I
в
I
в
I
Иногда такую прививку осуществляют многократно.
От разветвленных полимеров легко перейти к трехмерным “сшитым” по-
лимерам. Для этого достаточно увеличить содержание многофункциональных
соединений в цепи полимера. Цепи можно сшить также специальными отвер-
дителями, т.е. соединениями, содержащими активные группы, способные ре-
26
Глава 1
NH2
I
R
NH2
агировать с функциональными группами главной цепи или с концевыми груп-
пами. Классический пример --- отверждение эпоксидных смол:
СН3
• • - о -О - С -О - О- СН2~ СН-СН2
СНз 0
СНз +
--0-О-С-О-0-СН2-СН-СН2
СНз Ч°У
СНз
•-0-O-C-O_°_CH2-CH-CH2-NH
СНз он '
СНз I
• - O-Q- c-©-o-ch2-ch-ch2-nh
СНз ОН
Далее замещается второй атом водорода в диамине и образуется сетка.
Согласно классификации, приведенной в работе [202], существует несколь-
ко основных способов получения сетчатых полимеров:
1) Проведение химической реакции между двумя (или более) различными
функциональными концевыми группами, присоединенными к цепи неболь-
шой молекулярной массы. В результате формируется частая сетка с коротки-
ми цепями между узлами сшивки.
2) Химическое связывание высокомолекулярных соединений по концевым
группам с помощью низкомолекулярного сшивающего агента. В результате
формируется редкая сетка с протяженными линейными фрагментами между
узлами сшивки.
3) Образование сетки за счет сополимеризации двух- и полифункциональ-
ных мономеров. Примером такой сетки является система стирол-дивинилбен-
зол:
сн2-сн-сн2-сн-сн2---
- сн2- сн-сн2- сн- сн2—
4) Вулканизация полимерных цепей путем вовлечения в реакцию функци-
ональных групп, расположенных вдоль основной цепи. Реакция провидится
Краткие сведения о типах полимеров и их химическом строении 27
либо при использовании низкомолекулярного сшивающего агента, либо за
счет радиации и других типов воздействия на функциональные группы.
Добавим к этому и другие возможные (и уже реализованные на практике)
пути получения сетчатых систем.
5) Образование сеток за счет реакции двух (или более) разнородных по-
лимеров по функциональным группам, расположенным вдоль цепи каждого
из полимеров (т.е. в повторяющихся звеньях, а не по концам).
6) Синтез полимерных сеток с помощью реакции полициклотримериза-
ции. С этой целью используются [56, 79, 101, 152] олигомеры с концевыми
группами, способными к образованию циклов в ходе реакции. Примером та-
кой реакции является тримеризация бифункциональных олигомеров (или мо-
номеров), содержащих цианатные концевые группы. Естественно, что возмож-
ны и другие пути получения полимерных сеток.
В последнее время получен новый тип полимеров, которые назвали “ин-
терполимеры” [16,215]. Под интерполимером подразумевают систему, пост-
роенную из двух (или более) разнородных по химическому строению макро-
молекул, химически связанных между собой за счет функциональных групп,
расположенных в повторяющихся звеньях каждой макромолекулы. Схемати-
чески изображение интерполимера показано на рис.2.
Рис2 .С хематическое изображение интерполимера
Schematic representation of interpolymer
Конкретным примером такой системы является, например, продукт взаи-
модействия полистирола с политрихлорбутадиеном:
А1С13
•• • — СН2—СН—•• • + -СН2-СН=СС1-СС12--------*
• ••-ch2-ch=cci-cci-
•-СНг-СН----
28
Глава [
Получение интерполимеров открывает новые возможности модификации
структуры и свойств полимеров.
Другим типом “двухтяжевых” систем являются лестничные полимеры,
примером которых служит полифенилсилсесквиоксан [113]:
О о
I I
— Si-O-Si-O-"'
। ।
О О
— Si-0-Si-O- —
Глава II
УПАКОВКА МАКРОМОЛЕКУЛ
И ПЛОТНОСТЬ ПОЛИМЕРОВ
1. Метод инкрементов и основные
физические представления
Рассмотрев кратко химическое строение полимеров, перейдем к объем-
ному изображению макромолекул, что необходимо для понимания особенно-
стей структурообразования в полимерах. В основу такого рассмотрения по-
ложим представления, развитые А.И. Китайгородским в органической крис-
таллохимии [75]. Согласно этим представлениям, каждый атом описывается
сферой с межмолекулярным радиусом R. Величины этих радиусов определя-
ются по данным рентгеноструктурного анализа идеальных кристаллов орга-
нических веществ. При этом считается, что валентно несвязанные атомы, всту-
пающие в межмолекулярное (а не химическое) взаимодействие, касаются друг
друга по границам сфер. Это схематически изображено на рис.З. Тогда, если
Рис.З. Схематическое изображение двух атомов при межмолекулярном (физическом)
взаимодействии
Schematic representation of intermolecular (Van-der-Waals) interaction of two atoms
30
Глава II
соприкасаются два одинаковых атома, межмолекулярный радиус определится
из соотношения
R = l!2, (1)
где I - расстояние между центрами масс двух одинаковых валентно несвязан-
ных атомов, которые, однако, способны к межмолекулярному физическому
взаимодействию.
Согласно тем же представлениям, химическое взаимодействие двух ато-
мов всегда приводит к их спрессовке, т.к. длина химической связи d, всегда
меньше, чем сумма двух межмолекулярных радиусов:
+ (2)
Это отчетливо видно из рис.4, на котором схематически изображены два
химически связанных атома. Если известны межмолекулярные радиусы R,
для всех атомов, входящих в повторяющееся звено полимера, а также все длины
химических связей между этими атомами, можно легко рассчитать собствен-
ный (Ван-дер-Ваальсовый) объем повторяющегося звена и построить модель
этого звена (или большего фрагмента макромолекулы), в которой объем каж-
дого атома окантован сферой с межмолекулярным радиусом 7?,. На рис.5 по-
казана такая модель фрагмента цепи полиэтилена.
Рис.4. Схематическое изображение двух химически связанных атомов
Schematic representation of two atoms chemically bonded
Рис.5. Модель фрагмента цепи полиэтилена
Model of polyethylene chain fragment
Упаковка макромолекул и плотность полимеров
31
В табл.1 приведены межмолекулярные радиусы для некоторых широко
распространенных атомов, из которых построено большинство полимеров.
Ван-дер-Ваальсовые радиусы R различных атомов
Van-der-Waals radii R of different atoms
Атом R, нм Атом R, HM
С 0,180 Si 0,210
Н 0,117 Sn 0,210
О 0,136 As 0 200
N 0,157 S 0,180
F 0,150 P 0,190
CI 0,178 Pb 0,220
Вг 0,195 В 0,165
I 0,221
Длины химических связей d, для некоторых пар атомов
Chemical bond lengths </, for some pairs of atoms
Связь* dj, нм Связь* dj, нм Связь* d, , нм
C-C 0,154 C-F 0,134 O-F 0,161
C-C 0,148 C-F 0,131 O = N 0,120
C = C 0,140 C-Cl 0,177 O = S 0,144
C = C 0,134 C-Cl 0,164 O = P 0,145
C = C 0,119 C-Br 0,194 N-P 0,165
C-H 0,108 C-Br 0,185 N-P 0,163
C-0 0,150 C-l 0,221 N-P 0,158
С- О 0 J 37 C -I 0?05 S -S 02Ю
C -N Q 140 C-B Q 173 S - As Q 221
C-N 0,137 C-Sn 0,215 S = As 0,208
C = N 0,131 C-A s 0,196 Si-Si 0,232
C =N 0 J27 C - Pb 0,220 P- F 0,155
C-N 0,134 H-O 0,108 P-Cl 0,201
CaN 0,116 H-S 0,133 P-S 0,181
C-S 0,176 H-N 0,108 B-B 0,177
C-S 0,156 H-B 0,108 Sn-Cl 0,235
C-Si 0,188 o-s 0,176 As-Cl 0,216
C-Si 0,168 O-Si 0,164 As-As 0,242
* Если одна и та же пара атомов связана единичной связью, то более длинная связь соответствует
присоединению данного атома к алифатическому атому углерода; более короткая связь соответствует
присоединению того же атома к ароматическому атому углерода.
В табл.2 приведены длины связей для различных комбинаций атомов, так-
же характерных для большинства существующих полимеров .Зная эти вели-
чины, можно рассчитать объем повторяющегося звена практически любого
из полимеров. Чтобы проделать это, необходимо предварительно определить
собственный объем каждого атома, входящего в повторяющееся звено. Расчет
проводится по формуле
32
Глава II
Упаковка макромолекул и плотность полимеров
ДГ,=|лЛ3 -^^(JR-h,), (3)
/
где Д К, - инкремент собственного (Ван-дер-Ваальсового) объема данного ато-
ма; R - межмолекулярный радиус этого атома; Л, - высота шарового сегмента,
который отсекается на данном атоме соседним, химически связанным с ним
атомом. Величина Л( вычисляется по соотношению
Ван-дер-Ваальсовые объемы атомов
Van-der-Waals vol umes of atoms
R1 + J,2 - 7?2
(4)
где Ri - межмолекулярный радиус соседнего, валентно связанного атома; -
длина химической связи (см.рис.4).
Инкременты объемов различных атомов и атомных групп приведены в
таблице 3. Совершенно очевидно, что объем данного атома зависит от его
окружения, т.е. от вида атомов, которые химически к нему присоединяются.
Чем больше объем соседнего, химически связанного атома, и чем меньше длина
химической связи, тем больше спрессовывается данный атом.
Определив инкременты объемов Д Vt всех атомов, входящих в повторяю-
щееся звено полимеров, можно рассчитать относительную долю занятого объе-
ма в общем объеме полимерного тела. В случае полимеров расчеты удобно
вести исходя из молярных объемов повторяющегося звена, поскольку поли-
меры всегда полидисперсны (т.е. содержат макромолекулы различной дли-
ны), а также потому, что при большой длине макромолекулы влиянием конце-
вых групп можно пренебречь. Тогда собственный молярный объем будет ра -
вен Исобста = Na Д Vt , а общий молярный объем = М/р, где р - плотность
I
полимерного тела; М- молекулярная масса повторяющегося звена; NA - чис-
ло Авогадро. Многочисленные эксперименты и расчеты показывают, что во
всех случаях соблюдается условие Гсобств < Иобщ Таким образом, в первом
приближении объем полимерного тела можно разделить на две части: соб-
ственный (Ван-дер-Ваальсовый) объем атомов, который они занимают в твер-
дом теле, и объем пустот, который определится как разность между Иобщ
и Исобств Представляет большой интерес определение доли занятого объема
или, по терминологии, принятой в органической кристаллохимии, коэффици-
ент молекулярной упаковки к:
г собств.
М / р
(5)
5.0
8.1
4.7
9.0
13.1
4.5
8.7
12.8
8.4
Таблица 3
4.2
8.4
34
Глава II
Упаковка макромолекул и плотность полимеров 35
таблица 3 (продолжение)
таблица 3 (продолжение)
o' \|
20.3
8.4
6.5
6.3
6.0
7.2
8.1
7.9
5.8
10.6
14.7
14.6
1 ri3
к
11.0
О С
10.8
12.2
16.2
36
Глава II
таблица 3 (продолжение'}
Упаковка макромолекул и плотность полимеров
37
таблица 3 (продолжение)
Глава II
таблица 3 (продолжение}
Упаковка .макромолекул и плотность полимеров
39
таблица 3 (продолжение')
As S As . As Pfc, .As &
121 >С) ХТ|'/ 122 nz 123 124 'n'
sj 5
S 11.5 S 10.9 c* S 10.4 C 2.0
125 'r'1 126 (h'j 127 \H > 128 (Й)
CO о я S$j s|
° 4.7 S 3.2 N 35 B 3.0
129 Cx 'О) 4 z' ,C cs 130 '4°J zc cs 131 >у/ '^} cx 132 \°J
3.4 2.7 2.1 5.6
133 cx zH sx 134 \^p \^z ,c Si 135 , >/Si 'ч° I Px 136 's0J
5.2 3.3 0.5 5.4
137 Px <*, & 3.2 ,c P 138 4-^ 'x°j 2.4 139 'k°J 4 C 5.8 140 4 N 7.2
40
Глава П
таблица 3 (продолжение)
12.9
16.5
Упаковка макрамолекул и плотность полимеров
41
таблица 3 (продолжение)
15.3
163 's')
18.9
С 19.7
17.7
169
15.6
14.7
16.0
17.2
's')
As
20.3
20.3
27.6
24.9
174
29.5
33.0
5.1
14.8
19.6
42
Глава ll
таблица 3 (продолжение)
187 ip) 188 '""f'j
4 ✓ \ ✓
§ S
C 8.9 P 9.3
Упаковка макромолекул и плотность полимеров
43
таблица 3 (окончание)
206
207
210
Естественно, что величина к для одного и того же полимера будет зави-
сеть от температуры и от физического состояния, в котором полимер находит-
ся, поскольку от этого зависит величина р. Расчет, проведенный для большого
числа аморфных монолитных полимеров, находящихся в стеклообразном со-
стоянии, показал, что в первом приближении величина к является постоянной
и практически не зависит от химического строения полимера [41]. Переход от
полимеров простого химического строения к полимерам очень сложного хи-
мического строения не приводит к какому-либо существенному изменению
доли занятого объема (т.е. величины к).
В табл. 4 показано химическое строение и приведены численные значе-
ния коэффициентов молекулярной упаковки для некоторых стеклообразных
полимеров. Из этой таблицы видно, что величины к для каждого из них дей-
ствительно в первом приближении одинаковы. Чтобы более наглядно проде-
монстрировать этот экспериментальный факт, на рис.6 показана зависимость
плотности р для различных полимеров от отношения Л//Л^^ДИ,- Из рис.6
44
Глава II
ii
Значения коэффициентов молекулярной упаковки к
для ряда стеклообразных полимеров
Values of the coefficients of molecular packing к for some glassy polymers
Структурная формула повторяющегося звена полимера Объём звена. см3/моль Коэффициент упаковки к
СНз -СНз-С- СНз 41,6 0,678
н -снг-с- C=N 32,6 0,682
СНз -сн2-с- С-О-СНз о 58,5 0,684
СНз - СН2- с— с—о—с2н5 о 69,1 0,680
СНз -с-о-О-с-О-о- II 1 о СНз 144,3 0,679
”С-п-с-°-Оч уО-о- II II с ° ° 234,7 0,679
-c-0-=-o-OvX>-°- ° 0 о''о 263,1 0,680
-CHCHiPa-C-HN-CX NH- II II с О О \ О NH % 277,5 0,688
Упаковка макромолекул и плотность полимеров
45
Рис.6. Зависимость плотности р от Л/ / МдУ, ДИ,
i
Dependence of density p on MI N \ &Vj
i
отчетливо видно, что все экспериментально определенные величины р хоро-
шо укладываются на одну и ту же линейную зависимость от отношения массы
атомов к их объему. Тангенс угла наклона этой прямой и представляет, в соот-
ветствии с уравнением (5), коэффициент молекулярной упаковки, который в
случае аморфных монолитных систем выступает в роли универсальной кон-
станты. Если это так, то плотность полимера р может быть рассчитана по
уравнению
которое непосредственно вытекает из (5) при условии kcp = const. В случае
аморфных монолитных полимеров кср = 0,681.
Таким образом, изменение химического строения полимера не может су-
щественно повлиять на долю занятого объема в аморфном полимерном теле,
а сама величина плотности р зависит только от соотношения массы и объема
повторяющегося звена.
Совершенно ясно, что речь идет сейчас об истинных монолитных телах
аморфной структуры. Реально можно сформовать полимерное тело с любой
пористостью, и тогда коэффициент к бутст принимать самые различные зна-
46
Глава II
чения. Однако в этом случае понятие плотности упаковки, количественно оце-
ниваемой величиной к, теряет свой обычный смысл, и рассчитывать эту вели-
чину следует только для материала стенок пор. К этому вопросу мы вернемся
ниже, когда будем рассматривать параметры пористой структуры полимеров,
определяемые методом сорбции.
Для сополимеров уравнение (6) записывается в виде:
=_________+а2^2 +- + а„М„)__________________
Р-^д[а1(ХЛ'//)1+а2(Хд^)2+- + ал(Хд^)л]’ (
/ i i
где аь а2, .... ап - молярные доли компонентов 1,2,...,и; М\, М2, Мп-
молекулярные массы повторяющихся звеньев тех же компонентов; (^ ДИД],
(^ДИД2,..., - их Ван-дер-Ваальсовые объемы. '
i i
В более компактной форме соотношение (7) имеет вид:
к=п
kcPYakMk
®
£аА(£ДИД*
4=1 i
где ак, Mh, (^ ДИД* - соответственно молярная доля, молекулярная масса и
i
Ван-дер-Ваальсовый объем /t-го компонента.
Если мы хотим выразить плотность сополимера через плотности р1;
Р2,..., р„ гомополимеров на основе компонентов 1,2,..., п, то соотношение (7)
приобретает вид
ot]A/| + а2^2 +--- + <хлА/и
(при этом следует учесть, что cq + а2 + ... + а„ = 1).
В компактной форме соотношение (9) выглядит следующим образом:
к=п
Y<*kMk
4=1
к Р/г
(Ю)
Упаковка макромолекул и плотность полимеров_______47
Соотношения (7)—(10) могут использоваться и для расчета плотности со-
вместимых смесей полимеров.
Перейдем теперь к температурным зависимостям коэффициентов моле-
кулярной упаковки стеклообразных полимеров. Расчет значений k при разных
температурах проводится по формулам, вытекающим из соотношения (5)
к(Т) =--------, (Т < Т„); (11)
MVg[\ + aG(T-Tg)]’ 1 sh
к(Г) =--------, (Т>т± (12)
MVg[\+aL(T-Tg)]’ V
где - удельный объем полимера при температуре стеклования Tg; aG»aL-
коэффициенты объемного расширения полимера соответственно ниже и выше
температуры стеклования.
Расчеты по уравнениям (11) и (12) показывают, что температурные зави-
симости коэффициентов молекулярной упаковки имеют вид, изображенный
на рис.7. Примечательное свойство этих температурных зависимостей зак-
к
Рис.7. Температурные зависимости коэффициентов молекулярной упаковки к для ряда
полимеров: 1 .- поли-н-бутилметакрилат; 2 - поли-н-пропилметакрилат; 3 - поли-
этилметакрилат; 4 - полистирол; 5 - полиметилметакрилат; 6 - поликарбонат на основе
бисфенола А
Temperature dependences of the coefficients of molecular packing к for a series of polymers:
1 - poly(n-byty) methacrylate), 2 - poly(n-propy! methacrylate), 3 - polyt ethyl methacrylate),
4 - polystyrene, 5 - poly(methyl methacrylate), 6- polycarbonate based on
bisphenol A
48
Глава II
лючается в том, что коэффициент молекулярной упаковки действительно в
первом приближении одинаков для всех монолитных полимеров при любой
температуре ниже точки стеклования. Во втором, более точном приближении
коэффициент молекулярной упаковки одинаков для каждого полимера при его
температуре стеклования. Эта величина kg ~ 0,667.
Учитывая, что удельный объем при температуре стеклования Tg равен
Уг = \/рг =--
8 ' 8 kgM
(13)
где pg - плотность полимера при Tg ; подставляя (13) в (11) и (12), получаем
[l + aG(7’-Tg)]’
{T<Tg\
(И)
к(Т) =-----------,
[l + a^r-Tg)]
(Г> Tg).
(15)
Уравнения (14) и (15) можно использовать для получения соотношений,
описывающих температурные зависимости плотности полимеров р в стекло-
образном и высокоэластическом состояниях .Для этого подставим (14) и (15)
в уравнение (6):
_______g__________
[l + aG(r-rg)]y^AK,. ’
(T<Tgy,
(16)
кгМ
Р(П — /т т w Улг’ (Г>^- (17)
[l + ai(7’-7’g)]y^XAK/
Поскольку, как это будет видно из дальнейшего изложения, величины ко-
эффициентов объемного расширения aG и aL, а также температуру стеклова-
ния Tg можно рассчитать на основании химического строения повторяющего-
ся звена полимера, температурные зависимости плотности р(Т) также могут
быть рассчитаны по соотношениям (16) и (17).
В заключение отметим, что постоянство коэффициента молекулярной упа-
ковки к справедливо только для аморфных монолитных тел построенных из
Упаковка макромолекул и плотность полимеров
49
полимеров. В случае кристаллических полимерных тел картина существенно
меняется. Если провести расчет коэффициентов молекулярной упаковки для
идеальных полимерных кристаллов, воспользовавшись данными рентгено-
структурного анализа, можно убедиться, что коэффициенты молекулярной
упаковки кристаллических полимеров, в отличие от аморфных, принимают
самые разнообразные значения. Наименьшие величины к характерны для али-
фатических систем с объемистыми боковыми привесками, например, для поли-
4-метилпентена-1 и поли-н-бутиральдегида.Наибольшие коэффициенты упа-
ковки характерны для 1,4-транс-р-полиизопрена и полихлоропрена.
В табл.5 приведены в качестве примера кристаллографические значения
плотностей и коэффициенты молекулярной упаковки для ряда характерных
кристаллических полимеров. Хорошо видно, что значения к для них колеб-
лются в широких пределах. Таким образом, кристаллические полимеры име-
ют весьма широкую кривую распределения по коэффициентам молекулярной
упаковки (рис.8).
Рис 8. Кривая распределения коэффициентов молекулярной упаковки к для кристал-
лических полимеров
Curve of distribution of the coefficients of molecular packing к for crystalline polymers
Таблица 5
Коэффициенты молекулярной упаковки к для ряда кристаллических полимеРов
Coefficients of molecular packing к for a series of crystalline polymers
Название Тип элементарной ячейки Химическая формула р, г/см3 к
1 2 3 4 5
Полиэтилен Ромбическая Псевдомоноклинная Триклинная -сн2-сн2- 1,0°° 1,014 0,9й5 1.013 0,736 0,746 0,710 0,745
Полипропилен : -изотактический -синдиотактически^ Моноклинная Моноклинная —сн2~сн — СН3 0,9зй 0^10 0,693 0,674
1,2-поли(бутаДиен): -синдиотактический -изотактический Ромбическая Ромбоэдрическая -сн2- сн- \ сн II сн2 0,9й3 0,9й0 0,692 0,690
1.4-транс- поли(бутадиен) Псевдогексагональная -сн2-сн=сн-сн2- 1,020 0,733
1,4-цис-поли(бутадиен) Моноклинная - сн2- сн=сн-сн2- 1.0|0 0,726
1,4-цис-полиизопрен Моноклинная -сн2-сн=с-сн2- сн3 1000 0,725
Полихлоропрен Ромбическая -сн2-сн=с-сн2- CI 1,65*7 0,893
таблица 5 (продолжение)
1 2 3 4 5 Упаковка макромолекул и плотность полимеров
Полиэтилентерефталат Триклинная -0-СН2-СН2-0-С-О-С- z II II о о I,455 0,776
11олигексаметилен- терефталат Триклинная -0-(СН2)6-0-С-О-С- 0 о и3’ 0,652
Полиэтиленизофталат Триклинная -0-СН,-СН2-0-С'-О II 1 0 ? о 1,358 0,724
Полиэтиленадипат Триклинная -О-(СН2)2-О-С-(СН2)4-С- о О 1,274 0,782
Полиамид 6,6: а - изомер Р - изомер Триклинная Триклинная -C-(CH2)4-C-HN-(CH2)6-NH- II II о О 1,240 1,248 0,764 0,769
Полиамид 6,Ю Триклинная -C-(CH2)b-C-HN-(CH2)6-NH- II II о О I,157 0,740
to
таблица 5 (продолжение)
1 э 3 4 5
Полиамид 6 Моноклинная -C-HN-(CH2)5- 0 1,230 0,758
Полиамид 11 Триклинная -C-HN-(CH2)10- 0 1,192 0,789
Поли-4-метилпентен-1 Тетрагональная -сн2-сн- сн2 сн-сн3 сн3 0,813 0,598
Поливинилхлорид Ромбическая Моноклинная -сн2-сн- CI 1,440 1,455 0,680 0,687
Политетрафторэтилен Псевдогексагональная Гексагональная -cf2-cf2- 2,400 2,360 0,794 0,781
Поливинилфторид Гексагональная -сн2-сн- F 1,440 0,742
Поливиниловый спирт Моноклинная -сн2-сн- он 1,350 0,770
£}
ct
Ci
таблица 5 (окончание)
1 2 3 _ 4 5
Полиакрилонитрил Ромбическая -сн2-сн- C = N 1,110 0,677
Полиметилметакрилатиз отактический Псевдоромбическая СН3 -сн2-с- с-с-сн3 II о 1,230 0,719
Полистирол Ромбоэдрическая -сн2-сн- 0 1,120 0,711
Полиоксиметилен Гексагональная -сн2-о- 1,506 0,808
Полиэтиленоксид Гексагональная -СН2-СН2-О— 1,205 0,723
Полипропиленоксид Ромбическая -сн2-сн-о- сн3 1,102 1,154 0,663 0,694
Упаковка макромолекул и плотность полимеров
54
Глава //
2. Связь между свободным объемом полимеров, коэффициентом
молекулярной упаковки и пористой структурой
Прежде чем приступить к изложению связей между упомянутыми физи-
ческими характеристиками, необходимо кратко остановиться на понятии “сво-
бодный объем”. Имеются три определения свободного объема:
1) Свободный объем представляет собой разность между истинным мо-
лярным объемом VM тела и его Ван-дер-Ваальсовым молярным объемом
' = (18)
Величину Д V, полученную таким образом, часто называют “пустым объе-
мом” (empty volume). Естественно, что пустой объем зависит от температуры ,
т.к. от нее зависит молярный объем VM = М/р. Подставляя это соотношение в
(16) и (17), получаем
i
} + aG(T-Tg) t
(T<Tg)-
(19)
(T> Tg) . (20)
Соотношения (19) и (20) описывают температурные зависимости пусто-
го объема.
2) Свободный объем представляет собой разность между объемами тела
при абсолютном нуле и при заданной температуре; иными словами, свобод-
ный объем представляет собой избыточный объем, возникающий в результате
теплового расширения тела. Это определение свободного объема является
наиболее употребительным. Кроме того, данный свободный объем подразде-
ляют на флуктуационный свободный объем и объем расширения.
3) Свободный объем представляет собой разность между объемом поли-
мерного тела при заданной температуре и объемом идеального кристалла,
построенного из полимера того же химического строения. Это понятие сво-
бодного объема используется крайне редко.
Теперь перейдем к анализу связи между свободным объемом полимеров,
коэффициентом молекулярной упаковки и пористой структурой.
Пористая структура полимеров во многом определяет их свойства. По-
этому следует более подробно остановиться на методах оценки пористой
структуры полимеров и связи ее параметров с такими характеристиками ,как
коэффициент молекулярной упаковки и свободный объем полимера. Дело в
Упаковка макромолекул и плотность полимеров 55
том, что размеры микропор существенно зависят от метода их оценки и, есте-
ственно, что трактовка их природы и связи характеристик микропористой струк-
туры со свойствами полимеров существенно зависит от метода их определения.
Свойства многих блочных и пленочных полимеров во многом зависят от
плотности упаковки макромолекул, а для таких систем, как сор бенты, иониты
и др., которые применяются в гельхроматографии и для изготовления ионооб-
менников, наиболее существенное значение имеют суммарный объем пор,
распределение их по размерам, а также их удельная поверхность.
Приведем определение, данное в работе [68]: “Поры - это пустоты или
полости в твердых телах, обычно сообщающиеся друг с другом. Их форма и
размеры разнообразны и различны, и в значительной степени определяются
природой и способом получения адсорбентов”.
Обычно о характеристиках микропористой структуры судят по экспери-
ментальным данным о равновесной адсорбции, капиллярной конденсации
паров и вдавливании ртути (ртутная порометрия) [121]. В последнее время
находит применение метод аннигиляции позитронов [3,48, ПО, 123,134, 140,
155, 164, 187, 211], с помощью которого можно определять характеристики
микропористой структуры, когда размер пор соизмерим с молекулярными раз-
мерами. Такие микропоры недоступны для проникновения молекул сорбата,
и тем более, они недоступны проникновению ртути при использовании мето-
да ртутной порометрии.
Полимеры и материалы, изготовленные из них, обладают той особеннос-
тью (в отличие от минеральных сорбентов), что в процессе сорбции паров
органической жидкости или воды они набухают. В результате структура их
меняется и, применяя обычные методы расчета, нельзя оценить истинную
пористую структуру исходного материала. Разумеется, что в опытах по сорб-
ции можно использовать пары таких органических жидкостей, в которых по-
лимер не набухает. Тогда можно определять параметры пористой структуры
исходного материала, но такие случаи довольно редки [107].
Перед тем как перейти к сопоставлению параметров пористой структуры
со свободным объемом полимера,необходимо отметить ,что параметры пори-
стой структуры для одного и того же полимера могут быть существенно раз-
личными в зависимости от условий его синтеза и последующей перераб отки.
Так, например, пленка или волокна могут быть получены из раз личных ра-
створителей [81 ], а также из смеси растворителей с осадителем [97], и будут
иметь разную микропористую структуру и свойства. То же самое можно ска-
зать и о материалах, получаемых прессованием и литьем под давлением, а
также с помощью гидростатической экструзии При этом могут образовываться
и макропоры, суммарный объем которых может быть достаточно велик. При-
меняя же специальные методы синтеза, можно получать материалы на осно-
56
Глава ll
ве полимерных сеток, которые имеют гигантскую удельную поверхность и
чрезвычайно большие радиусы пор [115]. Естественно, что такие макропоры
не определяются плотностью упаковки макромолекул. Они могут быть обра-
зованы неплотной упаковкой более крупных образований, чем макромолеку-
лы, или могут быть вызваны проведением химического процесса образова-
ния сетки в специфических условиях [167].
Следует сделать еще несколько общих замечаний. Помимо макропор, в
полимерном теле присутствуют, как это было отмечено выше, микропоры,
размер которых соизмерим с размерами молекул сорбата. Естественно, что в
этом случае молекулы сорбата не могут проникать в такие микропоры (счита-
ется, что для проникновения молекул сорбата в поры, объем последних дол-
жен в несколько раз превышать объем проникающих молекул). Поскольку
молекулы сорбата могут быть различны, т.е. могут иметь разные размеры,то
параметры пористой структуры, определяемые из сорбционных данных, бу-
дут зависеть от типов и размеров молекул сорбирующихся веществ. Поэтому
введены такие термины, как “пористость по азоту” /‘пористость по бензолу”
и т.д. Интересно, что сорбционный метод определения пористой структуры
полимерных тел не может применяться в том случае, когда тело содержит
достаточно крупные макропоры. Это связано с тем, что в условиях полимоле-
кулярной адсорбции, когда происходит образование многих молекулярных
слоев на стенках макропор, их слияние становится затруднительным, то есть
капиллярная конденсация отсутствует. Тогда суммарный объем пор, рассчи-
танный по количеству проникшего в полимерное тело сорбата, будет меньше
истинного объема макропор.
Перед тем как перейти к анализу соотношений между физическими ха-
рактеристиками полимерного тела и его микропористой структуры, введем
ряд определений и обозначений:
5уд - удельная поверхность микропор,
1Е0 - суммарный объем пор,
И'о'™* _ максимальный объем пор, доступных для молекул сорбата любых
размеров (в расчете на 1 г вещества),
У г - свободный объем (в данном случае - объем расширения),
Eg- - “пустой объем” (см. выше),
Ут - удельный объем полимерного тела при данной температуре,
У о - удельный объем полимерного тела при абсолютном нуле,
Уцг - Ван-дер-Ваальсовый объем (в расчете на 1 г вещества),
Еид кр - удельный объем идеального кристалла или монолитного аморф-
ного полимера (под монолитным аморфным полимером подразумевается та-
кой, в поры которого не могут проникать никакие молекулы сорбата).
Запишем ряд соотношений, связывающих эти характеристики:
Упаковка макромолекул и плотность полимеров
57
Уе=Ут-Уо\ (21)
VF — Vy- Vцг (22)
Далее, свяжем эти характеристики с коэффициентом молекулярной упа-
ковки к (см. выше):
k^ViVIVT-, \ -k^--VFiVT (23)
Как было отмечено выше, существуют так называемые непористые сор-
бенты (например, кристаллические тела), в которые не могут без набуха-
ния проникать никакие молекулы сорбата. Естественно, что для таких тел
(Fomax -0. В то же время коэффициенты молекулярной упаковки кристаллов,
как свидетельствуют данные табл.5, находятся в пределах 0,64-0,89. Учиты-
вая, что коэффициент молекулярной упаковки по своему определению пред-
ставляет саб ой долю занятого (Ван -дер -Ваальсового) объема,можно сказать,
что доля пустого (но недоступного) объема составляет 1 - к = 0.11 -0,36. Этот
пустой объем недоступен для проникновения даже самых малых молекул сор-
бата; обозначим его через .Тогда объем идеального кристалла(или моно-
литного аморфного полимера Емон) можно записать как
у = у,,,+ у • у = у,,, у у (24)
у ид.кр. у И7 у Н’ у мои у W у н •
Объем реального полимерного тела (содержащего доступные для сорбата
микропоры) будет складываться из трех частей,
\'Т- Vw+ Ен+ 1Еотах. (25)
Тогда
IL'max = у у И/™х = у у (26)
уу О УТ ' ид.кр.’ гг 0 УТ у мои • х 2
(27)
Коэффициент молекулярной упаковки в монолитной части полимера оп-
ределится из соотношения
к=--------------------------------------
Ут - Л 0,,1ах
В случае оценки плотности упаковки макромолекул для реального полимер-
ного тела, содержащего доступные для молекул сорбата микропоры, коэффи-
циент молекулярной упаковки к следует рассчитывать по соотношению
/с_ Vw
VT-W^ '
(28)
где Ид - суммарный объем микропор (на 1 г вещества), определенный на ос-
новании сорбционных измерений.
Величина 1Еотах, которая представляет собой разность между удельным
объемом тела при данной температуре и объемом истинного монолитного тела,
58
Iлава II
по существу идентична фактору пористости Р = 1 / рк -1 / ри , где р,- - кажу-
щаяся плотность, ри - истинная плотность. При этом рк представляет собой
плотность тела при данной температуре, на которую влияют имеющиеся в
нем поры. Лучше всего кажущуюся плотность измерять для тел правильной
геометрической формы, т.к. не используя никаких растворителей, можно найти
величину р,. делением массы тела на его объем. Если измеряют кажущуюся
плотность тел неправильной формы, можно использовать пикнометреческий
или дилатометрический методы. Трудность здесь заключается в подборе жид-
кости, которая не смачивает поверхность данного тела и не проникает вглубь
него. Истинная плотность ри - это плотность монолитной части материала,
не содержащей пор. Наилучшим образом истинная плотность определяется
для идеального кристалла, поскольку ее можно рассчитать на основании па-
раметров кристаллической решетки. В случае аморфных и частично-крис-
таллических тел можно применять метод градиентных трубок, используя
жидкости, хорошо проникающие в поры. Следует, однако, иметь в виду, что
при создании градиента плотности в трубке используют смеси двух жидко-
стей, и каждая из них может обладать различной смачивающей и проникаю-
щей в поры способностью. Тогда картина искажается и определяемая плот-
ность не будет истинной.
Приведенные выше соотношения позволяют однозначно оценить набуха-
ет ли полимер в данном сорбате, который используется для оценки пористой
структуры полимера. Если в результате опыта оказалось, что величина И’()
больше, чем Wzomax, то это свидетельствует о том, что объем поглощенных
полимером паров больше объема содержащихся в нем пор, тс полимер набу-
хает в процессе сорбции.
Теперь рассмотрим экспериментальные и расчетные данные по определе-
нию параметров структуры полимеров и коэффициентов их молекулярной
упаковки. Эти данные представлены в табл.6. Для идеальных кристаллов по-
лиэтилена Eg = Ия и И’(|тах = 0. Коэффициент молекулярной упаковки является
достаточно большим. Для частично-кристаллического полиэтилена пустой
объем Ея больше, чем в случае идеального кристалла, и при этом часть его
доступна для проникновения малых молекул сорбата Однако суммарный объем
пор, определенный по сорбции метанола, равен всего 0,01 см3/г. Коэффици-
ент молекулярной упаковки для монолитной части такого полиэтилена суще-
ственно ниже, чем для идеального кристалла.
Полимеры, находящиеся в высокоэластическом состоянии, например по-
лиизобутилен, также обладают сравнительно небольшими величинами сво-
бодных объемов и являются практически непористыми сорбентами (Eg= Ен).
В противоположность этому, полимеры, полученные поликонденсацией
или полимеризацией в растворе, обладают огромными значениями 1-Ротзх При
Упаковка макромолекул и плотность полимеров
59
Таблица 6
Параметры пористой стдуклуры и коэффициенты молекулярной упаковки
ряда полимеров
Parameters of porous structure and voeflicicnts of molecul ar packing of a series of polymers
Полимер см3/г И^КС см3/г 0 см’/г Гн. см’/г к
Полиэтилен (степень кристалличности 100 %) -сн2-сн2- 0,26 •0 ~0 0,26 0,736
Полиэтилен (степень кристалличности < 100%) —сн2~ сн2~ 0.35 0 08 0 01 0 27 (1675
Полиизобутилен - сн2- С(СН3)2- 0,36 -0 -о 0,36 0,678
Полиметилиденфтал ид -сн9-с- оО° % полимеризация в растворе диметил- (|юрмамида полимеризация в расплаве 1,28 0,22 1,06 ~0 - 0,22 0,22 0,687 0,687
1 Полиарилат Ф-1 -c-0-c-o-04ZQ-o- 1 > с 0 ° о; > поликонденсация в растворе совола спрессован при 360 °C и давлении 312,5 МПа 0,82 0.24 0,58 ~0 0.31 0 0.24 0,24 0,688 0,688
таком способе синтеза поры образуются вследствие удаления растворителя,
распределенного в объеме синтезируемого полимера. Это видно из того, что
те же полимеры, полученные полимеризацией в расплаве, практически непо-
ристые, и значения Iдля них очень малы, а И/Отах= 0.
60
Глава 11
Образующиеся при синтезе поры могут закрываться при прессовании по-
лимера под большим давлением, и тогда пористый полимер становится непо-
ристым. При этом И'о во всех случаях меньше И/Отах, что свидетельствует об
отсутствии набухания.
Для всех полимеров значения Г„ близки к значениям этой величины, ха-
рактерной для плотности закристаллизованных образцов. Обращает на себя
внимание то, что независимо от способа получения коэффициент молекуляр-
ной упаковки аморфных и аморфно-кристаллических полимеров в их моно-
литной части в первом приближении одинаков и близок к средней величине
кср = 0,681, о чем говорилось выше Для кристаллического образца величин а
к существенно выше.
Рассмотрим еще один интересный вопрос, связанный с молекулярной упа-
ковкой, а именно - изменение объема системы в процессе полимеризации, т.е.
при переходе от мономера к полимеру.
Известно, что переход от мономерной жидкости к твердому стекло-
образному' полимеру сопровождается заметной контракцией, т.е. умень-
шением объема [76J. Удельный объем полимера V„ всегда меньше удельного
объема мономера Им, и их разность ДИ = Ип - 1Д, < 0. Одной из причин кон-
тракции является замена более длинных межмолекулярных связей, имеющихся
в жидких мономерах на более короткие химические связи, образующиеся меж-
ду молекулами мономера в полимере. При этом уменьшаются собствен-
ные Ван-дср-Ваальсовые объемы атомов вследствие их “спрессовывания”
(см. выше). Но это не единственная причина контракции. Из рассмотрения
экспериментальных значений удельных объемов следует, что име ется и дру-
гая причина контракции - более плотная упаковка полимерных це пей по срав-
нению с упаковкой мономерных молекул. Об этом свидетельствует тот факт,
что коэффициенты упаковки полимеров всегда больше, чем их мономеров
(*п > А'м)-
Представим общую контракцию АУо6|Ц в виде суммы двух величин. АИ] -
контракции, обусловленной заменой межмолекулярных связей на химичес-
кие, и АР 2 - контракции, обусловленной более плотной упаковкой цепей,
АИоб1Ц=ДГ1+АП2 (29)
и оценим каждое из слагаемых.
Для этого рассчитаем значения удельного объема полимера в предполо-
жении, что он имеет тот же коэффициент упаковки, что и мономер км ,те.
Упаковка макромолек\>л и плотность полимеров
61
где (^ЛЦ )П - Ван-дер-Ваальсовый объем атомов в повторяющемся звене
/
полимера; Л/- молекулярная масса звена. Рассчитанные таким образом зна-
чения У'п для некоторых полимеров представлены в табл. 7. Они всегда боль-
ше экспериментально измеренных значений удельных объемов полимера УП.
Разность между и Им представляет собой величину
ДИ^ДИ'-Д^, (31)
а остальная часть контракции вычисляется по формуле
Л1/2=А1/об,ц-Л1/1. (32)
Относительные доли контракции определяются из соотношений:
Ц] = ДР^ / Д/об1Ц ; (33)
а2=ДГ2/ДПоб1Ц. (34)
Из данных табл.7 следует, что во всех случаях меньшая доля контракции
обусловлена раскрытием двойных связей, большая - плотной упаковкой по-
лимерных цепей. При этом химическое строение мономера и соответствую-
щего полимера оказывает существенное влияние на величины Ц] и а2.
В ряду полиакрилатов и полиметакрилатов с увеличением объема боково-
го заместителя а2 возрастает, а затем уменьшается. Уменьшение эффекта плот-
ной упаковки цепей, по всей вероятности, обязано стерическим затруднени-
ям.
Таким образом, из изложенного выше следует: что понятия пористости и
плотности упаковки неадекватны. Пористость практически всегда отражает
пустоты, имеющие размеры больше молекулярных, т.е. достаточно крупные
пустоты. Что касается плотности упаковки самих макромолекул, то о ней можно
судить, рассматривая только непористую часть образца. Как было отмечено
выше, для анализа микропористой структуры полимеров предпочтительно
применение методов аннигиляции позитронов [3, 48, ПО, 123, 134, 140, 155,
164, 187, 211], с помощью которых можно получить качественную и количе-
ственную информацию о характеристиках субмикропор (2—15 А) в полиме-
рах.
Р ассмотрим результаты изучения процессов аннигиляции позитронов в
двух полимерах, которые являются хорошими моделями предельных харак-
теристик плотности упаковки макромолекулярных цепей. Одним из них яв-
ляется полиимид, характеризующийся высокоупорядоченной, квазикристал-
Изменения в объёме системы при полимеризации
Changes in volume of the system as a result of polymerization
to
Таблица 7
Полимер (мономер) см’т см’/г ’ п , см’/г общ см7г ДС, см’/г ди2 см’/г а I, % а г, %
I 2 3 4 5 6 7 8 9
сн3 -СН2-С- С-О-СНо II J О 1,068 0,855 0,968 0,213 0,080 0,133 37,6 62.4
сн3 -сн2-с- С-О-С,Нс и z 0 О 1.102 0,890 1,031 0,212 0,071 0,141 33,5 66.5
сн3 -сн2-с- С— О-С3Н7 II J ' 0 1.109 0,928 1,045 0,181 0,064 0,117 35,4 64,6
таблица 7 (окончание)
1 2 3 4 5 6 7 8 9
-сн2-сн- с-о-сн3 II J О 1,046 0,815 0,951 0,231 0,095 0,136 41.1 58,9
-сн2-сн- С-О-С2Н5 II * a О 1,082 0,873 1,000 0,209 0,082 0.127 39,2 60,8
-СН2-СН- С—О—С4На II 4 a о 1.098 0,952 1,036 0,146 0,062 0.084 42,5 57,5
- сн2-сн- о-с-сн3 II О 1,073 0,841 0,976 0,232 0,097 0,135 41,8 58,2
- СН2-СН- 1,104 0,942 1,028 0,162 0,076 0,086 46,9 53.1
Глава 11 Упаковка макромолекул и плотность полимеров
64
Глава I!
лической структурой, а вторым - поли-( 1 -триметилсилил-1 -пропин) (ПТМСП),
который, напротив, характеризуется малым коэффициентом молекулярной
упаковки.
Рассмотрим структурные изменения в ПТМСП, которые происходят при
его долговременной выдержке при комнатной температуре после синтеза.
Для сравнения приведем данные по аннигиляции позитронов и для ряда
других модельных полимеров. Химическое строение всех упомянутых выше
систем приведено ниже.
Поли-(1-триметилсилил-1-пропин)
СН3
[-с=с-]„
СН^ ^'"СНз
сн3
Полиизопрен
[-СН2-С=СН-СН2-]„
СН3
Полидиметилсилоксан
СН3
[-o-si-o-L
СН3
Полистирол
[-СН2-СН-]„
О
П ол итетрафторэтиле н
[~CF2-CF2-]„
Полиимид
О О
Il II
II И
о о
Наблюдение аннигиляции позитронов в ПТМСП проведено с помощью
методики регистрации спектров времени жизни позитронов (измерения про-
ведены С.А. Тишиным; данные не опубликованы). Измерения проводились
Упаковка макромолекул и плотность полимеров 65
на термостабилизированном спектрометре, реализующем традиционную бы-
стро-медленную схему регистрации, с отобранными и оптимизированными
по оригинальной методике [111] временными фотоумножителями.
Обработка экспериментальных спектров проводилась с помощью извест-
ных программ “Resolution" и “Positron FIT'.
В табл.8 приведены результаты выделения параметров долгоживущего
компонента при трехкомпонентном разложении спектров времени жизни по-
зитронов для ПТМСП, полиимида, полистирола, полидиметилсилоксана и
политетрафторэтилена. Видно, что ПТМСП обладает аномально большим
временем жизни атома орто-позитрония, с аннигиляцией которого посредством
pick-off- распада связывается происхождение долгоживущего компонента спек-
тра времени жизни в полимерах [3, 48, НО, 123, 134, 140, 155, 164, 187,211].
Ранее в полидиметилсилоксане и тефлоне наблюдались наибольшие значения
времени жизни долгоживущего компонента td [123, 164] в твердых полиме-
рах. Сравнение с результатами измерений на модельных полимерах показы-
вает (см.табл.8), что ни наличием ненасыщенной связи, ни наличием массив-
ной боковой группы или атома кремния в отдельности нельзя объяснить столь
высокое значение td для ПТМСП.
Таблица 8
Параметры долгоживущего компонента спектра времени жизни позитронов для ряда
полимеров, а также расчетные значения радиуса R и объёма Имикропор
Parameters of the longest component of positron lifetime spectrum for a series of polymers and
rated values of radius R and volume Hof micropores
Образец T/;+0,03, нс 1о ±0,25, % Я0,А Я, А Г, А’ £,эв
ПТМСП 5,78 38,4 6,76 5,10 416,5 0,41
Политетрафторэтилен 4,27 21,6 6,05 4,39 265,8 0,5 Г
Полиметилсилоксан 3,23 41,3 5,45 3,79 170,9 0,63
Полиимид 2,77 38,1 5,14 ' 3,48 132,1 0,71
Полистирол (атактический) 2,05 40,5 4,56 2,90 76,7 0,90
Можно сделать два предположения о причинах аномально высокого сред-
него времени жизни позитрония в ПТМСП.
Во-первых, молекулярное строение повторяющегося звена позволяет пред-
положить, что высокая концентрация массивных, малоподвижных боковых
групп создает пористую структуру с размером пор порядка величины Ван-
дер-Ваальсового объема -Si^CgHg бокового фрагмента.
Во-вторых, размер пор может быть связан с большим временем релакса-
ции синтезированного ПТМСП при комнатной температуре. Можно предпо-
66
Глава II
дожить, что образование и эволюция микрополостей большого размера дол-
жны быть обусловлены движением крупных сегментов макромолекул или даже
структурных фрагментов с большим временем перегруппировок.
Величина времени жизни атома орто-позитрония относительно pick-off-
аннигиляции позволяет оценить размер микрополости, в которой он локали-
зовался до аннигиляции [140]. Результаты расчетов также приведены в табл.8.
В соответствии с моделью [ 140] позитроний рассматривается в сферичес-
кой яме, окруженной слоем электронов толщиной А/?. Для волновых функций
в сферических координатах
W) =
(2тг7?о) 1/2 -y-sin(nr/ в яме;
0 вне ямы.
(35)
Вероятность пребывания позитрония за пределами плотности будет:
^(/?) = l-A + J_sin(2Z^) (36)
R0 2л Ro
где R = Rii AR.
Предполагая, что скорость аннигиляции орто-позитрония внутри элект-
ронного слоя составляет 0,5 нс1, усредненное по спинам значение скорости
распада будет:
Ао=1/тд=21Т(Д)
(37)
с константой AR =1,66 А, подобранной эмпирически для твердых тел.
Рассмотрим результаты измерения пористой структуры пленок ПТМСП в
результате их старения.
Исследования долговременной релаксации пленок ПТМСП проведены с
помощью измерения спектров времени жизни позитронов. Как видно из табл .9
Таблица 9
Долговременная релаксация ПТМСП поданным измерения параметров долгоживущего
компонента спектра времени жизни позитронов
(Т„ - время жизни промежуточного компонент а)
Long-term relaxation of РТМТР from the data of measurement of the longest component
parameters of positron lifetime spectrum (T„ - is lifetime of intermediate component)
Длительность старения,сутки T/, + 0.03, ис Л/т ±0,25, % Т„ ±0,080,нс
13 5,78 38,40 0,687
17 5,68 37,53 0,607
24 5,72 38,09 0,678
83 5,40 38,08 0,507
210 5,09 37,91 0,453
Упаковка макромолекул и плотность полимеров
67
Рис.9. Зависимость размеров R позитронийчувствительной микрополости от времени
выдержки tc при 25 °C для ПТМСП
Dependence of sizes R of the positron-sensitive microcavity to time of exposition tc
at 25 °C for PTMTP
и рис.9, на которых приведены некоторые характеристики разложения вре-
менного спектра на три компонента и расчетный радиус микропор Л,а также
длительность старения образцов, наблюдается уменьшение времени жизни
долгоживущего компонента с увеличением времени выдержки ПТМСП при
комнатной температуре. Интенсивность долгоживущего компонента практи-
чески не зависит от времени релаксации.
Наблюдаемый результат связывается с медленной структурной релакса-
цией, а не со “старением” (если под “старением” понимать появление разры-
ва основной цепи), так как последний процесс обычно сопровождается изме-
нениями интенсивности /0 (в качестве примера можно привести результаты
наблюдения долговременного старения полиэтилена методом измерения вре-
мени жизни позитронов, хотя в полимерах “старение” обычно очень специ-
фичный процесс).
Учитывая связь величины Тд с радиусом микропор в полимерах [140],
следует заключить, что при долговременной релаксации ПТМСП наблюдает-
ся уменьшение размеров пор (см.рис.9) и, вероятно, снижение подвижности
макромолекулярных цепей из-за уменьшения свободного объема.
Концентрация ловушек позитрония, как следует из неизменности величи-
ны /д, не зависит от времени выдержки в исследованном интервале времен.
Теперь остановимся на результатах исследования аннигиляции позитро-
нов в полиимиде.
68
Глава 11
Как показали проведенные измерения [48], процесс аннигиляции позит-
ронов в полиимиде существенно отличается от обычно наблюдаемого в боль-
шинстве полимеров. Аннигиляционный спектр в полимерах характеризуется
наличием, как правило, трех или четырех компонентов со средними времена-
ми жизни от 100 пс до 4 нс [54, 164, 187]. Однако для полиимида наблюдается
иная структура спектра, где обнаружен лишь один, причем короткоживущий,
компонент с то = 0,388 нс (рис .10) .Временное распределение хорошо аппрок-
симируется одной распадной прямой, угол наклона которой определяет сред-
нее время жизни.
Рис.10. Спектр времени жизни позитронов т в исходной полиимидной пленке
(N — число отсчетов в канале)
Positron lifetime spectrum т of the starting polyimide film (Here N is a number
of readings in a channel)
Величина времени жизни и структура спектра позволяют предположить,
что аннигиляция в полиимиде происходит из позитронного состояния (Ёз о б
разования атома позитрония, как это свойственно для металлов и полупро -
водников с высокой подвижностью электронов и упорядоченной кристалли-
ческой структурой.
В этом смысле полиимид образует уникальную для полимеров электрон-
ную структуру, характеризующуюся высокими показателями величин и сте-
пени однородности функции плотности электронов.
По отношению к взаимодействию с позитронами микроструктура исход-
ной (недеформированной) полиимидной пленки является бездефектной. Од-
нако после деформации временные спектры меняются (рис. 11 и табл. 10). Вме-
сто одного в деформированном образце наблюдаются два компонента: с мень-
Упаковка макромолекул и плотность полимеров
69
Рис.11. Времена жизни т и интенсивности компонентов (%) в спектрах для исходного
(I) и подвергнутых предварительному растяжению образцов полиимида, измеренных
при длительности отдыха 1 (И) и 24 (III) часа
Lifetimes т and intensities of components (%) in the spectra of the original sample (I)
and deformed samples of polyimide after recovery lasting for 1(11) and 24(111) hr
шим и большим временами жизни. После отдыха в течение 24 ч при комнат-
ной температуре наблюдается увеличение времени жизни обоих компонен-
тов и снижение интенсивности более долгоживущего. Характер происходя-
щих изменений позволяет предположить, что при деформации происходит
перестройка надмолекулярной структуры полиимида; межмолекулярные связи
разрушаются и образуются микродефекты - свободные объемы, достаточные
для локализации позитрона. Величина долгоживущего компонента Т2 в этом
случае должна отражать изменения среднего размера, а интенсивность /2 - кон-
центрацию таких дефектов. Аналогичные изменения в спектрах наблюдали при
образовании и отжиге дефектов в металлах и полупроводниках. Данные изме-
нения обычно анализируют с помощью модели захвата позитронов. Эта модель
качественно хорошо отражает наблюдаемые при деформации полиимида изме-
нения во временных спектрах Уменьшение времени жизни короткого компо-
нента, связанного с аннигиляцией в бездефектной части полимера, обуслов-
Таблица 10
Аннигиляционные характеристики полиимидной пленки
Annihilation characteristics of polyimide film
Образец Длительность восстановления , часы То. нс Т], пс Тг, пс А, % Скорость счета, k ioс
Исходный - 385±5 - - - -
Деформи- р овинный 1 - 294±30 440±17 59±5 0,60±0,15
Дсформи- р ованный 24 - 361±10 531±30 9±2 0,12±0,05
70
Глава II
лено высокой скоростью захвата в деформированном образце. После частич-
ного сокращения в процессе отдыха концентрация дефектов падает и время
жизни tj приближается к характерному для исходного полимера. При этом
уменьшается и интенсивность долгоживущего компонента Г, образованного
вследствие аннигиляции позитронов на дефектах. Рост времени жизни т2 мож-
но объяснить коагуляцией (объединением мелких дефектов в более крупные)
при отдыхе или быстрой релаксацией мелких пор, а, следовательно, увеличе-
нием среднего радиуса захвата.
Как показали оценки, концентрация микродефектов после частичной ре-
лаксации падает более чем в 7 раз, при этом величина индуцированного де-
формацией свободного объема уменьшается в4 раза [48]. Полученные значе-
ния указывают на то, что присутствуют оба процесса - слияние микродефек-
тов и релаксация наиболее мелких, хотя интенсивность последнего, видимо,
выше.
Таким образом, для исходной полиимидной пленки характерен одноком-
понентный спектр. В деформированном образце наблюдаются по крайней мере
два компонента во временных спектрах, связанные с аннигиляцией позитро-
нов из свободного и локализованного в микропорах состояний, образовав-
шихся при растяжении. В процессе релаксации структуры возрастает время
жизни и снижается интенсивность дефектного компонента.
Результаты, полученные с использованием модели захвата позитронов,
хорошо описывают наблюдаемые изменения временного распределения и по-
зволяют предположить, что структура свободного объема при релаксации из-
меняется не только в результате быстрой рекомбинации наиболее мелких пор,
но и вследствие их объединения с образованием долгоживущих микрополос-
тей большего размера.
На основании проведенного анализа в работе [48] предложена следующая
модель аннигиляции позитронов и связанный с ней механизм релаксационно-
го процесса: до деформации все позитроны аннигилируют из захваченного в
мелкие ловушки состояния с энергией связи, немногим превышающей теп-
ловую энергию; после деформации появляются достаточно протяженные (в
сравнении с длиной диффузии позитрона) области, в которых концентрация
мелких ловушек (размером ~10 нм) значительно снижается, одновр еменно
образуются разрыхленные области с глубокими центрами захвата по зитрона,
время жизни позитронов в которых больше; релаксация происходит таким
образом, что образованные при деформации поры рекомбинируют и, кроме
того, увеличиваются в результате слияния.
Такйм образом, измерив времена жизни позитронов, можно получить дан-
ные об изменениях в структуре незанятого объема ,происходящих после де-
формации полимерной пленки. Однако для трактовки полученной информа-
Упаковка макромолекул и плотность полимеров
71
ции необходимо детальное изучение природы компонентов сложного времен-
ного спектра аннигиляции, характерного для неравновесного состояния по-
лимера. Решить поставленную задачу с помощью одного из позитронных ме-
тодов не удалось [3, 110,156], поэтому было проведено [49] комплексное изу-
чение аннигиляции позитронов в деформированном полиимиде с помощью
измерения времени жизни позитронов и угловой корреляции аннигиляцион-
ного излучения.
В работе [49] описаны две серии экспериментов. В первой серии пленку
полиимида подвергали деформации растяжения на 20 °/о.Затем пленку осво -
бождалй из зажимов и она свободно релаксировала. Спектры времени жизни
на свободнорелаксирующей пленке снимали через каждые 1,5 ч. Параметры
угловых распределений определяли через каждый час в течение суток.
Во второй серии экспериментов изучали процесс релаксации напряже-
ния при деформаци и £q = 20 %. Характеристики угловых распределений оп-
ределяли для плен ки с закрепленными концами. Измерения проводили с по-
мощью устройства, позволяющего деформировать образцы непосредствен-
но в измерительной камере. Одновременно снимали сами кривые релаксации
напряжения (зависимости напряжения о от времени т), а также кривые вос-
становления (зависимости деформации е от времени т).
Полученные из спектров значения времени жизни позитронов приведены
в табл.11 и на рис .12 .Подобно описанным выше результатам двухкомпонент-
ного анализа, в структуре временного спектра, аппроксимированного тремя
компонентами, после деформации наблюдались изменения аннигиляционных
характеристик, которые затем постепенно релаксировали к характерным для
исходного образца полиимида величинам .
Таблица П
Изменение аннигиляционных характеристик полиимидной пленки в
зависимости от длительности релаксации после деформации на 20%
Change of annihilation characteristics of polyimide film depending on duration of
relaxation after deforming by 20 %
Длительность релаксации после дефор- мации , ч Время жизни 11 ± 1,5, % Угловая корреляция
ТСр±1, ПС Т] ±10, ПС FWHM ±0,05.. мрад г, ±0 07, мрад 0 ±0,07 , мрад //;±1,5, %
0 365 201 74,3 10,44 10,49 7,14 28,2
1 360 176 73,6 10,77 - — —
5 368 208 77 2 10 60 — - -
24 362 205 73,0 10,48 1064 7,14 34,7
240 364 200 74,1 10,43 10,72 6,95 32,3
Отожженный | 368 | 220 | 76,3 | - - - -
Примечание' тср т и /-> - характеристики спектров времени жизни позитронов; FWHM - полная
ширина на половине высоты полного спектра; Г, - FWHM первого гауссиана; 0р и /р -
характеристики параболического компонента спектра угловой корреляции.
72
Глава II
Рис. 12. Зависимости характеристик спектров времени жизни позитронов от длительности
релаксации свободной полиимидной пленки (обозначения см. в табл. 11)
Positron lifetime spectrum as a function of the relaxation duration for a freely relaxing
polyimide film (For designation see Table 11)
Были выделены три компонента: величина времени жизни первого ко-
роткоживущего компонента (170-220 пс) сильно зависела от длительности
релаксации; время жизни второго (388±10 пс), как показали проведенные ис-
следования [49], не зависит или слабо зависит от состояния образца, однако
наблюдаются заметные изменения интенсивности этого компонента. Харак-
теристики третьего компонента не изменялись в течение эксперимента.
В цитируемой работе проводились также эксперименты по измерению
угловой корреляции (наряду с измерением времени жизни позитронов). Не
останавливаясь на детальном анализе результатов этих измерений, отметим
лишь, что в экспериментах с закрепленными концами (в условиях релаксации
Упаковка макромолекул и плотность полимеров
73
напряжения) хорошо проявляется увеличение незанятого объема после де-
формации и последующая его медленная релаксация, происходящая за счет
снижения концентрации микропор.
В целом, методом позитронной диагностики выявлены изменения макро-
и микропараметров полиимидной пленки в процессах релаксации напряже-
ния и восстановления после деформации. Обнаружены немонотонные изме-
нения характеристик спектров времени жизни позитронов и угловых распре-
делений аннигиляционных фотонов в течение времени восстановления. Вы-
делено два интервала изменения позитрон-чувствительных свойств
полиимида, связываемых с “быстрыми” и “медленными” релаксационными
процессами, и обнаружены отличия в характере релаксации микропористой
структуры полимера в зависимости от условии де формации и ‘Ьтдыха ". №-
блюдаемые эффекты обусловлены образованием областей локального размо-
раживания молекулярной подвижности
Все эти экспериментальные факты свидетельствуют о том, что в процессе
релаксации напряжения происходит перестройка микропористой структуры
полимера, выражающаяся в перераспределении размеров микропор и их сли-
янии друг с другом. Таким образом, метод аннигиляции позитронов позволя-
ет не только оценивать микропористую структуру полимеров, но и следить за
ее изменением в процессе механического воздействия.
Глава III
ТЕМПЕРАТУРНЫЙ КОЭФФИЦИЕНТ
ОБЪЕМНОГО РАСШИРЕНИЯ
Тепловое расширение тел является следствием ангармоничности тепло-
вых колебаний частиц тела. В случае полимеров тепловое расширение имеет
ряд особенностей, связанных с различными физическими переходами поли-
мера по мере роста температуры. Для экспериментальной оценки темпера-
турного коэффициента объемного расширения определяют зависимость удель-
ного объема полимера от температуры. Схематически эта зависимость изоб-
ражена на рис. 13.
Такая зависимость в виде ломаной прямой характерна для многих поли-
меров вблизи температуры стеклования Tg. При температурах, лежащих ниже
температуры стеклования, эта зависимость более пологая, чем в интервале
температур выше точки стеклования. Таким образом, при условии Т< Т тем-
пературный коэффициент объемного расширения (который представляет со-
бой тангенс угла наклона дилатометрической зависимости) ниже, чем при ус-
ловии Т > Т В первом случае температурный коэффициент объемного рас-
ширения обозначается о^, а во втором - aL. В соответствии с этим удельный
объем полимерного тела может быть подсчитан по уравнениям
I/ = rg[l + aG(T-Tg)]> (7’<Tg); (37)
V=Vg[l + aL(T-Tg)l, {T>Tg)> (38)
где К,-удельный объем полимера при температуре стеклования; '/'-темпера-
тура.
Изображенная на рис. 13 дилатометрическая зависимость является весь-
ма упрощенной. На самом деле мы имеем дело не с ломаной прямой, а с кри -
вой, которая называется дилатометрической. Кривизна дилатометрических
зависимостей может выражаться двояким образом. Во-первых, переход из
Температурный коэффициент объемного расширения
75
Рис. 13. Схематическое изображение зависимости удельного объема Кот температуры Т
(дилатометрическая кривая) (скорость нагрева qx>q-j>qy>q^
Schematic representation of the dependence of specific volume К on temperature T
(dilatometric curve) (rate of heating q}>q^q^>q^)
стеклообразного состояния в высокоэластическое характеризуется не резким
изломом на дилатометрической зависимости, а плавным переходом одной ветки
дилатометрической кривой в другую. Это отчетливо видно из рис. 14, на кото-
ром приведена экспериментальная дилатометрическая кривая для полистиро-
ла, определенная вблизи температуры стеклования (105 °C) этого полимера.
Сама же температура стеклования определяется по пересечениюкасате лшых
к двум ветвям дилатометрической кривой. Во-вторых, если определить дила-
томстрическую кривую в широком интервале температур, можно убедиться,
что при температурах ниже точки стеклования она не является линейной на
всем своем протяжении, а обнаруживает отчетливо выраженную кривизну.
Рис. 14. Зависимость удельного объема v от температуры Т для полистирола
Dependence of specific volume v on temperature T for polystyrene
76
Глава III
Рис. 15. Зависимость термического коэффициента объемного расширения aG от темпе-
ратуры Т для полиметилметакрилата
Dependence of thermal coefficient of volumetric expansion a(; on temperature T for
poly(methyl methacrylate)
Характер этой кривой таков, что коэффициент объемного (или линейного)
расширения с понижением температуры уменьшается, т.е. эта величина не
является константой полимерного тела. На рис. 15 приведена эксперименталь-
ная температурная зависимость коэффициента линейного расширения для
полиметилметакрилата, определенная в широком интервале температур [154].
Легко убедиться, что величина aG снижается с уменьшением температуры,
т.е. дилатометрическая зависимость при Т < Tg не является прямой. Чтобы
подсчитать объем полимерного тела в данном случае недостаточно пользе -
ваться уравнением (37), а необходимо перейти к более общему' соотношению
где Од - термический коэффициент объемного расширения, зависящий от тем-
пературы; 10 - удельный объем полимера вблизи абсолютного нуля.
Зная эту зависимость, можно по уравнению (39) рассчитать удельный объем
полимерного тела при любой температуре Т.
Дилатометрические зависимости имеют не только практическое значе-
ние как зависимости, позволяющие отыскать температуру стеклования поли-
меров. Они имеют также и большое теоретическое значение. Во-первых, при
некоторых температурах, лежащих ниже температуры стеклования, могут на-
блюдаться изломы, не связанные с переходом полимерного тела из стеклооб-
разного состояния в высоко эластическое. Эти переходы происходят при тем-
Температурный коэффициент объемного расширения 77
пературах, лежащих ниже точки стеклования (т.е. внутри температурного ин-
тервала стеклообразного состояния), и имеют несколько иную природу, чем
основной переход. Таким образом, по дилатометрической кривой могут быть
определены температуры этих переходов. Во-вторых, согласно одной из кон-
цепций, развиваемой Бойером и Симхой, а несколько ранее сформулирован-
ной Флори, переход из стеклообразного состояния в высокоэластическое про-
исходит при той температуре, при которой доля свободного объема в полиме-
ре становится одинаковой и равной /с = 0,025. Согласно этой концепции,
справедливо следующее соотношение [205]
(ад -aG)Tg =0,113. (40)
Это соотношение является достаточно грубым приближением, поскольку
оно не учитывает кривизны дилатометрической зависимости. Учет этой кри-
визны позволил Симхе [154] уточнить концепцию свободного объема и опре-
делить его более точно. Однако даже и в этом случае концепция является лишь
достаточно грубым приближением, хотя она и позволяет описывать переход
из стеклообразного состояния в высокоэластичсское.
В табл.12 показаны экспериментальные значения aG для ряда стеклооб-
разных полимеров. Чем ниже температура стеклования полимера, тем выше
коэффициент термического расширения. Это согласуется с концепцией Сим-
хи-Бойера и с уравнением (40). Таким образом, теплостойкие полимеры, име-
ющие высокую температуру стеклования, обладают более низкой величиной
aG , а традиционные полимеры, размягчающиеся при низкой температуре,
имеют более высокий коэффициент термического расширения, который, сле-
довательно, зависит от химического строения полимера.
В работе [35] рассмотренные выше проблемы были детально исследова-
ны для сетчатых полимеров на основе эпоксидных смол. Отвержденные мо-
нолитные образцы получали, используя эпоксидную смолу ЭД-20, изо-ме-
Таблица 12
Расчетные с^р и эксперимента пъные э значения термических коэффициентов объём-
ного расширения и температуры стеклова1шя Г? для ряда стеклообразных полимеров
Calculated а^р and experimental aG3 values of thermal coefficients <f vd umft nc expansion and
the glass transition temperature Tg for a senes о fg hssy po piners
Полимер Г,, к Og.3'104, К?1 Ос.р ] о4, к 1
Полиметилметакрилат 378 2,69 2,55
Полиэтилметак рилат 338 2,99 2,84
Поли-н- пропилметак рилат 308 3,19 3,05
Поли-н-бутилметакрилат 293 3,34 3,22
Поли метилакр илат 293 3,03 2 ^0
П олиспрол 378 2 50',2.83 2.50
Ноликгр бонат на основе бшф енола А 423 - 2,27
78
Глава III
тилтетрагидрофталевый ангидрид как отвердитель, а также азелаинову ю кис-
лоту (для удлинения линейных фрагментов между узлами сетки) и олеиновую
кислоту (для получения “подвешенных” цепей). Оказалось, что коэффициен-
ты молекулярной упаковки для отвержденных сеток на основе эпоксидных
смол выше, чем для линейных полимеров. Это характерно для систем, не со-
держащих “подвешенных” цепей (разветвлений), т.е. при использовании в
качестве соотвердителей азелаиновой кислоты. При комнатной температуре
коэффициент молекулярной упаковки для них практически не зависит от со-
става сетки и его средняя величина составляет &ср=0,694, что несколько выше
средней величины кср = 0,681 для линейных стеклообразных полимеров. При
этом средний коэффициент молекулярной упаковки отвержденных сеток при
их температуре стеклования kg = 0,681, что также выше величины kg = 0,667,
характерной для линейных полимеров По данным этих измерений
(aL-aG)Ts =0,106. (41)
Расчеты и измерения также показали, что для отвержденных эпоксидных
смол доля свободного объема, возникающего вследствие теплового расшире-
ния, равна 0,078. Эта величина рассчитывается по формуле
/=-^-^- = aG7g, (42)
g
где Vg и 170 - удельные объемы полимера при температуре стеклования Т и
вблизи абсолютного нуля соответственно.
Эта же величина определяется соотношением
/ = (43)
где к0 и kg - коэффициенты молекулярной упаковки при температуре стекло-
вания Т и вблизи абсолютного нуля соответственно. Для линейных полиме-
ров величина / составляет 0,096.
Как было отмечено выше, тепловое расширение тел является следствием
ангармоничности тепловых колебаний частиц тела. На основании этого мож-
но представить, что коэффициент термического расширения состоит из вкла-
дов различных колебательных движений этих частиц.
Прежде всего необходимо учесть роль слабых дисперсионных взаимодей-
ствий. Следует заметить, что каждый атом обладает собственным дисперси-
онным взаимодействием, зависящим как от вида атома, так и от его ближай-
шего окружения, т.е. от тех атомов, которые химически с ним связаны.
В расчетной схеме [28, 43] для определения коэффициента термического
расширения сделано предположение, что вклады каждого атома пропорцио-
Температурный коэффициент объемного расширения
79
нальны доле Ван-дер-Ваальсового объема ДЕ каждого атома от общего Ван-
дер-Ваальсового объема £ ДИ, повторяющегося звена полимера.
/
Кроме того, совершенно необходимо учитывать влияние сильных межмо-
лекулярных взаимодействий, которые возникают при наличии в повторяю-
щемся звене полимера различных полярных групп. Сюда, в первую очередь,
относятся такие группы, как сложноэфирная —С—О—, нитрильная — C=N,
О
различные галогены, которыми замешаются атомы водорода (—CHCI—,
—CHF—, — CF3) и т.д. Эти группы приводят к диполь-дипольному взаимо-
действию различного типа.
Наиболее существенное влияние также оказывают полярные группы ,при -
водящие к возникновению водородных связей. К таким группам относятся
амидная —NH—С—, уретановая —NH— С—О—, гидроксильная —ОН, кислот
ная —С—ОН— О
II
О
Естественно, что энергия водородных связей так же, как и диполь -диполь -
ного взаимодействия, будет зависеть от химического строения полярных групп.
Казалось бы, что их вклад в коэффициент термического расширения должен
быть различен. Однако, если для каждого типа диполь-дипольного взаимо-
действия и водородных связей вводить различные параметры, характеризую-
щие энергию сильного межмолекулярного взаимодействия, то это не только
существенно усложнит расчетную схему, но и приведет к невозможности рас-
считывать коэффициент термического расширения для полимеров, содержа-
щих новые полярные группы. Поэтому в работах [28, 43] ограничились пер-
вым приближением, согласно которому вклад любого диполь-дипольного вза-
имодействия определяется одним и тем же параметром , не зависящим от
химического строения полярной группы.
Однако, поскольку Ван-дер-Ваальсовый объем каждой полярной группы
различен, то, как будет видно из дальнейшего, вклад каждой полярной гру и -
пы в коэффициент термического расширения также различен. Относительно
водородных связей также можно ограничиться одним значением параметра
Р/,, характеризующего энергию водородных связей.
Исключение составляет лишь класс полиамидов, который обладает спе -
цифическим поведением и требует нескольких параметров Рй, характеризую-
щих энергию водородных связей.
В результате получено соотношение для расчета термического коэффици -
ента объемного расширения для полимеров, находящихся в стеклообразном
состоянии, в виде.
80
Глава JII
Еа/А(//+Ер7
aG = --------------- (44)
IX
/
где а - парциальные коэффициенты объемного термического расширения,
обусловленного слабым дисперсионным взаимодействием /-го атома с сосед-
ними атомами; Л1Л- Ван-дер-Ваал ьсовый объем /-го атома; 0. -- параметры,
характеризующие вклад каждого типа специфического межмолекулярного вза-
имодействия (диполь-дипольное, водородные связи) в коэффициент терми-
ческого расширения.
Рассмотрим физический смысл параметров а., характеризующих слабое
дисперсионное взаимодействие.
Известно, что коэффициент объемного расширения описывается соотно-
шением
3R5,
tt'~ 2
У/ Гоа ’
где R - универсальная газовая постоянная; 3( - коэффициент ангармонизма,
1 А
(45)
; <р - потенциал взаимодействия /-го атома с соседними атома-
Г0,' д2(р
ми; у - гармоническая силовая постоянная, у, = —z-
ст
; г0 - расстояние
'о,/
между рассматриваемым /-м атомом и соседними атомами.
Для оценки коэффициента 5; и гармонической силовой постоянной у можно
воспользоваться потенциалом Леннард-Джонса
<р(г) = £)[(г0/г)12 -2(гд/г)6].
(46)
В уравнении (46) величина D характеризует энергию межмолекулярной
связи, а г() - равновесное расстояние между атомами, которые не связаны хи-
мически, а вступают в межмолекулярное взаимодействие. Тогда можно запи-
сать
72 О, 7560, 1 R 7 R
У/ = —т2-; 5, = —> ai=----------; =-----
16 Di 16 “/
(47)
Выражения (47) позволяют оценивать величины энергии дисперсионно-
го взаимодействия для каждого атома. Эти значения приведены в табл. 13 и
они показывают, что действительно величины D, соответствуют энергиям меж-
молекулярного взаимодействия, а не энергиям химической связи. Однако, если
эти величины оценить другими методами [66] (обозначим их D<}), то оказыва-
ется, что значения D. отличаются от D,u в несколько раз. Это происходит пото-
Температурный коэффициент объемного расширения
81
Таблица 13
Значения инкрементов а,, [J , и и Л; для различных атомов
и типов межмолскулярпого взаимодействия
Values of increments aj, p , u: and A, for various atoms and types
of intermolecidar interactions
Атом или тип межмолекулярного взаимодействия Сим вол а, Символ a, io3, K'1 b, -io3, A3K'‘
Углерод ас 0,00 ас 0,02 -
Водород ан 1,92 ан 19,98 -
Кислород в основной цепи ао.о 2,21 ^О.о 22,95 -
Кислород в боковой группе -О- = о ао,б ао.б' 1,54 0,77 @0.6' 16,00 8,00 -
Азот в основной цепи Щ/.о 0,83 8,62 -
Азот в боковой группе 0,61 'ZVo 6,35 -
Хлор И(’/ 0,39 Г/р/ 4,01 -
Фтор aF 0,66 aF 6,90 -
Сера в основной цепи а до 0,72 о 7 SO -
Сера в боковой цепи СЦ’.б 0,20 Q-S.6 2,04 -
Кремний в основной цепи 0,80 ^Si.o 8,30 -
Кремний в боковой группе as,в 0,27 а ч, б 2,80
Бор в карборанах ан -0,96 ан -10,00 -
Диполь - дипольное взаимодействие * Рд -5,31 bj - -55,4
Водородная связь ** Р/, -13,44 Ьн - -139,6
Тип замещения бензольных ядер *** пара- мета- орто- Рп Рм Ро 2 41 1.54 5,47 bn bM b0 - -25 0 16,0 57,0
Коэффициент для полидиенов р, 8,88 b* - 135,0
* Параметры 0,; и bd вводятся для каждого разветвления в основной или боковой цепи, они также
вводятся при наличии полярной группы любого типа , если в алифатических полимерах при одном
атоме углерода находятся две СН3- группы, или два атома F, С1,то инкремент bd не вводится.
Для фрагментов —СН— СН— СН— и —СН —
1111
С-О- О—С — C-S- S-C-
II II II II
О 0 0 о
вводится дополнительный инкремент bd = 51.
** Инкремент bh вводится при наличии водородной связи любого типа для всех полимеров, кроме
полиамидовн для последних инкременты [2 и bh содержатся в табл. 18.
Инкременты рп , рм , ро и Ьп ,ЬМ ,Ь0 вводятся при замещении ароматических ядер в пара-,
мета- и орто-положениях соответственно; количество этих инкрементов равно количеству
замещенных ядер. В случае структуры вводится 2[1П и2Ап.
82
Глава III
му, что в любом низкомолекулярном веществе или полимере каждый атом
находится в межмолекулярном взаимодействии не с одним атомом, а с не-
сколькими, с которыми он координирует. Тогда следует записать:
D,=zD°, (48)
где z - координационное число.
В этом случае общая усредненная энергия слабого дисперсионного взаи-
модействия определяется из соотношения z<D0>, где
(49)
7
Для сополимеров уравнение (44) приобретает вид:
ai(Xa,AI/ +ХР7)1 +а2(Еа/Л,< +Е₽р2 +••• +
а<7=-----'------------------------L----------->
7 7
+Р;)»
----J---- (50)
+ а„(£Д1<)„
где otj, а2, ..., ая- молярные доли компонентов 1,2, ..., и (не следует путать
эти величины со значениями ар находящимися в выражениях в скобках):
(^2а,Д^+ ХРу)1, , ..., +S₽;)« -наборинк-
' J ' j ' j
ремонтов а/; для компонентов 1,2,..., и сополимера, (2^)1 ...,
I /
(Z ЛV) )„ - Ван-дер-Ваальсовые объемы повторяющихся звеньев этих компо-
7
нентов.
В более компактной форме соотношение (50) можно записать в следую-
щем виде:
ХаЛ(£а,Д1/
а(7 = k~] "кТ~----------- (51)
Температурный коэффициент объемного расширения
83
Если желательно выразить термический коэффициент объемного расши-
рения aG сополимера через аналогичные коэффиценты аа р аа 2,ао п для
соответствующих гомополимеров, то подстановка (44) в (50) дает:
+a2aG.2&A,/ih +-- + anaG,n(^^/i)n
OG =-------------------------------------------L-----> {52)
а1(£Д^)1 +«2(^ДГ//)2 + ••• + ап(ХД^)«
i i i
где ар а, ..., аи - молярные доли компонентов 1,2, .п сополимера.
В более компактной форме соотношение (52) выглядит так:
Ёа^“с,А-(ЕЛ1<)А-
Щ; = --------L------• (53)
к=п
к=\ i
Следует отметить, что зависимость а сот состава сополимера а не явля-
ется линейной, а обнаруживает кривизну, зависящую от энергии межмолеку-
лярного взаимодействия компонентов и их Ван-дер-Ваальсового объема
(рис. 16).
Расчетные значения коэффициентов термического расширения ряда по-
лимеров, находящихся в стеклообразном состоянии, показаны в табл. 12. Во-
обще, следует иметь в виду, что несмотря на кажущуюся простоту этойфизи-
ческой характеристики, ее экспериментальное определение встречает доста-
Рис.16. Схематическое изображение зависимости термического коэффициента
объемного расширения ; от состава сополимера а
Schematic representation of dependence of the tliermal coefficient of volumetric
expansion aCron composition of copolymer a
84
Глава III
точные трудности. Именно поэтому в литературе можно встретить существен-
но различающиеся значения аа для одного и того же полимера. Все расчетные
и экспериментальные значения aG, приведенные в табл. 12, характеризуют
участок дилатометрической прямой, непосредственно примыкающий к тем-
пературе стеклования.
Что касается термического коэффициента объемного расширения а; в вы-
сокоэластическом состоянии, то, как было отмечено выше, он может быть
определен с помощью соотношения (40), хотя при этом и возможны суще-
ственные погрешности. В монографии Ван-Кревелена [214] приводится дру-
гое соотношение для оценки aL:
=ЧР/М , <54)
где £; = 10-10‘4 VM, VM- молярный объем (в расчете на повторяющееся звено
полимера); = Д' . Д[ ’, где A'zj - число Авогадро. -Ван-дер-Ва-
i i
альсовый объем повторяющегося звена; М - молекулярная масса повторяю-
щегося звена; р - плотность полимера.
Учитывая, что по данным Ван-Кревелена в высокоэластическом состоя-
нии Гм= 1,60 l';v, из соотношения (54) получается, что коэффициент объем-
ного термического расширения для полимеров, находящихся в высокоэласти-
ческом состоянии, одинаков и равен величине
aL «6,3-Ю^К-1.
Глава IV
ТЕМПЕРАТУРА СТЕКЛОВАНИЯ ПОЛИМЕРОВ
IV.1. Термомеханический и другие методы определения
температуры стеклования полимеров
Термомеханический метод исследования полимеров является одним из
наиболее распространенных методов экспериментального определения
температуры стеклования Т Этот метод был разработан В.А. Каргиным и
ТИ .Соголовой Сущность метода заключается в следующем. Полимерное тело
подвергается действию постоянной или переменной нагрузки и при этом фик-
сируется его деформация при каждой температуре и выбранном времени дей-
ствия силы. Известно, что если полимерный образец подвергается действию
постоянного напряжения, в нем развивается ползучесть. Графически это выг-
лядит так, как изображено на рис. 17. Чтобы проводить опыты в сравнимых
условиях, деформацию необходимо отсчитывать за строго постоянное время
наблюдения, которое может быть выбрано любым, но желательно таким, что-
бы отсчет деформации производился на втором, пологом участке кривой пол-
зучести. Проделав такой опыт при разных температурах, можно построить
температурную зависимость деформации, которая будет иметь в об щем слу-
чае вид, изображенный на рис. 18.
Прежде чем перейти к рассмотрению особенностей графика на рис. 18,
заметим, что аналогичная кривая может быть получена и в том случае, когда
на образец действует небольшая постоянная сила, а температура непрерывно
возрастает во времени. Такой вид термомеханических испытаний в настоя-
щее время наиболее распространен, причем температура, как правило, возра-
стает по линейному закону
Г — 7() + ой .
(55)
86
Глава II'
Рис. 17. Схематическое изображение процесса ползучести (зависимости деформации
с от времени t при постоянном напряжении)
Schematic representation of creep (dependence of deformation s on time t at
constant stress)
Рис. 18. Схематическое изображение термомеханической кривой (зависимости дефор-
мации е от температуры Г): I - стеклообразное состояние полимера, II - высоко-
эластическое состояние полимера, III - вязкотекучее сос тояние полимера
Schematic representation of thermomechanical curve (dependence of deformation e
on temperature T) on which I is the glassy state of polymer, II is the rubbery' state of polymer
and III is the viscous flow state of polymer
где Tq - температура, с которой начинается опыт; t - время; а -скорость роста
температуры со временем.
Стандартная скорость роста температуры обычно составляет от 1,5 до
4 град/мин. Такой способ термомеханического исследования более удобен в
аппаратурном отношении, хотя он дает меньшую информацию, чем первый
способ. Особенно это касается тех случаев, когда с помощью термомехани-
ческого метода исследуют тонкие структурные превращения, проходящие в
образце при его нагревании. В дальнейшем к этому' вопросу мы вернемся, а
сейчас рассмотрим термомеханическую кривую идеального полимера, в кото-
ром никаких структурных превращений при нагревании не происходит. Такая
Температура стеклования полимеров Х7
кривая изображена на рис. 18. и она получается в том случае, когда опыт начи-
нается с достаточно низких температур.
В области этих температур деформация образца мала и лишь незначи-
тельно увеличивается по мере роста температуры. При достижении опреде-
ленной для каждого полимера температуры деформация начинает быстро воз-
растать. Если нагрузка мала, возрастание деформации происходит нс беспре-
дельно, а довольно быстро заканчивается. На графике образуется плато,
получившее название “площадки высокоэластичности”. Протяженность этой
площадки по оси температуры может быть достаточно велика и, как будет
видно дальше, она зависит от молекулярной массы полимера. При дальней-
шем нагревании деформация вновь резко начинает возрастать и образец по-
лимера растекается.
Термомеханическая кривая, изображенная на рис. 18, отчетливо подраз-
деляет температурную область испытания на три интервала. В первом из них
полимер находится в твердом, стеклообразном состоянии (если он амор ^ный).
Деформация в этом состоянии весьма мала и слабо повышается с ростом тем-
пературы: полимер ведет себя во многом подобно тому, как ведут себя обыч-
ные низкомолекулярные вещества.
Первый резкий подъем на термомеханической кривой связан с переходом
полимера из твердого стеклообразного состояния в высокоэластическое. Это
состояние характеризуется тем, что полимер приобретает каучу ко подобные
свойства, т.е. обладает способностью к развитию больших обратимых дефор-
маций при действии небольшой силы. Это состояние характерно только для
полимеров, и оно не проявляется в случае низкомолекулярных твердых тел.
При дальнейшем нагревании резкое увеличение деформации связано с
появлением вязкотекучего состояния, при котором характерно вязкое течение
полимерного вещества. Соответствующие температуры переходов из стекло-
образного состояния в высокоэластическое и из высокоэластического в вязко-
текучее получили название “температура стеклования” и “температура теку-
чести”. Прежде чем рассмотреть природу каждого из физических состояний
полимеров, отметим ,что в зависимости от химического строения полимера,
т.е. от гибкости или жесткости его макромолекул, температура стеклования
может принимать самые разнообразные значения В настоящее время извест-
ны полимеры, у которых температура стеклования изменяется от -123 до 600 °C.
Примером первого из них является полидиметилсилоксан, который имеет сле-
дующую структурную формулу'
СН3
[-SHO-L
СН3
88
Глава IV
Примером второго является полинафтоиленбензимидалол следующего
химического строения
Синтез полимеров с возможно более низкой температурой стеклования
имеет важное значение для получения морозостойких каучуков. Синтез поли-
меров с возможно более высокой температурой стеклования имеет столь же
важное значение для получения твердых теплостойких полимеров, способ-
ных работать при повышенных температурах и нагрузках. Многочисленные
полимеры, применяющиеся в настоящее время, имеют температуры стекло-
вания, лежащие внутри этого огромного интервала. В табл. 14 представлены
некоторые из них, и рассматривая эти данные, легко представить, как влияет
химическое строение полимеров на их температуру стеклования.
Следует отметить, что переход из стеклообразного состояния в высоко-
эластическое совершается не при какой-то одной определенной температуре,
а происходит в некотором интервале температур, который может составлять
несколько десятков градусов. Это вполне понятно, поскольку рассматривае-
мый переход не является фазовым переходом (таким, например, как плавле-
ние), а представляет собой физический переход из одного состояния в другое
при сохранении одной фазы. В случае аморфных стеклообразных веществ
такой фазой является жидкость независимо от того, в каком агрегатном состо-
янии находится полимерное вещество - в стеклообразном (твердом), высоко-
эластическом или вязкотекучем.
В принципе каждый полимер, если он имеет достаточно высокую молеку-
лярную массу, может находиться в одном из трех физических состояний. Кау-
чуки отличаются от пластмасс лишь тем, что температура стеклования поли-
меров, из которых они получены, лежат ниже комнатной температуры, а для
пластмасс - выше комнатной. Однако, если каучук охладить до температур,
лежащих ниже температуры стеклования (например ,до -80 °C) ,то каучу копо-
добное тело станет твердым и будет вести себя аналогично тому, как ведет
себя обычный стеклообразный полимер.
И наоборот, если твердый стеклообразный полимер (например, полиме -
тилметакрилат) нагреть выше его температуры стеклования (выше 100 °C),
то он станет каучукоподобным и будет вести себя как обычный каучук при
комнатной температуре. Таким образом, разделение полимерных тел на кау-
чукоподобные и твердые стеклообразные является весьма условным и сложи-
лось исторически.
Температура стеклования полимеров
89
Таблица 14
Температура стеклования /^представителей раз.тичпых рядов полимеров
Glass transition temperature Tg of representatives of various series of polymers
Название Структурная формула Тд, к
1 2 3
1,4-полибутадисп - сн2— сн=сн-сн2- 170
Полидиметилсилокса н сн3 — Si—О— 1 сн3 150
1,4-поли изо прен —сн2—сн=с—сн2— СН3 202
Полнизобутилен СНз СН2-С- сн3 199
Поливинилмегиловый эфир - сн2- сн- о—сн3 258
Полинмпилэтнловый эфир —сн2—сн— О-С2Н5 248
Поливипилацегат —СН2~СН — О-С-СНо II о 298
Поли ви нилхлорид - сн2-СН— CI 355
Полиметмлметакрилат сн3 -сн2—с - с-о-сн3 II О 378
П оли э ти л м е г а кр млат СНз -сн2-с - C-O-C2HS II о 338
90
Глава IV
таблица 14 (продолжение)
1 2 3
Полигексилметакрилат сн3 -СН2-С- С — О—СдН-|о II ь О 268
Полиметилакрилат - СН2-СН- С-О-СНз о 293
Полибутилакралат -сн2- сн— с-о-С4Н9 О 233
Полистирол -сн2- сн- 6 378
Поливиниловый спирт —СН2— СИ- ОН 358
Полиэтилентерефталат -С-О-С-0-(СН2)2-0- о о 353
Полиакрилонитрил —СН2—СН — C = N 418
Пол иакр иламид —СН2—СН — z 1 с—nh2 II о 426
Поликарбонат на основе бисфенола А 0=0 А 1 о £> 1 £ 0-0-0 1 0 А 1 422
Сложный полиэфир на основе себацииовой кислоты и фенолфталеина 1 о 0 о хо )о ох 0 о 1 о 1 0 = 0 1 J5 сч I о 1 0=0 1 368
Температура стеклования полимеров
91
В газообразном состоянии полимер не может находиться, т.к. гораздо легче
разорвать цепи макромолекул на отдельные фрагменты, чем преодолеть сум-
марные силы межмолекулярного взаимодействия: полимерные цепи имеют
громадную длину и большое количество межмолекулярных связей между
соседними цепями, поэтому без разложения полимера перевести его в газо-
образное состояние не удается.
Выше было отмечено, что в каждом из трех физических состояний поли-
мер может находиться только в том случае, если его молекулярная масса дос-
таточно велика. Тогда при нагревании твердого пластика он переходит после-
довательно из твердого стеклообразного состояния в высокоэластическос, а
затем - в вязкотекучее (см. рис. 18). Если молекулярная масса полимера мала,
он из стеклообразного состояния переходит непосредственно в вязкотекучес,
атермомеханичсская кривая имеет вид, схематически изображенный на рис .19.
Такая кривая характерна также и для низкомолекулярных веществ. Таким
образом, высокоэластическое состояние характерно только для полимеров и
не проявляется у низкомолекулярных веществ.
Теперь необходимо определить, с какой молекулярной массы полимер на-
чинает обнаруживать высокоэластическое состояние. Для этого рассмотрим
серию термомеханических кривых полимергомологов различной молекуляр-
ной массы. На рис.20 схематически изображена такая серия; хорошо видно,
что с ростом молекулярной массы температура стеклования полимера внача-
ле растет, а затем практически перестает изменяться. Та молекулярная масса,
92
Глава IV
Рис. 19. Схематическое изображение термомехаиической кривой для полимера
с малой молекулярной массой: I - стеклообразное состояние полимера, II - вязкотекучсе
состояние полимера
Schematic representation of thermomechanical curve for a polymer with small
molecular mass on which 1 is the glassy state of polymer and 11 is the viscous flow S a e d'
Pnc.20. Схематическое изображение термомеханических кривых для полимеров разной
молекулярной массы: ЛС<Л<Л<Л/з<ЛС<^/5<А/6
Schematic representation of a series of thermomechanical curves for polymers
having different molecular таз8Л/|<Л/-><Л/з<Л/4<Л-Д<1''4,
с которой перестает наблюдаться это изменение и является молекулярной мас-
сой сегмента. С этой молекулярной массы полимер начинает проявлять высо-
коэластическое состояние Эго вполне понятно, т.к. сегмент и есть тот отре-
зок макромолекулы, концы которого могут перемешаться независимо друг от
друга. Следовательно, если макромолекула имеет длину большую, чем про-
тяженность сегмента,тепловое движение может осуществляться лишь в от-
дельных ее местах, не затрагивая макромолекулы в целом. Это и приводит к
Температура стеклования полимеров
93
появлению характерных каучука подобных свойств, когда для полимера ха-
рактерны большие обратимые деформации без приложения существенных
механических усилий. Это и есть высокоэластическое состояние.
Естественно, что если молекулярная масса полимера превосходит вели-
чину сегмента, то дальнейшее ее увеличение не может привести к росту тем-
пературы стеклования, т.к. движение сегментов как отдельных частей м акро-
молекул уже может вполне проявляться. Напротив, температура теку чести
полимера все время возрастает с увеличением молекулярной массы, посколь-
ку течение есть ни что иное, как перемещение отдельных макромолекул от-
носительно друг друга в целом. Понятно, что чем больше длина макромоле-
кулы, тем больше тепловой энергии необходимо затратить, чтобы перемес-
тить макромолекулы относительно друг друга, т.е. вызвать течение. Поэтому'
температура текучести в сев рсмя растет с увеличением.молекулярной массы
Отсюда совершенно ясно ,что с помощью тсрмомеханичсского метода
исследования можно определить величину механического сегмента. Эта ве-
личина будет соответствовать той молекулярной массе, при которой появля-
ется высокоэластическое состояние и превышение которой не может привес-
ти к увеличению темперагуры стеклования. На рис. 21 изображена зависи-
мость температуры стеклования полистирола от его молекулярной массы М п.
Эта зависимость подтверждает правильность сделанных выше рассуждений
Рис 21 Завизимосп темп.-ратуры стеклования 7g от молекулярной массы А4,
полистирола
1 Эерей сисе 6 Hie fc asst ransli' lont empert ure'/’ g on mi> eclar ma-ssV „for
polystyrene
94
Глава IV
и хорошо описывается рядом соотношений, из которых наибольшее распрос-
транение получили уравнение Флори
Tg = Tgao-a/M, (56)
где Tg;О9 - температура стеклования полимера при молекулярной массе, стре-
мящейся к бесконечности; а - параметр, и уравнение
где р - параметр.
Теперь рассмотрим экспериментальные и расчетные способы определения
величины механического сегмента, те. молекулярной массы Мс, начиная с
которой проявляется высокоэластическое состояние. Одним из удобных спо-
собов экспериментального определения величины механического сегмента,
как было отмечено выше, является термомеханический метод.
Рассматривая вновь рис.20, можно сказать, что молекулярная масса сег-
мента, определенная термомеханическим методом, равна М3 .
Величина сегмента зависит от химического строения макромолекулы, и
чем более жесткой является макромолекула, тем большей величиной сегмен-
та она обладает. Наименьшей величиной сегмента обладают гибкие макро-
молекулы, для которых вращение отдельных звеньев относительно друг дру-
га является достаточно свободным. В табл.15 приведены молекулярные мас-
сы сегментов макромолекул различных полимеров. Из этой таблицы хорошо
видно, что в зависимости от химического строения полимера его макромоле-
кула обладают совершенно различными размерами се гментов. Н ai6 олее ко-
роткий сегмент характерен для макромолекул полиизобутилена, а наиболее
длинный сегмент - для макромолекул полиарилата*. Наиболее жесткоцеп-
ные полимеры обладают чрезвычайно большой величиной механического
сегмента, и во многих случаях величина этого сегмента равна всей длине мак-
ромолекулы. Иными словами, в таких макромоле гулах нельзя переместить
их отдельные участки, не затрагивая макромолекулу в целом. Поскольку при
нагревании такого полимера его макромолекулы перемещаются относитель-
но друг друга целиком, то температуры текучести и стеклования для него со-
впадают, ибо по своему определению текучесть полимеров - это смещение
отдельных макромолекул относительно друг друга.
Следует обратить внимание еще на одну' интересную особенность, хоро-
шо видную из табл. 15. Разные полимер ы, имеющие, однако, совершенно оди-
Не следует отождествлять механический сегмент макромолекул который является пред-
метом нашего рассмотрения, с сегментом Куна, который характеризует гибкость изолированной
цепи и определяется по поведению макромолекул в растворе.
Температура стеклования полимеров
95
Таблица 15
Значения молекулярной массы сегмента Мс различных полимеров,
определенные т ермомеханнческнм методом
Values of molecular mass of a segment Mc for different polymers determined
by tile tliermoineclianical method
Полимер
П о л и и зо бутил ей сн3 -сн2-с- сн3 1000-1200
Поливинилхлорид -СН9-СН- 1 CI 12 000
Полистирол -сн2-сн- 6 30 000 40 000
Полиарилат Ф-1 O-c-0-Q4 zQ-o- /п р -с 0 ^/Ч ' Ох Р ° cf чо 50 000-70 000
наковый скелет цепи и отличающиеся только боковыми заместителями, об-
ладают различными длинами сегментов. На первый взгляд это противоречит
тому, что гибкость макромолекул проявляется нс вследствие их изгибания, а
в результате вращения отдельных звеньев относительно ординарных связей,
т.е. тех связей, которые связывают скелетные атомы (в данном случае - это
атомы углерода в полиизобутилене, полиэтилене, полистироле и т.д ). Однако
это противоречие является только кажущимся. Свобода (или заторможенность)
вращения отдельных групп относительно друг друга зависит не только от
скелета макромолекулы, но и от характера боковых заместителей. Чем более
объемистым и полярным является заместитель, тем при прочих равных усло-
виях вращение отдельных звеньев будет заторможенным. Таким образом,
следует говорить не только о скелетной жесткости, но и о жесткости кинети-
ческой связанной с заторможенностью вращение возникающей вследствие
наличия объемистых боковых заместителей. В этом отношении характерным
является полистирол, у которого в повторяющемся звене в качестве бокового
заместителя находитсяф енильное ядро.В рез ульяте этот полимер обладает
96
Глава IV
большим механическим сегментом по сравнению с другими полимерами, при-
веденными в табл. 15.
Теперь необходимо связать величину молекулярной массы сегмента с па-
раметрами химического строения полимера, а также с его характерными тем-
пературами - температурой стеклования и температурой текучести. Однако
прежде необходимо договориться о том, каким способом мы будем опреде-
лять эти температуры. Если такое определение выполняется термомехани-
ческим методом, то очень важно правильно выбрать способ нахождения этих
температур по тсрмомсханической кривой.
Теория тсрмомсханического метода, развитая В.А.Каргиным и ГД.Сло-
нимским, гласит о том, что температура стеклования и температура текучести
должны определяться так, как это схематически изображено на рис.22. Выби-
рается величина деформации е0, которая откладывается от оси абсцисс, а так-
же от высоты площадки высокоэластичности. Далее проводятся линии, па-
раллельные оси абсцисс, и точки пересечения этих линий с термомеханичес-
кой кривой и дадут искомые температуры стеклования и текучести. Таким
образом, согласно этому определению, температура стеклования - эта та тем-
пература, при которой деформация при действии той или иной нагрузки раз-
вивается на величину е0. Температура текучести - эта та температура, при
которой пластическая деформация (течение) развивается на ту же величи-
ну' £0. Величина е^ может быть выбрана произвольно, но она не должна быть
слишком большой, чтобы не превышать высоту площадки высокоэластично-
сти. На практике за величину е0 принимают определенный процент от высо -
ты площадки высокоэластичности.
Рис.22. Определение температур 7g и 7’f по выбранной величине е(1 па термомехани-
ческой кривой сошасно теории Каргина-Слонимского
Evaluation of temperatures Тъ and 7f according to the value c0 chosen on the
thermomcchanical curve in accordance with Kargin-Slonymskii’s theory'
Температура стеклования полимеров
97
Теория Каргина и Слонимского, основанная на рассмотрении деформации
модели полимерного тела с учетом изменения температуры, приводит к сле-
дуюгцеьгу соотношению:
IgM =\g АТ с+ (58)
С +(lf -tg)
где А/- молекулярная масса полимера; АТС - молекулярная масса сегмента;
Tj -температуратекучести; Tg-температурастеклования;В иС - параметры
полимера.
Уравнение (58) связывает величину молекулярной массы полимера с ве-
личиной молекулярной массы сегмента и с разностью температур текучести и
стеклования (7’у - Т„). Не останавливаясь пока на практическом значении этого
уравнения, отметим, что его параметры Ви Симеют ясный физический смысл.
Параметр В определяется из соотношения'.
S = lg^-, (59)
ПОЕО
где- о - постоянное напряжение, приложенное к образцу в процессе термо-
механического испытания; г0 - длительность воздействия этого напряжения;
е0 - деформация, при которой определяются величины 7g и Тj (ее смысл хо-
рошо виден из рис.22); i|()- предэкспоненциальный множитель в уравнении
Фульчера-Таммана, которое связывает вязкость системы с температурой. Это
уравнение выглядит следующим образом
л
1] = т]ое7 , (60)
где г| -вязкость системы при температуре Т,выраженной в градусах Кельви-
на; т|0 - интересующий нас предэкспоненциальный множитель; А и 7’0 - пара-
метры материала.
Таким образом, все величины, входящие в параметр В, определены. Кон-
станта С в уравнении (58) находится из соотношения
С = 0,434-4 (61)
Следует заметить, что уравнение Фульчера-Таммана хорошо описывает
область стеклообразного состояния вблизи температуры стеклования. Имен-
но эта область нас сейчас и интересует.
Теперь кратко рассмотрим практическое значение уравнения Каргина-
Слонимского . Это уравнение, если параметры Мс, В иС в нем известны, по-
зволяет определять молекулярную массу полимера без перевода его в раствор.
Для этого необходимо определить термомеханическую кривую этого поли-
мера, с ее помощью найти температуру текучести Ти стеклования Tg и под-
ставить их в соотношение (58). При этом параметры В и С не обязательно
98
Глава IV
Определение степени полимеризации Nc
Determination of polimerization degree Nc
Полимер Мс [174] мо Ас [174]
Полиэтилен 3460 28 124
Полиизобугилен 15625 56 279
Полистирол 38073 104 366
Полибу(адиен 5625 54 104
Полиизопрен 10000 68 147
Поливинилацетат 24287 86 282
Полиметилметакрилат 30246 100 302
определять с помощью уравнений (59) и (61). Для этого необходимо лишь
определить три термомеханические кривые полимера с известными молеку-
лярными массами М и решить систему из трех уравнений, составленных на
основе (58), в которой неизвестными будут Мс, В и С. Затем можно опреде-
лять молекулярную массу любого количества полимергомологов данного ряда.
Следует, однако, отметить, что уравнение (58) не позволяет рассчитать
величину Мс полимера, основываясь только на его химическом строении.
Величина механического сегмента макромолекулы Л/с может быть опре-
делена чисто расчетным путем на основании химического строения полиме-
ра. В работе [96] получено следующее выражение для расчета Мс (или соот-
ветствующей степени полимеризации Nc = MJ Л/о, где Мо - молекулярная
масса повторяющегося звена):
Nc = const ^(А^Д^)1'3 (62)
i
Величина Л) может быть рассчитана исходя из химического строения по-
вторяющегося звена с помощью аддитивных схем, поскольку с их помощью
оценивается температура стеклования Tg и Ван-дер-Ваальсовый объем звена
X (Na - число Авогадро). Константа в формуле (62) может быть выраже-
i
на через параметры полимерной системы, а также может быть найдена по
экспериментальным данным для эталонных систем. Оценки дают const = 0,21,
если калибровку проводить по полистиролу - данные работы [177], const =
= 0,24 если калибровку проводить по полистиролу -даянье работы[174,214].
В табл. 16 приведены значения Nc, найденные по данным работы [174] и
по формуле (62), с разными константами (Nc с const = 0,24 и Nc* с const =
= 0,21). При const = 0,21 различие значений, найденных по работе [174], не
превышает 10 %.
Температура стеклования полимеров
99
таблица 16 (продолжение)
Гй,к i см’/моль Nc М*
195 20,60 128 112
199 41,30 165 144
378 66,00 366 320
171 36,48 136 119
200 48,90 175 153
298 47,73 259 227
378 58,05 351 307
До сих пор мы рассматривали такие физические характеристики полиме-
ров, как температура стеклования, температура текучести, величина сегмен-
та макромолекулы, которые экспериментально определялись с помощью тер-
момеханического метода исследования полимеров.
При определении этим методом температурных интервалов, в которых
полимер находится в твердом (стеклообразном), высокоэластическом и вязко-
текучем состояниях, часто встречаются с определенными трудностями, в осо-
бенности, если речь идет о новом полимере.
Попытаемся в общих чертах разобрать возможные случаи деформа-
ционного поведения полимеров в условиях термомеханических испытаний.
Напомним, что в этих условиях образец находится под действием нагрузки
при возрастающей температуре. Чаще всего напряжение действует постоян-
но на протяжении всего опыта, а температура растет по линейному закону.
В принципе термомеханический метод исследования сразу позволяет оп-
ределить температурные интервалы всех трех физических состояний полиме-
ра. Однако уверенно говорить о наличии тех или иных физических состояний
и соответствующих им температурных интервалах можно лишь в том случае,
если известно, что исследуемый полимер ведет себя как “классический”, т.е.
дает классическую термомеханическую кривую, показанную на рис. 18. При
оценке свойств нового полимера, как будет видно дальше, даже совпадение
формы термомеханической кривой с классической еще не позволяет одно-
значно судить о температурных интервалах физических состояний и даже о
самих состояниях.
Прежде чем остановиться на этом, разберем некоторые методические воп-
росы. Наиболее часто возникает вопрос, каким способом определять точки
переходов по термомеханической кривой? Как было отмечено выше, теоре-
тически обоснован следующий способ: выбирается определенная величина
деформации £q, которая откладывается от оси температур и от площадки вы-
100
Глава IV
сокоэластичности. Точки стеклования и текучести будут соответствовать тем-
пературам, при которых развивается одна и та же величина s0 соответственно
высокоэластической и пластической деформаций.
Такой способ является наиболее правильным, но он применим тогда, ког-
да форма термомеханической кривой имеет классический вид с резкими за-
гибами кривых в переходных областях температур. Тогда изменение величи-
ны £0 не повлечет за собой больших сдвигов в определении Tg и Tj. Если
деформация развивается более плавно, то устанавливаемые точки переходов
7g и Гу будут весьма неопределенными.Они будут существенно зависить от
величины So (см. рис.22).
Поэтому на практике часто пользуются другим способом - определяют
величины и Гу по точкам пересечения касательных к двум соответствую-
щим ветвям термомеханической кривой (рис.23). В этом случае значения Tg и
Ту меньше зависят от формы термомеханической кривой, и для сравнитель-
ной оценки полимеров такой способ вполне оправдан.
Сравнивая термомеханические кривые ряда полимеров, можно определять
точку стеклования и как температуру, при которой деформация развивается
на величину, составлявшую определенный процент от высоты площадки вы-
сокоэластичности. Тогда для каждого полимера эта характерная деформация
будет принимать различные значения, поскольку высота площадки высоко-
эластичности тоже разная.
Выбор способа определения величин Tg и Гу зависит от формы термоме-
ханической кривой полимеров и в условиях сравнительной оценки их свойств
можно выбрать любой из способов. Важно лишь, чтобы сравнение несколь-
ких полимеров всегда проводилось одним и тем же способом.
Рис.23. Определение температур 7g и Т( но точкам пересечения касательных к двум
ветвям термомеханической кривой
Evaluation of temperatures 7'g and Tf by the points of intersection of tangents to
two branches ofthermomechanical curve
Температура стеклования полимеров
101
Теперь перейдем к описанию деформации полимеров в условиях термо-
механических испытаний. Очень часто, особенно в условиях первичной оцен-
ки, полимер испытывается в виде порошка. Порошок можно испьп ьшать только
в условиях сжатия, причем, поскольку он помещается в специальную ячейку,
сжатие близко к всестороннему. Прежде всего следует иметь в виду, что по-
рошкообразный полимер - это еще не полимерный материал, а только его
основа. В результате термомеханического испытания охарактеризовывается
то вещество, которое находится под пуансоном, передающим нагрузку на об-
разец . Поэтому мы получаем термомеханическую кривую не полимерного
материала, а порошка полимера. Поскольку порошок может быть не очень
плотно уложен в ячейку' (не по вине экспериментатора, а вследствие своих
свойств), а также в связи с тем, что в порошке могут оставаться небольшие
количества растворителя или по бочных продукт св реакции форма термоме-
ханической кривой может существенно искажаться. Поэтому оценка физи-
ческих состояний полимера на порошкообразных образцах носит самый гру-
бый характер, особенно когда речь идет о новом, неизвестном полимере.
Д аже, если термомеханическая кривая имеет классический вид (см. рис. 18)
и состоит из трех участков, следует воздержаться от утверждения, что поли-
мер обладает всеми тремя физическими состояниями, переходя из одного в
другое при нагревании. Нужно учитывать, что возрастание деформации в по-
рошкообразном образце может быть вызвано побочными причинами. Опре-
делив термомеханическую кривую, лучше сперва обратить внимание на пос-
леднюю ветвь кривой. Если эта ветвь находится в интервале температур, где
термическая или термоокислительная деструкция еще не проходит достаточ-
но глубоко, можно говорить о течении полимеров. Что бы у бедиться в том,
что развитие большой деформации (до 100 % при сжатии) вызвано течением,
а не глубокой деструкцией полимера, необходимо параллельно сделать тер-
могравиметрический анализ (получить термогравиметрическую кривую). Эго
особенно важно в случае теплостойких полимеров, для которых развитие боль-
шой деформации наступает в интервале температур 600-800 °C, и эта деформа-
ция, вызванная глубокой термической деструкцией полимера, может быть
ошибочно принята за течение. Нужно учитывать также, что в процессе термо-
механических испытаний помимо деструкции может происходить и структу-
рирование. Эти два процесса всегда сосуществуют при нагревании полимера,
но один из них протекает с гораздо большей скоростью и определяет направ-
ление всего процесса. Структурирование может проявляться в образовании
поперечных связей между цепями полимера, в циклизации и т.д. В результате,
начавшееся течение полимера будет приостановлено, и на термомехани-
ческой кривой появится площадка, аналогичная по форме площадке высоко-
эластичности для линейных полимеров. Поэтому наличие такой площадки
102
Глава! V
(а вернее, задержанной деформации) еще не позволяет утверждать, что поли-
мер в данном интервале земператур наход ится в высокоэластическом состоя-
нии.
Убедившись в том, что большая деформация на последнем участке тер-
момеханическопкрпвойвызвана течением полимера, следует попытаться пе-
ревести его в монолитный образец любым из существующих способов (на-
пример, горячим прессованием). После этого образец вновь подвергается тер-
момеханическим испытаниям. Если форма повторной зермомеханической
кривой близка к первоначальной (полученной для порошкообразного образ-
ца), химические изменения, возможные в процессе переработки, несуществен-
ны. Теперь лишь следует убедиться, что плато на термомеханической кривой
отражает высокоэластическое состояние, а не вызвано побочными причина-
ми. Одной из характерных черт высокоэластической деформации является ее
полная обратимосзъ, а так же способность проявляться при очень малых на-
грузках (напомним, что модуль эластичности обычно назри-чсгыре десятич-
ныхпорядка меньше модуля упругости). Обнаружив площадку на термоме-
ханической кривой, необходимо проверить обра пзмость деформациив этой
области температур. Проверка осуществляется периодическим нагружением
образца, причем нагрузка должна превышать первоначальную, иногда в не-
сколько раз. Если при приложении дополнительной нагрузки деформация
резко возрастает, а после снятия ее быстро восстанавливается, можно гово-
рить об обратимости. Это отражено на рис.24. Зная величину добавочной
на1рузки и вызванную ею дополнительную обратимую деформацию, легко
рассчитать модуль эластичное™ делением добавочной нагрузки на при-
ращение деформации (Езл = Ео/Ее.). Убедившись, что модуль эластичное™
имеет небольшую величину (до ~ 1 МПа), можно утверждать, что полимер в
зоне плато действительно находится в высокоэласзическом состоянии. Для
£
дм/
Рнс.24. Проверка обратимости деформации в зоне предполагаемого плато высоко-
эластичности (см. текст)
Checking of reversibility of deformation in the zone of the supposed rubbery-like
plateau (See text)
Температура стеклования полимеров
103
Рис.25. Рычажное приспособление с переменным плечом
для поддержания постоянного натяжения в ходе процесса
ползучести (схема)
Lever mechanism with variable arm for ensuring of
constant tension in the course of creep process (Scheme)
жесткоцепных полимеров модуль эластичности будет существенно выше
(~ 10 МПа), и здесь главное - проверить обратимость деформации в зоне пла-
то. При этом нередко оказывается, что часть дополнительной деформации
будет обратимой (высокоэластической), а часть - необратимой (пластичес-
кой). В этом случае модуль эластичности определяется делением До на при-
рост обратимой части деформации, иговорить об истинной высокоэластич-
ности вряд ли следует.
Все сказанное справедливо, когда термомеханические испытания прово-
дятся и при действии растягивающего напряжения. При растяжении побоч-
ные эффекты не могут гак сильно исказить истинную картину, как при сжа-
тии. Проводя термомеханические исследования при небольшой растягиваю-
щей нагрузке и получая классическую термомеханическую кривую, нужно лишь
убедиться в том, что плато отражает нспппгуто высокоэластичность. Провер-
ка осуществляется аналогичным образом прикладывается дополнительная
нагрузка и после снятия ее проверяется обратимость деформации. Если дефор-
мация обратима, она носит высокоэластический характер. При проведении
термомеханических испытаний на растяжение следует стремиться поддержи-
вать напряжение постоянным. Здесь, в отличие от опытов при сжатии, вегре-
чаются с некоторыми трудностями При деформации образца поперечное се-
чение его уменьшается и, следовательно, при действии постоянной силы на-
пряжение в нем возрастает. Чтобы компенсировать прирост напряжения
вследствие развивающейся деформации, нагрузку на образец передают через
рычажное приспособление с переменным плечом (рис.25). При разви-
104
Глава IV
тии деформации плечо уменьшается и нагрузка автоматически падает так,
что напряжение в образце остается постоянным.
Убедившись в том, что плато на термомеханической кривой отражает
высокоэластичность, можно уверенно говорить о переходе в высокоэласти-
ческое состояние. “Точка” перехода находится описанными выше приемами.
До сих пор мы рассматривали термомеханическую кривую аморфных
полимеров, имеющую классический вид и показывающую три физические
состояния и две переходные температурные области между ними. Следует
учитывать, что термомеханическая кривая кристаллических полимеров мо-
жет иметь аналогичный вид (см. рис. 18). Наряду' с термомеханическими ис-
следованиями обязательно нужно провести рентгеноструктурный анализ, что-
бы убедиться в аморфности образца. Не останавливаясь пока на переходах в
кристаллических полимерах, рассмотрим отдельные случаи поведения амор-
фных полимеров в термомеханических условиях испытаний.
Сразу можно заметить, что классические термомеханические кривые
встречаются не всегда. Отклонение формы термомеханической кривой от клас-
сической связано с разными причинами Например, часто термомеханичес-
кая кривая аморфного полимера имеет вид, показанный на рис.19. Совер-
шенно очевидно, что вязкотекучего состояния данный полимер не проявляет.
Очевидно и то, что деформация образца (в условиях сжатия) не доходит до 100 %,
и, следовательно, образец сгорает при высоких температурах, а оставшаяся его
часть находится под пуансоном и не дает ему продавливаться до конца.
Осторожно следует относиться и к температурному интервалу, в котором
деформация быстро возрастает. Если деформация вблизи установления гори-
зонтальной площадки полностью обратима, она носит высокоэластический ха-
рактер. Однако без дополнительных экспериментов нельзя утверждать, что по-
лимер переходит в высокоэластическое состояние и подъем кривой указывает
на температуру стеклования. В полне вероятно, что при этой температуре проис-
ходит интенсивная сшивка цепей полимера, который при этом деформируется,
но впоследствии теряет плавкость и растворимость. Поэтому наряду с термоме-
ханическими испытаниями обязателен термогравиметрический анализ.
Иногда полимер дает классическую термомеханическую кривую, последняя
ветвь которой расположена в области очень высоких температур (700-800 °C).
Ясно, что развитие больших деформаций в этой области вызвано не вязким
течением, а интенсивной термодеструкцией. В этом случае нельзя говорить о
температуре текучести и о переходе в вязкотекучее состояние. Особенно оче-
видно это в тех случаях, когда термомеханическая кривая имеет вид, пока-
занный на рис.26. Искажение площадки высокоэластичности (если таковая
существует для испытуемого полимера) вызвано протеканием термодеструк-
ции, выделением газообразных продуктов и тд.
Температура стеклования полимеров
105
Рис26 . Т ермомеханическая кривая полимера, претерпевавшего термическую
деструкцию в ходе измерения (схема)
Thermomechanical curve of polymer undergoing thermal degradation during
the course of measurement (Scheme)
8
Рис.27. Появление '‘отрицательной” деформации на термомеханической кривой
(см. текст)
Appearance of “negative” deformation on the thennomechanical curve (See text)
В условиях сжатия термомеханическая кривая может также иметь вид,
изображенный на рис.27. В этом случае понижение и даже появление “отри-
цательной” деформации вызвано увеличением высоты образца, в результате
чего пуансон, передающий нагрузку на образец, несколько поднимается. Это
явление может быть вызвано выходом остаточного растворителя при нагреве
порошкообразного или монолитного образца, выделением газообразных про-
дуктов деструкции и т.д. В таком случае определение истинной температуры
стеклования и текучести затрудняется.
Одна из причин появления отрицательной деформации может заключаться
в следующем. В процессе подготовки к термомсханическим испытаниям по-
рошкообразный полимер спрессовывается при комнатной температуре, кото-
рая, как правило, лежит гораздо ниже области стеклования. Следовательно,
полимер подвергается при этом вынужденно-эластической деформации. При
термомеханическом испытании температура повышается и, когда она дости-
106
Глава IV
Рис. 28. Снижение деформации на термомеханической кривой, определяемой в условиях
растяжения (см. текст)
Decreasing of deformation on the thermomechanical curve determining under
conditions of tension (See text)
гает интервала стеклования, вынужденно-эластическая деформация становит-
ся быстро обратимой. Образец расширяется и, если опыт проводится в усло-
виях сжатия при малой нагрузке, на термомеханической кривой появляется
“отрицательная” деформация. Чем выше давление, приложенное к порошко-
образному полимеру при таблетировании, тем больше отрицательная дефор-
мация и тем сильнее искажается форма термомеханической кривой. В этом
случае температура стеклования соответствует понижающейся, а не повы-
шавшейся ветви термомеханической кривой.
При термомеханическом испытании полимера на растяжение деформация
также иногда уменьшается с ростом температура, а не возрастает (рис.28).
Сокращение образца может происходить в результате сшивки, циклизации
и т.д., проходящей в процессе самого испытания.
Особенно часто наблюдаются случаи, когда термомеханическая кривая
состоит из двух (см. рис. 19), а не трех участков. Как правило, это связано с
тем, что полимер имеет недостаточно большую молекулярную массу, и не
достигая высокоэластического состояния, переходит из стеклообразного не-
посредственно в вязкотекучее состояние. Подъем соответствующей ветви тср-
момеханической кривой определяет температуру текучести, и можно сказать,
что температуры стеклования (размягчения) и текучести совпадают. Когда
вторая ветвь термомеханической кривой лежит в области высоких темпера-
тур, нужно убедиться, что в этой области деструкция еще не происходит и
резкое возрастание деформации действительно связано с течением полиме-
ра, а не с его разложением. В последнем случае размягчение и течение еще нс
достигнуты, и во всем возможном интервале температур полимер находится
в стеклообразном состоянии.
Когда переходы из стеклообразного состояния в высоко эластическое и из
высокоэластичсского в вязкотскучее хорошо выражены на термомсханичес-
Температура стеклования полимеров
107
Thermomechanical curve with “blurred” transition (Scheme)
кой кривой (деформация в температурных интервалах переходов быстро воз-
растает), точки переходов TgnTj легко определить. Однако часто термомеха-
ническая кривая имеет вид, показанный на рис.29. С помощью такой термо-
механической кривой невозможно определить температуру стеклования: пе-
реход сильно “размыт”. Поэтому следует использовать другие методы.
Переходы из стеклообразного состояния в высокоэластическое и из высоко-
эластического в вязкогеку чес, а также температуру плавления полимеров мож-
но определять такими методами, как дилатометрический, калориметрический,
оптический; целесообразно пользоваться также динамическими методами
исследования и т.д. (см. ниже).
Перейдем теперь к кристаллическим полимерам. В одних случаях форма
термомеханических кривых кристаллических полимеров отличается от формы
тех же кривых для аморфных полимеров, а в других случаях почти совпадает.
Только с помощью термомеханического метода исследования нельзя от-
ветить на вопрос, является полимер аморфным или кристаллическим. Пред-
варительно необходимо получить рентгенограмму образца, а затем сопоста-
вить данные рентгеноструктурного анализа с результатами термо-
механического исследования. Полимеры в кристаллической форме могут
получаться непосредственно в процессе синтеза и дальнейшей обработки.
Термомсханическая кривая для кристаллического полимера может иметь
вид, схематически показанный на рис.30. Если полимер достаточно глубоко
закристаллизован, высокоэластическое состояние практически полностью
подавляется и в широком интервале температур он не обнаруживает замет-
ных деформаций. При подходе к температуре плавления деформация резко
возрастает и достигает максимального значения.
Термомеханическая кривая на рис.30 соответствует случаю, когда высо-
коэластическая деформация полностью подавлена кристаллизацией В реаль-
108
Глава JI
Рпс.30. Термомеханическая кривая хорошо закристаллизованного полимера (схема)
Thermomechanical curve of well-crystallized polymer (Scheme)
Рис.31. Термомеханическая кривая аморфно-кристаллического полимера (схема)
Thermomechanical curve of semi-crystalline polymer (Scheme)
ных условиях в кристаллическом полимере содержатся аморфные области,
которые при нагревании ведут себя подобно аморфному полимеру. При тср-
момеханическом исследовании аморфно-кристаллических полимеров в ин-
тервале стеклования деформация будет возрастать с дальнейшим образова-
нием плато (рис.31). Однако площадка высокоэластичности не будет иметь
такую же высоту', как и для чисто аморфного полимера того же строения. Она
будет существенно ниже в зависимости от степени кристалличности. Во вся-
ком случае, для аморфно-кристаллического полимера можно определять тем-
пературу стеклования и температуру текучести.
Особо следует остановиться на полимерах, которые легко кристал-
лизуются в процессе самого термомеханического испытания. Тогда опыт луч-
ше всего вести с периодической нагрузкой*.
* Во всех случаях, когда термомсхапическим методом проводятся более тонкие исследова-
ния, нагрузку лучше прикладывать периодически, давая образцу отдых, т е. возможность вос-
станавливать деформацию после снятия па|рузки. Тогда все эффекты стеклования, кристал-
лизации и т.д. будут проявляться более отчетливо.
Температура стек'юванпя полимеров
109
Рис.32. Схематическое изображение термомеханической кривой аморфно-
кристаллического полимера, определяемой при периодическом изменении нагрузки
(см. текст)
Schematic representation of thermomechanical curve for a semi-crystalline
polymer determined at periodic variation of load (See text)
Быстрая кристаллизация аморфного полимера наступает при температу-
ре выше Tg. Поэтому аморфный полимер, способный к быстрой кристалли-
зации, в стеклообразном состоянии ведет себя подобно обычному аморфному'
полимеру, кристаллизующемуся трудно. При термомеханическом испытании
он дает малую деформацию вплоть до температуры стеклования. Пройдя эту
температуру, полимер развивает большую высокоэластическую деформацию
(рис.32, кривая 2). Однако появление кристаллизации при Т> Tg придает по-
лимеру жесткость, и если нагрузка прикладывается периодически, деформа-
ция быстро уменьшается (см. рис.32, кривая 2). После того, как полимер до-
статочно глубоко закристаллизуется, его деформация уже не будет отличать-
ся от деформации заранее закристаллизованного полимера (см. рис.32, кривая
1). Температуры текучести также будут совпадать (см рис .32).
Естественно, что поведение кристаллических полимеров в условиях тер-
момеханических испытаний не ограничивается рассмотренными выше слу-
чаями. Реальные полимеры могут давать самые различные формы термоме-
ханических кривых. Однако приведенные примеры, по-видимому, позволят
читателю ориентироваться во всем многоо фазии экспериментальных т ер
момеханических кривых и оценивать с их помощью физические состояния и
температуры переходов из одного состояния в другое.
Теперь кратко остановимся на дру гих способах экспериментальной оценки
температуры стеклования полимеров. Одним из них является дилатометри-
ческий, согласно которому измеряется зависимость удельного объема поли-
мера от температуры. Схематически эта зависимость изображена на рис. 13.
Такая зависимость в виде ломаной прямой характерна для многих полимеров
вблизи температуры стеклования. При температурах, лежащих ниже точки
стеклования, эта зависимость более пологая, чем при температурах, лежа-
щих выше точки стеклования. Это и является причиной излома , который на-
но
Глава IV
блюдается при температуре стеклования (см.рис.13). Естественно, что дила-
тометрические зависимости можно получать при разной скорости нагрева (или
охлаждения) образца. В результате оказывается, что если менять скорость
нагрева в достаточно широких пределах, то дилатометрические зависимости
будут изменяться так, как это схематически изображено на рис. 13. Чем выше
скорость нагрева образца, тем при более высокой температуре будет наблю-
даться излом и наоборот, чем меньше скорость нагрева, тем при более низкой
температуре будет наблюдаться излом, а сам удельный объем в точке излома
будет уменьшаться. Таким образом, температура стеклования зависит от ско-
рости нагрева или охлаждения образца, что связано с релаксационным меха-
низмом процесса застекловывания (см. ниже).
Следует отметить, что изменение скорости нагревания или охлаждения не
так существенно сказывается на характере дилатометрической кривой и на по-
ложении температуры стеклования, как изменение величины механического
напряжения на характере термомеханической кривой и на температуре стекло-
вания. Для того, чтобы в первом случае температура стеклования существенно
изменилась, необходимо изменить скорость нагревания или охлаждения на много
десятичных порядков, что экспериментально невозможно. Изменение же ско-
рости нагревания в десять или в сто раз приводит к изменению температуры
стеклования всего лишь на несколько градусов. Между тем, изменение
механического напряжения всего лишь на несколько процентов может привес-
ти к резкому увеличению или снижению температуры стеклования. Особую роль
при этом играет скорость механического воздействия.
Анализ этих экспериментальных фактов позволил Г.М. Бартеневу (53]
высказать точку зрения о том, что следует различать механическое и структур-
ное стеклование. Под структурным стеклованием понимается процесс пере-
хода из стеклообразного со юяния в высокоэластическое (и обратный про-
цесс) в отсутствии механического напряжения. Этот переход и его темпера-
тура связаны только с химическим строением полимера и точка стеклования
зависит только от скорости нагревания и охлаждения Механическое стекло-
вание связано с быстрым механическим воздействием на полимер и его пове-
дением как твердого тела при быстром действии механической силы. Когда
на полимерный материал действует механическая сила и одновременно из-
меняется температура, механическое стеклование может наступить при го-
раздо более низких температурах, чем структурное стеклование.
Помимо дилатометрического метода определения температуры стек-
лования получили распространение и другие методы, например, калори-
метрический В этом случае измеряется теплоемкость Q полимерного тела
при изменении температуры, причем зависимость Ср от Т имеет характерный
вид (рис.33).В области стеклообразного состояния теплоемкость слабо увели-
Температура стеклования полимеров
111
Рис.33. Температурная зависимость теплоемкости Ср (схема)
Temperature dependence of heat capacity Cp (Scheme)
чивается с ростом температуры, но при достижении Tg наблюдается скачок
теплоемкости в сторону ее увеличения. После прохождения переходной зоны
теплоемкость вновь увеличивается весьма слабо с ростом температуры.
Наибольшее распространение, по-видимому, получил динамический ме-
ханический анализ согласно которому измеряются температурные зависимо -
сти действительной Е' и мнимой Е" частей комплексного модуля упругости
Е* = Е' + iE", а также тангенса угла механических потерь tgSp = Е"/Е' (рис.34).
Температурная зависимость tgSE обнаруживает несколько максимумов, из ко-
торых наиболее интенсивный (и высокотемпературный) связан с переходом
из стеклообразного состояния в высокоэластическое.
Рис.34 .Т емпературная зависимость та п енса угла механических потери ( схем^
Temperature dependence of the mechanical loss -factor tg§E(Scheme)
112
Глава IV
Рис.35. Температурная зависимость тангенса угла диэлектрических потерь tgd£ (схема)
Temperature dependence of the dielectric loss-factor tgd£ (Scheme)
Аналогичным образом выглядит температурная зависимость тангенса угла
диэлектрических потерь tgiy. = где е" и е' - действительная и мнимая
части комплексной диэлектрической проницаемости е*= г' + /е"(рис.35).
Наконец, температура стеклования может быть определена по резкому
увеличению коэффициента диффузии. Не останавливаясь подробно на этих
методах экспериментального определения Т,„ отошлем читателя к специаль-
ной литературе [5, 51, 124].
IV.2. Механизм сзекловапня
Механизм процесса стеклования изучается на протяжении многих лет,
но единой точки зрения о механизме застекловывания не существует. Поэто-
му рассмотрим основные общепринятые точки зрения относительно этого
механизма. Заметим, что эти точки зрения нс противоречат друг другу.
Рассмотрим в первую очередь концепцию о релаксационном механизме
процесса застекловывания, которую выдвинули А П.Александров, Ю.С.Ла-
зуркин, П. П. Кобеко, Г И. Гуревич. Предварительно еще раз заметим, что пере-
ход в стеклообразное состояние и обратный переход в высокоэластическос
состояние не являются фазовыми переходами первого или второго рода (на-
помним, что, согласно определению Эренфеста, фазовым переходом первого
рода называется такой переход, при котором термодинамические функции
имеют резкий скачок при температуре перехода, а производная температур-
ной зависимости термодинамической функции имеет разрыв. Фазовым пере-
ходом второго рода называется такой переход, при котором температурные
Температура стеклования полимеров
ИЗ
зависимости термодинамических функций при температуре перехода изме-
няются плавно, а разрыв имеют лишь вторые производные этих функций).
Внешне некоторые температурные зависимости термодинамических фун-
кций в случае полимеров выглядят так, как это характерно для перехода вто-
рого рода. Например, температурная зависимость удельного объема (дилато-
метрическая кривая) имеет излом при температуре стеклования, по которому7,
собственно, и определяется эта температура (см. рис. 13). Однако это сход-
ство является чисто внешним, ибо рассматриваемый переход не связан с по-
явлением новой фазы, как в случае плавления или кристаллизации из распла-
ва, а является переходом из одного физического состояния в другое при со-
хранении единой жидкой фазы. Таким образом, твердое стекло по фазовому
состоянию представляет собой жидкость, так же как эластомер и текущий
полимер. Они различаются между собой лишь по агрегатному состоянию,
представляя единую жидкую фазу.
Теперь перейдем к рассмотрению релаксационного механизма застекло-
вывания Описание этого механизма начнем с низкомолекулярных веществ.
Каждая молекула такого вещества занимает определенное положение в про-
странстве и совершает колебательные движения вследствие тепловыхф лук-
туаций, т.е. в результате превышения температуры данной точки простран-
ства над средней температурой. Чтобы перейти в другое положение в про-
странстве, необходимо затратить какую-то энергию. Пусть кинетическая
единица (атом или молекула) находится в таком положении, когда ее потенци-
альная энергия минимальна. Этот случай, показанный на рис.36, соответству-
ет устойчивому равновесию. Для того, чтобы перевести кинетическую едини-
Рис.36. Нахождение кинетической единицы в минимуме потенциальной энергии (схема)
Location of kinetic unit in the minimum of potential energy (Scheme)
114
Глава IV
цу в новое состояние равновесия, необходимо преодолеть некоторый энерге-
тический барьер, который и является энергией перехода и называется потен-
циальным барьером. Таким образом, потенциальный барьер- это та энергия,
которую нужно затратить кинетической единице, чтобы перейти в новое поло-
жение. Рассмотрим из чего складывается этот барьер.
Каждая термодинамическая система характеризуется величиной свобод-
ной энергии или изобарно-изотермическим потенциалом F:
Р' =11 -TS,
(63)
где U-внутренняя энергия системы; Т- абсолютная температура; S - энтро-
пия.
Величина внутренней энергии Uскладывается из энергии поступательного
движения частиц, энергии их вращательного движения, а также энергии коле-
бательного движения частиц относительно своего положения равновесия. Что
касается энтропии, то, согласно определению Больцмана, энтропия равна
S = кБ In 1Г ,
(64)
где кБ - константа Больцмана; IV - термодинамическая вероятность суще-
ствования системы.
Понятие энтропии как статистической характеристики лучше всего про-
демонстрировать на примере различных конформаций макромолекул Возьмем
один из крайних случаев - полностью распрямленную цепь (рис.37,а). Есте-
ственно, что такая цепь может иметь только одну конф ормацию, а расстояние
Рис.37. Схематическое изображение выпрямленной (а) и скрученной (б) цепи полимера
(h - расстояние между концами цепи)
Schematic representation of straightened (a) and twisted (6) polymer chain
(Л is a distance between the chain ends)
u
Температура стеклования полимеров
115
между ее концами h будет равно полной длине макромолекулы. Вероятность
существования такой конформации чрезвычайно мала, поскольку она всего
лишь одна Если же взять цепь свернутую (см рис.37, б), то при одном и том
же расстоянии между' концами цепи сама цепь может принимать множество
различных конформаций, т.е. ее элементы могут располагаться в простран-
стве самым различным образом при сохранении одинакового расстояния меж-
ду концами цепи. Чем меньше это расстояние h (до определенного предела),
тем больше конформаций макромолекула может принять и тем больше веро-
ятность существования макромолекулы в таком (ввернутом )виде. Пэскольку
энтропия, согласно определению Больцмана пропорциональна логарифму’
термодинамической вероятности, то энтропия будет тем больше, чем меньше
расстояние между концами макро молекулы Согласно второму закону термо -
динамики, самопроизвольный процесс всегда происходит с увеличением энт-
ропии, и поэтому в случае гибких макромолекул тепловое движение приводит
к их скручиванию.
Возвращаясь к системе, построенной из малых молекул, заметим, что при
переходе кинетической единицы из одного состояния в другое будет меняться
как внутренняя энергия .так и энтропия Изменение свободной энергии опре -
делится из соотношения
Д/г = Д7/-7ДХ, (65)
где Д U - изменение внутренней энергии; AS' - изменение энтропии.
Вероятность того, что данная кинетическая единица находится в опре-
деленном положении в пространстве, зависит от ее энергии и абсолютной тем-
пературы. Эта вероятность определяется соотношением
И'=уое“£/7?7’, (66)
где Vq - собственная частота колебаний атомов или молекул; Е - энергия час-
тицы; R универсальная газовая постоянная; Т- абсолютная тем пература.
Уравнение (66) применяется в тех случаях,когда расчет ведет ся на 1 моль
вещества. Если же расчет ведется на одну молекулу, вместо R следует подста-
вить величину кр (константа Больцмана). Эти величины связаны соотношени -
ем кБ = R/Na? гДе = 6.O23-1O23 моль’1 (число Авогадро).
Хорошо известно, что собственная частота колебаний атомов в твердых
телах составляет ~1013 Гц, а время (период) одного колебания определяется
из соотношения т0 = 1 А'о Интересно, что время оседлой жизни атома в дан-
ном положении составляет 1()'10-10’11 с. Рассматривая переход полимера из
стеклообразного состояния в высокоэластическое, введем такую характерис-
тику, как время этого перехода. Это время тр определяется из соотношения
116
Глава IV
5
tp=W^> (67)
где Гр 0 - предэкспоненциальный сомножитель, который представляет собой
обратную величину от собственной частоты колебаний атомов; Up - энергия,
которую необходимо затратить, чтобы переход осуществился.
Чем меньше величина тр, тем быстрее осуществляется переход, т е. пере-
ход осуществляется тем легче, чем выше температура и чем меньше величина
энергии перехода Up.
Будем рассматривать высокоэластическое полимерное тело, которое ох-
лаждается с некоторой скоростью изменения температуры. Если при данной
температуре время перехода структурных элементов (кинетических единиц)
из одного состояния в другое мало, таюй переход осуществляется быстро и
изменение структуры будет происходить сразу же вслед за изменением темпе-
ратуры.
Можно сказать, что если время перехода тр гораздо меньше времени на-
блюдения тн, то переход будет осуществляться очень быстро и равновесное
состояние стру ктуры полимерного тела устанавливается очень легко вслед за
изменением температуры. При снижении температуры уменьшается тепловая
энергия, которая характеризуется произведением R Т. и время релаксации (см
уравнение (67)) существенно возрастает. Если, однако, оно остается меньше
времени наблюдения за системой, переход осуществляется достаточно быстро.
Дальнейшее снижение температуры приведет к резкому’ возрастанию тр и к вы-
полнению условия тп «• тр, а затем и тп < тр. Это соответствует случаю, когда
время наблюдения становится гораздо меньше, чем время перехода и, естествен-
но, что этот переход уже не удается наблюдать, хотя он и происходит. Переход
осуществляется настолько медленно, что наблюдение за шгмстановится затруд-
нительным. Поэтому полимерное тело становится твердым, те. переходит в стек-
лообразное состояние. Еще раз следует отметить, что на самом деле структура
полимерного тела при таком переходе нс является равновесной, она как бы за-
мораживается в том состоянии, которое непосредственно предшествует перехо-
ду. Если бы удалось проследить за процессом перехода чрезвычайно дл итс ль-
ное время, можно было бы заметить, что этот переход осу ществляется, хотя и
очень медленно. Таким образом, согласно этим представлениям переход из вы-
соко эластичного состояния в стеклообразное является релаксационным процес-
сом и связан с тем, что при определенной для каждого полимера температуре
время релаксации (время перехода) становится чрезвычайно велико и полимер-
ное тело начинает вести себя как твердое стеклообразное тело.
Эта теория перехода была впоследствии математически обоснована М.В.
Волькснштейном и О.Б.Птициным, которые провели математический анализ
Температура стеклования полимеров
117
релаксационного перехода из высокоэластического состояния в стеклообраз-
ное и обратно и установили условия, при которых такие переходы могут осу-
ществляться. Согласно этой теории
, ^р.о^р Up 1 ГбЯА
In— / +------— = 1g— , (Р8)
ЛГ2 RTg q
где q - скорость охлаждения системы.
Уравнение (68) показывает, что температура стеклования зависит от ско-
рости охлаждения полимера.
Теория Волькенштейна-Пгицина в ее первоначальном варианте не учи-
тывает кооперативного характера теплового движения кинетических единиц.
Как было показано впоследствии, такой учет совершенно необходим, т.к. вслед-
ствие большой длины и гибкости макромолекул локальное движение какой-
либо одной кинетической единицы затрагивает и своих соседей. Так, движе-
ние в пределах одного повторяющегося звена макромолекулы приводит к дви-
жению соседних звеньев, что и является причиной появления сегментальной
подвижности. Учет кооперативности теплового движения кинетических еди-
ниц различных размеров, проведенный Птициным и Шароновым, привел к
уточнению теории Волькенштейна-Птицина и позволил голучить разумные
величины энергии активации релаксационного процесса.
Анализ с помощью нелинейной модели [87, 89] полимерного тела пока-
зывает [90], что переход из стеклообразного состояния в высокоэластическое
должен происходить не в точке, а в некотором интервале температур, который
зависит от скорости нагревания или охлаждения q. Схематически это изобра-
жено на рис.38, где дано изображение термомеханической и дилатомстричес-
Рис.38. Схематическое изображение термомеханической (а) и дилатометрической (б)
кривых в зоне перехода из стеклообразного состояния в высокоэластическое (см.тскст)
Schematic representation of thermomechanical (a) and dilatometric (6) curves in the zone о f
transition from the glassy state to rubbery state (See text)
118
Глава IV
кой кривых в области перехода. Здесь Tgl - температура, при которой начи-
нается переход из высокоэластического состояния в стеклообразное при ох-
лаждении системы, a Tg 2 - температура, при которой этот переход заканчива-
ется.
Полученный в работе [90] результат показывает, что при малых скоростях
охлаждения температурный интервал стеклования вырождается и остается
только одна температура стеклования. В общем случае при анализе поведения
полимера в области Т< Tg, ।, большую роль играет скорость охлаждения. Если
скорость охлаждения велика, то температуры Tgl и Tg 2 существенно нс со-
впадают, т.е. переход в стеклообразное состояние происходит в более широ-
ком интервале температур.
На практике при увеличении скорости охлаждения (или нагревания) тем -
пераэурный интервал, в котором происходит процесс стеклования, действи-
тельно расширяется, но не скачкообразно, а достаточно плавно. При этом с
понижением скорости охлаждения (или нагревания) зависимость деформа-
ции от температуры в области перехода становится более крутой. Это видно
из рис. 39, на котором показаны термомеханические кривые для ряда отверж-
денных компаундов на основе эпоксидных олигомеров. В соответствии с по-
лученными теоретическими результатами температурный интервал стеклова-
ния расширяется с увеличением скорости нагревания (рис.40).
Рис.39. Термомеханические кривые, измеренные в условиях сжатия, для отвержден-
ного компаунда на основе эпоксидной смолы. Скорость нагрева, град/мин, равна: 0,62( 1);
1,07(2); 2,39(3); 4,61(4); 6,39(5); 9,44(6); 15,74(7). Состав композиции: ЭД-20 - 1 моль,
МТГФА - 0,6 моля, азелаииовая кислота - 0,7 моля
Thermomechanical curves of cured epoxy compound measured under conditions of
compression. Rate of heating 0,62(1), 1,07(2), 2,39(3), 4,61(4), 6,39(5), 9,44(6),
and 15,74(7) deg/min.Composition of specimen; ED-20 - 1 mole, MTHPA - 0,6 mole,
azelaic acid - 0,7 mole
Температура стеклования полимеров
119
Р ис40 .Зав исимостъ разности температурЛ? =Т&\ -Т6тот торости нагревания </'.(а) со -
ответствует композиции, указанной в подписи к рис.39; (б) соответствует композиции:
ЭД- 20 - 1 моль, МТГФА - 1,3 моля, олеиновая кислота - 0,7 моля
Dependence of temperature differences ДГ = 7’gj - on of heating rate q:
(a) correlates with the same composition as in figure 39; (a) corresponds to the composition
of specimen: ED-20 — 1 mole, MTHPA — 1 3 mole, oleic acid —0,7 mole
Выше мы рассмотрели одну из концепций перехода полимерного тела из
стеклообразного состояния в высокоэластическое и обратно. Эта концепция
основана на допущении релаксационного механизма процесса перехода, при-
чем время такого перехода (время релаксации) зависит от температуры и оп-
ределяется величиной энергетического барьера, который необходимо преодо-
леть, чтобы переход осуществился. Поскольку разные макромолекулы обла-
дают различной гибкостью или жесткостью, они характеризуются разной
величиной энергетического барьера, и отсюда -разными температурами стек -
лования. Чем более гибкой является макромолекула, тем меньшей величиной
энергетического барьера характеризуется система, построенная из таких мак-
ромолекул.
Концепция перехода полимерного тела из стеклообразного состояния в
высокоэластическое и обратно, основанная на релаксационном механизме
процессов перехода, не является единственной. Другой концепцией, объясня-
ющей эти переходы, является концепция С.Н.Журкова, основанная на призна-
нии большой роли межмолекулярных связей в полимерах. Разберем эту кон-
цепцию подробней.
В каждой полимерной макромолекуле имеются полярные группы, об-
ладающие различными силами взаимодействия друг с другом. Сйи о (разуют
связи, которые не являются химическими ,а имеют физическую природу и
называются межмолекулярными связями. Такие связи могут быть весьма ела-
120
Глава IV
быми, как, например, в полиизобутилене, полипропилене, полиэтилене и в
других аналогичных полимерах, но могут быть и очень сильными, как, на-
пример, в полиамидах, сложных полиэфирах и т.д. В первом случае речь идет
о слабых Ван-дер-Ваальсовых связях, энергия которых очень мала. Во вто-
ром случае речь идет о сильных водородных связях, возникающих из-за на-
личия амидных групп — NH— С — (полиамиды), и о сильном диполь-диполь-
О о
и II
ном взаимодействии за счет сложноэфирных групп — С — О—. Межмолеку-
лярные связи могут быть обусловлены наличием не только этих полярных
группировок, но и наличием других групп, способных к сильному межмоле-
кулярному взаимодействию. К таким группам относятся нитрильная — С — N
(например, в полиакрилонитриле), гидроксильные группы — ОН (например,
в поливиниловом спирте) и многие другие. Полярные группы в соседних мак-
ромолекулах при определенных условиях могут взаимодействовать друг с
другом, образуя поперечные мостики между ними, которые являются доста-
точно прочными. Напомним, что речь идет не о химических связях, а о связях
физического характера. Схематически это изображено на рис. 41.
Необходимо обратить внимание еще на одно очень важное обстоятель-
ство. Межмолекулярные связи, в отличие от химических связей, являются
достаточно слабыми. Энергия, которую необходимо затратить для их разру-
шения, приблизительно на порядок меньше энергии, которую необходимо зат-
ратить на разрушение химических связей. Напомним, что энергия разрыва хи-
мических связей составляет несколько десятков ккал/моль, а энергия разрыва
межмолекулярной связи составляет всего лишь несколько ккал/моль. Наиболее
Рис.41. Схематическое изображение сетки физических (межмолекулярных) связей
Schematic representation of a network of physical (intermolecular) bonds
Температура стеклования полимеров
121
сильными связями обладают амидные, сложноэфирные, гидроксильные и
некоторые другие группы. Однако энергия разрыва межмолекулярных свя-
зей, образованных этими группами, не превышает десяти ккал/моль (а фак-
тически эта энергия меньше).
Таким образом, межмолекулярные связи не являются стабильными, а не-
прерывно распадаются под действием тепловых флуктуаций и возникают в
новых местах. Они как бы перемещаются вдоль полимерной цепи и тем са-
мым связывают не строго определенные звенья макромолекул, а те звенья, в
которых они возникают в данный момент. Однако при каждой данной темпе-
ратуре количество одновременно существующих межмолекулярных связей
примерно одинаково для полимеров определенного химического строения.
Можно сказать, что при эквивалентном характере рассмотрения межмо-
лекулярные связи действуют в определенных местах макромолекул, обра'уя
своеобразную сетку и не давая возможности перемещаться им относительно
друг друга. Учитывая, что на самом деле межмолекулярные связи непрерыв-
но возникают и распадаются под действием тепловых флуктуаций, равнове-
сие можно сдвинуть в ту или иную стсро нуте .в сторону образования или
распада межмолекулярных связей Как и в случае обычной химической реак-
ции, при повышении температуры равновесие сдвигается в сторону распада
межмолекулярных связей, а при понижении температуры - в сторону их обра-
зования. При охлаждении полимерного тела, находящегося в высокоэласти-
ческом состоянии, количество образующихся межмолекулярных связей все
время возрастает При достижении определенной температуры количество
образовавшихся мостичных связей достигнет величины, достаточной для того,
чтобы закрепить отдельные макромолекулы относительно друг друга и не до-
пустить их трансляционной подвижности. При этом вибрационная подвиж-
ность остается.
Образование достаточно частой и прочной сетки межмолекулярных свя-
зей при охлаждении полимера до определенной температуры способствует тому,
что при этой температуре подвижность макромолекул в достаточной степени
утрачивается и полимерное тело в целом становится твердым, т.е. переходит в
стеклообразное состояние. Температура, при которой наблюдается это явле-
ние, и есть температура стеклования. При нагревании полимерного тела, на-
ходящегося в стеклообразном состоянии, равновесие сдвигается в сторону
распада межмолекулярных связей, и когда этот процесс распада зайдет дос-
таточно далеко, макромолекулы приобретут трансляционную подвижность и
полимерное тело размягчится, те. перейдет в высокоэластич еское состояние
Такова, согласно рассматриваемой концепции, природа переходов полимер-
ного тела из высокоэластического состояния в твердое стеклообразное и об-
ратно.
122
Глава IV
Чтобы такой переход осуществился, в каждом полимерном теле не-
зависимо от химического строения макромолекул должно образоваться опре-
деленное число межмолекулярных связей. Обозначим это число черезх. Кон-
центрацию активных групп в полимере, способных к образованию меж-
молекулярных связей, обозначим через N. Учитывая, что межмолекулярные
связи непрерывно распадаются и возникают в других местах, и что суще-
ствует равновесие между числом образовавшихся и распавшихся связей, мож-
но в данном случае для описания этого процесса применить обычное уравне -
ние химической кинетики, описывающее равновесный процесс:
х^2(У-2х). (69)
В левую часть уравнения входят число межмолекулярных связей, образу-
ющихся при данной температуре; в правой части уравнения в скобках показа-
но число оставшихся активных групп, способных к образованию таких свя-
зей. Двойка в скобках появляется потому, что на образование каждой связи
расходуются две активные группы. По этой же причине двойка появляется и
перед скобкой. Поскольку рассматриваемая нами реакция является равновес-
ной, к ней применим закон действующих масс, согласно которому отношение
концентрации образовавшегося вещества к концентрации исходного вещества
является постоянной величиной, равной константе равновесия. При этом сте-
хиометрические коэффициенты уравнения реакции входят в показатели сте-
пени соответствующих концентраций. Таким образом, в нашем случае
(AZ~ 2x)2 = kp , (70)
где кр - константа равновесия.
Согласно Аррениусу, температурная зависимость константы равновесия
описывается следующим соотношением
_А
kp=A*e~llT, (71)
где А * - стерический фактор; Е - энергия связи (в нашем случае - межмолеку-
лярной); R - универсальная газовая постоянная; Т- абсолютная температура.
Подставив (71) в (70), получаем
Е
x = (N -2х)2 A*eRT . (72)
Уравнение (72) связывает число образовавшихся межмолекулярных свя-
зей с числом активных групп, способных к образованию таких связей, и с
температурой.
Для того, чтобы выявить механизм процесса застекловывания полиме-
ров, необходимо блокировать часть полярных групп тем или иным способом,
Температура стеклования полимеров
123
чтобы выключить их из взаимодействия. Блокировку можно вести различ-
ным способом, например, путем небольшого изменения химического строе-
ния макромолекулы или введением малых молекул низкомолекулярных ве-
ществ, оказывающих пластифицирующее действие. Такое введение можно
осуществить путем сорбции полимером малых молекул сорбата, находящих-
ся в газообразном состоянии Допустим, что в потимерное тело ввели п таких
малых молекул и тем самым заблокировали такое же количество полярных
групп. В этом случае (72) запишется так:
Е_
х = (Н-2х-и)2А*е,{Т' , (73)
где п - число молекул растворителя.
Чтобы выполнилось условие равновесия, и количество образовавшихся
межмолекулярных связей вновь составило величину х, необходимо изменить
температуру, т.е. понизить ее, чтобы величина в скобках в правой части урав-
нения (73) уменьшилась. Поэтому в уравнении (73) вместо Т стоит 1\ .
Решим совместно систему уравнений (72) и (73) и в результате получим
следующее соотношение
В уравнении (74) Т соответствует температуре стеклования исходного,
непластифицированного полимера, а 7) температуре стеклования пластифи-
цированного полимера, поскольку по условию х - это число межмолекуляр-
ных связей, необходимых для офазования прочной пространственной сетки.
При образовании такой сетки полимер переходит в стеклообразное состояние.
Уравнением (74) можно пользоваться для описания процесса перехода
полимера из твердого в высокоэластическое состояние при введении в него
определенного количества пластификатора. Однако это уравнение можно су-
щественно упростить. Во-первых, будем считать, что количество межмолеку-
лярных связей значительно меньше, чем общее количество полярных групп,
способных к межмолекулярному взаимодействию (именно так и происходит в
действительности, как показали эксперименты: для образования прочной сет-
ки межмолекулярных связей и для отверждения полимера необходимо зафик-
сировать всего лишь менее 10 % полярных групп). Иными словами соблюда-
ется условие
N » 2х . (75)
Введем второе допущение, а именно, будем работать с малым количест-
вом молекул пластификатора (естественно, что такое допущение можно и не
вводить). Тогда запишем, что
124
Глава IV
n!N«A- (76)
Проведя логарифмирование уравнения (74) и учитывая, что при усло-
вии (76) 1п(1 - и / N) = -и / N, запишем
_Е Т-Тх
N R Т{Г
(77)
Если в полимер ввели малое количество молекул пластификатора, темпе-
ратуры стеклования исходного и пластифицированного полимеров не будут силь-
Т — Т !ГГ
норазличаться и можно допустить, что Т ~ Т\ и -1 = . При этих услови-
ях соотношение (77) будет выглядеть так
2а= N Ry1' (78)
Отсюда EN (79)
ИЛИ ДГ = ал , (80)
где 2RT2 а = . EN (81)
Уравнение (80) отображает правило мольных долей Журкова. Согласно
этому правилу, снижение температуры стеклования пластифицированного
полимера по сравнению с температурой стеклования исходного полимера про-
порционально молярной доли введенного пластификатора. Это правило озна-
чает, что независимо от химического строения пластификатора депрессия тем-
пературы стеклования одинакова, если вводятся равные молярные доли
пластификатора. Эго вполне понятно, т. к., согласно данной концепции, отверж-
дение полимера (переход из высокоэластического состояния в стеклообраз-
ное) происходит при образовании одинакового количества межмолекулярных
связей, и если каким-либо путем вывести часть полярных групп из работы,
то снижение температуры стеклования будет зависить только от количества
этих групп, а не от химического строения блокирующих молекул.
Эксперименты и расчеты, проведенные С.Н.Журковым с сотрудниках»,
приводят к значениям энергии мсжмолскулярного взаимодействия для раз-
личиях групп, показанным в табл. 17.
Из этой таблицы отчетливо видно, что энергия межмолекулярного взаи-
модействия имеет небольшие величины, на порядок меньшие, чем энергия
Температура стеклования полимеров
125
Таблица 17
Величины энергий межмолекулярного взаимодействия
д ня различных iio.Tnprri.ix групп
Values of energies ofintennolecular interaction for various polar groups
Полимер Группа Е. ккал/моль
Полиметилметакрилат 1 /С о о о I 2,25
Полиакрилонитрил - С= N 2 22
П олиизоб утилеп -сн3 0 67
Полиэтилен —СН,— Q 32
химических связей. Наибольшей энергией межмолекулярного взаимодействия
обладают водородные связи, а также связи, возникающие за счет взаимодей-
ствия карбонильных групп и нитрильной группы. Наименьшая энергия меж-
молекулярного взаимодействия (Ван-дср-Ваадьсовые силы) характерна для
-СН2_ групп.
Чем больше энергия мсжмолску.тярного взаимодействия, тем больше тре-
буется тепловой энергии для преодоления этих связей и тем ваше температура
стеклования полимеров, имеющих сильные межмолекулярные связи. Чем
меньше энергия межмолекулярного взаимодействия, тем меньше тепловой
энергии требуется для разрыва этих связей и тем при более низкой темпера-
туре полимер переходит из стеклообразного в высокоэластическое состоя-
ние. Такие полимеры, как полиэтилен, полиизобутилен, полипропилен и т.д.,
имеют низкие температуры стеклования, а такие полимеры, как сложные по-
лиэфиры, полиамиды, поливиниловый спирт, полиакрилонитрил и т.д., обла-
дающие сильным межмо.тетулярным взаимодействием, имеют и более высо-
кие температуры стеклования.
При таком рассмотрении, естественно, не учитывается скелетная жест-
кость самой макромолекулы, т.е. возможность или невозможность вращения
отдельных групп в основной цепи относительно ординарных связей. Между
тем этот фактор может быть решающим и оказывать огромное влияние на
температуру стеклования полимера. Следует обратить внимание еще на одно
очень важное обстоятельство. В случае застекловывания низкомолекулярных
жидкостей молекулы которых содержат полярные группы, все они могут всту-
пать во взаимодействие друг с другом, поскольку для этого нет никаких сте-
рических препятствий.
В случае застекловывания полимеров макромолекулы лишены такой воз-
можности вследствие их огромной длины. Только в идеальном кристалле по-
126
Глава IV
лимера, свободном от всяких нарушений и дислокаций, полярные группы в
повторяющихся звеньях могут полностью взаимодействовать друг с другом.
При застекловывании полимерной жидкости (те. при переходе из вязко-те-
кучего или высокоэластического состояния в стеклообразное) структура по-
лимера далека от идеального кристалла, и поэтому вследствие стерических
затруднений только часть полярных групп может вступать в мсжмолскуляр-
ное взаимодействие. Согласно опытам по измерению теплоемкости полимер-
ных тел при изменении температуры было найдено, что только небольшая
часть полярных групп вступает в межмолекулярнос взаимодействие при ох-
лаждении полимера до температуры стеклования. Количество таких групп
составляет —10 % от общего числа групп, способных к межмолекулярному
взаимодействию. Но даже этого небольшого количества взаимодействующих
групп оказывается достаточным для того, чтобы полимерное те по перешло
из высокоэластического состояния в стеклообразное в результате образова-
ния пространственной сетки межмолекулярных связей.
Образование таких связей при охлаждении полимера и их разрушение
при его нагревании может быть обнаружено прямыми методами физического
исследования. К таким методам прежде всего относится метод ИК-спектро-
скопии, с помощью которого действительно было показано, что при охлажде-
нии полимера до температуры стеклования или при нагревании полимера выше
этой температуры, происходит интенсивное образование или распад межмо-
лекулярных связей.
Столь подробное изложение данной концепции связано с тем обстоя-
тельством, что в дальнейшем, при описании расчетных методов оценки тем-
пературы стеклования полимеров Tg, будут рассмотрены различные вариан-
ты проявления межмолекулярного взаимодействия, что сказывается на рас-
четных значениях Tg. Действительно, межмолекулярное взаимодействие
между полярными группами, расположенными в соседних цепях, приводит к
образованию физической сетки межмолекулярных связей. Однако такое вза-
имодействие может осуществиться и между полярными группами, располо-
женными в одном и том же повторяющемся звене макромолекулы. Тогда эти
группы выключаются из взаимодействия между соседними цепями и темпе-
ратура стеклования понижается. Такой же эффект будет наблюдаться и при
аномальном присоединении звеньев в процессе полимеризации или сополи-
меризации (присоединение “голова к голове” или “хвост к хвосту”). В этом
случае межмолекулярнос взаимодействие осуществляется между полярны-
ми группами, расположенными в соседних повторяющихся звеньях одной и
той же цепи; естественно, что при этом они выключаются из взаимодействия
между соседними цепями, что приводит к снижению температуры стеклова-
ния. Все эти вопросы будут детально проанализированы ниже.
Температура стеклования полимеров
127
IV.3. Расчет температуры стеклования линейных полимеров
В данном разделе будем рассматривать расчетную схему для оценки тем-
пературы стеклования Tg, развитую в работах [6, 128]. Согласно этой схеме,
коэффициент молекулярной упаковки полимеров различного химического стро-
ения примерно одинаков при температуре стеклования каждого из полиме-
ров, причем это значение kg оценивается величиной kg ® 0,667 для линейных
полимеров. Вблизи абсолютного нуля коэффициент молекулярной упа-
ковки к0 также примерно одинаков для всех полимеров и составляет 0,731.
Эти измерения показывают, что переход из стеклообразного состояния в
высокоэластическое происходит при достижении определенной доли пустого
(empty volume)объема, равной fE= 1- kg= 0,333, а выражение для температу-
ры стеклования Tg может быть записано в виде:
Т
g
kQ/kg-l _ 0,0906
о<? «G
(82)
Следует отметить, что материал, излагаемый в данной главе,тесно свя-
зан с материалом, изложенным в главе III. Используя полученные в главе III
выражения для (р-г, следует записать:
(83)
, 1 \
где ’
1 _ R 1
Tg ~ 0,455 zD0
У —— ДГ,
i Т-Ры
,Z - координационное число /-го атома; DOi - энер-
2—1 /
гия связи /-го атома с соседними; ДИ, - инкремент Ван-дер-Ваальсового объе-
ма /-го атома.
Численные значения l/Z,£>0„ характерные для каждого атома и каждого
типа межмолекулярного взаимодействия, определены с помощью стати сги-
ческой обработки экспериментальных данных по методу “наименьших квад-
ратов”. Получающиеся в результате обработки экспериментальных данных
значения энергий связи, как отмечено выше, соответствуют энергиям межмо-
лекулярного взаимодействия. Наличие в полимерах полярных групп, приво-
дящих к сильному межмолекулярному взаимодействию у читынзется введе-
нием специальных инкрементов.
Дтя практических расчетов формулу (83) удобно переписать в виде:
(84)
128
Глава IV
где я, - набор инкрементов, характеризующих энергию слабого ди-
сперсионного взаимодействия в виде усредненного вклада каждого атома в
это взаимодействие; />, набор инкрементов, характеризующих энергию силь-
ного специфического межмолскулярного взаимодействия (диполь-дипольное,
водородные связи и др ).
Величины я, и bj связаны с параметрами а, следующими соотношениями:
«,=----------= -®1— (85)
Z:0/A-g-l 0,0906’
Ъ =__________= Р; (86)
1 k0/kg-l 0,0906
Используя соотношение (84), можно рассчитать температуру стеклова-
ния огромного количества полимеров. Это связано с тем обстоятельством,
что описываемый подход является “атомистическим”, те. каждый атом ха-
рактеризуется своим инкрементом а,(их величины приведены в табл. 13).
Что же касается специфических межмолекулярных взаимодействий (диполь-
дипольные, водородные связи), то они характеризуются своими инкремента-
ми bj, не зависящими от химического строения полярной группы .Так,напри-
мер, диполь-дипольные взаимодействия разных типов характеризуются од-
ним и тем же инкрементом bj = -55 1О'3 А3К-1. Несколько сложнее дело обстоит
с водородными связями в полиамидах, что связано со спецификой их влия-
ния на Tg в пределах данного класса полимеров (табл. 18)*.
В табл. 19 в качестве примера приведены экспериментальные и расчетные
значения температур стеклования ряда модельных полимеров, начиная от по-
лимеров простого химического строения до весьма сложных систем. Там же
приведены наборы параметров соотношения (84).
Развитый подход [6, 128] и полученное на его основе соотношение (84)
позволяет учитывать влияние типа присоединения звеньев в цепи полимера
на его температуру стеклования. Если присоединение нормальное (“голова к
хвосту”), как это схематически изображено на рис.42,а, то межмолекулярное
взаимодействие может осуществляться между соседними цепями полимера с
образованием физической сетки межмолекулярных связей (см рис.41) . Если
же часть звеньев имеет аномальное присоединение (например, “голова к го-
лове”), как это схематически изображено на рис.42,б, то мсжмолскулярнос
* Естественно, что такой подход имеет преимущество перед популярным методом расчета,
основанном на групповых вкладах [214], Если в полимере имеется группа, вклад которой в темпе-
ратуру стеклования неизвест ен, расчет становится невозможным. В том же подходе, который явля-
ется предметом нашего описания, вклад любой группы просто складывается из вкладов составля-
ющих ее атомов плюс вклад специфических взаимодействий, если эти группы полярны.
Температура стеклования полимеров
129
Значения инкрементов Ру и bj для амидных групп - NHCO -
Values of increments ру and бу for amide groups - NHCO -
Тип полиамида Ру-10’, а’к4 ЙуЮ3, А’К’1
Ароматические полиамиды -11,5 -119
Алифатические полиамиды всех типов* -20,4 -212
Алифатические - ароматические полиамиды, содержащие СН2-группы** -14,0 145
Алифатические - ароматические полиамиды на основе изо-, терефталевой кислоты и мета-, парафенилендиамина, содержащие четное число CHj-групп** -18,1 -188
Алифатические - ароматические полиамиды на основе изо-, терефталевой кислоты и мета-, парафенилендиамина, содержащие нечетное число СН>- групп** 16,3 -169
* Когда число (и) - CH2 — групп, приходящихся на одну — NHCO - группу, и > 5, вводятся
дополнительные инкременты йСщ=-23,0 в количестве m (i - 5) где т-- количество- NHCO -
групп .входящих в повторяющееся звено.
* * Когда число (и)- СН2 —групп в повторяющемся звене и > 6, вводят дополнительные инкременты
i>CH2= -23,0 в количестве (и - 6).
взаимодействие будет происходить между полярными группами, расположен-
ными в соседних звеньях в пределах одной и той же юпи .Естественно ,что
тогда эти полярные группы выключаются из образования физической сетки
между соседними цепями (см. рис.42,6). Поскольку сильные межмолекуляр-
Рис.42. Характер межмолекулярного взаимодействия в случае присоединения молекул
“голова к хвосту”(а) и “голова к голове”(б)
Nature of intermolecular interaction in case of molecules joined by the method
“head to end”(a) and “head to head”(6)
130
Глава J V
Таблица 19
Ван-дер-Ваальсовые объемы звеньев £Д1^- , величины £а/Д^ , н температуры
/ i j
стеклования Tg ряда полимеров
Полимер i
1 2
—сн2—сн=с—сн2— СН3 81,3
-СН2-СН=СС1-СН2- 79,0
сн3 —Si—О— 1 сн3 71,8
-сн2-сн- СН3 51,2
сн3 —сн2—с— СН3 68,3
—сн2—сн— с2н5 68,3
—СН2—СН — о-сн3 60,7
—сн2—сн— о—с2н5 77,8
-сн2-сн2- о-с4н9 111,9
—сн2—сн— CI 48,9
Температура стеклования полимеров
131
Van-der-Waals volumes of the repeating units , values and the glass
I ' J
transition temperatures for a series of polymers
Xa/AI<-lO’,A3K-' i Ybj - io5, л3к‘ J T
Расчет Эксперимент
3 4 5 6
319 81,5 203 200
279 81,5 219 225
478 0 150 150
239 -55,4 279 258;293
319 0 214 199
319 -55,4 259 248
295 -55,4 253 258
375 -55,4 244 248
534 -55,4 234 221
199 -55,4 341 355
132
Глава IV
1 2
- CHji— cci2- 63,7
сн3 -СН2-С- С-О-СНо II 3 О 96,4
сн3 -СН2-С- С-О- С2Н5 О 113,5
СНз -СН2-С- С-О-СдНо II 4 а О 147,6
СНз —сн2—с— С—О—Cg Н< з II ь О 181,8
—сн2— СИ- ОН 41,5
-С-0-С-О- (СН2)2—О— О о 165,6
—сн2—сн— CN 54,0
—С—(СН2)4—С—NH—(CH2)g—NH— о о 231,8
Температура стеклования полимеров
133
таблица 19 (продолжение)
3 4 5 6
238 0 268 256
422 -166,2 377 378
502,0 -166,2 338 338
661,0 -166,2 298 293
820,0 -166,2 278 268
309,0 -195,0 364 358
578,4 -136,4 375 353
183 -55,4 423 418
1121 -424 333 325
134
Глава IV
Температура стеклования полимеров
135
таблица J 9 (продолжение)
1 2
- С- (СН2)5-NH- О 115,9
- сн2- СН — c-nh2 II О 64,3
-сн2- СН — 6 109,8
СНз —сн2—с— 6 126,9
—СН?—СН — 1 с-он II О 60,2
1 о 1 О 1 0-0-0 □: 1 1 “ 0“ 1 о 1 0=0 1 238,8
1 о Q /°х //° 'о о 0 о 1 о 1 0 = 0 1 со CN I о т 0=0 1 451,1
1 о Q /°ч р ;о о 0 о 1 о 1 0 = 0 1 0 1 0 = 0 1 465,4
1 о Q о d 1 о 0=0 1 ох 0=0 1 437,3
3 4 5 6
560 -212 333 335
363 -212 426 426
320 -27,7 376 378
399 -83,3 402 435
356 -195,0 374 379; 439
771 -162,0 431 422
1481 -300,5 382 363
1163 -351,7 573 593
1060 -256,8 544 543
136
Глава IV
1 2
- С-О-С-HN-ZO-NH- II II г ° 0 <о ч 395,5
1 1 X 0 = 0 0=0 \ Z 1 о 1 0 СП 1 со X 1 X о-о-о 6 1 о о о=</ чо=о х z X 1 519,9
1 0=0=0 1 0 1 о 1 0 1 0=0=0 1 0 1 0 1 368,1
1 1 о 1 1 0=0 1 0 1 о 1 262,3
1 о 6 А/О z° Z о Ъ 1 о 1 0 = 0 1 0 1 0 = 0 1 389,7
ные взаимодействия учитываются в соотношении (84) параметрами bj, то ес-
тественно записать, что
^=^a,.Ar/+(l-Y)^/ (87)
' 7
где у - доля аномально присоединенных звеньев.
Согласно соотношению (87), температура стеклования полимера при на-
личии аномального присоединения звеньев будет пониженной. Ниже на кон-
кретном примере расчета это будет продемонстрировано.
Описанный выше эффект влияния типа присоединения звеньев на Tg по-
лимера распространяется и на полимерные системы, построенные из изоме-
ров. Так, например, полиамидофенилхиноксалины, проанализированные в
работе [83], имеют строение:
Температура стеклования полимеров 137
таблица 19 (окончание)
3 4 5 6
1066 -453,3 646 653
1331 -298,4 503 500
1133 -467 553 560
692 -76,8 426 420
1003 -326,1 575 583
о о 0 0
ПАФХ-1
ОО ° °
ПАФХ-2
В случае ПАФХ-2 полярные группы, представляющие собой фенилхи-
ноксалиновые циклы, находятся в тесном контакте друг с другом, и внутри-
молекулярное взаимодействие, возникающее за счет этих групп, проявляет ся
в пределах звена', тогда диполь-дипольное взаимодействзе между звеньями
соседних цепей нс реализуется.
Ik.
138
Глава IV
В случае ПАФХ-1 и ПАФХ-2 - суть изомеры с одинаковой брутто-фор-
мулой.
В работе [83] были проведены эксперименты и расчеты по определению
температур стеклования двух пар полимеров, строение которых приведено
выше. Поскольку сильное межмолекулярное взаимодействие, возникающее
за счет отдельных полярных групп,существенно влияет на 7'г,то естественно,
что когда это взаимодействие проявляется внутри звена, Tg таких полимеров
должна быть ниже, чем для полимеров полностью аналогичного строения,
но с таким расположением полярных групп, когда межмолскулярнос взаимо-
действие проявляется между звеньями соседних цепей.
Расчеты и эксперименты подтвердили, что действительно так и происхо-
дит. При расчете температуры стеклования для ПАФХ-1 = 4/>rr + 2Z>h +
+ 2b А + 2 — bd , а для ПАФХ-2 £ bj = 4ЬП + 2/>h . В результате, расчетная ве-
2 I
личина Tg для ПАФХ-1 составляет 272 °C, а для ПАФХ-2 - 214 °C. Соответ-
ствующие экспериментальные значения равны 275 и 210 °C.
При одинаковой брутто-формуле повторяющегося звена температура стек-
лования также зависит и от расположения отдельных ароматических фраг-
ментов. Так, например, полиимиды ПИ-1 и ПИ-2 имеют совершенно одина-
ковую брутто-формулу, но разное расположение ароматических циклов. При
расчете температуры стеклования ПИ-1 = 2/)d + 2АП , а в случае ПИ-2
2_jbj = 2Ad +5ЛП . В соответствии с этим Tg полиимида ПИ-1 составляет
}
252 °C, а ПИ-2 - 312 °C. Эксперимент дает примерно те же величины.
О О
-n^:o-°-cc|> -о-о-о-
II II
о о
ПИ-1
Следует отметить, что существенное внимание необходимо обращать на
случаи, когда обнаруживается заметное расхождение экспериментальных и
Температура стеклования полимеров
139
расчетных данных по температуре стеклования. Это может быть вызвано как
появлением полярной группы, обладающей особым влиянием на энергию меж-
молекулярного взаимодействия, так и, наоборот, выключением какой-либо
полярной группы из образования сетки физических связей между соседними
цепями полимера. В первом случае потребуется введение нового инкремен-
та bj, но нужно всегда помнить, что чем больше введено инкрементов в рас-
четную схему, тем ее предсказательная сила становится хеньше, и в предель -
ном случае когда каждый новый полимер требует введения нового инкремен-
та bj, предсказательная сила расчетной схемы становится равной нулю.
Для сополимеров соотношение (84) преобразуется к виду:
а1 (X AI'' )1 + а2 (Е Д^^2 + • • • +
Tg + +--+~У
i J ' J
+ал (Е )«
+ ’ (88)
' J
где а], а2, а„, - молярные доли компонентов 1, 2, ..., и; (Ед^)1>
)
(^AF;)2 , .... (^Aly Ван-дер-Ваальсовые объемы повторяющихся зве-
) I
ньев компонентов(90)(91) 1, 2, ...,и; (^а,ЛУ, + У. b j )?,
> i ' J
..., (ХщА>< + Е^;)я - наборы инкрементов для компонентов 1, 2, ...,и(сле-
' J
дует помнить, что ai + a2 + ... + а„ = 1).
Соотношение (88) всегда приводит к нелинейным зависимостям Tg от
состава сополимера.
Если мы хотим выразить Tg сополимера через температуры стеклования
соответствующих гомополимеров, то, используя соотношения (84) и (88), мож-
но записать
«1 (Е Д ГО1 + a2 <Е Д и >2 + • • + “я (X АРХ
А= (89)
«1 Ч,,----+ а2 ' 7,----+... + а„ _
7g.I yg.2 lg,n
140
Глава IV
где Tgj , Tg^, , Tgn - температуры стеклования гомополимеров, получен-
ных на основе компонентов 1,2, и.
В другой форме соотношение (89) можно записать следующим образом:
7“ = 7 Pi +т—Р2 +-"+7—Зп
g g,l ‘g,n ’ (90)
где
ai(ZA^)i
31 “ а1(ХД1<)1 +а2(Хдг,)2 +... + a„Q>;)„;
ai(ZA^)2
ft, =---------------!-------------------
ai£X)i +а2(£ДИ,)2 +... + а„(^Д1/)„ ;
«1(£А^)И
Зи “ аКХД^)! +а2(ХАЦ)2 +... + а„(£Д1<)„ '
/ / i
Таким образом, по своему смыслу коэффициенты рь р2, ..., р„, - суть
доли Ван-дер-Ваальсовых объемов звеньев 1,2, ..., п.
В более компактной форме соотношение (88) записывается в виде:
s\(ZA^)t
£afc(Z>A^
Z:=l I j
где ak-молярная доля к-го компонента; (]Сд[Л)д. и (^^ДГ' + ^Pj>j)k соот-
' ' J
ветственно Ван-дер-Ваальсовый объем и набор инкрементов для к-го компо-
нента; и - общее число компонентов в сополимере.
Соотношения (89) и (90) в компактной форме выглядят следующим обра -
зом: к=п
Т - ___L______
к=\ 1 g,k
Температура стеклования полимеров
141
Рис.43. Зависимости температуры стеклования 7g от состава а двойных сополиме-
ров, вычисленные по уравнениям (88) (кривая 1) и (94) (кривая 2)
Dependences of the glass transition temperature 7g on composition a for two-
components copolymers calculated from equations (88) (Curve 1) and (94) (Curve 2)
'g k=\lg,k
Зависимости (88)-(93) температуры стеклования Tg сополимера от его
состава не являются линейными, и в случае двойных сополимеров выглядят
так, как это схематически представлено на рис.43. Эти зависимости не учи-
тывают ослабления межмолекулярного взаимодействия в звеньях сополиме-
ра, хотя такое ослабление должно привести к снижению температуры стекло-
вания по сравнению с аддитивной величиной.
На практике “провалы” на зависимостях Tg от состава больше, чем рас-
считанные по соотношениям (88)-(93), и составляют ДГ-20 -30° [78]. Поэто -
му в работе [39] предложено модифицированное соотношение, учитываю-
щее это обстоятельство:
+a2(XAf/|)2 + +
ре =--------------------1----------1---------------------------->
+ Z^)1 +а2(£°1д,//+Ybjh +--- + ая(£Лд,/; + ^ЬР>" +
' J ' J ' J
+аи ) n
-э---------------------------------------- , (94)
+ [a1(l-a1) + a2(l-a2) + ... + a„(l-a„)]0,03
142
Глава IV
где все обозначения - те же, что и в формуле (91); 0,03 - эмпирический коэф-
фициент, учитывающий снижение сильных межмолекулярных взаимодей-
ствий при сополимеризации.
В более компактной форме соотношение (94) можно записать следующим
образом:
(95)
у1 ______________к =1__I___________________
S к=п к=п 3
Z ак (Е aiАИ/+ X bj к-+ °’03 Е “Д1 - ак)
Л=1 / у к=1
где (%, (Хд^к и (Еа>д,//+Е^у)* - соответственно молярная доля, Ван-дер-
' ' j
Ваальсовый Щзъем и набор инкрементов для к-го компонента сополимера.
Соотношения (94) и (95) для расчета температуры стеклования сополи-
меров не требуют знания экспериментальных температур стеклования гомо-
полимеров.
Другой вариант формулы для расчета температуры стеклования сополи-
меров может быть получен комбинацией уравнений (84) и (94):
ai(EAI/i)1 + аг(ЕА^)2 +•• +
* (2>')1 (1лс,)2 (Еа^)„
Cti i h (17 -----h ... + Ct„ — --h
1 rrt 4* rj-t II rri
7g,l 7g,2 Lg,n
(E A I-1 ) n
-------------------1_______________________, (96)
+ [а1(1-а1) + а2(1-а2) + ... + а„(1-а„)]-0,03
где Tgд, Tg 2,..., Tg „, - температуры стеклования гомополимеров, построен-
ных из компонентов 1, 2, ..., п. В более компактной форме уравнение (96)
записывается в виде:
к=п
Т =----------—---------------------- (97)
g к-п к-п
Еак —+°-°3 Zако -ак)
k=l Jg,k к=1
Теперь рассмотрим влияние разветвлений цепей на температуру стекло-
вания.
Температура стеклования полимеров
143
б
а
Рис.44. Различные типы разветвлений в полимерах (см. текст)
Different types of branchings in polymers (See text)
Разветвления цепей встречаются достаточно часто и являются либо ре-
зультатом побочных процессов, происходящих при синтезе линейных поли-
меров, либо создаются целенаправленно для модификации свойств Так на-
пример, ответвления могут содержаться уже в структуре мономеров типа
R'
сн2=сн; сн2=с
R R
где R-углеводородный радикал:—CnH2n+1;—О-СпН2п+1; — С-О~СпН2п+1
и т.д.
В общем случае количество возможных вариантов разветвлений цепи
может быть достаточно велико. Рассмотрим основные из них, схематически
изображенные на рис.44, где показана основная цепь и возможные варианты
ответвлений.
В простейшем (но менее вероятном) случае ответвления могут иметь оди-
наковую длину и располагаться в каждом повторяющемся звене (см. рис.44,а).
Другой вариант соответствует ответвлениям оди наковой длины, которые рав-
номерно распределены по основной цепи чере з определенное число звеньев
(см. рис.44,б). Третий вариант соответствует случаю, когда имеются ответв-
144
Глава IV
ления разной длины, но они равномерно распределяются вдоль основной цепи
через определенное число звеньев (см. рис.44,в). Далее возможен вариант,
когда имеются ответвления разной длины и они распределены вдоль основ-
ной цепи случайным образом (см. рис.44,г). Рассмотрим еще два варианта.
По одному из них ответвления разной длины распределены вдоль основной
цепи случайным образом, и в каждом ответвлении имеется еще по одному
ответвлению, имеющему различную длину (см. рис.44,д). Естественно, что
этот общий случай может включать и частные случаи, когда ответвления оди-
наковы и распределены вдоль цепи равномерно, а вторичные ответвления -
одинаковые или разные, и т.д. Еще один принципиальный случай - это раз-
ветвление в виде дерева (см. рис.44,е). Здесь появление каждого нового от-
ветвления влечет за собой появление еще одного ответвления.
Следует отметить, что такие системы в настоящее время реально синтези-
рованы [82], и они получили название “дендритные полимеры” или “дендри-
меры”. I
Во всех рассмотренных случаях ответвления могут иметь либо одну и ту
же химическую природу по отношению к главной цепи, либо различную при-
роду. Случай, когда ответвления имеют иную химическую природу по сравне-
нию с главной цепью полимера, соответствует привитым сополимерам .В та-
ких системах в большинстве случаев из-за термодинамической несовмести-
мости основной и привитой цепи происходит микрофазовое расслоение. При
этом каждая фаза может иметь, например, свою температуру стеклования,
которая, однако, во многих случаях отличается от температуры стеклования
индивидуальных компонентов. Поэтому рассчитывать величину Tg для при-
витых сополимеров можно только при полной совместимости исходного и
привитого полимеров. Однако можно решать и обратную задачу - по темпера-
турам релаксационных переходов в каждой' фазе оценивать ее состав, считая,
что в каждую из фаз включено определенное количество инородных звеньев .
В работе [24] проанализировано влияние разветвлений разных типов на
физические характеристики полимеров.
В гомополимерах ответвления могут присоединяться непосредственно к
основной цепи путем замещения одного из атомов или присоединяться через
развязку другой химической природы. Рассмотрим все эти варианты на конк-
ретных примерах. В качестве одного из модельных полимеров для такого рас-
смотрения выберем полиэтилен и его производные:
Структура I
—СН2—СН—(СН2 —СН2 )т—
(СН2)П
СНз
Температура стеклования полимеров
145
Рассмотрим влияние разветвлений на температуру стеклования Tg. Предва-
рительно заметив, что если т = 0, то имеем случай, изображенный на рис.44,а,
когда ответвления имеют одинаковую длину и расположены в каждом повторя-
ющемся звене. Рассмотрим ход расчета для этой структуры в деталях. Темпера-
тура стеклования рассчитывается по уравнению (84).
Для структуры I, изображенной выше,
ХАК = (51,3 + 17,1и +34,2m) А3;
=(185,34 + 80,25и+160ш)-10 3 А3К‘'.
При расчете значений У Ь, необходимо учитывать, что каждое ответв-
J
ление приводит к необходимости введения одного инкремента bj В резуль-
тате получаем
51,3+17,In + 34,2»! 10,
* 185,34 + 80,25//+160,5m (98>
Результаты расчета, проведенного по соотношению (98), показаны на
рис.45. Видно, что если ответвления находятся в каждом повторявшемся
звене (т = 0), то Tg - очень сильно зависит от числа CHj-rpyпп в ответвле-
нии. С уменьшением п Tg начинает резко возрастать, когда п < 5.
Рис.45. Зависимости температуры стеклования Те от п для структуры I. Числа у кри-
вых - значения т (см. текст )
Dependences of the glass transition temperature 7’g on n for structure I. Numbers at
curves denote m values (See text)
146
Глава IV
]1дя. случая т = 0 существует ряд разветвленных полимеров, свойства ко-
торых изучены экспериментально. Так, например, при т = 0 и и = 0 получаем
полипропилен, для которого расчетная величина Tg составляет 277 К, а экс-
периментальная 263 К. При т = 0 и п = 1 имеем полибутен-1, для которого
расчетная величина Tg составляет 258 К, а экспериментальная 248 К. Такая
же сходимость, характерная для данного метода, наблюдается и для других
полимеров при т = 0 ил = 2 и 6.
Если ответвления разрежены (т > 0), то влияние числа звеньев в ответв-
лении на Tg снижается, и, когда ответвления становятся достаточно редкими
(т = 10), длина ответвления практически не влияет на Tg (заметим, кстати,
что, когда т —> оо, это соответствует звездообразному полимеру).
Теперь рассмотрим случай, когда ответвления в полиэтилене распределены
случайным образом вдоль цепи, причем, эти ответвления имеют разную дли-
ну. Допустим, что величина т подчиняется случайному распределению
F(m) = l-em/mcp(m>0), (99)
где тср - средняя величина т.
Функция плотности распределения имеет вид
f(m) = _Le-m/n,‘P (100)
тср
Допустим также, что степень полимеризации ответвлений подчиняется
распределению Флори
q(n) = y2ne~'/n, (101)
1 _
где у = ; nw - средневесовая величина п.
Для дальнейших расчетов примем, что предельная величина п = 10. Тогда
число звеньев в ответвлении определится из соотношения
‘=пк , ,
»ome. = Е' ге '7 •
(102)
Формула для расчета температуры стеклования Tg, полученная на основе
уравнения (84) и с учетом всего изложенного выше, для химической структу-
ры I примет вид
Температура стеклования полимеров
147
51,3 4-17,1’2/+34,27]Г‘ j2₽Vyp
rg (К)=----------------------------------ю3, (юз)
185,3 + 80,252/2 у2 е“‘7 +160,5 2
/=1 /=1
о 1
где р=----.
тср
Результаты расчета, проведенного по формуле (ЮЗ), показаны на рис 46.
Характер зависимости Tg от nw аналогичен зависимости Tg от п, с той лишь
разницей, что зависимости Tgот nw более плавные.
В целом результаты проведенных расчетов [24] для разветвленного поли-
этилена показывают, что наибольшее влияние на температуру стеклования
оказывают короткоцепочечные разветвления, часто расположенные вдоль ос-
новной цепи.
Рис.46. Зависимости Т& от nw для структуры I. Числа у кривых - значения тср
(см. текст)
Dependences of Tg on nw for structure I. Numbers at curves denote mcp values
(See text)
148
Глава IV
Перейдем к следующему варианту разветвлений, когда в самом от-
ветвлении появляются новые ответвления. В этом случае структурная форму-
ла разветвленного полиэтилена имеет вид
Структура II
—СН2—СН— (СН2— СН2)П—
(СН2)Х
СН—(СН2)у—СН3
(СН2)г
СН3
Это соответствует схеме на рис.44,д. Для этого случая формула для рас-
чета Tg, полученная на основе уравнения (84), выглядит так
т (К) = 85,3+ 17,l(x + y + z)+ 34,2»; 3
’ 400,97 + 80,25(x + y + z) + 160,39w-110,8 ' (104)
Смысл обозначений х, у, z и m виден из структурной формулы разветв-
ленного полиэтилена. Заметим, что если х+у + г = 0и»г = 0, то это соответ-
ствует полимеру следующего химического строения
Рис.47. Зависимости 7gOT(x +у + 7.) для структуры II (а)и от» (1)и nw (2) для структу-
ры III (б). Числа у кривых - значения m (см. текст)
Dependences of 7g on (x+у+z) for structure II (a) and dependences of 7g on n (1) and nw (2)
for structure III (6). Numbers at curves denote m values (See text)
Температура стеклования полимеров
149
•••—сн2—сн—•••
СН-СНз
СНз
Для этого полимера расчетная температура стеклования составляет
294 К, а экспериментальная 302 К.
Анализ соотношения (104) показывает, что Tg зависит только от суммы
(х +y+z) и от величины т . На рис. 47,а показаны зависимости 7gOT(x+y + z)
при различной величине т , т.е. при различной частоте ответвлений, которая
определяется величиной \/т. Здесь влияние ответвлений примерно такое же,
как и в предыдущих случаях. Однако появление вторичных ответвлений при-
водит к более резкому возрастанию температуры стеклования, что осо баню
отчетливо проявляется при коротких и частых (т = 0) разветвлениях.
Рассмотрим разветвления в виде дерева. В случае полиэтилена с трех-
функциональным узлом химическое строение такой системы изображено ниже
Структура III
СН2 —СН— :
(СН2)„ (СН2)„
СН-(СН2)„-СН-(СН2)„-СН-(СН2)„—
(СН2)„ (СН2)„-СН-(СН2)„-СН—
—СН-(СН2)„-СН : :
: (СН2)„
При определении необходимо учитывать, что каждое новое ответ-
вление приводит к необходимости введения инкремента bj и его влияние ста-
новится наиболее ощутимым, когда имеются наиболее короткие ответвления.
Если эти ответвления имеют одинаковую длину (и = const), то формула для
расчета температуры стеклования, полученная на основе уравнения (84), при-
нимает вид
17,1л+ 11,0 з
80,25л-15,25
(Ю5)
Зависимость Tg от п , рассчитанная по формуле (105), показана на
рис.47,б (кривая 1). Хорошо видно, что при наличии разветвлений в виде
150
Глава IV
дерева /^системы меняется гораздо сильнее, чем во всех предыдущих случа-
ях. Когда ответвления становятся наиболее короткими (и = 1), Tg достигает
160 °C; это намного выше, чем Tg исходного полиэтилена. Причина столь резко-
го повышения Tg заключается в том, что при коротких разветвлениях хими-
ческое строение полимера существенным образом отличается от химическо-
го строения полиэтилена, при этом роль диполь-дипольных взаимодействий
возрастает.
Рассмотрим и другой вариант, когда распределение ответвлений по дли-
нам подчиняется распределению Флори. В этом случае формула для расче-
та Tg , полученная на основе (84),приобретает вид
17,1^д2у2е"‘у +110
т;(а:) =--------------------ю3, (Юб)
80,25 р2у2е г/-15,25
(=1
а результаты расчета, проведенного по формуле (106), показаны нарис.47,6 (кри-
вая 2). Можно отметить, что в случае наличия распределения ответвлений по
длинам зависимость Tg от nw более слабая, чем при ответвлениях одинако-
вой длины; естественно, что сравнение проводится при и = nw .
Перейдем теперь к рассмотрению разветвлений одинаковой химической
природы по сравнению с основной цепью, но присоединенных к ней через
развязку другого химического строения. В качестве примера возьмем струк-
туру разветвленного полимера, изображенную ниже
Структура IV
— сн2—сн—(сн2—сн2)т—
О
I
сн2
(СН2)И
СНз
Частный случай такой системы (т = 0) представляет набор виниловых
эфиров с разной величиной и . Для такого случая формула для расчета Tg
имеет вид
= 78,1 + 17,1» 1о3
г 289,4 +80,25и
и расчеты по соотношению (107) приводят к зависимости Tg от п, представ-
ленной на рис.48. Эта зависимость аналогична рассмотренным выше. При
Температура стеклования полимеров
151
Рис.48. Зависимость 7g от и для структуры IV
Depen cfence о f 7g on и for structure IV
этом и в данном случае наблюдается обычное для данного метода согласие
расчетных и экспериментальных значений Tg.
При наличии распределения по длинам ответвлений и их случайном рас-
положении вдоль цепи формула для расчета Tg приобретает вид
78,1 + 17,1 +34,27ХЖ7Р
Tg (*)=-----------тЬ-----------------------1 °3 • (108)
289,4 + 80,25 2у + '7 +160,5
<=1 7=1
В общем случае зависимость Tg от среднего числа СН2 -групп в ответвле-
ниях при различных значениях т выглядит так, как это показано на рис.49.
Характер этих зависимостей также аналогичен рассмотренным выше для раз-
ветвленного полиэтилена.
В заключение рассмотрим ряд органических стекол на основе полиме-
такрилатов. Общая формула таких систем выглядит следующим образом:
Структура V
СНз
•••—сн2—с—
с- о-СН2—(СН2)„- СНз
II
о
152
Глава IV
Рис.49. Зависимости Tg от nw для структуры IV. Числа у кривых указывают на значения
т (см. текст)
Dependences of Те on nw for structure IV. Numbers at curves denote m values
(See text)
Формула для расчета Tg полиметакрилатов, полученная на основе урав-
нения (84), имеет вид
W) =
113,85 + 17,1и з
355,0+80,25л
Зависимость Tg от длины бокового ответвления п показана на рис. 50
С ростом длины ответвления температура стеклования быстро уменьшается и
наблюдается переход от стекла к каучукоподобному телу.
Подводя итоги, можно сделать некоторые выводы относительно влияния
разветвлений на температуру стеклования Tg .
Первый вывод состоит в том, что с увеличением длины ответвлений свой-
ства полимеров изменяются незначительно и приближаются к свойствам гомо-
полимеров, имеющих такое же химическое строение, как и сами ответвле-
ния. Это справедливо в том случае, когда ответвления располагаются в каж-
дом повторяющемся звене. Если ответвления являются более редкими и
распределены вдоль основной цепи полимера случайным образом, то их вли-
яние на свойства более существенно, если они имеют химическую природу,
отличную от химической природы основной цепи.
Температура стеклования полимеров
153
Рис.50. Зависимости 7g отп дня структуры V
Dependences of 7g on n for structure V
Наибольшее влияние на физические свойства оказывают короткие раз-
ветвления. Для температуры стеклования влияние коротких разветвлений
формально учитывается введением в случае каждого нового разветвления
инкрементов hd в формуле (84). Это влияние связано с увеличением жесткос-
ти цепи, а также с появлением дополнительного межмолекулярного взаимо-
действия при введении ответвления, содержащего полярную группу. Следует
также отметить, что при наличии большого количества короткоцепочечных
ответвлений качественным образом меняется химическое строение полиме-
ра, и если короткоцепочечные разветвления реализуются в виде дерева, то
химическое строение получаемой системы лишь отдаленно напоминает хи-
мическое строение исходного полимера.
FV.4. Расчет т емпературы стеклования сетчатых полимеров
Из эксперимента известно, что температура стеклования, как и многие
другие свойства сетчатых систем, зависит от числа повторяющихся звеньев
между узлами сшивки т так, как это схематически изображено на рис.51.
Если сетка является редкой, то ее температура стеклования слабо зависит
от т. но когда число повторяющихся звеньев цепей между соседними узлами
существенно уменьшается, температура стеклования начинает резко возрас-
тать и принимает очень высокие значения. Опыты и расчеты показывают, что
начало резкого возрастания температуры стеклования наблюдается, когда чис-
ло звеньев в линейных фрагментах, соединяющих узлы, становится меньше
154
Глава IV
Рис.51. Схематическое изображение зависимости температуры стеклования Т~ сеток
от числа повторяющихся звеньев m в межузловых фрагментах
Schematic representation of dependence of the glassy transition temperature 7g of
networks on the number of repeating links m in linear fragments between cross-linked
points
4-5. Именно при такой величине m можно говорить о переходе от редких
сеток к частым.
Рассмотрим сначала два крайних случая'.
1) Сетка является чрезвычайно редкой, что соответствует случаю m —> оо.
В этом случае свойства сеток практически не отличаются от свойств линей-
ного полимера того же химического строения. Соответственно, температура
стеклования Tg может быть рассчитана по уравнению (84).
2) Сетка является исключительно частой и состоит лишь из одних узлов
(несколько ниже понятие “узел сетки” будет рассмотрено детально). В этом
крайнем случае размягчение сетки может произойти при нагревании только
за счет ее термического распада по химическим связям в узлах. Температура
размягчения такой сетки, а точнее - температура га чала интенсивной терми -
ческой деструкции, может быть рассчитана по уравнению (181).
Реально имеющиеся сетки в большинстве случаев отвечают промежу-
точному варианту', поскольку содержат достаточно протяженные, но конечные
линейные фрагменты между узлами. Учет влияния как этих линейных фраг-
ментов, так и узлов сетки на температуру стеклования, привел к следующему'
уравнению, которое является обобщением уравнений (84) и (181).
(£Д^)п.ф.
Т =------=-----'_________________ (109)
Я £>;ДГ,+1^)л+(Е^А'/,)у ’
' 7 '
Температура стеклования полимеров
155
где (^А^)п.ф. -Ван-дер-Ваальсовый объем повторяющегося фрагмента сет-
7
ки; )л - набор инкрементов для линейных цепей, входящих
' J
в повторяющийся фрагмент сетки; Q^A/,) - набор инкрементов для узла
сетки. '
Это уравнение учитывает как крайние случаи чрезвычайно редких и наи-
более частых сеток, так и промежуточные случаи. Действительно, если сетка
состоит практически только из линейных фрагментов, влиянием узлов можно
пренебречь и (^ А^АИДу—> 0; уравнение (109) переходит в (84). Напротив,
/
если сетка является наиболее частой, т.е. состоит из одних узлов, то уравне-
ние (109) переходит в (181). Во всех промежуточных случаях действуют оба
слагаемых, находящихся в знаменателе уравнения (109).
Теперь рассмотрим два важных методических вопроса. Первый вопрос
связан с оценкой Ван-дер-Ваальсового объема повторяющегося фрагмента
сетки. Проанализируем несколько возможных вариантов сеток, которые схе-
матически изображены на рис.52. Сетка, изображенная на рис.52,а, имеет
Рис.52. Схематическое изображение сеток различных типов: (а) сетка, построенная из
фрагментов полимерных цепей одинаковой химической природы, обладающая
четырех функциональным узлом; (б) сетка, построенная из <]рагмептов цепей
одинаковой химической природы, но обладающая трехфункциональным узлом;
(в) сетка, построенная из линейных цепей одной химической природы и сшивающих
мостиков другой химической природы, обладающая трехфункциональным узлом
Schematic representation of networks of different types' (a) network consisting of
fragments of polymer chains of tire same chemical structure and possessing tetra-functional
crosslinked point; (6) network consisting of fragments of polymer chains of tire same chemical
structure but possessing tri-functional crosslinked point; (в) network consisting of linear
chains of the same chamical structure and crosslinks of different chemical structure possessing
tri-functional crosslinked point
156
Глава IV
четырехфункциональный узел (из каждого узла выходит четыре цепи, при-
чем все цепи имеют одинаковое химическое строение). Для такой сетки Ван-
дер-Ваальсовый объем повторяющегося фрагмента сетки вычисляется как
(£Д'Оп.ф. = 2(»/ -2)(£дг/)л +4(ХД1<)* +(£AI<)v (110)
i I i ,
где (^А^Ол - Ван-дер-Ваальсовый объем повторяющегося звена в линей-
i
ных цепях, соединяющих узлы; (^ДЕ,)у - Ван-дер-Ваальсовый объем узла
i
w ч *
сетки; (2_Д^)л _ Ван-дер-Ваальсовый объем звена линейного фрагмента,
непосредственно примыкающего к узлу (это звено имеет несколько иное хи-
мическое строение по сравнению с ‘"нормальными” звеньями).
Другой вариант отображает случай, когда сетка имеет трехфункциональ-
ный узел, соединяющий линейные цепи одинакового химического строения
(см. рис. 52,6). Для такой сетки формула для расчета Ван-дер-Ваальсового объе-
ма повторяющегося фрагмента принимает вид:
3 *
(ЕД1<)п.ф. =(ш-2)-(Хд1/)л+3(Хд)/.)л+(Хд1>;)У; (ill)
i 1 i « /
где все обозначения - тс же, что в формуле (110).
Еще один тип сетки изображен на рис. 52,в. Эта сетка построена из сши-
ваемых цепей одной химической природы и сшивающих мостиков другой
химической природы. В этом случае формула для расчета Ван-дер-Ваальсо-
вого объема повторяющегося фрагмента сетки имеет вид:
(ЕД^)п.ф. =(/«1 -2)(ХДГ<)л,1 + 2(1ДЕ/);1 + ->
7 7 7
^+^¥^(ХД^)л,2+(1Д^)л,2+(1Д^)у, (112)
где (ХД^)л,1 - Ван-дер-Ваальсовый объем повторяющегося звена .линей-
7
ных фрагментов сшиваемых цепей, причем т, - среднее количество этих зве-
ньев, заключенных между узлами сетки; (X Д^ )л,1 - то же самое для звена,
7
непосредственно примыкающего к узлу (имеющего другое химическое стро-
ение по сравнению с “нормальными” звеньями),-Ван-дер-Ва-
Температура стеклования полимеров
157
альсовый объем повторяющегося звена линейного фрагмента сшивающих
мостиков, причем >?/2 _ количество таких звеньев; Д ) л 2 - то же для зве-
J
на, непосредственно примыкающего к узлу.
Теперь остановимся на понятии “узел сетки”. Для того, чтобы сформули-
ровать это понятие, в работе [30] была проведена калибровка уравнения (109)
ш ос нэве экспериментальных данных по температурам стеклования для мно-
гочисленных и хорошо охарактеризованных сетчатых систем. В результате
оказалось, что для наилучшего согласия расчетных данных с эксперименталь-
ными необходимо принять следующее определение узла: узлом сетки являет-
ся группа атомов, включающая атом, от которого начинается разветвление
цепей, плюс соседние химически связанные с ним атомы со своими ближай-
шими заместителями. Ниже показаны сетки различной химической приро-
ды, в которых узлы, согласно данному определению, обведены пунктиром.
Ориентируясь на данное определение узла и приведенные примеры, можно
достаточно легко идентифицировать узел в сетке любой химической приро-
ды. :
i
сн2
-----р_£ —!
! сн2 ]
I----------1 j I I
CH2-f-CH2-CH-CH2-tCH2-......—СН2тСН2—С—CH2-J-CH2—
ill I I I
;___CH2____; !______CH_2___-
CH2 CH2
Chfe—CH-+CH2—CH—CH2-+CH—CH2—CH—
! i
• •—СН2— CH-* CH2- CH- CH_2j- CH- CH2— CH - •
0 0 Q
______OH OH
! CH24cH-CH2- 0-O- O-CH2-CH- CH2— •
CH2—N4 ; г-л
n CH2-{-CH- CH2— O—O- o—ch2—ch- ch2—
III I
------OH 0H
158
Глава IV
I
СНз-ЗНСИз
СНз] О ; СНз
I I I I
Si-rO-Si-OtSi--
I ! I ।
СНз! О I СНз
।--1---J
СН3-Si-СНз
I
Ниже будут проанализированы различные варианты сеток и даны при-
меры расчета их температуры стеклования Tg. Сейчас остановимся на таком
вопросе, как оценка величины молекулярной массы усредненного межузло-
вого фрагмента. Такая оценка может быть проделана на основе химического
строения сетки и экспериментально определенной температуры стеклования
Для этого в уравнение (109) нужно подставить экспериментальную вели-
чину Tg и решить это уравнение относительно т. Проделаем этот анализ в
общем виде для различных типов сеток, изображенных на рис.52.
Сначала рассмотрим сетку, изображенную на рис.52,а; из каждого узла
этой сетки выходят четыре цепи, и при этом все межузловые фрагменты име-
ют одинаковое химическое строение повторяющегося звена. Тогда, рассмат-
ривая повторяющийся фрагмент сетки, на основании соотношения (ПО) и
уравнения (109) необходимо записать
2(щ-2)(£Д1<)л +4(Х ДК,)*,+(£ АС,)у
р ___________________1_________i_________[_____________
ё " 2(ш-2)(£Й!А1< + 4(£АГ' + '£bJh +(1^;АИ,)у ’ (ИЗ)
' J i 7 i
где (^^АС, + ^bj)Si - набор инкрементов для повторяющегося звена ли-
' j
нейных межузловых цепей; (^а,Д1’, + 2^/)д - то же, для крайних звеньев,
' 7
непосредственно присоединенных к узлам; (£КДИ,) - набор инкремен-
тов для узла сетки; остальные обозначения те же, что и в формуле (110).
Молекулярную массу линейных цепей между узлами Мс можно опре-
делить как
Мс = (т-Т)М + 2М* +уА/у, (И4)
где М- молекулярная масса “нормального” звена; А/ - молекулярная масса
звена, непосредственно примыкающего к узлу’, 4/у-молекулярная масса узла.
Температура стеклования полимеров
159
Обозначим:
(£ДК;)л = Л; 2(ХДИ0л+|(2;Д1/;)у=В; +
' / 2 1 ' j
nXM/.+Zb^+^(ZKj^y=D 015)
' J 2 '
Тогда соотношения (113) и (114) после некоторых преобразований при-
нимают вид:
15-1гО * 1
Мс =-----—А/+23/ + —Л-/,, (117)
TgC-A 2 у- ’
Теперь проанализируем сетку с трехфункциональными узлами, для кото-
рой химическое строение звеньев цепей, выходящих из узла, одинаковое
(см.рис.52,б). Для повторяющегося фрагмента такой сетки следует записать
(т-2) | (X ДИ,.)л +3(£ДК,)'л +(ХДК,.)у
rg =---------------'---------------------------i--------------,(118)
(W-2) |(^Д/ДГ1. +^bj)n +3£>,.ДК,. +Xbj)*n + £W)y
"" ' J i j i
где все обозначения те же, что и в формуле (113).
Тогда
2B.-TgDx
т-2--------21— ,
3 TgC-A
(И9)
где
/31=3(^Д1/-)>(2Д^)У; ^=3(SM^+S^)>(S^A^)y (120)
/ / 1 j i
Отсюда
* 2 2 А - / Т>] , 2
Мс = (т - 2).\/ + 23/ +—A/v =--—3/ + 2Л/ +— 3/v.mn
с 3 у 3 TgC-A 3 у (121)
Третий вариант на рис.52,в. Эта сетка состоит из линейных цепей, сши-
тых цепями другой химической природы; узел сетки является трехфункцио-
нальным. Для такой сетки зависимость Tg от состава имеет вид
160
Глава IV
(W1 -2)(2 ДН)^ + 2(2 ДИ,)‘, + |(2 ДИ)у +
Т =-----------------
в (щ2 -2)£>,.Д^ +Yb.iU + 2(2«,ДИ,. +(Е^АИ)у +
+ —2“ (Z Д,<) л.2 + (Z д Vt) л,2 +1 (X Д1< )у
_> ______-____'---------L-----------1------, (122)
+ +Е/’7)л,2 + (£а,Д^ +2>;)л,2
2 ' J ‘ 7
где ahi и »/2 - числа “нормальных” звеньев сшиваемых цепей и сшивающих
мостиков между соседними узлами. Все параметры с индексом * относятся к
звеньям, непосредственно примыкающим к узлам. Соотношение между тх и
ти2 находится исходя из мольного соотношения компонентов 1 и 2:
'"i /т2 = 91 /42,
где 91 и <72 ~ числа молей компонентов 1 и 2 соответственно. Подставляя это
соотношение в формулу (122), получаем
В2 -2АХ-А2 -Т {D2-2С-С2)-
тх -2 ------------------------------>
Tg(Cx+-~—C2) —
292
-2^(С1-^С2) + 2(4 + ^4)
-у___-___________________2-Я1__, (123)
-(4+^4)
292
где
4 =(Ед^к1; в2=2(2 дк,.);, +|(Х ДИ )у +(2 д^С +|(1ДИ)У;
i i i i i
4 = (2 ди, )л>2; q = (2ди, + 2 bj )лд;
i ' 7
d2 =2(2«<ди,- +2^)‘.л +(2нд»/1)у +(1«<д^ +ZM‘2;
i j > ‘ J
C2=^a^Vi+^bj)„2,
' 7
Температура стеклования полимеров
161
Введем обозначения:
В = В2 -2Л1 - А2 +2(Ai + — Л2);
<72
D* = D2~C2 + ^-С2',С* = С, +-^-С2\А* =С] +-^-А2. (124)
<?2 292 2<?2
Тогда получаем
В* -Т D*
т'~г-Т^7- ,125)
В сетке, построенной из разнородных фрагментов цепей, заключенных
между узлами, целесообразно определять два значения Мс (!Асл молекуляр-
ная масса фрагмента цепи между узлами для сшиваемых цепей и Мс2 то же
для сшивающих мостиков). При этом
Мс] = (»»] -2)Л/1 +2М,‘ +Л/у1; (126)
Л/с 2 = (т2 — 2)М2 + 2Л/2 + Л/у 2 = — 2) + 2Л/2 + у, 2 > (122)
где A/Vj - молекулярная масса той части узла, которая принадлежит сшивае-
мым цепям (на рис 52 р соответствует черным кружкам)',Л/у2 -молекуляр-
ная масса той части узла, которая принадлежит сшивающим мостикам (на
рис.52,в соответствует белым кружкам). При этом величина Л/у] +Л/у2 равна
молекулярной массе всего узлаЛ/у. Тогда, с учетом формулы (125), выраже-
ния (126) и (127) приобретают вид
МсЛ = л ,а/1+2Л/; +л/уД;
(?1 В —TqD *
Мс 2 =—Л/2(-;---J- + 1) + 2(Л/2 ~М2)+Му2 _
Я2 Tgc -А
(128)
(129)
Среднее значение молекулярной массыцепи между узлами сетки мож-
но рассчитать по соотношению
^=(^^+(1-^71/^, (130)
где а =91/(91 + 92)-
162
Глава IT
Рассмотрим в деталях ход расчета Tg и Мс для всех трех вариантов. Пер-
вый из них соответствует сетке с четырехфункциональным узлом и цепями
одинакового химического строения, выходящими из узла. В качестве простей-
шего примера проанализируем гипотетическую сетку на основе полиэтилена
Структура IV
i
.—с_...
I
СН2
_____(СН_22т-_2_
: ! СН2 :
—С-СН2-(СН2)т.24сН2-С-СН24(СН2)т.2-СН2-С—•
I । I । ।
(CH2U2
: СН, :
I I I
-С-СН2-(СН2)т-2-СН2-С-СН2-(СН2)^2-СН2-С—
СН2 :
Для такой сетки Ван-дер-Ваальсовый объем повторяющегося фрагмента
определится из соотношения
(£Д^)п.ф. =(Х^)у+2(2Д^)о('«-2), (131)
7 7 t
где (£ДР/)У - Ван-дер-Ваальсовый объем узла (ограничен пунктирными
7
линиями); - Ван-дер-Ваальсовый объем — СН2-группы.
При этом* 1
(ХАР/)у = AJ/C,1 +4ДГСдо+8ДСЯ;124 = 5,0+ 4-13,1+ 8-2,0 = 73,4 А3;
7
(Z Д^)о = Д^С,10 + 2ДСя>124 = 13,1 + 2 • 2,0 = 17,1 А3-
Тогда
(£ ДК, )п.ф. = 73,4 + 2 • 17,1(ш - 2) = 50 + 34,2п/.
7
Номера атомов соответствуют тем, которые даны в табл.З
Температура стеклования полимеров
163
Рис.53. Зависимость 7g от т для модельной сетки иа основе полиэтилена с
четырехфункциональным узлом
Dependence of Тъ on т for the model polyethylene network with tetra-functional
crosslinked point
Величина (^а,Д1^ + = 2(г7СДРсд0 +aff 2Д17/-/j24)(w ~2)~
' J
=2(0,021-13,1 + 19,98-2-2,0)(m - 2)-10’3 = (160,39m - 32O,78)-1O’3 A3K-].
Величина (^ А1,ДЕ, )v = K'f-(АГ'с^ + 4Д1/(2до) + Адд -8-Д1'7/д24 =
i
= |l,15(5,0 + 4-13,1)+ 2.307-8-2.0]-10’3 = 102,92-10"3 A’K'1.
Подставляя эти значения в уравнение (113), получаем
73 4 1
Те(К)=--------—-----------103. (132)
7 160,39»i-217,86
Соотношение (132) справедливо, когда т > 2. Зависимость Tg от т, рас-
считанная по этому' соотношению, показана на рис.53.
Если т = 1 или т = 0, картина изменяется. При т = 1 структура сетки VI
выглядит следующим образом
164
Глава IV
С--
: ( <ЭНтд :
Il I ' I
—c-qH2-c-cH2-c—
I I ill
: ---:
—-C—
Такая сетка состоит только из одних узлов, причем каждая—С Н2-группа
принадлежит двум соседним узлам.
Тогда
(1А^)п.ф. =(1М)У = ДИСЛ +-АИСД0 +уД^124 =
= 5,0 + 2-13,1 + 4-2,0 = 39,2 А3;
(2«/Д’<+2^)л=о;
' j
-Кс -(ДТсд + — Al'cjo) + Кн •~Д,/Я,124 =
i
= [1,15(5,10+ 2-13,1) +2307-4-2,0]-10-3 = 5435-10-3А3К'1.
Поскольку данная сетка не содержит линейных фрагментов и для нее
(^2 а, А К, + ^Ьу)л = 0, то размягчение такой сетки может произойти только
' J
в результате ее термической деструкции, т.е. распада по химическим связям.
Подстановка всех параметров в уравнение (113) дает
39 2 з
Те =Td = ^--Ю3 = 721К.
г 54,34
Если m = 0, то структура сетки VI принимает вид
I
— С—
I
i i i i
•—с-с -4— c-r c-c--
i i L4-.J i i
I
—-C—
I
Температура стеклования полимеров
165
т.е. имеет идеальную структуру алмаза. Для нее
(Ед^)п.ф. = д;/сд = 5-° Д3; = о;
i ' j
(£К,А Р')у = А'с Ссл = 1,15-5,0 10~3 = 5,75- ICT3 А3К.
/
Подстановка всех этих значений в уравнение (113) дает
5 0 з
Т„ =Td = —-103 =870К.
ё d 5,75
Таким образом, расчетная температура начала термодеструкции алмаза со-
ставляет --600 °C; из литературы известно, что алмаз полностью сгорает при
нагревании до 850-1000 °C.
Теперь рассмотрим поведение сетки
Структура VII
-------------1 । 1
—(CH2)m5-VCH2-CH-CH2T-(CH2)m.2-;-CH2-CH—
L.-cjk-.j
(CH2)m.2
—(СНД^СНг-СН-СН^ССН^^СНг-СН—
которая имеет трехфункциональный узел сшивки, обведенный пунктирными
линиями. Для такой сетки
(£ ДИ,)у = ДИС>6 + ЗДСсд0 + 7ДКНД24 = 9,0+ 3 13,1 + 7 • 2,0 = 62,3 А3;
i
(£ Д^)л = (ДКс,ю + 2ДИнд24)(т - 2) = (13,1 + 2 • 2,0)(ш -2) =
i
= (17,1m-34,2) А3;
(£А*аФ +1,5(ХДК,)Л = 62,3 + 1,5(17,1ш-34,2) =
= (26,65m+ 11) А3;
166
Глава IV
+Ybj )л + (£ ДЕ, )у = 1,5[(асДИс 10 + 2а н ДГЙ,124 )(т -2)]л +
+ [КС (AVc+ ЗДГсдо) + 7Кц AV// l24 ]у = {[1,5(0,021 13,1 + 2 • 19,98 2,0) х
х (т - 2)] +1,15(9,0 + 3 13,1)4-7 2,307 • 2,0} • 10“3 = (120,3m -152,76) • 10~3 Д3К'1.
Подставляя все полученные выше величины в уравнение (118), имеем
„ 25,65т + 11 , „з
Т„(К) =----------------10 61331
g 120,3m-152,76 ' u ’
Зависимость Tg от т показана на рис.54. Хорошо видно, что Tg резко
убывает с ростом т, т.е. с увеличением размера линейных межузловых фраг-
ментов.
Формула (133) справедлива при т > 2. Если т = 1, имеем сетку следую-
щего строения:
Рис.54. Зависимость Tg от т для модельной сетки на основе полиэтилена с трехфункди-
ональным узлом
Dependence of Т& on т for the model polyethylene network with tri-functional
crosslinked point
Температура стеклования полимеров
167
—СН -фг-СН-СНг-СН—
-С-Нг-'
•—СН-СН2-СН-СН2-СН--
Эта сетка состоит только из узлов сшивки, которые и являются се повто-
ряющимися фрагментами; эти фрагменты-узлы заключены в пространство,
обведенное пунктирными линиями.
Для такой сетки
(ЕДН)пф. = (ЕДИ)у =ДКС6+1,5ДКС.1О+Д/Я,124 =
i i
= 9,0 +1,5 • 13,1 + 3 2,0 = 34,65 А3;
(Е^Д'/,+Е^)л=о;
' j
(1>,ДЦ)у =^с(Д1/с,б + 1,5А1 сдо) + "З-Д^ядгл =
/
= [1,15(9,0 + 1,5-13,1) + 2,307 • 3 2,0] • 10'3 = 46,8 • 10~3 А3К.
Подставляя все полученные величины в уравнение (118), имеем
34 65 т
Д =Т,, =—---103 = 740К.
s d 46,88
Наконец, рассмотрим сетку' структуру VII при условии т = 0. Эта сетка
имеет вид:
I г — i
--- СН-гСН-1-СН—
-—ch-ch-ch—-
I I
Здесь разветвляющим атомом является каждый атом углерода, поэтому сле-
дует записать:
(ХД^)У =ДИС6+ДИЯ>124 = 9,0 + 2,0 = 11,0 А3;
i
(£п,ар;+Х^)л=о;
/ J
168
Глава IV
= ^K,\V,)y =КС -AVc!6 +ки -wH<nA =
i i
= (1,1 5 • 9,0 + 2,307 • 2,0) • 10-3 = 14,964 • 10'3 A3^1.
Подставляя эти значения параметров сетки в уравнение (118), получаем
Т„ =Td =-li2--103 = 735К.
g d 14,964
Теперь рассмотрим сетку третьего типа, когда сшиваемые цепи имеют
одно химическое строение, а сшивающие мостики - другое. В качестве при-
мера проанализируем реальную сетку на основе эпоксидной смолы следую-
щего химического строения (30]
Структура VIII
—-СН2|СН-СН2-О -Q-O-CH2-CH-CH2-O-Q--O
ОНг_____________________________он
^-О-СН^СН-СНг-О-О+О-СНг-СН-гСНг-Н-СН^СН— СН2
—CHrlCH- сн2-о-
------J I
он
г
сн2
6
I
--N—
Стехиометрический состав сетки соответствует 2 молям эпоксидного оли-
гомера на 1 моль отвердителя - диамина. Узел сетки отмечен пунктирными
линиями.
Для повторяющегося фрагмента сетки имеем:
(Ед^)пф. =(А^,144 +2ЛИС;56 + ДСс,21 + 4ДИн 124)+[2ДКс>39 +
i
+2ДКС4О +2ДКСд30 +2ДИСд32 +2ДГЯ125 + 6ДС//д24 + 2ДСс20 +
+4Д,/СД8 + 4ДИ/7124 + (2Д1/(9130 + 2ДИС;40 + ДПсдд + Д^сдзг +
+5ДСЯд24 + ДР7/;]25 + 2ДСС20 + 4ДССд8 + 4ДСЯд24)н1]л,1 +(4ДГ'С,18 +
+ AI с,19 +4АСЯд24 +уАИСд2 + —-2-ДИу 124)^2 =(0,9 + 2-14,6 + 10,2 +
+4 • 2,0) + [2 12,2 + 2 16,2 + 2 2,7 + 2 • 5,6 + 2 4,7 + 6 2,0 + 2 • 11,6 + 4 • 12.7 +
Температура стеклования полимеров 169
+4-2,0) +(2-2,7 +2-16,2 +12,2 + 5,6 + 5-2,0+4,7+2-11,6 + 4-127 + 4-20)т] +
+ (4-12,7 + 8,4+4-2,0 + |-12,6+-~2-2jD) = (300j6 +152 3)m А3;
+ £Ау)л,1 = «с(2ДИС;39 + 2ДИС;40 + 2ДИС;2о + 4ДИС18) +
«jY(2AI<7,125 +6Д1/АГ124 + 4Д1/ЯД24) + яЛ>,о(2Д1/?Д13о) + аО,з(2Д,/ОД32) +
+[лс(2Д</С,40 +Д1/С,39+ 2A,ZC,20 + 4ДИС18) + а//(5Д1/Я;124 + Д,/Я,125 +
+4ДИуу,124) + ,0(2^^0,130) + аО,з(ДКэ, 132 )l/w +bd '2+bh -1+bpf +
+(bd + bh+bM)m= {0,021(2 • 1-2,2+ 2 • 16,2 + 2 • 11,6 + 4 12,7) +19,98(2 • 4,7 +
+6 • 2,0 + 4 2,0) + 22,95 • 2 • 2,7 +16,0 • 2 • 5,6 + [0,021(2 • 16,2 +12,2 + 2 • 11,6 +
+4 • 12,7) +19,98(5 • 2,0 + 4,7 + 4 • 2,0) + 22,95 • 2 • 2,7 + 16,0 • 5,6] w + 2(-55,4) +
+ 2(-l 39,6) +16,0 + (-55,4 -13 9,6 +16,0)m} • 10 3 =
= (519,3 + 490,6m) 10 3 A3K4;
(£д1// +£йу)л,2 =ас(4Д,/С,18 + Д*/С,19 +уД1/С,12) +
' j
+ ан (4ДУЛгд24 +у • 2 Д 1^124) +ЬП = [0,021(4-12,7 + 8,4 +1 -12,6) +
+19,98(4 • 2,0 + 2,0) + (-25,6)] -10^=175,6 • 10“3 А3К4;
(£^|Д^)у = ^мД^М,144 + Д^С(2Д^С,56 + Af C,21) + 4 Д/77 Д24 =
/
= [2,52 • 0,9 +1,15(2 • 14,6 +10,2) + 2,307 • 4 2,0] • 10'3 = 66,1 10“3 А3К4.
Подставляя все вычисленные значения параметров сетки в уравнение
(122), получаем:
Tg(K)=--------Ж6 + 11523^-----10.3
г 519,3 +490,6m+ 175,6+ 66,1
При т = 0 имеем структуру отвержденной эпоксидной смолы на основе
диглицид илового эфира соответствующего бисфенола
но-0-^-он
170
Глава IV
Рис.55. Зависимость Гьраи от т для отвержденной эпоксидной смолы (см. текст)
Dependence of Т& расч on т for the cured epoxy resin (See text)
Для такой смолы расчетное значение расч = 395К; экспериментальное зна-
чение Т^.эксп = 396 К.
На рис.55 показана зависимость Tg от т , вычисленная по формуле (134).
При уменьшении линейного межузлового фрагмента сетки температура стекло-
вания возрастет, особенно интенсивно, когда т 0.
Теперь рассмотрим влияние особенностей строения полимерных сеток и
их дефектов на температуру стеклования Tg.
а) Влияние распределения сшивок в сетке
на температуру стеклования
Сначала проанализируем влияние распределения сшивок вдоль цепей на
температуру стеклования сетки. В качестве модельной сетки, хорошо изучен-
ной экспериментально, выберем систему на основе полистирола, сшитого ди-
винилбензолом. Структура такой сетки имеет вид:
Структура1Х •
-----------, f ।
•-(CH2-CHUiTCH2-CK-CH2tCH-(CH2-CHUi+CH2-CH-"-
о дхб'.х.ф о "Г””.'.'.
--- <СН2-СН)т.,Гснг-СН-СНДСН-(СН2~СН)т,ГСН2-СН-"-
О 6 О '-"СТ-
I
Температура стеклования полимеров
171
Эта сетка является трехфункциональной, т.е. из каждого узла сетки (окан-
тованного штриховыми линиями) выходят три цепи. Для нее
= APc,7 +2^IZC,10 + ^С,19 + 5А!/Я,124 =
i
= 8,7 + 2 13,1 + 8,4 + 5 • 2,0 = 53,3 A3;
(1>,)M = ^Ус,7 + Д^С,19 +5ДИС>18 +6Д1///д24 +(Д^С,10 + Д^С,7 +
/
+Д1+- 19 + 5ДКС lg +8ДИЯ 124 )(m -1) = 8,7 + 8,4 + 5 • 12,7 + 6 • 2,0+ (13,1 +
+8,7 + 8,4 + 5 12,7 + 8 • 2,0)(т -1) = (109,7да -17,1) А3;
(Z W)л,2 = 2ДИСД8 + 2Дд24 = 2 • 12,7 + 2 • 2,0 = 29,4 А3;
/
(2Х,.ДК)У =^с(ДИс,7 +2ДГСд0 +ДКСд9) + АГя5ДКНд24 =
L
= [1,15(8,7 + 2 • 13,1 + 8,4) + 2,307 • 5 • 2,0] 10-3 = 72,9 • 1 (Г3 А3^1;
+ X/P-I.1 = ас(Д^С,7 + Д^С19 + 5Д1/С,18) + ая6Д///д24 +
' У
+[й(2 (ДК^ iq + ДИ^ 7 + Д17с 19 + 5ДР^ 18 ) + а77 8ДИд7Д24](ш — 1) + 6^ / 2 +
+bd!2 {т -1) ={0Р21(8 7+8,4 + 5-12,7) + 19,98-6-2,0 + [0,021(13,1 +
+8,7 + 8,4 + 5 • 12,7) +19,98 • 8 • 2,0] (да -1) + (-55,4) / 2 + (-55,4) / 2 х
х(m-1)} 10-3 = (-8Q2+ 2940/1)- lO^A3^1;
(Х«, ДИ,- +1>Д,2 =«с-2ДИс,18 +й//2ДКЯ1124 +ЬП / 2 =
‘ J
= (0,021-2-12,7+19,98 - 2,0 - 25,6/2) - ИГ3 = 67,6 -10-3 A3K4.
Подставляя все вычисленные значения параметров сетки в уравнение
(122), получаем
64,6 + 109,7w 1()3
60,3+ 294,Ош
(135)
Формула (135) справедлива при условии т > 1. Зависимость 7^ от т, вы-
численная по формуле (135), показана на рис.56 (следует заметить, что когда
172
Глава IV
Рис.56. Зависимость Т от т для сетки на основе полистирола и дивинилбепзола
Dependence of Тъ on m for the polystyrene and divinylbenzcne network
m —> oo, мы получаем расчетную величину Tg для линейного полистирола,
равную 373 К).
Видно, что температура стеклования резко возрастает при уменьшении
числа звеньев полистирола между соседними сшивками, когда количество
этих звеньев (m - 1) —> 0.
При т = 0 структура сетки IX имеет следующее строение
сн2-сн-сн2-сн-сн2-сн---
Температура стеклования полимеров
173
что соответствует 100 %-й сшивке. Узел такой сетки окантован пунктирными
линиями. Для него
(^ГДР'Ду = 2- — ДИСд0 +Д1/с,7 +2'~А^Я,124 + Д^Я,124 + Д^С,19 ~
1 2
= 2- —-13,1 + 8,7 + 2- —-2,0+2,0+8,4 = 36,2 А3.
2 2
Величина С^а,^ + ^Ь})л-1 = 0,т.к. все атомы в сшиваемых цепях вхо-
) J
дят в состав узлов.
£ ДИ,)^ = + 2ДИЯД24 = 2 • 12,7 + 2 - 2,0 = 29,4 А3;
I
l^a^V, +ХАу)л,2 = аС ’2Д^С,18 + °я2Д^Я,124 + Ап /2 =
' J
= (0,021 -2-12,7 + 19,98 - 2 • 2,0 - 25,6 / 2) • 10~3 = 67,6 • КГ3 А3К4;
1 2
(2^/Af7) =^с(2- —AIZC,10 +Д^С,7 + Д^С,19) +-^/7 (2 'тД17//,124 +
> 2 2
J
1 2 -т.
+ ДИ//124) = [1,15(2-у-13,1 + 8,7 + 8,4) + 2,307(2-у-2,0+ 2,0]-10 =
= 48,57 -10-3 А-’К'1.
Подставляя все эти величины в уравнение (122), получаем
Т (К) = 36’^-—— Ю3 = 565 .
g 48,57 + 67,6
Теперь рассмотрим влияние распределения узлов вдоль цепей на темпе-
ратуру стеклования Tg. Анализ проведен в терминах степени сшивки а. Для
фрагмента сетки
определим значения всех параметров уравнения (122).
174
Глава IV
(^2 A1Z/)1 — 2A1zc,18 + 2AI7f/i24 + Al'cjo + ^'C,7 + A^C,19 + 3AIz/yi24 =
i
= 2 • 12,7 + 2 • 2,0 +13,1 + 8,7 + 8,4 + 3 • 2,0 = 65,6 A3,
(Ха,ДЦ+^, +S Wh =«с(2Л1/с,18) + ^(2А1/я,124) + 4/’п +
/ j / 2
+^с(ДИсдо +ДКС7 +AKc>19) + ^ArH]24 =[0,021-2-12,7 +
+19,98-2 • 2,0-^--25,6+ 1,15(13,1+8,7 +8,4) +2,307-3-2,0]-103 =
= 116,2-IO-3 A3K-!.
Теперь необходимо учесть, что в повторяющемся звене полистирола
— СН2тСН--------
6
I
: 2*
которое непосредственно примыкает к узлу сетки, группа — С Н2 — (отмечена
пунктиром) входит в состав узла. Тогда для этого звена полистирола имеем:
= ~^С,Ю + ^'с,7 +А^С,19 +5А1'С,18 +8Д^ЯД24 =
/ i
= 13,1 + 8,7 + 8,4 + 5 • 12,7 + 8 • 2,0 = 109,7 А3;
+ 5L^/A1Z)2 =ac(AlZC,7 + AIzc,19 + 5A1'C,18) +
' J '
+ая *"(’</ (2 + A?cAKCjl0 + Kfj 2Д ,24 =
= [0,021(8,7 + 8,4 + 5 -12,7) +19,98 • 6 • 2,0 - 55,4/2 +1,15 • 13,1 +
+ 2,307 • 2 • 2,0] -10-3 = 238,1 - IO-3 A3K'*.
Для “нормального” звена полистирола
сн2—<рн------
О
имеем (У ДИ )ПС =109,7 А3;
I ' 11L ’ ’
Температура стеклования полимеров
175
(2La! + X/J)|1C - °c(AJ'C’,10 + АП2,7 + Д^'С,19 + 5A17c,\8) +
' J
+ aH -8AK// I24 +^bd =[0,021(13,1 + 8,7 + 8,4 + 5-12,7) + 19,98-8-2,0-
-55,4/2]-IO-3 = 294,0-HF3 A 'K'1.
После подсчета всех параметров уравнения (122) проанализируем три
варианта.
I) Равномерное распределение сшивок
Для такого случая уравнение (122) приобретает вид
а(£Д^)1 +
а(£я,-Д1/- + £й,. +2Х-ДГ,-), +а(£«,-ДК,- + ^Ъ j+ИК i^V
+ (1-а)(£А1<)пс
+-------------------------,
+ (1 - 2а)(Х а,ЛР, + ^bj )пс
' J
(136)
где а - степень сшивки, равная Мт, характеризующая долю связанных фе-
нильных групп.
Подставляя в (136) значения всех параметров сетки, определенных выше,
получаем
Т W=а-65,6 + (1-а)109,7 1()3
г а-116,2 + а-238,1+(1-2а)294,0
(137)
Формула (137) действует лишь при условии 0 < а < 0,5. Если а > 0,5,
свободных звеньев полистирола не остается, и формула (137) переходит в
соотношение
Т g-^ + d-aWJi^ (]38)
g а-116,2 + (1-а)238,1
При а = 0 получаем расчетную температуру стеклования линейного по-
листирола.
Зависимость 'Л от степени сшивки а, определенная с помощью соотно-
шений (137)и (138), показана на рис.57, кривая 1.
176
Глава IV
Рис.57. Зависимость Т от степени сшивки а для сетки на основе полистирола и
дивинилбензола: 1 - равномерное распределение узлов сшивки; 2 - случайное
распределение узлов; 3 - наиболее неравномерное распределение узлов. Точки
относятся к экспериментальным значениям Т
Dependence of Т& on crosslinking degree a for the polystyrene and divinylbenzene
network: 1 - unifonn distribution of crosslinked points; 2 - irregular distribution of crosslinked
points; 3 - the most ununiform distribution of crosslinked points. The points denote Ts alues
found by experiment
Из рис.57 видно, что зависимость Tg от степени сшивки а состоит из двух
участков - с быстрым и медленным возрастанием Tg . Это связано со специ-
фическим влиянием узлов сетки на Tg, что видно из проведенного выше ана-
лиза.
2) Случайное распределение сшивок.
Как известно, функция распределения по расстояниям для случайного
поля точек, лежащих на прямой, имеет вид
= (139)
где / - расстояние между соседними точками, а в нашем случае - расстояние
между узлами; /ср среднее расстояние.
Температура стеклования полимеров
in
Переходя к степени полимеризации т, получаем
F(rn) = \-e mlmcp . (140)
Если узлы располагаются рядом друг с другом
•— CH2-CH-CH2-CH-CH2-CH-CH2-CHtCH2-}-CH-CH2-CH—
6 6 6 6 "’’"6 6
1 I I Г----1
— СНг-СН-СНг-СН-СНг-СН-СНг-СН+СНг-ГСН-СНг-СН----
6 '““'6 6
то количество — СН2~ групп, принадлежащих остаткам стирола (отмечены
пунктиром на схеме) и одновременно входящих в состав узла, будет меньше,
чем в том случае, когда узлы разделены линейными фрагментами:
-фСНгч-СН-СНг-СНтСНг+СН-СНг-СН-СНг-СН-СНг-СН--
''~6 6 '"'’'6 6 6 6
------ I ,---- I I
—-гСН2-{-СН-СН2-СН4СН2*СН-СН2-СН-СН2-СН-СН2-СН- —
'''6 '"’"'6 6
В результате значение a.(^iajNVl + ^bj + уменьшается на
< J '
величину, пропорциональную количеству рядом расположенных сшивок. Тог-
да на ту же величину увеличивается значение (1 - а)(^п,Д1/1 + Ь;)пс
' J
Поскольку нас интересуют узлы, расположенные рядом, F(m) при т = 1
составляет F(\) = 1 - г 1 , аучитывая, что тср = 1/а, получаем
F(l)=l-ea.
(141)
Тогда в соотношении (137) в знаменателе вместо а во втором слагаемом
следует подставить а - а (1- е‘а) = а/е“,а в третьем слагаемом - вместо
(1 - 2ц) нужно подставить 1 - 2а + а (1- е‘а) = 1 - а - а/еа.
178
Глава IV
Произведя такие подстановки, получаем
a(ZA^)i +
Tg (К) --------------------------
а£>,.ДК,. +Xbj +4<1>-АГ-' +%bJ +ХК^К)*2 +
, j i е i j
+ (l-a)(£AJZ)no
->--------------1-------------. (142)
+ (1-а-а/е“)(^а1Д1/,+^/>y)nc
' j
Подставляя в (142) все вычисленные выше значения параметров сетки,
получаем
Tg (К) =--------------------------------10з (143)
а116,2 + -^-238,1 + (1-а-а/е“)294,0
Формула (143) действует при условии (а + а/е“) < 1; это условно соответству-
ет а < 0,66
Если (а + а/е“) > 1, свободных звеньев полистирола не остается, и форму-
ла (143) переходит в соотношение (138).
Зависимость Tg от а, полученная с помощью (143) и (138), показана на
рис.57, кривая 2.
3) Наиболее неравномерное распределение узлов.
Такое распределение соответствует случаю, когда большое количество уз-
лов сшивки располагается рядом и образуют значительные последовательности
узлов, связанные линейными фрагментами полистирольных цепей (см. схему
на рис.58).
При таком расположении узлов количество звеньев полистирола, примы-
кающих непосредственно к узлу, становится малым и можно записать.
а(^ДК,)1+(1-а)(^ ДИ,)ПС
К} =--------------------------------------------------- (14*
aQ>,.AK,. +Z^AKi)1 + (1 -а)(^2+ £>у)пс ’
z j i i j
Подставляя в формулу (144) все значения параметров сетки, получаем
a65,6 + (l~a)109,7 3
1 о (А ) —----------------1U
* а116,2 + (1-а)294,0
(145)
Температура стеклования полимеров
179
Рис.58. Наиболее неравномерное распределение узлов сетки (схема)
Hypothetical case of the most ununiform distribution of crosslinked points
of network (Scheme)
Расчеты, проведенные по соотношению (145), приводят к зависимости
Tg от а, показанной на рис.57, кривая 3.
На этом же рисунке приведены экспериментальные данньЕ по зависимости
Tg от степени сшивки о, заимствованные из работы [160].
Экспериментальные точки хорошо укладываются на кривую 2; это свиде-
тельствует о том, что узлы в реальной сетке распределены случайным обра-
зом.
б) Влияние строения узла на температуру стеклования сетки
Рассмотрим еще один вопрос о влиянии химического строения сетки на
ее температуру стеклования. В данном случае будем анализировать сетки с
одинаковым химическим строением межузловых фрагментов, но с разным
строением узлов сшивки. Для такого анализа удобны полидиметилсилоксано-
вые сетки следующего химического строения [27]:
I СНЗЧ -'СК ХСН3! СН3 СН3 СН3
' ,Si Si, ' I 3 I I
—CHoT-CH/A A4CH2fCH2-Si-(O-Si)„.2-O-Si-CH2-”-
; X ! CH3 CH3 CH3
! ,ch2 ch3 !
^;ch2" (A)
180
Глава IV
! снзх
CHj-CH/Y
I
•”-СН2[
о
I
2-Si-O-SrCH2-rCH2—•
СН3 ЧСН3;
/Сн3; сн3 сн3 сн3
.Si-O-SL I I 3 I 3 I 3
XJH2fCH2-Sr(O-Si)fr2-OHSrCH2- -
I ! СН3 СН3 СН3
(Б)
При малых величинах п (частая сетка) синтез основан на применении
индивидуальных соединений с точно заданной величиной н, поэтому при близ-
кой к 100 %-ной конверсии величина Л/с в таких сетках задана с достаточной
точностью.
Прежде чем приступить к анализу влияния строения узлов на Tg, напом-
ним определение узла сетки с точки зрения его химического строения: узлом
сетки является группа атомов, включая атом, от которого происходит развет-
вление, плюс соседние химически связанные с ним атомы со своими ближай-
шими заместителями (при этом другой конец ответвления также должен вхо-
дить в соответствующий узел, в противном случае это будет просто разветв-
ление). Тогда для рассматриваемых сеток узлы имеют строение, заключенное
в пространство, ограниченное штриховыми линиями.
Определим для этих систем все значения инкрементов, входящие в урав-
нение (109):
•I
(£Д^)п.ф. =здг/с ,106 + ЗДИС
,109 +здгс
,109 + у[4ДГ/Сюб +
3
+ 2(и-1)ЗДКСД06] + 21ДКяд24 +—[12ДКя д24 +6(и-2)ДИяд24] +
3 3
ч-ЗДГодзз + — [ДИО135 +(п -2)ДИОД35] + ЗДИад72 +—[2ДИ5(Д73 +
+(и - 2)Д Vsiyn ] = (239,85 +108,15и) А3;
(Ха|Д^ +2^/) = ас{ЗД1/сдо9 +[4Д1/с,106 +2(н-2)Д1/Сд06]-—} +
' j 1
3
+ ан {6ДИЯд24 + [12Д^77,124 + 6(л - ,1241 “} +аО,о[Д^О,135 +
3 3
+ (и-2)ДИО1з5]-у+ оЛ[2ДИЛд73 +(п-2)ДИ5,Д72]-— .
Температура стеклования полимеров
181
Подстановка всех величин в данное соотношение приводит к значению
(^а;ДИ; + ^/>у) =(176,0+ 721,5и)-10-3 А3К'1 (при этом =0, так как
' J J
полимер не содержит отдельных полярных групп, приводящих к сильному
межмолекулярному взаимодействию).
Для узла сетки А имеем
(£-^/АЮу,Л = ^c(3AIZC,106+^Д1/С,109) + ^”я 15-Д1///124 +
/
+ Ко 3 Д дз5 + KSi • 3 ДГад72 = 2 52,3 • 10-3 A3K'>
Подстановка значений (]ГД^)п.ф., (Ха^^ +2Л». <2 К,ЛУ,)у>А В
/ , j i
уравнение (109) приводит к следующей зависимости температуры стеклова-
ния Tg сетки от степени полимеризации.
T(jC)=^W«.103 (146)
я 428,3 +721,5и
Графически эта зависимость показана на рис.59. Для сеток с четырех-
функциональными узлами (схема Б) легко показать, что расчетная зависи-
мость Tg от степени полимеризации п такая же, как и соотношение (146),
поскольку каждая величина в числителе и знаменателе этого соотношения
умножается на одно и то же число 4/3.
Рис.59. Зависимость Т от 1/п доя структур А и Б (см. текст)
Dependence <f Tg ond/nforA and Б structures (See text)
182
Глава IV
Теперь рассмотрим влияние строения узла полидиметилсилоксановых
сеток на их температуру стеклования. Исходя из химических соображений,
возможные узлы можно изобразить так, как это представлено ниже.
Для расчета Tg сеток, содержащих различные узлы, целесообразно опре-
делить сначала величины +Х^./)Л.ф. И (2Д10л.ф. ДЛЯ одного ли-
i J ‘
нейного фрагмента сетки, заключенного между двумя соседними узлами:
(Х^)лф. -2ДИу,д72 +4ДИСд06 + &VSjl72(n-2) + 2Д1/С106(и-2) +
i
+Д^О,135 + Д^О,135(и-2) + 12ДИЯд24 +6ДИ//д24(и-2) ;
(IX-U = (72,1л-0,5) А3-,
+2^у)л,Ф -ас[4ДРс, 106 +2ДИСдОб(я-2)] + аО0[ДКО1з5 +
> j
+^С,135(И “2)] + а//[12ДГ}/|24 +6ДТ/д д24(п “2)] + ал[2ДИ5Ц72 +
+ ^Ksi,i72(n ~ 2)];
(^а,ДИ, +2>,)л.ф. = (481и— 11,5) 10-3 А3К-1.
' J
Найденные значения (^Д^Ь.ф. и (^а,Д1<+^/>7)Лф Для всех рас-
сматриваемых сеток одинаковы. ' }
Ниже показаны возможные узлы полидиметилсилоксановых сеток, а так-
же соотношения, полученные на основе уравнения (109) и позволяющие рас-
считывать величину Tg в зависимости от числа повторяющихся звеньев п в
линейных фрагментах, соединяющих эти узлы.
i
СН3—Si-CH3
СИз! о ! СН3
Si-rO-Si-OfSi-- (!)
I ! I I
СНз1, О > СН3
CH3-si-CH3
Tg(K) = 144’2”+33’7ю3
7 962л+ 11,3
Температура стеклования полимеров
183
СНз; СНз | СНз
I I I I
Si-гО—Si—O-J-Si—
I । I I
CH3; О ; CH3
CH3-SHCH3
I
(2) Tg(K)
108,15л + 52,9 3
721,5л+ 46,5
СНз] СНз ; (рНз
•—Si-тО-Si-Of Si—
I 1 I I
СНз,1 CH2 ' СНз
I--1----J
CH3 CH2 CH3
---Si —O—Si— O—Si---
I I I
CH3 СНз СНз
(3) Tg(K)
72,1л + 66,11Q3
48 In+ 72,6 °
СНз; СНз ", СНз
I I I I
—Si-rO-Si-OfSi—•
I..! I I'..
СНз
(4) Tg(K)
СНз1. I ; СНз
СНт--'
СНз | СНз
---Si —O-Si-O-Si---
I I I
CH3 СНз СНз
72,ln+ 58,1 1q3
481л+ 61,2 °
СНз! СНз ' СНз
I I I I
—Si 4o-Si-OfSi—•
11 1 I
1 1 СНз] । ;'снз
СНз СНз
---Si—О-Si- О-Si---
I I I
CH3 CH3 CH3
(5) Tg(K)
72,1л + 49,31Q3
48 In+48,8
Согласно соотношениям, приведенным выше, зависимости Tg сеток от
числа п полидиметилсилоксановых звеньев представлены на рис.60. Хорошо
видно, что строение узла оказывает существенное влияние на Tg только при
184
Глава IV
значениях п = 1 + 4 (а = 0,25 - 1). С ростом расстояния между узлами роль
последних быстро понижается и уже при и = 10 температура стеклования для
всех сеток приближается к Tg полидиметилсилоксана, равной 150 К.
Рис.60. Зависимости Т сетчатых полидиметилсилоксанов от 1/п для разных типов
узлов. Обозначения узловых фрагментов 1-5 па рисунке соответствуют их
обозначениям в тексте
Dependences of Т& of networked polydimethylsiloxanes on 1/n for different
types of crosslinked fragments. Designations 1-5 of crosslinked fragments in the figure
correspond to their designations in the text
в) Влияние подвешенных цепей и других дефектов сетки
на температуру стеклования сетчатых полимеров
Среди множества различных вариантов подвешенных цепей рассмотрим
детально только два из них. Первый вариант является модификацией сетки I
на стр. 166, когда часть поперечных мостиков “разорвана” пополам:
I
-----С----
I
сн2
(CHzVz
_____:______ Г СН2 1 ___________
• - С -СН2| (CH2)m.2j СН2 - С - СН2 -г (CH2)m.2j СН2 - С -
: J I_______________СН2_____; ' :
(СН2)т.2
СН3
Температура стеклования полимеров
185
СН3
(CHjJm 2
,----
____:______ ; СН2 I ____________________________
- с —сн2ф (CH2)m.24 сн2 - с - сн2 4 (сн2)т.24 сн2 - с - -
—-4——1 1 сн2 ! —J—-
>---г-----1
В этом случае узел сетки сохраняется, но он становится трехфункцио-
нальным (в данном примере, как видно из схемц химическое строение узла
также остается неизменным).
Повторяющийся фрагмент данной структуры состоит из повторяющего-
ся фрагмента трехфункциональной сетки и подвешенной цепи. Тогда можно
записать для Ван-дер-Ваальсового объема
(X )п.ф. = (X А )у + 1=5(Х А )о (« - 2) + (X АП, )п.ц., (147)
lit i
где (ХдИ)у — Ван-дер-Ваальсовый объем узла сетки, ограниченного пу нк-
I
тирными линиями на схеме; (X А^ )0 - Ван-дер-Ваальсовый объем - СН2 _
i
группы; (X АГ) )п ц - Ван-дер-Ваальсовый объем подвешенной цепи.
При этом
(148)
/И
(Хд1;)п.ц. = (ХА1/,)0(у-2) + (ХАИ;)с„3
где (ХА^Оснз -Ван-дер-Ваальсовый объем концевой СН3—группы.
i
Величины (ХдТ,)у и (ХА^)О были вычислены ранее;
(ХА^)снз = АИсдз +ЗДК/Л124 =(17,2 + 3-2,0)= 23,2 А3.
/
Прежде чем приступить к дальнейшим расчетам, необходимо сделать одно
важное замечание. В принципе не может быть такой ситуации, когда сетка, не
имеющая подвешенных цепей, и сетка, их содержащая, имеют абсолютно
одинаковое химическое строение Иными словами, появление подвешенных
цепей всегда влечет за собой изменение в химическом строении сетки, что
сказывается на температуре стеклования и других свойствах.
186
Глава IV
Итак
(Е Л1')п.ф. = 73>4 +1,5 • 17,1(т- 2) + 17,1(у - 2) + 23,2 = (11,1 + 34,2т) А3;
+Х^./)л =1>3(<7c^1/c,io +ан ‘2ЛИЯ124)(т-2) +
' у
+ (асДИСд0 + а//2Л1/я 124)(-^--2) + (<7САИС13 + ая ЗДКЯд24)+6^ =
= [1,5(0,021 • 13,1 +19,98 • 2 2,0)(т - 2) + (0,021 13,1 +19,98 2 • 2,0)(у - 2) +
+ 0,021 • 17,2 + 19,98 -3-20 + (-54 4)] • 10“3 = (16Q 4т. - 336 2) Ю-3 А3К’1.
Подставляя все найденные выше величины в уравнение (109) .получаем
Т (К} =--------а(1121_+34,2т) + (1-а)(5,0 + 34,2т)___
s a(l 60,4т - 336,2 +102,9) +(l-a)(l 60,4т -217,86)
ИЛИ
Т(К) =-----3;0+34,2т + 6,1a---3
g 160,4т-15,44a-217,86
Соотношение (150) действует до a < 5, т.к. при m > 2 разрыв каждой линей-
ной цепи приводит к двум узлам сетки, из которых выходят подвешенные цепи.
Зависимость температуры стеклования Тл,от а,определенная с помощью
формулы (150), показана на рис.61. Видно, что Tg слабо возрастает с ростом
а, причем при увеличении числа звеньев между сшивками m наклон этой
зависимости убывает. На первый взгляд может показаться, что появление де-
фектов сетки в виде подвешенных цепей должно приводить к снижению а не
к повышению Tg. Однако следует иметь в вид}; что в данном конкретном слу-
чае появление подвешенных цепей не снижает количества узлов и даже не
изменяет их химического строения. Кроме того, появление каждого разветв-
ления требует введения инкремента bd что способствует повышению Tg.
В том же случае, когда в результате ‘"разрыва'’ цепей узел утрачивает свой
перво начальный смысл, т.е. не является больше узлом сетки, температура стек-
лования будет, естественно, снижаться. Такой вариант сетки изображен ниже:
СН3
(СН2)т 2
,------
___________ I СН2 1 ___________
- - С -СН2ф (сн2)т 24 сн2 -с - сн2 4 (сн2)т. 2| сн2 - с - -
Г” " (_____!
Температура стеклования полимеров
187
Рис.61. Зависимости Т от а (при разных т) для четырехфункциопальной поли-
этиленовой сетки с подвешенными цепями, когда число узлов остается постоянным
Dependences of Тъ on a (with different т) for tetra-functional polyethylene network
with dangled chains and constant numbers of crosslinked points
При таком “разрыве” линейных фрагментов сетки образуется линейная
цепь и для бывшего узла нужно при расчете использовать инкременты а, и hj
вместо К,, т.е.
+ ХМу = ас(А^С,1 +4ДИс,ю) +Ду -8Д^Я,124 =
' У
= 10,021(5,0 + 4 • 13Д) + 19,98 • 8 • 2,0] • 10“3 = 320,9 • 10-3 А3К4.
Тогда для фрагмента сетки, заключенного между двумя вертикальными
пунктирными линиями, следует записать:
(£ЛИ,)ф-73,4 + 17Дда-2) + 171(— -2)-2 + 23 2-2 = (17 2 + 34 2w) А3;
+Xbjh = {320,9 + 0,021{13,l(m-2) + 13,l(—— 2)-2 +
' J 2
+ 17,2-2]+19?8[2 -2р(да-2) +2 -2р(у-2) -2 + 6 -2р]} -Ю-3 =
= (80,2 + 160,4m) 10~3 А’К'1.
Подставляя все эти величины, а также ранее вычисленные значения па-
раметров сетки в уравнение (109), получаем
188
Глава IV
т (К} ._ а(17,2 + 34,2w) + (1 -а)(5,0 + 34,2от) дq3 <151)
г ’ а(80,2 + 160,4от) + (1 -а)(160,4от -217,86)
ИЛИ
5 +12,2а+ 34,2»! 3 (152)
1 а (A ) -------------------1 и
г 160,4от + 298,1а - 217,86
где а - доля бывших узлов сетки, из которых выходят 2 разорванные цепи.
Соотношение (152) действует при а < 0,25.
Зависимость Tg от а, вычисленная по соотношению (152), показана на
рис.62. Хорошо видно, что с увеличением доли узлов, из которых выходят две
разорванные цепи, температура стеклования быстро снижается .Это объясня -
ется тем, что место разветвления перестает играть роль узла и линейный фраг-
мент, заключенный между' соседними узлами, увеличивается вдвое.
Теперь рассмотрим трехфункциональную сетку с подвешенными цепями .
Схема такой гипотетической сетки на основе полиэтилена показана ниже:
Рпс.62. Зависимости Г от а (при разных от) для четырехфупкционалыюй поли-
этиленовой сетки с подвешенными цепями, когда образование таких цепей снижает
количество узлов сетки
Dependences of 7g on a (with different от) for tetra-functional polyethylene network
with dangled chains when the formation of them reduce a quantity of crosslinked points
Температура стеклования полимеров
189
i' । Г I
- (СН,) m.2- СН2-СН- СН2-(CH2U2+ СН2 -СН-
I 1 I I______________
I ?Нз I
! (СНг)т.2 1
СН3
СН3
(СН2)ш_2
сн2 ____________
• • - (СНг)т.2- сн2 -сн- СН2-(СН2)т.2| СН2 - СН-
В данном случае при разрыве поперечных цепей узел, из которого выхо-
дит подвешенная цепь .перестает играть свою роль у зла, и теперь он входит в
состав линейного фрагмента .который имеет разветвление. Для повторяюще-
гося фрагмента сетки, ограниченного двумя вертикальными пунктирными
линиями, имеем:
(£ Д 1<)ф = 62,3 +17,1 w - 34,2 +17,1(— - 2) + 23,2 = (25,65m +17,1) А3;
=<7с[Д,/С,6 + ЗА1/сдо + д,/с,ю -2) +
' j
+ Д1/С,10 (— ~ 2) + Д г/с,131 + аН Г7 Д VH, 124 + 2 Д ^,124 (т - 2) +
+ 2Д VH 124(у - 2) + ЗДИЯ 124] + bd = {0,021(9,0 + 3 13,1 + 13,l(m -2) +
+13,1(у - 2) +17,2] +19,98(7 • 2,0 + 2 • 2,0(ш - 2) + 2 • 2,0(у - 2) +
+ 3 -2 0] -55,4}-10-3 ^(243 + 120 3m)-10"3 А3К4 .
Подставляя все эти, а также найденные ранее, параметры сетки в уравне-
ние (109), получаем:
Т (К)_ а(25,65от + 17,1) + (1-а)(25,65да+11) 1q3 ц53)
' а(24,8 + 120,Зш) + (1-а)(120,Зот-152,76)
ИЛИ
Т 6,1а+ 25,65ш+11,0 10з (154)
g 177,56а + 120,3m-152,76
190
Глава IV
Рис.63. Зависимости Т от а (при разных т) для трехфункциональной полиэтиленовой
сетки с подвешенными цепями
Dependences of Т& on a (with different m) for tri-functional polyethylene network
with dangled chains
где а - доля структур с подвешенными цепями.
Зависимости Ts от а при разных значениях m показаны на рис.63, С рос-
том доли подвешенных цепей температура стеклования сетки быстро убыва-
ет, особенно при малых т.
Выше были рассмотрены гипотетические сетки на основе полиэтилена, в
которых фрагменты между узлами сшивок и подвешенные цепи имеют оди-
наковое химическое строение. Теперь рассмотрим систему, в которой подве-
шенные цепи имеют иное химическое строение по сравнению с цепями, обра-
зующими сетку. Такая система, полученная на основе эпоксидной смолы, ре-
ально синтезирована и исследована в работе [35]. Эпоксидная смола ЭД-20
отверждалась метилтетрагидро фталевым ангидридом, часть которого заме-
нялась олеиновой кислотой с целью получения сетки с подвешенными цепя-
ми. Детально с результатами расчетов и экспериментов, проведенных для та-
ких сеток, можно ознакомиться в работах [35, 125].
Помимо подвешенных цепей сетка может содержать и такие дефекты,
как изолированные петли. На рис.64 изображены различного типа дефекты в
виде разветвлений или изолированных петель. Узлами являются только те
места разветвления, где цепи, выходящие из них, присоединены к соседним
Температура стеклования полимеров
191
Рис.64. Схематическое изображение различных дефектов сетки (см. текст)
Schematic representation of different defects of network (See text)
цепям, т е. образуют сшивку. Такие структуры помечены знаком если же
из места разветвления выходит цепь, которая не образует сшивку, а представ-
ляет собой просто ответвление, или эта цепь образует изолированную петлю
за счет присоединения своим концом по месту выхода или в другом месте той
же цепи, то эти места также не являются узлами (они помечены знаком
При использовании соотношения (109) для расчета температуры стекло-
вания это необходимо учитывать. Если место разветвления - узел (в том
смысле, как это отмечено выше), при использовании соотношения (109) надо
брать инкременты X,. Если же место разветвления не играет роль узла, для
тех же атомов нужно подставить в соотношение (109) инкременты а, и bj.
Используя соотношение (109), можно путем сопоставления расчетных и
экспериментальных значений Tg оценить количество дефектов в реальной сет-
чатой системе. Проведем такой анализ в деталях на примере сетки, образо-
ванной на основе сополимера метилметакрилата и 1 -метокси-1 -винил-1 -сила-
2,7-диокса-4,5-(1,2-карборано)циклогептана. Химическое строение этого со-
полимера имеет вид [67]:
с—с
И ВюНю
’I
192
Глава IV
При термическом воздействии на сополимер цикл в структуре II раскры-
вается и образуется сетка следующего химического строения:
СНз
-сн2-с---------
С-О-СН3-
II 3
О
сн2-сн—
— Si—О—СН2—С~С—СН2—О —
I 2 \/ 2
О Вюню
Согласно данному выше определению узла, его химическое строение для дан-
ной сетки имеет вид
— +-сн2-сн—1--
I I I
Рассчитаем величины ^ДР( , +'^ibJ и ^Х^ДЦ для данной сет-
ки. ' ' 7 '
Для структуры I
(^ДР/Ь = ДРсд0 + ДРсдз + ДРсд +ДИС48 + ДРС41 +
+ 8ДР//Д24 + ДРо,129 + ^^0,139 = 96,8 А3.
Для структуры II
(^Д^)п = ДИСд0 + ДКС>41 + ДР,* + 2ДРС40 + 2ДРС117 +
I
+ЮДРяд24+10Д1/НД28+ЗДИО* + ДР&Д74 + 6ДР5]85 + 4ДР5д84 =
= 208,8 А3.
Инкременты Ван-дер-Ваальсового объема для атомов, отмеченных знаком “*”,
отсутствуют в табл.З, поэтому приведем их отдельно.
С
Si^CC^H ДР . =8,0 A3; Si^.'Pj^C ДР, * =1,95 Д-\
S
с
Температура стеклования полимеров
193
О
?|
O^'Si'^C AI<S7 = 30,1 А3
?!
О
Теперь рассчитаем величины ]Г atЛ К; + для идеальной сетки, структу-
ра которой изображена выше. ' J
Для структуры I
+ ZMl =ос(Д^С,10 + Af/C,13 + Д,/С,1 + Д[/С,48 + Д^С,41) +
' J
+ аН (8 А Ун ,124 ) + аО, 5^^0,129 + аО", 5^0,139 + = 255,8 10 3 А3К
Для структуры II
(^ОуДИ/ +^Ьу)п = ас(2ДИс,40 + 2Д,/СЛ7 + Д(/С,4|) +
' J
+г7Я(7Д1/Яд24 +1ОДКЯД28) + Й'в(бд,/Й,1«5 + 4ДК5д85) +bd =
= 401,0 -10—3 А3К-‘.
Рассчитаем значение (^ЗСуДГ7 )у для узла сетки, строение которого изобра-
жено выше. ’
(£К^У,)у = ^С(ДИСДО +ДКс,) + ^(ЗДРЯд24) +
7
+ К$(3/\УО.) + Ks,^yS/,) = 69,9 10-3 А3К-'.
Рассмотрим в общем виде зависимость температуры стеклования от хими-
ческого строения сетки, учитывая, что сетка имеет дефекты структуры и раз-
ветвления, изображенные на рис.64. Выше было отмечено, что если ответв-
ление не образует сшивку, то, хотя химическое строение узла в месте развет-
вления такое же, как и при образовании сшивки, этот узел перестает играть
свою роль и для него нужно при использовании соотношения (109) брать
инкременты не К,, а величины а, и bj. Тогда соотношение (109) переписыва-
ется в виде
ZA,<
Tg = (Z^ + Z.AA +т(Еа;.л^)у +(1-т)(Х^Д1< + 1^)у > (155)
i j i i j
194
Глава IV
где у - доля разветвляющихся мест, выполняющих функцию узлов. Сопос-
тавляя расчетную и экспериментальную температуру стеклования, можно най-
ти величину у из соотношения, вытекающего из выражения (155)
-£>,д^
_ 1 j/2 j
' ' J
где Tg,3 - экспериментальная температура стеклования.
В случае использования сополимеров для образования сетки, как это имеет
место в нашем случае, картина усложняется. Допустим, что один из компо-
нентов сополимера сетки не образует. Тогда в идеальном случае (100 % сшив-
(1-лХ^А^)! +«(ХД^)п
Tg ~ +2^)+(2^дк)У]и (156)
i j i j i
(n - доля структуры II).
Если часть мест разветвления не выполняет роль узлов, то
(1-И)(Хди,)1 +
т =---------------------------------------_>
2 (1 ~п)С^а^У, +^ьг>1 +«[(Еа/А,< +ЕА/)л +
' 7 ' 7
+ "(£ ДЕДц
+у(ХК,д^)у +(1 +Z^)1ii ’ °57)
где у - доля активных узлов.
Воспользуемся соотношением (157) для расчета доли активных узлову и
неактивных (1 -у). Предварительно рассчитаем величину (^т^АГу + ^Ау):
' 7
(£ХД^ +X^j )y,II =яс(А1/С,10 + Д^с* ) + аН (ЗД^Н,124) +
' 7
+ аО10(2ДГо*) + по8ДГ . + aSibV , = 490,4 • 10“3 А3К-'.
Подставляя найденные значения (^Д^Х, (^Д^)ц, (^<7,ДТ;
' ' ' 7
(^Й/Д^ +X/’./)ib +Х^)л В формулу (157), получаем
‘ 7 i i j
Температура стеклования полимеров
195
Т' (К) =-----96,8 +112,1 и----j оз
* 255,8-420,5иу + 635,6 п
(158)
Экспериментальные зависимости Tg от составов компонента II, заимство-
ванные из работы (20], показаны на рис.65. На этом графике также нанесены
рассчитанные зависимости Tg от содержания второго компонента при раз-
ных долях работающих узлову. Расчет проведен по форм)'ле (158). Из рисун-
ка видно, что, например, при доле компонента II20 % число активно работа-
ющих узлов составляет ~58 %, неактивных - 42 %; при содержании этого
компонента в количестве 10 % доля активных узлов составляет ~55 % и т.д.
Таким образом, полученные в работе [20] сетки хотя и являются частыми, но
обладают значительной дефектностью. При содержании стру ктурыП в сопо -
лимере в количестве 20 % доля золь-фракции составляет 18,5 %. Следова-
тельно, часть неработающих узлов содержится в разветвленном сополимере,
Рис.65. Зависимости Tg от доли n I-метокси-1-винил-1-сила-2,7-диокса-4,5+1,2-
карборапо)никло1сптана; доля активных узлов составляет: 1(1); 0,9(2); 0,8(3); 0,7(4);
0,6(5); 0,5(6); 0,4(7). Точки соответствуют эксперименту
Dependences of 7g on proportion n of 1-methoxy-l-vinyl-1-sila-2,7-dioxa-4,5-
(1,2-carborano)cycloheptane; the proportion of active crosslinked points: 1(1), 0,9(2), 0,8(3),
0,7(4), 0,6(5), 0,5(6), 0,4(7). The points denote the experiment values
196
Глава IV
который вымывается из системы. Оставшаяся часть неработающих узлов
(42 - 18,5 = 23,5 %) находится в составе сетки и образует структуру типа раз-
ветвлений или изолированных петель.
Приведем еще один пример оценки структуры сетки на основе сопостав-
ления расчетной и экспериментальной температур стеклования 7g [105]. В
качестве такой сетки рассмотрим эбониты, поскольку в них можно реализо-
вать различные типы сеток, а также потому, что накоплены значительные, но
противоречивые экспериментальные данные об их структуре и механических
свойствах. Проведем расчет Tg эбонита на основе полиизопрена. Будем исхо-
дить из того, что количество серы, взятое в реакцию, соответствует одному
атому серы на повторяющееся звено полиизопрена. Рассмотрим несколько
вариантов образования сетки, в состав которой могут входить структуры че-
тырех типов.
О --сн2-сн-с-сн2—; 2) --сн2-сн-с-сн2—;
S СН3 S СН3
I I
: S
I
I
3) -сн2-сн-с-СН,—-; 4) -СН2“СН=С - сн2_ •
II Iх
(S)„ СНз СНз
Структура 1 содержит моносульфидные мостики, структура 2 - дисуль-
фидные, структура 3 - полисульфидные, структура 4 представляет исходные
звенья полиизопрена. Для структуры 1 все атомы входят в состав узла сетки,
поэтому для нее а, ДР, + X bj) =0. Далее имеем
' j
= 2ДРсд0 + ДИС» +AJZC** + АРсдз+8ДР/УД24+
+Др£д57 = 92,2 А3;
(X^AIZ,)1 =^с(2АИСДо+АКс* +АИС« + ДСсдз) +
+ AT/ySAP^ J24 + АГ^ДР^ J57 =136,9-10 ^A3K'L
Температура стеклования полимеров
197
Поскольку инкременты Ван-дер-Ваальсового объема для некоторых ато-
мов, входящих в структуру сеток 1-4, отсутствуют в табл.З, приведем отдель-
но их значения
S
5|
С154_'С'>^-С №<, = 10,2 А3;
н
S
5|
С154.,^,154 с дг = 6,1 А3;
й
С
sajo^zio s Д|7. = 1^9 А3
Для структуры 1 температура стеклования Tg , вычисленная по уравне-
нию (109), совпадает с температурой начала интенсивной термической дест-
рукции Td (поскольку все атомы входят вузел сетки): Tg=Td = 673 К.
Следовательно, если вся сера расходуется на вулканизацию полиизопрена
таким образом, что образуются только моносульфидные связи, то Tg должна
совпадать с температурой начала интенсивной термодеструкции, и она состав-
ляет 400 °C. На практике эбониты имеют температуру стеклования 60-120 °C
[84], поэтому необходимо допустить, что помимо моносульфидных должны
возникать ди- и, возможно, полисульфидные мостики. Рассчитаем Tg сетча-
той системы, в которой присутствуют различные типы мостиков между цепя-
ми полиизопрена.
Для структуры 2
Q2 AP))2 = 2ДГС 10 + Д17,* + Д17.,** + ДИСд3 +8ДРя,124 +
/
+ 2Д^164 = 111,1 А3;
=-^с(2АГ/с,]о + +ДРС« +ДИС13) +
/
+ Кн 8ДКЯд24 + &S 2ДИуд64 = 172,8 -10“3 А3К‘1.
Для структуры 3
(^Д17,)з =2Д1/сл0 + Д17с* + ДН^** +ДИСд3 +8ДИ//Д24 +
I
+2ДН5д64+(и-2)Д^.;
<2Ж)з = (73,3 + 18» А3;
=172,8 НО-3 А3КЛ;
198
Глава IV
+Xbj^ =(n- 2)aSA^* J
i J
(£д,ДИ,)з =(141,7n-283,5)-10-3 A3K-‘.
Для структуры 4
(^ДИ,)4 = 2ДИС10 + AVC16 + AVCI5 ч-ДИсдз +8ДИ//Д24 = 81,5 A3;
+ ^bj)4~ ac(2AVcl0 +AVC16 +AVC I5 + ДИСд3) +
+ aH^AVH^u) + bd +Д, =400,7-IO-3 A3!*?1.
Найдем соотношение, связывающее Tg эбонита с мольными долями мос-
тиков, содержащих моно-, ди- и полисульфидные группы. Пусть рь р2, ...,
р„, - мольные доли узлов (мостиков), содержащих моно- ди- и п-сульфидные
группы. При заданном стехиометрическом соотношении серы S и полиизоп-
рена возникновение одной дисульфидной связи приводит к тому, что одно зве-
но полиизопрена не будет сшито; возникновение трисульфидной связи приво-
дит к двум несшитым звеньям полиизопрена и т.д. Следовательно, доля ри,
несшитых звеньев полиизопрена будет равна
Ри = Р2 + 2Рз +... + (и-1)Р„ ='£(/-1)р, • <159)
/=2
Тогда на основе изложенного выше и соотношения (159) получаем
Р^ХДИ,)! +р2(£ДИ,)2 + ... + Р„(£ДГ<)з +
Tg = рлх^д^)] +p2(1Xai/,)2 +р„[(Х^ди,)з +
/ / /
Н£ДТ<)42р, ('-1)
/=2(160)
+ (X О, ди, )3] + (X д, ДИ, + X bj )4'f р, (/ -1)
/ i j i=2
При этом учитывается, что £(/-1)Р, = 1 - Pi - Р2 - ... - р„ = 1 - р; .
i=2 <=1
Рассмотрим ряд частных случаев. Допустим, что сетка образована только
моно- и дисульфидными связями. Тогда
Pi(Z wh+р2 (S )2+а - Pi -р2)(Е ди,)4
т =________I_____________________________________ (161)
8 Р1(Х^Л1/,)1 + р2(Х^ДИ,.)2+(1-р1-р2)(Ха;ДИ1+ХЬ7)4
' ' ' J
Температура стеклования полимеров
199
Учитывая, что 02 = 1 - Pi _ Рз , окончательно имеем
(1-2р2)(^Л1/)1 + ₽2(^Д^)2 +р2(2Х)4
7 ---------------->----------i__________1____________(162)
8 (1-2₽2)(Х^Д1<)1 +₽2(ZW)2 +Р2(£>;ЛК +1>7)4 •
' < ' j
Соотношение (162) связывает Tg сетки эбонита с мольной долей Р2 ди-
сульфидных мостиков. Подставляя в него численные значения всех инкре-
ментов, получаем
Т<К} = ^_,2 + 8,2Р2_10з
g 136,9+ 299,7р2
(163)
Зависимость Tg от р2 показана на рис.66. С увеличением доли дисуль-
фидных связей Tg плавно снижается и при р2 = 0,5 (имеются толью дисуль-
фидные мостики) становится равной 60 °C.
Учитывая, что в литературе имеются данные об образовании мостиков
лишь с ограниченным числом атомов серы (среднюю величину принимают
равной 2,6 [84]), рассмотрим вопрос о влиянии доли мостиков разной длины
(вплоть до трех атомов серы) на Tg.
Обозначим число связанных звеньев тсв = т\ + т2 + т$ , где mi, т2 и
иг3 - числа звеньев, связанных моно-, ди- и трисулъфидными мостиками со-
Рис.66. Расчетная зависимость эбонитов, содержащих только моно- и дисульфидные
поперечные связи, от доли Р2 дисуль фщных связ ей
Calculated dependence Tg for ebonites containing only mono- and disulphide
crosslinks on proportion p2 of disulphide links
200
Глава IV
ответственно. Общее число звеньев, как связанных, так и свободных, равно
тсв + т2 + 2/и3 = ту + /п2 + + /и2 + 2/п3 = /и2 + 2/н2 + Зт3 . Тогда
--------
ту + 2/и2 + 3/и3 ’
₽3 =-------------------
/и1 + 2ш2 + 3/и3 ’
р2 =--------111.--.
ту + 2/и2 + 3/м3 ’
р4 + 2w3
ту + 2т2 + Зт3 '
Разделим числитель и знаменатель этих выражений на т3
pt = тх/т3 =______т2/т3
ту!т3 + 2т2/т3 +3 ’ ту !т3 + 2т21т3 + 3 ’
р3=__________!________ р т2/т3+1 (164)
ту/т3 + 2т2 /т3 +3 ’ ту/т3 + 2т2 !т3 + 3 '
Подставляя выражение (164) в соотношение (160), получаем
Т (К) -- 92-2wt/w3 + lll,]w2/w3 + 130,0 +81,5(/и2//н3 +2) ]q3
} 136,9ту!т3 + 172,8ш2/т3 + 239,5 + 413,25(ш2/т3 +2)
Теперь рассмотрим треугольник (рис.67), аналогичный треугольник) Гиб-
бса, на сторонах которого отложены доли мостиков с различными связями,
а изолинии представляют линии равных Tg. Из него видно, что Tg эбонитов
Рис.67. Треугольная диаграмма, характеризующая влияние состава сетки эбонитов
на Т. Числа у изолиний - значения Т
t J g
Tnangular diagram characterizing the influence of composition of network of
ebonites on 7^ ; the numbers at iso lines denote Tg
Температура стеклования полимеров 201
может изменяться в очень широких пределах при наличии моно-, ди и
трисульфидных мостиков. Максимальная температура размягчения (присут-
ствуют только моносульфидные связи), совпадающая в этом случае с темпе-
ратурой интенсивной деструкции, составляет 400 °C; минимальная величина
Tg = -10 °C характерна для сетки, содержащей только трисульфидные мостики.
Можно представить также случаи, когда серные мостики могут имеет раз-
личную длину и в реальной системе имеется определенное распределение по
длинам. Такие варианты были рассмотрены в работе [105], где температура
стеклования эбонитов рассчитывалась как зависящая от функции распределе-
ния по длинам поперечных мостиков, построенных из атомов серы. В резуль-
тате был сделан вывод о том, что в мостиках не может содержаться большое
количество атомов серы, и утверждение в литературе о том, что мостики обра-
зованы преимущественно дисульфидными цепочками, справедливо.
Глава V
ТЕМПЕРАТУРА ТЕКУЧЕСТИ АМОРФНЫХ
ПОЛИМЕРОВ
Еще раз отметим, что аморфный полимер в зависимости от температуры
может находиться в трех состояниях; стеклообразном, высокоэластическом и
вязкотекучем. Способы оценки температуры стеклования полимеров Tg на
основе химического строения повторяющегося звена изложены выше. Оцен-
ка по химическому строению полимера не только температуры стеклования
Tg, но и температуры перехода в вязкотекучсе состояние Гу, определяющих
протяженность температурного интервала высокоэластичности, крайне жела-
тельна. При этом знание величины Гу необходимо при переработке полиме-
ров.
Положение Гу относительно Tg полимера тесно связано с молекулярной
массой цепи Л/.
Попытка оценки величины Гу была предпринята в работе [96]. Она осно-
вана на использовании выражения, устанавливающего зависимость нулевой
(ньютоновской) сдвиговой вязкости т|о от молекулярной массы цспиЛ/[177|:
. pRTu2 [г Ме 015Т3 \ М 1,5
15 Me L м J 3 М
(166)
2 2,/ 22
где K=Lh N^/kj-Tv. М (b - длина сегмента Куна; NQ - число сегментов,
из которых состоит данная макромолекула; Q — коэффициент трения сегмента
включающий внешнее и внутреннее трение исходной макромолекулы; Ад -
кон-станта Больцмана), Ме - масса макромолекулы, начиная с которой суще-
ственно сказываются зацепления цепей на вязкость расплава; А -универсаль-
ная газовая постоянная.
Выражение (166), согласно работам [175,176], справедливо в области зна-
чений Л 4 <Л/ < 5Ме.
Температура текучести аморфных полимеров 203
Можно показать, что величина К от Л/ не зависит. Действительно, Ь2 =
= поа2 , где и0 - число повторяющихся звеньев в сегменте; а - характерный
размер повторяющегося звена; £ = Соио (Со - коэффициент трения для одного
повторяющегося звена). Тогда
К = (J)2Nl 1к.БТ11~М~ = Со//о«ой2Л\)/кЕТп2 М =
= ^(an0N0)2/k,Tn2(Man0N0)2 =Соа2/кБТл2М2 (167)
Таким образом, величина К зависит только от характеристик повторяю-
щегося звена и не зависит от молекулярной массы; при этом а ~ Д1у )2/3,
i
где XДУ, - Ван-дер-Ваальсовый объем повторяющегося звена.
i
Выражение (167) использовано в работе [84] для определения температу-
ры перехода в вязкотекучсе состояние 7у. Не приводя всего хода анализа, запи-
шем конечное выражение для оценки 7/:
+ ----В--------г
ТГ С .VI4<TV)M-I|’+T ' <|68)
где С - константа (С = 26); А' = М/Мс, М - молекулярная масса полимера,
Мс- молекулярная масса механического сегмента макромолекулы, те. такая
молекулярная масса, начиная с которой температура стеклования перестает
зависеть от 37; А = МфМё-. К = (Л0-5 -1 )3+1 /3 .
Как показывают данные работы [177], для большинства полимеров (по-
листирола, поли-а-метилстирола, поливинилацетата, полиизобутилена, поли-
изопрена) А ~ 2,4. Лишь только для полиэтилена А ~ 5,2-6,8 и для полибута-
диена А ~ 4,3. Расчеты для разных значений Л дают следующие константы В:
А = 2,4 -> В = 0,5;
А = 4,3 -+В - 1,57;
А = 5,2—» В =2,43;
А = 6,8 ->/>' = 4.48
Подставляя значения параметров А, В и С для общего случая в уравнение
(168), получаем
--- — 1 “I 111 7Т-
77 26 А/ и Д з 1
1 (- - )Ч |(2.4 ---)(15 -Гр + -
Мс | А// 3J
204
Глава V
Для того, чтобы воспользоваться уравнением (169), необходимо сначала
оценить величину Мс. Она может быть рассчитана с помощью уравнения (166)
с учетом того, что Мс = Nc Л40, где Nc - степень полимеризации механическо-
го сегмента макромолекулы; Л/о - молекулярная масса повторяющегося звена
полимера.
Величина Nc (или Л/с) определяет температуру перехода в высокоэласти-
ческое состояние полимера, когда Tg уже не зависит от молекулярной массы.
Однако при N < Nc (или М < Мс) полимер тоже стеклуется, но при этом Tg
будет уже зависеть от молекулярной массы. В таком случае Tg ~ Tf (но при
этом необходимо иметь в виду, что Tg, меньше истинной температуры стекло -
вания полимера).
Выражение (169) позволяет определить Ту и для данного случая.
Совершенно очевидно, что предельная температура перехода в вязкоте-
кучее состояние ограничивается температурой начала интенсивной термоде-
струкции Td (см. об этом подробно в главе VII). С этой точки зрения н е при
любой молекулярной массе полимер может быть переведен в вязкот екучее
состояние. Воспользовавшись выражением (169) и принимая '/) = Td, можно
определить максимальную степень полимеризации (или максимальную мо-
лекулярную массу), при которой полимер может быть переведен полностью в
вязкотекучее состояние (та бт. 20) Отнако получающиеся в этом случае темпе-
ратурные интервалы высокоэтастического состояния на 15 -20 °/«превосходят
реально наблюдаемые. Это связано с тем, что из-за полидисперсности синте-
тических полимеров часть цепей переходит раньше в вязкотекучее состояние
(фактически наблюдается наложение вязкотекучего и высокоэластического
состояний), что вызывает перегрузку более длинных цепей.
Таблица 20
Максимальные степени полимеризации Nmax, при которых полимер может быть
переведен в вязкотекучее состояние без деструкции
Maximum degree of polymerization Nmax at which the polymer can be transferred from the
glassy state to the viscous flow state without degradation
Полимер 7g / Td [84] max Nc N 1 * max
Полиэтилен 0,575 ~28 112 3136
Полиизобутилен 0,420 72 144 10368
Полистирол 0,612 18 320 5760
Поливицилацетат 0,716 -9 227 2043
П ол и м ети л метакр и л ат 0,506 40 307 12280
Поливинилхлорид 0,630 15 230 3450
Для иллюстрации сказанного проанализируем некоторые эксперименталь-
ные данные, приведенные в работе [109], в частности, по полиизобутилену и
поливинилхлориду. Анализ данных по полиизобутилену' с помощью выраже-
Температура текучести аморфных полимеров 205
ния (169) показывает, что при Л' > 10400 полимер не может быть переведен в
вязкотекучее состояние. Все остальные молекулы с более высокой степенью
полимеризации при попытке перевести их в вязкотекучее состояние неиз-
бежно бы только деструктировали (под действием либо механических напря-
жений, либо температуры). Последнее приводит к тому, что вследствие пере-
грузки длинных молекул имеет место их разрыв. Разорванные цепи могут
быть обнаружены после термомеханического анализа при определении мо-
лекулярно-массового распределения.
йиденная с по мощью выражения (169) 7урасч = 181 °C при N = 10400, в
то время как 7узксп -120 °C. Понижение 7у(7уРасч - 7узксп = б 1 °C) обусловле-
но тем, что из-за перехода более коротких молекул в вязкотскучее состояние
произошла перегрузка длинных молекул в ~50 раз, по данным [109]. Если
эксперимент проводится при нагрузке 0,07 МПа, то на длинные молекулы в
области Ту действует нагрузка —3,5 МПа.
Таким образом, если мы хотим с помощью термомеханического анализа
получить Ту. на которую не действуют трудно учитываемые привходящие фак-
торы в виде полидисперсности, необходимо ввести дополнительные ограни-
чения на степень полимеризации образцов, при этом поставив условие, что-
бы величина АТ = 7уРасч - '/уэксп была меньше погрешности эксперимента.
Степень полимеризации /V до которой полидисперсность не влияет на Ту,
можно оценить из эмпирического выражения
^7’„
ТГ^-ЯЗ. (170)
Влияние полидисперсности должно проявляться при динамических ме-
ханических испытаниях. В этом случае асимметрия температурной зависи-
мости тангенса угла механических потерь в области a-перехода (при Т < 7’а)
тоже связана с переходом в вязкотекучее состояние более коротких цепей, име-
ющих N<NC. Полимер, не имеющий низкомолекулярной компоненты (N> Nc~),
должен иметь симметричный максимум потерь.
Глава VI
ТЕМПЕРАТУРА ПЛАВЛЕНИЯ ПОЛИМЕРОВ
Температура плавления определяется как температура, при которой по-
лимер переходит из кристаллического состояния в вязкотекучее. В отличие
от низкомолекулярных веществ, где этот процесс совершается скачкообраз-
но, в случае полимеров плавление наблюдается в некотором температурном
интервале. Это происходит вследствие полидисперсности полимерных це-
пей, их разнозвенности и несовершенства образованных кристаллитов. Раз-
личают равновесную температуру плавления и экспериментальную. Равно-
весная температура плавления Тт° = где А//т - энтальпия плавле-
ния, \Sm - энтропия плавления. Равновесная температура плавления
определяется точкой фазового равновесия между монокристаллом полимера
и его расплавом. Поскольку совершенные монокристаллы из полимера полу-
чить практически невозможно, то равновесную температуру плавления опре-
деляют экстраполяционными методами, например, экстраполяцией зависи-
мости экспериментальной температуры плавления от размеров кристалли-
тов или от молекулярной массы полимера.
Температура плавления Тт является физической характеристикой .кото-
рая наиболее трудно поддается расчету. Речь идет о расчете на основании хи-
мического строения повторяющегося звена полимера. Рассмотрим два подхо-
да к решению этой задачи. Ошн из них основан на оценке отношения темпе-
ратуры стеклования Tg к температу ре плавления Тт . Следует заметить, что,
согласно правилу Бимсна [132], Tg/Tm ~ ИЗ. Однако детальный анализ боль-
шого количества полимеров самого разнообразного строения показал [172],
что это отношение варьируется в широких пределах, хотя действительно для
большой группы полимерных систем оно составляет —2/3.
Уравнение, связывающее отношение температуры стеклования Tg к тем-
пературе плавления Тт. получено в работе [42] на основании данных экспсри-
Температура плавления полимеров
207
мента, согласно которому коэффициент молекулярной упаковки кристалли-
ческого полимера при тсмпер;ггурс плавления приблизительно равен коэф-
фициенту молекулярной упаковки аморфного полимера того же строения при
температуре стеклования, те. плавление кристаллического полимера и пере-
ход из стеклообразного состояния в высокоэластическое аморфного полимера
происходит при достижении одного и того же значения доли пустого объема.
Соотношение для Т^Тт записывается в виде [42]
7g _ Tm Г "J -1 Zak, 1 a (171) Z(W) + Zb ' j
Здесь 5, = (Ао - &g)! к, (к, - парциальный коэффициент упаковки /-го атома);
у- - инкременты, учитывающие вклад сильных межмолекулярных взаимодей-
ствий; А = kg/ (к0 - kg) = 10,418; смысл всех остальных обозначений тот же,
что и в соотношении (82 ) качения Д и у. приведены в табл 21.
J Таблица 21
Значения параметров 8/и у/различных атомов н типов межмолекулярпого
взаимодействия
Values of parameters 8f and yf- of various atoms and types of intermolccular interaction
Атом или тип межмолекулярпого взаимодействия Условное обозначение 8, Y< . '0
Кремний 0,0840
'Углерод Зс 0,8685 -
Водород &н 0,0740 -
Кислород в основной цепи 3<э.о 0,0621 -
Кислород в боковой цепи 3q« 0,0963
Азот в основной цепи Змо -0,0212
Диполь диполгное взаимодействие 4<i - -0,0727
Водородная связь - -0,0188
п-замещение - - -0,190
Водородные связи в полиамидах: -
А? оматические полиамиды - - 0,422
Четные али(|втические по лиамиды - - 0,392
Четные - нечетные алифатические полиамиды - 0.392
Нечетные алифатические полиамиды - 0,467
А ли}) анческие -ароматические полиамиды , содержащие четное число -СНз-групп - - 0,445
Алифатические-ароматические полиамиды, содержащие нечетное число-СНз-групп , - 0,544
Алифатические-ароматические полиамиды па основе изо-, терефталевых кислот и р, т- фенниендиамипов, содержащие только -СНг- 1'руппы 0.445
208
Глава VI
Расчеты, проведенные по формуле (171), во многих случаях показывают
хорошее совпадение с экспериментом. Для повышения точности расчетов
желательно иметь групповые вклады + полимерных групп,
' 7
из которых построены многие полимеры. Эти групповые вклады получаются
суммированием величин о, Л К,- для атомов, входящих в данную группу, затем
к ним прибавляются величины уу, характерные для каждой полярной группы
и типа замещения в ароматических ядрах, а также небольшие поправки, по-
вышающие точность расчета. Эти поправки находятся путем сравнения экс-
периментальных и расчетных величин Тт для ряда полимерных стандартов.
Величины X (З/АИД + X Y; для Ряда атомных групп приведены в табл.22.
' 7
Использование этих величин позволяет с большей точностью рассчиты-
вать температуры плавления. Это демонстрирует табл.23, в которой для ряда
полимеров приведены экспериментальные значения Тт, а также расчетные,
полученные с использованием атомных и групповых вкладов.
Температура плавления сополимеров не может быть описана простым
соотношением, полученным на основе уравнения (171) с использованием пра-
вила аддитивности.
Другой подход [29] основан на рассмотрении повторяющегося звена по-
лимера как набора ангармонических осцилляторов.
Согласно [114], свободная энергия ангармонического осциллятора рав-
на:
2
где
Fq = kTln(2sh-^)- (о =
h ,. hm ч
о =----ctK-----) ’
2ш(о 2кТ
(m - масса атома, а - коэффициент упругости осциллятора, [3 - коэффициент
ангармонизма осциллятора, а - величина, характеризующая смещение поло-
жения равновесия гармонического осциллятора относительно нуля).
Величина а в выражении (172) находится из условия минимума свобод-
ной энергии по а, те. при
шсо2я-Ра2 -Р(у2)0 =0 . <173)
Рассматривается условие потери устойчивости данной системы, а крити-
ческая температура 7]., при которой устойчивость теряется, определяется вы-
ражением:
Температура плавления полимеров
209
где
. 2та та2 , ?
А =----(----у
П 2Р
Критическая температура является температурой фазового перехода,
т.е. в нашем случае плавления.
Таблица 22
Скорректированные значения величии Л J7/
для ряда фрагментов (см. текст)
Corrected values of quantities £3/ДР/ for a series of basic fragments (See text)
Химическое строение фрагмента и соседние, связанные с ним атомы 18;АИ, ' J
1 2
С -СНз- с 1,434
с -сн- с 1 СН3 2,795
СНз с -сн- с 1 СН3 4,302
с -сн- с С2Н5 4,228
с -сн- с 1 о-сн3 3,652
с -сн- с 1 CI 2,702
С —СС12— с 3,770
с -сн- с 1 О 7,889
сн3 с -сн- с 1 О 9,177
210
Глава VI
таблица 22 (продолжение)
1 2
с.. 1 0=0 1 с 1,576
с... .. -сн-.... 1 СН3 ..о 3,064
с... сн3 ... -с- 1 сн3 0 4,623
с... .. -сн-.... 6 ..о 8,175
с... -о--- .0 6,745
с... —— ..о 6,645
о.... .-сн2-... ..о 1,972
О... -0- • .0 7.028
О... •• - .0 6,928
О... _/СН3 ХСН3 . о 9,826
с... ... -о- .с 0.211
с... ... -0- с 0,168
с.... .. -NH-.... .с 0,153
S.... -0-- с 6.567
S... ..с 6,467
S.... -0-- ..S 6.671
-Температура плавления полимеров
211
таблица 22 (продолжение)
1 2
S.... -о-... ..S 6,571
S... •-0-- .N 6,724
S.... ..-о-.... .N 6,624
S.... ~о~ .О 6,849
S... О 6.749
с... ... -S-.... .С 1.338 '
С -N' о о II II •сх Ху II II о о м- с 12,000
С... ... -с-.... с
о о :у 9.455
С... ... -с- о'-Ъ .с 13.290
СРз
с... ...-С-.... .с 5,997
CF3
с... ~0- ..с 6.463
с... *~О“ ..с 6,263
с - ^,zN1K СХ z-c- с 8,630
с.... .—СН2— ... ...о 1 ,703
с... ... -о-.... с 0,1.30
с.... . -сн2-... ..N 1,573
212
Глава VI
таблица 22 (продолжение)
1 2
С.1:54 -C-O-J;50 С II 0 2,083
С 1'54. -С-О- 1137 с II О 2,039
о 1 0=0 1 И 1,856
с -Q- N 6,619
о -О- N 6,519
с.1.’37. -NHC- J.'54.c II 0 2,403
о -0- N 6,902
О -О- N 6,802
N -Q- N 6,776
N -Q- N 6,676
р I Z / \ 0=0 0=0 W 0=0 0=0 у о 16,646
о |' 0=0 0=0 I 6 9,882
Температура плавления полимеров
213
таблица 22 (окончание)
1 2
с 1 0=0 0=0 \_/ 1 с 9,600
о. -ocNH>- N с 8,912
S о / \ 0=0 0=0 У с 9,704
S. .... -crN>- с 8,734
СНз
О. -Si- 0 5,986
СН3
Si -0- Si 0,042
о. 1 0=0 1 0 2,541
с. —N — 1 СН3 с 2,049
N... “СО" ...N 10,685
С... ~~00_ ...с 10,172
С.. ... —cf2—.... ..с 2,048
Если воспользоваться потенциалом-Леннард-Джонса, с помощью кото-
рого определяются параметры уравнения (172), а также выражением для ко-
эффициента объемного расширения, то можно получить следующее соотно-
шение для оценки температуры плавления [29].
1 _ /________
7т ’
(175)
214
Глава VI
Таблица 23
Экспериментальные и расчетные температуры плавления Тт для ряда полимеров
Experimental and calculated values of melting point Tm fora series of polymers
Полимер Тт, эксперимент Тт , К вычислено по формуле/171) тт, к вычислено по формуле (175)
Полиэтилен 410 - 410
Полипропилен 449 451 421
Полибутен 405 413 418
Поли-4-метилпентен-1 508 500 504
Полиэтиленадипат 323 .334 321
Полиэтиленоксид 339 348 314
П ол и п роп ил е но кс ид 340,348 356 342
Полите1раметиленоксид 338; 309 311 349
Политетраметиленадипат 332 314 334
Политриметиленадипат 311 315 311
Полистирол 513; 523 509 -
Поли-а-винилнафталин 633 625 —
Полигексаметиле надии амид 539 539 —
Полигексаметиленсебацамид 499; 488 494 —
Полиамид-6 488; 499 496 —
Полиамид-11 467; 455 481 —
Поливинилизобутиловый эфир 438 497 —
ГI о л и мети j 1 м етак ри лат 433 465 —
Поливииилэтиловый эфир 417 398 —
Полидиметилсилоксап 234 234 —
где Kt = 18,52Я/(г£>() (R - универсальная газовая постоянная, z - координаци-
онное число, / *, - энергия межмолекулярного взаимодействия, возникающая
за счет /-го атома).
Количество значений А.', определяется числом атомов, образующих по-
вторяющееся звено. Но так как некоторые атомы входят в состав полярных
групп, обладающих сильным диполь-дипольным взаимодействием, водород-
ными связями и т.д., то последние можно учесть путем добавки к энергиям
дисперсионных взаимодействий £>, той доли энергии сильного межмолеку-
лярного взаимодействия, которое обусловлено вкладом /-го атома. Тогда
И т.д.,
(176)
5 К) 2 ~---------Г ’ I 3 ~
D, ’ D, + Mjf ’ D, + А£>"
где ДО^ - вклад /-го атома в диполь-дипольное взаимодействие; Д/)^' - то же
в водородное связывание и т.д.
Расчеты, проведенные по уравнению (175) на ЭВМ методом наимень-
ших квадратов,показали [29] ,что для полимеров .содержащих атомы угтсро -
да, водорода и кислорода в повторяющемся звене, достаточно знать парамет-
Температура плавления полимеров
215
Таблица 24
Численные значения Kj, /),• н А/), ,тля атомов водорода н кислорода*
Numeric values Kj, Д and АД for hydrogen and oxygen atoms
Элемент Условное обозначение Kj^-lo’, град 1 о,, ккал/м оль Д<7; , ккал/моль
Водород Кц К» 10,42 10,03 0.88 0.92 0,04
Кислород К О Ко 16,5 13,3 0,56 0,70 0,14
* Для атома углерода значение пренебрежимо мало
ры DИ , DdH, Do и Dq для удовлетворительного расчета температуры плав-
ления ряда полимеров исходя из химического строения повторяющегося зве-
на (табл. 24); значения D, вычислены при z = 4.
В табл. 23 приведены расчетные значения Тт для ряда полимеров, содер-
жащих в повторяющихся звеньях атомы углерода, водорода и кислорода. Не-
смотря на достаточно хорошую сходимость расчетных и экспериментальных
данных следует отметить, что данный метод имеет ограниченное применение.
Глава VII
ТЕМПЕРАТУРА НАЧАЛА ИНТЕНСИВНОЙ
ТЕРМИЧЕСКОЙ ДЕСТРУКЦИИ ПОЛИМЕРОВ
Будем рассматривать характеристику термостойкости полимеров, опре-
деляемую с помощью термогравиметрического анализа. С помощью такого
анализа определяются зависимости массы вещества от температуры при не-
прерывном ее повышении (термогравиметрические кривые). Как известно,
для большинства полимеров термогравиметрические кривые имеют вид, схе-
матически изображенный на рис.68. При оценке термостойкости полимера
будем использовать температуру начала интенсивной термодеструкции Тд
определенную по пересечению касательных к двум ветвям термогравимет-
рической кривой (см. рис.68).
Соотношение, приведенное в работах [88] для оценки температуры нача-
ла интенсивной термической деструкции Tj, получено на основании рассмот-
рения валентно-связанных атомов как набора ангармонических осциллято-
ров, которые образуют повторяющееся звено полимера. Здесь подход тот же,
что и при оценке температуры стеклования Tg и плавления Тт, но учитывается
энергия химических связей, а не энергия межмолекулярного взаимодействия,
хотя последняя существенно влияет и на энергию диссоциации химических
связей. Так, например, известно, что энергия распада С - С связей (углерод-
углерод) изменяется в пределах от 30 до 90 ккал/моль [64] в зависимости от
того, в какую группу входят атомы углерода, т.е. в зависимости от их валент-
ного окружения; то же самое относится и к другим парам валентно связанных
атомов (С - О, С - S, С - N и т.д.).
При нагревании полимера происходит изменение его объема, причем это
изменение складывается из двух частей: увеличения свободного объема и из-
менения длин химических связей. Анализ этих изменений привел к следую-
щей зависимости температуры начала интенсивной термической деструкции
Td от параметров химического строения полимера [88]:
Температура начала интенсивной термической деструкции полимеров 217
Рис.68. Схематическое изображение термогравимезрической кривой и способа
определения величины Td
Schematic representation of the thermogravimetric curve and the method of evalu-
ation of T. value
d
aX; /
_ = 42-t~------,
Td
(177)
где ax - парциальный коэффициент объемного расширения /-го атома, воз-
никающего за счет изменения длин химических связей; ДР} - Ван-дер-Вааль-
совый объем /-го атома; У, Д V, - Ван-дер-Ваальсовый объем повторяющего-
ся звена полимера. '
При этом
a
ЗАР
yX
(178)
где р - коэффициент энгармонизма, р = -
1 а3с
2 dd3
; у - силовая постоянная,
do
А.
dd2 ’
Y =
- потенциал химического воздействия. Все величины взяты для /-
го атома, химически связанного с другими атомами.
218
Глава l7II
Для оценки энергии диссоциации химических связей воспользуемся по-
тенциалом Морзе
ad) = £[(e'a(J-do>-l)~2 -1], (179)
где d- расстояние между химически связанными атомами; - равновесное
расстояние; Е - энергия диссоциации химических связей. С учетом выраже-
ния (179)
Тогда
где
3 t R ч йх> 4 йс/0Е '' (180)
1 (181)
К, =315—. (182)
В случае распада по С -Н -связям з naic ние d0 =0,108,Еср =396 Дж/моль,
величина а =0,0266 нм (
Значения величин К, приведены в табл 25 .
Таблица 25
Условные обозначения и числетшые значения констант К,
Conventional symbols and numeric values of constants Kj
Atom или группа Условное обозначение константы Ki-io3, A3 К ' Примечание
1 2 3 4
Углерод Кс 1,150 -
Углерод Кс 1,920 Действует для атома углерода, входящего в полярную группу
Водород К-н 2,307 -
Водород 0556 Действует для атома водорода , входящего в полярную группу
Кислород Ко 0,058 -
Кислород Као 1,572 Действует для атома кислорода входящего в молярную г руп пу
Азот IS-d 2,520 Действует для атома азота, входящего в полярную группу
Азот Kd 0,411 Действует для атома азота, входящего в состав гетероцикла
Сера Ks 1,900 -
Температура начача интенсивной термической деструкции полимеров 219
Таблица 25 (окончание)
1 2 3 4
Сера 6,300 Действует для атома серы, входящего в состав полярной группы
Фтор Ku 1,360
Хлор 2,500 Действует для атома хлора, входящего в полярную группу
Водородная связь за счет ОН-группы K()H 3,450 Действует при наличии водо- родной связи за счет ОН-группы
Водородная связь за счет NHCO-rpvnnu Kh л NHCO 2,200 Действует при наличии водородной связи за счет NHCO-группы
Поскольку атомы могут входить в состав полярных групп, обладающих
специфическим межмолекулярным взаимодействием, то вклад их в термо-
стойкость будет отличен от вклада,вносимого теми же атомами, обладающи-
ми лишь слабым Ван-дср-Ваальсовым взаимодействием. Например, полиме-
ры могут содержать следующие полярные группы:
-СН3;-С -;-С -О--С-ОН. -ОН С ^-N NH-С
К II II II
ООО О
N —N о
И К II
-NH- С-О-; - С С—; — S - Ci; - О-СН2--
II х и
о ° о
При расчете 7 а по уравнению (181) учет межмолекулярного взаимодей-
ствия проводится следующим образом.
Если рассматриваемый атом не входит в состав полярной группы, то его
параметр ^умножается на соответствующий Ван-дер-Ваальсовый объем. Если
же атом входит в состав полярной группы, то его параметр А', обозначается
или Ktd (h- водородная связь d- диполь-дипольное взаимодействие) и
К1. или j'd уМН0Жается только на соответствующий Ван-дср-Ваальсовый
объем, т.е. вклад атома в Ван-дер-Ваальсовос взаимодействие, как существенно
более слабое, не учитывается.
Примеры расчета величин Т / для представителей различных классов по-
лимеров приведены в табл.26.
Уравнение (181) позволяет нс только оценивать температуру начала ин-
тенсивной термической деструкции полимера при его нагревании в инертной
среде, но и даст возможность решать ряд других задач. Так. в течение некото-
рого времени дискуссионным был вопрос о том, может ли интенсивная тер-
220
Глава VII
Таблица 26
Расчетные и экспериментальные температуры начала интенсивной термической
деструюцш в инертной среде для ряда полимеров
Химическое строение повторяющегося звена - IO-1, А'/К
1 2
- сн2- сн2- 48,6
сн3 —СН2—СН— 76,5
- С-(СН2)5- NH- о 190,3
-с-(снг )4- c-nh-(ch2 )6-nh- О О 380,6
-0-(СН2)2-0-С-О-С- о о 222,0
—СН2—СН— 6 177,6
сн3 - сн2- с- С-О-СНз О 154,3
Рч ''о с/ ох 1 212,9
—CF2—CF2— 74,0
-сн2-сн- C=N 95,0
Температура начала интенсивной термической деструкции полимеров 221
Calculated and experimental values of temperatures of onset of intense thermal degradation Гд
in inert medium for a series of polymers
7ZK экспер. г^к. расчет
3 4 5
34,2 713 704
51,5 673 673
115,6 623; 633; 643 621
231,3 593; 653 621
149,1 663 672
109,9 633 618
97,0 623 629
126,1 563 592
57,2 773 773
54,1 570 569
222
Глава VII
1 2
z 1 0 О /) /°C °z 0 о i X 1 0 = 0 1 0 1 0=0 1 1 566,1
1 z / \ 0=0 0=0 Q о о 0=0 0=0 У 1 Q P о о 1 721,3
1 0=0 1 “l 0=0 1 о 1 о 0 A /< ° ° 0 1 о 1 532,8
1 ^o=z О 1 ^o=z 1 0 1 о 1 0 1 ^o=z О 1 ^o=z p 6 A /°C ° ° 0 1 648,1
^'NFKr^ss,_pij /0-°-0;c-n'-0 2 O.N,c- c c // \ // \ N NH N NH \_/ \_/ G> О 779,4
i о )' о я Г\ 0=0 о h' °А УЧ V । 497,6
। о 1 0 । 0-0-0 с? 1 ш" 0 1 о 1 0=0 0 0=0=0 0 0=0 1 657,7
Температура начала интенсивной термической деструкции полимеров 223
таблица 26 (окончание)
3 4 5
394,6 683 697
538,8 783 747
389,5 728,- 743 731
490,6 733; 728 757
576,0 743; 733; 773 739
380,7 793 765
437,4 663; 653; 658 665
224
Глава VII
модеструкция начинаться при температурах, лежащих ниже температуры стек-
лования Tg. Иными словами, должен ли полимер при нагревании сначала пе-
реходить из стеклообразного состояния в высоко эластическое или вязкотеку-
чее для того, чтобы была возможна термодеструкция, или последняя может
проходить и в стеклообразном состоянии?
Для многих полимеров температура начала интенсивной деструкции дей-
ствительно лежит выше температуры стеклования или плавления. Однако в
ряде случаев деструкция начинается и при температурах,лежащих ниже тем -
пературы стеклования или плавления. Последнее обусловлено тем, что устой-
чивость химической связи сильно зависит от диполь-дипольных взаимодей-
ствий и водородных свя зей. Рассмотрим это более подробно.
Если при одинаков ом скелете основной цепи полимер имеет в своем со-
ставе полярные группы, занимающие достаточно большой Ван-дер-Ваальсо-
вый объем по сравнению с другими атомами, не входящими в состав этих
групп, то такой полимер обладает низкой температурой термодеструкции.
Покажем это на примере полистирола
-сн2-сн-
В случае полистирола наличие объемистой фенильной группы, которая
является полярной, приводит к следующему выражению для расчета Td по
формуле (181):
Td (К) =------------------------------103 = 618
Кс -22,2 + KH-6,0 + Кс X'l.b + Kf! -10
(для того, чтобы получить это выражение, необходимо воспользоваться дан-
ными табл.З, в которой приведены Ван-дер-Ваальсовые объемы атомов).
Если формально рассматривать полистирол как замещенный полиэтилен,
в котором один атом водорода в каждом звене замещен на фенил, то можно
сделать вывод, что такое замещение приводит к снижению Td. Это снижение
может быть настолько большим, что интенсивная термическая деструкция
может начинаться до достижения температуры стеклования. Так ведут себя
полимеры с объемистыми боковыми заместителями, содержащими полярные
группы. Одним из них является полиметилиденфталид:
-сн2-с-
Оч /°
Температура начала интенсивной термической деструкции полимеров 225
Формула для расчета температуры начала интенсивной деструкции по-
лиметилиденфталида, полученное на основе (181), имеет вид
Td W ---------------d--------------~d-----~d----I О3 - 592
Кс-Tlfi + Kc -Ш + Ки-4 + К„ -8 + Ко -9,25
Наличие объемистой полярной группировки в боковой цепи приводит
к высокой температуре стеклования, которая для этого полимера составляет
390 °C .Эта величина Т ?цолучается как в результате расчета ,так и экспери -
мента [55]. На последнем следует остановиться более подробно, т.к. в тех
случаях когда интенсивная термодеструкция начинается раньше, чем дости-
гается температура стеклования, прямое определение Tg становится невоз-
можным . В связи с этим в работах [55] использован стедующий спос об опре-
деления Tg для полиметилиденфталида: был синтезирован ряд сополимеров
метилиденфталида со стиролом, метилметакрилатом и метилакрилатом раз-
ных составов, для которых определялась температура стеклования. На рис.69
показаны зависимости Tg от состава этих сополимеров. С ростом содержания
метилиденфталида Tg сначала возрастает, но когда Tg достигает таких значе-
ний, при которых начинается интенсивная термическая деструкция, этот рост
Рис.69. Зависимости Т& от молярной доли а стирола (1), метилметакрилата (2) и метил-
акрилата (3) для сополимеров этих компонентов с метилиденфталидом
Dependences of on the molar part a of styrene (1), methyl methacrylate (2) and
methyl acrylate (3) for the copolymers of these components with methylydenephthalyde
226
Глава I'll
прекращается. В этой области больших концентраций метилиденфталида раз-
мягчение сополимеров происходит за счет их разложения. Экстраполяция же
зависимостей Tg от состава на 100 % -ю концентрацию метилиденфталида
приводит к значению Tg для полиметилиденфталида, равному 390 °C. Таким
образом, интенсивная деструкция этого полимера начинается при 300 °C, и,
следовательно, этот процесс для полиметилиденфталида начинается при го-
раздо более низких температурах, чем расстекловывание.
Напротив, в случае слабо полярных полимеров (полиэтилен, полидиме-
тилсилоксан и др.) температуры стеклования и плавления лежат намного ниже
температуры термодеструкции. Можно подобрать такой случай (на примере
полигетероариленов), когда температуры стеклования и термической дестру к-
ции будут практически совпадать.
Таким образом, при анализе влияния химического строения полимеров
на их термические характеристики и при прогнозировании свойств полиме-
ров необходимо учитывать, что одни и те же группировки могут оказывать
противоположное влияние на различные термические характеристики.
Особо следует рассмотреть случай, когда деструкция полимера начина-
ется с распада по концевым группам. Если такой распад не приводит к обра-
зованию тех же концевых групп, то расчет величины следует проводить исхо-
дя из химического строения повторяющегося звена полимера, как это изло-
жено выше. Если же распад концевых групп, который происходит легче, чем
распад в основной цепи полимера, приводит к образованию тех же концевых
групп, то расчет следует проводить исходя из химического строения только
концевых групп. Характерным в этом отношении является распад полифор-
мальдегида
°" (сн2 - ov -
Расчет Tj по строению концевых групп (отмечены пунктиром) приводит
к следующему значению:
39,4
Tj 1 (X) = —-------------------103 = 447.
-25,1 + А^-4 + Х^-10,3
Это значение хорошо совпадает с экспериментальной величиной Т^, оп-
ределенной по первому изменению массы полиформальдегида в условиях тер-
могравиметрического анализа (рис .70). Этот анализ провод или дляоб ja зи ,
стабилизированного группами - СН2 - СН2 - О - , которые были введены в
полимер в количестве 2 %. Введение указанных звеньев, как известно [69],
предотвращает преждевременный распад полиформальдегида, проходящий
по концевым группам. Поэтому расчет температуры второго спада массы об-
разца проводится по структуре повторяющегося эвена. Предварительно еле-
Температура начала интенсивной термической деструкции полимеров 227
Рнс.7О. Термогравимстрическая кривая для полиформальдегида .стабилизированного
2 % групп -CIE -СЬЕ-О- .(Опыт проведен в среде аргона при скорости нагревания
5 град/мип)
Thermogravjmdric curve for pdyfomialddiyde slcb'iized by 2 '/oof groups
-CH 2 -CH 2-O- (The experunent was performed in medium of argon at rate of heatmeg
5 deg/min)
дует отмстить, что полиформальдегид - полярный полимер, звенья которого
обладают сильным межмолею'лярным взаимодействием. Тогда
^.2(К)
32,5
. . , ю3
Kdc -25,1 +-4 + Кд -3,4
= 583.
Эта величина также хорошо совпадает с экспериментальным значением,
определенным по термогравиметрической кривой (см. рис.70).
Таким образом, при анализе влияния химического строения на термичес-
кую дестру кцию полимера имеется возможность проводить сканирование по
различным полярным группам находящимся как на концах макромолекул
так и в повторяющихся звеньях. При этом может оказаться, что температура
распада этих групп является более низкой, чем температура начала интенсив-
ной термической деструкции полимера в целом. Дальнейшее сканирование
желательно проводить с учетом химических превращений, которые претер-
певают эти группы при нагревании по димера. Естественно, что такой расчет-
ный анализ не может полностью заменить экспериментальные исследования
228
Глава VII
процесса термической деструкции полимеров, который является очень слож-
ным.
В случае сополимеров уравнение (181) приобретает вид:
ai CL д ’Oi + а2 (X Д ) 2 + • • • + а„ (X Д ’Ом
Td =--'-------------------------------------- (183)
cqQXAiOi +а2(]>Хдг/)2 +...+а„(2ХдГ/)„ ’
i i i
где аь а2, а„ - молярные доли компонентов 1, 2, п, (^Al/.)j ,
I
(£М<)2, ...» - Ван-дер-Ваальсовые объемы этих компонентов;
/ i
(2ХД>91, (£^a^)2,..., (X KjAVj )„ - наборы инкрементов для компо-
' I у
нентов 1,2,..., и. В более компактной форме соотношение (183) записывается
в виДе к=п
Td=^-------1-------. (184)
ХМХ^Д^
k=i /
Если желательно выразить температуру начала интенсивной термичес-
кой деструкции через аналогичные величины для гомополимеров, построен-
ных из компонентов 1,2, .то, согласно уравнению (181) и соотношению
(183), можно записать
«1 (X Д )1 + “2 (£ Д )2 + • • • + ап (X Д )и
т“ <185)
он —-----+ а2 —------+... + а„ -----
1 ГТ1 Ха ГТ1 Н гр
1d,\ ld,2. ld,n
где T^i, Т^2, • ••, Tdn - температуры начала интенсивной термической дест-
рукции гомополимеров, синтезированных из компонентов 1,2, ..., п.
В более компактной форме соотношение (185) записывается в виде:
Х«нХд^)*
Td = -^1-. (186)
Sil—т—
t=l ld,k
Другая форма записи соотношения (183) выглядит так:
Температура начала интенсивной термической деструкции полимеров 229
1
Td
=₽17— + Р2^ + -+P«t; ,
Ld,\ ld,1 1d,n
(187)
ai(SA^)i
----------
k-1 i
а2(£АК)2
>
a„(LA^)«
P« =T^
k=l i
Глава VIII
ОПТИЧЕСКИЕ И ОПТИКО-МЕХАНИЧЕСКИЕ
СВОЙСТВА ПОЛИМЕРОВ
VIII. 1. Показатель преломления
Среди оптических свойств полимеров важнейшим является показатель
преломления. Эта характеристика самым непосредственным образом связана
с диэлектрической проницаемостью вещества. В общем случае в статическую
диэлектрическую проницаемость вносят вклад три молекулярных процесса:
ориентация постоянных моментов в поле, относительное смещение положи-
тельных и отрицательных ионов внутри молекулы и смещение электронов
относительно ядер. Эти три процесса описывают соответственно ориентаци-
онную, атомную и электронную поляризации.
В области звуковых частот диэлектрическая проницаемость обычно не
зависит от частоты. С возрастанием частоты ориентационная поляризация
начинает отставать от поля и в конце концов совсем перестает влиять на диэ-
лектрическую проницаемость. Этот переход обычно происходит в радиочас-
тотной области. При еще более высоких частотах, обычно в инфракрасной
области, наблюдается другой тип перехода, когда частота воздействия при-
ближается к собственным частотам колебаний ионов или превосходит их. В
оптической области вклад от ориентационной поляризации прене фсжимо мал;
вкладом от атомной поляризации также можно пренебречь. В этой об тис ти
измеряется не диэлектрическая проницаемость е, а показатель преломления
н, квадрат которого равен с при условии, что обе эти величины определены
при одной и той же частоте. Показатель преломления несколько изменяется с
частотой в оптической области, что связано с постепенным приближением к
собственным частотам колебаний электронов .лежащим в ультрафиолетовой
области.
Простейшим методом, в котором делается попытка учесть электростати-
ческие взаимодействия между молекулами, является метод локального поля
Оптические и оптико-механические свойства полимеров 231
Лорентца. Лорентцовскос вычисление напряженности локального поля со-
храняет свою силу и при оптических частотах в тех же условиях, что и в
статическом случае, если длина волны переменного поля велика по сравне-
нию с периодом решетки. Применительно к полимерам это означает, что длина
волны должна быть велика по сравнению с размерами элементарной ячейки
(или приближенно со средним расстоянием между атомами соседних макро-
молекул).
Изменение показателя преломления п в зависимости от плотности при
данной частоте с поправкой Лорентца подчиняется закону.
и2 - ] д/
= R. (188)
w2 +2 Р
Данное выражение представляет собой формулу' Лоренц-Лорентца, ко-
торое является оптическим аналогом формулы Клаузиуса-Моссотги (см. ниже)
В уравнении (188) М- молекулярная масса (повторяющегося звена в слу-
чае полимеров); р - плотность; R - молекулярная рефракция.
В случае стеклообразных полимеров величина р рассчитывается по фор-
муле (б), подстановка которой в уравнение (188) дает
2 .
----i-----= Я = , (189)
И +2 Хер ;
где А'л -- число Авогадро; /сср - средний коэффициент молекулярной упаковки
(для блочных монолитных тел /сср = 0,681, для пленок /:ср 0,695); т, - число
атомов /-го типа в повторяющемся звене; г, - удельная рефракция атомов /-го
типа; Л, - атомная масса; А/, - инкременты В ан-дер43 аальсовых объемов
атомов, входящих в повторяющееся звено полимера.
Молекулярная рефракция R является аддитивной величиной и складыва-
ется из рефракций R, отдельных атомов и инкрементов для типов химических
связей (двойная, тройная). Некоторые значения/?,, необходимые для расчета,
приведены в табл. 27, составленной по данным Эйзснлора для длины волны
0,5893 мкм (/)).
Для расчета показателя преломления п сополимеров у равнение (189) при-
нимает вид:
П — 1 ^ср (“1^1 + ^2^2 +••• + ) (190)
/72 +2 )1 +a2(^,^7i)2 +••• +1
232
Глава VIII
Таблица 27
Атомные рефракции ряда атомов в органических соединениях по Эйзенлору
Atom refractions of a series of atoms in organic compounds as per Eisenlore's data
Атомы, атомные группы и особенности структуры Символ R[) , см3/моль
Углерод Rc 2,418
Водород Rh 1,100
Кислород:
в ОН Ro- 1,525
в эфирах* Ro< 1,643
в СО Ro- 2,211
Хлор Ra 5,967
Бром RBr 8,865
Йод Rj 13,900
Двойная связь С = С Ri= 1,733
Тройная связь С = С Ri. 2,398
Азот:
в первичных аминах Rnj^c 2,322
во вторичных аминах RhN(C)2 2,502
в третичных аминах Rn(C)3 2,840
в имидах (третичных) Rc-n - c 3,776
в нитрилах** и £ 3,118
* А также в а-оксидах, причем па трехчленный оксидный цикл никакого инкремента не вводится
** Значения атомной рефракции азота в имидах и нитрилах включают инкременты для двойной
и тройной связей углерод-азот
где аь аз, аи - молярные доли компонентов 1, 2, ..., н; R2, Rn -
молярные рефракции гомополимеров на основе компонентов 1, 2, п\
(^Д^Ь, (^ДЦ)2, (£Д^)И - их Ван-дер-Ваальсовые объемы.
I 1 ।
В более компактной форме соотношение (190) можно записать в виде
к=п
2 j ^"ср
И __________________к=1___________
„2 , о к=п
к=\ I
(191)
Уравнения (189) и (190) позволяют с достаточной точностью оценить по-
казатель преломления полимеров и сополимеров на основе их химического
строения. Однако в случае полимеров и сополимеров с низкой температурой
Оптические и оптико-механические свойства полимеров
233
стеклования величина п часто бывает несколько заниженной. Это связано с
тем, что коэффициент молекулярной упаковки к для таких систем несколько
ниже среднего значения &ср. Поэтом)' для более точной оценки показателя
преломления желательно учесть температурную зависимость к, которая опи-
сывается уравнениями (14) и (15). Подстановка этих зависимостей в форму-
лу (189) дает
и2-1 ______Rkg_____
п2 +2 [l+Mr-^W^AK,.
(T<Tg);
(192)
------ =------:, (Т > Т„), (193)
и2+2 П +
i
где kg = 0,667.
Соответственно, используя формулу (191) для сополимеров, получаем:
к^-п
•>
-V-*—---------------, (Т<ту, (194)
[1 + ас(Г -Tg)]N
Л-1 i
к=п
2 _ < g'^Ja-kR-k
(195)
И 2 4- ? к=П S
[l + aL(T-Tg)]NA^k(Z^h
Ы i
В табл. 28 представлены показатели преломле ния дляр яда ад юрф ных го -
лимеров. Видно, что величина п зависит от химического строения полимера
и возрастает при переходе от алифатических полимеров к ароматическим.
Видно также, что для полимеров, находящихся при комнатной температуре
20
в высокоэластическом состоянии (Т> Tg), величины nD , рассчитанные с
учетом температурной зависимости коэффициента молекулярной упаковки,
т.е. с использованием формулы (192), лучше совпадают с эксперименталь-
20
ными величинами nD . При этом для расчета использовались значения тем-
пературы стеклования Tg, определенные по уравнению (84), и величины ко-
эффициентов объемного расширения о<; и (р, определенные по соотноше-
ниям (44) и (41).
234
Глава ГШ
Оптические и оптико-механические свойства полимеров
235
I
Таблица 28
л.
Молярные рефракции К , экспериментальные н расчетные "/драсч. значения
показателей преломления для ряда аморфных полимеров
Molar refractions R, experimental exp and calculated w£^calc values of the refractive
indices for a series of amorphous polymers
Полимер IX i сма/моль и20 "£),эксп. 20 ^Л,расч. Tg, расч., К
1 2 3 4 5
- сн2- сн— С-О-СН-, II д о 20,126 1,479 1.49/ 1,47 282
—сн2—сн— С-О- С2Н5 о 24,744 1,469 1,49 / 1,47 267
—СН2—СН— С-О-СдНо II 4 9 о 33,980 1.466 1,50 / 1,48 242
сн3 —СНт—С — 1 С—О—СНз II 3 о 24,744 1.490 1,490 377
СНз —СН-т—С — 1 С-О-СпНс II 2 ь о 29.362 1,485 1,50/ 1,49 338
СНз —сн2—с— С-О-С4Но II 4 9 о 38,598 1.483 1,50/ 1,49 287
таблица 28 (окончание)
1 2 3 4 5
СНз —СН2—С — с—о—Q II о 44,233 1,568 1,56 378
-сн9-сн- 6 33,343 1,591 1,60 376
сн3 -СНп-С- 6 37,961 1,587 1,59 401
-СНп-СН- 6-с| .38,210 1,610 1,62 356
—сн2—сн— О-СН3 15,497 1,467 1,49/ 1,47 253
—СН2—СН— 2 1 О-С-СНз II о 20,126 1,467 1,49 301
—сн2—сн— O-Q 34,986 1,578 1,58 353
1 0=0 1 о 1 О 1 „ о-о-о 6 1 о 1 69,983 1,585 1,58 431
1 о 1 0 о о У V 0 о 1 о 1 0=0 1 0 1 0=0 1 117,662 1,610 1,60 582
* В графе 4 даны два тиачения Лрао[ : первое из них определено по уравнению (189), а второе
по уравнению (193), т.е. с учетом температурной зависимости коэффициента молекулярной упаковки к.
236
Глава ПП
VIII.2. Коэффициент оптической чувствительности
по напряжению
Полимерные материалы нашли широкое применение в поляризационно-
оптическом методе исследования напряжений. Этот метод основан на обна-
руженном Д.Брюстером явлении двулучепреломления, возникающем в про-
зрачных оптически изотропных материалах при деформировании и обуслов-
ленным анизотропией показателя преломления в двух взаимно перпендику-
лярных направлениях.
В случае стеклообразных полимеров первым следствием прикладывае-
мой нагрузки является изменение межатомных расстояний и валентных углов
в полимерной цепи. Эти изменения определяют мгновенную упругую дефор-
мацию. Упругая деформация связана с подвижностью атомов, составляющих
звенья макромолекул внутри статистического сегмента макроцепи. При де-
формировании полимеров, находящихся в стеклообразном состоянии, возник-
новение двулучепреломления и его величина в основном обусловлены смеще-
нием электронных оболочек атомов и электронных облаков, образующих хи-
мические связи, а также искажением валентных углов, что приводит к
анизотропии поляризуемости элементарных звеньев макромолекул.
В стеклообразном состоянии двулучепрсломление иногда может быть
также связано с упругой ориентацией оптически анизотропных макромоле-
кул или их частей (например, подвижных боковых метильных групп в поли-
акрилатах и фторидных групп в полиметакриловых эфирах) вблизи их равно-
весного состояния. При этом возникает так называемая упругая составляю-
щая двулучепреломления, которая достигает своего максимального значения
практически мгновенно после приложения нагрузки. В случае идеальных
упругих тел общее двулучепреломление определялось бы упругой деформа-
цией, так как в этих условиях упруго деформированный полимер находился
бы в равновесном состоянии. Однако следует отметить, что поведение реаль-
ных полимерных тел отличается от упругого. Для них характерно изменение
деформации и величины двулучепреломления во времени даже в стеклооб-
разном состоянии.
Для оптически чувствительных полимерных материалов, находящихся в
стеклообразном состоянии, до определенных уровней напряжений применим
экспериментально установленный закон Вертгейма, связывающий оптичес-
кую разность хода 6 в любой точке полимерной модели, находящейся в плос-
конапряженном состоянии с разностью главных нормальных напряжений О]
и 02= действующих в плоской модели в той же точке, и толщиной модели d
S = Co(°l
(196)
Оптические и оптико-механические свойства полимеров
237
или Ди =«]-л2 =Q(°1-о2)^ ’ (197)
где Са - коэффициент оптической чувствительности по напряжению; Ди -
двойное лучепреломление; п\ и и2 - показатели преломления вдоль и поперек
оптической оси.
Вводя в уравнение (197) значение длины волны света получают соот-
ношение, связывающее двойное лучепреломление с порядком полос:
(198)
Л
где т - порядок полос для данной длины волны
Коэффициент Са обычно считают независящим от длины волны, но, как
показали более тщательные исследования, это не совсем правильно. В прак-
тике поляризационно-оптического метода для характеристики оптической чу в-
ствительности полимерных материалов использу юг и дру гую величину - цену
полосы материала по напряжениям , равную:
1,о
On --
c°d
В случае линейной зависимости между напряжением и деформацией двой-
ное лучепреломление можно выразить через деформации. Тогда зависимость
преобразуется к виду:
(199)
(201)
5-СЁ (£)-е2)б/, (200)
где ('< -коэффициент оптической чувствительности материала по деформа-
циям; £j и е2 - главные деформации.
Оба оптических коэффициента связаны между собой соотношением:
£ 1+н 4’°’
где ц - коэффициент Пуассона; Е - модуль упругости; - цена полосы
материала по деформациям.
Поскольку' величина двулучепреломления полимера в стеклообразном со-
стоянии в известных пределах пропорциональна анизотропии поляризуемо-
сти связей элементарного звена, увеличение анизотропии поляризуемости мак-
ромолекулы, а следовательно, и оптической чувствительности полимера, мо-
жет быть достигнуто введением в молекулу исходного мономера или олигомера
групп с большой анизотропией поляризуемости, таких как ароматические коль-
ца типа бензольных, нафталиновых, антраценовые карбонильных групп и
вообще молекулярных группировок, содержащих двойные или тройные свя-
зи, те. группировок, содержащих подвижные я-элекгроны.
В настоящее время имеется два подхода для численной оценки коэффи-
циента оптической чувствительности Са для полимеров на основании хими-
Глава VIII
238
ческого строения повторяющегося звена. Первый подход, предложенный в
работе [36], является эмпирическим.
В результате обработки большого количества экспериментальных дан-
ных в цитируемой работе предложено соотношение, связывающее Са с пара-
метрами химического строения повторяющегося звена полимера
где С, - инкременты, характеризующие вклады каждого атома и типа межмо-
лекулярного взаимодействия в коэффициент оптической чувствительности
(табл.29); - Ван-дер-Ваальсовый объем повторяющегося звена, скла-
i
дывающийся из объемов атомов, входящих в это звено', - число Авогадро',
П = 0,3544- 1()^*см2/кГ - универсальный параметр.
В табл. 30 приведены значения коэффициентов оптической чувствитель-
ности Со для ряда полимеров, находящихся в стеклообразном состоянии.
Таблица 29
Величины Су, характеризующие вклады каждого атома и з ила межмолекулярного
взаимодействия в коэффициент оптической чувсгвитс.гьности по напряжению
Values С, characterizing contributions of each atom and type of intermolecular interaction to the
stress-optical coefficient
Атом или тип межмолекулярного взаимодействия Символ Ci ю3, МПА'1 см’/моль
Углерод Сс -2,0492
Водород Сн -0,5227
Кислород в основной цени Со, о 3,1980
Кислород в боковой группе Со, 6 -0,7568
Азот в основной цепи Сн.О 7,1750
Азот в боковой группе Сн,ь 1,3030
Хлор Са -3,4760
Сера Cs -0,7900
Диполь-дипольное взаимодействие* cd -1,6000
Водородная связь ch -6,2100
п-замещение ароматических ядер** с„ 1,7000
* Коэффициенте^ используется для каждой полярной группы любой химической природы; если
две одинаковые группы находятся у одного и того же атома, то нужно вводить один коэффи-
циент Cd- Для фенильной ]руппы Сд - 2, ] 5*1О"3
** Коэффициент Сп вводится в случае п-замешения ароматических ядер-,количество вводимых
коэффициентов Справно количеству ароматических ядер., замещенных в п-положении
Оптические и оптико-механические свойства полимеров
239
Величина Са изменяется в очень широких пределах в зависимости от хими-
ческого строения полимера - от небольших отрицательных значений для по-
лиметилметакрилата и поли-а-метилстирола до очень больших положит еяь-
ных значений, характерных для ароматических полимеров. Высокая опти-
ческая чувствительность этих полимеров обусловлена их строением:
наличием большого числа конденсированных ядер, характеризуемых значи-
тельной анизотропией полярности. Соединения, насыщенные атомами азо-
та, серы и ароматическими ядрами, как это следует из табл. 30, обладают
наиболее высокими значениями Са. Большое влияние на оптическую чув-
ствительность оказывает также тип замещения ароматических ядер. Пара-
замещение способствует увеличению Са.
Наличие большого числа полярных С = О - групп приводит к снижению
оптической чувствительности. Обладая большой отрицательной анизотропи-
ей поляризуемости ,эта группа снижает общий положите тьнь ш эффект. То,
что С = О - группа является носителем отрицательного эффекта, отмечено в
работе [206] и подтверждено [ 100] при изучении оптико-механических свойств
полимеров, приведенных в табл.30.
На основании уравнения (202) можно провести количественную оценку'
вклада каждой группы в коэффициент оптической чувствительности. Этот
Таблица 30
Экспериментальные и расчетные значения коэффициентов оптической
чувствительности по напряжению Са для ряда стеклообразных полимеров
Experimental and calculated values of stress-optical coefficients Ca for a series of amorphous
polymers
Полимер С„- 10й, МПа' эксп. Со-106,Мпа’ расч.
1 2 3
—CHj—CH— 6 10,7 10,65
сн3 -сн2-с- C-CHo II J о -3,3 -2,80
сн3 -сн2-с- 6 -2,0 -4,57
240
Глава VIII
таблица 30 (продлжение)
1 2 3
-СНп-СН- д 1 СН3 15,7 22,30
—СН9—СН— а-д-С| 7,3 14,80
—сно—сн— д 1 CI 22,9 16,70
СНз -СН2-С- c-o-ch9-Q II 2 о 24,5 10,10
-СН2-СН- N 0-0 57,5 43,9
СНз -о-О-с-О-о-с- 1 н СН3 ° 111,0 105,0
O'C=N М'°'О 161,0 160,0
-c-Q-c-nh-Q. ,Q-nh- II п ° ° 0(> % 90 77,3
Оптические и оптико-механические свойства полимеров
241
таблица 30 (окончание)
1 2 3
У / 0-0 II \\ Z Z \ / 9 О=й“О А / 150,0 143,0
1 0 = 0 9 0 = 0 1 о о 6 А /°х ° ° 0 । о 1 93 83,0
± 6 о 1 0 о 0=0 6 1 0=0 1 77,4 66,4
0 о 1 т Z 1 0 = 0 1 0 1 0=0 1 62,0 60,1
вклад характеризуется значением / КАГ, , причем, поскольку это от-
ношение отрицательно, то меньшему абсолютному значению 21G /
/ i
соответствует большее значение Са. По вклад)' (^С, / А1^) 10 в ко-
7 /
эффициент оптической чувствительности отдельные группы располагаются
242
Глава VIII
Приведенная расчетная схема для определения коэффициента оптичес-
кой чувствительности дает возможность оценить ожидаемую оптическую чув-
ствительность полимера и имеет определенное значение для синтеза поли-
меров, пригодных для использования в поляризационно-оптическом методе
исследования напряжений (метод фотоупругости).
Для сополимеров уравнение (202) записывается в виде
а1СЕ С()1 +«2(Х^)2 + -+аи(^^)и
+ П, (202')
na «1(ЕД1О1 +а2(£д^)2 +- + аДЕд^)„
где аь а2, ..., а„ - молярные доли компонентов 1, 2, ..., «; (^СД,
(ZQ)2,..„ (^С,)„ -наборы констант для компонентов 1,2, ...,и; (^A^)j,
(Хд^)г, - Ван-дер-Ваальсовые объемы компонентов 1,2, ...,/7;
z i
- число Авогадро.
В более компактной форме уравнение (202') записывается в виде
к=п
к=\ i
NA ^kCLNV^k
(202'')
где 0^, Q^C;)fc и (^ДИ,)А. - соответственно молярная доля, набор констант
и Ван-дер-Ваальсовый объем Л-го компонента.
Если желательно выразить коэффициент оптической чувствительности
по напряжению для сополимеров через коэффициенты оптической чувстви-
тельности компонентов, уравнение (202') принимают вид
ai(Col -ПХ^Д^! +а2(Сп 2 -П)(£ Д1<)2 + ...+
а1(^Д17)1+а2(ХД1<)2+- +
+ а„(Саи-п)£д1/)и
+ ая(£ДГ,)„
(202"')
Оптические и оптико-механические свойства полимеров 243
где Са I, Со2 ,Са п - коэффициенты оптической чувствительности компо-
нентов 1, 2,..., п.
В более компактной форме уравнение (202"') имеет вид
Ca = k=l k=n--------!------, (202"")
k=l i
где Сад.- коэффициент оптической чувствительности для к-го компонента.
Теперь рассмотрим полуэмпирический метод оценки коэффициента оп-
тической чувствительности по напряжению предложенный в работе [91].
Дело в том, что описанная выше расчетная схема для определения Со, позво-
ляющая с достаточно высокой точностью определить Са полимера по хими-
ческому строению повторяющегося звена, не устанавливает связи между Са
и другими оптико-механическими показателями (модулем упругости, темпе-
ратурой стеклования и др.). Проведем сначала анализ в общем виде.
Рассмотрим тонкую пластину, лежащую в плоскости XOY. Поскольку дли-
на световой волны значительно меньше поперечных размеров о фазца, то его
можно рассматривать как бесконечную пластину Луч света падает в направ -
лении оси Z. Так как пластина тонкая, то согласно [86]
Uzz = (IIхх' + Uyy) , (203)
1-ц
где t/zz, Uxx, Uyy - смещения по соответствующим осям; ц - коэффициент
Пуассона.
Когда пластинка не деформирована, материал ее представляет изотроп-
ный диэлектрик с диэлектрической проницаемостью ео. При деформации пла-
стинки происходит изменение оптической симметрии среды, в результате чего
тело становится оптически анизотропным и может быть описано введением
тензора диэлектрической проницаемости е,£. Согласно [85]
£ik = e0S/i + alUik + a2Ulfiik >
где - тензор деформации; a^ и a2 - упругооптические постоянные.
В дальнейшем нас будет интересовать величина ezz, которую в соответ-
ствии с (204) можно записать
eZZ = £0 “ Й1 , № XX + ^уу) + а2~,-~(&ХХ + ~,--~иУУ )=
1-ц 1-ц 1-ц
= Eq + (а2 — — - <7| —)(UXY + Uyy ) (205)
1-ц 1-ц
244
Глава VIII
Пусть Gjy =0 (одноосная деформация). Тогда pU хх = ~Uyy и
£ZZ = £о +[я2(1-2ц)-о1ц][/хг'> (206)
(207)
(208)
Е у
®хх = ~ хх “И Uxx) = EUхх
1-ц~
где Е- модуль упругости.
Из системы уравнений (206) и (207) можно найти зависимость ezz от ;
£zz =£о+~[(1~2ц)я2.
Заменяя szz на n^z , a £g на (где nzz и «о - показатели преломления в
направлении Z и изотропного вещества соответственно), из выражения (208)
можем найти коэффициент оптической чувствительности Со, который, со-
гласно определению, будет равен
. _ Snzz
а я
8°хх
—[(1 -2Ц)472
(209)
Воспользовавшись для а} и а2 выражениями, полученными в работе [50],
согласно которой
1
Щ =6р(—)т --(Е0 -1)(е0 + 2);
ор 3
(210)
а2 =-Зр(——)г+ (Ео-1)(ео + 2),
ор 3
где р - плотность полимера, получим окончательное выражение:
А(и + 1)(„2 +2)(я2 _1)_3рЖ
2и0Е |_ 3 ор
(2П)
(212)
'о
1
6е
Оценим в формуле (212) член (де/др)т . Перепишем (^(у как
(213)
др дТ ор
При Т« Tg можно принять, что в определенном интервале температур
бе
коэффициент объемного расширения Од = const, - const < 0. в результа-
те выражение (212) можно привести к виду
Оптические и оптико-механические свойства полимеров
245
(214)
са =—-—(—)Т +п*,
2nQaGE дТ
которое является аналогом уравнения (202). В выражении (214)
п* (1 + Ц)(/702 - 1)(/72 + 2)
Зп0Е
где ц - коэффициент Пуассона; п0 - показатель преломления; Е - модуль уп-
ругости.
Так как согласно изложенному выше в случае аморфных полимеров
0,0962 , „г„ч
Од =------ (см. раздел IV.3), а для изотропного диэлектрика согласно [85]
е0 = -4—--, то выражение (214) окончательно примет вид;
2Ж
о
«0^
+ п*,
(215)
где С, = дг/дТ ; ДИ, - инкременты Ван-дер-Ваальсовых объемов атомов.
Величины С, практически не зависят от температуры и напряжения, а
также слабо меняются от температуры и напряжения в оптическом диапазо-
не частот [57, 106].
Для определения Сст будем исходить из следующих соображений. Так как
П* - величина нулевого порядка, а Со - величина 1-го порядка малости, то
С, можно представить как С, = С, 0 + ЗС,, где Ci0 - член нулевого порядка, а
ЗС, - член 1-го порядка малости. Тогда определение Со из выражения (215)
сведется к решению системы уравнений
Хс,.одр; SsQA't
^г+п'=0;^г=с^ (216)
Второе уравнение системы (216) использовано в работе [91] при расчете
значений инкрементов 8С,для различных атомов и типов межмолекулярного
взаимодействия. Калибровка метода проводилась на основании эксперимсн-
246
Глава VIII
Таблица 31
Значения 1шкрементов 5Cf для различных атомов и типов
межмолекулярного взаимодействия
Values of increments 8С, for different atoms and types of intermolecular interaction
Элемент или тип молекулярного взаимодействия Условное обозначение Величина 8С,-106, МПа 1
Углерод 5Сс -0,005
Водород ьс1Г -0,118
Кислород в основной цепи 5('о. о 2,660
Кислород в боковой группе 5COis -0,700
Азот в основной цепи SGv, 0 16,620
Азот в боковой группе 6C.V, 5 0,640
Сера 8G 0,740
Диполь-дипольное взаимодействие 8Q, 6,470
Водородная связь ЗСА -66,040
Пара-замещение зсп -0,730
Мега-замещение 8СИ -3,410
тальных значений Cn. hq, E и Tg для ряда хорошо изученных аморфных поли-
меров путем решения избыточной системы уравнений, построенной на осно-
ве соотношения (215). В результате получен набор инкрементов 8С„ пред-
ставленных в табл.31. Зная эти инкременты, можно сначала определить вели-
чину С'о по выражению (216), а затем найти Со из соотношения
С;-15,67g
WqF
(217)
Проведем такой анализ в деталях для полифенилхиноксалина
XC=N
I
— О—
N=Cx
имеющего наибольший коэффициент оптической чувствительности. Для этого
полимера
8С; ДР) = 86^-(4 Д + 4ДР(2 21 + 4Д1/(219 2Д1/с 20 "^20Д1^д§) +
+ ^Н,20^н, 124 + ^COoI\Vol3l + 43Су5ДКл?151 + 26Cd -
=-0,005(4-11,1+4-10,2+4-8,4+2-11,6+20-12,7)+ (-0J 18)-20-2,0 +
Оптические и оптико-механические свойства полимеров
247
+2,66 2,1 + 4 - 0,64 6,1 + 2 6,47 = 27,4 МПа-‘см3/моль
(£ДЦ) = 462,5А-\
/
27 4
Тогда С’ = = 0,0592 .
462,5
Подставляя в формулу (217) значение С„, 7^ = 563 К,л0=1,62 иЕ= 1900 МПа
получаем
0,0592-15,6-563 □
—-----------------ioJ
1,62-1900
= 169 К
Развитый в работе [91] полу эмпирический подход для оценки коэффици-
ента оптической чувствительности по напряжению Са полимеров позволяет
также рассчитывать модуль упругости линейного стеклообразного полимера.
Используя соотношение (217) для определения величины С'а , можно записать
, (У ЗС ДИ)-15,6-7’
Е = c;-!5,6-rg = н g
С^о £ДИ,)С>0 (218)
/
Подставляя в формулу (218) уравнение (84) для расчета Tg линейного по-
лимера, окончательно имеем
(£ЗС;Д1<)-15,6
Е =—; .
Са«о(Е<7/ЛГ//(219)
' У
Результаты расчета Е для представителей разных классов теплостойких
полимеров представлены в табл.32. Следует отметить, что модули упругости
стеклообразных полимеров при температурах ниже Tg отличаются друг от
друга незначительно (различие, например, в 2 раза не следует считать боль-
шим, т.к. такое различие может проявиться в результате испытания при раз-
ных скоростях деформирования, для образцов разной формы, для образцов
одного и того же полимера с разной предисторией получения и т.д.). П оэтому
к расчетной оценке модуля упругости линейных стеклообразных полимеров
следует относиться с осторожностью.
Коэффициент оптической чувствительности по напряжению Со является
фундаментальной характеристикой материалов, используемых в поляризаци-
онно-оптическом методе исследования напряжений. Согласно этом)' методу
из прозрачных о птигсс ки чу вс твит льных материалов изготавливается мо-
248
Глава VIII
Таблица 32
Оптико-механические характеристики ряда теплостойких ароматических полимеров
Строение повторяющегося звена Tg, к с' ХАП i МПа’1 - см3/моль
1 2 3
Z Z о ъ / \ Z Z 1 \ 563 27,4
О О у II II ос> 0 ° % 788 2017,0
1 0 = 0 1 "| 0 = 0 1 о 1 о р .О °z ° 0 1 593 12,940
9 O=w=O 6 1 о Q /°ч & о 0 о 1 о 1 543 10,600
-C-O-C-NH-04 zQ-nh- ° ° сДу 630 7,813
Оптические и оптико-механические свойства полимеров
249
Optico-inechanical properties of a series of heat-resistant aromatic polymers
ca- io6 МПа'1 E, МПа п
Расчет Эксперемент Расчет Эксперемент Расчет
4 5 6 7 8
169,0 161,1 1810 1900 1,62
120,7 111,2 3120 3100 1,62
88,1 92,7 2200 2190 1.60
72,8 72,8 1640 1630 1,63
80,2 77,4 1260 1260 1,64
250
Глава VIII
дель натурной конструкции, к которой затем прикладываются соответствую-
щие нагрузки. В результате в материале модели появляется двойное лучепре-
ломление и по картине полос можно судить о напряженно-деформированном
состоянии конструкции.
В случае объемных моделей весьма эффективен метод замораживания де-
формаций. Суть его состоит в том, что модель нагревается до температуры, при
которой материал модели переходит в высокоэластическое состояние; затем
модель нагружается и под нагрузкой охлаждается до комнатной температуры,
которая для обычных оптически-чувствительных материалов примерно на SO -
100 °C ниже температуры стеклования. Возникшие при нагружении модели де-
формации и оптическая анизотропия замораживаются. Дальше модель распи-
ливается на тонкие пластинки, которые затем исследуются.
Не останавливаясь на других разновидностях поляризационно-оптичес-
кого метода исследования напряжений, заметим, что успешное развитие этих
методов возможно только при создании новых полимерных материалов с тре-
буемыми оптико-механическими свойствами. Для решения различных задач
требуются материалы двух типов - упругие и вязкоупругие. При исследова-
ниях методом динамической фотоупругости полей напряжений в слоистых
упругих средах необходимы разномодульные полимерные материалы с соот-
ношением модулей упругости 2 ... 10, обладающие упругими свойствами,
как при статическом, так и динамическом нагружениях. В практике динами-
ческой фотоупругости такие материалы появились недавно [45].
Трудность получения таких материалов заключается в том, что все поли-
мерные стекла независимо от химического строения имеют примерно одина-
ковый модуль упругости порядка ~103 МПа. Казалось бы, что для получения
разномодульных материалов можно синтезировать полимеры, которые при
температуре испытания (например, комнатной) находились бы в переходной
зоне из стеклообразного в высокоэластическое состояние. Поскольку в это й
зоне модуль упругости резко снижается, всегда можно подобрать материал с
нужным модулем упругости. Однако материалы в переходной зоне обладают
чрезвычайно сильно выраженной вязкоупругостью в то время как для поля-
ризационно-оптического метода исследования напряжений для решения дан-
ной задачи требуются упругие материалы. Эти два несовместимых требова-
ния можно удовлетворить в сетчатых полимерах определенного химического
строения путем синтеза частых сеток, которые, обладая чрезвычайно боль-
шим модулем высокоэластичности, имели бы низкие температуры стеклова-
ния (значительно ниже комнатной). Современное состояние вопроса в облас-
ти прогнозирования свойств таких систем позволило осуществить предска-
зание строения частых сеток, которые удовлетворяли бы этим двум противо-
речивым требованиям [46, 47].
Оптические и оптико-механические свойства полимеров
251
При использовании изложенного выше подхода осуществлено прогнози-
рование температуры стеклования и модуля упругости, и в качестве конкрет-
ного объекта исследования выбраны сетчатые карбофункциональные крем-
нийорганические полиизоцианураты с различной, но малой длиной межуз-
лового фрагмента [45, 46] _
ьп3
< г - - - “|
1 '(А '
]О=С С=О[
^-СН3
R
R
где
СНз
I
R -NH-C-O-CH2-CH2-O-CH2- -Si—О
II I
[СНз In СНз
СНз
I
Si—СНг-О—СНз-СНг-О-С—NH—
II
О
О
Замечательным свойством данных материалов является то, что несмотря
на величины модулей, характерных для переходной зоны из стеклообразного
состояния в высокоэластическос, они обладают упругим поведением, как стек-
ла и резины, а не вязкоупругим поведением, как обычные материалы, находя-
щиеся в переходной зоне.
Сравним релаксационное поведение данных материалов и тех материа-
лов (упругого и вязкоупругого), которые обычно применяются в методе фото-
упругости [47]: эпоксидного олигомера ЭД-20, отвержденного ангидридом
полисебациновой кислоты (вязкоупругий материал) ,и олигомера ЭД 20 ,от -
вержденного мстилтетрагидрофталевым ангидридом (7'g =115 °C, упругий
материал). Нахождение переходной зоны (из стеклообразного в высокоэласти-
ческое состояние) вязкоупругого материала в области температур от-5до .34 °C
позволяет изменяя температуру испытания проводить сравнение релаксаци-
онных свойств этих полимеров при одинаковых значениях начального моду-
ля упругости.
Анализируя представленные на рис.71 данные, можно заключить, что
кривая релаксации напряжения для полиизоциануратной сетки имеет харак-
тер, близкий к релаксационной кривой для стеклообразного полимера. Абсо-
лютная величина спада напряжения после достижения участка малой скорос-
ти релаксации для образца сетчатого полиизоцианурата примерно такая же,
как и для стеклообразного материала (ЭД-20 + МТГФА), и значительно мень-
ше, чем для вязкоупругого полимера.
На рис .72 представлены зависимости механической /(/) -e(z)/g и опти-
ческой D(t) = m(t)lsd ползучести от времени: m(t) - порядок интерференци-
252
I "лава VIII
Рис.71. Релаксация напряжения дая образцов ЭД-20 + МТГФА (1), 1 юлиизоциануразной
сетки (2) и вязкоупругого материала (3), состав которого см. на рис.72
Stress relaxation for specimens ED-20 + MTHPA (1), polyisocyanurate network (2)
and viscoelastic material (3) the composition of which is indicated in Fig. 72
онных полос в момент измерения; о напряжение в образце; d - толщина
образца в направлении просвечивания.
Из рис.72,6 видно, что для вязкоупругого материала после нагружения
наблюдается плавный рост значений податливости. Однако для кремнийсо-
держащих сетчатых полиизоциануратов (см. рис.72,а)в отличие оттипично-
го вязкоупругого полимера, наблюдаемая ползучесть быстро затухает.
Таким образом, можно отметить, что д ля синтезированных в работах
[45,46] карбофункциональныхкремнийорганических полиизоциануратов ха-
рактерно упругое поведение, осложненное лишь слабыми признаками вязко-
упругости.
В табл. 33 приведены значения равновесных модулей упругости, получен-
ные в результате аппроксимации кривых релаксации напряжения с привлече-
Таблица 33
Оптико-механические характеристики сшитых мякродиизоцианатов
Optico-mechanical properties of macrodiisocyanafes networks
n Статическое нагружение Динамическое нагружение
E, МПа Од0 , МПа (при 22 °C), Ср , м/с Ед , МПа „1,0 , МПа
1,2 866 1,26 1980 4822 2,6
2,5 283 1,14 1700 3454 2,3
3,8 149 1,08 1270 1897 2,1
5,4 - 0,80 1000 1140 1.8
6,2 43 0,69 800 727 1,6
9,2 24 0,41 570 357 1,0
Оптические и оптико-механические свойства полимеров
253
Рис.72. Кривые механической 1(1) и оптической D(t) ползучести сетчатых полиизоциан-
уратов при п = 6,2 (см. структурную формулу в тексте), а = 1,4 МПа (а) и вязкоу! ipyroro
полимера на осиовеэпоксндного олигомера ЭД-20, отвержденного смесью полисебаци-
нового ангидрида (27,3 %) в присутствии азелаиповой кислоты (13,8 %) (б)
Mechanical I(t) and optical D(t) creep curves of network polyisocyanurates at n = 6,2
(See the structural formula in the text), a = 1,4 MPa (a) and viscoelasticpolymcr based on
epoxy oligomer ED-20 cured with the mixture of polysebacic anhydride (27,3 %) in the
presence of ozelaic acid (13,8 %) (6)
нием новых ядер релаксации (см. ниже), обеспечивающих, наряду с высоки-
ми коэффициентами корреляции, получение ряда физических характеристик
материала (количество микродефектов, начальная энтропия системы и др.).
В этой же таблице помещены величины цены полосы материала по напряже-
нию о))’0. Каквидноизтаблицы.врядуполиизощтануратов,различающихся
только количеством димсптлсилоксановых звеньев между узлами сетки, оп-
тическая чувствительность по напряжению сростом величины межузлового
фрагмента п возрастает. Очевидно, повышение оптической чувствительнос-
ти, несмотря на уменьшение концентрации групп с большой анизо тропией
поляризуемости (изоциануратный цикл, ароматические кольца), связано суве-
личением подвижности макроцепи, зависящей как от концентрации узлов в
сетке, так и отколичесгва дим стал силоксановых звеньев, обладающих низ-
кой потенциальной энергией вращения связи Si—С — Si [52].
254
Глава VIII
Динамические характеристики оптико-механических свойств полимеров
в значительной мере могут отличаться от статических из-за влияния времен-
ного фактора. Так, при действии кратковременных имульсных нагрузок про-
цессы, связанные с регистрацией в модели оптической картины полос, длят-
ся от нескольких микросекунд до сотен микросекунд. В этом случае обычные
квазистатические испытания на ползучесть и релаксацию напряжения не мо-
гут отражать сути происходящих при динамическом воздействии явлений,
протекающих в полимерном материале.
Анализ фотограмм показывает, что продольные и поперечные волны рас-
пространяются по стержню из сетчатых полиизо циануратов с постоянной ско-
ростью. Значения скорости распространения продольной волны Ср и дина-
мического модуля упругости Ед, рассчитанного по уравнению Ед = рС^, где
р - плотность материала, представлены в табл.33. Как видно, для данных
материалов наблюдается широкий диапазон значений скорости продольной
волны Ср, (500 ... 2000 м/с) и динамического модуля упругости ЕЛ (300 ...
5000 МПа).
Таким образом, оптически чувствительные материалы на основе сетча-
тых полиизоциануратов с короткими цепями между узлами сетки существен-
но отличаются от традиционно используемых, для которых при различном
статическом модуле упругости характерны практически одинаковые значения
динамического модуля, что препятствует их применению при решении ряда
динамических задач.
С целью оценки вязкоупругих свойств сетчатых полиизоциануратов в
условиях динамического воздействия рассмотрим импульсы картин полос
m(f) в различных сечениях / стержней. Для сопоставления значения импуль-
сов m(t) нанесены на один график (рис. 73)со сдвигом по времени, учитыва-
ющим скорость распространения волны в материалах. В отличие от типич-
ного вязкоупругого полимера (см. рис.73,б) для образцов сетчатых полиизо-
циануратов с п = 1 ... 9 характерно незначительное изменение формы
импульсов и их длительностей, (см. рис.73,а) наблюдаемое с увеличением
пройденного в стержнях расстоян ия, что свидетельствует о небольшой их
вязкоупругости при импульсном нагружении. Следовательно, оптически чу в-
ствительные сетчатые полиизо цианураты пригодны для исследования напря-
женно-деформированного состояния слоистых сред методом динамической
фотоупругости.
Выше были рассмотрены упругие полимерные материалы. Однако для
решения ряда задач требуются оптически-чувствительные материалы, обла-
дающие вязкоупругостью. Естественно, что вязкоупругое поведение наиболее
характерно для переходной зоны из стеклообразного состояния в высокоэла-
стическое.
Оптические и оптико-механические свойства полимеров
255
Рис .73 .Изменение импульсов картины полос »i(t) в различных сечениях / стержней из
сетчатого полиизоцианурата при и = 2,5 и / = 50 (1); 100 (2); 150 мм (3) (а) и вязко-
упругого полимера, состав которого см. на рис.72, при Z = 60(l); 110 (2); 160 мм (3)(б)
Change of impulses of pattern of bands m(t) in different sections / of network
polyisocyanurate rods at n = 2.5 and I =50 (1); 100 (2); 150 mm (3) (a) and viscoelastic
polymer at / = 60 (1); 110 (2); 160 mm (3) (6) tire composition of with is indicated in Fig.72
Существующие в настоящее время способы моделирования ползучести
поляризационно-оптическим методом (метод фотоползучести) основаны на
специальном подборе материалов, которые обладали бы наряду с высокой
оптической чувствительностью отчетливо выраженным вязкоупругим пове-
дением. Для решения этой задачи необходимо иметь сетчатые полимеры, на-
ходящиеся при температуре испытания в области перехода из стеклообраз-
ного в высокоэластическое состояние.
При этом вязкоупругость материалов будет возрастать по мере прибли-
жения температуры испытания к температуре стеклования Tg. Отсюда необ-
ходимо иметь ряд полимерных материалов с заданной 7\,, разноудаленной от
температуры испытания.
В работе [35] использовали описанную выше расчетную схему' определе-
ния Tg сетчатых полгмеров для прогнозирования термических и физических
.характеристи к вновь синтезируемых эпоксиангидридокислотных компаундов,
используемых для решения данной задачи.
Для того чтобы получить оптически чувствительные полимеры, различа-
ющиеся по своим термическим и оптико-механическим показателям, для син-
256
Глава ПИ
Рис.74. Зависимость цены полосы, приведенной к единичному напряжению т/а,
от времени действия нагрузки t. Состав композиций: 1) ЭД-20 - 1 моль, метилтетрагид-
рофталевыи ангидрид (МТГФА) - 1,4 моля, олеиновая кислота - 0,6 моля; 2) ЭД- 20 -
1 моль, МТГФА - 0,2 моля, азелаиновая кислота - 0,9 молей; 3) ЭД-20 - 1 моль,
МТГФА - 1,6 моля, олеиновая кислота - 0,4 моля
Dependence of material strip that is brought to unit stress m/а on time of load action t.
Formula of compositions: l)ED-20- 1 mole, methyltetrahydrophthalic anhydride (MTHPA)-
1,4 mole, oleic acid - 0,6 mole; 2) ED-20 - 1 mole, MTIfPA - 0,2 mole, azelaic acid -
0,9 mole; 3) ED-20 - 1 mole, МГНРА - 1,6 mole, oleic acid - 0,4 mole
теза модельных материалов были использованы эпоксидный олигомер
ЭД-20, отвердитель - изо-метилтетрагидрофталевый ангидрид, соотвердите-
ди - дикарбоновая азелаиновая кислота, монокарбоновая олеиновая кислота
в различных стехиометрических соотношениях с целью регулирования часто -
ты сшивки и молекулярной массы межузлового фрагментаМс.
В результате был получен набор сетчатых полимеров, у которых темпе-
ратура стеклования лежит вблизи комнатной температуры, и естественно, что
материалы на основе этих сеток обладали отчетливо выраженной вязкоупру-
гостью. Такие полимеры обладают способностью к ползучести в очень ши-
роком интервале абсолютных величин податливости и скоростей процесса.
Это приводит и к отчетливо выраженной зависимости цены полосы, приве-
денной к единичному напряжению, от времени (рис. 74).
Глава IX
ДИЭЛЕКТРИЧЕСКАЯ ПРОНИЦАЕМОСТЬ
ПОЛИМЕРОВ И ОРГАНИЧЕСКИХ
РАСТВОРИТЕЛЕЙ
Расчет диэлектрической проницаемости полимеров по их химическому
строению является важной задачей как с точки зрения направленного синте-
за полимеров с заданной диэлектрической проницаемостью, так и для оцен-
ки полярности (магнитного момента) повторяющегося звена полимера, что
имеет существенное значение и для предсказания растворимости полимера в
органических растворителях. Поэтому количественную оценку диэлектричес-
кой проницаемости полезно также проводить и для органических жидкостей,
являющихся растворителями полимеров. Следует сразу же заметить, что про -
блема расчета диэлектрической проницаемости органических жидкостей яв-
ляется более сложной, чем для полимеров. Это связано с тем обстоятельством,
что коэффициент молекулярной упаковки для аморфных позимсров пример -
но одинаков и мало зависит от химического строения полимера. Как отмече-
но выше, в первом приближении коэффициент молекулярной упаковки для
стеклообразных аморфных полимеров при комнатной температуре оценива-
ется величиной 0,681. В более точном приближении коэффициент молеку-
лярной упаковки примерно одинаков для всех полимеров при их температуре
стеклования Tg; эта величина составляет к,, = 0,667. Это позволяет, как будет
видно ниже, провести более точные расчеты диэлектрической проницаемос-
ти е для полимеров при комнатной температуре.
Что касается органических жидкостей, то для них коэффициент молеку-
лярной упаковки существенно зависит от химического строения; коэффици-
енты молекулярной упаковки для органических жидкостей колеблются в пре-
делах от 0,45 до 0,705. Это обстоятельство затрудняет расчет плотности (или
молярного объема) жидкости, которые необходимы для дальнейшего расчета
диэлектрической проницаемости.
258
Глава IX
Как известно, связь между вектором поляризации Р и вектором напря-
женности электрического поля Е в вакууме и в диэлектрике имеет вид:
D = Е + 4тсР = еЕ; где D - вектор электрической индукции. Теория приводит
к следующему выражению для диэлектрической проницаемости в случае не-
полярных диэлектриков:
е=1+—-----------
i
где и, - концентрация атомов, ионов или молекул Z-го типа; а - поляризуе-
мость этих структурных элементов, р, - фактор, учитывающий взаимодей-
ствие диполей друг с другом.
Как известно, диэлектрическая проницаемость однородного диэлектри-
ка £ показывает, во сколько раз уменьшается величина напряженности элект-
рического поля Е внутри диэлектрика по сравнению с величиной внешнего
поля £0:
Е = ^-.
£
Макроскопическое поле Е есть векторная сумма Е = Ео + Е; внешнего
поля Ео и поля Е„ обусловленного поляризацией вещества Р во внешнем поле:
Е, = -АпР.
В слабых полях поляризация пропорциональна макроскопическому' полю
Р = 7_Е, где х есть макроскопическая восприимчивость вещества.
Отсюда с учетом приведенных выше уравнений получаем хорошо извес-
тную связь макроскопических характеристик
£ = 1 + 4 к/ . (220)
При микроскопическом описании величина поляризации определяется
микроскопической поляризуемостью Хо (и/ - концентрация и а, -
поляризуемость структурных элементов вещества) и величиной локального
поля Елок.
Р = Хо^лок ,
где локальное поле равно
Елок -Е + ^Р.
Коэффициент деполяризации )5 для изотропного диэлектрика равен 4л/3.
Из приведенных выше соотношений можно найти связь между' макроскопи-
ческой х и микроскопической хо поляризусмостями:
Диэлектрическая проницаемость
259
Y= _Ь_.
i-Рь
Можно также получить формулу связи диэлектрической проницаемости
и микроскопической поляризуемости (форму'ла Клаузиуса -Моссотти)'.
е-1 4я 4л v
~ТХ" (22|>
Электронная поляризуемость <х1Л в области оптических частот равна:
а =—V—------------
зл ZL ? О’
т k &>0к -tt)D
где (Од = 3,2-1015 рад/с - частота, соответствующая D - линии натрия; и
/0А. суть частоты и силы осцилляторов для электронного спектра вещества,
связанного с переходом 0 —> к По порядку величины электронная поляризу-
емость аэл составляет 10’24 см3.
Соответствующий вклад в формуле (221) записывают в виде:
3 / Mi
где р - плотность; Л/ - молекулярная масса; R, - молярная рефракция /-го
структурного элемента (в случае полимеров расчет ведется на повторяющее-
ся звено).
Ориентационная поляризуемость а,Л„, в слабых полях, связанная с вра-
щением постоянных диполей d, равна:
d1
При комнатных температурах для диполей д' ^ |/ ) величина ad)1„ также
составляет приблизительно 10‘24см3. Соответствующий вклад в формулу Кла-
узиуса -Моссотти, связанный с наличием в структуре полярных групп, запи-
шем в виде.
где сумма поj идет по всем полярным группам стру кту рных элементов. Окон-
чательно формула (221) примет вид:
+ (222)
260
Глава IX
В случае полимеров расчет ведется на повторяющееся звено. Учитывая,
что плотность полимеров может быть рассчитана по уравнению (6), прихо-
дим к следующему выражению, удобном}7 для расчета диэлектрической про-
ницаемости
. (222')
е + 2
• i
Величина молярной поляризуемости Р является аддитивной и складыва-
ется из поляризуемо стей атомов, а также из инкрементов пол яри?}'с мости,
связанных с наличием различных типов химических связей (двойная, трой-
ная) и с другими особенностями строения молекул. Здесь картина та же, что и
в случае оценки молярной рефракции. Дтя неполярных диэлектриков диэ-
лектрическая проницаемость обусловлена только деформационной поляриза-
цией и, согласно соотношению Максвелла, практически совпадает с квадра-
том показателя преломления в области высоких частот е » и2. Для таких по-
лимеров (полиэтилен, политетрафторэтилен, полибутадиен и т. д.) молярная
рефракция R практически совпадает с молярной поляризацией Р.
Для полярных диэлектриков картина осложняется. Под действием элект-
рического поля у них происходит ориентация постоянных диполей. Эти дипо-
ли возникают за счет наличия полярных групп в полимере, таких, например,
как - ОН, - СО - COO - Cl, - NHCO -, - NHCOO - CN и т.д. Это при-
водит к тому, что величина поляризации Р для этих групп превышает величи-
ну рефракции/?.
Проведем анализ поправок, которые необходимо вводить в рефракцию
для расчета поляризуемости молекулы. Для полярных гру пп различной хими-
ческой природы величину поляризуемости запишем как
Д=Д+АД, (222”)
где /?, - молярная рефракция этой группы; А/?, - поправка на ориентацию
диполей. Величины этих поправок вычислены с помощью линейного регрес-
сионного анализа на основании сравнения показателей преломления и диэ-
лектрических проницаемостей большого числа полярных полимеров ,а само
вычисление проводилось путем использования формул (222') и (222"), комби-
нация которых дает следующее выражение
+2М)
Е -1 / j
е + 2 ~ (223)
Диэлектрическая проницаемость
261
Таблица 34
Величины Ай, для вычисления поляризуемости
Values Ай, for calculation of polarizability
Группа A/?z. см3 /моль Группа ARi, см3 /моль
-о- 3,557 -C-NH II О 21,000
—с— II О 5,371 — F 0,845
-о-с- II о 8,728 -CI 3,900
-о-с-о- 1 О 17,085 —cf2— 1,352
-он , 3,500 1 Л о о 10,300
—C=N 5,464
Результаты расчета приведены в табл.34, где даны численные значения
ДЯ, для различных полярных групп, наиболее часто встречающихся в поли-
мерах. Зная эти величины можно рассчитать диэлектрические проницаемос-
ти для большого числа органических полимеров различных классов. Резуль-
таты проведенных расчетов показывают хорошее совпадение с эксперимен-
тальными данными [133 ,214] (см .табл 34 а) .
Для более точного расчета диэлектрической проницаемости полимеров
при комнатной температуре желательно учитывать температурную зависи-
мость коэффициента молекулярной упаковки. Это относится в первую оче-
редь к полимерам, находящимся при комнатной температуре в высокоэласти-
ческом состоянии. Согласно работе [128] для этих полимеров температурная
зависимость к(Т) описывается соотношением
к„
кД) =---------—4--------
l + 356-l(P4(7-7g) g
При комнатной температуре имеем
----- kg______
£(298) =
1 +3 56 -10“4(298 -Tg) '
262
I 'лава IX
Таблица 34,а
Расчетные и экспериментальные значения диэлектрической проницаемости
для ряда полимеров
Calculated and experimental values of the dielectric constant for a series of polymers
Полимер £ расч. £ эксп. Ошибка, %
1 2 3 4
Политетрафторэтилен 1,98 2,00; 1,96; 2,01; 2,10 -1,0
Поли(4-метнл-1 -пентан) 2,27 2,13 6,6
Полипропилен 2,27 2,15; 2,20 3,2
Поли из об утиле н 2,23 2,23 0,0
Поли(винил циклогексац) 2,38 2,25 5,8
Поли( 1 -бутан) 2,25 2,27 -0,9
Полиэтилен 2,23 2,20; 2,30 1,4
Поли(а, а, а’, а' -тетрафтор-п-кенлилен 2,40 2,35 2,1
Полиизопрен 2,28 2,37 -3,8
Поли(о-метил стирол) 2,54 2,49 2,0
Поли( 1.4-бутадиен) 2,27 2,51 -9.6
Поли(/?-вин11л нафталин) 2,65 2,51 5,6
Полистирол 2,57 2,55; 2,60; 2,50 0,8
Поли(а-метил стирол) 2,54 2,57 -1,2
Г1<иш(циклогексид метакрилат) 2,70 2 58 4,6
ПолихлоротрпФторэтилен 2,70 2,80; 2,60 3,8
Поли(а-винил нафталин) 2,65 2,60 1,9
Поли [окси (2,6-днметпл-1,4-фенилен)! 2,77 2,75; 2,65; 2,60 0,7
Поли [1,1 -циклогексан бис (4-фенил) карбонат 2,97 2,60 14,2
Поли(гтксилилен) 2,58 2,65 -2.6
Поли(п-хлор строи) 2,88 2,82; 2,63; 2,65 2,1
Полпвинилбугнраль 2,70 2,69 0,4
Эпищеллюлоз а 2,71 2,70 0,4
ПолиСизобутнл метакрилат) 2,71 2,70 0,4
11оли(диметил силоксан) 2,75 2,75 0,0
Поли [окси (2,6-дифенил- 1.4-фепилеи)! 2,78 2,80 -0,7
Поли(м-хлор стирол) 2,88 2 80 28
ПолиСн-бутнл метакрилат) 2,64 2,82 -6,4
Поли(винилиден хлорид) 2,87 2,90; 2,92; 2,85 0,7
Бисфенол- А-поликарбонат 3,11 3,00; .3,05; 2,90 2,0
Поли (N-винил карбазол) 2,69 2,90 -7,2
Полн[1,1 -тган бис (4 -(jjeHiui) карб onai’i 3,18 2 90 9 6
Поли(3,4-дихлорстирол) 3,16 2,94 7,5
Поли(хлоро-п-ксилилен) 2,89 2,95 -2,0
Полнвннилхло fl 1Д 3,14 3,15; 3,05; 2,95 -0.3
Поли(1,4-циклогекснлцдеп днметилеп терефталат) 2.94 3 00 -2 0
Поли (тгил метакрилат) 2,84 2,80; 3,00; 2,90 1 ,4
Поли(оксп-2,2-дихлороме'11 цприметилен) 3,06 3,00 2,0
Поли(п-метокси-о-хлоро стирол) 2,94 3,08 -4,5
Диэлектрическая проницаемость
263
таблица 34,а (окончание)
1 2 3 4
Полиметилмстакрнлат 2,94 2,94; 3,15; 3,10 0,0
Г1оли[г11о(п-(|1енилен)] 2,99 3,10 -3,5
Полиоксиметилен 2,96 2,95; 2,85; 3,10 0,3
Полн(тсгр амстилен тер с^палат) 3,09 .3,10 -0,3
Поли(этнл а-хлора крила г) ,3,26 3,20,- 3,16,- .3,10 1,9
Поли[4,4,-пзо11рогпип1дсн дифенокси ди (4'феннлен)сульфон| 2,93 3,18 -7,9
Поли(чфирэ ф1 ркетон ) 3 оо 3 20 -6 2
П олп гекса мети ленсеб а цин амид 3,60 3 80-.3 20 -5,3
Поливиннлацетат 3,10 3,02; 3,30; 3,20; 3,22; 3,25 2,6
П олиэтиленк'рефталат 3,29 3,40; 3,50; 3,Ю; 3,25 1,2
Поли(п-гндрокеи бензоат) 3,25 3,28 -0,9
Поли [2.2'-(м-фенилеп)-5,5'-бибенз11М11- дазол] 2,72 3,30 -17,6
11 ол! 1 (метил а-х ло ра крилат) 3,47 3,45; 3,32; 3,40 0,6
Поли [4,4,-Дпфеноксиди(4'феш1лен) сульфон| 3,54 3,44 2,9
11 олн(гексамет11лен ад1 ш амид) 4,13 4,14; 4,10; 4,00; 3,50 -0,2
Поли [К N- (и п- окепдифенплен) пиромелитимид/ 3 39 3 50 -31
П олн[4,4'-суль|) а |днфенокепди(4 фени - лен) суды] юн] 372 3 80 -2-J
Полиакрилонитрил 3,01 3,26; 3.15; 3, 10,- 4,00 -2,9
Для сополимеров уравнение для расчета диэлектрической проницаем ос-
ам записывается в виде
£-1 ____________+a2jP2 + - + ая^п)_____________ (723')
£ + 2 ^4[а1(£Д1;)1+а2(^А1Д2+... + аи(^Д1Д„]"
i i i
где aj, a2, - молярные доли компонентов 1, 2, .... n сополимера;
(^2 AVt)], (JL')>,, (S AP^),, - Ван-дер-Ваальсовыс объемы этих же
компонентов; Рь ,..., Р,, -поляризуемости компонентов 1,2,..., п.
В более компактном виде уравнение (223') записываемся в виде
к=п
£~.!_ =_____^1________ (223")
+ 2 к '
к=\ i
264
Глава IX
где и (ЕАЮ&~ молярная доля и Ван-дер-Ваальсовый объем к-го
компонента соответственно; — его поляризуемость.
Если желательно выразить диэлектрическую проницаемость для
сополимера через диэлектрические проницаемости компонентов, то уравнение
(223') можно переписать в виде
е -1
е + 2
“I ^,4 (Z ДЬ + “2 (Е)2 + • • +
£1+2 ________г2+2 ________
«1(Е А*<)1 +а2(ЕД^^2 +...+
+ а«-^(Е^)и
____£» + /__
+ аи
(223"')
где еь е2, е„ - диэлектрические проницаемости компонентов 1, 2, п.
В более компактной форме уравнение (223"') записывается в виде
£ ~ 1 _ £=1 + 2 ,
е + 2 k=n
ЕаИЕА^
к=1 /
(223"")
где - молярная доля к-ro компонента, - его Ван-дер-Ваальсовый
объем.
Перейдем теперь к описанию расчетной схемы для оценки диэлектричес-
кой проницаемости органических жидкостей, которые являются растворите-
лями для полимеров.
Как было отмечено выше, коэффициент молекулярной упаковки для орга-
нических жидкостей существенно зависит от химического строения и не яв-
ляется постоянной величиной. Поэтому' расчет диэлектрической проницае-
мости по формуле (222) затруднен, поскольку плотность жидкости р не мо -
жет быть рассчитана с достаточной точностью. Однако это не основная
причина того, что диэлектрическая проницаемость жидкостей не может быть
оценена с помощью уравнения Клаузиуса-Моссотти с приемлемой точнос-
тью. Так, например, если рассчитывать величину е для такого растворителя
как н-пропиловый спирт и принять такую же величину ДА„ как и в полиме-
рах, то получаем следующие значения: ДА,= 3,3 см3/моль, рэксп = 0,799 г/см3,
Е АЕ, = 70,65 А3, Е ДА, = 17,579 см3/моль, Р = 21,079 см3/моль. Подставляя
Диэлектрическая проницаемость
265
Таблица 34,6
Зависимость А/?, от Ваи-дер-Ваальсового объема жидкостей
Dependence of A/?, on the Van-der-Waals volume of liquids
Класс жидкостей ДЯ/ = Л£А1<) i
Хлорированные соединения, содержащие 1 атом углерода &R'CI =-0,49£ Л Г- +43,8 i
Хлорированные соединения, содер- жащие больше 1-го атома углерода AR^ = -0,332^ А И; + 45,52 i
Хлорированные соединения с двойной связью при атоме хлора &Rc! =-О,325£ДГ;+33,5 i
Спирты l\ROH =-L708-10"3(^Ar,)2 + 7 +0,76 +0,343
Кислоты &R-COOH “3,7 5-10 rj)2 - i -0,825^Д1+ +57,0 i
Сложные эфиры kRcoo = 0,18£Д1+ + 20,2 r
Кетоны A/?ra =0,525£Al< +8,25 i
Простые эфиры &RO - 29,0
Альдегиды McotI =-l,67-10-3(XAK,r + +0,75 l^AK, +2,648 7
Нитрилы ARcn =O,525£AI+ +9,75 i
Митро соединения MiNO г =0 667^A1Z ,+ 3 64 i
эти величины в формулу (222) находим, что 8 = 2.17, что на порядок ниже экспе-
риментальной величины, равной 20,1. Такие же расчеты проведены и для ряда
других жидкостей и при этом результат аналогичен предыдущему: во всех слу-
чаях расчетная величина диэлектрической проницаемости существенно ниже
экспериментальной. Таким образом ,даже если бы была возможность рассчи-
тать плотность жидкости с хорошей точностью, это не привело бы к правильным
значениям диэлектрической проницаемости, вычисляемым по формуле (222).
266
Глава IX
Можно было предположить, что величина А/?, для одной и той же поляр-
ной группы, содержащейся в полимерах и низкомолекулярных жидкостях,
должна быть разной. Проведенные расчеты показывают, что это справедливо
не только при сравнении поведения органической жидкости и полимера, но и
при сравнении самих жидкостей, принадлежащих к одному и тому же классу.
Так, например, вклад в величину АЛ, от ОН -группы не является одинаковым
в ряду спиртов, а зависит от химического строения спирта. Во всех случаях
для жидкостей, принадлежащих к одному и тому же классу, вклад полярной
группы в величину А/?, возрастает с увеличением Ван-дер-Ваальсового объе-
ма жидкости. Такой анализ проделан на основе формулы (223), в которую
подставлялось усредненное значение коэффициента молекулярной упаковки
кср для жидкостей различных классов, а величины ^R, рассчитывались по
таблицам, приведенным в работах [28] и [128]. 1
При этом использовались экспериментальные значения диэлектрической
проницаемости £ и по формуле (223) вычислялись значения Л/?, для каждой
полярной группы. Всего было проанализировано 11 классов органических
жидкостей, которые приведены в табл.34,б. При этом для хлорированных со-
единений учитывалось влияние атома хлора на поляризацию, для спиртов -
группы ОН, для кислот - группы СООН и т. д.
На рисунке 74' показана зависимость /\ROHm Ван-дер-Ваальсового объема
молекулы спирта. Видно, что все то чки хорошо укладываются на единую кри-
вую, которая может быть аппроксимирована с помощью соотношения
Рис.74'. Зависимость поправки AR к поляризуемости от Ван-дер-Ваальсового объема
дляОН-группы
1
Dependence of correction A/? to polarizability on Van-der-Waalz volume ^2 Al',
for OH-group ,
4
Диэлектрическая проницаемость
267
МД>Н = -1,708- 1(Г3(£Д1;)2+(), 761£ Д1< +0,343.
/ /
Аналогичным образом получены соотношения для расчета величин А/?,
всех остальных классов жидкостей. Эти соотношения приведены в табл.34,6.
Зная их, легко рассчитать вклад каждой полярной группы в величину Л/?,.
Такие расчеты проделаны для большого количества органических жидкостей
(табл.35). Расчеты проводились с помощью соотношений, приведенных в
табл.34б; Ван-дер-Ваальсовые объемы и молярные рефракции определялись
по обычной методике [28, 128] .Проведенные расчеты показали достаточно
хорошее совпадение с экспериментальными величинами £, которое ранее не
достигалось другими способами. Таким образом, имеется возможность рас-
чета диэлектрической проницаемости полимеров и их растворителей, кото-
рый проводится на основании химического строения повторяющегося звена
полимера или молекулы органической жидкости.
Таблица 35
Значения Ван-дер-Ваальсового объема, поляризуемости, расчетных
н экспериментальных величин диэлектрической проницаемости
для ряда органических жидкостей
Values of the Van-der-Waals volume, polaryzability, calculated and experimental quantities of
d ielectric constant for a series of organic liquids
Жидкости XAI', А3 Р, см3/моль £ расч. £ эксп. Ошибка, %
1 2 3 4 5 6
и-Пентан 97,40 25,29 1,932 1,844 4,80
Изопентан 97,40 25,29 1,932 1,843 4,80
н-Гексан 114,00 29,31 1,945 1,890 2,90
Циклогексан 102,00 27,71 1,990 2,023 -1,60
н-Гептан 132,00 34,53 1,941 1,924 0,90
н-Октан 149,00 39,14 1,947 1,948 -0,05
н-Нонан 166,00 43,76 1,951 1,972 -1,20
н-Декан 183.00 48,38 1,950 1,991 -2,10
Бензол 88,24 26,31 2,261 2.284 -1,00
Толуол 105,00 30,93 2,240 2,379 (25°) -5,80
о-Ксилол 121,00 35,54 2,568 2,568 0,00
м-Кснлол 121,00 35,54 2,374 2,374 0,00
п-Ксилол 121,00 35,54 2,270 2,270 Q00
Этилбензол 122,00 35,54 2,220 2,412 -8,00
Нафталин 1 34,00 41,65 2,339 2,540 (85°) -7,90
И зопропилбе изол 139,00 40,16 2,207 2-380 -7,30
Стирол 118,00 35,08 2,256 2,430 (25°) -7,20
Хлорбензол 102,00 35,07 4,160 5,621 -26,00
Бромбензол 109,00 34,07 5,400 5,400 0,00
Этил б ром ид 68,02 19,20 9,380 9,390 -0,10
268
Глава IX
таблица 35 (продолжение)
1 2 3 4 5 6
1,2-Дибромэтан 89,83 26,97 4.780 4,780 0,00
Метилиодид 64,68 19,62 7,000 7,000 0,00
Этилиодид 81,75 24,24 7,820 7,820 0,00
Циклогексанол 110,00 32,79 14,790 15,000 -1,40
Метанол 36,51 11,90 32,660 32,630 -0,70
Этанол 53,58 16,52 26,610 24,300 8,80
н-Пропанол 70,65 21,14 21,440 20,100 6,01
н-Бутанол 87,72 25,75 17,580 17,100 2,28
Изобутанол 87,72 25,75 17,580 17,700 -1,10
н-Пентанол 105,00 30,37 14,630 13,900 4,80
н-Гексанол 122,00 34,99 12,500 13,300 -6,40
п-Октанол 150,00 44,23 10,450 10,340 0,80
Уксусная кислота 55,16 17,24 6,240 6,150 0,70
Масляная кислота 89,29 26,47 2,890 2,970 -2,60
Изовалериановая кислота 106,00 31,09 2,650 2,640 0,20
н-Валериановая кислота 106,00 31,09 2,650 2,660 -0,60
Метилформиат 57,29 21,82 9,640 8,500 13,10
Этил формиат 74,36 26,44 7,170 7,160 -0,10
Метилацетат 74,36 26,44 7,170 6,680 7,10
Этилацетат 91,43 31,05 6,090 6,020 1,00
н-Пропилацетат 109,00 35,67 5,450 5,690 -4,30
Этилпропионат 109,00 35,67 5,450 5,650 -3,60
н- Бутилацетат 126,00 40,29 5,080 5,010 1,20
Ацетон 64,84 21,44 22,550 20,700 8,90
Метилэтил кетон 81,91 26,05 18,920 18,510 2,20
Диэтилкетон 98,98 30,67 17,070 17,000 0,40
Циклогексанон 104,00 33,09 17,930 18,300 -2,00
Диэтиловый простой эфир 89,86 25,87 4,400 4,335 1,50
Дипропиловый простой эфир 124,00 - 3,410 3,390 (26°) 0,60
Диизопропиловый простой эфир 124,00 35,11 4,880 3,880 25,80
Дибутиловый простой эфир 158,00 - 3,030 3,060 (25°) -1,00
Диамиловый простой эфир 192,00 - 2,810 2,770 (25°) 1,40
Диизоамиловый простой эфир 192,00 - 2,810 2,820 0,30
Бутилэтиловый простой эфир 124,00 - 3,410 3,060 (25°) 11,40
Этаналь 47,77 23,410 21,100 10,90
Пропаналь 64,84 — 18,000 18,500 2,70
Бутаналь 81,91 - 14,720 13,400 9,80
Диэлектрическая проницаемость
269
таблица 35 (окончание)
1 2 3 4 5 6
Нитрометан 46,19 — 39,050 35,870 (30°) 8,90
Нитроэтан 63.26 — 30,540 28,060 (30°) 8,80
1-Нитропропан 80,33 — 27,080 23,240 (30°) 16 50
2-Нитропропан 80,33 — 26,760 25,520 (30°) 4 90
Нитрил уксусной кислоты 48,96 16,72 40,780 37,500 8,70
Нитрил пропионовой кислоты 66,03 21,34 25,240 27,200 -720
Нитрил бутановой кислоты 83,09 25,95 20,450 20,300 0,70
11итрил пентановой кислоты 100,00 30,57 18,110 17,400 4,10
Нитрил изогептановой кислоты 117,00 35,19 16 ,730 15 500 7 90
Глава X
РАВНОВЕСНЫЙ МОДУЛЬ
ВЫСОКОЭЛАСТИЧНОСТИ СЕТЧАТЫХ
ПОЛИМЕРОВ
Х.1. Расчетный способ оценки равновесного модуля
Для оценки равновесного модуля высокоэластичности Ех и молекуляр-
ной массы межузлового фрагмента Мс в случае сетчатых эластомеров с дос-
таточно редкими сшивками пользуются известным уравнением классичес-
кой теории высокоэластичности:
(224)
где р - плотность сшитого эластомера; R - универсальная газовая постоян-
ная; Т- абсолютная температура.
Применение уравнения (224) к частым сеткам,межузловой фрагмент ко-
торых содержит очень малое количество звеньев, вплоть до 1 и даже меньше,
приводит к существенным расхождениям экспериментальных и расчетных
знаний Е„.
Для того, чтобы уравнение (224) работало при описании свойств частых
сеток, в него вводят так называемый фронт-фактор Ф;
Однако введение непредсказуемого фронт-фактора в уравнение (224) не
изменяет ситуацию в лучшую сторон); поскольку, сопоставляя расчетные и
экспериментальные значения Е^,, можно лишь оценить этот фронт-фактор. В
связи с этим в работе [31] сделана попытка получения обобщенного соотно-
шения для оценки Е„, и Мс, действующего как для редких, так и для частых
сеток.
Равновесный модуль высокоэластичности сетчатых полимеров 271
Проведем в деталях анализ влияния большого количества узлов сетки на
равновесный модуль высокоэластичности. Предварительно заметим, что для
редких сеток Ван-дер-Ваальсовый объем узлов несоизмеримо меньше, чем
Ван-дер-Ваальсовый объем линейных межузловых фрагментов, и поэтому при
оценке сжимаемости сетчатой системы им можно пренебречь. В случае же
частых сеток этого сделать нельзя, т.к. суммарный Ван-дер-Ваальсовый объем
узлов примерно одинаков с суммарным Ван-дер-Ваальсовым объемом линей-
ных фрагментов и даже может превосходить его.
Рассматривая сетчатый эластомер как систему, состоящую из двух подси-
стем-упругой и поворотно-изомерной, проанализируем сначала последнюю.
В работе [28] было показано, что для определения коэффициента упругости
поворотно-изомерной подсистемы необходимо знать разность энергий пово-
ротных изомеров, которая следующим образом зависит от размеров “молеку-
лярных дефектов” if в полимере:
АА, =4О,<72(/^)2. (226)
где D, - усредненная энергия межмолекулярного взаимодействия, характер-
ная для атома данного типа; а - константа потенциала Морзе:
<р(Я) = /.;[(<< д(R-1)2 -1], (227)
где D - глубина потенциальной ямы; г0 - равновесное расстояние между ато-
мами в гармоническом приближении.
Для определения размера молекулярного дефекта if рассмотрим “ци-
линдр взаимодействия” (рис.75), понятие о котором было введено в работе
Рис.75. “Цилиндр взаимодействия” двух атомов водорода (см. текст)
“Cylinder of interaction” of two hydrogen atoms (See text)
272
Глава X
[91]. Его объем складывается из объемов взаимодействующих атомов ДЕ, и
собственно объема дефекта, характеризуемого о, ДЕ,
Ецк =2Д^+5* ДЕ, (228)
Еца =2ДЕ;+8,аД1< , (229)
где ДЕ, - Ван-дер-Ваальсовый объем /-го атома; величины 5,к и 8а , входя-
щие в соотношения (228) и (229), рассмотрении детально в работе [31]; при
этом Ец - объем цилиндра взаимодействия для кристаллического полимера,
Ец - то же, для аморфного полимера.
Отсюда величина дефекта равна
J/a _[/К . лК' дт/.
id = _« _ц_ = ——(230)
' 2S 2 S ’
где ДЕ, - часть Ван-дер-Ваальсового объема /-го атома, которая перекрывает-
ся Ван-дер-Ваальсовым объемом атома, химически с ним связанного , число
2 в знаменателе появляется вследствие того, что величина амплитуды пере-
скока /-го атома равна половине размера дефекта; - площадь цилиндра
взаимодействия.
Оценим величину ld. Для этого рассмотрим предельный случай, когда
радиус основания цилиндра взаимодействия равен Ван-дер-Ваальсовому объе-
му /-го атома. Тогда
//=(8,а-5,к)|я,. (231)
Для углеводородных полимеров /?, = RH ; размер дефекта - постоянная
величина, равная if =O,O53RH, где RH - Ван-дер-Ваальсовый радиус атома
водорода, равный 1,17 А. Подставив полученное значение if в выражение
(226), найдем величину вклада атома водорода в разность энергий поворот-
ных изомеров ДЕ/у = 4,56 кДж/моль. Найденное значение по порядку величи-
ны соответствует спектроскопическим данным относительно величины раз-
ности энергий поворотных изомеров.
Для дальнейшего анализа перепишем соотношение (226) в виде
Д/f, =4£),д2А2(-^-)2 . (232)
' ' ' R, '
где Rj - Ван-дер-Ваальсовый радиус /-го атома; if - размер дефекта, образу-
емого данным атомом.
Равновесный модуль высокоэластичности сетчатых полимеров 273
В работе [28] была сделана оценка величины аР„ причем оказалось,
что aR,~ 6.
Для того, чтобы оценить вклад поворотно-изомерной подсистемы в тем-
пературную зависимость модуля упругости полимера в переходной области и
в области высокоэластического состояния, необходимо найти величины if
для различных атомов и типов межмолекулярного взаимодействия.
Как показали проведенные расчеты [28], температуры плавления поли-
меров Тт, энергия межмолекулярного взаимодействия £)„ которая входит в
выражение (232), для атома данного типа зависит от того, входит ли он в
состав группы атомов, образующих водородную связь или диполь-диполь-
ное взаимодействие. Поэтому при расчете АЕ, по формуле (232) влияние ука-
занных типов специфического взаимодействия будет проявляться в основ-
ном через энергию межмолекулярного взаимодействия В, атома данного типа,
а инкременты 8* и 8* , как показывают расчеты, не зависят от влияния водо-
родных связей и диполь-дипольного взаимодействия.
В случае, когда повторяющееся звено состоит из набора атомов разного
типа, введем понятие эффективной разности энергии ДЕ.ф поворотных изо-
меров, которую можно найти следующим образом. Используем соотношение,
полученное в работе [28], для оценки модуля упругости аморфных полимеров
IX
Е = —------,
уА'7Л
; к,/,
(233)
где АГ, - Ван-дер-Ваальсовый объем /-го атома повторяющегося звена; S, -
Ван-дер-Ваальсовая поверхность /-го атома, через которую происходит меж-
молекулярное взаимодействие ; к, - коэффициент упругости связи /-го атома;
/, - характерный размер связи (см. рис.75).
Для высокоэластического состояния знаменатель выражения (233) равен
VW>
7 .---—, где кп - коэффициент упругости поворотно-изомернои подсисте-
I I
мы. Величина кг„ согласно работе [28],
(234)
RT
,EEix
где g, = exp(——-); ДЕ, - разность энергий поворотных изомеров. С учетом
RT
соотношения (234) знаменатель уравнения (233) примет вид
274
Глава X
(235)
RTl, ' '
Введем эффективную величин}’ </.м|, таким образом, чтобы она определя-
лась из условия
//<Л2 о ^эф) 9эф‘^эфХЛ^
дК/ =-------------‘. (236)
1 '/ Ьф
Так как 13ф, согласно соотношению (226), вычисляется по форм}1 ле
(Ф2
АЕэф
4ОэфЯ2
где Лэф = aRTm, а ^эф = (S) и /эф = (/), то условие (236) можно записать сле-
дующим образом:
т=тт
А^эф'/эф у
4аМта2{Г) Г
(237)
Левая часть выражения (237) известна, и в правой части (S) и (/) также
известны. Поэтому задача сводится к поиску решения уравнения типа Ь =
где х = AE3^/RTm, а b включает все известные члены уравнения (237). На ос-
новании этого можно определить, как влияют атомы различного типа на ДЕ,ф.
Если в качестве исходного, наиболее типичного атома, входящего в поли-
меры, взять атом водорода и соответствующую ему величину ХЕН, то появле-
ние в структуре повторяющегося звена атомов другого типа с ДЕ, приведет к
изменению ДЕ.ф, причем это изменение зависит от того ,с каким весовым
коэффициентом входит этот атом в повторяющееся звено. Величина ДЕэф бу-
дет либо возрастать, либо уменьшаться: ДЕ,ф > КЕН, если ДЕ) > А/:}/, и А/'^ф <
&ЕН, если ДЕ’, < ДЕ//.
Рассмотрим теперь, как влияет степень сшивки на модуль упругости сет-
чатого полимера в высокоэластическом состоянии. Представляя сетчатый по-
лимер как смесь линейных фрагментов и узлов, запишем
1/Ее = у/Ел + (1-у)/Еу
ИЛИ г
Ее=-------а-—, (238)
У + (1- 7) J
Еу
Равновесный модуль высокоэластичности сетчатых полимеров 275
где £л и Еу - модули для линейных фрагментов и узлов; у - концентрация
линейных фрагментов.
Поскольку формула (238) получена путем суммирования сжимаемо-
стей линейных фрагментов полимерной цепи и узлов, соответственно
у = (^ДЦ)Л/^Д1/ , где (^ДГ/)Л -Ван-дер-Ваальсовый объем линейного
1 у /
фрагмента, ^Д^ -Ван-дер-Ваальсовый объем повторяющегося фрагмента
i
сетчатого полимера. Что касается понятия узла сетки с рассматриваемых по-
зиций, то оно было дано выше: узлом сетки является группа атомов, включая
атом, от которого происходит разветвление, плюс соседние, химически свя-
занные с ним атомы со своими ближайшими заместителями.
(ЕА!<)л
Величина —--------может быть записана в виде
i
(^Д1<)л н(ЕАЦ)“
—L----— =---------I------------ (239)
‘ i i
где (^А^ )л - Ван-дер-Ваальсовый объем повторяющегося звена линейно-
i
го фрагмента; п - число повторяющихся звеньев, приходящихся на один узел.
Вводя обозначения £ = ЕЛ!ЕУ и Р = (^Д^)у А^)л > гдс Ал - модуль
упругости линейного полимера; Еу - модуль упругости сетчатого полимера,
состоящего из одних узлов; (Z Д^ )у ~ Ван-дер-Ваатьсовый объем узла сет-
i
ки, получаем следующее соотношение' .у ~-п 1(п + Р) и выражение (238) пре-
образуется к виду.
Е
—3- = у + (1-у)£ =
Ес
(240)
П , ре
п + Р п + Р
Так как полимер, построенный из одних узлов, имеет модуль, на много
десятичных порядков превышающий модуль линейного эластомера, то 1
276
Глава X
и для частых сеток с << 0. Поэтому вторым членом в выражении (240) мож-
но пренебречь. Отсюда получаем, что
Е„ _ "
Ес « + ₽'
(241)
Вернемся теперь к эффективным значениям /эф, <?эф: Дф и Л’эф. Учитывая,
что коэффициент упругости поворотно-изомерной подсистемы пропорциона-
лен среднестатистическому числу п звеньев, заключенных в линейных фраг-
ментах между узлами, можно записать соотношение (233) в виде
l^RT
Ея=-^---------, (242)
(А)ф ) <7 эф ^эф И
а для случая л0 = 1 и температуры Ту поворотно-изомерный модуль такого
гипотетического полимера равен
(£л)0 =
1^0
,,d ,2 0 е
1‘эф/ 7эф'Чф
(243)
Взяв отношение этих модулей, получаем
Ея _ Т А ,
(^л)о <?эф/7
ДТГздф о ^^эф
где <7эф =ехр(—г-) и <?эф = ехр(——).
Подставив данное выражение в соотношение (241), имеем
(244)
Ez и + Р 'Г <7эф
С£д)о п2 2q <уэф
(245)
Для практических расчетов необходимо учитывать, что повторяющийся
фрагмент сетки состоит из нФ/2 повторяющихся звеньев, и поэтому выраже-
ние (245) с учетом функциональности Ф необходимо записать в виде
Ес _2(/7 + р)7?эф
(Ея)0 фц2
(246)
Равновесный модуль высокоэластичности сетчатых полимеров 277
Проанализируем более детально уравнение (246), сопоставив его с урав-
нением (224) классической теории высокоэластичности. Легко видеть, что
эти уравнения аналогичны, причем уравнение (246) описывает модуль как
для редких, так и для частых сеток. Действительно, записав, что п - Мс / А/о,
из уравнения(246) получаем
_ 2 (А/С/Мо+Р) Т <?эф
ф (МСШО)2 то <Ьф
(247)
гдс.'Ц) - молекулярная масса повторяющегося звена.
Для редких сеток Р « Мс/М0, и тогда
Ес=(Еп)0
2 Т <?3°ф
ф (?эф ’
(248)
Сопоставляя формулы (224) и (248), находим, что
ЗрЯФГо^ф
(^л'о — п
2М о7эф
(249)
Если измерения модуля проводить при Т = 70 , то = <"Дф и уравнение
(246) упрощается, принимая вид
£с=(£л)о^т2. <25°)
Фи
При наличии больших фрагментов цепей между узлами (редкие сетки)
соблюдается условие Р « и, и уравнение (250) переходит в соотношение
7
£с=(^л)0 —
Фи
(251)
и, с учетом соотношений Tq /Т = д^ф / дэф и М$п =МС , получаем
(252)
т.е. соотношение (252) полностью совпадает с уравнением (224).
Соотношение (250) удобно для практических расчетов модуля частых се-
ток, причем величина (ЕДо может быть рассчитана либо по уравнению (249),
278
Глава X
либо определена на основе экспериментальных данных по равновесному мо-
дулю для редких сеток, когда п » 1.
Проделаем еще ряд преобразований. При Т = То , <7эф = <7эф выражение
(249) принимает вид Зр/?ФТ (£л)о - (253) 2Л/0
Тогда из соотношений (253) и (250) имеем
_ЗрЛТ(и + Р) м$п
Учитывая, что Мс = Mq-h, из (254) получаем
£c_W<^P> (255) Мсп
Разделим числитель и знаменатель формулы (255) на п:
ЗрЯТ(1 Д) (256) мс
ИЛИ ЗрЯ7’(1 Д) А/с = (257) £с
Для редкой сетки р « 1 и п » 1, поэтому 0 и обобщенное уравне-
ние (256) трансформируется в обычноеуравнение (224).
Для оценки величины Л/с в случае частых сеток можно воспользоваться
формулой (257). Предварительно для удобства ее можно преобразовать с уче-
том того, что и = MJMq.
ЗрЯГ(1 + ~°Р) К = (258) мс
Тогда из (258) имеем
Мс = . Зр77? + ^/Зр/?7'(Зр7?7' + 4£’сЛ'70Р) « г-.
Равновесный модуль высокоэластичности сетчатых полимеров 279
Для редких сеток 0 -> 0 и формула (259) переходит в уравнение (224).
В качестве примера рассмотрим модельные сетки на основе полидиме-
тилсилоксана. исследованные в работе [188]. Строение сетки имеет вид
I
О
СНз Г о I СНз
I I I
(О-81)т+1фО—Si- О-{-(Si—О)т+1
I । I । I
сн3 J СНз
(НзС- Si—СНз)т+1
О
I
Узел сетки заключен в пространство, обведенное пунктирными линия-
ми. Для такой сетки
(XAIOy =AI/S,J75 +4АСол35 =34,7 А3;
(^ДЦ)Л = m(ACai72 +2А1/С106 +4АИОдз5 + 6ДКуд24) =72,1 А3;
₽ = 34,7 /72,1 =0,481.
При расчете равновесного модуля высокоэластичности частых сеток по
уравнению (250), а также при расчете температуры стеклования Tg по форму-
ле (109) необходимо учитывать все детали химического строения сетки. Дело
в том, что когда т - 0 структура данной сетки приобретает вид
I
О
I
Н3С-Si-СНз :
сн3! о ;сн^ о
' । । । । ।
—-O+Si-H-O-Sr-OfSi-iO-Si-O—
] I । I । I । I
______!сн3|___ о___! сщ___о
Н3С-Si-СНз :
,----1--!
I1 о !
I I I
280
Глава Л'
СНз
В этом случае только часть звена —St—О— остается в качестве линейного
СН3
фрагмента между соседними узлами сетки, т.к. атом кислорода этого звена
входит в состав соседнего узла. Определив п как (^ДК, )* /(^Д1, где
СНз
(^2 Д^)л Ван-дер-Ваальсовый объем —SH группы, получим п = 0,99. Если
' СНз
т = 1, то п = 1,99 и тд. Это особенно важно учитывать при расчете температу-
ры стеклования 7’g, которая вычисляется по уравнению (109). В данном слу-
чае уравнение (109) трансформируется в формулу'
(£Д^у+/<ГЛИ,^+(£Д<л
т =-----'---------L————•------------ (260)
/ / /
где (Ея/Д^)л и - наборы инкрементов соответственно для стру ic-
СНз СН3
I ।
тур —Si—О—и—Si— (заметим, что в случае рассматриваемой сетки = 0),
СНз СНз '
Для расчета равновесного модуля высокоэластичности £с по уравнению
(250) необходимо сначала оценить величину (£'л)(). Как было отмечено выше,
это можно сделать двумя способами. Первый способ заключается в использо-
вании формулы (253), т.е. величина (£л)о определяется чисто расчетным пу-
тем. Подставляя в эту формулу р = 1,169 г/см3, Ф = 4,Л-70 = 74,15, 71 = 293 К,
находим, что (/'.’До = 230 МПа.
Второй способ заключается в использовании формулы (250) и экспери-
ментального значения £’с для редкой сетки. Если использовать эксперимен-
тальное значение Ес, полученное в работе [188], то на основании (250) нахо-
дим, что (/у,)о - 197 МПа. Расчеты £с по формуле (250) с использованием
этого значения (£л)о приводят к величинам равновесных модулей, приведен-
ным в табл.36. Видно, что приуменьшении расстояния между соседними уз-
лами сетки величина £с быстро возрастает и для очень частой сетки (и = 1)
Равновесный модуль высокоэластичности сетчатых полимеров 281
достигает больших значений. При этом температура стеклования остается
ниже комнатной.
Таблица 36
Величины Р, Ес и Tg для 11озшдимет11лс11локсановь1х сеток
Values of р, Ес and J' for polydimethylsiloxane networks
т Z7 Р £'с , МПа у;, к
0 0,99 0,482 148,0 183
I 1,99 0,241 61,5 166
2 2,99 0,160 38,2 161
3 3,99 0,120 27,6 1.58
4 4,99 0,096 21,6 156
6 6,99 0,069 15 0 154
9 9,99 0,048 10,3 153
X.2. Разномодульные и градиентные полимеры
В заключение данной главы рассмотрим применение расчетных схем,
позволяющих оценивать температуру стеклования Tg и равновесный модуль
высокоэластичности для создания полимерных материа нов с необычными
свойствами. Речь пойдет о получении упругих полимерных материалов двух
типов: 1) разномодульных, у которых модуль упругости является постоянным
для каждого образца, но меняется в очень широких пределах при переходе от
одного образца к другому ,2) градиентных ,у которых моду ль упругости плав -
но меняется в пределах одного и того же образца в данном направлении и при
этом в материалах отсутствуют какие-либо слои или границы раздела. В пос-
леднем случае необходимо получить плавный переход от резины к пластмассе
или наоборот в объеме одного и того же материала, что позволяет создать
сложные разномодульныс конструкции без применения традиционных мето-
дов соединения - склейки, сварки и т.д.
Сложность реализации этой идеи связана с двумя основополагающими
особенностями поведения полимерных материалов, которые требуют разъяс-
нения.
На рис.76 схематически изображена температурная зависимость модуля
упругости для типичного полимера.в области стеклообразного состояния мо-
дуль упругости, к сожалению, мало зависит от химического строения полиме-
ра и колеблется в пределах (2-3) 103 МПа. В этом заключается первая труд-
ность получения разномодульных и градиентных материалов .
В области перехода из стеклообразного состояния в высокоэластическое
модуль упругости резко падает в узком интервале температур (ДТ = 20-30°),
умсньштясь на несколько десятичных порядков (см рис 76) В связи с этим
на первый взгляд может показаться, что задача получения полимеров с раз-
личными модулями упругости лежащими ,например в интервале от 3103 МПа
282
Глава X
Рис. 76. Схематическое изображение зависимости модуля упругости/? от температуры Т
Schematic representation of dependence of elastic modulus E on temperature T
до 3 МПа, является простой: для этого нужно получить полимеры, у которых
температура стеклования Tg близка к комнатной (если разномодульные мате-
риалы должны работать при комнатной температуре). Однако, как хорошо
известно, материалы в переходной зоне обнаруживают ярко выраженное вяз-
ко-упругое поведение, и кроме того, их механические свойства резко меня-
ются при очень небольшом изменении температуры как в сторону ее пониже-
ния (переход к пластмассе), так и в сторону ее повышения (переход к резине).
В этом заключается вторая трудность получения разномодульных материа-
лов, которые наряду с широким интервалом изменения модуля упругости
должны обладать упругими, а не вязкоупругими свойствами, а кроме того,
они должны сохранять заданный градиент свойств в широком интервале тем-
ператур.
Теоретически широкого диапазона модуля упругости без применения ка-
ких-либо пластификаторов или наполнителей можно достичь, создавая час-
тосетчатые стру ктуры, содержащие объемистые жесткие у злы, связанныегиб-
кими линейными цепями регулируемой длины (рис.77).
Это вытекает из обобщенного уравнения (256) для оценки равновесного
модуля высокоэластичности Е,г для сетчатых систем.
Согласно уравнению (256), большого значения Ею можно достичь, пере-
ходя к частым сеткам с объемистыми узлами сшивки, когда и =1, а величина
b > 1. При этом, чтобы сохранить низкую температуру стеклования Tg, линей-
ные фрагменты между узлами должны быть очень гибкими. В качестве струк-
тур, содержащих жесткие объемистые узлы, связанные межузловыми гибки-
Равновесный модуль высокоэластичности сетчатых полимеров 283
Рис.77. Схематическое изображение структуры сетки, содержащей объемистые узлы
сшивки и короткие и гибкие цепочки в качестве межузловых фрагментов
Schematic representation of network consisting of balky crosslinked points and
short flexible chains as intercosslinked fragments
ми цепочками (- R -), были синтезированы [45,46] сетчатые полиизоцианура-
ты, химическое строение которых показано на стр. 251.
Роль узла (отмечен пунктиром) выполняет изоциануратный цикл, имею-
щий функциональность, равную трем.
В качестве межузловых фрагментов были использованы короткие крем-
нийорганичсские цепочки, строение которых показано на стр. 251.
В другом варианте получали сетчатые сополимерные полиизоциану-
раты [127].
Обшим принципом синтеза данных материалов является то ,что исполь-
зуя в качестве одного из исходных компонентов олигомерные диолы, взаимо-
действием их с диизоцианатом, в частности, с 2,4-толуилендиизоцианатом
по реакции уретанообразования получают сначала макродиизоцианаты:
СНз СНз
0=C=N-O О О 0-N=C=0
\ II II /
NH-C-O-R-O-C-NH
В качестве диолов можно использовать промышленные олигомерные
каучуки с концевыми гидроксильными группам, в частности, полиэфир
ПФ-ОП-15, представляющий собой олигомерный сополимер тетрагидрофу-
рана и окиси пропилена:
R-. {-[О-(СН2)4]т-(О-СН2-СН)п-}р
СН3
nin =19,7/1 7; р=12
284
I'лава Л'
Полученные из олигомерных каучуков макродиизоцианаты образуют сет-
чатые полимеры по реакции полициклотримеризации in situ, а также сополи-
меризуются с диизоцианатами любого химического строения (ароматически-
ми, алициклическими или алифатическими), способными с достаточной ско-
ростью образовывать сетчатые полиизоцианураты. Реально в этом случае
образуется смешанная сополимерная структура. Полимер одновременно мо-
жет содержать сетку, построенную из продуктов гомополициклотримсриза-
ции олигомера и диизоцианата, и сетку, являющуюся результатом взаимодей-
ствия олигомера и диизоцианата с образованием структур с произвольным
расположением связывающих цепочек.
С увеличением избытка диизоцианата по отношению к олигомерному
макродиизоцианату в исходной реакционной смеси возрастает содержание
жестких циклических структур в образующейся сетке. Такимоб разом, можно
по желанию изменить соотношение гибких (полиэфирных) и жестких (изоци-
ануратных циклов с примыкающими к ним циклическими ядрами диизоциа-
ната) фрагментов и этим регулировать в широких пределах механические свой-
ства. Для проверки действительной возможности такого регулирования свойств
сетчатых полимеров предварительно был проведен расчет величины 7’g ис-
следуемых сетчатых систем в зависимости от их химического состава с ис-
пользованием уравнения (250). Из рис.78 видно плавное уменьшение вели-
чины Tg с повышением доли гибкого каучукового фрагмента.
Проведенные расчеты равновесного модуля упругости по уравнению (250)
показали 1127|, что при низкой Tg (ниже комнатной) равновесный модуль Еа,
может принимать высокие значения, промежуточные между' величинами мо-
дулей для резины и пластмассы. Такие предварительные расчеты были прове-
дены для сеток с кремнийорганическими межузловыми фрагментами [45]. В
результате для рассматриваемой структуры получено выражением, связыва-
ющее Tg сетки с количеством диметилсилоксановых звеньев и в линейном
фрагменте:
751 + 108,15»
2450 + 721,5» •
(261)
Расчеты, проведенные по выражению (261), сведены в табл. 37, из кото-
рой видно, что температура стеклования лежит ниже комнатной и убывает по
мере роста величины п, приближаясь к температуре стеклования полидиме-
тилсилоксана при п = 44.
В табл.37 приведены также величины равновесных модулей, которые
принимают самые разнообразные значения в интервале от 3 до 870 МПа в
зависимости от величины и.
Проведенный синтез сетчатых полимеров с рассмотренной выше струк-
турой подтвердил правильность сделанных предположений и расчетов [45,
Равновесный модуль высокоэластичности сетчатых полимеров 285
Рис.78. Зависимость расчетной температуры стеклования Tg полиизопиануратных
полимеров на основе диизопианатов с различной структурой R в зависимости от
мольной доли а кау чука ПФ-ОГ1-15:
’) R: ЕО-сн2-О-]; 2)R: [-О-];
3) R: [-<н)-сн2-(н)-]; 4) R: f-(CH2)6-].
Dependence о!'calculated glass transition temperature of poly isocy anurate polymers
on the basis of diisocyanates with different structure R as dictated by mole fraction a of
rubber PP-OP-15
Таблица 37
Значения расчетных температур стекловании Tg
и равновесных модулей упругости Еотвержденных макродиизоцианатов
в зависимости от количества диметилсилоксановых звеньев
Values of calculated glass transition temperatures Tg and equilibrium modulus of elasticity E of
cured macrodiisocyanate depending on the number of dimethylsiloxane repeating inits
/7 , °C ft расч, МПй ^-эксп МПа
0 33 -
1 -2 884 886
2 -5 249 283
3 -41 123 149
6 -67 40 4.3
9 -80 22 24
19 -99 8 7
44 -112 3 3
286
Глава X
Рис.79. Логарифмическая зависимость модуля упругости Е от числа диметилсилокса-
новых единиц п в межузловом фрагменте
Logarithmic dependence of modulus of elasticity E on a number of dimethylsiloxane
units n in intercrosslinked fragment
46]. Так, например, £,, полученных сеток с кремнийорганическими межузло-
выми фрагментами изменяется в пределах от 3 до 3 - К)3 МПа (рис.79) в зави-
симости от дайны межузлового фрагмента.
Для таких материалов очень важным является процесс микрофазо-
вого расслоения, связанный с существенно различной поверхностной энер-
гией кремнийорганических цепочек (21 дин/см) и изоциануратных узлов
(35 дин/см). Процесс микрофазового расслоения был подтвержден методами
рентгеновской фотоэлектронной спектроскопии, электронной микроскопии
и динамического механического анализа.
Вообще говоря, процесс микрофазового расслоения может происходить
как в линейных блок-сополимерах или смесях полимеров, так и в сетках.
В этих случаях на температурной зависимости тангенса угла механичес-
ких потерь tg 3 наблюдается два отчетливо выраженных максимума.
На рис. 80 показана температурная зависимость тангенса угла механи-
ческих потерь tg 3 для блочных образцов рассматриваемых полиизоцианура-
тов с различной дайной межузловых кремнийорганических фрагментов. Низ-
котемпературный пик смещается в сторону низких температур при увеличе-
нии длины межузлового фрагмента, приближаясь к Tg полидиметилсилоксана.
Высокотемпературный максимум, связанный с расстекловыванием системы
в целом, практически не зависит от величины л, начиная с п = 2.
Таким образом, имеются достаточно отчетливо выраженные две темпе-
ратуры перехода, одну' из которых можно отнести к температуре расстекло-
Равновесный модуль высокоэластичности сетчатых полимеров 287
tg 8
Рис.80. Температурная зависимость тангенса угла механических потерь rgS ддя блочных
образцов полиизоциапуратных сеток с различной длиной межу зловых кремнийоргани-
ческих фрагментов и : 1 — 1,2; 2 — 2,1; 3 — 3,2; 4 - 5,4; 5 - 9,2; 6-22
Temperature dependence of the mechanical loss-factor zgS for bulky specimens of
polyisocyanurate networks with different length of intercrosslinked organosilicon frag-
mentsw: 1 - 1,2; 2-2,1; 3 - 3,2; 4-5,4; 5 - 9,2; 6-22
вывания полидиметилсилоксановой микрофазы в полимерной сетке, а вто-
рую - к температуре расстекловывания микрофазы, содержащей изоциану-
ратные узлы с примыкающими к ним развязками. Тем самым необходимо
признать наличие микроф азового расслоения в сисе ле ,с вжанного с нссов -
местимостью полидиметилсилоксановых фрагментов с изоциануратными уз-
лами и примыкающими к ним развязками.
Учитывая, что температуры переходов в обеих микрофазах смещены на-
встречу друг другу, можно расчетным путем определить состав микрофаз,
исходя из условия, что температура перехода должна совпадать с температу-
рой стеклования микрофаз данного состава. Поскольку температура перехода
в микрофазе, построенной в основном из кремнийорганических фрагментов,
находится выше температуры стеклования полидиметилсилоксана, то очевидно,
что в полидиметилсилоксановой микрофазе содержатся соседние фрагмен-
ты.
Для ответа на вопрос, каково строение этих фрагментов в работе [45]
проведены расчеты температуры стеклования структур, показанных ниже.
288
Глава А'
СН3 СН3
Hill I J I ° | ° I I I I II
тмн-гсуо-г-сн2- сн2-т-о+сн2-[ sh obsi-снло+сн,- сн,- o+c+nh^-
и ' II ' | '' | <11 < < I || I ||
о СНз СНз °
Расчет температуры стеклования проводился по формуле (84), причем
осуществлялся поэтапный расчет Tg полидиметилсилоксанового фрагмента
с различным и и примыкающих к нему отрезков цепей, отмеченных штри-
ховыми линиями. Эти отрезки ‘наращивались’’ до тех пор, пока расчетная
температура стеклования нс начинала совпадать с экспериментально опреде-
ленной температурой первого перехода. В результате оказалось, что совпаде-
ние расчетной и экспериментальной температуры перехода наблюдается в том
случае, когда полидиметилсилоксановые домены содержат отрезки цепей, от-
меченные двумя штриховыми линиями, при этом структура этих отрезков нс
зависит от величины п, те. от длины крсмнийорганической цепочки. Такой
прием оценки состава микродоменов может быть распространен на любые
объекты, характеризующиеся микрофазовым расслоением.
Если в системе происходит микрофазовое расслоение, то при расчете рав-
новесного модуля высокоэластичности понятие “узел сетки”, с точки зрения
его химического строения, должно быть расширено. Действительно, наличие
жесткой микрофазы (например, рассмотренной выше, включающей изоциа-
нуратные узлы с примыкающими развязками) приводит к тому, что сама эта
микрофаза играет роль узла сетки. При этом нужно лишь убедиться ,что тем-
пература стеклования этой микрофазы лежит выше комнатной. Наличие та-
кого “макроузла” приводит к значительному повышению величины [) в урав-
нении (250), что способствует увеличению равновесного модуля высокоэлас-
тичности.
В работе [45] такой анализ был проделан для системы, рассмотренной
выше, в результате чего было найдено, что температура стеклования жестких
доменов составляет 33 °C. а их Ван-дер-Ваальсовый объем равен 751 А3.
Учитывая, что Ван-дер-Ваальсовый объем повторяющегося звена поли-
диметилсилоксана равен 180,15 А3,величина 3 = 751/180,15 -4,2.
Экспериментально равновесные модули высокоэластичности определя-
лись [46] с помощью измерений кривых релаксации напряжения, которые
аппроксимировались с привлечением физически обоснованных ядер релак-
сации (см. ниже).
Механическое поведение сетчатых полиизоциа ну ратов, содержащих крсм-
нийорганичсские межузловые фрагменты, уже было продемонстрировано
выше (см. рис.71). На рис.79 показана зависимость модуля упругости поли-
изоциануратных сеток от числа повторяющихся звеньев полидиметилсилок-
сановых цепей, связывающих узлы. Модуль упругости таких сеток перекры-
Равновесный модуль высокоэластичности сетчатых полимеров 289
вает диапазон модулей, характерных для переходной зоны из стеклообразно-
го состояния в высокоэластическое. Однако, несмотря на это, механическое
поведение данных материалов является не вязкоупругим, как для всех поли-
меров в переходной зоне, а упругим, характерным для полимерных стекол.
Доказательством этого служит ход кривых релаксации напряжения, показан-
ный на рис.71.
Обратим внимание еще раз на механическое поведение полии чоциану-
ратной сетки с линейными кремнийорганическими фрагментами с п = 6,2.
При таких размерах линейных цепей начальное напряжение о0, примерно
совпадает с о0 для вязкоупругого материала (7 5 МПа) ,но механическое пове-
дение коренным образом отличается от него: напряжение быстро релаксирует
на небольшую величин}' в начальный момент времени, но затем спад напря-
жения практически прекращается, т е. материал ведет себя как упругий стек-
лообразный полимер*.
Перейдем теперь к анализу свойств градиентных матер ш л> в, получен-
ных на основ? олигомерного каучукаПФ-ОП-15 и 2,4-то оу илендиизоцианата
Для получения градиентных материалов производится плавная дозиров-
ка исходных компонентов, чем достигается направленное регулирование хи-
мического состава сетки в пределах одного и того же образца.
Таким путем в работах 119, 129] были получены о фазцы, в которых мо-
дуль упругости меняется от 4,5 МПа (что характерно для резин) до 2000 МПа
(что характерно для пластиков). При этом такое изменение проходило плавно
в пределах одного и того же материала без всяких границ раздела и промежу-
точных слоев.
На рис .81 в качестве примеров показана зависимость модуля упругости
от содержания 2,4-толуилендиизоцианата в исходной реакционной смеси.
Хорошо видно, что по длине образца модуль упругости меняется линейно и
таким образом осуществляется плавный переход от резины к пластмассе без
всяких границ раздела, как это было отмечено выше.
Для анализа характера механического го ведения полученных материа-
лов определяли кривые релаксации напряжения для микрообразцов, выре-
занных из исходного макрообразца в различных точках градиента (рис.82),
на котором для сравнения приведена также кривая релаксации напряжения
для вязкоупругого материала - эпоксидной смолы ЭД-20, отвержденной ан-
гидридом полисебациновой кислоты в присутствии азелаиновой кислоты, для
которой 7,, находится вблизи комнатной температуры ,те для того же мате -
’ Следует имен, ввиду, что абсолютно упругих полимерных материалов пс бывает (за исклю-
чением идеальных кристаллов) -Поэтому термин! i “упругое поведение” и “упругий материал
применительно к полимерам являются условными: под упругим поведением подразумевается та-
кое, при котором напряжение релаксирует очень медленно -
290
ГлаваХ
1____________I___________।____________I______
0 3 0 60 90 1мм
Рис.81. Зависимость модуля упругости IgE или Е от содержания 2,4-толуилепди-
изоцианата G , изменяющегося по длине образца / ,в его смеси с олигомерным
макро диизоцианатом
Dependece of modulus of elasticity IgE or E on content of 2,4-toluylenediisocyanate
G in its mixture with oligomer macrodiisocyanate; the content of G varies as per the length
of specimen /
Равновесный модуль высокоэластичности сетчатых полимеров 291
Рис.82. Кривые релаксации относительного напряжения о/о0ддя сетчатых полиизо-
циануратов, полученных из олигомерного макродиизоцианата и 2,4-толуиленди-
изоцианата при содержании последнего (вес %): 1 - 6; 2 - 46; 3 - 27; а также 4 - эпоксид-
ный олигомер ЭД-20, отвержденный метилтетрагидрофталевым ангидридом, 5 - эпоксид-
ный вязкоупругий полимер (состав указан в тексте)
Curves of relative stress relaxation c/c0 for network polyisocyanurates prepared from
oligomer macrodiisocyanate and 2.4-toluylenediisocyanate when the content of latter in
percent by weight is: 1 -6,2-46, 3 -27 respectively, 4-epoxy oligomer ED-20 cured with
methyltetrahydrophthalic anhydride; 5- epoxy viscoelastic polymer (The composition is
indicated in the text)
риала, релаксационное поведение которого показано на рис.71. Кривые ре-
лаксации напряжения строились в координатах “относительное напряжение” -
время; относительное напряжение вычислялась, как о/а0, где а - текущее ре-
лаксирующее напряжение, о() - начальное напряжение, которое развивается в
момент окончания “мгновенного” задания деформации.
Из этого рисунка видно, что в то время как относите льное напряжение
для обычного вязкоупругого материала быстро релакс пр уст практически до
нуля, для полученных в работах [19, 129] сетчатых полимеров наблюдается
более медленный спад напряжения, характерный для полимерных стекол или
резин с пос ждующим переходом к очень малой скорости релаксации напря-
жения.
Таким образом, несмотря на то, что определенная часть материала в пре-
делах одного и того же образца обладает величинами модуля, характерными
для переходной зоны .механическое поведение является упругим, каку сте-
кол или резин, а не вязкоупругим, характерным для всех полимеров в пере-
ходной области.
292 Глава X
Динамический механический анализ показывает, что tg 3 для получен-
ных сеток является очень малым, что характерно для упругих материалов,
несмотря на значения модуля накопления Д, характерного для переходной
зоны.
На основе сетчатых полиизоциануратов получены также пленки с гради-
ентом модуля упругости перпендикулярно плоскости пленки. Эти пленки, сфор-
мованные из частосетчатых полимеров, обладают хорошими механическими
свойствами (прочность 50 МПа и удлинение при разрыве 90 %). Таким обра-
зом, получение градиентных материалов позволяет избавиться от основного
недостатка частосетчатых полимеров - их хрупкости.
Глава XI
ОПИСАНИЕ РЕЛАКСАЦИОННЫХ
ПРОЦЕССОВ В ПОЛИМЕРАХ
XL1. Релаксация напряжения
К настоящему времени для описания процессов релаксации напряжения
и ползучести предложены различные варианты ядер в соответствующих урав-
нениях Больцмана-Вольтерры. Сводное описание этих ядер и их резольвент
имеется в монографии [112]. Ядра содержат три или четыре параметра, при-
чем, как правило, имеют дробную степень времени, так как только в этом
случае возможно описание экспериментальных данных по релаксации напря-
жения и ползучести с хорошим приближением.
Анализ существующих ядер показал, что при надлежащем выборе пара-
метров они с достаточной точностью передают ход релаксационных процес-
сов, однако физический смысл этих параметров не всегда ясен, хотя попытки
раскрытия физического смысла некоторых ядер предпринимались [74, 104].
В работе [71 предложен подход к получению ядер релаксации, основанный на
рассмотрении термодинамических функций и их изменении в ходе релакса-
ционного процесса
Предположим, что процесс релаксации напряжения происходит в резуль-
тате взаимодействия и диффузии кинетических единиц - релаксаторов. Ре-
лаксаторами могут быть различные атомные группы, повторяющиеся звенья,
более крупные фрагменты и целые сегменты макромолекул. К релаксаторам
относятся также отдельные элементы свободного (пустого) объема, те. мик-
рополости, концентраторы напряжения и т.д. Эти микрополости могут, взаи-
модействуя друг с другом, сливаться, перестраиваться и диффундировать в
полимерном материале в процессе релаксации, образуя такую структуру, ко-
торая способствует снижению релаксирующего напряжения. Тогда полимер-
ный материал можно рассматривать как состоящий из релаксаторов и нере-
лаксаторов. причем после ' мгновенного’' задания деформации подавляющая
294
Глава XI
часть материала образца состоит из релаксаторов, взаимодействующих меж-
ду собой с образованием нерелаксирующего материала. Возникновение час-
тиц двух сортов (релаксаторов и нерелаксаторов) и их диффузия приводят к
производству энтропии системы, которая возрастает в ходе релаксации на-
пряжения.
Производство энтропии (или скорость возникновения энтропии) опреде-
dS 1
ляется выражением
, где .S' - энтропия, t - время, Г' - объем системы.
Ядра релаксации получены в работе [7] при допущении, что движущей силой
процесса является производство энтропии системы (образца), которая возрас-
тает в процессе релаксации напряжения до максимального значения.
Как известно, если система состоит из двух типов частиц, энтропия сме-
шения .S’ этой системы, определяемая с помощью уравнения Больцмана .на-
ходится из выражения
т*1
- (262)
где т* - общее число частиц (в нашем случае число релаксаторов и нерелак-
саторов в единице объема); щ - соответственно число релаксаторов и
нерелаксаторов в единице объема; кк - константа Больцмана. Учитывая, что
мы имеем два вида частиц, можно записать
т !
(263)
(оу?/ )![(!- а)т ]!
где а - доля релаксаторов от общего числа частиц. Применяя приближенную
формулу' для вычисления факториала при большом т*, на основании (263)
полу'чаем
г. , . х/злл/ е
s = к Б , --------;-----1 , ---------------7.--------- • (264)
V2naw* (a??/*)“"' е ~ап' - а)т* | (1 - a е ^~а)т
После ряда преобразований и пренебрежения малыми членами имеем
.S = -кБт [alna+(l -a)ln(l -a)] . (265)
Величина а меняется со временем t от 1 до 0,5, так как при а = 0,5 энтро-
пия смешения принимает максимальное значение.
Предположим, что функция памяти (ядро в уравнении Больцмана-Воль-
терры) связана с энтропией обратной зависимостью типа
Описание релаксационных процессов в полимерах
295
7’(т) = Х0(1--1-)Г7’*(т)Л (266)
‘S ‘Smax о
где 7*(т) - переменная часть ядра. Тогда, подставляя в формул)' (266) выраже-
ние (265), получаем
7'(т)=-^-
kFm pallia+ (1 - a) In(l-a)
1
In 0,5
(267)
CO
где m j T (т)Л .
0
Теперь необходимо найти зависимость а от т (напомним, что a - доля
релаксаторов от общего числа частиц в системе). Как было отмечено, измене-
ние а со временем т может быть обусловлено двумя причинами: взаимодей-
ствием релаксаторов и переходом их в нсрелаксаторы; ди фф зией кинетичес-
ких сдиниц( заметим, что механизм, связанный с диффузией микродс|)ектов,
описан в работах [ 104, 119]). Рассмотрим эти причины.
Поскольку процесс взаимодействия релаксаторов является сложным, его
естественно описывать уравнением реакции п-ro порядка. Если при обычной
химической реакции, например, третий порядок наблюдается редко (так как
это требует активного соударения сразу трех молекул), то в данном случае
релаксаторы “конденсированы” в образце и элементарный акт процесса их
взаимодействия может включать сразу несколько релаксаторов (например,
слияние нескольких микрополостей в одну). При этом порядок реакции мо-
жет быть и дробным. Для такого случая действительно кинетическое уравне-
ние
de , „
— = kc , (268)
th
где k - константа скорости реакции; с - концентрация. Интегрируя (268) от
т = 0 до I, получаем
[1+Со 1 (п - 1)А"| -1
где с0 - начальная концентрация релаксаторов любого типа (примем для про-
стоты, что эти концентрации равны для разных типов релаксаторов). Тогда
296
Глава XI
4: 1 1
где к = кеа ; Р =------; и - порядок реакции.
л-1
Для получения ядра релаксации необходимо выражение (269) подставить
в формулу (267). При этом следует учесть, что к моменту' окончания развития
деформации доля релаксаторов составит не 1, а несколько меньшую величи-
ну; равную 1 - а(). Учитывая изложенное, имеем
?i(T) = -
Sq
_____1____
(1 + /Л/р)р
________
(1 + Гт/р)₽
(270)
Функция 7](т) имеет физический смысл только при условии, что
---------г->0,5. Таким образом, ядро 7) (г) содержит четыре параметра.
(1 + к т / Ру
А = tS° ; к* =ксд 1; Р = 1/(и —1) и ctg.
Согласно требованиям к ядрам релаксации, величина Од должна быть край-
не малой; в работе [7] величина otg была принята равной 1О'10 на основании
аппроксимации кривых релаксации напряжения для ряда полимеров.
При малых значениях А*т/р, т.е. на начальных стадиях процесса релакса-
ции, функция 7’Дт) значительно упрощается. Действительно, при малых //т/р
величина (1 + /[})Р = 1 + к\. Тогда
Описание релаксационных процессов в полимерах
297
(271)
После дальнейших преобразований имеем
w=-
______________1______________
(&*т + ар)[1п(/с*т + ар)-1]
1
In 0,5
(272)
Пренебрегая вторым слагаемым в фигурной скобке ввиду его малости,
запишем
Т = —--------------------------1--------------- (273)
4> «? (А + ц )[1п(А-‘т + ц У 1] '
Проинтегрируем соотношение (273), предварительно сделав некоторые
преобразования:
о
Sq г__________1__________j к т + а0
к^пцк* 20 к\ + а0 ^к\ + а0 е
е е
(274)
Получаем
ГТ|(1М1 = __^_1п1П(*',4~11°)
0 кБт{к е
Sp ]п 1п(£ z + <х0) — 1
кБтхк* 1па0-1
• (275)
t
Подставим значение |7](т)А в уравнение Больцмана:
о
О = Ор
1-jW
о
(276)
где о0 - начальное напряжение, которое развивается в момент окончания
“мгновенного” задания деформации.
В результате находим соотношение для описания релаксации напряже-
ния на начальном участке (при малых значениях t):
298
Глава XI
QqS'o lnln(£*f+a0)-l
кБтк' lna0-l
(277)
Если выражение (277) справедливо, то зависимость о от ln-^^ ? + ао)—1
lna0 -1
tSaSn
должна представлять прямую,тангенс угла наклона которой равен — * , а
кБтк
отрезок, отсекаемый на оси ординат — oq. На рис.83 показана такая зависи-
мость для полиметилметакрилата. Видно, что экспериментальные точки с
хорошей точностью укладываются на прямую.
0,9 1 1,1 1,2 1,3 1,4 1,5 1,6 1,7 In
1п(А- /+а0)— 1
lna0 —1
Рис.83. Зависимость а от In f+ ао)—1 (см пояснения в тексте) Релаксация
In a0 -1
напряжения в условиях одноосного сжатия, для ПММА при температуре 295 К ,
деформации 2,2 %
, г , ln(A:*t+an )-1 „ , .
Dependence of a on In-------—— (For explanation see text). Stress relaxation
lna0 -1
for PMMA-material under conditions of uniaxial compression when the temperature
is 295 К and the deformation is 2,2 %
Описание релаксационных процессов в полимерах
299
Рассмотрим теперь диффузионный механизм релаксации. При случайном
блуждании кинетических единиц количество мест, занимаемых ими к моменту
времени т, а следовательно, доля нерелаксаторов 1 — а определяется соотно-
шением [146]
(1-а) = л?/2, (278)
где 0 < b < 1; а - константа. В случае, если b = 1, соотношение (278) соответ-
ствует фиковской диффузии:
/ л
где / -размер образца; D - коэффициент диффузии, т.е. а = —(—)12
/ л
Подставляя соотношение (278) в (267), получаем
1
1
(279)
кБтг
ат* 1пат7 + (1-ат7)1п(1-ат7) 1п0,5
где у = Ь/2. Функция Т^т) имеет физический смысл только при условии, если
ат7 <0,5.
5
Ядро (279) содержит три параметра: А =--—; а и у. Оно представляет
собой функцию со слабой особенностью при т = 0.
При малых значениях ат1 функцию Т^т) можно упростить. В этом случае
соотношение (279) запишем в виде
Т2(т)=-
So 1 1
кБтг ат7(Inат7 -1) 1п0,5
(280)
Пренебрежем вторым слагаемым в квадратной скобке, тогда
Г2(т)=-
So 1
М»2 ат1In
(281)
Проинтегрируем соотношение (281), сделав предварительно ряд преоб-
разований:
|Г2(т)</т = -
о
Sp
' т1’7 (пт1
Г—---------d —
о ат7 ат7 е
0---In----- V
Сделаем замену переменной .
е е
300
Глава XI
Тогда: = X е V хе • V = т = а а
$T2(x)dx = 0 о *2 ( — J’ кБт2ау Х£у/у-1 dx. xlnx (282)
Далее имеем /Т2(Т)Л = 0 l/y-1 sqS кБт2а'/уу X2rl/y~2 [- dx. J Inx (283)
Учитывая, что Xj = 0, а х2 --, получаем
е
j Т2 (t)rft = - S°e h li| (-)
о кБт2а
£)1/vI/i-7
е
(284)
где li - интегральный логарифм.
Подставляя значение J'/2(t)A в уравнение Больцмана (276), находим
. о
соотношение для описания процесса релаксации напряжения на начальном
участке релаксационной кривой при условии, что аГ « 1:
o0V'V1
°-°о +- —
кБт2а гу
£)1/у-1Д-у
в
(285)
При выполнении соотношения (285) в координатах о
£)1/у-1Д-у
е
получаем прямую, тангенс угла наклона которой равен u--, а отрезок,
кБт2а1'Ь
отсекаемый на оси ординат, равен с0. Если лимитирующей стадией процесса
релаксации является взаимодействие релаксаторов и переход их в нерелакси-
Описание релаксационных процессов в полимерах 301
рующий материал, то должно работать ядро (270) или на начальном участке -
упрощенное выражение (277); если лимитирующей стадией процесса явля-
ется диффузия образовавшихся нерелаксаторов, то должно работать ядро (279),
а на начальном участке - упрощенное соотношение (285).
t
На рис. 84 показаны в качестве примера зависимости о от j7] (т)А и от
t о
|7’2(т)г7т. Видно, что в данном случае ядро 7'i(t) лучше передает процесс
о
релаксации напряжения для полиметилметакрилата.
Вернемся теперь к ядру релаксации, основанному на анализе кинетики
процесса взаимодействия релаксаторов и перехода их в нерелаксирующий
материал. Ядро (270) получено при условии, что процесс взаимодействия ре-
лаксаторов представляет собой одностороннюю необратимую реакцию и-го
порядка, т.е. считается, что провзаимодействующие релаксаторы переходят в
о о
что и на рис. 83)
t t
Dependences of a on J 7J (t)A (l)and j (т)Л (2) (Conditions оfexperiinent
0 0
are similar to those shown in Fig. 83)
302
Глава XI
нерелаксирующий материал таким образом, что данный процесс является нео-
братимым.
В работе [44] рассмотрен процесс взаимодействия релаксаторов в виде
обратимой двусторонней реакции н-го порядка, те. допускается, что нере-
лаксирующий материал в ходе процесса может вновь производить релаксато-
ры. Кинетическое уравнение такой реакции имеет вид
-~ = ки" -fc(l-a)". (286)
Уравнение (286) записано при условии, что константы скорости прямой
и обратной реакции одинаковы и равны величине к. Это приводит к тому, что,
когда система переходит в равновесие, доля релаксаторов и нерелаксаторов
становится одинаковой и равной 0,5. Уравнение (286) интегрируется до конца
только в отдельных частных случаях, например, при п = 2. В общем случае,
когда и является дробной величиной, интегрирование можно произвести только
численными методами. С целью нахождения зависимости степени превраще-
ния а от времени t в работе [44] применили численный метод Рунге-Кутта с
автоматическим выбором шага интегрирования. По найденным значениям
величин а, которые были рассчитаны при различном малом шаге по t ,опреде -
лялись с помощью ЭВМ значения интеграла от переменной части ядра
t
j 73* (т)</т , где
о
(а-а0)1п(а-а0) + (1-а + а0)1п(1-а + а0) 1п0,5 •
Таким образом, мы имеем три ядра релаксации 7) (г), 7’2(т) и Т’з(т), при-
чем
7i(T)=-r^--7i*(T); (288)
кБт\
Т’2« = -7^—^(т); (289)
кБт2
Т3(т)=--^—Т3\т), (290)
кБтЗ
Описание релаксационных процессов в полимерах
303
где Т* (т), 72*(т) и 73 (т) - переменные части ядер 7'1(т), 72(т) и Т3(т).
Эти переменные части ядер релаксации описываются соотношениями
(a-a0)ln(a-a0) + (l-a + a0)ln(l-a + a0) In 0,5 ’
1
а=------;---r ;
(1+fc т/₽)р
яту liWT7 + (1 - ат* )1п(1 - ат*) 1 гО 5
Переменная часть ядра 7’3(т) описывается соотношением (287). Подстав-
ляя в уравнение Больцмана (276) ядра 7’1(т). Т’2(т) и Г3(т), получаем:
a = ao_£o^j7’1‘(T)A; (293)
кБ»>1 о
о = а0 - J Т’г (Т)Л J (294)
кБ™2 о
а = а0 - -2^0- Jr3* (т)Л . (295)
кБ”Ч о
В уравнениях (293 -295)
mi =/и,* |Г1*(т)Л; (296)
о
00
т2=тЦТ2(т)дт; (297)
о
w3 =отз рТ’з (т)Л , (298)
о
304
Глава XI
где тх и w3 - количество неоднородностей (релаксаторов) в полимерном
материале, которые взаимодействуют друг с другом в ходе релаксационного
процесса; т2 - количество диффундирующих неоднородностей в материале
в процессе релаксации напряжения.
Если уравнения (293), (294) и (295) правильно передают ход эксперимен-
t
тальных кривых релаксации напряжения, то в координатах а - |Т’1*(т)А,
о
i t
а - jТ2 (-г)А и а - |т3*(т)<А должны образовываться прямые, тангенс угла
о о
наклона которых равен /^w, , а отрезок, отсекаемый на оси ординат-а0.
Чтобы воспользоваться уравнениями (293), (294) и (295), необходимо знать
г t t *
численные значения интегралов J 7’f (т)А , j Т2 (r)di и J Т2 . Эти значе-
0 0 0
ния зависят соответственно от двух пар параметров - к* и 0, а и у. Было про-
ведено [13,44] численное определение этих интегралов с помощью ЭВМ при
варьируемых параметрах к* и 0, а и у.
В табл.(38—40), приведены численные значения переменных частей (г),
Т2 (т) и 73 (т) ядер релаксации Т ,(т), Т 1л) и Т3(т), заимствованные из работ
[13,44]. Имея эти значения, можно проводить аппроксимацию кривых релак-
сации напряжения в широком интервале длительностей процесса t.
Для обработки экспериментальных данных, согласно уравнениям (293),
(294) и (295), методом наименьших квадратов целесообразно использовать
следующую процедуру. В память ЭВМ предварительно заносятся все значе-
/ t t
ния интегралов j7}* (т)А , JТ2 (T)di и j 7'3 (т)А в виде трех массивов. Каж-
0 ° 0 t t г
дый массив содержит значения интегралов J Т\ (т)А, j Т2 (т) А и j 73 (т) А
0 о 0
при разных выбранных временах t для каждой пары параметров системы к и
0, а и у (массив 1 соответствует табл.38, массив 2-табл.39, массив 3 - табл.40).
Каждая экспериментальная зависимость о(1) аппроксимируется уравнением
(293), (294) или (295) и автоматически выбираются такие значения пар пара-
метров к* и 0, а и у, при которых сумма квадратов отклонений эксперимен-
тальных значений от расчетных является минимальной, а коэффициент кор-
реляции - максимальным.
Описание релаксационных процессов в полимерах
305
Экспериментальная проверка ядер (270). (279) и (287) и всей процедуры
расчета в целом была проведена в серии работ [11. 12, 14, 38] на примере
полиметилметакрилата, полиоксадиазола, полиимида, полибензоксазола и
других полимеров. Таблица 38
Значения J7’, (i)Jt Values of j Tj* (x)<7x
о о
1 Р = 0,2 Р = 0,3 Р - 0,4 Р = 0,5 Р =0,6 Р - 0,7 Р =0,8
1 2 3 4 5 6 7 8
к* = 0,1
0.5 17,57 17.52 17,49 17,47 17,46 17,45 17.45
1 19,06 18.93 18,86 18,82 18,80 18,78 18.76
2 20,62 20,34 20.20 20,11 20,05 20,01 19,98
3 21,56 21,14 20,92 20,78 20,69 20,63 20,57
4 22,22 21,66 21,37 21,19 21,06 20,98 20,91
5 22,73 22,03 21,67 21,45 21,30 21.19 21,10
6 23,13 22,31 21,88 21,62 21,44 21,31 21,22
9 23,98 22,82 22,22 21,86 21,62 21,45 21.33
15 24,88 23,21 22,39 21,94 21,66 21,47 21,34
30 25,70 23.31 22,39 21,94 21,66 21,47 21,34
45 25,87 23.31 22.39 21,94 21.66 21,47 21,34
60 25,88 23.31 22,39 21,94 21,66 21,47 21,34
90 25,90 23,31 22,39 21,94 21,66 . 21,47 21,34
120 25,90 23.31 22,39 21,94 21.66 21,47 21,34
180 25,90 23,31 22.39 21.94 21 ,66 21,47 21,34
240 25,90 23,31 22,39 21,94 2 1,66 21,47 21,34
300 25,90 23,31 22,39 21,94 21,66 21.47 21,34
360 25,90 23.31 22,39 21,94 21.66 21.47 21,34
720 25,90 23,31 22,39 21,94 21,66 21.47 21,34
1440 25,90 23,31 22,39 21,94 21,66 21.47 21,34
2880 25.90 23,31 22,39 21,94 21,66 21,47 21,34
5760 25,90 23,31 22,39 21,94 21,66 21,47 21,34
10080 25,90 23,31 22,39 21,94 21.66 21.47 21,34
100000 25,90 23,31 22,39 21,94 21,66 21,47 21,34
к* -0,01
0,5 133.5 133,4 133,4 133,4 133,4 133,4 133,4
1 144,6 144,5 144,5 144,4 144,4 144,4 144,4
2 156,8 156,6 156,5 156,4 156,4 15.6 ,3 156.3
3 164.5 164.2 164.0 163,9 163,8 163 ,8 163,7
4 170 2 169 7 169 5 169 3 169 3 16 9,2 169,1
5 174,8 174.1 173,8 173,6 173,5 173,4 173,3
6 178,6 177,8 177 4 177, 1 177,0 176,9 176,8
9 187,3 186.0 185.4 185.0 184,8 184,6 184,4
15 198,6 196,5 195.4 194 ,7 194,3 193,9 193,7
30 214,7 210,3 208,1 20 6 7 205 8 205 1 204 6
45 223.1 217,5 214.2 212,1 210.7 209,7 208,9
60 230,3 222,0 217,6 215,0 213 .2 211,9 210,9
306
Глава XI
Описание релаксационных процессов в полимерах
307
таблица 38 (продолжение)
таблица 38 (окончание)
1 2 3 4 5 6 7 8
90 238,9 227,2 221,2 217,6 215,2 213,5 212,2
120 244,3 229,7 222,4 218,2 215,4 213,5 212,2
180 250.7 231,6 222,8 218,2 215,4 213,5 212.2
240 254,0 231.9 222,8 218,2 215,4 213,5 212.2
300 255,9 232,0 222,8 218,2 215,4 213,5 212,2
360 257,1 232,0 222,8 218,2 215,4 213,5 212.2
720 258,0 232,0 222,8 218,2 215,4 213.5 212.2
1440 258,1 232,0 222,8 218,2 215,4 213,5 212,2
2880 258,1 232,0 222,8 218,2 215,4 213,5 212,2
5760 258,1 232,0 222,8 218,2 215,4 213.5 212,2
10080 258,1 232,0 222,8 218,2 215,4 213,5 212.2
100000 258,1 232,0 222,8 218,2 215,4 213,5 212,2
А’* = 0,001
0,5 1027 1027 1027 1027 1027 1027 1027
1 1110 1110 1110 1110 1110 1110 1110
2 1201 1200 1200 1200 1200 1200 1200
3 1258 1257 1257 1257 1257 125.7 1257
4 1300 1299 1299 1299 1299 1299 1299
5 1334 1333 1333 1333 1333 1332 1332
6 1362 1361 1361 1361 1361 13 60 1360
9 1427 1426 1426 1425 1425 1 425 1425
15 1514 1513 1512 1511 1511 1511 1510
30 1644 1640 1639 1637 1637 16.36 16.36
45 1724 1719 1716 1714 1713 1712 1711
60 1784 1776 1772 1769 1768 1767 1766
90 1872 1860 1853 1849 1847 1845 1844
120 1936 1919 1910 1905 1901 1899 1897
180 2027 2001 1988 1980 1974 1970 1967
240 2093 2059 2041 2029 2022 2017 2013
300 2145 2101 2079 2065 2055 2049 2043
360 2188 2136 2109 2092 2081 2073 2067
720 2341 2244 2194 2163 2142 2127 2116
1440 2472 2308 2227 2182 2154 2135 2121
2880 2556 2319 2227 2182 2154 2135 2121
5760 2578 2319 2227 2182 2154 2135 2121
10080 2579 2319 2227 2182 2154 2135 2121
100000 2579 2319 2227 2182 2154 2135 2121
к* =0,0001
0,5 7900 7900 7900 7900 7900 7900 7900
1 8556 8556 8556 8556 8556 8556 8556
2 9256 9256 9256 9256 9256 9256 9256
3 9692 9692 9692 9692 9692 9692 9692
4 10010 10010 10010 10010 10010 10010 10010
1 2 3 4 5 6 7 8
5 10270 10270 10270 10270 10270 10270 10270
6 10480 10480 10480 10480 10480 10480 10480
9 10970 10970 10970 10970 10970 10970 10970
15 11620 11620 11620 11620 11620 11620 11620
30 12570 12570 12570 12570 12570 12570 12570
45 13170 13160 13160 13160 13160 13160 13160
60 13610 13600 13600 13600 13600 13600 13600
90 14270 14260 14260 14260 14260 14260 14260
120 14760 14750 14740 14740 14740 14740 14740
180 15470 15450 15440 15440 15440 15440 15440
240 16000 15980 15900 15950 15950 15950 15950
300 164.30 16390 16380 16360 16360 16350 16350
.360 16800 16760 16730 16720 16710 167 00 16700
720 18230 18130 18080 18050 18030 18020 18010
1440 19760 19560 19460 19390 19350 19.320 19300
2880 21360 20940 20720 20590 20500 20440 20390
5760 22930 2213 0 21170 21450 21280 21 150 21060
10080 24090 228 20 22170 21780 21520 213 40 21210
100000 24090 22820 22170 21780 21520 21340 21210
к* = 0,00001
0,5" 59840 59840 59840 59840 59840 59840 59840
1 65230 65230 65230 65230 65230 65230 65230
2 70910 70910 70910 70910 70910 70910 .70910
3 74420 74420 74420 74420 74420 74420 74420
4 76970 76970 76970 76970 76970 76970 76970
5 78990 78990 78990 78990 78990 78990 78990
6 80670 80670 80670 80670 80670 80670 80670
9 84500 84500 84500 84500 84500 84500 84500
15 89610 89610 89610 89610 89610 89610 89610
30 96900 96900 96900 96900 96900 96900 96900
45 101400 101400 101400 101400 101400 101400 101400
60 104700 104700 104700 104700 104700 104700 104700
90 109700 109700 109700 109700 109700 109700 109700
120 113300 1 13300 113300 113.300 11.3300 113300 113300
180 118600 118600 118600 118600 118600 118600 118600
240 122500 122500 122500 122500 122500 122500 122500
300 125700 125600 125600 125600 125600 125600 125600
360 128400 128400 128300 128300 128300 128300 128300
720 139000 139000 138900 1.38900 138900 138900 1 .38900
1400 150800 150600 150500 150500 150500 150400 150400
2880 163600 163200 163100 162900 162900 162800 162800
5760 177500 176800 176400 176200 176000 175900 175800
10080 189700 188300 187500 18 7100 186800 186600 186500
100000 189700 188300 187500 1 87100 186800 186600 186500
308
Глава XI
' , ' Таблица 39
Значения р2(г)Л Values of |7'2*(г)Л
о о
t а = 0,315 а = 0,284 а - 0,252 а = 0,220 а --- 0.189 а 0,158
1 2 .3 4 5 6 7
у - 0,1
0,5 0,144 0,189 0,247 0,323 0,421 0,556
1 0,236 0,317 0,425 0,567 0.750 1.003
2 0,375 0,523 0,722 0,984 1.326 1,799
3 0,494 0,693 0,977 1,353 1,844 2,527
4 0,576 0,843 1,205 1,691 2,327 .3,213
5 0,656 0,977 1,417 2,007 2,784 3,867
6 0,727 1,099 1,614 2,307 3,220 4,498
9 0,900 1,418 2,144 3.132 4,443 6,28.3
15 1,142 1,915 3,029 4,567 6,628 9.541
30 1,464 2,754 4,711 7,492 11.285 16,700
45 1,609 3,303 5,991 9,902 15,300 23.070
60 1,675 3,693 7,036 12,000 18.930 28.960
90 1,707 4,198 8,684 15,590 25,410 39 ,780
120 4,488 9,948 18,640 31.180 49,680
180 4,745 11,780 23,670 41.320 67,710
240 4.808 1.3,030 27,780 50.190 84,090
300 13,910 31,250 58,160 99,290
360 14,540 34,240 65,440 113,600
720 15,850 46,290 100,100 187.100
1440 56.880 146.400 301,100
2880 61,700 200,800 470.700
5760 252,000 707,900
10080 277,400 948,100
100000 1694,000
t 0-0,199 а - 169 а 0,149 а -0,1096 а - 0,0798 а 0.050
у = 0,2
0.5 0,521 0,660 0,860 1,127 1.601 2,577
1 0.837 1,081 1,4.31 1.91.3, 2,757 4,493
2 1,316 1.744 2,353 3,221 4,721 7.811
3 1,694 2.285 3,130 4,349 6,447 10,780
4 2,011 2,754 .3,819 5,368 8,031 13,530
5 2,286 3,173 4,448 6,310 9.514 16,1.30
6 2,531 3,555 5,029 7.195 10,920 18.620 ।
9 3,133 4.536 6.574 9,596 14,800 25,590
15 3,979 6,047 9,097 13,680 21,610 38,090
30 5,124 8,5.30 13,740 21,740 35,740 65.000
45 5,647 10,10 17,140 28,180 47.660 88.580
60 5.885 11.17 19,830 33,650 58.250 110.100
90 6,003 12,47 23.910 42.770 76.860 1 49,300
120 1.3.13 26.800 50,270 93.160 185,000
180 13,56 30,800 62,200 121.300 2 49 ,300
Описание релаксационных процессов в полимерах
309
таблица 39 (продолжение)
1 2 3 4 5 6 7
240 33,160 71,460 145,300 307,400
300 34,570 78,930 166,800 361,000
360 35,390 85,090 186,000 411,200
720 106.500 274,300 667,700
1440 117.800 382.800 1063.000
2880 493,300 1649,000
5760 567,100 2471,000
10080 3310,000
100000
t а = 0,126 <7 = 0,104 <7 = 0,0819 а = 0,0598 а = 0,378 а = 0J0157
у = 0,3
0,5 1,219 1,488 1,913 2,621 4,064 9,064
1 1,901 2,362 3,076 4,265 6,680 15,020
2 2,914 3,697 4,894 6,888 10,940 24,870
3 3,701 4,762 6 381 9,080 14 560 33,380
4 4,358 5.673 7,677 11,020 17,81 0 41,120
5 4 926 6,478 8 841 12,790 20 8 Ю 48,330
6 5,429 7 204 9,908 14,430 23,620 55,140
9 6,664 9,049 12,690 18,800 31,240 73.880
15 8 396 11,830 17,110 26,030 44,240 106 600
30 10,750 16,240 24,900 39,720 70,200 174,900
45 11,830 30,350 50,350 50,220 91,380 233,100
60 12,320 20,650 34,500 58.900 109,800 285,400
90 12,560 22,600 40.450 72,860 141,400 379,000
120 23,440 44,460 83,880 168,300 462,800
180 23,780 49,180 100,500 213,600 611800
240 51,430 112,600 251,300 744,400
300 52,390 121,700 283,800 865,600
360 52,690 128,700 312,400 978,300
720 147 500 436,300 1545 000
1440 568 700 2401.000
2880 669,000 365Q ООО
5760 5383,000
10080 7146,000
100000 12981,00
t а = 0,079 а = 0,0642 а = 0,0494 а = 0,0346 а - 0,0198 а = 0,005
Y = 0,4
0,5 2,456 3,007 3,858 5,370 8,928 29,87
1 3,712 4,583 5,928 8,310 13,890 46,81
2 5.526 6.904 9,030 12.790 21,560 73,33
.3 6,909 8,710 11,490 16,400 27,850 95,35
4 8.053 10,230 13,590 19,540 33,370 114,80
5 9,036 И ,560 15,450 22,340 38,360 132,70
6 9,902 12,740 17, 140 24,910 42.970 149,30
9 12,020 15,720 21,450 31,620 55,220 193,90
15 14,970 20,110 28 120 42,360 75,440 269,50
310
Глава XI
Описание релаксационных процессов в полимерах
311
таблица 39 (окончание)
1 2 3 4 5 6 7
30 18,990 26,900 39,410 61,830 114,100 420,50
45 20,870 30,870 46,970 76,130 114,400 544,70
60 21,750 33,380 52,530 87,580 170,100 654,10
90 22,210 36,050 60,110 105,300 212,900 845,60
120 37,060 64,860 118,700 248,400 1014,00
180 69,760 137,600 305,900 1306,00
240 71,490 150,300 352,000 1562,00
300 71,860 158,900 390,500 1792,00
360 164,900 423,400 2004,00
720 175,000 553,900 3047,00
1440 664,600 4571,00
2880 706,500 6734,00
5760 9665,00
10080 12595,00
100000
t а = 0,05 а = 0.0403 а = о.озоб а = 0,0209 <7 = 0,0113 <7=0,00158
7= 0,5
0,5 4,738 5,767 7,420 10,42 17,95 102,4
1 6,850 8,384 10,830 15,28 26,42 150,4
2 9,792 12,080 15,710 22,33 38,84 221,1
3 11,980 14,870 19,450 27,80 28,60 277,2
4 13,760 17,170 22,570 32,42 56,93 325,4
5 15,280 19,160 25,290 36,50 64,34 368,6
6 16,600 20,910 27,740 40,17 71,08 408,1
9 19,810 25,250 33,850 49,57 88,57 511,9
15 24,230 31,510 43,030 64,13 116,40 681,0
30 30,180 40,870 57,930 89,34 167,30 1003,0
45 32,950 46,140 67,480 107,00 205,50 1258,0
60 34,250 49,370 74,220 120,70 236.90 1476,0
90 34,920 52,580 82,960 141,10 287,60 1850,0
120 53,600 87,970 155,60 328,10 2169,0
180 92,330 174,80 391,20 27130
240 93,240 186,20 439,50 3178,0
300 193,00 478,00 3590,0
360 196,90 509,70 3965,0
720 620,30 575 7,0
1440 680,20 82 81,0
2880 1 1745,0
5760 16307,0
10080 20775 0
100000 34975,0
t I * Таблица 40
Значения Values of j^3 (х№
о о
t 0 = 0,2 0 = 013 0 = 0,4 0 = 0,5 0 = 0,6 0 = 0,7 0 = 0,8
1 2 3 4 5 6 7 8
к* = 0,1
0,5 17,5 70 17,52 17,49 17,47 17,460 17,45 17,45
1 19,060 18,94 18,87 18,83 18,800 18,78 18,77
2 20,740 20,47 20,32 20,23 20,180 20,13 20,11
3 21,700 21,29 21,06 2Q 93 2Q 840 2Q 78 2Q 73
4 22,370 21,82 21,52 21,35 21,230 21,15 21,10
5 22,880 22,20 21,84 21,62 21,480 21,38 21,32
6 23,290 22,48 22,06 21,80 21,640 21,53 21,46
9 24,170 23.02 22,45 22,12 21,910 21 ,77 21,68
15 25,150 23,50 22,73 22,31 22,060 2 1,90 21,79
30 26,140 23,79 22,85 22,38 22,104 21,93 21,82
45 26,400 23,81 22,85 22,38 22,105 21,93 21,82
60 26,470 23,81 22,85 22,38 22,105 21,93 21,82
90 26,516 23,81 22,85 22,38 22,105 21,93 21,82
120 26,520 23,81 22,85 22,38 22,105 21,93 21,82
180 26,521 23,81 22,85 22,38 22,105 21,93 21,82
240 26,521 23,81 22,85 22.38 22,105 21,93 21.82
300 26,521 23.81 22,85 22,38 22,105 21,93 21,82
360 26,521 23,81 22,85 2 2,38 22,105 21,93 21,82
720 26,521 23,81 22,85 22,38 22,105 21,93 21,82
1440 26,521 2 3,81 22,85 22,38 22,105 21,93 21,82
2880 26 321 23> 81 22,85 22 38 22 Ю5 21,93 21,82
5760 26,521 23,81 22,85 22,38 22,105 21,93 21,82
10080 26,521 23,81 22,85 22,38 22,105 21,93 21,82
100000 26,521 23,81 22,85 2238 22,105 21 93 21 82
к*-- = 0,01
0,5 133,50 133,40 133,40 133,40 133,40 133,40 133,40
1 144,69 1 44,53 144,50 144,48 144,47 144,46 144,45
2 157,73 157,44 157,36 157,29 157,26 157,23 157,21
3 165,29 165,17 165,02 164,91 164,85 164,80 164,77
4 171,35 1 70,79 170,57 170,42 170,33 170,26 170,21
5 175,93 175,23 174,94 174,74 174,62 174,54 174,47
6 179,75 178,91 178,54 178,30 178,15 178,04 177,96
9 188,68 187,39 186,79 186,42 186,17 186,00 185,87
15 200,54 198,35 197,29 196.63 196,20 195,89 195,67
30 217,70 213,32 211,11 209,76 208,87 208,26 207,81
45 227,25 220,82 216,55 215,57 214,29 213,41 212,78
60 23Д 75 225 43 221, 22 21§ 71 217 Ю 216 01 215 23
90 242,53 230,88 225,14 221,83 219,76 218,38 217,42
120 248,07 233,66 226,82 223,00 2 20,67 219,14 218,09
180 254,85 236 25 228,08 223,76 221,21 219 56 218 44
240 258,50 237,11 228,36 223,88 221,28 219,61 218,48
312
Глава XI
таблица 40 (продолжение)
1 2 3 4 5 6 7 8
300 260,62 237,41 228,42 223,90 221,29 219,61 218,48
360 261,90 237,52 228,43 223,91 221,29 219,61 218,48
720 265,01 237,69 228,45 223,91 221,29 219,61 218.48
1440 265,38 237,69 228,45 223,91 221,29 219,61 218,48
2880 265,39 237,69 228,45 223,91 221,29 219,61 218,48
5760 265,39 237,69 228,45 223,91 221,29 219,61 218,48
10080 265,39 237,69 228,45 223,91 221,29 219,61 218,48
100000 265,39 237,69 228,45 223,91 221Д9 219,61 218,48
к* 0,001
0,5 1027 1027 1027 1027 1027 1027 1027
1 1111,7 1111,6 1111,5 1111,4 1111,3 1111,3 1111,2
2 1209,9 1209,6 1209,4 1209,3 1209,1 1209,0 1208,2
3 1268,4 1268,0 1267,7 1267,5 1267,3 1267 2 1267 0
4 1311,2 1310,7 1310,3 1310,1 1309,8 13 09,7 1309.5
5 1345,4 1344,7 1344,3 1344,0 1343 8 1 343,6 1343,4
6 1373,9 1373,2 1372,7 1372,4 1372,1 1371,9 1371,7
9 1441,2 1440,0 1439,3 1438,9 1438,6 1438,3 1438,1
15 1532.3 1530,4 1529,3 1528,6 1528,2 1527,8 1527.5
30 1669,9 1666,1 1664,0 1662,7 1661,8 1661,2 1660.7
45 1752,6 1746,7 1743,5 1741,6 1740,3 1739,4 17 38,6
60 1813.0 1805,0 1800,6 1798,0 1796.3 1795,1 1 794,1
90 1902,3 1889,8 1 883,2 1879,2 1876,6 1874,7 1873,2
120 1966.9 1949,9 1940.9 1935,5 1932,0 1929,4 1927,5
180 2061,3 2035,3 2021,6 2013,4 2007,9 2004,0 2001.1
240 2128,4 2093,6 2075,3 2064,3 2056,9 2051,8 2048,0
300 2180,5 2137,0 2114,2 2100,4 2091,3 2084,9 2080,3
360 2222,8 2170,9 2143,7 2127,3 2116,5 2109,0 2103,5
720 2390,4 2293,4 2244,0 2215,0 2196,5 2184,0 2175,1
1440 2536,4 2373,2 2296,8 2255,0 2229,6 2212,9 2201,4
2880 2638,6 2405,2 2310,7 2262,7 2234,6 2216,7 2204,5
5760 2685,1 2409,8 2311,4 2262,9 2234,7 2216,8 2204,5
10080 2692,0 2409,9 2311,4 2262,9 2234,7 2216,8 2204.5
100000 2692,5 2409,9 2311,4 2262,9 2234,7 2216.8 2204,5
А* =0,0001
0,5 7900,0 7900,0 7900,0 7900,0 7900,0 7900,0 7900,0
1 8543,0 8543,0 8543,0 8543,0 8543,0 8543,0 8543,0
2 9257,0 9257,0 9257,0 9257,0 9257,0 9257,0 9257.0
3 9696,1 9696,1 9696,1 9696,1 9696,1 9696.1 9696,1
4 10020,0 10020,0 10020,0 10020,0 10020,0 10020,0 10020 0
5 10279,0 10279,0 10279,0 10279,0 10279,0 10279,0 10279,0
6 10496,0 10496,0 10496,0 10496,0 10496,0 10496,0 10496.0
9 11003,0 11003,0 11003,0 11003,0 11003,0 11003.0 11003,0
15 11682,0 11682,0 11682,0 11682,0 11682,0 1168 2,0 11682,0
30 12705,0 12700,0 12699,0 12699,0 12699,0 12699 0 12699,0
45 13318,0 133 ю р 13309 0 133080 133080 133080 13308 0
Описание релаксационных процессор, й полимерах
313
таблица 40 (окончание)
1 2 3 4 5 6 7 8
60 13768,0 13758,0 13756.0 13755,0 13754.0 13754.0 13753,0
90 14439,0 14425,0 14421,0 14419,0 14418,0 14417,0 14416,0
120 14932,0 14914,0 14909,0 1490.5,0 14903,0 14902.0 14901.0
180 15668,0 15642,0 15633,0 15627,0 15623.0 15621,0 15620,0
240 16208,0 16174,0 1 6161,0 16153,0 16148,0 16145.0 16142,0
300 16639 0 16597 0 16580,0 16569,0 16562.0 16558,0 16554.0
36С 16999,0 16949,0 16928,0 16914.0 16906.0 16900,0 16896,0
720 18531,0 18429.0 18381,0 18351,0 18332,0 18319,0 1830.3,0
1440 20172,0 19963.0 19860,0 19796,0 19755.0 19726.0 19704.0
2880 21886.0 21465,0 21252,0 2112?0 210370 20978 0 20934 0
5760 23592,0 22789,0 22382,0 22139 ,0 21983,0 21877.0 21801,0
10080 24827,0 23555,0 22936,0 22583,0 22363,0 22218,0 22117,0
100000 ,26080 0 ,24104 0 ,23243 11 22789,0 225200 22348,0 2223 0,0
к* = 0.00001
0.5 59840 59840 59840 59840 59840 59840 59840
1 65248 65248 65248 65248 65248 65248 65248
2 71358 71358 71.358 71358 71358 71358 711.5.8
3 74932 74931 74931 74 931 7493 1 74931 74931
4 77514 775.14 77514 7 7514 77514 77514 77514
5 79553 79553 79553 79553 79553 79553 79553
6 81247 81246 81246 81246 81246 81246 81246
9 85191 85190 85190 85190 85189 85189 85189
15 90455 90454 90453 9045.3 9045.3 90452 90452
30 98281 98278 98277 98277 98276 98276 98276
45 102922 102918 102916 102915 102915 102914 102914
60 106304 106300 106297 106295 106294 105294 106293
90 11 1.320 111313 111309 111307 111305 111304 111303
120 114985 114975 114970 114967 114965 114963 114962
180 120433 1 20417 120409 120404 120401 120398 120397
240 124422 124401 124390 124383 124379 124376 124373
300 127605 127577 127563 127554 127549 127545 127542
360 130268 130233 130216 130206 130199 130194 130190
720 141656 14158 1 141544 141522 141507 141496 141488
1440 154141 1539 80 153899 153850 15.3818 153795 15.3778
2880 167823 167475 167298 167192 167121 167071 167033
5760 182744 181990 181608 181.377 181223 1811L3 181032
10080 195357 193966 193256 192826 192538 192335 192184
100000 211967 209097 207617 206718 206120 205700 205393
Результаты расчета приведены в табл.41 и на рис. 85. Видно, что ядро 7] (т)
лучше передает ход релаксации напряжения, чем 7?(т). Коэффициент корре-
ляции при этом близок к единице. Следовательно, лимитирующей стадией
процесса на начальном участке кривой релаксации напряжения в данном слу-
чае является взаимодействие релаксаторов и переход их в нсрслаксируюший
материал. Из табл.41 видно также, что величина к*, пропорциональная кон-
314
Глава XI
i t
Рис.85. Зависимости с от jГ*(т)Л (а) и jT2(t)dt (б) для блочных образцов ПММА
о о
(деформация 2,2 %) при Т, К: 294 (1); 313 (2); 323 (3) и 333 (4)
t t
Dependences of c on j'/ffrja't (a) and |7’*(т)Л (6) for bulky specimens of
о 0
PMMA (the deformation is 2,2 %) at T, K: 294 (1), 313 (2), 323 (3) and 333 (4)
стайте скорости взаимодействия к, при разных температурах является посто-
янной. Основной вклад в скорость процесса релаксации вносит величина
/Sq, пропорциональная числу кинетических единиц /и,, вызывающих ре-
лаксацию. Эти величины закономерно уменьшаются с повышением темпера-
туры. Естественно, что для более точного определения параметров ядер Тi (т)
и Т2(т) нужны более длительные эксперименты.
В заключение следует рассмотреть один методический вопрос, который
связан с надежностью определения параметров релаксационного процесса
путем аппроксимации кривых релаксации напряжения. (
С этой целью рассмотрим графики зависимостей к* J/j* (г) А от Igt для
* о
ядра Т3 (т), показанные на рис.86. Видно,что для каждого выбранного значе-
ния константы скорости реакции к* кривые для разных значений [5 на началь-
ных участках сливаются, а затем начинают расходиться .Длительность релак-
Описание релаксационных процессов в полимерах
315
Таблица 41
Значения параметров /г*, р, А,. = с^оЦк^т^ к^т,JSq, а, у, ядер релаксации Т^т) л ТзСг),
на'шльнык напряжений og и коэффициентов корреляции г для полиметилметакрнлата
при деформации Eg = 2,2 %
Values of parameters /с*, p, А, = GoSo^ksmj), kg m, ISq, а, у, memory functions Тi(t) and 7j(t),
initial stresses ag, and correlation coefficients r for polymethylmethacrylate with deformation
equal to eq =2,2 %.
Т,К. 7’;(т); к* = 0,0001 мин4; 3 = 0,2 ТУт); а = 0,05 мин у = 0,5
А104, МПа По, МПа кБт{ So Г а2 , МПа Оо, МПа кБт2 So Г
294 1,6 49,64 310250 0,998 2,272 40,39 17,78 0,991
313 2,3 52,29 227350 0,999 3,197 39,33 12,30 0,995
323 1,8 38.25 212500 0,999 2,465 28,2 0 14,44 0,991
333 2,3 39,25 170650 0,999 3,177 26, 31 8,28 0,991
* Для атома углерода значение к* пренебрежимо мало
t
Рис.86. Зависимость к* (т)А от Igt, при к'= 0,1 (1); 0,01 (2); 0,001 (3); 0,0001 (4)
0 и 0,00001 (5)
г
Dependences of к* (x)dx on Igt when к* = 0,1 (1); 0,01 (2); 0,001 (3);
° 0,0001 (4) and 0,00001 (5)
сациоиного процесса, при которой наступает расхождение этих кривых, за-
висит от к*'.чем выше эта величина дем при меньших длительностях процес -
* ‘
са начинается расхождение зависимостей к |73 (т) А от t. Эта область t и к*
о
(заштрихована на рис.87) характеризует длительность релаксационного про-
цесса, которую необходимо реализовать в эксперименте для того, чтобы по-
лучить надежные значения величин 0, характеризующих порядок реакции п.
316
Глава XI
т.к. Р = 1/(и - 1). Эксперимент, проведенный при меньших временах /, не по-
зволяет надежно определить порядок реакции. С помощью такого экспе-
римента можно определить лишь константу скорости. Например, если кон-
станта скорости равна 0,1 мин1, то длительность релаксационного процесса,
реализуемая в эксперименте, должна превышать 2 мин, при к* = 0,01 мин1
эта длительность составляет уже 60 мин, и т.д.
Таким образом, с помощью графика на рис.87 можно определить мини-
мальное время, в течение которого необходимо вести эксперимент по релак-
сации напряжения для надежного определения параметров процесса. Есте-
ственно, что при снижении константы скорости процесса длительность экспе -
римента должна существенно увеличиваться.
Вся изложенная выше процедура аппроксимации кривых релаксации на-
пряжения а(/) справедлива для случая линейного механического поведения
полимерных материалов, когда параметры процесса не зависят от его дли-
тельности и величины деформации. Следует остановиться на возможности
описания нелинейных релаксационных процессов, которые для полимерных
материалов являются наиболее характерными даже при малых деформациях.
В настоящее время наиболее распространенным методом аппроксимации
кривых релаксации напряжения в нелинейной области механического пове-
дения является способ, основанный на главной ку сичной теории Ильюшина
[73]. Согласно [73], сначала проводится аппроксимация релаксационного
модуля Er(t) = о(/)/£о в линейной области вязкоупругости, а затем, путем ввс-
Рис.87. Логарифмическая зависимость минимальной длительности релаксационною
процесса t от к', при которой возможно надежное определение парамецж (1
Logarithmic dependence of minimum duration of relaxation process t on k* at which
reliable detennination of [5 parameter is possible
Описание релаксационных процессов в полимерах
317
дения еще одного параметра и использования того же самого ядра релакса-
ции, но с другими параметрами, аппроксимация релаксационных кривых в
нелинейной области. Другой способ заключается в использовании уравне-
ний, содержащих дробный показатель степени времени, причем этот показа-
тель считается зависящим от величины деформации [220], поддерживаемой
постоянной в ходе релаксационного процесса. Как в одном, так и в другом
случае, хотя и достигается хорошее согласие экспериментальных кривых с
расчетными физический смысл вводимых новых параметров не раскрывает-
ся.
В данном разделе рассмотрим подход к описанию кривых релаксации
напряжения в нелинейной области с помощью физически обоснованных па -
рамстров. входящих в ядро релаксации (270).
Отметим, что ядро (270) получено на основе расчета энтропии смешения
только для двух типов единиц - релаксаторов и нерелаксаторов; если же ко-
личество типов кинетических единиц,вн осящнх «ущественны! вклад в ре-
лаксационный процесс, больше двух, то появится несколько констант скоро-
стей взаимодействия ралаксаторов различиях типов и соответственно несколь-
ко величин к*, и это в конечном итоге приведет к появлению спектра констант
взаимодействия, что аналогично спектру времен релаксации. Однако много-
численные опыты показали, что для описания кривых релаксации напряже-
ния даже при значительной длительности процесса t достаточно одной кон-
станты скорости взаимодействия релаксаторов к*. Это ознатоет гризшние
факта, что хотя релаксаторы могут быть разных типов, но лишь один из них
вносит существенный вклад в ход релаксационного процесса. Поэтому в даль-
нейшем, переходя к описанию процедуры аппроксимации релаксационных
зависимостей в нелинейной области механического поведения полимеров,
будем пользоваться ядром 7’](т).
Идеология такой аппроксимации состоит в следующем [15]. Запишем
выражение для температурной зависимости константы скорости
к* ^kfocxp(-MJ/RT) , (299)
где к*0 - предэкспоненциальный множитель, &U - энергия активации про-
цесса взаимодействия; R -универсальная газовая постоянная; /’-абсолютная
температура. Известно, что в ходе деформирования полимеров их свободный
объем увеличивается (под свободным объемом в данном случае подразумева-
ем пустой объем, представляющий собой разность между истинным объе-
мом полимерного тела и Ван-дер-Ваальсовым объемом атомов, который они
занимают в полимерном теле). При сильном деформировании твердых (стек-
лообразных и кристаллических) полимеров свободный объем увеличивается
до достаточно большой величины, что существенно облегчает перескок ки-
318
Глава XI
нетических единиц из одного положения в другое. Это и приводит к вынуж-
денной эластичности, т.е. к вынужденному размягчению материала. Поэтому
если предположить, что энергия активации взаимодействия ралаксаторов сни-
жается с ростом механического напряжения, то при достаточно высоком его
значении это может повлечь за собой возникновение избыточного свободно-
го объема. На этой основе возникает выражение для температурной зави-
симости времени релаксации напряжения [1, 65]. Таким образом, запишем
,♦ ,* , XIX-Ъа*. * _ АГ/п-5Егеп,
к = А'оехр(---—Ч=Аоехр(---------°-- °), (300)
где Ег - релаксационный модуль; AU0 - начальная энергия взаимодействия
релаксаторов; ог - релаксирующее напряжение; е о -постоянная деформация.
5 - флуктуационный объем, в котором происходит элементарный акт взаимо-
действия релаксаторов.
При этом в линейной области механического поведения, когда напря-
жение еще не столь велико для образования избыточного свободного
объема, будем считать, что величина 5=0, т.е. константа скорости к*-
*
= к0 ехр(-А£70 /RT) и не зависит от механического напряжения. С ростом
задаваемой постоянной деформации Eq наступает момент, когда появляется
большой избыточный свободный объем, что существенно облегчает взаимо-
действие релаксаторов и приводит к ускорению релаксационного процесса.
Это и есть с рассматриваемых позиций переход к нелинейному' поведению. В
этом случае величина к* не является константой, а становится зависимой от
релаксационного модуля согласно выражению (300). Учет этого позволяет
провести аппроксимацию кривых релаксации напряжения в нелинейной об-
ласти и одновременно определить избыточный флуктуационный объем 5, в
котором происходит элементарный акт взаимодействия релаксаторов.
Прежде чем описать процедуру аппроксимации релаксационных кривых
с помощью предложенного подхода, перепишемуравнение Больцмана в виде
t
E(t) = Е0 -E0^T(T)dr, (301)
о
где Ео - начальный модуль, возникающий после “мгновенного” задания де-
формации; Т(т) - ядро релаксации. Опыт показывает, что наилучшая аппрок-
симация релаксационных кривых для твердых полимеров достигается при
применении ядра 7\(т), которым и будем пользоваться в дальнейшем. Под-
ставляя ядро T’i(t) в уравнение (301), получаем
Е(1) = £0 --^47f(T)A . (302)
kF,m\ о
Описание релаксационных процессов в полимерах
319
где - переменная часть ядра 7'| (т), описываемая уравнением (291).
Для случая релаксации напряжения в нелинейной области механическо-
го поведения
(303)
1
а =
* АГ/о -8Ег£о
ехр(------—------)т
____________Л 7______
р
Из сравнения уравнений (269) и (303) следует, что член кд ехр(-ДС7 /RT),
не зависящий от релаксационного модуля Ег, соответствует к* в уравнении
(269) для линейной области механического поведения. Таким образом, мож-
но записать
(304)’
Процедура аппроксимации состоит в определении значения 8, при кото-
ром минимально значение функции ф(8), являющейся суммой квадратов от-
клонений:
п
ф(5) — 2j(^/,pac4. — ^;,эксп.) ,
/=1
где п - количество экспериментальных точек; Etpac4 и Е, ^„ - вычисленные
по уравнению (302) и определенные экспериментально значения релаксаци-
онного модуля соответственно.
Алгоритм вычислений заключается в следующем. В компьютер после-
довательно в порядке возрастания значений деформации Eq вводятся значе-
ния релаксационных модулей для экспериментальных кривых релаксации
напряжения. Каждая вводимая кривая, кроме первой, сравнивается с усред-
ненной кривой, представляющей средние значения релаксационных модулей
из ранее введенных кривых. Если у вновь введенной кривой каждое значение
модуля при одном и том же значении длительности релаксации меньше, чем
у усредненной кривой, и среднее арифметическое относительных отклоне-
ний превышает 10 %, то такая кривая считается относящейся к нелинейной
области механического поведения. Тогда для усредненной кривой по методу,
320
Глава XI
описанному выше, вычисляются параметры релаксации для линейной облас-
ти и на основе их проводится аппроксимация для случая, относящегося к
нелинейной облаете.
Поиск минимума функции ф(б) проводится методом обратного перемен-
ного шага, а интегрирование ядра релаксации осуществляется методом Симп-
сона с заданной точностью (как правило 0,001 %).
В функцию а, входящую в подинтегралыюе выражение (302), входитре-
лаксационный модуль Е, ко торый, как и сама функция (304), зависит от вре-
мени т. Однако причисленном интегрировании ядра релаксации (270) релак-
сационный модуль представляется в диапазоне между двумя эксперименталь-
но определенными точками Г;И /(+| в виде постоянной величины,равной
Е/+ ] эксп , что, как будет показано ниже, не отражается на результатах аппрок-
симации.
Необходимо о гмететь, что из уравнений (270) и (302) следует, что в точке
т = 0 а - 1, а разность (а- с^)равна числу 0,9999999999, которое ЭВМ рас-
сматриваегкак 1 и выдает сообщение об ошибке, так как аргумент натураль-
ного логарифма обращается в 0. Чтобы обойти эту сложность, уравнение Боль-
цмана можно переписать в виде
/
Е(О = £’,-Я0/7'(т)Л,
О
где/|иЕ) соответствешюзначенияврсмениирелаксапионногомодулядля
первой точки экспериментальной кривой. Одновременно такое представле-
ние значительно снижает время работы программы, т ак как отпадает необхо-
димость интегрирования наиболее крутого участка релаксационной кривой.
XI.2. Процессы сорбции и набухания
Рассмотрешгьгй вьгше аппарат для описания релаксационных явлений в
полимерах применим и к таким процессам, как сорбция и набухание Дело в
том, что данные процессы проходят не только путем заполнения отдельных
пор в полимерном теле, но и вызывают конформационные перестройки мак-
ромолекул, т.е. сопровождаются релаксаг дюнными процессами .Эга идея была
высказана сравнительно давно в рядерабог [63,67,71], однако детальный
анализ этого процесса с учетом релаксаггнойного механизма сорбции и набу-
хания был проведен в работах [10, 72]. Так ой подход позволяет выявить о б -
ратные связи, развивающиеся в процессе сорбции паров полимерами Обрат-
ные связи обусловлены, как уже упоминалось, тем, что проникновение паров
низкомолекулярных жидкостей в полимер вызывает реконструкцию, подчас
серьезную, надмолекулярной организации, что в свою очередь влияет на ки-
нетику дальнейшей сор бцг ги и диффузии.
Описание релаксационных процессов в полимерах 321
Анализ проведем на основе решения системы дифференциальных урав-
нений [120]
r.v_^ = 0. v.v_Dfr_ = Q (305)
дх дх
где / - сила, действующая на диффундирующую частицу; v - количество
диффундирующих частиц; v - скорость диффундирующей частицы; D -
коэффициент диффузии; Р - осмотическое давление, равное (y/N)R T;N-
общее количество частиц в системе; R - универсальная газовая постоян-
ная; Т - абсолютная температура.
Из системы уравнений (305) с учетом выражения для Р находим
72(0 = — •— (306)
N f •
Поскольку полимер вязкоупругая среда, смещение диффундирующей
частицы в нем, определяемое скоростью ее движения v, зависит от кинетичес-
ких релаксационных параметров среды. Иными словами, необходимо связать
скорость диффундирующей частицы, которая при проникновении в вязкоуп-
ругое тело становится зависимой от времени v(t), с силой /, входящей в сис-
тему уравнений (305).
П роведсм анализ процессов сорб ции и наб ухания в об щемвпде ,исполь -
зуя наследственную теорию Больцмана-Вольтерры, и выбрав рассмотренные
выше ядра для описания ползучест полимерных тел.
Поскольку резольвенты от ядер (270) и (279) еще не найдены, для описа-
ния процесса ползучести полимеров можно воспользоваться теми же ядрами
(270) и (279), но с другими параметрами, чем те, которые пригодны для опи-
сания релаксации напряжения.
Процесс ползучести с применением ядер (270) в (279) описывается соот-
ношениями
£(О = ео[1 + |7](т)Лт] ; (307)
о
<0 =EO[1+J 7’2(тИт] , (308)
о
где e(Z) - деформация, развивающаяся к моменту времени t; щ упругая (мгно-
венная) деформация.
Если процесс лимитируется скоростью взаимодействия релаксаторов, то
ползучесть описывается уравнением (307), а если он лимитируется ди фузи-
ей неоднородностей в материале, то ползучесть описывается уравнением (308).
Проведем анализ процесса сорбции и набухания с помощью аппа-
рата, использованного в работе [72], с привлечением ядер 7'|(т) и 7’2(т).
322
Глава XI
Величина v составит
р(т)=Дф)/0], (309)
ат
где /0 - характерная начальная длина образца.
Воспользовавшись выражениями (307) и (308), запишем
v(t) = e0/07](t) (310)
или
v(t)=e0/0T2(t). (311)
Из соотношений (310) и (311) имеем
г(т)=^7’1(т) (312)
ЛЬ
ИЛИ
г(т)=МГ2(т); (313)
оЛ
где 5 - поперечное сечение образца; Е - мгновенный модуль упругости;
/ - сила, действующая на образец.
Подставляя соотношение (312) и (313) в выражение (306), получим
(314)
N SE
или
В(т)=—-^(т). (315)
,V Ж
Решая так же, как и в работе [72], уравнение диффузии с переменным
коэффициентом диффузии 7)(т) для пластины толщиной /, можно найти кине-
тическую зависимость относительного количества сорбированного вещества
\ X-1 1 f г г +1 2 R-R г* г / +1 2
А/(/)Ш(оо)= £-------{ехр[-(л——) •—/ (0)]-ехр[-(л—-—)2 х
„=о(2» + 1)2 I I
RT /мшг 1 < г / 2л + 1ч2 RT ,
<0)Ь
- ехр[-~f’(<>п, (316)
IN
Описание релаксационных процессов в полимерах
323
где /(/)=^[j7im + AJ SE Jo (317)
или (318)
При этом 0
/*(0) = —В] или f (0)=-^--В2 SE ЬЕ (319)
/(со) = —(/Ij+Bj) или f (°°) = -^;(^2 + 52), Ж SE (320)
где Ai = jTjCOA или А2 = |Г2(т)А .
о о
RT *
Так как---f (0 «1, то на основании формулы (316) можно записать
N
M(t) /(0)-/(0 (321)
Л/(со) /(0)-/(*>)'
Подставляя всоотношенис (321)выражения(317), (319) или(318), (320),
получаем соответственно f
|Л(т)Л
-^2. = °------ (322)
л/(оо) 4
ИЛИ /
[Т2(т)Л:
W) = о________ (323)
A<f(co) Л2
В случае процессов десорбции или синерезиса кинетические уравнения
приобретают вид
^ = 1-}г](т)Л (324)
и о
или
^ = 1-р2(т)Л, (325)
и о
где Л/о - начальная масса набухшего образца.
324
ГлаваХ!
м(оо)
или
WL
Л/(оо)
Учитывая, что ядра релаксации (270) и (279) содержат постоянные вели-
чины, которые можно вынести за знак интеграла, из выражений (322) и (323)
получаем
t
_ Jo______
7 , (326)
J Л (t)Jt
О
t
jr2*(T)dT
, (327)
jT’jCOt/T
о
где Т*(т) и 7’2*(т) - соответственно переменные части ядер ГДт) и Т2{т).
Для экспериментального подтверждения возможности описания процесса
сорбции с помощью формул (326) и (327) в работе [10] проведены опыты по
измерению кинетики на свободных пленках и покрытиях на основе отверж-
денной эпоксидной смолы ЭД-20.
На рис.88 показаны кинетические кривые сорбции, полученные на об-
разцах отвержденных эпоксидных смол как в виде свободных пленок, так и в
виде покрытий на алюминиевой подложке с различным видом ее обработки.
Результаты расчетов приведены в табл.42. Расчеты кинетических пара-
метров процесса сорбции проводили по уравнениям (326) и (327), используя
t t
табулированные значения интегралов j7] (т)<7т и ^Т2 (t)dz , приведенные
выше. Если уравнения (326) и (327) правильно описывают процесс сорбции,
t t
то графики в координатах Л/(/) - j?] (т)о'т илиЛ/(/)- $Т2 (т)Л должны пред-
ставлять собой прямые, выходящие из начала координат, а тангенс угла на-
клона их равен Л/(оо)/ Ау или Л/(оо)/ А2 . Этому требованию удовлетворяет
уравнение (325), что хорошо видно из рис.89. Экспериментальные точки хо-
t
рошо укладываются на расчетную прямую в координатах M(f) - jT2 (т)Ат с
выходом прямой из начала координат. Коэффициент корреляции кэлеблется
от 0,998 до 0,996. Параметры ядра Т2(т) приведены в табл.42.
Описание релаксационных процессов в полимерах
325
Рис.88. Кинетические кривые сорбции паров воды прир/р = 0,08: 1 - свободная пленка
ЭД-20; 2 - ЭД-20 на обезжиренной алюминиевой фольге; 3 - ЭД-20 на алюминиевой
фольге, обработанной у-аминоиронилтриэтоксисилапом
Kinetic curves of water vapour sorption at p/px = 0.08: 1 - free film of ED-20; 2 -
when ED-20 is on degreased aluminum foil; 3 - when ED-20 is on aluminum foil proccessed
withY-aminopropyltriethoxysilane
Описание кинетических кривых сорбции с привлечением ядра 1\ (т) по-
казало [10] худшее совпадение расчетных и экспериментальных значений А/(().
Напомним, что ядро '/((т) действует в том случае, если ход релаксационного
процесса лимитируется скоростью взаимодействия релаксаторов. Если же этот
процесс лимитируется их диффузией в материале (те. самодиффузисй), то
действует ядро Т. В случае сорбции паров низкомо пекулярных жидкостей ,
Кинетические параметры процесса сор Гщпг
Kinetic parameters of sorbing processes
Вид образца DIO’10, см7с Параметры ядра ТУт) (время в мин) Скорость изменения Mt!Ми пр и 1
а У 1 мни 60 мин
Свободная пленка 4,361 0,0346 0,4 0,0297 0,00395
Иокрьиис на обезжиренной поверхности алюминия 3,451 0,0209 0,5 0,0427 0,00414
Покрытие па обработанной новерхноси] алюминия 2,707 0,1040 0.3 0,065.3 0.004.3.3
326
ГлаваХ!
С *
Рис.89. Зависимость Л//Л/о от I Tj (т)</т
О
i
С *
Dependence of М,/Мд on 73 (т)dz
О
как показали проведенные расчеты [10], процесс хорошо описывается с по-
мощью ядра 7'2(т), т.е. лимитирующей стадией процесса, лежащего в основе
сорбции, является самодиффузия релаксаторов. В таблице также приведены
коэффициенты диффузии, рассчитанные по обычному уравнению Фика.
В табл.42 приведена скорость изменения относительного привеса массы
{г7[Л7(г)/Л7(°о)]}/<Й, которая вычисляется на основе соотношения (323)
(328)
И®)
где величина берется из табл.39. Зная параметры ядра 7, (т), легко рас-
считать v в различные моменты времени t.
Таким образом, метод описания кинетических кривых сорбции, унизы-
вающий релаксационный характер этого процесса, позволяет с использова-
нием новых ядер релаксации аппроксимировать кинетические зависимости с
хорошей точностью на всем их протяжении. Если же использовать для этих
целей уравнения Фика с постоянным коэффициентом диффузии, то это не
удается, и можно лишь аппроксимировать процесс на небольшом начальном
отрезке времени.
Глава XII
РАСТВОРИМОСТЬ ПОЛИМЕРОВ
ХИЛ. Плотность энергии когезии
органических жидкостей и полимеров.
Параметр растворимости Гильдебранда
Для предсказания растворимости полимера в различных органических
растворителях, а также для предвари тельной оценки совместимости полиме-
ров друг с другом или с пластификаторами част о пользуются такой характе-
ристикой, как параметр растворимости 5. Эта характеристика введена Гиль-
дебрандом для описания растворов неэлектролитов. Параметр растворим ос-
та Гильдебранда определяется из соотношения
5= (329)
где А£о = ДН0 -ЯГ; Д770 скрытая теплота испарения жидкости-,R -уни-
версальная газовая постоянная; Г абсолютная температура; К- мольный
объем жидкости.
Квадрат параметра растворимости представляет собой плотность энер-
гиикогезии жидкости, т.е. величину энергии когезии, деленную на мольный
объем:
S2=AE0/r. (330)
Эта представления распространены и на полимеры, причем оценки при-
водятся в расчете на повторяющееся звено. Трудность здесь заключается в
том, что экспериментально величину 5можно определить только для низко-
молекулярных жидкостей, испаряющихся без разложения. Для полимеров,
которые нельзя испарить без разложения, значения 5 определяются косвенны
328
Глава XII
ми методами или расчетным путем по инкрементам энергий для отдель-
ных атомов и групп атомов [141, 150,206].
Учет характера упаковки молекул в жидкостях и полимерах приводит к
следующему уравнению для расчета плотности энергии когезии [25] .
62=-2^ = -±_____________ (331)
i /
где ЛЕ* = к\Еа - энергия когезии жидкости или повторяющегося звена по-
лимера, уменьшенная во столько раз, во сколько Ван-дер-Ваальсовый объем
молекулы (или звена) меньше мольного объема; Аг - коэффициент .молекуляр-
ной упаковки жидкости или полимера.
Величина АТ?* является аддитивной и представляется в виде ХЕ* = уДЕ,
где ДЕ,*- вклад каждого атома и типа межмолекулярного взаимодействия в ДЕ*.
Значения ДЕ,* приведены в табл.43, в которой даны соответствующие по-
яснения. С их помощью можно рассчитать параметр растворимости 5 для
многих полимеров разнообразного химического строения.
Таблица 43
Значения для различных видов атомов и типов межмолекулярного взаимодействия
Values A/?z for different atoms and types of interinolecular interaction
Атом и тип межмолекулярного взаимодействия Условное обозначение •> кал/моль
1 2 3
Углерод ДА’с 550,7
Водород ле; 47,7
Кислород ле; 142,6
Азот 1205,0
Фтор Ojfp 24,2
Сера XE*S 1750,0
Хлор -222,7
Бром EE*Br 583,0
Йод ХЕ* 1700,0
Растворимость полимеров
329
таблица 43 (окончание)
1 2 3
Двойная связь ДЕ* -323,0
Ароматический цикл (скелет) ДЕ^ 713,0
Днполь-дипольиое взаимодействие де; 1623,0
Диполь-дипольное взаимодействие в неполярных апротонных растворителях: амидного типа типа диметилсульфоксида * <3* ♦ с $ 1623,0 2600,0
Водородная связь де; 3929,0
Специфические взаимодействия в присутствии группы = СС12 №,..сс12 2600,0
В напряженных трех-пятичленных циклах в присутствии атома О Мо.ц 2430,0
Изомерия углеводородного радикала АЕ* -412,0 ।
1) Инкремент ЛЕ1* вводится при наличии двойных связей ,не входящих в состав полярных групп.
2) Инкремент AFq вводится при наличии ароматических ядер в количестве, соответствующем числу
этих ядер.
3) Инкремент ДЕ^ вводится во всех случаях при наличии полярной группы любого типа; в случае
хлорированных соединений при наличии 2 и более атомов С1, присоединенных к одному и тому
же атому углерода, необходимо вводить 2 ЛЕ^ .
4) Инкремент ЛЕДф дг вводится при расчете S для диполярных апротонных растворителей амидного
типа; при этом обычное диполь-дипольное взаимодействие за счет полярных групп учитывается
введением соответствующею количества инкрементов.
5) То же самое, что и в предыдущем случае, но для растворителей типа диметилсульфоксида.
6) Инкремент ДЕ^ вводился при наличии водородной связи любого типа.
7) Инкремепт АЕ^СС/2 учитывает специфическое взаимодействие при наличии группы
С1
~С < ; ПрИ этом инкремент AEj не вводится
а
8) Инкремент ДЕО ц учитывает появление полярности в напряженных 3-5 членных циклах,
содержащих гетероатом О.
9) Инкремент ДЕ’* вводится при переходе от п.углеводородов к разветвленным.
330
Глава XII
Экспериментальные методы определения параметра растворимости 8 зак-
лючаются в следующем. Измеряется величина характеристической вязкости
1] полимера в наборе растворителей, которые обладают различными значени-
ями параметра растворимости. Далее строятся зависимости т] полимера от
параметра 3 растворимости того растворителя, в котором они были измерены.
Схематически это изображено на рис.90. Максимум такой зависимости
определяет параметр растворимости полимера, который в данном случае ра-
вен параметру растворимости жидкости, в которой характеристическая вяз-
кость данного полимера максимальна (см.рис.90).
Другим экспериментальным методом оценки величины 8 является изме-
нение равновесной степени набухания с последующим построением зависи-
мости этой величины от параметра растворимости жидкости, в парах которой
измерялось набухание. Эта зависимость аналогична изображенной на рис.90.
Следует отметить, что экспериментальные методы определения 8 трудоемки
и не всегда надежны. Для предварительной оценки 8 предпочтение можно
отдать расчетным методам.
Для предсказания растворимости полимеров иногда сравнивают расчет-
ное значение 8П для полимера с экспериментальными значениями 8р раство-
рителей. Если эти величины совпадают, можно надеяться на растворение по-
лимера в данных растворителях. Если же значения 8 для полимера и раство-
рителей различаются сильно, то растворение не происходит. Совпадение
параметров растворимости полимера и растворителя, однако, еще не гаранти-
рует растворения полимера в данном растворителе. Как показывает практика,
в случае совпадения величин 8 растворение наблюдается лишь в50% случаев
(см. ниже).
I’ll
Рис.90. Схематическое изображение зависимости характеристической вязкости [т|]
полимеров в разных растворителях от параметра растворимости растворителя 6р
Schematic representation of dependence of intrinsic viscosity [t|] of polymers in
different solvents on solubility parameter of solvent
Растворимость полимеров
331
В табл. 44 приведены параметры растворимости 8 для полимеров разно-
образного химического строения. Химическое строение оказывает весьма су-
щественное влияние на величину 8. Наличие в полимере сильных полярных
групп типа — ci, -С~О“> — С SN и ДР- приводит к возрастанию энергии
II
О
когезии, а следовательно, и величины 8. Особенно сильно увеличивают пара-
метр растворимости водородные связи, возникающие при наличии — ОН и
—HNC- групп, а также ароматические ядра.
О
Для сополимеров уравнение для расчета параметра растворимости запи-
сывается в виде
) 1 + а 2<Е )2 + • • •+а л(Е ) п
§2 =--_j--------!-----------!----
NА а1 (X )1 + а2(X)2 + + “и Д1< X
(331')
п
Таблица 44
Значения параметров растворимости для ряда полимеров
Parameters of solubility of a series of polymers
№№ un Наименование / см/моль 8, кал0,5/см'’5
расч. экспер. [54]
1 Полиметилметакрилат 5043 58,5 9.3 9,1; 9,5; 9,4
2 Полиэтил мета крилат 5689 69.0 9,1 8,95
3 Поли-и-пропил-мегакрилаг 6335 79,3 8.9 -
4 Поли-п-бутил-метакрилат 6981 89,6 8,8 -
5 Поли метилакрилат 4397 48,2 9.55 10,1
6 Полиэтилакрилат 5043 58,5 9,3 9,4
7 Поли ст и рол 5500 66,0 9,1 9,1; 8,6; 8,7
8 Поли изобутилен 2584 41.6 7,9 7,95; 7,8; 8,05
9 Полиакрилонитрил 4623 32,6 11,9 -
10 Полиэтилентерг^палат 10418 102,4 10,1
II Найлон 6,6 18210 139,2 11,4 -
12 Полиарилат фенолфталеина и изофталовой кислоты 27189 234,7 10,7 10,8
13 Полиарилатфенолфталеина и терефталевой кислоты 27189 234,7 10,7 10,7
14 Полиарилат фенолфталеина и 4,4‘ дифенилдикарбоновой кислоты 31397 279? 10 6 10 4
332
Глава XII
гдеаьа2, ..., а,,-молярные доли компонентов 1, 2, (£AJ<)i, (£&K)2,
i i
, (£Щ)п -Ван-дер-Ваальсовые объемы компонентов 1,2, ,
(^АЕ/ h, , - энергия когезии компонентов 1, 2, it, Na - число
i i
Авогадро.
В более компактной форме уравнение (33 Г) записывается в виде
к=п
'£ак(£ЛЕ*)к
S2 = -^=п z----------, (331")
±ak(£W,)lc
к=] i
где и (^ ^'i )k ~ молярная доля и Ван-дер-Ваальсовый объем А--го компо-
I
нента соответственно, (^ДЛ';)^ -его энергия когезии.
i
Если желательно выразить параметр растворимости через параметры ра-
створимости компонентов сополимера, следует записать
ai52(S W )1 + «232(Е h + • • •+а„52п (X Щ)п
32=---------------------'----------------------- (33 Г")
a,(£AE,)i +(Х2(£Д^)2 +... + а„(ХД^)л
i i i
где 8b 82, ..., 8„ - параметры растворимости компонентов 1, 2, п.
В более компактной форме это уравнение записывается в виде
к=п
Х^кХ^к
52=-^---------'----, (331"")
к=1 ;
где ак, (^ АИ, )к, 6к - молярная доля, Ван-дер-Ваальсовый объем и параметр
растворимости для к-го компонента соответственно.
Растворимость полимеров
333
XII.2. Критерий растворимости
Проблема предсказания растворимости полимеров является актуальной
в течение многих лет. Оцин из способов предварительной оценки раствори-
мости полимера - сопоставление величин параметров растворимости 8 Гиль-
дебранда для полимера 8П и растворителя 8р. При этом часто считается, что
если соблюдается условие 8П ~8р,то можно ожидать растворения полимера в
данном растворителе. Опыт показывает, однако, что с помощью такого сопо-
ставления можно лишь уверенно “отбросить” те растворители, в которых ра-
створение данного полимера происходить не будет. Это системы, для кото-
рых 8П» 8рили 8П« Зр. С помощью такой оценки удается значительно
сузить круг подлежащих проверке растворителей, в которых полимер может
растворяться. Оценки и опыт показывают [128], что, например, из 160 ра-
створителей можно таким способом сразу же для каждого полимера исклю-
чить из расемотрения 120-130 органических жидкостей, как явно непригод-
ных для растворения. В оставшихся растворителях, подчиняющихся усло-
вию 8П ~ 8р, примерно в половине из них полимер будет растворим.
Следовательно, соблюдение условия 6П = Зр нс может дать гарантию ра-
створимости полимера .Желательно иметь более точный способ предваритель-
ной оценки растворимости полимера по отношению к тем растворителям, для
которых соблюдается условие 8П = Зр. Рассмотрим в деталях критерий раство-
римости, предложенный в работах [32,95], который обладает достаточно вы-
сокой предсказательной силой.
Особенность проблемы растворения заключается в том, что здесь, в от-
личие от определения некоторых простейших свойств, необходимо учиты-
вать не только химическое строение, но также конкретную надмолекулярную
структуру полимера.
Действительно, хорошо известно, что кристаллический полимер раство-
ряется гораздо хуже, чем аморфный полимер того же химического строения.
Ориентированные образцы также хуже растворяются по сравнению с изот-
ропными образцами .Возможно, что и в случае изотропных образцов аморф-
ных полимеров надмолекулярная структура может быть различной, однако
этот вопрос до сих пор является предметом дискуссии. На международной
конференции в Лондоне в 1979 г. были представлены экспериментальные и
теоретические данные об отсутствии “нодульной” структуры в амор (]ных по-
лимерах, причем данные электронно-микроскопических исследований повер-
хности пленок и сколов были причислены к артефактам [142] .Трудно,одна-
ко, представить, что если поверхность пленки, полученной из раствора, и по-
верхность скола блочного образца, полученного из расплава, дают одну' и ту
же электронно-микроскопическую картину глобул, то эта картина является
следствием артефактов.
334
Глава XII
Было отмечено, что только метод нейтронного рассеяния может дать пря-
мую информацию по этому вопросу, хотя трактовка данных не является од-
нозначной. Эта дискуссия продолжалась и далее. На основании результатов
рентгеновского рассеяния утверждается [189], что имеется некоторый уро-
вень упаковочной регулярности и в аморфных полимерах.
При анализе проблемы растворимости в работе [32] исходили из модели
надмолекулярной структуры, развитой в работах [92,93], в которых надмоле -
кулярная структура аморфных полимеров моделируется в виде глобул, при-
чем в этих работах сделана попытка обосновать отсутствие большого периода
при малоугловом рентгеновском рассеянии. Принято также, что каждая гло-
була состоит из глобул-макромолекул [4,102]. Полагая, что те и другие глобу-
лы связаны друг с другом поясками связи, рассмотрим наи более характерный
элементарный акт растворения, те. распада частиц до отдельных глобуляр-
ных макромолекул, который схематически изображен на рис.91.
Рис.91. Схематическое изображение поверхности полимера и элементарного акта
растворения
Schematic representation of polymer surface and elementary event of dissolution
Здесь показан случай неровной поверхности (допустим, порошка или
пленки с шероховатостями), когда сила со стороны растворителя приложена
к пояску глобулы, по которому она связана с соседними глобулами, и отделе-
ние глобулы от полимерного тела и переход ее в растворитель происходит за
счет разрыва пояска связи.
Следует заметить, что при оценке растворимости полимеров необходимо
рассматривать изменение свободной энергии, т.е. учитывать как энергетичес-
кий, так и энтропийный члены. В данном рассмотрении ограничимся случа-
ем рассмотрения аморфных полимеров, построенных из макромолекулярных
Растворимость полимеров
335
глобул, сохраняющих свою форму в растворе. В этом конкретном случае кон-
формационный набор практически сохраняется и изменение энтропии при
переходе макромолекул в раствор будет немного отличаться от идеального
случая только вследствие различия в размерах молекул растворителя и мак-
ромолекулярных глобул. Поэтому в первом приближении изменением энтро-
пии при растворении в данном случае можно прен ебречь При анализе ра-
створимости кристаллических полимеров или аморфных неглобулярных по-
лимеров с гибкими макромолекулами энтропийная составляющая свободной
энергии может быть существенной, и рассматриваемый энергетический кри-
терий видоизменится.
Остановимся на тех физических допущениях, которые использованы при
определении условий растворения. При погружении образца полимера в ра-
створитель будут отрываться сначала те глобулы, которые находятся на повер-
хности образца. Рассмотрим силы, действующие на эти глобулы. На рис.92
изображено сечение глобулы и пояска связи рассматриваемой глобулы с ос-
тальными глобулами надмолекулярной структуры полимера. В момент по-
гружения полимера в растворитель образуется граница раздела твердое тело
(глобула) - жидкость. (5 разование единицы такой поверхности влючает
работу WA, определяемую процессом адгезионного смачивания;
Утл
Рис.92. Сечение глобулы и пояска связи глобулы с остальными глобулами надмолеку-
лярной структуры (схема)
Cross-section of tire globule and the belt whereby the globule is linked to the other
globules of supemiolecular structure (Scheme)
336
Глава XII
=YPn ~(Уп +YP), (332)
где уп и YP - поверхностное натяжение соответственно полимера и раствори-
теля; урП - межфазное натяжение. При этом представляет собой работу
адгезии, т.е. работу, необходимую для разделения поверхностей (восстанов-
ления исходного состояния).
Наличие такой работы вызывает появление сил, действующих на глобулу
надмолекулярной структуры и зависящих от величины и знака кривизны по-
верхностей, образующих глобулу и поясок связи. Эти силы приводят к отры-
ву глобулы от остальной части образца полимера. Но как только такой отрыв
происходит и глобула переходит в растворитель, возникает свежая поверх-
ность другой, прежде закрытой глобулы. Она также претерпевает смачивание
растворителем с возникновением тех же сил. При отрыве этой глобулы карти-
на повторяется - возникают свежие поверхности ранее экранированных гло-
бул, которые в результате действия сил смачивания последовательно перехо-
дят в растворитель.
Рассмотрим более подробно силы, действующие на глобулу и возникаю-
щие при ее адгезионном смачивании растворителем. Согласно изображению
на рис.92, на исходную глобулу надмолекулярной структуры и поясок связи
действуют две силы: сила, определяемая И'у!, т.е. поверхностным натяжением
растворителя и межфазным натяжением, приложенная к поверхности глобу-
лы надмолекулярной структуры, находящейся над пояском связи, и стремя-
щаяся прижать глобулу надмолекулярной структуры к полимеру, и сила, обус-
ловленная теми же характеристиками, приложенная к внешней поверхности
пояска связи и направленная внутрь растворителя, которая стремится выр-
вать глобулу из полимера (это происходит вследствие разного знака кривиз-
ны этих поверхностей). При определении второй силы можно допустить, что
поверхность, к которой приложена отрывающая глобулу сила, является по-
верхностью тора (заштрихована на рис.92). При этом напряжение силы сма-
чивания будет приложено к половине поверхности тора. Так как поперечные
размеры пояска связи значительно меньше большого радиуса пояска, то в даль-
нейшем все расчеты проводятся на характерных размерах пояска связи, поверх-
ность которого также принимается тороидальной. Определим условия раство-
рения полимера. На рис.93 изображен треугольник, вершины которого находят-
ся в центрах глобул надмолекулярной структуры. При этом А'Д=ДВ' = г, АД =
ДВ = R ,ДО - ОЕ - R y~RJ2, где г - малый радиус тора связи, R - радиус глобулы
надмолекулярной структуры, Ят - большой радиус тора связи.
На глобулу 1 будет действовать сила адгезионного смачивания, прижима-
ющая глобулу к полимеру, которая, согласно закону Лапласа, определяется из
соотношения:
Растворимость полимеров
337
Рис.93. Характерные расстояния между глобулами (см. текст)
Characteristic distances between globules (See text)
f = yp+yn~Y-pp =ltS!?+!^-2EL. (ззз)
™ T R 2
Кроме того, к поверхности пояска связи приложена сила /т, стремящаяся
оторвать глобулу от вещества
- 2лЛг sin-^(—у-)(ур + у п -урп) . <334)
2 г
0
Так как /С, = R/2, — -л/6 , то
А = - ^-)(у + уп - Урп) • (3 35)
г R
Чтобы произошел отрыв глобулы надмолекулярной структуры, необхо-
димо выполнение двух условий. Первое условие - сила, приложенная со сто-
роны растворителя на отрыв /т, должна быть больше силы /1Л, вызывающей
прижатие глобулы к веществу, т.е.
(УР+Гп-Грп)(3“)>0- (336)
Для глобул 8r/3R « 1 и поэтому условие (336) можно записать в виде
Ур+Уп>Урп. (337>
338
Глава XJI
Второе условие формулируется так: работа, совершаемая силами поверх-
ностного натяжения растворителя А, должна превыша й, энергию разрыва
межмолекулярных связей в пояске связи, т.е. необходимо, чтобы плотность
энергии когезии полимера о2 была меньше илиравнаработерастворителя
*2 „
по разрыву тора связи, отнесенной к единице объема тора связи 5р . Ве-
личину Л можно записать в виде
= (Jr ~ /гл)Ета^ > &
<=у(Гр+Г„-ГРпМЗ-^)^. 038)
Г
Таккак8т/ЗЛ « 1, выражение (338) примет вид
8;2=(ур + уп-урп)^. (339)
г 1 1 4кг
Итак, второе условие можно записать следующим образом:
^<3’р2. (340)
Преобразуем выражение (339)
Гр
где 5р - плотность энергии когезии растворителя; ор = 3y£n,ax/4тш ,
г*тах -максимальное расстояние между молекулами растворителя, на кото-
ром еще действуют силы взаимодействия, г* ~ характерный размер связи френ-
келевских роев растворителя, р = zmaxr* l£ma:tr • Обозначив p = 8„/82 и
учитывая, что, согласно работам [147—149],
Урп = УР + Уп - 2Ф(урГп)1/2, (342)
условие растворимости (340) можно записать следующим образом:
р<2рФ(—)1/2, (343)
УР
где р = о2 / 5р ; ог[ и 5р параметры Гильдебранда соо гвегственно для поли-
мера и растворителя; р константа; уп и ур -поверхностное натяжение поли-
мера и растворителя соответственно;
339
Растворимость полимеров
(344)
Здесь 17р и Г’п - соответственно мольные объемы растворителя и поли-
мера (в расчете на 1 звено). Заметим, что значения Ф порядка единицы.
Выражение (343) получено при условии, что набухания полимера не
происходит. Рассмотрим другой случай, когда полимер набухает. Это
означает, что в полость между глобулами А, В, С (см.рис.92) проникает
растворитель. В этом случае условие (343) формально можно записать в
таком же виде, однако в качестве отношения уп/ур будет фигурировать
эффективное значение (уп/ур)э которое по своей величине будет меньше
истинного значения уп/ур, поскольку проникновению растворителя в по-
лость между глобулами оказывает расклинивающее действие и снижает
силу /т, необходимую для отрыва глобулы В.
Этот вопрос детально рассмотрен в работе [32]. В результате для крите-
рия рас творимости получено следующее выражение
ц < 2рФ(Ф- д/ф2 -1 + а), (345)
где а = урп/ур; остальные параметры те же, что и в уравнении (343).
Напомним, что ц = 8„ /8р; р- константа; величина Ф вычисляется по фор-
муле (344). Из выражения (345) следует, что существуетвеличина amin = 1 - Ф2,
при которой возможна растворимость (например, при Ф = 0,95, ат,п = 0,1).
Поскольку двсегда положительно, растворимость возможнапри а1)ц), <а<1.
Максимальное значение ц,,мхбудет
ц,„0А. = 2рФ2. <346)
Таким образом, согласно критерию (345), растворимость будет наблю-
даться в том случае, когда правая часть критерия (345) будет больше левой
части, причем обе часта критерия вычисляются на основании химического
строения повторяющегося звена полимера и м олекулы растворителя.
Экспериментальная проверка критерия растворимое™ (345) проведена
на примере -300 систем полимер -растворитель. В качестве полимерных
объектов исследования в работе [32] выбраныкак традиционные аморфные
полимеры (полиметилметакрилат, полистирол, поливинил ацетат, полиизобу-
тилен, бутадиеновыйиизопреновыйкаучукиит.д.), так и ряд теплостойких
полимеров кардового строения (полиарилаты, ароматические полиамиды и
полиимиды, полифешыхиноксалин); в качестве растворителей были исполь-
зованы около 50 органических жидкостей.
340
Глава XII
Как отмечено выше, растворимость должна наблюдаться в том слдучае,
если расчетные значения 2рФ(Ф-у/ф2 -1 + а) оказываются большими или
равными значению р = 52 / 5р ; при этом величины 5П и Зр могут браться как
экспериментальные, так и расчетные.
Введенная выше величина р прямо не определяется. Однако, если вели-
чина р является почти постоянной, то вводя обозначение
р = Ф(Ф - з/ф2-1 + а) (347)
имеем
Р < 2рр, (348)
т.е. в координатах (р, р ) зависимость р = 2рР представляет собой прямую,
выходящую из начала координат. Выше ее должны располагаться точки, со-
ответствующие отсутствию растворимости, а ниже - ее наличию. Проведен-
ные расчеты для упомянутого количества пар показали (рис. 94), что точки,
соответствующие случаю нерастворимости (темные точки), располагаются,
как правило, выше упомянутой прямой, а точки, соответствующие случаю
растворимости (светлые точки), располагаются ниже ее. Таким образом, мас-
сив значений р делится на два поля, одно из которых является полем нера-
створимости, а другое - полем растворимости. Однако в каждое из этих по-
лей попадает небольшое количество “инородных” тощ к, доля которых со-
Рис.94. Зависимость р от р для различных пар полимер - растворитель (пояснения в тексте)
Dependence of ц on р for different pairs of “polymer - solvent’’ (for explanation see text)
Растворимость полимеров
341
ставляет 15 %. В целом количество темных и светлых точек приблизительно
^2 2
одинаково, а величина ц = оп / 5р колеблется около единицы.
Пунктирная прямая на рис.94, соответствующая зависимости ц = 2р(3,
проведена таким образом, чтобы количество инородных точек выше и ниже
прямой было минимальным и примерно одинаковым. При выполнении этих
условий величина 2р = 1,374. Из рис. 94 сразу видно, что если соблюдает-
ся только условие 5р = Sn, то растворение будет наблюдаться лишь в 50 %
случаев.
Следовательно, для систем, у которых соблюдается условие Зр ~ Зп, со-
гласно критерию (343) или (345), можно предсказать растворимость с более
высокой точностью, равной 85 %. Учитывая, что предварительно уже отбро-
шены явные нерастворители, для которых 8р » Зп и 8р « 5П, предсказатель-
ную сил}' критерия можно считать высокой.
Как правило, растворимость наблюдается в тех случаях, когда поверхно-
стное натяжение растворителя близко к поверхностном}' натяжению полиме-
ра; тогда межфазное натяжение мало и величина а также приобретает малые
значения. Если же при соблюдении равенства Sp ~ 3„ поверхностное натяже-
ние растворителя существенно меньше поверхностного натяжения полиме-
ра, отношение межфазного натяжения к поверхностном}' натяжению раство-
рителя велико, а приобретает большие значения, а величина Р - малые. Точки
на диаграмме (см.рис.94) попадают в левое верхнее поле. Таким образом, воз-
растанию величины р способствуют два фактора: равенство мольных объе-
мов растворителя и повторяющегося звена полимера и равенство поверхнос-
тного натяжения растворителя и полимера. В свою очередь, согласно рис.94,
повышенное значение Р способствует растворимости полимера.
Издавна считалось, что подобное должно растворяться в подобном, одна-
ко приемлемого определения подобных веществ пока не дано. С точки зрения
критерия растворимости (343) или (345) подобными веществами можно счи-
тать такие, которые имеют близкие плотности энергии когезии (параметр ра-
створимости Гильдебранда 3), мольные объемы и коэффициенты поверхност-
ного натяжения. В этом случае растворимость и совместимость, как правило,
наблюдаются.
И з рис. 94 видно, что существуют и отде льнье исключения. Так, в случае
полиарилата
О-с-о-О-с-О-о-
-со О р
342
Глава XII
согласно критерию (345), растворение в хлороформе происходить не долж-
но, а циклогексанол должен растворять полимер. На практике наблюдается
обратная картина - хлороформ хорошо растворяет многие теплостойкие по-
лимеры, а циклогексанол - нет. Именно эти исключения и составляют основ-
ную часть нарушения критерия (345). Видимо, в случае хлороформа раство-
рение происходит практически без набухания, что подтверждают предвари-
тельные эксперименты.
Другие возможные причины отклонений от критерия (345) будут проана-
лизированы ниже.
Процедура оценки растворимости полимера заданного химического стро-
ения в том или ином растворителе, согласно изложенным выше представле-
ниям, заключается в следующем. Для данного полимера и растворителя рас-
считываются величины параметра растворимости S по формуле (331). Затем
для полимера рассчитывается величина поверхностной энергии уп по форму-
ле (389) или (399, 400). Можно также рассчитать уп с помощью парахора по
формуле (372). Необходимый для этого мольный объем повторяющегося зве-
на полимера определяется как
!/=-----1----} (349)
^ср
где кср = 0,681. Если известно значение плотности полимера d„, то Уп =
где Л/- молекулярная масса повторяющегося звена.
Значение поверхностного натяжения для растворителя ур можно взять
как экспериментальное, так и рассчитанное по формуле (382). Величина Ф
рассчитывается по формуле (344). Необходимые для этого значения Ур и Уп
определяются из соотношений Ип = M/dn и JZp = M/dp, где dp и dn - плотности
растворителей и полимера. Затем определяете величина межфазного натяже-
ния урп по формуле (342), а далее величина а = ур„/ур. После этого рассчиты-
вается величина 1,347Ф(Ф-уФ2-1 +а) и полученное значение сравнивает-
ся с величиной ц = 82 / 8р ; если р. < 1,347Ф(Ф— д/ф2 -1 + а), то с вероятнос-
тью 85 % можно ожидать растворения полимера в данном растворителе.
Приведем пример расчета для системы полиметилметакрилат - бензол.
СНз
-СН2—С— Полимер |^J) Растворитель
С-О-СНз
II
О
Растворимость полимеров 343
Сначала необходимо определить параметры растворимости для полимера 8П
и растворителя 8р.
Для полимера (£ ДЕ* )п = 5ДЕс + 8AEJ/ + 2АЕ^ + АЕ^ = 5-550,7 + 8-47,7+
I
+ 2-142,6+ 1623 = 5043,3кал/моль = 21081 Дж/моль (все величины ДЕ* взяты
из табл. 43).
(^А^)п = А^СДО + А 17с,13 + А^Д + ДГ-С,49 + АГ'С’Л! + A1o,139 +
i
+A7O129 + 8АГЯ Д24 = 13,1 +17,2 + 5,0 +15,9 + 20,3 + 5,8 + 3,4 + 8 • 2,0 =
= 96,7 А3 (все номера атомов соответствуют номерам в табл.З).
Подставляя вычисленные значения (^ ДЕ* )п и (^ AI+ )п в уравнение
/ i
(331), получаем
52 =---21081 = 362 д^З 5 = 19 0 (Дж/смЗ)!/?
п 0,6023-96,7
Для растворителя
(£ АЕ* )р = 6АЕс + 6ДЕ^ + AEq = 6 • 550,7 + 6 • 47,7 + 713 =
= 4303 кал/см3 = 17988 Дж/см3.
(ХАИ/)Р =6ДКСд8+6ДИЯд24 =6-12,7 + 6-2,0 =88,2 А3.
С этими параметрами из уравнения (331) имеем
S2 17988 , ,
°р - 0 6023-88 2 = 338,6 Дж^гм3; 5р = 18>4 (Дж/см3)1/2
Видим, что 8П ~ 8р и можно предположить растворимость полиметилме-
такрилата в бензоле.
Теперь следует вычислить поверхностное натяжение полимера ираство-
рителя. Подставляя величины (^ АЕ, )п, (^ AI7, )р и т - 15 (число атомов в
/ i
повторяющемся звене полиметилметакрилата) в уравнение (389), получаем
(полиметилметакрилат относится к полярным группам 1):
Уп =0,0751-----2,1;^1 -- = 30,5 дин/см.
(96,7)2 151/3
344
Глава XII
Для бензола, согласно формуле (382), имеем
17988
у_ =0,0287—----- = 26,1 дин/см.
Р (88,2)2'3
Молярный объем бензола Гр = 89 см3/моль, молярный объем полимстилме-
такрилата, согласно формуле (349), равен
„ 0,6023-96,7 3,
И. =-----------= 85,5 смДмоль.
п 0,681
Теперь необходимо вычислить величину Ф по формуле (344):
4-(89 -85,5)1/3
ф =------------—----= 1
(89,/3 + 85,61/3)2 '
Затем вычисляется величина межфазного натяжения по формуле (342).
урп = 26,1 + 30,5 — 2 -1 (26,1 - 30,5)1/2 = 0,17 дин/см.
Величина а = урп/ур = 0,17/26,1 = 0,00656.
Подставляя все полученные значения параметров системы полимер - раство-
ритель в критерий (345), находим
= <1,374-1,0 • (1,0 - л/1,02 -1 + 0,00656);
h 52 338,6 ’
°Р
1,069 < 1,263
Поскольку левая часть критерия (345) меньше правой его части, то полиме-
тилметакрилат должен растворяться в бензоле, что и наблюдается на практике.
Приведем еще один пример расчета для более сложной системы поли-
мер - растворитель.
В качестве полимера выберем полипиромеллитимид анилинфталеина, в
качестве растворителя - нитробензол:
Для полимера
(^~* АГ, )п = ЗОДЕС +14/\2?уу + 6ATq + 2Д/тд/ + ЗАГ^ + 4Д/Д. =
Растворимость полимеров
345
= 30 550,7 + 14- 47,7 + 6 • 142,6 + 2 • 1205 + 3 • 1623 + 4-713 =
= 28175 кал/моль= 117733 Дж/моль;
(Z А1/-)п = 4АИс>64 + 8ДДс,19 + ИДИ^ д8 + + Д^С,34 + ^^С,49 +
/
+14 ДИуу 124 + 5Д I'7i J39 + ДVq 129 + 2ДИуу 1^4 =4 -14 J + 8 -8,4+ 14 -12,7 +
+2-10,2+ 7,9 +15,7+ 14-2,0+ 5-5,8 +3,4 + 2-0,9 = 407,6 А.
По уравнению (331) получаем
З2 =---117773— = 479 7 Дж/Смз; § = 21,9 (Дж/см3)1/2.
п 0,6023-407,6
Для растворителя
(^. ДД, )р = бДДуу + 5ДДуу + Д/Ду + 2Д/*.0 + АДф + ДИj = 6 • 550,7 +
+5-47,7 +1205 + 2 • 142,6 + 713 +1623 = 7369 кал/моль = 30802 Дж/моль;
(ХА1<)р =5ДИСд8 + ДИС21 +5ДИуу Д24 + А^ядзо + 2А^од40 =
= 5 -12,7+10,2 + 5-2,0 + 7,0 + 2-7,2 = 105,1 А3.
По уравнению (331) получаем
_2 30802
°р ~ 0 6023 105 1 = 486>6 Дж/см3; 8р = 22,06 (Дж/см3)1/2.
Видно: 8П = Зр, те. данный полиимид в принципе может растворяться в
нитробензоле.
Далее вычисляем поверхностное натяжение полимера и растворителя.
Число атомов в повторяющемся звене данного полиимида т = 52. Подставляя
все параметры полиимида в уравнение (389), получаем
117773
Уп =0,0751------—---------- = 43,1 дин/см.
(407,6)2'3 -521'3
Для нитробензола, согласно формуле (382), имеем
30R07
Ур =0,0287------— = 39,7 Дин/см.
(105,1)2'3
Молярный объем нитробензола Др = 103 см3/моль; молярный объем Дп поли-
имида, согласно формуле (349), равен
0,6023-207,6 3.
И. =------------— = 360,5 см3/моль.
0,681
Вычислим величину Ф по формуле (344):
346
Глава XII
4-(103-360,5)1/3
(1031/3 + 360,51/3)2
= 0,9576.
Величина межфазного натяжения урп, вычисленная по формуле (342), равна:
урп = 43,1+39,7-2-0,9576-(43,1-39,7)1/2 =3,58 дин/см.
Величина а = урп/ур = 3,58/39,7 = 0,0901.
Подставляя все значения параметров системы полимер - растворитель в
критерий (345), получаем
Зц. = 1Z22 <! 374.0 9576. (0 9576 _ Jo,95762 -1 + 0,0901)
S2 486,6 ’
°Р
0,986 < 1,149.
Согласно критерию (305), данный полиимид будет растворяться в нитро-
бензоле, что и наблюдается на практике [6].
XIT.3. Влияние молекулярной массы и
ориентации макромолекул на растворимость
Критерий растворимости (345) справедлив для случая изотропных амор-
фных полимеров, имеющих глобулярную надмолекулярную структуру.Кро-
ме того, данный критерий не учитывает влияние степени полимеризации по-
лимера на растворимость, хотя известно, что оно может быть существенным
при переходе к большим молекулярным массам. В работе [95] сделана по-
пытка учесть влияние типа надмолекулярной структуры и степени полиме-
ризации полимеров на его растворимость, а также установить связь между
параметрами теории Флори-Хаггинса и химическим строением полимера и
растворителя.
В критерий растворимости (345) входит константа р, которая описывает-
ся соотношением
еп ''п
(350)
eLx rn
где e'J|uz - максимальная относительная деформация межмолекулярных свя-
зей полимера в момент их разрыва и перехода полимера в растворитель; е₽ 1ах
- максимальная деформация жидкости, те. такая деформация, при которой
происходит нарушение сплошности; гр - характерный размер связи френке-
левских роев растворителя; гп - малый радиус глобулы связи для полимера.
Представим, что в области очень малых деформаций полимер и раство-
ритель ведут себя как упругие тела [77], характеризуемые модулями упругос-
ти Е„ и Ер соответственное. Тогда
Растворимость полимеров
347
3n = £nG4>J2/2; 3^=ep(£U)2/2 (351)
и величина ц запишется следующим образом:
т2 р / п ,2
и = ^(Чпах) (352)
82 Е (ер г
ир '-р'-^тах/
В момент отрыва глобулы напряжения в глобуле и растворителе равны,
т.е. о = ЕПг'/,ах = Ерг^ах, и поэтому выражение (348) примет вид
— <2(5. (353)
гр
Так как гп = п1ц,2ап, гр = пр2ар, где ап и ар соответственно размеры по-
вторяющихся звеньев полимера и растворителя, а пп и пр - число звеньев в
глобуле связи полимера и молекул растворителя соответственно, то, с учетом
поворотно-изомерной теории [58],
ап2 = (М)п/3ехр(^); (354)
I Лх
AF
«p2=(ZAK)2/3exp(-^), (355)
*
где AK, n и AVJjP -Ван-дер-Ваальсовые объемы /-х атомов, входящих в повто-
ряющееся звено полимера и молекулу растворителя соответственно; ДЕП и
А£р - разности энергий поворотных изомеров полимера и растворителя. С
учетом этого выражение (353) можно записать
<2(-^-)1/2
«п
(2><)Р
1/3
₽
(356)
АЕП-АЕ
где q =ехр(- -) .
2.К1
Величин}- пр найдем (с точностью до константы) из условия, что пр равно
числу молекул растворителя, покрывающих одним слоем гло & лу связи. Ъгда
«Р
«п
\2/3
аг;
Л^Р) ;
/ \2/3
I ДУп ]
6
и,/3 АИ
"п Г
(357)
n=t п
г
р
348
Глава Xll
Здесь ДИП =(£Д^)п Д^р = (^AIz/)p. Подставив выражение (357) в
/ /
формулу (356), получим
^<2(-^-)1/2(358)
«п
т.е. р = (6/л„/3)1/2, a q по смыслу равно ц (q = ц).
Учитывая, что не вся глобула связи омывается растворителем (часть ее
находится в глобуле - макромолекуле), запишем выражение для р в виде.
Р = (-^-)1/2С-’, (359)
Пп
где С - доля поверхности глобулы связи, омываемая растворителем. С - кон-
станта и ее величину можно определить из условия р = 0,687 при па = 24 [94]
(при степени полимеризации N = 104).
Тогда С = 2,1, а условие растворимости (358) примет вид
^(4т)1/2-Р-
Величина пп является функцией только степени полимеризации полиме-
ра. Отсюда следует, что чем больше молекулярная масса полимера, тем хуже
он растворяется.
Пусть;/,, = где 4 =0,24 IO 3 приУ = У0= 104 [94]. Тогда п'р = О,13У1/3
и условие растворимости (359) можно записать как
ОЙ1)
Поскольку расчеты всех констант в соотношении (360) были выполнены
для N = 104, а у реальных полимеров N может принимать различные значе-
ния, в более общем виде выражение (360) следует записать так.
1
5<1,374(—)1/6р, (361)
где 2,1/(104)1/6 = 1,374 - значение коэффициента при р в выражении (361) при
N= 104.
ВеличинаУд, соответствующая 2р = 1,374, получается из графика, приве-
денного на рис.94, который построен без учета молекулярных масс анализи-
руемых полимеров. В рамках данного подхода эта величина должна соответ-
ствовать некоторой средней степени полимеризации Nq, при которой необхо-
димо экспериментально оценивать растворимость полимера в различных
растворителях. Так как степень полимеризации N реальных полимеров мо-
жет отличаться от А'о. условие (361) окончательно примет вид
Растворимость полимеров
349
4<U74A)1/6₽. (362)
N
Таким образом, даже если полимер имеет глобулярную надмолекуляр-
ную структуру, условие его растворимости зависит от степени полимериза-
ции. Чтобы выполнялось условие растворимости в виде выражения (348),
необходимо выполнить приведение всех систем полимер - растворитель по
молекулярным весам, т.е. записать критерий растворимости как
ц*< 1,3740, (363)
где ц* = р.(У / У0)1/6 ( ц имеет тот же смысл, что и ранее т.е. ц = 8„ / 8р ).
Для иллюстрации влияния степени полимеризации на растворимость по-
лимеров на рис.95 показан график зависимости ц*/ц от (У/У0)1/6. Если
реальная степень полимеризации N < No, то растворимость улучшается, и
наоборот. Учет влияния молекулярной массы полимера в ряде случаев улуч-
шает предсказательную силу критерия.
До сих пор анализ критерия растворимости был проведен для аморфных
полимеров с глобулярной надмолекулярной структурой Однако хорошо из-
вестно, что такой фактор, как ориентация влияет на растворимость полиме-
ров. Например, поливиниловый спирт хорошо растворяется в воде, но волок-
на, сформованные из этого полимера, в воде достаточно устойчивы. Поэтому
критерий растворимости в виде (345) не может быть применен для предска-
зания растворимости полимерных тел, макромолекулы в которых вытянуты.
Рассмотрим, в связи с этим, как изменится критерий (345) при переходе к
фибриллярной струкзуре. Ограничимся случаем, когда соприкосновение фиб-
рилл происходит по образующим. Связь фибриллы с другими фибриллами
осуществляется через “циливдры связи” (ориентированный полимер) в от -
350
Глава XII
Л
Рис.96. Сечение фибрилл и схема приложения поверхностных сил
Cross-section of fibrils and schematic of surface forces applied
личие от “глобул связи”, детально рассмотренных выше. На рис .96 изображе -
но сечение фибрилл и дана схема приложения сил в этом сечении.
Сила /ф, прижимающая рассматриваемую фибриллу к другим, согласно
закону Лапласа, равна
4 = <з«>
к.
где 1R„ - расстояние между цилиндрами связи фибрилл (см.рис. 96); - дли-
на образующей цилиндра связи, равная длине фибриллы. Учитывая, что
R„ = А72, выражение для /ф запишем следующим образом:
/ф — Л)> (Ур "* Уп — У рп) • (365)
Здесь урп - коэффициент межфазного натяжения на границе полимер - ра-
створитель.
Сила /ц, приложенная к цилиндру связи со стороны растворителя и стре-
мящаяся оторвать фибрилл}', равна
г 1 т 0 Ур+Уп-Урп . .
/ц = 2лгф£ф sin - = л7,ф (ур + уп - ур„), (366)
где Гф - радиус цилиндра связи; 0 = 60° (см.рис.96).
Первое условие отрыва, согласно которому /ц - /ф > 0, имеет такой же
вид, как и в случае глобулярной структуры (см. выше)
Ур Уп -> Урп
Это условие выполняется всегда.
Второе условие отрыва связано с работой отрыва
С/ц /ф)^тахг >
(367)
Растворимость полимеров 351
и
совершаемой силами поверхностного натяжения растворителя, где £„юх -де-
формация при разрыве цилиндра связи. Эта работа должна превысить энер-
гию разрыва межмолекулярных связей.
Работу растворителя, затрачиваемую на разрыв цилиндров связи и отне-
сенную к объему двух цилиндров связи, запишем как
(0*р)2 = Л/(2ИЦ). (368)
Здесь Рц - объем цилиндра связи, равный Иц = тгг2Лф .
Воспользовавшись выражениями (.365) и (366), получил!
<s;>2=sjp„i^-2e. (369)
7р
где
(бр ) — /(ДтГГр ) ,
п _^итах 'р 2(л-1)
Г
стах Ф
Соответственно второе условие можно записать как
Sp-(Sp)2
или
И<2рф0, (370)
здесь ц и р имеют такой же смысл, как и в случае глобулярной надмолекуляр-
ной структуры.
Это условие отличается от условия (348) толь ко значением коэффициен-
та р. Отношение рф/р,я, где рга соответствует полимеру с глобулярной надмо-
лекулярной структурой, можно записать в виде
Рф _ 2(тс -1) ггл
Ргл 3 Гф
(гга - радиус связи глобулы, Гф - радиус цилиндра связи).
Так как, согласно оценкам работы [94] гт/г^ ~ (1/3)° , то соответственно
-^-«0,82
Ргл
Следовательно, условие растворимости полимеров с фибриллярной струк-
турой более жесткое, чем с глобулярной. Действует тот же критерий, но с
меньшим коэффициентом 2рф = 1,125:
ц < 1,1250 = 1,125Ф(Ф-з/ф2-1 + а) • (371)
Таким образом, полимер с фибриллярной надмолекулярной структурой
может не растворяться даже при условии растворимости полимера с глобу-
лярной структурой.
Глава XIII
ПОВЕРХНОСТНЫЕ СВОЙСТВА
ОРГАНИЧЕСКИХ ЖИДКОСТЕЙ И
ПОЛИМЕРОВ
Знание величины поверхностного натяжения и умение ее предсказывать
исходя из химического строения вещества важно, поскольку от поверхност-
ного натяжения жидкостей и поверхностной энергии твердых тел зависят мно-
гие их индивидуальные свойства, а также совместимость и растворимость
друг в друге. Среди существующих способов расчета поверхностного натяже-
ния наибольшее распространение получила аддитивная схема, основанная на
суммировании парахоров, характеризующих вклад отдельных атомов в по-
верхностное натяжение. Расчет проводится по формуле
где Р - общий парахор молекулы (или повторяющегося звена полимера),
Р, - парахоры, характерные для каждого атома.
Значения атомных парахоровРь а также инкременты, характеризующие
вклад различных типов связей (двойная, тройная) и колец (трехчленное, че-
тырехчленное и т.д.) в общий парахор вещества, приведены в табл.45.
Формула (372) позволяет с достаточно хорошей точностью рассчитывать
величину у для органических жидкостей и полимеров. Однако физический
смысл величин Р„ входящих в соотношение (372), не ясен. Это не позволяет
проанализировать влияние слабого Ван-дер-Ваальсового взаимодействия и
сильных полярных групп на формирование тех или иных поверхностных
свойств органических жидкостей и полимеров. Этот вопрос достаточно важен
потому, что анализ такого влияния на количественном уровне позволяет ре-
гулировать химическое строение веществ с целью придания им нужных по-
верхностных свойств.
Поверхностные свойства органических жидкостей и полимеров 353
7абяица 45
Значения атомных парахоров Р, н парахоров ряда инкрементов Р,
Values of atomic parachors P, and parachors for a series of increments Pj
Азом Р, Инкременты р,
с 4,8 Двойная связь 23,2
II 17,1
о 20,0 Тройная связь 46,4
о2* 60,0 3-х членный цикл 16,7
N 12,5
S 48,2 4-х членный цикл 11,6
F 27,5
С1 54,3 5-и членный цикл 8,5
Вг 68,0 6-и членный цикл 6,1
I 91,0
* Для двух атомов кислорода, входящих в сложно-эфирную группу — С — О —
О
XIIL1. Поверхностное натяжение
органических жидкостей
Рассмотрим подход к анализу влияния химического строения на величи-
ну поверхностного натяжения, предложенный в работе [34]. При таком ана-
> лизе исходили из следующих соображений. Как известно, молекулы жидко-
1 сти, находящиеся на поверхности, претерпевают иное межмолекулярное вза-
имодействие, чем в объеме. Если рассматривать объемную задачу (рис.97), то
* при гексагональной упаковке сферических молекул, находящихся в объеме,
у координационное число равно 12, а на поверхности - 9, т.е. их отношение
v составляет 4/3. Поскольку при увеличении координационного числа свобод-
' нал энергия системы понижается, молекулы стремятся перейти из поверхно-
стного слоя в объем. Поэтому для образования единицы новой поверхности
нужно затратить энергию.
Рассчитаем прежде всего количество молекул и, находящихся на единице
поверхности.
Легко показать [34], что при гексагональной упаковке (см.рис.97,б) вели-
чина п на 1 см2 поверхности составит
1016-0,2887
П=—
где г - радиус молекулы, выраженный в А.
При этом
г= -------3 = 0,6204(£ АК,)1/3
(373)
(374)
354
Глава XIII
Рис.97. Схема расположения и взаимодействия молекул в вертикальной (а) и
горизонтальной (б) плоскостях
Schematic representation of arrangement and interaction of molecules in the
vertical (a) and horizontal (6) planes
где ЛИ, Ван-дер-Ваальсовые объемы атомов, входящих в молекулу.
С учетом соотношений (373) и (374) имеем
1016
»=--- 7/з--°>75.
(£АЦ)2/3
(375)
Рассчитаем энергию когезии е*, приходящуюся на одну молекулу. Согласно
подходу для оценки параметра растворимости (см. выше), энергия коге-
зии, приходящаяся на один моль вещества, равна АЕ* / к, где АЕ* - моль-
i
ные инкременты, имеющие смысл энергии межмолекулярного взаимодействия
для каждого типа атома и отдельных полярных групп, входящих в молекулу'
или повторяющееся звено полимера (величины ДЕ* приведены в табл.43);
к - коэффициент молекулярной упаковки в объеме рассматриваемого веще-
ства.
Тогда
ХАЕ* ^ДЕ*
е* = -----= -4-— • 0,166 .
kNA ыо16
(376)
В формуле (376) величина е выражена в Эргах, а ДЕ* - в Джоулях.
Энергия когезии у*, которой обладали бы в объеме все молекулы, распо-
19 *
ложенные в поверхностном слое с площадью 1 см2, равна не и, согласно
соотношениям (375) и (376), равна
2Ж
у =0,125---------—
*(£ДГ<)2/3 '
(377)
Поверхностные свойства органических жидкостей и полимеров 355
Величина у* существенно превышает величину поверхностного натяже-
ния, т.к. для образования единицы новой поверхности нужно преодолеть не
всю энергию когезии, а только ее часть, поскольку в поверхностном слое мо-
лекула обладает ненулевым координационным числом. Из рис.97,а видно, что
для выхода молекулы из объема на поверхность нужно затратить примерно
1/4 энергии когезии, поскольку координационное число при этом изменяется
с 12 до 9. Кроме того, следует учитывать, что упаковка молекул в поверхнос-
тном слое не является идеальной, как это изображено на рис.97,6. Обозначим
реальный коэффициент упаковки молекул в поверхностном слое через к„- Для
дальнейших расчетов определим коэффициент упаковки Агпи в поверхностном
слое при идеальной гексагональной укладке шаров. Чтобы определить мак-
симально возможный занятый объем , умножим число молекул п в едини-
це поверхностного слоя на объем одной молекулы
И = 0,2887—ти-3 =1,2086 -1016 А3
г2 3
(г выражено в ангстремах). Общий объем поверхностного слоя 1/общ = 2-1016 А3,
и тогда /спи = И/Гобщ = 0,604.
Таким образом, коэффициент упаковки А'пи при идеальной укладке шаров
в поверхностном слое равен 0,604, в то время как в объеме соответствующий
коэффициент кОИ равен 0,740.
Следовательно, доля молекул, находящихся на поверхности, равна к^0,604,
где к„ - реальный коэффициент молекулярной у паковки в поверхностном слое
Тогда для величины у* получим
у* = 0,125------1----—-.
0,604(£Д1')2/3
(378)
Теперь учтем ту часть энергии когезии, которую нужно преодолеть, чтобы
молекула могла перейти из объема на поверхность. Эта доля а, обусловленная
изменением координационного числа при таком переходе, определится из со-
отношения
12_______________
0,740 0,604
12 -
0,740
3 k
= 1-2Дп_. 1225.
4 к
(379)
Тогда для поверхностного натяжения жидкостей имеем
356
Глава XIII
_ . 0-M74^ .. О.^„
Ур Уа Н0,74(ХД1/,)2/3 к
(380)
На основании соотношения (380) в работе [34] проведен расчет значений
к„/к для большого количества органических жидкостей различной природы.
Исходные данные и результаты этого расчета для представителей различных
классов жидкостей приведены в табл.46. Видно, что для всех органических
жидкостей величина кп/к < 1. При этом оказалось, что для многих органичес-
ких жидкостей, особенно однотипных, например, углеводородов, спиртов и
т.д., отношение к„/к приблизительно постоянно. Проведенные расчеты пока-
зали [34], что для углеводородов, перфторсоединений, галогенсодержащих
соединений, альдегидов, кетонов и нитросоединений средняя величина
(ки/к)ср = 0,851. Для спиртов и кислот (кп/к)ср = 0,954, а для нитрилов
(кп/к)ср = 0,910. Сами значения коэффициентов молекулярной упаковки в по-
верхностном слое также приведены в табл. 46.
Так как для отдельных рядов органических жидкостей величина к,/к вы-
ступает в роли константы, то соотношение (380) можно переписать в виде
2Ж
1'р = '4(ЕД1<)2'-’'
(381)
где
А =0,207-(Лгп/Лг)-(1-0,919 -Ап/А) .
Учитывая приведенные выше средние значения к„/к, можно записать сле-
дующее выражение для расчета величины поверхностного натяжения орга-
нических жидкостей
Vp=Aj-^-------27Т- (382)
(£ДП,)2/3
Для углеводородов, перфторсоединений, галогеносодержащих соедине-
ний, сложных эфиров, альдегидов, кетонов и нитросоединений (группа I)
Ai= 0,0287; для спиртов и кислот (группа II) Л2 = 0,0181; для нитрилов (груп-
па Ill) А3 = 0,0229. Напомним, что Д£* выражается в Джоулях, ДК,- в А’, и
тогда с указанными коэффициентами А, величина ур выражается в дин/см.
Поверхностные свойства органических жидкостей и полимеров 357
Таблица 46
Исходные данные и результаты расчета поверхностного натяжения для ряда
органических жидкостей
Initial data and results of calculation of the surface tension of a series of organic liquids
Соединение i Дж/моль (LAK,)2'3 , i А3 k у , дин / см
эксперимент расчет
1 2 3 4 5 6 7
Угле во до. эоды
н-Петпан 13903 21,30 0,510 0,453 16,03 17,960
Изопентан 12180 21,40 0,510 0,447 15,00 17,330
н-Гексан 16603 23,70 0,527 0,462 18,41 19,895
Циклогексан 16206 21,90 0,567 0,443 25,12 23,760
н-Гептан 19303 25,90 0,541 0,469 20,21 21,604
н-Октап 22003 28,20 0,549 0,472 21,75 22,700
Изооктан 20281 28,20 0,545 0,479 18,85 21,710
н-Нонан 24704 3030 0,558 0,479 22,91 23,940
н-Декан 27404 32,30 0,571 0,489 23,92 25,900
н-Тетрадекан 38209 39,90 0,589 0,506 26,96 28,770
Бензол 19186 19,80 0,597 0,501 28.78 29,760
Толуол 20687 22,30 0,592 0,489 28,53 28,680
н-Ксилол 23391 24,70 0,610 0,960 30,03 30,650
м-Ксилол 23391 24,70 0,600 0,515 26,63 29,740
о-Ксилол 23391 24,70 0,595 0,499 28,31 29,250
Этилбензол 23391 24,60 0,598 0,514 29,04 29,599
Изопропилбензол 24369 26,80 0,596 0,490 28,20 28,790
Перфторсоединения
н-Пер фтор п е нтан 12724 29,50 0,574 0,523 9,87 16,904
н-Перфтор гептан 15228 32,80 0,551 0,478 12,60 17,330
н- Пе р фторо ктан 17732 35,90 0,571 0,493 13,60 18,680
Галогенсодержащие соединения
Дихлорметан 14408 15,14 0,554 0,458 28,12 24,800
Хлороформ 13275 17 j60 0,555 0,424 27 Д6 23 000
Ч етыр ех хлор исты й углерод 12147 19,87 0,553 0,350 26,75 20,900
Этилхлорид 11453 15,52 0,527 0,423 22,18 20,500
1,1-Дихлорэтан 17109 17,94 0,544 0,475 24,75 23,800
1,2-Дихлорэтан 17109 17,94 0,579 0,455 32,23 28,010
1,1,1-Трихлорэтан 15976 20,24 0,555 0,440 25,77 23,700
Тетрахлорэтан 28416 22,40 0,607 0,518 36,04 34,230
Изобутил хлорид 15132 20,96 0,550 0,458 21,9 9 22 160
цис-Дих ло рэтнле н 15357 17,20 0,571 0,468 28,00 26,090
транс-Ди хлор- этилен 15357 17,20 0,563 0,484 25,00 । 25,195
Трихлорэтилен 18313 19,57 0,582 0,472 29,50 28,600
Тетрахлорэзилен 21268 21,70 0,605 0,481 32,26 30,720
Хлорбензол 23642 21,80 0,605 0,479 33,19 32,600
358
Глава XIII
таблица 46 (продолжение)
1 2 3 4 5 6 7
Бромбензол 27011 23,00 0,634 0,522 36,34 36,790
Иодбепзол 31688 24,60 0,661 0,558 37,65 41,753
Бромоформ 23379 20,73 0,650 0,609 31,68 38,000
Э1илбромид 14822 16,70 0,542 Д470 24,15 22,520
J ,2-Дибромэтан 23843 20,05 0,628 0,497 38,91 36,800
Этилиодид 19491 18,41 0,590 0,535 28,10 30,400
Спцрты
Гликоль 39840 15,50 0,659 1 0,625 1 46,49 51,900
Глицерин 59557 19,41 0,705 0,660 62,29 71,100
Цикло гексанол 33443 22,96 0,65 8 0,576 34,37 44,600
Метанол 20118 11,02 0,5 44 0,539 22,55 10,300
Этанол 22819 14,10 0,565 0,558 22,32 23,600
н-Пропанол 25511 17,20 0,563 0,547 23,70 23,930
н- Булано л 28219 19,77 0,578 0,562 24,57 29,200
Изобутиловый спирт 26497 19,77 0,573 0,564 22,98 28,300
н-Амиловый спирт 30919 22,30 0,587 0,559 25,68 30,820
н-Гексанол 33624 24,70 0,590 0,564 24,48 31,460
н-Октанол 39024 21,90 0,597 0,557 27,53 32,840
Кислоты
Уксусная 23015 14,60 0,589 0,566 27,42 30,450
Масляная 28420 20,00 0,587 0,556 26,96 30,700
Изовалериановая 29398 22,80 0,597 0,563 25,31 32,640
н-Валериановая 31120 22,50 0,591 0,555 27,35 31,680
Сложные Э( ЬИдЫ
Метилформ иат 13376 14,90 0,567 0,490 24,62 25,696
Этил формиат 16076 17,80 0,565 0,495 23,84 25,510
Метилацетат 16076 17,80 0,565 0,490 24,49 25,510
Этилацетат 18781 20,40 0,565 0,499 23,75 25,650
н-Пропилацетат 21481 22,87 0,573 0,505 24,28 26,770
Изопропилацетат 19758 22,90 0,567 0,498 22,35 25,350
Этилпропионат 21481 22,87 0,573 0,505 24,27 26,770
п-Бутилацетат 24181 25,20 0.577 0,506 25,20 27,430
Амилацетат 26882 27,40 0,584 0,511 25,88 28,470
Этилизовалериат 25159 27,00 0,570 0,504 2 3,77 26,350
Диэтилкарбонат 22075 24,20 0,593 0,502 26,44 28.550
Альдегиды и кетоны
Бензальдегид 27672 22,50 0,636 0,513 39,70 37,890
Ацетальдегид 12782 13,20 0,509 0,468 23,32 16,410
Ацетон 15483 16,30 0,534 0,478 24,60 21,299
Метилэтилкетон 18183 18,98 0,556 0,491 25,18 24,690
Диэтилкетон 20883 21,50 0,570 0,503 25,18 26,660
Циклогексанон 23186 22,10 0,575 0,450 35,62 27,850
Простые эфиры
Диэтиловый эфир 11796 20,20 0,521 0,411 16,49 18,270
1,4-Диоксан 25564 19,70 0,614 0,540 33,70 35,590
Поверхностные свойства органических жидкостей и полимеров 359
таблица 46 (окончание)
1 2 з 4 5 6
Амины, амиды
Анилин 30008 21,50 0,658 0,541 43,31 43,230
Пиридин 17514 18,50 0,595 0,403 37,25 29,700
Фзомамид 31739 12,30 0.640 0,585 58,35 43,700 .
Нитрилы
Ацетонитрил 17021 13,50 0,565 0,514 29,10 26,210
Пропионитрил 19725 16,40 0,565 0,546 27,25 26,390
н-Валер ионигр ил 25126 21,70 0,589 0,532 27,44 30,600
Капр о нитрил 27826 24,00 0,592 0,533 27,87 31,080
Метакр илонигр ил 20674 18,50 0,574 0,529 24,40 27,990
Нигр осоединения
Нитрометан 15913 12,86 0,518 0,428 36,98 14,700
Нитроэтан 18613 15,88 0,534 0,433 32,06 20,160
2-Нитропропан 19592 18,70 Ц 547 0,471 29,12 23,299
Нитробензол • 31199 22,30 0,615 0,506 43,35 36,530
Расчеты, проведенные по формуле (382), показывают достаточно хоро-
шее согласие расчетных и экспериментальных значений у, причем расхожде-
ния примерно такие же, как и при расчете с помощью парахора. Соотношение
(382) позволяет оценивать вклад отдельных полярных групп и специфическо-
го межмолекулярного взаимодействия в величину поверхностного натяжения.
Так, например, обычно интересуются вкладом водородных связей, вносимым
в формирование поверхностных свойств органических жидкостей. Оценим
такой вклад на примере спиртов и кислот. Для этого преобразуем соотноше-
ние (382) к виду
0,0181 v-, * *
где Y /\p*h - вклад в энергию когезии водородных связей; ^AEiq =
i I
- Ул^ - вклад в энергию когезии слабого дисперсионного взаи-
I „ i
модеиствия.
Для этанола
£ДЕ* =2ДЕс+6ДЕя+АЕо+£л£,* = 2-550,7 + 6-47,7+142,6+3929=
i i
= 5460 кал /моль = 22820 Дж/моль, а = 16423 Дж/моль. Тогда часть
поверхностного натяжения, обусловленная водородным связыванием,
определится как
360
Глава XIII
0,0181
= ~------77т 22 =20,9 дин/см.
(22А>9 7
i
Вклад, связанный со слабым дисперсионным взаимодействием, составляет
0,0181 О1
Ypq = (^Д1/)2/3 £= 8Д дан/СМ-
i
Общее поверхностное натяжение ур = ур/, +ypq = 29 дин/см, а вклад каж-
дой составляющей соответственно равен 72 и 28 %.
Аналогично для уксусной кислоты ур/, = 20,5, a ypq = 8,2 дин/см; вклад
каждой составляющей соответственно равен 71,0 и 29,0 %.
Такой анализ можно проделать не только для водородных связей, но и
для специфических взаимодействий других типов. Следует заметить, что по
методу Фоукса [143] получаются существенно иные вклады водородного
связывания в общую величину поверхностного натяжения. Так, для форма-
мида ур/, =19, а ур = 58,2 дин/см [143]. Доля поверхностного натяжения, обус-
ловленная водородным связыванием, составляет 33 %. По расчетам, прове-
денным с использованием данных табл.46, для формамида ^АЕ*;, = 16423 и
Z
22 АЕ* = 31739 Дж/мо ль. Тогда эта доля составляет /j^AE* г®Д2
или 52 %, что существенно больше, чем получается по Фоуксу.
Помимо оценки величины поверхностного натяжения у органических
жидкостей представляет интерес связать величину у с плотностью энергии
когезии жидкости 82 или с параметром растворимости 8. Этот вопрос нео-
днократно затрагивался в литературе; имеются эмпирические соотношения,
позволяющие установить такую связь. Например, известно соотношение [122]
8 = 4,1(у/И1/3)0’43 , (383)
где V - мольный объем жидкости. Физический смысл этого соотношения не
ясен и, кроме того, оно не описывает свойства всех органических жидкостей
Так, например, расчет по этому соотношению величин 8 для спиртов и орга-
нических кислот приводит к существенно заниженным значениям.
С помощью соотношения (382) можно получить зависимость, связываю-
щую величину параметра растворимости с поверхностным натяжением. Для
этого воспользуемся соотношением (331), по которому рассчитывается пара-
метр растворимости 8. Сначала преобразуем соотношение (381), ум-
ножив числитель и знаменатель на величину (22 )13 • Тогда получим
Поверхностные свойства органических жидкостей и полимеров 361
у = А—-------------
Ур
(384)
Подставляя соотношение (331) в формулу (384), находим
ур =Л-0,6023(£ЛИ,)1/382 . (385)
В соотношении (385) величины ДИ, выражены в ангстремах, S2- в Дж/см3.
Учитывая, что величина А имеет несколько отличные значения для разных
групп органических жидкостей, получаем соотношение
ур=/?/Хд/<)1/3-82, (386)
I
где Bj = 0,6023-Aj. Для жидкостей группы I = 0,0172, группы IIВ2 = 0,0109,
группы III Вт, = 0,0138. Для определения величины у по соотношению (386)
можно пользоваться как расчетными значениями 8, так и экспериментальны-
ми. Результаты расчета у, проведенного с использованием эксперименталь-
ных значений 8, приведены в табл.46. Видно достаточно хорошее согласие
экспериментальных величин уэксп и вычисленных значений урасч.
Можно решить также обратную задачу (которая более важна), те. по экс-
периментальному значению поверхностного натяжения у рассчитать значе-
ние параметров растворимости 8. С этой целью преобразуем соотношение
(386) к следующему виду (с учетом того, что
= (0,6023)’/6 Ур 1 / 2 (3 87)
в"2-k"6 г’/3
Не трудно увидеть, что соотношение (387) по форме близко к эмпиричес-
кой формуле (383). Однако сомножитель, стоящий перед величиной (у/1/1/3),
не является константой, а зависит от химического строения органических жид-
костей, поскольку от него зависит величина коэффициента упаковки £. В пер-
вом приближении можно принять, что для рассматриваемых выше групп орга-
нических соединений величины к колеблются в небольших пределах и можно
использовать среднее их значение. Проведенные расчеты показывают, что для
группы I органических жидкостей ксрл = 0,580, для группы II & 2 = 0,601, для
группы III кср з = 0,586. Тогда на основе формулы (387) запишем
(388)
где = 7,67; С*2 = 9,58 ; С3* = 8,56.
362
Глава XJII
XIII.2. Поверхностное натяжение полимеров
Перейдем теперь к расчету поверхностного натяжения твердых полиме-
ров. На первый взгляд кажется, что величину поверхностного натяжения по-
лимеров можно рассчитывать непосредственно по формуле (382), где У и
X А1< определяются из расчета на повторяющееся звено полимера. Однако
здесь возникают трудности, связанные как с цепочечным строением полиме-
ра, так и с некоторой неоднозначностью трактовки повторяющегося звена.
Поскольку величина входит в это соотношение в степени 2/3, а вели-
чина X АЕ* - в первой степени, то чем больше число звеньев п будет рас-
сматриваться в качестве кинетической единицы (длина всей цепи,длина сег-
мента), тем большее значение у будет получаться по формуле (382).
Вторая причина хорошо видна на примере полиэтилена и других по-
лимеров. Если для полиэтилена принять повторяющееся звено в виде
— (СН2 — СН2) —, то по соотношению (382) получаем у = 14,7 дин/см. Если
же принять повторяющееся звено в виде - (СН2) -, то у = 11,65 дин/см.
Такой же результат получается и в случае полиамидов. Например, для
полиамида 6
[~(CH2)5-C-NH-]n
О
и полиамида 6,6
[ - (СН2)4- С -NH-(CH2)6- NH-C -]„
О О
при использовании формулы (382) получаем у =45,9 дин/см (полиамид 6) и
у = 58,0 дин/см (полиамид 6,6). С точки зрения брутто-формулы химическое
строение этих полиамидов одинаковое (полиамид 6,6 содержит вдвое больше
тех же элементов, что и полиамид 6). Экспериментальные значения у для этих
полиамидов примерно одинаковы и колеблются в пределах 40—17 ди к/бм.
Изложенные выше вопросы детально проанализированы в работе (33].
В результате получено соотношение
<3“
/
где т - число атомов в повторяющемся звене полимера. Для неполярных по-
лимеров группы I (углеводородных, перфторполимеров, простых полиэфи-
Поверхностные свойства органических жидкостей и полимеров 363
ров) С]н = 0,1277; для полярных полимеров группы I (сложных полиэфиров,
полимеров, содержащих нитрогруппу и т.д.) С1п = 0,0751; для полимеров,
содержащих спиртовые, кислотные и амидные группы (все они являются по-
лярными и проявляют водородное связывание), С2 = 0,0476; для полимеров,
содержащих нитрильные группы (все они также полярные), С3 =0,0600. Если
полимер содержит только ароматические ядра (например, полистирол, поли-
фенилен и др.), то величины С4 = 0,1014.
Расчеты,проведенные по формуле (389) доказывают (табл 47) ,что в боль-
шинстве случаев наблюдается хорошее совпадение расчетных и эксперимен-
тальных значений поверхностного натяжения уп. Наибольшие расхождения
наблюдаются для полимеров, которые легко кристаллизуются; к ним относят-
Таблица 47
Исходные данные и результаты расчета поверхностного натяжения полимеров
Initial data and results of calculation of the surface tension of polymers
Полимер i 4 i Дж/моль ZAI', , А3 Уп эксп, [133, 214] Уп по уравнению (389) упио уравнениям (399)и(400)
дин/см
Полиэтилен 5401 34,2 31,0-35,7 36,0 35,6
Полигцгопилен 14885 51,3 29.0-29,6 38,8 39,6
Полиизобутилен 10801 68,6 27,0-33,6 28,8 34,8
К%чу к СКВ 9054 64,3 32,0 33,5 32,9
К^чу к СКИ 10030 81,5 31,0 29,0 28,6
Поливинил^ т<р ид 12087 39,3 28,0-36,7 43,2 44,3
Поливинилхлорид 11055 49,0 39,0-41,5 34,2 36,3
Поливинилиденфторид 5205 44,6 23,0-32,7 29,1 29,0
Политрифтор этилен 11891 49,7 22,0-23,9 36,3 38,9
Полихлор тр ифтор этиле н 10761 65,7 31,0 27,4 30,7
Политегр яЬторэтилен 5008 55,0 18,5-19,0 24,5 18,8
Пол ИСТф ол 22990 109,7 33.0-43,0 40,5 38,0
Поливиниловый СПИ [Г 23675 41,6 37,0 46,5 61,0
Полиакрил они рш 19324 54Q 44Q 42 6 50 0 '
II олиметилакри лат 18379 79,5 41,0 32,6 33,8
Полиамид 11 51562 201,2 31,0-33,0 34,9 31,4
Полиамид 6 .38059 116 0 40 С14 7 0 45 0 -39-5
Полиамид 6,6 76118 231,5 39,3-46,0 45,2 35,1
Полиамид 10,10 97723 368,3 28,5-32,0 36,1 29,1
Полиэтилентер ефталат 43547 169,9 41,0-47,0 38,1 352
Полиэтилакр илат 21080 96,7 35,0 30,4 31,6
Полиметил метакрилат 21080 96,7 39,0-40,2 30,4 31,6
П ол иэтил ме та крилат 23780 113,8 33,0 29,1 30,0
Поливинилацетат 18379 79,6 36,0 32,6 33,8
Поли фо рмальдегид 3297 27,2 3 6,0 29,1 36,0
Полиэтиленоксид 5997 43? 43,0 32,3 43,0
Полипропиленоксид 8698 60,9 32,0 33,2 31,9
364
Глава XIII
ся полиэтиленоксид и полиформальдегид, причем для них коэффициент мо-
лекулярной упаковки существенно превышает среднее значение.
Величину поверхностной энергии уп для полимеров удобно рассчитывать
с помощью параметра растворимости 5. Действительно, из уравнения (331)
имеем
(390)
7 i
Подставляя формулу (390) в уравнение (389), получаем
Q>I/,)l/2S2^
Yn = -'j ^1/2 (391)
или
lx W
1п=^2у----------= (392)
I т
где Dj = CjNA (Dj — параметр, как и Cj , зависящий от принадлежности поли-
мера к данной группе). С учетом значений С,, приведенных выше, можно
установить, что для неполярных полимеров (углеводородов, простых поли-
эфиров и т.д.) D1 н = 0,0769. Для полярных полимеров, содержащих сложноэ-
фирныс и другие полярные группировки. Dln = 0,0452. Для полимеров, со-
держащих гидроксильные, кислотные и амидные группы (сильное водород-
ное связывание) D^ = 0,0287; для полимеров, содержащих ширильные группы,
Оз = 0,0361. Для полимеров, содержащих только ароматические ядра (напри-
мер, полистирол, полифенилен и др.), О4 = 0,061. При этом если Ван-дер-Вааль-
совый объем выражен в А3, а плотность энергии когезии 52 в Дж/см3, то повер-
хностное натяжение, рассчитываемое по формуле (392), определяется в дин/см.
Недостаток описанной выше расчетной схемы заключается в том, что пс -
реход от одного ряда жидкостей к другому или от одной группы полимеров к
другой требует применения своего коэффициента Aj или Q в уравнениях (3 82)
и (389). Трудности, которые здесь возникают, связаны с тем, что молекулы
жидкости или повторяющиеся звенья полимеров могут обладать большим
объемом и относительная доля тех специфических групп ,по которым разде -
ляются вещества по данному признаку, будет мала. Например, нитрильные
группы могут содержаться не только в полиакрилонитриле, где их относи-
тельный вклад в Ван-дер-Ваальсовый объем и энергию когезии будет велик,
но и в других полимерных системах, где их вклад может быть весьма малым.
Кроме того, затруднения возникают при расчете поверхностной энергии со -
полимеров, звенья которых принадлежат к разным классам гомополимеров.
В связи с этим в работе [371 развита расчетная схема оценки поверхност -
ного натяжения органических жидкостей и полимеров, параметры которой
Поверхностные свойства органических жидкостей и полимеров 365
чявисят только от химического строения органической жидкости или поли-
мера, но не от принадлежности их к какому-либо классу.
В цитируемой работе величина кП выражена через соотношение
5 ~(£Д[/)2/3
(393)
Здесь 5* - плотность поверхностей энергии когезии, т.е. энергия когезии,
приходящаяся на единицу поверхности. Отметим, что эта величина отлича-
ется от обычной плотности энергии когезии, которая определяется соотно-
шением (331).
Между коэффициентом молекулярной упаковки жидкости на ее поверх-
ности к„ и плотностью поверхностной энергии когезии S имеется следую-
щая зависимость [37]:
кп = -0,4112 + 0,3O121g3* •
(394)
После подстановки (380) в формулу (394 ) получаем
0,0461-lgS* -0,063 „ 0,2768-lg3* -О,378ч
Гр = S -----------к-------с-------------к--------)
Подставляя в формулу (395) выражение (5) для к, окончательно имеем
(0,0461-lg5*-0,063)Л/
! (0,2768 1g 3*-0,378)3/
рЛ'дЕДИ,
/
(396)
Соотношение (396) позволяет с достаточной для практических расчетов
точностью оценивать поверхностное натяжение органических жидкостей не-
зависимо от того, к какому классу данная жидкость принадлежит. Результаты
расчета показывают [37], что в большинстве случаев наблюдается удовлетво-
рительная сходимость расчетных и экспериментальных данных (см. табл.46).
Перейдем теперь к оценке поверхностной энергии полимеров. Исходное
соотношение для получения уравнения (389) выглядит следующим об разом
[37].
к Я* к
у,, =а 0.1532-и--^-(1 -0.919/-), (397)
к ти' к
где а - постоянная зависящая только от принадлежности полимера к классу,
полярных или неполярных.
Примем ту же зависимость коэффициента молекулярной упаковки в по-
верхностном слое кп от 5*. что и в жидкостях. Коэффициент молекулярной
366
Глава XIII
упаковки полимеров в объеме, как неоднократно отмечалось выше, практи-
чески не зависит от химического строения полимеров и его средняя величина
А'ср = 0,681 для монолитных полимерных тел.
Используя зависимость кп от 5* в виде соотношения (394) и А’ср = 0,681, на
основании соотношения (394) получаем
8* * *
уп = а—— (0,067761-1g 8 — 0,0925)(1,5549 — 0,406? 1 -lgS ) (398)
т1
Проведенный анализ показал [3?], что величины а действительно при-
близительно одинаковы; для полярных полимеров = 2,09?, а для неполяр-
ных полимеров ctcp = 3,055.
Тогда формула для расчета поверхностной энергии полярных полимеров
выглядит так:
Упп = -П77(0,1421 1g8* - 0,194)(1,5549 -0,406? • 1g 8*), (399)
пг
а для неполярных полимеров
8* * *
Упн =—i7?(0,20?0-lg8 -0,2826)(1,5549-0,4067-lg8 ) (400)
/и1 3
Результаты расчета поверхностной энергии по формулам (399) и (400)
приведены в табл.47. Видно, что в большинстве случаев наблюдается хоро-
шее соответствие расчетных и экспериментальных значений поверхностной
энергии. Следует отметить, что экспериментальное определение поверхнос-
тной энергии твердых тел, в том числе и полимеров, затруднено, хотя имеют-
ся различные варианты метода оценки уп. Поэтому в литературе можно встре-
тить существенно различающиеся данные по поверхностной энергии для
одного итого же полимера.
Полученные в работе [37] соотношения (399) и (400) позволяют оцени-
вать поверхностную энергию полимеров любого химического строения безот-
носительно их принадлежности к какому-либо классу' химических соедине-
ний. Это снимает те же трудности, которые характерны для расчета поверхно-
стного натяжения органических жидкостей, отмеченные выше.
В заключение рассмотрим еще одну связь между поверхностной эн ерги-
ей органических жидкостей и полимеров и параметром растворимости 8, ве-
личина которого определяется из соотношения (331).
«у у £
Еде,
Учитывая, что 8 = —-----—- и подставляя это выражение в уравнение
(Ед^)
(331), получаем '
Поверхностные свойства органических жидкостей и полимеров 367
5*=52Лл(2Х),/3.
(401)
Далее, подставляя выражение (401) в формулу (400), и выполняя необхо-
димые преобразования имеем
Гр=^82(1Д^)1/3
0,09221g 6 + 0,01537 lg(£ А !<) - 0,073
________________________i_____________
k
0,5541g5 + 0,0923 lg(£ AI< ) -0,439
____________________l____________
k
(402)
Соотношение (402) связывает величину поверхностного натяжения жид-
костей ур с параметром растворимости 8. По указанному соотношению можно
найти величин}' 3, если известны экспериментальные значения поверхност-
ного натяжения жидкостей (оценка проводится методом подбора). Это важно
в связи с тем, что поверхностное натяжение жидкостей измеряется сравни-
тельно легко, а параметр растворимости, связанный со скрытой теплотой ис-
парения жидкости, более сложным образом. Особенно это трудно для полиме-
ров, поскольку для них параметр растворимости может быть определен толь-
ко косвенными методами - по измерению набухания в различных растворите-
лях, по вязкости растворов и т.д. Следует также отмстить, что возможность
оценки расчетным путем поверхностной энергии полимеров важно и потом}',
что их поверхностная энергия простыми соотношениями связана с энергией
когезии и энергией сублимации.
При расчете поверхностной энергии сополимеров или гомогенных сме-
сей могут возникать различные ситуации. Если компоненты сополимера отно-
сятся к одной группе веществ (согласно приведенной выше классификации),
то, поскольку величина С, для них одинакова, соотношение (389) примени-
тельно к сополимерам принимает вид
«|(ЕД£^1 + а2(£ЛЕ*)2 +...+
Уп = С ---------------------------------------------
7 [ai(EAl<)i+a2(^AI<)2+... + a„(^AI<);,]2/3(a1w1 +
I i t
+ а„(^АЕ*)п
1/3 '
+ Ct 2 ^2 + • • • + Ctfl m n )
368
Глава XIII
где eq, а2, а„ - молярные доли компонентов 1, 2, п;
i
(E^*)2> - - энергии когезии для компонентов 1, 2, и;
/ i
dA^)b (X Ы7)п - их Ван-дер-Ваальсовые объемы; т}.
i i i
т^, т„ - количество атомов в повторяющихся звеньях компонентов
1,2, ..., п. В более компактной форме соотношение (403) записывается в виде
A'=w
Е ak(Y^ih
k=i i
(404)
k=l i
Ik=n
Takmk
^=1
Если мы хотим выразить поверхностную энергию сополимера через по-
верхностные энергии гомополимеров у,,,ь уп>2, ..упи, полученных из компо-
нентов 1, 2, и, то уравнение (403) приобретает вид
уп = С ----------------------------------------—►
7 [а](Ед,/-)1+а2(ХД1<)2+--- + а»(ЕА1<)«]2/3х
+ a«Yn,„(EAK/)«/3wi/3
- -----------. (405)
x(a!^ +a2w2 +... + а„щ„)
В более компактной форме соотношение (405) можно записать следую-
щим образом:
Е^УпДЕД^М73
Уп =---—---------. (406)
Ё\ (ЕА^Л-
_к=1 <
Если все компоненты сополимера относятся к разным группам веществ
(значения Су для них разные), то соотношение (403) для такого сополимера
записывается так (на примере двойного сополимера):
‘ Ск=п
Хактк-
I W
сДЕ^ )i +a2(XA/V )2
Уп - (alQ,/ +а2С'jjl)-------!—^7“-----------------------------------ПТ (407)
[И) (X А1< )1 + а2(Е А,Э )2 I2 3 (“1'«] + «2ОТ2 ) '
Поверхностные свойства органических жидкостей и полимеров 369
или
aiYn.i(?A^)?/3w',/3/CA/ +
у п = (а, С,, + а, С „)----!----------------утт— -»
i i
+ IС jp
________i_______________
x(a1w1 +a2w2)13
(408)
В случае многокомпонентного сополимера могут встречаться различные
варианты: часть компонентов может принадлежать к одной и той же группе
веществ, а часть к другой группе. В общем случае следует записать
Су -щСр\ + a2Cy2 +... + а„Су (409)
где Cjp - величина Q для группы веществ, к которой принадлежит компо-
нент 1 (это может быть Сд, С,ц, Сущ и Czjv); Су.2,..., Су „ - то же самое для
компонентов Я • п Тогда с учетом (409) соотношение (404) записывается в
виде
к=п
Уп — ''У'.У-кС i .к
к=\
к=п
'£ak(£№*)k
_________к=\ i__________________
[к-н f/3
CLak”‘k)
_к=А / J А-=1
(410)
а соотношение (406) принимает форму
к=п
У„ = XakCj,k
к-Л
&*Уп.к(2Ж)ГМ/3/Cj,k
кА_____[_____________
к=п 32/3 к=п
X^k(Z^i)k (2>Л)1/3
кА i кА
(411)
Приведенные выше соотношения для расчета поверхностной энергии со-
полимеров не совсем удобны тем, что приходится каждый раз учитывать при-
надлежность полимера к какой-либо из групп. Поэтому для компьютерной
реализации метода удобно выразить величины Cj через поверхностные энер-
гии компонентов сополимера. Из уравнения (389) имеем
ulX.)27 3"'173
370 Глава Х1П
Тогда для сополимера получаем:
ynl(ZA^)?/3< ь2ст)ГМ/3
У п = “1-4:—;-----+ и 2----;---------+
OX), (М)2
УПИ(ХДК)»/3^/31 +а2(ХДЕ‘)2 +
+ a" (£ае‘)и ’[a1(£A^)i+a2(LA^)2 +
+ ... + a„(£AE‘)„
------------------------------------------Г7Т ’ (41 Г)
+ ... + а„(2,АТ;)п] (aj/Mj +а2от2 + ... + a„w„)1/J
где все обозначения те же, что в приведенных выше формулах. В более ком-
пактном виде соотношение (41 Г) записывается следующим образом:
к=п Уп,а(2А^)а/3^/3
Тп ~ X aA . „* .
a=i (Zae,)*
дл
.4=1 I
-------- (4П")
(Ea*wi)1/3
А=1
Приведем пример расчета поверхностной энергии для сополимера поливи-
нилового спирта с поливинилацетатом:
—сн2—сн- -сн2-сн-
I I
ОН О—С—СН3
О
Поливиниловый спирт (ПВС) Поливинилацетат (ПВА)
Для ПВС имеем
^ДЕ* =2ДЕс+4ДЕя+ДЕо+АЕ* =2-550,7 + 4-47,7 + 142,6 +
+’3929 = 5363,8 кал/моль = 22420,6 Дж/моль.
X АС; = АСС10 + ДИГ39 + 3AKw125 + АИ^дэд + ДСО132 =
= 13,1 +12,2 + 3 • 2,0 + 4,7 + 5,6 = 41,6 А3.
Число атомов т в повторяющемся звене ПВС равно 7.
Поверхностные свойства органических жидкостей и полимеров 371
Подставляя величины ^АЕ(- , ^АГг - и т в уравнение (389) и учи-
i i
тывая, что данный полимер относится к группе II полярных полимеров
(С2 = 0,0476), получаем
у =0,0476---22,2?’б,,д =46,5 дин/см.
(41,6)2/3-7 73
Для ПВА имеем
£АЕ‘=4ДЕс+6АЕ^+2АЕо+АЕ^ =4-550,7 + 6-47,7 + 2-142,6 +
+ 1623 = 4397 кал/моль = 18380 Дж/моль.
^АИ,- - АИСд0 + АИС39 + AKC4g +АИС13 +6АИ//Д24 + АКО129 + АИО139 -
i
= 13,1 + 12,2 + 15,9 + 17,2+6-2,0 + 3,4 + 5,8 =79,6 А3.
Число атомов т в повторяющемся звене ПВА равно 12.
Используя уравнение (389), и учитывая, что ПВА принадлежит к поляр-
ным полимерам группы 1 (С1п = 0,0751), находим
Уп =0,0751-------—-----— = 32,6 дин/см.
(79,6)273 • 12173
Для расчета уп сополимера воспользуемся сначала соотношением (407);
примем, что молярная доля ПВС а, = 0,4, а для ПВА а2 = 0,6. Подставляя все
параметры сополимера в соотношение (407), получаем
уп = (0,4 0,0476 + 0,6 0,0751) х
_________0,4-22420,6 + 0,6-18380_________
(0,4 • 41,6 + 0,6 • 79,6)2'3 - (0,4 • 7 + 0,6 • 12)1'3
= 37,0 дин/см.
Теперь воспользуемся соотношением (408):
уп = (0,4 • 0,0476 + 0,6 - 0,0 751)
0.4 • 46,5 41,62 7 3 • 717 3 / 0.0476 +
(0,4-42,6 + 0,6-79,6)273 х
+ 0,6-32,6-79,6273-121/3 /0,0751
х(0,4-7+0,6-12)1/3
= 37,0 дин/см.
Теперь рассчитаем поверхностную энергию тройного сополимера на основе
акрилонитрила, бутадиена и стирола (АБС-пластик).
372
Глава XIII
—сн2—сн— —сн2—сн=сн-сн2— —сн2—сн—
C=N о
ПАН ПБ ПС
Для ПАН
=ЗАЕс + ЗДЕ„ + XE*N + AEd =3-550,7 + 3-47,7+1205 + 1623 =
= 4623,2 кал/моль = 19325 Дж/моль.
А^- = ДГС10 + АГС6 + АИс71 + ЗАИ^дэд + ДК#;154 =
= 13,1 + 9,0 +15,9 + 3 - 2,0 +10 = 54 А3.
ПАН относится к полимерам 3-й группы, так как содержит нитрильную группу
(С3 = 0,060). Число атомов т в повторяющемся звене ПАН равно 7. Для него,
согласно формуле (389),
19325
уп =0,060———— = 42,4 дин/см.
542/3.71/3
Для ПБ
£ДЕ,‘ = 4ДЕ^ +6ДЕ^ + ДЕ* = 4-550,7 + 6-47,7-323 =
i
= 2166 кал/моль = 9054 Дж/моль
£ДР,- = 2ДКс>10 + 2ДЕС1б +6ДИ//Д24 = 2-13,1+2-13,1 + 6-2,0= 64,4 А3.
i
ПБ относится к неполярным полимерам 1 группы (С1н = 0,1277); для него
т = 10. Использование формулы (389) приводит к следующему результату’
уп =0,1277
9054
(64.4)2/3 -101/3
= 33,4 дин/см.
Для ПС
£ Д/д* = 8ДЕ(- + 8ДЕ^ + ДЕ*, = 8 • 550,7 + 8 - 47,7 + 713 =
i
= 5500 кал/моль = 22991 Дж/моль.
Хдг,- = ДКсдо + ДКС7 +ДКС19 +5ДГС18 +8ДИ/4124 =
i
= 13,1 + 8,7+8,4 + 5-12,7 + 8-2,0 = 109,7 А3.
Поверхностные свойства органических жидкостей и полимеров 373
ПС относится к IV группе полимеров (С4 = 0,1014), для него т = 16. Подста-
новка всех значений параметров ПС в формулу (389) дает
22991
уп =0,1014-------—-----— = 40,4 Дин/см
(109,7)2/3 • 161/3
При расчете поверхностной энергии сополимера примем, что доля звень-
ев ПАН а! =0,3, доля звеньев ПБ а2 =0,2 и доля звеньев ПС а3 =0,5. Тогда из
формулы (408) имеем
уп = (0,3 • 0,06 + 0,2 • 0,1277 + 0,5 • 0,1014) х
0,3-42,4-542/3 -71/3/0,06+0,2-33,4-64,42/3 101/3 /0,1277 +
(0,3 • 54 + 0,2 • 64,4 + 0,5 • 109,7)2/3 х
+ 0,5-40,4-109,72/3 -161/3/0,1014
х (0,3 -7 + 0,2 • 10 + 0,5 • 16)1/3
= 40,9 дин/см.
Глава XIV
СОВМЕСТИМОСТЬ ПОЛИМЕРОВ
Проблема совместимости полимеров в настоящее время является одной
из наиболее важных. Дело в том, что сейчас создание новых полимерных ма-
териалов идет, как правило, не путем синтеза новых полимеров, а путем со-
здания смесей известных полимеров. При этом речь идет не только о смесе-
вых композициях , в которые компоненты смеси вводятся в сравнимых коли-
чествах, но и о введении микродобавок полимеров, их поверхностной
модификации и т.д. Здесь же возникают и такие вопросы, как микрофазовос
расслоение, умение управлять составом и размерами микрофаз и т.д. Не имея
возможности рассмотреть многочисленные публикации, появившиеся в пос-
леднее время в этой области, проанализируем один из возможных путей пред-
сказания совместимости полимеров и оценки состава микрофаз.
Один из возможных путей решения вопроса о предсказании совмести-
мости полимеров заключается в использовании критерия (345), который пред-
назначен для анализа растворимости полимеров .При этом ,если один поли -
мер вводится в малых количествах в другой полимер, то первый из них рас-
сматривается при использовании критерия (345) как “полимер”, а второй -
как “растворитель”. В принципе возможны следующие варианты.
1. При использовании критерия (345) оказывается, что во всех случаях
левая часть критерия больше правой части, т.е. имеет место абсолютная не-
совместимость. Под всеми случаями подразумеваются такие, когда первый
полимер вводится в небольших ко л шест вах во второй и насб орот, второй по-
лимер вводится в небольших количествах в первый. Тогда критерий совмес-
тимости полимеров имеет вид:
- при введении первого полимера во второй:
Р1 =-^- > 1,374Ф(Ф - ^Ф2 -1 + «]) = 2рЗ, •, (412)
5п,2
Совместимость полимеров 375
- при введении второго полимера в первый:
g2 ----------
р2 = -^>1,374Ф(Ф-у/Ф2-l + a2) = 2pft2, (413)
5П,1
где 8пд и Sn 2 - параметры растворимости полимеров 1 и 2 соответственно;
ф_ (414)
(^п,11/3 +Г/п,21/3)2 ’
где Кп j и - молярные объемы полимеров 1 и 2 соответственно;
Ф = Уп,1;п,2/Уп,2; (415)
а2 = Уп.1;п,2^УпД >
где
Уп, и,л = Уп, 1 + Уп,_э - 2Ф(уп, 1 • У,, 2 )2, <416)
Уп1 и Уп2 _ поверхностные энергии полимеров 1 и 2 соответственно.
Критерии (412) и (413) означают, что полимеры являются абсолютно
несовместимыми.
2. При введении небольших количеств первого полимера во второй кри-
терий (345) показывает, что совместимость их наблюдается, т.е.
р. =-^<1,374Ф(Ф-л/Ф2-1 + п]) = 2р01. (417)
^.2
Однако при введении второго полимера в первый, может оказатся, что совме-
стимость не наблюдается, т.е.
ц2 = —> 1,374Ф(Ф--^Ф2 - 1+ #2) =2р 02 • (418)
5П,1
Этот, на первый взгляд, парадоксальный вывод на самом деле имеет место,
что будет продемонстрировано ниже. Здесь же следует заметить, что обычно о
совместимости двух полимеров судят по температуре стеклования смеси. Для
абсолютно совместимых полимеров характерно одна температура стеклова-
ния смеси, которая лежит между температурами стеклования исходных ком-
понентов. Для смесей абсолютно несовместимых полимеров наблюдаются две
температуры стеклования, каждая из которых соответствует температуре стек-
лования исходного компонента. При частичной совместимости, когда в каж-
дой микрофазе присутствуют оба компонента, но в разных количествах, про-
являются также две температуры стеклования, но по сравнению с температу-
рами стеклования исходных компонентов они смещены навстречу друг другу.
376
Глава XIV
Рис.98. Схематическое изображение зависимости температур стеклования Tg
от состава а смеси: 1 - частично совместимые полимеры; 2 - полностью совместимые
полимеры
Schematic representation of dependence of the glass transition temperature Tg
on composition of mixture a: 1 - partially compatible polymers; 2 - completely compatible
polymers
Для рассматриваемого случая, когда первый полимер совмещается со вто-
рым, а второй не совмещается с первым, зависимость температуры стеклова-
ния от состава имеет вид, схематически представленный на рис.98, кривая 1.
Объяснение такого хода кривой будет дано ниже на примере конкретной сис-
темы “полимер 1 - полимер 2”.
3. Этот случай соответствует абсолютной совместимости полимеров, т е.
первый полимер “растворяется” во втором, а второй -“растворяется” в пер-
вом. Критерий совместимости приобретает вид:
- при введении первого полимера во второй
М1 =^<1,374Ф(Ф-А/Ф2 -l + q)=2ppi; (4)9)
5п,2
- при введении второго полимера в первый
ц2 = < !’374ф(ф->/ф2-1 + «2) = W (420)
“п,1
При такой ситуации, как было отмечено выше, проявляется одна темпе -
ратура стеклования Tg, а зависимость Tg от состава смеси имеет вид, схема-
тически представленный на рис.98, кривая 2.
Теперь рассмотрим поведение ряда конкретных, хорошо изученных по-
лимерных смесей. Первая из них - смесь полистирола с поливинилметило-
вым эфиром:
Совместимость полимеров
377
— СНг-СН~ Полимер! — СНг-СН- Полимер 2
6 ?
СНз
Поведение этой смеси различных составов детально изучено в раде ра-
бот [131.153,168,198-200]. Зависимость температуры стеклования Tg этой
смеси от состава показана на рис.99.
Рис.99. Зависимость температуры стеклования Tg от молярной доли а полистирола
для смеси поливинилметиловый эфир-полистирол (2). Пунктирная кривая (1)
отображает зависимость Ts от а при условии полной совместимости компонентов
Dependence of th е glass transition temperature Tg on mole fraction a of polystyrene
for the mixture of polyvinylmethyl ether with polystyrene (2). Dotted curve (I) repre-
sents dependence of Tg on a on condition at components are completely compatible
Хорошо видно, что при увеличении содержания полистирола от 0 до 40
% величина Tg изменяется очень слабо и практически не зависит от состава
смеси. Эта зависимость не описывается уравнением (94), пригодным для ста-
тистических сополимеров и гомогенных смесей полимеров (кривая 1).
Проанализируем в деталях совместимость полистирола (ПС) и поливи-
нилметилового эфира (ПВМЭ) с помощью критерия (345). Сначала предпо-
ложим, что ПС является “растворителем” для ПВМЭ. Исходные характери-
стики, необходимые для использования критерия (345), приведены в тгб л48 .
Подставляя значения этих характеристик в критерий (420), получаем
378
Глава XIV
Таблица 48
Исходные данные для оценки совместимости ряда полимеров: па'шстнрол (ПС),
полившшлметиловьв'! эфир (ПВМЭ) и поли-2, 6-диметил-1, 4-фениленоксид (ИФО)
Initial data for estimating compatibility of a series of pointers: polystyrene (PS),
poly(vinylmethyl ether) (PVME) and poly(2,6-dimctliyl-l,4-phenylenc oxide) (PPO)
Физические характеристики ПС ПВМЭ ПФО
Параметр растворимости S , (кал/см3)1'2 9,12 7,54 8,93
Поверхностная энергия у , дин/см 41,6 33,4 44,4
Молярный объем V. см3/моль 97,08 60,7 103,75
Энергия когезии LAZi, , Дж/моль / 22988 8696 23587
Ван-дер-Ваальсовый объем . А3 / 109,8 60,7 117,4
Температура стеклования Те, К 373 245
n2 =0,684 <1,218 = 2pP2.
Таким образом, поскольку левая часть критерия совместимости меньше
его правой части ,то ПВМЭ хоро шо“ растворяете^" вПС.
Теперь предположим что ПВМЭ является “растворителем’" для ПС Тог-
да с помощью критерия совместимости (412) получаем
щ = Х462> Ц75 = 2р[31.
Поскольку левая часть критерия больше правой части, это означает, что
поливинилметиловый эфир не “растворяет” полистирол. Поэтому, когда ПС
вводится в ПВМЭ, следует ожидать микрофазовое расслоение. Однако, по-
скольку ПС является “растворителем” для ПВМЭ часть ПВМЭ будет совме-
щаться с ПС. В результате могут образовываться две микрофазы, одна из ко-
торых содержит ПВМЭ, а вторая смесь ПВМЭ с ПС. При возрастании кон-
центрации ПВМЭ во второй микрофазе совместимость этой микрофазы с
ПВМЭ будет улучшаться и при определенной концентрации ПВМЭ вторая
микрофаза будет совместима с ПВМЭ. Определим эту критическую концент-
рацию ПВМЭ. С этой целью запишем соотношение для расчета параметра ра-
створимости данной смеси полимеров. На основе уравнения (331) получаем
«(SMh +(!-«)(! (421)
^2 ______[_______________i_______
смеси ^[а(£Д1/)2 +(1 -а)(ХА1<)1] ’
i i
где а - молярная доля ПВМЭ в смеси' (^Д/<*)] и (£Д£[ )2 - молярные
/ '
энергии когезии для ПС и ПВМЭ соответственно', (^ДСД ] и (^ДГ,) 2 -
Совместимость полимеров
379
Ван-дер-Ваальсовые объемы повторяющихся звеньев ПС и ПВМЭ соответ-
ственно.
Подставляя в соотношение (421) характеристики полимеров, приведен-
ные в табл.48, имеем
с 2
°смеси
22988-14292а
418(6613 29,57а)
кал/см3.
(422)
Теперь оценим поверхностную энергию смеси на основе уравнения (410):
<£ а£(. )2 +
Усмеск ~ [«С, н + (1 - И)С4 ]-—---1--------------------►
4 [a(XA^)j +(l-a)(ZA^)i]2/3x
i i
+ (1-а)(£ДЕ*)1
------------L-----ПТ’ (423)
х[а/и2 + (l-a)wj ]
где С1„ и С4 - коэффициенты, входящие в уравнение (410) для ПВМЭ (непо-
лярный полимер) и ПС соответственно; пт2 - числа атомов в повторяю-
щихся звеньях ПС и ПВМЭ соответственно.
Подставляя все величины из табл.48 в соотношение (423), получаем
У смеси
(0,023 la+ 0,1046)
22988-14292a
(109,8 - 49,la)2/3(16-6а)1/3
(424)
Для дальнейшего анализа нам нужно оценить молярный объем смеси
Исмеси = a53,68 + (l-а)97,088. (425)
Используем соотношения (422), (424) и (425) для расчета левой и правой
частей критерия (345). При этом будем рассматривать двухкомпонентную
смесь, один из компонентов которой представляет собой ПВМЭ, а второй —
смесь (микрофазу) ПВМЭ/ПС с разной молярной долей аПВМЭ. Результаты
расчета показаны на рис. 100 в виде двух зависимостей обеих частей критерия
(345) от мо тарной доли ПВМЭ. Точка пересечения этих двух зависимостей
соответствует содержанию ПВМЭ в микрофазе, при котором наступит совме-
стимость ПВМЭ с этой микрофазой. Эта критическая концентрация акр = 0,62.
Ван-дер-Ваальсовый объем смеси с критической концентрацией ПВМЭ бу-
дет равен
(X А/лф = 60 7 -,0 62 +109 8 -,0 38 = 7,9 36 А3.
Теперь можно рассчитать температуру стеклования смеси с критической
концентрацией ПВМЭ. Для этого воспользуемся уравнением (94)
380
Глава XIГ
4
T
Д’.кр
“кр
Рис. 100. Зависимости р (2) и 2рр (1) от молярной доли а поливинилметилового эфира
внутри микрофазы (см. текст)
Dependences of р (2) and 2рр (1) on mole fraction a of poly(vinylmethyl ether)
within microphase (For explanation see text)
®кр [(£ AJ/ )2 - (X Дr,)l ] + £ AC, )j
i i i
(£АИ,)2 (Saf')i’I (2Х)!
' ~~----- + ----+ 2aKp(l-aKp)0,03
lg,2 1 gA 1 gA
(426)
Подставляя в соотношение (426) все параметры системы из табл.48 и учи-
тывая, что акр = 0,62, находим, что Tg,Kp = 284 К. Теперь можно найти зависи-
n гость температуры стеклования Tg от состава смеси, состоящей из ПВМЭ и
‘критическои'”смсси, т.е. такой, которая состоит из ПС и ПВМЭ с критичес-
кой концентрацией аК[ЛВМЭ , равнсйО 62 И спользуя для это'й цели уравне-
ние (94), получаем
о[(ЕАГ) 2-(ZAI^Kpl + (ZAI/i)Kp
(427)
где a - молярная доля ПВМЭ
Совместимость полимеров
381
©отношение (427) <п рж ал иво только в интервале а от 1 до 0 62 Кривая
построенная в соответствии с соотношением (427) .показана на рис.99 (кри-
вая 2) Видно \ito экспериментальные точки хорошо укладываются на эту
зависимость.
Для описания второй части зависимости Tg от состава смеси ПВМЭ/ПС
(внутри интервала а от 0,62 до 0) прежде всего необходимо определить Ван-
дер-Ваальсовый объем смеси, для которой Tg = 284 К:
£Д1/. = 0,62(£ АК,)2 + 0,38[0,62(£Д1<)2 + О,38(£ ДЦ), ]!
i i i I
XAC,=673A3.
(428)
Теперь можно описать зависимость Tg от состава смеси, один из компо-
нентов которой представляет собой ПС, а второй — микрофазу с Tg = 284 К
(см.выше). Для этой системы имеем
__________а(109,8-67,8)+ 67,8_______
8 “ ,109.8 67,8 67,8 ,п ,А
а(-----------)н----+ 2а(1-а)-0 03
373 248 248
где а молярная доля ПС на шкале от 1 до 0,38 Для того чтобы определить
реальную молярную долю ПС в смеси,следует записать
(1 -о)--0 38 +а' 0 62 ,
где а- молярная доля ПВМЭ и (1 — а) — молярная доля ПС в обшей смеси.
„ , (1-а)-0,38
Подставляя а = Л---— В соотношение (428), находим, что полу -
ченное соотношение хорошо описывает вторую часть зависимости Tg в интер-
вале а от 0 до 0,62 от состава смеси при высоком содержании ПС (см. рис.99).
Т акигобразом критерий растворимости (345) может быть с успехом при-
менен для описания зависимости температуры стеклования Tg такой смеси
полимеров, в которой один из полимеров хорошо “растворяет” второй поли-
мер, но этот второй полимер плохо “растворяет” первый.
Теперь проанализируем более простой случай, когда два полимера абсо-
лютно совместимы друг с другом.
В качестве примера тако"и смеси рассмотрим систему полистирол
(ПС) - поли(2,6-диметил-1, 4-фениленоксид) (ПФО), хорошо изученную в
ряде работ [139, 166., 169, 197, 203, 204, 209].
Предположим, что ПС является “растворителем” для ПФО. Применяя кри-
терий растворимости (345) с учетом всех физических характеристик компонен-
тов смеси (см .табл.48)., находим, что ц2 = / dj2 = 0,959; Ф = 1,0; у| 2 = 0,0456;
382
Глава XIV
Рис 101. Зависимость температуры стеклования Tg от молярной доли а пали(2,6- диметич-
1,4-фениленоксида) для смеси этого полимера с полистиролом
Dependence of the glass transition temperature Tg on mole fraction a of poly(2,6-
dimethyl-l,4-phenyleneoxyde) for the polymer blend with polystyrene
a = 0,001096; 2p02 = 1,328 (3| и 32 — параметры растворимости ПС и ПФО
соответственно). Поскольку ц2 < 2р02, то согласно критерию (345), ПС явля-
ется хорошим “растворителем” для ПФО, т.е. должна наблюдаться совмести-
мость. Теперь примем, что ПФО является “растворителем” для ПС, тогда
Pi = З2 /З2 = 1,043; Ф = 1,0;У1,2 = 0,0456; а = 0,00103; 2р₽! = 1,33. Видно, что
и в этом случае левая часть критерия (345) меньше правой части, т.е. совмес-
тимость также должна иметь место. Эксперименты показывают, что действи-
тельно, данная пара полимеров является абсолютно совместимой. Это при-
водит к тому, что температура стеклования смеси ПС/ПФО описывается той
же зависимостью от состава, что и для статистических сополимеров, те. со-
отношением (94). Эта зависимость показана на рис. 101, из которого видно,
что экспериментальные точки хорошо укладываются на расчетную кривую
Следует отметить, что рассматриваемый подход обладает тем преимуществом
перед рядом других подходов, что он не требует введения никаких “подго-
ночных” параметров. Иными словами, для такого анализа нужно знать толь-
ко химическое строение компонентов, на основании которого все физичес-
кие характеристики полимеров определяются расчетным путем.
Глава XV
ВЛИЯНИЕ КОНЦЕВЫХ ГРУПП НА СВОЙСТВА
ПОЛИМЕРОВ
Вопрос о влиянии молекулярной массы полимера и связанный с ним воп-
рос о роли концевых групп и их влиянии на свойства неоднократно затрагивал-
ся в литературе. Имеются, например, данные о зависимости температуры стек-
лования Tg и плавления Тт от молекулярной массы в виде кривых с насыщени-
ем, а также сведения о других свойствах, зависящих от молекулярной массы.
В качестве примера на рис.21 показана зависимость 7'гот среднечисло-
вой молекулярной массы полистирола. Существует ряд соотношений для опи-
’ сания таких зависимостей, например [144]
Ig = 7g(«) М ,
где Tg^—температура стеклования полимера при молекулярной массе М —»оо;
к— константа полимера.
Зависимости такого рода не описывают кривые Tg(M) на всем их протя-
- жении. Более сложная зависимость Tg(\T) имеет вид [145]
Tg -7g(oo)- К*
где К* и М* — подгоночные параметры. Естественно, что от молекулярной
массы зависит не только температура стеклования Tg, но и все другие свой-
ства. Поиски этих зависимостей представляют существенный интерес.
Особенно актуальна эта задача при анализе влияния молекулярной мас-
сы на свойства растущих цепей при очень малом числе звеньев и. При таком
анализе не всегда учитывается то обстоятельство, что химическое строение
[ концевых групп может существенно (а иногда и кардинально) отличаться от
строения повторявшегося звена полимера. Это приводит к тому, что при по-
i строении зависимости параметра какого-либо физического свойства от п каж-
дое новое значение и не равноценно предыдущему', ибо при переходе от п к
384
Глава AT
(л + 1) и т.д. меняется химическое строение усредненного звена. Лишь когда
п ~ 10-20, влияние концевых групп становится малым, и зависимости свойств
от п приближаются к значениям параметров, характерных для повторяюще-
гося звена полимера.
В работе [8] выполнено количественное описание зависимостей трех фи-
зических параметров —Ван-дер-Ваальсового объема, молярной рефракции и
мольной энергии когезии - от числа звеньев в полимерной цепи, начиная от
п = 1. При этом учитывалось влияние типа инициатора, применяемого при
полимеризации, на химическое строение концевых групп и вытекающие от-
сюда свойства димеров, тримеров и т.д. Расчеты проводили на примере четы-
рех полимеров — полиметилметакрилата (ПММА), полистирола (ПС), поли-
этиле нтерефталата (ПЭТФ) и поликарбоната (ПК) на основе бисфенола А.
Рассматривали следующие типы систем:
СНз СНз СНз
н3с-С-(СН2- С)Л.2-СН2-СН С-ОСНз С-ОСНз С-ОСНз О О О СНз ^Нз <рН3 ПММА-1
с=сн-(СН2- с )П.2-СН=С С-ОСНз С-ОСНз С-ОСНз о о о СНз СНз <рн3 ПММА-2
Н3С-С-(СН2-С )„.2-СН = С С-ОСНз С-ОСНз С-ОСНз и и II О О О ПММА-3
О СНз 9Нз
ф-С-(СН2-С )П.1-СН2-СН
С-ОСНз С-ОСНз
II II
о о
(СН2-
СНз О
Н /=s\
c)n-c-Q
ПММА-4
ПММА-5
С -ОСН з
II
о
Влияние концевых групп на свойства полимеров
385
Н3С - СН- (СН2- СН)„.2-сн2- СН2
ООО
НзС - сн- (СН2- СН)п.2-СН=сн
обо
о
Н £ -СН -(СН 2-СН) „-1-С-О
6 о
ПС-1
ПС-2
ПС-3
О О
О-С-(СН2-СН)„-С-О ПС'4
о
HO-[C-Q-C-O-(CH2)2-Oln-H ПЭТФ-1
о о
НО-[С0 -C-O-(CH2)2-d п-с0 о он ПЭТФ 2
о о о о
СНз
нЧо-0-с-0-о-£]п-он пк-1
СНз о
СНз
н 40 О -С-0-О-С] S-O-Q ПК-2
СНз о
СНз СНз
нЧо-£^ -с -0 -о -с] л-о-0-с-0-он пк-з
СНз о СНз
Для всех структур были проведены расчеты Ван-дерВаальсового объема
молярной рефракции и молярной энергии когезии 22 АЕ* В
i / i
качестве примера рассмотрим в деалях ход расчета этих характеристик для
структуры ПММА-1
386
Глава XV
(X^^)nMMA-l -3AJZC,13 +^С,1 +2Д^С,48 + 2z^ZC,41 + Д1/С,ю +
i
+^7c,6 + 18ДГ///д24 + 2A1zO,i29 + 2А1/одз9 + (Д1/СД0 + AlZC,l + А^СДЗ +
+AIZC,48 + А И’, 41 + 8А1/Яд24 + A1ZO,129 + ^/O,139)(n ~ 2) >
(£ AV, )пмма-1 = 3 • 17,2 + 5,0 + 2 • 15,9 + 2 20,3 +13,1 + 9,0 +18 • 2,0 +
i
+ 2 • 3,4 + 2 5,8 + (13,1 + 5,0 +17,2 +15,9 + 20,3 + 8-2,0 + 3,4 +5,8)(л -2) =
= 205,5 + (n- 2) -96,7 A3.
(У. AE; )пмма-| = ЮД/д-- + 18Д7Г// + 4Д£'(9 + 2AEj + (5AEc + 8A£// +
i
+ 2AEO + AA^)(/? — 2)!
A£*)пмма-1 = 10 •550,7 + 18 47,7 + 4 -142,6 + 2 1623 + (5 550,7 +
i
+8-47,7 + 2-142,6 +1623)(n — 2) = 10182 + (n - 2) • 5043 кал/моль =
= 42561 +(n —2)-21080 Дж/моль.
О')пмма-1 = 10^c +182?// + 23?O< + 2A0= + (5RC + 8R{{ + Ro< + R0=)(n - 2);
i
(Z2?i)nMMA-1^10-2,418 + 18-1300+2-l,643 + 2-2,211 + (5-2,418 +
/
+8 • 1,100 +1,643 + 2,21 l)(n — 2) = 51,688 + (n — 2) • 24,744 см3/моль
Помимо этих характеристик рассчитаем также теплоемкость в жидком
состоянии С1р . ДляПММА-1 имеем.
(^р)пММА -1 - Ср,с ’ (3Alzc,i3 + A1ZC,1 + 2Д1'с,48 + 2APq 41 + APq 10 +
+ A1ZC,6) + ср л 18ДИ# j 24 + Clp q • (2 AIZO J29 + 2AIZO J39) + JClp cx
X(A1ZC,1O +дг/с,1 +Дг/СДЗ +Д,/С,48 + A1ZC,41) +('lp,H '^VH,n4 +^p,O x
Х(Д1/<Э,129 + Д^О,139 )КЛ ~ 2)
(Ср )ПММА-! = 0,34565(3 17,2 + 5,0 + 2 15,9 + 2 • 20,3 +13,1 + 9,0) +
+0,62289-18 -2,0 + 0,92998(2 -3,4 + 2 • 5,8) + [0,34565(13,1 + 5,0 +17,2 +
Влияние концевых групп на свойства полимеров
387
+15,9 + 20,3) + 0,62289 • 8 2,0 + 0,92998(3,4 + 5,8)] (и - 2) =
= 91,8 + 43,2(и - 2) кал/(мольтрад).
Аналогичные расчеты проделаны и для всех остальных структур. Затем
полученные значения У ДЕ, , и С'р были пересчитаны на одно
I ' i
звено полимера простым делением на п и построены зависимости
др;) / п = дv, )0 (X де*) / п = (£ де* )0; (£ %) /« = (£ я, )0 и с'р /« = с1р0
I i I i i '
ar числа звеньев и. Эти зависимости представлены на рис. 102 для ПММА с
различными концевыми группами. В качестве примера также приводится
табл.49 со значениями (^ДИ;)0, (^АЕ*)0, (^А, )о и о, в зависимости
i i i
от и для ПММА-5
Из графиков на рис.102 итабл.49 видно, что зависимости Ван-дер-Вааль-
сового объема, энергии когезии, молярной рефракции и теплоемкости имеют
вид кривых, асимптотически приближающихся к значениям (У ДЕ,)П,
i
(2М*)о, (У А,)о и Ср0, характерным для повторяющегося звена поли-
I i
Значения ( £Alz, )п ,( У AE,)(i ,( )о ч
i i i
для I1MMA-5 при разных значениях степени полимеризации п
Values ( £)0> ( УЛ/?- )о* ( S)e and Ср 0 forPMMA-5 with different values of
the polymerization degree n
n (LAP, )o, A3 (£A£- )o, 7 кал/моль )o, i см3/моль кал/(м<ын 'Град)
1 297 0 17541 84 $2 125,0
2 197,0 11292 54,50 84,5
3 163,0 9209 44,70 70,7
4 146,5 8168 39,80 63,8
5 136,6 7543 36,60 59,6
7 115,6 6363 31,60 50,3
10 107,0 5828 29,07 46,8
15 100,3 5411 27,08 44,0
20 97,0 5203 26,08 42,7
388
Глава X V
(2Х)о,А3
Рис.102.Зависимости(£ДV, )о(а),(2ДЕ; )о(б),(^^- )о(в)и Ср,а (г)отстепени
I 1 i
полимеризации п для полиметилметакрилата. Номера кривых соответствуют
номерам образцов ПММА (см. текст)
Dependences of (£ ДР/ )о(а), (^А£,* )о(б), )o(B)and Ср0 (г) as a function
i i i
of degree of polymerization n for poly(methylmethacrylate). Numbers of curves
correspond!о the numbers of specimens PMMA (See text)
Влитие концевых групп на свойства полимеров
389
мера. При этом вид кривых неодинаков для различныхконцевых групп поли-
мера. В ряде случаев, когда концевые группы не вносят существенного вкла-
да в рассматриваемые характеристики, даже при малых п значения (£ А Гу)0,
i
(^Д)о и Ср 0 незначительно отличаются от асимптотических,
характерных для данной цепи. Если жеконцевая ipynna имеет значительный
Ван-дер-Ваальсовый объем, то ее вклад в физические характеристики стано-
вится весьма значительным и величины (^А^)о, и Cpfi
при малых п очень существенно отличаются от асимптотических. Приближе-
ние к асимптоте наступает (в зависимости от типа концевой группы) при
п = 10—20, что иногда принимают за величину сегмента цепи. Такая картина
характерна, например, для П ММ А-4, когда в качестве инициатора при поли-
меризации метилметакрилата используется перекись бензоила.
Анализируя полученные в работе [8] данные, можно выявить и некото-
рые другие закономерности влияния химического строеггия концевых групп
на физические характеристики полггмера. Так, на Ван-дер-Ваальсовый объем
усредненного повторяющегося звена полимера, естественно, основное влия-
ние оказьгвает объем концевых групп. То же самое можно сказать и о моляр-
ной рефракции. Рассмотрение рис. 102 подтверждает это заключение. Для
систем ПММА-1, ПММА-2 и ПММА-3 Ван-дер-Ваальсовый объем и моляр-
ная рефракция слабо зависят отв. Это хорошо видно из рассмотрения кривых
1—3 на рис. 102. То же самое для данных систем можно сказать и отностпельно
энергии когезии. Это связано с тем, что концевые группы в случаеПММА -1,
ПММА-2и ПММА-Згге обладаюткаким-либо специфическим межмолеку-
лярным взаимодействием и поэтому не прггводягк существенному измене-
нию энергии когезии. Наличие же объемистых концевых групп, существенно
отличных по химическому строешгю отповторяюгцегося звена полимера, при-
водиткрезкому изменению всехрассмогренньгххарактеристик (см .рггс .102.,
кривые 4 5) Все сказанное выше в равной степени относится и к системам на
основе полистрола.
По другому ведут себя системы,которые содержаткогщевыс группы хотя
и малого объема, но обладающие сильным спещгфическггм межмолекуггяр-
ным взаггмодействнем. Это могут быть, например, концевые ОН-группы в
случае поли.) пглентерефталата и поликарбоната. Такая специфика строения
концевых гр уппприводиткигтым закономерностям: характеристики, связан-
ные с объемом и молекулярнойрефракцией,изменяются с величиной п незна-
чительно •, эггер гия же когезии, где специфическое межм олекулярное взаг гм о -
действие играет очень существенную роль, изменяется весьма значительно с
390
Глава XI7
ростом п. Все это необходимо учитывать при анализе влияния степени поли-
меризации (особенно на начальных стадиях процесса) на измеряемые физи-
ческие характеристики.
Выше были проанализированы физические характеристики ряда поли-
меров в зависимости от длины цепи, которые являются исходными для оцен-
ки физических параметров полимерных тел. Так, по величине Ван-дер-Ва-
альсового объема (^ ACJq' и энергии когезии (^ДЕ, )0 можно рассчиты-
вать такой важный параметр вещества, как плотность энергии когезии или па-
раметр растворимости Гильдебранда. Представляет интерес найти зависимость
этого параметра 5 от п при переходе от мономера к димеру, тримеру и т.д.
Такие расчеты были проведены в работе [8] для систем ПММА-4, ПС-4,
ПЭТФ-1 и ПК-1. Результаты расчета в виде зависимости 5 от и показаны на
рис. 103. Можно видеть, что для некоторых систем (ПММА-4, ПС-4) незави-
симо от того, содержат ли они объемистые концевые группы или нет, пара-
метр растворимости существенно зависит от и. Это связано с тем, что нали-
чие концевых ОН-групп в этих полимерах приводит к возникновению водо-
родных связей, что требует введения инкрементов АЛ';*, учитывающих вклад
энергии водородных связей в общую энергию когезии. По мере увеличения п
вклад этих связей ослабевает и при п = 10-20 практически не проявляется.
Рис. 103. Зависимость параметра растворимости 3 от степени полимеризации п для
ПС-4 (1), ПММ/М (2), ПК-1 (3) и ПЭТФ-1 (4)
Dependence of solubility parameter 8 on the degree of polymerization и for
PS-4 (1), PMMA-4 (2), PC-1 (3) and PETP-1 (4)
Влияние концевых групп на свойства полимеров
391
Помимо параметра растворимости существенный интерес представляет
также рассмотрение влияния химического строения концевых групп на по-
верхностное натяжение.
На рис. 104 показана зависимость поверхностного натяжения от степени
полимеризации п для ПММА-5. Видно, что поверхностное натяжение убыва-
ет с ростом п, стремясь к предельному значению, характерному для ПММА.
Рис.104. Зависимость поверхностного натяжения у„ от степени полимеризации п для
ПММА-5
Dependence of surface tension yn as a function of the degree of polymerization n for
PMMA-5
Таким образом, рассмотрение влияния химического строения концевых
групп полимеров на их молекулярные характеристики и макроскопические
свойства показывает, что это влияние может быть весьма различным. Нали-
чие объемистьгх концевых групп сказывается прежде всего на Ван-дер-Вааль-
совом объеме “усредненного” звена, энергии когезии и молярной рефракции.
Присутствие небольших по объему, но обладающих специфическим межмо-
лекулярным взаимодействием групп (типа гидроксильных), оказывает основ-
ное влияние на энергию когезии и параметр растворимости, а также на по-
верхностное натяжение, мало влияя на Ван-дер-Ваальсовый объем и моляр-
ную рефракцию. Рассматривая зависимость каких-либо физических свойств
от длины цепи и, эти факторы совершенно необходимо учитывать.
Глава XVI
ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА
ПОЛИМЕРОВ
К теплофизическим свойствам относятся теплоемкость, температуро-
проводность и теплопроводность.
XVI. 1. Теплоемкость
Под теплоемкостью подразумевают количество тепла, которое нужно зат-
ратить на нагревание тела на 1°С. Различают молярную теплоемкость, если
речь идет о моле вещества, и удельную теплоемкость, если речь идет об 1 г
вещества. Теплоемкость при постоянном давлении Ср равняется скорости из-
менения энтальпии с ростом температуры, а теплоемкость при постоянном
объеме Су-скорости изменения внутренней энергии с ростом температуры.
В довольно широком интервале температур теплоемкость увеличивается
линейно с ростом температуры, причем температурный коэффициент роста
теплоемкости для твердых полимеров имеет среднюю величин)' З Ю'3. При
фазовом или физическом переходе полимера теплоемкость меняется скачком.
Например, при переходе из стеклообразного состояния в высокоэласти-
ческое наблюдается достаточно резкий скачок теплоемкости в сторону ее уве-
личения. После прохождения физического перехода теплоемкость вновь на-
чинает слабо увеличиваться с ростом температуры.
Теплоемкость полимеров зависит от их химического строения (табл.50).
Наиболее низкой теплоемкостью среди углеводородных полимеров обла-
дает полиэтилен и полиоксиметилен.
При замещении атомов водорода на полярные группы теплоемкость воз-
растает. При переходе от алифатических полимеров к ароматическ им наб лю-
дается существенное возрастание теплоемкости.
ГЬпытки расчета теплоемкости полимеров на основании химического
строения повторяющегося звена пре дпринимались неоднократно. Рассмот-
Теплофизические свойства полимеров
393
Таблица 50
Расчетные и экспериментальные значения молярных теплоемкостей для ряда полимеров
Calculated and experimental values of molar heat capacities for a series of polymers
Полимер Ср , кал/(мольтрад) С1р. кал/(мольтрад)
Расчет Эксперимент Расчет Эксперимент
Полиэтилен 11,02 10,4; 11,1; 11,8; 10,15 14,7 15,1
Полипропилен 16,9 15,7; 16,5 21,7 21,6
Полиизобутилен 22,8 22,4 28,8 26,4
Поли- 4- метилпеитен-1 34,6 33,6 - -
Полибутадиеи 20,0 21,0 26,3 24,3
Полиизопрен 25,8 25,7 33,3 31,3
Полистирол 32,4 30,5; 29,3 43,0 42,6
Полиоксиметилен 8,8 10,2; 8,9 13,2 15,0
Полиокситетрамстилен 26,4 28,2 34,1 35,7
Полиоксипропилен 19,8 19,7 26,0 26,5
11ол иви н ил хлор ид 16,2 14,4; 13,8; 16,2; 18,1 18,1
Полихлортрифторэ 1 илен 25,4 25,0; 23,3 - -
Полите трафгорэтиле 11 23,4 23,0; 21,1 23,0 23,0
Поливиниловый спирт 16.3 13,6; 15,4 - -
Поливинилацетат 27,2 30,2; 23,6 37,0 39,5
Полиметилметакрилат 33,1 33,0; 32,0 44,0 43,5
Полиакрилонитрил 15,5 15,9 - -
Полиамид-6 40,2 39,1; 39,2 59,4 57,8
Полиамид-6,6 81,1 79,0; 766 - -
Полиамвд-6.10 104,7 107 146,2 147,0
Полиэтилешер еф талат 52,9 52,0; 52,2 73,3 71,0
Поликарбонат па основе бисс])снола А 72,7 72,4; 71,0 97,6 97,8
Пол hi ip опиленсул ьфо! i 29,4 29,4 38,8 38,8
Поли-2, 6-диметил-1, 4-фениленоксид 35,1 36,4; 34,9 47,0 50,5
рим способ расчета, развитый в работе [22]. В цитируемой работе сделано
предположение, что молярная теплоемкость полимерного тела пропорцио-
нальна Ван-дер-Ваальсовому объему' атомов, входящих в повторяющееся зве-
но полимера. Иными словами
<429)
и
<4М|
I
где Csp и С!р — молярные теплоемкости полимера, находящегося, соответ-
ственно, в стеклообразном и высокоэластическом состояниях; С г и --
инкременты для каждого атома .имеющие смысл приведенной к единице Ван-
394
Глава XVI
s ; Таблица 51
Значения С? t н С? t /гтя различных атомов
Values С? / and Clp { for different atoms
Атом Условное обозначение Численное значение, кал/f моль-град) A3 Условное обозначение Численное значение, кал/(мольтрад) А3
Углерод CL 0,232030 d 0,345646
Водород Сн 0,714129 С'и 0,622889
Кислород Cso 0,634726 С'о 0,929977
Азот С» 0,314997 c'N 2,099874
Фтор CSF 0,543367 dF 0,444909
Хлор Cci 0,368819 c‘cl 0,284693
Сера Css 0,273109 C's 0,303031
дер-Ваальсовогообъема теплоемкости, действующие, соответственно, в стек-
лообразном и высокоэластическом состояниях;^ и А/ — параметры, равные
As = 0,77 калУ(мольтрад), А/ = 0,69 кал/(мольтрад).
Величины GJ, / и Cpi были получены на основании регрессионного ана-
лиза с помощью решения избыточной системы уравнений, полученной на ос-
нове соотношения (429) или (430) с использованием экспериментальных дан-
ных по теплоемкости для хорошо изученных полимеров (так называемых по-
лимерных стандартов—полиэтилена, полистирола, полиметилметакрилата и т.д ).
Полученные значения Срр и cj,,, для каждого атома приведены в
табл.51. С помощью этих значений и величин Ван-дер-Ваальсовых объемов
имеющихся в табл.З, легко рассчитать молярные теплоемкости Csp и С1р для
огромного количества полимеров. Удовлетворительная точность расчета видна
из табл.50.
XVI.2. Температуропроводность и теплопроводность
Температуропроводность — характеристика, которая описывает скорость
распространения температуры под действием теплового потока в нестацио-
нарных температурных условиях. Эта характеристика определяется из соот-
ношения
а = —, (431)
Срр
где X - теплопроводность; Ср - удельная теплоемкость при постоянном дав-
лении; р - плотность полимера.
Теплофизические свойства полимеров 395
Для твердых (стеклообразных и кристаллических) полимеров темпера-
туропроводность слабо уменьшается с ростом температуры. Однако, когда
.полимер переходит из стеклообразного состояния в высокоэластическое, тем-
пературопроводность резко снижается.
Температуропроводность зависит от химического строения полимера, а
также от степени кристалличности, молекулярной массы и давления. В табл .52
приведены значения температуропроводности для ряда полимеров различ-
ного химического строения.
Теплопроводность - это способность полимерных тел переносить тепло
от более нагретых элементов к менее нагретым. Коэффициент теплопровод-
ности X - это коэффициент пропорциональности между потоком тепла и гра-
. диентом температуры. Теплопроводность связана с распространением и рас-
сеиванием упругих волн, вызываемых тепловыми колебаниями частиц тела.
При температуре, стремящейся к абсолютному нулю, теплопроводность так-
Таблица 52
Температуропроводность а ряда полимеров при 293 К
Thermal conductivity a for a series of polymers at 293 К
Название Химическая формула а-Ю7,м2/с
Полиэтилен высокого давления -сн2-сн2- 1,40
Полиизопрен —сн2—сн=<р—сн2— СН3 0,90
Полиэтиленоксид -сн2-сн2- о- 0,90
Полндиметклсилоксан сн3 —Si—О— 1 сн3 1,08
ГЬлистирол —СН2—СН— 6 Q 99
Поливинилхлорид —СН2—СН — CI 1,21
Полиметилметакрилат сн3 —СН2—С — с-о-сн3 II J о 1,19
396
Глава XVI
же стремится к нулю. Теплопроводность полимеров зависит от их химичес-
кого строения в пределах одного и того же физического состояния. Для амор-
фных стеклообразных полимеров теплопроводность несколько ниже, чем для
кристаллических. На температурную зависимость теплопроводности влияет
химическое строение полимера. Для одного ряда полимеров наклон этой за-
висимости положительный, для другого - отрицательный. В области физи-
ческого перехода из стеклообразного состояния в высокоэластическое на бзю-
дается слабый максимум теплопроводности. В табл.53 приведены значения
теплопроводности для ряда полимеров различного химического строения, из
которой видно, каким образом химическое строение влияет на теплопровод-
ность. Особое значение имеет такое свойство полимеров, как анизотропия
теплопроводности. Это свойство характерно для ориентированных полимер-
ных систем, в которых теплопроводность различна вдоль и поперек оси ори-
ентации. Подробнее с теплофизическими свойствами полимеров можно позна-
комиться в работах [59, 61, 62].
Таблица 53
Теплопроводность X ряда полимеров при 293 К
Heat conductivity X for a series of polymers at 293 К
Название Химическая формула Вт/(м.К)
Полиэтилен: высокого давления низкого давления - сн2-сн2— 0,380 0,470
Полипропилен: атактический изотактический сн3 —СН2—СН— 0,175 0,230
Полистирол —СН2—СН — 6 0.130
Поливинилхлорид - сн2- CH- CI 0,170
Полиметилметакрилат сн3 —СН2—С — С—О—СНо II -3 О 0,190
Политетрафторэтилен -cf2-cf2- 0.250
П о лид иметил си ло кс ан СНз —Si—О— 1 СНз 0,167
Глава XVII
МОЛЕКУЛЯРНЫЙ ДИЗАЙН И
КОМПЬЮТЕРНЫЙ СИНТЕЗ ПОЛИМЕРОВ
С ЗАДАННЫМИ СВОЙСТВАМИ
В предыдущих разделах монографии продемонстрирована возможность
расчета основных физических свойств полимеров исходя из химического стро-
ения повторявшегося звена линейного полимера или повторяющегося фраг-
мента полимерной сетки. В настоящее время указанные выше расчеты вы-
полняются с помощью ЭВМ. При этом возможно решение прямой и обрат-
ной задач, которые являются основными, и ряда вспомогательных задач.
Прямая задача заключается в том, чтобы по данным о химическом строе-
нии повторяющегося звена полимера или фрагмента сетки рассчитать свой-
ства полимера.
Обратная задача (более сложная) состоит в предсказании химической фор-
мулы повторяющегося звена такого полимера, который обладал бы одной или
несколькими требуемыми характеристиками физических свойств.
Решение обеих задач описано в работах [9, 17, 26, 126].
При постановке и решении поставленных задач возможны два крайних и
ряд промежуточных подходов. Первый крайний подход заключается в том,
что в память ЭВМ заносится большой ряд полимеров различного химическо-
го строения, для которых указанные выше физические характеристики вы-
числены заранее. Тогда роль ЭВМ будет заключаться только в поиске нужной
структурной формулы в базе данных и выдаче требуемой информации при
решении как прямой, так и обратной задач. В случае такого подхода точность
выдаваемых характеристик близка к 100 %, но предсказательная сила для от-
сутствующих в базе данных структур равна нулю. Фактически такой подход
позволяет лишь создать базу данных.
Второй крайний вариант заключается в следующем. В память ЭВМ вво-
дится таблица значений Ван-дер-Ваальсовых объемов атомов, фрагмент ко-
торой в качестве примера приведен ниже.
398
Глава XVII
С Н О Н
^1 со| о| со|
ю] о| ш| о|
С^Сг^С С^Сг^-С с^с'г^с С^Ср^С
с^'с'^с ГнП с^Го'^с Го1
5 s|p 6 sj’ 7 8 sy
Ъ с с
Каждый из обведенных пунктиром атомов может, в принципе, валентно
связываться с другими атомами; такая возможность для приведенных выше
структур продемонстрирована в матрице (1 - возможность присоединения.
О - запрет):
Атом, № 12345678
1 111110 0 0
2 1111110 0
3 111110 10
4 1111110 0
5 111110 0 1
6 0 1 0 1 0 0 0 0
7 00100000
8 00001000
Таблица значений Ван-дер-Ваальсовых объемов атомов, представленная
в данной монографии, содержит свыше 200 значений объемов для атомов,
валентно связанных с самыми разнообразными атомами. Задача конструиро-
вания повторяющегося звена полимера из всего этого многообразия атомов,
даже при ограниченном их количестве в повторяющемся звене полимера, весь-
ма трудоемка для программирования, а ее решение на ЭВМ потребует слиш-
ком большого машинного времени ввиду возникновения огромного количе-
ства вариантов. В данном случае речь идет о решении обратной задачи, когда
с помощью компьютера требуется “синтезировать” голимеры с заданными
свойствами.
Изложенное выше привело к необходимости разработки промежуточного
(между описанными крайними) подхода к решению задачи прогнозирования
с помощью ЭВМ физических характеристик полимеров и их компьютерного
синтеза. Этот подход заключается в предварительном суммировании всех
инкрементов, входящих в исходные соотношения для расчета свойств, для
Молекулярный дизайн и компьютерный синтез полимеров
399
удельных фрагментов (“заготовок”), из которых затем с помощью ЭВМ осу-
ществляется конструирование повторяющегося звена полимера.
В варианте программы, описанной в работе [17], предусмотрено конст-
руирование полимеров, повторяющиеся звенья которых содержат только по
два фрагмента, имеющих возможность химически связываться друг с дру-
гом. По способу взаимных присоединений все рассмотренные фрагменты раз-
деляются на 8 классов:
Класс Строение Класс Строение
I V O^.Ri^O
II C^R^C VI o^rJ-^o
III с±^9“?.с2-ог^с VII N-^Ri^^N
IV c±2L&r_RiJ2-pj12Zc VIII C^RaJ^C
raeRv -СН2-;-СН-;-(СН2)4-ит.д.;Р2: ит.д.;
ОН
Представленная ниже матрица отражает возможности взаимных присое-
динений фрагментов, относящихся к разным классам (1- возможность присо-
единения, 0 - запрет):
Фрагмент, № I II III IV V VI VII VIII
I 1 0 0 0 0 0 0 0
II 0 1 0 0 0 0 0 0
III 0 0 0 0 I 0 0 0
IV 0 0 0 0 0 I 0 0
V 0 0 I 0 0 0 0 0
VI 0 0 0 1 0 0 0 0
VII 0 0 0 0 0 0 0 I
VIII 0 0 0 0 0 0 1 0
400
Глава XVII
Всего в работе [17] рассчитаны наборы инкрементов для 194 фрагментов
(“заготовок”) различного химического строения; некоторые из них в качестве
примера представлена в табл. 54. Пунктиром обведено химическое строение
самого фрагмента; показаны атомы, которые присоединяются к фрагменту', и
длины химических связей, выраженные в А.
Возможность присоединения одного из этих 18 фрагментов к другому,
учитывая их принадлежность к различным классам, продемонстрирована в
следующей матрице (1 - возможность присоединения, 0 - запрет):
Фрагмент, № 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
2 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
3 1 1 1 0 1 1 1 1 1 1 0 0 0 0 0 0 0 0
4 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0
5 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
6 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
7 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
8 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
9 1 1 1 0 1 1 1 1 1 0 0 0 0 0 0 0 0 0
10 0 0 0 0 0 0 0 0 0 1 1 1 1 0 0 0 0 0
11 0 0 0 0 0 0 0 0 0 1 1 1 1 0 0 0 0 0
12 0 0 0 0 0 0 0 0 0 1 1 0 0 0 0 0 0 0
13 0 0 0 0 0 0 0 0 0 1 1 0 0 0 0 0 0 0
14 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1
15 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1
16 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1
17 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0
18 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 1 0 0
Программа предусматривает как решение прямой задачи, заключающей-
ся в нахождении свойств полимера исходя из химического строения повторя-
ющегося звена, так и обратной задачи, состоящей в поиске таких звеньев,
полимер из которых обладал бы заданными физико-химическими свойства-
ми. При решении обратной задачи машина просчитывает параметр одного из
требуемых свойств, используя все возможные варианты комбинаций занесен-
ных в ее память “заготовок”. Для полимеров, свойство которых оказывается
в нужном интервале, рассчитывается параметр другого свойства, опять отби-
раются нужные полимеры и т.д.
Недостаток данной программы заключается в том, что построение повто-
ряющегося звена полимера из двух крупных “заготовок” не охватывает ог-
ромного количества возможных структур. Очевидно, чем меньше размер “за-
Молекулярный дизайн и компьютерный синтез полимеров
401
готовок”, используемых для построения повторяющегося звена, тем больше
вариантов полимеров можно получить за счет большого количества и разно-
образия “заготовок” в звене и рассчитать их физические свойства.
Поэтому А.А. Аскадским, Е.Г. Гальперн, А.Л.Чистяковым и И.В. Станке-
вичем [126] разработана программа машинного “синтеза” полимеров из наи-
более редких “заготовок”, т.е. таких, которые нельзя в принципе “размель-
чить”. Эти заготовки (базовые фрагменты) представлены в табл.55.
Для обозначения возможности присоединения друг к другу заложенных
в память машины этих мелках фрагментов, каждому концу “заготовок” при-
писываются определенные метки. Отметим, что используемые для построе-
ния повторяющегося звена фрагменты обладают только 20 различными кон-
цевыми группами и соответственно 20 различными метками (см .табл .55),
которые могут вступать друг с другом во взаимодействие в соответствии с
введенной в память ЭВМ матрицей.
В этой матрице не делается различия между данной С-С связи 1,54 А
(в алифатических фрагментах) и 1,48 А (в ароматических фрагментах), по-
скольку переход от одной связи к другой вызывает лишь небольшие измене-
ния Ван-дер-Ваальсового объема, что не сказывается существенно на точнос-
ти расчета физических свойств.
402
Глава XVI 1
Химическое строение и наборы инкрементов для различных фрагментов
Фрагмент № Класс Химическое строение фрагмента ZA^-, i X M
1 2 3 4 5
I I C^iCH2j—с 17,1 14
2 I cl^-'сн 1-bHlC ।1 ! |О-СН3 43,9 44
3 I ch3 1 1 С |-Ь5±С 1 1 C-O-CHol II л| £ J 79,6 86
4 VI c U7. Qi—c 82,1 76
5 I C-^-ICH 1 1 jOH 24,5 30
6 I CK54 C-O-(CH2)4-O-C r^-C II 24 II1 0 ol 124,9 144
7 I C.L54 c-o-Q-o-c II II 0 0 t54c 131,0 164
8 I 128,9 142
C±54 C-HN-(CH2)4- NH-C 0 0 151c
9 I C^ic-нгно I л NH—C 1 II [ o_ 136,5 162
Молекулярный дизайн и компьютерный синтез полимеров 403
Таблица 54
Chemical structure and sets of increments for various fragments
ZO^ + +h,)io3, AV Z W ' io3, AV Z(3;A^ + i + Yz)> A3 Z*z, i аХ/мапь i кал/моль z:g 103, i см3/моль) МПа4
6 7 8 9 10 11
80,2 24,3 1,434 4,618 646 -3,090
129,0 62,3 3,122 10,879 3058 -9,458
195,2 129,5 6,778 15,290 6020 -17,871
135,8 100,7 7,028 24,107 4208 -12,686
33,9 54,2 2,076 6,143 4718 -10,061
449 194,4 10,440 31,016 7502 -16,618
231,8 191,6 11,090 36,651 9126 -16,926
216,2 226,9 9,911 34,934 14334 -17,106
208,0 226,4 10,655 40,570 15958 -19,113
404
Глава XVI]
1 2 3 4 5
10 II 75,6 76
11 II Q_o_0 М8С 159,8 168
12 II С—1— о я 0=0 1 о 0" 1 I о 1 1 0 = 0 130,6 164
II 162,7 170
CU8 C-HN-fCH^NH-C £ °. ±*®с
14 ш с— jo-Q—Q-o 163,1 184
15 ш С-^0,Г81±50С 3,4 16
16 ш С±50|о—О-О С 87,5 108
17 IV С ±37 о 0 1 о , ULc 86,3 108
18 V о±50 |(cH2)4j±^-O 74,6 56
Мельчайшие базовые фрагменты для компьютерного синтеза полимеров
№ фрагмента Химическое строение фрагмента
1 2
1 С -СН2- с
2 С -СН- С 1 СН3
Молекулярный дизайн и компьютерный синтез полимеров
405
таблица 54 (окончание)
6 7 8 9 10 11
135,7 96,2 6,463 24,107 4208 -12,686
319,6 200,0 14,521 49,864 8559 -22,174
273,4 190,6 11,047 36,651 9126 -18,626
376,6 274,4 12,736 44,170 15626 -23,295
395,4 199,0 14,786 51,500 8701 -18,976
78 0,20 0,211 1,643 143 +3,198
259,7 103 7,363 27,393 4494 -6,290
273 103 7,288 27,393 4494 -7,990
321 104,9 6,318 18,472 2584 -12,378
Таблица 55
Smallest basic fragments for the computer synthesis of polymers
Длина связей с соседними атомами, А Метки Ван-Дер- Ваальсовый объем, А’ Молярная рефракция Rj, см3/моль Энергия когезии ДЕ’, кал/моль
левая правая левая правая
3 4 5 6 7 8 9
1,48; 1,54 1,48; 1,54 I 1 17,1 4,618 646
1,48; 1,54 1,48; 1,54 2 2 34,2 9,236 2915
406
Глава Xl'll
1 2
3 сн3 с -с- с 1 СН3
4 с -сн- с С2Н5
5 с -сн- с 1 о-сн3
б с -сн- с 1 CI
7 ' с - са2- с
8 с -сн- с 1 0
о X 1 о-о-О 1 о !
1 10 н 1 0=0 1 о 1
н с -с- с ОС)° %
12 с -с- с 0-Ъ
13 CF3 С -с- с 1 CF3
14 с ~0" с
.Молекулярный дизайн и компьютерный синтез полимеров
407
таблица 55 (продолжение)
3 4 5 6 7 8 9
1,54 1,54 2 2 51,3 13,854 1938
1,48; 1,54 1,48; 1,54 2 2 51,3 13,854 3561
1,48; 1,54 1,48; 1,54 2 2 43,9 10,879 3058
1,48; 154 1,48,1,54 2 2 31,9 9,485 1999
1,48; 1,54 1,48; 1,54 2 2 46,8 12,752 2705
1,48; 1,54 1,48; 1,54 2 2 92,6 28,725 4854
1,48; 1,54 1,48; 1,54 2 2 109,8 33,343 7123
1,48; 1,54 1,48; 1,54 2 2 18,6 4,629 693
1,48; 1,54 1,48; 1,54 2 2 108,9 32,797 7218
1,48; 1,54 1,48; 1,54 2 2 155,2 40,234 8966
1,48; 1,54 1,48; 1,54 2 2 81,1 14,694 1797
1,48 1,48 3 3 75,6 24,107 4208
408
Глава XVII
1 2
15 О 1 0 1 о
16 N-N С —С с- с
17 с “О^С- с
18 р 1 о I ю 1 °
19 С -СН- 0 1 СН3
20 СНз С -С- 0 1 СНз
21 С -СН- О о
22 р 1 ) о 1
23 р 1 0 ) о 1
24 р 1 о о
25 р 1 1 о
26 о -Q- о
27 сн3 о -Q- о хсн3
28 о । о 1 о
29 с -о- с
30 с -о- с
Молекулярный дизайн и компьютерный синтез полимеров
409
таблица 55 (продолжение)
3 4 5 6 7 8 9
1,48 1,48 3 3 75,6 24.107 4208
1,54 1,54 2 2 54,6 1.3,399 6084
1,48 1,48 3 3 100,3 33,127 8791
1,54 1,50 1 4 20,2 4,618 646
1,54 1,50 2 4 37,8 9,236 2915
1,54 1,50 2 4 54,9 13,854 3561
1,54 1.50 2 4 96,2 24,725 4854
1,48 1,37 3 5 78,8 24.107 4208
1,48 1,37 3 5 78.8 24,107 4208
1,50 1,50 4 4 23,3 4,618 646
1,37 1,37 5 5 82.1 24,107 4208
1,37 1,37 5 5 82,1 24,107 4208
1,37 1,37 5 5 115,5 33,343 8746
1,50 1,50 8 8 3,4 1,643 143
1,37 1,50 9 8 2,7 1,643 143
1,37 1,37 9 9 2,1 1,643 143
410
Глава XVII
1 2
31 С . -С-О-. 11 о ...с
32 С 1 0=0 1 о 1 ...с
33 с... .. -сн2-.. ...N
34 с.. .... -с-.... II 0 .N
35 с... ...N
36 о.. . . . -О- ..N
37 с.... . —NHC—.. II О ....С
38 о... --о-- ...N
39 о.. ... — О- ••• ...N
40 N.:. -о-- ..N
41 N... ... — Q— ...N
42 с... ... —NH—... ..С
43 S... -о- ..С
44 S... ' —““ - - - ..С
45 S... -о- ..S
46 S... ... ... ...S
47 S... ••-о- ..N
Молекулярный дизайн и компьютерный синтез попимеров
411
таблица 55 (продолжение)
3 4 5 6 7 8 9
1 54 1,50 2 8 25,1 6.272 2460
1,54 1,37 2 9 24,4 6,272 2460
1,54 1,37 1 6 18,6 4,618 646
1,54 1,37 2 6 20,1 4,629 2316
1,48 1,37 3 6 77,4 24,107 4208
1,48 1,37 3 6 77,4 24,107 4208
1,37 1,48; 1,54 11 14 28,4 8,231 5875
1,37 1,37 5 6 80,6 24,107 4208
1,37 1,37 5 6 80,6 24,107 4208
1,37 1,37 6 6 79,2 24,107 4208
1,37 1,37 6 6 79,2 24,107 4208
1,37 1,37 11 11 8,5 3,602 1253
1,76 1,48 7 3 76,8 24,107 4208
1,76 1,48 7 3 76,8 24,107 4208
1,76 1,76 7 7 78,0 24,107 4208
1,76 1,76 7 7 78,0 24,107 4208
1,76 1,37 7 6 78,6 24,107 4208
412
Глава XVII
1 1
48 S -О- N
49 О l' О 1 (Л
50 s -Q- о
51 С -S- с
52 о II С -S- с II о
53 о о II II С с II II о о
54 о о с -n^Z>CE>i- с II II о о
55 о 1 0=0 0=0 \_/ 1 о
56 о 1 А 0=0 0=0 ъ 1 о
57 ° с
58 о 0 0=0 0=0 1 о
Молекулярный дизайн и компьютерный синтез полимеров 413
таблица 55 (продолжение)
3 4 5 6 7 8 9
1,76 1,37 7 6 78,6 24,107 4208
1 76 1 37 7 5 80 Q 24,107 4208
1 76 1 37 7 5 80 Q 24,107 4208
1,76 1,76 12 12 16,5 8,00 1750
1 ,76 1 ,76 12. 12 26,1 8,870 3656
1 37 1,37 11 И 144,6 46,103 12542
1 37 1 37 11 11 190,2 61,441 15553
1 37 1 37 5 И 113,4 35,105 83.75
1 48 1,37 3 И 110,1 35,105 8375
1,37 1,48 3 5 103,4 33,126 8791
1,76 1,37 7 11 111,3 35,105 8375
414
Глава XVII
1 2
59 S 1 X V с
сн3
60 о. -Si- о
сн3
61 Si -о- Si
62 о. 1 0=0 1 о
63 с. —N— 1 сн3 с
64 N... ~00~ • ...N
65 С... ~ ...С
66 с.. ... —CF2—.... с
В качестве примера, иллюстрирующего конструирование повторяюще-
гося звена, на рис.105 представлено комбинаторное дерево, построение кото-
рого ведется из трех фрагментов (“заготовок”) с различными концевыми мет-
ками. Правило, согласно которому образование связи между концевымигруп-
пами или разрешено, или запрещено, отображено в матрице ихвзаимодействий.
Видно, что скаждым шагом присоединения очередного фрагмента к наращи-
ваемому звену увеличивается количество возможных вариантов повторяю-
щегося звена полимера.
Следует иметь в виду, что если в построении будут участвовать все зане-
сенные в память ЭВМ “заготовки’ ’ и при это м каждая из них м ожег присоеди-
няться к наращиваемому звену и одним, и другим концами, то на каждом
этапе присоединения следующего фрагмента возникает 2я вариантов повто-
ряющегося звена (где и—количество “заготовок”). Таким образом, эта проце-
дура представляет собой геометрическую прогрессию со знаменателем 2.
ЭВМ-программы д ля прогнозирования физических свойств полимеров
позволяютрешать, кроме прямой и обратной, и другие задачи. Например.
Молекулярный дизайн и компьютерный синтез полимеров
415
таблица 55 (окончание)
3 4 5 6 7 8 9
1,76 1,48 3 7 101,3 33,127 8791
1,64 1,64 13 13 71,6 - -
1,64 1,64 10 10 0,5 - -
1,50 1,50 4 4 24,8 4,629 2316
1,37 1,37 11 11 25,6 8,558 1899
1,37 1,37 6 6 124,8 39,445 7219
1,48 1,48 3 3 121,2 39,445 7219
1,48; 1,54 1,48; 1,54 1 1 27,1 4,898 599
можно определи!!, верхний и нижний интервалы физических характеристик,
которые в принципе способны давать органические полимеры; например, ниж-
нее значение температуры начала интенсивной термической деструкции орга-
нических полимеров /^составляет 150 °C, а верхний предел 575 °C. Таким
образом, получить органический полимер, который бы не начинал интенсив-
но разлагаться выше 575 °C, практически невозможно. Можно также оцени-
вать “совместимость” различных свойств в одном и том же полимере и т.д.
(см. ниже).
ЭВМ-программа, основанная на использовании мельчайших базовых
фрагментов, позволяетрассчитывать и “синтезировать” полимеры следую-
щих классов:
1. Полиолефины, виниловые полимеры и т.д.
2. Простые полиэфиры и полиэфиркетоны.
3. Сложные полиэфиры.
4. Полиамиды.
5. Полисульфоны, полиэфирсульфоны.
416
Глава XVII
6. Полиимиды.
7. Полиоксадиазолы.
8. Полибензимидазолы.
9. Полисульфиды.
10. Кремнийорганические полимеры.
11. Поликарбонаты.
При необходимости количество классов полимеров может быть расши-
рено. Для этого необходимо ввести в базу данных группу, которая определяет
принадлежность полимеров к выбранному классу; например, введение в базу
данных уретановой группы позволяет рассчитать свойства полиуретанов и про-
водить компьютерный синтез полиуретанов с заданными свойствами. Недо-
статком этой программы является возможность решения отмеченных выше
задач только для полимеров, которые содержат базовые фрагменты, занесен-
ные в память ЭВМ. И хотя количество полимеров в этом случае является огро-
мным, все же некоторые ограничения существуют. В связи с этим А.А. Аскад-
ским и А.Ф.Елинских [22] разработана ЭВМ-программа, с помощью которой
химическое строение полимера “набирается” не из “заготовок”, а из отдель-
ных атомов. В этом случае химическое строение повторяющегося звена запи-
сывается на экране дисплея в виде структурной формулы органического со -
Рис. 105. Схема построения комбинаторного дерева при конструировании повторяю-
щегося звена полимера (см. текст)
Schematic representation of construction of the combinatoric tree when constructing
a repeating unit of polymer (See text)
Молекулярный дизайн и компьютерный синтез полимеров 417
единения, как это делается химикам и на бумаге. Затем физические свойства
полимера данной структуры рассчитываются и выдаются пользователю прак-
тически мгновенно. При этом можно осуществлять молекулярный дизайн по-
лимера, изменяя его химическое строение, вводя различные группы и т.д., и
сразу получать его физические характеристики. Данная программа позволяет
ршать только первую (прямую) задачу, поскольку при компьютерном синтезе
полимеров из отдельных атомов (а не заготовок) количество вариантов столь
велико, что они не могут быть реализованы с помощью ЭВМ любой мощности.
Данная программа позволяет рассчитывать следующие свойства полиме-
ров и сополимеров, а также полимерных сеток:
1. Молекулярная масса повторяющегося звена.
2. Молярный объем при комнатной температуре.
3. Температурная зависимость молярного объема.
4. Зависимость молярного объема от степени кристалличности.
5. Ван-дер-Ваальсовый объем.
6. Плотность при комнатной температуре.
7. Зависимость плотности от степени кристалличности.
8. Температурная зависимость плотности.
9. Величина теплового расширения в заданном интервале температур.
10. Параметр растворимости Гильдебранда.
11. Поверхностная энергия.
12. Показатель преломления при комнатной температуре.
13. Температурная зависимость показателя преломления.
14. Диэлектрическая проницаемость при комнатной температуре.
15. Температурная зависимость диэлектрической проницаемости.
16. Дипольный момент.
17, Коэффициент оптической чувствительности по напряжению в стекло-
образном состоянии.
18. Температура стеклования.
19. Зависимость температуры стеклования от молекулярной массы.
20. Температура начала интенсивной термической деструкции.
21. Температура перехода полимера в вязкотекучее состояние в зависи-
мости от молекулярной массы.
22. Коэффициент объемного расширения в стеклообразном состоянии.
23. Коэффициент объемного расширения в высокоэластическом состоя-
нии.
24. Молярная теплоемкость при постоянном давлении при комнатной тем-
пературе в стеклообразном состоянии.
25. Молярная теплоемкость при постоянном давлении при комнатной тем-
пературе в высокоэластическом состоянии.
418
Глава XVII
26. Температурная зависимость молярной теплоемкости в стеклообраз-
ном и высокоэластическом состояниях
27. Удельная теплоемкость при комнатной температуре и постоянном дав-
лении в стеклообразном состоянии.
28. Удельная теплоемкость при комнатной температуре и постоянном дав-
лении в высокоэластическом состоянии.
29. Скачок молярной теплоемкости при переходе из стеклообразного со-
стояния в высокоэластическое.
30. Скачок удельной теплоемкости при переходе из стеклообразного со-
стояния в высокоэластическое.
31. Полная энергия межмолекулярного взаимодействия.
32. Энергия дисперсионного взаимодействия.
33. Энергия сильного диполь-дипольного взаимодействия и водородных
связей.
34. Доля энергии дисперсионного взаимодействия в полной энергии меж-
молекулярного взаимодействия.
35. Доля энергии диполь-дипольного взаимодействия и водородных свя-
зей в полной энергии межмолекулярного взаимодействия.
36. Молярная рефракция.
37. Молярная поляризуемость.
38. Полная энергия когезии.
39. Доля энергии когезии, обусловленная водородными связями.
40. Доля энергии когезии, обусловленная диполь-дипольным взаимодей-
ствием.
41. Доля энергии когезии, обусловленная дисперсионным взаимодействи-
ем.
42. Проницаемость по кислороду.
43. Проницаемость по двуокиси углерода.
44. Проницаемость по азоту.
45. Межфазное натяжение на границе полимер - растворитель.
46. Межфазное натяжение на границе полимер - полимер
47. Модуль упругости в области плато высокоэластичности.
48. Величина механического сегмента (молекулярная масса или степень
полимеризации, при которой начинает проявляться высокоэластичес-
кое состояние).
49. Энергия активации низкотемпературного у-перехода.
50 Ньютоновская вязкость полимерных расплавов.
51. Предсказание растворимости полимеров в органических растворите-
лях .
52. Влияние молекулярной массы на растворимость.
Молекулярный дизайн и компьютерный синтез полимеров
419
53. Влияние степени ориентации на растворимость.
54. Предсказание совместимости полимеров.
Для низкомолекулярных жидкостей в программе предусмотрен расчет
следующих свойств:
1. Молекулярная масса.
2. Молярный объем.
3. Ван-дер-Ваальсовый объем.
4. Плотность.
5. Параметр растворимости Гильдебранда.
6. Поверхностное натяжение.
7. Показатель преломления.
8. Диэлектрическая проницаемость.
9. Молярная теплоемкость в твердом состоянии.
10. Молярная теплоемкость в жидком состоянии.
11. Молярная рефракция.
12. Молярная поляризуемость.
13. Полная энергия когезии.
14. Доля энергии когезии, обусловленная водородными связями.
15. Доля энергии когезии, обусловленная диполь-дипольным взаимодей-
ствием.
16. Доля энергии когезии, обусловленная дисперсионным взаимодействи-
ем .
Перейдем теперь к принципам компьютерной реализации метода атом-
ных инкрементов. Предварительно заметим, что практическая значимость этого
метода для решения задачи поиска структур повторяющегося звена, обеспе-
чивающих необходимые физико-химические свойства полимера, вполне оче-
видна. Например, в рамках метода инкрементов можно рассчитать значения
характерных температур полимеров (стеклования, деструкции и плавлен! тя) и
найти структуры, удовлетворяющие требованиям по всему комплексу свойств
перечисленных ранее. Найденные структуры могут служить основой для
использования программ компьютерного планирования органического синте-
за (КПОС). Реализация вычислительной схемы метода инкрементов на ЭВМ
позволяет резко сократить временные затраты на выполнение такой задачи
поиска.
Рассмотрим подробнее вычислительную схему метода инкрементов на
примере расчета температуры стеклования полимера Tg по химической фор-
муле его повторяющегося звена. Расчет Tg проводится по формуле (84).
В алгоритме предусматривается: 1) анализ структуры по атомам и свя-
зям; 2) выделение групп атомов, ответственных за диполь-дипольные взаи-
модействия и водородные связи (включая различные случаи водородного свя-
420
Глава .VI7/
зывання, характерные для полиамидов); 3) определение орто-, мета- и пара-
замещений для ароматических ядер основной цепи; 4) определение двойных
связей “углерод-углерод” в основной цепи. При логической непротиворечи-
вости этих правил они допускают формализованное описание, обеспечиваю-
щее компьютерную реализацию алгоритма. Аналогичный анализ проводит-
ся и при расчете всех других свойств полимеров, сеток и органичес ких жидко-
стей.
В основе компьютерной реализации метода атомных инкрементов лежат
принципы модульности, развитого, дружественного Windows—интерфейса и
согласованности с программами по КПОС. Первые два принципа хорошо из-
вестны и общеприняты, поэтому остановимся подробнее на третьем. Структу-
ра целевого соединения, являющаяся исходным объектом для применения
КПОС, должна обеспечивать требуемые физико-химические свойства целе-
вого соединения. Ввиду того, что поиск такой структуры является одной из
задач в методе инкрементов, необходимо обеспечить согласованное описание
структур в методе инкрементов и в КПОС. Основными в программах по КПОС
являются таблица атомов и таблица связности, задающие набор атомов и типы
связей в структуре соединения. При реализации метода инкрементов было
признано целесообразным формировать такие таблицы для каждой обраба-
тываемой формулы повторяющегося звена. Особое внимание было обращено
на решение проблемы взаимодействия пользователя с программой. Для изоб-
ражения структуры звена полимера использовано планарное (двумерное) пред-
ставление расположения атомов, соединенных определенными типами свя-
зей. При необходимости с помощью таблиц атомов и связности устанавлива-
ется соответствие с линейными формулами Висвессера или с формой записи
но Моргану'. Важным представляется вопрос об однозначной интерпретации
изображаемой на дисплее структуры повторяющегося звена. Для этого в про-
грамме использован контроль наличия данных по атомам, группам атомов и
связям в базе данных.
Программа представляет совокупность модулей, обеспечивающих: 1) ре-
жим редактирования анализируемой структуры (ввод, удаление и перемеще-
ние символов атомов, связей, групп атомов, запись на диск и чтение с диска
файлов данных со структурой и т.п.); 2) анализ структуры, расчет параметров
и обработку структурных ошибок. Выделение модулей второй группы позво-
ляет сделать программу' легко переносимой между различными системами.
Практически программа используется начиная с 1994 года, вначале под уп-
равлением DOS, а в настоящее время— в среде Windows 95.
Тестирование программы проводилось на всех соединениях, для которых,
с одной стороны, известны экспериментальные данные и. с другой стороны,
были ранее проведены расчеты по методу инкрементов. Так, рассчитывались
Молекулярный дизайн и компьютерный синтез полимеров 421
свойства полиолефинов, виниловых, акриловых и диеновых полимеров, али-
фатических и ароматических полимеров (включая полиамиды), полиэфиров,
полиимидов, полисульфонов, полиэфиркето нов и т.д.. Показано, что програм-
ма удовлетворительно справляется с интерпретацией структуры повторяюще-
гося звена в соответствии с вычислительной' схемой метода инкремсн тсв и
<бе спечивает несб ходимую точюс ть расчета метода в 3 -э °/оЛримеры реше -
ния прямой,обратной и смешанной задач компьютерного синтеза полимеров
на основе описанных программ приведены в Приложениях 1-3.
Рассмотрим еще одну задачу, которую можно решать на основе описан-
ных выше ЭВМ-программ.
В настоящее время в связи с использованием полимеров в самых различ-
ных областях существенно усложняются требования к их физическим свой-
ствам . Возникают задачи, для решения которых необходимы полимеры, об-
ладающие комбинацией ряда физических свойств, одновременно попадаю-
щих в заранее заданные интервалы значений их характеристик. Возникает
вопрос о возможности совмещения свойств полимеров. В работе [23] описы-
вается способ его решения с помощью ‘"диаграмм совместимости", который
состоит в следующем: допустим, для большой группы полимеров (из экспе-
римента или из расчета) известны значения параметров двух каких-либо фи-
зических свойств. Построим диаграмму, на которой каждому полимеру соот-
ветствует определенная точка (рис. 106), где на оси X отложены значения па-
раметров первого свойства, а на оси Y — второго. Все эти точки помешаются
Рис. 106. Схематическое изображение области возможного существования двух свойств
полимеров (см. текст)
Schematic representation of the reyion of possible existence for two properties of
polymers (See text)
422
Глава XVII
внутри некоторой области, которую можно обвести контуром. Эта область
названа “областью совместимости” указанных физических свойств для дан-
ной группы полимеров. Следует обратить внимание на то, что плотность рас-
пределения точек по области может быть весьма неравномерна. Такие диаг-
раммы позволяют без затрат дополнительного труда оценивать возможность
существования полимеров с заданными свойствами. Действительно, пусть
заданы интервалы (Х2 —ХД и (Y2 — Y,) значений свойств, которыми должен
обладать полимер. Если область, определяемая этими интервалами, попадает
на диаграмме в область совместимости и там присутствуют некоторые точки,
то есть возможность существования такого полимера (случай 1). В противном
случае искать нужный полимер среди полимеров данных классов маловероят-
но (случай II).
Для построения диаграмм совместимости подобного рода можно вос-
пользоваться либо экспериментальными данными по различным физическим
свойствам полимеров, либо расчетными данными. Можно также воспользо-
ваться комбинацией этих данных.
Для расчета различных физических свойств полимеров удобно использо -
вагь программу для ЭВМ, предложенную в работе [17], поскольку с ее помо-
щью рассматриваются свойства полимеров, либо уже синтезированных, либо
тех, которые можно реально синтезировать. Эта программа и была использо-
вана в работе [23] для построения областей совместимости разнообразных
физических свойств полимеров. Среди этих свойств рассматривались: плот-
ность р, показатель преломления п, температура стеклования Tg, отношение
температуры стеклования к температуре плавления Tg /Тт, температура нача-
ла интенсивной термической деструкции Td, параметр растворимости Гиль-
дебранда 5 (плотность энергии когезии), коэффициент оптической чувстви-
тельности по напряжению Са.
В работе [23] с помощью ЭВМ построено 27 диаграмм, отражающих со-
вместимость любых из двух перечисленных физических свойств полимеров.
Формулы, с помощью которых можно проследить связь между двумя задан-
ными параметрами свойств, приведены в монографии [6].
На рис. 107 в качестве примера показаны диаграммы совместимости та-
ких свойств полимеров, как температура стеклования Tg и температура начч -
ла интенсивной термической деструкции Td, Tg и параметр растворимости 6.
показатель преломления п и 8. Рассмотрим некоторые особенности этих и
других подобных диаграмм.
Все области совместимости имеют разную плотность. Это означает, что
существуют подобласти совместимости, в которых находится основная масса
полимеров, и именно из этих подобластей легче всего выбрать химическое
строение полимеров, которые удовлетворяли бы двум заданным свойствам
Молекулярный дизайн и компьютерный синтез полимеров
423
400
300
200
-100
300
8, (кал / см3),/2
I
700 Tg °C -100
300
700 Tg °C
8, (кал/см3),й
14г в
1,40 1,45 1,50 1,55 1,60 1,65 п
Рис .107. Диаграммы совместимости Та и Tg (а), 8 и Tg (б), 8 и п (см. текст)
Combination diagrams of 7 j and Tg (a), 3 and Tg (6), 8 and n (See text)
На всех диаграммах можно проследить наличие отчетливо выраженных
зависимостей одного свойства от другого. Каждая из этих зависимостей ха-
рактеризует определенные ряды полимеров, например, полиэ фиры, поли ами-
ды, полиимиды и тд.
424
Глава XT7I
На каждой из диаграмм имеется точка, от которой отходят эти зависимо-
сти. Это связано с особенностью программы [17], в которой в качестве исход-
ных фрагментов из числа 194 заложено несколько гомологов —(СН2)Л— до
и = 20, и точка, из которой исходят эти зависимости, характеризует полиэти-
лен.
Остальные особенности, присущие каждой из диаграмм ,хорошо видны
на соответствующих рисунках. Имея эти диаграммы, можно прогнозировать
возможность получения полимеров, которые обладали бы необходимой со-
вместимостью одного из нескольких свойств. Например, если нужно полу-
чить полимеры с параметром растворимости 5 = 10 (кал/см3)1/2 и с Tg ~ 300 °C,
то это сделать легко, поскольку точка, соответствующая этим координатам,
попадает в наиболее плотную часть диаграммы рис. 107,6. Если же требуется
получить полимер, у которого при том же значении параметра растворимос-
ти температура стеклования Tg была бы ~ 500 °C, то это сделать труднее, а
при Tg = 600 °C нереально, поскольку' точка, соответствующая этим коорди-
натам, выходит за границы области совместимости. Такой анализ легко мо-
жет быть проделан для любой из представленных в работе [23] диаграмм, а
также из их совокупности, что позволяет прогнозировать возможность полу-
чения полимеров с комплексом заданных свойств. Естественно что если та-
кие диаграммы построить с помощью ЭВМ-программы, согласно которой
поли кер “ко к: тр уируется’ из мельчайших11 заготовок’, области совместимо-
сти полимеров существенно пополнятся точками,отображающими свойства
огромного числа полимеров.
Приложение 1
Пример решения прямой задачи оценки свойств
полимеров по их химическому строению
Решение прямой задачи по оценке свойств полимеров на основе их хи-
мического строения рассмотрим на примере природных гплимеров Особен -
ность этих полимеров заключается в следующем:
1) Данные п олимеры содержат циклические неароматические структуры
с большим количеством ОН-групп, способных к образованию водородных
связей.
2) В)дородные связи в этих полим ерах могут образовываться как между
соседними цепями так и в пределах одной цепи ц даже в пределах одного
повторяющегося звена. Схема таких взаимодействий показана на рис.41. На-
личие межцепных и внутрицепных (в том числе и внутризвенных) водород-
ных связей зависит от стерического фактора — расположения ОН-групп по
отношению к плоскости колец.
3) М ногие высоком) текулярные соединения ,в том числе и целлюлоза ,
при нагревании начинают претерпевать термическую деструкцию прежде,
чем наступает процесс расстекловывания, и в результате такая наиболее важ-
ная для полимеров характеристика, как температура стеклования, не может
быть определена экспериментально.
4) Многие природные высокомолекулярные соединения являются разнозвен-
ными, т е. содержат в своей цепи фрагменты различной химической природы.
В дани мразделе рассмотрим применение подхода разработанного для
оценки физических свойств полимеров на основе их химического строения,
к природным высокомолекулярным соединениям, которые обладают весьма
сложным, но хорошо изученным химическим строением.
В табл П-1-1 приведено химическое строение 16 фрагментов структур
гемицеллюлоз, наиболее часто встречающихся в структуре природных поли-
меров Обращает на себя внимание тот факт, что в ряде случаев, несмотря на
426
Приложение 1
одинаковую брутто-формулу, данные фрагменты, входящие в полимерные
цепи, приводят к разным физическим свойствам природных полимеров. Так,
например, для структур Фс1 и Фс2 брутто-формулы совершенно одинаковы,
но в структуре фрагмента Фс2 ОН-группы, расположенные по одну сторону
плоскости кольца, могут образовывать водородные связи между собой, и та-
ким образом они участвуют в образовании связей между соседними цепями.
Это обстоятельство сказывается на температуре стеклования и ряде других
свойств полимеров, содержащих эти фрагменты.
Рассмотрим более детально влияние химического строения упомянутых
природных полимеров на их температуру стеклования. Температура стекло-
вания рассчитывается по уравнению (84). При расчете температуры стекло-
вания по этому уравнению необходимо учитывать все тонкие особенности
структуры полимерного звена. Если водородные связи образуются между
цепями полимера (в результате чего происходит формирование физической
сетки за счет межмолекулярных связей), то в уравнение (84) необходимо вво-
дить параметр 6А = -140- НУ3 А3К'*. Если же ОН-группы в повторяющемся
звене расположены таким образом, что водородные связи образуются между
ними в пределах одного и того же звена, то сетка физических межмолекуляр-
ных связей между цепями не образуется (см рис.П-3-1,б) и дня этих групп в
уравнение (84) следует вводить не константы 6/,, а константы bd, которые вво-
дятся для каждого ответвления от главной цепи полимера.
Использование такого приближения приводит к значениям температу-
ры стеклования и всех других свойств, которые показаны в табл.П-1 -2. Хоро-
шо видно, что при одинаковой брутто-формуле температура стеклования струк-
туры Фс1 (541 К) значительно выше, чем структуры Фс2 319 К. При этом
меняется также такая важнейшая характеристика полимеров, как А//, т.е. мо-
лекулярная масса механич еского сегмента макромолекулы, при котором по-
является высокоэластическое состояние. Насыщение структуры гемицеллю-
лоз ОН- и СООН-группами приводит к резкому возрастанию температуры
стеклования, если эти группы способны образовывать водородные связи меж-
ду цепями полимеров. Если же водородные связи образуются внутри повто-
ряющегося звена, то температура стеклования резко снижается.
Рассмотрим кратко некоторые другие свойства. Плотности монолитных
образцов примерно одинаковы, причем наименьшей плотностью об ладает струк-
тура Фс5, в составе которой находится лишь одна ОН-группа. В результате это-
го такая структура обладает наибольшей температурой начала интенсивной тер-
мической деструкции Следует отметить, что в ряде случаев температура начала
интенсивной термической деструкции лежит ниже температуры стеклования
или близка к ней, что и является причиной того, что экспериментальное опреде-
ление Tg для природных полимеров весьма затруднено.
Приложение 1
427
Поверхностная энергия для рассчитанных 16 структур изменяется в
довольно широких пределах, причем наименьшая величина поверхностной
энергии характерна для структуры Фс5, содержащей наименьшее количество
ОН-групп, а наиболее высокое значение характерно для структуры Фс8, со-
держащей наибольшее количество этих групп.
Диэлектрическая проницаемость примерно одинакова, параметр раствори-
мости по сравнению с синтетическими полимерами достаточно высок, показа-
тель преломления примерно одинаков, а температурный коэффициент объем-
ного расширения в стеклообразном состоянии существенно зависит от хими-
ческого строения фрагмента и изменяется антибатно температуре стеклования.
Далее рассмотрим свойства гемицеллюлоз, построенных из указанных
выше фрагментов. Хим ический состав8 гемицеллюлоз и расположение в них
фрагментов приведен ы в табл.П-1-3. Основные свойства гемицеллюлоз дан-
ного состава показаны в табл.П-1-4.
Все свойства рассчитывались исходя из молярного содержания фрагмен-
тов. Видно, что температура стеклования гемицеллюлоз различной химичес-
кой структуры принимает значения от 332 до 517 К а температур а н ат ала
интенсивной термической деструкции колеблется от 517 до 580 К.
Из остальных характеристик, приведенных в табл.П-1-4, следует отме-
тить близость значений этих температур для гемицеллюлозы С4. Обращает
внимание чрезвычайно низкая проницаемость монолитных пленок гемицел-
люлоз по отношению к различным газам.
Экспериментальная проверка результатов расчетов проведена на основе
1,4-р-О-глюкоманнана, структура которого Ф8 показана в табл.П-1-3. Термо-
механическая кривая для исходного препарата, содержащего неконтролируе-
мое количество влаги, показана на рис.П-1-1. После сушки в вакууме при
температуре 80 °C в течение двух часов вид термомеханической кри вби не-
сколько видоизменился рис. П 1- 2) Три такой термин еской обрабо тке в ва
кууме меняется также вид дебаеграмм (рис П-1-3) доказывающих, что ис-
следуемые образцы глюкоманна были практически аморфными. Расчетное
значение температуры стеклования (356 К) с обычной для таких расчетов
точностью совпадает с экспериментальным значением, которое, судя по тер-
момеханической кривой (см.рис.П-1-2), составляет 333 К.
Не исключено, что после дальнейшей сушки температура стеклования
могла бы несколько увеличиться. 2-ой подъем дф ормации на термомехани-
ческой кривой происходит при температуре, близкой к температуре интен-
сивной термической деструкции и, вероятно, с ней связан.
Разумеется, результаты вса проведенных расчетов можно рассматривать
лишь как предварительные и их в дальнейшем необходимо связать не только
с первичной, но и со вторичной структурой гемицеллюлоз.
428
Приложение 1
Таблица П-1-1
Химические формулы фрагментов структур гемицеллюлоз
Chemical formulas of the fragments of hemicellulose structures
Обозначения фрагментов структур Химические формулы фрагментов структур
1 2
сн2он у|-- О.
ФС1 Г< >1 \?н у
6н
сн2он J——О
Фс2 ['< У] уН нсу
J О
Фс3 г< У] vH У
он
Фс4 соосн3 /1 °ч К У’ VH У
он
J °ч
Фс5 1 СООСНз /* J
ОН
у| О [ъ/ У]
Феб 1 \ он J оХ
н^\?н |/
ОН
Приложение 1
429
таблица П-]-1 (продолжение)
1 2
Фс7 соон J О /1 1 \ он НО^рИ у ' дн
Фс8 1 о r X он \ ОН -Л ОН
Фс9 1 о jx] 1 \ 6 Н J о\ 1/ \? НсКсОООНз/ ' ^ООСНз
ФСЮ 1 о г< я \?Н / уГ соон к \ нс^4сн з он
J
430
Приложение 1
таблица П-1-1 (продолжение)
1 2
ФС11 J 2Н но/ои V бНзОН ^Z° —°ч Г< >] уН HQ/
Фс12 СН2ОН J—- °к 1 \ 6н °/он нЬо °' СН2ОН
Фс13 /1 0 °н\ нзс0н \_2 соон
Фс14 сн2он у 1=0 он/1 Ч ОН/1 ч р° он ° он ']
Приложение 1
431
таблица П-1-1 (окончание)
Таблица П-1-2
Свойства фрагментов струкгур гемицеллюлоз
Properties of hemicellulose structure fragments
Обозначения фрагментов структур Значения свойсгв фрагментов структур
мм vm, см3/моль i Р, г/см3 Tg,K 7rf,K
ФС1 162 116 132 1,39 541 556
Фс2 162 116 132 1,39 319 556
ФсЗ 132 95 107 1,40 511 571
Фс4 190 135 152 1,41 459 585
Фс5 174 128 145 1,36 403 607
фс6 264 189 214 1,40 574 527
Фс7 308 212 240 1,45 641 520
Фс8 250 174 197 1,44 427 520
Фс9 348 256 290 1,36 436 570
Фс10 322 229 259 1,41 518 530
ФС11 324 233 263 1,39 431 511
Фс12 324 233 263 1,39 432 511
Фс13 322 229 259 1,41 518 530
Фс14 486 349 395 1,39 388 525
Фс15 454 324 367 1,40 552 517
Фс16 929 661 748 1,41 437 513
432
Приложение 1
таблица П-1-2 (продолжение,
Обозначения фрагментов структур Значения свойств фрагментов структур
у. дин/см ккал/моль аА, отн. ед. Hid . отн. ед. «г/. отн. ед. Е, отн. ед. Е, ккал/моль
Фс1 45.5 23,9 0.72 0,00 0,28 3,36 49.6
Фс2 45.5 23,9 0.72 0,00 0.28 3,36 49.6
ФсЗ 54.8 18,2 0.63 0.07 0,30 3.34 37.96
Фг4 46.4 21.5 0,54 0.11 0,35 3.49 46.15
Фс5 37.9 16,8 0.34 0.21 0,44 3.33 38.76
ф(*-6 53,0 35,2 0.66 0.03 0.31 3.34 85.22
Фг7 58,9 43,3 0.67 0,05 0.28 3,56 103.60
Ф<8 62.1 37 8 051 0,13 0 26 3.45 90 93
Ф,9 36 5 32 3 0.36 0,18 0,46 3.33 83.85
ФсЮ 49,9 39.7 0.58 0,09 0.33 3,43 90.52
Фс11 45,5 47,8 0.72 0.00 0.28 3,36 109,43
Фс12 45.5 47.8 0.72 0.00 0.28 3,36 109.43
Фе 13 49.9 39,7 0.58 0,09 0,33 3,43 90,52
Фг-14 45,5 71.7 0.72 0,00 0.28 3,36 158.65
Фс15 50.3 56.7 0.61 0.06 0,33 .3,40 137,3 3
Фс16 46,6 139.1 0.70 0.03 0.27 3,38 321.32
Обозначения фрагментов структур Значения свойств с|рагментов структур
Edd+h, | ккал/моль Ed, | ккал/моль 5, (кал/см3)1/2 п, отн. ед. ди, ккал/моль Ос' юЛ К"1
ФГ1 37,60 12,0 14,30 1,49 27,9 1.78
Фс2 22.20 12,0 14.30 1.49 27.9 3.01
ФсЗ 27,70 10.26 13.85 1,49 26,4 1.88
Фс4 30,18 15.97 12.65 1.48 23,7 2,09
Фг5 21,53 17.23 11.43 1.48 20.8 2,38
Феб 64.07 21.15 13.63 1.49 29.7 1.67
Фс7 80.77 22.83 14.29 1,48 33.1 1,50
Фс8 39.00 18.74 14.72 1.48 34,7 1,43
Фс9 48.40 .35.45 11.23 1.48 22,6 2.20
ФсЮ 63,65 26,87 13.16 1.48 26.8 1,85
Фс11 84,83 24.60 14.34 1,49 30,8 2,22
Фс12 84,83 24,60 14.34 1.49 30,8 2.22
Фс13 63,65 26,87 13.16 1.48 26,8 1,85
Фс14 70.17 36,61 14,34 1.49 29.8 1.66
ф(15 99,57 37.76 13,23 1,48 28 5 1.74
Фг16 157,40 69.68 14.51 1,49 31,9 1.55
Обозначения фрагментов структур Значения свойств фрагментов структур
аг1оЛ к-1 И/, отн. ед. Cs, кал/(мольтрад) кал^мольтрад) Л, см3/моль А/;
1 2 3 4 5 6 7
ФС1 4,46 467 52,7 67,6 33,37 79034
Фс2 4,46 487 52,7 67,6 33,37 46602
ФсЗ 4,71 431 41,5 54,0 27,23 56980
Фг4 5.25 435 57,6 76.1 38,12 82740
Фс5 5.97 376 52,3 69.5 36,59 65434
Приложение 1
433
таблица П-1-2 (окончание)
1 2 3 4 5 6 7
Феб 4.20 609 83,8 107.3 54,45 160900
Фс7 3.76 707 95,7 123,7 60,61 217850
ф 8 3 59 693 77 9 100 3 49 83 110198
Фс9 5,52 512 105,4 138,3 73,18 178375
ФсЮ 4.65 586 99,9 129.4 65.30 188853
Ф П ,4 04 678 106 1 13,4 5 66 74 219817
Ф 12 __ .4 04 678 106 1 134.5 66 74 219817
ф 13 4 65 586 99 9 129 4 65 30 188853
Фс14 418 750 159.5 201.3 100,11 245324
Фс15 437 700 142,1 182,7 92,57 318105
Ф <16 1 90 995 302 7 381 4 189 45 653567
Обозначения фрагментов структур Знач и ия свойств фрагментов структур
р, см3/моль G. Б д td Й.о Рса, , IE PN , П2 IE
Ф 1- 51 ,2 104 1.55 0,03 0,07 0,00
ф г 5 1.2 104 1,55 0,03 0,07 0,00
ФсЗ 41.4 132 1,39 0.04 0.09 0.01
Фс4 61.1 91 1,70 0.22 0,56 0,03
Ф-5 56 0 104 1 63 1 38 4 12 0 24
Ф6 82 9 108 1 99 0 11 Q 27 0 02
Ф 7 97 8 99 2 16 0 02 0 03 0 00
Фс8 78,3 113 1.93 0,00 0,00 0.00
Фс9 112.0 95 2,32 3,17 10,23 0.60
Ф-10 102 5 108 2 21 ,0 11 ,0 26 ,0 02
Ф И 102 3 95 2.2 1 ,0 03 .0 07 ,0 00
Фс1 2 1Q2 3 95 .2 21 .0.03 .0.07 0.00
Фс 13 102.5 108 2,21 0,11 0,26 0,02
Ф<44 1 53,5 98 2.72 0,03 0,07 0,00
Ф45 144.0 101 2,63 0.15 0,37 0,02
Ф 16 292 6 96 3 76 0 01 0 оз ооо
Примечание: ММ - молекулярная масса; Vm - молярный объем; 52 А Ван-дер-Вааль-
совый объем; р - плотность; Т& - температура стеклования; Tj - температура начала интенсив-
ной термической деструкци; у - поверхностная энергия; Е* - энергия когезии; а/, -отношение
энергии водородны х связей к полной энергии когезии; - отношение энергии диполь-диполь-
ного взаимодейств ия к полной энергии когезии; aj - отношение энергии дисперсионного взаи-
модействия к полной энергии когезии; е - диэлектрическая проницаемость; Е - полная энергия
межмолекулярного взаимодействия Edd+h~ энергия диполь-дипольного взаимодействия и водо-
родных связей; Ed -энергия дисперсионного взаимодействия; 8 - параметр растворимости; п -
показатель преломления; Д£/ - энергия активации низкотемпературного гамма-перехода; -
коэффициент термического расширения в стеклообразном состоянии; ol - коэффициент терми-
ческого расширения в высокоэластичном состоянии; д/ - степень полимеризации полимера при
наступлении высоко эластичного состояния; Cps - молярная теплоемкость в стеклообразном со-
стоянии; Ср - молярная теплоемкость в высокоэластичном состоянии ,/? -молярная рефракция;
М( - молекулярная масса полимера при наступлении высоко эластичного состояния; Р - поляри-
зуемость; Са -коэффициент оптической чувствительности по напряжению; ц - дипольный мо-
мент; Ро2»?со2> Pn2 ~ проницаемость соответственно по кислороду.,двуокиси углерода и азоту
(единица измерения проницаемости 1 ДЕ=О,45-1О-10 см" С"1-атм _1).
434
Приложение 1
Фрагментный состав и структурные формулы гемицеллюлоз
Условные обозначения гемицеллюлоз Фрагментный состав и наименование природного биополимера
1 2
С1 (Фс12)з - (Фс2)42 - (Фс1)13 ’ (Фс1 1)5 глюкоманнан осины [208]
С2 (Фс12)1 - (Фс2)в - (Фс1)з - (Фс12)з глюкоманнан ели [116]
СЗ (ФсЮ)19 - (ФсЗ)125 - (Фс6)5 4-0-метилглюкуроноксилан осины [117]
С4 Фс3 - ФСЮ— Фс3 - Фс6 - Фс8 -Фс15 арабиноглюкуроноксилан ели [118]
Приложение 1
435
Таблица П-1-3
Fragmentary composition and structural formulae of hemicelluloses
436
Приложение 1
1 2
С5 Фс7 - (Фс4)50 - Фс7 галактоуронан растений [130]
С6 (Фс9)з - (Фс5)21 - (ФсЗ)52 - (Фс13)|5- (Фс5)з ксилан березы [118]
« С7 Фс16- Фс14 арабиногалактан лиственницы [60]
С8 (фс2 ФС1 (Фс2)4- Фс1 - Фс2]48 1,4-0 Г) глюкоманнан [2]
Приложение 1
437
Примечание: химические формулы приведенных в таблице гемицеллюлоз и образец 1,4-p-D-
глюкоманнана представлены д.б.н. Щербухиным В.Д. и к.б.н. Колобовой А.В.
1
438
Приложение 1
Таблица П-1-4
Свойства гемицеллюлоз
Properties of hemicelluloses
Условные обозначе- ния гемицел- люлоз Свойства гемицеллюлоз
A3 P, г/см3 T К Td, К ОоЮЛ К'1 8, (кал/см3)|/2 Y> дин сКГ РО2, ДЕ Рсо?, ДЕ PN , 2 ДЕ
Cl 146 1,39 332 546 2,90 14,3 45,5 0,03 0,07 0,00
C2 160 1,39 367 538 2,62 14,3 45,5 0,03 0,07 0,00
C3 130 1,40 499 557 1,92 13,7 53,4 0,05 0,12 0,01
C4 211 1,41 517 530 1,83 13,6 53,3 0,05 0,11 0,01
C5 156 1,42 464 580 2,07 12,8 47,2 0,19 0,47 0,03
C6 154 1,38 435 566 2,21 12,8 47,0 0,21 0,54 0,03
C7 571 1,40 419 517 1,62 14,5 46,2 0,02 0,04 0,00
C8 1052 1,39 356 556 2,70 14,3 45,5 0,03 0,07 0,00
Примечание', обозначения свойств гемицеллюлоз соответствуют обозначениям, приведенным
в табл.П-1-2
Рис.П-1-1. Термомеханическая кривая исходного 1,4-р4Э-глюкоманнана
Thermomechanical curve of initial 1,4-p-D-glucomannan
Рис.П-1-2. Термомеханическая кривая высушенного 1,4-0-D- ппокоманнана
Thermomechanical curve of dry 1,4-P-D-glucomannan
Приложение 1
439
РисП-1-3. Дебаеграммы порошкообразных образцов исходного (а) и высушенного
(б) 1,4-Р-В-глюкоманнана
Debye patterns of powder-like initid (a) and dry (a) specimens of 1,4-PD-
glucomannan
Приложение 2
Пример решения обратной задачи
синтеза полимеров
Таблица П-2-1
Химические формулы полиэфиркетонов с температурой стеклования Tg выше 493 К
и температурой плавления Тт ниже 653 К
Chemical formulae of poly(ether ketones) with the glass transition temperature Tg above 493 К
and the melting point Tm below 653 К
Номера структур полиэфиркетонов Свойства полиэфиркетонов
т к Т * К 5, (кал/см3)1'2 Р, г/см3 £ , отн. ед. и , отн. ед.
1 2 3 4 5 6 7
1 495 632 9,78 1,24 2,95 1,62
2 495 632 9,78 1,24 2,95 1,62
3 498 652 9,63 1,20 2,83 1,59
4 498 652 9,63 1,20 2,83 1,59
5 495 632 9,78 1,24 2,95 1,62
6 498 652 9,63 1,20 2,83 1,59
7 495 632 9,78 1,24 2,95 1,62
8 498 652 9,63 1,20 2,83 1,59
9 495 632 9,78 1,24 2,95 1,62
10 495 632 9,78 1,24 2,95 1,62
II 498 652 9,63 1,20 2,83 1,59
12 498 652 9,63 1,20 2,83 1,59
Приложение 2
441
Обратная задача заключается в синтезе, т.е. нахождении химической фор-
мулы или формул полимеров, значения свойств которых должны лежать в
заданном интервале. Пусть требуется синтезировать полиэфиркегоны с тем-
пературой стеклования, лежащей выше 493 К, и одновременно с температу-
рой плавления, лежащей ниже 653 К (последнее важно для успешной пере-
работки). На значения остальных свойств (параметр растворимости,
.1 плотность, диэлектрическая проницаемость, показатель преломления)
ограничений нет.
Решение данной задачи будем искать с помощью программы, опериру-
ющей наиболее мелкими базовыми фрагментами. Выберем количество ба-
зовых фрагментов в повторяющихся звеньях, равное 7. В результате реше-
: ния данной задачи получаем химические формулы полиэфиркетонов, приве-
денные в табл. П-2-1 (было просчитано 450000 структур и из mix в нужный
интервал свойств попало 24 структуры).
Несколько расширим интервал свойств, введя для температуры стекло-
вания нижнюю 1раницу, равную 483 К. В этом случае из того же коли-
чества просчитанных структур было отображено 84 структуры полиэфирке-
тонов, химические формулы которых приведены в табл. П-2-2.
Химические формулы полиэфиркетонов
_________________8_________________
-СО—рС6Н4—О—рС6Н4—pht—С10Н6—тС6Н4-
-СО—рС6Н4—О—рС6Н4—pht—тС6Н4—С1ОН6-
-СО—рСбН4—О—рС6Н4—fl—С1ОН6—mC6H4-
-CO—рСбН4—О—pC6H4—fl—тС6Н4—СЮНб-
-СО—рС6Н4—О—рС6Н4—С10Н6—pht—тС6Н4-
-СО—рС6Н4—О—рС6Н4—С10Н6—fl— тС6Н4-
-СО—рС6Н4—О—рС6Н4—тС6Н4—pht—С10Н6-
-СО—рС6Н4—О—рС6Н4—mC6H4—fl—С10Н6-
-СО—С10Н6—pht—рС6Н4—О—рС6Н4—тС6Н4-
-СО—С10Н6—pht—тС6Н4—рСбН4—О—рС6Н4-
-СО—С10Нб—П—рСбН4—0—рСбН4—тС6Н4-
-СО СЮН6 fl-тСбНТ рС6Н4 О рСббНГ
442
Приложение 2
1 2 3 4 5 6 7
13 495 632 9,78 1,24 2,95 1,62
14 498 652 9,63 1,20 2,83 1,59
15 495 632 9,78 1,24 2,95 1,62
16 498 652 9,63 1,20 2,83 1,59
17 495 632 9,78 1,24 2,95 1,62
18 495 632 9,78 1,24 2,95 1,62
19 498 652 9,63 1,20 2,83 1,59
20 498 652 9,63 1,20 2,83 1,59
21 495 632 9,78 1,24 2,95 1,62
22 498 652 9,63 1,20 2,83 1,59
23 495 632 9,78 1,24 2,95 1,62
24 498 652 9,63 1,20 2,83 1,59
Примечание, условные обозначения см. в примечании к табл. П-2-2
Химические формулы полиэфиркетонов с температурой стеклования Tg выше 483 К
и температурой плавления Т,п ниже 653 К
Номера структур полиэфир- кетонов Свойства полиэфиркетонов
т 1 £ к тт, К 3, (кал/см’)|/2 Р, 17см3 Е , отн. ед. п. отн. ед.
1 2 3 4 5 6 7
1 489 626 9,71 1,24 2,96 1,62
2 489 626 9,71 1,24 2,96 1,62
3 493 648 9,55 1,19 2,83 1,58
4 493 648 9,55 1,19 2,83 1,58
5 489 626 9,71 1,24 2,96 1,62
6 493 648 9,55 1,19 2,83 1,58
7 489 626 9,71 1,24 2,96 1.62
8 493 648 9,55 1,19 2.83 1,58
9 489 626 9,71 1,24 2,96 1,62
Приложение 2
443
таблица II-2-1 (окончание)
8
-СО —СЮНб—рС6Н4—О—рС6Н4- pht mC6H4
СО—СЮНб—рС6Н4—ОрС6Н4—fl- тС6Н4
-СО—СЮНб—тС6Н4—pH—рСбН4—О—рСбН4—
-СО -С10Н6—mC6H4- -fl—рСбН4— О—рСбН4 -
-СО—тС6Н4—pH—рС6Н4 -О—рСбН4 - С10Н6—
-СО—тС6Н4--pH—СЮНб—рСбН4—О—рСбН4—
-СО—тСбН4—fl рСбН4 - О—рСбН4—СЮНб-
-СО—тСбН4 —fl—СЮНб—рС6Н4—О—рС6Н4-
-СО—тСбН4—рС6Н4—О -рСбН4—pht -C10II6-
-СО—тСбН4—рСбН4—О —рСбН4—fl- СЮНб-
-СО—тСбН4—СЮНб—pH—рСбН4—О--рС6Н4—
-СО—тСбН4—СЮНб—fl - рС6Н4—О—рС6Н4-
Таблица П-2-2
Cliemcal formulae of polyfether ketones) with the glass transition temperature Tg above 483 К
and the melting point T,„ below 653 К
Химические формулы полиэфиркетонов
__________________8________________
-СО—тС6Н4—О—рС6Н4—pH—тС6Н4—рСбН4-
-СО—тС6Н4—О—рСбН4—pH—рС6Н4—тС6Н4-
—СО—тСбН4—О—-рС6Н4—fl—тС6Н4—рС6Н4—
- СО—тСбН4—О—РС6Н4—fl—рС6Н4—тС6114-
—СО—тС6Н4—О—рС6Н4—тС6Н4—pH—рС6Н4
-СО—тСбН4—О—РС6Н4—тСбН4—fl—рС6Н4-
-СО—П1С6Н4—О—рСбН4—рС6Н4—pH—тС6Н4-
-СО—тСбН4—О—рС6Н4—рС6Н4—fl—П1С6Н4-
—СО—р С6 Н4—()—т С 6 Н 4—р Н—т С 6 Н 4—рС 6 Н 4-
444
Приложение 2
Приложение 2
445
1 2 3 4 5 6 7
10 489 626 9,71 1,24 2,96 1,62
11 493 648 9,55 1,19 2,83 1,58
12 493 648 9,55 1,19 2,83 1,58
13 489 626 9,71 1,24 2,96 1,62
14 493 648 9,55 1,19 2,83 1,58
15 489 626 9,71 1,24 2,96 1,62
16 493 648 9,55 1,19 2,83 1,58
17 495 632 9,78 1,24 2,95 1,62
18 495 632 9,78 1,24 2,95 1,62
19 489 550 9,71 1,24 2,96 1,62
20 498 652 9,63 1,20 2,83 1,59
21 498 652 9,63 1,20 2,83 1,59
22 493 578 9,55 1,19 2,83 1,58
23 495 632 9,78 1,24 2,95 1,62
24 498 652 9,63 1,20 2,83 1,59
25 495 632 9,78 1,24 2,95 1,62
26 489 550 9,71 1,24 2,96 1,62
27 498 652 9,63 1,20 2,83 1,59
28 493 578 9,55 1,19 2,83 1,58
29 495 632 9,78 1,24 2,95 1,62
30 495 632 9,78 1,24 2,95 ' 1,62
31 498 652 9,63 1,20 2,83 1,59
32 498 652 9,63 1,20 2,83 1,59
33 495 632 9,78 1,24 2,95 1,62
34 498 652 9,63 1,20 2,83 1,59
35 495 632 9,78 1,24 2,95 1,62
36 498 652 9,63 1,20 2,83 1,59
37 489 626 9,71 1,24 2,96 1,62
таблица П-2-2 (продолжение)
8
-СО —рС6Н4 —О —П1С6Н4—pht—рС6Н4—П1С6Н4-
-СО—рС6Н4—О—тС6Н4—fl—тС6Н4—рС6Н4-
-СО—рС6И4—О—тСб Н 4—fl—рС6 Н 4—тСб Н4-
-СО—рС6Н4—О—тС6Н4—тС6Н4—pht—pC6H4-
—СО—рСбН4—О—тСбН4—тС6Н4—fl—рС6Н4-
-СО—рС6Н4—О—П1С6Н4—pC6H4—phi—тС6Н4-
-СО—рС6Н4—О—тСбН4—рС6Н4—fl—тС6Н4-
-СО—рС6Н4—О—рС6Н4—pht—С10Н6—тС6Н4 -
-СО—рС6Н4—О—pC6H4—pht—тС6Н4—С10Н6-
-СО—pC6H4—О—рС6Н4—pht—тС6Н4—тСбН4-
-СО—рСбН4—О—рС6Н4—fl—С10Н6—тС6Н4-
-СО—рС6Н4—О—рС6Н4—fl—тС6Н4—С10Н6-
-СО—рС6Н4—О—рСбН4—fl—тС6Н4—тС6Н4-
-СО—РС6Н4—О—pC6H4—С10Н6—phb—тС6Н4-
-СО—рС6Н4—О—рС6Н4—С10Н6—fl—тС6Н4-
_ СО—рС6Н4—О—рС6Н4—тС6Н4—pht—С10Н6-
-СО—РС6Н4—О—рС6Н4—тС6Н4—pht—тС6Н4
-со—РС6Н4—О—рС6Н4—тС6Н4—fl—С10Н6-
-СО—рС6Н4—О—рС6Н4—тС6Н4—fl—тС6Н4-
-СО—С10Н6—pht—рС6Н4—О—рС6Н4—тС6Н4-
—СО—С10116—pht—тС6Н4—рС6Н4—О—рС6Н4-
-СО—С10Н6—11— рС6Н4—О—рС6Н4—тС6Н4—
-СО—С10Н6—fl—mC6H4—рС6Н4—О—рС6Н4-
-СО—С10 Нб —рСб Н4—О—рСб 114—pht—тСб Н 4
-СО—С10Н6—рС6Н4—О—рС6Н4—fl—тС6Н4-
—СО—С10Н6—тС6Н4—pht—рС6Н4—О—рС6Н4-
-СО —С 10Н6— пС6Н4—fl —[С6Н4—О — [С6Н4—
-СО—тС6Н4—pht—mC6H4—О—рС6Н4—рС6Н4-
446
Приложение 2
1 2 3 4 5 6 7
38 489 626 9,71 1,24 2,96 1,62
39 495 632 9,78 1,24 2,95 1,62
40 480 550 9,71 1,24 2,96 1,62
41 495 632 9,78 1,24 2,95 1,62
42 489 550 9,71 1,24 2,96 1,62
43 489 626 9,71 1,24 2,96 1,62
44 489 626 9,71 1,24 2,96 1,62
45 493 648 9,55 1,19 2,83 1,58
46 493 648 9,55 1,19 2,83 1,58
47 498 652 9,63 1,20 2,83 1,59
48 493 578 9,55 1,19 2,83 1,58
49 498 652 9,63 1,20 2,83 1,59
50 493 578 9,55 1,19 2,83 1,58
51 493 648 9,55 1,19 2,83 1,58
52 493 648 9,55 1,19 2,83 1,58
53 489 626 9,71 1,24 2,96 1,62
54 493 648 9,55 1,19 2,83 1,58
55 489 626 9,71 1,24 2,96 1,62
56 493 648 9,55 1,19 2,83 1,58
57 495 632 9,78 1,24 2,95 1,62
58 489 550 9,71 1,24 2,96 1,62
59 498 652 9,63 1,20 2,83 1,59
60 493 578 9,55 1,19 2,83 1,58
61 495 632 9,78 1,24 2,95 1.62
62 498 652 9,63 1,20 2,83 1,59
63 489 550 9,71 1,24 2,96 1,62
64 493 578 9,55 1,19 2,83 1,58
65 489 626 9,71 1,24 2,96 1,62
Приложение 2
447
таблица П-2-2 (продолжение)
8
-СО—mC6H4—pht—рС6Н4—О—тСбН4—рС6Н4-
-СО—mC6H4—pht—рС6Н4—О—рС6Н4—С10Н6-
-СО—П1С6Н4—pht—рС6Н4—О—рС6Н4—тС6Н4-
-СО—тС6Н4—pht—С10Н6—рС6Н4—О—рС6Н4
-СО—тС6Н4—pht—тС6Н4—рСбН4—О—рС6Н4-
-СО—тС6Н4—pht—рС6Н4—тС6Н4—О—рС6Н4-
—СО—тСбН4—pht—рС6Н4—рС6Н4—О—mC6H4-
-СО—тС6Н4—fl—тС6Н4—О—рС6Н4—рС6Н4-
-СО—mC6H4—fl—рС6Н4—О—тС6Н4—рСбН4-
—СО—mC6H4—fl—рС6Н4—О—рС6Н4—СЮНб-
-СО—тС6Н4—fl—рС6Н4—О—рС6Н4—тС6Н4-
—СО——тС6Н4—fl—С10Н6—рС6Н4—О—рС6Н4—
-СО—тС6Н4—fl—тС6Н4—рСбН4—О—рС6Н4-
-СО—тС6Н4—fl—РС6Н4—тС6Н4—О—рСбН4-
-СО—П1С6Н4—fl—рС6Н4—рС6Н4—О—тС6Н4—
-СО—тС6Н4—mC6H4—О—рС6Н4—pht—рС6Н4
—СО—П1С6Н4—П1С6Н4—О—рС6114—fl—рС6Н4-
-СО—тС6Н4—рС6Н4—О—тС6Н4—pht—рС6Н4
-СО—тС6Н4—рС6Н4—О—тС6Н4—11—рСбН4—
-СО—П1С6Н4—рС6Н4—О—рС6Н4—pht—С1ОН6-
-СО—тС6 Н 4—рСб Н4—О—рС6Н4—pht—тСб Н 4-
-СО—111С6Н4—рС6Н4—О—рС6Н4—fl—СЮНб-
-СО—тС6Н4—рС6Н4—О—рС6Н4—11—тС6Н4-
-СО—тС6Н4—С10Н6—pht—рС6Н4—О—рС6Н4-
-СО—mC6H4—СЮНб—fl—рС6Н4—О—рС6Н4-
—СО—mC6H4—тСбН4—pht—рС6Н4—О—рС6Н4
-СО—П1С6Н4—тС6Н4—(1—рС6Н4—О—рСбН4-
-СО—тС6Н4—рС6Н4—pht—тС6Н4—О—рСбН4-
448
Приложение 2
1 2 3 4 5 6 7
66 489 626 9,71 1,24 2,96 1,62
67 493 648 9,55 1,19 2,83 1,58
6s 493 64х 9,55 1,10 2,ИЗ ' 1,58
69 489 626 9,71 1,24 2,96 1,62
70 489 626 9,71 1,24 2,96 1,62
71 489 626 9,71 1,24 2,96 1,62
72 489 626 9,71 1,24 2,96 1,62
73 493 648 9,55 1,19 2,83 1,58
74 493 648 9,55 1,19 2,83 1,58
75 493 648 9,55 1,19 2,83 1,58
76 493 648 9,55 1,19 2,83 1,58
77 489 626 9,71 1,24 2,96 1,62
78 493 648 9,55 1,19 2,83 1,58
79 489 626 9,71 1,24 2,96 1,62
80 493 648 9,55 1,19 2,83 1,58
81 489 626 9,71 1,24 2,96 1,62
82 489 626 9,71 1,24 2,96 1,62
83 493 648 9,55 1,19 2,83 1,58
84 493 648 9,55 1,19 2,83 1,58
Примечание: 1. В химических формулах полиэфиркетонов использованы условные обозначе-
ния следующих базовых фрагментов:
-pht- Q О, -fl- Q_Q, -П1С6Н4- -fj- -рС6Н4--0-.
%
2. Tg-температура стеклования; Тт-температура плавления; 8 -параметр растворимости;
р - плотность; £ - диэлектрическая проницаемость; п - показатель преломления.
Приложение 2
449
таблица П-2-2 (окончание)
8
—СО—mC6H4—рСбН4—pht—рС6Н4—О—тСбН4
-СО—тСбН4—pC6H4—fl—mC6H4—O—pC6H4-
-СО—mC6H4—pC6H4—fl—рС6Н4—О—тС6Н4-
—СО—рС6Н4—pht—тС6Н4—О—рС6Н4—тС6Н4-
—СО—рС6Н4—pht—рС6Н4—О—П1С6Н4—тСбН4-
-СО—рС6Н4—pht—niC6H4—тС6Н4—О—рС6Н4
-СО—рС6Н4—pht—тС6Н4—рСбН4—О—тСбН4-
-СО—рСбН4—fl—тС6Н4—О—рС6Н4—тС6Н4—
-СО—pC6H4—fl—рС6Н4—О—тС6Н4—П1С6Н4—
—СО—pC6H4—fl—тС6Н4—тСбН4—О—рСбН4-
—СО—pC6H4—fl—тС6Н4—рСбН4—О—П1С6Н4—
—СО—рС6Н4—тС6Н4—О—рС6Н4—pht—тС6Н4-
-СО—pC6H4—тС6Н4—О—рС6Н4—fl--mC6H4-
—СО—рС6Н4—рС6Н4—О—тС6Н4—pht—тС6Н4-
-СО—рС6Н4—рС6Н4—О—тС6Н4—fl—тС6Н4-
—СО—рС6Н4—тС6Н4—pht—тС6Н4—О—рСбН4-
—СО—рС6Н4—тСбН4—pht—рСбН4—О—тС6Н4-
-СО—рС6Н4—тС6Н4—fl—тСбН4—О—рСбН4—
-СО—рСбН4—тС6Н4—fl—pC6H4—О—тС6Н4—
Приложение 3
Пример решения смешанной задачи -
анализ химического строения
фенолоформальдегидной смолы
Пример решения смешанной задачи синтеза полимеров относится к ана-
лизу химического строения одного из представителей сетчатых полимеров -
фенолоформальдегидной смолы, находящей широкое применение, в частно-
сти, при производстве прессованных древесных изделий.
Перед тем как проанализировать структуру сетчатого полимера на осно-
ве фенолоформальдегидной смолы, необходимо отмстить, что хотя фсноло-
формальдегидные смоля (ФФС) являются одними из старейших представи-
телей часто сетчатах полимеров, интерес к их структуре и свойствам сохраня-
ется до сих пор [201J. Имеется огромное количество работ, посвященных
анализу структуры и свойств ФФС, однако единого мнения о химическом
строении сетки, образованной этими смолами, до сих пор не существует. Это,
по-видимому, связано с тем обстоятельством, что в зависимости от хода реак-
ции отверждения ФФС структура сетки может быть различной. Другое об -
стоятельство ассоциируется с тем фактом, что в структуре сетки присутству-
ют ОН-группы, способные к образованию водородных связей. Эти водород-
ные связи могут проявляться как при межмолекулярном взаимодействии этих
групп, расположенных в соседних цепях ФФС, так и за счет того же взаимо-
действия между ОН-группами, расположенными в пределах одного и того же
повторяющегося звена.
Смысл таких понятий, как "межмолекулярное взаимодействие” и “внут-
римолекулярное взаимодействие” с позиций излагаемого подхода удобно по-
яснить с помощью упрошенной схемы, изображенной на рис.П-3-1. Рис.П-3-1 .а
демонстрирует случай, соответствующий межмолекулярному взаимодей-
ствию, приводящему к образованию сетки физических (водородных) связей
между цепями. Естественно, что образование этой сетки способствует повы-
шению температуры стеклования. Случай, показанный на рис П-3-1 б ,ото-
Приложение 3
451
Рис.П-3-1. Схематическое изображение сетки водородных связей между соседними
цепями (а) и в пределах одного повторяющегося звена (б)
S d iem;l ic representation of a network of hydrogen bonds (a) appearmg between
neighboring chains and (6) within tire same repeating unit
бражает ситуацию, когда две ОН-группы находятся в пределах одного и того
же звена линейного фрагмента ФФС и тогда водородное связывание проис-
ходит в пределах этого звена, а сетка физических связей между соседними
цепями не образуется. Этот вопрос был детально проанализирован выше на
примере полиимидофснилхиноксалинов и полиамидофенилхи ноксалинов. Ра-
зумеется, реально могут сосуществовать различные типы межмолекулярно го
взаимодействия (водородного связывания), оказывающих, как будет показа-
но ниже, существенное влияние на свойства ФФС.
В отвержденн ой ФФС, согласно имеющимся данным, могут присутст-
вовать следующие структуры (повторяющиеся фрагменты сетки):
Структура I:
I
-------сн -----
I I I
сн2-О-сн2--
i он I
Эта структура входит только в состав сетки, повторяющийся фрагмент
которой отмечен пунктиром.
Расчет температуры стеклования сетки проведем по соотношению (109)
Согласно определению химического строения узла сетки, структура узла
образована набором атомов, включая атом, от которого происходит развегв -
лснис, а также соседние атомы со своими заместителями. Для структуры 1
сетки строение узла заключено в пространство, обведенное пунктирными
линиями:
452
Приложение 3
м
... jQL...
।
он
Естественно, что весь узел сетки содержит три таких набора атомов. Тог-
да для структуры 1 имеем*:
(^ДЕ,)1 = 3(ДИС19 +уД1'сд2 + --2ДИЯд24) + 2ДИСд8 +
+ 2ДЕЯд24 + АИс, го + А^одзз + Af'//,125 = 3(8,4 + —-12,6 + — 2 - 2,0) +
+2- 1? 7+ 1] 65+.5 2+4 7-10)1 А3,'
Е Я;Д^ =<3с(2ДЕСд8 + Д1/С 20) + ая(2ДИЯд24 + ДИЯД25) +
' У
+ао5Д1/Од33 + 6/г =[0,021(2 -12,7+ 11,65)+ 19,93(2 -2,0 + 4,7)+ 703 -5? -
-139,6] 10-3 =71,6-10'3 А3К-’;
- Кс(ДУс,19 +~^/С,12) + КИ2 —AVн д24 =[1,15(8,4 +
+ у ‘ 12,6) + 2,307 • 2 • 1 • 2,0] • 103 =21,5• 103 А3К’’.
Подстановка значений (^а^ДЕ,^, (]Га;Д1++ )t и (^ГА^ДЕ,)) вурав-
' ' У '
нение (109) приводит к следующей величине температуры стеклования сет-
ки в случае идеальной структуры 1:
Т„ =----—-------103 =743 К. (432)
2 21,5-3 + 71,6
Полученная расчетная величина Tg существенно превышает интервал экс-
периментальных значений 7&чксп , приведенной в табл.П-3-1, и, следовательно,
реальная сетка ФФС по своей структуре будет отличаться от идеальной стру ктуры 1.
* Здесь и дальше номера атомов соответствуют их номерам в табл.3.
Приложение 3
453
Таблица П-3-1
Экспериментальные -значения характеристик отвержденной ФФС,
Experimental values of the characteristics of cured PFR
Наименование свойств Единица изц® ения Пределы изменении
Температура стеклования, 7'г , эксп. к 343-433
Плотность, р , эксп. Г/СМ’’ 1,24-1,33
Показатель преломления, И , эксп. отн. ед. 1,57-1,63
Коэффициент объемного расширения, О;, эк/п к-* (1.80 -2,25)-10 1
Молярная теплоемкость, С/ , эксп. , <ал/(моль-град) 29 48
Структура T.
-CH 2-i
i!hoz
СН2 I
ОН I
Такая возможная структура ФФС относится к линейному полимеру' (пун-
ктиром отмечено повторяющееся звено).
Для этой структуры
У Д1)" - 3ДК(д 19 + 2ДПС; is + — • 2Alzq 12 + Д1 с 20 + ^1'с, 40 +
+ 4-уДИЯд24 + 2ЛГ/л124 +2ДИЯд24 + 2AIzh J25 + Д1ЛОДЗЗ + ^f/<2,132 _
= 3-8,4 + 2-12.7+ —• 2-12,6 +1165+ 16.0 + 4 • —• 2,0 + 2 • 2,0 + 2 2,0 +
2 2
+2 • 4,7 + 5,2 + 5,6 = 123,05 А3;
У. «/ДЕ/ = <7С(ЗД17С19 + 2АЕС18 + у 2AIzCj2 + Д17с,20 + Д1/с,4о) +
+ аН (4 • — Л17Л/, 124 + 124 + 2 Д Г7-// 124 + 2Д1'я. 125 ) + аО 5 О J33 +
+ ЛУО j32) = [0,021(3 8,4 + 2 • 12,7 + у 2 • 12,6 +11,65 +16,0) +19,98(4 у х
x 2,0 +2-2,0+ 2-2,0+ 2-4,7)+ 7,03(5,2+ 5,6)|-10“3 = 505,4-10 3 A3K’1
Если обе ОН-группы участвуют в образовании водородных связей между це-
пями (как это показано на рис.П-3-1), то
454
Приложение 3
Zbj=^bh+bM ; Sb7 = (-139,6-2+16,0) IO-3 = -263,2 ИГ3 A3K ’.
j /
Подстановка всех значений параметров в уравнение (84) приводит к следую-
щей величине Tg идеальной линейной структуры 2.
------12X05----10з=508К (433)
g 505,4 - 263,2
Это значение, как и для структуры 1, выше экспериментального темпера-
турного интервала, в котором наблюдается a-переход для ФФС.
Структура 3:
--СН2-О-СН2 "
| ОН |
Эта структура образуют только линейные цепи, повторяющееся звено ко-
торых заключено между пунктирными линиями. Для структуры 3 имеем’.
(^ЛИДз =2AIzf-|9 +ДИс,20 + ЗДИС, is +~’2AI/cj2 + —•4ДИя,124 +
+ дря ,125 + ЗА[/Я ,124 + Д lo.i33 = 2 ’ 8,4 +11,65 + 3 • 12,7 + ~ 2 • 12,6 +
+ —-4 2,0 +4.7+3 • 2,0 + 5,2 = 99,05 А3’
2
(2а/Д1//)з = йс(2Д1/С,19 +Д1/С,20 + ЗДСсд8 + —-2ДИс,12) +
7
+ аИ(-- 4АИ/УД24 +ДИ//Д25 + ЗДИ/7,124) + аО,5 ’Д^ОДЗЗ ~
= [0,021(2 8,4 +11,65 + 3 12,7 + ± 2 • 12,6) + 19,98(у 4 • 2,0 + 4,7 +
+3 -2,0) +7,03 -5,2|-1(Г3 =331,9-10~3 А3К’’;
Xbj =bh +bA{; Ybj =(-139.6 +16.0)-НГ3 =-123.6-io^A’k-1,
./ ./
Подстановка всех этих значений параметров в уравнение (84) приводит к сле-
дующему значению Tg идеальной структуры 3:
(434)
Т =-------------И)3 =476 К .
g 331,9-123,6
Эта величина Tg несколько превышает температурный интервал а-пере-
хода для ФФС.
Приложение 3
455
Структура 4:
--СНг-О-СНг -
Эта структура образует сетку, повторяющийся фрагмент которой заклю-
чен между пунктирными линиями. Структура узла этой сетки имеет следую-
щий вид (заключена в пространство, обведенное пунктирными линиями):
Для структуры 4 имеем'
(^АС;)4 = 2А17СД9 +Д1/с,20 + ~'2Д,7’,12 +ЗАКСД8 +ЗАИАгд24 +
+~4АСя>]24 +АГа131 = 2-8,4 + 11.65 + у-2-12,6 + 3-12,7 + 3-2,0 +
+ --4-2,0 + 2,1 = 91,25 А3;
2
)4 = ас(ЗАГсд8) + ан (ЗАИ# 124) = (0,021-3-12,7 +
/
+ 19,98 • 3 • 2,0) 10“3 = 120,7 -10“3 А-3К/
3 11
(2L^zAI//)4 = ^c(2^l/c,19 +&l/C,20 +~'2'А1/СД2)+£Я у •4А1/Яд24 +
Ко&ИО;131 =[115(2- 8,4 +11,65 + у 2 • 12,6) + 2,307 - у 4 • 2,0 +
+ 0,058 2,1] • 10-3 = 56,5 -10 3 А’К'1.
Подстановка этих значений в уравнение (109) приводит к следующему значе-
нию /^идеальной сетки построенной из структуры 4:
Г =----------103=515К. (435)
g 56,5 + 120,7
Таким образом, температура такого частосетчатого полимера была бы
достаточно высокой по сравнению с Tg^za^.
456
Приложение 3
Структура 5.
сн2
I 'он
Эта структура с точки зрения брутто-формулы та же, что и структура 2,
но в данном случае водородное связывание происходит внутри одного повто-
ряющегося звена. Такая структура описана в работе [99].
Если водородные связи образуется между соседними ОН-группами, рас-
положенными в одном повторяющемся звене,то, как это было отмечено вы-
ше, физическая сетка между цепями, обусловленная водородными связями,
не образуется. Тогда при расчете температуры стеклования величина
j
рассчитывается следующим образом: поскольку эта структура - линейная,
то нужно учесть инкремент Ьи , учитывающий м-замещение в ароматичес-
тре-
ком ядре; кроме того, имеются два ответвления от ароматического ядра ,что
бует введения двух инкрементов bt! .Тогда = 6м + 26^= [16,0 -2-55,4 ]-10‘3 =
j
= -94.8-10‘3A3K-1. Подстановка значений , ^<т(А1/' и вуравне-
i ' J
ние (109) приводит к следующему значению температуры стеклования для
структуры 5:
- =——ю3 = зоо к.
8 505,4-94,8
(436)
Эта величина Tg лежит ниже температурного интервала a-перехода для
отвержденных ФФС.
Как видим, расчетные значения температуры стеклования ни для одной
из рассмотренных идеальных структур ФФС не попадают в интервал экспе-
риментальных значениятемпературы стеклования Т &эксп Это позволяет пред -
положить наличие более сложной структуры отвержденной ФФС, чем рас-
смотренные идеальные, что и отмечает большинство исследователей. Веро-
ятнее всего, реальная структура ФФС состоит из набора рассмотренных
идеальных структур.
С целью установления наиболее вероятной реальной структуры ФФС
рассчитаем, кроме Tg, такие характеристики отвержденных ФФС, как плот-
Приложение 3
457
ность р, показатель преломления п, коэффициент объемного расширения
и молярную теплоемкость Csp . Выбор этих характеристик обусловлен как их
важностью и чувствительностью к изменению структурной формулы веще-
ства, так и тем фактом, что с ростом температуры стеклования для разных
идеальных структур ФФС может н аблюдаться как рост, так и снижение значе-
нии остальных характеристик, что важно для поиска наиболее вероятной струк-
туры ФФС с помощью метода планирования эксперимента (см.ниже).
Расчетные значения плотности ФФС для структур 1-5, выполненные по
формуле (6), приведены в табл. П-3-2.
Таблица П-3-2
Расчетные значения Tg, р, п, и Ср для идеальных структур отвержденной ФФС
Calculated values Tg, р , п, and Cps for tbc ideal structures of cured PFR
Идеальная структура ФФС Д, ,К р, г/см1 П, огн. ед. «G-ioCtc1 Cps, кал/( моль-град)
СТру тктур а 1 743 1,253 1,621 1,30 3 19
Стру тъ р а 2 507 1,250 1,583 3,20 42,4
Структура 3 476 1.205 1,604 2,02 31,4
Структура 4 515 1,301 1,645 1,86 26,1
Структура 5 300 1,250 1,583 3.20 42.4
Расчеты показателя преломления n проведем во уравнению (189). в кото-
рое входит молярная рефракция, складывающаяся из рефракций отдельных
атомов и инкрементов идя разных типов связей (двойные, тройные).
Для структуры 1 имеем (значения R, взяты из [28]):
Д = 3- (7Д Ч— Ry 3"Д/ ) З- 27?(- + 2/\).- + Rq + Rq * + R^ + 37Д = 3(2,418 +
+ 1-2 (П8 + 1 ]0) + 2-2 (П8+2-1,10 + 2,418 + 1,525 + 1,10 + 3-1,733 =
= 31,459 см3/моль.
Для структуры 2:
R2 =4RC + 8R/f + 2-Ro< +3R* = 8-2,418+8-1,10+ 2-1,525+ 3-1,733 =
= 36,393 см3/моль.
Для структуры 3:
R3 =2Rc+6R!I +Ro.+3R* =7-2,418 + 6-1,10 + 1,525 + 3-1.733 =
= 30,25 см3/моль.
Для структуры! :
Д, -=77?с +5RH + RfJ< +.37Д - 7 • 2 418 + 5 • 1,10 +164.3+ 3 1 7.33 =
= 29,268 см3/моль.
Для структуры 5 величина 7? зеовпадает с величиной /С Значения вели-
чин п для различных структур, найденных с помощью уравнения (189), при-
ведены в табл.П-3-2.
458
Приложение 3
Расчеты молярной теплоемкости Ср в стеклообразном состоянии поли-
мера проводили по соотношению (429).
Величина 0$ напрямую связана с температурой стеклования Tg соотноше-
нием:
_ 0.096
(437)
Расчетные значения величин Ср и для всех пяти идеальных структур
представлены в табл.П-3-2.
Сопоставление расчетных значений характеристик ФФС для возможных
идеальных структур, приведенных в табл.П-3-2, с их экспериментальными
значениями (см табл П-3-1) позволяет сделать вывод о том, что отвержденная
ФФС не представляет собой идеальную сетку, а содержит набор структур, для
установления которого удобно воспользоваться методом планирования экспе-
римента для многокомпонентных системю построением симплексных реше-
ток и полиномиальных моделей “состав-свойство”. В качестве независи-
мьк переменных X таких моделей примем рассмотренные выше идеальные
структуры, сочетание которых и определит реальный структурный состав от-
вержденной ФФС: X! - структура 1, х2 — структура 5, х3 — структура 3 и х4 -
структура 4, а в качестве функций отклика Y — свойства отвержденной ФФС:
7^- температуру стеклования, К\ р—плотность, г/см3; и—показатель прелом-
ления, отн.ед; а<; — коэффициент объемного расширения, К'1 и Ср — моляр-
ная теплоемкость, кал/(моль град).
Как видим, вместо идеальной структуры 2 в качестве независимой пере -
менной рассматривается структура 5, отличающаяся наличием водородного
связывания групп-ОН внутри одного повторявшегося звена что, при прочих
одинаковых характеристиках, обеспечивает более низкое значение температу-
ры стеклования (см. табл.П-3-2).
Зависимость функций отклика Y от переменных X будем искать в виде
модели второго порядка [70]
l<i<4; 1<у<4, (438)
где
Р, Ру =4у,у-2у;-2у7 . (439)
План эксперимента и соответствующая ему симплексная решетка приве-
дены соответственно в табл.П-3-3 и на рис.П-3-2,а.
Как видно из диаграммы, точки плана эксперимента с одним индексом у,
находятся в вершинах тетраэдра, с двумя индексами у у — в середине ребра
Приложение 3
459
Таблица П-3-3
Матрица плана эксперимента и расчетные значеютя свойств ФФС
в точках плана эксперимента
Matrix of the experimental design and calculated values of PFRs
at the points of experimental d esign
Точка плана экспери- мента Координаты точек । плана эксперимента Расчетные значения ФФС '
X] Х2 Хз Х4 Т J S’ К Р. г/см3 И , отн. ед. аа- 104, К1 С ' -р , кал/(мольтрад)
У1 1 0 0 0 743 1,253 1,621 1,30 31,90
У2 0 1 0 0 300 1,250 1,583 3,20 42,40
Уз 0 0 1 0 476 1,205 1,604 2,02 31,40
У4 0 0 0 1 515 1,301 1,645 1,86 26,10
У12 1/2 1/2 0 0 410 1,251 1,600 2,34 37,2 0
У23 0 1/2 1/2 0 359 1,232 1,594 2 67 36,90
У13 1/2 0 1/2 0 582 1,232 1,613 1,65 31,65
У14 1/2 0 0 1/2 615 1,276 1,633 1,56 29,00
У24 0 1/2 0 1/2 364 1,272 1,614 2,64 34,2 5
У34 0 0 1/2 1/2 494 1,254 1,624 1,94 28,75
Рис.П-3-2. Обозначение откликов в точках симплексной решетки: а) точки плана
эксперимента, б) то же, контрольные
Designation of responses at the points of simplex lattice: a) experimental design
pointy 6) control design points
тетраэдра,имеющего вершины i и j .При этом значения функций отклика Tg,
р, п, ас и Ср в точках плана эксперимента у/, находящихся в вершинах тет-
раэдра, вычисляем до формулам (109), (6), (189), (429) и (437), а в промежу-
точных точках эксперимента, те. с двумя и большим числом индексов при р,
по формулам .
460
Приложение 3
- для температуры стеклования
+ «з(ЕА^)з +«4(2Д1/,)4 +
s а1[(£а,д^ + Z>7)1 +(ХА:/А^)11 + аз(Еа/АГ/> +EAj)3 +
' У ' 'У
+ а5(ХдИ/)5
—►--------------------------!---------------------------(440)
+ а4[(£а,д1< +X6j)4+(Z^<Ari)41 + a5(Za/Al// +£Л>5 ’
‘ J ' ' J
— плотности
_ 0,68 l(ct^AYj + oi-jA/^ + сцЛ/^ + )
Р ” ^[a1(XA^)i+a3(SAr<)3+a4(LA^)4 + a5(SA^)51 ’ (441)
! ! I I
- показателя преломления
л2-1_ 0,681(0,2?! + а3Л3 +а4Я4 + а5Т?5) /'4474
п2 + 2 (XА^)1 + «з(1Ж)з +“4(ЕА^)4 +«5(ЕД^)5] ’
i i i i
- молярной теплоемкости
CSP =а1(Х^,,дР;)1 +а3(2;с^,ДГ,)3 +a4(^C^,AI/)4 +
+ а5(Хс;<;А^)5+С0; (443)
Ср = сцСрд + а3Ср 3 + a4Csp 4 + а5Ср5 + Со .
Коэффициент объемного расширения вычисляем по соотношению (437),
причем величина Tg определяется по формуле (440). В соотношениях (440)-
(443) а, ,а3,04 и а5 молярные доли структур 1,3,4 и 5 соответственно; при
этом cq = xj,a3 = х3,а4 = и а5= х5
С использованием значений функций отклика в точках плана эксперимен-
та, приведенных в табл.П-3-3, и уравнений (439) для расчета коэффициента 0
модели второго порядка (438) находим для описания свойств отвержденной
ФФС полиномиальные модели, в которых исключены незначимые коэффици-
енты:
- температуры стеклования, К
Tg =743xi +300х2 +476х3 + 515х4 -446x^2 -116Х]Х3 -56Х]Х4 -
— 116х2х3-174х2х4; (444)
- плотности, г/см3
р = 1,253х, + 1,250х2 + 1,205х3 + 1,301х4 + 0,012xiX3 +
+0,018х2х3-0,014х2х4 ; (445)
Приложение 3
461
- показателя преломления, отн.ед.
« = 1,621*1 +1,583*2 +1,604*3 +1,646*4 - 0,009*!*2; (446)
- коэффициента объемного расширения, К'1
CLq — (1,3 *! + 3,2 *2 + 2,02*3 +1,86*4 + И 36*!*2 +
+ 0,24*2*3 +0,44*2*4 )-10“4; (447)
- молярной теплоемкости, кал/(моль град)
Ср = 31,9*! + 42,4*2 + 31,4*з + 26,1*4 + 0,2*!*2 . (448)
С использованием полученных полиномиальных моделей (444)-(448) и
формул (109), (6), (189), (429) и (437) в контрольных точках плана экспери-
мента, показанных на рис.П-3-2,б, (точка плана с тремя индексами yIjk нахо-
дится в середине грани тетраэдра с вершинами i,j и к) рассчитаны свойства
отвержденной ФФС.
Кж видно из табт.П-3-4, относительная погрешность прогноза свойств
отвержденной ФФС не превышает 1,7 %, что является вполне приемлемым
для использования полиномиальных моделей как с целью определения реаль-
ной структуры отвержденной ФФС, так и степени влияния типа идеальной
структуры на свойства ФФС. В частности, на рис.П-3-3 в качестве примера
показана графическая интерпретация полиномиальной модели температуры
стеклования ФФС (444) в области изменения соотношения идеальных струк-
тур 1, 3 и 5 (при отсутствия идеальной структуры 4). Как видим, изолинии
Рис.П-3-3. Изолинии температуры стеклования Т^ФФС в области идеальных
структур */, *2 и *з (при *4 = 0)
Isolines of the glass transition temperature Tg of PFRs in the region of ideal
structures xj , *2 and x3 (x4= 0)
462
Приложение 3
Погрешность прогноза свойств отвержденной ФФС
Контрольные точки плана эксперимента Координаты контрольных точек плана эксперимента Значения свойств ФФС в контрольных точках плана эксперимента
*1 *2 Хз Xj р/р'
У123 1/3 1/3 1/3 0 428 431 1.238 1,259
У124 1/3 1/3 0 1/3 436 442 1,265 1,266
134 1/3 0 1/3 1/3 559 558 1,253 1,254
^234 0 1/3 1/3 1/3 394 397 1,252 1,253
Примечания'.
1. В числителе приведены свойства ФФС, рассчитанные по формулам (I), (7), (8), (9) и (11),
а в знаменателе - по полиноминальным моделям (444) - (448).
2. Относительную погрешность прогноза свойств ФФС определяют по формуле
С У ijk У ijk
^Уук----------;--------юо,
У ijk
температуры стеклования ФФС представляют собой прямые, что говорит об
аддитивности вклада отдельных идеальных структур в формирование свойств
искомой реальной структуры ФФС, и, кроме того, температура стеклования
ФФС преимущественно определяется относительным содержанием идеаль-
ных структур 1 и 5.
Поиск искомой реальной структуры отвержденной ФФС заключается в
определении такого соотношения между содержанием идеальных структур 1,
3,4 и 5, при котором выполняются ограничения на экспериментальные значе-
ния свойств смолы, приведенные в табл.П-3-1. Допустимый набор таких иде-
альных структур О.х (при xi= 1,0 < xz < 1, i = 1 - 4) устанавливается в сле
дующей последовательности. При фиксированном содержании структу-
ры 4 (х4 = const) для каждого исследуемого свойства смолы Tg, р, п, а<; и Csp
в плоскости изменения переменных Xi, х2 и Хз определяются области допус-
тимых наборов трех остальных идеальных структур, а последующим совме-
щением этих областей получают искомое значение QA]J4=cons(.
Пример такой процедура при х4 = О показан на рис.П-3-4. Как ви-
дим , такие показатели отвержденной ФФС , как показатель преломления
(рис.П-3-4,в) и молярная теплоемкость (рис.П-3-4,д) не оказывают влияния
на формирование области допустимого набора идеальных структур ЛАА4=0
(рис.П-3-4,г). По-вцдимому, влияние этих характеристик ФФС сказывается
Приложение 3
463
Таблица П-3-4
Errors of the predicted properties of cured PFRs
Значения свойств ФФС в контрольных точках плана эксперимента Относительная погрешность прогноза свойств ФФС, %
11 /п' С//С/' зт; 1 8р. 1 8л 1 6aG 1 8С/
1,603 1,602 2,240 2,2 35 35,23 35,23 -0,7 -1,7 0,1 0,2 0
W7 1,615 2, 200 2,2 00 33,47 33 47 -1,4 -0,1 о,1 0 0
1623 1624 1, 720 1,714 29 80 29,80 ,0 2 -0,1 0,1 0,4 0
1,611 J 611 2.440 3436 33,3 33,3 -0,8 -0,1 0 0,2 0
идеальных структур ФФС Г^прих^ = 0 .а) область допустимого набора струкгур £. о
для температуры стеклования отвержденной ФФС; б) то же, плотности Q р х_Х4=ов) то
же, показателя преломления <Г ХЛу ог) то же, коэффициента объемного расширения
12° ; д) то же, молярной теплоемкости <2С ; е) область допустимого набора
идеальных структур ФФС flxx=о с координатами вершин 7) (0,35; 0,34; 0,31; 0,00);
Т2(0 34; 0,36-, О ЗГ, 0 рО), Г 3(0 5,0 45,0 РО, О РО)
Schematic representation of the search procedure for an admissible set <lx of
ideal PFR structures forx^=0. Showing domains of the admissible sets of structures: (a)<2‘rxw=e
for the glass transition temperature <f curd PFR ;(6)QP =/or the density,(в) fl" =/or
the refractive index; (r) £lG for the volume expansion coefficient; (д) flc tot the
molar heat capacity; (e) for the ideal PFR structures with the coordinates of vertices T}
(0,35; 0,34; 0,31; 0,00); Л (0,34; 0,35; 0,31; 0,00); 73 (0,55; 0,45; 0,00; 0,00)
464
Приложение 3
только при учете идеальной структуры 4 и усиливается с увеличением содер-
жания последней (рис.П-3-5).
Область допустимого набора идеальных структур отвержденной ФФС
£1Х определяется последовательным перебором областей fiXA4=Const ПРИ варь-
ировании содержания структуры х4 с шагом 0,01 в пределах от 0 до 1. В ре-
зультате получается, что искомая область представляет собой неправиль-
ную фигуру, координаты вершин которой приведены нарис.П-3-6. Располо-
жение областей О.х в факторном пространстве идеальных структур ФФС
позволяет сделать вывод о том, что реальная структура отвержденной ФФС
может быть представлена только сочетанием линейных (структуры 3 и 5) и
сетчатых (структура 1 и 4) фрагментов полимерной цепи, что подтверждает-
ся и другими исследованиями [99].
Для количественной оценки свойств ФФС, содержащей различные типы
структур, подставим в исходные соотношения (440)-(443) все вычисленные
нами параметры. В результате получаем следующие выражения для расчета
температуры стеклования Tg, плотности р, показателя преломления п, коэф-
фициенты объемного расширения <1с, и молярной теплоемкости Ср .
гр _ ct) • 101,1 + ct; 123,05 + ct3 99,05 + а4 -91,25 . (449)
8 ~ а,-136,1 + а5- 410,6 + а3- 208,3 + а4-177,2 ' ’ 1 ?
0,6
Рис.П-3-5. Влияние идеальной структуры 4 на изменение области допустимого
набора идеальных структур fl* показателя преломления (I) и молярной теплоемкости
(II) отвержденной ФФС: а) область допустимого набора идеальных структур ФФС il*
при х4 = 0; б) то же, при х4 = 0,2; в) то же, при х4 = 0,4
Effect of the ideal structure 4 on the change of the domain Qx of admissible
set of ideal structures of cured PFRs for the refractive index (I) and for the molar heat
capacity (II) at x4 = 0 (a); 0.2 (6); 0.4 (в)
Приложение 3
465
0,681(01)-112 +а5-136 +а3-106 +а4-105)
P ~ 0,6023(g) -101,l + g5 -123,05 + a3 99,05+ g4 -91,25) ’
n2 -1 _0,681(g) -31,459 + g5 • 36,393 + g3 • 30,25 + g4 - 29,268) iq3 . Д51
„2 + 2 ~ 0,6023(g) -101,1 + g5 -123,05 + g3 -99,05 + g4 -91,25)
дд =0,096/Ту, (452)
Ср =gj-31,9 + a5-42,4 + g3 -31,4 + g4-26J , (453)
где gi, g3, g4 и g5 - молярные доли структур 1,3,4 и 5 соответственно.
Одно из возможных соотношений между относительным содержанием
идеальных структур, отвечающим области допустимого набора (рис.П-3-6)
имеет вид: д> = 0,345, д5 = 0,345, д3 = 0,2 и д4= 0,11.
Подставляя эти величины в соотношения (449)-(453) получаем для ре-
альной сетки: Tg = 429 К, р = 1,248 г/см3, п = 1,6057, = 2,238-Ю'4 К'1,
Csp = 34,8 кал/(моль-град); хорошо видно (см. таблЛ-З-З), что все эти вели-
чины попадают в интервал экспериментальных значений.
Учитывая наличие узкой области допустимого набора идеальных струк-
тур отвержденной ФФС (см. рис.П-3-6), можно заключить, что установлен-
ная нами реальная структура сетки является близкой к наиболее вероятной.
Рис.П-3-6. Координаты вершин облаете допустимого набора идеальных структур
отвержденной ФФС: Т\ (0,35; 0,34; 0,31; 0,00); 72 (0,34; 0,35; 0,31; 0,00);
73 (0,55; 0,45; 0,00; 0,00); Г4 (0,00; 0,22; 0,13; 0,65)
Coor dhates о {vertices о ft he dunaiii о fa dnissfbb se to fi'da Is true tire s
fijor cured PFRs Л' ,(0 35 ,0 34,0 3F,0 Q0) 2(0 34 ,0 35',0 3F,0 Q0)-,r3 (0 55 ,0 45',
0,00; 0,00); Г4 (0,00; 0,22; 0,13; 0,65)
466 Приложение 3
Таким образом, подход, описанный в разделе IV.4, совместно с методом
планирования эксперимента позволяет, во-первых, сделать определенные вы-
воды о строении сложных частосетчатых систем, основываясь на сравнении
экспериментальных и расчетных характеристик сетки. Во-вторых, рассмот-
ренная задача анализа структуры фенолоформальдегидной смолы относится
к смешанной задаче, так как на первом этапе решается прямая задача - опре-
деляются свойства идеальных структур ФФС по их химическому строению,
а на втором этапе исследований рассматривается решение о фатной задачи -
производится поиск химической структуры ФФС, обеспечивающей требуе-
мый комплекс свойств такой смолы. Такой комплекс свойств в данном случае
задается их экспериментальными значениями, приведенными в табл.П-3-1.
Отметим также применение в данном примере отличного от описанного в
Приложении 2 подхода к решению обратной задачи синтеза полимеров.
Приложение 4
Применение подхода к многокомпонентным
сополимерам
Как правило, анализ структуры и свойств сополимеров проводился для
систем, содержащих два компонента, гораздо реже—для тройных сополиме-
ров. В данном приложении рассмотрим применимость описанного в моно-
графии подхода к анализу структуры и свойств сополимеров, содержащих от
трех до пяти сополимеров.Одновременно сопоставим экспериментальные и
расчетные значения физических характеристик как для гомополимеров, так и
для многокомпонентных сополимеров на их основе.
Для анализа свойств многокомпонентных сополимеров в работе [39] выб-
раны шесть мономеров, которые были использованы в реакциях полимериза-
ции и сополимеризации: метилметакрилат (ММА), бутилметакрилат (БМА),
бутилакрилат (БА), н-нонилакрилат (НА), 2-этилгексилакрилат (ЭГА) и н-геп-
тилакрилат (ГА). Гомополимеры и сополимеры на основе этих соединений
являются полностью аморфными высокомолекулярными веществами, кото-
рые легко образуются в блоке и удобны для исследования термических и оп-
тических характеристик. В табл. П-4-1 приведен состав пяти синтезирован-
ных в работе [39] сополимеров. Расчет температуры стеклования Tg много-
компонентных сополимеров проведен по уравнениям (94) и (96). Первое из
этих уравнений для расчета температуры стеклования сополимеров не требу-
ет знания экспериментальных температур стеклования гомополимеров. Урав-
нение (96) содержит величины температур стеклования для гомополимеров,
причем при расчете Tg сополимеров использовались экспериментальные зна-
чения Tg для соответствующих гомополимеров.
Плотность рассчитана по соотношениям (7), (454) и (455). Первое из них
является приближенным, полученным на том основании, что коэффициент
молекулярной упаковки примерно одинаков для всех полимеров и при ком-
натной температуре равен кср = 0,681 для блочных образцов. Однако для по-
468
Приложение 4
Таблица П-4-1
Состав сополимеров
Composition of copolymers
Сополимер Мольные доли в сополимере звеньев
ММА БМА БА НА ЭГА ГА
1 0,333 0,333 0,333 - - -
2 0,250 0,250 0,250 0,250 - -
3 0,250 0,250 0,250 - 0,250 -
4 0,200 0,200 0,200 0,200 0,200 -
5 0,250 0,250 0,250 - - 0,250
лимеров и сополимеров, находящихся при комнатной температуре в высоко-
эластическом состоянии, при расчете плотности желательно учесть темпера-
турные зависимости коэффициента молекулярной упаковки к, так как реаль-
ный коэффициент упаковки для них тем больше отличается от средней величи-
ны кср, чем ниже температура стеклования. Уравнения (454) и (455) получе-
ны с учетом этого обстоятельства и позволяют, как будет видно ниже, более
точно рассчитать плотность полимеров и сополимеров. Для последних имеем
0,667 £алА4
Pg =------------------, 293 <Tg- (454)
[1 + aG (293 - Tg )]Na X (£ AI< )t
i
Таблица ГТ 4-2
Расчетные и экспериментальные характеристики гомополимеров*
Г омополимер р, г/см3 7^, К И г
ПММА 1.17-1.23 1,17; 1,17 378 377 1,49: 1.55 1,49 3,10 2.94
ПБМА 1,06 300 1Л8 2,82
1,09; 1,06 287 1,50 2.71
ПБА 1.04 221 1.47 -
1,11; 1,05 242 1.50 2,76
ПНА - 211 —
1,04; 0,97 219 1,51 258
ПЭГА -
1,05:0,99 242 1.51 2.60
ПГА - 220 . —
1,06; 0,99 225 1,51 2,63
Примечания: 1. * - в числителе приведены экспериментальные значения физических характеристик,
в знаменателе - расчетные величины. В случае расчетных значений р первое из них получено по
уравнению (7), а второе -по уравнениям (454) и (455} 2. р- плотность; Тя-температура стеклования;
п - показатель преломления; £ - диэлектрическая проницаемость ,б -параметр раствори мостг.
Приложение 4
469
к=п
0,667
р?° =---------------------------------- 293 >Тд (455)
к=п ’ s
[1 + aL (293 - Tg )]#л X аИЕ А['Н
к=1 i
Кроме температуры стеклования и плотности для сополимеров рассчита-
ны такие характеристики, как показатель преломления п, коэффициент опти-
ческой чувствительности по напряжению Са , температура начала интенсив-
ной термической деструкции Td, диэлектрическая проницаемость е, параметр
растворимости 5, поверхностная энергия у. Расчеты проводили соответствен-
но по уравнениям (190), (183), (202'), (223'), (33 Г) и (410). Прежде всего сопо-
ставим расчетные и экспериментальные величины свойств гомополимеров.
Эти данные приведены в табл П-4-2. В большинстве случаев наблюдается
хорошее совпадение экспериментальных данных с расчетными. Отдельно сле-
дует остановиться на расчете такой характеристики гомо- и сополимеров, как
плотность р, для которых в таблице приводится два расчетных значения. Пер-
вое из них определено с помощью ур авнения (7), а второе - по соотношениям
(454) и (455), которые учитывают температурную зависимость коэффициен-
та молекулярной упаковки. В случае стеклообразного полимера (полиметил-
метакрилат) расчетное значение плотности, полученное по уравнению (7),
хорошо совпадает с экспериментальным зн^тением. Для гомополимеров с низ-
кими температурами стеклования, которые при комнатной температуре нахо-
дятся в высокоэластическом состоянии, учет температурной зависимости ко-
Calculated and experimental characteristics of homopolymers
5, (Дж/см3)"2 у, дин/см aG •IO4, К4
18.6 39.0 40.2 2.70 623 -3.30
19,0 31,0 2,55 629 -2,85
17.7-18.4 — ~ —
18,2 27,2 3,05 627 16,6
18,2-18.4 30,7 2,80 ~~
Ц4 28,0 3,05 627 322
— — — —
17,5 25,3 3,05 626 40.5
— 29,2 —
17,5 25,7 3,05 626 39,4
— — — -
17,7 26,1 3,05 626 38,1
у - поверхностная энергия' i*g - термический коэффициент объемного расширения в стеклооб-
разном состоянии; температура начала интенсивной термической деструкции; Са - коэффици-
ент оптической чувствительности по напряжению.
470 Приложение 4
эффициснта молекулярной упаковки приводит к более низким значениям
плотности по сравнению с теми, которые определяются на основании средней
величины кср. При этом данные значения плотности хорошо совпадают с эк-
спериментальными величинами.
Температура стеклования, определенная по уравнению (84), с обычной
для таких расчетов точностью совпадает с экспериментальными значениями.
Показатель преломления для гомо полимеров рассчитан по уравнению (188), а
также по уравнению (193), которое учитывает температурную зависимость
коэффициента молекулярной упаковки. Последнее обстоятельство позволяет
рассчитать показатель преломления с большей точностью.
Что касается таких характеристик, как параметр растворимости, поверх-
ностная энергия, температура начала интенсивной термической деструкции,
термический коэффициент объемного расширения в стеклообразном состоя-
нии и коэффициент оптической чувствительности по напряжению, то эти ха-
рактеристики совпадают с экспериментальными с обычной для таких расче-
тов точностью
Свойства многокомпонентных сополимеров приведены в табл. П-4-3. Зцесь
совпадение расчетных и экспериментальных характеристик примерно такое
же, как и для гомополимеров. Плотности, рассчитанные по уравнению (7) с
использованием среднего значения коэффициента молекулярной упаковки,
Таблица П-4-3
Физические характеристики сополимеров*
Physical characteristics of homopolymers
Сополимер Ван-дер- Ваальсовый объем, А3 р, г/см3 П G, в Tg, к Td, К
1 125 1.09 1,12; 1,08 1,50 153 _29g.. 273; 268 tel 627
2 148 1.04 1 09; 1,03 1,50 21,6 237 247; 243 tell 627
3 143 i1.»/ 1,09; 1,04 Р50 213 233 258,255 570 627
4 158 1.02 1,08; 1,02 L50 25,2 -2.40 244; 240 610 627
5 139 _. LQ6 1,10; 1,04 1,50 2L0 243 25J 247 570 627
Прмечания". 1. * - в числителе приведены экспериментальные значения.,» в знаменателе - расчетные^
для р первое значение получено из уравнения (7), а второе - (455); для Tg первое значение
рассчитано по соотношению (94). а второе - по соотношению (96) с использованием
экспериментальных величии 7^ для гомополимеров.
2. р - плотность; и - показатель преломления; Са- коэффициент оптической чувствительности по
напряжению; Tg - температура стеклования; Tj - температура начала интенсивной термической
деструкции.
Приложение 4
471
юсколько выше; чем экспериментально определенные величины. Это обус-
эвлено тем, что все сополимеры имеют температуру стеклования ниже ком-
натной. Значения плотности р, вычисленные по соотношению (455), которое
считывает температурную зависимость коэффициента молекулярной упаков-
ки, несколько ниже, чем вычисленные по равнению (7) ц хорошо совпадают
с экспериментальными величинами р
Показатель преломления п для всех сополимеров приблизительно одина-
ков, что связано с близкими значениями показателя преломления для всех
(указанных выше гомополимеров. Коэффициент оптической чувствительнос-
ти по напряжению также почти одинаков для всех сополимеров, за исключе-
нием сополимера 1 с повышенным содержанием звеньев ММА, обладающих
отрицательным значением С о .
Температура стеклования сополимеров, приведенная в табл. П-4-3, рас-
считана по двум уравнениям - (94) и (96). Первое из них не требует знания
экспериментальных величин температуры стеклования гомополимеров, а во
втором такие значения используются. В целом наблюдается хорошее совпаде-
ние расчетных и экспериментальных величин Td . При использовании экспе-
риментальных значений Tg для гомополимеров и подстановки их в уравнение
(96) в подавляющем большинстве случаев соответствие расчета эксперимен-
ту несколько улучшается. Что касается температуры начала интенсивной тер-
мической деструкции Tg, то хорошее совпадение наблюдается для сополиме-
ра 4, для которого ошибка составляет 2,7 %. Для других сополимеров откло-
нение расчетных значении от эксперим тт альных для этой характеристики
было на 50° в сторону' увеличения, что составляет 8 %. Причина этого пока не
ясна Следует лишь иметь в виду, что такая характеристика, как температура
начала интенсивной термической деструкции, в большей степени, чем дру-
гие, зависит от наличия примесей и других причин. Расчетные значений этой
характеристики определяются для идеальной полимерной системы.
Таким образом, возможность прогнозирования различных характеристик
многокомпонентных сополимеров вполне н в идна При этом использован
ный подход не требует знания каких-либо величин, которые нужно опреде-
лять экспериментально. Весь прогноз осуществляется только на основании
химического строения многокомпонентного сополимера.
Приложение 5
Влияние сильного межмолекулярного
взаимодействия, возникающего между двумя
разнородными полимерами, на их совместимость
При смешении двух и более полимеров может наблюдаться ситуация, ког-
да между цепями этих полимеров возникает дополнительное сильное межмо-
лекулярное взаимодействие, которое не проявляется между макромолекулами
каждого из полимеров, взятых в отдельности. Это могут быть водородные
связи или сильные диполь-дипольные взаимодействия. Анализ влияния этих
взаимодействий на температуру стеклования будет проведен ниже.
В серии работ, посвященных анализу совместимости полимеров и свой-
ствам получаемых смесей, основное внимание уделяется именно этим специ-
фическим взаимодействиям, возникающим между макромолекулярными це-
пями смешиваемых полимеров. Схема такого взаимодействия продемонстри-
рована на следующем примере
Кратко рассмотрим примеры таких взаимодействий и эксперименталь-
ные методы их определения. Смеси сополимера стирола с винилфснилгек-
сафторметилкарбинолом с такими полимерами, как поликарбонат на основе
бисфенола А, полибутилметакрилат, поли-2,6-димстил-1,4-фснилсноксид
могут служить примером этого интересного явления |209]. Введение гидро-
Приложение 5
473
ксильных групп в указанный сополимер приводит к тому, что возникают во-
дородные связи между сополимером и поликарбонатом, что приводит к рез-
кому улучшению совместимости. На термограммах, полученных методом ДСК
для смесей полистирола с поликарбонатом отчетливо проявляются две тем-
пературы стеклования, что свидетельствует о несовместимости этих полиме-
ров. Картина резко меняется, когда вместо полистирола в смесь вводится со-
полимер указанного выше строения: на термограммах проявляется одна тем-
пература стеклования (что свидетельствует о совместимости), при этом
температура стеклования закономерно увеличивается с ростом концентрации
поликарбоната. Смеси сополимера указанного выше строения с полиэтиле-
ноксидом (ПЭО), который является кристаллическим полимером, также про-
являют этот эффект [210]. Кристалличность ПЭО изменяется в смеси таким
образом, что температура плавления снижается. Образование водородных
связей между ПЭО и сополимером изучалось в зависимости от температуры.
По мере повышения температуры водородные связи между сополимером и
ПЭО диссоциируют, но при охлаждении вновь восстанавливаются. Даже в
случае кристаллического полимера, такого как ПЭО, наличие водородного
связывания между' цепями смешиваемых полимеров приводит к улучшению
их совместимости, подавлению кристаллизации и образованию однофазной
системы.
Поведение смесей сополимера стирола с винилфенилгексафторметилкар-
бинолом изучалось с такими полимерами, как п оливинилапетат полиметил-
метакрилат , по лиэтилметакри лат, поли-н-бу ти лметакри лат, по лиметилвини-
ловый эфир, поли-2,6-диметил-1,4-фениленоксид, поликарбонат на основе
бисфенола А, сополимер стирола с акрилонитрилом, а также с аморфными и
кристаллическими сложными полиэфирами и полиамидами [193]. Для этих
систем водородное связывание влияет на совместимость компонентов, что бы-
ло подтверждено измерением температуры стеклования, а также методами
ИК-спектроскопии с Фурье-преобразованием. В работе [165] проведено де-
тальное исследование совместимости этого сополимера с рядом алифатичес-
ких полиамидов, таких как найлон-6,12 и Ы,М’-диметилзамещенныи наилон-
6,12. Вновь критерием совместимости считали единую температуру стекло-
вания для смеси, и при этом использовали такие полимеры, для которых
температура стеклования значительно отличалась от таковой для полистиро-
ла и сополимера. Так, например, для найлона-6,12 температура стеклования
составляет 46 °C, а температура плавления оценивается величиной 206-215 °C;
для того чтобы оценить влияние кристалличности на совместимость, помимо
найлона-6,12 был использован Ы,М’-диметилзамещенный найлон-6,12, а так-
же сополимеры с различным его содержанием. Основные выводы, получен-
ные по результатам работы [165], заключаются в том, что найдено образова-
474
Приложение 5
ние водородных связей между компонентами смеси, которые диссоциируют
при нагревании и возникают вновь при охлаждении; при этом введение не-
большого количества карбинольных групп в полистирол приводит к улучше-
нию совместимости таких несовместимых полимеров, как полистирол и по-
лиамид. Следовательно, рассматриваемый сополимер может служить компа-
тибилизатором .
В работе [192] изучена совместимость замещенных фенольных конден-
сационных смол с полиметилметакрилатом (ПММА). Изучались следующие
смолы:
где R=H, /-бутил, NO2, CI.
В этой работе вновь было показано, что образование водородных связей
между компонентами смеси играет существенную роль в совместимости. Были
определены термодинамические параметры диссоциации водородных связей,
такие как энтальпия и энтропия. Наибольшее влияние оказывают такие заме-
стители, как NO2 и CI. Все смеси данных фенольных смол с полиметилме-
такрилатом обнаруживали единую температуру стеклования, что говорило о
хорошей их совместимости. При этом зависимости температуры стеклова-
ния от состава соответствовали трем различным случаям:
1. Температура стеклования смесей лежит выше средневесовой темпера-
туры стеклования.
2. Температура стеклования смесей всегда ниже, чем средневесовая тем-
пература стеклования.
3. Зависимость температуры стеклования от состава носит ^образный
характер по отношению к средневесовой зависимости.
Авторы работы [192] предлагают описывать поведение смеси, соответ-
ствующее случаям 1 и 2, следующим соотношением: (456)
Ts =Il'17gl+^2rg2+qirlIV2,
где Tgi и Tg2—соответственно температуры стеклования полимеров I и 2 ; Hj и
W2 — их массовые доли; параметр q может быть интерпретирован, как вклад
водородных связей, которые могут рассматриваться в качестве псевдосшивок.
При этом величина q характеризует интенсивность водородного связывания.
Из проведенного эксперимента найдено, что когда в качестве замести-
телей используются NO2 и /-бутил, значения q — отрицательны и примерно
одинаковы по абсолютной величине. Если заместителями являются Н и CI,
Приложение 5
475
то значения q положительны (все сказанное справедливо при замещении аро-
матического ядра в пара-положении).
В результате, смеси фенольных смол с /-бутил- и МО2-группами с поли-
метилметакрилатом показывают поведение, соответствующее второму слу-
чаю. Когда замешение отсутствует, поведение системы соответствует третье-
му случаю. Наконец, когда заместителем является CI, поведение смеси соот-
ветствует первому случаю.
Для третьего случая 5-образная форма кривой зависимости температуры
стеклования Tgor состава может быть описана соотношением:
^Tgi +^2rg2
+ q^\w2,
(457)
которое является более общим по сравнением с выражением (456).
В дальнейшем [190] соотношение (457) было применено для описания
зависимости температуры стеклования от массовой доли компонентов для
смесей замещенных фенольных смол (смвыше) с такими полимерами, как
полиэтилметакрилат и полиметилметакрилат, причем в качестве заместите-
лей в фенольной смоле использовали F и /-бутил, а также изучали поведение
смесей на основе незамещенной фенольной смолы.
Получили все три описанных выше случая поведения смесей и нашли все
параметры уравнения (457 ).
В работах [170] и [171] изучали явление водородного связывания между
макромолекулами в смесях полимеров методом ИК-спектроскопии с Фурье-
преобразованием, а также с применением теории Patterson and Robard [ 173] и
получением диаграмм с нижней критической температурой растворения. Изу-
чалась растворимость и совместимость полиэтилоксазолина
(-CH2-N-CH2-)n
С2Н5С=О
который является изомером поли-М,М’-диметилакриламида
(-СН2-СН-)„
O=CN(CH3)2
Для оценки совместимости полиэтилоксазолина с другими полимерами
были проведены две серии экспериментов. В первой серии получали пленки
на основе смесей пар полимеров, одним из которых был полиэтилоксазолин.
Во второй серии экспериментов получали комплексы на основе тех же пар
полимеров. Эти комплексы получались смешением полимерных растворов с
последующим отделением осадка, его сушкой в вакууме в течение длительно-
го времени до постоянной массы. Было найдено, что состав комплекса отли-
чается от состава исходной смеси, причем состав комплекса соответствует
476
Приложение 5
соотношению полярных групп, приводящих к сильному мсжмолскулярному
взаимодействию.
Температура стеклования смесей полиэтилоксазолина с полиакриловой
кислотой лежит ниже аддитивных значений, в то время как для комплексов
температура стеклования находится выше их. Авторы связывают это с обра-
зованием сетки физических связей в случае комплексов.
Изучалась также совместимость поливинилметилового эфира с сополи-
мерами стирола с метилметакрилатом [136]. Найден критический состав со-
полимера, при котором наблюдалась совместимость его с поливинилметило-
вым эфиром. Такой сополимер должен содержать около 60 % (молярных) поли-
стирола. ГЬлучены фазовые диаграммы и так называемые ' окна совмести-
мости” для данных смесей.
Помимо темпера1уры стеклования изучали также термостабильность сме-
сей [221] (на примере смесей поли-п-гидроксистирола с поливинилпирроли-
доном ис полиэтиленоксазолином). Было найдено, что в процессе нагревания
небольшая потеря массы полигидроксистирола (6 %) в температурном интер-
вале 200—250 °C вызвана реакцией сшивания, приводящей к образованию
простых эфирных связей. Отмечают также важность образования водород-
ных связей между цепями полимеров.
Интсрмолскулярныс комплексы могут образовываться непосредственно
в процессе полимеризации [135]. Эго, например, проявляется при фотополи-
меризации акриловой кислоты, в которой растворен полиэтиленоксид. Тем-
пература стеклования комплексов не только выше, чем температура стекло-
вания смесей, но и выше, чем температура стеклования исходных компонентов.
Эти комплексы растворяются в таких растворителях, как диметилформамид
и диметилсульфоксид, и набухают до определенной степени в воде и метано-
лс, однако не набухают в диоксане. Если уменьшить количество групп, спо-
собных к образованию водородных связей, путем сополимеризации акрило-
вой кислоты с метилметакрилатом, способность к комплексообразованию со-
храняется, хотя она до некоторой степени снижается.
Термическое окисление также зависит [191] от образования водородных
связей между цепями полимеров (например, для смесей поливинилметило-
вого эфира и модифицированного полистирола). Модифицированный поли-
стирол представлял собой сополимер стирола, содержащий 2,5 % (молярных)
гексафтор-2-пропилстирола, т.е. он содержал гидроксильные группы, склон-
ные к образованию водородных связей. Эго обстоятельство обеспечивало улуч -
шейную совместимость компонентов смеси, как неоднократно отмечалось
выше. Однако, в добавление к этому, фенольные группы в сополимере приво-
дили к тому, что устойчивость системы к термоокислению повышалась, т.е.
эти группы работали как антиоксиданты. Это выразилось в том, что увсаичи-
Приложение 5
477
вался индукционный период термоокисления поливинилметилового эфира,
а сама скорость процесса снижалась. Комплексообразование, описанное выше,
наблюдалось также между поли-М,М’-диметилакриламвдом и фенолоформаль-
дегидной смолой [218]. Осаждение комплексов проводили из растворов в аце-
тоне, этилацетате и диоксане. При этом состав этих комплексов, как и ранее,
соответствовал молярному соотношению компонентов. Температура стекло-
вания комплексов была выше температуры стеклования исходных компонен-
тов. Полидиметилакриламид образовывал также комплексы с п-метоксифе-
нолоформальдегидной смолой, и при этом температура стеклования суще-
ственно возрастала по сравнению с температурой стеклования каждого из
компонентов [216, 217].
В работе [162] были синтезированы взаимопроникающие полимерные
сетки (ВПС) на основе совместимых полимеров - поли-1-гидрокси-2,6-ме-
тилфенилена и полиметилметакрилата. Использовали два типа сшивающих
агентов при различных температурах: гексаметилентетрамин и 1,3-диоксо-
лан. Интенсивность водородного связывания в смесях и в ВПС, как показано
в цитируемой работе, опрделяется изменением температуры сшивания и
уменьшением концентрации групп, способных к образованию водородных
связей. Изменение концентрации этих групп достигали использованием со-
полимера метилметакрилата и стирола. С помощью ИК-спектроскопии с
Фурье-преобразованием показано, что для сохранения совместимости в дан-
ных смесях необходимо поддерживать величину межмолекулярного взаимо-
действия, обусловленного водородными связями, не ниже определенной кри-
тической величины.
Водородное связывание оказывает влияние на фазовое поведение смесей
полимеров [163]. В качестве объектов такого исследования выбраны сополи-
меры метилметакрилата со стиролом и фенолоформальдегидной смолой, в
которой гидроксильные группы частично метилированы. Построены диаграм-
мы совместимости и найдены так называемье “окна совместимости”,когда в
зависим осг и от температуры и содержания одного из компонентов наблюда-
ется полная совместимость и микрофазовое расслоение.
В работе [222] изучали совместимость смесей гомополимеров с сополи-
мерами. Первая пара представляла собой смесь поли-4-гидроксистирола с
сополимером н-бутилакрилата с t-бутилметакрилатом. Указанные смеси со-
вместимы, когда содержание бутилакрилата в сополимере равно 64 % и выше.
В торая пара представляла ссб ой поли-t -бутила^илат и сополимер стирола с
4-гидроксистиролом. “Окно совместимости” для этой пары было при содер-
жании 4-гидроксистирола в сополимере между 28 и 66 % (молярных). Темпе-
ратура стеклования совместимых смесей поли-1-бутилакрилата и сополимера
стирола с 4-гидроксистиролом существенно ниже аддитивных значений, и,
478
Приложение 5
что самое интересное, она очень слабо зависит от состава смеси, находясь
примерно на уровне температуры стеклования самих сополимеров. Такое
поведение может быть объяснено только образованием водородных связей
между компонентами смеси, что подтверждено экспериментально методами
ИК-спекгроскопии.
Дальнейшее развитие этих идей можно найти в работе [161], где изуча-
лось водородное связывание в ВПС, полученных из совместимой полимерной
смеси поли-1-гидроксил-2,6-метиленфенилена и полиметилметакрилата. Бы-
ли использованы также сополимеры метилметакрилата и стирола, для того
чтобы уменьшить количество карбонильных групп. Для получения ВПС при-
меняли различные сшивающие агенты, такие как гексаметилентетрамин и
1,3-диоксолан. Реакцию сшивания проводили при различных температурах, в
результате чего варьировали интенсивность водородного связывания в ВПС.
Совместимая смесь двух упомянутых полимеров обнаруживала термообрати-
мость в отношении водородного связывания. Полу-ВПС и ВПС, приготовлен-
ные при температуре выше температуры стеклования смеси, не обнаружива-
ли исходного количества водородных связей после охлаждения до комнатной
температуры; когда же вместо полиметилмстакрилата использовался его со-
полимер со стиролом (т.е. приуменьшенном количестве карбонильных групп),
полу-ВПС не образовывали единую фазу при значительном уменьшении кон-
центрации этих групп. Однако полу-ВПС и ВПС, синтезированные при срав-
нительно низких температурах (ниже температуры стеклования смеси), со-
храняли большое количество водородных связей по сравнению с теми, кото-
рые были синтезированы при высоких температурах, и единую фазу [196].
В работе [186] авторы обратились к новым объектам, таким как полибен-
зимидазолы и поли-4-винилпиридин. Основным объектом исследования был
поли-2,2'-(м-фенилен)-5,5'-бибензимидазол
Все смеси разных составов обнаруживали единую температуру стеклова-
ния, что свидетельствовало о совместимости компонентов, и при этом темпе-
ратура стеклования лежала выше аддитивных величин. Авторы объясняют
это, как и в предыдущих работах, образованием водородных связей, происхо-
дящим по схеме:
(-СН-СН2—)„
Приложение 5
479
Образование в (дородных связей подтверждено экспериментальным ме-
тодом ИК-спектроскопии с Фурье-преобразованием.
Основной практический вывод, который может быть сделан на основе
цитируемых работ; заключается в том что дчя улучшения совместимости мож-
но в один из компонентов ввести небольшое количество функциональных групп,
например, гидроксильных, которые приводят к возникновению водородных
связей между полимерами — компонентами смеси. При этом могут образовы-
ваться домены различного размера и для уменьшения их размера достаточно,
например, внести 4,4 % (молярных) гидроксильных групп в полистирол для
того, чтобы он начал совмещаться с поли-п-бутилакрилагом. Предваритель-
ные исследования этих областей релаксации и размера доменов методом ЯМР
было проведено в работе (158]. В дальнейшем в серии работ [159, 178, 195,
207, 223] эти исследования были продолжены.
Изучены комплексы на основе поли-4-гидроксистирола и поли-М.М'-ди-
метилакрипамида методом ЯМР 13С с кросс-поляризацией с вращением об-
разца под магическим углом. Размер неоднородностей, определенный с помо-
щью этого метода, оказался = 2,5 нм [207]. Далее такое исследование было
проведено на ВПС. Было найдено, что размеры агрегатов в этом случае мень-
ше 2,2 нм [195].
Методом ЯМР исследовано влияние микротакгичности ПММА на его
совместимость с сополимером стирола с винилфенолом [159]. Оказалось, что
синдиотактический ПММА лучше совместим с указанным сополимером и
образует единую фазу в широком интервале составов смеси на основе обоих
метилметакрилатов; как показано методом ЯМР, размер микронеоднородно-
стей для большинства композиий составляет около 2 нм. Подобного рода ис-
следования были распространены также на поли-4-винилфенилдиметилси-
ланол и его сополимеры со стиролом [178,185]. В данном случае также обна-
ружили существенное влияние водородного связывания между фенольной и
фос фатнои группами, что алло показано методами ИК-спскгроскопии, ЯМР
31Ри ЯМР 13С [224].
В работе [137] изучены смеси полиэтиленоксида и полибутилметакрила-
та с модифицированным полибутилметакрилатом, который содержал груп-
пы, способные к образованию водородных связей [179-184 ] ведение 4гид-
рокси-4,4-бис-3-фторметилбутильной группы
СН3
—Si—О
(СН2)3
F3C-C-CF3
480
Приложение 5
даже в силоксановый полимер, который обладает низкой поверхностной энер-
гией, приводит к возможности получения совместимых смесей с полиэтиле-
ноксидом и полибутилметакрилатом.
В работе [151] оценивается параметр взаимодействия полимер - поли-
мер на основе изучения диффузии растворимости водного пара в смесях. Во
всех случаях этот параметр взаимодействия отрицательный, и при введении
в полимер гидроксильных этот параметр становится более отрицательным,
что указывает на лучшую совместимость. Найдены корреляции между коэф-
фициентом диффузии и удельным свободным объемом полимера.
Отмечается [194], что функционализация полистирола за счет введения в
него фторалкилкарбинольных или гидроксильных групп улучшает совмести-
мость полистирола с другими полимерами и повышает его термостабильность,
устойчивость к окислению и замедляет горение, что проявляется в его сме-
сях с другими полимерами.
Изучена совместимость ряда полиамидов с другими полимерами [128,
138, 157]. В качестве объекта исследования в [138] выбран сульфонирован-
ный поли-л-фснилентерефталамад
[-HN-O-NH-CO-0-CO-]n
SO3H
На основе его смесей с поливинилпирролидоком, поли-4-винилпириди-
ном и поливиниловым спиртом получены так называемые молекулярные ком-
позиты, в которых жесткоцепной сульфонированный полиамид служил арми-
рующим элементом. Найдено существенное влияние водородного связывания
между элементами молекулярного композита. При этом для смесей с поливи-
нилпиридином и ЛВС температура стеклования лежала ниже аддитивных зна-
чений, что, по мнению авторов, свидетельствует о небольшом числе контак-
тов между макромолекулами.
Сделана также попытка модификации найлона-6 путем приготовления
его смесей с небольшими количествами фенолоформальдегиднойсмолы [157].
Добавление фенолоформальдегидной смолы в количестве 1—2 % приводит к
увеличению модуля упругости и уменьшению поглощения влаги. При этом
образуются сферолиты более крупных размеров. Увеличение содержания
фенолоформальдегидной смолы в композиции не вызывает улучшения свойств
и на этом основании сделан вывод, что совместимость найлона-6 с феноло-
формальдегидной смолой весьма ограничена и лежит в пределах до 3 % фе-
нолоформальдегидной смолы. Целью работы [128] явилось изучение меха-
низма полимераналогичных реакций между политрихлорбутадиеном (ПТХБ)
и алифатическими диаминами: t-бутиламином, диэтиламином и триэтилами-
ном, которые моделируют химические процессы, протекающие в отдельных
Приложение 5
481
фрагментах полимерных цепей в ходе взаимодействия ПТХБ с разветвлен-
ным полиэтиленимином.
Методами ИК-Фурье и электронной спектроскопии было показано, что
при взаимодействии поли-1,1,2-трихлорбутадиена с аминами параллельно с
реакциями замещения аллильного хлора на аминогруппу и дегидрирования
происходит образование водородно-связанных ионных комплексов с перено-
сом заряда. Вклад каждого из этих процессов в общую конверсию функцио-
нальных групп полимера зависит от природы амина (степени N-замещения) и
типа растворителя. Первичный и вторичный амины более склонны к образо-
ванию с полимером стабильных водородно-связанных комплексов, тогда как
третичный амин вызывает преимущественно дегидрохлорирование полимера
и образование различных по протяженности полиеновых последовательнос-
тей. В диоксане преобладает дегидрохлорирование, в хлороформе - замеще-
ние и комплексообразование.
Таким образом, чтобы более объективно предсказать совместимость по-
лимеров друг с другом, а также провести анализ причин совместимости, боль-
шое внимание следует уделять не только свойствам индивидуальных компо-
нентов, но и специфическому межмолекулярному взаимодействию, возника-
ющему между' ними.
Как показывают результаты многочисленных работ, улучшения совмести-
мости полимеров можно добиться несколькими путями:
1. Подбором полимерных пар или модификацией полимеров, которые про-
водятся для того, чтобы между разнородными полимерными цепями возника-
ло сильное межмолекулярное взаимодействие (например, водородное связы-
вание) . Этот вопрос детально будет проанализирован ниже.
2. Второй путь заключается в проведении химических реакций между'
компонентами смеси, что, в конечном итоге, приводит к получению так назы-
ваемых интерполимеров, детально описанных в [215].
3. Третий путь заключается в введении в систему компатибилизаторов,
т. е. низкомолекулярных и высокомолекулярных соединений, обладающих фун-
кциональными группами, приводящими к усилению специфического межмо-
лекулярного взаимодействия между цепями. В качестве компатибилизаторов
могут применяться также сополимеры.
Рассмотрим б о лее детально вопросы, связанные с у сиянием специфичсс -
кого межмолекулярного взаимодействия между' сшиваемыми полимерами.
Если такое взаимодействие не возникает, то температура стеклования од-
нородной смеси совместимых полимеров рассчитывается по уравнению (96).
В него входят температуры стеклования гомополимеров на основе компонен-
тов 1 и 2 .которые при оценке температуры стеклования смеси могут браться
как расчетные, так и экспериментальные. В другой форме уравнение (96) за-
482
Приложение 5
писывается в виде (94), в которое входят наборы констант для компонен-
тов 1 и 2, связанных с энергией межмолекулярного взаимодействия.
Теперь предположим, что водородное связывание появляется тогда, ког-
да полимер 2 добавляется к полимеру 1. В этом случае константа blt = -140 •
10’3А3К-1, характеризующая вклад водородных связей, должна быть введена
в величину (^а,Д17+^йу)| для компонента 1. В этом случае из уравнения
(94) имеем ' /
а,(ХА1<)1+а2(ХД1<)2
8 +2026/,)!+а2(Ха1ДЦ)2+ 2aja2-0,03
' 7 ' J
поскольку часть повторяющихся единиц полимера 1 оказывается связанной
водородными связями с компонентом 2. Это уравнение получено исходя из
того, что два повторяющихся звена связаны водородными связями, причем
одно из них принадлежит полимеру 1, а второе - полимеру 2. Принимая во
внимание уравнение ( 96 ) и соотношение 04 = (1 - а2), следует записать
(1-а2)(^А^.)1+а2(ХДК,.)2
Tg = * 1 2 3 ’ (459)
------------------+ 2(1 -a2)a2bh +^2-‘т---+ 2(1-а2)а2 0,03
где Tg । и Tg£ — температуры стеклования (рассчитанные или эксперимен-
тальные) для компонентов 1 и 2 соответственно.
Теперь проанализируем зависимость температуры стеклования от соста-
ва смеси, которая отображается уравнениями (458) и (459). Будем рассматри-
вать три различных случая:
1) Ван-дер-Ваальсовый объем повторяющихся звеньев полимеров 1 и 2
примерно одинаков: (^A^)j «QTAI7,^.
/ i
2) Ван-дер-Ваальсовый объем повторяющегося звена полимера 1 значи-
тельно меньше, чем полимера 2: (^Д17), «(^ДС,)2 .
/ /
3) Ван-дер-Ваальсовый объем повторяющегося звена полимера 1 значи-
тельно выше, чем полимера 2: (^ДИ, )2 » (^ДИ|)2 .
Исходные величины (£ДИ,),, (£ДИ,)2 , Tg<\, Tg^ , (Ха/Д1/1 + 22 Ёи
' ' ' j
(^ai^i для смесей, соответствующих всем трем упомянутым слу-
' j
Приложение 5
483
Таблица П-5-1
Исходные величины параметров дня расчета температуры стеклования совместимых
смесей гомополимеров
Initial values of parameters for calculation of the glass transition temperature of compatible
mixtures of homopolymers
Рису- нок Номера кривых на рис. П-5-1 (W’ i А3 ом)2, 1 А3 ОХ-ДК; +!>;)], I i А3К-‘ +Xbi)2 - i i А’К"’
1 ПО 115 293 260
П-5-1,а 2 250 260 665 588
3 350 360 931 814
4 450 460 1197 1041
1 105 240 279 543
П-5-1,б 2 105 340 279 769
3 105 440 279 995
4 105 540 279 1222
1 225 115 598 260
П-5-1,в 2 340 115 904 260
3 440 115 1170 260
4 540 115 1436 260
Примечание: Tg] , = 376 K, Tg2 = 442 K.
чаям, приведены в таблице П-5-1. На рис. П-5-1,а показаны зависимости тем-
пературы стеклования смесей от содержания компонента 2 для случая, когда
ДИ,)] = (^ ДК;)2, причем эти величины варьируются в широком интер-
вале- Когда Ван-дер-Ваальсовый объем повторяющихся звеньев небольшой,
величина (^а,ДК, также невелика, причем рассматриваются слу-
‘ j
чаи, когда температура стеклования примерно одинакова для обоих компо-
нентов смеси. Поскольку величина bh, характеризующая влияние водород-
ных связей на Tg, является достаточно большой, из уравнений (458) и (459)
можно видеть, что чем меньше Ван-дер-Ваальсовый объем, тем больше тем-
пература стеклования превышает величину, соответствующую обычному слу-
чаю, при котором дополнительное специфическое взаимодействие между' ком-
понентами отсутствует. Это отражается на ходе зависимостей Tg от , пред-
ставленных нарис.П-5-1,а. Когда Ван-дер-Ваальсовый объем повторяющихся
звеньев обоих компонентов увеличивается, эффект дополнительного взаимо-
действия снижается и температура стеклования смеси лишь немного превы-
шает величину Tg, полученную по уравнениям (94) и (96).
Рис.П-5-1,б соответствует случаю, когда (£Д1у) ] «(^ДИ,)2 . Здесь
/ /
температура стеклования смеси захетно превышает величины Tg,рассчитан-
484
Приложение 5
Приложение 5
485
ную по соотношению (458). Следует заметить, что даже если дополнитель-
ное водородное связывание отсутствует, температура стеклования смеси пре-
вышает величину Tg, рассчитанную просто исходя из молярных долей компо-
нентов.
Рис. П-5-1, в описывает ситуацию, когда » (X АГ/ )2 • Вэтом
случае влияние дополнительного водородного связывания снижается, посколь-
ку суммарная энергия дисперсионного взаимодействия заметно превышает
энергию водородных связей. Подобная ситуация возникает и в тех случаях,
когда избыточное межмолекулярное взаимодействие между компонентами при
их смешении является диполь-дипольным взаимодействием, возникающим
благодаря присутствию полярных групп в повторяющихся звеньях. В этом
случае величина blt в уравнениях (458) и (459) заменяется на величину :
aidAP/h+MZAJ/.h
Т ------------------------------------------------------(460)
МХа/АГ,+ Xbj + 2a:>Mi +а2(£а1АК,- + 2>7)2 + 2а,а20,03 ’
i j i J
(1-а3)(ХДИ;)1 +а2(ХАИ/)2
Tg = (ёдИ); ! (t^)i •(461)
(1 -а2)—-----+ 2(1 -a2)a2bd +а2 —'---+ 2(1-аг)а2 0,03
ГЛ,1 ’ Гг,2
Рнс.П-5-1. Зависимость температуры стеклования Tg от концентрации второго
компонента а2 для совместимых смесей двух полимеров при возникновении
водородного связывания между ними: а) (ХЛК )i чХлчь ;б) Ха,;)! «Онг;
в) (ХДИ)1 »(ХА11)2 ; номера на кривых соответствуют номерам кривых, приведенных
в табл.П-5-1. Кривые 1 4 соответствуют ситуации, когда дополнительного
водородного связывания между компонентами не появляет ся
Dependence of the glass transition temperature J'g on concent rat ion of
second component a2 for compatible mixtures of two polymers when formation of hydrogen
bonding between them take place: a) (XA|/i)i ® lXA,'lh; 6) (XAI'th « (XA,/tH’
i i I _ i
в) (£д 1/) i » <XAI Th ;numberoncurvesa>rrespondto the numbersin the curves 1 - 4 correspond
to the situation when no additional hydrogen bonding between components taken place
486
Приложение 5
Когда (^ЛЦ)] ~ ДИ; )2, влияние дополнительного межмолекулярно-
z i
го взаимодействия на зависимость Tg от состава также проявляется, но оно
существенно ниже, чем при возникновении водородных связей между ком-
понентами (рис.П-5-2,а). Причина заключается в том, что величина Ь,<, ха-
рактеризующая влияние диполь-дипольного взаимодействия на температуру
стеклования, по абсолютной величине ниже, чем величина 6/,.
Если Ван-дер-Ваальсовый объем компонента 2 значительно больше, чем
компонента 1, положительное отклонение величин Tgor средних значений
также имеет место, однако эффект нс так велик, как в случае возникновения
водородных связей (рис.П-5-2,б). Если Ван-дер-Ваальсовый объем повторя-
ющегося звена компонента 1 значительно выше, чем компонента 2, влияние
дополнительного межмолекулярного взаимодействия не столь велико и зави-
симости температур стеклования от состава довольна близки к таковым, ко-
торые рассчитаны по уравнению (94), т.е. без учета дополнительного межмо-
лекулярного взаимодействия между компонентами. Это хорошо видно из
рис.П-5-2 ,в.
Известно, что помимо зависимостей температур стеклования от состава
смесей с максимумами (кривая 1) и минимумами (кривая 2) довольно часто
появляются зависимости S-образной формы (кривая 3) (рис.П-5-3). Это свя-
зывается с тем фактом, что один из компонентов смеси представляет собой
сополимер, содержащий ограниченное число полярных групп, способных к
образованию водородных связей. Рассмотрим эту ситуацию более детально.
Предположим, что сополимер содержит ограниченное число повторяющих-
ся звеньев, способных к водородному связыванию (сополимер 2). Например,
сополимер 2 может быть представлен в виде
-(СН2-СН)т-(СН2-СН),-
о о
I
он
(*) (**)
Молярную долю повторяющихся единиц (**) обозначим через [1. Предпо-
ложим, что контр-полимер (полимер 1) способен к образованию водородных
связей с повторяющимися звеньями (**), содержащимися в сополимере 2. С
этими обозначениями уравнения (458) и (459) могут быть записаны в виде:
Рис.П-5-2. То же самое, что ина рис. П-5-1, но при появлении дополнительного диполь-
дипольного взаимодействия между компонентами
Similar to Fig. П-5-1 situation but on occurrence of additional dipole-dipole
interaction between components
Приложение 5
487
488
Приложение 5
Рис.П-5-3. Схематическое представление типичных отклонений температуры
стеклования смесей совместимых полимеров от средних значений (см. текст)
Schematic representation of typical deviation of the glass transition temperature
for mixtures of compatible polymers from its average values (see text)
(l-a2)Q>^)1 +
T -----------------------'-------------------->
2 (1-«2)(Еа/АИ/ +£Z>y)i + 2(l-a2)a22a2Pfy, +
' j
+ аг(Х
,__________________(462)
+ а2£>,Д1< + 2>7)2 + 2(1-а2)а2-0,03 ’
' 7
(1 -а2)(ХД^), +
Т =------------------------1----------------------->
s QX), ОЖ)2
(I - а2) -4---+ 4(1 - а2 )а2 а2 + а, -----+
+ а2(£ДИ,)2
+ 2(1-а2)а2 -0,03 ' (463)
Как и выше, рассмотрим ряд случаев:
1) Ван-дер-Ваальсовый объем повторяющихся звеньев примерно одинаков.
Исходные величины всех параметров,необходимые для расчета, представ-
лены в табл.П-5-2. При расчете будем варьировать содержание компонента
(»♦) для сополимера 2. Результаты расчета показаны на рис.П-5-4,а. Можно
видеть, что доля компонента (**), способного к образованию водородных свя-
Приложение 5
489
Таблица П-5-2
Исходные величины параметров для расчета температуры стеклования совместимых
(Tgi, = 376 К Tg2= 442 К)смесей гомопотнмера (1) и сополимера (2)
О 0,2 0.4 0.6 0.8 1,0 С/-2
Рис.П-5-4. Зависимость температуры стеклования Tg от концентрации второго
компонента а2 для совместимых смесей гомополимера и сополимера (см. текст): а)
водородное связывание между компонентами имеет место; 6) диполь-дипольное
взаимодействие между компонентами имеет место
Dependence of the glass transition temperature 7'e on concentration of sec-
ond component a2 for compatible mixtures of homopolymer and copolymer (See text) when:
a) hydrogen bonding between components exists; 6) dipole-dipole interaction between
components prevails
490
Приложение 5
зей, является достаточно умеренной и при этом образуются 5-образные зави-
симости температуры стеклования смесей от состава. Эти зависимости на-
блюдались в целом ряде работ [180, 192,193, 209].
2) Этот случай соответствует образованию сильных диполь-дипольных
взаимодействий между сополимером 2 и полимером 1.
Соответствующие уравнения для расчета температуры стеклования вы-
текают из (462) и (463):
(1-а2)(£ДР/)1 +
Т =-------------------------------------------->
8 (1-а2)<Ха/Л1// + 2(1-а2)а22а2Р^ +
+ а2(£ДС,)2
—>-----------------1------------------ ; (454 \
+ а2 (X я, Д )2 + 2(! - а2)а2 0,03
' У
(1-а2)(ХД^)1 +
-4-------+ 4(1 - а2 )a2a2p/)rf + а, --+
J g,l 7g,2
+ а2(Х
---------1---------- (465)
+ 2(1-а2)а2 -0,03
Результаты расчетов, полученные с помощью уравнений (464) и (465),
показаны на рис.П-5-4,б. Расчетные зависимости Tg от доли сополимера 2
быпи получены при различных долях компонента (**) в сополиме ре Следует
заметить, что появление 5-образной формы зависимости Tgoi состава появ-
ляется только в тех случаях, когда сополимер 2 содержит значительную долю
компонента (♦*)• Однако во всех случаях эти зависимости отличаются от та-
ковых, полученных без учета возникновения дополнительного диполь-диполь-
ного взаимодействия между компонентами смеси. Расчетные данные отно-
сительно изменений температур стеклования, возникающих в результате
избыточного межмолекулярного взаимодействия между компонентами, по-
казывают, что эти взаимодействия играют существенную роль. Сами же ве-
личины Tg отличаются от таковых, когда никакого дополнительного межмо-
лекулярного взаимодействия не возникает. Отметим также то важное обсто-
ятельство, что все использованные уравнения (458}-(465) для анализа зави-
симости температуры стеклования от состава смеси не содержат ни одного
Приложение 5
491
“подгоночного” параметра, и, таким образом, все расчеты проводятся только
на основании химического строения компонентов смеси.
Рассмотрим поведение ряда конкретных смесей двух совместимых поли-
меров . Первая из них представляет собой смесь поли-н-бутилметакрнлата
(ПБМА) с сополимером стирола с 4-винилфенилдиметилсиланолом (Ст-
ВФДМС) [180]’.
-(СН2-СН)„-(СН2-СН)„-
6 о
I
CH3-Si-CH3
ОН
Сначала необходимо рассчитать температуры стеклования для сополи-
мера Ст-ВФДМС при разном содержании ВФДМС. Такие расчеты продела-
ны по уравнению (94) и их результаты представлены в табл.П-5-3. Видно
хорошее согласование расчетных и экспериментальных данных. Для даль-
нейших расчетов температуры стеклования смесей ПБМА с данным
сополимером нам необходимо иметь значения + Х^)пмба и
-вфдмс , которые определяются с помощью уравнения (94).
' j
Также необходимо определить эти же значения для сополимеров всех соста-
вов. Эти величины также представлены в табл П-5-3. Они были использова-
ны для дальнейшего расчета температуры стеклования ряда смесей ПБМА и
Ст-ВФДМС различных составов. Результаты расчетов показаны на рис.П-5-5
Таблица П-5-3
Параметры сополимеров стирола с винилфенидтиметилсиланолом
Штате tors о fcopo tymc rs of styrene with vinylphenyldimethylsilanol
I Обозначения Доля ВФДМС, мол- % Tg ЭКСП, 1 42 Tg расч, । °C Е ДР,-, А3 но3, А3к~‘
ПС 100 105 ЮЗ ПО 293
ПБМА 100 29 19 J 4о
ПВФДМС-2 1,9 97 —ГОЗ— —т— 295
11ВФ ДПС-4 4,1 98 103 113 301
ПВФДИС 9 8 7 98 103 иб 309
ПВФцМС-Н 11,4 99 103 11/" 311 "
ПВФДМС-18 18.2 101 104 122 324"
ПВФДМС-34 33,9 115 109 133 348
ПВФДМС-60 60,0 121 122 151 382
ПВФ ДПС-100 100 - 153 178 418
492
Приложение 5
Рис.П-5-5. Зависимость температуры стеклования Тх от молярной доли сополимера
а2 для совместимых смесей ПС с ВФДМС/ПБМА: а) ПВФДМС-34, б) ПВФДМС-9.
1 - расчетная кривая, 2 - экспериментальная зависимость, 3 - линейная зависимость
Dependence of the glass transition temperature Tg on mole fraction of copoly-
mer a2 for compatible mixtures of PS with VPIJMS/PBMA: a) P VPDMS-34,6) PVPDMS-9.
1 - calculated curve; 2 - experimental dependence; 3 - linear dependence
для двух смесей в виде зависимостей температуры стеклования от состава.
Эти зависимости для совместимых смесей, содержащих Ст-ВФДМС-34 и Ст-
ВФДМС-9, были получены с помощью уравнения (462). Наблюдается вполне
Приложение 5
493
хорошее согласие с экспериментальными данными для каждой смеси
(см.рис.П-5-5, а и б).
Кроме того, проанализируем зависимость Tg от состава для смеси ПБМА
и Ст-ВФДМС-9 с помощью уравнения (463), причем введем эксперименталь-
ные значения Tgв это уравнение. В результате получаем хорошее согласова-
ние расчетных и экспериментальных данных (см.рис.П-5-5,в). Заметим, что
температуры стеклования смесей лежат ниже средних величин, когда содер-
жание модифицированного полистирола (т. е., Ст-ВФДМС) в смеси мало. Когда
же содержание этого компонента в смеси увеличивается, температуры стек-
лования начинают превышать средние значения. Такое превышение объясня-
ется образованием большого количества водородных связей между компо-
нентами при их смешении.
Теперь рассмотрим поведение другой смеси, полученной при смешении
поли-(2,2'-м-фенилен-5,5'-бибензимидазола) (ПБИ)
и поли-4-винилпиридина:
-[СН2-СН]-
Эги смеси исследованы в работе [186]. Было показано, что водородные
связи образуются между остатками 4-винилпиридина и NH-группами в ПБИ.
По этой причине образуются гомогенные смеси на основе этих двух полиме-
ров. Экспериментальные зависимости температуры стеклования от состава
этих смесей, заимствованные из работы [186], показаны на рис.П-5-6. Темпе-
ратуры стеклования смесей превышают величины Tg для исходных компонен-
тов. Расчетные зависимости Tg от состава, определенные с помощью уравне-
ния (463), также показаны на рис.П-5-6. Из этого рисунка видно, что экспери-
ментальные точки хорошо ложатся на расчетную кривую, когда молярная доля
ПБИ в смеси не превышает 0,3. Отклонение экспериментальных точек от рас-
четной кривой при высоком содержании ПБИ связывается с тем, что поли-4-
винилпиридин начинает деструктировать при нагревании выше 375 °C. Еще
раз отметим, что все расчеты проводятся по уравнениям, которые не содержат
"подгоночных параметров ". Таким о бразом. все результать i получаются только
на основе химического строения компонентов смеси.
Сделаем некоторые заключения из проведенного выше анализа. В случае
совместимых смесей двух гомополимеров зависимость температуры стекло-
вания от состава смеси может принимать различные формы. Если Ван-дер-
494
Приложение 5
Рис.П-5-6. Зависимость температуры стеклования Tg от состава <ь для совместимых
смесей поливини.чпиридина и полибензимидазола . 1 - расчетная кривая,
2 - экспериментальная зависимость, 3 - линейная зависимость
Dependence of the glass transition temperature Tg on composition a2 for
compatible mixtures of polyvinylpyridine with polybenzimidazole. 1 - calculated curve;
2 - experimental dependence; 3 - linear dependence
Ваальсовые объемы повторяющихся звеньев гомополимсров примерно оди-
наковы и никакого дополнительного сильного межмолекулярного взаимодей-
ствия между компонентами не возникает, температура стеклования лежит
ниже средних значений. Если Ван-дер-Ваальсовый объем звена гомополиме-
ра 1 существенно ниже, чем гомополимера 2, положительное отклонение в
температурах стеклования от средних величин имеет место даже в том слу-
чае, если дополнительные межмолскулярные взаимодействия отсутствуют.
Если же такие взаимодействия проявляются, всегда имеет место положитель-
ное отклонение величн Tg от средних значений.
В случае смесей гомополимера с сополимером, содержащим ограничен-
ное количество полярных групп, способных к сильному межмолекулярному
взаимодействию с другим компонентом (диполь-дипольное взаимодействие.
Приложение 5
495
водородные связи), зависимости Tg от состава носят 5-образный характер.
Все упомянутые особенности поведения таких совместимых смесей могут
быть описаны с помощью приведенных выше уравнений, которые позволя-
ют оценивать свойства смесей на основе химического строения компонен-
тов. Разумеется, что данный подход не может претендовать на всеобщность и
требуются дальнейшие исследования для учета различных специфических
взаимодействий и тонких особенностей структуры компонентов смесей на
их термомеханическое поведение.
SUMMARY
Chapter I
Brief Information on Types of Polymers and their
Chemical Structure
Polymers are classified into cafbochain polymers whose macromolecules of
the backbone consist of atoms of carbon, heterochain polymers whose macromol e-
cules of tire backbone contain, along with atoms of carbon, heteroatoms of oxygen,
nitrogen, sulfur etc., and high molecular compounds with the conjugate system of
bonds. Chemical structure of any representative of a class of polymers is character-
ized by structural formula of the repeating unit. Availability of a different number
of such repeating units and different methods of their addition with one another in
a macromolecule affects both existence of a great number of isomers and the vari-
ety of chemical structure and names of polymers. Thus, the monosubstituted unsat-
urated hydrocarbons, where the arrangement of the substituent R is irregular rela-
tive to the plane of ordinary bonds of the backbone, are referred to as irregular or
atactic polymers, and those having their substituent?? with its directions alternating
at regular intervals, are referred to as stereoregular ones. Furthermore, if substitu-
ents are arranged on one side of the main bonds, tire stereoregular polymers are
referred to as isotactic, and if they are arranged on both the sides, the stereoregular
polymers are referred to as syndiotactic ones. In case of polymers svnthesized from
disubstituted monomers, the synthesis of macromolecules from cis-isomers leads
to production of erithrodiisotactic polymers, and in case of synthesis of macromol-
ecules from trans-isomers it leads to production of threodiisotactic polymers. In
case of synthesis of high molecular compounds from several monomers they obtain
a copolymer, which is referred to as alternating polymer if repeating units in the
chain alternate continuously, and it is referred to as a random copolymer if the
Summary
497
addition of repeati ng unis is draotic. A block-copolymer is produced when sepa-
rate blocks of links are connected together in the chain.
Depending on configuration of a macromolecule chain polymers are divided
into linear, branched and cross-linked ones. In the chapter the principal methods of
production of such polymers are given. Also highlighted is a great interest aroused
in the so-called dendric polymers pertaining to branched polymers (Fig. 1), inter-
polymers (Fig.2), and ladder polymers [113].
Chapter II
Packing of Macromolecules and Polymers Density
For understanding tire features of structural formation in polymers it is neces-
sary to proceed to the three-dimensional presentation of polymer macromolecules.
According to [75], each atom of such a macromolecule can be presented as a sphere
with intermolecular radius R. In case of intermolecular interaction valency-unbonded
atoms contact one another along the borders of the spheres (Fig. .3), whereas in
chemical interaction of such atoms their spheres overlap one another (they get com-
pressed) (Fig. 4), the length of chemical bond between two atoms d, always being
less than the sum of their intermolecular radii (2). They determine the increment of
their own (Van-der-Waals) volume of such atoms AF, (Table 3) from sizes of inter-
molecular radii (Table 1) and lengths of chemical bonds (Table 2) by formulas (3)
and (4). In its turn, by summing up the increments of atoms own volumes in the
polymer repeating unit and by multiplying them by Avogadro number NA, their own
(occupied) molar volume of repeating unit is obtained. The value of Vown is always
less than the total molar volume of the same unit Vt()tab which is equal to the rela-
tion between molecular mass of the repeating unit AY and the density of polymer p.
The ratio between these volumes determines the value of the molecular packing
coefficient к (5), which in tire case of amorphous polymers as opposed to crystal-
line polymers (Table 5, Fig.8) varies within a narrow range (Table 4) and, on the
average, is equal to Aavg= 0.681. Equations (14) and (15) preassign dependence о f
the molecular packing coefficient on temperature.
A relationship exists between the packing coefficient, porous structure and
various types of free volume of polymers. Tire so-called empty volume represents a
difference between the true molar volume of substance and its Van-der-Waals mo-
lar volume (18). The porous structure of a polymer is determined by its macro- an d
microporosity which, in its turn, depends both on tire method of polymer synthesis
and methods of determining the characteristics of porosity: total volume of pores,
their size distribution as well as specific surface of pores. Equations (23), (27), and
(28) predetermine the relationship between molecular packing coefficient, empty
volume and porosity of polymer.
498
Summary
The connection between molecular packing and volume variation of the sys-
tem in the process of polymerization referred to as contraction lias been analyzed.
Depicted in the book is the influence of a monomer and an associated polymer on
the amount of contraction of chemical structure as well as on the factors caused by
reduction of Van-der-Waals volumes of atoms owing to their ‘ compressing" and
the factor caused by a greater packing density' of polymeric chains as compared to
packing of monomer molecules, the factor of greater packing density of polymeric
chains making the largest contribution to contraction of the system.
The characteristics of microporous structure of polymers are determined by the
methods of equilibrium adsorption, capillary condensation, mercury' porosimetry
and, recently, by the positron annihilation method. The last mentioned method pos-
sesses a number of important advantages. First, no disruption is caused to the mi-
crostructure of polymer as a result of its swelling (during sorption) or deformation
of pores underpressure (during mercury' pressing into pores) and, second, it allows
to determine characteristics of the micropores commensurate to molecul c sizes and
beyond the reach of other methods of measurements. In particular, it Iras been found
by the positron annihilation method that in the process of stress relaxation, rear-
rangement of the microporous structure of polymer takes place. The rearrangement
is expressed in terms of recombination of the finest pores and their merging with
formation of long-living microcavities of a greater size.
Chapter III
Thermal Coefficient of Volumetric Expansion
Thermal expansion of bodies is a consequence of anharmonicity of thermal
oscillations of their particles. The thermal expansion of polymers has a number of
peculiarities connected with various physical transitions occurring in a polymer as
temperature is increased. The thermal expansion of polymers is characterized by
the thermal coefficient of volumetric expansion determined from a dilatometric
curve (Fig. 13) for the areas below (coefficient u<;) and above (coefficient a./) the
glass transition temperature 7g. Coefficient Og, as a rule, is not a cmis'mit value. К
depends on temperature (Fig. 14) and can be calculated more precisely from Equa-
tion (39). Since thermal expansion of bodies is a consequence of anharmonicity of
thermal fluctuations of their particles, it is natural to assume that the thermal coef-
ficient of polymer volumetric expansion consists of tire contributions made by var-
ious oscillatory' movements of such particles. In particular, as it is calculated the
role of weak dispersion interaction and strong intennolccular interactions stipulat-
ing the dipole-dipole interaction (ester and nitrile groups, halogens, substituting
hydrogen atoms etc.) and the initiation of hydrogen bonds (amide,urethane,hy-
droxyl, acid groups, etc.) is taken into account.
Summary
499
In the calculation scheme [28, 43] to account for weak dispersion interactions
during determination of the thermal coefficient of volumetric expansion, it w as
assumed that die contribution of each atom is proportional to a fraction cf its Van-
der-Waals in tlie total Van-der-Waals volume of repeating unit and, th t he f'rst ap-
proximation, the contribution of any dipole-dipole interaction is determined b у tire
same parameter flj irrespective of chemical structure of the group. Contribution of
hydrogen bonds is also estimated by one parameter 0A characterizing the energy of
hydrogen bonds. With due regard for these parameters, whose values are given in
Table 13, the thermal coefficient of volumetric expansion for the polymers in the
glassy state is determined from Equation (44), and for copolymers it is determined
from Equation (50) or (51). A presentation of thermal coefficient of volumetric
expansion of a copolymer in terms of the known similar coefficient о f appropriate
homopolymers is given by Equation (52) or, in a more compact form,b у E quation
(53). At the same time thermal coefficient of volumetric expansionfor polymers in
the rubbery state is, as a rule, constant and equal to =6.3 JO-4 K-1.
Chapter IV
Glass Transition Temperature of Polymers
Glass transition temperature of polymers 7gis most often determined from a'
thermomechanical curve which is built in accordance with the resit ts of test s on
polymer samples under a constant or varying loading determining deformation о f
samples at each fixed temperature and selected time of force action. A similar curve
is obtained in case when a small constant force acts on a sample while the temper-
ature grows continuously with time. The temperature area of the test on a typical
thermomechanical curve for amorphous polymeric bodies is subdivided into three
intervals (Fig. 18): in the first interval the body is in the glassy state, in t h sccon d
interval it is in the rubbery state, and in tire third interval it is in th e viscous fl ow
state. The corresponding temperatures <f transitionfromthe {Jassy state to the
rubbery state and from the rubbery state to t he viscous flow s ate have received the
names of “the glass transition temperature Tg’ an d t h tempera dire о f tansi ilo n
into the viscous state Tf” respectively. These temperatures ontlie thennomedtarii-
cal curve are determined either fromtlr ed f onnahon sothat istak enbeforehand
under the scheme presented in Fig.22, orfromtlie pouts tf irtersedicn of the
tangents to two appropriate branches of tiro curve (Fig. 23). In Table 14 a range of
glass transition temperature fora series of pd ymers is shown as well as the influ-
ence of chemical structure of polymers upon T. I s ton Id te pom i ebu t that from
the thermomechanical curve it is also possibletodet ermine amechanical segment,
i.e. tire value of molecular mass Mc beginning wi th which tie rubbery state of poly-
mer occurs (in Fig. 20 the molecular mass ofth e mebanical segmeh is eqial t oV/ ) .
500
Summary
The value of the mechanical segment can be determined according to the Kaigin-
Slonymsku's formula (58) using three thermomechanical curves of a polymer with
the known molecular mass Mor by the calculation according to the formula (62) on
the basis of the polymer chemical structure.
We should focus our particular attention on tire correct interpretation of the
thermomechanical curve obtained. In particular, as a proof of development of a
large deformation of the polymer in the area of high temperatures owing to its flow
rather than degradation, it is necessary to simultaneously apply the thermogravim-
etry method of research. To confirm the presence of the area of the rubbery state of
a polymer a reversible character of its deformation should be proved, whereas dur-
ing analysis of the thermomechanical curve some important information can be
obtained from preliminary X-ray analysis of a polymer. Along with the thermome-
chanical method of determination of the glass transition temperature and the tem
perature of transition into the viscous state of the polymer, dilatometric methods
and dynamic mechanical analysis, as well as the method of measurement of the
diffusion coefficient of the polymer are used.
There are two basic points of view that do not contradict each other as to the
mechanism of the glass transition process of polymers, namely - the relaxation
mechanism and formation of a physical network of intennolecular bonds Accord-
ing to the first conception, tire relaxation mechanism of glass transition is based on
the assumption of a possibility of transition of the polymer kinetic unit from one
energy level which is typical for the glassy state of a polymer to another energy
level that is peculiar to the rubbciy state of a polymer. The time of such transition
(the time of relaxation) depends on the temperature and is determined by the value
of the energy bamer which it has to overcome in older that such a transition can
take place. The more flexible is the macromolecule the smaller is the value of the
energy barrier of the system constnicted of such macromolecules. According to the
second concept, the leading role in the glass transition process is given to intermo-
lecular bonds in polymers: at cooling a polymeric body that is in its robbery state
the quantity of emerging intennolecular bonds grows, and with a certain tempera-
ture which is the glass transition temperature tire number of such cross-linkage
bonds becomes sufficient to fix separate macromolecules in relation to each other
and to prevent their translation mobility (Fig.41). At tins temperature the mobility
of macromolecules is lost to a sufficient degree and hence the polymeric body be-
comes solid, i.e. it transits into the glassy state. As will be shown below the display
of intermolecular interaction is necessary' to be taken into account when the calcu-
lated glass transition temperature of polymers is determined on the basis of tlieir
chemical structure.
The glass transition temperature of linear polymers is determined from Equa-
tion (84) or, with regard to influence of links connection type in a chain of a poly-
Summary 501
mer (“head-to-head” or “head-to-tail”), from Equation (87). For copolymers Equa-
tion (84) takes the form (95) or (97) if the glass transition temperatures of ho-
mopolymers are known. A good agreement between calculated and experimental
values Tg is observed (Table 19). The described approach is known as “atomic” and
has an essential advantage over the wide-spread method of calculation based on
contributions from separate groups of atoms [214]. Actually, if in polymer there is
a group whose contribution to the glass transition temperature is unknown, the
calculation of Tg by the method of group contributions becomes impossible. In the
approach that is the subject of our description, on the contrary, the contribution of
any group is simply summed up from contributions of atoms in such a group and
from contributions of specific interactions, if these groups are polar. Consideration
of influence of various types of branchings of the macromolecule chains (Fig.44)
upon the glass transition temperature of polymer shows that, under frequent and
regular arrangement of branches with an increase in their length, the properties of
polymers vary insignificantly and approach the properties of homopolymers hav-
ing chemical structure identical to that of the branchings themselves. If the branch-
ings are less frequent and they are distributed along the backbone of a polymer in a
random way, their chemical nature being different from that of the backbone, then
the influence upon the properties of polymer is more significant. However, the
greatest influence upon the glass transition temperature of a polymer is rendered by
short branchings, a great number of which brings about qualitative changes into the
chemical structure of a polymer. During calculation of the glass transition temper-
ature of polymer networks it is necessary to take into account the number of repeat-
ing units between cross-linked points /и; their number being reduced in high-cross-
linked netwoiks, the glass transition temperature begins to grow sharply (Fig.54).
For such polymers the glass transition temperature is determined by the number of
functional connections of a cross-linked point, its structure, distribution of cross-
links in a network, and by presence of defects of a network in the form of dangled,
branched chains or isolated loops. Equation (109) has been obtained that takes into
account on a par with chemical structure of a polymer the influence from both
linear fragments and cross-linked points upon the glass transition temperature of a
polymer. Using Equation (109) for various types of polymer cross-linked points
and different chemical structure of linear fragments between them some examples
of calculation of glass transition temperature are given, the value of the tempera-
ture approaching the experimental values of Tg. Taking polystyrene and divinyl-
benzene networks as examples, the influence of cross-links distribution within the
network upon Tg has been shown: at the identical degree of cross-linking a glass
transition temperature increases during transition from the most irregular distribu-
tion of cross-links to the random one and then to their regular distribution (Fig.57).
On the example of polydimethylsiloxane netwoiks having an identical structure of
502
Summary
linear fragments, but different structure of cross-linked points it has been shown
that the action of structure of the said points upon Tg becomes essential when the
number of polydimethylsiloxane units is reduced to n = 1-4 (Fig.60). By using a
hypothetical polyethylene-based network it is found out that, for instance, such a
type of network defect as a dangled chain decreases the glass transition temperature
of polymer networks (Figs.62 and 63).
Chapter V
Temperature of Transition into the Viscous Flow State for
Amorphous Polymers
The value of amorphous polymer temperature of transition into the viscous
flow state Tf determines the scope of rubber}' state temperature range which should
be known to process polymers. Using Expression (166) determining the ratio be-
tween zero (Newtonian) shear viscosity and the molecular mass Formula (168) was
obtained to determine a polymer Tf from its chemical structure. Temperature of
transition into the viscous flow state Tj being dependent upon a polymer molecular
mass Л/, at Л/ < Mc being molecular mass of a mechanical segment) equation
Tg= T} will be valid and glass transition temperature Tg will depend upon the
polymer molecular mass as well (it should be noted that the actual glass transition
temperature of the polymer will not be reached). On the other hand, the molecular
mass value determining the value of Ту is limited by the temperature of onset of
intense thermal degradation Td (see Chapter VII), thus, a polymer cannot be trans-
ferred into a viscous flow state at any temperature. Limiting values ofAf allowing
to completely transfer a polymer into a viscous flow state are determined from
Expression (168) and given in Table 20. It must be noted that the rubbery state
temperature ranges obtained by tliis calculation are 15-20 % higher than the actual-
ly observed values of those ranges. It is explained by tire fact that due to poly disper-
sity of polymer chains some of them come to a viscous flow state earlier than oth-
ers, which causes the longer chains overstress and breakage. The broken chains can
be found from the results of analysis of molecular mass distribution in the polymer.
Chapter VI
Melting Point of Polymers
A polymer melting point Tm is defined as a temperature at which the polymer
comes from its ciystaline state into a viscous flow state. Due to polydispersity of
polymeric chains, different number of links in them and imperfection of crystallites
formed, tliis process is smooth within a certain temperature range, ratherthan abrupt.
There exist two approaches to calculation of T„, based on the chemical structure of
Summary
503
a polymer repealing unit. In the first approach Equation (171) connecting the glass
transition temperatures and melting point of polymer has been obtained from the
experimental fact that both melting of a crystaline polymer and transition from the
glassy state to the rubbery state of a polymer with the same chemical structure take
place at reaching an approximately identical percentage of empty vo lume. In order
to increase the accuracy of Tm calculation it is expedient to take into consideration
the experimental values of parameters 8 and у for different atoms and types of
intermolecular interaction (Table 21); during which these parameters are related to
the partial packing coefficients for different atoms and constants of polymers. The
use of those parameters ensures an acceptable accuracy of melting point calculation
(Table 23). The other approach derives Equation (175) connecting Tm to the polymer
chemical structure from consideration of a polymer repeating unit as a set of anhar-
monic oscillators. In spite of a sufficiently good fitting of calculated and experimental
values of Tm (Table 23), we should mention a limited applicability of tliis approach.
Chapter VII
Temperature of Onset of Intense Thermal Degradation of
Polymers
The most important characteristic о fpo Vmer (term al stability is the tempera
ture of onset of intense thermal degradation 7’jdetcmuned at a crossing of tangents
to two branches of a thermogravimetric curve (Fig.68). As in calculation of the
glass transition temperature Tg and melting point Tm Equation (177) was obtained
from consideration of valence-connected atoms of a polymer repeating unit as a set
of anharmonic oscillators, however the energy of chemical bonding is taken into
consideration rather than the energy of intermolecular interaction, though the latter
may substantially influence the dissociation energy of chemical bonds. The last
circumstance is considered in Equation (181) with the help of coefficients Kh their
numerical values being different for the same atom depending upon its presence or
absence in a polar group (Table 25). The calculated values of Td as specified in
Table 26 for representatives of various polymer classes satisfactorily fit their ex-
perimental values. The use of Equation (181) confirmed a possibility for polymer
thermal degradation taking place at temperatures below their respective tempera-
tures of glass transition or melting point. This found an experimental and calculat-
ed verification for polymethylidenephthalide (55). Such a peculiarity in polymer
behaviour can be explained by presence of voluminous lateral subsitituents con-
taining polar groups, as distinct from, say, weakly polar polymers whose tempera-
tures of glass transition and melting point are much lower than their tlerma 1 cfegra-
dation temperature. A situation is also possible (as for polyheteroaiylenes) when
the temperatures of glass transition and thermal degradation actually coincide.
504
Summary
A peculiar case takes place when a polymer degradation starts with its decom-
position at end groups. If such a decomposition does not lead to formation of the
same end groups, the value of Td is calculated from chemical structure of a repeat-
ing unit; otherwise the value of Td is calculated only from the structure of end groups.
The consideration of these two cases in analysis of polyformaldehyde thermal
decomposition has shown a good fit between calculated and experimental values of
temperature of onset of intense thermal degradation for such a polymer (Fig.70).
Chapter VIII
Optical and Optico-Mechanical Properties of Polymers
The most important optical property of polymers is their refraction index n,
which is closely connected with the matter dielectric constant e. In the general case
the static dielectric pennittivity originates from orientational, atomic and electronic
polarization. In optical range of frequencies the contribution from orientational and
atomic polarization can be neglected. In this case tire square of refraction index n
will be equal to e provided both values have been determined at an identical fre-
quency. Using the Lorenz-Lorentz“s equation (188), Equations (189) and (190)
were obtained for calculation of the refraction index of polymers and copolymers
respectively on the basis of their chemical structure. Atomic refractions R used in
those relations are specified in Table 27 whereas Table 28 gives calculated and
experimental values of refraction index. As we can see, the relation between mo-
lecular packing index I'and temperature according to Equations (192)—(195) being
taken into consideration increases the accuracy of n calculation for polymers with
low glass transition temperature values (second values of nca/cin Column4, Table 28).
Another important property of polymers in the glassy state is stress-optical
coefficient Cc used in optical polarization method of studying strain state of mate-
rials and products. Sometimes strain-optical coefficient C'£ is used. Those coeffi-
cients are connected by the relation (201).
Empirical and semiempirical approaches to numerical evaluation of stress-op-
tical coefficient Ca based on chemical structure of a polymer repeating unit are
discussed.
Under the empirical approach in relation (202) the influence of chemical struc-
ture parameters of a polymer repeating unit is taken into consideration using the
values of increments C, obtained by calibration and characterizing the contribution
of each atom and each type of intennolecular interaction into a polymer strcss-
optical coefficient (Table 29). Table 30 where the calculated and experimental val-
ues of Co coefficient are given shows that this value is widely varied depending
upon tire polymer chemical structure. According to Equation (202), tire contribu-
tion of separate atomic groups into the value of Ca coefficient has been evaluated.
Summary
505
The results of calculation are given on page 238. As we can see, the largest negative
anisotropy of polarizability is inherent in C = 0 polar group. A similar equation for
copolymers has the form (202').
At the same time an advantage of relation (215) obtained on the basis of semiem-
pirical approach is in determining a connection between stress-optical coefficient
and modulus of elasticity, refraction index and glass transition temperature of poly-
mers. This relationship allows to calculate not only Ca value, but, for instance, the
modulus of elasticity E of a glassy polymer from its chemical structure with an
acceptable degree of accuracy (see Table 32).
The relations obtained allowed to analyze the contribution оf particular a toms
and polar groups into the values of the Ca coefficient and modulus of elasticity E
and to obtain for the dynamic photoelasticity method a polymer possessing an ex-
tremely high modulus of elasticity at glass transition temperature lower than room
temperature. A polymer of that kind based on a carbcfun choral organoslicon pd y-
isocyanurate network lias short chains between cross-linked points as distinct from
the polymers traditionally used in the dynamic photoelasticity method. Traditional
polymers have practically identical values of dynamic modulus of elasticity under
different static values of modulus of elasticity, which precludes their application to
solution of dynamic problems using the dynamic photoelasticity method (Table 33).
Chapter IX
Dielectric Constant of Polymers and Organic Solvents
The calculation of dielectric constant £ for polymers on the basis of their chem-
ical structure is an important task from the point of view of the directed synthesis of
polymers with the preset dielectric constant. On the other hand, it is necessary to
know the values of г for the polymer and organic solvent in order to predict solubil -
ity of the polymer in the solvent.
Expression (222') for calculation of dielectric constant of a substance from its
chemical structure has been obtained on the basis of the Glausius-Mossotti formula
(221).
There are some peculiarities in application of Expression (222') for calculation
of the value of dielectric constant of non-polar, polar dielectrics and organic liq-
uids. For glassy amorphous polymers, the molecular packing coefficient at room
temperature is evaluated to be 0,681, while at the polymer glass transition temper-
ature it is 0,667. For nonpolar dielectrics, i.e, polymers with polar groups absent in
their chemical structures (polyethylene, polybutadiene, polytetrafluorethylene, etc ),
molar polarization P actually coincides with molar refraction P of a polymer re-
peating unit. In this case, as well as for evaluation of molar refraction, the v акте о f
molar polarizability P is additive consisting of polarizabilities of atoms and of polar-
506
Summary
izability increments connected with various types of chemical bonds and other pe-
culiarities of molecular structures.
For polar dielectrics the value of polarization P is larger than the value of re-
fractionfl of a polymer repeating unit due to orientation of constant dipoles emerg-
ing from the presence of polar groups like -OH, -CO-, -COO-, -Cl, -NHCO-,
NHCOO-, -CN, etc. Using the linear regression analysis and Expression (223), as
well as refraction and dielectric constants of a large number of polar polymers,
molar refraction corrections A/? have been obtained for the most frequent polar
groups in polymers (Table 34). These data were used to calculate dielectric con-
stants for a large number of various classes of organic polymers. The results of
calculation fit well with experimental values of e (Table 34a). In some cases, espe-
cially for polymers being in the rubbery state at room temperature, the accuracy of
£ calculation increases the share of the molar packing coefficient dependence upon
temperature k(T). For copolymers the value of £ is calculated according to Expres-
sion (223") or (223"").
For organic liquids the value of the molecular packing coefficient varies in a
wide range (0.45 to 0.705) which makes calculation of density and molar volume
necessary to define £ of the liquid too difficult. At the same time calculation shows
that this factor is not critical for accurate prediction of £ of organic solvents. It has
been shown that the value of molar refraction correction Afl for the same polar
group contained in polymers and in low-molecular liquids must be different. It is
true not only for comparison of behaviour of organic liquids and polymers but also
for comparison of the liquids themselves belonging to the same class. Thus, ii e
contribution into the value of AT? from an OH-group is not identical in the row of
alcohols, it depends upon the chemical structure of an alcohol. In Table 34b for
eleven classes of liquids the relations between AT? and Van-der-Waals volume of
organic solvent molecules are given The application of these relations led to calcu-
late values of the dielectric constant for organic solvents. As we see, a sufficiently
good agreement is observed between calculated and experimental values of £ for
organic liquids, which was not attained before (Table 34a).
Chapter X
Equilibrium Rubbery Modulus for Polymer Networks
To evaluate the equilibrium robbery modulus E'x and the molecular mass of
linear chains between cross-linked points Mc in the case of elastomer networks
with sufficiently rare cross-links the equation of the classic rubber elasticity theory
has been used (224). The application of this equation to liigh cross-linked polymers
causes a substantial divergence between experimental and calculated values of E^,
andA/c. Expression (223) for modulus of elasticity of amorphous polymers as ob-
Summary
507
tained in paper [103] may be used to some degree of approximation only for wide
cross-linked polymers. The explanation lies in the fact that for wide cross-links
Van-der-Waals volume of cross-linked points is umneasurably less than Van-der-
Waals volume of linear fragments between them and, therefore, it may be neglected
at evaluation of compressibility of a cross-linked system That cannot be done in
the case of high cross-linked networks because the total Van-der-Waals volume of
cross-linked points is roughly equal to the total Van-der-Waals volume of linear
fragments and may even exceed it.
Regarding a polymer network, on the one hand, as a system consisting of two
subsys terns — e las tic and rotational- isomeric ones and on the other hand, by pre-
senting a polymer network as a mixture of linear fragments and cross-linked points
(238) we obtain Expression (250) forcaiculation of the equilibrium rubbery modu-
lus for the polymer networks Ec depending on their chemical structure. Transfor-
mation of (250) leads to Expression (259) for determination of the molecular mass
Mcof polymer networks. U sing (250) the modulus of rubber elasticity Ec has been
calculated for a model network on the basis of polydimethylsiloxane, its value in-
creasing with increasing distance between the cross-linked points and reaching
/-^148 MPa at n = 1,in this case glass transition temperature remains lower than
room temperature (Table 36).
Calculation schemes for evaluation of glass transition temperature T& and the
equilibrium rubbery modulus Ex were used to create polymer materials with un-
conventional properties - materials with their modulus of elasticity varying in a
very wide range for particular samples remaining constant for a single sample, and
gradient materials whose moduli of elasticity can vary in a very w ide range along
the length of the same sample without any noticeable boundaries. A theoretical
basis for obtaining such a polymer is Expression (256) w'hich states that the value
of £oo reaches its highest limits during transition to high cross-linked networks
with bulky cross-linked points when n = 1 and fl > 1. To maintain a low glass
transition temperature Tg and to somewhat regulate the value of Еж in a wide range,
the linear fragments between cross-linked points have to be very flexible. As repre-
sentatives of such structures containing rigid bulky cross-linked points connected
by flexible chains -R- polyisocyanurates were synthesized, their chemical struc-
ture being shown on page 251. In this case isocyanuric cycle serves as a cross-
linked point, short organosilicion chains being linear fragments. Calculations ac-
cording to (250) showed (Table 37) that for such a polymer at glass transition tem-
perature under room temperature depending on the length of linear fragments n the
rubbery' modulus varies by almost three orders of magnitude; a high predictive
capability of Expression (250) should be noted.
Gradient materials are obtained by directed regulation of chemical composi-
tion of a network within the same sample with corresponding smooth variation of
508
Summary
modulus of elasticity from values typical for rubbers to values typical for plastics,
without any boundaries.
It is shown taking poly isocyanurates as an example that to determine the prop-
erties of such unconventional materials one should take into consideration the pro-
cess of microphase separation caused by a substantially different surface energy' of
organosilicon chains (21 dyne/cm) and isocyanurate cross-linked points (35 dynes/
cm). The presence of microphase separation process is confirmed by appearance of
two distinct maxima in the curve of the polymer mechanical loss factor versus tem-
perature (Fig.80).
Chapter XI
Description of Relaxation Processes in Polymers
Recently some variants of memory functions in the corresponding Boltzmann -
Volterra equations have been proposed for description of processes of stress relax-
ation and creep in polymers (112). The analysis of the existing memory' functions
shows that they express the course of relaxation processes with a sufficient accura-
cy provided appropriate parameters have been selected, but the physical meaning
of those parameters is not always clear. In this respect more appropriate approach
to obtaining relaxation memory functions seems to be based on consideration of
thermodynamic functions and their variation in the course of relaxation process
[7]. Such an approach is based on presenting the process of strain relaxation as a
result of interaction and diffusion of kinetic units - relaxers, which may be various
atomic groups, repeating units, particular elements of free (empty) volume, i.e.
microcavities, stress concentrators, etc. In this respect a polymeric material can be
treated as consisting ofrelaxers and non-relaxers. Interaction between relaxers and
non-relaxers and their mutual diffusion leads to production of entropy S in the
system which increases during stress relaxation due to transition of relaxers into
non-relaxers and their intermixing caused by mutual diffusion. Memory functions
were obtained issuing from tire assumption that the driving force of relaxation pro-
cess is production of system entropy, which grows during stress relaxation and
reaches its maximum value
Relaxers transform into non-relaxers because of their interaction and diffu-
sion. The description of process of interaction between relaxers is based on a kinet-
ic equation of the n-tli order (268). By integration and subsequent transformations
of this equation a memory function Ti(t) is obtained caused by an irreversible tran-
sition of relaxers into non-relaxers due to interaction between relaxers. If a revers-
ibility of such a transition is assumed, when a non-relaxing material can produce
relaxers in the course of the process, then the kinetic equation (268) takes the form
(286), and a corresponding memory function I\(t) is described by Equation (290).
Summary
509
Under diffusion mechanism of relaxation a random roaming of relaxers is con-
sidered whereas the percentage of non-relaxers is described by relation (278). Sub-
stituting relation (278) for (267) and having made necessary transformations we
can find Expression (289) for determination of a corresponding memory function
T2(t). By substitution of the memory functions obtained into Boltzmann equation
(276)we о btain relations (293)- (295) which describe the stress relaxation in the
poly merfor time t. Numerical values of integrals in these expressions are specified
in Tables 38—40. An experimental test for obtained memory functions conducted
on polymethylmetacrylate, polyoxadiazole, polybenzoxazole and other polymers
showed the efficiency of calculation procedure anda high pre diction capability of
the equations obtained.
The discussed approach to description of relaxation phenomena in polymers
may be applied to such processes as sorption and swelling because they take place
not onlyby filling pores in a polymerbody but also cause a confomidional rear-
rangement of macromolecules, thus, are accompanied by relaxation processes. A
relative amount of soibed matter A/(Z)A/(oo) is determined from Equation (316)
obtained by solution of the diffusion equation (306) in which, in turn, the velocity
of particle diffusing in a polymer body Э is determined according to (310) or (311)
depending on the type of the relaxation mechanism. Equation (310) corresponds to
the interactive mechanism, whereas Equation (311) is used to describe diffusion of
kinetic units In the final form a relative amount of soibed matter is described by
Equation (326) if the sorption process is determined predominantly by interaction
of polymer relaxers, whereas sorption process limited by diffusion of relaxers is
described by Equation (327). The acceptability of Equations (326) and (327) was
confirmed by the experiments devoted to measurement of kinetics during sorption
of free films and coatings on the basis of cured epoxy resin ED-20 (Fig.88). The
best fit between calculated and experimental values is reached by using memory
function T2(t). This shows that the limiting stage of the process serving as a base
for sorption is the process of relaxer diffusion (self-diffusion). The use of memory
function obtained pennits to approximate sorption kinetic curves with a good fit for
almost all of their period which is not possible when applying the Fick equation
with a constant diffusion coefficient for this aim. In this case sorption process can
be approximated with a sufficient accuracy only in a small initial time period.
Chapter XII
Solubility of Polymers
The problem of polymer solubility prediction has remained urgent for many
years. One way of preliminary evaluation of a polymer solubility is a juxtaposition
of the values of Hildebrand solubility parameter 8 as defined by relation (329) for a
510
Summary
polymer 3p and for a solvent 8S. If the values of 3p and S, are in a substantial diver
gence one may firmly predict that this polymer is insoluble in the given organ -
ic liquid. Experimental determination of 5 value is made only for low molecu-
lar liquids which are evaporate without decomposition. For polymers which
cannot be evaporated without decomposition the value of 8p is determined by
indirect methods or by calculation, which is preferable because of difficulties
in realization of experiments. Calculation formula (331) for determination of
solubility parameter 5 for liquids and polymers has been attained allowing for
the type of molecular packing, cohesion energy components AE‘, as typical for
each atom and type of intermolecular interaction of a polymer repeating unit
(Table 43). A similar relation for copolymers is written as Equation (331") or,
if the values of solubility parameters for homopolymers are known or calculat-
ed, as Equation (331""). Table 44 shows calculated and experimental values of
solubility parameters for polymers of diverse chemical structures. As we see,
the value of 3 increases when strong polar groups are present such as -Cl, -
COO—, —CN. The solubility parameter is especially enhanced by hydrogen
bonds emerging when —OH and —HNCO— groups are present as well as aro-
matic rings.
When solubility is predicted for a particular polymer a sufficient number of
polymers remain for which 3p but tire polymer would solve only in nearly a half
of them.
To create a solubility criterion having a sufficiently high predictive capability
the supermolecular structure of the polymer should be accounted for. Such a solu-
bility criterion taking account of the globular supermolecular structure has been
obtained for amorphous polymers, both unswelling (criterion (343)) and swelling
(criterion (345)). Both parts of the solubility criterion are calculated taking into
consideration chemical structures of the polymer and the solvent.
It follows from those equations that a polymer is, as a rule, soluble if the poly-
mer and the solvent have similar values of cohesion energy density, molar volumes
and coefficients of surface tension. The results of an experimental test for solubil-
ity criterion (345) made on approximately 50 organic liquids and near 300 poly mer-
solvent systems are shown in Fig.94. As we see, the share of polymer-solvent sys-
tems not meeting criterion (345) (black dots are lower than the dotted line, whereas
white points are over it) is very small, not exceeding 15 % of the total number of the
systems studied.
Appearance of “foreign points" may be caused by non-swelling of a polymer in
a given solvent (in this case criterion (343) should be used), or non-accounting for
the degree of polymerization of the polymer, or presence of another type of super-
molecular structure. For these cases respective solubility criteria have been ob-
tained which show that the larger a polymer molecular mass is (criterion (359)) the
Summary
511
less soluble is an amorphous polymer with globular structure. The presence of a
fibrillary supermolecular structure (criterion (371)) decreases the solubility of a
polymer having an identical chemical structure but different in the presence of a
globular supermolecular structure.
Chapter XIII
Surface Properties of Organic Liquids and Polymers
The value of surface tension у of liquids and polymers determines many of
their individual properties, among them mutu alcompa tih i'tyan dmu U al solubility
Among the existing methods of surface tension cd cd afionth e one most cften used
is the additive calculation scheme from Expression (372) based on summation of
parachors P, characterizing the contribution of individual atoms into surface ten-
sion. Equation (372) pennits to calculate the value of у to a sufficient accuracy but
due to physical uncertainty of the P, values it does not allow to analyze the influ-
ence of weak Van-dcr-Whals interaction and polar groups upon formation of one or
another surface property of organic liquids and polymers. At the same time perfor-
mance of such a quantitative analysis would permit to regulate the chemical struc-
ture of a substance in order to impart necessary surface properties to it
F or cal cul at ion of surface tension of organic liquids Expression (382) was ob-
tained which accounts for chemical structure of a liquid and, thus, allowing to eval-
uate contn bu ion о fpa rtc ular polar groups and specific intermolecular interaction
into the val ue of у,. F or example ,the contribution of hydrogen bonds into the value
of yi for alcohols and acids is as great as 72 %, whereas for lormamide it equals
only 33 %.
A similar expression for calculation of surface tension for polymers у[ has the
form (389). A disadvantage of those equations is a necessity to take into consider-
ation a particular material belonging to a certain group of substances depending on
the presence of hydrocarbon, ester, alcohol, acid, amide, nitrile etc. groups It is
taken into account by different meanings of ^inequation (382) on calculation of yi
for liquids and of Qin equation (389)on calculation of yp for polymers. These
drawbacks are absent in formulas obtained fordeterminationof surface tension of
liquids and polymers issuing from their chemical structure only. The value of sur-
face tension for organic liquids is calculated according to Equation (396), whereas
for polar and non-polar polymers according to Equations (399) and (400) respec-
tively. These formulas possess a high predictive capability (see Table 46 for liq-
uids, Table 47 for polymers) and as distinct from Equation (372) allow to connect
the value of surface tension to the solubH typarame pro f the substance or to cohe-
sion energy density (402). For copolymers the suface tension is better calculated
according to Expression (411") via surface tensions of homopolymers.
512
Summary
Chapter XIV
Miscibility of Polymers
The problem of miscibility is one of the most important for polymers. That is
due to the fact that new polymer materials are developed, as a rule, not by way of
syndtesis of new polymers but mostly by mixing of the existing ones. In the course
of this not only mixed compositions are created where the components are present
in comparable quantities, but also microadditions of polymers are introduced, sur-
face of components is modified, etc. Under such circumstances new problems arise,
such as microphase separation, skill of control of microphase composition and di -
mensions, etc.
One of possible approaches to the problem of miscibility prediction of poly-
mers is based on the idea of “solubility” of one polymer in another and application
of Equation (345) for obtaining the miscibility criterion of polymers aimed at eval-
uation of polymer solubility. As a result of analysis performed for the case of total
incompatibility between polymers when one homopolymer does not “solve” in an-
other, and the second one does not solve in the first, that criterion has the form
(412) and (413). Under partial miscibility when one homopoly mer does not “solve”
in another, but the second one does “solve” in the first, or vice versa, the miscibility
criterion is given by inequalities (417) and (418). Under absolute miscibility of
polymers when the first and the second polymers solve in one another the corre-
sponding criterion lias the form (419) and (420). Taking as examples systems with
partial (one polymer is polystyrene, another is polyvinylmethyl ether) and absolute
(one polymer is polystyrene, another is poly-2, 6-dimethyl-l,4-phenyleneoxide)
miscibility a high predictive capability of the criterion obtained lias been shown in
the whole range of ratios between mixture components (Figs.99,101). Il should be
noted that the discussed approach has an advantage over several other approaches
in that it does not require an introduction of “fitting” parameters, but only a knowl-
edge of chemical structure of the components.
Chapter XV
Influence of End Groups upon Properties of Polymers
Much attention is paid to evaluation of the role of a polymer molecular mass
and the role of end groups connected to it in formation of polymer properties. This
problem is especially important under analysis of action of the molecular mass
upon the properties of growing chains when the number n of links in a molecular
chain is small. The proposed relations between polymer properties and its molecu-
lar mass are, as a rule, of empirical nature and do not permit to determine the action
of particular atomic groups, type of intennolecular interaction and relative volume
Summary
of end groups upon tire polymer properties. Such an evaluation performed for four
polymers - poly methylmetacry late, polystyrene, polyethyleneterephthalate and poly-
carbonate - allowed to find out some important regularities in variation of Van-der-
Waals volume, molar refraction, molar cohesion energy, solubility parameter, sur-
face tension and heat capacity of polymers depending on the number of repeating
units in a polymer chain, beginning with n = 1. The analysis of the calculations
made (their results being cited in Table 49 and Figs. 101-104) shows that the ap-
proximation to asymptotic values of polymer properties under study does depend
on the type of end group and on the average is reached at the number of repeating
units in a polymer chain n = 10-20. At the same time the action of chemical
structure of end groups upon molecular characteristics and properties of poly-
mers can prove to be significant and quite different. Thus, the presence of
bulky end groups first of all affects Van-der-Waals volume of an “averaged”
unit, molar refraction and heat capacity of a polymer. At the same time the
presence of some groups (hydroxyl < у pd smdl tn vd ume, b it possessing a
specific intermo lecular interaction cn'tica Ik te Ik upon mo hr co lesion energy,
surface tension and solubility parameter, but is insignificantf or Van-der-Waal s
volume and molar refraction. Therefore, it has been shown how important the
chemical 4 ructure of end groups is in stud у of regd ard epend ence cf pd ymer
properties upon their molecular mass.
Chapter XVI
Thermo-physical Properties of Polymers
Thermo-physical properties of polymers are their heat capacity, temperature
cord udi vity’ and heat conductivity.
In a relatively wide temperature range heat capacity grows linearly with an
increase in temperature. During transition from a glassy state to a rubbery state heat
capacity jumps to al arger vd ue. Af ter such a ph ysical transition a weak increase of
heat capacity begins again together with growing temperature.
Heat capacity of polymers depends upon their chemical structure (Table 50).
Thus, when hydrogen atoms are substituted for polar groups, their heat capacity
increases. A substantial growth of heat capacity is observed in transition from ali-
phatic to aromatic polymers. Formulas for calculation of polymer heat capacity on
the basis of their chemical structure have been obtained from tire assumption that
contribution of each atom of a polymer repeating unit into heat capacity is propor-
tional to its Van-der-Waals volume. Molar heat capacity under the constant pressure
Cp for a polymer in a glassy state Cs is calculated according to Equation (429), and
for a polymer in a robbery state Cp according to Equation (430). The accuracy of
molar heat capacity calculation Cp and Cp can be seen from Tabic 50 .
514
Summary
Temperature conductivity a is determined from relation (431) and serves as a
characteristic of temperature propagation in a substance under the action of thermal
flow at unstationary thermal conditions. For solid (glassy and crystalline) polymers
temperature conductivity weakly decreases with the growing temperature. Howev-
er, when a polymer transits from its glassy state into a rubbery state, a sharp fall in
temperahire conductivity is observed. Temperature conductivity depends upon the
degree of crystallicity, molecular mass, pressure and, as it follows from Table 52,
chemical structure of polymers.
Heat conductivity X characterizes the capacity of polymer bodies to transfer
heat from more heated elements of the body to less heated. Heat conductivity of
polymers depends on their physical and phase states as well as on chemical struc-
ture of polymers (Table 53). Within the same physical state the action ofthe chem-
ical structure of a polymer upon its heat conductivity versus temperature may be
different. For one series of polymers the slope of this curve is positive^ whereas for
another series it is negative. A weak maximum of heat conductivity is observed in
the area of physical transition from the glassy state to the rubbery one.
Chapter XVII
Molecular Design and Computer Synthesis of Polymers with
Predetermined Properties
In the preceding chapters of this monograph it has been demonstrated that the
basic physical properties of polymers can be calculated issuing from chemic als Imc-
ture of repeating unit in a linear polymer or a repeating fragment of a polymer
network. Nowadays such calculations are made on computers. In that way it is
possible to solve principal and inverse problems, which are the basic ones, and
some auxiliary problems.
The principal problem is to calculate properties of a polymer from the data of
chemical structure of a polymer repeating unit ora fragment of a polymer network.
The most complicated inverse problem is to predict chemical structure of a
repeating unit or network fragment of a polymer that would meet the required set of
properties.
Solution of such problems by a simple exhaustion of a large number of polymers
and their properties contained in a database obtained by increment methods, other
calculation method or experimentally does not have any predictive capacity for
structures which are absent in the computer memory. For efficient solution of the
principal and inverse problems in computer-aided synthesis of polymers the frag-
ment method has been proposed; then a computer designs a repeating unit of the
polymer of those fragments. A part of the 194 fragments calculated in [ 17] is shown
in Table 54, the possibility of their mutual connection being given by a matrix on
Summary
515
page 400. This approach was later improved in the basic fragment method when
computer-aided synthesis of polymers is performed from smaller basic fragments
than tire above-discussed fragments which cannot be “comminuted” in principle.
Tire list of basic fragments for computer-aided synthesis of polymers is given in
Table 55, the matrix of their combinations along 20 different end marks being shown
on page 401. In Fig. 105 a combinatory tree is presented illustrating the design of a
recurrent polymer repeating unit of three basic fragments with different end marks.
A computer program based on the use of tiny basic fragments permits to calculate
and “synthesize” polymers of the following classes: 1. Polyolefins, vinyl polymers,
etc. 2. Polyethers and polyetherketones. 3. Polyesters. 4. Polyamides. 5. Polysul-
fones, polyethersulfones. 6. Polyimides. 7. Polvoxadiazoles. 8. Polybenzimidazoles.
9. Polysulphides. 10. Organosilicon polymers. 11. Polycarbonates. If necessary the
number of polymer classes can be enlarged. For this purpose it is necessary’ to
introduce the group which defines belonging of polymers to a selected class into
the database. For instance, urethane group being introduced into the database al-
lows to calculate properties of pd yurdh anes and t о perf orm comprt er-ad d syn-
thesis of polyurethanes with the presetproperties.
The capacities of the chemical fragmert method, are, nevertheless, limited to
solution of the above-mentioned problems only for polymers whose basic frag-
ments are contained in a computer database. This disadvantage is eliminated in the
method of structural formulas of chemical compounds, when chemical structure of
a polymer is “composed” not of blank fragments, but of individual atoms. In this
case chemical structure of a repeating unit is composed on the display screen in the
form of a structural formula of a chemical compound, just like a chemist draws it on
paper. This allows to perform molecular design of a polymer, varying its chemical
structure, introducing various atoms and atomic groups and to obtain physical char-
acteristics of a substance being synthesized from the very beginning.
This program allows to calculate the following properties of polymers, copol-
ymers and polymer networks:
1. Molar volume. 2. Temperature dependence of molar volume. 3. Van-der-
Waals volume. 4. Density. 5. Temperature dependence of density. 6. Variation of
volume in a given temperature range. 7. Coefficient of thermal expansion in glassy
state. 8. Coefficient of thermal expansion in rubbery state. 9. Glass transition tem-
pcrature. 10. Melting point. 11. Temperature of onset of intense thermal degrada-
tion. 12. Solubility parameter. 13. Oxygen permcabili ly. 14. Nitrogen permeability.
15. CO2-permeabilit5'. 16. Cohesion energy. 17. Share of dispersion interaction in
cohesion energy. 18. Ratio of dipole-dipole interaction in cohesion energy. 19. Ratio of
H-bonds in cohesion energy. 20. Surface tension. 21. Interface tension at boundary
between polymer and solvent. 22. Interface tension at boundary between polymer-1
and polymcr-2.23. Prediction of solubility of polymers in organic solvents .24 .Pre -
516
Summary
diction of compatibility between polymers. 25. Molar heat capacity in glassy state.
26. Molar heat capacity in rubbery state. 27. Specific heat capacity in glassy state.
28. Specific heat capacity in rubbery' state. 29. Jump of heat capacity at transition of
polymer from glassy state to rubbery state. 30. Refractive index. 31. Temperature de-
pendence of refractive index. 32. Molar refraction. 33. Stress-optical coefficient. 34.
Energy of intermolecular interaction. 35. Energy of dispersion interaction. 36. Energy
of dipole-dipole interaction and H-bonds. 37. Ratio of dispersion interaction in total
energy of intermolecular interaction. 38. Ratio of dipole-dipole interaction and H-
bonds in total energy of intermolecular interaction. 39. Activation energy of gam-
ma-transition. 40. Dielectric constant. 41. Polarizability. 42. Degree of polymeriza-
tion for rubbery state occurrence. 43. Molecular mass of polymer for rubbery state
occurrence. 44. Rubber elasticity modulus infield of plateauforlinearpdymers.
45. Equilibrium modulus of rubber elasticity for polymer networks. 46. Tempera-
ture of transition of polymer in viscous-flow state versus molecular mass. 47. Sol-
ubility of polymer versus molecular mass. 48. Solubility of polymer versus degree
of orientation. 49. Calculation of properties of copolymers depending on their com-
position. 50. Properties of polymers versus degree of their crystallinity.
For low molecular liquids the program envisages calculator) cf thefdl owing
properties:
1. Molecular mass. 2. Molar volume. 3. Van-der-Waals volume. 4. Density. 5.
Solubility parameter . 6. Dielectric constant. 7. Surface tension. 8. Refractive in-
dex. 9. Molar heat capacity in glassy state. 10. Molar heat capacity in liquid state.
11. Specific heat capacity in glassy state. 12. Specific heat capacity in liquid state.
13. Molar refraction. 14. Molar polarizability 15. Cohesion energy. 16. Ratio of
dipole-dipole interaction in cohesion energy. 17. Ratio of dispersion interaction in
cohesion energy. 18. Ratio of H-bonds in cohesion energy.
Examples of solution of the principal, inverse and mixed problems in comput-
er-aided polymer synthesis on the basis of the above-discussed programs can be
found in Appendices 1 and 3.
ЛИТЕРАТУРА
1. Александров А.П. Морозостойкость высокомолекулярных соединений //Тр.
1 и II конф, по высокомолекулярным соединениям. М.-Л.: Изд АН СССР
1945. С.49-59.
2. Анулов О.В., Барышникова Г.В., Шиманова Н.И.,Местечкина ИМ, Щербу-
хин В.Д. Глюкоманнан корней интродуцированного на Урале Eremurus
fuscus //Прикл. биохим. и микробиология. 1995. Т.31. №1. С.87-91.
3. Арифов П.У., Тишин С.А., Шевченко А.В., Тишин В.А. Наблюде-
ние молекулярного движения в полимерах методом аннигиляции
позитронов//Высокомолек. соединения. 1987. Б29. №1. С.59-63.
4. Аскадский А А. Физико-химия полиарилатов М.:Химия. 1968. 214 с.
5. Аскадский АА. Деформация полимеров. М.:Химия. 1973. 448 с.
6. Аскадский АА. Структура и свойства теплостойких полимеров. М.:Химня.
1981. 320 с.
7. Аскадский АА. Новые возможные типы ядер релаксации//Механ. композит,
материалов. 1987. №3. С.403-409.
8. Аскадский АА. Учет роли концевых групп при анализе свойств полиме-
ров//Высокомолек. соединения. 1989. А31. №10. С.2141-2148.
9. Аскадский А А. Компьютерный синтез полимеров с заданными свойства-
ми//Механ. композит, материалов. 1990. № 6. С.963-977.
10. Аскадский АА., Арсламбеков В.А., Андрющенко ТА., Матвеев Ю.И.,
Блюменфельд А.Л. Оценка кинетических параметров сорбции и
набухания полимеров с учетом релаксационного механизма данных
процессов//Высокомолек. соединения. 1989. А31. №8. С.1616-1623.
11. Аскадский АА., Банявичюс Р.Б., Вихаускас З.С., Марма А.И., Блюмен-
фелъд А.Л. Аппроксимация кривых релаксации напряжения для
полиоксадиазола и полиимида с помощью новых ядер релаксации //
Высокомолек. соединения. 1988. АЗО. №8. С.1684-1689.
518
Литература
12. Аскадский АА., Белошенко В А., Бычко КА., Шапошникова В.В., Само-
рядов А.В., Коврига О.В., Салазкин С.Н., Сергеев В А., Слободина ВГ.,
Генин Я.В. Гидростатическая экструзия ароматических поликетонов И
Высокомолек. соединения. 1994. А36. №7. С.1143-1147.
13. Аскадский А.А., Блюменфельд А.Л., Гальперн Е.Г., Чистяков А.Л.
Интегралы от функций влияния, учитывающих изменение энтропии в
процессе релаксации напряжения, и анализ с их помощью эксперимен-
тальных данных//Высокомолек. соединения. 1988. АЗО. №4. С.886-894.
14. Аскадский А.А., Бычко К.А., Коврига О.В., Цуцуран С.В., Шлейфман Р.Б.,
Курашев В.В. Механические релаксационные свойства поликапроамида,
полученного анионной полимеризацией е-капролактама в присутствии
трехмерных полифункциональных активаторов // Высокомолек.
соединения. 1991. АЗЗ. №11. С. 2477-2486.
15. Аскадский А А., Валецкий М.П. Аппроксимация релаксационных кривых
в нелинейной области механического поведения с помощью новых ядер
релаксации И Механ. композит, метериалов. 1990. №3. С.441-446.
16. Аскадский А А., Воинцева И.И. Парные полимеры // Высокомолек. соеди-
нения. 1987. А29. №12. С.2654-2669.
17. Аскадский А.А., Гальперн Е.Г., Матвеева Т.П., Чистяков А.Л., Слоним-
ский Г.Л. Поиск полимеров с заданными физико-химическими
свойствами с помощью ЭВМ // Высокомолек. соединения. 1987. А29.
№11. С.2433-2440.
18. Аскадский А.А.,Гальперн ЕГ., Суров Г.В.,Булатов В.В., Слонимский ГЛ.,
Коршак В.В. Возможности сочетания различных физических свойств
полимеров // Высокомолек. соединения. 1989. БЗ1. №4. С.308-317.
19. Аскадский А А., Голенева Л.М., Бычко КА. Градиентные разномодульные
полимерные материалы//Высокомолек. соединнения. 1995. А37. №5.
С, 829-841.
20. Аскадский А А., Казанцева В.В., Мельник О А., Бычко КА., Сахарова А А.,
Слонимский Г.Л., Фрунзе ТМ. Определение числа разветвлений и дру-
гих дефектов в случае частосетчатой структуры полимера //
Высокомолек. соединения. 1988. АЗО. №6. С.1285-1293.
21. Аскадский А А., Китайгородский А.И., Слонимский Г.Л. О механических
зацеплениях полимерных цепей//Высокомолек. соединения. 1975. А17.
№10. С.2293-2300.
22. Аскадский А.А., Клинских А.Ф. ЭВМ-программа для расчета свойств
полимеров и их растворителей И Пластические массы. 1998. №4.
С.29-33.
23. Аскадский А А., Клинских А. Ф. Компьютерный дизайн полимеров и метод
атомных инкрементов//Высокомолек. соединения. 1999. А41.№1. С.83-85.
519
Литература
24. Аскадский АЛ., Коврига О.В. О влиянии разветвлений на физические
характеристики полимеров И Высокомолек. соединения. 1991. АЗЗ. №9.
С.1945-1955.
25. Аскадский А А., Колмакова Л.К., ТагерА. А., Слонимский Г.Л.,Коршак В.В.
Об оценке плотности энергии когезии низкомолекулярных жидкостей и
полимеров//Высокомолек. соединения. 1977. А19. №5. С.1004-1013.
26. Аскадский АА., Кондращенко В.И. О структуре отвержденных фенол -
формальдегидных смол//Высокомолек. соединения. 1997. А39. №10.
С.1625-1634.
27. Аскадский А.А., Литвинов ВМ., Жданов АА., Слонимский Г.Л. Анализ
структуры модельных полидиметилсилоксановых сеток // Высокомолек.
соединения. 1985. А27. №11. С.2408-2414.
28. Аскадский А.А., Матвеев Ю.И. Химическое строение и физические
свойства полимеров. М.:Химия. 1983. 248с.
29. Аскадский А А., Матвеев Ю.И., Слонимский Г.Л., Коршак В.В. Влияние
энергии межмолекулярного взаимодействия различных типов связей
на температуру плавления полимеров // Докл. АН СССР. 1978. Т.238.
№3.С.592-595.
30. Аскадский АА., Матвеев Ю.И., Пастухов А.В., Розенберг БА., Понома-
рева Т.П., Щеголевская НА.,Маршалкович А.С. О расчете температуры
стеклования сетчатых полимеров и определении молекулярной массы
фрагмента цепи между узлами сетки // Высокомолек соединения .1983.
А25. №1. С.56-71.
31. Аскадский А А., Матвеев Ю.И., Матвеева Т.П. Обобщенное уравнение
для оценки равновесного модуля высокоэластичности и величины Мс,
действующее для редких и частых сеток // Высокомолек. соединения.
1988. АЗО. №12. С.2542-2550.
32. Аскадский А А., Матвеев Ю.И., Матевосян М.С. О предсказании раство-
римости полимеров // Высокомолек. соединения. 1990. А32. №10.
С.2157-2166.
М.Аскадский А.А., Матевосян М.С., Слонимский Г.Л. Деп. ВИНИТИ.
№6949-В.Деп. ИНЭОС 2 октября 1985г. М. 1985 26с.
34. Аскадский А.А., Матевосян М.С., Слонимский Г.Л. Расчетная схема для
определения поверхностного натяжения органических жидкостей и
полимеров исходя из их химического строения // Высокомолек.
соединения. 1987. А29. №4. С.753-760.
35. Аскадский А А., Пастухов А.В., Маршалкович А.С. Прогнозирование не-
которых физических характеристик и получение оптически чувстви-
тельных эпоксидных полимеров//Высокомолек. соединения. 1984. А26.
№1. С.160-171.
520 Литература
36. Аскадский А А., Прозорова С.Н., Слонимский Г.Л. Оптико-механические
свойства ароматических теплостойких полимеров // Высокомолек
соединения. 1976. А18. №3. С.636-647.
37. Аскадский А А., Размадзе Т.Р. Универсальная расчетная схема для оценки
поверхностного натяжения органических жидкостей и полимеров //
Высокомолек. соединения. 1991. АЗЗ. №5. С.1141-1148.
38. Аскадский А А., Размадзе Т.Р., Салазкин С.Н., Сергеев В А., Саморядов А.В.,
Бычко КА., Коврига О.В., Бабчиницер Т.М., Варада Раджулу А.
Механические свойства материалов на основе поликетона и его смесей
с полисульфоном//Высокомолек. соединения. 1991. АЗЗ. №6. С. 1239-1250.
39. Аскадский АА., Сахарова АА., Мельник О А., Казанцева В.В. Структура
и свойства многокомпонентных сополимеров на основе ряда акрилатов
и метакрилатов//Высокомолек. соединения. 1997. А39. №7. С. 1160-1165.
40. Аскадский А А., Слонимский Г.Л. Универсальная расчетная схема для
определения температуры стеклования полимеров // Высокомолек.
соединения^ 1971. А13. №8. С.1917-1919.
41. Аскадский АА., Слонимский Г.Л., Китайгородский А.И. Об изменении
плотности упаковки макромолекул при физических превращениях в
полимерах//Высокомолек. соединения. А16. №2. С.424^130.
42. Аскадский А А., Слонимский ГЛ., Матвеев Ю.И., Коршак В.В. Упаковка
макромолекул в полимерах и температура стеклования // Докл. АН СССР.
1975. Т.224. №3. С.612-615.
43. Аскадский АА., Слонимский Г.Л., Матвеев Ю.И., Коршак В.В. Упаковка
макромолекул и температура стеклования полимеров // Высокомолек.
соединения. 1976. А18. №9. С.2067-2074.
44. Аскадский А А., Суров Г.В., Немчинов В.В., Блюменфег1ьдА.Л.,Вихаускас З.С.
Ядро релаксации, учитывающее обратимый характер взаимодействия
релаксаторов//Высокомолек. соединения. 1989. АЗ 1. №6. С. 1320-1327.
45. Аскадский АА., Суров Г.В., Панкратов В.А., Френкель Ц.М.,Жданов А.А.,
МакароваЛ.И,,Маршалкович А.С., Радченко Л.Г. Механические свой-
ства разномодульных полимерных стекол И Высокомолек. соединения.
1990. А32. №7. С.1517-1527.
46. Аскадский АА., СуровГ.В., Панкратов ВА., Френкель Ц.М., Макаров Л.И.,
Жданов АА., Благодатских П.В., Пастухов А.В. Синтез и свойства
карбофункциональных кремнийорганических диолов, макроди-
изоцианатов и сетчатых полимеров на их основе // Высокомолек.
соединения. 1990. А32. №7. С. 1528-1534.
47. Аскадский А.А., Суров Г.В., Маршалкович А.С., Двалишвили В.В.,
Панкратов В.А., Френкель Ц.М. Новые разномодульные оптически
чувствительные полимерные материалы для метода динамической
фотоупругости И Механ. композит, материалов .1991. №.1. С .3 -8.
Литература
521
48. Аскадский А.А., Тишин С А., Казанцева В.В., Коврига О.В. О механизме
деформации теплостойких ароматических полимеров на примере
полиимида//Высокомолек. соединения. 1990. А32. №12. С.2437-2445.
49. Аскадский А А., Тишин С А., ЦаповецкийЛГП., Казанцева В.В., Коврига О.В.,
Тишин В А. Комплексный анализ механизма деформационных и
ралаксационных процессов в полиимиде // Высокомолек. соединения.
1992. А34. №12. С62-72.
50. Бараш Ю.С. О макроскопическом описании действующего поля в неко-
торых диэлектриках // Жури, экспер. и теор. физики. 1980. Т.79. №6.
С.2271—2281.
51. Бартенев ГАЛ., Зеленев Ю.В. Физика и механика полимеров. М.:Высш.
школа. 1983. 391с.
52. Бартенев Г.ЛЛ. Френкель CJJ. Физика полимеров. И Под ред. д.ф.-м.н.
А.М.Ельяшевича. Л.:Химия. 1990. 432с.
53. Бартенев ГАЛ., Бартенева А.Г. Релаксационные свойства полимеров.
М.:Химия. 1992. 383с.
54. Варисов А.З., Кузнецов Ю.Н., Прокопьев Е.П., Филипьев А.П. Физика и
химия превращений позитронов и позитрония в полимерах // Успехи
химии. 1981. Т.50. №10. С-1892-1923 .
55. Виноградова С.В., Салазкин С.Н., Челидзе Г.Ш., Слонимский Г.Л., Аскад-
ский А.А., Бычко К.А., Комарова Л.И., Журавлев И.В., Коршак В.В.
Сополимеры метилиденфталида//Пластич. массы. 1971. №8. С.10-13.
56. Виноградова С.В., Панкратов В.А., Френкель ЦАЛ., Ларина Л.Ф., Кома-
рова Л.И., Коршак В.В. Исследование полициклотримеризации
изоцианатов//Высокомолек. соединения. 1981. А23. №6. С. 1238-1243.
57. Волькенштейн М.В. Молекулярная оптика. М.-Л. :Гос. изд. техн, итеорет. лит.
1951. 744с.
58. Волькенштейн А-1.В. Конфигурационная статистика полимерных цепей.
М.-Л.Изд-воАкад. наукСССР, Ленингр. отд-ние. 1959. 466с.
59. Вундерлих Б., Баур Г. Теплоемкость линейных полимеров. / Пер. с англ, и
нем. канд. хим. наукЮ.К.Годовского/М.:Мир. 1972. 238с.
60. Гемицсдлюпозы/Подред. В. С Громова и М. С. Дудкина Рига:3инатне. 1991.488с.
61. Годовский Ю.К. Тепло физические методы исследования полимеров.
М.:Химия. 1976. 216с.
Ы.ГодовскийЮ К. Теплофизика полимеров. М.:Химия. 1982. 280с.
63. Гуль В.Е. Роль молекулярных сил в механизме набухания высокополимеров.
II. Кинетика набухания//Коллоид, журнал. 1953. Т. 15. №3. С. 170-177.
64. Гурвич Л.В., Караченцев Г.В., Кондратьев В.Н., Лебедев Ю.А., Медве-
дев В А., Потапов В.К.,Ходеев Ю.С. Энергии разрыва химических свя-
зей. Потенциалы ионизации и сродство к электрону. М:На/ка .1974 351с.
522________________________Литература_____________________________
65. Гуревич Г.И. О законе деформации твердых и жидких тел // Жури. техн.
,, 4 физики. 4947, Т.17. №12 CJ491-15U2. Л v 1О-.
66. Пашевскии Вл. Конформации органических молекул. М..Химия. 1974.432с.
67. Догадкин БА., Гуль BE. Изменение релаксационных свойств при набух-
^нии^ву^канизированного каучука И Докл. АН СССР. 1950. т.70. №6.
68. ДубининЛМ. Поверхность и пористость адсорбентов // В кн: Основные
проблемы теории физической адсорбции. Труды Первой Всесоюз. конф,
по теор. вопросам адсорбции, М.:Наука. 1970. С.251-269.
69. Ениколопов Н.С., Вольфсон С А. Химия и технология полиформальдегида.
М.:Химия. 1368. 279с.
70. Зедгинидзе И.Г. Планирование эксперимента для исследования многокомпо-
нентных систем. М.:Наука. 1976. 390с.
71. Зубов П.И. Механизм образования белковых студней: Дисс дхк М :НИХФИ
им. Л.Я.Карпова. 1949. 174с.
72. Зубов П.И.,Матвеев Ю.И., Аскадский АА., Андрющенко ТА. К вопросу о
набухании полимеров в парах низкомолекулярных жидкостей на при-
73. Ильюшин АА.Мо^Юря Б^£)с^ры математической теории термовязко-
74. КаЗанОвЧ^1 к вопросу о кинетике релаксационных процессов в реаль-
ных полимерных телах // Докл. АН СССР. 1970. Т. 195. №2.С.402-405.
75. Китайгородский А.И. Органическая кристаллохимия. М:Изц-воАНСССР
1955. 558с.
76. Клименков В.С., Каргин В А., Китайгородский А.И. Оплотностяхупаковок
высокополимерных соединений И Журн. физ. химии. 1953. Т.27. №8.
77. Корн^сльР Упругость и прочность жидкостей. М.-Л.:Гос. изд. техн-
теорет. лит. 1951. 108с.
78. Коршак В.В. Разнозвенность полимеров. М.Наука. 1977. 301с.
79. Коршак В. В-, Виноградова. С.В.^Слонимский Г.Л..Панкратов В.А
Аскадский А А., Френкель ЦМ, Ларина ЛЕФ., Бычко КА. Исследование
свойств полиизоциануратов, полученных полициклотримеризацией
ароматических и алифатических диизоцианатов // Высокомолек.
соединения. 1981. А23. №6. С.1244-1251.
80. Коршак В.В., Аскадский АА. О количестве возможных хими-
ческих полимерных индивидов, возникающих под влиянием
разнозвенности полимеров И Докл. АН СССР. 1986. Т.289. №6.
С.1412-1416.
Литература
523
81. Коршак В.В., Слонимский Г.Л., Аскадский А А., Русанов А.Л.. Лекае Т.В..
Матевосян М-С Исследование механическихсвойств пленок сополиме-
ров регулярного строения’ содержащих фенилхиноксалиновые и
нафтоилен-бензимидазольные фрагменты//Высокомолек. соединения.
1989. АЗ 1 №1. С.34-39.
82. Красовский ВТ., Садовский НА., Горбацевич О.Б., Музафаров AM.,
МЯкушев ВД-, Ильина МН-, Дубовик ИИ-, Стрелкова Т-В-, Пап-
ков В.С. Полиаллилкарбосилановые дендримеры с флуоресцентной
„ Т. меткой//Высокомолек. соединения. 1994-А36.№4. С-714-720.
83. Кронгауз Е.С., Беломоина НМ., Аскадский А А., Казанцева В.В., Бычка КА.,
Слонимский Г.Л., Коршак В.В. Сравнительное изучение свойств
полиимидофенилхиноксалинов и полиамидофенилхиноксалинов //
Высокомолек. соединения. 1986. А28. №7. С .1473-1478.
84. Кузьминский А.С., Боркова Л В. Вулканизация и механические свойства
эбонитов // Журн. физ. химии. 1958. Т.32. №9. С.2051-2060.
85. Ландау ЛД.,Лифшцц ЕМ. Теоретическая физика. Т.8. Электродинамика
сплошных сред. / Изд. 2-е, испр и доп. ЕМЛифшицеми Л.ППитаевским.
М Нж.кя. 1982. 623ct „
86. Ландау лД., Лифшиц ЕМ. Теоретическая физика. Т.7. Теория упру-
о_ . . гости • / Изд • 4-е ’И$ПР • и доп Наука • 1987 • 246с •
87. Матвеев Ю.ТГ., Аскаоскии лл. О появлении дискретных спектров времен
релаксации//Высокомолек. соединения. 1981. А23. №6. С.1347-1357.
88. Матвеев Ю. И., Аскадский А. А., Журавлева И. В., Слонимский Г.Л..
Коршак В.В. О влиянии химического строения полимеров на их
термостойкость //Высокомолек. соединения. 1981. А23. №9. С.2013-2026.
89. Матвеев Ю.И., Аскадский А.А. Спектры времен релаксации полимеров в
области малых деформаций // Высокомолек. соединения. 1982 . А24 .
90. Матвее Аскадский АА. Влияние скорости изменения температуры
и деформации на процесс стеклования полимеров // Высокомолек. со-
единения . 1985. А27. №11. С .2357-2362.
91. Матвеева Т.П., Матвеев Ю.И., Аскадский А.А. Определение упругих
характеристик полимеров исходя из химического строения повторя-
ющегося звена И Механ. композит, материалов. 1986. №2. С.201-206.
92. Матвеев Ю.И., Аскадский АА. Об образовании надмолекулярных структур
в аморфных полимерах И Высокомолек. соединения. 1986. А28. №7.
С.136э-1372.
93. Матвеев Ю.И., Аскадский АА. Фазовое состояние полимеров как след-
ствие образования надмолекулярной структуры однополостными гипер-
болоидами связи//Высокомолек. соединения. 1989. А31.№3. С617-622.
524 Литература
94. Матвеев Ю.И., Аскадский А А. Расчетный способ оценки[размеров эле-
ментов надмолекулярной структуры полимеров // Высокомолек.
соединения. 1989. A3I. №3. С.526-532.
95. Матвеев Ю.И., Аскадский А А. Влияние физических характеристик и типа
надмолекулярной структуры полимера на его растворимость //
Высокомолек. соединения. 1994.А36. №3. С.436-443.
96. Матвеев Ю.И., Аскадский АА. Определение температуры перехода в
вязкотекучее состояние полимеров // Высокомолек. соединения. 1993.
А35. №1. С.63-67.
97. Матевосян М.С., Аскадский А А., Слонимский Г Л. Механические релакса-
ционные свойства пленок полимеров в зависимости от предыстории их
получения из растворов // Высокомолек. соединения. 1987. А29. №4.
С.761-767.
98. Музафаров AM., Ребров ЕА., Папков В.С. Объемнорастущие поли-
органосилоксаны. Возможности молекулярного конструирования в
высокофункциональных системах // Успехи химии. 1991 т.60. №7.
С.1596-1612.
99. Николаев А.Ф. Синтетические полимеры и пластические массы на их
основе. / Изд. 2-е, испр. и доп. М.-Л.:Химия. Ленингр. отд-ние. 1966.768с.
100. НоркинаР.С., Соколов С.И., Щеголевская НА. О полимерных материалах
для поляризационно-оптического метода определения напряжений- IV
Использование объемной контракции и рефракции при изучении
кинетики реакции в структурированных системах //Изв. Вузов. Химия
и хим. технология. 1964. Т.7. №4. С.645-650.
101. Панкратов В А., Аскадский А А., Пацурия ММ., Кочергин Ю.С., Виногра-
дова С.В., Слонимский Г.Л., Коршак В.В. Исследование структуры и
физико-механических свойств сшитых полимеров на основе
олигомерных дицианатов // Высокомолек. соединения. 1977. А19. №1.
С. 142-148.
102. Слонимский Г Л., Коршак В.В., Виноградова С В.,Китайгородский А И,
Аскадский А.А., Салазкин С.Н., Белавцева Е.М. Физико-химичес-
кие пути регулирования надмолекулярных структур и механических
свойств аморфных полиарилатов на основе фенолфталеина и его
производных//Высокомолек. соединения. 1967. А9. №2. С.402-408.
103. Слонимский Г.Л., Коршак В.В., Мжельский А.И., Аскадский АА., Вы-
годский АА., Виноградова С.В. Пластики на основе ароматических
полиимидов // Докл. АН СССР. 1968. Т.182. №4. С.851-854.
104. Слонимский ГЛ., Шестопал В.О. Статистическое описание релакса-
ционных процессов в полимерах //Высокомолек. соединения. 1978. А20.
№8. С.1712-1721.
Литература
525
105. Слонимский Г.Л., Аскадский АА. О размягчении частосетчатых по-
лимерных тел и о механизме их деформативности // Высокомолек. соеди-
нения. 1986. А28. №5. C.1014-107f
106. Сперанская ТА., Тарутина Л.Н. Оптические свойства полимеров.
Л.:Химия. Ленингр. отд-ние. 1976. 136 с.
107. Тагер АА., Цилипоткина М.В., Суворова А.И. Определение удельной
поверхности и объема пор твердых полимерных сорбентов // Докл.
АН СССР. 1958. Т.120. №3. С.570-572.
108. Тагер А А., Колмакова Л.К. Параметр растворимости, методы его оцен-
ки, связь с растворимостью полимеров // Высокомолек. соединения.
1980. А22. №3. С.483-496.
109. Тейтельбаум Б Я. Термомеханический анализ полимеров. М.:Наука. 1979.
234с.
ПО. Тишин С А., Тишин ВА., Шипилевский БА. Наблюдение структурных
превращений при модификации эпоксидных полимеров методом
измерения угловых распределений аннигиляционных фотонов // Механ.
композит, материалов. 1990. №1. С.3-8.
111. Тишин С А. Отбор ФЭУ-36 и ФЭУ-37 по временному разрушению // Приб.
и техн, эксперимента. 1990. №5. С. 171-174.
112. Уржумцев Ю.С., Максимов РД. Прогностика деформативности поли-
мерных материалов. Рига:3инатне. 1975. 416 с.
113. Успехи в области синтеза элементоорганических полимеров. С.143-231./
В.В.Коршак, Н.М.Козырева, А. И. Кирилин и др.; Подред. В.В.Коршака;
АН СССР Ин-т элементоорган. соединений им. А. Н. Несмеянова.
М.Иаука. 1988. 319с.
114. Фейнман Р. Ф. Статистическая механика: Курс лекций. / Пер. с англ.
Н.М.Плакиды и Ю.Г.Рудого; Под. ред. проф. Д.Н.Зубарева. 2-е изд.
М..Мир. 1978.407с.
115. Цилипоткина М.В., Тагер АА.,Нечаева О В.,Васильев А В ..Быкова Л М.,
Булыгина С.М., Васнев В.А., Салазкин СН. Пористая структура
полиарилагов//Высокомолек. соединения. 1972. А14. №9. С.2053-2057.
116. Шарков В.И., Куйбина Н.И., Соловьева ЮЛ. Выделение и свойства
глюкоманнана из гемицеллюлоз древесины ели обыкновенной // Журн.
прикл. химии. 1965. Т.35. №5. С.1119-1128.
117. Шарков В.И., Куйбина Н.И., СоловьеваЮЛ. Исследование ксилоуронида
из древесины осины И Журн. прикл. химии. 1967. Т.40. №11. С.2609-2611.
118. Шарков В.И., Куйбина Н.И. Химия гемицеллюлоз. М . Леей. пром-сть.
1972. 440с.
119. Шестопал В.О. Диффузия вакансий и реологические свойства металлов
при высоких температурах // Физ.тверд тела. 1970. Т.12.№1 .С291-294.
526 Литература
120. Эйнштейн А. Собрание ночных трудов. ТЗ. Работы по кинетической теории,
теории излучения и основам квантовой механики. М. Наука. 1966.631с.
121. Экспериментальные методы в адсорбции и молекулярной хроматогра-
фии. / Под ред. А.В.Киселева и В.П.Древинга. М.:Изд-во Моск, ун-та.
173.433с.
122. Энциклопедия полимеров. Т.1. 1043с. М.:Сов. Энциклоп. 1972.
123. Arifov Р., Vasserman S., Tishin S. Study of Elastomer Structure Microdefects
by Positron - Annihilation // Phys. Stat. Sol. A-Appl. Res. 1987. V.102,
№.2. P.565-570.
124. Askadskii A A. Physical Properties of Polymers // Encyclopedia of Fluid
Mechanics. V 9. Polymer Flow Engineering. N.P Cheremisinoff, Editor.
1990. P.103-147.
125. Askadskii AA. Analysis of the Structure and Properties of High- Crosslinked
Polymer Networks. Chemistry Reviews. V. 16, part 3. Harwood Academic
Publishers. 1992. 120 p.
126. Askadskii A A., Galpern E.G., Stankevich I.V., Chistyakov A.L. Computer
Simulation of Polymers with Predetermined Physical Properties //J. Chem.
Biochem. Kinet. 1992 V.2. №. 4. P.225-232.
127. Askadskii A A., ShvorakA.E., Frenkel TJvf., Babtshinitzer TJvL.Bychko C A.,
Kovriga О. V., Pankratov VA., Rajulu A.V. Structure and Mechanical Pro-
perties of the Compositions of Polyisocyanurate Polymers with Low Mole-
cular Weight Rubbers//J. Appl. Polymer Sci. 1995. V.55 №8.P.1173-1183.
128. Askadskii AA. Physical Properties of Polymers, Predication and Control.
Gordon and Breach Publishers, Amsterdam. 1996. 333p.
129. Askadskii AA. GolenevaL.M. Computer design, syntesis and investigation of
gradient-modulus polymers // Macromolecular Symposia “Nano-Structures
in Polymer Systems”. 1966. V.106. №.9. April P.9-17.
130. Aspinal G.O. Constitution of Plant Cell Wall Polysacharides // Encyclopedia
of Plant Physiology I Ed. By W. Tanner, E. A. Loewus- Berlin etc:. Springer
\fcrl. 1981. V13B. Plant Carbohydrates. 11: Extracellular Carbohydrates, P.5-8.
131. Bank M., Leffmgwell J., Thies C. The Influence of Solvent upon the
Compatibility of Polystyrene and Polyfvinyl methyl ether) // Macro-
molecules. 1971. V.4 №.l. P.43^16.
132. Beaman R.G. Relation between (Apparent) 2ND- Order Transition
Temperature and Melting Point//J. Polym. Sci. 1952 V.9 №.5. P.470-472.
133. Bicerano J. Prediction of Polymer Properties. New-York; Marcel Dekker,
Inc. 1996. 528 P.
134. Calleja F. J. B.. Serna J., Vicente J., Segovia MA. Structural Implications on
Position Lifetimes in Lamella Polyethylene with Chain Defects // J. Appl.
Phys. 1985. V58. №.l. P.253-259.
Литература
527
135. Chen F.-L.,Pearce EM,Kwei T.K. Intermacromolecular complexes by insitu
polymerization//Polymer. 1988 V.29. №.12. P.2285-2289.
136. Chien Y.Y., Pearce EM., Kwei T.K. Miscibility of Poly (vinyl methyl ether)
with Styrene - Methyl Methacrylate Copolymers // Macromolecules. 1988.
V.21. №.6. P1616-1619.
137. Chu E.YPearce EM., Kwei T К., Yeh T F., Okamoto Y. Blends of Poly
(ethylene oxide) and Poly (butyl methacrylate) with Modified Poly dimethyl-
siloxane - containing Hydrogen - Bonding Donor Groups // Makromol.
Chern., Rapid Commun. 1991. V. 12. №.1. P. 1—4
138. Dai Y K.,Chu E.Y.,Yu ZS.,Pearce EAd., Okamoto Y.,Kwei T K. Molecular
Composites from Sulfonated Poly (p-phenylene terephthalamide) // J. Poly m.
Sci.: Part A:Polym. Chem. 1994. V.32. P.397-400.
139. De Araujo MA., Stadler R. Cantow H.- J. Composition and Molecular -
Weight Dependence of the Glass Transition in Polystyrene -Poly(2,6-dirnethyl-
phenylene ether) Blends // Polymer. 1988. V29. №.12. P.2235-2243.
140 .Eldrup M ,,Lightbood D.,.Sherwood J .N .The Temperature - Dependence of
Positron Lifetimes in Solid Pivalic Acid// Chem. Phys. 1981. V.63 .№..1-2.
P.51-58.
141 .Fedors R F. Method for Estimating both Solubility Parameters and Molar
Volumes of Liquids 11 Polymer Eng. and Sci. 1974. V.14 .№.2 P.147-154.
142. Flory PJ. Levels of Order in Amorphous Polymers // Faraday Disc. Chem.
Soc . 1979.№68.P.14-25.
143 .Fowkes FM. Attractive Forces at Interfaces // Indust .Eng Chem. 1964 .V 56 .
№.12. P.40.
144. Fox T. G., Flory P.J. 2ND - Order Transition Temperatures and Related
Properties of Polystyrene. 1 .Influence of Molecular Weight//J Appl .Phys .
1950. V21 .№.6. P.581-591.
145 .Fox T G ,,Loshaek S. Influence of Molecular Weight and Degree of Cross-
linking on the Specific Volume and Glass Temperature of Polymers // J.
Polym. Sci. 1955. V.15. №.80. P.371-390.
146. Gaylord R.J., Joss B., Bendler J. T., Di Marzio E.A. The Continuous -
Time Random Walk Description of the Non - equilibrium Mechanical
Response of Crosslinked Elastomers // Brit. Polymer. J. 1985. V.17. №.2.
P. 126-128.
147. Girifalco LA.,GoodR J A Theory for the Estimation of Surface and Interfacial
Energies. 1. Derivation and Application to Interfacial Tension // J. Phys.
Chem. 1957. V61. №.7. P.904-909.
148 .Good R. J. ,Girifalco LA.,Kraus GA .Theory for Estimation of Surface and
Interfacial Energies. 2. Application to Surface Thermodynamics of Teflon
and Graphite // J. Phys. Chem. 1958. V.62. №.11. P. 1418-1421.
528
Литература
149. GoodR.J., Girifalco L.A. A Theory for Estimation of Surface and Interfacial
Energies. 3. Estimation of Surface Energies of Solids from Contact Angle
Data// J. Phys. Chem. 1960. V.64. №.5. P.561-565.
150. Gordon J.L. Cohesive - energy' density // Encyclopedia of Polymer Science
and Technology. V.3. New-York, John Wiley and Sons, Inc. 1965. P.833-862.
151. Gsell T.C., Pearce EM., Kwei T.K. Miscibility and Segmental Mobility in
Hydrogen-Bonded Polymer Blends//Polymer. 1991. V9. P.1663-1674.
152. Gupta AM., Macosko C.W. Characterization and Modeling of Rigid Branched
Polycyanates//Macromolecules. 1993. V26. №.10. P.2455-2463.
153. Halary J.-L., Ben Larbi F.C., Oudin P., Monnerie L. Molecular Mobility in
Polystyrene - Poly (methyl vinyl ether) Blends as Studied by Fluorescence
Polarization//Macromol. Chem. 1988. V.189. №.9. P.2117-2124.
154. Haldon RA., Simha R. Multiple Transitions in Polyalkyl Methacrylates //
J. Appl. Phys. 1968. № 39. P. 1890-1898.
155. Hamielec A.E., EldrupM., Mogensen ОE., Jansen P. Positron: Annihilation
Techniques (PAT) in Polymer. Science and Engineering // J. Macromol.
Sci. 1973. V9. №.2. P.305-337.
156. Ни C.K., Gruzalsk G.R. Temperature Dependence of Positron - Annihilation
at Disfocations in PB (CD) // Phys. Rev. 1983. В 27. №.l. P.1-8.
157. Huang W., Zhu KJ., Pearce EM., Kwei T.K. The Modification of Nylon 6 by
a Phenol - Formaldehyde Resin // J. Appl. Sci. 1993. V.48. P.563-573.
158. Jong L., Pearce EM., Kwei Т.К., Dickinson L.C. Miscibility Study of Poly
(styren - co-vinylphenol) with Poly(n-butyl methacrylate) by NMR //
Macromolecules. 1990. V.23. №.24. P.5071-5074.
159. Jong L., Pearce EM., Kwei T.K. NMR Study of Hydrogen Bonded Polymer
Blends: Influence of tire Tacticity of Poly (methyl methacrylate) on Its Miscibility
with Poly (styrene-co-vinylphenol) // Polymer. 1993. V.34. №1. P.48-55.
160. Kanig G. Zur Theorie der Glastemperatur von Vemetzten Polymeren //
J. Polym. Sci., part C. 1967. P. 1957-1964.
161. Kim H.-L. Kwei T.K., Pearce EM. Hydrogen Bonding Interaction in
Interpenetrating Polymer Network // in Contemporary Topies in Polymer
Science. V.6. (B.Culberison, Ed.) Plenum. New York. 1989. P.687-697.
162. Kim H.-L., Pearce EM., Kwei T.K. Miscibility Control by Hydrogen Bonding
in Polymer Blends and Interpenetrating Networks // Macromolecules. 1989.
V22. №8. P.3374-3380.
163. Kim H.-L., Pearce EM., Kwei T.K. Phase Behaviour of Copolymer Blends //
Macromolecules. 1989. V.22. №.8. P.3498-3500.
164. Kindi P., Reiter G. Investigations on the Low - Temperature Transitions
and Time Effects of Branched Polyethylene by the Positron Lifetime
Technique //Phys. Stat. Sol. A -Appl. Res. 1987. V.104. №2. P.707-713.
Литература 529
165. Kotzev D.L., Pearce E.A4., Kwei T.K. Studies on the Compatibility of
Poly (styrene - co - vinyl - phenyl hexafluorodimethyl carbinol) with Nylon
6,12 and N,N’ - Dimethyl - Substituted Nylon 6,12 // J. Appl. Polym. Sci.
1984. V.29. P.4443-4448.
166. Kryszewski AL, Jachowicz J., ALalanga M.. Vogl О. Blends of Head - to -
head Polystyrene with Poly (2,6 - dimethyl - 1,4 - phenylene oxide) //
Polymer. 1982.V.23. №.2. P.271-276.
167. Kun K., Kunin R. Pore Structure of some Macroreticular Ion Exchange
Resins // J. Polymer Sci. 1964. В 2.P587.
168. Kwei T.K., Nishi T., Roberts R.F. A Study of Compatible Polymer
Mixtures // Macromolecules . 1974 ,V7 ,№_5 .P 667-674 .
169. Kwei T.K., Frisch H.L. Interaction Parameter in Polymer Mixtures //
Macromolecules. 1978. V.ll. №.6. P.1267-1271.
170. Kwei T.K., Pearce E.M., A'Hn B.Y. Effect of Hydrogen Bonding on tire Lower
Cfiicd SdufionTemperature cf a Polymer Mixture // Macromolecules.
1985. V.18. №.11. P.2326,2327.
171. Kwei T.K.. Pearce EM., Ren F., Chen J.P. Hydrogen Bonding in Polymer
Mixtures // J. Polym Sci.: PartB: Polym. Pliys 1986. V24. №.7. P.1597-1609.
172. Lee IT.A., Knight G.J. Ratio of the Glass Transition Temperature to the Melting
Point in Polymers // Brit. Polymer J. 1970. V.2. №.2. P.73-80.
173. Lin P., Clash C., Pearce EM., Kwei T.K. Solubility and Miscibility of Poly
(ethyl oxazoline) // J. Polym. Sci.: PartB: Polym Phys. 1988. V26. P.603-619.
174. Lin Y.-H. A General Linear - Viscoelastic Theory for Nearly Monodisperse
Flexible Linear Polymer Melts and Concentrated Solutions and Comparison
of Theory and Experiment // Macromolecules. 1984. V.17. №12. P.2846-
2856.
175. Lin Y.-H. Comparison of Experiment and the Proposed General Linear
Viscoelastic Theory. 2. Stress Relaxation - Line Shape Analysis //
Macromolecules. 1986. V.19. №.1. P.159-168.
176. Lin Y.-H. Comparison of Experiment and the Proposed General Linear
Viscoelastic Theory. 3. Zero - Shear Viscosity and Steady - State
Compliance // Macromolecules. 1986. V. 19. №-.l P.168-173 .
177. Lin Y.-H. Linear Viscoelasticity and Translational Diffusion of Linear Flexible
Polymers //Macromolecules. 1991. V.24. №. 19. P.5346- 5350.
178. Lu S., Pearce EM., Kwei T.K. Synthesis and Characterization of
(4 - vinylphenyl) Dimethylsilanol Polymer and Copolymers // Macro-
molecules. 1993. V26. №.14. P.3514-3518.
179. Lu S„ Pearce E.M., Kwei T.K. Synthesis and Characterization of Novel
4 - Vinyl -phenylmethylphenylsilanol Polymer and Its Styrene Copoly-
mers // j. Polym. Sci.: Part A: Polym. Chem. 1994. V.32 .P2597-2601.
530
Литература
180. Lu S., Pearce EM., Kwei T.K. Novel Silanol - Containing Polymers and their
Blends// J. Macrom. Sci.: Pure. Appl. Chem. 1994. V.A 31. №.11. P.1535-
1560.
181. Lu S., Melo MM., Zhao J., Pearce EM., Kwei T.K. Organic - Inorganic
Polymeric Hybrids Involving Novel Poly (hydroxy - methylsiloxane) //
Macromolecules. 1995. V.28. №.14. P.4908-4913.
182. Lu S., Pearce EM., Kwei T.K. Effect of Casting Solvent on the Heterogeneity
ofFilms of Polymer Blends//Polymer Eng. Sci. 1995. V35. №.13. P.1113-1116.
183. Lu S., Pearce EM., Kwei T.K. Blends and Interpolymer Complexex of
Poly(styrene-co-4-vinylphenylmethylphenylsinol) and Poly(N-vinylpyrro-
lidone) // Polym. Adv. Tech. 1996. V.7 P.553-559.
184. Lu S., Pearce EM., Kwei T.K. Miscibility Window for the Blends of
Poly(Styrene-co-4-vinylphenylmethylphenylsilanol) with Poly(n-butyl
methacrylate) // J. Polym. Sci.. Part A: Polym. Chem. 1996. V34 P.3163-3172.
185. Luo D., Pearce EM., Kwei T.K. Miscibility of Stereoregular Poly(methyl
methacrylates) with Poly(styrene-co-p-(hexafluoro-2-hydroxy-2-propyl)
styrene) //Macromolecules. 1993. V.26. №.23. P.6220-6225.
186. Makhija S., Pearce EM., Kwei T.K., Liu F. Miscibility Studies in Blends of
Polybenzimidazoles and Poly(4-vinyl pyridine) // Polym. Eng. Sci. 1990.
V.30. №.13. P.798-801.
187. Malhotra B.D.,PethrickR.A. Positronium Annihilation Studies of Polycarbo-
nate, Polyethersulfone and Polysulfone // Eur. Polym. J. 1983. V.19. №.6.
P.457-459.
188. .Mark J.E., Sullivan J.L. Model Networks of End - Linked Polydimethyl-
siloxane Chains. 1. Comparisons Between Experimental and Theoretical
Values of Elastic Modulus and Equilibrium Degree of Swelling // J. Chem.
Phys. 1977. V66. № 3. P.1006 - 1011.
189. Miller R.L., Boyer R.F. Regularities in X - Ray - Scattering Patterns from
Amorphous Polymers // J. Polymer Sci., Polymer Phys. Ed. 1984. V.22.
№.12. P.2043-2050.
190. Ogawa K., Tanaka F., Tamura J., Kadowaki К., Okamura K. Correlation
between the Glass Transition Temperatures of Polymer Mixtures and
Intermolecular Force Parameters // Macromolecules. 1987. V.20. №.5.
Pl 174-1176.
191. Park FL, Pearce EM., Starnes W.IL,Kwei T.K. Thermal Oxidation of Blends
of Poly (vinyl methyl ether) and Modified Polystyrenes // J. Polym. Sci.:
Part A: Polym. Chem. 1990. V.28. P.1079-1089.
192. Pennacchia.J.R., Pearce EM., Kwei T.K.,Bulkin В J., ChenJ.-P. Compatibility
of Substituted Phenol Condensation Resins with Poly (methyl metha-
crylate) //Macromolecules. 1986. V.19. №.4. P.973-977.
Литература
531
193. Pearce ЕМ., Kwei Т.К., Min В. Y. Polymer Compatibilization through Hydrogen
Bonding// J. Macromol. Sci.-Chem. 1984. V A21. №.8,9. P.1181-1216.
194. Pearce EM., Kwei T.K. Studies on and with Functionalized Polystyrenes //
J. Macromol. Sci.-Chem. 1991. V. A28. №.11,12. P.1207-1212.
195. Pearce EM., Kwei T.K. Hydrogen Bonding Interaction in Polymer Blends //
Polym. Solutions, Blends and Interfaces. 1992. P.133-149.
196. Pearce EM., Kwei T.K., Lu S. Miscible Blends through Hydrogen Bonding:
Effects on Polymer Properties//Polym. Adv. Techn. 1994. V.5. P.600-602.
197. Prest WM., Porter RS. Rheological Properties of Poly (2,6 - dimethylphenylene
oxide) Polystyrene Blends // J. Polymer. Sci.: Part A-2: Polym. Phys. 1972.
V.10. №.9. P.1639-1647.
198. Schmidt-Rohr K., Clauss J., Spiess H.W. Correlation of Structure, Mobility
and Morphological Information in Heterogeneous Polymer Materials by
Two-Dimensional Wideline-Separation NWR Spectroscopy // Macro-
molecules. 1992. V25. №.12. P.3273-3277.
199 .SchneiderH A ,,BreknerNJ. Thermodynamic Aspects of the Glass-Transition
in the Compatible Blend Poly (styrene) - Poly (vinylmethyl ether). // Polymer
Bull. 1985. V.14. №.2. P.173-178.
200. Schneider H.A., Leikauf B. Glass-Transition Temperatures of PVME-PS
Blends Influence of the Molecular Weight of PS // Thermochim. Acta. 1987.
V.114. №.l. P.165-170.
201. Schuerman B.L. Computer simulation on Novolak-Phenol Formaldehyde Resin //
Nano-Structure and Self-assemblies in Polymer Systems, Int. Conference.
St-Petersburg-Moscow, May 18-26. 1995. OL70.
202. Shefer A., Gottlieb M. Effect of Cross-Links on the Glass Transition
Temperature of End-Linked Elastomers // Macromolecules. 1992. V25.
№.15. P.4036M042.
203. Shultz A.R., Gendron BM. Thermo-Optical and Differential Scanning
Calorimetric Observations of Mobility Transitions in Polystyrene - Poly
(2,6 - dymethyl - 1,4 - phenylene oxide) Blends // J. Appl. Polymer Sci.
1972. V16. №.2. P.461-471.
204. Shultz AS., Young A.L. Glass Transitions of Styrene Alpha-Methylstyrene
Statistical Co-Polymers and of theirBlends with PPO Resin// J Appl Polym.
Sci. 1983. V.28. № 5. P.1677-1684.
205. Simha R. On a General Relation Involving Glass Temperature and Coefficients
of Expansion of Polymers // J. Chem Phys. 1962. V37. №.5. P.1003.
206. Small P.A. Some Factors Affecting the Solubility of Polymers // J Appl.
Chem. 1953. V.3. №.2. P.71-80
207. Suzuki T., Pearce EM., Kwei T.K. Hydrogen-bonded Polymer Complexes //
Polymer. 1992. V33. №.1. P.198-201.
532
Литература
208. Timell Т.Е. Wood Hemicellulose // Adv. In Carbohydrate Chern. New York:
1964. V19. P.247-302.
209. Ting S.P., Pearce EM., Kwei T.K. Compatibility Studies of Poly(styrene-co-
vinylphenyl hexafluorodimethyl carbinol) with Bisphenol A Polycarbonate,
Poly (butyl methacrylate) and Poly(2,6-dimethyl-l,4-phenylene oxide) //
J Polym. Sei., Polym. Lett. 1980. V.18. №.3. P.201-209.
210. Ting S.P., Bulkin В J, Pearce EM. Compatibility Studies of Styrene and
Hydroxal Contaning Styrene Copolymers with Poly(ethylene oxide) // J.
Polym. Sci.: Part A : Polym. Chem. 1981. V.19. №.6. P.1451-1473.
211. Tisin SA., BakaevE.S., Tisin VA.,SipilevskijBA. Strukturund Eigenschaften
von mit modifizierten Aerosilen // Plaste und Kautschuk. 1991. Heft 4.
S.45-49.
212. Tomalia DA., Noylor AM., Goddard WA. Starburst Dendrimers. Molecular-
Level Control of Size, Shape, Surface-Chemistry, Topology and Flexibility
from Atoms to Macroscopic Matter // Angew. Chem. 1990. V29. №.2.
P.138-175.
213. Vaidya MM., Levon K., Pearce EM. Miscibility of Poly(s-caprolactone), a
Semicrystalline Polymer, with Amorphous Poly(styrene-4-hydroxystyrene)
Copolymers И J. Polym. Sci. Part: B.PolymPhys. 1995. V.33. P.2093-2108.
214. Van Krevelen D. ft'. Properties of Polymers /Third Edition Elsevier, Amsterdam.
1990. P.875.
215. Vointseva 1.1., Askadskii AA. Interpolymers (Paired Polymers). Chemistry'
Reviews, Soviet Scientific Reviews. V.16. Part 2. Harwood Academic
Publishers. Chur-Reading. Paris, Philadelphia, Tokyo, Melbourne. 1991.
P.86.
216. Wang L.F., Pearce EM., Kwei T.K. Polymer Association in Styrene-Acrylie
Acid Copolymers // J. Polym. Sci.: PartC.: Polym. Lett. 1990. V.28. P.317-321.
217. Wang L.F., Pearce EM., Kwei T.K. Glass Transitions in Hydrogen-Bo tided
Polymer Complexes // J. Polym. Sci.: Part B.: Polym. Phys. 1991. V29.
P.619-626.
218. Yang T.P., Pearce E.M., Kwei T.K. Yang NL. Complexation of Poly(N,N'-
dimethylacrylamide) and Phenol Formaldehyde Resins // Macromolecules.
1989. V22. №.4. P.1813-1818.
219. Yang X., Painter P C., Coleman MM., Pearce EM., Kwei T.K. Equilibrium
Constants and Predictions of Miscibility Windows and Maps for Polymer
Blends Involving p-(hexafluoro-2-hydroxy-2-propyl) styrene with Metha-
crylate and Acetoxy Groups //Macromolecules. 1992. V25. № 8. P2156-2165.
220. Yee A.F., Bankert R.J., Ngai K.L., Rendell R.W. Strain and Temperature
Accelerated Relexation in Polycarbonate //J. Polymer Sci 1988. V26. №. 12.
P.2463-2483.
Литература 533
221. Zhu K.J., Pearce ЕЛ., Kwei T.K. Thermostability of Poly (p-hydroxy styrene)
Blends with Poly(vinyl pyrrolidone) and Poly(ethyl oxazoline) // J. Appl.
Poly. Sci. 1989. V37. №.2. P573-578.
222. Zhu K.J., Chen S.F., Ho T., Pearce EM., Kwei T.K. Miscibility of Copolymer
Blends //Macromolecules. 1990. V.23. №.l. P.150-154.
223. ZhuangH., Pearce EM., Kwei T.K. Miscibility Studies of Poly(styrene-co-4-
vinylbenzenephosphonic acid diethyl ester) with Poly(p-vinylphenol) //
Macromolecules. 1994. V.27. №.22. P.6398-6403.
224. Zhuang H., Pearce EM., Kwei T.K. Self-Association in Poly(styrene-co-4-
viny Ibenzenep hosp home acid) and Miscibility of Its Blends // Polymer. 1995.
V.36. №.11. P.2237-2241.
предметный указатель
Азот 31,81
Алмаз 165
Альдегиды 358
Амиды 358
Аминогруппа 481
Амины 358
Ангармонический осциллятор 208
Ангидрид полисе бациновой
кислоты 289
Анизотропия:
-показателя преломления 236
-поляризуемости 236
Анилинфталеин 91
Аннигиляция позитронов 66
Арабиногалактан 436
Арабиноглюкуроноксилан 434
Артефакты 333
Барьер:
-потенциальный И 4
-энергетический 114
Бензол 342
Бисфенол 169, 384
Бисфенол-А-поликарбонат 262
Бор31,81
Бромбензол 267
Бром 31
Бутаналь 268
н-Бутанол 268
t-бутил 475
Бутилакрилат 467
Бутилметакрилат 467
н-Бутилацетат 268
Ван-дер-Ваальса:
-объем атома 273,32
-поверхность атома 273,32
-радиус атома 272,32
Вектор:
-напряженности электрического
поля 258
-поляризации 258
-электрической индукции 258
Величина-.
-дефекта 272
-механического сегмента 94
-характеристической
вязкости 275
Взаимодействие:
-внутримолекулярное 450
-диполь-дипольное 79,120
-межмолекулярное
(физическое) 29, 450
-межмолекулярное сильное 79
-дисперсионное слабое 78
-дисперсионное собственное 78
-химическое 30
Водород 31,81
Время:
-наблюдения 116
-перехода из стеклообразного
сосгоянияввьюокоэластичное 115,116
-релаксации 65, 116
Время жизни:
-атома, оседлой 115
-долгоживущего компонента 65
-позитрона 65
-промежуточного
Предметный указатель
535
компонента 66 Высоташарового сегмента 32 Вязкость: 1,3-Диоксолан 477 Диффузия: -кинетических единиц 293,299
-сдвиговая, ньютоновская 202 -системы 97 Галактоуронан 436 Г алогенсодержащие соединения 357 Гексагональная упаковка353 Гексаметилентетрамин 477 н-Г ексан 267 н-Гекс анол 268 Гемицеллюлоза 427 н-Гептан 267 н-Гептил акр 1 ыат 467 Главные: -релаксаторов 293,299 -фиковская299 Диэлектрики 260 Диэлектрическая проницаемость 287 Д иэтиламин 480 Длина: -волны 231 -межмолекулярного радиус 29 -сегмента Куна 202 -химической связи 30 Домены 288 3 акон (правило): -аддитивности 208
-деформации 237 -нормальные напряжения236 Глобула334 Глюкоманнан 436 Группа: -Вертгейма 236 -действующих масс 122 - Ильюшина 316 -Лапласа 336,350 -Максвелла 260
-амидная 79, 120, 363 -4-гидроксил-4,4-бис-3- фторметилбутильная 479 -гидроксильная 79,120 -кислотная 79, 363 -нигрильиая 79, 120, 363 -полярная79 -сложноэфирная 79, 120, 363 -спиртовая 363 -уретановая 79 -фторалкилкарбинольная 480 Двойное лучепреломление (двулучепреломление) 236 Дебаеграмма 427 н-Декан 267 Дендримеры 144 Деструкция: -мольныхдолей,Журкова 124 Заместитель боковой 95 Замещение: -мета 81 -орто 81 -пара 81 Звено полимера: -диметилсилоксановое 284 -повторяющееся 21 -элементарное 20 Изобутанол 268 Изомеры 21,136 Изопентан 267 Изопропилбензол 267 Инкремент: -линейных цепей 155 -сильного межмолекулярного
-термическая 101 -термоокислительная 101 Дефекты в виде: взаимодействия 81 -слабого межмолекулярного взаимодействия 81
-изолированной петли 190 -подвешенных цепей 190 -разветвлений 190 1,2-Дибромэтан 268 Диизоцианат 283 Димер 390 N ДЧ’-диметилакриламид 479 М^’-димепшзамещенный найлон-6, 12 473 -собственного (Ван-дер- Ваальсового) объема 30 -узла сетки 155 -фрагментов 399 Интенсивность долгоживущего компонента 69 ИодЗ! Испытания: -ренттеноструктурные 104
536
Предметный указатель
-с периодической нагрузкой 108
-термогравиметрические 101
-термомеханические 101
Каучук:
-бутадиеновый 339
-изопреновый 339
-синтетический 21
Кетоны 358
Кинетическая единица 113,293
Кислород 31,81,207
Кислота:
-азелаиновая 291
-н-валериановая 268
- изовалериановая 268
-изо-,терефталевая 19
-изофталевая 91
-масляная 268
-полиакриловая 467
-себациановая91
-уксусная 360
Компатибилизатор 481
Константа:
-Больцмана 114
-потенциала Морзе 271
-равновесия 122
-скорости реакции 295
-универсальная газовая 115
Контракция 60
Конформация макромолекул 114,320
Координационные связи 20
Коэффициент:
-энгармонизма осциллятора 208
-весовой 274
-деполяризации 258
-диффузии 299
-линейного расширения,
термический 76
-молекулярной упаковки 44,45,32,365
-объемного расширения,
парциальный 80
-объемного расширения,
термический 76
-оптической чувствительности по
деформациям 237
-оптической чувствительности по
напряжению 237
-Пуассона 237
-ростатеплоемкости 392
-силовой 217
-трения повторяющегося звена 202
-трения сегмента 202
-упаковки молекул в
поверхностном слое 355
-упаковки, парциалЛый 207
-упругости осциллятора 208
-упругостиповоротно-изомерной
подсистемы 273
-упругости связи 273
-уравнения реакции,
стехиометрический 122
Кремний 31,81
Кривая-.
-восстановления 73
-дилатометрическая 74
-релаксации напряжения 73,319
-с насыщением 383
-термогравнметрическая 101, 102
Критерий:
-растворимости 338,339
-совместимости 374
Кс ил ан 436
о-Ксилол 267
м -Ксилол 267
п-Ксилол 267
Макродювоцианаты 283
Макромолекула-
-линейная 24
-разветвленная 25
-гибкая 94
-жесткая 94
Мета-,парафеннлецдиамин 129
4-0 -метнлглюкур онокснлан 434
Метилацетат 268
Метилметакрилат 345
Метилформиат 268
Метилиодид 268
Метилзтилкетон 268
Метод
-аддитивных схем 98
-адсорбции, равновесной 55
-аннигиляции позшронов 55
-атомных инкрементов 419
-градиентных трубок 58
-диаграмм совместимости 419
-дилатометрический 107,58
-динамических испытаний 107
-динамического механического
анализа 286
-ДСК 473
-замораживания деформаций 248
Предметный указатель
537
-ИК-спектроскопии 473
-калориметрический 107,110
-капиллярной конденсации 55
-локального поля Лорентца 230
-малоуглового рентгеновского
рассеяния 334
-наименьших квадратов 127
-нейтронного рассеяния 334
-о братного переменного шага 320
-оптический 107
-пикнометрический 58
-планирования эксперимента 457
-поликонденсации 58
-полимеризации в растворе 58
-поляризационно-оптический 247
-регистрацииспектров времен
жизни позитронов 64
рентгеновской фотоэлектронной
спектроскопии 286
-ртутной порометрии 55
-Рунге-Кутта, численный 302
-Симпсона 320
-термогравиметрический 101,102
-термомеханический 85
-фотоползучести 255
-фотоупругости 250,251
-ФоуксаЗбО
-Фурье-преобразования 479
-экстраполяционный 206
-электронной микроскопии 286
-электроннойспектроскопии 481
-ЯМР, ЯМР^С.ЯМР31 Р 479
Механизм:
-застекловывания,
релаксационный 112
-набухания, релаксационный 320
-процесса стеклования 112
-релаксационного процесса 70
-сорбции, релаксационный 320
Микродомены 288
Микрофаза 374
Модель:
-аннигиляции позитронов 70
-полиномиальная 458, 460
Модификация физическая 23
Модуль:
-высокоэластичности,
равновесный 270
-линейных фрагментов 275
накопления 292
-релаксационный 316
-узлов 275
-упругости 102, 111,252, 275
-эластичности 102
Молекулярная масса:
-звена, примыкающего к узлу 158
-линейной цепи между узлами 158
-межузлового фрагмента 270
-нормального звена 158
-повторяющегося звена 46
-полимера 383
-сегмента 92
сшивающих мостиков 161
-узла 158
-цепи 202
Молярные доли компонентов 46
Момент”
дипольньп'т 433
-магнитный 257
Мономеры:
-двухфункциональные 26
-дизамещенные 22
-полифункциональные 26
Мышьяк 31
Н адмолекулярная организация 320
Найлон 6,473
Натяжение'.
-межфазное 336
-поверхностное 336
Нафталин 267
Непредельные углеводороды:
-дизамещенные 22
-монозамещенные 22
Нерелаксаторы 317
Н игрил359
Н итрил кислоты:
-бутановой 269
-нзогептановой 269
-пентановой 269
-пропионовой 269
-уксусной 269
Нитробензол 344
Нитрометан 269
1-Нитропропан 269
2-Нитропропан 269
Нитросоединения 359
н-Нонан 267
н-Н онилакрилат 467
Объем-
ат см а32
1 Указатель преломления 230
Ползучесть:
-механическая 251
-оптическая 251
-полимера293
Полиакриламид 364
Полиакрилонитрил 125
Полиамид 19, 57, 91, 129, 339, 207,
415, 362
Полиамид 6,6-а-изомер 51,362
Полиамнд6,6-р-изомер51
Полиамцдофенилхиноксалины 136
Полиарилат Ф-1 95
Полиацетилен 20
Полибензимидазолы 416
Полибензоксазол 305
Полибутадиен 98
1,2-полибутадиен 50
1,4-полибутадиен 50,89
Поли(1-бутан) 262
Полибугилакрилат 90
Поли-н-бутилакрилат473,491,473,447
Йоли-1-б.утилметакрилат485
оли-н-бутир альдегид 47
Полибугилметакрилат 472
Поли(н-бутил метакрелат) 262
Поливинилацетат 89,98,339,370,473
Поливинилбутираль 262
П о Л1<винилиден хлорид) 262
Поли (винил циклогексан) 262
Полн-а-винилнафталин 214
Поли-Р-винилнафталин 262
Полира-винил карбазол) 262
Поливинилпирролидон 476, 480
Поли-4-винилпиридин 478,480,493
Поливинилфторид 52
Поливинилхлорид 52,89,95
Полигексаметиленадипамид 214
Полигексаметиленсебацамид 214
Полигексаметиленсебацинамид 263
Полигексаметилентерефталат 51
Полигексилмегакрилат 90
Поли(п-гидроксибензоат) 263
Поли-1 -гцпрокси-2,6-метилфенилен477
Поли-1 -гидроксил-2,6-метилен-
фенилен 478
Поли-4-гцпроксистирол 477
Поли-п-гцдроксисшрол 476
Полцдиметилсилоксан 19, 64, 87,89,284
Поли-Ы,ЬГ-диметилакриламид475
538 Предметный указатель
-Ван-дер-Ваальсовьш, повторяю-
щегося фрагмента сетки 155
-Ван-дер-Ваальсовый, полимера 384
-Ван-дер-Ваальсовый, свободный 32
-дефекта 27 2
-идеального кристалла 56
-концевых групп 389,383
-мольный (жидкости) 327
-общий молярный 32
-повторяющегося звена 32
-пустой 32
-расширения 54
-свободный 54
-собственный (истинный)
молярный 32
-удельный 47
-флуктуационный 54,263
-цилиндравзаимодействия 271
н-Октан 267
н-Октанол 268
Олово 31
Осмотическое давление 321
Ос1яж
-частоты 259
Парахор:
-атома 352
-молекулы 352
н-П ентан 267
н-Пентанол 268
Перекись бензола 389
Периодколебания 115
Перфторсоедииение 357
П промеллитовый ди ангидрид 91
Плавления:
-температура 206
-энтальпия 206
-энтропия 206
Плотность:
-истинная 58
-кажущаяся 58
-полимера 32
-энергии когезии 327,338
Площадь (поверхность):
-Ван-дер-Ваальсовая, атома 273
-тора 282
-цилиндравзаимодействия 272
Подвижность:
-вибрационная 121
-трансляционная 121
539
___________________________Предметный указатель
11оли(диметил силоксан) 262
П оли-2,6-диметил-1,4-фениленоксцд 473,
378
П оли[4,4-диф енокси ди
(4-фенилен)сульфон] 263
Поли(3,4-дихлорстирол) 263
П олпизобутилен 20,89,95,98,125,339
Поли(гаобутил метакрилат) 262
Полиизопрен 64
1,4-полиизопрен 89
Поли[4,4-изопропилен дифеноксп ди(4-
фенилен)сульфон] 263
1,4- полиизоцианураты 250,283
Полиимид64, 339, 305,138
Поликарбонат416
Поликарбонатнао снове
бисфенола А 9 0, 384
П оли(п-ксилен) 262
Полимеры:
алифатические 31
-аморфно-кристаллические 60,108
-аморф ные45 ,60 ,108,334
- ар ом этические 31
-атактические, нерегулярные 22
-виниловые 416
-гетепоцепные 19
-глобулярные, аморфные 335,346
-гомополимеры 46
-градиентные 285,289
- дендритные 24 144
-диеновые 421
-жесткоцепные 94
-звездообразные 146
-изотактические 22
-изотропные, аморфные 346
-интерполимеры 27
-карбоцепные 19
-кардового строения 339
-каучукоподобные 88
-кремнийорганические 416
-кристаллические 49
-лестничные 28
-монолитные, аморфные 56
-неглобулярные, аморфные 335
-непо лярные 364,379
•опт ически чувствительные 236
-ориентированные 349
-полярные 343,364
-разветвленные 24
-разнозвенные 425
-разномодульные 285
-с сопряженной системой связей 20
- сетч( тие 26 450
-синдиотактические 22
-стеклообразные 88,289
-стереорегулярные 22
-теплостойкие 33 9
-трео-диизотакгические 23
-хелатные 20
-частично-кристаллические 58
-элементоорганические 19
-эритро-диизотакгические23
П олиметилакрилаг 90
Поли-4-метилпентен 47
П оли-4-метилпенген -1 214
Полиметилиденфталвд 59
П олиметилметакрилат 473
Поли-а-метилстирол 243,262
Поли(метил а-хлор акрилат) 263
Поли(о метил стир 04262
Поли(4-метил-1-пентан) 262
П олигнф тоиленбензимидазол88
Ноли н пропилметакпилат 47
олиоксадиазол 20,305
Поли (МЬГ-(п,п-оксидифенилен)
пиромелитимид! 263
П олиоксиметилен 53
Поли[оксн(2,6-дифенил-
1,4-фенилен)] 262
Поли(окси-2,2-
дихлормнтилтриметилен) 262
Полиолефины 415
Полипиромеллитимид
анилинфталеина 344
Полипропилен 19
П олипропиленоксвд 53
П олистирол 53,64
Полисульфиды 416
Полисульфоны 415
П олн[4,4-сульфон диф енокси ди(4-
фенилен)сульфон] 263
Политетраметиленадипат 214
П о литетр аметиленоксад 214
Поли(тетраметилен терефталат) 263
Политетрафторэтилен 64,52
Поли(о,а,а,и-тстрафтор-п-ксилилен) 262
Поли-трет-бутилакрилат 477
Политр иметнленадипат 214
Поли[тио(п-фенилен)| 263
Поли(1 -триметилсилил-1 -пропин) 64
540
Предметный указатель
Политрихлорбутадиен 27
Поли-1,1,2-трихлорбугадиен 481
Полиуретаны 19,416
Полифенил 20
Полифенилен 363
Поли[2,2-(м-фенилен)-5,5-
бибензимцдазол] 263, 478, 493
П олифенилсилсесквиоксан 28
Полифенилхиноксалин 339
ПолиформальдегидЗбЗ
Полифосфонитрилхлорцд 20
Полихлоропрен 49,50
Полихлоротрифторэтилен 262
Поли(хлоро-п-ксилилен) 262
Поли(п-хлор стирол) 262
Поли(м-хлор стирол) 262
П оли[1,1 -циклогексан
бис(4-фенил)карбонат] 262
Поли(1,4-циклогексилидеидиметилен
терефталат) 262
Поли(циклогексил метакрилат) 262
Поли[ 1,1 -этан бис(4-фенил)
карбонат] 262
Полиэтилен 19, 59, 98
П олиэтиленадипат 51
Полиэтиленизофталат 51
Полиэтиленоксазолнн 476
Полиэтиленоксцд 19,363,473
П олиэтилентерефталат 19,51,90
Полиэтилмстакрплат47,89,473
Полпэтилоксазолин 475
Поли(этила-хлоракрилат) 263
Полиэфиры 90,283
Полиэфирысложныена основе:
-себациановой кислоты и
фенолфталеина 90
- изофталевой кислоты и
фенолфталеина 91
Полиэфиркетоны 415,441
П ол! гэфирсуль ф оны 415
Поли(эфир эфир кетон) 263
Поляризация:
-атомная 230
-вещества 258
-деформационная 260
-макроскопическая 258
-микроскопическая 258
-ориентационная 230
-структурных элементов 258
-электронная 230
Поляризуемость 258,433
ПоправкаЛоренгца 231
Пор:
-распределение по размерам 55
-суммарный объем 55
-удельная поверхность 55
Потенциал:
-Леннард-Джонса 213
-Морзе 218
-химического воздействия 218
Присоединение звеньев:
-аномальное (“головак голове”,
“хвост к хвосту”) 21, 128
-нормальное
(“голова к хвосту”) 21, 128
Проницаемость 433
Пропаналь 268
н-Пропаналь 268
н-Пропилацетат 268
Процесс:
-адгезионного смачивания 336
-десорбции 323
-набухания 320
-полимеризации 59
-равновесный 122
-синерезиса 323
-сополимеризации 24
-сорбции 320
-микрофазового расслоения 286
-ползучести 293
-релаксации напряжения 293
Работа адгезии 236
Радиус:
-захвата 70
-межмолекулярный 32
Разветвление цепей 143
Размер:
-кристаллитов 206
-повторяющегося звена,
характерный 202
-связиф ренкелевских роев
растворителя, характерный 338, 346
-связи, характерный 273
Разность:
-главных нормальных
напряжений 236
-хода, оптическая 236
Распределение Флори 146
Расслоение микрофазовое 144
Растворимости:
Предметный указатель
541
-критерий 339 -стирола с акрилонитрилом 473
-параметр Гильдебранда 327 -стирола с винилфенилдиметил-
Реакция: силанолом 479
-полициклотримеризации 27, 284 -стирола с 4- винилфениддиметил-
-уретанообразования 283 силанолом491
-тример изации 27 -стирола с винилфенолом 479
Резольвенты ядер 293 -стирола с винилфенилгексафтор-
Релаксаторы 293 метилкарбинолом 472
Релаксация: -стиролас гексафтор-2-
-микродефектов 70 пропилстиролом 476
-напряжения 293 -стиролас 4-гцдроксистиролом 477
Рефракция: -стирола с давинплбензолом 26
-атомов, по Эйзенлору 232 -стироласметилметакрилатом 476
-атомов, удельная 231 -чередующийся 23
-молекулярная 231 Сорбат 56,57
-молярная 259 Сорбент56,57
Свинец 31 Спектр времени жизни позитронов 66
Связи: Спирт:
-Ван-дер-Ваальсовые 120 -поливиниловый 1Я 52 90
-внутризвенные 425 Стеклование:
-внутрицепные 425 -механическое 110
-водородные 33,120,425 -структурное!10
-глобулы 350 -температурный интервал 118
-межмолекулярные 119 Степень:
-межцепные 425 -полимеризации 346
-обратные 320 -сшивки 175
-физические 120 Стерический фактор 122,425
-цилиндра 49, 349,350 Стерическое препятствие 125
Сегмент: Стирол 267
-Куна 202 Структур а полимера:
-механический 94 -глобулярная 333,350
-макромолекулы 66 -макропористая 56
-молекулярная масса 93,94 -микропористая 55,56
Сера 31, 81 -надмолекулярная 333
Сетки: -модульная333
-редкие 154 -пористая 55
-частые 154 -фибриллярная 349
Симплексная решетка 458 Структурный элемент 116
Скорость: Су бмикр опоры 61
-диффундирующей частицы 321 Сульфонированный'.
-нагревания системы 118 -полиамид 480
-охлаждения системы 118 -поли-п-фениле1ггерефталамцд 480
Совместимость полимеров 472 Схема:
Совол 59 -аддитивная 352
Сополимер: -метода инкрементов 420
-блок-сополимер 23 Тангенс угла:
-и-бутил акр плата с -диэлектрических потерь 112
третбутилметакрилагом 477 -механических потерь 111
-привитый 25 Температура:
-статистический 23 -абсолютная 114
542
Предметный указатель
-интенсивной термической
деструкции 154,224
-критическая 208
-начала ингенсивнойтермической
деструкции 427
-плавления 206, 421
-размягчения сетки 154
-расстекловывания 286
-растворения,
нижняя критическая 475
-стеклования 47,85,426
-текучести 87, 202
-фазового перехода 213
Теория (концепция дравило):
-Patterson and Robard 475
-Волькенштейна-Птицина 117
-Журкова 119
•Ильюшина 316
-поворотно-изомерная 347
-Симхи-Бойера 77
-Флори-Хаггинса 346
Теплоемкость 392
Термодинамическая:
-вероятность 114
-функция 112
Тефлон65
2,4-толуилендиизоцианат 283
1,4-транс-Ь-полиизопрен 49
1,4-транс-полибугадиен 50
Транс-изомер 22
Толуол 267
t-бутиламин 480
Треугольник Гиббса 200
Тример 390
Тризтиламин 480
Углеводор оды:
•замещенные, предельные 21
-монозамещенные, непредельные 22
УглеродЗ!, 81
Узел:
-изоциануратный 286
-сетки 156
-сшивки 26, 153
-трехфункциональный 149,156
•четырехфункциональный 156
Уравнение:
-Арениуса 122
•Больцмана 114
-Больцмана-Вольтерры 293
-Каргина-Слонимского 97
-Клаузиуса-Моссотти 231,259
-Лоренц-Лорентца 231
-Фика 326
-Флори 94
-Фульчера-Таммана 97
-химической кинетики 122
Фазовый переход:
-второгорода 112
-первого рода 112
Фактор пористости 58
Фенолоформальдегидная смола 20,450,480
Фенолфталеин 91
Фенольные смолы 474
Фибриллы 349
Физическое состояние:
-высокозластическое 48
-вязкотекучее 87
-стеклообразное 43,48
Формамид 360
Формула Висвессера 420
Фосфор 31
Фрагменты:
-базовые 441
-линейные 153
-межузловые, усредненные 158
-сетки, повторяющиеся 156
Фронт-фактор 270
Фтор 31,81
Функция:
-отклика 458
-памяти 294
Хлор 31,81
Хлорбензол 268
Хлороформ 342
Цепи полимеров:
-главная 24
-кремнийорганнческие 283
-линейные 24
-разветвленные 24
•сшитые 26
Циклогексан 267
Циклогексанол 342, 268
Циклогексанон 268
Цилиндр взаимодействия 271
Цис-изомер 21
1,4-цис-полнбугадиен 50
1,4-цнс-полиизопрен 50
Частота:
-D-линии натрия 259
-колебаний, собственная 115
Предметный указатель
543
Число:
-Авогадро 32
-звеньев цепи 384
-координационное 82
-повторяющихся звеньев
в сегменте 202
Эластомеры сетчатые 270
Энергия:
-активации низкотемпературного
гамма-перехода 433
-активации процесса
вз аим ©действия 317
-ангармонического осциллятора,
свободная 208
•взаимодействия релаксаторов 318
-внутренняя 114
-водородных связей 79
-вращательного движения 114
-диполь-дипольного
взаимодействия 79
-дисперсионного
взаимодействия 433
-диссоциации
химических связей 218
-когезии 327,365, 384
-межмолекулярного
взаимодействия 79
-перехода 114
-поверхностная 286
-поворотных изомеров, разность 273
-поступательного движения 114
-свободная 114
•связи 122
-сублимации 322
-тепловая 116
-частицы 115
Энтропия 115,294
Эпоксидная смола 26,245
Этаналь 267
Этанол 359
Этил целлюлоза 262
Этилбензол 267
Этилбр о мид 267
2-этилгексил акр плат 467
Этилиодид 268
Этилпропионат 268
Этилформиат 268
Эфир:
-бутнэтиловый, простой 268
-диамиловый, простой 268
-дибутиловый, простой 268
-диглицидиловый 169
-диизоамиловый,простой 268
-диизопропиловый, простой 268
-дипропиловый,простой 268
-диэтиловый, простой 268
-поливинилизобутиловый 214
-поливинилметиловый 476
-полиметилвпниловый 473
-поливинилэтиловый 89
-простой358,416
-сложный 358, 416
Ядро:
-ароматическое 364
-переменная часть 295,319
-релаксации 296, 299, 302,318
Научное издание
Аскадский Андрей Александрович,
Кондращенко Валерий Иванович
КОМПЬЮТЕРНОЕ
МАТЕРИАЛОВЕДЕНИЕ ПОЛИМЕРОВ
т. 1. Атомно-молекулярный уровень
«Научный мир»
119890. Москва, Знаменка, 11/11
Тел./факс (095) 291-28-47.
E-mail: naumir@ben.irex.ru. Internet: www.rfbr.ru
ЛР № 030671 от 09.12.95 г.
Гигиеническое заключение
Нэ 77.99.6.953.П.3619.6.99 от 29.06.1999 г.
Подписано к печати 10.01.2000
Формат 60x90/16
Гарнитура Таймс. Печать офсетная. Усл. печ. л. 34
Тираж 500 экз. Заказ № 123
Издание отпечатано в типографии
ООО “Галлея-Принт”
Москва, 5-ая Кабельная, 26