/



















































































































































































































































































































































































































































































































































































































































































































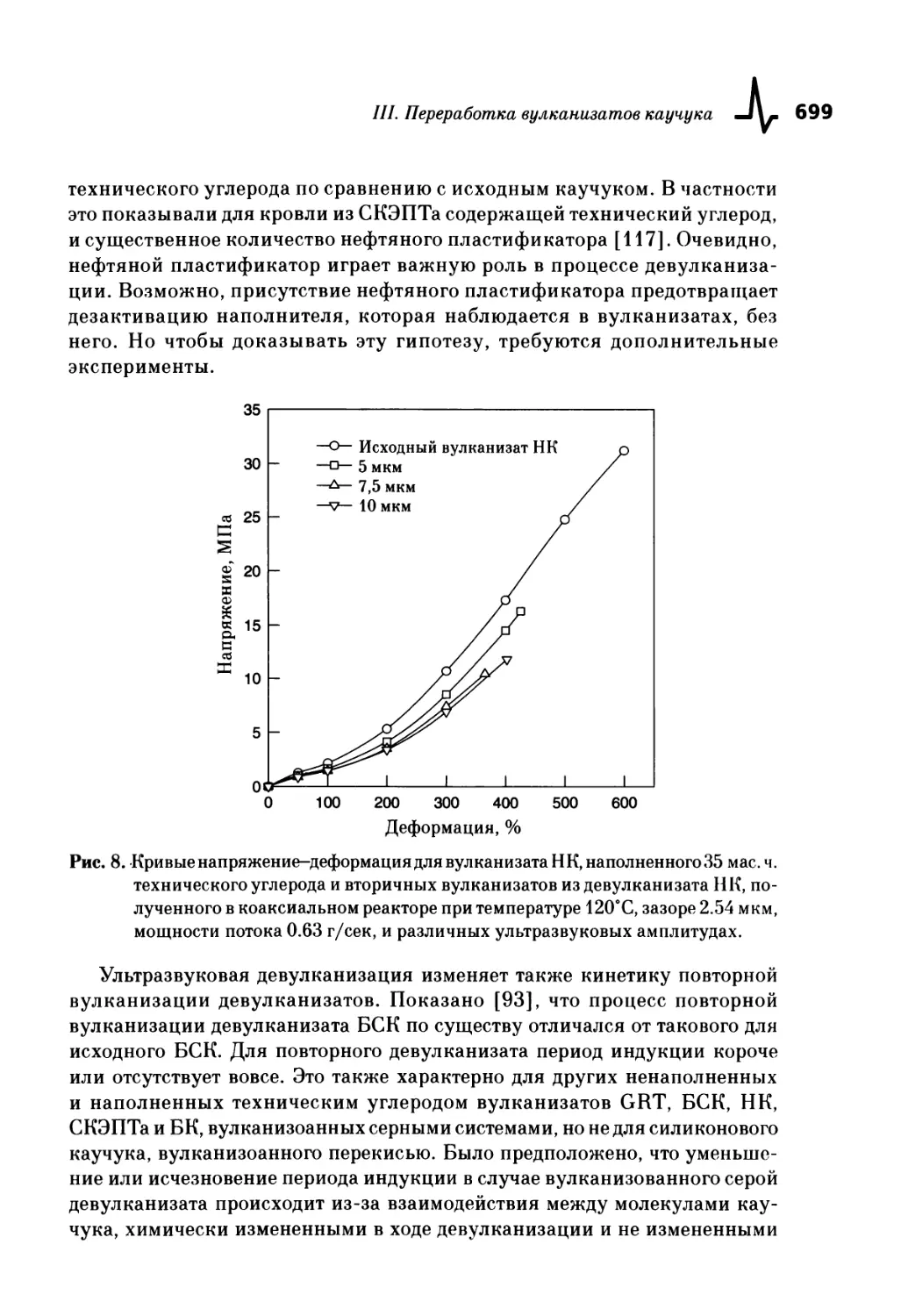





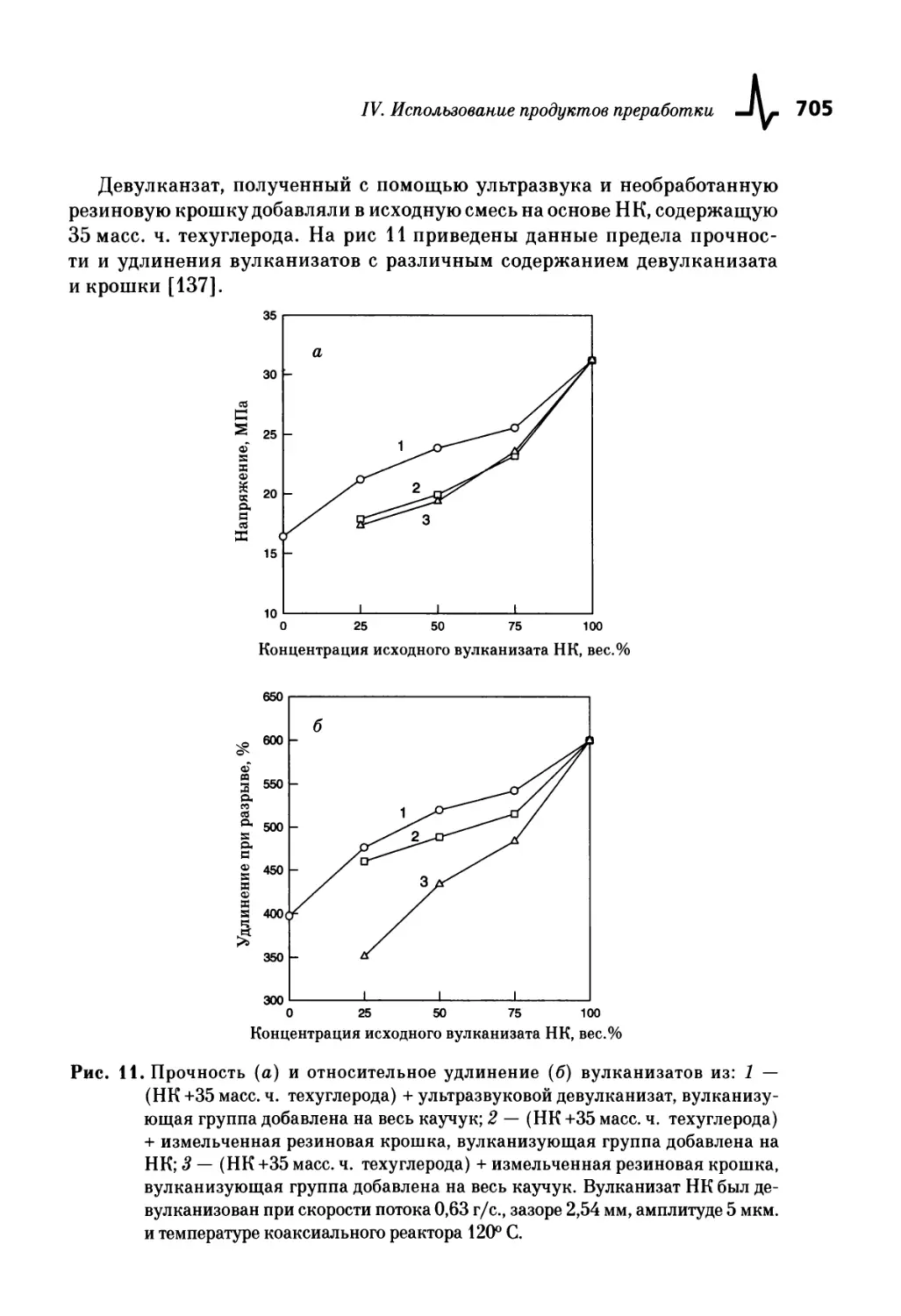




































































Текст
л
Издательский Дом
ИНТЕЛЛЕКТ
КАУЧУК И РЕЗИНА.
НАУКА И ТЕХНОЛОГИЯ
Монография под редакцией
Дж. Марка, Б. Зрмана, Ф. Эйрича
КАУЧУК И РЕЗИНА.
НАУКА И ТЕХНОЛОГИЯ
Монография под редакцией
Дж. Марка, Б. Эрмана, Ф. Эйрича
Перевод с английского
под редакцией А.А. Берлина и ЮЛ. Морозова
л
Издательский Дом
ИНТЕЛЛЕКТ
ДОЛГОПРУДНЫЙ
2011
Дж. Марк, Б. Эрман, Ф. Эйрич (ред.)
Каучук и резина. Наука и технология. Монография. Пер. с англ.: На-
учное издание / Дж. Марк, Б. Эрман, Ф. Эйрич (ред.) — Долгопрудный:
Издательский Дом «Интеллект», 2011. — 768 с.
ISBN 978-5-91559-018-1
Книга, каждая глава которой написана специалистом в соответствующей об-
ласти, содержит основы современных представлений о науке и технике в области
эластомеров. Весь материал изложен четко и ясно и вместе с тем на высоком науч-
ном уровне. Книга будет полезна широкой аудитории специалистов: студентам
старших курсов и аспирантам, инженерам, работающим в резиновой, шинной про-
мышленности и в производстве синтетического каучука, а также во всех отраслях,
где используются эластомерные материалы и изделия. Несомненный интерес вы-
зовет она у разработчиков резиновых изделий и научных работников.
Учебно-справочное руководство выдержало уже три издания на английском
языке, став наиболее авторитетным источником в мировой литературе.
Для химических и химико-технологических факультетов, технических универ-
ситетов.
Science
and
Technology
RUBBER
Third Edition
Edited by
James E. Mark
Department of Chemistry
The University of Cincinnati
Burak Erman
Department of Chemical and Biological Engineering
Кос University
Istanbul. TUrkey
Frederick R. Elrlch
Brooklyn, New York
AMSTERDAM • BOSTON • HEIDELBERG • LONDON
NEW YORK • OXFORD • PARIS • SAN DIEGO
SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO
ISBN 978-5-91559-018-1
© 2005, Elsevier Inc. All rights reserved
© 2011, 000 Издательский Дом «Интеллект»,
перевод на русский язык, оригинал-макет,
оформление
СОДЕРЖАНИЕ
Предисловие редакторов русского перевода... 8
Предисловие к третьему изданию 10
Предисловие ко второму изданию....................................... 10
Предисловие к первому изданию.............. 11
Глава 1
Уцоугость резины: основные положения и поведение
Введение............................./... 13
Ik Упругость одиночной молекулы 13
III. Упругость трехмерной сетки полимерных молекул................17
IV. Сравнение с экспериментом.................................... 22
V. Континуальная теория упругости резины........................ 23
VI. Напряжения второго порядка .................................. 33
VII. Упругое поведение при малых деформациях 34
VIII. Некоторые нерешенные проблемы эластичности резины.. 38
Глава 2
Полимеризация: синтез эластомеров
I. Введение................................................. 39
II. Классификация и кинетическое рассмотрение реакций полиме-
ризации .................................................... 40
III. Полиприсоединение/поликонденсация............................44
IV. Цепная полимеризация по свободнорадикальному механизму 47
V. Эмульсионная полимеризация................................... 55
VI. Сополимеризация.............................................. 66
VII. Цепная полимеризация по катионному механизму... 72
VIII. Цепная полимеризация по анионному механизму 82
IX. Стереоспецифическая цепная полимеризация и сополимеризация
на координационных катализаторах............................. 93
X. Привитая и блок-сополимеризация 104
Глава 3
Характеристика структуры эластомеров
I. Введение.... ИЗ
II. Химический состав........................................... 114
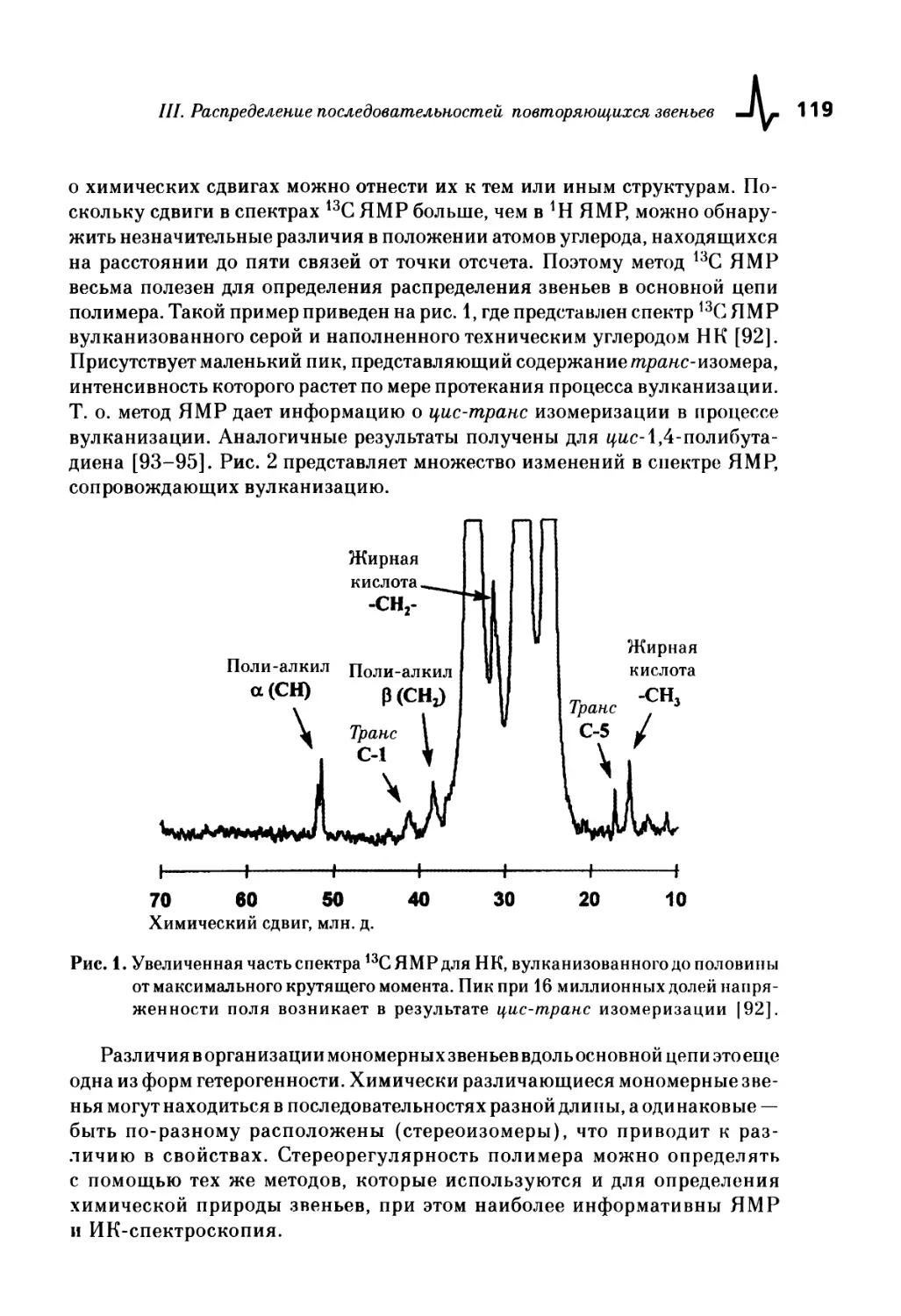
III. Распределение последовательностей повторяющихся звеньев ... 117
4 -’V- Содержание
IV. Архитектура цепи.... 120
V. Стеклование и другие физические релаксационные процессы . 139
VI. Морфология....... 144
Глава 4
Молекулярные основы высокоэластичности
I. Введение......................................................... 162
II. Структура идеальной сетки.. 163
III. Простые молекулярные теории 165
IV. Более продвинутые молекулярные теории 174
V. Феноменологические теории и молекулярная структура . 178
VI. Набухание сеток и восприимчивых гелей 179
VII. Энтальпийная и энтропийная составляющие высокоэластичности:
зависимость сила-температура 182
VIII. Прямое определение молекулярных размеров 184
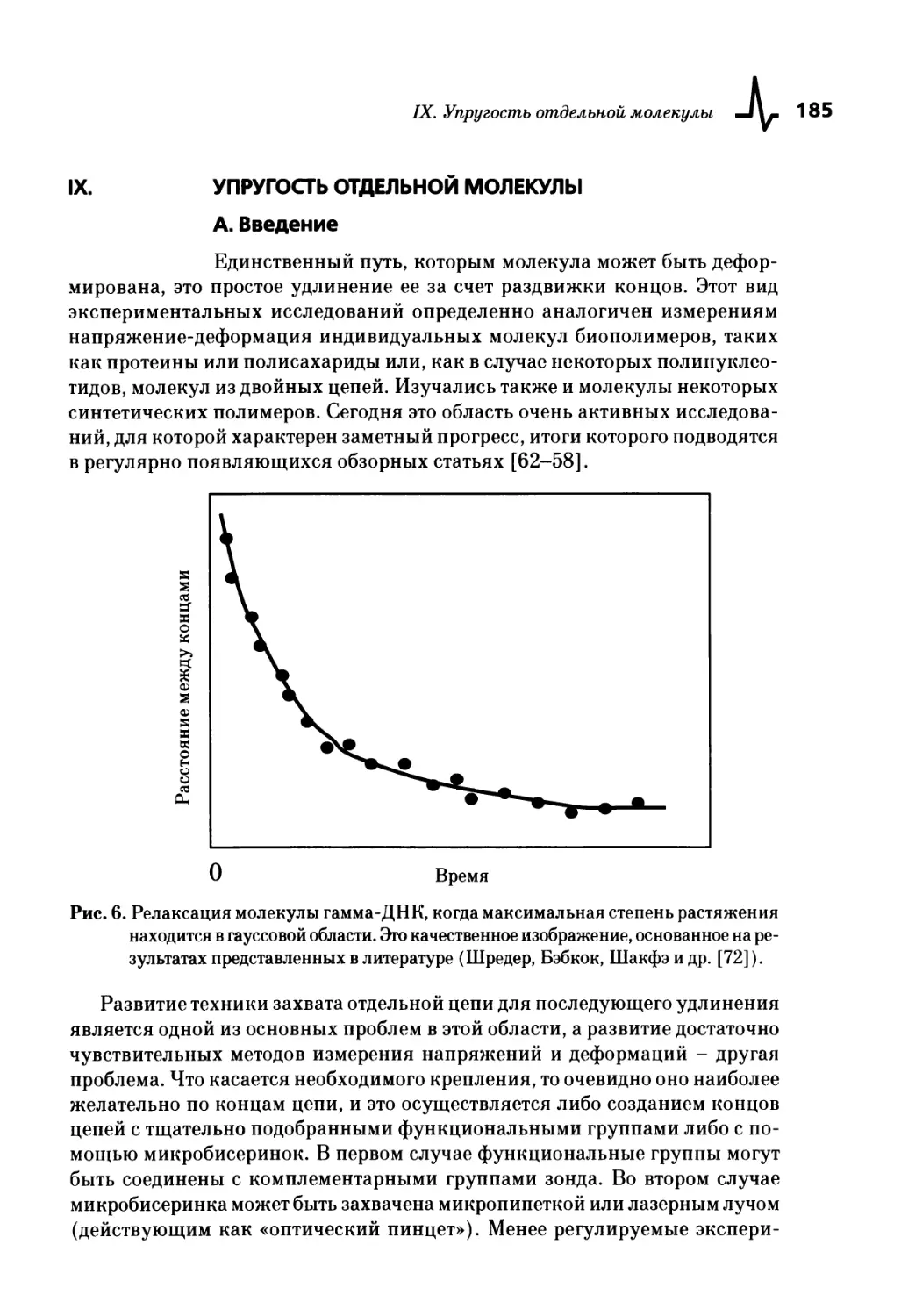
IX. Упругость отдельной молекулы 185
Глава 5
Вязкоупругие свойства каучука
I. Введение... 188
II. Определение измеряемых величин, J(t), G(t) и G*(со) и спектры
L(log X) и H(log т).. 189
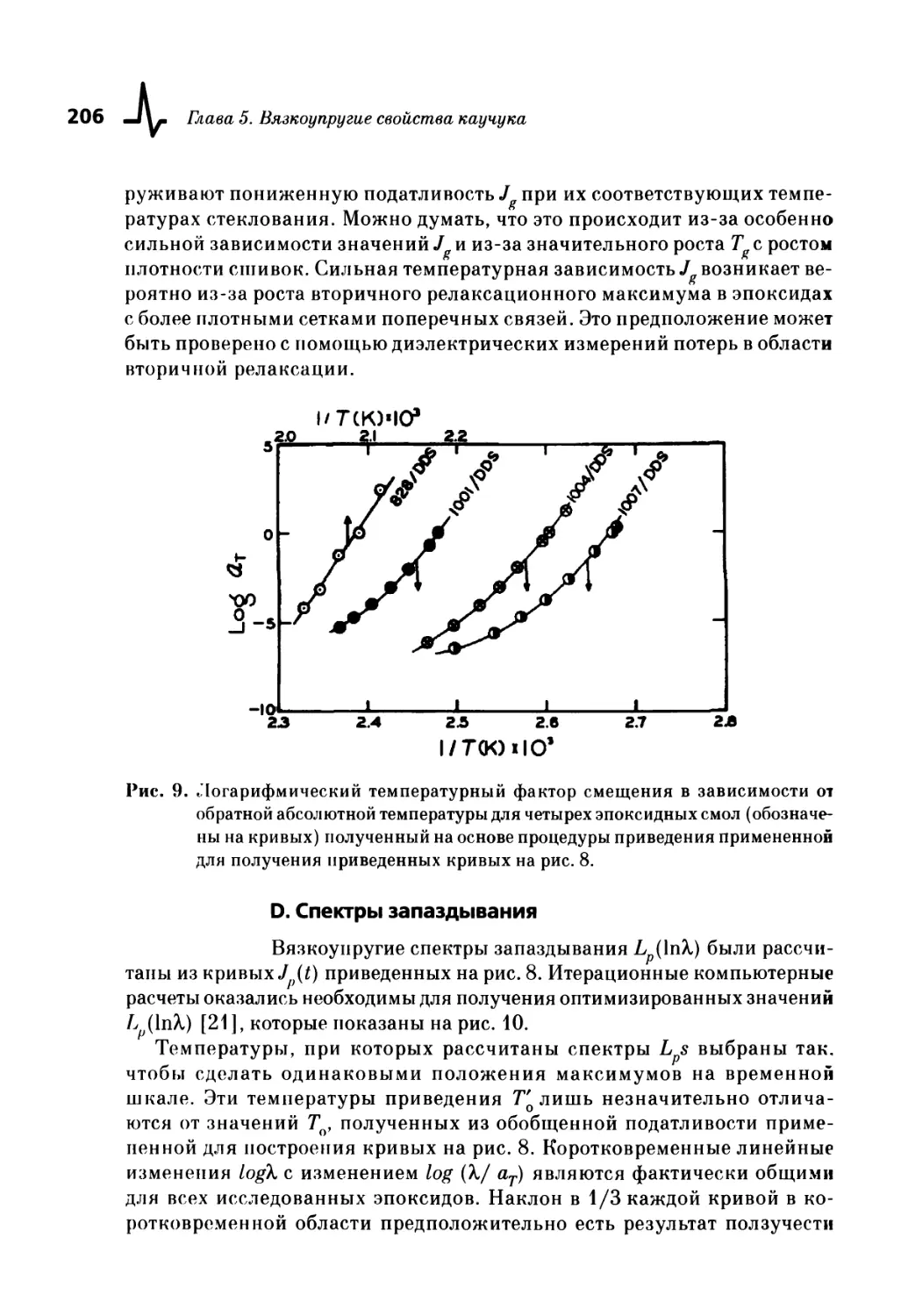
III. Температура стеклования 195
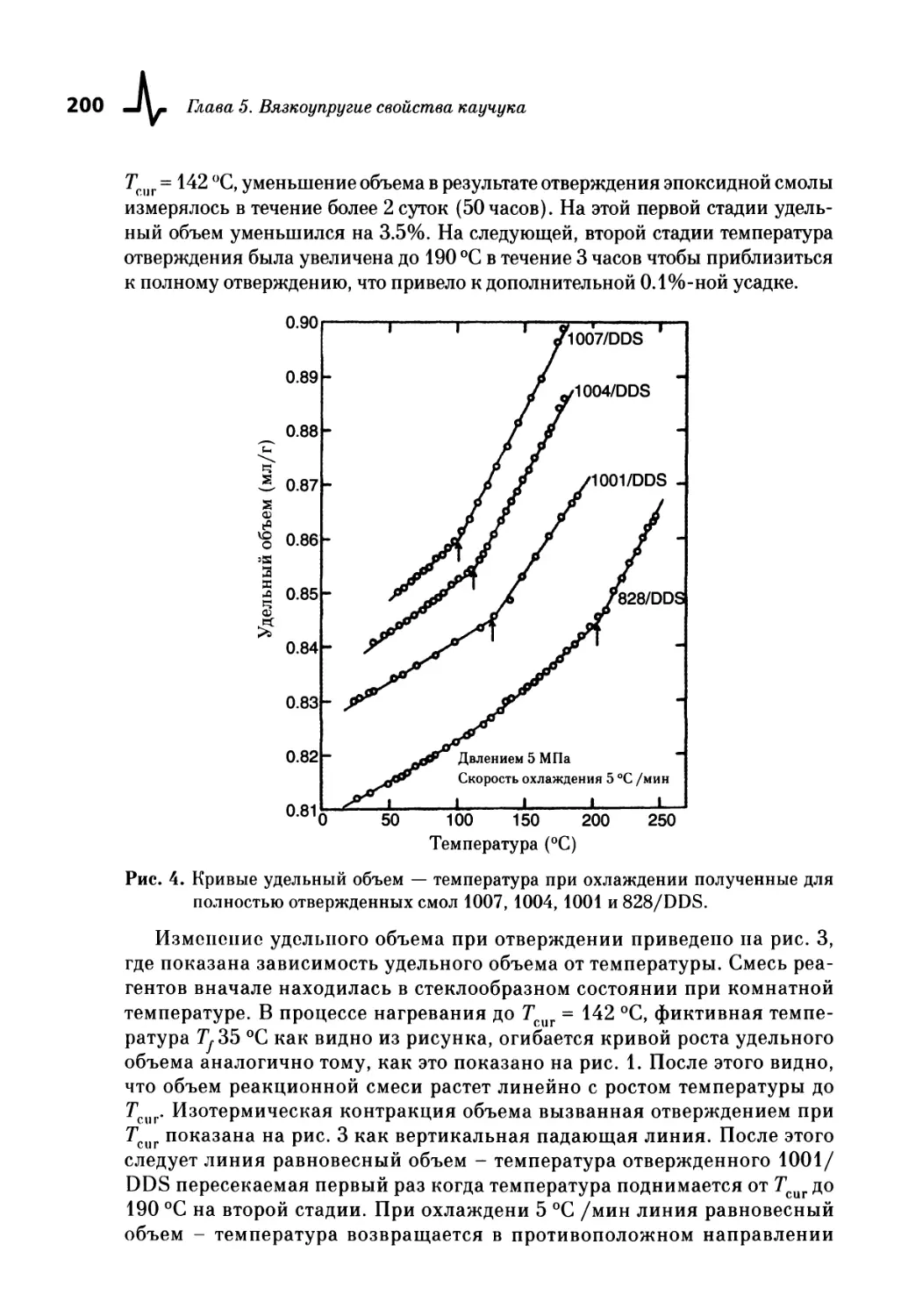
IV. Изменение объема в процессе вулканизации .. 197
V. Вязкоупругие свойства при температуре выше . 202
VI. Вязкоупругие свойства других модельных эластомеров. 208
VII. Расчет энергии раздира эластомеров из данных по их вязкоупругости 219
VIII. Теоретическая интерпретация вязкоупругих механизмов и их аномалий 224
IX. Приложение: список обозначений 242
Г л а в а 6
Реологические свойства и переработка невулканизованного каучука
I. Введение......................................................... 245
II. Основные положения механики 250
III. Реологические свойства... 253
IV. Граничные условия 277
V. Механохимические процессы. 281
VI. Реологические измерения. 285
VII. Технология переработки 293
VIII. Инженерный анализ переработки. 308
Глава 7
Вулканизация
1. Введение... 322
II. Определение вулканизации.......................................... 323
III. Влияние вулканизации на свойства вулканизатов 324
IV. Характеристика процесса вулканизации.............................. 326
V. Вулканизация серой без ускорителей... 329
VI. Вулканизация серой с ускорителями.. 332
VII. Вулканизация производными фенолов, бензохинонов
или бисмалеимидов 353
VIII. Вулканизация под действием оксидов металлов 357
IX. Вулканизация под действием органических пероксидов.................360
X. Динамическая вулканизация.......................................... 366
Содержание -lb- 5
Глава 8
Усиление эластомеров дисперсными наполнителями
1. Введение.................................................370
II. Производство наполнителей... 371
III. Морфологическое и физико-химическое описание наполнителей. ... 373
IV. Смесь как нанокомпозит эластомера и наполнителя......... 386
V. Механические свойства наполненных резин 392
Глава 9
Наука о рецептуростроении резин
I. Введение................................................ 405
II. Полимеры................................................ 406
III. Системы наполнителей 422
IV. Системы стабилизаторов...... 435
V. Вулканизующие системы 442
VI. Специальные ингредиенты смесей 450
VII. Разработка композиций................................... 455
VIII. Получение композиций.................................... 460
IX. Требования к безопасности окружающей среды в рецептуростроении 461
X. Выводы..... 464
Глава 10
Прочность эластомеров
I. Введение................................................ 466
II. Возникновение разрушения... . 467
III. Предельные прочности и растяжимости 474
IV. Разрушение при многоосном напряжении 477
V. Распространение трещины..................................481
VI. Разрыв при растяжении 491
VII. Повторное нагружение: механическая усталость............ 497
VIII. Поверхностное озонное растрескивание 500
IX. Абразивный износ 501
Глава 11
Химическая модификация полимеров
I. Введение................................................. 506
II. Химическая модификация полимеров по основной цепи и концевым
группам................................................. 507
III. Этерификация и гидролиз полимеров...................... 510
IV. Гидрирование полимеров 513
V. Дегалогенирование, отщепление и реакции галогенирования в
полимерах.................................................. 515
VI. Другие реакции присоединения к двойной связи 518
VII. Реакции окисления полимеров 521
VIII. Функционализация полимеров 522
IX. Другие химические реакции полимеров 523
X. Блок и привитая сополимеризация......................... 523
Глава 12
Смеси эластомеров
I. Введение................................................ 540
II. Взаиморастворимые смеси эластомеров. 542
III. Взаимонерастворимые смеси эластомеров.... 550
6 -*v- Содержание
IV. Заключение............................................... 564
Приложение 1: Обозначение названий эластомеров 565
Глава 13
Термопластичные эластомеры
I. Введение................................................. 567
II. Синтез термопластичных эластомеров 573
III. Морфология термопластичных эластомеров.................. 581
IV. Свойства и влияние структуры............................. 603
V. Термодинамика фазового разделения........................ 612
VI. Термопластичные эластомеры в приповерхностных областях. 618
VII. Реология и переработка................................... 627
VIII. Практическое применение 631
Глава 14
Технология шин
I. Введение. . 633
II. Типы и характеристики шин 634
III. Основы конструкции шины 636
IV. Конструирование шины 639
V. Материалы шины 653
VI. Испытания шин............................................ 669
VII. Производство шин 673
VIII. Выводы 679
Глава 15
Вторичная переработка резины
I. Введение. . 680
II. Восстановление шин 681
III. Переработка вулканизатов каучука 682
IV. Использование продуктов переработки...................... 700
V. Пиролиз и сжигание резин................................. 712
VI. Заключение 713
Список литературы. 715
Каучук
u резина
Журнал
издается с ноября 1927 г.
Периодичность
6 номеров в год
Все об эластомерах.
Наука, технология производства, области применения.
Обзоры,
информация о российских и международных конференциях и
выставках, анонсы новых книг.
Содержание журнала
«Rubber Chemistry and Technology».
Журнал переводится на английский язык издательством RAPRA ,
переводы публикуются в журнале
«International Polymer Science and Technology».
В редакции журнала можно
оформить подписку и приобрести комплекты
и отдельные номера журналов за последние годы.
Индекс журнала в каталоге «Роспечать» 70429
Телефон: +7 (499) 245 69 82
e-mail: kir@kired.ru; сайт: www.kired.ru
ПРЕДИСЛОВИЕ РЕДАКТОРОВ
РУССКОГО ПЕРЕВОДА
Эластомерные материалы (каучуки, резины, термоэла-
стопласты) и изделия из них непременный атрибут современной цивилизации,
без которого невозможно себе представить сегодняшнюю жизнь. Присущая им
высокоэластичность — быстрое и обратимое деформирование под нагрузкой на
сотни процентов в широком интервале температур — определяет возможность
функционирования почти всего эластичного, что нас окружает.
Натуральный каучук (НК) оказался первым эластомером, широкое рас-
пространение которого, в отличие от тоже природных целлюлозы и белков,
целиком и полностью явилось заслугой человечества, благодаря которому
НК прошел путь от экзотической игрушки южноамериканских аборигенов до
огромного (более 8 млн. тонн) производства, за счет чего возникла и сущест-
вует шинная промышленность, производство РТИ и огромный ассортимент
других резиновых и латексных изделий.
Природный полиизопрен явился основой известной триады XIX века —
НК, гуттаперчи и эбонита — прообразом современной синтетической поли-
мерной триады — СК, термопластов и реактопластов.
Природное происхождение НК, ранний (начало XIX века) инженерный
и научный интерес к нему сделали НК, а затем и другие каучуки, одними из
наиболее детально изученных высокомолекулярных (как впоследствии было
установлено) объектов. Во многом это определяет устоявшийся характер
науки о каучуке и резине.
Сам факт появления Ш издания предлагаемой книги (первое издание
1978 года) подтверждает вышесказанное. Ее авторы - самые известные
ученые и специалисты в своих областях, в основном из США, а также из
Франции, Англии и Турции.
Форма написания книги — отдельные независимые друг от друга главы,
имеет очевидные достоинства. Основное преимущество таких «сборных»
книг — мобильность: главы можно добавлять, трансформировать, обновлять,
Предисловие редакторов русского перевода 9
что, очевидно, и делалось. Другое принципиальное преимущество: подго-
товленный читатель может каждую главу рассматривать и использовать как
отдельную исчерпывающую книгу по данному вопросу.
Высокое качество книги полностью оправдало избранный подход. В книге
рассмотрен широкий круг тем, охватывающий все важнейшие проблемы
в области эластомеров — от синтеза и модификации каучуков — до вторич-
ной переработки резины.
Глубокое понимание природы высокоэластичности, получения, структуры
и свойств каучуков, сырых резиновых смесей, вулканизатов, ТЭП, механизмов
вулканизации совершенно необходимо современным инженерам и ученым,
профессионально занимающимся эластомерами и изделиями из них. В этой
книге они все это найдут. Книга такого охвата и уровня необходима в России. За
последние 15-20 лет ничего подобного о каучуке и резине из иностранных авто-
ров у нас не переводилось и собственных отечественных не издавалось. Конечно,
в одной, даже объемной книге невозможно отразить все многообразие проблем
каучука и резины. В данном издании предпочтение отдано фундаментальным
физическим, химическим и физико-химическим вопросам. Технология каучука
и резины и изделий из них отражены лишь отчасти.
Третье предлагаемое здесь издание вышло в США в 2005 году. Казалось
бы текст должен был несколько устареть. Ничего подобного. В том-то и смысл
приглашения в авторский коллектив крупных ученых, что они предлагают
читателям устоявшийся, не устаревающий «классический» материал.
Следуя давней, еще советской традиции отметим, что в книге отечест-
венные авторы и их работы не нашли должного отражения. В частности это
касается работ по синтетическому каучуку, где заслуги советских ученых
и инженеров огромны. Много в СССР и России было сделано в области вул-
канизации, понимания природы высокоэластичности, физики эластомеров.
Это находит свое отражение в книгах советских и российских авторов, ко-
торые, кстати, зарубежных работ и авторов не чураются. Мы предоставили
переводчикам, также известным ученым, право сделать краткие дополнения
к переведенным главам. Кто-то сделал это, кто-то посчитал излишним.
Предлагаемая книга по своей тематике и глубине анализа проблем кау-
чука и резины является уникальной и, безусловно, будет полезна всем, кто
профессионально занимается высокомолекулярными соединениями, прежде
всего эластомерными материалами — студентам, аспирантам, инженерам и
ученым соответствующих специальностей.
Книгу перевели: главы 1 и 10 — Б.Л. Бухин и М.Ф. Бухина; главу 2 —
[И.А. Тугорский|, В.А. Шершнев и В.А. Сагомонова; главу 3 — М.Ф. Бухина;
главы 4, 5, 6 и 12 — В.Н. Кулезнев и Е.Ю. Талдонова; главу 7 — В.А. Шер-
шнев; главу 8 — Ю.А. Гамлицкий и К.С. Сдобнов; главу 9 — В.А. Шершнев
и М.В. Дудник; главу 11 — |И.А. Туторский|и Ю.Л. Морозов; главу 13 —
А.А. Канаузова, Т.Т. Рахматулин и Ю.Л. Морозов; главу 14 — Б.Л. Бухин;
главу 15 — Д.Р. Разгон, Ж.В. Перлина и Ю.Л. Морозов.
ПРЕДИСЛОВИЕ
К ТРЕТЬЕМУ ИЗДАНИЮ
Основная цель этого нового издания — актуализировать
материал II издания, которому сейчас более 10 лет. В результате 14 глав
предыдущего издания были переработаны и расширены, и новая глава по
вторичной переработке резин была добавлена.
И мы опять надеемся, что эта книга, представляющая собой широкий
обзор в области эластомеров и их механических свойств будет полезна по-
лимерной науке и инженерному сообществу в целом.
ПРЕДИСЛОВИЕ
КО ВТОРОМУ ИЗДАНИЮ
Цель нового издания этой книги во многом та же, что описано
в Предисловии к I изданию, а именно, широкий обзор эластомеров и кау-
чукоподобной эластичности. Опять-таки необходимо подчеркнуть единый
подход, идущий от химических аспектов, таких как синтез и отверждение
эластомеров, через теоретические вопросы и характеристики равновесных
и динамических свойств, к конечному применению (включая производство
шин и РТИ).
Хотя материал был поделен на те же 14 глав, успехи, достигнутые в нашей
области с 1978 года, потребовали дополнения значительным количеством
нового материала. В результате этого обновления главы стали на 20-30%
длиннее.
За последние 15 лет часть авторов первого издания оставили нас, ушли
на пенсию или занимаются другими исследованиями или вовсе не исследо-
ваниями. Из 23 авторов второго издания примерно половина — новые.
Редакторы, соавторы и издатели надеются, что это новое издание будет
с энтузиазмом встречено читателями, интересующимися полимерной на-
укой и общими инженерными проблемами, прежде всего, специалистами,
работающими в области эластомеров.
ПРЕДИСЛОВИЕ
К ПЕРВОМУ ИЗДАНИЮ
Непреходящий постоянный успех учебного курса, основан-
ного на книге профессора Мортона «Технология резины», изданного Отде-
лением резины Американского химического общества, побудило Комитет
по образованию Отделения выпустить второй, более продвинутый курс.
Отделение поручило подготовить набор глав для студентов высших учебных
заведений и аспирантов, обратив особое внимание на непрерывающиеся
взаимосвязи в области синтеза, структуры, физики и механики эластоме-
ров с проблемами технологии резины и резиновой промышленности. Этот
сборник, с разной глубиной раскрывающий наиболее важные аспекты науки
и технологии каучука и резины, написан рядом авторов — ведущих авто-
ритетов в своих областях. Он интересен не только тем, кто хочет расширить
свое формальное образование, модернизировать его и повысить свой опыт
в этой области, но и тем, кто интересуется необычной химией и физикой,
а также выдающимися свойствами и реальной полезностью эластомеров.
Достаточно высокий уровень представления материала — смеси теории,
экспериментальных данных и практического опыта должен представлять
ценность для студентов, практиков, исследователей и менеджеров.
Глубокое убеждение редактора, базирующееся на многолетнем препо-
давании химии полимеров в Политехническом институте состоит в том, что
наиболее успешный путь преподавания и обучения предмету «полимеры»
заключается в постоянном обращении к специфическим особенностям
макромолекул. Для эластомеров, в частности, наиболее целесообразно объ-
яснять уникальное свойство высокоэластичности гибкостью длинноцепных
макромолекул и сетчатой структурой, а изменения, связанные с большими
деформациями, ключевой ролью, которую играет внутренняя вязкость,
как функция температуры и скорости деформации. Следование авторов
этому подходу неизбежно приводит к некоторым повторениям, но в то же
время позволяет изложить вопросы синтеза и структуры, эластичности
и течения, совмещения, наполнения и вулканизации с разных точек зрения;
такая структура книги дает возможность обходиться без частого обращения к
литературе других глав. По той же причине допускается некоторое варьирова-
ние номенклатуры, особенно если это отражает различные подходы в разных
источниках.
Немного о построении и содержании глав этой композиционной книги,
представляющей собой некую комбинацию информации и инструкции.
Первые 10 глав продвигают читателя от введения к особенностям синтеза,
12 -*v- Предисловие к первому изданию
механического поведения, течения, основным стадиям наполнения, сме-
шения и вулканизации, и далее к теориям и измерению эластичности, имея
в виду строгий материаловедческий подход. Следующие 3 главы посвящены
широкому обсуждению вопросов совмещения, модификации и термоэлас-
топластам. Последняя глава (в нашем издании предпоследняя) по причинам
ограниченности объема оказалась единственной из первоначально запла-
нированных производственных глав. Возможно, эта глава станет предтечей
следующего тома.
Все главы, представляют ли они теорию, механизм, или воззрения авторов
на внутреннюю логику поведения материалов, служат также дополнитель-
ным источником данных и гидом по цитируемой литературе и предметом
дальнейшего самообразования. Если это так, то эта книга является не
только основой нового курса, но также и инструментом и инструкцией для
студентов, преподавателей, а также работников во всех областях полимеров
и материаловедения. Таково было намерение всех авторов, чье напряженное
и терпеливое сотрудничество сделало эту книгу возможной.
Особая благодарность д-ру A. Gessler и Exxon Corporation, Linden, New
Jersy и д-ру E. Kontos, Uniroyal Chemical Division, которые задумали идею
этого второго курса и природу данной книги, а также д-ру Н. Remsberg,
Korlisle Tire and Rubber Company, позднее Президенту Комитета по образо-
ванию, без чьей твердой поддержки и постоянного понимания, эта попытка
ничем бы не закончилась. Доктора Gessler, Kontos и Remsberg были очень
изобретательны при комплектовании авторского коллектива и безжалостны
при анализе ранних вариантов рукописей.
ГЛАВА
1
УПРУГОСТЬ РЕЗИНЫ: ОСНОВНЫЕ
ПОЛОЖЕНИЯ И ПОВЕДЕНИЕ
А.Н. ДЖЕНТ
Университет Акрона. Акрон, Огайо, США
I. ВВЕДЕНИЕ
Уникальное и наиболее важное свойство эластомеров, ко-
торое, собственно и определило их название, это способность подвергаться
большим упругим деформациям, иначе говоря, растягиваться и возвра-
щаться к своей первоначальной форме обратимым путем. Теории, объяс-
няющие это характерное свойство высокоэластичности, прошли через три
различные фазы: ранняя разработка молекулярной модели, основанной на
экспериментальных наблюдениях известных особенностей каучукоподобных
полимеров; затем обобщение этого подхода, с привлечением соображений,
заимствованных из механики сплошных сред, которая не учитывает молеку-
лярную структуру; и в настоящее время, критическая переоценка основных
предпосылок, на которых основаны эти две количественные теории. В этой
главе кратко изложено теоретическое рассмотрение и показано как можно
вполне успешно рассчитать наблюдаемое упругое поведение каучукоподоб-
ных материалов. Затем, из-за его технической важности, в некоторых деталях
обсужден специальный случай малых упругих деформаций. В заключение
внимание привлечено к некоторым аспектам упругости резины, которые
еще недостаточно понятны.
II. УПРУГОСТЬ ОДИНОЧНОЙ МОЛЕКУЛЫ
Вещество каучукоподобно, только если оно состоит из
длинных гибких, подобных цепям молекул. Поэтому сами молекулы
должны иметь основную цепь («становой хребет»), состоящую из мно-
гих неколлинеарных единичных валентных связей, вокруг которых
в результате теплового движения возможно быстрое вращение. Повторя-
ющиеся единицы молекул некоторых представителей каучукоподобных
полимеров показаны на рис. 1; тысячи таких единиц связаны вместе
14 -*V" Глава 1. Упругость резины: основные положения и поведение
в цепь, образующую типичную молекулу эластомеров, приведенных на
рис. 1. Такие молекулы быстро и непрерывно изменяют свою форму при
обычной температуре из-за броуновского движения. В отсутствие напря-
жений конформация молекул случайна, но они приобретают до некоторой
степени ориентированную конформацию, если к их концам приложена
растягивающая сила (рис. 2). Один из первых вопросов для рассмотре-
ния это соотношение между приложенным растягивающим усилием f
и средним расстоянием между концами цепи г, усредненным по времени
или по большому числу цепей в один момент времени.
н
н-СхН
I /н
с=с
Н / \ /Н
--с /С
'н н
цис-1,4-пол ибутадиен
Н Н
hk I /Н Н\ 1/Н
С. /С
п ол и изобутиле н пол ид и метилеилоксан
цис-i, 4-пол иизопрен
Рис. 1. Повторяющиеся единицы молекул некоторых обычных каучукоподобных
полимеров
Рис. 2. Случайная (а) и ориентированная (б) конформации цепи
Изолированные цепи имеют сильно различающиеся конформации1*,
определяемые тремя факторами: статистикой случайных процессов;
преимуществом для некоторых последовательностей расположения
связей из-за стерических и энергетических ограничений внутри моле-
кулы; исключением некоторых гипотетических конформаций, которые
требуют, чтобы части цепи занимали один и тот же объем в пространстве.
Кроме того, по пространственным соображениям в концентрированных
Хотя термины конформация и конфигурация используются как взаимозаменяемые,
первый имеет специальное значение в органической стереохимии и им обозначают спе-
цифические стереоструктуры. Термин конформация используется здесь для обозначения
конфигурации молекулы, которая появляется при вращении единичных валентных связей
в основной цепи.
II. Упругость одиночной молекулы 15
растворах или в твердом состоянии предпочтительны кооперативные
конформации
Флори [1] показал, что для изолированных цепей в твердом состоянии
исключение некоторых конформаций, занимающих один и тот же объем,
точно сбалансировано внешним (отталкивающим) окружением подобных
цепей и что поэтому для твердого состояния фак-
тор исключения некоторых конформаций можно
игнорировать. Прямое наблюдение размеров
единичной цепи в твердом состоянии методом
неупругого нейтронного рассеяния дает вели-
чину, полностью согласующуюся с размерами
невозмущенной цепи, полученными для разбав-
ленных растворов в тета растворителях2^ [2], хотя
внутримолекулярные эффекты могут искажать
локальную случайность конформации цепи.
Флори также выдвинул убедительные при-
чины для заключения о том, что расстояние
между концами цепи г в твердом состоянии
Рис. 3. Модельная цепь со
свободно сочлененными
связями
описывается Гауссовым
распределением для достаточно длинных цепей, даже если их короткие
участки относительно жесткие и негибкие [1]. Из этого следует, что соот-
ношение усилие растяжения — смещение для длинных цепей становится
простым линейным
/ = Лг
(1)
где/ — растягивающая сила, г — среднее расстояние между концами цепей
и А — величина, обратная среднеквадратичной величине расстояния между
концами г02для ненапряженной цепи
А = ЗкТ/г% (2)
где к — постоянная Больцмана и Т — абсолютная температура.
Если заменить реальную молекулу гипотетической цепью, состоящей из
большого числа п жестких свободно сочлененных звеньев, каждое длиной I
(Рис. 3), тогда
Го=п/2 (3)
В этом случае г02не зависит от температуры, поскольку предполага-
ется полностью случайное расположение звеньев. Растягивающая сила /
в уравнении (1) увеличивается тогда исключительно по энтропийному
2) Это растворители (плохие), в которых отталкивание между различными сегментами
полимерной молекулы уравновешивается отталкиванием между сегментами полимерной
молекулы и молекулами растворителя.
16 Глава 1. Упругость резины: основные положения и поведение
механизму, т. е. из-за тенденции цепи принимать максимально случайную
конформацию, а не из-за какого-либо энергетического преимущества одной
конформации перед другой. Тогда сила /прямо пропорциональна абсолют-
ной температуре Т.
Для реальных цепей, состоящих из большого числа п главных валентных
связей вдоль основной цепи, каждая длиной I
Л=С„п12 (4)
где коэффициент Сж представляет степень отклонения поведения реальной
молекулы от свободно сочлененной модели. Найдено, что варьируется
от 4 до 10, в зависимости от химической структуры молекулы и также от
температуры, поскольку энергетические барьеры упорядочения случайных
связей более легко преодолеваются при высоких температурах [1]. Таким
образом величина С°УЧ может рассматриваться как эффективная длина
связи реальной цепи и как мера «жесткости» молекулы.
Уравнение (1) достаточно точно только для относительно коротких рас-
стояний г, меньших, чем примерно одна треть полной длины растянутой
цепи [2]. К сожалению, отсутствуют хорошие результаты для натяжения
в реальных цепях при больших расстояниях между концами. Таким обра-
зом, мы должны возвращаться к модели цепи со свободно сочлененными
связями, для которой
f=(kTll)L-x{rlnl) (5)
где Л-1 обратная функция Ланжевена. Разложение этого уравнения по сте-
пеням r/nl имеет вид
/ = (3Zc7>/nZ2)[l+(3/5)(r/nZ)2 (
+ (99/175)(r/n/)4 +(513/875)(r/nZ)6 + -
Это дает полезное указание на то, где можно ожидать значительные от-
клонения от уравнения (1).
Согласно уравнению (5) кривая, описывающая соотношение между
натяжением и расстоянием между концами цепи, круто поднимается, когда
растяжение цепи приближается к предельному (рис. 4), в противополож-
ность Гауссовому распределению, представленному уравнением (1), которое
перестает описывать поведение цепи при г > (54) nl. Кривая, описывающая
экспериментальное соотношение между напряжением растяжения и удлине-
нием для резины подобным образом, круто поднимается при больших удли-
нениях. Действительно, в сеточной теории упругости резины использование
уравнения (5) вместо уравнения (1) дает хорошее соответствие эксперимен-
тальному соотношению напряжение-деформация (см. следующий раздел).
III. Упругость трехмерной сетки полимерных молекул
Л
17
Деформация, при которой начинается малое, но значимое отклонение между
наблюдаемым напряжением и тем, что предсказано по теории малых де-
формаций по уравнению (1), дает величину эффективной длины I свободно
соединенных связей для ре-
альной молекулярной цепи.
Это предоставляет прямую
экспериментальную меру
жесткости молекулы. Для
единственного полимера,
который пока был исследо-
ван этим методом, цисЛД-
полиизопрена [3], получен-
ная величина относительно
велика, порядка 5-15 связей
в главной цепи.
Уравнение (5) также было
r/nl
Рис. 4. Соотношение натяжение-смещение кон-
цов для цепи со свободно сочлененными связями,
согласно ур. (5); —Гауссово приближение, со-
гласно ур. (1) [37]
использовано для вычис-
ления силы, при которой
молекула каучука при дефор-
мации наполненной резины
отделяется от частиц усилива-
ющего наполнителя, например технического углерода [4]. На этом пути было
получено общее полуколичественное описание размягчения наполненных
резин под действием напряжения (эффект Маллинза), см. рис. 5.
III. УПРУГОСТЬ ТРЕХМЕРНОЙ СЕТКИ ПОЛИМЕРНЫХ
МОЛЕКУЛ
Чтобы образовать связное твердое тело и предотвратить тече-
ние молекул эластомера как жидкости, необходимо образование некоторой
постоянной структуры. Этому требованию отвечает введение малого числа
межмолекулярных химических связей (поперечных связей) для создания
свободной трехмерной молекулярной сетки. Обычно предполагается, что
такие поперечные связи образуются в наиболее вероятных позициях, так что
длинные участки молекул между ними имеют тот же самый спектр расстоя-
ний между концами, какой имело бы аналогичное множество несвязанных
молекул. Под действием броуновского движения каждая секция молекул
может иметь такой же, как и раньше набор конформаций, но теперь они
подчиняются условию, что их концы лежат в местах поперечных связей.
Упругие свойства такой молекулярной сетки будут рассмотрены позже.
Сначала рассмотрим другой тип взаимодействия между молекулами.
Полимеры с высоким молекулярным весом при запутывании молекул
образуют зацепления с расстоянием между ними (в твердом состоянии),
18 JV‘ Глава 1. Упругость резины: основные положения и поведение
Рис. 5. Ориентационное размягчение
наполненного техническим углеродом
вулканизата бутадиен-стирольного со-
полимера (25:75); ---, кривая напряже-
ние-деформация для ненаполненного
вулканизата [5]
характерным для конкретной молекулярной структуры [6]. Некоторые
типичные значения молекулярного веса Ме между зацеплениями показаны
в Таблице 1. Следовательно, даже в отсутствие каких-либо постоянных межмо-
лекулярных связей для полимерных
расплавов с высоким молекулярным
весом будет характерно переходное
каучукоподобное поведение.
В резине с поперечными связями
многие из этих зацеплений посто-
янно локализованы (рис. 6), тем
более чем выше степень поперечного
сшивания. Если рассматривать их
как полный эквивалент поперечных
связей, то эффективное число цепей
сетки на единицу объема N можно
определить как сумму двух вели-
чин N и N, т. е. числа зацеплений
и химических поперечных связей,
соответственно, где
Ne=pNjMe, Nc=pNjMc
ир — плотность полимера, АЛ — чис-
ло Авогадро, МG и Мс — значения
среднего молекулярного веса между
зацеплениями и поперечными связя-
ми, соответственно. Эффективность
зацеплений в ограничении отде-
льных цепей является, однако, несколько неопределенной, в частности, когда
число химических связей относительно мало [7-9]. Более того, соотношение
между силой и удлинением для цепи с зацеплениями будет отличаться от того
же соотношения для цепи с поперечными связями [10], причем оно будет бо-
лее жестким в начале и нелинейным по форме. Поэтому эффективное число А
для молекулярной цепи, которая лежит между фиксированными точками
(т. е. между поперечными связями и молекулярными зацеплениями), яв-
ляется несколько условной величиной, даже когда химическая структура
сетки полностью определена.
Удобно определять упругое поведение сетки в терминах плотности
энергии деформации W на единицу недеформированного объема (упруго-
го потенциала). Энергия деформации w для отдельной цепи определяется,
исходя из уравнения (1), как
w = Ar2/2
III. Упругость трехмерной сетки полимерных молекул -'V 19
Таблица 1. Представительные величины среднего молекулярного веса Ме между
зацеплениями для расплавов полимеров3
Полимер Ме Полимер Ме
Полиэтилен 4 000 Полиизобутилен 17 000
цис-1,4 Полибутадиен 7 000 Полидиметилсилоксан 29 000
^ис-1,4 Полиизопрен 14 000 Полистирол 35 000
л| Получено из измерений вязкости
Рис. 6. Схема постоянно локализованного зацепления [37]
Для статистической сетки из N таких цепей при сложной деформации,
характеризуемой тремя степенями удлинения Х3 (отношениями дефор-
мированных и недеформированных размеров) в трех главных направлениях
(рис. 7), Ж определяется так [И]:
W = NAr2(tf +Х^+^-3)/б (8)
где rf2 — среднеквадратичное расстояние между концами цепи (места по-
перечных связей или эквивалентные сочленения) в недеформированном
состоянии. Подобие уравнений (7) и (8) очевидно, особенно если принять
^=(rf2/3)(x12+x22+V)-
Для статистического поперечного сшивания можно принять, что
rf2 эквивалентно г02, соответствующему среднеквадратичному расстоянию
между концами несвязанных цепей той же самой молекулярной длины.
Поскольку?! обратно пропорционально г02, см. уравнение (2), единственный
молекулярный параметр, остающийся в уравнении (8), это число /Уупругих
эффективных цепей на единицу объема. Таким образом, предсказывается
зависимость упругого поведения молекулярной сетки при средних дефор-
20 Глава 1. Упругость резины: основные положения и поведение
мациях только от числа молекулярных цепей, а не от их гибкости. При этом
предполагается, что они достаточно длинны, чтобы подчиняться Гауссовой
статистике.
Хотя обычно предполагается, что rf2 и г02 эквивалентны при температуре
образования сетки, они могут сильно отличаться при других температурах
из-за температурной зависимости г02 для реальных сеток, см. уравнение (4).
Действительно, температурная зависимость упругих напряжений каучуко-
подобных сеток широко применялась для изучения температурной зависи-
мости г02, см. [1,9].
Рис. 7. Недеформированное (а) и деформированное (б) состояния
Другой причиной, из-за которой rf2 и г02 могут различаться, является
изменения сетки после ее формирования. Например, когда сетка при
набухании поглощает жидкость, г2 для набухшей сетки будет возрастать
в отношении Xs2b сравнение с её первоначальной величиной, где Xs
линейная степень набухания. В то же время число цепей на единицу
объема сократится в отношении Xs-3. Следовательно, плотность энергии
деформации при данной деформации будет меньше для набухшей сетки
в отношении Xs-1.
Из общего соотношения для упругой энергии, уравнение (8), следует,
что упругие напряжения, требуемые для поддержания любой заданной де-
формации, могут быть получены посредством виртуального рассмотрения
работы (рис. 7)
= ЭИЛ/ЭХ1
с аналогичным соотношением для t2 и t3. Из-за практической несжимаемости
каучукоподобных материалов по сравнению с их легкой деформируемостью
другими способами, первоначальный объем приблизительно сохраняется при
деформации. Поэтому степени удлинения связаны простым соотношением
ХЛА3=1 (9)
III. Упругость трехмерной сетки полимерных молекул 21
В результате получаем соотношение напряжение - деформация
=Х1(ЭИг/ЭХ1)-р, и аналогично
где р означает возможное гидростатическое давление (которое не влияет
на несжимаемое тело). Следовательно, только разности напряжений могут
быть описаны явно
-t2 = (№4г2/з)(А-1 - ) (10)
Для простого растяжения, например в направлении 1, мы положим =
X и Х2 = Х3 = X-1/2 [из уравнения (9)], и /2 = /3 = 0. Следовательно
<(=<1) = ^№4г2/з)(Х2-X”1) (И)
Естественно выразить этот результат в терминах растягивающей силы/,
действующей на испытуемый образец поперечного сечения Ао в недеформи-
рованном состоянии, где//Л0 = t/X
Соответствующее соотношение
показано на рис. 8. Оно иллюстриру-
ет общую особенность эластического
поведения каучукоподобных тел; хотя
поведение составляющих цепей под-
чиняется линейному соотношению
сила-растяжение [уравнение (1)],
поведение сетки нелинейно. Эта осо-
бенность возникает из-за геометрии
случайно ориентированных цепей.
Действительно, степень нелинейности
зависит от типа приложенной деформа-
ции. При простом сдвиге предсказыва-
ется, что соотношение будет линейным
с наклоном (модуль сдвига G), опреде-
ляемым так
А (»!♦«)
Рис. 8. Соотношение сила-растяжение
для простого растяжения; Ли-
нейное соотношение для бесконечно
малых деформаций [37]
tl2 = Gy,G = JVAr2/3 (12)
где у величина сдвига, т. е. dx/dy.
Поскольку каучукоподобные
материалы фактически несжимаемы
в твердом состоянии, величина коэффициента Пуассона близка к 0,5.
Поэтому модуль Юнга Е определяется как 3G с хорошим приближением;
однако предсказываемое соотношение между напряжением и деформа-
цией растяжения е (= X - 1) линейно только для очень малых удлинений
22 -*V Глава 1. Упругость резины: основные положения и поведение
(рис. 8), так что модуль Юнга применим только для растяжения или
сжатия в несколько процентов.
Все соотношения для напряжений, приведенные выше, выводятся из
уравнения (8). Таким образом, они справедливы только для небольших
деформаций сетки, т. е. для деформаций, которые достаточно малы, для
того, чтобы можно было принять линейность между натяжением цепей
и расстояниями г между их концами [уравнение (1) ]. К сожалению, нет со-
ответствующего простого выражения, сформулированного для W, используя
уравнение (5), т. е. соотношения для больших деформаций составляющих
сетку цепей, в котором бы снова появлялся параметр молекулярной жестко-
сти. Вместо этого, чтобы получить близкую аппроксимацию поведения сетки
резины при больших деформациях, могут быть использованы различные
аппроксимации рядами, как в уравнении (6) [12].
IV. СРАВНЕНИЕ С ЭКСПЕРИМЕНТОМ
Описание эластичности резины, данное в предыдущем
разделе, в общем достаточно успешно, но некоторые несоответствия все
же обнаруживаются. Первое состоит в том, что наблюдаемые напряжения
выше, чем предсказанные, например, по уравнению (И); дополнительный
вклад часто называют членом С2. Этот вклад относительно велик при малых
деформациях (хотя это всегда меньшая часть наблюдаемых напряжений);
роль его уменьшается при возрастании деформаций. Он также уменьшается,
когда сетка расширяется при набухании в инертной жидкости, и становится
равным нулю при степени набухания около 5. Следовательно, «С2-напряже-
ние» появляется как отражение не-гауссовой характеристики сетки цепей,
которая важна только при малых величинах расстояния между концами
цепей г. Действительно, Томас [13] показал, что величина напряжения от
С2 и его сложная зависимость от типа и степени деформации и от степени
набухания может вся быть аккуратно описана простым дополнительным
членом в соотношении для энергии деформации w для одиночной цепи сетки,
и уравнение (7), принимает вид
2w = Ar2+Br~2 (13)
Второй член явно становится незначимым при больших г.
Дальнейшее доказательство, имеющее отношение к физической природе
несоответствий, дается двумя другими наблюдениями: для С2не выявлено
сильной зависимости от температуры и, следовательно, эта величина не свя-
занна с энергетикой конформаций цепи, а тесно коррелирует с тенденцией
полимерной цепи к образованию молекулярных зацеплений. Например,
те полимеры, которые имеют высокую плотность зацеплений в твердом
состоянии (см. Таблицу 1) образуют каучукоподобные сетки, для которых
компонент напряжения, связанный с С2, относительно велик [9].
V. Континуальная теория упругости резины -*и- 23
В заключение отметим, что отсутствуют доказательства того, что поведе-
ние изолированных цепей в тета растворителях не согласуется с Гауссовой
статистикой, поэтому представляется, что расхождения, связанные с С2,
начинают возникать, когда молекулярные цепи связываются в сетку.
Все это подтверждает связь «С2-напряжений» с поведением цепей сетки,
входящих в зацепления (рис. 6), и особенно с различиями в ограничениях
на доступные конформации для этих и других цепей сетки [7-9]. Прагер и
Фриш [10] показали, что поведение цепей, входящих в модельные зацеп-
ления, определяется отличающейся статистикой; их заключения полностью
согласуются с тем, что известно о напряжениях, определяемых С2.
Второе расхождение между теорией и экспериментом было обнаружено,
когда Гауссова часть измеренных напряжений была сравнена с теоретиче-
ским результатом для идеальной сетки. Между плотностью эффективных
цепей, вычисленной по наблюдаемым напряжениям и по химии поперечного
сшивания, были получены численные различия до 50%. Это расхождение
может быть обусловлено ошибкой в теоретическом анализе, данном здесь.
Джеймс и Гут [14], основываясь на несколько отличных теоретических ис-
ходных позициях, получили напряжения, составляющие только половину
полученных по уравнению (10).
Третье и главное расхождение, указанное ранее, относится к большим
деформациям, когда сетка цепей перестает подчиняться Гауссовой стати-
стике, даже приближенно. Значительный успех в этом случае был достигнут
при использовании для определения напряжения в сетке уравнения (5)
вместо уравнения (1).
Несмотря на эти расхождения простая трактовка упругости резины,
описанная в этом разделе, оказывается замечательно успешной в объясне-
нии упругих свойств резин при средних деформациях до, примерно, 300%
от длины до деформации (в зависимости от длины и гибкости и, следова-
тельно, от растяжимости составных частей цепи). Корректно предсказаны
главные формы соотношений напряжение — деформация для разнообраз-
ных деформаций, приближенные численные величины напряжений для
полимеров различной химической структуры, а также влияние на упругое
поведение температуры и набухания резины в инертной жидкости. Также
предсказаны новые напряжения второго порядка, не имеющие аналогов
в классической теории упругости, которые будут рассмотрены позже. В
целом, выше представлены главные достижения в понимании свойств по-
лимерных материалов.
V. КОНТИНУАЛЬНАЯ ТЕОРИЯ УПРУГОСТИ РЕЗИНЫ
Общая трактовка соотношений напряжение-деформация
каучукоподобных твердых тел была разработана Ривлиным [15,16] в един-
ственном предположении, что материал изотропен при упругом поведении
24 -*V Глава 1. Упругость резины: основные положения и поведение
в недеформированном состоянии и несжимаем. Совершенно удивительно
заметить, какие далеко идущие заключения следуют из этих элементарных
предположений без рассмотрения молекулярной структуры.
Соображения симметрии позволяют предположить, что соответствующее
описание деформации можно дать тремя инвариантами деформации, опре-
деленными следующим образом
Л = X?Хо + Х2Х3 + А,3 А,? - 3
J, =^1^2^3 “1
где Х2, А3 — главные степени удлинения (отношения длин в растянутом
и нерастянутом состояниях, рис. 7). Более того, для несжимаемого материала
/3 тождественно равно нулю и, следовательно, остаются только две независи-
мые меры деформации, Jx и J2. Следовательно, плотность энергии деформации
W (упругий потенциал) является функцией только этих двух переменных:
W = f(Jx,J2) (14)
Далее, считая, что соотношения напряжение — деформация при малых
деформациях линейны, получим, что прежде всего Wдолжно быть функ-
цией второго порядка от деформаций ev е2, е3. Следовательно, простейшая
возможная форма для функции энергии деформации такова
W = CtJi+C2J2 (15)
где С\ и С2 коэффициенты упругости, а их сумма 2(Ci + С2) эквивален-
тна модулю сдвига G для малых деформаций. Уравнение (15) впервые
предложено Муни [17] и часто называется уравнением Муни-Ривлина.
Заслуживает внимания, что первый его член соответствует соотноше-
нию, полученному исходя из молекулярной теории упругости резины,
а именно уравнению (8), если коэффициент ^идентифицировать как
Nar2/& = (l/2)7V/cT(r/r0)2.
При разложении уравнения (15) в степенной ряд по деформациям е,
где е = X - 1, появляются члены с е2 и е3. Следовательно, это неизбежно дает
хорошее согласие с экспериментом при малых деформациях, скажем до
величин е до 10 и 20%, где более высокие степени е пренебрежимо малы.
Однако значительные разочарования возникают при применении этого
соотношения к большим деформациям для величины е в 100% или более,
где оно теряет силу. Скорее неудачно, что экспериментальное соотношение
напряжение-деформация при простом растяжении оказывается в согласии
с уравнением (15) вплоть до умеренно больших деформаций. Это случайное
согласие возникает потому, что специальная функция энергии деформации
V. Континуальная теория упругости резины -*v* 25
для резины, обсуждаемая далее, зависит от деформации таким образом, что
два соотношения напряжение-деформация подобны по форме при растя-
жении. Соотношения для других типов деформации совершенно различны,
даже для небольших деформаций [18].
А. Соотношения напряжение-деформация
Напряжения могут быть определены через производные от
функции энергии деформации W:
t^XfldW/d^-p (16)
Переписывая уравнение (16) в терминах частных производных 3W/dJ{ и
dW/dJ2 получим
= 2[X? Э W/dJj - (1Д? )э W/dJ2 ] - р, t2wt3 аналогично (17)
Функции dW/dJi и ЭЖ/ЭУ? соответственно обозначим как и VK2.
Экспериментальные измерения показывают, что приблизительно
постоянно. Однако второй член далеко не константа, даже при умеренных
деформациях. Хорошее согласие получается, когда он выражен как лога-
рифмическая функция J2 [19]:
W = + C2'Zn[(/2 + 3)/3] (18)
где С2 константа. Эта форма второго члена дает достаточно хорошее чис-
ленное согласие с предсказанием по дополнительному слагаемому Томаса
в функции энергии деформации для одиночной цепи, см. уравнение (13),
и проще по форме.
Величины С\ и С2 близки по величине, от 0,25 до 0,5 МРа, для типичных
мягких вулканизатов. Однако, тогда как С\ приблизительно пропорциональ-
но числу цепей сетки на единицу объема, С2 скорее является константой,
независимой от степени сшивания, и таким образом относительно более
значимой для слабо сшитых материалов. Как отмечено ранее, по-видимому,
это отражает физические ограничения, накладываемые на молекулярные
цепи. Они подобны представленным в модели «трубка» для ограничений
конфигураций в конденсированном состоянии [9]. Это ограничения,
важность которых уменьшается с ростом деформации или с возрастанием
разделения цепей.
Затруднение деформирования при больших деформациях
Резину становится труднее деформировать при больших деформациях,
вероятно из-за того, что длинные гибкие молекулярные цепи, из которых
состоит материал, не могут растягиваться неограниченно. Функция энергии
26 -*V- Глава 1. Упругость резины: основные положения и поведение
деформации (упругий потенциал), рассматриваемая до этого момента, не
учитывала эту особенность и не могла описывать поведение при больших
деформациях. Затруднение деформирования может быть введено простой
модификацией первого члена в уравнении (18), включением максимально
возможной величины для инварианта деформации обозначенной Jm [20]:
iy = -C1JJn(l-J1/Jm)+C27n[(J2+3)/3] (19)
Уравнение (19) сводится к уравнению (18), когда деформации относи-
тельно малы, т. е. отношение J J J' мало. Следовательно, уравнение (19)
вероятно наиболее простая возможная функция энергии деформации,
которая описывает упругое поведение с хорошей аппроксимацией полного
диапазона деформаций [21]. Оно требует трех настроечных параметров, два
из которых связаны с модулем сдвига G для малых деформаций:
G = 2[C1+(С2/3)] (20)
Молекулярная теория каучукоподобной эластичности предсказывает,
что первый коэффициент Q пропорционален числу Nмолекулярных цепей,
которые создают трехмерную сетку. Второй коэффициент С2 по-видимому
отражает физические ограничения на молекулярные цепи, подобно тому,
как это представлено в модели «трубка» [9], и в принципе доступен для
вычисления. Третий параметр Jm не является в действительности незави-
симым. Когда цепи длинные и гибкие, он определяется приблизительно
как ЗХт2, где \т максимальная степень удлинения, усредненная для всех
цепей. Но величина Хт2обратно пропорциональна числу N статистических
цепей в недеформированном состоянии [И]. Поэтому Jm оказывается об-
ратно пропорциональным Сг Таким образом, полный диапазон упругого
поведения определяется только двумя фундаментальными молекулярными
параметрами.
Значительный успех также был достигнут в описании наблюдаемого
упругого поведения резин с помощью функции энергии деформации, ко-
торая записывается прямо в терминах степеней удлинения Xj, Х2, Х3, вместо
инвариантов деформации и J2 [22]. Хотя экспериментальные результаты
могут быть описаны этим способом экономично и аккуратно, примененные
функции являются эмпирическими и числовые параметры, использованные
как настроечные константы, по-видимому, не имеют никакого прямого фи-
зического смысла в терминах молекулярной структуры материала. С другой
стороны, молекулярная теория упругости, дополненная простым негауссо-
вым членом, молекулярное происхождение которого, в принципе, достаточно
понятно, по-видимому, позволяет рассчитать наблюдаемое поведение при
малых и средних деформациях со сравнимой точностью.
При средних деформациях величина J2 часто достаточно велика для
того, чтобы членами, включающими W2, можно было бы пренебречь. Мно-
V. Континуальная теория упругости резины -'V- 27
гие соотношения напряжение-деформация теперь выводятся, используя
это приближение, для иллюстрации выполнения некоторых вычислений
и определения при каких условиях деформации становятся нестабильными.
Нестабильность интересна с теоретической точки зрения, поскольку она
возникает внезапно при хорошо определенных деформациях, и они часто
не определяются на основе классической теории упругости. Более того,
сравнение наблюдаемого возникновения нестабильности с предсказания-
ми по различным функциям энергии деформации Издает, по крайней мере
в принципе, критический тест для оценки предлагаемых форм для IV. С прак-
тической точки зрения нестабильные состояния крайне нежелательны из-за
высокой неоднородности, приводящей к преждевременному разрушению.
Раздувание тонкостенной трубки
Раздувание трубки описывается степенью удлинения Х4 в окружном на-
правлении и Х2 в осевом направлении, Х3 при толщине стенки h становится
равной поскольку объем резины остается постоянным. Внутреннее
давление Р приводит к возрастанию напряжений в окружном и осевом
направлениях:
= Х^Х2гР/Л
t2=X\X2rP/2h (21)
где г радиус трубки в недеформированном состоянии.
Из уравнения (21) полагая, что напряжение /3 = 0, величина некоторого
неопределенного давления определяется как:
p = -2Wjtf% (22)
(в тонкостенной трубке большого радиуса давление раздувания Р много
меньше, чем напряжения и /2, которые оно создает, следовательно, при оп-
ределении р величиной Р можно пренебречь в сравнении с напряжением /3).
Вводя этот результат для р в уравнения (21) и (22), получаем соотношение
между степенью удлинения Х2 и степенью расширения и (= внутрен-
него объема трубки:
2^=(р2 + 1)/2р2 (23)
Отсюда получим соотношение между давлением раздувания Р и внут-
ренним объемом трубки в виде:
Pr/h(\ = 2(р2 - 1)[2р/(р2 +1)]1/3/р2 [1 -(Л/4)] (24)
Это соотношение показано на графике рис. 9 для различных величин
лимитирующей меры деформации Jm. Давление раздувания проходит через
28 -*V* Глава 1. Упругость резины: основные положения и поведение
максимум при степени расширения объема между 58 и 66% в зависимости
от величины, принятой для J . Эта особенность наводит на мысль, что боль-
шие раздувания будут нестабильны. Действительно, тонкостенные трубки
подвергаются поразительно неоднородной деформации при критическом
давлении раздувания, что показано схематически на рис. 10. Одна часть
трубки становится сильно надутой как пузырь или аневризма, в то время
как остальная часть лишь слабо раздута. Две стабильные зоны деформации,
которые могут существовать совместно при том же самом давлении разду-
вания после достижения критического состояния, показаны схематически
горизонтальной разделяющей линией на рис. 9. Однако, когда Jm бесконечно
велико, аневризма неограниченна и разрушение должно наступить немед-
ленно при достижении критического давления.
Рис. 9. Соотношение давление - объем для тонкостенной трубки по уравнению (24)
с использованием различных максимально возможных значений Х[Т). Верти-
кальная штриховая линия обозначает наступление нестабильности. (Е = 66^)
Раздувание тонкостенного сферического баллона
В этом случае, если радиус баллона увеличится в X раз, то возникнет рав-
номерное двухосное растяжение X, при этом толщина стенки уменьшится
в отношении 1/Х2 при неизменности объема резины. Окружные напряжения
Z1 и ^эквивалентны и определяются, исходя из уравнения (17), как
tx=t2= 2Wt (V - Г4 )/[1 - (/, /4)] (25)
а напряжение /3 = 0 и W2 = 0. Давление раздувания Р теперь определяется
так:
Pr/AC1=4(X-,-r7)/[l-(J1/4)]
(26)
V. Континуальная теория упругости резины
г
29
где г и h радиус и толщина стенки недеформированного баллона. В этом
случае потенциальная нестабильность появляется при радиальном расшире-
нии от 38 и до 50% в зависимости от величины, выбранной для Jm, т. е. даже
раньше, чем при раздувании трубки. На практике деформация становится
достаточно сложной (рис. 11). Баллон остается приблизительно сферическим
по форме, но одна его часть слабо растянута, в то время как остальная часть
растянута сильно. Два состояния деформации напоминают две деформации,
которые предсказывались при определенном давлении после достижения
критической точки.
А
Рис. 11. Раздувание тонкостенного сферического резинового баллона. Сплошная
линия построена по уравнению (26) при Jm = «»[37].
Раздувание толстостенной сферической оболочки
Внутреннее давление Р, необходимое для раздувания малой сферической
полости в центре толстого блока, может быть определено интегрированием
вкладов от концентрических оболочек (тонкостенных баллонов), описан-
ных в предыдущем разделе. Результат для бесконечно растяжимой резины
(Ущ = ее) таков [23]
(27)
30 -* V Глава 1. Упругость резины: основные положения и поведение
где Хо степень двухосного растяжения на поверхности полости. Это соотно-
шение не дает максимума и, следовательно, не обозначает, что деформация
нестабильна. Однако, при больших Хо давление Р асимптотически приближа-
ется к постоянной величине около 5Ср т. е. около 5G/2, где G модуль сдвига
при малых деформациях. Для типичных каучукоподобных материалов, для
которых G около 0,5 МПа, максимальное давление поэтому около 1,2 МПа
или около 12 бар. Любая малая полость будет сильно расширятся при этом
более чем скромном давлении накачивания. Поэтому в мягких каучукопо-
добных телах при таких давлениях накачивания или эквивалентных трех-
осных растяжениях будут наблюдаться внутренние разрушения. Показано,
что на практике во всех каучукоподобных телах, когда они перенасыщены
газами или жидкостями при давлении или трехосном растяжении, близ-
ком к 5G/2, развиваются внутренние разрушения [23, 24].
Заметим, что начальный радиус сферической полости не входит в урав-
нение (27). Следовательно, предсказывается, что полости любого размера
раздуваются одинаково. Однако, мы пренебрегли вкладами поверхностной
энергии, которые будут создавать тенденцию к стабилизации малых полос-
тей. Когда они будут приняты во внимание, обнаружится, что только полости,
имеющие радиусы, большие, чем около 100 нм, будут резко расширяться
при малых давлениях, предсказываемых уравнением (27). Наличие внут-
ренних разрушений наводит на мысль, что вулканизованная резина должна
содержать много первоначальных полостей этого эффективного размера
или больших.
Поверхностная нестабильность сжатых или изогнутых блоков
Био [25] показал, что поверхность упругого полупространства становится
нестабильной при критической величине степеней удлинения Х1 Д3в двух
перпендикулярных направлениях на поверхности. Критическим условием
является
Х3 = 0,2956 (28)
Когда блок подвергнут одноосному сжатию, параллельному поверхнос-
ти, при свободно разрешенном растяжении в двух других направлениях,
тогда Х3= I/X^^h уравнение (28) дает для Х1 критическую величину
0,444. Поэтому предсказывается, что большой блок резины при простом
сжатии становится поверхностно нестабильным при сжатии 55,5%.
(Бьюти [26] заметил, что различные формы деформации коробления и
раздувания обычно встречаются перед этим в зависимости от толщины
блока.) Если блок подвергается равномерному двухосному сжатию, па-
раллельному поверхности, тогда Х3 = и критическое сжатие становится
равным 33;3%.
V. Континуальная теория упругости резины 31
Рис. 12. Схема изгиба блока,
показывающая сгибы, которые
появляются при больших дефор-
мациях сжатия
Когда толстый упругий блок (кубоид) изгибается, то внутренняя поверх-
ность становится сжатой, в то время как степень деформации Х3 вдоль шири-
ны существенно не изменяется. Таким образом, из уравнения (28) получим,
что нестабильность должна ожидаться на внутренней поверхности, когда
X,! станет равной 0,554, т. е. когда поверхность сожмется примерно на 46%.
В эксперименте острые складки или сгибы появляются внезапно на
внутренней поверхности изгибаемого блока при критической степени
изгибания [27], см. рис. 12. Однако, критическое сжатие внутренней
поверхности значительно меньше, чем предсказанное по теории Био, 35%
вместо 46%. Неизвестно почему нестабиль-
ность появляется настолько раньше, чем
предполагается. Хотя поведение резины
описывается более сложными функци-
ями энергии деформации, чем простые,
принятые в этой теории, непохоже, что это
различие должно бы дать такой большой
эффект.
Резиновые детали часто подвергаются
довольно жестким изгибным деформациям,
например в шинах. Складки или сгибы на
внутренней стороне могут оказаться неза-
меченными. Более того, они представляют
линии концентрации высоких напряже-
ний и места возможных разрушений. При
скольжении мягкой резины по жесткой
поверхности также появляются складки («волны Шалломаха») [28]. По-
видимому, они являются описанными Био складками, вызванными сжатием
поверхности при трении.
Сопротивление сжатого блока внедрению индентора
Когда блок подвергается достаточно большому одноосному сжатию в
плоскости поверхности, он становится нестабильным по отношению к малым
внедрениям. Грин и Зерна [29] представили соотношение между силой N и
глубиной внедрения d как
Af/G = (8/3)7?1/2d3/2/(X) (29)
где G модуль сдвига материала полупространства, R радиус индентора и/ (X)
функция степени одноосного сжатия X, определенная соотношением
/(1) = (х9 + Х6 +ЗХ3 -1)/V(X3 +1) (30)
32 ""•V- Глава 1. Упругость резины: основные положения и поведение
Рис. 13. Зависимость силы N (при малой величине внедрения) от равномерной
двуосной деформации е, параллельной поверхности полупространства,
7V0 - сила при е = 0 [29]
Зависимость силы 2V (при малой фиксированной величины внедре-
ния) от равномерной двуосной деформации е, параллельной поверхнос-
ти полупространства (где е = X- 1), рассчитанная по уравнению (29),
представлена на графике рис. 13. (2V0 — величина для первоначально
недеформированного блока, когда/(Х) = 2.) Видно, что сопротивление внед-
Рис. 14. Схема загиба и петли, по-
являющихся при кручении растя-
нутого резинового стержня [30]
рению резко уменьшается при возрастании
деформации сжатия, становясь нулевым
при деформации сжатия 0,333 в согласии с
результатами Био.
Нестабильность длинного резинового
стержня при кручении [30]
Другое нестабильное состояние встре-
чается, когда длинный резиновый стер-
жень подвергается большому кручению.
Внезапно появляется загиб на одном из
участков стержня, рис. 14, затем несколь-
ко загибов образуют петлю, т. е. закручен-
ный участок стержня.
Минимизация полной упругой энергии
деформации показывает, что стержень
будет становиться нестабильным при кри-
V. Континуальная теория упругости резины 33
тической величине кручения: часть стержня будет раскручиваться и обра-
зовывать устойчивое кольцо, в то время как остальная часть стержня будет
становиться несколько более растянутой. На основе этой схемы может быть
выведен простой критерий для начала «загибов». Для нео-Гуковского мате-
риала, описываемого уравнением (8), условие образования загиба таково:
4(1 - 1Д3) = -(а2ф2 Д)+2л (а2ф2 Д)/[я - (оф/Х1/2)] (31)
где ф — критическая величина кручения, при которой однородное кручение
становится нестабильным, X — степень удлинения и а — радиус стержня.
Измеренная величина для стержней различного радиуса, растянутых до ве-
личины 250%, оказалась в удовлетворительном согласии с уравнением (31).
Это показывает, что внезапное образование петель в закрученном резиновом
стержне является действительно следствием упругой неустойчивости.
VI. НАПРЯЖЕНИЯ ВТОРОГО ПОРЯДКА
Функция упругой энергии деформации (упругий потенциал)
для резины описывает большие деформации и приводит к соотношениям
напряжение - деформация, являющимися нелинейными по структуре,
как это следует ожидать при малых деформациях, и напряжения не будут
прямо пропорциональны деформации, а будут зависеть от квадратов и более
высоких ее степеней. Замечательный пример этой особенности больших
упругих деформаций представляют нормальные напряжения Z22, Z33,
которые необходимы для создания деформации простого сдвига величины
у (вдобавок, конечно, к напряжению простого сдвига) [15, 16]. Показано,
что эти напряжения возрастают пропорционально у2.
t22 = -2(W1+W2)/2
Рис. 15. Напряжения, необходимые для поддержания деформации простого сдвига,
равной у. Нормальное напряжение установлено равным нулю [37]
Напряжения представлены схематично на рис. 15 и 16 для двух раз-
личных вариантов произвольного гидростатического давления р, выбран-
ного так, чтобы дать соответствующее отсчетное (нулевое) напряжение.
34
Глава 1. Упругость резины: основные положения и поведение
На рис. 15, например, нормальное напряжение /нв направлении сдвига
установлено равным нулю; это условие осуществляется вблизи передней
и задней поверхности блока при сдвиге. На рис. 16 равным нулю установлено
нормальное напряжение /33; соответствующее условие осуществляется на
боковой поверхности блока при сдвиге. Оказывается, что в каждом случае
для простой сдвиговой деформации необходимо приложить сжимающее на-
пряжен ие t22. В его отсутствие имелась бы тенденция возрастания толщины
блока при сдвиге.
Рис. 16. Напряжения, необходимые для поддержания деформации простого сдвига,
равной у. Нормальное напряжение ^установлено равным нулю [37]
Когда дополнительная деформация является неоднородным сдвигом,
как при кручении, возникают нормальные силы (соответствующие напря-
жениям t22 на рис. 15 и 16), изменяющиеся отточки к точке по поперечному
сечению (рис. 17). Точный закон их распределения зависит от конкретной
формы функции энергии деформации, принятой для резины, т. е. от вели-
чин и W2, которые имеют место для дополнительного деформированного
состояния [31].
VII. УПРУГОЕ ПОВЕДЕНИЕ ПРИ МАЛЫХ ДЕФОРМАЦИЯХ
При малых деформациях резина является линейным упругим
телом. Из-за высокого модуля всестороннего сжатия, около 2000 МН/м2,
и в сравнении с модулем сдвига величиной от примерно от 0,2 до 5 МН/м2 она
может рассматриваться как относительно несжимаемая. Следовательно, уп-
ругое поведение при малых деформациях может быть описано единственной
упругой константой G. Коэффициент Пуассона фактически равен !4, а модуль
Юнга Е определяется как 3G с хорошим приближением.
Широкий диапазон величин G может быть получен варьированием струк-
туры эластомера, т. е. изменением химии поперечных связей, разбавлением
VI. Напряжение второго порядка
35
маслом и изменением содержания наполнителя; однако, мягкие материалы
с модулем сдвига, меньшим чем около 0,2 МН/м2, оказываются слишком
мягкими и редко применяются. Также особо жесткие материалы, полученные
путем сшивания до высокой степени, оказывают-
ся хрупкими и нерастяжимыми. Практический
диапазон модулей сдвига при изменении степени
сшивания и разбавления маслом лежит от 0,2 до
1 МН/м2. Ужесточение наполнителями поднимает
верхний предел до 5 МН/м2; но те наполнители,
которые создают особо заметное ужесточение,
приводят и к появлению эффектов размягчения
при напряжениях, подобно показанному на рис.
5. Таким образом, модуль становится в какой-то
степени количественно неопределенным.
Обычно модуль, жесткость или твердость ре-
зин характеризуют измерением степени упругого
Рис. 17. Схема цилин-
дрического бруса при
кручении, показываю-
щая распределение нор-
мального напряжения t22
(соответствующего — t22
на рис. 15 и 16) по попе-
речному сечению бруса
[И]
внедрения жесткого штампа заданного размера
и формы при определенных условиях нагружения.
Величину твердости из этих измерений определяют
по различным нелинейным шкалам [32]. Соот-
ветствующие величины модуля сдвига G для двух
обычных шкал твердости даны на рис. 18.
Многие резиновые изделия в нормальных
условиях подвергаются довольно малым дефор-
мациям, редко превосходящие 25% растяжения
или сжатия или 75% при простом сдвиге. Хорошая аппроксимация для
соответствующих напряжений может быть получена обычным упругим
анализом, предполагая линейные соотношения. Рассмотрим здесь один
важный частный случай деформации: сжатие или растяжение тонких
резиновых блоков, прикрепленных по главным поверхностям к жестким
пластинам (рис. 19). Общий анализ таких деформаций был рассмотрен
в [34].
Удобно предположить, что деформация проходит в две стадии: чистое
однородное сжатие или растяжение на величину е, требующее однородного
сжимающего или растягивающего напряжения at = Ее, и сдвиговая дефор-
мация, восстанавливающая точки в плоскостях закрепленных поверхностей
в их исходных позициях в этих плоскостях. Для цилиндрического блока
радиуса а и толщины h соответствующее напряжение сдвига t, действующее
на поверхностях крепления на радиальном расстоянии г от оси цилиндра
определяется так
t = Eer/h
36 -*v Глава 1. Упругость резины: основные положения и поведение
Это сдвиговое напряжение связано с соответствующим нормальным
напряжением или давлением, определяемым как
ст2 =/?е(а2//г2)[1-(г2/а2)] (32)
Распределения этих напряжении показаны схематически на рис. 19.
Хотя в углах блока расчетные напряжения не должны оказаться соответ-
ствующими реальным, поскольку допущение о деформации простого сдвига
в этих точках сингулярности не может быть справедливым, результат, по-
видимому, обеспечивает удовлетворительные приближения в главной части
закрепленных поверхностей [42].
Полная сила сжатия ^определяется интегрированием суммы нормальных
напряжений а1 + а2по поверхности закрепления в форме
F/na2e = E'[l + (a2/2^2)] = Е' (33)
Ясно, что из-за ограничений, создаваемых закрепленной поверхностью,
эффективная величина модуля Юнга Е' для тонкого блока большого ради-
уса, представленная правой частью уравнения (33), много больше, чем ре-
альная величина Е. Действительно, для
отношения a/h, большего, чем примерно
10, значительный вклад в наблюдаемые
перемещения возникает от объемного
сжатия или расширения, поскольку Е'
делается таким большим, что становится
сравнимым с модулем объемного сжатия
[33] (рис. 20).
Более аккуратное рассмотрение
сжатия закрепленного блока проведено
Нортоном и др. [34] без введения пред-
положения о том, что на закрепленных
углах имеет место деформация простого
сдвига. Полученный результат имеет ту
же самую форму, что и уравнение (33),
но с заменой выражения в квадратных
скобках в правой части на [1,2 + (а2/2Л2)]. Однако, этот член не сводится
к правильной величине 1 для высокого блока, т. е. когда a/h мало, а экви-
валентен уравнению (33) для тонких блоков большого радиуса, когда a/h
велико. Он должен, поэтому, рассматриваться как лучшее приближение для
блоков промежуточного размера.
Когда тонкий закрепленный блок подвергается растяжению, в его
центральной области возникает состояние приблизительно равномерного
трехосного растяжения. Величина напряжения в каждом направлении
Рис. 18. Зависимость между модулем
сдвига G и твердостью: —, по Шор А;—-.
по международной шкале твердости
(IRH) [5]
VII. Упругое поведение при малых деформациях
Л
37
определяется из уравнения (32) как напряжение растяжения или отрица-
тельное давление g2 при г = 0, т.е. величиной Eea^/h^. Под этим напряже-
нием, направленным наружу, малая полость в центральной области блока
будет расширяться неограниченно при критической величине напряжения
около 5Е/6. Следовательно, если внутри
закрепленного блока присутствуют поло-
сти, то для них предсказывается неограни-
ченное расширение, т. е. разрушение, при
критической деформации растяжения е ,
приблизительно определяемой как
ес =5Л2/ба2
и при соответствующей критической
величине приложенной растягивающей
нагрузки, полученной подстановкой
этой величины е в уравнение (33). Что-
бы избежать внутреннего разрушения
этого вида необходимо, следовательно,
ограничить среднее напряжение растя-
жения, прилагаемое к тонким закреп-
ленным блокам, величиной, меньшей,
примерно, чем Е/3.
С другой стороны, при сжатии могут
поддерживаться весьма большие на-
пряжения. Предел напряжения может
быть вычислен в предположении, что
максимальное сдвиговое напряжение,
возникающее на углах закрепления, не
должно превышать G\ т. е. максимальная
сдвиговая деформация не должна пре-
вышать 100%. Это приводит к величине
для допускаемой полной деформации
сжатия h/Ъа, соответствующей средне-
му напряжению сжатия порядка Е для
дисков с a/h между 3 и 10. Это вычис-
ление проведено в предположении, что
приближенный анализ напряжений,
описанный ранее, справедлив для углов
блоков, а это наверняка неправильно.
Действительно, местные напряжения
в этих областях сильно зависят от де-
талей формы свободной поверхности
рядом с углом.
Рис. 19. Схема закрепленного ре-
зинового блока при малом сжа-
тии. Распределение нормальных Q
и сдвиговых t напряжений, действу-
ющих на закрепленных поверхно-
стях, представлены на верхней части
диаграммы [5]
Рис. 20. Зависимость между эффек-
тивным модулем Юнга Е для закреп-
ленных блоков и отношением радиуса
к толщине a/h [5]
38 Д-
Глава 1. Упругость резины: основные положения и поведение
VIII. НЕКОТОРЫЕ НЕРЕШЕННЫЕ ПРОБЛЕМЫ
УПРУГОСТИ РЕЗИНЫ
Мы возвращаемся теперь к некоторым особенностям упру-
гой реакции каучукоподобных материалов, которые все еще не полностью
поняты.
Как обычно предполагается, молекулярная сетка включает в себя цепи
с широким распределением молекулярных длин. Количественно имеется
тенденция преобладания цепей малой длины. Влияние этого разнообразия
на упругое поведение сетки, в частности при больших деформациях, неиз-
вестно. Соответствующая проблема имеет отношение к упругости коротких
цепей. Их поведение неизбежно негауссово и анализ их конформационной
статистики вероятно, будет трудным. Тем не менее, этот анализ представ-
ляется необходимым выполнить, чтобы получить возможность правильно
объяснять поведение реальных сеток
Желательно также более детально рассмотреть топологию сетки, включая
функциональность поперечных связей, их распределение в пространстве
и образование петель. Влияние взаимодействия между цепями в конденсиро-
ванном состоянии, по-видимому, может быть удовлетворительно рассчитано
для несшитых полимеров с помощью модели «трубки», но ее применимость
к сеткам кажется неполной. Однако проблемой, заслуживающей наиболь-
шего внимания, является реакция высоконаполненных эластомеров на
напряжение. Наполненные эластомеры не являются вполне упругими; со-
отношения «напряжения-деформации» для них являются необратимыми
(см. рис. 5), и поэтому неуместно описывать их реакцию на напряжение
с использованием функции энергии деформации (упругого потенциала).
Более того, они, по-видимому, становятся анизотропными при растяжении
и в некоторой степени - после разгрузки. В настоящее время еще неясны
как происходящие при деформировании молекулярные процессы, так
и подходящая для их описания математическая структура.
ГЛАВА
2
ПОЛИМЕРИЗАЦИЯ:
СИНТЕЗ ЭЛАСТОМЕРОВ
РОДЕРИК П. КВИРК
Полимерный институт им. М. Мортона
Университет Акрона
Акрон, Огайо
ДИННА Л. ГОМОХАК ПИККЕЛЬ
Исследовательские лаборатории
Химическая компания Истман
Кингспорт, Теннесси
I. ВВЕДЕНИЕ
Создание синтетического каучука сыграло особую роль в химии
полимеризации. Произошло это потому, что попытки синтезировать каучук
были сделаны задолго до понимания природы реакции полимеризации. Такие
попытки начались вскоре после изящной аналитической работы Вильямса
[1], который в 1860 г. ясно продемонстрировал, что каучук из Гевеи состоит из
изопрена. Затем, Бушарда [2] в 1879 г. действительно получил каучукоподобное
вещество из изопрена (которое он приготовил пиролизом каучука), используя
нагревание и хлористый водород. Тильдон [3] повторил этот процесс в 1884 г.,
но использовал изопрен, полученный пиролизом скипидара (живицы), для
того, чтобы показать, что необязательно использовать «материнское вещество»
каучука. За этими исследованиями вскоре последовали работы Кондакова
(1900 г.) [4] с 2,3-диметилбутадиеном, работа Тиле (1901) [5] спипериленом и,
наконец, работа Лебедева (1910 г.) [6] с бутадиеном. Следует упомянуть, что по-
чти одновременно и, по-видимому, независимо друг от друга в 1910 г. Гаррисом
в Германии [7], Мэтьюсом и Стренджем [8] в Англии была открыта эффективная
полимеризация изопрена под действием натрия.
Хотя все эти попытки реально имели благородную цель, но применяемые
приемы следует жестко оценить с точки зрения вклада в науку, так как пре-
вращение простых молекул диенов в «коллоидное» вещество, каким тогда
считали каучук, было далеко от его осмысления химической наукой.
40 “*V- Глава 2. Полимеризация: синтез эластомеров
Фактически промышленные производство синтетического каучука уже
было организовано, по крайней мере, в Германии и России до того, как
Штаудингер установил основы своей макромолекулярной теории строения
каучука в 20-е гг. [9]. Такие современные эластомеры, как полихлоропрен
и поли (алкиленсульфиды) уже производились в промышленном масштабе
в 1930-31 гг. еще до того, как Карозерс с сотрудниками впервые изучили
полимеризацию хлоропрена [10].
Следовательно, не развитие теории полимеризации привело к получе-
нию синтетического каучука, а, скорее, наоборот. Основой этого явилось
новое учение об органических макромолекулах, которое предложил Шта-
удингер, и оно быстро распространилось в 30-40-е гг., указав путь синтеза
громадного количества новых полимерных материалов, включая синтети-
ческие волокна, пластики и новые эластомеры. Это новое развитие теории
включает классические исследования поликонденсации, выполненные
Карозерсом и Флори и установление принципов управления свободнора-
дикальными цепными реакциями полимеризации Шульцем, Флори, Майе
и другими [11, 12].
В результате пути развития учения о синтетическом каучуке и макро-
молекулярной химии пересеклись и вышли на широкую дорогу [13].
Следовательно, сегодня создание нового или модификация уже известного
эластомера требует того же типа молекулярной архитектуры, которая при-
менима к любому другому полимеру и основана на понимании принципов
реакции полимеризации.
II. КЛАССИФИКАЦИЯ И КИНЕТИЧЕСКОЕ РАССМОТРЕНИЕ
РЕАКЦИЙ ПОЛИМЕРИЗАЦИИ
Исторически полимеры подразделяются на два широких
класса: конденсационные полимеры и аддиционные (полимеризационные
полимеры) [11, 14, 15]. Флори [И, с. 37] определил их как «конденсаци-
онные полимеры, в которых молекулярная формула структурной единицы
(или единиц) содержит некоторые атомы, присутствующие в мономере,
из которого он состоит, и часть, которых может быть удалена химическим
путем, и полимеризационные полимеры, в которых молекулярная формула
структурной единицы (или единиц) идентична формуле мономера, из ко-
торого получен полимер».
Таким образом, примером конденсационного полимера является полиэ-
фир, полученный реакцией конденсации между гликолем и дикарбоновой
кислотой (с выделением воды), тогда как полимеризационный полимер
представлен, например, полистиролом, полученным присоединением мо-
номерных молекул стирола.
Хотя эти ранние определения основаны на цепной структуре полимеров,
они тесно связаны, как было упомянуто, с типом образования. Вскоре стало
II. Классификация и кинетическое рассмотрение реакций полимеризации -’V 41
ясно, что такая классификация имеет серьезные недостатки, т. к. так назы-
ваемые поликонденсаты могут быть получены в результате реакции аддици-
онной полимеризации. Например, хотя найлон-6 может быть получен реак-
цией поликонденсации е-аминокапрновой кислоты [16], в настоящее время
он синтезируется путем реакции раскрытия цикла е-капролактама [17],
и этот процесс оказывает большое влияние на свойства получаемого по-
лимера, в основном за счет различной молекулярной массы конечного
продукта.
Вследствие чрезвычайно большого размера макромолекул, что придает им
необычные свойства, наиболее разумно классифицировать реакции синтеза
полимеров в соответствии с тем, как они определяют молекулярный размер
и распределение по размерам макромолекул, т. е. в терминах механизма по-
лимеризации. Поэтому имеются только два процесса, посредством которых
синтезируются макромолекулы [18-24]: (1) ступенчатая полимеризация
(поликонденсация и полиприсоединение) и (2) цепная полимеризация.
А. Полиприсоединение/поликонденсация
Отличительной чертой механизма ступенчатой полимери-
зации [18-24] является то, что все молекулярные частицы в системе могут
реагировать друг с другом с образованием высокомолекулярных частиц.
Как показано в уравнении (1), где Р. —частицы со среднечисленным числом
мономерных звеньев в цепи, равным I, Р. — частицы со среднечисленным
числом мономерных звеньев в цепи, равным j, Pi+j — частицы со средне-
численным числом мономерных звеньев в цепи, равным j + ь. Кинетическая
последовательность этого механизма роста полимера состоит в том, что длина
цепи увеличивается монотонно со степенью реакции, т. е. со временем ре-
акции, как показано на рис. 1а.
Pi + Pj^Pi+j
(1)
Ступенчатые реакции полимеризации делятся на два класса [23, 24]:
Поликонденсация: рост полимерной цепи протекает путем реакции кон-
денсации между молекулами различной степени полимеризации. При этом
также образуется побочный низкомолекулярный продукт (АВ).
A-R-A +B-R-B -> A-R-R-B + АВ (2)
Полиприсоединение: рост полимерной цепи происходит за счет реакций
присоединения между молекулами всех степеней полимеризации без выде-
ления низкомолекулярного вещества.
A-R-A +B-R-B -> A-R-AB-R-B (3)
42 -*v- Глава 2. Полимеризация: синтез эластомеров
Здесь А и В - функциональные концевые группы, которые реагиру-
ют друг с другом. Примерами поликонденсации могут быть образование
полиэфиров (1) и полиамидов (2), где А и В (1) гидроксильные и карбо-
ксильные группы и (2) аминные и карбоксильные соответственно, которые,
соединяясь, отщепляют молекулу воды [16-19]. С другой стороны реакция
полиприсоединения [ур. (2)] может иллюстрироваться с помощью реакции
диизоцината с гликолем с образованием полиуретанов. В этом случае ника-
кие побочные продукты не образуются.
% конверсии
Рис. 1. Изменение молекулярной массы с конверсией (%) для ступенчатой поли-
меризации (А), цепной полимеризации (В), и полимеризации с живыми
цепями без передачи и обрыва цепи (С)
Процессы полимеризации, показанные в ур. (2) и (3), фактически
представляют известные реакции малых молекул, при этом единственным
различием являются минимальные требования бифункциональное ши каж-
дой молекулы для формирования полимера, которая позволяет продукту
каждой реакции участвовать в дальнейших реакциях. Как правило, фун-
кциональные группы сохраняют реакционную способность независимо от
длины цепи [11, с. 75], так что эти реакции подчиняются таким же кинети-
ческим правилам, как и для простых молекул; однако, в противоположность
реакции полиприсоединения, реакции поликонденсации характеризуются
обратимостью (например, гидролиз или «деполимеризация»), как результат
возможного накапливания побочного продукта (например, воды), и это
следует учитывать. В общем, из-за малой константы равновесия реакций
поликонденсации [20] получение высокомолекулярных полимеров требует
удаления низкомолекулярных побочных продуктов [18].
II. Классификация и кинетическое рассмотрение реакций полимеризации -’V 43
В обоих предшествующих типах реакций молекулярной массой поли-
мера управляют два фактора: стехиометрия и глубина реакции [22]. Таким
образом, очевидно, что избыток одного типа концевых групп будет контро-
лировать достигаемую длину цепи, и она может быть оценена, если известно
исходное молярное соотношение функциональных групп. С другой стороны,
с равным количеством двух типов концевых групп конечная длина цепи
теоретически безгранична.
В. Цепная полимеризация
Отличительной чертой различия роста цепи при цепной
полимеризации [21, 22-24] является то, что рост цепи происходит только
при присоединении мономера к реакционному центру, присутствующему
на растущей полимерной молекуле, как показано в ур. (4), где Рп* — цепь
полимера с активным центром (*) и степенью полимеризации п,М — моно-
мерная единица, а Рп+* — цепь полимера с активным центром (*) и степенью
полимеризации п+1
р;+м->р;+1 (4)
Этот тип полимеризации включает присоединение мономеров к растущей
цепи, которое инициируется некоторыми видами активных частиц (ини-
циаторов). В таких реакциях присоединения могут участвовать кратные
связи или циклы. Активные частицы, инициирующие такие цепные реак-
ции, должны представлять собой радикал, электрофил, нуклеофил, либо
металлоорганическое соединение и должны быть способны разрывать одну
из связей мономера. Следовательно, эти процессы полимеризации могут
происходить по различным механизмам, зависящим от природы частиц,
способных удлинять цепь, а именно: свободных радикалов, катионов, ани-
онов и координационных соединений. Это иллюстрируется следующими
уравнениями для реакций двойных связей с различными типами иници-
ирующих частиц.
R. + С = С —► к—с —С.
/ \ II
\ / । 1
Н+А +рс =С\ —► н— с — С+А- —►••• —►
\ / I I
А' М+ + С = С —► А— С— С‘М+—► - —►
Свободнорадикальная
Катионная
Анионная
Координационная ' 4 1 1
R-Met + ЧС = СХ —► R— С — С-------Met ——►
44 -*v- Глава 2. Полимеризация: синтез эластомеров
В этих уравнениях точная природа инициирования, а также природа
частиц, продолжающих цепь, может изменяться от преимущественно
ковалентной для металлорганических соединений переходных металлов
в координационной полимеризации до ионных пар или свободных ионов
в ионной полимеризации в зависимости от структуры продолжающих цепь
частиц, противоиона, растворителя и температуры.
Значительное различие между ступенчатым и цепным процессами поли-
меризации состоит в том, что в последнем каждая макромолекула образуется
с помощью «цепной реакции», которая инициируется на каждой ступени
роста макромолекулы. Таким образом, при любом данном времени в течение
полимеризации присутствующие реагирующие частицы состоят только из
растущих цепей, молекул мономеров и стабильной полимерной цепи, обра-
зовавшейся ранее при обрыве цепных реакций. Эти растущие цепи могут
быть очень коротко живущими (т. е. свободные радикалы или свободные
ионы), но могут достигнуть очень большой длины цепи в течение короткого
времени жизни, как показано на рис. 1 (В). С другой стороны, они могут
иметь очень долгое время жизни (т. е. живые полимеры [22-24]), и в этом
случае длина цепи может увеличиваться как прямая функция времени ре-
акции, как показано на рис. 1 (С). Следовательно, в отличие от ступенчатой
полимеризации, молекулярная масса продуктов цепной полимеризации
может быть как прямо зависящей от времени или глубины реакции, так
и не подчиняться этому положению [см. рис. 1 (А)].
III. Полиприсоединение/поликонденсация
Хотя, как было показано ранее, полиприсоединение и по-
ликонденсация как термины отсутствуют в ранних исследованиях синтеза
каучука, именно они явились одними из основных методов, примененных
для полимеризации, благодаря их относительной простоте. Потому не уди-
вительно, что первые синтетические смолы и пластики, такие как фенол-
формальдегидные и полиэфирные, относятся к конденсационному типу.
Концепция соединения вместе реакционных концевых групп для построения
больших молекул является очень простой для понимания и описывается
простым математическим анализом.
Как установлено ранее, кинетика полиприсоединения и поликонден-
сации следует общим правилам простых монофункциональных реакций
[18, 20, 22, 25], так как реакционная способность сохраняется независимо
от длины цепи [11, с. 75]. Единственной новой чертой является увеличе-
ние молекулярных размеров, и это поддается математическому анализу
[И, с. 91]. При рассмотрении типа реакций, определяемых уравнениями (2)
и (3) в нормальном случае, где число групп А и В является равным, длина
цепи легко предсказывается как функция степени реакции. Таким образом,
если р представляет долю концевых групп, прореагировавших к данному
III. Полиприсоединение/поликонденсация 45
времени, тогда среднечисленное значение единиц на цепь (Хп) определяется
выражением 1/(1 -р). Таким образом,
Мп=М0/(\-р) (5)
где Мп — среднечисленная молекулярная масса полимера и MQ — молеку-
лярная масса повторяющейся единицы цепи. Следствие этой простой зави-
симости имеет большое значение. Например, когда 50% функциональных
групп прореагировало, среднечисленная степень полимеризации равна 2.
Чтобы получить полимер с требуемыми свойствами необходима молекуляр-
ная масса, по меньшей мере, 10000; это означает, что степень превращения
функциональных групп должна превышать 99% для повторяющейся еди-
ницы с молекулярной массой 100 г (Хп = 100). Очевидно, что сравнительно
малое число реакций будет подчиняться этому жесткому требованию из-за
протекания побочных реакций.
Так как этот тип полимеризации является случайным процессом, в кото-
ром все молекулы имеют равную вероятность прореагировать, распределение
молекулярных масс соответствует наиболее вероятному и биномиальному
распределению, которое связано со степенью полимеризации следующими
соотношениями:
Кх=гр'~1(1-р)2 (6)
Nx = p*-\l-p) (7)
где W — весовая доля х-мера (цепей, имеющих х — единиц) и Nx — мольная
доля х — мера. Это функция распределения может быть применена для рас-
чета Mw, средневесовой молекулярной массы, так как Mw = M0XxWx. Можно
показать, что следующее суммирование приводит к соотношению
МЮ = |^(МО) (8)
1-р
которое означает, что
Мю/Мп=1 + р (9)
Следовательно, отношение средневесового к численному значению длины
цепей в этих системах последовательно увеличивается с увеличением глубины
реакции, достигая предельного значения 2.
Таким образом, мы видим, что полиприсоединение и поликонденсация
характеризуются следующими особенностями:
1. Все молекулы имеют равную вероятность прореагировать.
2. Скорости полимеризации существенно определяются концентрациями
и реакционной способностью функциональных групп.
46 -'V Глава 2. Полимеризация: синтез эластомеров
3. Длина цепи является монотонной функцией глубины реакции и,
следовательно, времени реакции.
4. Достижение высоких молекулярных масс требует высокой степени
конверсии (р —> 1).
В случаях, когда, по меньшей мере, один из мономеров имеет более
двух функциональных групп, возникают дополнительные разветвления
цепей, в конечном итоге, приводящие к образованию молекулярных
сеток [И, с. 347], т. е. гелеобразованию. Это, естественно, усложняет
распределение по молекулярным размерам, но не влияет на кинетику
полимеризации.
Рассмотренные соотношения длины цепей и глубины реакции позволя-
ют описать такую ступенчатую полимеризацию, как синтез поли (алкилен
сульфидов) из дигалогенов и полисульфидов натрия (поликонденсация) или
образование уретановых полимеров из гликолей и диизоционатов (полипри-
соединение). Полисульфидная реакция протекает в суспензии дигалогена
в водном растворе полисульфида, с применением ПАВ для стабилизации
образующейся полимерной суспензии.
Уретановые полимеры представляют интересную иллюстрацию ожидае-
мых величин молекулярных масс для данного типа полимеризации, которая
может быть описана следующим образом:
НО—Р—ОН +OCN—R—NCO ---------► о о
II II
НО-^Р—О—С—NH—R—NH—С—О-^Р—ОН
х
(Ю)
Необходимо отметить, что Р в уравнении (10) представляет низко-
молекулярной полимер сложного или простого полиэфира (MW 2000),
так что это действительно реакция «удлинения цепи». Оказалось, что
реакции между изоцианатными группами и гидроксилами протекают
до высокой степени конверсии, т. е. приблизительно до 98% (р = 0.98).
Следовательно, значение х в уравнении (10) должно быть около 50, и
конечная молекулярная масса уретанового полимера около 100 000. Та-
кая высокая молекулярная масса зависит исключительно от того факта,
что реакция протекает быстро до завершения, т. е. когда концентрация
реакционноспособных функциональных групп уменьшается приблизи-
тельно до 10-2 моля.
IV. Полимеризация по свободнорадикальному механизму
Л
47
IV. ЦЕПНАЯ ПОЛИМЕРИЗАЦИЯ ПО
СВОБОДНОРАДИКАЛЬНОМУ МЕХАНИЗМУ
А. Общая кинетика
Общая кинетика по этому механизму включает обычные
три стадии любой цепной реакции, т. е. инициирование, рост и обрыв цепи
[26]. Инициирование протекает путем образования свободных радикалов
вследствие гомолитической диссоциации слабых связей (например, в пере-
кисях или азосоединениях) или действия облучения. Реакции обрыва для
виниловых полимеров могут протекать либо рекомбинацией радикалов, либо
диспропорционированием, или комбинацией обеих реакций (обсуждается
ниже).
Инициирование
R. +М------► R_M.
Рост цепи
кр
м7. +м--------------------------------► Му+1.
Обрыв цепи
jt
Му • + Mfc • —► нереакционноспособные цепи
(*tc + ^td)
где I = инициатор, М = мономер, Я* = первичный свободный радикал,
и М.» = растущий свободный радикал.
Рекомбинация
RO-[-CH2-CH ] СН2СН’ + RO-J-CH2—CH j сн2сн •
XX XX
^tc
ro J сн2 - сн X сн2сн - снсн2 -Хен - СН2 L OR
И I I L j
X X X X
48 -*V Глава 2. Полимеризация: синтез эластомеров
Диспропорционирование
^td
ro-[-ch2-ch|-ch2ch . + ro|ch2-ch^ch2ch . —►
XX XX
RO-[CH2-CH-]-CH2CH2 + RO-j-CH2—CH-j-CH=CH
XX X X
Эта последовательность стадий ведет к следующей простой кинетической
обработке [26]:
Скорость инициирования Я,=2А:;[7] (Н)
Скорость роста цепи Rp = Kl)[Mj-]W (12)
Скорость обрыва цепи R^Zk^M^ (13)
Полагая стационарные условия реакции, в которых скорость образования
радикалов равна скорости их исчезновения, т. е. = Rt
[м..] = /сА’1/2Ш,/2 (14)
и
Rp = kp^l2k;'i2[M}[ir (15)
Уравнение (15), таким образом, иллюстрирует зависимость общей скоро-
сти полимеризации от концентрации инициатора и мономера. Зависимость в
степени 1 /2 скорости от концентрации инициатора, по-видимому, является
универсальной чертой свободнорадикального механизма и может приме-
няться как диагностический тест для оценки этого механизма.
Другим важным аспектом свободнорадикальной полимеризации явля-
ется зависимость среднечисленной степени полимеризации от концентра-
ции инициатора и мономера, как показано в уравнении (16). Сравнение
с уравнением (15) показывает, что увеличение скорости полимеризации
посредством увеличения концентрации инициатора увеличивает скорость
полимеризации, но уменьшает степень полимеризации Хп, что соответствует
среднечисленному значению мономерных звеньев на цепь.
Xn=/cpA:,+,/V/2[^][^r,/2 (16)
Общей чертой свободно-радикальной цепной полимеризации является
следующая особенность. Из-за высокой реакционной способности растуще-
IV. Полимеризация по свободнорадикальному механизму -*v- 49
го макрорадикала он может существовать очень короткое время, в лучшем
случае несколько секунд. Это выражается очень низкой стационарной кон-
центрацией растущих цепных радикалов (около 10-8 М в гомогенной среде).
За это короткое время, однако, каждый растущий радикал еще имеет воз-
можность присоединить тысячи мономерных единиц. Следовательно, длина
цепи макромолекул, образованных в системах, не связана прямо с глубиной
реакции, т. е. степенью конверсии мономера в полимер [смотри ур. (16)
и рис. 1 ]. В любое время полимеризации реакционная смесь содержит только
мономер, малую концентрацию растущих цепей и образовавшийся полимер,
который обычно имеет высокую молекулярную массу.
Чтобы четко проиллюстрировать природу свободнорадикальной полиме-
ризации, следует измерить значения индивидуальной константы скорости
роста и обрыва цепи. Значения этих констант скорости можно получить,
используя измерения нестационарных состояний, такие как методы враща-
ющегося сектора и эмульсионную полимеризацию [26]. Недавно рабочая
группа ИЮПАК по «Моделированию кинетики и процессов полимеризации»
рекомендовала проводить анализ молекулярно-массового распределения
полимеров методом инициирования полимеризаци и пульсирующим лазером
(PLP), чтобы определить значения константы роста [27,28]. Значения кон-
стант роста и обрыва цепи показаны в Таблице 1 [29-35]. Таким образом,
хотя стадия роста цепи является очень быстрой реакцией (на несколько
порядков выше, чем скорость ступенчатых реакций концевых групп, обсуж-
даемых в параграфе III), её скорость всё же на несколько порядков ниже,
чем скорость стадии обрыва цепи, т. е. реакции двух радикалов. Высокое
значение отношения kjk , приводит к очень низкой стационарной концен-
трации растущих радикалов (10-8М) в этих системах.
Таблица 1. Константы скорости роста и обрывав радикальной полимеризацииа)
Мономер /ср при 60 °C (литр*моль-1’Сек-1) /собрпри 60°С (IO 7) (литр-моль *«ceK *)
Стирол 176 (340) [29] 3.6
Метилметакрилат 367 (830) [33] 1.0
Метилакрилат 2100 0.5
Винилацетат 3700 7.4
Бутадиен 100 (320) [34] ~100 (700) [34]
Изопрен 50
Хлоропрен (1270) [35]
а) Данные взяты из работ [30-32]; данные в скобках получены при полимеризации при ини-
циировании пульсирующим лазером
50 -* V- Глава 2. Полимеризация: синтез эластомеров
Первичный контроль длины цепи определяется соотношением индиви-
дуальных стадий [см. ур. (16)]. Однако, могут протекать реакции «передачи
цепи» поскольку одна цепь обрывается и инициируется новая цепь, не влияя
на скорость полимеризации, и эти реакции определяют длину цепи. Передача
цепи обычно включает гомолитический разрыв наиболее чувствительной
связи в молекуле растворителя, мономера, примеси, и т. д., с образованием
растущего радикала, например
может быть определено следующими соотношениями:
Передача на мономер М}*+М—к'г- ->Mj +М* (17)
Передача на растворитель M..+S—>Mj +5« (18)
S'+M—k-^SM> (19)
Следовательно, длина цепи полимера, получаемого к любому данному мо-
менту времени, может быть выражена как отношение скорости роста к сумме
всех реакций, приводящих к обрыву цепи, как показано в уравнениях (20)
. =____________W‘][M]_______________
" (ktc+2kld)[Mrf +ktrM[Mj*\ [М ] + к^[мг} [5]
ИЛИ
1 _ + ^td )Rp + ktrM + ktrS [5]
X» " кр[М]2 kp kp[M]
(20)
гдеХ; — среднечисленная степень полимеризации, klc — константа скорости
обрыва рекомбинацией и ktd — константа скорости обрыва диспропорцио-
нированием.
В. Молекулярно-массовое распределение
Распределение длин цепей при свободно радикальной поли-
меризации может быть определено из простой статистики. Таким образом,
для полимера, полученного к любому данному моменту времени, это распре-
IV. Полимеризация по свободнорадикальному механизму -*\г 51
деление является наиболее вероятным и определяется отношением скорости
роста цепи к обрыву цепи,
Wx = xpx~x(i-р)2 (21)
где р — вероятность роста, (1 -р) — вероятность обрыва (диспропорцио-
нированием или передачей). Это выражение идентично выражению (6) за
исключением различного значения р. В отличие от уравнения (6), однако,
оно выражает мгновенную длину цепи полимера, а не кумулятивное значе-
ние для всего полученного полимера.
Из уравнения (21) следует, что среднечисленные и средне весовые длины
цепи Хп и Xw выражаются уравнением (22),
так как р должно всегда быть близко к единице для высокомолекулярных
полимеров. Следовательно, это снова означает, что
Xw/Xn = 2 (23)
Значение отношения Xw/Xn для всего полимера может быть, конечно,
много больше в зависимости от изменения значения р с увеличением кон-
версии. Необходимо, однако, отметить, что это справедливо только, когда
растущие цепи обрываются диспропорционированием или передачей, а не
рекомбинацией. В последнем случае может быть показано [И, с. 335, 336],
что распределение прироста цепи намного уже, т. е.
Xw/Xn = \5 (24)
Таким образом, кинетика свободно-радикальной полимеризации харак-
теризуется следующими особенностями:
1. Скорость прямо пропорциональна корню квадратному из концентра-
ции инициатора.
2. Молекулярная масса обратно пропорциональна корню квадратному
из концентрации инициатора
3. Время жизни растущей цепи является коротким (несколько секунд),
но при этом достигается высокая молекулярная масса, что приводит
к образованию высокомолекулярного полимера на начальной стадии
реакции.
4. Не существует никакой прямой зависимости между степенью превра-
щения и длиной цепи.
5. Мгновенная длина цепи является статистической, но кумулятивное
значение может быть значительно шире вследствие изменений в от-
носительных скоростях роста и обрыва цепи.
52
Глава 2. Полимеризация: синтез эластомеров
С. Особый случай полимеризации диенов
Поскольку полидиены всё еще являются основой промыш-
ленности синтетического каучука, важно рассмотреть особенности, которые
проявляют диены в свободнорадикальной полимеризации. Несмотря на то,
что этот вид полимеризации играет и будет играть главную роль в промыш-
ленном производстве различных полимеров, он не подходит для полиме-
ризации в массе или в растворной полимеризации диенов. Это следует из
кинетических особенностей свободнорадикальной полимеризации диенов,
как показано в Табл. 1. Таким образом, относительно высокое отношение
kJкр (по сравнению с другими мономерами) приводит к очень низкой моле-
кулярной массе и очень низкой скорости полимеризации для полидиенов,
полученных в гомогенных системах, как показано в Табл. 2.
Из представленных данных видно, что даже в случае этих некаталитичес-
ких реакций термической полимеризации, когда молекулярная масса должна
была бы быть максимальна по сравнению с каталитическими системами, она
напротив является слишком низкой, по меньшей мере, на порядок величины.
Эти системы также усложняются конкурирующей реакцией Дильса-Альдера,
приводящей к образованию низко молекулярных соединений, т. е. «масел».
Поэтому неудивительно, что первые исследователи не видели перспективы
для этого механизма полимеризации бутадиена, изопрена и т. д. ни при чисто
термическом инициировании, ни при использовании свободнорадикальных
инициаторов, таких как перекиси. Вместо этого они обратились к натриевой по-
лимеризации, которая, хотя также слишком медленна и трудновоспроизводима,
по крайней мере, позволяла получить высокомолекулярные каучукоподобные
полимеры диенов. Позже в 30-гг., когда появилась эмульсионная полимери-
зация, было обнаружено, что, хотя она протекает по свободнорадикальному
механизму, она приводит как к высокой скорости реакции, так и к высокой
молекулярной массе, т. е. благоприятна для получения синтетического каучука.
Характерные особенности эмульсионной полимеризации, которые приводят к
такому удивительному результату, обсуждаются в разделе V.
Таблица 2 . Термическая полимеризация диенова).
Температура (°C) Время (ч) Изопрен 2,3-диметилбутадиен
Выход (%) МВ, каучук Выход (%) МВ, каучук
Масло Каучук Масло Каучук
85 100 7.9 16.3 4600 - - -
85 250 - - - 2.7 19.6 3500
85 900 - 35.3 5700 - 49.7 3500
145 12.5 54.7 15.6 4000 11.1 15.6 2100
а) Данные взяты из работы [37].
IV. Полимеризация по свободнорадикальному механизму -* V 53
D. Контролируемая радикальная полимеризация
Революция в химии свободнорадикальной полимеризации
началась в 80-гг., с пионерским патентом Соломона, Риццардо и Кациоли
[38]. Эти ученые обнаружили, что радикальная полимеризация мономеров,
таких как стирол и алкил (метил) акрилат, может быть контролируемой,
если проводить её в присутствии стабильных нитроксильных радикалов,
как показано ниже.
N--О—CHCHjrbCH—CH^R \| О»+ R-t-CH2CH+^CH2CH.
CO2R CO2R ~CO2R <3O2R
Было обнаружено, что эта контролируемая полимеризация, протекаю-
щая в присутствии устойчивых нитроксильных радикалов, таких как тет-
раметилпиридинилокси радикалы (TEMPO), показанные выше, приводит
к получению полимера с контролируемой молекулярной массой, узким моле-
кулярно-массовым распределением, строением и составом блоксополимеров
и возникновению концевых функциональных групп [38-42]. Ключевые
требования для этого типа контролируемой полимеризации заключаются:
(а) в наличии термически лабильной связи, которая подвергается обрати-
мому гомолизу с образованием реакционно-способных радикалов, которые
могут осуществлять инициирование или рост цепи в полимеризации вини-
ловых мономеров; (Ь) в одновременном образовании устойчивых радикалов,
которые быстро и обратимо рекомбинируют с растущими радикалами, но
не присоединяются к виниловым мономерам; (с) в равновесном состоянии
между радикалами и ковалентными частицами, что облегчает образование
последних. Для систем этого типа обычно отношение концентрации актив-
ных радикальных частиц к неактивным должно быть меньше 10-5 [43]. Это
подтверждает, что большая часть времени жизни цепи затрачивается на
неактивное состояние. Хорошие системы должны содержать оптимальное
количество нитроксида, необходимое для протекания полимеризации с
достаточной скоростью [44]. Необходимо отметить, что может также про-
текать соединение радикала с радикалом, но оно сводится к минимуму при
низкой концентрации растущих радикалов (около 10-8М). Из-за всё ещё
54 ~*V Глава 2. Полимеризация: синтез эластомеров
наблюдающейся реакции обрыва, очевидно, неправильно называть эту
полимеризацию «живой», хотя этот тип контролируемой радикальной
полимеризации часто обозначается в литературе этим термином. Кине-
тика устойчивой свободнорадикальной полимеризации контролируется
стабильным радикальным эффектом, который четко был объяснен Фи-
шером [45, 46].
Тщательное и широкое исследование этой полимеризации, иницииро-
ванной нитроксидами (также обозначаемой как стабильная свободноради-
кальная полимеризация) позволило установить оптимальные условия для
контролируемой радикальной полимеризации различных виниловых моно-
меров [47,48]. Исследованные параметры включают структуру нитроксида
и другие добавки, контролирующие спонтанную полимеризацию мономе-
ра, такого как стирол. Необходимо отметить, что вместо алкоксиаминных
инициаторов может быть применена смесь обычных свободнорадикальных
инициаторов, таких как азосоединения или перекиси.
Применение этого метода к 1,3-диенам приводит к возникновению
проблем. Было показано, что полимеризация замедляется, а затем вообще
прекращается из-за накопления избытка свободного нитроксида [44]. Эф-
фективный процесс контролируемой полимеризации изопрена при 145°С
потребовал введения сахара-восстановителя, такого как глюкоза в присут-
ствии бикарбоната натрия, для реакции с избытком нитроксида [44]. Через
4 часа был получен полиизопрен с Мп = 21000 и Mw/Mn = 1.33 с выходом
25%. Реакция полистирола, ограниченного TEMPO, с бутадиеном или
изопреном приводит к образованию соответствующих диблоксополимеров,
что оценивается методом протонного магнитного резонанса и сканирующей
электронной микроскопии [49]. Никаких доказательств образования гомо-
полимера полистирола или полидиена нет.
Альтернативная процедура уменьшения концентрации избыточных нит-
роксильных радикалов описана Гакером и сотр [50]. Они применяли ини-
циатор, показанный ниже, для успешного воздействия на контролируемую
полимеризацию изопрена. Сообщалось, что соответствующий нитроксид,
имеет атом водорода в a-положении, который может отщепляться на стадии
диспропорционирования, тем самым препятствуя накоплению избыточного
нитроксида. С применением этой системы инициирования нитроксидом
стало возможным получение широкого круга полиизопренов с контроли-
руемой молекулярной массой, высоким содержанием 1,4-микроструктуры
и полидисперсностью, лежащей в интервале 1,07 для малых молекулярных
масс (около Мп = 5000) до 1,20 для среднечисленной молекулярной массы
100000. Однако, требуемыми условиями реакции были температура 130°С
и время реакции 48 часов. Точно также получали хорошо охарактеризо-
ванные сополимеры изопрена и стирола (717^=17000, Мш/Мп=1,1-1,2) или
(метил) акрилата при 120°С.
IV. Полимеризация по свободнорадикальному механизму 55
Для контролируемой радикальной полимеризации было разработано
несколько других методов применимых для синтеза эластомеров [47, 48].
Одной из других наиболее важных систем контролируемой радикальной
полимеризации является радикальная полимеризация с передачей атома
(ATRP) [51]. Катализатор переходного металла (Mt) участвует в окисли-
тельно-восстановительном равновесии посредством обратимой передачи
атома, часто галогена, от стабильной частицы (инициатор или полимерная
цепь), как показано ниже
Рп—X + М/нУлигянд)—►
Рп • + X-Mrm+I (лиганд)
Несмотря на то, что эффективным является широкий круг солей пере-
ходных металлов, наиболее широко были исследованы соли меди.
V. ЭМУЛЬСИОННАЯ ПОЛИМЕРИЗАЦИЯ
А. Механизм и кинетика
Полимеризация в водных системах, которая так широко
развита технологически, представляет особый случай свободнорадикальной
цепной полимеризации в гетерогенной системе [52-58]. В большинстве
систем эмульсионной полимеризации применяется не растворимый в воде
мономер с ПАВ и свободнорадикальным инициатором. Хотя можно думать,
что полимеризация не растворимых в воде мономеров в эмульгированном состо-
янии просто включает прямое превращение дисперсии мономера в суспензию
полимера, это не отражает реального положения вещей, что подтверждается
следующими особенностями истинно эмульсионной полимеризации:
1. Эмульсия полимера или латекс имеет на несколько порядков меньший
размер частиц, чем эмульгированный мономер.
56
Глава 2. Полимеризация: синтез эластомеров
2. Скорость полимеризации на 1 или 2 порядка больше, чем скорость
полимеризации неразбавленного мономера.
3. Молекулярная масса эмульсионного полимера на 1 или 2 порядка
больше, чем полимера, полученного путем полимеризации в массе.
Из этого очевидно, что механизм эмульсионной полимеризации гораздо
сложнее полимеризации мономера в блоке в высокодисперсном состоянии.
В действительности, очень малые размеры частицы латекса относительно
размеров исходной мономерной эмульсии указывает на существование
особого механизма образования таких полимерных частиц.
Механизм эмульсионной полимеризации, как первоначально предло-
женный Харкинсом [59], может быть понят проверкой компонентов этой
системы, как указано на рис. 2, для типичного водонерастворимого мономера
как стирол (растворимость 0,07 г/л) [60].
R
Полимер-
мономерная
частица
ю,3см'3
Свободный
радикал
?????????????
Мономер
ААААААААААААА
Набухшая мицелла мыла
~ю,в см-3
Рис. 2. Локализация компонентов при эмульсионной полимеризации.
Из рисунка видны места локализации мономера, которые соперничают
между собой за доступные радикалы. Таким образом, на начальной стадии
мономер находится в трех местах: растворенный в водном растворе, как
эмульгированные капли, и внутри мицелл мыла. Как растворенный мономер,
так и относительно крупные мономерные капли невыгодны для реакции
с радикалами инициатора (исключение, пожалуй, составляет случай мономе-
ров хорошо растворимых в воде). Большое число мицелл мыла, содержащих
поглощенный мономер, однако представляет статистически благоприятное
V. Эмульсионная полимеризация -* V 57
местоположение для инициирования полимеризации. Таким образом, неудиви-
тельно, что большинство полимерных цепей генерируется внутри мицелл мыла,
набухшего в мономере. Очень большое число (~1015/мл) очень малых полимер-
ных частиц полученных, таким образом, стабилизируется адсорбционными
монослоями мыла, что уменьшает количество молекулярно растворенного мыла,
разрушая мицеллы мыла на ранних стадиях полимеризации (-10% конверсии
в обычном рецепте). Так как все имеющиеся в наличии мыла распределены
и перераспределены на поверхности растущих частиц, количество мыла явля-
ется главным фактором, контролирующим размер частиц латекса.
Поэтому на второй стадии эмульсионной полимеризации, доступные места
для мономера состоят из растворенного мономера, капель свободного мономера
и мономера, поглощенного многочисленными полимерными частицами. Как
и прежде первые два этих положения вносят малый вклад, тогда как полимер-
но-мономерные частицы обеспечивают главное местоположение для реакции
с радикалами инициатора, диффундирующими из водной фазы. Основная
реакция полимеризации протекает внутри очень большого числа латексных
частиц, которые изолированы друг от друга электростатическим отталкиванием
и продолжают оставаться насыщенными мономером [61], диффундирующим из
капель мономера. Именно этот аспект приводит к уникальным характеристикам
этой системы [62]. Таким образом, поскольку радикалы инициатора вступают
в полимерно-мономерные частицы и инициируют цепь, последняя может
продолжать расти с имеющимся в наличии мономером, пока другой радикал
не войдет в эту частицу. Таким образом, скорость обрыва цепи действительно
контролируется скоростью входа радикалов в частицу и это главным образом
увеличивает время жизни растущих цепей и, следовательно, длину цепи. Более
того, так как растущие цепи все расположены в различных частицах, они не
в состоянии ограничивать друг друга, что приводит к высокой концентрации
растущих цепей и, следовательно, к более высокой скорости.
Таким образом, в системах эмульсионной полимеризации возможно
одновременное достижение гораздо более высоких скоростей и много более
высокой молекулярной массы, чем в гомогенных системах. Сравнение ки-
нетических особенностей полимеризации в блоке и эмульсионной полиме-
ризации стирола дано в Табл. 3.
Таблица 3. Сравнение методов свободнорадикальной полимеризации.
Гомогенный Эмульсионный
Концентрация мономера (М) 5 5°>
Концентрация радикала(М) 10“8 10"6
Скорость полимеризации при 60 °C (%/ч) -2 100
Молекулярный вес (Мп) 105 107
а) Внутри латексных частиц
58 -*V- Глава 2. Полимеризация: синтез эластомеров
Очевидно, что главное различие состоит в том, что в эмульсионных систе-
мах возможно повышение стационарной концентрации растущих радикалов
на 2 или 3 порядка, а не за счет увеличения скорости обрыва, наблюдаемого
в гомогенном растворе [см. ур. (16)]!
Ситуация, описанная ранее, т. е. когда радикалы, вступающие в отдельные
латексные частицы, инициируют и обрывают растущие цепи, относится
к идеальной эмульсионной полимеризации, согласно теории Эварта-Смита
[62]. При этих условиях концентрация растущих цепей в единице объема
латекса легко предсказуема, так как в любое данное время половина частиц
будет содержать растущую цепь. Другими словами, число растущих цепей
равно половине числа частиц. Так как число последних порядка 1018 М
в литре, концентрация растущих цепей равна 10"6 молей по сравнению с 10-8 М
для гомогенной полимеризационной системы. Из-за того, что растущие
цепи находятся в окружении такого большого количества мономера (внутри
мономер-полимерных частиц) неудивительно, что скорость эмульсионной
полимеризации на 1 или 2 порядка выше, чем скорость полимеризации
в блоке, как показано в Табл. III. Более того, эта высокая концентрация ра-
дикалов не влияет на время жизни радикалов, т. е. на длину цепей, которая
управляется исключительно доступностью другого свободного радикала для
обрыва цепи, и, таким образом, временем между успешным вступлением
радикалов в частицу. Для данной скорости инициирования время между
вступлением радикала зависит от числа латексных частиц, т. е. время жиз-
ни радикала (и молекулярная масса) увеличиваются с увеличением числа
частиц. Согласно теории Эварта и Смита, число полимерных частиц зависит
как от концентрации инициатора, так и от концентрации ПАВ,
/Voc[/]2/5[5]3/5 (25)
где [/] — концентрация инициатора и [5] — концентрация ПАВ.
Эта ситуация, конечно, относится только к идеальному случаю, как было
определено раньше. Если растущие цепи внутри латексных частиц подвер-
гаются побочным реакциям, которые нейтрализуют активность радикалов
в частице до вступления следующего радикала, или, если скорость обрыва
цепи невелика, когда 2 радикала занимают одну и ту же частицу, тогда число
растущих цепей в данный момент времени будет соответственно меньше или
больше половины числа частиц. Последний случай (более чем один радикал
на частицу) может встречаться, например, если размер частицы достаточно
велик и скорость обрыва цепи также низкая. Скорость и молекулярная масса
тогда управляются другими правилами, чем интервал между вступлением
активных радикалов. Диагностически эта ситуации могут отличаться от
идеального случая из-за влияния добавленного инициатора на скорость
полимеризации после того, как образование частиц заканчивается. Таким
образом, в любом случае увеличение концентрации инициатора будет приво-
дить к большей скорости вступления радикалов в частицу и, следовательно,
V. Эмульсионная полимеризация -•V- 59
увеличению числа радикалов, приходящихся на одну частицу, что приво-
дит к увеличению скорости полимеризации. Наоборот, в идеальном случае
увеличение частоты проникновения радикалов в частицы не должно влиять
на скорость, так как частицы будут всё еще содержать радикалы только по-
ловину времени, не смотря на то, что период роста цепей будет короче, что
приведёт к более низкой молекулярной массе.
Идеальный случай теории Смита и Эварта действительно предполагает
более элегантный метод для получения абсолютного значения константы
скорости роста цепи к для систем эмульсионной полимеризации, как по-
казано в ур. (26), где 7V — число частиц в единице объема.
Rp = kp[M]N/2 (26)
Ур. (26) приводит к решению для кр из доступных знаний скорости/?^, кон-
центрации мономера в мономер-полимерных частицах [Л/] и числе частиц [7V].
Этот метод применялся к нескольким мономерам, и может быть особенно
полезен в случае диенов, где в классическом методе фотоинициирования
возникают трудности. Некоторые из этих результатов показаны в Табл. 4
в форме кинетических параметров. Для сравнения включены результаты,
полученные для фотоинициированной полимеризации стирола.
Таблица 4. Константы скорости роста цепи при эмульсионной полимеризации.
Мономер кр (литр-моль ^сек при 60 °C Ер (ккал-моль 4р(хЮ-7) (литр-моль !- •сек-1) Работа
Бутадиен 100 9.3 12 [31]
Изопрен 50 9.8 12 [31]
2,3-диметилбутадиен 120 9.0 9 [63]
Стирол 280 7.9 4 [31]
Стирол 176 7.8 2.2 [30]
Видно, что совпадение данных является достаточно хорошим, учитывая
значительное различие использованных экспериментальных методов. Недавнее
изучение эмульсионной полимеризации бутадиена показало, что константа
скорости роста цепи даже выше, чем оценивалась ранее (см. Табл. 1) [34].
Данные Табл. 4 доказывают, что причиной низких значений скоростей
и низких молекулярных масс, появляющихся в гомогенной свободноради-
кальной полимеризации этих диенов, является не только низкая константа
скорости полимеризации роста цепи, а в большей степени высокое значение
константы скорости обрыва цепи (как показано в Табл. 1). Следовательно,
в специальных условиях эмульсионной полимеризации, где скорость обрыва
цепи контролируется скоростью вступления радикалов в частицы, становится
60 -* V Глава 2. Полимеризация: синтез эластомеров
возможным достигать как высоких скоростей, так и высоких молекулярных
масс. Именно это позволяет применять эмульсионно-полимеризационные
системы для получения синтетических каучуков на основе диенов.
В. Бутадиен-стирольный каучук
1, Кинетика и молекулярные массы
Наиболее успешным методом, разработанным для полиме-
ризации синтетического каучука общего назначения, была эмульсионная
сополимеризация бутадиена и стирола (БСК), которая все еще является
основным, применяемым сегодня процессом [54, 64-69]. Общие принципы
сополимеризации будут обсуждены в последующем разделе, но здесь следует
проверить другие основные особенности этой системы. Типы применяемых
рецептов показаны в Табл. 5 [67].
Приведенные рецепты являются типичными, так как они часто применяют-
ся. Необходимо отметить, что инициатором при 50°С полимеризации (горячая
полимеризация) является персульфат, тогда как в рецепте при 5°С (холодная
полимеризация) инициатор состоит из окислительно-восстановительной систе-
мы, содержащей гидропероксид-железо (II)-сульфоксилат-ЭДТА. В последнем
случае инициирующие радикалы образуются реакцией гидропероксида с ионом
железа, концентрация которого контролируется комплексообразующим агентом
ЭДТА; сульфоксилат необходим, чтобы превратить окисленные ионы железа (III)
обратно в ионы железа (II). Соль фосфата служит стабилизирующим электроли-
том для латекса. В обоих рецептах тиол действует как агент передачи цепи, для
того чтобы предотвратить образование слишком высокомолекулярного полиме-
ра, возможно в эмульсионных полимеризационных системах (см. Табл. 6).
Он действует аналогично растворителю в ур. (18) и (19), за исключе-
нием того, что связь серы с водородом крайне подвержена атаке растущим
полимерным радикалом, который, таким образом, отрывает атом водорода,
образуя радикал RS •, инициирующий рост новой цепи:
Р• + RS-Н —> Р-Н + RS • (27)
RS- + M—RS-M- 28)
Эти тиолы, которые известны как «регуляторы», имеют константы пере-
дачи больше единицы, т. е. klr/kp может быть 3-4, так что достаточно малых
их количеств для уменьшения молекулярной массы от нескольких милли-
онов до нескольких тысяч. Полимеры на основе диенов могут подвергаться
реакциям сшивания при полимеризации, что приводит к образованию не-
растворимого гель-каучука, когда молекулярная масса становится слишком
высокой. Следовательно, тиолы необходимы как модификаторы для предо-
твращения гелеобразования и сохранения обрабатываемости каучука. Также
необходимо остановить реакцию на промежуточном уровне конверсии, чтобы
минимизировать нежелательное гелеобразование (см. Табл. 7).
V. Эмульсионная полимеризация
61
Таблица 5. Типичные рецепты эмульсионной полимеризации при получении БСКа).
БСК-1000 ь> БСК-1500ь>
Температура полимеризации (°C) 50 5
Время (ч) 12 12
Степень превращения (%) 72 60-65
Ингредиенты
Бутадиен 71 71
Стирол 29 29
Вода 190 190
Мыла (жирных или канифольных кислот) 5 4.5-5
Персульфат калия 0.3 -
н-додекантиол 0.5 -
т-додекантиол - 0.2
Гидрокпероксид п-ментанас) - 0.08
Фосфат натрия (Na3PO4*10H2O) - 0.5
Сульфат железа (II) (FeSO4*7H2O) - 0.4
Сульфоксилат формальдегида натрия - 0.10
Этилендиаминтетраацетат натрия (ЭДТА) - 0.06
а) Весовые доли. Данные взяты из работы [67].
Ь) Обозначения промышленных сортов определены Международным институтом производите-
лей синтетического каучука для «горячего» и «холодного» БСК соответственно.
с) Или гидропероксид пинена.
Таблица 6. Типичные свойства эмульсионного БСКа).
Свойство Горячий БСК Холодный БСК
Содержание стирола 24 24
Молекулярный вес
Средневязкостный 1.5-4.0 х 105 2.8 х 105
Средневесовой - 5х 105
Среднечисленный 0.3-1.0х105 1.1-2.6 хЮ5
Данные взяты из работы [67].
62 -*!/• Глава 2. Полимеризация: синтез эластомеров
Таблица 7. Параметры сшивания для полибутадиенаа).
Температура (°C) Относительная скорость сши- вания, гс (х104)Ь) %w первичных цепей в гель-точке (х104)
60 1.98 2.15
50 1.36 3.13
40 1.02 4.18
0 (кал.) 0.16 26.3
а) Данные взяты из работы [20].
ь) г = Где и к?, соответственно, константы скорости сшивания и скорости роста цепи.
Вскоре после Второй мировой войны американская промышленность
синтетического каучука начала производить «холодный» SBR (БСК), из
которого были получены превосходные шинные резины особенно, что
касается сопротивления износу. Последующее изучение показало, что
снижение температуры от 50 до 5°С оказывает незначительное или совсем
не оказывает влияния на микроструктуру полидиеновых единиц (цис-
1,4 по отношению к транс-\,к и) или состав сомономера, но оказывает
заметное влияние на молекулярно-массовое распределение (Табл. 7).
Также было показано, что реакция сшивания, т. е. присоединение ра-
стущих цепей к полимерным двойным связям (главным образом 1,2)
существенно уменьшается при этих низких температурах, таким образом,
уменьшая тенденцию гелеобразования при любой данной молекулярной
массе [70].
Табл. 7 показывает, что максимальные молекулярные массы полибу-
тадиена достигаются при различных температурах полимеризации, пред-
шествуя гелеобразованию, по критической средневесовой длине цепи, хш
или длине первоначальных цепей в гель точке. Таким образом, видно, что
можно увеличить длину цепи в 9 раз, без гелеобразования, уменьшением
температуры полимеризации от 50 до 0°С. Следовательно, количество
агента передачи цепи (тиола) также может быть уменьшено и это улуч-
шает общее распределение по длинам цепей, избегая образования очень
низкомолекулярной фракции, которая образуется при быстрой реакции
тиола на ранних стадиях полимеризации. Более того, эта возможность
получения безгелевого каучука БСК высокой молекулярной массы при
уменьшении температуры полимеризации достигается получением по-
лимера с высоким значением вязкости по Муни (-100), который может
быть пластифицирован дешевыми нефтяными маслами («маслонапол-
ненный» каучук) и еще сохранять свои высокие механические свойства.
Поэтому методом холодной полимеризации БСК получается более 85%
эмульсионного БСК [68].
V. Эмульсионная полимеризация -*v- 63
2, Микроструктура цепи
Как можно было бы ожидать, эмульсионные полимериза-
ционные системы не изменяют основной механизм свободнорадикальной
полимеризации, по отношению к структуре мономерного звена. Последняя,
конечно, не зависит от типа применяемого свободнорадикального инициа-
тора, вследствие «свободной» природы растущей цепи концевого радикала.
Температура полимеризации оказывает некоторое влияние, как следует из
данных Табл. 8, но не в очень большой степени.
Можно видеть, что содержание боковых 1,2 винильных групп гораздо
менее чувствительно к температуре, тогда как содержание транс-1,4 групп
увеличивается с понижением температуры, за счет содержания tyuc-1,4
структуры. Последние почти исчезают при низкой температуре и тогда
полимер сохраняет высокое содержание транс-\Д структур, около 80%.
Следовательно, этот тип полибутадиена достаточно стереорегулярный, чтобы
кристаллизоваться при охлаждении [72, 73]. Однако, введение стирола как
сомономера достаточно, чтобы разрушить регулярность цепи, необходимую
для кристаллизации. Более того желателен именно полибутадиен с высоким
содержанием цис-1,4 структур, а не транс-1,4 структуры, т. к. последний
имеет точку плавления около 150°С и не является эластомером при темпера-
туре эксплуатации. Как видно из Табл. 8, возможность образования полимера
с высоким содержанием цис-1,4 структуры при достаточно высокой темпе-
ратуре полимеризации представляет довольно отдаленную перспективу.
Таблица 8. Структура цепи эмульсионного полибутадиена и БСКа)
Температура поли- меризации (°C) Изомер (% вес)
цис-1,4 транс-1.4 1.2
Полибутадиен
-33 5.4 78.9 15.6
5 13.0 69.9 16.5
50 19.0 62.7 18.8
70 20.8 59.4 19.8
БСК
-33 5.4 80.4 12.7
5 12.3 71.8 15.8
50 18.3 65.3 16.3
70 20.0 63.0 17.3
100 22.5 60.1 17.3
а) Данные взяты из работы [71].
64 -'V- Глава 2. Полимеризация: синтез эластомеров
Следовательно, из рассмотренного ясно, что малое влияние температуры на мик-
роструктуру бутадиеновых звеньев не будет реально влиять на свойства БСК.
С. Эмульсионная полимеризация хлоропрена
1. Кинетика
Единственным другим диеном, который широко применяет-
ся для промышленной эмульсионной полимеризации, является хлоропрен
(2-хлор-1,3-бутадиен) [64, 74-77]. Наличие атома хлора, по-видимому,
сообщает заметную реакционную способность этому мономеру, т. к. он по-
лимеризуется много более быстрее, чем бутадиен, изопрен или другие диены
(см. Табл. 1 и 4); кр (35°С) = 595 л моль-1сек-1 [35]. В действительности,
хлоропрен даже более чувствителен к самопроизвольной свободной полиме-
ризации, чем стирол, и требует мощных ингибиторов для стабилизации [78].
Он полимеризуется чрезвычайно быстро в эмульсионных системах, так что
скорость необходимо внимательно контролировать.
Для эмульсионной полимеризации хлоропрена с персульфатом калия
как популярным инициатором могут применяться различные рецепты
[79, 80]. Основной рецепт, в котором проявляются некоторые интересные
особенности этого мономера, показан в Табл. 9. Два аспекта этого рецепта
особенно существенны: применение мыла из древесной смолы и присутствие
элементной серы. Мыла из смолы известны как замедлители в эмульсионной
полимеризации, т. к. являются наиболее полиненасыщенными жирными
кислотами. Полная конверсия может быть достигнута за несколько часов.
С насыщенными жирными кислотами реакция почти полностью завершается
[79] в течение одного часа при 40°С.
Таблица IX. Основной рецепт для неопрена GN а)
Ингредиент Доля по весу
Хлоропрен 100
Вода 150
N древесная канифоль 4
Сера 0.6
Гидроксид натрия 0.8
Персульфат калия 0.2-0.1
Стабилизатор латексав 0.7
а) Температура, 40°С; время, несколько часов; конверсия 90%;
Данные взяты из работы [81].
Ь) Натриевая соль продукта конденсации нафталинсульфокислоты
с формальдегидом.
V. Эмульсионная полимеризация JV 65
Сополимеры серы с хлоропреном [81,82] образуют ди- и полисульфидные
связи в цепи (как показано в формуле (29)).
СН2=С—СН=СН2 + S8 ->
Cl
-Ьсн2 /CH24-OTSx-bCH2 zCH24-„ (29)
с=с с=с
с/ \ с/ \
Латекс затем обрабатывается хорошо известным ускорителем вулканиза-
ции, тетраэтилтиурамдисульфидом, который посредством обмена серными
связями, разрушает сшитый полихлропреновый гель и делает его раствори-
мым и перерабатываемым, как схематически показано в ур. (30).
-Ьсн2 сн2 -Ь sx 4-сн2 сн2 -Ь
с=с с=с
с/ \ с/ \
(Et)2NCS4CN(Et)2
(30)
-f-CH2 CH2+mSy—SCN(Et)2
C=C
cr хн
Таким способом, достигается цель, аналогичная применению тиолового
агента передачи цепи в полимеризации БСК. В действительности, получены
новые сорта полихлоропрена [75, 77, 83] с применением тиолов и других
агентов передачи цепи. Установлено [80], что тиолы приводят к более узко-
му молекулярно-массовому распределению для хлоропрена по сравнению
с бутадиеном или изопреном, благодаря сильно замедленной скорости их
расхода в присутствии хлоропрена. Эти сорта меркаптанового регулирования
представляют обычные стандартные сорта [77].
2. Структура цепи
Другой особенностью эмульсионной полимеризации хло-
ропрена, которая отличает ее от полимеризации других диенов, является
тот факт, что она приводит преимущественно к транс-1,4 микроструктуре
цепи. Таким образом, даже при температуре полимеризации окружающей
среды полихлоропрен содержит свыше 90% транс-1,4 единиц, как показано
в Табл. 10, что иллюстрирует влияние температуры полимеризации на стерео-
регулярность цепи [87]. Снижение температуры полимеризации приводит
к более стереорегулярному транс-1,4 полихлоропрену. Из-за более высокой тем-
пературы плавления кристаллов транс-1,4 полихлоропрена (Тп = 105°С) [88],
66 Jv* Глава 2. Полимеризация: синтез эластомеров
по сравнению с соответствующей температурой для цис-1,4 полиизопрена из
каучука гевеи (-20°С), даже полимер, содержащий менее 80% транс-структу-
ры кристаллизуется легче при охлаждении или при растяжении. Температура
плавления эмульсионного полихлоропрена обычно лежит в интервале 40-50°С
[75]. Следовательно, эмульсионный полихлоропрен является единственным
латексным полимером, который напоминает природный каучук, в том, что он
является существенно стереорегулярным, чтобы кристаллизоваться при растя-
жении [74, 75], что выражается в повышении разрывной прочности ненапол-
ненных вулканизатов без усиливающих агентов, также как в случае каучука
гевеи. Это делает возможным применение хлоропрена в различных резиновых
изделиях, обладающих превосходной стойкостью к маслам и растворителям
(из-за высокой полярности), так же как и высокой прочностью.
Таблица 10 Влияние температуры полимеризации на микроструктуру цепи
полихлоропренаа).
Температура (°C) Изомерная микроструктура цепи i (%)
Транс-1,4 1,2 Изомеризо- ванная 1,2е) 3,4 Цис-1,4
Общая Инверсионная1*)
-150 -100 2.0 - - - -
-40 97.4 4.2 0.8 0.6 0.5 0.8
-20 97.1 4.3 0.9 0.6 0.5 0.8
0 95.9 5.5 1.2 1.0 1.1 1.8
20 92.7 8.0 1.5 0.9 1.4 3.3
40 90.8 9.2 1.7 0.8 1.4 5.2
90 85.4 10.3 2.3 0.6 4.1 7.8
а) Данные взяты из работы [75, 84-86].
Ь)4,1 присоединение
с> -СН2-С=СНСН2С1.
VI. СОПОЛИМЕРИЗАЦИЯ
А. Кинетика
Сополимеризация включает одновременную цепную полиме-
ризацию смеси из двух или более мономеров [89-92]. Из общего кинетичес-
кого рассмотрения, которое управляет этими цепными реакциями, как опи-
сано раньше, имеются дополнительные особенности, а именно относительное
участие различных мономеров в росте цепи. Этот новый параметр — самый
важный, т. к. он контролирует состав сополимера. Системы, включающие
VI. Сополимеризация
Л
67
более двух мономеров, трудно описать подобным образом; возможность отно-
сительно легко описать существует в случае пары мономеров [91, 93-95].
При цепной полимеризации двух мономеров независимо от данного
механизма растущая цепь всегда может иметь возможность реагировать
с одним из двух мономеров. Более того, имеется два типа растущей цепи,
зависящие от типа мономерной единицы, имеющей растущий конец. Таким
образом, могут быть описаны четыре типа элементарных актов роста цепи,
как следует для любой цепной полимеризации двух мономеров, предпола-
гая, что реакционная способность конца цепи зависит только от конечного
мономерного звена цепи (концевая модель) [89]:
м*+мх- кц (31)
М\ + М2- kj-2 -+м2 (32)
м*2+м2- к 22 -+м2 (33)
M2+Mt- ^21 +м* (34)
где М* и Мх представляют растущие цепи и мономер соответственно, а звез-
дочка относится к двум типам мономера в смеси. Видно, что эти 4 реакции
роста цепи приводят к 4 константам скорости роста, как показано ниже.
Следовательно, скорость расхода каждого мономера может быть выражена
следующими уравнениями.
d[M,]/dt = МК] W ]+MM2*] [Mi ] (35)
d[M2]/dt = kX2\M*] [M2]+fc>2[М2 ] [М2] (36)
d[Mt] d[M2] к^М'^М^+к^М'^М^ (37)
Т. к. эти выражения представляют относительную скорость расхода двух
мономеров, которая определяет состав цепи, она может быть выражена урав-
нением (37), полученным делением уравнения (35) на уравнение (36).
Очевидно, чтоур. (37) достаточно неудобно для прямого использования.
Однако, можно значительно упростить его с использованием ранее описан-
ной постадийной обработки. Это делается из предположения, что скорость
описываемая ур. (32) равна скорости в ур. (34), что приводит к равенству
4^217 17K71J
68 -*V Глава 2. Полимеризация: синтез эластомеров
которое после подстановки в ур. (37), дает ур. (38), после соответствующих
перестановок:
d[M2]
(38)
где = kn/ki2 и r2 = k22/k2V Параметры и r2 известны как относительные
реакционные способности мономера, т. к. они выражают относительную
реакционную способность каждого из двух типов концов растущих цепей
их собственного мономера по сравнению с другим мономером. В действи-
тельности это может быть рассмотрено как выражение тенденции гомопо-
лимеризации для каждого типа мономера по отношению к пересечению
с другим сомономером.
Ур. (38), которое выражает отношение мгновенного состава сополиме-
ра к преобладающей концентрации мономера, может быть применено для
того, чтобы определить значения гх и г2. Многие такие значения приве-
дены в работах [91, 94, 96, 97]. Типичные значения этих параметров для
полимеризации стирола показаны в Табл. 11, что говорит о разнообразии
вариантов. Относительная реакционная способность действительно
выражает относительную реакционную способность каждого мономера,
проявляемую по отношению к стирольным радикалам в сравнении с
реакцией с мономером стирола.
Таблица 11 Относительная реакционная способность мономеров при свобод-
норадикальной сополимеризации со стиролом (М{)а).
Мономер (М2) Г1 Г2 Относительная реактивность (1 /rt)
Малеиновый ангидрид 0.097 0.001 10.3
2,5-дихлоростирол 0.268 0.810 3.73
Метилметакрилат 0.585 0.478 1.71
Метилакрилат 0.871 0.148 1.15
Винилиденхлорид 1.839 0.087 0.54
Диэтилмалеат 6.07 0.01 0.16
Винилацетат 18.8 0.02 0.05
а) Данные взяты из работы [96].
Таким образом, значения г1 и г2 позволяют сделать некоторые выводы об
ожидаемом составе сополимера при любом данном отношении мономеров.
Например, из Табл. 11 вытекает, что сополимер стирола и малеиного
ангидрида (г}г2 —> 0) должен быть в значительной степени чередующимся
VI. Сополимеризация -’V- 69
[90], т. к. мало вероятна последовательность двух стирольных звеньев
и совсем невероятно соединение двух звеньев малеинового ангидрида.
Также очевидно, крайне трудно получить сополимер стирола и винил-
ацетата, т. к. последний мономер фактически исключен из сополимери-
зации со стиролом.
Из ур. (38) видно, что состав сополимера не обязательно соответствует
количеству сомономера в смеси, а зависит от значений и г2. Конечно,
в желаемой системе сомономеры должны входить в сополимер в соотноше-
ниях их концентраций в исходной смеси, т. е. где.
d[M2] [М2]
(39)
Это определяется как «азеотропная» сополимеризация [90] по аналогии с
дистилляцией смеси двух совместимых жидкостей. Ур. (37), которое при-
менимо в условиях, где
г2[Л/2]+[М,] 1 ’
например, где = r2 = 1. В этом случае ур. (39) может применяться для всех
отношений сомономеров, т. е. два типа растущих цепей не обнаруживают
существенного преимущества для любого из двух мономеров; это случай
статистической сополимеризации [90]. Также ур. (40) должно быть спра-
ведливо при ri = г2и [Л/J = [М2], т. е. в случае, когда азеотропная сополи-
меризация, происходит только при эквимолярном соотношении в смеси. В
общем ур. (40) справедливо, когда
= (41)
[М2] (q-l)
Это означает, что любая сополимеризация относится к азеотропному типу
при соотношении сомономеров, указанном в ур. (41). Однако, это также
означает, что азеотропная сополимеризация возможна только, когда оба
значения и г2или больше, или меньше 1.
Важно обратить внимание, что эта кинетическая обработка пригодна
для любого цепного механизма полимеризации, т. е. свободнорадикаль-
ной, катионной, анионной и координационной. Однако в случае ионного
механизма тип применяемого инициатора и природа растворителя могут
влиять на значения г1 и г2. Это происходит из-за того, что растущий
конец цепи в ионных системах обычно связан с противоионом так, что
структура и реакционная способность таких концов цепей, как можно
ожидать, определяется инициатором и растворителем. Это будет обсуж-
дено в разделе VIII С.
70 Глава 2. Полимеризация: синтез эластомеров
В. Эмульсионная сополимеризация диенов
Три системы, которые применяют для сополимеризации
промышленных синтетических каучуков, представляют собой бутадиен-
стирольный (SBR), бутадиен-акрилонитрильный (NBR) и хлоропреновый
каучуки с различными сомономерами.
1. Бутадиен-стирольный каучук (БСК)
Было проведено большое количество исследований по реак-
ционной способности в такой сополимеризации, как в гомогенных, так и в
эмульсионных системах, и средние значения г были получены для бутадиена
и стирола:
rfi=l,6, г5=0,5 [69,98,99]
Эти значения применимы к растворной и эмульсионной полимеризации,
предположительно потому, что оба мономера не являются водорастворимыми
и чувствительными к температуре. Поэтому очевидно, что бутадиен должен
встраиваться в цепь существенно быстрее его содержания в шихте, и что
каждый участок образующегося полимера содержит большее количество
стирола. Это подтверждается изменением состава сополимера с конверсией,
как показано в Табл. 12. Видно, что при высокой конверсии количество вновь
образующегося полимера обогащено стиролом с сопутствующей потерей
каучукоподобных свойств, несмотря на то, что суммарный или интеграль-
ный состав еще соответствует низкому содержанию стирола. Это требует
проведения реакции при конверсии не свыше 60%. [64]
Таблица 12. Состав сополимеров БСКа).
Степень превращения (%) Стирол в сополимере (% вес)
Дифференциальный Интегральный
0 17.2 -
20 18.8 17.9
40 20.6 18.7
60 23.3 19.7
80 29.5 21.2
90 36.4 22.5
95 45.0 -
100 (100) (25.0)
а) Весовые соотношения мономеров, 75/25 бутадиен/стирол , 50 °C. Данные взяты из ра-
боты [99].
VI. Сополимеризация 71
Необходимо также отметить, что значение г для этой системы не позво-
ляют проводить азеотропную полимеризацию, что предполагает ур. (39).
Согласно распределению стирольных мономерных звеньев в сополимере
произведение реакционных способностей мономеров, rBrs = 0,8 близко
к 1, что соответствует «идеальной» сополимеризации [90], которая будет
соответствовать статистическому распределению стирольных звеньев
в цепи. Для «идеальной» сополимеризации [90] относительные скоро-
сти расхода двух мономеров не зависят от концевого звена цепи, как это
следует из ур. (42).
1 коп ксо
гв= — , следовательно —— = (42)
rs kBS
Сообщается, что среднечисленное количество стирольных звеньев
в последовательностях равно 1.2, что определено гельпроникающей хро-
матографией высокого разрешения продуктов озонолиза. [100] Наблюда-
емые распределения последовательностей мономерных звеньев находятся
в соответствии с расчетными значениями, основанными на относительной
реакционной способности мономеров [101].
2. Бутадиен-акрилонитрильный (нитрильный каучук)
Согласно Хофману, [102] значения относительной реакци-
онной способности этой пары мономеров при 50°С в эмульсионной поли-
меризации
ГВ = °-4> rAN= °-04
и они иногда уменьшаются при низкой температуре, но в незначительной
степени. На эти отношения несомненно влияют заметная растворимость
акрилонитрила в воде по сравнению с бутадиеном.
Предыдущие значения г приводят к следующей ситуации. Согласно
ур. (39), азеотропный сополимер образуется, когда содержание акрило-
нитрила находится в пределах от 35 до 40% весовых (или мольных) долей,
так что постоянный состав поддерживается в течение полимеризации. Если
содержание акрилонитрила ниже этого значения, сополимер относительно
обогащен акрилонитрилом, содержание которого прогрессивно уменьша-
ется с увеличением конверсии. Однако, если содержание акрилонитрила
выше азеотропного, сополимер содержит меньше акрилонитрила, чем
в исходной смеси, но содержание акрилонитрила увеличивается с конверсией.
Т. к. промышленные нитрильные каучуки содержат от 10 до 40% нитрила,
это обстоятельство имеет большое практическое значение. По отношению
к распределению сомономерных звеньев в сополимере, отношение реакци-
онной способности мономеров гвг^ = 0,016, близко к 0, что соответствует
чередующемуся распределению мономерных звеньев [90].
72 -* V Глава 2. Полимеризация: синтез эластомеров
3. Хлоропрен
Полихлоропрен вообще получается промышленно как го-
мополимер, хотя малое количество сомономера включается в некоторые
сорта этих эластомеров. Существуют две веские причины для получения
небольшого количества сополимеров хлоропрена. Первая причина состоит
в том, что гомополимер, как показано ранее, имеет высокое содержание
транс-1,4 структуры в цепи и поэтому более способен к кристаллизации при
растяжении, как и натуральный каучук, что приводит к высокой прочности.
Он также имеет другие улучшенные механические свойства. Вторая причи-
на состоит в том, что хлоропрен не очень восприимчив к сополимеризации
по цепному радикальному механизму, что иллюстрируется значениями г
приведеными в Табл. 13. Таким образом, за исключением 2,3-дихлор-1,3-бу-
тадиена хлоропрен недостаточно способен к сополимеризации с другими мо-
номерами. Следовательно, неудивительно, что небольшое число промышленно
доступных сополимеров хлоропрена содержит только незначительное число
сомономеров, которые используются для модификации свойств эластомеров.
Таблица 13. Относительная реакционная способность моно-
меров при сополимеризации с хлоропреном а>.
Мономер (М2) Г1 Г2
Стирол 5.98 0.025
Изопрен 2.82 0.06
Акрилонитрил 5.38 0.056
Метилметакрилат 6.33 0.080
2,3-дихлоро-1,3 бутадиен 0.31 1.98
а) Данные взяты из работы [96,103].
VII. ЦЕПНАЯ ПОЛИМЕРИЗАЦИЯ ПО КАТИОННОМУ
МЕХАНИЗМУ
А. Механизм и кинетика
В этих цепных реакциях присоединения активные центры,
имеющие катионную природу, инициируются сильными кислотами или
протонными кислотами или кислотами Льюиса [21,104,111]. Т. к. большин-
ство этих процессов ионной полимеризации проводятся только в неводных
растворителях с низкой диэлектрической постоянной [109], мало вероятно,
что активные центры являются свободными ионами аналогично свободным
радикалам. Большинство активных центров, которые можно рассматривать
как частицы, обеспечивающие рост цепи, пердставлены ниже набором кати-
VI. Сополимеризация
Л
73
онных центров, один или несколько из которых могут выступать в качестве
активного центра роста цепи, особенно в более полярных растворителях
[112]. К сожалению, имеется очень мало информации о точной природе
растущих центров в катионных системах.
R-х =5==* R° Хв R° Хв -W—- RB + Хв
ковалентный контактная разделенная свободные
ионная пара ионная пара ионы
м V м *р2 М к D3 М к/
▼ ▼ и ▼
Это, главным образом, происходит из-за присущих этим системам экспе-
риментальных трудностей, вызванных высокой реакционной способностью
и чувствительностью к примесям, особенно к следам воды.
Наиболее часто применяемыми инициаторами катионной полимери-
зации являются кислоты Льиюса, такие как А1С13, BF3, SnCl4, TiCl4, хотя
также могут применяться сильные кислоты, такие как H2SOZj,. Катионная
полимеризация ограничивается виниловыми мономерами с электроно-
донорными или электроно-делокализующими заместителями, например
изобутиленом, винилалкиловым эфиром, виниламином, стиролом и другими
сопряженными углеводородами. Эти виды полимеризации характеризуются
очень высокими скоростями при очень низких температурах, например изо-
бутилен полимеризуется почти мгновенно при -100°С под действием А1С13.
Присутствие доноров водорода, таких как вода или протонные кислоты,
являющихся сокатализаторами, обычно является необходимым условием,
как было показано в случае изобутилена [ИЗ]. На этой основе предложен
механизм Эванса-Поляни [114] для следующей последовательности реакции
полимеризации изобутилена моногидратом BF3.
Инициирование
СН3
I
bf3-h2o + сн2 =С(СН3)2-----► сн3 - Си BF3OHB (43)
сн3
Рост цепи
СН, СН,
। m в ।
СН3—CSBF3OH +СН2=С—►-—►
СН3 СН3
сн
сн3
с —
I
сн3
сн,
I
сн9— с—
I
сн3
сн,
|в в
•СН9--eSBF3OH
I
г СН3
(44)
74
Глава 2. Полимеризация: синтез эластомеров
Обрыв цепи
СНз
СН—<р---CH*
сн3
12
“Г
сн3
СН2—(ри BF3OHB
сн3
сн3 сн3
I 3 I 3
сн3
сн3
сн=с
/СН3
^СНз
>СН2
сн3
+ СН3—с----СН2-С------сн2—(jT+ BF3.OH2
СНз
сн3
сн3
сн3
х
X
(45)
Передача цепи на мономер также является существенной стадией в ка-
тионной полимеризации изобутилена, и именно эта реакция контролирует
молекулярную массу. [116]:
Передача цепи
СН3—с----СН, — с —
I |
сн3 сн3
СН,
I
СН2—сш BF3OHB
сн3
X
сн2^=с
^tr
сн3
сн3
сн3
—с
сн3
:н2—с
сн3
сн2 — с
СН
сн3
(CH3)3CBBF3OHE1
2
(46)
Этот механизм подтверждается данными ИК и ПМР спектроскопии, кото-
рые показывают присутствие полимерных концевых групп, предполагаемых
по этому механизму. [105, 106, 117-119], т.е
СН3
(СН3)3С —
—СН2 — С = СН2
СН3
—СН2=С
СН3
VII. Цепная полимеризация по катионному механизму -* V- 75
Более того, применение изотопного комплекса BF3-D2O показало [118],
что полимер содержал дейтерий, хотя инициатор превратился в BF3-H2O.
Этот механизм действительно включает инициирование присоединени-
ем протона (Ур. 43) из кислот Бренстеда, образуемых кислотами Льюиса
и соинициатором, к мономеру и последующему ограничению роста цепи
передачей протона к аниону инициатора (ур. (45)) или передачи цепи
к мономеру (ур. (46)). Поэтому рост цепи происходит в течение краткого
времени жизни карбениевого иона, а инициатор или новый карбениевый
ион постоянно регенерируется. Можно ожидать, что стадия передачи цепи
не оказывает влияния на скорость, т. к. триметилкарбениевый ион должен
быстро реинициировать рост цепи, но длина цепи будет уменьшаться. Необ-
ходимо снова подчеркнуть, что в виду низкой диэлектрической постоянной
применяемого растворителя мала вероятность того, что активными центрами
роста цепи в этих системах являются свободные ионы, поэтому противоионы
изображены как связанные с карбениевым ионом на каждой стадии реакции.
Это упрощенный механизм не отражает действительной природы растущих
катионных центров [112].
Предыдущий упрощенный механизм поддается кинетическому анализу,
с применением постадийного метода, как в случае свободнорадикального
механизма. Используя обозначения НА, для кислотного инициатора, мы
запишем
Инициирование НА + М ► нм + А (47)
Рост цепи НМ4 ’ А” + М *р + _ ——► Н[М]ХМ+ А (48)
Обрыв цепи Н[М]ХМ+А“ kt ► Мх+1 + НА (49)
Передана Н[М]ХМ+Д" +М &tr ► MX4-1 4- НМ +А“ (50)
Скорость инициирования R = ^[НА][М] (51)
Скорость обрыва цепи
Я^МНМ^А-]
(52)
76
Глава 2. Полимеризация: синтез эластомеров
Скорость роста цепи
Яр = Лр[НМхМ+Л-][М] (53)
Скорость передачи к мономеру
7?tr = /ctr[HMxM+A-][M] (54)
Здесь ^считается реакцией первого порядка, т. к. противоион А” рассмат-
ривается специфически связанным с карбениевым ионом, как показано
в ур. (45), а не является отдельным центром. Применяя стационарное рас-
смотрение, мы приравниваем значения R- и 7?t и таким образом получаем
ур. (55) для стационарной концентрации растущих цепей.
[НМхМ+А“] = [НА] [М] (55)
Следовательно,
Rp = -d[M]dt = (kp kt/kt) [HA] [M]2 (56)
Здесь снова скорость роста цепи фактически является скоростью поли-
меризации, т. к. расход мономера на стадии инициирования незначителен.
В отличие от свободнорадикального случая здесь скорость имеет первый
порядок по концентрации инициатора, очевидно, из-за первого порядка
стадии обрыва цепи.
Экспериментальное подтверждение предыдущей кинетической схемы
получено для катионной полимеризации стирола [120] и винилалкильных
эфиров [121], где обнаружено, что скорость полимеризации действительно
зависит в первой степени от концентрации инициатора и квадрата концент-
рации мономера. Однако, необходимо отметить, что эта простая кинетическая
схема не является общей для всех видов катионной полимеризации. Даже
стационарное рассмотрение не применимо для многих процессов катионной
полимеризации [108].
Молекулярная масса полимера может снова быть выражена исходя из
Хп, среднечисленного значения числа мономерных звеньев в цепи, которое
может быть определено здесь как отношение скорости роста цепи к сумме
скоростей всех процессов, приводящих к обрыву цепи, включая передачу
цепи. Следовательно, из ур. (52-54)
х /сДНМхМ+А~][М]
п Rt + Rtr kt [НМ,М+А" ] + ktT [НМ,М+А" ] [М]
или
yxn = klr/kp+kt/kp[M]
(57)
VII. Цепная полимеризация по катионному механизму -* V 77
С помощью ур. (57) можно определить относительный вклад стадий обрыва
и передачи цепи. Таким образом, если klT в большей мере связана с kv моле-
кулярная масса фактически не будет зависеть от концентрации мономера,
но если возможно обратное положение, то хп будет прямо пропорциональ-
но концентрации мономера [Л/]. Следовательно, это отношение поддается
простому экспериментальному определению, т. е. графической зависимости
1/А^0 (обратное начальное значение Хп) от 1/[М]° (обратное значение на-
чальной концентрации мономера). Было обнаружено, что полимеризация
стирола под действием SnCl4B дихлорэтане [120] и винилалкиловых эфи-
рах под действием SnCl4 в м-крезоле, обнаруживает преобладание обрыва
цепи над передачей, т. е. Хп а [М]. Однако, для полимеризации изобутилена,
катализируемой Т1С14в н-гексане [122], наблюдаемая молекулярная масса
полимера не зависит от концентрации мономера, т. е., по-видимому, преоб-
ладает передача цепи.
Интересно сравнить природу индивидуальных стадий в катионной поли-
меризации с индивидуальными стадиями свободнорадикального механизма.
Можно ожидать, что стадия обрыва цепи в катионной полимеризации тре-
бует большей энергии, чем стадия роста, т. к. включает разрыв a-связи, по
сравнению с низко энергетической атакой растущим карбениевым ионом
я-связи мономера. Если действительно стадия обрыва имеет более высокую
энергию активации по сравнению с ростом, тогда повышение температуры
должно приводить к увеличению отношения реакции обрыва к росту, таким
образом, приводя к низкой концентрации растущих цепей. Полученный
численный результат должен, таким образом, уменьшать скорость полиме-
ризации и молекулярную массу, т. е., кажущуюся «отрицательную» общую
энергию активации для полимеризации. Это, конечно, может реализоваться
частично или полностью, если энергия активации стадии инициирования
является достаточно высокой. Однако, в большинстве случаев это не так,
и тогда высокие скорости (и высокие молекулярные массы) в действитель-
ности получаются при пониженной температуре (около -100°С) [115]. Ки-
нетическое изучение показывает, что константа скорости роста ионных пар
(5-6)*108л*моль_1сек_1 для полимеризации изобутилена с EtAlCl2B смеси
гексан/метил хлорида (60/40, об/об) при-80°С [123].
В. Бутилкаучук
Единственным важным промышленным эластомером, по-
лучаемым методом катионной полимеризаций, является бутилкаучук, т. е.
сополимер изобутилена и изопрена. Последний мономер вводится в отно-
сительно малых количествах (~1,5 мольных % [76]), для того чтобы обеспе-
чить ненасыщенность для серной вулканизации. Суспензионный процесс
с алюминий хлоридом от -98 до -90°С в метилхлориде описан следующим
образом [115, 116, 124]. В этом процессе полимеризация протекает почти
78 “*V Глава 2. Полимеризация: синтез эластомеров
мгновенно, и для контроля реакции необходимо всестороннее охлаждение
жидким этиленом.
95.5-98.5% Изобутен
4.5-1.5% Изопрен
0.2% А1С13 Суспензия
Юоб.% раствора в СН3С1В ----> крошки
in СН3С1 сополимера
-100°С
испарительный
чан
(горячая вода)
Фильтрация
и сушка
полимера
Рекуперация
мономера
и растворителя
Молекулярные массы различных сортов бутилкаучука находятся в ин-
тервале 300,000-500,000 и они очень чувствительны [124] к температуре
полимеризации выше-100°С. Например, повышение температуры полиме-
ризации до 25°С приводит к 5- или 10-кратному уменьшению молекулярной
массы, преимущественно из-за кинетических факторов, обсужденных ранее.
Распределение по молекулярной массе бутилкаучука может быть широким,
напримерMw/Mn = 3-5 [115], преимущественно из-за гетерогенной природы
процесса полимеризации.
Изопрен применяется как сомономер при получении бутилкаучука (0,5-
2,5 мольных %) [115], потому что отношение реакционных способностей
изобутилена к изопрену более благоприятно для включения диенов, нежели
изобутен-бутадиеновой пары [124]. Так, для первой пары г (изобутилена) =
=2,5 иг (изопрена) = 0,4 [124]. Следует заметить, однако, что, как обсужда-
лось ранее, такие значения г могут заметно зависеть от природы инициатора
и растворителя, используемых при полимеризации. Полученные значения
применимы для промышленных процессов получения бутилкаучука, как
описано ранее. Было показано, что изопреновые единицы вступают в цепь
преимущественно в 1,4 конфигурации [125].
С. Процессы живой катионной полимеризации
Контролируемые/живые процессы полимеризации алкил-
винильных эфиров были описаны в 1984 г., используя инициирующую
систему Н1/12 в неполярных растворителях [126]. Полимеры, полученные
этим методом полимеризации, имели контролируемые молекулярные массы,
узкое молекулярно-массовое распределение и среднечисленное значение
молекулярной массы, линейно увеличивавшееся с конверсией. Вскоре после
этого Фост и Кеннеди [127] описали живую катионную полимеризацию
изобутилена инициированием смесью трихлорида бора с кумил ацетатом
в смеси хлорированных растворителей с н-гексаном для осуществления
гомогенной полимеризации. Последующие исследования обнаружили
многообразие живущих катионных полимеризационных систем [123, 128,
VII. Цепная полимеризация по катионному механизму -'v- 79
129]. Для контролируемой/живой полимеризации изобутилена вообще
применяют третичные галоиды в комбинации с сильными кислотами Лью-
иса как соинициаторами (BF3, SnCl4, TiCl4, EtAlCl2H Ме2А1С1) [123, 128,
129]. Ключевым ингредиентом в большинстве этих систем является ловушка
протонов, например, такая как ди-трет-бутилпиридин, необходимый для
подавления инициирования протонами. Общие черты живых полимери-
зационных систем аналогичны системам с контролируемой радикальной
полимеризацией, т. е. с преобладанием нереакционноспособных неактивных
центров (ковалентных связей, R-Х или Р-Х) в равновесии с малой концент-
рацией реакционно-способных растущих центров (катионные ионные пары
Р+Х“), как показано ниже.
г © ©
R-X + А(кислота Льюиса) _____ R , ХА
© © © ©
R , XAU + мономер —2—► Р , ХА
© А
Р ,ХА° ► Р-Х+А
активная форма скрытая форма
D. Другие виды катионной полимеризации:
гетероциклические мономеры
Хотя бутилкаучук до сих пор является наиболее важным
промышленным эластомером, синтезируемым методом катионной поли-
меризации, некоторые гетероциклические мономеры позволяют получить
эластомерные материалы с помощью этого же метода. Эпихлоргидрин мо-
жет полимеризоваться до высокой молекулярной массы с использованием
комплексного катализатора, образованного из триалкилалюминиевого
соединения и воды, как показано в ур. (58) [64,130-132]. Для сополимери-
зации с оксидом этилена катализатор получается из триалкилалюминиевого
соединения, воды и ацетилацетона [64, 130].
R3A1/H2O
СН2—СНСН2С1 -------------► -Е-ОСН2—chJ—
'o' L | J n
CH2C1
(58)
Предполагаемый механизм этой полимеризации иллюстрируется ур.
(59) где координация двух алюминиевых центров позволяет объяснить
стереохимический механизм этой полимеризации. Средняя молекулярная
масса гомополимера — 500000, тогда как эквимолярный сополимер оксида
этилена имеет молекулярную массу приблизительно 1,000,000 [64].
80 -*v- Глава 2. Полимеризация: синтез эластомеров
(59)
Катионная полимеризация тетрагидрофурана применяется в промыш-
ленности для получения а,ш-дигидроксиполи (тетраметилен оксида) (РТМО
гликоль). Хотя этот полимер не используется сам по себе как эластомер,
его применяют в качестве одного из эластомерных блок-компонентов для
получения сегментированных термопластичных полиуретановых [133]
и термопластичных полиэфирных [134] эластомеров. Катионная полиме-
ризация тетрагидрофурана (ТГФ) является живой полимеризацией при
соответсвующих экспериментальных условиях [135-139], т. е. она не ингиби-
руется никакой стадией обрыва, что очень сильно напоминает аналогичные
процессы анионной полимеризации, которые обсуждаются в разделе VIII.
Однако, эти полимеризационные процессы усложняются тем фактом, что
предельная температура, при которой свободная энергия полимеризации
равна 0, приблизительно составляет 83±2°С в объеме раствора мономера
[140]; поэтому полимеризация является обратимой и часто наблюдается
неполная конверсия особенно в присутствии добавленного растворителя.
Например, в равновесных условиях в блоке при 20°С конверсия достигает
72%; в смеси 37,5 об% метиленхлорида конверсия достигает только 27%.
Эти факторы ограничивают возможность получения политетрагидрофурана
с контролируемой молекулярной массой и узким молекулярно-массовым
распределением, которое часто связывают с процессом живой полимериза-
ции. По мере приближения к равновесию распределение полимера по моле-
кулярным массам становится шире вплоть до статистического значения 2.
Для полимеризации тетрагидрофурана в промышленности в качестве
катализатора обычно применяется фторсульфоновая кислота, как показано
вур. (60) [139].
О FSO3H Н2О
(л+1) | ► ____». НО Ч-СН2СН2СН2СН2О4^СН2СН2СН2СН2ОН
(60)
VII. Цепная полимеризация по катионному механизму 81
Механизм этой катионной полимеризации достаточно отличается от
полимеризации изобутилена (Ур. (43-46)) тем, что растущим концом цепи
является промежуточный оксониевый ион, в котором положительный заряд
локализуется на атоме кислорода в большей степени, чем на атоме углерода,
как показано далее [136, 139, 141]:
Инициирование реакции
(61)
Рост цепи
НО(СН
SO3FQ
НО(СН2^-ЕОСН2СН2СН2СН2Ч— °вз
SO3FB
(62)
Интересным аспектом этой полимеризации является наблюдение, что
ковалентный сложный эфир находится в равновесии с оксониевым ионом
(ур. (63)) и что оба этих центра могут участвовать в реакции роста цепи
с мономером [142].
НО(СН2^Ч OCH2CH2CH2CH24-Os
SO3FB
HO(CH2^-f OCH2CH2CH2CH2-WCH2)4SO3F
(63)
Полимеры а,ш-дигидроксиполитетраметиленоксида (РТМО гликоль)
получаемые в промышленности имеют молекулярную массу в интервале
600-3000 с молекулярно-массовым распределением в интервале 1,2-1,6, что
согласуется с равновесной природой этой полимеризации [139].
82
Глава 2. Полимеризация: синтез эластомеров
VIII. ЦЕПНАЯ ПОЛИМЕРИЗАЦИЯ ПО АНИОННОМУ
МЕХАНИЗМУ
А. Механизм и кинетика
Анионный механизм предполагается для таких процессов по-
лимеризации, которые инициируются металлорганическими соединениями
щелочных металлов, причем металл является сильно электроположительным
по отношению к углероду или другому атому на конце растущей цепи [21,
143-151]. Однако, аналогично обсуждению активных центров катионной
полимеризации, разнообразные активные частицы могут включаться в ка-
честве центров роста цепи в анионной полимеризации, как показано ниже
[150]. Однако в противоположность катионной полимеризации имеются
экспериментальные доказательства вовлечения многих этих частиц в неко-
торых условиях полимеризации [145, 147, 148].
(R-M)n R-М RB мн
агрегаты ковалентная контактная
ионная пара
м 1 г М 1 г М 1 Г
RB II м= , ==^ RB + м0
разделенная свободные
ионная пара ИО1 ны
М 1 *р г М 1 Г
Хотя способность щелочных металлов, таких как натрий, инициировать
полимеризацию ненасыщенных молекул известна давно (ранние данные
относятся к работам Мэтьюса и Стрэнжа [8] и Гарриса [7] около 1910 г.
для полимеризации диенов), механизм во многом оставался неясным из-за
гетерогенного характера этого типа катализа. Новаторские работы Хиг-
гинсона и Вудинга [152] по гомогенной полимеризации стирола амидом
калия в жидком аммиаке, а также работы Робертсона и Мариона [153] по
полимеризации бутадиена с натрием в толуоле показывают важную роль
растворителя в участии реакции передачи цепи.
Истинная природа гомогенной анионной полимеризации была выясне-
на только путем последовательного изучения растворимых ароматических
комплексов щелочных металлов, таких как нафтил-натрий. Известно, что
эти активные центры являются анион-радикалами [154-158] с одним не
спаренным резонансно стабилизованным электроном и высокой энергией
сольватации, поэтому они химически эквивалентны растворимому на-
трию. Они инициируют полимеризацию по механизму передачи электрона
[145, 148] так же как в случае металлов, за исключением того, что реакция
является гомогенной, и поэтому включает более высокую концентрацию
инициатора. Механизм полимеризации инициирования щелочными
металлами (или их растворимыми комплексами) может быть описан на
примере стирола [145, 148].
VIII. Цепная полимеризация по анионному механизму -'V 83
Инициирование
Na + СН2=СН [ СНг _ сн ] -NaB (64)
С6Н5 С6Н,
2[ CH2=Ch]-№s ---------NaE QCH—СН, — СН, -СН^ Na0
I I I
сбн5 с6н5 сбн5
(65)
Рост цепи
NaS БСН—СН2—СН2 —СНБ Na3 + (2и) СН2 = СН—► ” —►
СбН5 С6Н5 С6Н5
NaS БСН—СН2—l-CH—СН2-НСН2 — СН -]—СН2 —СНБ
I 2 | 2 п | |
С6Н5 С6Н5 С6Н5 С6Н5
□
Na
(66)
Таким образом, первая стадия в реакции инициирования (ур. (64))
включает обратимую реакцию передачи электрона от щелочного металла
к мономеру стирола с образованием стирольного радикал-аниона; в быст-
рой последующей стадии два радикал-аниона соединяются с образованием
дианиона, который может начинать рост полимерной цепи с двух концов.
В случае растворимых металлоароматических комплексов общая реакция
инициирования протекает крайне быстро из-за высоких концентраций
радикал-анионов (~10-3М) и мономера (~1М), как и последующая реак-
ция роста цепи. Однако, в случае инициирования щелочными металлами
стадия передачи электрона намного медленнее из-за гетерогенной природы
реакции, так что образование радикал-анионов много медленнее. В дей-
ствительности, имеются доказательства, что в таких случаях вторая стадия
передачи электрона может протекать между металлом и радикал-анионом
с образованием дианиона, в большей степени, чем соединение радикал-
анионов [153]. В любом случае конечным продуктом является дианион, т. е.
бифункциональная растущая цепь.
Однако, именно исследования гомогенных систем, инициированных на-
фтил- натрием в полярных растворителях, показали особую природу анион-
ной полимеризации, т. е. что стадия обрыва цепи может быть при некоторых
84 —Глава 2. Полимеризация: синтез эластомеров
обстоятельствах подавлена. Это привело к концепции «живых» полимеров
[159]. Поскольку они являются гомогенными системами, стехиометрия ре-
акции становится очевидной, т. е. две молекулы нафтил-натрия генерируют
одну цепь. Более того, т. к. все цепи инициируются быстро, и все цепи имеют
равные возможности для роста, распределение их по молекулярной массе
становится очень узким, приближаясь к распределению Пуассона [160]. Эти
аспекты неясны в процессах полимеризации, инициированных металлами,
т. к. здесь отмечено медленное инициирование в течение продолжительного
периода времени, что приводит к большому различию в размерах растущих
цепей и, следовательно, в распределении по размерам.
Полимеризация, инициированная передачей электрона от металла или
от ароматического радикального аниона, представляет только один анион-
ный механизм. Возможно рассмотреть отдельно эти типы полимеризации,
направленно инициированные металлорганическими соединениями. Среди
последних литийорганические соединения, возможно, являются лучшим
примером, т. к. они растворимы в разнообразных растворителях и относи-
тельно стабильны. Более того, именно эти металлорганические, соединения
применяются в промышленности для получения синтетических эластоме-
ров [161, 162]. Механизм этих процессов в некоторой степени проще, чем
в случае нафтил-натрия, т. к. отсутствует стадия передачи электрона. Поли-
меризация протекает по схеме:
Инициирование
RBLiB + СН,— СН -------► R— СН, — CHBLiS
| | (67)
СбИ С6Н5
Рост цепи
к-сн2^нВи= + — к-[-сн2-СН^СН-CHBLi=
С6Н5 С6Н5 С6Н5 С6н5
(68)
Обрыв цепи примесями или целевой обрыв
R-f—сн2—СН-J—сн2—СНВ Li® + Н2О--►R-f-CH2— СН -^СН— СН2 + LiOH
с6н5 С6Н5 С6Н5 С6Н5
(69)
VIII. Цепная полимеризация по анионному механизму -*и- 85
Следовательно, каждая литийорганическая молекула генерирует одну
цепь, и не протекают реакции обрыва растущих цепей или передачи цепи
в отсутствии случайных примесей, таких как вода или кислоты. Побочные
реакции можно предотвратить, не повышая температуру.
В отличие от нафтил-натрия, требующего присутствия высоко сольвати-
рующих растворителей, таких как ТГФ, литийорганические системы могут
применяться с различными полярными и неполярными растворителями,
такими как простые эфиры или углеводороды. Однако, скорость в последнем
случае ниже, чем с первыми растворителями. Следовательно, если реакция
инициирования (ур. (67)) протекает много медленнее, чем реакция роста
цепи, молекулярно-массовое распределение может быть значительно рас-
ширено [163]. Это не снижает важность живой полимеризации, которая,
как было показано [164], применима в этих системах независимо от типа
растворителя и в отсутствии побочных реакций.
Отсутствие реакций обрыва и передачи цепи в гомогенной анионной
полимеризации может привести к новым путям синтеза. Так, при продол-
жении роста цепи возможно синтезировать блокполимеры последователь-
ным добавлением нескольких мономеров [165, 166]. Другой возможно-
стью является синтез линейных цепей с различными функциональными
концевыми группами за счет реакции конца анионной полимерной цепи
с различными электрофильными агентами, например, с СО2 с образованием
карбоксильной группы [167]. Кроме того, реакции сшивания полимерных
цепей многофункциональными электрофильными реагентами приводит
к образованию звездообразных полимеров [168-171]. Конечно, эти возмож-
ности представляют значительный промышленный интерес.
Ввиду необычного механизма анионной полимеризации, особенно от-
сутствия реакции обрыва и передачи цепи, кинетика может быть описана
различно по сравнению с другими механизмами. Можно при подходящей
экспериментальной технике отдельно изучить скорость реакции иници-
ирования и роста цепи [172, 173], т. к. устойчивые металлорганические
концы цепей присутствуют в концентрациях [10-3-10-5М], которая легко
измеряется методом УФ и видимой спектроскопии [174]. Конечно, реакция
роста представляет значительно больший интерес при условии, что иниции-
рование является полным. Кинетика гомогенной анионной полимеризации
была широко освещена в работах по оценке природы противоиона и роли
растворителя.
Обнаружено, что, в соответствии с ур. (66) и (68), реакция роста цепи имеет
всегда первый порядок по отношению к концентрации мономера независимо
от системы растворителей или противоинов. Однако, в противоположность
предыдущим уравнениям было обнаружено, что зависимость константы ско-
рости роста ниже первого порядка по отношению к концентрации растущих
86 Глава 2. Полимеризация: синтез эластомеров
цепей, и порядок сильно зависит от природы растворителя и противоина
[148, 172-175]. Сильно сольватирующие растворители, такие как простые
эфиры и амины, обеспечивают намного большие скорости, чем неполярные
растворители, и влияют на кинетику этих полимеризационных процессов
совершенно иначе, чем углеводородная среда, т. к. более диссоциированные
ионные частицы, такие как разделенные ионные пары и свободные ионы,
выступают в качестве частиц инициирующих рост цепи [148]. Применение
лития как противоиона в углеводородной среде необходимо при анионном
синтезе эластомеров, следовательно, последующее обсуждение будет сосре-
доточено на кинетике этих процессов.
Можно было бы ожидать, что кинетика полимеризации, инициирован-
ной литийорганическими соединениями в углеводородных растворителях,
будет упрощена благодаря ожидаемому соответствию между концентрацией
инициатора и концентрацией анионных частиц, вызывающих рост цепи из-за
отсутствия реакций обрыва и передачи цепи. Однако, несмотря на интенсивное
изучение, нет общего согласования многих кинетических аспектов этих про-
цессов. Усложнением является тот факт, что литийорганические соединения
связаны в агрегаты в углеводородных растворах, и степень агрегации зависит
от структуры этих соединений, их концентрации, растворителя и температуры
[147,176-178]. В общем, простые алкиллитиевые соединения ассоциирова-
ны в гексамеры или тетрамеры в углеводородных растворах.
Кинетика инициирования полимеризации стирола и диенов литийорга-
ническими соединениями вообще имеет дробный порядок по концентрации
алкиллитиевого инициатора (например, 1 /4 или 1/6). Это может быть понято
следующим образом:
Инициирование
V
(RLi)„ л RLi (70)
ki
RLi + СН,=СН --------► R—СН,—CHLi
| | (71)
с6н5 с6н5
=/c.[RLi] [7И] (72)
[RLi] =/f1/"[(RLi)n]1/" (73)
= fcz/C1/'I[(RLi)„]1/« [J/]
(74)
VIII. Цепная полимеризация по анионному механизму -* V 87
Таким образом, предполагается, что неассоциированные алкиллитиевые
соединения (образованные диссоциацией агрегатов, ур. (70)) реагируют с
мономером на стадии инициирования так, что скорость инициирования
может быть выражена ур. (72)
Равновесная концентрация неассоциированного алкиллития может быть
выражена ур. (73). Когда это выражение для [RLi] подставляется в ур. (72),
получается ур. (74).
Хорошим примером кинетического поведения является система бутил-
литий-стирол в бензоле, в которой наблюдался кинетический порядок,
равный 1/6 по концентрации н-бутиллития, согласующийся с преимущес-
твенно гексамерной степенью ассоциации н-бутиллития [179]. Однако,
это ожидаемое соответствие между степенью ассоциации алкиллитиевого
соединения и дробным кинетическим порядком реакции инициирова-
ния по концентрации алкиллития наблюдалось не всегда [147]. Одним
из предположений этого различия является то, что, возможно, только
неассоциированная алкиллитиевая молекула может инициировать по-
лимеризацию. С некоторыми реакционноспособными инициаторами,
такими как вторичный бутиллитий в растворе гексана, начальная ско-
рость инициирования имеет первый порядок по отношению к концен-
трации алкиллития в предположении, что агрегаты могут реагировать
непосредственно с мономером для инициирования полимеризации [180].
Дальнейшим усложнением является кросс-ассоциация инициатора
с инициируемой полимерной цепью; в общем кросс-ассоциированные
частицы обнаруживают степень ассоциации и реакционную способность,
отличающуюся от алкиллитиевого инициатора [181, 182]. Как результат
кросс-ассоциации только начальные скорости инициирования могут быть
применены для определения кинетического порядка от концентрации
инициатора. К сожалению, это рассмотрение не всегда подтверждается.
Интересно заметить, что существует обратная зависимость между общей
реакционной способностью алкиллития как инициатора и степенью
агрегации [183], т. е. вторичный бутиллитий (тетрамер)>н-бутиллитий
(гексамер) [180].
Кинетика реакции роста цепи при полимеризации стирола и диенов под
действием литийорганических соединений в неполярных растворителях,
например углеводородах, также является предметом интенсивного изуче-
ния. Для полимеризации стирола наблюдался кинетический порядок 1/2
по концентрации концов цепей (ур. 75).
^ = W/2l(PsLi)2]1/2[Jw] (75)
(PsLi)2 2 PsLi
88 -*Ц" Глава 2. Полимеризация: синтез эластомеров
PsLi + СН2^=СН —£-► Ps—СН2—CHLi
С6Н5 кн5
Поскольку установлено, что литий-полистирол ассоциирован преимуще-
ственно в димеры в растворе углеводородов [147], наблюдаемый кинетический
порядок может быть объяснен (ур. 76,77) аналогично кинетике инициирования
(ур. 70-74). Это объяснение основано на предположении, что только неассоци-
ированные (диссоциирвоанные) концы цепей являются активными.
Кинетический порядок реакции роста относительно концентрации кон-
цов поли (диенил) литиевой цепи для полимеризации диенов, иницииро-
ванных алкиллитием, изменяется от 1/4 до 1 /6 для бутадиена и от 1/2 до 1/4
для изопрена [147, 172, 173. 184]. Однако, попытки связать эти значения
кинетического порядка с предполагаемой высокой степенью ассоциации
поли (диенил) литиевых концов цепи усложнены из-за противоречивых
физических данных, полученных на одинаковых приборах различными
исследователями, показавшими, что такие концы цепей ассоциированы
в димеры и тетрамеры в углеводородной среде [150]. Эти физические измере-
ния включают вязкость растворов, криоскопию и измерение светорассеяния
[185]. Следовательно, эти наблюдения ставят под вопрос всю общую теорию
нереакционной способности ассоциированных комплексов и предполагают, что
возможно контролировать прямое взаимодействие между мономером и ассоции-
рованным концом полимерной цепи [186-192,144 с. 128]. Другим усложнением
этого положения являются результаты Феттерса и сотрудников, показавшими,
что агрегаты высокого порядка находятся в равновесии с димерами [193].
В заключение необходимо отметить, что молекулярные массы и их рас-
пределение следуют правилу, первоначально обсужденному для живых
полимеров [194]. Это означает, что независимо от применяемых растворите-
лей и противоинов, если отсутствуют реакции передачи и обрыва цепи или
побочные реакции, и если реакция инициирования является более быстрой
по отношению к реакции роста цепи, то молекулярно-массовое распределе-
ние будет подчиняться распределению Пуассона, т. е.
+ (78)
гдеХп — среднечисленная величина мономерных единиц, &Хш — средневе-
совое число мономерных едениц. Это означает, что, в принципе, полимерная
цепь из 100 единиц должна иметь отношение Xw/Xn — 1,01. Это, конечно,
невозможно проверить экспериментально и можно предполажить, что ре-
альное распределение должно быть в некоторой степени шире из-за несо-
вершенного смешения реакционной смеси. Однако, обычно в этих системах
определяется значение 1,05 для Xw/Xn [185, 195].
VIII. Цепная полимеризация по анионному механизму -*и- 89
В. Микроструктура цепей полидиенов
Щелочные металлы, в отличие от систем Циглера-Натта,
обычно не полимеризуют несопряженные олефины, и неизвестно, при-
водят ли они к какой-либо тактичности, но они действительно влияют
на микроструктуру цепи полидиенов. Так, отношение цис-1,4 и транс-
1,4 присоединения к 1,2 и 3,4-присоединению (для полиизопрена) может
быть заметно изменено природой противоиона, так же как и раствори-
теля. После открытия возможности литиевой полимеризации изопрена,
дающей высокое содержанию цис-1,4 структуры [196] (характерной для
натурального каучука) было проведено много исследований этого эф-
фекта [147, 197-200]. Таблица 14 показывает некоторые эти результаты
для анионной полимеризации изопрена и бутадиена. Из этих данных
следует, что стереоспецифический полиизопрен с высоким содержанием
цис-1,4 структуры получается только в случае лития в углеводородных
растворителях, наиболее высокому содержанию цис-микроструктуры
также благоприятствует высокое отношение мономера к концам растущих
цепей [199-201]. Другие растворители и/или противоионы приводят к
резкому изменению микроструктуры цепи с образованием участков цепи
структур 1,2 и 3,4. Подобный эффект наблюдался с бутадиеном и другими
диенами [147, 150, 197]. Однако, в случае бутадиена достигалось значи-
тельно меньшее максимальное содержание цис-1,4 структуры, чем для
полиизопрена; типичные промышленные полибутадиены, полученные
в углеводородном растворе с бутиллитиевыми инициаторами, имеют
микроструктуру в интервале содержания цис-1,4 структуры 36-44%,
транс-1,4 48-50% и 1,2 микроструктуры — 8-10% [198]. Влияние по-
лярных растворителей или более электроположительных щелочных
металлов приводит к получению полибутадиена с высоким содержанием
1,2 микроструктуры.
Заметная чувствительность стереохимии анионной полимеризации к
природе противоиона и растворителя может быть связана со структурой
концов растущих цепей. Последняя включает связь углерод-металл, которая
может иметь различные характеристики, изменяясь от ассоциированных
частиц с ковалентным характером до ионных частиц [150]. Присутствие
более электроположительного металла и/или растворителя сольватирующего
катион, такого как простые эфиры, может влиять на изменения в природе
карбанионного конца цепи: (а) степень ассоциации концов цепей может
уменьшаться или исключаться; (б) взаимодействие катиона с анионом может
уменьшаться сольватацией катиона; (в) более ионная связь углерод-металл
будет увеличивать делокализацию я-электронов; (г) полярные растворители
будут промотировать ионизацию, образуя ионные пары и свободные ионы.
Прямое доказательство этих эффектов было получено из измерений в кон-
90 “'V Глава 2. Полимеризация: синтез эластомеров
центрированных растворах [205, 206], и 13С ЯМР спектроскопии [147,
207], УФ и видимой спектроскопии [147,184, 208] и измерений электроли-
тической проводимости [145].
Таблица 14 Микроструктура полимеров, полученных методом анионной
полимеризации.
Растворитель Микроструктура цепи (моль%)
Катион Цис-1,4 Транс-1,4 1,2 3,4 Работа
Бутадиен
Гексана) Li+ 30 62 8 - [200]
Циклогексан Ь) Li+ 68 28 4 - [200]
Нет Li+ 86 9 5 - [200]
Тетрагидрофуран (ТГФ) Li+ 6 6 88 - [202]
Пентан Na+ 10 25 65 - [202]
ТГФ Na + 0 9.2 90.8 - [203]
Пентан K+ 15 40 45 - [202]
Пентан Rb+ 7 31 62 - [202]
Пентан Cs+ 6 35 59 - [202]
Изопрен
Циклогексан Ь) Li + 94 1 - 5 [201]
Циклогексан а) Li + 76 19 - 5 [201]
Нет Li + 96 0 - 4 [200]
ТГФ Li + 12е 29 59 [204]
Циклогексан Na + 44е 6 50 [199]
ТГФ Na + 11е 19 70 [199]
Циклогексан K+ 59f 5 36 [199]
Циклогексан Cs+ 69е 4 27 [199]
а) Отношение мономер/инициатор ~17.
Ь) Отношение мономер/инициатор 5х104.
с) Общее содержание цис- и транс-форм.
Контроль структуры цепи и молекулярной массы, осуществляемый при ли-
тийорганической полимеризации бутадиенов, конечно, представляет большой
технологический интерес [161,162,209]. Так получены (1) полибутадиеновые
VIII. Цепная полимеризация по анионному механизму -’V- 91
эластомеры с различной структурой цепи [162,198,209] и функциональными
концевыми группами [210]; (2) жидкие полибутадиены [211], (3) бутадиен-
стирольные сополимеры (растворные ДССК) [69, 161, 162, 209], (4) стирол-
диеновые триблоксополимероы (термоэластопласты) [212].
С. Сополимеры бутадиена
Возможности, появляющиеся в анионной полимеризации
бутадиена и стирола с помощью литийорганических инициаторов, при-
вели ко многим новым разработкам. Первой является синтез бутадиен-
стирольных сополимеров, необходимый для создания альтернативы или
улучшения БСК, полученного эмульсионной полимеризацией, благодаря
контролю молекулярной массы в анионной полимеризации. Поведение
бутадиена (или изопрена) при сополимеризации со стиролом представ-
лено в Табл. 15. Как показано ранее, в отличие от свободнорадикальной
полимеризации, эти анионные системы заметно чувствительны по от-
носительной реакционной способности к типу растворителя (аналогич-
ный эффект наблюдался для противоионов щелочных металлов). Так,
в неполярных растворителях бутадиен (или изопрен) преимущественно
полимеризуются первоначально до полного исчезновения стирола, тогда
как обратный эффект наблюдается в полярных растворителях. Это припи-
сывается большому влиянию сольватации на структуру литий-углеродных
связей, которые становятся значительно более ионными в таких средах,
как это обсуждалось ранее [144]. Образующийся полимер, получаемый
сополимеризацией в углеводородной среде, описанный как переходный
блоксополимер, состоит из блока полибутадиена с небольшим количес-
твом введенного стирольного сомономера, за которым следует сегмент,
содержащий бутадиен и стирол, а затем блок полистирола. Структура
схематически представлена ниже:
[бутадиен]-[Б/С]-[стирол]
Данные Табл. 15 иллюстрируют проблемы, встречающиеся при такой
сополимеризации. Применение полярных растворителей приводит к по-
лучению статистического стирол-диенового полимера желаемого состава,
но в то же время и к увеличению содержания боковых винильных групп
(1,2 или 3,4) в диеновых единицах (см.таб. XIV). Это не вполне желательно,
т. к. такие цепные структуры приводят к увеличению температуры стек-
лования (Г.), и поэтому к потере каучукоподобных свойств. Для решения
этой проблемы применяются два метода: (1) применение ограниченного
количества полярных добавок, таких как тетрагидрофуран, чтобы установить
разумный компромисс между структурой диена и распределением последо-
вательностей мономера [218], и (2) добавление малых количеств третичных
алкоксидов калия [219].
92 -* V Глава 2. Полимеризация: синтез эластомеров
Как упомянуто выше, живая природа растущих цепей анионной поли-
меризации делает этот механизм особенно подходящим для синтеза блок-
сополимеров путем последовательного добавления различных мономеров.
Такие сополимеры заметно отличаются по свойствам от простых сополимеров
и будут обсуждены отдельно (в секции X).
Таблица 15. Относительная реакционная способность мономеров при сополиме-
ризации стирола и диенов в присутствии литийорганических
соединений.
Мономер 1 Мономер 2 Растворитель Г1 Г2 Работа
Стирол Бутадиен Толуол 0.004 12.9 [213]
Стирол Бутадиен Бензол 0.04 10.8 [214]
Стирол Бутадиен Триэтиламин 0.5 3.5 [214]
Стирол Бутадиен Тетрагидрофуран 4.0 0.3 [214]
Стирол Изопрен Бензол 0.26 10.6 [215]
Стирол Изопрен Тетрагидрофуран 9.0 0.1 [216]
Бутадиен Изопрен Гексан 1.72 0.36 [217]
D. Полидиены с концевыми функциональными
группами
Другой характеристикой этих гомогенных типов анион-
ной полимеризации, как упомянуто выше, является возможность синтеза
полимерных цепей, имеющих реакционно-способные концевые группы.
Сообщалось, что функционализация концов цепей высокомолекулярного
полибутадиена и растворных эластомеров (сополимеров бутадиена со стиро-
лом) производными кетона Михлера, 4,4-бис (диэтиламино) бензофенона,
позволяет получать протекторные резины с низким сопротивлением каче-
нию и хорошим сопротивлением скольжению на мокрой дороге [210]. Эти
эффекты наблюдались несмотря на низкую концентрацию концов цепей в
этих полимерах (молекулярная масса >100,000) [162].
Получение жидких короткоцепных бифункциональных полимеров мето-
дом анионной полимеризации представляет значительный технологический
интерес, имеет важное значение и привлекает внимание в последние годы
из-за реализации технологии получения простых и сложных полиэфиров,
применяемых в уретановых полимерах. Такие жидкие «телехелевые» полиди-
ены позволяют получать посредством реакции удлинения цепи или сшивания
непосредственно жидкоформуемые полидиеновые сетки [161,162].
В прямом методе получения телехелевых (бифункциональных) поли-
диенов используется дилитиевый инициатор, который растворим в угле-
IX. Стереоспецифическая цепная полимеризация... -'V 93
водородах [220, 221]. Наиболее целесообразным методом приготовления
такого дилитиевого инициатора является реакция 2 молей алкиллитиевого
соединения с дивинильным соединением, которое не гомополимеризуется.
Из-за ассоциации литийорганических соединений в углеводородной среде
[176-178], многие системы не работают, т. к. в результате этой ассоциации
образуются нерастворимые сетчатые структуры [221]. Добавление оснований
Льюиса может решить проблему растворимости дилитиевого инициатора, од-
нако, такие добавки приводят к высокому содержанию микроструктур 1,2 и 3,4
(см. Табл. 14). Единственным исключением является продукт, полученный
путем добавления 2 эквивалентов втор-бутиллития к 1,3-бис (1-фенилэтинил)
бензолу, как показано в ур. (79) [222,223]. Хотя дилитиевый инициатор растворим
в углеводородной среде, получается полимер с бимодальным ММР; мономодаль-
ное распределение может быть получено в присутствии алкоксидов лития или
добавлением оснований Льюиса [224, 225]. Этот инициатор также применялся
для получения телехелевых полимеров с высоким выходом [226].
IX. СТЕРЕОСПЕЦИФИЧЕСКАЯ ЦЕПНАЯ ПОЛИМЕРИЗАЦИЯ
И СОПОЛИМЕРИЗАЦИЯ НА КООРДИНАЦИОННЫХ
КАТАЛИЗАТОРХ
А. Механизм и кинетика
Термин катализаторы Циглера-Натта относится к широкому
кругу инициаторов полимеризации, обычно получаемых из смеси солей пере-
ходных металлов IV-VIII группы и основных металлалкилов II или III групп
[21, 227, 228]. Этот вид полимеризации основан на выдающемся открытии
Циглера и других [229] и заключается в том, что смесь четыреххлористого титана
и алкил алюминия полимеризует этилен при низком давлении и температуре;
94
Глава 2. Полимеризация: синтез эластомеров
и на не менее ярком открытии Натта [230], что катализаторы Циглера могут сте-
реоспецифически полимеризовать моноолефины с образованием тактических
кристаллических полимеров. Эти системы могут включать многие комбинации
каталитических компонентов, не все из которых каталитически активны или
стереоспецифичны. Однако, мы коснемся только полимеризационных про-
цессов, включающих промышленные эластомеры, такие как полиизопрен,
полибутадиен [231-233], и этилен-пропиленовые сополимеры [234-238].
Механизм полимеризации алкенов с применением катализаторов Циг-
лера-Натта описан как координационный [239] или полимеризационный
процесс внедрения [240]. Координационная терминология предполагает,
что растущая полимерная цепь связана с атомом переходного металла, и что
внедрение мономера в связь углерод-металл предшествует, и предпочтитель-
но активируется координацией мономера с центром переходного металла.
Т. к. координация мономера может как быть, так и не быть специфической
особенностью этих процессов полимеризации, терминология внедрения ак-
центирует внимание на том, что эти реакции включают ступенчатое внедрение
мономера в связь между атомом переходного металла и последним углеродным
атомом растущей цепи. Важно заметить, что связывание атома углерода и пе-
реходного металла происходит в основном как ковалентное [240] в противопо-
ложность анионным металлорганическими центрам, таким как органощелочные
металлические центры, которые являются сильно ионными.
Типичные растворимые катализаторы для сополимеризации этилена
и пропилена образуются из смеси солей ванадия с алкилалюминий хло-
ридами, например VCl4c либо A1R2C1, либо A1RC12, где R — алкильная
группа. Возможная гексакоординированная металлическая структура
для образующегося активного катализатора показана ниже:
Важными чертами активного центра в соответствии с общей моделью
Арлмана и Косей [241] являются: (1) алкилированный ванадиевый центр,
т. е. связь R-V; (2) свободные орбитали на ванадии, представленные квадра-
том в структуре, которые могут служить участками связывания внедряемого
мономера; степень окисления ванадия +3 [242-244].
IX. Стереоспецифическая цепная полимеризация и сополимеризация... -* V 95
Образование активного каталитического центра по реакции между пе-
реходным металлом и алюминийорганическим производным схематически
показано в ур. (80). Снижение до низшего валентного состояния может
сопровождать эту реакцию алкилирования, т. к. это обычно считается, что
активные каталитические центры, имеют степень окисления +3 [236].
Образование активного центра
ci R
I l.l I
V--□ + -А1--R--► V---□ + -А1-С1
(80)
Стадии цепной полимеризации алкенов, в которой участвуют катализа-
торы данного типа, показаны в уравнениях (81-85) [236].
Инициирование
R
I
V—□ +
сн,
I
СНСНз
rch2chch3
-► V—□
(81)
Рост цепи
rch2chch3
V—□ +
сн2
снсн3
rch2chch3
_ | СН2
-► v......||
СНСН3
(82)
СН3
RCH,CH
I СН2
V......||
СНСНз
СН3 Я =Н
RCH2CH...сн2
V....СНСНз
(83)
▼
|нз rch2chch3
rch2chch2chch3 -------------- □ 1н
I I I
v---П V--СНСНз
Обрыв цепи
P-V ------►
неактивная полимерная неактивный каталитический (84)
цепь
центр
96 “•V Глава 2. Полимеризация: синтез эластомеров
Спонтанная передача
СН3
сн3
сн3
РСН2СНСН2СНСН3
СН~!СН--СНСН2Р
V—□
V...н
(85)
сн3
V__Н + СН3СН=СН--СНСН2Р
Следует отметить, что стадия координации мономера, показанная на
схеме (82), может не быть определяющей стадией, как обсуждалось ранее.
Важной особенностью этого механизма, которая влияет на стереоспеци-
фичность полимеризации олефинов для этого типа растворимых катали-
заторов, является тот факт, что внедрение мономера в связь переходного
металла с углеродом происходит при вторичной реакции внедрения,
т. е. более замещенный атом углерода двойной связи в мономере связыва-
ется с переходным металлом [245]. Наоборот, первичный механизм вндре-
ния, в результате которого должна образоваться связь переходного металла
с менее замещенным атомом углерода при двойной связи [Ti-CH2-CHR-P]
мономера реализуется в полимеризации, при применении типичных
гетерогенных катализаторов, например, галогенидов титана и алкила-
люминиевых соединений [228].
Одна из моделей, предлагаемых для объяснения стереоспецифичности рас-
творимых катализаторов на основе ванадия, постулирует, что имеет место мини-
мизация стерических эффектов в четырех-центровом переходном состоянии для
включения мономера (см. ур. 83), которая ответственна за стерео-специфичность
полимеризации [244,245]. Таким образом, считается, что транс-конфигурация
минимизирует стерические эффекты в переходном состоянии, и это приводит
к синдиотактической конфигурации полимерной цепи, как показано ниже.
благоприятная
транс-конфигурация
'СН2 Н
более затруднительная
^ас-конфигурация
IX. Стереоспецифическая цепная полимеризация и сополимеризация... -*v- 97
В общем, кинетика полимеризации алкенов с применением катализаторов
типа Циглера-Натта усложняется многообразием активных центров, старением
катализатора и эффектами дезактивации, многообразием процессов передачи
цепи и часто относительно высокой скоростью полимеризации [228, 234].
В. Этилен-пропиленовые каучуки
Сополимеризация пропилена с этиленом усложняется не-
благоприятным соотношением реакционной способности для пропилена
и других мономеров с этиленом, как показано в Табл. 16. В общем, менее
затрудненный этиленовый мономер предпочтителен в процессе сополиме-
ризации Циглера-Натта почти на два порядка величины для некоторых
комбинаций катализаторов. Для получения однородных сополимеров
необходимы непрерывные процессы и неполная конверсия пропиленовых
сомономеров [236]. Другим аспектом промышленного применения этилен-
пропиленовых каучуков является включение третьего диенового сомономера,
который приводит к ненасыщенности конечного полимера, облегчая реакции
сшивания пероксидами и позволяющее осуществить серную вулканизацию;
эти тройные сополимеры называются СКЭПТ (ЭПДК) в противоположность
бинарным сополимерам, которые называются СКЭП (ЭПК). Следующие
несопряженные диеновые мономеры применяются в промышленности,
т. к. они дают структуры, содержащие ненасыщенные связи в боковых под-
весках, а не в основной цепи, таким образом предотвращая окислительный
разрыв цепи:
СН3
сн2=сн—сн2—сн=сн—сн3
дициклопентадиен этилиден-норборнен 1,4-гексадиен
Таблица 16. Относительная реакционная способность мономеров при сополи-
меризации этилена (Мх) и пропилена (М2) на катализаторах Циглера-Натта.
Катализатор Сокатализатор Температура (°C) rl r2
VC14 A1(C2H5)2C1 -10 13.7 0.021
21 3.0 0.073
VOC13 Al(i-C4H9)2C1 30 16.8 0.052
V(acac)3a) Al(i-C4H9)2C1 30 16 0.04
y-TiCl3 A1(C2H5)2C1 60 ~8 0.05
а) Ацетилацетонат ванадия
98 -*\r Глава 2, Полимеризация: синтез эластомеров
В составах более чем 150 видов эластомеров СКЭПТ входит от 40 до
90 мольных % этилена и от Одо 4 мольных % диена [236]. Таким образом,
структура типичного СКЭПТ эластомера с этилиденнорборненом как третьим
мономером может быть представлена следующей структурой.
СН3
—ЬСН2 СН2Ч---[- СН-СН2Ч—
59 40
СНСН3
Состав СКЭПТ эластомера регулируется применением соответству-
ющего соотношения подачи мономера (см. ур. (38)), чтобы получить
желаемый состав в непрерывном процессе полимеризации. В основном
избыток сверх требуемого количества пропилена возвращается в рецикл.
Молекулярная масса этилен-пропилендиенового сополимера контроли-
руется первоначально реакциями передачи цепи с добавками молеку-
лярного водорода (ур.86,87), как обычно в других видах полимеризации
Циглера-Натта [228].
^tr
P-V + Н2 --------► Р-Н + V-H
V-Н +СН2 =СНСН3 > V-CH----СН3
СН3
(86)
(87)
В последние 20 лет произошла революция в области катализаторов
Циглера-Натта и связанных с ними других видов катализаторов для
полимеризации олефинов. Причина её заключается в открытии одно-
точечного гомогенного металлоценового катализа, который проявляет
высокую активность и способность легко включать более стерически
затрудненные сомономеры с этиленом с получением более однородной
полимерной цепи [246, 247]. Металлоценовые катализаторы содержат
один или два циклопентадиенильных кольца, координированных с пере-
ходным металлом, таким как титан, цирконий или гафний. Высокая ак-
тивность металлоценового катализатора означает, что процесс может быть
проведен без необходимости в стадии удаления катализатора. Структура
высокоактивного одноцентрового металлоценового катализатора показана
ниже. Необходимо отметить, что металлоценовый катион выступает как
каталитически активная частица. Для этого типа катализатора, сочетаю-
IX. Стереоспецифическая цепная полимеризация и сополимеризация... 99
щегося с различными противоинами, отношение реакционной способности
для полимеризации этилена с пропиленом составляет г (этилен) = 1,35
и г (пропилен) = 0,82, что соответствует почти статистичекой сополимериза-
ции [248]. Эти результаты можно сравнить с параметрами сополимеризаци и
стандартных катализаторов Циглера-Натта (Табл. 16).
(C6H5)3C® B(C6F5)°
I \i(CH3)2 ------------►
(CH3)2Si—
C(CH3)3
С(СН3)3
С. Полидиены
Вскоре после открытия катализаторов Циглера-Натта, было
обнаружено, что аналоги катализаторов переходных металлов могут также
влиять на стереоспецифическую полимеризацию диенов [249]. Широкий
интервал стереорегулярных полибутадиенов может быть получен с этими
катализаторами, как показано в Табл. 17. Стереохимия полимеризации за-
висит от солей переходных металлов, металлалкилов, температуры, добавок
и стехиометрии компонентов. Промышленные полибутадиены с высоким
содержанием цис-1,4-микроструктуры получаются с использованием ши-
рокого круга катализаторов переходных металлов, среди которых наиболее
важными являются производные кобальта, никеля, неодима и титана, ана-
логично указанным в Табл. 17.
Механизм стереоспецифической полимеризации 1,3-диенов также рас-
сматривается как полимеризация внедрения, а упрощенные представления
стереоселективности для цис- (ур. (88)) и транс- (ур. (89)) расположения
показаны ниже [231].
цис-стереоспецифичность
син-п-ыинлл
100 -’v- Глава 2. Полимеризация: синтез эластомеров
транс-стереоспецифичность
Как показано в этих уравнениях, основным фактором, определяющим
стереохимию расположения, является тип координации центра переходного
металла с мономером с образованием либо син- или анти- л-алллильного
типа промежуточного соединения. В общем, координация с двумя двой-
ными связями 1,3-диенов в s-транс-конфигурации (см. ур. 89) является
менее распространенной, чем s-цис-конфигурация, показанная в ур. (88)
[231]. Эта интерпретация усложняется фактом, что син и анти- л-алллиль-
ные комплексы находятся в равновесии. Эти простые механистические
представления основаны на том, что стереохимия полимеризации диенов
может изменяться добавлением доноров электронов, таких как N (С2Н5)3,
Р (ОС6Н5)3, С2Н5ОН. Таким образом, добавка этих доноров электоронов
изменяет стереохимию от высоко цис-стереоспецифической до высоко
транс-стереоспецефической для бутадиена с катализаторами, такими как
Со (асас)2/А1 (С2Н5)2С1 [231]. Это объясняется предположением, что донор
электоронов занимает одно из двух координационных мест, требуемых для
цис-присоединения (см. ур. (88)), которое заставляет мономер координи-
роваться только с одним центром (ур. (89)).
Последнее достижение в катализе стереоспецифической полимери-
зации диенов лежит в области катализаторов на основе редкоземельных
металлов или лантанидов, особенно комплексов неодима [251, 252].
Преимуществами этих систем является высокая стереоспецифичность,
высокая активность, контроль молекулярной массы и отсутствие геле-
образования [251].
IX. Стереоспецифическая цепная полимеризация и сополимеризация... -* V 101
Таблица 17. Микроструктура полидиенов, полученных полимеризацией,
инициированной переходными металлами а).
Соли переходных металл ов/ал килы металлов Микроструктура цепи (моль%)
Цис-t,4 Транс-t/i 1,2 3,4
Изопрен
TiCl4/AlR3 (1/1) 97 3b)
a-TiCl3/Al(C2H5)3 98-100
Ti(OR)4/AIR3 (1/7-8) 36
Бутадиен
TiI4/Al(i-C4H9)3 (1/4-5) 92-93 2-3 4-6
Ni(oitTaHoaT)/ A1(C2H5)3/BF3 (1/17/15) 96-97 2-3
СоС12/A1(G2H5)2 Cl / пиридин-Н2О (1/1000/100) 98 1 1
NdCl3/Al(i-C4H9)3-nLc> 97 2.7 0.3
VC13/A1(C2H5)3 99-100
Co(acac)3/ A1R3/ CS2 99-100'1’
а) Данные взяты из работы [231].
ь> Работа [250].
с) L-донор электрона, такой как в тетрагидрофуране или пиридине.
d) Синдиотактический
D. Полиалкеномеры
Циклические олефины участвуют в очень необычном типе
полимеризации с раскрытием цикла в присутствии некоторых катализаторов
переходных металлов [253-257]. Это иллюстрируется в ур. (90) для мета-
тезисной полимеризации с раскрытием цикла (РЦМП) [255] циклооктена
с образованием полиоктеномера. Удивительно, что в полимере сохраняется
двойная связь, т. е. это не полимер, полученный обычным путем присоеди-
нения. Общепринятый механизм для этого типа полимеризации предпола-
гает, что активным центром роста цепи является металл-карбеновое проме-
жуточное соединение, которое участвует в реакции циклоприсоединения
с циклоалкеном с образованием промежуточного четырехчленного цикла
интермедиата, т. е. металлоциклобутана [256].
Owci6
---► —[-сн2сн =СНСН2СН2СН2СН2СН2Ч—
C2H5A1C12 (90)
С2Н5ОН
102 -’v- Глава 2. Полимеризация: синтез эластомеров
В металлоциклобутане происходит раскрытие цикла с образованием новой
металлокарбеновой цастицы, инициирующей рост цепи, как показано ниже
в схеме для полимеризации циклопентена, где Mt представляет переходный ме-
таллический центр, □ — четырехугольник представляет незаполненную орбиталь,
которая доступна для координации с двойной связью мономера, Рп — растущую
полимерную цепь со среднечисленным числом мономерных единиц, равным п.
Как показано, эти процессы являются обратимыми полимеризационными
процессами, в которых достигается равновесное распределение мономера, цик-
лических олигомеров, и образуется полимер с высокой молекулярной массой.
Схема полимеризации для метатезиса с раскрытием цикла
Возможная последовательность реакций образования металл карбенов
показана в ур. 91, где [ W] — вольфрамовый каталитический центр с сопутс-
твующим лигандами, особо не указанными [256].
I I
[W]—С1 +-----А1---СН2СН3-----►[W]---СН2СН3-----► [W] =снсн3
ci J1 С1
-НС1
▼
[W]=CHCH3
(91)
IX. Стереоспецифическая цепная полимеризация и сополимеризация... -'и- 103
Хотя широкий круг соединений переходных металлов может катализиро-
вать эти процессы полимеризации с раскрытием цикла, наиболее активные
катализаторы основаны на производных молибдена, вольфрама и рутения. Эти
соединения часто используются с металлоорганическими сокатализаторами,
аналогично другим катализаторам переходных металлов для полимеризации
олефинов и диенов, описанным в предыдущих разделах. Каталитическая
система WCl6/EtAlCl2/EtOH, описана как промышленно используемый тип
катализатора [253]. В общем, стереохимия полимеризации изменяется с ката-
лизатором и временем реакции. Полимеризующиеся мономеры, важные для
синтеза эластомеров, включают циклопентен, циклооктен и 1,5-циклооктадиен;
необходимо отметить, что циклогексен не полимеризуется по этому методу, воз-
можно из-за того, что отсутствует напряжение цикла, необходимое для развития
полимеризации [257]. Другим мономером промышленного значения является
норборнен, который очень реакционноспособен; однако, образующийся поли-
мер имеет относительную высокую температуру стеклования (Т = 35°С для 80%
транс полимера) [254], но она может быть снижена до -60°С использованием
пластификаторов [258].
Так как метатезисная полимеризация путем раскрытия циклов является
обратимой полимеризацией, обычно получается равновесное распределе-
ние молекулярных масс циклических олигомеров и высокомолекулярных
полимеров [256], например, полиоктеномер обычно содержит от 10 до 15%
циклических олигомеров и от 85 до 90% полимеров [253]. При малом вре-
мени реакции и высокой концентрации мономера при контроле кинетики
образуется полимер относительно высокой молекулярной массы; молеку-
лярная масса уменьшается с увеличением времени реакции. Равновесие
процесса также уравновешивает конфигурацию двойных связей в полимере,
и в конечном счете получается равновесное распределение конфигураций.
Контроль молекулярной массы в метатезисной полимеризации с раскрытием
цикла достигается добавлением ациклических алкенов, которые реагируют
с растущими цепями, обрывая цепь и генерируя новый металлкарбеновый
инициатор, как показано в ур. (92). Промышленно доступные полиоктано-
меры (Вестенамеры, Хюлс, AG) имеют средневесовые молекулярные массы
приблизительно 105г/моль, различное содержание транс-двойных связей
(от 62 до 80%) итемпературу стеклования от-75 до -80°С [253]. Интересной
особенностью физических свойств полиоктеномеров является их способность
к кристаллизации при растяжении [254]. Описанные выше промышленные
полимеры имеют приблизительно от 8 до 30% кристалличности для образцов
с содержанием транс-двойных связей от 62 до 80%, соответственно [253].
□
/Н
=С
н
н
Mt =
+ сн2=снсн2сн3-
Mt
+ сн3сн2сн
Ри
(92)
104 -'V Глава 2. Полимеризация: синтез эластомеров
X. ПРИВИТАЯ И БЛОК-СОПОЛИМЕРИЗАЦИЯ*)
Идея привитой [259-261] или блоксополимеризации [262],
возможно, впервые возникла как способ модификации природных поли-
меров, как целлюлоза, хлопок, каучук или шерсть. Графт- или привитая
сополимеризация по аналогии с ботаническим термином относится к росту
цепей различного химического состава на «стволе» линейной макромолеку-
лы. Наоборот, термин блоксополимеризация относится к специфическому
случаю роста полимерной цепи, от конца линейной молекулы, приводя таким
образом к сложной линейной макромолекуле, состоящей из двух или более
«блоков» различной химической структуры. Важность этих типов полимер-
ных структур определяется тем фактом, что полимерные цепи различной
химической структуры, которые обычно несовместимы и образуют отдельные
фазы (потому что малая энтропия смешения недостаточна, чтобы перекрыть
обычно положительную энтальпию смешения) химически связываются друг
с другом. Это приводит к микрогетерогенности, которая может иметь большое
влияние на механические свойства этих гетерофазных систем, по сравнению
с двумя гомополимерами или физической смесью двух полимеров [263].
Как можно было бы ожидать, привитая и блоксополимеризация могут
осуществляться посредством каждого из трех известных механизмов, т. е.
радикального, катионного и анионного, каждый из которых характеризуется
своими особыми характеристиками [264]. Следовательно, эти механизмы
использовались во всех случаях, где это было возможно для данных полимера
и мономера. Примеры, рассматриваемые ниже, будут касаться преимущес-
твенно эластомеров.
А. Привитая сополимеризация по
свободнорадикальному механизму
Этот тип сополимеризации наиболее широко применялся
для получения привитых сополимеров, т. к. он представляет собой наиболее
простой метод и может быть использован для широкого круга полимеров и
мономеров. Он не очень удобен для синтеза блоксополимеров, что очевидно
из рассмотрения используемых методов. Это можно проиллюстрировать
следующим образом:
1. Химическое инициирование
Это наиболее распространенный метод для привитой сопо-
лимеризации эластомеров посредством свободных радикалов. Свободные
радикалы (Г) генерируются тем же типом инициаторов, которые приме-
няются для свободнорадикальной полимеризации и сополимеризации
(см. раздел IV).
*) См. также главы 12. 13.
X. Привитая и блок-сополимеризация -*\г 105
В общем, эти радикалы образуются в присутствии полидиеновых эласто-
меров и мономеров; следовательно, возможны несколько реакций этих ради-
калов, полученных при действии инициатора, как показано в ур. (93-96).
I ----► I.
k
I . + М ---1-М .
(93)
I. + -f-CH2CH=CHCH2^- 2 » I-H + —4-СНСН=СНСН2—
(94)
I. + —f-CH2CH=CHCH2]— кз » —f-CH2CH-CHCH24-
I
I- + -|-СН2СНН—
сн=сн2
Л3
—► -Ч-СН2СНЧ-
| (96)
• СН-СН2-1
Конкуренция между инициатором полимеризации мономера (ур. (93))
и реакциями образования полимерных радикалов зависит от реакционной
способности инициирующего радикала. Так, привитой сополимер не об-
разуется при полимеризации мономера стирола с полибутадиеном, либо
полиизопреном, когда для генерации радикалов применяется азобисизо-
бутиронитрил. Хорошая эффективность прививки отмечена для радикалов,
генерированных из пероксида бензоила [265, 266].
Этот результат также показывает, что растущие полистирольные ради-
калы не отрывают водород от этих полидиенов с образованием полимерных
радикалов. Конкуренция между присоединением радикалов-инициаторов
к двойной связи в полидиене (ур.95,96) и отрывом водорода (ур.94) также
зависит от инициатора [267]. Так, трет-бутоксирадикал ((СН3)3СО) обладает
необычным преимуществом в отрыве водорода по сравнению с алкильными
радикалами, как показано в Табл. 18. Было показано, что для радикалов пе-
роксида бензоила различие между скоростью отрыва водорода от полидиена
(ур. 94), по сравнению с присоединением инициатора радикала к мономеру
стирола (ур. 93), т. е. &от/&пр5, составляет 1,2 для полиизопрена и 0,63 для
полибутадиена [265]. Для присоединения инициирующих радикалов
106 -*v- Глава 2. Полимеризация: синтез эластомеров
к двойной связи полидиенов показано, что прививка облегчается высоким
содержанием 1,2-микроструктуры в полидиене [268, 269]; скорость присо-
единения к винильпым боковым цепям (ур. 96) больше, чем присоединение
к двойной связи главной цепи (ур. 95).
Таблица 18. Реакционная способность радикалов к отрыву водорода по срав-
нению с присоединением к двойной связиа)
Радикал ^отр./^прис.
t-(CH3)3CO* 30
ROO« 1,0
Н3С • 0,25
RS< Исключительно присоединение
а) Данные взяты из работы [267]
Ь) Отношение констант скорости отрыва водорода [см. ур.(94)] и
присоединения к двойной связи [см. ур. (95) и (96)]
Образование привитых сополимеров на полимерных радикалах по
ур. (93-96) показаны в ур. (97,98), где Р* представляет полимерные радика-
лы основной цепи. Наконец, для того чтобы контролировать молекулярную
массу привитых цепей, добавляют агенты передачи цепи, такие как длин-
ноцепочечные алкилтиолы (см. ур. 27) [270].
р. + иМ ------► Р—[М]„—м • (97)
привитой сополимер
Р* + I—[М]п—М* ------► привитой сополимер (98)
Так как все эти реакции привитой сополимеризации протекают одно-
временно всегда возможно образование гомополимера в ходе реакции при-
вивки путем реакции инициатора радикала с мономером (ур. (93)) и также
передачи цепи на растущую цепь с центрами, отличающимися от основной
цепи полимера (тоесть мономер, растворитель, инициатор). Следовательно,
в общем привитой сополимер будет засорен как исходным гомополимером,
так и гомополимером, полученным из мономера [259].
Этот тип привитой сополимеризации применялся для прививки моно-
меров стирола и метилметакрилата к натуральному каучуку [271] непосред-
ственно в латексе [272, 273]. Подобные методы разработаны для прививки
ранее упомянутых мономеров и многих других виниловых мономеров,
к синтетическим каучукам, типа БСК, что привело к получению широкого
круга различных эластомеров, усиленных пластиком, и ударопрочных
пластиков, усиленных каучуком [270, 274]. В этом случае прививка может
X. Привитая и блок-сополимеризация -* V 107
также протекать путем «сополимеризации» мономера с ненасыщенными
связями (главным образом, винильными) в полимере, как указано ранее
[см. ур. (96)]; таким образом:
RM/ + vwCH2 —СНлм *wCH2 — СНиал
СН9=СН —> RMX— СН2— СН-
Эта реакция может также привести к поперечному сшиванию полимерных
цепей, что должно контролироваться.
2. Другие методы
Другие методы генерирования свободных радикалов могут
быть также применены для инициирования привитой полимеризации с элас-
томерами, как природными, так и синтетическими. Они включают облучение
полимер-мономерных смесей УФ светом [275], излучением высоких энергий
[276, 277] и механическим сдвигом. Последнее представляет особенный
интерес из-за уникального механизма, и интенсивно исследуется.*
Так, эластомеры при действии некоторых механических сдвиговых усилий
претерпевают гомолитический разрыв связи с образованием цепных свобод-
ных радикалов. Последние в присутствие кислорода могут затем подвергаться
различным реакциям, приводящим к стабилизации разорванных цепей или
реакциям с другими цепями с образованием разветвленных или сшитых поли-
меров [278,279]. При пластикации смеси различных эластомеров межполимеры
образуются путем взаимодействия радикалов, образованных из сополимеров.
В дальнейшем такие механохимические процессы имеют место, когда эла-
стомеры пластифицируются в присутствии полимеризующихся мономеров.
Цепные радикалы инициируют полимеризацию и приводят к образованию
блок- и привитых структур. Это показано для натурального каучука [274]
и других эластомеров [281, 282]. Необходимо отметить, что живая анионная
полимеризация [283] и контролируемая/живая радикальная полимеризация
[284], такая как радикальная полимеризация с передачей атома, применяется
для получения хорошо охарактеризованных привитых сополимеров. Очень
удобным методом является сополимеризация определенных макромономеров,
дающих полимеризуемые цепные концевые группы с другим мономером, для
того чтобы получить привитой сополимер со статистическим распределением
хорошо охарактеризованных привитых ветвей [285].
В. Блоксополимеры, получаемые по анионному
механизму
Анионный механизм является наиболее подходящим для син-
теза блоксополимеров, т. к. многие из этих систем являются живыми полиме-
рами, как описано ранее [144, 150, 165, 194, 262, 263]. Таким образом, можно
* См. также главу 11.
108 -*v- Глава 2. Полимеризация: синтез эластомеров
применять органощелочные инициаторы, чтобы получить блок-сополимеры
в гомогенном растворе последовательным добавлением мономеров, где каждый
блок имеет предсказуемую молекулярную массу, основанную на стехиометрии
мономер-инициатор, так же как и очень узкое молекулярно-массовое распреде-
ление (Пуассона) [185]. Можно ожидать, что такие блоксополимеры являются
очень чистыми из-за отсутствия любых побочных реакций при полимеризации
(таких как обрыв цепи, передача цепи на мономер, разветвление).
Полистирол Полибутадиен Полистирол
Рис. 3. Структура термопластичных эластомеров из триблоксополимеров АВА
(по Мортону [291 ]).
Литийорганические инициаторы особенно удобны в этом отношении,
т. к. они растворимы в большинстве растворителей [178] и могут иниции-
ровать полимеризацию широкого круга мономеров, таких как стирол и его
гомологи, 1,3-диенов, алкил-метакрилатов, винилпиридинов, циклических
оксидов и сульфидов и циклических силоксанов [143, 144, 150, 262]. Син-
тезированы различные блок-сополимеры из этих мономеров, некоторые
X. Привитая и блок-сополимеризация -’и- 109
в промышленном масштабе, но выдающимся достижением в этой области
является синтез триблоксополимеров типа АВА, и это заслуживает отде-
льного рассмотрения [263].
Триблоксополимеры типа АВА, которые являются наиболее распро-
страненными в промышленности, представлены стирол-диен-стирольными
сополимерами, полученными последовательной полимеризацией, иниции-
рованной соединениями алкиллития, как показано в ур. (99-101) [263,286].
Поведение этих блоксополимеров связано с особыми характеристиками
блок- (или привитых) сополимеров и основано на общей несовместимости
различных блоков [287]. Так, для типичных термоэластопластов [263] поли-
стирол ьные концевые блоки (~ 15,000-20,000MW) сегрегируют в отдельную
фазу, которая образует микродисперсию внутри матрицы, состоящей из
цепей полидиена (50,000-70,000 MW) [288-290]. Схематическое представ-
ление этой морфологии показано на рис. 3.
Это фазовое разделение происходит в расплавленном или набухшем со-
стоянии и приводит при температуре эксплуатации к возникновению сетки
эластичных полидиеновых цепей, связанных друге другом стеклообразными
полистирольными микродоменами. Следовательно, эти материалы ведут
себя как фактически сшитые эластомеры при температуре эксплуатации, но
при этом полностью термопластичны и жидкие (текучие) при повышенной
температуре.
C4H9Li
PsLi + т СН2=СН-СН=СН2--»>
(100)
PS —fCH2CH=CHCHd— CH2CH=CHCH2Li
m-1
Ps-PBDLi
Ps-PBDLi + nCH2=CH
ROH
->> Ps-PBD-PsLi ----->. Ps-PBD-Ps
(101)
110 -’V Глава 2. Полимеризация: синтез эластомеров
Важно отметить, что морфология блок-сополимеров АВА зависит
в первую очередь от относительного состава компонентов блока [165, 287].
Например, если содержание стирола увеличивается, морфология изменяется
от сферических полистирольных доменов до цилиндрических; дальнейшее
увеличение содержания стирола приводит к ламелярному выстраиванию
обеих фаз и в конечном итоге к инверсии фаз с образованием непрерывных
полистирольных фаз. Свойства триблоксополимеров АВА зависят и изме-
няются с составом.
Рис. 4. Микрофотография ультратонкой пленки триблоксополимера стирол-изо-
прен-стирол, полученная методом просвечивающей электронной микроско-
пии. (MW 16200-75600-16200). хЮОООО.
X. Привитая и блок-сополимеризация -’V- 111
Такие термопластичные эластомеры очень перспективны технологи-
чески, т. к. они могут формоваться при нагревании подобно термопластам
и проявлять свойства вулканизатов каучука после охлаждения. Их структу-
ра, морфология, механические свойства широко изучаются [291-294 J. Элек-
тронные микрофотографии типичных стирол-изопрен-стирольных (СИС)
триблок-пленок показаны на рис. 4, а прочностные свойства ряда таких
триблоксополимеров — на рис. 5. Необычно высокая разрывная прочность
таких эластомеров, большая, чем у обычных вулканизатов, объясняется как
заметной регулярностью сетки, как показано на рис. 5, так и способностью
поглощать энергию полистирольными доменами, которые переходят в теку-
чее состояние и изменяют форму (текут и деформ и роуются) под действием
больших напряжений [291].
Степень растяжения (X)
Рис. 5. Прочностные свойства триблоксополимеров стирол-изопрен-стирол (Из
Мортона [291]).
Это интересное поведениетриблоксополимеров АВА не является уникаль-
ной чертой строл-диеновых структур и может проявляться в случае других
аналогичных химических структур. Так, термопластичные эластомеры, полу-
ченные из других триблоксополимеров, где диены замещены на циклические
сульфиды [295], циклические силоксаны [296] или алкилакрилаты [297];
поли (алкилметакрилат) ные концевые блоки также исследованы [297].
Более того, преимущество ряда различных типов живой полимеризации
с переходными металлами, катионными или радикальными центрами роста
цепи определяют новые механизмы синтеза блок-сополимеров типа АВА
с использованием широкого типа мономеров [263].
112 -’v- Глава 2. Полимеризация: синтез эластомеров
Важно отметить, что любая молекулярная архитектура, которая образует
термопластичный блок, химически связанный с эластомерным блоком, ко-
торый в свою очередь связан с другим термопластичным сегментом, должна
проявлять свойства термоэластопластов. Например, привитые на эласто-
мерную цепь термопластичные ветви ведут себя как термоэластопласты
[285, 298]. Другими примерами являются полимеры сегментированного
типа - [АВ] п - с чередующимися жесткими и мягкими сегментами. Такой
широкий круг сегментированных полиэфиров и полиуретанов со сложными
или простыми полиэфирными мягкими сегментами проявляют свойства
термоэластопластов [263, 298, 299].
ПРИМЕЧАНИЕ ПЕРЕВОДЧИКОВ
Авторы данной главы пользуются исключительно источника-
ми американской и другой зарубежной литературы и не цитируют известные
работы советских и российских ученых в области свободнорадикальной
и эмульсионной полимеризации, анионной и ионно-координационной
полимеризации (работы школы академика Медведева С.С., ленинградских
ученых Института синтетического каучука).
ГЛАВА
3
ХАРАКТЕРИСТИКА СТРУКТУРЫ
ЭЛАСТОМЕРОВ
С. М. РОЛАНД
Исследовательская лаборатория Военно-морских сил
Отделение химии, код 6120
Вашингтон
I. ВВЕДЕНИЕ
Структурные характеристики полимеров на ранних стадиях
их изучения были ориентированы на свойства растворов и их связь с моле-
кулярным весом [1-4]. Затем получили развитие спектроскопия и хрома-
тография и стали доступны и другие характеристики [5-10]. В этой главе
описываются различные методы характеристики структуры полимеров. Их
описание, особенно в части, относящейся непосредственно к эластомерам,
существенно обновлено по сравнению с предыдущими изданиями. Большая
часть этой информации отсутствует в открытой литературе. Обсуждение
классических методов анализа осталось, в основном, без изменения, но
и текст и ссылки обновлены. Данные об использовании ядерного магнитного
резонанса (ЯМР, NMR), малоуглового рассеяния нейтронов (МУРН, SANS)
и физической релаксации существенно расширены и включено несколько
новых тем, в том числе результаты по ориентации, дефектности структуры
и смесям эластомеров.
Знание химического состава, распределения последовательностей пов-
торяющихся звеньев, а также молекулярно-весового распределения, коли-
чества и типа разветвлений позволяет сделать заключение о механических
свойствах (гл. 4), реологии (гл. 5 и 6), поведении при вулканизации (гл. 7),
усилении наполнителями (гл. 8) и химической реакционноспособности
(гл. И) эластомеров. Однако соотношения между основными молекуляр-
ными характеристиками и свойствами готовых материалов в лучшем случае
имеют полуколичественный характер, корреляция никогда не гарантирует
наличия причинной связи. Как и в предыдущих изданиях, методы опре-
деления молекулярного веса рассматриваются более подробно, чем спек-
троскопические. Это связано с идеей, согласно которой больше внимания
114 Глава 3. Характеристика структуры эластомеров
следует уделять тем методам, при использовании которых могут возникать
общие проблемы, и по которым отдельные исследователи могут предлагать
разную интерпретацию.
II. ХИМИЧЕСКИЙ СОСТАВ
Химический элементный анализ полимеров часто может
быть проведен с помощью тех же методов, что и для органических низкомо-
лекулярных веществ [1, 2, 9, 11-14]. Это, в частности, так при применении
метода сжигания образцов. Так, С, Н и N могут быть определены на образцах
массой порядка миллиграмма путем полного сжигания образца с последу-
ющим анализом выделяющегося газа методом газовой хроматографии. То-
чность при этом составляет 0,3%. Сера и галогены также легко определяются
после сжигания путем титрования сульфатов или SO2 для определения серы,
или путем потенциометрического титрования с AgNO3 для определения га-
логенов после обработки газов, например, NaOH и гидразин сульфатом. При
определении серы необходимо отслеживать возможное влияние присутствия
азота. Использование рентгеновских методов позволяет количественно
и быстро определять содержание таких малых количеств металлов, как одна
миллионная доля (ppm).
С помощью элементного анализа можно определить только то, какие
атомы присутствуют в образце. Определение химической структуры требует
часто использования спектроскопических методов. Отдельные элементы
структуры полимеров поглощают или испускают излучение определенных
частот, характерных для их химической структуры. Переходы в связях
основной цепи могут фиксироваться на инфракрасных или Рамановских
спектрах, электронные переходы, характерные для ненасыщенных связей,
могут наблюдаться в ультрафиолетовой или видимой части спектра; атом-
ные ядра, обладающие магнитным моментом, могут обнаруживаться, и их
положение фиксироваться в экспериментах по магнитному резонансу.
Для получения основной информации используют инфракрасную (ПК, IR)
спектроскопию [8-10,15,16] (обычно — Фурье ИК-спектроскопию, FTIR).
Это прямой метод, доступный неспециалистам. Для измерений используют
тонкие пленки эластомера, которые получают отливкой или формованием
образцов весом 20-30 мг между слоями полиэфирной или алюминиевой
фольги (желательно, покрытой тефлоном). Необходимо исключить окисле-
ние образца, если формование проводится при повышенных температурах.
В большинстве случаев используют ИК-диапазон средних длин волн
от 4000 до 400 см-1. Идентификацию образцов проводят путем сравнения
спектров с эталонами с использованием широко известных справочников
[8, 17]. Если образец вулканизован, приготовление образцов затруднено.
Можно использовать получение срезов на микротоме или спектры, полу-
ченные при отражении, обычно методом нарушенного полного внутреннего
II. Химический состав Jb 115
отражения (НПВО, ATR) [18-22]. Реже используют фотоакустическую
Фурье-спектроскопию [23,24]. Этот метод использует измерение звука, воз-
никающего в результате селективной абсорбции модулированных световых
волн. В результате абсорбции поверхность образца нагревается, что приво-
дит к нагреванию приповерхностного воздуха, в результате чего возникают
изменения давления, т. е. звуковые волны. Инфракрасная спектроскопия
позволяет исследовать образцы весом менее миллиграмма [16, 25-36].
Рамановская спектроскопия дает аналогичную информацию, но допол-
нительную к ИК-спектроскопии, т. к. колебания, которые неактивны в инф-
ракрасной области, обычно активны в Рамановской, и наоборот. Например,
связи углерод-сера легко обнаруживаются при Рамановских измерениях
[5, 10]. Т. к. свет рассеивается от образца, легко получать спектры вул-
канизованных образцов. Интерференцию, связанную с флуоресценцией,
легко избежать, используя источник света с большими длинами волн [37].
Рамановская спектроскопия весьма перспективна для исследования про-
странственных структур [38].
В то время как Рамановская и ИК-спектроскопия используются для
«обнаружения отпечатков пальцев» (т. е. для идентификации химической
структуры), распространение спектроскопии на видимую и ультрафиоле-
товую (УФ, UF) область используется, в основном, для количественного
анализа [39-41]. Коэффициенты затухания для сопряженных ненасыщен-
ных структур очень велики.
Для использования рассмотренных выше спектральных методов как
количественных необходима их калибровка. А измеряемая методом ядер-
ного магнитного резонанса (ЯМР, NMR) [9, 10, 42-44], в частности, на
протонах, 0H ЯМР)’ интенсивность абсорбции прямо пропорциональна
количеству атомов водорода; следовательно, отношение интенсивностей
можно использовать для определения количества химически различимых
атомов водорода в образце. Протоны резонируют при определенной харак-
теристической частоте в зависимости от своего химического окружения
(«химический сдвиг»), поэтому можно идентифицировать специфическую
химическую природу протонов.
Химическую информацию можно получить и методом ЯМР на углероде 13,
,3С ЯМР. В то время как измерения методами 13С ЯМР производят, в основ-
ном, в растворе, что позволяет улучшить разрешение, 1Н и 13С ЯМР может быть
использован и для твердых образцов, таких как эластомеры. Это возможно в
том случае, когда молекулярная подвижность в образце достаточна для ус-
реднения зависимой от ориентации части химического сдвига (анизотропии
химического сдвига, АХС, CSA) химически идентичных атомов. Химический
сдвиг в диапазоне, характерном для 13С ЯМР, значительно больше, чем для
’Н ЯМР, поэтому использование 13С ЯМР дает лучшее спектральное раз-
решение. Однако ядерный эффект Оверхаузера и другие релаксационные
процессы приводят к отклонению интенсивности абсорбции, определяемой
116 Глава 3. Характеристика структуры эластомеров
методом 13С ЯМР, от прямой пропорциональности количеству атомов углеро-
да. Таким образом, если не использованы специальные приемы, стандартный
жидкостной метод 13С ЯМР не является количественным.
Это также справедливо при использовании твердотельного варианта ме-
тода 13С ЯМР [43] для получения спектров высокого разрешения твердых
некаучукоподобных образцов. Используя сочетание вращения под маги-
ческим углом (ВМУ, MAS) и кросс-поляризацию протонов (путем облуче-
ния на радиочастотах с высокой энергией протонных резонансов) можно
избежать эффекта расширения линий от анизотропии химического сдвига
и диполь-дипольного взаимодействия 13С-протон. Относительно высокое
разрешение 13С ЯМР спектров может быть получено, но интенсивности аб-
сорбции не будут пропорциональны количеству атомов углерода в образце.
Метод ЯМР позволяет получить не только химическую информацию, но и
данные о регулярности полимера и длине последовательностей, о коротких
разветвлениях и кристаллизации.
Используя элементный анализ, ИК и ЯМР методы возможно оценить
структуру повторяющихся звеньев полимера. В некоторых случаях, таких
как малая концентрация повторяющихся звеньев или функциональных
групп, или для других, кроме С и Н атомов, количественное определение
связано с большими трудностями. Альтернативный анализ основан на
химической реакционноспособности конкретных групп [45]. Например,
достоверные сведения о кислотных группах можно получить путем титрова-
ния основаниями; олефины в ряде случаев можно определить количественно
с использованием озона и галогенов; гидроксильные группы можно этерифици-
ровать воздействием ангидридов и обратным титрованием оценить содержание
гидроксильных групп. В ряде случаев можно использовать приемы, разрабо-
танные для низкомолекулярных вещества. Конечно, низкие скорости реакций,
связанные с низкими концентрациями, и проблемы с доступностью подходящих
растворителей как для реагента, так и для полимера, создают определенные
трудности. Краткие испытания для оценки результатов в функции времени или
температуры позволяют обычно установить пригодность метода.
Пиролиз [46-50] образцов может приводить к образованию характерных
фрагментов, которые могут быть проанализированы методом газовой хрома-
тографии (ГХ, GC) [9] или масс-спектроскопии (МС, MS) [51]. Поскольку
соотношение получающихся фрагментов и исходного полимера достаточно
сложно, этот метод используется обычно лишь для нерастворимых образцов
и образцов, для которых другие методы неприменимы. Комбинированный
метод — сочетание пиролитической газовой хроматографии и масс-спектро-
скопии используют для изучения летучих компонентов натурального каучу-
ка [52]. При озонолизе [9] ненасыщенный образец, вступая в химическую
реакцию, образует нестабильный промежуточный озоноид, который при
дальнейших реакциях позволяет провести химическую идентификацию.
Озонолиз эластомеров обычно используют в сочетании с ГХ [53-561. При
III. Распределение последовательностей повторяющихся звеньев -’V- 117
использовании вторичной ионной масс-спектроскопии (ВИМС, SIMS)
поверхность образца облучают пучком ионов с последующим изучением
вторичных ионов методом масс-спектроскопии [57, 58]. Метод ВИМС на-
шел различные применения для эластомеров, включая анализ поверхности
[59-61] и изучение взаимодействия с техническим углеродом [62, 63]. Пи-
ролиз также используется для идентификации химической структуры при
рутинных анализах. При использовании метода термогравиметрического
анализа (ТГА, TGA) [64,65] образуются летучие, при этом остаток позволяет
определить содержание технического углерода или других наполнителей.
Электронный парамагнитный резонанс (ЭПР, EPR) или электронный
спиновый резонанс (ЭСР, ESR) может быть использован для определения
типа и количества свободных радикалов. Эта информация важна при изучении
химических процессов, сопровождающих деструкцию и разрушение полимер-
ных материалов [66-71]. Метод ЭПР также можно использовать при изучении
технического углерода или других наполнителей в полимерах [72-74].
III. РАСПРЕДЕЛЕНИЕ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
ПОВТОРЯЮЩИХСЯ ЗВЕНЬЕВ
Большинство эластомеров содержит множество добавок
(в большинство рецептов входит более дюжины ингредиентов), также как
и возможные примеси. Вдобавок и сам полимер может представлять собой
сополимер, состоящий из различных мономерных единиц, или каучуковая
часть может быть смесью двух и более каучуков. Чтобы определить содер-
жит ли эластомер добавки или примеси и (или) гетерогенна каучуковая
часть, необходимо разделить компоненты, т. е. провести фракционирование.
Множество методов позволяет разделить компоненты с последующей их
идентификацией (см. Табл. 1). Методы осаждения и/или растворения просто
используют различную растворимость компонентов [75]. Если молекуляр-
новесовое распределение широкое, метод фракционирования используют
вместе с одним из более сложных методов, описанных ниже. Если гетеро-
генность внутримолекулярная, может быть необходимо фракционирование
в поперечном поле («ортогональное» фракционирование), которое требует,
по меньшей мере, использования двух механизмов фракционирования.
Метод фракционирования путем элюирования при подъеме температуры
(ФЭПТ, TREF) [76,77] был разработан для измерения распределения компо-
нентов в частично закристаллизованных полимерах. Полимер расплавляется
на субстрате по мере подъема температуры в пределах интервала плавления.
Таким образом разделение основано на различии в кристаллизуемости фрак-
ций. Аналогичный метод, основанный на использовании сверхкритических
жидкостей [78], также может дать информацию о распределении последова-
тельностей повторяющихся звеньев, т. к. кристаллизуемость более длинных
последовательностей выше.
118 -*v- Глава 3. Характеристика структуры эластомеров
Таблица 1 Методы характеристики состава
Метод Принцип работы
Элементный анализ Анализ продуктов разложения
Поглощение инфракрасных волн 1 < X (|хм) < 16 Характеристические частоты колебаний
Рамановское рассеяние Характеристические частоты колебаний
Ядерный магнитный резонанс (ЯМР) Характеристические энергии переходов некоторых ядер с магнитным моментом (1Н,2Н, 13С, 129Хеидр.)
Поглощение ультрафиолетового (УФ) и видимого света 0,2 < X (р,м) < 0,8 Характеристические энергии переходов электронов
Определение функциональных групп Анализ на известные реакции химических частей
Пиролиз/озонолиз Пиролиз с идентификацией фрагментов методами хроматографии или масс-спект- роскопии
Электронный парамагнитный резонанс (ЭПР) Энергия спиновых переходов неспаренных электронов, зависящая от химического окружения
Масс-спектроскопия (МС) Масса фрагментов, отражающая химиче- ский состав
Масс-спектроскопия вторичных ионов (ВИМС, SIMS) Характеристика ионов, эмитирующих с поверхности
Эксклюзионная хроматография (ЭХ, SEC) [9, 79-81], которую обычно
называют гельпроникающей хроматографией (ГПХ, GPC), использует
различия гидродинамического объема компонентов. Более детально
этот метод рассмотрен ниже. Наиболее простой является абсорбцион-
ная хроматография [82, 83]. В этом методе разделение осуществляется
за счет различия в удерживании разных повторяющихся единиц или
функциональных групп в результате их взаимодействия с неподвижной
поверхностью. Другие методы основаны на различиях в скорости седи-
ментации [84, 85] и явлении диффузии-абсорбции (тонкослойная хро-
матография, ТСХ, TLC) [86, 87]. Термическая диффузия — основа для
фракционирования потока в термическом поле (ФТПП, TFFF) [88-91],
будет рассмотрена далее.
Разное расположение мономерных звеньев является источником раз-
личия химических сдвигов и скалярных слияний (расщеплений) пиков
в спектрах ЯМР. Используя селективные правила и эмпирические данные
III. Распределение последовательностей повторяющихся звеньев 119
о химических сдвигах можно отнести их к тем или иным структурам. По-
скольку сдвиги в спектрах 13С ЯМР больше, чем в ЯМР, можно обнару-
жить незначительные различия в положении атомов углерода, находящихся
на расстоянии до пяти связей от точки отсчета. Поэтому метод 13С ЯМР
весьма полезен для определения распределения звеньев в основной цепи
полимера. Такой пример приведен на рис. 1, где представлен спектр 13С ЯМР
вулканизованного серой и наполненного техническим углеродом НК [92].
Присутствует маленький пик, представляющий содержание транс-изомера,
интенсивность которого растет по мере протекания процесса вулканизации.
Т. о. метод ЯМР дает информацию о цис-транс изомеризации в процессе
вулканизации. Аналогичные результаты получены для ^ис-1,4-полибута -
диена [93-95]. Рис. 2 представляет множество изменений в спектре ЯМР,
сопровождающих вулканизацию.
Жирная
кислота
-СН2-
I-------1--------1--------1-------1--------1-------1
70 60 50 40 30 20 10
Химический сдвиг, млн. д.
Жирная
кислота
Рис. 1. Увеличенная часть спектра 13СЯМРдля НК, вулканизованного до половины
от максимального крутящего момента. Пик при 16 миллионных долей напря-
женности поля возникает в результате цис-транс изомеризации |92].
Различия в организации мономерных звеньев вдоль основной цепи это еще
одна из форм гетерогенности. Химически различающиеся мономерные зве-
нья могут находиться в последовательностях разной длины, а оди паковые —
быть по-разному расположены (стереоизомеры), что приводит к раз-
личию в свойствах. Стереорегулярность полимера можно определять
с помощью тех же методов, которые используются и для определения
химической природы звеньев, при этом наиболее информативны ЯМР
и ИК-спектроскопия.
120 -*v Глава 3. Характеристика структуры эластомеров
Когда длина последовательностей достаточно велика, т. е. в них входит
больше шести-восьми мономерных звеньев, для ее прямого определения
метод ЯМР непригоден, нужны другие методы. Может быть предложен
метод пиролиза в сочетании с последующим совместным применением
газовой хроматографии и масс-спектроскопии (ГХ-МС, GC-MS); при этом
длинные последовательности определяют как фрагменты пиролизата.
Однако получаемые данные усложнены и трудны для интерпретации
[96-99].
Рис. 2. Увеличенная часть спектра 13С ЯМР 1{ис-1,4-полибутадиена после вулкани-
зации. Стрелками показаны новые пики, возникающие в результате вулкани-
зации. Первые 7 представляют углерод, входящий в метильные, а 18 других —
в метиленовые последовательности [94].
IV. АРХИТЕКТУРА ЦЕПИ
Вязкоупругие свойства полимеров при повышенных
температурах в большой степени определяются средним молекулярным
весом Мш, присутствием длинных разветвлений и молекулярно-весовым
распределением (МВР) [100-105]. Даже свойства вулканизованных
эластомеров могут отражать длину исходных молекулярных цепей, т. к. их
концы, количество которых обратно пропорционально^, представляют
собой дефекты.
IV. Архитектура цепи -* V 121
А. Молекулярный вес и его распределение
Распределение молекулярных цепей по длинам обычно
представляют в виде зависимости молярной или средневесовой доли молекул
фракции от ее молекулярного веса. Различные средние молекулярные веса
представляют определенные позиции распределения молекулярных цепей
по длинам (молекулярно-весового распределения)
(о
i=l / i=l
где j = 1,2 и 3 это среднечисленный молекулярный вес М, средневесовой мо-
лекулярный весMw, иг-средний (среднеседиментационный) молекулярный
вес М2 соответственно. Для монодисперсного распределения Мп = Mw = М2.
Для статистического МБР Mw /Мп = 2 и Мz /Мп =1,5, а для распределения
Шульца-Цимма Мz + Мп =2 Mw и 1 < Мz / Mw < 2. На рис. 3 представлено
типичное молекулярно-весовое распределение.
Рис. 3. Типичное молекулярно-весовое распределение. Мгпх = X иг М*+а/Iw. M-
где а — показатель степени в уравнении Марка-Хувинка [13].
Среднечисленный молекулярный вес М , определяет обобщенные
(коллигативные) свойства (т. е. свойства, которые зависят только от числа
растворенных молекул) растворов полимеров. Измерение понижения тем-
пературы плавления (криоскопия) или повышения температуры кипения
(эбулоскопия) может, в принципе, давать ту же информацию о макромолеку-
122 -*и- Глава 3. Характеристика структуры эластомеров
лах, что и о низкомолекулярных веществах [106-108]. Это прямые методы,
которые дают абсолютные результаты и не требуют калибровки. Изменение
температуры плавления или кипения А71 описывается соотношением
АТ/кс = М;'+ААс + А2с2 + - (2)
где с — концентрация, 4. — вириальные коэффициенты, к — константа,
зависящая от растворителя, температуры и типа прибора. Количество
молей макромолекул, соответствующее массовой концентрации, необходи-
мой для получения идеального раствора, очень мало. Соответственно мало
и изменение температуры плавления или кипения АТ. Для Мп = 105 г/моль
АТ = 10‘5 -10“4оС. Поэтому эти классические методы не применяются при
Мп больше 104 г/моль. Более того, оба эти метода требуют большого времени
и точность их мала.
Для образцов с относительно низким молекулярным весом используют
метод осмометрии давления паров (ОДП, VPO) [107,108]. Этот метод основан
на понижении давления паров растворителя в присутствии растворенного
полимера. Различие в равновесном давлении паров приводит к различию
в скорости конденсации на двух термисторах, расположенных в камере
с насыщенными парами растворителя. Один термистор покрыт раство-
рителем, а другой — раствором полимера. Большее количество раствора
конденсируется на растворителе, повышая его температуру. Измеряется
возникающая вследствие этого разность температур, и с помощь, калибров-
ки можно определить М . Метод ОДП не требует большого времени и с его
помощью можно определить значения Мп порядка 5.104 г/моль. Существуют
соответствующие серийные приборы.
Мембранная осмометрия [107-109] основана на понижении актив-
ности (уменьшении свободной энергии) растворителя при растворении
вещества, что дает возможность непосредственно определить Мп. Когда
раствор полимера приводится в контакт с чистым растворителем, градиент
концентрации нарушается вследствие диффузии. Если между раствором
и чистым растворителем поместить полупроницаемую мембрану полимер
задерживается, а растворитель проходит сквозь мембрану. Равновесие не
может быть достигнуто, но если раствор находится в замкнутом объеме, со
стороны раствора повышается давление, которое постепенно препятствует
течению растворителя. Величина этого «осмотического» давления, П, бу-
дет зависеть только от количества присутствующих молекул полимера, по
крайней мере при отсутствии взаимодействия полимер-полимер (т. е. при
малых концентрациях раствора). Уравнение, связывающее эти величины,
аналогично уравнению (2) для изменений температур плавления и кипе-
ния. Измерения при нескольких концентрациях с и экстраполяция вели-
чины П/с на с = 0 позволяет получить значения М . Для раствора вещества
с Мп = 10° г/моль концентрацией 1% давление при 25 °C около 260 Па.
IV. Архитектура цепи -* V 123
Обычно этот метод дает хорошие результаты для полимеров с молекулярным
весом от 104до 106г/моль.
Часто встречающаяся трудность при использовании этого метода заклю-
чается в том, что зависимость П/с от с может быть нелинейной при больших
концентрациях. Если это происходит вследствие агломерации, решить
проблему можно, изменив растворитель или температуру. Чтобы получить
измеримые значения осмотического давления при больших Мп„ необходимо
увеличить интервал концентраций с. Для хороших растворителей второй
вириальный коэффициент А2уменьшается с ростом Мп, (А2 ~ Мп “°’2), но не
так быстро как необходимо увеличивать с. Поэтому при измерении больших
Мп часто встречается нелинейность. Чтобы линеаризировать данные строят
зависимость (П/с)1/2от с. Самые большие трудности при использовании
этого метода возникают из-за несовершенства мембран. Иногда полимер
диффундирует через мембрану при стационарных значениях проницаемости
для молекул с М < 5 • 103г/моль. Это может приводить к ошибкам до 10%
и более, так что распределение искажается со стороны малых молекулярных
весов.
Светорассеяние это также прямой (абсолютный) метод определения мо-
лекулярного веса (> 103г/моль) [9,107,108,110-116]. (Мырассматриваем
только статическое светорассеяние, используемое для получения информа-
ции о структуре. Однако неупругое светорассеяние это мощный метод для
изучения динамики полимерных молекул. При использовании калибровки
этот метод также позволяет определять молекулярный вес [111]). Свет,
проходящий через вещество, рассеивается благодаря наличию областей
с разным коэффициентом преломления п; поэтому полимерные молекулы,
растворенные в растворителе с другим коэффициентом преломления, могут
рассеивать свет. Пренебрегая вкладом самого растворителя, (который мал
по сравнению с рассеянием на макромолекулах) изолированные молекулы
полимера (интерференция между частицами отсутствует), размеры которых
меньше длины волны света (внутримолекулярная интерференция отсутству-
ет), могут рассеивать свет с интенсивностью, определяемой по формуле
/(9)=4^(Wkc (з)
X Na \ ос )т
где q ^(4тс/Х)$т(0/2), 0 — угол рассеяния, /0 — интенсивность света,
рассеянного при 0 = 0, X — длина волны света, TVA — число Авогадро. п()
и п — показатели преломления растворителя и раствора соответственно.
Изменения показателя преломления при изменении концентрации раствора
(Эп/Эс) т приблизительно равно отношению разности показателей прелом-
ления полимера пП0Л и растворителя пок плотности полимера р: (Эп/Эс)т =
= (п - По)/р. Обычно для растворов полимеров (Эп/Эс) т< 0,2 мл/г.
124 Д.
Глава 3. Характеристика структуры эластомеров
Выражение в скобках в уравнении (3) в каждом данном эксперименте
постоянно, т. е.
R(q) н I(q)/l0 =КМс
(4)
где R(q) — релеевский коэффициент. Полимерные цепи достаточно велики
(> Х/10), так что свет, рассеянный от разных концов молекулы, имеет разный
сдвиг фаз. Поэтому уравнение (4) модифицируют введением структурного
фактора Р (q)
R(q) = KMcP(q)
(5)
Для малых углов рассеяния структурный фактор можно аппроксимировать
в виде
P(q)~ 1-|?Ч2 + - (6)
О
что справедливо для молекул любой формы. Sg это среднеквадратичный
радиус инерции молекул полимера. Угол можно считать малым если вы-
полняется условие q « Зависимость R (q) от q2 при малых q линейна, ее
пересечение с осью ординат дает значение молекулярного веса, а отношение
наклона к этой величине — радиус инерции. Поскольку обратная величина
релеевского коэффициента более чувствительна к малым изменениям, чем
сама величина /?, уравнение (5) обычно записывают в форме
Кс
= м-'[14а!
V 3 *
(7)
(при этом использовано соотношение 1 + х = [1 - х]~\ справедливое для
малых х). Тогда строят зависимость R(q)~x от q2 из которой, экстраполяцией
на q = 0 получают значения молекулярного веса и Sg.
Изложенные выше расчеты сделаны в предположении независимости
рассеяния от каждой частицы. При обычных концентрациях растворов поли-
меров, даже если видимого помутнения не наблюдается, волны, рассеянные
от отдельных молекул, могут интерферировать. Такая интерференция между
частицами может приводить к отклонению зависимости интенсивности рас-
сеяния от с от линейности. Для корректировки концентрационного эффекта
принимается степенной ряд в виде
(8)
где А2 называют вторым вириальным коэффициентом. Заметим, что при
рассмотрении молекулярно-массового распределения, М будет средневе-
IV. Архитектура цепи -* V 125
совым молекулярным весом, а полученное значение радиуса инерции —
среднеквадратичным значением z-среднего. Применение уравнения (8)
требует экстраполяции на с = 0 в дополнение к экстраполяции на нулевой
угол. Обе эти экстраполяции производят с помощью зависимостей Цимма
(рис. 4). Правильное построение этих зависимостей позволяет получить
и радиус инерции и вириальный коэффициент, в согласии со значениями Мш,
полученными для полимера. Отклонения от линейности при малых углах
означает наличие артефактов: агрегации, геля, пыли и т. д. [117]. Проблема
очистки каучуков особенно серьезна из-за возможности удаления части
полимера вместе с загрязнениями. Используя лазерные лучи с малой пло-
щадью сечения иногда можно смотреть как бы «между» частицами геля
или пыли, которые в этом случае периодически обнаруживаются в виде
«зубцов» большой интенсивности. Это в некоторой степени может частич-
но облегчить задачу очистки растворов. Для коррекции влияния крупных
примесей можно рекомендовать параллельное измерение динамического
светорассеяния [118].
Использование нескольких детекторов (отЗ до 18) способствует облегче-
нию экстраполяции на нулевой угол. В принципе, детектор рассеянного света
может быть расположен при достаточно малых углах, так что экстраполяция
интенсивности рассеяния на q = 0 становится ненужной, по крайней мере,
для не слишком больших молекулярных весов. Если детектор рассеянного
света придан к хроматографу, и концентрация раствора достаточно низка,
минимизируется необходимость экстраполяции на нулевую концентрацию.
Конечно, в этом случае нельзя получить радиус инерции и вириальный ко-
эффициент. Однако в этом случае определение молекулярного веса методом
светорассеяния можно завершить быстро.
Информация о структуре может также быть получена методом малоугло-
вого рассеяния нейтронов (МУРН, SANS) [119-121]. Рассеяние нейтронов
это следствие их взаимодействия с ядрами вещества. Отличия от светорассе-
яния связаны, в основном, с разницей в длинах волн, а не коэффициентов
преломления. Нейтронные сечения атомов обычно выражают через соот-
ветствующие функции корреляции. Интенсивность упругого когерентного
рассеяния пропорциональна преобразованию Фурье для функции парной
корреляции. Угловое распределение интенсивности когерентного рассеяния
дополняется интерференцией от структурных элементов, что позволяет
получить информацию о структуре и конформации. Разрешимы размеры
от 1 нм до 1 Jim, т. е. метод можно рассматривать как дополнительный по
отношению к электронной микроскопии.
Неупругое когерентное и некогерентное рассеяние пропорционально
пространственному и временному преобразованиям Фурье для функций
парной корреляции и самокорреляции соответственно. Аналогично динами-
ческому светорассеянию эти методы используются для изучения динамики
полимерных молекул [122-125].
126 -*v Глава 3. Характеристика структуры эластомеров
Рис. 4. Зависимости Цимма для раствора серийного поливинилхлорида в тетрагид-
рофуране: а — очищенный раствор, для которого получаются ожидаемые линей-
ные зависимости R (q)~\ б — исходный материал, для которого зависимости
R (q)~x криволинейны [117].
Сечения атомов углерода и кислорода по отношению к рассеянию ней-
тронов полностью когерентны. Большое некогерентное сечение и малое
когерентное имеют атомы водорода и обратное характерно для дейтерия.
Одним из преимуществ метода рассеяния нейтронов является возможность
использования меток (обычно замена водорода на дейтерий) для селектив-
ного контрастирования. Такие метки оказывают минимальное химическое
действие и позволяют определять < S > и Mw в тех случаях, когда неприменим
метод светорассеяния. Наличие пыли также не создает проблем при исполь-
IV. Архитектура цепи -*v- 127
зовании этого метода. Недостатком является высокая стоимость получения
образцов с метками.
Область, в которой метод рассеяния нейтронов дает информацию,
которую нельзя получить другими методами, это определение размеров
молекулярных цепей в блочных образцах. Образцы могут иметь толщину
порядка нескольких миллиметров, при этом рассеивается до 50% нейтро-
нов. Среднеквадратичный радиус инерции и Мш можно определить для
дейтерированных образцов полимера, смешанных с аналогом без меток.
Несмотря на наложение и взаимопроникновение молекул анализ Цимма
в этом случае не требуется. Единственное требование — одинаковые М
и МБР обоих изотопов. Независимый от модели анализ проводят с исполь-
зованием аппроксимации Гинье, справедливой для малых углов:
/(д) = /ое.гр(-|д2.^1 (9)
\ о )
Эластомеры обычно имеют гибкие цепи, так что можно ожидать, что
форм-фактор единичной цепи будет иметь Дебаевскую форму для Гауссового
клубка [см. ур. (6)] [111].
Р(9) = ^2-[еМ"^)_(1_«2/?в)] (10)
Результаты, полученные для полиизопрена, показаны на рис. 5 [126].
Прямые измерения методом рассеяния нейтронов подтвердили гипотезу
Флори об идеальном поведении полимерных молекул в блоке. Другая область
применения МУРН для эластомеров это исследование микроскопических
аспектов деформации сеток [127-129] и влияния наполнителей, таких как
технический углерод и силика, на деформацию сеток [130-132].
Два источника нейтронов это ядерные реакторы и ускорители частиц.
Оборудование для изучения рассеяния нейтронов в Северной Америке
имеется в ряде лабораторий*). Обычно получение допуска к работе включает
систему предложений о работе без права собственности на результаты и затем
исследования с правом собственности на платной основе. Такое оборудование
есть также в Европе, Азии и Австралии.
В дополнение к методам рассеяния, такие методы как криоскопия,
эбулиометрия и мембранная осмометрия дают абсолютные значения мо-
лекулярного веса независимые от наличия разветвлений. Методы, резуль-
таты которых зависят от размера молекул в растворе, такие как измерение
характеристической вязкости, эксклюзионная (или гельпроникающая)
хроматография (ЭХ, SEC или ГПХ, GPC), седиментация и т.д. требуют
*> National Institute of Standards and Techno/ogy, Oak Ridge National Laboratory, The Los
Alamos Neutron Science Center, Argonne National Laboratory, Chalk River (Canada) Neutron
Beam Laboratory, and the University of Missouri (Columbia).
128 -*v- Глава 3. Характеристика структуры эластомеров
Рис. 5. МУ PH от смеси дейтерированного и обычного полиизопрена (Mw = 10* г/моль),
построенная с использованием аппроксимации Гинье [ур. (9)]. Сплошная
линия — Дебаевский форм-фактор для Гауссовою клубка [ур. (10)] [126].
калибровки, и результаты зависят от геометрии молекул, в частности, от
наличия длинноцепных разветвлений (ДСР, LCB).
Еще один параметр, который можно отнести к параметрам, зависящим
от молекулярного веса, это относительная вязкость, определяемая как отно-
шение вязкости раствора Т| к вязкости растворителя Т|о. (Табл. 2)
Вязкость, пригодную для оценки М > 103 г/моль, можно определять, из-
меряя время течения через капилляр (диаметром * 1 мм), обычно под дейс-
Таблица 2. Вязкость раствора.
Вязкость Относи- тельная Удельная Приведенная Внутренняя Характери- стическая
Опреде- ление Пге|=П/П0 nsp = nrel-1 ^red “ Bsp/C ninh = ln(nre,)/C [П ] = limnn.d C->0
Т| = вязкость раствора, Т|о= вязкость растворителя.
твием силы тяжести. Широко распространены и доступны автоматические
приборы. Вискозиметры ротационного и колебательного типа используют
тогда, когда требуются одинаковые, хорошо определенные испытания или
малые скорости сдвига. Отношение двух вязкостей, Т|/г|0, называется отно-
сительной вязкостью; разность между этой величиной и 1 это «удельная»
IV. Архитектура цепи -* V 129
вязкость. Значения «удельной» вязкости, деленные на концентрацию
и экстраполированные на нулевую концентрацию (с —> 0), дают значения
характеристической вязкости [т|]. Ее связь с молекулярным весом [4]
[т|] = Ф^г2^3/2//И (И)
где (г2) среднеквадратичное расстояние между концами цепей и Ф — константа
(= 2,6.1021 если г выражено в см). Уравнение (И) можно переписать в виде
[т1]е = *М1/2 (12)
Значок 0 означает, что уравнение относится к 0-условиям. К постоянно
для достаточно больших молекулярных весов (~ 5.103 г/моль). Работа
в 0-условиях не всегда возможна из-за трудностей контроля температуры
и тенденции к осаждению вследствие ограниченной растворимости. Удобной
альтернативой является использование соотношения Марка-Хоувинка
[107, 108, 133, 134]
[п]=Ж (13)
где Мп< Mv< Mw (см. Рис. 3) и для каучукоподобных полимеров а может
принимать значения от 0,5 до 0,8. Как К, так и а зависят от полимера, рас-
творителя и температуры и могут быть определены эмпирически, используя
полимеры с известным молекулярным весом. Для хороших растворителей
величина [т|] меняется в пределах ± 30% и с достаточной точностью может
быть определена эмпирически [135, 136].
Удобный и весьма распространенный метод определения молекулярного
веса (< 107 г/моль) это эксклюзионная хроматография (ЭХ, SEC) или, как ее
обычно называют, гелъпроникающая хроматография (ГПХ, GPC) [9, 79-81,
137]. Разбавленный раствор полимера пропускают через колонну, заполненную
малыми (например, 5 jim) пористыми частицами. Поры имеют размер того же
порядка, как и молекулы полимера в растворе. При прохождении раствора через
колонну молекулы полимера диффундируют в поры и из пор. Доступность поры
определяется гидродинамическим объемом растворенных молекул. Маленькие
молекулы попадают во множество пор, поэтому для них время элюции из ко-
лонны больше. В действительности, поток может быть остановлен на несколько
минут при небольшом искажении хроматограммы. Измеряемая величина
это элюированный объем V, который является функцией размера молекул
в растворе. Основываясь на калибровке в идентичных условиях в идентичных
колоннах с использованием стандартных образцов с узким МВР, из данных по
Ve можно прямо получить значения молекулярного веса.
Эмпирически известно, что если различные полимеры имеют одинаковые
значения Ve в одной и той же колонне в идентичных условиях, то произве-
дение [т|]М постоянно при условии, что [т|] также определяли в одинако-
вых условиях (температура и растворитель). Это следует из соотношения
130 -*v Глава 3. Характеристика структуры эластомеров
Эйнштейна-Симхи, в соответствии с которым величина [т|]7И пропорцио-
нальна гидродинамическому объему. Гидродинамический объем зависит от
температуры и растворителя и грубо его можно считать равным половине
объема, рассчитанного из радиуса инерции^. Используя «универсальный
калибровочный метод» молекулярный вес полимера можно определить
с помощью известных стандартов для различных полимеров [138-140].
Такие стандарты молекулярных весов можно получить на коммерческой
основе и определить молекулярный вес изучаемого полимера по формуле
М„ =
*и\
(14)
где индексы U и S относятся к неизвестному и стандартному полимеру со-
ответственно.
Удерживаемый объем, мл
Рис. 6. Хроматограммы для смеси линейного и разветвленного сложного полиэфира,
полученные с использованием различных детекторов: светорассеяния (СР,
LS), вискозиметра (В, DV), рефрактометра (Р, DRI) [146]. В области больших
молекулярных весов (меньших значений удерживаемого объема) наибольшие
значения дает СР; чувствительность в области больших значений удержи-
ваемого объема максимальна для Р.
Разрешение калибровочной кривой ухудшается при больших и малых
молекулярных весах, так что большое различие в молекулярных весах
приводит к незначительной разнице Ve Заметим, что добавки, такие как
антиоксиданты, также могут вымываться, так что их можно спутать с низ-
комолекулярной фракцией полимера. Чтобы повысить разрешение конк-
ретного эксперимента, проводимого методом ГПХ, возможны различные
усовершенствования [107, 108, 141]. Проблемы разрешения минимизиру-
ются при использовании современных колонн, однако уменьшение размера
IV. Архитектура цепи -*v 131
частиц в колонне может приводить к сдвиговому разрушению образцов. Ге-
терогенность образца, например, в случае смеси или наличия разветвлений,
обостряет проблему разрешения метода. Детекторы, используемые в методе
ГПХ, это дифференциальные рефрактометры и детекторы в УФ и видимой
областях, сигнал которых пропорционален концентрации раствора. Исполь-
зуются также вискозиметры и детекторы светорассеяния, сигнал которых
зависит как от концентрации, так и от молярной массы (см. выше). Чтобы
облегчить разделение перекрывающихся значений удерживаемого объема
используют несколько детекторов (рис. 6.) [142-146]. Так, элюент из ко-
лонны проходит сначала через ячейку для измерения светорассеяния или
вискозиметр, а затем попадает в рефрактометр. Последний служит детекто-
ром концентрации и стоит обычно последним в ряду детекторов, поскольку
противодавление наиболее легко может нарушить его работу.
Потенциально существенная ошибка метода ГПХ связана с расширением
пиков, происходящим из-за неодинаковости структуры пор, и поведения
потока, проходящего через колонну. Разрешение колонны можно охарак-
теризовать отношением размывания элюированного объема для монодис-
персного образца к пику объема удерживания. При повторном пропускании
образца через тот же прибор значения элюированного объема для образцов
с экстремально широким МБР могут сдвигаться на величину, которая со-
ответствует до 50% от значений молекулярного веса. Тип расширения пика
связан с диффузией, наличием смеси, влиянием концентрации, иногда,
частично, с зависимостью гидродинамического объема от концентрации.
Другой источник ошибок в случае образцов очень больших молекулярных
весов (например, М~ 106г/моль или больше) это деструкция (разрыв цепей)
в результате прохождения через колонну [141, 147].
Классический метод фракционирования, связанный с растворимостью
или нерастворимостью, применительно к МБР и композиционному распре-
делению основан на различиях в растворимости разных частей образца. Это
эмпирический и длительный метод, при котором необходимо характеризовать
разделенные фазы («срезы») по мере их появления. Однако фракционирование
можно проводить, используя минимум оборудования, и для многих полимеров
это единственная возможность получения узких фракций по молекулярному
весу и составу, которые могут служить стандартами для хроматографии или
вискозиметрии. Метод фракционирования путем элюирования (вымывания)
при подъеме температуры (ТПФ, TREF), рассмотренный выше, представляет
собой автоматизированный вариант фракционирования.
Термин фракционирование в потоке, находящемся в поле (ФПП, TFFF)
[107, 108, 148-150] обозначает целый набор хроматографических методов,
позволяющих получать однофазные образцы при потоке через узкие каналы.
В принципе, ФПП может давать абсолютные значения молекулярного веса,
но обычно применяют калибровку, аналогичную используемой при работе
методом ГПХ. Внешнее поле должно быть приложено перпендикулярно на-
132 -*v- Глава 3. Характеристика структуры эластомеров
правлению ламинарного потока раствора в канале. Параболический профиль
скоростей потока в канале приводит к разделению молекул в соответствии с их
положением относительно стенок канала. Молекулы, имеющие более слабое
взаимодействие с полем, остаются дальше от стенок, поэтому элюируют быстрее.
В качестве внешнего поля может быть использовано электрическое, магнитное
или температурное. При фракционировании в потоке, находящемся в темпе-
ратурном поле [88-91], устанавливается температурный градиент в десятки
градусов, вызывающий миграцию растворенного полимера, конкурирующую
с броуновским движением, и аккумуляцию его на холодной стенке. Коэффици-
ент переноса, определяющий термическую диффузию, зависит от химического
состава и молекулярного веса. Для данного растворенного вещества меньшие
молекулы, имеющие больший коэффициент диффузии, мигрируют к центру
и потому элюируют быстрее. В дополнение к разделению, основанному на разме-
ре молекул, этот метод может быть использован для измерения коэффициентов
диффузии молекулярных цепей [151, 152].
Рис. 7. Собственное время релаксации (пропорциональное вязкости расплава!
1<ис-1,4-полиизопрена в зависимости от молекулярного веса. Пропорцио-
нальность по Роузу при Mw~ 103 г/моль сменяется зависимостью от Af^3’7 [158а].
ФПП это метод, не требующий больших затрат времени и обеспечиваю-
щий хорошее разрешение. Одно из преимуществ метода, что не требуется
субстрат и канал достаточно большой, и поэтому не засоряется гелем. Поэто-
IV. Архитектура цепи 133
му он чаще всего используется для полимеров, содержащих гель, и может
применятся для образцов с молекулярной массой до 107 г/моль. Существуют
и серийные приборы.
Один из видов масс-спектрометрии (MALDI-TOF) также дает абсолютные
значения молекулярного веса. Это времяпролетная масс-спектрометрия,
идентифицирующая частицы, полученные десорбционной лазерной иони-
зацией в специальных средах [153-155]. Образец диспергируют в среде,
абсорбирующей УФ-излучение (например, транс-коричная кислота или
2,5-дигидроксибензойная кислота). Лазерное УФ-облучение индуцирует
испарение ионизированных полимерных цепей, которые затем идентифи-
цируются с помощью времяпролетного масс-спектрометра. Для измерения
этим методом пригодны образцы со сравнительно узким МВР. Имеются
соответствующие серийные приборы. Используют также альтернативные
ионизационные методы, такие как электроспреевая ионизационная масс-
спектрометрия, когда ионизация проводится в аэрозоле. Этот метод с успехом
применяется для некоторых полимеров [156]. Оба метода могут применяться
для анализа и полидисперсных образцов и смесей полимеров [157].
В то время как седиментация основана на силе тяжести, которая служит
движущей силой для разделения, вращение образцов (до 70 000 рад/мин) ус-
коряет процесс [9]. При ультрацентрифугировании для определения концент-
рационных профилей используют оптические приборы [158]. При равновесном
центрифугировании необходимы меньшие скорости вращения (<104 рад/мин).
При этом могут быть определены абсолютные значения средневесового моле-
кулярного веса (> 5.102г/моль). Однако время равновесия может достигать
дней, поэтому метод используется редко. В биологических исследованиях ис-
пользуется неравновесная седиментация, в этом случае результаты получаются
значительно быстрее, но они менее точны. Ультрацентрифугирование - один из
немногих методов, позволяющих получить значения z-среднего молекулярного
веса. Этот метод также позволяет получить информацию о гомогенности образца.
Существуют серийные центрифуги для аналитических исследований.
Методы, рассматривавшиеся выше, требовали использования рас-
творов полимеров. Нерастворимые образцы можно исследовать непо-
средственно с помощью измерения вязкости расплавов. При достаточно
низких скоростях сдвига (по сравнению со скоростями броуновского
движения полимерных молекул) вязкость расплава не зависит от скорости
сдвига («Ньютоновское течение»), Т| = Т|ои соотношение вязкость—Mw
при заданной температуре позволяет получить достоверные значения
Mw (на рис. 7. представлены соответствующие данные, полученные из
диэлектрических измерений). Учитывая высокий молекулярный вес
эластомеров, наибольшая трудность заключается в достижении условий,
соответствующих нулевой скорости сдвига. При комнатных температурах
Т|о > 107 Па.с. Заметим, что «холодное течение» невулканизованного кау-
чука обычно занимает неделю или больше. Очевидный путь уменьшить
Таблица 3. Методы характеристики молекулярного веса и его распределения
Метод Измеряемая величина Принцип работы Пределы г/моль
Мембранная осмометрия Среднечисленный молекулярный вес, вириальный коэфициент Осмотическое давление вследствие диф- фузии растворителя через мембрану, неп- роницаемую для полимера
Осмометрия давления пара Среднечисленный молекулярный вес Понижение давления пара < 5 104
Криоскопия Среднечисленный молекулярный вес Снижение температуры замерзания < 5 104
Эбулиометрия Среднечисленный молекулярный вес Повышение температуры кипения < 104
Рассеяник света (PC, LS) Среднечисленный молекулярный вес, вириальный коэфициент, радиус инер- ции Интенсивность рассеяния света пропор- циональна молекулярному весу раство- ренного вещества. Угловое распределение связано с размером частиц > 103
Рассеяние нейтронов Среднечисленный молекулярный вес, ви- риальный коэфициент, радиус инерции Интенсивность рассеяния пропорциональ- на поперечному сечению ядра
Характеристическая вязкость Характеристическая вязкость. Среднечис- ленный молекулярный вес по вязкости Вязкость раствора зависит от молекуляр- ного веса полимера > 103
Хроматография запрещен- ных размеров (ХЗР, SEC) или гель-проникающая хро- матография (ГПХ, GPC) Молекулярный вес и его распределение Проникновение полимера из потока рас- твора в пористую стационарную фазу < 107
Фракционирование потока в поле (ФПП, FFF) Молекулярный вес и его распределение Внешнее поле влияет на разделение рас- творенного вещества.в потоке < 107
Ультрацентрифугирование Среднечисленный молекулярный вес Градиент концентраций в сильном гравитаци- онном поле связан с молекулярным весом > 5 102
Седиментация Молекулярный вес Седиментационный коэффициент Скорость диффузии в сильном гравитаци- онном поле связан с молекулярным весом > 5-102
Вязкость расплава Динамическая и стационарная вязкость Эмпирическая корреляция вязкости и Мш -
Глава 3. Характеристика структуры эластомеров
IV. Архитектура цепи -* V 135
вязкость это проводить измерения при высоких температурах. Однако,
особенно для ненасыщенных каучуков, за время достижения равновесия
может проходить деструкция каучука. Ситуация ухудшается если полимер
имеет длинноцепные разветвления.
Б. Длинноцепные разветвления
Длинноцепные разветвления (ДЦР, LCB), которые определяют
как разветвления, молекулярный вес которых всего в несколько раз меньше
молекулярного веса между зацеплениями, вполне обычны для каучуков. Их на-
личие, прежде всего, приводит к росту вязкости; их вводят в некоторые серийные
каучуки для уменьшения хладотекучести (т. е. ползучести при хранении при
комнатной температуре). Наличие ДЦР в каучуке может приводить к необхо-
димости использовать полимеры с низким Мп для сохранения вязкости, обес-
печивающей необходимый уровень технологических свойств. Однако наличие
множества концов цепей может приводить к ухудшению свойств вулканизатов,
особенно таких, как теплообразование и прочность. Наличие разветвлений
изменяет и другие, кроме вязкости, свойства полимеров, что может служить
способом характеристики степени разветвленности [159]. Например, природа
разветвлений влияет на геометрию и размер молекулярных цепей (рис. 8.).
Наличие разветвлений приводит к уменьшению характеристической
вязкости [см. ур. (И)]. Это уменьшение характеризуется отношением со-
ответствующих среднеквадратичных радиусов инерции для разветвленного
и неразветвленного полимеров, имеющих одинаковые Мш.
Для полимеров с ДЦР параметр g меньше единицы и иногда может быть
определен как отношение среднеквадратичных значений Sgbr/SgUn. Можно
непосредственно провести расчет g для заданной структуры разветвлений,
но трудно определить радиусы инерции экспериментально. Характеристи-
ческую вязкость легко измерить, но соотнести g к g', а, следовательно, и к
структуре разветвлений, достаточно сложно. Pac4eTg' требует привлечения
теории разбавленных растворов с рассмотрением типа разветвлений в явной
форме. Альтернативный путь это оценка g с использованием модельных
полимеров. Прямое определение абсолютного уровня разветвленности по
данным характеристической вязкости невозможно, т. к. результаты зависят
от природы разветвленности, включая функциональность узлов и их архи-
тектуру (случайная, звездообразная, дендритная и др.). Однако относитель-
ные величины g могут отражать относительную степень разветвленности.
Прямое сопоставление величин g может быть сделано с использованием
радиусов инерции, определенных методом светорассеяния [см. ур. (6)].
136 -*v- Глава 3. Характеристика структуры эластомеров
Экспериментально определенные в 0-растворителях значения добычно сов-
падают с расчетными за весьма примечательными исключениями [160].
Гомополимеры
Звездообразный
Гребеневидный
Блоксополимеры
Рис. 8. Пространственная структура полимерных цепей зависит от природы и типа
архитектуры молекул [159а].
Для обнаружения разветвлений можно использовать гель-проникаю-
щую хроматографию (ГПХ) [161-170]. Одновременное использование
детекторов, дающих абсолютные значения молекулярного веса, позволяет
оценить ДЦП [171]. Можно также использовать постоянство произведения
[т|] М для образцов с фиксированной величиной элюированного объема Ve.
При этом данные ГПХ получают для гомогенной фракции образца с неиз-
вестными разветвлениями, и рассчитывают отношение характеристической
вязкости такого образца к значениям [т|] для линейного полимера с таким
же Ve. Полученные таким образом значения Мсопоставляют с значениями,
рассчитанными непосредственно из данных по [т|]. Нужно помнить, что
использование такого метода требует, чтобы фракции были гомогенны.
Эмпирическое соотношение для образцов с широким ММР дает среднее
значение
// _ ^v,itn х ( Mw
^w,LS
(16)
IV. Архитектура цепи -•V 137
где Мщ — молекулярный вес, определенный из данных по [т|] в предположе-
нии, что полимер линейный, MwLS — значения Mw, полученные методом све-
торассеяния, и значение (Мш/Mv) Gpcполучено из величины элюированного
объема в предположении отсутствия разветвлений. Например, для стандарта
разветвленного полиэтилена (NBS 1476),g" = 0,4 при использовании значе-
ния [т|] = 1,04 дцл/г, определенного для раствора в декалине (Мш/Mv) Gpc =
= 1,16, 125 000 г/моль. Количество разветвлений, соответствующих па-
раметру разветвленности 0,4, зависит от природы разветвлений. Для случай-
ных тетрафункциональных разветвлений, значениеg = 0,4 при заданном Mw
соответствует 20 концам цепей на средневесовую цепь.
Усовершенствованный метод ГПХ предполагает одновременное под-
ключение аппаратуры для измерения светорассеяния и вязкости (см. выше,
раздел IV А). Калибровка, дающая возможность измерять зависимость про-
изведения [т|]Мот времени элюирования для конкретных полимерных об-
разцов, вместе с измерениями [т|] Вг и MwBr позволяет рассчитать значение# —
фактора как отношение измеренной характеристической вязкости к значе-
нию [т|], рассчитанному для соответствующего линейного полимера (т. е.
полимера с тем же временем элюирования).
Если имеются хорошо охарактеризованные образцы, для оценки разветвлен-
ности можно использовать калибровку вязкоупругих свойств неразбавленных
образцов. Разветвленность влияет на вязкость расплава, так же как и на модуль
и на другие вязкоупругие свойства. Наличие разветвлений низкого молекуляр-
ного веса, ниже некоторого значения, которое всего в несколько раз отличается
от молекулярного веса между зацеплениями, приводит к уменьшению вязкости
расплава, т. к. в этом случае размеры разветвленных молекул меньше размеров
неразветвленных при одинаковом молекулярном весе. Однако присутствие
достаточно длинных разветвлений, которые сами могут создавать зацепления,
приводит к росту вязкости и вязкость разветвленного полимера становится
больше вязкости его линейного аналога с тем же Mw [159,172-174].
Для таких каучуков, как НК (или синтетический 1{ис-1,4-полиизопрен)
[175], полибутадиен (как 1,2-,таки 1,4-изомеров) [176], наличие ДЦРпри-
водит к увеличению основного времени релаксации и влияния температуры
на вязкость. Поэтому, сравнивая кажущуюся энергию активации, или, что
более обычно, если уравнение Аррениуса несправедливо, отношение времен
релаксации неизвестного полимера и линейного образца того же полимера
позволяет сделать вывод о ДЦР (рис. 9). Для таких полимеров, как цис-1,4-
полиизопрен (см. рис. 7), у которых дипольный момент параллелен моле-
кулярной цепи, диэлектрические измерения температурной зависимости
собственного времени релаксации позволяют оценить наличие ДЦР.
Заметим, что не для всех полимеров наличие ДЦР изменяет температурную
зависимость свойств. Например, для линейного и разветвленного полиизобути-
лена [174,177],полидиметилсилоксана [178] и гидрированного полибутадиена
[176] наличие ДЦР не изменяет температурную зависимость их свойств.
138 -*v* Глава 3. Характеристика структуры эластомеров
В. Гель
В случае наличия геля, нормальное течение невозможно
(материал, по-существу, является твердым). В этих случаях может быть
использован золь-гель анализ. Если в исследуемом образце создать задан-
ную, контролируемую сетку поперечных связей (например, с помощью
пероксида или ионизирующего облучения), исходный молекулярный вес
можно определить по содержанию геля. Но это требует знания скоростей
сшивания и разрыва цепей. Если сшивание продолжается после точки
гелеобразования, соотношение геля и золя непосредственно связано
с МВР образца, что, в принципе, может быть использовано для опреде-
ления МВР [179].
Рис. 9. Обратные температурные зависимости главных времен релаксации для
образцов разветвленного 1,4-полиизопрена (трехлучевые звездообразные
разветвления) разного молекулярного веса Mw (г/моль): 284 000 (S284).
342 000 (S342) и 854 000 (S854), нормализованные относительно времени
релаксации линейного цис- 1,4-полиизопрена. Избыточная энергия акти-
вации в результате присутствия ДЦР составляет 400 J на моль зацеплений,
образованных разветвлениями [175].
V. Стеклование и другие физические релаксационные процессы 139
Сетки с плотностью только немного больше, чем в точке гелеобразования,
имеют минимальную прочность. Поэтому при проведении золь-гель анализа
необходимо избегать разрушения образовавшейся сетки. Если содержание геля
экстраполируется на отрицательные значения плотности сетки, необходимые
для образования геля, это значит, что исходный материал содержал гель. Хотя
течение материала, содержащего гель, не является линейным вязкоупругим
течением, слабосшитый каучук можно вальцевать и проводить смешение в за-
крытых смесителях, возможно также измерение его вязкости по Муни. К тому
же в растворах полимеров может проходить ассоциация, в результате которой
образуются достаточно устойчивые агрегаты [180, 181]. Это обычное явление
для полимеров, способных к кристаллизации (например, этиленпропиленовые,
поливинилхлорид, полиуретаны). Обратимые ассоциаты, которые обычно изме-
няются при изменении растворителя или температуры, существенно отличаются
от геля, образуемого ковалентными поперечными связями.
V. СТЕКЛОВАНИЕ И ДРУГИЕ ФИЗИЧЕСКИЕ
РЕЛАКСАЦИОННЫЕ ПРОЦЕССЫ
При измерении динамических свойств полимеров (напри-
мер, методами динамической или диэлектрической спектроскопии или
динамического светорассеяния или динамического рассеяния нейтронов)
наблюдается максимум поглощения энергии, проявляющийся при измене-
нии временных параметров эксперимента (например, частоты приложен-
ного механического или электрического напряжения). В пределах малых
изменений отклика системы (например, деформации), когда поведение
системы можно считать линейным, релаксация прямо отражает равновесное
броуновское движение. Такое соответствие следует из флуктуационно-дис-
сипационной теории, первоначально разработанной для объяснения тепло-
вого шума в проводниках электричества. Гигантские размеры полимерных
молекул обеспечивают огромное количество степеней свободы, поэтому
движение занимает широкую область на временной шкале (рис. 10). Самым
большим временам соответствуют перемещения центров масс молекул,
и этот процесс называют конечной или глобальной динамикой. При мень-
ших временах релаксационные процессы отражаютдвижение все меньших
частей молекулярных цепей. Плато высокоэластичности отражает наличие
нестационарной сетки зацеплений, распад которой приводит к возникнове-
нию пика модуля потерь (а также диэлектрических потерь для полимеров
с дипольным моментом, параллельным цепи) [182]. При частотах, лежащих
за пределами плато высокоэластичности, зацепления не влияют на движения
цепи. В зоне размягчения модуль сильно зависит от частоты, и поведение
системы описывается Роузовской динамикой. (Теория Роуза первоначально
бала разработана для разбавленных растворов полимеров, но с небольшими
изменениями может быть применена к расплавам, не содержащим зацеп-
140 Jlr Глава 3. Характеристика структуры эластомеров
лений). При более высоких частотах, вблизи температуры стеклования,
в релаксационный процесс включаются участки цепи, слишком короткие,
чтобы поведение было гауссовым (т. е. расстояние между концами для таких
участков не описываются гауссовым распределением). Такие «суб-роузов-
ские моды движений» при высоких частотах непосредственно переходят
в локальную сегментальную динамику.
динамика клетки
к
S
X
ф
р?
X
ф
Сб
р?
ф
Он
X
л
X
у
ф
плато §
высокоэластичности *
6
Igt (с) -----------------------------►
Рис. 10. Схема областей движения и соответствующих частот для цис- 1,4-полиизо-
прена (71/^ = 5.105 г/моль) при -40 °C.
Локальная сегментальная динамика связана с переходом в стеклооб-
разное состояние при температуре стеклования Т . Только в переходной
области вязкоупругого спектра, используя соответствующие методы, можно
одновременно, при одной и той же температуре, измерять моду движе-
ния, связанную с молекулами и локальную сегментальную моду. Т. к. обе
моды имеют различную температурную зависимость [183-185], в области
стеклования наблюдается нарушение принципа температурно-временной
суперпозиции. Поэтому обобщенную кривую для молекулярной динамики
можно построить так, что она будет проходить от конца области размягчения
до конечной релаксации; возможно и построение обобщенной кривой для
локальной динамики. Однако в области размягчения форма вязкоупругого
спектра меняется с температурой (рис. И).
Все времена релаксации зависят от температуры и давления, однако только
глобальные движения (вязкость, конечное время релаксации, стационарная
обратимая податливость) являются функциями Mw (и в меньшей степени
МВР). Примеры различных динамических мод для цис- 1,4-полиизопрена
представлены на рис. 10. При частотах выше частот, соответствующих локаль-
ной сегментальной релаксации, или при температурах ниже Т , особенно на
спектрах диэлектрической релаксации, можно наблюдать вторичные (мелкомас-
штабные) релаксационные процессы. В полимерах многие из этих вторичных
(мелкомасштабных) процессов связаны с движением боковых подвесок. При
этом наиболее медленные вторичные релаксационные процессы рассматривают
V. Стеклование и другие физические релаксационные процессы 141
как процессы Джохари- Гольдштейна, связанные с движением всех атомов,
входящих в повторяющиеся звенья цепи или центров молекул для материалов
с малымМ . Релаксация Джохари-Гольдштейна это предшественник основного
физического перехода — стеклования.
Рис. 11. Обобщенная кривая для действительной (модуль запаса, сплошные значки)
и мнимой части (модуль потерь, незаполненные значки) комплексного модуля для
для цис-1,4-полиизопрена (Mw = 5.105 г/моль); температура приведения 7^=
= -10 °C. В области размягчения можно проследить за изменением хода кри-
вой модуля потерь. Эти изменения хорошо видны на частотных зависимостях
тангенса угла потерь, которые имеют четко выраженный пик; при изменении
температуры эксперимента от -66 до -48 °C высота пика уменьшается [183].
Tg эластомеров ниже комнатной температуры, обычно ниже -30 °C. Иначе
эластомеры при комнатной температуре не имели бы гибкости, связанной с их
высокоэластичностью. Температура стеклования это переход второго рода*\
т. к. в отличие от плавления и кипения, на температурной зависимости объема
нет разрыва при Т , разрыв есть только на температурной зависимости изме-
нений объема (т. е. коэффициента термического расширения, рис. 12) [186].
Экспериментально Tg это просто температура, при которой локальные сегмен-
тальные движения становятся медленными по сравнению с временной шкалой
эксперимента. Таким образом, Tg это функция как частоты воздействия, так
Дискуссию о природе стеклования см. в книгах: Бухина М.Ф., Курлянд С.К. Морозо-
стойкость эластомеров. М.: Химия 1989.176 с. и Bukhina M.F.,Kurlyand S.K. Low Temperature
Behavior of Elastomers. Leiden — Boston: ///VSP///. 2007. 187 pp.
142
—Глава 3. Характеристика структуры эластомеров
и временных параметров конкретного наблюдаемого релаксационного процесса.
Существует ряд свойств, по изменению которых можно определять Т. Присут-
ствие сомономеров в главной цепи приводит к изменению Т, изменяется Tg
и в зависимости от состава эластомерного материала. Возможны как пластифи-
кация (снижение Tg), так и антипластификация (повышение Tg).
Рис. 12. Температурная зависимость удельного объема полистирола (Mw= 1,1.10* г/моль)
при различных давлениях. На вставке представлена зависимость от дав-
ления [186].
При неизотермических измерениях желательно достигать Tg из равно-
весного состояния (т. е. при охлаждении), т. к. свойства в стеклообразном
состоянии могут меняться в результате физического старения. Физическое
старение относится к процессам структурной релаксации, протекающим при
температурах существенно ниже Т , когда вклад локального сегментального
движения отсутствует. Оно связано с медленным достижением равновесных
значений плотности, вследствие чего меняются физические свойства, включая
смещение температуры стеклования. О мере такого отклонения от равновесия
можно судить по фиктивной температуре 7j, которую называют также структур-
ной или равновесной температурой. это температура, при которой энтальпия
вещества приняла бы равновесное значение. Т. е., по разнице между фиктивной
и действительной температурами можно судить о структурной релаксации.
Наиболее распространенный метод определения Tg это измерение тепло-
емкости (С^). Обычно для измерений используют метод дифференциальной
сканирующей калориметрии (ДСК, DSC). При изменении температуры образца
V. Стеклование и другие физические релаксационные процессы -IV 143
с заданной скоростью измеряют тепловой поток. Более старый и менее прак-
тичный вариант это метод дифференциального термического анализа (ДТА,
DTA), при котором задается тепловой поток, а измеряется изменение темпера-
туры. В обоих методах разрыв в дифференциальном отклике свидетельствует
о термическом переходе. В методе ДСК величину Tg фиксируют обычно по
середине изменений теплоемкости. Эго почти эквивалентно альтернативному
определению, когда фиксируют температуру пересечения экстраполяции базо-
вой линии с прямой, соответствующей тангенсу угла максимального наклона.
[65]. Измерения методом ДСК рекомендуется делать при охлаждении, чтобы
поддерживать равновесие. В противном случае физическое старение может
привести к появлению эндотермического пика при стекловании. В этом случае
фиктивная температура более информативна, чем кажущаяся температура
перехода. Графический метод определения Tf показан на рис. 13.
Рис. 13. Кривая ДСК для вулканизованной эпоксидной смолы. Чтобы определить
фиктивную температуру следует построить параллелограмм на низкотем-
пературной стороне эндотермического пика с горизонтальными сторонами,
параллельными измеренной теплоемкости. Начало пика определяет высо-
котемпературную границу параллелограмма, а нижнюю (т. е. Tf) выбирают
так, чтобы площадь параллелограмма была равна площади пика [186а].
Модулированный (или переменный) ДСК (МДСК, MDSC) [187-189] это
метод, в котором обычная постоянная скорость нагрева (или охлаждения)
модулируется переменной составляющей. Измеряемый тепловой поток раз-
144 -* ъ- Глава 3. Характеристика структуры эластомеров
деляется на две компоненты: обратимую (независимую от времени), которая
пропорциональна теплоемкости, и необратимую (зависящую от времени),
появляющуюся в результате кинетических эффектов. Это позволяет разде-
лить обратимые тепловые процессы, такие как стеклование, и необратимые,
такие, как релаксация энтропии и кристаллизация. МДСК позволяет фикси-
ровать очень слабые переходы, которые невозможно определить с помощью
обычного ДСК, проанализировать мультиплетные эндотермы и отделить
кристаллизацию, существовавшую в образцах перед экспериментом, от
возникшей в процессе измерения. Метод позволяет также получить точные
данные о теплоемкости в процессе квази-изотермических экспериментов
VI. МОРФОЛОГИЯ
Анализ морфологии эластомеров включает не только харак-
теристику молекулярной структуры самого полимера и ингредиентов, но
может распространяться и на надмолекулярные размеры. Такая структура
включает структуру сетки наполнителя (см. гл. 8), фазовую морфологию сме-
сей полимеров и термопластичных эластомеров и любых агрегатов (класте-
ров) в каучуках, содержащих ионные группы (например, в карбоксилатных
каучуках). На рис. 14 представлен обзор экспериментальных методов. Ниже
обсуждены некоторые избранные аспекты морфологии каучуков.
А. Ориентация
Высокоэластическая деформация возникает в результате
ориентации сегментов молекулярной цепи, и степень такой ориентации
определяет механические свойства. Наиболее простой способ количествен-
ной характеристики ориентации это измерение двойного лучепреломле-
ния, определяемого как разница показателей преломления в двух взаимно
перпендикулярных направлениях, Ап = пц- Двойное лучепреломле-
ние зависит как от анизотропии звеньев цепи, так и от их ориентации.
Дп = Дп0/2(ф) (17)
В случае двойного лучепреломления ориентационная функция это второй
полином Лежандра от угла ориентации ф:/2(ф) = <3 соз2ф - 1 >/2, где скобки
< > означают усреднение по всем цепям. Двойное лучепреломление идеально
ориентированной цепи Дп0, (которое иногда называют характеристическим
двойным лучепреломлением) можно рассчитать из оптической поляризу-
емости звеньев цепи, на практике измерения двойного лучепреломления
дают только относительные величины/2(ф). Тот факт, что напряжение о
пропорционально тому же самому ориентационному фактору, обуславливает
справедливость соотношения
Дп = Са
(18)
VI. Морфология -IV 145
о.оос 1^ 0.001 Диамет /10 0.01/ р частиц, 1 юг олл микропы ( I03 1/1( [д)/А ,4 1< (1 мм) ) 100 1,000 10,000
Эквивалентные размеры |б°|4р|20|1;2|в|з номер сита (США)
Обычные атмосферные дисперсоиды — г. Обл [ака Дождь *
kuM и ту ман Изморось
Типичные частицы и газообразные дисперсоиды ОКИ Н-Ви Техническ * углерод I Взвесь част & цинка в лоидна^ илика ж — Атмосф русы-Н Конта ийсерноки * । испа^ L Kpacil I пигме> иц . озд^хе йнс И Щелоч ДЫ» Г ерная пыл ктные од слотные р ^ения । 1и, гты | | Порошки 'ектофунги! [змельченн |— слюда , 1НОЙ t | L MyJ при раз ь Бактерии- Удобр •ение IK
(мельченн! 'аспыленнь! । уголь 1ИДОВ 1ЭЯ р Пыльца-»* са и 1моле яй извести! 1Й
Фильтры
Полимерные структуры енты (латист нЛЧ (<S2>1 ический бок ( «огИм/ _ Максим при р Крис альная дли астяжении талличесю ламели Н Лап дельные фг ке 1на »| 'е н жсы ► Сфере 1зы в смесь >литы ► 1
(крис и а — (омены г талличесю морфные)
Методы анализа Широ1 дифр< Малоугло «угловой, 1кция эле'н 1 „ н— вой рентге не 1 рентген, тронов 1 Н н, дифрак! 1 фракция г (йтронов । £ 1ия электр< 1 леторассея 1 1 )НОВ Невооруж гла; :енный _ —►
Электр микро* юнная * ( скопия 1 'оптическг микроскоп 1Я _ ИЯ
Рис. 14. Характеристика частиц.
146 Глава 3. Характеристика структуры эластомеров
где С оптический коэффициент напряжения [190]. Это соотношение исполь-
зуется для определения напряжения полимеров при течении. Поскольку кон-
станта С не зависит от густоты сетки, соотношение (18) может использоваться
и для сеток, но оно может не выполняться при очень больших деформациях,
что связано с негауссовым поведением молекулярных цепей (предельная
растяжимость). В области перехода в стеклообразное состояние уравнение
(18) также может не выполняться из-за различных вкладов в величину
Дп0 разных мод движений, связанных с движением цепи и локальных сег-
ментальных [191]. Величина двойного лучепреломления непосредственно
связана с деформацией, а не с напряжением, поэтому соотношение (18)
неприменимо в случае обратимой ползучести (крипа) [192], когда напря-
жение равно 0, а деформация эластомера сохраняется. Как видно на рис. 15,
то же явление имеет место для двойных сеток, образованных при сшивании
предварительно сшитых и ориентированных образцов [193, 194].
Рис. 15. Фотографии образцов натурального каучука, с двойной сеткой (а, в)
и обычного вулканизата (б). Напряжение в обоих образцах равно нулю,
но образец с двойной сеткой, прозрачен в скрещенных поляроидах из-за
наличия внутренней ориентации (в). Соотношение между напряжением
и двойным лучепреломлением не выполняется, как и в случае обратимой
ползучести несшитого каучука.
Для частично-кристаллического материала двойное лучепреломление
это сумма Ап от ориентированной аморфной фазы и от кристаллических
областей. Имеется также вклад двойного лучепреломления формы, возни-
кающего вследствие искажения световых волн при изменении показателя
преломления на границе между аморфной к кристаллической фазами.
Другой стандартный метод измерения ориентации полимеров это инф-
ракрасный дихроизм. Это отношение величин абсорбции ИК-излучения
с взаимно перпендикулярной поляризацией. ИК дихроизм дет ту же информа-
цию, что и двойное лучепреломление видимого света [т. е./2(ф) ]. Основное
преимущество этого метода — селективность по отношению к химическому
составу. Он позволяет определить ориентацию специфических групп и ком-
понентов в матрице [195-197]. Такой анализ требует знания ориентации
моментов колебательных переходов к оси молекулярной цепи. Естественно,
следует избегать полос, где налагаются разные колебания. В образец может
VI. Морфология -IV 147
быть добавлено небольшое количество дейтерированного полимера, чтобы
можно было исследовать образцы большей толщины, еще удовлетворяющие
закону Беера (поглощательная способность < * 0,7). Это создает возможность
одновременно измерять напряжение в образце [198]. Дополнительно для
характеристики ориентации используют метод поляризационной Раманов-
ской спектроскопии [199-201].
При использовании поляризованного флуоресцентного излучения об-
разец освещают поляризованным светом и измеряют интенсивность эмис-
сии при поляризации параллельно и перпендикулярно падающему свету.
Эксперимент позволяет получить как/2(ф), так и 4-й полином Лежандра
/4(ф) = <35 соз4ф - 30 сов2ф + 3>/8, который представляет собой функцию,
уменьшающуюся с ф сильнее, чем /2(ф). Поэтому ее вклад наиболее заме-
тен в том конце функции распределения ориентации, который относится
к большим ориентациям. Для одноосных распределений этих двух функций
достаточно, чтобы адекватно описать ориентацию [202].
Еще один метод для характеристики ориентации это метод ЯМР на дей-
тронах. Дейтроны имеют спин с квантовым номером 1 и поэтому ядерный
квадрупольный момент. Взаимодействие между квадрупольным моментом и
двумя возможными ЯМР-переходами (результат наличия трех Зеемановских
уровней для ядер со спином 1) приводит к изменению энергетического уров-
ня двух переходов, в результате в спектре ЯМР образуется дублет. Разделение
составляющих дублета пропорционально величине/2(ф), (которая зависит
от ориентации осей химических связей дейтрона относительно направления
внешнего магнитного поля); что позволяет прямо определить ориентацию.
Ориентация сшитого эластомера прямо отражает конфигурационную эн-
тропию и конформационную внутримолекулярную энергию молекулярных
цепей. Однако, как впервые было показано с помощью ЯМР на дейтронах для
силиконового каучука [203,204], незакрепленные в сетку молекулы-зонды
и часть молекулярных цепей оказываются ориентированными благодаря
их присутствию в ориентированной матрице. Этот эффект «нематической
связи» вызван межмолекулярными взаимодействиями (исключая объемное
взаимодействие и анизотропные силы) [195,205]. Ориентация эффективна
только локально, поэтому ее вклад в величину напряжения пренебрежимо
мал [206], и цепи сохраняют свои изотропные размеры [204].
Широкоугловое (под большими углами) рентгеновское рассеяние так-
же может быть использовано для оценки ориентации, но этот метод более
информативен для частично кристаллических полимеров. В принципе,
методы рассеяния позволяют определить все ориентационные функции,
однако экспериментальные возможности ограничивают их применение
для определения /4(ф). С использованием мощных синхротронных источ-
ников рентгеновского излучения можно измерять ориентацию в процессе
деформации. Эти возможности были использованы для изучения поведения
аморфной фазы НК при растяжении (рис. 16) [207, 208].
148 Глава 3. Характеристика структуры эластомеров
Рис. 16. Общая ориентированная часть, кристаллическая фракция и ориентированная
аморфная фракция вулканизованного серой образца НК, медленно рас-
тянутого при комнатной температуре. Верхние кривые соответствуют рас-
тяжению, а нижние — сокращению. При деформации > 200% начинается
ориентационная кристаллизация, приводящая к уменьшению ориентации
аморфной фазы [208].
Б. Смеси
Хотя многие каучуки могут быть смешаны с образованием
систем, кажущихся гомогенными, основная часть таких смесей имеет морфо-
логию, соответствующую существованию раздельных фаз. Чтобы получить
эластомер с хорошими свойствами достаточно достижения хорошего дис-
VI. Морфология 149
пергирования компонентов. Настоящая термодинамическая совместимость,
предполагающая совмещение компонентов на сегментальном уровне, очень
редка, но важно определить ее наличие. Размеры фаз в гетерогенных смесях
зависят от смешиваемости компонентов и, в меньшей степени, от условий
смешения. Распределение частиц по размерам можно определить, используя
электронный микроскоп и программное обеспечение для анализа изображе-
ний [209, 210]. При плохом контрасте изображения возможна недооценка
размеров, связанная со слабой различимостью периферийных участков.
При исследовании микротомных срезов ошибки могут возникать из-за того,
что толщина срезов может быть меньше размера частиц [211]. В принципе,
размеры дисперсной фазы можно определить методом малоуглового рассея-
ния рентгеновских лучей или нейтронов. Благодаря сравнительно большому
объему образца, выделяемого падающим пучком, методы рассеяния вносят
меньшие искажения, чем микрография. Однако из-за проблем, присущих этим
методам при получении структурной информации, можно получить данные
только в ограниченных пределах обратного (сопряженного) пространства.
Более того, однозначное определение распределения частиц по размерам из
кривых рассеяния невозможно. Для распределения можно получить лишь
«эквивалент рассеяния», основанный на принятых предположениях о форме
частиц и типе распределения. На рис. 17 представлены распределения частиц
о
Радиус (А)
Рис. 17. Размеры дисперсной фазы в смеси, содержащей 5% цис-\,4-полибутадиена
в полихлоропрене, определенные путем анализа изображения, полученного
методом просвечивающей микроскопии (ТЕМ, треугольники) и численных
результатов, полученных при совместном использовании малоуглового
нейтронного и рентгеновского рассеяния (кружки); сплошная кривая —
логнормальное распределение, лучше всего описывающее распределение
по размерам, полученное методами рассеяния [212].
150 -*\r Глава 3. Характеристика структуры эластомеров
по размерам для г{ис-1,4-полибутадиена в полихлоропрене, определенные
с помощью просвечивающего электронного микроскопа и малоуглового
рентгеновского рассеяния [212]. Просвечивающая сканирующая рентге-
новская микроскопия, основанная на химической специфичности рентге-
новской абсорбции создает возможности для исследования морфологии, но
для этого требуются синхротронные источники рентгеновских лучей [213].
Рис. 18 демонстрирует возможности этого метода для изучения морфологии
смесей; удовлетворительное разрешение достигается без протравливания
образцов.
Рис. 18. Микрофотографии (Юрм2) смеси полибутадиена и сополимера полиизобути-
лена с 4-метилстиролом (30:70), содержащей 20 вес. ч. технического угле-
рода, полученные методом просвечивающей сканирующей рентгеновской
микроскопии при возрастающей энергии фотонов: (а) — минимальная энер-
гия, видны только частицы технического углерода; (б) — большая энергия,
виден также ненасыщенный полимер; (в) и (г) — наиболее высокие энергии
фотонов, виден полиизобутилен в обратном контрасте. Более темные области
на микрофотографиях указывают на положение вещества с более высокой
абсорбцией, светлые области более прозрачны [213].
Простейший способ определения гомогенности смеси это калориметрия.
Наличие двух переходов, соответствующих Tg компонентов указывает на
разделение фаз. Однако наличие только одного перехода еще не означает тер-
модинамической совместимости компонентов, особенно если их Tg близки.
С другой стороны, спектроскопические измерения могут свидетельствовать
VI. Морфология -*и- 151
о наличии двух раздельных релаксационных пиков даже в случае термодина-
мическиЬ совместимой смеси [214-217]. Этот эффект можно рассматривать
как «динамическую гетерогенность», которая возникает, если сильно разли-
чается свойственная компонентам подвижность. На движение компонентов
влияет их химическая структура и окружение. Последнее усредняется
в гомогенных системах, в результате смесь обычно имеет единую Т . Однако,
когда подвижность компонентов сильно различается, могут наблюдаться два
перехода, как это показано на рис. 19. Калориметрический переход при
для динамически гетерогенных смесей обычно очень широк [218].
Log]0№]
Рис. 19. Изотермические кривые диэлектрических потерь для термодинамически
совместимой смеси 25% 1,2-полибутадиена (ПВЕ) с НК. Несмотря на
гомогенную морфологию, соответствующая подвижность компонентов
различается, поэтому на спектрах наблюдается два пика.
Как уже отмечалось, если Tg компонентов близки, единая смеси не
может служить свидетельством ее гомогенности. В действительности из-
быточный объем при смешении, и специфические взаимодействия могут
вызвать аномальное поведение таких систем. При этом Tg может быть ниже
(как это показано для смесей полихлоропрена с эпоксидированным поли-
изопреном [219]) или выше (как для смесей полиэпихлоргидрина с поли-
винилметилэфиром [220]) чем Т для каждого компонента. В этом случае
эффективно использование 129Хе ЯМР [221], т. к. ксенон легко поляризуется
и даже наличие ван-дер-ваальсового взаимодействия приводит к заметному
химическому сдвигу. При растворении в гетерогенной смеси полимеров
в спектре 129ХеЯМР наблюдается две полосы если размеры доменов от-
152 —• vr Глава 3. Характеристика структуры эластомеров
дельных компонент достаточно велики (относительно времени диффузии
ксенона). С другой стороны, наличие одной полосы свидетельствует о
совместимости и указывает на верхнюю границу размеров доменов. Этот
метод особенно эффективен для каучукоподобных материалов, т. к. в этом
случае спектральные линии узкие. Для исследования фазовой морфологии
метод 129ХеЯМР используется в различных лабораториях [222-226]. На
рис. 20 показаны изменения 129ХеЯМР спектров при нагревании образцов
смеси 1,4-полиизопрена и 1,4-полибутадиена выше нижней критической
температуры растворения (НКТР, LCST).
Химический сдвиг (млн. д)
Рис. 20. Спектры ЯМР для 129Хе, растворенного в гомогенной смеси (нижняя кривая)
и в течение разного времени после нагревания выше НКТР, когда прояв-
ляется фазовое разделение. Самая верхняя кривая получена для образцов
отдельных полимеров, одновременно находящихся в измерительной ячейке
ЯМР, но физически разделенных [224].
Одна из проблем со смесями, которые содержат фазовые границы раздела,
это получение равномерного распределения ингредиентов. Например, распре-
деление вулканизующих агентов может быть искажено диффузией из одной
фазы в другую. Особенно важна эта проблема, если параметры растворимости
компонентов существенно различаются. Предложены разные методы оцен-
ки степени поперечного сшивания в компонентах смеси [227]. Поскольку
вулканизация влияет на поведение при стекловании, особенно при высоких
степенях поперечного сшивания, информацию о распределении поперечных
связей можно получить при измерении Т* [228, 229] или локальной сегмен-
VI. Морфология
Л
153
тальной релаксации [230]. Однако при малых степенях поперечного сшивания
чувствительность этих методов мала, причем требуется, чтобы Tg существенно
различались. Если степень поперечного сшивания настолько мала, что суще-
ственная часть полимера остается несшитой, золь-гель анализ потенциально
может дать оценку относительного сшивания компонентов [231 ]. Были попытки
использовать просвечивающую электронную микроскопию набухших образцов,
чтобы исследовать распределение поперечных связей [227]. ЯМР-визуализация
может обнаружить пространственное изменение плотности поперечных связей.
Однако, хотя этот метод был использован для изучения окисления каучуков
[231], для изучения смесей его пространственное разрешение слишком грубое
Химический сдвиг (млн. д)
Рис. 21. ^ЯМР-спектры набухших эластомеров на основе НК. Спектры наверху
получены для образцов с плотностью поперечных связей в 4,5 раза боль-
шей, чем у образцов, спектры которых расположены внизу. Резонансные
полосы при 5,2 млн. д относятся к протонам в цепи олефинов и могут быть
использованы для определения относительной плотности поперечных
связей [227].
Чтобы исследовать распределение поперечных связей в смесях исполь-
зовали ЯМР набухших образцов [233, 234]. Набухание увеличивает под-
вижность молекулярных цепей, и изотропное движение ядер усредняет ло-
кальные поля, что приводит к сужению спектральных линий. Это позволяет
охарактеризовать отдельные резонансные процессы. Наличие поперечных
связей ограничивает подвижность и увеличивает ее анизотропию, поэтому
некогерентное усреднение становится менее полным, в результате линии
уширяются (рис. 21). Для получения количественных результатов требуется
эмпирическая корреляция с плотностью поперечных связей.
154
Глава 3. Характеристика структуры эластомеров
Кристаллизация в совместимых смесях может приводить к вытеснению
некристаллизующегося компонента, и его концентрация в аморфной фазе
увеличивается. И наоборот, если он может входить в элементарную ячейку,
он захватывается и соответственно изменяется средний объем элементар-
ной ячейки [235]. В НК возможен также переход от а- к Р-ламелям [236]
(рис. 22). Эти ламели имеют одинаковые элементарные ячейки, но Р-ламе-
ли имеют большую свободную энергию складчатой поверхности. Поэтому
некристаллизующийся компонент смеси предпочтительно размещается на
складчатой поверхности кристалла.
Температура (°C)
Рис. 22. Эндотермы плавления НК (верхние кривые) и его смеси с 1,2-полибута-
диеном (ПВЕ) в соотношении 50:50 (нижние кривые) после кристалли-
зации при температуре -25 °C в течение времени, указанного у кривых.
НК первоначально кристаллизуется в виде более стабильных а-ламелей,
Р-ламели образуются при больших степенях кристаллизации. Совмести-
мая смесь кристаллизуется преимущественно с образованием Р-ламелей,
имеющих более низкую температуру плавления [236].
В. Кристаллизация*)
Высокоэластичность, т. е. способность к большим обратимым
деформациям, подразумевает отсутствие кристалличности, и это обычно так
для недеформированных эластомеров. Однако для некоторых эластомеров
*> Подробно о кристаллизации эластомеров см. в книгах: Бухина М.Ф. Кристаллизация
каучуков и резин. М.: Химия 1973. 239 с. и Бухина М.Ф., Курлянд С.К. Морозостойкость
эластомеров. М.: Химия 1989. 176 с., а также Bukhina, M.F.,Kurlyand S.K. Low Temperature
Behavior of Elastomers. Leiden - Boston: ///VSP///. 2007. 187 pp. (Прим, nep.)
VI. Морфология -IV 155
при комнатной температуре может быть характерна очень небольшая степень
кристаллизации. Это, например, этиленпропиленовые каучуки с высоким
содержанием этилена, эпихлоргидриновый и пропиленоксидный каучуки.
Кристаллиты в этих материалах могут вести себя как усиливающие напол-
нители. Многие термопластичные эластомеры содержат кристаллические
домены, которые служат обратимыми поперечными связями [237].
Если основная цепь каучука достаточно регулярна, он кристаллизуется
при низких температурах. Это, в основном, влияет на поведение каучуков
при хранении. При кристаллизации неориентированного НК через стадию
образования ламелей, формируются сферолиты. Как указывалось выше,
при этом могут образовываться два типа ламелей, имеющих одинаковую
элементарную ячейку: более стабильные а-ламели и медленнее образующи-
еся Р-ламели [245], различающиеся не только скоростью роста и свободной
энергией складчатой поверхности, но и толщиной и морфологией. Благодаря
более высокой свободной энергии складчатой поверхности Р-ламелей, на
ней легче размещаются некристаллизующиеся части каучука [238]. Это
аналогично изменению морфологии 1{ис-1,4-полиизопрена в присутствии
смешивающегося второго компонента (см. рис. 22) [236].
Хотя эластомеры в основном аморфны, ориентационная кристал-
лизация наблюдается для таких каучуков, как цис-\,4-полибутадиен,
бутилкаучук и НК. Ориентационная кристаллизация, обнаруженная
200 лет назад [239], приводит к увеличению модуля и большинства
прочностных свойств эластомеров; во многих случаях она очень важна
при эксплуатации [240-243]. Для НК способность к ориентационной
кристаллизации прямо коррелирует с прочностными свойствами [244].
При ориентационной кристаллизации НК образуются ряды зародышей-
ламелей, которые растут перпендикулярно направлению растяжения
[238, 245]. Последующая вторичная кристаллизация может быть очень
медленной [246] и скорость ее не зависит от деформации растяжения
[247]. Однако скорость образования рядов зародышей очень сильно ра-
стет с ориентацией, вызывая быструю кристаллизацию [244-250]. Время
ориентационной кристаллизации при комнатной температуре меньше
60 миллисекунд [246]. Это существенно затрудняет измерение скорости
ориентационной кристаллизации для эластомеров разного состава.
Необходимым условием ориентационной кристаллизации является высо-
кая регулярность основной цепи полимера и этим обусловлены преимущест-
ва в эксплуатации НК по сравнению с синтетическим цис-1,4-полиизопреном
[251,252]. Усиливающие наполнители влияют на ориентационную кристал-
лизацию [253-256], влияют на нее и другие ингредиенты. Денатурированные
протеины и другие нерастворимые в углеводородах примеси, содержащиеся
в НК, в первую очередь влияют на скорость ориентационной кристаллиза-
ции. Например, было показано, что экстрагирование в ацетоне [257] и де-
протеинизация [258] НК приводят к уменьшению скорости кристаллизации,
156 -* v- Глава 3. Характеристика структуры эластомеров
Рис. 23. Зависимость относительного гистерезиса (отношения диссипированной
механической энергии к общей энергии деформации) от деформации для
образцов из четырех марок НК, растянутых при комнатной температуре до
различных степеней деформации. Ориентационная кристаллизация при де-
формациях 250% и выше приводит к более заметному гистерезису [262].
Рис. 24. Зависимость времени релаксации (времени, за которое происходит падение
напряжения) от деформации для образцов из четырех марок НК. При
меньших деформациях время релаксации определяется вязкоупругими свой-
ствами, при больших напряжениях начинается кристаллизация [262].
VI. Морфология Jv- 157
предположительно вследствие уменьшения областей зародышеобразования.
Степень кристаллизации, достижимая в отсутствии ориентации, определя-
ется микроструктурой основной цепи, особенно длиной i{uc-l,4 последова-
тельностей, а не присутствием некаучуковых составляющих [249].
Присутствие примесей влияет, прежде всего, на скорость зародышеоб-
разования, а не на скорость роста [260, 261]. И степень кристаллизации,
и ее зависимость от деформации не так важны в отношении прочностных
свойств эластомеров. Однако, минимальная деформация, требуемся для
кристаллизации, определяет концентрацию напряжения, необходимую для
начала кристаллизации в устье трещины, так же, как и ее пространственное
распространение (см. гл. 10).
Методы изучения ориентационной кристаллизации включают дифрак-
цию рентгеновских лучей [207, 208, 263-265], двойное лучепреломление
в видимом свете [266, 267], ИК и Рамановскую спектроскопию, дилатомет-
рию [269, 270], электронную микроскопию [268], ЯМР [271] и измерение
механических свойств [193, 262, 272]. Ориентационная кристаллизация
проявляется как в увеличении гистерезисных потерь (рис. 23), так и в уве-
личении времени падения напряжения (рис. 24). Однако форма кривой
напряжение-деформация при растяжении не обнаруживает с очевидностью
начало кристаллизации [207, 208, 262].
Г. Дефекты
Хорошо известно, что эластомеры, как практически все твер-
дые тела, имеют предварительно существующие «естественно наблюдаемые»
дефекты [263]. При увеличении локальных напряжений такие дефекты
оказывают влияние на поведение эластомеров при разрушении. В последнее
время озабоченность их существованием возросла в связи с потенциальной
возможностью ухудшения барьерных свойств эластомерных пленок. Эти
свойства являются критическими при использовании латексных пленок для
таких изделий, как хирургические перчатки и презервативы для блокирова-
ния проникновения частиц субмикроскопического размера, ответственных
за СПИД, гепатит и другие вирусные заболевания.
Прохождение частиц с размерами вирусов через неповрежденные види-
мым образом пленки натурального латекса непосредственно наблюдалось
в лаборатории [275, 276] и косвенно — на практике [277]. Свидетельство
существования таких дефектов в НК было получено с помощью электрон-
ного микроскопа [278], а также из данных по абсорбции воды; ее быстрое
начальное поглощение предполагает существование капиллярных ка-
налов [279].
Наличие внутренних дефектов влияет на поведение эластомеров при
разрушении, и это может быть использовано для их характеристики.
Присутствие дефектов видимого размера между 5 и 70 микрон влияет
158 JV Глава 3. Характеристика структуры эластомеров
на прочность эластомеров [280-283]. Следовательно, измеряя прочность
образцов с нанесенными надрезами различных размеров, можно оценить
наибольшие размеры внутренних дефектов. Чтобы получить размер
внутренних трещин, данные экстраполируют на значения прочности
в отсутствие надрезов. Если каучук способен к ориентационной крис-
таллизации, предварительные надрезы должны быть малыми, так чтобы
перед разрывом могла осуществиться кристаллизация основной массы
материала [284]. Для линейного упругого материала прочность изме-
няется пропорционально корню квадратному из размера трещин [282],
или, в более общем случае, энергия разрыва пропорциональна размеру
трещин [285]. В Таблице 4 представлены размеры дефектов, полученные
из данных по энергии деформации при разрыве для резин на основе НК
разных марок [244] (см. гл. 10).
Усталостное разрушение эластомеров представляет собой рост внут-
ренних дефектов, поэтому изучение усталостной долговечности (т. е. числа
циклов деформации до разрушения) может служить мерой размеров внут-
ренних дефектов [286-288]. В Таблицу 4 включены также размеры дефектов,
полученные из данных по усталостной долговечности для тех же резин на
основе НК [244].
Определенные таким образом значения размеров внутренних дефек-
тов представляют собой только эффективные размеры, соответствующие
данной концентрации напряжения [289]. Эта концентрация напряжения
зависит от формы трещины (т. е. ее остроты), и от диссипативных свойств
самого материала [290]. В то время как состав и параметры эластомерного
материала, в частности, плотность сетки поперечных связей, влияют на
прочностные свойства, их влияние на размеры внутренних дефектов более
ограниченно [287, 288, 291]. Измеренные размеры дефектов не зависят от
температуры [281], но могут изменяться при изменении типа техничес-
кого углерода [292]. Влияние степени диспергирования ингредиентов на
прочность эластомеров обусловлено, очевидно, механизмом, связанным
с наличием дефектов [293].
Таблица 4 . Характерные размеры дефектов в натуральном каучуке.
Характерные размеры дефектов |лм
Марка НК Энергия деформации Усталостная долговечность
Г ваюла 29 26
SMR-10 29 31
SMR-L 26 17
Депртеинезированный НК 16 10
VI. Морфология -IV 159
Д. Зацепления
Упругое поведение эластомеров при больших деформациях
отражает влияние топологических взаимодействий, которые известны
как зацепления. Зацепления ограничивают подвижность молекулярных
цепей, подавляя внутренние движения. Наличие псевдо-сетки зацеп-
лений ответственно за появление характерного плато на зависимости
механических свойств от времени для невулканизованного каучука.
В то время как протяженность плато (вдоль оси времени или частоты) оп-
ределяется молекулярным весом, его высота О^отражает концентрацию
и эффективность зацеплений. Действительная часть динамического моду-
ля (модуль запаса или накопления) мало зависит от частоты и приблизи-
тельно пропорционален концентрации зацеплений ve. Такая зависимость
для расплава имеет исключительно энтропийную природу и не зависит
от различия в энергиях для разных конформеров. Молекулярный вес
отрезка цепи между зацеплениями М = p/G0^ (где р плотность) зависит
от химической структуры и поэтому может сдужить характеристикой
полимера. Взаимодействие зацеплений определяет реологию несшитых
полимеров, влияя на вязкость, динамический модуль и обратимую по-
датливость. Наличие зацеплений влияет и на свойства вулканизатов.
В развитии уточненной теории высокоэластичности их роль является
ключевой (см. гл. 4). Прочностные свойства гибкоцепных пластиков
также зависят от количества зацеплений.
Существуют различные методы определения величины Ме и других
характеристических значений молекулярного веса, связанных с взаимо-
действием зацеплений. Т. к. действительная часть динамического модуля
(модуль запаса) в области плато высокоэластичности все же слабо зависит
от частоты, высота плато дает лишь порядок значений G°N. Простые соотно-
шения, эмпирическое [294]
G«=3.56Gmai (19)
или основанное на феноменологической теории [295]
G«=4.83G_ (20)
дают соотношение между GQNn максимальным значением модуля потерь.
К сожалению, оба они дают существенно заниженные величины для поли-
дисперсных полимеров из-за негомогенного расширения дисперсии. Более
надежная оценка Снисходит из интегрирования дисперсии модуля потерь,
отделяя начало вязкого течения
(% =^J2G"(<0)d/n® (21)
160 -•V Глава 3, Характеристика структуры эластомеров
Здесь данные в области высоких частот экстраполируются (например,
в предположении степенного закона [296,297]), чтобы избежать наложения
данных, относящихся к переходной области.
Как было показано выше, метод ЯМР может быть использован для
определения концентрации химических поперечных связей, но наличие
физических зацеплений не влияет на вид спектров. Т. к. эти зацепления
вносят вклад в упругие свойства вулканизатов (см. гл. 4), разница между
данными по плотности сетки, полученными методам ЯМР и из измерений
модуля может служить количественной оценкой концентрации зацепле-
ний. Большинство применений этого метода направлено на оценку роли
взаимодействий полимер — наполнитель в формировании жесткости
эластомеров [95].
Рис. 25. Зависимость безразмерной величины модуля на плато от отношения шага
(сегмента) Куна lk к межцепному расстоянию (упаковочной длине) р. Вместе
с различными расчетными данными (квадраты, кружки, ромбы) включены
экспериментальные данные для расплава (+) и концентрированных рас-
творов (х) [307].
Физическая природа зацеплений и их зависимость от химической
структуры рассматривалась многими исследователями. Заключения,
что контурная длина цепи в единице объема определяет величину топо-
логических ограничений, сделаны, в основном, на основе скейлинговых
соображений [298-301]. Однако, поскольку имеется две скейлинговые
длины, относящиеся к этому процессу, а именно межцепное расстояние
(упаковочная длина) и длина шага (сегмента) Куна, использование толь-
ко скейлинговых соображений не дает однозначного решения проблемы
[302]. Должны быть введены некоторые идеи относительно природы
VI. Морфология
-\r 161
зацеплений, приводящие к количественным соотношениям между хими-
ческой структурой полимера и модулем в области плато. Были предло-
жены различные определения, касающиеся взаимодействия зацеплений,
включая фиксированное число бинарных контактов на зацепление [303],
фиксированное число бинарных контактов на объем зацепления [304]
и фиксированное число пучков (стрендов) на объем зацепления [305,
306]. Различаясь количественно, все эти подходы правильно предсказы-
вают тот экспериментальный факт, что увеличение объема, занимаемого
молекулярной цепью, и ее подвижности приводит к уменьшению модуля
на плато [300]. Последние результаты компьютерного моделирования
представлены на рис. 25.
ГЛАВА
4
МОЛЕКУЛЯРНЫЕ ОСНОВЫ
ВЫСОКОЭЛАСТИЧНОСТИ
БЮРАК ЭРМАН
Химико-биологический департамент Кок-Университета,
Румелифенери Йоли, Стамбул, Турция
ДЖЭЙМС Е. МАРК
Департамент Химии Университета Цинцинатти,
Цинцинатти, Огайо
I. ВВЕДЕНИЕ
Высокоэластичные материалы состоят из относительно
длинных полимерных цепей, обладающих значительной степенью гиб-
кости и подвижности и которые соединены в сетчатую структуру. Условие
гибкости и подвижности связано с их очень высокой деформируемостью.
Под действием приложенных внешних напряжений длинные цепи могут
изменять конформацию, изменение это происходит относительно быстро
благодаря высокой подвижности цепей. Требование того, чтобы цепи были
связаны в пространственную сетку, связано с требованием придания сетке
свойств твердого тела, где цепи были бы ограничены в возможности течения
друг относительно друга под действием внешних напряжений. Как следс-
твие этого, типичный каучук может быть растянут до 10 раз по сравнению
с первоначальной длиной. После удаления внешней силы он быстро вос-
станавливает свои прежние размеры при практически полном отсутствии
остаточных напряжений. В результате таких уникальных механических
свойств каучуки нашли важнейшие применения от автомобильных шин
и до клапанов сердца, уплотнителей в реактивных самолетах и космических
аппаратах.
В обычных твердых телах, таких как кристаллические или аморфные
стеклообразные, приложенная извне сила изменяет расстояния между сосед-
ними атомами, отчего возникают межатомные или межмолекулярные силы.
В таких материалах расстояния между двумя атомами изменяются не более
чем на доли ангстрема, если деформация является обратимой. При больших
II. Структура идеальной сетки 163
деформациях атомы перемещаются друг относительно друга, вследствие
чего наблюдается или течение или разрыв. В то же время реакция каучуков
оказывается почти полностью внутримолекулярной.
Внешняя сила воздействует на длинные цепи через поперечные связи,
приводя к изменению конформации, и каждая цепь действует подобно
пружине в ее отклике на внешнюю силу. Молекулярный механизм высоко-
эластичности был разработан в начале 1930-х годов. Развитые тогда ста-
тистические механические теории, описывающие механическое поведение
каучуков были даны в работах Гута и Джеймса [1], Уолла [2], Флори [3],
и Флори и Ренера [4]. Современные представления о молекулярных основах
высокоэластичности создались в основном благодаря этим ранним теориям.
Они дали идеализированную картину высокоэластической деформации,
но в то же время создали основу для более развитых молекулярных теорий
высокоэластичности, которые учитывают эффекты межмолекулярных за-
цеплений, присутствующих в реальных сетках. Развитие идей в этой области
от первых работ до настоящего времени было представлено в нескольких
монографиях (например, Трилор [5], Марк и Эрман [6], и в самой последней
монографии Эрмана и Марка [7]).
В этой главе мы прежде всего обсудим структурные особенности сеток,
которые влияют на накопление напряжений в процессе деформации. Мы
обсудим простые классические модели эластичности и отклонения от этих
простых моделей. В особенности мы проведем различие между двумя клас-
сами моделей, (1) модель стесненности (the constraint models), которая пред-
полагает, что общая упругая энергия сетки равна сумме энергий отдельных
цепей сетки, и (2) модель захваченных узлов (the trapped entanglement mo-
dels) , которая предполагает, что зацепления, которые фиксируются на стадии
поперечного сшивания, делают дополнительный вклад в упругую энергию
сетки. Мы также дадим молекулярную интерпретацию коэффициентов,
входящих в феноменологические теории. Набухшие гели и восприимчивые
гели (responsible gels) находятся среди наиболее широко исследованных
систем в минувшие несколько лет, и последние работы в этой области также
будут рассмотрены. Мы затем рассмотрим термоупругое (сила-температура)
поведение сеток. Заключительная тема включает результаты экспериментов
по рассеянию нейтронов (что позволяет непосредственно определить раз-
меры цепей как в недеформированной, так и в деформированной сетке),
а также недавнее экспериментальное изучение упругости отдельных изо-
лированных цепей.
II. СТРУКТУРА ИДЕАЛЬНОЙ СЕТКИ
Сетка возникает путем связывания полимерных цепей,
возникшие связи могут быть физическими или химическими. Физические
связи могут быть образованы за счет (1) адсорбции цепей на поверхности
164
—Глава 4. Молекулярные основы высокоэластичности
тонкодисперсных частиц наполнителя, (2) образования мелких кристал-
литов, (3) коалесценции ионных групп, или (4) коалесценции блоков
стеклообразных полимеров в блоксополимерах. Эти физические связи, как
правило, не постоянны и могут исчезать при набухании или при повышении
температуры. Соответствующие сетки рассматриваются как «физические»
или «термообратимые» и в данной главе не рассматриваются. Читатель
может обратиться к книге Бурхарда и Росс-Мурфи [8] для дополнительной
информации о таких материалах.
Химические связи могут быть образованы случайным соединением
сегментов в уже сформированных цепях, путем актов статистической сопо-
лимеризации или путем соединения цепей с функциональными группами
на концах. Серная вулканизация, пероксидная вулканизация, облучение
ионизирующими излучениями также являются известными методами со-
здания случайных сшивок. Сополимеризация мономеров, среди которых
по крайней мере один имеет три или более активных центров, также ведет
к образованию случайных сшивок. Образование сеток путем соединения
по концам индивидуальных цепей ф -функциональными связями является
наиболее подходящим методом образования хорошо охарактеризованных
структур, где функциональность ф связи определяется числом цепей участ-
вующих в создании узла. Сетка называется совершенной, если ее узлы имеют
функциональность по крайней мере 3, она не имеет свободных концов (цепи
соединенные с сеткой лишь одним концом), и она не имеет петель (цепи, оба
конца которых присоединены к одному узлу). Свойства идеальных сеток об-
суждаются в этом разделе. Читатель может обратиться к Эрману и Марку [7]
и к оригинальной работе Флори [9, 10], где рассматриваются свойства не-
идеальных сеток.
Структура идеальных сеток может быть охарактеризована двумя пере-
менными: циклическим рангом (cycle rank) Е, и средней функциональнос-
тью ф. Циклический ранг определяется числом цепей, которые должны
быть удалены, чтобы превратить сетчатую структуру в ветвистую. Другие
три параметра, часто применяемые для характеристики сетки являются
(1) число цепей (цепей между узлами) V, (2) число узлов р, и (3) молеку-
лярный вес М цепей между узлами сетки. Они могут быть вычислены из £
и ф с помощью уравнений
v = t——
[1--
V Ф
2v
ц ф
(1>
(2)
III. Простые молекулярные теории -* v 165
(3)
где р — плотность, VQ — объем сравнения сетки (reference volume), NA— чис-
ло Авогадро.
Циклический ранг определяет связность сетки и является единственным
параметром, определяющим эластичность сетки, как это будет показано
в следующем разделе, посвященном простым молекулярным теориям.
В других исследованиях, учитываются также зацепления, захваченные
в процессе вулканизации, дополнительно к химическим сшивкам [11].
Модель фиксированных зацеплений будет рассмотрена позже.
В типичном эластомере число связей в основной цепи сетки колеблется от
примерно 100 до 700 [12]. Сетки с цепями короче, чем 100 связей обладают
малой растяжимостью. Те, у которых число связей более 700 могут иметь
очень большую растяжимость, но они слишком слабы, чтобы выполнять
функции материала, несущего нагрузку. Можно создать бимодальные сетки
путем сшивания по концам как очень коротких, так и очень длинных цепей,
чтобы получить сетку со значительной жесткостью [13].
III.
ПРОСТЫЕ МОЛЕКУЛЯРНЫЕ ТЕОРИИ
Основное допущение простых теорий высокоэластичности
гласит, что упругий потенциал (elastic free energy) сетки равен сумме упру-
гих потенциалов отдельных цепей. В этом разделе сначала анализируется
эластичность отдельной цепи, а затем следует простая теория эластичности
сеток. Поправки к теории следуют из взаимного влияния макромолекул, что
не учитывается простой теорией и они рассматриваются в разделе 4.
А. Эластичность изолированной цепи
Химическая структура полимерной цепи определяет ее
статистические свойства, такие как средние размеры в пространстве и ее
гибкость. Эти параметры в свою очередь, определяют различные свойства
сетки, состоящей из таких цепей. Детальное представление об изолированной
цепи является поэтому важным.
Короткий отрезок цепи поли (диметилсилоксана) (ПДМС) показан
в качестве примера на рис. 1, а. Атомы кремния и кислорода чередуются попе-
ременно вдоль основной цепи при наличии групп СН3 образующих боковые
группы. Структура основной цепи между i-1 и i+2 связями основной цепи
показаны на рис. 1, б. Длины связей Z- и углы между связями ф, показанные
на рисунке сохраняются приблизительно постоянными. Вращение вокруг
166 -* V Глава 4. Молекулярные основы высокоэластичности
связей может осуществляться относительно легко вокруг связей основной
цепи. Угол поворота вокруг i-той связи обозначен на рисунке буквой ф.
Большое количество поворотов может происходить вокруг связей основ-
ной цепи, в результате чего цепь может принимать разные конформации в
пространстве, одна из которых показана на рис. 1, в. Величина г определяет
мгновенное значение вектора расстояния между концами цепи.
Рис. 1. а — короткая последовательность поли
(диметилсилоксановой) цепи; б — параметры
структуры основной цепи; в — типичная про-
странственная конформация; г — типичное
распределение конформационной энергии
для разных углов поворота ф{
В большинстве молекулярных
теорий высокоэластичности ин-
дивидуальная цепь представлена
моделью свободносочлененной
цепи или цепи со свободным
вращением вокруг связей. В дей-
ствительности, однако, вращение
вокруг каждой связи заторможено
благодаря наличию барьера вра-
щения вокруг цепи и сил отталки-
вания между соседними атомами
вдоль цепи. Энергия, которая оп-
ределяется углом поворота вокруг
связи основной цепи показана на
рис. 1, г. Зависимость, показанная
на этом рисунке, демонстриру-
ет три минимума, относящиеся
к трем «изомерным состояниям»
или трем «изомерным миниму-
мам». Естественно, что число
изомерных состояний может быть
меньше или больше 3, в зависи-
мости от строения цепи. Три ми-
нимума, показанные на рис. 1,г,
разделенные интервалами в 120°
характеризуют транс (£), гош
(g+) и гош (g-) состояния. Фор-
ма цепи изменяется непрерывно
и быстро, поскольку каждая связь
флуктуирует вокруг изомерного
минимума с амплитудой порядка
+60° и иногда может перескочить
через энергетический пик в другой
изомерный минимум. Частота пе-
реходов из одного изомерного минимума в другой составляет величину порядка
одной обратной наносекунды при достаточно высоких температурах и зависит
в основном от температуры и величины энергетического барьера показанного
III. Простые молекулярные теории -*\г 167
на рис. 1, г. Эти переходы определяют динамику цепи и частично ее температуру
стеклования. С другой стороны равновесные свойства цепи зависят от энергети-
ческого уровня изомерных минимумов также как от их расположения. Транс-
состояние цепи со статистической конформацией показанное на рис. 1, г более
предпочтительно, чем g+ и g- состояния. Среднее число связей участвующих
в Z,g+ ng- состояниях в статистической механике определяется больцмановским
распределением [14,15]. В соответствии с правилами статистической механики
число каждого типа изомеров в цепи остается практически неизменным, когда
цепь растягивается за ее концы.
Изменение радиуса-вектора расстояния между концами цепи происходит
за счет перераспределения изомерных состояний вдоль цепи. Поскольку число
каждого типа изомерных состояний остается постоянным, общая внутренняя
энергия цепи остается постоянной в процессе растяжения. Упругость цепи,
возникающая при перераспределении изомерных состояний, рассматривается
как энтропийная упругость и большая часть упругости сетки является поэтому
энтропийной. Если часть работы деформации затрачивается на изменение
относительной вероятности изомерных состояний, валентных углов, и длин
связей в цепи, происходит изменение внутренней энергии, приводящее
в результате к «энергетическому» вкладу в величину упругости. Взаимосвязь
энтропийной и энергетической составляющей с молекулярной структурой
сетки рассматривается в следующих разделах.
Вектор г соединяющий концы цепи принимает разные значения вследс-
твие вращений вокруг простых связей. Для цепей с более чем 50 связями
в основной цепи, вероятность Ж(г) dxdydz того, что один конец вектора г
находится в начале координат, а другой конец в бесконечно малом объеме
dV = dxdydz удовлетворительно выражается функцией Гаусса [16]
\3/2
W(r)dxdydz
dxdydz
(4а)
где (г2 является средне-квадратичным расстоянием между концами цепи,
а индекс 0 указывает, что цепь находится в невозмущенном или так называе-
мом тета-состоянии [9]. Сейчас надежно установлено, что цепь в ненабухшем
твердом полимере находится в невозмущенном состоянии. Уравнение (4а)
выражает вероятность распределения величины вектора г. Менее детальная
форма выражения распределения w(r) показывает, что величина г вектора
г имеет определенное значение независимо от направления вектора. Так
вероятность того, что расстояние между концами цепи находится в пределах
от г до г + dr независимо от направления, может быть выражена
\3/2
3
tr(r)dr =
4лг2с?г
(4Ь)
168 -*v Глава 4. Молекулярные основы высокоэластичности
Схематическое изображение гауссовой функции по ур. (46) дано пунктир-
ной кривой на рис. 2. Абсцисса дана в приведенных единицах, полученных
делением расстояния между концами на контурную длину цепи, ордината
дана в безразмерном виде умножением w (г) на контурную длину.
Для цепей состоящих из менее, чем 50 связей, таких как например ко-
роткие цепи в бимодальных сетках, например, распределение отличается
заметно от гауссового. Среди различных выражений w(r) для коротких
цепей можно отметить ряды Хермита [16], распределение Фиксмана-Албе-
на и моделирование Монте-Карло [17]. Распределение Фиксмана-Албена
представлено формулой
iv(r)dr ос exp^-ar2 + b2r4 ^nr2dr (4с)
где а и b — коэффициенты. Это распределение и результаты моделирования
Монте-Карло для ПДМС с 20 связями в основной цепи сравнивается с гаус-
совым распределением на рис. 2.
Рис. 2. Распределение расстояний между концами цепи ПДМС с 20 свя-
зями в осовной цепи длиной 1 = 1,64 А. Распределение Фиксмана-
Албена (кривая точек) и кривая моделирования по Монте-Карло
(сплошная кривая) сравниваются с гауссовым приближением
(пунктир).
Молекулярные теории сеток, которые будут представлены в следующих
параграфах, основываются на гауссовом представлении о поведении цепей
образующих сетку. Со ссылкой на вид функции распределения, эти теории
представлены как «гауссовы теории».
Упругий потенциал Ае1 гауссовой цепи связан с функцией распределения
Ж(г) термодинамическим уравнением [7]
III. Простые молекулярные теории -* V 169
AvX=C[T}-kTlnW{r) (5)
где С(Т) является функцией только температуры Т,&к — константа Больц-
мана. Подставляя (4а) в уравнение (5) получим
Л1 = ^*(П+
(6)
Здесь А*(Т) является функцией только температуры. Уравнение (6) выра-
жает упругий потенциал гауссовой цепи с концами, закрепленными на рас-
стоянии г. Средняя величина силы, необходимой для того чтобы удерживать
два конца на таком удалении друг от друга, представлена термодинамическим
уравнением [14]
(7)
(8)
Где уравнение (8) получено подстановкой уравнения (6) в уравнение (7).
Индекс Т означает дифференцирование при постоянной температуре.
Уравнение (8) показывает, что изолированная цепь ведет себя подобно
линейной пружине с модулем З/сТ1 /(г2) Некоторые экспериментальные
результаты, касающиеся изолированной цепи и предсказанные форму-
лой (8) приведены в разделе IX.
В. Упругий потенциал сетки
Общий упругий потенциал сетки Д4е1 по сравнению с неде-
формированным состоянием рассчитывается суммированием уравнений (6)
относящихся к v цепям сетки [6]:
(9)
Зу/стГ г2
(Ю)
170 Глава 4. Молекулярные основы высокоэластичности
где (г2) = Er2 /v есть средне-квадратичные векторы цепей в недеформиро-
ванной сетке. Подставляя
(r2) = (x2)+(jz2)+(z2) (И)
в уравнение (10) и учитывая тот факт, что размеры цепей изотропны в не-
деформированном состоянии
(12)
можно получить
(13)
Отношения средне-квадратичных размеров в уравнении (13) явля-
ются отношениями микроразмеров. Чтобы выразить упругий потенциал
сетки в величинах, соответствующих макроскопическому (лабораторно-
му) состоянию деформации, следует сделать допущение, связывающее
микроразмеры цепи с макромасштабной деформацией. Эта взаимосвязь
с макродеформацией, которой подвергается сетка, является главным
направлением исследований высокоэластичности. Было предложено
несколько моделей для этой цели, которые рассматриваются в следу-
ющих разделах. Перед этим, однако, мы рассмотрим макродеформацию,
напряжения и модули сетки.
С. Приведенные напряжения и упругие модули
Макродеформация может быть рассмотрена по аналогии с
деформацией прямоугольной призмы, со значениями относительной длины
Хг, по осям х, у и z соответственно, как
X 7 у 7 Z 7 V 7
у — у — 1
^х~ J У~Т ^z~ г
(14)
где Lx0, Ly0, Lz0 относятся к сторонам призмы до деформации, a Lx, Ly, Lz — со-
ответствующие стороны в деформированном состоянии. С целью упрощения
мы рассматриваем здесь сухие сетки, т. е. сетки в отсутствие растворителя как
на стадии формирования, так и в процессе деформирования. В то же время
III. Простые молекулярные теории 171
мы рассматриваем влияние набухания в разделе VI. Также мы рассматриваем
только одноосное растяжение. Истинное напряжение т т. е. сила, отнесенная
к сечению деформированного образца, возникающее в результате одноосного
растяжения сетки может быть рассчитана по уравнениям Трилора [5, 7]
2 dAz4ei
т = 2Т
(15)
где индекс i указывает направление силы приложенной в одном направле-
нии, a j указывает одно из двух других направлений, в которых действует
гидростатическое давление. Выражение в квадратных скобках получено при
постоянной температуре и постоянном объеме, на что указывают индексы
Т и V. Отсылаем читателя к Трилору [5] и Огдену [18] по поводу других
напряженных состояний.
«Инженерное» напряжение о, определяемое как сила, отнесенная к единице
педеформированного сечения, может быть получено из уравнения [15] как
(16)
Приведенная сила [/*] определяется в виде:
(17)
В предельном случае малых деформаций приведенная сила равна модулю
сдвига образца, т. е.
G = lim [/*]
x->iL J
(18)
Выражения, приведенные в этом разделе и подробно проанализирован-
ные в работе Эрмана и Марка [17], являются обобщенными выражениями.
В следующем разделе мы введем две модели сеток, которые используются
в простых теориях высокоэластичности, чтобы связать микро- и макроско-
пическую деформации модели аффинной и фантомной сеток.
1. Модель аффинной сетки
Одно из ранних предположений, касающихся микродефор-
маций в сетках, состояло в том, что узлы сетки движутся аффинно (линейно)
с макродеформацией. Отсюда следует, что расстояния между узлами сетки
меняются также аффинно, поэтому
НО. НО.
(19)
172 “* V- Глава 4. Молекулярные основы высокоэластичности
Подставляя ур. (19) в ур. (20) получим с учетом ур. (1)
М>,affine = + + -3)
(20)
Более тщательный статистический анализ [9] дает дополнительный
член, связанный с изменением объема -\\kT(V/VQ) в ур. (20). Этот член
не появляется в упрощенном уравнении представленном здесь.
Истинное напряжение для одномерного случая может быть получено
подстановкой ур. (20) в ур. (15):
Модуль сдвига Gaj аффинной сетки может быть получен из ур. (17) и (18)
в виде
где 70 — объем в процессе формирования сетки.
2. Модель фантомной сетки
Мгновенное значение вектора г, соединяющего узлы на
концах отрезка цепи сетки может быть записано в виде суммы средне-квад-
ратичных значений г и мгновенных флуктуаций Дг от этого среднего, т. е.
г = г + Дг
(23)
В соответствии с моделью фантомной сетки флуктуации Дг независимы
от деформации и среднее значение г меняется аффинно с макроудлинением.
Возводя в квадрат обе части ур. (23), и усредняя по всем цепям, получим:
(г2) = (г2) + ((Дг)2)
(24)
Средняя величина г-Дг была приравнена нулю при получении уравне-
ния (24) т.к. эта величина может быть равновероятно положительной или
отрицательной как следствие флуктуаций величины Дг. И тогда среднее
из всех возможных случаев исчезает. Уравнение (24) применимо как к де-
формированному, так и к недеформированному состоянию. Это может быть
записано в терминах и в виде:
III. Простые молекулярные теории
Л
173
(25)
Уравнение (25) получено с использованием выражений:
(26)
Эти два выражения получены из модели фантомной сетки, как это пока-
зано в выводах уравнений приведенных в литературе [6, 12].
Соединив уравнение (26) с уравнением (25) и подставив полученное
выражение в ур. (13) получим
М.,phantom + *, +М -3) (27)
Сравнивая выражения для упругого потенциала аффинной и фантомной
моделей сетки, видим, что они различаются только на фронт-фактор. Выра-
жения для упругого потенциала моделей более близких к реальным сеткам,
чем аффинная и фантомная, приведены в следующем разделе.
Истинное напряжение в соответствии с моделью фантомной сетки полу-
чается подстановкой ур. (27) в ур. (15):
(28)
и модуль сдвига Gph получается из уравнений (17) и (18) в виде:
(29)
Ч)
Уравнение (29) показывает, что модуль пропорционален величине цик-
лического ранга £ и что модуль не зависит ни от каких других структурных
параметров. Число фиксированных зацеплений в сетке не изменяет величину
циклического ранга. Возможный вклад этих фиксированных зацеплений
в величину модуля не может быть, следовательно, следствием топологии
фантомной сетки.
174 -* V Глава 4. Молекулярные основы высокоэластичности
IV.
БОЛЕЕ ПРОДВИНУТЫЕ МОЛЕКУЛЯРНЫЕ ТЕОРИИ
Модели представленные в предыдущих разделах относятся
к числу простых в том смысле, что они не учитывают вклады межмолекуляр-
ных эффектов (таких как зацепления, захваченные при образовании сетки).
К числу теорий, учитывающих вклады зацеплений, относятся (1) подход
Дима и Эдвардса [19] в терминах топологических инвариантов, (2) модель
скользящих связей (the slip-link model) [20,21], (3) модель стесненных узлов
и стесненных цепей (the constrained-junction and constrained-chain models)
[22-27], (4) модель захваченных зацеплений (the trapped entanglement
model) [11, 28]. Модели скользящих связей, стесненных узлов и стесненных
цепей могут быть проанализированы с помощью обычного подхода насколь-
ко это можно видеть из работы Эрмана и Марка [7]. В иллюстративных
целях мы представим здесь более детально модель стесненных узлов. Затем
мы обсудим модели захваченных зацеплений.
Модель стесненных узлов была предложена для того чтобы объяснить
падение модуля упругости сетки при растяжении. Впервые она была введена
Ронка и Аллегра [22] и Флори [23]. Модель предполагает, что флуктуации
узлов снижаются до уровня меньше чем флуктуации в фантомной сетке бла-
годаря наличию зацеплений и что растяжение увеличивает интервал флукту-
аций снова до уровня фантомной сетки. Как следует из второй части ур. (26),
флуктуации в фантомной сетке существенны. Для тетрафукциональной
сетки средне-квадратичные флуктуации узлов достигают половины средне-
квадратичного расстояния между концами отрезков цепи. Интенсивность
стеснения этих флуктуаций оценивается параметром к определяемым как
(30)
Где ((ДЯ)2) и ((Д«)2) означают соответственно средне-квадратичные
флуктуации узлов в фантомной сетке и домене зацепления (entanglement
domain). Если интервал флуктуаций уменьшается до нуля из-за наличия
зацеплений, к становится бесконечно большим. Если эффект зацеплений
незначителен, то к = 0. Параметр к пропорционален числу узлов в объеме
занятом данным отрезком цепи сетки. Отсюда:
k=/(^;%/v0) он
где I— константа пропорциональности, Для сетки с тетрафункциональными
узлами к может быть записано как
K = /(^d/2)3/2((r2)o/M)3/2(^/Vo)-,/2 (32)
IV. Более продвинутые молекулярные теории
Л
175
где d — плотность стеки, а М — молекулярный вес цепи между поперечными
связями
Упругий потенциал модели со стесненными узлами дается выраже-
нием:
М 4wZ(*(2-i)+£
1=1 ъ
3
+ £>, -/«(1 + В,)- 1п(1 + О,)]
(33)
где
Bi = к2 (х2 - 1)(х2 + к) Di = Х2к-1В;
(34)
Истинное напряжение приодноосном растяжении следует из уравнений (15)
и(33)как
* = ~ (Х-Х-2)+|[ХАГ(х2)-Х-2АГ(Х’1)]
(35)
где
аг(х2)=В В(В + 1)-1 + к-1(Х2В + в)(в + кХ“2)~‘
(36)
в = Д- = в[(Х2-if -2(Х2+к)-11 (37)
ЭХ L
Приведенная величина силы дается выражением:
^кТ
*0
£ х-х
(38)
з
Модуль сдвига модели со стесненными узлами получается в пределе при
малых деформациях как
GPh
(39)
откуда следует, что для ненулевых значений параметра к модуль сдвига
в модели стесненных узлов больше, чем модуль сдвига в фантомной сетке.
Для аффинного предела к °° и модуль сдвига равен
(40)
176
Глава 4. Молекулярные основы высокоэластичности
Уравнение (40) показывает, что при малых деформациях модуль сдвига
аффинной сетки возрастает неопределенно по сравнению с модулем фан-
томной сетки, когда функциональность узлов приближается к 2.
Модель скользящих связей включает эффект зацеплений вдоль кон-
тура цепи в величину упругого потенциала. В соответствии с механизмом
действия скользящей связи изображенного на рис. 3, связь соединяет две
соседние цепи и может перемещаться на расстояние а вдоль контура цепи.
Упругий потенциал в соответствии с этой моделью запишется как
A^=^-NkT<
el 2 с
yi2 . у (1 + Л)\2
+ /о^(1 + т]Х?)
(41)
а
Рис, 3. Схематическое изображение
скользящей связи с ее возможными дви-
жениями вдоль отрезка цепи сетки, опре-
деляемыми расстоянием а и ее замыкание
в положение поперечной связи.
сильных стеснений узлов, тогда ка
где Nc и Ns — число химических
связей и скользящих узлов соответс-
твенно, а т| = 0.2343. Первый член
правой части ур. (41) отражает вклад
упругого потенциала фантомной
сетки. Эффект зацеплений входит
как следующий вклад и является
пропорциональным числу скользя-
щих узлов.
Упругий потенциал модели со
стесненными узлами, аналогичной
модели со скользящими связями,
является суммой упругого потенци-
ала фантомной сетки и потенциала
за счет стесненности узлов. Упругие
потенциалы как модели со сколь-
зящими связями так и модели со
стесненными узлами уменьшаются
до уровня модели фантомной сетки
когда эффект зацеплений уменьша-
ется до нуля. Одно важное различие
между двумя моделями состоит в
том, что упругий потенциал модели
со стесненными узлами становится
равным таковому в модели аффин-
ной сетки в пределе бесконечно
упругий потенциал модели со сколь-
зящими связями может превосходить таковой для аффинной деформации,
как это следует из ур. [41].
IV. Более продвинутые молекулярные теории 177
А. Вклад захваченных зацеплений в величину
модулей
Циклический ранг сетки указывает на число цепей, кото-
рые должны быть разорваны, чтобы сетка стала подобной ветвистому дереву.
Модули моделей фантомной, со скользящими связями, со стесненными
узлами, все пропорциональны величине циклического ранга. Циклический
ранг не зависит от числа фиксированных зацеплений в сетчатой структуре.
Сетка, в которой цепи сильно перепутаны, имеет тот же циклический ранг,
что и сетка без зацеплений. Следовательно, модели, названные выше, ка-
тегорически отрицают вклад захваченных зацеплений в величину модуля.
Однако же значительное количество экспериментальных данных показывает,
что определенная часть фиксированных зацеплений дает вклад в величину
модуля [И, 28]. Вклады в величину модуля определяются широко извест-
ным уравнением Лэнгли
G = Gch+TeG°N (42)
Здесь модуль G представлен суммой модуля за счет химических связей Gch
и члена, определяемого захваченными зацеплениями TeG^ , где Те (назы-
ваемое фактором захвата Лэнгли) представляет собой долю захваченных
зацеплений, которая делает вклад в величину модуля, a G^ является мо-
дулем на плато и связан с молекулярным весом Ме между зацеплениями
согласно уравнению
го рНТ
В соответствии с соображениями, основанными на модели стесненных узлов,
член Gch должен быть равен модулю фантомной сетки, в которую сделан
вклад за счет зацеплений.
Экспериментальные определения вкладов сверх тех, что предсказывает
модель фантомной сетки, взятой как базовая, оказываются противоречи-
выми. Данные Реннара и Оппермана [29], полученные для сетки ПД МС,
сшитого по концам макромолекул, представлены точками на рис. 4, по-
казывают, что вклады от фиксированных зацеплений значительны при
малых степенях сшивания по концам молекул, но несущественны когда
цепи, входящие в сетку оказываются короче. Данные Эрмана и др. [30]
касающиеся сетки статистически сшитого поли(этилакрилата) попадают
на сплошную линию и показывают, что модули, измеренные при предель-
но больших деформациях, находятся в пределах допускаемых моделью
стесненных узлов.
178 Глава 4. Молекулярные основы высокоэластичности
Рис. 4. Экспериментальные результаты Реннара и Оппермана [29] показаны линией
точек. Совпадение значений модуля с расчетом по фантомной сетке на-
блюдается для больших степеней сшивания. Результаты Эрмана, Вагнера
и Флори [30] попадают на прямую линию и полностью согласуются с моделью
фантомной сетки.
V. ФЕНОМЕНОЛОГИЧЕСКИЕ ТЕОРИИ И МОЛЕКУЛЯРНАЯ
СТРУКТУРА
Упругий потенциал согласно как простым, так и продви-
нутым теориям представляется симметричными функциями трех относи-
тельных деформаций Хх , Ху и Xz. Можно также выразить зависимость
упругого потенциала от удлинения в рамках других трех переменных,
которые в свою очередь являются функциями и Xz. В феноменоло-
гических теориях механики сплошной среды, где рассматривается только
наблюдаемое поведение материала и не учитываются обусловливающие
его молекулярные механизмы деформации, эти три функции могут быть
записаны как
12=^у+^+^2 W
/3=едм
о х у Z
АЛ1 -
(45)
VI. Набухание сеток и восприимчивость гелей 179
В наиболее общей форме упругий потенциал может быть записан в виде
степенного ряда
Mi= X Q4/1-3)i(72-3)J(/3-l)fc (46)
где Cijk являются феноменологическими коэффициентами. Простой случай
фантомной или аффинной сетки может быть выражен первым членом ряда
Mi =Goo(A-3) (47)
Упругий потенциал для твердого тела Муни-Ривлина может быть получен
из ур. (46) как
Mi=Goo(A-3)+С010(/2-3) (48)
Приведенное напряжение следует из уравнений (15) и (17) как
[/»| = 2С,+^ (49)
Л
где С\ = С100/70и С2 = С010/70. Для больших деформаций приведенное напря-
жение равно 2Ср что может быть определено как модуль в модели фантомной
сетки. Для малых деформаций 2С2 может быть получен, приравняв ур. (49)
к ур. (39) для модели стесненных узлов. Таким образом
2G=GpA (50)
Другие работы в рамках феноменологического подхода цитируются в
работах Трилора [5], Огдена [18], Эрмана и Марка [7] и Марка [31].
VI. НАБУХАНИЕ СЕТОК И ВОСПРИИМЧИВЫХ ГЕЛЕЙ
В предыдущем рассмотрении предполагалось, что сетка
образуется в сухом состоянии и испытывается также в сухом состоянии.
В последнее время, однако, много внимания было уделено набуханию сеток
и их фазовым переходам в зависимости от разного взаимодействия в сис-
теме сетка-растворитель. Крупномасштабные переходы, инициированные
малыми изменениями параметров окружающей среды, привлекли внимание
180
Глава 4. Молекулярные основы высокоэластичности
к возможному использованию набухших гелей в области технологии вос-
приимчивых материалов. Переходы включали растворители, отжимаемые
гелями, например при понижении температуры. Происходящая в резуль-
тате усадка («синерезис») широко известна как «коллапс геля» и показана
схематически как инициированное температурой изменение на рис. 5.
В дальнейшем будут специально рассмотрены системы с заряженными
группами, поскольку присутствие зарядов облегчает объемные переходы в
набухших гелях.
Рис. 5. Гель, отжимающий растворитель при понижении температуры, что сопро-
вождается усадкой («синерезисом») обычно называемой коллапсом геля.
Изменение свободной энергии при набухании сетки является суммой
изменения упругого потенциала ДАе1, изменения свободной энергии сме-
шения AAmix и вклада ионных групп AAj:
(52)
где ДАе1 может определяться любым выражением, взятым из модели, AAmix
есть свободная энергия смешения, а ДА^ вклад ионных групп входящих в состав
цепи. Общая величина химического потенциала Др^ растворителя в набухшем
полимере для модели стесненных зацеплений может определяться как
^^- = /п(1-р2) + р2 +Х^2 1 +—/i(X2)
RT V 2/ 2 42 '
-iv\
— — (53)
где v2 — объемная доля полимера, % — параметр взаимодействия Флори, Vt —
мольный объем растворителя, Мс — молекулярный вес отрезка цепи в сетке,
и X — относительная длина в случае набухания определяемая как:
VI. Набухание сеток и восприимчивость гелей -•и- 181
х=
V У/3
Vo)
(54)
щ\\ +хУрг2
Здесь х —число повторяющихся единиц в отрезке цепи сетки, пх —число
молекул растворителя, п2 —общее число цепей в системе, i —число ионных
групп в отрезках цепей, v — число цепей, и v20 — объемная доля цепей
в момент формирования сетки.
Приравнивая химический потенциал нулю, получим взаимосвязь между
равновесной степенью набухания и молекулярным весом Мс. Уравнение для
М h получено для тетрафункциональной фантомной сетки в виде
1/3
Mc-ph =----------------( V \(v
Zn(l-v2)+y2 + XP2 -М ~
V А ДР20
(55)
где v2 — выражает равновесную степень набухания.
Для аффинной модели сетки молекулярный вес между поперечными
связями определяется как
=-------------------- ( v Y
ln(l-v2)+v2+%vl -w —J-
V k0/V А Ду20
(56)
С другой стороны, выражение для химического потенциала может быть ре-
шено для v2 , что позволит рассчитать равновесную степень набухания сетки.
Решение уравнения показывает, что степень набухания возрастает с ростом
расстояния между узлами сетки. Доминирующие силы, действующие в процессе
набухания гелей, не содержащих ионных групп, это Ван-дер-Ваальсовы силы,
водородные связи, и силы, возникающие за счет энтропии цепей.
Если цепи в сетке содержат ионные группы, возникают дополнительные
силы, влияющие на способность к набуханию. Трансляционная энтропия
противоионов, кулоновские взаимодействия и возникновение мультиплетов
ионных пар являются силами, порождающими интересные особенности
ион-содержащих гелей. Эти явления были детально изучены Хохловым
с сотрудниками [32-35]. Выражение для свободной энергии сеток в работах
этой группы следующее:
“ АДтих + ^Mel + AArans + ^Coulomb (57)
где AAtrans и AACou|omb — это вклады в упругий потенциал сетки трансля-
ционной энтропии противоионов и свободной энергии кулоновских взаи-
182
Глава 4. Молекулярные основы высокоэластичности
модействий. Несколько интересных особенностей следуют из применения
ур. (57). Цепь, входящая в сетку полиамфолитного геля, содержит как
положительные, так и отрицательные заряды. Жидкая фаза в набухшем
полиамфолитном геле может содержать дополнительные противоионы.
Теоретические и экспериментальные публикации о таких гелях сделаны
недавно Нисато и Кандау [36]. В ион-содержащих гелях, в случае, когда
ионные группы полностью диссоциированы, происходит дополнительное
набухание геля благодаря стремлению свободных противоионов занять
как можно больший объем. В другом экстремальном случае, называемом
состоянием иономера, противоионы группируются у противоположно заря-
женных мономерных единиц, образуя ионные пары с последующим образо-
ванием мультиплетов. Это снижает осмотическое давление в геле и приводит
к коллапсу. Условия образования ионных пар и физические и химические
факторы, обусловливающие набухание геля и его коллапс сформулированы
Хохловым и Филипповой [37].
VII. ЭНТАЛЬПИЙНАЯ И ЭНТРОПИЙНАЯ СОСТАВЛЯЮЩИЕ
ВЫСОКОЭЛАСТИЧНОСТИ: ЗАВИСИМОСТЬ
СИЛА-ТЕМПЕРАТУРА
Основная составляющая упругости сеток возникает благодаря
«энтропийной упругости» индивидуальных цепей. Это являлось основным
положением прежних молекулярных теорий высокоэластичности [7]. Более
внимательное рассмотрение статистики индивидуальных цепей показывает,
что поворотные изомерные состояния соответствующие каждому углу поворота
цепи обладают неодинаковой энергией и растяжение цепи или изменение тем-
пературы может сместить их от одного изомерного минимума к другому, более
энергетически выгодному. Это порождает энергетический вклад в упругость
цепи. Поэтому общая величина силы действующей на сетку может быть запи-
сана как сумма энтропийного вклада fs и энергетического вклада fe.
f = fs+fe <58>
Температурно-силовая зависимость позволяет количественно оценить
относительные вклады энтропийной и энергетической составляющей в
упругость сетки.
При одноосной деформации энергетическая составляющая в общей
величине упругой силы [5-7, 31, 38, 39] представлена термодинамически
точным уравнением
Л = T\dln(f/T)
f~ L dT JtT
(59)
VII. Энтальпийная и энтропийная составляющие высокоэластичности... -*Ь- 183
Индексы L и V означают дифференцирование при постоянной длине
и объеме. Чтобы произвести дифференцирование, показанное в ур. (59) необ-
ходимо получить выражение для общей упругой силы/. Можно использовать
выражение, данное в ур. (28) для модели фантомной сетки. Приравнивая
правую часть ур. (59) к ур. (28) получим
/ dT
(60)
Уравнение (60) важно потому, что правая часть относится к микровели-
чинам (г2 } , а левая часть является отношением энергетической компоненты
силы к общей величине силы, т. е отношением макровеличин. Необходи-
мо подчеркнуть, что ур. (60) получено с использованием молекулярной
модели. Экспериментальное определение силы при постоянном объеме
весьма непросто. Поэтому используется выражение для силы, измеренной
при постоянной длине и давлении р или при постоянных аир. Вот эти
выражения
Л = грМ//П1 РГ
/" L dT \l,p p-i)
fe = ТГЭ/П(//Г)] _PT
f dT 3
(6i)
(62)
где P — коэффициент термического расширения сетки. Необходимо,
однако, заметить, что оба эти уравнения получены на основе уравнения
состояния для простой молекулярной модели и их нельзя рассматривать
как основанных исключительно на экспериментальных данных. Значе-
ния энергетической составляющей для некоторых типичных эластомеров
приведены в Табл. 1.
Таблица 1. Некоторые типичные значения отношения /,//
Эластомер fe/f
Натуральный каучук 0.18
Цис-1,4-пол ибутадиен 0.13
Поли (диметилсилоксан) 0.20
Эластин 0.26
184
Глава 4. Молекулярные основы высокоэластичности
VIII. ПРЯМОЕ ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНЫХ РАЗМЕРОВ
До недавнего времени информация о размерах цепей
в эластомерной сетке (как в деформированном, так и в недеформированном
состоянии) основывалась на результатах полученных на основе макромас-
штабных измерений и/или из молекулярной модели. Представление о том,
что цепи принимают случайные конформации был основано на совпадении
значений модуля, определенного экспериментально, с предсказанными
соответствующими теориями. Аналогично то, что деформации цепей оказы-
ваются промежуточными между предсказаниями аффинной и фантомной
моделей, следовало из соответствия значений модуля измеренных макро-
скопически и предсказанных теоретическими моделями. Развитие техники
спектроскопии за последнее десятилетие сместило однако акценты в сторону
молекулярной картины сетки. С помощью этих экспериментов размеры цепи
и ее превращения под действием внешней деформации стали наблюдаемы
непосредственно на молекулярном уровне и выводы на основе макроско-
пически измеряемых величин перестали быть необходимы. Малоугловое
нейтронное рассеяние оказалось сегодня самым мощным средством для
определения размеров цепей и их превращений при растяжении.
Техника нейтронного рассеяния и его применение для исследования
полимеров в твердом состоянии и в растворах, для полимерных смесей
и сеток описано в нескольких обзорных статьях [40-45]. Наиболее общий
вывод из этих исследований состоит в том, что размеры цепи входящей
в сетку идентичны их невозмущенным размерам в блоке несшитого полиме-
ра. За этими результатами последовало обнаружение [46] того, что радиус
инерции цепи ПДМС в сетке равен таковому в твердом несшитом состоянии.
Влияние (1) набухания и (2) одноосного растяжения сетки на размеры
цепи интенсивно изучались начиная с 1980 [46-55], и привели к следу-
ющему заключению: (1) изменение формы цепи в общем попадает между
предсказаниями аффинной и фантомной моделей и (2) сильно выражено
влияние разбавления системы в процессе образования сетки на изменение
размеров цепи. Малоугловое нейтронное рассеяние было также применено
для изучения поведения сетки в процессе релаксации после наложения по-
стоянной одноосной деформации [56]. Результаты измерения динамического
нейтронного рассеяния Хиггинса и др. [57-59] показали, что сегменты цепей
сетки диффундируют хаотично, и энергия активации этих перемещений
оказывается меньше, чем энергия активации движения центра масс целой
цепи. Измерения Ивэна и сотрудников [60,61] на сетках ПДМС с мечеными
узлами показали, что флуктуации узлов существенны и примерно адекватны
описываемым на основе фантомной модели сетки. Их результаты показывают
также, что движения узлов имеют диффузный характер, подобный тому, как
это описывает модель Рауза и что движения узлов намного более медленны,
чем движения дейтерированных свободных концов цепи.
IX. Упругость отдельной молекулы -*ъ- 185
IX. УПРУГОСТЬ ОТДЕЛЬНОЙ МОЛЕКУЛЫ
А. Введение
Единственный путь, которым молекула может быть дефор-
мирована, это простое удлинение ее за счет раздвижки концов. Этот вид
экспериментальных исследований определенно аналогичен измерениям
напряжение-деформация индивидуальных молекул биополимеров, таких
как протеины или полисахариды или, как в случае некоторых полинуклео-
тидов, молекул из двойных цепей. Изучались также и молекулы некоторых
синтетических полимеров. Сегодня это область очень активных исследова-
ний, для которой характерен заметный прогресс, итоги которого подводятся
в регулярно появляющихся обзорных статьях [62-58].
Рис. 6. Релаксация молекулы гамма-ДНК, когда максимальная степень растяжения
находится в гауссовой области. Эго качественное изображение, основанное на ре-
зультатах представленных в литературе (Шредер, Бэбкок, Шакфэ и др. [72]).
Развитие техники захвата отдельной цепи для последующего удлинения
является одной из основных проблем в этой области, а развитие достаточно
чувствительных методов измерения напряжений и деформаций - другая
проблема. Что касается необходимого крепления, то очевидно оно наиболее
желательно по концам цепи, и это осуществляется либо созданием концов
цепей с тщательно подобранными функциональными группами либо с по-
мощью микробисеринок. В первом случае функциональные группы могут
быть соединены с комплементарными группами зонда. Во втором случае
микробисеринка может быть захвачена микропипеткой или лазерным лучом
(действующим как «оптический пинцет»). Менее регулируемые экспери-
186 -*ъ- Глава 4. Молекулярные основы высокоэластичности
менты могут быть выполнены путем просто физической или химической
адсорбции цепи, когда другая часть ее таким же образом адсорбирована на
зонде. Зонд во всех случаях представляет собой кантилевер, по-существу
такой же, как в атомно-силовом микроскопе. Степень его перемещения есть
мера деформации (в масштабе нанометров), а степень отклонения есть мера
деформирующей силы (обычно в пределах пиконьютонов, pN).
Полезность таких экспериментов для области высокоэластичности по-
казана в следующем разделе.
Рис. 7. Релаксация молекулы той же что на рис. 6, когда максимальная степень
растяжения находится за пределами гауссовой области. Качественное изоб-
ражение, основанное на экспериментальных результатах приведенных в
литературе (Перкинс и др. [69-71]).
IX. Упругость отдельной молекулы 187
В. Соотношение гауссовых и негауссовых эффектов
Проведенные эксперименты по деформации макромолекулы
дают также ценную информацию о сокращении растянутой индивидуальной
молекулы [62,69-72]. В этих исследованиях молекулы были помечены так,
чтобы они могли быть видны во флуоресцентном микроскопе. Кроме того, эк-
сперименты были проведены в растворителе таком как вода, что было сделано
в опытах с изолированной молекулой ДНК показанных на рис. 6 [62].
Цепи были растянуты до желаемого удлинения и затем оставлены для
релаксации. Точками показаны экспериментальные результаты, а кривая
построена на основании ожидаемого поведения, когда сокращающая цепь
сила/пропорциональна расстоянию между концами г сохраняющемуся на
данный момент сокращения. Результаты показывают, что для умеренных
деформаций растянутых цепей ДНК они все еще находятся в гауссовой
области, для которой ур. (8) / = 13/с7’/^г2^ lr предсказывает наблюдаемую
пропорциональность.
В других экспериментах на той же самой системе, цепи ДНК были в дейс-
твительности растянуты близко к пределу их растяжимости [70]. Результаты
для этого более сложного случая схематически показаны на рис. 7. При
больших удлинениях цепи очевидно находятся в негауссовой области, что
подтверждается более интенсивным первоначальным падением сжимающей
силы. Это затем сопровождается гауссовой областью падения /, поскольку
удлинение достаточно мало, для того чтобы реализовалась гауссова область
(примерно две трети от полного растяжения [7]).
Хотя собственно сами эти исследования очень интересны, однако следует
заметить, что они не дают ответа на многие нерешенные вопросы из области
высокоэластичности касающиеся взаимодействий между цепями, образую-
щими эластомерную сетку.
ГЛАВА ВЯЗКОУПРУГИЕ СВОЙСТВА КАУЧУКА
5
К.Л. НГАИ
Военно-Морская Исследовательская Лаборатория,
Вашингтон, О. К.
ДОНАЛЬД ДЖ. ПЛАЗЕК
Университет Питтсбурга, Питтсбург, Пенсильвания
I. ВВЕДЕНИЕ
Большинство резин получают из сшивающихся высоко-
молекулярных линейных полимеров с низкой температурой стеклования
[1-6]. Большой молекулярный вес необходим, чтобы обеспечить высокую
растяжимость конечного эластомера, а низкая температура стеклования
необходима, чтобы обеспечить упругость. Такие исходные вещества пред-
ставляют собой совокупность линейных молекул, которые в конечном счете
способны течь друг по отношению к другу и являются поэтому вякоупругими
жидкостями [1, 5, 7]. Они вязкоупруги, что характеризуется их механи-
ческим откликом, зависящим от времени, и отражающим беспорядочные
изменения конфигураций молекул. После того как произошло достаточное
химическое сшивание, образуется молекулярная сетка химических связей
(резина), что превращает полимер в вязкоупругое твердое тело, которое уже
не течет. Подобно исходному полимеру его вязкоупругие свойства сильно
зависят от времени или частоты, от температуры, давления, присутствия
растворителя или наполнителя. Среди вязкоупругих твердых тел резина
обладает уникальной способностью сохранять целостность даже после того
как подвергнута большим напряжениям или деформациям, хотя вязко-
упругие свойства сильно зависят от уровня напряжений или деформаций
[3, 5, 8-10]. Однако для достаточно малых напряжений и деформаций вяз-
коупругие свойства сохраняются постоянными и преобладает состояние
линейной вязкоупругости [1]. В этой главе мы опишем линейные вязкоуп-
ругие свойства каучука и их зависимость от разных параметров таких как
густота пространственной сетки, химическая структура и молекулярный
вес, основываясь на экспериментальных данных полученных в основном
II. Определения измеряемых величин и спектры 189
на серии хорошо охарактеризованных и полностью сшитых эпоксидных
смол на основе бисфенола А [ 11-14], а также на некоторых полибутадиенах
и фторэластомерах [15-17]. Некоторые особенности нелинейного вязкоуп-
ругого поведения также будут рассмотрены.
Поскольку каучуки как правило являются аморфными полимерами,
именно температура стеклования означает тот предел ниже которого струк-
тура перестает быть равновесной. Также как и в случае других аморфных
полимеров молекулярная сегментальная подвижность определяет темпе-
ратуру стеклования каучука [1, 5, 7, 18-20]. Переход от стеклообразного
к каучукоподобному состоянию происходит тогда, когда хаотично ориен-
тированные полимерные цепи между сшивками приобретают способность
к вязкоупругому отклику. Имеется много общих черт, также как много
различий в вязкоупругом поведении аморфных полимеров и каучуков.
Лучшее понимание обеих систем может быть достигнуто путем сравнения
и интерпретации на основе теоретических моделей.
II. ОПРЕДЕЛЕНИЯ ИЗМЕРЯЕМЫХ ВЕЛИЧИН ДГ)( G(t)
И G*(co) И СПЕКТРЫ £(log X) И Н(1од т)
Вязкоупругость представляет собой зависимый от времени
механический отклик и обычно характеризуется величиной податливости при
ползучести, релаксацией напряжений или динамическими механическими из-
мерениями. Поскольку время является переменным, дополнительным к дефор-
мации и силе, для получения однозначных характеристических зависимостей
из этих измерений одно из переменных должно сохраняться постоянным.
А. Ползучесть и восстановление
В экспериментах по сдвиговой ползучести напряжение сдвига оо созда-
ется в предварительно отрелаксированном материале и поддерживается
постоянным, тогда как возникающая деформация сдвига y(Z) монотонно
растет со временем t.
При достаточном времени наблюдения скорость ползучести постепен-
но уменьшается стремясь к пулю, и y(Z) достигает предела, если образец
является твердым вязкоупругим телом. Напротив, если материал является
вязкоупругой жидкостью, скорость ползучести уменьшается до конечной
постоянной величины. Достигается установившееся вязкоупругое состо-
яние и y(Z) возрастает неограниченно. Эксперимент по ползучести имеет
вторую стадию, когда напряжение делают нулевым после определенного
периода ползучести. Частично или полностью деформация, накопленная
в процессе ползучести при этом восстанавливается как функция времени для
вязкоупругого твердого тела или жидкости. Для вязкоупругой жидкости та
часть деформации, которая является постоянной и невосстанавливающейся,
190 “* V Глава 5. Вязкоупругие свойства каучука
отражает вклад вязкого течения в общую величину деформации накопленной
в процессе ползучести. Поскольку вязкоупругое твердое тело не течет, вся
деформация накопленная в процессе ползучести является обратимой.
Когда деформация или скорость деформации достаточно малы, упругий
отклик при ползучести оказывается линейным. В этом случае, когда вели-
чина деформации делится на постоянное напряжение возникает необычная
кривая податливости при ползучести; иначе говоря в каждый момент вре-
мени существует только одно значение этого отношения, т. е. у(0/ао = J (0.
Необычная функция податливости при сдвиговой ползучести J(t) (Па-1
или см2/дин, 1 Па-1 = 0,1 см2/дин) полученная для аморфного полимера
включает обычные составляющие
J (0 = У (0/ сто = Jg + + t/r\ = Jr (z) + Z/l] (1)
где Jd — запаздывающая податливость; \y(t) — нормализованная функция
памяти, которая равна нулю при t=0 и равна единице когда t = <»; а Т| (Па*с
или Пуаз) является сдвиговой вязкостью (10 Пуаз = 1 Па*с). Величину Jg
называют податливостью стекла, представляющую долговременной предел
деформации, который увеличивается так быстро, что его зависимость от
времени не может быть наблюдаема в обычно достижимых эксперименталь-
ных пределах, даже при температуре стеклования, когда большинство мо-
лекулярных движений замедлено. Член £/т| отражает непрерывную вязкую
деформацию. Jr(t) это обратимая податливость сдвига которая может быть
определена при измерении ползучести при восстановлении. Для вязкоуп-
ругого твердого тела Т| практически является бесконечностью, поскольку
существует молекулярная сетка, предотвращающая всякую непрерывную
деформацию. Аналогом ур.(1) для вязкоупругого твердого тела является
Эксперименты по ползучести и восстановлению являются единственной
характеристикой вязкоупругого поведения которую можно легко интерпре-
тировать, т.к. согласно ур. (1) вклады в величину податливости при ползу-
чести являются аддитивными по отношению к деформации. Молекулярные
процессы происходящие при этом являются просто коротко- и длиномасш-
табными конфигурационными переориентациями, а также вязкое течение,
отражающее постоянно растущее смещение центров тяжести соседних моле-
кул. Также было показано, что растворитель в случае растворов полимеров
вносит аддитивный вклад в деформацию ползучести [21].
Имея все вязкоупругие функции важно отметить предельные величины
или формы которые качественно независимы от молекулярной структуры.
Для вязкоупругой жидкости = Jg, = 0], a Jr(t)
-Z/T| = J' + Jd = Js- Последнее предельное значение Js называется обратимой
сдвиговой податливостью при установившемся процессе. Она отражает
II. Определения измеряемых величин и спектры -* V 191
максимальную обратимую деформацию в расчете на единицу напряжения
и отражает в свою очередь максимальные конфигурационные перестройки
достижимые при существующем напряжении.
Для вязкоупругого твердого тела Jr(t) = J(0,lim^J(0 = Jgn =
= Jg+ Обозначение Je отражает равновесную податливость твердого
тела.
В. Релаксация напряжений
После того как достигнута постоянная величина дефор-
мации в предварительно отрелаксированном материале, возникшее на-
пряжение сдвига уменьшается с течением времени до нуля вязкоупругой
жидкости и до предельного равновесного значения для вязкоупругого
твердого тела. Сдвиговый модуль при релаксации напряжений (Па или
дин/см2)
G(0 = ®(0/YO =Се +[ЭД-<?е]<Р(0 (3)
где уо есть созданная постоянная дефоромация, ф(0 функция релаксации
уменьшающаяся от (р(0) = 1 при t = 0 до (р(°°) = 0 при t = и Ge — равно-
весный модуль имеющий конечное значение для вязкоупругого твердого
тела и равный нулю для вязкоупругой жидкости. Зависящие от времени
напряжения, обусловленные разными молекулярными механизмами
являются неаддитивными и поэтому трудно или вообще невозможно
выделить и охарактеризовать каждый из них индивидуально. Тем не ме-
нее G(t) можно связать с J(t) через интегральное уравнение , с помощью
которого одна функция может быть рассчитана из другой численными
методами [22].
С. Динамические механические измерения
К образцу прилагаются синусоидальные напряжения или
деформации вплоть до достижения установившихся значений напряжени
и деформаций с фиксированным углом сдвига фаз между сигналами входа
и выхода. В случае синусоидальной деформации сдвига
у(г) = Уо5^сог (4)
где уо амплитуда деформации и со угловая частота, тогда напряжение осцил-
лирует синусоидально как
а(0 = ао sin((Qt+ 8) (5)
Поскольку напряжение всегда приводит к деформации, угол сдвига фаз
всегда положительный. Используя тригонометрическую формулу для синуса
192 Глава 5. Вязкоупругие свойства каучука
суммы двух углов, ур (5) может быть переписано в применении к модулю
накопления и модулю потерь как
o(z) = у0 [G'(co)sm(coZ) + G"(co)cos(coZ)] (6)
где G’(со) = (со/уо) cos8 и G” (со) = (оо/уо) sin8. Здесь G’ является мерой
упругой энергии накапливаемой и обратимо получаемой, a G” есть мера
энергии диссипированной в тепло в процессе циклических деформаций.
Отношение G”(со)/G’ (со) есть tanS. Хотя это и не пишется обычно в явном
виде, 8 является функцией со. Комплексный динамический модель сдвига
G*(co) определяется как
G * (со) = G'(co) + iG"(co) = (q0 /у0 )exp(iS) (7)
Можно определить [1] из общего выражения для расчета напряжений
при любой предистории развития деформаций что G’ и G” связаны с релак-
сационным модулем G(t) в ур. (3) следующим образом:
G'(co) = Ge +cojo [G(z)-Ge]smcoZ (8)
G"((o) = <oJ”[G(«)-Ge]cos®< (9)
Снова в ур. (8) и (9) модуль Ge равен нулю для жидкости но в то же вре-
мя существует равновесный модуль для вязкоупругого твердого тела. Для
жидкости можно показать что lim 0С'(ю) = и lim 0C"(<w) = ем/- Для
твердого тела lim^gG^tw) = Ge и = cof^[G(0 - Ge]dt.
Аналогично вышеприведенному покажем, что синусоидальная дефор-
мация по отношению к приложенному синусоидальному напряжению при
постоянной частоте может быть выражена в терминах податливости накоп-
ления (в фазе) Г (со) и податливости потерь J” (со) (на 90 °C отстающей от
фазы) в виде
y(z) = q0 [ J'(co)sm(coZ) - ./"(co)cos(coZ)] (10)
где J'(со) = (/q/ct^cosS и = (/^o^sinS. Комплексная динамическая сдви-
говая податливость J* (со) определяется как
J* (<о) = /'((»)- = (у0 /о0 )exp(-i8) (11)
Знак минус в уравнениях (10) и (11) появился как следствие того что
деформация отстает от напряжения. Можно показать что для вязкоупругой
жидкости динамическая податливость при ползучести связана с податливо-
стью ползучести J (t) с помощью односторонних преобразований Фурье:
J'(co) = Js-со£ [Js - J(t) + t/v^sin(ntdt
(12)
II. Определения измеряемых величин и спектры
Л
193
/"(со) = 1/сот| + ®Jo [/9 - J(t)+tlT\\costotdt (13)
Соответствующие соотношения для вязкоупругого твердого тела
/'(со) = Je -cojQ [/е -J(t)]sin(Otdt (14)
/"(со) = J~[Je - J(t)\cos(otdt (15)
Из уравнений (7) и (И) немедленно следует, что
Z*(co) = l/G*(co) (16)
Для жидкости низкочастотные пределы равны:
и lim (/"(w) = 1/ам/. Для твердого тела они составляют lim
ИПт^Н<»)=<<[4-7(0И.
D. Спектры запаздывания
Спектр запаздывания Л(Х) где X является временем запаз-
дывания, характеризует вклад в податливость при ползучести /(Z), дина-
мической податливости накопления /'(со) и динамической податливости
потерь /"(со) в соответствии со следующими формулами (для вязкоупругой
жидкости; для вязкоупругого твердого тела Т| в условиях опыта равна бес-
конечности в следующих уравнениях),
J(t) = Jg + "j L(i-e-i/K)dlnX + - (17)
—ОО Т)
/»=/,
dink
(18)
/"(со) =
7 LcoX
£ (1 + со2Х2)
1
dlnk + —
СОТ]
(19)
Интегралы, включающие L, касаются только обратимой деформации
которая в большинстве случаев выражает ориентацию молекул в отличие
от других механизмов. Постоянная деформация характеризуется членами
содержащими коэффициент вязкости Т|. Из ур. (17) можно видеть, что дол-
говременной предел дает обратимую податливость.
/s=/r(oo) = = + J Ldlnk
(20)
194
Глава 5. Вязкоупругие свойства каучука
Таким образом податливость в установившемся состоянии Js есть сумма
податливости в стекле и предельной замедленной податливости,
Jd = j Ldlnk (21)
Е. Спектры релаксации
Релаксационный спектр Н(т) где т - время релаксации, ха-
рактеризует вклад в модуль сдвига G(0, динамический модуль накопления
6"(со) и динамический модуль потерь G"(co) в соответствии со следующими
формулами (для вязкоупругого твердого тела; для вязкоупругой жидкости
Ge = 0 в следующих уравнениях),
G(t) = Ge +^He-t/xdlnz (22)
Gz(co<) — Ge +
На)2т
(1 + (O2T2)
(23)
Han
dim
(24)
В пределе при co—>°o yp. (23) приводит к формуле,
Gg=Ge+^ffdlriT:
где G есть модуль стекла.
(25)
F. Податливость растяжения (объемная), модуль
растяжения (объемный)
Несмотря на то, что выше были приведены вязкоупругие
функции и их взаимосвязи для сдвиговой деформации, они также могут
быть применены для деформации растяжения и для объемной деформации.
Аналогами J(t), J'(co), J"(co), G'(co) и G"(w) в случае деформации растя-
жения являются податливость при растяжении D(t), податливость накоп-
ления при растяжении ZT(co), податливость потерь при растяжении
модуль растяжения E(t), модуль накопления при растяжении Е'(со), и модуль
потерь при растяжении £"((0) соответственно. Для объемной деформации
соответствующими величинами являются объемная податливость B(t).
объемная податливость накопления В'(со), объемная податливость потерь
III. Температура стеклования -* V 195
модуль объемной деформации K(t), объемный модуль накопления
Л'(со), объемный модуль потерь А^"(со). Все уравнения в предыдущем тексте
справедливы для растяжения (объемной деформации) после того как все
величины будут изменены соответственно от сдвига к растяжению (к объ-
емной деформации). Можно показать что три податливости и три модуля
связаны следующими уравнениями
3 9
(26)
£(«) = —^+—(27)
Если образец совершенно несжимаем так, чтоK(t) » G(t),nB(t)
мы получим более простые формулы:
£(<) = 3G(<) (28)
Z)(z) = J(z)/3 (29)
Эти уравнения позволяют связать вязкоупругие функции при деформа-
ции сдвига с соответствующими величинами при растяжении. При наличии
высокой податливости каучуки оказываются практически несжимаемыми
и приведенная взаимосвязь между модулями и податливостями при сдвиге
и при растяжении выполняется хорошо. Однако при малой податливости,
приближающейся к Jg, коэффициент Пуассона jx (равный при деформации
растяжения -d\nw/d\nl, где w — ширина образца, а I его длина) оказывается
меньше чем 0,5. При этом уравнения (28) и (29) более не соблюдаются. Для
стекла |i * 0.25. В случае, когда G(t) = K(t), E(t) = 2.25G(t).
III. ТЕМПЕРАТУРА СТЕКЛОВАНИЯ
Температура стеклования Т является функцией скорости
охлаждения и давления при котором она определяется. При заданной ско-
рости охлаждения и давлении, Tg определяет соответственное состояние
характеризующее вязкоупругие свойства и области применения некристал-
лических полимеров [5,20]. Это параметр, являющийся характеристикой ма-
териала. Так например, если Темного выше чем температура эксплуатации,
то полимер является твердым стеклообразным и может быть пригодным для
применения в качестве инженерного пластика. Если Tg существенно ниже,
полимер является каучукоподобным и может быть использован в резиновой
промышленности. Поскольку употребляется выражение «переход из каучу-
196 -•v Глава 5. Вязкоупругие свойства каучука
Рис. 1. Схематическое изображение изменения объема Vи энтальпии Нс изменением
температуры. Верхняя линия охлаждение от состояния равновесной жидкости
при достаточно большой скорости Qv Линия в средине — охлаждение при
меньшей скорости Q2. Тонкие линии — экстраполяция линий стекла к более
высоким температурам. Их пересечение с равновесной линией жидкости
(толстая пунктирная линия) соответствует температурам стеклования Tg(Q{)
и Tg (Q2). Направленная вниз точечная стрелка отражает изотермическое
физическое старение в течение определенного времени. Самая нижняя линия
отражает нагревание со скоростью Q2 застеклованного полимера для восста-
новления термического равновесия при повышенной температуре.
коподобного состояния в стеклообразное состояние» заметим, что на самом
деле такой переход не существует, поскольку и каучук и стекло находятся
в одинаковом термодинамическом состоянии, различна только молекулярная
подвижность. Кинетические изменения при переходе от каучукоподобного
к стеклообразному состоянию отражаются в изменениях энтальпии Н или
объема V. Это видно например на рис. 1 где при фиксированной скорости
охлаждения как Я так и V уменьшаются при движении по равновесной
кривой до тех пор пока приближение жидкости к ее равновесной структуре
становится столь медленным что она не успевает приходить в соответствие
с уменьшающейся температурой. При всех более низких температурах
удельный объем превышает равновесное значение и наблюдаемое поведе-
ние соответствует стеклообразному состоянию. При более низкой скорости
охлаждения Q2 система приближается к равновесию при более низкой тем-
пературе и тогда наблюдается более низкое значение Tg. Точка пересечения
равновесной линии жидкости с неравновесной линией стекла определяется
III. Температура стеклования -*v 197
как Tg. Таким образом /^характеризуется убывающей функцией зависимости
от скорости охлаждения.
После прекращения охлаждения при температуре ниже Т энтальпия
и объем медленно но непрерывно уменьшаются приближаясь к их равновесным
значениям. Во время этого снижения другие кинетические свойства такие как
скорость ползучести уменьшаются. Такое замедление кинетических процессов
называется физическим старением и на рис. 1 отмечено точечной линией. Эти
эффекты обратимы, в том смысле что они исчезают если материал нагреть до
температуры выше Tg, где быстро достигаются равновесные свойства [23-27].
Скорость объемного сжатия сильно уменьшается при падении температуры
и становится невозможным достигнуть равновесия при температурах ниже чем
примерно /^-25 °C. Если физическое старение заканчивается после времени г
и стекло в процессе отжига нагревается при постоянной скорости, V сначала
увеличивается вдоль линии более плотного стекла, следуя ниже равновесной
линии объема и наконец возвращается к равновесию при температуре несколько
выше чем Tg (Q2)- Объяснение этого поведения не является предметом рассмот-
рения этой главы но может быть найдено в литературе [5,26,27]. Температура,
при которой линия стекла пересекается с равновесной линией называется
фиктивной температурой /^отожженного стекла.
IV. ИЗМЕНЕНИЕ ОБЪЕМА В ПРОЦЕССЕ ВУЛКАНИЗАЦИИ
Полимер, независимо от того, переплетены или нет его
цепи, является вязкоупругой жидкостью. Для того чтобы превратить его
в вязкоупругое твердое тело, полимерные цепи должны быть химически
связаны друг с другом с помощью вулканизующих агентов с образованием
молекулярной сетки. Этот процесс называется отверждение (curing) или
вулканизация (vulcanization). Объем уменьшается в процессе вулкани-
зации потому что Ван-дер-Ваальсовы «связи» заменяются более корот-
кими ковалентными связями. Поскольку Tg сильно зависит от объема,
необходимо точно знать как меняется объем в процессе вулканизации,
величину объема достигаемую после завершения вулканизации, а также
его зависимость от плотности сшивки и других параметров. Кроме объем-
ного сжатия интересно знать изменение других параметров как функции
времени вулканизации £сиг таких как (1) увеличение фиктивной темпе-
ратуры Tj [25-28], (2) увеличение степени вулканизации и количество
гель-фракции, (3) увеличение вязкости и (4) уменьшение равновесной
податливости Je. Такие закономерности полученные экспериментально —
редки. Полезно поэтому привести результаты экспериментального изу-
чения отверждения серии эпоксидных смол на основе диглицидилового
эфира бис-фенола А отличающихся длиной исходных линейных молекул
[11-14]. Некоторые основные параметры этих марок Epon Resins 828,
1001F, 1004F, 1007F приведены в Табл. 1.
198 -*v- Глава 5. Вязкоупругие свойства каучука
Таблица 1 Свойства эпоксидных смол
Материал г/моль n f Растворимая фракция, %
Epon 828 380 0.14 -14 2.0 0.3 420
Epon 1001F 996 2.31 31 1.9 1.3 910
Epon 1002F 1342 3.52 40 1.8 1.9 ИЗО
Epon 1004F 1450 4.85 56 1.7 2.8 1520
Epon 1007F 2600 70 1.4 9.0 2870
ОН
Их структурные формулы следующие:
о—сн2—сн—сн2
п
о—сн^—сн—сн2
сн,
где п — среднее число повторяющихся единиц в молекуле эпоксидной смолы,
а/ — функциональность.
Отвердителями являются диамин 4,4' — метилен дианилина (MDA)
Н
н
и 4,4'-диамино дифенил сульфон (DDS).
Усадка Epon 1001F в результате его реакции в стехиометрическом соотноше-
нии с диамином DDS при двухстадийном отверждении представлена на рис. 2.
Реакционная смесь сначала нагревается со скоростью 5 °C /мин до 142 °C под
давлением 5 МПа. После достижения постоянной температуры отверждения
IV. Изменение объема в процессе вулканизации 199
Рис. 2. Изменение удельного объема и температуры при отверждении эпоксидной
смолы 1001F/DDS под давлением 5 МПа.
Рис. 3. Значения удельного объема из рис. 2 представленные как функция тем-
пературы.
200 Глава 5. Вязкоупругие свойства каучука
ТС||Г = 142 °C, уменьшение объема в результате отверждения эпоксидной смолы
измерялось в течение более 2 суток (50 часов). На этой первой стадии удель-
ный объем уменьшился на 3.5%. На следующей, второй стадии температура
отверждения была увеличена до 190 °C в течение 3 часов чтобы приблизиться
к полному отверждению, что привело к дополнительной 0.1%-ной усадке.
Рис. 4. Кривые удельный объем — температура при охлаждении полученные для
полностью отвержденных смол 1007, 1004, 1001 и 828/DDS.
Изменение удельного объема при отверждении приведено па рис. 3,
где показана зависимость удельного объема от температуры. Смесь реа-
гентов вначале находилась в стеклообразном состоянии при комнатной
температуре. В процессе нагревания до Т = 142 °C, фиктивная темпе-
ратура 7} 35 °C как видно из рисунка, огибается кривой роста удельного
объема аналогично тому, как это показано на рис. 1. После этого видно,
что объем реакционной смеси растет линейно с ростом температуры до
Tcur. Изотермическая контракция объема вызванная отверждением при
ТС11Г показана на рис. 3 как вертикальная падающая линия. После этого
следует линия равновесный объем - температура отвержденного 1001/
DDS пересекаемая первый раз когда температура поднимается от Tcur до
190 °C на второй стадии. При охлаждени 5 °C /мин линия равновесный
объем - температура возвращается в противоположном направлении
IV. Изменение объема в процессе вулканизации -•V 201
вплоть до температуры стеклования Т полностью отверженного 1001/
DDS когда линия объема претерпевает излом чтобы следовать по линии
стекла как это показано на рис. 1.
Кривые охлаждения удельный объем — температура полученные для
полностью отверженных смол 1007, 1004, 828/DDS приведены па рис. 4.
Закономерное снижение удельного объема с ростом плотности сшивки
наглядно отражает рост Т с ростом плотности сшивки. Температуры стек-
лования для полностью отвержденных 1007, 1004, 1001 и 828/DDS равны
соответственно 101, 112, 127, и 204 °C.
Рис. 5. Кривые удельный объем — температура при охлаждении полученные для пол-
ностью отвержденной смолы 1001/DDS при разных скоростях охлаждения.
Зависимости удельного объема и температуры стеклования от скорости
охлаждения в общем виде объяснены рис. 1. Температура стеклования
определяется из пересечения равновесной кривой с кривой стекла экс-
траполированной до высокой температуры. Эта процедура определения Т
убедительно показана на рис. 5 на кривых удельный объем-температура
для полностью отвержденных смол 1001/DDS. Зависимость Tg от Q также
показана на рис. 5.
202
—Глава 5. Вязкоупругие свойства каучука
V. ВЯЗКОУПРУГИЕ СВОЙСТВА ПРИ ТЕМПЕРАТУРЕ ВЫШЕ Тд
А. Изотермические измерения частотных
и временных зависимостей
Вязкоупругий отклик равновесных каучуковых сеток может
быть исследован измерением модулей сдвига или растяжения, или податли-
востей как функции времени, или соответствующих динамических модулей
или податливостей как функции частоты. Как было показано в разделе II
значения одних вязкоупругих функций могут быть пересчитаны в другие
вязкоупругие функции.
Рис. 6. Кривые приведенной податливости при сдвиговой ползучести Jp (/), см2/дин
для Epon 1007/DDS при семи температурах (цифры на кривых) в логариф-
мических координатах.
Эпоксидные смолы марки Epon 828, 1001, 1002, 1004, 1007 предельно
вулканизованные стехиометрическими количествами DDS являются при-
мерами хорошо охарактеризованных сеток. Следовательно, исследования
механических свойств этих объектов дадут возможность заглянуть вглубь
природы вязкоупругости каучуковых сеток. Податливость при сдвиговой
ползучести J(t) этих смол была исследована при температуре выше темпе-
ратуры стеклования [11, 12, 14]. Согласно статистической теории высоко-
эластичности [1-5, 29-33] равновесный модуль Ge пропорционален произ-
ведению Тр где р — плотность при температуре Т, и поэтому равновесная
V. Вязоупругие свойства при температуре выше 7^ Jv 203
податливость Je пропорциональна (Гр)-1. Поэтому следует ожидать что./(£)
будет пропорциональна (Гр)-1 и/(£) Тр является величиной, которая должна
сравниваться при разных температурах. В действительности приведенная
податливость при ползучести
Jp(t) = J(t)Tp/ToPo (30)
произвольно откладывается на графике в зависимости от времени, где
То — температура отсчета, а р плотность при температуре То. Это служит
для того, чтобы можно было получить из этих кривых действительные
значения J(t) измеренные при температуре отсчета То. Наглядные дан-
ные приведены на рис. 6, где Jp (t) определялась для Epon 1007/DDS при
семи температурах в интервале между 99.8 и 127.3 °C при выбранной Г
= 100.7 °C. Значения Jp(t) меняются от уровня стеклообразного состоя-
ния чуть выше 10-10см2/дин вплоть до каучукоподобной податливости
близкой к 10-7см2/дин.
В. Температурная зависимость
Традиционно принято, что температурная зависимость мод
(движений) или механизмов реализации вязкоупругости пропорциональ-
на одному и тому же мономерному коэффициенту трения £() [1, 5, 7, 34].
Для каучуковых сеток моды вязкоупругости состоят из двух групп: моды
с короткими временами запаздывания, ответственные за изменение
объема и температуры стеклования, и моды с большими временами за-
паздывания, характерные для молекулярных прядей между сшивками,
ответственных за высокоэластическую деформацию и за Je. Таким обра-
зом, времена запаздывания Х(Т) всех вязкоупругих мод составляющих J
(t) при любой температуре Токазываются связанными с теми же модами
при температуре отсчета То с помощью одного и того же умножающего
фактора следующего вида
ат=^0(Т)/^(Т0) (31)
Это означает, что величина (t) измеренная при (Т, t) эквивалентна из-
меренной при (То, t/a^, как это видно из рис. 6, где значения податливости
сдвигаются в сторону меньших времен вдоль логарифмической шкалы
с ростом температуры. Такая температурно-временная эквивалентность вяз-
коупругих функций рассматривается как простая термореологическая. На
практике проверка наличия температурно-временной эквивалентности эк-
спериментальных данных выполняется путем последовательного смещения
величин полученных при Т, сначала для Тближайшей к То вдоль оси log(t)
путем подбора значения -logaTтак чтобы обеспечить взаимное перекрытие
кривых с образованием в результате обобщенной кривой. Величины для
204
Глава 5. Вязкоупругие свойства каучука
Т > То смещаются в сторону больших времен, тогда как величины, измерен-
ные при Т < TQ смещаются в сторону коротких времен. Хорошо построенная
приведенная кривая показывает, что вязкоупругий отклик является терморе-
ологически простым [35]. Он означает, что logJp(t) при TQ распространяется
на весь температурный интервал. Фактор смещения Ор который был применен
для смещения кривых податливости при ползучести для того чтобы получить
единую приведенную кривую включает и температурную зависимость. Фактор
приведения о^подходит и для аналитических форм зависимостей среди которых
часто выбирают уравнение Вильямса-Лэндела-Ферри [1],
logaT =—С^(Т — Tq)/{C2+T — (32)
Или эквивалентное уравнение Фогел я-Фулчера-Таммана-Гессе (ФФТГ) [1J,
ат = Аехр[сЦТ-Тх)\ (33)
Для выбранной температуры отсчета То, А - еау^СДТо-Т’»)]. Пара-
метр С/2.303 равен произведению (\С2, двух констант в уравнении ВЛФ, а
Тж = То -С2 . По уравнениям ВЛФ или ФФТГ температурной зависимости
^обобщенная кривая может быть построена для другой выбранной темпе-
ратуры отсчета Т.
Рис. 7. Приведенная кривая податливости при сдвиговой ползучести Jp(t) сдвинута
по шкале приведенного времени //а7для наложения на кривую при темпе-
ратуре отсчета 100.7 °C.
Такое построение выполнено для величин Jp(t) Epon 1007/DDS на рис. 6.
Приведенная кривая протяженностью в 10 порядков по временной шкале
при выбранной температуре отсчета То= 100.7 °C показана на рис. 7.
Подобного рода проверка принципа температурно-временной эквива-
лентности удивительно надежно подтвердила его достоверность и на других
молекулярных сетках в полимерах [14-16].
Кривые J (t) для других исследованных эпоксидных смол Epon 1004,
1002,1001,8z8/DDS были успешно приведены при выбранных температурах
V. Вязоупругие свойства при температуре выше Tg Jv 205
отсчета и результаты показаны на рис. 8 как функции приведенного времени
1/аТв двойных логарифмических координатах. Этот сравнительный график
был построен таким образом, чтобы все приведенные кривые пересекались в
одной точке со значением податливости logJ (t) = -8.5. Выбранные значения
температур отсчета Тобыли: 100.7,110.7,118.6,130 и 205 соответственно для
смол Epon 1007, 1004, 1002, 1001/DDS.
Рис. 8. Сравнение приведенных кривых податливости при сдвиговой ползучести
эпоксидных смол Epon 828,1001,1002,1004,1007/DDS в логарифмических
координатах при температурах отсчета указанных на кривых. Значения
температур близки к соответствующим температурам стеклования.
Значения фактора сдвига по шкале времен аТ которые были получены
смещением кривых податливости при сдвиговой ползучести для построения
обобщенных кривых, показанных на рис. 8, приведены на рис. 9. Логарифм
ат изображен в зависимости от обратной абсолютной температуры. Получен-
ная температурная зависимость может быть описана как уравнением ВЛФ
(32), так и уравнением ФФТГ (33) за исключением наиболее плотной сетки
828/DDS, которая описывается уравнением Аррениуса.
С. Равновесная податливость Je
Из рис. 8 можно видеть, что равновесная податливость Je
монотонно снижается при переходе от 1007/DDS к 828/DDS как и сле-
довало ожидать исходя из основ теории высокоэластичности, поскольку
концентрация цепей сетки увеличивается и молекулярный вес отрезков
цепей Мх между сшивками уменьшается в том же порядке. Значения Мх,
рассчитанные как pRTJe приведены в Табл. 1. Они замечательно близки к
значениям молекулярных весов исходных эпоксидных смол.
Как видно из рис. 4, порядок величин плотности в первую очередь
определяется плотностью сшивок. Однако эпоксидные сетки с меньшей
плотностью сшивок и меньшей удельной плотностью неожиданно обна-
206 JV" Глава 5. Вязкоупругие свойства каучука
руживают пониженную податливость Jg при их соответствующих темпе-
ратурах стеклования. Можно думать, что это происходит из-за особенно
сильной зависимости значений Jg и из-за значительного роста Tgс ростом
плотности сшивок. Сильная температурная зависимость/^возникает ве-
роятно из-за роста вторичного релаксационного максимума в эпоксидах
с более плотными сетками поперечных связей. Это предположение может
быть проверено с помощью диэлектрических измерений потерь в области
вторичной релаксации.
Рис. 9. Логарифмический температурный фактор смещения в зависимости от
обратной абсолютной температуры для четырех эпоксидных смол (обозначе-
ны на кривых) полученный на основе процедуры приведения примененной
для получения приведенных кривых на рис. 8.
D. Спектры запаздывания
Вязкоупругие спектры запаздывания Lp(lnX) были рассчи-
таны из кривых Jp(t) приведенных на рис. 8. Итерационные компьютерные
расчеты оказались необходимы для получения оптимизированных значений
Lp(lnX) [21], которые показаны на рис. 10.
Температуры, при которых рассчитаны спектры Lps выбраны так.
чтобы сделать одинаковыми положения максимумов на временной
шкале. Эти температуры приведения 71'лишь незначительно отлича-
ются от значений То, полученных из обобщенной податливости приме-
ненной для построения кривых на рис. 8. Коротковременные линейные
изменения logk с изменением log (X/ аг) являются фактически общими
для всех исследованных эпоксидов. Наклон в 1/3 каждой кривой в ко-
ротковременной области предположительно есть результат ползучести
V. Вязоупругие свойства при температуре выше -'V 207
Андраде [36, 37], в коротковременной области обычно используется
для определения Jg. Нельзя исключить того, что действительный наклон
может отличаться при переходе от сетки к сетке. Тем не менее, за преде-
лами области ползучести Андраде разные сшитые смолы обнаруживают
довольно симметричный максимум logLp, высота которого увеличи-
вается с ростом молекулярного веса структурных единиц сетки, т. е.
с ростом длины прядей молекулярной сетки. Более рыхлые молекулярные
сетки не неожиданно проявляют способность в большей степени дисси-
пировать механическую энергию превращая ее в тепловую. Увеличиваю-
щаяся высота пика logLp ясно указывает, что энергия разрушения должна
обнаруживать аналогичное увеличение и это действительно имеет место
[38]. Две эпоксидные смолы с наиболее рыхлыми сеткам и обнаруживают
начало появления второго долговременного пикаЛр(1пХ), который должен
появляться тогда, когда в величину Je делают вклад как сетка зацеплений
так и сетка поперечных связей. Для того чтобы обнаружились оба пика
сетка поперечных связей должна быть намного более рыхлой чем сетка
зацеплений.
Рис. 10. Спектры запаздывания в зависимости от приведенного времени в логарифми-
ческих координатах для четырех эпоксидных смол, обозначенных на рисун-
ке. Температуры, при которых даны спектры, выбраны так, чтобы привести
к одному времени положения максимумов.
208 JV" Глава 5. Вязкоупругие свойства каучука
VI. ВЯЗКОУПРУГИЕ СВОЙСТВА ДРУГИХ МОДЕЛЬНЫХ
ЭЛАСТОМЕРОВ
Вязкоупругие свойства серии предельно вулканизованных
эпоксидных смол отличающихся густотой сетки и присущие им закономер-
ности были рассмотрены в некоторых деталях в предыдущем разделе. На
качественном уровне эти свойства присущи всем пространственного сшитым
эластомерам, хотя количественно они зависят от строения макромолекул
и от химической природы сетчатого полимера и сшивающего агента. От-
сюда возникает необходимость рассмотреть вязкоупругие свойства других
модельных сетчатых систем и взаимно их сопоставить.
А. Фторэластомеры марки «Вайтон» (15)
Исходным материалом является сополимер винилиденфто-
рида и гексафторпропилена с молекулярным весом около 200000. Вулкани-
зующая группа: на 100 частей сополимера 3 — высокоактивная магнезия, 3 —
гидроксид кальция и 1-4 — вулканизующие агенты. Были исследованы три
образца фторсополимера «Вайтон» разной степени поперечного сшивания
полученных от фирмы Дюпон. Средние молекулярные веса отрезков цепей
сетки Мс равнялись: 2700 (образец 10-В), 4100 (10-А), и 7800 (11-А). Эти
характеристики вместе с другими показателями свойств, приведены в Таб-
лице 2.
1. Исследование сдвиговой ползучести
Исследование торсионального сдвига позволило получить
значения сдвиговой податливости при ползучести J(t) которые менялись
в интервале от стеклообразного состояния 10"10см2/дин (10“9Па-1) до близких
к равновесным значениям порядка обычных 10-7см2/дин (10-6Па-1). Зна-
чения приведенной сдвиговой изотермической податливости, рассчитанной
из ур. (30) соответствуют принципу температурно-временной суперпозиции
и хорошо очерченные обобщенные кривые показаны в логарифмических
координатах на рис. 11 в зависимости в зависимости от приведенного
времени t/aT, где а^есть обычный фактор смещения по шкале времен.
Наиболее плотно сшитый образец имеет наибольшую Т= -19.6 °C как
и следовало ожидать. Помимо увеличения Tg рост плотности сшивки приво-
дит к снижению равновесной податливости Je. Такое снижение конечно
оправдано с точки зрения классической теории высокоэластичности, из
которой известно, что Je обратно пропорциональна концентрации упру-
гих элементов, т. е. числу цепей в единице объема сетки. Приближение
к различным значениям Je для трех изученных фторэластомеров наглядно
показано на рис. 11.
VI. Вязоупругие свойства других модельных эластомеров 209
Таблица 2 .
Образец Плотность при 25 °C, г/см3 logGe Паа> logGe Паь> Мс, г/мольа> Мс, г/мольс
11-А 1.837 5.76 5.80 7790 7220
10-А 1.833 6.05 5.94 4060 5220
10-В 1.830 6.22 6.17 2720 3070
Образец Cv кПа С2, кПа Log2(Ct + С2) d> г/ге>
11-А 150 280 5.93 3.01
10-А 300 200 6.00 2.42
10-В 620 0 6.09 2.13
а' По данным сжатия набухших образцов
Ь| Оценочные данные по кривой ползучести при То = -20 °C
' Оценочные данные на основе Je (ползучесть)
d* и С2 (Па) — константы уравнения Муни-Ривлина. (Уравнение см. [1])
' Индекс набухания (вес.) в метилэтилкетоне при 25 °C
Рис. 11. Приведенная сдвиговая ползучесть Jp(t) в зависимости от приведенного
времени в логарифмических координатах. Температура отсчета Т равна
-20.0 °C и одинакова для всех кривых. Экспериментальные точки показаны
только для образца 11-А.
2. Температурная зависимость факторов смещения
Горизонтальные сдвиги по логарифмической шкале времен
необходимые для совмещения кривых полученных при разных температурах
определяются величиной логарифма фактора смещения. Таким образом,
значения факторов смещения отражают принципиальную зависимость от
температуры вязкоупругих механизмов деформации. Оказалось возможным
представить температурно-временные зависимости для трех образцов еди-
210 —• V- Глава 5. Вязкоупругие свойства каучука
ной кривой построенной по уравнению ФФТГ (33) в котором есть только
один параметр Тж, который отражает изменения Tg, зависящей от степени
сшивания. Совпадение расчета и эксперимента показано на рис. 12. Важна
атмосфера в которой проводятся измерения т.к. в этих условиях происходит
абсорбция влаги тогда как образцы испытанные в условиях даже небольшо-
го вакуума (около 10-2тор = 1.3 Па) могут считаться относительно сухими.
Константа С в уравнении ФФТГ равна 990 °C для всех исследованных образ-
цов. Разница Д между Т и Тж составляет 32.8 °C для 11-А, 31.3 °C для 10-А
и 30.4 °C для 10-В. В пределах погрешности эксперимента А как оказывается
является постоянной что и наблюдается обычно для гомологических рядов
полимеров или растворов полимеров в пределах ограниченного интервала
концентраций [37,39].
3. Спектры запаздывания
Спектры запаздывания Lp были рассчитаны из кривых
J (I) на рис. 10. Результаты, после приведения к То = -20 °C показаны на
рис. 13, где главный максимум сосредоточения механизмов запаздывания
смещается к большим временам параллельно с ростом Т при увеличении
степени сшивки. Логарифмические кривые в области коротких времен
показывают наклон в 1/3 для всех трех образцов фторэластомеров что
предположительно является следствием развития ползучести Андраде
[36, 37], J(t) =J+ Р^1/3 в области коротких времен использована для
определения Jg.
Существенная особенность, которую следует отметить, это наличие дол-
говременного или терминального пика, высота которого растет с уменьше-
нием плотности сшивки. Образец 10-В с наибольшей плотностью сшивки
обнаруживает лишь малозаметное плечо в долговременной области, тогда
как образец 11-А с плотностью сшивки примерно в три раза меньшей обна-
руживает четкий долговременной терминальный пик означающий большие
вязкоупругие потери при больших временах по-видимому за счет участия
сетки зацеплений.
В перспективе может возникнуть подобный эффект, если различные
сетки будут исследованы при температурах соответственных состояний
(обычно температуры равноудаленные от соответствующих Г ). Такое
сопоставление спектров запаздывания сделано на рис. 14. При вы-
бранных температурах отсчета разность &=TQ-Tg оказывается равной
4.2, 5.3, 6.1 °C соответственно для 11-А, 10-А, 10-В, а участки спектра
в области коротких времен совершенно совпадают. Учитывая различия
в ординатах кривых на рис. 14 мы можем сделать вывод о том, что при
log (Х/аг) = 10 образец 11-А будет развивать ползучесть примерно в 100 раз
быстрее чем образец 10-В.
VI. Вязоупругие свойства других модельных эластомеров -*и- 211
Рис. 12. Логарифм фактора смещения в зависимости от разности температур. Ок-
ружающая среда в которой проводились испытания была либо влажной что
обусловливало абсорбцию влаги образцом, либо являлась вакуумом, что
делало образец более сухим. Тто = -57, -54, -50, -48 °C соответственно для
образцов 11-А (воздух), 10-А (вакуум), 10-В (воздух), 10-В (вакуум).
4. Динамические механические свойства
Если известны спектры запаздывания в широком времен-
ном диапазоне, оказывается возможным рассчитать в числовом выраже-
нии компоненты комплексной динамической сдвиговой податливости
Л(со) -//''(со) по уравнениям (18) и (19). Имея значения/'(со) и/''(со) мож-
но с помощью ур. (16) в буквенном выражении рассчитать динамические мо-
дули накопления G'p(со) и потерь G"p(со). Полученные результаты для f(со) и
212 Jv- Глава 5. Вязкоупругие свойства каучука
Рис. 13. Спектры запаздывания в зависимости от приведенного времени \/аТ (се
кунды) в логарифмических координатах. Температура отсчета То = -20 °C
для всех кривых.
Рис. 14. Спектры запаздывания в зависимости от приведенного времени в логариф-
мических координатах приведенные к соответствующим температурам
приведения так, чтобы показать соответствие первичных максимумов
в области коротких времен в переходной области.
67'(со) приведены на рис. 15 и 16 соответственно. Температуры отсчета выбран-
ные на этих рисунках одинаковы с аналогичными температурами для спектров
запаздывания на рис. 14 за исключением незначительного (1 °C) изменения для
образца 10-В. Кривые могут быть получены как полуколичественноезеркаль-
VI. Вязоупругие свойства других модельных эластомеров -'V 213
ное изображение кривых Jp (t). Предельное низкочастотное значение является
в то же время равновесной податливостью Уе, получаемой как долговременной
предел кривой Jp (t) для вязкоупругого твердого тела. Небольшое изменение
в форме первичного максимума потерь является отражением значительного
вклада степени поперечного сшивания в вязкоупругие потери в этой области
диапазона частот. Поскольку количество энергии диссипированной за один
цикл деформации пропорционально G" представляет интерес получить зна-
чение приведенного модуля потерь также как и тангенса потерь который яв-
ляется мерой относительной величины потерь. Рис. 16 показывает, что потери
в низкочастотной области в слабосшитом образце много больше при температу-
рах соответственных состояний. В области главного максимума потерь все три
кривые приблизительно накладываются друг на друга, а имеющиеся различия
не вполне ясны.
Рис. 15. Сравнительное изображение кривых приведенной динамической податли-
вости накопления (см2/дин=10 Па-1) в зависимости от приведенной частоты
соа^с-1) в логарифмических координатах. Приведенные температуры отсче-
та обеспечивают соответствие потерь в переходной области и сопоставляют
первичные максимумы тангенса потерь.
Однако же кривые тангенса потерь (рис. 17) показывают, что образец 11-А
с наименьшей концентрацией цепей в единице объема характеризуется
наибольшей относительной энергией диссипации в области первичного
максимума потерь. Ясно что сравнительная оценка потерь энергии оди-
накова как в случае первичного максимума потерь так и в низкочастот-
ной области, т. е. образец 10-В с наиболее плотной молекулярной сеткой
характеризуется наименьшим относительным количеством диссипиро-
ванной энергии.
214 —• V- Глава 5. Вязкоупругие свойства каучука
Рис. 16. Зависимость приведенных динамических модулей потерь (дин/см2 = 0.1 Па)
от приведенной частоты в логарифмических координатах. Приведенные
температуры сравнения обеспечивают соответствие потерь в области раз-
мягчения и сопоставляют первичные максимумы тангенса потерь.
Рис. 17. Зависимости тангенса потерь от приведенной частоты в логарифмических
координатах приведенные к температурам отсчета, что указывает на поло-
жение первичных максимумов.
VI. Вязоупругие свойства других модельных эластомеров V 215
В. Полибутадиеновые эластомеры с уретановыми
сшивками*)
Были исследованы вязкоупругие свойства следующих об-
разцов: три полибутадиеновых эластомера с уретановыми сшивками (ТВ-1,
ТВ-2, ТВ-3) с разной степенью сшивки; также аналогичным образом сшитый
бутадиенстирольный сополимер (HTBSR) и два полибутадиеновых полиме-
ра со статистической сшивкой полученной с помощью пероксида дикумила
(РВ-1 и РВ-2). В других местах текста образцы ТВ-1, ТВ-2, и ТВ-3 могут
иметь обозначения соответственно как НТРВ-1, НТРВ-2 и НТРВ-3.
Исследования торсионального сдвига были проведены на полибутадие-
новых полимерах с уретановыми сшивками при температурах в интервале от
-68 до 25 °C. Средние значения молекулярных весов цепей сетки Мс равны
3400, 5200 и 8300 соответственно для ТВ-1, ТВ-2 и ТВ-3. Кривые приве-
денной сдвиговой податливости Для всех трех образцов приведены
на рис. 18. Температуры отсчета взяты 7.4, 0.0 и 17.0 °C соответственно для
ТВ-1, ТВ-2 и ТВ-3 и таким образом обеспечивается суперпозиция в области
коротких времен в области первичного максимума потерь. Наиболее рыхлая
сетка сшитого уретаном ТВ-3 имеет наибольшую Je = 2.5-10-6 Па-1. Имеется
плато промежуточное между податливостью стеклообразного состояния J
(не достигнутой в наших опытах) и величиной ее уровень около 2.0-10-7 Па *
для всех трех образцов. Плотность сетки не влияет на форму температурноза-
висимого отклика вплоть до и даже включая промежуточное плато на кривых
Jp(t). Только часть переходной области близкой к Je оказывается подверженной
такому влиянию. Фактор смещения, который был принят для построения при-
веденных кривых ат неожиданно оказался подчиняющимся аррениусовской
температурной зависимости для всех трех образцов, а не зависимости соот-
ветствующей ВЛФ как это имело место для Epons и Vitons. Такой переход к
аррениусовской зависимости связан с природой сшивающих узлов [16].
Приведенные спектры запаздывания Lp рассчитанные из кривых сдвиговой
ползучести Jp^/ay) приведены на рис. 18, а также в логарифмических коорди-
натах на рис. 19 в зависимости от приведенного времени запаздывания Х/ат.
Выбранные температуры отсчета привели к совмещению коротковременных
частей спектра указывая на идентичность сеток с разной плотностью. Вклады
в терминальный пик напротив отражают топологические различия сеток.
Наиболее слабо сшитые сетки заметно более диссипативны при относительно
больших временах т. е. на этом отрезке шкалы времен реализуются механизмы
делающие вклад в долговременной или терминальный пик L. Объективной
мерой ширины плато на кривых Jp(t/ay) является расстояние между пиками
кривых/^ (Х/оу). Терминальные пики и их удаление от пика альфа-релаксации
необычайно велико (от 12 до 13 порядков по времени). Обычно спектральные
*> Химия образовния этих эластомеров подробно рассмотрена в “Жидкие углеводородные
каучуки” — Могилевич М.М., 1\ров Б.С., Морозов Ю.Л., Уставщиков Б.Ф. — М.: Химия,
1983. - с. 200.
216 -»V- Глава 5. Вязкоупругие свойства каучука
Рис. 18. Зависимость приведенной сдвиговой податливости Jp(t) (Па-1) от приве-
денного времени t/aT (в секундах) в двойных логарифмических коорди-
натах для трех полибутадиеновых эластомеров сшитых по концам цепей
уретанами. Время в секундах. Температуры отсчета при приведении вы-
браны так чтобы достигалась суперпозиция при малых временах дисперсии
первичного размягчения.
Рис. 19. Приведенные спектры запаздывания Ля как функция времени запаздывания
Х/ат в двойных логарифмических координатах для трех образцов поли-
бутадиеновых эластомеров сшитых уретанами по концам цепей. Отклик
приведен к температурам соответственных состояний этих эластомеров: 74,
О, 17 °C для области перехода первичного размягчения.
пики занимают до полдюжины порядков по шкале времен, и удаление терми-
нального пика от альфа-пика находится в сильной зависимости от плотности
сетки [ 1, 40]. Тем не менее обратная зависимость вязкоупругой диссипации
в области больших времен от плотности сетки впервые отмоченная в случае
отклика сеток натурального каучука [1,41], подтверждена феноменологически
на примере сшитых уретанами полибутадиеновых эластомеров, даже если их
вязкоупругие спектры не вполне объяснимы на молекулярном уровне.
VI. Вязоупругие свойства других модельных эластомеров 217
С. Сравнение различных эластомеров
Зависимости приведенной сдвиговой податливости J^t/ay) от
приведенного времени log(t/ат) в двойных логарифмических координатах для
нескольких эластомеров при О °C приведены на рис. 20. В числе исследованных
следующие эластомеры: (1) сшитый полиуретанами полибутадиеновый элас-
томер ТВ-2, описанный в подразделе В; (2) фторэластомер со статистической
сеткой Viton 10-А описанный в подразделе А; (3) два полибутадиеновых элас-
томера РВ-1 и РВ-2 статистически сшитых с помощью пероксида дикумила
(соответственно 0.8 и 0.3 г на 100 гполимера) [16]; (4) бутадиенстирольный со-
полимер [16] аналогичный сшитым уретанами полибутадиеновым эластомерам.
В то же время мы не будем рассматривать различные вязкоупругие функции
полученные на натуральном каучуке с разными плотностями сшивок [40-42].
Релаксация напряжений, динамические механические свойства и ползучесть
были исследованы на аналогичной серии образцов натурального каучука соот-
ветсвенно Тирионом и Шассэ [41], Ферри с сотр. [42] и Плазеком [40].
Рис. 20. Приведенная податливость при сдвиговой ползучести Jp(t) (Па-1). В зависи-
мости от времени запаздывания t/a^ в двойных логарифмических координа-
тах для пяти эластомеров при То=0 °C. Сплошная линия (НТРВ-2), пунктир
с длинными черточками (Viton), пунктир с короткими черточками (РВ-2),
линия точки-тире (РВ-1), линия точек (HTSBR).
Значения равновесной сдвиговой податливости Je измеренные в области
больших времен все равны 1.0-10-6 Па-1 за исключением податливости в РВ-1
которая неожиданно оказалась несколько выше. Тот факт, что переходная
218 Jv- Глава 5. Вязкоупругие свойства каучука
область для РВ-1 оказалась в области более коротких времен чем в случае
РВ-2 может быть объяснено большим значением отношения цис к транс
равного 0.80 по сравнению с 0.67, что обусловливает более низкую Т.
Тот факт, что скорость ползучести эластомера Viton приблизительно на пять
порядков величины меньше в переходной области, чем в случае РВ-2 объ-
ясняется вероятно влиянием Т которая на 70 °C выше. Наиболее неожи-
данно то, что переходная область сшитых уретанами эластомеров находится
в области удивительно коротких времен, так например промежуточное плато
у ТВ-2 достигается вблизи log(t/ay)= -12 несмотря на тот факт что его Т
почти на 13 °C выше чем в образце РВ-2. В то же время плато последнего
достигается в области времен около log(t/a^= -6, т. е. почти на семь порядков
позже. Следовательно материал который больше превышает его Т ползет
медленнее в переходной области.
Рис. 21. Приведенная податливость при сдвиговой ползучести Jp(t) в зависимости
от приведенного времени запаздывания t/a^ в двойных логарифмических
координатах для четырех эластомеров при То = ^Сплошная линия (НТРВ-
2), пунктир с длинными черточками (Viton), пунктир с короткими черточ-
ками (РВ-2), линия точек (HTSBR).
Однако же кривые податливости при ползучести при их соответствующих Т
показывают (рис. 21) что в пределах ошибки измерений переходные облас-
ти располагаются в одинаковом диапазоне на шкале времен. Характерно,
что протяженность всех четырех кривых J при уровне податливости
1.0-10-8Па-1 оказывается лишь немного больше одного порядка по шкале
времен. Относительная неточность определения Т равной ±1.5 °C может
быть причиной такой протяженности. До тех пор, пока не будут получены
более точные относительные значения Tg, мы можем предположительно
считать, что при Tg все полимеры при одинаковой скорости находятся далеко
в пределах переходной зоны. Такое заключение кажется обоснованным если
считать что малые смещения цепей сетки определяют как скорость ползу-
VII. Расчет энергии раздира эластомеров... -•и- 219
чести сразу после расстекловывания так и жидкостную структуру полимера
тепловые движения в которой определяют величину Т.
В Табл.З приводятся некоторые параметры, характеризующие комплекс
физических свойств эластомеров. Сравнение эластомеров друг с другом
можно также проводить основываясь на этих параметрах.
D. Другие исследования вязкоупругости
Исследования линейной и нелинейной вязкоупругости
эластомеров имеют давнюю историю. Вместо того чтобы делать обзор работ
выполненных в прошлом различными исследователями пользовавшимися
разными методиками мы предпочли представить вязкоупругие свойства
эластомеров измеренные, как нам кажется, точными методами с использо-
ванием хорошо охарактеризованных образцов. Мы надеемся, что эти данные
помогут увидеть наиболее общие вязкоупругие свойства эластомеров и их
зависимость от таких переменных как плотность сшивок и температура
стеклования. Сохраняются конечно различия между группами эластоме-
ров, которые обусловлены влиянием молекулярной структуры полимера
и природы сшивающего агента. Тем более читатель должен ознакомиться
с предыдущими работами в этой области. Здесь мы приведем лишь некото-
рые из них [40-53].
VII. РАСЧЕТ ЭНЕРГИИ РАЗДИРА ЭЛАСТОМЕРОВ
ИЗ ДАННЫХ ПО ИХ ВЯЗКОУПРУГОСТИ.
При высоких температурах (T»Tg) и малых скоростях
деформации, когда деформация практически равновесна, природа и про-
чность связей в основной цепи полимера играют определяющую роль при
разрушении (раздирании) эластомера. Лэйк и Томас [54] создали хорошую
теорию определяющую энергию раздира T0(Jm-2) в условиях близких к рав-
новесным, и зависящую от энергии связи и плотности сшивок в эластомере.
Подбор таких значений попавших как оказалось в интервал 40-80 Jm-2 был
произведен Джентом и Тобиасом [55]. При реальных скоростях и темпера-
турах раздира значения Т оказываются выше на порядки величины.
Смит [56], Маллинс [57] и Мэллер с Краусом [58] на качественном уровне
связали энергию раздира эластомеров и их зависящие от времени механи-
ческие свойства. Подробный анализ процесса разрушения эластомеров дан
в главе 10 этой книги. Здесь же мы хотим показать как скоростная зависи-
мость энергии раздира, которую трудно определить может быть рассчитана
из более легко определяемой функции податливости при ползучести. В про-
цедуре такого расчета все взаимосвязи структуры и вязкоупругих свойств
которые были выявлены для эластомеров могут быть использованы для
создания более упругих каучуков.
Таблица 3. Физические свойства модельных эластомеров,
Эластомер Tg(»C) logtf H/m2 togG' H/m2 re/V> xIO^cm-3 M* M-R Ml хим. Sw. G x!0”5H/m2 C2 x10-5H/m2 log2(C,+C2) S.I5 MS
ТВ-10 0.9577 -81n 6.10 6.13 5.11 2730 3140 1860 1760 4.25 1.3 6.04 2.95 32.9
ТВ-20 0.9557 -80n 5.97 6.0o 3.69 5230 6200 2590 2370 2.2 3.0 6.02 3.18 32.9
3.19
ТВ-33 0.9535 -80" 5.60 5.60 1.60 9700 5930 5930 1.20 1.30 5.70 4.53 14.2
РВ 0.896 -98k 5.7,
0.8% Di-cup 40-50-10m РВ 0.904 -93n 5.96 6.0, 3.66 3780 2600 2.75 3.30 6.08 3.25 35.0
0.3% Di-cup 5.96 3.71 3.27
36-54-10m
SBR-1 (SI) 0.962J (0.955)n -65n 5.96 5.90 3750 3740 2980 3.1 1.45 5.96 26.1
Viton 11A 1.837 -24.2a 5.763 5.80 2.36h 7790 7220 1.5 2.8 5.93 3.01 24.0
Viton 10A 1.833 -22.7a 6.045 5.9Z1 4.54h 4060 5220 3.0 2.0 6.0 2.42 28.8
Viton 10B 1.830 -19.6a 6.218 6.17 6.76h 2720 3070 6.2 0 6.09 2.13 37.3
220 -*v- Глава 5. Вязкоупругие свойства каучука
VII. Расчет энергии раздира эластомеров... -’V 221
Вклад необратимых вязкоупругих процессов в энергию раздира при
наименьших скоростях деформации должен явиться следствием самых
медленных масштабных молекулярных движений. С ростом скорости де-
формации энергия раздира должна монотонно возрастать поскольку более
быстрые короткомасштабные перемещения цепей включаются в вершине
растущей трещины.
Поскольку функция распределения времен запаздывания (спектр запаз-
дывания L) определяет набор механизмов вязкоупругости при движении по
шкале времен [17] наиболее простой вариант получения энергии раздира
из совокупности вязкоупругих свойств материала может быть
f(C) = (34)
гдеj\ и с1 — константы пропорциональности, а С (м/с) — есть скорость раз-
дирания. Необходимо допустить что с1/С = у-1 (сек), где у — деформация
растяжения в вершине растущей трещины. Нет однако оснований априорно
утверждать что все вязкоупругие механизмы одинаково эффективны в их
вкладе в энергию раздира. Применение уравнения (1) показывает что это
не так. Простое интегрирование спектра L приводит к пропорциональному
росту рассчитанной при относительно коротких временах (больших ско-
ростях) что является слишком малым. Средневесовая функция необходима
чтобы увеличить роль механизмов находящихся в начале переходной зоны;
см. следующий текст.
Примечания к Таблице 3.
ТВ (или НТРВ) = полибутадиен с гидроксильными концевыми группами модифицированный
толуилендиизоцианатом с отношением NCO/OH от 2 до 0, удлиненный 1,4-бутандиолом и
сшитый триметилолпропаном.
SBR-1 = бутадиенстирольный сополимер с гидроксильными концевыми группами, модифи-
цированный, пластифицированный и сшитый как ТВ.
РВ = полибутадиен сшитый пероксидом дикумила
Vitons =фторэластомеры
а> DSC 10°С/мин
Ь) Измерения при сжатии набухших образцов
с) оценка по кривой податливости То= -20 °C для Вайтон, То= 0 °C для других эластомеров
d) по константе С1 уравнения Муни-Ривлина
е) из молекулярного веса исходных веществ.
f) оценка по значению Je из кривой ползучести
h) измерения выполнены Адамсом и Гу-Гуофаном
к) оценка из положения переходной области размягчения на шкале времен
т) содержание цисчпранс-внннл
п) измерено в Акронском институте полимеров. ДСК скорость 5 °C /мин
р) эффективная концентрация цепей сетки в моль/см3. Из данных по сжатию. С\ и С2 в Па.
ч) S. I. = индекс набухания весовые доли. Вайтон в метилэтилкетоне, остальные в толуоле.
г) модули Юнга из кривой напряжение-деформация
222 -*v- Глава 5. Вязкоупругие свойства каучука
Наилучшее уравнение имеющее предсказательную силу для расчета
энергии раздира следующее
Т(С) = kf{ Л(/пс2Х)[1 + с3 ехр(с^ lnc2^ - с5 )2Jdln X (35)
где:
— масштаб связывающий скорость раздира и скорость растяжения
с2 — совмещает шкалу времен запаздывания и скорости раздира
с3 — определяет величину средневесовой функции
с/( — определяет ширину средневесовой функции
сГ) — определяет положение средневесовой функции, которое зависит от положения
коротковременного пика.
J\ — первичный масштабный фактор
к — фактор зависящий от равновесной податливости эластомера который равен
(5.0 -logJJ
Попытки улучшить совпадение результатов рассчитанных из ур. (35)
с результатами определения энергии раздира при температуре приведения
дали следующие значения ct=1000; с2, 100; с3, 10; с4, 0.020; а/р 7.5*109для
всех исследованных эластомеров. Значение с5 меняется от полимера к поли-
меру как это показано в Табл. 4 где представлены свойства исследованных
эластомеров. Значение с- было определено путем фиксации пика средневзве-
шенной функции в размере 5.0 десятичных порядков для больших времен по
сравнению с коротковременным пиком L (1пХ). Равновесная податливость
и значения энергии раздира вблизи нее также приведены в Табл.4.
Таблица 4. Константы некоторых модельных эластомеров.
Epon 28-1 есть Epon 828 фирмы Шелл сшитый стехиометрическими количествами метилен-
диамина. Остальные эластомеры описаны в Табл. 3.
Эластомер log/e, Па-1 р (25°С), г/см3 ^5
НТРВ-1 -79 -6.14 45 0.9577 1760 25.56
НТРВ-2 -79 -6.01 50 0.9557 2370 25.24
НТРВ-3 -79 -5.63 60 0.9535 5930 23.35
HTSBB-1 -65 -5.95 60 0.955 2980 25.42
Viton 10В -17 -6.19 45 1.830 3070 26.53
Viton ЮЛ -19 -5.98 55 1.833 5220 23.35
Viton 11А -22 -5.85 75 1.837 7220 23.дек
РВ (0.3%) -93 -5.99 45 0.904 2600 24.92
Epon 28-1 164 -7.21 10 1.194 270 29.48
VII. Расчет энергии раздира эластомеров... —'V 223
Рис. 22. Спектры запаздывания L(lnX) (в Па-1) в зависимости от времени запазды-
вания X (сек) в двойных логарифмических координатах для четырех
пространственносшитых полимеров. Температура отсчета для всех поли-
меров является их температурой стеклования.
Протяженные спектры запаздывания четырех сетчатых полимеров, иссле-
дованных выше, приведены на рис. 22 при соответствующих температурах
стеклования. Их свойства в коротковременной части спектра одинаковы,
но огромное различие наблюдается в длинновременной части спектра Х/ат.
Эпоксидная смола Epon 828 предельно сшитая стехиометрическим количес-
твом 4,4-метилендианилина имеет наиболее плотную сетку Мх = 270 и она
обнаруживает единственный узкий максимум в спектре L(X) и никаких
молекулярных движений в долговременной части шкалы. Фторэластомер
Viton 10А обладает существенно более рыхлой сеткой Мх и следовательно
больший по величине главный максимум L(X) определяющий наличие
значительно большей Je. Заметен небольшой длинновременной максимум
свидетельствующий что ячейки сетки достаточно велики чтобы стал заметен
вклад сетки зацеплений. Значительное количество долговременных движе-
ний отражает спектр сшитого изоцианатами полибутадиена с гидроксиль-
ными группами па концах и бутадиенстирольного сополимера имеющего
промежуточное плато на J(t). Такое механическое поведение не наблюдается
на других эластомерах и не нашло объяснения. Возможно влияние микрофа-
зового расслоения но не было получено никаких подтверждений для такого
предположения. Однако наличие совокупности весьма долговременных
молекулярных процессов в соответствии с гипотезой положенной в ос-
нову ур. (35) должно обеспечить большие энергии раздира при обычных
скоростях раздира. Безусловно результаты интегрирования средневесовой
224 -*v- Глава 5. Вязкоупругие свойства каучука
функции Л(Х) от больших до коротких времен показанные на рис. 23
и совпадение расчетных и экспериментальных данных является обна-
деживающими
Рис. 23. Измеренные (точки) и рассчитанные (кривые поур. 35) значения энергии
раздира для четырех аморфных пространственносшитых полимеров обо-
значенных на рисунке.
VIII. ТЕОРЕТИЧЕСКАЯ ИНТЕРПРЕТАЦИЯ ВЯЗКОУПРУГИХ
МЕХАНИЗМОВ И ИХ АНОМАЛИЙ
А. Нарушение термореологически простых
закономерностей в случае низкомолекулярных
полимеров
Резина является вязкоупругим твердым телом получаемым
пространственным сшиванием линейного полимера, который в исходном
состоянии является вязкоупругой жидкостью. Несмотря на это различие
имеются и общие проблемы в физике процесса стеклования и в вязкоупругих
механизмов в области диссипативного размягчения (называемой переходной
VII. Расчет энергии раздира эластомеров...
Л-
225
зоной между стеклообразным и высокоэластическим состоянием по терми-
нологии Ферри [1). Проблема в этой области станет понятной если сравнить
свойства чистой эпоксидной смолы Epon 1001F [14] со свойствами предельно
сшитой Epon 1001F | DDS рассмотренной в разделе V. Температура стекло-
вания Epon 1001F равна 31 °C, что существенно ниже 127°С — температуры
стеклования предельно сшитой Epon 1001F/DDS/. Рост Т при переходе
от олигомера к резине может быть объяснен (по крайней мере частично)
уменьшением удельного объема которое может быть оценено при сопостав-
лении рис. 4 и 5. Однако не так просто объяснить различия в температурной
зависимости положения областей диссипативного размягчения этих двух
материалов. Как ранее показано на рис. 6-8 кривые приведенной сдвиговой
податливости Jp(t) Epon 1001F/DDS измеренные при разных температурах
были приведены к обобщенной кривой при температуре отсчета TQ.
Такая термореологическая упрощенность области диссипативного размяг-
чения Epon 1001F/DDS не была обнаружена в случае исходной Epon 1001F.
Сдвиговая вязкость Т| была определена в интервале температур от 30 до
77°С. Обратимая сдвиговая податливость Jr(t) (определяемая ур. (1)) была
получена при девяти температурах в интервале от 30.2 до 53.5 °C как пока-
зано на рис. 24.
Рис. 24. Обратимая сдвиговая податливость Jr(t) как функция времени в логарифми-
ческих координатах при девяти температурах указанных на рисунке. Огром-
ное исчезновение долговременных вязкоупругих молекулярных процессов
очевидно при приближении температуры к Tg.
226 -’V Глава 5. Вязкоупругие свойства каучука
Установившееся значение обратимой сдвиговой податливости Js =
= указывает на сильную температурную зависимость. Колоссальное
десятикратное падение Js показано на рис. 24 при понижении температуры на
20 °C по направлению к Tg. Это значит, что кривые logJr(t) не могут быть при-
ведены к обобщенной кривой. Термореологическая сложность обнаруженная
исходной смолой Epon 1001F аналогична таковой для многих других низко-
молекулярных полимеров [18,19,37,59,60-67]. Удобным примером является
низкомолекулярный полистирол с молекулярным весом 12.300 взятый для
сравнения потому что его значение Js при высоких температурах почти оди-
наково с чистым Epon 1001F. Значения Jr(t) этого практически монодиспер-
сного образца полистирола приведены на рис. 25. Падение Js при понижении
температуры приближающейся к Т количественно совпадает в обоих случаях.
Такое аномальное, но в то же время весьма общее вязкоупругое поведение
низкомолекулярных полимеров несомненно противоречит предположению
о том, что времена релаксации всех релаксационных механизмов или мод просто
смещаются по шкале времен и пропорциональны мономерному коэффициенту
трения £о [1]. Это предположение, если бы оно было правильным, означало
бы что полимер термореологически прост, что очевидно не имеет места. Это
представляет фундаментальный интерес при изучении молекулярной дина-
мики полимеров. К сожалению, вероятно из-за трудности дать объяснение,
это обстоятельство было проигнорировано при исследовании вязкоупругости
полимеров в последние несколько десятилетий.
Рис. 25. В двойных логарифмических координатах зависимость обратимой сдвиговой
податливости Jr(t) от времени для образца практически монодисперсного по-
листирола с молекулярным весом 12.300 при шести температурах указанных
на рисунке. Драматично резкое исчезновение долговременных вязкоупругих
молекулярных процессов очевидно при приближении температуры к Tg.
VIII. Теоретическая интерпретация вязкоупругих механизмов... 227
1. Модель кооперативности (Coupling model)
Насколько нам известно, в настоящее время только одно
объяснение может быть дано нарушению термореологической упрощен-
ности в случае низкомолекулярных полимеров [18, 19, 61, 68, 69] и оно
основывается на рассмотрении модели кооперативности [70-77]. Детальный
анализ этой модели и выводы, которые могут быть сделаны на ее основании,
выходит за рамки этой главы. Здесь дается лишь краткое описание наиболее
существенных моментов. Релаксация сегментов определяющая «релаксацию
структуры» и закономерности стеклования и сопровождающаяся взаимной
укладкой молекул и их взаимодействием, является кооперативным про-
цессом, включающим сложные, зависящие от времени, движения многих
молекул. В то же время собственно сегменты участвуют в конфигурацион-
ных перестройках характеризуемых примитивным временем релаксации
и примитивной функцией релаксации (primitive relaxation function):
ф(<) = е;гр(-</тОа)
(36)
не все попытки перестроек здесь могут быть удачны из-за стесненности воз-
никающей благодаря межмолекулярному взаимодействию или спариванию
(coupling) отдельных сегментов. Первоначально удачные (неудачные) они
соответственно могут быть названы быстрыми (медленными) движениями
отдельных сегментов. Поскольку полимолекулярная динамика является
стохастической, в следующий промежуток времени быстрые (медленные)
движения становятся медленными (быстрыми). Немедленными параллель-
ными следствиями этой сложной полимолекулярной динамики являются:
(1) требуется большее количество порядков времени сегментальной релак-
сации для ее завершения (растягивается шкала времени), та оказывается
много больше чем тоа. (2) Процесс релаксации динамически неоднородный,
это означает что в любой момент времени имеются и быстрые и медленные
молекулы и они меняются ролями при движении по шкале времени в пре-
делах полного сегментального процесса релаксации (а-процесс) [72, 78].
Функция корреляции Кольрауша,
ф(/) = ехр[-(//та)1-"] (37)
где экспонента (1 -п) — есть доля единицы, а та — характеристическое время
релаксации сегментов, является вероятно одной из функций с наименьшим
количеством параметров которая однако прослеживает протяженность
времени сегментальной релаксации на несколько порядков. Шкала вре-
мен растягивается с ростом п. Замедление означает что та » тОа. Априори
соотношение между та и неизвестно. Однако модель кооперативности
предлагает взаимосвязь между этими двумя временами релаксации. При
достаточно коротких временах примитивная релаксация преобладает, про-
текая беспрепятственно. Только после некоторого, довольно хорошо опре-
228 -'и* Глава 5. Вязкоупругие свойства каучука
деленного времени t полимолекулярная динамика замедляет релаксацию.
Отсюда следует, что корреляционной функцией является ур. (36) для t < tc
и для t > tc плавно переходит в ур. (37) вблизи tc. Время перехода tc не зависит
от температуры, его значение определяется потенциалом межмолекулярного
взаимодействия. Для полимеров t » 2*10-12c [76, 77,80]. Этот переход пред-
сказывается теоретически [70, 72, 73], что обусловлено строгим анализом
простых моделей [74, 75] и подтверждается экспериментально [77, 80, 81].
Используя преимущества этого общего свойства перехода корреляционной
функции и полагая квазинепрерывность ее двух частей при tc мы получим
соотношение,
<38>
между двумя временами релаксации через параметр кооперативности п
который входит в дробную экспоненту корреляционной функции Коль-
рауша (ур. 37). Это соотношение было использовано, чтобы аномальные
свойства обусловленные существованием сложной полимолекулярной
динамики объяснить исходя из соответствующего нормального и по-
нятного показателя для процесса простой и независимой релаксации
[18,77,81].
В полимерах не имеющих боковых групп, появление вторичного макси-
мума релаксации при частотах больших чем частоты сегментальной релак-
сации является загадочным. Примеры этого включают полибутадиен (ПБ)
и даже полиизопрен (ПИ). Существование вторичной релаксации в ПБ
хорошо известно [82,83], но в ПИ она была обнаружена лишь подавно [84].
Результаты диэлектрической релаксации ПБ и ПИ демонстрирующие нали-
чие и сегментальной и вторичной релаксации представлены на рис. 26 и 27.
Одинаково загадочным является и возникновение вторичной релаксации в
жестких низкомолекулярных стеклах таких как толуол и хлорбензол [85-87].
Поскольку отсутствуют внутренние степени свободы, такая вторичная ре-
лаксация может происходить за счет некоторых локальных перемещений
всей молекулы. Такая вторичная релаксация относится к типу релаксации
Джохари-Голдстейна (ДжГ) [85-87]. Она по-видимому универсальна, су-
ществуя во всех стеклообразных веществах и может рассматриваться как
предшественница первичной структурной релаксации. Некоторые критерии
отличающие ДжГ-релаксацию от остального «огородного» разнообразия
обнаруженных вторичных релаксаций основывается на тесной взаимо-
связи ее проявлений с проявлением первичной релаксации. Примитивная
или независимая релаксация в модели кооперативности (ур. 36) является
предшественницей кооперативной (т. е. межмолекулярно спаренной)
а-релаксации (ур. 37) и это влечет за собой движение всех частей молекулы,
одновременно являясь локальным процессом. Таким образом, эти признаки
примитивной релаксации являются общими с ДжГ-релаксацией и таким
VIII. Теоретическая интерпретация вязкоупругих механизмов... 229
образом можно ожидать, что время независимой релаксации тОа практи-
чески близко к наиболее вероятному времени релаксации Тр ДжГ-релак-
сации при всех температурах и давлениях. Уравнение аналогичное ур. 38,
то« = (1С )П (та ) ~П позволяет рассчитать тОа если известны та и п в уравнении
Кольрауша (ур. 37) что соответствует временной зависимости а-релаксации.
Для полимеров и низкомолекулярных жидкостей tc приблизительно равно
2*10-12с. Кажется замечательным, что соотношение
Хоа(ЛР) » Тр(Т,Р) (39)
как оказалось справедливо количественно для многих стеклообразных
веществ [87-91]. Соответствие между двумя временами релаксации иллю-
стрируется рис. 26 и 27 для ПБ и ПИ. Универсальная ДжГ-релаксация дала
жизнь примитивной релаксации модели кооперативности.
Log10v (Hz)
Рис. 26. Результаты измерения изотермических диэлектрических потерь 1,4-полибута -
диена обнаруживающие проявление ДжГ-релаксации в области переохлаж-
денного жидкого состояния выше Tg. Компьютерное построение вписывается
в пик а-релаксации в виде в виде сплошной линии. Значение п определенное
таким образом приведено на рисунке. Каждая стрелка указывает на опре-
деленную величину при некоторой температуре определяющее положение
частоты независимой релаксации vOa= 1/21?^ где рассчитано по ур.
(38). ▲, о, V, — результаты полученные при температурах -97.5, -95, -92.5,
-91.2 соответственно.
Особенно интересно отметить, что ДжГ-релаксация была обнаружена
в несшитом Epon 828 и ее время релаксации Тр находится в хорошем
согласии с тОа [87, 91].
230
—Jлава 5. Вязкоупругие свойства каучука
। 11111Н| । ггнnq । 11|||||| । Iimiq । Iiiiiiij -niiuii|-ri ниц i riiiuq • 11 iinnf
1 1 Hlllll_I 11 mill i j mud I 11 Hili inniii_iiiniii iinmi iniini iiuiiii
W4 103 10-2 10-1 10° 101 102 103 104 105
Frequency (Hz)
Рис. 27. Спектры диэлектрических потерь ПИ. Результаты при 216.0 К (О), 211.15 К(п),
208.15 К (о), и 204.15 К (V) получены с помощью IMass Time Domain Di-
electric Analizer. Все другие данные в пределах от 10 Гц до 100 кГц были
получены с помощью емкостного моста CGA-83. Видно хорошее совпаде-
ние данных CGA-83 при 216.7 К (♦), 212.7 К (), 208.7 К (•), и 204.7 К (▼)
с данными IMass при 216.0 К (О), 211.15 К (□), 208.15 К (о), и 204.15 К (V)
соответственно, когда последние перемещены горизонтально на величину
• определенную по температурной зависимости ФФТГ для частоты а-релак-
сации для того чтобы учесть небольшие различия в температуре. Другие
восемь спектров получены только при помощи GGA-83. Спектры которые
обнаруживают a-пик потерь соответствуют (справа налево) температурам
236.7 К (*), 232.7 К (А), 228.7 К (+), и 224.7 К (д) Нижние три кривые CGA-
83 которые обнаруживают P-пик потерь были определены при температурах
(сверху вниз) 169.7 К (Д), 181.7 К (+), и 200.7 К (*). Вертикальные стрелки
показывают места рассчитанных частот от примитивной релаксации . При
тем перату paxvOa= (слева направо) 212.7 К (), 208.7 К (•), и 204.7 К
(▼). Положения этих можно сопоставить с положением вторичных ре-
лаксационных пиков при этих температурах.
Протяженность релаксации, Аер, ДжГ-релаксации [92] во всех стек-
лообразных веществах изменяется в процессе нагревания при переходе
температуры стеклования таким же образом как меняется энтальпия Н,
энтропия 5 и объем V. Производная от Аер по температуре A£^/dTувеличи-
вается от низких значений при температуре ниже Tg до больших значений
при температуре выше Т отражая такие же закономерности изменения
удельной теплоемкости Ср и коэффициента теплового расширения, которые
VIII. Теоретическая интерпретация вязкоупругих механизмов... -*V 231
являются производными соответственно dH/dTи dV/dT [92]. Угол поворотов
ДжГ-релаксации и отсюда Ае^ весьма вероятно зависят от удельного объема
и энтропии. Таким образом, скорость изменения Ае^ с температурой анало-
гична термодинамическим величинам. Более того, хотя времена релаксации
Тр всех (3-релаксаций включая ДжГ-релаксацию обнаруживают аррениусов-
скую зависимость в стеклообразном состоянии, фактическая температурная
зависимость при температурах выше Tg в области равновесного объема не
является продолжением аррениусовской температурной зависимости из об-
ласти ниже Tg. Это более похоже на температурную зависимость типа ФФТГ,
которая слабее чем зависимость та [93-95]. Тот факт, что Тр характеризуется
также температурной зависимостью ФФТГ в области выше Т согласуется
с ур. (38) и (39) поскольку температурная зависимость тоа рассчитанная
по ур. (38) из температурной зависимости та согласно ВФТГ не является
аррениусовской.
Интересно то, что протяженность релаксации Аер также как время ДжГ-
релаксации Тр обнаруживает изменения при Tg. Несмотря на тот факт, что
быстрая ДжГ-релаксация возникает задолго до медленной а-релаксации. Это
обстоятельство позволяет предположить, что ДжГ-релаксация чувствительна
к величине удельного объема (или его доли — свободного объема/) и/или
энтропии 5 (или конфигурационной энтропии 5С). В частности коэффици-
ент трения в процессе релаксации Тр зависит от V(f) и/или 5(5с). Благодаря
соответствию между двумя временами релаксации (ур. (39)), аналогичное за-
ключение может быть сделано по поводу коэффициента трения процесса тоа.
2. Объяснение термореологической сложности
Благодаря ДжГ-релаксации мы в предыдущей части смогли
сделать заключение о том, что время примитивной локальной сегментальной
релаксации тОл или ее коэффициент трения зависят от удельного объема V
(или долинювободного объема/) и/или от энтропии 5 (или конфигурационной
энтропии Sc). Поскольку примитивная релаксация также как ДжГ-релакса-
ция определяется локальными процессами, включающими мономер целиком
и соответственно их коэффициент трения может быть идентифицирован как
мономерный коэффициент трения £0 [1], т. е.
г0 « [е (40)
Мы спешим подчеркнуть, что в литературе [1] обычно связывают
с коэффициентом трения процесса тл и временами релаксации всех остальных
долговременных вязкоупругих молекулярных механизмов (как например
полная релаксация или спектр запаздывания). Эта общепринятая взаимо-
связь с тл не принята в рамках модели кооперативности (МК) в которой
вместо этого трактуется как коэффициент трения тОа [18, 59-61, 65, 68,
69], хотя зависимость от V(f) и/или от5(5с) сохраняется.
232 “* V- Глава 5. Вязкоупругие свойства каучука
Основная цель МК состоит в том чтобы учесть межмолекулярную коопе-
ративность в процессе локальной сегментальной релаксации и в процессе
перехода тОа в та . Из ур. (38) и (40) следует, что
" ft»]''1"” 1'1)
Экспонента 1/( 1 — тг) больше чем единица. Отсюда та зависит от V(f)
и/или от S(SC), а также от температуры и давления намного сильнее чем
ТОа ИЛИ Тр .
Переходная зона на термомеханической кривой низкомолекулярных
полимеров, имеющих однако зацепления, часто трактуется на основе моде-
ли Рауза модифицированой применительно к нерастворенному полимеру
[1]. Основной предпосылкой модели является то, что учитываются только
координаты одной цепи, поэтому движение цепи по Раузу не может быть
кооперативным и времена релаксации согласно модели xRi пропорциональ-
ны мономерному коэффициенту трения [18], т. е.
т» - С. <42>
Таким образом, время релаксации сегментального процесса ха харак-
теризуется более сильной температурной зависимостью, чем по модели
Рауза в переходной области, хотя первая короче чем последняя. Это различие
в температурной зависимости сразу дает объяснение разрыву термореоло-
гической упрощенности у низкомолекулярных полимеров (см. рис. 24 и 25)
[19, 37, 61]. Это также помогает объяснить наблюдаемое снижение Js ког-
да температура снижаясь приближается к Tg, объяснение в соответствии
с работой Нгаи, Плазека и Бэро [69].
Несоответствие между температурными зависимостями ха и xHi (ур. 41
и 42) определяется величиной параметра кооперативности п. У полистиро-
ла п больше (= 0.63) чем у полиизобутилена (= 0.45) [19, 96, 97]. Отсюда
можно предположить, что степень нарушения термореологической упро-
щенности окажется меньше у полиизобутилена (ПИБ), чем у полистирола
(ПС). Это предположение, также как и другие предсказанные различия
в вязкоупругости ПИБ и ПС были подтверждены экспериментально [68,
69]. Когда разбавитель с низкой Т вводится в ПС степень межмолекуляр-
ной кооперативности в чистом ПС снижается и параметр кооперативности
п уменьшается. Следовательно, в растворах ПС разрыв термореологической
упрощенности оказывается меньше и снижение податливости в установив-
шемся режиме Js при понижении температуры подавляется. Такое изме-
нение вязкоупругих свойств при переходе от чистого ПС к его растворам
подтверждено экспериментально [98], при этом вязкоупругий спектр ПС
в растворе похож на спектр ПИБ [99]. Позже мы рассмотрим динамику
узлов в пространственно сшитом полимере и ее изменение при добавлении
растворителя (при набухании). Динамика узлов также хорошо описывается
VIII. Теоретическая интерпретация вязкоупругих механизмов... -* v 233
моделью кооперативности и характеризуется параметром кооперативности
узлов rtj [100, 101]. Уменьшение яупри разбавлении было установлено при
экспериментальном исследовании динамики узлов [100, 101] аналогично
изменениям динамики сегментов ПС при добавлении разбавителя.
В. Термореологическая упрощенность эластомеров
В противоположность исходному чистому Epon 1001F
(см. рис. 24) кривые сдвиговой податливости Jp(t) эластомера Epon 1001F/
DDS были успешно приведены к хорошо очерченной обобщенной кривой
Jp^t/dy) при температуре отсчета То (см. рис. 6-8). Термореологическая
сложность и сильное падение Js с понижением температуры обнаруженные
в чистом Epon 1001F, отсутствовали в Epon 1001F/DDS. Такое изменение
вязкоупругих свойств в результате пространственной сшивки в полимере
можно объяснить следующим образом.
Полимерные пряди в Epon 1001F/DDS привязаны обоими концами
с помощью агента сшивания и очевидно, что их релаксация не может быть
описана моделью Рауза. Более того, узлы поперечного сшивания находятся
в условиях значительного стеснения. Эти стеснения приводят к межмолеку-
лярному спариванию движений узлов. Как показано в следующей главе D
динамика узлов хорошо описывается в рамках модели кооперативности с по-
мощью убывающей экспоненциальной функции Кольрауша ехр[ -(t/Xj )x~nj ]
при наличии значительного по величине параметра rtj [100, 101]. Здесь
есть время релаксации узлов замедленное в результате межмолекулярного
спаривания. По аналогии сур. (38) для сегментальной релаксации свя-
зано со временем примитивной релаксации узлов т0/ соотношением:
=[гМ'/"") ,43)
В модели кооперативности все времена примитивной релаксации вклю-
чая тОл и т0/ пропорциональны мономерному коэффициенту трения .
Отсюда из ур. (43) получим температурную зависимость в свою очередь
зависящую от rtj
ъ - к,/”-’ 1441
Поскольку узлы являются межмолекулярно спаренными и характе-
ризуются весьма большим значением rij возникают крупномасштабные
моды i полимерных прядей закрепленных на обоих концах узлами, и ко-
торые теперь характеризуются ненулевым параметром кооперативности
и очевидно не могут быть описаны в рамках модели Рауза. Температурная
зависимость их времен релаксации 06 ]1^1”я‘J не отличается от та
поскольку сравнимо с rtj и п. Таким образом, термореологическая
сложность вязкоупругого отклика пространственных сеток сводится
234 -*V- Глава 5. Вязкоупругие свойства каучука
к минимуму и не может быть обнаружена в процессе температурно-вре-
менного суперпозиционирования.
Кажется, что узлы сетки могут быть более эффективными в стеснении вяз-
коупругих механизмов поскольку являются более масштабными (т. е. боль-
шие длины волн и большие времена запаздывания). Наибольший эффект
должен проявляться по отношению к моде у которой масштаб перемещения
соответствует молекулярному весу между узлами, Мх, и которая определяет
равновесную податливость сетки. Мы рассмотрим сейчас эту проблему путем
повторного анализа податливости при ползучести ряда предельно вулкани-
зованных эпоксидных смол. Чтобы облегчить сравнение мод отрезков цепей
между узлами, мы вычтем/ из каждой приведенной податливости J(t) при
ползучести Epon 828F/DDS, 1001F/DDS, 1004F/DDS, 1007F/DDS. Раз-
ность ~ Jg) отложена в зависимости о приведенного времени на рис. 28.
Для податливости, уровень которой около 4 ГПа-1, разность - Jg) имеет
форму степенного закона и обусловлена исключительно сегментальным
движением. Отметим, что такое подчинение степенному закону согласуется
в области коротких времен с падающей экспоненциальной формой
полученной для податливости обусловленной сегментальным запаздыванием
на примере нескольких полимеров [61,96,98,102] и для которой Д/а равно
примерно 4 ГПа-1. Значения - Jg) выше, чем 4 ГПа-1 обусловлены вяз-
коупругими модами, масштаб которых больше, чем масштаб сегментальных
движений. В случае сеток наибольший масштаб вязкоупругих мод определя-
ется величиной М . На рис. 28 фактор смещения и температура отсчета для
828/DDS такие же как на рис. 8. Для трех других эпоксидов потребовалось
дополнительное горизонтальное смещение чтобы их режим степенного за-
кона наложился на аналогичную зависимость у 828/DDS. Таким образом,
при скорректированных температурах отсчета все сетки имеют одинаковое
время сегментального запаздывания.
Теперь можно сопоставить вязкоупругие отклики четырех сеток. Одно
очевидное различие между ними состоит в том, что равновесная податли-
вость уменьшается с уменьшением М, как следствие исчезновения длин-
номасштабных вязкоупругих мод. Менее заметно смещение вязкоупругих
мод при одинаковом уровне податливости в направлении больших времен
с ростом плотности сетки. Величина смещений неодинакова но увеличивается
с податливостью (то же с ростом масштаба вязкоупругих мод). Две горизон-
тальные стрелки на рис. 28 иллюстрируют эту тенденцию. Такое поведение
легко объяснимо на основе модели кооперативности. Действительно вяз-
коупругая мода i с фиксированным масштабом перемещения оказывается
более стесненной (и, следовательно характеризуется большим значением
параметра спаривания nJ в плотной сетке 828/DDS, чем в более рыхлой
VIII. Теоретическая интерпретация вязкоупругих механизмов... -* V 235
сетке 1007F/DDS. Из уравнения модели кооперативности т, ~п,)
следует что т. в 828/DDS более чувствителен к изменению температуры, чем
это наблюдается в случае 1007F/DDS.
Рис. 28. Сравнение кривых [Jp (t)- Jg] построенных в зависимости от приведенного
времени t/aT для предельно сшитых Epon 828/DDS (о), 1001/DDS (О),
1004/DDS (Д) и 1007/DDS (V). Фактор смещения и температура отсчета
для Epon 828/DDS такие же как показанные на рис. 8. Для остальных трех
эластомеров горизонтальные смещения -0.72, -1.18, -1.5 были проведены,
чтобы обеспечить суперпозицию данных для коротковременной области
соответствующей сегментальной релаксации.
С. Изменение времени сегментальной релаксации
и времени релаксации Джохари-Голдстейна
с изменением густоты сетки.
Мы уже отмечали в разделе VIII. 1 что ДжГ-релаксация об-
наруженная в несшитом Epon 828 и ее время релаксации Тр находятся в хо-
рошем согласии с тОл. Мы также показали, что параметр кооперативности п
сегментальной релаксации повторяющихся единиц вблизи узлов сопоставим
с параметром кооперативности п л самих узлов (см. также следующий раздел D).
В результате наблюдается рост среднего значения п при сегментальной ре-
лаксации с ростом плотности сетки. Согласно ур. (38) такое увеличение п
обусловливает сопутствующий рост времени сегментальной релаксации та
и отсюда рост температуры стеклования /^который безусловно наблюдается
в этом случае. С другой стороны, тОл не меняется если плотность сетки не
слишком высока, чтобы оказывать влияние на удельный объем и конфигу-
236 “* V- Глава 5. Вязкоупругие свойства каучука
рационную энтропию. Можно думать, исходя из равенства ^оа(^)~тр(^)
(ур. (39)) что после пространственного сшивания Epon 828 время ДжГ-ре-
лаксации не будет заметно отличаться от времени найденного в чистом Epon
828, тогда как та и температура стеклования существенно возрастут. Изохро-
ны [103] и изотермы [104] диэлектрической релаксации Epon 828 показали,
что действительно меняется мало и подчиняется приблизительно той же
аррениусовской температурной зависимости, тогда как Т увеличивается
значительно с ростом времени вулканизации. При температуре стеклова-
ния Т, та оказывается одинаковым для всех образцов по определению,
но интервал раздвижки log[xa(Tg)] - log[x$(Tg)] увеличивается с ростом
времени вулканизации [103, 104]. Такая закономерность предсказывается
моделью кооперативности с помощью ур. (38) которое демонстрирует рост
/og[Ta(7^)]-Zog[Tp(7^)] с ростом п который обусловлен ростом плотности
сшивки, при этом 4a(Tg)^^(Tg) /ур. (39)/.
D. Динамика узлов
1. Экспериментальные данные
Динамика движений (релаксации) узлов в сетчатых по-
лимерах оставалась не вполне ясной вплоть до «пришествия» результатов
измерения спин-решеточной ЯМР-релаксации в полимерах, содержащих
изотоп фосфора 31Р, и измерений выполненных на примере сеток на основе
политетрагидрофурана с узлами из трис (4-изоцианатофенил)-тиофосфата
[100]. Закономерности релаксации узлов были изучены в сетках с моле-
кулярным весом цепей между узлами Мс в пределах от 250 до 2900. Доми-
нирующий механизм ядерной 31Р спин-решеточной релаксации был изучен
в широком интервале температур и был описан удовлетворительно с помощью
спектральных функций полученных с помощью Фурье-преобразований
убывающей экспоненциальной корреляционной функции Кольрауша
Cj (0 = )1-n' ] (46)
где Tj и rij соответственно время корреляции узлов и параметр кооператив-
ности. В температурном интервале измерений в области существенно выше
Tg [100] было сделано допущение что характеризуется температурной
зависимостью аррениусовского типа
х}=£ехр^Пт} (47)
Основываясь на этом Ши и др. [100] рассчитали параметр кооперативности
па предэкспоненциальный множитель и энтальпию активации Еа для
ряда сетчатых полимеров с различной плотностью пространственной сетки
а также набухшего образца (рис. 29).
VIII. Теоретическая интерпретация вязкоупругих механизмов... -*и- 237
2. Интерпретация на основе модели кооперативности
Было обнаружено, что nj возрастает с ростом густоты
сетки и уменьшается при добавлении растворителя при постоянной
густоте сетки [100]. Такие закономерности nj находятся в соответствии
с интерпретацией nj как параметра кооперативности поскольку межмо-
лекулярные стеснения возрастают с ростом густоты сетки. Наблюдается
существенный рост энтальпии активации Еа с ростом густоты сетки что
компенсируется сопутствующим уменьшением предэкспоненциального
множителя ^оо. С другой стороны, при набухании сетчатого полимера
с Мс = 650, значение Еа уменьшается при одновременном увеличении .
Экспериментальные данные касающиеся пр Еа , и исследованных
сетчатых полимеров приведены на рис. 30 и 31.
Рис. 29. Параметр кооперативности в динамике узлов определенный из экспери-
ментальных данных Ши и др. [100] для четырех полимерных сеток
с разными молекулярными весами Ме. Темные треугольники и кружки
отражают результаты 31Р ЯМР-исследований при поперечной и про-
дольной поляризации соответственно. Темные квадраты для набухших
образцов с Мс = 650. Линии проведены, чтобы облегчить отслеживание
закономерности.
Аррениусовская температурная зависимость подразумевает то же самое
времени примитивной релаксации узлов т0/что в явной форме может быть
записано как
XQj = x^exp{EalRT)
(48)
238 -* v- Глава 5. Вязкоупругие свойства каучука
Рис. 30. Темные перевернутые треугольники и светлые кружки - кажущаяся энтальпия
активации Еа полученная из 31Р ЯМР-измерений при поперечной и про-
дольной поляризации соответственно [100. Темный квадрат — Еа для на-
бухших образцов с Мг = 650. Светлые перевернутые треугольники и темные
кружки — Еа рассчитанная по ур. (49) по значениям Еа и nj для опытов
при поперечной и продольной поляризации соответственно. Светлый квад-
рат есть Еа рассчитанная из Еа и nj для набухшего образца. Темный ромб
и светлый треугольник есть Еа определенная непосредственно из экспери-
мента для образца с Ме = 250 с использованием поперечной и продольной
поляризации соответственно.
Взаимосвязь между Ту и дублирующая ур. (38) была выражена ур. (43).
Это последнее порождает две отдельных зависимости,
Е* = Еа/(1-п/) <49>
И
(50)
Эти уравнения качественно объясняют экспериментально наблюдаемый рост
Еа и уменьшение тто с ростом плотности сшивки на основе одновременного
роста пГ Количественно можно рассчитать ЕП и тто по ур. (49) и ур. (50)
соответственно имея экспериментальные значения пр Еа , и тто для каждой
сетки и /с=2-10"12с [106]. Рассчитанные значения Еа и т^ показанные на
рис. 30 и 31, соответственно, оказываются приблизительно постоянными
(Еа» 27 кДж/моль и тто 10-14с) показывая что все узлы сетчатого полимера
VIII. Теоретическая интерпретация вязкоупругих механизмов... 239
имеют приблизительно одинаковую подвижность, их движения не замедля-
ются межмолекулярным спариванием до различной степени в зависимости
от плотности сетки. Приблизительно постоянная величина Еа» 27 кДж/моль
может быть интерпретирована как барьерная энтальпия и 10"14с как
величина обратная некоторой вероятной частоте перескока.
Рис. 31. Темные перевернутые треугольники и светлые кружки - кажущийся пред-
экспоненциальный множитель определенный Ши и др. [100] из их эк-
спериментальных результатов исследования четырех сетчатых полимеров с
разными молекулярными весами цепей между узлами Мс, применяя попе-
речную или продольную поляризацию соответственно. Темные квадраты —
для набухших образцов с Мс = 650. Светлые перевернутые треугольники
и темные кружки означают рассчитанное по ур. (50) из значений
при поперечной и продольной поляризации соответственно при t = 2-10-12 с.
Светлые квадраты означают рассчитанное из значений и nd для
набухшего образца.
3. Аналогия модели стесненных узлов Флори
(модели эластичности) и модели кооперативности
при интерпретации динамики узлов
Применение модели кооперативности к анализу динамики узлов обна-
руживает по-существу аналогию с применением модели Флори стеснен-
ных узлов [33, 107, 108] к анализу природы высокоэластичности сеток.
Расчет кривой напряжение-деформация реальной сетки требует анализа
отклика данной цепи на деформацию всего образца которая классически
описывается двумя крайностями: аффинной [109] и фантомной [3, 29, 30]
240 -*и- Глава 5. Вязкоупругие свойства каучука
моделями. Реальная сетка обнаруживает зависимость упругого напряжения
от деформации, которая варьирует между предсказаниями либо аффинной
либо фантомной моделями сеток. Это не удивительно, поскольку узлы
в реальной полимерной сетке флуктуируют от положений соответствующих
аффинному смещению, в то же время помехи со стороны соседних цепей
уменьшают величину этих флуктуаций по сравнению с теми которые до-
ступны в фантомной сетке [31, 110]. В модели стесненных узлов Флори
и сотр., флуктуация узлов ограничена доменом стеснений налагаемых сте-
рическими препятствиями создаваемыми соседними сегментами причем
размер и расположение этих доменов изменяется в процессе деформации.
Флори ввел параметр к, определяемый числом узлов с объеме, занимаемом
одним отрезком цепи сетки и являющийся мерой жесткости локальных
стеснений по сравнению с теми, которые возникают в фантомной сетке.
Вклад в величину силы со стороны стеснений является нулевым, если к = 0.
Таким образом, те же доводы приведенные Флори в пользу важности учета
локальных стеснений узлов (обусловленных взаимодействием соседних
цепей) в его анализе упругости полимерных сеток, подтверждают необ-
ходимость использования моделей, таких как модель кооперативности,
которая убедительно объясняет влияние таких стеснений на динамику
при описании процесса релаксации узлов.
Модель стесненных узлов Флори и модель кооперативности учитыва-
ют влияние стеснений на узлы в полимерных сетках. Они подчеркивают
различные проявления влияния на узлы сетки стеснений, создаваемых
окружающими цепями. Флори рассматривал последующие ограничения
возможных конфигураций в сетке, которые влияют на энергию упругости.
Модель кооперативности концентрирует внимание на путях, которыми
межмолекулярные стеснения замедляют движения узлов сетки и приводят
к изменению корреляционной функции узлов идеальной сетки фантомных
цепей С h(t). Учитывая что фантомные цепи могут проходить сквозь друг
друга мы ожидаем, что С h(t) должна экспоненциально зависеть от времени
ехр (-^/туо). Следуя основному физическому принципу лежащему в основе
модели кооперативности, стеснения узлов превратят Cph(t) в Cj(t) как это
показано в ур. (46) для времен больше чем t = 2‘10-12с.
Согласно модели кооперативности величина п возрастает с ростом жес-
ткости стеснений по сравнению с налагаемыми фантомной сеткой. Таким
образом п имеет сходство с параметром к в модели стесненных узлов. Упру-
гие напряжения в модели стесненных узлов fc отличаются от таковых для
фантомных цепей f h в идеальной сетке (отсутствие свободных концов).
Отношение fc/fph является мерой вклада в величину напряжения со стороны
локальных стеснений флуктуаций узлов от вклада фантомных цепей. Подоб-
ным образом в модели кооперативности мы можем пользоваться логарифмом
отношения Zog(T00/T00) и отношением Е*а/Еа как мерой влияния стесне-
Таблица 5. Аналогия между моделью Флори стесненных узлов в теории высокоэластичности и моделью
кооперативности в динамике узлов.
Характеристика сетки Ожидаемый эффект Модель Флори Модель кооперативности 31Р ЯМР эксперимент
Л п Е*а/Еа-\ п Е*а/Еа- 1
Повышенная плотность сетки Жестче фиксация узлов выше выше выше выше выше выше выше
Разбавитель Пониженная интенсив- ность стеснений ниже ниже ниже ниже ниже ниже ниже
Повышенная функциональность узлов Большая стесненность узлов выше выше выше выше
Растяжение Облегчение стеснений ниже ниже ниже ниже
VIII. Теоретическая интерпретация вязкоупругих механизмов..
NJ
Р*
242 -* V" Глава 5. Вязкоупругие свойства каучука
ний на релаксацию узлов. Обычно является величиной порядка 10-14с
(см. также рис. 31). Это, вместе с 10"14с показывает, что отношение
намного меньше единицы. Отсюда, следуя ур. (49) и (50) как Zog(T00/T00) так
и разность ( Еа / Еа -1) увеличиваются с ростом п или с ростом жесткости
стеснений, и обе величины исчезают при п = 0 (что соответствует фантомной
сетке). Такие зависимости п аналогичны соответствующим зависимостям
величины fc в модели Флори.
Показанные аналогии подтверждаются зависимостями от плотности
сетки, концентрации разбавителя, функциональности узлов и макродефор-
мации стеснений узлов в модели Флори вместе с n, ) и ( Еа / Еа -1)
в модели кооперативности. Результаты такого сопоставления модели Флори
и модели кооперативности суммированы в Табл. 5.
IX. ПРИЛОЖЕНИЕ: СПИСОК ОБОЗНАЧЕНИЙ
А (Lrp В в* В' В" с 62 cphw Cj(t) С1 эмпирическая константа в уравнении ФФТГ фактор смещения объемная податливость комплексная объемная динамическая податливость объемная податливость накопления объемная податливость потерь эмпирическая константа в уравнении ФФТГ, или скорость раздирания коэффициенты в уравнении ВЛФ удельная теплоемкость корреляционная функция идеальной сетки фантомных цепей корреляционная функция узлов константа
С2 сз Сг) ь п* D' I)" Е Е* е: Е” Е* f Л fc fph константа константа константа константа податливость при растяжении комплексная динамическая податливость при растяжении податливость накопления при растяжении податливость потерь при растяжении модуль Юнга (при растяжении) комплексный динамический модуль при растяжении модуль накопления при растяжении модуль потерь при растяжении энергия активации примитивной релаксации измеренная или кажущаяся энергия активации доля свободного объема или функциональность константа пропорциональности упругое напряжение в модели Флори упругое напряжение в фантомной модели
IX. Приложение: список обозначений 243
G модуль свига
<М G* G' G’ Ge & J J(t) J* J' J" Jd Im jm Js релаксационный модуль при сдвиге комплексный динамический модуль сдвига модуль накопления при сдвиге модуль потерь при сдвиге равновесный модуль модуль сдвига в стеклообразном состоянии релаксационный спектр при сдвиге или энтальпия податливость при сдвиге податливость при сдвиговой ползучести комплексная динамическая податливость при сдвиге сдвиговая податливость накопления сдвиговая податливость потерь замедленная сдвиговая податливость равновесная сдвиговая податливость сдвиговая податливость стеклообразного состояния приведенная сдвиговая податливость установившаяся сдвиговая податливость модуль объемной деформации спектральная функция
К к* К' К" k L ьр(0 n ni nj P Q R S sc T Tf объемный модуль комплексный динамический объемный модуль объемный модуль накопления объемный модуль потерь коэффициент пропорциональности спектр запаздывания при сдвиге приведенный спектр запаздывания при сдвиге средний молекулярный вес отрезка цепи между узлами параметр кооперативности сегментальной релаксации параметр кооперативности i-той моды пряди цепей между узлами сетки параметр кооперативности релаксации узлов сетки давление скорость нагревания или охлаждения газовая константа энтропия конфигурационная энтропия температура фиктивная температура
T. r, T T. t tc ^cure температура стеклования температура отсчета температура в уравнении ФФТГ энергия раздира энергия раздира вблизи равновесия время время перехода к кооперативной динамике время вулканизации
244 JV- Глава 5. Вязкоупругие свойства каучука
V удельный объем
Р коэффициент Андраде
у0 заданная сдвиговая деформация
y(Z) деформация сдвига
ДеР релаксация напряжения ДжГ-релаксации
Д угол фаз между напряжением и деформацией
£0 мономерный коэффициент трения
X время запаздывания
Т| вязкость при сдвиге
к параметр Флори в модели стесненных узлов
р плотность
с0 заданное напряжение сдвига
q(Z) напряжение сдвига
т время релаксации
т7 время релаксации узлов сетки
та время релаксации сегментов
тоа время примитивной релаксации в модели коопертивности
Тр время вторичной релаксации Джохари-Голдстейна
то/ время примитивной простой релаксации узлов сетки
т/й время релаксации i-той моды модели Рауза
т^ префактор
тоо* префактор
со частота в рад/с
ГЛАВА
6
РЕОЛОГИЧЕСКИЕ СВОЙСТВА
И ПЕРЕРАБОТКА
НЕВУЛКАНИЗОВАННОГО КАУЧУКА
ДЖЭЙМСЛ. УАЙТ
Департамент Полимер Инжениринг
Университет Акрона
Акрон, Огайо
I. ВВЕДЕНИЕ
А. Общая характеристика
Изготовление резиновых деталей обычно включает процессы
смешения и переработки невулканизованных твердых резиновых смесей,
а иногда растворов и эмульсий с помощью сложного оборудования. Легкость
или трудность изготовления зависит от того, как эти резиновые системы ре-
агируют на приложенные напряжения и деформации, от их реологических
свойств (от rheos — «течь» и logos — «наука о...»). Целью этой главы является
описание реологических свойств и переработки невулканизованных эласто-
меров и резиновых смесей. Мы также рассмотрим некоторые приложения
реологии к переработке. Предмет этой главы — наука и технология перера-
ботки — рассмотрен более детально в книге автора [W16].
В. Реологические свойства
Изучение реологических свойств и переработки эластомеров
и их растворов и резиновых смесей восходит в промышленности к 1820-м.
Патентная литература, мемуары и обзоры начала 19-го столетия содержат
много дискуссий, касающихся течения и переработки натурального каучука
и гуттаперчи [В 17, В29, С6, С7, G12-14, Н4-11]. Основные свойства и методы
переработки каучука связаны с именами Томаса Хэнкока, Чарльза Макин-
тоша, Эдвина Шаффе, Чарльза Гудьира, Ричарда Брумана, Чарльза Хэнкока
и других, в том числе давно забытых. Однако только начиная с развития
трехмерной теории вязкоупругости Людвигом Больцманом [В26] в Венском
246 -'V- Глава 6. Реологические свойства и переработка...
университете в 1874г., понимание реологических свойств высокоэластичных
материалов стало достаточным для того чтобы можно было проводить раци-
ональные исследования. Потребовалось, однако, еще полстолетия прежде
чем Бруно Марцетти [М8-11] из ПиреллиРиеШ SpA в Италии и (в 1930-х)
Дж. Р. Скотт [S5,6] из Британской исследовательской ассоциации произво-
дителей каучука (BRMRA), Джон X. Диллон [D7-9] изФайерстонтайрэнд
раббер Ко., Мелвин Муни [М40, М42, М46] из Ю.С. раббер Ко.(последние
два в США) предприняли исследование деформации и течения невулкани-
зованного каучука. В каждом случае мотивацией исследований было, по-ви-
димому, стремление расширить базу приборов для контроля качества, чтобы
обеспечить удовлетворительную перерабатываемость. К счастью каждый из
четырех был тщательным, наблюдательным и вдумчивым исследователем.
Марцетти интерпретировал экструзию каучука через цилиндрическую
головку как течение жидкости с вязкостью, зависящей от скорости сдвига.
Такая точка зрения была подтверждена Скоттом, Диллоном и Муни. Скотт
[S5] был первым, кто понял, что каучук наполненный большим количеством
маленьких частиц обнаруживает предел текучести, ниже которого течение
отсутствует. Это наблюдение было подтверждено Диллоном и Джонстоном
[D9]. Муни впервые получил количественную зависимость вязкости от
скорости сдвига на примере каучука и впервые измерил величину упругого
восстановления, а Диллон и Купер [D8] опубликовали первые исследования
переходной области в начале течения.
Послевоенные исследования реологических свойств невулканизованных
эластомеров определялись мыслью о том, что они являются вязкоупруги-
ми. Исследования в этом направлении были начаты Лидерманом [L4, L5]
в Исследовательском текстильном институте Принстонского университета,
который повторил работу Больцмана [В26] цитированную ранее. В конце
40-х Тобольский и сотрудники [А2, АЗ, Dll, Т5] в Принстонском универ-
ситете провели широкие исследования релаксации напряжения в полии-
зобутилене. В последующие годы исследования линейной вязкоупругости
были выполнены на широком круге полимеров в области температур, где
они обнаруживали высокоэластичность. Тобольский наметил программу
установления связи вязкоупругости с молекулярными параметрами, такими
как молекулярный вес и температура стеклования. Эта работа, выполненная
в конце 40-х и в 50-х была обобщена в монографии Тобольского в 1950 [Т5].
В 60-х Бернстейн, Керсли и Цапас [В14, В15] провели широкие иссле-
дования вязкоупругости полиизобутилена при больших деформациях
в нелинейной области и предложили трехмерные уравнения состояния,
описывающие его поведение. В последующие годы результаты исследова-
ний полиизобутилена и натурального каучука в нелинейной области были
интерпретированы аналогичным образом [М24, М34].
I. Введение Jb 247
Предположение Скотта [S5] о том что высоконаполненные эластомеры
должны имеет предел текучести было подтверждено Захаренко [Z1] и ее
сотрудниками, а также Виноградовым и др. [V3, V4] а позднее и другими
авторами [МЗЗ, М37, 010, Sil, Т7, W28]. Пределы текучести были также
найдены и последующими исследователями на примере термопластов
содержащих многие наполнители, например тальк [С8], диоксид титана
[М2, М29, Т1], карбонат кальция [М2, S25, Т1] а также техуглерод [L14,
М3, Т1]. Маллинс и Уорлоу [М51, М52] из BRMRA нашли, что каучук вы-
соконаполненный техуглеродом, обладает свойствами сильно зависящими
от времени (тиксотропия). Их результаты были подтверждены и дополнены
другими авторами [М37]. Начиная с конца 70-х, были предприняты попытки
[15, LIO, М34-36, S26, W9, W22, W24] создать трехмерные уравнения со-
стояния, выражающие для этих материалов предел текучести, тиксотропные
и вязкоупругие характеристики.
С. Приборы для контроля качества
Первые 40 лет 20-го столетия стали свидетелями огромно-
го роста производства продукции на основе каучука особенно в шинной
промышленности. Ужасающая неоднородность природного каучука, при-
меняемого в 19-м столетии, была уменьшена появлением плантационного
каучука из Малайзии в 1910 (см. выступление Литчфилда [L14]). С 1920-х
усилия ученых, связанных с плантациями, и ученых из промышленности
были направлены на развитие контроля качества. Здесь мы особенно отметим
усилия Марцетти [М8-11] из Пирелли, Вильямса [W29] из Файерстона,
Гриффита [G18] из Данлоп Раббер Со., Ван-Россема и Ван-дер-Мейдена
[VI] из Нидерландского правительственного института каучука (NGRI),
Каррера [К2, КЗ] из Б.Ф. Гудрич Со., Диллона [7-9] из Файерстоун, Муни
[М40, М46], Хэкстра [Н18], также из (NGRI) и Баадера [В1] из Конти-
нентал Гуммиверке. В их работах применялись капиллярные приборы
[D7, D8, G18, М8-11], реометры с компрессионным течением между па-
раллельными дисками [Bl, Н18, К2, КЗ, VI], и ротационные реометры
«диск-диск» [М40, М46].
В конце 30-х И.Г. Фарбениндустри начала промышленное производство
эмульсионного бутадиенстирольного сополимера (SBR), синтетический ка-
учук под маркой Буна S [К10]. После сравнения многих приборов [Н1 ], они
остановились на Дефо (компрессионное течение) предложенном Баадером
[В1 ] из Континентал гуммиверке. Этот прибор использовался, начиная с 30-х
и до конца мировой войны, для испытаний и оценки качества немецкого
Буна S. В американской правительственной программе синтетического
каучука [W4] для развития SBR обозначенного как GR-S был принят дис-
ковый сдвиговый вискозиметр Муни [М40].
248 -*v- Глава 6. Реологические свойства и переработка...
В послевоенный период вискозиметр Муни по-видимому сохранял
главенствующее положение, хотя новое поколение приборов позволяющее
оценивать вязкоупругие характеристики все чаще бросало ему вызов. Муни
[М42, М46] и сам понял необходимость обеспечить измерение эластиче-
ского восстановления в его дисковом приборе, также Баадер [В1] включил
измерение восстановления в первоначальный метод Дефо. Мало внимания
уделялось проявлению вязкоупругости в переработке вплоть до 60-х. Ис-
следователи из ЮС Раббер Униройал [Т8, T9, W25] и Гудрич независимо
пришли к пониманию важности учета релаксации напряжений. Гудрич
Ко. В 70-х ввела заводской двухконусный прибор типа «папа-мама» для
релаксации напряжений известный как DSR [Н14, М31]. В дальнейшем
Раббер анд Пластик Рисерч Ассошиэйшен (RAPRA наследница BRMRA)
сконструировала модифицированный пластометр сжатия для этой цели
[В16, N7].
В 80-х Байер АГ, продолживший работы И. Г. Фарбениндустри по синте-
тическому каучуку, создал два новых прибора в соответствии с программой
руководимой Коопманом [К13-К18]. Они оба были: вискозиметр Муни
с возможностью измерения релаксации напряжений [К17] и усовершенс-
твованный прибор Баадера (Дефо) [К13-18]. Байеровские исследователи
предпочитали Дефо. Вискозиметр Муни с релаксацией напряжений произво-
дится теперь фирмой Альфа Технолоджис [G21 ]. Новый Дефо производится
фирмой Хааке по лицензии Байера.
Альфа Технолоджис выпускает прибор, который измеряет линейную
механическую динамическую вязкоупругость.
D. Переработка
Первая успешная попытка создания технологии переработки
каучука была предпринята Чарльзом Макинтошем [НИ, М4, W7] в 1820-х,
когда приготовлялись растворы каучука в летучем растворителе, которыми
покрывалась поверхность ткани с целью придания ей водозащитных свойств.
Фирма Макинтоша в Манчестере и фирма Томаса Хэнкока в Лондоне были
первыми крупными производствами. В 1930-х два американца Эдвин Чаффи
[С6, G14, W16, W31] и Чарльз Гудьир [G12, G14] создали технологии валь-
цевания, каландрования и вулканизационного формования. Это привело
к тому, что такие компании как Фаррел Фаундри и Машин и Бирмингам
Айрон Фаундри (после 1927 г. объединены в Фаррел Бирмингам, позднее
Фаррел Корп.) начали производство вальцев и каландров [Fl, Wil, W13].
Эти технологии были экспортированы в Европу. В 1870-х Гарбургер Айзен
и Бронзеверке [М5] (с 1960 — часть Круппа) начали производство калан-
дровых валков. Создание шнекового экструдера для гуттаперчи и каучука
в 1870-х [G15, Н15, S4] привело к образованию новых машиностроительных
фирм, особенно таких как Фрэнсис Шоу и Ко. [W14] в Манчестере, Анг-
I. Введение -»U 249
лия, и Джон Ройл в Паттерсоне, Нью-Джерси. Немецкая промышленность
переработки каучука сконцентрировалась в Ганновере [М5], с наиболее
крупной фирмой (с 1870) Континентал Гуммиверке. Две известные фирмы,
сосредоточившие внимание на производстве оборудования для переработки
каучука, включая экструзию и каландрование, создались в Ганновере. Это
Пауль Тростер Машиненфабрик (в 1892) [М5, W12] и Германн Машиненбау
[М5] (1896), возникшие обе в последнем десятилетии 19 столетия.
20-е столетие стало свидетелем подъема шинной промышленности
и развития применения органических ускорителей вулканизации. Эти два
обстоятельства отвечали требованиям автомобильной промышленности
и привели к созданию закрытого смесителя вместо открытых вальцев. Пер-
воначально Вернер и Пфляйдерер были лидирующей компанией [Н16, К4,
W13, W15], но их нежелание следовать патенту Фернли X. Бэнбери привело
к развитию технологии на их закрытом смесителе [В12, В13, Н16, К4, W13]
фирмой Бирмингам Айрон Фаундри и, после 1927, фирмой Фаррел
Бирмингам (сейчас Фаррел Корп.) [К4, Wil, W13].
Заводская система, основанная на закрытых смесителях, шнековых
экструдерах, каландрах и вулканизационных прессах сохранялась в основ-
ном неизменной в последние полстолетия. Закрытые смесители получили
существенные улучшения, такие например, как зацепляющиеся роторы,
предложенные Фрэнсис Шоу и Ко. [С16] и Вернер энд Пфляйдерер [L3],
и валки с переменным зазором [Р1], предложенные Помини-Фаррел SPA.
Была создана усложненная система компьютерного контроля. Прежние
экструдеры с горячей запиткой были заменены экструдерами с подачей
холодных ингредиентов и с усложненной конструкцией, включая экструдер
с оштифтованным цилиндром (pin barrel extruder) [G7, Н12, Н13, М18, W16]
вместе с системой комплексного контроля.
Е. Моделирование потока при переработке
Начало моделирования потока при переработке восходит, ве-
роятно, к появлению уравнений Навье-Стокса [S22] и их ранним решениям
[LI, S24]. Моделированию Рейнольдсом в 1886 [R6] течения смазочного
масла в подшипниках оказало огромное влияние на развитие моделирова-
ния потоков вязкой жидкости в узких зазорах. Специальные исследования,
касающиеся потока каучука в операциях по переработке и учет неньютонов-
ского характера течения началось с 1930 г. работой Муни [М39] и Диллона
и Джонстона [D9]. Эти работы впрочем, тесно связаны с визкозиметрией и их
лучшим продолжением стал анализ Ардичвили 1938 г течения между [А10]
и вокруг [АН] валков каландра, за которым последовало моделирование
неньютоновского потока Гаскеллом [G2] для решения этих проблем.
В 1950-х значительное внимание было уделено моделированию процесса
экструзии в шнековом экструдере исследователями в Гудьир Тайер Энд Раб-
250 -•V- Глава 6. Реологические свойства и переработка...
бер Ко. [РЮ], Дюпон [С1] и Байер АГ [М20, М21]. В 60-х довольно сложные
модели неизотермического, неньютоновского течения в зоне дозирования
шнека экструдера были опубликованы Гриффитом [G17], и Замодицем
и Пирсоном [Z2]. В моделировании переработки полимеров в 1960-х до-
минировали работы Дж. Р.А. Пирсона из Кэмбриджа, чьи первые работы
по шнековой экструзии мы только что цитировали. Пирсон [Р2-6] в серии
работ, опубликованных в начале 1960-х, показал, как гидродинамическая
теория смазки может быть применена к расчету головки. Впоследствии
он также показал как теория мембран может быть применена к экструзии
пленки через кольцевой зазор и к раздувному формованию [Р5, Р6].
В 1970-х внимание было обращено на моделирование литья под давлени-
ем термопластов, причем первые работы были посвящены либо изотермиче-
скому заполнению формы [R8, W8] или неизотермическому заполнению
очень простых форм [К1]. Начиная с конца 1970-х общедоступное про-
граммное обеспечение для моделирования неизотермического процесса
литья под давлением было создано работами С. Остина и его фирмой
Молдфлоу, Австралия и К.К. Вангом с сотрудниками в Корнельском
университете.
70-е годы ознаменовались также первым компьютерным применением
метода конечных элементов к расчету отдельных операций переработки
полимеров, при этом ведущую роль в применении метода к разбуханию
экструдата и к покрытию проволоки сыграли работы Таннера с сотрудни-
ками [СЗ, Т2]. Впоследствии доступные компьютерные программы осно-
ванные на применении метода конечных элементов для анализа механики
ньютоновских и неньютоновских жидкостей были созданы различными
исследователями, среди которых следует отметить М. Крошэ из Университета
Лувен-ле-Нев и его Полифлоу. Крошэ [С22] опубликовал обзор достигнутого
прогресса в области применения метода конечных элементов к решению
проблем вязкоупругих жидкостей.
Вплоть до 1980-х большинство работ по моделированию процессов
в полимерах относилось к термопластам. Специализированное оборудование
для резиновой промышленности привлекало мало внимания. Только в конце
1980-х появилось практическое моделирование закрытых смесителей [СЮ,
СИ, Н20, Н21, К6-8, W19] и pin barrel extruders [В31, В32, В34, К22]. Оно
основывалось преимущественно на теории моделирования смазок.
II ОСНОВНЫЕ ПОЛОЖЕНИЯ МЕХАНИКИ
В этом разделе мы рассмотрим основные идеи классической
реологической мысли. Мы будем считать, что эластомеры деформируются
как сплошная среда и подчиняются формализму механики сплошных сред
[Т10]. Мы начнем с развития представлений о природе внешних сил и тен-
зора напряжений.
II. Основные положения механики -JU 251
Представление о тензоре напряжений в материале исходит из необходи-
мости показать влияние приложенных сил на величину деформации. При-
ложенная сила F действующая на тело может быть представлена как сумма
контактных сил действующих на поверхности и сил в самом теле f таких как
гравитация, которые действуют непосредственно на элемент массы. Можно
написать [Т10] (рис. 1)
F = ^ttAat- =^tda+^pfd7 (1)
i t
где t сила отнесенная к единице поверхности (вектор напряжения) дей-
ствующая на элемент поверхности Да. и ф показывает что интегрирование
производится по всей поверхности.
Рис. 1. Вектор напряжений.
Представление о тензоре напряжений основано на том, что t связано
с единичным вектором п нормальным к поверхности с помощью урав-
нения
t = GH
(2)
где G есть массив девяти величин
°11
G- G21
°31
°12 °13
°22 °23
°32 °33
(3)
известный как тензор напряжений или матрица. G может рассматриваться
как тензор второго порядка или диадик Гиббса
” = (4)
j
Концепция вектора напряжений и тензора напряжений была развита
Коши в 1820-е. Направление компоненты напряжения есть i. Если на-
правление компоненты напряжения i оказывается перпендикулярным
252
—Глава 6. Реологические свойства и переработка...
плоскости (Gfji = j), напряжения называются нормальными напряжениями.
Когда I направлено по касательной к поверхности j Ф j) напряжения
называются напряжениями сдвига.
Применение теоремы дивергенции к уравнению (1) позволяет полу-
чить:
= = JV-odV (<>)
F = J[V-G + pf]dV (6)
где V - дельта-оператор
п д д д / п \
V = e4 —+ е2 —+ е3— (7)
CfXj 6^2
Полная динамика деформируемого тела требует учета контактных сил.
гравитационных сил pf с инерционными силами. Для макроскопической
массы М
Р 47+(v’v)v
ol
F = -^JpW7 = J[Va+pfp7 (8)
При этом для макроскопического «контрольного объема» фиксированного
в пространстве, через который может двигаться масса [Т10]
J^(PV) ^ + fpv(v-n) da = J[V-G + pf] dV (9)
Отсюда следует, что в точке внутри тела [Т10]
= V-G + pf (Ю)
Уравнение (10), которое определяет баланс сил в точке внутри тела из-
вестно как закон движения Коши.
Компоненты тензора напряжения не являются независимыми друг от
друга. С помощью баланса крутящих и угловых моментов аналогичных
тем, которые привели к ур. (10), можно показать, что тензор напряжений
является симметричным [Т10]; т. е.
а = ат или оц=0л (И)
III. Реологические свойства JU 253
Таким образом, вне-диагональные компоненты ур.(4) которые являются
напряжениями сдвига взаимосвязаны:
°12 ~ °21
°13 - °31
°23 ~ °32
Основная проблема реологии состоит в выражении а в терминах дефор-
мации и кинематики материала. Закономерности деформирования сплош-
ной среды могут быть, поэтому, определены путем решения ур. (10).
Если тело не подвергается действию внешних сил, компоненты напря-
жения превращаются в эквивалентные нормальные компоненты гидроста-
тического давления
о = -р1, 1 =
(12)
гдер давление. В более общем виде, когда имеются внешние силы, мы можем
выразить тензор напряжений, через давление и внешний тензор напряже-
ний Р через уравнение
о = -р1 + Р
(13а)
Отдельно для нормальных напряжений
а_й=-р+Р_й
(13в)
и для напряжений сдвига
(13с)
при этом нет необходимости делать различие между (5,7 и PtJ если i * j
NI.
РЕОЛОГИЧЕСКИЕ СВОЙСТВА
1
0
0
0
1
0
0
0
1
°у=^, (»*>)
А. Чистые каучуки
1. Общие задачи
Этот раздел мы начнем с общего обзора экспериментальных
исследований реологических свойств невулканизованных эластомеров
и аналогичных материалов. Затем мы попытаемся обобщить эти данные
с точки зрения теории вязкоупругости. Начнем с линейной теории вязкуп-
ругости, в рамках которой наблюдается общее согласование точек зрения
реологов. Затем мы рассмотрим нелинейную теорию, в рамках которой на-
блюдается значительно меньшее совпадение взглядов.
254
—Глава 6. Реологические свойства и переработка...
2. Экспериментальные исследования
Экспериментальные работы в целом делятся на две части —
изучение свойств при малых деформациях и исследования установившегося
сдвигового течения. Значительная часть посвящена попыткам установить
связь свойств с молекулярной структурой.
а. Исследование малых деформаций
В течение 1940-х и начала 50-х Лидерман [L4, 5], Тобольский [А2, АЗ,
Dll, Т5], Ферри [F3] и другие провели широкие исследования свойств
эластомеров и других подобных материалов при малых деформациях. Эти
исследования включали ползучесть (деформация при постоянном напря-
жении), релаксацию напряжения после заданной деформации и также
осциллирующие деформации. Эти и другие методики эксперимента были
описаны детально в монографии Ферри [КЗ]. Исследования показали, что
все эти деформации могут быть представлены в рамках принципа суперпо-
зиции Больцмана [В26] [См ур. (43)]. Тобольский с соавторами предпри-
нял значительные усилия для описания релаксационных характеристик
эластомеров, особенно полиизобутилена. Напряжение падает до нуля со
скоростью зависящей от температуры и молекулярного веса (рис. 2). Они
выразили процесс релаксации через серию экспонент или в виде спектра
времен релаксации. Рассмотрим падение напряжений сдвига g(Z), после-
довавшее после наложения деформации у0. Можно выразить модуль сдвига
при релаксации G(t) следующим образом
Я^ = б?(<) = £С/е-^ (14)
Yo i=i
G(t) = ^H(^e~l/xdlnT
В Табл. 1 приведены значения модулей G., и времен релаксации т. для
смокед-шитса при 100°С.
Таблица 1. Значения G(t) для линейной
вязкоупругости каучука SMR5-NR при 100°С.
1 (Па) Мс)
т 2,199 230
т - 1 9,166 37.7
т - 2 33,290 7.31
т - 3 59,200 0.925
АП -4 59,000 0.095
III. Реологические свойства JU 255
Рис. 2. Релаксация напряжений из публикации Эндрюса и др. [А2, АЗ].
256 Д.
Глава 6. Реологические свойства и переработка...
Тобольский с сотр. установили, что можно получить кривую G(t) в очень
широком интервале по шкале времен путем построения приведенной кривой
(«master curve») на основе данных, полученных при разных температурах.
Конкретно они нашли что:
С(«,7’) = -^-б?(^/ат) (15)
Ps^s
уравнение, которое взаимосвязывает показатели релаксации напряжения
при разных температурах Тгде ^зависящий от температуры фактор смеще-
ния по шкале времен. Здесь р плотность a р5 и Ts соответствующие значения
при стандартной температуре, характерной для каждого полимера. Фактор
смещения может быть представлен универсальной зависимостью в виде
функции Т-Т. Значение Ts связано с температурой стеклования. Это было
показано Тобольским и Ферри с их соответствующими сотрудниками, но
сегодня связывается в основном с именами Вильямса, Лэнделла и Ферри
[W30].
Тобольский с сотрудниками нашел, что релаксационный спектр Я(т)
полиизобутилена может быть представлен комбинацией «клина» и «ящика»,
т. е. в виде:
Я(т) = -^ KTj (16а)
л/Т
Я(т) = Я0 х2<х<хт (16в)
Здесь //() и А не зависят от молекулярного веса, тогда как тт обнаруживает
сильную зависимость. Конкретно:
В то же время проводились и эксперименты с осциллирующей деформа-
цией особенно в работах Ферри и сотрудников [W3]. Оказалось, что напря-
жение при этом состоит из двух компонентов — один «в фазе» с деформацией
и другой «не в фазе». Это было выражено в виде уравнения
a(i) = G'(®)Y(i)+n'(co)^ (18)
at
Где Т|’(со) означает динамическую вязкость. Можно также выразить комп-
лексную вязкость т|*(со):
Т] * (со) = 7[С7со]2+(л')2
(19)
III. Реологические свойства JU 257
В последующие годы функции G(t) и Я(т) исследовались и другими
учеными такими как Ниномия [N4], Богю и др. [В23] и особенно Масуда
и др. [М12, М13] с целью выяснения влияния молекулярно-весового распре-
деления. Присутствие высокомолекулярной фракции полимера приводило
к замедленной релаксации б(т). Влияние на Я(т) выражалось в том, что Я(т)
уменьшалась медленнее при больших т. Было обнаружено, что при больших
временах в образцах с узким молекулярно-весовым распределением «ящик»
в ур. (16в) заменяется на более сложную функцию, включающую «острие»
(spike). Ниномия [N4] и Богю с сотр. [В23] попытались найти такие виды
функций, которые могли бы учесть ширину молекулярно весового распре-
деления в функции Я(т)
б. Сдвиговая вязкость и нормальные напряжения
Измерение сдвиговой вязкости эластомеров и расплавов полимеров
было начато Муни [М41] в 1936 и продолжалось вплоть до 1940-х. Первые
измерения были направлены на изучение зависимости сдвиговой вязкости
от молекулярного веса и скорости сдвига.
Было обнаружено, что сдвиговая вязкость уменьшается с ростом скорости
сдвига, однако имеет некоторое асимптотическое значение т|о при малых
скоростях сдвига. Фокс и Флори [F5] нашли, что начальная сдвиговая
вязкость т|о линейного полимера зависит в степени 3.4 от молекулярного
веса. Это впоследствии было подтверждено рядом исследователей [В39, F6,
G20, Р13, Р14]. Прежде всего, было обнаружено, что начальная сдвиговая
вязкость разветвленного полимера ниже, чем линейного при одинаковом
молекулярном весе [S2]; однако впоследствии Краус и Грувер [К20] показа-
ли, что в большей мере, чем молекулярный вес на величину вязкости влияет
топология макромолекул, так специально синтезированные крестообразные
и Y-образные полимеры обнаруживают более быстрый рост т|о примерно
пропорциональный шестой степени.
Зависимость вязкость-скорость сдвига при больших скоростях сдвига так-
же определяется молекулярно-весовым распределением. В средине 1960-х
Виноградов и Малкин [V6] нашли, что кривая т|/т|о - Т|оу не меняет свое
положение при изменении температуры для всех полимеров. Они также
предположили, что это есть зависимость универсальная для всех полимеров.
Впоследствии, однако, это не подтвердилось. В последующие годы в ряде лабо-
раторий [МЗО, 09, Y3] была обнаружена сильная зависимость этой кривой от
молекулярно-весового распределения. Величинат|/т|0падаетзначительнобыс-
трее, когда молекулярно-весовое распределение сужается. Ямани и Уайт [Y3]
обобщили результаты, полученные на широком круге полимеров, таких как
полистирол, полипропилен, полибутен-1, построив обобщенную кривую,
которая позволила прогнозировать влияние ширины молекулярно-весового
распределения на зависимость вязкость-скорость сдвига.
258 “’V Глава 6. Реологические свойства и переработка...
Корреляции, приведенные в предыдущем разделе, распространяются на
линейные гомополимеры и не применимы к длинноцепным разветвленным
полимерам, таким как полиэтилен низкой плотности.
log о
Рис. 3. Сдвиговая вязкость чистых каучуков: натуральный каучук и бутадиен-
стирольный (Бриджстоун-Файерстон, Дураден 706) в зависимости от на-
пряжения сдвига.
Кокс и Мерц [С20] установили существенный экспериментальный факт,
что зависимость неньютоновской вязкости от скорости сдвига имеет тот же
вид что и зависимость комплексной вязкости от частоты, т. е.
Т](у)=Т]*(<о) (20)
Нормальные напряжения первоначально обнаруженные Вайссенбер-
гом [W3], заметившим эффект поднятия по стержню жидкой суспензии
мыл в углеводородах и растворах полимеров, начали исследоваться на
примере термопластов в 1960-х [С9, К9]. Уайт и Токита [W24] обна-
ружили их возникновение в чистых эластомерах под действием сдвига
в дисковом реометре Муни. Еще Вайссенбергом их возникновение было
связно с наличием упругости расплавов и отмечена чувствительность
III. Реологические свойства JU 259
к ширине молекулярно-весового распределения. Уайт и Кондо [W20]
установили зависимость коэффициента разности нормальных напряже-
ний Yi ПРИ малой скорости сдвига от молекулярного веса. Они предпо-
ложили, что зависимость должна быть порядка 6.5-7.0 что согласуется
с их собственными экспериментами и с теми, которые опубликованы
в литературе. В статье 1987г. Ода, Уайт и Кларк [02] связали нормальные
напряжения полистиролов с разным молекулярно-весовым распреде-
лением с напряжением сдвига. Было установлено, что первая разность
нормальных напряжений Nx и ее зависимость от напряжения сдвига не
зависит от температуры:
NX=A<5*2 (21)
Величина Nx была больше при одинаковом о12 для полимера с широким
молекулярно-весовым распределением но возрастала не так быстро с ростом
напряжения сдвига (т. е. А было большим, а а маленьким).
3. Теория линейной вязкоупругости
В конце 1940-х такие экспериментаторы как Лидерман
н Тобольский обнаружили, что свойства эластомеров и расплавов термо-
пластов могут быть описаны в рамках теории вязкоупругости предложенной
Больцманом [В26] в 1870-х. Это было хорошо обосновано в монографиях
Лидермана [L5] и Марка с Тобольским [М7], которые были опубликованы
в тот период. В следующие два десятилетия это направление было развито
в монографиях Тобольского [Т5] и, позднее, Ферри [F3].
Больцман в своей теории основывался на рассмотрении линейных сред
с неполной памятью. Рассмотрим материал, в котором при деформации е
в нулевой момент времени возникает напряжение
g=G(0y. (22)
где G(t) функция модуля, падающая во времени. Если реакция этого ма-
териала линейная, напряжение возникающее в серии последовательных
деформаций ур у2, у3... в моменты времени тр т2, т3... окажется равным
в момент времени t
a = G(t-tl)yi+G(t-t2)y2+G(t-t3 )у3 + (23)
= Г G(t-s)dy(s)= Г G(t-s)—ds> (24)
J-°o J-oo fa
где интеграл относится к пределу, для которого приложенная деформация
непрерывна. Уравнение (21) известно как интеграл суперпозиции Больц-
мана.
260 -* V Глава 6. Реологические свойства и переработка...
Если в нулевой момент времени задана постоянная скорость сдвига, то
возникают напряжения
о= J^G(s)dsjy (25)
Для больших времен интеграл соответствует начальной сдвиговой вяз-
кости Г]о т. е.
По=/ос(«И» (26>
Другой, представляющий интерес, способ деформации — это наложе-
ние синусоидальной сдвиговой деформации yosin wt. Напряжение а теперь
можно выразить как
a = co^jo G(s)smcosds]yosjnco^4-co^Jo G(s)coscosds]y0 cosco (27)
т. e. возникают компоненты «в фазе» и «вне фазы» [Ср. ур. (18) ]. Уравнение
(27) обычно записывают в виде
а(/) = G'(co)у0 sin (at + G"(co)y0 cos co/ (28)
где
G'(co) = cojo G(s)sin Qtsds (29)
G"(co) = cojo G(s)cos(&sds (30)
G'((to) известен как модуль накопления, a G”((&) модуль потерь. Уравнение
(28) часто записывают в виде
а(/) = G * (co)sfn(co/ + 8) (31)
Где 8 угол сдвига фаз, a G*(tp) известен как комплексный модуль. Очевидно,
G'(co) = G * (co)cos5 (32a)
G"(<o) = G*(®)sira5 (326)
что приводит к
G* = >/(G')2+(G")2
(33)
III. Реологические свойства 261
tanb = G"/G' (34)
Величина tanb известна как тангенс угла потерь.
Выражения для модуля потерь и тангенса угла потерь позволяют считать,
что Gn(w) в ур. (28) связан с диссипативными потерями. Это должно быть
очевидно из сравнения входовой величины y0sinotf и выходящей компо-
ненты напряжения в coswt уравнения (18), где вводится динамическая
вязкость Г|г (со)
Т|' = (в*) = |о G(s)cos (tisds (35)
Стало общепринятым представлять спектр G(t) или как дискретный
или как непрерывный, состоящий из набора экспонент как это показано
в ур. (14). Исторически причиной использования экспонент было то, что
экспонента характерна для модели предложенной Максвеллом [М14]
в 1860-х еще до публикации Больцмана [В26]. Эта модель привела к форме
дифференциального уравнения, имеющего вид
d<5 „dv 1
---= G— --G
dt dt т
Решение yp. (36) имеет вид
a = Jo Ge S)/Ty(s)ds (37)
Это означает, что G(t) может быть выражен как сумма экспонент, т. е. так,
как представлено в ур (14). Использование ур. (14) для выражения G(t)
приводит к значению начальной ньютоновской вязкости
По = J~GO)rfs = = ^H(x)dx (38)
Для гармонических колебаний модуль накопления и модуль потерь могут
теперь быть выражены как
с'(«-)=Е^Й- о9»
1 + (О Т;
<39ь>
l + arcf
а динамическая вязкость Т|'(®)
1 + (0 х ~
262 -'V Глава 6. Реологические свойства и переработка...
Напишем также и выражение для комплексной вязкости Т|* (со)
П*(Ю) = 1[(6'(Ю))2 +(G"(<o))2 J72 (39d)
Если со 0 , Т|г(со), и Т| * (со) имеют то же выражение, что и сдвиговая вязкость
при установившемся режиме, тогда
По = п(0) = п'(о) = И * (0) (40)
Приведенные выше формулы соответствуют одному измерению, и в них
входит напряжение сдвига. Все это можно выразить и в трех измерениях.
Это конечно же было сделано в оригинальной статье Больцмана [В26].
Уравнение (21) может быть представлено через тензор напряжений Q
и давление р\
a = -pl + 2j G(t-s)A(s)ds (41)
где
d = |[w + (Vv)7’] (42)
d известно как тензор скорости деформации, а I единичный тензор. Это может
быть показано «интегрированием по частям» что эквивалентно:
a = -pl + 2j Ф(£-$)у($)с/$ (43)
где
Ф(() = _^£М и G(oo) = 0 (44а)
dt
Мы можем записать Ф(0 как
ф(<)=Х^е’(/т' (44Ь)
где у тензор малых деформаций, измеренный при внезапном течении, т. е.
а = -р\ + 21 Ф(г)у(z)dz (45)
4. Теория нелинейной вязкоупругости
Серьезная разработка специальной теории нелинейной вяз-
коупргости была произведена Зарембой [Z5] в 1903 г. приблизительно 30 лет
спустя после классической публикации Больцмана [В26]; однако современ-
ный период разработки теории начался с работы Олдройда [Об] в 1950 г.
III. Реологические свойства JU 263
В 1950-х ряд специальных теорий нелинейной вязкупругости были разви-
ты Олдройдом [07, 08], Де-Виттом [D6], Ривлиным и Эриксеном [R10],
и Ноллом [N5]. Лодж [L16] и Ямамото [Y2] сделали попытку создания не-
линейной теории на молекулярной основе. Полностью в обобщенном виде
теория вязкоупругости при больших деформациях была создана Грином
и Ривлином [G16] и Ноллом [N6] в конце десятилетия. Эта обобщенная те-
ория была применена для моделирования различных специальных случаев,
таких как вязкое ламинарное сдвиговое течение [С16, С20, R9] и распро-
странение волны [С14].
Несмотря на общее признание теории линейной вязкоупругости Боль-
цмана, конкретные формы нелинейной теории не сразу стали общеприня-
тыми. Обобщенное выражение нелинейной теории, дополненное сущест-
венно в работах Грина, Ривлина, Нолла, Колемана, Эриксена и Трусделла
не получило развития в работах следующих поколений ученых. Более того,
начиная с середины 1960-х, роль этой теории стала уменьшаться. Начиная
с 1960 г. возрастало количество публикаций с новыми теориями. Некоторые
из этих теорий были феноменологическими, другие до некоторой степени
основывались на молекулярных моделях.
С начала 1970-х мощные вычислительные средства и высокопроиз-
водительные компьютеры становятся все более доступными. В области
динамики ньютоновских жидкостей это привело к огромному росту наших
знаний в этой области. Для нелинейных вязкоупругих жидкостей, где не
было общепринятых конститутивных уравнений, вместо этого начался
процесс моделирования по многим разным моделям с массой различных
прогнозов свойств потока и с числовыми несогласованиями, обусловлен-
ными спецификой каждой модели. В 1990-х теория нелинейной вязко-
упругости и механики текущих сред во многих случаях рассматривалась
как ненаучная.
Необходимо, однако, попытаться найти какие-либо рациональные корни
в этих работах, чтобы можно было построить рациональную нелинейную
теорию. Основное положение Грина и Ривлина [G16] состояло в том, что
тензор напряжений является общей наследственной функцией, отража-
ющей предысторию деформаций. Тензор напряжений был выражен как
бесконечный ряд интегралов:
о = -Р1+J~0(z)e(z)dz+J“, Z2 )e(z( )e(z2) (46)
+ Z(zl' )[tr e(Zl )] *^2 )]dztclz2 +...
где e(z) мера нелинейных деформаций а Ф(г), 4f(zi,z2) и Z(zifz2) функции
памяти. Здесь z время, отсчитываемое назад от настоящего момента. Де-
формация е также отсчитывается в обратном направлении. Величина <&(Z)
264 -*V" Глава 6. Реологические свойства и переработка...
есть функция линейной вязкоупругой релаксации по ур. (45). Опыты, про-
веденные Запасом и Крафтом [Z4], показали что существует только одна
функция релаксации напряжения. Уайт и Токита [W26] показали, что это
эквивалентно приведению ур. (46) к виду
а = -р1 +1 -кс] dz (47)
Где m(z) зависит от инвариантов с-1 тензора фингеровской деформации, или
с тензора деформации Коши. Этот вид уравнения состояния присутствует
у разных авторов, включая Бернштейна, Керсли с Запасом [В14, В15],
и Вагнера [W1]. Другие авторы [В20, В22, В24, С2] пришли к тем же фор-
мам уравнений состояния, но использовали другие инварианты такие как
инварианты тензора скорости деформации. Общее выражение ур. (47) часто
цитируют как характеризующее жидкость БКЗ (по именам Бернштейна,
Керсли и Запаса, которые были первыми предложившими его).
Одна из специфических форм ур. (47) которая успешно была применена
для описания нелинейного вязкоупругого поведения эластомеров, возникла
благодаря работам Богю с сотр. [В22, В24, С9]. Эта форма использовалась
и Мидлманом [М24] и Монте с Уайтом [М34]:
m(z) = (48)
i ^ieff
при следующих значениях параметров
(49а)
‘49ь>
nd = 2trd2 = 2<£,<£, (49<0
здесь d тензор скорости деформации в ур. (43) для чистых эластомеров.
В работе этих последних авторов описание линейного вязкоупругого пове-
дения с помощью G(t) для образца натурального каучука SMR-5 при 100 °C
было проведено с применением параметров, приведенных в Табл. 1. Параметр
а в ур. (49а) взят равным 0.7. Сравнение расчетных данных по ур. (47-49)
с экспериментальным определением сдвиговой вязкости при установившем-
ся течении, сдвиговой вязкости, в переходном режиме, переходной сдвиговой
вязкости при старте потока, и релаксации напряжения при остановке потока
приведены на рис. 4. Совпадение вполне удовлетворительное.
III. Реологические свойства JU 265
у (sec’1)
b
time (sec)
time (sec)
Рис. 4. Сравнение реологической модели [уравнения (47)-(49)] с эксперименталь-
ными данными для натурального каучука, а — вязкость в установившемся
режиме, b — вязкость в переходном режиме в начале течения, с — релаксация
напряжений после остановки потока.
266 -*ь- Глава 6. Реологические свойства и переработка...
Криминале, Эриксен и Филби [С21] (см. также Уайт [W5] показали
что для сдвигового потока большой продолжительности общие уравнения
состояния типа (46) и (47) могут быть приведены к виду
о =-pl + 2r|d + 4T2d2 -Ч*! (50а)
где
— = ^-d + (v-V)d-Vvd-dVv (50b)
dt v '
Здесь Т| сдвиговая вязкость, % и Т2 первая и вторая разность нормальных
напряжений. Они являются функциями trd2, следа, т. е. суммы, диагональ-
ных компонентов в d*d [См. ур. (49с)],
Т| = т] (trd2 Jt, = % (trd2 )т2 = Т2 (trd2) (51)
При малых значениях d и tr d2 сдвиговая вязкость в ур. (51) переходит
в величину вязкости, выражаемую в теории линейной вязкоупругости [С15,
W5],T.e.
Т| jo G(s)ds (52а)
и величина % переходит в ее первый момент
2jo~sG(s)ds (52b)
Для течения при простом сдвиге, т. е.
v1 = у ^2ei + 0е2 + 0ез (53)
Ур. (50) позволяет описать поле напряжений, т. е.
ct12 = t]Y (54)
Oi! - о22 = % у2, о22 - ст33 = Ч*2 у2
где Т|, , 4*2 , зависят от у2 Таким образом, можно рассчитать как на-
пряжения сдвига, так и нормальные напряжения. Результаты, изложенные
в этом параграфе важны, поскольку они создают основу для понимания
вискозиметрии при сдвиговом течении вязкоупругих расплавов полимеров
а также эластомеров.
III. Реологические свойства JU 267
Имеется много литературы, посвященной второму классу теорий, ос-
нованных на обобщении уравнения Максвелла (36). Это работы Зарембы
[Z5], Олдройда [06-8], Де-Витта [D6], Нолла [N5] и работы предшеству-
ющие появлению ур. (46). Последующие работы Уайта и Метцнера [W22],
Джонсона и Сегалмана [Л], а также Фан-Тьена и Таннера [Р7, Р8] развили
это подход. Этот класс уравнений состояния в общей форме выглядит сле-
дующим образом
а = -р! + Р
(55)
+Z а7 (Vv’P)+6(р) = 2Tl(tr d2 )d+c(tr d2
Здесь P тензор экстра напряжений, а &, 6, т| и с функции tr d2 как это показа-
но. Эти формулы значительно более произвольны по характеру написания,
чем подход представленный в ур. (46) и (47).
Формулы (47) и (55) подверглись критике со стороны Леонова [L9, L11]
и других авторов, как не проверенные на согласование со вторым законом
термодинамики. Для ньютоновских жидкостей такая проверка требует
положительной сдвиговой вязкости. Для линейной вязкоупругой среды
можно показать, что функция модулей релаксации должна быть всегда
положительной, что удовлетворяет второй закон. С предъявлением таких
требований к ур. (47) и (55) такой ясности нет. Леонов намеревался развить
нелинейные вязкоупругие реологические модели, основанные на принципах
термодинамики.
Б. Композиции
7. Общие задачи
Экспериментальное исследование течения каучуковых ком-
позиций началось еще в 1930-х [D9, S5], но только в конце 1980-х началось
действительно широкое исследование этой проблемы. Приемлемые реологи-
ческие модели также относятся к 1980-м; однако по этому вопросу появилось
лишь несколько работ, по которым не было и всеобщей согласованности.
В этом разделе мы сначала проанализируем экспериментальные работы.
Затем обратимся к примененным реологическим моделям. Центральным
вопросом является существование предела текучести и тиксотропных явле-
ний в каучуковых композициях.
2. Экспериментальные данные
Начнем этот раздел с описания установившегося сдвигового по-
тока. Затем перейдем к обсуждению характеристик, зависящих от времени.
268 -*v- Глава 6. Реологические свойства и переработка...
В 1931 г. Скотт [S5] работая на пластометре сжатия обнаружил что
композиции, содержащие большое количество дисперсного наполнителя
обнаруживают предел текучести; позже он предложил уравнение
<т = У + АУ (56)
чтобы показать зависимость напряжение сдвига - скорость сдвига при
напряжениях выше, чем предел текучести Y. Такой подход был поддержан
последующими работами Диллона и Джонстона [D9] выполненными на
капиллярном вискозиметре. Диллон и Джонстон также употребляли ур. (56).
Ур. (56) обычно именуется уравнением Гершеля и Бакли [Н17], которые
ранее применили его для характеристики растворов полимеров. Скотт-Гер-
шель-Бакли является предпочтительным наименованием уравнения (56).
Направление это мало обсуждалось в литературе в последующие 25 лет
и только в 1960-х внимание возобновилось. Так в 1962 г. Захаренко и др.
[Z1 ] в Москве сообщили результаты исследования сдвигового потока саже-
наполненной каучуковой композиции. В 1972 Виноградов и др. [V8] также
в Москве получили аналогичные результаты для других саженаполненных
каучуковых смесей и указали на существование предела текучести. В то же
время аналогичные результаты были получены на тальксодержащих поли-
пропиленовых композициях Чепманом и Ли [С8], и для смесей полиэтилена
и диоксида титана Минагавой и Уайтом [М29].
Начиная с 1980 г. проводились широкие исследования сдвиговой вязкос-
ти саженаполненных каучуковых композиций и аналогичных наполненных
расплавов полимеров. Пределы текучести в саженаполненном полистироле
в сдвиговом потоке были обнаружены Лобэ и Уайтом [15] в 1979 и Танакой
и Уайтом [Т1 ] в 1980 на примере полистирола, наполненного карбонатом
кальция и диоксидом титана а также техуглеродом. Начиная с 1982 г. Уайт
и сотр. нашли предел текучести в композициях на основе бутадиенстироль-
ного сополимера [Ml, М37, S12, S18, Т7, W29], полиизопрена [МЗЗ, М37,
S12, S18], полихлоропрена [S18], и этиленпропиленового тройного сополи-
мера [010, S18]. Характерные кривые вязкость — напряжение сдвига для
саженаполненных каучуков показаны на рис. 5, а, Ь. Уайт и др. показали,
что эти данные описываются как ур. (56), так и следующим выражением
c=rVp-i-^ <57)
1 + Ву
В целом было показано, что объемная доля 0.15 мелкодисперсных частиц
достаточна для возникновения предела текучести. Как величина предела
текучести Y так и вязкость увеличиваются с ростом содержания частиц
(рис. 5, а и Ь) и с уменьшением размера частиц (рис. 5, с). Наибольший Y
в смесях с ISAF и меньший в смесях с FEF. Он оказывается наименьшим
в смесях с SRF. Порядок тот же, как и при влиянии размера частиц (у ISAF
наименьшие частицы). Большие значения предела текучести и вязкости
III. Реологические свойства Jb 269
были обнаружены в смесях с карбонатом кальция [S25]. Для каучуковых
композиций величина предела текучести оказывается порядка 50-100 кПа,
а показатель степени в степенном законе п равен около 0.2. В Таблице 2
приводятся данные по величинам У, А, В, К и п в уравнениях (56) и (57).
Таблица 2. Параметры У, А, В, К и п уравнений (56) и (57) моделирующих
течение каучуковых композиций.
Материал Тип сажи Объем фракции У (кПа) А=А/В (кПа-сл) А (кПа-с) в (с1-") п
NR №220 0.2 119 134.5 73 0.54 0.21
№550 0.2 79 105 97 0.93 0.20
№762 0.2 62 89 1,268 1.43 0.20
SBR №220 0.2 130 200 1,77 8.85 0.225
№550 0.2 82 187 7,13 3.82 0.21
№762 0.2 62 140 12,97 92.65 0.255
NR №330 0.2 63 103 587 5.68 0.175
№330 0.3 138 104 330 3.17 0.18
№326 0.2 65 108 4,5 41.5 0.2
SBR-1500 №330 0.2 60.3 132 704 5.32 0.22
№330 0.3 158 141 1,03 7.31 0.19
№326 0.2 13 142 586 41.33 0.195
CR №330 0.2 56 ИЗ 178 15.8 0.22
EPDM №330 0.2 57.5 176 500 31.3 0.17
Недавно Осанайе и др. [010] провели обширные эксперименты по ползу-
чести сажекаучуковых смесей при очень малых напряжениях. Было установ-
лено, что при некоторой величине напряжений течение вообще отсутствует.
Значение предела текучести, определенное в работах этих авторов было
несколько ниже по сравнению величинами опубликованными ранее.
Результаты, полученные Осанайе [010], были впоследствии подтверж-
дены на примере содержащих частицы композиций на основе эластомеров
или расплавов полимеров [А10, К8а, L13].
Исследование переходных режимов течения сажекаучуковых компози-
ций было впервые предпринято Маллинсом и Уорл оу [М51, М52] в 1950 г.
Они обнаружили существование сильных временных тиксотропных эф-
фектов в сажекаучуковых композициях, которые не наблюдались в чистых
каучуках. Если в течение определенного времени чистый эластомер (1)
270 -*V Глава 6. Реологические свойства и переработка...
Shear stress о12 (Ра)
III. Реологические свойства -JU 271
Shear stress, o12 (Pa)
Рис. 5. Кривые вязкость-напряжение сдвига композиции EPDM. Проценты —
объемные (а). Кривые вязкость-напряжение сдвига композиции на основе
бутадиенстирольного каучука. Цифры — объемные доли (Ь). Кривые вяз-
кость-напряжение сдвига, демонстрирующие влияние размера частиц (с).
272 “*V Глава 6. Реологические свойства и переработка...
подвергается сдвигу и течение затем останавливается (2), а потом начи-
нается снова (3), то в течение короткого времени материал помнит свою
предысторию деформации и в потоке возникают напряжения меньшие
чем те, которые были в начальный момент. Если период отдыха был более
1 минуты, вся предыстория оказывается забытой и возникающие в потоке
напряжения равны исходным. Такая картина не воспроизводится в сажекау-
чуковых композициях. Вторичное течение, как и в случае с чистым каучуком
начинается при меньших напряжениях. Снова, но уже через существенно
больший промежуток времени, восстанавливаются напряжения в потоке.
Однако в отличие от чистых каучуков композиция продолжает накапливать
«структуру» непрерывно. Чем больше время «отдыха» тем большие напря-
жения требуются, чтобы снова началось течение. Такая же закономерность
описана в работе Монтэ и др. [М37]. Типичные примеры влияния времени
отдыха на величину переходных напряжений в начале течения показаны на
рис. 6. С ростом времени отдыха возникающие перенапряжения становятся
все больше и больше. При достаточном времени отдыха они могут превысить
имевшиеся в первоначальном образце.
Лоуб и Уайт [15] исследовали релаксацию напряжений после наложения
деформаций в саженаполненном полистироле и нашли, что напряжения
падают не до нуля, а до определенного напряжения приблизительно равно-
го пределу текучести в ур. (56) и (57). Монте и др. [М37] нашли такие же
эффекты в сажекаучуковых композициях. Это показано на рис. 7. Эти же
авторы нашли аналогичные закономерности и в релаксации напряжений
после остановки сдвигового течения.
Было много работ по определению динамической вязкости Т|'(со) и ком-
плексной вязкости Т| * (со) сажекаучуковых смесей и других наполненных
систем. Накаджима и др. [N2] и последующие исследователи нашли, что
правило Кокса-Мерца [С19] упоминавшееся ранее для расплавов гибкоцеп-
ных полимеров не соблюдается для наполненных расплавов полимеров или
эластомеров. Обычно получается неравенство ц*(со) > ц(у). Комплексная
вязкость оказывается существенно выше.
3. Теория вязкопластичных жидкостей
Возникновение теории течения материалов имеющих предел
текучести, отмечено работой Шведова [S8] в Одесском университете в 1890 г.
Шведов спроектировал и построил прибор с коаксиальными цилиндрами
для измерения сдвиговой вязкости суспензий желатина. Для режима уста-
новившегося течения он получил уравнение
<т = ^ + т1в'У (58)
которое позднее было вторично открыто Бингамом [В18, В19] и обычно
носит его имя.
III. Реологические свойства -JU 273
log t (sec)
Рйс. 6. Влияние отдыха на развитие напряжений в начале течения: вязкость в переход-
ном режиме НК содержащего объемную долю 0.2 техуглерода №326 при
100 °C (а); вязкость в переходном режиме SBR с 0.2 (об.) техуглерода №326
при 300°С (Ь).
274 “’V Глава 6. Реологические свойства и переработка...
Рис. 7. Релаксация напряжений в композиции техуглерод — натуральный каучук
после остановки потока. Объемные доли наполнителя: 0.2(a) и 0.3(6).
В трехмерной форме пластическое тело Бингама было описано в работе
Хохенемзера и Прагера [Н19] в 1932 г. при использовании критерия плас-
тичности Мизеса (см. Рейнер [R4] и Прагер [Р15]). Использовали девиатор
тензора напряжений Т следующего вида
a = -pjI + T
(59а)
где
tr Т - + Т22 + ?зз - 0
(59b)
III. Реологические свойства -JU 275
Принимая предел текучести по Мизесу
trT2 = 2K2 (60)
Выражение для Т показано выше и предел текучести определяется из
ур.(60):
Т=^Т+2М (61>
Здесь Т2 есть проекция скалярного произведения Т«Т, a d тензор скорости
деформации по ур. (29в). Олдройд [ОЗ] показал что ур. (49) может быть
написано в более простой форме
2У
T = -^=Td + 2nBd (62)
d2
Олдройд [05] был первым, кто выразил вязкость через значение предела
текучести и неньютоновскую вязкость. Он написал
T = -7====d + 2T]B(tr d2)d
V2trd2 '
(63)
где Т|в зависит от tr d2. Уравнение Скотта-Гершля-Бакли (56) может быть
выражено как
T = ^==d+2^[2trd2](n’1)/2d (64)
а ур. (57) как
Т =
2У ! А
+[1 + B[2lrdz]"4'1'2
(65)
Параметры уравнений состояния (64) и (65) приведены в Табл. 2.
4. Модель пластичной вязкоупругой жидкости
Представление о материале, имеющем предел текучести и при
напряжениях выше предела текучести текущем как вязкоупругая жидкость,
было также сформулировано Шведовым [S8]. Он исследовал переходный
режим течения и отметил, что желатин течет как вязкоупругий материал
при напряжениях выше предела текучести. Это дало ему возможность мо-
дифицировать линейную модель Максвелла
276 -*V Глава 6. Реологические свойства и переработка...
^=с^_£(а_г) (66)
dt dt V
и предложить ее для описания реологических свойств суспензий жела-
тина.
В противоположность Максвеллу, у Шведова не оказалось последо-
вателей. После того как прошло почти столетие, о его работе вспомнили
лишь благодаря монографии Рейнера [R2, R3]. Только после публикации
Хохенэмзера и Прагера [Н19] и особенно после работы Уайта [W9] уже в
1979 был отмечен в обзорах подход Шведова к решению проблемы. Особо
было подчеркнуто, что ур. (66) является дифференциальным уравнением
и его решение выглядит следующим образом
о = (б7>
J0 dsK '
Это можно рассматривать как частный случай более общей формы:
а = У + Jo G(t-s)^-ds (68)
Используя аналогию с ур. (61) Уайт предложил для материала, те-
кущего как вязкоупругая жидкость при напряжениях выше предела
текучести,
Т= , Y Т+Н
П77 (69'
J-trT2
v2
где Н функционал памяти. Было показано, что это эквивалентно следующему
T = -^L=H + H (70)
V2trH2
Для трехмерной деформации в значение Т входит интегральная форма
нелинейной вязкоупругости что показано в работах Уайта [W9], Уайта и
Танаки [W23], и Уайта и Лобэ [W21]. Они все соответствуют обобщенной
форме ур. (70) куда входит (ср. ур. (47)):
H=J0~m(z) c"j(tr с‘‘
dz
(71)
где m(t) функция памяти. Исаев и Фан [15] также предложили свою форму
записи типа ур. (70), но используя модель Леонова [L9] как основу для
выражения Н.
IV. Граничные условия -* V 277
5. Тиксотропная пластичная вязкоупругая жидкость
Проблема уравнений состояния, учитывающих тиксотро-
пию, является довольно старой. В 1930-х Фрейндлих и Джонс [F9] наряду
с другими связывали тиксотропию в суспензиях малых частиц с наличием
предела текучести.
Выражения, приведенные в предыдущих разделах, показывают связь
предела текучести с вязким и вязкоупругим течением, но не с тиксотропией.
Есть возможность включения тиксотропии в комплекс свойств пластичной
жидкости, допустив зависимость У от предыстории tr d2. Такой подход был
применен по отношению к жидкостям, обладающим вязкоупругостью выше
предела текучести в работе Суецугу и Уайта [S26] а позднее в работе Монтэ
и Уайта [М35].
Другой подход к описанию тиксотропного разрушения и восстановления
структуры учитывает величину напряжений, скорость деформации и внут-
реннюю структуру. Линейные теории такого типа начали разрабатываться
в работах Гудива и Уитфилда [GIO, G11 ].
Последней работой такого типа явилась работа Монтэ и Уайта [М35],
в которой сравниваются реологические свойства каучуковых композиций
с предсказаниями теории и она здесь цитируется. Монтэ и Уайт дали формулу
для функции предела текучести (ср. ур. (49с)):
y(nd,i)=Kf +(р1П‘/2 +p2nd)f^an‘/2e-“n^ (t-s)ds (72)
где m(z) имеет то же вид что и в ур. (48а) и (49). Модуль Gt имеет то же
значение что и Gt° , т. е. то же значение что и для чистого каучука, a взята
равной /(ф)тр, где Тр величина для чистого каучука и /(ф) интенсивность
роста тр в случае неактивного наполнителя.
Выражения (70)-(72) были проверены экспериментально на примере
натурального каучука SMR-5 содержащего 0.2 об. доли техуглерода N-326.
Результаты для сдвиговой вязкости в установившемся режиме приведены на
рис. 8, а и для восстановления сдвиговых напряжений в переходном режиме
в начале течения на рис. 8, с. В целом совпадение хорошее.
Другие выражения довольно сильно отличающиеся от приведенных
в этом разделе даны Леоновым [L10]. В них также объединяются тиксо-
тропные свойства и предел текучести, но такое сочетание базируется на
термодинамическом подходе.
IV. ГРАНИЧНЫЕ УСЛОВИЯ
В настоящее время общепринятым считается то, что текущие
ньютоновские жидкости прилипают к твердой поверхности, как это предположе-
но Голдштейном [G9]. Это предположение носило спорный характер вплоть до
1840-х, в чем можно убедиться, читая работы Стокса того периода [S22-S24].
278
—Глава 6. Реологические свойства и переработка...
Time (sec)
Рис. 8. Сравнение с экспериментом реологических моделей представленных уравне-
ниями (70)-(72) на примере сажекаучуковых смесей (а). Сдвиговая вязкость
в установившемся режиме (Ь). Изменение вязкости в режиме сдвиг-отдых-
сдвиг в потоке (с).
IV. Граничные условия -*v 279
Известна долгая история развития представлений о граничных условиях
каучука и каучуковых композиций на стали и других металлических поверх-
ностях. Мелвин Муни в своем выступлении по случаю награждения медалью
Гудьира отметил [М46], что потенциальная возможность проскальзывания
каучука по стальной поверхности составляла его первый интерес, когда ему
было предложено компанией Ю.С. Раббер в 1928 г. заняться перерабатывае-
мостью невулканизованного каучука. Конечно, это привлекало его внимание
на протяжении всей его карьеры и явилось предметом статьи, которую он
опубиковал [М45] в год (1959), когда он уходил на пенсию. Опыт Муни
в области каучука привел к тому, что он явился первым ученым, попытав-
шимся создать приборную технику позволявшую измерить скольжение
материала в процессе течения. В статье опубликованной в 1931 г. [М39]
он описал методы оценки скольжения как в капиллярном реометре, так
и в реометре с коаксиальными цилиндрами. Часто цитируемый капилляр-
ный анализ Муни будет описан в разделе VI, D. В статье 1936 года [М41]
посвященной определению сдвиговой вязкости натурального каучука
в вискозиметре с коаксиальными цилиндрами, Муни тщательно проделал
канавки в цилиндре и в роторе, чтобы исключить проскальзывание на их
поверхностях. Это было также сделано в 1934 г. в приборе с вращающимся
диском, предназначенном для оперативного контроля качества [М40]. Муни
был не единственным в тот ранний период, кто интересовался проскальзы-
ванием. Такое же внимание было уделено Гарнером [G1] по отношению
к пластометру сжатия Вильямса.
В 1940-х этому вопросу уделялось мало внимания, однако в 1950-х мы
находим, что Муни и Блэк [М47] обсуждают проблему проскальзывания при
экструзии через щелевую головку. В статье 1954, Декер и Рот [D4] приводят
результаты нескольких экспериментов надисковом реометре Муни. Они на-
шли, что измеренная величина вязкости зависит от глубины насечек на диске
и от природы металла, покрывающего поверхность. Муни [М43, М45] провел
в 1950-х широкие исследования скольжения, в основном путем сравнения
результатов полученных в вискозиметре Муни с гладким ротором и ротором
с насечками. Он обнаружил, что проскальзывание возникает при введении
мыл в каучук, особенно в бутадиен-стирольный сополимер эмульсионной
полимеризации. Он провел эксперименты, когда ротор покрывался слоем
политетрафторэтилена, которые показали что крутящий момент при этом
сильно снижается, что по-видимому связано с проскальзыванием. В 1958 г.
в обзоре посвященном реологии эластомеров Муни [М44] представил про-
скальзывание как хорошо установленный факт и привел результаты работы
цитированной выше, где он пытался сделать результаты количественными,
вычисляя скорость проскальзывания.
За два десятилетия после ухода Муни на пенсию в литературе не поя-
вилось почти ничего по этому вопросу; однако в 1980 Турнер и Моор [Т12]
280 “•V Глава 6. Реологические свойства и переработка...
из Авон Раббер вернулись к этому вопросу. Эти авторы сконструировали
ротационный вискозиметр с регулируемым давлением для исследования
реологических характеристик и характеристик проскальзывания в потоке
каучука и композиций на его основе. Таким образом, можно было сравнивать
свойства при одинаковом давлении. Они впервые сопоставили результаты
измерений на чистом бутилкаучуке в этом приборе, и тогда оказалось, что
зависимости момент-скорость одинаковы для всех трех роторов; однако
когда перешли к высоконаполненному нитрильному каучуку с большой
дозировкой пластификаторов и наполнителей, оказалось что момент на
гладком роторе гораздо меньше чем на шероховатом. Это было принято как
доказательство проскальзывания. Проскальзывание было также обнару-
жено на примере этилен-пропиленового тройного сополимера с большим
наполнением техуглеродом и маслом. Было обнаружено, что скорость
проскальзывания повышается с температурой, но уменьшается с ростом
дР(МРа)
Рис. 9. Влияние давления на вращающий момент в биконическом реометре.
Аан и Уайт [А1 ] показали что карбоновые кислоты и амиды ини-
циируют проскальзывание в расплавах полиэтилена и полипропилена
вероятно за счет миграции на поверхность и образования слоя с малой
вязкостью. Это также характерно для саженаполненных смесей, но кар-
тина с композициями, содержащими полярные частицы, оказывается
более сложной.
В 1988 Монте с соавторами [М37] в Акронском университете опублико-
вал результаты эксперимента на модернизированном вискозиметре Муни,
V. Механохимическе процессы -*!/• 281
который был многоскоростным, и где можно было менять давление как в ра-
боте Турнера и Моора [Т12]. Были получены зависимости момент-скорость
ротора при разных заданных давлениях для чистых каучуков и для компо-
зиций. Было найдено, что при давлениях менее 0.5 МПа (500 кПа) момент
существенно понижается, указывая на наличие проскальзывания (рис. 9).
Если вначале задать низкое давление, а затем его существенно повысить,
вращательный момент внезапно увеличивается, как показано на рис. 10.
Это указывает на быстрое возникновение адгезионной связи полимера
с поверхностью ротора. Если давление снять, момент медленно уменьшается
(рис. 10). Возникновение проскальзывания можно наблюдать на резиновых
колпачках снятых с ротора; оно начинается на внешней части ротора и рас-
пространяется внутрь. Эти исследования были продолжены Ханом с сотр.
[НЗ] и Уайтом с сотр. [18] из той же лаборатории. Эти авторы нашли, что
величина критического давления, при котором начинается проскальзывание,
по-видимому, составляет около 0.2 МПа и не зависит от природы металла
поверхности за исключением политетрафторэтилена (ПТФЭ) где это выра-
жено наиболее сильно. Если, однако, работать при малом давлении, медь
и латунь обеспечивают наиболее высокие напряжения сдвига, далее следует
алюминий, затем сталь и наконец ПТФЭ. Как правило проскальзывание на-
чинается при одинаковом критическом давлении как на гладком роторе так
и на роторе с насечками, однако в области проскальзывания более высокие
значения напряжений сдвига возникают на роторе с насечками.
Брзосковский с соавт. [ВЗО, ВЗЗ, В35] из Акронского университета
провели работу по экструзии через головку, выполненную из пористого
металла. Когда давление воздуха достигало 0.2 МПа, происходило огромное
падение градиента давления вдоль оси головки. Падение могло достигать
90%. Экспериментальное исследование, показывающее влияние падения
давления воздуха на каучуковую композицию, показано по стадиям на
рис. И. Когда давление воздуха падает ниже 0.2 МПа, осевое давление
возрастает значительно. Эти результаты весьма надежно указывают на роль
проскальзывания.
V. МЕХАНОХИМИЧЕСКИЕ ПРОЦЕССЫ
А. Деструкция индивидуальных эластомеров
Размягчение натурального каучука при «мастикации» было
открыто Хэнкоком [НИ] примерно в 1820 г., и он же установил, что растворы
мастицированного каучука при любой концентрации характеризуются более
низкой вязкостью, чем растворы исходного каучука. Однако вплоть до того
как гипотеза цепных макромолекул была общепринята и не ранее появления
работ Коттона [С27] и Бюссэ с Каннингхемом [В37, В38] стала понятной
роль кислорода (также как и механизм его действия) в процессе «мастика-
282 Д,
Глава 6. Реологические свойства и переработка...
BR 60°С
0.5 rpm
Т
25 Mooney
1
_ One MPa
Рис. 10. Влияние предыстории развития давления на изменение момента в ротацион-
ном реометре.
IV. Граничные условия -*и- 283
Air in
Air out
------►
Rubber in
Rubber out
7777777Y7777777777777777
Air in
Air out
Zone A Zone В (air zone)
In the air zone air is blown between the
flowing rubber and the die walls
| Rubber
Рис. 11. Влияние давления воздуха в головке изготовленной из пористого металла
на поток каучука. В зоне В воздух подается в зазор между потоком пористой
стенкой головки.
284 “*V Глава 6. Реологические свойства и переработка...
ции» каучука. Результаты этих исследователей были позднее подтверждены
работой Пайка и Уотсона [Pl 1]. По-существу все пришли к убеждению, что
натуральный каучук распадается с образованием свободных радикалов под
действием напряжений и что возникшие радикалы стабилизируются в ре-
зультате реакции с кислородом: R-R' HanP^eH“e у р* 4- R* +О2 >RO2
Одно из важных наблюдений было сделано Бюссэ, отметившим свое-
образное влияние температуры на деструкцию. При низких температурах
деструкция обусловлена приложенными напряжениями сдвига и если
температура возрастает при сохранении общей кинематики процесса
пластикации, то деструкция полимера уменьшается. Если температура
возрастет и далее, то интенсивность деструкции, пройдя через минимум,
начинает возрастать вновь. Это обусловлено конкурирующими процессами
уменьшения вязкости и увеличения скорости взаимодействия с кислородом
с ростом температуры.
Обычно находят, что молекулярный вес мало уменьшается при пласти-
кации в среде азота. Бюссэ и Каннингхэм [В38] обнаружили, что введение
акцепторов свободных радикалов приводит к снижению молекулярного веса
при пластикации в среде азота. Пайк и Утсон предположили, что акцепторы
свободных радикалов играют роль подобную кислороду. Количественное
подтверждение этого предположения получено в работе Аурей и др. [А13],
которые связали результаты спектрального и радиохимического анализа кон-
цевых групп со снижением молекулярного веса в процессе деструкции.
Влияние пластикации на молекулярно-весовое распределение было изу-
чено Анжиром [А5] и последующими исследователями. Было обнаружено,
что высокомолекулярные фракции деструктируют быстрее и образующийся
полимер имеет более узкое молекулярно-весовое распределение.
Хотя большинство исследований деструкции под действием напряжений
было проведено на натуральном каучуке, исследовались и другие эластоме-
ры (синтетический цис- 1,4-полиизопрен, цис-1,4-полибутадиен, бутадиен-
стирольный сополимер) [В27, F4]. Фолт [F4] исследовал влияние деструк-
ции при пластикации на реологические свойства эластомеров, особенно
полибутадиенов и полиизопренов. Наибольшая деструкция наблюдалась
в цис-1,4- полиизопрене, что увязывалось с (1) кристаллизацией происхо-
дящей под действием напряжений в процессе деформирования, что, в свою
очередь, приводит к возникновению особенно высоких напряжений и (2)
с резонансной стабилизацией образующихся радикалов.
В. Смеси каучуков
Анжир и Уотсон [А7, А9] обнаружили что привитые сополи-
меры могут быть получены при пластикации смесей эластомеров в отсутствие
кислорода. Они исследовали смеси натуральный каучук - полихлоропрен,
натуральный каучук - бутадиен-стирольный сополимер, полихлоропрен-
V. Механохимическе процессы -*и- 285
бутадиен-стирольный сополимер. Предполагался следующий механизм
взаимодействия:
А _А напряжение у
В — В’ HanP^eHue ) д е
А*+В*------->А-В
С. Эластомеры набухшие в мономерах
Анжиром и Уотсоном [ А8, W2] было показано, что если элас-
томер набух в виниловом мономере (стирол, хлорстирол, акриловая кислота,
метилакрилат, метакриловая кислота, метилметакрилат, винилпиридин,
метилвинилкетон и др.) то пластикация в отсутствие кислорода может при-
вести к образованию блоксополимеров. Этот процесс может осуществляться
по следующему механизму:
р _ О' напряжение ч р р г
д<+м—>вм*
RM*+M---->RMM*
RMM • +М--> RMn+1 M •
Анжир и др. [А4, С5] показали что этот механизм применим и к стеклооб-
разным виниловым пластикам, если они перерабатываются в высокоэла-
стическом состоянии.
VI.
РЕОЛОГИЧЕСКИЕ ИЗМЕРЕНИЯ
А. Реометры с параллельными пластинами
и сэндвичевый
Вероятно в принципе простейший тип реометра может быть сконстру-
ирован путем наслоения исследуемого материала между двумя или тремя
параллельными плитами разделенными на расстояние Я при движении од-
ной плиты относительно другой со скоростью V (рис. 12). Скорость сдвига у
равна V/Н. Для обычных жидкостей это не применяется, но для эластомеров
и их композиций вполне применимо. Приборы такого типа были созданы
и применялись Захаренко и др. [Z1], Мидлманом [М24], Голдштейном [G8],
286 -Jv- Глава 6. Реологические свойства и переработка...
Фурутой [F11], Лобэ и Уайтом [L14], Токи и Уайтом [Т7], Монтэ-и др. [М37],
Осанайе и др. [010], и К. Дж. Кимом и Уайтом [К8а]. Приборы (с термо-
камерой) могли быть помещены в установку для измерения напряжений
и приводиться в движение с заданной
скоростью ^определяющей постоянную
скорость сдвига. Они могли быть при-
менены для исследования ползучести,
когда применялись подвешенные грузы.
Это обеспечивало проведение опыта при
постоянном напряжении. При малых на-
пряжениях необходимо компенсировать
вес центральной плиты, которая порож-
дает гравитационные силы [010]. При
очень малых нагрузках можно точно
определить предел текучести сажекау-
чуковых смесей. Осанайе и др. [010]
Рис. 12. Сэндвичевый реометр. проводили измерения при напряжениях
ниже предела текучести.
Скорость сдвига у есть отношение скорости движения плиты V к рас-
стоянию между плитами, т. е.
y = V/H
Напряжения сдвига о12 равны
F _ F
2А~ 2^-VWt)
(73)
(74)
Член VWt отвечает за уменьшение активной поверхности лицевой части
сэндвича. Ширина сэндвича W и t время эксперимента.
В. Биконический реометр
Концепция реометров конус-плоскость и биконических
реометров была развита в 1940-х (рис. 13). Прибор конус-плоскость при-
менялся в работах Фримена и Вайссенберга [F10] и предназначался для
жидкостей со средним значением вязкости. Он был основой их «реогони-
ометра» который измерял также нормальные напряжения. Биконический
реометр был создан в то же время Пайпером и Скоттом [Р12] из BRMRA
и с самого начала предназначался для каучука. Аналогичные приборы были
предложены Турнером и Мором [Т12], и Монтэ и др. [М37, М38]. В этих
последних приборах давление регулировалось благодаря подаче каучука
в реометр через заправочное приспособление, работающее под давлением.
VI. Реологические измерения -•v- 287
Значительное преимущество прибора с геометрией конус - плоскость
состоит в том, что при малом угле между ними скорость сдвига постоянна
по всему зазору между конусом и плоскостью. Она может быть выражена
формулой:
Q Q Q
у =---=------=-----
Л(г) rtana tana
где h(r)— расстояние по вертикали между
конусом и плоскостью и а — угол между о с
J J J Рис. 13. Ьиконическии реометр,
конусом и плоскостью.
Напряжения сдвига в биконическом приборе могут быть рассчитаны,
зная момент М по следующей формуле
М = 2|оЯ2лг2о12</г = ^у-а12 (76)
В приборах Турнера и Мора [Т12] и Монтэ и др. [М38] давление в рабочем
узле поддерживалось внешним резервуаром. Это позволяло проводить из-
мерения в сдвиговом потоке при постоянной скорости сдвига и создавалась
возможность определения условий, при которых начиналось проскальзы-
вание в каждой данной композиции.
С. Вискозиметр типа «диск-диск»
Диск, обеспечивающий деформацию сдвига в вискозиметре,
был предложен Муни в 1934 г. [М40] и вискозиметр Муни сыграл заметную
роль в резиновой промышленности, поскольку применялся для контроля
качества образцов резиновой смеси и значит для контроля технологического
процесса (рис. 14). Он близок к прибору с параллельными дисками, зада-
ющими деформацию сдвига, который широко применялся для исследования
растворов и расплавов полимеров.
Скорость сдвига между двумя дисками на расстоянии радиуса г равна
y(r) = rQ/fe (77)
где h расстояние между дисками, a Q скорость вращения движущегося
диска. Крутящий момент при этой геометрии прибора равен
М = J 2яг2о12с?г (78)
где о12 меняется в зависимости от радиуса г. Как было показано рядом
исследователей, ур. (77) может быть подставлено в ур (78) и интеграл реша-
288
—Глава 6. Реологические свойства и переработка...
ется по отношению к скорости сдвига у(Я), и тогда на внешнем периметре
диска получим
Р+ЗУ2ЛП ,7Q1
<^4—(79)
где
S = dlogM/d log Cl
Для степенной жидкости S~ п.
б
Рис. 14. Вискозиметры типа «диск-диск»: параллельные диски (а), Муни (б).
Для вискозиметра типа «диск-диск» крутящий момент на диске может
быть выражен как
М = 2 ]о 2лг2(Уъаг +2nR1H(GQr)
R
(80)
поверхность периферия
где Н толщина диска. Ур. (80) может быть решено для крутящих напря-
жений сдвига на внешнем радиусе диска, при этом получается выражение
аналогичное ур. (73):
(81)
где функция /’дана в работе Накаджима и Харрел [N3].
Вискозиметр со сдвигающими дисками для текущего контроля качества
всегда имел одну скорость вращения диска (2 об/мин), хотя Муни [М42]
еще в 1940-м создал многоскоростной прибор. Применение ур. (81) для
определения G0Z(/?) и сдвиговой вязкости пригодно и для работы на много-
скоростном приборе.
В течение длительного времени проявлялся значительный интерес к из-
мерению вязкоупругих свойств полимеров с помощью этого прибора. Муни
VI. Реологические измерения -* V 289
[ М42] привел результаты измерения эластического восстановления, Коопман
и Крамер [К16] из Байер АГ создали усовершенствованный вискозиметр
Муни, позволивший измерять релаксацию напряжения после остановки
течения. Такой прибор позднее был проанализирован Монтэ и др. [М37].
Монсанто [G21] и позднее Альфа Технолоджис стали выпускать серийный
прибор для контроля качества, аналогичный усовершенствованному Коо-
перманом и Крамером.
D. Капиллярный реометр
Капиллярный реометр, один из старейших и широко
применяемых приборов для измерения вязкости жидкостей (рис. 15),
применялся для исследования почти всех видов сложных жидких сред
основателями современной реологии на
протяжении 1920-х-1930-х. Его приме-
нение для исследования каучука описано
в работах Марцетти [М8-11] и Диллона
с сотр. [D7-9] и с тех пор он продолжает
широко использоваться.
Основная идея прибора состоит в том, что-
бы связать потерю давления при экструзии
через трубку малого диаметра D и длины L с
напряжениями сдвига на стенке капилляра Л Л_ т(.
Рис. 15. Капиллярный реометр,
и скорость экструзии со скоростью сдвига на
стенке. Общее падение давления рт в канале есть сумма давлений внутри
канала Др и вторичное падение на входе и на выходе из канала Дре:
(82)
Можно связать Др с напряжениями сдвига на стенке (о12)^ с помощью
простого баланса сил:
n£L(o12)w =(л£2/4)Др (83)
что позволяет написать
(g12)w=W4L (84а)
/*r = APe+4(Gi2)wi/^
(84b)
Как отмечено Муни и Блэком [М47] и позже Бэгли [В8], величина &Ре
может быть большой по сравнению с Др . Путем применения набора капил-
ляров с разными L/D можно определить из наклона зависимости рт
от L/D что и называется поправкой Бэгли [В8]. Скорость сдвига на стенке
290 -'V- Глава 6. Реологические свойства и переработка...
должна поддерживаться постоянной, но в этом следует убедиться, подбирая
капилляры с одинаковым диаметром и меняя Q.
Несомненной в ур. (84) является мысль о том, что диаметр резервуара,
предшествующего капилляру, намного больше, чем сам капилляр. В этом
случае потери давления в самом резервуаре могут не учитываться. Поправки
такого рода были рассмотрены Метцгером и Ноксом [М22].
Скорость сдвига на стенке капилляра может быть рассчитана способом,
предложенным Вайссенбергом и соавторами [Е4], (см. также Муни [М39]).
Заметим что ур. (83) применимо к случаю телескопического течения при
каждом значении радиуса и мы видим, что напряжения сдвига меняются
линейно с изменением радиуса
<’l2(H = (a12)w(r/ в) (85)
Это позволяет нам переписать выражение для скорости экструзии как
Q = [ 2ТСГ0!dr = кВ f(o,2)w
Jo 8(°12)wJ I dr)
которое мы интегрируем по частям и предполагаем совершенное прилипание
материала к стенке. Дифференцируя 32^/tcD3 по (о12)ш и применяя прави-
ло Лейбница дифференцирования интегралов, можем получить выражение
для скорости сдвига на стенке капилляра
f 1 _ГЗ^' + Л32<2
I dr Jw V 4д JtlD3
где
dZTi(a12)w
б/Ц32<2/л£>3)
(87а)
(87b)
Применение уравнений (84) и (87b) позволяет оценить функцию вязкости
п(т).
Муни модифицировал ур. (86) для случая течения с проскальзыванием
и показал, как можно определить проскальзывание на стенке капилляра.
В общем виде вместо ур. (86) можно написать
_ ря , тс/?2 те/)3 r(o12)wf rf'OiV
Q = 2nrv*dr =-------n + —------г— ----------L Hz (ooa)
Jo ' 4 s 8(o12)Jo dry
(88b)
лД3 D (o12)wJ I dr)
VI. Реологические измерения
Л
291
Интеграл в ур. (88в) зависит только от напряжений сдвига на стенке
капилляра, так что
(89)
Это предусматривает эксперименты с постоянным D&p/4L и с перемен-
ным диаметром D, что означает и переменное давление.
Капиллярные реометры широко использовались для контроля качества в
резиновой промышленности начиная с 1920-х [D7, D8, G18, М8-11]. С конца
1970-х они были запатентованы Монсанто. Основная проблема с прибором
в том, что он дает значения вязкости даже при больших скоростях сдвига,
когда вязкость уже мало чувствительна к молекулярному весу и его распре-
делению. Кроме того, чистые эластомеры в таких приборах могут оказаться
в области нестабильного течения.
Е. Реометр сжатия
Основная теория течения неньютоновских жидкостей
в реометре сжатия была создана Скоттом [S5], который рассматривал те-
чение степенной жидкости (рис. 16). Предполагается, что течение в основ-
ном является ламинарным сдвигом и жидкость перемещается радиально
к наружной стороне сближающихся дисков. Учитывая непрерывность при
Согласно Скотту, как уже отмечалось ранее, связь между напряжениями
сдвига и скоростью сдвига определяется степенным законом:
( Н\др
I 2 ) dr
(92)
292 -'V Глава 6. Реологические свойства и переработка...
(93)
(94)
F = 2r
Можно рассчитать поле скоростей vr(z). Используя ур. (90) это может быть
пересчитано в скорость сближения плит (-dH/dt).
Сжимающая сила F связана с р(г) выражением
F = -j/nrp(r)dr
Это приводит к
2га+ 1Y KRn+3 1 ( dH У
п J га + 3 fj2n+1 dt )
Скорость сдвига и напряжение сдвига на внешнем радиусе диска равны
2д + 1 f R 1 (95)
(%)
Реометры сжатия исследовались начиная со времен Вильямса и др.
[W29] и Ван-Россемаи Ван-дер-Мэйдена [VI]. В 1920-х Файерстон [W29]
и Гудрич [К2, КЗ] применили пластометр сжатия в качестве прибора для
контроля качества. Впоследствии, в 1930-х Баадер [В1-7] и Континентал
Гуммиверке [В1-7] сконструировали аналогичный прибор и назвали его
Дефо для контроля качества, который использовался в И. Г. Фарбениндустри
во время войны для тестирования синтетического каучука Буна S. После
войны интерес к Дефо упал и вискозиметр Муни (раздел VI, С) победно
пришел к всеобщему признанию. Коопман с сотрудниками в Байер АГ
[К14-17] сконструировали улучшенный Дефо, который производится Хааке
Месстехник в Карлсруэ.
F. Реометр растяжения
Большое разнообразие приборов было создано в качестве
реометров растяжения. Первым, такого типа, был прибор Баллмана [ВИ],
он имел вертикальный образец, растягиваемый ускоряющимся движением
зажимов. Скорость растяжения при этом
1 dL
L dt
(97)
где L длина образца. Если она постоянна во времени, то величина L(t)
должна возрастать как
VII. Технология переработки -*v 293
L(0 = L(O)e^z (98)
Напряжение при этом
а = F(t)/nR2(t) (99)
где И — радиус образца. Другой метод растяжения состоит в том, что образец,
погруженный в ванну с силиконовой жидкостью, зафиксирован на одном
конце и растягивается за другой под действием груза [С12], движущегося
зажима [V5] или с помощью валков [И]. По одной из методик применяли
две пары валков [М17]. В эксперименте с грузом применяли эксцентрик,
чтобы обеспечить постоянство напряжения. В методике с фиксированным
зажимом, а с другого конца с валками, скорость растяжения определялась
как
V
(100а)
Lj
а в случае применения двух валков
Те =7^ (100в)
Li/
Если использовали один из зажимов ускоряющийся и с погружением
в ванну, применимо ур. (98).
Все описанные выше эксперименты предназначались для термопластов.
Только Коттон и Тиль [С19] предложили прибор для одноосного растяжения
предназначенный для эластомеров. В приборе экструдированная полоска ка-
учука помещалась на два шкива. Оба конца опускались вниз проходя между
парами шкивов с насечкой, которые вытягивали концы полоски вниз.
VII. ТЕХНОЛОГИЯ ПЕРЕРАБОТКИ
А. Закрытые смесители
1. Общие положения
Первая стадия переработки каучука состоит в смешении
ингредиентов резиновой смеси в смесительном аппарате. В начальный пе-
риод развития промышленности этот процесс осуществлялся на открытых
двухвалковых вальцах [С7, G14]. Начиная с 20-х годов прошлого столетия
начался переход, давно завершенный, к смешению в закрытых смесителях
конструкции в основном предложенной Бенбери [В12, В13] (см также Уайт
[W13] и начатый первоначально нынешней Фаррел Корп. [W11 ]). Совре-
менный закрытый смеситель состоит из смесительной камеры содержащей
два ротора, вращающихся навстречу друг другу. В центре верхней части
294 -*v- Глава 6. Реологические свойства и переработка...
смесительной камеры имеется канал, через который поступают ингредиенты.
На протяжении большей части цикла смешения в канале находится плунжер,
который вдавливает ингредиенты в камеру. На дне смесительной камеры
имеется затвор, который в конце цикла открывается и смесь вываливается
наружу. Такой аппарат показан на рис. 17.
Роторы закрытого смесителя имеют оригинальную конструкцию, разра-
ботанную Бенбери [В12], они разделены и имеют два искривленных гребня,
которые прокачивают материал в противоположных направлениях. В после-
дующие годы было предпринято немало усилий, чтобы улучшить конструк-
цию закрытых смесителей. Лаш и Фрэй [L12] из Вернер анд Пфлайдерер,
Тисон и Компер [Т13] из Гудьир, Сато и др. [S1 ] из Бриджстоун/Кобэ Стил
предложили смесители, у которых на валках было не два, а четыре гребня.
В настоящее время большинство закрытых смесителей с разделенными
роторами имеют четыре ребра (рис. 18).
Закрытый смеситель с существенно отличающейся конструкцией сме-
сительной камеры был сконструирован Куком [С17] из Фрэнсис Шоу анд
Ко. и Лаш и Стромером [L3] из Веренр анд Пфлайдерер. Эти смесители
имеют зацепляющиеся противоположно направленные роторы (рис. 19).
В этих смесителях оба ротора должны вращаться с одинаковой угловой
скоростью. Смеситель фирмы Фрэнсис Шоу «Интермикс» был выпущен
в продажу первым и привлек значительное внимание. Через некоторое время
закрытые смесители с зацепляющимися роторами стали доминирующими
в резинотехнической промышленности Европы и Японии.
Пассони [Р1 ] из Помини предложил конструкцию зацепляющихся ро-
торов которые могли перемещаться в направлении, перпендикулярном их
осям с тем чтобы можно было регулировать зазор между ними.
Закрытые смесители с разделенными роторами выпускались компанией
Фаррел Ко. в Соединенных Штатах и в Англии. Кобэ Стил и ее филиал Ко-
белко-Стюарт Боллинг выпускали такие машины в Японии и Соединенных
Штатах, Вернер энд Пфлайдерер Гуммитехник (недавно поглощенная фир-
мой Крупп) производила закрытые смесители в Германии, а Техинт-Помини
в Италии. Мицубиси Хэви Индастриз по лицензии Вернер анд Пфлайдерер
Гуммитехник выпускала закрытые смесители с разделенными роторами
в Японии. Смесители с зацепляющимися роторами производились фирмой
Фрэнсис Шоу энд Ко. в Англии (теперь поглощена фирмой Фаррэл). Вернер
анд Пфлайдерер Гуммитехник (и их лицензиат Мицубиси Хеви Индастриз)
производили такие закрытые смесители в Германии и Японии. Промини
производила закрытые смесители с регулируемым зазором в Италии.
2. Основные исследования
Вплоть до недавнего времени было мало фундаментальных
исследований закрытых смесителей. В 1979 Фрекли и Ван Идрис [F8]
опубликовали первое исследование по визуализации потока в закрытом
VII. Технология переработки
-\г 295
Рис. 17. Закрытый смеситель Бэнбери и конструкция роторов (Из Banbury
[В12, В13]).
296 -* V Глава 6. Реологические свойства и переработка...
а
смесителе с незацепляющимися роторами с помощью стеклянного окошка,
расположенного перпендикулярно осям роторов. Более широкие исследо-
вания были позже опубликованы компаниями Бриджстоун и Кобе Стил
[А12, S1] направленные на оптимизацию конструкции роторов. В этих
работах были применены перемещающиеся роторы в прозрачной камере
из поликарбоната. Водные растворы полимеров были взяты в качестве ис-
следуемой жидкости. Более поздние
исследования Мина и Уайта [М27,
М28], Морикавы с сотр. [М48-50]
и Кима и Уайта [К5, Кб, W19] были
выполнены на аппаратах, подоб-
ных вышеназванному. Фрекли
и Ван Идрис обратили внимание
не только на движение материала
в закрытом смесителе, но также срав-
нили поведение разных эластомеров
и проследили полностью весь цикл
смешения (рис. 20). Чо и др. [С12]
сравнили работу роторов Бэнбери
с двумя гребнями и роторов Тисона-
Компера с четырьмя гребнями при
исследовании вулканизации в пото-
ке. Роторы с четырьмя гребнями пе-
ремешивали быстрее. Сравнительно
недавно исследования визуализации
потока были опубликованы Тохом
и др. [Тб].
Основное, что было установлено
в этих исследованиях - материал
в смесителе, как с двух- так и с
четырехгребневыми роторами со-
вершает круговые движения вокруг
смесительной камеры (рис. 21). На-
туральный каучук имеет тенденцию
к налипанию на роторы, большинс-
тво синтетических каучуков разди-
рается на куски. Добавление техуглерода или масел в камеру смесителя
было исследовано методом визуализации потока для широкого круга элас-
томеров. Раздир эластомеров с образованием крошки препятствует процессу
смешения с техуглеродом, что осуществляется более равномерно в случае
натурального каучука. Скорость абсорбции масел определяется площадью
поверхности эластомера и развивается быстрее при дроблении каучука
в крошку и медленно в случае натурального каучука.
b
Рис. 18. Роторы закрытого смесителя
конструкции Сато и др. [S1] из Бридж-
стоун и Кобэ Стил.
VII. Технология переработки -*v- 297
Рис. 19. Зацепляющиеся роторы закрытого смесителя конструкции Кука (Из Коле-
мана и Нолла [С16]).
298 -*!/ Глава 6. Реологические свойства и переработка...
Рис. 20. Визуализация потока в закрытом смесителе.
Рис. 21. Циркуляция материала в закрытом смесителе.
VII. Технология переработки 299
Относительно недавно Ким и Уайт [К9] сравнили методом визуализации
процесс смешения в смесителе конструкции Р.Т. Кука [С17] с зацепляю-
щимися и с разделенными роторами. Гомогенизация закладки, введение
техуглерода и абсорбция масел оказались лучшими в случае зацепляющися
роторов. Колихиран и Уайт [К19] исследовали введение силикагеля, талька
и техуглерода в бутадиенстирольный каучук в двух смесителях - в Бэнбери
с двухгребневыми роторами и в смесителе Кука с зацепляющимися роторами.
Диспергирующее смешение осуществляется гораздо быстрее в смесителе
с зацепляющимися роторами.
В. Шнековая экструзия
1. Общие сведиения
Шнековые экструдеры впервые вошли в резиновую про-
мышленность в конце 19 столетия благодаря фирмам Фрэнсис Шоу, Джон
Ройл, Пауль Тростер, в которых эти машины впоследствии составили основу
производства (рис. 22). Первые шнековые экструдеры запитывались в го-
рячем виде лентами резиновой смеси подогретой на вальцах соединенных
с экструдером. Шнеки имели глубокую нарезку и малое отношение длины
к диаметру.
Рис. 22. Шнековый экструдер.
В 1930-х Пауль Тростер Машиненфабрик [W12] выпустила шнековый
экструдер с холодной запиткой, в который подавались холодные ленты
резиновой смеси.
Экструдер с холодной запиткой имел мелкие каналы шнека и значительно
большее отношение длины к диаметру. Первый экструдер с холодной запит-
кой производил экструдаты с широким распределением по температуре, ко-
торые искривлялись при выходе из головки. Поэтому задачей, стоящей перед
экструзией с холодной запуткой, было получение термогомогенизированного
продукта на шнеке экструдера, который бы давал экструдат с однородным
температурным полем и который бы не искривлялся на выходе. Начиная
с 1960-х, было предложено несколько усовершенствований экструдеров
с холодной запиткой. Во-первых Майлефер [Мб] и Гэйер [G3] (Юниройал
Инк.) создали конструкцию запорного шнека. Это означало установку вто-
рого ребра, которое изолировало менее текучий материал и предотвращало
его уход со шнека до тех пор, пока он не размягчался. Промышленный
300 -*v- Глава 6. Реологические свойства и переработка...
выпуск был налажен в Соединенных Штатах фирмой NRM (по лицензии
Юниройал) под названием «Пластискрю». Предназначенный для контроля
полноты плавления гранул термопластов его конструктором Майлефером,
а также Гэйером для изолирования участков резиновой смеси, подвергшихся
скорчингу, этот шнек, примененный в экструдерах с холодной запиткой,
обеспечил более равномерное температурное поле, но при развитии более
высоких температур.
Впоследствии было предложено много новых конструкций специальных
секций в шнековых экструдерах с холодной запиткой таких, например как
секция сдвига Тростера и разные конструкции Ленэна и Менгеса [L8, Ml9]
обеспечившие ряд улучшений процесса.
В 1970-х Менгес и Хармс [Н12, Н13, М18] из Института Кюнстофферар-
байтунг и из Юниройал предложили оштифтованный экструдер с холодной
запиткой (рис. 23). Оказалось, что это дает существенное улучшение одно-
родности экструдата. Сначала лицензия на эту машину была получена фир-
мой Пауль Тростер Машиненфабрик, а затем другими машиностроительны-
ми фирмами включая Герман Берсторф Машиненбау и Круп Гуммитехник
в Германии, Технит-Помини в Италии, Накатозоки в Японии и Фаррел
Корп. в Соединенных Штатах.
Рис. 23. Шнековый экструдер с оштифтованным цилиндром (pin barrel screw
extruder).
2. Основные исследования
Наиболее ранние исследования течения эластомеров в шне-
ковых экструдерах обнаруживаются в работах Вила [V3] и Пижотт [РЮ].
Результаты Пижотт особенно интересны. Всего несколько исследований
в основном направлении появилось в последующие годы. Характеристики
потока и подача в голодном режиме в шнековых экструдерах с отсосом были
проанализированы Брзосковским и др. [В36], и Вергнесом и др. [V2]. Более
глубокое исследование работы в голодном режиме экструдеров с холодной
запиткой было проведено Куботой и др. [К21], которые показали, что степень
VII. Технология переработки -*v- 301
наполнения пропорциональна давлению на выходе. Течение резиновых
смесей с маркерами потока в шнековых экструдерах было начато Менгесом
и Лэненом [М19] с целью оценить смесительный и гомогенизирующий эф-
фект разных конструкций шнеков. Эти исследования заключались в запитке
в экструдер равновязкостных белых и темных лент смесей, содержащих
вулканизующую группу. После достижения установившегося режима,
экструдер останавливали, нагревали и обеспечивали вулканизацию. Шнек
затем удалялся, и резиновая смесь отслаивалась и нарезалась пластинками.
Исследования с маркерами потока, работы оштифтованных экструдеров
с холодной запиткой были впервые опубликованы изобретателями Менгесом и
Хармсом [М18] и в последние годы Ябуситой и др. [Y1], и Шинном и Уай-
том [S11 ]. В целом было установлено, что секции со штифтами в цилиндре
перемешивают гораздо лучше (смешение окрашенных и белых смесей),
чем обычные секции шнека (рис. 24). В исследованиях Ябуситы и др [Y1]
и Шина и Уайта [S11] сравнивались также обычные шнеки со шнеками
имеющими проточки в гребнях, но не имеющими штифтов. Такие шнеки
были промежуточными по гомогенизирующей способности. Авторы [S11,
Y1] показали, что создание проточек (slices) в гребнях шнека снижает его
способность как подающего насоса. Они также установили, что создание штиф-
тов мало влияет на нагнетающую способность шнека. Шин и Уайт сравнили
целый ряд резиновых смесей и показали, что хотя при установке штифтов всегда
улучшается гомогенизация, уровень гомогенизации меняется существенно от
смеси к смеси. Вероятно, именно эта гомогенизирующая способность связана
с формированием температурных полей в больших экструдерах, что позво-
ляет экструдерам с оштифтованным цилиндром (pin barrel extruders) обес-
печивать однородность экструдатов при большой производительности.
С. Экструзия через головку
1. Общие положения
Конструкция головок всегда была проблемой с самого на-
чала создания технологии экструзии. Патентная литература средины 19-го
столетия [В17, В29, G15] обнаруживает значительное внимание созданию
цилиндрических, угловых головок и головок для изоляции проволоки
в плунжерных и шнековых экструдерах. В этом же столетии уделялось много
внимания созданию листовальных головок, включая изобретения разного
рода контрольных систем для поддержания равнотолщинности [L6, L12, Р9,
Bl, В7]. Такие головки имели или (1) конвергирующие (дивергирующие)
секции которые превращали цилиндрический поток на выходе из экструдера
в щелевой или (2) систему каналов (manifold), которая распределяет поток
поперек головки, обеспечивая равномерную подачу расплава в головку
(рис. 25). Их часто называют Т- образными головками.
302 -’V- Глава 6. Реологические свойства и переработка...
Ь
Рис. 24. Эксперименты с маркерами потока в экструдере с оштифтованным цилиндром
согласно Ябусита и др. [Y1]. Поперечный срез полоски вулканизованного
каучука извлеченного из пространства между первым и вторым разрезом
(экструдер работал без применения pin). Условия эксперимента Тв= 80 °C,
Ts = 80°C, N = 10мин-1 (а). Поперечный срез полоски вулканизованного
каучука извлеченной из пространства первого разреза (экструдер работал без
применения pin). Условия эксперимента: Тв = 80°С, Ts 80°С, N = 10 мин-1 (6).
VII. Технология переработки “* V 303
Головки, которые могут соэкструдировать протектор и боковину или
протектор/боковину/тканевую основу привлекли большое внимание,
начиная с 1930-х [ЕЗ, F2, L7, S3, S14] (рис. 26). В целом сегодня не-
сколько видов шнековых экструдеров сконструированы в варианте,
обеспечивающем подачу сразу нескольких индивидуальных компонентов
в одну головку.
Рис. 25. Головка конструкции «ласточкин хвост» для производства листового экс-
трудата.
Было много успешных продвижений в создании профильных головок.
Была разработана технология, которая позволяла проводить экструзию
искривленных секций [G5, М25, М26]. Работа Готтлера [G5] и Миллера
с сотр. [М25, Мб] описывает разные технологии экструзии кривых труб
и профилей.
Резиновые смеси часто содержат резаные волокна. Важно регулировать
ориентацию волокон в экструдатах, проходящих через головку. В качестве
примера можно назвать экструзию камеры или рукава, где волокна должны
быть ориентированы по окружности, а не по оси. Разнообразные методы были
предложены, чтобы достичь этого. Сюда входили разнообразные головки
с вращающимся дорном [С5, D10], и головка радиально-лучевой конструк-
ции, предложенная Готтлером и др. [G4, G6] из Монсанто.
2. Основные исследования
Начало экспериментальных работ по исследованию течения
эластомеров через каналы относится к работам Мацетти [М8-11] в 1920-х.
Любой объективный обзор современного состояния знаний в этой области
должен осветить обширную литературу, касающуюся течения термопластов.
Исследования термопластов включали как исследование качества экструда-
та, так и его разбухания, а также двулучепреломление и движение маркеров
потока с целью изучения движений жидкости внутри канала. Типичными
работами в этом направлении явились работы Бэгли и Биркса [В9],
Балленджера и Уайта [В 10], а также Хана и Дрекслера [Н2] в которых
304 -*и- Глава 6. Реологические свойства и переработка...
получена информация о наличии циркуляции потока внутри канала,
о поле напряжений в текущем расплаве, и причинах появления неста-
бильностей в процессе экструзии. Полиэтилен низкой плотности (ПЭНП)
характеризуется наибольшими циркуляционными течениями в потоке на
входе в канал. Однако подобный эффект найден также и для полистирола
[В10].
Рис. 26. Головка для соэкструзии протектор/боковина.
Обычно каучук перерабатывается в виде высоконаполненной композиции
и не является прозрачным. Это не позволяет активизировать исследова-
ния резиновых смесей за исключением измерения разбухания экструдата
и оценки его качества. Однако стоит сказать все-таки о том, что относится
к проблеме.
Первые заметные исследования течения резиновых смесей в каналах
были выполнены Муни и Блэком [М47], которые обнаружили значительные
потери давления на входе в канал (см. обсуждение в разделе YI, D). Анало-
VII. Технология переработки -»v- 305
гичные результаты на расплавах термопластов были получены впоследствии
Бэгли [В8]. Уайт и Кроудер [W17] обнаружили значительные потери дав-
ления в резиновых смесях на выходе из канала. Их результаты показали,
что эти эффекты более значительны, чем в термопластах.
Экспериментальные исследования Ма и др. [М3] показали, что добавле-
ние малых частиц, включая техуглерод, подавляет большие циркуляционные
вихри обнаруженные в ПЭНП. Исследования Ма и др. [Ml] и Сонга и др.
[S18] проведенные на резиновых смесях не обнаружили подтверждения
наличия циркуляционных потоков на входе в канал.
Наиболее важный эффект введения техуглерода и других наполнителей
в эластомеры и термопласты состоит в снижении разбухания экструдата
и усадки и улучшении качества его поверхности, т. е. проявляется тенденция
к устранению искривления экструдата [S12, W17].
D. Прессование
1. Общие положения
Прессование представляет собой не одну технологию,
а целый набор технологий (рис. 27). Они простираются от компрессионного
прессования, когда куски или листы помещаются в открытую форму, кото-
рая затем смыкается, окружая загрузку. Когда изделие сформовано (а для
резины свулканизовано) форма раскрывается и изделие вынимается. При
инжекционном формовании материал инжектируется под давлением в за-
крытую форму. Когда изделие сформовано и свулканизовано, форма снова
открывается. Литьевое (трансферное) формование является комбинацией
инжекционного и компрессионного формования, когда резиновая смесь
инжектируется в форму из резервуара. Раздувное формование предполагает
изготовление преформы, которая помещается затем в саму форму. Префор-
ма затем раздувается (посредством горячего воздуха/пара или какой-либо
жидкости, возможно в гибкой оболочке) и заполняет форму.
Эти технологии возникли в первой половине 19-го столетия и были раз-
виты братьями Хэнкок (Томасом и Чарльзом в Англии) и братьями Гудьир
(Чарльз и Генри в Соединенных Штатах). Большая часть этих изобретений
описана в автобиографиях и патентах Томаса Хэнкока [Hl 1] и Чарльза Гудь-
ира [G14] опубликованных в средине 19-го столетия. Хэнкок [Н4-7] описал
процессы инжекционного формования, раздувного формования (включая
применение гибкой оболочки, изготовленной из вулканизованной резины,
с находящимся внутри горячим воздухом, обеспечивающим давление)
и формования пеноматериалов с применением гуттаперчи, натурального
каучука и их смесей.
20-е столетие стало свидетелем быстрого развития двух конкретных видов
прессования в технологии резины. Первый состоял в формовании автопок-
рышек, второй — в формовании резино-технических изделий. Технология
306 “• ir Глава 6. Реологические свойства и переработка...
VII. Технология переработки -*и- 307
d
Descending
parison
t
Inflating
Inflated and cooling
Рис. 27. Различные технологии прессования: компрессионное формование (а);
литьевое (трансферное) прессование (Ь); литье под давлением (с); раздув-
ное формование (d), репродуцировано с разрешения В.А. Холмс Уокер,
«Polymer Conversion» Halsted Press, London, 1975.
308 “* V Глава 6. Реологические свойства и переработка...
производства шин состояла из двух процессов на заключительной стадии
формования. Первый — это получение заготовок на барабане [F17, G19,
М16], второй — прессование заготовок с получением конечной автопок-
рышки с одновременной вулканизацией [DI, S9, Т11].
В производстве резино-технических изделий можно увидеть широкий
круг производственных операций, включая компрессионное формование,
литьевое прессование и шнековое литье под давлением.
2, Основные исследования
Базовые исследования механизмов течения при прессовании
резиновых смесей отсутствовали. Практически все исследования были пос-
вящены термопластам. Большинство работ были посвящены исследованию
заполнения литьевой формы.
Давно стало очевидным, что имеется два механизма заполнения формы.
Это медленное распространение фронта или впрыск струи в форму. Ода
и др. [01] показали что струйное заполнение формы есть результат того, что
полимер или резиновая смесь характеризуются малым разбуханием экс-
трудата. Очень часто сложные закономерности возникновения разбухания
связаны с температурой расплава и ее колебаниями в разных опытах. Исаев
с соавторами [13,14,16] провел широкие исследования изменения давления
и заполнения формы резиновыми смесями, когда фронт медленно двигался
вдоль полости формы.
VIII. ИНЖЕНЕРНЫЙ АНАЛИЗ ПЕРЕРАБОТКИ
А. Общие положения
Имеется много путей применения реологии эластомеров
к процессам переработки. Это благодаря тому, что переработка в большинстве
случаев — это просто течение и формование резиновых смесей. Наиболее
очевидная польза, это установление причин трудностей, возникающих при
заводском производстве. Часто нарушение выпуска стандартной продукции
может быть сведено к общей причине — высокий уровень вязкости или уп-
ругая память смеси. Это конечно и история о том, как первые специалисты
реологи в области каучука нашли свое применение и, в значительной сте-
пени, о том, чем они заняты и в настоящее время.
В. Теория подобия
1, Общие положения
Одной из наиболее плодотворных идей для реолога-поли-
мерщика в промышленности является теория подобия. В этом традицион-
ном виде инженерного анализа законы сохранения (сила-момент, энергия
ит. д.) выражаются в безразмерной форме, и возникающие безразмерные
VIII. Инженерный анализ переработки -*v- 309
величины являются предметами рассмотрения. Были случаи применения
в 19-м столетии такого подхода к моделированию бассейнов для кораблей,
или к течению жидкости по трубам. Конечно основная идея применения
теории подобия в инженерии была развита Осборном Рейнольдсом как раз
для последнего применения. Эта идея была далее развита в начале 20-го
столетия Вильгельмом Нуссельтом. Безразмерные соотношения, предло-
женные этими авторами, были использованы в качестве руководящей идеи
при проектировании трубопроводов, теплообменников и колонн с насадкой.
Многочисленные пособия описывают этот предмет и являются основной
частью университетского инженерного образования [В21, Е2].
К сожалению, результаты работ Рейнольдса-Нуссельта по теории подобия
неприменимы напрямую к расплавам полимеров и к переработке каучуков
по двум важным причинам. Во-первых, они основаны на поведении при
течении ньютоновских жидкостей и, во-вторых, они не учитывают вязкого
диссипативного тепловыделения.
Ранние попытки применения теории подобия к анализу изотермического
течения вязкоупругих жидкостей были сделаны Карлом Вайссенбергом в
период с 1928 по 1948 г. Вайссенберг [W3] предположил, что обратимая
эластическая деформация жидкости характеризует эластичность таким же
образом, как числа Рейнольдса характеризуют инерцию. Большие обрати-
мые деформации характеризуют значительные вязкоупругие силы. Рейнер
[R4] позднее отметил, что основная безразмерная величина, характеризую-
щая проявления вязкоупругости есть «число Деборы», которое может быть
представлено как отношение характеристического времени релаксации Tch
к времени ожидания t. Большое значение t приданном xch или малое значение /
могут, согласно Рейнеру, определять вязкоупругий отклик. Более количе-
ственное выражение интуитивной идеи Вайссенберга и Рейнера появилось
тогда, когда была создана современная теория нелинейной вязкоупругости.
Уайт [W6], Уайт и Токита [W26], затем Мейсснер и др. [М23] опубликовали
работы, которые позволили ввести эти идеи в рамки формального рассмот-
рения. Этот подход должен быть применим к эластомерам и, в значительной
мере, к чистым каучукам, но не к резиновым смесям.
Начнем с закона движения Коши, ур. (10) и с общего реологического
уравнения состояния (47), из которых получим
(101)
Характеристическая скорость Uи длина L вводятся в это уравнение, и все выра-
жение переводится в безразмерную форму. Эти безразмерные величины суть:
310 -*и* Глава 6. Реологические свойства и переработка...
где тт максимальное время релаксации (смур. (14)) a Gm соответствующий
модуль. Здесь LUp/GmTrn число Рейнольдса, означающее соотношение
инерциальных и вязких сил; U2 / gL есть число Фруда, означающее соот-
ношение инерциальных и гравитационных сил; TmU/L известно как число
Вайссенберга, а тт /t есть число Деборы, оба выражают соотношение вязкоуп-
ругих и вязких сил. Остальные величины являются численными параметрами
вязкоупругости, определяющими детальный характер функционала.
Как правило, инерциальные силы незначительны в расплавах полимеров
и в эластомерах, также как гравитационные силы и поэтому их можно не
учитывать. После этого остаются числа Вайссенберга, Деборы и отношения
параметров вязкоупругости. Если рассмотреть их применение к процессам
переработки, при которых материал течет через какое-то оборудование, то t
должно быть определено как время нахождения в оборудовании и
tm/t = [tm/(L11/L7)] = tmL7/L11 (103)
Здесь является характеристической длиной в направлении потока.
Число Деборы оказывается эквивалентным числу Вайссенберга с учетом
характеристической длины в направлении потока. Таким образом, наши
безразмерные величины сводятся к
*mU/L, \/хт, Gl/Gm,/Gj (104)
Предыдущий параграф приводит к некоторым важным заключениям.
Во-первых, характеристические скорость и длина появляются только в одной
безразмерной величине, и виде отношения U/L. Это означает, что для любой
конкретной вязкоупругой жидкости должно быть динамическое подобие
между большими и малыми геометрически подобными изотермическими
системами только если скорости отмасштабированы по длине (scaled with
length). Более того, если в такой системе возникают нестабильности или измене-
ния в режиме, они должны происходить при критическом значении безразмер-
ного параметра вида тш, где характеристическое время дается выражением
т = тт/*’[т| /тт, , ау] (105)
Здесь функция Сможет быть довольно сложной, так что т может отличаться
заметно от материала к материалу (с разными численными параметрами
вязкоупругости) даже если они обладают одинаковыми тт.
2. Резиновые смеси
Теперь мы обратимся к резиновым смесям и более конкретно
к пластично-вязким и пластично-вязкоупругим жидкостям.
Теория подобия в применении к пластичным жидкостям, обладающим
пределом текучести, впервые была развита Олдройдом [04] и впослед-
VIII. Инженерный анализ переработки -•и* 311
ствии Прагером [Р15]. Олдройд предложил новую безразмерную величину
в виде:
YL
x\U
(106)
Эта величина была впоследствии названа числом Бингама.
Для пластично-вязкоупругих жидкостей безразмерные величины урав-
нения (102) должны быть заменены на
YL U X; G:
------ Т — __— _— п
xmGmU’ т L’ хт’ Gm’ J
(107)
Как и в случае вязкоупругих жидкостей в ур. (100) и пластично- вяз-
ких жидкостей в ур. (106), динамическая идентичность включает системы
с одинаковым U/L. Закономерности течения как вязкоупругих, так и
пластичных жидкостей объединяются безразмерными величинами со-
держащими U/L.
Следует уделить внимание и тиксотропным эффектам. Предыдущая
формулировка динамической идентичности должна основываться на экви-
валентных периодах деформации.
3. Неизотермические эффекты
Рассмотрение в предыдущих параграфах ограничено изо-
термическими системами, с которыми мы, как правило, не встречаемся.
Следует, поэтому, проанализировать уравнение энергии [В21], которое
может быть записано как
pc ^+(v-V)T =/cV2r+ o:Vv
L dt
(108)
Вводя характеристические скорость Uи длину L и разность температур 0 мож-
но показать, что безразмерные величины в этом уравнении приобретают вид
LUpc/k, x\U2/kQ (109)
Также как и связанные с вязкоупругостью величины (102)
или параметры вязкоупругой пластичности в (107) соответственно. Здесь
LUpc /к известно как число Пекле и характеризует отношение конвектив-
ных тепловых потоков к теплопроводности, тогда как T\U2 //сО является
числом Бринкмана, отражающего отношение вязкостного разогрева к теп-
лопроводности [В21].
Число Бринкмана является величиной огромной важности в анализе
процессов переработки полимеров, поскольку он характеризует способность
системы отводить тепло, возникающее в результате вязкой диссипации.
312 -*V- Глава 6. Реологические свойства и переработка...
Полезно переписать его в виде
9 z \ z \ 2
^- = L2f—V-) (НО)
/с0 \k£))\L)
Если мы сравним малые и большие системы, характеризуемые постоян-
ной величиной U/L, как это следует ожидать из принципа динамического
подобия, число Бринкмана увеличивается пропорционально//2. Чем больше
система, тем больше тенденция вязкостного разогрева превосходить тепло-
проводность и увеличивать температуру. Другой способ охарактеризовать
ситуацию, это сказать, что большие системы стремятся к адиабатическому
состоянию. Поэтому применение теории подобия в изотермических условиях
больше подходит к системам с малыми размерами. Для больших систем мы
также должны учитывать вязкостную диссипацию.
Возникновение эффектов, описанных выше, может быть рассмотрено
и с другой точки зрения. Нагрев за счет вязкостной диссипации пропорцио-
нален объему системы L3, тогда как теплоотдача пропорциональна площади
поверхности, изменяющейся пропорционально L2. Когда мы переходим
к большим системам, площадь поверхности у единичного объема уменьшает-
ся, затрудняя теплопередачу и система становится более адиабатической.
Последний важный момент, который уже упоминался, но заслуживает
теперь более обстоятельного рассмотрения, это невозможность подобия
при обоих постоянных — числе Вайссенберга (или числе Бингама) и числе
Бринкмана т. е. при постоянных
tU/L, x\U2lk& (Hl)
Единственный способ, которым можно свести к одному масштабу, при
постоянном числе Бринкмана, это сохранять постоянной характеристичес-
кую скорость U в то время как L возрастает, что приведет к уменьшению
числа Вайссенберга. Весьма часто неизотермические эффекты доминируют,
и тогда следует главным образом обеспечивать равенство (110).
С. Моделирование закрытых смесителей
Попытки моделировать течение в закрытых смесителях
прослеживаются к началу 1950-х [В25, Р16], однако заметное продвижение
вперед не произошло вплоть до конца 1980-х когда появился ряд незави-
симых исследований на эту тему [СЮ, СИ, D2, D3, Н20, Н21, К6-8, W19,
Y4]. Два направления развались при этом: первое основывалось на теории
смазок [Н20, Н21, К6-8, W19], а второе — на полных уравнениях движения
и применении метода конечных элементов [СЮ, СИ, Y4].
В первом из этих направлений первые работы основывались на модели-
ровании в декартовых координатах [D2, Кб-8, W19] и напоминали теорию
работы подшипников. В последнее время в этом направлении были взяты за
313
VIII. Инженерный анализ переработки
основу выражения в цилиндрических координатах [D3, Н20-22] по существу
направленные на решение ряда уравнений
А др д ( Эи
° = + Пт- Г~Г
az дг\ аг
1 f*2 л
g, = —- rvdr
(112а)
(112b)
и интегрирующие поле скоростей, чтобы получить уравнения для потоков
qz и qQ определяемых выражениями
1 Г*2 л
q7 = тг- гъ.аг
4z
(113а)
(113b)
Потоки qz и qQ сбалансированы в соответствии с
(114)
Рис. 28. Определение составляющих
частей потоков FL, Fc, Flr, Flv в закрытом
смесителе.
которые затем решаются численно.
Оказывается необходимым решить вышеуказанные уравнения, как при
граничных условиях, обеспечивающих течение (1) между стенками камеры
и ротором, так и (2) в пространстве
между роторами, чтобы опреде-
лить граничные условия. Решение
ур. (ИЗ) дает поле давлений, ко-
торое может быть преобразовано
в поле потоков. Прежде всего,
было установлено, что оба ротора
работают как червячные насосы,
нагнетающие в противополож-
ных направлениях и создающие
циркуляционные потоки в камере
смешения. Ким и др. [К6-8, W19]
и Хью и Уайт [Н20, Н22] попы-
тались представить поле потоков,
которое соответственно отражает
составляющие потоки являющиеся
продольными вдоль роторов (FL)
и по окружности вокруг роторов (Fc), подающий поток между роторами
(Flr) и «рассеивающий» поток направленный поверх гребней ротора (Fd)
(рис. 28). Они рассчитали эти зависимости для разной геометрии роторов,
314 -*V Глава 6. Реологические свойства и переработка...
включая многие из тех, что описаны в патентной литературе. Типичные
результаты собраны в Табл. 3, где роторы Бэнбери с двумя ребрами сравни-
вались с четырехгребневыми роторами Лаша и Фрэя [L2], Тисона и Компера
[Т13], и Сато с соавт. [S1 ].
Таблица 3. Характеристики потоков в двух- и четырехреберных роторах
закрытых смесителей, взятые из патентной литературы
Изобретатель Компания Год Fl+F* F' Fd \QL/Ql\ ^circ (мин)
Бэнбери [В12] W&P Birmingham Iron Foundry 1916 0.321 0.674 0.005 0.31 2.7
Лаш и Фрэй [L2] Werner& Pfleiderer 1943 0.295 0.689 0.016 0.21 3.7
Тисон и Компер [Т13] Goodyear Tire& Rubber 1966 0.375 0.615 0.010 0.78 2.3
Сато с соавт [S1] Rubber Kobe Steel 1981 0.398 0.588 0.014 0.50 2.0
Роторы Сато в сравнении с роторами Бэнбери обеспечивали на 20%
более быструю циркуляцию и на 180% более быструю циркуляцию поверх
вершин гребней. Ким и Уайт [К10] моделировали потоки в смесителе с за-
цепляющимися роторами.
D. Моделирование экструзионного процесса
на шнеке
Моделирование потоков в одношнековом экструдере относит-
ся к 1920-м, причем первые работы относились главным образом к насосам
для смазочных масел. В 1950-х потоки в шнековых экструдерах стали пред-
метом интереса к течению во многих промышленных лабораториях. Первая
работа в этом направлении была в значительной степени повторением работ
по червячным насосам 1920-х годов.
Простейшая модель ньютоновской жидкости, основанная на гидродина-
мической теории смазок, начинается с решения уравнений
0 = -^- + п^- (И5а)
дхх 3^2
0 = _^ + n^S. (115в)
Эх3 3^2
где направление 1 вдоль шнека, направление 2 перпендикулярно поверхнос-
ти шнека, а 3 — перпендикулярно ребру шнека. Распределение скоростей
и г)3 для случая, когда начало координат закреплено на шнеке:
VIII. Инженерный анализ переработки
Л
315
(^2) = 7С^^СО5Ф
Н2 ( др ( х2 ( х2 У
2nljrJ [//J
ъ3 (x2) = -nDN sin ф
(116a)
(116b)
где Я глубина канала шнека, N— скорость шнека, D — диаметр шнека, а ф —
угол нарезки шнека. Значение др/дх3 было определено исходя из гранич-
ных условий при отсутствии утечки по гребню шнека:
гя / \
Jo B3(x2)dx2=0
(И7)
Объем подачи по шнеку
<? = WJo W2)^2
1 ™ х H*W др
= —nDNcos6------—
2 Y 12т| дхх
(118)
Примерно в 1960-х были сделаны попытки моделировать потоки в шне-
ковом экструдере, как неньютоновских жидкостей, так и жидкостей теку-
щих в неизотермическом режиме. Особенно заметно это было представлено
в работах Гриффита [G17] и Замодица с Пирсоном [Z2]. Они сформули-
ровали основные принципы модели смазки в декартовых координатах
с уравнениями движения вида:
°=<+^ Эть ох2 _ (119а)
0="^+^ d'Do Эх2 (119b)
и уравнением энергии (без учета конвекции тепла)
д2Т
0 = /с^у + п
3^2
2
Э1Ц
Эх2
(120)
Неньютоновская вязкость может быть записана в виде (как это следует
из теории Криминале-Эриксона-Филби [С20], ур. (50) и (51):
П = ^(Т)/2
(121а)
316 -*v- Глава 6. Реологические свойства и переработка...
где вязкость жидкости описывается степенным законом
ЭП1V
Эх )
(121b)
Гриффит [G17] и Замодиц с Пирсоном [Z2] одновременно решили уравнения
(119), (120) и (121) с целью получения характеристик шнека как червячного
насоса. Оказалось, что уменьшение экспоненты п в степенном уравнении, когда
жидкость становится «более неньютоновской» снижает производительность
насоса (рис. 29). Аналогично увеличение вязкости и увеличение температурной
зависимости вязкости путем увеличения В(Т) также снижает производитель-
ность насоса. Приведенные выше выражения могут быть усовершенствованы
добавлением члена /дхх в левую часть ур. (119а).
Рис. 29. Влияние неньютоновской вязкости на характеристики производительности
шнека.
Резиновые смеси имеют предел текучести, так что действительное их по-
ведение в шнековом экструдере должно быть более сложным по сравнению
с тем, как это показано выше. Если резиновая смесь обладает пластичностью,
тогда уравнения (119а) и (119в) должны быть заменены:
VIII. Инженерный анализ переработки -’V 317
др Эо12 дх} Эх12 (122а)
др Эст32 Эх3 Эх2 (122b)
или
др
П|2 — *^2
ох]
др „
°32 ~ х2 Ч +
</Х3
(122с)
(122d)
Рассмотрим случай, когда нет головки: градиент давления Эр/Ъхх равен
нулю, и напряжения сдвига одинаковы по всей глубине канала шнека. Когда
О12 превосходит У, начинается простое сдвиговое течение. Это в свою оче-
редь создаст градиент давления Эр/Эх3 благодаря движению ребер шнека
одновременно с возникновением циркуляционного потока. Если имеется
градиент давления Эр/Э^ тогда в соответствии с ур. (122с) напряжения
сдвига должны меняться в поперечном сечении канала, достигая максиму-
ма на поверхности рабочего цилиндра, и могут достичь низких значений
в средине поперечного сечения канала. Если эти напряжения окажутся ниже
предела текучести, образуется твердая пробка в средней части поперечного
сечения. Однако, как это следует из работ Мергеса и Ленена [М19, L8] и Брзо-
сковского [В36, К17], возникает лишь циркуляционное течение, а пробка
не обнаружена. Это объясняется низким значением предела текучести.
Течение в экструдере с оштифтованным цлиндром (pin barrel extruder)
недавно было моделировано Брзосковским и др. [В31, В32, В34] на основе
гидродинамической теории смазок примененной для ньютоновской жидко-
сти. В качестве основы они поместили координатную систему в канал шнека
и установили баланс потоков в виде (эквивалентном ур. (114)):
dtp | Эд3 Q
Эл?! Эх3
где потоки записаны в виде
г» / X , „ н3 др
я, = и (х2 )dx2 = —U2H------------—
1 J0 ,v 21 2 2 2 12г)Э^,
[H , А , 1 rr „ Н3 др
= п3 \х? )ах2 = —IkH----------
•/з Jo 3V 2) 2 2 3 12пЭХз
(123)
(124a)
(124в)
318 -'V Глава 6. Реологические свойства и переработка...
(dP!dz)=\S
0=0.13
(dP/dz) = 1.4
0=0.28
Рис. 30. Картина потока в экструдере с оштифтованным цилиндром.
Уравнение (121) решается численно методом конечных разностей. Рису-
нок 30 показывает компьютерную картину потоков вокруг штифтов.
Кумазава и др. [К22] рассчитали температурные поля вокруг шнека,
введя тепловой баланс
pcq^ = -[Us(T-Ts) + ub(T-Tb)] + (ci2)b^NCoS^
+ (п32 )b itDN sin ф -
dp
dxx
(125)
VIII. Инженерный анализ переработки -*\г 319
где Т есть усреднение температуры по скоростям в объеме («сир mixing»
velocity averaged) температуры Ts и Ть это температуры шнека и стенки ци-
линдра, a Us и Ub коэффициенты теплопроводности шнека и цилиндра.
Возможно провести моделирование полных полей скоростей и температур
в поперечном сечении канала экструдера, не ограничивая расчет средни-
ми величинами потоков. Так можно обобщить уравнения (119а), (119b)
и (120) в виде
Эр Э Эъ^
-^- + Т|—1
Эх1 Эх2 L Эх2
Э Эъ1
(126а)
. dp Э d'Do
иХд dx2 do2
(126b)
(126с)
Это позволяет оценить роль гребней шнека и штифтов, но эта проблема
гораздо более сложно решаемая.
Е. Моделирование экструзионного процесса
в головке
Попытки моделировать течение расплавов полимеров
и резиновых смесей в головках связаны с развитием вискозиметрии вообще.
Первые значительные исследования моделирования потока в головках были
проведены Пирсоном [Р2-5], который применил ту же теорию смазок, что
описана в двух предыдущих разделах, т. е. в виде уравнений (123а), (123b),
(124а) и (124b).
Рассмотрим следующий простой анализ, примененный к Т-образным
головкам и головкам типа «ласточкин хвост». В головке существует толь-
ко поток вызванный давлением. Для ньютоновской жидкости ур. (115а)
и (115в) можно найти простое решение (если система координат помещена
в центр канала головки)
Н2( Эр "j
2т| дх3 J
(127b)
320 ^\r Глава 6. Реологические свойства и переработка...
Это определяет выражения для потоков
= 2j0 v, {x2)dx2 = (128а)
9з = 2 £Я/2Чз (*2 )^2 = тУ “TN (128в)
1 ZT| у См7д )
Потоки могут менять положение, в связи с чем qx и должны быть сба-
лансированы согласно ур. (123). Это приводит к выражению
’ + ’ =о
у J у J
(129)
определяющему поле давлений в головке и поле потоков полученных из
ур.(128).
Определение геометрии головки эквивалентно определению Н (дц, z3). Это
позволяет произвести компьютерный расчету? (хр х3) и qj (xv х%) для разных
конструкций головок. Общая проблема состоит в том, чтобы определить,
что функция Н (хр х3) делает q^L, х3) постоянным, т. е. поток на выходе из
головки независим от его положения вдоль стенок.
Вышеназванные проблемы необходимо сформулировать в более общем
виде, используя ур. (119а) и (119в) и учитывая также неньютоновский ха-
рактер течения согласно ур. (121а). Потоки q^ и #3 зависят от Н и градиентов
давления в ином виде в случае неньютоновской жидкости. В этом случае
ур. (123) оказывается применимым, а ур. (124) следует заменить на
у ) ОД73 \ ^*^3 /
(130)
где Sравно
ггЗ
5 = (131)
12т|
и где Г| есть эффективная (неньютоновская) вязкость.
Заметим, что резиновые смеси являются пластическими материалами
и что напряжения имеют низкие значения в центре поперечного сечения.
Когда напряжения снижаются до уровня ниже предела текучести У, градиент
скорости падает до нуля и в центре образуется твердая пробка.
Можно также получить информацию о температурных изменениях.
В первом приближении, если рассматривается адиабатический процесс, ло-
гика приводит к ур. (125) позволяющем предполагать, что рост температуры
в потоке идущем через головку есть
VIII. Инженерный анализ переработки 321
Т-То= — (132)
рс
где Т есть усредненная температура («сир mixing» temperature).
В более общем виде необходимо решить ур. (130) совместно с
pcQi^ = -C/(r-7;n)+91^- (133)
dx} dx{
при этом полагая, что вязкость не зависит от температуры. Шах и Пирсон
[S10] рассмотрели этот тип проблем и показали, что нестабильности могут
привести к тому, что qx станет зависеть от z3.
Профили скоростей и температур могут быть определены в поперечном
сечении с помощью ур. (126), хотя это снова трудная задача.
F. Моделирование заполнения формы
Единственная имеющаяся литература по формованию кау-
чука — это заполнение формы при литье, т. е. заполнение формы данной
геометрии через литник. Подход к моделированию похож на тот, что рассмат-
ривался нами в применении к головкам в предыдущем разделе. Заполнение
формы изотермической ньютоновской жидкостью математически может
быть описано ур. (129), а в случае неизотермического заполнения ненью-
тоновской жидкостью с дополнительным привлечением ур. (130). Такой
процесс был моделирован Исаевым с сотр. [12-4, 16, 115, 116]. Решение
в принципе базировалось на литье под давлением термопластов за исключе-
нием необходимости учета кинетики вулканизации в процессе заполнения
формы. Тепловое моделирование включало учет теплопередачи в поперечном
направлении (потолщине).
ГЛАВА ВУЛКАНИЗАЦИЯ
7
ОБЕРТ ДЖ. КОРАН
Коран Консалтинг,
Лонгбоат Кей, Флорида, США
I. ВВЕДЕНИЕ
Наиболее часто используемые резиновые изделия, такие как
шины и РТИ, не могут быть произведены без вулканизации. Невулканизо-
ванный каучук обычно менее прочен, не сохраняет форму после больших
деформаций и может быть очень липким. Иными словами, невулканизован-
ный каучук имеет ту же консистенцию, что и жевательная резинка.
Первый производственный метод вулканизации был реализован Чар-
льзом Гудьиром. Его процесс (нагревание натурального каучука с серой)
был впервые опробован в Сприндфильде, шт. Масса чузетс, в 1841г.1) Томас
Гэнкок применил по-существу тот же процесс годом позже в Англии. С тех
далеких дней продолжался прогресс в усовершенствовании этого метода
и получения изделий из вулканизованного каучука. Годы спустя натураль-
ный каучук дополнился многими синтетическими каучуками. То же самое
касается серы, вместе с которой были использованы многие вещества в каче-
стве дополнительных вулканизующих систем. Эта глава представляет собой
общий обзор науки и технологии вулканизации. При этом упор делается
на вулканизацию «высокодиеновых» каучуков общего назначения (нату-
ральный каучук [НК], бутадиенстирольный каучук [БСК] и бутадиеновый
каучук [ПБ]) серой в присутствии органических ускорителей.
Ускоренная серная вулканизация этих каучуков наряду с другими каучу-
ками, вулканизуемыми по очень близкой технологии (этиленпропиленовый
каучук [СКЭПТ], бутилкаучук [БК], галогенбутилкаучуки, нитрильный
каучук [БНК]) занимает более 90% всей вулканизации. Рассмотрены
также некоторые аспекты вулканизации другими вулканизующими
Примечание переводчика: Автор ошибается, т.к. по более точным данным это событие
имело место в 1839 г.
Глава адаптирована из статьи «Вулканизация» А. Дж. Корана в «Энциклопедии полимерной
науки и техники», второе изд., том 17, 1989. John Wiley & Sons, Inc. Раздел «Динамическая
вулканизация», 1994, Ac. Press, Inc.
II. Определение вулканизации -*v- 323
агентами, такими как органические пероксиды, фенольные и хиноидные
производные.
Кроме того, рассмотрена также динамическая вулканизация (ДВ). ДВ
представляет собой сшивание одного полимера в смеси полимеров в са-
мом процессе смешения, когда эти полимеры находятся в расплавленном
состоянии. Этот процесс используется при получении композиций типа
термоэластопластов из смесей каучуков с пластиками.
II. ОПРЕДЕЛЕНИЕ ВУЛКАНИЗАЦИИ
Вулканизация — это процесс, относящийся к каучукоподобным
или эластомерным материалам, которые характеризуются быстрым возвраще-
нием к исходной форме после механической деформации. Вулканизация уве-
личивает усилие, необходимое для растяжения образца и уменьшает величину
деформации, остающейся после удаления деформирующей силы. Таким
образом, вулканизация увеличивает эластичность и снижает пластичность.
Она сопровождается образованием поперечно сшитой молекулярной сетки
из исходных линейных молекул каучука (рис. 1).
Рис. 1. Образование сетки.
Согласно теории каучукоподобной эластичности [1], сила, сопротив-
ляющаяся деформации, пропорциональна числу узлов сетки, удержи-
вающих полимерные цепи (в единице объема эластомера). Полимерной
324 -•V Глава 7. Вулканизация
цепью называется отрезок линейной цепи полимера между двумя узлами
сетки. Увеличение числа узлов или поперечных связей (сшивок) ведет
к увеличению числа таких отрезков цепей. В невулканизованном линейном
высокомолекулярном полимере (выше его температуры плавления) узлы
образуются только зацеплениями цепных молекул.
Вулканизация, таким образом., представляет собой процесс образования
химических узлов в сетке путем создания поперечных связей между поли-
мерными цепями. Поперечной связью может быть несколько атомов серы
в виде короткой цепи, один атом серы, углерод-углеродная связь, полива-
лентный органический радикал, ионный кластер или поливалентный ион
металла. Процесс обычно осуществляется нагреванием каучука, смешанного
с вулканизующими агентами, в форме под давлением.
III. ВЛИЯНИЕ ВУЛКАНИЗАЦИИ НА СВОЙСТВА
ВУЛКАНИЗАТОВ
При вулканизации происходят большие изменения на моле-
кулярном уровне. Длинные молекулы каучука (молекулярный вес обычно
в пределах 100-500 тысяч дальтон) связываются друге другом узлами (попе-
речными связями), расположенными вдоль полимерных цепей при среднем
расстоянии между узлами, соответствующему значению молекулярного
веса между поперечными связями от 4 до 10 тысяч дальтон. В результате
образования сетки каучук становится нерастворимым в растворителях
и его нельзя перерабатывать способами, которые основаны на текучести,
т. е. в смесителе, в экструдере, на вальцах, на каландре или в процессах
формования или литья. Т. о., очень важно, чтобы вулканизация проходила
только, когда резиновое изделие приобретает его конечную форму.
Основное влияние вулканизации [2-4] на механические свойства показаны
схематично на рис. 2. Следует отметить, что статический модуль возрастает при
вулканизации в большей степени, чем динамический. (Здесь понятие статиче-
ского модуля более корректно заменить равновесным модулем при медленном
растяжении, т. е. модулем при малой скорости растяжения. Динамический
модуль обычно измеряется при воздействии малого растяжения по синусои-
дальному закону при частотах от 1 до 100 Гц.) Динамический модуль включает
вязкую и упругую составляющие, тогда как статический модуль в основном
является мерой только упругой компоненты реологических параметров.
Гистерезис снижается с увеличением числа поперечных связей. Гистере-
зисом называется отношение зависимой от скорости, или вязкой, составля-
ющей к упругой компоненте деформации. Он является также мерой энергии
деформации, которая не накапливается в эластической сетке, а превраща-
ется в тепловую энергию. Вулканизация, таким образом, восстанавливает
эластичность взамен вязкого или пластического поведения. Значения этих
свойств возрастают при небольшом количестве поперечных связей, но затем
III. Влияние вулканизации на свойства вулканизаторов -*и- 325
они снижаются при дальнейшем образовании сетки. Сопротивление раздиру,
усталостная выносливость и прочность относятся к разрушающим видам
энергии. Значения этих свойств возрастают уже при малом содержании по-
перечных связей, но снижаются при их увеличении. Свойства, относящиеся
к энергии разрыва, возрастают с увеличением как числа цепей сетки, так и
гистерезиса. Поскольку гистерезис уменьшается с увеличением числа цепей
сетки, то свойства, связанные с энергией разрушения, достигают максимума
при некоторой промежуточной плотности сетки.
Свойства
вулканизата
Рис. 2. Свойства вулканизатов в зависимости от степени вулканизации.
Следует отметить, что свойства представленные на рис. 2, не являются
функцией только плотности поперечных связей. На них также влияет тип
поперечных связей, природа полимера, вид и количество наполнителя и т.д.
Реверсия. Термин реверсия в широком смысле относят к разрушению
структуры сетки при термическом старении в отсутствии кислорода. Его
обычно применяют при вулканизации изопреновых каучуков серой. Ре-
версия может быть связана со слишком большим временем вулканизации
(перевулканизация) или тепловым старением толстых образцов. Наиболее
заметна она при температурах свыше 155 °C. Реверсия имеет место у вул-
канизатов, содержащих большое количество полисульфидных поперечных
связей. Механизм реверсии сложный и достаточно хорошие представления
о химических изменениях, которые происходят в процессе реверсии нату-
рального каучука, представлены в [5].
Иногда термин «реверсия» применяют к другим типам не окислительной
деструкции, особенно в отношении резин не из изопренового каучука. На-
326 -*v- Глава 7. Вулканизация
пример, термическое старение БСК (бутадиен-стирольный каучук), которое
приводит к увеличению плотности сетки и жесткости, называется реверсией,
т.к. оно может быть результатом перевулканизации.
Переработка, тепловая обработка
Рис. 3. Влияние переработки на изменение тепловых параметров.
Степень
вулканизации
Рис. 4. Влияние тепловых процессов (переработка) на сопротивление преждевре-
менной вулканизации.
IV.
ХАРАКТЕРИСТИКА ПРОЦЕССА ВУЛКАНИЗАЦИИ
Важные характеристики процесса вулканизации зависят
от времени до начала сшивания, скорости образования поперечных связей
и степени сшивания в конце процесса. Должно быть достаточное ингибиро-
IV. Характеристика процесса вулканизации -*v- 327
вание или сопротивление преждевременной вулканизации для того, чтобы
обеспечить смешение, формование, течение и прессование до вулканизации.
После этого образование поперечных связей должно происходить быстро
и при контролируемой степени сшивания (рис. 3 и 4).
Сопротивление преждевременной вулканизации (илискорчингу) обычно
измеряется временем при данной температуре, которая требуется для начала
образования поперечных связей. Это оценивается по резкому увеличению
вязкости. Обычно используется вискозиметр Муни [4]. В течение этого
испытания полностью смешанная, но невулканизованная резиновая ком-
позиция, содержится в нагреваемой камере. В эту смесь погружен враща-
ющийся диск. Вязкость непрерывно измеряется как функция времени (по
сдвиговому усилию, которое необходимо для поддержания вращающегося
диска при постоянной скорости). Температура выбирается такой, чтобы
охарактеризовать более жесткие условия переработки (экструзия, калан-
дрование и т.д.).
Как скорость вулканизации после окончания периода скорчинга, так и
окончательная степень вулканизации измеряются устройством, который
называется кьюрометр (вулкаметр). Многие исследователи работали над
его совершенствованием [6]. Наиболее распространенными кьюрометрами
являются реометры с колеблющимся диском типа разработанного компанией
Монсанто около 1965 г. Совершенствование этих реометров с колеблющимся
диском, главным образом, благодаря усилиям Р.В. Вайз, явилось началом
современной вискозиметрии, кото-
рая стала стандартной методикой
в промышленности. До внедрения
вулкаметров нужно было измерять
механические свойства образ-
цов резин, каждый из которых
вулканизовался в течение опре-
деленного времени при данной
температуре.
Для измерения вулканиза-
ционных характеристик образец
помещается в закрытую камеру
(рис. 5).
Погруженный в резиновую
смесь металлический диск осу- Рис. 5. Реометр с колеблющимся диском,
ществляет колебательные сину-
соидальные движения в плоскости, перпендикулярной его оси. Вулка-
низация измеряется по увеличению крутящего момента, необходимого
для поддержания заданной амплитуды колебания (т. е. угла деформации)
при данной температуре. Крутящий момент пропорционален модулю
эластичности при малой деформации. Поскольку этот крутящий момент
Камера
328 “'V Глава 7. Вулканизация
измеряется при высокой температуре вулканизации, то доля в нем вяз-
костной составляющей минимальна. Таким образом, можно принять, что
увеличение крутящего момента в процессе вулканизации пропорциональ-
но числу поперечных связей в единице объема вулканизата. Крутящий
момент автоматически записывается во времени, давая так называемую
реометрическую кривую, реограмму, или кривую вулканизации.
Системы измерения
крутящего момента
Испытуемый
образец v
Уплотнитель
Смыкающиеся
плиты
Нижняя полуформа
(колеблющаяся)
Верхняя полуформа
(стационарная)
Система колебательных
движений
Рис. 6. Реометр с движущейся формой
В настоящее время внедряются новейшие образцы вулкаметров, схема
одного из которых представлена на рис. 6. Камера значительно меньше
и отсутствует ротор. В этом типе вулкаметра половина формы (верхняя часть)
стационарна, а другая половина колеблется. Эти приборы получили название
реометров с подвижной формой. Образец стал значительно меньше, и тепло-
передача происходит быстрее. Поскольку здесь нет ротора, то температура
V. Вулканизация серой без ускорителей -*и- 329
в полости камеры и образца может меняться быстрее. В каждом случае (ко-
леблющийся диск или подвижная форма) крутящий момент автоматически
откладывается от времени. Такая зависимость показана на рис. 7.
Вулканизационная кривая представляет достаточно полную картину
кинетики образования поперечных связей и даже исчезновения поперечных
связей (реверсия) для данной резиновой смеси. В некоторых случаях вместо
реверсии может быть зафиксировано продолжительное плато или увеличе-
ние степени вулканизации. Поэтому вулкаметр широко используется для
контроля качества и однородности резиновых смесей (называемых также
резиновыми композициями).
Рис. 7. Геометрическая вулканизационная кривая.
Вулкаметрия сначала использовалась как исследовательский прибор
для изучения вулканизации. А затем, его стали применять для контроля
однородности резиносмешения на заводах. Приборы с программируемым
температурным режимом для вулкаметрии используются для улучшения
рецептуростроения в промышленности. Зависимость температура-время
вулканизации в промышленных условиях может быть сопоставлена с усло-
виями вулканизации в полостях кьюрометра. Образец для испытания может
быть свулканизован в вулкаметре при тех же условиях, которые применя-
ются на заводе. Как степень вулканизации, так и температура могут быть
одновременно описаны как функции времени.
V.
ВУЛКАНИЗАЦИЯ СЕРОЙ БЕЗ УСКОРИТЕЛЕЙ
Изначально вулканизация проводилась при использовании
элементной серы в количестве 8 масс. ч. на 100 масс. ч. каучука (м. ч.). На
это требовалось 5 часов при 140°С. Дополнительное введение оксида цинка
330 -Jv- Глава 7. Вулканизация
снижало это время до 3 час. Использование ускорителей при таких малых
концентрациях, как 0,5 м. ч. сразу снизило время до таких величин как
1-3 мин. Таким образом, вулканизация эластомеров серой без ускорителей
не представляет существенного практического значения. (Как исключение
в производстве формованных продуктов из жесткой резины, или эбонита,
используют 30 или более м. ч. серы с малым количеством ускорителя или
без него.) Несмотря на то, что неускоренная серная вулканизация не пред-
ставляет практического интереса, ее химизм явился предметом большого
количества исследований и изучения.
Химия неускоренной вулканизации противоречива. Очень много мед-
ленных реакций протекает в течение длительного периода вулканизации.
Некоторые исследователи предполагают свободно-радикальный механизм
(Схема!) [7-9].
СНз
~сн—с=сн~
I
н
СНз
•sx- • I
~СН—с=сн~ + HSX-
Каучук
СНз
~сн—с=сн~
СН:
~сн—с=сн~
Поперечная связь S, СН3
СН—С^ + ~*сн—с=сн
V. Вулканизация серой без ускорителей 331
Другие исследователи предлагают ионный механизм (Схема 2) [10]:
R—Sx—Sy—R -------► RS^ + RSy
сн3
—CH2—C=CH—CH2~~
CH3
—CH2 —C— CH—CH2~~
\ <fH3
\~CH2—C=CH—CH2“
\ Перенос H~
—CH2 —C—CH2—CH2—
Sx CH3
R + —CH2 —C— CH—CH —
CH3 CH3
~CH2-C-CH=CH- + -CH2-C-CH-CH-
(RSX+) (RSX+)
Промежуточные продукты
сн3 сн3
—СН2-С-СН-СН2™ and —СН2-С-СН-СН2™
\+/ +.....................
Y
sx
R
были предложены в схеме 2 для объяснения того факта, что модельные
продукты реакции образуют как ненасыщенные, так и насыщенные соеди-
нения, причем атомы серы связаны с вторичными и третичными атомами
углерода.
332 Глава 7. Вулканизация
VI. ВУЛКАНИЗАЦИЯ СЕРОЙ С УСКОРИТЕЛЯМИ
Органические ускорители не применялись до 1906 г. (более
65 лет после открытия Гудьиром и Генкоком неускоренной вулканизации
(рис. 8)), когда Оенслагером было открыто действие анилина на серную
вулканизацию [И]. Это произошло, по крайней мере, частично вследствие
изобретения пневматических шин и автомобилей на рубеже столетий.
Однако анилин слишком токсичен для использования в резиновых ком-
позициях. Менее токсичный продукт его реакции с дисульфидом углерода,
тиокарбанилид, был предложен в качестве ускорителя в 1907 г. Дальней-
шие исследования привели к гуанидиновым ускорителям [12]. Продукты
реакции сероуглерода и алифатических аминов (дитиокарбаматы) были
впервые использованы как ускорители в 1919 г. [13]. Они были и остаются
до сих пор наиболее активными ускорителями, как по скорости сшивания,
так и по степени образования поперечных связей. Однако, большинство
дитиокарбаматных ускорителей характеризуются отсутствием, либо малым
сопротивлением подвулканизации, и поэтому во многих производственных
процессах их использование не возможно. Первые ускорители замедленного
действия были предложены в 1925 г. на основе 2-меркаптобензтиазола (МВТ)
и 2-бензотиазолдисульфида (или2-2'-дитиобисбензотиазол) (ДБТД) [14-16].
Это по времени совпадает с внедрением конструкций шин с кордными слоями
(1920-1930), что способствовало массовому производству автомобильных шин.
Еще большее замедляющее действие и последующую быструю вулканизацию
обеспечило в 1937 г. внедрение первого промышленного бензотиозолсульфе-
намидного ускорителя [17-18]. Большее замедление реализовано в 1968 г.
с появлением чрезвычайно эффективного ингибитора подвулканизации (PVI).
Это соединение называется N-циклогексилтиофтальимид (ЦТФ), малые кон-
центрации которого были использованы с бензотиозолсульфенамидными уско-
рителями. Вся история развития быстрой вулканизации, но с лучшим контролем
подвулканизации, или скорчинга, иллюстрируется рисунками 8, 9,10.
Серно-ускорительная вулканизации наиболее широко распространен-
ный метод. Для многих сфер применения это единственный быстрый способ
сшивания, который, в частности, может обеспечить замедленное действие
при переработке, формовании, прежде, чем образуется устойчивая вулкани-
зационная сетка. Он используется для вулканизации натурального каучука
(НК), синтетического изопренового каучука (СКИ), бутадиенстирольного
каучука (БСК), нитрильного каучука (БНК), бутилкаучука (БК), хлорбу-
тилкаучука (ХБК), бромбутилкаучука (ББК) и этиленпропилендиеного
каучука (СКЭПТ). Реакционноспособный участок у всех этих эластомеров
может быть представлен как
Н
I
>с—с=сн—
VI. Вулканизация серой с ускорителями -*и- 333
Типичный состав вулканизующих систем для одного из вышеуказанных
эластомеров содержит 2-10 м. ч. оксида цинка, 1-4 м. ч. жирной кислоты
(например, стеариновой), 0,5-4 м. ч. серы и 0,5-2 м. ч. ускорителя. Оксид
цинка и жирная кислота в вулканизующей системе являются активаторами.
Жирная кислота с оксидом цинка образует соль, которая создает комплекс
с ускорителями и продуктами реакции между ускорителями и серой (уско-
рители классифицированы и представлены в Таблице 1).
Год
1850-
— Серная вулканизация
Вулканизующий
—Г агент
Вулканизация
без ускорителей
1900-
Анилин
“ Тиокарбанилид
Вулканизация
с ускорителями
Дитиокарбаматы
Дифенилгуанидин
2-Меркаптобензотиазол
\ 2-Бензотиазолилдисульфид
2-Алкиламинотиобензотиазолы
Органические
ускорители
1950-
Вулканизация
(сульфенамиды)
с ускорителями
с индукционным
периодом
— N -циклогексилтиофталимид
Ингибитор
преждевременной
вулканизации
Рис. 8. История серной вулканизации.
Часто применяются смеси ускорителей. Широко применяется ускоритель
бензотиазольного типа с небольшим количеством дитиокарбамата (тиурама)
или аминным ускорителем. Эффективность действия смеси ускорителей
двух разных типов может состоять в том, что каждый активирует другой,
и получается более чем ожидаемая скорость сшивания. А смешение ускори-
телей одного и того же типа приводит к промежуточным или усредненным
результатам.
334 -*Ь- Глава 7. Вулканизация
Таблица 1. Ускорители серной вулканизации.
Соединения Сокращения Структура
Бензотиазолы
2- Меркаптобензотиазол МВТ (OJL с—sh
Дибензотиазолилдисульфид ДБТД я П 1 сл 1 СЛ 1 $
Бензотиазолсульфенамиды
N-циклогексилбензотиазол- 2-сульфенамид САЦ И Z 1 и 1
Ь1-трет-бутилбензотиазол-2- сульфенамид CAT Y Г3 Ю1 C-S-N-C-CH3 сн3
2 - морфолилтиобензотиазол САМ COjT /c“s“4 р
N- дициклогексилбензотиазол- 2-сульфенамид ДЦБС я n ' 1 и бЪ
Дитиокарбаматы
Тетраметилтиураммоносульфид тмтм СНЗХ i I /СН3 /N—С—S—С—N' СНЭ СНз
Тетраметилтиурамдисульфид тмтд CHj^ i i /СНз /N—с—s—s— с—NC' CHj CHj
Диэтилдитиокарбамат цинка ZnflTK (сгн5^ Й \ к
Амины
Дифенилгуанидин дфг /А\ /F?\ <О/ NH—С—NH-/02
Ди-о-толилгуанидин Д-о-ТГ ^-NH-C-NH-^ ^"^СНз at;
VI. Вулканизация серой с ускорителями -*!/• 335
Рис. 9. Химия синтеза ускорителей.
Здесь следует отметить, что существует настоятельная необходимость
снижения применения ускорителей на основе вторичных аминов, которые
могут реагировать с оксидами азота с образованием нитрозоаминов, кото-
рые предположительно являются канцерогенными. Эта проблема особенно
актуальна для ускорителей дитиокарбаматного типа. Предлагаемые уско-
рители, которые не дают канцерогенных нитрозоамидных производных,
включают дибензиламинные производные дитиокарбаматов и стерически
затрудненные амины.
336
Глава 7. Вулканизация
Различные типы ускорителей характеризуются вулканизационными
параметрами, которые различаются как сопротивлением подвулканизации,
так и скоростью сшивания. На рис. 11 представлена картина характеристик
ускорительных систем. В пределах групп или типов могут существовать
различия при выборе индивидуальных ускорителей. В группе бензотиа-
золсульфенамидов сопротивление подвулканизации и время вулканизации
возрастают в ряду: CAT или САЦ, САМ, ДБЦС.
Время, мин. при 140°С
Рис. 10. Совершенствования в развитии серной вулканизации натурального каучука.
Влияние малых количеств ингибиторов преждевременной вулканизации
(PVI), (N-циклогексилтиофтальимид) также представлено на рис. И. Этот
ингибитор [19] часто используется, чтобы контролировать сопротивление
преждевременной вулканизации при отсутствии или малом влиянии на
скорость сшивания [4]. До внедрения N-циклогексилтиофтальимида как
PVI использовались замедлители кислотной природы, напр. салициловая
кислота, ацетилсалициловая кислота, фталевый ангидрид и бензойная
кислота. Эти добавки улучшили сопротивление преждевременной вулкани-
зации, но также сильно снижали скорость образования поперечных связей
после периода индукции. Другой замедлитель подвулканизации в прошлом
N-нитрозодифениламин менее активен и в настоящее время не используется
по токсикологическим соображениям.
А. Химия серно-ускорительной вулканизации
Основное направление реакций серно-ускорительной вул-
канизации следующее [4,20-24]: ускоритель реагирует с серой с образо-
ванием мономерных полисульфидов следующей структуры Уск.-8-Уск.,
VI. Вулканизация серой с ускорителями 337
где Уск. — это органический радикал ускорителя (напр., бензотиазил-).
Мономерные полисульфиды реагируют с каучуком с образованием полисуль-
фидов, напр., каучук-Эх-Уск. В этой реакции образуется 2-меркаптобенз-
тиазол (МВТ) если ускоритель является производным бензотиазола и если
эластомер — натуральный каучук. (В БСК МВТ связывается с эластомерной
молекулярной цепью вероятно, как тиоэфир, каучук-Э-Уск.) Если МВТ
является ускорителем в натуральном каучуке, то он сначала исчезает, а затем
появляется в виде BT-S-Sx-S-BT и каучук-Эх-Уск. В итоге каучуковые
полисульфиды реагируют либо напрямую, либо через промежуточные соеди-
нения с образованием поперечных связей типа каучук-Эх-каучук. Можно
предложить следующую схему реакции (см. Схема 1).
[инициатор]
(-R2NH)
| мвт
Таким образом, очевидны различия между вулканизацией с ускорителями
и без ускорителей. (Большая эффективность образования поперечных связей
и большая скорость сшивания получаются при вулканизации с ускорителями).
Но очевидны также другие отличия. Результаты, полученные на основе модель-
ных реакций с вулканизующими компонентами, показывают, что сера присо-
единяется к каучуковому углеводороду практически полностью по аллильным
участкам [25]. Этого нет в случае серной вулканизации без ускорителя, т. о.:
Глава 7. Вулканизация
338
Рис. И. Вулканизационные характеристики различных ускорителей и их ком-
бинаций.
(BTSSSxYck.)
Вероятнее, что вместо промежуточного восьмичленного цикла, показанного
выше, можно предположить промежуточное соединение при сульфуризации
через шестичленный цикл:
VI. Вулканизация серой с ускорителями -*и- 339
(каучук)
(BTSS 8хУск.)
Эго подобно тому, что предложено в [26] относительно действия дитиокар-
баматного ускорителя.
Другие авторы предполагают, что цинк может участвовать в сульфу-
ризации [27]. Во всяком случае, поперечные связи образуются по ряду
направлений, например:
Используя метод ЯМР С-13 твердофазной спектроскопии Koenig и др.
[28] получили много данных по химической структуре серосодержащих
боковых групп в сетке. Следующая схема показывает возможные типы
структур, которые могут быть предложены для мест присоединения атомов
серы каучуковыми молекулами:
340 -*V- Глава 7. Вулканизация
СН3
С =сн
- сн/ \н-
I
Sx
СН3
С =сн
-СНХ \н2-
I
Sx
I
Ale
Bic
СНз уСН2 —
с = снх
-снх
I
Sx
I
Bit
СН3
I
— СН2—с — сн=сн —
I
Sx
I
Ненасыщенные
А2
СН3
I
- СН2- С—СН—СН —
I
Sx
I
Насыщенные
А2
Sx
I
CH2
с =сн
-сн2х хсн2-
С1с
Koenig и сотр. провели большую работу по условиям, которые изменяют
относительные количества разных типов этих присоединений. Например,
как В1с, так и Bit виды полисульфидов увеличиваются с увеличением со-
держания технического углерода (для видов № 110, № 220, № 326, № 330,
№550 и №765) [29].
Б. Вулканизация с ускорителями замедленного
действия
Если образование поперечных связей происходит по сво-
бодно-радикальному механизму, то ингибирующее действие может быть
результатом гашения радикалов мономерными полисульфидами, которые
образуются при реакции между ускорителем и серой. Если полимерные
политиильные радикалы (предшественники поперечных связей) быстро
гасятся по обменной реакции прежде чем они образуют поперечные связи,
то образование связей будет заторможено пока не исчерпаются мономерные
полисульфиды [4]. Это иллюстрируется следующими схемами:
---Sf + каучук
А7
Ас — Sx ”Sy—Ac
Sj—Sy—Ac + Ac—Sx*
VI. Вулканизация серой с ускорителями 341
2 Ac—Sx-------► Ac—Sx— Sx—Ac
(каучуках'-"
Ac-SH + ।—Sx—Sy—Ac
KQ $► Kc
Таким образом, одна из теорий ингибирующего действия заключается
в гашении свободных радикалов предшественников поперечных связей
мономерными полисульфидами. Оказалось, что если смешать бисалкилпо-
лисульфиды с невулканизованной резиновой смесью, то в результате наблю-
дается больший индукционный период. Также было показано, что продукты
реакции ускорителя с серой (Уск. Эх-Уск.) являются ингибиторами обра-
зования поперечных связей. Те вещества, которые ускоряют образование
предшественников поперечных связей (каучук-Эх-Уск.) ингибируют обра-
зование поперечных связей [25]. Следует отметить, что предложены другие
механизмы для ингибирующего действия. В случае ускорения реакции
бензотиазолсульфенамидами ускоритель расходуется в автокаталитической
реакции с образованием 2-меркаптобензотиазола (МВТ). Скорость этого
расхода приблизительно пропорциональна количеству МВТ. Существует
строгое доказательство, указывающее на следующие реакции, протекающие
в системах с сульфенамидными ускорителями:
BT-S-NHR + BT-SH -----» BT-S-S-BT + RNH2
Сульфенамидный МВТ ДБТД
ускоритель
Если МВТ выделить из системы в момент его образования, то результатом
будет существенное увеличение безопасной переработки. Это тот же слу-
чай, когда присутствует ингибитор преждевременной подвулканизации —
N-циклогексилтиофтальимид (PVI). Это соединение [19] и ему подобные
быстро реагируют с МВТ с образованием 2-алкилдитиобензотиазолов,
R-S-S-BT, которые являются активными ускорителями, но реагируют
быстро с сульфенамидным ускорителем:
L-S-R + BT-SH—BT-S-S-R + L-H
Ингибитор МВТ 2-алкилдитиил неактивное
преждевременной бензотиазол соединение
вулканизации
здесь L — «живая группа» ингибитора преждевременной вулканизации
(т. е., N-фтальимидо- для PVI).
342 Глава 7. Вулканизация
Нельзя переоценить важность контроля скорчинга. Современные шинные
заводы не смогут выжить без хорошего контроля сопротивления преждев-
ременной вулканизации или безопасности переработки, как он обычно на-
зывается. Эта безопасность необходима для быстрого проведения процессов
переработки при высоких температурах (экструзия, каландрование и т.д.)
заготовок для прессования (т. е. деталей шины).
Механизм ингибирующего действия и кинетические особенности реакции
были изучены и изложены в обзорах [24-36].
В. Роль цинка в вулканизации с ускорителями
бензотиазольного типа
Увеличение концентрации жирных кислот и, следователь-
но, увеличение концентрации ионов цинка приводит к увеличению общей
скорости ранних реакций (в течение периода ингибирования), что ведет
к образованию каучук-8х-Уск.. Однако, это приводит к уменьшению
скорости образования поперечных связей, но к увеличению степени сши-
вания [37]. Увеличение скорости ранних реакций может быть объяснению
следующей схемой:
где хелатные формы ускорителя являются более реакционноспособными,
чем свободный ускоритель в ранних реакциях:
Где I-S---ионизованная форма линейной серы. Она может быстро об-
разоваться в реакции между серой и любым количеством инициирующих
фрагментов. Другие исследователи полагают, что присутствие иона цинка
может увеличивать скорость сульфурирования через образование комп-
лексов типа:
L
I
Ac-Sx-Zn-Ac
I
L
где L — это лиганд, такой как молекула амина [27, 36].
Пониженная удельная скорость образования поперечных связей и повы-
шенная степень поперечного сшивания вследствие присутствия ионов цинка
VI. Вулканизация серой с ускорителями 343
с ускорителем вулканизации бензотиазольного типа может быть объяснена
следующей схемой [37]
Каучук - Sx - Sy-4-S—
^=±Каучук- Sx
очень медленно
медленно
каучук —Sx- + «Sy
''s-
[Каучук]
быстро
Поперечная связь
Каучук Sx-Sy-Каучук
[Каучук]
быстро
1
Каучук — S—С
ч
[Каучук]
быстро
Каучук - Sx Каучук
Поперечная связь
Каучук — Sy
не предшественник
поперечной связи
новый предшественник
поперечной связи
Образование хелатной структуры с цинком изменяет положение S-S связи
с большей склонностью к разрыву. Поскольку прочная связь может разо-
рваться, скорость снижается. Хотя скорость сшивания меньше, степень
образования поперечных связей увеличивается, т.к. необходимо меньшее
количество атомов серы для каждой поперечной связи. Иными словами,
поперечные связи становятся менее сульфидными.
Присутствие соединений цинка может также промотировать сниже-
ние сульфидности поперечных связей в течение высокотемпературного
старения вулканизата, т. е. во время реверсии [38]. В некоторых случаях
соединения цинка действительно промотируют разрушение поперечных
связей [23].
Г. Пути достижения специфических характеристик
вулканизации
В течение многих лет большие трудности вызывал независи -
мый контроль двух основных вулканизационных характеристик, сопротив-
ление подвулканизации (безопасная переработка) и скорость образования
поперечных связей. В случае натурального каучука (НК) при выборе быс-
трой вулканизующей системы для того, чтобы сократить время вулканиза-
ции в прессе, сильно страдала безопасность переработки. Если выбирали
344 Глава 7. Вулканизация
ускорительную систему замедленного действия, то скорость вулканизация
в прессе была существенно ограничена. Внедрение высокоэффективного
ингибитора преждевременной вулканизации N-циклогексилтиофтальимида
(PVI) изменило оба этих показателя, так как стало возможным существенно
улучшить сопротивление подвулканизации с малой или совсем неизме-
ненной скоростью сшивания [39]. Таким образом, скорость образования
поперечных связей регулируется выбором ускорителя. Например, умеренно
активный ускоритель замедленного действия третбутилбензотиазолсуль-
фенамид (CAT) может быть частично заменен малым количеством второго
ускорителя (например, от 0,1 до 0,2 м. ч. тетраметилтиурамдисульфида
[ТМТД] или тетраметилтиураммоносульфида [ТМТМ]) для достижения
существенно увеличенной скорости вулканизации; однако сопротивление
подвулканизации заметно снижается. В этом случае сопротивление подвул-
канизации может быть восстановлено добавлением 0,05-0,25 м. ч. PVI без
заметного снижения скорости сшивания [4,40].
В действительности увеличение концентрации CAT приводит к увели-
чению скорости вулканизации с очень малым изменением сопротивления
подвулканизации. Однако увеличение концентрации ускорителя обычно
бывает значительно больше и тогда концентрация серы может быть снижена
для того, чтобы твердость и жесткость сохранялись постоянными (постоян-
ная плотность поперечных связей). Относительно большие изменения
концентрации серы и ускорителя приводят к изменениям эксплуатаци-
онных свойств вулканизатов (см. следующий параграф по свойствам
вулканизатов).
В смесях, содержащих только синтетический каучук, напр., бутадиенсти-
рольный (БСК) или бутадиеновый (СКД), влияние замены вулканизующих
систем может быть не так заметно, как это было в случае НК. Однако если
даже небольшие количества НК содержатся в смеси, то влияние изменения
вулканизующих систем на параметры процесса вулканизации схожи с их
влиянием в смесях из одного НК.
Одной из вулканизационных характеристик, которая требует контроля,
является реверсия, проявляющаяся в смесях, содержащих натуральный
каучук. Существует несколько подходов к снижению доли реверсии. Мож-
но использовать доноры серы или увеличивать отношение концентрации
ускорителя к концентрации серы. Можно проводить вулканизацию при
пониженных температурах в течение более длительного времени. Однако эти
подходы приводят к проявлению эффектов, которые надо компенсировать.
Другой подход заключается в использовании добавок, таких как некоторые
бисимиды, напр., М,М'-м-фениленбисцитроконимид или N,N'-м-фенилен-
бисмалеимид [41-43] или триалкоксисилилалкилполисульфиды, напр.,
бис- (триэтоксисилилпропил) -тетрасульфид [44].
VI. Вулканизация серой с ускорителями -*v- 345
Д. Влияние на адгезию к латунированной стали
Адгезия между каучуком и латунированной сталью (напр.,
стальной корд для радиальных шин) явилась предметом широкого изу-
чения и спекуляции. Латунирование в настоящее время является основ-
ным методом, обеспечивающим адгезию между натуральным каучуком
и сталью в шинном корде. В течение многих лет были предложены различ-
ные мнения относительно его механизма, но по одному аспекту адгезии
натурального каучука к латунированному корду достигнуто согласие:
реальный адгезив между НК и латунированным кордом образуется in
situ в процессе вулканизации как межфазный слой сульфидов и оксидов
меди [45,46].
Адгезионный слой между каучуком и кордом образуется, как полагают,
при взаимодействии между медью и вулканизующей группой. В результате
критическим параметром является оптимизация вулканизующей системы
в смысле адгезии. Изменение в составе композиции латунного покрытия
на стальном корде или изменение его толщины требует изменения вул-
канизующей группы, для того, чтобы поддержать оптимальный уровень
адгезии. Обзоры по проблеме адгезии латунированного стального корда
к натуральному каучуку опубликованы Ван Оойем, который провел много
работ в этом направлении [46]. Он представил модель адгезии каучука
к латуни, в которой слой сульфида меди образуется на латунированной
поверхности до образования поперечных связей. Тонкая пленка сульфида
меди облагает хорошей адгезией и когезией. В дополнении к этому пленка
является пористой так, что молекулы каучука могут быть захвачены ей. При
этом не требуется, чтобы пленка возникала одновременно с образованием
поперечных связей в процессе вулканизации; наоборот требуется, чтобы
пленка сульфида меди полностью сформировалась до начала сшивания. По-
этому адгезия между латунированной сталью и натуральным каучуком часто
может быть улучшена при применении замедлителя подвулканизации, PVI
[4] или при применении ускорителя замедленного действия, как например,
М-дициклогексилбензотиазол-2-сульфенамид (ДЦБС) [47].
Разрушение пленки на границе каучука и сульфида меди встречается
редко. Обычно имеет место когезионное разрушение сульфидной пленки
или адгезионное в слое ниже сульфидной пленки.
Сульфидирование латунированной поверхности не происходит вследс-
твие ее взаимодействия с элементной серой, а является результатом взаимо-
действия между поверхностью латуни и продуктами реакции ускорителей
с серой, которые могут быть представлены общей структурой Уск.-8у-Уск.
и Уск.-8у-Н, где Уск. является остатком ускорителя (напр., бензотиазо-
лильная группа). Величина у возрастает с ростом концентрации серы по
отношению к ускорителю, используемому в вулканизующей группе. Обычно
346 -*V Глава 7. Вулканизация
повышенное содержание серы и высокое отношение серы к концентрации
ускорителя благоприятствует адгезии каучука к латуни.
Выбор ускорителя также влияет на качество адгезии между каучуком
и кордом. Ускоритель не должен образовывать устойчивого медного ком-
плекса, который растворяется в каучуке. Это может вызвать коррозию
латунного покрытия. В этом отношении бензотиазолы и их сульфенамиды
гораздо лучше дитиокарбаматов. Д ЦБС является хорошим сульфенамидным
ускорителем для адгезии каучука к латуни.
Е. Влияние на свойства вулканизатов
Увеличение концентрации серы и ускорителя обеспечива-
ет повышенную плотность поперечных связей и поэтому более высокие
значения модулей, жесткости, твердости и т.д. Однако, когда отношение
количества ускорителя к количеству серы возрастает, то увеличивается
доля моносульфидных поперечных связей в смесях на основе натурального
каучука (также называемых резиновой композицией). Большее количес-
тво ускорителя (по отношению к сере) приводит к увеличению боковых
групп типа — 8Х-Уск., которые присоединены и «висят» на молекулярных
цепях каучука. Повышенные отношения концентрации серы к концент-
рации ускорителя способствуют как образованию более полисульфидных
поперечных связей, так и большего содержания серы, присоединенной
к молекулярным цепям каучука в форме серосодержащих шестичленных
гетероциклических колец вдоль молекулярных цепей. Дополнительно вдоль
основной полимерной цепи образуются сопряженные олефиновые двойные
связи. Указанные особенности показаны на рис. 12. Эти изменения сетчатой
структуры вулканизата без сомнения являются ответственными за изменение
свойств вулканизатов в результате коррекции в рецепте вулканизующей
системы [48-53].
Влияние изменений концентраций ускорителя и серы на свойства вулка-
низатов было изучено на примере следующего рецепта (весовые части): нату-
ральный каучук — 100; технический углерод N-330 — 50; N-изопропил-М' -
фенил-п-фенилендиамин (антиоксидант n-N-ДФА) — 2; оксид цинка — 5;
стеариновая кислота — 3; пластификатор — 3; сера — переменно; N-цикло-
гексилбензотиазол-2-сульфенамид (САЦ) — переменно [4].
Влияние изменений концентраций ускорителя на 300% модуль (это
жаргон для значения напряжения при 300% растягивающего удлинения),
термоокислительное старение и усталостную выносливость (рост трещин
при многократном изгибе по Де Матиа) показаны на рис. 13.
Влияние на величины 300% модуля показаны диагональными линиями
с отрицательным наклоном. Они параллельны и показывают, что напря-
жение при 300% удлинении возрастает с увеличением либо концентрации
серы, либо концентрации ускорителя. Зависимости сохранения разрывного
VI. Вулканизация серой с ускорителями 347
удлинения после старения в горячем воздухе (100°С в течение двух дней)
показывают, что окислительное старение в присутствии n-N-ДФА зависит
только от концентрации серы. Повышенное содержание серы приводит
к ухудшению характеристик старения в связи с повышенным числом точек
сульфурирования цепей. Это предполагает, что введение серы вдоль цепей
может активировать разрыв цепей в реакциях с кислородом [54]. Другая
точка зрения заключается в том, что сера препятствует действию антиокси-
данта (в данном случае n-N-ДФА) [55].
Рис. 12. Типы поперечных связей и модификация цепей.
Зависимости для усталостной выносливости являются сложными. Испы-
тания проводятся так, что образцы испытывают приблизительно одинаковое
растяжение; однако, возникает вопрос, должны ли быть испытания проведе-
ны при одинаковом растяжении или при одинаковой энергии растяжения
[56, 57]. Для некоторых случаев, когда растяжения ограничены условиями
изготовления изделий, испытания на усталость должны быть сопоставлены
при одинаковой амплитуде удлинения. Для других применений, где удлине-
ния не лимитированы, испытания должны быть проведены при одинаковой
энергии удлинения. Все точки на кривой должны быть сопоставлены при
приблизительно одинаковом удлинении за цикл; однако, если мы интер-
полируем кривые усталостной выносливости, но только при постоянных
348 -*\r Глава 7. Вулканизация
значениях модулей, мы получим значения, соответствующие приблизитель-
но одинаковой энергии растяжения за цикл. При выборе повышенных
значений модулей будут наблюдаться более высокие величины энергии
растяжения.
Рис. 13. Свойства вулканизатов: — 300% модуль (МПА); —, усталостная выносливость
при многократном изгибе по Де Матиа (кц х 10-1); -О-О-О-О- процент со-
хранения разрывного удлинения после двух дней старения при 100°С.
Рассматривая группу кривых усталостной выносливости в целом, или
при приблизительно одинаковых значениях энергии в цикле, можно придти
к заключению о следующем: высокое содержание как серы, так и ускорите-
ля приводит к ухудшению усталостной выносливости. Однако, при выборе
подходящего отношения концентрации серы к концентрации ускорителя
могут быть получены большие величины модулей вулканизатов с некоторой
оптимизацией усталостной выносливости. Усталостная выносливость при
VI. Вулканизация серой с ускорителями -* V 349
приблизительно одинаковой энергии растяжения за цикл может быть про-
иллюстрирована рассмотрением величин модулей 13,8 МПа вдоль контурной
линии. Таким образом, можно составить Таблицу 2.
Таблица 2 . Усталостная выносливость при постоянном значении 300% модуля.
М. ч. серы/м. ч. сульфенамида Усталостная выносливость (кц)
6 400
5 500
4 530
3 550
2 550
1 400
0,6 350
0,4 270
0,3 250
0,2 190
Для значения энергии растяжения, соответствующей величине модуля
300% в 13,8 МПа максимальная усталостная выносливость (измеренная по
методу Де Матиа) достигается при 2,5 кратном превышении содержания серы
к ускорителю. Другие оптимальные значения различных модулей 300% могут
быть получены из кривых. Некоторые из них представлены в Таблице 3.
Таблица 3. Оптимизированные значения усталостной выносливости.
Напряжения при 300% удлинении, 300% модуль (МПа) Оптимальное отношение концентрации серы к концентрации ускорителя Оптимизированная усталостная выносливость (кц)
6,9 3,50 800
10,4 3,00 800
13,8 2,50 550
15,5 1,00 300
17,2 0,45 120
19,0 0,27 70
Эти оптимальные отношения и значения усталостной выносливости
следует рассматривать как универсальные. Различные рецепты, различные
350 -*V- Глава 7. Вулканизация
типы противостарителей, различные типы наполнителей, различные кон-
центрации противостарителей и наполнителей, различные базовые полиме-
ры, различные виды испытаний на усталость и др. приводят к различиям
оптимальных соотношений концентраций серы и ускорителя и различиям
оптимальных значений усталостной выносливости. Тем не менее были от-
мечены представленные здесь тенденции в общем виде.
Низкие значения усталостной выносливости при малых содержаниях
серы, но высоких содержаниях ускорителя, относятся к высоким концент-
рациям присоединенных фрагментов ускорительных групп [58] и высоким
концентрациям моносульфидных поперечных связей. Моносульфидные
поперечные связи не способны к обмену, перегруппировке или разрыву,
чтобы снизить напряжения без разрушения основных цепей.
С другой стороны полисульфидные поперечные связи способны
к перегруппировке под напряжением [2, 4, 59]. Перегруппировка попе-
речной связи происходит в две стадии: (1) разрушение, (2) образование
новой связи.
Последние данные [60] показывают, что только разрыв слабых полисуль-
фидных поперечных связей необходим для упрочнения вулканизационной
сетки. Таким образом, необходимо только достаточное количество слабых
поперечных связей в сравнении со связями вдоль основной цепи для того,
чтобы резина была прочной. Во всяком случае, если используются умеренно
высокие концентрации серы (по отношению к ускорителю) усталостная
выносливость улучшается преимущественно из-за присутствия достаточ-
но слабых или перегруппировывающихся полисульфидных поперечных
связей.
Даже если используются повышенные концентрации серы (с поддержа-
нием постоянных значений модуля), усталостная выносливость снижается.
Возможно, что это связано с большим количеством циклических модифи-
каций в цепях с одновременным высоким содержанием серы.
Композиции на основе натуральногьо каучука, вулканизованные
с большим содержанием ускорителя и малым содержанием серы, названы
EV и полу-EV вулканизующими группами. «EV» означает эффективную
вулканизацию, т.к. сера используется эффективно при образовании попе-
речных связей. В среднем, поперечные связи более короткие, чем в случае
обычной вулканизации: они содержат больше моносульфидных и меньше
полисульфидных связей, т. е. их среднее значение серосодержания ниже.
Хотя EV вулканизаты плохи для усталостной выносливости, они часто
используются благодаря прекрасному сопротивлению реверсии (сопротив-
лению безкислородному термическому старению или перевулканизации)
и хорошему сопротивлению термоокислительному старению. Считается, что
сопротивление реверсии или термически инициированному распаду попе-
VI. Вулканизация серой с ускорителями 351
речных связей является результатом внутренней стабильности малосерных
(дисульфидных и особенно моносульфидных) поперечных связей. Полу-EV
вулканизаты (когда отношение концентрации серы к концентрации уско-
рителя является промежуточной величиной) представляют собой удачный
компромисс, когда имеет место довольно хорошая усталостная выносливость
до старения, сохраняемая после старения.
Вместо использования большого количества ускорителя для получения
EV и полу- EV вулканизатов иногда выгодно заменить некоторое количество
серы так называемыми донорами серы. Примером является тетраметилтиу-
рамдисульфид (ТМТД) и 4,4'-дитиодиморфолин (ДТДМ).
Этот тип вулканизующей системы был описан Мак Коллом [57]. Он обна-
ружил, что разумный баланс уровней ускорителя, серы и ДТДМ позволяет
получить хорошие вулканизационные характеристики, хорошую терми-
ческую стабильность, хорошую усталостную выносливость и превосходное
ее сохранение.
Ж. Серно-ускорительная вулканизация различных
ненасыщенных каучуков
Долгие годы основные исследования по серно-ускоритель-
ной вулканизации проводились на натуральном каучуке, как на модельной
матрице. Натуральный каучук был первым эластомером, поэтому усилия
для понимания вулканизации концентрировались на работах по натураль-
ному каучуку. Даже в последние годы большинство работ, опубликованных
по изучению вулканизации, относились к этому каучуку. Это объясняется
традицией проведения исследований на натуральном каучуке и тем обсто-
ятельством, что основные базовые данные касались натурального каучука.
Следует отметить, что главным фактором, обеспечившим традиционные
исследования по вулканизации натурального каучука была Британская
Ассоциация производителей и исследователей каучука (BRPRA), в на-
стоящее время называемая Малазийской Ассоциацией производителей
и исследователей каучука (-MRPRA). Естественно, что эта Ассоциация
занималась в основном натуральным каучуком.
Большинство работ, цитированных в предыдущих разделах, относится
к натуральному каучуку. Однако, некоторые исследования посвящены
вулканизации 1,4-бутадиеновых полимеров [51, 61, 62]. Другие базовые
работы были выполнены по вулканизации этилен-пропилен-диенового
каучука (ЭПДК) [63, 64].
Химия серно-ускорительной вулканизации ПБ, БСК и ЭПДК имеет много
общего с вулканизацией натурального каучука: Перед образованием попе-
речных связей происходит сульфурирование каучука производными реакций
ускорителя с серой —полисульфидами (Уск.-8Х-Уск.) с образованием макро-
352 -'v- Глава 7. Вулканизация
молекулярных полисульфидных промежуточных соединений (каучук-8х-
Уск.). Однако, если в случае ускорителя бензотиазолсульфенамидного типа
при серной вулканизации натурального каучука в процессе образования
каучук-SX-BT выделяется МВТ в результате реакции каучука с BT-SX-BT,
то в случае ПБ и БСК МВТ не выделяется и остается неэкстрагируемым воз-
можно потому, что он связывается в виде макромолекулярного тиоэфира ка-
учук-S-BT (БТ это 2-бензотиазолильная группа). Как и в случае натураль-
ного каучука, средняя длина поперечной связи (ее степень сульфидности, т. е.
значение х в поперечной связи, каучук-Бх-каучук) возрастает с ростом
отношения концентрации серы к концентрации ускорителя (S/Уск.)
в резиновой смеси. Однако, в случае ПБ или БСК степень сульфидности
не столь существенна к отношению S/ Уск., как в натуральном каучуке.
Модельные изучения вулканизации ЭПДК (когда этилиденнорборнен
использовался как модель ЭПДК) [50, 51] показали, что степень по-
лисульфидности поперечных связей в ЭПДК, вероятно, соответствует
изменению в соотношении S/Уск. подобно смесям из натурального
каучука. Разница здесь состоит в том, что отсутствуют доказательства
существования модельных моносульфидных связей, тогда как модельные
дисульфидные поперечные связи более вероятны, чем в случае вулкани-
зации натурального каучука.
Реверсия (определяемая как потери поперечных связей в течение без-
кислородного термического старения вулканизата) представляет собой
проблему, в большей степени связанную с натуральным каучуком или
с синтетическими изопреновыми полимерами. Она может иметь место при
более жестких условиях в бутадиеновом каучуке; в БСК, вместо размягчения,
наблюдающегося при безкислородном старении натурального каучука, про-
исходит повышение твердости (так называемый рост модуля) в течение об-
ширной перевулканизации. В случае натурального каучука и синтетических
полиизопреновых каучуков поперечные связи имеют большую тенденцию к
увеличению полисульфидности, чем в ПБ и БСК. Поперечные связи высокой
степени полисульфидности более чувствительны к тепловым воздействиям,
чем их менее сульфидные аналоги в ПБ и БСК; они с большей вероятностью
подвергаются разрушению, чем образуют циклические модификации
цепей. Но причины образования поперечных связей высокой степени
сульфидности в изопреновых каучуках по сравнению с бутадиеновыми
весьма иллюзорны. Хотя это может быть отнесено к метильным группам,
как заместителям вдоль цепи изопреновых полимеров, отсутствующим
в цепях полибутадиена.
Роль цинка гораздо значительнее при вулканизации изопреновых кау-
чуков, чем при вулканизации ПБ и БСК. Опять причина для различий не
известна, но наиболее значимой является отличие, связанное с присутствием
метильных групп только в изопреновых каучуках.
VI. Вулканизация серой с ускорителями -*v 353
И. Некоторые рецепты серно-ускорительных систем
Примеры рецептов даны в Таблице 4. Эти рецепты не на-
правлены на полное решение проблем создания композиций. Без сомнения
необходимы варианты для учета особых требований.
Таблица4. Рецепты серно-ускорительных вулканизующих систем®).
НК БСК Нитрильный (БНК) Бутил (БК) эпдк
1 2
Оксид цинка 5,00 5,00 3,00 2,00 3,00 5,00
Стеариновая кислота 2,00 2,00 0,50 0,50 2,00 1,00
Сера 2,50 1,80 0,50 0,25 2,00 1,50
ДТДМ6» - - - 1,00 - -
CAT6» 0,60 1,20 - - - -
ДБТД6» - - 2,00 - 0,50 -
МБТ6» - - - - - 0,50
ТМТД6» - - 1,00 1,00 1,00 1,50
Условия вулканизации8)
Температура (°C) 148 153 140 140 153 160
Время (мин.) 25 30 60 60 20 20
а) концентрация в м. ч.
б) ДТДМ, 4,4'-дитиодиморфолин; CAT, М-трет-бутилбензотиазол-2-сульфенамид; ДБТД,
2,2'-дитиобисбензотиазол (2-бензотиазолдисульфид); МБТ, 2 - меркаптобензотиазол; ТМТД,
тетраметилтиурамдисульфид.
в) условия меняются в зависимости от других особенностей композиций.
VII. ВУЛКАНИЗАЦИЯ ПРОИЗВОДНЫМИ ФЕНОЛОВ,
БЕНЗОХИНОНОВ ИЛИ БИС-МАЛЕИМИДОВ
Диеновые каучуки, такие как натуральный каучук, БСК, ПБ,
могут быть вулканизованы при действии фенольных соединений [65-68],
в которых присутствуют заместители (обычно ди-) в виде -СН2-Х групп,
где X является группой -ОН или атомом галогена. Высокодиеновый каучук
может быть также подвергнут вулканизации динитрозобензолом, который
образуется in citu при окислении хинондиоксима [69-73] с помощью окис-
ляющего агента, напр., пероксида свинца.
Атака молекул каучука вулканизующей системой может быть наглядно
представлена подобно реакции, которая предложена для сульфурирования
354
Глава 7. Вулканизация
этих молекул при действии серно-ускорительной системы. Схема реакции
для этих двух типов вулканизации следующая:
Вулканизация фенольными вулканизующими агентами.
Вулканизация бензохинондиоксимом.
Как показано выше, требованиями к химическим структурам для этих
типов вулканизации являются наличие в молекулах эластомеров аллиль-
ных атомов водорода. Реагирующие вещества вулканизующей системы
должны содержать структуры для присоединения протона и электрона в
подходящем пространственном расположении. Это дает возможность для
следующей перегруппировки, где А — акцептор протона и В — акцептор
электрона:
355
VII. Вулканизация производными фенолов, бензохинонов или...
Каучук
н
Вулканизующий
агент
Это объясняет тот факт, что данный вид вулканизации не требует двойной
связи как таковой, т. е. без аллильного водорода в молекуле эластомера
[4,74]. (Необходимо отметить, что фенольный вулканизующий агент может
также действовать по несколько иному механизму с образованием поперечн
ых связей, которые содержат хромановые структуры [75], причем аллильный
водород также необходим.)
Другим вулканизующим агентом для высокодиеновых каучуков является
л-фениленбисмалеимид. Для инициирования реакции обычно используется
катализирующий ее источник свободных радикалов как дикумилпероксид
или бензотиазилдисульфид (ДБТД). Схема реакции для этого типа вулка-
низации, заимствованная из [76], следующая:
356
Глава 7. Вулканизация
Хотя с малеимидным вулканизующим агентом часто используется
источник свободных радикалов, при достаточно высоких температурах
вулканизации малеимиды реагируют с каучуком без этих источников. Это
происходит следующим образом:
I I
Это другой пример реакции, которая в разных вариантах называется
псевдореакцией Дильса-Альдера, еновой или «реакцией без механиз-
ма» [77]. Она подобна реакции, описанной для атаки молекул каучука
фенольными производными или для in citu образующихся нитрозоп-
роизводных из хиноидов (например, бензохинондиоксима). Она также
близко соотносится со схемой сульфирования, предложенной для серно-
ускорительной вулканизации. Сравнение серно-ускорительной, феноль-
ной, хиноидной и малеимидной вулканизации может быть представлено
следующим образом:
Примерные рецепты для вулканизации фенольными производными, бен-
зохинон диоксимом и .м-фениленбисмалеимидом представлены в Таблице 5.
Вулканизаты, полученные с этими типами вулканизующих систем, исполь-
зуются, в частности, когда необходима их термическая стабильность.
VIII. Вулканизация под действием оксидов металлов 357
Таблица 5. Рецепты вулканизации фенольными вулканизующими агентами,
производными хинона или малеимидова).
Бутилкаучук БСК БНК
1 2 1 2
Оксид цинка 5,00 5,00 - - -
Пероксид свинца (РЬ3О4) - 10,00 - - -
Стеариновая кислота 1,00 - - - -
Фенольное производное (SP-1056)6) 12,00 - - - -
Бензохинондиоксим (GMF) - 2,00 - - -
л-фениленбисмалеимид (HVA - 2)в) - - 0,85 0,85 3,00
2- бензотиазилдисульфид (ДБТД) - - 2,00 - -
Дикумилпероксид - - - 0,30 0,30
Условия вулканизацииг)
Температура (°C) 180 153 153 153 153
Время (мин.) 30 20 25 25 30
содержание в м. ч.
б) Скенектеди кемикалс
в) Дю Пон
г) Условия изменяются в зависимости от других аспектов композиции.
VIII. ВУЛКАНИЗАЦИЯ ПОД ДЕЙСТВИЕМ ОКСИДОВ
МЕТАЛЛОВ
Хлорбутадиеновые или хлоропреновые каучуки (ПХП), называемые
также неопреновыми каучуками, обычно вулканизуются под действи-
ем оксидов металлов. ПХП может быть представлен следующей стру-
ктурой:
2—С = СН-СН2Т-
п
Вулканизующим агентом обычно является оксид цинка, наряду с которым
используется оксид магния. ПХП может вулканизоваться в присутствии
одного оксида цинка; однако оксид магния необходим для улучшения
сопротивления подвулканизации. Полагают, что в реакцию вовлекается
358 -*V- Глава 7. Вулканизация
аллильный атом хлора, который образуется в результате полимеризации
в положении 1,2 небольшого количества мономера:
Механизм, который предложен для вулканизации ПХП оксидом цинка
и оксидом магния следующий [3]:
ZnCl2 + MgO —► ZnO + MgCl2
Большинство ускорителей, используемых в серно-ускорительной вулка-
низации других высокодиеновых каучуков, не пригодны для металлоксидной
вулканизации ПХП. Исключением является использование так называемой
смешанной вулканизующей системы для ПХП, в которой металлоксидная
вулканизация связана с серно-ускорительной вулканизацией. Наряду
с оксидами металлов используются тертаметилтиурамдисульфид (ТМТД),
]\,]>Г-ди-о-толилгуанидин (ДОТГ) и сера. Это желательно для высокой
эластичности или хорошего сохранения размеров изделий. Ускорителем,
который широко используется при металлооксидной вулканизации, явля-
VIII. Вулканизация под действием оксидов металлов 359
ется этилентиомочевина (ЭТМ) или 2-меркаптоимидозолин. Дальнейшее
использование ЭТМ для вулканизации ПХП до некоторой степени вызывает
опасение, поскольку она может быть канцерогенной. Похожее соединение —
тиокарбанилид, старый ускоритель серной вулканизации описан для вул-
канизации ПХП. Предложены также другие производные ЭТМ [78, 79].
Механизм вулканизации ЭТМ, предложенный в [80], следующий:
Примеры рецептов для металлоксидной вулканизации представлены
в Таблице 6. Следует отметить, что в одном случае стеарат кальция был
использован вместо оксида магния для улучшения характеристик сопро-
тивления старению [81].
360 -*v Глава 7. Вулканизация
Таблица 6. Вулканизующие системы для хлоропренового каучукаа).
ZnO 5,00 5,00 5,00
MgO 4,00 - 4,00
Стеарат кальция - 5,50 -
Стеариновая кислота - - 1,00
ТМТМ - - 1,00
ДОТГ - - 1,00
ЭТМ 0,5 0,5 -
Сера - - 1,00
Условия вулканизации6)
Температура (°C) 153 153 153
Время, мин. 15 15 15
а) содержание в м. ч. на 100 м. ч. неопрена W
б) условия изменяются в зависимости от других аспектов композиции.
IX. ВУЛКАНИЗАЦИЯ ПОД ДЕЙСТВИЕМ ОРГАНИЧЕСКИХ
ПЕРОКСИДОВ
Большинство эластомеров можно вулканизовать под дей-
ствием органических пероксидов [82,83]. Используются диацилпероксиды,
доалкилпероксиды и перэфиры. Хорошие вулканизаты получаются с ди-
трет-бутилпероксидом и дикумилпероксидом, но первый слишком летуч
для общего применения. Широко используется дикумилпероксид, однако
вулканизаты имеют запах ацетофенона, который является побочным продуктом
процессов вулканизации. Другими нелетучими пероксидами того же класса, ко-
торые не выделяют ацетофенон, являются 1,1-бис (третбутилперокси) -3,3,5-три-
метилциклогексан и 2,5-диметил-2,5-бис (третбутилперокси) -гексан. Последнее
соединение особенно подходит для вулканизации при высоких температурах
(180°С), поскольку оно более термически стабильно, чем другие.
Следует отметить, что ингредиенты кислого типа (жирные кислоты, неко-
торые виды технического углерода и кислые силикаты) могут катализировать
нерадикальный распад пероксидов с большим количеством побочных продук-
тов. Другие ингредиенты композиции, такие как антиоксиданты, могут снижать
эффективность сшивания путем гашения или изменения природы свободных
радикалов, прежде чем они смогут прореагировать с полимером.
Пероксиды являются вулканизующими агентами для тех эластомеров,
которые не содержат участков цепей способных взаимодействовать с другими
IX. Вулканизация под действием органических пероксидов 361
типами вулканизующих агентов. Они используются для этиленпропилено-
вых каучуков (ЭПК), этиленвинилацетатныхсополимеров (ЭВА), некоторых
вальцуемых уретановых каучуков и силиконовых каучуков. Они обычно
не используются для вулканизации бутилкаучука (сополимер изобутилена
с изопреном), вследствие тенденции к разрыву цепей, большей, чем попе-
речное сшивание, если этот полимер подвергнут действию пероксида.
Эластомеры производные изопрена и бутадиена активно сшиваются
пероксидами; но многие свойства вулканизатов хуже, чем серно-ускори-
тельных вулканизатов. Однако, пероксидные вулканизаты этих диеновых
каучуков могут быть предподчительны, где требуется повышенное сопро-
тивление тепловому старению и остаточному сжатию.
А. Пероксидная вулканизация ненасыщенных
углеводородных эластомеров
Инициирующей стадией пероксидной вулканизации явля-
ется распад пероксида с образованием свободных радикалов:
Пероксид —> 2R-,
где R — это алкокси, алкил или ацилокси-радикал, в зависимости от типа
используемого пероксида. (Дибензоилпероксид дает бензоилоксил радика-
лы, а дикумилпероксид — кумилоксил и метильные радикалы [84]). Если
эластомером является производные изопрена, то следующей стадией будет
либо отрыв атома водорода из аллильного положения полимерной молекулы
или присоединение пероксидного радикала к двойной связи полимерной
молекулы [85-87].
II .II
R. + AAzCH2—С=С^---► ^СН—С=С^ + RH
II т.
R- + ~СН2—С=С------► ~СН2—(р—(j!-vv
Для изопренового каучука отрыв водорода преобладает над присоеди-
нением радикала. Два полимерных свободных радикала могут затем соеди-
ниться с образованием поперечной связи
-wCH—C==C-vv
2-v^CH—С=С-~ —►
~СН—С=С^
362
—Глава 7. Вулканизация
Поперечные связи могут также образоваться по цепной реакции,
в которой происходит присоединение полимерных свободных радикалов
к двойным связям [82, 88].
~сн—с=с~
-wCH—C=C'WV
~vCH
-wCH—C==C'V^ ~CH—С=С-'Л-
I . I
~C—C—CH2~ ~c—(j!H—CH2~
+ —► +
^CH2—C=C-~ ~vCH—C=C~v-
В этом случае поперечное сшивание происходит без гибели свободных ра-
дикалов так, что процесс может повторяться до обрыва при рекомбинации
свободных радикалов. Рекомбинация может происходить между двумя по-
лимерными радикалами с образованием поперечной связи или в результате
побочных процессов: полимерный радикал может соединяться с радикалом
пероксида. Если полимерный радикал разлагается с образованием виниль-
ной группы и нового полимерного радикала, то результатом является разрыв
полимерной цепи.
Несколько мономерных радикалов теряется при рекомбинации с поли-
мерными радикалами, если в качестве вулканизующих агентов использу-
ются диалкилпероксиды. Если же эластомер выбран правильно, то реакция
разрыва цепей не так существенна [82-88]. Эффективность поперечного
сшивания натурального каучука дикумилпероксидом может быть оценена
почти равной 1,0. Один моль поперечных связей образуется на каждый моль
пероксида; поперечное сшивание в основном происходит путем рекомби-
нации полимерных радикалов [89, 90]. Одна молекула пероксида дает два
мономерных свободных радикала, которые реагируя с каучуком образуют
два полимерных радикала, и их рекомбинация приводит к образованию
одной поперечной связи.
В случае ПБ или БСК эффективность значительно больше, чем 1,0,
особенно, если удалены антиоксиданты. В этом случае реализуется цепная
реакция. Это можно объяснить стерическими соображениями. В каучуках на
основе бутадиена двойные связи очень реакционноспособны. Присоединение
радикалов к двойным связям приводит к образованию высокореакционно-
IX. Вулканизация под действием органических пероксидов -*V> 363
способных радикалов, которые с большей вероятностью присоединяются
к другим двойным связям полимера. Цепное присоединение более веро-
ятно в бутадиеновых каучуках, чем в присутствии препятствующих этому
метильных групп в изопреновых каучуках.
Можно ожидать, что нитрильный каучук также вулканизуется с эффек-
тивностью большей, чем 1,0; Однако, хотя двойные связи в нитрильном
каучуке высоко активны, эффективность сшивания несколько меньше,
чем 1,0 [88].
Б. Пероксидная вулканизация насыщенных
углеводородных эластомеров
Насыщенные углеводородные полимеры также сшиваются
под действием органических пероксидов, хотя эффективность его снижена
за счет разветвления. Полиэтилен сшивается дикумилпероксидом с эффек-
тивностью 1,0, насыщенный ЭПК дает эффективность около 0,4, тогда как
бутилкаучук вообще не может быть вулканизован. Для полиэтилена схема
реакции подобна ненасыщенным эластомерам.
Пероксид —> 2R-
R- + —СН2—СН2-► RH + —СН2—СН—
2 —СН2—СН--►
—СН2—СН—
I
—СН2—СН—
Однако в разветвленных полимерах происходят другие реакции
Здесь, несмотря на то, что пероксид расходуется, поперечные связи
между цепями не образуются, а средний молекулярный вес полимера даже
снижается за счет разрыва цепей.
364 -*Ь- Глава 7. Вулканизация
Сера, или так называемые соагенты [88, 91], могут быть использованы
для подавления деструкции. Примерами соагентов являются ли-фенилен-
бисмалеимид, высоковинильный полибутадиен с большим содержанием
1,2-звеньев, триаллилцианурат, диаллилфталат, этилендиакрилат и др.
Механизм их действия может быть следующий:
Поперечные связи
В. Пероксидная вулканизация силиконовых каучуков
Силиконовые каучуки могут быть представлены формулой
СН3 п
где R — может быть метил, фенил, винил, трифторпропил или 2-цианэтил.
Силиконовые каучуки, содержащие винильные группы, могут быть вулка-
низованы диалкилпероксидами, таким как дикумилпероксид. Насыщенные
силиконовые каучуки требуют диацилпероксидов, таких как бис-(2,4-
дихлорбензоил) пероксид. В случае насыщенных силоксановых каучуков
механизм заключается в отрыве атома водорода, с последующей рекомби-
нацией полимерных радикалов с образованием поперечных связей. Не про-
IX. Вулканизация под действием органических пероксидов -*Ь- 365
дуктивное использование пероксида происходит в результате рекомбинации
полимерных радикалов с низкомолекулярными свободными радикалами,
образованными при разложении пероксидного вулканизующего агента.
Введение винильных групп повышает эффективность сшивания [92, 93].
Вулканизация часто происходит в две стадии. После предварительной
вулканизации в форме проводят высокотемпературную (напр., 180 °C)
поствулканизацию на воздухе. Эта высокотемпературная поствулканизация
позволяет удалять кислотные производные, которые могут катализиро-
вать гидролитический распад вулканизата [94]. Высокая температура
приводит также к образованию дополнительных поперечных связей
следующего типа [95]:
Г. Пероксидная вулканизация уретановых эластомеров
Уретановые эластомеры, пригодные для пероксидной вулка-
низации, обычно готовятся из олигоэфирадипинатов с концевыми гидрок-
сильными группами и 4,4'-дифенилметандиазоцианата (МДИ). Типичная
структура представляется следующим образом
Атомы водорода могут отщепляться от ариленовых метиленовых групп,
а также от адипинатных структур. Хотя этого обычно достаточно, но эффек-
тивность вулканизации можно повысить введением мочевиновых структур
в полимерную цепь [96].
Д. Рецепты для пероксидной вулканизации
Примеры начальных рецептов даны в Таблице 7. Выда-
ющимися свойствами пероксидных вулканизатов являются малое остаточное
сжатие и высокая термическая стабильность сетки.
366 -* V Глава 7. Вулканизация
Таблица 7. Рецепты для пероксидной вулканизации а).
НК БСК эпк Силиконовый каучук Вальцуемый уретан
Дикумилпероксид 1,0 1,0 2,7 - 2,0
Бис(2,4-дихлорбензоил) пероксид - - - 1,0 -
Триалилцианурат - - 1,5 - -
Условия вулканизации6*
Температура (°C) 150 150 160 115,250в> 153
Время, мин. 45 45 30 141,440"» 45
а) содержание в м. ч.
б) условия меняются в зависимости от других аспектов композиции
в) температура и время поствулканизации на воздухе
X. ДИНАМИЧЕСКАЯ ВУЛКАНИЗАЦИЯ*)
Динамической вулканизацией (ДВ) называется вулкани-
зация или поперечное сшивание одного полимера, находящегося в рас-
плавленном состоянии, при смешении с другим полимером или другими
полимерами. Сначала полимеры тщательно перемешиваются, а затем, при
дальнейшем смешении, один из полимеров становится поперечно сшитым,
тогда как другие полимерные материалы остаются не сшитыми. В этом
процессе образуется дисперсия поперечно сшитого полимера в матрице
или непрерывной фазе несшитого полимера. Если диспергированный
сшитый материал является эластомером, а непрерывная фаза или матрица
является расплавом пластика, тогда композиция может быть использована
как ударопрочная термопластичная смола, а в случае достаточно большого
содержания каучука в композиции она может быть лучше использована как
термопластичный эластомер (ТПЭ), или термоэластопласт (ТЭП).
Фишер [97] использовал процесс для получения композиций, содержа-
щих частично вулканизованный каучук. Позднее было установлено, что
могут быть получены улучшенные и очень прочные эластомерные компо-
зиции ЭПДК и полипропилена методом динамической вулканизации, если
каучук полностью вулканизован [98].
Процесс ДВ для термоэластопластов может быть описан следующим об-
разом: после полного смешения пластика и каучука в расплаве добавляются
вулканизующие агенты. Вулканизация каучуковой фазы происходит, когда
*) Раздел, названный «Динамическая вулканизация» представляет собой
копию, изданную в 1994 г. издательством Academic Press, Inc.)
X. Динамическая вулканизация -*Ь- 367
смешение продолжается. После извлечения из смесителя охлажденная компо-
зиция может быть раскрошена, экструдирована, таблетирована, использована
влитье под давлением и т.д. Такая композиция рассматривается как дисперсия
очень маленьких частиц вулканизованного каучука в термопластичной матрице
пластика. Подобные композиции изготавливаются в промышленном масштабе
в непрерывном процессе с использованием двухшнекового экструдера.
Динамическая вулканизация дает следующие преимущества в сравнении
со смесями, которые не вулканизованы динамически: пониженные усадки,
улучшенные предельные свойства, улучшенная усталостная выносливость,
улучшенное сопротивление действию горячих масел, улучшенная стабиль-
ность фазовой морфологии, большая прочность расплава и т.д.
А. Композиции ЭПДК с полиолефинами
Динамическая вулканизация смесей ЭПДК с полиолефина-
ми (ПП или ПЭ) описана в [98]. Варьировали соотношение каучук-пластик
и степень вулканизации. В некоторых случаях каучук вначале вулкани-
зовался в прессе, затем размалывался до различных по размерам частиц.
Измельченные частицы резины смешивались затем с расплавом полипропи-
лена. Было установлено, что предельные свойства (прочность и разрывное
удлинение) изменяются обратно пропорционально размеру частиц резины.
Поскольку, наименьшие размеры частиц вулканизованного каучука полу-
чаются при динамической вулканизации (не при измельчении вулканизо-
ванного каучука), то более долговечные композиции были получены при
динамической вулканизации.
Для значительного улучшения усадки при напряжениях необходимо
образование только небольшого числа поперечных связей. Однако сопро-
тивление разрыву непрерывно улучшается, когда степень сшивания в ка-
учуковой фазе возрастает. Композиции могут быть вулканизованы серой с
ускорителем, метилолфенольными производными (катализируемых SnCl2)
или другими вулканизующими агентами [99].
С возрастанием концентрации полиолефиновых пластиков композиции
становятся менее подобными резине и более подобными пластику. Модули,
жесткость, усадка при напряжении и прочность возрастают.
Б. Композиции БНК с полиамидами
Отличные эластомерные композиции БНК с нейлоном были
получены путем динамической вулканизации в процессе тщательного
смешения в расплаве смесей БНК с различными нейлонами. В этом случае
влияние вулканизующих агентов было осложнено тем фактом, что некоторые
нитрильные каучуки склонны к самовулканизации при температурах сме-
шения. Могут быть использованы сера, фенольные производные, малеимиды
368 -'V- Глава 7. Вулканизация
или пероксидные вулканизующие агенты. Термопластичные эластомерные
композиции, полученные динамической вулканизацией смесей нейлона
с БНК, обладают высоким сопротивлением к действию горячих масел. Как
и в случае смесей ЭПДК с полиолефинами, увеличение содержания каучука
в композиции снижает жесткость, но повышает сопротивление остаточному
сжатию.
В. Другие эластомерные композиции, полученные
динамической вулканизацией
В дополнение к композициям ЭПДК с полиолефинами
и БНК с полиамидами применяют большое количество других комбинаций
каучуков с пластиками для получения термопластичных вулканизатов ме-
тодом динамической вулканизации [100].
Наилучшие композиции получаются, когда поверхностные энергии
каучука и пластика близки, когда зацепление молекулярных цепей кау-
чука мало и когда пластик кристаллический. Необходимо также, чтобы ни
пластик, ни каучук не разлагались в присутствии других компонентов при
температурах, необходимых для смешения в расплаве. Кроме того, в каж-
дом случае необходима вулканизующая система для каучука в условиях
смешения расплава.
Г. Технологические особенности
Мотивацией для развития термоэластопластов является по-
ниженная стоимость процессов переработки термоэластопластов. Однако
недостатки в достижении действительно каучукоподобных свойств тормозят
внедрение технологии термоэластопластов. Тем не менее, реализованные
промышленные композиции на основе полипропилена и полностью вул-
канизованного ЭПДК обладают многими выдающимися свойствами поли-
уретана и термоэластопластов полиэфирного типа, даже улучшенными по
остаточной деформации и динамической выносливости. Можно перечислить
следующие области применения этих материалов: ролики, воздуходувки,
диафрагмы, герметизирующие устройства, трубопроводы, валики, бам-
перы, аппретированные уплотнители, экраны, всасывающие колпачки,
гибкие детали, работающие на сдвиг, вибрационные изоляторы, пробки,
соединительные детали, катки, маслостойкие линии для литья, зажимные
устройства, покрытия рукавов, вакуумные трубки, втулки, защитные муфты,
виброизоляторы, трубопроводы, различные рукава (напр., гидравлические,
сельскохозяйственные для распыления, распылители красок, водновоз-
душные разбрызгиватели для растений, шахтные рукава и т.д.), изоляторы
проводов, изоляторы кабелей, детали для снижения напряжений, обогре-
вательные рубашки и т.д.
X. Динамическая вулканизация -* V 369
ПРИМЕЧАНИЯ ПЕРЕВОДЧИКА
Автор пользуется ограниченными сведениями по механизму
вулканизации серой с ускорителями, заимствованными, главным образом,
из публикаций английских и американских авторов. В частности, опи-
сывая роль активатора вулканизации, он не пользуется представлениями
о гетерогенных и топохимических реакциях, протекающих в присутствии
твердых частиц оксида цинка, как активатора. Мало сведений приведено
о действии двойных систем ускорителей. Обширная литература, опублико-
ванная по этим и другим особенностям серно-ускорительной вулканизации,
советскими и российскими авторами, а также некоторыми индийскими
исследователями отсутствует в прилагаемом списке литературы. В этой
связи, не вдаваясь в подробности указанной публикации и обратив только
особое внимание на реакции адсорбции и хемосорбции соединений серы
с ускорителями, а также участков молекулярных цепей полидиенов на
поверхности оксида цинка, считаю необходимым привести следующий
перечень основных публикаций:
1. Б.А. Догадкин, А.А. Донцов, В.А. Шершнев. Химия эластомеров. М. «Химия», 1981, 376 с.
2. А.С. Кузьминский, С.М. Кавун, В.П. Кирпичев. Физико-химические основы получения,
переработки и применения эластомеров. М. «Химия». 1976, 368 с.
3. А.А. Донцов Процессы структурирования эластомеров. М. «Химия», 1978, 288 с.
4. В.А. Dogadkin, V.A. Shershnev. Rubber Chem. Technol., 35,1, 1962.
5. V.A. Shershnev. Vulcanization of polydiene and other hydrocarbon elastomers. Rubber Chem.
Technol., v.55, №3, p. p.543-574, 1982.
6. Б.А. Догадкин, В.А. Шершнев. Вулканизация каучуков в присутствии органических уско-
рителей. Успехи химии, 1961, т. XXX, вып. 8, с. 1013-1049.
ГЛАВА
8
УСИЛЕНИЕ ЭЛАСТОМЕРОВ
ДИСПЕРСНЫМИ НАПОЛНИТЕЛЯМИ
ЖАН-БАПТИСТ ДОННЕ
Высшая Национальная Школа Химии,
Мюлуз, Франция
ЭММАНУЭЛЬ КУШТОДЕРО
Отдел производства пневматических шин «Мишлен»,
Клермонт-Ферран, Франция
I. ВВЕДЕНИЕ
Усиление эластомеров дисперсными наполнителями интен-
сивно изучалось в прошлом, особенно в 1960-70-ые годы. Первая причина
этого заключается в существенном изменении механических свойств, которое
происходит при усилении наполнителями: множество полезных применений
эластомеров не могли бы осуществиться без использования дисперсных
наполнителей. Другая причина, как нам кажется, заключается в «тайне»
механизма усиления, который очень интересует многих ученых и остается
сегодня, несмотря на их попытки, не понятым до конца.
Необходимо определить точно, что такое усиление, потому что это слово
имеет очень разные значения, когда применяется к термопластам, реак-
топластам и эластомерам. Недопонимание главным образом обусловлено
тем фактом, что усилением называется повышение уровня показателей ме-
ханических свойств, но то, что рассматривается как механические свойства,
очень различно при рассмотрении разных материалов и изделий.
Для пластиков усиление приводит к повышению модуля и твердости.
Эффект дисперсных наполнителей совершенно ясен — они замещают часть
матрицы: таким образом модуль становится выше, но в то же время удлине-
ние при разрыве уменьшается.
Ситуация для эластомеров сильно отличается: использование усиливающих
наполнителей вызывает одновременно повышение модуля и удлинения при
разрыве. Любопытно, что замещение части деформируемой матрицы твердыми
II. Производство наполнителей -*v 371
объектами не уменьшает ее деформируемость. Возрастание этих двух антаго-
нистических свойств характеризует усиление эластомера. Этот удивительный
парадокс, хотя и не изучен до конца, объясняет способность усиленных эласто-
меров обеспечивать уникальные свойства материала и изделий и подтверждать
свое преимущество в различных технологических областях.
II. ПРОИЗВОДСТВО НАПОЛНИТЕЛЕЙ
А. Неусиливающие наполнители
Как будет показано далее, размер частиц наполнителя является,
возможно, одним из самых важных свойств для усиления. Так, дисперсные
наполнители, полученные измельчением минералов или грубым разделени-
ем за счет осаждения, обычно являются неусиливающими из-за их размера:
их частицы слишком велики. Такие наполнители могут быть использованы
в эластомерах, но придают им всего лишь небольшое увеличение модуля, при
этом происходит значительное падение в прочностных свойствах.
В. Усиливающие наполнители
1. Технический углерод
Большинство из исследований технического углерода после
его получения посвящено проблеме увеличения показателя прочности (то
есть качества усиления). Таким образом, проводимые химические модифи-
кации были тесно связаны с различными теориями усиления.
В 1960-х взаимодействие технический углерод — эластомер рассматрива-
лось как результат образования химической связи [1,2] между кислотными
функциональными группами на поверхности технического углерода и ос-
новными натурального каучука [3,4]. Так, много исследований было посвя-
щено увеличению активности техуглерода окислением его поверхности [4]:
кислородом при высоких температурах, пероксидом водорода, озоном,
азотной кислотой. Вид примененного окисления определяет число и тип
полученных функциональных групп; следует отметить, что такие методы
химической модификации применяются в промышленном масштабе для
специальных видов техуглерода (чернила, пигменты).
В начале 1980-ых Данненберг предложил механизм молекулярного
проскальзывания [5], и процессы модификации после получения повер-
нулись в сторону химической прививки полимерных цепей к поверхности
техуглерода [6].
В недавнее время необходимость в композитах с низким гистерезисом
заставила возобновить исследования химической модификации. Были
предложены другие методы модификации: функционализация [7], покрытие
поверхности техуглерода белой сажей [8] и окисью алюминия [9].
372 Глава 8. Усиление эластомеров дисперсными наполнителями
2. Кремнекислотные наполнители
Применение кремнекислоты в резиновых смесях не может
рассматриваться как новое явление, потому что этот наполнитель исполь-
зовался в рецептах резины с начала 20-го века [10]. Кремнекислота с немо-
дифицированной поверхностью не является усиливающим наполнителем,
если говорить строго, потому что смеси, усиленные ей, проявляют гораздо
более низкие механические свойства, особенно модуль, прочность и сопро-
тивление истиранию. Таким образом, кремнекислотные наполнители не
использовались как усиливающие наполнители отдельно, а главным образом
вместе с техуглеродом.
Два главных прорыва преобразовали этот доступный продукт в усилива-
ющий наполнитель, который смог достичь всех свойств смесей, содержащих
техуглерод, и вместе с тем низкого гистерезиса, что представляет большой ин-
терес для применения в шинах. Первый шаг был сделан в 1970-ых Вольффом,
который предложил специальный силановый связывающий агент — ТЭСПТ
[11]. Второй шагбыл сделан в 1990-ых с появлением патента Р. Раулина, в кото-
ром представлено использование специальной осажденной кремнекислоты,
эластомеров и условий смешения для достижения усиления [12].
а. Осажденные марки кремнекислоты (осажденная кремнекислота)
Марки кремнекислоты, используемые как усиливающие наполнители,
получаются в основном осаждением [13, 14]. Процесс, по сути, состоит в
получении стеклообразной кремнекислоты путем плавления в щелочной
среде чистого песка и основной соли. Затем эта кремнекислота солюбилизи-
руется в воде при высокой температуре и осаждается. Полученная суспензия
кремнекислоты затем фильтруется, промывается и сушится.
Для того чтобы получить усиливающие марки кремнекислоты, основное
внимание нужно уделить рецептам разделения (для получения мелких частиц)
и условиям сушки (для обеспечения высокой дисперсности) [14-16].
Также стоит отметить, что белая сажа может быть очень легко химически
модифицирована путем добавления и прививания групп в течение или после
ее получения [17].
б. Пиролитическая кремнекислота
Данные марки кремнекислоты получается путем высокотемпературного
разложения в присутствии кислорода силана или других метилгидридных
аналогов (SiHMe3, SiH2Me2...):
n SiH4 + 2n O2 n SiO2 + 2n H2O
После выхода из печи пиролитическая кремнекислота получается
в рыхлой форме и из-за высокой температуры ее образования имеет очень
устойчивую морфологию и мало поверхностных силанольных групп по срав-
II. Производство наполнителей -’V 373
нению с осажденной кремнекислотой. Это придает высокую дисперсность
н реакционную способность пиролитическим маркам, но из-за их высокой
цены они редко используются в резиновой промышленности.
3. Новые усиливающие наполнители
Совсем недавно много исследований было проведено для
нахождения новых усиливающих систем. Эти системы моделируют ком-
позиты с кремнекислотой и характеризуются применением связывающего
агента для химического соединения эластомерных цепей с поверхно-
стью наполнителя. Были запатентованы многие усиливающие системы:
□ксигидроксид и оксид окиси алюминия [18, 19], оксиды титана [20]
н нитрид/карбид силикона [21].
III. МОРФОЛОГИЧЕСКОЕ И ФИЗИКО-ХИМИЧЕСКОЕ
ОПИСАНИЕ НАПОЛНИТЕЛЕЙ
Как будет показано позже, морфология и физико-химичес-
кие свойства усиливающих наполнителей имеют огромную важность пото-
му, что они прямо определяют их усиливающую способность. Их описание
ранее было основано преимущественно на морфологических свойствах
(площади поверхности и структуре), но из-за использования белой сажи как
усиливающего наполнителя, в настоящее время существует настоятельная
потребность в описании дисперсности и химии поверхности.
А. Описание морфологии наполнителя
1. Морфология наполнителя
Важно подчеркнуть то, что действительная морфология
техуглерода, используемого в производстве резины, оставалась неизвест-
ной десятилетиями, несмотря на то, что он традиционно использовался
в резиновой промышленности. Это объясняется чрезвычайно малым
размером составляющих его объектов; они меньше 0,1 мкм и могут быть
рассмотрены только с помощью просвечивающей электронной микро-
скопии1).
а. ТЕМ
Как обнаружено с помощью просвечивающей электронной микроскопии
(ТЕМ), техуглерод существует в виде нерегулярных цепеподобных развет-
вленных агрегатов частично сплавленных сфер [22].
В настоящее время прямое наблюдение и исследование структуры и свойств частиц,
агрегатов и агломератов технического углерода эффективно осуществляется методами зондо-
вой микроскопии (см., напр.: Бинниг Г., Рорер Г. Сканирующая туннельная микроскопия —
от рождения к юности. — УФН, 1988, т. 154, вып. 2, с. 261-278). Это замечание относится
и к дальнейшему тексту.
374 -*V- Глава 8. Усиление эластомеров дисперсными наполнителями
Агрегаты являются мельчайшими дисперсионными единицами техугле-
рода и практически не разрушаются в обычных условиях использования,
следовательно, агрегаты должны рассматриваться как действительные
усиливающие объекты.
Цепеподобная разветвленная структура агрегатов делает их очень рыхлы-
ми и их эффективный объем гораздо больше, чем объем всех частиц самого
агрегата. Это наблюдение первостепенной важности, потому что эффектив-
ный объем агрегата будет больше (или меньше) его собственного объема
в смесях и будет определять, какая часть резины может деформироваться,
а какая не может. Это свойство агрегатов обычно называется структурнос-
тью и в основном определяется другими методами (смотри далее раздел
о структуре); несколько очень интересных исследований было проведено
в прошлом по классификации форм агрегатов техуглерода (объемные, эл-
липсоидальные, линейные и т.д.) [23].
Первичные частицы в виде сфер, которые составляют агрегаты, частично
сплавлены вместе и никогда не существуют сами по себе. В любом случае, их
размер представляет большую важность потому, что он определяет действи-
тельную поверхность взаимодействия между техуглеродом и эластомерной
фазой: чем меньше размер первичных частиц, тем выше степень взаимо-
действия. Распределение первичных частиц по размерам было составлено
после анализа снимков ТЕМ, но площадь поверхности техуглерода обычно
более эффективно определяется адсорбционными методами (смотри далее
в главе о площади поверхности).
При очень сильном увеличении возможно непосредственно наблюдать
внутреннюю структуру первичных частиц техуглерода. Они состоят из пе-
рекрывающихся графитовых слоев, которые имеют локальную квазикрис-
таллическую турбостратную структуру с расстоянием между слоями
приблизительно 0,35 нм, что близко к чистому графиту 0,332 нм).
б. STM, AFM
Для описания техуглерода была применена сканирующая туннельная
микроскопия (STM) [24-26]. Исследования методом ТЕМ показали, что
поверхность первичных частиц техуглерода представляет собой перекры-
вающиеся графитовые слои в виде лукоподобной структуры (рис. 1).
Поверхность техуглерода является чрезвычайно упорядоченной, и грани-
цы графитовой решетки должны быть определены как химически активные
зоны, хотя ранее считались «аморфными» зонами [27].
Атомно-силовая микроскопия (AFM) также была использована для
трехмерного описания агрегатов техуглерода [28].
в. Кремнекислотные наполнители
Исследование кремнекислоты методом просвечивающей электронной
микроскопии представляет сложную задачу. Кремнекислота, особенно
III. Морфологическое и физико-химическое описание наполнителя
К
375
полученная методом осаждения, предрасположена к реагломерации
в процессе получения и довольно часто неустойчива при сильном уве-
личении.
Усиливающая кремнекислота, наблюдаемая методом ТЕМ, имеет морфо-
логию очень близкую к техуглероду; она состоит из маленьких цепеподобных
разветвленных агрегатов. Тем не менее, идентификация первичных частиц
кремнекислоты вызывает гораздо больше трудностей из-за того, что они
более сильно сплавлены.
< 6нМ ' ---- »
Data taken Fri Jul 19 11:3В: 16 1991
Buffer гШЗЗОХ.СОе). Rotated O’. XY axee (nm). Z axis Cnml
Рис. 1. Снимок STM поверхности техуглерода в атомном разрешении. (По Бики [2])
a---------- 6 нм ---------►
2. Площадь поверхности
а. Введение
Площадь поверхности является, возможно, самой важной морфоло-
гической характеристикой усиливающих наполнителей потому, что она
соответствует величине границы раздела, то есть величине взаимодействия
между эластомером и поверхностью наполнителя.
Как показано посредством просвечивающей электронной микроско-
пии [23, 29], площадь поверхности прямо связана с размером первичных
частиц, поэтому Американское Общество Испытания Материалов (ASTM)
выбрало этот параметр для номенклатуры техуглерода [30]. Если говорить
более точно, номенклатура ASTM включает 4 знака, первый относится
к скорости вулканизации (N для нормальной, S для медленной), затем
3 цифры, первая из которых соответствует диаметру первичных ча-
стиц [31].
376 -*v- Глава 8. Усиление эластомеров дисперсными наполнителями
Номен- клатура noASTM Диаметр первичных частиц (нм) Предыдущая номенклатура Перевод
900-999 201-500 МТ: Medium Thermal Средняя термическая
800-899 101-200 FT: Fine Thermal Высококачественная термическая
700-799 61-100 SRF: Semi-Reinforcing Furnace Полуусиливающая печная
600-699 49-60 GPF: General Purpose Furnace Печная общего назначения
500-599 40-48 FEF: Fine Extrusion Furnace Высококачественная экструзионная печная
400-499 31-39 FF: Fine Furnace Высококачественная печная
300-399 26-30 HAF: High Abrasive Furnace Высокоизносостойкая печная
200-299 20-25 ISAF: Intermediate Super Abrasive Furnace Улучшенная высоко- износостойкая печная
100-199 11-19 SAF: Super Abrasive Furnace Сверхизносостойкая печная
000-099 1-10 - -
Диаметр частиц может быть определен только посредством ТЕМ, это
сложно и дорогостояще, поэтому площадь поверхности наполнителей обычно
определяют различными адсорбционными методами. (По Янцену и Краусу
[32] и Янцену [33].)
б. Адсорбция азота (БЭТ)
Адсорбция азота и определение площади поверхности по методу БЭТ являет-
ся, наверно, самым широко применяемым методом для характеристики площади
поверхности усиливающих наполнителей [34-36]. Этот метод очень чувствитель-
ный, надежный и может быть применен ко всем усиливающим наполнителям
потому, что либо совсем никак, либо очень слабо связан с химией поверхности.
Главным недостатком метода БЭТ является то, что полученная площадь
поверхности включает микропоры, поверхность которых не может быть достиг-
нута эластомерными цепями, размеры которых гораздо больше, чем размеры
молекул азота [37]. Так, многие сейчас используют метод Т-диаграммы, который
допускает определение площади сетки, исключая микропоры [35].
в. Адсорбция ЦТАБ
Адсорбция цетилтриэтиламмонийбромида (ЦТАБ) широко исполь-
зуется в промышленности техуглерода [32, 34, 38]. Этот метод состоит
III. Морфологическое и физико-химическое описание наполнителя 377
в приготовлении суспензии известной массы техуглерода в водном растворе
ЦТАБ известной концентрации. Техуглерод затем фильтруется и количество
адсорбированного ЦТАБ определяется титрованием оставшегося ЦТАБ
в фильтрате. Площадь поверхности техуглерода затем вычисляется из ко-
личества адсорбированного ЦТАБ с использованием ранее определенной
калибровки относительно стандартного техуглерода.
На величину площади поверхности, определенной по методу ЦТАБ,
микропоры не влияют из-за размера молекулы ЦТАБ, но на нее могут пов-
лиять химия поверхности и примеси [39]. Поэтому существует тенденция
к избавлению от этого метода и замене его методом Т-диаграммы (по БЭТ),
который является более надежным.
г. Адсорбция йода
Определение площади поверхности с использованием йода производит-
ся точно так же, как и ЦТАБ, за исключением того, что адсорбированное
вещество это 12 вместо ЦТАБ. Этот метод может быть применен только
к техуглероду [40].
Так как йод частично адсорбируется и частично реагирует с двойными
связями на поверхности, йодный метод чрезвычайно чувствителен к любым
поверхностным функциональным группам, модификациям или загрязне-
ниям [41]. Поэтому этот метод, ранее широко используемый в промыш-
ленности техуглерода, сейчас заменяется методом ЦТАБ или Т-диаграммы
(по БЭТ).
3. Структура
Структуру довольно легко определить на основе ТЕМ изо-
бражений, однако гораздо сложнее описать ее количественно. Тем не менее,
определение структуры является задачей первостепенной важности потому,
что структура определяет действительный объем наполнителя в смеси и, сле-
довательно, уровень увеличения деформации деформируемой фазы (смотри
«Механические свойства наполненных резин»).
а. ТЕМ измерения
Были предприняты некоторые попытки использовать ТЕМ измерения
для определения структуры наполнителей. Но, несмотря на хорошо прове-
денные исследования [42,43], анализ ТЕМ изображений, которые являются
двухмерными, не приводит к 3-мерной картинке.
б. Абсорбция ДБФ
На практике определение структуры наполнителей производят с помо-
щью очень простого и легкого метода: абсорбции дибутил фталата (DBPA)
[44]. Этот метод заключается в наполнении пустот внутри и между агрега-
тами техуглерода дибутилфталатом, который является вязкой жидкостью.
Исторически объем ДБФ определялся путем образования твердого шарика,
378 Д.
Глава 8. Усиление эластомеров дисперсными наполнителями
состоящего из техуглерода и ДБФ, ручным способом. Теперь эта точная
процедура делается автоматически с помощью абсортометра «Банбери»,
в который помещается заранее известное количество сухого техуглерода.
Затем ДБФ добавляется капля за каплей и структурное число определяется,
когда крутящий момент достигает заданного значения.
Измерения по методу ДБФ чрезвычайно надежны и могут быть легко
и быстро проведены. Тем не менее, измерения по этому методу чувствительны
к химии поверхности, и значения, полученные для наполнителей разной
природы, не могут быть прямо сопоставлены. Для белой сажи вместо ДБФ
также может быть использован диоктилфталат [14].
Значения по методу ДБФ также зависят от процесса образования шарика
для гранулированного наполнителя. Это важно из-за того, что ДБФ измеряет
общий объем пустот: внутриагрегатные пустоты, которые являются нужным
параметром, а также межагрегатные, которые по сути отражают процесс
образования шарика в случае гранулирования. Таким образом, техуглерод
в рыхлом или слабо утрамбованном шарике будет иметь более высокий
показатель абсорбции ДБФ, точно так же распыленно-высушенная белая
сажа будет иметь более высокое значение, чем гранулированная, даже если
их агрегаты абсолютно одинаковы.
в. Абсорбция ДБФ сжатого образца
Очень часто абсорбция ДБФ проводится с техуглеродом, предварительно
подвергнутым уплотнению под высоким давлением в цилиндре, например,
24000 фунт/кв. дюйм (165 МПа) четырежды. Абсорбция ДБФ сжатого об-
разца (или CDBPA) эквивалентна обычной абсорбции ДБФ для низкострук-
турных марок техуглерода и значительно ниже для высокоструктурных.
Предполагается, что процедура уплотнения под высоким давлением вы-
зывает разрушение агрегатов в процессе смешения, но из-за того, что ДБФ
занимает и внутри- и межагрегатные пустоты, трудно установить точно,
обусловлено ли падение показателя абсорбции ДБФ уплотнением напол-
нителя под воздействием давления [45] либо фактическим разрушением
агрегатов.
г. Ртутная порометрия
Структура наполнителя также может быть легко определена ртутной
порометрией [46-48]. Наполнитель помещается в небольшую камеру,
и ртуть вводится в пустоты под возрастающим давлением. Кривая интрузии
(проникновения) дает объем ртути, введенной в поры для каждого прило-
женного давления. Обычно кривые интрузии дают хорошо определяемый
шаг при высоком давлении, которое соответствует заполнению мельчайших
пор (пустот внутри агрегатов и между первичными частицами). Величина
шага дает структурный индекс, который исключает межагрегатные пустоты,
и таким образом является более репрезентативным для внутренней струк-
III. Морфологическое и физико-химическое описание наполнителя -’V- 379
туры наполнителя. Ртутная порометрия также позволяет проводить прямое
определение площади поверхности наполнителя, исключая микропоры [49]
и может быть применена для любого дисперсного наполнителя [50].
4. Распределение размеров агрегатов
Распределение размеров агрегатов является последней морфологической
характеристикой усиливающих наполнителей. Эта характеристика очень
редко используется, несмотря на огромный интерес к информации о разме-
ре агрегатов, который прямо влияет на расстояние между усиливающими
частицами в смеси, и, следовательно, на увеличение деформации. Эта уди-
вительная ситуация обусловлена тем фактом, что это определение особенно
сложно произвести [33].
а. ТЕМ/AI измерение
Просвечивающая электронная микроскопия с последующим анализом
снимка (ТЕМ/AI) использовалась в течение долгого времени для получения
распределения размеров агрегатов техуглерода и белой сажи [51]. Такие
исследования очень дорогостоящи, потому что они нуждаются в измерении
размеров по меньшей мере нескольких тысяч агрегатов, чтобы точно уста-
новить распределение размеров. Тем не менее, при использовании TEM/AI
агрегаты рассматриваются и измеряются как двумерные проекции, что,
возможно, максимизирует их размер.
б. Фотометрия осадка в дисковой центрифуге
Более надежным методом установления распределения размеров аг-
регатов, возможно, является использование седиментометра с дисковой
центрифугой (с просвечиванием в видимой или рентгеновской области)
[52]. Этот аппарат состоит из непрозрачного диска с полостью, который
может вращаться на очень высокой скорости. Сначала во вращающийся
диск вводится среда седиментации, а затем очень маленькое количество
суспензии наполнителя впрыскивается на ее поверхность. Седиментация
регистрируется просвечиванием светом или рентгеновскими лучами.
Распределение размеров агрегатов техуглерода [53] и белой сажи может
быть легко получено с использованием этого метода. Его главным недостат-
ком является, в отличие от ТЕМ/AI, то, что он не может предоставить инфор-
мацию о реальных размерах агрегатов, потому что в расчетах предполагается
самая маленькая площадь проекции, то есть сферической частицы.
в. Красящая сила (сила окраски)
Пришедшее из промышленности чернил и пигментов измерение кра-
сящей силы является очень простым методом оценки среднего размера
агрегатов техуглерода [54, 55]. Это испытание состоит в приготовлении
пасты из известного количества техуглерода, белого порошка (оксида титана
или цинка) и масла (например, ДБФ). Затем измеряется ее отражательная
380 -'V Глава 8. Усиление эластомеров дисперсными наполнителями
способность. Показано, что значения красящей силы грубо коррелируют со
средним размером агрегатов и могут рассматриваться как показатель широты
распределения частиц по размерам [33, 56].
Б. Дисперсность
Хотя наличие взаимосвязи между распределением размеров
агрегатов наполнителя и износостойкостью точно установлено, относительно
мало исследований было проведено по описанию дисперсности наполнителя.
Такая ситуация обязана главным образом тому факту, что дисперсность тех-
углерода обычно признавалась удовлетворительной, отчасти потому, что она
действительно высока, но скорее из-за того, что все аппараты для смешения
были разработаны под техуглерод.
Использование белой сажи пролило новый свет на эту область, потому что,
в противоположность техуглероду, дисперсность является одним из ключевых
свойств при получении смесей, усиленных кремнекислотой [14,48, 57].
Очевидно, что на дисперсность наполнителя главным образом влияют
взаимодействия между агломератами и/или агрегатами, другими словами —
для разделения двух объектов нужна сила и энергия. Для техуглерода
эти взаимодействия обусловлены главным образом ван-дер-ваальсовыми
силами, которые очень слабые по сравнению с водородными связями, су-
ществующими в кремнекислоте. К тому же процесс гранулирования имеет
большое влияние на дисперсность: любое действие, приводящее к большему
уплотнению наполнителя, увеличивает взаимодействие между частицами
наполнителя и, таким образом, уменьшает его дисперсность.
а. Отражающая способность
Наиболее часто используемая техника оценки дисперсности наполнителя —
это изучение отражающей способности смесей со строго определенным
составом. Для оценки распределения наполнителей были разработаны не-
которые аппараты, использующие калиброванный набор вышеупомянутых
смесей (Dispergrader). Однако такая оценка главным образом обнаруживает
дефекты диспергирования в размерах нескольких десятков микрон, и пря-
мое сравнение смесей, содержащих техуглерод и кремнекислоту, должно
проводиться с большим вниманием. В любом случае необходимо сделать
смесь, что означает выбор рецептуры, смесителя и условий смешения, так что
результат не может рассматриваться как измерение внутренней дисперсности
наполнителя, но только лишь как отражение дисперсности наполнителя
в данной смеси с определенным набором условий смешения.
б. Лазерная гранулометрия
Недавно был запатентован еще один метод для определения дисперсности
наполнителя. Он состоит в непрерывном измерении дисперсности напол-
III. Морфологическое и физико-химическое описание наполнителя -* V 381
нителя лазерной гранулометрией в процессе ультразвуковой дисаггломе-
рации [18]. Эта методика может быть применена к любому наполнителю,
а полученная характеристика является его внутренним свойством. Однако
использование воды как среды дисаггломерации может быть проблематич-
ным из-за ее высокой полярности по сравнению с эластомерами.
В. Физхимия наполнителя
По сравнению с морфологией химия наполнителя изучалась
лишь поверхностно, отчасти из-за сложности таких исследований, но более
вероятно из-за того, что с 1970-х усиление повсеместно рассматривалась
как физическое взаимодействие между эластомером и наполнителем. Та-
ким образом, описания химии техуглерода главным образом датируются
1960-ыми, и, начиная с этого времени, к описанию поверхности техугле-
рода было применено несколько новых технических методов. Ситуация
для белой сажи несколько другая, потому что усиление кремнекислотой
является следствием химической реакции между силаном и поверхностью
кремнекислоты. В области усиления эластомеров было опубликовано мало
исследований, вероятно потому, что поверхность кремнекислоты уже была
хорошо охарактеризована для других применений.
Что касается физико-химических характеристик, исследования огра-
ничиваются определением распределения поверхностной энергии, которое
будет обсуждено первым.
1. Поверхностная энергия
Усиление эластомеров техуглеродом в основном рассматривается как
следствие адсорбции полимерных цепей на поверхности техуглерода. По-
этому знание поверхностной энергии техуглерода имеет первостепенную
важность. Однако опубликовано очень мало исследований по измерению
поверхностной энергии техуглерода, что может быть легко объяснено, если
учесть сложность таких измерений для высоко гетерогенной и неровной
поверхности.
Иммерсионная калориметрия позволяет определить поверхностную
энергию техуглерода [58, 59] или кремнекислоты [60]. Этот метод может
применяться для различных жидкостей или растворов низкомолекулярных
эластомеров, но имеет главный недостаток в том, что дает значение средней
поверхностной энергии (-50-70 мДж/м2), в то время как поверхностная
адсорбция полимерных цепей по-видимому происходит преимущественно
на участках с самой высокой поверхностной энергией.
Такая ситуация оправдывает применение обратной газовой хромато-
графии (IGC) для описания поверхностной энергии наполнителя. Данная
техника может использоваться в 2 различных вариантах: «бесконечного»
и «конечного» разбавления.
382 -*V Глава 8. Усиление эластомеров дисперсными наполнителями
В методе «бесконечного» разбавления очень малые количества алканов
с возрастающим числом атомов углерода адсорбируется на поверхности техугле-
рода [61,61а, 62] или кремнекислоты [60]; из их времен удержания возможно
подсчитать рассеивающий компонент поверхностной энергии ys d. Из-за очень
низкого покрытия поверхности в процессе эксперимента это значение соответ-
ствует высокоэнергетическим участкам поверхности [24]. Согласно этому методу
поверхностная энергия растет с площадью поверхности техуглерода; так как
техуглерод с высокой площадью поверхности является более усиливающим, то
этот результат должен быть признан обнадеживающим. Полученные значения
поверхностной энергии могут достигать 300-500 мДж/м2, что не может считаться
реалистичным для углеродных поверхностей. К тому же, данное вычисление, по
всей видимости, также чувствительно к химической модификации поверхности
технического углерода, даже если применяемые датчики характеризуют лишь
дисперсную часть поверхностной энергии.
«Конечное» разбавление является более мощным методом, так как дает
возможность получить полное распределение участков поверхности по
энергии для техуглерода [24,63] или кремнекислоты [64,65]. В этом методе
поверхность полностью покрывается образцом, и распределение вычисляется
за счет специальной последующей обработки десорбционного сигнала.
При использовании данной методики для техуглерода получается при-
близительно такое же распределение поверхностной энергии, отличающееся
только количеством участков адсорбции. Распределение участков поверх-
ности по энергии получается особенно широким, от участков с высокой
(-100 мДж/м2) до участков с низкой («10 мДж/м2) энергией. Средние значе-
ния согласуются с данными, полученными иммерсионной калориметрией.
Наконец, следует упомянуть, что метод «конечного» разбавления, схожий
с обратной газовой хроматографией, может быть применен к изотерме ад-
сорбции азота и позволяет описать наноразмерные неровности поверхности
любого наполнителя [66].
2. Химия поверхности
а. Техуглерод
Примеси. Из-за процесса производства поверхность техуглерода содер-
жит некоторые органические и минеральные примеси [67, 68].
Органические примеси состоят в основном из полиароматических уг-
леводородов (ПАУ) [69]. Они соответствуют составу сырья, частично не
подвергнувшегося конверсии, которое реадсорбировалось на техуглероде.
Эти ПАУ присутствуют в очень небольшом количестве, и для их удаления по
причине их прочной адсорбции на техуглероде экстракция должна прово-
диться в аппарате Сокслета в сильном растворителе (толуоле) и при высокой
температуре (8042). Однако было показано, что органические примеси не
оказывают значительного влияния на усиление техуглеродом [24].
III. Морфологическое и физико-химическое описание наполнителя 383
Минеральные примеси появляются во время стадий гашения и грану-
лирования в процессе производства техуглерода. Уменьшение темпера-
туры техуглерода и отработанных газов достигается главным образом путем
погружения в большую массу воды. Дополнительная вода используется также
в процессе гранулирования техуглерода. Даже если эта вода очищена, оставши-
еся минеральные соли попадают на поверхность техуглерода и из-за высокой
температуры восстанавливаются в основные соли. Минеральные примеси
техуглерода могут быть легко удалены путем солюбилизации в воде, как и при
определении так называемого «pH техуглерода», для чего делается суспензия
техуглерода в воде, затем вода фильтруется и измеряется ее pH. Минеральные
примеси, вообще говоря, не изменяют усиливающие свойства техуглерода,
но они оказывают значительное влияние на скорость вулканизации, которая
увеличивается с увеличением значения pH водной суспензии техуглерода.
Кислородсодержащие функциональные группы. Кислородсодержащие
функциональные группы на поверхности техуглерода были обнаружены
в начале 1950-х [70] и полностью описаны Боэмом в 1960-е [71]. В то время
взаимодействие между техуглеродом и натуральным каучуком рассматри-
валось как следствие химических реакций между поверхностными кислот-
ными группами техуглерода и щелочными, присутствующими в структуре
натурального каучука [71а].
Рис. 2. Химические функциональные группы на поверхности техуглерода (По Вики [2]).
Определение количества функциональных групп на поверхности техугле-
рода состоит в приготовлении суспензии заданного количества техуглерода
в растворах известной концентрации различных оснований: NaHCO3,
Na2CO3, NaOH в воде и EtONa в этаноле [72]. Затем техуглерод отфильтро-
вывается, и количество прореагировавших кислотных групп получают из
титрования оставшегося основания в фильтрате (рис. 2).
В 1960-ых техуглерод изготовляли в основном канальным способом,
и кислотные функциональные группы в таких марках присутствовали в ко-
384 —Глава 8. Усиление эластомеров дисперсными наполнителями
личестве около 10’3экв/г, что позволяло относительно легко проводить их
определение, но в настоящее время в печных марках техуглерода количес-
тво поверхностных кислотных групп составляет около 10‘5экв/г, и поэтому
требуются особые способы определения [73] либо должны использоваться
жесткие условия реакции [24]. Очевидно, что такие тонкие определения
должны проводиться с техуглеродом, который ранее был очищен для того,
чтобы разрушить щелочные минеральные примеси, которые затрудняют
любое определение кислотных групп, присутствующих на поверхности
техуглерода в малом количестве.
Это наблюдение позволяет считать, что кислотные группы на поверхности
техуглерода возникают в основном за счет окисления поверхности в произ-
водственных процессах и, возможно, в течение сушки после гранулирования.
Следовательно, кислотные группы могут рассматриваться как изменение
поверхности техуглерода.
Эта точка зрения подтверждается тем фактом, что окисленный техугле-
род с содержанием поверхностных кислотных групп примерно 10-2экв/г
проявляет очень низкую усиливающую способность.
Двойные углеродные связи, содержание атомов водорода. Все проводи-
мые в прошлом химические исследования по техуглероду фокусировались
на химических примесях или функциональных группах, возникших за счет
частичного окисления поверхности техуглерода, а не на его собственной
поверхностной реакционной способности.
В настоящее время поверхность техуглерода не может рассматриваться
как химически инертная. Ее реакция с йодом или кислородом, ее структура,
подтвержденная снимками сканирующей туннельной микроскопии [24], пока-
зывает наличие большого количества реакционноспособных двойных связей.
Согласно Медалии и Краусу [41] такие двойные связи могут реагировать
с серой [74-76], олефинами [77] и радикалами [3,78] образуя химическую
связь между поверхностью техуглерода и полимером, но их прямое количе-
ственное определение не было произведено. Некоторые авторы наблюдали
строгую корреляцию между содержанием атомов водорода в техуглероде и его
усиливающей способностью. Содержание и реакционная способность ато-
мов водорода, находящихся на концах графитовых слоев, были определены
с помощью изотопного обмена, и согласуются с усиливающей способностью
техуглерода [79, 81]. Этот метод является особенно сложным, и поэтому
обычно используется массовое содержание водорода; эти значения также
отлично коррелируют с усиливающей способностью техуглерода [81].
б. Кремнекислотные наполнители
Силанолы. Из-за разнообразного и длительного использования кремне-
кислоты для других целей, химия ее поверхности гораздо лучше известна,
чем у техуглерода [13].
III. Морфологическое и физико-химическое описание наполнителя
Л
385
Химия поверхности кремнекислоты определяется, главным образом,
содержанием на поверхности силанолов Si—ОН; силанолы могут быть изо-
лированными 0 = f^i—ОН или сдвоенными O=Si=(OH)2. В основном они
сильно ассоциированы за счет водородных связей (рис. 3).
Рис. 3. Изолированные, сдвоенные и ассоциированные силанолы.
Для пирогенных марок кремнекислоты содержание силанола примерно
2 Si-OH/нм2 [82,83], но для марок осажденной кремнекислоты содержание
может достигать от 6 до 10 и более Si-OH/нм2 [84]. Важно подчеркнуть,
что такое высокое содержание силанольных групп нельзя рассматривать
буквально: если рассматривать длину связи и расположение атомов крем-
ний-кислород, то содержание 2-3 Si-OH/нм2соответствует полному
покрытию поверхности силанольными группами [13]. Такие высокие зна-
чения можно объяснить существованием цепей поликремниевой кислоты
-[Si (ОН)2] -O-Si (0Н)3и тем, что площадь поверхности, измеряемая
по методу БЭТ, не учитывает поры очень маленького размера, в которых
расположены некоторые силанольные группы.
Содержание силанольных групп на поверхности по существу не может
рассматриваться как индикатор усиливающей способности кремнекислоты;
действительно, из-за своего размера связующий агент, использующийся для
соединения поверхности кремнекислоты с эластомером, не может взаимо-
действовать с более чем 2 или 3 поверхностными силанольными группами
[85,86]. Далее, большое число силанольных групп будут образовывать боль-
ше ассоциатов и, таким образом, уменьшать их реакционную способность.
Другие поверхностные функциональные группы. Химия поверхности
кремнекислоты не может использоваться для определения содержания сила-
нольных групп на поверхности. Особенно это касается марок кремнекислоты,
полученных осаждением, так как некоторые другие химические вещества
могут изменить реакционную способность ее поверхности.
Например, из-за неполного гидролиза силиката на поверхности может
образовываться =Si-O-Na, из-за неполного промывания кремнекислоты
после фильтрования на ее поверхности может адсорбироваться сульфат.
К тому же некоторые соли, используемые при производстве кремнекислоты,
могут также входить в структуру поверхности и вносить изменения в реак-
ционную способность [17].
386 -* V Глава 8. Усиление эластомеров дисперсными наполнителями
Очевидно, что, как говорилось ранее в разделе о производстве белой
сажи, поверхность кремнекислоты также может быть модифицирована
другими химическими веществами, добавляемыми в процессе или после ее
получения. В любом случае, такая модификация изменяет реакционную
способность силанольных групп лишь за счет увеличения [87] или умень-
шения их кислотности.
IV. СМЕСЬ КАК НАНОКОМПОЗИТ ЭЛАСТОМЕРА
И НАПОЛНИТЕЛЯ
Даже с учетом того, что термин нанокомпозит обычно не ис-
пользуется при обсуждении усиления дисперсными наполнителями, следует
особо отметить, что смешение твердых веществ, обладающих усиливающими
свойствами, с эластомерами не сводится к арифметической сумме свойств
обоих ингредиентов, взятых по отдельности, а приводит к синергизму, что
позволяет достичь новых свойств.
Термин наполнитель является, вообще говоря, неадекватным, потому
что дисперсное твердое вещество используется не только для заполнения
пустот, но и для уменьшения стоимости эластомерного изделия. Действи-
тельно, эластомер и усиливающий наполнитель должны рассматриваться как
2 неразделимые части, имеющие равное значение в композите. По меткому
высказыванию Папирера «техуглерод и каучук это свадьба века».
Данное вступление значит гораздо больше, чем просто семантические
рассуждения, оно подчеркивает важность комплексного подхода, включа-
ющего взаимодействие между усиливающим наполнителем и эластомерной
матрицей.
А. Диспергирование, размер агрегатов
и расстояния между ними
1. Диспергирование
а. Диспергирование наполнителя в каучуковой матрице
Качество распределения дисперсного наполнителя в эластомерной мат-
рице имеет первостепенное значение для механических и эксплуатационных
свойств изделий [88-92].
В недавнее время опять вернулись к рассмотрению описания дисперс-
ности наполнителя в связи с трудностями, с которыми столкнулись при
диспергировании кремнекислоты в каучуке [93, 94].
Обычно распределение наполнителя достигается в смесителе «Банбери»
и происходит в 2 этапа. На первом этапе наполнитель распределяется в чистом
полимере в форме гранул или субгранул (то есть агломератов) [95], а затем
на втором этапе субгранулы постепенно разрушаются до агрегатов по типу
отшелушивания слоев лука [96]. Эта вторая фаза, которая разрушает агломе-
IV. Смесь как нанокомозит эластомера и наполнителя 387
раты и определяет расстояния между действительными агрегатами, гораздо
длиннее по времени и требует высоких затрат механической энергии.
Как обсуждалось ранее, дисперсность и, таким образом, распределение
зависит главным образом от силы взаимодействия между агломератами
и/или агрегатами, которая является прямым следствием их поверхностной
энергии. Влияние поверхностной энергии было четко установлено за счет
использования различных матриц; когда поверхностная энергия матрицы
возрастает и приближается к поверхностной энергии наполнителя, диспер-
гирование облегчается.
б. Описание распределения частиц
Описание распределения наполнителя особенно сложно из-за того, что
должно проводиться в очень широком диапазоне масштабов [97]: от мик-
роскопических нераспавшихся агломератов, которые являются дефектами
и будут уменьшать срок эксплуатации изделия, до наноскопических рас-
стояний между агрегатами, очень сильно влияющих на уровень усиления
[98]. Действительно, описание распределения наполнителя проводится
с использованием большого количества аналитических методов: оптической,
электронной [30,99] и атомно-силовой микроскопии [100,101], атакже диф-
ракции рентгеновских лучей [23,102,103] и нейтронов. Главная сложность
заключается в том, чтобы составить общую картину с использованием самых
различных данных, так как каждый из этих методов дает полное представ-
ление только об одном из направлений описания диспергирования.
в. Влияние свойств наполнителя
Помимо поверхностной энергии наполнителя, которая имеет большое
значение, на диспергирование также оказывают влияние его морфологи-
ческие свойства.
На диспергирование сильно влияет площадь поверхности наполнителя:
чем больше площадь поверхности, тем хуже распределение [104]. Этот
результат объясняется, возможно, тем фактом, что наполнитель с большой
площадью поверхности обычно имеет маленький размер агрегатов, которые
будут более сильно взаимодействовать друг с другом.
Структура наполнителя также имеет определенное влияние на диспер-
гирование: высокоструктурные марки лучше распределяются в смеси. Этот
факт хорошо установлен и объясняется тем, что более «открытые» струк-
туры агрегатов вызывают меньшее число контактов друг с другом в сухом
состоянии.
2. Размеры объектов в смеси
Как уже упоминалось ранее, наполнитель находится
в смеси в виде распределенных агрегатов с различными размерами. Расчет
этого распределения потенциально интересен, но рассчитанный результат
388
—Глава 8 '. Усиление эластомеров дисперсными наполнителями
в смесях может быть не достигнут. Действительно, диспергирование может
быть не завершено, и часть агломератов может остаться в смеси, смещая
действительный размер усиливающих объектов к более высокому значе-
нию. С другой стороны, из-за высоких сдвиговых усилий, возникающих во
время смешения, может происходить некоторое разрушение агрегатов,
что приводит к уменьшению действительных размеров усиливающих
объектов по сравнению с ожидаемыми на основании расчета. Поэтому,
рассматривая способ существования наполнителя в смеси, мы будем ис-
пользовать термин объект, который может относиться как к агрегатам,
так и агломератам.
Очевидно, что описание распределения размеров агрегатов в смеси имеет
свои особенности. Некоторые наблюдения, сделанные с помощью просвечи-
вающей электронной микроскопии, проводились на тонких микротомиро-
ванных образцах, но такие методы анализа применимы к небольшому ко-
личеству агрегатов и позволяют провести только качественную оценку [23].
Прямое определение распределения объектов в смеси также проводилось с
использованием дифракции рентгеновских лучей [105] или нейтронов, но
такой подход строго ограничен высокой концентрацией объектов напол-
нителя и их индексом рефракции, которые относительно близки к индексу
рефракции каучука. Еще один способ определить распределение размеров
объектов — это выделение наполнителя из смеси термическим или катали-
тическим разложением полимера, но эти процессы сильно влияют на размер
объектов из-за возможной реагломерации частиц.
Представления о нахождении распределения объектов наполнителя
в смеси постоянно развиваются. Но в любом случае, существует общепри-
нятое мнение, что размер агрегатов техуглерода уменьшается в процессе
смешения [29,106], даже если применяются методы анализа, позволяющие
избежать влияния артефактов.
Стоит также вспомнить, что метод адсорбции ДБФ сжатого образца, как
уже обсуждалось ранее, был разработан с учетом возможного разрушения
агрегатов. Данные, касающиеся этой проблемы, были опубликованы после
исследования этим методом кремнекислоты, но та же самая проблема раз-
рушения агрегатов представляется по-прежнему вероятной.
3. Расстояния
Нахождение расстояний между объектами в смеси взаимо-
связано с проблемой определения размеров объектов и имеет те же самые
трудности и такой же недостаток экспериментальных данных.
Тем не менее, следует заметить, что обычные дозировки наполнителя,
используемые в эластомерах (около 20% по объему), очень близки к макси-
мальному содержанию, которое может быть введено в эластомерную матрицу.
Это содержание очень мало по сравнению с компактным расположением
сфер наполнителя, но, как обсуждалось ранее, агрегаты наполнителя имеют
IV. Смесь как нанокомозит эластомера и наполнителя -*Ь- 389
весьма открытую структуру; кроме того, максимальная дозировка сильно
зависит от структуры наполнителя.
На примере простых моделей было показано, что расстояния между
объектами находятся в диапазоне нескольких десятков нанометров, что
согласуется с фактом наличия электропроводности у смесей, содержащих
техуглерод, который подразумевает, что расстояния между объектами доста-
точно мало, чтобы допускать туннельную проводимость [107].
Кроме того, было показано, что оптимум загрузки наполнителя соот-
ветствует одинаковым межагрегатным расстояниям, независимо от размера
агрегатов или их структуры [108]. Этот очень интересный результат под-
черкивает не вызывающее сомнений влияние межагрегатных расстояний
в усилении эластомеров.
Б. Взаимодействие наполнитель-эластомер
1. Техуглерод
а. Адсорбция эластомера
Сетка наполнителя. Из-за высокой поверхностной энергии техугле-
рода эластомерные цепи сильно адсорбированы на его поверхности. Эта
адсорбция, даже если она ограничена небольшой частью эластомерных
цепей, называемых «хвостами», существенно снижает их подвижность
[103, 109, 110].
Как упрощение — что не совсем корректно — можно считать, что «хвосты»
имеют пониженную температуру перехода [111,112]. Точная толщина этого
слоя еще обсуждается [109, 112а, ИЗ], но обычно упоминаются значения
в 1-5 нм [114]. В любом случае, очень существенно, что такая толщина
соответствует, по меньшей мере, 3-15% от общей эластомерной фазы [114].
Принимая во внимание структуру техуглерода и неровность его поверхности,
было предположено, что значения толщины должны достигать 30%.
Более точный подход учитывает, что эластомерные цепи имеют градиент под-
вижности по направлению от поверхности техуглерода к массе эластомера.
Высокие площадь поверхности и дозировка техуглерода, используемого
в усилении эластомеров, обеспечивают такие маленькие расстояния между
усиливающими объектами, что почти любая эластомерная цепь контактирует
как минимум с одним агрегатом [108]. Также из-за того, что статистический
размер полимерных цепей находится в диапазоне межагрегатных рассто-
яний, соседние объекты, возможно, удерживаются вместе за счет цепей,
адсорбированных на обоих этих агрегатах.
Связанные агрегаты техуглерода составляют сетку наполнителя.
Количество связанного каучука. Существование сетки наполнителя лег-
ко доказывается измерением количества связанного каучука в резине [115,
117]. Это очень специфическое измерение, которое производится с сырыми
смесямй. Суть эксперимента заключается в определении доли каучука, ко-
390 -’v- Глава 8. Усиление эластомеров дисперсными наполнителями
торый не может быть экстрагирован хорошим растворителем [118]. Неболь-
шое количество резиновой смеси, предварительно взвешенное, помещается
в толуол и подвергается экстракции при комнатной температуре. Образцы
набухают, но обычно не растворяются, окружающий их растворитель регу-
лярно обновляется для обеспечения оптимальной экстракции, а образцы
взвешиваются для контроля над процессом экстракции. После 1 или 2 недель
экстракция завершается, и образцы высушиваются и взвешиваются. Разница
в массе соответствует растворимой части эластомера, то есть цепям, которые
были слабо адсорбированы на поверхности техуглерода.
При увеличении температуры экстракции количество связанного каучу-
ка уменьшается, и выше примерно 80°С образцы полностью растворяются,
показывая исчезновение непрерывной сетки агрегатов техуглерода с эласто-
мерными цепями.
Так как количество связанного каучука характеризует адсорбцию элас-
томера на поверхности наполнителя, оно сильно зависит от дозировки на-
полнителя, доступной поверхности и структуры, а это параметры, которые не
могут быть измерены независимо [119]. Тем не менее, при данном содержа-
нии, площади поверхности и структуре техуглерода количество связанного
каучука зависит от поверхностной энергии техуглерода [120].
б. Химическое связывание поверхностью
До 1970-х в основном считалось, что усиление эластомеров техуглеродом
имеет химическую природу [121]. Предполагалось, что кислотные группы
на поверхности техуглерода при взаимодействии с основными функцио-
нальными группами натурального каучука образуют сильную ковалентную
связь, которая и отвечает за усиление.
В 1970-х печной способ производства постепенно заменил канальную
технологию. Но даже притом, что печные марки техуглерода имеют в 10 раз
меньше поверхностных кислотных групп, их усиливающая способность
остается неизменной или увеличивается. С другой стороны, синтетические
эластомеры, которые, очевидно, имеют больше основных функциональных
групп, чем натуральный каучук, также прекрасно усиливаются техуглеро-
дом. К тому же окисление поверхности техуглерода или получение привитого
техуглерода [4] приводит к уменьшению усиливающей способности.
Тем самым стало очевидно, что химическая реакция кислотных групп
поверхности техуглерода с основными функциональными группами нату-
рального каучука не отвечает за усиление. Поэтому недавно разработанный
механизм «молекулярного проскальзывания», предложенный Данненбергом
и основанный на идее молекулярной адсорбции, был сразу и полностью
принят [5, 122].
Эти наблюдения не позволяют сделать полное опровержение теории
химического усиления. Образование химической связи за счет кислот-
но-основной реакции отвергается, но по данным Медалии и Крауса [41]
IV. Смесь как нанокомозит эластомера и наполнителя -*\г 391
в смесях, содержащих техуглерод, могут происходить другие химические
реакции. Например, разрушение эластомерной цепи во время смешения
может приводить к образованию радикалов, которые могут реагировать
с поверхностью техуглерода [123]; также должно быть рассмотрено прямое
связывание серой эластомера и техуглерода [77].
2. Кремнекислотные наполнители
а. Реакция кремнекислота-силан
В отличие от техуглерода для достижения усиления эластомеров, содер-
жащих кремнекислоту, необходимо использовать связывающий агент. На-
иболее широко распространенным связывающим агентом является ТЭСПТ,
триэтоксисилилпропилтетрасульфид.
ТЭСПТ является бифункциональной молекулой с триэтоксисилильной
функциональной частью, реакционноспособной по отношению к сила-
нольным группам кремнекислоты, и полисульфидной, которая реагирует с
эластомерными цепями.
Рис. 4. Содержание ТЭСПТ около 1 Si (OEt)3Ha нм2, что примерно соответствует
полному покрытию поверхности кремнекислоты. Таким образом, для осаж-
денных марок кремнекислоты с содержанием 6-10 Si-OH/нм2 множество
поверхностных силанольных групп остается непрореагировавшими.
Из-за того, что температуры реакций несколько отличаются, реакция
ТЭСПТ с поверхностью кремнекислоты происходит, главным образом,
в процессе смешения, в то время как реакция полисульфидной части имеет
место в течение вулканизации [85, 86] (рис. 4).
б. Адсорбция полимера
Когда связывающий агент используется для образования ковалентной
связи между поверхностью кремнекислоты и эластомерными цепями, он
также ограничивает адсорбцию полимера из-за экранирующего эффекта.
392 -*v- Глава 8. Усиление эластомеров дисперсными наполнителями
Таким образом, в соединениях кремнекислота-силан-эластомер сетка на-
полнителя будет гораздо более слабой, чем в системах техуглерод-эластомер.
Так или иначе, она остается качественно такой же, и подвижность эласто-
мерной цепи также ограничивается ближайшими соседними агрегатами на
поверхности кремнекислоты [124].
Очевидно, что при добавлении любого связывающего агента адсорб-
ция полимера всё равно будет происходить [125, 126], а из-за высокой
полярности кремнекислоты некоторое взаимодействие между ее агрега-
тами будет иметь место и будет образовывать добавочную сетку напол-
нитель-наполнитель. Эти эффекты не будут происходить в системах,
усиленных кремнекислотой, когда используется адекватное количество
связывающего агента.
Определение количества связанного каучука также применимо к резино-
вым смесям, содержащим кремнекислоту, даже с учетом того, что многочис-
ленные возможные взаимодействия, естественно, затрудняют интерпретацию
полученных результатов [127, 128].
V. МЕХАНИЧЕСКИЕ СВОЙСТВА НАПОЛНЕННЫХ РЕЗИН
А. Механические свойства резиновых смесей
Увеличенный срок эксплуатации, ожидаемый от усиле-
ния резин дисперсными наполнителями, относится, естественно, только
к вулканизатам. Введение усиливающих наполнителей сильно изменяет
вязкость сырых смесей, придавая пластические свойства, необходимые для
их переработки.
1. Вязкость
Общепринято, что эластомеры с объемной долей наполнителя ф
имеют вязкость согласно [129]:
Т| = По (1 + 2.5-ф + 14.1ф2)
Следует вспомнить что, это отношение было установлено для твердых
сферических частиц, имеющих какое-либо взаимодействие друг с другом
и/или окружающей их средой. Как уже подробно обсуждалось в этой статье,
усиливающие системы весьма далеки от этого, агрегаты техуглерода или
кремнекислоты являются высоко структурированными, и эластомерные
цепи сильно адсорбируются поверхностью техуглерода.
2. Окклюдированный каучук
Приблизительная оценка и более точная корректировка вза-
имодействия эластомера с техуглеродом или кремнекислотой очень сложна.
С другой стороны, ее можно упростить, если рассматривать действительный
V. Механические свойства наполненных резин 393
объем агрегата в смеси, поэтому было предложено использовать уточненное
значение объемной доли наполнителя фс, включающее в себя влияние струк-
туры наполнителя, получаемое из метода адсорбции ДБФ [130-132].
1 +
1+0.02139 DBP
1.46
Для Т] = »70 • (1 + 2.5-& +14.1*0С2)
Значение фс отражает «фактический» размер агрегатов наполнителя в сме-
си, оно включает, разумеется, сам объект наполнителя плюс значительный
объем полимера, который не участвует в деформации за счет экранирования
неровностями агрегатов. Эту часть полимера, которая не деформируется,
обычно называют окклюдированной [131, 133, 134].
Однако окклюдированный каучук нельзя путать с той частью полимера,
молекулярная подвижность которой изменяется за счет адсорбции. Окклю-
дированный каучук, который захватывается в агрегатах фрактальной струк-
туры, представляет собой только ту часть объема, молекулярное движение
которой замедлено.
Количество окклюдированного каучука и вязкость смеси возрастают
с увеличением структурности наполнителя и его содержания. Существует
и обратная зависимость: эффективная площадь поверхности наполнителя
влияет на вязкость сырых смесей.
3. Зависимость вязкости от сдвига, неньютоновские
свойства
Присутствие в смеси усиливающих наполнителей также уве-
личивает неньютоновскую составляющую поведения эластомеров. Данный
эффект возникает в основном из-за того, что введение наполнителей в эласто-
меры уменьшает объем деформируемой фазы. Как будет обсуждено далее, это
уменьшение не ограничивается действительным объемом наполнителя, но
также должно включать в себя существование окклюдированного каучука.
Таким образом, когда наполненные смеси подвергаются сдвиговым усилиям,
из-за более низкого объема, доступного для деформации, фактическая вели-
чина деформации и ее скорость гораздо выше, чем у ненаполненных смесей
[1, 134]. Это явление обычно называют эффектом усиления деформации.
Очевидно, что усиление деформации характерно не только для усиленных
систем, но и для любого наполненного полимера.
Влияние наполнителя не ограничивается увеличением неньютоновской
составляющей поведения эластомеров. При очень низких скоростях сдвига
у наполненных сырых смесей появляется также дополнительное увеличение
вязкости, которое не может быть объяснено усилением деформации. Этот
эффект обычно приписывается существованию сетки наполнителя: прямое
связывание усиливающих объектов адсорбированными цепями эластомера
обеспечивает увеличение силы, необходимой для разрушения. Очевидно, что
394 -’V Глава 8. Усиление эластомеров дисперсными наполнителями
это влияние может наблюдаться только при очень низких значениях дефор-
мации, потому что очень небольшое увеличение межагрегатных расстояний
незамедлительно вызывает десорбцию связывающих эластомерных цепей.
Б. Механические свойства вулканизатов
7. Введение
Стадия вулканизации для чистых эластомеров обеспечива-
ет создание серных связей между эластомерными цепями и связывает их
в пространственную сетку. Считается, что на вулканизацию наличие усили-
вающих наполнителей значительно не влияет, и она преобразует пластичную
резиновую смесь в вязкоупругий вулканизат.
Для смесей, содержащих кремнекислоту, высокая температура вулкани-
зации обуславливает химическую реакцию полисульфидной части связы-
вающего агента с эластомером, что обеспечивает образование ковалентной
химической связи между каучуком и поверхностью кремнекислоты.
Как и в сырых смесях, усиление деформации, обусловленное ограничен-
ным объемом фактически деформируемой фазы, остается главным резуль-
татом введения наполнителя. Для каждой конкретной макроскопической
деформации действительная деформация полимерной матрицы будет всегда
гораздо выше, и очевидно, что она зависит от объема и структуры наполни-
теля, которая определяет объем окклюдированного каучука.
Уравнение, учитывающее вязкость, обычно записывается для модуля
Юнга (или модуль сдвига) G* [129, 132, 135]:
G* = Gq (1 + 2.5 фс+14.1ф2)
где G*o — модуль сдвига ненаполненной вулканизованной матрицы при том
же значении деформации сдвига. Очевидно, что можно изменять модуль путем
изменения условий вулканизации, обеспечивающих большее или меньшее
количество серных поперечных связей и, таким образом, более высокие или
низкие значения G*. В любом случае, как видно из уравнения, вулканизация
изменяет значение G*o, но практически не влияет на усиление как таковое.
2. Свойства при малых деформациях, определение
динамических вязкоупругих свойств
а. Эффект Пейна
Усиленные вулканизованные образцы проявляют явные вязкоупругие
свойства, которые обычно исследуются с помощью динамических испыта-
ний. В таких экспериментах образец подвергается периодической синусои-
дальной сдвиговой деформации у (при заданной частоте (О и температуре Т).
Динамический модуль сдвига образца G* является комплексным и может
быть записан как сумма модуля накопления G' и модуля потерь G"
V. Механические свойства наполненных резин -•м- 395
g‘(y)=g'(yHg"(y)
Динамический модуль накопления G' с увеличением у изменяется
следующим образом: при очень низкой деформации сдвига G' является
постоянным (участок 1 на рис. 5), затем сильно падает (участок 2 на рис. 5)
и выходит на плато (участок 3 на рис. 5). Такое изменение модуля накоп-
ления обычно называется эффектом Пейна [136].
Рис. 5. Схематическое изображение изменения модуля накопления G' и модуля
потерь G"
Такой характер изменения G' соотносится с ходом кривой изменения G"
которая проходит через максимум. Угол сдвига фаз напряжения и дефор-
мации 5 определяется как:
Такое изменение значений G' и G" в интервале амплитуды деформации от
0,1 до 0,5 имеет крайне важное значение, так как эта область соответствует
наиболее распространенному применению наполненных резиновых изделий,
например, таких как протектор шин [137].
Модуль потерь G" нельзя путать с гистерезисными потерями, выраже-
ние которых полностью зависит от условий испытания изделия: гистере-
зисные потери пропорциональны G" (при постоянной деформации) или
С”/G*2~ tan(5)/G' (при постоянном напряжении) или tan(5) (при посто-
янной энергии).
Для напряжения также важно то, что наполненные эластомеры являются
сложными термореологическими системами: изменение модулей накопле-
396 “'V Глава 8. Усиление эластомеров дисперсными наполнителями
ния и сохранения не подчиняется одинаковым законам для разной частоты
и температуры [138].
б. Механизм
Эффект Пейна общепринято считать механическим следствием про-
грессирующей деструкции сетки наполнителя под действием деформации
сдвига.
Применимость эффекта Пейна к сетке наполнителя строго подтвержда-
ется тем фактом, что комплексный модуль паст техуглерода, полученных из
техуглерода и низкомолекулярных масел, практически не изменяется при
очень низкой сдвиговой деформации [139]. Очевидно, что при увеличении
деформации комплексный модуль для паст техуглерода падает очень бы-
стро и гораздо медленнее для наполненных резин по причине постепенной
десорбции эластомерных цепей.
Модель молекулярного проскальзывания Данненберга, которая ниже
будет обсуждена более детально, дает хорошее схематическое представление
о молекулярном механизме, ответственном за эффект Пейна. Цифры О. ®
и ® относятся к рисунку 5.
О При равновесии эластомерные цепи адсорбированы на поверхнос-
ти наполнителя (состояние ©). Увеличение деформации вызывает
прогрессирующее удлинение сегментов эластомерной цепи, которые
связывают частицы наполнителя (состояние О)- Очевидно, что это
удлинение гораздо выше макроскопической деформации из-за эффек-
та усиления деформации. При очень низких значениях деформации
энергия макроскопической деформации запасается в удлиненных
цепях как упругая энергия и, таким образом, может быть полностью
возвращена, когда деформация уменьшается: модуль потерь G" низок
и является константой.
® При более высоких деформациях необходимо разложить цикл испы-
тания на составные части. В течение первого удлинения запасенная
упругая энергия превышает энергию адсорбции, и эластомерные цепи
постепенно десорбируются с поверхности наполнителя (состояние 0),
черной звездочкой помечены участки десорбции)
V. Механические свойства наполненных резин
397
Эта десорбция удлиняет полимерные сегменты, которые связы-
вают непосредственно взаимодействующие частицы наполнителя.
Во-первых, модуль накопления (?', который обратно пропорционален
длине связывающих цепей, уменьшается; во-вторых, часть упругой
энергии, запасенной в деформированных цепях, тратится на моле-
кулярную подвижность и механические потери, что проявляется
в возрастании G"
Уменьшение деформации, которое следует сразу же после первого
удлинения при более высоком проценте деформации, не приводит к
точно такому же первоначальному состоянию ф; действительно, за
короткое время цикла динамической деформации адсорбция не мо-
жет достичь равновесного состояния и остается незавершенной, как
показано состоянием (D.
Таким образом, в течение фазы О эластомерные цепи, связыва-
ющие частицы, подвергаются циклам адсорбции-десорбции между
«псевдоравновесными» состояниями (D и (D:
Очевидно, что десорбция эластомера происходит постепенно из-за
очень широкого распределения межагрегатных расстояний, которое
вызывает также и широкое распределение связывающих сегментов
эластомера. Это объясняет равномерное понижение модуля накоп-
ления G' для смесей и вид кривой для более коротких молекул, таких
как масла.
398 Глава 8. Усиление эластомеров дисперсными наполнителями
О Постепенно под действием десорбции происходит усреднение длин
сегментов эластомера, связывающих частицы наполнителя. Такая
гомогенизация длин и стабилизация модуля будут обсуждены детально
в следующем разделе.
в. Гистерезис
Из приведенного механизма следует, что на изменение G' и G” прямо
влияют межагрегатные расстояния [108] и сила адсорбции цепей эластомера
на поверхности наполнителя.
г. Межагрегатные расстояния
Любое изменение в составе смеси или ее переработке, которое затрагивает
распределение расстояний между агрегатами, будет влиять на гистерезис:
чем меньше среднее расстояние, тем выше гистерезисные потери и наоборот
[140, 141].
Повышение степени наполнения, несомненно, сокращает межагре-
гатные расстояния. При уменьшении среднего размера агрегатов напол-
нителя (при одинаковой степени наполнения) количество усиливающих
объектов увеличивается и уменьшается среднее расстояние между агрега-
тами. Повышенная структурность обеспечивает (при одинаковой степени
наполнения) более низкие межагрегатные расстояния. Таким образом,
повышенные степень наполнения, площадь поверхности (то есть меньший
размер агрегатов) или структурность придают смесям более высокий уровень
гистерезисных потерь.
С другой стороны, смеси с низким гистерезисом могут быть получены
путем подбора режима смешения таким образом, чтобы увеличить меж-
агрегатные расстояния. Увеличение времени смешения, 2-стадийный режим
или методы с приготовлением маточной смеси используются для получения
смесей с уменьшенным гистерезисом [23, 104].
д. Адсорбционная прочность
Связывающий агент, покрывающий поверхность наполнителя, сильно
уменьшает адсорбцию эластомера и, таким образом, снижает гистерезис
смесей. Это частично объясняет низкое значение гистерезиса для смесей,
содержащих кремнекислоту [140].
Для снижения гистерезиса смесей также применяются эластомеры
с повышенным содержанием функциональных групп, такие эластомеры
имеют особые химические группы, которые реагируют с поверхностью на-
полнителя. Эти реакции снижают гистерезис за счет уменьшения количества
несвязанных полимерных цепей и экранирования поверхности наполнителя
[142, 143].
III. Морфологическое и физико-химическое описание наполнителя Jv- 399
3. Свойства при больших деформациях
а. Наблюдения
При больших деформациях динамические испытания вязкоупругих свойств
не могут быть произведены с большой точностью из-за нагрева образца в ходе
эксперимента. Поэтому свойства наполненных резин при больших деформациях
исследуются при помощи одноосного растяжения [144] (рис. 6).
Рис. 6. Зависимости напряжения и модуля Юнга от деформации для резин с усили-
вающими наполнителями (по Медалии и Краусу [41]).
При проведении динамических испытаний с целью установления зависимости
напряжение-деформация явное уменьшение модуля Юнга Е + о/е наблюдается
при увеличении деформации (от 0 до £ ~ 1). Это соответствует ранее описанным
фазам О и ®, даже если фазу О наблюдать затруднительно (см. рис. 5).
При более высоких процентах деформации (е > 1) модуль Юнга возрастает
и достигает «псевдомаксимума» только перед самым разрывом образца.
Самым значительным явлением, обнаруженным при динамических испы-
таниях, стал эффект понижения напряжения, также называемый эффектом
Маллинза1) [145, 146]. В этом эксперименте образец резины растягивается до
величины деформации е4 и возвращается в состояние с нулевой деформацией,
а затем растягивается снова. Для деформации ниже е4 кривая о-е образца
располагается значительно ниже первоначальной и совпадает с ней вновь
при достижений Снижение напряжения зависит от начального уровня
деформации, эффект может быть частично уменьшен за счет термической
обработки, но он не может быть нейтрализован полностью (рис. 7).
1> Существенно раньше Маллинза этот эффект был тщательно исследован и подробно описан
Г.А. Патрикеевым [Общая химическая технология, т. 2. Под ред. С.И. Вольфковича. Государ-
ственное научно-техническое издательство химической литературы. - М. - Л., 1946 г., 491 с.
(В этой книге есть глава XXII «Технология каучука и резины», написанная Г.А. Патрикеевым.
В этой главе на с.с. 407 - 409 в разделе «Изменение структуры резины при растяжении» описано
явление размягчения и приведены графики).]
400 -* V Глава 8. Усиление эластомеров дисперсными наполнителями
Рис. 7. Схематическое изображение эффекта понижения напряжения.
Рис. 8. Кривая напряжение-удлинение для резин: ненаполненной, содержащей
графитизированный и усиливающий техуглерод.
б. Интерпретация экспериментальных данных
Парадокс усиления дисперсными наполнителями заключается в одно-
временном повышении модуля и относительного удлинения. Этот факт четко
виден при сравнении кривых напряжение-деформация для ненаполненного
эластомера и эластомеров, содержащих техуглерод (рис. 8).
Возрастание модуля является логическим следствием усиления дефор-
мации, обусловленного заменой части деформируемой эластомерной фазы
жесткими частицами наполнителя: для макроскопической деформации
III. Морфологическое и физико-химическое описание наполнителя -* V 401
Е локальная деформация эластомерных цепей, связывающих частицы на-
полнителя, гораздо выше. Усиление деформации должно также вызывать
точно такое же уменьшение относительного удлинения, которого, однако,
не происходит: в этом и заключается парадокс.
Если данный вопрос и остается по-прежнему нерешенным, один из путей
обойти этот парадокс заключается в том, чтобы рассматривать совместное
распределение напряжения.
В начале 1960-х Вики был, вероятно, первым, кто предложил рассматри-
вать техуглерод как часть полифункциональной сетки [1, 2]. В его модели
агрегаты техуглерода были химически связаны эластомерными цепями
и составляли то, что Медалиа и Краус описывают как «гигантскую мульти-
функциональную структурированную» сетку [41].
Даже с учетом того, что модель Вики пытается объяснить усиление мо-
лекулярной причиной, по-прежнему трудно аргументировать то, что она
обеспечивает массовое локальное распределение напряжений: когда самая
короткая цепь достигает своей конечной растяжимости, действительно, не
очевидно, что большая часть напряжения распределяется на другие цепи.
Кроме того, модель Вики предполагает химическую связь эластомерных
цепей с наполнителем, что является как минимум спорным и дискутируется
(смотри предыдущий текст). В любом случае, следует упомянуть, что разру-
шение цепи в процессе удлинения было показано с помощью ESR [147].
В конце 1960-х, Данненберг предложил абсолютно новый подход
к пониманию усиления, представив модель «молекулярного проскальзы-
вания», которую мы ранее использовали для иллюстрирования эффекта
Пейна [5, 118]1J. В противоположность Вики, Данненберг предполагает,
что взаимодействие между эластомером и техуглеродом обусловлено,
главным образом, адсорбцией, а не химической связью. Из-за низкой
энергии и обратимости адсорбции может происходить продолжительное
изменение контактов эластомер-наполнитель и, таким образом, усред-
нение длин сегментов, которое обеспечивает локальное распределение
напряжений [41, 122].
О Гораздо раньше Данненберга, в 1944 г., Александров и Лазуркин [Александров А.П.,
Лазуркин А.С. Прочность аморфных и кристаллизующихся каучукоподобных полимеров //
ДАН СССР. 1944. Т. 45, № 7. С. 308-311] предложили модель, аналогичную той. что рассмат-
ривает Данненберг.
402 -* V Глава 8. Усиление эластомеров дисперсными наполнителями
Цепи разной длины
Цепи усредненной длины
Стоит отметить, что концепция «гигантской мультифункциональной
структурированной» сетки, объединенная с моделью «молекулярного сли-
пания», предлагает возможный механизм усреднения длин связывающих
цепей и дает более удовлетворительное объяснение парадокса усиления.
Таким образом, возрастание модуля во время фазы © обусловлено распре-
делением напряжения между связывающими цепями.
Очевидно, что полученное усреднение прямо зависит от максимальной
деформации, для которой резина предназначена. Другими словами, длины
сегментов цепи выравниваются для любого значения деформации ниже
максимального, при более высокой деформации должно происходить новое
усреднение [148]. Это соответствует так называемому эффекту понижения
напряжения, о котором говорилось ранее [146].
Распределение напряжения за счет усреднения длин сегментов ограничива-
ется, естественно, количеством связывающих сегментов между усиливающими
объектами; когда все связанные цепи достигают приблизительно одной длины,
модуль находится у своего максимума. Дальнейшее увеличение деформации
вызывает общее уменьшение количества самых коротких цепей за счет их раз-
рушения, и, как следствие, уменьшается количество связывающих сегментов
(фаза О). Каждая оставшаяся цепь должна выдерживать увеличенную нагруз-
ку, что вызывает их массовое отслоение от частиц наполнителя и макроскопи-
ческое разрушение. Выделение наполнителя под действием деформации было
доказано прямым наблюдением с помощью ТЕМ [149]
11 Изложенная концепция усиления содержит в себе существенные противоречия. Ос-
новное связано с наличием на поверхности частиц активного наполнителя слоя толщиной
до 5 нм с пониженной подвижностью и повышенной жесткостью. Совершенно не ясно,
как в таком «псевдостеклообразном» состоянии может проходить адсорбция и десорбция с
поверхности отдельно взятой макромолекулы по модели Данненберга. Кроме того, многие
наблюдаемые на эксперименте закономерности объяснены не строго, а умозрительно. Более
реалистичная модель и теория явления усиления изложены в работах [Гамлицкий Ю.А.,
III. Морфологическое и физико-химическое описание наполнителя -*и* 403
В. Применение
Как говорилось во введении, усиление эластомеров может
считаться особым явлением, потому что оно увеличивает срок эксплуатации
изделия. И для того, чтобы дать некоторую практическую иллюстрацию
различных идей, которые были обсуждены ранее, мы представим некоторые
результаты усиления техуглеродом материалов, подвергающихся износу,
например усиленных резин для протекторов шин.
Рис. 9. Зависимость индекса износостойкости от содержания техуглерода и площади
поверхности. (По Соне [108].)
Содержание техуглерода в резине, площадь его поверхности и структу-
ра в основном увеличивают износостойкость. Для очень высокой степени
наполнения или площади поверхности наблюдается значительное падение
износостойкости (рис. 9), этот эффект можно объяснить недостаточным
распределением [108]. Поверхностная активность техуглерода, что следует из
Басс Ю.П. К описанию явления усиления наполненных эластомеров. - Инженерно-фи-
зический журнал, 2003, т. 76, № 5, с. 101-105; Гамлицкий Ю.А., Швачич М.В. Прочность
резины. Модель и расчет. Высокомолекулярные соединения, Серия А, 2005, т. 47, № 4, с.
660-668; Yu. A. Gamlitsky and M.V. Shvachich. Bubber Strength: Model and Calculation.
Polymer Science, Series A, vol. 47, No. 4, 2005, pp. 396-402]. которые позволяют получать
достаточно строгие количественные оценки.
404
Глава 8. Усиление эластомеров дисперсными наполнителями
115
& 110
® 105
с 100
Сб
g 95
1 90
I 85
ЕС
S 80
75
0.200 0.220 0.240 0.260 0.280 0.300 0.320 0.340 0.360
Содержание водорода, %
Рис. 10. Зависимость износа протектора от содержания водорода втехуглероде [81].
140
Сб
Я
О.
Я
§ 120
х
5S
О
S но
о
* 100
ЕС
я
S
на основе
маточной смеси
/ О
/ q'-'-Q.
,• / О на основе
/ / ° сухой смеси
/
е///
а'
I ISAF I I SAF I I спец, марки I
90
80 ЮО 120 140 160 180 200 220 240
Площадь поверхности по ЦТАБ (м2/г)
Рис. 11. Сравнение износостойкости резин на основе сухих и маточных смесей (По Соне,
Ишигуро, Акимото и др. [104].)
содержания водорода, также имеет значительное влияние на износостойкость [81 ]
(рис. 10). Распределение техуглерода также влияет на сопротивление ис-
тиранию, и максимум, наблюдаемый для высоких значений содержания
техуглерода и площади поверхности, может быть сильно смещен за счет
использования специальных методов смешения, таких как, например, при-
готовление маточных смесей [104, 150] (рис. И).
I.
ГЛАВА
9
НАУКА О РЕЦЕПТУРОСТРОЕНИИ
РЕЗИН
БРЕНДАН РОДЖЕРС и УОЛТЕР ВЕДДЕЛЛ
Экссон Мобил Хемикал Компани
Хьюстон, Техас
ВВЕДЕНИЕ
Терминомрецептуростроение широко пользуются в шинной
н резиновой промышленности. Рецептуростроение - это наука о материалах,
касающаяся модификации каучуков, эластомеров или смесей полимеров
и других материалов с целью оптимизации свойств конечных изделий для
выбранных областей применения или установленных параметров эксплу-
атации. Поэтому рецептуростроение - это комплексная наука, требующая
знаний в таких направлениях как физическая, органическая и неоргани-
ческая химия, химия полимеров и кинетика химических реакций. Научный
сотрудник, имея дело с созданием резиновых композиций, сталкивается с оп-
ределенными задачами и ограничениями. Требования к эксплуатационным
свойствам изделий диктуют выбор ингредиентов. Такие материалы должны
быть экологически безопасны, не наносить вреда здоровью и удовлетворять
требованиям по технике безопасности, перерабатываться на производствен-
ном оборудовании и быть рентабельными.
Резины, как композиции, обладают уникальными свойствами, не прояв-
ляемыми другими материалами: способностью гасить колебания, высокой
эластичностью и износостойкостью. Поэтому резины нашли применение
в таких изделиях как шины, конвейерные ленты, элементы фундаментов
зданий, детали двигателей автомобилей, широкий спектр изделий бытового
назначения. Ингредиенты для формирования смесей каучуков могут быть
разбиты на 5 категорий:
1. Полимеры
2. Наполнители
Натуральный каучук, синтетические
полимеры
Технический углерод, каолин, крем-
некислотные наполнители, карбонат
кальция
406 -* V Глава 9. Наука о рецептуростроении резин
3. Стабилизирующие системы Антиоксиданты, антиозонанты, воска
4. Компоненты вулканизационных Сера, ускорители, активаторы
систем
5. Специальные компоненты Вторичные компоненты, такие как
пигменты, масла, смолы, вспомога-
тельные вещества для переработки,
короткие волокна
Каждый класс материалов рассмотрен в настоящей главе.
II. ПОЛИМЕРЫ
Мировое потребление каучука составляет около 18 мил-
лионов тон, причем примерно 46% приходится на натуральный каучук,
остальное — синтетические каучуки, где 18% составляет бутадиен-стироль-
ный. Синтетические каучуки (47%) включаютют полибутадиеновый каучук
и полимеры специального назначения такие, как полиуретаны, галоген-
содержащие полимеры, полисилоксаны и полиакрилаты. Традиционно, рост
потребления синтетического и натурального каучуков совпадает с ростом
валового национального продукта Северной Америки, Европейских стран
и северо-западных стран Тихоокеанского бассейна. [1,2]
А. Натуральный каучук
Общее мировое потребление натурального каучука разделя-
ется на шинную промышленность (75%), детали автотранспорта (5%), неав-
тотранспортные изделия (10%) и изделия различного назначения такие, на-
пример как медицинские и медико-санитарные (10%). С 1960-х гг качество
изделий из натурального каучука улучшилось преимущественно благодаря
внедрению стандартов, определяющих марки каучука. Натуральный каучук
производится трех основных типов: каучуки технической классификации,
каучуки визуальной оценки и каучуки специального назначения.
Американская ассоциация испытаний и материалов (ASTM) описала
шесть основных категорий натурального каучука технического назначения,
которые производятся и поставляются 34 килограммовыми блоками [3] (Таб-
лица 1). Эти шесть основных категорий натурального каучука технического
назначения более детально разработаны странами производителями. Стан-
дартный Малазийский каучук (SMR), Стандартный Индонезийский каучук
(SIR) и Тайский каучук технического назначения (TTR) представляет до-
ступный ассортимент каучуков. Например, Стандартный Малазийский кау-
чук CV имеет две марки каучука постоянной вязкости: SMR CV50 и CV60 [2].
Марки SMR10 и SMR20 представляют марки каучука со стабилизированной
вязкостью (SMR lOCV и SMR 20CV).
II. Полимеры 407
Таблица 1. Спецификации натурального каучука технического назначения.
Свойства Категории каучука
L CV 5 10 20 50
Примеси (% максим.) 0,050 0,050 0,050 0,100 0,200 0,500
Зола (% максим.) 0,60 0,60 0,60 0,750 1,00 1,50
Летучие вещества (%) 0,80 0,80 0,80 0,80 0,80 0,80
Азот (%) 0,60 0,60 0,60 0,60 0,60 0,60
Пластичность 30 - 30 30 30 30
Индекс сохранения пластичности 60 60 60 50 40 30
Индекс цвета 6,0 - - - - -
Вязкость по Муни - 60 - - - -
Таблица 2. Международные типы натурального каучука и их спецификация.
Тип Натуральный каучук Описание
1 Ребристый «смокед-шит» Коагулированные листы, высушенный и окурен- ный латекс. Доступно 5 видов (RSS1-5)
2 Белый и светлый креп Коагулюм натурального жидкого латекса, валь- цованного для получения крепа
3 Плантационный корич- невый креп Свежие сгустки и другие высококачественные отходы, собранные на плантации
4 Компокреп Сгустки, сборы с дерева и срезы «смокед-шит», вальцованные в креп
5 Тонкие листы коричне- вого крепа Неокуренные листы, влажные пластины, комья и другие сборы с плантаций и мелких хозяйств
6 Толстый листовой креп Влажные пластины, комья и неокуренные лис- ты, вальцованные для получения крепа
7 Плоский корообразный креп Все виды сборов натурального каучука, включая сборы с земли
8 Чистый окуренный листовой креп Вальцованный окуренный каучук, исключитель- но из ребристых окуренных листов
Ассоциация производителей каучука представляет перечень стандартов
качества и упаковки для категорий латексов натурального каучука.
В Таблице 2 представлены восемь типов каучука, входящие в их спе-
цификации. Коагулированный латекс отлистован, высушен и упакован
в кипы до 113,5 кг. Классификация производится визуально. Лабора-
408 -*V- Глава 9. Наука о рецептуростроении резин
тории обеспечения качества имеют системы визуальных стандартов
оценки [4].
Третья категория натурального каучука - специальные материалы, кото-
рые включают жидкий низкомолекулярный каучук, привитые сополимеры
метилметакрилатом, маслонаполненный натуральный каучук, депротени-
зированный натуральный каучук, эпоксидированный натуральный каучук,
и натуральный каучук высокой степени перерабатываемости.
Потребление НК увеличилось, главным образом, в производстве совре-
менных радиальных шин. Бернард и сотрудники [5] сравнили потребление
натурального каучука в радиальных шинах повышенной проходимости
с аналогичными диагональными и отметили следующее увеличение:
Натуральный каучук (%) Диагональные Радиальные
Протектор 47 85
Обкладочные (прорезиненные)
слои каркаса 70 100
Боковина 43 58
Причины этого увеличения были отнесены к росту прочности сырых
резин, адгезии между компонентами, повышению сопротивления разди-
ру, более низким температурам в шинах при динамических воздействиях
в процессе эксплуатации, и пониженным сопротивлением качению шины
с увеличением экономичности автомобильного топлива.
Рост потребления натурального каучука связан с расходом примерно
21 кг на шину радиальной конструкции против приблизительно 9 кг — на
диагональную грузовую шину. Смеси из натурального каучука также на-
ходят применение в производстве высококачественных конвейерных лент,
где требуемые качественные характеристики такие же, как и в протекторах
грузовых шин. Низкие гистерезисные потери, высокая прочность на разрыв
и хорошая износостойкость необходимы для обоих видов изделий.
Б. Синтетические эластомеры
Классификация синтетических каучуков определена Меж-
дународным Институтом Производителей синтетического каучука (IISRP).
Для бутадиен-стирольного каучука, полиизопрена и полибутадиена установ-
лена номерная градация, по которой классифицируют основные свойства
полимера [6]. Например, серия IISRP 1500 указывает на то, что бутадиен
стирольный каучук получен методом низкотемпературной эмульсионной
полимеризации (т. е. ниже 10°С) и не содержит каких-либо пигментов. Се-
рия полимеров 1700 описывает маслонаполненный бутадиен-стирольный
каучук, полученный методом холодной эмульсионной полимеризации.
Таблица 3 представляет основные серийные номера, применяемые IISRP.
Система нумерации для стереорегулярных эластомеров, полученных методом
полимеризации в растворе, приведена в Таблице 4.
II. Полимеры -*v- 409
Таблица 3. Классификация синтетических каучуков по IISRP,
Кл ассификацион - ный номер Описание
1000 Бутадиен-стирольный каучук высокотемпературной эмуль- сионной полимеризации (температура полимеризации выше 38°С), без пигментов.
1500 Бутадиен-стирольный каучук низкотемпературной эмуль- сионной полимеризации (температура полимеризации ниже 10°С), без пигментов.
1600 Бутадиен-стирольный каучук низкотемпературной полимери- зации/маточная смесь с техническим углеродом/ 14 массовых частей масла (максимум)
1700 Маслонаполненный БСК низкотемпературной эмульсионной полимеризации
1800 БСК низкотемпературной эмульсионной полимеризации/ маточная смесь с техническим углеродом/ более 14 массовых частей масла
1900 Эмульсионные смоло-каучуковые маточные смеси
Таблица 4 . Стереорегулярные эластомеры, полученные методом растворной
полимеризации по IISRP.
Бутадиен и сополимеры Изопрен и сополимеры
Полимер без добавок 1200-1249 2200-2249
Маслонаполненный 1250-1299 2250-2299
Маточная смесь с техническим углеродом 1300-1349 2300-2349
Маточной смеси с маслом и техническим углеродом 1350-1399 2350-2399
Латекс 1400-1449 2400-2449
Другие 1450-1499 2450-2499
Шинная промышленность потребляет приблизительно 60% мирового
производства синтетических каучуков. Наибольший объем потребления
составляет бутадиен-стирольный каучук (БСК), составляющий 65% синте-
тического каучука, используемого в шинах. Полибутадиен (ПБ) занимает
второе место по производимой продукции [1, 2]. Таблицы 5-7 отражают
потребление синтетического каучука по группам продукции. Бутадиен-сти-
рольный каучук нашел широкое применение в протекторах шин благодаря
своим свойствам сцепления и сопротивлению проскальзыванию на мокрой
410 -* V Глава 9. Наука о рецептуростроении резин
Таблица 5. Потребление синтетических каучуков.
Шины 60%
Детали автомобилей 10%
Технические неавтомобильные изделия 9%
Композиции на основе термоэластопластов 6%
Обувь 4%
Конструкционные изделия 3%
Кабель и электропровод 2%
Адгезивы 1%
Прочие изделия 5%
Таблица 6. Потребление бутадиен-стирольного каучука в США.
Изделие Процент от общего потребления
Легковые шины 50
Восстановление протектора шин 13
Грузовые шины 8
Шины специального назначения (авиация, землеройные ма- шины и т.д.) 4
Технические изделия автомобилей 7
Изделия прочего назначения (бытового назначения, медицинское оборудование, строительство) 18
Таблица 7. Потребление полибутадиена в США.
Изделие Процент от общего потребления
Легковые шины 45
Грузовые шины 28
Протекторные заготовки для восстановления шин 4
Шины специального назначения (авиация, землеройные ма- шины и т.д.) 1
Технические изделия 2
Изделия прочего назначения (полимерные смеси, полимерные модификаторы с полистиролом или тройной сополимер стирол- акрилонитрил-бутадиен) 20
II. Полимеры 411
поверхности при сохранении хорошей стойкости к истиранию. Полибутадиен
часто используют в протекторах, боковинах и некоторых деталях каркаса
шин благодаря его хорошей стойкости к истиранию, износостойкости про-
тектора и повышенному сопротивлению разрастанию трещин. Полибутадиен
может также смешиваться с натуральным каучуком и есть много данных,
что такие композиции имеют улучшенные усталостные свойства и сопро-
тивление разрастанию трещин [7].
Таблица 8. Микроструктура полибутадиена.
Катализатор Доля изомеров, +/-1%
Цис-, % Транс-, % Винил-, %
Li 35 55 10
Ti 91-94 2-4 4
Со 96 2 2
Nd 98 1 1
Ni 96-98 0-1 2-4
Перед рассмотрением характеристик эластомеров, необходимых для
задания каких-либо параметров поведения шин, необходимо выделить два
из них, с помощью которых материаловед может описать полимер: его мак-
ро- и микроструктура.Макромолекулярная структура полимера включает
молекулярный вес, а также распределение поперечных связей, разветвление
цепей полимера и образование кристаллитов. Распределение мономеров
в цепи полимера определяет его микроструктуру. Бутадиен может принимать
одну из трех конфигураций, как показано на рисунке 1. Эти молекулярные
или стереохимические конфигурации могут быть описаны так:
винил-(1,2)
транс- (1,4)
цис-( 1,4)
Третий и четвертый углеродный атомы находятся в боковых группах;
первый и второй - составляют основную цепь.
Атомы водорода присоединены к двойной углерод-углеродной связи
полимерной цепи с противоположных сторон от двойной связи.
Два атома водорода присоединены к двойной углерод-углеродной связи
полимерной цепи по одну сторону от двойной связи.
Таблица 8. показывает вличние каталитических систем на микрострук-
туру полимера [10].
Соотношение количеств каждого из трех стереоизомеров в полимерах,
таких как полибутадиен, может сильно влиять на свойства конечных ма-
териалов. Например, растворные полимеры с литиевым катализатором,
содержащие приблизительно 36% tyuc-конфигурации, легко перераба-
тываются, тогда как полимеры с Ti и Ni катализаторами (92% цис-кон-
фигурации) перерабатываются при заводских температурах труднее, но
412 -* V Глава 9. Наука о рецептуростроении резин
в этом случае получаются резины с лучшей стойкостью к абразивному
износу. Полибутадиен с высоким содержанием (93%) транс-изомеров
- жесткий, кристаллический материал при комнатной температуре. По-
либутадиен с высоким содержанием винильных групп проявляет хорошие
свойства сцепления и торможения на мокрой поверхности в протекторах
шин [7-10].
цис- транс- винил-
Рис. 1. Микроструктура полимера: возможные конфигурации бутадиеновых звеньев
в бутадиен-стирольном каучуке и полибутадиене*\
Нордсайк [9] предложил серии эмпирических правил, которыми можно
пользоваться при создании резин высокого качества. Создавая ряд смесей
из полибутадиена и бутадиен-стирольного каучука, Нордсайк получил
серии композиций, в которых Тс возрастала с -100 до -30 °C. Он отметил
следующие положения:
1. При увеличении Т наблюдается практически линейное снижение
сопротивления износу.
2. Сцепление шин с влажной дорогой улучшается практически линейно
с увеличением Т резин.
3. Добавление модификатора к катализатору в процессе получения
полибутадиена растворной полимеризацией с литием приводит к уве-
личению содержания 1,2-звеньев бутадиена в каучуке и увеличению
Тс. При этом падает сопротивление истиранию и растет сцепление
с влажной дорогой.
4. Введение стирола приводит к улучшению дорожных свойств и сни-
жению износостойкости. Существует линейная зависимость между
содержанием стирола и 1,2-полибутадиеном. Приблизительно две
единицы 1,2-полибутадиена дают тот же эффект сцепления с дорогой,
что и одна единица стирола.
5. Включение звеньев 3,4- в полиизопрен приводит к увеличению Тс
резин и соответствующему улучшению сцепления, а увеличение
процентного содержания 1,2- или 3,4-пипериленовых звеньев
в полипипериленах приводит к увеличению Тс, вызывая снижение
сопротивления истиранием, но улучшение сцепления.
1
Правильнее говорить о расположении соседних групп -СН2 в основной цепи
полимера относительно двойной связи. (Примечание переводчика к рис.1).
II. Полимеры -'V 413
Таблица 9. Характеристика идеализированной протекторной композиции: по за-
висимости tan 8 от температуры.а)
Температурный интервал (°C) Характеристика Рабочий параметр
от -60 до -40 Тс Истирание
-20 - Низкотемпературные свойства
+20 - Сцепление с мокрой поверхностью
ОТ 4-40 ДО 4-60 - Сопротивление качению
ОТ 4-80 ДО 4-100 - Теплообразование
Данные, приведенные Нордсайком [9].
Эти условия позволили разделить зависимости tan8 от температуры на кривой
для протекторной смеси в интервале от -100 до +100 °C на зоны, которые харак-
теризуют рабочие параметры протектора шин (Табл. 9). Такие целевые свойства
сделали возможным развитие концепции «комплексной резины», т. е. каучук мо-
жет быть синтезирован таким образом, чтобы совместить сопротивление качению,
сцепление и изнашиваемость протектора без потери в работе всей шины.
Дэй и Футамура [11] оценили влияние различия содержания 1,2-бута-
диеновых и стирольных звеньев в бутадиен-стирольном каучуке на свойс-
тва резин. Кратко: 1 — увеличение содержания звеньев стирола приводит
к увеличению предела прочности на разрыв резины; 2 — увеличение содер-
жания винильных звеньев приводит к потере как сопротивления раздиру,
так и разрывного удлинения; 3 — при одинаковых ^содержание винильных
и стирольных звеньев не влияет на гистерезисные свойства.
Затем Брентли и Дэй провели исследование по сравнению работы шин на
основе бутадиен-стирольного каучука эмульсионной и растворной полиме-
ризации [12]. Авторы отметили, что полимеры растворной полимеризации,
у которых более узкое молекулярно массовое распределение и более низкая
Тс по сравнению с аналогичными полимерами эмульсионной полимеризации
имеют более низкие гистерезисные свойства резин. Они затем показали, что
растворный бутадиен-стирольный каучук с таким же содержанием стирола,
как и в эмульсионном, показывает более низкое сопротивление качению,
улучшенное сцепление с сухой дорогой и меньшую изнашиваемость протек-
тора. Эмульсионный бутадиен-стирольный каучук, однако, имеет тенденцию
к проявлению лучших тормозных свойств, сцепления резины и управления
на мокрой дороге. Позже Керн и Футамура развили эту работу по оценке
влияния содержания винильных звеньев в растворном бутадиен-стирольном
каучуке и повторным его сравнением с эмульсионным БСК [13]. Несмотря
на то, что работа была проведена для легковых шин, многие из этих принци-
пов могут быть приемлемы для шин других областей применения, таких как
легкие грузовые и тяжелые грузовые шины.
414 -*V Глава 9. Наука о рецептуростроении резин
Таблица 10. Сравнение бутадиен-стирольного каучка растворной и эмульси-
онной полимеризации.
Свойства Эмульсионный БСК Растворный БСК
Вязкость (Ml1+4 при 100°С) 50 57
Время до оптимума вулканизации (мин при 150°С) 40 25
Разрывная прочность (МПа) 26 21
Разрывное удлинение (%) 400 300
Эластичность по отскоку (%) 48 61
Результаты испытаний авторов предоставлены в Таблице 10. Из этих дан-
ных видно, что среднечисленный молекулярный вес, или Мп, промышленного
эмульсионного бутадиен-стирольного каучука, такого как IISRP 1500 или 1712,
обычно колеблется от 90000 до 175000. Исходный молекулярный вес полимера,
полученного методом растворной полимеризации с анионными литиевыми ка-
тализаторами, может, напротив, быть повышен до 250 000 без гелеобразования.
Кроме того, эмульсионный бутадиен-стирольный каучук содержит только около
92% углеводорода каучука из-за присутствия примесей производственного
процесса; растворные же полимеры содержат почти 100% углеводорода. Как
следствие, авторы делают вывод, что среднечисленный молекулярный вес может
быть рассмотрен в качестве ключевого параметра полимерной макроструктуры,
в частности это касается гистерезисных характеристик протектора. Поэтому
различия в макроструктуре эмульсионных и растворных полимеров определяют
большинство свойств протекторов шин.
При рассмотрении растворных полимеров следует учитывать, что микро-
структура полимера в наибольшей степени определяет свойства протектора
шины. Таблица 11 показывает влияние повышения содержания винильных
звеньев от 10 до 50% в полибутадиене в составе протектора на сцепление
с дорогой, сопротивление качению и изнашиваемость [12]. Соответствующее
ухудшение изнашиваемости и улучшение сопротивления качению согласуется
с эмпирическим правилом Нордсайка [9], который объяснил такие тенденции
в изменении свойств шин влиянием температуры стеклования полимера.
Таблица 12 показывает, как микро- и макроструктура полибутадиена,
т.е. молекулярный вес, Мш и Мп, полидисперсность и разветвленность могут
влиять на перерабатываемость полимера [14]. Исследование полибутадиенж.
полимеризованного с помощью кобальтовых и неодимовых катализаторов,
показало взаимосвязь между полидисперсностью, или молекуляно-весовыя
распределением, и возрастанием релаксации напряжений. Рост релакса-
ции напряжений, измеренного с помощью вискозиметра Муни, приводит
к большим трудностям при переработке смесей, контроле размера заготовки,
«нерва» и усадки экструдата или кландрованного листа [15].
II. Полимеры -* V 415
Таблица 11. Влияние содержания винильных звеньев в полибутадиене на
свойства шин.
Содержание винильных звеньев 10% 50%
Температура стеклования -90 °C -60 °C
Свойства покрышкиа)
Сцепление с влажной дорогой 100 120
Оценка сопротивления качению 100 95
Оценка изнашиваемости покрышки 100 90
а) Чем выше оценка, тем лучше.
Таблица 12. Макроструктура и вязкость по Муни [14].
Катализатор Образец полимера Mw Мп Mw/Mn MLl+4 Релаксация напря- жения по Муни
Кобальт 1 338 156 2,17 47 4,50
2 318 131 2,43 45 7,50
3 321 125 2,57 46 9,00
4 303 108 2,81 44 14,00
Неодим 1 353 186 2,10 50 5,00
2 381 103 3,70 42 8,00
3 347 87 3,99 44 9,00
4 368 86 4,28 42 10,00
Галогенированный бутил-каучук (ГБК) обычно используется во внутренних
слоях шины и в белых боковинах. Эти эластомеры являются наилучшими для
удерживания воздуха в шине благодаря низкой воздухопроницаемости, а также
высокой стойкости к старению и усталостной выносливости. Хлорированный
(ХБК) и бромированный (ББК) производные бутил-каучука (БК) могут быть
смешаны с другими эластомерами для улучшения кинетики вулканизации
и адгезии между бутилкаучуком и эластомерами общего назначения [16].
Попытки использования полимера на основе галогенированного изо-
бутилена, как основного полимера в протекторных смесях ограничены
несмотря на то, что такие протекторные композиции хорошо проявляют
себя при использовании в зимних условиях и имеют хорошее сцепление
с дорогой. Новый полимер изобутилена, модифицированный п-метилстиро-
лом, затем бромированный, открывает возможность использовать полностью
насыщенную основную цепь с целью обеспечения сопротивления старению
и в то же время улучшения совместимости с эластомерами общего назначе-
ния, такими как натуральный и бутадиен-стирольный каучуки.
416 Глава 9. Наука о рецептуростроении резин
Обычно в рецептуре протектора шины смешивается более одного типа
каучука. Примером является протектор грузовой шины, который должен
обладать не только высокой прочностью, но и хорошей усталостной вынос-
ливостью. В легковых шинах для протекторов может быть использовано до
четырех различных полимеров, т. е. в 100 масс, частях каучука в протек-
торной смеси, может быть 25 частей эмульсионного бутадиен-стирольного
каучука, 25 частей растворного бутадиен-стирольного каучука, 30 частей
полибутадиена и 20 частей натурального каучука. Если рассмотреть типы
растворных бутадиен-стирольных каучуков по группам, каждая с пониже-
нием температуры стеклования на 10 °C, то существуют, по крайней мере,
девять групп специальных видов растворных промышленно доступных
БСК в дополнение к ряду имеющихся полимеров по схожим химическим
процессам синтеза для нужд шинной промышленности [7, 16].
Температура стеклования
Рис. 2. Влияние Тс на сцепление шины с дорогой (Из Saito [18]).
На рисунке 2 показано влияние Тс на тормозные свойства шин на мокрой
дороге. Если требуется улучшение сцепления шин с влажной поверхностью
дороги с минимальным влиянием на сопротивление качению, то влиять
на изменение Тс лучше путем увеличения содержания винильных звеньев
в ПБ, чем стирольных. Напротив, если изнашиваемость является наиболее
важным фактором, то Тс можно отрегулировать путем изменения содержа-
ния стирольных звеньев. Поэтому оптимальная Тс может быть получена
регулированием содержания либо винильных звеньев, либо стирольных
для того, чтобы достигнуть требуемого сцепления шины с мокрой дорогой,
сопротивление качению и изнашиваемости. Показано [17-19], что улучше-
ние износостойкости приводит к ухудшению сцепления (рис. 3).
Промышленные полимеры с высоким молекулярным весом производят
маслонаполненными для облегчения переработки и это одновременно дает воз-
можность готовить резиновые изделия с лучшими механическими свойствами,
II. Полимеры -'V 417
чем изделия из тех же каучуков, но с более низким молекулярным весом [20].
Таблица 13 отражает выборку категорий эмульсионного БСК. Ароматические
масла могут повышать температуру стеклования соответствующего полимера
без масла. Нафтеновые масла проявляют тенденцию к смещению температуры
перехода ниже таковой для маслоненаполненного каучука.
Рис. 3. Взаимоотношение между износом протектора к тормозным показателям.
(По Оберстеру, Боутону и Валайтесу [19]).
Таблица 13. Маслонаполненные эмульсионные БСК [20].
Полимер по IISRP Номинальная вязкость по Муни (MLl+4) Стирол (%) Тип масла Содержание масла (PHR)
1707 50 23,5 Нафтеновое 37,5
1712 50 23,5 Ароматическое 37,5
1720 40 23,5 Нафтеновое 50,0
1721 55 40,0 Ароматическое 37,5
Таблица 14. Классификация масел по ASTM и IISBP для маслонаполненных
эластомеров [6].
Тип Асфаль- тены Содержание полярных ком- понентов (%) Содержание насыщенного углеводорода (%) Категория Константа вязкости
101 0,75 25,0 20,0 Высоко арома- тическое >0,900
102 0,50 12,0 20-35 Ароматическое 0,900
103 0,30 6,0 35-65 Нафтеновое 0,875
I 104А 0,10 1,0 65,0 Парафиновое >0,820
I 104Б 0,10 1,0 65,0 Парафиновое 0,820 макс
418 Глава 9. Наука о рецептуростроении резин
Первичная функция масла в каучуке - упрощение переработки, т. е.
облегчение смешения в закрытом смесителе, повышение однородности
композиции по ее вязкости и улучшение последующего этапа переработки —
экструзии. Специальные масла, используемые для получения маслонапол-
ненных эластомеров, выделены в пять основных групп, которые представ-
лены в Таблице 14.
Содержание акрилонитрила (%)
Рис. 4. Содержание акрилонитрила и способность к маслопоглощению бутадиен-
нитрильными каучуками.
Натуральный каучук, БСК и СКД составляют самую большую долю пот-
ребления полимеров, поэтому некоторые другие каучуки такие, как бутадиен-
нитрильный (БНК), полихлоропрен, бутил и этилен-пропилен-диеновый
эластомер (СКЭПТ) из-за их экономического значения будут рассмотрены
в меньшей степени [2, 6].
Нитрильный каучук (БНК) — сополимер акрилонитрила и бутадиена.
Наиболее важное его свойство — это маслостойкость; поэтому он нашел
широкое применение в таких изделиях, как гидравлические рукава, детали
двигателей машин, где необходима маслостойкость. На рисунке 4 показа-
но влияние содержания акрилонитрильных звеньев на поглощение масла
(масло IRM 903). С другой стороны, БНК обладает плохими эластическими
свойствами при пониженной температуре, что препятствует его применению
в изделиях, работающих в холодном климате.
БНК легко подвергается деструкции на вальцах или в резиносмесителе.
Обычно пептизаторы не нужны, но добавки для предотвращения гелеобра-
II. Полимеры -* V 419
зования необходимы, если температура смешения превышает 140 °C. Из-за
низкой прочности сырых смесей не достигается достаточных сдвиговых уси-
лий в процессе смешения для облегчения введения технического углерода
типа SAF или ISAF. Это объясняет также плохую перерабатываемость на
вальцах. Существенными компонентами для БНК являются антиоксидан-
ты, т.к. БНК легко окисляется на воздухе при повышенных температурах.
Наиболее эффективен полимеризованный 2,2,4-триметилилгидрохинолин.
Антиозонанты и воска не эффективны в составе композиций на основе БНК.
Полихлоропрен получают из ацитилена:
НС^СН — НОС-СН=СН, -УЯ н,с=с—сн=сн,
I
Ацетилен С1
Хлоропрен
(1)
Н2С=С—сн=сн2 Полимеризация> [ —СН2—СН=С—СН2— ]„
С1 С1
Полихлоропрен
или бутадиена
С1—СН2—СН=СН —СН2—С1
Н2С=СН—СН=СН2 сь +
Бутадиен О—СН2—СН—СН=СН2
С1
С1—СН2—СН=СН—СН2—С1 о—сн2—сн—сн=сн,
С1
С1—СН2—СН—СН=СН2 -Я2Я
С1
СН2=С—СН = СН2 + NaCl + Н2О
С1
Хлоропрен
Ацетилен реагирует с образованием винил-ацитилена, который затем
хлорируется до хлоропрена. Далее хлоропрен полимеризуется, образуя
полихлоропрен, который содержит примерно 85% транс-, 10% цис- и 5%
420 -'V- Глава 9. Наука о рецептуростроении резин
винил-хлоропрена. Из-за большого содержания /пргшс-звеньев полихлоро-
прен легко кристаллизуется.
В зависимости от вида полихлоропрен может вулканизоваться с помощью
оксида цинка или магния. Тетраметилтиурам дисульфид может служить
как замедлитель. По маслостойкости полихлоропрен уступает БНК, но тем
не менее значительно превосходит натуральный каучук, БСК или ПБ. Как
и БНК, полихлоропрен нашел широкое применение там, где требуется вы-
сокая маслостойкость, т. е. в таких изделиях как сальники, уплотнители,
рукава, ремни передач автомобильных двигателей.
Бутилкаучук — это сополимер изобутилена и изопрена:
СН3
I
сн2=с + сн2=с—сн=сн2-------------->
I I
СН, СН3
Изобутил Изопрен
(3)
СН3
-f СН2 — С->7?СН2— С=сн — сн2—
СН3 сн3
Бутил-каучук
Изобутилен и изопрен находится в соотношении приблизительно 50/1.
Хлорбутил и бромбутил каучук получают галогенированием бутил каучука.
Бутил и галобутил каучук непроницаемы для воздуха, обладают низкой
способностью к абсорбции воды, хорошей тепло- и озоностойкостью. Как
отмечено ранее, они нашли широкое использование в производстве слоев
радиальных шин, покрытий и изоляции высоковольтных электрических
кабелей, рукавах радиаторов и автомобильных двигателей.
В высоко-прочных на разрыв композициях с бутил каучуком обычно
используется технический углерод марки FEF или GPE. Вулканизующие
системы обычно основываются на ускорителях тиазольного типа таких,
как дисульфид меркаптобензтиазола (ДБТД) и тиурамовых ускорителях—
тетраметилтиурам дисульфид (ТМТД). В композициях с низкой разрывной
прочностью используют каолиновые и кремнекислотные усиливающие
наполнители вместо технического углерода.
Путем сополимеризации этилена и пропилена получают эластомеры,
которые практически инертны из-за отсутствия двойных углерод-угле-
родных связей (СКЭП). Такие полимеры сшиваются пероксидной или
радиационной вулканизацией. Для улучшения реакционной способности
этилен-пропиленового сополимера вводят от 1 до 10% третьего мономера,
получая тройной сополимер, или этилен-пропилен-диеновый сополимер
II. Полимеры -*v 421
(СКЭПТ). Диеновые мономеры, используемые в СКЭПТ: 1,4-гексади-
ен, дициклопентадиен, этилиденнорборнен. Введение ненасыщенного
мономера такого, как этилиденнорборнен, позволяет применять серные
вулканизующие системы.
СКЭПТ обладает хорошей влагоустойчивостью, стойкостью к окислению
озоном и кислородом. Поэтому он применяется в областях, где требуется
хорошая влагоустойчивость и температуростойкость. Кровельные мате-
риалы, оболочки высоковольтных электрических кабелей и специальные
автомобильные рукава производят с использованием СКЭПТ.
Смотрите Таблицу 15 сокращений некоторых эластомеров.
Таблица 15. Номенклатура отдельных эластомеров [6].
AU (ПЭУ) Полиэфируретан (сложный эфир)
BR (ПБ) Полибутадиен
BUR (ББК) Бромированный сополимер изобутилена с изопреном (бромбутил-каучук)
СПБ (ХБК) Хлорированный сополимер изобутилена с изопреном (хлорбутил-каучук)
СРЕ (ХПЭ) Хлорированный полиэтилен
СВ (ПХП) Полихлоропрен
CSM (ХСПЭ) Хлорсульфированный полиэтилен
ЕАМ (ЭВА) Сополимер этилена и винилацетата
EPDM (СКЭПТ) Тройной сополимер этилена, пропилена и диена с неболь- шой частью ненасыщенных звеньев в цепи
ЕРМ (СКЭП) Этилен-пропиленовый сополимер
EU (ПЭУ) Полиэфируретан (простой эфир)
HNBR (Гидриро- ванный БНК) Гидрированный бутадиен-акрилонитрильный каучук (вы- соконасыщенный нитрильный каучук)
IIR (БК) Бутил каучук
IR (СКИ) Синтетический полиизопрен
NBR (БНК) Бутадиен-нитрильный каучук
SBR (БСК) Бутадиен-стирольный каучук
Е-SBR (СКС) Эмульсионный бутадиен-стирольный каучук
S-SBR (ДССК) Растворный бутадиен-стирольный каучук
Х-NBR (СКН-26-1) Карбоксилированный бутадиен-нитрильный каучук
Х-SBR (СКС-30-1) Карбоксилированный бутадиен-стирольный каучук
YSBR (СБС) Блоксополимеры стирола и бутадиена
422 -'v- Глава 9. Наука о рецептуростроении резин
III. СИСТЕМЫ НАПОЛНИТЕЛЕЙ
Наполнители, или усиливающие агенты — технические
углерод, каолин и кремнекислоты вводят в резиновые смеси с целью улуч-
шения свойств, таких как прочность и сопротивление износу. Технология
технического углерода также сложна, как и наука о полимерах, и существует
большое количество видов технического углерода, каждый из которых при-
дает специфические свойства композиции. Правильный выбор технического
углерода очень важен для создания полимерных композиций и свойств
получаемых резин. Таблица 16 показывает основные классы технического
углерода для резиновых смесей по стандарту ASTM D1765-05 [21].
Таблица 16. Типы технического углерода.
Тип Обозначение по ASTM Размер частиц, нм Основное назначение
SFR N762 61-100 Детали шин, кроме протектора
GPF N660 49-60 Детали шин, кроме протектора
FEF N550 40-48 Детали шин, кроме протектора
FF N475 31-39 Детали шин, кроме протектора
HAF N330 26-30 Протектор и другие детали
ISAF N220 20-25 Протектор
SAF N 110 11-19 Протектор
А. Свойства технического углерода
Технический углерод качественно характеризуется рядом
свойств: размером частиц (площадь поверхности), распределением частиц
по размерам, структурностью (агрегаты частиц), активностью поверхности
(химические функциональные группы такие, как карбоксильные и кетон-
ные). Ключевые свойства технического углерода могут быть перечислены
так (Таблица 17):
Йодное число Мера площади поверхности (размер частиц). Чем больше йодное число, тем меньше размер частиц.
Адсорбция дибутилфталата (ДБФ) Мера структурности или размер агрегатов технического углерода. Чем выше значение адсорбции ДБФ, тем выше структурность.
Цветовой тон Оптическая плотность, значение которой увеличивается с уменьшением размера частиц.
Адсорбция (ЦТАБ)+) Значение удельной площади поверхности с поправкой на влияние микропор.
*> Цетил-три-метиленаммронийбромид.
III. Системы наполнителей Jb 423
Таблица 17. Свойства технического углерода.
Обозначение ио ASTM Йодное число Адсорбция дибутил- фталата (ДБФ) Показатель адсорбции ДБФ с предваритель- ной тренировкой образца Общая абсорбция азота STSA*> Степень оптической плотности
N 110 145 ИЗ 97 127 115 123
\ 115 160 из 97 137 124 123
N 120 122 114 99 126 ИЗ 129
N 121 121 132 111 122 114 119
; N 125 117 104 89 122 121 125
N 134 142 127 103 143 137 131
N 219 118 78 75 123
N 220 121 114 98 114 106 116
N 231 121 92 86 111 107 120
N 234 120 125 102 119 112 123
N299 108 124 104 104 97 ИЗ
N 326 82 72 68 78 76 111
N 330 82 102 88 78 75 104
N 339 90 120 99 91 88 111
N 343 92 130 104 96 92 112
N 347 90 124 99 85 83 105
N 351 68 120 95 71 70 100
N 358 84 150 108 80 78 98
N375 90 114 96 93 91 114
N472 250 178 114 270 145
N 550 43 121 85 40 39
N 630 36 78 62 32 32
N 650 36 122 84 36 35
N 660 36 90 74 35 34
N 762 27 65 59 29 28
N 772 30 65 59 32 30
N 990 43 37 8 8
N 991 35 37 8 8
*> Площадь внешней поверхности по многоточечной адсорбции азота (ASTM D5816).
424 -'V- Глава 9. Наука о рецептуростроении резин
Терминология, применимая к техническому углероду, представлена
в Таблице 18. Дополнительные сведения можно получить в стандартах ASTM
D1566-04 по общим показателям создания композиций [22] и D3053-04 —
специфичным для технического углерода [23].
Таблица 18. Описание характеристик технического углерода.
Печной технический углерод Тип технического углерода, получаемый инжекцией определенных марок нефтяного сырья в высокоскоро- стной поток сжигаемых газов при заданных условиях, т.е. от N 110 до N 762.
Термический технический углерод Тип технического углерода, получаемого при термиче- ском разложении газообразных углеводородов, т.е. N 990, N991.
Микроструктура Микроструктура технического углерода описывает распо- ложения углеродных атомов внутри частицы технического углерода.
Частица Малая сферическая часть агрегата технического углерода, получаемая при его разрушения. Размер частицы измеря- ется электронной микроскопией.
Агрегат Масса частиц коллоидных размеров в наименьшей диспер- гируемой единице.
Агломерат Расположение, или кластер, агрегатов.
Структура Мера отклонения агрегата технического углерода от сфе- рической формы.
Йодное число Вес йода, в граммах, абсорбированного килограммом тех- нического углерода. Мера величины поверхности частицы. Чем меньше размер частиц, тем больше йодное число.
Адсорбция дибутил- фталата (ДБФ) Объем дибутилфталата, в кубических сантиметрах, абсор- бированного 100 граммами технического углерода. ДБФ — мера структурности агрегатов технического углерода.
Цветовой тон Отношение коэффициента отражения контрольной пасты к коэффициенту отражения испытуемой, содержащей смесь оксида цинка, пластификатора и технического углерода.
Адсорбция ЦТАБ Мера удельной площади поверхности частиц, скорректи- рованная на наличие микропор. ЦТАБ (цетилтримети- ленаммоний бромид) выделяется из мелких пор и, таким образом, лучше отражает долю площади поверхности в контакте с полимером.
Площадь поверхности по азоту Мера общей площади поверхности по количеству газообраз- ного азота, способного покрыть всю поверхность, включая поры без граничных слоев от органических функциональ- ных групп.
III. Системы наполнителей JU 425
Таблица 18. (Продолжение)
Адсорбция ДБФ с предварительной тре- нировкой образца То же что и показатель адсорбции ДБФ, но перед испы- танием образец подвергается серии подпрессовок (4 раза, ~12 кг). Это позволяет определить изменения показателей технического углерода под влиянием последующих про- цессов смешения.
Гранулы (Таблетки) Масса прессованного технического углерода, сформирован- ная для снижения запыленности, облегчения переработки и улучшения течения.
Содержание пыли Количество пыли, присутствующей в гранулированном техническом углероде; этот показатель должен быть ми- нимальным.
Твердость гранул Мера нагрузки, в граммах, необходимая для разрушения определенного числа гранул. Контролируется по количест- ву гранулирующего агента. Для наилучшей долговечности гранул композиции распределение гранул по твердости должно быть узким. Примерами гранулирующих агентов являются лигносульфонаты и мелассы (черная патока).
Зола Остаток после сжигания технического углерода при 550 °C в течение 16 часов; первичная оценка качества охлаждаю- щей заводской воды.
Обесцвечивание то- луола Углеводороды, экстрагируемые толуолом из технического углерода; может быть использовано как мера времени пребывания в печи.
Содержание водорода и кислорода Остаточное содержание водорода и кислорода, оставшееся после получения технического углерода; содержится — в виде фенольных, лактонных, карбоксильных, хинонных и гидроксильных функциональных групп. Такие группы могут значительно влиять на кинетику вулканизации и усиливающий потенциал технического углерода.
По опытным данным увеличение размера агрегатов технического угле-
рода и структурности приводит к улучшению усталостной выносливости
и сопротивления росту трещин резин. Уменьшение размера частиц ведет
к улучшению сопротивления износу и раздиру, но падению эластичности,
росту гистерезиса и теплообразованию. Влияние типа и количества техни-
ческого углерода на поведение протекторных резин изучали Гесс и Кламп
[24]. Они исследовали 16 типов технического углерода в трех протекторных
смесях с переменным содержанием масел. Авторы определили некоторые
критерии относительно влияния технического углерода на гистерезисные
свойства резин. Сюда входит степень наполнения, размер агрегатов, пло-
щадь поверхности, распределение агрегатов по размерам, нерегулярность
426 -*v- Глава 9. Наука о рецептуростроении резин
агрегатов (структурность), поверхностная активность, дисперсность и рас-
пределение фаз в гетерогенной полимерной системе.
По результатам испытаний шин с различными типами технического
углерода были отмечены следующие особенности:
1. Снижение содержания технического углерода уменьшает сопротив-
ление качению шины. При постоянном содержании технического
углерода увеличение содержания масла ведет к росту сопротивления
качению и также улучшает сцепление с дорогой (при малом содер-
жании масла увеличение его количества может снизить гистерезис за
счет улучшения диспергирования технического углерода).
2. Увеличение дисперсности технического углерода ведет к росту как
сопротивления качению, так и сцепления с дорогой.
3. Увеличение ширины распределения агрегатов по размерам при
постоянстве площади поверхности, определяемой по ДБФ, снижает
сопротивление качению шины.
4. Технический углерод для резин протекторного типа можно выбрать
по заданным эксплуатационным параметрам сопротивления качению,
сцепления с дорогой, износа и пр.
Содержание технического углерода (масс, ч.)
Рис. 5. Влияние содержания технического углерода на свойства композиции.
На рисунке 5 показаны общие тенденции влияния наполнения техни-
ческим углеродом протекторных смесей на физические свойства резин.
Если содержание технического углерода увеличивается, то наблюдается
повышение теплообразования и твердости резин, а в шинах — улучшение
сопротивления качению и сцепления с влажной дорогой. Однако, сопро-
III. Системы наполнителей Jb 427
тивление разрыву, перерабатываемость смеси и сопротивление истиранию
проходят через оптимум, после которого эти свойства ухудшаются.
Используя результаты работы Гесса и Клампа по улучшению сопротив-
ления качения шины, Свор с сотрудниками разработали новую технологию
технического углерода серии № 200, которая, по их данным, в лучшей степе-
ни характеризует эксплуатационные свойства шины, а основные принципы
приемлемы для ряда конструкций шин [24].
В попытках предсказать направления будущих исследований по техно-
логии технического углерода литературные данные по исследованию взаи-
модействия технический углерод-эластомер показывают его большой потен-
циал в прогнозировании свойств резин. Ле Бра показал, что карбоксильные,
фенольные, хинонные и другие функциональные группы на поверхности
технического углерода взаимодействуют с полимером, и доказал наличие
химической сшивки между этими компонентами в вулканизатах [25]. Айала
и сотрудники [26, 27] определили величину параметра взаимодействия кау-
чука с наполнителем непосредственно с помощью испытаний вулканизатов.
Авторы установили, что соотношение q/п, где Q — угол наклона кривой
напряжение-удлинение, определяет взаимодействие технического углеро-
да с полимером; а величина п равна отношению динамического модуля Е’
при 1 и 25% амплитудах деформации, является мерой взаимодействия
наполнитель-наполнитель. Этот параметр взаимодействия отражает вклад
взаимодействия технический углерод-полимер и снижает влияние физи-
ческого фактора, связанного с образованием сетки. Использование этого
параметра позволяет сделать ряд заключений:
1. Величина ъ/п представляет собой хорошую меру взаимодействия
технический углерод-полимер для ряда полимеров: БСК, БК, НК
и БНК.
2. Более высокие значения <з/п были получены для БСК и БНК, т.к. аро-
матические структуры в БСК и полярные -CN группы в БНК опре-
деленно влияют на взаимодействие технический углерод-каучук.
3. Анализ высушенной поверхности технического углерода позволил
обнаружить наличие ряда углеводородных групп, что соответствует
ранее проведенным исследованиям [25]. Эти группы способны реа-
гировать с другими функциональными группами.
Учитывая наличие органических функциональных групп на поверхности
технического углерода, Вольф и Горл изучили реакционную способность
органосиланов, таких как бис(3-триэтоксисилилпропил)тетрасульфан
с печным техническим углеродом [28]. Авторы пришли к выводу, что такие
группы как карбоксильные, лактольные, хинонные и кетонные реагируют
с этокси-группами бис(3-триэтоксисилилпропил)тетрасульфана, образуя
подвески на поверхности технического углерода:
428 -*Ц- Глава 9. Наука о рецептуростроении резин
-карбоксил
-хинон
-фенол
-кетон
X—Si (OR)3
карбоксил-
хинон-
фенол-
кетон-
О —R
— О — Si — X
О—R
— О О—R
><
—о7 X
(4)
Поверхность
технического углерода
Активированная поверхность
технического углерода
По данным анализа экстрактов и свойств композиций с техническим
углеродом, обработанным органосиланами, Вольф и Горл сделали вы-
воды:
1. Технический углерод способен связывать определенное количество
триалкоксисилана.
2. Количество связанного органосилана соответствует площади повер-
хности частиц технического углерода и количеству кислород-содер-
жащих функциональных групп.
3. Триэтоксисилильная группа является реакционноспособной ча-
стью силана, образующего ковалентную связь с техническим угле-
родом.
4. Реакция бис (3-триэтоксисилилпропил) тетрасульфана с техническим
углеродом позволяет снизить гистерезис резин.
Эта работа стала фундаментальной для многих новых технологий тех-
нического углерода. Они распадаются на два направления: пост-процессы
модификации — в этом случае обрабатывается поверхность технического
углерода для улучшения его свойств, и процессы модификации при произ-
водстве технического углерода, когда вводятся другие компоненты опять же
для улучшения основных свойств наполнителя [29].
Примеры пост-процессов модификации: окисление поверхности с ис-
пользованием озона, пероксида водорода или азотной кислоты. Такие под-
ходы используют в производстве проводящих видов технического углерода.
Проводятся также исследования реакций с солями диазония, обработки
плазмой и прививки полимера.
Процессы модификации при производстве включают присоединение
металлов, получение инверсионных видов технического углерода, или
наноструктурных технических углеродов, или двухфазных наполнителей
технический углерод-кремнекислотные наполнители.
III. Системы наполнителей JU 429
Б. Кремнекислотные наполнители
Введение в резиновые смеси кремнекислотных наполнителей
дает ряд преимуществ таких, как улучшение сопротивления раздиру, сниже-
ние теплообразования и увеличение адгезии в многокомпонентных изделиях,
например, шинах. Два основных свойства кремнекислотных наполнителей
определяют их применение в резиновых композициях — это предельный
размер частиц и степень гидратации. Другие физические свойства такие, как
pH, химический состав и абсорбция масел имеют вторичное значение.
Кремнекислотные наполнители по сравнению с техническим углеродом
сходных размеров частиц не обладают той же усиливающей способностью,
хотя этот недостаток в значительной степени устраняется, если использовать
совместно с кремнекислотными наполнителями агенты сочетания. Вагнер
установил, что введение кремнекислотного наполнителя в протекторную
смесь приводит к снижению сопротивления износа протектора даже при
улучшенных гистерезисных свойствах и сопротивления раздиру [30]. Поте-
ри сопротивления износу можно скорректировать использованием агентов
сочетания [31].
Химические свойства кремнекислотных наполнителей можно охаракте-
ризовать следующим образом:
1. Аморфный кремнезем состоит из атомов кремния и кислорода,
формирующих тетраэдрическую структуру в трехмерной решетке..
Размер частиц колеблется от 1 до 30 нм, площадь поверхности — от
20 до 300 м2/г. В частицах отсутствует дальний порядок, существует
только домены ближнего порядка, беспорядочно расположенные
относительно соседних доменов.
2. Концентрация силанольных групп на поверхности (силанольные
группы -Si-O-Н) влияет на степень поверхностной гидратации.
3. Различают три типа силанольных групп — изолированные, спаренные
(две гидроксильные группы при одном атоме кремния) и вицинальные
(у соседних атомов кремния), как показано на рисунке 6.
4. Кислотность поверхности определяется гидроксильными группами на
поверхности кремнекислотного наполнителя и является промежуто-
чной между кислотностью Р-ОН и В-ОН. Кислотность может влиять
на процесс пероксидной вулканизации, в случае же серной — сильного
влияния не наблюдается. Таким образом, взаимодействие системы
каучук-наполнитель зависит от этих факторов.
5. Поверхностная гидратация, вызванная абсорбцией (поглощением)
паров воды, определяется концентрацией силанольных групп на
поверхности. Высокая степень гидратации отрицательно влияет на
конечные физические свойства резин. Кремнекислотные наполнители
гигроскопичны, поэтому они должны храниться в сухом месте.
430 -*v- Глава 9. Наука о рецептуростроении резин
Таблица 19 Влияние гидратации поверхности на свойства силиката.
Кремнекислотный наполнитель А Кремнекислотный наполнитель В
Площадь поверхности (м2/г) 152,0 152,0
Потери при 150°С (% Н2О) 6,8 0,5
Скорчинг по Муни 14,0 16,0
Время t_90 по реометру 27,0 47,0
Разрывная прочность (МПа) 18,3 21,7
Относительное удлинение 480 480
Модуль при 300% удлинении (МПа) 7,0 9,5
Истираемость по Пико 67,0 103,0
Si
I
---Si—ОН
I
о
I
---Si—ОН
I
Вицинальные
---О
---Si—ОН
---О
Изолированные
Спаренные
Рис. 6. Типы силанольных групп на поверхности кремнекислотного наполнителя.
Чтобы показать влияние поверхностных гидроксильных групп на
свойства резины, Вагнер [30] выбрал ряд кремнекислотных наполнителей
с различной площадью поверхности, гидроксилированных до различных
степеней и затем добавил по 50 масс. ч. их в смеси из БСК (Таблица 19).
Автор сделал вывод о том, что снижение содержания силанольных групп
вследствие увеличения абсорбированной воды, снижает время вулканиза-
ции, разрывную прочность, а также сопротивление истиранию.
В общем, кремнекислотные наполнители обладают большей усилива-
ющей способностью в более полярных каучуках таких, как БНК и ПХП,
чем в неполярных — БСК и НК. Недостаток усиливающей способности
кремнекислотных наполнителей в НК и БСК может быть устранен использо-
ванием силановых агентов сочетания. Существенное преимущество агентов
сочетания состоит в том, что их молекулы бифункциональны, т. е. способны
химически реагировать как с кремнекислотным наполнителем, так и прямо
или косвенно с полимером посредством участия в реакциях вулканизации
и процессах серного сшивания.
111. Системы наполнителей JU 431
Таблица 20. Группы кремнекислотных наполнителей.
Площадь поверхности (м2/г) 90-130 130-180 180-220
Традиционные Каркас шины. Не шинные внутрен- ние и наружные детали. Протектор шин. Каркас и не шин- ные наружные де- тали. Протектор шин и нешинные наруж- ные детали, стой- кие к истиранию.
Полувысокодиспер- сные Каркас шины. Не шинные наружные детали. Протекторы шин. Каркас. Протекторы шин
Высокодисперсные Каркас шин. Про- текторы шин Протекторы шин Высоко качествен- ные протекторы шин.
Использование кремнекислотных наполнителей в резинах дает следу-
ющие преимущества: снижение теплообразования в случае частичной
замены доли технического углерода и улучшение сопротивления раздиру,
порезам, отслаиванию и скалыванию. Однако, если наполнение приближа-
ется к 20%, то сопротивление истиранию, например, протекторных резин,
снижается настолько, что данная рецептура становится уже не целесообраз-
ной. Использование силанольных агентов сочетания открывает возможность
избежать такого снижения рабочих параметров резин. Поэтому, с целью
повышения эффективности кремнекислотных наполнителей уместно обсуж-
дение свойств и химии агентов сочетания, особенно, силанольных.
Кремнекислотные наполнители можно разделить на три группы, или
класса: стандартные, или обычные; полу-высокодисперсные (полу-ВД),
или легкодиспергируемые кремнекислотные наполнители; и последняя
группа включает высокодисперсные наполнители, или ВДКН (Таблица 20).
Структура силанольных групп на поверхности трех типов кремнекислотных
наполнителей требует разъяснений, но можно ожидать, что ВДКН будут
иметь большие концентрации сдвоенных групп, тогда как традиционные
кремпенкислотные наполнители будут содержать большее количество изо-
лированных силанольных групп [10, 31].
В. Химия силанольных агентов сочетания
Существует три силанольных агента сочетания промышлен-
ного значения: меркаптопропилтриметоксисилан (А189), бис(триэтоксиси
лилэтилтолуилен)полисульфид (Y9194) и бис(З-триэтоксисилилпропил)
тетрасульфан (TESPT), все они обладают схожими свойствами. Промыш-
ленное обозначение указанно в скобках. Характеристика агента сочетания
TESPT встречается в литературе чаще других, однако, ниже будет рассмот-
рено действие всех трех продуктов в качестве агентов сочетания [31].
432 -*V Глава 9. Наука о рецептуростроении резин
TESPT — бифункциональный полисульфид органосилана; вводится как
агент сочетания с целью повышения усиливающих свойств кремнекислот-
ных наполнителей в резинах. Использования агентов сочетания дает ряд
преимуществ:
- снижение теплообразования и гистерезисных потерь резин, содержа-
щих кремнекислотные наполнители;
- увеличение значения модуля 300% удлинения и разрывной про-
чности, опять же, в смесях, содержащих кремнекислотные напол-
нители;
- повышение усиливающего эффекта каолина и мела;
- сопротивление реверсии в склонных к ней вулканизующих системах;
- улучшение показателя стойкости к абразивному износу согласно DIN.
Механизм усиливающего действия агента сочетания можно разбить на
2 этапа:
1. Реакция гидрофобизации, в которой агент сочетания взаимодействует
с кремнекислотным наполнителем;
2. Образование поперечных связей между модифицированным кремне-
кислотным наполнителем и каучуком.
Силанилизация поверхности кремнекислотного наполнителя может
протекать достаточно быстро; хотя в TESPT системах реакция, как пра-
вило, происходит in situ в закрытом смесителе при температурах от 150 до
160 °C. Несмотря на то, что на поверхности кремнекислотного наполнителя
имеется избыток силанольных групп и такая реакция протекает быстро,
все же необходима высокая температура из-за стерических затруднений
у силилпропильных групп в TESPTe.
Как отмечено ранее, на поверхности кремнекислотного наполнителя
имеется три типа силанольных групп: изолированные гидроксильные
группы, спаренные группы (две ОН-группы присоеденины к одному атому
кремния) и вицинальные группы (см. рис.6). Реакция силанизации изобра-
жена на схеме (5).
С2Н5О
ОС2Н5
Si---ОН + С2Н5О —Si—(СН2)3 —S4—(СН2)3—Si—ОС2Н5
Si---ОН
— С2Н5ОН
С2Н5О
ОС2Н5
Кременекислотный
наполитель
TESPT
III. Системы наполнителей -JU 433
— С2Н5ОН
Si--О. OC2H5
Si
Si--^(CH2)3—S—S
si—°\ ДСН2)3—S—S
Si
(5)
Si------О
XOC2H5
Промежуточный продукт реакции
кремнекислотный наполнитель/ TESPT
Теперь промежуточный продукт наполнитель/силан может взаимодей-
ствовать с аллильными позициями ненасыщенных связей полимерной цепи.
Как известно, вулканизация каучука происходит в присутствии ускорителя,
например, такого как сульфенамид, с серой, оксидом цинка и стеариновой
кислотой с образованием сульфурирующего агента [32]. На схеме реакций
6 приведены несколько упрощенные реакции вулканизации. По заверше-
нии реакции подвеска ускорителя (т. е. каптакса) отщепляется после об-
разования сшивок. Остаток ускорителя, каптакса, в свою очередь является
ускорителем и продолжает участвовать в дальнейших реакциях сшивания
пока продолжается вулканизация.
Как предположил Вольф, в кремнекислотных усиливающих системах,
содержащих TESPT, промежуточный продукт реакции кремнекислотный
наполнитель/TESPT действует вместо серы, и механизм реакции схож, но
в этом случае сшивающим агентом является полисульфид. Вольф показал,
что дисульфид меркаптобензтиазола (ДБТД) взаимодействует с тетрасуль-
фоновыми группами с образованием двух молей полисульфида, схема (6).
Частица кремнекислотного наполнителя находится с одной стороны и
меркаптобензтиазолил — с другой. Подвеска полисульфидной группы на
поверхности наполнителя теперь образует сшивку с полимером, почти по
такому же механизму, который имеет место в случае связанных с каучуком
промежуточных продуктов, которые превращаются с образованием попе-
речных связей. Вольф [33] предположил, что МВТ реагирует с аллильной
позицией двойной связи каучука и, таким образом, высвобождается МВТ
и образуется связь каучук-кремнекислотный наполнитель схема (7).
Описанные условия смешения кремнекислотных наполнителей с аген-
тами сочетания позволяют использовать их в производстве:
- обувных подошв;
- опор двигателей, где резины, содержащие натуральный каучук и агент
сочетания/кремнесодржащий наполнитель, обеспечивают необходи-
мые гистерезисные свойства;
434 -*V- Глава 9. Наука о рецептуростроении резин
- в протекторах шин, где опять же важны гистерезисные свойства;
- в производстве ряда других изделий, например, мячей для
гольфа [31].
(6)
+ Промежуточный продукт TESPT/SiO2
2
С2Н5О
:—S—Sx-S—CH2-CH2-CH2-Si—О-
С2Н5О
(7)
С—
II
с
С2Н5О
CH-SX-S—СН2-СН2-СН2—Si—О-
? Сгн5о
+ МБТ
п
IV. Системы стабилизаторов -*v- 435
Г. Другие системы наполнителей
Ряд дополнительных систем наполнителей будут описаны не
так подробно не в связи с их усиливающими свойствами, а в связи с их ши-
роким потреблением. Это каолин (гидратированный алюмосиликат), слюда
(калий алюмосиликат), тальк (силикат магния), мел (карбонат кальция)
и диоксид титана.
Свойства каолина так же, как и кремнекислотных наполнителей, могут
быть улучшены благодаря обработке поверхности силанольными агентами
сочетания. Тиоалкилсиланы взаимодействуют с поверхностью с образова-
нием подвесок тиольных групп, которые могут реагировать с полимером,
либо через водородные связи, Ван-дер-Ваальсовы связи, либо поперечные
связи с другими реакционноспособными группами:
I—О—(СНД —SH
Каолин —
(8)
L-O—(СН2)Х —SH
Такой каолин улучшает сопротивление раздиру, увеличивает модули,
улучшает адгезию между компонентами в многокомпонентных композициях
и сопротивление старению.
Карбонат кальция используется как дешевый наполнитель в резиновых
изделиях при статических условиях эксплуатации, например, ковровых
подложек. Диоксид титана нашел широкое применение при производстве
светлых изделий, таких как белые боковины шин, где внешний вид изделия
имеет большое значение.
IV. СИСТЕМЫ СТАБИЛИЗАТОРОВ
Ненасыщенность эластомеров является причиной их уни-
кальных вязкоэластических свойств. Наличие же двойных углерод-углерод-
ных связей делает эластомеры восприимчивыми к действию кислорода, озона
и термической деструкции. Исчерпывающий обзор окисления эластомеров
и роль антиоксидантов и антиозонантов представлен в [34].
А. Деструкция каучука
Окисление эластомеров ускоряется рядом факторов таких,
как температура, содержание соединений тяжелых металлов, серы, действие
света, влаги, набухание в маслах и растворителях, динамическая усталость,
присутствие кислорода и озона. Существует три фактора, препятствующие
деструкции эластомеров — это тип полимера, вулканизующая система
и система противостарителей.
436 -* v- Глава 9. Наука о рецептуростроении резин
Термоокислительная стабильность напрямую зависит от вулканизующей
системы. Пероксидная вулканизующая система позволяет предупредить
реверсию благодаря отсутствию серы и образованию углерод-углеродных
поперечных связей. Характерной чертой эффективных вулканизующих
систем (ЭВ) является низкое содержание серы (0,0-0,3 масс, ч.) и высокое
содержание ускорителей и доноров серы, что подобным образом обеспечивают
высокую термостабильность и стойкость к окислению. Но такие системы дают
низкую усталостную прочность благодаря преимущественному содержанию мо-
носульфидных связей. Обычные вулканизующие системы, характеризующиеся
высоким количеством серы и малой концентрацией ускорителя, обладают низ-
кой теплостойкостью и стойкостью к окислению, поскольку полисульфидные
связи термически нестойки и легко окисляются. Такие вулканизующие системы,
однако, позволяют получать резины с лучшей усталостной выносливостью.
Полу-эффективные вулканизующие системы, являющиеся промежуточными
между ЭВ и обычными системами, представляют компромисс между стойкостью
к окислению и усталостной выносливостью изделий.
Процесс старения протекает по двум основным механизмам.
1. Сшивание'. Ди- или полисульфидные связи распадаются до моно-
сульфидных. Повышается жесткость, снижается усталостная вы-
носливость, материал становится более жестким. Такой механизм
преобладает при окислении БСК, СКЭПТ, БНК и полихлоропрена.
2. Деструкция: Полимерные цепи разрушаются, вызывая размягчение
материала и снижение сопротивления истиранию. Такое поведение
характерно для натурального каучука.
Деструкция ненасыщенных полимеров — автокаталитический процесс, сво-
боднорадикальная цепная реакция, которую можно разбить на три стадии:
Инициирование
О2, свет
RH (эластомер) -----> R.HOO (свободный радикал) (9)
тепло е
Развитие реакционной цепи
R. + О2-----> ROO
ROO + R'H------> ROOH + R'.
ROOH----> RO + ОН
• •
(10)
RO + R"H---> ROH + R".
pH + R"H---> R". + H2O
IV. Системы стабилизаторов -•и- 437
Обрыв реакционной цепи
R. + .R
r -I- ROO —» ПРОДУКТЫ реакции,
• . не содержащие свободных радикалов ' '
ROO + OOR
• •
Скорость реакции увеличивается с повышением температуры, как
и любой химический процесс. Рост температуры эксплуатации ускоряет
деструкцию каучука. Скорость реакции окисления подчиняется уравнению
Аррениуса.
Ультрафиолетовое излучение инициирует образование свободных ра-
дикалов на незащищенной поверхности эластомерного изделия, что при-
водит к образованию слоя окисленного каучука. Тепло, влага или высокая
влажность могут привести к образованию трещин на поверхности, которые
впоследствии могут разрушить материал. Такое разрушение поверхности
изделия более заметно у ненаполненных резин, чем у резин, наполненных
техническим углеродом.Таким образом, резины без технического углерода
(белые боковины шин), требуют большего содержания не меняющих цве-
та антоксидантов по сравнению с наполненными техническим углеродом
материалами.
Тяжелые металлы переменной валентности такие, как железо, марга-
нец и медь катализируют окисление эластомеров. Соединения марганца
или меди, олеаты и стеараты, хорошо растворимы в каучуке, тем самым,
облегчая быстрое окисление полимера. Противостарители, производные
пара-фенилендиаминов, используют для того, чтобы снизить активность
таких ионов металлов.
Главной причиной разрушения резиновых изделий является разраста-
ние трещин. Рост трещин под действием циклической нагрузки приводит
к снижению усталостной выносливости. Усталостные трещины образуются
в зонах с повышенным напряжением. Атака озоном индуцирует образование
поверхностных трещин, которые затем разрастаются в результате изгиба.
Инициированные озоном трещины можно наблюдать в виде крейзов на
поверхности боковин старых шин. Озон быстро реагирует с двойными уг-
лерод-углеродными связями ненасыщенных эластомеров с образованием
озонидов. Под действием растяжения озониды легко разлагаются, вызывая
разрыв полимерных цепей и снижение молекулярного веса. Снижение мо-
лекулярного веса является причиной образования поверхностных крейзов
и трещин:
438
Глава 9. Наука о рецептуростроении резин
О
R — СН = СН — R —R —нс СН—R_^2> +
0-0 (!2)
О
Ненасыщен-
ный эластомер Озонид R
ОН
Продукты
разложения
Смеси несовместимых полимеров обладают улучшенной усталостной
выносливостью. Например, натуральный каучук и полибутадиен прояв-
ляют хорошую усталостную выносливость, сопротивление образованию
и разрастанию трещин благодаря возникновению гетерогенных полимерных
фаз; рост трещины в фазе одного полимера приостанавливается на границе
раздела фаз с другим полимером.
Смеси натурального каучука и полибутадиена используются в боковинах
шин, которые подвержены изгибам, а также в протекторах шин, которые
имеют выступающий рисунок и, как следствие, имеют зоны повышенных
напряжений в основании блоков протектора.
Таким образом, использование противостарителей являются важным
фактором защиты эластомерных композиций от действия этого широкого
ряда атмосферных, химических и эксплуатационных факторов, приводящих
к старению материала.
Б. Применение противостарителей
При выборе противостарителей руководствуются следую-
щими критериями:
1. Обесцвечивние и изменение цвета: Обычно фенольные антиоксиданты
не изменяют цвет, а амины — наоборот. Таким образом, в эластомеры,
наполненные техническим углеродом, вводят более активные амин-
ные антиоксиданты, где обесцвечивание не является столь важным
фактором.
2. Летучесть: Как правило, чем больше молекулярный вес антиоксидан-
та, тем он менее летуч. Несмотря на это пространственно затрудненные
фенолы более летучи по сравнению с аминами той же молекулярной
массы. Таким образом, необходимо учитывать этот фактор при введе-
ние антиоксидантов в резиновую смесь, если потеря массы материала
должна быть исключена.
IV. Системы стабилизаторов
Л-
439
3. Растворимость: Низкая растворимость противостарители приво-
дит к выцветанию его на поверхность материала, что снижает его
защитное действие. Поэтому растворимость противостарителей, а
особенно антиоозонантов, определяет их эффективность. Противо-
старители должны быть растворимы вплоть до 2,0 масс, ч., должны
быть способны к миграции к поверхности, но не должны раство-
ряться в воде и других растворителях (например, в гидравлических
жидкостях), чтобы предотвратить экстракцию противостарителей
из резины.
4. Химическая стабильность: Противостарители должны быть сами
достаточно длительно стойки к действию тепла, света, кислорода
и растворителей.
5. Концентрация: Для большинства противостарителей характерна опти-
мальная концентрация, соответствующая наибольшей эффективности,
после которой лимитирующим фактором становится растворимость.
Пара-фенилендиамины обладают хорошим противоокислительным
действием при содержании от 0,5 до 1,0 масс. ч. и защитой от действия
озона — от 2,0 до 5,0 масс. ч. При содержании свыше 5,0 масс. ч. пара-
фенилендиамины имеют тенденцию к выцветанию.
6. Защита окружающей среды, здоровья и техника безопасности: Для
облегчения транспортировки и во избежание пыления и попадания
в дыхательные пути противостарители должны быть непылящими,
если находятся в свободном потоке в воздухопроводе.
С. Типы противостарителей
1. Антиоксиданты, не изменяющие цвета: Этот класс антиоксидантов
делится на четыре группы: фосфиты, пространственно затрудненные
фенолы, пространственно затрудненные бисфенолы и гидрохиноны.
Затрудненные бисфенолы, такие как 4,4’-тиобис (6-трет-бутил-м-
крезол), наиболее действенны из всех четырех групп. Из-за низкой
молекулярной массы пространственно затрудненные фенолы склонны
к летучести. Фосфиты используют в качестве стабилизаторов синте-
тических каучуков, а гидрохиноны, такие как 2,5-ди-трет-амилгид-
рохинон используют в адгезивах:
(СН3)3С С(СН3)3
но \О/ s—\О/ он
СН3СН3
СН3СН2(СН3)2С
он
С(СН3)2СН2СН3
он
4,4’-тиобис (6-7претп-бутил-л£-крезол)
2,5-ди-тпретп-амилгидрофенол
440 “*V Глава 9. Наука о рецептуростроении резин
2. Антиоксиданты, изменяющие цвет: Два класса изменяющих цвет
или обесцветивающихся антиоксидантов нашли широкое применение
- это полимеризованные дигидрохинолины и дифениламины:
СН3
Антиоксиданты
класса дифениламинов
Полимеризованный!,2-дигидро-
2,2,4-триметилхинолин
Дигидрохинолины отличаются друг от друга степенью полимеризации,
что определяет их миграционную способность и продолжительность
защитных свойств. Они хорошие антиоксиданты общего назначения,
а также эффективны по отношению к солям тяжелых металлов, таких
как ионы никеля и меди — катализаторам окисления. Полимер-
ная природа дигидрохинолинов снижает их летучесть и миграцию
в вулканизатах. Благодаря этому наблюдаются минимальные потери
противостарителей, вызванные вымыванием или диффузией; продол-
жительность действия и стабильность при повышенных температурах
возрастают. Дифениламинные антиоксиданты придают материалу
улучшенную усталостную выносливость.
3. Антиозонанты: Пара-фенилендиамины (ПФД) единственный класс
антиозонантов широкого применения. Их общая структура:
R — NH-/Q)— NH — R
Они не только обеспечивают защиту резиновых изделий от озона, но
также улучшают их усталостную выносливость, стойкость к воздей-
ствию кислорода, тепла и ионов металлов. Существует три основные
категории парафенилендиаминов, которые можно описать так:
а. Диалкил-ПФД: Обе замещающие R-группы — это алкилы, на-
пример, диизопропил-п-фенилендиамин. R-группы могут быть
от СЗ вплоть до С9. Противостарители типа диалкил-ПФД имеют
высокую склонность к подвулканизации резиновых смесей по
сравнению с другими классами ПФД, а так же мигрируют быст-
рее других противостарителей из-за своей низкой молекулярной
массы. Они недостаточно устойчивы.
б. Алкил-арил-ПФД: Одна R-группа — ароматическое кольцо, другая —
алкильная. Наиболее широко используемый ПФД этого класса
V. Вулканизирующие системы
441
— ЬМ,3-диметилбутил-ЬГ-фенил-п-фенилендиамин. Этот анти-
озонант проявляет хорошие защитные свойства в динамических
и статических условиях, когда используется совместно с восками,
способствует стабильной переработке смесей в процессе вулкани-
зации, обладает низкой миграционной способностью, что позволяет
ему быть более стойким и подходящим для изделий длительного
использования.
в. Диарил-ПФД: Третий класс ПФД содержит в своей структуре две
ароматические группы, это — дифенил-п-фенилендиамин или ди-
Р-нафтил-п-фенилендиамин. Они менее активны по сравнению с ал-
кил-арил-ПФД и имеют тенденцию к выцветанию на поверхности,
что делает их непригодными для многих областей применения.
4. Воска'. Воска — дополнительный класс материалов, используемых для
улучшения стойкости резин к озонному разрушению преимуществен-
но в статических условиях. Воска, входящие в рецептуру эластомеров,
делятся на две категории. Микрокристаллические воска имеют точку
плавления от 55 до 100°С, их получают из остатков тяжелого масля-
ного сырья после перегонки нефти. Парафиновый воск имеет точку
плавления от 35 до 75°С, получается из легкого масляного дистиллята
сырой нефти.
Свойства восков представлены в Таблице 21. Воска защищают резины от
озонолиза в статических условиях путем образования барьерного слоя на их
поверхности. Воск непрерывно мигрирует из массы каучука, поддерживая
постоянную концентрацию на поверхности. Микрокристаллические воска
мигрируют к поверхности медленнее, чем парафиновые, из-за высокой
молекулярной массы и разветвленной структуры. К тому же, микрокристал-
лические воска лучше защищают при повышенных температурах эксплуа-
тации в то время, как парафиновые — при низких. Это связано со скоростью
миграции воска к поверхности изделия.
Таблица 21 Свойства парафиновых и микрокристаллических восков.
Микрокристаллические воска Парафиновые воска
Молекулярный вес 500-800 340-430
Точка плавления (°C) 55-100 35-75
Длина главной углеродной цепи С-25 С-60
Структура Разветвленные молекулы Линейные молекулы
Необходимо отметить, что в динамических условиях защитная восковая
пленка разрушается, после чего антиозонантная система в составе резиновой
композиции будет действовать как основной стабилизатор по защитному
442 “*V Глава 9. Наука о рецептуростроении резин
механизму. Воска используют для защиты от действия озона при хранении
готовых изделий, например, шин в складских помещениях.
Суммарно, можно пользоваться несколькими эмпирическими правилами
для создания противостарительных систем в эластомерных композициях:
1. Краткосрочная защита в статических условиях обеспечивается пара-
финовыми восками.
2. Микрокристаллические воска обеспечивают длительную озоностой-
кость, если конечное изделие находится в условиях хранения.
3. Необходим критический уровень выцветания воска для образования
защитной пленки в статических условиях защиты от озона.
4. Оптимизированные смеси восков и ПФД обеспечивают длительную
защиту изделий как в статических, так и динамических условиях
в широком интервале температур.
5. Избыток выцветающего воска может отрицательно сказаться на
усталостной выносливости из-за того, что толстый слой воска может
разрушиться под действием деформации, и трещина может проник-
нуть в изделие.
V. ВУЛКАНИЗУЮЩИЕ СИСТЕМЫ
Вулканизация, назвнная в честь Вулкана, римского бога
огня, описывает процесс, в результате которого мягкая резиновая смесь
превращается в высококачественные инженерные изделия. Вулканизу-
ющая система состоит из трех или четырех компонентов эластомерной
композиции и предназначена для образования поперечных связей между
соседними полимерными цепями в ней. Типичная вулканизующая сис-
тема состоит из трех компонентов: (1) активаторы; (2) вулканизующие
агенты, обычно, сера; и (3) ускорители. Химия вулканизации описана
в нескольких местах данной монографии. Необходимо, однако, рассмот-
реть каждый из этих компонентов с учетом получения изделий заданного
практического назначения.
А. Активаторы
Активаторы вулканизующей системы, состоящие из оксида
цинка и стеариновой кислоты, исследованы в гораздо меньшей степени,
чем остальные компоненты резиновой смеси. Соотношение стеариновой
кислоты и оксида цинка 2.0 и 5.0 масс, ч., соответственно, общеприняты
в резиновой промышленности; такое содержание активирующей системы
соответствует оптимальным физическим свойствам получаемого материала,
когда она используется в комбинации в широком диапазоне классов и типов
ускорителей, а также соотношений ускоритель/сера. Для того чтобы понять,
почему оксид цинка предпочтительнее других оксидов металлов, было про-
К Вулканизирующие системы -'V 443
ведено сравнительное изучение действия оксидов магния, кальция, железа,
свинца, цинка и диоксида титана. Все оксиды металлов стандартизированы
по ASTM D3184 [35]; смесь номер 1А (ненаполненная) и 2А (наполненная
техническим углеродом) были также отнесены к смесям Американского
Химического Общества (АХО) 1 и 2 соответственно (Табл. 22). Результаты
исследований представлены в Таблицах 23-24 [36].
Таблица 22. Рецепты смесей 1А и 2А по стандарту ASTM D3184 (смеси 1 и 2
по АХО).
Смесь 1 Смесь 2
Натуральный каучук 100 Натуральный каучук 100
Оксид металла 6 Оксид металла 5
Стеариновая кислота 0,5 Стеариновая кислота 2
Сера 3,5 Технический углерод (IRB 5) 35
МБТ 0,5 Сера 2,25
СА-Т 0,7
Таблица 23. Влияние типа оксида металла на свойства резин по базовому
рецепту 1 по АХО.
Рецепт Оксид металла 1 MgO 2 СаО 3 TiO2 4 FeO 5 ZnO 6 PbO
Элктроотрицательность катиона 1,2 1,о 1,5 1,9 1,6 1,8
% свободной серы 2,69 2,51 2,60 2,74 1,43 1,03
Разность величины крутящих момен- тов по реометру Монсанто при 150 °C [M„-ML(dH-M)] 14,0 17,0 19,0 24,0 29,0 43,0
Разрывная прочность (МПа) 4,84 6,37 3,09 4,40 14,80 20,00
Относительное удлинение (%) 731 695 817 530 667 634
Модуль при 300% удлинении (МПа) 0,90 1,14 0,58 1,85 2,96 2,2
Твердость по Шору при 21 °C 32 34 26 24 38 42
Сопротивление раздиру по ASTM die В (кН/м) 38 53 11 21,5 68 67
Зависимость электроотрицательности шести металлов исследованных
оксидов от крутящего момента реометра (MH-ML) показала, что за пределами
диапазона электроотрицательности от 1,6 до 1,8 оптимальные свойства вул-
канизатов не достигаются. Электроотрицательность — мера сродства атома
444
Глава 9. Наука о рецептуростроении резин
металла к притяжению электрона. Исходя из приведенных данных, можно
сделать вывод, что в случае металлов с электроотрицательностью менее, чем
1,55 сдвиг ионной связи с серой приводит к снижению электрофильности
предпоследних атомов серы в комплексах:
X—Sx—S“— М+2—S“— Sx—X
R—H—Sy—X
Наоборот, для металлов с электроотрицательностью более чем 1,85, как
для железа, более ковалентный характер M+-S- связи с пониженнным
разделением заряда неблагоприятно влияет на образование аминных или
карбоксилатных лигандов по отношению к иону металла, как в
что в свою очередь будет снижать растворимость сульфидирующего реа-
гента и в итоге снизит его активность, т. е. приведет к ухудшению свойств
вулканизатов.
Таким образом, цинк является наиболее подходящим для участия в фор-
мировании сульфидирующего комплекса. Координация внешних лигандов
(ROO-, R’2 NH:) у атома цинка вызывает ослабление связи между XS-Sx ...
и ...S -SX группами, поэтому увеличивается вклад полярных форм:
xs—s; —Zn2+ —s;—sx
Этот эффект оказывают лиганды с вакантными 4р орбиталями и рас-
пределяющими положительный заряд металла. В результате растет нук-
леофильность XSSx- связи в сульфидирующем комплексе и снижаться
электрофильность XSy+. Это же справедливо и для комплексов Cd2+
и РЬ2+, которые имеют вакантные /^-орбитали, свободные для присоедине-
ния координированных лигандов. В случае комплексов Mg2+ и Са2+ такого
распределения самопроизвольно не происходит, и эти металлы не способны
в дальнейшем образовывать конфигурацию инертного газа, как в более
стабильных металлоорганических производных. Токсичность CdO и РЬО
предотвращает их употребление, поэтому ZnO нашел широкое практическое
использование в резиновой промышленности; количество оксида, вводимого
в смесь, определяется назначением изделия.
V. Вулканизирующие системы -* V 445
Таблица 24. Влияние типа оксида металла на свойства резин по базовому
рецепту 2 по АХО.
Рецепт Оксид металла 1 MgO 2 СаО 3 TiO2 4 FeO 5 ZnO 6 PbO
Элктроотрицательность катиона 1,2 1,0 1,5 1,9 1,6 1,8
% свободной серы 0,39 0,35 0,72 0,68 0,15 0,15
Разность величины крутящих момен- тов по реометру Монсанто при 150 °C [MH-ML(dH-M)] 24,0 23,5 18,5 29,5 61,5 54,0
Разрывная прочность (МПа) 14,21 20,28 12,03 15,41 25,63 26,38
Относительное удлинение (%) 631 592 595 565 492 502
Модуль при 300% удлинении (МПа) 2,94 4,86 2,91 2,53 11,04 8,9
Твердость по Шору при 21 °C 39 46 40 39 57 57
Сопротивление раздиру по ASTM die В (кН/м) 30 61 22 31,5 161 140
Таблица 25. Влияние содержания жирных кислот в вулканизатах.
Рецепт Жирная кислота Масс.ч. 1 2 3 4 5 6
Стеариновая Олеиновая
1,0 2,0 3,0 1,0 2,0 3,0
Плотность сетки (рейтинг) 100 94 106 75 80 89
Энергия активации (КДж моль1) 131,5 101,5 97,6 135,1 114,2 110,5
Разрывная прочность (МПа) 27,5 26,8 26,9 28,5 28,0 26,4
Относительное удлинение (%) 545 535 538 591 576 551
Твердость по Шору 52 53 50 50 52 52
Сопротивление раздиру по ASTM die В (кН/м) 72 112 103 72 94 80
Разрывная прочность после старения (МПа) 17,5 18,1 21,3 15,8 16,8 17,3
Как часть изучения роли оксидов металлов, была проведена сравнитель-
ная оценка олеиновой и стеариновой кислот, они вводятся в смесь в 1,0, 2,0
и 3,0 масс. ч. по рецепту ASTM № А2 (по АХО — №2).
Данные, представленные в Таблице 25, подчеркивают несколько мо-
ментов:
446 -’V Глава 9. Наука о рецептуростроении резин
1. Увеличение содержания жирной кислоты снижает энергию активации
вулканизации, причем в наибольшей степени этот эффект проявляется
стеариновой кислотой.
2. Соотношение стеариновая кислота/ZnO в активирующей системе
приводит к увеличению плотности поперечных связей в сравнении с
системой с олеиновой кислотой.
3. Сопротивление старению и раздиру вулканизатов, полученных с ис-
пользованием комплекса стеариновая кислота/ZnO, выше таковых,
полученных с применением олеиновой кислоты.
Эффективность стеариновой кислоты в активации вулканизации явля-
ется функцией ее растворимости в эластомере, молекулярного веса и точки
плавления.
Б. Агенты вулканизации
В резиновой промышленности нашли широкое применение
три вулканизующих агента: сера, нерастворимая сера и пероксиды. Химия
пероксидов была рассмотрена в главе 7. Ромбическая сера - самая распро-
страненная форма серы, используемая в резиновой промышленности; она
соответствует приемлемым условиям гигиены труда и методам эксплуатации,
а также не требует каких-либо особых видов обработки и условий хранения.
Сера растворима в натуральном каучуке до уровня 2,0 масс. ч. Сверх этого
содержания необходимо использовать нерастворимую серу для предотвра-
щения миграции серы на поверхность смеси, т. е. выцветания серы.
В. Ускорители
Ускорители — это вещества, используемые как для ускоре-
ния процесса серного сшивания резиновых — смесей, так и для повышения
плотности сшивания.
Вторичные ускорители при добавлении к основным повышают скорость
вулканизации и степень сшивания, причем комбинации первичного и вто-
ричного ускорителей могут быть совершенно произвольными. Особенностью
таких бинарных систем является синергизм. Если комбинация ускорителей
синергична, то она проявляет более мощный эффект, чем аддитивный эффект
индивидуальных компонентов.
Ускорители легко классифицируют по одному из двух параметров:
7. Скорость вулканизации: Ультраускорители включают дитиокарбаматы
и ксаптогенаты. Полуультраускорители включают тиурамы и амины.
Быстрые ускорители — тиазолы и сульфенамиды. Система со средней ско-
ростью — дифенилгуанидин. Медленные ускорители — тиокарбанилид.
2. Химическая классификация: Большинство ускорителей принадлежат
одной из восьми представленных групп.
V. Вулканизирующие системы
Л
447
Альдегидамины
Тиомочевины
Гуанидины
Тиазолы
Сульфенамиды
Дитиокарбаматы
Тиурамы
Ксантогенаты
При выборе системы вулканизации необходимо учитывать тип эластомера,
тип и количество оксида цинка и жирной кислоты, скорость вулканизации,
требуемую усталостную выносливость и условия эксплуатации. Рекомендуется
избегать использования ускорителей, образующих нитрозоамины.
Тип эластомера влияет на скорость вулканизации, а также на образующу-
юся сетку поперечных связей. Натуральный каучук вулканизуется быстрее
бутадиен-стирольного каучука. Вулканизующие системы с тиурамными
ускорителями, например, тетраметилтиурам дисульфид, обладают корот-
ким индукционным периодом и быстрой скоростью сшивания в сравнении
с системами с дифенилгуанидином.
Сульфенамидные ускорители представляют самый широкий класс ус-
корителей:
Циклогексилбензтиазол сульфенамид
(сульфенасмид Ц, САЦ)
Трет-бутил-2-бензтиазол сульфенамид
(сульфенамид Т, CAT)
Механизм и химия вулканизации были рассмотрены ранее. Поэтому более
разумно определить основные принципы активности ускорителей, таких как
сульфенамид. Остановимся на трех параметрах, заслуживающих пояснения:
1. Прочность связи сера-азот: Сульфенамиды распадаются на мер-
каптобензтиазол, и аминные фрагменты в процессе образования
сульфидирующего комплекса, а амины образуют лиганды с ионом
цинка. Энергия связи должна быть достаточно низкой с тем, чтобы не
препятствовать образованию активных фрагментов ускорителя или
сульфидирующего агента.
2. Стереохимия аминных фрагментов: Если стерически блок аминного
лиганда, координированный с ионом цинка, очень большой, то он может
препятствовать образованию активного сульфурирующего агента. Это
проявляется в увеличении индукционного периода, изменении скорости
вулканизации и, в конечном счете, изменении физических свойств.
3. Основность аминного фрагмента: Повышение основности аминного
фрагмента сульфенамида приводит к увеличению скорости вулкани-
зации. Амины с высокой основностью имеют тенденцию к снижению
сопротивления скорчингу (Табл. 26). Дальнейшее пояснения по вул-
канизации следует отнести к главе 7.
448 -’V Глава 9. Наука о рецептуростроении резин
Таблица 26. Влияние основности (рКь) аминных фрагментов сульфенамида
на скорчинг смесиаб).
Радикал R рКь свободного амина Скорчинг по Муни t-10 при 135 °C Индекс скорости вулканизации
3,3 29,8 1,00
3,7 32,7 2,05
СН3 — NH —С-СН3 СН3 4,2 31,8 2,10
—N О 6,2 42,4 2,32
£ £ о о 1 6,2 45,2 2,57
а) Смесь: 100 масс. ч. БСК-1500, 50 технического углерода HAF, 4,0 ZnO, 2,0 стеариновой кис-
лоты, 10,0 масла, 1,5 анитоксиданта, 1,75 серы, эквимолярное содержание ускорителя.
б) Индекс скорости вулканизации t35-t10npn 135 °C по Муни [36].
Г. Ингибиторы подвулканизации и агенты,
предотвращающие реверсию
Индукционный период, или сопротивление скорчингу смеси,
могут быть откорректированы введением ингибиторов. N-циклогексилтиоф-
талимид (ЦТФ) лучший из всех многотонажных ингибиторов, используемых
в резиновой промышленности. Для того чтобы понять механизм действия ЦТФ,
а также химию других специальных ингибиторов, таких как вещества класса
тиосульфонамидов, читатель может ознакомиться с работой Морита [37].
Сопротивление реверсии смеси, особенно смесей на основе натурального
каучука, привлекло недавно большое внимание из-за широкого набора тре-
бований, которые включают ускорение переработки смеси в производстве,
переработки при повышенных температурах и, что особенно важно, удлине-
ние срока службы изделий. В промышленном масштабе получили широкое
применение три вида агентов предотвращения реверсии:
1. Как показано ранее, полу-ЭФ система представляет собой комплекс,
предназначенный для получения вулканизатов, содержащих баланс
V. Вулканизирующие системы
Л
449
моносульфидных и полисульфидных поперечных связей в оптимуме
вулканизации. Если полисульфидные связи надо сохранить в течение
длительного периода, то во время реверсии должны образоваться
новые взамен разрушенных. При использовании обычных систем ус-
корителей такие возможности ограничены. Сохранение полисульфид-
ной сетки в процессе вулканизации требует использование двойных
вулканизующих систем, каждая из которых действует независимо
друг от друга. Это принцип равновесных вулканизующих систем (РВ).
В этом случае бис (3-триэтоксилилпрорил) тетрасульфон (TESPT)
вводится как слабый донор серы [38] (Рис. 7).
2. Бис (цитроконимидометил) бензол с промышленным названием
Perkalink 900 вводится в смеси исключительно как замедлитель ре-
версии. Подразумевается, что он образует шестичленное кольцо на
полимерной цепи через реакцию Дильса-Альдера (Рис. 8). Образую-
щаяся в пределе поперечная связь термически стабильна и заменяет
серные связи, которые распадаются во время реверсии [39].
3. Натрий гексаметилен-1,6-бистиосульфид дигидрат при ведении
в вулканизующую систему распадается, и образуются гексаметилен-
1,6-дитиильные группы в дисульфидных или полисульфидных попе-
речных связях. Это называется гибридной поперечной связью. При
продолжительном времени вулканизации или за счет аккумуляции
тепла при эксплуатации изделия гексаметилен-полисульфидные
поперечные связи укорачиваются, образуя моносульфидные термо-
450 -*V" Глава 9. Наука о рецептуростроении резин
стабильные связи. При содержании до 2,0 масс. ч. наблюдается малое
влияние на индукционный период или время скорчинга композиции,
и на другие ее механические свойства [39].
+ Na’SO3- S-(CH2) - S—SO3Na+ 2Н2О
Гексаметилен-1,6-бис (тиосульфат)
двунатриевая соль, дигидрат
В Таблице 27 представлены принятые промышленные обозначения
некоторых ускорителей.
Цепь полиизопрена
Рис. 8. Предпологаемый механизм предотвращения реверсии с помощью бис
(цитраконимидометил) бензола (ВЦИМБ).
VI. СПЕЦИАЛЬНЫЕ ИНГРЕДИЕНТЫ СМЕСЕЙ
В дополнение к четырем основным компонентам резиновой
смеси (полимеры, наполнители, системы стабилизаторов и вулканизующие
системы) существует ряд вторичных веществ, таких как вспомогательные
вещества для переработки, смолы, окрашивающие пигменты (например,
диоксид титана, используемый в белых боковинах шин). Рассмотрим их
кратко для руководства при выборе этих ингредиентов для практических
рецептур резиновых смесей.
VI. Специальные ингредиенты смесей -•V 451
Таблица 27. Сопоставление сопротивления подвулканизации некоторых уско-
рителей промышленного значенияа).
Ускоритель6) Обозна- чение Время подвулканиза- ции (мин)
Для смеси 1 по ACS Для смеси 2 по ACS
Диизопропилбензотиазол ДИПБТ 63 21
Циклогексил-2-бензтиазолсульфенамид СА-Ц 26 15
Третбутил-2-бензтиазолсульфенамид СА-Т 60 23
Оксидиэтилен бензтиазил-2-сульфенамид СА-ДЭ (САМ) 60 24
Диметил морфолин бензтиазол-2-суль- фенамид СА-М 60 27
Бензтиази л-N,N-диэтилтиокарбамил сульфид - 26 15
Меркаптобензтиозол МБТ 12 9
Бензтиазил дисульфид ДБТД 73 12
Тетраметилтиурам моносульфид тмтм 25 12
Тетраметилтиурам дисульфид тмтд 13 6
Тетраэтилтиурам дисульфид тэтд 18 9
Дибутилдитиокарбамат цинка Zn-ДБДТК 5 4
Диметилдитиокарбамат цинка Zn дмдтк 6 4
Дифенилгуанидин ДФГ 16 10
а) Данные получены для смеси 2 по АХО (ASTM D3184, 2а), но содержащей 50 масс. ч. HAF.
б) Необходимо избегать использования ускорителей, образующих нитрозоамины.
А. Масла
Масла в составе рецептуры резиновой смеси являются прежде
всего вспомогательными веществами для переработки. Масла принадлежат
к одному из трех основных классов: парафиновые, нафтеновые и аро-
матические. Выбор масла для включения его в рецептуру очень важен.
Если масло несовместимо с полимером, то оно мигрирует из смеси,
снижая впоследствии физические и поверхностные свойства и адгезию
между компонентами в изделиях, таких как шины. Совместимость мас-
ла с полимером зависит от его свойств — вязкости, молекулярного веса
и строения молекул. В Таблице 28 представлены физические свойства масел
трех типичных классов.
452 -*v- Глава 9. Наука о рецептуростроении резин
Таблица 28. Физические свойства масел трех классов, используемых в рези-
новой промышленности.
Физические свойства Метод ASTM Парафиновые Нафтеновые Ароматические
Удельный вес D1250 0,85-0,89 0,91-0,94 0,95-1,0
Точка потери текучести (°C) D97 от -17,8 до -13 от -40 до -7 от +5 до +32
Коэффициент преломления D1747 1,48 1,51 1,55
Анилиновая точка D611 200-260 150-210 95-150
Плотность по API D287 28-34 19-28 10-19
Молекулярный вес D2502 320-650 300-460 300-700
Содержание ароматики (%) 15 44 68
Анилиновая точка является мерой содержания ароматики в масле. Это
точка, при которой масло растворяется в анилине. Таким образом, чем ниже
анилиновая точка, тем выше содержание ароматики. Все три класса масла
содержат большое количество циклических углеродных структур; разница
заключается в содержании насыщенных и ненасыщенных колец. Качест-
венно масла могут быть охарактеризованы так:
- Ароматические масла содержат большое количество ненасыщенных
колец, ненасыщенных нафтеновых колец и разветвления из алкильных
насыщенных углеводородных цепей. Преобладающая структура —
ароматическое кольцо.
- Нафтеновые масла содержат большое число насыщенных колец
и малое количество ненасыщенных.
- Парафиновые масла содержат большое количество нафтеновых колец,
но также много алкильных разветвлений, ненасыщенных углеводо-
родных разветвлений и, что наиболее важно, несколько нафтеновых
групп на одну молекулу. Чистые парафины из очищенной нефти
конденсируются в виде воска.
Выбор масла для определенного полимера зависит от наличия в нем по-
лярных групп таких, как -CN в БНК и -С1 в ПХП. Эффективность масла
в смеси определяется наличием водородных связей и Ван-дер-ваальсовых
сил. В таблице 29 приведены основные позиции, определяющие выбор
масла для данного полимера. Эта таблица вынужденно краткая и поэтому
существует множество исключений. Необходимо отметить ключевые пара-
метры: растворимость масла в полимере, тенденция масла к обесцвечиванию
изделия или окрашиванию других компонентов в изделии [36].
VI. Специальные ингредиенты смесей -•V- 453
Таблица 29. Правила выбора масла для ряда эластомеров промышленного
значения.
Масла Полимер Примеры применения изделий
Нафтеновые Этилен-пропиленовый каучук Этилен-пропилен-диеновый каучук Полихлоропрен БСК ПБ Уплотнители и уплотняющие составы Адгезивы Обычные резиновые изделия
Парафиновые Натуральный каучук Полиизопрен синтетический Бутил каучук БСК Полихлоропрен Материалы для текстильной промышленности, Уплотнители и уплотняющие составы
Ароматические Натуральный каучук БСК Полибутадиен Шины Детали автомобилей
Б. Пластификаторы
Хотя масла, воска и жирные кислоты можно назвать плас-
тификаторами, но в резиновой промышленности термином пластификатор
наиболее часто называют класс материалов, включающий сложные эфиры,
сосновую смолу и низкомолекулярный полиэтилен.
Фталаты — наиболее часто используемые сложные эфиры. Дибутилфта-
лат (ДБФ) склонен придавать мягкой композиции липкость (клейкость),
диоктилфталат (ДОФ) менее летуч, а композиции получаются более жестки-
ми из-за его высокой молекулярной массы. Полимерные эфиры такие, как
полипропиленадипинат (ППА), используются там, где необходима низкая
летучесть одновременно с хорошей теплостойкостью.
Несмотря на тенденцию к снижению общего потребления, сосновая
смола хорошо совместима с натуральным каучуком, обеспечивает хорошее
распределение наполнителей в нем, повышает такие свойства метериала,
как усталостную выносливость и адгезию компонентов, что очень важно
для долговечности шины. Другие пластификаторы, вводимые в небольших
количествах: фактис (вулканизованное серной вулканизующей системой
растительное масло); соли жирных кислот, такие как стеарат цинка, который
может также действовать как пептизатор; канифоль; низкомолекулярный
полипропилен; органосиланы, такие как диметилполисилоксан.
В. Химические пептизаторы
Пептизаторы действуют либо как катализаторы окисления,
либо как акцепторы радикалов, которые в основном дезактивируют сво-
бодные радикалы, образующиеся в процессе смешения. Это предотвращает
454 -*V Глава 9. Наука о рецептуростроении резин
рекомбинацию полимерных радикалов, приводит к снижению молекулярной
массы полимера и, как следствие, понижению вязкости смеси. Такое размяг-
чение полимера облегчает введение ряда ингредиентов согласно рецептуре.
Примеры пептизаторов: пентахлоротиофенол, фенилгидразин, некоторые
дифенилсульфиды и ксилил меркаптан. У каждого пептизатора есть своя
оптимальная концентрация, при которой он наиболее эффективен.
Пептизаторы, такие как пентахлоротиофенол, обычно вводят в количес-
тве от 0,1 до 0,25 масс. ч. Это позволяет значительно облегчить переработку,
снизить энергию, затрачиваемую на смешение, и улучшить однородность
смеси. Высокое содержание пептизатора может неблагоприятно сказыва-
ется на свойствах материала, так как избыток продолжает катализировать
деструкцию полимера в процессе эксплуатации изделия.
Г. Смолы
Смолы делят на три категории: (1) облегчающие переработку,
(2) смолы для повышения клейкости и (3) смолы-вулканизующие агенты.
Смолы также довольно произвольно классифицируют на углеводородные,
нефтяные и фенольные.
Углеводородные смолы имеют высокую температуру стеклования, та-
ким образом, при температурах переработки они плавятся, что позволяет
улучшить вязкость расплава. Однако, они затвердевают при комнатной
температуре, что позволяет поддерживать жесткость и модули материала.
В ряду углеводородных смол ароматические используют как усиливающие
агенты, алифатические — улучшают клейкость, а промежуточные смолы
проявляют оба эти качества. Кумарон-инденовые смолы — пример таких
систем. Такие смолы способствуют:
1. Улучшению разрывной прочности в результате затвердевания при
комнатной температуре.
2. Повышению усталостной выносливости в результате улучшенного
диспергирования наполнителей и смачивания их поверхности.
3. Замедлению роста трещин из-за рассеивания напряжений в вершине
трещины (в результате снижения вязкости материала).
Нефтяные смолы — побочные продукты перегонки нефти. Выпуска-
ется ряд видов аналогично углеводородным смолам. Алифатические смо-
лы, содержащие олигомеры изопрена, используются в качестве веществ,
повышающих клейкость, тогда как ароматические смолы, содержащие
также большое количество дициклопентадиена, относят в большей степени
к усиливающим системам.
Фенольные смолы бывают двух типов: реактивные и нереактивные.
Нереактивные смолы существуют в виде алкил-фенил-формальдегидных
олигомеров, где пара-алкильные группы могут быть от С4 до С9. Такие смолы
VII. Разработка композиций -•V- 455
используют в качестве веществ, повышающих клейкость. Реактивные смо-
лы содержат свободные метилольные группы. В присутствии метиленовых
доноров, например гексаметилентетрамина, образуется сетка поперечных
связей, что дает возможность реактивным смолам действовать в качестве
усиливающих смол и промоторов адгезии.
Д. Короткие волокна
Короткие волокна могут вводиться в композиции с целью
повышения прочности материала. Они могут перерабатываться как другие
ингредиенты композиции. Короткие волокна включают нейлон, полиэфи-
ры, стекловолокно, арамид и целлюлозу. Введение коротких волокон, как
усиливающих компонентов, зависит от области применения изделия. Основ-
ные преимущества коротких волокон: улучшенная разрывная прочность,
улучшенная усталостная выносливость и сопротивление росту трещин,
повышенная жесткость, увеличенная жесткость компонентов или изделий,
повышенная стойкость к порезам и отслоениям, например, в покрышках
шин.
VII. РАЗРАБОТКА КОМПОЗИЦИЙ
Выше рассмотрен ряд материалов, входящих в резиновые
композиции с целью достижения различных механических свойств. Компо-
зиции могут быть разработаны одним из двух методов. Модельная рецептура
может быть получена из литературных источников поставщиков сырья или
других промышленных источников, например, отраслевых журналов. Такие
рецептуры соответствуют необходимым физическим свойствам материала и
эксплуатационным требованиям. В процессе оптимизации может быть от-
корректировано содержание вводимых компонентов, например, количество
ускорителя для получения требуемого индукционного периода вулканиза-
ции или технического углерода с целью получения требуемой разрывной
прочности. Когда требуется подобрать более сложный комплекс свойств,
и нет модельных рецептур, используют более эффективный метод анализа
Тагучи или многоплановый регрессионный анализ.
Ряд ингредиентов в рецептуре можно оптимизировать одновременно при
использовании компьютерных программ. При этом необходим ряд рецептур
для использования в заданных экспериментах [40]. Независимо от метода
или выбранной модели, необходим ряд простых шагов, предшествующих
экспериментальной работе:
1. Формулировка задач работы
2. Определение вариантов анализируемых рецептур
3. Выбор подходящего метода анализа для накопления эксперименталь-
ных данных
456 -JV‘ Глава 9. Наука о рецептуростроении резин
4. Исследование данных с учетом ранее опубликованных данных
и знаний об активности и характеристиках изучаемого сырья
5. Статистические закономерности данных (распределение данных,
ошибка опыта и т.д.)
Выбранная модель должна повлечь за собой:
1. Определение ключевых свойств модели, таких как разрывная про-
чность, усталостная выносливость и гистерезис.
2. Выбор подходящего метода, например, бивариантный факториал или
трех-, четырех-, пятивариантную множественную регрессию.
3. Вычисление коэффициентов множественной регрессии из накоплен-
ных экспериментальных данных. Коэффициенты могут быть вычис-
лены уравнениями регрессии, которые могут быть линейными,
свойство = аХ +bY + С
в случае простой факториальной модели, либо полинома второго
порядка, где взаимодействия компонентов рецептуры могут быть
выражены следующим образом:
свойство = аХ + bY + cZ + dX? + eft + fZ? + gXY + hXZ + jYZ + С
Здесь «свойство» — зависимая переменная, может быть, например, мо-
дулем, а X, Y и Z — независимые переменные, например, содержание
масла, технического углерода и серы, a, b, с, d и т.д. — коэффициенты
соответствующих зависимых переменных, а С — индивидуальная
константа для соответствующей модели. Очевидно, что возможны
и другие уравнения, вид которых зависит от цели исследования.
4. Построение графических зависимостей, отображающих общий харак-
тер данных и повышенную роль взаимодействия между компонентами
в композиции.
5. Расчет оптимального содержания ингредиентов.
6. Если требуется, разработку точной технологии смеси для подтверж-
дения рассчитанного оптимального содержания ингредиентов.
Имеется большой набор построения экспериментальных исследований
и поэтому рекомендуем обратиться к прилагаемым литературным источни-
кам для получения дальнейшей информации.
В Таблицах 30-35 представлены модельные рецептуры, на которых может
быть основана дальнейшая работа по оптимизации составов композиций.
Вдобавок, возможно использование рецептур из промышленных публика-
ций, таких как Малазийская Ассоциация Производителей и Исследователй
каучука [41, 42].
VII. Разработка композиций -* V 457
Таблица 30. Примеры шинных рецептур.
Протектор Боковина Обрезинка проволоки Прорези- ненный слой Внутренняя обкладка
Система полимеров НК ПБ БСК эмуль. БСКраств. НК ПБ НК НК ПБ БК ХБК ББК НК
Система наполнителей SAF ISAF HAF FF FEF GPF HAF HAF GPF
Вулканизу- ющая система Полуэф- фективная Адаптиро- ванная к полимер- ной системе Обычная система Обычная система Полуэф- фективная Адаптиро- ванная к полимерной системе
Прочие компоненты Масла Воска Антиокси- данты Воска Промоторы адгезии Промоторы адгезии -
Таблица 31. Модельная рецептура протектора грузовой шины.
Натуральный каучук 100,00
Технический углерод (N220) 50,00
Пептизатор 0,20
Парафиновый воск 1,00
Микрокристаллический воск 1,00
Ароматическое масло 3,00
Полимеризованный дигидротриметилхинолин 1,00
Стеариновая кислота 2,00
Оксид цинка 5,00
Сера 1,20
СА-Т 0,95
ДФГ 0,35
Ингибитор (если требуется) 0,25
458 “'V Глава 9. Наука о рецептуростроении резин
Таблица 32. Модельная рецептура боковины.
Натуральный каучук 50,00
Полибутадиен 50,00
Технический углерод (N330) 50,00
Пептизатор 0,20
Парафиновый воск 1,00
Микрокристаллический воск 1,00
Ароматическое масло 10,00
Полимеризованный дигидротриметилхинолин 1,00
Диметилбутилфенил-п-фенилендиамин 3,00
Стеариновая кислота 2,00
Оксид цинка 4,00
Сера 1,75
СА-Т 1,00
ДФГ 0,35
Ингибитор (если требуется) 0,25
Таблица 33. Модельная рецептура прорезиненных слоев каркаса.
Натуральный каучук 70,00
Полибутадиен 30,00
Технический углерод (N660) 55,00
Пептизатор 0,20
Парафиновый воск 1,00
Микрокристаллический воск 1,00
Ароматическое масло 7,00
Полимеризованный дигидротриметилхинолин 1,00
Диметилбутилфенил-п-фенилендиамин 2,50
Стеариновая кислота 2,00
Оксид цинка 4,00
Сера 2,50
ДБТД 0,80
VII. Разработка композиций -*V 459
Таблица 34. Модельная рецептура наружного слоя конвейерной ленты.
Натуральный каучук 100,00
Технический углерод (N330) 50,00
Пептизатор 0,20
Парафиновый воск 1,00
Микрокристаллический воск 1,00
Ароматическое масло 5,00
Полимеризованный дигидротриметилхинолин 1,00
Диметилбутилфенил-п-фенилендиамин 2,50
Стеариновая кислота 2,00
Оксид цинка 4,00
Сера 2,50
СА-Т 1,00
Таблица 35. Подковровый слой.
Бутадиен-стирольный каучук 100,00
Карбонат кальция 150,00
Регенерат (как наполнитель) 20,00
Каолин 25,00
Оксид железа 4,00
Бикарбонат натрия 10,00
Порообразующий агент 1,00
Пептизатор 0,20
Парафиновый воск 1,00
Микрокристаллический воск 1,00
Ароматическое масло 60,00
Стеариновая кислота 1,50
Оксид цинка 3,00
Сера 3,00
МБТ 1,50
ТМТД 0,50
ДФГ 0,50
460 -*v- Глава 9. Наука о рецептуростроении резин
VIII. ПОЛУЧЕНИЕ КОМПОЗИЦИЙ
В производстве современных шин и основных изделий,
резиновые смеси готовят в закрытых резиносмесителях. Закрытые рези-
носмесители включают камеру, в которую вводятся ингредиенты смеси.
В камере расположены два ротора, которые создают большие сдвиговые
усилия, диспергирующие наполнители и другие компоненты сырой смеси в
полимере. Благодаря этим сдвиговым напряжениям получаются однородные
высококачественные композиции. После определенного времени смешения
смесь подается на вальцы или в экструдер, где завершается процесс смеше-
ния и смесь профилируется в форму, удобную для дальнейшей работы. Как
альтернатива, смесь может быть превращена в гранулы.
В зависимости от сложности рецептуры, объема резиносмесителя
и назначения композиции цикл смешения может быть разделен на последо-
вательные стадии. Для всех композиций на основе натурального каучука,
содержащих 50 масс. ч. технического углерода, 3 масс. ч. ароматического
масла, антиоксиданты и полуэффективную вулканизующую систему
типичный цикл смешения в резиносмесителе Бенбери может быть пред-
ставлен так:
Стадия 1 Введение натурального каучука; если требуется, введение пептизатора.
Обработка на вальцах при 165 °C.
Стадия 2 Диспергирование технического углерода, масел, антиоксидантов, оксида
цинка, стеариновой кислоты и разнообразных пигментов, например,
огнезащитных, при 160 °C
Стадия 3 Если требуется снизить вязкость смеси, проводят повторное смешение
в закрытом резиносмесителе в течение 90 секунд при 130 °C
Стадия 4 Введение в смесь вулканизующей системы и смешение при температуре
не превышающей 115 °C.
Компьютерный мониторинг переменных закрытого резиносмесителя,
таких как потребляемая мощность, температурный градиент по всей сме-
сительной камере и время смешения, дает возможность на современных
смесителях производить стабильные высоко качественные смеси в больших
объемах. Готовая смесь затем транспортируется либо в экструдеры для по-
лучения профилей экструдата, на каландры для листования, либо для литья
под давлением.
Смесь может быть приготовлена на вальцах, если это требуется по
физическим свойствам композиции. Смешение на вальцах занимает
более продолжительный период, требует больше энергии и позволяет
получать смеси меньшего объема. Однако, температурный режим в про-
цессе смешения понижается, что является преимуществом, если в составе
смеси имеются системы ультраускорителей. Смешение на двухвалковых
вальцах осуществляется за счет сдвиговых деформаций, вызываемых
IX. Требования к безопасности окружающей среду в рецептуростроении 461
вращением валков с различной скоростью (фрикцией). Это соотношение
скоростей валков можно варьировать и устанавливать в зависимости от
типа смеси. Чем больше фрикция, тем больше сдвиговое напряжение
и интенсивность смешения.
IX. ТРЕБОВАНИЯ К БЕЗОПАСНОСТИ ОКРУЖАЮЩЕЙ
СРЕДЫ В РЕЦЕПТУРОСТРОЕНИИ
Как дополнение к производству изделий с целью удов-
летворения требований потребителей, необходимо рассмотреть влияние
этого производства на окружающую среду. Воздействие производства на
окружающую среду должно рассматриваться с двух позиций: (1) при-
менение изделий и его длительное воздействие на экологию, (2) техника
безопасности и влияние на здоровье человека, как в производстве, так и
работе изделий.
Примером роли качества изделия на его использование и воздействие на
окружающую среду яв ляется сопротивление качению шины и его влияние
на потребление моторного топлива. Снижение сопротивления качению
приводит к снижению потребления моторного топлива. Это незамедлитель-
но сказывается на образовании выхлопных газов, таких как угарный газ,
углекислый газ и оксиды азота.
Наружная область шины, которая включает протектор и боковину,
ответственна приблизительно за 75% сопротивления качению легковых
радиальных шин. Улучшение гистерезисных свойств протектора приводит
к снижению сопротивления качению и, как следствие, к экономии автомо-
бильного топлива. Внешняя область шин и, в частности, рецептура самого
протектора, также влияет на долговечность шины. Шины длительного
использования (включая восстановление протектора) удлиняют время,
когда отработанные шины уже перейдут в категорию твердых отходов для
системы утилизации.
Критически важным для жизненного цикла шины является способ-
ность сохранять давление воздуха. Внутренние слои шины делают на
основе галобутилкаучуков, которые характеризуются очень низкой вла-
го- и воздухопроницаемостью. Таким образом, шины, созданные с хоро-
шим подбором компонентов, могут снижать скорость преждевременного
разрушения, т. е. откладывать их переход в твердые или измельченные
отходы.
Таблица 36 показывает, как неправильный выбор полимера внутреннего
слоя может привести к быстрому снижению рабочих свойств шины. Замеще-
ние хлорбутил-каучука на натуральный каучук или бутил-каучук приводит
к более быстрому снижению давления воздуха в шине и ухудшению других
рабочих свойств [43].
462
—Глава 9. Наука о рецептуростроении резин
Таблица 36. Взаимосвязь между содержанием ГБК/БК и проницаемостью
и выносливостью при ступенчатом нагружении.
Содержание каучука в слое ГБК/БК об.% Проница- емость слоя (хЮ8) при 65 °C Равновесное внутрикаркасное давление МПа Время до разрушения при ступенчатом нагружении, час
100 ББК 65,2 3,0 0,032 61,5
75 ББК/25НК 48,3 4,2 0,063 56,9
65 ББК/35 НК/20 БК*' 42,2 5,9 0,063 40,2
60 БСК/40 НК/20 БК*> 16,1 6,9 0,090 31,5
*} БК в виде регенерата камер, содержащий -50% по весу БК. Рассматривается как наполнитель.
Улучшение конструкции шины позволило снизить уровень шума. Это
имеет важное значение для охраны окружающей среды. Оптимальное дав-
ление шины на поверхность уменьшает разрушение дорожных покрытий
и мостов. Улучшение всех этих характеристик, таких как, сопротивление
качению, долговечность, производимый шум и давление шины на повер-
хность входят в характеристики всех шин от малых легковых до больше-
грузных шин и землеройных машин. В современных радиальных шинах
используют от 60 до 80% натурального каучука как полимерной составля-
ющей композиций. Поскольку натуральный каучук получают из растений,
он является идеальным возобновляемым ресурсом, и, таким образом, как
биотехнологический материал предпочтительнее синтетических полимеров,
если возможно достижение эквивалентных свойств изделий.
Шины — одни из наиболее долговечных технических изделий, произво-
димых сегодня. Они представляют собой упругие и долговечные композиты
из текстиля, стали, технического углерода, натурального каучука и синтети-
ческих полимеров. Качества, которыми обладают шины и другие резиновые
технические изделия, делают их изделиями высокой важности и поднимают
проблему утилизации. Шины и другие резиновые изделия, например кон-
вейерные ленты и гидравлические рукава, не биоразлагаемы и не могут быть
повторно использованы как стекло, алюминий или пластик. Определены
четыре потенциальных направления для использования твердых отходов:
1. Теплотворная способность шин выше (35 МДж/кг) теплотворной
способности угля (24 МДж/кг). С учетом хорошо разработанного
оборудования, шины можно сжигать для обогрева в цементных печах
для обжига и сушки.
2. Шины могут быть сожжены в печах энергетических станций для по-
лучения электрической энергии.
3. Измельченные шины начали применяться в различных целях при
производстве асфальта.
IX. Требования к безопасности окружающей среду в рецептуростроении —'V 463
4. Шины с хорошей технологией сборки могут быть использованы
в различных целях в строительной промышленности в качестве опор,
могут быть применены в энергосберегающем домостроении, для ма-
гистральных усиливающих насыпей и для предотвращения эрозии
почвы.
Эти четыре способа утилизации представляют наилучшие варианты при-
менения измельченных шин и резиновых изделий. Ожидается, что в будущем
появится ряд новых применений измельченных резиновых изделий.
В итоге специалисты материаловеды должны предусмотреть решения
по выбору материалов с позиций затрат энергии на производство изделия,
на характеристики во время эксплуатации и, как завершение, методы
утилизации. Управление по охране окружающей среды предлагает также
ограничения, которые специалист по материалам должен учитывать при
конструировании. Поскольку большинство резиновых изделий содержит
примерно от 6 до 20 различных компонентов, то не только сами ингредиен-
ты должны быть чистыми и безопасными, но и любые побочные продукты,
которые образуются в процессе производства шин, также должны быть
безопасны как для человека, так и для окружающей среды.
Ароматическая составляющая технического углерода и масел рассмат-
ривается как представляющая опасность. Были собраны данные, которые
показали, что технический углерод стабилизирован и не является опасным
для рабочих. Смолы для вулканизации и повышения клейкости, антиок-
сиданты, антиозонанты и ускорители вулканизации должны быть также
проанализированы, для того, чтобы убедиться, что материалы и любые
примеси не вредят здоровью и отвечают стандартам безопасности. На все
материалы должны быть предоставлены данные по их опасности для здо-
ровья и токсичности.
Химикаты, образующие нитрозоамины, являются предметом анализа на
подозрительные вещества, которые должны быть изъяты из производства
резиновых изделий, несмотря на то, что до сих пор еще не принят соответству-
ющий законопроект. Нитрозоамины могут образовываться, когда в составе
вулканизующей группы содержатся ускорители, содержащие вторичные
амины. Эти изменения в выборе ускорителя имеют очень большое значение
во всей резиновой промышленности.
Природа растворителя и ограничения по летучести имеют большое
значение в производстве синтетического каучука, а также шинной про-
мышленности. Ограничения на наличие следов примесей, разработанные
федеральным актом чистоты воздуха в США, основаны на представленных
опасностях, а не только на наиболее доступных их оценках.
В итоге, специалист-материаловед должен уметь оценивать в стандартах
безопасности влияние применяемых реагентов как на окружающую среду,
так и на здоровья человека.
464 -'V Глава 9. Наука о рецептуростроении резин
X. ВЫВОДЫ
В этой главе были рассмотрены типы и свойства эластомеров
в композициях с рядом наполнителей или усиливающих систем таких, как
технический углерод и наполнители с улучшенными свойствами благодаря
использованию новых компонентов, в виде силанольных агентов сочетания.
Были также рассмотрены другие компоненты, такие как вулканизующие
системы и системы антиоксидантов. Задача современного исследователя-
материаловеда в шинной и резиновой промышленности — использование
материалов с целью улучшения действующего производства и разработки
новых материалов. Существует четыре основных положения, определяющих
этот процесс развития:
1. Рабочие показатели: Изделие должно удовлетворять ожиданиям
потребителей.
2. Качество: Изделие должно быть надежным и иметь хороший внешний
вид; соответствующий контроль производства должен гарантировать
постоянство и стандартность изделий.
3. Влияние на окружающую среду: Изделие должно быть экологически
безвредным при производстве, применении и утилизации.
4. Стоимость: Система должна давать рыночную стоимость, доступную
для покупателя.
В достижении этих изделий наука о рецептуростроении резин выросла
из «черной магии», какой она была в начале 20 века, до сложной науки,
требующей знаний современной химии, физики и математики.
ПРИМЕЧАНИЯ ПЕРЕВОДЧИКА
Авторы пользуются некоторым количеством сведений, недо-
статочно разъясняемых читателю. Например, рейтинговые сравнительные
оценки полимеров и некоторых ингредиентов (рис. 2, рис. 3).
Излагая представления о механизме действия активатора вулкани-
зации оксидов металлов (глава V, стр. 442-446), авторы предлагают
классифицировать их по значениям электроотрицательности метал-
лов. По их представлениям активность при вулканизации характе-
ризуется цифрами этих величин 1,6-1,8, куда попадают только ZnO
и РЬО. Сюда должен быть включен CdO, о котором авторы упоминают, что
он токсичен и поэтому не исследовался. РЬО также токсичен, но он вклю-
чен в Таблицу (Табл. 23-24 ). Это одно из объяснений активирующего
действия металлов при вулканизации. Однако, электроотрицательность
в указанных авторами пределах имеют такие металлы, как Ga, Ge, Cr,
In, Sn, W, Mo, V (см. Л. Полинг. Общая химия. M., Мир. 1974, стр. 168),
оксиды которых активаторами не являются.
X. Выводы Jb 465
Другая точка зрения состоит в объяснении роли оксидов металлов в связи
с их каталитической активностью и способностью адсорбции и хемосорбции
вулканизующих веществ на поверхности. На основе представлений об этой
роли оксидов металлов, изложенных О.В. Крыловым (см. Крылов О.В. Ка-
тализ не металлами. Л. Химия, 1967, 240 с.: Киселев В.Ф., Крылов О.В. Ад-
сорбционные процессы на поверхности полупроводников и диэлектриков,
М. Наука, 1978,256 с.), было предложено оценивать активирующее действие
ряда оксидов металлов в реакциях вулканизации на примерах вулканизации
дисульфидами — ТМТД, ТЭТД, ДБТД, ДТДМ (см. V.A. Shershnev. Rub.
Chem. Tecnol, 1982, v. 55, № 3, pp. 543-551; Манько E.H., Шершнев B.A.,
Догадкин Б.А., Коллоидн. Журн., 1971, т. 33, №4, с. 562; там же 1973, т. 35,
№ 2, с. 271; Каучук и резина, 1969, № 6, с. 14; Догадкин Б.А., Донцов А.А.,
Шершнев В.А. Химия эластомеров, М. Химия, 1981, с. 260-269; Кузь-
минский А.С., Кавун С.М., Кирпичев В.П. Физико-химические основы
получения, переработки и применения эластомеров. М. Хмия, 1976,
с. 124-140; 166-181). Оксиды исследованных металлов были разделены на
две категории: первая категория — ширина запрещенной зоны (Цв) более
3,3 — MgO — 8,7; СаО — 7,5; GeO2 — 6,0; Ga2O3 — 4,6; In2O3 — 3,5; вторая
категория — U в интервале 2,3 — 3,3 — ZnO — 3,3; CdO — 2,5; PbO — 2,3;
Bi2O3 — 2,9. Активирующее действие проявляют, хотя и в разной степени,
оксиды второй категории. Определяющим является ширина запрещенной
зоны оксида как полупроводника, характеризующая энергию возбуждения
электронов. При этом оксид металлов должен быть способен адсорбировать
и хемосорбировать на своей поверхности вулканизующий агент. На повер-
хности оксида происходит хемосорбция молекул вулканизующих веществ,
в частности, указанных дисульфидов с разрывом химической связи в них.
В серно-ускорительных системах этот же эффект имеет место при хемо-
сорбции сульфидирующих комплексов на оксидах металлов. На оксидах
с более высокими значениями Uтакая адсорбция и хемосорбция с разрывом
химических связей ди- и полисульфидов не происходит. Тоже наблюдается
и для оксидов с меньшими значениями U, но эти оксиды включают металлы,
которые способствуют развитию окислительных реакций и поэтому их роль
оценить однозначно не удается.
Таким образом, представление о диссоциативной адсорбции более полно
описывает активирующее действие оксидов металлов. Эти реакции имеют
признаки топохимических реакций и вписываются в общую концепцию
гетерогенных реакций при вулканизации. Жирные кислоты способствуют
лучшему диспергированию оксидов и вулканизующих систем. При обра-
зовании солей с оксидами эти реакции продолжаются на их поверхности.
Реакции стеариновой кислоты с оксидом цинка исследованы и имеют все
признаки топохимической реакции: скорость ее имеет максимум в зависи-
мости от времени и степени превращения реагентов.
ГЛАВА
10
ПРОЧНОСТЬ ЭЛАСТОМЕРОВ
А. Н. ДЖЕНТ
Университет Акрона.
Акрон. Огайо.
I. ВВЕДЕНИЕ
Разрушение является высокоизбирательным процессом:
только малое число молекул, составляющих образец для испытаний или
деталь, действительно подвергаются разрыву; огромное большинство их не
участвуют в разрушении. Например, из 1026 цепных молекул на кубичес-
кий метр, что типично для эластомера, только около 1018/м2, попадающих
в плоскость разрыва, будут действительно разрушены. Более того, они будут
все разрушены не одновременно, а последовательно, так как разрушение
распространяется по образцу с конечной скоростью.
Таким образом, первым вопросом, возникающим при изучении прочности
эластомеров (так же как и других материалов), является такой — где и при
каких условиях начинается разрушение? А также, какой закон определяет
рост трещины, после того, как она возникла? В этой главе книги ищем ответ
на эти вопросы, сначала в общем виде, а затем при конкретном рассмотрении
важнейших форм разрушения эластомеров в процессе эксплуатации. Не бу-
дут рассматриваться различные сложные проблемы прочности композитных
структур, таких как пневматические шины, которые включают разрушение
адгезионных связей на поверхностях между компонентами, так же как
и разрушение самих компонентов.
Прежде всего, мы рассмотрим возникновение разрушения в дефектах
или ослабленных местах, где действующие напряжения существенно выше.
Скорость развития трещины после её возникновения будет рассмотрена
позже. Естественно, что это зависит от уровня локальных напряжений, но
также и от характера изменений этих напряжений во времени. Для примера,
быстрый рост трещины может иметь место, если напряжения часто возни-
кают и исчезают, в то же время трещина может расти очень медленно, если
напряжение поддерживается постоянным и никогда не исчезает. Это явление
ускорения роста при динамическом напряжении называется механичес-
II. Возникновение разрушения -’V 467
кая усталость или динамический рост трещины. Оно будет рассмотрено
в разделах У(Д) и VII.
Поскольку при изменении размеров резина является высокоэластичным,
или, в более общем смысле, квазиупругим материалом и поскольку её ме-
ханические свойства зависят от скорости деформации и температуры, то не
удивительно, что прочность также зависит от скорости, с которой возникает
напряжение, и от действующей температуры. Эти эффекты будут рассмотрены
в разделах V(B) и VI(А). Другие составляющие влияния окружающей среды,
прежде всего деструктивное действие озона, будут рассмотрены в разделе VIII.
В последнем разделе дано краткое рассмотрение абразивного износа.
II. ВОЗНИКНОВЕНИЕ РАЗРУШЕНИЯ
А. Дефекты и концентраторы напряжений
В каждом твердом теле имеются дефекты или ослабленные точки, возник-
шие в результате неоднородности конструкции или структуры. Кроме этого,
из-за наличия острых углов, царапин, щербинок, порезов и внедрившихся
частиц примесей или других острых включений действующие напряже-
ния увеличиваются (концентрируются) в соответствующих регионах тела
и именно там они значительно превышают средние значения. Разрушение
начнется в таких местах, где локальные напряжения превышают критиче-
ский уровень и мелкий дефект дает старт росту трещины.
Фактор концентрации напряжений, т. е. отношение напряжения в верши-
не острого дефекта Gt к действующему напряжению растяжения G, определя-
ется соотношением Инглиза для упругого твердого тела, предполагающего
прямую пропорциональность между напряжениями и деформациями [1]:
o(/g = 1 + 2(//f)1/2 (1)
где I — глубина краевого дефекта, а г — радиус вершины дефекта в недефор-
мированном состоянии. Если дефект полностью закрыт, то он, грубо говоря,
эквивалентен краевому дефекту глубины 1/2 (рис. 1). Таким образом, кра-
евые дефекты более серьезные концентраторы напряжения, чем закрытые
дефекты того же самого размера, и они являются более обычными источни-
ками разрушения, чем включения. Но так бывает не всегда. Гетерогенность
композитов показывает, что зарождающиеся трещины усталостного разру-
шения являются внутренними [2]. Кроме того, некоторые типы трещин не
могут образовываться вблизи свободной поверхности (см. раздел IV).
Когда радиус вершины много меньше, чем глубина дефекта, то кажется
вероятным, что для сильных концентраторов напряжений, приводящих
к разрушению, уравнение (1) может быть упрощено до
о = (а/«)/21'« (2)
468 -*v- Глава 10. Прочность эластомеров
Рис. 1. Напряжения вблизи трещины глубиной I и с радиусом вершины г.
Рис. 2. Разрушающие напряжения для образцов с разрезами длиной Z, экспониро-
ванных в озоне [4].
Отсюда следует, что разрушающее напряжение, обозначаемое как Gb,
будет изменяться обратно пропорционально глубине дефекта I (в пропорции
1 /Z1/2). Это предположение было проверено для хрупких полимеров, т. е. по-
лимеров в стеклообразном состоянии [3], и для каучукоподобных материалов
при озонном растрескивании (рис. 2). В обоих случаях было показано, что
разрушающее напряжение Gb меняется в зависимости от глубины трещины
или разреза бритвой, сделанного на одном краю испытываемого образца,
в соответствии с уравнением (2). Однако, для эластомеров, разрушаемых
только механическим напряжением, удлинение при разрыве, как правило,
много больше, чем следует из предположения о линейном соотношении меж-
ду напряжением и деформацией, и уравнение (2) дает относительно плохое
приближение. Но даже и при этом, экстраполируя измеренные величины
разрушающего напряжения для различных глубин краевого разреза на раз-
рушающее напряжение для испытываемого образца, вообще не имеющего
разрезов, можно получить характеристику глубины дефекта в материале.
II. Возникновение разрушения -'и- 469
Рис. 3. Усталостная долговечность
Nобразцов с начальным надрезом
глубины /, подвергнутых повтор-
ным растяжениям до обозначен-
ных удлинений.
(На самом деле полученная величина есть глубина разреза, эквивалентного
по мощности концентрации напряжений естественному дефекту, который
может быть меньшим и более острым или большим и затупленным, чем
разрезы ножом, эквивалентные по мощности концентраторам).
Как для резин, так и для стекол полученная этим способом величина
около 40 ± 20 мкм. Такая же величина была получена при экстраполяции
напряжения, измеренного при озонном растрескивании (см. рис. 2), на
величину, наблюдаемую при испытании образца, не имеющего начального
разреза, и при экстраполяции величины
усталостной долговечности образца с вне-
сенным разрезом на усталостную долговеч-
ность образца без разреза (рис. 3). Во всех
этих случаях, естественно, была получена
та же самая величина исходного размера
дефекта. Более того, результат существен-
но независим от использования того или
иного конкретного эластомера и состава
резиновой смеси, даже если эти факторы в
значительной степени изменяют характер
влияния размера надреза на разрушающее
напряжение или усталостную долговеч-
ность, что будет обсуждено позднее. Таким
образом, всё разнообразие процессов раз-
рушения начинается с исходного дефекта,
эквивалентного острому краевому разрезу
глубиной 40 мкм.
Точная природа этих участков возник-
новения разрушения еще неизвестна. Они
могут состоять из случайных царапинок
на поверхности прессформ или режущих
инструментов, но даже при очень большой тщательности приготовления
образцов для испытаний (например, при прессовании между полирован-
ными стеклами) разрушающее напряжение не возрастает существенно.
По-видимому, чтобы инициировать разрушение, частички пыли и грязи
или другие неоднородности, близкие по эффективности к дефектам пресс-
формы, должны присутствовать в достаточном количестве. Только когда
размер образца для испытаний снижается до 10“8м3или меньше, наблю-
дается значительное возрастание прочности, свидетельствующее о том, что
мощные концентраторы напряжений присутствуют только в концентрации
Ю^/м3 или меньшей. Конечно, если бы мог быть найден способ исключить
их, или, по крайней мере, снизить эффективную остроту этих естественных
дефектов, то могло бы быть достигнуто существенное возрастание прочности,
470 -*V- Глава 10. Прочность эластомеров
и даже более поразительное возрастание усталостной долговечности, что
будет обсуждено позже.
Однако, в настоящее время, они, по-видимому, являются неизбежным
следствием процессов, используемых в производстве эластомерных мате-
риалов и изделий.
Б. Критерии разрушения по напряжению и энергии
Уравнения (1) и (2) ставят несколько других вопросов. Что
такое радиус г естественного дефекта? Какова величина разрушающего
напряжения Gt в вершине, где дефект начинает расти как трещина? При
сравнении экспериментальных соотношений, таких как показаны на рис. 2,
с результатами по уравнению (2) может быть определено только произве-
дение g^1/2, но не каждую величину по отдельности. Однако маловероятно,
чтобы величина г превышала 1 мкм для острого разреза, и следовательно,
может быть вычислено, что напряжение в вершине Gt больше, чем 200 МПа,
если принять величину произведения GtH/2 равной 0,2 МНм-3/2, что ти-
пично для разрушения при механических напряжениях. Для озонного
растрескивания это произведение имеет величину 900 Нм-3/2 (см. рис. 2)
и следовательно напряжение в вершине в этом случае предположительно
1 МПа или больше.
Однако мы должны признать, что раздир, который начинает распростра-
няться от начального разреза или дефекта, будет сам по себе быстро образо-
вывать характерную вершину радиуса г, независимо от остроты исходного
концентратора напряжений [5]. Поэтому более уместно рассматривать про-
изведение G^1/2 как характеристику разрушения материала. Действительно,
Ирвин [6, 7] предположил, что разрушение наблюдается для различных
форм образца и для различных условий нагружения при характерной вели-
чине «фактора интенсивности напряжений» Кс, определенного как
^=(^/2)0,^ = ^ (3)
и выраженного в зависимости от действующего напряжения Gb по уравне-
нию (2).
Альтернативный, но эквивалентный взгляд на характеристику крити-
ческого напряжения для разрушения был предложен Гриффитом [8, 9] для
упругих тел и применен Ирвиным [6, 7] и Орованом [10] к таким твердым
телам, которые в целом упруги, даже если вокруг вершины трещины обра-
зуется пластическая зона. Гриффит предположил, что дефект распростра-
няется в напряженном материале только тогда, когда снижение запасенной
энергии упругой деформации Wпри этом больше, чем величина свободной
энергии, необходимая для образования при разрушении новой поверхнос-
ти. Ирвин и Орован установили, что реально расход энергии, затраченной
II. Возникновение разрушения -*и- 471
на местную пластическую деформацию во время роста трещины, обычно
значительно превышает истинную поверхностную энергию; однако только
при условии, что полная затраченная энергия пропорциональна величине
поверхности, созданной при разрушении, соотношения Гриффита все еще
могут быть применены.
Критерий разрушения Гриффита имеет следующую форму
-(ЭЖ/дЛ) > 6с/2 (4)
где Л площадь поверхности образца, которая возрастает при росте трещины,
и Gc количество энергии на единицу поверхности материала, требуемое для
раздира. Фактор 2 возникает от перехода от разрываемой площади к пло-
щади двух вновь образованных поверхностей. Производная вычисляется
при постоянной длине образца, так что приложенные силы не совершают
работы на развивающейся трещине. (Пример, где это не имеет места, пос-
кольку трещина будет развиваться только тогда, когда приложенные силы
производят необходимую работу, будет приведен в разделе 2Г.)
В оригинальном исследовании Гриффита вместо обобщенной энергии
разрушения GJ'L. была использована поверхностная свободная энергия,
отнесенная к единице площади поверхности разрушения Поэтому его ре-
зультаты подразумевали термодинамическую обратимость. В противополож-
ность этому Gc просто представляет собой энергию, диссипированную при
разрушении. Однако величина Gc может быть принята как характеристика
прочностных свойств материала, независимая от метода испытания, только
при условии, что энергия диссипирована в непосредственной окрестности
вершины трещины и независима от общей формы образца и от способа,
которым силы приложены к его краю. Вероятность этого подтверждена кри-
тическими экспериментами на различных материалах, включая эластомеры.
Были использованы образцы, для которых соотношение между разрывным
напряжением <зь и интенсивностью освобождения энергии деформации G.
определенной по уравнению (4), может быть или подсчитано или измерено
экспериментально [И, 12]. Далее будут рассмотрены два важных случая.
В. Образец для испытаний на растяжение
Как показано на рис. 4, тонкая полоска толщины t с разрезом
глубины I на одном краю подвергается растяжению до разрыва. Влияние
разреза на уменьшение полной запасенной упругой энергии при заданном
растяжении может быть приближенно подсчитано при рассмотрении малой
треугольной области вокруг разреза в недеформированном состоянии (по-
казана штриховкой на рис. 4) и остальной части образца, не измененной
наличием разреза, с запасенной упругой энергией деформации Uна единицу
объема. Уменьшение энергии деформации из-за наличия разреза окажет-
472 “* V Глава 10. Прочность эластомеров
ся равным kl2tU, где к численная константа, величина которой зависит от
приложенного напряжения (к определяется приближенно как я/(1 + е)1/2,
где е — деформация растяжения [13]). Таким образом, при возрастании
энергии растяжения к уменьшается от величины я при малых деформациях
до величины, близкой к 1, при больших деформациях.
Следовательно, для растянутого образца — (dW/ЭЛ) = kltUи (SA/dZ) = 2Z.
Уравнение (4) принимает форму [И]
2klU> Gc (5)
Заметим, что разрывное напряжение Gb не представлено явно в этом
критерии разрушения. оьесть напряжение, при котором плотность энергии
деформации Uудовлетворяет уравнению (5). Поэтому оно зависит от упру-
гих свойств материала и начальной длины разреза, также как от энергии
разрушения Gc. Для материала, подчиняющегося линейным соотношениям
между напряжением растяжения о и деформацией е, запасенная энергия U
определяется как Ее2/2 или g2/2E, где Е модуль Юнга. Напряжение и уд-
линение при разрыве определяются так
ob=(Gc£/^)1/2 <6>
eb=(Gc/n/E)1/2 (7)
где к принимает величину я для линейного упругого материала. Уравнения
(6) и (7) получены Гриффитом [9].
При сравнении уравнений (3) и (6) мы видим, что фактор интенсивности
критического напряжения Кс и энергия разрушения или критическая интен-
сивность освобождения энергии деформации Gc соотносятся друг с другом
и с разрушающим напряжением в вершине трещины как
Kl = EGC = = (n/2)UtEr (8)
где Ц плотность энергии деформации в вершине трещины. Следовательно
Gc=(n/2)Utr (9)
Таким образом, энергия разрушения Gc есть произведение энергии,
требуемой для разрыва единицы объема материала в вершине трещины,
т. е. в отсутствии порезов или внешних дефектов, и эффективного диаметра
в вершине, как указано Томасом [14]. Эти два фактора могут быть рассмот-
рены как независимые компоненты энергии разрушения: «собственная»
прочность [7 и характерная геометрия образовавшейся трещины, представ-
ленная величиной г. Поскольку в величину Кс входит также модуль упруго-
сти Е, то для материалов, подобных эластомерам, с сильно различающимися
//. Возникновение разрушения
Л
473
значениями модуля, она не может рассматриваться как подходящая мера
прочности (как Gc).
Уравнение (5) имеет более общее применение, чем уравнение (6), поскольку
оно не ограничивается линейными упругими материалами. Оно устанавливает
критерий для разрыва при растяжении высокоэластичного материала, имею-
щего надрез на одном краю длины Z, в терминах
энергии разрушения Gc. Два важных примера
образцов для испытаний этого типа следующие:
(1) образец для испытаний вулканизованной
резины на раздир (ASTM D624-54) и (2) ти-
повой образец для испытаний на растяжение,
который имеет малую случайную щербинку,
вызванную, например, несовершенством по-
верхности прессформы или штампа при изго-
товлении образца.
Некоторые особенности уравнений (6) и (7)
заслуживают внимания. Для данной величины
энергии разрушения Gc жесткий материал
с высоким значением модуля Юнга Е будет
иметь более высокое разрывное напряжение
и меньшее удлинение при разрыве, чем более
Рис. 4. Образец для испытаний
на растяжение.
мягкий материал. Эти корреляции хорошо известны в шинной промышлен-
ности. Менее известно влияние размера начального разреза или дефекта на
разрывное напряжение и удлинение при разрыве. Окончательно, если критерий
разрушения по уравнению (5) применим для начального дефекта глубины Z, то
он будет существенно превышен в процессе разрушения. В результате трещина
будет двигаться через образец, катастрофически ускоряясь.
Г. Образец для испытаний на раздир.
Этот образец для испытаний, показанный на рис. 5, имеет
два участка («руки», I), находящиеся в состоянии простого растяжения,
и область II, фактически ненагруженную. Если «руки» достаточно широки
или они усилены нерастяжимыми лентами, то их растяжение при усилии
раздира Сбудет пренебрежимо малым. Тогда работа разрушения GCA4 прямо
определяется приложенной силой F, действующей на расстоянии 2AZ, где AZ
есть ступень разрыва. Соответствующая площадь ступени разрыва есть ZAZ,
где Z - толщина полоски1 \ Приравнивая приложенную работу к требуемой
для раздира, получим критерий разрушения
В действительности раздир имеет тенденцию распространяться под
углом 45° к направлению по толщине, т. е. под прямым углом к главному
напряжению растяжения, и следовательно раздир имеет ширину около ^2Л
вместо Z [15, 16].
474 -’V Глава 10. Прочность эластомеров
F>Gct/2 (Ю)
Поскольку усилие раздира в этом случае является прямой мерой энергии
разрушения Gc и оно независимо от упругих свойств материала и от длины
раздира, этот образец для испытаний особенно подходит для изучения вли-
яния состава эластомера и условий испытаний на Gc [17-22].
Рис. 5. Образец для испытаний на раздир.
Важно установить, что энергия разрушения Gc не является постоянной
величиной для конкретного материала; она сильно зависит от температуры и
скорости раздира, т. е. от скорости, с которой материал деформирован перед
разрывом в вершине раздира, см. раздел V( Б). Однако некоторые критичес-
кие величины могут быть установлены. Наименьшая возможная величина
это, конечно, удвоенная величина свободной энергии поверхности, около
50 мДж/м2 для жидких углеводородов и полимеров [23]. Величины этого
порядка действительно наблюдались для разрушения, вызванного озоном,
когда действие приложенных сил направлено только на разделение молекул,
уже разрушенных химической реакцией, см. раздел VIII.
Другая критическая величина есть та, что необходима для разрушения
всех молекул, пересекающих плоскость разрыва, в отсутствии любых других
процессов, поглощающих энергию. Найдено, что этот минимум энергии,
требуемый для механического разрушения, составляет около 50 Дж/м2,
см. следующий раздел. Наконец, имеются значительно большие величины,
обнаруженные в обычных экспериментах на разрушение, меняющиеся от
100 до 100.000 Дж/м2. Это будет описано в разделе V.
III. ПРЕДЕЛЬНЫЕ ПРОЧНОСТИ И РАСТЯЖИМОСТИ
Предельная величина для энергии разрушения эластомеров
была впервые установлена Лейком и Линдлеем при изучении роста усталост-
ных трещин [24]. Путем экстраполяции они нашли, что для распространения
трещины было необходимо минимальное количество механической энергии,
около 50 Дж на м2 поверхности раздира. Мюллер и Кнаусс непосредственно
измерили предельно низкие энергии раздира, применяя низкие скорости
раздира, высокие температуры и уретановую эластомерную композицию,
III. Предельные прочности и растяжмости -*v- 475
сильно набухшую в потоке жидкости [25]. При этих условиях, близких
к равновесным, они получили нижний предел для энергии раздира около
50 Дж/м2, аналогичный полученному Лейком и Линдлеем экстраполяцией.
Позднее предельные прочности при раздире были измерены для нескольких
эластомеров с различной степенью структурирования. Снова величины
оказались около 20-100 Дж/м2, много меньшие, чем энергии раздира,
полученные в обычных экспериментах по раздиру, варьирующиеся от 103
до примерно 105Дж/м2в зависимости от скорости раздира, температуры
и эластомерной композиции [26]. В действительности, это соответствует
только 1 фунту силы для раздира, проходящего через пластину толщиной
в несколько дюймов.
Однако они много больше, чем можно ожидать на основе только прочнос-
тей связей С-С. Например, около 2 X 1018 молекул пересекают случайно
выбранную поверхность разрушения площадью 1 м2, а энергия диссоциа-
ции связи С-С около 5 X 10“19 Дж. Следовательно, по этим данным можно
ожидать, что энергия разрушения будет составлять только около 1 Дж/м2,
вместо наблюдаемой величины около 50 Дж/м2.
Большое расхождение было отнесено Лайком и Томасом [27] засчеттого,
что эластомеры являются длинноцепочными полимерами: многие связи
в молекулярной цепи должны быть напряжены одинаково, чтобы разо-
рвать одну из них. Таким образом, чем больше в молекуле длина между
узлами сетки поперечных связей, тем больше энергия, необходимая для
разрыва молекулярной цепи. С другой стороны, когда цепи длинные,
меньшее число их пересекает случайно выбранную плоскость разру-
шения. Эти два фактора не уравновешиваются; эффект сетки есть пред-
сказанная зависимость предельной энергии разрушения б0от среднего
молекулярного веса Мс цепи между точками поперечных связей, пред-
ставляемая в форме [27]
G0 = aMc,/2 (И)
Две других особенности молекулярной цепи должны быть приняты
во внимание, по крайней мере в приближенном виде; наличие физи-
ческих зацеплений между цепями на характеристическом расстоянии
вдоль каждой цепи молекулярного веса Ме, и наличие концов молекул,
которые образуют часть сетки, не несущую нагрузку. Уравнение (11)
принимает форму
Go = а[(1/Л/с)+(1/7Ие)]’1/2 [1 - 2(7ИС/7И)] (12)
где Ммолекулярный вес полимера до образования поперечных связей [27].
Константа а отражает плотность полимера, массу, длину и эффективную
гибкость мономерных единиц, а также энергию диссоциации связи С-С,
предположительно являющейся наиболее слабым звеном в молекулярной
476 -*и- Глава 10. Прочность эластомеров
цепи. Если взяты разумные значения для этих величин [27], то а оказы-
вается около 0,3 Дж/м2 (г/г-моль)-1/2. Таким образом, для характерной
молекулярной сетки, принимая Мс = Ме = 15000 и М = 300000, получим
предел энергии разрушения около 25 Дж/м2 в удовлетворительном согласии
с экспериментом, принимая во внимание неопределенности и приближения
в теории. Кроме того, предсказанное возрастание энергии разрыва с ростом
молекулярного веса отрезков цепи Мс между поперечными связями пред-
ставляется правильным; возрастание плотности поперечных связей (более
короткие цепи сетки) приводит к меньшему пределу энергии разрушения
[20-22]. Однако, так как разрывная прочность Gb также включает модуль
упругости Е [ур. (6) ] и Е растет при образовании поперечных связей, предел
прочность при разрыве показывает общий рост при возрастании количества
поперечных связей.
Предельные величины прочности и растяжимости при растяжении могут
быть рассчитаны по уравнению (6), используя усредненную величину
предела энергии разрушения 50 Дж/м2, «естественный» размер дефекта
40 мкм (предположив его независимость от состава резины), и типичную
величину модуля Юнга Е для резины 2 МПа (что соответствует твердо-
сти по Шору А около 48°). Результатом будет оь 0 = 0.9 МПа и е ь 0 = 0,45.
Эти величины действительно близки к экспериментальным «пределам
прочности», т. е. напряжениям и деформациям, ниже которых усталост-
ная долговечность действительно бесконечна в отсутствие химических
воздействий [24].
Для прочности резины при растяжении и, как следствие, для прочности
на раздир был обнаружен удивительно большой эффект как результат специ-
фического распределения молекулярных весов Мс в сетке [28]. Когда малая
доля коротких цепей, около 5 мол %, включается в сетку длинных цепей,
разрывная прочность значительно выше, чем для других смесей. По-види-
мому, подобно этому, предельная прочность также выше. Это замечательное
увеличение прочности может быть результатом перестройки сетки, т. е.
способности подвергнуться неафинным деформациям, так что внутренние
концентрации напряжений минимизируются. Какова бы ни была причина,
это явление имеет и научный и практический интерес.
Низкие величины энергии разрушения, только примерно на один порядок
величины выше предельного уровня, могут быть получены при измерении
сопротивления резины разрезанию острым ножом и бритвенным лезвием.
Эффекты трения были минимизированы путем растягивания образца при
разрезании. При добавлении энергии, внесенной растягивающей силой,
к внесенной при разрезе, было найдено, что полная энергия разрушения
остается постоянной и низкой, порядка 300 Дж/м2 [29]. На эту величину
влияет острота лезвия, она ниже для острого лезвия, что и можно ожидать
согласно уравнению (9).
IV. Разрушение при многоосном напряжении -*v- 477
IV. РАЗРУШЕНИЕ ПРИ МНОГООСНОМ НАПРЯЖЕНИИ
Хотя по разрушению эластомеров при комплексном на-
пряжении было выполнено относительно мало работ, но как следует из
дальнейшего, из них могут быть извлечены некоторые общие заключения
относительно разрушения при специфических напряженных состояниях.
А. Сжатие и сдвиг
По-видимому, эластомеры не разрушаются по плоскостям
сдвига. Вместо этого, разрушение развивается под углом 45° к направлению
сдвига (Рис. 6), т. е. под прямым углом к соответствующему главному напря-
жению растяжения [15,16], при сдвиговом напряжении теоретически рав-
ном разрывной прочности [9]. Действительно, общее условие для разрыва,
по-видимому, состоит в достижении определенного напряжения растяжения
Gt в вершине имеющегося дефекта и это условие может достигаться даже
тогда, когда оба действующих напряжения являются сжимающими, при
условии, что они неодинаковы [9]. Когда все напряжения сжатия одинаковы,
т. е. при однородном трехосном сжатии, эластомер будет только уменьшаться в
объеме. Случаи разрушения при таких условиях нагружения неизвестны.
Для одноосного напряжения теория хрупкого разрушения предсказыва-
ет, что разрушающее напряжение, см. уравнение (6), в восемь раз больше,
чем для растяжения при росте трещины в косом направлении [9]. Однако,
однородное напряжение сжатия, достигается не так легко. При этом трение
на нагруженной поверхности тонкого сжатого блока обычно предохраняет
эластомер от свободного расширения в боковом направлении, и реализуются
комплексные условия напряже-
ния. Когда локальные напряжения
растяжения достаточно высоки,
внешние выпуклые поверхности
могут разрушаться снаружи, но
эти местные разрывы не распро-
страняются очень далеко внутрь,
поскольку внутренняя область в
значительной степени находится Рис. 6. Раздир при сдвиговых напряжени-
в состоянии трехосного сжатия. ях (схематически) [15] и [16].
Вместо этого, раздир распространя-
ется в окружном направлении и, в конечном счете, вызывает отрыв кольца
резины от боковой части блока, оставляя основную часть блока неповреж-
денной, но с более узким центральным поперечным сечением [30]. Таким
образом, резиновый блок при сжатии эффективно сопротивляется разру-
шению, но его жесткость может быть значительно снижена после многих
циклов нагружения из-за потери внешних областей резины.
478 -’V- Глава 10. Прочность эластомеров
Разрушение при простом сдвиге еще более сложно. Приближенное опи-
сание для межслойной трещины, начинающейся на одном краю, приводит
к соотношению, аналогичному уравнению (5):
G = kUt (13)
Здесь константа к первоначально равна 0,4 и затем изменяется между
0,2 и 1,0 по мере увеличения длины трещины [31].
Б. Равномерное двухосное растяжение
Совершенно удивительно, что разрушающее напряжение
при равномерном двухосном растяжении оказывается значительно более
высоким, примерно на 20 или 30%, чем при одноосном растяжении [32,33].
Удлинение при разрушении ниже, но запасенная упругая энергия выше.
Следует заметить, что пластины для испытаний, подвергнутые двухосно-
му растяжению, не имеют надреза по краю в желаемой точке разрушения
в центральной области пластины, тогда как образцы для одноосных испы-
таний обычно вырезаются из пластин в форме тонких полосок. Поэтому
увеличение напряжения, вызванное разрезанием, будет иметь место только
при одноосных испытаниях.
Эксперименты с достаточно хрупкими резиновыми пластинами, в которые
намеренно введены начальные трещины того же самого размера в образцы
как для одноосного так и для двухосного нагружения, показали, что раз-
рушающее напряжение примерно от 20 до 30% выше, чем при одноосном
растяжении [33]. Однако результаты оказались совместимыми при единой
величине энергии разрушения (около 150 Дж/м2). Различие между этими
двумя испытаниями состояло в том, что при росте трещин в пластине при
равномерном двухосном растяжении освобождается только половина энер-
гии деформации, запасенной в её площади, тогда как при трещине, растущей
в одноосно нагруженном образце, освобождается вся энергия. Как следствие,
энергия деформации должна быть значительно больше, в два раза или более,
чтобы вызвать разрушение при равномерном двухосном растяжении.
В. Трехосное растяжение
Под действием давления раздувания Р малая сферическая
полость внутри резинового блока расширяться упруго от её начального ради-
уса г() до нового радиуса Хг0. Аналогичным образом, она будет расширяться
в той же степени, когда поверхность блока будет подвергнута однородному
трехосному растяжению -Р, т. е. отрицательному гидростатическому дав-
лению (рис. 7), при условии, что сама резина не изменяет объема. Когда
расширение полости мало, оно пропорционально Р и определяется как
1= 1 + ‘iP/^E, где Е — модуль Юнга резины. Когда расширение полости
IV. Разрушение при многоосном напряжении -•V 479
велико, оно увеличивается быстрее, чем по прямой пропорциональности.
Действительно, для резины, подчиняющейся нео-Гуковскому уравнению
состояния для упругих тел (см. гл. 1), такое расширение становится неоп-
ределенно большим при конечной величине приложенного растяжения,
определяемого согласно [34 — 36] как
-Р = 5Е/6
(14)
Конечно, начальная полость взорвется, когда растяжение её стенки достиг-
нет разрушающего растяжения резины, и сформируется большой раздир,
величина которого определяется энергией, требуемой для его роста. Для
больших предшествующих деформированию пустот растягивающее напря-
жение для взрывного раскрытия будет много меньше, чем предсказывает
уравнение (14) [37]. И для малых предшествующих пустот имеются две
дальнейших сложности: должна быть принята во внимание фактическая
поверхностная энергия полости [37-38]
и кроме того, взрывные напряжения ста-
новятся настолько большими, что резина
вокруг полости перестает следовать со-
отношениям упругости, справедливым
только для малых и средних деформаций
[39]. Однако показано, что в удивитель-
но широком диапазоне значений началь-
ного радиуса г0, примерно от 0,5 мкм до
1 мм, уравнение (14) хорошо аппрок-
симирует предсказанные напряжения
разрушения [39]. Рис. ? Раздувание полости при
Было установлено, что при трехосном трехосном растяжении.
нагружении, определяемом уравнени-
ем (14), резиновые образцы почти неизменно подвергаются внутренней
кавитации. Поэтому это явление должно быть рассмотрено в согласии
с теорией больших упругих деформаций как следствие упругой нестабиль-
ности, а именно, неограниченного упругого раздувания уже существовавших,
предшествующих полостей, слишком малых для легкого обнаружения. Это не
должно обычно включать энергию разрушения, поскольку это принципиаль-
но трансформация потенциальной энергии (от нагружающего устройства)
в энергию деформации. Очевидно, резина содержит много предшествующих
пустот критического размера от 0,5 мкм до 1 мм (но не больше, поскольку
они должны были бы раскрыться при более низких напряжениях).
Критическое напряжение, предсказываемое уравнением (14), зависит
только от модуля упругости, а совсем не от прочности эластомера. В соот-
ветствие с этим для кавитационных напряжений в резиновых блоках с огра-
ничениями деформации при растяжении (рис. 8 и 9) [35] и вблизи жестких
480 -*Ь- Глава 10. Прочность эластомеров
включений, в точках, где имеет место трехосное растяжение (рис. 10 и 11)
[40], были установлены их точная пропорциональность Е и независимость от
прочности на раздир эластомера, что оказалось в согласии с доминирующей
ролью упругости, скорее, чем с критерием разрыва для разрушения.
Сс
, 1JJ и,
' । шТ
Ос
Рис. 8. Кавитация в привулканизован-
ном блоке (схематично).
Рис. 9. Зависимость критического для
возникновения кавитации приложенно-
го напряжения в привулканизованном
блоке (см. рис. 8) от модуля Юнга Е
эластомера [35].
Рис. 10. Кавитация вблизи жесткого
включения (схематично).
Е(МПа)
Рис. 11. Зависимость критического
для возникновения кавитации прило-
женного напряжения вблизи жесткого
включения (см. рис. 10) от модуля Юнга
Е эластомера [40].
Возникновение кавитации вблизи малых жестких включений более
затруднено [41] вероятно потому, что объем той части резины, которая
подвергнута критическому трехосному растяжению, слишком мал, чтобы
V. Распространение трещины JV" 481
содержать относительно большие предшествующие пустоты. А для расши-
рения полости, меньшей, чем приблизительно 0,5 |1м в диаметре необходимы
большие напряжения.
Если эластомеры могли бы быть изготовлены без каких либо микрополо-
стей, больших чем, скажем, радиуса 10 нм, их сопротивление возникновению
кавитации могло бы быть гораздо выше. Это кажется маловероятной идеей
для разработки, однако уравнение (14) остается все же важным общим
критерием разрушения для эластомеров. Оно предсказывает удивительно
низкое критическое трехосное напряжение, порядка только нескольких
атмосфер, для мягких низкомодульных эластомеров. В этих случаях следует,
по-видимому, полностью избегать условий трехосного напряжения.
Кавитация является практически важной проблемой, когда эластомеры
используются для хранения газов высокого давления [42]. Если газ растворя-
ется в резине и мигрирует, наполняя существующие пустоты, он будет нахо-
диться при внешнем (высоком) давлении. Когда внешнее давление внезапно
исчезает, полости раскрываются в соответствии с уравнением (14).
V. РАСПРОСТРАНЕНИЕ ТРЕЩИНЫ
А. Общие положения
Возникновение разрушения, по-видимому, является процес-
сом, одинаковым для всех эластомеров, однако распространение трещины
существенно различается. Можно сопоставить три основных картины рас-
пространения трещины или раздира, наблюдающиеся в трех характерных
типах эластомерных композиций:
1. Аморфные эластомеры, подобные БСК
2. Эластомеры, подобные натуральному каучуку и неопрену, которые
кристаллизуются при растяжении, хотя бы в вершине трещины, где
местные напряжения особенно высоки
3. Усиленные эластомеры, содержащие большие количества, около 30%
по объему, хорошо распределенного усиливающего наполнителя,
такого как технический углерод
Для эластомеров первой группы характерно простейшее поведение при
раздире и поэтому оно описывается первым. Для этих материалов, как только
разрушение инициировано, раздир распространяется с интенсивностью, за-
висящей от двух принципиальных факторов: интенсивности освобождения
энергии деформации G и температуры Т. Первая величина количественно
представляет интенсивность, с которой энергия деформации преобразуется
в энергию разрушения по мере продвижения трещины вперед. Она опреде-
ляется соотношением, аналогичным уравнению (4):
С = -2(АЖ/А4) (15)
482
Глава 10. Прочность эластомеров
Здесь W означает полную энергию деформации образца и А площадь по-
верхности (которая, конечно, возрастает по мере продвижения трещины).
Даже если трещина стационарна, поскольку критическая величина Gc
возникновения разрыва не достигнута, уравнение (15) все еще может быть
полезным для определения интенсивности G, с которой происходит осво-
бождение энергии из деформированного образца. Для растянутой полоски
с краевым надрезом это дает
G = 2itlU (16)
по аналогии с уравнением (5), а для образца для испытания на раздир
G = 2F/t (17)
по уравнению (10).
Б. Вязкоупругие эластомеры
Рис. 12. Зависимость энергии разруше-
ния от температуры и скорости раздира
для ненагюлненного БСК [17].
Экспериментальные соотношения между энергией разру-
шения G, скоростью раздира и температурой испытаний показаны как
трехмерная диаграмма на рис. 12 для резины на основе БСК. Видно, что
энергия разрушения выше при высоких скоростях раздира и низких темпе-
ратурах, и наоборот. Это напоминает
зависимость энергии диссипации
в вязком материале от скорости дефор-
мации и температуры. Действитель-
но, если разделить скорость раздира
на соответствующую молекулярную
сегментальную подвижность при тем-
пературе испытания фг то величины,
полученные для различных темпера-
тур, образуют единую обобщенную
кривую, как показано на рис. 13 [43].
На этом рисунке значения скорости
умножены на фактор
= Фт* /Фт (18)
приводящий их к эквивалентной
скорости раздира при температуре стеклования полимера Tg, равной -57°С
для БСК, данные для которого приведены на рис. 12.
Кроме того, для пяти других аморфных эластомеров, двух бутадиен-сти-
рольных сополимеров с низким содержанием стирола (Т = -72 и -78 °C)
и трех бутадиен-акрилонитрильных сополимеров, имеющих величины Tg
-30, -38 и -56 °C, величины энергии разрушения G, рассчитанные таким же
образом, также попадают на ту же единую кривую, возрастая со скоростью
V. Распространение трещины
Л
483
раздира аналогично диссипации внутренней энергии процесса, контро-
лируемого вязкостью [43]. Можно заключить, что энергия разрушения G
приблизительно одинакова для всех ненаполненных аморфных слабо
структурированных эластомеров при условии равенства сегментальной
подвижности и что зависимость прочности
при раздире от температуры усиливается
единственно в результате соответствующих
изменений сегментальной подвижности.
Таким образом, внутренняя диссипация
энергии определяет сопротивление раздиру
таких эластомеров: чем больше диссипация,
тем больше прочность при раздире. Эта точка
зрения будет также обсуждаться при рас-
смотрении изменений разрывной прочности
эластомеров (см. раздел VI). В данном случае
Рис. 13. Зависимость энергии
разрушения от приведенной
скорости раздира (обобщенная
кривая) для шести ненапол-
ненных эластомеров; темпера-
тура приведения - температура
стеклования Tg [43].
это поразительно проявляется в пропорцио-
нальности между энергией разрушения G и
прямой мерой диссипации энергии, а именно
модулем потерь при сдвиге G", для тех же са-
мых шести эластомеров (рис. 14) [43].
Для простых эластомеров с С-С связями
[44] фактор приведения использованный
для построения обобщенной кривой по результатам, представленным на
рис. 12, такой же, как при построении рис. 13. Показано, что он точно со-
ответствует универсальной форме соотношения ВЛФ, используемого при
описании эквивалентности скорости и температуры [45]:
logaT=-il.5(T-Tg)/(52+T-Tg) (19)
При низких скоростях раздира, около 10-20м/с, и при Т вклад вязких по-
терь становится пренебрежимо малым и прочность на раздир снижается до
малой пороговой величины, GQ. При росте скорости раздира прочность на
раздир растет пропорционально (/?аг)0’24, пока она не достигает значения
около 1000 X Gq, т. е. большей почти на три порядка величины. Таким об-
разом, в широком диапазоне скорости и температуры прочность простых
каучукоподобных твердых тел может быть выражена как
G/Go = l + 2.5xl04(/?ar)0'24 (20)
где Goопределяется структурой молекулярной сетки, см. уравнения (И)
и (12), [44].
При высокой скорости раздира около 1 м/с и при Tg прочность на раздир
предельно высока, приблизительно 106Дж/м2. В тоже время эластомеры
становятся кожеподобными, и, в конце концов, стеклообразными. Дей-
484 -*v- Глава 10. Прочность эластомеров
ствительно, при еще более высоких скоростях и низких температурах они
будут разрушаться подобно типичным полимерным стеклам, в процессе
разрушения которых образуется и распространяется узкая волосная трещи-
на; за ней следует бегущая трещина [46]. Энергия разрушения этого процесса
относительно мала, около 500-1000 Дж/м2, в сравнении с высоковязким, но и
высокодеформируемым эластомером, так что кривая, показанная на рис. 13,
при высоких скоростях повернется резко вниз, чтобы выйти на этот уровень.
Рис. 14. Зависимость энергии разрушения от модуля потерь при сдвиге для шести
ненаполненных эластомеров [43].
Почему прочность на раздир так сильно зависит от скорости раздира
и температуры, т. е. от вязкоупругих свойств полимера? Эта замечательная
особенность была объяснена Кнауссом [47] как следствие «запаздываю-
щей» эластичности. Его трактовка предполагает, что фактор интенсивности
напряжения, определяемый уравнением (8), существенно неизменен при
изменении скорости и температуры, так что
K* = E(t)Go = E(t = ~)G (21)
где Е(0 означает модуль растяжения Е при времени t после нагружения.
Е (I = оо) означает равновесный модуль. Энергия G, полученная из квази-
равновесного твердого тела далеко от конца трещины, затрачивается в конце
трещины в разрушающемся материале, имеющем зависящий от времени
модуль E(t). Таким образом, ожидается, что энергия разрушения G возра-
стает со скоростью раздира R приблизительно пропорционально возрастанию
модуля упругости Е со скоростью деформации
G/Go = E(t = 6/R)/E(t = ~) (22)
где 8 есть характеристическое расстояние перед концом трещины, на котором
создается высокая концентрация напряжения, когда конец трещины продви-
гается. Эта простая картина дает хорошую интерпретацию наблюдаемого воз-
растания энергии разрушения со скоростью и температурой, но величина 8,
полученная прямым сравнением G и Е, слишком мала, около 1 А, чтобы
быть физически оправданной [44]. Расхождение может возникнуть из-за
V. Распространение трещины
г
485
прерывистости раздира на микроскопическом уровне, т. е. он может иметь
место в форме «задержка - движение». При надрезании резины с контро-
лируемой скоростью острым ножом величина сопротивления разрезанию
с изменением скорости разрезания и температуры может быть скорректиро-
вана по уравнению (22), принимая величину 8 примерно равной диаметру
острия лезвия [29, 48, 49].
Прочность на раздир эластомеров с серными связями выше и она зависит
от температуры сильнее, чем ожидается по уравнению (19) [44]. Это может
быть приписано тому, что скорее идет разрушение слабых полисульфидных,
чем более сильных С-С связей в сетке цепей. Как результат, возникают
зависящие от температуры вторичные процессы, поскольку прочность S-S
связей падает с повышением температуры. Таким образом, прочность про-
изводственных резиновых смесей отражает не только внутреннюю диссипа-
цию энергии в результате процессов, связанных с вязкостью [44], но также
отделение от частиц наполнителя, изменение радиуса вершины раздира [48]
и разрушение слабых поперечных связей [44].
В. Эластомеры, кристаллизующиеся
при деформации
Как показано на рис. 15, прочность при раздире эластоме-
ров, кристаллизующихся при деформации, значительно увеличивается
в том диапазоне скоростей раздира и температуры, где кристаллизация при
растяжении происходит в вершине тре-
щины. Однако, при высокой температуре
кристаллизация становится термодина-
мически запрещенной, поскольку даже
высокая температура плавления крис-
таллов в сильно растянутом эластомере
оказывается превышенной. Наоборот,
при низкой температуре и высокой
скорости раздира молекулярная реор-
ганизация кристаллов не может иметь
место в короткое время растяжения пока
продвигается вершина трещины. Таким
Рис. 15. Зависимость энергии раз-
рушения от температуры и скорости
раздира для эластомера, кристаллизу-
ющегося при растяжении (НК) [17].
образом, упрочняющий эффект кристал-
лизации, вызванной напряжением, огра-
ничен определенной областью скоростей
раздира и температуры, как показано на
рис. 15. Вне этой области материал обла-
дает только прочностью, связанной с его вязкостными характеристиками,
зависящими от разности между температурой испытания и температурой
стеклования, Т — Т£.
486 JV" Глава 10. Прочность эластомеров
Высокая прочность кристаллизующегося при растяжении материала
обусловлена явно выраженной диссипацией энергии при растяжении
и сокращении, связанной с образованием и плавлением кристаллов при
неравновесных условиях [50]. Как будет показано ниже, частицы усили-
вающих наполнителей оказывают подобное упрочняющее действие, и они
также вызывают заметное возрастание диссипации энергии. Является ли это
единственной причиной для упрочняющего влияния кристаллов и наполни-
телей, и есть ли другие упрочняющие включения, такие как жесткие области
в блок-сополимерах, сегменты с водородными связями и т.п., еще не ясно.
Г. Усиление наполнителями
Значительный усиливающий эффект достигается при
добавлении мелкодисперсных наполнителей, таких как технический уг-
лерод или силика (двуокись кремния) к резиновой смеси. Они вызывают
десятикратный рост прочности при раздире и разрывной прочности, когда
в состав смеси добавлено, например, 40% по весу технического углерода. Но
это упрочняющее действие ограничено определенной областью скоростей
раздира и температуры, которая зависит от типа наполнителя и эластомера
[17, 51] (рис. 16). Вне этой области эффективности наполнитель не увели-
чивает наблюдаемую прочность даже в близкой к этой степени
Рис. 16. Энергия разрушения для
аморфного эластомера (БСК), уси-
ленного 30 вес. % высокодисперс-
ного технического углерода [18].
Заметное увеличение прочности на
раздир в некоторых условиях связано
с явно выраженным изменением в характе-
ре процесса раздира. Он изменяется от раз-
дира с относительно гладкой поверхностью
(с неровностями порядка 0,1-0,5 мм), до
прерывистого раздира с участками задерж-
ки и движения. Такой раздир отклоняется
от прямого пути и даже поворачивается
в направлении, идущем параллельно прило-
женному напряжению, пока через образец
не прорвется новый раздир. Эта форма
раздира была названа узловатый раздир
[17]; пример показан на рис. 17. Типичное
изменение силы при раздире показано на
рис. 18, а; оно может быть сравнено с соот-
ветствующим графиком для ненаполненного материала на рис. 18, в. Пик
силы раздира на участке «задержка» достигает значительной величины, но
сила во время катастрофического «движения» падает до много меньшего
уровня, который только примерно вдвое больше уровня, достигаемого при
непрерывном раздире ненаполненного эластомера. Действительно, при
создании препятствий для отклонения раздира от прямого пути усилие
V. Распространение трещины Jv- 487
раздира много меньше, и только в два или три раза превышает усилие
раздира для соответствующего ненаполненного материала (см. рис. 18, б).
Такие препятствия создаются или путем ограничения пространства близко
расположенными металлическими направляющими, или путем растяжения
[52] образца в направлении раздира, или созданием предварительного рас-
тяжения в том же направлении [53].
Ш////////Ш
I--------------1
ЮО мм
Рис. 17. «Узловатый» раздир в эластомере, усиленном техническим углеродом [52].
Расстояние (мм)
Рис. 18. Усилие раздира для (а) наполненного эластомера без ограничений; (б) тот
же материал с раздиром по первоначальному пути; (в) соответствующий
ненаполненный эластомер с ограничениями и без них [52].
Таким образом, увеличение прочности при раздире путем введения на-
полнителей может быть двух видов: малым (не больше, чем в два или три
раза) увеличением собственной прочности и основным, связанным с откло-
нением порядка нескольких миллиметров пути раздира, что эффективно
при специальных условиях скорости раздира, температуры и молекулярной
ориентации. Первый эффект может быть обусловлен повышением энергии
диссипации в наполненном материале, что было рассмотрено в предыдущем
разделе. Второй приписывается снижению сопротивления раздиру на боко-
488 -*V- Глава 10. Прочность эластомеров
вом пути, параллельном направлению растяжения. Если сопротивление раз-
диру значительно снижается в направлении такого бокового пути, то раздир
будет сворачивать на боковой путь и может быть относительно безвредным.
Парадоксально, что резина усиливается, если её прочность снижается в не-
котором направлении — в таком, где это не приводит к катастрофическому
разрушению. Этот механизм усиления подтверждается двумя наблюдениями:
измерения прочности при раздире в направлении растяжения показывают
заметное снижение при возрастании растяжения, а согласно вычислениям,
энергия, необходимая для поворота трещины в этом направлении, удивитель-
но велика (40% или более того, что необходимо для продолжения движения
в первоначальном направлении, когда образец сильно растянут) [54]. Таким
образом, когда прочность при раздире в направлении растяжения падает
до 40% или ниже прочности при первоначальном направлении трещины,
можно ожидать, что она повернет в сторону.
Д. Повторное приложение напряжения:
динамическое распространение трещины
Хотя показано, что раздир аморфного эластомера происхо-
дит стационарно при скорости, контролируемой энергией G, достаточной
для разрушения (как показано на рис. 13 и 14), кристаллизующиеся при
растяжении эластомеры не подвергаются непрерывному раздиру при ма-
лых величинах G, например для натурального каучука меньших чем около
104 Дж/м2 (см. рис 15). Тем не менее, когда малые напряжения прилагаются
повторно, трещина будет расти ступенчато с шагом Д/ на один акт прило-
жения напряжения, даже если соответствующая величина G много ниже
критического уровня [5].
Экспериментально наблюдались четыре различных закона роста (рис. 19),
соответствующие четырем уровням напряжения [55, 56]:
1. G < Go: при раздире трещина не растет, она появляется только при
химическом воздействии (озоном)
2. Gq<G < Gy шаг роста Д/, пропорционален G - Go
3. GX<G < Gc: шаг роста Д/, пропорционален Ga
4. G ~ С: катастрофический раздир
Переходная величина G между одним законом роста трещины и дру-
гим, обозначенная выше как Gv была найдена равной 400 Дж/м2. Еще не
выдвинуты объяснения ни для формы этих экспериментальных законов
роста, ни для переходов между ними. Таким образом, они пока должны рас-
сматривается как эмпирические соотношения для шага роста Д/ на каждое
приложение напряжения.
Обычно на практике для приближенного описания роста трещины
в широком диапазоне изменения величины G (но большей, чем пороговая
И Распространение трещины -•ь- 489
величина б?0) применяют соотношение «Парижский закон», которое может
быть представлено в форме
(23)
где величина константы & оказывается около 1 А на каждое приложение
напряжения для многих резиновых смесей, а экспонента а принимает
различные значения для различных эластомеров, изменяющиеся от 2 для
смесей на основе натурального каучука,
представленных прерывистой линией на
рис. 19, до величин от 4 до 6 для некристал-
лизующихся эластомеров, таких как БСК
и цис-транс-полибутадиен.
Рост трещины в резинах на основе на-
турального каучука приводит только к воз-
растанию деформации; если деформация
поддерживается постоянной, трещина не
растет дальше, пока усилия недостаточны,
чтобы вызвать катастрофический раздир.
Причиной этого является образование в
Log G (Дж/м2)
Рис. 19. Зависимость шага роста
трещины А/ на каждое приложе-
ние напряжения от энергии G.
необходимой для разрушения
для вулканизатов натурального
каучука [24].
сильно напряженном материале кристал-
лической области в вершине трещины,
эффективно препятствующей дальнейше-
му раздиру. Это объясняет замечательную
особенность роста трещины в эластомерах,
кристаллизующихся при деформации: шаг
роста при повторяющихся напряжениях
становится предельно малым, если испытываемый образец не релаксирует
полностью между каждым приложением напряжения [55]. В этих условиях
кристаллическая область не плавится; она остается неповрежденной и пре-
дохраняет от дальнейшего роста трещины при повторном приложении напря-
жения. В результате механическая усталостная долговечность (обсуждаемая
в следующем разделе) значительно увеличивается, если образец никогда не
релаксирует до ненапряженного состояния (см. рис. 33). Действительно,
разрушение в этих условиях скорее есть следствие химического воздействия
обычно атмосферного озона [55], чем механического разрыва.
Для аморфных эластомеров характерен больший рост трещины при пе-
риодическом приложении напряжения, чем при постоянном напряжении;
при этом зависимость дополнительного роста трещины за цикл напряжения
от энергии G, необходимой для разрушения, имеет существенно тот же вид,
что и для натурального каучука. Принципиальное различие выявляется
в третьей области, показатель степени а в уравнении (23) для БСК около
четырех, в отличие от двух для НК [56].
490 -*и- Глава 10. Прочность эластомеров
Эндрюс выдвинул общее объяснение для замедления роста трещины
в аморфных эластомерах (и полного прекращения раздира в эластомерах,
кристаллизующихся при деформации) после того, как было приложено
напряжение. Оно базируется на изменениях напряжения во времени
в вершине трещины [57, 58]. В результате концентрация напряжений
в растущей вершине меньше для вязкоупругого или для диссипирующего
энергию материала, чем можно было бы предполагать из чисто упругого
рассмотрения. Рост трещины соответственно медленнее. Эта концепция,
обычно связываемая с Кнауссом, описана кратко в разделе V (Б).
Было установлено, что кислород в окружающей атмосфере способствует
росту трещины, вероятно путем окислительных реакций разрыва цепей,
катализируемых механическим разрушением. Найдено, что минимальная
энергия несколько больше для экспериментов, производимых в вакууме
[55, 59, 60]. Когда в рецептуру смеси включаются антиоксиданты, резуль-
таты, полученные в атмосфере, содержащей кислород, приближаются
к полученным в вакууме.
Е. Термопластичные эластомеры
Физические характеристики термопластичных эластомеров
определяются фундаментальной несовместимостью различных полимеров.
Термопластичные эластомеры содержат трехблочные молекулы, общей
структурой А-Б-А, где А означает стеклообразную полимерную после-
довательность, например полистирол, а Б означает гибкую полимерную
последовательность, например полибутадиен. Конечные последователь-
ности А не смешивается с полимером В, поэтому они стремятся совместно
образовать малые участки стеклообразного полимера, изолированные от
эластомерной матрицы. Более того, поскольку последовательность А на
каждом конце трехблочной молекулы обычно становится частью различных
стеклообразных участков, образуется сетка эластомерных последовательно-
стей, связанных вместе малыми жесткими участками, диаметром 10-30 нм.
Материал этого рода ведет себя как типичный высокоэластичный, по край-
ней мере ниже температуры стеклования полимера А и выше температуры
стеклования полимера В.
Прочность на раздир для характерного термопластичного эластомера
Кратон 1101 (Химическая компания Шелл) вполне сравнима с хорошо уси-
ленным аморфным или кристаллизующимся при растяжении эластомером
и близка к 20 Дж/м2 при комнатной температуре. Эта удивительно высокая
величина определяется пластическим течением твердых участков при вы-
соких местных напряжениях, достигающих уровня разрушения. Действи-
тельно, такой деформационный процесс, по-видимому, существенный для
этих материалов, имеет способность диссипировать энергию деформации,
также как это происходит в любом жестком материале.
VI. Разрыв при растяжении
Л-
491
VI.
РАЗРЫВ ПРИ РАСТЯЖЕНИИ
А. Влияние скорости и температуры
На рис. 20 показаны некоторые соотношения для разруша-
ющего напряжения Gb ненаполненного вулканизата БСК в зависимости от
скорости растяжения при различных температурах [61]. Небольшой фактор
коррекции (Т /Т) был применен к измеренным величинам, чтобы учесть
изменения модуля упругости с температурой. Коррелированные величины
обозначены как оь'.
Экспериментальные соотношения образуют параллельные кривые,
хорошо совмещаемые при горизонтальном перемещении. Прочность при
данной температуре, таким образом, эквивалентна прочности при другой
температуре при условии, что скорость скорректирована путем умножения
на коэффициент, зависящий от разно-
сти температур. (При использовании
логарифмической шкалы для скорости
растяжения этот постоянный коэффи-
циент эквивалентен постоянному гори-
зонтальному перемещению). Как и для
энергии разрушения G (см. V(B)), было
найдено, что этот коэффициент есть от-
ношение сегментальных подвижностей
при двух температурах фг^/фт [61] и легко
подсчитывается по уравнению ВЛФ (19).
Таким образом, с использованием соот-
ветствующих величин фактора сдвига к
соотношениям, полученным при разных
температурах, может быть построена
обобщенная кривая для температуры
Рис. 20. Зависимость напряже-
ния при разрыве оь от скорости
деформации ё для вулканизата
БСК [61].
приведения Т, в качестве которой здесь
для удобства принята Tg, Обобщенная кривая для разрывной прочности,
полученной по показанным на рис. 20 соотношениям, приведена на
рис. 21.
Таким образом, сегментальная подвижность является первоначальной по
отношению к изменениям разрывной прочности с температурой, так же как
к изменениям энергии разрушения. Более того, обобщенная кривая имеет
такую же форму, как и для характеристик, контролируемых вязкостью: она
резко поднимается с возрастанием скорости растяжения до максимальной
величины при высоких скоростях, когда сегменты не двигаются и матери-
ал разрушается как хрупкое стекло [62]. Удлинение при разрыве сначала
растет с ростом скорости растяжения, отражая возрастающую прочность,
а затем падает при более высоких скоростях, так как сегменты становятся
неспособными реагировать достаточно быстро (рис. 22).
492
Глава 10. Прочность эластомеров
Разрыв растягиваемого образца может рассматриваться как катастрофи-
ческий раздир в вершине случайного дефекта. Успех принципа приведения
ВЛФ для энергии разрушения G при раздире означает, что он будет также
сохраняться для прочностных свойств при растяжении. Действительно, Gb
и eh могут быть рассчитаны по соответствующей величине G для каждой
скорости и температуры, используя соотношение, аналогичное уравнениям
(6) и (7). Скорость растяжения в вершине трещины будет, однако, много
больше, чем скорость растяжения целого образца и это расхождение по
скорости должно быть принято во внимание [63].
Рис. 21. Обобщенная кривая для разрыв-
ного напряжения <зь в функции скорости
удлинения ё, построенная с использо-
ванием соотношения ВЛФ, уравнения
(18) и (19); температура приведения Т
(-60°С). [61].
-20 -16 -12 -8 -4 О
Log ёот(1/с)
Рис. 22. Обобщенная кривая для разрыв-
ного удлинения еь в функции скорости
удлинения ё; температура приведения Tg
(-60°С) [61].
В дополнение отметим, что при дифференцировании уравнения (5) ста-
новится ясным смысл величины U. Она представляет собой скорее энергию,
которую можно получить из деформированного материала, чем энергию,
затраченную на его деформирование. Для материала способного к дисси-
пации энергии, энергия, доступная для разрушения, есть только часть той,
что приложена. Поэтому такой материал будет вдвое более прочным при
испытаниях на растяжение или при любом другом процессе разрушения,
в котором энергия раздира подводится не прямо, а за счет изменения дефор-
мации в другом месте.
Б. Огибающая разрывов
Альтернативное представление данных по разрывам при
нагружении в широком диапазоне температур и скоростей растяжения
получается по графической зависимости разрушающего напряжения оь от
соответствующего удлинения при растяжении еь [64]. Результаты по рас-
тяжению, на которых основаны рис. 20-22, перестроены таким образом на
рис. 23. Они образуют общую кривую, названную огибающая разрывов, ко-
VI. Разрыв при растяжении -*и- 493
торая имеет характерную параболическую форму. Движение по этой кривой
в направлении против часовой стрелки соответствует возрастанию скорости
растяжения или уменьшения температуры, хотя эти две переменные не
разделяются. Таким образом, на низшей
части кривой разрушающие напряжения и
удлинения оба малы. Эти условия осущест-
вляются при низкой скорости деформации
и при высоких температурах. Наоборот,
верхняя часть кривой соответствует высо-
ким разрушающим напряжениям и низкой
растяжимости. Эти условия осуществля-
ются при высоких скоростях деформации
и низких температурах, когда материал
реагирует как стеклообразный.
Есть два принципиальных преиму-
щества представления данных в форме
«огибающей разрывов». Первое, ясная
Рис. 23. Огибающая разрывов для
вулканизата БСК [64].
индикация максимально возможного разрывного удлинения еь тагдля мате-
риала. Найдено, что это хорошо коррелирует со степенью структурирования,
определяемой молекулярным весом между поперечными связями Af как
предсказывает теория эластичности:
^=1 + еЬ.таХж^/2
(24)
Второе, огибающая разрывов может быть обобщена для того, чтобы
охватывать различные степени структурирования, как будет рассмотрено
ниже. Поэтому она будет применяться для выявления различий между из-
менениями в структурировании и другими изменениями, которые влияют
на зависимость от скорости удлинения и температуры, но не обязательно
влияют на М. Примером этого является пластификация и добавление уси-
ливающих наполнителей [65].
В. Влияние степени сшивания
Зависимость разрушающего напряжения при возрастании
степени поперечного сшивания от нуля обычно проходит через острый
максимум. Пример показан на рис. 24. Этот максимум обусловлен, главным
образом, не изменением собственной прочности, а изменением вязкоупру-
гих свойств с изменением плотности сетки поперечных связей. Например,
он значительно меньше выражен при низкой скорости растяжения [66]
и отсутствует у набухших образцов [67]. Поэтому Бюхе и Дудек [66] и Смит
и Чу [68] сделали вывод, что в условиях упругого равновесия этот максимум
будет отсутствовать.
494
Глава 10. Прочность эластомеров
При построении огибающей разрывов для материалов, имеющих различ-
ную степень поперечного сшивания, используют две различные схемы при-
ведения. Первая, показанная на рис. 25, состоит в установления масштаба
удлинения при разрыве еь в долях от его максимальной величины, которая,
конечно, зависит от степени поперечного сшивания [ур. (24)]. Разрывное
напряжение оь также пересчитывают в истинное напряжение при разрыве,
вместо того, чтобы использовать номинальное, как делали до сих пор. (Но-
минальное растягивающее напряжение определяется как сила растяжения,
деленная на недеформированное поперечное сечение образца. Оно обычно
используется в литературе, относящейся к деформации и разрушению элас-
томеров). Эта схема приведения вполне успешно применима для широкого
диапазона плотности сетки поперечных связей (см. рис. 25) [65].
Log ъе
Рис. 24. Зависимость разрывной про-
чности вулканизата БСК от степени
поперечного сшивания, представленной
величиной ve [66]. Пунктирная кривая:
авторская оценка предельной прочности
в условиях отсутствия диссипации.
Рис. 25. Огибающая разрывов для ма-
териала Viton A-HV с разной степенью
поперечного сшивания (еь тах изменя-
ется от 2,5 до 18). [65].
Второй метод состоит в изменении масштаба по оси напряжения путем
деления номинального напряжения при разрыве на меру плотности сетки
поперечных связей [69]. Этот метод, по-видимому, также дает разумные
данные для различных сшитых материалов.
Г. Эластомеры, кристаллизующиеся
при деформации
В то время как для аморфных эластомеров характерно
плавное уменьшение разрывной прочности при повышении температуры
(см. рис. 20), для кристаллизующиеся при деформации эластомеров обычно
VI. Разрыв при растяжении
Л
495
характерное© внезапное снижение при критической температуре Тс (рис. 26)
[70-73]. Эта температура сильно зависит от степени поперечного сшивания,
как показано на рис. 26. Это явно связано с разрушением кристаллов при вы-
соких температурах; однако, хотя основная часть образца является аморфной
выше Т, высокодеформированный материал в вершине дефекта вероятно
продолжает быть кристаллическим. Томас и Вайттле [72] провели параллель
между падением прочности при критической температуре Тс и аналогичным
резким падением прочности при критической глубине 1С краевого разреза
(рис. 27), измеренной при комнатной температуре.
Рис. 26. Зависимость прочности при раз-
рыве от температуры для натурального
каучука, вулканизованного пероксидом
дикумила (ПКД) [72].
Рис. 27. Зависимость прочности при
разрыве от глубины первоначального
краевого разреза для натурального кау-
чука, вулканизованного 2% пероксида
дикумила [72].
Два других аспекта критической температуры заслуживают внимания.
Первый, по существу тот же самый, что и для смесей, усиленных наполни-
телями. Второй, сильное влияние типа поперечных связей; она выше для
длинных полисульфидных связей и ниже для углерод-углеродных связей.
Очевидно, что лабильность связей это важный фактор для развития ориен-
тационной кристаллизации.
Д. Диссипация энергии и прочность
Общая корреляция между разрывной прочностью и темпе-
ратурным интервалом (Т-Т) от температуры испытания Тдо температуры
стеклования Т известна в течение многих лет. Это рассмотрено с разделе
VI (Б) [74]. Пример показан на рис. 28, где величины прочности полиурета-
новых эластомеров с величиной Т меняющейся от-67 до-17°С приведены
на графике в зависимости от Т- Tg [65]. Все результаты ложатся на единую
кривую, показывающую еще раз, что сегментальная вязкость определяет
наблюдаемую прочность.
496 -* v- Глава 10. Прочность эластомеров
Более прямая демонстрация четкой связи между диссипацией энергии
и прочностью была дана Грошем и др. [75]. Они показали, что существует
прямое соотношение между плотностью энергии f/b, требуемой для разру-
шения эластомеров, и плотностью энергии, диссипируемой при растяжении
почти до удлинения при разрушении Ud. Это связь сохраняется независимо
от механизма потери энергии, т. е. для наполненных и ненаполненных,
кристаллизующиеся при деформации и аморфных эластомеров (рис. 29).
Их эмпирическое соотношение имеет вид
(7b=410t7d2/3 (25)
Ub и Ud (и Wb и IV р рис. 29) выражены в джоулях на кубический метр. Те
материалы, которые требуют наибольшую энергию для разрушения, т. е.
наиболее прочные эластомеры, это именно те, в которых большая часть
энергии диссипирует перед разрывом.
Рис. 28. Зависимость прочности полиуретановых эластомеров при растяжении
от T-Tg(Tg меняется от-67 до-17°С) [65].
Рис. 29. Зависимость между работой разрушения (Жь) и дисипированной энергией (Жа)
при растяжении почти до разрывного удлинения [75].
VII. Повторное нагружение: механическая усталость -»и- 497
VII. ПОВТОРНОЕ НАГРУЖЕНИЕ: МЕХАНИЧЕСКАЯ
УСТАЛОСТЬ
При повторяющихся деформациях растяжения трещины появ-
ляются, как правило, на краю образца и прорастают через него в возрастающем
темпе. Это процесс известен как усталостное разрушение. Он рассматривается
количественно в терминах ступенчатого раздира от начальной щербинки или
дефекта [76,77] следующим образом: в каждый момент, когда проходит дефор-
мация, энергия G в форме энергии деформации, становится достаточной, чтобы
вызывать рост раздира в виде малой щербинки на краю образца. Величина G
для растянутого образца определяется уравнением (5). Соответствующий шаг
роста AZ предположительно подчиняется уравнению (23), т. е. является пропор-
циональным Ga, так что закон роста трещины становится таким
AZ/Z“=(2Zcf/)“B'An (26)
где п есть число раз приложения деформации, а к численная константа,
близкая к 2 (см. раздел II В). Глубина трещины послеNциклов деформации
получается интегрированием
^l-a) _z(l-a) =(2l{Uy- B,N (27)
где 10 начальная глубина щербинки. Пример роста трещины показан на
рис. 30; он согласуется полностью с уравнением (27) при а = 2,0.
Рис. 30. Рост трещины на краю образца из вулканизата натурального каучука при
повторных растяжениях до деформации 46% [26].
Если трещина растет до величины, во много раз большей её начальной
глубины, так что I » окончательным разрушением, тогда соответ-
ствующая усталостная долговечность может быть определена подстановкой
I = ©о в уравнение [27], что приводит к
i/N = (2kU)aB'l£~l) (28)
498 V- Глава 10. Прочность эластомеров
Это количественный прогноз для усталостной долговечности N в терми-
нах энергии деформации Uи двух характеристик материала, экспоненты а,
определяющей рост трещины, и характеристики размерности S', которые
могут быть определены в отдельных экспериментах, как описано выше
(см. раздел У(Д). Измеренные величины усталостной долговечности для
образцов с начальными разрезами различной длины (см. рис. 3) и для
воздействующих деформаций различной величины оказались в хорошем
согласии с предсказанными по уравнению (28) [55, 76, 77].
Рис. 31. Зависимость усталостной долговечности от размера первичного разреза
для образцов из натурального каучука и БСК при повторных растяжениях
до деформации 50% [55].
Примеры зависимости усталостной долговечности от размера начального
разреза показаны на рис. 3 и 31. Времена жизни образцов, не содержа-
щих преднамеренно внесенных разрезов, представлены горизонтальными
штриховыми линиями на рис. 3, они могут быть интерпретированы как
ступенчатый раздир от гипотетической щербинки или дефекта около 20 мкм
глубины, как показано выше. Особо заслуживает внимания то, что близкий
размер получается для натуральных дефектов как для кристаллизующихся
при деформации, так и некристаллизующихся эластомеров путем такой
экстраполяции, хотя для некристаллизующегося эластомера (БСК) за-
кон роста трещины в основной области раздиров отличается. Показатель
степени а в соотношении для усталостной долговечности (28) становится
равным 4 вместо 2. Найдено, что измеренная усталостная долговечность
для вулканизатов ненаполненного БСК находится в хорошем согласии
с этим соотношением для широкого диапазона глубин начального разреза
и амплитуд деформации [55].
Различные законы роста трещины для кристаллизующихся при де-
формации и некристаллизующихся эластомеров приводят к совершенно
различным соотношениям для усталостной долговечности. Для некристал-
лизующегося эластомера усталостная долговечность значительно больше
зависит от размера начального дефекта (см. рис. 31) и величины прило-
женной деформации, так что такие эластомеры являются, в основном, дол-
VII. Повторное нагружение: механическая усталость -*ь- 499
гоживущими при малых деформациях и при несущественных надрезах, но
много более короткоживущими при более жестких условиях. Усталостная
долговечность также существенно снижается при высоких температурах,
как результат резкого увеличения скорости роста трещины при уменьшении
собственной вязкости (рис. 32). В противоположность этому, гистерезис,
связанный с ориентационной кристаллизацией, сохраняется при условии,
что температура не становится такой высокой (около 100°С для натурального
каучука), что кристаллизация больше не происходит. Поэтому усталост-
ная долговечность натурального каучука не сильно зависит от умеренного
подъема температуры.
Температура °C
Рис. 32. Зависимость усталостной долговечности от температуры для образцов из
натурального каучука и БСК, при повторных растяжениях до деформации
175% [26].
Более поразительное различие обнаружено между кристаллизующимися
при деформации и некристаллизующимися эластомерами, когда напряжение
не релаксирует до нуля во время каждого цикла. Как показано на рис. 33,
усталостная долговечность вулканизатов натурального каучука сильно
возрастает, когда минимум деформации поднимается от нуля до, примерно,
100%, поскольку при минимальной деформации кристаллический барьер
раздиру у кончика трещины не исчезает. В результате рост дефектов фак-
тически останавливается, пока полная приложенная деформация не станет
слишком большой, около 400%. Ничего сравнимого с этим эффектом упроч-
нения при увеличении уровня минимальной деформации не наблюдается
для некристаллизующихся эластомеров.
Соответственно, для пороговой величины энергии раздира Go, ниже ко-
торой не происходит рост трещины при механическом разрушении, имеется
минимум деформации растяжения е0, ниже которой дефекты нормального
размера при усталостном нагружении не растут. Вычислением по уравне-
нию (7) и по прямым наблюдениям [24] найдено, что для типичных эласто-
500 —* V Глава 10, Прочность эластомеров
меров этот механический предел усталости лежит в пределах от 50 до 100%
удлинения. При удлинении ниже этого уровня предел усталости бесконечен
в отсутствие химического воздействия, например от озона в окружающей
атмосфере.
Рис. 33. Зависимость усталостной долговечности от минимума растяжения emin для
образцов из натурального каучука, Де — дополнительная деформация при
повторных растяжениях [78].
Усиливающие наполнители сильно повышают прочность эластомеров
при раздире и растяжении, но не вызывают эквивалентного улучшения при
росте трещин и в усталостных свойствах. При заданной подведенной энергии
деформации, измеренные значения долговечности заметно больше, но при
сравнении при равных доступных уровнях энергии они совсем не намного
больше. Таким образом, размер начального дефекта и порог энергии раз-
дира Go оказываются почти такими же, как для ненаполненного материала.
Шаг роста трещины очевидно слишком мал для заметного отклонения пути
раздира и, следовательно, «усиление» по этому механизму для усталостного
разрушения не так заметно.
VIII. ПОВЕРХНОСТНОЕ ОЗОННОЕ РАСТРЕСКИВАНИЕ
В содержащей озон атмосфере у растянутых образцов не-
насыщеных эластомеров образуются поверхностные трещины, которые
растут в длину и глубину и, в конце концов, разрывают образец. Даже когда
трещины очень малы, они могут вызвать серьезное снижение прочности
и усталостной долговечности. Напряжение растяжения, которое необходи-
мо приложить для появления озонных трещин, может быть приближенно
вычислено по уравнению (6). Энергия разрушения G оказывается только
около 0,1 Дж/м2 [4] и составляет малую долю от энергии, необходимой для
IX. Абразивный износ -*v 501
«разрушения» жидкой среды, т. е. около удвоенной поверхностной энергии
для углеводородной жидкости [23]. Разрыв молекул, очевидно, легко про-
исходит при реакции с озоном и для его начала не требуется механической
энергии. Принимая величину 2 МПа как характерную для модуля Е мяг-
кой резины, и величину 40 мкм для эффективной глубины I случайного
поверхностного дефекта, по уравнению (6) получим, что критическое
напряжение растяжения для озонного растрескивания около 50 кПа,
и критическая деформация около 5%. Эти находятся в достаточно хорошем
согласии с экспериментально наблюдаемыми минимальными величинами
при озонном воздействии.
Когда уровень напряжения поднимается выше минимальной величины,
многочисленные слабые повышения напряжения становятся эффективными
и образуется большое число трещин. Фактически большое число малых вза-
имно взаимодействующих трещин является менее вредными, чем несколько
широких отдельных трещин, которые развиваются в глубокие надрезы, так
что наиболее вредные условия создаются как раз при напряжениях выше
критического.
Скорость, при которой растет трещина, когда условия критической
энергии удовлетворены, зависит от двух факторов: скорости распростра-
нения озона в вершине трещины и скорости сегментального движения
в зоне трещины. Когда один их этих процессов достаточно мал, он становится
контролирующим скорость. Общая скорость R роста трещины определяется
приблизительно как
У?-1 (sec/m) = 8 х lO1^1 +1.2 х 105Си (29)
где фг (с-1) собственная частота броуновского движения молекулярных сег-
ментов при температуре ^определяемая соотношением ВЛФ [ур. (18) и (19),
при приведении к ф?{? (где = 0,1 с-1 [45]), и С (мг/л) есть концентрация
озона в окружающем воздухе [79].
Для типичной уличной атмосферы величина С порядка 10-4 мг/л, тогда
второй член в уравнении (30) является преобладающим для величин фг,
больших чем примерно 104с-1, что имеет место при температурах больше
чем на 25°C выше Tg [79].
IX. АБРАЗИВНЫЙ ИЗНОС
А. Механизмы износа
Абразивный износ это отрыв маленьких частиц эластомера
под действием сил трения, когда происходит скольжение между поверхно-
стью эластомера и контртелом. Подходящей мерой скорости износа является
отношение Л/р, где Л — объем резины, удаленной при истирании, на единицу
нормальной нагрузки и на единицу пути скольжения, ар, — коэффициент
502 —* V Глава 10. Прочность эластомеров
трения. Это отношение, называемое истираемость, дает представление
о величине этого объема на единицу энергии, диссипированной при сколь-
жении. Обобщенная кривая для зависимости истираемости от скорости
скольжения, приведенная к подходящей температуре с помощью соотноше-
ния ВЛФ [ур. (18) и (19)], показана на рис. 34. Истираемость уменьшается
с ростом скорости, проходит через минимум, а затем увеличивается снова
при высоких скоростях, когда материал ведет себя как стеклоподобный. Это
Log Vq мс 1
Ts
Рис. 34. Зависимость истира-
емости Л/ji от скорости сколь-
жения V, приведенная к 20°С,
для вулканизатов бутадиен-
стирольного (SBR) и бута-
диен-нитрильного (NBR)
каучуков [80].
поведение напоминает зависимость изменения
обратной величины энергии разрушения Ub от
скорости деформации (соотношение обратное,
поскольку высокая истираемость соответству-
ет низкой прочности). Действительно, Грош
и Шалломах [80] нашли общее соответствие
между А/11 и 1/С7ь. Для этого сравнения были
определены величины энергии разрушения
J7b при высоких скоростях растяжения, око-
ло 10.000% в секунду, чтобы привести их
в соответствие с измерениями истираемости,
выполненными при скорости скольжения
10 мм/с. Это соотношение масштабов означа-
ет, что размер элементов резины, включенных
в деформацию и износ, был порядка 0,1мм, что
сравнимо с размером абразивных неровностей
на конкретных поверхностях в экспериментах.
Более того, коэффициент пропорциональности
С между истираемостью и энергией разруше-
ния, около 10’3, близок для всех исследованных
ненаполненных эластомеров. Величина С пред-
ставляет собой объем Л истертой резины на единицу энергии, подведенной
путем трения к материалу, для которого, чтобы вызвать разрушение при
растяжении, необходимо подвести единицу энергии Ub на единицу объема. Это
можно рассматривать как меру неэффективности разрушения от тангенциаль-
ных поверхностных усилий; большие объемы деформированы, но только малые
объемы удаляются. Очевидно, это отношение аналогично для всех переходов из
высокоэластического в стеклообразное состояние, и для всех эластомеров.
Было показано, что истираемость Л/р., вообще говоря, вдвое больше для
эластомеров, наполненных техническим углеродом, чем для соответствую-
щих ненаполненных эластомеров. Это удивительное наблюдение, установив-
шее, что «усиленный» материал изнашивается быстрее, может быть частично
объяснено в терминах измерения прочности на раздир, рассмотренной в пре-
дыдущем разделе. Было сделано заключение, что при условии относительно
гладкого раздира собственная прочность усиленного материала не особенно
IX. Абразивный износ -*и- 503
высока; было установлено, что она сравнима с прочностью ненаполненного
эластомера. Измерения истираемости, рассмотренные здесь, предполагают,
что при истирании она фактически несколько ниже.
Рис. 35. Схема отдельного поверхностного гребня, подвергнутого действию силы
трения (раздира) [81].
Log [подводимая работа (Дж • м 2)]
Рис. 36. Зависимость скорости износа А от величины подводимой работы трения для
НК, БСК, д, □ и изомеризованного (некристаллического) НК, о. Сплошная
линия представляет характеристики роста трещины А/ (= АС) тех же самых
материалов при повторных нагружениях [81].
Саутерн и Томас [81] с использованием простой теоретической схемы
сопоставили степень износа А с сопротивлением резины росту трещины.
Рисунок поперечных гребней возникает при стационарном износе, из-
вестном как рисунок износа Шалломаха. Единичный гребень показан на
рис. 35. Сила трения F тянет в поперечном направлении верхнюю часть
гребня и создает раздир резины в направлении, обозначенном пунктирной
линией, направленной под углом 0 к поверхности. Энергия разрушения G,
требуемая для раздира в этом направлении, определяется как F (1 + cos0).
Поэтому трещина будет продвигаться вперед на расстояние А/, определяе-
мое уравнением (23). Это приводит к потере толщины резины на величину
AZ(sin0). Следовательно
A = B'lG$Fa(\ + cosQ)a sinG (30)
504 -* V Глава 10. Прочность эластомеров
где а либо 2, либо 4 в зависимости от типа эластомера. Угол 6 может быть
установлен прямым наблюдением пути, по которому рисунок истирания
движется по поверхности при износе. Все другие величины в уравнении
(30) могут быть определены по измерениям роста трещины. Следовательно,
теория не включает никаких настроечных констант. В этой связи замеча-
тельны ее успехи в подсчете скорости износа некоторых ненаполненных
эластомеров при жестких условиях истирания (рис. 36).
Однако должны быть отмечены две трудности. Согласие неудовлетво-
рительно для резины на основе ненаполненного натурального каучука,
который изнашивается значительно быстрее, чем могли бы предсказать
измерения роста трещины (см. рис. 36). Можно было предположить, что
в этом материале кристаллизация не может развиваться в условиях истира-
ния, т. е. при быстродействующих напряжениях сжатия и сдвига. Поэтому
не проявляется высокое сопротивление росту трещины, связанное с крис-
таллизацией. Износ усиленной резины также много медленнее, чем мог бы
быть предсказан на основе измерений роста трещины. Необходимы даль-
нейшие работы для получения ответов на эти вопросы, имеющие большое
практическое значение.
Б. Химические эффекты
При мягких условиях истирания химические изменения в
эластомере становятся важными для износа [82-84]. Величина изнашивания
остается малой, но частицы, образующиеся при износе, часто склеиваются и
агломерируют, образуя большие частицы, размером в несколько миллимет-
ров. Действительно, tyuc-полиизопрен и этиленпропиленовый сополимер,
для которых разрыв молекул в условиях сдвига становится особенно явно
выраженным, образуют при истирании жидкий смолистый поверхнос-
тный слой. В противоположность этому, структурная деструкция цис-
полибутадиена не проявляется. Продукты износа остаются химически
неизменными и не слипшимися. Очевидно, что различные эластомеры
подвергаются различным химическим изменениям и это обуславливает
различные типы износа.
Во время износа могут происходить две реакции: окислительная де-
струкция как результат нагревания трением в контактной зоне и механо-
химическая деструкция, инициированная разрушением химических связей
деформациями сдвига. Имеющиеся доказательства свидетельствуют в пользу
второго процесса. Например, в отсутствие кислорода износ tyuc-полиизопрена
изменяется и становится похожим на износ tyuc-полибутадиена, тогда как
износ этиленпропиленового сополимера не изменяется [84]. Результаты
находятся в соответствии с реакциями этих материалов при взаимодействии
со свободными радикалами.
IX, Абразивный износ -•и- 505
Оказалось неожиданным, что продукты химических изменений эласто-
мера способны вызывать быстрый износ металлов, применяемых для ис-
тирания [85-87]. Например, когда лезвие ножа используется как скребок
для истирания резины, оно само изнашивается и становится тупым, а объем
истертого металла становится значительно большим, когда разрушенные
молекулы эластомера образуют относительно стабильные свободные ради-
калы [87]. Таким образом, износ металла объясняется прямым химическим
воздействием реакционноспособных частиц, вероятно свободных радикалов,
во время трения [87]. Подобный износ наблюдался и для полимеров в дру-
гих физических состояниях [88]; представляется, что разрыв молекул при
скольжении является совершенно общим явлением.
БЛАГОДАРНОСТИ
Этот обзор первоначально был подготовлен в процессе работы
по программе исследований прочности, поддержанной грантом от Иссле-
довательского Бюро Военно-Морских сил (контракт N00014-85-K-0222;
Ответственный офицер др. Р.С. Миллер). Обзор основан, в основном, на пяти
предшествующих работах [89-93]. Автор обязан Э.Х. Эндрюсу, Т.Л. Смиту,
А.Г. Томасу и другим авторам, цитируемым в тексте, за многие полезные
дискуссии, а Р.А. Паден за подготовку многих диаграмм.
ГЛАВА
11
ХИМИЧЕСКАЯ МОДИФИКАЦИЯ
ПОЛИМЕРОВ
А. ХАЛАСА
Исследования и разработки
Гудъир шайр энд раббер компании
Акрон, Огайо
Ж. МАССИ
Лексмарк Интернэйшл
Лексингтон, Кентукки
R ЦЕРЕЗА
Факультет химии и технологии полимеров
Сауз Бэнк Политехника
Лондон, Англия
I. ВВЕДЕНИЕ
Термин каучук и эластомер охватывают те полимеры, кото-
рые обладают каучукоподобностью, имеют высокие эластические свойства
при температуре эксплуатации. Однако, многие полимеры, которые сами
не являются каучуками, могут быть химически модифицированными
в относительно малой степени, чтобы проявлять вязкоупругость. Напри-
мер, введение небольшого количества атомов хлора и сульфоксидных
групп в полиэтилен препятствует образованию кристаллических областей,
и материал становится эластомером в широком температурном интервале.
По этим группам возможна вулканизация; при этом получаются обратимо
деформируемые, стойкие к растворителям материалы. С другой стороны,
можно с помощью химической реакции превратить макромолекулярные
цепи каучука таким образом, что они теряют свою вязкоупругость и ста-
новятся термопластичными при температурах эксплуатации (например,
циклизация полиизопрена). Пограничная линия между вязкоупругим
и пластическим состоянием относительно трудно определяется. Общим
II. Химическая модификация полимеров по основной цепи и концевым группам -* v 507
для каучуков и пластиков является их макромолекулярная природа.
Это позволяет лучше понимать структуру на молекулярном уровне (пла-
стики, модифицированные каучуком, и пластифицированные каучуки).
За несколькими исключениями, модификация, которая проводилась на
полимере одного вида, при определенных условиях может быть выполнена
на другом полимере. Одинаковый принцип химической модификации
будет придавать некие новые свойства материалам, которые желательно
получить без потери уже имеющихся свойств, с тем чтобы получить ве-
щество, во-первых, полезное и, во-вторых, подтверждающее правильность
модификации.
На заре полимерной химии и технологии химическая модификация,
успешно применяемая к одному полимеру, могла быть использована для
целого круга химически подобных веществ (например, этерификация цел-
люлозы), и такие реакции были действительно распространены на аналогич-
ные материалы. Сегодня число доступных гомополимеров выросло до сотен,
а число статистических сополимеров до тысяч; поэтому число возможных
процессов химической модификации, включая блок и привитые полимеры,
достигают астрономических цифр. Только часть этих потенциальных сис-
тем была оценена (и часто неглубоко, чтобы получить патентную защиту),
так что эта область химии полимеров широко открыта для исследований
и разработок. Из-за своей широты и глубины область химической модифи-
кации полимеров в данной главе может быть рассмотрена только в общих
чертах и могут быть описаны только наиболее важные реакции. Однако,
для сохранения интереса читателей этот широкий обзор содержит более
глубокое обсуждение некоторых областей, которые представляют интерес
для химика-резинщика и которые, по мнению авторов, имеют значитель-
ный потенциал для дальнейшего развития. Главными являются принципы,
лежащие в основе химической модификации полимеров, и частные условия
химических реакций умышленно опускаются. Ряд подходов к химической
модификации каучуков рассматриваются в деталях в других главах этой
книги, и читатель отсылается к соответствующим главам по химии, вулка-
низации, характеристикам, блоксополимерам и т.д.
II. ХИМИЧЕСКАЯ МОДИФИКАЦИЯ ПОЛИМЕРОВ
ПО ОСНОВНОЙ ЦЕПИ И КОНЦЕВЫМ ГРУППАМ
Свойства полимеров зависят от многих факторов, включая
взаимодействия концов цепи с субстратом, таким как технический угле-
род или кремнеземные наполнители, глинистые минералы и карбонат
кальция. С этой точки зрения известно много работ, которые выполнены
по модификации концов цепей, особенно проведенной путем анионной
полимеризации с металлами первой или второй группы [1, 2, 3], как
показано ниже.
508 Глава 11. Химическая модификация полимеров
Этот подход стал возможным по реакции концов цепи, содержащих
металл, с активным галоидным соединением, как показано дальше, та-
ким как тетрахлорид олова или тетрахлорид кремния. Эти типы реакций
приводят к увеличению молекулярной массы, так же как к улучшению
взаимодействия с наполнителем, что проявляется в улучшенных свой-
ствах полимера [4].
Модификация полимеров с помощью введения полярных групп, таких
как аминосилоксановые, может быть проведена с помощью анионного ката-
лиза или так называемой «живой полимеризации», а также с функциональ-
ными инициаторами, или с цепными функциональными мономерами, как
было показано несколькими группами исследователей. Функциональными
инициаторами являются циклические амины, связанные с алкильными
группами, а функциональные мономеры основаны на стироле и дизопро-
пенил бензоле [5-9].
Совсем недавно получен американский патент, принадлежащий компа-
нии Goodyear Tire and Rubber Company, в котором говорится, что полярные
функциональные мономеры могут быть сополимеризованы с сопряжен-
ными диенами и винил ароматическими соединениями для того, чтобы
модифицировать полимерную цепь. Функциональные мономеры, такие как
3-(2-пирролидиноэтил) стирол
II. Химическая модификация полимеров по основной цепи и концевым группам 509
или 3-(пирролидино-2-метилэтил) а-метилстирол:
были сополимеризованы в растворе и эмульсии с бутадиенстирольными
сополимерами в колчестве от 1 до 10%, что улучшает свойства полимера
и приводит к низким гистерезисным потерям и лучшему сцеплению шин
с мокрой дорогой.
510 -*v- Глава 11. Химическая модификация полимеров
III. ЭТЕРИФИКАЦИЯ И ГИДРОЛИЗ ПОЛИМЕРОВ
Процессы химической модификации, обсуждаемые в этом
разделе, в историческом и научном плане так тесно связаны с уникальным
полимером, целлюлозой, что хотя последний применяется преимущественно
как волокно, а не как эластомер, обсуждение здесь процессов модификации
целлюлозы вполне уместно. За исключением того факта, что некоторые про-
изводные целлюлозы, подобно этилцеллюлозе, при пластификации могут
быть в достаточной степени эластифицированы, возможности модификации
основного полимера хорошо демонстрируются на самом жестком и высоко-
кристаллическом веществе — целлюлозе. Более того, ввиду ожидаемого
дефицита углеводородов целлюлоза может вскоре начать играть новую роль,
как сырье для получения других полимеров.
Целлюлоза, химически идентифицируемая как Р~1,4-глюкан, явля-
ется широко распространенным природным полимером, составляющим
непрерывную структуру клеточных мембран растений. Для ознакомления
с общими свойствами и химией самой целлюлозы читатель отсылается к
стандартным учебникам и недавним обзорам.
Большая часть ранней истории химической модификации целлюлозы
связана с попытками обнаружить растворитель для нее, т. к. ее макромо-
лекулярная структура не было понята в то время. В 1844 г. Мерсер обна-
ружил и реализовал в промышленном масштабе взаимодействие щелочи
с волокнами целлюлозы; этот процесс все еще используется под названием
«мерсеризация». Начальный продукт реакции, щелочная целлюлоза, не
является продуктом химической модификации, но представляет собой фи-
зическую форму, в которой вода и ионы натрия проникают в макромолеку-
лярную структуру и снижают уровень водородных связей с последующим
набуханием волокон. Начальный продукт, целлюлозу I превращают в цел-
люлозу II путем сложной физико-химической модификации на конечной
стадии промывки. Степень и скорость набухания в этом процессе зависит
от источника целлюлозы, и если волокна растягиваются во время реакции,
достигается оптимум взаимодействия. Целлюлоза набухает во многих других
растворах неорганических солей [13], из которых хлорид цинка, как было
найдено, имеет наиболее широкое применение. Водные растворы тиомоче-
вины, резорцина, хлораль гидрата и сульфоната бензола также приводят
к ограниченному набуханию целлюлозы. Во всех случаях уменьшение фи-
зического взаимодействия может быть зафиксировано с помощью изучения
содержания кристаллической фазы на рентгенограммах.
Полная растворимость целлюлозы в медноаммиачных растворах, от-
крытая в 1857 г. Швейцером, привела к развитию производства волокон,
но так же как в случае щелочной целлюлозы, регенерированный полимер
является химически тем же самым. Регенерация целлюлозы растворами
ксантата, открытая Кроссом и Беваном в 1893 г, является другим процессом,
III. Этерификация, гидролиз полимеров 511
который применяется до сих пор; он составляет основу для производства
целлофана.
Первый процесс химической модификации целлюлозы, открытый
Брэконном в 1833 г, был осуществлен с получением нитрата целлюлозы из
широкого круга целлюлозных материалов. Продукты представляли собой
легко воспламеняемые порошки, которые могут растворяться в концентри-
рованной уксусной кислоте с получением светлых жестких лаков. (Отметим
превращение волокна в пленку путем химической модификации). В 1847 г.
глубоко нитрованная целлюлоза, нитроклетчатка, была открыта Ботгером
и Шонбейном, и в 1870 г. Шуценберг получил ацетилированную целлю-
лозу, используя горячий уксусный ангидрид как реакционную среду. Эти
реакции протекают по обычному механизму [14], а именно этерификации
гидроксильных групп в основной целлюлозной матрице:
н ОН СН2ОН
(1)
В этих ранних работах использовался очень широкий круг органических
кислот для получения сложных, смешанных и простых эфиров целлюлозы [15].
Типичным примером, представляющим значительный промышленный ин-
терес, является ацетилированная целлюлоза. Так же как во всех процессах
этерификации макромолекулярных материалов доступность гидроксильных
групп к этерифицирующему агенту имеет первостепенное значение. Реакция
(1) соответствует полной этерификации, процессу, который, вероятно, никогда
не может быть осуществлен. Идентификация этерифицированных продуктов,
следовательно, зависит не только от содержания ацетильных групп, но так же и
от локализации этих групп в основной макромолекулярной цепи. Оба эти фак-
тора оказывают влияние на метод получения и условия этерификации [14].
Хотя многие реакции этерификации [14] основаны на неорганичес-
ких кислотах, для нерастворимых гидроксильных соединений, подобных
целлюлозе, взаимодействие с сероводородом является наиболее важным.
Для получения набухшей щелочной целлюлозы, которая (после старения)
реагирует с сероуглеродом с образованием натриевой соли ксантогената
целлюлозы, обычно используется гидроксид натрия:
.C=S
О' О S'
I I (2)
лллДеллюлозаллдл + CS2-хллюЦеллюлозапмг
512 “*V" Глава 11. Химическая модификация полимеров
Целлюлоза может быть регенерирована прядением (или экструдировани-
ем пленки) в кислотной ванне, содержащей соли, такие как сульфат натрия
или цинка [13]. При прядении или экструзии макромолекулы ориентиру-
ются в направлении потока, давая вискозное волокно с высокой прочностью
или целлофан. Протекание макромолекулярной ориентации при прядении
является очень важным, и это применяется в химической модификации
многих полимеров.
Простые эфиры целлюлозы составляют другую важную группу произ-
водных, полученных из щелочной целлюлозы стандартными реакциями эте-
рификации между гидроксильными группами и алкилгалоидами. Свойства
простых эфиров зависят от глубины реакции, т. е. степени этерификации.
В общем, этилцеллюлоза является водонерастворимым термопластичным
материалом, тогда как метиловый эфир, этилгидроксиэтилцеллюлоза и
карбоксиметилцеллюлоза растворимы в холодной воде и могут применяться
как вязкоэластичные загустители и адгезивы.
Для получения синтетических гидроксиполимеров гидроксильные
группы могут вводиться путем сополимеризации основных мономеров с
гидроксилсодержащими мономерами. Эти группы могут использоваться для
этерификации [16], но относительно высокая стоимость мономеров ограни-
чивает их широкое применение в прямой сополимеризации. Вместо этого
вводят группы, требующие полного или частичного гидролиза эфирных групп
в гидролизуемом полимере, таком как поли (винилацетат). Полный гидролиз
дает поли (виниловый спирт) (ПВА), водорастворимый полимер, применяемый
в качестве стабилизатора и модификатора вязкости для водных систем:
^сн2—СН—сн7—сн^
1’1 (3)
он он
ПВА имеет уникальное значение как усиливающее волокно в комбина-
ции со слабыми материалами, такими как шерсть мериноса при прядении
деликатных волокон, из которых он может быть в последующем удален про-
мывкой водой. Главная часть полученного полимера реагирует с альдегидом
с образованием соответствующего поливинилформаля, поливинилацеталя
и поливинилбутираля:
ОН CZ (4)
jLat + RCHO---> + Н2О
Имеются различные сорта каждого из этих материалов, отличающиеся
молекулярной массой и степенью замещения. Эти полимеры применяются
как компоненты систем с уникальными адгезионными свойствами, напри-
мер, в производстве безопасных многослойных стекол (поливинилбутираль
и смешанные производные) и металл-металл-адгезивов для крепления ме-
IV. Гидрирование полимеров -•V- 513
талла к металлу (поливинилормаль), отвержденных фенольными и другими
смолами. Реакции поливинилового спирта с кислотами или альдегидами
протекают как обычная этерификация, способом, применяемым для синтеза
полимеров и сополимеров, которые не могут быть легко получены путем
стандартной полимеризации (например, когда не позволяет относительная
реакционная способность мономеров).
Природный и синтетический каучуки в общем не содержат в достаточном
количестве гидроксильные группы для использования в реакции этерифика-
ции. Концевые гидроксильные группы могут быть введены в синтетические
каучуки как фрагменты катализаторов или инициаторов и применяться для
реакций сочетания или удлинения цепи.
IV. ГИДРИРОВАНИЕ ПОЛИМЕРОВ
Любой полимер с ненасыщенными углеводородными груп-
пами в главной цепи или в боковых группах может быть прогидрирован.
Ранние исследования по гидрированию эластомеров концентрировались на
деструктивном гидрировании с последующей потерей макромолекулярной
структуры. Это выходит за пределы данной главы, так что читатель отсыла-
ется к соответствующим источникам [12, 17, 18]. Наиболее свежие работы
по гидрированию приводили к получению превосходных продуктов, таких
как полиэтилен, полученный гидрированием поли (1,4-бутадиена) [19,20],
и чередующийся сополимер этилена и пропилена — каучук, полученный
гидрированием полиизопрена.
-(-сн2—с=с—сн2-> -(-СН2—СН2СН2—СН2-)-„ (5)
-f-CH2—с=с—сн2-у- -/-сн2—с—сн2—сн24-
г । I Т । | (6)
\ СН3 ’ ' СНз ' п
Эти реакции были выполнены с использованием гетерогенного ката-
лиза.
Для создания новых полимеров путем гидрирования были использованы
гомогенные растворимые катализаторы переходных металлов. Катализато-
ры гомогенного гидрирования обычно получаются из катализаторов типа
Циглера-Натта, основанных на органических солях никеля или кобальта,
восстановленных в присутствии алюминийорганически или литийоргани-
ческих соединений. Эти катализаторы применяются для получения насы-
щенных эластомеров гидрированием ненасыщенных эластомеров. Конечный
полимер имеет вязкоупругие свойства, сильно отличающиеся от свойств
ненасыщенных исходных полимеров. Например, гидрирование на 99%
поли(1,2-бутадиена) приводит к образованию полибутена [21], который
имеет более низкую температуру стеклования, чем исходные полимеры.
514 -•V- Глава 11. Химическая модификация полимеров
Интересно заметить, что гидрирование не влияет на молекулярномассовое
распределение и архитектуру основной цепи.
Легкость гидрирования и достигаемая степень насыщения отражают
микроструктуру полимера. Гидрирование ненасыщенных эластомеров,
содержащих различные 1,4-1,2 цис-транс структуры, обычно протекает
блочным путем. Это происходит из-за различающейся реакционной способ-
ности различных двойных связей. В общем, двойные связи 1,2-структуры
в четыре раза более реакционноспособны, чем двойные связи 1,4-структуры,
цис-1,4 звенья более реакционноспособны, чем транс-1,4 звенья. Чембер-
лен и сотрудники [22] показали, что гидрирование 1,2 звеньев протекает по
статистическому механизму, в то время как при гидрировании 1,4 звеньев
наблюдается другой механизм.
Полное гидрирование поли (1,4-бутадиена), цис- или транс-структуры при-
водят к получению полиэтилена с низкой точкой плавления около 115 °C. Счита-
ется, что этот линейный полиэтилен является материалом с низкой плотностью.
Неопубликованные данные, полученные при гидрировании на 99% цис-поли
(1,4-бутадиена) показывают, что гидрирование протекает с образованием бло-
ков. При гидрировании 40 или 50% поли(1,4-бутадиена) (содержание цис —
98%) на никелевом катализаторе образуется полимер с температурой плавления
+98 °C и температурой кристаллизации -13 °C определенной методом дифферен-
циальной сканирующей калориметрии (ДСК). Это подтверждает тот факт, что
гидрирование цис-поли-1,4-бутадиена имеет блочный характер с образованием
блоков полиэтилена и блоков поли(^ис-1,4-бутадиена).
Полибутадиены, полученные на анионных катализаторах в присутствии
полярных модификаторов, имеют смешанную структуру, состоящую из
цис-1,4, транс-1,4 и 1,2 звеньев. Гидрирование этих полимеров приводят
к интересным продуктам. Как упомянуто ранее гидрирование 1,2 звеньев
протекает в 3-4 раза быстрее по сравнению с 1,4 звеньями. Из-за этого
различия в реакционной способности при гидрировании полибутадиена,
содержащего от 40 до 50% 1,2 звеньев, получается полимер, содержащий
часть полиэтилена с температурой плавления от 85 до 95 °C и каучукопо-
добную часть с температурой стеклования -62 °C.
Блоксополимеры могут быть также гидрированы с образованием уни-
кальных продуктов. Гидрированные триблоксополимеры поли(стирол-бу-
тадиен-стирол) (SBS) промышленно доступны и поставляются компанией
Shell под названием Кратон G. Средний блок обычно имеет смешанную
микроструктуру, представляющую смесь поли(1,2-бутадиена) и поли (1,4-
бутадиена). Образующийся продукт представляет собой гидрированный
ненасыщенный полимер, который проявляет более высокую термо- и окис-
лительную стойкость, по сравнению с исходным триблокполимером SBS.
Подобные процессы применялись другими исследователями [23] для гид-
рирования двублочных поли (1,4-бутадиен-1,2-бутадиена) [24] и трехблочных
поли (1,4-бутадиен-1,4-изопрен-1,4-бутадиен). При гидрировании этих диблок
V. Дегалогенирование, отщепление и реакции галогенирования в полимерах -*v- 515
и триблоксополимеров образуются термопластические эластомеры с крис-
таллическими и аморфными сегментами. Все эти материалы обнаруживают
кристалличность, растворимость и спектр динамических механических потерь,
отличающиеся от соответствующих свойств ненасыщенных материалов.
Был разработан другой метод получения насыщенных каучуков [25]
с использованием восстановления диимидом. Этот метод может применяться
для достижения высокой степени насыщенности в зависимости от типа ис-
пользуемого реагента; однако, при реализации этого метода могут протекать
побочные реакции. Генерирование диимида из п-толуол сульфонил гидридов
приводит к образованию кислотных фрагментов, которые могут вызвать
циклизацию некоторых ненасыщенных эластомеров.
V. ДЕГАЛОГЕНИРОВАНИЕ, ОТЩЕПЛЕНИЕ
И РЕАКЦИИ ГАЛОГЕНИРОВАНИЯ В ПОЛИМЕРАХ
А. Дегидрохлорирование поли(винилхлорида)
Дегидрохлорирование поли (винилхлорида) является предме-
том многих исследований, особенно при разработке ПВХ полимеров и сополи-
меров с повышением стабильности. Подобно многим полимерным реакциям
дегидрохлорирование является сложным процессом. Виниленовые группы,
получающиеся отщеплением НС1 от соседних атомов углерода в цепи
АЛ^СН 2—СНС1—СН 2—СНС1 —> НС1 + СН = СН — СН 2СНС1^
(7)
могут быть результатом свободнорадикальных или ион-радикальных стадий.
Присутствие малых количеств структур «голова к голове», «хвост к хвосту»
и других конфигурационных нерегулярностей в структуре основной цепи ПВХ
приводит к более сложным стадиям отщепления при термической деструкции
в чистом виде или в присутствии катализаторов, таких как хлорид алюминия.
Образование циклических структур, представляющих главный процесс в тече-
ние дегидрохлорирования, влияет на распределение атомов хлора в полимерной
основной цепи. Бромид водорода может эффективно термически отщепляться
от поливинилбромида, т.к. дегидрогалогенирование является термической
реакцией, сложность которой увеличивается от хлорида к бромиду.
В. Термическое отщепление
Процесс термического отщепления может применяться к наиболее «на-
сыщенным» группам виниловых полимеров при контролируемом пиролизе
от 600 до 700 С, с образованием поливиниленовых соединений, например,
путем отщепления уксусной кислоты от поливинилацетата. Тщательный
контроль температуры может привести к достижению бифункциональной
реакции и/или внутримолекулярной циклизации. Это разработано в про-
516 Глава 11. Химическая модификация полимеров
мышленном масштабе при относительно высокой температуре в случае
полимеризации метакриламида выше 65 °C, с образованием полимера
с существенной долей имидных групп:
Н
сн31 сн3
АЛМк | S' । X. |
с I с
I н I
С=О С=О + NH3
(8)
Н
Полимеры и сополимеры мультифункциональных виниловых мономеров
(применяемый термин, позволяет охватить присутствие галогена или других
реакционно способных групп дополнительно к винильным группам, т. е.
более одной полимеризуемой группы), такие как а-хлоракриловая кислота,
часто подвергается частичной лактонизации и гидролизу в течение полиме-
ризации. Нагревание в спиртовом растворе или электролизуемом спиртовом
растворе приводит, например, к введению двойных лактамовых циклов во
время кислотного гидролиза поли (а-ацетоаминоакриловой кислоты)
NHAc СООН NH-------------С=О
"«С—СН,—С -------»лл»С—СН,—С"" + АсОН
I I I NHAc
СООН NHAc СООН
(9)
NH------С=О
I I
---» С—СН,—С + АсОН
I I
СО------NH
Изменение цвета полиакрилонитрила, являющееся следствием реакции
отщепления такого же типа, которая протекает как внутри, так и межмолеку-
лярно, образует поперечносшитые нерастворимые циклические продукты:
Н
Н гн Н СН2 I
•ммСН,— CHnwCH,—СН*м-/1\ / С*«
1 1 , Y I
CN CN III
CN /СН, Н
Л*«СН2 N--СН2 N
I I
_с, ,сн
I СН,
н
(10)
V. Дегалогенирование, отщепление и реакции галогенирования в полимерах -'V 517
Контролируемое нагревание волокон полиакрилонитрила в механически
напряженном состоянии также вызывает отщепление азотсодержащих
продуктов с образованием углеродного волокна с высокой прочностью, ко-
торое может рассматриваться как конечный продукт в химических реакциях
отщепления. Разработаны углеродные волокна из целлюлозных материалов,
лигнина, различных интерполимеров и смесей. Структура этих продуктов
представляет, главным образом, трехмерные углеродные сетки, частично
кристаллические и частично графитоподобные и аморфные.
С. Галогенирование полимеров
Галогенирование и гидрогалогенирование эластомеров
широко описано в литературе [26]. Основными проблемами этих реакций
являются циклизация и разрыв цепи, которые протекают параллельно ре-
акции галогенирования. Они затрудняют характеристику образующихся
продуктов. Несмотря на это некоторые продукты могут быть получены в про-
мышленном масштабе. Хлорирование поли(1,4-бутадиена) для получения
продукта, аналогичного ПВХ, описано несколькими исследователями [27].
В этом процессе протекают многообразные побочные реакции и деструк-
ция цепи. Хлорирование бутилкаучука приводит к конечным продуктам,
которые применяются в шинной промышленности, как внутренние слои
автомобильных камер.
Этилен-пропилен-диеновые сополимеры (EPDM) вследствие получе-
ния их статистической полимеризацией являются аморфными, и поэтому
легко галогенируются. EPDM был хлорирован для улучшения его свойств
и возможности совулканизации с другими каучуками. Хлорированию под-
вергался третий мономер — дициклопентадиен с образованием аллильных
хлоридов [28]. Таким способом был хлорирован EPDM и образующиеся
продукты имели улучшенные свойства.
Каучуки EPDM модифицируются 1,2-присоединением N-хлоротио-
сульфонамидами по своим олефиновым участкам [29-31]. Такие процессы
присоединения могут быть проведены в растворе или без растворителя
в смесителе. Реакция без растворителя облегчается добавлением карбо-
новых кислот [32,33] или некоторых солей металлов слабых кислот [34].
Модифицированные EPDM представляют интерес из-за своей способности
совулканизоваться в озоностойких смесях с полидиеновыми каучуками.
Менее исследованы как модификаторы N-хлоротиокарбоксоамид и имиды,
которые также реагируют с EPDM, образуя модифицированные продукты,
совулканизуемые в смесях с полибутадиенами [35].
В отсутствие кислорода хлорирование полиэтилена с добавкой или без
добавки катализатора может контролироваться для получения продуктов
с различным содержанием хлора. Процесс хлорирования — статистический,
так что хлорирование полиэтилена до того же самого содержания хлора, что
518 JV Глава 11. Химическая модификация полимеров
и в поли (винилхлориде) (60%), дает продукт, который химически отли-
чается от ПВХ, будучи полностью совместимым с ним. Это беспорядочное
хлорирование полиэтилена разрушает его кристалличность. При степени
хлорировании, соответствующей полной потере кристалличности, хлори-
рованный продукт становится растворимым при комнатной температуре.
Бромирование полиэтилена протекает аналогично с образованием каучуко-
подобного полимера при 50% содержании брома.
Как хлорирование, так и бромирование полипропилена и полиизобу-
тилена приводит к разрушению главной цепи с потерей многих полезных
свойств. Деструкции при хлорировании, однако, можно избежать при низких
температурах ограничением реакции до 2%-го превращения. В этом случае
образуется полезный промышленный продукт.
Добавление хлористого водорода к ненасыщенным эластомерам также
привлекает значительное внимание. Интенсивные работы были проведены
по гидрохлорированию Гевеи [поли(гргс-1,4-изопрена)] и Балаты [поли
(77гранс-1,4-изопрена)], начиная с 1940 г [36,37]. Как ^ис-1,4, так и транс
цис-1,4-полиизопрены легко присоединяют хлористый водород, согласно
правилу Марковникова, при незначительной циклизации.
D. Циклизация полимеров
Катионная циклизация ненасыщенных полимеров, таких
как поли(цис-1,4-изопрен), поли(3,4-изопрен), поли(1,2-бутадиен) и поли
(1,4-бутадиен), обычно приводит к образованию циклических смолообраз-
ных продуктов, не имеющих промышленного значения. Обширный обзор по
этому вопросу опубликован Шульцем и др. [38]. Циклизация ненасыщен-
ных эластомеров, таких как полиизопрен, может быть проведена в твердом
состоянии, в растворе или даже в латексе. Процесс включает превращение
линейных макромолекул цепи в более короткие цепи, содержащие моно-,
ди-, три- и тетрациклические группы, распределенные беспорядочно по
цепи. Циклизация полиизопрена увеличивает температуру стеклования
до 20-30 °C. Механизм циклизации эластомеров зависит от применяемого
в процессе катализатора.
VI. ДРУГИЕ РЕАКЦИИ ПРИСОЕДИНЕНИЯ К ДВОЙНОЙ
СВЯЗИ
А. Производные этилена
Кроме только что рассмотренного присоединения галогенов
и гидрогалогенов по двойной связи, имеется много других реагентов, кото-
рые реагируют с ненасыщенными полимерами по свободнорадикальному,
ионному или радикал-ионному механизмам. Первостепенное значение
имеет присоединение производных этилена к полидиенам. Одной из первых
VI. Другие реакции присоединения к двойной связи 519
реакций натурального каучука, изученных детально, является соединение
с малеиновым ангидридом [39]. В зависимости от условий реакции и присут-
ствия или отсутствия свободнорадикальных инициаторов могут иметь место
одна или более из четырех основных реакций с образованием продуктов,
показанных ниже (стрелка показывает, где происходит присоединение
и образование новых связей).
1. Внутримолекулярное присоединение двойной связи в полиизопре-
новой цепи:
— СН ^С —СН2аллг
I хсн2—сн2
(11)
2. Межмолекулярное присоединение к двойной связи в различных по-
лимерных цепях. В эту группу могут быть включены статистически
возможные реакции между далеко разделенными двойными связями
внутри одной молекулы:
(12)
3. Присоединение к а-метиленовому атому углерода полиизопреновой
цепи:
СН3 Н
\ /
с=с
ЛЛЛЛСН 2 СН****
(13)
520 -*v* Глава 11. Химическая модификация полимеров
4. Межмолекулярное присоединение к а-метиленовым атомам углерода
в соседних цепях (или удаленных а-метиленовых атомов углерода
в той же самой макромолекуле):
(14)
В общем, скорость реакции увеличивается с повышением температу-
ры и в присутствии кислорода или свободнорадикальных инициаторов,
но при тех же самых условиях ускоряются межмолекулярные реакции,
приводящие к образованию геля. Аналогичные реакции имеют место
с гуттаперчей, синтетическим поли(цис-1,4-изопреном) и поли(цис-1,4-
бутадиеном). Многие исследователи применяют вальцы и другое пла-
стицирующее оборудование как удобный путь смешения малеинового
ангидрида с каучуками при повышенной температуре, но при исполь-
зовании этого оборудования процесс осложняется механохимическими
реакциями. Продукты реакции натурального каучука, содержащие от 5
до 10% связанного малеинового ангидрида, могут быть вулканизованы
по стандартному серному рецепту, однако больший интерес представляет
возможность создания поперечных связей с использованием оксидов
кальция, магния и цинка.
Соединения, реагирующие аналогично через активацию двойной связи
(исключая здесь блок или привитую сополимеризацию) включают малеи-
новую кислоту, N- метил-малеимид, хлормалеиновый ангидрид, фумаровую
кислоту, у-кротолактон, р-бензохинон и акрилонитрил. Другие полимеры
с ненасыщенной основной цепью, такие как полибутадиен, сополимеры
бутадиена со стиролом и с акрилонитрилом, и бутил-каучук реагируют подоб-
ным образом. Реакция восстановления с ПВХ является механохимической
по природе (обсуждается ниже).
VII. Реакции окисления полимеров -*v- 521
В. Реакция Принса
Еще одним примером присоединения к полимеру с ненасы-
щенной главной цепью является реакция Принса между этиленовыми угле-
водородами и соединениями, содержащими альдегидные группы. Кирхгоф
в 1923 г. описал реакцию натурального каучука в растворе бензола с водным
формальдегидом в присутствии серной кислоты. Общая реакции альдегида,
RCHO, с полиизопреном в присутствии органических или неорганических
кислот или ангидридов солей металлов представлена ниже:
х
Н\ нс I ^СНОН
л»^С=С^>т + С/ —(15)
I н I х- I хн
СН3 R СН3
В отсутствии таких катализаторов реакция приводит к сдвигу двойной связи
в большей степени, чем к отщеплению:
^снон
алл1»С = + R — >С — С™™' (16)
I н нН н
сн3 СН2
Эти реакции могут быть проведены в растворе, дисперсии или в твердой фазе
[39]; в последнем случае опять возникает затруднение отличить реакцию
Принса от механохимических реакций, инициированных разрывом цепи
при пластикации.
Другие альдегидные соединения, такие как глиоксаль и хлораль, также
реагируют аналогичным путем с полиизопренами и ненасыщенными кау-
чуками [например, поли(^ис-1,4-изопреном), поли(г{ис-1,4-бутадиеном)]
и сополимерами изобутилена и изопрена. Применение сильных кислот
или кислот Льюиса, вызывает осложнения, т.к. сами кислоты при со-
ответствующих условиях катализируют циклизацию и цис-транс изо-
меризацию, и эти реакции могут протекать одновременно с реакциями
присоединения.
VII. РЕАКЦИИ ОКИСЛЕНИЯ ПОЛИМЕРОВ
Неконтролируемое окисление каучука является вредным
по влиянию на физические свойства. Реакции окисления протекают легко
с ненасыщенными группами полимеров и часто упоминаются в общем виде
как эпоксидирование; однако, окисление при контролируемых условиях
может приводить к полезным продуктам, таким как эпоксидированный
натуральный каучук, производимый Ассоциацией производителей каучука
522 -*V* Глава 11. Химическая модификация полимеров
[38-41]. Натуральный каучук в форме латекса обрабатывается перекисью
водорода, растворенной в уксусной кислоте. Это приводит к образованию
натурального каучука, эпоксидированного на 50%. Этот каучук проявляет
очень интересные физические свойства; в нем превосходно распределя-
ется техуглерод. Аналогично неводное эпоксидирование синтетического
полиизопрена может быть достигнуто с использованием либо перекиси
водорода, либо гипохлорида в трет-бутаноле. Контролируемая степень эпок-
сидирования обычно приводит к очень интересным продуктам. Например,
эпоксидированный на 25% синтетический полиизопрен представляет собой
эластомер с вязкоупругими свойствами, аналогичными свойствам неэпокси-
дированного материала, но лучше диспергирует техуглерод. Это обеспечивает
высокие модули и прочность, однако, высокая степень эпоксидирования
(от 60 до 75%) приводит к образованию смолообразного материала, который
не является каучуком.
VIII. ФУНКЦИОНАЛИЗАЦИЯ ПОЛИМЕРОВ
Полимеры с насыщенной основной цепью, такие как поли-
стирол, полиэтилен и полипропилен, могут быть функционализирован-
ны. Функционализация полстирола привлекает значительное внимание,
т.к. он представляет собой уникальный исходный полимер с ароматичес-
кими кольцами, который способен подвергаться многим нуклеофильным
и электрофильным реакциям. Смола, относительно недавно появившаяся
на рынке, это — сульфированный полистирол. Данная смола приме-
няется в качестве ионообменного материала и носителя для катализа-
торов.
Электрофильное замещение полистирола по реакции хлор-металлирова-
ния приводит к образованию функциональных групп хлора. Это открывает
возможности получения многих производных полистирола. На основе хлор-
металл и рованного полистирола могут быть получены такие производные,
как четвертичные аммониевые или фосфониевые соли. Аналогично могут
быть получены производные простых и сложных эфиров, сульфонамидов,
силанов и кетонов замещением атомов хлора на хлорметаллированный
полистирол. В случае полистирола, было показано, что функционализация
концов цепи может быть осуществлена, если на концах цепей находятся
металлы I группы (литий и калий).
Japanese Synthetic Rubber Company, и Nippon Zeon сообщили, что у элас-
томеров, полученных анионной полимеризацией, которые функционально
ограничены активным литием, цепи могут обрываться кетоном Михлера,
бензофеноном и различными енамидными группами. Более того, эти цепи
могут содержать на концах кремний или олово. Функционализация концов
X. Блок и привитая полимризация 523
цепи не изменяет вязкоупругость полимерных цепей, но резко улучшает
взаимодействие эластомера с наполнителем, и, следовательно, уменьшает
гистерезисные потери.
IX. ДРУГИЕ ХИМИЧЕСКИЕ РЕАКЦИИ ПОЛИМЕРОВ
Прямое замещение атомов водорода в ароматических коль-
цах, таких как стирол или аллильного водорода в поли (1,4-бутадиене)
или в поли(1,4-изопрене), может быть выполнено через металлирование
с металлорганическими соединениями I группы, таких как литий, натрий
и калий. Обычно выход реакции в этих случаях невысокий, и продукт яв-
ляется нерастворимым; однако, применение хелатных диаминов с литийор-
ганическими соединениями увеличивает выход, и продукт становится
растворимым в циклогексане. Например, полистирол металлируется
с высоким выходом, образуя полиэтилированный полистирол, в который
введено несколько функциональных групп. Аналогично, полиизопрен
и полибутадиен успешно металлируется либо s-бутиллитием, либо трет-
бутиллитием в присутствие тетраметилэтилендиамина (ТМЕДА) при
50°С. В случае полиизопрена разрыв цепи и уменьшение молекулярной
массы происходит при высокой температуре и времени металлирования.
Во многих случаях эти литийфункциализированные полимеры приме-
няются для получения привитых и блок сополимеров. Они обсуждаются
более детально в разделе X (В).
X. БЛОК И ПРИВИТАЯ СОПОЛИМЕРИЗАЦИЯ
А. Влияние на структуру и свойства полимеров
Некоторые наиболее существенные изменения в структуре
и свойствах полимеров могут быть расширены за счет блок- или привитой
сополимеризации (см. главу XIII). Термин блоксополимер применяется
к макромолекулам, имеющим последовательности с различными химиче-
скими (или физическими, например, тактическими) структурами, обычно
представляемые как А, В и С. Последовательности имеют молекулярную
массу, которая сообщает им полимерные свойства, даже если их разделять.
Способ, которым эти последовательности расположены, определяет тип
блоксополимера. Диблоксополимер изображается как АВ, указывая, что
сегмент с химическим составом А связан с сегментом В. Другие возможные
типы блоксополимеров включают три блока АВА и АВС, они могут быть
линейными или разветвленными; линейные структуры называются блок-
сополимерами,
АААААА • ВВВВ • • ВВВВВ • ААААА
(17)
524 “'V Глава 11. Химическая модификация полимеров
а разветвленные структуры называются привитыми сополимерами (18),
м? «мм»ААА AAA»*»-**»* «w •nwBBBBB • • • BBB'Ww %
в В А А
в (а) В или А (Ь) А
в В А А (18)
в В А
i В i А
1 А А
Такие полимерные последовательности могут быть соединены вместе как
блок или как привитые сополимеры с различными свойствами, хотя по
опыту авторов, главные различия между свойствами привитых и блок со-
полимеров того же состава выражаются только в растворе или в расплаве.
Например, натуральный каучук может быть блоксополимеризован с поли-
метилметакрилатом, или мономерный метилметакрилат может быть привит
к натуральному каучуку. Посредством номенклатуры можно отличить
одну структуру от другой включением буквы b (блок) и£ (графт) для блок
и привитых сополимеров. Соответственно, названия специфических после-
довательностей, которые вводятся, будут таковы — натуральный каучук-Ь-
поли (метил метакрилат) и натуральный каучук-g-пол и (метил метакрилат)
в случае приводимых выше примеров. Структуры этих двух макромолекул
представлены (17) и (18), где ...АААА... представляет натуральный каучук,
а ...ВВВВВ... поли(метил метакрилат).
Число и порядок последовательностей могут быть более сложными. Блок
сополимеры обычно получаются свободнорадикальной или живой полиме-
ризацией. В этих процессах могут получаться полимеры, которые состоят
из чистых блоков А, связанных с чистыми блоками В без межфазной зоны
смешанных структур А и В. Получение блоксополимеров не ограничивается
мономерами А и В, но может также включать сегменты статистических
сополимеров. Например, блок статистического сополимера АВ может
быть связан с блоком полимера А или В. Более того, точка присоедине-
ния блоков может быть или на конце, или посередине полимерной цепи.
Некоторые примеры различных возможных типов блоксополимеров
показаны ниже:
- АААААА ВВВВ ААААА - или - АААААААААААА -
В В
В В (19)
В В
В В
X. Блок и привитая полимризация -’V- 525
~ АВАВАВ АААААА ~ или ~ АВАВВАВВ АААААААА ~
А В
А В
~ ABABA ВВВВВВВ ~ д п
о
(20)
Когда последовательности получаемых сегментов являются статисти-
ческими сополимерами, может быть введен префикс «со», и в этом случае
основной составляющий мономер предшествует минимально составля-
ющему. Основная цепь бутадиен-стирольног каучука с привитым стиролом,
содержащим малое количества акриловой кислоты, будет обозначена как
поли[(бутадиен-со-стирол)-(стирол-со-акриловая кислота)] и может быть
схематически представлена как
***ААВАВАААВВАВААААВВА™г
I 1
в в
в в
в с
с в
в с
в в
с в
в в
1 I
где А представляет бутадиен, В — стирол и С — акриловая кислота.
В. Синтез блок сополимеров
Для синтеза блок сополимеров может быть использовано
несколько методов. Используя живую полимеризацию мономер А гомополи-
меризуется с образованием блока А; затем добавляется мономер В, который
реагирует с активными сегментами концов цепи А с образованием блока
В. При тщательном контроле условий реакции этот процесс может приводить
к образованию широкого круга хорошо охарактеризованных блоксополи-
меров. Такие ионные процессы обсуждены более детально в следующем
параграфе. Механохимическая деструкция обеспечивает очень широкий
и простой путь получения полимерных свободных радикалов. Когда каучук
подвергается механическому сдвигу [40], например, во время пластикации,
происходит уменьшение молекулярной массы в результате физического
526 -*V* Глава 11. Химическая модификация полимеров
разрыва части макромолекул. При этом разрыве цепей образуются радика-
лы А и В, которые затем рекомбинируют с образованием блоксополимеров.
Этот метод не предпочтительный, т.к. обычно он приводит к смеси трудно
характеризуемых блоксополимеров.
Используя живую полимеризацию, компания Shell осуществила про-
мышленное производство нескольких поли(стирол-бутадиен) и поли
(стирол-изопрен) блоксополимеров, известных в промышленности как
Кратон 1101 и Кратон G. Эти блоксополимеры нашли широкое приме-
нение в производстве подошв для обуви и промышленности адгезивов.
Их физические свойства зависят от макроструктуры и микроструктуры этих
блоксополимеров.
С. Примеры
В качестве основного примера возьмем три мономера: бута-
диен, стирол и акрилонитрил и посмотрим, как они могут блоксополимери-
зоваться вместе механохимическим путем. Из большого числа теоретических
возможностей для обсуждения может быть выбрано 11; они могут быть
получены пластикацией, как показано ниже:
1. бутадиен-стирольный сополимер с акрилонитрильным мономером
2. полиакрилонитрил, пластифицированный смесью мономеров бута-
диена и стирола
3. бутадиен-акрилонитрильный сополимер со стиролом
4. полистирол со смесью полибутадиена и акрилонитрила
5. стирол-акрилонитрильная смола со смесью мономеров стирола
и бутадиена
6. полибутадиен со смесью мономеров стирола и акрилонитрила
7. бутадиен-стирольный каучук с полиакрилонитрилом (хорошо плас-
тифицированным)
8. стирол-акрилонитрильная смола с полибутадиеном
9. бутадиен-акрилонитрильный каучук с полистиролом
10. стирол-бутадиеновая смола с высоким содержанием стирола с акри-
лонитрильным мономером
11. стирол-бутадиеновая смола с высоким содержанием стирола с поли-
акрилонитрилом (пластифицированным)
(Все вышеупомянутые реакции, за исключением 2 и И описаны в па-
тентной литературе или имеют промышленное значение). В каждом
случае продукт должен быть физически и химически отличен по составу
структурных последовательностей, и все свойства должны также зависеть
от относительного соотношения исходных компонентов. Во всех механо-
химических реакциях некоторые из исходных полимеров или сополиме-
ров остаются неизменными, главным образом, их низкомолекулярная
X. Блок и привитая полимризация -’и* 527
фракция, которая неэффективно подвергается сдвиговым напряжениям,
а некоторые гомополимеры могут быть образованы из полимеризующихся
мономеров реакциями передачи цепи. Изменение условий пластикации
сильно влияет на выход и скорость реакции; и менее влияет на хими-
ческую природу продукта. Присутствие бутадиена как одного из состав-
ляющих (либо полимера, либо мономера) будет вызывать увеличение
содержания геля при продолжительной пластикации. Процессы 3, 5, 6, 9,
как известно, дают продукты, в которых каучуковая фаза диспергирована
в смолообразную матрицу; они являются альтернативными методами для
получения сополимера типа А-В-СЛ Присутствие доли блок или приви-
того сополимера в системе помогает стабилизации дисперсии каучуковой
фазы в смолообразной матрице, действуя на фазовой границе как «мыло»,
т. е. компатибилизирующий агент.
D. Другие методы эффективных механохимических
реакций
Механохимическая деструкция является термином, приме-
няемым для описания цепного разрыва основной полимерной цепи пос-
редством сдвиговых напряжений при переработке. Раннее считалось, что
этот тип процесса приводит к разрыву углерод-углеродной цепи, что обычно
вызывает резкое изменение реологических свойств. В начале 1950-х гг.
Уотсон и сотрудники [41, 42] показали, что в отсутствии кислорода, ради-
калы, получаемые механическим сдвигом, могут быть использованы для
инициирования полимеризации виниловых мономеров с образованием
блоксополимеров. Например, вибропомол натурального каучука ниже
температуры стеклования позволил осуществить в лабораторных условиях
блоксополимеризацию натурального каучука с метилметакрилатом с превра-
щением выше 86%. Аналогичные результаты были достигнуты со стиролом
и с акрилонитрилом.
Этот метод был использован для присоединения антиоксидантов к не-
насыщенным полимерам. Новый подход Скотта в 70-х г. [43], основанный
на применении метода, предложенным Уотсоном, состоит в атаке двойных
олефиновых связей замещенными аллилмеркаптанами и дисульфидами по
реакциям Караша.
Механохимические реакции могут протекать при переработке, когда по-
лимер превращают в конечный продукт. Разрыв связи может протекать как
в насыщенной, так и в ненасыщенной основной цепи полимера. Например,
при переработке на экструдере при разрыве главной цепи в полипропилене
На практике было показано, что ряд мономеров, которые не полимеризуются по
свободно-радикальному механизму в интервале температур 10-50°С, могут быть блоксо-
полимеризованы пластицированием по механизму ионного инициирования путем гетеро-
литического разрыва.
528 “* V Глава 11. Химическая модификация полимеров
получаются длинные свободные радикалы, которые образуют макроалкиль-
ные пероксидные радикалы. Эти пероксиды ответственны за наблюдаемое
уменьшение вязкости расплава. В отсутствии кислорода эти моноаллильные
макрорадикалы могут быть использованы для прививки к основной цепи
новых мономеров или таких полимеров, как полипропилен и полиэтилен.
Таким способом были получены продукты присоединения малеинового
ангидрида к полипропилену, что придает ему улучшенные окрашиваемость,
гидрофильность и адгезию.
Е. Ионные механизмы
Ионные механизмы для получения блоксополимеров яв-
ляются очень важными инструментами химика-синтетика-полимерщика.
Особенностью многих гомогенных процессов в растворе является то, что
обрыв цепи может быть достигнут при тщательном контроле условий экспе-
римента. В действительности, теоретически возможно неопределенное время
жизни активных концов цепей, и это привело к термину «живые» полимеры.
Полимерные карбанионы могут продолжать расти после дальнейшего при-
соединения мономера.
Путем изменения состава мономерной композиции блоксополимеризация
легко инициируется, и этот процесс может повторяться. Главным преиму-
ществом этого типа синтеза по сравнению со свободнорадикальными процес-
сами является способность контролировать длину цепи последовательностей
регулированием концентрации инициирующих центров и мономеров на
каждой стадии блоксополимеризации.
Анионная блоксополимеризация применяет литийорганические ини-
циаторы, которые имеют широкое распространение из-за своей хорошей
растворимости, в том числе в углеводородах [44]. Литийорганические со-
единения действуют как инициаторы посредством прямой атаки молекул
мономеров органическими анионами, последующей быстрой реакцией
в отсутствии соединений с активным атомом водорода без передачи или об-
рыва цепи. При тщательном проведении эксперимента реакция позволяет
осуществлять точный контроль молекулярной массы и (узкого) молекулярно
массового распределения.
Удобство этого метода привело к разработке многих промышленных
продуктов, включая термопластичные эластомеры, основанные на трехблоч-
никах из стирола, бутадиена и изопрена. Инициаторы, применяемые в этих
системах, представляют собой литийорганические соединения, растворимые
в углеводородах. Для получения мультиблоксополимеров в некоторых случа-
ях применяются растворимые в углеводороде дилитиевые инициаторы. Для
получения термопластичных эластомеров типа АВА применяются различные
методы. Все они обсуждены подробно во второй главе. Краткое обобщение
этих методов приводится ниже.
X. Блок и привитая полимризация -*v* 529
1. Трехстадийный процесс с монофункциональными
инициаторами
В этом методе применяющемся, например, для синтеза блок-
сополимеров, поли(стирол-Ь-бутадиен-Ь-стирол) (СБС), полистирольныйблок
образуется при использовании n-бутиллития в качестве инициатора в арома-
тических растворителях. Затем добавляется бутадиеновый мономер, реагиру-
ющий с концом цепи полистирол-литий с образованием пол и (бутадиен )ового
блока. Если реакция обрывается на этой стадии, получается поли(стирол-Ь-
бутадиен)овый сополимер, который не имеет термопластичных свойств. Далее
добавляется мономерный стирол, чтобы получить трехблочный СБС. Процесс
получения СБС очень тщательно контролируется для избежания образования
двухблочника, т.к. присутствие любого заметного количества СБ резко умень-
шает термопластичные свойства СБС.
2. Двухстадийный процесс с бифункциональными
инициаторами
Были разработаны несколько промышленных процессов,
использующих бифункциональные инициаторы, основанные на раствори-
мых литийорганических соединениях. Эти соединения могут инициировать
полимеризацию «в обе стороны». Бифункциональные инициаторы удобны
в случае АВА блок сополимеров, где В может инициировать А, но А не
может инициировать В. Эти бифункциональные инициаторы удобны для
получения СБС. Эластомерный бутадиеновый блок полимеризуется в ра-
створе гексана. Добавляемый мономерный стирол также растворим в гексане.
Этот метод также удобен для получения трехблочников с углеводородными
средними блоками и полярными концевыми блоками, такими как поли
(метакрилонитрил-Ь-изопрен-Ь-метакрилонитрил).
3. Монофункциональное инициирование и сочетание
В этом двухстадийном процессе В последовательно сополи-
меризуется с А, и затем две цепи сочетаются, образуя блоксополимер ABBA.
Этим методом получен трехблочный СБС с применением метилендихлорида
в качестве связующего агента. Недостатком является образование радикал-
анионов, которые могут приводить к образованию смесей трехблочников
с многоблочниками.
4. Конусообразные (тапированные) блоксополимеры
Этот метод применяется для образования блоксополимеров,
которые состоят из двух сегментов существенно гомополимерной структуры,
разделенной блоком «тапирующего» сегмента беспорядочного сополимер-
ного состава. Они обычно получаются за счет использования различий ско-
ростей реакций мономеров. При индивидуальной полимеризации в гексане
бутадиен реагирует в шесть раз медленнее, чем стирол: однако, когда стирол
530
Глава 11. Химическая модификация полимеров
и бутадиен сополимеризуются в растворе угдеводорода, такого как гексан,
бутадиен полимеризуется в шесть раз быстрее, чем стирол. Это приводит
к тапированию стирола в реакции сополимеризации. Для более детального
описания этого метода читатель отсылается к главам 2 и 13.
F. Синтез привитых сополимеров
Синтез привитых сополимеров более разнообразен, но тем не
менее может быть разделен на группы связанных процессов: 1. передача на
полимер; 2. сополимеризация по ненасыщенным группам; 3. окислительно-
восстановительная полимеризация; 4. облучение высокими энергиями;
5. фотохимический синтез; 6. металлирование с использованием активного
органолития с хелатными диаминами.
1. Передач на полимер
В свободнорадикальной полимеризации передача цепи
является важной реакцией. Может иметь место передача цепи на мономер,
растворитель, меркаптан или на растущие цепи. Когда происходит реакция
передачи цепи на другую цепь, она образует радикалы, которые действуют
как центры дальнейшего роста цепи и прививки (см. главу 2 для дополни-
тельных деталей).
Р + Р —♦ PH + Р*
R* + Р —» RH + ₽• (22)
Р. + Р. _ PH + р.
Реакция протекает посредством передачи атома водорода или галогена
(в случае галогенированных полимеров) от макромолекулы Р к растущей
цепи Р*(или к избытку свободнорадикального инициатора R*, который
обрывает цепь). Реакционная способность теперь сосредоточена на макро-
молекуле, на которую передана цепь. Она в свою очередь инициирует сопо-
лимеризацию, т. е. рост привитой боковой цепи вновь введенного второго
мономера. Явление прививки происходит с большинством мономер-поли-
мерных систем, особенно систем, инициированных перекисью бензоила,
при высоких концентрациях полимера и инициатора.
Самый простой метод заключается в растворении полимера в подходящем
растворителе; далее добавляют перекисный инициатор, который отрывает
водородный радикал и генерирует радикал полимерной цепи; затем до-
бавляется свежий мономер для прививки к этому центру. Этот метод был
применен при прививки метилакрилата на натуральный каучук и синте-
тический полиизопрен. Таким образом, получаются некоторые промыш-
X. Блок ii привитая полимризация -•V- 531
ленно используемые продукты, такие как смолы АБС. Однако, эластомеры
не получаются этим путем из-за образования частиц микро- и макрогеля,
которые ухудшают физические свойства. Во многих случаях прививки
к латексу получают термопласт, а не каучук.
2. Сополимеризация на ненасыщенных группах
Другие методы (например, большинство методов, связанных
с ремонтом кузова автомобиля) основаны на процессе сополимеризации
полиэфира со стиролом (усиленной различными типами тканевых сеток
или стекловолокна), путем привитой сополимеризации стирола на основ-
ные цепи ненасыщенных полиэфиров относительно низкой молекулярной
массы. В общем, для значительного выхода привитого полимера необхо-
дима высокая концентрация боковых винильных групп в основной цепи
полимера. Для пластиков, усиленных стекловолокном, с этой точки зрения
выбраны полиэфирные смолы. В натуральном каучуке всегда присутствует
небольшие количества таких групп на молекулу1), они, несомненно, участ-
вуют в нормальной прививке. Содержание боковых винильных групп может
быть увеличено пластикацией ненасыщенных каучуков в атмосфере азота,
т.к. резонансные структуры рекомбинируют, как показано ниже.
СН3
™~с=сн—сн-2
СНз
*™с—сн=сн2
СНз
•сн2—с=сн™*
СНз
сн2=с—сн™*
СНз
СНз
СНз
™с — сн2 — с=сн™*
*™С=СН — СН
сн
сн2
2 — СН™*
с—СНз
сн2
etc.
(23)
Для получения макромолекулярных инициаторов для синтеза блоксо-
полимеров, например, поли(метилметакрилат-Ь-акрилонитрил)а и поли
п Около 0,4% ненасыщенных групп являются боковыми винильными
группами в усредненном образце светлого крепа экстрагированного аце-
тоном.
532 -•V- Глава 11. Химическая модификация полимеров
(метилметакрилат-Ь-стирол)а, применяется прямое введение пероксидных
групп в основную цепь полимера, такого как поли (метилметакрилат). Для
введения эпоксидных кольцевых структур в натуральный каучук может
быть применено также озонирование с тщательным контролем степени
озонолиза.
СИ’ СН3х О
•~СН2—С=СН—сн,.»» °\ Хсн—СН2"" (24)
О---О
При проведении реакции до около 4% по доступным двойным связям в рас-
творителе, таком как толуол, при низкой температуре после продувания азо-
том эффективность прививки может быть повышена добавкой в атмосфере
азота мономерного метилметакрилата (реагирующего при 80°С в запаянных
ампулах). При этом образуются две ММА цепи, связанные кислородом через
открытый мостик -О-О-. Этот метод может быть применен к изопреновым
и бутадиеновым сополимерам.
5. Окислительно-восстановительная полимеризация
Процессы окислительно-восстановительной полимери-
зации являются наиболее популярными методами реакций прививки и
широко используются с инициирующими системами, основанными на
окислении иона железа и восстановлении иона церия. В окислительно-
восстановительной полимеризации гидроперекисные или аналогичные
группы восстанавливаются до свободного радикала плюс ион, в то время
как ион металла окисляется до состояния с высшей валентностью, и в то
же время добавляется мономер. Когда восстанавливаемая группа присо-
единяется к полимерной цепи свободнорадикальные центры прививки,
полученные таким образом на макромолекуле, действуют как инициаторы
привитой сополимеризации.
'~^СН2 — СН™ 4- Fe2
О —ОН
—----->™СН2—СН™ + Fe3 + + ОН"
amine of z । /ot-\
aldehyde I (^o)
О
Этот метод применяется для прививки метилметакрилата к латексу
натурального каучука. (Действительно, свежий латекс содержит неболь-
шое количество гидроперекисных групп в макромолекуле, которые могут
принимать участие в реакции прививки). Повторно центрифугированный
концентрат латекса смешивают с метилметакрилатом, и добавляют раствор
тетраэтиленпентамина, вслед за малым количеством раствора сульфата же-
леза. Гомогенизированная смесь выдерживается часто в течение ночи [45].
Привитой сополимер отделяется коагуляцией. Так как практически все
свободнорадикальные центры образуются в основной цепи каучука, очень
X. Блок и привитая полимризация -’V 533
малое количество свободного поли (метилметакрилата) остается в привитой
системе; с другой стороны некоторые цепи каучука остаются без прививки,
т.к. не все цепи имеют гидроперекисные группы. Высокий выход привитого
полимера получается с применением мономера, растворенного до равновесия
с латексными частицами перед добавлением амина и иона железа в качестве
инициатора. Сообщалось, что пропускание кислорода (воздуха) через латекс
в течение нескольких часов уменьшает содержание непривитого натураль-
ного каучука в продуктах полимеризации, но затем необходимо продувание
азотом, чтобы предотвратить растворение кислорода, действующего как
ингибитор реакции.
Гидроксиполимеры могут быть привиты путем окислительно-восстано-
вительной полимеризации с использованием водорастворимых перекисей,
таких как перекись водорода в сочетании с ионами железа. Радикалы • ОН
отщепляют атомы водорода от гидроксигрупп в полимере, инициируя свобод-
норадикальные центры прививки на основной цепи. Этот метод применяется
с производными крахмала, целлюлозы, но образуется значительное коли-
чество гомополимера из исходных гидроксильных радикалов параллельно
с отрывом атома водорода. Путем введения небольшого количества гидрок-
сильных групп в сополимерный синтетический каучук может быть повышена
эффективность прививки гомополимера. Смеси сульфата аммония железа
и аскорбиновой кислоты применяют для окислительно-восстановительных
систем. Во многих патентах сообщается предпочтительное использование
ионов церия, которые легко окисляются гидроксильными группами в ра-
дикал-ионных реакциях.
R—ОН + Се3+ R—ОН + Се2’
(26)
R—ОН + ОН —» R—О’ + Н2О
Преимущество этой реакции заключается в том, что только гидроксилы
на полимере превращаются в R-0 свободные радикалы так, что гомополимер
не получается и наблюдается образование чистого привитого полимера.
4. Метод облучения высокими энергиями
При действии излучений высоких энергий в вакууме, напри -
мер с применением источника 60Со, происходит деструкция главной цепи
натурального каучука и других полиизопренов.
сн3 сн3
I I (27)
«'~сн2—С=СН—СН2—СН2—С=СН—CH2~w->
Значительное количество энергии излучения затрачивается на отрыв
атомов водорода.
534
Глава 11. Химическая модификация полимеров
СН3 СН3
JVWCH2—с=сн— сн2- + -СН2 —С=СН—СН/^
(28)
СН3 СНз
™сн2—с=сн—сн2™^>-Сн2-С=СН —сн™
Облучение натурального каучука в присутствии виниловых мономеров
преимущественно приводит к синтезу привитых сополимеров, но всегда при-
сутствует некоторое количество блоксополимеров. Синтез, инициированный
действием излучения, может быть проведен в растворе, в контакте с жидким
мономером (или без разбавления), в контакте с мономером в паровой фазе,
в эмульсии или в суспензии. Каучук может быть предварительно облучен
в отсутствии воздуха с образованием свободных радикалов для последующего
добавления мономера. Однако время жизни этих радикалов непродолжи-
тельно в результате их подвижности в каучуковой матрице. Излучение при
очень низких температурах делает возможным применение метода радика-
лов-ловушек для различных натуральных и синтетических каучуков. Пла-
стики и полимеры с кристаллической фазой легче образуют радикалы при
облучении с последующей прививкой радикалами-ловушками. Облучение
можно провести на воздухе для введения пероксидных групп.
СН2—С=СН — СН2 . —>^СН2—С=СН—СН2—О—О-
^или I
СН3 СН3 СНз
сн2—С=СН—СН2—ООН + R- сн2—с=сн—сн2—о—о—сн2—с=сн
(29)
СН, СН3
I I
—сн,—с=сн—сн —> сн,—с=сн—сн
/ I А—
СН, сн,
I 3 ИЛИ I 3 (30)
СН2—С=СН—СН + RH сн2—с=сн—СН
ООН о
сн, о
I I
сн,—с—сн—сн
X. Блок и привитая полимризация
-\г 535
Эти группы могут быть также использованы для инициирования прививки
любым обсужденным ранее методом.
Вследствие своей простоты предпочтительна прививка в латексе; к на-
туральному каучуку этим путем прививаются метилметакрилат, стирол,
акрилонитрил и винилхлорид [46].
Другая возможность состоит в облучении смешанных сеток для реком-
бинации разных последовательностей разорванных цепей. Этот способ мо-
жет быть применен к сеткам натурального каучука и поли(винилхлорида)
с получением привитых и блок сополимеров с высокими выходами без реге-
нерации мономера. Этот же метод был применен для прививки полихлоро-
прена на дисперсию синтетического полиизопрена и на полибутадиеновые
вулканизаты различных рецептур.
5. Фотохимический синтез
Макромолекулы, содержащие фоточувствительные группы,
которые поглощают энергию УФ-частот часто деструктируют с образованием
свободных радикалов. Процессы деструкции, как правило, являются доста-
точно медленными, но при добавлении фотосенсибилизаторов, таких как
ксантон, бензил, бензоин и 1-хлороантрахинон, скорость может быть увели-
чена до такой степени, что может происходить привитая сополимеризация
с метилметакрилатом или другими мономерами. Это может быть проведено
в случае натурального каучука в фазе латекса при достаточно высоком выходе
привитого сополимера. Натуральный каучук-z/-полистирол и поли (бутадиен-
?/-стирол) могут быть получены УФ-облучением этих сенсибилизированных
латексно-мономерных дисперсий. Комбинация фотохимического синтеза
и инициатора окислительно-восстановительного типа также может быть
реализована (одноэлектронное окисление). В этом случае обеспечивается
прививка с минимальным образованием гомополимера.
Атомы брома основной цепи полимера могут легко отщепляться под
действием УФ-облучения, образуя свободнорадикальные центры для пос-
ледующей реакции прививки. Бромирование может быть инициировано
фотохимически
или агентом передачи цепи, таким как четырехбромистый углерод, который
может быть применен на стадии полимеризации для введения лабильных
групп.
536 -*v- Глава 11. Химическая модификация полимеров
Вг2
—СН- + СВг3 СНВг + -СВг3 etc.
(32)
Ряд специальных сенсибилизаторов, таких как бромбутилкаучук, ценных
благодаря своей огнестойкости, могут действовать как полимеры с основной
цепью, пригодной для широкого круга реакции прививки.
Синтез блоксополимеров в прежние годы был основан на УФ-облучении
поли(метилвинилкетона) в присутствии акрилонитрила. Первоначальной
стадией деструкции является:
н н н н
с—с--с —> сн2- + -с—СН3—
III I
н с=он с=о
сн3 сн3
(33)
Эта реакция деструкции, в дополнение к различным реакциям окисления, ока-
залась ценной в связи с возможностью получения фотодеструктируемых плас-
тиков как части компании по уменьшению количества пластиковых отходов
из упаковки. Хотя спроса на фотодеструктируемые резины пока нет, введение
небольшого количества винилкетона в сополимер или гомополимер каучука
открывает путь к синтезу фотодеструктируемых блоксополимеров.
Описаны многие другие методы синтеза блок и привитых сополимеров,
но уже много сказано, чтобы понять масштаб этих реакций и потенциал,
который полностью еще не используется. Свойства многих привитых и блок-
сополимеризационных систем еще только оцениваются для применения,
но потенциально способны к расширению диапазона свойств каучуков.
6. Металлирование с использованием активированного
орагнолития с хелатирующими диаминами
Ненасыщенные эластомеры могут быть легко металлирован-
ны с активированными литийорганическими соединениями в присутствии
хелатирующих диаминов или алкоксидов калия или натрия. Например,
полиизопрен, полибутадиен, стирол-бутадиеновые сополимеры или сти-
рол-изопреновые сополимеры могут быть металлированны с комплексом
н-бутиллитий-TMEDA (1/1 или 1/2) с образованием аллильных или бен-
зильных анионов. Образующийся аллильный анион может действовать как
инициирующий центр роста определенных разветвлений или комбиниро-
ванных полимерных частиц. Эти полимеры могут включать полистирол,
который может образовывать жесткие домены, или полибутадиен, который
образует мягкие домены.
X. Блок и привитая полимризация -’V- 537
Исследования в этой области привели к получению нескольких комби-
нированных полимеров [47, 48]. Метод металлирования является удобным
и многосторонним методом, т.к. может быть применен для любых полимер-
ных материалов, которые содержат небольшое количество двойных связей.
Например, к этилен-пропиленовому каучуку успешно привит норборнен.
Подобная реакция проводится на полимерных материалах, которые содержат
ароматические циклы, такие как полистирол, поли-а-метилстирол, а также
на полипропиленоксиде.
В общем, полимерные материалы, которые содержат боковые груп-
пы, аллильные группы основной цепи или кислотные атомы водорода,
могут быть металлированны с литийорганическими соединениями
в присутствии хелатирующих диаминов. На них также могут быть приви-
ты ионнополимеризуемые мономеры с образованием комбинированных
материалов.
G. Основные свойства полимеров
Хотя свойства блок и привитых сополимеров обсуждены
в главах 3, 5 и 13, некоторые свойства сополимеров особенно близкие
к материалу данной главы будут кратко рассмотрены здесь.
Свойства привитого натурального каучука с метилметакрилатом не могут
быть оценены, если этот сополимер не отделен от гомополимера. Применяе-
мые методы основаны на фракционном осаждении, селективном растворении
или комбинации этих методов. Для уточнения читателя отсылаем к главе 3.
В связи с тем, что во многих случаях технологи имеют дело с производс-
твенными материалами, мы рассмотрим только свойства блок и привитых
сополимеров с неудаленным гомополимером.
Присутствии двух химически различных полимерных последовательно-
стей в одной и то же цепи приводит к действию макромолекулы как ПАВ, т. е.
она способствует совмещению двух гомополимеров в смеси путем накопления
на межфазной поверхности, помогая постепенному переходу от одной фазы
к другой, и таким образом, уменьшая межфазную энергию. Преобладающим
случаем на практике является микрофазовое разделение, но макрофазовое
разделение в связи с этим прекращается. В плотно упакованном полистироле
и сополимерах АБС, полученных реакциями прививки, диспергированная
каучуковая фаза в стеклообразной матрице и диспергированная стеклообраз-
ная фаза внутри частиц каучука в равной мере предотвращают образование
отдельных фаз привитыми цепями сополимера, которые на молекулярном
уровне имеют каучуковые сегменты, ассоциированные с частицами каучука,
и пластические сегменты, ассоциированные со стеклообразной фазой. В этом
отношении имеется мало различий в свойствах между привитым и линейным
блоксополимером - существенной особенностью является присутствие двух
типов последовательностей в одной макромолекуле.
538 -’V Глава 11. Химическая модификация полимеров
Блоксополимерныетермоэластопласты обязаны своим свойствам именно
этой структуре посредством полистирольных концевых блоков, образующих
микрофазу с гомополимерным полистиролом, которая диспергирована внут-
ри непрерывной фазы полибутадиена, образованной из полибутадиеновых
сегментов центральной последовательности в блоксополимере типа СБС.
(Чтобы это получить, общий объем полибутадиеновых сегментов должен
превышать общий объем полистирольных сегментов. В обратном случае
продукт проявляет свойства высокопрочного полистирола; см. главу 13).
Хотя полистирольные «структуры» действуют как физические попереч-
ные связи при низкой температуре, при температуре обработки выше
температуры размягчения полистирола оба типа сегмента проявляют
пластичность, позволяя материалу экструдироваться, перерабатываться
методом инжекционного формирования и т.д. При охлаждении домены
полистирола снова становятся жесткими и восстанавливают свое влияние
на свойства материала.
Когда блок и привитые сополимеры диспергируются в растворителях,
растворы имеют свойства, которые зависят от того будет или не будет по-
лимер полностью растворим. Если растворитель является «хорошим» для
обоих сегментов, например хлороформ в случае привитого сополимера
натурального каучука и поли (метилметакрилата) [50], тогда оба типа сег-
ментов развернуты и пленки, отлитые из разбавленных растворов, будут
промежуточными по свойствам двух гомополимеров (проявятся свойства
усиленных каучуковых пленок). Если растворитель является хорошим для
каучука, но плохим для поли (метилметакрилата), например, петролейный
эфир, то растворы обнаруживают типичную мутность для привитого или
блок сополимеров и отлитая пленка высоко эластична. Когда растворитель —
ацетон, хороший растворитель для поли (метилметакрилата), но не рас-
творитель для каучука, отлитые пленки являются пластиком с высоким
сопротивлением раздиру. Смотрите главу 10.
При прививке на полимер в таких условиях, когда определяющей являет-
ся его физическая форма, исходные свойства субстрата обычно доминируют,
а прививка лишь улучшает их. Например, субстрат может находитсья в виде
волокна целлюлозы, нейлона, терилена, привитых с различными мономер-
ными системами. Природа реакции прививки на этих волокнах обычно
такова, что на субстратном полимере образуется поверхностное покрытие;
поэтому поверхностные характеристики, такие как окрашиваемость, обычно
улучшаются в прививаемых системах.
Очень сомнительно, чтобы любые смеси двух полимеров или химически
различных сополимеров, с термодинамической точки зрения были полностью
совместимыми. Даже большинство блок- и привитых сополимерных систем
проявляют микрофазовое разделение, которое типично для них. Химическая
модификация, проведенная гомогенной реакцией, как обсуждено в преды-
X. Блок и привитая полимризация
539
дущей части этой главы, будет в общем случае приводить к образованию
единой полимерной фазы. Однако, необходимый выбор не ограничен этими
двумя подходами, т.к. химическая модификация может быть проведена
после блок или привитой сополимеризации или наоборот. Опубликовано
очень немного работ по такому последовательному использованию этих
двух физически различных путей химической модификации полимеров, так
что есть значительный резерв для создания новых процессов модификаций
давно известных каучуков, так же как вообще для превращения старых
полимеров в новые.
ГЛАВА
12
СМЕСИ ЭЛАСТОМЕРОВ
СУДХИН ДАТТА
Экссон Мобил Кемикал Ко.
Бэйтаун, Техас
I. ВВЕДЕНИЕ
Смеси эластомеров имеют и технологическое и коммерчес-
кое значение, поскольку они позволяют потребителю обеспечить свойства
смесевого и вулканизованного эластомера, которые недостижимы на основе
единственного промышленного эластомера. Эти, в принципе улучшаемые
свойства, включают физические, химические и технологические. Они кор-
релируют между собой, отличаясь от свойств индивидуальных эластомеров
и изменяясь с составом смеси. Технология смесей эластомеров в значи-
тельной мере и сосредоточена на выборе индивидуальных эластомеров и на
создании смесей, с целью достичь комбинации заданных свойств. Эта глава
показывает некоторые примеры применения смесей эластомеров. Эмпири-
чески возникшие пути создания новых смесей эластомеров сопровождаются
возникновением немалых сложностей.
Получение смесей является экономичным процессом, обеспечивающим
получение материала с комплексом свойств недостижимых в одном эласто-
мере. В эластомерах, состоящих из одного мономера, с одним способом
соединения в макромолекуле (например, 1,2-полибутадиен), нет других
способов влияния на свойства кроме смешения. В случае эластомеров, яв-
ляющихся сополимерами (например, бутадиенстирольный каучук [SBR])
изменения в составе макромолекул, такие как образование блоксополимера
вместо статистического сополимера, также эффективны. Однако возмож-
ность внутримолекулярных изменений ограничена возможностями синтеза.
Межмолекулярные изменения в составе или в характере распределения
мономеров не имеют системных или синтетических ограничений.
Теоретически смеси эластомеров позволяют достичь широкой вариации
свойств. Комбинации эластомеров могут привести к изменению в свойствах
либо за счет различий в самих компонентах, либо за счет усиления или вул-
канизации составляющих компонентов. Взаиморастворимые смеси эласто-
I. Введение -JU 541
меров состоят из одной эластомерной фазы с однородным распределением по-
перечных связей и частиц наполнителей, обнаруживая средне-взвешенные
свойства по отношению к свойствам компонентов. Взаиморастворимые смеси
являются обычно применяемыми, хотя это признается очень редко. Анализ
таких смесей, особенно после вулканизации, затруднителен. Современная
аналитическая техника обладает немногим большими возможностями, чем
классические приемы селективного осаждения невулканизованной смеси
эластомеров из раствора [1].
Обычным примером взаиморастворимых смесей являются этилен-
пропиленовые сополимеры разного состава, которые включают полукри-
сталлические сополимеры с высоким содержанием этилена и аморфные
с малым содержанием этилена. Такие смеси сочетают высокую прочность
кристаллических полимеров и выгодные низкотемпературные свойства
аморфных полимеров. Химические различия взаиморастворимых смесей
этиленпропиленовых и бутадиенстирольных сополимеров могут также
возникать за счет различий в распределении и типе реакционноспособных
точек вулканизации в эластомерах. Неоднородное распределение диенов,
которые являются реакционноспособными точками вулканизации в смесях
этилен-пропилен-диеновых эластомеров, могут привести к образованию двух
раздельных взаимопроникающих вулканизационных сеток.
Взаимонерастворимые смеси обнаруживают дополнительные более
сложные закономерности благодаря неоднородной фазовой структуре двух
эластомерных компонентов. Две раздельные фазы обычно имеют разное
сродство к наполнителям и пластификаторам, а также разную склон-
ность к вулканизации в присутствии вулканизующих агентов. Изменение
свойств эластомеров за счет неравномерного распределения наполнителей
и вулканизующих агентов является, однако, наиболее общим явлением
в применении смесей взаимонерастворимых эластомеров [2]. Инженерные
свойства эластомеров (такие как прочность, гистерезис) в вулканизованных
смесях зависят не только от эластомера как такового, но также от количества
и природы наполнителей и пластификаторов, а также от степени вулкани-
зации. Во взаимонерастворимых смесях количество этих добавок в каждой
фазе может регулироваться изменением вязкости и химической природы
эластомера, или химией поверхности наполнителя, химической природой
пластификатора и последовательностью введения компонентов, также как
техникой смешения. Большой объем практических приемов (vide infra) было
предложено для достижения термодинамически метастабильного, между-
фазного распределения ингредиентов в смесях. В результате вулканизации
такое распределение фиксируется и обеспечивает достижение желаемых
инженерных свойств смеси.
Имеется два обзора по эластомерным смесям. Во-первых, обзор Гесса
с сотрудниками [3], касающийся применения, анализа и свойств взаимоне-
542 -* V Глава 12. Смеси эластомеров
растворимых эластомерных смесей. Второй — обзор Роланда [4] содержит
дополнительно материал по физике смешения несовместимых смесей поли-
меров и последние достижения в области методов анализа. Другие обзоры
Кориша [5] и Мак-Донела [6] посвящены специфическим аспектам смесей
эластомеров. Эти обзоры посвящены несовместимым смесям эластомеров.
В этой главе мы дополним их информацией о смесях совместимых (взаимо-
растворимых) эластомеров.
II. СМЕСИ ВЗАИМОРАСТВОРИМЫХ ЭЛАСТОМЕРОВ
А. Термодинамика
Распространение термодинамики на смеси эластомеров
обсуждается Роландом [4]. Взаиморастворимые смеси обычно образуются
при смешении эластомеров с близкими значениями трехмерных [7] пара-
метров растворимости. Примером таких смесей являются смеси сополимеров
(например, этилен-пропиленовых или бутадиенстирольных сополимеров),
когда полимерные компоненты имеют разный состав, микроструктуру
и молекулярный вес. Если силы взаимодействия между полимерными ком-
понентами смеси почти нацело дисперсионные, взаимная растворимость
достигается лишь при очень близком совпадении трехмерных параметров
растворимости Хансена [7].
Взаиморастворимые смеси эластомеров отличаются от соответству-
ющих смесей термопластов в двух основных аспектах. Во-первых, не-
обходимость обеспечения высокой эластичности в эластомерах требует
создания полимеров с большим молекулярным весом и ограниченной
полидисперсностью. Это снижает возможное взаиморастворение раз-
нородных эластомеров за счет взаимодиффузии низкомолекулярных
компонентов смеси. Во-вторых, эластомеры пластифицируются в обыч-
ном процессе компаундирования добавлением технологических масел.
Присутствие пластификатора приводит к увеличению свободного объема
компонентов смеси и стабилизирует в некоторой степени смеси разно-
родных эластомеров.
В. Кинетика
Образование взаиморастворимой смеси каучуков замедляет
скорость кристаллизации [8а, б], если один из компонентов способен крис-
таллизоваться. Это явление ответственно за наблюдаемое снижение теплот
плавления, что коррелирует со степенью фазовой однородности смесей
эластомеров [9]. Кроме того, и закономерности плавления полимера могут
измениться во взаиморастворимой смеси. Стабильность жидкого состоя-
ния при образовании взаиморастворимой смеси снижает относительную
термическую стабильность кристаллической части и снижает равновесную
II. Смеси взаиморастворимых эластомеров -* V 543
температуру плавления [10а, б]. Такая депрессия точки плавления невелика
во взаиморастворимой смеси при наличии только дисперсионных взаимо-
действий между компонентами.
С. Анализ
1. Температура стеклования
Основной эффект взаиморастворимости в смеси разнородных
эластомеров выражается в изменении температуры стеклования. Посколь-
ку во взаиморастворимых смесях изменения конформаций полимерных
цепей незначительны, плотность сетки зацеплений должна быть средне-
взвешенной по отношению к плотностям сеток зацеплений полимерных
компонентов.
2. Спектры ядерного магнитного резонанса
Ядерный магнитный резонанс (NMR) был применен для
исследования гомогенности взаиморастворимых смесей, что отражено
вобзоре Ченга [11а] и Роланда [116]. Если компоненты смеси имеют разные
Т, NMR позволяет оценить фазовую структуру смеси в результате быстрого
уменьшения протон-протонного спаривания при расхождении ядер [Ив].
Для смесей эластомеров с почти равными ^применялся MAS NMR, когда
один из компонентов был почти полностью дейтерирован [12]. Другой
метод являлся перекрестной поляризацией MAS 13С NMR [13]. Перенос
спин-поляризации от протонов атомов 13С дейтерированного компонента
может произойти, если эти углеродные атомы находятся в непосредственной
близости (нанометры) к протонам.
3, Кристалличность
Изменения в степени кристалличности также привлекались
для изучения гомогенности смесей эластомеров [14]. Моррис [14а] изучал
скорость кристаллизации цис-1,4-ЕН в смесях с SRR. При любом составе
смеси скорость кристаллизации RR уменьшалась при большей гомогенности
смеси. Сиркар и Ламонд [146] также изучали изменения в кристалличности
RR в его смесях с NR, IR, ЕРМ, CIIR, NRR, CR (рис. 1). Природа смеши-
ваемых компонентов оказывает значительное влияние, поскольку более
совместимые смеси (меньший размер доменов) больше теряют в кристал-
личности RR.
4. Взаимодиффузия
Езаимодиффузия между двумя полимерами является про-
явлением их термодинамической совместимости. Адгезия между приведен-
ными в контакт листами каучука также отражает взаимодиффузию поли-
мерных цепей [15а]. Если контактирующие листы изготовлены на основе
544 -•и- Глава 12. Смеси эластомеров
несовместимых полимеров, взаимодиффузия не происходит1. Натуральный
каучук (NR) и 1,2-полибутадиен (1,2-RR) смешиваются даже при условии
большого молекулярного веса [156, в]. Если NR приведен в контакт с 1,2-BR
их взаимодиффузия проходит самопроизвольно. Если листы сколько-нибудь
различаются по рассеивающей способности, взаимодиффузия увеличивает
рассеяние (рентгеновских лучей или нейтронов) при исследовании таких
склеенных листов. Разнообразные спектроскопические методы [15 г-ж]
были использованы для определения областей взаимодиффузии.
FVMQ FKM
- т
NBR ECO
АСМ
АЕМ
CSM CM VMQ
CR
EPDM PVMQ
160
NR
SBR
IR
BR
CIIRBIIR
HR
HR EPM
70 100 125 150 175 200 225 250
Service Temperature, °C
Рис. 1. Эластомеры по их стойкости к ароматическим растворителям (ордината)
и температуре (абсцисса).
1 > Взаимодиффузия между двумя полимерами может быть двух видов: а) диффузия частей
молекул, т. е. их сегментов и б) диффузия макромолекул. Первая происходит между любыми
двумя полимерами даже при их полной несовместимости и приводит к образованию межфазного
слоя сегментальной совместимости (растворимости) толщиной 0.5-15 нм в течение нескольких
секунд или минут. Чем ближе по полярности полимеры, тем больше сегментальная раствори-
мость, тем больше адгезия, достигающая максимального значения при образовании смешанной
сетки зацеплений в этом слое. Так создается возможность совулканизации несовместимых
полимеров (В.Н. Кулезнев. «Смеси полимеров». Химия, 1980 г.). Второй тип диффузии в меж-
фазном слое — диффузия макромолекул, скорость которой больше зависит от молекулярной
массы, чем от соотношения полярностей. Поэтому олигомеры в основном взаиморастворимы,
а полимеры взаимонерастворимы. При обычных температурах, с учетом полифракционного
состава, каучуки могут быть взаиморастворимы в пределах нескольких процентов (А.Е. Чалых,
В.К. Герасимов, Ю.М. Михайлов. «Диаграммы фазового состояниия полимерных систем».
М. Янус-К, 1998). Прим, перев.
II. Смеси взаиморастворимых эластомеров -*!/ 545
5. Механические свойства
Взаиморастворимые смеси должны обладать большей ме-
ханической однородностью, чем сопоставимые многофазные структуры.
Совместимые смеси каучуков, которые реагируют химически, уплотняются
и характеризуются повышенной плотностью энергии когезии. Они должны
обладать улучшенными механическими свойствами, однако это наблюдалось
только при температуре ниже Tg [16].
Таблица 1. Компоненты смесей этилен-пропиленовых сополимеров,
отличающихся молекулярным весом и кристалличностью.
Образец Состав С2 вес. % Вязкость ML (1+4), 125 °C
А 60 41
Bi 74 72
В2 76 247
В3 78 1900
В« 68 189
В5 84 291
D. Сополимеры с градиентом состава
Существенным достижением [17] последнего десятилетия
было применение взаиморастворимых смесей EPDM, отличающихся по
составу. Смеси были составлены так, чтобы сочетать вязкоупругие свойства,
такие как скорость экструзии или адгезию с физическими свойствами, та-
кими как прочность, путем изменения релаксационных характеристик. Это
достигалось выбором компонентов, имеющих различные средние молекуляр-
ные веса или различия в кристалличности (благодаря протяженным последо-
вательностям этилена) или имеющих оба эти различия. Пример компонентов
таких смесей приведен в табл. 1. Смеси компонента «А» с компонентами
«В» взаиморастворимы и были получены смешением гексановых растворов
эластомеров. Рис. 2 и 3 показывают влияние на прочность смешанных, но
невулканизованных смесей компонентов показанных в табл. 1. Смеси на
рис. 2 есть смеси компонентов с разным молекулярно-весовым распреде-
лением, тогда как компоненты на рис. 3 отличаются и по составу молекул
и по кристалличности. Различия в распределении и кристалличности ска-
зываются и на других вязкоупругих свойствах, таких как адгезия при отсла-
ивании. Рисунки 4 и 5 показывают результаты определения аутогезии, как
силы необходимой для отслаивания смесей друг от друга, смесей, свойства
которых показаны на рис. 2 и 3 соответственно. Адгезия возрастает с ростом
546 JV" Глава 12. Смеси эластомеров
молекулярного веса и композиционной неоднородности в смеси. Это можно
видеть из сравнения данных для В1 и В2 на рис. 4 и В4 и В2 на рис. 5. Малое
возрастание кристалличности и молекулярно-весового распределения при
смешении увеличивает адгезию, замедляя последнее без заметного влияния
на первую. Дальнейшее увеличение также сильно тормозит перепутывание
цепей необходимое для обеспечения аутогезии.
Рис. 2. Изменение прочности невулканизованных компаундированных смесей
этилен-пропиленовых сополимеров в зависимости от их различий в моле-
кулярно-весовом распределении.
Рис. 3. Изменение прочности невулканизованных компаундированных смесей эти-
лен-пропиленовых сополимеров, отличающихся составом и кристалли-
чностью.
II. Смеси взаиморастворимых эластомеров -* V- 547
0.5
0 ---------------1---------------1--------------1---------------1-----------------1
60 70 80 90 100 110
Wt % A in Blend
Рис. 4. Изменение адгезии по отслаиванию невулканизованных компаундированных
смесей этилен-пропиленовых сополимеров отличающихся молекулярно-
весовым распределением.
о ---------1----------1--------1--------1-----------1-------1---------1--------1---------1
60 65 70 75 80 85 90 95 100 105
Wt % A in Blend
Рис. 5. Изменение адгезии по отслаиванию невулканизованных компаундированных
смесей этилен-пропиленовых сополимеров отличающихся составом и кри-
сталличностью
Неоднородные вулканизационные сетки во взаиморастворимых смесях
эластомеров оказывают сильное влияние на прочность и удлинение при
разрыве. Эти сетки характеризуются межмолекулярным распределением
плотности сшивок и имеют в каждом компоненте разные концентрации
участков молекул, способных к сшиванию. Различия в количестве диена
548 “’V Глава 12. Смеси эластомеров
в цепи (5-этилиден-2-норборнена) с сополимерах EPDM [18,19] или разни-
ца в количестве ненасыщенных концевых групп способных к вулканизации
в силоксановых полимерах характерны для этих смесей [20]. Такие сетки
обеспечивают рост, как удлинения, так и прочности при большом удлинении
по сравнению с вулканизатами той же вязкости, но с однородной сеткой.
Особо отметим, что неоднородные сетки обнаруживают нелинейный рост
модулей растяжения при больших удлинениях. Эта нелинейность есть следс-
твие неаффинной деформации сетки при больших удлинениях, что приводит
к постоянному перераспределению напряжений в процессе деформации
в сторону слабосшитого компонента смеси, который наилучшим образом
аккомодирует с деформацией. Смеси компонента А с разными количе-
ствами полимера В в табл. 2 получали смешением гексановых растворов,
затем в них вводились ингредиенты и они вулканизовались. Оба полиме-
ра смеси аморфны, но полимеры А отличаются по молекулярным весам
и содержат около 3% способного к вулканизации диена (ENB). Поли-
мер В содержит меньше диена - порядка 0.7% ENB. Прочность смесей,
полученных смешением всех А с разными количествами В показана на
рис. 6. Во всех случаях, когда малые количества (<25%) В содержатся
в смеси, прочность выше в смесях, чем в матричном полимере А, даже
если степень вулканизации ниже в смеси, чем в А, из-за пониженного
общего содержания диена.
Рис. 6. Прочность смесей, приведенных в табл. 2, отличающихся плотностью сшивки.
II. Смеси взаиморастворимых эластомеров -*!/ 549
Таблица 2 . Компоненты смесей этилен-пропиленовых сополимеров отлича-
ющиеся плотностью сшивки.
Образец Состав С2 вес. % Состав ENB вес. % Вязкость ML (1+4), 125 °C
А1 57.0 3.2 20
^2 60.2 2.9 32
Аз 60.3 2.8 41
А4 59.4 2.6 51
А5 60.5 3.2 67
В 64 0.9 2100
Е. Отдельные полимеры
1. Смеси IR - BR
Смеси 1,4- IR с преимущественно 1,2- BR по-своему уникаль-
ны поскольку они являются взаиморастворимыми, химически разнородными
гомополимерами с большим молекулярным весом [21а-е], хотя в них и нет
дипольных или специфических взаимодействий между компонентами [21в].
С ростом концентрации 1,4-единиц в BR уменьшается и его смешиваемость с IR.
Определение параметра взаимодействия в смесях Ши BR с разными количест-
вами 1,2 и 1,4 показывает, что обменная энтальпия между Ши BR становится бо-
лее эндотермической с ростом единиц присоединенных в положение 1,4 [216, г].
Различия в структуре очевидны из значений коэффициентов теплового рас-
ширения компонентов. Различия в коэффициентах теплового расширения Ш
и BR сильно уменьшаются с ростом содержания 1,2-единиц.
2, Смеси эпоксидированных PI-CPE
Внутримолекулярное эпоксидирование 25 мол. % PI обеспе-
чивает получение взаиморастворимых смесей с СРЕ, содержащего 25 вес. %
хлора [22а, б]. Причина смешиваемости состоит здесь в специфических
взаимодействиях между эпоксидной группой и хлором.
F. Реакционноспособные эластомеры
Образование взаиморастворимых смесей за счет химической
реакции между компонентами смеси было изучено Колеманом с сотрудни-
ками [23 а]. Химическая реакция во взаиморастворимой смеси приводит
к негативной свободной энергии, необходимой для компенсации невыгод-
ных i-j контактов между разнородными эластомерами. Взаиморастворимые
смеси были образованы путем создания слабых водородных связей между
550 Глава 12. Смеси эластомеров
2,3-диметилбутадиен - 4-винилфенольным сополимером и карбонильной
группой таких полимеров как EVA и полиалкилметакрилатами. Степень
стабилизации под влиянием Н-связей регулировалась концентрацией фе-
нольных остатков в ненасыщенном сополимере. Выбор стабилизирующих
химических связей между эластомерами имеет большое значение. Прочная,
локализованная химическая связь должна приводить к потере вязкоупругих
свойств невулканизованным эластомером. Слабая водородная связь подвижна
и допускает течение в ответ на приложенное напряжение в смеси эластомеров.
III. ВЗАИМОНЕРАСГВОРИМЫЕ СМЕСИ ЭЛАСТОМЕРОВ
А. Получение
Взаимонерастворимые смеси могут быть приготовлены из
любых двух разнородных эластомеров. Они могут быть получены непосред-
ственно в процессе сополимеризации, смешением растворов полимеров или
суспензий, или механическим смешением. Все эти способы рассмотрены
Коришем [5]. В противоположность смесям других полимеров, эластомеры
могут быть смешаны либо непосредственно, либо в виде маточных смесей.
Непосредственное смешение состоит в том, что необходимые ингредиенты
(например, наполнители, пластификаторы, вулканизующие агенты) вво-
дятся одновременно со смешением несовместимых полимеров. В методе
смешения через маточные смеси, ингредиенты вводятся отдельно в каждый
индивидуальный полимер в различных операциях компаундирования.
Компаундированные эластомеры затем смешиваются друг с другом. Смеси
полимеров, полученные через маточные смеси обеспечивают большую опре-
деленность в первоначально заданном распределении ингредиентов между
фазами, таких как наполнители, пластификаторы и вулканизующие агенты
по сравнению со смесями полученными непосредственным смешением.
В. Формирование фазовой структуры смеси.
Процесс образования фазовой структуры определяется дву-
мя противоположными факторами. Во-первых, это течение вязкоупругих
эластомеров, присутствие пластификаторов, длительный период вылежки
(дни) между моментом смешения и вулканизации, также как стадии пе-
реработки с малой деформацией сдвига (например, каландрование), все
это способствует созданию равновесной фазовой структуры. В то же время
медленные диффузионные процессы в высокомолекулярных эластомерах,
присутствие дисперсного наполнителя и наступающее, в конце концов,
химическое структурирование приводят к возникновению устойчивой не-
равновесной морфологии. Токита [25а] и Авгеропулос [256] исследовали
смешение EPDM с диеновыми каучуками — NR или BR в зависимости от
их относительной вязкости.
III. Взаимонерастворимые смеси эластомеров 551
Параметры фазовой структуры смесей эластомеров зависят от (1) спосо-
ба смешения, (2) реологических свойств смешиваемых компонентов, и (3)
от межфазного взаимодействия. Также как и в смесях полимеров, вообще,
эластомер с меньшей вязкостью стремиться образовать непрерывную фазу
[26а, б]. Непрерывность обеих фаз наблюдается только в случае эластомеров с
равными значениями вязкости1). Вязкоупругие силы, развивающиеся в про-
цессе получения компаундированной смеси из двух реологически различных
эластомеров, оказываются, в основном, ответственными за формирование
фазовой структуры. Межфазное натяжение, возникающее за счет различия
в химическом составе эластомеров, является менее важным [26в].
С. Аналитические исследования
1. Микроскопия
Фазово-контрастная световая микроскопия широко приме-
няется для исследования ненаполненных бинарных смесей эластомеров.
Этот метод основан на разности показателей преломления полимеров и
рассмотрен в обзоре Крузе [27а]. Каллан и др. [27б-г] показали пригодность
метода для широкого круга смесей включающих NR, SBR, BR, CR, NBR,
EPDM, HR, CIIR. Результаты этих экспериментов приведены в табл. 3,
где приводится перечень измеренных удельных поверхностей дисперсных
фаз почти 50 комбинаций полученных в Бэнбери смесей 75/25 с участием
8 различных эластомеров. Смеси IIR-CIIR, SBR- BR в списке отсутствуют
из-за низкого контраста. Можно видеть, что NBR обнаруживает наибольшую
гетерогенность во всех смесях за исключением смесей с CR.
Просвечивающая электронная микроскопия (ТЕМ) применима как
к наполненным, так и ненаполненным смесям эластомеров. Однако во многих
полимерных фазах контраст в ТЕМ отсутствует. Если полимеры существенно
отличаются по ненасыщенности, можно достичь контраста обработкой смеси
тетрахлоридом осмия или тетрахлоридом рутения. Эти окислы селектив-
но окисляют двойные связи и концентрирование атомов металлов в фазе
с большей ненасыщенностью обеспечивает контраст в электронном пучке
в ТЕМ. Контраст в случае ТЕМ-анализа смесей диеновых каучуков достига-
ется методом отверждения серой, развитом Ронингером [28а] и применен-
ным Смитом и Андрисом [286]. Небольшой образец каучука погружается
в расплавленную смесь серы, ускорителя и стеарата цинка. Селективная
абсорбция солей цинка в фазу SBR делает ее более темной по сравнению
с BR при просвечивающей электронной микроскопии.
При изменении соотношения каучуков в смеси отО до 100% происходит переход от одной
однофазной системы к другой. В промежутке, поэтому, обязательно происходит обращение фаз
и если вязкости каучуков близки, то обращение фаз происходит в интервале от 30 до 70 об.%.
Маловязкий компонент может образовывать непрерывную фазу уже при малом его содержании
в смеси. Прим перев.
552 Глава 12. Смеси эластомеров
Таблица 3. Среднее значение удельной поверхности (в мкм2) дисперсной фазы
в ненаполненных смесях эластомеров (75:25).
Дисперсная Фаза 25% Матрица (75%)
NR CR BR SBR NBR EPDM IIR CIIR
NR — 45 1.5 1.2 300 1.5 2.0 3.2
CR 35 — 40 2.5 1.5 25 20 15
BR 0.7 4.5 — — 15 2.2 2.1 2.5
SBR 0.5 2.7 — — 20 2.1 12 10
NBR 400 1.3 17 30 — 250 100 225
EPDM 3.5 75 2.8 2.6 225 — 2.0 1.5
IIR 3.0 15 3.0 4.2 75 1.0 — —
CIIR 2.2 25 2.3 2.5 85 1.2 — —
Сканирующая электронная микроскопия требует более простых опера-
ций при подготовке образцов, чем ТЕМ. Методика контраста с помощью
тетрахлоридов осмия или рутения применяется и для SEM и пригодна как
для исследования больших образцов, так и для пленок. Атомный силовой
микроскоп (AFM), продолжающий идею сканирующего туннельного
микроскопа (STM) может работать на уровне атомного разрешения. В его
простейшем виде AFM [29а-г] работает как «миниатюрный профилометр
поверхности» и дает топографическое изображение. Потенциальные воз-
можности этих методов включают большее разрешение, более простую
подготовку образцов и большую пригодность при разных методах достиже-
ния контрастности. Цифровой анализ снимков обеспечивает определение
размера частиц по микрофотографиям [29д-ж] в пределах разрешения до
нескольких ангстрем.
2. Температура стеклования
Наиболее обычный метод определения степени однород-
ности смеси эластомеров, это определение температур переходов от каучу-
коподобного состояния к стеклообразному [30]. В случае эластомеров эти
температуры ниже комнатной. Определение температур стеклования не
дает информации о структуре смесей. Возникновение четких переходов,
соответствующих компонентам смеси указывает на существование гетеро-
фазной структуры. Возможный источник ошибки с том, что вулканизация
имеет тенденцию увеличивать Tg из-за ограничения подвижности цепей; это
может быть воспринято как смешиваемость.
III. Взаимонерастворимые смеси эластомеров 553
3. Исследование с помощью магнитного резонанса
NMR резонанс был применен для оценки размеров фазовых
частиц с разрешением менее 50 мкм во взаимнонерастворимых смесях, со-
держащих полибутадиен. Бутера и др. [31а] усовершенствовали 13С NMR —
метод, применив вращение образца под «магическим углом» в смесях, со-
держащих дейтерированный и протонированный полимеры. Сравнительно
недавно Кёниг и др. [31б-д] применили NMR для определения эффектив-
ности вулканизации в смесях эластомеров.
4. Световое, рентгеновское и нейтронное рассеяние
Роланд и Боом [32а] применили SAXS и SANS в сочетании с
ТЕМ для оценки размеров доменов полибутадиена (5% вес.) в матрице CR.
Роланд и др. взяли разные маточные смеси, содержащие дейтерированный
и протонированный полибутадиены соответственно. Каждая смесь переме-
шивалась на вальцах для изучения коалесценции частиц полибутадиеновой
фазы. Коалесценция (оценивалась по SANS) была минимизирована прове-
дением смешения при низкой температуре или существенным увеличением
молекулярного веса дисперсной или непрерывной фазы, чтобы обеспечить
значительное различие в вязкости эластомеров. Предварительные резуль-
таты SANS-исследования IR смешанного с дейтерированным 1,2-BR были
недавно опубликованы [326, в]. Все методики определения рассеяния были
затруднены присутствием наполнителей, которые приводили к более широ-
кому рассеянию, чем рассеяние самой фазовой структуры. Это накладывает
жесткие ограничения на широкое применение этих методик к большинству
эластомеров.
D. Распределение наполнителей, вулканизующих
агентов и пластификаторов между фазами смеси
Как при комнатной температуре, так и при температуре
переработки, эластомеры являются вязкими жидкостями с устойчивыми
диффузионными характеристиками. В несовместимых смесях это приво-
дит к изменению размера и формы частиц диспергированного эластомера
и миграции наполнителей, пластификаторов и вулканизующих агентов
из одной фазы в другую. Эти изменения ускоряются в процессе перера-
ботки и в присутствии пластификаторов и замедляются завершающим
процессом вулканизации. Сохранение положительных свойств мета-
стабильной смеси, которые часто достигаются только при определенной
фазовой морфологии и распределении наполнителей/пластификаторов,
требует тщательного контроля, как условий переработки, так и условий
вулканизации.
554 -*V- Глава 12. Смеси эластомеров
1. Миграция вулканизующих агентов и
пластификаторов в смесях эластомеров
В начале процесса смешения вулканизующие агенты скон-
центрированы в непрерывной фазе [33а]. Поскольку вулканизующие
агенты растворимы в эластомере, может происходить их миграция через
границу раздела фаз [33б-д]. Благодаря повышенной растворимости серы
в эластомерах, содержащих диеновые или серусодержащие мономеры,
или большего сродства ускорителей к каучукам, содержащим полярные
группы, при вулканизации возникает значительная разница в плотности
вулканизационной сетки в разных фазах. Более того, возросшая скорость
вулканизации в эластомерах, содержащих диеновые или стирольные группы,
могут привести к исчерпанию вулканизующих агентов в этой фазе, приводя
к еще большей миграции вулканизующей группы (принцип Ле-Шателье).
Для большинства смесей эластомеров эти эффекты взаимосвязаны, приводя
к значительному дисбалансу вулканизации в разных фазах [ЗЗе]. Плюс к это-
му граничные слои двух эластомеров, примыкающие к межфазной границе,
могут по составу отличаться от эластомера в объеме, благодаря значительной
миграции вулканизующих агентов и пластификаторов в эти области из окру-
жающей среды. Перенос вулканизующих агентов в одну фазу за счет обеднения
другой, создают морфологию, в которой граничные слои отделяют обедненный
вулканизующими агентами и слабо сшитый эластомер от находящегося рядом
обогащенного вулканизующими агентами и плотно сшитого эластомера.
Таблица 4 . Коэффициенты диффузии вулканизующей группы в смесях эласто-
меров
Вулканизующая группа От До DxlO7cm2/c
Ускоритель (TDDC) ПН BR 12.66
EPDM 1.09
CR 1.08
SBR 0.58
NR 0.70
Сера ПН SBR 4.73
SBR & 50 PHR 17.2
№700 CB
NR 2.82
Амеронген [34а] опубликовал один из первых обзоров миграции вул-
канизующих агентов в гетерофазных смесях эластомеров, исследованной
с помощью оптических и радиохимических методов. Позднее появилась
III. Взаимонерастворимые смеси эластомеров -*v- 555
более детальная работа Гардинера [34б-г] применившего оптический анализ
для изучения диффузии вулканизующих агентов через границы раздела
в смесях, состоящих из комбинации эластомеров CIIR, IIR, EPDM, CR, SBR,
BR, NR. Гардинер измерил градиент диффузионного потока для интервала
концентраций в зависимости от расстояния и от времени. Его результаты
определения диффузии ускорителя и серы из IIR к другим эластомерам
приведены в табл. 4.
2. Совулканизация
Удовлетворительные свойства вулканизованной смеси кау-
чуков зависят от уровня совулканизации между фазами. Совулканизация
это образование единой сетчатой структуры, включающей сшитые макромо-
лекулы обоих полимеров. Степень вулканизации находится на одинаковом
уровне в обеих фазах и между фазами. Шершнев [35а], а еще раньше Ренер
и Уэй [356] сформулировали практические требования для обеспечения
совулканизации компонентов в смеси эластомеров. Они следующие:
1. Смешение маточных смесей должно сопровождаться очень короткой
по времени высокотемпературной вулканизацией [35в].
2. Агенты вулканизации должны быть химически присоединены к эла-
стомеру.
3. Ускорители имеют параметр растворимости близкий к параметру
менее активного эластомера [35ж-к].
4. Нерастворимые вулканизующие группы, которые не могут мигри-
ровать.
5. Неполярные вулканизующие агенты, которые распределяются равно-
мерно и обладают одинаковой активностью по отношению к разнород-
ным эластомерам (например, пероксиды или реакционноспособные
смолы).
Гардинер [346—г], Вудс и Давидсон [36] улучшили совулканизацию
смесей, применив нерастворимый, содержащий свинец — вулканизующий
агент для менее ненасыщенного эластомера. Смеси с растворимыми вул-
канизующими агентами из этих же эластомеров были значительно хуже.
Коран [35е] совулканизовал смесь EPDM-NR путем модификации EPDM
малеиновым ангидридом. Модифицированный EPDM мог теперь сшиваться
оксидом цинка с образованием сетки ионных связей. Миграция вулканизую-
щих агентов была изучена Боувиком и Дэ [35в] в бинарных смесях NR, BR,
SBR. Скорости вулканизации уменьшались в этом же порядке, а миграция
вулканизующих агентов была наибольшей в смеси NR-SBR.
Степень вулканизации в фазах смеси может быть определена по измене-
нию высоты пика потерь [37а] и из депрессии точки замерзания набухших
сеток. Гонибал и Мак-Гилл [376] применили метод депрессии точки замер-
зания для набухших смесей NR-BR для определения плотности сшивки
556 “*V Глава 12. Смеси эластомеров
в отдельных полимерных фазах. Величина депрессии точки замерзания рас-
творителя в набухшем вулканизате зависит от объемной доли растворителя
в эластомерной фазе.
3. Перенос наполнителя между фазами
Распределение наполнителя (например, сажи, силикагеля)
и различных технологических добавок может быть неоднородным. Межфаз-
ный перенос наполнителя наблюдался как в смесях с участием насыщенных,
так и диеновых эластомеров. Миграция происходит из-за большего смачи-
вания наполнителя одним из полимеров в смесях, полученных смешением
расплавов1). Каллан и др. [38а] обнаружил значительный перенос сажи от
NR к BR и силикагеля от BR к NR как в растворе, так и в латексных смесях.
Значительный перенос сажи от малоненасыщенного эластомера (IIR
и EPDM) к высоконенасыщенному (например, диеновому) эластомеру
осуществляется, несмотря даже на смешение в виде маточных смесей
[38а]. Это можно наблюдать на смесях EPDM или IIR с NR или SBR.
В процессе механического смешения происходит хемосорбция нена-
сыщенного полимера на саже, что предотвращает дальнейший перенос
частиц сажи. Во всяком случае, сажа концентрировалась почти полностью
в фазе NR, когда вводилась в предварительно смешанные эластомеры.
Перенос сажи от EPDM к NBR мог стать обратным, если EPDM был об-
работан первичным амином [386]. Амины сильно хемосорбируются на
поверхности сажи и такая обработка позволяет фазе EPDM сохранить
значительное количество наполнителя. Распределение сажи показано
на рис. 7, а для смеси EPDM с NBR и сравнивается с рис. 7, б для той же
смеси, обработанной амином.
Каллан и др. [38а, б] применили метод селективного набухания (vide
infra) для оценки относительного сродства сажи к различным эластоме-
рам. Данные были получены для бинарных смесей 50/50 и сажу вводили
примерно в 25 комбинаций разных эластомеров. Удерживание сажи
в каждой фазе было оценено по ТЕМ-микрофотографиям смеси. Ряд по
уменьшению сродства к саже разных эластомеров выглядел как SBR>BR,
1) Твердые частицы наполнителя не обладают способностью к диффузии в вязкой матри-
це каучука. При введении наполнителя в смесь в процессе смешения частицы контактируют
то с одним то с другим каучуком, оставаясь там, где они прочнее удерживаются (т. е. лучше
смачиваются данным каучуком). Частицы сажи лучше смачиваются диеновыми каучуками,
т. е. полибутадиеновым или бутадиенстирольным, в этом плане к ним близок полиизопрен. Чем
больше величина каучук-сажевого геля, тем больше сродство сажи к каучуку, тем интенсивнее
частицы переходят в эту фазу в процессе смешения. Если сродство каучуков к саже примерно
одинаково, то частицы концентрируются преимущественно в менее вязком каучуке, который
деформируется более интенсивно, что обеспечивает лучшее смачивание частиц. Поэтому при
введении маловязкого НК или СКД в маточную смесь сажи и высоковязкого СКЭПТ, сажа
переходит в НК или СКД. Если вязкость СКД больше, чем СКЭПТ, то сажа остается в СКЭПТ.
(В.Н. Кулезнев. «Смеси полимеров». М. Химия 1980 г.). Прим, перев.
III. Взаимонерастпворимые смеси эластомеров
Л
557
CR, NBR>NR>EPDM>CHR>HR. Коттон и Мерфи [38] в дальнейших
экспериментах определили распределение частиц семи разных сортов
сажи в предварительно полученных смесях SBR-BR и SBR-NR. Резуль-
таты приведены в табл. 5. Разные сорта сажи отличаются по структуре
поверхности и по размеру частиц: удельная поверхность меняется от 43
до 136 м2/г. Для всех смесей SBR-NR сажа преимущественно концент-
рируется в фазе SBR.
а б
Рис. 7. Микрофотографии распределения сажи №234 всмеси70:30 NBRhEPR:EPR —
сополимер с 42 мол.% этилена (a); EPR содержит 43 мол.% этилена
и 0.9 мол.% присоединенного первичного амина (б).
Таблица 5. Распределение сажи в фазах смеси 50:50 натурального каучука и SBR
при общем содержании сажи 45 вес. ч.
Тип сажи Удельная поверхность (СТАВ) м2/г Связанный каучук, % Сажа в вес. ч.
NR SBR
№560 43 17.0 41.2 48.8
№347 87 17.0 33.9 56.1
№339 95 30.2 31.1 58.9
№234 120 30.4 30.0 60.0
Vulcan (ЮН) 136 33.1 27.2 62.8
№326 84 22.5 31.9 68.1
№330 81 25.0 34.0 66.0
558 “*V Глава 12. Смеси эластомеров
Е. Анализ величины межфазного переноса
1. Микроскопия
Электронная микроскопия является наиболее обычным
методом определения распределения наполнителей в гетерофазных смесях
эластомеров. Диас и др. [39] использовали время пролета во вторичной
масс-спектрометрии (TOF-SIMS) для одновременного определения фазовой
морфологии и диффузии вулканизующей группы в смеси BIMS-диеновый
эластомер.
2. Селективное набухание
Метод селективного набухания тонких срезов смесей EPDM
и ПКбыл впервые применен Каланом и др. [40а]. В смесях с EPDM фаза IIR
поглощала больше растворителя; следовательно, домены IIR меньше и вы-
глядят светлее в ТЕМ. Гесс и др. [406] применили тот же метод селективного
набухания для анализа распределения сажи при малом содержании напол-
нителя. Уанг и др. [40с] усовершенствовали метод селективного набухания,
применив два разных растворителя - каждый для своей фазы.
3. Контрастирование
Контрастирование с помощью летучих реакционноспособ-
ных окислов металлов, таких как OsO4 и RuO4 является предпочтительным
методом создания фазового контраста для ТЕМ-исследования. Оно приме-
нимо для смесей эластомеров, отличающихся ненасыщенностью, таких как
смесь NR-EPDM.
4. Селективный пиролиз
Этот метод применим к смесям, содержащим полимеры
с существенно разными температурами термодеструкции. Он был применен
для анализа распределения сажи в смесях NR-SBR и NR-BR [41а, б].
5. Анализ связанного каучука по количеству саже-
каучукового геля
Связанный каучук, это эластомер нерастворимый в раствори-
телях благодаря его хемосорбции на саже в процессе смешения. Он выделя-
ется в результате набухания невулканизованного полимера в растворителе
в течение длительного времени. Растворимая часть низкомолекулярного
полимера, несвязанного с сажей при этом удаляется. Этот метод был впервые
применен Калланом [38а] для многих смесей эластомеров.
6. Механические потери
Величина тангенса угла потерь (при для наполненных
эластомеров ниже, чем для ненаполненных [42а, б]. Это объясняется ростом
динамического упругого модуля в наполненной смеси с высокотемператур-
II. Смеси взаиморастворимых эластомеров -*v- 559
ной стороны пика при Tg. Эффект зависит от содержания наполнителя. Маити
и др. [42в] использовали этот эффект снижения тангенса потерь при 7^ для
оценки распределения сажи в смеси несовместимых эластомеров.
F. Компатибилизация
Компатибилизация сильно несовместимых эластомеров
применялась весьма ограниченно [43 а-г]. Компатибилизация это введение
малых количеств межфазного агента, что служит лишь для стабилизации
большой межфазной поверхности при высокой степени дисперсности частиц
в смеси. Количество и состав межфазных агентов не имеет целью изменить
свойства самих контактирующих фаз. Свойства смеси эластомеров опреде-
ляются интенсивными свойствами, такими как энергия когезии, плотность
поперечных связей и хемосорбция компонентов частицами наполнителя, т. е.
свойствами, на которые не влияет присутствие компатибилизатора. Однако
в бинарных смесях эластомеров с большой разницей параметров раствори-
мости, таких как полиолефиновые эластомеры с полярными эластомерами,
свойства определяются большими размерами частиц и малой межфазной
адгезией. Такие смеси эластомеров существенно улучшаются при добавле-
нии компатибилизатора.
Сетуа и Уайт [44 а, б] применили СМ (хлорированный полиэтилен)
в качестве компатибилизатора для улучшения гомогенности бинарных
и тройных смесей CR, NBR и EPR. Смеси NBR-EPM и CR-EPDM гомогени-
зировались быстрее, если добавлялись малые количества СМ. Присутствие
компатибилизатора снижает как время необходимое для смешения, что
наблюдается при прямой визуализацией потока, так и уменьшает размер
частиц диспергируемого полимера, что видно благодаря SEM. Аржунян
и др. [44в] использовали сополимер этилена и акриловой кислоты, а также
EPR привитый акрилатами в качестве компатибилизатора в смесях EPDM-
CR. Введение компатибилизатора приводило к снижению размера частиц
дисперсной фазы EPDM при одновременном увеличении сопротивления
разрыву и раздиру смеси.
Интенсивные свойства компонентов смеси, которые определяют свойства
ее вулканизата улучшаются, если компатибилизатор составляет значитель-
ную долю в смеси эластомеров. Дэвисон и др. [45] описал образование ком-
патибилизированной смеси полиалкилакрилата и специально полученного
привитого сополимера акрилата на EPDM, которая после вулканизации
стала стойкой к растворителям. Акрилаты, привитые на EPDM, это сопо-
лимеры с участием метил-, этил-, и м-бутилакрилатов. Такие смеси после
вулканизации обнаруживают прекрасные свойства — прочность и модули,
аналогичные исходному эластомеру. Коттону и Мэрфи [38г] удалось син-
тезировать привитый сополимер непосредственно в процессе смешения.
Предварительно обработанный амином EPDM смешивался с NBR с обра-
560
Глава 12. Смеси эластомеров
зованием привитого сополимера в результате реакции амидирования амина
с нитрилом. Реакция прививки каталитически ускорялась в присутствии
кислого фосфитного пластификатора в фазе NBR. Компатибилизатор спо-
собствует образованию очень маленьких доменов дисперсной фазы. Рис. 8, а
и 8, б представляют микрофотографии дисперсии EPDM и привитого ами-
нами EPDM соответственно, в матрице NBR. И в этом случае, также как на
рис. 7, видно удерживание частиц сажи в объеме фазы EPDM. Вулканизация
смеси привитого аминами EPDM и NBR неполярным пероксидом, который,
скорее всего, равномерно распределяется между фазами смеси, приводит
к получению смеси с прекрасной стойкостью к растворителям и высокой
температуростойкостью.
а б
Рис. 8. Микрофотографии дисперсной структуры смеси 70:30 NBR и ЕР-эласто-
мера: этилен-пропиленовый эластомер - сополимер с 42 мол. % этилена (а);
этилен-пропиленовый сополимер включает 43 мол.% этилена и 0.9 мол.%
привитого первичного амина (б).
G. Свойства несовместимых смесей
В случае, когда не требуется истинная взаиморастворимость
для обеспечения комплекса свойств смеси, оказывается необходимой хо-
рошая адгезия между несовместимыми фазами. Несовместимые смеси,
удовлетворяющие этому критерию, обнаруживают значительные возмож-
ности изменения реологических, прочностных свойств и истираемости по
сравнению с совместимыми смесями.
1. Переработка
Смешение часто применяется для улучшения технологичес-
ких свойств каучуков. Это улучшение может включать либо понижение вяз-
II. Смеси взаиморастворимых эластомеров -*ъ- 561
кости, либо получение материала менее склонного к образованию разрывов
текущего расплава. Во-вторых, проявление эластических эффектов, таких
как разбухание струи на выходе из головки тоже может регулироваться
смешением. Авгеропулос и др. [46а] показали, что менее вязкая фаза в
бинарной смеси стремится стать непрерывной. Этот эффект ускоряется при
сдвиге, поскольку морфология смеси зависит от приложенных напряжений.
Вблизи стенки скорость сдвига оказывается максимальной, и маловязкий
компонент накапливается у стенки [466]. Введение лишь нескольких про-
центов EPDM во фторэластомер или PDMS в SBR [46в] снижает вязкость
при установившемся течении. Это происходит в результате накопления
маловязкого компонента у поверхности канала.
Большая часть информации в этой области происходит или из практи-
ческого опыта либо недостоверна. Теоретические расчеты вязкости смесей
эластомеров [46ж] имеют ограниченное значение, поскольку: (1) неодно-
родная фазовая структура смеси легко меняется в ответ на приложенные
напряжения и (2) неоднородное распределение наполнителей и пластифи-
каторов определяет течение. Эти изменения структуры в смесях эластомеров
обусловливают аномальные реологические свойства, совершенно отличные
от ожидаемой аддитивности по отношению к свойствам эластомерных ком-
понентов.
2. Модули
В микрогетерогенной смеси особенности фазовой структуры
в общем не оказывают значительного влияния на вид кривой нагрузка —
удлинение. В противоположность ожиданию, что непрерывная фаза должна
оказывать наибольшее влияние, кривая нагрузка — удлинение ненаполнен-
ной смеси EPDM-BR оказалась независимой от того являлась ли фаза BR
непрерывной или дисперсной [47а].
Характер распределения сажи оказывает значительное влияние на вели-
чину модулей. Мэйнеке и Тафтаф [476] показали, что рост неоднородности
распределения сажи приводит к снижению модулей смеси. Унос части сажи
из одной из фаз понижает ее модули больше, чем увеличение модулей фазы
с большим содержанием сажи. Этот эффект связывается с нелинейной за-
висимостью модулей каучука от содержания сажи [47в, г].
3. Липкость и адгезия
Адгезия есть поверхностное явление, обусловленное пе-
репутыванием цепей на поверхности. Адгезия разнородных эластомеров
до (липкость) и после вулканизации (совулканизация) часто достигается
только при смешении разнородных эластомеров с одним общим полимером.
Однако состав поверхности и, следовательно, показатели адгезии могут
быть изменены, не прибегая к созданию высокой концентрации данного
эластомера в смеси.
562 -* V- Глава 12. Смеси эластомеров
Смеси компонентов, отличающихся по вязкости, имеют тенденцию
к концентрированию маловязкого компонента на поверхности во время
переработки. Наиболее обычный способ увеличения липкости и совулкани-
зации смесей эластомеров сводится к тому, чтобы один из эластомеров был
преобладающей фазой в каждой контактирующей смеси. Моррисей [48]
показал, что липкость разнородных эластомеров, находящихся в смеси с NR
улучшается монотонно с увеличением количества NR в смеси. Увеличение
доли одного из эластомеров в обеих смесях обеспечивает возможность со-
единенных поверхностей обеспечить монолитизацию.
4. Гкстерезис
Основное применение смеси эластомеров находят при изго-
товлении боковин автомобильных шин. Снижение гистерезисных потерь
(«сопротивления качению») является основной задачей конструирования.
Понижение гистерезиса данного эластомера требует снижения дозировки
сажи или увеличения плотности химической сшивки. Такие изменения от-
рицательно влияют на другие аспекты производства. Смешение эластомеров
обеспечивает пониженный гистерезис с минимальным количеством таких от-
рицательных эффектов. Гистерезис наполненного эластомера, содержащего
зоны различной концентрации сажи оказывается меньше, чем в эластомере с
равномерным содержанием сажи. Смеси разнородных эластомеров с разной
способностью удерживать частицы сажи дают возможность легко и реально
создать неоднородность распределения сажи.
5. Разрушение
Улучшенные прочностные свойства могут быть обеспечены
смешением эластомеров, включая комплекс свойств, превосходящий чистые
компоненты. Важный аспект фазового структурообразования смеси каучуков —
это природа межфазных связей. Механическая однородность морфологии
с химическими связями на межфазной границе обычно обеспечивает превос-
ходные свойства. В смесях SBR и хлорбутилкаучука, например, увеличение
динамической выносливости было достигнуто созданием химических связей на
межфазной границе. Аналогично этому, введение межфазных агентов улучшает
прочностные свойства смесей EPDM-силиконовый каучук [49а].
Наблюдается улучшение стойкости к истиранию, которое связывают с
селективным переходом сажи в фазу BR в смесях NR-BR и SBR-BR. Пер-
вые исследования влияния распределения сажи на истираемость смесей
NR-BR были проведены Краковским и Тинкером [50а]. Было обнаружено,
что сопротивление истиранию протектора непрерывно возрастает с ростом
содержания сажи в фазе BR, на что указывали исследования методом ТЕМ.
Тсэ и др. [506] показали, что в дисперсии BIMS в матрице BR можно увели-
чить сопротивление многократным деформациям, если среднее расстояние
между поперечными связями в BIMS окажется меньше 60 нм.
II. Смеси взаиморастворимых эластомеров 563
Растрескивание под действием озона было исследовано на примере смесей
NR-EPDM [49г-е]. Эндрюс [49ж] показал, что малые микрообласти EPR
в смеси EPR-NR создают барьер, который тормозит рост озоновых трещин.
Амбеланг и др. [49з], обнаружил значительную роль присутствия малых
частиц ЕРМ в смесях EPDM-SBR. Мэтью [496] показал, что сажа улучшает
стойкость к озону смеси NR-EPDM. Улучшение наблюдалось в смесях с
равномерным распределением сажи по фазам смеси, полученной методом
обращения фаз. Это происходит потому, что (1) увеличивается эффект уси-
ления фазы EPDM и (2) присутствие сажи в EPDM увеличивает объем этой
фазы. В смесях BIMS-BR озонное растрескивание может быть замедлено
уменьшением размера частиц BIMS [506].
Транспортные характеристики в смесях полимеров представляют
интерес, как с точки зрения практического применения, так и в качестве
возможности оценить фазовую структуру смеси. Исследование влияния
состава смеси на ее проницаемость в зависимости от природы каучуков
приведено в [49и]. Проницаемость смеси эластомеров зависит от коли-
чества непрерывной фазы и от морфологии дисперсной фазы. Вытянутая
структура частиц дисперсной фазы, особенно когда она является волок-
нистой или слоистой, приводит к снижению проницаемости благодаря
тому, что искривляется, т. е. удлиняется путь, который должен проходить
пенетрант [49к].
Н. Применение
1. Смеси ненасыщенных эластомеров
Наиболее обычные смеси ненасыщенных эластомеров это те,
которые применяются в различных частях автомобильных шин. В табл. 6
приводится перечень важнейших компонентов шины и типичные смеси для
их изготовления. Большая часть литературы по смесям эластомеров касается
именно этих важнейших применений. Мы покажем основные принципы,
положенные в основу создания этого перечня смесей.
Таблица 6. Эластомерные смеси в автомобильных покрышках.
Компонент Легковые шины Грузовые шины
Протектор SBR-BR NR-BR или SBR-BR
Брекер NR NR
Каркас NR-SBR-BR NR-BR
Боковина NR-BR или NR-SBR NR-BR
Герметизирующий слой NR-SBR-IIR NR-IIR
564 -*V- Глава 12. Смеси эластомеров
2. Смеси насыщенных и ненасыщенных эластомеров.
Применение смесей полиолефиновых эластомеров, таких
как 11R и EPDM, как наиболее существенных компонентов смесей ненасы-
щенных эластомеров, представляет собой быстро развивающуюся область.
Мури и др. [51] провели сравнение свойств смесей EPDM-NR и BIMS-NR
как материалов для боковин. Во многих областях применения насыщен-
ный эластомер рассматривается в качестве полимерного антиоксиданта
для диенового каучука. Существует убеждение, что высокомолекулярные
полиолефины предпочтительны для такого применения из-за из ограничен-
ной интердиффузии и более устойчивой фазовой морфологии. Некоторые
преимущества в части прочностных свойств и сопротивления истиранию
могут возникать вследствие интердиффузии высокомолекулярных цепей
разнородных эластомеров через фазовую границу. Значительный прогресс
был достигнут в модификации структуры полиолефиновых эластомеров с
целью увеличения их совместимости с ненасыщенными эластомерами. Тсэ
и др. [506] показали, что смеси насыщенного и ненасыщенного эластоме-
ров, не содержащие компатибилизатор, возможны, если первый содержит
значительные количества (>12%) стирольных групп. В перспективе это
представляется важной областью исследований с появлением в результате
новых методов синтеза полиолефинов.
IV. ЗАКЛЮЧЕНИЕ
Получение и применение смесей эластомеров является техно-
логической потребностью. Совместимые смеси применяются широко, но это
не всегда достоверно, поскольку разделение вулканизованных эластомеров
экспериментально невозможно. Несовместимые смеси требуют высокого
уровня дисперсности дисперсной фазы и межфазной адгезии, характерной
для всех смесей полимеров. Плюс к этому они требуют тщательно контроли-
ровать распределение наполнителя между фазами и регулировать плотность
сшивки каждого компонента. Это необходимо для обеспечения механической
однородности вулканизованных эластомеров. Сегодняшний критерий ка-
чества североамериканских автомобильных шин требует от протектора про-
бега 80 000 миль при износе менее 0.4 дюйма. От вулканизованной кровли
требуется 35 лет работы в погодных условиях при минимальном изменении
удлинения и прочности. Техническая сложность анализа и применения
смесей эластомеров привела к секретности многих рецептур и применений.
Несмотря на трудности анализа и пробелы в понимании, применение смесей,
содержащих эластомер, продолжает быть активной и растущей областью
исследований. Отчасти стимулом здесь является возможность направленного
синтеза многих прежних эластомеров. Эти новые технологии синтеза вклю-
чают новые катализаторы и саму методику синтеза при производстве этилен-
V. Обозначения названий эластомеров -* V 565
полиолефиновых эластомеров, полимеризацию с передачей цепи в случае
акрилатов, и анионную полимеризацию в случае диеновых эластомеров.
Получение эластомеров с узким молекулярно-весовым и композиционным
распределением в результате этих синтезов делает преимущества смешения
более очевидными и доступными для применения1^
V. ОБОЗНАЧЕНИЯ НАЗВАНИЙ ЭЛАСТОМЕРОВ
АВВ акрилатбутадиеновый каучук
АСМ сополимер этилакрилата и сомономера (акриловый каучук)
ANM этилакрилат-акрилонитрильный сополимер (акриловый каучук)
BIMS бромированный изобутиленпараметилстирольный каучук
вв бутадиеновый каучук (полибутадиен)
вив бромбутиловый каучук
СНВ хлорбутиловый каучук
CFM полихлортрифторэтилен (фторкаучук)
см хлорополиэтилен (предыдущее обозначение СРЕ)
со эпихлоргидриновый гомополимерный каучук (полихлормети- локсиран)
св хлоропреновый каучук (полихлоропрен)
CSM хлоросульфополиэтилен
ЕАМ этиленэтилакриловый сополимер (например, Vamac)
ECO сополимер оксиэтилена (оксиран) и хлорометилоксирана
ENM или H-NBB предполагаемый код для гидрированного NBB
ENB эпоксигидрированный NB
EPDM этилен-пропилен-диеновый терполимер
ЕРМ этилен-пропиленовый сополимер
EVM этилен-винилацетатный сополимер (ранее EVA или EVAC)
FMQ метилсиликоновый каучук с фторгруппами (предыдущее обо- значение FSI)
FPM /FPM каучук, имеющий фтор- и фтороалкильные или фтороалкокси замещающую группу
п Свойства резин на основе смесей каучуков зависят главным образов от
вводимых ингредиентов и прежде всего от вулканазующей группы и режима
вулканизации. Наполнители и пластификаторы, как это видно из статьи
С. Датта, также играют важную роль. За последние 40 лет в литературе прак-
тически не появилось ни одной кривой свойство-состав смеси полимеров,
полученной для смеси каучуков невулканизованной или ненаполненной,
а ведь такая кривая отражает взаимодействие полимеров, их сродство. Эта
особенность отличает смеси каучуков от двух других групп полимерных
смесей: смесей термопластов и смесей реактопластов. В статье естественно
приводятся примеры влияния химического сродства каучуков на свойства
их смеси, но это влияние невелико по сравнению с ролью ингредиентов
и технологии получения смеси. Прим, перев.
566
Глава 12. Смеси эластомеров
HR изобутилен изопреновый каучук (бутилкаучук)
IB изопреновый каучук (синтетический каучук)
MQ (PVMQ) метилсиликоновый каучук (с винильными и фенильными кон-
NBB акрилонитрил бутадиеновый каучук (нитрильный каучук)
NR изопреновый каучук (натуральный каучук)
PUR общее обозначение уретановых каучуков
Q общее обозначение силиконовых каучуков
SBR стирол-бутадиеновый каучук
TM полисульфидный каучук
TOR транс-полиоктенамер
VMQ метилсиликоновый каучук с винильными группами
ГЛАВА ТЕРМОПЛАСТИЧНЫЕ ЭЛАСТОМЕРЫ
13
БРАЙАН П. ГРЕДИ
Школа химического инжиниринга и материаловедения
Университет Оклахомы
Норман, Оклахома
СТЮАРТ Л. КУПЕР
Факультет химического и биомолекулярного инжини-
ринга
Университет штата Огайо
Колумбус, Огайо
I. ВВЕДЕНИЕ
ТПЭ — быстро растущий сегмент полимерной промыш-
ленности. До 2007г. ожидался годовой прирост 5%, в это же время общая
потребность в США в этих материалах должна была достигнуть 1,5 млрд,
фунтов при общей ежегодной продаже на ~ 3 млрд. $/год [1]. Большую
долю в этом росте обеспечивает замена других типов материалов, а также
рост потребности в так называемых «мягких на ощупь» («soft touch»)
поверхностях. Примерно за 10 лет с выхода второй редакции этой книги,
произошёл важный технический прорыв в этой области: значительный
рост производства полиолефиновых (ПО) ТПЭ за счёт внедрения метал-
лоценового (МЦ) катализатора.
Первейшее преимущество ТПЭ перед традиционной резиной — лёгкость
(и, соответственно, низкая стоимость) переработки, возможность широко
варьировать свойства, и возможность вторичной переработки и вторич-
ного использования. Помимо очевидных выгод для окружающей среды,
брак/отходы ТПЭ могут перерабатываться вторично. Недостатки ТПЭ по
сравнению с резиной: относительно высокая цена сырья, общая для всех
ТПЭ невозможность разбавления их дешёвыми наполнителями типа тех-
углерода, плохая хим- и температуростойкость. Последнее свойство делает
невозможным использование ТПЭ в автошинах.
568 Глава 13. Термопластичные эластомеры
Чтобы иметь право называться ТПЭ, материал должен иметь три суще-
ственные характеристики:
1. Способность к растяжению до достаточно больших деформаций
и к возвращению после снятия нагрузки к форме, близкой к перво-
начальной;
2. Перерабатываемость в расплаве при повышенных температурах;
3. Отсутствие значительной ползучести.
Рис. 1. Диаграмма зависимости модуля от температуры для сополимеров. На всех
рисунках штриховые линии обозначают поведение чистых материалов. За-
штрихованная область показывает, в каких пределах может варьироваться
поведение в зависимости от относительных количеств компонентов А и В.
а — статистический сополимер A-В; б — блок-сополимер А и В с чрезвычайно
короткими блоками; в — сегментированный блок-сополимер с неполным раз-
делением фаз; г — Сегментированный блок-сополимер с полным разделением
фаз. Бонарт (Bonart) [220].
Почти во всех случаях ТПЭ являются сополимерами, то есть в цепи полимера
присутствуют хотя бы два мономера. ТПЭ должен, в общем, иметь такой вид
зависимости модуля от температуры, как показано на рис. 1, в. Область плато
должна включать температуру эксплуатации материала. Плато может сдвигать-
ся вниз или вверх в зависимости от состава или свойств сомономеров, что даёт
изготовителю широкую возможность варьировать эластичность.
Структурные характеристики большинства ТПЭ имеют некоторое сход-
ство. Сомономеры обычно имеют длинные последовательности, что делает
I. Введение JL 569
материал блок-сополимером. Сомономеры почти всегда разнородны,
что приводит к микрофазовому разделению на уровне нанометрической
шкалы длин, что означает, что эти материалы правильно именовать на-
номатериалами (эти материалы появились задолго до того, как термин
вошёл в моду!). Движущая сила фазового разделения всегда имеет эн-
тальпийную природу, и обычно на 1-2 порядка слабее, чем первичные
валентные связи. Кристалличность, водородные связи, ионные, и ван-
дер-Ваальсовские силы — все приводят к микрофазному расслоению
в этих системах.
Две фазы в этих системах имеют различные свойства. Одна фаза, мягкая,
содержит компонент, который находится выше температуры стеклования
(Tg) и температуры плавления (Тт), так что его цепи имеют высокую степень
подвижности. Другая фаза, твёрдая, содержит жёстко зафиксированные
цепи, поэтому температура эксплуатации материала должна быть ниже её
Tg или Тт. Относительное количество двух фаз контролирует физические
свойства ТПЭ, определяя, какая фаза изолированная или непрерывная. Воз-
можность легко варьировать эти параметры через стехиометрию позволяет
использовать ТПЭ в широком спектре применений, как упомянуто ранее.
Структуры большого количества полимеров попадают в категорию
ТПЭ. Структуры некоторых общеизвестных ТПЭ показаны на рис. 2. Shell
Development Company разработала первый коммерчески доступный ТПЭ
в начале 1960-х, который стал родоначальником семьи материалов КРАТОН
(KRATON). Обычно это трёхблочники — ПС-ПБ-ПС (СБС), ПС-ПИ-ПС
(СИС), или ПС-ПЭБ-ПС (СЭБС). Разделение фаз проявляется вследствие
несовместимости твёрдого и мягкого сегмента. Полистирольные домены
служат в качестве твёрдой фазы, поскольку Tg полистирола примерно
100°С. Полидисперсность этих ТПЭ обычно невелика, потому что их полу-
чают анионной полимеризацией. Концевые стирольные блоки скрепляют
полимер и обеспечивают материалу необходимую прочность, в то время как
мягкий сегмент даёт эластичность. Примерно 50% от всех производимых
ТПЭ — это трёхблочники СБС, СИС или СЭБС.
Другая крупная группа ТПЭ, занимающая ~ 30% рынка ТПЭ, основана
на полиолефинах. Три основных материала составляют эту группу: сопо-
лимеры этилена и пропилена (ЭП, ЕР), сополимеры пропилена и более
высоких а-олефинов, таких как 1-бутен и 1-октен, и сополимеры этилена
и а-олефинов. В двух последних случаях, главным компонентом является
пропилен или этилен. Между обогащёнными этиленом или пропиленом
материалами есть разница в эластичности и точке размягчения; обога-
щённые этиленом материалы более эластичны, но при этом размягчаются
при температуре на ~ 50°С ниже, чем обогащённые пропиленом. Эти
типы материалов в достаточной степени важны, чтобы получить общую
аббревиатуру — ТПО, что означает термопластичные эластомерные оле-
570 -'V- Глава 13. Термопластичные эластомеры
тп/п-8
п~100
1 Н рн2“"
)с ==с
—сн2 хн
ССН2-
I
сн
II
сн2
п= 1-5
т-20
х-20
HS ss
д
тп/(п+1)-20
п-1
х-100
СНз
[CH2CH2fcT сн2с —
ss
F=°
O'Na+
HS
СНз
СН2С —
4=о
он*
Рис. 2. Структуры коммерчески значимых ТПЭ: HS — твёрдый сегмент, SS — мяг-
кий сегмент, а — СБС; б — МДИ-БД-ПТМО полиуретан; в — сополиэфир
ПТМТ и ПТМО; г — сополиамид найлона 66 и ПТМО; д — статистический
сополимер иономер Е-МАА, нейтрализованный натрием.
I. Введение JU 571
фины. Особый пример — семейство материалов Ингейдж (Engage) [3],
являющихся сополимерами этилена и а-олефинов. МЦ-катализаторы,
упомянутые ранее, позволяют лучше контролировать длину фрагментов
цепей обычных ЭП-сополимеров. В общем, ЭП-сополимеры и смеси со-
полимеров имеют немного более высокую стоимость и лучшие свойства,
нежели трехблочные сополимеры.
ЭП-ТПЭ отличаются от сшитых аналогов, которые не являются тер-
мопластами, поскольку не способны вторично перерабатываться. Очень
важным термопластичным эластомером является смесь полипропилена,
полиэтилена или ЭП-сополимера с этилен-пропилен-диеновым (ЭПДК,
EPDM) каучуком. Последний способен сшиваться в термореактивный ма-
териал; однако, эти материалы могут перерабатываться как термопласты,
если способный сшиваться компонент присутствует в достаточно низкой
концентрации, чтобы образовывать изолированную фазу. Переработка
в расплаве приводит к формированию химических связей в пределах
изолированной фазы каучука; процесс имеет название динамической
вулканизации. Коммерческий пример этого типа материалов — Сантопрен
(Santoprene) [4] фирмы AES. Другие смеси неспособных к сшиванию
ТПЭ со способными к сшиванию материалами также коммерчески ис-
пользуются. Эти материалы классифицированы как смеси эластомеров
и рассматриваются в главе 12.
Полиуретановые эластомеры — сополимеры с жестким сегментом, ко-
торый содержит ароматические кольца, и мягким сегментом, содержащим
простой или сложный полиэфир. Полиуретановые эластомеры — часть се-
мейства материалов, именуемых сегментированными блок-сополимерами,
которые характеризуются как материалы с чередованием твёрдых и мягких
сегментов, повторяющихся много раз в одиночной полимерной цепи. Мик-
рофазовое разделение происходит из-за несовместимости жёстких и мягких
сегментов. В некоторых случаях жесткий сегмент может также кристаллизо-
ваться. Сегментированные блок-сополимеры имеют общую формулу (АВ) х,
в то время как трёхблочный сополимер имеет общую формулу АВА. Полиуре-
таны обычно изготовляются из ароматического диизоцианата, олигомерного
диола и низкомолекулярного диола. Низкомолекулярный диол называется
удлинителем цепи, потому что он связывает сегменты АВ вместе. Типичный
полиуретан на основе 4,4’-дифенилметандиизоцианата (МДИ, MDI), поли
(тетраметиленоксида) (ПТМО, РТМО) и бутандиола (БД, BD) показан на
рис. 2. Коммерческий пример полиуретана — это семейство материалов
МДИ-БД (MDI-BD) фирмы Dow Chemical Company под коммерческим
названием Пеллетан (Pellethane) [5]. Полиуретаны относительно дороги
и нашли применение для высококачественных эластомерных изделий
и пен. Приблизительно 15% рынка термопластичных эластомеров занимают
полиуретаны.
572 -*V- Глава 13. Термопластичные эластомеры
Другой класс сегментированных блок-сополимеров — сегментированные
блок-сополимеры, содержащие ароматический сложноэфирный жёсткий
сегмент и мягкий сегмент на основе простого или сложного олигоэфира.
Кристаллизация жёсткого сегмента является движущей силой фазового
разделения в таких системах. Сополиэфир, полученный из поли (бути-
ленгликольтерефталата) и поли (тетраметиленоксида) (4ГТ-ПТМО, 4GT-
РТМО) из семейства высококачественных ТПЭ Хайтрел (Hytrel) [6] фирмы
DuPont, также показан на рис. 2. Эти материалы являются маслостойкими
и устойчивыми к более высоким температурам, чем другие термопластичные
эластомеры, что делает их более подходящими для таких областей, как детали
двигателей автомобилей.
Два других термопластичных эластомера показаны на рис. 2. Сополи-
амидные ТПЭ сопоставимы по структуре с сополиэфирными. Движущей
силой фазового расслоения в этих материалах также служит кристаллизация.
Эти материалы имеют особенно высокую химстойкость и хорошие свойства
при низких температурах. Коммерческий пример сополиамида — ПЕБАКС
(РЕВАХ) [7], поставляемый на рынок фирмой Atofina. Сополиамиды кон-
курентоспособны с полиуретанами и сополиэфирными ТПЭ.
Последними ТПЭ, которые мы будем обсуждать, будут иономеры. Ионо-
меры — материалы, в которых малая мольная доля мономеров, обычно
меньше 10%, содержит ионные функциональные группы.
Эти материалы не сегментированы подобно большинству других обсуж-
даемых в этой главе материалов. Ионные группы распределены беспоря-
дочно по полимерной основе. Несовместимость между ионными группами
и неполярной полимерной основой ведет к формированию обогащённых
ионами доменов. Коммерческий пример иономера — Сурлин (Surlyn) [8]
от DuPont, показан на рис. 2.
Эта глава построена следующим образом: общие концепции синтеза,
обсужденные в Главах 2 и 11, будут расширены обсуждением того, что
связано с методами, важными в синтезе ТПЭ. Большая часть главы будет
посвящена морфологии ТПЭ, так как именно морфология определяет их
физические и механические свойства. Это обсуждение будет сопровож-
даться определенными примерами, иллюстрирующими влияние струк-
туры на физические свойства и термодинамику фазового разделения,
проведено детальное обсуждение морфологии, а также температурного
поведения. Будет обсуждаться реология и переработка этих материалов.
Наконец, будут приведены некоторые области применения термоплас-
тичных эластомеров.
Акцент будет делаться на темах, которые являются общими для всех
ТПЭ; однако будет обсуждаться также специфика некоторых коммерче-
ски важных материалов. Эти обсуждения не будут исчерпывающими; для
получения дальнейшей информации заинтересованный читатель может
воспользоваться ссылками, имеющимися в этой главе.
II. Синтез термопластичных эластомеров -*и- 573
II. СИНТЕЗ ТЕРМОПЛАСТИЧНЫХ ЭЛАСТОМЕРОВ
А. Ступенчатая полимеризация: полиуретаны,
блоксополиэфиры, полиамиды
Полиуретаны, сополиэфиры и сополиамиды производятся
путём ступенчатой полимеризации. В ступенчатой полимеризации при-
менительно к ТПЭ молекула, содержащая две реакционноспособные фун-
кциональные группы одного типа (например, изоцианатные), реагирует
с другой молекулой, содержащей две реакционноспособные функцио-
нальные группы другого типа (например, гидроксильные); в результате
образуется полимер. Как было более подробно обсуждено в Главе 2, при
ступенчатой полимеризации для получения высокомолекулярного продукта
требуется чрезвычайно высокая степень превращения (>99%). Вообще-
то, свойства ТПЭ довольно слабо зависят от молекулярного веса, так что
полидисперсность обычно не очень важна, хотя важно достижение опре-
делённого молекулярного веса. Для достижения высокого молекулярного
веса решающим является регулирование соотношения функциональных
групп; формула показывает случай стехиометрического дисбаланса двух
реагирующих функциональных групп:
_ 1 + г
Пп ”1 + г-2гт>
где г — степень начального дисбаланса функциональных групп (всегда
меньше !),/> — глубина превращения, и п — среднечисловая степень поли-
меризации. Даже для достижения умеренного молекулярного веса не может
допускаться нарушение стехиометрии более чем на несколько процентов.
Перед началом краткого описания синтеза полиуретанов следует сооб-
щить читателю, что полиуретаны в общем разделяются на три класса: пены,
покрытия и ТПЭ. Эта глава касается только последних, и морфологическая
разница между термопластичными полиуретанами (ТПУ) и прочими состо-
ит в том, что это линейные полимеры, и длины их сегментов больше. При
синтезе сшитые материалы получают с применением воды; принимая это во
внимание, следует исключить воду из реакции получения ТПУ. В книгах по
этой теме [9-12] рассмотрены все три типа полиуретанов, иногда без чёткого
различия между различными применениями.
Полиуретаны могут синтезироваться в растворе [13] или в блоке [14].
Полиуретаны растворной полимеризации обычно имеют более однородное
распределение твёрдых и мягких сегментов. Полиуретаны, полимеризован-
ные в блоке, имеют более высокий молекулярный вес, что частично связано
с побочными реакциями, которые вызывают сшивание [15]. Большая часть
полиуретанов выпускаемых в промышленности, получают в блоке при
574 Глава 13. Термопластичные эластомеры
температурах от 80°С до 120°С. Реакция изоцианата и гидроксила является
весьма экзотермической, из чего следует, что надлежит отводить теплоту
из реагирующей смеси таким образом, чтобы температура сохранялась
ниже температуры деструкции 140°С. Вообще, более высокие температуры
приводят к большему количеству побочных реакций и к сшиванию. Для
получения полностью линейных полиуретанов необходимо придерживаться
температурного режима не выше 50°С [16]. Для полимеризации в блоке
используются два метода, «одностадийный» и преполимерный. В «одно-
стадийном» методе все компоненты смешиваются вместе; в преполимерном
методе сначала дают прореагировать диизоционату и олигомерному диолу,
затем добавляется удлинитель цепи. Часто олигомерный диол и диизоцианат
несовместимы, так что реакция происходит на границе раздела между двумя
компонентами, что может приводить к большим композиционным откло-
нениям по ходу реакции. Для растворной полимеризации необходим общий
растворитель для диола, диизоцианата и полимера. Относительно полярный
органический растворитель типа М,М-диметилацетамида или диметилсульфок-
сида вполне подойдёт. При полимеризации полиуретанов в растворе обычно
используется аналог преполимерного метода, описанного ранее при синтезе
ПУ в блоке. В отличие от реакции в блоке, при растворной полимеризации
используют оловоорганический катализатор. Оловоорганические катализаторы
обычно не используются при полимеризации в блоке, так как неполное уда-
ление катализатора приведет к недостаточной гидролитической стабильности
конечного продукта.
Другой часто встречающийся термопластичный эластомер, получаемый
ступенчатой полимеризацией сополиэфир ПЭ ТЭП. Общая схема реакции
близка к получению полиуретанов, за исключением реагентов для образо-
вания жёсткого сегмента — вместо диизоцианатов используются диэфиры,
при этом для достижения высоких степеней превращения должен быть
удален низкомолекулярный побочный продукт. Сополиэфиры получают в
расплаве путём трансэтерификации, описанной Вициэпе (Witsiepe) [17]. На
первом этапе олигомерный диол, удлинитель цепи и диэфир смешиваются
и реагируют при повышенной температуре («200°С) в присутствии титано-
вокислого катализатора с получением преполимера. Высокая температура
служит для проведения реакции и её завершения, а также для удаления
низкокипящего побочного продукта. Полимеры синтезируют при темпе-
ратуре до 250°С и пониженном до величины менее 1 мм. рт. ст. давлении,
что вызывает вторичную переэтерификацию. Удлинитель цепи удаляется
как побочный продукт на этой второй стадии. Температура должна сохра-
ниться ниже 260°С для предотвращения существенной деструкции [18].
Завершение реакции отслеживается по вязкости реакционной смеси. Как
правило, в результате такой процедуры получаются молекулярные массы
приблизительно 25 000 г/моль.
II. Синтез термопластичных эластомеров -* V 575
Для формирования амидных связей и получения сополиамидов можно
использовать различные пары функциональных групп [19]. Они включают
реакции между карбоновыми кислотами и диаминами, хлорангидридами
и диаминами, карбоновыми кислотами и изоцианатами. Последняя пара
особенно удобна для получения сополиамидов с ароматическими жесткими
сегментами. Сополиамиды обычно производятся со сложноэфирными либо
амидными связями между амидным жёстким сегментом и мягким сегмен-
том. Для получения высокомолекулярного материала необходимы высокие
температуры.
Б. Анионная полимеризация: диен-стирольные
сополимеры (СБС)
Анионная полимеризация используется для получения
стирольных блок-сополимеров и позволяет получать полимеры с чрез-
вычайно узким молекулярно-весовым распределением, как блоков, так
и всего полимера в целом. Узкое МБР весьма удобно для фундаментальных
исследований полимеров, и это внесло большой вклад в изучение анионной
полимеризации, что намного более важно, чем их коммерческая значимость.
Фактически, стирольные блок-сополимеры — единственные полимеры,
получаемые в больших количествах путём анионной полимеризации.
Чрезвычайно узкая полидисперсность весьма наглядно видна в следующем
выражении для индекса полидисперсности:
Zk = i+_
(«х+1)2
где nw — средневесовая степень полимеризации и пх — среднечисловая
степень полимеризации. При этом учитываются реакции, в которых не про-
исходит ни реакции передачи цепи, ни обрыва цепи. Это легко достигается
путём использования низких температур и чистых компонентов. Также
предполагается чрезвычайно высокая скорость инициирования относитель-
но скорости роста.
Трёхблочные сополимеры СБС или СИС производятся под высоким ва-
куумом, который служит для устранения кислорода и воды из реакционной
смеси [20]. Помимо общих особенностей, обсужденных в Главе 2, которые
применимы к любому анионному синтезу, важно произвести полидиеновый
блок с высоким содержанием 1,4-звеньев, чтобы температура стеклования
мягкой фазы была достаточно низкой. Это обычно достигается путём исполь-
зования относительно неполярного растворителя, например циклогексана
или толуола, который содействует получению 1,4-структуры. СЭБС полу-
чается путём гидрирования СБС.
Для получения диен-стирольных трёхблочных сополимеров обычно
могут использоваться три метода анионной полимеризации: трёхстадийный
576 -'V- Глава 13. Термопластичные эластомеры
процесс с монофункциональным инициатором, двухстадийный процесс
с монофункциональным инициатором и дифункциональным связующим
агентом, и двухстадийный процесс с дифункциональным инициатором [21].
Во всех случаях лучшими инициаторами служат литийорганические соеди-
нения. В первом процессе сначала полимеризуется стирол, за ним следует
бутадиен, и затем добавляется дополнительное количество стирола для
получения трёхблочного полимера. Бутадиен быстро инициируется,
будучи добавлен к первичным стирольным блокам; однако необходимо
небольшое количество полярного растворителя, чтобы инициировать ани-
онную полимеризацию конечного стирола. Во втором процессе стирольные
и диеновые блоки полимеризуются раздельно, затем объединяются с помо-
щью бифункционального связующего агента, в результате чего образуются
симметричные трёхблочники. Ключевое значение в этом методе имеет
точное управление стехиометрией таким образом, чтобы не формировались
двухблочники. Даже небольшое количество двухблочников чрезвычайно
неблагоприятно влияет на механические свойства. Наконец, может исполь-
зоваться бифункциональный инициатор, чтобы инициировать диеновый
блок, с последующим образованием стирольных концевых блоков. Этот
метод несовершенен вследствие трудности подбора подходящего бифунк-
ционального инициатора.
В. Каталитическая полимеризация
Этилен-пропиленовые сополимерные ТПЭ и другие этилен-
а-олефиновые сополимеры изготовляются на катализаторах Циглера-Натта
и на металлоценах (МЦ). Полимеризация на катализаторах Циглер-Натта
представляет собой реакции, катализируемые смесью металлорганических
галоидных соединений, например, А1 (С2Н5)2С1 и соли переходного метал-
ла, например, TiCl4. Эта полимеризация обычно даёт кристаллизующийся
стереоспецифический полимер. Например, в сополимерах пропилена с
а-олефинами, полипропиленовые кристаллиты являются жёсткой фазой для
ТПЭ. Длинные блоки одного компонента, необходимые для разделения фаз,
являются результатом соотношений специфической реакционноспособно-
сти компонентов. Несмотря на промышленную значимость полимеризации
Циглер-Натта, её кинетика не вполне ясна. Это связано как с частой гетеро-
генностью этой реакции, так и с возможностью множественных механизмов.
Полимеризация Циглер-Натта обсуждена более подробно в Главе 2. Смеси
этилен-пропиленовых (ЭП) сополимеров с изотактическим полипропиленом
часто получаются в процессе двухступенчатой реакции. Первый реактор
содержит только мономер пропилена, в то время как второй реактор содер-
жит и пропилен, и этилен. Другие смеси изготовляются путём смешения
чистых компонентов друг с другом в расплаве. Условия смешения оказывают
большое влияние на конечные свойства. Для регулирования характеристик
II. Синтез термопластичных эластомеров 577
материала могут использоваться компатибилизаторы, которыми зачастую
являются трёхблочные сополимеры.
Металлоценовая полимеризация имеет характеристики, подобные поли-
меризации Циглер-Натта, но, как можно видеть из названия, катализаторы
весьма различны. МЦ-катализаторы имеют два общих свойства, которые
приводят к получению стереоспе-
цифических продуктов: во-первых,
катализатор является твёрдым, во-
вторых, хиральным. Такие особен-
ности являются следствием наличия
катиона металла, лежащего между
двумя отрицательно заряженными
циклопентадиенильными анионами
(сэндвич-структура). Рисунок3 де-
монстрирует типичный МЦ-катали-
затор — 1,Г-этиленди-Т|5-инденил-
цирконий дихлорид. Модификация
этой структуры может состоять в
том, чтобы заменить один из цикло-
пентадиенильных анионов кольцом,
содержащим атом азота, и затем
ограничить это кольцо, связав азот
через связывающий атом, обычно
кремний. Так же, как в полимериза-
ции на катализаторах Циглер-Натта,
требуется соединение с лабильными
группами алканов; одно из типично
используемых соединений — мети-
лалюмоксан. Недостатком процесса
Рис. 3. Структура 1, Г-этиленди-Т|5-ин-
денилциркония дихлорида, обычного
металлоценового катализатора. Показан
только один энантиомер (зеркальный
изомер, антипод); оба энантиомера дают
изотактический продукт для этого конк-
ретного катализатора.
является то, что, количество требуемого алюминия в МЦ-катализаторах на-
много выше, чем в катализаторах Циглер-Натта. Несмотря на более высокую
стоимость, МЦ-полимеризация имеет свою долю на рынке из-за возможности
лучше контролировать число дефектов, что, в свою очередь, может использо-
ваться для регулирования свойств сополимера. Так же, как и полимеризация
на катализаторах Циглер-Натта, МЦ-полимеризация может использоваться
для получения нестереоспецифических полимеров из-за изменений в молеку-
лярно-весовом распределении и в реакционноспособности относительно других
типов полимеризации.
Г. Свободно-радикальная сополимеризация
Свободно-радикальная сополимеризация применяется для
получения иономеров, которые используются в качестве ТПЭ в промышлен-
578 “* V Глава 13. Термопластичные эластомеры
них масштабах. Имеются два типа иономерных ТПЭ: сополимеры этилена
и метакриловой кислоты, и сополимеры этилена и акриловой кислоты.
Мольная доля кислотного мономера обычно составляет 5% или меньше.
Разница в свойствах между двумя типами сополимеров невелика; сополи-
меры этилена и метакриловой кислоты являются несколько более стойкими
к образованию ангидридов, которые могут сшивать полимер. При обычном
методе получения используют высокое давление («1500 атм) и высокие
температуры («130°С), подобно методу, используемому для полиэтилена
низкой плотности. В отличие от полимеризации на катализаторах Циглер-
Натта, кинетика процесса хорошо известна (см. Главу 2). Катион нейтра-
лизации после синтеза — водород; однако в таком материале не происходит
разделение фаз на агрегаты наноразмера, необходимое для повышения
прочности ТПЭ. Поэтому в промышленности применяется нейтрализация
солью соответствующего металла. Полимер и соль смешиваются друг с другом
в экструдере или на вальцах [22]. В материалах промышленного значения
не все водородные атомы заменены катионами металла, иначе получается
материал со слишком высокой вязкостью; обычно степень нейтрализации
в большинстве иономеров составляет около 50%.
Д. Молекулярный вес и структура цепей
Для некоторых термопластичных эластомеров очень трудно
определять молекулярный вес и микроструктуру цепей. Почти все методы
определения молекулярного веса основаны на возможности растворения
полимера в растворителе таким способом, чтобы полимерные цепи вели себя
индивидуально. Так как ТПЭ обычно составляются из двух разнородных
компонентов, хороший растворитель для одного из компонентов может быть
недостаточно хорошим для другого компонента, что ведет к соединению
частей однородных компонентов от различных цепочек. Следовательно,
молекулярный вес является скорее суммарным, чем истинным. Даже если
эта проблема преодолевается, методы, при которых измеряют молекулярный
вес относительно стандарта, типа гельпроникающей хроматографии (GPC,
ГПХ), не могут быть использованы для определения истинного молекуляр-
ного веса, потому что константа пропорциональности зависит от состава
сополимера.
Для полиуретанов и сложных полиэфиров для контроля степени реакции
и конечного молекулярного веса может использоваться анализ концевых
групп, как титрованием, так и с помощью инфракрасной спектроскопии, если
стехиометрия реакции управляется должным образом. Этот метод работает
хорошо только в том случае, если молекулярный вес ниже «20,000 г/моль.
Мембранная осмометрия и рассеяние света могут давать достоверные значе-
ния молекулярного веса, если используется соответствующий растворитель;
однако, эти измерения занимают длительное время. Различные растворители
II. Синтез термопластичных эластомеров 579
должны быть проверены таким образом, чтобы можно было получить данные
без существенных отклонений, указывающих на агрегацию. Ультрацентри-
фугирование почти никогда не используется, потому что, помимо других про-
блем, для термопластичных эластомеров тета-растворитель не существует по
определению. Молекулярный вес, рассчитываемый по измерениям вязкости,
также будет зависеть от состава сополимера, так как радиус вращения (Rg)
полимера обычно является функцией состава. Методы фракционирования
также обычно непригодны, потому что эффективность фракционирования
зависит не только от распределения по молекулярным весам, но также и
от распределения по составу. Наиболее часто используется ГПХ (GPC), и
значения молекулярного веса обычно приводятся относительно стандарта
и для конкретного растворителя.
Определение молекулярного веса полиуретанов с использованием ГПХ
было подробно изучено в [23]. Были опробованы три различных метода
стандартизации молекулярного веса, и обнаружилось, что метод несколь-
ких детекторов, при котором используются включенные последовательно
ячейка для определения коэффициента преломления и УФ-спектрометр,
обеспечивает наиболее точные результаты. УФ-спектрометр был исполь-
зован для расчета в каждой точке хроматограммы изменений производной
коэффициента преломления по концентрации, что позволяет учесть эффект
изменения состава сополимера с молекулярным весом. Авторы нашли, что
использование нормальных стандартов полистирола для калибровки ГПХ,
дает верхний предел фактического молекулярного веса. Однако авторы не
решились называть эти значения абсолютными. Смежная проблема — мо-
лекулярный вес линейных преполимеров, если используется постадийная
схема реакции; процедура определения молекулярного веса и распределения
мягких и жестких сегментов блокированных по концам диизоцианатов,
с использованием ГПХ и метода двойного детектирования была недавно
представлена в [24].
Для трёхблочных сополимеров точные молекулярные веса измеряются
легко. Поскольку эти материалы получаются путём анионной полимериза-
ции, теоретический молекулярный вес обычно очень близок к фактическому.
В зависимости от метода полимеризации, блок чистого стирола или чистого
диена может быть удален и молекулярный вес может быть измерен до по-
лучения триблока. Наконец, стандарты молекулярных весов полистирола,
полибутадиена и полиизопрена имеются в продаже, из чего следует, что
измерения абсолютного молекулярного веса этих блоков легко выполнить,
используя ГПХ.
Микроструктура цепи оказывает очень большое влияние на свойства
ТПЭ. Как упомянуто ранее, СБС и СИС необходимо получать с высоким
содержанием 1,4-звеньев в эластичном блоке. Свойства ТПО также весьма
негативно реагируют на любые отклонений микроструктуры от идеальной
580 -'V Глава 13. Термопластичные эластомеры
«голова-хвост», чистой изотактической или синдиотактической микро-
структуры. Такие свойства, как тактичность (регулярность молекулярной
структуры), цис-транс изомеризация и содержание сополимеров, обычно
исследуют, используя ЯМР. Позиции и интенсивности пиков используются
для количественного определения микроструктуры с высокой степенью точ-
ности. Состав сополимера также может быть определен с применением ЯМР.
Чтобы определить характеристики микроструктуры некоторых полимеров
можно также использовать инфракрасную спектроскопию.
В сегментированных блок-сополимерах очень важной характеристикой
является средний молекулярный вес жестких и мягких сегментов. Число
исследований различных типов ТПЭ, в которых изучали этот параметр,
слишком велико, чтобы перечислить их здесь, хотя анионные блок-сопо-
лимеры заслуживают особого упоминания из-за их важности в прояснении
фундаментальной термодинамической информации. В общем, чем выше
молекулярная масса блоков, тем более законченным будет разделение фаз.
Однако следует отметить, что полное разделение фаз не всегда желательно,
так как, в конечном счете, когда разделение фаз станет более полным, по-
низится прочность. Еще одним параметром, который особенно важен как
в сегментированных блок-сополимерах, так и в ТПО, является распределение
(в противоположность среднему числу) длин жестких и мягких сегментов.
В ТПО жесткими сегментами обычно являются кристаллические домены про-
пилена или этилена, и изучение распределения кристаллитов по размерам —
важная задача, и не только для ТПЭ.
Для блок-сополимеров ступенчатой полимеризации характеристика
распределения по размерам мягких сегментов довольно тривиальна, так
как полностью сформированный мягкий сегмент обычно является одним из
компонентов, вступивших в реакцию. Жесткий сегмент формируется в ходе
полимеризации, и требуются более сложные подходы для его измерения.
Для изучения влияния распределения длин жестких сегментов применялись
два подхода: первый состоит в том, чтобы синтезировать жёсткие сегменты
с известным распределением длин [25], а второй — в попытках измерить
длины жестких сегментов. Конечно, это необходимо для всех коммерческих
материалов. Для определения распределения длины сегмента использовал-
ся ряд экспериментальных методов. Один из методов состоит в том, чтобы
химически расщепить структуру в точке, где жесткий и мягкий сегменты со-
единяются друг с другом, и анализировать полученные фракции, используя
гельпроникающую хроматографию или высокоэффективную жидкостную
хроматографию (ВЭЖХ, HPLC) [26-29].
Для определения длины жёсткого сегмента в полиуретановых цепочках
МДИ-ПТМО, удлинённых этилендиамином, использовались 13С ЯМР [30]
и масс-спектроскопия [31]. Для получения распределения молекулярного веса
жёсткого сегмента при идеальных и неидеальных условиях синтеза использо-
валось моделирование процессов Маркова методом Монте-Карло [32-37].
III. Морфология термопластичных эластомеров -* V 581
III. МОРФОЛОГИЯ ТЕРМОПЛАСТИЧНЫХ ЭЛАСТОМЕРОВ
А. Общие характеристики
Несмотря на широкое разнообразие химических структур
двухфазных термопластичных эластомеров, число вариаций основной
морфологии в коммерчески важных материалах на удивление мало. Если
мы предполагаем, что А — первоначально второстепенный (меньший)
компонент и В — главный компонент, тогда семь равновесных морфологий,
получающихся при увеличении содержания А, будут таковы [38]:
1. Изолированные сферы А в непрерывной матрице В;
2. Гексагонально упакованные изолированные цилиндры А в непрерыв-
ной матрице В;
3. Чередование ламелей А и В;
4. Гексагонально упакованные изолированные цилиндры В в непрерыв-
ной матрице А;
5. Изолированные сферы В в непрерывной матрице А.
Другие, более сложные морфологии могут образовываться в блок-сопо-
лимерах анионного синтеза; полный обзор этой темы лежит за пределами
этой главы, и заинтересованный читатель может обратиться к обзорам [39,
40] и монографиям [41,42] на эту тему. Электронные микрофотографии для
каждой из фаз блочного полимера СБС показаны на рис. 4-6. Однако, мор-
фология в реальных материалах не совсем такая, как на микрофотографиях.
Ранее была определена общая морфология (сферы, цилиндры, и т.д.), в тоже
время остаются специфические вопросы относительно формы доменов, их
размеров и расположения в пространстве. Более детально с этими вопросами
мы разберёмся в этой главе несколько позже.
Морфология оказывает большое влияние на физические свойства. Мягкая
фаза обычно является протяженной средой и обеспечивает эластомерное по-
ведение материала. В материалах, которые имеют кристаллические жесткие
сегменты, типа сополиэфиров и полиуретанов, обе фазы по существу непре-
рывны. Термопластичные эластомеры почти никогда не имеют мягкие сегменты
как изолированную фазу и жесткие сегменты в качестве непрерывной среды.
Наиболее общая морфология для ТПЭ показана на рис. 7 для триблочных
сополимеров СБС. Изолированная жесткая фаза действует как межмолеку-
лярные точки связи для эластомерной мягкой фазы. Обычно изолированные
жесткие домены имеют размеры 1-20 нм. Способность кристаллизоваться резко
изменяет морфологию; кристаллические области формируют прямоугольные
тонкие слои материала, называемые ламелями и следовательно не находятся
в изолированных доменах, а формируют большей частью непрерывную струк-
туру. Как во всех полимерах, ламелли могут организовываться в сферолиты,
немного удивительно, что сегментационная природа ТПЭ не препятствует
формированию этих больших сверхструктур.
582 -’V Глава 13. Термопластичные эластомеры
Поверхность раздела между жестким и мягким сегментом часто рассмат-
ривается как отдельная фаза. Влияние поверхности раздела на свойства
ТПЭ считается очень важным, хотя это и не вполне понятно. Более вы-
сокая стоимость ТПЭ относительно обычных полимеров обосновывается
различными способами для разных материалов, но, в общем, есть свойство,
которое оправдывает дополнительные затраты - это прочность. Для раз-
рушения ТПЭ требуется много энергии. Высокая прочность достигается,
в частности, путем протекания в материале процессов снятия напряжений,
и в ТПЭ большинство этих процессов происходят на поверхности раздела
жесткой и мягкой фаз, наличие которой приводит к отклонению в развитии
трещин или их разветвлению [43]. Адгезия между поверхностью раздела и
полимерной матрицей особенно важна для повышения прочности в смесях
ЭП-сополимеров, так как разрушение в смесях зачастую происходит из-за
недостаточной межфазной адгезии.
Рис. 4. ТЭМ сферических бутадиеновых доменов в плёнке СБС (80% стирола), полу-
ченной из раствора в толуоле. Такие же картины получились как при нор-
мальном, так и при параллельном сечении, что подтверждает периодичность
бутадиеновых доменов. Мацуо (Matsuo) [221]).
Как было показано ранее, кинетические соображения играют чрезвычай-
но важную роль в морфологии ТПЭ. Прогнозирование точной морфологии
любого ТПЭ без определённого знания истории образца невозможно. Чтобы
получить материал с морфологией, приближённой к равновесной, обра-
III. Морфология термопластичных эластомеров 583
зец ТПЭ можно отжечь при температуре выше температуры размягчения
жесткого сегмента, но ниже температуры перехода порядок-беспорядок.
Полидисперсность по длинам блоков может также давать морфологию,
которая существенно отклоняется от основной. Наконец, многие про.-
мышленно произведенные детали имеют ориентированные микрообласти,
остающиеся после переработки. Последняя часть этого раздела содержит
введение в аналитические методы, обычно используемые для характеристики
морфологии ТПЭ. Краткое описание каждого метода дается с акцентом на
характеристики, важные для ТПЭ. Важные примеры каждого метода также
обсуждены, так что читатель может понять, как каждый метод используется
для характеристики морфологии ТПЭ.
Рис. 5. ТЭМ цилиндрических микродоменов в экструдированном и отожжённом
образце СБС, микрофотография сделана перпендикулярно направлению
экструзии. Электронная микрофотография параллельно направлению экс-
трузии показала слоистую структуру. Длугош (Dlugosz) и др. [222]).
Б. Изучение морфологии
1. Трансмиссионная (просвечивающая) электронная
микроскопия (ТЕМ, ТЭМ)
В трансмиссионной электронной микроскопии образец бомбардируется
электронами, и количество электронов, прошедших через образец, пропорци-
584 -•и- Глава 13. Термопластичные эластомеры
онально его толщине и электронной плотности; оно измеряется как функция
положения. Если электронные плотности не идентичны для каждой фазы
ТПЭ, число электронов, прошедших через две фазы может быть различено
и, следовательно, будет создан контраст. ТЭМ может использоваться на
образцах с чрезвычайно хорошо упорядоченной морфологией, где масштаб
длины вариаций электронной плотности больше, чем разрешающая способ-
ность метода. Если морфология не упорядоченная, то среднее число доменов
одного типа, через которые проходит одиночный электрон, не изменяется
с положением. Можно уменьшать толщину пленки, но существует практи-
ческий предел уменьшения толщины. Так как разность электронной плот-
ности между атомами, которые составляют полимеры, обычно очень мала,
то почти всегда добавляется вещество с высокой электронной плотностью,
которое предпочтительно связывается с одной из двух фаз или мигрирует к
ней. Типичные составы, используемые для такого протравливания — OsO4
и RuO4.
Рис. 6. ТЭМ ламеллярных доменов в плёнке СБС (40% стирола), полученной из
раствора в циклогексане: перпендикулярное сечение (а); параллельное се-
чение с ламеллярными слоями, ориентированными своими поверхностями
параллельно поверхности плёнки (б).
Стирол-диеновые блок-сополимеры представляют собой оптималь-
ный материал для ТЭМ. Если они подготовлены должным образом, то
эти материалы могут быть почти на 100% сепарированы по фазам с по-
мощью контрастирующих агентов, которые локализуются почти исклю-
чительно в диеновой фазе. Морфология обычно хорошо упорядочена,
и индивидуальные размеры доменов достаточно велики. Примеры элект-
ронных микрофотографий СБС блок-сополимеров показаны на рис. 4-6.
ТЭМ — вероятно наиболее важный метод характеристики, используемый для
определения морфологии в блоке, так что в большинстве статей по этой теме
III. Морфология термопластичных эластомеров -*v- 585
публикуются ТЭМ-микрофотографии именно этих систем. Обзорные статьи
и книги, на которые мы сслылались ранее, содержат тщательное описание
ТЭМ-исследований блок-сополимеров.
Рис. 7. Фазовое разделение в триблочных сополимерах СБС, обычно используемых
как ТПЭ. Изолированные твёрдые стирольные домены образуют жёсткую
фазу, которая выполняет роль как межмолекулярной точки связи, так и
наполнителя. Непрерывная фаза бутадиена придаёт полимеру эластомерные
характеристики.
Изображения сополиэфиров [44] и полиуретанов [45] также были полу-
чены. Эти два материала много лет не были в фокусе исследований с помощью
электронной микроскопией из-за трудностей в исключении артефактов.
Именно по этой причине нужно рассматривать опубликованные результаты
по этим системам критически. За последние 5 лет вышел ряд статей Вини
(Winey) и др. [46-48], описывающих результаты исследования иономеров
с помощью ТЭМ. Для обеспечения необходимого контраста применяются
атомы тяжелого металла, и для получения изображения этих систем исполь-
зуется ТЭМ с высокой разрешающей способностью. Было найдено намного
более широкое разнообразие морфологии совокупности ионов, включая
сферы и вакуоли, чем предполагалось.
2. Инфракрасная и Рамановская спектроскопия
В инфракрасной спектроскопии луч инфракрасного света
пропускается через образец, и свет поглощается, если его частота такая же,
как частота собственных колебаний. Этот метод чувствителен к природе мо-
586 -’V- Глава 13. Термопластичные эластомеры
лекулярной связи между атомами; закон Бира (Beer) связывает поглощение
(4) колебаний и их абсолютную концентрацию:
А = Etc (3)
где £ — коэффициент поглощения, t — длина пути, и с — концентрация.
Как показано ранее, инфракрасная спектроскопия может использо-
ваться для исследования микроструктуры цепей. Помимо химической
характеристики, инфракрасная спектроскопия может помогать в описании
таких взаимодействий между цепями, как водородная связь или кристал-
лизация. Водородная связь - вторичная связь водородных атомов с атомом,
содержащим неспаренные электроны. Водородные связи обычно имеют
прочность порядка 3 ккал/моль, что на полпорядка меньше, чем прочность
ковалентной связи, но больше, чем простых Ван-дер-Ваальсовских вза-
имодействий [49]. Водородная связь чрезвычайно важна в полиуретанах
и сополиамидах и возникает между водородом уретана или амида и кис-
лородом карбонила. Так как водородная связь имеет место, прежде всего,
между жесткими сегментами, это обеспечивает сильную движущую силу для
фазового разделения и кристаллизации жёсткого сегмента.
Таблица 1 Идентификация полос функциональных групп в ТПЭ, содержащих
водородные связи.
Материал Свободная Связанная Ссылки
С=О 1720 см"1 1685 см-1 59,61
Полиуретан: гексаметилен диизоцианат/бутандиол 1700 см-1
N-H 3440 см-’ 3320 см-1
С=О 1740 см"1 1700 см’1 55
Полиуретан: 2,6 толуилен диизоцианат/бутандиол/ПТМО
N-H 3460 см"1 3300 см"1
С=О 1733 см"1 1703 см-1 53,56
Полиуретан: МДИ/бутандиол/ПТМО
N-H 3420см-1 3320 см-1
С=О 1680см-1 1636 см-1 60,62
Полиамид: Найлон 11 1645 см-1
N-H 3450 см-1 3300 см-1
III. Морфология термопластичных эластомеров -* v 587
Водородная связь вызывает сдвиг к более низкому волновому числу (более
низкой энергии) в колебании связей, как в водородно-донорной, так и в водород-
но-акцепторной группах. При этом прочность первичных связей уменьшается
из-за образования водородной связи. В типичном термопластичном эласто-
мере, который может иметь водородные связи, колебания функциональных
групп, участвующих в водородной связи, разбиваются на полосы, называющи-
еся связанной и свободной. В Таблице 1 указаны волновые числа, которые были
определены как связанные и свободные полосы для С=О и N-Н колебаний для
ряда полимеров. Имеется маленькое, но значимое влияние типа полимера на
точное волновое число. Две полосы некоторых С=О связей относятся к упоря-
доченным (кристаллическим) и беспорядочным состояниям.
Так как водородные связи являются чрезвычайно важными, а исследования
с помощью инфракрасной спектроскопии относительно недороги и просты,
по этой части было опубликовано множество статей. Много исследований
посвящалось попыткам связать количество водородных связей со степенью
фазового разделения, с использованием закона Бира. Харткок (Harthcock) [50]
исследовал карбонильные группы в ориентированных модельных полиурета-
нах с монодисперсными длинами жёстких сегментов. Детальная морфологиче-
ская модель, которая связывает расположение жестких сегментов с частотой
инфракрасных колебаний, показана на рис. 8. Подобное исследование было
предпринято Луо (Luo) и др. [51] для полиуретанмочевин. Также изучались
изменения в водородных связях из-за варьирования структур жесткого [52]
и мягкого сегментов [53]. Метод Фурье-спектроскопии (FTIR) помог в уста-
новлении взаимосвязи структура — механические свойства для сегментиро-
ванных полиуретанов [54]. Термическое поведение водородной связи детально
исследовалось в работах [55-57]. Ряд статей, опубликованных Пэйнтером
и Коулмэном (Painter & Coleman) [58-62], о полиамидах и полиуретанах пока-
зывает, что многие из исследований термического поведения были основаны на
неправильном представлении относительно природы водородных связей. Эти
авторы показали, что количественный анализ N-Н области был невозможен
из-за большой разницы между коэффициентами затухания для связанных
и свободных полос, наряду с большим изменением коэффициента затухания
связанных N-Н полос как в функции температуры. Простой анализ [55],
обычно используемый для описания зависимости коэффициента затухания от
температуры не мог быть использован, потому что частота колебаний N-Н свя-
зей сдвигалась, что показывало, что прочность водородных связей изменялась.
Поэтому читателю следует быть очень осторожным при интерпретации статей,
в которых количественно анализируется расширенная N-Н область.
Рамановская спектроскопия обычно даёт дополнительную информацию
к инфракрасной спектроскопии, но из-за исторически сложившихся экспери-
ментальных трудностей в выполнении этих опытов Рамановские исследования
ТПЭ проводились много реже, чем инфракрасная спектроскопия. За последние
10 лет в связи с разработкой мощных лазеров и систем CCD-детектирования,
Глава 13. Термопластичные эластомеры
588
Рамановские эксперименты стали намного более простыми, хотя стоимость
Рамановского спектрометра все еще намного больше, чем Фурье-спектрометра.
Рамановская спектроскопия имеет два видимых преимущества перед инфра-
красной спектроскопией: во-первых, сигнал может измеряться дистанционно
с помощью волоконной оптики, что делает возможным использование Раманов-
ской спектроскопии для управления технологическим процессом, во-вторых,
излучение лазера можно сфокусировать на пятно размером приблизительно
1 микрон, что позволяет исследователям отобразить очень маленькие площади
поперечного сечения. Пространственно-разрешающая способность микро-Ра-
мановской спектроскопии была использована, чтобы исследовать композици-
онную неоднородность в полиуретанах [63].
1701 см 1 (упорядоченная)
1732 см 1 (свободная)
1708 см-1/1716 см-1
(«дальняя»
разупорядоченность)
Межфазная
1716 см-1-1724 см-1
(«ближняя»
1732 см-1 (свободная)
Рис. 8. Морфологическая модель, разработанная Харткоком (Harthcock), которая
показывает соотнесение к локальному окружению волновых чисел колебаний в
расширенной С=О области поглощённой интенсивности (Harthcock [50])
Другое применение инфракрасной спектроскопии использует линейно
поляризованное инфракрасное излучение, чтобы получить информацию
об ориентированных образцах. Такой эксперимент называется измерением
III. Морфология термопластичных эластомеров -* v- 589
инфракрасного дихроизма. Абсорбция будет максимальная, когда вектор
электрического поля и вектор дипольного момента имеют одно направление.
Абсорбция будет нулевая, когда они перпендикулярны. Рассматривается
только одноосная ориентация, потому что такая ситуация отражена в лите-
ратуре. R, дихроический коэффициент, определяется следующим образом:
я=А (4)
А
где: Лп — абсорбция, когда направление ориентации и направление поля-
ризации параллельны друг другу и А± — абсорбция, когда два направления
перпендикулярны. Относящаяся к этому геометрия показана на рис. 9.
Пусть/(0) представляет функцию распределения ориентации, то есть,/(0)
dQ — часть оси цепи, находящаяся в пределах dQ в окрестности угла 0. Эта
функция может быть разложена в ряд полиномов Лежандра:
Л0)=^Ё^^(рЛсо50)(рЛсо50) (5)
Следует рассматривать только многочлены с четными номерами, так как
предположение об одноосной симметрии означает, что средние величины
нечетных степеней cos0 равны нулю. Исходя из рис. 9 после довольно об-
ширных математических преобразований можно показать, что:
/Р( з(со?е(-1 (я-1)(яо+2)
(рг(»»е))=—-—=(л+2)(яо_() (в)
Rq определяется как 2cot2a, где a — угол между вектором момента диполя
и осью цепи. <P2(cos0)) — однопараметрическая мера ориентации, обычно
приводимая в литературе для одноосно ориентированных образцов, a - функ-
ция рассматриваемых колебаний, известная для ряда колебаний, исполь-
зуемых при изучении термопластичных эластомеров [64]. Инфракрасный
дихроизм может использоваться для того, чтобы следить за ориентацией
каждой фазы независимо, если для каждого домена могут найтись непере-
крывающиеся полосы, для которых известно а. В полиуретанах, например,
связанная полоса карбонила обычно используется, чтобы контролировать
ориентацию жёсткого сегмента, в то время как свободная полоса карбонила
или ориентированные полосы СН используются для отслеживания ори-
ентации мягких сегментов [65-67]. Ориентация жёсткого сегмента почти
всегда меньше чем ориентация мягкого сегмента при той же самой степени
вытяжки. В полукристаллических и более высоко упорядоченных поли-
уретанах жесткие сегменты вначале располагаются поперёк направления
растяжения, а позже выстраиваются в направлении удлинения. Морфоло-
гически, при низких удлинениях радиальные лучи сферолитов ориенти-
590 -*и- Глава 13. Термопластичные эластомеры
руются в направлении растяжения, что означает, что оси цепей становятся
ориентированными перпендикулярно к этому направлению. При более
высоких удлинениях жёсткие сегменты удаляются из лучей сферолитов
и располагаются в направлении удлинения. В релаксационных эксперимен-
тах мягкие сегменты имеют тенденцию релаксировать очень быстро к почти
невозмущенному состоянию, в то время как твёрдые сегменты релаксируют
намного более медленно, особенно при высоких напряжениях. Поперечная
ориентация жестких сегментов обратима, в то время как удалённые из сфе-
ролитов жесткие домены не могут быть возвращены к их первоначальному
окружению без нагрева образца. Были сделаны измерения дихроизма для
4ГТ-ПТМО сополиэфиров, которые показали ту же самую отрицательную
ориентацию при низких удлинениях, за которой следовала положительная
ориентация при более высоких удлинениях [68]. Измерения дихроизма
при более высоких температурах в МД И-БД полиуретанах показали изме-
нения в поведении; начало положительной ориентации проявляется при
намного меньших удлинениях, а ориентация жёсткого сегмента при том
же коэффициенте вытяжки становится намного больше. Эти результаты
интерпретировались как ослабление когезии доменов жестких сегментов
при повышенных температурах [69]. Поведение при растяжении меняется
также, когда полиуретаны подвергаются гидролизу [70] или пластифици-
руются [71].
Рис. 9. Геометрия одноосно растянутого образца.
3. Широкоугловое рентгеновское рассеяние (ШУРР, WAXS)
Когда электронная плотность в образце изменяется пери-
одически через определенное расстояние и это расстояние имеет тот же
самый порядок величин, что и длина волны рентгеновских лучей, рентге-
новские лучи будут рассеиваться образцом когерентно. Соответствующие
III. Морфология термопластичных эластомеров —’V 591
этой периодичности пик или пики будут появляться на картине рассеяния
(рентгенограмме), если периодичность имеет место достаточное количест-
во раз. Ширина и число пиков будут пропорциональны отклонению этого
повторяющегося расстояния от его среднего значения, а также зависеть от
количества повторений этой периодичности. ШУРР позволяет измерять
вариации электронной плотности на расстояниях порядка ангстремов,
что соответствует межатомным расстояниям. Поэтому ШУРР использу-
ется для изучения термопластичных эластомеров с кристаллическими
жесткими или мягкими сегментами.
Фундаментальное соотношение, которое связывает повторяющееся
расстояние изменений электронной плотности и угол рассеяния — закон
Брэгга (Bragg) [72]
nk = 2dsinQ (7)
где п — целое число, X — длина волны излучения, 20 — угол между пада-
ющим и исходящим излучением, и d — повторяющееся расстояние между
кристаллографическими плоскостями. Дальнейшие подробности относи-
тельно кристаллографического анализа полимеров весьма усложнены и в
этой главе не приводятся. Заинтересованный читатель может справиться об
этом в работах Александера (Alexander) [73].
Детальные кристаллографические исследования проводились для сопо-
лиэфиров, сополиамидов и полиуретанов. Что касается первого, жёсткие
кристаллиты 4ГТ поли(бутиленгликольтерефталата)в сополиэфирах иден-
тичны жёстким кристаллитам гомополимера 4ГТ в недеформированном
состоянии. В процессе растяжения ТПЭ в его жестком сегменте обнару-
живаются различные кристаллические формы из-за образования транс-
конфигурации метиленовых последовательностей [74, 75]. С помощью
электронной микроскопии [76] и малоуглового рентгеновского рассеяния
(МУРР) [77] было показано, что толщина ламеллей меньше, чем средняя
длина жёсткого сегмента. Это означает, что должно иметь место складывание
цепей. Хотя элементарная ячейка нечувствительна к доле мягких сегментов
или их составу, общее количество (доля) кристаллитов в жёстких сегментах
уменьшается при увеличении доли мягких сегментов, как показано на рис.
10. Однако, доля единиц 4ГТ, являющихся кристаллическими, увеличи-
вается при возрастании доли мягких сегментов [78]. Эти материалы также
демонстрируют ориентационную кристаллизацию при растяжении [79].
При исследованиях методом ШУРР было установлено, что сополиами-
ды имеют многие особенности, идентичные особенностям сополиэфиров.
Кристаллическая структура жестких сегментов (нейлона) такая же, как
в гомополимере [80]. Возможность появления складчатых кристаллов
зависит от длины жесткого сегмента. Для малых длин постулируется
модель бахромчатых мицелл, в то время как при больших длинах цепи
складываются [81-83]. При достаточно низких температурах (нижеО°С),
592 “’V Глава 13. Термопластичные эластомеры
мягкий сегмент, если это ПТМО поли(тетраметиленоксид), кристаллизу-
ется [84]. Наконец, сегменты ПТМО проявляют способность к ориента-
ционной кристаллизации, которая обратима, если образец нагреть чуть
выше комнатной температуры. Кристаллизация достаточно велика, так
что имеет место остаточная деформация [85].
Рис. 10. Взаимосвязь весовой доли кристаллитов, Wc, и весовой доли мягких сегмен-
тов, Ws, в сополиэфире 4ГТ-ПТМО. А — получено измерениями плотнос-
ти, • — получено методом ШУРР; незакрашенные символы — получено
методом ДСК.
Особенности кристаллизации как жёстких, так и мягких сегментов
в полиуретанах аналогичны особенностям кристаллизации в сополиэфирах
и сополиамидах. Существенная разница между полиуретанами и предыду-
щими двумя материалами состоит в том, что не для всех товарных ТПУ имеет
место кристалличность жёсткого сегмента; для полиуретанов на базе МДИ
характерна кристалличность, в то время как для полиуретанов на базе ТДИ —
нет. В жёстких сегментах МДИ-БД в неориентированном состоянии были об-
наружены две кристаллические структуры, отличные от структуры, обнару-
женной в ориентированном состоянии [86,87]. В других исследованиях было
установлено влияние структуры удлинителя цепи на кристалличность [88].
Различия между полиуретанами, в которых жёсткие сегменты имеют крис-
таллиты со складчатыми или вытянутыми цепями, интенсивно изучались
Эйзенбахом (Eisenbach) и др. [89].
В сополимерах изотактического пропилена (и-ПП, iPP) с а-олефи-
нами также обнаружены дифракционные максимумы, характерные для
кристаллитов и-ПП. Можно полагать, что увеличение содержания а-оле-
фина уменьшает процент кристалличности. При смешении сополимера
с и-ПП гомополимером, имеет место сокристаллизация; подобное явление —
III. Морфология термопластичных эластомеров -»v- 593
редкость среди полимеров. Как полагают, совместная кристаллизация спо-
собствует улучшению механических свойств, обнаруженному у такой смеси
[90]. В противоположность гомополимеру в этих смесях не обнаружено
крупных сферолитов, а форма кристаллов скорее моноклинная, чем
смектическая [91].
4. Малоугловое рентгеновское рассеяние (МУРР, SAXS)
ШУРР позволяет измерять рассеяние рентгеновских лучей
на расстояниях относительно далеких от первичного луча, в то время как
МУРР позволяет измерять рассеяние очень близко к первичному лучу.
МУРР дает сведения о длинах скорее порядка нанометров, чем ангстремов,
и этот метод используется для исследования двухфазной морфологии ТПЭ.
Другими словами, ШУРР характеризует внутрифазную, а МУРР межфаз-
ную морфологию.
Для того чтобы делать измерения близко к первичному лучу, необходимы
точные системы коллимации. Кроме того, рассеяние под малыми углами
обычно очень слабо. Используется и точечная, и линейная коллимация;
в системах блочной коллимации требуются математические преобразования
данных от линейной к точечной коллимации. В течение последних десяти-
летий разработка и развитие источников синхротронного излучения и улуч-
шенных детекторов привела к быстрому росту количества экспериментов по
рассеиванию, производимых в реальном времени.
Интерпретация кривых МУРР более трудна, чем кривых ШУРР.
В ШУРР атомы настолько малы, что могут рассматриваться как точечные
рассеиватели. В МУРР размеры индивидуальных доменов не являются
несущественными в масштабе длин волн рентгеновских лучей, и рассея-
ние ими надлежит учитывать. Рассеяние, связанное с индивидуальными
доменами, называется рассеянием, определяемым фактором формы,
в то время как рассеяние, связанное с пространственным расположением
доменов, называется рассеянием, определяемым фактором структуры.
Общее рассеяние может рассматриваться как результат рассеяния этих
двух видов тогда и только тогда, когда домены сферически-симметрич-
ны. Для сферически-симметричной двухфазной системы с однородными
электронными плотностями в каждой фазе и одной фазой, дискретно
распределённой в массе другой, интенсивность рассеяния может быть
представлена как [92]:
j^; = nVo2(pi-Po)202(g/?)5(g) (8)
где I(q) — интенсивность рассеяния при векторе рассеяния q(q = 4Ttsin0/k),
Ie(q) — рассеяние на единичном электроне, V— облученный объем, п — чис-
594 -»v- Глава 13. Термопластичные эластомеры
ленная плотность дискретных доменов, (p^Pq) — разность электронной
плотности между двумя фазами, ф(<?/?) — рассеяние, определяемое фактором
формы, и рассеяние, определяемое фактором структуры. Для ряда обычных
форм ф(#/?) было рассчитано [93].
5(^), вообще говоря, не является простой функцией, разве только
система очень хорошо упорядочена. При этом на рентгенограмме МУРР
должны появиться множественные пики. Относительное расположение
этих пиков может использоваться для экспериментального определения
плотности упаковки доменов. Множественные пики показывают только
ТПЭ, полученные методом анионной полимеризации, и, как правило,
только лабораторные образцы. Метод МУРР широко использовался при
исследованиях ТПЭ, описанных в литературе. Список работ, посвящён-
ных этим исследованиям, можно найти в ранее приведённых обзорных
статьях
Для коммерчески важных ТПЭ чаще получаются невыразительные рен-
тгенограммы; наиболее обычная рентгенограмма МУРР для ТПЭ это очень
широкий одиночный пик. Для вычисления расстояния между доменами
может использоваться закон Брэгга; конечно, эти результаты искажаются
рассеянием, связанным с фактором формы. Для анализа этих данных обычно
используют два количественных подхода. Один состоит в том, чтобы разра-
ботать морфологическую модель, рассчитать рентгенограмму рассеивания,
и изменять регулируемые морфологические параметры модели, пока предска-
занная рентгенограмма не совпадёт с экспериментальной. Второй подход —
разложить данные в ряд Фурье и подсчитать функцию радиального распре-
деления для электронной плотности. Описание этих подходов находится
вне контекста данного обзора; заинтересованному читателю стоит изучить
монографии по этой теме [94, 95].
Таблица 2 . Относительное положение пиков, определяемых фактором структуры,
для обычных случаев хорошо упорядоченной морфологии.
Структура Относительное положение пиков
Простые кубически упакованные сферы 1,4 2,4 3,4 4,4 5,4 6,4 8,4 9,...
Центрированные кубически упакованные сферы 1,^2,43,44,45,4 6,47,48,...
Гранецентрированные кубически упакованные сферы 1,1.155,1.633,1.915, 2,...
Ромбовидно упакованные сферы 1,1.633,1.915, 2.309, 3.416,...
Гексагонально упакованные цилиндры 1,43,44,47,49,...
Ламелли 1,2, 3, 4, 5,...
III. Морфология термопластичных эластомеров
595
iMv
МУРР может использоваться для исследования граничной области между
двумя фазами. Метод даёт однопараметрическую меру толщины межфазной
области, если допускается некоторый профиль распределения концентрации.
В двухфазной системе с резкими границами раздела, рассеяние под более
высокими углами будет выражаться (после удаления фона):
= 2л(Р1-р0)2£ (9)
где 5— полная площадь межфазной поверхности. Присутствие диффузной
поверхности раздела приводит к более быстрому уменьшению интенсивнос-
ти, чем предсказанная выше зависимость <?’4. Руланд (Ruland) [96] показал,
что уравнение (9) преобразуется, как показано ниже:
—>ooL eV*/ J
Функция Н2^) была рассчитана для сигмовидных и линейных [97]
градиентов концентрации. Из-за ошибок, связанных с определением фона,
использование МУРР для изучения свойств поверхности раздела должно
рассматриваться скорее как относительное, нежели абсолютное.
Исследования ТПЭ с применением МУРР, весьма многочисленны.
Поскольку информация МУРР часто неоднозначна, наиболее эффективно
проводить совместно с МУРР различные другие морфологические иссле-
дования, такие как электронная микроскопия или малоугловое рассеяние
нейтронов. Здесь мы рассмотрим лишь три интересных примера, чтобы дать
понять читателю, что дают эксперименты МУРР для ТПЭ.
Методом МУРР подробно исследовались деформированные триблочные
сополимеры СБС [98-101]. Послетого, как образцы отрелаксировали, были
получены дифрактограммы, показывающие, что деформация существенно
влияет на морфологию [102]. Использование синхротронного излучения
наряду с двумерными детекторами позволяет измерять интенсивность
в процессе вытяжки образца. В одном таком исследовании [103] в непрерыв-
ную фазу полибутадиена были внедрены короткие цилиндрики полистирола.
Было обнаружено, что деформация однородна в меридиональном направлении
до 3-х кратного удлинения, что соответствует точке перегиба кривой напряже-
ние-деформация. При увеличении коэффициента вытяжки до 4 цилиндры вы-
строились своими продольными осями параллельно направлению растяжения.
Вплоть до коэффициента вытяжки 8 цилиндрики не разрушались.
МУРР был использован для того, чтобы показать, что МД И-БД уретаны на
основе простых полиэфиров имеют меньшие межфазные слои, чем уретаны на
базе сложных полиэфиров [104]. Трудность правильного удаления фона под-
596 -*и- Глава 13. Термопластичные эластомеры
робно обсуждалась в [ 105,106], однако полученная толщина межфазного слоя,
имеющая профиль линейного градиента, была приблизительно вдвое больше
для сложноэфирных полиуретанов, нежели для полиуретанов на основе простых
полиэфиров, при использовании для расчета двух различных процедур. Абсо-
лютная величина чисел сомнительна, так как другие авторы нашли существенно
большие значения межфазных слоев для подобных материалов [107,108].
Малоугловое рентгеновское рассеяние использовалось для регистрации
зависящих от температуры морфологических изменений в сложных сополи-
эфирах. [109-111]. Для замедления кристаллизации жесткого сегмента, 4 ГТ
был заменен на политетраметиленизофталат (4ГИ). Было установлено, что,
как и в системах 4ГТ, скорость кристаллизации слабо влияет на морфологию
этих сополиэфиров. При фиксированной температуре Брэгговский период
увеличился при уменьшении содержания твёрдых сегментов, а большой пе-
риод примерно пропорционален обратной величине недоохлаждения. Отжиг
при температурах вблизи температуры плавления приводил к перестройке
морфологии за счет плавления дефектных кристаллитов и рекристаллиза-
ции с образованием более совершенных кристаллитов, что сопровождалось
необратимым увеличением Брэгговского периода.
5. Малоугловое рассеяние нейтронов (МУРН, SANS)
Единственное различие между МУРР и МУРН состоит в том,
что контраст, приводящий к рассеянию нейтронов, связан с изменениями
плотности рассеяния, а не электронной плотности. Плотность рассеяния
является функцией числа нуклонов (а не атомного номера!) и изменяется
сложным путём. Поскольку разность в плотности рассеяния между водо-
родом и дейтерием велика, то для создания контраста, необходимого для
применения МУРН, используется изотопное замещение. Возможно, что
наиболее известным использованием МУРН в полимерной науке была экспе-
риментальная проверка того факта, что полимерные цепи в блоке принимают
конформацию невозмущенных статистических клубков [112, ИЗ].
МУРН может использоваться для того, чтобы изучать те же самые объ-
екты, для которых применяется МУРР, то есть: размер домена, расстояние
между смежными доменами, межфазные размеры, ит.д., и во множестве
исследований МУРН использовали именно таким образом. Однако МУРН
имеет возможность, которой не имеет МУРР: если небольшая часть це-
пей в системе содержат дейтерий, а остальная — водород, или наоборот,
то возможно исследовать рассеивание от индивидуальных цепей. Поэтому в
двухфазных системах с изотопным замещением рассеяние будет формировать-
ся из двух источников: рассеяние цепей, как межцепное, так и от одиночных
цепей, и межфазный контраст. Подгоняя плотности рассеяния двух фаз путём
частичного маркирования одной или обеих фаз, возможно исключить рассеяние
из-за межфазного контраста (который дает информацию, подобную МУРР)
и изучать только рассеяние одиночной цепи. Так как степень смешения фаз
III. Морфология термопластичных эластомеров -»v- 597
несущественна (полагая, что никаких изменений объёма при смешении не
происходит) и состав чистых фаз известен для большинства ТПЭ, подгонка кон-
трастов осуществляется относительно просто. Были также разработаны методы
для выделения рассеяния одиночной цепи в системах, где контраст выровнен,
путём вычитания междоменного рассеяния, с использованием результатов либо
МУРР, либо МУРН непомеченных образцов [114].
Для двухфазной системы, где межфазное рассеяние было устранено,
и только одна фаза была частично помечена, интенсивность когерентного
рассеяния может быть представлена как:
(ДР)2тга0
Рт^А
ViVd(\-vd)NS{q)
(И)
где АР — разность плотности длины когерентного рассеяния нейтронов
между полностью водородным и полностью дейтериевым материалами,
Vj — объёмная доля помеченной фазы; vd — объёмная доля дейтериевого
материала в помеченной фазе; NA — число Авогадро, т(} и N— молекулярный
вес мономера и число повторяющихся звеньев, соответственно, и S(q) —
функция рассеяния одиночной цепи. Более полный обзор теоретических
и экспериментальных исследований с применением МУРН к полимерам
можно найти в монографиях на эту тему [115-117].
Влияние температуры и состава на конформацию цепей исследова-
лось для МДИ-БД-ППО полиуретанов [118], 4ГТ-ПТМО сополиэфиров (
4ГТ — полибутиленгликольтерефталат, ПТМО — политетраметиленоксид),
и МДИ-БД-ПТМО полиуретанов (МДИ — дифенилметилен-4,4’ диизоци-
анат, БД — бутандиол) [119-122]. При комнатной температуре Rg мягких
сегментов в ПУ ТПЭ приблизительно на 25% больше, чем в блоке, в то
время как для сополиэфиров увеличение составляет лишь «10%. Радиус
инерции мягкого сегмента уменьшается при повышении температуры
выше комнатной для всех материалов, кроме МДИ-БД-ПТМО в молярном
соотношении 7:6:1. Подтверждение смешения фаз было обнаружено при
достижении достаточно высоких значений температуры для всех материалов,
кроме 7:6:1-материала; это доказывается увеличением Rg мягкого сегмента
с ростом температуры. Измерения МУРН для 4ГТ-сегментов показывают,
что в сополиэфирах имеет место значительное складывание цепи, /^жёстких
сегментов в сополиэфирах резко возрастал с увеличением температуры, что
указывало на изменение числа складок цепи и/или степени фазового
разделения. В 7:6:1-полиуретане Rg жёстких сегментов уменьшался при
увеличении температуры; удовлетворительно объяснить это явление
авторы не смогли. Измерения размеров всей цепи в сополиэфирах пока-
зали, что цепь, первоначально сжатая, распрямляется при повышении
температуры.
598
—Глава 13. Термопластичные эластомеры
В ламеллярных стирол-диеновых двухблочных сополимерах исследо-
вания МУРН показали, что Rg сегмента сжимается на 70% от невозмущен-
ного значения параллельно поверхности раздела и расширяется до 160%
от невозмущенного значения перпендикулярно к поверхности раздела
[123, 124]. Эти значения были получены как для стирольных, так и для
диеновых блоков. Изучение растяжения СИС-блоксополимеров, имеющих
сферические стирольные домены, показало, что деформация в направлении
растяжения была больше, чем аффинная, а деформация перпендикулярно
к направлению растяжения — значительно меньше [125].
6. Ядерный магнитный резонанс (NMR, ЯМР)
Твердотельный ЯМР даёт возможность получать широкий
диапазон разнообразных характеристик ТПЭ, включая как статические, так
и динамические свойства, а также информацию об ориентации. Как было
упомянуто ранее, ЯМР может использоваться для получения информации
о микроструктуре цепей, такой как тактичность и последовательность
фрагментов. Применительно к ТПЭ ЯМР использовался для исследования
пространственных взаимодействий между атомами, а также относительной
подвижности отдельных сегментов. Маркирование дейтерием существенно
расширяет возможности ЯМР. При использовании импульсного режима
может изучаться время релаксации сегментов в различных временных
масштабах. Однако, при таких больших возможностях, информация, по-
лученная этим методом, хотя и является существенной и важной, не столь
велика, как можно было ожидать. Это следствие чрезвычайной трудности
интерпретации спектров.
ЯМР измеряет изменение спинового магнитного момента ядер. Сильное
магнитное поле (~105 Гаусс) вдоль одной оси, обычно принимаемой за на-
правление z, вызывает распределение всей совокупности ядерных спинов,
направленных параллельно магнитному полю. Энергия поляризованного
электромагнитного излучения в области радиочастот (102MHz) с вектором
магнитного поля, перпендикулярным к оси z, вызывает переходы к более вы-
сокому энергетическому состоянию спинов. В типичных ЯМР экспериментах
измеряют зависимость напряжения, которое пропорционально релаксации
ядер от более высокого энергетического состояния до более низкого, от час-
тоты электромагнитного излучения. Однако чаще, чем частоту, используют
химический сдвиг, который определяется как:
у(образец) — у(стандарт)
-----------:-------- (
V (стандарт)
где v — частота, а стандарт — материал, который содержит те же типы
атомов. Только ядра с ненулевыми спиновыми квантовыми числами могут
изучаться этим методом. Для ТПЭ наиболее обычными ядрами, для которых
выполняется это требование, являются 1Н, 2D, и 13С.
III. Морфология термопластичных эластомеров -»v- 599
Химический сдвиг, который появляется вследствие электронного экра-
нирования со стороны атомов окружающей среды, как правило, в твёрдых
телах является анизотропным. В твердотельном ЯМР могут проявляться три
других анизотропных взаимодействия: диполь-дипольные взаимодействия,
которые являются взаимодействиями через пространство с другими ядрами;
спин-спиновое взаимодействие, которое представляет собой взаимодейс-
твием через связь двух спинов; и квадрупольное взаимодействие, которое
имеет место, когда квантовое число спина большее, чем 1/2, оно обычно
затемняет спектр, если появляется. В растворе, из-за быстрого движения
молекул, диполь-дипольные и квадрупольные взаимодействия обычно ис-
чезают, и остаётся только изотропный химический сдвиг и спин-спиновое
взаимодействие. В большинстве случаев при использовании твердотельного
ЯМР создают экспериментальные условия, при которых некоторые из этих
специфических трудностей уменьшаются или устраняются.
Для удаления анизотропных взаимодействий используется методика, на-
зываемая «вращение под магическим углом» (ВМУ, MAS). Взаимодействия
в твердом теле имеют приблизительную угловую зависимость (3cos20 - 1),
где 0 — угол между осью z и образцом. Если образец вращают вокруг оси,
и ось находится под магическим углом относительно магнитного поля
(0 ® 54.7°), то большая часть анизотропии ичезает. Если частота вращения
меньше, чем характеристическая частота усредненного взаимодействия, то
будут появляться побочные полосы частот вращения, целочисленно кратные
скорости вращения. Для определения истинных свойств, твердотельные
ЯМР спектры обычно получаются при двух или больше частотах вращения.
Другие взаимодействия могут быть удалены при использовании радиоим-
пульсов. Нарушение связи протонов высокой энергии удаляет протонное
диполь-дипольное и спин-спиновые взаимодействия для таких ядер, как 13С.
Кросс-поляризация усиливает намагниченность спинов редких ядер, воз-
никающую от соседних распространенных ядер. Ядра 13С часто усиливают,
используя 1Н.
Радиочастотные импульсы используются также для определения времени
релаксации. В ТПЭ были определены три времени релаксации, и каждое из
них чувствительно к различным явлениям. 7\ — время спин-решеточной
релаксации в лабораторной системе отсчета. Это время релаксациии сово-
купности спинов от неравновесного распределения, созданного импуль-
сом, к равновесному Больцмановскому распределению. 7^ чувствительно
к молекулярным движениям с частотами в диапазоне 106-109 Гц. Т2, время
спин-спиновой релаксации, которая связана с установлением равновесия
между ядерными спинами в пределах системы. Измерения спин-спиновой
релаксации также дает сведения о движениях, с частотами в диапазоне
106-109Гц; однако низкочастотные движения (102-103Гц) также влияют
на Т2. Обычно в твердых полимерах Т2на 1-3 порядка меньше, чем 7\. Т\ ,
600 -'V- Глава 13. Термопластичные эластомеры
время спин-решеточной релаксации во вращающейся системе отсчета, дает
сведения о движениях с частотами порядка 103-104 Гц. Для измерений Тх
обычно используют кросс-поляризацию.
а
О
II
-N NCO(CH2)4OC-
4 О(СН2)4|-
1 J 27.6
Рис. 11. Структура полиуретанов, основанных на пиперазине. Маркирование дей-
терием только на фрагментах пиперазина 1 и 5, или только на
кольцах 2 и 4, или только на кольце 3 (а). Доля динамически ус-
редненного материала (рМА как функция температуры: • пиперазин
в центре (третье кольцо), 0 во втором и четвертом кольце, о во вне-
шнем (первое и пятое кольцо) (б).
Были проведены измерения Г2(Я) для ряда блок-сополимеров СБС
с содержанием стирола от 14 до 30% [126]. Было обнаружено три различных
Т2 релаксационных процесса: быстрая релаксация, обусловленная матери-
III. Морфология термопластичных эластомеров -*v- 601
алом жёсткого сегмента, средняя релаксация, обусловленная межфазным
материалом, и медленная релаксация, обусловленная материалом мягкого
сегмента. для межфазной области примерно на два порядка больше, чем
для жесткого сегмента, в то время как Т2для мягкого сегмента приблизи-
тельно на один порядок больше, чем для межфазной области. Т2 для мягкой
бутадиеновой фазы было меньше, чем для чистого каучука, что указывает
на ограничение подвижности бутадиена при сополимеризации. Во всех
случаях в фазе стирола было обнаружено меньшее количество материа-
ла, чем можно было предсказать; однако в фазе бутадиена обнаружилось
приблизительно стехиометрическое количество материала. Расчеты для
межфазных областей показали, что межфазные области состоят, в основном,
из сегментов бутадиена; таким образом, существенное количество стирола
должно быть растворено в бутадиене. Расчётная толщина межфазных об-
ластей составляет 20А, что приближенно согласуется с результатами МУРР
для подобных систем [127,128].
ЯМР может использоваться для изучения двух фаз и межфазной области
ТПЭ при маркировке дейтерием. Шписс (Spiess) и др. [129,130] проана-
лизировали ряд модельных полиуретанов с монодисперсными жёсткими
сегментами на основе пиперазина (рис. И). Синтез очень тщательно уп-
равлялся таким образом, чтобы в жестком сегменте были помечены только
определенные части. Чтобы количественно определить относительную долю
подвижных фрагментов пиперазина, которая была названа долей динами-
чески усредненного компонента (ФМА), использовали отношение площадей
узкого и широкого сигнала ЯМР. Доля динамически усреднённого компо-
нента зависит от положения фрагмента пиперазина в жестком сегменте, как
показано на рис. 11. Основываясь на исследовании материалов, содержащих
только единичные фрагменты пиперазина, начальное повышение динами-
чески усредненного компонента, наблюдающееся на трёх кривых между
250°К, и 300°К, было приписано фрагментам пиперазина, находящимся
в растворённых жестких сегментах. Основываясь на этом предположении,
авторы вычислили, что только 15% жестких сегментов растворяется в мягких
сегментах, в то время как остальные 85% содержались в пределах доменов
жестких сегментов.
Для изучения молекулярных движений в 4ГТ-ПТМО, где два внутренних
углерода РТМО были заменены дейтерием, использовалась квадропольная
характеристика дейтерия [131]. Предыдущие 13С ЯМР эксперименты того
же автора [132, 133] показали, что 4ГТ-группы можно рассматривать как
молекулярные якоря (анкеры), потому что масштаб времени движения
этих групп намного более медленный. Форма кривой ЯМР, полученная
при импульсном воздействии, использовалась для изучения трехсвязных
типов движения последовательностей метилена в ПТМО. Была рассчитана
энергия активации этого движения, 5,8 ккал/моль, которая оказалась чуть
602 -*Ц* Глава 13. Термопластичные эластомеры
большей, чем для вращения одной С-С связи в бутане (3,7 ккал/моль).
Были представлены шесть различных транс-гош конформационных пе-
реходов и сопоставлены с данными ЯМР; четыре из этих возможностей
были устранены, так что остались только две, как показано на рис. 12. Ав-
торы доказывают, что оба оставшихся механизма могут вносить свой вклад
в наблюдаемое движение.
а
Рис. 12. Возможные механизмы транс-гош конформационных изменений ПТМО
в 4ГТ-ПТМО. Обнаружено, что только (д) и (е) согласуются с результатами
ЯМР.
IV. Свойства и влияние структуры -* V 603
Для МДИ-БД-ПТМО полиуретанов и МДИ-ПТМО коммерческого по-
лиуретана, цепь которого удлинена этилендиамином (ЭД, ED), был изме-
рен ряд времен релаксации, чтобы дифференцировать межфазную область
и фазы жёстких и мягких сегментов [134-136]. Было установлено, что из-
мерения 7^ (С) иТ1р(С) показывают различия между жесткими сегментами,
растворёнными в мягкой фазе, и жёсткими сегментами в жесткой фазе для
материала с цепью, удлинённой бутандиолом (БД, BD), в то время как изме-
рения Т1р(С) для материала с цепью, удлинённой этилендиамином, позволя-
ют различать карбонилы, находящиеся во всех трех областях. Одиннадцать
процентов уретановых групп растворены в мягких сегментах материала
с соотношением МДИ-БД-ПТМО 7:6:1, в то время, как только 20% блоков
МДИ были кристаллическими. Для ЭД-материала было обнаружено, что
50% жестких сегментов способны к свободному вращению; это наводит на
мысль о том, что степень фазового разделения в этом материале меньше, чем
в МДИ-БД-материале. Поскольку поли (уретанмочевины) обычно имеют
лучшее фазовое разделение, чем полиуретаны [137], этот результат был,
вероятно, обусловлен такими факторами, как методика подготовки препа-
рата и исходные материалы. Влияние изменения температуры изучалось
для системы, где в качестве мягких сегментов вместо ПТМО использовали
полиметиленадипинат. Было показано, что с ростом температуры доля
подвижного компонента фазы жесткого сегмента увеличивается. Степень
увеличения зависит от состава полиуретана, этот эффект авторы связывают
с дефектностью жестких доменов [138].
IV. СВОЙСТВА И ВЛИЯНИЕ СТРУКТУРЫ
А. Общие характеристики.
В таблице 3 приведены свойства некоторых типов термо-
пластичных эластомеров и других эластичных полимерных материалов.
ТПЭ обычно можно растягивать до высоких удлинений без разрушения, они
обладают высокой разрывной прочностью, т. е. являются очень прочными
материалами. Как упоминалось ранее, термопластичный эластомер должен
возвращаться к начальной форме после удаления напряжения. Предельное
удлинение, после которого ТПЭ будет возвращаться к первоначальному
размеру после снятия напряжения, обычно не так велико, как у традици-
онных резин. При высоких удлинениях после снятия напряжения ТПЭ
часто сохраняют некоторое остаточное удлинение, называемое остаточной
деформацией. Как обсуждалось в разделе по инфракрасной спектроско-
пии, под действием больших напряжений домены могут течь. Энергия
разрушения этих материалов (площадь под кривой напряжение-дефор-
мация) обычно во много раз больше, чем у обычных вулканизованных
резин.
604 —Глава 13. Термопластичные эластомеры
Таблица 3. Сравнение свойств типичных ТПЭ и других каучукоподобных
полимеров.
Материал Относи- тельная цена Прочность при разрыве (МПа) Относительное удлинение при разрыве (%) Температура эксплуатации (°C) Твёрдость (Шор А)
Бутадиен- стирольный каучук 1 15 (усиленный) 500 (усиленный) высокая 35-100
Натуральный каучук 1 30 (усиленный) 500 (усиленный) высокая 30-100
Силиконовый каучук 1,2 5(усилен- ный) 150 высокая 40-100
Полиэтилен 1 10 высокое -10-50 100
Фторэластомер 1,5 10 200 высокая 50-90
СБС 2 25 800 -20-80 50-90
Смесь ПП-ЭПК 1,5* 20 500 0-110 70
Полиуретан 6 50 600 -20-80 50-100
Полиэфирный ТЭП 7 40 600 -40-150 >100
Иономер 5 15 500 -20-100 50-90
Эффект Маллинза [139], называемый также размягчением под напряже-
нием, проявляется в большинстве ТПЭ. Если образец ТПЭ вначале дефор-
мируется, а затем освобождается от нагрузки, то для вторичной деформации
потребуется меньшее напряжение. Этот эффект количественно выражается
рассеянной энергией (гистерезисными потерями), которая является разно-
стью площадей под кривыми напряжение-деформация для этапа нагружения
и следующего за ним этапа разгрузки. Эффект Маллинза может приводить
к теплообразованию в материале, которое, в большинстве случаев, является
нежелательным. При этом способность ТРЕ к рассеиванию энергии связана
с его прочностью и твердостью.
Разрушение полимера включает инициирование, медленный рост трещины
и катастрофическое ее распространение. Максимальное значение прочности
в термопластичных эластомерах достигается в большей степени из-за замед-
ления катастрофического распространения трещины после ее возникнове-
ния, чем от предотвращения инициирования или медленного роста трещины.
В таблице 4 приводятся механизмы, по которым может осуществляться усиле-
ние двухфазного материала. Жесткие домены в ТПЭ одновременно действуют
и как наполнитель, и как межмолекулярные связи. Жесткие домены являются
эффективными наполнителями, если их объемная доля превышает 0.2, размер
!) в несколько раз больше.
IV. Свойства и влияние структуры
605
составляет менее 100 нм, а температура размягчения намного превышает тем-
пературу испытания [ 140]. Влияние доменов как наполнителя является одним
из условий достижения максимальной прочности, свойственной большинству
ТПЭ. Другим условием является наличие поверхности раздела между жестки-
ми и мягкими сегментами; предполагается, что в смесях полимеров [141] самые
прочные материалы образуются из двух полимеров, которые находятся на грани
совместимости. Эти результаты наводят на мысль, что образование широких гра-
ничных зон приводит к улучшению свойств двухфазных блочных сополимеров.
Таблица 4. Процессы усиления в полимерах.
Матрица Рассеивание энергии вблизи вершины трещины
Кристаллизация, вызванная напряжением
Процессы ориентации
Частицы наполнителя Повышенное рассеяние энергии
Отклонение и раздвоение трещин
Кавитация
Деформация доменов
Из-за двойственной функции жестких доменов (усиливающий наполни-
тель и поперечная связь) молекулярный процесс деформации ТПЭ совер-
шенно не подчиняется Гауссовой статистической теории сеток, применяемой
для описания поведения традиционных вулканизатов. Зацепления, которые
служат эффективными поперечными связями, играют важную роль в фор-
мировании поведения ТПЭ. Зависимость напряжение-деформация для боль-
шинства ТПЭ описывается эмпирическим уравнением Муни-Ривлина:
<1з>
\МС X Д V)
где а — напряжение (сила/единицу поперечного сечения), R — универсаль-
ная газовая постоянная, Т— абсолютная температура, X — степень удлине-
ния, а Мс — средний молекулярный вес отрезка цепи между поперечными
связями. С2 — эмпирическая константа, которая зависит от материала. Оп-
ределение зависимости напряжение-деформация и набухания показывает,
что в СБС Мс ближе к молекулярной массе между зацеплениями, чем к длине
мягкого сегмента [142]. Влияние наполнителя определяется количественно
уравнением Гута-Смолвуда (Guth-Smallwood):
^ = (1 + 2.5ф + 14.1ф2) (14)
606 -*и- Глава 13. Термопластичные эластомеры
rjifiEF/E— отношение модулей наполненного и ненаполненного эластомера,
а (р — объемная доля наполнителя. Комбинация этих двух уравнений дает
разумные значения для Мс в СБС сополимерах; однако имеются возражения
против использования такого подхода [143-145]. Для объяснения характера
деформационного поведения термопластичных эластомеров были предло-
жены и более сложные теории [146-148].
Деформация (Х-1
Рис. 13. Влияние температуры и скорости растяжения на кривую напряжение-
деформация для термопластичных эластомеров, о — напряжение, То — тем-
пература приведения, Г — температура эксперимента, X — степень удлине-
ния, Е — деформация. Петрович и Фергюсон [15] (Petrovic & Ferguson).
Обычный характер изменения деформационных свойств ТПЭ при увели-
чении температуры показан на рис. 13. ТПЭ становится более твердым при
приближении температуры к Т мягкого сегмента и при этой температуре
резко возрастает хрупкость. Обычно в процессе эксплуатации Tg мягкого
сегмента не достигается, так как эта температура значительно ниже ком-
натной. При повышении температуры модуль и прочность типичного ТПЭ
уменьшаются из-за размягчения жестких доменов. При температуре полного
размягчения (плавления или расстеклования) жестких доменов происходит
резкое уменьшение модуля, и материал больше не может использоваться
как термопластичный эластомер. Однако двухфазная структура может со-
храняться и в расплаве. Точная температура полного размягчения зависит
от природы жесткого блока. Для ее снижения можно использовать плас-
IV. Свойства и влияние структуры 607
тификаторы или другие добавки, однако они могут также способствовать
нарушению доменной структуры.
Химическая стойкость многих ТПЭ ниже, чем у традиционных резин.
Полиуретаны, полиэфиры, полиамиды очень восприимчивы к окислению,
особенно при повышенных температурах. Для улучшения химической
стойкости в товарные продукты вводят антиоксиданты и другие добавки.
Технический углерод может быть добавлен для улучшения стойкости
к ультрафиолетовому излучению, если цвет материала не имеет значения.
Низкая гидролитическая стойкость полиамидов и полиуретанов на основе
сложных полиэфиров обусловлена тем, что сложноэфирная связь может быть
разрушена водой. Для всех ТПЭ характерно то, что некоторые органические
растворители могут их разрушить, если один или оба из блоков распадутся
в специфическом растворителе. Более полярные ТПЭ обладают высокой
стойкостью к действию многих обычно применяемых масел и смазочных
материалов.
Б. Механические свойства
В связи с существенными различиями в структуре разных
типов ТПЭ, закономерности влияния структуры на механические свойства не
могут быть универсальными для всех материалов. Однако, некоторые общие
характеристики сохраняются для большинства ТПЭ и будут рассмотрены
в этом разделе. Относительное количество жестких и мягких сегментов вли-
яет на механическое поведение ТПЭ. С увеличением содержания жестких
сегментов материал будет изменяться от гибкого эластомера до прочного
жесткого пластика. Это иллюстрируется рис. 14, на котором представ-
лено изменение деформационных кривых в зависимости от содержания
стирола в бутадиенстирольных ТПЭ. Аналогичные данные имеются для
блоксополимера полибутилентерефталата с политетраметиленоксидом
(4ГТ-РТМО), полиэфиров и полиуретанов. При переходе твердой фазы
в ТПЭ от дискретной к непрерывной наблюдается качественное измене-
ние формы кривой растяжения этих материалов. Предел текучести при
содержании стирола около 40% интерпретируется как доказательство
образования непрерывной фазы полистирола; обычно существование
предела текучести в ТПЭ подразумевает наличие непрерывной жесткой
фазы. Изменение относительных количеств жестких и мягких сегментов
в материале может быть достигнуто или их путем изменения длины жес-
ткого, или длины мягкого сегмента, или обоих. Влияние длины сегмента
было исследовано при постоянных относительных количествах жесткого
и мягкого сегментов. В стирол-диеновых триблоксополимерах прочност-
ные свойства не зависят от длины сегмента пока полистирол имеет доста-
точный молекулярный вес для формирования прочных жестких доменов
(>8000) [149].
608 -'V Глава 13. Термопластичные эластомеры
Однако молекулярный вес блоков будет влиять на кинетику фазового
разделения в системах стирол-диен, которая может, в свою очередь, затра-
гивать механические свойства.
Удлинение, %
Рис. 14. Зависимость деформационно-прочностных свойств СБС от содержания
стирола.
В полиуретанах, полиэфирах и полиамидах основным фактором, опре-
деляющим механических свойств материала, является молекулярный вес
каждого блока. Было установлено, что для получения материала с оптималь-
ными свойствами длина эластичного блока должна находиться между 1000
и 5000. Ниже этого значения материалы не способны к фазовому разделению.
Более низкие значения молекулярного веса также препятствуют способности
мягких сегментов к ориентационной кристаллизации (кристаллизации при
растяжении), что обеспечивает важный механизм упрочнения. Наконец,
при более низких значениях молекулярного веса мягких сегментов и при
постоянном общем количестве этих сегментов требуются короткие жесткие
домены, что может приводить к существенному подавлению их кристалли-
зации. При повышении молекулярного веса мягких сегментов возникают
практические проблемы. Так как в промышленности эти материалы синте-
зируются методом блочной полимеризации (без растворителя), повышение
молекулярного веса эластичных (мягких) сегментов приводит к значительно-
му увеличению вязкости в процессе полимеризации. При этом затрудняется
смешение и выделение образующегося полимера, а также достижение необ-
ходимой высокой конверсии. Кроме того, высокий молекулярный вес блоков
IV. Свойства и влияние структуры -*V 609
способствует фазовому разделению, что снижает фракционную конверсию.
Так как все методы ступенчатой полимеризации для достижения большого
молекулярного веса требуют высокой фракционной конверсии, использование
исходных высокомолекулярных сегментов затруднитрельно. Влияние длины
блока жестких сегментов также важно. Увеличение длины блока жестких сег-
ментов приводит к лучшему фазовому разделению и лучшим свойствам. Однако,
более длинные блоки жестких сегментов вызывают затруднения при синтезе.
Некоторые из этих трудностей могут быть преодолены путем использования
способа растворной полимеризации, который в дальнейшем может стать, а
может и не стать синтезом промышленного масштаба.
Суммарный молекулярный вес в ТПЭ может меняться. Величина молеку-
лярного веса, пока он выше некоторого порогового значения, незначительно
влияет на механические свойства. Он становится важным в двух случаях.
Если используемые в полиблочных сополимерах мягкие сегменты имеют
низкую функциональность, то молекулярно-весовое распределение будет
весьма широким, и свойства будут существенно хуже [150]. Большинство
имеющих промышленное значение мягких сегментов имеют исходную функ-
циональность около двух, поэтому этот показатель обычно не важен. Однако
более важным, особенно в полиуретанах, является возможность побочных
реакций, которые могут приводить к образованию поперечных связей. Не-
большое количество поперечных связей может фактически улучшать [151]
свойства за счет, как предполагается, упрочнения жесткого домена. Большие
количества поперечных связей нежелательны, так как могут затруднять
фазовое разделение и увеличивать хрупкость ТПЭ.*
Влияние полидисперсности жестких и мягких сегментов было исследо-
вано в различных полиуретанах [25, 152, 153]. Полидисперсность мягких
сегментов существенно не изменяет механические свойства, в то время как
материалы с монодисперсными жесткими сегментами имеют более высокий
модуль и повышенную прочность при растяжении. Из этих исследований
следует важный вывод о том, что молекулярно-весовое распределение мягких
сегментов не управляется синтезом, что было недавно доказано с использо-
ванием новой аналитической методики [154]. Улучшенные механические
свойства могут быть связаны с более совершенным фазовым разделением, как
было показано методом МУРР [155]; плотность упаковки жестких сегментов
может также играть определенную роль. Состав ТПЭ является параметром,
который можно легко менять, и который может оказывать существенное
влияние на их свойства. В бутадиенстирольных триблоксополимерах это
может быть замена центрального блока на изопрен или конечных блоков
на а-метилстирол. В первом случае механические свойства не меняются за
исключением того, что предел прочности СБС-блоксополимеров зависит,
а СИС-блоксополимеров не зависит от содержания стирола. Замена а-ме-
1) а также делать невозможной повторную переработку ТПЭ.
610 -'V- Глава 13. Термопластичные эластомеры
тилстирола приводит к образованию более жесткого полимера, по крайней
мере частично, из-за более высокой температуры стеклования (160°С про-
тив 100°С) нового конечного блока. Несмотря на заметные отличия в Tg для
изопрена и бутадиена, существенного различия в механических свойствах
ТПЭ не установлено [156]. Эти различия подчеркивают важность жесткой
фазы в определении механических свойств материала.
В сегментированных блок-сополимерах типа полиуретанов или поли-
эфиров можно легко менять состав жесткого и мягкого блоков. Изменение типа
жесткого сегмента приведет к изменению кристаллического состояния жесткой
фазы. Например, можно получить сополиэфир, используя смесь тетраметилен-
терефталата и другой двухосновной кислоты, типа тетраметиленизофталата.
Как показано у Витсайпа (Witsiepe) [ 17], конечные свойства материала, содер-
жащего смешанные жесткие блоки лучше, а влияние на твердость и жесткость
меньше, если общее их количество невелико. Недостатком использования смеси
кислот является ограничение и значительное снижение скорости кристал-
лизации. В полиуретанах при использовании симметричных диизоцианатов
образуется более прочный ТПЭ. Присутствие заместителей в ароматическом
кольце способствует снижению прочности полиуретанов. В полиуретанах на
основе сложного полиэфира и бутандиола, диизоцианаты по степени увеличе-
ния прочностных свойств располагаются в ряд: МДИ > гексаметилендиизоциа-
нат > изофорондиизоцианат > толуилендиизоцианат (ТДИ) [157].
Тип мягкого сегмента также играет важную роль в формировании фи-
зических свойств полиблочных сополимеров. Кристаллизующиеся при
растяжении мягкие блоки обеспечивают создание более жестких материалов
с высокими значениями прочности при растяжении и сопротивления раз-
диру [158]. Так как объединение в блочный сополимер изменяет кинетику
кристаллизации и слегка понижает температуру плавления, ненапряженный
эластомер может не содержать закристаллизованных мягких сегментов.
Кристаллизация, которая происходит при растяжении, приводит к большой
остаточной деформации в материале. Подъем на кривой зависимости нагруз-
ка-удлинение при высоких удлинениях часто принимается за доказательство
кристаллизации, но наиболее прямое и убедительное доказательство — это
данные рентгеноструктурного анализа (ШУРР). Мягкие блоки, кристалли-
зующиеся при деформации, имеют температуру плавления немного выше
рабочей температуры, а более высокий молекулярный вес мягкого сегмента
способствует кристаллизации при деформации. Политетраметиленоксид
и поликапролактон являются двумя общеизвестными мягкими сегментами,
которые могут кристаллизоваться при деформации.
Тип мягкого сегмента также влияет на движущую силу фазового разделения.
Однако улучшенное фазовое разделение не обязательно приводит к улучшен-
ным свойствам. Так, мягкие блоки простого полиэфира [55] или полибутадиена
способствуют более полному фазовому разделению, чем сложные полиэфиры,
IV. Свойства и влияние структуры -*v- 611
но материалы на основе сложных полиэфиров имеют лучшие механические
свойства. Наиболее вероятное объяснение этого результата - недостаточная
межфазная адгезия в системах с хорошим фазовым разделением. [159].
В. Термические и химические свойства
Реакция на термические и химические воздействия окру-
жающей среды в значительной степени является результатом основной
химической структуры материала. Как показано во введении, недостато-
чная устойчивость к химическому или тепловому воздействию ограничи-
вает использование ТПЭ, особенно по сравнению с обычными резинами.
К сожалению, трудно, если не невозможно, существенно изменить тепловые
и химические характеристики материала. Использование небольших ко-
личеств антиоксидантов или наполнителей может несколько улучшить эти
свойства, но злоупотребление ими может привести к большому изменению
в механическом поведении материала.
Чувствительность к гидролизу является ключевой проблемой во многих
областях применения. Сложноэфирная связь в сополимере полибутилен-
гликольтерефталат-политетраметиленоксид (4ГТ-ПТМО) чувствительна
к гидролизу; однако, она хорошо защищена, так как большая часть сложного
эфира входит в кристаллическую структуру. Добавка небольшого количества
(1-2%) блокированного ароматического поликарбодиимида существенно
увеличивает продолжительность эксплуатации этого материала в горячей
воде или паровой среде [160]. Полиуретаны, особенно содержащие мягкие
сложноэфирные блоки, восприимчивы к гидролитическому воздействию.
Однако, полиуретаны со сложноэфирными мягкими сегментами более
стойкие к маслам, неполярным органическим растворителям и термической
деструкции. Иономеры будут набухать при испытаниях в воде. Действитель-
но, промышленный гидрированный перфторсульфоновый иономер (Nafion)
используется как мембранный разделитель в хлор-щелочных ячейках.
Диен-стирольные сополимеры и полиолефиновые ТПЭ нечувствительны
к действию воды.
Температуростойкость ТПЭ зависит, в основном, от химической струк-
туры. Максимальная рабочая температура обычно приблизительно на
40°С ниже температуры плавления или стеклования жесткого сегмента.
В процессе эксплуатации ТПЭ из-за гистерезисных потерь может произойти
чрезмерное теплообразование и локальная температура материала может
быть намного выше номинальной. Изменение максимальной рабочей тем-
пературы приводит к изменению структуры жесткого блока. Использование
а-метил стирола в диен-стирольном трибл оксополи мере или удлинителя цепи
этилендиамина в полиуретанах может существенно расширить интервал
рабочих температур в этих ТПЭ. Минимальная рабочая температура обычно
приблизительно на 10°С превышает Tg мягкого сегмента. При более низкой
612 -*V" Глава 13. Термопластичные эластомеры
температуре материал становится ломким. Если мягкий сегмент способен
кристаллизоваться, то низкие температуры могут вызывать кристаллиза-
цию и соответствующее увеличение жесткости и хрупкости. Использование
сополимерного или смесевого мягкого сегмента устраняет эту проблему, но
ценой подавления ориентационной кристаллизации при более высоких
температурах.
V. ТЕРМОДИНАМИКА ФАЗОВОГО РАЗДЕЛЕНИЯ
В этом разделе будут представлены теоретические основы
и предположения относительно термодинамики фазового разделения в бло-
чных сополимерах. Большинство теорий рассматривает четыре фактора, ко-
торые влияют на фазовое разделение блоксополимеров: (1) параметр взаимо-
действия Флори-Хаггинса (Flory-Huggins) х, (2) степень полимеризации N,
(3) конструкционные параметры типа числа блоков и линейность или
звездообразная структура блок-сополимера и (4) весовая концентрация f
одного из компонентов. Термодинамические теории удобно разделяют
фазовое состояние на три типа: высокая степень сегрегации (ВСС, SSL),
низкая степень сегрегации (НСС, WSL), и промежуточная область сегре-
гации (ПОС, ISR). В ВСС материал в состоянии устойчивого равновесия
содержит относительно чистую фазу А, отделенную от относительно чистой
фазы В. В НСС обе фазы тесно перемешаны. ПОС является по существу
областью, которая образуется из-за конечного молекулярного веса, при
бесконечном молекулярном весе ПОС не существует. Поэтому вызывает
сомнение, должна ли рассматриваться ПОС как часть термодинамического
фазового пространства.
Переход между ВСС и НСС называется переходом порядок-беспорядок
(ППБ, ODT), который также называется переходом микрофазового разде-
ления (ПМР, MST). Аналогичный переход в низкомолекулярных соедине-
ниях называется переход «твердое тело — жидкость». Читатель должен быть
предупрежден о переходе порядок-порядок (ППП, ООТ), при котором от
одного типа к другому (например, от сфер к цилиндрам) изменяется мор-
фология. Это может происходить при изменении температуры для блочных
сополимеров анионной полимеризации при очень специфической длине
блоков [161]. Если температура ППБ ниже 7^ или Тт жесткого сегмента, то
материал является однофазным выше температуры размягчения жесткого
сегмента. Если температура ППБ выше Т или Тт жесткого сегмента, то
материал будет существовать как двухфазный расплав. Фазовое состояние
ТПЭ при температуре выше температуры размягчения имеет существенное
влияние на реологические свойства, так как однофазные смеси имеют вяз-
кость намного меньше, чем двухфазные.
Было потрачено много усилий на развитие теорий для диблок- или три-
блоксополимеров, многие из этих теорий основаны на построении фазовых
V. Термодинамика фазового разделения —*V 613
диаграмм, которые в свою очередь обеспечивают хорошую проверку теорий.
Следует отметить, что строгое применение этих теорий к ТПЭ, используемым
в коммерческих целях, ограничено, поскольку почти во всех системах про-
исходит торможение из-за замедления кинетических процессов. Наиболее
строгие теории были разработаны для блоков с монодисперсной длиной,
например, для ТПЭ, полученных методом анионной полимеризации. В не-
которых случаях эти теории были перенесены на многоблочные сополимеры,
имеющие распределение по размерам блоков. И далее, аморфные системы
дают существенно более ясную картину, чем кристаллические.
В НСС топология цепей является невозмущенной; например, величина Rg
имеет порядок N1/2- Однако вероятность обнаружения сегментов А или В
на расстоянии г от определенной точки зависит не только от/, но также от
того, находится ли исходная точка на сегменте А или сегменте В. Если исход-
ная точка находится на сегменте А, то вероятность обнаружения другого сег-
мента А на близком расстоянии становится намного выше, а на расстоянии,
сопоставимом с радиусом вращения цепи, выше вероятность обнаружения
сегмента В. Эта концепция называется эффектом дырочной корреляции
[162]. При очень больших расстояниях вероятность обнаружения сегмента
А уменьшается до объемной доли сегментов А в НСС. Как правило, в ВСС
цепи не принимают их невозмущенную конформацию. Вероятность обна-
ружения сегмента на расстоянии г зависит в значительной степени от типа
морфологии и может также зависеть от направления, в то время как в НСС
вероятность является изотропной.
Теоретические разработки Гельфанда и Вассермана (Helfand
и Wasserman) [163-167] содержат все необходимые компоненты для закон-
ченного описания фазового разделения при строгих пределах сегрегации;
фактически, теория была слегка изменена так, чтобы рассматриваемые
результаты были надежными в количественном отношении [168]. Теория
включает три энергетических компонента: (1) проигрыш в энтропии, свя-
занной с локализацией разных сегментов, возникающий из-за концентра-
ции АВ связей на поверхности раздела, (2) проигрыш в конформационной
энтропии, возникающий из-за удлинения цепей и (3) энтальпия, связанная
с перемешиванием сегментов А и В. Уравнение записано для функции, ко-
торая пропорциональна плотности вероятности того, что один конец цепи из
N сегментов находится в точке г0, а другой в г. Результирующее уравнение
идентично по форме уравнению диффузии, описывающему зависимость от
времени, в котором дифференциал по времени заменен дифференциалом по
количеству сегментов. Завершение анализа включает решение этого урав-
нения с соответствующими граничными условиями. Полное аналитическое
решение уравнения не было получено, но были рассчитаны численные реше-
ния. Эта теория была первоначально разработана для диблоксополимеров,
а затем распространена на триблочные системы.
614 -*v Глава 13. Термопластичные эластомеры
Предполагалось, что толщина межфазного слоя (£) должна быть при-
близительно равна:
Z = 0.816axV2 (15)
где а — статистическая длина сегмента. Это уравнение предполагает, что тол-
щина граничного слоя не зависит от молекулярного веса. Было установлено,
что в пределе, когда N —> ©о, период доменной структуры D, определенный
путем численного анализа, имеет размерность:
О~а№/,4х,/7 (16)
В этих пределах энтропия, связанная с локализацией связей, незначитель-
на по сравнению с энтропией, связанной с натяжением цепи. Численный
прогноз предсказанной периодичности был сделан для диблок- и три-
блоксополимеров для цилиндрической, сферической и пластинчатой мор-
фологии. Совпадение теоретических предположений и экспериментально
наблюдаемых результатов было хорошим в основном для цилиндрических
и пластинчатых систем, тогда как для сферических систем оно было хуже.
Вследствие того, что сферы изолированы, изменения в размере домена могут
происходить только путем перемещения сегментов через несовместимое
с ними окружение, что обеспечивает существенную диффузионную устой-
чивость к изменениям в морфологии. Таким образом, морфология имеет
тенденцию не изменяться сразу после формирования, и соответствие между
теорией и экспериментом оказывается недостаточным [169]. Для расчета
фазовых диаграмм был использован численный метод, включающий вы-
числение основной морфологии (сферы, цилиндры, и т.д.) как функцию
состава. Было предсказано, что основной тип морфологии практически не
зависит от температуры.
Многочисленные исследования блочных сополимеров в ВСС были за-
вершены намного раньше появления теории элементов, лежащей в основе
подхода, разрабатываемого Гельфандом и Вассерманом. В НСС экспери-
ментальные данные соответствуют теории поля осредненных величин, пер-
воначально разработанной Лейблером (Leibler) для диблоксополимеров
с монодисперсными блоками [170]. Этот метод использует аппроксимацию
беспорядочно распределенной фазы (АБРФ, БРА) [171] для расчета свобод-
ной энергии через параметр порядка 4х(г), который описывает среднее откло-
нение от равномерного распределения в любой точке г. Было установлено,
что термодинамическое состояние материала зависит только от %Nn/точно
так же, как в подходе Гельфанда и Вассермана. Эта модель теоретически
предсказывает образование только трех двухфазных морфологий: объем-
но-центрически-кубически упакованных (оцк, Ьсс) сфер, гексагональных
плотно упакованных цилиндров (гуц, hep) и ламеллей. Поскольку % зависит
от температуры, можно изменять морфологию от сфер до цилиндров или
V. Термодинамика фазового разделения -*ь- 615
ламеллей, просто изменяя температуру, в отличие от теории Гельфанда
и Вассермана, которая предсказывает, что основная морфология не зависит
от температуры. Критическая точка (переход второго порядка) предсказана
при %N = 10.495 для симметричного диблоксополимера. Это также соответ-
ствует минимальному значению %/V, разделяющему упорядоченное и беспо-
рядочное состояния. Так как точка расслоения смеси двух гомополимеров
идентичного молекулярного веса соответствует х^ = 2, соединение двух
индивидуальных цепей концами означает, что для фазового разделения
индивидуальные цепи в диблоксополимере должны быть более чем в 2.5 раза
длиннее, чем в гомополимерах.
Теория Лейблера намечает экспериментальный метод для подтверждения
ее заключений. Структурный фактор рассеяния излучения разупорядочен-
ной фазой был предложен в виде:
5(«) Я
(17)
где F — безразмерная функция, которая связана с Дебаевской функцией
корреляции для Гауссовой цепи. Лоренцев пик предсказан для разупоря-
доченной фазы. Позиция максимума пика при д* не изменяется с темпера-
турой. Периодичность флуктуаций концентрации определяется как 2л/д*,
что примерно равно радиусу инерции цепи. Из-за изменений х высота пика
является функцией температуры. Вообще, было найдено, что х зависит от
температуры следующим образом:
Х = у + Я (18)
где А и В — константы, определяемые опытным путем. Согласно этому ана-
лизу, график зависимости обратной величины максимума интенсивности от
обратной температуры в экспериментах МУРР и МУРН должен быть ли-
нейным, когда полимер находится в беспорядочном состоянии. Отклонение
от линейности будет отмечать температуру перехода порядок-беспорядок,
в то время как экстраполяция к нулевой интенсивности позволит вычислить
температуру спинодального распада. Согласно этой теории ППБ (ПМР) мо-
жет также быть определен по температуре, где д* изменяется как функция
температуры, поскольку предполагается, что в беспорядочном состоянии д*
от температуры не зависит.
Показано, что НССтеория, разработанная Лейблером, оказалась ошибоч-
ной из-за отклонений от правила, лежащего в основе условия теории осред-
ненных величин. На рисунке 15 показаны экспериментальные результаты,
полученные для диблоксополимера полиэтиленпропилена/полиэтилэтилена
(ПЭП-ПЭЭ), который удовлетворяет представлениям теории Лейблера без
616 Глава 13. Термопластичные эластомеры
подгоночных параметров, поскольку ПМР и % были рассчитаны из рео-
логических измерений [172]. Теория поля осредненных величин не дает
качественного описания поведения этого материала. Другие исследования
показали, что RPA аппроксимация [173] и предположение о Гауссовом
клубке [174, 175] неточны около ПМР.
1О3 Т-’, К'1
Рис. 15. Зависимость обратной величины максимума интенсивности МУРН I от
температуры инверсии для диблоксополимера ПЭП-ПЭ (РЕР-РЕЕ)
и сравнение с теорией.
При первоначальном анализе принималось, что эта теория не должна
применяться около критической точки. Теории поля осредненных вели-
чин игнорируют флуктуации концентрации на расстояниях, отличных
от q = g*. Вблизи критической точки флуктуации большого размера
становится все более и более важными. Была разработана модификация
этой теории, включающая флуктуации концентрации [176]. Теория флук-
туаций не прогнозирует существования критической точки, скорее для
всех композиций прогнозируется фазовый переход первого рода. Ниже
приведена зависимость величины хМ которая отделяет упорядоченную
V. Термодинамика фазового разделения 617
фазу от разупорядоченной, от молекулярного веса для симметричного
диблоксополимера:
X/V = 10.495 + 41.022AT1/3 (19)
Минимальное значение %N, соответствующее разупорядоченной фазе,
наблюдается еще при/ = 1/2. Теория флуктуаций прогнозирует ПМР при
немного более низкой температуре, чем теория Лейблера. Образование
фазы в виде ламеллей и гексагонально плотно упакованных цилиндров
(гуц, hep) непосредственно из беспорядочного состояния более достижимо,
что, по-видимому, подтверждается экспериментом, скорее, чем образование
объемно сферически-упакованных (оцк, Ьсс) сфер, как в теории Лейблера.
Довольно простой структурный фактор, представленный в уравнении (17),
сохраняется; однако х заменяется на xeff, который сложным образом зависит
от температуры, состава и молекулярного веса. При температурах, немного
превышающих ПМР, предполагается образование частично упорядоченной
морфологии. На рисунке 15 также приводится сравнение эксперимен-
тальных данных с рассчитанными по теории флуктуаций без каких либо
подгоночых параметров. Теория флуктуаций достаточно хорошо описывает
качественную форму экспериментальной кривой; однако, количественное
соответствие все еще не найдено.
Ранее обсужденные теории были разработаны для монодисперсных ди-
блоксополимеров, которые не являются ТПЭ. Однако Лейблеровская теория
поля осредненных величин была расширена за счет включения полидиспер-
сности [177] и включения в рассмотрение триблок-, звездчатых- и графтсо-
полимеров [178,179]. В первом случае поправка на полидисперсность имеет
тенденцию понижать %N соответствующую ПМР. Как следовало ожидать
из сходства между смесями и диблоксополимерами, фазовое разделение
триблоксополимеров будет наблюдаться при более высоком значении хА,
чем в аналогичных диблоксополимерах. Эта теория прогнозирует монотон-
ное возрастание критического значения хАпо мере увеличения симметрии
трехблочного сополимера, до максимума, равного приблизительно 18 для
симметричного трехблочника. Как ни удивительно, минимальное значение
ХЛГ, которое отделяет области порядка и беспорядка в трехблочных сополи-
мерах, не обязательно соответствует критической точке.
Усовершенствованная теория поля осредненных величин для триблок-
сополимеров аналогична представлениям, развиваемым Лейблером. Второй
параметр, т, определяет асимметрию трехблочника. Если блочный сополимер
обозначен как АВА, тогда, начиная от центра блока В, т — это доля звеньев А,
идущих в одном направлении по цепи, деленная на полное число блоков А
в сополимере. По условию, т всегда меньше чем 0.5, поэтому т = 0 характе-
ризует диблоксополимер, в то время как т= 0.5 — симметричный триблок-
сополимер./определяется как общая доля блоков А в триблоксополимере.
618
Глава 13. Термопластичные эластомеры
Для всех значений т критическая точка рассчитывается при определенном
составе и %/V. При f = 0.5 критическая точка не наблюдается как в случае
симметричных материалов, скорее состав сополимера в критической точке
является функцией т. Упорядоченную фазу после ПМР образуют оцк (Ьсс)
сферы, за которыми следуют гуц (hep) цилиндры и, наконец, ламели. Исклю-
чение составляет критическая точка. Экспериментальные методы и анализ,
которые используются для диблоксополимеров, могут также использоваться
и для триблоксополимеров. Представляется, что #* мало зависит от темпе-
ратуры, а форма кривой рассеяния качественно отличается только крутым
подъемомпри малых# [180]. Разность температур ПМР для диблоксополи-
мера ПЭЭ-ПЭП и триблоксополимера ПЭП-ПЭЭ-ПЭП составила 72°С, что
мало отличается от величины около 61 °C, прогнозируемой по теории поля
осредненных величин [181].
VI. ТЕРМОПЛАСТИЧНЫЕ ЭЛАСТОМЕРЫ
В ПРИПОВЕРХНОСТЫХ ОБЛАСТЯХ
А. Общая характеристика
Двухфазность ТПЭ влияет также на те области приложе-
ния, которые чувствительны к поверхностным или граничным свойствам.
Как правило, содержание компонента на поверхности может существенно
отличаться от общего объемного содержания. Наличие межфазной грани-
цы приводит к другому термодинамическому рассмотрению; может также
изменяться морфология по сравнению с существующей в основном объеме
образца. Изменения состава и морфологии зависят от условий обработки,
которые использовались для образования поверхности, а также от при-
роды другой поверхности. Энергетически эффективной движущей силой
этих различий является стремление минимизировать энергию поверхности
раздела фаз. Первичным параметром, характеризующим поверхностные
свойства материала, является поверхностное натяжение или поверхностная
энергия.
Хотя часто эти два термина используются взаимозаменяемо, строго говоря,
они не являются идентичными. Поверхностное натяжение определяется как:
(20)
где А — свободная энергия Гельмгольца всей системы, Q — энергия на по-
верхности раздела фаз, V— объем системы, Т— температура, и — число мо-
лей f-го компонента. Эта производная определена, исходя из предположения,
что окружающая среда является вакуумом. Более высокое поверхностное
натяжение означает более сильное сопротивление формированию большей
площади поверхности. Выражение для поверхностной энергии идентично,
VI. Термопластичные эластомеры в поверхностях -* V 619
только свободная энергия Гельмгольца для поверхности заменяет свободную
энергию Гельмгольца для системы. Для полимеров, способных деформиро-
ваться, разница между двумя выражениями невелика, и обычно игнори-
руется [182]. Поверхностное натяжение определяется экспериментальным
путем или для полимерных расплавов или путем определения угла смачива-
ния. Первый способ требует экстраполяции к твердому телу, в то время как
в последнем случае поверхностное натяжение не может быть измерено непо-
средственно и должно быть рассчитано по полуэмпирическим уравнениям.
Однако, значения, определенные двумя методами, обычно достаточно близки
[183]. В «Справочнике по полимерам» (Polymer Handbook) приводятся
значения поверхностной энергии для многих сегментов, наиболее часто
входящих в ТПЭ. Большинство полимеров имеет поверхностные энергии в
диапазоне 30-45 дин/см при комнатной температуре. Исключением из этого
простого обобщения являются фтор — и кремнийсодержащие полимеры,
поверхностные энергии которых значительно ниже.
В этом разделе описаны аналитические методы, которые используются
для характеристики атомного состава или морфологии поверхностей ТПЭ.
Измерение поверхностного состава ТПЭ может иметь один существенный
недостаток. Так как мягкая фаза подвижна при комнатной температуре,
состав поверхности может изменяться в зависимости от среды, в которой
сделано измерение. Многие аналитические методы исследования осущест-
вляются в сверхвысоком вакууме, (СВВ, UHV), который не является ре-
альной атмосферой для применения ТПЭ. Обычно различия в поверхности
ТПЭ, подвергнутой воздействию сверхвысокого вакуума или атмосферному
воздействию, невелики; существенные различия появляются, если поверх-
ность подвергается воздействию жидкости типа воды. Для преодоления этой
проблемы были разработаны различные методики, и некоторые из них будут
обсуждены в этом разделе. Для получения количественных результатов ана-
литическими методами исследования состава поверхности также требуется
очень чистая поверхность. Незначительные количества добавок, обычно
присутствующих в коммерческих материалах, могут также существенно
изменять состав поверхности [184]. Наконец, некоторые методы могут сами
изменять поверхность в процессе измерения.
Другим чрезвычайно важным параметром любой методики исследова-
ния поверхности является глубина проникновения (dp), характеризующая
расстояние от исследуемой поверхности. В таблице 5 приведены приблизи-
тельные глубины проникновения для методов исследования поверхности
ТПЭ, обсуждаемых в этом разделе. Хотя, как видно из таблицы 5, глубина
проникновения изменяется в широких пределах, изменение состава мож-
но проследить от поверхности почти непрерывно до глубины в несколько
тысяч ангстрем. С другой стороны, некоторые методы, обсужденные в этом
разделе, определяют морфологию вблизи поверхности. Исследования по-
620 “* v- Глава 13. Термопластичные эластомеры
верхности ТПЭ менее многочисленны, чем исследования объемных свойств,
что в основном говорит о не слишком большой важности поверхностных
свойств ТПЭ для большинства областей их практического применения. Од-
нако исключения из этого заключения определенно существуют, например
в области медицины.
Таблица 5. Глубины проникновения для поверхностно-чувствительных методов
анализа.
Метод Глубина проникновения
Инфракрасная спектроскопия нарушенного полного внутреннего отражения 1,000-10,000 А
Рентгеновская фотоэлектронная спектроскопия 10-100 А
Статическая масс-спектроскопия вторичных ионов 1-ЮА
Динамическая масс-спектроскопия вторичных ионов Различная
Нейтронная отражающая способность Различная
Атомно-силовая микроскопия Поверхность
Б. Исследование поверхностей
1. Сканирующая электронная микроскопия (СЭМ, SEM)
В СЭМ электроны отражаются от поверхности, и измерение
осуществляется на той же самой стороне поверхности, где находится ис-
точник излучения; в ПЭМ (просвечивающая электронная микроскопия)
электроны проходят через образец и регистрируются с другой стороны.
Измерения методом СЭМ выполняется в условиях сверхвысокого вакуума
в основном 10-7-10“9тор. Возможно, наиболее распространенным в науке о
полимерах является использование СЭМ для исследования поверхности сколов.
Морфология поверхности сколов дает информацию о механизме разрушения.
При исследовании ТПЭ, например, этот метод не предназначен для определения
где, внутри фазы или по межфазной границе, происходит разрушение, слишком
малы домены и имеется большое количество смешанной фазы.
В системах с хорошо разделенными фазами, то есть в блоксополимерах,
синтезируемых методом анионной полимеризации и выделенных из раство-
ра, информация относительно состава поверхности может быть получена
микротомированием перпендикулярно к поверхности и затем направле-
нием электронов параллельно поверхности среза. Верхняя (или нижняя)
часть отпечатка содержит информацию о составе свободной поверхности.
Первоклассное иследование на ламеллярном стирол-изопреновом блок-
сополимере с 52% стирола, отлитом из раствора в толуоле [185], показало,
что вообще ламелли ориентированы параллельно межфазной границе
VI. Термопластичные эластомеры в поверхностях -’V 621
воздух — полимер, что соответствует более ранним исследованиям
[186,187] и исследованиям других сополимеров [188,189]. Микрофото-
графия стирол-изопренового диблоксополимера с ламеллями, параллель-
ными межфазной границе, представлена на рис. 16. Блок полиизопрена
с более низкой поверхностной энергией (темный компонент на рис. 16)
всегда размещался на межфазной поверхности между полимером и ваку-
умом. На некоторых микрофотографиях обнаружена перпендикулярная
ориентация ламеллей относительно поверхности, что также показано на
рис. 16. Однако, и в этом случае, как видно на фотографии, на поверхности
раздела воздух-полимер расположена тонкая пленка полиизопрена, то
есть, на межфазной поверхности был зафиксирован блок с более низкой
поверхностной энергией. Очень похожая система, но с кристаллизую-
щимся блоком, ведет себя по-другому, то есть состав поверхности зависит
от ориентации кристаллитов [190]. Этот тип взаимодействия на поверх-
ности может использоваться в блочных сополимерах, синтезированных
методом анионной полимеризации, для создания на поверхности весьма
оригинальной морфологии, когда очень тонкие пленки отливаются из
раствора [191, 192].
Рис. 16. Электронная микрофотография изопрен-стирольного двухблочного сополи
мера (MW = 524,000 г/моль, 52% стирола) на свободной поверхности: Ла-
мелли ориентированы параллельно поверхности (а); Ламели ориентированы
перпендикулярно поверхности (б)
622 -*!/• Глава 13. Термопластичные эластомеры
2. Инфракрасная спектроскопия нарушенного полного
внутреннего отражения (ИКНПВО, ATR)
В ИК НПВО луч инфракрасного света полностью отра-
жается внутри специально обработанного пропускающего инфракрасное
излучение материала, который имеет высокий показатель преломления.
Для изготовления ИК НПВО-призм обычно используют германий, кремний
и ZnSe. Поскольку показатели преломления полимера и призмы различны,
затухающая волна проникает через полимер, если он находится в тесном
контакте с призмой. Инфракрасное излучение взаимодействует с молеку-
лярными колебаниями таким же образом, как и в обычной инфракрасной
спектроскопии. Амплитуда затухающей волны убывает по экспоненте от
поверхности, поэтому глубина проникновения произвольно принята как
точка, где амплитуда снижается до 1/е (37%) от первоначального значения.
Глубина проникновения зависит от отношения коэффициентов преломле-
ния полимера и призмы, угла падения, и частоты излучения следующим
образом [193]:
(21)
где v — частота, п2 и пх — коэффициенты преломления полимера и призмы
соответственно, а 0 — угол падения инфракрасной волны. Зависимость глу-
бины проникновения не так проста, как предлагает это сотношение, потому
что коэффициенты преломления зависят также от частоты. Для неизотроп-
ных материалов, dp также зависит от направления вектора электрического
поля излучения относительно поверхности призмы. ИК НПВО-спектроско-
пия универсальная методика и теоретически может использоваться почти
в любой среде. Последнее свойство сделало ее очень важным методом для
описания характеристик ТПЭ, используемых в области медицинской ап-
паратуры.
Хотя ИК НПВО использовалась для определения количественных изме-
нений в составе поверхности ТПЭ [ 194], сопутствующее полезное свойство —
способность контролировать in situ процессы типа реакционно-инжек-
ционного формования [195] и адсорбции протеина на полиуретановой
подложке [196]. В последнем случае влияние скорости сдвига на кинети-
ческие процессы адсорбции протеина и десорбции из фосфатированного
буферного физраствора (ФБР, PBS) изучались в специально разработан-
ной проточной ячейке. Очень тонкая пленка из промышленного поли-
уретана Biomer (МДИ-ЭД-ПТМО) отливалась из раствора на призму из
германия. Толщина пленки была меньше глубины проникновения, так
что за концентрацией протеина можно было следить после вычитания
VI. Термопластичные эластомеры в поверхностях -•v- 623
инфракрасного поглощения полимера и фосфатированного буферного
раствора. Было установлено, что увеличение граничной скорости сдвига
не влияет на скорость адсорбции протеина, но скорость десорбции сни-
жается. В более поздних исследованиях, использующих по существу тот
же самый аппарат [197], спектральные изменения, соответствующие
конформационным изменениям протеина, наступали после адсорбции
протеина на полиуретановой подложке.
3. Рентгеновская фотоэлектронная спектроскопия
(РФС, XPS)
РФС (также называемая электронной спектроскопией для
химического анализа) проводится в вакууме и используется для определе-
ния относительного количества разных атомов на поверхности материала.
Может быть идентифицирован любой химический элемент, кроме водорода
и гелия; метод РФС также чувствителен к образованию химических по-
перечных связей. В РФС поверхность облучена низкоэнергетическими
рентгеновскими лучами (обычно алюминиевый или магниевый Ка), что
приводит к выбросу электронов внутреннего уровня. В качественном
отношении глубина проникновения ограничена неупругим рассеянием
выбиваемого фотоэлектрона, перемещающегося к поверхности. В коли-
чественном отношении, d — чрезвычайно сложная функция, которая
зависит от энергии рентгеновских лучей и структуры материала; обычно
глубина проникновения в полимер порядка нескольких ангстрем. Число
выбиваемых фотоэлектронов, которые достигают детектора, снижается эк-
споненциально с увеличением расстояния. d определяется как глубина, на
которой число испускаемых электронов достигает 37% от первоначального
значения; она пропорциональна cosO, где 0 — угол эмиссии относительно
нормали к поверхности образца.
Наиболее распространенным является использование рентгеновской
фотоэлектронной спектроскопии для количественного определения доли
каждой фазы на поверхности термопластичных эластомеров. Путем изме-
нения угла испускания можно исследовать композицию при различных
глубинах проникновения. Исследования с использованием РФС после-
довательно показали, что на свободной поверхности будет преобладать
компонент с низким значением поверхностной энергии. Даже маленькая
разность в поверхностной энергии обеспечивает сильную движущую силу
для обогащения поверхности компонентом с более низкой энергией. Влияние
скорости испарения растворителя и общего молекулярного веса изучалось
на симметричном полиметилметакрилат-полистироле (ПММА-ПС), отли-
том из толуола. Разность поверхностной энергии этих двух компонентов —
менее, чем 1 дин/см. Количество полистирола было больше на поверх-
ности пленки, полученной медленным испарением. Доля полистирола на
поверхности увеличивалась с увеличением общего молекулярного веса
624 -’v- Глава 13. Термопластичные эластомеры
и достигала постоянного значения, приблизительно 90%, когда общая
степень полимеризации была приблизительно 1000 [198]. Более позднее
исследование на отожженных пленках тех же самых материалов показало,
что слой полистирола полностью закрыл поверхность при степени полиме-
ризации более, чем 2500 [199]. В обоих исследованиях было установлено,
что избыток полистирола на поверхности Фр8 удовлетворяет следующему
уравнению:
ФР5 =a-b/V“,/2 (22)
где а и b — эмпирические константы. Эта форма уравнения согласуется с тео-
рией поля осредненных величин разработанной Фредриксоном (Fredrickson)
применительно к блочным сополимерам на поверхности [200]. Было дано
физическое истолкование константам, относящееся к различиям в поверх-
ностной энергии и способности поверхности изменять энергетическое взаи-
модействие между двумя полимерами. Было также установлено, что избыток
полистирола на поверхности уменьшается с увеличением температуры, что
соответствует теоретическим предсказаниям [201].
Основным недостатком этой методики является необходимость исполь-
зования сверхвысокого вакуума. Как было сказано ранее, поверхностный
состав исследуемых в сверхвысоком вакууме образцов может существенно
отличаться от их состава при нормальных условиях, особенно если мате-
риал погружен в жидкость, например, в областях применения, связанных
с контактом с кровью. Для количественного определения поверхностного
состава на жидкой межфазной границе были разработаны новейшие методы,
использующие сушку вымораживанием и измерения при температурах ниже
температуры стеклования мягкого сегмента. При исследовании различных
полиуретанов наблюдался избыток компонента с низкой поверхностной
энергией, когда спектры получали в нормальных условиях, тогда как при
использовании метода сушки вымораживанием в избытке был высокоэнер-
гетический компонент [202]. Это общее правило соблюдалось для любого из
двух случаев, был ли компонент с низкой поверхностной энергией мягким
или твердым сегментом. Это исследование демонстрирует возможность по-
верхностной перестройки, которая может происходить при любом изучении
полимерных поверхностей.
4. Масс-спектроскопия вторичных ионов (ВИМС, SIMS)
Когда поверхность бомбардируется пучком высокоэнер-
гетических первичных ионов, атомные столкновения между пучком
и твердым телом вызывают выброс вторичных ионов, которые могут быть
охарактеризованы с помощью масс-спектрометра. Эти вторичные ионы дают
информациею о типах атомов и молекул, присутствующих на поверхности.
Обычно для ВИМС-анализа используются ионы О2+, О“, и Аг+; первый ион
VI. Термопластичные эластомеры в поверхностях -*!/• 625
является наиболее общеупотребительным. Эксперименты ВИМС могут быть
как динамическими, так и статическими. При статической ВИМС интенсив-
ность ионного потока низкая (меньше чем 5 нА/см2), поэтому поверхность
протравливается очень медленно. Глубина проникновения для статической
ВИМС — приблизительно 10 А. Для динамической ВИМС токи в пучке
намного выше и последовательные слои быстро стравливаются в процессе
испытания. Этот метод, в конечном счете, приводит к образованию выемки
в образце из-за травления. Атомные разновидности могут быть охарактери-
зованы количественно как функция расстояния от поверхности, если могут
быть измерены отношения скоростей травления. В процессе исследования
методом ВИМС поверхность образца может стать заряженной, потому что
большинство полимеров являются непроводящими; этот заряд должен быть
удален в течение эксперимента. Для исследований методом ВИМС требуется
сверхвысокий вакуум.
ВИМС является чрезвычайно чувствительным методом (даже к миллион-
ным долям) и широко используется в полупроводниковой промышленности.
ВИМС и рентгеновская фотоэмиссионная спектроскопия (РФС) являются
дополняющими методами. Хотя они обеспечивают сходную информацию
[203], но разрешение по глубине в статической ВИМС существенно выше,
чем в РФС. Однако количественное использование ВИМС, как и РФС
в науке о полимерах в настоящее время не разработано. ВИМС использо-
валась для изучения поверхности полиуретанов [204, 205] и ламеллярного
диблоксополимера ПММА-ПС [206]. В последнем случае использовалась
динамическая ВИМС для того, чтобы показать, как после отжига ламелли
располагаются параллельно поверхности. Было показано, что толщина
ближайшего к свободной поверхности слоя, также как и слоя, лежащего
около поверхности полимерной подложки, составляет примерно половину
толщины внутренних слоев. Экспериментальные данные соответствовали
модели, которая учитывала стадию образования ламеллей в диблоксополи-
мере, и результаты были сравнимы с полученными методом МУРР для того
же самого материала.
5. Атомно-силовая микроскопия (ACM, AFM)
Концепция атомно-силовой микроскопии чрезвычайно
проста. По существу очень маленький зонд (размером 50-100 нм) пере-
двигается по поверхности («ощупывает», сканирует её) таким образом,
что сила давления зонда на поверхность сохраняется постоянной; для
исследования ТПЭ используется обычный метод «обстукивания», когда
на основное «ощупывание» накладываются вертикальные колебания. По
существу, измеряется прогиб образца под действием зонда как функция
координаты. Взаимодействие между зондом и поверхностью исследовано
недостаточно, но получаемое АСМ-изображение, повидимому, не сильно
зависит от характеристик зонда. Разрешающая способность АСМ находится
626 -'V Глава 13. Термопластичные эластомеры
в диапазоне 1 нм. Основным преимуществом метода АСМ является диапазон
сканирования, который допускает исследование поверхностей различного
качества и характеристику свободной поверхности при большой боковой
поверхности, т. е. толщины образца.
Рис. 17. Схематическая модель полиуретана с монодисперсными длинами жёстких
сегментов, показывающая сферолитную структуру жёстких доменов и ори-
ентацию цепи сегментов внутри сферолитов.
С использованием метода АСМ были получены изображения различных
ТПЭ, включая полиуретаны [207, 208], термопластичные эластомеры на ос-
нове полипропилена [209], сополиамиды [210] и синтезируемые анионной
полимеризацией блочные сополимеры [211,212]. Не только возможность ви-
зуализации топографических различий каждого образца, но также различия
между жесткими и мягкими сегментами, и, следовательно, форму этих обра-
зований на межфазной границе позволяет определить метод АСМ. Контрас-
тность обеспечивается разностью модулей жестких и мягких сегментов; эта
разность позволяет АСМ проводить различие между двумя компонентами.
И наоборот, образец может быть нагрет и одновременно получено изобра-
жение для выявления изменений в морфологии по мере того, как структура
размягчается [213]. В композициях, где кристаллизация является главной
движущей силой фазового разделения, полученные методом АСМ результаты
качественно идентичны результатам, полученным для стандартных частично
кристаллических полимеров. В композициях, где дело обстоит не так, на-
пример, полиуретанах, морфология может сильно отличаться. Например,
в композиции с монодисперсным распределением длин жестких доменов,
со сферолитной суперструктурой, ориентация осевой линии цепей такая
VIL Реология и переработка -* V 627
же, как в обычной сферолитной структуре, то есть, в направлении, танген-
циальном по отношению к радиусу, но складывания цепей не происходит,
как показано на рис. 17 [214].
VII. РЕОЛОГИЯ И ПЕРЕРАБОТКА
Некоторые особенности реологии эластомеров рассмотрены
в главах 5 и 6. Теоретическое развитие и методы определения характеристик,
изложенные в тех главах, в равной степени применимы и к ТПЭ. Однако
между обычным каучуком и ТПЭ имеются фундаментальные различия
в реакции на действие напряжения. Конечно, самое большое отличие заклю-
чается в том, что ТПЭ текут при высоких температурах, а сшитый каучук — нет.
В этом разделе будут обсуждены некоторые общие черты и различия между
этими двумя типами материалов. Кроме того, будут также представлены
методы переработки, используемые для ТПЭ. Технология переработки этих
материалов ничем не отличается от технологии переработки любого другого
термопластичного материала, и заинтересованный читатель может получить
информацию в монографиях по этой теме [215, 216].
При температуре эксплуатации и обычных частотах воздействия реоло-
гические свойства находящегося в твердом состоянии ТПЭ и традиционной
резины аналогичны, тогда как при больших деформациях ТПЭ, в отличие
от резины, в большинстве случаев будут иметь остаточную необратимую
деформацию. Существенные различия между ними появляются только при
изменении температуры и/или частоты. Если температура понижается или
частота повышается, качественные изменения свойств этих двух материа-
лов очень схожи. Сопротивление обоих материалов действию напряжения
возрастает вплоть до температуры стеклования Т мягкого сегмента в ТПЭ
или высокоэластичной матрицы в традиционной резине. Ниже этой темпе-
ратуры и обычный вулканизат, и ТПЭ ведут себя как однофазный хрупкий
стеклообразный полимер. По мере повышения температуры наблюдается
качественное различие в поведении двух материалов. Обычный вулканизат
продолжает вести себя как эластомер, пока не будет достигнута температура
деструкции. В ТПЭ жесткие домены в конце концов разупрочняются и текут,
что позволяет перерабатывать материал как обычный термопласт. Режимы
переработки будут тесно связаны с температурой перехода микрофазового
разделения, так как при ПМР вязкость заметно уменьшается.
Существенно различаются динамические механические свойства ТПЭ
и традиционных резин. Уравнение ВЛФ (WLF) (см. главу 5) обычно не
применяется для ТПЭ, так как двухфазность делает материал сложным ре-
ологическим объектом. Динамические механические исследования исполь-
зуются в качестве макроскопической характеристики фазового разделения,
поскольку материал с более сильным фазовым разделением будет иметь
628 -* V Глава 13. Термопластичные эластомеры
больший температурный интервал между переходами мягкого и жесткого
сегментов и модулями на плато высокоэластичности.
На рисунке 18 [217] показана зависимость Е' и тангенса 3 от содержания
жестких сегментов для ряда полиуретанмочевин. Эта зависимость нагляд-
но подтверждает, что чем больше содержание жесткой фазы, тем лучше
фазовое разделение. Рисунок 18 также представляет основные характерис-
тики, определяемые всеми динамическими механическими методами для
ТПЭ: температуру стеклования гибкого сегмента, которая ниже комнатной
температуры и температуру плавления жесткого сегмента, которая выше
комнатной температуры. Нормально область работоспособности ТПЭ это
область плато между двумя температурными переходами. Как следует из
диаграммы, можно сдвигать верхнюю температуру перехода путем увели-
чения перемешивания фаз, которое обычно достигается путем уменьшения
длины жестких сегментов.
Температура (°C)
Рис. 18. Зависимость динамического модуля упругости (Е') и тангенса 3 (Е'/Е")
от температуры для полиуретанмочевины МДИ-ЭД-ПТМО (Молекуляр-
ный весПТМО = 1000). Мольное соотношение компонентов составляет:
3:2:1 (---------); 2:1:1 (---); 1,3:0,3:1 (-).
Реологические измерения могут также дополнять определение темпе-
ратуры перехода микрофазового разделения независимо от экспериментов
по рассеянию. При помощи этого метода динамический модуль упругости
и модуль потерь определяются как функция частоты при различных тем-
пературах выше и ниже ПМР. При проведении этих исследований должен
VII. Реология и переработка -• V 629
использоваться один уровень напряжений, так как показано, что ниже
ПМР динамические механические свойства существенно зависят от напря-
жения. Если частота достаточно высокая, можно использовать уравнение
ВЛФ, позволяющее совместить все данные на одной кривой зависимости
модуля от приведенной частоты. Частота, при которой кривые начинают
расходиться, является критической частотой (сос) материала. Критические
частоты не обязательно идентичны для динамического модуля упругости
и модуля потерь, но вообще отличаются меньше чем на порядок величины.
После того, как сос определена, динамический модуль упругости или модуль
потерь измеряется как функция температуры при постоянной частоте, кото-
рая значительно ниже критической. Температура, при которой происходит
разрыв непрерывности на кривой для модуля, представляет собой ПМР
температуру, как показано на рис. 19. Авторы рекомендуют использо-
вать динамический модуль упругости, потому что разрыв непрерывности
в этом случае более резкий. Этот метод основывается на том, что поведение
динамического модуля упругости и модуля потерь при низких частотах
различно для разупорядоченных и упорядоченных систем. Поведение
разупорядоченной системы идентично нормальному установившемуся
поведению однофазных материалов, то есть, G' ~ со2 и G"~ со, в то время
как в упорядоченной системе оно зависит от морфологии и ориентации
материала; для макроскопически неориентированной ламеллярной сис-
темы, G' ~ G" ~ со0’5.
Вязкость ТПЭ сильно зависит от того, где, выше или ниже ПМР темпе-
ратуры, находится система. Например, промышленно важные материалы,
такие как бутадиен-стирольные трёхблочные сополимеры имеют темпера-
туру ПМР выше приемлемой температуры переработки; поэтому порядок
величины вязкости расплава блочного сополимера выше, чем для гомополи-
меров того же молекулярного веса. При высоких скоростях сдвига типичные
значения вязкости составляет 104 пуаз; порядок величины может возрастать
с уменьшением скорости сдвига.
Обычно ТПЭ перерабатываются как любой другой термопласт, то есть,
литьем под давлением в форму методом впрыска, экструзией, каландро-
ванием и т.д. Некоторые ТПЭ, например, иономеры, имеют склонность
прилипать к поверхности, так что может быть необходимо специальное
оборудование и способы для выемки деталей, полученных литьем под
давлением. Температура переработки должна быть выше температуры
размягчения жесткого сегмента, но даже двухфазные расплавы могут
перерабатываться на обычном оборудовании. Вязкость термопластичных
эластомеров при температурах выше ПМР довольно низкая по сравнению
с вязкостью других термопластов из-за низкого общего молекулярного
веса. Действительно, для некоторых однофазных материалов с низким
общим молекулярным весом типа полиэфиров из-за низкой вязкости не
630
Глава 13. Термопластичные эластомеры
может использоваться формование с раздувом [218]. Сформировавша-
яся морфология и свойства материала сильно зависят от метода перера-
ботки.
7
GJ= 10.0 с
6
2% амплитуда
деформации
5
сч
ж 4
S
о
? 3
о
2
Упорядоченный
переход
_ микрофазного
разделения
о
1
Геометрическое
температурное ।___।
разрешение
01 - —1----------------1------------------
275 280 285 290 295 300
Т, °C
Рис. 19. Температурная зависимость G', динамического модуля при сдвиге, при
частоте 1,6 Гц для двухблочного сополимера ПЭП-ПЭЭ. Рассчитанная
температура ПМР составляет 291±1°С.
Реакционно-инжекционное формование (РИФ, RIM)*) является спе-
циализированным методом, очень распространенным в производстве по-
лиуретанов. Два жидких потока, один из которых содержит диизоцианат, а
другой диолы, смешиваются ударным смешением при высоком давлении.
Для создания высокого давления обычно используются поршневые насосы
высокого давления, а также требуется точное дозирование компонентов. Ди-
изоцианат может заранее прореагировать до ударного смешения с некоторым
количеством многоатомного спирта в соответствии с методом получения пре-
полимера, обсужденным ранее. Смешанный материал быстро впрыскивается
в закрытую форму таким образом, чтобы поток был ламинарным. Материал
быстро отверждается, в результате получают готовую деталь.
#) Подбробнее в кн.: Любартович С.А., Морозов Ю.Л., Третьяков О.Б. Реакционное фор-
мование полиуретаов. — М.: Химия, 1990. — 288 с.
VIII. Практическое применение -*v- 631
VIII. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ
Как было показано ранее, наиболее важным классом термо-
пластичных эластомеров являются диенстирольные трёхблочные сополиме-
ры. В отличие от большинства других ТПЭ, их можно смешивать с большим
количеством добавок без существенного влияния на свойства. Почти всегда
при практическом применении фактическое содержание трёхблочного со-
полимера составляет менее 50%. Масла используются как технологические
добавки, не приводящие к существенной потере свойств, если не пластифи-
цируются полистирольные домены. По этой причине отдается предпочтение
нефтяным маслам. Использование инертных наполнителей типа глин или
мела приводит к снижению стоимости конечного материала. В отличие от
традиционных резин, инертные наполнители не оказывают существенного
влияния на механические свойства ТПЭ. Термопласты типа полиэтилена
или полипропилена используются для улучшения стойкости к действию
растворителей и могут увеличивать верхнюю температуру эксплуатации.
Гомополимер полистирола используется как технологическая добавка,
которая также увеличивает весовую долю твердой фазы и обуславливает
упрочнение материала.
Полимеры «Кратон» (KRATON Polymers), самый большой мировой про-
изводитель бутадиен-стирольных блоксополимеров, производит четыре типа
материалов. Композиции на основе СБС используются в обувных подошвах и
как модификаторы свойств асфальта. В последнем случае применения блок-
сополимер является дополнительным компонентом (-10%), но улучшает
эластичность при низкой температуре и увеличивает температуру размяг-
чения асфальта. Изопрен-стирольные блоксополимеры (СИС) используют
почти исключительно как клея, тогда как СЭБС-триблоксополимеры приме-
няются как конструкционные материалы. В адгезивах, склеивающихся под
действием давления, для получения материала с большей клейкостью необ-
ходимо, чтобы содержание полистирола было низким, для клеев-расплавов
липкость адгезива может быть нежелательной и приемлемы более твердые
материалы. Отсутствие двойной связи в СЭБС-сополимерах обеспечивает
им существенное улучшение сопротивления УФ излучению и высоким
температурам, делая возможным использовать СЭБС-сополимеры вместо
СИС и СБС в тех областях применения, где это сопротивление необходимо.
Наконец, путем малеинизации в СЭБС вводятся кислотные группы, которые
улучшают адгезию к различным подложкам. Эти малеинизированные мате-
риалы часто используются в виде компаундов, содержащих неорганический
наполнитель. Практическое применение термопластичных полиолефинов
(ТПО), полиуретанов, полиэфиров и полиамидов существенно отличается
от применения триблоксополимеров. В этих материалах используются
только небольшие количества добавок, так как большие количества любой
добавки имеют тенденцию существенно ухудшать их механические свой-
632 Глава 13. Термопластичные эластомеры
ства. Области применения этих полиблочных сополимеров определяются
их преимуществом по сопротивлению истиранию, сопротивлению раздиру
и твердости. ТПО немного дороже термопластов массового потребления;
а сегментированные блочные сополимеры значительно дороже их и, сле-
довательно, их применение должно оправдывать увеличенную стоимость
улучшением механических свойств. В автомобильной промышленности ТПЭ
используются для изготовления внутренних и наружных деталей. Например,
ТПЭ применяют для изготовления чехлов, уплотнителей для мест стыка, для
некоторых шлангов, а также для наружных бамперов и некоторых панелей.
Другие промышленные области применения включают рукава, систему
привода велосипеда и кабели.
Область медицинской аппаратуры несомненно является очень важным
потребителем ТПЭ, хотя по объему этот рынок невелик. Одной из наиболее
значимых областей применения является использование полиуретанов при
пластических операциях на сосудах. Полиуретаны (также как полиамиды
и полиэфиры) входят в состав материала надувного баллона, вводимого
в сосуды, который затем раздувается и увеличивает сечение артерии для
улучшения кровотока. Дополнительную информацию о применении по-
лиуретановых ТПЭ в медицинской аппаратуре можно найти в монографии
по этой теме. Один из ее авторов является также одним из авторов данного
обзора [219]. Спортивные товары - весьма значительная группа товаров
широкого потребления, производимых из ТПЭ. Обувь, включая лыжные
ботинки и футбольную обувь, часто содержит значительное количество
ТПЭ. Особенно большой областью применения ТПЭ является изготовление
подошвы для спортивной обуви. Спортивные изделия типа лыж и покрытие
мячей для игры в гольф - области, где также используются ТПЭ. Полиуре-
таны применяются в качестве покрытия дорожек и открытых площадок.
Наконец, ТПЭ начинают использоваться в производстве снаряжения для
открытого воздуха типа тентов и палаток из-за их низкой водопроницае-
мостии и высокой прочности.
ГЛАВА
14
ТЕХНОЛОГИЯ ШИН
БРЕНДАН РОДЖЕРС и УОЛТЕР ВЕДДЕЛЛ
Эксонмобил Химическая компания
Хьюстон, Техас
I. ВВЕДЕНИЕ
В истории человечества колесо является одним из наиболее
важных изобретений, поскольку оно нашло применение в широкой области
приложений, таких как транспортная техника, строительное оборудование
и детали машин. Подобно многим изобретениям, колесо было развитием
таких более ранних устройств как каток, используемый для передвижения
тяжелых объектов и датируемый бронзовым веком, отстоящим от нашего
времени более чем на 5000 лет. Колесные транспортные средства суще-
ствовали в Шумере в 3500-х, в Ассирии в 3000-х и в центральной Европе
в 1000-х годах до нашей эры. Четырехколесные повозки с поворачивающей-
ся передней осью, используемой для управления, были отмечены в 1500-х
годах до нашей эры.
С появлением в Месопотамии быстрых лошадей из азиатских степей
тележка была приспособлена для военных целей. Было использовано ко-
лесо со спицами, и тогда же появилась первая «шина», состоящая сначала
из кожаного, а затем из медного и железного обода, предохраняющего от
повреждений деревянное колесо.
Следующее наиболее важное событие произошло, вероятно, в 1846 году,
когда Томпсон получил патент на упругую воздушную трубку, фиксируемую
на колесе, которая уменьшает усилие тяги экипажа, облегчает движение
и снижает шум. Эта идея была существенно усовершенствована в 1880-х
годах, когда была разработана первая пневматическая шина для трехко-
лесных велосипедов.
Открытие вулканизации Чарльзом Гудьиром в 1839 г. и индустриализа-
ция Европы и Северной Америки дали возможность шине эволюционировать
от обрезиненного холста, покрывающего резиновую трубку, к комплексному
композиту из ткани, стали и эластомера.
634 -lb Глава 14. Технология тин
II.
ТИПЫ И ХАРАКТЕРИСТИКИ ШИН
В терминах категорий продукции и понимания потребителей
пневматические шины разделяются по существу на девять типов, которые оп-
ределяются применением транспортных средств. Имеются шины для скоро-
стных автомобилей, легковых автомобилей и легких грузовых автомобилей,
у которых вес с грузом, как правило, не превышает 7250 кг. В таких шинах
для усиления используется значительное количество тканей. Большие шины,
такие как шины для тяжелых грузовых автомобилей, сельскохозяйственных
машин, строительного оборудования и больших самолетов, представляют
собой сочетание стальной проволоки и усиливающих тканей. Кроме того,
имеется ряд специальных шин, который включает шины, используемые
на вилочных подъемниках, легких самолетах, легком производственном
оборудовании и машинах для гольфа.
Положительный угол наклона
Стабилизирующий момент М
Плоскость дороги
Боковая сила F
Центр контакта
шины
V(S>5
X
X
X
+х
Продольная сила F*
Направление движения
колеса
Положительный угол увода
Плоскость колеса
X
X
Момент сопротивления
качению М
Момент = FxR^ + MvC0Sy + A4zsin,
Опрокиды-
вающий момент М
Нормальная сила Fz
(вертикальная нагрузка)
Рис. 1. Силы и моменты шины, действующие вщентре контакта шины. (Воспроизве-
дено с разрешения. © 1988 34-ая Биккендальская лекция Общества инже-
неров транспортников).
В соответствии с конструкцией и применением все пневматические шины
должны обеспечивать фундаментальный ряд функций:
1. Способность нести нагрузку
2. Смягчение ударов и демпфирование
II. Типы и характеристики шин -’и- 635
3. Передача тяговых и тормозных моментов
4. Создание боковых сил
5. Стабильность размеров
6. Сопротивление истиранию
7. Реакция на управление
8. Низкое сопротивление качению
9. Минимальный шум и минимальные вибрации
10. Заданная долговечность.
Демпфирующие характеристики, упругие свойства резины и уникаль-
ная деформируемость и восстанавливаемость в совокупности делают шины
единственным изделием, которое удовлетворяет всем этим функциям.
Направление вперед
(работоспособность)
Рис. 2. Функции шины.
По существу три параметра определяют функции шины. Это (1) основное
назначение автомобиля; (2) механические свойства и такие характеристики,
как сопротивление износу и прочность конструкции; (3) эстетичность, ком-
фортабельность и характеристики движения, такие как точность управления
автомобилем.
Механические свойства шины описываются характеристиками её
реакций на приложение нагрузки, момента и управляющего воздей-
ствия, что проявляется в возникновении внешних сил и прогиба. Такие
механические свойства являются взаимосвязанными и, следовательно,
конструкторские решения, действующие на один фактор, будут влиять
и на другие факторы позитивно или негативно. Результат представлен
комплексной системой сил, действующих на катящуюся шину автомо-
биля (Рис. 1).
На рис. 1 показана система осей координат с началом в центре контакта
шины с дорожной поверхностью. Ось х — пересечение плоскости колеса
636 Jb Глава 14. Технология тин
и плоскости дороги с положительным направлением вперед, ось z — пер-
пендикуляр к плоскости дороги с положительным направлением вниз.
Следовательно, нормальная сила, действующая на шину, является поло-
жительно направленной, а реакция на вертикальную нагрузку, которая
является давлением дороги на шину, рассматривается как отрицательная.
Ось у лежит в плоскости дороги, её направление определяется так, чтобы
система координат была правой и ортогональной. Представленная система
осей точно соответствует системе осей для автомобиля, где шина установлена
на правой передней позиции. В этом случае автомобиль осуществляет левый
поворот с положительным направлением стабилизирующего момента (Afz)
и отрицательным вектором боковой силы. Силы, действующие на шину,
могут быть разделены на три фундаментальных вектора: вертикальные
силы контролируют впечатление от автомобиля и комфорт, боковые силы
осуществляют контроль движения автомобиля, продольные или тяговые
силы контролируют такие характеристики движения, как сопротивление
качению (рис. 2).
III. ОСНОВЫ КОНСТРУКЦИИ шины
Шина по существу является резинокордным композитом.
Все шины имеют слои усиливающих нитей корда, уложенные поперечно от
борта к борту. На верхней части слоев имеется брекер, расположенный под
протектором. Корд брекера обладает низкой растяжимостью и выполнен из
стали или ткани в зависимости от назначения шины. Нити корда брекера
ориентированы под относительно малым углом между 15° и 25° и служат
как ограничители для 90-градусных слоев каркаса.
А. Конструкция шины
В шине находится ряд специализированных компонентов,
которые служат для обеспечения результата, отвечающего требованиям
к конструкции и внешнему виду. Например, легковые шины высокой рабо-
тоспособности могут иметь найлоновый защитный слой, также называемый
покровным слоем, который расположен над пакетом брекера. Найлоновый
покровный слой ограничивает и контролирует увеличение размера шины,
вызываемого центробежными силами, возникающими при высоких скоро-
стях. Заворот слоев, образующий форму, в которой слои каркаса оберты-
ваются вокруг бортовой проволоки и заворачиваются на боковую стенку,
закрепляет слои каркаса на бортовом кольце и усиливает нижнюю часть
области боковой стенки. Обертка носка, которая является усиленным най-
лоном резиновым компонентом, защищает носек борта от разрывов во время
монтажа и демонтажа. Выше носка бортовая лента дополнительно защищает
область борта от истирания о фланцы обода при эксплуатации (Рис. 3).
Рис. 3. Поперечное сечение легковой шипы высокой работоспособности.
III. Основы конструкции шины
ш
638 -»L- Глава 14. Технология шин
Б. Компоненты шины
Шина является сборкой ряда частей или деталей, каждая из
которых имеет специфические функции в эксплуатации и работоспособности
изделия. Рис. 4 иллюстрирует ключевые компоненты грузовой шины. Далее
приводятся части шины:
Протектор Компонент, сопротивляющийся износу шины в контакте с дорогой. Он должен также осуществлять тяговое усилие, сцепление с мокрой дорогой и хорошие характеристики управления с минимальной генерацией шума и низким теплообразованием. Компоненты протектора могут со- стоять из смесей натурального, полибутадиенового (ПБ) и бутадиенстирольного (БСК) каучуков, смешанных с тех- ническим углеродом, силикой, маслами и вулканизующими химикатами.
Плечевая зона протектора Верхняя часть боковой стенки; влияющая на рассеивание тепла от протектора и на свойства шины, определяющие управление.
Подканавочный слой Также называемый «подушка»; резина, обеспечивающая хорошую адгезию между брекером и протектором, рассеи- вание тепла и низкое сопротивление качению.
Боковина покрышки Защита каркаса от боковых царапин, контроль характери- стик движения системы автомобиль - шина, также помогает поддерживать протектор. Резины состоят из натурального каучука, ПБ и БСК с техническим углеродом и рядом масел и органических химикатов.
Защита от бордюра Выступ на резине боковой стенки по окружности шины, защищающий её от царапанья по бордюру.
Борта Нерастяжимые петли стальной проволоки, которые закреп- ляют слои, и также закрепляют шину на колесе так, что она не будет скользить или качаться на ободе.
Компоненты бортовой зоны Наполнительный шнур; бортовая лента, которая защищает компоненты проволочного борта; оберточная лента, которая защищает нижнюю часть боковой стенки и крыльевая лента, которая помогает удерживать борт на месте.
Слои Слои текстильные корда или металлокорда, протянутые от борта до борта и тем самым служащие как первичный усиливающий материал в каркасе шины.
Брекер Слои текстильного корда или металлокорда, располо- женные под протектором и служащие для ужесточения каркаса. Брекер улучшает износостойкость, реакцию на управление и сопротивление повреждениям, а также защищает слои корда каркаса от опасных воздействий дороги.
IV. Конструирование шины -’V- 639
Резиновый клин под
краем брекера
Герметизирующий
слой
Резина с высоким уровнем адгезии в области боковой стенки
между брекером и каркасом, улучшающая износостойкость
протектора и прочность.
Бутилкаучук или галопроизводные таких полимеров, кото-
рые удерживают сжатый воздух внутри шины.
Рис. 4. Компоненты радиальной грузовой шины.
IV. КОНСТРУИРОВАНИЕ ШИНЫ
По существу шина является композицией различных эласто-
меров, нитей, тканей и стального корда. Термин структура шины означает
число, положение и размеры различных компонентов, используемых в этой
композиции. Основными компонентами, определяющими работоспособ-
ность шины, являются слои каркаса, конструкция борта, брекер, бокови-
на, герметизирующий слой и протектор. Бортовая лента, крыльевая лента
и покровные слои, которые являются полосками обрезиненной ткани, рас-
положенными в бортовой и коронной зонах шины, называются вспомога-
тельными компонентами, потому что они защищают основные компоненты,
минимизируя концентрации напряжений [1,2].
640 JV Глава 14. Технология шин
А. Наименования и размеры
Терминология, используемая для описания размеров шины
и обода, показана на Рис. 5. Эти обозначения обычно используются всей
шинной промышленностью для описания факторов размера, высоты и
клиренса проема колеса в добавление к вычислению изменяющихся ве-
личин, таких как нагрузочная способность и число оборотов на единицу
пути [3, 4, 5]. Кроме того, должны быть указаны следующие определения:
Отношение Н/В Вычисляемая величина, выражающая отношение высоты профиля шины к его ширине. Величина Н/В, равная 70, означает, что высота профиля шины составляет примерно 70% от его ширины.
Угол нитей корда Угол между направлением нити корда и окружным направлением в шине; преобладающий фактор влияния на форму или контур шины. В радиальных шинах угол нитей корда каркаса равен 90°
Наружный диаметр Диаметр новой ненагруженной шины, смонтирован- ной на ободе.
Ширина профиля Ширина профиля новой шины, исключая боковые ребра, надписи и декоративные элементы.
Статический радиус Расстояние от дорожной поверхности до осевой ли- нии обода нагруженной шины.
Минимальный двойной интервал Минимальное расстояние, рекомендуемое для опти- мальной работоспособности, между осевыми линия- ми ободов сдвоенных колес.
Длина площади контакта Ширина площади контакта Контурная площадь контакта Фактическая площадь контакта Длина площади контакта нагруженной шины. Ширина площади контакта нагруженной шины. Полная площадь контакта нагруженной шины. Площадь протектора, исключая канавки, определен- ная по отпечатку нагруженной шины. Насыщенность рисунка - процентное отношение фактической пло- щади к контурной.
Асимметрия Конструкция шины, в которой рисунок протектора с одной стороны от центральной линии отличается от рисунка с другой стороны.
Уровень нагруженности Максимальная нагрузка, которую шина может} нести при данных условиях и определенном внут- реннем давлении. Для определения нагрузочной способности шины также используется термин «Индекс нагрузки». Уровни нагрузки определе- ны таблицами Tire & Rim Association. Таблица 1 иллюстрирует величины нагрузки для одинарной смонтированной радиальной грузовой шины сред- него размера.
IV. Конструирование шины -*v 641
Используются три основных системы обозначения размеров шины:
1. Шины обычных размеров, устанавливаемые на ободах с плоской базой,
обычно камерные шины
2. Шины обычных размеров, устанавливаемые на ободах с наклоном
полок 15е или бескамерные
3. Метрические шины, также установленные на ободах с наклоном полок
15е; бескамерные
Кроме того, буква «R» в обозначении размера означает «радиальные»,
«D» или «- » — диагональные и «МЬ» означает шины для горных и лесных
работ. Буква «Р» означает шину для легковых автомобилей и «ЬТ» шину
для легких грузовиков.
I
Ширина
Ширина
нагруженной
шины (LS)
Рис. 5. Размеры шины и обода.
Контурная
площадь
контакта
Например, обозначение шины «LT235/75R15»
имеет следующее зна-
чение:
LT Легкогрузовой автомобиль
235 Приблизительная величина ширины профиля шины в миллиметрах, при
монтаже на требуемый обод
Соотношение Н/В
75
642 -lb Глава 14. Технология шин
R Радиальная конструкция
15 Номинальный диаметр обода в дюймах
Шина тяжелых грузовых автомобилей будет иметь обозначение типа 11 R24,5
для обычных бескамерных шин, а низкопрофильные метрические шины
обычно могут быть обозначены как 295/75R22,5. Так для обычной беска-
мерной шины:
11 Номинальная величина ширины сечения в дюймах
R Радиальная конструкция
24,5 Диаметр обода в дюймах
Шины обычного размера имеют стандартное соотношение Н/В, равное
80. Начальным шагом конструирования шины является определение тре-
буемого размера. Он определяется диаметром обода, колесным проемом,
эксплуатационными нагрузкой и скоростью, внутренним давлением. Для
метрических шин тяжелых грузовых автомобилей эти факторы определя-
ются по уравнению
L = (6.075 х 10’5)К х Р° 7 х 1 (Dt + 5d) (1)
где L — нагрузка при 100 км/час (кг); Р — давление (кПа); Sd — фактор
размерности, ширина профиля, исправленная по Н/В; Dr — диаметр обода
и К — постоянная, зависящая от скорости автомобиля. Используя уравне-
ние (1), разработанное Tire & Rim Association, инженер-шинник может оп-
ределить оптимальный размер шины для конкретных условий применения.
Обычно требования к нагрузке известны, так что по уравнению (1) можно
определить требуемое рабочее давление. Этот процесс затем используется
для выбора индекса размера и нагрузки. В процессе конструирования часто
используются стандарты шинной промышленности [5, 6]. Спецификация
нагрузок приведена в таблицах 1 и 2.
Таблица 1 Величины нагрузки, установленные Tire & Rim Association для
одинарной смонтированной радиальной грузовой шины среднего
размера [3].
Ранг нагрузки Норма слойности Пример размера шины Предел нагрузки на шину (фунт) Внутреннее давление
F 12 295/75R22,5 5.070 90
G 14 295/75R22,5 5,780 100
Н 16 295/75R22,5 6,610 120
Н 16 285/75R24,5 6,780 120
J 18 385/65R22,5 9,370 120
L 20 425/65R22,5 11,400 120
IV. Конструирование шины -* V 643
Таблица 2. Цифровой код индекса нагрузки для несу-
щей способности шины по European Tire and Rim Technical
Organization ETRTO [4].
Индекс нагрузки Несущая способность шины (кг)
144 2800
145 2900
146 3000
149 3250
152 3550
155 3875
160 4500
Б. Конструирование прессформы
Конструирование прессформы шины начинается с определе-
ния всех измерений накачанной шины требуемого размера. По накачанной
шине и изменению размеров шины при накачивании рассчитываются пред-
варительный контур слоя и размеры прессформы (Рис. 6). Как только стали
известны граничные размеры прессформы, расположение контура слоя,
глубина и ширина протектора, могут быть установлены контуры протектора,
плечевой зоны, боковой стенки и компонентов борта. Эти размеры и контуры
разрабатываются с использованием вычислительной техники (Рис. 7).
Первичный интерес в конструировании шины лежит в определении зон
брекера и обода и натяжения нитей корда брекера и каркаса. Поскольку
радиальная шина имеет многослойный брекер, эта совокупность слоев рас-
сматривается как пакет. Жесткость пакета слоев брекера радиальной шины
является функцией угла и калибра нитей корда брекера, толщины брекера
и резины. Рис. 8 иллюстрирует конфигурацию четырехслойного брекера
радиальной грузовой шины. С эмпирической точки зрения увеличение жес-
ткости пакета брекера при сохранении его ширины приводит к улучшению
износостойкости протектора.
Жесткость брекера шины может быть количественно оценена пара-
метром «жесткость по Гафу (Gough)» (5), который измеряет жесткость
при изгибе в плоскости слоя, состоящего из жесткой проволоки, корда
и резины. Высокая жесткость брекера радиальной шины по короне мо-
жет значительно улучшить износостойкость протектора при установке
твердого основания для резины протектора [7, 8]. При тщательном
исследовании Гаф определил, что для прогнозирования относительной
644 JV Глава 14. Технология шин
износостойкости протектора может быть использована простая балочная
модель особенностей конструкции шины. С учетом сдвигового и изги-
бающего моментов в расчете, было получено эмпирическое уравнение,
определяющее параметр S для простой пластины
S = P/d (2)
где нагрузка Р есть сила, приложенная к центральной зоне пакета слоев или
пластины и создающая прогиб d.
Рис. 6. Границы контура слоя.
В шине жесткость по Гафу есть функция окружного и сдвигового моду-
лей композита.
S = (Е х G)/(C\ х Е) + (С2 х G) (3)
S— жесткость по Гафу, Е— окружной модуль резинокордной пластины, G —
сдвиговой модуль резинокордной пластины, Сх и С2 — константы, завися-
щие от размера шины. По существу уравнение (3) может быть упрощено до
модели простой поддерживающей балки длиной L с упругими константами
EuG, прогибаемой на расстоянии d силой Р
S = PI?/4&EI + 2PL/%AG (4)
IV. Конструирование тины -*v- 645
где А — есть площадь поперечного сечения, в котором приложена сила Р,
а I — момент инерции балки.
Результаты компьютерного анализа влияния угла брекера на жесткость
по Гафу четырехслойного пакета слоев показаны на рис. 8, б.
Центральная
плоскость
шины
Наружный
диаметр
прессформы
Ширина п !
протектора •
Диаметр обода
Контур
слоя
Ширина профиля
прессформы
Радиус верхней
части боковой
стенки
Радиус
боковой
стенки
части боковой
стенки
Радиус
плечевой зоны
средней части
Радиус нижней
। - Ширина
| основания
• прессформы
Рис. 7. Полость прессформы, разработанная по CADAM.
Для конфигурации четырехслойного брекера «правый/ правый/левый/
правый» угол корда (0) второго и третьего слоев варьируются от 10° до 26°
Требуемая жесткость по Гафу достигается вычислением оптимальной струк-
тура пакета слоев брекера.
Для анализа формы поперечного сечения накаченной и деформированной
нагрузкой шины и напряженного состояния в зоне брекера используются
такие методы расчета структуры как метод конечных элементов (МКЭ).
Такое исследование дает как количественный анализ, так и качественное
сравнение уровня оптимальности конфигурации брекера. На рис. 9 пока-
зана грузовая шина для тяжелых условий работы в нагруженном и нена-
груженном состояниях. Частота сетки конечных элементов конструируется
так, чтобы сохранить особенности геометрии поперечного сечения и в то же
время сохранить полное число узлов сетки.
При анализе пакета брекера и определении уровня плотности энергии
деформации оценка может производиться в трех состояниях шины; на-
646 -Jb Глава 14. Технология шин
качанная шина, нагруженная шина в сечении, противоположном центру
площади контакта, нагруженная шина в центре площади контакта. Рис. 10
иллюстрирует эти три случая. Он показывает, что плотность энергии де-
формации (в мегапаскалях) в накачанной шине подобна той, что имеет
место в сечении противоположном центру площади контакта нагруженной
шины. Но в центре площади контакта плотность энергии деформации по
углу брекера возрастает от 0,01-0, 27 МПа до 0,05-0,51 МПа. В реальных
шинах эти зоны высокой деформации соответствуют типичным регионам,
чувствительным к разрушениям.
#4 Подпротекторный
слой
#2&3 Рабочий слой
#1 Разделенный
переходный слой
V 1400
1200
>& 1000
Л
О 800
С
Й 600
I 400
Каркас =90°
Радиальная грузовая шина
295/75R22.5
Брекер #1=65° (правый)
#2=0 (правый)
#3=0 (левый)
#4=21°(правый)
I -----—j ,
10 12 14 16 18 20 22 24 26
Угол нитей корда (6)
Рис. 8. Типичный четырехслойный брекер грузовой шины (а). Изменение жесткости
по Гафу (б).
Подобно зоне брекера в шине, область борта также доступна конечно-
элементному анализу. Подгонка деталей сетки к борту позволяет выполнять
анализ деформаций концов слоев как в накачанной, так и в нагруженной
шине, делающей полный оборот. Рассматривая рис. 11, возможно количест-
венно оценить по МКЭ деформации, вызываемые давлением, и циклические
деформации в шине без изготовления и испытания шин.
IV. Конструирование шины 647
В. Натяжение корда
Выбор материала корда для брекера и слоев корда и соот-
ветствующего их расположения в шине приводит к необходимости дальней-
шего конечноэлементного анализа. При рассмотрении накачанной шины
без брекера выясняется, что слои корда, т. е. единственный слой каркаса
радиальной шины или многослойная диагональная конструкция, будут
принимать конфигурацию, которая минимизирует деформацию композита.
Соответствующее расположение нитей корда определяется как нейтраль-
ный контур. Опоясанность шины ограничивает диаметр накачанной шины
и нейтральный контур или линия слоя такой системы соответственно из-
меняется.
Рис. 9. Структура конечных элементов при расчете тяжелой грузовой шины.
/
Контур слоя определяется как показано на рис. 12. Принципы опреде-
ления контура слоя были разработаны Парди (Purdy), пионером — матема-
тиком, который вывел основные математические уравнения для пути корда
и свойств шины [9].
Типичные графики натяжения корда в накачанной грузовой шине по-
казаны на рис. 13. В накачанном, но ненагруженном состоянии натяжение
648 -»L- Глава 14. Технология шин
корда в брекере сосредоточено у центральной линии, и натяжение в слое
каркаса наибольшее в точке, соответствующей положению боковой стенки.
Однако при приложении нагрузки к накачанной шине и возникающем при
этом прогибе натяжение корда возрастает на краях брекера в стороне от
центральной линии, а также в зоне борта. Как было сказано раньше, эти
две области в конструкции шины имеют тенденцию быть зонами разру-
шения (рис. 14).
Рис. 10. Конечноэлементный анализ зоны борта, показывающий плотности энергии
деформации (в МПа): накачивание (а); нагружение, 180° от центра контакта (б);
нагружение, в центре контакта (в). \
Г. Конструирование рисунка протектора
Рисунок протектора влияет на способность шины передавать
тяговые, тормозные и боковые силы при движении в широком диапазоне
дорожных и внедорожных поверхностей. Конструкция рисунка протектора
по существу есть разделение гладкого протектора на малые элементы или
блоки. Эти элементы располагаются как повторяющийся рисунок выемок,
ребер, грунтозацепов, щелей и канавок. Рисунок в первую очередь описы-
вается в терминах длины, ширины, процентной доли выемок в различных
IV. Конструирование шины -’V 649
элементах и величины углов. Элементы протектора по существу определяют
тяговые характеристики, оптимальную величину насыщенности рисунка и
минимальное шумообразование [1, 10, 11].
Рис. 11. Конечноэлементный анализ зоны борта (а). Напряжение на конце слоя в за-
висимости от расстояния от центра контакта (б).
Для грузовых шин рисунок протектора может быть классифицирован по
пяти основным типам (рис. 15):
1. Ребра дорожного рисунка
2. Комбинация «ребра / грунтозацепы» дорожного рисунка, где ребро
может быть на внутреннем или на внешнем краю плечевой зоны
650 J\- Глава 14. Технология шин
(грунтозацепы будут, соответственно, противоположны ребрам, на
внешнем или внутреннем краю).
3. Грунтозацепы дорожного рисунка
4. Универсальный рисунок
5. Внедорожный рисунок
Рис. 12. Определение контура слоя. Уравнения Парди определяют накачанную или
«естественную» форму для «тонкой пленочной» структуры: С — кривизна
линии слоя, рс — расстояние от оси колеса до центра линии слоя, рм — рас-
стояние от оси колеса до самой широкой точки слоя.
Рисунки из ребер с конструктивными элементами, принципиально
расположенными в окружном направлении, являются наиболее обычным
типом рисунка протектора, они показывают хорошую работоспособность
на всех четырех позициях автомобиля в летних условиях. На тяжелых
грузовиках они используются почти исключительно на направляющих
и ведущих осях, из-за их характеристик бокового сцепления и однород-
ности износа. Комбинация «ребра / грунтозацепы» находит применение
во всесезонных шинах, для которых требуется баланс между хорошей
износостойкостью протектора, тяговыми характеристиками и сцепле-
нием на мокрой дороге. На ведущих осях тяжелых грузовых машин, где
тяговые характеристики являются первым требованием, и где происходит
IV. Конструирование шины -'V- 651
быстрый износ протектора как результат скольжения, вызванного мо-
ментом, требуются грунтозацепы для дорожного рисунка. Для условий
движения по бездорожью рисунок протектора строится в шахматном
порядке для обеспечения бокового и продольного сцепления. Канавки
должны быть больше и глубже, а наклон их стенок должен предотвращать
захват камней.
Длина нити корда от центральной
плоскости шины (см)
Рис. 13. Накачанная и ненагруженная шина.
Длина нити корда от центральной
плоскости шины (см)
Рис 14. Накачанная и нагруженная вертикальной силой шина.
Отношение фактической зоны контакта к полной площади отпечатка
протектора уменьшают в тех случаях, когда сцепление с мокрой дорогой
становится более важным. Таблицы 3 и 4 иллюстрируют процент насыщен-
ности для различных рисунков протектора шины.
652 Jb Глава 14. Технология шин
б
Рис. 15. Основные рисунки протектора: ребра дорожного рисунка (а); ребра/грунто-
зацепы дорожного рисунка (б); универсальный рисунок (смешанныеусло-
вия) (в); внедорожный рисунок (г).
Ряд дополнительных терминов, используемых для описания рисунка
протектора шины:
Амплитуда канавки В извилистой канавке величина колебаний относительно центрального направления по пути канавки.
Прорези Малые индивидуальные щели в протекторе, обычно добав- ляемые к конструкции протектора для улучшения тяговых характеристик.
Длина шага Длина каждого повторяющегося элемента в рисунке протектора. Переменная длина шага может быть использована для уменьше- ния шума. Шаг может быть аналогом длине волны синусоиды.
Щель Углубление в готовой шине, соответствующее выступу в вул- канизационной форме, который формирует часть рисунка протектора,. ч
Ребро, выбрасывающее камень Часть ребра протектора, сконструированная так, чтобы выбра- сывать камни благодаря изгибанию стенки шины. Выступы, расположенные на 75% ниже полной глубины канавки, пре- дохраняют от проникновения небольших камней к основанию канавки, откуда они не могут быть выброшены.
К Материалы шины -*и- 653
Таблица 3. Насыщенность рисунка протектора и условия эксплуатации [1,8].
Тип условий эксплуатации Насыщенность рисунка Относительная глубина протектора Управляемость Износ
Дорожная шина, Направляющая 70-75 4-4- 4- 4-4-4-
Дорожная шина ведущей оси 70-80 4-4-4- 4-4- +++ +
Дорожная шина прицепа 75-85 4- 4- 4-4-4-4-
Универсальная шина 60-70 4-4-4- 4-4-4- 4-4-
Внедорожная шина 55-65 4-4-4-4-4- 4-4-4- 4-4-
Таблица 4. Классы рисунков протектора автомобильных шин.
Кате- гория Конструкция Применение Насыщенность Удобство управления
1. Центральное ребро внешние ребро и блоки Высокий пробег Высокая 4-4-4-4-4-
2. Блоки Всесезонные Средневысокая 4-4-4-4-
3. Блок и ребро Сцепление Средняя 4-4-4-
4. Центральная канавка внешние блоки Сцепление, высокая работоспособность Ниже средней 4-4-4-
5. Направление - асимметричное - симметричное Высокая работоспособность Ниже средней 4-4-
V. МАТЕРИАЛЫ ШИНЫ
Шинное инженерное дело основывается на изучении на-
пряжений, возникающих внутри шины, и включает такие вопросы, как
деформирование её компонентов в процессе изготовления шины, напря-
жения в шине, смонтированной на колесе нагруженного и движущегося
автомобиля, количественная оценка этих напряжений и их минимизация
путем эффективного распределения нагрузок и правильного выбора мате-
риалов. Следовательно, группа шинных инженеров - технологов стремится
быть междисциплинарной командой, состоящей из инженеров - механиков,
научных работников - компьютерщиков, инженеров - химиков, химиков и
математиков. Чтобы понимать технологию шины необходимо иметь знания
о функциях каждого из типов материала, используемого в структуре шины.
654 JU Глава 14. Технология шин
Это могло быть по существу рассмотрено в двух аспектах: системы усиления
шины и рецептуростроение резины.
А. Усиление шины
Шина является композитом текстиля-стали-резины; ме-
таллокорд и текстильные корда усиливают резину и являются первичной
структурой, несущей нагрузку в шине. Поскольку для работоспособности
требуется сопротивление усталости, разрывная прочность и деформируе-
мость, семь главных материалов смогли стать подходящими для применения
в шине: хлопок, вискоза, найлон, полиэфир, сталь, стекловолокно и арамид;
последние три материала нашли главное применение в каркасе и брекере
шины.
Наука об усилении шины применяет специализированную терминоло-
гию, которую инженер-шинник должен понимать:
Вес латуни Обычно 3,65 г/кг корда, толщина латунного покрытия по- рядка 0,3 микрона.
Разрывная прочность Напряжение при разрыве в процессе растяжения.
Корд Структура, содержащая две или более стренг, при использо- вании как крученая пряжа или как готовый продукт.
Денье Частота нитей (EPI) Волокна Вес корда, выраженный в граммах на 9000 метров. Число нитей корда на дюйм ширины ткани. Линейные макромолекулы, ориентированные вдоль оси нити.
Филаменты Наименьшие протяженные элементы текстиля или стали в стренге.
Поперечные нити Легкие нити, образующие прямой угол с основой (также обозначается как «уток), которые предназначены для под- держания целостности ткани.
Специфическая нагрузка (LASE) Длина свивки Нагрузка при определенной величине удлинения или де- формации. Осевое расстояние элемента или стренги, требуемое для пол- ного оборота вокруг корда.
Кручение слоя Закручивание шинной пряжи, требуемое число поворотов на дюйм; крученая пряжа с двух или более катушек затем скручивается снова в корд; например, если две найлоновых нити корда с денье 840 скручиваются вместе, образуется конструкция найлонового корда 840/2; если три полиэфир- ных нити корда с денье 1300 скручиваются вместе, они дают корд конструкции 1300/3.
Шаг нитей корда Расстояние между нитями корда в ткани; большой шаг опи- сывает ткань с низкой частотой нитей.
V. Материалы шины 655
Сопротивление разрыву Текс Кручение Прочность корда, часто выражаемая в граммах на денье. Вес корда, выражаемый в граммах на 1000 м. Число оборотов на единицу длины корда или пряжи; направ- ление кручения может быть или по (кручение «S») или против (кручение «Z») часовой стрелки; кручение придает корду долговечность и сопротивление усталости, хотя разрывная прочность может снизиться.
Основа Уток Пряжа Нити корда в шинной ткани, расположенные по её длине. Тонкие нити, расположенные поперек основы. Совокупность филаментов.
Нити и стальной корд являются основным усиливающим и несущим
нагрузку материалом в шине. Следовательно, уместно рассмотреть свойства
этих материалов с позиции применения в шинах.
Б. Металлокорд
В шинах используется металлокорд различных конфигу-
раций, но это всегда стренды покрытой латунью проволоки, скрученные
так, чтобы получить корда различных характеристик в зависимости от
их применения. Шинный металлокорд изготовляется из прутков вы-
сокоуглеродистой стали, которые сначала протягиваются до диаметра
приблизительно 1,2 мм. После этого наносится латунное покрытие,
а затем производится окончательное протягивание до 0,15-0,40 мм. Затем
эти филаменты скручиваются и образуют корд, который конструируется
и оптимизируется в соответствии со специфическими рабочими требова-
ниями [12].
Шинный металлокорд изготавливается из высококачественной стали,
необходимой из-за требований к работоспособности, которые предъявля-
ются к шинам. Состав типового металлокорда показан в таблице 5. Клю-
чевыми механическими свойствами, определяющими металлокорд или
проволоку, являются их разрывная прочность, удлинение и изгибная
жесткость. Конструкция шинного корда обычно определяется структурой,
длиной свивки и направлением свивки. Полное описание металлокорда
дается формулой
(NxF) xD + (NxF) x£) + (FxjD) (5)
где N — число стренг, F — число филаментов и D — номинальный диаметр
филаментов (в миллиметрах).
Как пример, спецификация металлокорда может иметь такой вид
(1 х 4) х 0.175 + (6 х 4) х 0.175 + (1 х 0.15) (6)
656 -JU Глава 14. Технология шин
Когда Nили Нравны 1, обозначение упрощается и (6) приводится к
4 х 0.175 + (6 х 4) х 0.175 + 0.15 (7)
Для описания металлокорда используются некоторые дополнительные
соглашения:
1. Если диаметр D двух или более частей в обозначении одинаков, то
диаметр указывается только для конца обозначения.
2. Диаметр спиральной обвивки указывается отдельно.
3. Когда самая глубокая стренга или проволока такая же, как смежная,
то определение проволоки может быть в дальнейшем упрощено
путем указания только суммы одинаковых компонентов. Тогда (7)
упрощается
7x4x0.175 + 0.15 (8)
4. Последовательность или порядок в обозначении проволоки следует
порядку производственного процесса. Длина и направление свивки
корда в (8) могут быть описаны, как показано на рис. 16.
Рис. 16. Длина и направление свивки. (С разрешения Bekaert Corporation).
Некоторое число эмпирических правил определяет конструкцию про-
волоки для применения в шинах. Например, если проволока используется
в слоях каркаса, а не в брекере, она будет подвергаться большему числу из-
гибаний. Следовательно, усталостная работоспособность будет более важна.
Если проволока используется в брекере, тогда жесткость становится первым
конструктивным параметром. Таким образом, могут быть определены клю-
чевые свойства конструкции; они показаны в таблице 6.
V. Материалы шины 657
Таблица 5. Состав шинного металлокорда.
Элемент Состав (%) Функция
Углерод 0,65 прочность
Хром 0,05 прочность
Медь 0,02 прочность
Марганец 0.60 раскисление
Кремний 0.25 раскисление
Сера 0,03 обрабатываемость
Таблица 6. Конструкция шинного корда.
Параметр конструкции Конструктивный фактор
Жесткость Число стрендов в корде, калибр
Прочность Калибр, число стрендов в корде, состав при разрыве стального прутка
Усталость Калибр филаментов
Удлинение Кручение, свивка
Чем толще корд, тем он жестче. Более тонкие корда имеют большее со-
противление усталости. Более толстые корда находят применение в больших
шинах, таких как шины тяжелых грузовых машин и шины для землеройного
оборудования.
Проволока находит применение для усиления шины в брекерах шин,
в каркасе шин для тяжелых условий работы (т. е. в шинах для больших
грузовых машин), в бортовых кольцах и бортовой ленте (которая защищает
борт от повреждений ободом колеса).
В. Механизм адгезии резины к латуни
Тонкий слой латуни на металлокорде является основным
адгезивом, используемым для крепления стали к резине. Качество этой сис-
темы крепления, возникающее при вулканизации, например, радиальных
шин, будет влиять на работоспособность металлокордного слоя каркаса или
брекера в шине и, в конечном счете, на прочность изделия. Хотя механизм
образования связи резины с металлокордом очень сложен, может быть сделан
краткий обзор для понимания этой проблемы.
Натуральный каучук, используемый, как правило, для резин, покры-
вающих проволоку, образует прочное крепление с латунью в результате
образования межфазовой пленки сульфида меди (CuS) при вулканизации.
658 JU Глава 14. Технология шин
Зоны сульфида меди создаются на поверхности латунной пленки благодаря
реакциям, проходящим при вулканизации. Такие зоны имеют высоко удель-
ную площадь поверхности и образуются внутри покрывающей проволоку
резиновой смеси, прежде чем вязкая полимерная фаза превращается в сши-
тую поперечными связями сетку эластомера. Таким образом, полимерные
молекулы оказываются запертыми в кристаллической решетке сульфида
меди (рис. 17). Важными факторами, определяющими эту связь, является
образование сульфида меди, когезионная прочность, адгезия к латун-
ному субстрату и скорость вторичных реакций коррозии под пленкой
сульфида меди.
Рис. 17. Межфазные пленки сульфида меди в связи резина - латунь [7].
Сульфиды цинка и железа не создают связи, потому что они не растут
достаточно быстро во время вулканизации, не образуют пористых зон и,
следовательно, не могут сращиваться с полимером. Первейшим условием
является образование зон сульфида меди перед возникновением попереч-
ных связей, но на образование связей может отрицательно влиять снижение
времени подвулканизации резиновой смеси.
Механическая стабильность зон сульфида меди является существенной
для сохранения долговременной прочности адгезии резины к проволоке.
Однако, имеет место коррозия адгезионных связей, катализатором которой
являются ионы Zn2+, диффундирующие через межфазные слои CuS. Это при-
ведет, в конце концов, к образованию избытка ZnS или ZnO/Zn (ОН)2 При
отсутствии влаги этот процесс идет медленно. Однако Zn2+будет мигрировать
к поверхности с соответствующим падением механической взаимосвязи зон
CuS и резины и последующей потерей адгезии.
Миграция ионов Zn2+является функцией электрической проводимости
латунного покрытия. Добавление ионов Со2+ или Ni2+снижает эту прово-
димость.
Соли кобальта в обкладочной резиновой смеси ускоряют вулканизацию
смесей с высоким содержанием серы, которые могут при этом использо-
V. Материалы шины -*v 659
ваться. Рост плотности поперечных связей приводит к увеличению силы
выдергивания проволоки из резины. Но более важно, что соли кобальта
образуют при вулканизации ионы Со2+ на поверхности раздела с латунью
и это будет влиять на образование сульфидов меди.
Сульфид цинка /
Сульфид меди
Окись цинка с медью
Латунь
Сульфид меди
Окись цинка
с ионами Со2+ и Со3+
Окись цинка
с зонами ионов меди
Латунь
Рис. 18. Образование сульфида меди при низкой концентрации кобальта: отсутствие
кобальта позволяет образоваться сульфиду цинка (а); ионы кобальта в
пленке окиси цинка препятствуют миграции ионов цинка и образованию
сульфида цинка (б).
Различия в эффективности между кобальтовыми промоторами ад-
гезии определяются легкостью, с которой могут быть образованы ионы
Со2+ Например, цинк или латунь реагируют много легче с комплексами
деканоата бора, чем с нафтенатом или стеаратом кобальта. Ионы Со2+
и Со3+ внедряются в пленку ZnO прежде, чем там образуется пленка
сульфидов. И ди- и тривалентные ионы кобальта снижают электриче-
660 -JU Глава 14. Технология шин
скую проводимость слоя ZnO, тем самым снижая диффузию ионов через
полупроводящую пленку.
Диффузия по зонам металлической меди к поверхности из-за окисления по
схеме R-Sx не так эффективна как миграция ионов Си2+ вдоль зернистых гра-
ниц слоя ZnO. Следовательно, если используется соль кобальта, то образование
сульфида меди на поверхности корда будет ускоряться, поскольку образование
ZnS будет затруднено (рис. 18). Этот обзор по необходимости краток и читатель
может получить дополнительную информацию в работах [13,14].
Г. Вискоза
Первой синтетической нитью для шины была вискоза.
Первоначально целлюлоза обрабатывается гидроксидом натрия, чтобы
образовалась щелочь целлюлозы. Затем она измельчается и оставляется
стареть на воздухе, где она окисляется и её молекулярный вес уменьшается,
что позволяет провести последующие операции размешивания. Обработка
дисульфидом углерода дает ксантогенат целлюлозы, который затем раство-
ряется в гидроксиде натрия, в результате чего образуется вискоза. Материал
подвергается дальнейшему гидролизу и затем пропускается через фильеру,
чтобы образовалась нить. Эта нить проходит через ванну с серной кислотой
и сульфатом натрия, где вискозные нити в дальнейшем коагулируют.
Далее следуют промывка, отбелка и скручивание в нить корда. Вис-
козные нити могут быть растянуты до 100% от их первоначальной длины,
чтобы кристаллизация при ориентации создала высокопрочную вискозу,
подходящую для шин.
Целлюлоза Щелочь целлюлозы
Ксантогенат целлюлозы
Регенерат целлюлозы
(9)
V. Материалы шины -*v- 661
Д. Найлон
Найлон является полиамидом, характеризующимся присутс-
твием амидных групп - (CO-NH) - в главной полимерной цепи. Существует
большое количество найлоновых полимеров, но только два из них нашли
применение в шинах: найлон 6,6 и найлон 6.
Найлон 6,6 получается при реакции конденсации между адипиновой
кислотой и гексаметилендиамином.
H2N—(СН2)6—nh2 + НООС—(СН2)4—соон
Гексаметилендиамин Адипиновая кислота (10)
--> -f NH—(СН2)6—NH—СО—(СН2)4—СО-);
Полимерная цепь найлона 6,6
«6,6» в наименовании полимера обозначает шесть углеродных атомов гек-
саметилендиамина и шесть углеродных атомов адипиновой кислоты.
Найлон 6 производится из капролактама путем полимеризации с раз-
рывом циклов. Так как капролактам содержит шесть углеродных атомов
и используется только один мономер, то полимер именуется найлон 6.
О
/СН2^С^
СН, \ --->[—NH—(СН2)5—СО—NH—(СН2)5—СО—]„
Капролактам Полимерная цепь найлона 6
(И)
После стадии полимеризации материал пропускается через фильеру,
чтобы образовались филаменты, охлаждается, а затем скручивается, чтобы
образовать прядь. После этого производится растяжение до 500%, ориен-
тирующее полимерную цепь, создающее кристаллические зоны в полимере
и увеличивающее прочность при растяжении (рис. 19).
Е. Полиэфир
Подобно найлону, имеется ряд полиэфиров, пригодных для
коммерческого использования в шинах. Наиболее важным из них является
полиэтилентерфталат (РЕТ). Также подобно найлону, полиэфир образуется
путем конденсационной полимеризации, но с мономерами этиленгликолем
и диметилтерфталатом.
662 JU Глава 14. Технология шин
НО—СО2—СН2—ОН + Н3СООС-/ QVcooch3
Этиленгликоль
Диметилтерфталат
(12)
--> [о—(СН2)2—ООС—(О)-СО]П
Полиэтилентерфталат
Полимеризованный материал затем продавливают через фильеру,
чтобы образовать филаменты около 0,025 мм в диаметре. Эти филаменты
охлаждают, скручивают в прядь и вытягивают, чтобы получить требуемую
ориентацию и кристалличность.
Циклогексан + воздух
Адипиновая кислота + аммиак
Адипиновая кислота
Адипиннитрил + водород
Гексаметилендиамин
Найлон соль
I
Полимеризация
Скручивание расплава
I
Растяжение
I
Найлоновое волокно
Рис. 19. Производство найлона 6,6.
Ж. Стекловолокно
Подобно металлокорду, стекловолокно является неоргани-
ческой нитью; по составу это кальций-алюмоборосиликатное стекло (табл. 7).
Оно производится путем смешивания песка, каолина, известняка и буры,
плавления смеси при 3000eF и протягивания филаментов из стекла через
платино-родиевый фильтр. Затем филаменты обрабатывают адгезивным
подслоем и собирают в прядь с низким кручением.
V. Материалы шины
Л
663
Таблица 7. Состав стекловолокна.
Двуокись кремния 53%
Окись кальция 21%
Окись алюминия 15%
Окись бора 9%
Окись магния 0.30%
Другие окиси 1,7%
3. Арамид
Другим классом волокон, которые находят применение
в шинах, является арамид. Кевлар — это торговое название полимера, который
нашел наиболее интенсивное применение среди арамидов. Арамид подобен
найлону тем, что содержит аминные связи - (CO-NH) -, но он производится
путем сополимеризации терфталатной кислоты в полиэфире пр — фенилен-
диамине. По этой причине ароматический полиамид назван арамид.
Таблица 8 иллюстрирует эволюцию в ряду усиливающих материалов,
которые использовались в шинах.
Таблица 8. Тенденции в применении кордов.
Время Корд
1900-1956 Хлопок
1939-1975 Вискоза
1950 до сих пор Найлон
1965 до сих пор Полиэфир
1970 до сих пор Металлокорд
1975- 1985 Стеклокорд
1980 до сих пор Арамид
1990 до сих пор Комбинированные корда
664 -J\, Глава 14. Технология шин
И. Конструкция шинного корда
Чтобы использовать весь ряд нитей для применения в ши-
нах, пряди должны быть скручены и превращены в корд. Сначала прядь
скручивается на определенное число поворотов на дюйм. Затем с двух
или более катушек крученая прядь скручивается в корд. Как правило,
направление второго кручения противоположно первому, это называ-
ется сбалансированное кручение. Имеется много причин для кручения
корда:
1. Кручение придает корду жесткость и сопротивление усталости, хотя
разрывная прочность может быть снижена.
2. При отсутствии кручения силы сжатия могли бы вызывать коробление
внешних филаментов корда.
3. Дальнейшее увеличение кручения корда уменьшает коробление фила-
ментов из-за роста растяжимости их пучков.
При росте кручения прочность достигает максимума, а затем начинает
снижаться. Это может стать понятным при рассмотрении влияния напряже-
ний на корд при увеличении кручения. При росте кручения угол подъема
спирали (угол между осями филаментов и корда) также возрастает (рис. 20).
Поэтому растягивающие напряжения, нормальные к оси корда, производят
наибольшее силовое растаскивание филаментов. Причина снижения про-
чности также очевидна из этого рисунка. Когда кручение корда возрастает,
то возрастает и сила в направлении оси волокна, вызывая снижение общей
разрывной прочности.
Таблица 9 Применение шинных кордов.
Тип волокна Прочность (г-Денье) Удлинение при разрыве, % Модуль (г-Денье) Относительная долговечность Применение шин
Вискоза 5,0 16 50 300 Легковые
Найлон 9.0 19 32 1200 Тяжелые и внедорожные
Полиэфир 6,5 18 65 400 Легковые, грузовые, сельскохоз.
Стекло- волокно 9,0 4,8 260 5 Легковые
Проволока 3,8 2,5 200 3 Грузовые, внедорожные
Арамид 20,1 4 350 400 Легковые
V. Материалы шины
Л
665
В зависимости от областей применения в дополнение к кручению могут
варьироваться все параметры корда (табл. 9). Как правило, корд из трех
прядей имеет наивысшую прочность. После скручивания прядей нити корда
собирают в ткань, используя редкие поперечные нити. Эти нити называются
уток. Этот процесс образования ткани вводит дополнительное конструк-
ционное переменное - число нитей корда на дюйм (EPI) вытканной ткани.
Ткань с большей частотой нитей корда имеет большую прочность при вдав-
ливании или большее сопротивление внедрению. Ткань с меньшей частотой
нитей корда имеет больший шаг корда (расстояние между нитями корда)
и имеет лучшее сопротивление раздиру из-за большего количества резины
вокруг кормов.
Напряжение Напряжение
в нити в корде
Рис. 20. Геометрия корда.
Высокое кручение
Поперечное
напряжение
Напряжение
в корде
Напряжение
в нити
К. Производство ткани
Наиболее критической стадией в изготовлении корда или
ткани для использования в шинах является обработка ткани, которая состоит
в добавлении адгезива при условиях, контролируемых по времени, темпера-
туре и натяжению. Этот процесс придает ткани следующие свойства:
1. Адгезия для крепления к резине
2. Оптимизация для применения в шинах таких физических свойств
корда как прочность, жесткость, ползучесть и удлинение
666 Глава 14. Технология шин
3. Стабилизация ткани
4. Выравнивание свойств поставок от различных источников снабжения
тканями
Процесс состоит в пропускании ткани через ряд зон, которые могут быть
обозначены следующим образом:
1. Зона нанесения адгезива или первая зона пропитки
2. Первая зона сушки
3. Первая зона тепловой обработки
4. Вторая зона пропитки
5. Вторая зона сушки и затем зона тепловой обработки
6. Зона окончательного охлаждения
Чтобы получить оптимальные свойства корда, такие как прочность, пол-
зучесть, усадка и модуль, на обрабатываемую ткань в течение различного
времени должны воздействовать определенные температуры и натяжения.
Температура и натяжения, в частности, определяют соотношение кристалли-
ческой и аморфной зон в нити и ориентацию кристаллитов, которые, в свою
очередь, определяют физические свойства корда. Например, при нагревании
полиэфир имеет тенденцию возвращаться к своей неориентированной форме
и корд усаживается. Вытягивание корда в первой зоне нагревания и затем
контролируемая релаксация во второй зоне тепловой обработки, то есть
релаксация напряжения, будут контролировать вытяжку.
Таблица 10. Соотношение свойств корда и режимов натяжения и температуры.
Свойства корда Изменение происходит при первом росте Изменение происходит при втором росте
времени натяжения температуры времени натяжения температуры
Разрывная прочность Немного уменьшается Уменьшается Немного уменьшается Уменьшается
Нагрузка при удлинении 5% Уменьшается Уменьшается Уменьшается Уменьшается
Удлинение при разрыве Возрастает Возрастает Возрастает Возрастает
Усадка Уменьшается Уменьшается Уменьшается Уменьшается
Покрытие резиной Возрастает Возрастает Возрастает Возрастает
Усталость Уменьшается Уменьшается Уменьшается Уменьшается
Механические свойства (SCEF) Уменьшается Уменьшается Уменьшается Уменьшается
Пористость Уменьшается Уменьшается Уменьшается Уменьшается
V. Материалы тины 667
Другой переменной является увеличение рабочей температуры, которая
может уменьшить разрывную прочность корда и модуль, но исправить уста-
лостную прочность. Общее соотношение между свойствами корда и рабочими
температурами и натяжениями показано в таблице 10.
Заметим, что не все свойства корда ведут себя одинаково при изменении
производственных условий. Следовательно, необходимо определить произ-
водственные условия, которые оптимизируют свойства корда по требованиям
к применению в шине. Когда два или больше диаметрально противополож-
ных свойства должны быть оптимизированы, следует использовать более
сложные математические методы.
Проволока или стекловолокно, являющиеся высокомодульными неорга-
ническими кордами для брекера, обрабатываются не так, как текстильные
корда. Металлокорд покрыт латунью на металлургическом заводе и, следо-
вательно, может быть использован непосредственно в каландрах. Стекло-
волокно обрабатывается адгезивной пропиткой и затем прямо используется
в ткацких операциях.
Л. Функции адгезивов
Имеются три аспекта образования адгезии шинного корда
к эластомеру при обработке: молекулярный, химический и механический.
Молекулярное крепление происходит благодаря адсорбции химикатов
адгезива из адгезивной пропитки или эластомерного покрытия на по-
верхности волокна путем диффузии и может быть описано с привлече-
нием представлений о водородных связях и Ван дер Ваальсовых силах.
Химическое крепление достигается химическими реакциями между
адгезивом, тканью и резиной, т. е. поперечным сшиванием эластомера
и образованием сетки в смоле. Механическая адгезия является функцией
качества покрытия корда слоем резиновой смеси. Чем больше покрытие,
тем лучше адгезия.
Для адгезии, в первую очередь, важны такие свойства нитей как реакци-
онная способность, характеристики поверхности и отделка. Вискоза имеет
много реагирующих гидроксильных групп. Реакционная способность най-
лона меньше, но он содержит высоко полярные амидные связи. Полиэфир
относительно инертен. Поэтому адгезивные системы должны быть разрабо-
таны для каждого типа нитей.
Независимо от вида нитей, каждая адгезионная система должна удов-
летворять ряду жестких требований:
1. Высокая скорость образования адгезии
2. Сочетаемость с многими типами резиновых смесей
3. Отсутствие вредного влияния на свойства корда
4. Теплостойкость
668
Глава 14. Технология шин
5. Стойкость к старению
6. Хорошая клейкость
7. Механическая стабильность
Адгезионное крепление между резиной и кордом осуществляется в цикле
вулканизации шины. Скорость образования адгезива должна обеспечивать
максимальную адгезию в момент снятия давления в вулканизационном
цикле.
Л. Рецептуростроение
Принципы разработки рецептур резиновых смесей были
рассмотрены ранее в тексте книги. Они включали фундаментальные ха-
рактеристики полимеров, систем наполнителей и основ вулканизации
в контексте разработки смесей для применения в шинах. Рецептура смеси
состоит из четырех основных компонентов: полимерная сетка, наполнитель,
или состоящие из частиц усиливающие системы, стабилизирующие системы
и вулканизующие системы (рис. 21). В дополнение в рецептуру может быть
включен ряд второстепенных материалов, таких как смолы, технические
масла и короткие усиливающие волокна [15, 16, 17].
Рис. 21. Основные компоненты резиновой смеси.
В радиальных шинах используются, в основном, четыре типа эластоме-
ров:
1. Натуральный каучук
2. Бутадиен-стирольный сополимер
3. Полибутадиен
4. Бутилкаучук (полиизобутилен с примерно 2% изопрена) и галогени-
зированный бутилкаучук
Наполнитель, или объединенная усиливающая система состоит из
технического углерода, каолина и силики. Оптимизация этих материалов
VI. Испытания шины Jb 669
в рецептуре зависит от компонента шины, для которого данная резина пред-
назначена, например протектор или боковина.
Стабилизирующая система или система антиоксидантов защищает
смеси от старения и окисления и улучшает долговременную прочность
шины.
Разработка вулканизующей системы является, вероятно, наиболее ин-
тересным аспектом в конструировании рецептуры смеси для применения
в шине. Для разработки таких систем важно знание активности ускорителя,
кинетики реакций и природы образующихся поперечных связей.
VI. ИСПЫТАНИЯ ШИН
Разработав новую конструкцию шины для определенных
технических требований, шинный инженер должен подвергнуть её ряду
испытаний [3, 4]. Эти испытания разделяются на две категории:
1. Механические характеристики шины
- Нагрузочная характеристика смонтированной и вертикально на-
груженной шины, нагрузочная способность и индекс нагрузки
- Свойства управляемости, такие как стабилизирующий момент,
характеристики движения на повороте и боковая и тангенциальная
жесткости шины
- Тяговые характеристики и поведение на мокрой дороге
- Сопротивление качению, которое влияет на топливную экономич-
ность автомобиля
- Шум
2. Прочность
- Износ протектора, который включает медленный износ, быстрый
износ и неравномерность износа
- Усталостное сопротивление каркаса
- Разогрев шины при динамических условиях нагружения
- Сопротивление протектора и боковой стенки сколам, порезам и
раздиру
Все испытания разделяются на три фазы: лабораторные испытания,
полигонные испытания и коммерческая оценка.
А. Лабораторные испытания
Лабораторным испытаниям шин предшествуют испытания
сырых материалов. Для сырых смесей и резин протектора и боковины,
обкладочной резины для проволоки и герметизирующего слоя такие ис-
пытания должны включать определение кинетики вулканизации, разрыв-
ной прочности, прочности на раздир, упругости и динамических свойств
670 J\- Глава 14. Технология шин
(т. е. модуля запаса и модуля потерь). Усиливающие элементы, такие
как корд, также испытывают на разрывную прочность, но, кроме того,
на поведение при ползучести, стабильность (поведение при усадке)
и сопротивление усталости. Многие из физических испытаний смесей
в современной лаборатории производятся с помощью робототехники.
Смеси вулканизуют при определенных условиях, образцы вырезают
в соответствии с типом испытаний, которые должны быть сделаны. Затем
роботы на испытательном оборудовании нагружают образцы, полученные
данные собираются компьютером и становятся доступными для инжене-
ров, проводящих испытания.
Материалы, которые соответствуют заданным физическим свойствам,
направляются на изготовление шин. Испытания этих шин зависят от на-
значения, для которого они были сконструированы. Поэтому уместно ввести
другую серию определений:
Вертикальная сила Нагрузка, действующая на шину весом автомобиля и груза.
Боковая сила Сила, действующая на шину при движении автомо- биля на повороте или с поперечным наклоном.
Коэффициент боковой силы Отношение величин боковой силы и вертикальной нагрузки.
Угол увода автомобиля Угол между направлением движения автомобиля и направлением качения управляемых колес.
Относительный коэффициент бокового увода Отношение величины боковой силы, соответству- ющей определенному углу увода, и вертикальной нагрузки. Более жесткая резина протектора приводит к улучшению этой величины.
Стабилизирующий момент Реакция от увода колеса, которая помогает шине и автомобилю вернуться к нейтральной позиции при завершении маневра.
Однородность Мера способности шины катиться без вибраций, обычно оценивается дисбалансами, вариациями радиальной и боковой сил и конусностью.
Дисбаланс Вес шины распределен по её окружности; непра- вильный баланс возникает из-за нерегулярности размеров компонентов. На дисбаланс может также влиять нерегулярность ширины стыков компонентов и неправильное наложение частей компонентов.
Радиальное биение Разность между максимальным и минимальным измерением расстояния от поверхности протектора на центральной плоскости шины до плоскости, па-
VI. Испытания шины JU 671
раллельной оси вращения колеса, за оборот шины, смонтированной на ободе и накачанной.
Боковое биение Разность между максимальным и минимальным измерением расстояния от поверхности боковой стенки на её максимальной ширине до плоскости, параллельной центральной плоскости шины, за оборот шины, смонтированной на ободе и нака- чанной.
Вариация силы Периодические изменения вертикальной или боко- вой силы нагруженной и свободно катящейся шины, повторяющиеся при каждом обороте.
Гармоники Составляющие вариации силы, имеющие форму синусоиды по окружности шины. Один период колебаний за оборот шины описывается как 1-ая гармоника. Когда наблюдаются два периода, это описывается как 2-ая гармоника. 1-ая гармони- ка вариации боковой силы возникает обычно от стыков протектора. Радиальная гармоника может возникать из-за нерегулярного расположения слоев брекера.
Конусность Тенденция шины тянуть автомобиль в одну или в другую сторону; она вызывается смещением ком- понентов от центра или со своего места при сборке шины.
Сопротивление качению Работа, затрачиваемая на качение шины. При- чиной её возникновения являются, в основном, гистерезисные свойства материалов шины и она прямо влияет на топливную экономичность ав- томобиля
Индекс скорости Буквенный индекс, определяющий скоростные возможности шины. Буква входит в обозначение размера шины. Например, шина 195/75SR14 имеет индекс «S». Таблица рангов скоростей и соответству- ющих индексов опубликована Tire & Rim Association (таблица И).
Основой лабораторного оборудования, сконструированного для измерения
вышеперечисленных свойств шины, является, как правило, стенд с беговым
барабаном с соответствующими измерительными устройствами, такими как
датчики и инфракрасные термометры. Результаты измерений вводятся непо-
средственно в компьютер для анализов в реальном времени и собираются на
рабочем месте шинного инженера. Многие из этих стендов контролируются
компьютерами, моделирующими условия работы. Например, авиационные
шины могут испытываться в комплексном цикле качения, взлета и посадки.
672 JU Глава 14. Технология шин
Таблица 11 Категории скорости качения шины.
Символ скорости Максимальная скорость (км/час) Типичное применение
D 50 Сельскохозяйственные тракторы
L 120 Коммерческие грузовики
М 130 Коммерческие грузовики
S 180 Легковые и легкогрузовые автомобили
т 190 Легковые автомобили
н 210 Роскошные легковые автомобили
V 240 Автомобили высокой работоспособности
Z 270 Спортивные автомобили
W выше 270 Автомобили сверхвысокой работоспособности
Б. Испытательный полигон
Наиболее объективным методом определения поведения
шины является оценка её работоспособности в дорожных испытаниях.
Полигонные испытания позволяют оценивать все типы шин, такие как
легковые, грузовые, внедорожные и сельскохозяйственные, в точно
фиксированных условиях. Промышленные полигоны будут в основном
располагать следующими испытательными треками и дорожными мар-
шрутами:
1. Высокоскоростные треки, кольцевые или овальные
2. Дороги, моделирующие основные магистрали
3. Гравийные и неотремонтированные дороги
4. Булыжные дороги
5. Маршруты, где происходят разрезы, сколы и разрывы шин
6. Мокрые и сухие площадки для испытания на занос, змейки и слалом-
ные курсы для визуальных оценок и испытаний на управляемость
7. Специальные треки для испытания на долговечность сельскохозяйс-
твенных шин
8. Стеклянные участки дороги для контроля отпечатков шины
Например, испытательный полигон фирмы Гудрич площадью 7250 ак-
ров включает пятимильное высокоскоростное кольцо, восемь миль дорог,
моделирующих основные магистрали, гравийные и каменные дороги для
испытания различных типов шин, площадки для испытания на занос
VII. Производство шин -*v- 673
с оборудованием для разбрызгивания воды и стеклянный участок дороги для
наблюдения за отпечатком шины и оценки распределения воды.
В. Коммерческая оценка
Когда новая конструкция шины будет оценена в лаборатории
и на испытательном полигоне как соответствующая целям по работоспособ-
ности, наступает следующая стадия цикла разработки продукции — ком-
мерческая оценка. Определенное количество грузовых шин для шоссейных
дорог передают для коммерческой эксплуатации, и их работоспособность
постоянно контролируется. Собранные по таким испытаниям данные вклю-
чают износ протектора, однородность износа, прочность каркаса и оценки
водителей. Шины эксплуатируются как в коротких рейсах, так и дальних
перевозках. Затем собранные данные накапливаются в компьютерах для
детального анализа, вычисления регрессионных соотношений и оценки
работоспособности. Участие заказчика является одним из ключевых пара-
метров в этих исследованиях.
VII. ПРОИЗВОДСТВО шин
Производство шин состоит из шести основных процессов:
1. Смешение эластомеров, технического углерода и химикатов для об-
разования резиновой смеси
2. Изготовление тканей и металлокорда и покрытие их резиной в про-
цессе каландрования
3. Шприцевание протекторов, боковин и других резиновых компонентов
4. Сборка компонентов на сборочном оборудовании
5. Вулканизация шины при нагреве и под давлением
6. Окончательная обработка, инспекция и упаковка
Процессы, вовлеченные в производство шины, показаны на рис. 22.
А. Смешение
Сначала полимеры дробятся в закрытом смесителе, где к поли-
меру может быть также добавлен пептизатор. Эта стадия по существу есть фаза
снижения молекулярного веса полимера. После первоначальной деструкции
полимера к нему могут быть добавлены технический углерод, химикаты
и масла в количествах, соответствующих рецептуре смеси. Обработка полимера
и смешение происходит обычно при высокой температуре около 180°С.
Смеси могут быть также приготовлены в открытых вальцах, но это тре-
бует значительно большего времени, вес заправок меньше и, следовательно,
процесс менее эффективен, чем применение закрытых смесителей.
Балансировка
Сырые материалы
Окончательная
инспекция
Стенд для измерения
силовой . ородности
Сборка борта
Смеситель
Бенбери
Производство
ткани
Металлургическая
промышленность Производство
проволоки для
борта, брекера
и каркаса
Текстильная
промышленность
Каучуковая
плантация
Химическая
п ром ы ш лен ность
Рис. 22. Процесс производства шины.
Машина для
разрезания
ткани иод
углом
Визуальная
инспекция
Каландр
для ткани
Вулканиза-
тор и пульт
контроля
информа-
1рентгеноскопи
ческий анализ
674 JI- Глава 14. Технология шин
Каландр для
металлокорда
Машина для
разрезания
металлокорда
VII. Производство шин -*v- 675
Степень переработки в обоих типах оборудования зависит от «фрикцион-
ного соотношения» или разности между операционной скоростью передних
и задних валков (или роторов для закрытого смесителя). Кроме того, ход
переработки определяют зазор, состояние поверхности ротора, давление
и скорость.
Процесс смешения разработан так, чтобы получить однородное смешение
всех материалов смеси в соответствии с рецептурой. Для каждой заправки
имеется определенные период смешения, температура, скорость вальцевания
или смешения и последовательность добавления материалов. Хотя общие
правила приготовления смесей для смешения на вальцах и смешения в за-
крытых смесителях подобны, смешение на вальцах вытеснено закрытыми
смесителями из-за эффективности, автоматизации, качества и однородности.
Например, закрытые смесители могут контролироваться компьютерами,
позволяющими отслеживать расход мощности, время смешения, темпе-
ратуры отдельных заправок и контролировать температурный градиент
в смешиваемой смеси в любой точке цикла смешения.
Последовательность добавления в закрытый смеситель типична:
(1) полимеры и пептизатор, (2) пластификаторы, технический углерод или
силика и масла, (3) баланс наполнителей и антиоксидантов и (4) компоненты
вулканизующей системы.
Организация цикла смешения определяется набором эмпирических
правил:
1. Держать сильно слипающиеся смолы отдельно от сухих порошков.
2. Удерживать температуры заправок выше точки размягчения твердых
полимеров.
3. Предохранять от утечки содержащиеся жидкости.
4. Использовать сдвиговые свойства резины для ускорения смешения.
5. Избегать преждевременной вулканизации и последующего образова-
ния вулканизованных частиц и крошки.
Эти пять правил должны быть дополнены снижением внутреннего трения
путем применения пластификаторов, адекватным измельчением каучука,
введением компонентов вулканизующей системы на конечной стадии
цикла смешения и использованием, где это возможно, маточных смесей.
После смешения смеси могут быть раскатаны в листы и охлаждены водой
или пропущены через установку для гранулирования и затем охлаждены
воздухом.
Б. Каландрование
Каландрование является формующей операцией, в кото-
рой резиновая смесь раскатывается или раскладывается ровным слоем
на ткани. Каландр это сверхмощная машина, оборудованная тремя или
676 Jb Глава 14. Технология шин
большим числом хромированных стальных валков, которые вращаются
в противоположных направлениях (рис. 23). Валки нагреваются паром
или циркулирующей водой; редукторы обеспечивают возможность валкам
вращаться с различными скоростями подобно валкам вальцев. Ткань или
слой металлокорда проходят через валки каландра, и смесь распределяется
на обеих сторонах материала, полностью покрывая его.
Наложение
резины на ткань
а
Промазка
Вращение
против
часовой
стрелки
Вращение
по и против
часовой
стрелки
Рис. 23. Наложение резины на ткань (а). Профилирование четырехвалковым гра-
вированным каландром (б).
Количество смеси, наложенное на ткань или металлокорд, определяется рас-
стоянием между валками и контролируется специальными датчиками. Каждая
нить корда изолирована со всех сторон резиновой смесью. Окончательно обра-
ботанное или каландрованное металлокордное полотно разрезается по длине
и под нужным углом для сборки шины. Ключевым требованием к смесям для
операции каландрования является минимальная усадка, оптимальная адгезия
компонентов и достаточное сопротивление подвулканизации, чтобы дать воз-
можность долгое время работать при высоких температурах.
В. Шприцевание
Многие из компонентов шины, такие как протектор, боко-
вины и наполнительный шнур бортового крыла формуются путем шприци-
вания невулканизованной или «сырой» резиновой смеси. Шприцмашины
(экструдеры) в системе шинного производства являются обычными систе-
мами винтового типа, которые разделяются на две категории:
Системы горячего питания
Системы холодного питания
Полосы сырой смеси поступают с вальцов в приемник
шприцмашины и затем в её ствол.
Сырая смесь поступает прямо в шприцмашину.
VII. Производство шин -•V 677
Шприцмашины состоят из ствола, в который запитывают материал. За-
тем сырая смесь выдавливается вперед по специально сконструированному
винту к фильтрующей системе, где любой посторонний материал удаляется,
и затем проходит через мундштук, где формируется требуемый профиль
смеси.
Винт шприцмашины состоит из трех первичных зон: питания, сжатия
и дозирования. В зоне питания, расположенной под питателем шприц-
машины, винтовые потоки хорошо организуются в пространстве, чтобы
оптимизировать поток смеси. Резина проходит в зону сжатия, где смесь
нагревается из-за деформации сдвига, затем в дозирующую зону, где смесь
продолжает нагреваться, чтобы снизить вязкость, и окончательно в мундштук
для формирования профиля.
Рис. 24. Шприцмашина горячего питания.
Мундштук Вакуум-отсос —Питатель —\
Мии тадАжгуунД
Рис. 25. Шприцмашина холодного питания.
Шприцмашина горячего питания (рис. 24) обычно имеет короткий ствол
и винт с короткими зонами сжатия и дозирования, поскольку смесь всегда
горячая и имеет малую вязкость, когда поступает в бункер. Для ствола от-
ношение длины к диаметру обычно лежит в пределах 4:1-6:1.
Шприцмашина холодного питания (рис. 25) имеет много большее отно-
шение длины к диаметру, поскольку из-за необходимости снижения плас-
тичности сырой смеси разогрев и подъем давления требуется осуществлять
в профиле шприцмашины. Типичное отношение длины к диаметру 24:1.
Современные шприцмашины холодного питания также контролируются
компьютером, который позволяет регулировать распределение температуры
в стволе, контролировать давление и скорость загрузки и течения смеси. Это
обеспечивает точный контроль прохода смеси через мундштук, качество
поверхности выдавливаемой смеси и пригодность к дальнейшей обработки
продукта шприцевания.
678 -lb Глава 14. Технология шин
Важен периодический контроль расстояния между винтом и стволом
для обеспечения минимального обратного давления и закручивания или
турбулентности потока в канале.
Г. Сборка шины
Все компоненты шины, т. е. шприцованные части, каланд-
рованные слои каркаса и брекера, и борт собираются на сборочном станке.
Обычно радиальные шины собираются на плоском барабане. Сборка начи-
нается с наложения на цилиндрический барабан герметизирующего слоя.
За ним следует слой каркаса и другие дополнительные компоненты, пре-
дусмотренные шинными инженерами в производственной спецификации.
Затем устанавливают борта, и на них заворачивают слой каркаса. Сборка
переносится на станок второй стадии, где накладывают брекер и протектор.
Сборочный барабан складывается и «сырая шина», показанная на рис. 26,
перемещается к вулканизационным прессам.
Рис. 26. Сырая радиальная шина.
В процессе вулканизации сырая шина формуется в высококачественный
инженерный продукт. В вулканизационной форме смесь перетекает, создавая
рисунок протектора и требуемую толщину боковины. Хорошее качество пото-
ка зависит от пластичности невулканизованной резины. Чтобы течь, смесь не
должна подвергаться подвулканизации. Поток должен быть создан, прежде чем
вулканизация начнется, иначе могут возникнуть искажения. Необходимый
поток создается эффективным использованием наполнителей в смеси, регули-
рованием уровня ускорителей и применением пластификаторов.
VIII. Выводы JU 679
Д. Окончательная инспекция шины
Изготовленная шина должна соответствовать следующим
критериям:
1. Все компоненты шины должны успешно пройти процессы подготовки
до сборочного станка.
2. Компоненты должны поступить совместно к сборочному станку без
помех для обеспечения производительности оператора сборочного
станка.
3. Качество и уровень работоспособности шины должны соответствовать
требованиям потребителя.
Качество гарантируется последней стадией производственного цикла.
Здесь продукт проверяется на соответствие требованиям потребителя. Про-
верка качества включает:
1. Шерохование шины и снятие выпресовок от вулканизационной формы.
2. Визуальная инспекция каждой шины для обнаружения дефектов.
3. Рентгеновский контроль расположения нитей корда в слоях.
4. Статистическая выборка шин для испытаний на прочность, неодно-
родность и динамический дисбаланс. Эти испытания включают многие
описанные ранее тесты, такие как конусность, радиальное биение
и вариации боковой силы.
После того, как шина прошла инспекцию, она готова для отправки на
склад для распределения.
VIII. ВЫВОДЫ
Во многих отношениях шина является инженерным чудом.
Геометрически шина есть тор. С позиций механики шина - гибкий контейнер
высокого давления. Структурно шина - композит высокой работоспособно-
сти, изготовленный из тканей, стали и ряда органических и неорганических
химикатов.
Шинная технология является комплексной комбинацией науки и инже-
нерного искусства, которые объединяют различные дисциплины. В разра-
ботке шины существенны знания в области геометрии шины, динамического
поведения шины, химии составляющих шину материалов и технологии
композитных структур. Результатом является широкий ряд продуктов,
который удовлетворяет производителей транспортных средств, также как
потребности окончательных потребителей в оптимальной работоспособности
при различных условиях эксплуатации.
ГЛАВА
15
ВТОРИЧНАЯ ПЕРЕРАБОТКА РЕЗИНЫ
АВРААМ И. ИСАЕВ
Институт полимерного инжиниринга
Университета Акрона, Акрон, Огайо
I. ВВЕДЕНИЕ
Производство шин и других резиновых изделий предусмат-
ривает процесс вулканизации, т. е. необратимую реакция между эластомером,
серой и другими химикатами, при котором образуются поперечные связи между
молекулярными цепями и происходит формирование трехмерной химичес-
кой сетки. Вулканизованный эластомер представляет собой нерастворимый
термоустойчивый материал, что делает невозможной его прямую переработку
и повторное использование шин и других резинотехнических изделий. Поэто-
му, проблемы окружающей среды, вызванные накоплением изношенных шин
и других резиновых изделий стали серьезными в последние годы.
Фактически, Чарльз Гудьир, открывший процесс серной вулканизации
более 150 лет назад [ 1 ], был также первым, кто предпринял усилия по пере-
работке резиновых отходов методом измельчения [2]. Даже после стольких
лет усилий по разработке эффективной технологии переработки резиновых
отходов, это остается проблемой для резиновой промышленности.
Согласно недавнему обзору Совета по изношенным шинам Ассоциации
изготовителей резин, приблизительно 281 миллион старых шин были накоп-
лены только в 2001 г. в Соединенных Штатах [3]. Рынок потребляет прибли-
зительно 77.6% общей суммы [3], в то время как остальное добавляется к уже
существующему запасу в 300 миллионов старых шин, расположенных вокруг
Соединенных Штатов [4]. Эти шины создают серьезные проблемы для окружа-
ющей среды: опасность возникновения пожаров, размножения грызунов, змей,
москитов, и других вредителей, представляя угрозу для здоровья населения [5].
В Соединенных Штатах в 2001г. приблизительно 53% использованных шин
были сожжены, и только 19% общего количества было переработано в GRT
(измельченную шинную резину) для использования в резиновой промышлен-
ности [4]. Проблемой в резиновой промышленности стало образование более
чем 150000 тонн отходов производства не шинных резиновых изделий [7].
II. Восстановление шин JU 681
Изношенные шины и резинотехнические изделия, сделанные на основе
высококачественных каучуков, представляют большой потенциальный
источник сырья для резиновой промышленности. Главные причины, сдер-
живающие применение вторичного сырья являются: строгие требования
к качеству резиновых изделий, и следовательно к качеству вторичного сырья;
использование других материалов вместо каучука (например, пластмассы в
некоторых случаях); высокие затраты на переработку отходов из-за строгих
инструкций по защите окружающей среды; сравнительно высокий уровень
трудовых затрат и следовательно, высокая стоимость восстановленного кау-
чука [8]. Однако, ужесточающееся законодательство, ограничивающее захо-
ронение отходов требует поиск экономичного и экологически чистого метода
переработки шин и отходов резинового производства. Недавняя агрессивная
политика автомобильной промышленности нацелена на увеличение исполь-
зования переработанных пластмассовых и резиновых материалов [9-11].
Это может служить примером растущего спроса на такие технологии.
Главная цель этой главы состоит в анализе современного состояния про-
блемы переработки старых шин и отходов резинового производства, включая
существующие методы и новые технологии измельчения, восстановления и
девулканизации, и также возможности использования в изделия продуктов
переработки. Девулканизация - процесс превращения вулканизованных
отходов с использованием механической, тепловой, или химической энергии,
в состояние, в котором они могут быть смешаны, обработаны и вулканизо-
ваны снова [12]. Строго говоря, девулканизация может быть определена
как процесс разрушения полностью или частично, поли-, ди-, и моносуль-
фидных поперечных связей сформированных при вулканизации [13].
Девулканизацию перекисного и смоляного вулканизатов можно рассмат-
ривать как процесс разрушения углерод- углеродных или других, более
сильных межмолекулярных связей. Однако, в существующей концепции,
девулканизация определена как процесс разрушения химической сетки по
макромолекулярным цепям [14].
Множество методов [11,13-17] было опробовано в попытке найти более
эффективные пути переработки шин. Эти методы включают восстановление,
размол, измельчение, микроволновые и ультразвуковые процессы, пиролиз
и сжигание. Технологии использования продуктов переработки также раз-
виваются, в том числе для производства резиновых изделий и добавок для
термопластов, в качестве добавок в асфальт и цемент.
II. ВОССТАНОВЛЕНИЕ ШИН
Восстановление — один из способов переработки шин, позво-
ляющий экономить энергию. Чтобы произвести одну новую грузовую шину
требуется приблизительно 83 литра нефти, а для восстановления только
26 литров. Стоимость восстановленной шины может быть на 30-50% мень-
682 Глава 15. Вторичная переработка резины
ше чем новой [17, 18]. Приблизительно 24.2 миллионов восстановленных
шин были проданы в Северной Америке в 2001 г., при этом объем продаж
составил больше, чем 2 миллиарда долларов. Восстановлению подвергаются,
главным образом грузовые (средние и крупногабаритные) шины, шины для
внедорожных транспортных средств, и авиационные шины. [18]. Однако,
высокие трудовые затраты и перспектива ужесточения правил техники
безопасности сдерживают бизнес [17].
III. ПЕРЕРАБОТКА ВУЛКАНИЗАТОВ КАУЧУКА
А. Технологии регенерации
Регенерация — процедура, при которой вулканизованные от-
ходы превращают, используя механическую и тепловую энергию, а также хи-
микаты, в пластичный продукт, который может быть смешан, обработан, и под-
вергнут повторной вулканизации. Принцип процесса — девулканизация [12],
в результате которой происходит разрушение межмолекулярных связей
химической сетки типа углерод-сера и/или сера-сера. При этом имеет место
уменьшение размеров цепей [14].
Много различных процессов регенерации [8, 13, 15-17, 19-22] при-
менялись в попытке решить проблему рециркуляции резиновых отходов.
Вообще, резиновые отходы, в большинстве случаев являются сырьем для
девулканизации. Варнер [13] и недавно Адхикари и другие. [23] и Исаев
[24] представили обзоры существующей литературы, в которых регенерация
является наиболее важным процессом рециркуляции резин.
Различные процессы регенерации [7,8,14,23-32] использовались в разные
годы в зависимости от характеристики отходов и экономики. Водный, паровой
(влажный или сухой), и механический или термомеханический процессы — в
настоящее время обычно используются для регенерации.
Паровой процесс [7,8,13, 24-27,29-32] осуществляют в автоклаве, обо-
рудованном мешалкой для непрерывного перемешивания. Водный процесс
может происходить в кислотной или щелочной среде, в то время как сухой
процесс использует только пар. Если необходимо, при регенерации в авто-
клав добавляется пластификатор нефтяного происхождения. Сухой процесс
имеет преимущество перед водным из-за меньшего количества вредных
выбросов, содержащих вредные вещества в воде и воздухе.
Механический или термомеханический процесс [25, 32-34] использу-
ется для непрерывной регенерации отработанных шин. Резиновая крошка
(обычно 30 меш) смешанная с различными пластификаторами подвергается
интенсивной механической обработке в одночервячном экструдере при
высокой температуре.
Резиновые отходы, содержащие натуральный и синтетический каучук,
могут быть регенерированы сухим методом с использованием пластифика-
III. Переработка вулканизатов каучука -* V 683
торов нефтяного происхождения молекулярного веса между 200 и 1000 и
состоящих из ароматических соединений и парафинов. Состав пластифи-
каторов запатентован [35].
Недавно появилось сообщение о новой технологии девулканизации отхо-
дов эластомеров [36] с испбльзованием продукта, названного Де-линк [37].
В этом процессе, 100 или более частей мелкой крошки размером 40 меш сме-
шивают на вальцах с 2-6 частями Де-линка. Состав Де-линка не раскрыт.
Простой процесс регенерации резины в присутствии продукта раститель-
ного происхождения являющегося возобновляемым ресурсом (RRM), был
предложен в [38-40]. Главным составляющим этой добавки является диал-
лилдисульфид, другие составляющие (RRM,) различные серосодержащие
химические вещества (моно-, ди- и полисульфиды и тиолы).
Известно, что серные вулканизаты натурального каучука (НК) могут
быть переработаны при 200-225°С, используя дифенилдисульфид |41 ].
Недавно, об эффективности различных дисульфидов в качестве регене-
рирующих агентов при регенерации вулканизатов НК и СКЭПТа было
сообщено в [42]. В то время как девулканизация НК наблюдалась при
200°С с уменьшением густоты сетки на 90%, было показано, что такая
же степень девулканизации для серных вулканизатов СКЭПТа была
достигнута в присутствии дифенилдисульфида при температуре 275 ° С
в течение 2 часов. В то же самое время, перекисные вулканизаты СКЭПТа
показывали уменьшение густоты сетки приблизительно на 40% при тех
же самых условиях.
Другой химический метод был недавно предложен в [43]. Метод осно-
ван на использовании растворителя 2-бутанола как девулканизующего
агента для серных вулканизатов при высокой температуре и давлении. Это
требуется, для того чтобы молекулярный вес каучука был сохранен, а его
микроструктура значительно не изменена в течение процесса девулканиза-
ции. Однако, этот процесс — чрезвычайно медленный и требует отделения
девулканизата от растворителя.
Кроме органических химикатов, резина может быть девулканизована
посредством неорганических составов. Крошка отработанных шин и вул-
канизованных отходов шинного производства размером 10-30 меш была
десульфирована в растворителе типа толуол, бензин, бензол, циклогексан
и т.д., в присутствии натрия [44]. Щелочной металл разрушает моно-,
ди-, и полисульфидные связи набухшего вулканизата при температуре
в пределах до 300°С в отсутствии кислорода. Однако, этот процесс не мо-
жет быть экономичным, потому что требует набухания резиновой крошки
в органическом растворителе, где металлический натрий в жидком состоянии
должен провзаимодействовать с сульфидными связями вулканизационной
сетки. Кроме того, растворитель может причинять вред окружающей среде.
Была также предложена технология регенерации с использованием окиси
684 -'V- Глава 15. Вторичная переработка резины
железа и фенилгидразина в качестве катализатора [45] и трибутилхлорида
одновалентной меди [46].
В зависимости от назначения готовых изделий, наполнители могут быть
добавлены к девулканизату на стадии переработки. Девулканизат подвер-
гается обработке, очистке, после чего ему придается законченная форма
в виде листов, рулонов и т.д..
Таким образом, химический процесс регенерации — возможный метод
девулканизации резин. Однако, этот процесс протекает медленно и создает
дальнейшие проблемы с удалением растворителей и наличием отходов.
Некоторые процессы включают сложные химические реакции и требуют
дополнительных мер безопасности.
Б. Технология поверхностной обработки резиновой
крошки
Технология поверхностной обработки [47-49] с использо-
ванием растворителя предусматривает изменение поверхности резиновых
частиц крошки. Это — разновидность ранее известной технологии [43].
Процесс осуществляют в температурном диапазоне между 150 и 300°С
и давлении по крайней мере 3.4 МПа в присутствии растворителя группы
спиртов и кетонов. Среди различных растворителей, 2-бутанол показал
наилучшую способность к девулканизации серных вулканизатов на основе
бутадиен стирольного каучука (БСК). Продолжительность процесса — более
20 минут.
Эти данные были получены с маленькими количествами резиновой
крошки в колонке для газовой хромотографии. Растворитель, подходящий
для этого процесса должен иметь критическую температуру приблизительно
200-350°С. В результате растворяется поверхность резиновой крошки, затем
крошка должна быть отделена от растворителя. При этих условиях проис-
ходит предпочтительное разрушение S-S и С-S связей с незначительным
разрушением главных цепей. Полученный продукт был подвергнут вулка-
низации как в чистом виде так и в смесях с НК и показал удовлетворитель-
ное сохранение механических свойств. Однако, этот процесс был проверен
только в лабораторном масштабе.
В. Технология дробления и тонкого измельчения
резиновых отходов
Использование отработанных вулканизатов часто требует
уменьшения размера частиц. Один из наиболее широко используемых
методов для уменьшения размеров частиц резиновых отходов является
процесс дробления. Этот метод был изобретен и опубликован Гудьиром при-
близительно 150 лет назад [2]. Теперь есть три метода дробления резиновых
III. Переработка вулканизатов каучука -*v- 685
отходов: дробление при положительной температуре, криогенное дробление и
гидравлический размол [50-52]. Есть множество способов раздробить шины.
Для первичного дробления целых шин использют гильотину, крекер-вальцы,
молотковую дробилку и фрезерование с использованием шероховального
станка. Процесс первоначально предусматривает уменьшение кусков до
2.5-5 см2. При этом магнитный сепаратор и сепаратор волокна удаляют
металл и фрагменты текстильного корда. Дальнейшее измельчение отходов
может быть осуществлено с использованием криогенного размола [8].
Для получения тонко измельченной крошки куски резины охлаждают
в жидком азоте ниже их температуры стеклования и затем измельчают
в молотковой дробилке. В результате получают резиновый порошок с разме-
рами частицы от 30 до 100 меш. Однако, криогенное измельчение шинной
резины считается экономически не эффективным из-за большого количе-
ства жидкого азота или других криогенных жидкостей, необходимых, чтобы
заморозить резину [25]. Процесс может быть экономически оправданным
для отходов на основе дорогого фторкаучука. Вместе с тем криогенное
измельчение обеспечивает высокое качество крошки вследствие меньшей
деструкции каучукового вещества и высокой степени очистки крошки от
металла и текстильного корда [7].
Из-за высокой стоимости криогенного дробления чаще используется ме-
ханическое дробление и измельчение при положительной температуре с ис-
пользованием набора мощного оборудования. Используя этот относительно
недорогой метод, можно получать как крошку с размером частиц 30 меш, так
и относительно крупную крошку. Кроме того, многократный размол может
использоваться, чтобы далее уменьшить размер частиц. При использовании
валкового оборудования частицы имеют развитую поверхность и хорошо
совмещаются с исходной резиновой матрицей [32]. Минимальный размер
частиц, который может быть получен этим методом 40 меш. Процесс, однако,
сопровождается существенным теплообразованием. Высокая температура
может вызвать деструкцию каучукового вещества. При отсутствии должного
охлаждения при хранении может произойти возгорание.
Другие предложенные процессы рециркуляции включают механические
и термо-химические методы, которые только дробят резину и не девулка-
низируют ее. Процесс, использующий влажный метод размола позволяет
достигать размера частиц крошки приблизительно 200 меш [53]. Резиновая
крошка с таким размером частиц имеет высокую удельную поверхность
и может быть девулканизована без использования химикатов только при
минимальном нагревании и механической обработке. Процесс влажного
размола обеспечивает получение самых маленьких частицы, в пределах от
400 до 500 меш. Преимущество маленьких частиц состоит в том, что они
позволяют получать хорошую обработку и гладкую поверхность при даль-
нейшей экструзии и каландровании [53].
686 -*v- Глава 15. Вторичная переработка резины
Методы тонкого измельчения с применением червячного экструдера были
первоначально предложены для пластмасс. Процесс получения порошка
полимера в одночервячном экструдере основан на использовании одновре-
менного сжатия и сдвига при определенных температурах, которые зависят
от типа полимера [54]. Основанный на этом методе, способ измельчения
резиновых отходов был также предложен в [55,56]. Полученные резиновые
частицы имели развитую поверхность и уникальную удлиненную форму.
Недавно, этот процесс был усовершенствован для измельчения резин
в червячном экструдере для получения частиц размером от 40 до 1700 мкм
[57-60]. Схема процесса измельчения в одночервячном экструдере показана
на Рис. 1, а, б [57].
а
Рис. 1 Схема одношнекового экструдера для измельчения резины (а); геометрия
фрагмента канала шнека с переменной глубиной нарезки (б) [57].
Как показано на рис. 1, а, экструдер состоит из трех зон: подачи (зона
загрузки 1), сжатия (Зона 2) и измельчения (Зона 3). Зона сжатия пос-
тепенно сужается, чтобы создать степень сжатия 5. Экструдер оснащен си-
III. Переработка вулканизатов каучука 687
стемой водного охлаждения для снижения температуры выделяющейся при
измельчения резины. Экспериментальные исследования показали, что при
измельчении вулканизатов в экструдере в результате трения существенно
повышается температура, что приводит к частичной деструкции каучуко-
вого вещества [57]. Резиновая крошка подается в зону загрузки экструдера
и поступает в зону сжатия, где она подвергается высокому сжимающему
усилию и сдвигу. При одновременном действии сжатия и сдвига из-за вра-
щения винта, происходит измельчение и образуется резиновый порошок,
имеющий меньший размер частиц. Одновременно происходит поверхностное
окисление резиновых частиц и увеличение доли мелких частиц. Полученные
частицы имеют шероховатую, грубую поверхность и пористую структуру.
Густота сетки и доля геля уменьшены по сравнению с исходной резиной.
Это указывает на частичную девулканизацию. Полученную таким образом
резиновую крошку можно формовать в изделия при высокой температуре
и давлении в течение по крайней мере 1 часа [61, 62]. В таблице 1 пока-
зана зависимость удлинения при разрыве, предела прочности и густоты
сетки при формовании вулканизатов из частиц крошки размером от 250 до
425 меш, полученной под действием давления и сдвига из резины на основе
натурального каучука. Приблизительный состав резины был 54% SMR-20,
27% технического углерода, 11% ароматического масла, и 8% вулканизую-
щей группы. Температура и давление при вулканизации составляли 157°С
и 11 МПа соответственно. Образцы, полученные без применения вулкани-
зующих агентов обладали наихудшими свойствами среди всех образцов,
полученных из резинового порошка. В образце F1 масло, остатки вулкани-
зующих агентов и золь фракция были удалены экстракцией в толуоле. Это,
согласно авторам, увеличивает густоту сетки, приводя к улучшению свойств
образца. С другой стороны, образцы F2 и F3, полученные с добавлением
серы показали более низкие физико-механические свойства чем образец F1
из-за меньшего размера частиц и увеличенной густоты сетки при повтор-
ной вулканизации. Кроме того, образцы F1-F3 характеризовались более
низкими прочностными свойствами по сравнению с исходной резиной, что
говорит о недостаточном сжатии резиновых частиц для достижения свойств
исходного вулканизата.
Частицы, полученные другими методами размола, могут также
формоваться при высоком давлении и температуре, как показано
Морин и др. [63] и Трипати и др. [64]. В частности в этих статьях
резиновые частицы, полученные из резин на основе различных кау-
чуков различными методами размола, были сформованы в плиты без
добавок различных кислот и химикатов. Влияние времени, давления,
и температуры на механические свойства пластин были изучены.
В частности на рис. 2 показано влияние увеличения температуры на
механические свойства пластин из крошки размером 80 меш на ос-
688
Глава 15. Вторичная переработка резины
нове смеси каучуков НК и БСК, полученных при давлении 8,5 МПа
в течение 1 часа [63]. Эти данные показывают важность температуры
процесса. Ниже приблизительно 80°С, этот процесс не работает. Самый
высокий предел прочности приблизительно 4 МПа был достигнут при
достаточно высоком удлинени (приблизительно 800%). Механизм кон-
солидации частиц в этом процессе — результат разрыва связей с образо-
ванием радикалов, которые реагируют с другими радикалами, и таким
образом образуются поперечные связи и материал в целом.
Таблица 1 Свойства образцов из измельченных резиновых отходов [62].
Система ревулканизации М%) о6(МПа) v (моль/м3)
Код образца Сера (масс, ч.) Сульфенамид (ТББС)
F1 - - 360 10.3 50.4
F2 1.0 0.5 350 7.0 73.9
F3 1.0 - 320 8.2 69.5
Исходный - - 470 16.5 66.9
Рис. 2. Влияние температуры формования на механические свойства пластин, изго-
товленных из частиц резины на основе смеси каучуков НК и БСК размером
80 меш в течение 1 часа при давлении 8.5 МПа [63].
III. Переработка вулканизатов каучука -*v- 689
Г. Технологии девулканизации
1. Микроволновый метод
Микроволновая технология также была предложена для
девулканизации резин [15, 65]. Этот процесс создает высокую температуру
очень быстро и однородно по массе вулканизата. Метод использует управ-
ляемое количество микроволновой энергии для девуканизации серных
вулканизатов каучука, содержащих полярные группы или компоненты
с которыми волны могут взаимодействовать и затем снова вулканизоваться
с получением изделий, требующих существенных физических свойств. Сетка
вулканизата состоит из связей углерод-углерод, углерод-сера, и сера-сера.
Предполагалось, что самые слабые связи сера-сера и сера-углерод факти-
чески разрушились. Однако, материал, который можно использовать в мик-
роволновом процессе должен быть достаточно полярен, чтобы воспринять
энергию в количестве достаточном для обеспечения высокой температуры,
необходимой для девулканизации. Этот метод для серийного производства
требует дорогого оборудования.
Недавно, для изучения изменений, наблюдающихся в вулканизатах при
девулканизации с использованием микроволновой обработки использовался
метод термогравиметрии [66]. Установлено, что степень деструкции цепей
полимера при микроволновой обработке позволяет получать девулкани-
зат более высокого качества для повторного использования в резиновых
смесях.
2. Ультразвуковой метод
Многочисленные публикации в литературе в последнее время
посвящены изучению влияния ультразвука на растворы полимеров [67-72],
и на расплавы полимеров в процессе экструзии [73-78]. Существенные
усилия были предприняты, чтобы понять механизм воздействия ультразвука
на жидкости [79, 80] и деструкцию полимера в растворе [81].
Применение сверхзвуковых волн для девулканизации резин — при-
влекательная область изучения. Большинство источников указывает, что
резина вулканизуется ультразвуком скорее, чем девулканизуется. Впервые
деструкция резины с использованием ультразвуковой энергии была описана
Окуда и Хатано [82]. Это был периодический процесс, в котором вулкани-
зат деструктировали ультразвуковыми волнами с частотой 50kHz в течение
20 минут. Было показано, что при этом разрушились связи углерод-сера
и сера-сера, но не связи углерод-углерод. Свойства повторного вулканизата
были близки к свойствам исходного.
Недавно, новый непрерывный процесс был реализован для девулка-
низации резины как эффективный способ переработки использованных
шин и отходов РТИ [83-119]. Эта технология основана на использовании
мощного ультразвука. Ультразвуковые волны определенных уровней,
690 -*V- Глава 15. Вторичная переработка резины
в присутствии давления и высокой температуры, могут быстро разрушать
трехмерную сетку резины. Процесс ультразвуковой девулканизации очень
быстр, прост, эффективен, не требует растворителей и химических доба-
вок. Девулканизация протекает очень быстро, в течение секунд, при этом
разрушаются преимущественно серные связи. Процесс также применим
для разрушения поперечных связей в перекисных вулканизатах каучука
и пластмасс. Схематическое изображение реакторов для выполнения этого
процесса показана на рис. 3. Первоначально, так называемый коаксиальный
реактор для девулканизации резины (см. рис. 3, а) был разработан в нашей
лаборатории. Реактор состоит из одночервячного экструдера и ультразвуко-
вого генератора. Имеющий форму конуса ультразвуковой генератор имеет
внутренние полости для подачи охлаждающей воды. Резиновая крошка
подается в экструдер специальным питателем. Таким образом, количество
подаваемой резины регулируется изменением подачи. В состав установки
входят излучатель, ультразвуковой преобразователь, вибратор. Вибратор со-
здает колебания с частотой 20 кГц и различными амплитудами. Ультразвуко-
вой излучатель установлен на внешней поверхности экструдера. Выпуклый
наконечник вибратора соответствует вогнутой поверхности генератора так,
чтобы акустические параметры вибратора и генератора были однородными.
Акустические параметры процесса контролируются.
Позже, (см. рис. 3, б) и (см. рис. 3, в) ультразвуковые установки были усо-
вершенствованы. В цилиндрический реактор помещены два сверхзвуковых
излучателя в форме рожков прямоугольного сечения, охлаждаемых водой.
Излучатели размещены друг против друга. В реакторе размещены также два
бронзовых рестриктора, которые блокируют поток крошки и направляют
поток через зазор между вращающимся червяком и наконечником рожка.
В секции девулканизации большой диаметр обеспечивает подачу сходяще-
гося потока крошки к зоне девулканизвции. В желобчатом (Ж) цилиндриче-
ском реакторе проделаны два винтовых канала на внутренней поверхности,
через которые резиновые потоки проходят к зазору между вращающимися
червяками и наконечниками рожков.
Согласно лицензии Университета Акрона Компания Massillon, Штат Огайо,
построила опытный образец установки для сверхзвуковой девулканизации шин
и резиновых изделий [120, 121]. Сообщалось, что грузовые шины, содержа-
щие 15% и 30% ультразвукового девулканизата, наполненного техуглеродом
вулканизата БСК, прошли предварительные динамические испытания на
выносливость [121].
Широкие исследования ультразвуковой деструкции резины и некото-
рые предварительные исследования ультразвуковой деструкции сшитых
пластмасс изложены в [83-122]. Показано, что этот непрерывный процесс
позволяет перерабатывать различные типы резин и термопластов. Вследствие
ультразвуковой девулканизации резина становится мягкой, способной к пов-
торной обработке, формованию и вулканизации тем же самым способом как
III. Переработка вулканизатов каучука
Л-
691
температуры Головка Рупор Ускоритель
и давления экструдера
а
б
в
Рис. 3. Схема коаксиального реактора (а), цилиндрического реактора (б) и цилиндри-
ческого желобчатого (Ж) реактора (в) для девулканизации резины.
692 “'V Глава 15. Вторичная переработка резины
и исходная резиновая смесь. Эта новая технология успешно использовалась
в лаборатории для девулканизации резиновой крошки на основе (GRT)1)
[85, 86, 90, 102, 105, 120], ненаполненного и наполненного НК [99, 103],
каучука гваюлы [122], ненаполненного и наполненного БСК [87,93,95-97,
99], ненаполненных и наполненных резин на основе силиконового каучука
перекисной вулканизации [98, 100, 104, 111-113, 115], ненаполненного
и наполненного СКЭПТа [110, 117], ненаполненного полиуретана [116],
ненаполненных резин на основе бутилкаучука смоляной вулканизации [118],
резиновой крошки из отработанных шин и камер [119], фторкаучука, вспене-
ного полиэтиленвинилацетата, сшитого полиэтилена [83,84]. После повторной
вулканизации резиновые образцы характеризуются хорошими механическими
свойствами, которые в некоторых случаях являются сопоставимыми или пре-
вышающими таковые исходных вулканизатов [95,100,110].
Рис. 4. Зависимость (начального) давления в зоне девулканизации различных реак-
торов от амплитуды ультразвука при скорости потока 0,63 г/с и от скорости
потока при амплитуде 10 р,м и зазоре 2 мм в процессе девулканизации GRT.
Ультразвуковые исследования позволили установить: влияние парамет-
ров давления, потребления мощности, длительности обработки, температуры,
величины потока и амплитуды ультразвука на эффект девулканизации;
GRT — Ground Tire Rubber — измельченная шинная резина (крошка резины на основе
НК, БСК, и полибутадиена).
III. Переработка вулканизатов каучука -’V 693
структурные изменения, происходящие в различных каучуках; реологиче-
ские свойства и кинетику процесса девулканизации; механические свойства
повторного вулканизата и влияние конструкции реактора. На рис. 4 показана
зависимость начального давления в зоне девулканизации GRT при величи-
не потока 0.63 г/с, от амплитуды (до 10 мкм) при толщине материала 2 мм.
Начальное давление зоны девулканизации было существенно уменьшено, по-
скольку амплитуда ультразвука была увеличена. Ультразвук облегчил поток
резины не только из-за сокращения трения в присутствии ультразвуковых
волн но также и из-за имеющей место девулканизации, поскольку частицы
GRT поступили в зону девулканизации. Цилиндрический реактор показывал
более высокое давление в зоне девулканицации, чем коаксиальный реактор,
цилиндрический (Ж) реактор показывал самое низкое давление при низ-
кой амплитуде ультразвука и мощности потока 0.63 г/с. Цилиндрический
реактор имел сходящуюся зону перед зоной девулканизации. Поток GRT
был по существу блокирован в зоне девулканизации при низкой амплитуде
ультразвука. Однако, при амплитуде ультразвука 10 мкм, входное давление
зоны девулканизации для коаксильного и цилиндрического реакторов было
почти одинаковым из-за сокращения эффекта при высокой амплитуде. Са-
мая высокая мощность потока, достигнутая в цилиндрических реакторах
была 6.3 г/с. Мощность потока 6.3 г/с в коаксиальном реакторе не могла
быть получена из-за перегрузки ультразвукового генератора. Кроме того,
в цилиндрическом (Ж) реакторе при увеличении потока до 6.3 г/с, размер
промежутка, должен быть увеличен до 3.5 мм, и ультразвуковая амплитуда
до 6 мкм из-за перегрузки ультразвукового генератора. Естественно, что
начальное давление в зоне девулканизации повышается с увеличением мощ-
ностью потока как это показано на рис. 4 для всех трех реакторов. Однако,
при высокой мощности потока цилиндрический реактор имел начальное
давление в зоне девулканизации ниже, чем в других реакторах при ампли-
туде ультразвука 10 мкм.
Различие в характеристиках давления от мощности потока среди этих
трех реакторов, при толщине девулканизуемого материала 2 мм, было
возможно связаны с различием в потреблении мощности и различиях в ус-
ловиях сжатия. R цилиндрических реакторах GRT в зоне девулканизации
был подвергнут давлению и сдвигу, в то время как в коаксиальном реакторе
только давлению.
Сравнение кривых растяжения вулканизатов, изготовленных из девул-
канизата GRT произведенного в этих трех реакторах при максимальной
мощности потока показано на рис. 5. Образец повторного вулканизата,
полученный после цилиндрического реактора, имеющего мощность потока
6.3 г/с, показывает предел прочности 8.7 МПа, удлинение при разрыве 217%,
и модуль при удлинении 100% 2.6 МПа. Образец повторного вулканизата,
полученный после цилиндрического (Ж) реактора с мощностью потока
694 -*V" Глава 15. Вторичная переработка резины
6.3 г/с, показывает предел прочности 8.3 МПа, удлинение при разрыве 184%,
и модуль при удлинении 100% 3.3 МПа. Образцы, полученные в цилинд-
рических реакторах были более высокого качества, чем образцы из коакси-
ального реактора. Кроме того, механические свойства образца позволили
использовать в цилиндрическом реакторе более высокую мощность потока —
которая не могла быть достигнута в коаксиальном реакторе. Эти свойства
позволяют использовать девулканизат в изделиях с высоким уровнем тех-
нических требований, например в шинах [7]. Образцы, с более низкими
показателями свидетельствуют о более высокой степени девулканизации
наряду с существенной деструкцией молекулярных цепей.
Рис. 5. Кривые растяжения вулканизатов, изготовленных из девулканизатов GRT,
произведенных тремя реакторами при максимальной скорости потока.
Предполагается, что процесс ультразвуковой девулканизации основан на
кавитации. В данном случае акустическая кавитация происходит в твердом
теле. Это отличается от известного явления кавитации, происходящего в
жидкостях, подвергнутых быстро изменяющемуся давлению высокой амп-
литуды, создаваемым мощным ультразвуком [123]. В течение отрицательной
половины цикла жидкость подвергается растяжению, а в течение положи-
тельной половины цикла сжатию. При наличии газонаполненного пузыря
в жидкости, происходит попеременное увеличение и уменьшение поверх-
ности контакта жидкости с газом. Пузырь может разрушаться внезапно в
течение сжатия. Это разрушение известно как кавитация и может сопро-
вождаться почти мгновенным выбросом сравнительно большого количества
Ш. Переработка вулканизатов каучука 695
энергии. Величина энергии, выброшенной таким образом, зависит от вели-
чины акустической амплитуды давления и, следовательно, интенсивности
акустического воздействия.
Хотя присутствие пузырей облегчает начало кавитации, это явление мо-
жет также происходить в жидкостях без газа, когда акустическая амплитуда
давления превышает гидростатическое давление в жидкости. В отрицатель-
ной части цикла давления, жидкость находится в состоянии растяжения,
при этом силы связи между соседними молекулами ослабевают и пустоты
формируются в слабых точках структуры жидкости. Эти пустоты растут
в размере и затем разрушаются, таким же образом, как и газонаполненные
пузыри. Кавитация в жидкостях без газа может быть вызвана наличием
различных дефектов в ее структуре, вызванных, например, присутствием
примесей.
Известно, что воздействие ультразвука на полимер, вызывает кавитацию
пузырей [67,124]. Формирование и разрушение пузыря играют важную роль
в деструкции полимеров. Большинство физических и химических эффектов,
вызванных ультразвуком, обычно приписывается кавитации, а именно, росту
и очень быстрому разрушению микропузырей под действием ультразвука.
Интенсивная волна удара, исходящая от пузыря взрывающегося вследствие
кавитации - несомненно, причина наиболее серьезных реакций. Эта волна
удара способна привести к разрушению макромолекул, находящихся на ее
пути. Деструкция возникает в результате эффекта распространения ультра-
звука в растворе. В любой среде существуют неравномерности, пустоты
и колебания плотности. Полагается, что они стимулируют кавитацию, ведя
к молекулярному разрыву. В твердых полимерах микропустоты ответственны
за кавитацию, когда они подвергнуты гидростатическому давлению в виде
импульса. Одна из главных причин наличия микропустот в полимерах это
разрушение межмолекулярных связей при механическом и тепловом воз-
действии, что показали обширные исследования [125].
Применительно к резине кавитация обычно аналогична эффекту фор-
мирования и неограниченного роста пустот в насыщаемых газом резиновых
образцах после внезапного снятия давления. [126, 127]. Вообще, это имеет
более широкий смысл и может быть понято как явление, связанное с фор-
мированием и динамикой образования пустот в непрерывных средствах
[88,102]. В науке о материалах, например, это означает способ разрушения,
характеризованный формированием внутренних пустот [128]. В акустике,
кавитация обозначает явления, связанные с динамикой поведения пузырей
в облученных ультразвуком жидкостях [129].
Исследования изменения структуры каучука, под действием ультозвука
показывают, что разрыв химических поперечных связей сопровождается
частичной деструкцией цепей каучука [85, 86, 96, 97]. Механизм девулка-
низации резины при ультразвуковой обработке до сих пор до конца не понят,
696 -’V Глава 15. Вторичная переработка резины
в отличие от механизма деструкции линейных полимеров [124]. Особенно не
ясны механизмы, управляющие преобразованием механической ультразву-
ковой энергии в химическую. Однако, показано, что девулканизация резины
при ультразвуковой обработке требует местной концентрации энергии, в то
время как однородно распределенная ультразвуковая энергия не способна
к девулканизации каучука [88, 89, 130].
Известно, что некоторые количества вакуолей или маленьких пузырей
присутствуют в резине на основе любого типа каучука [131]. Формирование
пузырей может определяться пустотами соответствующего размера исход-
ного материала [126]. Предложенные модели [88-90, 101, 102] были осно-
ваны на механизме разрушения сетки, вызванной кавитацией, которая
создана высокой интенсивностью ультразвуковой волны в присутствии
давления и высокой температуры. Вакуоли под действием ультразвука
пульсируют с амплитудой, зависящей главным образом от соотношения
между окружающими и ультразвуковыми давлениями (акустическая
кавитация).
Известно, что, в отличие от пластмасс, связи в резине разрушаются в том
случае, когда они полностью вытянуты [132, 133]. Ультразвуковое поле со-
здает высокочастотные напряжения сжатия и растяжения в сетке. Поэтому,
эффекты высокоэластичности резины были включены в описание динамики
кавитации [101,102]. Девулканизация резиновой сетки может происходить
прежде всего вокруг пульсирующих пустот из-за высокого уровня напря-
жения, произведенного мощным ультразвуком [102].
Вообще, разрыв в цепях полимера приводит к образованию макрора-
дикалов [134, 135], существование которых было подтверждено спектро-
скопически при помощи радикальных акцепторов типа дифенилпикрил-
гидразина (ДФПГ). Очевидно, в отсутствии акцепторов радикалов мак-
рорадикалы способны взаимодействовать с образованием макромолекул
меньшего размера, зависящего от размера объединяющихся фрагментов
[136].
Было показано [95], что при некоторых условиях девулканизации пре-
дел прочности повторного вулканизата БСК был намного выше, предела
прочности исходного вулканизата, а удлинение при разрыве фактически
не изменилось. В частности рис. 6 показывает кривые напряжение — де-
формация необработанного вулканизата и повторного вулканизата БСК
изготовленного из девулканизата полученного при различных величинах
ультразвуковых амплитуд А. Девулканизат получен с использованием
коаксиального ультразвукового реактора, изображенного на рис. 3, а, тем-
пературе 120°С, скорость червяка 20 об/мин, и мощности потока 0.63 г/с.
Диаметр ультразвуковой головки 76.2 мм. В отличие от обычных результатов
по механическим свойствам девулканизата, полученного с использованием
различных методов, приведенные данные довольно неожиданны. Было вы-
III. Переработка вулканизатов каучука
Л
697
двинуто предположение, что улучшение механических свойств повторного
вулканизата БСК является следствием возникновения бимодальной сетки,
которая образуется в процессе повторной вулканизации резины обрабо-
танной ультразвуком. Превосходящие свойства повторного вулканизата
каучука также наблюдались в случае ненаполненного вулканизата СКЭПТа
[110] и силиконового каучука [100]. Повторный вулканизат НК также име-
ет хорошие свойства, в частности удлинение при разрыве такое же, как
у исходного вулканизата НК, а прочность составляет приблизительно 70%
от исходной. Интересно, что вызванная напряжением кристаллизация,
типичная для первоначального вулканизата НК осталась неизменной
в повторном вулканизате, как показано на рис. 7, где подъем кривых
напряжение-деформация наблюдается и в исходном и в повторном вул-
канизате. Эти образцы были девулканизованы в коаксиальном реакторе
при различных мощностях потока и ультразвуковых амплитудах и под-
вергнуты повторной вулканизации с рецептом, состоящим из 2.5 мае. ч.
ZnO, 0.5 мае. ч. стеариновой кислоты и 2 мае. ч. серы.
Рис. 6. Кривые напряжение-деформация ненаполненных исходного вулканизата
и повторного вулканизата БСК, полученного из резины девулканизован-
ной в коаксиальном реакторе при различных величинах ультразвуковой
амплитуды.
Наполнители играют интересную роль в процессе девулканизации. Ри-
сунок 8 показывает кривые напряжение-деформация для исходного и пов-
торного вулканизатов НК, наполненного 35 мае. ч. технического углерода.
Исходный вулканизат был получен с использованием 5 мае. ч. ZnO, 1 масс. ч.
698 -*v- Глава 15. Вторичная переработка резины
стеариновой кислоты, 1 мае. ч. сульфенамида Ц и 2 мае. ч серы. Повторный
вулканизат содержал 2.5 мае. ч. ZnO, 5 мае. стеариновой кислоты, 0.5 мае. ч.
сульфенамида Ц и 2 мае. ч. серы.
Деформация, %
Рис. 7. Кривые напряжение- деформация для вулканизатов ненаполненного НК из
девулканизатов обработанных ультразвуком в коаксиальном реакторе при
различных мощностях потока и амплитудах, толщине обрабатываемого ма-
териала 2.54 мм и температуре 120°С.
Эксперименты показали, что у наполненного техническим углеродом
вулканизата НК, после девулканизации механические свойства повтор-
ного вулканизата ухудшаются, и уровень ухудшения зависит от условий
девулканизации. Это очевидно из рис. 8. Было выдвинуто предположение,
что сверхзвуковая девулканизация вызывает частичную дезактивацию
наполнителя из-за разрыва макромолекулярных цепей, связанных
с поверхностью технического углерода. Во многих случаях, этот эффект
ведет к низким свойствам девулканизовнной наполненной техническим
углеродом резины. Для корректировки свойств девулканизованная
ультразвуком резина был смешана с НК [137]. Смесь обладала сущест-
венно лучшими свойствами. Некоторое количество нового технического
углерода было добавлено в девулканизованную резину. Показано, что
вторичный вулканизат, содержащий добавленный технический углерод
обеспечивает получение более высоких упруго-прочностных свойств, чем
таковой не содержащий дополнительный технический углерод. В некото-
рых случаях наблюдалось даже улучшение физико-механических свойств
при введении в девулканизованную резину дополнительного количества
IIL Переработка вулканизатов каучука -* V 699
технического углерода по сравнению с исходным каучуком. В частности
это показывали для кровли из СКЭПТа содержащей технический углерод,
и существенное количество нефтяного пластификатора [117]. Очевидно,
нефтяной пластификатор играет важную роль в процессе девулканиза-
ции. Возможно, присутствие нефтяного пластификатора предотвращает
дезактивацию наполнителя, которая наблюдается в вулканизатах, без
него. Но чтобы доказывать эту гипотезу, требуются дополнительные
эксперименты.
Деформация, %
Рис. 8. Кривые напряжение-деформация для вулканизата НК, наполненного 35 мас.ч.
технического углерода и вторичных вулканизатов из девулканизата НК, по-
лученного в коаксиальном реакторе при температуре 120°С, зазоре 2.54 мкм,
мощности потока 0.63 г/сек, и различных ультразвуковых амплитудах.
Ультразвуковая девулканизация изменяет также кинетику повторной
вулканизации девулканизатов. Показано [93], что процесс повторной
вулканизации девулканизата БСК по существу отличался от такового для
исходного БСК. Для повторного девулканизата период индукции короче
или отсутствует вовсе. Это также характерно для других ненаполненных
и наполненных техническим углеродом вулканизатов GRT, БСК, НК,
СКЭПТа и БК, вулканизоанных серными системами, но не для силиконового
каучука, вулканизоанного перекисью. Было предположено, что уменьше-
ние или исчезновение периода индукции в случае вулканизованного серой
девулканизата происходит из-за взаимодействия между молекулами кау-
чука, химически измененными в ходе девулканизации и не измененными
700 -*V- Глава 15. Вторичная переработка резины
исходными цепями каучука, заканчивающегося сшиванием. Показано, что
приблизительно 85% ускорителя остались в девулканизованном ультразву-
ком девулканизате БСК [93].
Девулканизат, получнный с помощью ультразвука, состоит из золя
и геля. Гель характеризуется мягкостью и низкой густотой сетки, значи-
тельно более низкой, чем исходный вулканизат. Из-за присутствия золя
девулканизованная резина может течь и формоваться. Густота сетки и золя
и геля, как было установлено, у девулканизовнной ультразвуком резины,
коррелирует с кривой для исходного вулканизата. [91, 100, 138]. Рисунок
9 иллюстрирует зависимость доли геля от густоты сетки девулканизовнного
GRT, полученного в трех реакторах. Доля геля и густота сетки GRT были 0.82
и 9.9х10-2 кмоль/м3, соответственно. Для каждого реактора, зависимость
доли геля от густоты сетки была описана кривой, схожей с кривой исходного
вулканизата, вид которой был независим от условий обработки, мощности
потока и амплитуды. Смещение корреляции долей геля и густотой сетки,
полученной в цилиндрических реакторах в область более низких густот
сетки чем в коаксиальном реакторе, указывая на лучшую эффективность
девулканизации. Возможно, дополнительный эффект сдвига, вызванный
вращением червяка в цилиндрических реакторах оказывал положительное
влияние на улучшение эффективности девулканизации.
IV. ИСПОЛЬЗОВАНИЕ ПРОДУКТОВ ПЕРЕРАБОТКИ
А. Общие замечания
Есть технические ограничения в девулканизации некоторых
резин, для которых повторная вулканизация практически невозможна [11].
Частичная девулканизация отходов резин приводит к падению физических
свойств. Во многих случаях, это может ограничивать объемы применения
регенерата в ответственных изделиях, таких как легковые шины. В то же
время регенерат можно использовать в значительных количествах в качестве
добавки к дешевым каучукам в малоответственных изделиях.
Согласно Франта [12], регенерат не может использоваться в смесях для
протекторов шин, поскольку при этом снижается их прочность.. Однако, это
положение не касается применения регенерата вместе с дополнительными
химикатами. Значительные количества регенерата используются в карка-
сах и боковинах шин, особенно если они изготавливаются из натурального
каучука; в каркасах радиальных шин регенерат не применяется. С другой
стороны, регенерат применяют в смесях для обрезинки бортовых колец
и боковин. Известно много областей применения продуктов переработки
резин вне резиновой промышленности. Так, резиновая крошка используется
как наполнитель для асфальтовых смесей при ремонте дорог и в материа-
лах для спортивных покрытий. Резиновая крошка может применяться как
IV. Использование продуктов преработки -*и- 701
наполнитель в различных материалах [139-144], в том числе в пластмассах
[139, 145]. Однако, проблема совместимости с матрицей и размер частиц
наполнителя, а также неоднородность в связях между этими двумя компо-
нентам, необходимо учитывать. Резиновые изделия, содержащие резиновую
крошку, имеют низкое удлинение при растяжении из-за слабой связи между
резиновой крошкой и каучуковой матрицей. Этот недостаток может быть
частично исключен путем добавки регенерата.
Рис. 9. Зависимость содержания геля от густоты сетки в девулканизатах GRT, полу-
ченных в различных условиях девулканизации с использованием коакси-
ального и цилиндрических реакторов.
Б. Использование в новых шинах
Шины являются сложным композитом, состоящим из про-
тектора, брекера, каркаса, бортового кольца. В производстве шин использу-
ется несколько типов каучуков и технического углерода. Следовательно, GRT
является смесью вулканизатов различных каучуков и типов технического
углерода. Соответственно при использовании дробленого вулканизата GRT
и девулканизата GRT в производстве новых шин необходимо рассмотреть
ряд факторов. Очевидно, измельченные отходы могут использоваться как
наполнитель для сырого каучука, а девулканизат GRT может использо-
ваться в смесях с сырым каучуком. Этот рынок оценивался приблизительно
20 тыс. т. крошки в 2001 году [3].
До недавнего времени, полагали, что только несколько процентов рези-
новой крошки может использоваться в новых шинах. Совет по Управлению
702 -*v- Глава 15. Вторичная переработка резины
отходами шин сообщил, что 5% переработанных шин используются в новых
шинах для Форда марки «Виндстар» [3]. Хотя никакая другая информация
относительно количества резиновой крошки, используемой в новых шинах
в открытой литературе не приводится, существует вероятность использова-
ния до 10% крошки переработанных шин в составах новых шин [17]. Недав-
но, сообщалось, что дорожные испытания грузовых шин, содержащих 10%
крошки показали, что износ шины почти равняется износу для аналогичных
новых шин [146]. Увеличение количества резиновой крошки в шинах растет,
но вероятно, что результаты не будут доступны в течение ряда лет.
В. Смеси исходных и вторичных резин
Резиновая крошка, получаемая из отходов шин может ис-
пользоваться в качестве добавки к сырому каучуку в производстве резиновых
смесей. Однако, в этом случае, совместимость с матрицей — существенная
проблема. Резиновые изделия, содержащие резиновую крошку, характери-
зуются более низким удлинением из-за недостаточной связи с каучуковой
матрицей. В таблице 2. показано влияние частиц GRT различных размеров,
включенных в состав НК, на его механические свойства. Определены предел
прочности, удлинение при разрыве и сопротивление раздиру до и после ста-
рения для вулканизата без добавок и вулканизатов, содержащих частицы
GRT различных размеров. Вулканизаты А, В, С, и Д содержали 30 мае. ч.
частиц GRT размером в диапазоне 650-450,300-215,205-160, и 150-10 мкм
соответственно. Контрольный образец — вулканизат НК без добавок. Рецепт
вулканизации содержал 6 масс. ч. ZnO, 0.5 мае. ч стеариновой кислоты,
3.5 мае. ч. серы, и 0.5 мае. ч. ускорителя. Добавление GRT в НК ухудшает
физико-механические свойства вулканизатов, причем чем больше размер
частиц крошки, тем существенне ухудшение физико-механических свойств.
Однако, вулканизат НК, содержащий GRT, показал лучшее сохранение
свойств после старения. Показано, что меньшие частицы содержат меньшее
количество каучука, но большее количество наполнителей и металлов.
Переработанная шинная резина в форме больших частиц крошки так-
же используется для создания дренажного слоя [148]. GRT смешивают
с полиуретанами (ПУ) для производства мягких покрытий для детских
площадок и спортивных сооружений. Покрытие укладывается на гравий,
бетон или асфальт.
Продукты переработки резин, как было показано выше, общепри-
нято добавлять к различным полимерам. Были изготовлены смеси ПУ
и шинной крошки различного размера и девулканизата, полученного
ультразвуковым методом и изучены их механические свойства [149].
В девулканизат была введена вулканизующая система. Для сравнения
вулканизующая система была добавлена в смеси на основе ПУ с рези-
новой крошкой.
Таблица 2. Свойства вулканизатов НК, наполненных GRT [147].
Свойства Композиция
Контроль А В С D
До старения После старения До старения После старения До старения После старения До старения После старения До старения После старения
Прочность (МПа)‘> 14.0 8.8 (63) 2.2 1.6 (71) 4.2 2.5 (60) 7.3 3.3 (45) 8.0 2.5 (31)
Удлинение при разрыве (%) 1175 770 (66) 430 230 (53) 620 360 (58) 780 410 (53) 860 400 (47)
Раздир (кН/м)*) 28.2 20.3 (72) 12.34 9.7 (78) 18.5 10.6 (57) 23.8 11.5 (48) 21.2 9.7 (46)
Золь, % 2.1 2.2 5.8 6.4 5.2 6.2 4.9 6.0 4.3 6.6
% увеличения золя после ста- рения — 4.8 — 10.3 — 19.2 — 22.4 — 53.5
*) Цифры в скобках показывают % сохранения свойств после старения при 100°С 36 часов.
IV. Использование продуктов преработки -'Ь- 703
704 -*V- Глава 15. Вторичная переработка резины
На рисунке 10 показаны результаты определения прочности и удлине-
ния композитов ПУ с ультразвуковым девулканизатом (а) и измельченным
вулканизатом (б) (вулканизующая группа вводилась на резиновую часть
композита). Из представленных данных следует, что композит с 25% де-
вулканизата превосходит контрольный образец по физико-механическим
показателям, а смесь с 50% сопоставима с исходной. Для измельченной ре-
зины только при концентрации введенной крошки 25% свойства ПУ и смеси
сопоставимы. Можно полагать, что девулканизат, полученный с помощью
ультразвука, лучше совмещается с ПУ
Деформация, %
Деформация,%
Рис. 10. Кривые растяжения композитов: ПУ — ультразвуковой девулканизат (а);
ПУ — измельченный вулканизат (б). Условия девулканзации: скорость
потока 1,26 r/сек., зазор 3 мм., амплитуда 7,5 мкм., температура коакси-
ального реактора 120° С.
IV. Использование продуктов преработки -*и- 705
Девулканзат, полученный с помощью ультразвука и необработанную
резиновую крошку добавляли в исходную смесь на основе НК, содержащую
35 масс. ч. техуглерода. На рис И приведены данные предела прочнос-
ти и удлинения вулканизатов с различным содержанием девулканизата
и крошки [137].
Концентрация исходного вулканизата НК, вес.%
Концентрация исходного вулканизата НК, вес.%
Рис. 11. Прочность (а) и относительное удлинение (б) вулканизатов из: 1 —
(НК +35 масс. ч. техуглерода) + ультразвуковой девулканизат, вулканизу-
ющая группа добавлена на весь каучук; 2 — (НК +35 масс. ч. техуглерода)
+ измельченная резиновая крошка, вулканизующая группа добавлена на
НК; 3 — (НК +35 масс. ч. техуглерода) + измельченная резиновая крошка,
вулканизующая группа добавлена на весь каучук. Вулканизат НК был де-
вулканизован при скорости потока 0,63 г/c., зазоре 2,54 мм, амплитуде 5 мкм.
и температуре коаксиального реактора 120° С.
706 -'V Глава 15. Вторичная переработка резины
Вулканизаты смесей на основе НК и девулканизата имеют намного
лучшие физико-механические свойства, чем вулканизаты, содержащие
резиновую крошку. С увеличением содержания девулканизата, получен-
ного с помощью ультразвука в исходной смеси на основе НК свойства
вулканизатов падают в существенно меньшей степени чем при добавлении
необработанной крошки. Был определен модуль при удлинении 100% для
вулканизата, содержащего 35 мае. ч. технического углерода [137]. Смеси
с ультразвуковым девулканизатом характеризовались более высоким
значением модуля, чем смеси с резиновой крошкой.
Г. Смеси термопластов и вторичных резин
Технология совмещения полимеров появилась как полезный
инструмент их приспособления к потребностям конечных пользователей.
Так, получило развитие получение смесевых термопластичных эластомеров
(ТПЭ). Эти ТПЭ становятся все более и более важными из-за их эластичности,
легкой перерабатываемости и более низкой стоимости.
Смешивание отходов резины с термопластами важно с точки зрения
использования отходов и сокращения стоимости изделия. Большее внима-
ния было сосредоточено на производстве смесей резиновой крошки с тер-
мопластами, которые можно затем повторно размягчать и перерабатывать
в различные формованные и прессованные изделия [150, 151]. Механиче-
ские свойства таких составов зависят от концентрации GRT, типа матричного
полимера, и адгезии GRT к матричному полимеру, а так же от размера частиц,
их дисперсности и взаимодействия между GRT и матрицей [152]. Вообще,
адгезия между GRT и матричным полимером и размер частиц GRT —
два главных фактора, управляющие механическими свойствами таких ма-
териалов. Методы динамической вулканизации могут использоваться для
улучшения свойств смесей девулканизатов, полученных с помощю ультра-
звука [153-156]. Динамическая вулканизация — процесс вулканизации
эластомера находящегося в пластичном состоянии в смеси с расплавом
термопласта [157].
Использование GRT, вместо каучука приводит к существенному ухудше-
нию механических свойств этих материалов. Было показано [158-160], что
GRT отрицательно влияет на большинство физических свойств полимера,
причем степень ухудшения, возрастает с увеличением количества и размера
частиц GRT. Было выполнено несколько исследований с целью улучшения
адгезии между частицами GRT и матрицей полимера.
Так, изучалось влияние различных добавок, улучшающих совмести-
мость полиэтилена (ПЭ) и GRT различного состава, с целью повышения
адгезии ПЭ и GRT [139, 161-163]. Авторы установили, что было до-
IV. Использование продуктов преработки 707
стигнуто частичное восстановление свойств материала путем смешения
в расплаве, где каждый компонент (GRT и пластик) сначала совмещался
с модификатором, прежде чем осуществлялось фактическое смешивание
[150]. Среди различых добавок, улучшающих совместимость, использова-
ли эпоксидированный НК [163], этиленакриловый сополимер [139, 161,
162], этиленгликоль, полиметилметакрилат [150,162]. Также сообщалось,
что частицы GRT меньшего размера облегчают переработку расплавов.
Увеличение энергии взаимодействия для линейного полиэтилена низкой
плотности (ПЭНП) и соединений на его основе выше, чем для полиэтилена
высокой палотности (ПЭВП) и его соединений. Предполагают, что низкая
полярность и/или низкая кристалличночть матричного полимера улучшают
совместимость. [152].
Модификация поверхности частицы GRT была изучена для улучшения
совместимости GRT и полимера. Поверхностная модификация может быть
осуществлена путем химической обработки, например, хромовой кислотой
[164], термическим окислением [165] или с помощью механической обра-
ботки. Использование привитого малеиновым ангидридом и хлорированного
GRT улучшило физические свойства смесей GRT-СКЭПТ — полиакрилат
и GRT-ПЭВП-ПВХсответственно [166,167]. Было найдено [168], что по-
верхностная модификация GRT позволила улучшать производство смесей.
Влияние криогенной GRT (приблизительно 250 микрон) из старых шин
на некоторые механические свойства ненасыщенной полиэфирной смолы
были исследованы в [169].
Обработку высокой энергией, включая плазму [170, 171], коронный
разряд и электроннолучевую радиацию [151] использовали, чтобы из-
менить поверхность GRT. Окисление поверхности GRT, проведенное
в плазме и автоклаве в атмосфере кислорода, позволило улучшить адгезию
между GRT и полиамидом [170]. Эпоксидная смола, с введенными в нее
частицами шинной крошки, модифицированной плазменной поверхност-
ной обработкой была также изучена. Было исследовано влияние коронно-
го разряда на GRT и на взаимодействие этого GRT с термопластами [162].
Фотоэлектронная рентгеновская спектроскопия показала, что обработка
коронным разрядом увеличила содержание кислородных групп на поверх-
ности GRT. Для повышения эффективности процесса девулканизации был
построен новый усовершенствованый ультразвуковой реактор с двумя из-
лучателями. В новом реакторе были получены смеси полипропилена (ПП)
и GRT с лучшими свойствами. Это, очевидно, было вызвано усилением
механохимических взаимодействий в материале. Механические свойства
композитов полипропилен-GRT, полипропилена-девулканизата GRT
и полипропилена-повторного вулканизата в пропорции 40:60 показаны
в таблице 3.
708 -*и- Глава 15. Вторичная переработка резины
Таблица 3. Механические свойства смесей полипропилен-GRT (ПП-GRT),
полипропилен-девулканизат GRT (ПП-ДСИТ) и полипропи-
лен-повторный вулканизат GRT (ПП-IIGRT) [154].
Смесь Прочность, МПа Модуль Юнга, МПа Удлинение при разрыве, %
ПП-GRT 6,7 116 16,6
ПП-ДОКТ 2 пропуска, 5 мкм 6,6 98 21,5
2 пропуска, 7,5 мкм 6,5 100 20,6
2 пропуска, 10 мкм 5,15 102 7,6
1 пропуск, 5 мкм 6,9 110 19,3
1 пропуск, 5 мкм 6,6 104 17,4
1 пропуск, 10 мкм 7,0 116 21,0
более ранняя работа [150] 5,2 108 20,7
ПП-RGRT 2 пропуска, 5 мкм 6,8 109 13,2
2 пропуска, 7,5 мкм 6,7 110 13,3
2 пропуска, 10 мкм 5,9 111 7,0
1 пропуск, 5 мкм 6,4 110 11,8
1 пропуск, 5 мкм 6,6 110 13,3
1 пропуск, 10 мкм 7,2 122 14,8
более ранняя работа [150] 6,7 110 18,0
Кроме того, GRT был смешан с девулканизованным ультразвуком GRT
и ПЭВД в смесителе типа Брабендер и в двухшнековом экструдере [152].
Эти смеси были подвергнуты динамической вулканизации. Смешанные
ранее ПЭВД и GRT проходили через зону ультразвуковой девулканизации
экструдера и затем подвергались динамической вулканизации. Смеси, полу-
ченные в двухшнековом экструдере, имеют лучшее относительное удлинение
и прочность, чем любые другие смеси. Реологические свойства этих смесей
были также изучены.
Новые смеси GRT и вторично переработанного ПЭВД от использованных
контейнеров для молока были изучены и запатентованы в [173,174]; иссле-
довано влияние размера частиц GRT и их концентрации на механические
и реологические свойства.
Д. Модификация цемента вторичной резиной
Производство наполненных каучуком цементов — область,
в которой возможно дальнейшее расширение использования GRT [175-182].
Преимущества использования GRT в цементе - увеличение сопротивления
образованию трещин при замораживании-оттаивании, ударостойкости, ки-
IV. Использование продуктов преработки -'V 709
слотостойкости, понижении© теплопроводности. Однако, введение резиновых
частиц в бетон снижает его сопротивление сжатию и изгибу. Относительно
влияния размера резиновых частиц на сопротивление сжатию, имеются
противоречивые результаты. Согласно Топку [183] и Элдин и др. [184], со-
противление сжатию бетона снижается после введения грубых резиновых
частиц, в то время как введение тонких частиц увеличивает сопротивление
сжатию. Напротив, испытания в [185] показали противоположные результа-
ты. Это противоречие можно объяснять различием в типе резины, геометрии
частиц и способе, которым частицы были получены.
Влияние формы резиновых частиц на механические свойства, работоспо-
собность и химическую стабильность наполненного резиной цемента было
изучено в [175]. Образец, содержащий резиновые крошки, был способен
остановить трещину и предотвратить катастрофическое разрушение, в то
время как образец, содержащий гранулированные резиновые частицы был
неспособен остановить трещину. Испытания показали слабую связь между
гранулированными частицами и матрицей. Фактически, многими исследо-
ваниями в этой области было показано, что связь между частицами резины
и цементом слаба. Были сделаны попытки улучшить связь с резиновыми
частицами [176, 180, 181]. Некоторое улучшение было достигнуто добав-
кой воды [180], воды с четыреххлористым углеродом и латексной смесью
[186], что привело к увеличению адгезии в наполненном крошкой цементе.
Несколько поверхностных модификаций также были предложены, включая
обработку серной и азотной кислотой, чтобы химически окислить крошку
и внедрить полярные группы. Вопреки ожиданию, модификация азотной
кислотой привела к уменьшению адгезиии [181]. С другой стороны, обра-
ботка серной кислотой улучшила адгезию крошки к бетону [187]. Показано,
что, используя комбинацию химических и поверхностных методов моди-
фикации, гидрофильность резиновой поверхности может быть улучшена
с помощью обработки кислотой [188,189], вследствие того, что резина обыч-
но содержит стеарат цинка, который мигрируя на поверхность обеспечивает
гидрофобность. Кислотной обработкой стеарат цинка может гидролизовать-
ся с образованием стеариновой кислоты. Цинковые ионы преобразуются
в ионы натрия путем обработки NaOH. Эти преобразования создают раство-
римый стеарат натрия. Было найдено [190], что введение резиновых частиц
вело к уменьшению сопротивления сжатию и изгибу из-за их вытеснения.
Однако, обработка резиновых частиц перед смешиванием бифункциональ-
ным сочетающим агентом типа гамматриметоксимеркаптосилана, улучшает
связь и ведет к увеличению податливости [190]. Было предложено расширить
это исследование, чтобы найти агенты сцепления для адгезии и улучшения
наполненного резиной цемента и бетона. Эти материалы могут использовать-
ся в перспективных программах строительства шоссе, тротуаров, звуковых
барьеров и другом транспортном строительстве.
710 -'V Глава 15. Вторичная переработка резины
Е. Асфальт, модифицированный переработанной
резиной
Асфальт может быть смешан с шинной крошкой, чтобы изме-
нить его свойства. Это остается самым большим рынком для вторичной ре-
зины. В 2001 г. 200 тыс. тн. или приблизительно 12 миллионов шин, исполь-
зовались в Соединенных Штатах для этих целей [3]. Главным образом, они
были использованы в Калифорнии, Аризоне, и Флориде. Другие государства
также начинают признавать выгоды модифицированного асфальта [3].
Сообщалось, что промышленность асфальта может поглощать до 40% шиных
отходов [191].
Использование крошки в асфальте имеет преимущества в прцессе
строительства дорог и по долговечности их эксплуатации. Они включают
увеличенную деформативность, сопротивление заносу и снижение шумо-
образования. Недостатки модифицированного крошкой асфальта — его
стоимость и возможность вредных выделений в воздух. Утверждают, что
асфальт с резиновой крошкой увеличивает стоимость дорожного стро-
ительства примерно до 50% по сравнению с обычным асфальтом [192].
Требование дополнительного горячего смешивания в процессе обработки
резинового асфальта может сопровождаться выделением вредных веществ
в воздух.
Два процесса используются в подготовке резиносодержащего асфальта:
сухой и мокрый. В обоих процессах прменяют частицы GRT размером от
6.35 мм до 40 меш. В мокром процессе битум смешивается с GRT и затем
добавляется в горячую смесь. В сухом процессе, GRT смешивается с напол-
нителем и затем вводится в асфальт.
Асфальт, смешанный с GRT использовался в течение весьма продол-
жительного времени. В зависимости от типа шины, состав GRT может
включать различную резину. Эта сшитая резина в основном не растворяется
в асфальте. Смеси показывают улучшение основных свойств асфальта за
счет свойственных резине характеристик и рассматриваются, как дисперсия
нерастворенных набухших резиновых частиц, действующих как эластичная
составляющая.
Смешивание GRT с асфальтом началось в 1960-х Макдональдом, за-
патентовавшим материал, который состоял из 25% отходов и асфальта,
смешанного при 190°С в течение 20 минут [193]. Макдональд продолжил
эту работу, применительно к реальным дорожным испытаниям и в 1968,
фирма Sahmaro Petroleum and Asphalt осуществили применение смеси GRT
и асфальта. Смесь, состоящая из гравия, минерального наполнителя, битума
и резиновой крошки, изготавливается в смесителе барабанного типа. Этот
материал применяется для верхнего слоя дорог. Распределенный по по-
верхности дороги материал уплотняется катками [194]. После этого было
предложено много разновидностей этого основного процесса горячего сме-
IV. Использование продуктов преработки -*v- 711
шения GRT и асфальта; в большинстве случаев GRT заменяет минеральный
наполнитель. Однако, этот метод существенно не изменяет свойства асфальта.
Таким образом, процесс смешения GRT и битума до изготовления основ-
ной смеси наиболее эффективен в улучшении свойств покрытий. Обычно
используется от 15 до 30% резины [195].
Ограниченный объем работы был сделан по оценке характеристик смесей
GRT и асфальта. Смеси, как правило, готовятся при температурах 150-205°С
в течение от 0.5 до 2 часа. Смесь становится вязкой и имеет консистенцию
суспензии с диспергировнными резиновыми частицами. При комнатной
температуре конечный состав — жесткий эластичный резиноподобный
материал. Этот период часто упоминается как время вываривания или
реакции. Было предположено в [195], что упругое качество смеси вызвано
механическим действием нерастворенных резиновых частиц, наполняющих
асфальт, который сам изменен частью резиновых частиц, которые распались.
Согласно Робертсу и Литтону [193] при смешении в течение 2 часов, в от-
личие от смешения в течении 0.5 и 1 часа, значительно улучшается упругое
восстановление материала. При увеличении времени смешения уменьша-
ется количество твердой резины в смеси и увеличивается доля распавшейся
резины [196]. Это является очень интересным предметом для дальнейшего
исследования: может ли увеличение растворимых асфальтом фракций
резины, которое происходит при более длительных временах смешивания
быть причиной повышения упругих свойств. Введение GRT в количестве
10, 20, и 30 вес. % значительно увеличило температуру размягчения
и восстанавливаемость асфальта с увеличением вязкости для всех трех
смесей [197].
Влияние состава асфальта и температуры на реологические свойства были
изучены в [198]. Содержание резиновой крошки, размер частиц и состав
асфальта являются главными факторами, воздействующими на реологию
резинового асфальта. Управляя этими переменными, покрытия с улучшен-
ным сопротивлением разрушению и разнашиванию могут быть произведены.
Наконец, отходы шин, используемые как модификатор асфальта, улучшают
качество покрытия и безопасность, являясь превосходным и рентабельным
модификатором при шоссейном строительстве [199].
Ж. Использование резиновой крошки в почве
Запатентован метод улучшения почвы с использованием
шиной крошки позволяющий уменьшать отрицательные воздействия,
связанные с ее уплотнением [200]. Упругость торфа — не прямой фактор
упругого характера крошки, а скорее результат увеличения его проницаемос-
ти [201]. Поверхностные характеристики твердости были оценены в [202].
Крошка значительно уменьшила твердость почвы, улучшила ее структуру
и водосодержание [203].
712 “* V Глава 15. Вторичная переработка резины
3. Изделия, сделанные из переработанной резины
Составы, содержащие девулканизат или крошку, могут
использоваться различными изготовителями, чтобы делать разнообразные
резиновые изделия. В частности эти составы могут использоваться или уже
используются для производства подошв обуви, труб, транспортерных лент,
технических резиновых лепных украшений, автомобильных ковриков,
брызговиков, полов в стойлах для домашнего скота, детских площадок,
железнодорожных переездов, покрытий для ступеней подъездов домов раз-
личных профилей, аккумуляторных батарей, и других формовых резиновых
изделий. С тех пор как перерабатывают наполненный техническим углеродом
каучук, изделия из продуктов его переработки имеют черный цвет и не могут
использоваться для изготовления цветных изделий, если дополнительные
меры по изменению цвета не были приняты. Очевидно, для каждого такого
использования крошка и девулканизат должны подвергнуться обширному
испытанию.
V. ПИРОЛИЗ И СЖИГАНИЕ РЕЗИН
А. Получение углеводородов и технического
углерода
Еще один метод, подходящий для использования старых
шин — пиролиз [204, 205]. Пиролиз — термическое разложение резины
в отсутствии воздуха и кислорода, для того чтобы произвести нефть (мас-
ла) и газы для повторного использования нефтехимическими отраслями
промышленности. Техуглерод и другие твердые продукты, остающееся
после пиролиза могут использоваться как наполнители. Пиролиз обыч-
но выполняется в котлах, автоклавах, ротационных печах, и в аппа-
ратах с псевдокипящим слоем. Гидрогенизация может также быть
выполнена в специальном реакторе [206]. Широкие исследования
в области пиролиза шинной резины с получение жидких углеводородов
и технического углерода были проведены в 1960-х и 1970-х годах и приве-
ли к строительству завода для пиролиза шинных отходов в 1970-х [207].
С тех пор, существенные исследования были выполнены по развитию
пиролиза [208] и использованию нефти (масла) и технического углеро-
да [209, 210]. Однако, эти попытки, оказались в то время экономически
невыгодными из-за низкой цены сырой нефти. Кроме того, заводы по
пиролизу, как полагают, производят токсические выбросы как побочный
продукт процесса [211].
В течение последних 20 лет, были выполнены многочисленные исследо-
вания и различные процессы пиролиза были опробованы [212-214]. Однако,
несмотря на это прогресс в области пиролиза шинных отходов был весьма
незначительным, главным образом из-за отсутствия широкого рынка для
VI. Заключение J\- 713
вторичных нефти (масла) и технического углерода полученных посредством
процесса пиролиза.
Б. Использование шин в качестве топлива
Шинная резина может быть преобразована в энергию методом сжигания.
Этот процесс реализован множеством резиновых компаний. Используются
резиновые чипсы различного размера и формы, содержащие или не содер-
жащие металлический корд. Оптимальный размер чипсов — приблизительно
5 х 10см. Чем больше размер, тем больше содержание металла и тем менее
вероятно, что эти шины могут использоваться как сырье. Старые шины ис-
пользуются как топливное дополнение для угля или газа в печах для произ-
водства портландцемента, извести, и стали. Это уменьшает на 25% количество
уголя потребляемого отраслями промышленности цемента. Старые шины
могут также быть сожжены, чтобы произвести электричество. Полученное из
шин топливо может уменьшать эмиссию серы на электростанциях и может
улучшать эффективность сгорания, регулируя надлежащую стехиометрию
в комбинации с различными углями, деревянными отходами, и домашним
мусором. Для получения топлива в Соединенных Штатах в 2001 было ис-
пользовано 115 миллионов старых шин [3]. Однако, при сжигании резины
в качестве топлива, ценные резиновые материалы теряются. Фактически для
изготовления 1 фунта синтетического каучука требуется 60000 квтч энергии
[215]. Напротив, калорийность шин при сжигании составляет от 13000 до
16000 квтч, т. е. не намного выше, чем калорийность горения угля, который
намного более дешев. Кроме того, сжигание шин для получения энергии
приводит к загрязнению воздуха [216].
VI. ЗАКЛЮЧЕНИЕ
Старые шины и резинотехнические изделия представляют
проблему международного значения. Данная работа описывает некоторые
направленя, доступные для её решения. Развиваются многочисленные техно-
логии. Среди них, в дополнение к известным методам размола, появляются
непрерывные методы измельчения в одно и двухчервячных экструдерах,
которые могут служить как возможные варианты получения резинового
порошка в качестве сырья для современных и будущих технологий. Они
включают девулканизацию, поверхностную обработку, ультразвуковую
девулканизацию и использование резиновой крошки для создания компо-
зитов с другими материалами. Некоторый успех был достигнут в развитии
ультразвуковой технологии, которая рассматривается как один из многообе-
щающих методов, делая девулканизат, подходящим для создания резиновых
изделий как из 100% переработанной резины, так и в качестве добавки
к каучуку, исходным и переработанным пластмассам, асфальту, бетону,
714 Д.
Глава 15. Вторичная переработка резины
и цементу. Должно быть усовершенствовано оборудование, что является
необходимым для создания высокотехнологичной продукции. Это сделает
процесс экономически целесообразным. Главное требование для промыш-
ленной реализации этого процесса — создание мощного ультразвукового
генератора, способного к действию в непрерывно режиме под высоким дав-
лением и при высокой температуре. Ясно, что имеется недостаток научного
понимания различных процессов, управляющих рециркуляцией резины.
Развитие научных технологий и процессов для рециркуляции резины
и использование переработанной резины в различных конечных продуктах,
значительно уменьшило бы всемирное потребление энергии, обеспечило
возобновление каучука из резиновых отходов и уменьшило загрязнение
окружающей среды.
ЛИТЕРАТУРА
Глава 1
1. P.J. Flory, «Statistical Mechanics of Chain Molecules» Wiley-Interscience, New York, 1969.
2. J.P. Cotton, B. Farnoux, and G.J. Jannink, J. Chem. Phys. 57, 290 (1972).
3. M.C. Morris, J. Appl. Polm. Sci. 8, 545 (1964).
4. F. Bueche, J. Appl. Polym. Sci. 4, 107 (1960); 5, 271 (1961).
5. A.V. Tobolsky and H.F. Mark (Eds.), «Polymer Science and Materials» Wiley, New York,
1971, Chap. 13.
6. L.J. Fetters, DJ. Lohse, and W.W. Graessley, J. Polym. Sci.: Part B: Polym. Phys. 37,1023 (1999).
7. N.R. Langley, Macromolecules 1, 348 (1968).
8. T.A. Vilgis, in «Elastomeric Polymer Networks» J.E. Mark and B. Erman (Eds.),
Prentice-Hall, Englewood Cliffs, NJ, 1992, Chap. 5.
9. W.W. Graessl ey, «Polymeric Liquids and Networks: Structure and Properties» Taylor and
Francis Books, New York, 2004.
10. S. Prager and H.L. Frisch, J. Chem. Phys. 46, 1475 (1967).
11. L. R.G. Treloar, «The Physics of Rubberlike Elasticity» 3rd ed., Clarendon Press, Oxford, 1975.
12. E.M. Arruda and M.C. Boyce, J. Meeh. Phys. Solids 41, 389 (1993).
13. A.G. Thomas, Trans. Faraday Soc. 51, 569 (1955).
14. H.M. James and E. Guth,/. Chem. Phys. 11, 455 (1943);/. Polym. Sci. 153 (1949).
15. R.S. Rivlin, Philos. Trans. Roy. Soc. (London) Ser. A 241,379 (1948).
16. R.S. Rivlin, in «Rheo/ogy, Theory and Application» F.R. Eirich (Ed.), Academic Press, New
York, Vol. 1,1956, Chap. 10.
17. M. Mooney, /. Appl. Phys. 11, 582 (1940).
18. R.S. Rivlin and D.W. Saunders, Philos. Trans. Roy. Soc. (London) Ser. A 243, 251 (1951).
19. A.N. Gent and A.G. Thomas, /. Polym. Sci. 28, 625 (1958).
20. A.N. Gent, Rubber Chem. Technol. 69, 59 (1996).
21. E. Pucci and G. Saccomandi, Rubber Chem. Technol. 75, 839 (2002).
22. R.W. Ogden, «Non-Linear Elastic Deformations» Ellis Harwood, Chichester, UK, 1984; Dover
Publications, Mineola, NY, 1997, Chap. 7.
23. A.N. Gent and P.B. Lindley, Proc. Roy. Soc. (London) A 249, 195 (1958).
24. A.N. Gent and D.A. Tompkins, /. Appl. Phys. 40, 2520 (1969).
25. M. Biot, «Mechanics of Incremental Deformations» Wiley, New York, 1965.
26. M.F. Beatty, in «Finite Elasticity» R.S. Rivlin (Ed.), AMD Vol. 27, American Society of
Mechanical Engineers, New York, 1977, p. 125.
27. A.N. Gent and I.S. Cho, Rubber Chem. Technol. 72, 253 (1999).
28. A. Schallamach, Wear 17, 301 (1971).
29. A.E. Green and W. Zerna, «Theoretical Elasticity» 2nd ed., Clarendon Press, Oxford, 1975,
Section 4.6, p. 135.
716 -’v- Литература
30. A.N. Gent and K.C. Hua, Int. J. Non-Linear Meeh. 39, 483 (2004).
31. R.S. Rivlin, J. Appl. Phys. 18, 444 (1947).
32. A.L. Soden, «А Practical Manual of Rubber Hardness Testing» Maclaren, London, 1952.
33. A.N. Gent, Rubber Chem. Technol. 67, 549 (1994).
34. J.M. Horton, G.E. Tupholme, and M.J.C. Gover, ASME, J. Appl. Meeh. 69, 836 (2002).
35. J.E. Mark and B. Erman, «Rubberlike Elasticity: A Molecular Primer» John Wiley & Sons,
New York, 1988.
36. J.E. Mark and B. Erman (Eds.), «Elastomeric Polymer Networks» Prentice-Hall, Englewood
Cliffs, NJ, 1992.
37. A.N. Gent, J. Polym. Sci. Poly. Symp. 28, 625 (1958).
Глава 2
1. G. Williams, Proc. R. Soc. London 10, 516 (1860).
2. G. Bouchardat, C. R.H.Acad. Sci. 89,1117 (1879).
3. W.A. Tilden,/. Chem. Soc. 45, 411 (1884).
4. I. Kondakow, J. Prakt. Chem. 62, 66 (1900).
5. J. Thiele, Justus Liebig's Ann. Chem. 319, 226 (1901).
6. S.V. Lebedev, Zh. Russ. Fiz.-Kim. Ova. Chast. 42, 949 (1910).
7. C.D. Harries, Justus Liebig's Ann. Chem. 383, 190 (1911).
8. F.E. Matthews and E.H. Strange, British Patent 24,790 (1910).
9. H. Staudinger, Chem. Ber. 53, 1073 (1920).
10. W.H. Carothers, J. Williams, A.M. Collins, and J.E. Kirby, J. Am. Chem. Soc. 53, 4203 (1931).
11. P.J. Flory, «Principles of Polymer Chemistry» Cornell Univ. Press, Ithaca, NY, 1953, Chap. 1.
12. H. Morawetz, «Polymers: The Origins and Growth of a Science» Wiley, New York, 1985.
13. M. Morton, Rubber Plast. Age 42, 397 (1961).
14. W.H. Carothers, J. Am. Chem. Soc. 51, 2548 (1929).
15. W.H. Carothers, Chem. Rev. 8, 353 (1931).
16. D. Braun, H. Cherdron, and W. Kern, «Practical Macromolecular Organic Chemistry»
Harwood Academic Publishers, New York, 1984, p. 255.
17. S.R. Sandler and W. Karo, «Polymer Syntheses» 2nd ed., Academic Press, San Diego, 1992, p. 127.
18. G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), «Comprehensive Polymer
Science» Vol. 5: «Step Polymerization» Pergamon Press, Oxford, 1989.
19. J.A. Moore (Ed.), «Macromolecular Synthesis, Collective Volume 1» Wiley, New York, 1978.
20. J.H. Saunders and F. Dobinson, in «Comprehensive Chemical Kinetics» C.H. Bamford and C.
F.H. Tipper (Eds.), Elsevier, New York, Vol. 15, 1976, Chap. 7.
21. G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), «Comprehensive Polymer
Science» Vol. 3: «Chain Polymerization I;» Vol. 4: «Chain Polymerization II» Pergamon Press,
Oxford, 1989.
22. G. Odian, «Principles of Polymerization» 4th ed. Wiley-Interscience, New York, 2004.
23. S.A. Penczek, J. Polym. Sci.: Part A: Polym. Chem. 40, 1665 (2002).
24. A.D. Jenkins, P. Kratochvil, R. F.T. Stepto, and U.W. Suter, PureAppl. Chem. 68,2287 (1996).
25. D.H. Solomon (Ed.), «Step-Growth Polymerizations» Dekker, New York, 1972.
26. G.C. Eastmond, in «Comprehensive Chemical Kinetics» Vol. 14A: «Free Radical
Polymerization» C.H. Bamford and C.F.H. Tipper (Eds.), Elsevier, New York, 1976, p. 1.
27. R.G. Gilbert, Pure Appl. Chem. 64, 1563 (1992).
28. R.G. Gilbert, PureAppl. Chem. 68, 1491 (1996).
29. M. Buback, R.G. Gilbert, R.A. Hutchinson, B. Klumperman, F.D. Kuchta, B.G. Manders, K.
F. O’Driscoll, G.T. Russell, and J. Schweer, J. Macromol. Chem. Phys. 196, 3267 (1995).
30. M.S. Matheson, E.E. Auer, E.B. Bevilacqua, and E.J. Hart, J. Am. Chem. Soc. 71, 497, 2610
(1949); ibid., 73, 1700, 5395 (1951).
31. M. Morton, P. Salatiello, and H. Landfield, J. Polym. Sci. 8, 215 (1952).
32. M. Morton and S.D. Gadkary, 130th Meet., Am. Chem. Soc., Atlantic City, N. J., 1956;
S.D. Gadkary, M.S. Thesis, Univ, of Akron, Akron, Ohio, 1956.
33. S. Beuermann, M. Buback, T.P. Davis, R.G. Gilbert, R.A. Hutchinson, O.F. Olaj, G.T. Russell,
J. Schweer, and A.M. van Herk, Macromol. Chem. Phys. 198, 1545 (1997).
Список литературы к главе 2 -*Ъ- 717
34. Р.А. Weerts, A.L. German, and R.G. Gilbert, Macromolecules 24, 1622 (1991).
35. R.A. Hutchinson, M.T. Aronson, and J.R. Richards, Macromolecules 26, 6410 (1993).
36. G.V. Schulz, Z. Phys. Chem., Abt., В 43, 25 (1939).
37. G.S. Whitby and R.N. Crozier, Can. J. Res. 6, 203 (1932).
38. D.II. Solomon, E. Rizzardo, and P. Cacioli, U.S. Patent 4,581,429 (1986).
39. M.K. Georges, R. P.N. Veregin, P.M. Kazmeier, and G.K Hamer, Macromolecules 29,5245 (1993).
40. C.J. Hawker, J. Am. Chem. Soc. 116, 11185 (1994).
41. T. Fukuda, T. Terauchi, A. Goto, Y. Tsuhii, T. Miyamoto, and Y. Shimizu, Macromolecules
29, 3050(1996).
42. C.J. Hawker, A.W. Bosman, and E. Harth, Chem. Rev. 101, 3661 (2001).
43. T. Fukuda and A. Goto, in «Controlled/Living Radical Polymerization» K. Matyjaszewski
(Ed.), ACS Symposium Series 768, American Chemical Society, Washington, D. C., 2000, p. 27.
44. B. Keoshkerian, M. Georges, M. Quinlan, R. Veregin, and B. Goodbrand, Macromolecules 31,
7559 (1998).
45. H. Fischer, Macromolecules 30, 5666 (1997).
46. H. Fischer, J. Polym. Sci., Part A: Polym. Chem. 37, 1885 (1999).
47. K. Matyjaszewski (Ed.), «Controlled/Living Radical Polymerization» ACS Symposium Series
768, American Chemical Society, Washington, D. C., 2000.
48. K. Matyjaszewski (Ed.), «Controlled/Living Radical Polymerization» ACS Symposium Series
685, American Chemical Society, Washington, D. C., 1998.
49. M.K. Georges, G.K. Hamer, and N.A. Listigovers, Macromolecules 31, 9087 (1998).
50. D. Benoit, E. Harth, P. Fox, R.M. Waymouth, and CJ. Hawker, Macromolecules 33,363 (2000).
51. K. Matyjaszewski and J. Xia, Chem. Rev. 101, 2921 (2001).
52. G.W. Poehlein, in «Encyclopedia of Polymer Science and Engineering» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 6, 1986, p. 1.
53. I. Piirma (Ed.), «Emulsion Polymerization» Academic Press, New York, 1982.
54. D.C. Blackley, «Emulsion Polymerization» Applied Science Publishers, London, 1975.
55. D.H. Napper and R.G. Gilbert, in «Comprehensive Polymer Science» G.C. Eastmond,
A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon, Oxford, Vol. 4, 1989, p. 171.
56. R.G. Gilbert, «Emulsion Polymerization: A Mechanistic Approach» Academic Press, San
Diego, 1995.
57. K. Tauer, in «Encyclopedia of Polymer Science and Techno/ogy» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 6, 2003, p. 410.
58. P.A. Lovell and M.S. El-Aasser (Eds.), «Emulsion Polymerization and Emulsion Polymers»
Wiley, New York, 1997.
59. W.D. Harkins, J. Am. Chem. Soc. 69, 1428 (1947).
60. J.L. Gardon, in «Polymerization Processes» C.E. Schildknecht (Ed.), Wiley-Interscience,
New York, 1977, Chap. 6.
61. M. Morton, S. Kaizerman, and M.W. Altier, J. Colloid Sci. 9, 300 (1954).
62. W.V. Smith and R.H. Ewart, J. Chem. Phys. 16, 592 (1948).
63. M. Morton and W.E. Gibbs, J. Polym. Sci., Part A 1, 2679 (1963).
64. W. Hofmann, «Rubber Techno/ogy Handbook» Hanser Publishers, Munich, West Germany,
1989.
65. C.M. Blow (Ed.), «Rubber Techno/ogy and Manufacture» Hewnes-Butterworths, London,
1971.
66. J.A. Brydson, in «Developments in Rubber Techno/ogy-2» A. Whelan and K.S. Lee (Eds.),
Applied Science Publishers, London, 1981, p. 21.
67. R.G. Bauer, in «Kirk-Othmer Encyclopedia of Chemical Techno/ogy» 3rd ed., Wiley, New
York, Vol. 8,1979, p. 611.
68. H.N. Sun and J.P. Wusters, in «Kirk-Othmer Encyclopedia of Chemical Techno/ogy» 4th ed.,
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 4, 2004, p. 365.
69. M. Demirors, in «Encyclopedia of Polymer Science and Techno/ogy» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 4, 2003, p. 229.
70. M. Morton and P.P. Salatiello, J. Polym. Sci. 6, 225 (1951).
71. J.L. Binder, Ind. Eng. Chem.. 46, 1727 (1954).
718 -*\г Литература
72. К.Е. Beu, W.B. Reynolds, C.F. Fryling, and H.L. McMurry, J. Polym. Sci. 3, 465 (1948).
73. A.W. Meyer, Ind. Eng. Chem. 41, 1570 (1949).
74. P.B. Johnson, Rubber Chem. Tech. 49, 650 (1976).
75. C.A. Stewart, Jr., T. Takeshita, and M.L. Coleman, in «Encyclopedia of Polymer Science and
Engineering» J.I. Kroschwitz (Ed.), Wiley, Vol. 3, 1985, p. 441.
76. D.C. Blackley, «Synthetic Rubbers: Their Chemistry and TechnoZogy» Applied Science
Publishers, Essex, 1983, p. 175.
77. R. Musch and H. Magg, in «Polymeric Materials Encyclopedia» J.C. Salamone (Ed.), CRC
Press, Boca Raton, FL, Vol. 2,1996, p. 1238.
78. D.I. Christie, R.G. Gilbert, J.P. Congalidis, J.R. Richards, and J.H. McMinn, Macromolecules
34,5158(2001).
79. M. Morton, J.A. Cala, and M.W. Altier, J. Polym. Sci. 19, 547 (1956).
80. M. Morton and I. Piirma, J. Polym. Sci. 19, 563 (1956).
81. AM. Neal and LR. Mayo, in «Synthetic Rubber» G.S. Whitby (Ed.), Wiley, New York, 1954, p. 770.
82. W.E. Mochel and J.H. Peterson, J. Am. Chem. Soc. 71, 1426 (1949).
83. C.A. Hargreaves, in «Polymer Chemistry of Synthetic Elastomers» J.P. Kennedy,
E. Tornqvist (Eds.), Wiley-Interscience, New York, 1968, p. 233.
84. M.M. Coleman and E.G. Brame, Jr., Rubber Chem. Tech. 51, 668 (1978).
85. M.M. Coleman, D.L. Tabb, and E.G. Brame, Jr., Rubber Chem. Tech. 50, 49 (1977).
86. J.R. Ebdon, Polymer 19, 1232 (1978).
87. R. Petiaud and Q.T. Pham, J. Polym. Sci., Polym. Chem. Ed. 23, 1333 (1985).
88. R.R. Garrett, C.A. Hargreaves, and D.N. Robinson, J. Macromol. Sci. Chem. A 4,1679 (1970).
89. A.E. Hamielec, J.F. MacGregor, and A. Penlidis, in «Comprehensive Polymer Science»
G.C. Eastmond, A. Ledwith, S. Russo, P. Sigwait (Eds.), Pergamon, Oxford, Vol. 3,1989, p. 17.
90. G. Odian, «Principles of Polymerization» 4th ed., Wiley-Interscience, New York, 2004, Chap. 6.
91. G.E. Ham (Ed.), «Copolymerization» Wiley-Intersceince, New York, 1964.
92. D.A. Tirrell, in «Encyclopedia of Polymer Science and Engineering» 2nd ed., J.I. Kroschwitz
(Ed.), Wiley, New York, Vol. 4, 1986, p. 192.
93. D.A. Tirrell, in «Comprehensive Polymer Science» G.C. Eastmond, A. Ledwith, S. Russo, and
P. Sigwait (Eds.), Pergamon, Oxford, Vol. 3, 1989, p. 195.
94. F.R. Mayo and C. Walling, Chem. Rev. 46, 191 (1950).
95. F.R. Mayo and F.M. Lewis, J. Am. Chem. Soc. 66, 1594 (1944).
96. R.Z. Grccnley, in «Polymer Handbook» 4th ed., J. Brandrup and E.H. Immergut (Eds.),
Wiley-Interscience, New York, 1999, H-181.
97. G.C. Eastman and E.G. Smith, in «Comprehensive Chemical Kinetics» C.H. Bamford and
C. F.H. Tipper (Eds.), Elsevier, New York, Vol. 14A, 1976, p. 333.
98. C.A. Uraneck, in «Polymer Chemistry of Synthetic Elastomers» Part I, J.P. Kennedy and
E. Tornqvist (Eds.), Wiley-Interscience, New York, 1968, p. 158.
99. C.F. Fryling, in «Synthetic Rubber» G.S. Whitby (Ed.), Wiley, New York, 1954, p. 257.
100. Y. Tanaka, H. Sato, and J. Adachi, Rubber Chem. Tech. 59, 16 (1986).
101. Y. Tanaka, H. Sato, Y. Nakafutami, and Y. Kashiwazaki, Macromolecules 16, 1925 (1983).
102. W. Hofmann, Rubber Chem. Tech. 37, 1 (1964).
103. R. Greenley, J. Macromol. Sci. Chem. 14, 445 (1980).
104. J.P. Kennedy and B. Ivan, «Designed Polymers by Carbocationic Macromolecular
Engineering: Theory and Practice» Hanser Publishers, Munich, 1992.
105. J.P. Kennedy and E. Marechai, «Carbocationic Polymerization» Wiley-Interscience, New
York, 1982.
106. J.P. Kennedy, «Cationic Polymerization of Olefins: A Critical Inventory» Wiley-Interscience,
New York, 1975.
107. M. Sawamoto and T. Higashimura, in «Encyclopedia of Polymer Science and Engineering»
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, 1989, Supplemental Volume, p. 399.
108. G. Odian, «Principles of Polymerization» 4th ed., Wiley-Interscience, New York, 2004,
Chap. 5.
109. A. Gandini and H. Cheradame, in «Encyclopedia of Polymer Science and Engineering»
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 2, 1985, p. 729.
Список литературы к главе 2 -* V 719
110. К. Matyjaszewski (Ed.), «Cationic Polymerizations: Mechanisms, Synthesis, and Applications»
Marcel Dekker, New York, 1996.
111. J.E. Puskas and G. Kaszas, in «Encyclopedia of Polymer Science and Techno/ogy»
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 5, 2003, p. 382.
112. S. Winstein, E. Clippinger, A.H. Fainberg, and G.C. Robinson, J. Am. Chem. Soc. 76, 2597
(1954).
113. R. G.W. Norrish and K.E. Russell, Trans. Faraday Soc. 48, 91 (1952).
114. A.G. Evans and M. Polanyi, J. Chem. Soc. 252 (1947).
115. E.N. Kresge, R.H. Schatz, and H.C. Wang, in «Encyclopedia of Polymer Science and
Engineering» J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 8, 1987, p. 423.
116. R.N. Webb, T.D. Shaffer, and A.H.Tsou, in «Encyclopedia of Polymer Science and
Techno/ogy» J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 5, 2003, p. 356.
117. D.C. Pepper, Sci. Proc. R. Dublin Soc. 25, 131 (1950).
118. F.S. Dainton and G.B. B.M. Sutherland,/. Polym. Soc. 4, 347 (1949).
119. I. Puskas, E.M. Banas, and A.G. Nerheim, J. Polym. Sci., Symp. 56, 191 (1976).
120. D.C. Pepper, Trans. Faraday Soc. 45, 404 (1949).
121. D.D. Eley and A.W. Richards, Trans. Faraday Soc. 45, 425, 436 (1949).
122. P.H. Plesch, J. Chem. Soc. 543 (1950).
123. L. Sipos, P. De, and R. Faust, Macromolecules 36, 8282 (2003).
124. J.P. Kennedy, in «Polymer Chemistry of Synthetic Elastomers» J.P. Kennedy and E.
Tornqvist (Eds.), Wiley-Interscience, New York, 1968, Part I, p. 291.
125. H.Y. Che and J.E. Field, J. Polym. Sci., Part В 5, 501 (1957).
126. M. Miyamoto, M. Sawamoto, and T. Higashimura, Macromolecules 17, 265 (1984).
127. R. Faust and J.P. Kennedy, Polym. Bull. (Berlin) 15, 317 (1986).
128. S. Hadjikyriacou, M. Acar, and R. Faust, Macromolecules 37, (2004) in press.
129. K. Matyjaszewski (Ed.), «Cationic Polymerizations: Mechanisms, Synthesis, and Applications»
Dekker, New York, 1996.
130. R.W. Body and V.L. Kyllingstad, in «Encyclopedia of Polymer Science and Engineering»
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 6, 1986, p. 307.
131. E.J. Vandenberg, J. Polym. Sci. 7, 525 (1969).
132. E.J. Vandenberg, in «Coordination Polymerization» C.C. Price and E.J. Vandenberg (Eds.),
Plenum Press, New York, 1983, p. 11.
133. W. Meckel, W. Goyert, W. Wieder, and H.G. Wussow, in «Thermoplastic Elastomers» 3rd ed.,
G. Holden, H.R. Kricheldorf, and R.P. Quirk (Eds.), Hanser Publishers, Munich, 2004, p. 15.
134. R.K. Adams, G.K. Hoeschele, and W.K. Witsiepe, in «Thermoplastic Elastomers» 3rd ed.,
G. Holden, H.R. Kricheldorf, and R.P. Quirk (Eds.), Hanser Publishers, Munich, 2004, p. 183.
135. S. Inoue and T. Aida, in «Ring-Opening Polymerization» K.J. Ivin and T. Saegusa (Eds.),
Elsevier, New York, Vol. 1, 1984, p. 185.
136. P. Dreyfuss, «Poly (tetrahydrofuran)» Gordon and Breach, New York, 1982.
137. S. Penczek, P. Kubisa, and K. Matyjaszewski, Adv. Polym. Sci. 68/69, 1 (1985).
138. S. Penczek and P. Kubisa, in «Comprehensive Polymer Science» G.C. Eastmond, A. Ledwith,
S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford, Vol. 3, 1989, p. 751.
139. P. Dreyfuss, M.P. Dreyfuss, and G. Pruckmayr, in «Encyclopedia of Polymer Science and
Engineering» J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 16, 1989, p. 649.
140. M.P. Dreyfuss and P. Dreyfuss, J. Polym. Sci. A-1 4, 2179 (1966).
141. G. Pruckmayr and T.K. Wu, Macromolecules 11,662 (1978).
142. K. Matyjaszewski, P. Kubisa, and S. Penzcek, J. Polym. Sci., Polym. Chem. Ed. 13, 763
(1975).
143. R.P. Quirk, in «Encyclopedia of Polymer Science and Techno/ogy» 3rd ed., J.I. Kroschwitz
(Ed.), Wiley-Interscience, New York, Vol. 5, 2003, p. 111.
144. M. Morton, «Anionic Polymerization: Principles and Practice» Academic Press, New York,
1982.
145. M. Szwarc, «Carbanions, Living Polymers and Electron Transfer Processes» Interscience,
New York, 1968.
146. P. Rempp, E. Franta, and J.E. Herz, Ado. Polym. Sci. 86, 145 (1988).
720 -*v Литература
147. R.N. Young, R.P. Quirk, and L.J. Fetters, Adi?. Polym. Sci. 56,1 (1984).
148. M. Szwarc, Adv. Polym. Sci. 49,1 (1983).
149. M. van Beylen, S. Bywater, G. Smets, M. Szwarc, and D.J. Worsfold, Adv. Polym. Sci. 86, 87
(1988).
150. H.L. Hsieh and R.P. Quirk, «Anionic Polymerization» Dekker, New York, 1996.
151. M. Szwarc, «Ionic Polymerization Fundamentals» Hanser, Cincinnati, 1996.
152. W. C.E. Higginson and N.W. Wooding, J. Chem. Soc. 760 (1952).
153. R.E. Robertson and L. Marion, Can. J. Res., Sect. B. 26, 657 (1948).
154. D.E. Paul, D. Lipkin, and S.I. Weissman, J. Am. Chem. Soc. 78, 116 (1956).
155. B.J. McClelland, Chem. Rev. 64, 301 (1964).
156. M.T. Jones, in «Radical Ions» E.T. Kaiser and L. Kevan (Eds.), Wiley, New York, 1968.
157. M. Szwarc and J. Jagur-Grodzinski, in «Ions and Ion Pairs in Organic Reactions» M. Szwarc
(Ed.), Wiley, 1974, p. 1.
158. N.L. Holy, Chem. Rev. 74, 243 (1974).
159. M. Szwarc, M. Levy, and R. Milkovich, J. Am. Chem. Soc. 78, 2656 (1956).
160. P.J. Flory, J. Am. Chem. Soc. 62,1561 (1940); see also Ref. 11, p. 336.
161. H.L. Hsieh, R.C. Farrar, and K. Udipi, СНЕМТЕСНЮ& (1981).
162. I.G. Hargis, R.A. Livigni, and S.L. Aggarwal, in «Developments in Rubber TechnoZogy-4»
A. Wheland and K.S. Lee (Eds.), Elsevier, Essex, UK, 1987, p. 1.
163. H.L. Hsieh and O.F. McKinney, J. Polym. Sci., Polym. Lett. 4, 843 (1966).
164. M. Morton, A. Rembaum, and J.L. Hall, J. Polym. Sci. A 1, 461 (1963).
165. R.P. Quirk, D.J. Kinning, and L.J. Fetters, in «Comprehensive Polymer Science» G. Allen
and J.C. Bevington (Eds.), Pergamon Press, Oxford, Vol. 7, 1989, p. 1.
166. N. Hadjichristidis, S. Pispas, and G.A. Floudas, «Block Copolymers: Synthetic Strategies,
Physical Properties, and Applications» Wiley-Interscience, New York, 2003.
167. R.P. Quirk, in «Comprehensive Polymer Science» First Supplement, S.L. Aggarwal and
S. Russo (Eds.), Pergamon Press, Oxford, 1992, p. 83.
168. H.L. Hsieh, Rubber Chem. Tech. 49,1305 (1976).
169. B.J. Bauer and L.J. Fetters, Rubber Chem. Tech. 51, 406 (1978).
170. S. Bywater, Adv. Polym. Sci. 30, 89 (1979).
171. N. Hadjichristidis, M. Pitsikalis, S. Pispas, and H. latrou, Chem. Rev. 101, 3747 (2001).
172. S. Bywater, in «Comprehensive Chemical Kinetics» C.H. Bamford and C.F.H. Tipper (Eds.),
Elsevier, New York, Vol. 15, 1976, p. 1.
173. A.H. M Iler, in «Comprehensive Polymer Science» Vol. 3: «Chain Polymerization I»
G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford, 1989,
p. 387.
174. S. Bywater, A.F. Johnson, and D.J. Worsfold, Can. J. Chem. 42, 1255 (1964).
175. M. Morton, in «Vinyl Polymerization» Part II, G. Ham (Ed.), Marcel Dekker, New York, 1969,
p. 211.
176. J.L. Wardell, in «Comprehensive Organometallic Chemistry: The Synthesis, Reactions and
Structures of Organometallic Compounds» G. Wilkinson, F. G.A. Stone, and E.W. Abel
(Eds.), Pergamon Press, Oxford, Vol. 1, p. 43.
177. T.L. Brown, Acc. Chem. Res. 1, 23 (1968).
178. B.L. Wakefield, «The Chemistry of Organolithium Compounds» Pergamon Press, Oxford,
1974.
179. D.J. Worsfold and S. Bywater, Can. J. Chem. 38, 1891 (1960).
180. S. Bywater and D.J. Worsfold, J. Organomet. Chem. 10, 1 (1967).
181. F. Schue and S. Bywater, Macromolecules 2, 458 (1969).
182. M. Morton, R.A. Pett, and L.J. Fetters, Macromolecules 3, 333 (1970).
183. C.M. Selman and H.L. Hsieh, J. Polym. Sci., Polym. Lett. Ed. 9, 219 (1971).
184. S. Bywater and D.J. Worsfold, in «Recent Advances in Anionic Polymerization» T. E.
Hogen-Esch and J. Smid (Eds.), Elsevier, New York, 1987, p. 109.
185. L.J. Fetters and M. Morton, Macromolecules 7, 552 (1974).
186. J.B. Smart, R. Hogan, P.A. Scherr, M.T. Emerson, and J.P. Oliver, J. Organomet. Chem. 64,
1 (1974).
Список литературы к главе 2 721
187. P.D. Bartlett, S.J. Tauber, and W.P. Weber, J. Am. Chem. Soc. 91, 6362 (1969).
188. P.D. Bartlett, C.V. Goebel, and W.P. Weber, J. Am. Chem. Soc. 91, 7425 (1969).
189. R.N. Young, L.J. Fetters, J.S. Huang, and R. Krishnamoorti, Polym. Int. 33, 217 (1994).
190. S. Bywater, Polym. Int. 38, 325 (1995).
191. L.J. Fetters, J.S. Huang, and R.N. Young, J. Polym. Sci.: Part A: Polym. Chem. Ed. 34, 1517
(1996).
192. S. Bywater, Macromol. Chem. Phys. 199, 1217 (1998).
193. J. Stellbrink, J. Allgaier, L. Willner, D. Richter, T. Slawecki, and L.J. Fetters, Polymer 43,
7101 (2002).
194. R.P. Quirk and B. Lee, Polym. Int. 27, 359 (1992).
195. M. Morton and L.J. Fetters, J. Polym. Sci., Macromol. Rev. 2, 71 (1967).
196. F.W. Stavely and coworkers, Ind. Eng. Chem. 48, 778 (1956).
197. L.E. Foreman, in «Polymer Chemistry of Synthetic Elastomers» Part IГ, J.P. Kennedy and
E. Tornqvist (Eds.), Wiley, New York, 1968, p. 491.
198. E.W. Duck and J.M. Locke, in «The Stereo Rubbers» W.M. Saltman (Ed.), Wiley-
Interscience, New York, 1977, p. 139.
199. S. Bywater, in «Comprehensive Polymer Science» Vol. 3: «Chain Polymerization I» G. C.
Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford, 1989, p.
433.
200. M. Morton and J.R. Rupert, in «Initiation of Polymerization» F.E. Bailey, Jr. (Ed.), ACS
Symposium Series 212, American Chemical Society, Washington, D. C., 1983, p. 283.
201. D.J. Worsfold and S. Bywater, Macromolecules 11, 582 (1978).
202. A.V. Tobolsky and C.E. Rogers, J. Polym. Sci. 40, 73 (1959).
203. A. Rembaum, et al., J. Polym. Sci. 61, 155 (1962).
204. S. Bywater and D.J. Worsfold, Can J. Chem. 45, 1821 (1967).
205. M. Morton and L.J. Fetters, J. Polym. Sci., Part A 2, 3311 (1964).
206. M. Morton, L.J. Fetters, R.A. Pett, and J.F. Meier, Macromolecules 3, 327 (1970).
207. E.R. Santee, Jr., L.O. Malotky, and M. Morton, Rubber Chem. Tech. 46, 1156 (1973).
208. T. Hogen-Esch, Adv. Phys. Org. Chem. 15,153 (1977).
209. A. Halasa, Rubber Chem. Tech. 54, 627 (1981).
210. N. Nagata, T. Kobatake, H. Watanabe, A. Ueda, and A. Yoshioka, Rubber Chem. Tech. 60,
837 (1987).
211. R. Luxton, Rubber Chem. Tech. 54, 596 (1981).
212. G. Holden and D.R. Hansen, in «Thermoplastic Elastomers» 3rd ed., G. Holden,
H.R. Kricheldorf, and R.P. Quirk (Eds.), Hanser Publishers, Munich, 2004, p. 45.
213. R. Ohlingerand F. Bandermann, Makromol. Chem. 181, 1935 (1980).
214. M. Morton and L.K. Huang, unpublished data; L.K. Huang, Ph. D. Dissertation, The
University of Akron, Akron, Ohio, 1979; cited in Ref. [144].
215. F.R. Ells, Ph. D. Dissertation, The University of Akron, Akron, Ohio, 1963; cited in
[144].
216. Y.L. Spirin, A.A. Arest-Yakubovich, D.K. Polyakov, A.R. Gantmakher, and S.S. Medvedev,
J. Polym. Sci. 58,1161 (1962).
217. D. J.T. Hill, J. H. O’Donnell, P. W. O’Sullivan, J.E. McGrath, I.C. Wang, and T.C. Ward,
Polym. Bull. 9, 292 (1983).
218. T.A. Antkowiak, A.E. Oberster, A.F. Halasa, and D.P. Tate, J. Polym. Sci., Part A-1 10,1319
(1972).
219. C.F. Wofford and H.L. Hsieh, J. Polym. Sci., Part A-1 7, 461 (1969).
220. M. Fontanille, in «Comprehensive Polymer Science» Vol. 3: «Chain Polymerization I»
G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford, 1989,
p. 376.
221. F. Bandermann, H.D. Speikamp, and L. Weigel, Makromol. Chem. 186, 2017 (1985).
222. L.H. Tung and G.Y. Lo, in «Advances in Elastomers and Rubber Elasticity» J. Lal and J.
E. Mark (Eds.), Plenum Press, New York, 1986, p. 129.
223. T.E. Long, A.D. Broske, D.J. Bradley, and J.E. McGrath, J. Polym. Sci., Polym. Chem. Ed. 27,
4001 (1989).
722 -*V Литература
224. R.P. Quirk and J.J. Ma, Polym. Inter. 24, 197 (1991).
225. R.P. Quirk, T. Yoo, Y. Lee, J. Kim, and B. Lee, Ado. Polym. Sci. 153, 67 (2000).
226. Unpublished work of R.P. Quirk and D. Xu, The University of Akron.
227. F. Ciardelli, in «Comprehensive Polymer Science» First Supplement, S.L. Aggarwal and
S. Russo (Eds.), Pergamon Press, Oxford, 1992, p. 67.
228. J. Boor, Jr., «Ziegler-Natta Catalysts and Polymerizations» Academic Press, New York,
1979.
229. K. Ziegler, E. Holzkamp, H. Breil, and H. Martin, Angew. Chem. 67, 541 (1955).
230. G. Natta, J. Polym. Sci. 16, 143 (1955).
231. L. Porri and A. Giarrusso, in «Comprehensive Polymer Science» Vol. 4: «Chain Polymerization
II» G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford,
1989, p. 53.
232. W. Cooper, in «The Stereo Rubbers» W.M. Saltman (Ed.), Wiley-Interscience, New York,
1977, p. 21.
233. Ph. Teyssie, P. Hadjiandreou, M. Julemont, and R. Warin, in «Transition Metal Catalyzed
Polymerizations» R.P. Quirk (Ed.), Cambridge University Press, Cambridge, UK, 1988, p.
639.
234. P. J.T. Tait and LG. Berry, in «Comprehensive Polymer Science» Vol. 4: «Chain Polymerization
II» G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford,
1989, p. 575.
235. F.P. Baldwin and G. Ver Strate, Rubber Chem. Tech. 42, 709 (1972).
236. G. Ver Strate, in «Encyclopedia of Polymer Science and Engineering» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 6, 1986, p. 522.
237. S.C. Davis, W. Von Hellens, H.A. Zahalka, and K.P. Richter, in «Polymeric Materials
Encyclopedia» J.C. Salamone (Ed.), CRC Press, Boca Raton, 1996, p. 2264.
238. J. W.M. Noordermeer, in «Encyclopedia of Polymer Science and Techno/ogy» J.I. Kroschwitz
(Ed.), Wiley-Interscience, New York, Vol. 6, 2003, p. 178.
239. E.J. Vandenberg, in «Encyclopedia of Polymer Science and Engineering» J.I. Kroschwitz
(Ed.), Wiley-Interscience, New York, Vol. 4, 1986, p. 174.
240. P. Pino, U. Giannini, and L. Porri, in «Encyclopedia of Polymer Science and Engineering»
J.I. Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 8, 1987, p. 147.
241. E.J. Arlman and P. Cossee, J. Catal. 3, 103 (1964).
242. P. Corradini, G. Guerra, and R. Pucciariello, Macromolecules 18, 2030 (1985).
243. A. Zambelli and G. Allegra, Macromolecules 13, 42 (1980).
244. A. Zambelli and C. Tosi, Adv. Polym. Sci. 15, 31 (1974).
245. P. Corradini, V. Busico, and G. Guerra, in «Comprehensive Polymer Science» Vol. 4: «Chain
Polymerization II» G.C. Eastmond, A. Ledwith, S. Russo, and P. Sigwait (Eds.), Pergamon
Press, Oxford, 1989, p. 29.
246. H.H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger, and R.M. Waymouth, Angew. Chem.
Int. Ed. Engl. 34, 1143 (1995).
247. E.P. Wasserman, in «Encyclopedia of Polymer Science and Techno/ogy» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 7, 2003, p. 35.
248. M. Galimberti, N. Mascellani, F. Piemontesi, and I. Camurati, Macromol. Rapid Commun. 20,
214 (1999).
249. S.E. Home, Jr., J.P. Kichl, JJ. Shipman, V.L. Folt, and C.T. Gibbs, Ind. Eng. Chem. 48,784 (1956).
250. S. Horne, Jr., in «Transition Metal Catalyzed Polymerizations: Alkenes and Dienes» Part B,
R.P. Quirk (Ed.), Harwood, 1983, p. 527.
251. H.L. Hsieh and H.C. Yeh, Rubber Chem. Tech. 58, 117 (1985).
252. M. Kerns, S. Henning, and M. Rachita, in «Encyclopedia of Polymer Science and Techno/ogy»
J.I. Kroschwitz (Ed.), Wiley-Interscience, Vol. 5, 2003, p. 317.
253. A. Dr xler, in «Handbook of Elastomers» 2nd ed., A.K. Bhowmick, H.L. Stephens (Eds.),
Marcel Dekker, New York, 2001, p. 697.
254. E.A. Ofstead, in «Encyclopedia of Polymer Science and Engineering» J.I. Kroschwitz (Ed.),
Wiley-Interscience, New York, Vol. 11, 1988, p. 287.
255. B.M. Novak, W. Risse, and R.H. Grubbs, Ado. Polym. Sci. 102, 47 (1992).
Список литературы к главе 2 -*v 723
256. K.J. Ivin, «Olefin Metathesis» Academic Press, New York, 1983.
257. A.J. Amass, in «Comprehensive Polymer Science» G.C. Eastmond, A. Ledwith, S. Russo, and
P. Sigwait (Eds.), Pergamon Press, Oxford, Vol. 4, 1989, p. 109.
258. A.K. Bhowmick, C. Stein, and H.L. Stephens, in «Handbook of Elastomers» 2nd ed.,
A.K. Bhowmick and H.L. Stephens (Eds.), Dekker, New York, 2001, p. 775.
259. P. Dreyfuss and R.P. Quirk, in «Encyclopedia of Polymer Science and Engineering»
J.L Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 62, 1986, p. 551.
260. P.F. Rempp and P.J. Lutz, in «Comprehensive Polymer Science» G.C. Eastmond, A. Ledwith,
S. Russo, and P. Sigwait (Eds.), Pergamon Press, Oxford, Vol. 6, 1989, p. 403.
261. R.P. Quirk, Rubber Chem. Tech. 57, 557 (1984).
262. G. RiessandG. Hurtrez, in «Encyclopediaof Polymer Science and Engineering» J.I. Kroschwitz
(Ed.), Wiley-Interscience, New York, Vol. 2, 1985, p. 324.
263. G. Holden, H.R. Kricheldorf, and R.P. Quirk (Eds.), «Thermoplastic Elastomers» Hanser
Publishers, Munich, Germany, 2004.
264. G. Odian, «Principles of Polymerization» 4th ed., Wiley-Interscience, New York, 2004, p. 752.
265. G.G. Cameron and M.Y. Qureshi, J. Polym. Sci., Chem. Ed. 18, 2143 (1980).
266. A. Brydon, G.M. Burnett, and G.G. Cameron, J. Polym. Sci., Chem. Ed. 11, 3255 (1973).
267. C. Walling, PureAppl. Chem. 15, 69 (1967).
268. D.J. Stein, G. Fahrbach, and H.J. Adler, Angew. Makromol. Chem. 38, 67 (1974).
269. B.J. Schmitt, Angew. Chem. Int. Ed. Engl. 18, 273 (1979).
270. D.M. Kulich, P.D. Kelley, and J.E. Pace, in «Encyclopedia of Polymer Science and
Engineering» J.L Kroschwitz (Ed.), Wiley-Interscience, New York, Vol. 1, 1985, p. 388.
271. C. S.L. Baker, in «Handbook of Elastomers» A.K. Bhowmick and H.L. Stephens (Eds.),
Marcel Dekker, New York, 2001, p. 61.
272. D.S. Campbell, in «Natural Rubber Science and Techno/ogy» A.D. Roberts (Ed.), Oxford
University Press, Oxford, 1988, p. 679.
273. P.W. Allen, in «Chemistry and Physics of Rubberlike Substances» L. Bateman (Ed.),
MacLaren, London, 1963, p. 97.
274. H. A.J. Battaerd and G.W. Tregear, «Graft Copolymers» Wiley-Interscience, New York,
1967.
275. W. Cooper, P.R. Sewell, and G. Vaughan, J. Polym. Sci. 41, 167 (1959).
276. E.G. Cockbain, T.D. Pendle, and D.J. Turner, J. Polym. Sci. 39, 419 (1959).
277. B. Gupta and N. Anjum, Adv. Polym. Sci. 162, 35 (2003).
278. R.J. Ceresa, «Block and Graft Copolymers» Butterworth, London, 1962, p. 65.
279. M. Pike and W.F. Watson, J. Polym. Sci. 9, 229 (1952).
280. D.J. Angier and W.F. Watson, J. Polym. Sci. 18, 129 (1955); Trans. Inst. Rubber Ind. 33, 22
(1957).
281. D.J. Angier, R.J. Ceresa, and W.F. Watson, J. Polym. Sci. 34, 699 (1959).
282. R.J. Ceresa and W.F. Watson, J. Appl. Polym. Sci. 34, 699 (1959).
283. N. Hadjichristidis, S. Pispas, M. Pitsikalis, H. latrou, and D.J. Lohse, in «Encyclopedia of
Polymer Science and Techno/ogy» J.L Kroschwitz (Ed.), Wiley-Interscience, New York, Vol.
6, 2003, p. 348.
284. K.A. Davis and K. Matyjaszewski, Adi?. Polym. Sci. 159, 1 (2002).
285. R.P. Quirk, D.L. Gomochak Pickel, and G.O. Schulz, in «Thermoplastic Elastomers»
G. Holden, H.R. Kricheldorf, and R.P. Quirk (Eds.), Hanser Publishers, Munich, Germany,
2004, p. 323.
286. N.R. Legge, Rubber Chem. Tech. 60, G83 (1987).
287. F.S. Bates and G.H. Fredrickson, Annu. Rev. Phys. Chem. 41, 525 (1990).
288. D.J. Meier, J. Polym. Sci., Part C26, 81 (1969).
289. E. Helfand.Acc. Chem. Res. 8, 295 (1975).
290. L. Leibler, Macromolecules 13, 1602 (1980).
291. M. Morton, in «Encyclopedia of Polymer Science and Techno/ogy» N. Bikales (Ed.), Wiley,
New York, Vol. 15,1971, p. 508.
292. G. Holden, E.T. Bishop, and N.R. Legge, J. Polym. Sci., Part C26, 37 (1969).
293. T.L. Smith and R.A. Dickie, J. Polym. Sci., Part C26, 163 (1969).
724 -’V Литература
294. I.W. Hamley (Ed.), «Developments in Block Copolymer Science and Techno/ogy» Wiley,
New York, 2004.
295. M. Morton, R.F. Kammereck, and L.J. Fetters, Br. Polym. J. 3, 120 (1971).
296. M. Morton, Y. Kesten, and L.F. Fetters, Polym. Prepr., Am. Chem. Soc., Div. Polym. Chem.
15,175 (1974).
297. R. Jerome, in «Thermoplastic Elastomers» G. Holden, H.R. Kricheldorf, and R.P. Quirk
(Eds.), Hanser Publishers, Munich, Germany, 2004, p. 444.
298. A.K. Bhowmickand H.L. Stephens (Eds.), «Handbook of Elastomers» 2nded., Marcel Dekker,
New York, 2001.
299. S. Abouzahr and G.L. Wilkes, in «Processing, Structure and Properties of Block
Copolymers» M.J. Folkes (Ed.), Elsevier, London, 1985, p. 165.
Глава 3
1. J.V. Dawkins (Ed.), «Developments in Polymer Characterization» Vol. 1-5, Elsevier, New
York, 1986.
2. C. Booth and C. Price (Eds.), «Comprehensive Polymer Science» Vol. 1: «Polymer
Characterization» Pergamon, New York, 1989.
3. H. Yamakawa, «Modern Theory of Polymer Solutions» Harper, New York, 1971.
4. P.J. Flory, «Statistical Mechanics of Chain Molecules» Oxford Univ. Press, New York, 1969.
5. Y. Tanaka, Rubber Chem. and Technol. 64, 325 (1991).
6. D. Campbell and J. White, «Polymer Characterization» Chapman and Hall, New York, 1989.
7. F.P. Baldwin and G. Ver Strate, Rubber Chem. Technol. 44, 709 (1972).
8. S.L. Hsu, «Handbook of Vibrational Spectroscopy: A Companion for Polymer Scientists»
Wiley, New York 2004.
9. B.H. Stuart, «Polymer Analysis» Wiley, New York, 2002.
10. J.L. Koenig, «Spectroscopy of Polymers» 2nd ed., Elsevier, New York, 1999.
11. D. Braun, «Simple Methods for Identification of Plastics» 3rd ed., Hanser, New York, 1996.
12. J. Mitchell (Ed.), «Applied Polymer Analysis and Characterization» Vol. 2, Hanser, New York,
1992.
13. E. Collins, J. Bares, and F. Billmeyer, «Experi xents in Polymer Science» Wiley, New York,
1973.
14. W. Tyler, Rubber Chem. Technol. 40, 238 (1967).
15. H. Ishida, Rubber Chem. Technol. 60, 497 (1987).
16. R. Messerschmidt and M. Harthcock (Eds.), «Infrared Microspectroscopy» Practical
Spectroscopy Series, Vol. 6, Marcel Dekker, New York, 1988.
17. http://www. spectroscopynow. com
18. I. Fuhrmann and J. Karger-Kocsis, J. Appl. Polym. Sci. 89, 1622 (2003).
19. S.R. Shield and G.N. Ghebremeskel, J. Appl. Polym. Sci. 88, 1653 (2003).
20. K.D. McCarley and A.L. Bunge, Internal. J. Pharmaceutics 250, 169 (2003).
21. F. H.A. Rodrigues, E.F. Santos, J. P.A. Feitosa, N. M. P.S. Ricardo, and R.C. M. de Paula,
Rubber Chem. Technol. 74, 57 (2001).
22. S. Sen, C. Mabuni, and D. Walsh, J. Appl. Polym. Sci. 82, 672 (2001).
23. M. C.P. Peckj, M.A. Samus, R.C. Killgoar, and R.O. Carter, Rubber Chem. Technol. 64, 610
(1991).
24. W.H. Waddell and J.R. Parker, Rubber Chem. Technol. 65, 836 (1992).
25. L. Stebounova, B.B. Akhremitchev, and G.C. Walker, Rev. Sci. Instr. 74, 3670 (2003).
26. F. Keilmann.Z Biol. Phys. 29, 195 (2003).
27. P. J. de Dumas, Phys. 7V104, 359 (2003).
28. F. Keilman, Vibr. Sped. 29, 109 (2002).
29. D.L. Wetzel and S.M. LeVine, Science 285, 1224 (1999).
30. D.V. Palanker, D.M. Simanovskii, P. Huie, and T.I.J. Smith, Appl. Phys. 88, 6808 (2000).
31. K.D. Kempfert, B. Coel, P. Troost, and D.S. Lavery, Amer. Lab. 33, 22 (2001).
32. C.A. Froud, I.P. Hayward, and J. Laven, Appl. Sped. Ы, 1468 (2003).
33. A. Ghanbari-Siahkali, K. Almdal, and P. Kingshott, Appl. Sped. 57, 1482 (2003).
34. A. Szep, B. Marosfoi, G. Bertalan, P. Anna, and G. Marosi, Macromol. Symp. 202, 269 (2003).
Список литературы к главе 3 -'V 725
35. Y. Maeda, Н. Yamamoto, and I. Ikeda, Macromolecules 36, 5055 (2003).
36. L. Berger, H.H. Kausch, and C.J.G. Plummer, Polymer 4A, 5877 (2003).
37. J.F. Raboltand D.B. Chase (Eds.), «Fourier Transform Raman Spectroscopy: From Concept
to Experiment» Academic Press, New York, 2000.
38. T.W. Zerda, G. Song, and W.H. Waddell, Rubber Chem. Technol. 76, 769 (2003).
39. F. Grum, in «Visible and Ultraviolet Spectrophotometry» A. Weissberger and B. Rossiter
(Eds.), Physical Methods of Chemistry, Part IIIB, Vol. 1, Wiley (Interscience), New York,
1972, p. 207.
40. V.M. Raubner, R. Jordan, O. Nuyken, T. Lippert, M. Hauer, B. Schnyder, and A. Wokaun,
Applied Surface Science 197, 786 (2002).
41. D. Braun, R. Rettig, and W. Rogler, Angewandte Makromolekulare Chemie 211,165 (1993).
42. H. Cheng, in «Modern Methods of Polymer Characterization» H. Barth and J. Mays (Eds.),
Chemical Analysis, Vol. 113, Wiley, New York, 1991, p. 409.
43. R. Kinsey, Rubber Chem. Technol. 63, 407 (1990).
44. H.T. Wang, T. Bethea, and H. Harwood, Macromolecules 26, 715 (1993).
45. W.T. Smith and J. Patterson, Anal. Chem. 58, 102R (1986).
46. T. Hammond and R. Lehrie, Brit. Polym. J. 21, 23 (1989).
47. D. Vitalini and E. Scamporrino, Polymer SS, 4597 (1992).
48. G.N. Ghebremeskel, J.K. Sekinger, J.L. Hoffpauir, and C. Hendrix, Rubber Chem. Technol.
69,874 (1996).
49. T. Yamada, T. Okumoto, H. Ohtani, and S. Tsuge, Rubber Chem. Technol. 64, 708 (1991).
50. H.R. Schulten, B. Plage, and R.P. Lattimer, Rubber Chem. Technol. 62, 698 (1989).
51. R. Lattimer, Rubber Chem. Technol. 63, 298 (1990).
52. V.P. Hoven, K. Rattanakaran, and Y. Tanaka, Rubber Chem. Technol. 76, 1128 (2003).
53. S. Kawahara, Y. Inomata, T. Kakubo, Y. Tanaka, K. Hatada, K. Ute, and N. Miyatake, Rubber
Chem. Technol. 71, 277 (1998).
54. Y. Tanaka, H. Sato, and J. Adachi, Rubber Chem. Technol. 60, 25 (1987).
55. Y. Tanaka, H. Sato, and J. Adachi, Rubber Chem. Technol. 59, 16 (1986).
56. G.H. Miller and R.H. Tobias, Rubber Chem. Technol. 51, 977 (1978).
57. http://www. simsworkshop. org/default. nclk
58. A. Benninghoven, F.G. Rdenauer, and H.W. Werner, «Secondary Ion Mass Spectrometry:
Basic Concepts, Instrumental Aspects, Applications, and Trends» Wiley, New York, 1987.
59. W.J. Van Ooij, M. Nahmias, and A. Brown, Surface and Interface Analysis 61, 539 (1988).
60. W.J. Van Ooij and V. Rangarajan, Rubber Chem. Technol. 61, 594 (1988).
61. D. Briggs, Brit. Polym. J. 21, 3 (1989).
62. P. Betrand, L.T. Weng, W. Lauer, and R. Zimmer, Rubber Chem. Technol. 75, 627 (2002).
63. P. Betrand and L.T. Weng, Rubber Chem. Technol. 72, 384 (1999).
64. A.K. Sircar, Rubber Chem. Technol. 65, 503 (1992).
65. A.K. Sircar, in «Thermal Characterization of Polymeric Materials» 2nd ed., E.A. Turi (Ed.),
Academic Press, New York, 1997.
66. S. Capancioni, K. Schwach-Abdellaoui, W. Kloeti, W. Herrmann, H. Brosig, H.H. Borchert,
J. Heller, and R. Gurny, Macromolecules 36, 6135 (2003).
67. D. Hauck, G. Fink, C. Chwatinski, P. Blumler, B. Blumich, and K. Unseld, Kauch. Gummi
Kunstat. 50, 392 (1997).
68. O. Hinojosa, J.C. Arthur, and Y. Nakamura, JPolym. Sci. CPolym. Symp. 37, 27 (1972).
69. K.L. Devries, J. Polym. Sci. CPolym. Symp. 32, 325 (1971).
70. R. Partridge, M. C.R. Symons, and J.L. Wyatt, J. Chem. Soc.—Faraday Trans. 89,1285 (1993).
71. M.D. Pace and C.M. Roland, Polymer 32, 1027 (1991).
72. C. Brosseau, P. Molinie, F. Boulic, and F. Carmona, J. Appl. Phys. 89, 8297 (2001).
73. H. Tang, Z.Y. Liu, J.H. Piao, X.F. Chen, Y.X. Lou, and S.H. IA,J.Appl. Polym. Sci. 51,1159 (1994).
74. H. Hommel, A. Touhami, and A.P. Legrand, Makromol. Chem.—Macro. Chem. Phys. 194,
879 (1993).
75. G. Ver Strate, C. Cozewith, and S. Ju, Macromolecules 21, 3360 (1988).
76. L. Gilet, M. Loustalot, and J. Bounoure, Polymer 4605 (1992).
77. L. Wild, Adv. Polym. Sci. 98,1 (1991).
726 JV Литература
78. A. Dekmezian, T. Morioka, and C. Campo, J. Pol. Sci. Phys. 28, 1908 (1990).
79. T. Zimina, A. Fell, and J. Castledine, Polymer 33, 4129 (1992).
80. A. Dekmezian, T. Morioka, and C. Campo, J. Polym. Sci. Polym. Phys. 28, 1908 (1990).
81. Z. Grubsic-Gallot, J.P. Lingelser, and Y. Gallot, Polymer Bull. 23, 389 (1990).
82. G. Glockner, in «Comprehensive Polymer Science» C. Booth and C. Price (Eds.), Vol 1:
«Polymer Characterization» Pergamon, New York, 1989, p. 313.
83. S.H. Xu, P.S. Wells, Y.M. Tao, K.S. Yun, and J.F. Porcher, ACSSymp. Series 748, 96 (2000).
84. H. Yamada, I. Manas-Zloczower, and D.L. Feke, Powder Technol. 92, 163 (1997).
85. C.B. Stanley and H.H. Strey, Macromolecules 36, 6888 (2003).
86. J. Tacx, J. Ammerdorffer, and A. German, Polymer 29, 2087 (1988).
87. S. Mori and M. Mouri, Anal. Chem. 61, 2171 (1989).
88. G. Stegeman, A.C. Vanasten, J.C. Kraak, H. Poppe, and R. Tijssen,i4naZ. Chem. 66,1147 (1994).
89. S.H. Lee and A. Molnar, Macromolecules 28, 6354 (1995).
90. L. Lewandowski, M.S. Sibbald, E. Johnson, and M.P. Mallamaci, Rubber Chem. Technol. 73,
731 (2000).
91. J. Janca, I. A. Ananieva, A. Y. Menshikova, and T.G. Evseeva, J. Chromatography В 800,33 (2004).
92. M. Mori and J.L. Koenig, J.Appl. Polym. Sci. 70, 1391 (1998).
93. A.M. Zaperand J.L. Koenig, Macromol. Chem. 189, 1239 (1988).
94. R.S. Clough and J.L. Koenig, Rubber Chem. Technol. 62, 908 (1989).
95. J.L. Koenig, Rubber Chem. Technol. 73, 385 (2000).
96. C. Tosi, Adv. Polym. Sci. 5, 451 (1968).
97. T. Yamada, T. Okumoto, H. Ohtani, and S. Tsuge, Rubber Chem. Technol. 63, 191 (1990).
98. J. Tulisalo, J. Seppala, and K. Hastbacka, Macromolecules 18,1144, (1985).
99. J.C. Hu, Anal. Chem. 53, 942 (1981).
100. W. Tuminello, W. Buck, and D. Kerbow, Macromolecules 26, 499 (1993).
101. W. Graessley and G. Ver Strate, Rubber Chem. Technol. 53, 842 (1980).
102. M. Doi, in «Structure and Properties of Polymers» E.L. Thomas (Ed.), Wiley-VCH,
Weinheim, Germany, 1993.
103. K.L. Ngai and D.J. Plazek, Rubber Chem. Technol. 68, 376 (1995).
104. S.H. Wasserman, J. Rheology 39, 601 (1995).
105. J.C. Majeste, C. Carrot, and P. Stanescu, Rheo. Acta. 42, 432 (2003).
106. R.U. Bonnar, M. Dimbat, and F.H. Stross, «Number-Average Molecular Weights:
Fundamentals and Determination» Interscience, New York, 1958.
107. H. Barth and J. Mays, «Modem Methods of Polymer Characterization» Wiley, New York, 1991.
108. A. Cooper (Ed.), «Determination of Molecular Weight» Chemical Analysis Series, Vol. 103,
J. Wiley, New York, 1989.
109. J. Brown and P. Verdier, J. Res. Natl. Bur. Stand. A 76, 161 (1972).
110. T. Tanaka (Ed.), «Experimental Methods in Polymer Science» Academic Press, San Diego, 1999.
111. B.J. Berne and R. Pecora, «Dynamic Light Scattering» Wiley, New York, 1976, Chap. 8.
112. A. Boothroyd and L.J. Fetters, Macromolecules 24, 5215 (1991).
113. G.V. Schulz, K. Altgelt, and H.J. Cantow, Rubber Chem. Technol. 30, 805 (1957).
114. E. Casassa, Polym. J. 3, 517 (1972).
115. A.R. Shultz and W.H. Stockmayer, Macromolecules 2, 178 (1969).
116. H. Suzuki, C. Leonis, and M. Gordon, Makromol. Chem. 172, 227 (1973).
117. J. Vavra, J. Lapcik, and J. Sabados, J. Polym. Sci. A-2 5, 1305 (1967).
118. H. Lindner and O. Glatter, Macromol. Symp. 162, 81 (2000).
119. J.S. Higgins and H.C. Benoit, «Polymers and Neutron Scattering» Clarendon Press, Oxford, 1997.
120. G.D. Wignail, in «Physical Properties of Polymers Handbook» J.E. Mark (Ed.), Amer. Inst.
Phys., Woodbury, NY, 1996, Chap. 22.
121. D.J. Lohse, Rubber Chem. Technol. 67, 367 (1994).
122. A. Arbe, J. Colmenero, B. Farago, M. Monkenbusch, U. Buchenau, and D. Richter, Chem.
Phys. 292, 295 (2003).
123. D. Richter, J. Appl. Cryst. 36, 389 (2003).
124. P. Stepanek, T.L. Morkved, K. Krishnan, T.P. Lodge, and F.S. Bates, PhysicaA314,411 (2002).
125. B.K. Annis, M.H. Kim, R. Alamo, and M. Pyda, J. Polym. Sci. Polym. Phys. 39, 2852 (2001).
Список литературы к главе 3 -• V 727
126. A.Z. Akcasu, G.C. Summerfield, S.N. Jahsan, C.C. Han, C.Y. Kim, and H. Yu, J. Polym. Sci.
Polym. Phys. 18, 863 (1980).
127. W. Gronski, F. Forster, W. Pyckhout-Hintzen, and T. Springer, Makromolekulare Chemie—
Macromol. Symp. 40, 121 (1990).
128. G. Boue, J. Bastide, M. Buzier, A. Lapp, J. Herz, and T.A. Vilgis, Coll. Polym. Sci. 269,195 (1991).
129. S. Westerman, W. Pyckhout-Hintzen, D. Richter, E. Straube, S. Egelhaaf, and R. May,
Macromolecules 34, 2186 (2001).
130. A. Botti, W. Pyckhout-Hintzen, D. Richter, V. Urban, E. Straube, and J. Kohlbrecher, Polymer
44, 7505 (2003).
131. S. Westermann, M. Kreitschmann, W. Pyckhout-Hintzen, D. Richter, E. Straube, B. Farago,
and G. Goerigk, Macromolecules 32, 5793 (1999).
132. Y.M. Zhang, S. Ge, B. Tang, T. Koga, M.H. Rafailovich, J.C. Sokolov, D.G. Peiffer, Z. Li,
A.J. Dias, K.O. McElrath, M.Y. Lin, S.K. Satija, S.G. Urquhart, H. Ade, and D. Nguyen,
Macromolecules 34, 7056 (2001).
133. Y. Du, Y. Xue, and H.L. Frisch, in «Physical Properties of Polymers Handbook» J.E. Mark
(Ed.), Amer. Inst. Phys., Woodbury, NY, 1996, Chap. 17.
134. M. Kurata and Y. Tsunashima, in «Polymer Handbook» 4th ed., J. Brandrup, E.H. Immergut,
and E.A. Grulke (Eds.), Wiley, New York, 1999, Chap. 7.
135. D. Van Krevelen, «Properties of Polymers» 3rd ed., Elsevier, Amsterdam, 1990.
136. C.Y. Kuo, T. Provder, and M.E. Koehler, J. Liq. Chromatography 13, 3177 (1990).
137. C.Y. Kuo and T. Provder, ACSSymp. Series 352, 2 (1987).
138. Z. Grubisic, P. Rempp, and H. Benoit, J. Polym. Sci. 21, 753 (1967).
139. J.E. Puskas and R. Hutchinson, Rubber Chem. Technol. 66, 742 (1993).
140. C. Kuo, T. Provder, and M.E. Koehler, ACSSymp. Series 521, 231 (1993).
141. J.G. Rooney and G. VerStrate, in «Liquid Chromatography of Polymers and Related Materials
III» J. Cazes (Ed.), Marcel Dekker, New York, 1981.
142. T. Provder, M.W. Urban, and H.G. Barth (Eds.), «Chromatographic Characterization of
Polymers: Hyphenated and Multidimensional Techniques» Adv. Chem. Series 247, Amer.
Chem. Soc., Washington, D. C., 1995.
143. Y. Brun, J. Liq. Chromatography & Related Technol. 21, 1979 (1998).
144. D.J. Nagy, Amer. Lab. 35, 38 (2003).
145. B.L. Wang, S. Mukataka, E. Kokufuta, M. Ogiso, and M. Kodama, J. Polym. Sci. Polym. Phys.
38,214(2000).
146. S.T. Balke and Т.Н. Mourey, J. Appl. Polym. Sci. 81, 370 (2001).
147. E. Meehan, J.A. McConville, and F.P. Warner, Polym. Internal. 26, 23 (1991).
148. M. Nguyen and R. Beckett, Polymer International 30, 337 (1993).
149. M.E. Schimpf, K. Caldwell, and J.C. Giddings, «Field-Flow Fractionation Handbook» Wiley,
New York, 2000.
150. S. Lee, C.H. Eum, and A.R. Plepys, Bull. Korean Chem. Soc. 21, 69 (2000).
151. A. C. van Asten, W.T. Kok, R. Tijssen, and H. Poppe, J. Polym. Sci. Polym. Phys. 34, 297
(1996).
152. M. Nguyen, R. Beckett, L. Pille, and D.H. Solomon, Macromolecules 31, 7003 (1998).
153. H. Creel, Trends Polym. Sci. 1, 336 (1993).
154. M. W.F. Nielsen, Mass. Spectrom. Rev. 18, 309 (1999).
155. D. Cho, S. Park, K. Kwon, T. Chang, and J. Roovers, Macromolecules 34, 7570 (2001).
156. P. Vana, L. Albertin, L. Barner, T.P. Davis, and C. Barner-Kowollik, J. Polym. Sci. Polym.
Polym. Chem. 40, 4032 (2002).
157. G.E. Kassalainen and S.K.R. Williams, Anal. Chem. 75, 1887 (2003).
158. T.M. Laue, in «Concise Encyclopedia of Polymer Science and Engineering» J.I. Kroschwitz
(Ed.), Wiley, New York, 1990, p. 1225.
158a. D. Boese and F. Kremer, Macromolecules 23, 829 (1990).
159. G.S. Grest, L.J. Fetters, J.S. Huang, and D. Richter, in «Advances in Chemical Physics» XCIV,
I. Prigogine and S.A. Rice (Eds.), Wiley, New York, 1996.
159a. J.E. Puskas, Polymer News 27, 325 (2002).
160. P.A. Small, Adv. Polym. Sci. 18, 1 (1975).
728 -*V Литература
161. S. Podzimek, T. Vlcek, C. Johann, J. Appl. Polym. Sci. 81, 1588 (2001).
162. S. Grcev, P. Schoenmakers, and P. ledema, Polymer 45, 39 (2004).
163. E. Zagar and M. Zigon, Macromolecules 35, 9913 (2002).
164. M. Jayakannan, J. L.J. Van Dongen, G.C. Behera, and S. Ramakrishnan, J. Polym. Sci. Polym.
Chem. 40, 4463 (2002).
165. A. Lederer, D. Voigt, C. Clausnitzer, and B. Voit, J. Chromatography A 976, 171 (2002).
166. M. Parth, N. Aust, and K. Lederer, Macromol. Symp. 181, 447 (2002).
167. L. Hong, X.L. Wang, and X.Z. Tang, J. Appl. Polym. Sci. 85, 2445 (2002).
168. P.J. DesLauriers, D.C. Rohlfing, and E.T. Hsieh, Polymer 43, 159 (2002).
169. S. Podzimek and T. Vlcek, J. Appl. Polym. Sci. 82, 454 (2001).
170. A.M. Striegel and M.R. Krejsa, J. Polym. Sci. Polym. Phys. 38, 3120 (2000).
171. S.T. Balke and Т.Н. Mourey, J. Appl. Polym. Sci. 81, 370 (2001).
172. G. Kraus and J.T. Gruver, J. Polym. Sci. A 3, 105 (1965).
173. K. L Ngai and C.M. Roland, J. Polym. Sci. Polym. Phys. 35, 2503 (1997).
174. C.G. Robertson, C.M. Roland, C. Paulo, and J.E. Puskas, J. Rheol. 45, 759 (2001).
175. P.G. Santangelo and C.M. Roland, J. Non-Cryst. Solids 235, 709 (1998).
176. J.M. Carella, J.T. Gotro, and W.W. Graessley, Macromolecules 19, 659 (1986).
177. P.G. Santangelo, C.M. Roland, and J.E. Puskas, Macromolecules 32, 1972 (1999).
178. C.M. Roland and P.G. Santangelo, J. Non-Cryst. Solids 307, 835 (2002).
179. G. G.A. В hm and J.O. Tveekrem, Rubber Chem. Technol. 55, 575 (1982).
180. M. Okabe, K. Mitsui, H. Oranaka, M. Jahahashi, and H. Masuda, Polymer J. 24, 653 (1992).
181. J. Horsky and M. Bohdanecky, Eur. Pol. J. 26, 1190 (1990).
182. J.P. Runt and J.J. Fitzgerald (Eds.), «Dielectric Spectroscopy of Polymeric Materials»
American Chemical Society, Washington, D. C., 1997.
183. P.G. Santangelo and C.M. Roland, Macromolecules 31, 3715 (1998).
184. D.J. Plazek, EC. Chay, K.L. Ngai, and C.M. Roland, Macromolecules 28, 6432 (1995).
185. C.M. Roland, K.L. Ngai, P.G. Santangelo, X.H. Qiu, M.D. Ediger, and D.J. Plazek,
Macromolecules 34, 6159 (2001).
186. C.M. Roland and R. Casalini, J. Chem. Phys. 119, 1838 (2003).
186a. D.J. Plazek and Z.N. Frund, J. Polym. Sci. Polym. Phys. 28, 431 (1990).
187. R. Androsch and B. Wunderlich, J. Polym. Sci. Polym. Phys. 41, 2039 (2003).
188. J.M. Hutchinson, J. Therm. Anal. Calorimetry 72, 619 (2003).
189. K. Kanari and T. Ozawa, Thermochimica Acta 399, 189 (2003).
190. E. Riande and E. Saiz, «Dipole Moments and Birefringence of Polymers» Prentice-Hall,
Englewood Cliff, NJ, 1992.
191. P.H. Mott and C.M. Roland, Macromolecules 31, 7095 (1998).
192. P.H. Mott, A. Rizos, and C.M. Roland, Macromolecules 34, 4476 (2001).
193. C.M. Roland and M.L. Warzel, Rubber Chem. Technol. 63, 285 (1990).
194. P.H. Mott and C.M. Roland, Macromolecules 33, 4132 (2000).
195. I.S. Zemel and C.M. Roland, Polymer 33, 4522 (1992).
196. D. Keroack, Y. Zhao, and R.E. Prud’homme, Polymer 40, 243 (1999).
197. A.K. Kalkar, H.W. Siesler, F. Pfeifer, and S.A. Wadekar, Polymer 7251 (2003).
198. R.M. Kannan and J.A. Kornfield,/. Rheol. 38, 1127 (1994).
199. M.J. Citra, D.B. Chase, R.M. Ikeda, and K.H. Gardner, Macromolecules 28, 4007 (1995).
200. L. A. Archer and G.G. Fuller, Macromolecules 27, 7152 (1994).
201. D.I. Bower, E. L.V. Lewis, and I.M. Ward, Polymer 36, 3473 (1995).
202. D.I. Bower, J. Polym. Sci. Polym. Phys. 19, 93 (1981).
203. B. Deloche and E.T. Samulski, Macromolecules 14, 575 (1981).
204. P. Sotta, B. Deloche, J. Herz, A. Lapp, D. Durand, and J.C. Rabadeux, Macromolecules 20,
2769 (1987).
205. J.F. Tassin, A. Baschwitz, J.Y. Moise, and L. Monnerie, Macromolecules 23, 1879 (1990).
206. M. Doi and H. Watanabe, Macromolecules 24, 740 (1991).
207. S. Toki, I. Sics, S.F. Ran, L.Z. Liu, and B.S. Hsiao, Polymer kk, 6003 (2003).
208. S. Toki, I. Sics, S.F. Ran, L.Z. Liu, B.S. Hsiao, S. Murakami, K. Senoo, and S. Kohjiya,
Macromolecules 35, 6578 (2002).
Список литературы к главе 3 729
209. М. H.Y. Cheng and Е.В. Nauman, J. Polym. Sci. Polym. Phys. 42, 603 (2004).
210. X.W. Qu, S.R. Shang, G.D. Liu, S.W. Zhang, Y. Zhang, and L.C. Zhang, J. Appl. Polym. Sci.
91,1685 (2004).
211. E.R. Weibel, in «Principles and Techniques of Electron Microscopy» M.A. Hoyat (Ed.), Van
Nostrand, New York, Vol. 3, 1973, Chap. 6.
212. C.M. Roland and G.G.A. В hm, J. Poly. Sci. Poly. Phys. 22, 79 (1984).
213. D.A. Winesett, H. Ade, A.P. Smith, S.G. Urquhart, A.J. Dias, and P. Stevens, Rubber Chem.
Technol. 76, 803 (2003).
214. J.B. Miller, K.J. McGrath, C.M. Roland, C.A. Trask, and A.N. Garroway, Macromolecules 23,
4543(1990).
215. C.M. Roland and K.L. Ngai, Macromolecules 24, 2261 (1991).
216. A. Alegria, J. Colmenero, K.L. Ngai, and C.M. Roland, Macromolecules 27, 4486 (1994).
217. K.L. Ngai and C.M. Roland, Macromolecules 28, 44033 (1995).
218. C.M. Roland, Macromolecules 20, 2557 (1987).
219. K.J. McGrath and C.M. Roland, Rubber Chem. Technol. 67, 629 (1994).
220. A. Alegria, C. Elizetxea, I. Cendoya, and J. Colmenero, Macromolecules 28, 8819 (1995).
221. J.B. Miller, Rubber Chem. Technol. 66, 455 (1993).
222. S.K. Brownstein, J. E.L. Roovers, and D.J. Worsfold, Magn. Reson. Chem. 26, 392 (1988).
223. M. Tomaselli, B.H. Meier, P. Robyr, U.W. Suter, and R.R. Ernst, Chem. Phys. Lett. 205,145
(1993).
224. J.H. Walton, J.B. Miller, C.M. Roland, and J.B. Nagode, Macromolecules 26, 4052 (1993).
225. J.H. Walton, J.B. Miller, and C.M. Roland, J. Polym. Sci. Polym. Phys. 30, 527 (1992).
226. K.J. McGrath and C.M. Roland, Rubber Chem. Technol. 67, 629 (1994).
227. A.J. Tinker, Rubber Chem. Technol. 68, 461 (1995).
228. Y.F. Shutilin, Polym. Sci. USSR 27, 2386 (1985).
229. A.J. Tinker, Rubber Chem. Technol. 63, 503 (1990).
230. C.M. Roland, Macromolecules 27, 4242 (1994).
231. N.P. Shvydkaya, L.A. Shumanov, L.P. Semenova, and A.S. Lykin, Kauch. Rezina 11, 19
(1980).
232. P. Blunder and B. Blumich, Rubber Chem. Technol. 70, 468 (1997).
233. M. J.R. Loadman and A.J. Tinker, Rubber Chem. Technol. 62, 234 (1989).
234. P.S. Brown, M. J.R. Loadman, and A.J. Tinker, Rubber Chem. Technol. 65, 744 (1992).
235. D.W. Tomlin and C.M. Roland, Polymer 34, 2665 (1993).
236. I.S. Zemel and C.M. Roland, Polymer 33, 3427 (1992).
237. A.K. Bhowmick and H.L. Stephens (Eds.), «Handbook of Elastomers» 2nd ed., Marcel Dekker,
New York, 2001.
238. B.C. Edwards, J. Polym. Sci., Polym. Phys. 13, 1387 (1975).
239. J. Gough, Proc. Lit. and Phil. Soc. Manchester, 2nd Ser. 1, 288 (1805).
240. J.H. Magill, Rubber Chem. Technol. 68, 507 (1995).
241. A.N. Gent and L.Q. Zhang, Rubber Chem. Technol. 75, 923 (2002).
242. G.R. Hamed and N. Rattanasom, Rubber Chem. Technol. 75, 935 (2002).
243. P.G. Santangelo and C.M. Roland, Rubber Chem. Technol. 74, 69 (2001).
244. I.S. Choi and C.M. Roland, Rubber Chem. Technol. 69, 591 (1996).
245. E.H. Andrews, P.G. Owen, and A. Singh, Proc. Royal Soc. London A 324, 79 (1971).
246. J.C. Mitchell and D.J. Meier, J. Polym. Sci. Polym. Phys. 6, 1689 (1968).
247. W.R. Krigbaum and R.J. Roe, J. Polym. Sci. A 2, 4391 (1964).
248. A. Ziabicki and L. Jarecki, Coll. Polym. Sci. 256, 332 (1978).
249. G.K. Elyashevich, Adv. Poly. Sci. 43, 205 (1982).
250. C.M. Roland and M.F. Sonnenschein, Polym. Eng. Sci. 31, 1434 (1991).
251. M.J. Brock and M.J. Hackathorn, Rubber Chem. Technol. 45, 1301 (1972).
252. W. Cooper and R.K. Smith, J. Polym. Sci. A 1, 159 (1963).
253. S. Trabelsi, P.A. Albouy, and J. Rault, Macromolecules 36, 9093 (2003).
254. J.E. Mark, Prog. Polym. Sci. 28, 1205 (2003).
255. M.A. Sharaf, A. Kloczkowski, and J.E. Mark, Rubber Chem. Technol. 68, 601 (1995).
256. G.R. Hamed and B.H. Park, Rubber Chem. Technol. 72, 946 (1999).
730 -* V Литература
257. A.N. Gent, J. Polym. Sci. 18, 321 (1955).
258. D.R. Burfield, Polymer 25, 1823 (1984).
259. D.R. Burfield and Y. Tanaka, Polymer 28, 907 (1987).
260. A.N. Gent, J. Polym. Sci. 18, 321 (1955).
261. A.N. Gent, Inst. Rubb. Ind. 30, 139 (1954).
262. I.S. Choi and C.M. Roland, Rubber Chem. Technol. 70, 202 (1997).
263. G.R. Mitchell, Polymer 25, 1562 (1984).
264. R. Oono, K. Miyasaka, and K. Ishikawa, J. Polym. Sci. 11, 1477 (1973).
265. S. Trabelsi, P.A. Albouy, and J. Rault, Macromolecules 36, 7624 (2003).
266. L. R.G. Treloar, Trans. Faraday Soc. 43, 284 (1947).
267. G.R. Taylor and S.R. Darin, J. Appl. Phys. 20, 1075 (1955).
268. T. Shimizu, M. Tsuji, and S. Kohjiya, Mat. Sci. Res. Internal. 4, 117 (1998).
269. A.N. Gent and L.Q. Zhang, Rubber Chem. Technol. 75, 923 (2002).
270. A.N. Gent and L.Q. Zhang, J. Polym. Sci. Polym. 39, 811 (2001).
271. T. Kameda and T. Asakura, Polymer 44, 7539 (2003).
272. A.N. Gent, Trans. Faraday Soc. 50, 521 (1954).
273. C.M. Roland and C.R. Smith, Rubber Chem. Technol. 58, 806 (1985).
274. C.M. Roland, Rubber World 208, 15 (1993).
275. R.F. Carey et al., Sexually Transmitted Diseases 19, 230 (1992).
276. V. Kiernan, New Scientist, March 23, p. 7 (1996).
277. The New York Times, March 22, 1994, p. C3.
278. S.G. Arnold, J.E. Whitman, C.H. Fox, and M.H. Cottier-Fox Nature 335, 19 (1988).
279. K.F. Gazeley, A. D.T. Gorton, and T.D. Pendle, in «Natural Rubber Science and Techno/ogy»
A.D. Roberts (Ed.), Oxford Univ. Press, Oxford, 1988, Chap. 3.
280. F. Bueche, Rubber Chem. Technol. 32, 1269 (1959).
281. M. Braden and A.N. Gent J. Appl. Poly. Sci. 3,100 (1960).
282. A.N. Gent, in «Science and Techno/ogy of Rubber» F.R. Eirich (Ed.), Academic Press, New
York, 1978, Chap. 10.
283. C.M. Roland and J.W. Sobieski, Rubber Chem. Technol. 62, 683 (1989).
284. A.G. Thomas and J.M. Whittle, Rubber Chem. Technol. 43, 222 (1970).
285. C. L.M. Bell, D. Stinson, and A.G. Thomas, Rubber Chem. Technol. 55, 66 (1982).
286. A.N. Gent, P.B. Lindley, and A.G. Thomas, J. Appl. Poly. Sci. 8, 455 (1964).
287. G.J. Lake and P.B. Lindley J. Appl. Poly. Sci. 9, 1233 (1965).
288. G.J. Lake Prog. Rubber Technol. 45, 89 (1983).
289. H.W. Greensmith J. Appl. Polym. Sci. 8, 1113 (1964).
290. G.R. Hamed, H.J. Kim, and A.N. Gent, Rubber Chem. Technol. 69, 807 (1996).
291. G.R. Hamed, Rubber Chem. Technol. 56, 244 (1983).
292. E.S. Dizon, A.E. Hicks, and V.E. Chirico, Rubber Chem. Technol. 46, 231 (1973).
293. W. May, Trans. Inst. Rubber Ind. 40, T109 (1964).
294. V.R. Raju, E.V. Menezes, G. Marin, and W.W. Graessley, Macromolecules 14, 1668 (1981).
295. R.S. Marvin and J. Oser, J. Res. Natl. Bur. Stand. В 66, 171 (1962); 67, 87 (1963).
296. R.H. Colby, L.J. Fetters, W.G. Funk, and W.W. Graessley, Macromolecules 24, 3873 (1991).
297. C.M. Roland, J.K. Kallitsis, and K.G. Gravalos, Macromolecules 26, 6474 (1993).
298. W.W. Graessley and S.F. Edwards, Polymer 22, 1329 (1981).
299. L.J. Fetters, D.J. Lohse, and W.W. Graessley, J. Polym. Sci. Polym. Phys. 37, 1023 (1999).
300. N. Heymans, Macromolecules 33, 4226 (2000).
301. L.J. Fetters, D.J. Lohse, C.A. Garcia-Francok, P. Brant, and D. Richter, Macromolecules 35,
10096 (2002).
302. D. Richter, B. Farago, R. Butera, L.J. Fetters, J.S. Juang, and B. Ewen, Macromolecules 26,
795 (1993).
303. F. Brochard and P.G. de Gennes, Macromolecules 10, 1157 (1977).
304. R.H. Colby, M. Rubinstein, and J.L. Viovy, Macromolecules 25, 996 (1992).
305. T.A. Kavassalis and J. Noolandi, Macromolecules 21, 2869 (1988); 22, 2709 (1989).
306. Y.H. Lin, Macromolecules 20, 3080 (1987).
307. R. Everaers, S.K. Sukumaran, G.S. Grest, C. Svaneborg, A. Sivasubramanian, and K. Kremer,
Science 303, 823 (2004).
Список литературы к главе 3 -*v- 731
Глава 4
1. Е. Guth and Н.М. James, Ind. Eng. Chem. 33, 624 (1941).
2. F.T. Wall, J. Chem. Phys. 10, 132 (1942).
3. P.J. Flory, J. Chem. Phys. 10, 51 (1942).
4. P.J. Flory and J. Rehner, J. Chem. Phys. 11, 512, 521 (1943).
5. L. R.G. Treloar, «The Physics of Rubber Elasticity» Clarendon Press, Oxford, 1975.
6. J.E. Mark and B. Erman, «Rubberlike Elasticity. A Molecular Primer» Wiley-Interscience,
New York, 1988.
7. B. Erman and J.E. Mark, «Structures and Properties of Rubberlike Networks» Oxford
University Press, New York, 1997.
8. W. Burchard and S.B. Ross-Murphy (Eds.), «Physical Networks: Polymers and GeLs»
Elsevier, London, 1990.
9. P.J. Flory, «Principles of Polymer Chemistry» Cornell Univ. Press, Ithaca, NY, 1953.
10. P.J. Flory, Macromolecules 15, 99 (1982).
11. W.W. Graessley, Adv. Polym. Sci. 47, 67 (1982).
12. P.J. Flory, Proc. R. Soc. London A 351, 351 (1976).
13. J.E. Mark, Br. Polym. J. 17, 144 (1985).
14. P.J. Flory, «Statistical Mechanics of Chain Molecules» Interscience, New York, 1969.
15. W.L. Mattice and U.W. Suter, «Conformational Theory of Large Molecules: The Rotational
Isomeric State Model in Macromolecular Systems» John Wiley & Sons, Inc., New York, 1994.
16. P.J. Flory and D.Y. Yoon, J. Chem. Phys. 63,5358 (1974); D.Y. Yoon and P.J. Flory, J. Chem.
Phys. 63, 5366 (1974).
17. B. Erman and J.E. Mark, J. Chem. Phys. 89, 3314 (1988).
18. R.W. Ogden, «Nonlinear Elastic Deformations» Dover Publications, New York, 1997.
19. R.T. Deam and S.F. Edwards, Philos. Trans. R. Soc. London Ser. A 280, 317 (1976).
20. R.C. Ball, M. Doi, S.F. Edwards, and M. Warner, Polymer 22, 1010 (1981).
21. S.F. Edwards and T. Vilgis, Polymer21, 483 (1986).
22. G. Ronca and G. Allegra, J. Chem. Phys. 63, 4990 (1975).
23. P.J. Flory, J. Chem. Phys. 66, 5720 (1977).
24. P.J. Flory and B. Erman, Macromolecules 16, 800 (1982).
25. A. Kloczkowski, J.E. Mark, and B. Erman, Macromolecules 28, 5089 (1995).
26. A. Kloczkowski, J.E. Mark, and B. Erman, Comput. Polym. Sci. 5, 37 (1995).
27. B. Erman and L. Monnerie, Macromolecules 22, 3342 (1989).
28. N.R. Langley, Macromolecules 1, 348 (1968).
29. N. Rennar and W. Oppermann, Coll. Polym. Sci. 270, 527 (1992).
30. B. Erman, W. Wagner, and P.J. Flory, Macromolecules 13, 1554 (1980).
31. J.E. Mark, Rubber Chem. Technol. 46, 593 (1973).
32. Y. Osada and A.R. Khokhlov (Eds.), «Polymer Gels and Networks» Marcel Dekker, New
York, 2002.
33. O.E. Philippova, Polymer Science Ser. C. 42, 208 (2000).
34. A.R. Khokhlov, in «Responsive Gels: Volume Transitions I» K. Dusek (Ed.), Springer Verlag,
Berlin, 1992, p. 125.
35. A.R. Khokhlov and O.E. Philippova, in «Solvents and Self-Organization of Polymers» S.E. Webber
et al. (Eds.), Kluwer Academic Publishers, NATO ASI Series, Vol. 327,1996, p. 197.
36. G. Nisato and S.J. Candau, in «Polymer Gels and Networks» Y. Osada and A.R. Khokhlov
(Eds.), Marcel Dekker, New York, 2002, p. 131.
37. A.R. Khokhlov and O.E. Philippova, in «Polymer Gels and Networks» Y. Osada and A.
R. Khokhlov (Eds.), Marcel Dekker, New York, 2002, p. 163.
38. P.J. Flory, Trans. Faraday Soc. 57, 829 (1961).
39. P.J. Flory, A. Ciferri, and C.A.J. Hoeve, J. Polym. Sci. 45, 235 (1960).
40. R. Ullman, Anna. Rev. Mater. Sci. 10, 261 (1980).
41. J.S. Higgins and H.C. Benoit, «Polymers and Neutron Scattering» (Oxford Series on Neutron
Scattering in Condensed Matter, 8), Oxford Univ. Press, Oxford, 1997.
42. A. Maconnachie and R.W. Richards, Polymer 19, 739 (1978).
43. C. Picot, in «Static Dynam: Properties Polym. Solid State» Reidel, Dordrecht, 1982, p. 127.
732 -*v- Литература
44. L.H. Sperling, Polym. Eng. Sci. 24, 1 (1984).
45. D.J. Lohse, Polym. News 12, 8 (1986).
46. M. Beltzung, C. Picot, P. Rempp, and J. Herz, Macromolecules 15, 1594 (1982).
47. J.A. Hinckley, C.C. Han, B. Moser, and H. Yu, Macromolecules 11, 836 (1978).
48. J. Bastide, R. Duplessix, and C. Picot, Macromolecules 17, 83 (1984).
49. J. Bastide and F. Boue, PhysicaA 104, 251 (1986).
50. J. Bastide, M. Buzier, and F. Boue, Polym. Motion Dense Sy st. 112 (1988).
51. J. Bastide, F. Boue, and M. Buzier, in «Molecular Basis of Polymer Networks» A.
Baumgartner and C. Picot (Eds.), Springer Verlag, Berlin, 1989, p. 48.
52. F. Boue, B. Farnoux, J. Bastide, A. Lapp, J. Herz, and C. Picot, Europhys. Lett. 1,637 (1986).
53. M. Beltzung, C. Picot, and J. Herz, Macromolecules 17, 663 (1984).
54. S.B. Clough, A. Maconnachie, and G. Allen, Macromolecules 13, 774 (1980).
55. H. Yu, T. Kitano, C.Y. Kim, E.J. Amis, T. Chang, M.R. Landry, J.A. Wesson, and C.C. Han, in
«Advances in Elastomers and Rubber Elasticity» J. Lal and J.E. Mark (Eds.), Plenum Press,
New York, 1986, p. 407.
56. F. Boue, J. Bastide, M. Buzier, A. Lapp, J. Herz, and T.A. Vilgis, Coll. Polym. Sci. 269, 195
(1991).
57. G. Allen, P.N. Brier, G. Goodyear, and J.S. Higgins, Faraday Symp. Chem. Soc. 6,169 (1972).
58. G. Allen, J.S. Higgins, and C.J. Wright, Faraday Symp. Chem. Soc. 7, 348 (1973).
59. G. Allen, M.J. Kirkham, J. Padget, C. Price, Trans. Faraday Soc. 67, 1278 (1971).
60. B. Ewon and D. Richter, Festkoerperprobleme 27, 1 (1987).
61. R. Oeser, B. Ewen, D. Richter, and B. Farago, Phys. Rev. Lett. 60, 1041 (1988).
62. S. Chu, Science 253, 861 (1991).
63. A. Janshoff, M. Neitzert, Y. Oberdorfer, and H. FYichs, Angew. Chem. Int. Ed. 39,3213 (2000).
64. H.L. Granzier and G.H. Pollack (Eds.), «Elastic Filaments of the Cell» Kluwer Academic,
New York, 2000.
65. T. Hugel and M. Seitz, Makromol. Rapid Commun. 22, 989 (2001).
66. T. Strick, J.F. Allemand, V. Croquette, and D. Benisimon, Phys. Today 53, 46 (2001).
67. W. Zhang and X. Zhang, Prog. Polym. Sci. 28, 1271 (2003).
68. G.W. Slater, Y. Gratton, M. Kenward, L. McCormick, and F. Tessier, Soft Mater. 1,365 (2003).
69. T.T. Perkins, D.E. Smith, and S. Chu, Science 264, 819 (1994).
70. T.T. Perkins, S.R. Quake, D.E. Smith, and S. Chu, Science 264, 822 (1994).
71. T.T. Perkins, D.E. Smith, R.G. Larson, and S. Chu, Science 268, 83 (1995).
72. C.M. Schroeder, H.P. Babcock, E. S.G. Shaqfeh, and S. Chu, Science 301, 1515 (2003).
73. R.B. Case, Y.P. Chang, S.B. Smith, J. Gore, N.R. Cozzarelli, and C. Bustamante, Science305,
222 (2004).
74. S. Cui, C. Liu, Z. Wang, X. Zhang, S. Strandman, and H. Tenhu, Macromolecules 37,946 (2004).
Глава 5
1. J.D. Ferry, «Viscoelastic Properties of Polymers» 3rd ed., Wiley, New York, 1980.
2. P.J. Flory, «Principle of Polymer Chemistry» Cornell University Press, Ithaca, NY, 1953.
3. L. R.G. Treloar, «The Physics of Rubber Elasticity» 3rd ed., Clarendon, Oxford, 1975.
4. J.E. Mark and B. Erman, «Rubberlike Elasticity: A Molecular Primer» Wiley-Interscience,
New York, 1988.
5. J.E. Mark et al., «Physical Properties of Polymers» 3rd ed., Cambridge Univ. Press,
Cambridge, 2004.
6. A.N. Gent, in «Science and Techno/ogy of Rubber» 2nd ed., Academic Press, New York, 2004,
Chap. 10.
7. D.J. Plazek and G. Berry, in «Glass: Science and Techno/ogy, Vol. 3, Viscosity and Relaxation»
D.R. Uhlmann and N.J. Kreidl (Eds.), Academic Press, New York, 1986, Chap. 7.
8. A.R. Payne and R.E. Whittaker, Rubber Chem. Technol. 340 (1971).
9. L. Mullin, J. Appl. Polym. Sci. 2, 257 (1959).
10. T.L. Smith, in «Treatise on Materials Science and Techno/ogy» Academic Press, New York, 1977.
11. I.-C. Choy and D.J. Plazek, J. Polym. Sci.: Part B: Polym. Phys. 24, 1303 (1986).
Список литературы к главе 4 -*и- 733
12. D.J. Plazek and I.-С. Choy, J. Polym. Sci.: Part B: Polym. Phys. 27, 307 (1989).
13. D.J. Plazek and Z.N. Frund, Jr., J. Polym. Sci.: Part B: Polym. Phys. 28, 431 (1990).
14. D.J. Plazek and In-Chul Chay, J. Polym. Sci.: Part B: Polym. Phys. 29, 17 (1991).
15. D.J. Plazek, I.-C. Choy, F.N. Kelley, E. von Meerwall, and L.-J. Su, Rubber Chem. Technol.
56, 866 (1983).
16. D.J. Plazek, G.-F. Gu, R.G. Stacer, L.-J. Su, E. von Meerwall, and F.N. Kelley,/. Mater. Sci.
23,1289(1988).
17. D.J. Plazek and M. Rosner, Rubber Chem. Technol. 71, 679 (1998).
18. K.L. Ngai, D.J. Plazek, and R.W. Rendell, Rheol. Acta 36, 307 (1997).
19. K.L. Ngai and D.J. Plazek, Rubber Chem. Technol. Rubber Reviews 68, 376 (1995).
20. D.J. Plazek and K.L. Ngai, in «Glass Transition Temperatures» in «Handbook of Polymer
Properties» J.E. Mark (Ed.), American Institute of Physics, College Park, MD, 1996, p. 139.
21. E. Riande, H. Markovitz, D.J. Plazek, and N. Raghupathi, J. Poly. Sci. Symp. 50, 405 (1995).
22. LL. Hopkins and R.W. Hamming, J. Appl. Phys. 28, 906 (1957).
23. L. C.E. Struik, «Physical Aging in Amorphous Polymers and Other Materials» Elsevier.
Amsterdam, 1978.
24. A.J. Kovacs, J.J. Aklonis, J.M. Hutchinson, and A.R. Ramos,/. Polym. Sci.:Polym. Phys. Ed.
17, 1097 (1979).
25. G.R. McKenna, in «Comprehensive Polymer Science, Vol. 2, Polymer Properties» C. Roolh
and C. Price (Eds.), Pergamon, Oxford, 1989, p. 311.
26. I.M. Hodge, Macromolecules 16, 898 (1983).
27. C.T. Moynihan et al. Ann. N.Y. Acad. Sci. 279, 15 (1976).
28. A.Q. Tool, /. Am. Cer. Soc. 29, 240 (1946).
29. H.M. James and E. Guth, /. Chem. Phys. 15, 669 (1947).
30. H.M. James and E. Guth, /. Chem. Phys. 21, 1039 (1953).
31. P.J. Flory, Proc. Roy. Soc. London A 351, 351 (1976).
32. P.J. Flory, Polymer 20,1317 (1979).
33. P.J. Flory and R. Erman, Macromolecules 15, 800 (1982).
34. M. Doi and S.F. Edwards, «The Theory of Polymer Dynamics» Clarendon Press, Oxford, 1986.
35. F.R. Schwarzl and A.J. Stavorman, /. Appl. Phys. 23, 838 (1952).
36. D.J. Plazek,/. Colloid Sci. 15, 40 (1960).
37. D.J. Plazek and V.M. O’Rourke, /. Polym. Sci., A-2 9, 209 (1971).
38. J.D. LeMay, R.J. Swetlin, and F.N. Kelley, in «Characterization of Highly Crosslinked Polymer»
ACS Series No. 243, American Chemical Society, Washington, D. C., 1984, p. 177.
39. G.C. Rerry and T.G. Fox, Adi?. Polym. Sci. 5, 261 (1968).
40. D.J. Plazek, /. Polym. Sci., A-2 4, 745 (1966).
41. P. Thirion and R. Chasset, in «Proceedings of the Conference on Physics of Non-Crystalline
Solids» North Holland, Amsterdam, 1965; Rev. Gen. Caoutchouc. 41, 271 (1964).
42. J.D. Ferry, R.G. Manke, E. Maekawa, Y. Oyanagi, and R.A. Dicke, /. Phys. Chem. 68, 3414
(1964).
43. H. Leaderman,/. Polym. Sci. 16, 261 (1955).
44. A.R. Payne, /. Appl. Polym. Sci. 3, 127 (1960).
45. H. Leaderman,/. Polym. Sci. 59, S42 (1962).
46. R.A. Dicke and J.D. Ferry, /. Phys. Chem. 70, 2594 (1966).
47. R.H. Valentine, J.D. Ferry, T. Homma, and K. Ninomiya, /. Polym. Sci.: Part A-2 6,479 (1968).
48. A.R. Payne and R.E. Whittaker, Rubber Chem. Technol. 44, 340 (1971).
49. J.F. Saunders and J.D. Ferry, Macromolecules 7, 681 (1974).
50. A.N. Gent, in «Science and Techno/ogy of Rubber» J.E. Mark and R. Erman (Eds.),
Academic Press, New York.
51. Y. Isono and J.D. Ferry, Rubber Chem. Technol. 57, 925 (1984).
52. K. Arai and J.D. Ferry, Rubber Chem. Technol. 59, 592 (1986).
53. K. Arai and J.D. Ferry, Rubber Chem. Technol. 59, 605 (1986).
54. G.J. Lake and A.G. Thomas, Proc. Roy. Soc., London 300, 108 (1967).
55. A.N. Gent and R.H. Tobias, /. Polym. Sci. Polym. Phys. Ed. 20, 2051 (1982).
56. T.L. Smith, /. Polym. Sci., A-2 1, 3597 (1963).
734 -*v- Литература
57. L. Mullins, Trans. Inst. Rubber Ind. 35, 213 (1959).
58. H.K. Mueller and W.G. Knauss, J. Appl. Meeh. 38, 483 (1971).
59. K.L. Ngai, D.J. Plazek, and S.S. Deo, Macromolecules 20, 3047 (1987).
60. K.L. Ngai, A. Sch nhals, and E. Schlosser, Macromolecules 25, 4915 (1992).
61. D.J. Plazek, C. Bero, S. Neumeister, G. Floudas, G. Fytas, and K.L. Ngai, J. Colloid Polymer
Sci. 272,1430 (1994).
62. R.W. Gray, G. Harrison, and J. Lamb, J. Polym. Sci. Polym. Phys. Ed. 14, 1361 (1976).
63. R.W. Gray, G. Harrison, and J. Lamb, Proc. R. Soc. London 356, 77 (1977).
64. J. Cochrane, G. Harrison, J. Lamb, and D.W. Phillips, Polymer 21, 837 (1980).
65. K.L. Ngai, A. Sch nhals, and E. Schlosser, Macromolecules 25, 4915 (1992).
66. A. Sch nhals and E. Schlosser, Physica Scripta T49A, 233 (1993).
67. D.J. Plazek, J. Rheology 40, 987 (1996).
68. D.J. Plazek, X.D. Zheng, and K.L. Ngai, Macromolecules 25, 4920 (1992).
69. K.L. Ngai, D.J. Plazek, and C. Bero, Macromolecules 26, 1065 (1993).
70. K.L. Ngai, Comments Solid State Phys. 9, 127 (1979).
71. K.L. Ngai, A.K. Rajagopal, and S. Teitler, J. Chem. Phys. 88, 5086 (1988).
72. K.L. Ngai and R.W. Rendell, in «Relaxation in Complex Systems and Related Topics» lan A. Campbell
and C. Giovannella (Eds.), NATO ASI Series, Vol. 222, Plenum, New York, 1990, p. 309.
73. K.L. Ngai, S.L. Peng, and K.Y. Tsang, Physica A 191, 523 (1992).
74. K.Y. Tsang and K.L. Ngai, Phys. Rev. E54, 3067 (1997).
75. K.L. Ngai and K.Y. Tsang, Phys. Rev. E60, 4511 (1999).
76. K.L. Ngai and R.W. Rendell, in «Supercooled Liquids, Advances and Novel Applications»
J.T. Fourkas, D. Kivelson, U. Mohanty, and K. Nelson (Eds.), ACS Symposium Series, Vol.
676, American Chemical Society, Washington, D. C., 1997, p. 45.
77. K.L. Ngai, IEEE Transactions in Dielectrics and Electrical Insulation 8, 329 (2001).
78. K. Schmidt-Rohr and H.W. Spiess, Phys. Rev. Lett. 66, 3020 (1991).
79. R. Kohlrausch, Pogg. Ann. Phys. 12, 393 (1847); Pogg. Ann. Phys. 91, 56, 179 (1854).
80. J. Colmenero, A. Arbe, G. Coddens, B. Frick, C. Mijangos, and H. Reinecke, Phys. Rev. Lett.
78,1928 (1977).
81. K.L. Ngai, in «Disorder Effects on Relaxational Properties» R. Richert and A. Blumen (Eds.),
Springer Verlag, Berlin, 1994, Chap. 3.
82. A. Kudlik, S. Benkhof, T. Blochowicz, C. Tschirwitz, and E. R ssler, J. Mol. Structure 479,
210 (1999).
83. R. Casalini, K.L. Ngai, C.G. Robertson, and C.M. Roland, J. Polym. Sci. Polym. Phys. Ed. 38,
1841 (2001).
84. C.M. Roland, M.J. Schroeder, J.J. Fontanella, and K.L. Ngai, Macromolecules 37, 2630 (2004).
85. G.P. Johari and M. Goldstein, J. Chem. Phys. 53, 2372 (1970).
86. G.P. Johari, Annals New York Acad. Sci. 279, 117 (1976).
87. K.L. Ngai and M. Paluch, J. Chem. Phys. 120, 2857 (2004).
88. K.L. Ngai, J. Phys.: Condens. Matter 15, SI 107 (2003).
89. K.L. Ngai and M. Paluch, J. Phys. Chem. В 107, 6865 (2003).
90. S. Correzi, M. Beiner, H. Huth, K. Schr ter, S. Capaccioli, R. Casalini, D. Fioretto, and
E. Donth, J. Chem. Phys. 117, 2435 (2002).
91. K.L. Ngai and S. Capaccioli, Phys. Rev. E69, 03150 (2004).
92. G.P. Johari, G. Power, and J.K. Vij, J. Chem. Phys. 116, 5908 (2002); G.P. Johari, G. Power,
and J.K. Vij, J. Chem. Phys. 117, 1714 (2002); G. Power, G.P. Johari, and J.K. Vij, J. Chem.
Phys. 119, 435 (2003).
93. M. Paluch, C.M. Roland, S. Pawlus, J. Ziolo, and K.L. Ngai, Phys. Rev. Lett. 91,115701 (2003).
94. J. Koplinger, G. Kasper, S. Hunklinger, J. Chem. Phys. 113, 4701 (2001).
95. A. Arbe, J. Colmenero, D. Richter, J. Gomez, and B. Farago, Phys. Rev. E. 60, 1103 (1999).
96. K.L. Ngai, D.J. Plazek, and I. Echeverria, Macromolecules 29, 7937 (1996).
97. K.L. Ngai, D.J. Plazek, and A.K. Rizos, J. Polym. Sci. Polym. Phys. 35, 599 (1997).
98. K.L. Ngai, D.J. Plazek, and V.M. O’Rourke, Macromolecules 30, 5450 (1997).
99. K.L. Ngai and D.J. Plazek, Macromolecules 35, 9136 (2002).
100. J.-F. Shi, L.C. Dickinson, W.J. MacKnight, and J.C.W. Chien, Macromolecules 26, 5908
(1993).
Список литературы к главе 5 -1\г 735
101. K.L. Ngai and C.M. Roland, Macromolecules 27, 2454 (1994).
102. B.E. Read, J. Non-Cryst. Solids 131, 408 (1991).
103. I. Alig and G.P. Johari, J. Polym. Sci. Part B: Polym. Phys. 31, 299 (1993).
104. M. Beiner and K.L. Ngai, to be published.
105. K.L. Ngai, C.M. Roland, and A.F. Yee, Rubber Chem.& Tech. 66, 817 (1993).
106. At the time of writing Reference 101, this value of tQ has not yet been determined directly by
experiment, and values about 10 times longer were used.
107. P.J. Flory, Rubber Chem. Technol. 52, 110 (1979).
108. P.J. Flory, Polym. J. 17, 1 (1985).
109. L. G.R. Treloar, Rep. Prog. Phys. 36, 755 (1973).
110. G. Ronca and G. Allegra, J. Chem. Phys. 63, 4990 (1975).
Глава 6
Al. S. Ahn and J.L. White, Int. Polym. Process. 18, 243 (2003); J. Appl. Polym. Sci. 90, 1555
(2003); Ibid. 91,651 (2004).
A2. R.D. Andrews, N. Hofman-Bang, and A.V. Tobolsky, J. Polym. Sci. 3, 669 (1948).
A3. R.D. Andrews and A.V. Tobolsky, J. Polym. Sci. 7, 221 (1952).
A4. D.J. Angier, R.J. Ceresa, and W.F. Watson, J. Polym. Sci. 34, 699 (1958).
A5. D.J. Angier, W.T. Chambers, and W.F. Watson, J. Polym. Sci. 25, 129 (1957).
A6. D.J. Angier, E. Farlie, and W.F. Watson, Trans. Inst. Rubber Ind. 34, 8 (1958).
A7. D.J. Angier and W.F. Watson, J. Polym. Sci. 20, 235 (1956).
A8. D.J. Angier and W.F. Watson, J. Polym. Sci. 25, 1 (1957).
A9. D.J. Angier and W.F. Watson, Trans. Inst. Rubber Ind. 33, 22 (1956).
A10. T. Araki and J.L. White, Polym. Eng. Sci. 38, 616 (1998).
All. G. Ardichvili, Kautschuk 14, 23 (1938).
A12. G. Ardichvili, Kautschuk 19, 41 (1938).
A13. T. Asai, T. Fukui, K. Inoue, and M. Kuriyama, Paper III-4 presented at International Rubber
Conference, Paris, June 1982.
A14. G. Ayrey, C.G. Moore, and W.F. Watson, J. Polym. Sci. 19, 1 (1956).
Bl. Th. Baader, Kautschuk 14, 223 (1938).
B2. Th. Baader, Kautsch. Gummi 3, 159 (1950).
B3. Th. Baader, Kautsch. Gummi 3, 205 (1950).
B4. Th. Baader, Kautsch. Gummi 3, 245 (1950).
B5. Th. Baader, Kautsch. Gummi 3, 279 (1950).
B6. Th. Baader, Kautsch. Gummi 3, 323 (1950).
B7. Th. Baader, Kautsch. Gummi 3, 361 (1950).
B8. E.B. Bagley, J. Appl. Phys. 28, 624 (1957).
B9. E.B. Bagley and A.M. Birks, J. Appl. Phys. 31, 556 (1960).
B10. T.F. Ballenger and J.L. White, J. Appl. Polym. Sci. 15, 1949 (1971).
Bll. R.L. Ballman, Rheol. Acta k, 137 (1965).
B12. F.H. Banbury, U.S. Patent 1,200,070 (1916).
B13. F.H. Banbury, U.S. Patent 1,227,522 (1917).
B14. B. Bernstein, E.A. Kearsley, and L.J. Zapas, Trans. Soc. Rheol. 7, 391 (1963).
B15. B. Bernstein, E.A. Kearsley, and L.J. Zapas, J. Res. Natl. Bur. Stand., Sect. В 68, 103
(1964).
B16. J.P. Berry, R.W. Sambrook, and J. Beasley, Plast. Rubber Process, p. 97 (1977).
B17. H. Bewley, English Patent 10,825 (1845).
B18. E.C. Bingham, J. Wash. Acad. Sci. 6, 177 (1916).
B19. E.C. Bingham, «Fluidity and Plasticity» McGraw-Hill, New York, 1922.
B20. R.B. Bird and P.J. Carreau, Chem. Eng. Sci. 23, 427 (1968).
B21. R.B. Bird, W.E. Stewart, and E.N. Lightfoot, «Transport Phenomena» Wiley, New York,
1960.
B22. D.C. Bogue, I EC Fundam. 5, 253 (1966).
B23. D.C. Bogue, T. Masuda, Y. Einaga, and S. Onogi, Polym. J. 1, 563 (1970).
736 -'V Литература
В24. D.C. Bogue and J.L. White, «Engineering Analysis of Non-Newtonian Fluids» NATO
Agardograph.
B25. W.B. Bolen and B.E. Colwell, SPEANTEC Tech. Pap. b, 1008 (1958).
B26. L. Boltzmann, Sitzungsber, Kaiserl. Akad. Wiss. Wien 70, 275 (1874).
B27. C. Booth, Polymer 471 (1963).
B28. H.H. Bowerman, E.A. Collins, and N. Nakajima, Rubber Chem. Technol. 47, 307 (1974).
B29. B.A. Brooman, English Patent 10,582 (1845).
B30. B. Brzoskowski, S. Montes, J.L. White, and N. Nakajima, J. Non-Newtonian Fluid Meeh. 31,
43 (1989).
B31. B. Brzoskowski and J.L. White, Int. Polym. Process. 5, 191 (1990).
B32. B. Brzoskowski and J.L. White, Int. Polym. Process. 5, 238 (1990).
B33. B. Brzoskowski, J.L. White, and B. Kalvani, Kunststoffe 80, 922 (1990).
B34. R. Brzoskowski, J.L. White, W. Szydlowski, N. Nakajima, and K. Min, Int. Polym. Process.
3,134 (1988).
B35. R. Brzoskowski, J.L. White, W. Szydlowski, F.C. Weissert, N. Nakajima, and K. Min, Rubber
Chem. Technol. 60, 945 (1987).
B36. R. Brzoskowski, J.L. White, F.C. Weissert, N. Nakajima, and K. Min, Rubber Chem. Technol.
59,634 (1986).
B37. W.F. Busse, Ind. Eng. Chem. 24, 140 (1932).
B38. W.F. Busse and R.N. Cunningham, Proc. Rubber Technol. Conf. London, p. 288 (1938).
B39. W.F. Busse and R. Longworth, Trans. Soc. Rheol. 6, 179 (1962).
Cl. J.F. Carley, R.S. Mallouk, and J.M. McKelvey, Ind. Eng. Chem. 45, 974 (1953).
C2. P.J. Carreau, Trans. Soc. Rheol. 16, 99 (1972).
C3. B. Caswell and R.I. Tanner, Polym. Eng. Sci. 18, 416 (1978).
C4. R.J. Ceresa, Polymer 1, 477 (1960).
C5. L.C. Cessna, U.S. Patent 3,651,187 (1972).
C6. E.M. Chaffee, U.S. Patent 16 (1836).
C7. E.M. Chaffee, U.S. Patent 1939 (1841).
C8. F.M. Chapman and S.L. Lee, Soc. Plast. Eng. J. 26, 37 (1970).
C9. I.J. Chen and D.C. Bogue, Trans. Soc. Rheol. 16, 59 (1972).
CIO. J.J. Cheng and I. Manas-Zloczower, Polym. Eng. Sci. 29, 701, 1059 (1989).
Cl 1. J.J. Cheng and I. Manas-Zloczower, J. Appl. Polym. Sci. Appl. Polym. Symp. 44, 53 (1989).
C12. J.W. Cho, J.L. White, and L. Pomini, Kautsch Gummi Tech 50, 496 (1997); J.W. Cho,
J.L. White, and L. Pomini, Kautsch Gummi Tech 50, 728 (1997).
C13. F.N. Cogswell, Plast. Polym. April, 109 (1968).
C14. B.D. Coleman, M.E. Gurtin, and I. Herrera, Arch. Rat. Meeh. Anal. 19, 1, 239, 266 (1965).
C15. B.D. Coleman and H. Markovitz, J. Appl. Phys. 35, 1 (1964).
Cl6. B.D. Coleman and W. Noll,Arc/i. Rat. Meeh. Anal. 3, 289 (1959).
C17. R.T. Cooke, British Patent 431,012 (1935).
Cl8. F.H. Cotton, Trans. Inst. Rubber Ind. 5, 487 (1931).
C19. G. Cotton and J.L. Thiele, Rubber Chem. Technol. 51, 749 (1978).
C20. W.P. Cox and E.H. Merz, J. Polym. Sci. 28, 619 (1958).
C21. W.O. Criminale, J.L. Erickson, and G.L. Filbey, Arch. Rat. Meeh. Anal. 1, 410 (1958).
C22. M. Crochet, Rubber Chem. Technol. 62, 426 (1989).
DI. B. Darrow, U.S. Patent 1,282,767 (1918).
D2. B. David, T. Sapir, A. Nir, and Z. Tadmor, Int. Polym. Process. 7, 204 (1992).
D3. B. David, A. Nir, and Z. Tadmor, Int. Polym. Process. 5, 155 (1990).
D4. G.E. Decker and F.L. Roth, India Rubber World 128, 399 (1953).
D5. J.S. Deng and A.I. Isayev, Rubber Chem. Technol. 64, 296 (1991).
D6. T. de Witt, J. Appl. Phys. 26, 889 (1955).
D7. J.H. Dillon, Physics (NY) 7, 73 (1936).
D8. J.H. Dillon and L.V. Cooper, Rubber Age (NY) p. 1306 (1937).
D9. J.H. Dillon and N. Johnston, Physics k, 225 (1933).
D10. H.J. Donald, U.S. Patent 3,279,501 (1966).
Dll. R.A. Duneli and A.V. Tobolsky. J. Chem. Phys. 17, 100(1949).
Список литературы к главе 6 -*Ъ- 737
El. S. Eccher and A. Valentinotti, Ind. Eng. Chem. 50, 829 (1958).
E2. E. R.G. Eckert and R.M. Drake, «Heat and Mass Transfer» McGraw-Hill, New York, 1959.
E3. J.E. Eilersen, U.S. Patent 3,099,859 (1963).
E4. R. Eisenschitz, B. Rabinowitsch, and K. Weissenberg, Mitt. Deutsch. Materialpruf. Son-derh,
9,9 (1929).
Fl. F. Farrel III, «Solid Men of the Place Gave Strong Roots» Newcomen Society, New York,
1956.
F2. P. Fay, U.S. Patent 2,569,373 (1951).
F3. J.D. Ferry, «Viscoelastic Properties of Polymers» Wiley, New York, 1969.
F4. V. Folt, Rubber Chem. Technol. 42,1294 (1969).
F5. T.G. Fox and P.J. Flory, J. Am. Chem. Soc. 70, 2384 (1949).
F6. T.G. Fox, S. Gratch, and E. Loeshaek, in «Rheo/ogy» Academic Press, New York, Vol. 1, 1956.
F7. L.C. Frazier, U.S. Patent 3,485,692 (1969).
F8. P.K. Freakley and N.Y. Wan Idris, Rubber Chem. Technol. 52, 134 (1979).
F9. H. Freundlich and A.D. Jones, J. Phys. Chem. 40, 1217 (1936).
F10. S.M. Freeman and K. Weissenburg, Nature 161, 324 (1948).
Fll. I. Furuta, V.M. Lobe, and J.L. White, J. Non-Newtonian Fluid Meeh. 1, 207 (1976).
Gl. T.L. Garner, India Rubber J. 78, (1929).
G2. R.E. Gaskell, J. Appl. Meeh. 73, 334 (1950).
G3. P. Geyer, U.S. Patent 3,375,549 (1968).
G4. L.A. Goettler and A.J. Lambright, U.S. Patent (filed Feb. 2, 1976) 4,056,591 (1977);
U.S. Patent (filed July 25,1975) 4,057,610 (1977).
G5. L.A. Goettler, A.J. Lambright, R.I. Leib, and P.J.D. Mauro, Rubber Chem. Technol. 54,
277 (1981).
G6. L.A. Goettler, R.I. Leib, and A.J. Lambright, Rubber Chem. Technol. 52, 838 (1979).
G7. H.J. Gohlisch, W. May, F. Ramm, and W. Ruger, Extrusion of Elastomers, in «Plastics
Extrusion Techno/ogy» F. Hensen (Ed.), Hanser, Munich, 1988.
G8. C. Goldstein, Trans. Soc. Rheol. 18, 357 (1974).
G9. S. Goldstein (Ed.), «Modern Developments in Fluid Dynamics» Clarendon Press, Oxford,
1938.
G10. C.F. Goodeve, Trans. Faraday Soc. 35, 342 (1939).
Gil. C.F. Goodeve and G.W. Whitfield, Trans. Faraday Soc. 34, 511 (1938).
G12. C. Goodyear, U.S. Patent 3633 (1844).
G13. C. Goodyear, U.S. Patent 5536 (1848).
G14. C. Goodyear, «Gum Elastic and Its Varieties with a Detailed Account of Its Applications and
Uses and the Discovery of Vulcanization» Privately printed, New Haven, CT, 1855.
G15. M. Gray, British Patent 5056 (1879).
G16. A.E. Green and R.S. Rivlin, Arch. Rat. Meeh. Anal, 1, 1 (1957).
G17. F.M. Griffith. Ind. Eng. Chem. Fundam. 1, 180 (1962).
G18. R.W. Griffiths, Trans. Inst. Rubber Ind. 1, 308 (1926).
G19. F.F. Grosevnor, U.S. Patent (filed Aug. 17, 1916) 1,254,685 (1918).
G20. J.T. Gruver and G. Kraus, J. Polym. Sci. A. 2, 797 (1964).
G21. H.G. Gurhin and W. Spreutels, Kautsch. Gummi Kunstst. 43, 431 (1990).
Hl. H. Hagen, Kautschuk 15, 88 (1939).
H2. C.D. Han and L.H. Drexler, J. Appl. Polym. Sci. 17, 2329 (1973).
H3. M.H. Han, J.L White, N. Nakajima, and R. Brzoskowski, Kautsch. Gummi Kunstst. 43,1060 (1990).
H4. C. Hancock, English Patent 11,147 (1846).
H5. C. Hancock, English Patent 11,208 (1847).
H6. C. Hancock, English Patent 11,575 (1847).
H7. C. Hancock, English Patent 11,874 (1847).
H8. T. Hancock, English Patent 4768 (1823).
H9. T. Hancock, English Patent 7344 (1837).
НЮ. T. Hancock, English Patent 9952 (1843).
ПН. T. Hancock, «Personal Narrative of the Origin and Progress of the Caoutchouc or India Rubber
Manufacture in England» Longman, Brown, Green, Longmans & Roberts, London, 1857.
738 “*V Литература
Hl 2. E.G. Harms, Eur. Rubber J. 6, 23 (1978); Kunststoffe 74, 33 (1984).
H13. E.G. Harms, G. Menges, and R. Hegele, German Offenlegungsshift 2,235,784 (1974);
U.S. Patent 4,178,104 (1979); U.S. Patent 4,199,263 (1980).
H14. E.R. Harrel, J. P, Porter, and N. Nakajima, Rubber Chem. Technol. 64, 254 (1991).
H15. H. Herrmann, «Schneckenmaschinen in der Verfahrenstechnik» Springer, Berlin, 1972.
H16. H. Herrmann, in «Kunststoffeein Werkstoff macht Karriere» W. Glenz (Ed.), Hanser, Munich,
1985.
H17. W.H. Herschel and R.S. Bulkley, Proc. ASTM26, 62 (1926).
H18. J. Hoekstra, Physics k, 295 (1933); Kautschuk 9, 750 (1933).
H19. K. Hohenemser and W. Prager, Z. AMM12, 216 (1932).
H20. B. Hu and J.L. White, Int. Polym. Process. 8, 18 (1993).
H21. B. Hu and J.L. White, Rubber Chem. Technol. 66, 257 (1993).
H22. B. Hu and J.L. White, Kautsch Gummi Kunstst 49, 285 (1996).
II. Y. Ide and J.L. White, J. Лрр/. Polym. Sci. 22, 1061 (1978).
12. A.I. Isayev, Injection Molding of Rubber Compounds, in «Injection and Compression Molding
Fundamentals» A.I. Isayev (Ed.), Dekker, New York, 1987.
13. A.I. Isayev, Injection Molding of Rubbers, in «Comprehensive Polymer Science» G. Allen,
J.C. Bevington, and S.L. Aggarwal (Eds.), Pergamon Press, Oxford, Vol. 7, 1989.
14. A.I. Isayev and Y.H. Huang, Adv. Polym. Technol. 9, 167 (1988).
15. A.I. Isayev and X. Fan, J. Rheol. 34, 35 (1990).
16. A.I. Isayev, M. Sobhanie, and J.S. Deng, Rubber Chem. Technol. 61, 906 (1988).
JI. M.W. Johnson and D. Segalman, J. Non-Newtonian Fluid Meeh. 2, 255 (1977).
KI. M.R. Kamal and S. Kenig, Polym. Eng. Sci. 12, 294 (1972).
K2. E. Karrer, Ind. Eng. Chem. Anal. Ed. 1, 158 (1929); Ind. Eng. Chem. 21, 770 (1929).
КЗ. E. Karrer, J.M. Davies, and E.O. Dieterich, Ind. Eng. Chem., Anal. Ed. 2, 96 (1930).
K4. D.H. Killeffer, «Banbury, The Master Mixer» Palmerton, New York, 1962.
K5. J.K. Kim, International Seminar on Elastomers, J. Appl. Polym. Sci., Appl. Polym. Symp. 50,
145 (1992).
K6. J.K. Kim and J.L. White, Nihon Reoroji Gakkaishi 17, 203 (1989).
K7. J.K. Kim and J.L. White, Int. Polym. Process. 6, 103 (1991).
K8. J.K. Kim, J.L. White, K. Min, and W. Szydlowski, Int. Polym. Process, k, 9 (1989).
K8a. K.J. Kim and J.L. White, Polym. Eng. Sci. 39, 2189 (1999).
K8b. P.S. Kim and J.L. White, Rubber Chem. Technol. 67, 871 (1994).
K9. P.S. Kim and J.L. White, Kautsch Gummi Kunstst 49, 10 (1996).
K10. P.S. Kim and J.L. White, Rubber Chem. Technol. 69, 686 (1996).
Kll. R.G. King, Rheol. Acta 5, 35 (1966).
K12. E. Konrad, Angew. Chem. 62,491 (1950).
K13. R. Koopmann, Kautsch. Gummi Kunstst. 36, 108 (1983).
K14. R. Koopmann, Polym. Testing 5, 341 (1985).
K15. R. Koopmann, Kautsch. Gummi Kunstst. 38, 281 (1985).
K16. R. Koopmann and H. Kramer, J. Testing Eval. ASTM 12, 407 (1984).
K17. R. Koopmann and J. Schnetger, Kautsch. Gummi Kunstst. 39, 131 (1986).
K18. G. Kraus and J.T. Gruver, J. Polym. Sci. A 3, 105 (1965).
K19. C. Koolihiran and J.L. White, J. Appl. Polym. Sci. 78, 1551 (2000).
K20. K. Kubota, R. Brzoskowski, J.L. White, F.C. Weissert, N. Nakajima, and K. Min, Rubber
Chem. Technol. 60, 924 (1987).
K21. T. Kumazawa, R. Brzoskowski, and J.L. White, Kautsch. Gummi Kunstst. 43, 688 (1990).
LI. H. Lamb, «Hydrodynamics» 6th ed., Cambridge University Press, London, 1932.
L2. A. Lasch and K. Frei, German Patent 738,787 (1943).
L3. A. Lasch and E. Stromer, German Patent 641,685 (1937).
L4. H. Leaderman, Ind. Eng. Chem. 35, 374 (1943).
L5. H. Leaderman, «Elastic and Creep Properties of Filamentous Materials and Other High
Polymers» Textile Found., Washington, D. C., 1943.
L6. L.J. Lee, J.F. Stevenson, and R.M. Griffith, U.S. Patent 4,425,289 (1984).
L7. P.W. Lehman, U.S. Patent 2,096,362 (1937).
Список литературы к главе 6 739
L8. J.P. Lehnen, Kunststofftechnik 9, 3, 90, 114, 198, 352 (1970).
L9. A.I. Leonov, Rheol. Acta 15, 85 (1976).
LIO. A.I. Leonov, J. Rheol. 34, 155 (1990).
Lil. A.I. Leonov, J. Non-Newtonian Fluid Meeh. 42, 343 (1990).
L12. G.H. Lewis, U.S. Patent 1,252,821 (1918).
L13. L.L. Li and J.L. White, Rubber Chem. Technol. 69, 628 (1996).
L14. P.W. Litchfield, «Industrial Voyage» Doubleday, Garden City, NY, 1954.
L15. V.M. Lobe and J.L. White, Polym. Eng. Sci. 19,617 (1979).
L16. A. Lodge, Trans. Faraday Soc. 52, 120 (1956).
Ml. C.Y. Ma, J.L. White, F.C. Weissert, A.I. Isayev, N. Nakajima, and K. Min, Rubber Chem.
Technol. 58,815 (1985).
М2. C.Y. Ma, J.L. White, F.C. Weissert, and K. Min, Polym. Composites 6, 215 (1985).
М3. C.Y. Ma, J.L. White, F.C. Weissert, and K. Min, J. Non-Newtonian Fluid Meeh. 17, 275 (1985).
M4. C. Macintosh, English Patent 4804 (1823).
M5. H. Mahn, H. Orth, K.H. Roitzsch, and W. Woeckener, in «Kunststoffe ein Werkstoffe macht
Karriere» W. Glenz (Ed.), Hanser, Munich, 1985.
M6. C. Maillefer, Swiss Patent 363,149 (1962).
M7. II. Mark and A.V. Tobolsky, «Physical Chemistry of High Polymeric Systems» 2nd ed..
Interscience, New York, 1950.
M8. B. Marzetti, India Rubber World 68, 776 (1923).
M9. B. Marzetti, Chim. Ind. Appl. 5, 342 (1923).
M10. B. Marzetti, Atti Della Reale Acad. Naz. Lincei 32, 399 (1923) ; Rubber Age 19, 454 (1924).
MH. B. Marzetti, Rubber Age 20, 139 (1925).
M12. T. Masuda, K. Kitagawa, T. Inoue, and S. Onogi, Macromolecules 116 (1970).
M13. T. Masuda, Y. Ohta, and S. Onogi, Macromolecules k, 763 (1971).
M14. J.C. Maxwell, Philos. Trans. R. Soc. 157, 249 (1866).
M15. J.M. McKelvey, Ind. Eng. Chem. 45, 982 (1953).
M16. F.M. McLaughlin, U.S. Patent 2,325,001 (1943).
Ml7. J. Meissner, Rheol. Acta 8, 78 (1969).
M18. G. Menges and E.G. Harms, Kautsch. Gummi Kunstst. 25, 469 (1972); 27, 187 (1974).
M19. G. Menges and J.P. Lehnen, Plastverarbeiter 20, 31 (1969).
M20. W. Meskat, Kunststoffe 41, 417 (1951).
M21. W. Meskat, Kunststoffe 45, 87 (1955).
M22. A.P. Metzger and J.R. Knox, Trans. Soc. Rheol. 9, 13 (1965).
M23. A.B. Metzner, J.L. White, and M.M. Denn, AIChEJ. 12, 863 (1966); Chem. Eng. Prog. 62,12,
(1966).
M24. S. Middleman, Trans. Soc. Rheol. 13, 125 (1969).
M25. W.H. Miller, U.S. Patent 3,069,853 (1991).
M26. W.H. Miller, C.C. Lee, and J.F. Stevenson, Int. Polym. Process. 6, 253 (1991).
M27. K. Min and J.L. White, Rubber Chem. Technol. 58, 1024 (1985).
M28. K. Min and J.L. White, Rubber Chem. Technol. 60, 361 (1987).
M29. N. Minagawa and J.L. White, J. Appl. Polym. Sci. 20, 501 (1976).
M30. W.W. Minoshima, J.L. White, and J.E. Spruiell, Polym. Eng. Sci. 20, (1980).
M31. S.R. Moghe, Rubber Chem. Technol. 39, 247 (1976).
M32. W.D. Mohr and R. S Mallouk, Ind. Eng. Chem. 51, 765 (1959).
M33. S. Montes and J.L. White, Rubber Chem. Technol. 55, 1354 (1982).
M34. S. Montes and J.L. White, Kautsch. Gummi Kunstst. kA, 731 (1991).
M35. S. Montes and J.L. White, Kautsch. Gummi Kunstst. kk, 937 (1991).
M36. S. Montes and J.L. White, J. Non-Newtonian Fluid Meeh., 49, 277 (1993).
M37. S. Montes, J.L. White, and N. Nakajima, J. Non-Newtonian Fluid Meeh. 28, 183 (1988).
M38. S. Montes, J.L. White, N. Nakajima, F.C. Weissert, and K. Min, Rubber Chem. Technol. 61,
698(1988).
M39. M. Mooney, J. Rheol. 2, 210 (1931).
M40. M. Mooney, Ind. Eng. Chem., Anal. Ed. 6, 147 (1934).
M41. M. Mooney, Physics 7, 413 (1936).
740 -* V Литература
М42. М. Mooney,/. ColloidSci. 2, 69 (1947).
M43. M. Mooney, personal communication (ca. 1965); unpublished U.S. Rubber Company report
(1953).
M44. M. Mooney, in «RheoZogy» F.R. Eirich (Ed.), Academic Press, New York, Vol. 2, 1958.
M45. M. Mooney, Proc. Int. Rubber Conf. (Washington), p. 368 (1959).
M46. M. Mooney, Rubber Chem. Technol. 35, xxvii (1962).
M47. M. Mooney and S.A. Black, J: Colloid Sci. 7, 204 (1952).
M48. A. Morikawa, K. Min, and J.L. White, Adv. Polym. Technol. 8, 383 (1988).
M49. A. Morikawa, J.L. White, and K. Min, Kautsch. Gummi Kunstst. 41, 1226 (1988).
M50. A. Morikawa, K. Min, J.L. White, Int. Polym. Process 4, 23 (1989).
M51. L. Mullins, J. Phys. Colloid Chem. 54, 239 (1950).
M52. L. Mullins and R.W. Whorlow, Trans. Inst. Rubber Ind. 27, 55 (1951).
№1. N. Nakajima, Rubber Chem. Technol. 54, 266 (1981); 55, 937 (1982).
№2. N. Nakajima, H.H. Bowerman, and E.A. Collins, J. Appl. Polym. Sci. 21, 3063 (1977).
№3. N. Nakajima and E.R. Harrel, Rubber Chem. Technol. 52, 962 (1979).
№4. K. Ninomiya, J. Colloid Sci. 14, 49 (1959).
№5. W. Noll, J. Rat. Meeh. Anal, k, 3 (1955).
№6. W. Noll, Arch. Rat. Meeh. Anal. 2, 197 (1958).
№7. R.H. Norman, Plast. Rubber Int. 5, 243 (1985).
01. K. Oda, J.L. White, and E.S. Clark, Polym. Eng. Sci. 16, 585 (1976).
02. K. Oda, J.L. White, and E.S. Clark, Polym. Eng. Sci. 18, 25 (1978).
03. J.G. Oldroyd, Proc. Cambridge Philos. Soc. 43, 100 (1947).
04. J.G. Oldroyd, Proc. Cambridge Philos. Soc. 43, 382 (1947).
05. J.G. Oldroyd, Proc. Cambridge Philos. Soc. 45, 595 (1949).
06. J.G. Oldroyd, Proc. R. Soc. A 200, 523 (1950).
07. J.G. Oldroyd, Q.J. Meeh. Appl. Math b, 271 (1951).
08. J.G. Oldroyd, Proc. R. Soc. A 245, 278 (1958).
09. S. Onogi,T. Masuda, I. Shiga, and F. Costaschuk, U. S.-Japan Seminar on Polymer Processing
and RheoZogy, Appl. Polym. Symp. 20, 37 (1973).
010. G. Osanaiye, A. I. Leonov, and J.L. White, J. Non-Newtonian Fluid Meeh., 49, 87 (1993).
Pl. G. Passoni, U.S. Patent 4,775,240 (1988).
P2. J. R.A. Pearson, Trans. J. Plast. Inst. 30, 230 (1962).
P3. J. R.A. Pearson, Trans. J. Plast. Inst. 31, 125 (1963).
P4. J. R.A. Pearson, Trans. J. Plast. Inst. 32, 239 (1964).
P5. J. R.A. Pearson, «Mechanical Principles of Polymer Melt Processing» Pergamon Press, New
York, 1966.
P6. J. R.A. Pearson and C.J.S. Petrie, Plast. Polym. 38, 85 (1970); J. Fluid Meeh. 40, 1 (1970);
J Fluid Meeh. 42, 609 (1970).
P7. N. Phan Tien, J. Non-Newtonian Fluid Meeh. 22, 259 (1978).
P8. N. Phan Tien and R.I. Tanner, J. Non-Newtonian Fluid Meeh. 2, 353 (1977).
P9. W.R. Phillips, U.S. Patent 3,195,183 (1965).
PIO. W.T. Pigott, Trans. ASME 73, 947 (1951).
PH. M. Pike and W.F. Watson, J. Polym. Sci. 9, 229 (1952).
P12. G.H. Piper and J.R. Scott, J. Sci. Inst. 22, 206 (1945).
P13. R.S. Porter and J.F. Johnson, Proc. 4th Int. Rheol. Cong. 2, 467 (1965).
P14. R.S. Porter and J.F. Johnson, Chem. Rev. 66, 1 (1966).
P15. W. Prager, «Mechanics of Continue» Ginn, Boston, 1961.
P16. W. Prager, in Appendix to the article by J.T. Bergen in «Processing of Thermoplastic
Materials» E.C. Bernhardt (Ed.), Reinhold, New York, 1959.
Pl7. W. Prager and P. Hodge, «Theory of Perfectly Plastic Solids» Wiley, New York, 1951.
Rl. V.C. Ratliff, U.S. Patent 2,720,679 (1955).
R2. M. Reiner, «Deformation and Flow» Lewis, London, 1948, and later editions.
R3. M. Reiner, «Lectures on Theoretical RheoZogy» North Holland, Amsterdam, three editions
(1943), (1949), (1960).
R4. M. Reiner, Phys. Today 17, 62 (1964).
R5. O. Reynolds, Philos. Trans. R. Soc. London, Ser. A 174, 935 (1883).
Список литературы к главе 6 741
R6. О. Reynolds, Philos. Trans. R. Soc. London, Ser. A 177, 157 (1886).
R7. J.G. Richardson, U.S. Patent, 3,761,553 (1973).
R8. S. Richardson, J. Fluid Meeh. 56, 609 (1972).
R9. R.S. Rivlin, J. Rat. Meeh. Anal. 5, 179 (1956).
RIO. R.S. Rivlin and J.L. Ericksen, J. Rat. Meeh. Anal. 4, 323 (1955).
SI. N. Sato, M. Miyaoka, S. Yamasaki, K. Inoue, A. Koriyama, T. Fukui, T. Asai, K. Nakagawa,
and T. Masaki, U.S. Patent 4,284,358 (1981); U.S. Patent 4,300,838 (1981).
S2. J.R. Schaefgen and P.J. Flory, J. Am. Chem. Soc. 70, 2709 (1948).
S3. G.H. Schanz, U.S. Patent 2,807,833 (1957).
S4. G. Schenkel, «Kunststoffe Extruder-Technik» Hanser, Munich, 1963.
S5. J.R. Scott, Trans. Inst. Rubber Ind. 7, 169 (1931).
S6. J.R. Scott, Trans. Inst. Rubber Ind. 10, 481 (1935).
S7. G. Schramm, Kaustch. Gummi Kunstst. 40, 756 (1987).
S8. T. Schwedoff, J. Deo Phys. 9, 34 (1890); Phys. Z. 1, 552 (1900).
S9. F.A. Seiberling and C.A. Carlton, U.S. Patent 2,032,508 (1936).
S10. Y.T. Shah and J.R.A. Pearson, Chem. Eng. Sci. 29, 1485 (1974).
SI 1. K.C. Shin and J.L. White, Rubber Chem. Technol. 66, 121 (1993).
S12. КС. Shin, J.L. White, R. Brzoskowski, and N. Nakajima, Kautsch. CummiKunstst. 43,181 (1990).
S13. A. H.P. Skelland, «Non-Newtonian Fluid and Heat Transfer» Wiley, New York, 1967.
S14. R.W. Snyder and J.I. Haase, U.S. Patent 1,952,469 (1934).
S15. M. Sobhanie, J.S. Deng, and A.I. Isayev; J. Appl. Polym. Sci., Appl. Polym. Symp. kb, 115
(1989).
S16. M. Sobhanie and A.I. Isayev, Rubber Chem. Technol. 62, 939 (1989).
S17. L.E. Soderquist, U.S. Patent 2,296,800 (1942).
S18. HJ. Song, J.L. White, K. Min, N. Nakajima, and F.C. Weissert, A/гл Polym. Technol. 8,43 (1988).
S19. J.F. Stevenson, AIChEJ. 18, 540 (1972).
S20. J.F. Stevenson, Extrusion of Rubber and Plastics, in «Comprehensive Polymer Science»
G. Allen, J.C. Bevington, and S.L. Aggarwal (Eds.), Pergamon Press, Oxford, Vol. 7, 1989.
S21. J.F. Stevenson and W.H. Miller, U.S. Patent 5,067,885 (1991).
S22. G.G. Stokes, Trans. Cambridge Philos. Soc. 8, 287 (1845).
S23. G.G. Stokes, «Report on Recent Researches in Hydrodynamics» Report of the British
Association (1846).
S24. G.G. Stokes, Trans. Cambridge Philos. Soc. 9, (1851).
S25. Y. Suetsugu and J.L. White, J. Appl. Polym. Sci. 28, 1481 (1983).
S26. Y. Suetsugu and J.L. White, J. Non-Newtonian Fluid Meeh. 14, 121 (1984).
Tl. H. Tanaka and J.L. White, iPolym. Eng. Sci. 20, 949 (1980).
T2. R.I. Tanner, U. S.-Japan Seminar on Polymer Processing and RheoZogy, Appl. Polym. Symp.
20, 201 (1973).
T3. R.H. Taylor, India Rubber World 112, 582 (1945).
T4. R.H. Taylor, J.H. Fielding, and M. Mooney, «Symposium on Rubber Testing» ASTM, New
York, 1947, p. 36.
T5. A.V. Tobolsky, «Properties and Structure of Polymers» Wiley, New York, 1960.
T6. M. Toh, T. Gondoh, T. Mon, S. Haren, and Y. Murakami, International Seminar on Elastomers,
J. Appl. Polym. Sci., Appl. Polym. Symp. 50, 133 (1992).
T7. S. Toki and J. L White, J. Appl. Polym. Sci. 27, 3171 (1982).
T8. N. Tokita, personal communication (1975).
T9. N. Tokita and J.L. White, J. Appl. Polym. Sci. 10,1011 (1966).
T10. C. Truesdell and R.A. Toupin, The Classical Field Theories, in «Handbuch der Physik» Vol.
III/l, Springer, Berlin, 1960.
Til. L.G. Turk, U.S. Patent 3,782,871 (1974).
T12. D.M. Turner and M.D. Moore, Plastic Rubber Proc., (Sept./Dec.), 81 (1980).
T13. D. Tyson and L. Comper, U.S. Patent 3,230,581 (1966).
V I. A. van Rossem and H. van der Meijden, Kautschuk 3,369 (1927); Rubber Age (NY} 23,443 (1928).
V 2. B. Vergnes, N. Bennani, and C. Guichard, Int. Polym. Process. 1, 19 (1986).
V 3. G.R. Vila, Ind. Eng. Chem. 36, 1113 (1944).
V 4. G.V. Vinogradov, Pure Appl. Chem. 39, 115 (1974).
742 Литература
V 5. G.V. Vinogradov, V.D. Fikham, R.D. Radushkevich, and A. Ya Malkin, J. Polym. Sci. A-2 8,
657 (1970).
V 6. G.V. Vinogradov and A.Y. Malkin, J. Polym. Sci. A-2 4, 135 (1966).
V 7. G.V. Vinogradov and A.Y. Malkin, «RheoZogy of Polymers» Mir, Moscow, 1980.
V 8. G.V. Vinogradov, A.Y. Malkin, E.P. Plotnikova, O.Y. Sabasi, and N.E. Nikolayava, Int.
J. Polym. Mater. 2, 1 (1972).
Wl. M.H. Wagner, Rheol. Acta 15, 136 (1976).
W2. W.F. Watson, Trans. Inst. Rubber Ind. 34, 237 (1958).
W3. K. Weissenberg, Nature 159, 310 (1947).
W4. G.S. Whitby (Ed.), «Synthetic Rubber» Wiley, New York, 1954.
W5. J.L. White, J. Appl. Polym. Sci. 8, 1129 (1964).
W6. J.L. White, J. Appl. Polym. Sci. 8, 2339 (1964).
W7. J.L. White,/. Inst. Rubber Ind. (Rubber Ind.) 8, 148 (1974).
W8. J.L. White, Polym. Eng. Sci. 15, 44 (1975).
W9. J.L. White, JNon-Newtonian Fluid Meeh. 5, 177 (1979).
W10.J.L. White, International Seminar on Elastomers, J. Appl. Polym. Sci., Appl. Polym. Symp.
50, 109 (1992).
Wll.J.L. White, Int. Polym. Process. 7, 2 (1992).
W12.J.L. White, Int. Polym. Process. 7,110 (1992).
W13.J.L. White, Rubber Chem. Technol. 65, 527 (1992).
W14.J.L. White, Int. Polym. Process. 7, 290 (1992).
W15.J.L. White, Int. Polym. Process. 8, 2 (1993).
W16.J.L. White, «Rubber Processing: TechnoZogy, Materials, and Principles» Epic Press, Hanser,
Munich, 1995.
W17.J.L. White and J.W. Crowder, J. Appl. Polym. Sci. 18, 1013 (1974).
W18.J.L. White, M.H. Han, N. Nakajima, and R. Brzoskowski, J. Rheol. 35, 167 (1991).
W19.J.L. White and J.K. Kim, J. Appl. Polym. Sci., Appl. Polym. Symp. 49, 59 (1989).
W20.J.L. White and A. Kondo, J. Non-Newtonian Fluid Meeh. 3, 41 (1977).
W21.J.L. White and Y. Lobe, Rheol. Acta 21,167 (1982).
W22.J.L. White and A.B. Metzner, J. Appl. Polym. Sci. 7,1867 (1963).
W23.J.L. White and H. Tanaka, J. Non-Newtonian Fluid Meeh. 8, 1 (1981).
W24.J.L. White and N. Tokita, J. Appl. Polym. Sci. 9, 1921 (1965).
W25.J.L. White and N. Tokita, J. Appl. Polym. Sci. 11,321 (1967).
W26.J.L. White and N. Tokita, J. Appl. Soc. Japan 22, 719 (1967); 24, 436 (1968).
W27.J.L. White and N. Tokita, J. Appl. Polym. Sci. 12, 1589 (1968).
W28.J.I. White, Y. Wang, A.I. Isayev, N. Nakajima, F.C. Weissert, and K. Min, Rubber Chem.
Technol. 60, 337 (1987).
W29.I. Williams, Ind. Eng. Chem. 10, 324 (1923).
W30.M.L. Williams, R.F. Landel, and J.D. Ferry, J. Am. Chem. Soc. 77, 3701 (1955).
W31.R.F. Wolf, India Rubber World, Aug. 1, p. 39 (1936).
Y l. Y. Yabushita, R. Brzoskowski, J.L. White, and N. Nakajima, Int. Polym. Process. 6, 219 (1989).
Y 2. M. Yamamoto, J. Phys. Soc. Japan 11, 413 (1956).
Y 3. H. Yamane and J.L. White, Polym. Eng. Rev. 2, 167 (1982).
Y 4. H.H. Yang and I. Manas-Zloczower, Int. Polym. Process. 7, 195 (1992).
Zl. N.V. Zakharenko, F.S. Tolstukhina, and G.M. Bartenev, Rubber Chem. Technol. 35, 326
(1962).
Z2. H. Zamodits and J.R.A. Pearson, Trans. Soc. Rheol. 13, 357 (1969).
Z3. L.J. Zapas, J. Res. Natl. Bur. Stand., Sect. A 70, 525 (1966).
Z4. L.J. Zapas and T. Craft, J. Res. Natl. Bur. Stand., Sect. A 69, 541 (1965).
Z5. S. Zaremaha, Bull Acad. Sci. Cracow, June, p. 594 (1903).
Глава 7
1. P.J. Flory, «Principles of Polymer Chemistry» Cornell Univ. Press, Ithaca, NY, 1953,
Chap. 11.
Список литературы к главе 6 -*и- 743
2. L. Bateman, C.G. Moore, М. Porter, and В. Saville, in «The Chemistry and Physics of Rubber-
Like Substances» L. Bateman (Ed.), John Wiley & Sons, Inc., New York, 1963, Chap. 19.
3. W. Hofmann, «Vulcanization and Vulcanizing Agents» Maclaren and Sons Ltd., London,
1967.
4. A.Y. Coran, in «Science and Techno/ogy of Rubber» F.R. Eirich (Ed.), Academic Press, New
York, 1978, Chap. 7.
5. N.J. Morrison and M. Porter, Rubber Chem. Technol. 57, 63 (1984).
6. G.E. Decker, R.W. Wise, and D. Guerry, Rubber Chem. Technol. 36, 451 (1963); A.I. Juve,
P.W. Karper, L.O. Schroyer, and A.G. Veith, Rubber Chem. Technol. 37, 434 (1964), and
references therein.
7. E.H. Farmer and F.W. Shipley, J. Polym. Sci. 1, 293 (1946).
8. E.H. Farmer, J. Chem. Soc. p. 1519 (1947).
9. E.H. Farmer, J. Soc. Chem. Ind. 66, 86 (1947).
10. L. Bateman, C.G. Moore, and M. Porter, J. Chem. Soc. p. 2866 (1958).
11. G. Oenslager, Ind. Eng. Chem. 23, 232 (1933).
12. M. Weiss, U.S. Patent 1,411,231 (1922).
13. S. Malony, U.S. Patent 1,343,222 (1920).
14. C. Bedford, U.S. Patent 1,371,922-4 (1921).
15. L. Sebrell and C. Bedford, U.S. Patent 1,522,687 (1925).
16. G. Bruni and E. Romani, India Rubber J. 62,63 (1921).
17. E. Zaucker, M. Bogemann, and L. Orthner, U.S. Patent 1,942,790 (1934).
18. M.W. Harmon, U.S. Patent 2,100,692 (1937).
19. A.Y. Coran and J.E. Kerwood, U.S. Patent 3,546,185 (1970).
20. R.H. Campbell and R.W. Wise, Rubber Chem. Technol. 37, 635 (1964).
21. R.H. Campbell and R.W. Wise, Rubber Chem. Technol. 37, 650 (1964).
22. P.L. Hu and W. Scheele, Kautsch. Gummi 15, 440 (1962).
23. N.J. Morrison and M. Porter, Rubber Chem. Technol. 57, 63 (1984).
24. P. Ghosh, S. Katare, P. Patkar, J.M. Caritjers, V. Venkatasubramanian, and K.A. Walker,
Rubber Chem. Technol. 76, 592 (2003).
25. T.D. Skinner, Rubber Chem. Technol. 45, 182 (1972).
26. P.J. Nieuwenhuizen, J. Reedijk, M. Van Duin, and W.J. McGill, Rubber Chem. Technol. 70,
368 (1997).
27. F. Ignatz-Hoover, A.R. Katritzky, V.S. Lobanov, Rubber Chem. Technol. 72, 318
(1999).
28. J.L. Koenig, Rubber Chem Technol. 73, 385 (2000).
29. M. Mori and J.L. Koenig, Rubber Chem Technol. 68, 551 (1995).
30. R.T. Armstrong, J.R. Little, and K.W. Doak, Rubber Chem. Technol. 17, 788 (1944).
31. A.Y. Coran, Chemtech 23, 106 (1983).
32. R.T. Leib, A.B. Sullivan, and C.D. Trivette, Rubber Chem. Technol. 43, 1188 (1970).
33. P.N. Son, Rubber Chem. Technol. 46, 999 (1973).
34. A.B. Sullivan, L.H. Davis, and O.W. Maender, Rubber Chem. Technol. 56, 1061 (1983).
35. R. Ding, A.I. Leonov, and A.Y. Coran, Rubber Chem. Technol. 69, 81 (1996).
36. A.V. Chapman and M. Porter, in «Natural Rubber Science and Techno/ogy» A.D. Roberts
(Ed.), Oxford University Press, Oxford, 1988.
37. A.Y. Coran, Rubber Chem. Technol. 38, 1 (1965).
38. R.W. Layer, Rubber Chem. Technol. 65, 211 (1992).
39. C.D. Trivette, Jr., E. Morita, and O.W. Maender, Rubber Chem. Technol. 50, 570 (1977).
40. H. Roebuck, K.M. Davies, Plast. Rubber Process, p. 63, June (1977).
41. R.N. Datta, A. H.M. Schotman, A. J.M. Weber, F. G.H. Van Wijk, P. J.C. Van Haeren,
J.W. Hofstraat, A.G. Talma, and A.G. V.D. Bovenkamp-Bouwman, Rubber Chem. Technol.
70, 129 (1997).
42. R.N. Datta, A.G. Talma, and A.H.M. Schotman, Rubber Chem. Technol. 71, 1073 (1998).
43. R.N. Datta and M.S. Ivan, Rubber World 212, 24 (1995).
44. E.-W. Tan and S. Wolff, U.S. Patent 4,517,336 (1985).
45. S. Buchan, «Rubber to Metal Bonding» Crosby Lockwood & Sons, London, 1959.
744 Литература
46. W. J. van Ooij, Rubber Chem. Technol. 52,605 (1979); Rubber Chem. Technol. 57,421 (1984).
47. K.D. Albrecht, Rubber Chem. Technol. 46, 981 (1973).
48. M.L. Studebaker, Rubber Chem. Technol. 39, 1359 (1966).
49. C.G. Moore, L. Mullins, and P. McL. Swift, J. Appl. Polym. Sci. 5, 293 (1961).
40. C.G. Moore and B.R. Trego, J. Appl. Polym. Sci. 5, 299 (1961).
51. T.D. Skinner and A.A. Watson, Rubber Chem. Technol. 42, 404 (1969).
52. R.M. Russell, T.D. Skinner, and A.A. Watson, Rubber Chem. Technol. 42, 418 (1969).
53. P.M. Lewis, NR Technology 17, 57 (1986).
54. C. R. and 0. Lorenz, Ind. Eng. Chem., Prod. Res. Dev. 2, 279 (1963).
55. C. L.M. Bell and J.L Cuneen, J. Appl. Polym. Sci. 11, 2201 (1967).
56. A.N. Gent, P.B. Lindley, and A.G. Thomas, J. Appl. Polym. Sci. 8,455 (1964).
57. E.B. McCall, J. Rubber Res. Inst. Malaya 22, 354 (1969).
58. D.S. Campbell, J. Appl. Polym. Sci. 14,1409 (1970).
59. W. Cooper, J. Polym. Sci. 28, 195 (1958).
60. P.S. Brown, M. Porter, and A.G. Thomas, «The Role of Crosslink Breakage in Determining the
Strength of Vulcanizates» International Rubber Conference, Harrogate, preprint 18a, 1987.
61. J.R. Wolfe, T.L. Pugh, and A.S. Killian, Rubber Chem. Technol. 41,1329, 1339 (1968).
62. E.C. Gregg, Jr. and S.E. Katrenick, Rubber Chem. Technol. 43, 549 (1970).
63. J. H. M. van den Berg, J.W. Beulen, E. F.J. Duynstee, and H.L. Nelissen, Rubber Chem. Technol.
57, 265 (1984).
64. J. H. M. van den Berg, J.W. Beulen, J. M.H. Hacking, and E.F.J. Duynstee, Rubber Chem.
Technol. 57, 725 (1984).
65. S. van der Meer, Rev. Gen. Caoutch. Plast. 20, 230 (1943).
66. C. Thelamon, Rubber Chem. Technol. 36, 268 (1963).
67. A. Giller, Kaut. Gummi Kunstst. 19, 188 (1966).
68. M. Van Duin and A. Souphanthong, Rubber Chem Technol. 68, 717 (1995).
69. J. Rehner and P.J. Flory, Rubber Chem. Technol. 19, 900 (1946).
70. R.F. Martell and D.E. Smith, Rubber Chem. Technol. 35, 141 (1962).
71. A.B. Sullivan,/. Org. Chem. 31, 2811 (1966).
72. C. S.L. Baker, D. Barnard, and M. Porter, Rubber Chem. Technol. 35, 141 (1962).
73. L.M. Gan and C.H. Chew, Rubber Chem. Technol. 56, 883 (1983).
74. F.P. Baldwin, P. Borzel, C. A, Cohen, H.S. Makowski, and J.F. Castle, Rubber Chem. Technol.
43, 522 (1970).
75. A. Greth, Kunststoffe^, 345 (1951).
76. P. Kovacic and P.W. Hein, Rubber Chem. Technol. 35, 528 (1962).
77. H. M.R. Hoffmann, Angew. Chem., Int. Ed. Engl. 8, 556 (1969).
78. K. Mori and Y. Nakamura, Rubber Chem. Technol. 57, 34 (1984).
79. H. Kato and H. Jujita, Rubber Chem. Technol. 55, 949 (1982).
80. R. Pariser, Kunststoffe 50, 623 (1960).
81. R.O. Becker, Rubber Chem. Technol. 37, 76 (1964).
82. L.D. Loan, Rubber Chem. Technol. 40, 149 (1967).
83. P.R. Dluzneski, Rubber Chem. Technol. 74, 451 (2001).
84. J. Scanlan and D.K. Thomas, J. Polym. Sci., Part A 1, 1015 (1963).
85. E.H. Farmer and S.E. Michael, J. Chem. Soc. p. 513 (1942).
86. E.H. Farmer and C.B. Moore/. Chem. Soc. p. 131 (1951).
87. E.H. Farmer and C.B. Moore/. Chem. Soc. p. 142 (1951).
88. L.D. Loan, /. Appl. Polym. Sci. 7, 2259 (1963).
89. D.K. Thomas, /. Appl. Polym. Sci. 6, 613 (1962).
90. K.W. Scott, /. Polym. Sci. 58, 517 (1962).
91. L.P. Lenas, Rubber Chem. Technol. 37, 229 (1964).
92. F.M. Lewis, Rubber Chem. Technol. 35, 1222 (1962).
93. D.K. Thomas, Polymer 7, 243 (1966).
94. C.W. Roush, J. Kosmider, and R.L. Baufer, Rubber Age 94, 744 (1964).
95. W. Hofmann, «Vulcanization and Vulcanizing Agents» Maclaren and Sons Ltd., London,
1967, p. 242.
Список литературы к главе 7 V 745
96. P.G. Вогк and C.W. Roush, in «Vulcanization of Elastomers» G. Alliger and I.J. Sjothun
(Eds.), Reinhold, New York, 1964, p. 366.
97. W. Fischer, U.S. Patent 3,758,643 (1973).
98. A.Y. Coran and R. Patel, Rubber Chem. Technol. 53, 781 (1980).
99. A.Y. Coran and R.P. Patel, «Thermoplastic Elastomers» 2nd ed., G. Holden, N.R. Legg,
R. Quirk, and H.E. Schroeder (Eds.), Hanser, Munich, 1996, Chap. 7, p. 153.
100. A.Y. Coran, R. Patel, and D. Williams, Rubber Chem. Technol. 55, 116 (1982).
(лава 8
1. F. Bucche, J. Appl. Polym. Sci. 5, 271 (1961).
2. F. Bueche, J. Appl. Polym. Sci. 107 (I960).
3. J.-B. Donnet and G. Heinrich, Bull. Soc. Chim. Fr. p. 1609 (1960).
4. J. Le Bras and E. Papirer, J. Appl. Polym. Sci. 22, 525 (1983).
5. E.M. Dannenberg, Rubber Chem. Technol. 48, 410 (1975).
6. J.-B. Donnet, E. Papirer, and A. Vidal, Grafting of Macromolecules onto Carbon Blacks.
«Chem. and Physics of Carbon» P.L. Walker, Jr. and P.A. Thrower (Eds.), Marcel Dekker.
New York, 1975.
7. N. Tsubokawa and M. Hosoya, ReactivePolym. 14, 33 (1991); N. Tsubokawa and M. Hosoya.
Reactive Polym. 14, 95 (1991).
8. M.J. Wang, T.A. Brown, W.J. Patterson, and R.A. Francis, International Rubber
Conference 1997, Kuala Lumpur, 6-9 October 1997.
9. EP1034222, E. Custodero, L. Simonot, and J.-C. Tardivat, Carbon black coated with an alu-
minous layer and method for obtaining same (1997).
10. A. Voet, J.C. Morawski, and J.B. Donnet, Rubber Chem. Technol. 50, 342 (1977).
11. S. Wolff, International Rubber Conferences, 14-17 October 1975, p. 295; S. Wolff, Kautchuk
Gummi Kunstoffe 34, 280 (1981).
12. EP0501227 and R. Rauline, Rubber compound and tires based on such a compound (1991).
13. A.P. Legrand, in «The Surface Properties of Silicas» A.P. Legrand (Ed.), J. Wiley and Sons,
New York, Vol. 1, 1998, pp. 1-18.
14. EP0157703, Y. Chevallier, and J.C. Morawski, Precipitated silica with morphological
properties, process for producing it and its application, especially as a filler (1984); Y. Che-
vallier, and J.C. Morawski, EUCHEM (1986).
15. N.A. Shabanova and I.V. Silos, Colloid J. 58, 256 (1996).
16. EP0890602, W.L. Hergenrother, J.C. Oziomek, and M. William, Addition of salts to improve
the interaction of silica with rubber (1997).
17. Y. Bomal, Y. Chevallier, and P. Cochet, Novel method for preparing precipitated silica, novel
aluminium-containing precipitated silicas, and use thereof for reinforcing elastomers, patent
W09630303, (1995).
18. WO9928376, E. Custodero, L. Simonot, and J.-C. Tardivat, Reinforcing aluminous filler and
rubber composition comprising such a filler (1997).
19. EP0810258, E. Custodero, and J.-C. Tardivat, Diene rubber composition containing alumina
as reinforcing filler and use in tire treads (1996).
20. EPl 114092, E. Custodero, L. Simonot, and J.-C. Tardivat, Rubber composition for tyre, based
on diene elastomer and a reinforcing titanium oxide (1999).
21. W02002053634, L. Simonot, T. Chartier, and E. Custodero, Rubber composition made
with diene elastomer and a reinforcing silicon carbide (2002); W02004003067, L. Simonot,
A. Lapra, A. Veyland, and E. Custodero, Rubber composition based on diene elastomer and a
reinforcing silicon nitride (2002).
22. W.M. Hess, G.C. McDonald, and E. Urban, Rubber Chem. Technol. 46, 204 (1973).
23. W.M. Hess, Rubber Chem. Technol. 64, 386 (1991).
24. E. Custodero, «Caract risation de la surface des noirs de carbone, nouveau mod le de surface
et implications pour le renforcement» Ph. D. Thesis, Universit de Haute Alsace, 1992.
25. N. Probst and J.B. Donnet, «Differentiation of Carbon Black Particularities by Scanning
Tunneling Microscopy» 2nd International Conference on Carbon Black, 1993, pp. 261-263.
26. J.-B. Donnet and E. Custodero, C.R. Acad. Sci., Ser. 2 314, 579 (1992).
746 "'V" Литература
27. J.-В. Donnet and E. Custodero, Carbon 30, 813 (1992).
28. J.-B. Donnet and T.K. Wang, Analusis 22, M24 (1994); J.-B. Donnet, E. Custodero, and
T.K. Wang, Kautsch. Gummi Kunstst. 49, 274 (1996).
29. C.R. Herd, G.C. McDonald, and W.M. Hess, Rubber Chem. Technol. 65, 107 (1992).
30. ASTM D2663.
31. ASTMD1765.
32. J. Janzen and K. Kraus, Rubber Chem. Technol. 44, 1287 (1971).
33. J. Janzen, Rubber Chem. Technol. 55, 669 (1982).
34. G. Kraus and J. Jansen, Kautchuk Gummi Kunstoffe 31, 569 (1978).
35. W.A. Wampler, The Carbon Aggregate 5, 2 (1997).
36. R.W. Magee, Rubber Chem. Technol. 68, 590 (1995); R.W. Magee, The Carbon Aggregate, 2,
1 (1995).
37. R.H. Bradley, I. Sutherland, and E. Sheng, J. Colloid Interface Sci. 179, 561 (1996).
38. M. Bole, A. Kodre, J. Grdadolnik, S. Pejovnik, and J.O. Besenhard, Carbon 36, 1207 (1998).
39. W.A. Wampler, M. Gerspacher, and C.P. O’Farrell, The Carbon Aggregate 4, 3 (1997).
40. B.R. Puri and R.C. Bansal, Carbon 3, 227 (1965).
41. A.I. Medalia and G. Kraus, Reinforcement of Elastomers by Particulate Fillers, in «Science
and Techno/ogy of Rubber» 2nd ed., J.E. Mark, B. Erman, and F.R. Eirich (Eds.), Academic
Press, San Diego, 1994.
42. W.M. Hess and G.C. McDonald, Rubber Chem. Technol. 56, 892 (1983).
43. T.C. Gruber, T.W. Zerda, and M. Gerspacher, Carbon 37, 1209 (1993).
44. D. Creeden, The Carbon Aggregate 4, 1 (1997).
45. R.E. Dollinger, R.H. Kallenberg, and M.L. Studebaker, Rubber Chem. Technol. 40,1311 (1967).
46. R. Pirard, B. Sahouli, S. Blacher, and J.P. Pirard, J. Colloid InterfaceSci. 217, 216 (1999).
47. L. Moscou, S. Lub, and O.K.F. Bussemaker, Rubber Chem. Technol. 44, 805 (1971).
48. L.R. Evans and W.H. Waddell, Kautchuk Gummi Kunstoffe 48, 718 (1995).
49. D.R. Milburn, B.D. Adkins, and B.H. Davis, in «Characterization of Porous Solids» K.K. Un-
ger (Ed.), Elsevier, Amsterdam, 1988, pp. 501.
50. l.B. Paul, T.N. Roy, and P.N. Mukherjee, Indian Journal of Technology 20, 441 (1982).
51. D. Goritz, J. В hm, and B. Freund, International Rubber Conference 1997, Kuala Lumpur,
6-9 October 1997, pp. 201-205.
52. T. Allen, Photocentrifuges, Powder Technol. 50, 193 (1987).
53. A.C. Patel and K.W. Lee, Elastomerics 122, 14 (1990); A.C. Patel and K.W. Lee,
Elastomerics 122, 22 (1990).
54. A.I. Medalia and L.W. Richards, J. Colloid Interface Sci. 40, 233 (1972).
55. A.I. Medalia and L.W. Richards, J. Colloid Interface Sci. 40, 233 (1972).
56. A.I. Medalia, E.M. Dannenberg, F.A. Heckmann, and G.R. Cotten, Rubber Chem. Technol.
46,1239 (1976).
57. P. Cochet, P. Barruel, L. Barriquant, J. Grobert, Y. Bomal, and E. Prat, Meeting Rubber Div.
Amer. Chem. Soc. Orlando, 26-29 October 1993, pp. 1-43; P. Cochet, P. Barruel, L. Barri-
quand, J. Grobert, Y. Bomal, and E. Prat, Rubber World, pp. 20-24 (1994).
58. R.A. Beebe, M.H. Polley, W.R. Smith, and C.B. Wendell, J. Am. Chem. Soc. 69, 2294 (1947).
59. M.L. Gonzalez-Marlin, B. Janczuk, L. Labajos-Broncano, and J.M. Bruque, Langmuir 13,
5991 (1997).
60. J. -B. Donnet, T.K. Wang, Y.J. Li, H. Balard, and G.T. Burns, Rubber Chem. Technol. 73,634
(2000).
61. S. Wolff, E.H. Tan, and J.B. Donnet, Kautch. Gummi Kunstoffe 47, 485 (1994).
61a. J.-B. Donnet and C.M. Lansinger, Kautchuk Gummi Kunstoffe 45, 459 (1992).
62. S. Wolff and M.J. Wang, Rubber Chem. Technol. 65, 329 (1992).
63. M.J. Wang and S. Woffi, Meeting Rubber Div. Amer. Chem. Soc., Detroit, 8-11 October, (1991);
M.J. Wang, S. Wolff, and J.B. Donnet, Rubber Chem. Technol. 64, 714 (1990).
64. M.J. Wang, S. Wolff, and J.B. Donnet, Rubber Chem. Technol. 64, 559 (1991).
65. J. Jagiello, G. Ligner, and E. Papirer, J. Colloid Interface Sci. 137, 128 (1990).
66. A. Lapra, E. Custodero, and N. Simon, Kautschuk Gummi, Kunststoffe. 57, 52 (2004).
67. B. Schubert, F.P. Ford, and F. Lyon, Encyclopedia of Industrial Chemical Analysis 8,1 (1969).
Список литературы к главе 7 747
68. M.L. Deviney, Adv. Colloid Inter/. Sci. 2, 237 (1969).
69. G.T. Taylor, T.E. Redington, and M.J. Bayley, Am. Ind. Hyg. Assoc. J. 41, 819 (1980).
70. D.S. Villars, J. Am. Chem. Soc. 69, 214 (1947).
71. H.P. Boehm, Angew. Chem. 78, 617 (1966); H.P. Boehm, Farbe Lak 79, 419 (1973).
71a. J.-B. Donnet, G. Heinrich, and G. Riess, Rev. Gen. Caoutch. Plast. 38, 1803 (1961);
J.-B. Donnet, G. Heinrich, and G. Riess, Rev. Gen. Caoutch. Plast. 41, 519 (1964).
72. H.P. Boehm, Carbon 32, 759 (1994).
73. P. Bertrand and L.T. Weng, Rubber Chem. Technol. 72, 384 (1998).
74. D. Rivin, Rubber Chem. Technol. 36, 729 (1963); D. Rivin, Rubber Chem. Technol. 307
(1971).
75. J.E. Lewis, L.R. Deviney, Jr., and C.F. McNabb, Rubber Chem. Technol. 43, 449 (1970).
76. E. Papirer, V.T. Nguyen, and J.B. Donnet, Carbon 16, 141 (1978).
77. D. Rivin, J. Aron, and A.I. Medalia, Rubber Chem. Technol. 41, 330 (1968).
78. W.F. Watson, Ind. Eng. Chem. 47, 1281 (1955).
79. E. Papirer, A. Voet, and P.H. Given, Rubber Chem. Technol. 42, 1200 (1969).
80. E. Papirer, J.B. Donnet, and J. Heinkele, J. Chim. Phys. 68, 581 (1971).
81. W.M. Hess, J.A. Ayala, P.C. Vegvari, and F.D. Kistler, Kautsch. Gummi Kunstst. 41, 1215
(1988); M. Soeda and Y. Kurata, Nippon Gomu Kyokaishi 68, 616 (1995).
82. S. Laufer, J. Molec. Struct. 60, 409 (1980).
83. B. Humbert, J. Non-Crystalline Solids 191, 29 (1995).
84. M. Zaborski, A. Vidal, G. Ligner, H. Balard, E. Papirer, and A. Burneau, Langmuir 5, 447
(1989).
85. A. Hunsche, U. G rl, A. M Her, M. Knaack, and T. G bel, Kautchuk Gummi Kunstoffe 50, 881
(1997).
86. U. Gori, A. Hunsche, A. M Iler, and H.G. Koban, Rubber Chem. Technol. 70, 608 (1997).
87. N. Cardona-Martinez and J.A. Dumesic, J. Catal. 125, 427 (1990); J. Uytterhoeven and J.
J. Fripiat, Bull. Soc. Chim. Fr. 788 (1962).
88. G.R. Cotten, Meeting Rubber Div. Amer. Chem. Soc. Houston, 25-28 October 1983, p. 38.
89. J.M. Funt, Rubber World (February), 21 (1986).
90. M. Gerspacher and C.P. O’Farrell, Rubber Plastics News 23, 85 (1993).
91. F. Borno and J.C. Morawski,XGS, Rubber Div., Houston, October, (1983).
92. B.R. Richmond, IRC 1993 Conference Proceedings, Orlando, 26-29 October 1993, Paper 158.
93. Y. Bomal, P. Cochet, B. Dejean, I. Gelling, and R. Newell, Kautchuk Gummi Kunstoffe 51,
259 (1998).
94. Y. Bomal, P. Cochet, B. Dejean, and J. Machurat, Rubber World, 6, 33 (1993).
95. B.B. Boonstra and A.I. Medalia, Rubber Age 92,892 (1963); B.B. Boonstra and A.I. Medalia,
Rubber Age 93, 82 (1963).
96. S. Shiga and M. Furuta, Rubber Chem. Technol. 58, 1 (1985).
97. L. Nikiel, M. Gerspacher, H. Yang, and C.P. O’Farrell, 157th ACS Rubber Division Meeting
Dallas, 4-6 April 2000, Paper 31.
98. A.Y. Coran and J.B. Donnet, Rubber World 65, 1016 (1993); A.Y. Coran, and J.B. Donnet,
Rubber Chem. Technol. 65, 998 (1993).
99. W.M. Hess, C.R. Herd, and E.B. Sebok, Kautchuk Gummi Kunstoffe 47, 328 (1994).
100. S. Maas and W. Gronski, Kautchuk Gummi Kunstoffe 47, 409 (1994).
101. A. Lapra, «Caracterisation moleculaireet proprietes mecaniques des reseaux elastomeres SBR
renforces par la silice» Universitd Paris VI, 1999.
102. F. Ehrburger-Dolle, M. Hindermann-Bischoff, F. Livet, F. Bley, and C. Rochas, Langmuir
17 329 (2001).
103. A.P. Legrand, N. Lecomte, A. Vidal, B. Haidar, and E. Papirer, J. Appl. Polym. Sci.: Appl.
Polym. Symp. 46, 2223 (1992).
104. K. Sone, M. Ishiguro, H. Akimoto, and M. Ishida, Rubber World 206, 29 (1992).
105. R.J. Young, D. H.A. Al-Khudhairy, and A.G. Thomas, J. Mater. Sci. 21, 1211 (1986).
106. G.C. McDonald and W.M. Hess, Rubber Chem. Technol. 50, 842 (1977).
107. A.I. Medalia, Rubber Chem. Technol. 59, 432 (1986).
108. K. Sone, Int. Polym. Sci. Technol. 26, 60 (1999).
748 -*и- Литература
109. J. O’Brien, Е. Cashell, G.E. Wardell, and V.J. McBrierty, Macromol. 9, 653 (1976); V.J. Mc-
Brierty and J.C. Kenny, Kautchuk Gummi Kunstoffe 47, 342 (1994).
110. T.A. Vilgis and G. Heinrich, Macromol. 27, 7846 (1994).
111. S. Kaufmann, W.P. Slichter, and D.D. Davis, J. Polym. Sci. A 9, 829 (1971).
112. F. De Candia, M. Carotenuto, L. Gargani, L. Guadagno, and E. Lauretti, Kautchuk Gummi
Kunstoffe 49, 99 (1996).
112a. J. Leisen, J. Breidt, and J. Keim, Rubber Chem. Technol. 72, 1 (1998).
113. M.E. Semaan, L. Nikiel, and C.A. Quarles, Carbon 39, 1379 (2001).
114. G. Kraus, Fortschr. Hochpolym. Forsch. 8, 155 (1971).
114a. Z. Rigbi, Kautchuk Gummi Kunstoffe 46, 36 (1993).
115. P.B. Stickney and B.D. Fa\b, Rubber Chem. Technol. 37,1299 (1964); G. Kraus, Rubber Chem.
Technol. 38, 1070 (1965); L.L. Ban, W.M. Hess, and L.A. Papazian, Rubber Chem. Technol.
47, 858 (1974).
116. D.S. Villars, J. Polym. Sci. 21, 257 (1996).
117. L.L. Ban, W.M. Hess, and L.A. Papazian, Rubber Chem. Technol. 47, 585 (1974).
118. E.M. Dannenberg, Rubber Chem. Technol. 59, 512 (1986).
119. B. Meissner, J. Appl. Polym. Sci. 18, 2483 (1974); B. Meissner, J. Appl. Polym. Sci. 50, 285
(1993).
120. S. Wolff, M.J. Wang, and E.H. Tan, Rubber Chem. Technol. 66, 163 (1993).
121. W.B. Wiegand, Trans. Inst. Rubber Ind. 1, 141 (1925).
122. E.M. Dannenberg and J.J. Brennan, Rubber Chem. Technol. 39,597 (1966); E.M. Dannenberg,
Trans. Inst. Rubber Ind. 42, T26 (1966).
123. J. Le Bras and E. Papirer, Rubber Chem. Technol. 52, 43 (1979).
124. H. Hommel, A.-P. Legrand, H. Balard, and E. Papirer, Makromol. Chem. 194, 879 (1993).
125. F. Borno, Makromol. Chem., Macromol. Symp. 23, 321 (1989).
126. H.G. Killian, H. Schenk, and S. Wolff, Colloid Polym. Sci. 265, 410 (1987).
127. M. Ashida, K. Abe, and T. Watanabe, Int. Polym. Sci. Technol. k, T42 (1977).
128. M. Strauss, H.G. Killian, B. Freund, and S. Wolff, Colloid Polym. Sci. 272, 1208 (1994).
129. E. Guth and O. Gold, Phys. Rev. 53, 322 (1938).
130. A.I. Medalia, Rubber Chem. Technol. 47, 411 (1974).
131. G. Kraus, J. Polym. Sci. В 8, 601 (1970).
132. A.I. Medalia, Rubber Chem. Technol. 46, 877 (1973).
133. A.I. Medalia, J. Colloid InterfaceSci. 32, 115 (1970).
134. A.I. Medalia, Rubber Chem. Technol. 47, 411 (1974).
135. H.M. Smallwood, J. Appl. Phys. 15, 758 (1944).
136. J.A.C. Harwood, L. Mullins, and A.R. Payne, J. Appl. Polym. Sci. 9,3011 (1965); J. A.C. Har-
wood and A.R. Payne, J. Appl. Polym. Sci. 10,315 (1966); J. A.C. Harwood, A.R. Payne, and J.
F. Smith, Rubber Chem. Technol. 43, 687 (1970).
137. A.I. Medalia, Rubber Chem. Technol. 64, 481 (1991).
138. B. Du perray and J.-L. Leblanc, Kautsch. Gummi Kunstst. 35, 298 (1982).
139. A.R. Payne, in «Reinforcement of Elastomers» G. Kraus (Ed.), Wiley Interscience, New York,
1965, Chap. 3.
140. J.O. Harris and R.W. Wise, in «Reinforcement of Elastomers» G. Kraus (Ed.), Wiley
Interscience, New York, 1965.
141. M.J. Wang, S. Wolff, and E.H. Tan, Rubber Chem. Technol. 66, 178 (1993).
142. N. Nagata, T. Kobatake, H. Watanabe, A. Ueda, and A. Yoshioka, Rubber Chem. Technol. 60,
837 (1987).
143. F. Tsutsumi, M. Sakakibara, and N. Oshima, Rubber Chem. Technol. 63, 8 (1990).
144. I. Soos, Int. Polym. Sci. Technol. 9, T84 (1982); I. Soos, Int. Polym. Sci. Technol. 10, T77
(1983); I. Soos, Int. Polym. Sci. Technol. И, T4 (1984).
145. L. Mullins and N.R. Tobin, J. Appl. Polym. Sci. 9, 2993 (1965).
146. L. Mullins, Rubber Chem. Technol. 42, 339 (1969).
147. A.B. Sullivan and R.W. Wise, Proc. 5th Int. Rubber Conf. 235 (1967).
148. A.N. Gent, J. Appl. Polym. Sci. 18, 1397 (1974).
149. W.M. Hess, F. Lyon, and K.A. Burgess, Kautsch. Gummi Kunstst. 20, 135 (1967).
Список литературы к главе 8 -'V 749
150. M.J. Wang, Р. Zhang, К. Mahmud, Т. Lanoye, and V. Vejins, Tire Technology International
58, 54, December? (2002).
Глава 9
1. Worldwide Rubber Statistics, 2003» International Institute of Synthetic Rubber Producers,
Houston, TX, 2003.
2. S. Datta, in «Rubber Compounding, Chemistry and Applications» B. Rodgers (Ed.), Marcel
Dekker Inc., New York, 2004.
3. ASTM D2227-96: Standard specification for natural rubber technical grades, 2002.
4. The Rubber Manufacturers Association, «The International Standards of Quality and Pack-
aging for Natural Rubber Grades, The Green Book» The International Rubber Quality and
Packaging Conference, Office of the Secretariat, Washington, D. C., January 1979.
5. D. Bernard, C. S.L. Baker, and I.R. Wallace, «NR TechnoZogy» Vol. 16, 1985, pp. 19-26.
6. «The Synthetic Rubber Manual» 14th ed., International Institute of Synthetic Rubber
Producers, Houston, TX, 1999.
7. S.M. Mezynski and M.B. Rodgers, «Heavy Duty Truck Tire Materials and Performance»
Kautschuk Gummi Kunststoffe, Frankfurt, Vol. 46, 1993, pp. 718-726.
8. B.D. Simpson, «The Vanderbilt Rubber Handbook» 12th ed., R. Babbit (Ed.), R.T. Vanderbilt
Co., Inc., Norwalk, CT, 1978.
9. K.H. Nordsiek, «The ‘Integral Rubber’ Concept—An Approach To An Ideal Tire Tread Rub-
ber» Kautschuk Gummi Kunstsoffe, Frankfurt, Vol. 38, 1985, pp. 178-185.
10. M.B. Rodgers, W.H. Waddell, and W. Klingensmith, «Rubber Compounding» in «Kirk-Oth-
mer Encyclopedia of Chemical TechnoZogy» 5th ed., John Wiley & Sons, New York, 2004.
11. G.L. Day and S. Futamura, Paper 22 presented at a meeting of the Rubber Division,
American Chemical Society, New York, 1986.
12. H.L. Brantley and G.L. Day, Paper 33 presented at a meeting of the Rubber Division,
American Chemical Society, New York, 1986.
13. W.J. Kern and S. Futamura, «Solution SBR as a Tread Rubber» ACS Rubber Division, Quebec,
Paper 78,1987.
14. N. Kumar, A. Chandra, and R. Mukhopadhyay, International J. Polymer Science 34, 91
(1996).
15. W.H. Waddell, M.B. Rodgers, and D.S. Tracey, «Natural Rubber» ACS Rubber Division
Meeting, Grand Rapids, MI, Paper A, 2004.
16. W.H. Waddell, M.B. Rodgers, and D.S. Tracey, «Tire Applications of Elastomers, Part 1.
Treads» Rubber Division Meeting, Grand Rapids, MI, Paper H, 2004.
17. S.M. Mezynski and M.B. Rodgers, «Radial Medium Truck Tire Performance and Materials»
ACS Rubber Division, Akron Rubber Group, 1989.
18. A. Saito, International Polymer Science & Technology 26, T/19 (1999).
19. A.E. Oberster, T.E. Bouton, and J.K. Valaites, Die Angewandte Mackromolekulare Chemie
29/30 (Nr 367), 291 (1973).
20. W.A. Schneider, F. Huybrechts, and KH. Nordesik, Kautschuk GummiKunstsoffe44,528 (1989).
21. ASTM D1795-04. Standard Classification System for Carbon Blacks Used in Rubber
Products, 2004.
22. ASTM D1566-04. Standard TerminoZogy Relating to Rubber, 2004.
23. ASTM D3053-04. Standard TerminoZogy Relating to Carbon Black, 2004.
24. W.M. Hess and W.K. Kemp, Rubber Chem. Technol. 56, 390 (1983).
25. J. LeBras and E. Papirer, Rubber Chem. Technol. 52, 43 (1979).
26. J.A. Ayala, W.M. Hess, F.D. Kistler, and G. Joyce, Paper 42 presented at a meeting of the
Rubber Division, American Chemical Society, Washington, D. C., 1990.
27. J.A. Ayala, W.M. Hess, A.O. Dotson, and G. Joyce, Rubber Chem. Technol. 63, 747 (1990).
28. S. Wolff and U. Gori, «The Influence of Modified Carbon Blacks on Viscoelastic Compound
Properties» Presented at International Rubber Conference, 1991.
29. W.A. Wampler, T.F. Carlson, and W.R. Jones, in «Rubber Compounding, Chemistry and
Applications» B. Rodgers (Ed.), Marcel Dekker Inc., New York, 2004.
30. M.P. Wagner, Rubber Chem. Technol. 55. 703 (1976).
750 V Литература
31. A. Blume, H.D. Luginsland, W. Meon, and S. Uhrlandt, in «RubberCompounding, Chemistry
and Applications» B. Rodgers (Ed.), Marcel Dekker Inc., New York, 2004.
32. L. Bateman, C.G. Moore, M. Porter, and B. Saville, in «Chemistry and Physics of Rubber-Like
Substances» John Wiley, New York, 1963.
33. S. Wolff, Rubber Chem. Technol. 55, 967 (1983).
34. S. W. Hong, in « Rubber Compounding, Chemistry and Applications» B. Rodgers (Ed.), Marcel
Dekker Inc., New York, 2004.
35. ASTM D3184-89. Standard Methods for Evaluation of and Test Formulations for Natural
Rubber, 1989.
36. W.W. Barbin and M.B. Rodgers, in «Science and Techno/ogy of Rubber» 2nd ed., J.E. Mark,
B. Erman, F.R. Eirich (Eds.), Academic Press, New York, 1994.
37. E. Morita, Rubber Chem. Technol. 53, 393 (1980).
38. S. Wolff, Presented at a meeting of the Rubber Division, American Chemical Society, Las
Vegas, NV, 1990.
39. »Rubber Chemicals» Flexsys Technical Bulletin, 1998.
40. R.J. Del Vecchio, «Understanding Design of Experiments» Hanser, Cincinnati, OH, 1997.
41. Malaysian Rubber Producers’ Research Association, «The Natural Rubber Formulary and
Property Index» Imprint of Luton, England, 1984.
42. W.H. Waddell, R.S. Bhukuni, W.W. Barbin, and P.H. Sandstrom, in «The Vanderbilt Rubber
Handbook» 13th ed., R.F. Ohm (Ed.), R.T. Vanderbilt Company, Norwalk, CT, 1990.
43. A. Niziolek, Nelsen, and R. Jones, Kautschuk Gummi Kunststoffe, 53, 358 (2000).
Глава 10
1. C.E. Inglis, Trans. Inst. Naval Architects (London) 55, 219 (1913).
2. R.J. Eldred, J. Polym. Sci. В 10, 391 (1972).
3. J.P. Berry, in «Fracture: An Advanced Treatise» Vol. 7: «Fracture of Non-metals and Com-
posites» H. Liebowitz (Ed.), Academic Press, New York, 1972, Chap. 2.
4. M. Braden and A.N. Gent, Kautsch. Gummi 14, WT157 (1961); E.H. Andrews, D. Barnard,
M. Braden, and A.N. Gent, in «The Chemistry and Physics of Rubberlike Substances»
L. Bateman (Ed.), Wiley, New York, 1963, Chap. 12.
5. A.G. Thomas, J. Polym. Sci. 31, 467 (1958).
6. G.R. Irwin, «Fracturing of Metals» Am. Soc. Metals, Cleveland, 1948.
7. G.R. Irwin, J. Appl. Meeh. 24, 361 (1957).
8. A.A. Griffith, Philos. Trans. R. Soc. (London), Ser. A 221, 163 (1921).
9. A.A. Griffith, «Proceedings, 1st International Congress on Applied Mechanics, Delft, 1924»
pp. 55-63.
10. E. Orowan, Rep. Progr. Phys. 12, 185 (1949).
11. R.S. Rivlin and A.G. Thomas,/. Polym. Sci. 10, 291 (1953).
12. A.G. Thomas, J. Appl. Polym. Sci. 3,168 (1960).
13. H.W. Greensmith, J. Appl. Polym. Sci. 7, 993 (1963); G.J. Lake, in «Proceedings, Interna-
tional Conference on Yield Deformation Fracture Polymers, Cambridge» Institute of Physics,
London, 1970, p. 53.
14. A.G. Thomas, J. Polym. Sci. 18, 177 (1955).
15. W.G. Knauss, Int. J. Fracture Meeh. 6,183 (1970).
16. A. Ahagon, A.N. Gent, H.-W. Kim, and Y. Kumagai, Rubber Chem. Technol. 48, 896 (1975).
17. H.W. Greensmith and A.G. Thomas, J. Polym. Sci. 18, 189 (1955).
18. H.W. Greensmith, J. Polym. Sci. 21, 175 (1956).
19. A.G. Veith, Rubber Chem. Technol. 38, 700 (1965).
20. A. Ahagon and A.N. Gent, J. Polym. Sci., Polym. Phys. Ed. 13, 1903 (1975).
21. A.N. Gent and R.H. Tobias, J. Polym. Sci., Polym. Phys. Ed. 20, 2051 (1982).
22. A.K. Bhowmick, A.N. Gent, and C.T.R. Pulford, Rubber Chem. Technol. 56, 226 (1983).
23. H. Tarkow, J. Polym. Sci. 28, 35 (1958).
24. G.J. Lake and P.B. Lindley, J. Appl. Polym. Sci. 9, 1233 (1965).
25. H.K. Mueller and W.G. Knauss, Trans. Soc. Rheol. 15, 217 (1971).
Список литературы к главе 8 -*v- 751
26. H.W. Greensmith, L. Mullins, and A.G. Thomas, in «The Chemistry and Physics of Rubber-
like Substances» L. Bateman (Ed.), Wiley, New York, 1963, Chap. 10.
27. G.J. Lake and A.G. Thomas, Proc. R. Soc. (London) A 300, 108 (1967).
28. J.E. Mark and M.-Y. Tang, J. Polym. Sci., Polym. Phys. Ed. 22, 1849 (1984).
29. G.J. Lake and O.H. Yeoh, Int. J. Fracture Meeh. 14, 509 (1978); Rubber Chem. Technol. 53,
210(1980).
30. A. Stevenson, Int. J. Fracture Meeh. 23, 47 (1983).
31. P.B. Lindley and S.C. Teo, Plast. Rubber Mater. Appl. 4, 29 (1979).
32. R.A. Dickie and T.L. Smith, J. Polym. Sci. A-2 7, 687 (1969).
33. C.W. Extrand and A.N. Gent, Int. J. Fracture Meeh. 48, 281 (1991).
34. A.E. Green and W. Zerna, «Theoretical Elasticity» Sect. 3.10. Oxford Univ. Press. London.
1954.
35. A.N. Gent and P.B. Lindley, Proc. R. Soc. (London) A 249, 195 (1958).
36. G.H. Lindsay, J. Appl. Phys. 38, 4843 (1967).
37. M.L. Williams and R.A. Schapery, Int. J. Fracture Meeh. 1, 64 (1965).
38. A.N. Gent and D.A. Tompkins, J. Polym. Sci. A-2 7, 1483 (1969).
39. A.N. Gent and C. Wang, J. Mater. Sci. 26, 3392 (1991).
40. A.E. Oberth and R.S. Bruenner, Trans. Soc. Rheol. 9, 165 (1965).
41. A.N. Gent and B. Park, J. Mater. Sci. 19, 1947 (1984).
42. B.J. Briscoe and S. Zakaria, Polymer 31, 440 (1990).
43. L. Mullins, Trans. Inst. Rubber Ind. 35, 213 (1959).
44. A.N. Gent and S.-M. Lai, J. Polym. Sci.: Part B: Polym. Phys. 32, 1543 (1994).
45. J.D. Ferry, «Viscoelastic Properties of Polymers» 2nd ed., Wiley, New York, 1970.
46. R.P. Kambour, J. Polym. Sci., Macromol. Rev. 7, 1 (1973).
47. W.G. Knauss, in «Deformation and Fracture of Polymers» H.H. Kausch, J.A. Hassell, and
R.I. Jaffee (Eds.), Plenum Press, New York, 1974, pp. 501-540; J.M. Bowen and W.G. Knauss,
J. Adhesion 39, 43 (1992).
48. G.J. Lake and O.H. Yeoh, J. Polym. Sci. 25, 1157 (1987).
49. A.N. Gent, S.-M. Lai, C. Nah, and C. Wang, Rubber Chem. Technol. 67, 610 (1994).
50. E.H. Andrews, J. Appl. Phys. 32, 542 (1961).
51. D. De and A.N. Gent, Rubber Chem. Technol. 69, 834 (1996).
52. A.N. Gent and A.W. Henry, in «Proceedings, International Rubber Conference, London,
1967» Maclaren, London, 1968, pp. 193-204.
53. R. Houwink and H.H.J. Janssen, Rubber Chem. Technol. 29, 4 (1956).
54. A.N. Gent, M. Razzaghi-Kashani, and G.R. Hamed, Rubber Chem. Technol. 76, 122 (2003).
55. G.J. Lake and P.B. Lindley, Rubber J. Int. Plast. 146, 24; 146, 30 (1964).
56. G.J. Lake and P.B. Lindley, J. Appl. Polym. Sci. 10, 343 (1966).
57. E.H. Andrews, / Meeh. Phys. Solids 11, 231 (1963).
58. E.H. Andrews and Y. Fukahori, J. Mater. Sci. 12, 1307 (1977).
59. A.N. Gent and M. Hindi, Rubber Chem. Technol. 63, 123 (1990).
60. A.N. Gent, G.L. Liu, and T. Sueyasu, Rubber Chem. Technol. 64, 96 (1991).
61. T.L. Smith, J. Polym. Sci. 32, 99 (1958).
62. F. Bueche, J. Appl. Phys. 26, 1133 (1955).
63. F. Bueche and J.C. Halpin, J. Appl. Phys. 35, 36 (1964); J.C. Halpin, Rubber Chem. Technol.
38, 1007 (1965).
64. T.L. Smith, J. Polym. Sci. A 1, 3597 (1963).
65. T.L. Smith, «RheoZogy» F.R. Eirich (Ed.), Academic Press, New York, Vol. 5,1969, Chap. 4.
66. F. Bueche and T.J. Dudek, Rubber Chem. Technol. 36, 1 (1963).
67. L.M. Epstein and R.P. Smith, Trans. Soc. Rheol. 2, 219 (1958).
68. T.L. Smith and W.H. Chu, J. Polym. Sci. A-2 10, 133 (1972).
69. R.F. Landel and R.F. Fedors, in «Proceedings 1st International Conference on Fracture,
Sendai» Vol. 2, p. 1247, Japan. Soc. Strength and Fracture of Materials, Tokyo, 1966.
70. В. B. S.T. Boonstra, India Rubber World 121, 299 (1949).
71. J. A.C. Harwood, A.R. Payne, and R.E. Whittaker, J. Appl. Polym. Sci. 14, 2183 (1970).
72. A.G. Thomas and J.M. Whittle, Rubber Chem. Technol. 43, 222 (1970).
752 Литература
73. A.N. Gent and L.-Q. Zhang, Rubber Chem. Technol. 75, 923 (2002).
74. A.M. Borders and R.D. Juve, Ind. Eng. Chem. 38, 1066 (1946).
75. K.A. Grosch, J. A.C. Harwood, and A.R. Payne, Nature 212, 497 (1966).
76. P.B. Lindley and A.G. Thomas, in «Proceedings, Fourth International Rubber Conference,
London, 1962» pp. 428-442.
77. A.N. Gent, P.B. Lindley, and A.G. Thomas, J. Appl. Polym. Sci. 8, 455 (1964).
78. S.M. Cadwell, R.A. Merrill, C.M. Sloman, and F.L. Yost, Ind. Eng. Chem.,Anal. Ed. 12,19 (1940).
79. A.N. Gent and J.E. McGrath, J. Polym. Sci. A 3, 1473 (1965).
80. K.A. Grosch and A. Schallamach, Trans. Inst. Rubber Ind. 41,80 (1965).
81. E. Southern and A.G. Thomas, Plast. Rubber Mater. Appl. 3, 133 (1978).
82. G.I. Brodskii, N.L. Sakhnovskii, M.M. Reznikovskii, and V.F. Evstratov, Sou. Rubber Technol.
18, 22 (1960).
83. A. Schallamach, J. Appl. Polym. Sci. 12, 281 (1968).
84. A.N. Gent and C.T.R. Pulford, J. Appl. Polym. Sci. 28, 943 (1983).
85. G.A. Gorokhovskii, P.A. Chernenko, and V.A. Smirnov, Sou. Mater. Sci. 8, 557 (1972).
86. Y.A. Evdokimov, S.S. Sanches, and N.A. Sukhorukov, Polym. Meeh. 9, 460 (1973).
87. A.N. Gent and C.T.R. Pulford, J. Mater Sci. 14, 1301 (1979).
88. G.V. Vinogradov, V.A. Mustafaev, and Y.Y. Podolsky, Wear %, 358 (1965).
89. E.H. Andrews, «Fracture in Polymers» American Elsevier, New York, 1968.
90. A.N. Gent, in «Fracture: An Advanced Treatise» Vol. 7: «Fracture of Non-metals and Com-
posites» H. Liebowitz (Ed.), Academic Press, New York, 1972, Chap. 6.
91. F.R. Eirich and T.L. Smith, in «Fracture: An Advanced Treatise» Vol. 7: «Fracture of Non-
metals and Composites» H. Liebowitz (Ed.), Academic Press, New York, 1972, Chap. 7.
92. A.N. Gent and C.T.R. Pulford, in «Developments in Polymer Fracture—1» E.H. Andrews
(Ed.), Applied Science Publishers, London, 1979, Chap. 5.
93. G.J. Lake and A.G. Thomas, Strength, in «Engineering with Rubber» 2nd ed., A.N. Gent
(Ed.), Hanser Publishers, Munich, 2001, Chap. 5.
Глава 11
1. D.M. Bielinsk, L.A. Sulsarki, H.M. Stanley, and R.A.J. Pethrick, Appl. Plym. Sci. 56, 853
(1995).
2. M. Yamato and M. Oahu, Progress in Organic Coating27, 277 (1996).
3. D.N. Schulz, A.F. Halasa, and A.E. Oberster, J. Polym. Sci. Chem. Ed. 12, 153 (1974).
4. C.A. Urneck and J.N. Short, J. Appl. Polymer Sci. 14, 1421 (1970).
5. A.F. Halasa, J. Polym. Sci. Polym. Ed. 19, 1357 (1981).
6. A. Yoshioka, A. Ueda, H. Watanabe, and N. Nagata, Nippon Kagaka Kaisha 341.
7. A. Uda Brain, Proceedings of the International Institute of Synthetic Rubber Producers, 33rd
Annual Meeting, 1992, pp. 65-83.
8. G. Bohm and D. Graves, U.S. Patent 4,788, 229 (1988).
9. A.F. Halasa and Wen-ling Hsu (to The Goodyear Tire and Rubber Co.), U.S. Patents
6,693,160 and 6,753,447 (2004).
10. Reference deleted in page proofs.
11. H.L. Hsieh and R.P. Quirk, «Anionic Polymerization: Principles and Practical Applications»
Marcel Dekker, New York, 1996.
12. J.E. Hall, T.A. Antikowiak, European Patent 0693 505 (1955).
13. V.C. Haskell, in «Encyclopedia of Polymer Science and TechnoZogy» H.S. Mark etal. (Eds.),
Wiley-Interscience, New York, Vol. 3, 1965, p. 60.
14. G.N. Bruxelles and N.R. Grassie, in «Encyclopedia of Polymer and TechnoZogy» H.S. Mark
et al. (Eds.), Wiley-Interscience, New York, Vol. 3, 1965, p. 307.
15. B.P. Rouse, in «Encyclopedia of Polymer and TechnoZogy» H.S. Mark et al. (Eds.), Wiley-
Interscience, New York, Vol. 3,1965, p. 325.
16. W. Jarowenko, in «Encyclopedia of Polymer Science and TechnoZogy» H.S. Marke/aZ. (Eds.),
Wiley-Interscience, New York, Vol. 3, 1965, p. 787.
17. J. Wichlatz, in «Chemical Reaction of Polymers» M. Fetters (Ed.), Interscience, New York,
1964, Chap. 2.
Список литературы к главе 9 V- 753
18. A. Yakubchik, В. Tikhominov, and V. Sumilov, Rubber Chem. Technol. 35, 1063 (1962).
19. H. Rachapudy, G. Smith, V. Raju, and W. Graessley, J. Polym. Sci. Phys. Ed. 17, 1211
(1979).
20. A.F. Halasa, Rubber Chem. Technol. 54, 627 (1981).
21. A.F. Halasa and J.M. Massie, «Kirk-Othemer Encyclopedia of Chemical TechnoZogy» 4th ed.,
Vol. 8, John Wiley & Sons, Inc., New York, 1993.
22. Y. Chamberline, J. Pascoult, H. Razzouk, and H. Cheradem, Makromol. Chem. Rapid Commun
2,322 (1981).
23. A.F. Halasa, L.E. Vescelius, S. Futamura, and J. Hall, «ШРАС Symposium on Macromol-
ecules» Vol. 28, Amherst, MA, 1982.
24. A.F. Halasa (to The Goodyear Tire & Rubber Co.), U.S. Patent 3,872,072 (1985) and U.
S. Patent 4,237,245.
25. H. Harwood, D. Russell, J. Verthe, and J. Zymons, Macromol. Chem. 163, 1 (1973).
26. M. Poutsma, in «Methods of Free Radical Chemistry» S. Huyser (Ed.), Dekker. New York,
Vol. 1, 1969, Chap. 3.
27. J. Bevington and L. Ratt, Polymer 16, 66 (1975).
28. L. Schoen, W. Raajien, and W. Van’twout, Br. Polym. J. 7,165 (1975).
29. R.J. Hopper (to The Goodyear Tire & Rubber Co.), U.S. Patent 3,915,907 (1975).
30. R.J. Hooper, Rubber. Chem. Technol. 49, 341 (1976).
31. R.J. Hooper, R.D. Mcquateand, T.G. Hutchin, Preprints, International Conference on Ad-
vances in the Stablization and Controlled Degradation of Polymers, Lucerne, Switzerland,
May 23-25, 1984.
32. R.J. Hooper (to The Goodyear Tire & Rubber Co.), U.S. Patent 4,820,780 (1989).
33. R.J. Hooper (to The Goodyear Tire & Rubber Co.), U.S. Patent 4,910,266 (1990).
34. D.A. White, R.S. Auda, W.M. Davis, and D.T. Ferrughlli (to Exxon Chemical Co.), U.S. Patent
4,956,420(1990).
35. R.J. Hooper (to The Goodyear Tire & Rubber Co.), U.S. Patent 4,017,468 (1977).
36. A. Staudinger, Rubber Chem. Technol. 17, 15 (1944).
37. M. Gordon and J. Tyler, J. Polym. Chem. 3, 537 (1953).
38. D.N. Schults, S. Turner, and M. Golub, Rubber Chem. Technol. 85, 809, (1983).
39. J.I. Cunneenand M. Porter, in «Encyclopedia of Polymer Science and TechnoZogy» H.S. Mark
et al. (Eds.), Wiley-Interscience, New York, Vol. 2, 1965, p. 502.
40. R.J. Ceresa, in «Encyclopedia of Polymer Science and TechnoZogy» H.S. Mark et al. (Eds.),
Wiley-Interscience, New York, Vol. 2, 1965, p. 502.
41. C. Avery and W. Watson, J. Appl. Polym. Sci. 19, 1 (1956).
42. R.J. Ceresa and W. Watson, J. Appl. Polym. Sci., 101, 1 (1959).
43. G. Scott, Polym. Eng. Sci. 24, 1007 (1984).
44. W.H. Janes, in «Block Copolymers» D.C. Allport and W.H. Janes (Eds.), Applied Science
Publishers, London, 1973, p. 62.
45. J.E. Morris and B.C. Sekher, Proceedings of the International Rubber Conterence, Washing-
ton, D. C., 1959, p. 277.
46. E.G. Cockbain, T.D. Pendle, and D.T. Turner, J. Polym. Sci. 39, 419 (1959).
47. A.F. Halasa, Adv. Chem. Ser. 130, 77 (1974).
48. J. Folk, R. Sclott, D. Hoey, and J. Pendeltor, Rubber Chem. Technol. 46, 1044 (1973).
49. A.F. Halasa, G. Mitchell, M. Stayer. D.P. Tate, and R. Koch, J. Polym. Sci. Chem. Ed. 14,297 (1976).
50. F.M. Merrett, Trans. Faraday Soc. 50, 759 (1964).
Глава 12
1. R. German, R. Hank, and G. Vaughan. Rubber Chem. Technol. 40, 569 (1967).
2. The sampling of polymer blends in «Polymeric Compatibilizers» by S. Datta and D.J. Lohse,
Hanser-Gardner, 1996, that >93% of polymer blends are combinations of thermoplastic and
elastomers. However, automotive tires (5000kt/yr in 1995), a blend of elastomers, are the
largest application for any immiscible polymer blend.
3. W.M. Hess, C.R. Herd, and P.C. Vergari. Rubber Chem Technol. 66, 329 (1993).
4. C.M. Roland. Rubber Chem. Technol. 62. 456 (1989).
754 Литература
P.J. Corish, in «Science and Techno/ogy of Rubber» F.R. Eirich (Ed.), Academic Press, New
York, 1978, Chap. 12.
6. E.T. McDonel, K.C. Baranwal, and J.C. Andries, in «Polymer Blends» D.B. Paul and S. New-
man (Eds.), Academic Press, New York, Vol. 11, 1978, Chap. 19.
7. (a) C.M. Hansen, «The Three Dimensional Solubility Parameter and Solvent Diffusion Co-
efficients» Danish Technical Press, Copenhagen, 1967. (b) C.M. Hansen and A. Beerbower,
Solubility Parameters, in «Encyclopedia of Chemical Techno/ogy, Supplement Volume» 2nd
ed., Wiley, New York, 1971.
8. (a) J.P. Runt and L.M. Martynowicz, Adv. Chem. Ser. 211, 111 (1985). (b) H.D. Keith and
F.J. Padden, J. Appl. Phys. 36, 1286 (1964).
9. A. Ghijsels, Rubber Chem. Technol. 60, 278 (1977).
10. (a) T. Nishi and T.T. Wang, Macromolecules 8, 909 (1975). (b) B. Rim and J.P. Runt, Mac-
romolecules 17, 1520 (1984).
11. (a) H.N. Cheng, in «Polymer Analysis and Characterization IV» H.G. Barth and J. Janca
(Eds.), John Wiley and Sons, New York, 1992, p. 21. (b) C.M. Roland, Rubber Chem. Technol.
62,456 (1989). (с) B. Albert, R. Jerome, P. Teyssie, G. Smyth, N.G. Boyle, and V.J. McBrierty,
Macromolecules 18,388 (1985). (d) C.M. Roland and C.A. Trask, Polym. Mater., Sci. Eng. 60,
832 (1989).
12. D.L. Vander Hart, W.F. Manders, R.S. Stein, and W. Herman, Macromolecules 20, 1726
(1987).
13. (a) J. Schaefer, M.D. Sefcik, E.O. Stejskal,and R.A. McKay, Macromolecules 14,188 (1981).
(b) T.R. Steger, J. Schaefer, E.O. Stejskal, R.A. McKay, and M.D. Sefcik, Annals NY Acad.
Sci. 371, (1981).
14. (a) M.C. Morris, Rubber Chem. Technol. 40, 341 (1967). (b) A.K. Sircar and T.G. Lamond,
Rubber Chem. Technol. 46, 178 (1973).
15. (a) C.M. Roland and G.G.A. Bohm, Macromolecules 18, 1310 (1985). (b) C.M. Roland,
Rubber Chem. Technol. 61, 866 (1988). (c) C.M. Roland, Macromolecules 20, 2557 (1987).
(d) J. Klein, Prilos. Mag. 43, 771 (1981). (e) J. Klein, D. Fletcher, and L.J. Fetters, Nature
(London) 304,526 (1983). (f) Y. Kumugai, K. Watanabe, T. Miyasaki, and J. Hata, J. Chem.
Eng. Jpn. 12, 1 (1979). (g) T. Gilmore, R. Falabella, and R.L. Lawrence, Macromolecules 13,
880(1980). (h) T. Hashimoto, Y. Tsukahara, and H. Kawai, Macromolecules 14, 708 (1981).
(i) J.E. Anderson and J.-H. Jou, Macromolecules 20, 1544 (1987). (j) G.C. Summerfield and
R. Ullman, Macromolecules 20, 401 (1987).
16. L.W. Kleiner, F.E. Karasz, and W.J. MacKnight, Polym. Eng. Sci. 19, 519 (1979).
17. (a) S. Datta and E.N. Kresge, U.S. Patent 4,722,971 (1988). (b) S. Datta, L. Kaufman, and
P. Ravishankar, U.S. Patent 5,571,868 (1996).
18. F. Morrar, L.L. Ban, W.G. Funk, E.N. Kresge, H.C. Wang, S. Datta, and R.C. Keller,
U.S. Patent 5,428,099 (1996).
19. (a) Z.M. Zhang and J.E. Mark, Polym. Sci. Polym. Phys. Ed. 20, 473 (1982). (b) J.E. Mark
and A.L. Andrady, Rubber Chem. Tech. 54, 366 (1981).
20. (a) D.L Livingston and R.L. Rongone, Proc. 5th, Int. Rubboer Conf., Brighton, England,
Paper No. 22 (1967). (b) G.M. Bartenev and G.S. Kongarov, Rubber Chem. Technol. 36, 668
(1983). (с) P.A. Marsh, A. Voet, L.D. Price, T.J. Mullens, and L. Kongarov, Rubber Chem.
Technol. 41, 344 (1968). (d) R. Couchman, Macromolecules 20, 1712 (1987). (e) T.K. Kwei,
E.M. Pearce, J.R. Pennacchia, and M. Charton, Macromolecules 20, 1174 (1987). (f)
M.-J. Brekner, H.A. Schneider, and H.-J. Cantow, Polymer 29,78 (1988). (g) M. Aubin and R.
E. Prud’homme, Macromolecules 21, 2945 (1988).
21. (a) C.M. Roland, J. Polym. Sci., Polym. Phys. Ed. 26, 839 (1988). (b) C.A. Trask and C.
M. Roland, Polym. Comm. 29,332 (1988). (c) C.M. Roland, Macromolecules20,2557 (1987).
(d) C.M. Roland and C.A. Trask, Rubber Chem. Technol. 62, 456 (1989). (e) C.A. Trask and
C.M. Roland, Macromolecules 22, 256 (1988).
22. (a) I.R. Gelling, NR Technol. 18, 21 (1987). (b) A.G. Margaritis, J.K. Kallitsis, and N.
K. Kalfoglou, Polymer 28, 2122 (1987).
23. (a) M.M. Coleman, G.J. Pehlert, and P.C. Painter, Macromolecules 29, 6820 (1996).
(b) G.J. Pehlert, P.C. Painter, B. Veytsman,and M.M. Coleman,Macromolecules30,3671 (1997).
Список литературы к главе 10 V" 755
(с) М.М. Coleman, G.J. Pehlert, X. Yang, J. Stallman, and P.C. Painter, Polymer37,4753 (1996).
(d) G.J. Pehlert, X. Yang, P.C. Painter, and M.M. Coleman, Polymer 37, 4763 (1996).
24. P.J. Corish and B.D.W. Powell, Rubber Chem. Technol. 47, 481 (1974).
25. (a) N. Tokita, Rubber Chem. Technol. 60, 292 (1977). (b) G.N. Avgeropoulos, R.C. Weissert,
P.H. Biddison, and G.G.A. Bohm, Rubber Chem. Technol. 49, 93 (1976).
26. (a) M. Takenaka, T. Izumitani, and T. Hashimoto, Macromolecules 20, 2257 (1987).
(b) H. Yang, M. Shibayama, and R.S. Stein, Macromolecules 19, 1667 (1986). (c) J. Kumaki
and T. Hashimoto, Macromolecules 19, 763 (1986).
27. (a) J.E. Kruse, Rubber Chem. Technol. 46, 653 (1973). (b) J.E. Callan, W.M. Hess, and C.
E. Scott, Rubber Chem. Technol. 44, 814 (1971). (с) C.E. Scott, J.E. Callan, and W.M. Hess.
J. Rubber Res. Inst. Malays. 22, 242 (1969). (d) J.E. Callan, B. Topcik, and F.P. Ford, Rubber
World 161, 60 (1965).
28. (a) F.H. Roninger, Jr., Ind. Eng. Chem., Anal. Ed. 6, 251 (1933). (b) R.W. Smith and J.
C. Andries, Rubber Chem Technol. KI, 64 (1986).
29. (a) E.W. Stroup, A. Pungor, V. Hilady, and J.D. Andrade, Polym. Prepr. ACS., Dir. Polym.
Chem. 34, 86 (1993). (b) W. Stocker, B. Bickmarm, S.N. Magonov, and H.J. Cantow. Ultra -
microscopy 42, 1141 (1992). (c) Y.H. Tsao, S.X. Yang, and D.F. Evans, Langmuir 8. 1188
(1992). (d) E. Hamada and R. Kaneko, Ultramicroscopy 42, 184 (1992). (e) J.E. Sax and J.
M. Ottino, Polymer 26, 1073 (1985). (f) T. Nishi, T. Hayashi, and H. Tanaka, Makromol.
Chem., Macromol. Symp. 16, 91 (1988). (g) J. Kruse, Rubber Chem. Technol. 46, 653 (1973).
(h) D. Vesely and D.S. Finch, Makromol. Chem., Macromol. Symp. 16, 329 (1988).
30. (a) R. Buchdahl and L.E. Nielsen, J. Polym. Sci. 15, 1 (1955). (b) M.C. Morris, Rubber
Chem. Technol. 40, 341 (1967). (с) H. K. de Decker and D.J. Sabatine, Rubber Age 99, 73
(1967). (d) L. Bohn, Rubber Chem. Technol. 41,495 (1968). (e) W. Scheele, Kautsch. Gummi
Kunstst. 24,387 (1971). (f) A.R. Ramos and R.E. Cohen, Polym. Eng. Sci. 17,639 (1977). (g)
K.A. Mazich, M.A. Samus, P.C. Kilgoar, Jr., and H.K. Plummer, Jr., Rubber Chem. Technol.
59, 623 (1986).
31. (a) R.J. Butera, J.B. Lando, and B. Simic-Glavaski, Macromolecules 20, 1724 (1987).
(b) M.R. Krejsa and J.L. Koenig, Rubber Chem. Technol. 64, 635 (1991). (c) M.R. Krejsa and J.
L. Koenig, RubberChem. Technol. 65,956 (1992). (d) S.R. Smith and J.L. Koenig, Macromolecules
24, 3496 (1991). (e) S.N. Sarkarand R.A. Komoroski, Macromolecules 25,1420 (1992).
32. (a) C.M. Roland and G.G.A. Bohm, J. Polym. Sci., Polym. Phys. Ed. 22, 79 (1984).
(b) C.A. Trask and C.M. Roland, Macromolecules 22, 256 (1989). (c) C.M. Roland and G.
G.A. Bohm, J. Polym. Sci., Polym. Phys. Ed. 22, 79 (1984).
33. (a) J.L. Leblanc, Plast. Rubber Process. Appl. 2, 361 (1982). (b) G.J. Van Amerongen, Rubber
Chem. Technol. 37,1065 (1964). (c) V.A. Shershnev, Rubber Chem. Technol. 66, 537 (1982).
(d) M.G. Huson, W.J. McGill, and R.D. Wiggett, Plast. Rubber Process. Appl. 6, 319 (1985).
(e) R.F. Bauer and A.H. Crossland, RubberChem. Technol. 61,585 (1988). (f) A.K. Bhowmick
and S.K. De, Rubber Chem. Technol. 63, 960 (1980). (g) R.L. Zapp, Rubber Chem. Technol.
46, 251 (1973).
34. (a) G.J. Amerongen, Rubber Chem. Technol. 37, 1065 (1964). (b) J.R. Gardiner, Rubber
Chem. Technol. 41, 1312 (1968). (c) J.R. Gardiner, Rubber Chem. Technol. 42, 1058 (1969).
(d) J.R. Gardiner, Rubber Chem. Technol. 43, 370 (1970).
35. (a) V.A. Shershnev, Rubber Chem. Technol. 66, 537 (1982). (b) J. Rehner, Jr. and P.E. Wei,
Rubber Chem. Technol. 42,985 (1969). (c) A.K. Bhowmick and S.K. De, Rubber Chem. Tech-
nol. 63, 960 (1980). (d) K.C. Baranwal and P.N. Son, Rubber Chem. Technol. 47, 88 (1974).
(e) K. Hashimoto, M. Miura, S. Takagi, and H. Okamoto, Int. Polym. Sci. Technol. 3,84 (1976).
(f) A.Y. Coran, RubberChem. Technol. 61,281 (1988). (g) R.W. Amidon and R.A. Gencarelli,
U.S. Patent 3,674,824 (1972). (h) R.P. Mastromatteo, J.M. Mitchell, and T.J. Brett, Rubber
Chem. Technol. 44,1065 (1971). (i) Sumitomo Chemical Company, British Patent 1,325,064
(1973). (j) M.E. Woods and T.R. Mass, U.S. Patent 3,830,881 (1974).
36. M.E. Woods and J.A. Davidson, Rubber Chem. Technol. 49, 112 (1976).
37. (a) M.G. Huson, W.J. McGill, and P.J. Swart, J. Polym. Sci., Polym. Lett. Ed. 22,143 (1984).
(b) D. Honiballand W.J. McGill, J. Polym. Sci., Polym. Phys. 26,1529 (1988). (c) L.D. Loan,
Rubber Chem. Technol. 40, 149 (1967).
756 Литература
38. (a) J.E. Callan, W.M. Hess, and C.E. Scott, Rubber Chem. Technol. 44,814 (1971). (b) J.E. Cal-
lan, W.M. Hess, and C.E. Scott, Rev. Gen. Caoutch. Plast. 48, 155 (1971). (с) A.K. Sircar
and T.J. Lammond, Rubber Chem. Technol. 46, 178 (1973). (d) G. Cotton and L.J. Murphy,
Rubber Chem. Technol. 61, 609 (1988).
39. A.J. Dias and A. Galuska, Rubber Chem. Technol. 69, 615 (1996).
40. (a) J.E. Callan, B. Topcik, and E.P. Ford, Rubber World 151, 60 (1951). (b) W.M. Hess,
P. A. Marsh, and F.J. Eckert, presented at a meeting of the Rubber Division, American Chemi-
cal Society, Miami Beach, FL, 1965. (c) Y.F. Wang and H.C. Wang, Rubber Chem. Technol.
70, 63 (1997).
41. (a) W.M. Hess, R.A. Swor, and P.C. Vegvari, Kautsch. Gummi Kunstst. 38, 1114 (1985).
(b) W.M. Hess and V.E. Chirico, Rubber Chem. Technol. 50, 301 (1977).
42. (a) A. Medalia, Rubber Chem. Technol. 51, 437 (1978). (b) W.P. Fletcher and A.N. Gent,
Br. J. Appl. Phys. 8, 1984 (1957). (c) S. Maiti, S.K. De, and A.K. Bhowmick, Rubber Chem.
Technol. 65, 293 (1992).
43. (a) M.E. Woods and T.R. Mass,71dy. Chem. Ser. 142,386 (1975). (b) E.T. McDonel, K.C. Ba-
ranwal, and J.C. Andries, in «Polymer Blends» D.R. Paul and S. Newman (Eds.), Academic
Press, New York, Vol. 2, 1978, p. 263. (c) L. Leibler, Makromol Chem. Macromol Symp. 16,
1, (1982). (d) J. Noolandi and K.M. Hong, Macromolecules 16, 482 (1982).
44. (a) D.K. Setua and J.L. White, Kautsch. Gummi Kunstst. 44, 821 (1991). (b) D.K. Setua
and J.L. White, Poly. Eng. & Sci. 31, 1742 (1991). (с) P.A. Aijunan, R.B. Kusznir, and A.
Dekmezian, Rubber World 21 (1997).
45. J.A. Davison, W. Nudenberg, and Y. Rim, U.S. Patent 4,316,971 (1982).
46. (a) G.N. Avgeropoulos, R.C. Weissert, P.H. Biddison, and G.G.A. Bohm, Rubber Chem.
Technol. 49, 93 (1976). (b) C.M. Roland and M. Nguyen, J. Appl. Polym. Sci. 36,141 (1988).
(c) A.C. Pipkin and R.l. Tanner, Ann. Rev. Fluid Meeh. 9,13 (1977). (d) W.M. Hess, P.C. Veg-
vari, and R.A. Swor, Rubber Chem. Technol. 68, 350 (1985). (e) W.M. Hess, R.A. Swor,
and P.C. Vegvari, Kautsch. Gummi Kunstst. 38,1114 (1985). (f) R.F. Heitmiller, R.Z. Maar,
and H.H. Zabusky, J. Appl. Polym. Sci. 8, 873 (1964). (g) S. Uemura and M. Takayanagi,
J. Appl. Polym. Sci. 10, 113 (1966).
47. (a) H. Yang, M. Shibayama, and R.S. Stein, Macromolecules 19,1667 (1986). (b) E. A. Meinecke
and M.I. Taftaf, Rubber Chem. Technol. 37,1190 (1988). (c) A.I. Medalia, Rubber Chem. Technol.
61,437 (1978). (d) E.A. Meinecke and M.I. Taftaf, Rubber Chem. Technol. 61, 534 (1988).
48. R.T. Morrisey, Rubber Chem. Technol. 44, 1029 (1971).
49. (a) J.M. Mitchell, Rubber Plast. News, June 3, 1985, p. 18. (b) N.M. Matthew, J. Polym. Sci.
Polym. Lett. Ed. 22, 135 (1984). (c) W. von Hellens, Rubber Division, American Chemical
Society, Detroit, MI, Fall 1989. (d) F.C. Cesare, Rubber World 201,14 (1989). (e) W. Hong,
Rubber Plast. News 19, 14 (1989). (f) W. von Hellens, D.C. Edwards, and Z.J. Lobos,
Rubber Plast. News 20, 61 (1990). (g) E.H. Andrews, J. Polym. Sci. 10, 47 (1966). (h) B.
Ambelang, F.H. Wilson, Jr., L.E. Porter, and D.L. Turk, Rubber Chem. Technol. 42, 1186
(1969). (i) J. Barrier, Rubber Chem. Technol. 28, 814 (1956). (j) J. Kinning, E.L. Thomas,
and J.M. Ottino, Macromolecules 20, 1129 (1987).
50. (a) F.J. Krakowski and A.J. Tinker, Elastomerics 122, 34; 122, 24 (1990). (b) M.F. Tse,
K.O. McElrath, H.-C. Wang, Y.F. Wang, and A.L. Tisler, Paper 24, 153rd Rubber Division
Meeting, ACS, Indianapolis, IN, May 5-8, 1998.
51. H. Mouri and Y. Tonosaki, Paper 65, 152rd Rubber Division Meeting, ACS, Cleveland, OH,
October 21-24, 1997.
Глава 13
1. »Thermoplastic Elastomers—2nd Edition» Market Research Report by Freedonia Group, 2003.
2. http://www. kraton. com
3. http://www. dupont-dow. com/Products/Engage/engage. asp
4. http://www. santoprene. com/home. htrnl
5. http://www. dow. com/engineeringplastics/prod/na/pel. htm
6. http://plastics. dupont. com/NASApp/myplastics/Mediator? id=75
7. http://www. atofinachemicals. com/techpol/pebax
Список литературы к главе 12 —* Ь- 757
8. http://www. dupont. com/packaging/products/surlyn. html
9. К. Uhlig, «Discovering Polyurethanes» Hanser, Munich, 1999.
10. M. Szycher, «Handbook of Polyurethanes» CRC Press, Boca Raton, FL, 1999.
11. D. Randall and S. Lee (Eds.), «The Polyurethanes Book» Wiley, New York, 2003.
12. G. Oertel (Ed.), «Polyurethane Handbook: Chemistry-Raw Materials-Processing-Applica-
tion-Properties» Hanser Publishers, New York, 1994.
13. D.J. Lyman, Journal of Polymer Science 45, 49 (1960).
14. W. Meckel, W. Goyert, and W. Wieder, in «Thermoplastic Elastomers—2nd Edition»
G. Holden, N.R. Legge, R.P. Quirk, and H.E. Schroeder (Eds.), Hanser Publishers, New
York, 1996, Chap. 2.
15. Z.S. Petrovic and J. Ferguson, Progress in Polymer Science 16, 695 (1991).
16. C. Hepburn, «Polyurethane Elastomers» Applied Science Publishers, New York, 1982, Chap. 1.
17. W.K. Witsiepe, ACS Advances in Chemistry Series 129, 29 (1973).
18. G.K. Hoeschele, Chimia 28, 544 (1974).
19. R.G. Nelb and A.T. Che, in «Thermoplastic Elastomers—2nd Edition» G. Holden.
N.R. Legge, R.P. Quirk, and H.E. Schroeder (Eds.), Hanser Publishers, New York, 1996.
Chap. 9.
20. M. Morton, «Anionic Polymerization: Principles and Practice» Academic Press, New York.
1983.
21. A. Noshay and J.E. McGrath, «Block Copolymers» Academic Press, New York, 1977, Chap. 6.
22. C.W. Lantman, W.J. MacKnight, and R.D. Lundberg, Annual Review of Material Science 19.
295 (1989).
23. D.C. Lee, T.A. Speckhard, A.D. Sorenson, and S.L. Cooper, Macromolecules 19. 2383
(1986).
24. N. Aust and G. Gobec, Macromolecular Materials and Engineering 286, 119 (2001).
25. C.D. Eisenbach, T. Heinemann, A. Ribbe, and E. Stadler, Angewandte Makromolekulare
Chemie 202, 221 (1992).
26. H. Suzuki and H. Ono, Bulletin of the Chemical Society of Japan 43, 682 (1970).
27. H. Suzuki and H. Ono, Bulletin of the Chemical Society of Japan 43, 687 (1970).
28. Y. L csei, O. Facosko, and R.F. Pape, Polymer Bulletin 9, 81 (1983).
29. S.A. Dombrowski, I. Goodman, and A.F. Johnson, Makromol. Chem., Rapid Commun. 7, 274
(1986).
30. A. Kaji and M. Murano, Polymer Journal 22, 1065 (1990).
31. D.J. Yontz and S.L. Hsu, Macromolecules 33, 8415 (2000).
32. H.K. Frensdorff, Macromolecules 4, 369 (1971).
33. E. Sorta and L.A. Melis, Polymer 19, 1153 (1978).
34. T.A. Speckhard, J.A. Miller, and S.L. Cooper, Macromolecules 19, 1550 (1986).
35. T.A. Speckhard, J.A. Miller, and S.L. Cooper, Macromolecules 19, 1558 (1986).
36. T.A. Speckhard, J.G. Homan, J.A. Miller, and S.L. Cooper, Polymer 2$, 758 (1987).
37. T.A. Speckhard, J.G. Homan, J.A. Miller, and S.L. Cooper, Polymer 28, 768 (1987).
38. F.S. Bates and G.H. Fredrickson, Annual Review of Physical Chemistry 41, 525 (1990).
39. G.H. Fredrickson and F.S. Bates, Annual Review of Materials Research 26, 501, (1996).
40. T.P. Lodge, Macromolecular Chemistry and Physics 204, 265 (2003).
41. I.W. Hamley (Ed.), «Developments Block Copolymer Science and TechnoZogy» Wiley, West
Sussex, 2004.
42. I.W. Hamley, «The Physics of Block Copolymers» Oxford University Press, Oxford, 1998.
43. T.L. Smith, Journal of Polymer Science-Polymer Physics Edition 12, 1825 (1974).
44. S. Chen, T. Cao, and Y. Jin, Polymer Comm. 28, 314 (1987).
45. C. Li and S.L. Cooper, Polymer 31, 3 (1990).
46. J.A. Laurer and K.I. Winey, Macromolecules 31, 9106, (1998).
47. K.I. Winey, J. Laurer, and B.P. Kirkmeyer, Macromolecules 33, 507, (2000).
48. B.P. Kirkmeyer, A. Taubert, J.S. Kim, and K.I. Winey, Macromolecules 35, 2648 (2002).
49. M.M. Coleman, J.F. Graf, and P.C. Painter, «Specific Interactions and the Miscibility of
Polymer Blends» Technomic Publishing Company, Lancaster, PA, 1991, Chap. 3.
50. M.A. Harthcock, Polymer 30, 1234 (1989).
758 -•и- Литература
51. N. Luo, D.N. Wang, and S.K. Yang, Polymer 37, 3045 (1996).
52. C. S.P. Sung and N.S. Schneider, Macromolecules 8, 68 (1975).
53. R.W. Seymour, G.M. Estes, and S.L. Cooper, Macromolecules 3, 579 (1970).
54. J.L. Sormana and J.C. Meredith, Macromolecules 37, 2186 (2004).
55. V. W. Srichatrapimuk and S.L. Cooper, Journal of Macromolecular Science-Physics B15, 267
(1978).
56. J.T. Koberstein, I. Gancarz, and T.C. Clarke, Journal of Polymer Science: Part B: Polymer
Physics 24, 2487 (1986).
57. N. Luo, D.N. Wang, and S.K. Ying, Macromolecules 30, 4405 (1997).
58. M.M. Coleman, D.J. Skrovanek, S.E. Howe, and P.C. Painter, Macromolecules 18,302 (1985).
59. D.J. Skrovanek, S.E. Howe, P.C. Painter, and M.M. Coleman, Macromolecules 18,1676 (1985).
60. D.J. Skrovanek, P.C. Painter, and M.M. Coleman, Macromolecules 19, 699 (1986).
61. M.M. Coleman, D.J. Skrovanek, and P.C. Painter, MakromolecularChemisty, Macromolecular
Symposia5,21 (1986).
62. M.M. Coleman, K.H. Lee, D.J. Skrovanek, and P.C. Painter, Macromolecules 19, 2149 (1986).
63. H. Janik, B. Fblys, and Z.S. Petrovic, Macromolecular Rapid Communications 24, 265 (2003).
64. C.W. Myers and S.L. Cooper, Journal of Applied Spectroscopy 48, 72 (1994).
65. R.W. Seymour, A.E. Allegrezza Jr., and S.L. Cooper, Macromolecules 6, 896 (1973).
66. A.E. Allegrezza Jr., R.W. Seymour, H.N. Ng, and S.L. Cooper, Polymer 15, 433 (1974).
67. C.B. Wang and S.L. Cooper, Macromolecules 16, 775 (1983).
68. A. Lilaonitkul, J. West, and S.L. Cooper, Journal of Macromolecular Science-Physics B12,563
(1976).
69. H.W. Siesler, Polymer Bulletin 9, 471 (1983).
70. J.R. Schoonover, D.G. Thompson, J.C. Osborn, E.B. Oder, D.A. Wrobleski, A.L. Marsh,
H.C. Wang, and R.A. Palmer, Polymer Degradation and Stability 74, 87 (2001).
71. D.K. Graff, H.C. Wang, R.A. Palmer, and J.R. Schoonover, Macromolecules 32, 7147 (1999).
72. W.L. Bragg, «The Crystalline State» Macmillan, New York, 1933.
73. L.E. Alexander, «Х-ray Diffraction Methods in Polymer Science» Krieger Publishing Com-
pany, Malabar, FL, 1985.
74. I.H. Hall and M.G. Pass, Polymer 17, 807 (1976).
75. I.J. Desborough and LJ. Hall, Polymer 18, 825 (1977).
76. R.J. Celia, Journal of Polymer Science C42, 727 (1973).
77. M.A. Vallance and S.L. Cooper, Macromolecules 17, 1208 (1984).
78. G. Perego, M. Cesari and R. Vitali, Journal of Applied Polymer Science 29, 1157 (1984).
79. E.V. Konyukhova, V.M. Neverov, S.N. Chvalun, and Y.K. Godovsky, Polymer Science Series
4 46,61 (2004).
80. G.R. Hatfield, Y. Guo, W.E. Killinger, R.A. Andrejak, and P.M. Roubicek, Macromolecules
26,6350(1993).
81. Y.C. Yu and W.H. Jo, Journal of Applied Polymer Science 54, 585 (1994).
82. Y.C. Yu and W.H. Jo, Journal of Applied Polymer Science 56, 895 (1995).
83. Y.C. Yu and W.H. Jo, Journal of Applied Polymer Science 64, 2155 (1997).
84. J.P. Sheth, J. Xu, and G.L. Wilkes, Polymer 44, 743 (2003).
85. S. Warner, Journal of Elastomers and Plastics 22, 166 (1990).
86. R.M. Briber and E.L. Thomas, Journal of Macromolecular Science-Physics B22, 509 (1983).
87. R.M. Briber and E.L. Thomas, Journal of Polymer Science-Polymer Physics Edition 23, 1915
(1985).
88. J. Blackwell, M.R. Nagarajan, and T.B. Hoitink, Polymer 23, 950 (1982).
89. C.D. Eisenbach and E. Stadler, Colloid and Polymer Science 273, 352 (1995).
90. H.W. Starkweather, Journal of Applied Polymer Science 25, 139 (1980).
91. E.N. Kresge, Journal of Applied Polymer Science: Appl. Polym. Symp. 39, 37 (1984).
92. A. Guinier and G. Fournet, «Small-Angle Scattering of Х-rays» John Wiley and Sons, New
York, 1955.
93. J.S. Pederson, Advances in Colloid and Interface Science 70, 171 (1997).
94. O. Glatter and O. Kratky (Eds.), «Small Angle X-ray Scattering» Academic Press, New York, 1982.
95. H. Brumberger (Ed.), «Modern Aspects of Small-Angle Scattering» Kluwer, New York, 1995.
Список литературы к главе 12 759
96. W. Ruland, Journal of Applied Crystallography 4, 70 (1971).
97. C.G. Vonk, Journal of Applied Crystallography 6, 81 (1973).
98. R. S gu la and J. Prud’homme, Macromolecules 11, 1007 (1978).
99. R. S gu la and J. Prud’homme, Macromolecules 14, 197 (1981).
100. R. S gu la and J. Prud’homme, Macromolecules 21, 635 (1988).
101. T. Pakula, K. Saijo, H. Kawai, and T. Hashimoto, Macromolecules 18, 1294 (1985).
102. T. Pakula, K. Saijo, and T. Hashimoto, Macromolecules 18, 2037 (1985).
103. S. Polizzi, P. В secke, N. Stribeck, H.G. Zachmann, R. Zietz, and R. Bordeianu, Polymer 31.
639 (1990).
104. Z. Ophir and G.L. Wilkes, J. Polym. Sci.: Polym. Phys. Ed. 18, 1469 (1980).
105. D. Ttyagi, J.E. McGrath, and G.L. Wilkes, Polymer Engineering and Science 26. 1371 (1986).
106. R.J. Roe, Journal of Applied Crystallography 15, 182 (1982).
107. R. Bonart and E.H. Muller, Journal of Macromolecular Science-Physics. B10. 345 (1974).
108. J.T. Koberstein and R.S. Stein, Journal of Polymer Science-Polymer Physics Edition 21.2181 (1983 >.
109. R.A. Phillips, J.M. McKenna, and S.L. Cooper, Journal of Polymer Science Part B-Polym*r
Physics 32, 791 (1994).
110. R.A. Phillips and S.L. Cooper, Macromolecules 28, 5734 (1995).
111. R.A. Phillips and S.L. Cooper, Journal of Polymer Science Part В-Polymer Physics 34.
(1996).
112. D.G. Ballard, J. Schelten, and G. Wignail, European Polymer Journal 9. 965 (1973».
113. J.P. Cotton, D. Decker, H. Ben it, B. Farnoux, J. Higgins, G. Jannink, R. Ober. C. Picot, and .1.
des Cloiseaux, Macromolecules 7, 862 (1974).
114. S.N. Jahshan and G.C. Summerfield, Journal of Polymer Science-Polymer Physics Edition 18.
2415 (1980).
115. A.R. Rennie, R.C. Oberthur, «Practical Small Angle Neutron Scattering» Tavlorand Francis,
New York, 1994.
116. J. Higgins and H. Benoit, «Polymers and Neutron Scattering» Clarendon Press. London, 1994.
117. B.J. Gabrys, «Applications of Neutron Scattering to Soft Condensed Matter» Taylor and
Francis, New York, 2000.
118. S. Naylor, N.J. Terrill, G.E. Yu, S. Tanodekaew, W. Bras, S.M. King, C. Booth, A.J. Ryan,
Polymer International 44, 371 (1997).
119. J.A. Miller, S.L. Cooper, C.C. Han, and G. Pruckmayr, Macromolecules 17, 1063 (1984).
120. J.A. Miller, G. Pruckmayr, E. Epperson, and S.L. Cooper, Polymer 25, 1915 (1985).
121. S.L. Cooper and J.A. Miller, Rubber Chemistry and Technology 58, 899 (1985).
122. S.L. Cooper, J. A. Miller, and J.G. Homan, Journal of Applied Crystallography 21, 692 (1988).
123. H. Hasegawa, T. Hashimoto, H. Kawai, T.P. Lodge, E.J. Amis, C.J. Glinka, and C.C. Han,
Macromolecules 18, 67 (1985).
124. H. Hasegawa, H. Tanaka, T. Hashimoto, and C.C. Han, Macromolecules 20, 2120 (1987).
125. R.W. Richards, G. Welsh, European Polymer Journal 31, 1197 (1995).
126. H. Tanaka and T. Nishi, Journal of Chemical Physics 82, 4326 (1985).
127. T. Hashimoto, M. Shibayama, and H. Kawai, Macromolecules 13, 1237 (1980).
128. T. Hashimoto, M. Shibayama, and H. Kawai, Macromolecules 13, 1660 (1980).
129. J.A. Kornfield, H.W. Spiess, H. Nefzger, H. Hayen, and C.D. Eisenbach, Macromolecules 24,
4787 (1991).
130. A.D. Meltzner, H.W. Spiess, C.D. Eisenbach, and H. Hayen, Macromolecules 25, 993 (1992).
131. L.W. Jelinski, J.J. Dumais, and A.K. Engel, Macromolecules 16, 492 (1983).
132. L.W. Jelinski, Macromolecules 14, 1341 (1981).
133. L.W. Jelinski, J.J. Dumais, P.I. Watnick, K.A. Engel, and M.D. Sefcik, Macromolecules 16,
409 (1983).
134. D.T. Okamoto, S.L. Cooper, and T.W. Root, Macromolecules 25, 1068 (1992).
135. D.T. Okamoto, S.L. Cooper, and T.W. Root, Macromolecules 25, 3301 (1992).
136. D.T. Okamoto, E. M. O’Connell, S.L. Cooper, and T.W. Root, Journal of Polymer Science, Part
В Polymer Physics 31, 1163 (1993).
137. C. S.P. Sung, C.B. Hu, and C.S. Wu, Macromolecules 13, 111 (1980).
138. N.J. Clayden, C. Nijs, and G. Eeckhaut, Macromolecules 31, 7820 (1998).
760 -*v* Литература
139. L. Mullins, J. Phys. Chem. 54, 239 (1950).
140. T.L. Smith, in «Encyclopedia of Materials Science and Engineering» M.B. Bever (Ed.),
Pergamon Press, New York, 1986, p. 1341.
141. D.R. Paul, Polym. Mater. Sci. Eng. 50, 1 (1984).
142. G. Holden, E.T. Bishop, and N.R. Legge, Journal of Polymer Science 26, 37 (1969).
143. D.J. Meier, Polymer Preprints 14, 280 (1973).
144. R.J. Gaylord and D.J. Lohse, Polymer Engineering and Science 18, 359 (1978).
145. R.J. Gaylord, in «Multiphase Polymers» S.L. Cooper and G.M. Estes (Eds.), ACS Advances
in Chemistry Series 176, Washington, D. C., 1979, Chap. 12.
146. J.K. Bard and C.I. Chung, in «Thermoplastic Elastomers» G. Holden, N.R. Legge, and
H.E. Schroeder (Eds.), Section 1, Hanser Publishers, New York, 1987, Chap. 12.
147. H. Veenstra, P. C.J. Verkooijen, B. J. J. van Lent, J. van Dam, A. P. de Boer, and A.P. H.J. Ni-
jhof, Polymer 41, 1817 (2000).
148. D.J. Read, P. I.C. Teixeira, R.A. Duckett, J. Sweeney, and T.C.B. McLeish, European Physical
Journal E8, 15 (2002).
149. G. Holden and N.R. Legge, in «Thermoplastic Elastomers—2nd Edition» G. Holden, N.R. Legge,
R.P. Quirk, and H.E. Schroeder (Eds.), Hanser Publishers, New York, 1996, Chap. 3.
150. T.A. Speckhard and S.L. Cooper, Rubber Chemistry and Technology 59, 405 (1986).
151. W. Nierzwicki, Journal of Applied Polymer Science 30, 761 (1985).
152. L.L. Harrell, Macromolecules 2, 607 (1976).
153. J.A. Miller, S.B. Lin, K. K.S. Hwang, K.S. Wu, P.E. Gibson, and S.L. Cooper,Macromolecules
18, 32 (1985).
154. J.T. Mehl, R. Murgasova, X. Dong, D.M. Hercules, and H. Nefzger, Analytical Chemistry 72,
2490, (2000).
155. D.J. Blundell, G. Eeckhaut, W. Fuller, A. Mahendrasingam, and C. Martin, Polymer 43,5197
(2002).
156. M. Morton, in «Thermoplastic Elastomers—2nd Edition» G. Holden, N.R. Legge, R.P. Quirk,
and H.E. Schroeder (Eds.), Hanser Publishers, New York, 1996, Chap. 4.
157. M.V. Pandya, D.D. Deshpande, and D.G. Hundiwale, Journal of Applied Polymer Science 32,
4959 (1986).
158. L. Morbitzer and H. Hespe, Journal of Applied Polymer Science 16, 2697 (1972).
159. N.S. Schneider and W. Matton, Polymer Engineering and Science 19, 1122 (1979).
160. M. Brown, G.K. Hoeschele, and W.K. Witsiepe (to DuPont), U.S. Patent 3,835,098
(September 10, 1974).
161. S. Sakarai, S. Okamoto, and K. Sakurai, in «Developments in Block Copolymer Science and
Techno/ogy» I.W. Hamley (Ed.), Wiley, West Sussex, 2004.
162. P.G. DeGennes, «Scaling Concepts in Polymer Physics» Cornell University Press, Ithaca,
NY, 1979.
163. E. Helfand, Macromolecules 8, 552 (1975).
164. E. Helfand and Z.R. Wasserman, Macromolecules 9, 879 (1976).
165. E. Helfand and Z.R. Wasserman, Macromolecules 11, 960 (1978).
166. E. Helfand and Z.R. Wasserman, Macromolecules 13, 994 (1980).
167. E. Helfand, in «Developments in Block Copolymers» I. Goodman (Ed.), Applied Science, New
York, 1982, Chap. 4.
168. M.D. Whitmore, in «Encyclopedia of Materials: Science and Techno/ogy» Elsevier Science,
Oxford, 2001.
169. R.J. Meier, in «Thermoplastic Elastomers—2nd Edition» G. Holden, N.R. Legge, R.P. Quirk,
and H.E. Schroeder (Eds.), Hanser Publishers, New York, 1996, Chap. 11.
170. L. Leibler, Macromolecules 13, 1602 (1980).
171. P.G. DeGennes, J. Phys. (Paris) 31, 235 (1970).
172. F.S. Bates, J.H. Rosedale, and G.H. Fredrickson, Journal of Chemical Physics 92,6255 (1990).
173. B. St hn and F. Stickel, Macromolecules 25, 5306 (1992).
174. F.S. Bates and M.A. Hartney, Macromolecules 18, 2478 (1985).
175. B. Holzer, A. Lehmann, B. St hn, and M. Kowalski, Polymer 32, 1935 (1991).
176. G.H. Fredrickson and E. Helfand, Journal of Chemical Physics 87, 697 (1987).
Список литературы к главе 13 -*и- 761
177. L. Leiblerand II. Benoit, Polymer 22, 195 (1981).
178. М. Olvera de la Cruz and I.C. Sanchez, Macromolecules 19, 2501 (1986).
179. A.M. Mayes and M. Olvera de la Cruz, Journal of Chemical Physics 91, 7228 (1989).
180. J.T. Koberstein, T.P. Russell, D.J. Walsh, and L. Pottick, Macromolecules 23, 877 (1990).
181. M.D. Gehlsen, K. Almdal, and F.S. Bates, Macromolecules 25, 939 (1992).
182. M.D. Lelah and S.L. Cooper, «Polyurethanes in Medicine» CRC Press, Boca Raton, FL, 1986.
183. S. Wu, in «Polymer Handbook» J. Brandrupand E.H. Immergut (Eds.), John Wilev and Sons,
New York, 1989.
184. B.J. Tyler, B.D. Ratner, D.G. Castner, and D. Briggs, J. Biomed. Mater. Res. 26, 273 (1992).
185. H. Hasegawa and T. Hashimoto, Macromolecules 18, 590 (1985).
186. T. Hashimoto, A. Todo, H. Itoi, and H. Kawai, Macromolecules 10, 377 (1977).
187. T. Hashimoto, M. Shibayama, and H. Kawai, Macromolecules 13, 1237 (1980).
188. S.H. Anastasiadis, T.P. Russell, S.K. Satija, and C.F. Majkrzak, Journal of Chemical Physics
92, 5677 (1990).
189. X. Chen, J.A. Gardella Jr., and P. L Kumler, Macromolecules 25, 6631 (1992).
190. H. Hasegawa and T. Hashimoto, Polymer 33, 475 (1992).
191. R.A. Segalman, K.E. Schaefer, G.H. Fredrickson, E.J. Kramer, and S. Magonov, Macromol-
ecules 36, 4498 (2003).
192. M.J. Fasolka and A.M. Mayes, Annual Review of Materials Research 31, 323 (2001).
193. H. Ishida, Rubber Chemistry and Technology 60, 497 (1987).
194. C. S.P. Sung and C.B. Hu, in «Multiphase Polymers» S.L. Cooper and G.M. Estes (Eds.),
Advances in Chemistry Series, 176, American Chemical Society, Washington, D. C., 1980.
195. H. Ishida and C. Scott, Journal of Polymer Engineering 6, 201 (1986).
196. W.G. Pitt and S.L. Cooper, Biomaterials 7, 340 (1986).
197. T.A. Giroux and S.L. Cooper, Journal of Colloid and Interface Science 146, 179 (1991).
198. P.F. Green, T.M. Christensen, T.P. Russell, and R. J r me, Macromolecules 22, 2189 (1989).
199. P.F. Green, T.M. Christensen, T.P. Russell, and R. J r me, Journal of Chemical Physics 92,
1478 (1990).
200. G.H. Fredrickson, Macromolecules 20, 2535 (1987).
201. P.F. Green, T.M. Christensen, and T.P. Russell, Macromolecules 24, 252 (1991).
202. T. Kajiyama and A. Takahara, J. Biomat. App. 6, 42 (1991).
203. L. Sabbatini and P.G. Zambonin, Journal of Electron Spectroscopy and Related Phenomena 81,
285 (1996).
204. Y. Deslandes, G. Pleizier, D. Alexander, and P. Santerre, Polymer 39, 2361 (1998).
205. A.G. Shard, M.C. Davies, S. J.B. Tendler, D.E. Jackson, P.N. Lan, E. Schacht, and M.D.
Purbrick, Polymer 36, 775 (1995).
206. G. Coulon, T.P. Russell, V.R. Deline, and P.F. Green, Macromolecules 22, 2581 (1989).
207. A. Aneja and G.L. Wilkes, Polymer 7221 (2003).
208. K. Gisselfalt and B. Helgee, Macromolecular Materials and Engineering 288, 265 (2003).
209. H. Schonherr, W. Wiyatno, J. Pople, C.W. Frank, G.G. Fuller, A.P. Gast, and R.M. Waymouth,
Macromolecules 35, 2654 (2002).
210. J. Krijgsman, D. Husken, and R.J. Gaymans, Polymer 7573 (2003).
211. B.K. Annis, D.W. Schwark, J.R. Reffner, E.L. Thomas, and B. Wunderlich, Makromol. Chem.
193, 2589 (1992).
212. A. Knoll, R. Magerle, and G. Krausch, Macromolecules 34, 4159 (2001).
213. C. Vasilev, H. Heinzelmann, and G. Reiter, Journal of Polymer Science Part В-Polymer Physics
42, 1312 (2004).
214. J.T. Garrett, C.A. Siedlecki, and J. Runt. Macromolecules 34, 7066 (2001).
215. T. Osswald, «Polymer Processing Fundamentals» Hanser Publishers, New York, 1998.
216. P.J. Corish, «Concise Encyclopedia of Polymer Processing & Applications» Elsevier, New
York, 1991.
217. C.B. Wang and S.L. Cooper, Macromolecules 16, 775 (1983).
218. R. K. Adams and G. K. Hoeschele, in«Thermoplastic Elastomers—2nd Edition» G. Holden, N. R. Legge,
R.P. Quirk, and H.E. Schroeder (Eds.). Hanser Publishers, New York, 1996, Chap. 8.
219. N. M.K. Lemba, K.A. Woodhouse, and S.L. Cooper, «Polyurethanes in Biomedical Applica-
tions» CRC Press, Boca Raton. FL. 1997.
762 “* V Литература
220. R. Bonart, Polymer 20, 1389 (1970).
221. M. Matsuo, Jpn. Plast. 2, 6 (1968).
222. J. Dlugosz, A. Keller, and E. Pedemonte, Kolloid Z.Z. Polym. 242, 1125 (1970).
Глава 14
1. T.L. Ford and F.S. Charles, «Heavy Duty Truck Tire Engineering» SAE’s 34th L. Ray
Buckingdale Lecture, SP729, 1988.
2. J.A. Davison, «Design and Application of Commercial Type Tires» SAE’s 15th L. Ray
Buckingdale Lecture, SP344, 1969.
3. The Tire & Rim Association, Inc. Year Book, 175 Montrose West Ave., Suite 150, Copley, OH
44321.
4. European Tyre and Rim Technical Organization, Standards Manual, 32/2 Avenue Brug-
mann—B1060 Brussels, Belgium.
Engineering Data Book, «Over-The-Road Truck Tires» The Goodyear Tire & Rubber Com-
pany, 2001.
6. F.J. Kovac, «Tire Techno/ogy» 5th ed., The Goodyear Tire & Rubber Company, 1978.
7. V.E. Gough, Rubber Chem. Technol. 44, 988 (1968).
8. S.K. Clark, «Mechanics of Pneumatic Tires» U.S. Department of Transportation, Washington,
D. C., 1982.
9. J.E. Purdy, «Mathematics Underlying the Design of Pneumatic Tires» 2nd ed., Hiney Print-
ing, Akron, OH, 1970.
10. J.W. Fitch, «Motor Truck Engineering Handbook» 4th ed., Society of Automotive Engineers,
Warrendale, PA, 1994.
11. W.ll. Waddell, M.B. Rodgers, and D.S. Tracey, «Tire Applications of Elastomers, Part 1.
Treads» Rubber Division Meeting, Grand Rapids, MI, Paper H, 2004.
12. Bekaert Corporation, Steel Cord Cata/ogue, Akron, OH, 1991.
13. W. J. van Ooij, Rubber Chem. Technol. 57, 421 (1984).
14. J. Duddey, in «Rubber Compounding, Chemistry and Applications» Marcel Dekker Inc., New
York, 2004.
15. B. Rodgers and W.H. Waddell, Rubber Compounding, in «Kirk-Othmer Encyclopedia of
Chemical Techno/ogy» 5th ed., John Wiley & Sons, New York, 2004.
16. H. Long, «Basic Compounding and Processing of Rubber» Rubber Division, American Chemi-
cal Society, Lancaster Press, Lancaster, PA, 1985.
17. K.C. Baranwal and H.S. Stephens, «Basic Elastomer Techno/ogy» Rubber Division,
American Chemical Society, Akron, OH, 2001.
Глава 15
1. C. Goodyear, inventor, U.S. Patent 3,633 (1844).
2. C. Goodyear, inventor, U.K. Patent 2,933 (1853).
3. U.S. Scrap Tire Markets 2001, Scrap Tire Management Council.
4. Scrap Tire Stockpile Abatement, May 2002, Rubber Manufacturers Association.
R.H. Snyder, «Scrap Tyres: Disposal and Reuse» Society of Automotive Engineers, Inc.,
Warrendale, PA, 1998.
6. P. Hous, H. Bartelds, and E. Smit, Rubber World 212, 36 (1995).
7. W. Klingensmith and K. Baranwal, Rubber World 218, 41 (1998).
8. V.M. Makarov and V.F. Drozdovski, «Reprocessing of Tyres and Rubber Wastes» Ellis Hor-
wood, New York, 1991.
9. Scrap Tire News, 13, 2, 6 (1999).
10. Scrap Tire News, 13, 6, 7 (1999).
11. J. Pryweller, European Rubber Journal 181, 17 (1999).
12. I. Franta (Ed.), «Elastomers and Rubber Compounding Materials» Elsevier, New York, 1989.
13. W.C. Warner, Rubber Chem. Technol 67, 559 (1994).
14. C.P. Rader (Ed.), «Plastic, Rubber and Paper Recycling» American Chemical Society,
Washington, D. C., 1995.
Список литературы к главе 13 -*V- 763
15. S.R. Fix, Elastomerics 112, 38 (1980).
16. P.P. Nicholas, Rubber Chem. Technol. 55, 1499 (1982).
17. M. Myhre and D.A. MacKillop, Rubber Chem. Technol. 75, 429 (2002).
18. Fact Sheet, Tire Retread Information Bureau (2002).
19. A.A. Phadke, A.K. Bhattacharya, S.K. Chakraborty, and S.K. De, Rubber Chem. Technol. 56,
726 (1983).
20. B. Siuru, Scrap Tire News 11, 14 (1997).
21. A. Accetta and J.M. Vergnaud, Rubber Chem. Technol. 55, 961 (1982).
22. D. De, S. Maiti, and B. Adhikari, J. Appl. Polym. Sci. 73, 2951 (1999).
23. B. Adhikari, D. De, and S. Maiti, Prog. Polym. Sci. 25, 909 (2000).
24. A.I. Isayev, in «Rubber Techno/ogist’s Handbook» J.R. White and S.K. De (Eds.), RAPRA,
Shawbury, U. K., 2001, Chap. 15, pp. 511-547.
25. B.D. LaGrone, Conservation and Recycling^, 359 (1986).
26. B. Bowers, D. Barber, and R. Allinger, Oct., 1986, Paper #82 presented at the meeting of the
ACS Rubber Division, Atlanta, GA.
27. K. Knoerr, Oct., 1995, Paper #5 presented at the ACS Rubber Division Meeting, Cleveland, OH.
28. P.M. Lewis, NR Technology 17, 57 (1986).
29. R. Schaefer, Oct., 1986, Paper #79 presented at the ACS Rubber Division Meeting, Atlanta, GA.
30. R. Schaefer and R. Berneking, Oct., 1986, Paper #80 presented at the ACS Rubber Division
Meeting, Atlanta, GA.
31. E.M. Solov’ev, V.B. Pavlov, and N.S. Enikolopov, Intern. Polym. Sci. Technol. 14, 10 (1987).
32. J.A. Szilard, «Reclaiming Rubber and Other Polymers» Noyes Data Corporation, London,
1973.
33. W. Kongensmith, Rubber World 203, 16 (1991).
34. J.J. Leyden, Rubber World 203, 28 (1991).
35. J.G. Bryson, U.S. Patent4,148,763 (1979).
36. R. Kohler and J. O’Neill, Rubber World 216, 32, 34 (1997).
37. B.C. Sekhar and V.A. Kormer, European Patent Application, EP 0 690 091 Al (1995).
38. D. De, B. Adhikari, and S. Maiti, J. Polym Material 14, 333 (1997).
39. D. De, S. Maiti, and B. Adhikari, Kautchuk Gummi Kunststoffe 53, 346 (2000).
40. D. De, A.K. Ghosh, S. Maiti, and B. Adhikari, Polym. Recycling4, 15 (1999).
41. K. Knorr, Kautchuk Gummi Kunststoffe 47 1, 54 (1994).
42. M. A.L. Verbruggen, L. van der Does, J. W.M. Noordermeer, M. van Duin, and HJ. Manuel.
Rubber Chem. Technol. 72, 731 (1999).
43. L.K. Hunt and R.R. Kovalak, inventors, Assignee Goodyear Tire and Rubber Companv,
U.S. Patent 5,891,926 (1999).
44. R.D. Myers, P. Nicholson, J.B. MacLeod, and M.E. Moir, U.S. Patent, 5,602,186 (1997).
45. N. Kawabata, B. Okuyama, and S. Yamashita, J. Appl Polym Sci. 26, 1417 (1981).
46. N. Kawabata, T. Murakami, and S. Yamashita, Nippon Gomu Kyokaishi 52, 768 (1979).
47. D.A. Benko and R.N. Beers, inventors, Assignee Goodyear Tire and Rubber Company,
U.S. Patent 5,380,269 (2002).
48. D.A. Benko and R.N. Beers, inventors, Assignee Goodyear Tire and Rubber Company,
U.S. Patent 6,387,965 (2002).
49. D.A. Benko and R.N. Beers, inventors, Assignee Goodyear Tire and Rubber Comapny,
U.S. Patent 6,462,099 (2002).
50. A.A. Hershaft, Environ. Sci. Technol. 6, 412 (1972).
51. A.A. Hershaft, Elastomerics 109, 39 (1977).
52. A. Ratcliffe, Chem. Eng. 79, 62 (1972).
53. J. Lynch and B. LaGrone, «Ultrafine Crumb Rubber» Paper #37 presented ata meeting of the
Rubber Division, ACS, Atlanta, GA, October 1986.
54. N.S. Enikolopian, Pure Appl. Chem. 57, 1707 (1985).
55. K. Khait and J.M. Torkelson, Polym. Plast. Technol. Eng. 38, 445 (1999).
56. K. Khait, Paper #24 presented at the ACS Rubber Division Meeting, Chicago, IL, 1994.
57. E. Bilgili, H. Arastoopour, and B. Bernstein, Rubber Chem. Technol. 73, 340 (2000).
58. E. Bilgili, B. Berstein, H. Arastoopour, AIChESymp. Ser. 95, 83 (1999).
764 “* V- Литература
59. Е. Bilgili, Н. Arastoopour, and В. Bernstein, Powder Technol. 115, 265 (2001).
60. E. Bilgili, H. Arastoopour, and B. Bernstein, Powder Technol. 115, 277 (2001).
61. H. Arastoopour, D.A. Schocke, B. Bernstein, and E. Bilgili, U.S. Patent 5,904,885 (1999).
62. E. Bigili, A. Dybek, H. Arastoopour, and B. Bernstein, J. Elast. Plastics 35, 235 (2003).
63. J.E. Morin, D.E. Williams, and R.J. Farris, Rubber Chem. Technol. 75, 955 (2002).
64. A.R. Tripathy, J.E. Morin, D.E. Williams, S.J. Eyles, and R.J. Farris, Macromol. 35, 4616
(2002).
65. D.S. Novotny, R.L. Marsh, F.C. Masters, and D.N. Tally, U.S. Patent 4,104,205 (1978).
66. T. Kleps, M. Piaskiewicz, and W. Parasiewicz, J. Thermal Analysis Calorimetry 60, 271
(2000).
67. A.M. Basedow and K. Ebert, Adi?. Polym. Sci. 22, 83 (1987).
68. G.J. Price, in «Advances in Sonochemistry» T.J. Mason (Ed.), J Al Press Ltd., Greenwich, CT,
Vol. 1, 1990, p. 231.
69. G.J. Price, D.J. Norris, and P.J. West, Macromolecules 25, 6447 (1992).
70. G. Schmid and O. Rommel, Z. Elektrochem 45, 659 (1939).
71. G. Schmid, Physik. Z. 41, 326 (1940).
72. H. H.G. Jellinek and G. White, J. Polymer Sci. 7, 21 (1951).
73. A.I. Isayev, C. Wong, and X. Zeng, SPEANTEC Tech. Papers 33, 207 (1987).
74. A.I. Isayev, C. Wong, and X. Zeng, Adi?. Polym. Technol. 10, 31 (1990).
75. A.I. Isayev, Proceeding of the 23rd Israel Conference on Mechanical Engineering, Technion,
Haifa, Paper #5.2.3, 1990.
76. A.I. Isayev and S. Mandelbaum, Polym. Eng. Sci. 31, 1051 (1991).
77. S.L. Peshkovsky, M.L. Friedman, A.I. Tukachinsky, G.V. Vinogradov, and N.S. Enikolopian,
Polym. Compost, 126 (1983).
78. R. Garcia Ramirez and A.I. Isayev, SPEANTEC Tech. Papers 37, 1084 (1991).
79. K.S. Suslick, Scientific American 260, 80 (1989).
80. K.S. Suslick, S.J. Doctycz, and E.B. Flint, Ultrasonics 28, 280 (1990).
81. G. Gooberman, J. Polymer Sci. 42, 25 (1960).
82. M. Okuda and Y. Hatano, Japanese patent application 62,121,741 (1987).
83. A.I. Isayev, inventor, Assignee the University of Akron, U.S. Patent 5,258,413 (1993).
84. A.I. Isayev and J. Chen, inventors, Assignee the University of Akron, U.S. Patent 5,284,625
(1994).
85. A.I. Isayev, J. Chen, and A. Tukachinsky, Rubber Chem. Tech. 68, 267 (1995).
86. A. Tukachinsky, D. Schworm, and A.I. Isayev, Rubber Chem. Tech. 69, 92 (1996).
87. V. Yu Levin, S.H. Kim, A.I. Isayev, J. Massey, and E. von Meerwall, Rubber Chem. Technol.
69, 104 (1996).
88. A.I. Isayev, S.P. Yushanov, and J. Chen, J. Appl. Polym. Sci. 59, 803 (1996).
89. A.I. Isayev, S.P. Yushanov, and J. Chen, J. Appl. Polym. Sci. 59, 815 (1996).
90. A.I. Isayev, S.P. Yushanov, D. Schworm, and A. Tukachinsky, Plast. Rubber and Compos.
Process, andAppl. 25, 1 (1996).
91. S.P. Yushanov, A.I. Isayev, and V.Y. Levin, J. Polym. Sci.: Part B: Polymer Physics 34, 2409
(1996).
92. A.I. Isayev, S.P. Yushanov, S.H. Kim, and V. Yu Levin, Rheol. Acta 35, 616 (1996).
93. V. Yu Levin, S.H. Kim, and A.I. Isayev, Rubber Chem. Technol. 70, 120 (1997).
94. S.T. Johnston, J. Massey, E. von Meerwall, S.H. Kim, V. Yu Levin, and A.I. Isayev, Rubber
Chem. Tech. 70, 183 (1997).
95. A.I. Isayev, S.H. Kim, and V. Yu. Levin, Rubber Chem. Tech. 70, 194 (1997).
96. V. Yu Levin, S.H. Kim, and A.I. Isayev, Rubber Chem. Tech. 70, 641 (1997).
97. S.P. Yushanov, A.I. Isayev, and S.H. Kim, Rubber Chem. Technol. 71, 168 (1998).
98. B. Diao, A.I. Isayev, V. Yu Levin, and S.H. Kim, J. Appl. Polym. Sci. 69, 2691 (1998).
99. M. Tapale and A.I. Isayev, J. Appl. Polym. Sci. 70, 2007 (1998).
100. B. Diao, A.I. Isayev, and V. Yu Levin, Rubber Chem. Technol. 72, 152 (1999).
101. V.V. Yashin and A.I. Isayev, Rubber Chem. Technol. 72, 741 (1999).
102. V.V. Yashin and A.I. Isayev, Rubber Chem. Technol. 73, 325 (2000).
103. C.K. Hong and A.I. Isayev, J. Appl. Polym. Sci. 79, 2340 (2001).
Список литературы к главе 13 -*и* 765
104. S.E. Shim and A.I. Isayev, Rubber Chem. Tech. 74, 303 (2001).
105. J. Yun, J.S. Oh, and A.I. Isayev, Rubber Chem. Technol. 74, 317 (2001).
106. A.I. Isayev, in «Rubber Techno/ogist’s Handbook» J.R. White and S.K. De (Eds.), RAPRA
Techno/ogy Ltd., U. K., 2001.
107. A.I. Isayev, in «Encyclopedia of Materials: Science and Techno/ogy» K. H.J. Buschow (Ed.),
Elsevier, Amsterdam, Vol. 3, 2001.
108. C.K. Hong and A.I. Isayev, Rubber Chem. Technol. 75, 617 (2002).
109. C.K. Hong and A.I. Isayev, Rubber Chem. Technol. 75, 133 (2002).
110. J. Yun and A.I. Isayev, Rubber Chem. Technol. 76, 253 (2003).
111. S.E. Shim, S. Ghose, and A.I. Isayev, Polymer 43, 5535 (2002).
112. S.E. Shim, J.C. Parr, E. von Meerwall, and A.I. Isayev, J. Phys. Chem. В 106, 12072 (2002).
113. S.E. Shim, A.I. Isayev, and E. von Meerwall, J. Polym. Sci.: Part B: Polym. Phys. 41, 454
(2003).
114. J. Yun, A.I. Isayev, S.H. Kim, and M. Tapale, J. Appl. Polym. Sci. 88, 434 (2003).
115. S.E. Shim, and A.I. Isayev, J. Appl. Polym. Sci. 88, 2630 (2003).
116. S. Ghose and A.I. Isayev, J. Appl. Polym. Sci. 88, 980 (2003).
117. J. Yun and A.I. Isayev, Polym. Eng. Sci. 43, 809 (2003).
118. W. Feng and A.I. Isayev, J. Appl. Polym. Sci. (in press).
119. W. Feng and A.I. Isayev, Paper presented at the ACS Rubber Division Meeting, Columbus,
OH, October 2004.
120. T. Boron, P. Roberson, and W. Klingensmith, Tire Technology International 96, 82 (1996).
121. T. Boron, W. Klingensmith, C. Forest, and S. Shringarpurey, 156th Meeting of the ACS
Rubber Division, Orlando, FL, 1999, Paper #136.
122. E. A. Gonzalez de Los Santas, F. Sorieno-Corral, Ma J. Lozano Gonzalez, and R. Cedillo-Garcia,
Rubber Chem. Technol. 72, 854 (1999).
123. J. Blitz, «Fundamentals of Ultrasonics» 2nd ed., Butterworth, London, 1967, Chap. 8.
124. K.S. Suslick, «Ultrasound: Its Chemical, Physical and Bio/ogical Effects» VCH, New York,
1988.
125. S.N. Zhurkov, V.A. Zakrevskii, V.E. Korsukov, and V.S. Kuksenko, 1972, Soviet Physics
Solid State 13, 1680(1972).
126. A.N. Gent and D.A. Tompkins, J. Appl. Phys. 40, 2520 (1969).
127. A.N. Gent, Rubber Chem. Technol. 63, G49 (1990).
128. M.B. Bever (Ed.), «Encyclopedia of Materials Science and Engineering» Pergamon Press,
Oxford, Vol. 4, 1986, p. 2934.
129. F.R. Young, «Cavitation» McGraw-Hill Co., London, 1989, Chap. 2.
130. A.I. Isayev, J. Chen, and S.P. Yushanov, in «Simulation of Materials Processing: Theory,
Methods and Application» S.F. Shen and P. Dawson (Eds.), Balkema, Rotterdam, 1995,
pp. 77-85.
131. A.I. Kasnerand E.A. Meinecke, Rubber Chem. Technol. 69, 424 (1996).
132. A.J. Kinloch and R.J. Young, «Fracture Behavior of Polymers» Applied Science Publishers,
London, 1983.
133. H.H. Kausch, «Polymer Fracture» Springer-Verlag, Berlin, 1987.
134. M. Tabata and J. Sohma, Chem. Phys. Lett. 73, 178 (1980).
135. M. Tabata and J. Sohma, Eur. Polym. J. 16, 589 (1980).
136. J.P. Lorimer, in «Chemistry with Ultrasound» T.J. Mason (Ed.), Elsevier, New York, 1990,
Chap. 4.
137. C.K. Hong and A.I. Isayev, J. Mater. Sci. 37, 385 (2002).
138. A.P. Yushanov, A.I. Isayev, and S.H. Kim, Rubber Chem. Technol. 71, 168 (1998).
139. P. Rajalingam and W.E. Baker, Rubber Chem. Technol. 65, 908 (1992).
140. N.P. Chopey, Chem. Eng. 80, 54 (1973).
141. D. Gibala and G.R. Hamed, Rubber Chem. Technol. 67, 636 (1994).
142. D. Gibala, K. Laohapisitpanich, D. Thomas, and G.R. Hamed, Rubber Chem. Technol. 69,115
(1996).
143. R.H. Wolk, Rubber Age 104, 103 (1973).
144. A.N. Theodore, R.A. Pett, and D. Jackson, Rubber World 218, 23 (1998).
766 -* V Литература
145. H.J. Radusch, T. Luepke, S. Poltersdorf, and E. Laemmer, Kautschuk Gummi 43, 767
(1990).
146. K. Fukumori, M. Matsushita, H. Okamoto, N. Sato, Y. Suzuki and K. Takeuchi, JSAEReview
23, 259 (2002).
147. A.K. Naskar, P.K. Pramanik, R. Mukhopadhyay, S.K. De, and A.K. Bhowmick, RubberChem.
Technol. 73, 902 (2000).
148. Scrap Tire News 15, No. 10, Oct. 2001, pp. 1-3.
149. S. Ghose and A.I. Isayev, Polym. Eng. Sci. 44, 794 (2003).
150. J. R.M. Duhaime and W.E. Baker, Plast. Rubb. Comp. Proc. Appl. 15, 87 (1991).
151. P. Rajalingam, J. Sharpe, and W.E. Baker, Rubber Chem. Technol. 66, 664 (1993).
152. R.D. Deanin and S.M. Hashemiolya, Polym. Mater. Sci. Eng. 57, 212 (1987).
153. T. Luo and A.I. Isayev, J. Elastomers and Plastics 30, 133 (1998).
154. J.S. Oh and A.I. Isayev, Rubber Chem. Tech. 75, 617 (2002).
155. C.K. Hong and A.I. Isayev, J. Elast, and Plast. 33, 47 (2001).
156. H. Michael, H. Scholz, and G. Mennig, Kautchuk Gummi Kunststoffe 52 510 (1999).
157. A.Y. Coran and R.P. Patel, in «Thermoplastic Elastomers» 2nd ed., G. Holden, N.R. Legge,
R. Quirk, and H.E. Schroeder (Eds.), Hanser Publishers, New York, 1996.
158. N.C. Hilyward, S.G. Tong, and K. Harrison, Plast. Rubb. Proc. Appl. 3, 315 (1983).
159. A.A. Phadke, S.K. Chakraborty, and S.K. De, Rubber Chem. Technol. 57, 19 (1984).
160. A.A. Phadke and S.K. De, Polym. Eng. Sci. 26, 1079 (1986).
161. K. Oliphant and W.E. Baker, Polym. Eng. Sci. 33, 166 (1993).
162. P.K. Pramanik and W.E. Baker, J. Elast. Plast. 27, 253 (1995).
163. P.K. Pramanik and W.E. Baker, Plast. Rubb. Comp. Proc. Appl. 24, 229 (1995).
164. D. Briggs, Surf. Interface Anal. 2, 107 (1980).
165. D. Briggs, Euro. Polym. J. 14, 1 (1978).
166. A.K. Naskar, S.K. De, and A.K. Bhowmick, J. Appl. Polym. Sci. 84, 370 (2002).
167. A.K. Naskar, A.K. Bhowmick, and S.K. De, J. Appl. Polym. Sci. 84, 622 (2002).
168. F.J. Stark Jr., A. Leigton, and D. Wagner, Rubber World 188, 36 (1983).
169. E.L. Rodriguez, Polym. Eng. Sci. 28, 1455 (1988).
170. A. Chidambaram and K. Min, SPEANTEC Tech. Papers 40, 2927 (1994).
171. R.H. Campbell and R.W. Wise, Rubber Chem. Technol. 37, 635 (1964).
172. Z. Xu, N.S. Losure, and S.D. Gardner, J. Avd. Mater. 30, 11 (1998).
173. A.Y. Coran and F. Howard, U.S. Patent 5,889,119 (1999).
174. F. Howard and A.Y. Coran, Paper presented at the ITEC, September, Akron, OH, 2000.
175. D. Raghavan, H. Huynh, and C.F. Ferraris, J. Mater. Sci. 33, 1745 (1998).
176. D. Raghavan, K. Tratt, and R.P. Wool, Mater. Research Soc. Sym. 344, 177 (1994).
177. H. Goldstein, Civ. Eng. 65, 60 (1995).
178. N.N. Eldin and A.B. Senouci, ASCEJ. Const. Eng. Mgmt. 188, 561 (1992).
179. N.N. Eldin and A.B. Senouci, Cem. Concr. Agg. 15, 74 (1993).
180. N.N. Eldin and A.B. Senouci, ASCEJ. Mater. Civ. Eng. 5, 478 (1993).
181. B.I. Lee, L. Burnett, T. Miller, B. Postage, and J. Cuneo, J. Mater. Sci. Lett. 12, 967 (1993).
182. F. Shutov, G. Ivanov, H. Arastoopour, and S. Volfson, Polym. Mater. Sci. Eng. 67, 404
(1992).
183. l.B. Topcu, Cement Concrete Research 25, 304 (1995).
184. N.N. Eldin and A.B. Senouci, Cement Concrete Compos. 16, 287 (1994).
185. N.A. Ali, A.D. Amos, and M. Roberts, in Proceed. Intern. Conf. Concrete 2000, University of
Dundee, U. K., 2000, pp. 379-390.
186. H. Rostami, J. Lepore, T. Silverstram, and I. Zandi, Proceed. Intern. Conf. Concrete 2000,
Dundee, U. K., 1993, pp. 391-399.
187. J.A. Lepore and M.W. Tantala, In Proceed. Concr. Inst. Australia, Concrete 97, 1997, pp.
623-627.
188. N. Segre and I. Joekes, Cement Concrete Research 30, 1421 (2000).
189. N. Segre, P. J.M. Monteiro, and G. Sposito, J. Coll. Interface Sci. 248, 521 (2002).
190. D. Raghavan, J. Appl. Polym. Sci. 77, 934 (2000).
191. Anonymous, Biocycle 34, 9 (1993).
Список литературы к главе 15 -’V 767
192. J.L. McQuillen Jr., Н.В. Takallou, R.G. Hicks, and D. Esch,A5C£V. Transport. Eng. 114, 259
(1988).
193. F.L. Roberts and R.L. Lytton, Transportation Research Record 115, 216 (1987).
194. R.H. Renshaw, «Rubber in Roads» Plastics and Rubber Institute, South African Section,
S. Africa, 1985, p. 1.
195. H.B. Takallou and M.B. Takallou, Elastomerics 123, 19 (1991).
196. I.S. Schuller, Ph. D. Dissertation, Texas A&M University, 1991.
197. H. Al-Abdual-Wahhab and G. Al-Amri, J. Mater. Civil Eng. 3, 189(1991).
198. T.C. Billiter, R.R. Davison, C.J. Glover, and J.A. Bullin, Petrol. Sci. Technol. 15, 205 (1997).
199. M. Rouse, Rubber World, p. 23, May (1995).
200. R.C. Malmgren, N. Parviz, P.N. Soltanpour, and J.E. Cipra, U.S. Patent 5,014,562 (1991).
201. G. Logsdon, Biocycle 31, 44, 84 (1990).
202. J.N. Rogers III and D.V. Waddington, Agronomy J. 84, 203 (1992).
203. P.H. Groenevelt and P.E. Grunthal, Soil and Tillage Res. 47, 169 (1998).
204. M. Moore, Crain's Tire Business 9, 15 (1991).
205. W. Kaminsky and H. Sinn, in «Book Recycling and Recovery of Plastics» J. Brandrup.
M. Bittner, W. Michaeli, and G. Menges (Eds.), Hanser Publishers, Munich, 1996.
206. A.M. Mastral, R. Murillo, M.S. Callen, and T. Garcia, Resources Conserv. Recycling 29, 263
(2000).
207. S. Kawakami, K. Inoue, H. Tanaka, and T. Sakai, in «Thermal Conversion of Solid Wastes
and Biomass» J.L. Jones and S.B. Radding (Eds.), Symposium Series 130, ACS Publishers,
Washington, D. C., 1980.
208. J.A. Conesa, A. Fullana, and R. Font, Energy Fuel 14, 409 (2000).
209. C. Roy, A. Chaala, and H. Darmstadt, J. Anal. Appl. Pyrol. 51, 201 (1999).
210. M.R. Beck and W. Klingensmith, ACSSympos. Series 609, 254 (1995).
211. J.D. Osborn, Rubber World p. 34, May (1995).
212. W. Kaminsky and C. Mennerich, J. Anal. Appl. Pyrolysis 58, 803 (2001).
213. P.T. William, Chem. Review 12, 17 (2002).
214. P.T. William and A.J. Brindle, J. Anal. Appl. Pyrolysis 67, 143 (2003).
215. P.T. William and F. Ferrer, Resources Conserv. Recycling 19, 221 (1997).
216. A.M. Mastral, M.S. Callen, R. Murillo, and T. Garcia, Environ. Sci. Technol. 33, 4155 (1999).
Научное издание
Заявки на книги присылайте по адресам:
zakaz@id-intellect.ru
solo@id-intellect.ru
id-intellect@mail.ru
тел. (495) 579-96-45
факс (495) 579-96-70
В заявке обязательно указывайте
свои реквизиты (для организаций) и почтовый адрес!
Подробная информация о книгах на сайте
http://www.id-intellect.ru
КАУЧУК И РЕЗИНА.
НАУКА И ТЕХНОЛОГИЯ
Монография под редакцией
Дж. Марка, Б. Эрмана, Ф. Эйрича
Компьютерная верстка — ТВ. Андреева
Художник — С.Ю. Биричев
Ответственный за выпуск - Л.Ф. Соловейчик
Формат 70x100/16. Печать офсетная.
Z' лл Гарнитура Ньютон.
W Печ. 48 л. Зак. № 1518
Бумага офсетная №1, плотность 80 г/м2
Издательский Дом «Интеллект»
141700, Московская обл., г. Долгопрудный,
Промышленный пр-д, д. 14,
тел. (495) 617-41-85
Отпечатано в ООО «Чебоксарская типография №1»
428019, г. Чебоксары, пр-т И. Яковлева, д. 15
НАИБОЛЕЕ АВТОРИТЕТНОЕ
И ШИРОКО ИСПОЛЬЗУЕМОЕ
РУКОВОДСТВО ПО
ЭЛАСТОМЕРАМ В МИРОВОЙ
ЛИТЕРАТУРЕ
ДЖЕЙМС Е. МАРК, Департамент химии
Университета Цинциннати США
БЮРАН ЭРМАН. Химико-биологический
департамент Кок-Университета,
Тумия
ФРЕДЕРИК Р. ЭЙРИЧ, Политехнический
университет, Бруклин, Нью-Йорк,
США
j-CuTTEP ВЕДДЕЛЛ, Эксон Мобил Кемикал Компани. США
БГлй д и П. ГРЕДИ. Школа химического инжиниринга и
материаловед к ния, Университет Оклахомы, США
СУДхмНДАПА, Эксон Мобил Кемикал Компани, США
А. ДЖЕ НТ. Университет Акрона, США
Ж< -БАПТИСТ ДОННЕ, Высшая национальная школа
химии, Мюлуз, Франция
ABFUM И. ИСАЕВ, Институт полимерного инжиниринга
университета Акрона, США
РОДЕРИК П. КВИРК. Полимерный институт им
М. Мортона. Университет Акрона. США
ОБЕРГ Я КОРАН. Коран Консалтинг, США
СТЮАРТ Я. КУПЕР. Факультет химического и
биомолекулвриого инжиниринга, Университет
штата Огама, США
ЭМманзлЬ КУШТОДЕРО. Отдел производства
^еаматичесмих шин «Мишлен», Франция
Ж МАССИ, Jk-снарк Иитэрнэйшл, США
к нгди. Ваемиа-морская исследовательская
«абераторня, США
ДИННА Л. ГОМОХАК ЛиКЕЛЬ, Химическая компания
Истмен. США
ДиНДЛьД ДЖ. ПЛАЗЕК. Университет Пипсбурга, США
БРЕиДДм РОДЖЕРС, Эксон Мобил Кемикал Компани,
США
С. РОЛАНД Исследовательская лаборатория Военно-
морских сил. США
ДЖЕЙКС Л. УАЙТ, Департамент Полимерного
Инжиниринга, Университет Акрона, США
R ДЕРЕЗА, Факультет химии и технологии полимеров
Сауз Бэнк Политехника, Великобритания
А ХАЛАСА, Гудьир Тайр энд Раббер Компани. США
ISBN 978-5-91559-018-1
www.id-intellect.ru
9
785915
590181